/































































































































































































Author: Самсонов Г.В.
Tags: общие сведения о металлоидах (неметаллах) химия химические процессы химические соединения
Year: 1969




Text
АКАДЕМИЯ НАУК
УКРАИНСКОЙ ССР
ИНСТИТУТ ПРОБЛЕМ
МАТЕРИАЛОВЕДЕНИЯ
Самсонов НИТРИДЫ
«Н А У К О В А ДУМКА»
КИЕВ— 1 969
54
С17
УДК 546.171.1
TECHNISCfiE
INFORMATIONSBIBLIOTHEK
HANNOVER
2—5-2
91—68М
Григорий
Валентинович
Самсонов
НИТРИДЫ
Печатается по постановлению ученого
совета Института проблем материалове-
дения.
Редактор Э. Е. Гриценко.
Оформление художника И. М. Гаврилюка.
Художественный редактор Е. И. Муигтенко.
Технический редактор А. М. Колодиева.
Корректоры Л. Г. Морозова, Л. Н. Рееета.
БФ 03843. Зак. № 272. Изд. № 521. Бумага
№ 1, 70x90716- Печ. физ. листов 24,0. Усл.
печ. листов 28,08. Уч.-изд. листов 26,02.
Тираж 1800. Подписано к печати 6/1II 1969 г.
Цена 2 руб. 83 коп.
Издательство «Наукова думка», Киев, Ре-
пина, 3.
Киевская книжная типография № 5 Коми-
тета по печати при Совете Министров
УССР, Киев, Репина, 4.
ПРЕДИСЛОВИЕ
Принципиальными основами технического про-
гресса являются развитие энергетики, автомати-
зации производства и разработка новых мате-
риалов. Такие новые материалы должны удов-
летворять сложным требованиям современной
техники, обладая либо отдельными, специфиче-
скими, достаточно четко выраженными свойства-
ми, либо комплексами свойств.
Особо важное значение приобретает разра-
ботка материалов, которые могли бы эксплуати-
роваться при экстремальных — очень высоких,
либо очень низких — значениях температур, дав-
лений, скоростей, напряжений, радиационных по-
токов, газовых потоков.
Среди таких материалов наиболее перспек-
тивными являются тугоплавкие металлы, их
сплавы, а также соединения тугоплавких метал-
лов с неметаллами — бором, углеродом, азотом,
кремнием и т. п., многие из которых уже успеш-
но используются в электронике, атомной энер-
гетике, технике полупроводников и диэлектри-
ков, космической технике, современном машино-
строении, металлургии, химической промышлен-
ности, электротехнике и других отраслях.
Особый интерес представляют соединения
металлов и неметаллов с азотом, так называемые
нитриды, среди которых многие обладают высо-
кой огнеупорностью, диэлектрическими и полу-
проводниковыми свойствами, способностью пере-
ходить к сверхпроводимости при относительно
высоких температурах, высокой химической ус-
тойчивостью в различных агрессивных средах, из
носостойкостью, другие же — каталитической ак-
тивностью, низкими температурами плавления,
низкими значениями твердости, смазочными свой-
ствами.
Исследование нитридов начато в первой
трети прошлого века работами главным об-
разом французских и немецких естествоиспыта-
телей, но особенно расширилось в начале XX ве-
ка в связи с общим развитием химии и
потребностями металлургии и других отрас-
лей техники.
Значительный вклад в исследование мето-
дов получения, свойств и областей применения
3
нитридов был сделан русскими учеными, из которых следует в первую
очередь упомянуть И. И. Жукова, выполнившего ряд принципиально
важных и во многом основополагающих работ по нитридам в первой
четверти XX века.
Крупные работы в этом направлении были сделаны за рубежом
Г. Брауэром, Р. Юца, Г. Хэггом, Н. Шенбергом, Е. Гебхардтом, К- Аг-
те, Е. Фридерих, Л. Зиттигом. В СССР работы по синтезу и исследова-
нию нитридов переходных металлов и некоторых неметаллов были вы-
полнены в Физико-химическом институте им. Л. Я. Карпова под руко-
водством Б. Ф. Ормонта, по электронографическому исследованию ни-
тридов — в Институте кристаллографии АН СССР под руководством
3. Г. Пинскера. В последние годы расширились работы по нитридным
огнеупорам (Д. Н. Полубояринов, Н. И. Воронин, С. Г. Тресвят-
ский), разносторонние исследования проведены в Институте проблем
материаловедения АН УССР.
Однако проблема изучения и применения нитридов еще далеко
не решена — всеми предыдущими исследованиями были лишь намече-
ны наиболее перспективные направления дальнейших более углублен-
ных работ.
Сведения о нитридах чрезвычайно распылены в разнообразных ори-
гинальных работах, вышедших за последние полтора века, что затруд-
няет их использование и создание единого представления об этом важ-
ном классе неорганических соединений.
В настоящей книге автор ставил своей целью в какой-то степени
восполнить этот пробел и содействовать дальнейшему изучению
и расширению использования в практике этих соединений, ознакомле-
нию с ними широких кругов металлургов и химиков.
Естественно, что эта первая попытка не может быть лишена недо-
статков.
Автор считает своим долгом отметить тот вклад в изучение нитри-
дов, который был сделан его талантливой ученицей Т. С. Верхоглядо-
вой, безвременно ушедшей из жизни, а также выразить глубокую бла-
годарность П. В. Гельду за большой труд по рецензированию книги
и ценные замечания, которые позволили существенно улучшить книгу,
а также всем советским и зарубежным ученым, которые предоставили
много оттисков своих работ, существенно облегчив написание этой
книги.
ВВЕДЕНИЕ
Среди соединений, образуемых элементами пе-
риодической системы с неметаллами, соединения
с азотом — нитриды — занимают особое место
по способности атома азота принимать элект-
роны партнера с достройкой до стабильной в
энергетическом отношении конфигурации элек-
тронов s2p6 или отдавать электрон партнеру с
образованием стабильной конфигурации sp3.
В первом случае образующиеся соединения обла-
дают четко выраженной ионной связью, во вто-
ром— типичной металлической, причем в обоих
случаях им сопутствует большая или меньшая до-
ля ковалентной связи, которая становится прева-
лирующей или практически единственной в сое-
динениях азота с неметаллами. Таким образом,
большая часть нитридов характеризуется гете-
родесмичностью химической связи с широкими
пределами доли различных типов этой связи,
чему соответствует изменение характера и зна-
чений физических и химических свойств. Так,
если нитриды неметаллов являются преимуще-
ственно ковалентными соединениями класса ди-
электриков, то нитриды переходных металлов с
дефицитом азота против стехиометрического ко-
личества и некоторые нитриды предельного сос-
тава (CrN и др.) обладают полупроводниковыми
свойствами со смешанным ионно-ковалентно-ме-
таллическим типом связи, а нитриды щелочных
и щелочноземельных металлов представляют
ионно-ковалентные соединения. В то же время
большая часть нитридов переходных металлов
предельного состава по азоту является типичны-
ми металлическими веществами.
Относительная простота электронных пере-
группировок самого атома азота и электронного
строения нитридов делает их удобными «модель-
ными» объектами для изучения химической свя-
зи. Многообразие нитридных фаз с широкими
пределами изменения характера связи и физи-
ко-химических свойств обеспечивает применение
нитридов в различных областях техники и от-
крывает широкие потенциальные возможности
создания нитридных фаз и материалов на их
основе с заранее заданными свойствами.
5
Большой интерес представляет механизм образования нитридов, осо-
бенно при действии азота на простые вещества. По развиваемым в на-
стоящей монографии представлениям, молекула азота построена по типу
sp3:sp3, где стабильные «реконфигурации связаны между собой спарен-
ными электронами. Это обеспечивает высокую прочность и известную хи-
мическую инертность молекулярного азота, а также быструю рекомбина-
цию атомарного азота в молекулярный. Поэтому для образования нитри-
дов необходимо либо нарушение парноэлектронной связи, что требует
достаточно сильных возбуждений термического, электрического характе-
ра, либо высокая акцепторная способность атомов азотируемого веще-
ства, либо, наконец, передача азотируемым веществом электронов азоту с
образованием «2р6-конфигураций и нарушением при этом схемы строения
молекулярного азота по типу: стабильная конфигурация — парноэлек-
тронная связь — стабильная конфигурация. Таким образом, изучение
процессов азотирования простых веществ дает возможность сделать
важные выводы о природе химической связи в нитридах и тех энерге-
тических изменениях, которые наступают при ликвидации молекулы
азота и образовании нитридных фаз.
При образовании нитридов из сложных соединений ионного типа
(путем их термической диссоциации) нередко образуются нитриды также
ионного типа, который не присущ стабильному состоянию и является ме-
тастабильным. Это относится к нитридам переходных металлов, кото-
рые при получении обычными способами в стабильном состоянии име-
ют составы, не характерные для ионных соединений, а при термической
диссоциации некоторых соединений приобретают составы, характерные
для ионных веществ.
Особенности электронного строения нитридов обусловливают заме-
чательные их свойства — высокую сверхпроводимость некоторых из
них, высокое значение термо-э. д. с., большие значения ширины запре-
щенной зоны, необычайно низкие или необычайно высокие значения
твердости и т. п., что само по себе вызывает необходимость детального
изучения природы этих соединений. Однако, несмотря на накопленный
материал о свойствах, строении и особенностях процессов получения
нитридов, пока по существу нет работ, которые позволили бы рассмот-
реть все данные с единой точки зрения, в частности с точки зрения
электронного строения нитридов. Хотя имеющиеся в настоящее время
представления по этому вопросу носят преимущественно качественный
характер, тем не менее использование указанных представлений много
может даты в понимании природы нитридов.
В данной монографии автор поставил своей задачей не только на-
иболее полно собрать имеющиеся сведения о нитридах, но и подчерк-
нуть интересные или труднообъяснимые особенности нитридных фаз,
процессов их образования.
ГЛАВА I
СТРУКТУРА
И СВОЙСТВА
НИТРИДОВ
1. ЭЛЕКТРОННАЯ И КРИСТАЛЛИЧЕСКАЯ СТРУК-
ТУРЫ НИТРИДОВ
Проблема электронного строения нитридов явля-
ется частью крупнейшей проблемы электронной
структуры соединений металлов и неметаллов, с
одной стороны, с неметаллами, с другой, и в на-
стоящее время известные шаги в ее решении
сделаны в основном в части рассмотрения соеди-
нений переходных металлов с неметаллами. Это,
естественно, связано теснейшим образом с разви-
тием представлений об электронной структуре
переходных металлов.
Характерным для нитридов переходных ме-
таллов является образование ими фаз внедре-
ния, т. е. фаз с простыми структурами, построен-
ными по типу внедрения атомов неметалла в
кристаллическую структуру металла. Условием
образования таких фаз является удовлетворение
правилу Хэгга, согласно которому отношение
радиусов атома металла Rm# и атома неметалла
Rx должно соответствовать критерию
Rx : RiAe 0,59.
Как следует из данных табл. 1, для нитри-
дов переходных металлов отношение Rx-Rms
всегда меньше критической величины Хэгга.
Таблица 1
Отношение для нитридов переходных
металлов
7
Основой структуры внедрения является решетка металла, в тетра-
эдрические или октаэдрические поры которой внедрены атомы неметал-
ла; при этом может наступить некоторое искажение решетки. Фазы
внедрения с составом Ме4Х образуют обычно КГЦ-решетку (К12), с
составом МезХ — гексагональную компактную (Г12), с составом МеХ—
—КГЦ (К12), либо КОЦ (К8), либо простую гексагональную (Г8)
[777]. Так как число октаэдрических пор в решетках Г12 и К12 равно
числу металлических атомов, а число тетраэдрических пор в два раза
больше, то при размещении атомов неметалла в октаэдрических порах
предельным составом является МеХ, а при занятии тетраэдрических
пор таким предельным составом является МеХ2.
Внедрение атомов неметалла в решетки атомов переходных метал-
лов является не простым размещением атомов неметалла в порах решет-
ки металла с образованием соединений типа клатратов включения
[778], а сопровождается образованием сильных химических связей
между атомами металлов и неметаллов, принципиально изменяющих
их химическую индивидуальность и физические свойства.
Выяснению природы таких химических связей было посвящено боль-
шое количество работ, особенно за последние 20—25 лет. Одна из пер-
вых попыток понять природу и особенности химической связи в фа-
зах внедрения предпринята А. X. Брегером [779]. Он указывал, что в-
осуществлении химической связи принимают участие как d-, s-валент-
ные электроны переходного металла, так и S-, р-валентные электроны
неметаллов, которые образуют резонирующие связи по шести направле-
ниям (в случае октаэдрической координации) практически равномер-
но, ибо все шесть соседей в решетке типа NaCl находятся на одинако-
вых расстояниях от центрального атома. Следовательно, по этим пред-
ставлениям, необходимо отдать предпочтение так называемым атомным
связям, т. е. преимущественно ковалентным связям, характеризующим-
ся распределением электронной плотности, подобным распределению
в алмазе (электронная плотность между ближайшими атомами имеет
конечное значение, не отличающееся по порядку величины от максиму-
мов электронной плотности соответствующих атомов). В этой же работе
указано, что на ковалентную связь накладывается значительная доля
ионной связи, возрастающая при переходе от С к N и О, а также (в изо-
электронных структурах) — от V к Ti. Ниже будет показано, что такая,
точка зрения, малоизвестная ранее, находит в настоящее время много-
численные подтверждения.
Большую известность получила точка зрения на природу химичес-
кой связи в фазах внедрения, развитая Я. С. Уманским [777, 781] и за-
ключающаяся в том, что при внедрении неметаллов в решетки, образован-
ные атомами металлов, происходит «металлизация» атома неметалла,
т. е. передача им всех или определенной части валентных электронов
в некоторый электронный коллектив, общий для атомов металла и не-
8
металла, и обусловливающий преимущественно металлический харак-
тер связи между атомами. Предполагают, что валентные электроны не-
металлов частично переходят на незаполненную электронную d-подгруп-
пу атомов переходных металлов, что приводит к повышению энергии
связи в кристаллической решетке, приближая их по свойствам к эле-
ментам с более застроенными оболочками и большими энергиями свя-
зи, как W и Мо. Так как потенциал ионизации азота больше, чем угле-
рода, электроны атомов азота, по-видимому, мало участвуют в застрой-
ке d-оболочки металла, и силы связи при образовании нитридов почти
не увеличиваются [78]. Хотя в настоящее время от этих представлений
практически полностью отказались, они были отправным этапом в
развитии теории фаз внедрения.
Автор настоящей работы сделал попытку придать некоторую коли-
чественную оценку возможности и степени перехода валентных элект-
ронов атомов неметалла на d-оостояния атомов переходных металлов
[782], предполагая, вслед за Я. С. Уманским, что вероятность такого пе-
рехода в общем должна увеличиваться с понижением ионизационного
потенциала атома неметалла. Вероятность перехода валентных элект-
ронов атомов данного неметалла на d-состояния металла повышается,
очевидно, с понижением главного квантового числа валентных d-элект-
ронов (т. е. пропорционально \/N, где N — главное квантовое число),,
благодаря возможности занятия электроном места на d-оболочке с наи-
большим освобождением энергии, а также с понижением числа элект-
ронов на d-оболочке изолированного атома переходного металла,(про-
порционально 1/п, где п— число электронов на d-оболочке), вследствие
стремления к заполнению этой оболочки. Следовательно, вероятность
заполнения электроном атома неметалла вакантных мест на d-оболоч-
ке атома металла определяется произведением вероятности частных со-
бытий, т. е. числом [fnN.
Корреляция физических свойств фаз внедрения и указанного кри-
терия, названного «акцепторной» способностью атома переходного ме-
талла, вполне удовлетворительна и позволила с определенных позиций
объяснить природу многих свойств фаз внедрения. Данному вопросу
посвящены наши многочисленные работы, некоторые из них выполнены
совместно с В. С. Нешпором и другими сотрудниками (см., например,
[783—790]). Следует отметить, что эти корреляции не потеряли своего
значения и в настоящее время, однако в критерий \lrtN вкладывается
совершенно иной смысл, чем следующая из представлений Уманского1
способность атомов переходного металла «акцептировать» валентные
электроны «металлизирующегося» атома неметалла.
В работе Партэ [791] указывается на необходимость трехмерных,
связей в решетках фаз внедрения и родственных им фаз для объясне-
ния наблюдающихся высоких значений физических свойств и плотной
упаковки структурных элементов наименьшего размера.
»>
По Рэндлу [792], при внедрении неметалла в переходной металл
связь Me—Me ослабляется, осуществляющие эту связь в металле элек-
троны частично переходят на связи Me — X. Принимается, что связи
в решетках фаз внедрения являются преимущественно ковалентными
(как и у Бретера), причем для фаз внедрения, образованных переход-
ными металлами первого ряда, четыре электронные пары резонируют
по шести связям (образуемым тремя 2р-орбитами неметаллических
атомов). Такие связи являются «электронно-дефектными», подобными
связям в гидридах бора (боранах), и характеризуются тем, что парно-
электронная структура связи должна оставлять неиспользованными
стабильные связывающие орбитали. В таком случае несколько атом-
ных орбиталей могут комбинироваться для образования связываю-
щей орбитали с минимумом энергии — такая комбинированная орби-
таль содержит только одну электронную пару, но может эффективно
связывать более чем два атома. Таким образом возникает представле-
ние о полусвязях, которые образуются, когда один элемент должен
иметь больше стабильных связывающих орбит, чем валентных электро-
нов (т. е. должен быть в общем случае металлом), а другой компонент
должен иметь относительно мало связывающих орбит и представлять
собой в общем случае неметалл; при этом электроотрицательности обо-
их компонентов не должны существенно различаться. Этим трем усло-
виям отвечают фазы внедрения типа МеХ со структурой NaCI, где Х =
= С, N и иногда О (если нет значительной разницы электроотрица-
тельности; фтор всегда слишком электроотрицателен и подобного типа
фаз не образует). С увеличением порядкового номера переходного ме-
талла в группе все большее число электронов металла участвует в свя-
зях Me—Me и меньшее число их передается на связь Me—X, что обус-
ловливает, по мнению Рэндла, трудность образования, например, пере-
ходными металлами VI группы фаз типа МеХ со структурой NaCI.
В работе Энгеля [793] развиты представления о том, что каждый
атом имеет фиксированную и четко определенную картину квантовых
•состояний, которая существенно! неизменяема; при любых химических
превращениях, аллотропических изменениях наблюдаются переходы
электронов из одного квантового состояния в другое, но без изменения
общей квантовой картины, являющейся неизменной для данного атома.
В известной мере Энгель подходит к представлениям о наборах стабиль-
ных электронных конфигураций, о которых будет указано ниже. Рас-
сматривая соединения переходных металлов с неметаллами, Энгель
полагает, что наиболее энергетически выгодна ионизация атомов не-
металлов с передачей части валентных электронов на d-состояния ато-
мов переходных металлов (spd-гибридизации он считает маловероятны-
ми по энергетическим соображениям, так как смешанные связи являют-
ся обычно более слабыми [794]). Таким образом, взгляды Энгеля
практически ничем не отличаются от представлений Уманского и пред-
зо
оставлений, развитых в наших прежних работах. На тех же позициях
находится Кисслинг [795], утверждая, что передача электронов атома-
ми неметалла к атомам металла усиливает связь Me—Me, которая и
является определяющей для фаз внедрения по сравнению со связью
Me—X. Подобные взгляды развивает и Робинс [796], который считает,
однако, что определяющей для образования фаз внедрения является
связь Me—X. Робинс полагает, что главной характеристикой фаз внед-
рения есть отношение концентрации электронов связи к координацион-
ному числу атомов металла.
Томан [948] также считает, что в соединениях с неметаллами 3d-
переходные металлы уменьшают заполненность sp-полосы, захваты-
вая из нее электроны, причем эта способность к захвату понижается в
направлении Cr>Mn>Fe>Co. У никеля наименьшая способность зах-
ватывать электроны практически равна нулю.
В последнее время развиваются представления, основанные на из-
менении функции плотности состояний у поверхности Ферми при из-
менении концентрации валентных электронов на атом [797—802]. Пока-
зано, что экстремумы на кривой плотности состояний соответствуют
некоторым экстремальным значениям электрофизических, термодинами-
ческих и других свойств. Особенно интересна работа Дэмпсея [802], в
которой хотя и принимается точка зрения Уманского об акцепторном
характере d-элементов, а также донорной роли неметаллов, все же ука-
зывается на существенную роль ковалентной связи между атомами пе-
реходных металлов и ее практическое отсутствие между атомами ме-
таллов и неметаллов. Обращается внимание на существование особо
стабильных зон, содержащих около 6 электронов (5,5—6,5 электронов),
которым соответствуют экстремальные свойства соединений металлов
с неметаллами.
Не имея возможности подробно рассмотреть все указанные пред-
ставления, следует отметить, что в большинстве из данных теорий при-
нимается, что при образовании соединений типа фаз внедрения атомы
неметаллов передают часть своих валентных электронов на d-состоя-
ния атомов переходного металла. Степень передачи отражается на ха-
рактере зависимости плотности состояний от числа валентных электро-
нов на атом. Экстремальным значениям плотности состояний отвечают
особые, экстремальные свойства вещества.
Возможен и несколько иной подход к рассмотрению электронной
структуры и свойств соединений металлов и неметаллов с неметаллами
£803]. При образовании твердого тела из изолированных атомов валент-
ные электроны последних частично локализуются у остовов атомов, а
частично переходят в нелокализованное состояние. Локализованная до-
ля валентных электронов образует довольно широкий спектр конфигу-
раций, различающихся по своей энергетической устойчивости, т. е. по за-
пасу свободной энергии, так что наряду с весьма устойчивыми конфигу-
11
рациями, которым отвечает минимум свободной энергии, появляются
менее и весьма неустойчивые. Допуская, что статистический вес наибо-
лее устойчивых в энергетическом отношении электронных конфигура-
ций атомов существенно превышает статистический вес неустойчивых,
можно приписать каждому атому состояния, отвечающие ограниченно-
му числу наиболее устойчивых конфигураций, называя последние ста-
бильными. Между стабильными конфигурациями и нелокализованной
частью валентных электронов осуществляется обмен, отвечающий за
связь между стабильными конфигурациями, а следовательно, и осто-
вами атомов друг с другом. Между нелокализованными электронами
осуществляется сильное отрицательное взаимодействие, которое приво-
дит к отталкиванию друг от друга стабильных конфигураций и остовов
атомов, вызывающее разрыхление кристаллической решетки и ослаб-
ление связи между атомами. Эти исходные положения позволяют рас-
смотреть с единой точки зрения особенности электронного, кристалли-
ческого строения и свойств элементов и их соединений. Все химические
элементы целесообразно подразделить на три основные группы по при-
знаку конфигурации валентных электронов их изолированных атомов:
1) s-элементы, имеющие внешние s-электроны при полностью за-
полненных или полностью незаполненных, более глубоко лежащих элек-
тронных оболочках;
2) ds- и /ds-элементы, атомы которых имеют недостроенные d- или
/-оболочки;
3) sp-элементы, имеющие валентные sp-электроны.
К первой группе относятся непереходные металлы, ко второй —
переходные металлы, к третьей — неметаллы и так называемые полу-
металлы.
Переходные металлы. Максимальное число электронов на d-обо-
лочке переходных металлов равно 10, а на /-оболочке—14. При обра-
зовании металлического кристалла из изолированных атомов d-пере-
ходных металлов локализованная часть валентных электронов может,
как отмечалось, образовывать любые конфигурации, среди которых наи-
более устойчивыми в энергетическом отношении для d-элементов яв-
ляются d° (тождественная нижележащей 52р6-конфигурации), d6 и
d10. Энергетическая устойчивость таких конфигураций показана в ра-
ботах по теории поля лигандов [804, 805]. Энергия стабилизации крис-
таллическим полем в современной теории катализа равна нулю при
числе электронов на d-оболочке, равном 0, 5, и 10 [806]. В работах
[807, 808] показана сферическая симметрия указанных конфигураций,
чему должен соответствовать минимум свободной энергии. В работе
[809] стабильность d5- и d ^-конфигураций установлена с точки зрения
их энергетической выгодности по оптическим термам, причем сделан
вывод о большей устойчивости d5-кoнфигypaций. Наибольшая устой-
чивость полузаполненных d- и /-состояний показана и в работе [5].
12
Устойчивые или стабильные конфигурации являются основой ковалент-
ной связи. По Полингу [810, 811], максимальная ковалентность наблю-
дается в середине каждого периода системы элементов.
По данным рентгеноспектрального исследования установлено рас-
пределение валентных электронов (при переходе атомов из изолиро-
ванного состояния в состояние металлического кристалла) на обеспе-
чивающие ковалентную связь (локализованные электроны) и коллекти-
визированные (нелокализованные электроны) (табл. 2).
Таблица 2
Распределение валентных электронов атомов переходных металлов
и статистический вес атомов с ^-конфигурациями в металлических
кристаллах
Металл
Число
валентных
электронов
(d+s)
Локализован • Нелокализоваи-
ные електроны ные электроны
Литература
Статистический
вес атомов с
^-конфигура-
циями
Sc
Y
La
Ti
Zr
Hf
V
Nb
Ta
Cr
Mo
W
3
3
3
4
4
4
5
5
5
6
6
6
~2,6
~3,7—3,9
~3,5
~1,4
~1,1—1,3
~2,5
~0,95—1,05
[812, 813]
[814]
[815]
*
18
22
23
43
52
55
63
76
81
73
88
94
* По данным M. И. Корсунского и Я. Е. Генкина.
Полагая, что атомы, имеющие в изолированном состоянии число
электронов на d-оболочке nd <15, образуют преимущественно только ста-
бильные конфигурации d° и d5 локализованных электронов, может быть
•определен статистический вес d5-кoнфигypaций атомов в металлических
кристаллах циркония, ниобия и хрома.
Исходя из этих результатов, приближенно может быть оценен ста-
тистический вес d ^конфигураций для остальных переходных металлов
с nd^5 (с использованием критерия \/Nn).
Подобные данные могут быть получены при определении по значе-
нию константы Холла количества носителей тока в элементарной ячей-
ке на один атом, принимая однозонную модель (что, в свою очередь,
оправдано локализацией части валентных электронов у остовов атомов
переходных металлов) и считая носители тока нелокализованной частью
валентных электронов. Соответствующий расчет, выполненный В. А. Гор-
13
батюком (КПП) и в работе [1037] привел к близким результатам. Так,,
статистический вес ^-конфигураций, по его данным, оказался рав-
ным: для Ti — 44%, Nb — 76%, Mo — 86%, Та —78%. Расхождение-
наблюдалось для хрома (83%), ванадия (78%), вольфрама (81%), что
объясняется неточным определением константы Холла.
Таким образом, с увеличением числа d-электронов в валентной обо-
лочке изолированных атомов возрастает статистический вес атомов,
обладающих d ^конфигурациями в металлических кристаллах. С ростом
главного квантового числа валентных d-электронов увеличивается энер-
гетическая устойчивость d ^конфигураций.
Для d-элементов с 5Znd<10 можно предположить, наличие атомов-
с двумя наиболее стабильными электронными конфигурациями d5 и d 10,
причем статистический вес атомов, обладающих d ^-конфигурациями,
возрастает с увеличением nd.
Существенным различием металлических кристаллов с nd<.5 и
nd>5 является то, что для nd<.5 характерна значительная доля нелока-
лизованных электронов, в то время как для металлов с nd^5 электро-
ны, освобождающиеся при образовании d5-кoнфигypaций, практически
полностью используются на образование d10-KOH(|)HrypanKft. Это, в
частности, видно из значений количества носителей тока на атом, рав-
ных для Fe — 0,1, Со — 0,19, Ru — 0,04, Pd — 0,13, Pt — 0,5 [1037], что
указывает на значительную «мобилизацию» всех электронов на построе-
ние стабильных конфигураций.
Аналогичная картина наблюдается для /-элементов, где металличес-
кие кристаллы элементов с nf^.7 представляют комбинации атомов с
f° и f ^конфигурациями и значительным числом нелокализованных
электронов, а с и/>7— комбинации f7- и f ^-конфигурации с малым
числом свободных электронов, причем наиболее устойчивым является
полузаполненное /7-состояние, а энергетическая устойчивость соответст-
венных /-состояний повышается с ростом главного квантового числа
/-электронов.
Во всех случаях возрастание статистического веса, энергетической
устойчивости стабильных конфигураций и соответственное уменьшение
статистического веса нелокализованных электронов вызывает увеличе-
ние сил связи, твердости, температур плавления.
Непереходные металлы. При объединении атомов s-металлов в
кристаллы, по-видимому, для металлов, имеющих я'-валентный элект-
рон, происходит объединение в пары s2, имеющие сферическую симмет-
рию и соответственно высокую устойчивость, в случае же $2-металлов—
нарушение пар для возможности организации связи между атомами.
Энергетическая устойчивость s ^конфигураций уменьшается с ростом
главного квантового числа валентных s-электронов. Статистический
вес s ^конфигураций резко уменьшается при возможности перехода
части s-электронов на полностью вакантные глубоколежащие d- или;
14
f-оболочки (например, у калия, серебра). Для металлов подгруппы:
бериллия возможны р-переходы с преобразованием «^конфигура-
ций в sp-. Вследствие уменьшения энергетической устойчивости любых
sp-конфигураций с ростом главного квантового числа валентных sp-
электронов (см. ниже), этот 5->р-переход особенно четко выражен у
бериллия, более слабо у магния и у кальция практически полностью
подавляется переходом части s-электронов на энергетически устойчи-
вые d-состояния, что далее развивается у стронция, бария, радия.
Неметаллы. Для атомов неметаллов, имеющих в изолированном
состоянии валентные sp-электроны, характерно стремление к образова-
нию наиболее энергетически устойчивых sp3- и 52р6-конфигураций.
Если з2р6-конфигурация может быть действительно названа стабиль-
ной, выгодной с точки зрения энергетического состояния атома, то sp3-
конфигурация не является устойчивой в изолированных атомах, возни-
кает под действием поля кристаллической решетки [809] и может быть
названа квазистабильной. Энергетическая устойчивость однотипных
sp-конфигураций всегда понижается с ростом главного квантового чис-
ла валентных sp-электронов (например, в группе углерода — умень-
шение ширины запрещенной зоны, твердости, температуры плавления
от алмаза к германию, вырождение 5р3-конфитураций у олова при обыч-
ных температурах, у свинца — при любых, сильная делокализация элек-
тронов, появление высокой пластичности).
Все элементы, расположенные слева от группы азота (Be, В, С, их
аналоги в соответствующих группах), стремятся вследствие s->-p-nepe-
ходов и взаимного обмена электронами приобрести максимальный ста-
тистический вес наиболее энергетически устойчивых 5р3-конфигураций;
напротив, элементы, расположенные справа от азота (О, F и их анало-
ги в группах), стремятся к достройке до стабильной конфигурации s2p6.
Азот и его аналоги по группе занимают промежуточное положение. С
одной стороны, атом азота, имеющий в изолированном состоянии конфи-
гурацию валентных электронов s2p3, способен к преобразованию: s2p3->
sp4—*-sp3-(-p с образованием 5р3-конфигураций и передачей партнеру
одного р-электрона, а с другой — может принимать от партнера по
соединению электроны с образованием 52р6-конфигураций. Эта особен-
ность атома азота определяет электронное строение его соединений с
металлами — нитридов и соединений азота с неметаллами.
Исходя из этих представлений, могут быть высказаны некоторые
общие, а также конкретные соображения об электронном строении нит-
ридов и их физических и химических свойствах.
Нитриды непереходных металлов. Низкие ионизацион-
ные потенциалы атомов щелочных металлов вызывают преимущественно
передачу их валентных электронов атомам азота с образованием нитри-
дов состава МезМ, являющихся соединениями ионного типа. Наряду с
этим возможна передача атомами азота легкоподвижного р-электрона
15
;атомам щелочных металлов с образованием азидов MeN3, причем, на-
пример, для натрия, калия и т. п. даже более вероятно образование ази-
да, чем нитрида, несмотря на уменьшающийся в этом направлении иони-
зационный потенциал. Это объясняется тем, что начиная с калия про-
исходит заполнение глубоколежащих d-состояний, устойчивость которых
возрастает с ростом главного квантового числа, что вызывает переход
легкоподвижного электрона от атома азота к атому щелочного металла.
При образовании азидов (MeN3) определяющим фактором являет-
ся группировка из атомов азота, поэтому частоты колебаний линейной
М3-группы в различных азидах отличаются друг от друга незначитель-
но [824].
Аналогичные фазы образуют металлы подгруппы меди, атомы ко-
торых могут преимущественно передавать валентные электроны ато-
мам азота с образованием комбинации d10- и $2р6-стабильных состояний
с менее выраженным характером ионной связи по сравнению с нитри-
дами щелочных металлов (так как у последних комбинируются две
52р6-конфигурации, а у нитридов меди и др. d10- и $2р6-конфигурации,
т. е. конфигурации, типичные для металлической и ионной связей). Это,
.в частности, обусловливает полупроводниковые (а не диэлектрические)
свойства нитридов металлов подгруппы меди, а также появление куби-
ческой структуры (а не тетрагональной или гексагональной).
Состав нитридов бериллия, магния и щелочноземельных металлов
отвечает формуле Me3N3, которая указывает на передачу валентных
электронов металла атомам азота с образованием £2-комбинаций (для
Be) или 82р6-конфигураций (для всех остальных) с з2р6-конфигурация-
ми атомов азота. Вследствие появления у кальция возможности перехо-
да части валентных электронов на вакантные d-состояния усложняется
азотный комплекс с передачей части электронов атомов азота кальцию,
что вызывает образование пернитрида Ca3N4. Повышение энергетичес-
кой устойчивости d-состояний вызывает у стронция появление еще боль-
шего числа нитридных фаз, в том числе субнитрида Sr3N, обладающего
металлическим характером (аналогичная субнитридная фаза имеется и
у бария).
В нитридах металлов подгруппы цинка образуются, как и в нитри-
дах металлов подгруппы меди, комбинации двух стабильных d10- и s2p6-
конфигураций (Me3N3) с соответствующей этим комбинациям кубичес-
кой решеткой.
Нитриды переходных металлов. При образовании нит-
ридов переходных металлов предполагается образование у атомов азота
как s2p6-, так и 8р3-конфигураций, соотношение статистических весов
которых зависит от особенностей партнера по соединению. По мере
повышения статистического веса d5-кoнфигypaций атомов переходного
металла в металлическом кристалле должна уменьшаться возможность
передачи нелокализованных электронов атомам азота с образованием
16
последними $2р6-конфигураций, т. е. возрастает статистический вес d5- и
^реконфигураций и уменьшается статистический вес d&- и «2р6-конфи-
гураций атомов металла и азота, входящих в состав нитрида.
В том же направлении должны возрастать те свойства нитридных
фаз, рост которых связан с повышением статистического веса наиболее
энергетически устойчивых стабильных конфигураций и понижением ста-
тистического веса нелокализованных электронов, но до определенного
предела, так как в случае очень высокого статистического веса ^-конфи-
гураций атомов металла атомы азота не могут приобрести $2р6-конфигу-
рации, а их легкоподвижные электроны (сверх «реконфигурации) вызы-
вают сильное разрыхление решетки.
Существенные результаты по изучению характера химической связи
в нитридах, особенно переходных металлов, получены при рентгеноспек-
тральных исследованиях [227, 228, 836—841, 971]. В [228] указывается о
смещении относительного максимума электронной плотности в нитриде
титана к атому азота и появлении сильной доли ионной связи [840] в от-
личие от карбида титана, где предполагается равноправная принадлеж-
ность электронного коллектива и к металлу и к неметаллу. Аналогичные
выводы делают в [227]. При замещении атомов азота в нитриде титана
атомами углерода резко изменяется (начиная с 30 %-ной концентрации
углерода в C + N) картина химических связей, которая существенно от-
клоняется от аддитивности [837]. Рентгеноспектральное исследование
нитрида ванадия в сравнении с окисью ванадия V2O5 позволяет говорить
о большей доле ковалентных связей в нитриде [836].
На основе рентгеноспектрального изучения нитридов хрома, выпол-
ненного в работе [815], пришли к выводу о переходе части валентных
электронов хрома к азоту в процессе образования нитридов, а также
о большем Me—Me взаимодействии в Cr2N по сравнению с CrN и силь-
ном Me—N взаимодесйтвии в решетке CrN.
Авторы [839] указывают на отчуждение части валентных электро-
нов от атома металла в нитриде ниобия.
Подобные взгляды развиты Юм-Розери [409], который считает, что
введение в железо азота приводит к акцептированию части электронов
железа атомами азота и освобождению соответственных Зй-состояний
для связи; при этом возникает ГПУ-структура сплавов железа с азотом.
Из результатов рентгеноспектральных исследований [841] следует
вывод о переходе части валентных электронов в нитридах от атомов пе-
реходных металлов к атомам азота, что хорошо согласуется с предполо-
жением о локализации в части нитридов, особенно тех металлов, у кото-
рых низкий вес стабильных ^-состояний, валентных электронов этих
металлов в «2р6-состояниях азота.
Это касается в первую очередь нитридов переходных металлов, яв-
ляющихся четко выраженными донорами нелокализованной части ва-
лентных электронов (титана, циркония, гафния и т. п.). Для нитридов
- '2
17
переходных металлов, меньше проявляющих донорные свойства (об-
ладающих высоким весом атомов со стабильными конфигурациями),
можно говорить об образовании атомами азота s2pe- и «^-конфигураций.
Это относится, например, к мононитриду тантала, рентгеноспектральное
исследование которого обнаружило черты сходства с Та2О3 («^-конфи-
гурации атомов азота) и с ТаС, ТаВ2 («реконфигурации атомов азо-
та) [971].
Стремление атомов переходных металлов к передаче части валент-
ных электронов атомам азота особенно четко проявляется при получе-
нии нитридов осторожным разложением соединений, где атомы металла
уже имели типичную ионную форму, например, комплексных хлорамми-
ачных, фтораммиачных и других подобных соединений. При этом обра-
зуются метастабильные соединения составов TiNi,i6, ZrNi>33, TaNi^r (см.
стр. 77).
Информацию о характере химической связи в нитридах можно по-
лучить из рассмотрения динамики тепловых колебаний кристаллической
решетки, как это сделано в [1029] при обработке данных по низкотемпе-
ратурной теплоемкости нитридов титана и ванадия. Показано, что силы
связи между неближайшими соседями — атомами решетки — умень-
шаются при переходе от карбидов к нитридам и далее к окислам ти-
тана и ванадия. Это соответствует меньшим энергиям атомизации, тем-
пературам плавления, твердости нитридов по сравнению с карбидами
и вызывается уменьшением роли коллективизированных электронов d-
полосы в осуществлении связи между атомами в решетке. Также под-
тверждается частично ковалентный характер связи в нитридах и других
подобных соединениях.
Нитриды неметаллов. Образование нитридов неметаллов и
вообще соединений неметаллов с азотом обычно связано с минимизацией
энергии системы в результате образования устойчивых sp-конфигураций
(со стремлением к образованию наибольшего статистического веса ато-
мов с наиболее устойчивыми «р3 * *- и «^-конфигурациями).
Такие общие соображения об электронной структуре нитридов на-
ходят свое конкретное отражение в особенностях кристаллической струк-
туры и физико-химических свойств отдельных нитридных фаз.
2. ФИЗИЧЕСКИЕ СВОЙСТВА НИТРИДОВ
Нитриды непереходных металлов [844]. Нитриды «-элемен-
тов построены в основном по типу соединений с высокой долей ионной свя-
зи, обусловленной передачей валентных электронов атомами металла
атомам азота с образованием при этом высокого статистического веса
«2р6- либо diQ- и «2р6-конфигураций (у лития и бериллия — также «г-кон-
фигураций). Это обусловливает полупроводниковые свойства нитридов с
18
таким набором стабильных конфигураций. Так, нитрид лития Li3N до
360° С практически не проводит электрического тока, а при более высо-
ких температурах, вследствие термического возбуждения стабильных
конфигураций и частичной делокализации электронов, начинает прово-
дить ток, приобретая свойства типичного полупроводника. Важно, что
температурная кривая проводимости при 446° С изменяет направление,
что связано с нарушением при 350—360° С одного типа стабильных кон-
фигураций (вероятно, s2), а при 446° С — другого (вероятно, s2p6). К со-
жалению, электрофизические свойства нитридов прочих щелочных
металлов неизвестны, но можно сделать предположение, что металлич-
ность нитридов должна увеличиваться с ростом главного квантового
числа валентных.«‘-электронов как в связи с уменьшением вероятности,
образования устойчивых «^конфигураций из «‘-электронов, так и вслед-
ствие перехода части s-электронов на d-состояния. Это, в частности,,
наблюдается для нитридов щелочноземельных металлов, у которых на-
чиная со стронция появляются субнитриды с металлическим типом
связи. В то же время нитриды бериллия и магния являются типичными
диэлектриками, что связано с образованием наиболее энергетически
устойчивых s2- и «2р6-конфигураций, на которых локализована значи-
тельная доля валентных электронов и для нарушения которых требуют-
ся высокие термические или иные возбуждения.
Металлы подгруппы меди и цинка образуют два типа стабильных кон-
фигураций в нитридах: di0 и s2p6. Это вызывает, как и у нитридов ще-
лочных и щелочноземельных металлов энергетическое обособление
атомов в решетке, однако меньшее, чем у последних, так как энергети-
ческая устойчивость ^‘“-конфигураций значительно меньше чем «2р6-кон-
фигурации. Такие вещества — полупроводники, но с меньшим удельным
электросопротивлением, с меньшей шириной запрещенной зоны. Они не-
являются подобно нитриду лития изоляторами при обычных температу-
рах. Так, удельное электросопротивление нитрида меди составляет при
обычной температуре всего 6-102 ом-см.
Энергетическая обособленность атомов со стабильными конфигура-
циями вызывает малую термическую' устойчивость нитридов «-металлов:
они разлагаются при нагревании с выделением азота. Легче разлага-
ются те нитриды, у которых наибольшее число валентных электронов
локализовано в наиболее энергетически устойчивых стабильных конфи-
гурациях. Та же энергетическая обособленность вызывает относительно
малую химическую устойчивость нитридов s-металлов, обычно повышаю-
щуюся с увеличением «металличности» нитридных фаз.
Преимущественно ионный характер связи между атомами металлов,
и неметаллов, а также наличие ковалентной связи между атомами ме-
таллов либо атомами азота (в азидах) позволяет считать эти соедине-
ния построенными по ионно-ковалентному типу с очень малой долей ме-
таллической связи в некоторых из них.
2*
19
Нитриды переходных металлов. Наиболее изучены фи-
зические свойства нитридов переходных металлов. В основном установ-
лена корреляция значений этих свойств с критерием 1/Л7г, о котором ука-
зывалось выше. Очевидно, истинный физический смысл этого критерия
заключается не в оценке «акцепторной способности», а в показателе сум-
марного статистического веса стабильных конфигураций атомов. Так, вы-
сокое значение l/Nn для титана означает малую локализацию валент-
ных электронов в стабильных конфигурациях, а низкие значения крите-
рия для металлов триады железа и платиноидов — высокую степень
локализации валентных электронов в стабильные состояния любых ти-
пов, т. е. малый статистический вес нелокализованных электронов.
При рассмотрении с этой точки зрения электрофизических свойств
нитридов переходных металлов IV—VI групп периодической системы
(табл. З, рис. 1) видно, что удельное электросопротивление закономерно
повышается с ростом статистического веса стабильных конфигураций,
что вызвано уменьшением концентрации носителей тока и трудностью
Таблица 3
Физические свойства нитридов переходных металлов
Нитрид Содержание азота Коэффициент термо-э. д. с. а, мкв-град Удельное электро- сопротив- ление Q, мком см Коэффициент Холла R 10*, см’/к Теплопроводность х, кал/см-сек-град Микротвер- дость ЯМе’ кг/мм*
вес. % ат %
TlNpeg 22,4 49,8 —7,78±1,1 26±2 —0,67±0 0,046 ±0,003 1994± 137
ZrN0t98 13,2 49,8 —4,78±0,5 28±3 — 1,3 ±0,2 0,049 ±0,002 1520 ±85
HfN0>86 6,4 46,5 —2,96±0,6 33±5 —4,2 ±0,5 0,0517±0,006 1640±161
VN0,338 8,5 25,3 —5,3 ±1,2 123±10 4-0,9 ±0,1 — 1900± 102
VN0,74 16,8 42,4 — — — — 1520±115
VNo.93 20,5 48,3 —4,6 ±0,8 85 ±4 +0,45±0,16 0,0270±0,07 —
NbN0>46 6,6 31,7 — — — — 1720± 100
NbN0>5 7,1 33,5 —4,57±0,7 142±6 ±1,9±0,4 0,0200±0,08 —
NbN0i96 12,7 49,1 —1,65±0,1 85±2 —0,47±0,2 — 1396 ±26
NbN100 13,1 50,0 —2,24 78±4 +0,52±0,19 0,009 ±0,002 —
Та*<0.45 3,4 31,2 —2,17±0,4 263±22 —0,46±0,1 0,0240±0,005 —
4,3 36,6 — — — — 1220±120
TaNlt01 7,3 50,3 —1,6 ±0,3 128±15 —3,61±0,9 0,0205 ±0,009 1060±72
<-'rN0,47 11,2 32,0 — — — — 1571 ±49
CrN0,497 11,8 33,2 —2,3 ±0,2 79±5 —0,72±0,1 0,0519±0,004 —
<-'Г^0,926 20,0 48,1 —92±4 640±40 —265 ±25 0,0300±0,003 —
CrNo,99 21,0 49,8 — — —. — 1093 ±93
MoN0.5 6,8 33,5 ±2,18±0,5 19,8±7 4-2,83±1,2 0,0427±0,007 630 ±86
20
возбуждения на проводимость электронов, входящих в состав все более
энергетически стабильных конфигураций. По тем же причинам в ука-
занном направлении уменьшается коэффициент термо-э. д. с. В работе
[816] проведен анализ значений некоторых электрофизических свойств
нитридов на основе рассмотрения величины б=и_и2-— п+и2+ , где
п-, и_ и п+, и+ — соответственно концентрация и подвижность электро-
нов и дырок. Для металлов IV группы значение величины б очень мало,
что говорит о примерно равном вкладе электронов и дырок в проводи-
мость (заметим, что у этих металлов при-
мерно одинаковые доли локализованных
и нелокализованных электронов). У тита-
на статистический вес 4/5-конфигурации
равен 43% и б = +0,05, что свидетель-
ствует о некотором превалировании элек-
тронной проводимости, а у циркония —
52% и б = —0,09, что свидетельствует о
некотором превалировании дырочной про-
водимости. При переходе к нитридам ме-
таллов IV группы (Ti, Zr, Hf) значение б
возрастает и имеет положительный знак,
Рис. 1. Зависимость удельного
электросопротивления и коэф-
фициента термо-э. д. с. нитри-
дов переходных металлов от
критерия |=1/Л’п.
т. е. увеличивается электронная проводи-
мость, что связано не только с передачей
части нелокализованных электронов ме-
талла атому азота с образованием по-
следним высокого статистического веса
«^-конфигураций, но и, вероятно, с неко-
торой делокализацией ранее (в металлическом кристалле) локализован-
ных в стабильные конфигурации электронов. При переходе к металлам
V—-VI групп разность б резко возрастает и имеет отрицательный знак,
т. е. увеличивается дырочный вклад в проводимость. Это следует сопо-
ставить с ростом статистического веса стабильных с/5-состояний. Нит-
риды же этих металлов имеют преимущественно электронную проводи-
мость (судя по знаку коэффициента Холла и разности б), что указывает
на малое участие в переносе тока стабильных конфигураций металлов и
на сильную делокализацию валентных электронов азота с переходом их
в слабо связанное состояние и одновременное образование «реконфигу-
раций атомами азота. Таким образом, при этом образуется высокий
статистический вес стабильных d5- и «реконфигураций. Однако одно-
временно образуется высокий статистический вес нелокализованных
электронов, что вызывает высокие доли электронной части сопротивле-
ния и теплопроводности (табл. 4, рис. 2), за исключением нитридов
тантала и хрома, обладающих высокой долей решеточной теплопро-
водности [470]. Это хорошо согласуется с полупроводниковым Харак-
тером нитрида CrN. С (разрыхляющей ролью слабо связанных электро-
21
нов азота можно сопоставить уменьшение твердости нитридов при
переходе от металлов IV к металлам V и VI групп периодической сис-
темы [147].
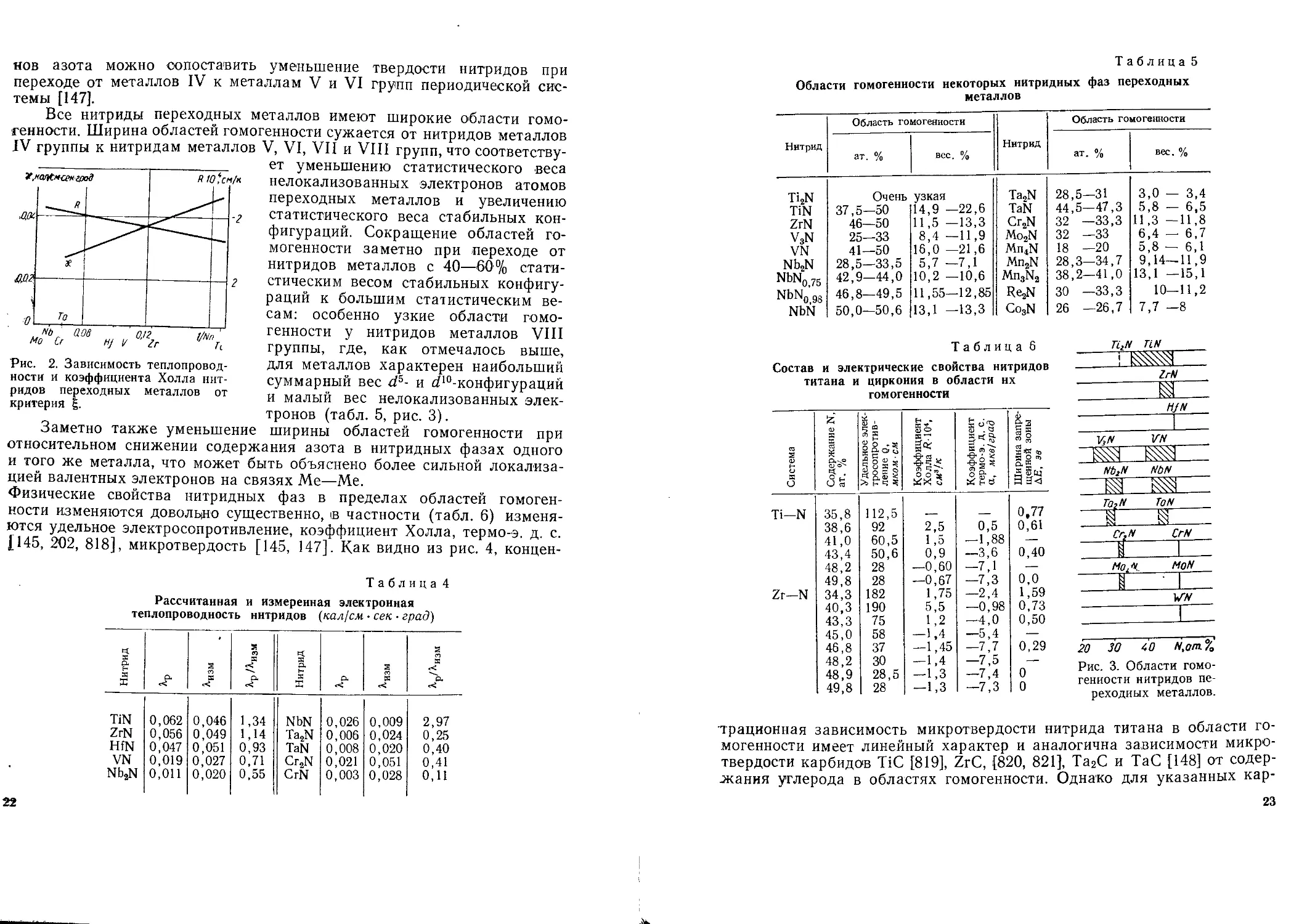
Все нитриды переходных металлов имеют широкие области гомо-
генности. Ширина областей гомогенности сужается от нитридов металлов
IV группы к нитридам металлов V, VI, VII и VIII групп, что соответству-
ет уменьшению статистического веса
нелокализованных электронов атомов
переходных металлов и увеличению
статистического веса стабильных кон-
фигураций. Сокращение областей го-
могенности заметно при переходе от
нитридов металлов с 40—60% стати-
стическим весом стабильных конфигу-
раций к большим статистическим ве-
сам: особенно узкие области гомо-
генности у нитридов металлов VIII
группы, где, как отмечалось выше,
для металлов характерен наибольший
суммарный вес г/5- и ^‘“-конфигураций
и малый вес нелокализованных элек-
тронов (табл. 5, рис. 3).
ширины областей гомогенности при
Рис. 2. Зависимость теплопровод-
ности и коэффициента Холла нит-
ридов переходных металлов от
критерия
Заметно также уменьшение
относительном снижении содержания азота в нитридных фазах одного
и того же металла, что может быть объяснено более сильной локализа-
цией валентных электронов на связях Me—Me.
Физические свойства нитридных фаз в пределах областей гомоген-
ности изменяются довольно существенно, в частности (табл. 6) изменя-
ются удельное электросопротивление, коэффициент Холла, термо-э. д. с.
[145, 202, 818], микротвердость [145, 147]. Как видно из рис. 4, концен-
Таблица 4
Рассчитанная и измеренная электронная
теплопроводность нитридов (кал/см сек град)
TiN
ZrN
HfN
VN
Nb2N
0,062
0,056
0,047
0,019
0,011
0,046
0,049
0,051
0,027
0,020
1,34
1,14
0,93
0,71
0,55
NbN
Ta2N
TaN
Cr2N
CrN
0,026
0,006
0,008
0,021
0,003
0,009
0,024
0,020
0,051
0,028
2,97
0,25
0,40
0,41
0,11
22
Таблица 5
Области гомогенности некоторых нитридных фаз переходных
металлов
Нитрид Область гомогенности Нитрид Область гомогенности
ат. % вес. % ат. % вес. %
Ti2N Очень узкая Ta2N 28,5—31 3,0 — 3,4
TIN 37,5—50 14,9 - -22,6 TaN 44,5—47,3 5,8 — 6,5
ZrN 46—50 11,5 - -13,3 Cr2N 32 —33,3 11,3 —11,8
V3N 25—33 8,4 - -11,9 Mo2N 32 —33 6,4 — 6,7
VN 41—50 16,0 - -21,6 Mn4N 18 —20 5,8 — 6,1
Nb2N 28,5—33,5 5,7 - 7,1 Mn2N 28,3—34,7 9,14—11,9
NbN0,75 42,9—44,0 10,2 - 10,6 Mn3Na 38,2—41,0 13,1 —15,1
NbN098 46,8—49,5 11,55- 12,85 Re2N 30 —33,3 10—11,2
NbN 50,0—50,6 13,1 - 13,3 CoaN 26 —26,7 7,7 —8
Таблица 6
Состав и электрические свойства нитридов
титана и циркония в области нх
гомогенности
TitN TLN
ГИГ
ZrN
HfN
1
V3N VN
NbaN NbN
To,N Ton
CraN CrN
MON
1
। Система i Содержание N, ' ат. % । Удельное элек- । тросопротив- ление Q, j мком-см 1 Коэффициент ; Холла R-104, ; смг/к Коэффициент термо-э. д. с. а, мкв/град Ширина запре- I щеиной зоны 1 дг, зв 1
Ti—N 35,8 112,5 0,77
38,6 92 2,5 0,5 0,61
41,0 60,5 1,5 —1,88 —
43,4 50,6 0,9 —3,6 0,40
48,2 28 —0,60 —7,1 —
49,8 28 —0,67 —7,3 0,0
Zr-N 34,3 182 1,75 —2,4 1,59
40,3 190 5,5 —0,98 0,73
43,3 75 1,2 —4,0 0,50
45,0 58 —1,4 —5,4 —
46,8 37 —1,45 —7,7 0,29
48,2 30 —1,4 —7,5 —
48,9 28,5 — 1,3 —7,4 0
49,8 28 —1,3 —7,3 0
20 30 40 Ы,ат.7о
Рис. 3. Области гомо-
генности нитридов пе-
реходных металлов.
"грационная зависимость микротвердости нитрида титана в области го-
могенности имеет линейный характер и аналогична зависимости микро-
твердости карбидов TiC [819], ZrC, [820, 821], Та2С и ТаС [148] от содер-
жания углерода в областях гомогенности. Однако для указанных кар-
23
бидов экстраполяция линии микротвердости на содержание угле-
рода, равное нулю, дает приблизительно значения микротвердости со-
ответствующих металлов, в то время как для нитрида титана этого не
наблюдается. Следовательно, в кристаллических решетках нитридов
(в отличие от карбидов) характер электронного строения, химической
связи, распределения электронной плотности принципиально отличает-
ся от таковых в металлах, видимо, в результате появления известной
Рис. 4. Концентрационная зависимость микротвердости в области гомо-
генности Ti—N.
Рис. 5. Изменение электросопротивления нитридов титана и циркония в
областях гомогенности:
I —Zr-N; 2-Ti-N.
доли ионной связи, увеличивающейся по мере снижения содержания
азота в пределах областей гомогенности. Последнее связано с усили-
ем связей Me—Me, локализацией на них значительной части валентных
электронов, уменьшением перекрытия полос валентных электронов ме-
талла и азота и соответственным появлением энергетических разрывов.
Изменение электросопротивления нитридов титана и циркония в об-
ластях гомогенности (рис. 5) также отличается от хода сопротивления
соответствующих карбидов, являясь нелинейным. Появление энерге-
тической щели в решетке нитрида титана с неполным содержанием
азота, предположенное в [822, 823], подтверждается изменением элек-
тросопротивления при высоких температурах (рис. 6) [14’5]. Нитрид
титана с содержанием азота, близким к стехиометрическому, обнаружи-
вает линейный рост электросопротивления с температурой при уменьше-
нии содержания азота до 48,4 ат. % появляется максимум сопротивле-
ния при 1800°С. При дальнейшем снижении содержания азота сопро-
тивление возрастает с одновременным смещением максимума в область
более низких температур. Ширина энергетической щели с уменьшением
содержания азота увеличивается. Для нитридов титана и цирко-
24
ния при уменьшении содержания в них азота в пределах областей
гомогенности происходит рост ширины запрещенной зоны (см. табл. 6)
[832].
Ширина запрещенной зоны нитридов титана и циркония линейно из-
меняется с увеличением содержания азота (рис. 7). Некоторое отклоне-
ние от линейности для содержания азота ниже 40% объясняется легким
загрязнением кислородом, атомы ко-
торого могут занимать свободные
Рис. 6. Концентрационно-температурная зависимость электросопротив-
ления нитрида титана в области гомогенности:
1 — 36,6; 2 — 38,6; 3 — 41,0; 4 — 43,4; 5 — 48,4; 6 — 49,8 ат.% N.
Рис. 7. Коцентрациоиная зависимость изменения ширины запрещенной
зоны в области гомогенности:
1 — Ti—N; 2 — Zr—N.
места в решетке ненасыщенного нитрида. Это предположение подтвер-
ждается наличием примесного характера электропроводности у ZrN при
34 ат.% N, в то время как при других содержаниях азота этого не наб-
людается. Необходимо отметить, что величина запрещенной зоны и ско-
рость ее изменения для нитрида циркония больше, чем для нитрида
титана, что связано с большим весом ^-конфигураций у циркония
и большей локализацией электронов на связях Zr—Zr, чем на свя-
зях Ti—Ti.
На рис. 8 показано изменение коэффициента Холла и термо-э. д. с.
нитридов титана и циркония в области гомогенности.
В то время как большинство нитридов переходных металлов с пре-
дельным содержанием азота обладает металлическими свойствами, у
нитрида хрома CrN четко выражены полупроводниковые свойства [326—
328]. Тем не менее и этот нитрид при низких температурах имеет метал-
25.
лическую (рис. 9, участок ав), при повышении температуры — примес-
ную (участок вс) и далее — собственную проводимость (участок cd),
характерную для полупроводников. Вероятно, при повышенных темпера-
турах происходит возбуждение состояний азота и передача одного элек-
трона атома азота атому хрома с образованием хромом стабильных
.^^-конфигураций, а азотом «/^-конфигураций с соответствующим знер-
а б
Рис. 8. Изменение коэффициента Холла (а) и коэффициента термо-э. д. с.
(б) нитридов титана и циркония в области гомогенности:
1 — Ti—N, 2 — Zr—N.
•ляется полупроводником, по-видимому, в связи с тем, что атомы
хрома образуют большой статистический вес с110«2-конфигураций, а
освободившийся электрон азота осуществляет металлургическую прово-
.димость.
При замещении в нитриде титана части атомов азота атомами кис-
лорода (при ~25—25 мол. % ПО) достигается минимальное значение
сопротивления, коэффициента Холла (табл. 7) и термо-э. д. с. [174] при
сохранении металлического характера проводимости. При нагревании
происходит переход от металлической проводимости к полупроводнико-
вой, температура такого перехода для всех содержаний ПО приблизи-
тельно одинакова и составляет около 1200°С [818]. Ширина запрещенной
зоны у них также одинакова и составляет 4,8—5 эв, что характерно для
ионных соединений, для которых эв. Предполагается, что в при-
оутствии кислорода атомы и азота и кислорода образуют преимущест-
венно $2р6-конфигурации, отвлекая на них нелокализованные электроны
атомов титана, которые в этом случае приобретают ^-конфигурацию,
т. е. образуются типично ионные соединения.
В работе [825] исследована температурная зависимость электро-
физических свойств нитридов титана и ванадия предельного состава.
26
Электросопротивление и термо-э. д. с. изменяются линейно с температу-
рой, что свидетельствует о металлической проводимости, хотя и смешан-
ного типа.
Небольшие значения коэффициентов термо-э. д. с. во всем темпера-
турном интервале измерений также подтверждают металлическую при-
роду их электропроводности. Подобно металлам, зависимость удельного
Таблица 7
Электрические свойства сплавов
TiN—TiO
Состав, мол. % Удельное элек- тросопротив- ление Q. МКОМ-см Коэффициент Холла Я-10*, , см3/к Коэффициент термо-э. д. с. а, мкв/град
TiN TiO
100 0 26 —0,67 —9,3
90 10 17,9 -0,4 —7,1
79,6 20,4 13,1 —0,17 —6,4
73,1 26,9 11,3 —0,48 —0,75
65 35 12,1 —0,82 —
62,7 37,3 12,7 —1,36 —8,6
47,6 52,4 14,2 —1,64 —
46,7 53,3 — —1,70 —13,4
46 54 14,3 —2,02 -—
41,8 58,2 27 —2,64 —
Рис. 9. Температурная зависимость
электропроводности CrN.
электросопротивления этих соедине-
ний от температуры может быть
описана обычным выражением вида
Q=QoX(l+aO, где а —темпера-
турный коэффициент электросопротивления, Qo — значение электросо-
противления при 0° С. Аналогичным выражением может быть описана
температурная зависимость термо-э. д. с. е = еоХ(1+₽0- Соответствую-
щие данные приведены в табл. 8.
Таблица 8
Температурная зависимость некоторых
электрофизических свойств нитридов титана и ванадия
предельного состава
Нитрид g й о • « О S ‘»01 » темпера- турный ин- тервал примене- ния Q, и а, °C et, мкв- град р-1о4, град 1
TiN 24 4,4 0—1200 —3,7 2,06
VN 88 0,7 0—1200 —6,2 1,16
280—1200
280—1200
27
Сравнение моттовских [826] значений о/Л402, (Л1— молекулярный
вес, & — характеристическая температура), проведенное для металли-
ческого титана и различных соединений титана и ванадия, показывает,
что у титана и его диборида TiBa температурная зависимость о/Л!©2
аналогична, а у TiC и TiN довольно сильно отличается от них (рис. 10).
Это указывает на отклонение характера связи в TiC и TiN по сравне-
нию с металлическим титаном. Малое значение температурного коэф-
Рис. 10. Температурная зависимость
приведенной электропроводности титана
и его соединений с азотом, бором и угле-
родом.
фициента электросопротивления у
нитрида ванадия (0,7 град-1) объяс-
Таблица 9
Коэффициенты термоэлектрической
добротности тугоплавких карбидов и
нитридов
Соедине- ние Z, град'-1 Соединение Z, град 1
TiC ZrC HfC VCo,88 NbCo,99 TaC Cr3C, Сг7С3 Cr23Ce Mo2C 2,1 • 10“5 1,1-IO-5 1,0-10“5 0,9-10“6 2,1 • 10“6 5,0-10“6 2,2-10“6 3,1 • 10“e 3,0-10“7 7,6-10“ 6 wc w2c ScNq,98 TiNo,98 ZrNo,98 HfNo,86 VNo,93 NbNi.oo TaNi.oi CrNo,93 0,9-10“4 3,0-10“6 0,8-10“4 1,2-10“5 4,0-10“6 1,2-Ю-6 2,2-10“6 3,1-10“7 2,3-10~7 l,4-10“4
высокого стати-
няется возможностью образования
у атомов ванадия
стического веса ^-конфигураций за счет привлечения электронов атомов
азота, а у атомов азота — соответственно высокого статистического
веса «/^-конфигураций, трудно возбуждаемых при росте температуры.
Низкое значение температурного коэффициента электросопротивления
нитрида ванадия связано с высокой точкой перехода к сверхпроводи-
мости (7,50° К), которая также обусловлена «инертностью» стенок
энергетических «каналов», образуемых атомами с высоким статистиче-
ским весом стабильных конфигураций.
Значения термоэлектрических коэффициентов добротности 2 = а2/(Д
(а-термо-э. д. с., q — удельное электросопротивление, X — теплопровод-
ность) карбидов переходных металлов в сравнении с нитридами при-
ведены в табл. 9. Для большинства нитридов коэффициент доброт-
ности невелик (10-7—10~5 град-1), что согласуется с металлическим
характером их электропроводности. Более высокое значение Z наблю-
дается только для CrN, что обусловлено его полупроводниковым харак-
тером. Из рис. 11 [827] видно, что с ростом критерия ^~\/Nn или с
28
уменьшением статистического веса ^-конфигураций атомов переходно-
го металла термоэлектрическая добротность заметно возрастает, что
вызвано повышением электронного вклада в теплопроводность (и
уменьшением решеточного вклада, обусловленного электронами, лока-
лизованными в стабильные конфигурации). Интересно, что у нитрида
скандия — металла, для которого характерно образование высокого
Рис. 11. Зависимость коэффициентов добротности карбидов и нитридов переход-
1ных металлов от критерия il/Nn:
□ — карбиды МеС; О — нитриды MeN; А — W2C; А—М02С; v—СгтСз; X — Сг3С2.
Рис. 12. Зависимость работы выхода при термоэмиссии нитридов 'переходных ме-
таллов при температуре 1700° С от критерия 1/Nn.
статистического веса ^-состояний, малого статистического веса ^-со-
стояний и сильная делокализация валентных электронов — коэффици-
ент добротности приближается к таковому для полупроводнико-
вого нитрида хрома. Очевидно, это является следствием разных при-
чин и в общем характеризует нитрид скандия как типичный полу-
металл.
Термоэмиссионные свойства. Работа выхода при термоэмиссии
определяется соотношением статистических весов и энергетических ха-
рактеристик локализованной и нелокализованной частей валентных
электронов, повышаясь с увеличением веса и энергетической устойчи-
вости стабильных конфигураций локализованных у атомов электронов
[845]. Работа выхода нитридов переходных металлов (табл. 10) воз-
растает от нитрида титана к нитридам тантала и ниобия (рис. 12)
[170, 252, 846—849].
Сравнение значений работы выхода нитридов и карбидов показы-
вает, что в последнем случае они меньше, что объясняется образовани-
ем в нитридах наряду с «реконфигурациями атомов азота (с соответ-
ствующим освобождением электрона и передачей его в коллектив),
также «2р6-конфигураций, образование которых связано со значитель-
ной локализацией валентных электронов как переходных металлов, так
и азота. Нитриды переходных металлов пока не нашли практического
использования в катодной электронике, так как их термоэлектрические
29
Таблица 10
Термоэмиссня нитридов
Нит- рид Работа выхода ф, эв Посто- янная Ричард- сона А, Примечания Лите- ратура
TIN 2,92 120,4 [170]
3,75 — Эффективная работа выхода при
2000°К, измеренная в порошке [846]
ZrN 2,92 120,4 — [170]
3,90 — Эффективная работа выхода при
1900°К, измеренная в порошке [846]
VN 3,56 -— Эффективная работа выхода при
1600°К [252]
NbN 3,92 — То же при 1950°К [846]
TaN 4,00 — То же при 1600°К [252]
ThN 3,1 — Фотоэмиссия [848]
UN — — Температурная зависимость эффек-
тивной работы выхода дается
выражением:
Фг=3,1+2,1Х10-4Г [849[
войства не очень высоки и, кроме того, они диссоциируют в вакууме-
[ри нагреве.
Сверхпроводимость. В настоящее время установлено, что большая
асть нитридов переходных металлов является сверхпроводниками [275,
304, 1001, 1013]. Соответствующие данные
Таблица 11 по сверхпроводимости нитридов представ-
Сверхпроводимость нитридов лены в табл. 11. _ Обоаптает внимание, что в обшем
итрид TK> °K Нитрид T OK возрастают при повышении веса стабиль- к ’ ных (^-конфигураций атомов соответст-
>cN fN FiN JrN IfN /N JbN lb2N ста( <1,40 <1,40 4,85 8,90—10,7 6,20 8,20 15,60 <1,20 СИЛЬНЫХ K( TaN Ta2N CrN MoN Mo2N w2n Th3N4 UN шфиг вующих Переходных мехаллов (для мо- <1,20 нонитридов), за исключением нитридов <1,20 тантала, хрома и вольфрама. При умень- <1,28 щении относительного содержания азота Ч’о в нитридной фазе понижаются. Если- <1,28 считать, что условиями сверхпроводи- <1,20 мости, с одной стороны, является образо- <1,20 вание атомами «энергетических каналов», устойчивость «стенок» которых обеспечи- вается сильной локализацией электронов /рациях, которые обладают пониженной способ-
)стью к рассеиванию электронов проводимости, а с другой стороны,
>разование электронами проводимости куперовских пар, также обла-
1ющих пониженной способностью к рассеиванию, то рост Тк с увеличе-
нием веса стабильных конфигураций в металле хорошо объясняется..
Так же объясняются вообще высокие 7’к нитридов по сравнению с кар-
бидами, боридами и другими подобными соединениями [275]. Действи-
тельно, в случае образования нитридов металлов IV группы имеется высо-
кое число нелокализованных электронов, которые частично локализуются
на «^-конфигурациях азота и ^-конфигурациях металла. При переходе
к металлам V и VI групп локализация валентных электронов у атомов
металлов выше, что вызывает образование высокого статистического
веса ^-конфигураций металла и «^-конфигураций азота. При этом
обеспечивается высокая стабильность «стенок» энергетической щели
[876] при умеренной концентрации носителей. В случае тантала, хрома
и вольфрама такие «стенки» также образуются, но с одновременным
образованием пониженного числа носителей тока, что приводит к сни-
жению 7k. Поэтому большой интерес представляет высокая 7’к твердых
растворов NbN—NbC, у которых при составе (NbC)o,28—(NbN)o,72
точка перехода составляет 17,9° К [828]. При этом обеспечивается вы-
сокий статистический вес ^-конфигураций атомов ниобия, «/^-конфигу-
раций атомов углерода и азота наряду с оптимальной концентрацией
носителей тока. По теории Бардина, Купера и Шриффера, температура
перехода к сверхпроводимости определяется выражением
kT* = 1,14 (Й®)с₽ exp (- 1 /# (0) • V),
где k — константа Больцмана; (Йа>)ср— средняя энергия фононов, рас-
сеивающих электроны у поверхности Ферми (пропорциональная деба-
евской температуре); N (0) —нормальная плотность электронных
энергетических состояний у поверхности Ферми; V — постоянная элект-
рон-фононного взаимодействия. Можно утверждать, что температура
перехода в сверхпроводящее состояние в основном определяется мно-
жителем N (О') • V или, учитывая приблизительное постоянство V, плот-
ностью состояний. В работе [828] действительно показано соответствие
между 7k и плотностью состояний для ряда нитридов, а также твердых
растворов NbC—NbN (рис. 13). Наилучшие результаты дает корреля-
ция 7’к с теоретическими кривыми плотности состояний, полученных
Бильтцем, а также Эрном и Свитендиком. Авторы предположили, что
главная роль в формировании свойств веществ принадлежит сильно
локализованным связям между атомами металла и неметалла, т. е.
ковалентным связям.
При изменении содержания азота в области гомогенности Тк изме-
няется, причем для нитрида циркония — монотонно (рис. 14) [275, 1069],
а для нитрида ниобия максимальная Тк отвечает нестехиометрическому
для нитрида ниобия максимальная Тк отвечает нестехиометрическому
составу NbN (рис. 15). Уменьшение 7’к при снижении содержания азота
в области гомогенности или определенной ее части согласуется с умень-
шением 7’к в нитридных фазах при снижении в них содержания азота
31
(например, MoN и M02N). Это объясняется усилением связей Me—Me
и локализацией на них подавляющей части валентных электронов с со-
ответствующим сокращением концентрации электронов проводимости и
появлением энергетических разрывов. В частности, нитрид хрома явля-
20- M>N
N(0) -2-1 0 1 Е,эв
ZrC(4,5) ToC(6) NbC(7) NbNfS) d - эл./omo"
Рис. 13. Изменение критической температуры перехода к
сверхпроводимости Тк в ряду ZrB—ZrC—NbC—NbN—
CrN:
1 — расчетные кривые изменения плотности состояний; 2 — по Биль-
цу; 3 — по Эрну и Свитенлику; 4 — по Коста и Конте; 5 — по Па-
перу; 6 — по Демпсею.
ется полупроводником. Энергетические разрывы достигают больших
значений в нитридах тантала, вольфрама, а также полунитридах Nb2N
и Mo2N. В случае изменения 7\ в области гомогенности нитрида нио-
32
бия снижение Т\ начиная с некоторых содержаний азота к стехио-
метрическому составу (в области от 0,95 до 1,00 ниобия в NbN) может
быть объяснено появлением при этих содержаниях азота сильных свя-
зей Me—Me и N—N и началом образования энергетической щели.
Авторы [1001] исследовали концентрационную зависимость Тк в
областях гомогенности нитридов титана TiNi-x, ванадия VNi-х, опре-
делили значение Тк для мононитрида иттрия. Значения Тк в областях
Рис. 14. Изменение точки перехода к сверхпроводимости в
области гомогенности ZrN.
Рис. 15. Изменение точки перехода к сверхпроводимости в
области гомогенности NbN.
гомогенности нитридов титана и ванадия, как и для нитридов циркония
и ниобия, понижаются с уменьшением содержания азота.
Магнитные свойства. Первое исследование магнитных свойств нит-
ридов некоторых переходных металлов было проведено в [829]. Показан
довольно слабый парамагнетизм нитридов титана и циркония (ум для
T1N + 48-10-6, для ZrN + 60 • 10~6). Установлено, что нитрид хрома яв-
ляется ферромагнетиком [331], нитриды Mn4N, MnsN2, Fe4N — сильными
парамагнетиками с эффективным магнитным моментом соответственно
1,2; 3,94; 2,22 магнетонов Бора [140], нитриды урана — очень слабыми
магнетиками с магнитной восприимчивостью: UN+7,73; U2N3+13,17
3-272 33
[508]. В ряду мононитридов лантаноидов
возрастает [478]:
LaN CeN PrN
4-60 4-296 4-4460
магнитная восприимчивость
NdN
4-5850
При растворении карбида титана в нитриде титана резко умень-
шается магнитная восприимчивость сплава [830], как это показано на
рис. 16.
Итак, в настоящее время не накоплен сколько-нибудь значительный
экспериментальный материал по магнитным свойствам нитридов пере-
Рис. 16. Магнитная восприимчи-
вость сплавов TiN—TiC.
Термические свойства.
ходных металлов и не предложено каких-
либо интерпретаций имеющихся данных.
Можно лишь отметить, что парамагнетизм
нитридов лантаноидов возрастает с повы-
шением статистического веса стабильных
^-конфигураций атомов лантаноидов. Умень-
шение магнитной восприимчивости нитрида
титана при растворении в нем карбида мо-
жет быть связано с понижением статисти-
ческого веса э2р6-конфигураций и возрас-
танием статистического веса $р3-конфигу-
раций при одновременной достаточно силь-
ной делокализации валентных электронов
и титана и азота.
Некоторые данные по магнитным свой-
ствам нитридов урана и плутония при низ-
ких температурах приведены в [831].
Выше показано, что для большинства нит-
ридов превалирующую роль играет электронная составляющая тепло-
проводности, за исключением нитридов хрома, тантала и ванадия, у
которых вследствие значительной локализации валентных электронов
на ковалентных связях превалирует решеточная составляющая тепло-
проводности.
Термическое расширение нитридов переходных металлов исследо-
вали в [781, 225] (табл. 12).
Довольно высокие значения коэффициента термического расшире-
ния нитридов мало отличаются от значений коэффициента термиче-
ского расширения соответствующих переходных металлов [781]. Это
говорит о том, что силы связи в нитридах типа MeN незначительно пре-
вышают силы связи в переходных металлах и в 2—3 раза меньше силы
связей в соответственных карбидах. Очевидно, что ответственна за это
разрыхляющее действие нелокализованная часть валентных электронов.
Наблюдается, хотя и не четко выраженное, уменьшение коэффициента
34
термического расширения нитридов с ростом локализации валентных
электронов в ^-конфигурациях металла и «^-конфигурациях азота.
Термическая устойчивость нитридов при высоких температурах ис-
следовалась во многих работах. На рис. 17 приведены, по данным [833],
расчетные кривые упругости диссоциации нитридов при разных темпе-
ратурах. Авторы [834] исследовали диссоциацию нитридов в вакууме
10~4 мм рт. ст., результаты показаны на рис. 18. Подробный обзор.
устойчивости нитридов в ваку-
уме приведен в моногра-
фии [835].
Исследования испарения
нитридов в вакууме показали,
что нитриды диссоциируют на
металл и азот
MeN(TB) = Me(TB) + yN2. (1,1)
В работе [203] определяли
скорости испарения и давле-
ния паров над нитридами ти-
тана и циркония. При темпе-
ратурах 2000—2240° К ско-
рость испарения титана дости-
гает значительной величины,
поэтому давление диссоциа-
ции расчитывали по схеме
TiN(TB) = Tira3 {-|n2. (1,2)
Нитрид циркония в тех
же условиях диссоциирует по
схеме (I, 1). Данные по ско-
рости испарения и давлению
Таблица 12
Термическое расширение нитридов переходных,
металлов
Нитрид Температурный интервал, °C Коэффициент термического расширения ало*, град”1 Порис- тость образ- ца, % Литера- тура
TiN 20—270 8,87±0,4 .— [7811
7i^o,98 20—1100 9,5 11 [225]
ZrN 20—270 10,2±0,5 — - [7811
ZrNo,92 20—1100 7,24 4,4 [225]
HfNo,86 20—1100 6,9 3,1 [225]
VN 17—190 8,35±0,74 — [78Ц*
VNo,33 20—1100 8,1 10 [225]
VNo,93 20—1100 9,2 4,5 [225]
NbN 20—270 10,1±0,2 — [781]*
NbNo.5 20—1000 3,26 9,2 [225]
NbNi.o 20-1000 Ю,1 3,7 [225]
TaNo,45 20—1000 5,2 5,4 [225]
TaNi.oi 20—790 3,6 7,8 [225]
CrNo,49 20—1100 9,41 6,1 [225]
CrN0,92 20—800 2,3 lo Q [225]
850—1040 7,5 [225]
M°N0_5 20—790 4,5 [225]
20—1100 6,2 [225]
Загрязнены углеродом.
диссоциации TiN и ZrN при-
ведены в табл. 13.
Из табл. 14 [74] видно, что диссоциация нитридов затрудняется с
уменьшением локализации валентных электронов в стабильные конфи-
гурации и существенно облегчается при сильной локализации, дохо-
дящей до появления энергетических разрывов между атомами метал-
лов и азота. Для полупроводниковых нитридов хрома, молибдена и
вольфрама упругость диссоциации наибольшая при наименьших темпе-
ратурах. Обращает внимание относительно высокая упругость диссо-
циации нитрида ванадия, которая может быть сопоставлена с низким
3*
35
термическим коэффициентом электросопротивления этого нитрида, отно-
сительно высокой температурой перехода к сверхпроводимости и други-
ми характеристиками, указывающими на высокий статистический вес
стабильных конфигураций атомов ванадия и азота в этом соединении и
на малый вес нелокализованной части валентных электронов.
Оптические свойства. Оптические свойства нитридов переходных
металлов изучены мало. Имеющиеся данные [340] по коэффициенту
Рис. 17. Расчетные кривые упругости диссоциации нитридов [833]
Рис. 18. Поведение некоторых нитридов в вакууме.
излучения нитридов при длине волны излучаемого света Z.=650 ммк
приведены в табл. 15.
Диффузия азота в переходные металлы. Обязательным условием
диффузии является образование в результате диффузионного процесса
системы, более устойчивой в энергетическом отношении, чем смесь ис-
ходных компонентов, т. е. обладающей меньшей свободной энергией.
Это условие может быть выполнено, если в результате диффузии воз-
растает статистический вес атомов, обладающих стабильными конфи-
гурациями локализованной части валентных электронов. При диффу-
зии азота возможна как передача его атомами одного электрона ато-
мам металла с образованием «/^-конфигурации либо прием атомами
азота валентных электронов металла с образованием «^-конфигура-
ции. Так как часть атомов азота может отдавать легкоподвижный
электрон, то энергия активации диффузии азота в переходные металлы
несколько выше, чем энергия активации при диффузии в те же металлы
36
углерода, но ниже, чем бора. Это объясняется тем, что в последнем
случае наиболее энергетически выгодным состоянием атомов бора яв-
ляется их объединение с освобождением части электронов и образова-
нием .^-конфигураций (табл. 16).
Скорость диффузии обычно возрастает с повышением энергии ак-
тивации, так как высокие значения энергии активации соответствуют
Таблица 13
Скорость испарения и давление диссоциации
TIN nZrN
TIN ZrN
«а к
• N о X • ’я
ЙЕ 23 ЙЕ
Ьм а. § ь» (STS-
1987 1,510 0,650 2236 0,534 0,915
2017 1,972 0,854 2259 1,299 2,397
2050 3,129 1,366 2318 2,979 5,583
2058 3,360 1,469 2333 2,714 4,977
2155 15,963 7,146 2344 3,020 5,536
2157 20,510 9,178 2451 14,981 28,96
2212 33,254 15,08 2466 16,127 31,04
2241 70,555 32,21
образованию на поверхности не очень
высокого статистического веса атомов со
стабильными конфигурациями и относи-
тельной легкости их нарушения для про-
должения и развития процесса диф-
фузии.
Таблица 14
Упругость диссоциации
нитридов [74]
Нитрид Температура (°C) при давлении
1 мм pm. cm. 10-1 мм pm. cm. 10-2 1 мм pm. cm. 10—3 мм pm. cm.
ScN 1880 1734 1607 1495
YN 1990 1838 1704 1587
TIN 2912 2689 2489 2331
ZrN 2603 2390 2205 2046
VN 1242 1127 1024 933
NbN 1946 1770 1620 1491
TaN 1921 1746 1599 1460
CrN 719 646 582 533
Mo2N 329 283 243 209
w2n 344 296 256 221
Th3N« 2401 2206 2037 1890
u3n4 2267 2070 1950 1753
LaN 2010 1855 1721 1602
CeN 2196 2029 1884 1756
Практически при низких температурах образуются твердые раст-
воры азота в металлах, при более высоких температурах диффузия
приобретает реакционный характер, т. е. сопровождается образованием
нитридных фаз, что часто используется для создания нитридных покры-
тий на металлах.
Прочностные свойства. Прочность нитридов переходных металлов
при обычных и высоких температурах исследована мало [140, 147, 851].
Между микротвердостью и множителем уравнения Линдемана, опреде-
ляющим частоту колебаний линейных осцилляторов, имеется известная
корреляция (рис. 19) [852]. Это понятно, исходя из близости процессов,
происходящих при плавлении и измерении твердости, на что в свое
время указывалось Борном, а в применении к фазам внедрения—в [852,
853]. В работе [858] сделана попытка связать твердость с энергией ато-
37
Таблица 15
Коэффициенты излучения нитридов переходных металлов
Нитрид 800 °C 900°C 1000 °C 1100°C 1200°C 1300°C 1400 °C 1500°C 1600°C 1700°C 1800°C 1900°C 2000°C
TiN 0,82 0,81 0,81 0,80 0,80 0,80 0,79 0,79 0,79 0,79 0,78 0,78 0,78
ZrN 0,73 0,73 0,73 0,74 0,74 0,75 0,75 0,75 0,75 0,75 0,76 0,76 0,76
HfN 0,84 0,84 0,84 0,84 0,84 0,84 0,84 0,84 0,84 0,84 0,84 0,84 —
V3N 0,82 0,82 0,82 0,82 0,82 0,82 0,82 0,82 0,82 — — —
VN 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77 0,77
Nb2N 0,82 0,82 0,82 0,82 0,82 0,82 0,82 0,82 0,82 0,82 — — —
NbN 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83
Ta2N 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 0,83 — —
TaN 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79 0,79
Cr2N 0,69 0,69 0,69 0,69 0,69 0,69 0.6Э 0,69 0,69 — — — —
CrN — — — 0,60 0,58 0,55 0,53 0,51 0,48 0,46 0,44 0,42 0,38
мизации полными и свободными удельными поверхностными энергия-
ми. Твердость низших нитридных фаз существенно выше твердости
мононитридов, что может быть
Таблица 16
Энергия активации при диффузии
некоторых неметаллов в переходные
металлы, кдж)моль [850]
Ме- талл Бор Угле- род Азот Кис- лород
Ti 128,12 79,97 141,51 140,26
Zr 144,44 171,66 164,12 124,77
Hf — — 238,65 233,83
Nb 247,02 133,98 161,6 115,87
Ta 200,97 104,67 184,22 121,42
Cr — 117,23 43,63 —
Mo 184,4 347,5? —
W — 257,49 — —
объяснено по аналогии с представле-
ниями [854, 781] существенным усиле-
нием связей Me—Me при уменьшении
относительного числа внедренных ато-
мов азота в фазах предельного со-
става. В работе [147] показано, что с
уменьшением содержания азота в пре-
делах области гомогенности нитрид-
ных фаз переходных металлов твер-
дость понижается. Уменьшение про-
исходит в большем темпе, чем для
карбидов, что объясняется появлени-
ем у нитридов доли ионной связи,
увеличивающейся с понижением со-
держания азота в пределах области
гомогенности, т. е. с увеличением
локализации электронов на связях
Me—Me и с возрастанием статистического веса «2ре-конфигураций ато-
мов азота. Уменьшение твердости сопровождается появлением и ростом
энергетического разрыва между состояниями атомов металла и азота,
что внешне проявляется в росте ширины запрещенной зоны.
Для фаз предельного состава по содержанию азота твердость за-
кономерно уменьшается с повышением веса стабильных конфигураций
атомов переходных металлов (рис. 20). При этом происходит пониже-
ние статистического веса «2р6-конфигураций и повышение статистиче-
ского веса «реконфигураций атомов азота, т. е. одновременное повы-
38
шение симметрии распределения электронной плотности, чему всегда
отвечает понижение твердости [851]. Необходимо отметить, что фактор
симметрии более сильный, чем фактор веса нелокализованных элект-
ронов, поэтому, например, для нитрида титана, несмотря на большой
вес нелокализованных электронов и вызываемое ими сильное разрых-
Рис. 19. Соотношение между числами микротвердости нитридов переходных
металлов и множителя уравнения Линдемана.
Рис. 20. Зависимость микротвердости нитридов переходных металлов от кри-
терия
ление решетки, твердость все же наиболее высокая из-за резкой асим-
метрии распределения локализованных электронов.
Наличие относительно высокого веса нелокализованных электро-
нов в нитридах переходных металлов, участвующих в статистическом
обмене с локализованными электронами, обусловливает не только их
меньшую твердость, чем других аналогичных соединений (карбидов,
боридов, силицидов), но и меньшую хрупкость [855, 856]. В данном слу-
чае это совершенно аналогично снижению хрупкости и повышению
пластичности в ряду С (алмаз) — Si—Ge—Sn (белое) — Pb. В
этом ряду уменьшается локализация валентных электронов на стабиль-
ных «/^-конфигурациях, обусловливающих жесткие, направленные свя-
зи, и увеличивается вес нелокализованных электронов, обеспечиваю-
щих связь с локализованными электронами, входящими в стабильные
конфигурации и, следовательно, с остовами атомов. Иначе говоря, у
нитридов переходных металлов большая способность узлов кристалли-
ческой решетки к перемещению при деформировании без разрыва меж-
атомных связей, т. е. без разрушения материала.
Проведенное нами совместно с В. В. Стасовской изучение абразив-
ной способности некоторых нитридов показало, что абразивная способ-
ность уменьшается пропорционально уменьшению твердости, довольно
заметно уступая при этом абразивной способности карбидов и особен-
но боридов тех же металлов [1068].
Нитриды переходных металлов разрушаются ультразвуком [776].
39
Нитриды неметаллов. Соединения неметаллов с азотом ха-
рактеризуются преимущественно ковалентным типом химической связи и
соответствующими этому типу физическими свойствами неметалличе-
ского или полуметаллического характера. Наибольший интерес пред-
ставляют соединения с азотом неметаллов и полуметаллов III и IV
групп. Соединения переходных металлов III группы с азотом являются
полупроводниковыми соединениями типа AmBv. При их образовании
атомы компонентов А(А = В, Al, Ga, In, Т1), имеющие в изолированном
состоянии х2р-конфигурацию валентных электронов, приобретают более
устойчивую «реконфигурацию в результате з->р-перехода, а затем, при-
соединяя к себе электрон атома азота — квазистабильную зр3-конфигу-
рацию, в то время как атом азота, так-
же теряя электрон, приобретает sp3-
конфигурацию, [857]. Полупроводники
типа AniBv аналогичны по структуре
и свойствам полупроводникам группы
углерода (алмаза). Очевидно, при этом
наиболее стабильными конфигурация-
ми партнеров А и В должен обладать
кубический нитрид бора—боразон, что
и вызывает большую ширину его за-
прещенной зоны, высокую твердость и
температуру плавления, а обособлен-
ность стабильных конфигураций —
диспропорционирование при нагреве и
инконгруэнтное плавление. При заме-
не бора алюминием и т. п. при неиз-
менном втором компоненте азоте,
закономерно понижаются твердость,
Таблица 17
Физические свойства нитридов
переходных металлов III группы
периодической системы
Нит- рид Кристалличес- кая структура Темпе- ратура плав- ления, °C Шири- на зап- рещен- ной зо- ны, эв Твер- дость по Моо- су
BN Кубическая типа сфа- лерита 3000 ~10 10
A1N Гексагональ- ная типа вюртцита 2200 3,8 9
GaN То же 1500 3,25 —
InN » » 1200 — —
температура плавления и ширина за-
прещенной зоны (табл. 17). При этом уменьшается энергетическая
обособленность конфигураций атомов; так, если BN при нагревании раз-
лагается, то GaN испаряется молекулярно [616]. Все элементы А, кроме
бора, образуют с азотом соединения, обладающие не решеткой сфале-
рита, а вюртцита, по-видимому, вследствие резкого различия в энерге-
тической стабильности соответственных электронных конфигураций. Это
приводит к акцепторной роли азота с достройкой его электронной
«2р3-конфигурации до стабильного состояния s2p6- образования
высокого статистического веса этих состояний обоими атомами
(А и В).
Имея в виду более высокую стабильность 52р6-конфигураций по
сравнению с sp3, понятно, почему соединения со структурой вюрицита
имеют большую ширину запрещенной зоны по сравнению с соединения-
40
ми, кристаллизующимися в форме сфалерита, а также относительно
высокие значения температур плавления и твердости (AIN, GaN, InN).
При переходе от азота к фосфору резкое снижение стабильности
возможной «2р6-конфигурации приводит к отдаче «2р6-конфигурацией
компоненту А одного электрона, образованию «реконфигураций обо-
ими атомами и соответственно — структуры сфалерита.
Соблюдается четкая корреляция между микротвердостью и шири-
ной запрещенной зоны полупроводников AinBv (рис. 21), а также кор-
реляция между температурой плавления и микротвердостью (рис. 22).
Обращает внимание, что плавное понижение твердости соединений бо-
ра, алюминия и индия нарушается несколько более высоким ходом
микротвердости ряда соединений галлия, т. е. относительно меньшей
Рис. 21. Соотношение между микротвердостью и шириной запрещен-
ной зоны полупроводников типа AnlBv.
Рис. 22. Соотношение между микротвердостью и температурой плав-
ления полупроводников типа ArnBv.
ширине запрещенной зоны соединений AniBv галлия соответствуют
относительно более высокие значения микротвердости, чем для соеди-
нений других металлов А. Подобное же отступление наблюдается и для
температур плавления соединений галлия, которые имеют относительно
высокие значения, также как и значения микротвердости. Повышение
твердости и температуры плавления может быть связано с появлением
для этих соединений наряду с «реконфигурациями также определенно-
го статистического веса конфигураций типа «2р6. Это, по-видимому, дол-
жно было бы усиливаться для соединений индия. Однако вследствие появ
41
ления у индия полностью вакантной 4[-оболочки возможно нарушение sp-
конфигураций в результате частичных переходов валентных электронов
на ^-состояния. Для алюминия образование х2р6-конфигураций наиболее
полно выражено для A1N, как уже указывалось выше, в соединениях
же алюминия с Р, As, и т. п. переход электронов от А1 к фосфору и
другим компонентам В затруднен в результате относительно близких
энергетических состояний sp-электронов обоих компонентов. Для гал-
лия, имеющего более низкое энергетическое состояние sp-электронов,
возобновляются переходы от него электронов к компоненту В. Таким
образом, возможно образование атомами компонентов одновременно и
sp3- и э2р6-электронных конфигураций, соотношение статистических
весов которых может колебаться в широких пределах —• от практически
100% конфигураций sp3 (и образованием структуры сфалерита) до
практически 100% конфигураций s2p6 (с образованием структуры вюрт-
цита). Поэтому для атомов галлия в его соединениях типа AniBv воз-
можен высокий статистический вес э2р6-конфигураций, можно ожидать
для этих соединений при определенных условиях полиморфных пре-
вращений.
3. ТЕРМОДИНАМИКА НИТРИДОВ *
Термодинамические свойства нитридов приведены в справочниках и
обзорных работах [47, 112, 862]. Поэтому в настоящем разделе ос-
новное внимание обращено на относительную термодинамическую ус-
тойчивость нитридов в различных средах, определение температурного
диапазона термодинамической устойчивости и равновесных значений
парциального давления азота над нитридами, возможность осущест-
вления реакций между нитридами и другими соединениями, а также
на оценку ожидаемых свойств продуктов реакций.
В табл. 18 и 19 приведены термодинамические свойства некоторых
нитридов, а на рис. 23 представлена зависимость изобарно-изотермиче-
ских потенциалов реакций образования нитридов от температуры.
Расчет теплоемкости некоторых нитридов. В процессе окисления
тугоплавких соединений аналогично тому, как это наблюдается при
окислении металлов и сплавов, образуется многослойная окалина, со-
стоящая из нескольких окисных фаз, между которыми при высоких
температурах возможно протекание разных обменных реакций, значи-
тельно изменяющих состав окалины и условие диффузии через нее
компонентов реакции.
Выяснение термодинамической стабильности' тугоплавких соедине-
ний в различных средах и определение термодинамической возможности
* Раздел написан Р. Ф. Войтович.
42
Рис. 23. Зависимость изобарно-изотермического потенциала реакций образова-
ния нитридов от температуры.
протекания окислительно-восстановительных реакций в слое окалины
состоит в определении изобарно-изотермических потенциалов соответ-
ствующих реакций при разных температурах.
Эти величины могут быть рассчитаны несколькими методами, из
которых наибольшего внимания заслуживает метод определения тер-
модинамических величин, в котором используют значение теплоемкос-
тей Ср при различных температурах, теплот образования АЯ^98 и
абсолютной энтропии S°29g компонентов реакции в стандартном состоя-
нии [863], а также метод, в основе которого лежит использование при-
веденных термодинамических потенциалов [864].
43
Термодинамические
Соединение 0 л^298 _ 0 д298 Фаза 1 •da’du2
кдж/моль ккал/моль । дж/моль град кал/моль • град
Li3N 196,78 47,0 Твердая Разлагается
K3N 73,69 17,6 .— .— В ___
Cu3N —74,53 — 17,8 .— — в Разлагается
Ag3N —255,39 —61,0 .— — в В
^е3^2 563,96 134,7 50,24 12,0 в 2473
— —. — .— Жидкая Разлагается
MgsN2 461,80 110,3 87,92 21,0 Твердая (а) 823
— — — — Твердая (₽) 1061
— —— — Твердая (у) Разлагается
CaCN2 351,69 84,0 — — Твердая
Ca3-^2 439,61 105,0 106,34 25,4 В 1468
Sr3N2 391,05 93,4 123,51 29,5 В 1303
Ba3N2 363,83 86,9 152,40 36,4 в Разлагается
^п31^2 22,19 5,3 — —. в В
Cd3N2 — 161,61 —38,6 •— .— в в
ScN 284,70 68,0 37,68 9,0 в 2923
YN 299,36 71,5 46,05 11,0 в
LaN 299,36 71,5 — »
CeN 326,57 78,0 48,99 11,7 в
SmN 314,01 75,0 — —. Твердая
GdN 314,01 75,0 — .— В
DyN 314,01 75,0 —- — в
ErN 314,01 75,0 —. в
YbN 314,01 75,0 —. в
LuN 314,01 75,0 — —. в
Th3N4 1297,91 310,0 178,78 42,7 в 2373
UN 286,80 68,5 75,36 18,0 в 2903
891,79 213,0 121,42 29,0 в
PuN 32,66 7,8 в
TiN 336,62 80,4 30,31 — »
ZrN0,56 218,97 52,3 39,36 9,4 в —
Zr3N2 22,19 5,3 — — в
Zr^0,69 268,37 64,1 40,61 9,7 в —
ZrN0,74 281,77 67,3 40,61 9,7 в —
Zr^0,89 319,45 76,3 38,93 9,3 в —
ZrN 365,51 87,3 38,93 9,3 в 3223
HfN 369,44 88,24 54,85 13,1 в 3255
VN 251,21 60,0 37,26 8,9 в 2323
Nb2N 255 61,1 — — в —•
44
Таблица 18
свойства нитридов
—Л^превр
кдж/моль
Теплоемкость Ср=а-[-ЬТ-[-сТг
0,46
0,92
19,324
! ккал/моль а ь-ю* с-10 -5
\ дж/моль-град кал/моль-град 1 ! дж/моль-град 1 1 кал/моль -град дж/моль-град кал/моль-град
—. 49,11 11,73 96,30 23,0 — —
— —. —
— 90,85 21,7 — — — —
— — —— — —
— 30,65 7,32 128,95 30,8 — —
— - ——- — —
0,11 86,96 20,77 46,892 11,20 — —
0,22 84,029 20,07 44,631 10,66 —
28,50 — — — — — —
—- — — —— ___
— 85,58 20,44 92,11 22,0 — —
— — — — — — —
— — —
— 79,55 19,0 94,20 22,5 — —
— — — —— — — —
— — — — — — —
— — __ — __
— 46,47 11,1 — —
— 46,89 11,2 — —. — —
— — — — — — —_
-— — — — — — —
— — — —- — — —
— — — — — — —
— — — — — — —
— — — —
— 116,309 27,78 133,14 31,8 — —
— 44,38 10,6 29,31 7,0 — —
— — — — — — —
— — — — — — —
— — — — —• — —
— — — — — — —
— 90,602 21,64 108,857 26,00 — —
— — — — — — —
— — — — — — —
— — — — — — —
— 46,473 11,10 7,034- 1,68 —7,201 —1,72
—• 41,20 9,84 9,29 2,22 —.
— 45,804 10,94 8,79 2,1 —9,253 —2,21
—• — — — — — —
Темпера-
турный нн
тервал, “К
298—800
273—373
298—800
298—823
823—1061
1061—1300
298—800
273—700
298
298
298—800
298—3000
298—1700
298—2000
298—1600
45
Соединение д//298 s 0 6298 Фаза о А А <Р С
1 кдж/моль i ккал/моль дж/моль-град кал/моль-град
NbN 237,81 56,8 43,96 10,5 Твердая 2323
Ta2N 270,89 64,7
TaN 247,02 59,0 51,08 12,2 3363
Cr2N 105,51 25,2 75,36 18,0
CrN 118,07 28,2 33,49 8,0 » Разлагается
Mo2N 69,50 16,6 87,92 21,0
w2n 72,01 17,2 »
Mn3N 130,63 31,2 »
Mn25N 115,56 27,6 — — Твердая
Mn5N2 201,80 48,2 192,17 45,9 »
Mn3N2 — .—
Re2N —4,19 —1,0 »
Fe8N 11,30 2,7
Fe4N 10,89 2,6 156,17 37,3 943
Fe2N 3,77 0,9 101,32 24,2
Co3N —8,37 —2,0 — — » Разлагается
№3N —0,84 —0,2 »
BN 254,14 60,7 15,37 3,67 » 3273
— —-• — — Жидкая 5340
AIN —379,32 —90,6 86,96 20,77 Газообразная
320,29 76,5 20,93 5,0 Твердая Разлагается
GaN 104,67 25,0 46,47 И,1 »
InN 19,26 4,6 42,71 10,2
Si3T\j4 749,44 179,0 96,30 23,0 » 2173
65,31 15,6 167,47 40,0 » Разлагается
PN 1 —84,57 —20,2 211,18 50,44 Газообразная —
~(PN)n 70,34 16,8 — — Твердая
p3n5 316,94
AsN SbN —29,31 —312,75 —7,0 —74,4 — — Газообразная » __
Значения последних для многих соединений и элементов в газооб-
разном и конденсированном состояниях сведены в специальных табли-
цах [864]. Последний метод чрезвычайно прост, так как состоит в
непосредственном расчете изобарно-изотермического потенциала Д-Z®,
реакций по известным данным теплот образования и приведенных тер-
модинамических потенциалов компонентов реакции.
46
Продолжение таблицы 18
ЬНпревр Теплоемкость Ср—а~ЬТ+сТ2 Температурный интервал, °К
С О X ккал/моль а Ь105 с-10—5
"о л» л о кал/моль-град 'а & л о кал/моль-град док/моль град кал/моль-град
. 36,383 8,69 22,609 5,40 — — 298—600
.— — —- — — —
. 32,364 7,73 32,657 7,80 — — 298—800
63,81 15,24 28,47 6,8 — — 273—800
41,20 9,84 16,33 3,9 — — 273—800
— — 46,85 11,19 57,778 13,80 — — 298—800
92,779 22,16 127,70 30,5 — — 298—800
— 127,907 30,55 160,773 38,40 — 298—800
— — 94,203 22,5 94,20 22,5 — — 298—800
— 112,37 26,84 34,16 8,16 — — 273—1000
— — 62,43 14,91 25,50 6,09 — 273—1000
. — 7,62 1,82 15,16 3,62 — — 298—1200
— 30,14 7,20 4,44 1,06 — 1,76 —0,42 298—2500
— — 22,90 5,47 32,66 7,80 — — 293—900
— — 70,464 16,83 98,81 23,6 — — 298—900
— — 31,150 7,44 3,768 0,90 —2,303 —0,55 298—2000
— — — — — — — — —
149,05 35,6 — — .— 298
33,66 8,04 2,26 0,54 —3,48 —0,83 298—2000
— — 34,583 8,26 . 1,758 0,42 —3,601 —0,86 298—2000
Однако до настоящего времени значения приведенных термодина-
мических потенциалов имеются лишь для нескольких нитридов и кар-
бидов, что исключает возможность использования метода для широкого
класса соединений. Поэтому расчет AZ°7. реакций с участием боридов
и силицидов, а также большей части карбидов, нитридов и сульфидов
47
-272
Таблица 19
Термодинамические потенциалы образования нитридов
Реакция -az»2„ AZT=a+67TgT+cT Температур- ный интер- вал, °К
a i>10‘ clO"
кдж/моль ккал/моль кдж/моль ккал/моль кдж/моль ккал/моль кдж/моль ккал/моль
3F'(tb)+1/ 2^2(Г) = — — 198,75 —47 ,47 142,35 34,0
=LisN(TB) ЗВе(тв)+^2(г)= 513,306 122,601 —563,962 —134,7* 298—1000
169,984 40,6
=Be3N2(TB)
3Mg(TB)+N2(r)= =Mg3N2(TB)a 399,722 95,472 —458,873 —109,6 — — 198,496 47 41 298—923
3^®(ж)+^2(г)= — — —485,25 —115,97 — — 227,343 54,3 923—1061
=Mg3N2(TB)p ЗМ£(ж)+М2(Г)= — — 484,622 —115,75 1061—1300
226,464 54,09
^S3N2(TB)^
3Ca(TB)+N2(r);= 377,231 90,1 —439,614 —105,0 — — 209,34 50,0 298—1000
=Ca3N2(TB) 3Sr(TB)+N2(r) = — — —382,67 —91,4 .
213,11 50,9
=Sr3N2(TB) 3^а(тв)+^2(г)= 292,636 69,895 —364,252 —87,0 298—1000
240,322 57,4
=Ba3N2(TB) ^(тв)"!"1/2^2(г)= 270,677 64,65 —301,868 —72,1
104,67 25,0 298—1000
=LaN(TB) C^TB)-!"1/2^2(r) = 295,379 70,55 —326,57 —78,0
104,67 25,0 298—1000
=CeN(TB)
3Th(TB)+2N2(r)== 1187,665 283,669 —1299,583 —310,4 — — 375,556 89,7 298—2000
=lh3N4(TB) U(TB)+1/sN2(I.)= 259,971 62,093 —286,796 —68,5 90,016
21,5 298—2000
=UN(TB)
3Цтв)+2^2(г)= — — 1151,4 —275,0 — — 342,9 81,9
"~~Цз^4(тв)
T’(TB)a+1/4N2(r)= 308,291 73,634 —335,991 —80,25 — — 92,947 22,2 298—1155
=™<tb)
TitTBlg+V г^2(г)— — —338,503 —80,85 — — 95,375 22,78 1155—1500
=™(TB) 80,353 —364,252 —87,0 93,387 22,305 298—1135
Zr(TB)a+1/' 2N2(r)= 336,422
=ZrN(T0j Zr(TB)p+1^N2(r)= — — — — -— — — — —
=ZrN(TB) Hf(TB)+V 2^2(г)— 340,806 81,4 —340,806 —81,4 — — — — 298
=HfN(TB) ^(TBj+’/^r)— 223,797 53,453 —251,208 —60,0 —7,327 —1,75 110,113 26,3 298—2000
=VN(TB) Та(тв)+^ 2N2(r)— 218,183 52,112 —246,603 —58,9 —28,889 —6,9 166,844 39,85 298—2240
=TaN(TB) 2Сг(тв)+1'/2^2(г)== 85,227 20,356 —108,647 —25,95 —24,074 —5,75 138,164 33,0 298—1400
=Cr2N(TB) Сг(тв)+1/2^2(г)= 94,287 22,52 —127,28 —30,4 —19,26 —4,6 158,39 37,83 298—1300
=CrN(TB) V gC^N^gj+V 4N2(r)= 49,915 11,922 —66,989 —16,0 —12,037 —2,875 87,085 20,8 298—1400
=CrN(TB) 2Mo(TB)+1'/2N2(r)= 50,095 11,965 —72,013 —17,2 —19,259 —4,6 121,208 28,95 298—1500
=Mo2N(tb) 5Mn(TB)+N2(r)= — — —69,501 —241,87 —16,6 —57,77 — — 87,923 152,4 21,0 36,4 1500—2000
=Mn5N2(r) ЗМп(ж)+И2(Г)= — — —191,76 —45,8 — — 149,47 35,7 —
=Mn3N2(TB) 4Fe(TB)+1/2N2(r)= —4,032 —0,963 —0,837 —0,2 48,651 11,62 —104,042 —24,85 298—950
=Fe4N(TBj А1(тв)+1/2^2(г)= 294,621 70,369 —322,384 —77,0 — — 93,156 22,25 298—923
=A1N(TB) А\ж)+1^2(г)— — — —265,862 —63,5 — — 115,137 27,5 1800—2000
=A1N{TB) B(TB)+1/2N2(r) = — — —108,857 —26,0 — — 40,612 9,7 1200—2300
=BN(TB) В(ж)+’^2(Г)= — — —656,909 —156,9 — — 223,994 53,5 2500—3000
=bn(tb) 3Si(TB)+2N2(r)— 653,312 156,041 —753,624 —180,0 — — 336,619 80,4 298—1680
=Si3N4(TB) =Si3N4(TB) — — —893,463 —790,468 —213,4 —188,8 — — 419,517 412,4 100,2 98,5 1680—1800 1800—2000
возможен лишь первым методом при известных уравнениях температур-
ной зависимости теплоемкости.
Для большего числа тугоплавких соединений в литературе отсут-
ствуют соответствующие значения степенных рядов теплоемкости. По-
этому были рассчитаны значения теплоемкостей нитридов простейшего
типа (табл. 20), используя метод Дебая [863].
Значения теплоемкостей нитридов при постоянном объеме (Су)
определяли интерполированием табличных значений функций Дебая
3CD для значений рассчитанных величин & /Т при всех взятых для рас-
чета температурах.
Характеристическую температуру элемента определяли по фор-
муле Линдемана
8=135/
где 7ПЛ — температура плавления элемента, М — атомный вес, V —
атомный объем.
Согласно теоретическим представлениям, развитым в [863, 865],
характеристическую температуру для элемента в соединении рассчи-
тывали по формуле
0' = 0
где 0 — характеристическая температура свободного элемента, Т'пл и
Т’пл — соответственно абсолютная температура плавления соединения
и элемента.
Значения температур плавления Тпл и молярных (атомных) объе-
мов V металлов, азота и нитридов, а также значения рассчитанных ха-
рактеристических температур представлены в табл. 20.
Используя величины 0' и суммируя найденные по графику
/ 0 \
3CD=fy—J значения атомных теплоемкостей, определяли теплоемкость
Cv тугоплавких соединений при данной температуре. Переход к значе-
ниям теплоемкостей при постоянном давлении осуществлялся с по-
мощью приближенной формулы
Ср = Cv fl + 0,0214 Cv
Опытные значения Ср для карбида титана сопоставляли со значе-
ниями, рассчитанными по описанной выше методике. Совпадение тео-
ретических и экспериментальных значений Ср оказалось удовлетвори-
тельным (рис. 24). В области высоких температур, выше 673° С, наблю-
далось полное соответствие рассчитанных и экспериментальных дан-
ных, наблюдаемое максимальное расхождение в области температур
273—473° К составляло около 5% Ср.
50
Рассчитанные значения теплоемкостей Ср для перечисленных в
табл. 20 нитридов при различных температурах приведены в табл. 21.
а на рис. 24 графически показана эта зависимость для нитридов. Тем-
пературная зависимость этих величин может быть представлена урав-
нением
Ср = а ф ЬТ-10~3Т — с.105-Т~2.
В табл. 22 представлены значения констант уравнений температур
ной зависимости теплоемкости нитридов,
Таблица 20
Таблица 21
Характеристические температуры
некоторых элементов и соединений
Характеристи ческая
температура, °К
Значения теплоемкости ср
нитридов при различных
температурах,
кал • град~'- моль~1
Be-
щест- пл’
во *
V, см
для ме- для ме-
талла в металла
соответ-
ствую-
щем
соеди-
нении
в соот-
ветст-
вующем
соеди-
нении
Sc 1673 14,99 44 —
Hf 2273 13,62 202 — —
Th 1973 21,18 144 — —
и 1406 12,49 141 —— —
n2 63 17,3 587 — —
ScN 2923 — — 578 754
HfN 3255 — — 241 796
ThN 2903 — — 175 752
UN 2923 — — 204 754
Таким образом, при отсутствии
экспериментальных данных тепло-
емкости метод Дебая может быть
успешно использован для оценки
теплоемкости нитридов простейше-
го типа.
Располагая необходимыми тер-
модинамическими величинами,мож-
Тем- пера- тура, °K ScN HfN ThN UN
73 1,21 4,00 4,93 4,57
173 6,49 7,97 8,64 8,34
273 9,10 9,95 10,22 10,18
373 10,46 10,88 11,10 11,03
473 11,18 11,46 11,62 11,57
573 11,64 11,78 11,92 11,89
673 11,97 12,07 12,19 12,16
773 12,32 12,28 12,39 12,36
873 12,42 12,43 12,56 12,53
973 12,59 12,58 12,71 12,68
1073 12,78 12,71 12,85 12,84
1173 — — 12,99 13,01.
1273 13,03 12,95 13,11 13,10
1473 13,28 13,17 13,35 13,33
1673 13,53 13,38 13,58 13,57
1873 13,76 13,59 13,81 13,79
2073 13,98 13,78 14,02 14,01
2273 14,19 — 14,24 14,23
2473 14,42 14,17 14,46 14,44
2673 14,64 — 14,67 14,66
2873 14,85 14,56 14,39 14,86
3073 — — — 15,07
но проводить термохимический расчет взаимодействия в системах нит-
рид — газообразная фаза, определив тем самым стабильность туго-
плавких соединений в различных средах.
4*
51!
Расчет приведенных термодинамических потенциалов некоторых
нитридов. Простой и удобный метод рассмотрения термодинамических
равновесий в системе тугоплавкое соединение — газообразная фаза и
оценка изобарно-изотермических потенциалов реакций взаимодействия
компонентов таких систем состоит в непосредственном расчете изобар-
но-изотермических потенциалов реакции, в использовании данных теп-
лот образования в стандартном состоянии AH°29S и приведенных тер-
модинамических потенциалов компонентов реакции. Значения послед-
Рис. 24. Температурная зависимость теплоемкости некоторых нитридов:
□ —HfN; О —ScN ; ^ThN; X — UN; ф — TiC (расчетное);-------— TiC (экспери-
ментальное).
них для элементов и многих соединений в конденсированном и газооб-
разном состояниях приведены в специальных таблицах [864]. Однако
имеются значения приведенных термодинамических потенциалов лишь
для некоторых карбидов и нитридов совершенно отсутствуют значения
этих величин для боридов и силицидов.
Как известно, приведенный термодинамический потенциал
(табл. 23) Ф* образуется путем прибавления энтальпийной функции
ууО __ ууО
т ? Т1 к обоим частям уравнения:
ZT — H°T
т т т - = — SoT
(1.3)
с получением
^0 _ rjO
* Ар-----П у
ФГ — -----р---'
Н°т —
Т 1
(1.4)
52
где индекс 0 обозначает стандартное состояние; Т — температура, °К,
а за температуру 1\ обычно принимается 298,15°К. Для конденсиро-
ванных фаз Ф* может быть получено из
т Фт = ^Н°т — Н^ Л Таблица 22 Значения постоянных а, Ь, с Значени уравнения Ср=а4-Ь-10—3 Т—с-105Т—2 ПО d (1.5) Таблица 23 я приведенных термодинамических Ф° — Н° тенциалов Фт= —- Т торых нитридов при различных мпературах, кал град~х- моль~у
Соеди- нения а Ь- 10s неко О105 те
ScN HfN ThN UN или, испо лоемкосп ♦ 1 Фт = у За с ратуру о( 298,15°К. мя станд турами с тем испот дующим Для 11,59 11,68 11,68 11,62 льзуя 4, т Ti ганда эычно Связ1 артнь сущее [ьзова эбразе хими 1,23 1,07 1,19 1,23 значе т 0 этнуЮ прин э меж ми т :твляе ния Л эм: ческо 2,08 рвд 1,49 298°К 500°К 1000°К 1500°К 20С0°К
1,31 1,30 Li3N Be3N2 Mg3N2 Ca3N2 ние теп- Ba3N2 LaN CeN Th3N* C°dT UN TiN 1 ZrN н Щ HfN (l.o) VN NbN темпе- TaN имается Cr2N ду дау- moN2n емпера- Mn5N2 тся ny- Mn3N2 r° еле- Fe,N 298 Fe4N AIN 70 w0 r/0 Щ -T — “298 1 “298 nt T 1 T й реакции ДФ* с 32,9 12,0 19,5 25,6 34,2 13,6 16,0 43,2 13,4 6,5 8,8 10,85 8,9 10,7 6,4 18,0 5,2 21,0 47,6 33,0 24,5 36,9 9,2 7О 7 пределя 79,8 20,3 23,1 28,6 38,1 13,9 17,2 45,8 14,6 8,2 10,4 10,1 11,7 10,5 20,0 6,0 22,9 52,5 36,3 26,4 40,8 9,5 но “о ется 23,2 34,6 40,2 49,5 17,4 21,4 59,5 19,2 12,7 14,7 14,4 16,3 17,6 27,4 7,3 29,9 33,3 52,6 13,2 гаким 33,7 43,5 71,7 23,8 16,2 18,2 18,0 20,2 22,3 34,9 7,8 35,7 же 44,4 82,5 27,7 23,6 25,8 32,8 8,0 (1,7) эбразом,
как и ДН или AZ.
ДФг — 2/Фг продуктов
реакций — S/Фг исходных компонентов.
(1.8)
53
Так как . &z°T ДФг = -у jr—1 (1-9)
и bZ°T= —RTlnK, (1,10)
то R\nK = 4- &Фт (1,Н)
и . Д2° дя^ . &Т соединения — р р "F 2ФНсходных компонентов. (1,12)
Таким образом, используя соотношения (I, 12), можно определить
значение приведенного термодинамического потенциала тугоплавкого
соединения, если известны значения изобарно-изотермических потенци-
алов &Ф°т реакции образования соединения при различных темпера-
турах и величины теплот его образования &H°29S в стандартном со-
стоянии.
Значения ДФ°г реакции образования нитридов (табл. 23) были
определены из уравнений температурной зависимости их изобарно-изо-
термических потенциалов, данных в [862]. Значения приведенных тер-
модинамических потенциалов соответствующих металлов и азота были
взяты из [864].
Располагая необходимыми данными AZ?r и соединений, а
также значениями приведенных термодинамических потенциалов соот-
ветствующих металлов и азота, рассчитывали значения Ф* нитридов
в соответствии с уравнением (I, 12).
Знание приведенных термодинамических потенциалов некоторых
нитридов позволяет значительно упростить расчеты термодинамических
потенциалов реакций, протекающих с участием этих соединений, не
прибегая к трудоемкому методу определения этих величин, используя
термодинамические соотношения (I, 3) по известным значениям теплот
образования Д//°298 и абсолютных энтропий S“9g , а также теплоемкос-
ти ср компонентов реакции в широкой области температур.
Термодинамическая стабильность нитридов в различных средах.
Стабильность тугоплавких соединений является важным фактором, оп-
ределяющим их успешное использование в качестве высокотемператур-
ных материалов.
На более высокую стабильность указывает большое положитель-
ное значение стандартной свободной энергии реакции взаимодействия
тугоплавкого соединения с данной газовой фазой. Однако некоторое
взаимодействие наблюдается даже при положительном значении AZ°r
благодаря тому, что продукты реакции могут существовать с активнос-
54
тями, меньшими единицы, например, при растворении их в исходных
компонентах. При большом положительном значении \Z°T реакция
становится практически невозможной.
Следует принять во внимание также кинетику реакций. В некото-
рых случаях взаимодействие с окружающей средой является настолько
слабым, что допускает использование материала в данных условиях,
хотя с термодинамической точки зрения он является нестабильным. Та-
ким образом, термодинамика, являясь надежным средством предска-
зания потенциально стабильных в данных условиях соединений, не
дает возможности получить исчерпывающую оценку стабильности сое-
динений. Рассмотрим некоторые закономерности термодинамической
стабильности нитридов в среде воздуха, углерода и в вакууме.
а. Стабильность нитридов в среде воздуха. Ни-
же рассмотрены термодинамические условия протекания реакции вза-
имодействия нитридов с воздухом при давлении, равном 1 ат (0,2 ат О2
и 0,8 am Ns), и приведены результаты термодинамического расчета
равновесий в системах нитрид—кислород и определения равновесия
давления газообразных продуктов реакции взаимодействия ряда нитри-
дов TiN, ZrN, VN, BN, S13N4 с кислородом воздуха.
Для расчета термодинамических величин были использованы при-
веденные термодинамические потенциалы, табулированные для элемен-
тов и соединений в газообразном и конденсированном состояниях в
специальных таблицах [864]. Расчет приводится в соответствии с урав-
нением (I, 11).
Для реакций, идущих с получением газообразных продуктов, кон-
станта равновесия К может быть представлена как
(1.13)
Р 02
где р—парциальное давление газообразного продукта реакции, р0 —
парциальное давление кислорода воздуха при давлении воздуха р=
= 1 ат. Уравнение (I, 11) принимает вид
_RlnK = -t (1,14)
Р02 1
Поскольку р воздуха составляет 0,2 ат (р„ =0,8 ат), задача
(J2 IN 2
сводится к определению давления газообразных продуктов реакции
Pn(no) из известных табличных значений Д//“д8 и Ф* компонентов
реакции.
Возможные реакции взаимодействия нитридов TiN, ZrN, VN, BN и
Si3N4 с кислородом воздуха (0,2 ат) представлены в табл. 24, из ко-
торой видно что взаимодействие нитридов с кислородом может проте-
кать как с образованием азота, так и его окисла NO.
55
Таблица 24
Равновесное давление N2 или NO реакций окисления некоторых b нитридов log
Реакция —a ь-10—3
2ZrN+2O2=2ZrO2+N2 5,0 79
2TiN+2O2=2TiO2+N2 11,5 66
2TiN+=/3O2=Ti3O6+N2 7,5 54
2TiN+3/2O2=Ti2O3+N2 6,5 48
2TiN+O2=2TiO+N2 1,0 22
ZrN+3/2O2=ZrO2+NO 5,0 35
TiN+O2=TiO+NO 1,5 28
TiN+3/2O2=TiO2+NO 3,0 20
TiN+s/4O2=V2Ti2O3+NO 3,0 22
TiN+4/8O2=i/8Ti3O6+NO 0,0 7
2VN+3/2O2=V2O6+N2 14,5 64
2VN+2O8=VA+N2 8,5 56
2VN+3/2O2=V2O3+N2 1,0 26
V2Si3N4+3/2O2=3/2SiO2+N2 5,5 5
2BN+3/2O2=B2O3+N2 2,0 38,5
2VN+O2=2VO+N2 5,2 54
^SisN^/A^/iSiOa+NO 5,0 46,3
VN+’/4O2=1/2V2O6+NO 6,1 27
vn+3/a=v2va+no 5,0 24,5
VN+5/4O2=i/2V2O3+NO 2,7 20,5
VN+O2=VO+NO 5,0 8
Рассчитанные значения равновес-
ных давлений газообразных продуктов
реакции в соответствии с уравнением
(I, 12) при различных температурах
(от 298 до 2000° К) представлены
в табл. 24 уравнениями темпера-
турной зависимости давлений N2
и NO.
Нитриды характеризуются более
высокой термодинамической стабиль-
ностью (большими отрицательными
значениями &.Z0 ), чем соответствую-
щие карбиды при низких температу-
рах, но значительно меньшей свобод-
ной энергией образования соответ-
ствующих окислов. Поэтому нитриды
быстро разрушаются при нагревании
на воздухе (или в кислороде) с обра-
зованием более стабильных соедине-
ний — окислов. Учитывая, что термо-
динамическая стабильность нитридов,
окислов и азота уменьшается с рос-
том температуры, значение свободной
энергии реакции взаимодействия нит-
рида с кислородом также уменьшается
с увеличением температуры.
, Из рассмотрения кривых log p=f(T) (рис. 25, 26) следует, что
изменение свободной энергии реакции и давление азота и окиси азота
уменьшаются с ростом температуры, т. е. термодинамическая стабиль-
ность нитридов на воздухе увеличивается. При некоторых определенных
температурах для данной реакции окисления нитрида AZ°(, =0, т. е.
протекание процесса становится термодинамически маловероятным.
Оценка величин этих температур показывает, что они лежат в области
очень высоких значений (более 6000° К), далеко превосходящих обыч-
ные температурные области экспериментальных исследований. Следова-
тельно, при обычных температурах процесс окисления нитридов характе-
ризуется высокой термодинамической вероятностью и протекает самопро-
извольно в сторону образования более стабильных продуктов реакции.
Рассчитанные давления газообразных N2 и NO не учитывают за-
щитного характера образующегося слоя окалины. Тем не менее в ус-
ловиях постоянного газовыделения образующийся слой окислов посте-
пенно разрыхляется и наблюдаемое давление N2 и NO может иногда
быть очень близким к рассчитанным значениям.
56
Как видно из рис. 25, 26, термодинамическая возможность проте-
кания реакции окисления нитридов с получением соответствующего
окисла и газообразного азота гораздо больше термодинамической воз-
можности реакций окисления, продуктами которых является NO. Дей-
ствительно, окись азота характеризуется в отличие от других соедине-
ний сравнительно большим отрица-
тельным тепловым эффектом обра-
зования и высоким значением энтро-
пии. Это определяет более низкое
значение величины свободной энер-
гии реакций, продуктом которых
является NO.
Рис. 25. Температурная зависимость изобарно-изотермических потенциалов
реакций окисления TiN и ZrN:
а) 1 — 2ZrN+2O2=2ZrO2+N2; 2 — 2TiN+2O2==2TiO2+N2; 3 — 2TiN+73O2 = 2/3Ti3O5+N2;
4—2TiN+3/0O2=Ti2O3+N2; 5 — 2TiN+O2=2TiO+N2;
б) 1 — ZrN+O2=ZrO2+NO; 2 — TiN+O2 = TiO + NO; 3 - TiN+3/2O2=TiO2+NO; 4-
TiN+s/402=V2Ti203+NO; 5 - TiN+'/3O2='/3Ti3Os + NO.
Для данного нитрида, продуктами окисления которого могут быть
несколько различных окислов, термодинамическая возможность явля-
ется максимальной для реакций, идущих с образованием высших окис-
лов данного металла. В среде кислорода, несмотря на то, что
A^°zrN>A2°TiN, нитрид титана термодинамически более стабилен, чем
нитрид циркония, благодаря более низкому значению свободной энер-
гии образования ТЮ2 по сравнению с ZrC>2.
Из рассмотрения термодинамического равновесия в системах нит-
рид — кислород и величин давления газообразных продуктов реакции
следует, что в интервале температур 298—2000° К нитриды малоустой-
чивы в кислороде и могут подвергаться окислению с образованием со-
ответствующих окислов и газообразных продуктов N2 и NO.
57
Реакции взаимодействия нитридов с кислородом, продуктами кото-
рых является азот, характеризуются более высокими значениями отри-
цательной свободной энергии, чем реакции, протекающие с образова-
нием NO. Поэтому при анализе продуктов реакции окисления нитри-
дов, как правило, обнаруживается только азот.
Стабильность нитридов в вакууме. Нитриды харак-
теризуются более высокими значениями свободной энергии образова-
Рис. 26. Температурная зависимость изобарно-изотермических потенциалов
реакций окисления VN, BN и S13N4:
a)/—2VN + »/2О2 = V2O5 + N2; 2-2VN + 2O2 = V2O4 + N2; 3-2VN + 3/2O2 = V2O3 +
+ N2; 4—Si„N4 + 3O2 = 3SiO2 4- 2N2; 5—2BN-J- 3/2O2 = B2O3 + N2; 6 —2VN+02=2VO+N,.
б) I-Si3N4 + 5O2 = 3SiO2 + 4NO; 2— VN+ ’/Юг =’/2V2O5+ NO; 3-VN -H/2O2 =
V2O4 + NO; 4 — VN + S/4O2 = V2V2O3+NO; 5-VN + O2 = VO + NO;
стабильности соответствующих карбидов при 298° К, однако значения
абсолютных величин энтропии образования нитридов выше, что и опре-
деляет резкое уменьшение свободной энергии образования нитридов с
ростом температуры.
Вследствие относительно низкой термодинамической стабильности
при высоких температурах нитриды характеризуются высокими значе-
ниями давления диссоциации азота, и этот фактор в большей степени,
чем величины их температур плавления, ограничивает возможность ис-
пользования их в качестве тугоплавких материалов.
58
В табл. 25 представлены рассчитанные значения равновесных дав-
лений азота при диссоциации некоторых нитридов
&Z°T
log Pn2 = 2,303-«Т ’
где р „ — равновесное давление диссоциации азота, AZ° — изменение
^2
свободной энергии образования нитрида.
На рис. 27 даны температурные зависимости равновесных давлений
азота. Температурная зависимость logp—1/Т может быть описана
уравнением вида Iogp=a4~b/T. В табл. 26 представлены значения а и
b соответственно для всех рассмотренных нитридов.
Таблица 25
Значения парциальных давлений азота при диссоциации некоторых нитридов
Реакция 298° К 500° к 1000° к 1500 °к 2000° к
2Li3N^6Li+N2 6,49-IO-65 5,65-IO-57
Be3N2^*3Be-|-N2 1,24-IO-90 1,26-10-51 2,85-Ю-21 — —
MgsN2<?3Mg-|-N2 9,95-10—71 3,18-IO-38 3,33-10~14 9,06-10—6 —
CagNa^SCa-pNa 8,45-10~67 1,08-IO-35 9,55-10—13 — —
Ba3N2^3Ba-|-N2 5,55-IO-52 3,3-IO-26 3,4-10-7 — —
2LaN^2La-|-N2 1,51-IO-95 7,95-IO-53 2,59-Ю-21 — —
2CeN^2Ce-|-N2 3.39-1O-104 5,55-IO-58 6,84-10-24 — —
9,75-10-105 9,31-IO-59 7,67. Ю-25 1,55-Ю-13 6,97-IO-8
2UN^?2U+N2 8,53-IO-92 3,31-io-51 2,87-10-21 2,74-Ю-11 2,67-Ю-6
2TiN^2Ti+N2 1,01-10“108 3.58-10-61 4,25-Ю-26 2,51 -IO-14 —
2ZrN^2Zr+N2 1,41-IO-118 1,18-Ю-67 5,3-Ю-39 3,03-10—16 —
2VN5:2V-)-N3 4,01 -IO-79 9,62-10-43 9,46-10-18 3,83-10—9 7,21 -Ю-5
2NbN£2Nb+N2 7,24-Ю-75 3,36-Ю-41 1,06-Ю-16 9,08-10-9 4,94-Ю-5
2TaN^?2Ta-j-N2 3,72-10-77 6,18 -IO-43 4,23-Ю-18 4,75-Ю-10 3,9-10-®
2Cr2N^?4Cr-|-N2 1,4-IO-30 9,06-IO-16 3,48-IO-5 1,96-Ю-2 —
2CrN^2Cr-|-N2 9,34-IO-34 3,42-Ю-15 1,64-Ю-3 1,95-Ю —
4CrN^t2CrNTN2 2,03-10—36 2,68-10—17 4,53-Ю-4 7,02 —
2Mo2N^4Mo-|-N2 2,83-10-18 1,55-10-8 1,27-Ю-1 1,8-10 3,55-Ю6
Mn5N 2^5Mn +N2 4,07-10—28 6,31 -10—14 — — —
MnsN2^;3Mn-|-N2 1,67-10—26 4,97-IO-13 — .— —
2FeN^t2Fe-|-N2 4,65-103 1,42-Ю4 4,3104 — —
2Fe4N?8Fe+N2 2,58-10 4,69-Ю2 1,94-Ю4 —
2A1N£2A1+N2 6,22-10—104 2,6-10—58 1,17-Ю-24 — 1,39-Ю-2
2BN^2B+N2 2,4-10~82 2,34-IO-46 7,55-Ю-20 4,6-Ю-4 3,61 -ю-2
59
-20-
-CO-
-60-
-do\
-too
logP„,
ЮЛ
_2Fe,N
~ 2FeJ
t/TtO3
2M0.H
Mn3H2
Mn.B-i,
2Cr2N
2CrN
CCrN
BasH2
2Li,N
COjN,
Нд,Пг
2Nb№
2ToN
2VN
2BN
веЛг
2UN
2LoN
2AIN
20eff
V2Th&
2 TiN
2ZrN
Рис. 27. Температурная зависимость парциальных давлений азота при диссо-
циации нитридов.
Как следует из рис. 27, термодинамическая стабильность нитридов
металлов уменьшается с увеличением порядкового номера металла,
образующего нитрид. Наиболее высокая термодинамическая стабиль-
ность характерна для мононитридов циркония и титана. Они имеют
наиболее низкое значение равновесного давления азота и поэтому мо-
60
гут применяться в вакууме. Несколько меньшей стабильностью обла-
дают мононитриды редкоземельных элементов и актиноидов, нитриды V
группы переходных металлов. Нитриды VI группы довольно неустой-
чивы. Например, для M02N парциальное давление азота достигает
1 ат при температуре 895° С. Нитриды VIII группы ЕегН и Fe4N ха-
рактеризуются большими значениями равновесного давления азота и
являются термодинамически нестабильными во всей рассмотренной об-
ласти температур.
Нитриды, давление разложения которых более 10-6 ат, неста-
бильны и легко летучи. Из данных табл. 26 следует, что все нитриды,
как правило, нестабильны при температуре 2000° К.
В процессе разложения большей части нитридов выделяется га-
зообразный азот и образуется твердый раствор азота в металле;
другие нитриды, например UN, образуют газообразный азот и пары
металла.
Таблица 26
Значение констант а и Ь уравнения температурной
Ь
зависимости log р=а+-^- парциальных давлений
азота при диссоциации некоторых нитридов
Соедине- ние a bio—3 Соедине- ние a bio-3
2Li3N —43,2 6,8 2TaN 7,6 25
Be3N2 10 30,4 2Cr2N 5,8 10,4
Mg3N2 11 24,4 2CrN 10 12,8
CagNa 12 24 2Mo2N 7,4 10
Ba3N3 13,4 19,6 Mn5N2 8,4 10,8
2LaN 10,6 30,8 Mn3N2 8,4 10
2CeN 12 35,2 2Fe2N 5 0,4
V2Th3N4 9,8 34,4 2Fe4N 6 1,2
2UN 10,8 30,4 2A1N 9,6 34
2TiN 9,2 34,9 (298—1000° C)
2A1N 20 46
2ZrN 10 38,4 (1000—2000° K)
2BN 12,8 28,8
2VN 9,2 26
2NbN 8 24,4
Значения парциальных давлений азота при диссоциации нитридов
при данной температуре можно также определить из рис. 27. Для опре-
деления давления диссоциации нитрида при данной температуре необ-
ходимо соединить точку D на крайней левой вертикали, соответствую-
щей температуре абсолютного нуля, с точкой на линии \Z°T для инте-
61
ресующего нитрида и продолжить эту прямую до пересечения с правой
шкалой давления. Точка пересечения даст значение парциального дав-
ления азота над нитридом при данной температуре.
Стабильность нитридов в среде углерода. При
высоких температурах нитриды характеризуются более низкой термо-
динамической стабильностью, чем соответствующие карбиды, вследст-
вие чего они взаимодействуют с углеродом с образованием соответству-
ющих карбидов либо карбонитридов и выделением газообразного азота.
Несмотря на важность подобных сведений, до настоящего времени
равновесия в системах нитрид — углерод рассмотрены мало. В работе
[112] приведены некоторые термодинамические данные реакций взаи-
модействия ряда нитридов (AIN, UN, SijN4, TiC) с углеродом при тем-
пературе 2000° К-
В табл. 27 представлены результаты термодинамического расчета
равновесий в системе нитрид—углерод при температурах 298, 500, 1000,
1500, 2000° К и возможные предполагаемые реакции между нитридами
и углеродом (но без учета возможного образования карбонитридов).
Рассмотрим кратко основные закономерности процессов взаимо-
действия нитридов с углеродом.
• Термодинамическая стабильность нитридов при 298° С выше тер-
модинамической стабильности соответствующих карбидов, однако зна-
чения абсолютных величин энтропии нитридов выше, что и определяет
резкое уменьшение величины свободной энергии образования нитридов
с ростом температуры.
При взаимодействии нитридов с углеродом образуются соответству-
ющие карбиды и выделяется газообразный азот. Термодинамическая
возможность протекания реакции и ее изменение с ростом температуры
зависит от результирующего баланса изменения с температурой термо-
динамической стабильности продуктов и исходных компонентов реак-
ции. Таким образом, можно ожидать, что в области низких температур
реакции взаимодействия нитридов с углеродом будут термодинамически
маловероятными. С ростом температуры процесс взаимодействия нит-
ридов с углеродом протекает самопроизвольно, т. е. нитриды в среде
углерода термодинамически нестабильны, и направлен в сторону обра-
зования соответствующего карбида и газообразного азота.
Действительно, полученные нами данные расчета изобарно-изотер-
мического потенциала реакции взаимодействия нитридов с углеродом
(рис. 28, см. табл. 27) показывают, что давление азота растет с увели-
чением температуры.
Из рассмотрения прямых logp—\/Т следует, что изменение сво-
бодной энергии реакции и соответственно давление азота увеличива-
ются с ростом температуры, т. е. термодинамическая стабильность ни-
тридов в присутствии углерода уменьшается. Из рассмотрения усло-
вия AZ°r = 0 (/(=1) (условие невозможности протекания реакции)
62
Рнс. 28. Температурная зависимость равновесного давления азота
взаимодействия нитридов с углеродом (номера кривых соответ-
ствуют номерам реакций в табл. 27).
Таблица 27
Равновесное давление азота реакций взаимодействия ряда нитридов с углеродом при различных температурах
Реакция Значения pjq**, am Коэффициенты уравнения Температур- ный интервал, °к
298°K 500°K 1000°К - | 1500°К | 2000°К а ' °
M&N2+®/2C = =3/2Mg2C3+N2 1,26-IO-90 4,90-IO-50 3,09-Ю-20 1-106 — 54 32,2 298—1000
3,8 1000—1500
Mg3N2+6C=3MgC2+N2 1,23-ю- 23 1,23-Ю-10 2,09 — .— 11 9,72 —
C&gNg 4“ 6C=ЗСаСц+N 2 5,25-10- 33 5,25-Ю-14 1,70-10 — — 15,25 14,22 —
1/2Th3N4+3/2C= 5,37-Ю-55
=3/2ThC+N2 1,05-10—97 3,16-Ю-28 1,12-Ю-12 2,29-Ю-7 7 32,23 —
1/2ThsN4+3C=
=s/2ThC2+N2 4,37-IO-58 4,27-Ю-30 6,03-Ю-11 1,45-Ю-4 2,46-Ю-1 7 19,2 —
2UN+2C=2UC+N2 1,59-10—59 1,05-Ю-31 3,39-Ю-11 1,1-Ю-4 1,99-Ю-1 8,2 20,15 —
2UN+3C=U2Cs+N2 7,59-10—56 9,33-IO-30 1,59-Ю-10 2,14-Ю-4 2,09-Ю-1 8,15 19,92 —
2UN+4C=2UC2+N2 2,46-IO-57 1,78-Ю-30 1,1-Ю-10 8,13-Ю-5 2,82-Ю-1 9,2 20 —
2TiN4-2C=2TiC+N2 3,63-IO-43 1,55-Ю-22 2,63-Ю-7 2,95-Ю-2 — 9,22 15,23 —
2ZrN+2C=2ZrC+N2 8,32-IO-55 3,89-Ю-27 3,09-Ю-11 2,63-Ю3 — 29,75 23,45 298—500
— — — 15,23 500—1000
— — — — — 5,52 1000—1500
2VN+C=V2C+N2 4,57-10~65 2,95-Ю-16 — — — 59,98 30,68 —
2VN+2C=2VC+N2 6.17-10-61 1,7-Ю-33 5,5-Ю—12 3,47-10-6 8,32-Ю-3 8 20,5 —
2NbN+C=2Nb05C+Na 1,07-Ю-44 2,24-Ю-23 4,9-Ю-8 3,63-Ю-3 — 6,35 25,85 —
2NbN+2C=2NbC+N2 5,25-10—27 7,94-Ю-13 9,12-Ю-2 2,04-10 — 4,35 10,46 —
2TaN+C=TaaC+N2 1,48-Ю-52 2,24-Ю-28 2,09-Ю-11 2,19-Ю-5 1,1-Ю2 7,25 10,83 —
2TaN+2C=2TaC+Na 2,57-IO-21 6,76-Ю-10 6,92-Ю-2 2,14-10 2,69-Ю2 4,45 9,22 —
у 2Cr2N+24/23C= =4/23Cr23C6+N2 1,12-10-17 6,31-Ю-8 4,27-Ю-1 1,15-10 1,77 10,2 1,76 +0,53 298—500 500—100 1000—1500
— 3,31-10 3,63-102 —• 6,00 5,00 —
2Cr2N+12/7C= 1,66-ю-’2 1,38-10
=4/7Cr7C3+N2 2Cr2N+8/3C== 1,1-IO-20 2,51 -Ю-9 3,09-10“1 2,88-10 — 6,2 8,00 —
=4/зСг3С24-Ы2 12,4 11,6
2CrN+12/a3C= =2/23Cr23Ce-bN2 2,57-IO25 2,88-IO-12 4,27 2,69- Ю5 ——
10,5
2CrN+6/,C= =2/7Cr7C3+N2 1,41-Ю-24 1,35-Ю-10 3,72-10 1,85-106 14,25
12,45
2CrN+4/3C= 7,59-10—29 6,31-Ю-13 3,55 4,57-Ю5 -— 15
=2/2Cr3C2+N2 1,29-IO-22 6.17-10-24 4 47.Ю-10 2,57-Ю-1 1 23-Ю2 6,92-Ю6 14,63 8,43 —
2Mo2N+2C=2Mo2C+Na Mn6N2+5/3C=5/3Mn3C+N2 2,19-Ю-11 — — — 7,92 8,30 —
Mn6N2+16/7C= 8,7-IO-32 1,35-10-22 1,12-Ю-13 — — 14,52 13,45 —
=s/7Mn7C3+N2 Mn3N2+C=Mn3C+N2 1,78-Ю-11 — — — 6,00 8,45 —
Mn3N2+9/ 7C= 5 88 -10—13 — — — 12,45 12,45 —
=3/7Mn7C3+N2 2Fe4N=8/3C=8/3Fe3C+N2 2Fe4N+4C=4FeaC+N2 o,9o*1U 1,91 -Ю-9 1,7-IO-6 5,13-Ю-2 4,57-Ю-1 1-Ю4 7,41 -Ю3 — — 8,2 8, 2 5,75 4,98
e/3Fe2N+4/3C= 1,35-Ю-1 1,51-Ю2 3,31-104 — — 7,98 +2,5 —
=4/з?езС+^2 2,13-Ю-3 1,7-101- 8,7 1,55 —
2BN+1/2C=1/2B4C+N2 2AlN+3/2C=1/2AliC3+Na V Si3N4+3/2C=3/2SiC+N 9,12-Ю-91 1,86-Ю-44 2,45-Ю—50 1,95-Ю-23 1,2-Ю-19 5,89-Ю-8 8,32-10“3 4,68-10 14,46 0,5 30,2, 15,7 0,3 — 5 298—1500 3 1500—2000
0 01
V су <ТР -г ' •<;?«! I
7 ч в 7 Т + q "f S= w -s 9 ? ° ?
следует, что при некоторых вполне определенных температурах реакции
взаимодействия нитридов с углеродом становятся термодинамически
маловероятными. Рассчитанные нами соответствующие температуры,
отвечающие условию \Z°T = 0, различны для всех нитридов и увеличи-
ваются обычно с уменьшением валентности металла, образующего нитрид.
Таким образом, из анализа прямых log р—следует, что тер-
модинамическая стабильность нитридов в среде углерода уменьшается
с ростом порядкового номера группы металла, образующего нитрид.
Из рис. 28 видно, что по своей стабильности по отношению к угле-
роду все нитриды образуют несколько определенных групп. Наиболее
высокой термодинамической стабильностью характеризуются нитриды
TI13N4, A1N. Реакции взаимодействия нитрида Th3N4 с углеродом ха-
рактеризуются наиболее низкими значениями величины давления азота
в широкой области температур, благодаря чему TI13N4 может приме-
няться в восстановительных средах вплоть до температур 2500° К и
выше. Несколько меньшей стабильностью характеризуются нитриды
переходных металлов четвертой и пятой групп VN, UN и ZrN, еще
меньшей стабильностью отличаются нитриды Sis-N4, NbN и TiN. Из
всех перечисленных нитридов наибольшей термодинамической устойчи-
востью по отношению к углероду обладает, таким образом, нитрид то-
рия Th3N4, а все остальные перечисленные нитриды могут взаимодейст-
вовать с углеродом начиная с температуры 1500° К.
Значительной неустойчивостью отличаются нитриды шестой и седь-
мой групп периодической системы. Стабильность нитридов уменьшает-
ся в такой последовательности: CrN, Mo2N, MgsN2 и Cr2N. Мононитрид
хрома более устойчив, чем его низший нитрид Cr2N. Наиболее стабилен
в этой группе нитрид Ca3N2.
В отличие от нитридов предыдущей группы для перечисленных ни-
тридов при 1000° С и выше процесс взаимодействия с углеродом может
оказаться термодинамически вероятным (р при 1000° С» 1 ат).
Последнюю группу нитридов составляют нитриды железа Fe4N и
Fe2N, которые характеризуются отсутствием какой бы то ни было
стабильности в присутствии углерода и при сравнительно низких тем-
пературах (выше 500° С) могут взаимодействовать с углеродом. Нитрид
железа Fe2N термодинамически нестабилен в присутствии углерода
даже при температурах порядка 300° К.
Таким образом, из рассмотрения термодинамических данных для
систем нитрид — углерод следует, что при температурах, превышаю-
щих 1500° К, все рассмотренные нитриды, за исключением Th3N4, тер-
модинамически нестабильны и интенсивно могут взаимодействовать с
углеродом. Температуры начала взаимодействия (или, точнее, интен-
сивного взаимодействия) различны и значительно меняются от одной
группы нитридов к другой.
66
4. КЛАССИФИКАЦИЯ НИТРИДОВ
Анализ свойств и электронной структуры нитридов позволяет предло-
жить их классификацию, основанную на единых принципах [859, 860].
Эта классификация, естественно, вытекает из классификации элементов
периодической системы на s-, ds-, fds-металлы и .sp-элементы (неме-
таллы и полуметаллы).
А первому классу относятся нитриды электроположительных ме-
таллов I и II групп периодической системы, атомы которых имеют
внешние 5-электроны при полностью вакантных или полностью застро-
енных глубоколежащих оболочках (в состоянии изолированных ато-
мов). Эти нитриды имеют составы, отвечающие обычным валентностям,
характеризуются передачей атомами металла валентных электронов
атомам азота с образованием при этом стабильных 52р6-конфигураций
обоими компонентами (металлом и азотом) либо d10- и з2р6-конфигу-
раций, что обусловливает их ионный характер, проявляющийся внешне
в гидролизе с выделением аммиака, высоком электросопротивлении,
полупроводниковых свойствах. Наряду с сильными и четко выражен-
ными ионными связями Me—N этим нитридам свойственна и ковалент-
ная составляющая связи, главным образом, между атомами металла
(в нитридах) и атомами азота (в азидах). Этот класс нитридов по типу
химической связи может быть назван классом и о н н о-к о в а л е и т-
н ы х нитридов.
Следует отметить, что соотношение этих двух типов связей в ни-
тридах указанного класса довольно сильно различается, у некоторых
нитридов появляются и признаки металлической связи, например, у
нитридов щелочноземельных металлов.
Ко второму классу относятся нитриды, образуемые sp-элементами,
т. е. неметаллами и полуметаллами. Для них характерно образование
обоими компонентами стабильных sp-конфигураций, которые по мере
роста их энергетической стабильности обособляются друг от друга в
энергетическом отношении. Это вызывает появление энергетических раз-
рывов между атомами неметалла и азота и соответствующие полупро-
водниковые или диэлектрические свойства. В основном им отвечают
обычные валентные формулы, связи между атомами в кристаллических
решетках — направленные, жесткие, ковалентного типа,— что вызывает
отсутствие у них областей гомогенности. Этот класс нитридов может
быть назван классом ковалентных нитридов.
К третьему классу относятся нитриды переходных металлов с до-
страивающимися d- и /-электронными оболочками. Нитриды данного
класса характеризуются более или менее широкими областями гомо-
генности, преимущественно металлическими свойствами, высокой элект-
ропроводностью, высокими температурами плавления, твердостью. Ни-
триды этого класса образуются из элементов со значительным выделе-
5* 67
к
нием тепла и являются результатом такого перераспределения валент-
ных электронов металла и азота, которое приводит к образованию мак-
симального статистического веса атомов, обладающих устойчивыми
конфигурациями локализованной части валентных электронов. Для
нитридов переходных металлов характерно наличие сильной ковалент-
ной связи между атомами металла, а также преимущественно метал-
лической связи между атомами металла и азота, что позволяет отнести
их к классу ковалентн о-м еталлических нитридов.
Диапазон изменения физических свойств внутри этого класса ни-
тридов чрезвычайно широк. Наряду с преимущественно металлоподоб-
ными нитридами внутри этого класса есть группа нитридных фаз, у
которых преобладает ковалентная связь, а также появляется известная
доля ионной связи. Такие фазы обладают полупроводниковыми свойст-
вами, но с характеристическими свойствами, уступающими свойствам
типично ковалентных нитридов.
Приведенная классификация носит условный характер, так как
невозможно провести резкие и четкие границы между какими-либо
классами нитридов. Свойства, электронное строение и типы химической
связи нитридов изменяются дискретно, т. е. при переходе от нитрида
к нитриду существует единая общая линия и направление изменения
электронного строения и свойств, отвечающая такому же изменению
свойств химических элементов, образующих нитриды [861].
ГЛАВА II
СПОСОБЫ
ПРИГОТОВЛЕНИЯ
НИТРИДОВ
Важнейшими способами приготовления нитридов
являются: 1) непосредственное действие азота,
аммиака или других газообразных азотсодержа-
щих соединений на элементы или их гидриды;
2) восстановление окислов в присутствии азота,
аммиака; 3) термическая диссоциация соедине-
ний, содержащих данный элемент и азот; 4) оса-
ждение нитридов из газовой фазы.
Непосредственное взаимодейст-
вие элементов с азотом осуществляется
действием азота, аммиака или других газообраз-
ных азотсодержащих соединений на порошки ме-
таллов и неметаллов либо на компактные метал-
лы, либо на их гидриды.
Реакция взаимодействия между элементами
(Э) и азотом описывается уравнением
2Э -4- N2 2ЭЫ.
Несмотря на высокую энергию диссоциации
молекулы азота (225 ккал!моль [734]), его ис-
пользование для азотирования предпочтительнее,
чем аммиака. Это объясняется тем, что образую-
щийся при диссоциации аммиака водород, хотя и
является восстановителем, удаляющим с поверх-
ности окисные пленки, может образовывать со
многими элементами, особенно металлами, отно-
сительно устойчивые гидриды. Поэтому для уда-
ления водорода и замещения его азотом необхо-
дима дополнительная затрата энергии, что
выражается в более высоких температурах обра-
зования нитридов. Например, при азотировании
переходных металлов действием аммиака обра-
зуются сложные фазы внедрения азота и водо-
рода в пространства между металлическими
атомами [735], так называемые нитридогидриды,
удаление из которых водорода проходит при бо-
лее высоких температурах, чем из обычных ги-
дридов. Так, при действии азота на порошок
циркония нитрид циркония ZrN образуется в
течение 1—2 ч при температуре 1200° С, а при
действии аммиака нитрид циркония с мак-
симальным содержанием азота может быть
получен лишь при двухчасовой выдержке при
1400° С [178]. То же наблюдается при использо-
69
I
®ании вместо металлов их гидридов. Так, при 500—800° С, когда гидрид
титана достаточно устойчив, содержание азота в продукте азотирования
в два раза меньше, чем в порошке негидрированного титана, азотируе-
мого при тех же условиях. При 1000° С, когда гидрид титана заметно
диссоциирует, насыщение азотом проходит более интенсивно и макси-
мальное насыщение азотом достигается при 1200° С в течение
2—4 ч, т. е. при тех же условиях, что и для чистого титана. Отсюда сле-
дует, что в общем азотирование гидридов происходит легче, чем обра-
ботка чистых металлов аммиаком, т. е. в условиях, когда легко обра-
зуются сложные фазы внедрения типа нитридогидридов.
Во многих случаях азотирование предпочтительнее проводить ам-
миаком, который способен с некоторыми металлами образовывать ами-
ды, легко превращающиеся в соответствующие нитриды [6]. Так, ще-
лочные металлы уже при обычной температуре и особенно быстро при
нагревании вытесняют один атом водорода из молекулы аммиака с
образованием амидов металлов
2 Me + 2 NH3 2 MeNH2 + Н2.
Щелочноземельные металлы образуют амиды с большим трудом
и только при нагревании.
При нагреве амиды разлагаются с образованием нитридов
3 NaNH2 - Na3N ф 2 NH3,
причем амиды щелочных металлов разлагаются гораздо труднее, чем
щелочноземельных.
Механизм реакции азотирования в общем случае сводится к диф-
фузии азота в глубину металла или неметалла (с образованием твер-
дого раствора). По достижении определенной температуры обычная
гетеродиффузия переходит в реакционную диффузию с образованием
нитридных фаз. Скорость образования нитрида лимитируется скоростью
реакции и диффузионной передачей азота через слой уже образовав-
шегося нитрида. Механизм процесса азотирования аналогичен меха-
низму процесса окисления [76], однако, как правило, скорость азотиро-
вания «.меньше скорости окисления. Это может быть объяснено тем, что
в то время как кислород, изолированный атом которого имеет конфи-
гурацию валентных электронов s2p4, стремится к достройке до стабиль-
ной конфигурации s2p6 как единственно возможной (с превращением
при этом в ион О2-), у атома азота (конфигурация валентных элект-
ронов s2p3) имеется как тенденция к достройке до конфигурации s2ps,
так и вероятность отдачи одного электрона с образованием зр3-кон-
фигураций, что и служит фактором, замедляющим диффузию по срав-
нению с кислородом [1070]. По той же причине константа скорости ре-
акции азотирования увеличивается по мере понижения статистического
веса нелокализованных электронов (табл. 28) [178].
то
ив
- j .. Т а б л и ц а 28
Константы скорости реакций азотирования порошков некоторых
переходных металлов, г[см3- сек
Темпе’ ратура. °C Ti—N Zr-N V-N Nb—N Та—N Сг—N
500 5,68 10-6 8,22-10-е 1,7-10~5 2,21-10-5 2,47-10—2 1,08.10-'
600 2,20-10-5 4,1.10-5 2,95-10-5 3,79-10-5 7,28-10—2 2,51-10-'
700 1,65-10—5 6,8-10-5 3,72-10-5 8-Ю-5 1,58.10-' 5,27-10-'
800 3,33 10~5 8,3-10-5 5,8 -10~5 1,1 -ю-4 1,5-10-3 3,39-10-'
900 6,0-10—5 5,2-10-5 1,8-Ю-4 1,4-10—4 1,1 -Ю'2 7,24-10-'
1000 4,1 10—4 1,01-ю-4 2,3-Ю-4 2,3-Ю-4 3,35-Ю'4 5,88-10-'
1100 7,47-10—4 2,2-IO-4 — 4,3-10—4 8,25-Ю'4 6,28-10-'
1200 7,47-Ю-4 2,2-Ю-4 3,71-Ю-4 6,11.10-3 — 4,55-10-'
Следовательно, для ускорения процесса образования нитридов не-
обходимо максимальное измельчение частиц порошка. Однако, когда
имеется склонность к поверхностному окислению, одновременно с из-
мельчением возрастает относительное содержание кислорода в по-
рошке и азотирование затрудняется. Таким образом, в каждом от-
дельном случае должен выбираться некоторый оптимальный размер
частиц.
Большое значение имеет максимальное развитие реакционной по-
верхности при образовании в процессе азотирования нитридов с кова-
лентным типом связей и следующей отсюда невысокой скоростью пере-
мещения атомов азота (BN, A1N). В данном случае для наи-
большего развития поверхности используются специальные «подклад-
ки» или «носители» из веществ, обладающие высокой свободной по-
верхностью. К такому типу веществ относятся мел, фосфат кальция и
многие другие вещества, которые разлагаются при нагревании с выде-
лением газообразных составляющих, вызывающих сильное их разрых-
ление, образование тонкой ажурной структуры с высокой поверхностью.
Подкладкой может служить при определенных условиях и сам полу-
чаемый нитрид при образовании им очень тонких частиц (например,
получение нитрида алюминия азотированием порошка алюминия, сме-
шанного с нитридом; то же при получении нитрида бора).
Аналогичные меры принимают в тех случаях, когда температура
азотирования выше температуры плавления металла и реакционная
поверхность резко ограничивается зеркалом расплава, как это наблю-
дается при получении нитридов галлия и индия. При этом азотируемый
металл перемешивается с веществом, активно разлагающимся при на-
гревании, например с углекислым аммонием. При разложении послед-
71
него выделяются аммиак и СОг, разрыхляющие и перемешивающие
расплав, облегчая доступ азота; кроме того, аммиак производит допол-
нительное азотирующее действие.
С другой стороны, азотирование некоторых относительно легко-
плавких металлов при образовании расплава происходит быстрее, чем
в твердом состоянии. Азотирование при этом происходит скачкообраз-
но, приблизительно при температуре плавления металла. Это объясня-
ется нарушением при плавлении тончайшей и труднопроницаемой для
азота пленки нитрида. Очевидно, образование нитрида происходит в
короткий промежуток времени плавления до образования сплошного
зеркала расплавленного металла.
Если теплота плавления азотируемого металла существенно мень-
ше теплоты образования нитрида (табл. 29), то реакционная масса
резко разогревается, например при азотировании металлических церия
и лантана, а также алюминия.
Т а б л и ц а 29
Значение отношений теплот образования нитридов к теплотам плавления
соответствующих металлов
Этот фактор также обусловливает высокую скорость реакции и,
вероятно, должен учитываться во всех случаях азотирования, когда на
поверхности металла образуется труднопроницаемая для газов пленка.
Такого рода пленки могут нарушаться при азотировании под давле-
нием, как это наблюдается, например, при получении нитрида алюми-
ния из алюминиевого порошка [590]. Кроме того, азотирование под
давлением часто позволяет интенсифицировать процесс образования
нитридных фаз (скорость реакции азотирования при этом увеличивается
приблизительно пропорционально корню квадратному из давления).
Кроме указанного примера азотирования алюминиевого порошка
[590], данный метод использовали при азотировании ниобия в [295].
Установка для азотирования ниобия под давлением до 240 ат показа-
на на рис. 29.
Сверхвысокие давления используют только в отдельных случаях,
например при получении кубической модификации нитрида бора (бора-
72
зона) из гексагонального BN типа графита. Для этого используются
давления порядка 40—70 тыс. ат и аппаратура, аналогичная устройст-
вам для получения искусственного алмаза.
Как показано в [1047], большое значение имеет при непосредствен-
ном азотировании порошков скорость отвода тепла из реакционного
пространства. При малой скорости отвода температура образца резко
повышается и скорость реакции азотирования возрастает, т. е. проис-
Рис. 29. Установка для азотирования ниобия под давлением:
/—стальная труба; 2 — стальной фланец; 3— стальная крышка; 4— уплотнительное кольцо; 5 —
соединительный колпачок; 6 — корундовая труба; 7 — керамическая труба из пифагоровой массы;
8 ~ молибденовая лента; 9 — центрирующая деталь нз латуни; 10 — пружинящая медная пластинка
для контакта; 11— подвод тока; 12— изолирующая втулка; 13 — кабель подвода тока; 14— контей-
нер с веществом; 15 — термопара в защитной трубке; 16 — соединительный колпачок с вводом для
термопары; 17 — платиновая проволока; 18 — охлаждающая рубашка.
ходит тепловое воспламенение, в результате которого порошок сильно
спекается или расплавляется, что, в свою очередь, ухудшает газопро-
ницаемость и условия азотирования. Установлено, что добавки нитри-
дов к металлическим порошкам весьма существенно повышают темпе-
ратуру воспламенения их в азоте, сглаживают температурные эффекты
в момент воспламенения, способствуют улучшению газопроницаемости
загрузки и технологических свойств промежуточных и конечных про-
дуктов азотирования.
Получение нитридов тугоплавких металлов, алюминия и магния
азотированием порошков в промышленных условиях [843] осуществля-
лось непрерывным методом, т. е. непрерывной подачей порошка в зону
нагрева печи в распыленном (взвешенном) состоянии. Это позволяет
примерно в 50 раз повысить производительность печей против периоди-
ческой загрузки, а также получать нитриды не в форме спеков, а в
форме порошков, что практически исключает последующее измельче-
ние и удешевляет процесс.
73
В последнее время пробуют получить нитриды распылением ме-
таллов в плазменной струе с применением азота. Так, в [950] синтези-
ровали нитриды магния и титана обработкой металлов в плазменной
струе азота. В полученных продуктах содержится 30—40% соответ-
ствующих нитридов. При аналогичной обработке вольфрама и молиб-
дена нитриды не образуются. Принципиально таким же методом могут
быть получены сложные нитриды — при обработке боридов плазменной
•струей образуются нитриды металлов или боронитриды; сложные сое-
динения с азотом образуются и при обработке силицидов и карбидов
[951, 952].
Восстановление окислов в присутствии азота
с образованием нитридов происходит по реакции
МеО ф Me (X) + N2 MeN + МеО (ХО),
где Me — металл-восстановитель, X — неметаллический восстановитель
(углерод, кремний, бор и т. п.).
Обычным восстановителем при этом является углерод. Однако при
восстановлении окислов карбидобразующих металлов наряду с нитри-
дом образуется и карбид, который может давать с нитридом непре-
рывный ряд твердых растворов, как это происходит при получении
нитрида титана, где образуется твердый раствор TiN—TiC (либо TiN—
—TiC—TiO) [ПО]. Это ограничивает возможности использования мето-
да в основном получением технических нитридов, если загрязнение угле-
родом не имеет существенного значения для их дальнейшего исполь-
зования.
Особенности получения нитридов восстановлением окислов метал-
.лов углем при одновременном азотировании исследованы на примере
нитридов титана и ниобия [1047]. Показано, что с повышением темпе-
ратуры и понижением давления азота содержание карбидов в равно-
весных твердых растворах МеС—MeN повышается. Поэтому для полу-
чения нитридов титана и ниобия, минимально загрязненных углеродом,
реакции необходимо проводить при возможно низких температурах и
при повышенных давлениях азота. Существенное влияние на скорость
восстановления и азотирования оказывает диффузия окиси углерода и
азота в порах уплотненной шихты (степень этого влияния зависит от
радиуса пор и геометрических размеров брикетов уплотненной шихты).
Реакция восстановления окисла металла углеродом и насыщение вос-
становленного металла азотом происходит главным образом внутри
пор шихты, т. е. связана с удалением окиси углерода и притоком азота
к месту реакции. Если скорость циркуляции окиси углерода и азота в
порах больше скорости реакции, то будут иметь место действительные
скорости реакции. Замедленная диффузия окиси углерода и азота че-
рез узкие поры вызывает большое парциальное давление окиси углеро-
да и малое давление азота, что замедляет реакцию образования нитри-
74
.да, смещает ее в сторону образования карбида. Установлено, что
механизм переноса окиси углерода и азота в порах шихты и промежу-
точных продуктов реакции определяется кнудсеновским режимом, для
которого характерен малый диаметр пор по сравнению с длиной сво-
бодного пробега молекул, а также малый коэффициент диффузии,
в результате чего создается большой градиент давлений окиси углерода
и азота по сечению брикета шихты. В кнудсеновских условиях поток
молекул через капилляр длиной Дх и радиусом г описывается урав-
нением
dn ___ 8 лг3 Др
di Дх ’
где
dn
dt
— поток молекул в секунду; т — масса молекул; Д’ — констан-
С -г
та Больцмана; Т — температура; —— градиент давления в поре.
Рассчитанные величины радиуса пор уплотненных исходных шихт
при получении нитридов титана и ниобия оказались равными 400—
500 А при общей пористости 47%, а рассчитанные радиусы пор про-
межуточных продуктов реакции находятся в пределах 3000—5000 А
при общей пористости 60—80%, причем с увеличением времени реак-
ции величины радиусов пор и общая пористость возрастают. Исходя
из этого, показано, что скорость потока молекул окиси углерода и азо-
та в порах промежуточных и конечных продуктов реакции при одина-
ковых градиентах давлений на несколько порядков больше, чем в порах
исходной шихты. Из-за малой скорости кнудсеновского потока молекул
в порах исходной шихты на первом этапе реакции создается наиболь-
шее парциальное давление окиси углерода и замедление реакции, а
образующаяся при реакции внешняя корка промежуточного продукта
или нитрида на поверхности брикета практически не создает дополни-
тельного градиента давления, и фронт реакции с течением времени пе-
ремещается к центру брикета, т. е. реакция не идет по всему объему
одновременно, а перемещается от периферии к центру, причем скорость
роста толщины корки (слоя) нитрида подчиняется линейному закону
\х—кх, где т — время.
Учет равновесных и кинетических особенностей прохождения реак-
ций восстановления — азотирования позволил установить оптимальные
режимы получения нитридов титана и ниобия, которыми являются со-
ответственно температуры 1250 и 1400° С, давление азота 4- Ю5 н/м2,
скорость тока азота 0,18 м/сек, продолжительность нагрева 3 и 4—5 ч
(для шихт, гранулированных в шарики диаметром 13 мм). Нитриды
ниобия и титана, получаемые при этих условиях, содержат соответ-
ственно 12,9 и 21,5% азота при отсутствии углерода в нитриде ниобия
ш содержании его 0,5—0,7% в нитриде титана.
75
В качестве восстановителя используют не только углерод и прочие-
неметаллы, но и металлы — кальций, магний или их гидриды. Авторы
[192] разработали способы получения нитридов титана, циркония и тан-
тала по схемам
ТЮ3 (Та2О5) + СаН2 Ti (Та) -J- Н2О ф СаО,
2Ti (Та) + N2(2NH3) -> 2TiN(TaN) -f- (ЗН2)
или
ZrO2 + 2Mg - Zr 4 2MgO,
2Zr + N2(NH3) - 2ZrN + (H2).
Данный метод подробно проанализирован применительно к полу-
чению нитрида циркония [251]. Показаны известные трудности его
использования, связанные с тем, что по мере восстановления двуокиси
циркония магнием и перехода в область твердого раствора кислорода
в цирконии величина энергии связи существенно возрастает и удаление
остаточного кислорода требует выбора соответствующего режима вос-
становления и его тщательного контроля. Процесс восстановления про-
ходит, по-видимому, через стадию образования нитрида магния Mg3Nz-
Температуры восстановления окислов при использовании в качест-
ве восстановителей металлов или их гидридов обычно 800—1200° С, уг-
лерода — выше (например, для образования нитрида титана —
1600—1700° С).
В некоторых случаях восстановление окислов с образованием ни-
тридов осуществляется непосредственно аммиаком, водород которого-
играет роль восстановителя. Например, таким способом можно полу-
чить нитрид меди
3 Cu2O + 2NH3 2Cu3N + ЗН2О.
Согласно [1024], указывается на возможность резкого понижения
температур восстановления и азотирования при использовании свеже-
осажденных гидроокисей, которые обрабатываются смесями аммиака
и водорода.
Термическая диссоциация осуществляется с примене-
нием соединений, содержащих одновременно металл и азот. Так могут
использоваться аминохлориды, например
TiCl4-4NH3 - TiN + NH3 + НС1
либо комплексные фтораммиачные соединения типа (NH4)X MeFy [274J,.
при разложении которых (при 300—800° С) образуются соответствую-
щие нитридные фазы. Данный метод дает возможность получать нитри-
ды AIN, VN, NbN, Ta3Ns, CrN, U3N4, Fe2N, однако нитриды марганца,.
76
'бериллия, цинка, титана, циркония, лантана таким способом получить
либо невозможно, либо трудно.
Так как металл входит в состав соединений уже в ионизированном
состоянии, то при осторожном разложении нередко образуются нитри-
ды, приближающиеся к составу, который определяется ионной состав-
ляющей связи этих соединений. Это, в частности, наблюдалось при
образовании нитрида титана из аминохлоридов, когда удавалось полу-
чить нитрид состава TiNme, а также при приготовлении нитрида тан-
тала из (NH4)2TaF7, которое приводит к образованию Ta3N5. При
непосредственном азотировании металлов образование такого рода
метастабильных фаз с гипертрофированной долей ионной связи прак-
тически никогда не наблюдается.
К этой же группе методов получения относится термическое раз-
ложение амидов и имидов, описанное выше, а также термическая дис-
социация высших (по содержанию азота) нитридных фаз с образова-
нием низших. Необходимо отметить, что в последнем случае довольно
трудно получить нитрид определенного фазового состава, т. е. чистые
нитридные фазы без примесей высших фаз (в случае нитридов переход-
ных металлов, высшие же нитриды непереходных металлов — азиды —
разлагаются с образованием фаз строго определенного состава).
Осаждение нитридов из газовой фазы использо-
валось давно в различных вариантах, а в настоящее время приобретает
особо важное значение в связи с возможностью получения таким обра-
зом монокристаллических и чистых нитридов.
Примером этого метода может служить взаимодействие хлоридов
или оксихлоридов металлов с аммиаком, которое может быть пред-
ставлено следующими схемами:
МеСЦ + NH3 - MeN {- НС1,
МеОС13 + NH3 MeN + H2O + HC1.
Эти реакции проходят обычно при
температурах порядка 800° С. Таким спо-
собом получали нитрид титана [736], ва-
надия [737], хрома и других переходных
металлов [257].
Ниже (табл. 30) приведены условия
осаждения нитридов при взаимодействии
хлоридов с азотно-водородной смесью.
Проведено термодинамическое иссле-
дование условий осаждения нитридов
из газовой фазы для осаждения нитри-
да титана на железе [157]
Таблица 30
Условия осаждения
нитридов из газовой фазы
Ннтрнд Исход- ный хлорид Азотиру- ющий агент Температура осаждения, °C
TiN TiCl4 ЗМ^-рНз 1100—1700
ZrN ZrCl4 1100—2700
ZrN ZrCl4 n2 2800—3000
HfN HfCl4 3N2“|~^2 1100—2700
VN VC14 3N24-H2 1100—1600
TaN TaCl5 n2 2400—2600
TiCl4 + 2Н2 + VaNa £ TiN + 4НС1,
TiC 14 + 2 Fe -I- x/a N2 TiN + 2FeCl2.
(II, 1>
(II,2>
(П,3>
Зависимость убыли свободной энергии от температуры выражается
соответственными уравнениями
AFX = 96100 — 6,7 7,
Д72 = 7500 — 13,45т,
bFs = 51300 — 34,27.
Полнота прохождения реакций (II, 1) и (II, 2) возрастает с повы-
шением температуры осаждения, реакция (II, 3) выше 1100° С стано-
вится невыгодной.
Приготовление изделий из нитридов принципи-
ально возможно различными методами, из которых основными являют-
ся: 1) спекание заготовок, спрессованных из предварительно получен-
ных порошков нитридов; 2) горячее прессование порошков нитридов;.
3) реакционное спекание; 4) литье изделий из нитридов.
Спекание заготовок, спрессованных из порошка нитрида, может
осуществляться в среде азота, азотсодержащих восстановительных
газов или в вакууме. В последнем случае происходит некоторая потеря
азота (порядка нескольких десятых процента), однако при определен-
ных температурных режимах эта потеря может быть снижена до мини-
мума при одновременном получении достаточно плотных (пористость
0—2%) изделий [232]. Особенно мало изменяются при спекании в ваку-
уме нитриды титана, циркония, ванадия, ниобия, тантала; напротив,,
нитриды хрома и молибдена теряют в вакууме значительные количества
азота и спекать их таким способом невозможно. Лучшие результаты
дает спекание в защитной среде азота.
Горячее прессование порошков нитридов дает удовлетворительные
результаты, однако спекаемое изделие при использовании обычно при-
меняемых графитовых прессформ значительно загрязняется углеродом
и требуются специальные мероприятия для предотвращения этого за-
грязнения (использование токонепроводящих прессформ из нитридов,
тщательная обмазка внутренней поверхности прессформ нитридом бора
или алюминия и т. п.).
Горячее прессование должно проводиться в защитной (содержащей-
азот) либо нейтральной (аргон) газовой среде [232].
Реакционное спекание — совмещение процессов образования ни-
тридов и их спекания — часто дает наиболее благоприятные результа-
те
ты [740]. В этом случае вследствие образования новых фаз и повышен-
ной активности атомов или атомных комплексов резко интенсифициру-
ются процессы, приводящие к усадке и уплотнению изделий по срав-
нению с обычным спеканием предварительно спрессованных заготовок
из порошков заранее полученных соединений. Удельный объем образу-
ющейся при азотировании фазы больше удельного объема исходного
металла, что приводит к снижению пористости спекаемой заготовки за
счет чисто объемного фактора. Из сравнения атомных объемов метал-
лов и молекулярных объемов их нитридов (табл. 31) следует, что уве-
Таблица 31
Сравнение атомных объемов
металлов и молекулярных
объемов их нитридов
Нитрид Атом- ный объем метал- ла, см9 Моле- куляр- ный объем нитри- да, см9 Измене- ние объема, %
TIN 10,6 11,4 +7,5
ZrN 14,03 14,8 +5,5
HEN 13,64 13,9 +1,9
V3N 8,36 10,8 +29,3
VN 8,36 10,7 +28,0
Nb2N 10,8 12,9 +19,2
NbN 10,8 12,8 +18,5
Ta2N 10,9 12,2 +12
TaN 10,9 12,7 +16,5
Cr2N 7,23 10,1 +39,5
CrN 7,23 10,8 +49,3
Mo2N 9,40 13,6 +45
Таблица 32
Режимы получения компактных образцов
Одновременное азоти- рование и спекание Дополнительное спе- кание методом горя- чего прессования
Соеди- о > * «и Р.О й «
неине д ® Й « а°
д Q •- ah' Рчс х Темпе тура, в? КС 3* 3 55 СЙ =5 Темпе тура. КС * 3 * СЙ X
TiN 2,0 1200 240 150 2300 3
ZrN 1,8 1200 240 150 2300 3
HfN 1,8 1200 240 150 2300 3
V3N 1,8 900 480 100 1850 3
VN 2,3 1300 240 100 1800 3
Nb2N 1,8 900 480 100 1850 3
NbN0.75 2,0 1100 480 — — —
NbN0>97 1,8 1100 480 — — —
NbN 2,3 1200 240 100 1800 3
Ta2N 3,0 900 900 480 1850 3
TaN 3,0 1200 240 100 1800 3
Cr2N 1,8 1300 240 — — —
CrN 2,0 950 240 — — —
Mo2N 1,8 700 480 — — —
личение объема обычно значительно, и если оно не превышает порис-
тости заготовки, спрессованной из металлического порошка, то должно
приводить к уплотнению брикета.
Однако одно лишь реакционное спекание не позволяет получить
достаточно низкую остаточную пористость (она обычно не ниже 10—
15%), что является следствием сильных распирающих усилий, вызван-
ных образованием новых фаз (по Раубу и Плате [738]). Поэтому после
реакционного спекания необходимо дополнительное спекание горячим
прессованием [739], которое приводит к получению изделий с низкой
остаточной пористостью. При горячем прессовании происходит загряз-
нение изделия за счет материала прессформы, так как возможности
79
проникновения этих загрязнений в предварительно спеченный брикет
резко сокращаются по сравнению с горячим прессованием порошка.
В табл. 32 приведены рекомендованные в работе [739] режимы такого
двойного процесса — реакционного спекания с последующей горячей
подпрессовкой.
Для активирования процесса спекания порошков нитридов, в част-
ности при горячем прессовании, иногда вводят в их состав небольшие
металлические добавки, которые создают возможность перекристалли-
зации частиц через жидкую фазу и частично после этого удаляются
испарением при высоких температурах спекания. Примером может слу-
жить спекание изделий из нитридов урана.
Что касается литья изделий из нитридов, то оно практически не
используется, за редким исключением, например получения изделий
из нитридов урана.
ГЛАВА III 1. НИТРИДЫ ЩЕЛОЧНЫХ МЕТАЛЛОВ
I
НИТРИДЫ
МЕТАЛЛОВ
I ГРУППЫ
ПЕРИОДИ-
ЧЕСКОЙ
СИСТЕМЫ
Рис. 30. Участок диа-
граммы состояния систе-
мы литий—азот.
Нитриды лития. В системе литий—азот уста-
новлено существование нитрида состава Li3N,
имеются отдельные указания о существовании
нитрида (азида) LiN3 [1]. Соединение Li2N, яко-
бы образующееся при действии азота на нитрид
Li3N, не подтверждено
при проведении данной
реакции в работе [2]. Диа-
грамма состояния систе-
мы литий—азот исследо-
вана в (3] только на уча-
стке Li—Li3N (рис. 30).
В этой работе не удалось
установить характер взаи-
модействия на участке,
прилегающем непосредст-
венно к литию. Исследо-
вание проводилось с не-
сколько загрязненными
железом сплавами, что, по
мнению авторов, вызвало
понижение точки плавле-
ния нитрида до 815°С
(вместо часто упоминаю-
щегося в литературе зна-
чения 845° С).
Обычным методом приготовления нитрида
лития Li3N является действие азота на металли-
ческий литий на холоду или при нагревании [24].
Даферт и Миклауз [4] установили, что нитрид ли-
тия образуется при действии сухого азота на ли-
тий в течение нескольких часов. При нагревании
реакция ускоряется и особенно энергично проте-
кает при температуре 450—460° С (сопровожда-
ется воспламенением) [1,5—9]. В работе [14] Li3N
также получали взаимодействием лития с азотом
при температуре 450° С. Франкенбургер [10], ис-
следовавший кинетику взаимодействия лития с
азотом, установил, что скорость реакции в зави-
симости от температуры находится либо в кине-
тической, либо в диффузионной области. Наибо-
лее полно изучил данный вопрос Клинаев [11],
результаты которого позволяют отнести реакцию азотирования лития
к топокинетическим реакциям, характеризующимся резкой фазовой
границей между исходной и вновь образующейся фазами. Кинетика
реакции азотирования лития при температуре 25° С описывается топо-
кинетическим уравнением Колмогорова—Ерофеева
а = 1 — е-^п,
где а — доля прореагировавшего вещества ко времени т, k — постоян-
ная, п — целочисленный показатель.
На скорость образования нитрида, по данным [11], существенное
влияние оказывают добавки, например при азотировании при 25° С
добавка к литию калия (0,18%) ускоряет реакцию, а добавки магния
(1,13%) или алюминия (0,53%) —замедляют ее. Примеси натрия
(1,45%) и кальция (0,38%) не оказывают заметного влияния на ско-
рость образования нитрида лития.
Кислород и водород — ингибиторы реакции взаимодействия лития
с азотом. Так, присутствие в азоте кислорода более 14 об. % или водо-
рода более 3,5 об.% полностью предотвращает азотирование, независи-
мо от чистоты исходного металла.
По данным [1059], нитрид лития рекомендуется получать действием
на литий азота под давлением в течение 1—2 ч. Температуру постепенно
повышают до 170—175° С, давление азота — до 6—8 ат, выдерживают
при этих условиях 4—6 ч, после чего продукт охлаждают и выгружают.
Выход нитрида лития составляет 98—99%, отношение содержания
Li : N в получаемом нитриде 3,0±0,1, нитрид получается в форме тем-
но-красных или коричневых пластинок, температура плавления 845° С,
плотность при 20° С — 1,390 г/см? (см. также [1071]).
При действии воздуха на литий образуется смесь нитрида (75%)
и кислородных соединений лития неопределенного состава [12]. Приме-
си к литию оказывают существенное влияние на скорость взаимодейст-
вия его с воздухом при температуре 20° С (рис. 31). Повышение влаж-
ности воздуха (рис. 32) и температуры также увеличивают скорость
азотирования-окисления. Выше 60° С возрастает устойчивость лития
по отношению к воздуху (при наличии защитной пленки, состоящей в
основном из нитрида лития).
Нитрид лития, полученный взаимодействием лития с азотом при
температуре 450—460° С, представляет губчатое вещество от черно-
голубого до фиолетово-черного цвета, иногда в проходящем свете име-
ет рубиново-красную окраску (таким же цветом обладают смеси LisN
и Li2O).
Нитрид лития имеет гексагональную структуру с периодами ре-
шетки, приведенными в табл. 34.
82
Нитрид лития на воздухе быстро изменяется (поэтому хранится в
среде азота), при действии 'воды разлагается с образованием гид-
роокиси и аммиака
Li3N +- ЗН2О -> 3LiOH + NH3.
При нагревании в водороде нитрид лития переходит в гидрид ли-
тия с образованием аммиака. Реакция проходит через промежуточные
стадии образования амида LiNH2 и
имида Li2NH. При нагреве до тем-
Рис. 31. Влияние примесей на взаимодействие лития с воздухом.
Рис. 32. Взаимодействие лития с воздухом различной влажности:
/ — влажность 100%; 2 — влажность 50%; 3 — влажность 30%; 4 — сухой воздух
пературы 800° С разъедает железо, никель, медь, платину, кварц, фар-
фор [13].
С нитридами других металлов нитрид лития дает соединения типа
Li3N-MeN [236]. Взаимодействие нитрида лития с азотом при темпера-
турах 400—500° С и давлении 300 ат не приводит к образованию выс-
ших нитридов лития [2].
В работе [241] изучена растворимость нитрида лития в расплав-
ленных солях (табл. 33).
При температурах до 360° С нитрид лития практически не проводит
электрического тока, но при повышении температуры примерно до
550° С его электропроводность быстро увеличивается [14], причем в
пределах 350—549° С кривая зависимости х от — состоит из двух отрез-
ков, пересекающихся в точке, отвечающей температуре 446° С, и
х—12,3-10 6 ом~х• слг1. Температурная зависимость электропроводно-
сти описывается выражением х = 5,28-10~2 ехр (—6196/7') + 4,ЗХ
Х107 ехр (—21700/7'), характерным для ионной проводимости. При
электролизе Li3N при 480—550° С на катоде выделяется литий, на ано-
де — азот, а на электродах появляется э. д. с. поляризации, что доказы-
6*
85
вает наличие в Li3N иона №~ Основные свойства нитрида лития при-
ведены в табл. 34.
Нитрид натрия. В отличие от лития натрий взаимодействует только
с азотом, переведенным в атомарное состояние электрическим разрядом,
Таблица 33 Растворимость Li3N в расплавленных солях облучением паров натрия в атмосфере азота <[15], или действием азота на гидрид
Солевой расплав Температу- ра, °C Растворимость Li3N, молей на 1 моль рас- плава натрия [9]. В обоих слу- чаях получается нитрид со- става Na3N. Более изучен азид нат-
КС1 —58,3 мол. % LiCl LiCl2 — 28 мол. % LiF LiCl LiCl — 23 мол. % LiF — 21 мол. % LiH LiBr 495—635 535—610 620—695 495—585 590—665 0,063—0,080 0,161—0,220 0,128—0,168 0,175—0,236 0,11—0,126 рия NaN3, образующийся только косвенными путями [15, 16]. Нитрид NaN3 получается пропусканием закиси азота над рас- плавленным амидом натрия
2 NaNH2 + N2O -> NaN3 4- NaOH 4- NH3.
Эта реакция, по данным [9], протекает при температуре 100—160° С
в течение 9,5 ч с выходом около 90%. NaN3 образуется также по ре-
акциям
NaNO3 % 3 NaNH2 - NaN3 + 3 NaOH % NH3 (175°C),
N2H4 • H2O + C2H5NO2 % NaOH -> NaN3 + C2H5OH + 3 H2O,
Na %- NH4N3 4" NH3 (жидкий) -* NaN3 4- 2 NH3 4- H.
Нитрид натрия Na3N устойчив при комнатной температуре, на
воздухе и в водороде разлагается водой, при нагревании до 300° С дис-
социирует, легко реагирует с хлором, фосфором и серой. Разбавленные
кислоты разлагают нитрид с образованием аммиака и соответствующих
солей натрия.
Азид натрия NaN3 — белое негигроскопическое вещество, при ком-
натной температуре растворяется в воде с образованием электропро-
водного раствора (при 17° С в 100 ч. Н2О растворяется 41,7 ч. NaN3)
[15, 24]. При высоких температурах NaiN3 разлагается водой по схеме
3 NaN3 4- 3 Н2О -> 3 NaOH 4- NH3 -J- 4N2.
Растворяется в неводных растворителях: бензине, спирте, бензоле.
В 100 ч. безводного спирта при 16° С растворяется 0,315 г NaN3, при
0° С — 0,22 г NaN3.
При нагревании до 275° С NaN3 разлагается без плавления.
«4
=(
S
со
св
S
Расчетные данные по [1017].
85
В работе [22] изучены монокристаллы азида NaN3. Монокристаллы
размером 4X6X1 мм получали в специальной конвекционной трубке.
Изучение полос двойного лучепреломления монокристаллов показало,
что они пересекаются под углом 120° и представляют плоскости в кри-
сталле под некоторым углом к оси с с индексом (ПО). Появление плос-
костей связывается с явлениями скольжения, комбинации скольжения
и двойникования, многократного двойникования.
Облучение у-лучами с дозой 107 р вызывает появление глубоких
выемок вдоль этих линий пересечения, а также пирамидальных ямок
травления.
Обзор свойств нитрида и азида натрия см. также в [1017].
Нитриды калия. Аналогично натрию калий не взаимодействует с
молекулярным азотом даже под давлением и при высоких температу-
рах [15]. При действии на калий азота, активированного электрическим
разрядом, образуются два соединения калия с азотом: нитрид K3N и
азид KN3, причем нитрид образуется в значительно меньших количест-
вах, чем азид [15].
Обычным методом получения нитрида калия является нагревание
гидрида калия КН в струе азота [6, 9], а также нагревание азида калия
в вакууме [16].
Азид калия KN3 получают действием калия на раствор NH4N3 в
жидком аммиаке по реакции [17, 15]
К К NH4N3 - KN3 + NH3 К 1/2Н2;
при действии KNO3 на жидкий аммиак [18, 15]
KNO3 K2NH8->KN3 + 3H2O;
пропусканием закиси азота при 270—280° С над амидом калия
[19, 20, 15]
2 KNH2 + N2O KNS + NH3 + КОН.
В последнем случае образуется азид KN3 с высоким выходом.
Нитрид K3N образуется из элементов по экзотермической реакции.
При нагревании с азотом или окисью ртути нитрид калия теряет азот,
с водой реагирует с образованием КОН и NH3 [9], при нагревании с
фосфором и серой дает фосфиды и сульфиды калия [21, 9]. При взаимо-
действии с разбавленными кислотами нитрид калия образует аммиак и
соответствующую соль калия. Нитрид калия — малоустойчивое соеди-
нение и в чистом виде выделяется с большими трудностями.
Нитрид (азид) KN3 представляет собой блестящие бесцветные кри-
сталлы тетрагональной структуры. Хорошо растворяется в воде [15, 24]:
t, СС 0 10,5 15,5 17 100
KN3 (г иа 100 г НгО) 41,4 46,5 48,9 49,6 105,7
86
В воде гидролизуется с образованием едкого калия и аммиака
KN3 4- Н2О КОН 4- NH3.
В присутствии платиновой черни азид калия разлагается водой по
схеме
3KNg + ЗН2О зкон 4- 4N2 + NHa.
Азид калия хорошо растворяется в жидком аммиаке, метиловом и
этиловом спиртах (в 100 г этилового спирта при 0° С растворяется
0,16 г KN3), менее растворим в бензоле [15, 16].
Раствор азида калия не реагирует с йодом, но в присутствии ма-
лых количеств CS2 (0,04—0,08 моля на 1 моль KN3) идет энергичная
реакция
2 KN3 + 2I2 -> 2 KI2 + 3 N2.
В работе [22] монокристаллы KN3 размером 6x6x3 мм получали
путем испарения насыщенного водного раствора в продолжение не-
скольких месяцев. Как и в кристаллах NaN3, были обнаружены полосы
двойного лучепреломления, пересекающиеся под углом 90° и отвечаю-
щие плоскостям с индексом (112). Причины их образования, а также
результаты изменения под действием у-лучей те же, что и для моно-
кристаллов NaN3.
Азид KN3 — взрывчатое вещество, плавится с разложением.
Нитриды рубидия. В системе рубидий—азот имеется два соедине-
ния: Rb3N и RbN3. Нитрид рубидия Rb3N получают нагреванием гидри-
да RbH в струе азота [6, 9], а также разложением азида RbN3 при
температуре порядка 340° С [23].
Азид RbN3 получают взаимодействием аммиака с карбонатом, ги-
дроокисью рубидия или реакцией между сульфатом рубидия и азида
бария BaNe [23].
Нитрид рубидия Rb3N красного цвета, устойчив при обычной тем-
пературе на воздухе и в водороде, при нагревании в среде водо-
рода переходит в гидрид RbH, легко реагирует с хлором, фосфором и
серой. При взаимодействии RbN3 с разбавленными кислотами образу-
ется соответствующая соль рубидия и аммиак [25].
Азид RbN3 хорошо растворим в воде: при 16° С растворимость
составляет 51,7%. Растворимость в абсолютном спирте — 0,182 г на
100 г спирта, нерастворим в эфире [24]. При нагревании переходит в
Rb3N, а при нагревании в вакууме разлагается до металла и азота, что
используется для получения рубидия высокой чистоты [23]. В отличие
от азидов лития, натрия и калия не взрывается.
Нитриды цезия. Нитрид цезия CsN получается нагреванием гидри-
да CsH в струе азота или осторожным нагревом азида CsN3 (при
87
340° С). Азид CsN3 образуется действием аммиака на карбонат це-
зия [24]
Cs2CO3 + 2 NH3 2 CsN3 + Н2О -f- СО2
либо при взаимодействии между сернокислым цезием и азидом бария
BaN6 [9, 23].
Нитрид цезия CsN3 представляет собой порошок серо-зеленого
цвета, в сухом воздухе устойчив, во влажном — разлагается с выделе-
нием аммиака, очень гигроскопичен. Растворимость при 16° С составля-
ет 307,4 г на 100 г воды и 1,037 г на 100 г абсолютного спирта, нераст-
ворим в эфире [24]. В водороде при обычной температуре устойчив, а
при нагревании переходит в гидрид CsH. Нитрид рубидия CsN3 легко
реагирует с серой, фосфором и хлором; с разбавленными кислотами
взаимодействует с образованием солей и аммиака [25].
При нагревании разлагается на нитрид Cs3N и азот, при ва-
куумном нагреве полностью теряет азот, переходя в металличе-
ский цезий. Азид CsN3, как и соответствующий азид рубидия, не
взрывается.
Свойства нитридов и азидов щелочных металлов приведены в
табл. 33; термодинамические характеристики даны в [1017].
2. НИТРИДЫ МЕТАЛЛОВ ПОДГРУППЫ МЕДИ
Нитриды меди. Диаграммы состояния металлов подгруппы меди с азо-
том не изучены. Азот не растворяется ни в твердой, ни в жидкой меди,
по крайней мере до температур в 1400° С, при которых проводились ис-
следования [26, 27]; не обнаружено взаимодействия меди с азотом до
900° С [28].
Тем не менее в системе медь—азот обнаружено существование
трех соединений Cu3N, CuN3 и Cu(N3)2, получаемых косвенными ме-
тодами.
Нитрид Cu3N получается пропусканием аммиака над тонкоизмель-
ченными СиО, Си2О или CuF2 при температуре 250—280° С [29]. Пред-
положения об образовании нитридов меди при пропускании аммиака
над медью при температурах 900—1000° С не были подтверждены.
Хрупкость меди в этих условиях объясняется не образованием химиче-
ских соединений, а влиянием на медь водорода, получающегося при
диссоциации аммиака.
Cu3N образуется также при действии аммиака на гидроокись
меди [1]
6 NH3 4- 3 (4 СиО • Н2О) = 2 Cu3N ф 3 Cu2O + 2 N2 ф 12 Н2О.
88
Азид меди CuN3 получают восстановлением сульфата меди с
помощью KHSO3 при добавлении азида натрия [30].
Нитрид меди Cu3,N представляет собой темно-зеленый порошок,
устойчивый на воздухе в обычных условиях, но разлагающийся при
нагревании в вакууме до 450° С. Cu3N имеет кубическую структуру ти-
па ReO3, пространственную группу Pm3m(O'h) с одной формульной
единицей в элементарной ячейке [31, 32]. Нитрид меди Cu3N— полу-
проводник с электросопротивлением 6 • 102 ом • см при комнатной тем-
пературе и шириной запрещенной зоны 0,23 эв [33]. На рис. 33 показа-
на температурная зависимость электросопротив-
ления Cu3N.
Легко растворяется в кислотах, при раство-
рении в НС1 образуются полухлористая медь и
хлористый аммоний [34], бурно разлагается в кон-
центрированных H2SO4 и HNO3. При 400° С в
токе кислорода окисляется с сильным разогре-
вом, при 450° С разлагается в вакууме [24].
Азид меди CuN3 — малоустойчивое соеди-
нение, образующееся с большим ростом сво-
бодной энергии /\Е°, составляющим по данным
[35], для реакции Cu+3/2N2=CuN3. 71219 кал.
Азид CuN3 имеет тетрагональную кристалли-
ческую структуру, пространственная группа C®/i
с восемью формульными единицами в элементар-
Рис. 33. Температурная
зависимость электросо-
противления Zn3N2 и
Cu3N.
ной ячейке [30]. Структура состоит из ионов меди
и линейных групп из атомов азота (Ni — в середине, N2 — снаружи),
которые расположены в виде цепочек в направлении (111). Кратчайшие
расстояния Си—Cu = Ni—Ni = 3,36; Си—Ni = 2,795; Си—N2=2,23; 3,28 и
3,56; N—N2=3,56 А.
Ранее структура CuN3 считалась ромбоэдрической [36].
Авторы [24] указывают на существование еще азида двухвалентной
меди Cu(N3)2, который получается по реакции
Си (NO3)2 + 2 NaN3 = Си (N8)2 + 2 NaNO3
либо при взаимодействии тех же реагентов в гидратированном со-
стоянии в спиртовом растворе, либо разложением соединения
Cu(N3)2-2NH3 [37]. Азид в зависимости от метода получения имеет вид
черно-коричневого порошка или непрозрачных игл того же цвета (кри-
сталлизуется в ромбической системе). Плохо растворим в воде и орга-
нических растворителях, легко — в кислотах, СН3ООН и NH4OH, в
водном растворе гидразина восстанавливается до CuN3. Легко взрыва-
ется, отличается сильными детонационными свойствами, детонируя в
6 раз сильнее азида свинца и в 450 раз сильнее гремучей ртути [24].
89
Свойства нитридов и азидов металлов подгруппы меди
Л
=г
х:
Л
Н
Нитриды серебра. Азот
не растворяется ни в твер-
дом, ни в жидком серебре
до 1300° С, однако косвен-
ными методами получены
нитрид серебра Ag3N и азид
AgN3 [26].
Нитрид серебра Ag3N
образуется при длительном
хранении аммиачных раст-
воров окиси серебра или
разложением аммиачного
раствора Ag2O спиртом, а
также ацетоном и осажде-
нием аммиачного раствора
AgCl твердой щелочью [39].
При растворении Ag2O в
концентрированном раство-
ре аммиака и последующем
отстаивании получаются
препараты Ag3N, загрязнен-
ные примесью серебра и
Ag2O. Загрязненные сереб-
ром препараты Ag3N обра-
зуются и при выдержива-
нии соединения Ag2N-2NH3
над серной кислотой [24].
Нитриды серебра могут
быть также получены при
взаимодействии паров се-
ребра с аммиаком при
1280° С [6].
Азид серебра AgN3 по-
лучается взаимодействием
азотнокислого серебра с
сернокислым гидразином
пли гидразонковой кисло-
той [1, 9] либо взаимодейст-
вием азотнокислого сереб-
ра с азидом натрия [24]
AgNO3 +- NaNs =
= AgN3 4- NaNOs.
99
Нитрид Ag3N имеет вид черных хлопьев или черных блестящих
кристаллов, нерастворим в воде, растворим в разбавленных минераль-
ных кислотах, с концентрированными кислотами реагирует со взрывом
[24], на воздухе в сухом и влажном состоянии устойчив, разлагается
при комнатной температуре медленно. Устойчив на холоду против дей-
ствия щелочей, в вакууме при комнатной температуре разлагается
быстро [39]. Важной особенностью Ag3N является способность взры-
ваться (гремучее серебро Вертело), он разлагается со взрывом на воз-
духе при 165° С, а также в соприкосновении с различными твердыми
веществами, при трении, ударе.
Нитрид серебра Ag3N имеет кубическую гранецентрированную ре-
шетку, в которой атомы азота занимают октаэдрические поры в
ГЦК-решетке атомов серебра [39].
Азид серебра AgN3 — бесцветное вещество, кристаллизующееся
в виде игл (ромбическая псевдотетрагональная решетка, ромбически
искаженный тип KN3), при нагревании до температуры 170—180° С
окраска меняется на серо-фиолетовую. Легко растворим в водном раст-
воре аммиака, в цианистом калии, трудно растворим в воде (0,01 вес. ч.
на 100 вес. ч. воды) и азотной кислоте. Взрывчатое вещество, взрывает-
ся при ударах, а также при нагревании примерно до 300° С.
Нитриды золота. Азот не растворяется в твердом и жидком золоте
до температуры 1300° С [26]. Известны два нитрида золота. Один из
них — гидратнитрид Au3N • 5НаО — получается при действии аммиака
на окись золота с последующим кипячением в воде образовавшегося
продукта. При взаимодействии АиО с аммиаком образуется нитрид
двухвалентного золота Au3N2 [1, 9]
ЗАиО + 2 NH3 = Au3N2 ф ЗН2О.
Оба нитрида — взрывчатые вещества. В табл. 35 приведены основ-
ные свойства металлов подгруппы меди.
ГЛАВА IV
НИТРИДЫ
МЕТАЛЛОВ
II ГРУППЫ
ПЕРИОДИ-
ЧЕСКОЙ
СИСТЕМЫ
1. НИТРИДЫ БЕРИЛЛИЯ, МАГНИЯ И ЩЕЛОЧНО-
ЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Нитриды бериллия. Система Be—N2 не исследо-
вана, установлено лишь существование в ней ни-
трида Ве3Нг и азида Be(N3)2. Нитрид Be3N2 полу-
чается при взаимодействии порошкообразного
бериллия с азотом при температуре 500° С и при
900° С в случае использования кускообразного
бериллия [6]. Реакция образования нитрида идет
медленно (от 24 до 60 ч), для ее ускорения до-
бавляют к азоту водород (2—6%), являющийся
катализатором данной реакции.
Кинетика реакции взаимодействия бериллия
с азотом исследована в работах Гульбранзена и
Эндрю (41], которые установили, что скорость ре-
акции при температурах до 850° С мала, при
950° С реакция протекает в два раза медленнее
реакции окисления бериллия. В результате про-
веденных опытов [41] по азотированию при
76 мм рт. ст. в пределах 725—925° С был обнару-
жен параболический закон азотирования с энер-
гией активации Be3N2 75000 кал/моль. При азо-
тировании бериллия образуются весьма плот-
ные, газонепроницаемые пленки, сильно затруд-
няющие процесс азотирования [76].
Согласно [42], азотирование чушкового бе-
риллия при пропускании 64-кратного количества
азота проходит при 700°С на 56%, при 800° С —
на 70%, при 900° С почти полностью заканчива-
ется даже в отсутствие водорода. Азотированием
бериллиевого порошка при 1000° С в течение 12—
18 ч смесью азота с 5% водорода был получен
порошок нитрида бериллия, содержащий 95—
98% Be3N2, 1—5% ВеО и 0,05—0,2% Ве2С [42].
Нитрид бериллия можно получать азотиро-
ванием металла аммиаком: из технического тон-
коизмельченного бериллия после азотирования в
токе сухого NH3 в продолжение 3 ч при 850° С
и последующего трехкратного азотирования при
1000° С получаются продукты, содержащие 94—
95% Be3N2. При использовании более чистого ме-
талла этим методом можно получить и более чи-
стый нитрид [24]. Аналогичный метод получения
92
нитрида бериллия азотированием порошка металла аммиаком при тем-
пературе 1000° С описан Бусевым [6].
Чиотти [43] азотированием порошка бериллия аммиаком в продол-
жение 5 ч при 800° С и 17 ч при 900° С получал продукты, содержащие
соответственно 46,4 и 43,2% азота (по сравнению с 50,89% азота в
ВезИг)- После обработки этих продуктов в вакууме при 2000° С полу-
чался рентгенографически чистый нитрид бериллия.
Перспективным методом получения нитрида бериллия технической
чистоты является восстановление окиси бериллия углем в токе азота
[9], а также нагревание карбида бериллия Ве2С в токе азота при
1250° С или в токе аммиака при 1000° С [42].
Азид бериллия Ве(Нз)2 получают нагреванием соединения
Ве(СН3)2, замороженного при температуре жидкого азота с избытком
эфирного раствора HN3, однако выделить его из водных растворов
весьма трудно, вследствие склонности к гидролизу.
Растворимость азота в бериллии невелика [44].
Нитрид бериллия ВезЫ2 представляет в чистом состоянии белое
вещество, в загрязненном — порошок серого цвета. Кристаллизуется в
кубической системе по типу анти-М.п20з, плавится при температуре
2200° С, разлагается с выделением азота при температурах выше
2240° С (упругость диссоциации достигает 1 ат при 2400° С), в вакууме
возгоняется при 2000° С [45].
По данным [961], нитрид бериллия испаряется при температурах
1640—1950° К конгруэнтно, одновременно происходит его разложение
по реакции ВезН2 (тв)=ЗВе (газ)+.N2 (газ); давление азота (ат)
определяется уравнением
lgpN2= - 1,898-104?"1 + 6,213.
Энтальпия диссоциации равна —136±5 ккал!моль, что хорошо со-
гласуется с принятыми в литературе данными (—137,8+1,5 ккал/моль).
Лэнгмюровская энтальпия активации для образцов с пористостью око-
ло 18% равна 430,2 + 6,8 ккал)моль, что на 22% выше равновесной эн-
тальпии. Коэффициент испарения Be3N2 равен 0,001. Нитрид бериллия
обладает высокой твердостью, в присутствии добавок А120з и некото-
рых других приобретает способность фосфоресцировать [6].
Теплоемкость Be3N2 выражается рядом Ср = 15,45+19,64 • 103Т—-
—4,80-105Т 3 кал/град• моль [47].
На воздухе нитрид бериллия вполне устойчив, очень медленно
разлагается кипящей водой с выделением аммиака, разбавленными
растворами галоидных кислот разлагается с образованием галоидных
солей бериллия, концентрированная НС1 взаимодействует с Be3N2 при
температуре 700—800° С с образованием безводного ВеС12
Be3N2 + 8НС1 = ЗВеС12 + 2NH4C1.
93
прессованием в графитовых
Таблица 36
Влияние добавок Be3N2 на меха-
нические свойства бериллия [42]
Присад- ка Ве3 N2, % Предел проч- ности, кг/мм2 Относительное удлинение, %
6 00° с 800°С 600°С 800°С
0 14,3 4,4 18,2 6,6
5 17,5 —. 3,4 —
10 ~14—15 —. ~2,3 —
20 ~14—15 6,1 ~2,3 6,4
При кипячении с кислородсодержащими кислотами разлагается
медленно. При температурах до 500° С ВезМ2 устойчив против окисле-
ния сухим кислородом, при температуре 1000° С быстро окисляется
[42, 45].
Азид бериллия быстро разлагается во влажном воздухе, в пламени
вспыхивает, не детонирует [44, 9].
Изделия из нитрида бериллия приготовляют спеканием, горячим
прессформах при температуре 1800—
1900° С, давлении 140 кг)см2 с получением
плотности, близкой к теоретической [42].
Добавление нитрида бериллия к бе-
риллию приводит к понижению пластич-
ности бериллия, увеличению его проч-
ности при повышенных температурах
(табл. 36).
Нитриды магния. В системе маг-
ний—азот установлено существование ни-
трида Mg3N2 и азида Mg(N3)2, причем
Mg3N2 существует в одной кристалличе-
ской форме [1025]. Образование нитрида
магния было обнаружено Сент-Клер Де-
вилем и Кароном [48] при возгонке маг-
ния на воздухе. Нитрид магния при этом
образуется в форме бесцветных игольчатых кристаллов, быстро разла-
гающихся водой с образованием окиси магния и выделением аммиака.
Первое систематическое исследование процесса образования нитрида
магния было проведено Бциглебом [49] нагреванием опилок магния в
токе аммиака или азота в фарфоровых лодочках. Замечено, что в состав
нитрида переходит кремний из лодочки, поэтому рекомендовалось ис-
пользовать для получения нитрида лодочки, уже бывшие в употреблении.
В этой же работе нитрид магния, загрязненной кремнием, получался
нагревом силицида магния в токе азота.
Хартунг [50] получил нитрид магния при нагреве порошка магния
в среде аммиака и установил, что реакция взаимодействия между
магнием и азотом начинается при температуре 450° С, протекает наи-
более интенсивно при 600—700° С, при 90ОО?С образовавшийся нитрид
диссоциирует. Ч
Мургулеску и Чисмару [2'44] показали, /что при 500—580° С магний
азотируется азотом при атмосферном давлении. Температурная зави-
симость скорости реакций имеет яараб.одйческий характер, а энергия
активации процесса составляет 31,7 ккаА^оль.
Мерц [51] нашел, чтр йитрид магния тлегко получается при простом
нагревании магния в стеклянной пробир&е на воздухе в пламени газо-
вой горелки. При этом, как и в опытах [49], обнаруживается переход
94
7
кремния из стекла в состав нитрида, сопровождающийся помутнением
и почернением стекла.
Кайзер [52] получал нитрид магния действием азота на нагретые
сульфид и хлорид магния, а также на гидрид магния, и обнаружил,
что гидрид магния легко переходит в нитрид, а последний — обратно в
гидрид
3MgH2 + 2N2 = Mg3N2 + 2NH3,
2Mg3N2 + 12H2 = 6MgH2 4NH3.
По мнению Смитса и Пашковецкого [53, 54], для приготовления
нитрида магния лучше применять не азот, а аммиак. Пропусканием
сухого аммиака над магнием, нагретым до 600° С, получали Mg3N2 и
в [6]. Напротив, Нойманн с сотрудниками [55] придерживается мнения,
что при азотировании магния аммиаком происходит сильная адсорбция
нитридом аммиака, что задерживает его диссоциацию и участие в про-
цессе нитридообразования.
Кирхнер [56] получал поверхностные слои из нитрида магния на
компактных магниевых образцах при нагревании их на воздухе.
По данным Эйдмана и Мозера [57], нитрид магния можно полу-
чать при нагреве на воздухе порошка магния в смеси с легкоокисляю-
щимися металлами, а также в смеси с карбидами некоторых металлов.
Например, смесь равных частей магния и железа, нагретых в открытом
тигле, образует продукт, содержащий около 36% нитрида магния. По
этим же данным, при нагреве порошка магния в плотнозакрытом тигле
с очень небольшим отверстием в крышке образуются два слоя: верх-
ний — из окиси магния и нижний — из нитрида. Эйдманн [58] наблю-
дал образование нитрида магния при нагреве до красного каления по-
рошка металлического магния с нитридами бора и кремния, цианидами
щелочных и щелочноземельных металлов.
Чарвази [59] получал нитрид магния нагреванием смеси магниевых
опилок с углем сначала в среде водорода, а затем на воздухе.
Менер [60] и Нойбергер [61] приготовляли нитрид магния аналогич-
ным методом: нагревом смеси магния и сажи в среде азота.
Фурнасос [62] обнаружил, что нитрид магния получается при вза-
имодействии магния с цианидами, причем реакция типа
2KCN + 3Mg = Mg3N2 + 2К % 2С
протекает весьма энергично.
Из всех перечисленных, часто весьма остроумных методов получе-
ния нитрида магния практическое применение имеют только методы,
основанные на обработке нагретого магния азотом или аммиаком. Од-
нако до сих пор не выработаны оптимальные температурные условия
проведения этих процессов.
95
Большинство исследователей предлагают относительно высокие
температуры азотирования магния: в пределах от 700 до 900—950° С.
Так, по данным Жукова [63], взаимодействие магния с азотом с обра-
зованием нитрида начинается при 780—800° С, по данным Нойманна,
Крегера и Куна [64] рекомендуется получать нитрид магния азотиро-
ванием порошка магния, очищенного магнитной сепарацией от железа
в токе обескислороженного азота в течение 4—5 ч при температуре
800—850° С (содержание азота в таком нитриде составляет 27,3—
27,6%, что почти точно соответствует расчетному содержанию азота в
MgaN2, равному 27,74%). Портер [65] указывает, что азотирование маг-
ния аммиаком, предварительно подогретым до 300° С, или азотом, по-
догретым до 400° С, следует проводить при 900—1000° С. Дэвис [66]
для получения нитрида магния в токе азота или аммиака рекомендует
применять температуру, находящуюся в пределах между температурой
сублимации и кипения магния для того, чтобы, возгоняющийся магний
мог разрушать слой нитрида с целью непрерывного азотирования.
Штаккельберг и Паулюс [46] получали нитрид магния при нагревании
магния в продолжение 4 ч в токе аммиака при температуре 850° С с
последующим 1,5 ч нагреванием в токе азота для удаления адсорбиро-
ванного аммиака. В этой же работе описан способ приготовления мо-
нокристаллов нитрида магния, заключающийся в быстром нагреве кус-
ка магния до 1050°С (т. е. почти до точки плавления, равной 1107° С)
в медленном токе азота в железной лодочке. При этом на поверхности
куска металла и на стенках лодочки образуются прозрачные бесцвет-
ные иглы кристаллов нитрида магния, имеющие габитус шестигранных
призм с трехгранной пирамидой или плоскостью на свободном конце.
Направление игл (111), боковых граней (211), пирамиды или концевой
площадки (Ш).
Митчелл [68] получал довольно чистый нитрид магния (с примесью
0,9% MgO) пропусканием азота над опилками магния в железной
лодочке сначала при 650—700° С в течение 3—4 ч, с последующим по-
вышением температуры до 950° С и выдержкой при этой температуре
в течение 12 ч.
Мургулеску и Чисмару [244] показали, что при температурах 500—
580° С магний азотируется азотом при атмосферном давлении. Скорость
реакции подчиняется параболическому закону, энергия активации рав-
на приблизительно 31700 кал/моль.
Авторы [244] исследовали азотирование компактного магния при
давлении азота от 7,5 до 30 ат, при температурах от 575 до 635° С в
течение длительного времени (более 115 ч). В этой работе, как и в
[271], показано, что азотирование с поверхности протекает медленно,
образуется нитрид магния, близкий по составу к стехиометрическому.
Опыты по получению нитрида магния были повторены нами сов-
местно с Т. В. Дубовик и В. С. Полищуком [242]. Магниевые опилки
96
размером 0,1—0,2 мм азотировали в фарфоровой лодочке, находящей-
ся в реакторе и окруженной азотом. Как показывают полученные дан-
ные (рис. 34), заметное поглощение азота магнием начинается при тем-
пературе 500—550° С, заканчиваясь образованием нитрида (с содержа-
нием 26,5% азота) за 4 ч при 800° С и за ’/2 ч при 900° С. Найдено
значение периода решетки нитрида магния а=9,92 А, что хорошо
совпадает с табличными данными для MgsNa.
Анализ механизма процесса азотирования магния приводит к сле-
дующим выводам. Резкое ускорение процесса азотирования при 700° С
можно объяснить в соответ-
ствии с данными работы [75],
согласно которой, несмотря
на относительно невысокую
энергию активации при азо-
тировании магния, диффу-
зия азота в магний происхо-
дит медленно (примерно в
два раза медленнее, чем
окисление, хотя энергия ак-
тивации окисления почти в
Рис. 34. Зависимость степени азотирования магния
от температуры и времени:
1 — 15; 2 — 30; 3 — 60; 4 — 120; 5 — 240 мин.
два раза выше энергии ак-
тивации азотирования маг-
ния). Это объясняется обра-
зованием на магнии плотной
газонепроницаемой пленки. Очевидно, до температуры 700° С пленка
существенно тормозит процесс азотирования. При 700° С происходит
расплавление магния, нарушение целостности пленки и процесс азотиро-
вания резко ускоряется. Азотирование при температуре 900—1000°С
фактически происходит уже с расплавленным магнием, поверхность азо-
тирования которого ограничена зеркалом расплава. Поэтому азотирова-
ние в общем проходит хуже. При температуре 800° С еще не успевает
образоваться зеркало расплавленного магния, а пленка нитрида уже
разрушена, следовательно, 750—800° С можно считать оптимальной
температурой процесса азотирования.
Указанные особенности процесса образования нитрида магния при-
водят к выводу о необходимости введения в порошок магния перед азо-
тированием достаточно инертного наполнителя, который препятство-
вал бы спеканию порошка магния и образованию на нем сплошной
нитридной пленки. В качестве такого наполнителя был использован
[242] предварительно полученный нитрид магния; для нормального хода
процесса количество нитрида должно составлять 30—40%. Такой метод
позволяет получать нитрид достаточно высокого качества по составу.
Кинетика азотирования магния подробно изучена в [75]. Исполь-
зовали чистый магний, содержащий 0,01 % Fe, 0,005% Мп, 0,005% А1,
97
0,001% Si, 0,0001% Си, 0,005% Pb, и азот, тщательно очищенный про-
пусканием над медной стружкой при 400° С через колонку с Mg(C104)2
для удаления О2 и Н2О. На рис. 35 показана изотерма азотирования
магния, которая имеет три участка: начальный, участок азотирования
и участок, на котором начинает сильно сказываться испарение магния.
Влияние температуры азотирования в пределах 415—485° С на рис. 36,
из данных которого выведено уравнение температурной зависимости
константы азотирования
К = 2,2 • 104 ехр (22300/ЯТ) [мг/см2- ч].
Рис. 35. Азотирование магния при 465°С (pNj = 100 мм рт. ст.):
I — начальный период азотирования; II — период азотирования; III — период испарения.
Рис. 36. Температурная зависимость азотирования магния (pN2 = 100 мм рт. ст).
Сравнение с константой скорости окисления магния К =
= 6,2 • 1012 ехр (50500/7??') показывает, что энергия активации процесса
азотирования в два раза меньше энергии активации процесса окисле-
ния (рис. 37). Однако скорость азотирования во много раз меньше ско-
рости окисления, что объясняется (в пределах второго кинетического
участка процесса азотирования) образованием плотной, газонепроница-
емой пленки нитрида на магнии. Увеличение давления азотирования
вызывает повышение константы скорости азотирования (рис. 38).
Сравнение кинетики азотирования и окисления магния было вы-
полнено также в [272]. Показано, что различия в процессах связаны с
тем, что трещинообразование в окисленном слое происходит легче, чем
в азотированном. И при окислении, и при азотировании вначале обра-
зуется стабильный во времени слой, дальнейшее увеличение которого
до некоторой критической толщины вызывает появление трещин.
В работе [247] также изучена кинетика азотирования мелких по-
рошков магния марок М-3 (99,57% Mg) и М-4 (99,60% Mg) с при-
месью железа (0,01—0,023%). Размер частиц порошка М-3 составлял
в среднем 250—300 мк, а М-4 — около 50—100 мк. Установлено, что
при 400° С в обоих случаях временная зависимость азотирования под-
чиняется логарифмическому закону, а при 450° С — параболическому.
98
В табл. 37 приведены кинетические характеристики процесса азотиро-
вания порошков магния.
Отмечается, что прохождение процесса азотирования по затухаю-
щим кривым обусловлено защитными свойствами пленки из нитрида
магния. Порошок М-4 более реакционноспособен, чем М-3, вследствие
магния. Порошок М-4 более реакцион-
носпособен, чем М-3, вследствие очень
Рис. 37. Температурная зависимость констант реакции окисле-
ния и азотирования магния.
Рис. 38. Зависимость скорости азотирования магния от давле-
ния при температуре 465° С.
Таблица 37
Кинетические характеристики процесса азотирования порошков
магния
Марка порош- ка Темпе- ратура, °C Уравнение скорости азотирования Константа скорости реакции К, мг/см2-ч Энергия актива- ции, ккал/моль
ЛА Q 400 Am*=0,0056 Igx** 0,0056
in-о 450 \т = —0,0315+0,0733т— | 49,97
—0,00932т2 0,07425
ЛА Л. 400 Ат =0,00165 IgT 0,00165
i*!-^ 450 Ат =0,00664+0,0021т + | 42,54
+0,000355т2 0,01485
* Am — привес, иа/сек2.
** т — время азотирования, ч.
развитой поверхности (удельная поверхность М-4 равна 3500, а
М-3 — 616 см2/г); об этом, в частности, свидетельствует более низкая
энергия активации азотирования порошка М-4.
Проведенные в этой же работе опыты по окислению порошков маг-
ния на воздухе показали, что образуются одновременно MgO и MgaNg.
7* 99
По данным [246], азотирование магния существенно ускоряется
при добавке к нему в качестве катализатора 5% KHF2.
Азид магния Mg(N3)2 получается растворением магния в азотисто-
водородной (гидразонковой) кислоте [1, 9].
Нитрид магния Mg3N2 — рыхлый порошок желто-оранжевого цвета,
сильно флюоресцирующий при облучении ультрафиолетовыми лучами
[68]. Mg3N2 имеет кубическую гранецентрированную решетку, аналогич-
ную решеткам стибнида и висмутида магния, а также нитрида берил-
лия, с периодом, составляющим по разным данным от 9,93 до 9,95 А.
В элементарной ячейке нитрида содержится 16 формульных единиц.
По данным Митчелла [68], нитрид магния существует в трех аллотроп-
ных модификациях а, (3 и у, температура a^p-превращения составляет
823° К (теплота превращения 220 кал/моль), а температура р^у-пре-
вращения— 1061° К (теплота превращения 260 кал/моль).
Изменение теплосодержания отдельных модификаций в пределах
от 350 до 1200° К определяется уравнениями
Д#а =—7125 + 22,817’ ]- 3,65 10"3Т2,
Д/7р = — 10020 + 29,607’,
\НУ --= — 9695 + 29,547’.
По данным Келли [38], молярная теплоемкость различных моди-
фикаций нитрида Mg3N2 может быть вычислена из уравнений
С₽а = 20,77 4-11,20- lO^V (298 — 823 К),
= 20,07+ 10,66 10~37’ (823 — 1061°К),
СРУ 28,50 (1061 — 1300°К).
Удельное электросопротивление нитрида равно 2-106 ом • см [63].
В сухом воздухе нитрид магния устойчив, температура диссоциа-
ции в этих условиях равна 1500° С [1]. Во влажном воздухе нитрид
быстро разлагается с выделением аммиака [69, 24]
Mg3N2 + 6Н2О = 3Mg(OH)2 + 2NH3,
поэтому хранение его возможно только в запаянных ампулах [64].
Вайтхауз [70] нашел, что нитрид магния восстанавливается водя-
ным газом до металлического магния с одновременным образованием
угля и цианамидов, сухой водород не восстанавливает нитрид до метал-
лического магния.
юо
В сухом кислороде нитрид магния горит с сильным накаливанием
и светоизлучением. С концентрированными и разбавленными кислота-
ми и хлористым фосфором реагирует по следующим схемам:
MgsN3 + 8НС1 = 3MgCla 4- 2NH4C1,
Mg3N2 4- 8HNO, = 3Mg(NO3)2 4- 2NH4NO3,
5Mg3N2 4- 6PC1S = 15MgCl2 -4 2P3N6.
Нитрид магния не реагирует с этиловым спиртом, спиртовым раст-
вором глицерина, щавелевой кислотой и фенолом [71].
Взаимодействие нитрида магния с окисью углерода и углекислым
газом изучали Фихтером и Шолли [72], которые установили, что при
температуре 1000—1250° С эти реакции описываются следующими
уравнениями:
Mg3N2 4- ЗСО = 3MgO 4- ЗС ф N2,
2Mg3N2 4- ЗСО2 = 6MgO 4- ЗС 4- 2N2,
причем реакция взаимодействия нитрида магния с СО2 проходит актив-
нее, чем с СО.
Азид магния Mg(N3)2 при нагревании разлагается со взрывом [1].
Физические свойства нитрида магния Mg3N2 приведены в табл. 38.
В настоящее время исследуются полупроводниковые свойства нитрида
магния, однако его практическое использование в этой области весьма
проблематично в связи с малой химической стойкостью.
Более реально применение нитрида магния в качестве дегидрати-
рующего агента в неорганическом и органическом синтезе и резиновой
промышленности [65]. Известно отрицательное влияние образования
нитрида магния в магниевых сплавах [73], где он увеличивает порис-
тость и резко снижает механические свойства, особенно ударную вяз-
кость, как при комнатной, так и при высоких температурах, однако за-
метного влияния на обрабатываемость сплавов давлением и процессы
их старения не оказывает.
Нитриды кальция. Участок диаграммы состояния системы каль-
ций—азот в области сплавов, богатых кальцием, показан на рис. 39 [77].
В системе кальций—азот установлено наличие трех соединений—
Ca3N2, Ca3N4 и CaN6.
Ca3N2 существует в трех модификациях: а, р и у [1025, 1072].
Низкотемпературная модификация черного цвета p-Ca3N2 образу-
ется при температуре выше 350° С взаимодействием азота и аммиака
с металлическим кальцием, переходит при температуре 700° С в
a-Ca3N2, причем переход возможен только в одном р—направлении
[263]. Взаимодействие кальция с азотом начинается уже при комнатной
температуре, при нагревании реакция протекает очень быстро и со
101
вспышкой. Обычной температурой получения p-Ca3N2 является 450° С,
при которой кальций выдерживают в токе очищенного азота в течение
3__4 ц [24]. Присутствие кислорода в кальции резко замедляет процесс
азотирования и повышает температуру начала азотирования вследствие
плохой газопроницаемости пленки из окиси кальция. Напротив, нали-
чие даже небольшой примеси натрия содействует азотированию, так
как натрий препятствует образова-
Рис. 39. Участок диаграммы состояния
системы Са—N.
зуется азотированием кальция при
нию на кальции сплошной пленки
нитрида. При азотировании кальция
аммиаком наряду с нитридом по-
лучается также некоторое количе-
ство гидрида кальция.
Найдено, что p-Ca3N2 имеет
псевдогексагональную структуру;
плотность 2,67 г! см3.
Среднетемпературная коричне-
вая модификация a-Ca3N2, сущест-
вующая в температурном интервале
700—1050° С, имеет кубическую ре-
шетку типа цнтц-Мп2О3, плотность
2,64 г/см3 [1072].
Впервые на существование вы-
сокотемпературной желтой модифи-
кации y-Ca3N2 указал Муассан [264,
265], затем подтвердили другие
авторы [77, 266, 268, 269]; подроб-
но она исследована в [270]. Фаза
y-Ca3N2 имеет ромбическую струк-
туру, плотность 2,63 г/см3. Обра-
температурах выше 1050° С с после-
дующим быстрым охлаждением, т. е. конденсацией из паровой фазы.
Особенно интенсивно идет образование y-Ca3N2 при введении подогре-
того кальция в азот при 1200—1300° С и быстром (практически мгновен-
ном) охлаждении. Хорошие результаты дает азотирование кальция в
лодочке из обожженной А12О3; проведение процесса в лодочках из дру-
гих материалов вызывает образование побочных продуктов (в железных
и стальных — цианамида кальция, а в лодочках из кремнезема — сили-
ката кальция). При нагревании до 900° С происходит внезапный пере-
ход y-Ca3N2 в a-Ca3N3. Этот переход, как и переход возможен
только в одном направлении, т. е. у->а.
Нитрид кальция устойчив при нагревании и плавится без разло-
жения при температуре 1195° С. Очевидно, это относится к y-Ca3N2
или скорее всего к смеси а- и у-фаз, которую наиболее часто получали
в ранних работах при азотировании кальция (например, высокотемпе-
102
ратурному продукту азотирования, имеющему желто-золотистый цвет,
приписывалась кубическая решетка типа «нти-Мп20з [29]). Жуков [63]
принимал, что верхней границей температурной устойчивости нитрида
кальция отвечает температура 1250° С.
Молярная теплоемкость нитрида кальция (,р-СазП2), по Келли [38],
в интервале 298—800° К выражается температурной зависимостью
Ср = 20,44 + 22,0 • 10~3Г кал/моль-град-, несколько иное выражение да-
ется в [76].
Нитрид кальция разлагается водой с образованием Са(ОН)2 и
аммиака, разлагается также слабыми минеральными кислотами, не-
растворим в спирте.
По данным [241], растворимость нитрида кальция Ca3N2 в LiCl из-
меняется в пределах температур 625—720° С от 0,068 до 0,085 молей
нитрида на 1 моль расплава LiCl, что существенно меньше раствори-
мости нитрида лития в расплавленных солях (см. стр. 84).
Нитрид Ca3N4 (пернитрид) получается нагреванием в вакууме
амида кальция [6], а соединение CaNe — азид, получается только из
водных растворов.
Нитриды стронция. В системе стронций — азот известны следую-
щие соединения: Sr2N (субнитрид), Sr3N2, Sr3N4 (пернитрид) и SrN6
(азид).
Субнитрид стронция Sr2N получается разложением в вакууме при
давлении порядка 10-2 мм рт. ст. и температуре 400—500° С нитрида
Sr3N2 в течение 1—2 ч [81]. В свою очередь нитрид Sr3N2 образуется
при нагревании металлического стронция в среде азота или аммиака,
азотирование начинается при 380° С. Этот нитрид может быть получен
также разложением азида SrN6 в вакууме.
Методы и условия получения пернитрида стронция Sr3N4 подробно
исследованы в работе [82]. При действии на стронций при нормальном
или пониженном давлении аммиака (~10 мм рт. ст.) образуется гек-
самин стронция Sr(NH3)6 золотистого цвета, который самопроизвольно
разлагается при нормальной температуре с выделением аммиака и
водорода, превращаясь в соединение Sr(NH2)2 белого цвета, последнее
при 500° С разлагается с выделением аммиака, давая желтоокрашен-
ный SrNH. При нагреве в высоком вакууме эта последовательность
реакций подавляется прямым разложением гексамина до пернитрида
3Sr(NH3)6 = Sr3N4 + 14NH3 + 6Н2
или
3Sr (NH2)2 = Sr3N4 + 2NH3 + 3H2.
Второй путь предпочтительнее, так как происходит более быстрая
откачка меньших объемов газов, но он двухстадийный и менее произ-
водительный по весовому выходу.
103
Свойства нитридов щелочноземельных металлов
Таблица 38
Характеристика Be3Na Mg3N2 GC-CajiNj P-CaaN, T-Ca,N3 CaN, Sr2N Sr3N3 Sr3N4 SrNe=Sr(N3)2 Ba2N BagNz BaN3 BaN,= ==Ba(Na)a
Содержание азота, вес. % 50,89 27,24 18,9 18,9 18,9 67,69 7,40 । 9,64 17,57 48,93 4,85 9,25 16,94 38,12
Кристаллическая структура Кубичес- кая Кубическая гранецент- рированная Псевдо- тетраго- нальная Кубическая типа анти- Мп2О3 [80] Ромбическая [270] — Псевдо- тетраго- нальная 1761 — Ромбическая Псевдогексаго- нальная [46] Моноклинная [88]
Периоды решетки, д: 6,22 кХ [89]
а 8,15[46] 9,93 [46] 3,56 [80] 11,42 [80] — — — — — 11,82[36] — — —
ь — —. — — — — — 11,47 — — —. 22,99 кХ
с 4,12 — — — — 6,08 — — — 7,02 «X
с/а — — 1,157 — — ,— — — — — — — — 105°14
Удельный вес 2,72 2,74 [24] 2,67 (270] 2,64 [270] 2,63 [264, — — — — — — — — 2,936 [9]
Температура плавления, °C 2200 .— — 1195 265] — — 1027 — — — 1000 — —
Температура разложе- ния, °C Температура диссоци- ации в °C при давлении, мм рт. ст.: 2240 1250 [63] 400—500 IO—2 мм рт.ст. 169 [1] 400—500 (10~2 мм рт. ст.) 219—225 [1,9]
1 — 1602 — •— —— — —— —— — — — — —-
10~1 — 1472 — —— .— — — — — — — —
Ю~г 1359 __ — — — — — — — —
10-3 760 2400 1260 — — — — — — — — — —
Теплота образования, ккал/моль 134,7± 1,5 110,24 [68] 105,0± 3,0 [76] — t — —75,8 [9] — 92,2 [84] — —49,0 [84] 53,4±2 [81] 89,9 [9] — 43,6 [85]
Энтропия, кал/г-град 40,6 47,2 25,4 [76] —. — — — — — — — — — —
Удельное электросопро- тивление, ОМ‘СМ — 2.106 [63] (298° К) — — — — — — — [81] 100 —•
Таблица 39
Температурная зависимость
молярной магнитной вос-
приимчивости нитридов
стронция
Температу- ра, °C Молярная магнитная восприимчивость, Х10»
Sr,N2 Sr3N
20 279 155
100 250,3 145
200 250,3 119
мент (в магнетонах бопа
Перед азотированием сырой стронций очищают вакуумной пере-
гонкой при температуре 675—870° С, после чего в специальной аппара-
туре действием на стронций аммиака получают гексамин, разлагаю-
щийся в течение двух дней до амида. Последний разлагается в ваку-
уме, однако при температуре 400° С продукт разложения содержит
около 65% пернитрида, дальнейшее повышение температуры приводит
к его разложению с образованием свободного стронция. Максимальное
содержание пернитрида, которое удалось получить в продуктах [82],
не превышает 70%, остальное — примесь SrNH.
Азиды SrNa или Sr(N3)2 аналогично азидам бария получаются пре-
паративными методами.
Субнитрид стронция — порошок черного цвета. Измерения его маг-
нитной восприимчивости, проведенные в работе [83] (табл. 39), привели
к выводу о его металлической природе. Метал-
лическая связь в решетке обеспечивается од-
ним из четырех электронов атомов стронция,
некомпенсированным их связью с трехвалент-
ным азотом.
Повышенный парамагнетизм Sr3N2, не со-
гласующийся с представлениями, согласно
которым это соединение должно быть диамаг-
нитным, объясняется [83] присутствием в пре-
паратах примеси железа, содержание которой,
по-видимому, резко снижалось при вакуумном
получении из Sr3N2 субнитрида Sr2N.
Измерения магнитных свойств пернитрида
[82] показали незначительный парамагнетизм
этого соединения: эффективный магнитный мо-
) —0,30 при 90° К до —0,60 при 673° К. Полу-
ченные данные [82] свидетельствуют о солеобразном, ионном характере
Sr3N4. Выдвинуты два предположения, объясняющие аномально малую
величину Цэфф: а) наличие антипараллельных и взаимно компенсирую-
щихся спинового и орбитального моментов (при этом на взаимодейст-
вие должно мало влиять повышение температуры), б) существование
зависящего от температуры равновесия: N®- (диамагнитный)
(парамагнитный), сильно сдвинутого в сторону димерного иона. Авторы
считают второе предположение менее вероятным.
Субнитрид стронция реагирует с водой по уравнению [81]
Sr2N % 4Н2О = 2Sr (ОН)3 % NH3 +
Минеральными кислотами разлагается быстро с образованием соответ-
ствующих солей.
Нитрид стронция Sr3Na устойчивое соединение, плавится при тем-
пературе 1027° С, в вакууме разлагается с образованием субнитрида.
104
Азид стронция SrNs — непрочное соединение, легко теряющее азот
с образованием Sr3N2, разлагающееся при нагревании до 169° С со
взрывом. При ударе дает слабую вспышку, сопровождающуюся выде-
лением пламени. Азид образуется по эндотермической реакции с по-
глощением тепла в количестве 49 ккал]моль.
Нитриды бария. В системе барий—азот обнаружено существование
четырех соединений — субнитрида Ba2N, нитрида Ba3N2, пернитрида
BaN2 и азида Ва(Пз)г- Имеются также указания о существовании пер-
нитрида бария Ba3N4 [999], который получают разложением азида ба-
рия Ba(Ns)2.
Субнитрид бария Ba2N получается, аналогично субнитриду строн-
ция [81], разложением Ba3N2 в вакууме (1()-3 мм рт. ст.) при темпера-
туре 450—500° С в течение 1—2 ч по реакции 4Ba3N2 = 6Ba2N + N2 +
+ 35,2 кал.
Нитрид бария Ba3N2 приготовляют нагреванием металлического
бария на воздухе при температурах 260—600° С (наиболее удобно про-
водить азотирование при 450° С в течение 3—4 ч) [24].
Пернитрид бария BaN2 получают [81] взаимодействием нитрида
Ba3N2 с азотом при температуре 400—500° С и давлении 200—320 ат, а
также разложением в тех же условиях азида бария [81, 85].
Азид Ba(N3)2 образуется при взаимодействии гидроокиси бария с
гидразонковой кислотой HN3 [1, 24, 86, 87]
2NaN3 + 2H2SO4 = 2NaHSO4 + 2HN3,
2HNS + Ba(OH)2 = Ba(N3)2 + 2H2O.
Субнитрид бария (порошок черного цвета), как и субнитрид строн-
ция, представляет собой металлоподобное соединение с удельной элект-
ропроводностью порядка 10“2 ом~1 • см-1 [81, 83]. Предполагается, что
металлический характер обусловливается лишним электроном, остаю-
щимся после образования связи между двумя атомами бария (4 элект-
рона) и атомом азота (3 электрона). При этом, однако, совершенно
не учитывается расход электронов на связь между атомами бария в
кристаллической решетке, что и вызывает относительно высокое элект-
росопротивление соединения при сохранении отрицательного коэффи-
циента электропроводности, характерного для металлов.
Субнитрид бария разлагается кислотами и водой с образованием
гидроокиси бария и аммиака.
Нитрид бария Ba3N2 — черный кристаллический порошок с псев-
догексагональной структурой, устойчив в обычных условиях, разлага-
ется при нагревании в вакууме, реагирует с водородом (при 300° С),
окисью углерода (при 700—750° С), разлагается водой на холоду с
образованием гидроокиси бария и аммиака. Пернитрид Ba3N4 разла-
гается водой, HNO3, воздухом [999].
105
Азид бария неустойчив, разлагается со взрывом при 219—225° С,
при ударе в обычных условиях дает слабую вспышку с пламенем,
имеет моноклинную кристаллическую структуру типа КС1О4.
2. НИТРИДЫ МЕТАЛЛОВ ПОДГРУППЫ ЦИНКА
Рис. 40. Температурная за-
висимость поглощения цин-
ком азота.
Нитриды цинка. Азот не растворяется в твердом и жидком цинке [89—
91]. Цинк образует с азотом два соединения — нитрид Zn3N2 и азид
Zn(N3)2 [ZnN6].
Нитрид цинка Zn3N2 получают накаливанием цинка (или паров
цинка) в токе аммиака в течение 17 ч при температуре 500° С, затем в
течение 8 ч при 550° С и 16 ч при 600° С (см.
также [1073]). При таком режиме цинк пере-
ходит в нитрид без плавления; около 2% цин-
ка отгоняется (теряется с током аммиака) [24,
92]. При получении нитрида при 600° С он
медленно разлагается, что вызывает образова-
ние продукта, обедненного азотом, которому
может быть приписана формула ZnsN2-«Zn
[93—95]. По данным Хартунга [50], который
нагревал цинковую пыль в аммиаке в течение
0,5 ч, температурная зона образования стехио-
метрического нитрида очень узка (рис. 40), по-
этому необходимо точно соблюдать темпера-
турный режим. Так, Бентли и Стерн [97], при
азотировании цинковой пыли аммиаком при
650° С (в течение 30 мин) получали нитрид с выходом 36,8%. Этим объяс-
няется примесь свободного цинка в нитриде, получаемом по вышепри-
веденному режиму [92], которая, однако, отгоняется при 600° С.
Другим методом приготовления нитрида Zn3N2 является разложе-
ние амида цинка при 200° С [96]
3Zn (NH2)2 = Zn3N2 + 4NH3,
а также нагревание цианида цинка с азотнокислым аммонием [62]
3Zn(CN)2 4- 12NH4NO3 = Zn3N2 % 6СО2 + 14N3 + 24Н2О.
По данным [98], нитрид цинка приготовляют нагреванием смеси
нитрида кальция с порошком цинка при 700—800° С.
Нитрид цинка образуется при электролизе воды с суспензией хло-
ристого аммония, при использовании цинкового анода и платинового
катода [99]. Образующаяся губчатая масса (удельный вес 4,6, что
ниже плотности нитрида, равной 6,22 г/см3) выделяет при нагревании
106
азот и некоторое количество водорода, что позволяет предполагать об-
разование соединения типа амида, а не чистого нитрида.
Некоторые работы [100—101] посвящены получению нитрида цинка
пропусканием электрической искры между цинковыми электродами в
среде азота. Установка, использованная для приготовления нитри-
да цинка в [101], показана на рис. 41. В стеклянную трубку А помеща-
ют платиновый катод В, впаянный в стекло. Подвижной анод С изго-
товлен из цинка или другого метал-
ла, нитрид которого необходимо
получить. Расстояние между анодом
и катодом регулируют вращени-
ем пробки D, снабженной винто-
вой нарезкой. Пробка уплотняется
ртутью в колпаке S. Боковые труб-
ки F и G используют для собирания
нитрида, образующегося в нижней
части реакционной трубки. В ниж-
нюю часть трубки А подается
жидкий азот и устанавливается дуга.
Пары цинка и азота нагревают, про-
исходит образование нитрида, кото-
рый быстро охлаждают избытком
жидкости, окружающей дугу. Приме-
няют смесь 1 объема азота с 9 объе-
мами аргона, что увеличивает ско-
рость расхода цинкового электрода.
Азид цинка Zn(N3)2 аналогично
Рис. 41. Установка для получения нитри-
да цинка.
Рис. 42. Электросопротивление нитридов
некоторых металлов [33].
азидам щелочноземельных метал-
лов получают косвенными методами [1, 9].
Нитрид цинка Zn3N2 — порошок черно-серого цвета * имеет куби-
ческую решетку типа анга-Мп20з, с периодом решетки 0,743±0,005 А
[102]. Молярная теплоемкость в интервале 298—800° К может быть вы-
числена по уравнению Ср—19,934-20,80 • 10~3Г (кал/моль-град) [38].
По данным [ЮЗ], она составляет:
Температура, °C
25 100 300 419,6
Ср, кал/моль-град 26,10 27,62 31,84 34,46
Нитрид является полупроводником (рис. 33, 42) с сопротивлением
при комнатной температуре 4,5 • 103 ом • см и шириной запрещенной зо-
ны 0,09 эв (рис. 42, [33]).
При нагревании в вакууме нитрид разлагается на цинк и азот при
300° С. при атмосферном давлении — при 700—750° С. Разлагается во-
* Нитрид, приготовленный разложением амидов, имеет зеленоватую окраску.
107
дой с образованием аммиака и гидроокиси цинка [96], разбавленными
кислотами с образованием аммиака и солей цинка. На воздухе доволь-
но устойчив [92].
Азид — сильно эндотермическое, неустойчивое соединение, более
взрывчатое, чем даже азиды щелочноземельных металлов [1].
Нитриды кадмия [1026]. Азот не растворяется до температуры
400° С ни в твердом, ни в жидком кадмии [89]. Как и для цинка, для
кадмия известны нитрид Cd3N2 и азид Cd(N3)2.
Получение нитрида кадмия Cd3N2 азотированием металлического
кадмия аммиаком затрудняется вследствие высокой летучести окиси
кадмия и ее трудной восстановимости до металла. Поэтому для приго-
товления нитрида кадмия обычно используют разложение амида в ва-
кууме при 180° С в течение 36 ч [106, 107, 24]
3Cd (NH2)3 = Cd3N2 + 4NH3.
Повышение температуры разложения вызывает диссоциацию нитрида.
Были попытки получить нитрид кадмия электролизом аналогично
нитриду цинка [99].
Нитрид кадмия — черное кристаллическое вещество с кубической
структурой, изоморфной а«тц-Мп20з, параметр решетки « = 10,79±
Таблица 40
Свойства нитридов металлов подгруппы цинка
Характеристика Zn3N2 Zn(N3)2 CdsN, Cd(N3)2 Hg„N2 | Hg,(N,)2
Содержание азота, вес. % 12,41 Кубиче- 56,22 7,67 Кубиче- 43,28 4,45 17,32
Кристаллическая струк- тура ская [Ю2] — ская [Ю2] — — —
Периоды решетки, д 9,743 [Ю2] — 10,7, [Ю2] — — —
Удельный вес пикнометрический 6,22 [Ю2] — 6,85 [Ю2] — — —
рентгенографический Температура разложе- 6,60 [102] Взрывает- 7,67 [Ю2] Взрывает- — —
ния, °C Теплота образования, ккал/моль 700—750 ся 320 СЯ — —
5,3 [Ю4] —50,8 [Ю5] —38,6 [9] —106,2 [9] — -100 [84]
Теплоемкость, кал/моль -град 26,10 [ЮЗ] — —
Удельное электросопротив- ление, ом • см 4,5.103 [33] (20°С)
108
±0,02 А, в элементарной ячейке 16 атомов [102]. Весьма неустойчив,
при нагревании разлагается при 320° С, на воздухе разлагается с обра-
зованием окиси, водой разлагается со взрывом.
Азид Cd(N3)2 еще менее устойчив, чем нитрид, и взрывается при
нагревании.
Нитриды ртути. Ртуть образует с азотом нитрид Hg3N2 и азиды
Hg(N3)a и Hg2(N3)2. Соединение Hg3N2 получают пропусканием аммиа-
ка над нагретой до 100—150° С желтой окисью ртути или косвенным
путем из соединения NHg2J [108, 109, 6].
Азиды получают также косвенными методами, в частности быстрым
осаждением из высококонцентрированных водных растворов Hg(NO3)2
или Hg2(NO3)2 с помощью NaN3. После центрифугирования остатки
Hg(N3)2 или Hg2(N3)2 многократно промывают спиртом и высушивают
в вакууме [1074].
Нитрид Hg3N2 — порошок бурого цвета, крайне взрывчатый,
азиды — белого цвета, несколько менее взрывчаты, легко диссоциируют.
Исследование ИК-спектров азидов, выполненное в работе [1074],
показало, что в Hg(N3)2 азидные группы взаимно «скручены» (симмет-
рия С2), а в Hg2(N3)2 образуют высокосимметричную транс-форму (С2/1).
Свойства нитридов металлов подгруппы цинка приведены в
табл. 40.
ГЛАВА V
НИТРИДЫ
ПЕРЕХОДНЫХ
МЕТАЛЛОВ
1. НИТРИДЫ РЕДКОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
(СКАНДИЯ, ИТТРИЯ И ЛАНТАНОИДОВ)
Нитриды скандия. Наиболее полное исследова-
ние условий образования и некоторых свойств
нитрида скандия ScN проведено в работе Фри-
дериха и Зиттиг [110].
При попытке получить нитрид скандия в
токе азота восстановлением окиси скандия уг-
лем при температуре выше 1300° С образуется
продукт, содержащий только 15% нитрида. При
добавлении в исходную шихту 10% окиси желе-
за содержание нитрида несколько возрастает.
Еще более благоприятное влияние оказывает
добавление в исходную шихту 10% смеси
ЫагСОз+гУгС, что повышает содержание нит-
рида в продукте восстановления до 95—96%
(22,6% азота и 4,5 5с20з, нерастворимой в
НС1).
Чистый нитрид получают при температуре
1700—1800° С в токе хорошо очищенного азота
по реакции [110]
Sc2O3 ф ЗС + N2 -> 2ScN + ЗСО.
Температурный режим проверен в [456, 467],
в которых химически чистую окись скандия сме-
шивали с сажей и восстанавливали в токе азота
при температурах 1400—2000° С.
Как следует из данных табл. 41, азотиро-
вание начинается при температуре 1400° С. Од-
нако резкое повышение содержания азота про-
исходит при 1600° С, для полного восстановле-
ния Sc2Os необходима температура 1900—
2000° С. Получающиеся при этом продукты име-
ют состав, выражаемый формулой ScNo,964~o,97o>
продукты загрязнены примесями углерода и кис-
лорода. Применение в составе шихты углекис-
лого аммония в качестве разрыхлителя, облег-
чающего доступ азота, несколько активизирует
процесс азотирования при температурах 1600—
1700° С. Но так как для полного восстановления
окиси скандия температуру необходимо подни-
мать до 1800—2000° С, то в конечном счете до-
бавка углекислого аммония нецелесообразна.
110
Продукт состава ScNo,97o
имеет кубическую гране-
центрированную решетку с
периодом ячейки а=4,499 ±
±0,002 А. Следует отме-
тить, что полученному нит-
риду соответствует лучший
состав, чем нитрида, образо-
ванного по методике [111],
однако, он все же представ-
ляет твердый раствор с кон-
центрацией около 1,2% ScC
в ScN.
В этой же работе [467]
изучены возможности при-
готовления нитрида непо-
средственным азотировани-
ем металлического скандия.
Результаты азотирования
металлического скандия
(табл. 42), свидетельству-
ют, что взаимодействие
скандия с азотом начинает-
ся при температуре 500° С,
при 1000° С содержание азо-
та достигает значения,
практически не меняющего-
ся при дальнейшем повы-
шении температуры. Сумма
содержаний скандия и азо-
та не достигает значений,
превышающих содержания
скандия в исходном метал-
ле, т. е. кислород, находя-
щийся в металле, пол-
ностью остается в продукте
азотирования. Отношение
содержаний скандия и азо-
та, без учета присутствия
кислорода, отвечает форму-
ле ScNo,922, однако практи-
чески в этом случае обра-
зуется оксинитрид, отвечаю-
щий приблизительной фор-
ГО Д' я. (8ОЭг(’’НН) яояавр -ott о) кинаничгаоэ ВТГЛйЗоф KBHSOICOA s e O> O> Q> Hlz° £ £ Q О CO GO GO
Т а б л 1 : + N2=2ScN + 3CO (время реакции 2 ч) ! состав, % С добавкой (NH4)2COe । фЦ19оэ+ +n±>s bwwXd 'O^S еваЭд QOSOq tn^Or-j я 3S — 2,48 — — — — — 76,75 20,58 1,8 1,1 0,7 0,40 99,53 76,15 21,07 2,15 1,38 0,77 0,30 99,64 76,52 21,80 0,70 He об- 0,70 He об- 99,02 нару- нару- 76,80 22,15 0,42 тТже 0,42 Тоже 99,37 76,52 22,62 0,25 » » 0,25 » » 99,39
Состав продуктов реакции ЗсгОз+ЗС S g ф О' S S S X Без добавки (NH4)2CO9 1 .ta9°O + bwwAq •o^s £BS0g QOSOq tn 9Oq я aS — 16,71 ————— 75,68 20,59 0,87 0,07 0,80 2,54 99,48 76,89 21,18 0,70 Не об- 0,70 0,39 99,16 нару- жен 76,81 22,05 0,40 Тоже 0,40 Не об- 99,26 нару- жен 76,41 22,75 0,23 » » 0,23 То же 99,39
С о ‘BdAivdanwai оо оо о 2 о о о о о 2 Tf СО ОО О g
При наличии в образцах Sc2O, содержание последней входит в сумму.
111
муле ScNo,85 Oo.oe- Оксинитрид имеет кубическую гранецентрированную
решетку с параметром а = 4,503±0,005 А.
Нитрид скандия устойчив на воздухе до температуры 600° С, после
полностью заканчивающееся при
чего начинается быстрое окисление,
1200° С (табл. 43) [466].
Таблица 42
Результаты азотирования метал-
лического скандия *
Режим азоти- рования Химический состав продуктов азотирова- ния, %
Темпе- ратура, °C Время, ч Sc N Сумма
500 1 1,18
600 1 — 1,26 —
700 1 .— 1,83 —.
800 1 — 3,22 —
900 0,5 — 8,85 —
900 1 — 9,25 .—
900 2 — 9,45 —
900 4 — 14,75 —
1000 1 77,98 20,11 98,09
1000 2 78,19 20,18 98,37
1000 4 78,48 20,15 98,63
1100 1 77,27 20,68 97,95
1200 0,5 77,71 20,70 98,41
1200 1 78,05 20,63 98,68
1200 2 77,30 20,75 98,05
1300 0,5 77,98 20,45 98,43
1300 1 77,43 20,85 98,28
1300 4 77,05 20,75 98,80
1400 0,5 77,85 20,42 98,27
1400 1 78,02 20,85 .98,87
* Расчетный состав ScN:Sc—
76,24%, N—23,76%, сумма—100%
Таблица 43 Изменение содержания азо- та в нитриде скандия при окислении на воздухе *
«Г ч о
X X
СО о S3?
о «Я у
S . о S < о о п а Ч,
о ° U « У С ел
300 1 22,15 0
300 2 21,98 0
400 1 21,95 0
400 2 22,02 0
500 1 22,10 0
500 2 22,13 0
600 1 22,05 0
600 2 21,86 0
700 1 13,14 40,02
700 2 9,02 58,9
700 4 5,87 73,3
700 6 2,05 90,7
800 1 1,16 94,7
800 2 0,65 97,0
900 1 0,99 98,1
900 2 0,42 95,5
1000 1 0,57 97,4
1000 2 0,49 97,7
1100 1 0,13 99,4
1100 2 Не об- 100
Нару-
жен
1200 1 То же 100
1200 2 » » 100
* Содержание в исходном по-
рошке N—22,12%.
Химические свойства изучали на порошках нитрида скандия состава
ScNo,97o [466, 612] и на компактных образцах, приготовленных горячим
прессованием нитрида в графитовых пресс-формах в среде аргона по
режимам, приведенным в табл. 44. Как видно из этих данных, содер-
жание азота в процессе спекания практически не изменялось, оста-
точная пористость образцов составляла от 14 до 30%. Данные по ус-
112
тойчивости порошка нитрида скандия в концентрированных и разбав-
ленных минеральных кислотах (табл. 45) показывают, что нитрид
полностью разлагается всеми кислотами, за исключением серной, ко-
Таблица 44
Режим спекания нитрида
скандия горячим прессова-
нием*
Температура спекания, °C Время спека- ния, мин Содержание азота в спечен- ном образце, %** Остаточная по- ристость, %
2000 4 21,78 30,1
2200 2 21,96 24,0
2200 4 21,84 23,1
2400 4 22,02 14,4
* Горячее прессование порош-
ка нитрида проводили под давле-
нием 150 кг/см2. ** В исходном
порошке содержится 21,98% N.
Таблица 46
Таблица 45
Поведение порошка нитрида
скандия в кипящих кислотах
Время, ч Неразложившаяся часть, %
H2SO. (1,84) H2SO. (1:1)
1 16,89 0,52
2 3,84 0,36
3 2,82 0,42
Примечание. Нитрид скандия
в кипящих HNO,(1:1), HN0s(l,19),
НС1 (1:1) не разлагается.
Поведение порошка нитрида
скандия в кипящих раство-
рах щелочи
Таблица 47
Поведение компактных об-
разцов из нитрида скандия
в кипящих кислотах
Время, ч Неразложившаяся часть, %
10%-иый NaOH 20%-ный NaOH
1 71,94 26,01
2 71,37 18,49
4 68,93 9,59
24 — —
Примечание. Нитрид
скандия в 40%-ной NaOH ие раз-
лагается.
Время, ч Неразложившаяся часть, %
H2SO. (1,84) H2SO4 (1:1)
1 99,40 97,50
2 99,23 96,47
4 99,14 96,24
Примечание. Нитрид
скандия в кипящих HNOS (1,4),
HNO3 (1:1), НС1 (1,19) НС1 (1:1) не
разлагается.
торая также разлагается довольно быстро. Нитрид стоек в холодной и
горячей воде, раствором NaOH разлагается, причем скорость и полнота
разложения возрастают с повышением концентрации раствора щелочи
(табл. 46). Компактные образцы нитрида (табл. 47) ведут себя в ки-
пящих минеральных кислотах аналогично порошку, только в серной
а-272 113
кислоте разлагаются очень медленно. Резко возрастает стойкость ком-
пактных образцов в кипящих растворах NaOH, причем тенденция к
снижению стойкости при увеличении концентрации щелочи заметно
сглаживается (табл. 48).
При комнатной температуре на компактные образцы нитрида скан-
дия заметно действует только азотная кислота (табл. 49).
Таблица 48
Поведение компактных об-
разцов из нитрида скандия
в кипящих растворах ще-
лочи
Неразложившаяся часть образца, %
Время,
ч 10%-ный NaOH 20%-ный NaOH 40%-ный NaOH
1 92,03 86,25 80,04
2 90,23 81,50 78,05
4 86,11 78,43 74,90
Таблица 49
Поведение компактных образцов нитри-
да скандия в кислотах и растворах ще-
лочи при комнатной температуре (24 ч)
Реагент Неразложив- шаяся часть образца. % Реагент 1 Неразложив- шаяся часть образца. %
H2SO4 (1,84) 100 10%-ный NaOH 99,34
H2SO4 (1:1) 100 20%-ный NaOH 99,00
HNO3 (1,4) 21,8 40%-ный NaOH 98,65
HNO3 (1:1) 6,7 — —
HC1 (1,19) 100 — —
HC1 (1:1) 89,9 — —
Для определения физических свойств [466] были использованы об-
разцы состава ScNq,97o, приготовленные горячим прессованием.
Микротвердость нитрида скандия, измеренная при нагрузке 50 г,
равна 1170+150 кг/мм2, температура плавления, определенная по ме-
тодике, описанной в [468], равна 2550+50° С. Коэффициент термиче-
ского расширения нитрида, определенный на кварцевом дилатометре,
в интервале температур 20—1070° С составляет 8,68-10~6 град~1.
Удельное электросопротивление измеряли на нескольких образцах
одного и того же состава, но различной пористости, полученные резуль-
таты экстраполировали на беспористое состояние с использованием
аппроксимационной формулы, которая для исследованных образцов
имела такой вид:
= Qn(i — п)2-8,
где рк и рп — удельное сопротивление соответственно беспористого и
пористого образцов, п — пористость в долях единицы. При комнатной
температуре удельное электросопротивление нитрида скандия состав-
ляет 25,4—0,9 мком • см. Зависимость удельного электросопротивления
от температуры, определенная по методу [469], представлена на рис. 43.
114
В интервале температур от 20 до 1000° С электросопротивление линейно
возрастает с температурой и может быть описано выражением
= 23 ± 0,095/.
Температурный коэффициент электросопротивления при комнатной
температуре составляет + 3,8 • 10-3 грасН1.
Сравнивая определенные в настоящей работе значения физических
свойств нитрида скандия состава ScNo.szo с литературными данными,.
следует отметить, что определенная в
работе [466] температура плавления
удовлетворительно совпадает с уста-
новленной ранее (2550 и 2650° С соот-
ветственно) . Нитрид скандия менее
тугоплавкий, чем нитриды металлов
IV группы периодической системы, но
более тугоплавкий, чем нитриды пере-
ходных металлов V, VI групп. Уста-
новленное в [466] значение удельного
электросопротивления на порядок
меньше, чем сообщается в [112], где
приводится цифра 308 мком-см. Ли-
нейное увеличение электросопротивле-
ния нитрида скандия с температурой
указывает на металлический характер
этого соединения.
Термический коэффициент элек-
тросопротивления нитрида скандия
Рис. 43. Температурная зависимость
электросопротивления и термо-э. д. с.
нитрида скандия.
занимает промежуточное положение между значением таковых для
нитридов титана ( + 2,48) и циркония ( + 4,3) и существенно выше та-
кового для нитридов тантала и ванадия ( + 0,7 и +0,03).
Термоэлектродвижущая сила нитрида скандия имеет отрицатель-
ное значение и убывает с ростом температуры, что, согласно [469], ука-
зывает на преимущественно электронный характер проводимости этого
соединения. Обращает внимание высокое для металлических провод-
ников абсолютное значение термо-э. д. с. нитрида скандия (от —20 до
—40 мкв/град) в области температур 100—600° С при низком удельном
электросопротивлении. Это указывает на перспективность применения
нитрида скандия в качестве материала отрицательной ветви термо-
электрического преобразователя тепловой энергии в электрическую.
Сравнивая электрические свойства металлического нитрида скандия с
таковыми для полупроводникового нитрида хрома [326], следует отме-
тить, что важная для оценки коэффициента полезного действия термо-
элемента величина а2/р (где а •— термо-э. д. с., Q — удельное электро-
сопротивление) для нитрида скандия больше, чем для нитрида хрома
8*
115
CrN (соответственно 1,45 и 1,27 в относительных единицах при комнат-
ной температуре). Величина термо-э. д. с. нитрида скандия наибольшая
из всех значений термо-э. д. с. для металлически проводящих нитридов
переходных металлов.
Теплопроводность нитрида скандия состава ScNo.gyo, измеренная
методом стационарного теплового потока при комнатной температуре
[470], составляет 0,27 вт/см -град. Известно, что теплопроводность твер-
дого тела, обусловленная тепловой диффузией свободных электронов,
связана с электросопротивлением соотношением Видемана—Франца
Хр/7’=2,45-10~8 вт-ом-град'1. Для ScN с использованием установ-
ленных значений тепло- и электропроводности получается Кр/Т=
= 2,34 • 10~® вт • ом • град-1, т. е. теплопроводность нитрида скандия
целиком обусловлена свободными электронами, а роль решеточной
теплопроводности по сравнению с электронной ничтожно мала.
Нитрид скандия ScN — темно-голубой порошок, его физические
свойства приведены в табл. 50. При обычной температуре ScN устой-
чив, при прокаливании на воздухе окисляется до 5с20з (низшие окис-
ли скандия не установлены), причем окисление происходит с большим
трудом. Легко растворим в соляной, азотной и серной кислотах, при
нагревании с едким натром и сплавлении с КОН выделяется аммиак и
происходит разложение нитрида скандия; легко гидролизуется водой.
Опыты, проведенные по изучению поведения нитрида скандия на
нити накаливания электролампы, показали, что он полностью испаря-
ется в течение 1 ч [ИО].
Нитриды иттрия. При восстановлении окиси иттрия углем в токе
азота образуется нитрид, содержащий при температуре восстановления
1300° С только частично нитрид иттрия, при более высоких темпера-
турах образуется карбид иттрия, поэтому приготовление чистого нитри-
да иттрия затруднительно.
Чистый нитрид иттрия получен в [113] действием азота на гидрид
иттрия YH2 при 900° С.
В работе [485] предлагается получать нитрид иттрия гидрирова-
нием компактного металла при низкой температуре (350—400° С), ко-
торое разрыхляет поверхность, с последующей обработкой азотом при
небольшом избыточном давлении 2—3 кг/см2 при температуре 1400—
1600° С (см. также [1075]).
Нитрид иттрия разлагается водой с образованием гидроокиси и
выделением аммиака.
По данным [1001], мононитрид иттрия не является сверхпроводни-
ком по крайней мере выше 1,4° К-
Нитриды лантана. Первые опыты по получению соединений лан-
тана с азотом были предприняты Фридерихом и Зиттиг [110]. Нитрид
лантана LaN был получен ими нагреванием окиси лантана с углем в
токе азота. Образование нитрида лантана начинается при температуре
116
иитпмпов педкоземельных металлов
ческие и » свойства (Я cS6Z) .01-х чхэониь -иииПцэон ввнхиилщу +60 [478] +315 [959] +5616 О Ю oo g 2 О о о о ~ е-*’(Y'T F 'oj ТС’00 F—lO г*—.1^0 "» -1 00 сс 00 00 <© 00 Ch оо О’оо 00 оо СО 00 СО 00 Й 00 , °0 сП 00 _ 00 .—> Ю СО СЧ 00 00 00 Ь- 00 О 00 СО 00 04 00 -+-+-Е +~+^+-+-+^+_+_
5 3 СЬ д S £ gg со « (Э о95) 'oHHairatfioau -ooodiMQtfe эоичи’эй'х o'r—) ° r— °O r-> OI •'t _ "«r ^7 —Р а—. г—> 1—» , _ От*ос^оо£2а::’йооОоо2<Р>с’<=:>(-ъ.§5оо0оо*со “С’ ^52^оос?оосчооОо>2Гс>£?Зо0оо~(*>&оо г.ю'* 12L?22J2L1X^21. "X+2L&X.
'wj 'uid о 2-0) g I 1 1 1 1 1 1 1 1 1 1 ) 1 1 1
г—1 О Щ) 'Uld iVW £-01 §§§§111111111111
Q а. Б •uzj ‘wd ww Z-0>
S. с f ‘UID 'Uid ww (—01 •*7 GO Ю Ch £g[gsillllllllll[ r~< _ r-> oq
ад В 'ш? ‘и/d ww i О о О to 0O СП —• O’ 1 ОО СП О 1 — —< 04 с+ 1 1 ! 1 1 1 1 1 1 1 Г
я 1 S S а. [8Ztl (Я „002—001) gotf? •кинэ4ипювб олоюэь •Hwdai хиэи'пиффео^ 9-10-6 30-10-6 13-io—6 i
[Zill grow • Qvd0/VVH •BHuodxHg
[ЗИ1 4V0K/VtWi ‘кннваосвЦро Bxoiruai О ю — О «)- + <+ «7 1 1 1 1 1 ! 1 1 1 1 1 [ Г- Ь- Г-
Do ‘кин -sirapiru EdXiBdaUKsj О „+ 1 1 1 1 1 1 1 ! 1 1 1 1 1 1 1 л
ические л и •• g з квяэ -ohHdianoHMHii ~gsp1 1 1 1 1 1 1 1 1 1 1 1 1 1
аллохим свойства г BBMO0OH9JXH9d ’” - , ”Lh * . 71LO Z2.UD S-00 S 2 2 ? 2 2 — 2 S 2 S? 2 8 2 cn 2 S 2 м S 2 г" [~~, со ^.оо ’"'.сп Г"1.,^ L^-Ch ZILo ZLo 7".о ZL’— TL’—' Z7
I S у 'иххашас! йоисЬц Z7O го?74 сп <£> м* , -,О г_-| оа^_, ег,г __, »ar- -t ’S',—, <»г—. Д, 1 ©^, »22o2S2S2S2S2oo2S2S2S2^2 7".to LJ.IO ZLlO u_Lrt* ьТ-т?' LLrf* L~□TJ’ L-Lrj’ LZ-тГ L-L’sF ^7-
летри- карак- гики % 'ООН вюее эинежйэЬ'оэ ф со Ci г- СО —_ о о со со с> сь о С4 — ioc4’’fooo^eo<ro<0CT’»-* 00 Ю «Ф —• —« Ch OO O' Ю rr СОоОСОСОООГ'Г-Г'-Г'-Г-'-*--
Стехио> ческие : терис- ээе ynHdKifXnsiroyv Г— СО со «ф СО О ch Ch .-ч о СО CN OJ Th xt< £г5 о ю 1Л ю 00 rF r- СЧ uo GO Ю LO о СЧ^ОСЧС^иОСЬСЧСЛОО 00 xF CD — 04 О OO —. СЧ r- S’ ю О <O b- N b' b- 00 oo oO oo
S X Kt s z z z z z о co aj t- <Z> >-_)(_> D. ZcgwObQZMHZ'U
Под давлением азота, *» Слабый парамагнетизм, не зависящий от температуры.
117
1300° С, а при 1500—1700° С образует карбид лантана [114], поэтому
приготовленные этим способом продукты представляют смеси нитрида
с карбидом.
Чистый нитрид лантана получают нагреванием опилок или стружки
металлического лантана в токе азота [115]. Опилки лантана, приготов-
ленные в среде сухого гелия или азота, помещают в молибденовую ло-
дочку, устанавливаемую в печь. Сначала опилки дегазируют, откачивая
реакционное пространство до давления в 10—5 мм рт. ст. при комнатной
температуре, затем впускают азот и включают обогрев печи. Темпера-
туру в течение 1 ч доводят до 600° С, при этой температуре начинается
реакция. Далее температуру повышают до 750° С и выдерживают от 2
до 4 ч, затем температуру поднимают до 900° С, выдерживают от 1 до
2 ч. Полная гомогенизация продукта достигается при 20 ч выдержке
при 900° С.
Аналогичным методом получали нитрид лантана в ранних работах
[П6, 117], однако азотирование проводили при несколько болёе высоких
температурах (на 100° С выше, чем по данным [115]).
В обзоре [24] сообщается, что азотирование опилок металлического
лантана при температуре красного каления в токе азота проходит
медленно: содержание азота в продукте азотирования через 15 мин
4,4%, через 30 мин — 6,0%. Полностью азотирование с получением
LaN (9,18 % N) заканчивается через 2,5 ч.
Нитриды лантана и лантаноидов получали азотированием метал-
лов аммиаком, причем для эталонирования при последующих рентгено-
съемках к металлам подмешивали КО (РЗМ. + ЗКС1) [119]. Смеси по-
мещали в кварцевую трубку печи в корундовой лодочке и нагревали
в токе аммиака (полученного испарением жидкого аммиака) в течение
3—4 ч при 700° С. Образованный при этом нитрид практически точно
соответствовал формуле и имел параметр решетки а=5,301А, что до-
вольно близко к значению п = 5,295 А, [115].
В работе [473] были повторены опыты по получению нитрида лан-
тана непосредственным азотированием металлического лантана. Исход-
ным материалом служили стружки 98,5%-ного La, приготовленные в
среде аргона. Азотирование проводилось аммиаком и азотом в реак-
торе, описанном в [178].
Результаты взаимодействия металлического лантана с азотом
(рис. 44) свидетельствуют о том, что до температуры 700° С азотирова-
ние лантана азотом идет медленно, при 700° С содержание азота сразу
возрастает до значения, близкого к стехиометрическому, при дальней-
шем повышении температуры и выдержек содержание азота практиче-
ски не меняется. Обращает внимание, что температура полного насы-
щения лантана азотом с образованием LaN в пределах точности ее
определения соответствует температуре плавления металлического лан-
тана (826°С), причем образование нитрида сопровождается сильным
118
разогревом реакционной массы, аналогично получению нитрида церия
(см. стр. 122). Показано, что для церия и лантана характерно высокое
отношение теплоты образования нитридной фазы к теплоте плавления
металла (см. табл. 29), что, очевидно, и является причиной сильного
разогрева реакционной массы при температуре плавления металла, а
отчасти объясняет и весьма быстрое и полное азотирование, наступа-
ющее при плавлении лантана. При плавлении, вероятно, разрушается
плохопроницаемая для газов нит-
ридная пленка, что также ускоряет
азотирование; в момент плавления
атомы металла находятся в наибо-
лее активном состоянии.
При использовании в качестве
азотирующего агента аммиака ход
насыщения металлического ланта-
на азотом существенно отличен от
предыдущего случая (см. рис. 44),
В первую очередь, гораздо более
высокой скоростью азотирования
при низких температурах, а также
достижением предельного содержа-
ния азота уже при 600—700° С. Это,
Рис. 44. Температурная зависимость со-
держания азота в лантане (ч) (время
азотирования 1 ч):
/ — расчетное содержание лантана в LaN;
2 — обработка лантана аммиаком; 3 — обра-
ботка лантана азотом.
очевидно, связано с разрыхлением
Металлического лантана в резуль-
тате образования и разложения
гидридных фаз, которые, по дан-
ным [474], получаются уже при низ-
ких температурах, а также имеют
кубические решетки либо идентичные, либо близкие по типу к ре-
шетке LaN. Продукты взаимодействия с азотом, полученные при темпе-
ратурах 800° С и выше, а также продукты взаимодействия с аммиаком,
полученные при 600° С и выше, имеют решетку типа NaCl с периодом
а=5,302±0,002А.
По данным Гринтала [475], для нитрида лантана, полученного дей-
ствием жидкого аммиака на порошок лантана при низких температу-
рах, обнаружена ромбическая структура с периодами решетки: а = 5,32,
в=5,30, с=5,25 А. Пикнометрический удельный вес этого нитрида
оказался равным 4,69, что значительно ниже удельного веса нитрида
LaN. Очевидно, продукт, полученный Гринталом, представляет вто-
рую нитридную фазу лантана с более высоким содержанием азота,
чем в LaN.
По данным Келли [475], стандартная свободная энергия AF реакции
LaN = La + */2N2 в итервале температур 25—800° С составляет ДК=
= 72100—25,0 Т, кал/моль.
119
Нитрид лантана представляет собой серый до темно-серого по-
рошок. Согласно [126], при температурах выше 1,8° К LaN не обнару-
живает сверхпроводимости. Однако в недавно проведенной работе
[479], где сверхпроводимость нитрида лантана изучали более тщательно,
показано, что LaN является сверхпроводником с размытой областью
перехода между ~ 1,35° К (по магнитным измерениям) и -~-4°К (по
началу аномалии теплоемкости). Существование этой области отно-
сится за счет наличия двух модификаций нитрида лантана, что в из-
вестной степени подтверждает вышеприведенные данные Гринтала.
Удельная теплоемкость LaN определена в [480].
Нитрид лантана химически нестоек, разлагается на влажном воз-
духе с выделением аммиака, несколько более устойчив в воде на хо-
лоду, а при нагревании в воде быстро разлагается с выделением боль-
шого количества аммиака [131]. При нагревании на воздухе окисляется
до окиси лантана La2Os • LaN, легко растворяется в минеральных кис-
лотах и растворах щелочей. Нитрид лантана устойчив в течение дли-
тельного времени в средах азота, аргона и СО2 [473].
Тигли, изготовленные из LaN, быстро разлагаются в результате
гидролиза [120]. Исследователи [492], изучив влияние редкоземельных
металлов на поведение азота в жидких железе и сталях, отмечают роль
образования при этом нитрида лантана.
Нитриды церия. В системе церий — азот установлено существова-
ние одного химического соединения состава CeN.
Фредерих и Зиттиг [ПО] пытались получить нитрид церия восста-
новлением окиси церия углем в среде азота по реакции
2СеО2 + 4С + N2 = 2CeN + 4СО.
Однако ни при 1250° С, ни при более высоких температурах до 1600° С
в продуктах восстановления не обнаруживали сколько-нибудь заметные
количества нитрида церия.
Матиньон [121] приготовлял нитрид церия восстановлением СеО2
магнием или алюминием в среде азота либо восстановлением хлорида
церия натрием в азоте.
Фурнасос [62] получал нитрид церия взаимодействием порошка
металлического церия с расплавленным цианистым калием.
Нитрид церия может быть получен действием азота на металли-
ческий церий [122]. Реакция азотирования при низких температурах
идет медленно [123], но при 850° С проходит очень энергично и сопро-
вождается сильным разогреванием. При использовании чистого от
кислорода азота образуется продукт черного цвета с бронзовым от-
ливом.
Некоторые исследователи получали нитрид церия действием азота
на гидрид церия при температуре 800—900° С [124]. Нитрид церия при-
готавливают также при нагревании карбида церия в азоте или в ам-
120
миаке при 1250° С, причем реакция с азотом идет быстрее, чем с амми-
аком [125], что объясняется образованием труднопроницаемой пленки
нитрида. По данным [1], церий поглощает азот и при комнатной тем-
пературе, но очень медленно.
Как сообщается в [129], нитрид церия получали, как и нитриды
других лантаноидов, азотированием смесей Се+ЗКС1 аммиаком в тече-
ние 3—4 ч. Однако в [478] данный метод получения нитрида церия не
Рис. 45. Зависимость содержания азота в продуктах азотирования метал-
лического церия и выхода нитрида от температуры (время азотирования
1 ч):
1 — расчетное содержание азота в CeN; 2 — содержание азота в продуктах взаимо-
действия церия с азотом; 3— выход прн взаимодействии с азотом; 4— содержание
азота в продуктах взаимодействия церия с аммиаком; 5 — выход прн взаимодействии
с аммиаком.
удалось воспроизвести — продукты азотирования содержали лишь 50—
60% азота против его содержания в Се, остальной церий оставался в
металлическом состоянии. Наоборот, хорошо азотировался аммиаком
компактный электролитический церий при 800° С.
Подробное изучение условий образования нитрида церия проведе-
но в [467, 476] методом азотирования 98%-ного металлического церия
в токе азота и аммиака в кварцевом реакторе с двойными стенками,
между которыми пропускали азот для предотвращения попадания кис-
лорода воздуха в реакционное пространство. Как показали результаты
азотирования металлического церия в токе хорошо очищенного азота
(рис. 45), до 700° С церий с азотом практически не взаимодействует, а
при 800° С образуется CeN (в течение 0,5 ч), причем дальнейшее повы-
шение температуры до 1100° С не оказывает влияния на состав про-
дукта азотирования. Рентгеновский анализ показал, что продукт азо-
тирования имеет решетку типа NaCl с периодом ячейки а = 5,023 А, что
хорошо совпадает с табличными данными.
121
л
Образование нитрида церия «скачком» при температуре 800° С
хорошо согласуется с данными работы [123], в которой, однако, тем-
пература образования нитрида равна 850° С. Совершенно очевидно, что
эта температура в пределах точности ее определения совпадает с тем-
пературой плавления металлического церия (815° С). Следует пола-
гать, что азотирование происходит с большой скоростью при плавлении
церия, по-видимому, вследствие уничтожения тончайшей и труднопро-
ницаемой для азота пленки нитрида [125] и относительно высокой упру-
гости паров церия при температуре плавления. Наблюдавшийся в [123]
и нами сильный разогрев реакционной массы при образовании нитрида
церия может быть объяснен малой теплотой плавления церия в сравне-
нии с теплотой образования нитрида (см. табл. 29). Этот фактор также
обусловливает высокую скорость реакции и, вероятно, должен учиты-
ваться во всех тех случаях азотирования, когда на поверхности азоти-
руемого металла образуется труднопроницаемая для газов пленка.
Следует отметить сходство наблюдаемого эффекта с явлением бурного
роста зерен при спекании изделий из торированного вольфрама.
Азотирование церия в токе аммиака начинается уже при темпера-
туре 100° С. Содержание азота резко возрастает при 200° С, содержа-
ние азота, равное расчетному, в CeN достигается при 700° С, далее
сколько-нибудь существенно не изменяясь. В этом случае церий гидри-
руется водородом аммиака, согласно [477], время, необходимое для
гидрирования церия, при 200° С минимально, т. е. скорость гидриро-
вания наибольшая. Гидрид церия, по-видимому, предотвращает обра-
зование сплошной пленки нитрида на металлическом церии. Кроме
того, замена водорода в решетке азотом выгодна в термодинамиче-
ском отношении (теплоты образования гидрида и нитрида церия равны
соответственно 42,3 и 78 ккал/моль). Образование активных атомов це-
рия при диссоциации гидрида церия не имеет существенного значения,
так как давление диссоциации гидридов церия, по крайней мере до 700—
800° С, невысоко.
Согласно [485], нитрид церия получали гидрированием компактного
металла при температуре 350—400° С с последующей обработкой азо-
том при 1400—1600° С.
В сухом воздухе и углекислоте при обычной температуре нитрид
церия устойчив, во влажном воздухе довольно быстро окисляется [62].
При действии водорода на нитрид церия при 100—400° С (особенно при
200° С) CeN разлагается с образованием гидрида церия и аммиака
CeN ЗН2 = СеН3 4- NH3.
При действии на церий смеси азота и водорода образуется нитрид
церия, который по вышеприведенному уравнению разлагается с обра-
зованием аммиака. Это дает возможность рассматривать нитрид церия
122
как своеобразный катализатор процесса связывания азота с водородом
в аммиак.
Взаимодействие нитрида церия с водой проходит весьма энергич-
но, сопровождается сильным разогревом до 700—800° С [131]
2CeN 4- 4НаО = 2СеО2 + 2NH3 + Н2.
CeN медленно растворяется в водном растворе КОН с образова-
нием гидроокиси церия и аммиака, при взаимодействии с разбавлен-
ными кислотами растворение идет активно с образованием солей це-
рия и аммония
2CeN + 4H2SO4 = Се2 (SO4)3 Г (NH4)2SO4.
Растворение в некоторых случаях сопровождается настолько боль-
шим разогревом, что часть выделяющегося аммиака разлагается на
азот и водород.
По данным [126], CeN при температуре выше 1,8° К не обнаружи-
вает сверхпроводимости. Стандартная свободная энергия реакции
CeN = Ce +‘/гИг, по данным Келли [475], равна 78000—25,0 Т кал/моль.
Авторы [959] исследовали изменение электросопротивления CeN
при низких температурах (до 300° С). При этом обнаружен нормальный
ход сопротивления без каких-либо аномалий типа магнитного порядка-
беспорядка. Установлено, что парамагнетизм нитрида церия обуслов-
лен ионом Се3+; %=%о + С(Г—0), где %о = 315'1О_6, 0 = 85° К, С = 2,75
магнетонов Бора.
Нитриды празеодима. Нитрид празеодима PrN получают азотиро-
ванием празеодима при 900° С, действием аммиака при температурах
до 1000° С на карбид празеодима, нагреванием празеодима в токе ам-
миака, а также восстановлением окиси празеодима магнитом в среде
азота [6, 9].
Эти исследования были продолжены М. Д. Лютой и А. Б. Гонча-
рук, которые показали, что особенно быстро происходит образование
нитрида при действии на металлический празеодим аммиака. Компакт-
ные образцы из металлического празеодима размером 10X5X5 мм
интенсивно взаимодействуют с аммиаком начиная с температуры 200° С,
когда содержание азота в продукте азотирования достигает 6,5%, а
после одночасового действия аммиака при 400°С празеодим полностью
переходит в нитрид стехиометрического состава, причем образец рас-
сыпается в порошок или мелкие кусочки нитрида. Азотирование амми-
аком при температуре 600°С приводит к переходу металла в нитрид
с растрескиванием образцов, а при 700° С — к получению компактных
образцов нитрида с сохранением формы исходного металлического об-
разца. С дальнейшим повышением температуры азотирования аммиа-
ком содержание азота в образцах празеодима резко снижается и со-
ставляет при 800° С — 1,5%, а при 1000° С — 1,8%. Быстрое азотирова-
123
ние при низких температурах может быть связано с разрыхляющим
действием одновременно образующегося гидрида. При высоких темпе-
ратурах скорость разложения гидрида выше скорости его образования,
что вызывает резкое сокращение скорости азотирования и практиче-
ское прекращение гидрирования.
Нитрид празеодима — вещество черного цвета, неустойчивое на
воздухе при обычных условиях, быстро гидролизуется [131]. По дан-
ным [989], точка Кюри PrN 0 =—11° К, магнитный момент 3,57 магне-
тонов Бора.
Нитриды неодима. Нитрид неодима NdN получают методами, пе-
Рис. 46. Температурная зависимость электросопротивле-
ния нитрида неодима:
1 — экспериментальная кривая; 2 — доля сопротивления, обуслов-
ленная рассеянием на фононах; 3 — доля сопротивления, обус-
ловленная разупорядочением спинов.
ных режимах. В [1] указывается на получение мононитрида неодима
действием очень чистого аммиака на металлический неодим при тем-
пературе 700° С в течение 3—4 ч. Химические свойства нитрида неодима
аналогичны свойствам нитрида празеодима.
Т. С. Верхоглядова показала, что при азотировании неодима азо-
том нитрид образуется за 2 ч при 800° С и за 15 мин при 1000° С. Инте-
ресно, что неоднократно удавалось получать, особенно при темпера-
турах 1050—1200° С, нитриды празеодима и неодима со значительно
большим содержанием азота, чем требуется по формуле MeN. Этим
продуктам отвечают условные формулы MeNi,7-i,8 (содержание азота
до 14—14,5 вес. °/о). Так как атомы празеодима и неодима имеют в
изолированном состоянии конфигурации f-электронов 4/3 и 4/4 соответ-
ственно, которые могут легко передаваться партнеру с переходом в
устойчивое /°-состояние, то полученные экспериментальные результаты
кажутся логичными.
124
Авторами работы [1075] показано, что при азотировании ком-
пактных образцов металлического неодима аммиаком взаимодейст-
вие начинается при 300° С, а при 500° С образуется компактный обра-
зец нитрида неодима. Повышение температуры азотирования металла
вызывает резкое снижение содержания азота. Причины повышения
температуры азотирования неодима аналогичны таковым при азотиро-
вании компактного празеодима (см. выше).
Исследователи [485] получали нитрид неодима гидрированием
компактного металла при 350—400° С с последующей обработкой азо-
том при 800—1600° С. В работе [481] исследовано электросопротивление
и термо-э. д. с. нитрида неодима (а —5,126 А) при низких температурах.
Температурная зависимость электросопротивления (рис. 46, кривая 1)
является выражением интегрального сопротивления: q =>gi + q2 + (?з, где
Рис. 47. Температурная зависимость термо-э. д. с. NdN.
Qi — доля электросопротивления, обусловленная дефектами и загряз-
нениями; р2 — доля электросопротивления, обусловленная рассеянием
на фононах; р3 — доля электросопротивления, обусловленная разупо-
рядочением спинов. На том же графике нанесены расчетные значения
Q2 (кривая 2) и рз (кривая 3). При абсолютном нуле pi = 84 мком • см,
а значение ро=12 мком • см. Термо-э. д. с. нитрида во всей этой области
температур отрицательна (рис. 47) и при температуре 60° К а состав-
ляет —0,17 мкв/град.
В этой же работе сделан вывод о полуметаллической природе нит-
рида неодима и подтверждается наличие в NdN, как и в других нит-
ридах лантаноидов [482], магнитной сверхструктуры. Точка Кюри NdN
Тс «27° К [480].
Нитриды самария. Методы получения нитрида самария SmN ана-
логичны методам получения нитрида празеодима. Кроме того SmN
приготавливали действием аммиака при температуре 700° С в течение
3—4 ч на металлический самарий [119].
125
Исследователи [128] приготовляли нитрид самария следующим
образом. Стружку самария в молибденовой лодочке помещали в трубу
вакуумной печи, в которой одновременно находилась стружка титана
(в качестве поглотителя кислорода). Трубу откачивали до давления
Ю—5 мм рт. ст., выдерживали при этом давлении в течение 4—6 ч. Азот
из баллона проходил через подогретый титан. Закрытую систему вы-
держивали 12—16 ч при 800° С. Продукт азотирования собирали в
наполненный сухим аргоном приемник.
Ианделли [129] получал нитрид самария нагреванием металличе-
ского самария (99,2—99,7%) при 1000° С в молибденовой лодочке в
среде азота. Металлический самарий приготавливали вакуумной дис-
тилляцией самария при реакции алюминотермического восстановления
окиси 5т20з при 1500° С. Реакция азотирования самария идет гораздо
медленнее, чем лантана, церия, празеодима и неодима, и содержание
в продуктах азотирования 94—95% SmN достигается только через 15—
20 ч. Азотирование затруднено образованием на каждой частице ме-
талла плотной газонепроницаемой пленки нитрида.
Нитрид самария — твердый порошок, быстро гидролизуется на воз-
духе, причем предполагалось, что гидролиз идет по реакции
SmN -j- ЗН2О = Sm(OH)3 % NH3.
Однако экспериментально определенный привес образцов не совпадает
с рассчитанным по этому уравнению. Предположение, что это вызы-
вается задержкой аммиака в твердой фазе, также не подтвердилось.
Рентгеновский анализ продуктов, находившихся на воздухе в течение
одной недели, показал их аморфность. Детальные исследования как
процесса гидролиза нитрида самария, так и процесса обезвоживания
гидроокиси, установили, что при гидролизе нитрида образуются гидра-
тированные гидроокиси Sm(OH)3 • 2,8Н2О и др.
Нитрид самария имеет кубическую гранецентрированную решетку
с параметром элементарной ячейки, по данным различных авторов:
а=5,0481 ±0,0008 [128], 5,039 + 0,003 [129], 5,046 + 0,001 А [119].
При нагревании нитрида самария до температуры 1600° С в ва-
кууме 10-5 мм рт. ст. не обнаружено его испарения или диссоциа-
ции [128].
Нитриды европия. Единственное соединение европия с азотом EuN
получено азотированием металлического европия аммиаком в течение
3—4 ч при 700° С [119], а также непосредственным действием азота на
европий по тем же режимам, которые описаны выше для нитри-
да самария [128]. Нитрид европия — вещество черного цвета, по
химическому поведению подобен другим нитридам редкоземельных
металлов. В [119] для установления возможности существования двух-
валентного европия в нитриде последний разлагали водой, однако не
126
было замечено выделения водорода с продуктами разложения ни для
нитрида европия, ни самария и иттербия.
Нитриды гадолиния. Нитрид гадолиния GdN получают азотирова-
нием металлического гадолиния чистым аммиаком в течение 3—4 ч
при температуре 700° С [119]. Эндтер [130] рентгенографически обнару-
жил GdN в поверхностном слое, образующемся при нагревании гадо-
линия на воздухе.
Нитрид гадолиния — вещество черного цвета с кубической струк-
турой, химические и физические свойства почти не изучены. Нитрид
гадолиния — ферромагнетик с точкой Кюри 69° К и ферромагнитным мо-
ментом в 7 магнетонов Бора, равным парамагнитному моменту. Под
давлением до 100 кбар изменение точки Кюри составляет для GdN
дТс!дР= +0,08°±0,04° К [1007]. В работе [1008] исследовано спонтан-
ное намагничивание этого нитрида.
Нитриды тербия, диспрозия и гольмия. Сведения о нитридах этих
металлов весьма ограничены. TbN, DyN и HoN приготавливали дейст-
вием аммиака в течение 3—4 ч при температуре 700° С на соответствен-
ные металлы [119]. Попытка получить нитрид диспрозия восстановле-
нием окиси диспрозия углем в среде азота не дала положительных
результатов; образуемый продукт не был однофазен [ПО].
В работе [483] нитриды тербия и гольмия получали из чистых
металлов (99,5%), которые сначала гидрировали, а затем гидриды
азотировали аммиаком при температурах 850—1100° С. Химический
анализ показал, что препараты содержат более 95% нитридов.
Свойства этих нитридов почти не изучены, в [119] приведены их
кристаллические структуры.
По данным [153], TbN и HoN — ферромагнетики с точками Кюри
соответственно 18 и 43° К; магнитный момент HoN 8,0± 1,0 магнетонов
Бора [475]. На основе изучения магнитных свойств TbN в [484] делается
вывод о том, что электрон переходит от №~ к ТЬ3+, оставляя азоту
антипараллельный спин.
Нитриды эрбия. При восстановлении окиси эрбия углем в среде
азота
Ег2О3 4- ЗС -{- N2 —> ErN 4- СО
получается продукт, содержащий около 50% ErN, а при добавлении
к шихте 10% смеси соды с углем (см. нитриды скандия, стр. ПО) при
температуре 1250° С образуется почти чистый нитрид.
Как сообщается в [119], нитрид эрбия приготавливали действием
аммиака при 700° С в течение 3—4 ч на металлический эрбий.
Нитрид эрбия ErN — вещество серовато-белого цвета, на воздухе
постепенно разлагается с выделением аммиака, его разложение пол-
ностью заканчивается за несколько дней.
127
Физические свойства нитрида эрбия не изучены (определена только
его кристаллическая структура [119]).
Нитриды тулия. Единственное упоминание о нитриде тулия TuN
имеется в [119]. Образуется он, аналогично другим нитридам редкозе-
мельных металлов, действием аммиака на металлический тулий при
700° С.
Нитриды иттербия. Первые попытки получения нитрида иттербия
YbiN принадлежат Фридериху и Зиттиг [ПО], которым, однако, не уда-
лось приготовить чистый нитрид восстановлением окиси иттербия уг-
лем в среде азота при температурах 1100—1600° С.
В работе [119] нитрид иттербия, как и нитриды других лантанои-
дов, получали действием аммиака на металлический иттербий при
700° С.
Эйк с сотрудниками [128] приготавливал нитрид иттербия анало-
гично нитридам самария и европия (стр. 126) действием азота на ме-
таллический иттербий, однако реакция образования нитрида иттербия
в отличие от нитридов самария и европия протекала настолько мед-
ленно, что пришлось отказаться от такого метода. Был использован
метод приготовления гидрида иттербия с последующим его превра-
щением в нитрид. Стружку иттербия обрабатывали в течение 12 ч при
температуре 500° С чистым водородом, полученным разложением гид-
рида урана UH3. Образовавшийся гидрид YbH2 нагревали в токе азота,
очищенного прохождением над активированной медью и титановой
стружкой, при температурах 600—1000° С в течение 12 ч. Даже при
очень длительных выдержках и температурах азотирования порядка
1000° С (что на 200° С выше температуры плавления иттербия) реакция
не проходит полностью. При попытке отогнать металлический иттер-
бий от нитрида в вакууме 10 ~5 мм рт. ст. оказалось, что при достаточ-
но длительных выдержках при 1400° С в дистиллат переходят металл
и нитрид. Таким образом, нитрид иттербия легко испаряется в вакууме,
что отличает его от нитридов некоторых других лантанидов, в частности
самария.
Нитрид иттербия имеет кубическую структуру типа NaCl с пара-
метром решетки а = 4,786±0,001 А по [119] или а = 4,7852±0,0008 А
по [125].
В работе [119] не обнаружено выделения водорода при действии
на нитрид иттербия воды, что свидетельствует об отсутствии нитрида
двухвалентного иттербия.
Нитриды лютеция. Нитрид лютеция LuN получается действием ам-
миака на металлический лютеций при температуре 700° С [119].
Структуры и физические свойства нитридов редкоземельных ме-
таллов. Все нитриды редкоземельных металлов, включая скандий и
иттрий, кристаллизуются в кубической гранецентрированной решетке
типа NaCl и отвечают составу MeN. Соединения других составов в си-
128
стеме редкоземельных металлов с азотом не обнаружены. Образова-
ние мононитридов редкоземельных металлов с простой решеткой хо-
рошо согласуется с величинами отношений радиуса атома азота к ра-
диусам атомов редкоземельных металлов, составляющими в соответст-
вии с данными табл. 51 от 0,34 до 0,43, что отвечает условию Хэгга
образования простых структур типа фаз внедрения.
Зависимость периодов решеток нитридов от порядкового номера
редкоземельных элементов (рис. 48) [119] обнаруживает эффект лан-
танидного сжатия. X од этой зависи-
Таблица 51 мости аналогичен ходу изменения пе-
Отношения радиуса атома азота к
радиусам атомов редкоземельных ме-
таллов
Sc 0,43 Pm 0,39 Ho 0,40
Y 0,39 Sm 0,38 Er 0,40
La 0,37 Eu 0,34 Tu 0,40
Се 0,38 Gd 0,39 Yb 0,36
Рг 0,38 Tb 0,40 Lu 0,40
Nd 0,38 Dy 0,40
сходство в типах химической
риодов решеток окислов редкоземель-
ных металлов, что свидетельствует о
значительной роли ионной связи в
нитридах. О том же говорит и отсут-
ствие аномалий периода решеток для
европия и иттербия, которые в самих
металлах и соединениях металлическо-
го типа (гексаборидах и дикарбидах
[486]) имеют повышенное значение в
связи с валентностью Ей и Yb мень-
шей +3, проявляемой в металлическом
состоянии. Некоторая аномалия для
нитрида гадолиния, совпадающая с
подобной же аномалией для окиси
гадолиния (также подчеркивающая
связи в решетках этих соединений),
обусловлена влиянием полузанятой электронной конфигурации Gd+3
(7 электронов из 14 на 4/-уровне). Последняя наряду с конфигурациями
лантана (4/°) и лютеция (4/14) наиболее устойчива и обеспечивает точ-
ную трехвалентность иона гадолиния в отличие от других лантаноидов,
где смешанная валентность придает им некоторые признаки металлич-
ности.
По данным магнитных измерений, выполненных Эссеном и Клеи-
мом [478], лантан в нитриде лантана также полностью находится в
форме ионов La+3, в то время как ориентировочное содержание ионов
Ме3+ в нитриде церия составляет приблизительно 11%, а в нитриде
празеодима — около 62%. Остальные атомы металлических компонен-
тов находятся в форме ионов Ме4+.
С высоким содержанием ионов Ме4+ в нитриде церия связаны из-
вестные отклонения от нормального хода кривой лантаноидного сжа-
тия, наблюдавшиеся в некоторых работах, в частности, с этой точки
зрения рассматривается размер элементарной ячейки CeN в работе
Ианделли [129].
q__пуп
129
a — окислов; б — нитридов лантаноидов.
Недостаточность данных о физических свойствах нитридов редко-
земельных металлов существенно затрудняет обсуждение природы их
электронного строения. Однако уже из
предыдущих соображений следует, что
они являются соединениями ионного
типа, что обусловлено высокими иони-
зационными потенциалами азота, вы-
зывающими сильную отрицательную
поляризацию остовов атомов азота, как
было показано, в частности, работами
Клемма [119] на основе расчета адди-
тивного участия в межатомных рассто-
яниях решетки радиуса иона N~3.
У нитридов скандия и иттрия степень
поляризации должна быть значитель-
но меньшей, чем в решетках нитридов
лантана и лантаноидов, и можно ожи-
дать для этих двух нитридов меньшей
доли ионной связи. От ScN до CeN
асимметрия распределения электрон-
ной плотности возрастает, если судить
по увеличению упругости паров и теп-
лот образования в соответствии с пред-
ставлениями, изложенными в [4], что
। свидетельствует о повышении доли
L ионной компоненты связи. Несомненно,
одновременно должен изменяться характер проводимости, связанный
с изменением ширины энергетических разрывов, свойственных, особен-
но для нитридов второго полупериода лантаноидов, полупроводникам,
в данном случае типа AInBv.
Эти соображения подтверждаются и другими данными по физи-
ческим свойствам нитридов лантаноидов. Так, Дау [487] показал, что
отношение электросопротивлений /?MeN Яме возрастает при переходе
от церия к празеодиму от 1,66 до 30 (при 25°С), что, очевидно, связа-
но с большей относительной делокализацией валентных электронов
празеодима при образовании им связи с азотом и стремлении перейти
в устойчивое [°-состояние. До 500° С для нитрида церия обнаружен ход
сопротивления, типичный для металлов с ростом отношения (/?MeN:
• ^Ме)5оо° c/^?MeN:^?ме) 25о° с приблизительно в четыре раза. Для пра-
зеодима этот рост практически отсутствует (рис. 49).
Следует отметить рост магнитной восприимчивости от нитрида лан-
тана к нитриду неодима, установленный в [478]. Характерно, что осо-
бенно резкое возрастание х наблюдается при переходе от нитрида церия
130
к нитриду празеодима, что соответствует резкому изменению электро-
сопротивления (рис. 50).
Подробное исследование магнитных свойств нитридов редкоземель-
ных металлов проведено в [488]. Магнитные моменты мононитридов
оказались близкими к магнитным моментам трех положительных ионов
редкоземельных металлов, что, как и в случае
сульфидов Me2S3 [489], указывает на значитель-
ную компоненту ионной связи в этих соедине-
ниях. Зависимость магнитных моментов нитридов
от порядкового номера редкоземельных металлов
аналогична таковой для
сульфидов. Характерно,
что особенно резкое воз-
растание магнитной вос-
приимчивости наблюда-
ется при переходах от
CeN к PrN, от SmN к
EuN и от TuN к YbN, что
соответствует резкому из-
менению электросопро-
тивления.
Более детальное ис-
следование электрических
свойств нитридов редко-
земельных металлов, про-
веденное в [491], действи-
тельно обнаружило нали-
чие энергетических щелей
Рис. 50. Зависимость от-
ношения T?prN : Лрг от
содержания азота (п —
атомные доли).
Рис. 49. Температурная
зависимость отношения
^CeN ^Се.
(по краям поглощения, найденным на кривых оптического пропускания)
для нитридов лантоноидов: DyN—2,60—2,90 эв, ErN—2,40—2,78 эв,
HoN—1,70—1,88 эв. Электросопротивление в пределах от 80 до 1500° К
носит металлический характер, что объясняется большой концентрацией
носителей тока — электронов, которая вызвана отклонениями от сте-
хиометрии по содержанию азота [491]. Таким образом, эти нитриды яв-
ляются дефектными вырожденными (за счет избытка электроположи-
тельных атомов лантаноидов) полупроводниками с широкой энергети-
ческой щелью. Для исследовавшихся в той же работе ScN и YN
определенного скачка поглощения не обнаружено, что очевидно, обу-
словлено высокой донорной способностью этих металлов и образо-
ванием типично металлической связи, осуществляемой коллективизиро-
ванными электронами металла и азота. Подобные выводы подтверждают
и рентгеноспектральные исследования ScN [960].
Данные по электрическим свойствам нитридов лантаноидов согла-
9*
131
суются с данными, полученными Дидченко и Гортсема [488], хотя по-
следние проводили измерения на нитридах стехиометрического состава.
Это несогласие должно быть устранено дальнейшими исследованиями,
однако нам кажется, что представления, развитые в работе [491], более
предпочтительны. Однако, естественно, нельзя эти представления рас-
пространять на нитриды скандия
и иттрия, являющиеся типичными
металлическими фазами.
Рис. 51. Зависимость электросопротивления нитридов от порядкового
номера лантаноидов.
Рис. 52. Температурная зависимость электросопротивления спрессован-
ных порошков нитридов лантаноидов.
Нитрид иттербия, по данным [488], отличается от нитридов прочих
лантаноидов отрицательным температурным коэффициентом электро-
сопротивления, характерным для полупроводников.
На рис. 51 показана зависимость электросопротивления нитридов
редкоземельных металлов от порядкового номера при комнатной тем-
пературе [488].
На рис. 52 приведена температурная зависимость электросопротив-
ления спрессованных порошков некоторых нитридов. Данные для Се
не поддаются рациональной интерпретации, что же касается нитридов
лантана и празеодима, то они обнаруживают в области низких темпе-
ратур типичную металлическую проводимость. Авторы [478] усматри-
вают в поведении CeN отражение высокого содержания в нем ионов
Се4+. Полуметаллический характер мононитрида церия обсуждается
в [490].
132
Р
Характерно, что при переходе от LaN к CeN коэффициент терми-
ческого расширения возрастает очень резко — от 9 до 30-10“6, а далее
у нитрида празеодима вновь довольно сильно понижается — до 13-10“®
[478], что соответствует ослаблению связей Се—Се и усилению связей
Се—N, о чем говорилось выше.
2. НИТРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ IV ГРУППЫ
Нитриды титана. Из диаграммы состояния системы титан—азот, по-
строенной по данным [132] (рис. 53), следует, что титан образует с азо-
том нитрид, отвечающий формуле TiN, обладающий широкой областью
гомогенности, а также нитрид, соответствующий е-фазе.
Рис. 53. Диаграмма состояния системы титан—азот.
В системе обнаружены четыре фазы: р, а, е и 6. Перитектоидная
P-фаза содержит (при 2020±25°С) около 1,4% N и образуется при
реакции между a-фазой и расплавом: a-J-Ж^Р- При этой же темпе-
ратуре a-фаза гомогенна в пределах 6,5—7,4% N, образуется по пери-
тектической реакции d+Ж^а при 2350±25°С. е-Фаза, которой припи-
сывают условную формулу TiaN, образуется по перитектической реак-
ции а+d^e при температурах 1000—1100° С, гомогенна в пределах
6,8—8,9% N, имеет тетрагональную решетку с параметрами элементар-
ной ячейки: а=4,92 кХ; с=55,61 кХ, с/а= 1,05 [132].
Растворимость азота в a-Ti при температуре перитектоидной ре-
акции лежит в пределах 6,5—7,4%. Добавление азота к титану вызы-
вает повышение температуры a^p-превращения, т. е. азот стабилизи-
рует a-титан. Теоретическое рассмотрение температурной зависимости
растворимости азота в титане от соотношения атомных радиусов титана
133
и азота, а также электронного строения образующихся твердых раст-
воров проведено в [133, 134]. Энергия активации диффузии азота в
«-фазе составляет 68 000 ккал/моль [1119].
Хольмберг [235] установил, что предельная растворимость азота
в а-титане составляет около 17 ат. % (TiNo,2o). Им обнаружена нитрид-
ная фаза TbN (е-фаза), которой ранее приписывалась формула TisN.
Эта фаза имеет тетрагональную решетку типа антирутила (пространст-
венная группа P^lmnm) с двумя формульными единицами в элемен-
тарной ячейке с параметрами решетки а = 4,9452, с = 3,0342 А.
Растворение азота в титане линейно изменяет параметры решетки
а-титана: больше, чем при растворении кислорода, и несколько меньше,
чем при растворении углерода [205]. Следовательно, измерение пара-
метра кристаллической решетки может служить надежным способом
определения малых содержаний азота в титане.
Область гомогенности нитрида титана TiN, по данным [132], со-
ставляет 10—22,6% N. По данным Эрлиха [135], нижний предел об-
ласти гомогенности соответствует сплаву с 10,9 вес. % N или формуле
TiNo,42, а по данным [235], по-видимому, более надежным — формуле
TiNo,6o (14,8 вес. % N). Верхняя граница области гомогенности этого
соединения может быть принята 50 ат.% N, т. е. соответствует формуле
TiNt.o [132]. Однако необходимо отметить, что в ранних работах (Бре-
тера [136], а затем Мюнстера и Загеля [137]) указывается на более
высокий верхний предел области существования этого нитрида, соот-
ветствующий, согласно [136], формуле TiNi,i6, что относительно близко
к составу валентного соединения Ti3N4, которое образовалось бы при
ионном характере связи между Ti4+ и N3-. Следовательно, образование
нитрида титана с содержанием азота более 50 ат. % является призна-
ком склонности титана к образованию с азотом ионных связей, что
обусловлено высоким ионизационным потенциалом азота. Чисто кри-
сталлохимически нитридные фазы титана с избытком азота (или недо-
статком титана) следует рассматривать, по Ормонту, как твердые
растворы азота в нитриде, построенные по принципу вычитания. Пра-
вильность представлений о нитридах с избытком азота, как о твердых
растворах вычитания, Брегер подтвердил анализом интенсивностей ин-
терференционных линий на рентгенограммах методом, принятым в ра-
ботах Уманского и Хидекеля о карбиде титана и Уманского о карбиде
ванадия [138]. Образование таких фаз Брегер связывает со способом их
получения, состоящим в диссоциации аминохлоридов типа TiC14-4NH3,
которая при низких температурах проходит не до конца, так что часть
избыточных атомов азота остается в решетке и удаляется лишь после
нагревания до высоких температур порядка 1500° С. К этому утвержде-
нию следовало бы добавить, что образованию фаз с избытком азота,
имеющих признаки ионной связи, содействует ионное состояние титана
в аминохлориде.
134
Рентгеноспектральное исследование нитрида титана предельного
состава и в области гомогенности [227—229] показало, что при увели-
чении содержания азота в области гомогенности нитрида титана про-
исходит изменение характера химической связи в направлении увеличе-
ния ее металлической компоненты. Межатомное взаимодействие в нит-
риде осуществляется при весьма малом участии d-орбит титана с
мобилизацией преимущественно внешних s- и p-орбит обоих атомов. Этот
вывод согласуется с современными представлениями о природе элек-
тронной структуры нитрида ти-
тана (см. стр. 17), если под
металлической связью подразу-
мевать связь, осуществляемую
обменом между нелокализо-
ванными электронами и элек-
тронами, входящими в состав
стабильных конфигураций, об-
разуемых электронами, лока-
лизованными у атомов титана
и азота. В связи с относитель-
Рис. 54. Зависимость периода решетки нитрида
титана от содержания в нем азота.
но низким статистическим
десом ^-конфигураций атомов титана (43%) для него велика
доля электронов, находящихся в нелокализованном состоянии (преиму-
щественно 4з-электронов). Кроме того, атомы азота, имеющие в изо-
лированном состоянии з2р3-конфигурацию, стремятся приобрести ква-
зиустойчивую зр3-конфигурацию с передачей одного р-электрона в не-
локализованное состояние. Таким образом, электроны d-оболочки титана
в металлической связи участия практически не принимают. С этой
точки зрения увеличение металлической компоненты связи при повы-
шении содержания азота в области гомогенности нитрида титана может
быть интерпретировано, как повышение статистического веса нелокали-
зованных в стабильные конфигурации электронов с одновременным
отвлечением электронов с ковалентных связей между атомами титана.
Очевидно, что ионная доля связи в нитриде титана обусловлена не-
которой вероятностью привлечения атомами азота электронов титана с
образованием электронных з2р6-конфигураций. Статистический вес по-
следних особенно велик в препаратах нитрида титана, полученных из
аминохлорида, где эти конфигурации образуются атомами хлора и
атомами азота и сохраняются в нитриде.
С повышением азота в области гомогенности нитрида титана из-
меняются как параметр кристаллической решетки и плотность (рис. 54,
табл. 52), так и все физические и химические свойства, что очень су-
щественно для его практического использования.
Свойства нитрида титана предельного состава, отвечающего фор-
муле TiN, подробно описаны в справочниках [139, 140], а в области го-
135
Таблица 52 Изменение плотности и молярного объема ни- трида титана в области гомогенности [135] могенности — приведены в табл. 53. Поэтому в настоящем разделе остановимся только на некоторых свойствах, имею- щих принципиальный харак- тер.
Состав Плотность, г/см3 Молярный объем, см3/моль
Физические свойства. Нит-
TiN] оо 5,21s 11,87 рид титана в порошкообразном
TiNo,7o 11,31 состоянии — желто-коричнево-
TiN0>44 4,870 11,10 го цвета, после спекания в ком-
пактном состоянии приобретя-
ет характерную золотисто-латунную окраску. В компактном состоянии
хорошо шлифуется, полируется.
Термические свойства нитрида титана в пределах температур 298—
1800° К исследованы Нэйлором [141], согласно данным которого
Нт — Н298,16 = 11,9 1 7~ + 0,0004т2 ---------4585 [ккал/моль],
Ср — 11,91 + 0,000947’ — [кал/град -моль].
По данным Сатона [142], удельная теплоемкость, измеренная на не-
чистых препаратах и исправленная для чистого нитрида в пределах
0—500° С, составляет:
Ср = 0,1410 4- 1,918-10-4Т —1,127-IO”7?2.
Теплоемкость при низких температурах, по данным [143], такова:
Т, °К 52,5 60,5 69,7 78,5 94,9 125,2 145,8 165,7
Ср, кал/моль 0,527 0,809 1,189 1,582 2,331 3,704 4,577 5,358
Т, °К 185,7 206,0 226,3 246,0 266,1 286,2 298,16
Ср, кал/моль 6,073 6,705 7,286 7,768 8,234 8,642 8,86*
Исследователи [но], определяя давление паров нитрида титана
отмечали его трудную испаряемость даже при температурах, близких
к точке плавления. Результаты количественных исследований, выпол-
ненных Келли [150], и приведенные по сводке Дэшмана [74] в табл. 14,
показывают, что вблизи точки плавления упругость диссоциации нитрида
не превышает 1 мм рт. ст. Данные по упругости паров нитрида титана
приведены также в [45].
Поллард и Вудворд [158] определяли давление диссоциации нит-
рида титана на препаратах, полученных наращиванием из газовой
* Получено экстраполяцией.
136
Таблица 53
Физические свойства нитрида титана в пределах области
его гомогенности (145, 147]
Условная фор- мула Содержание N Термо-э.д.с. а, мкв/град Коэффици- ент Холла Я-10*, см* (к Удельное электросоп- ротивление Q, МКОМ-СМ Микротвер- дость нм t кг/ммг
вес. % ат. %
TiN0,557 14,0 35,8 — — 112,5 —
TiN0,59 14,7 37,1 + 0,9 +2,5 — 1200±137
™0,6 14,9 37,5 +0,5 — 91,2 —
™0,63 15,7 39,1 — — — 1440±182
TiN0,65 15,9 39,5 —0,75 + 1,9 70,6 —
TiN0,69 16,7 40,8 —1,88 + 1,5 60,5 —
T'N0,745 17,8 42,7 —3,6 + 0,9 50,6 —
TiN0>79 18,7 44,3 —4,3 — — —
TiN0i84 19,7 45,8 —5,34 — — —
TiN0,85 19,9 46,1 — — — 163О±Ю1
TiN0i88 20,4 46,8 — —0,37 — —
TiNo91 21,0 47,6 — — 31 —
TiN0,92 21,4 47,9 — — — 1780±51
TiNo,95 21,7 48,7 —7,1 —0,63 — —
™0.97 22,2 49,3 — — — 1900± 182
^'^0.98 22,4 49,8 —7,38 —0,67 26 1994±137
фазы на вольфрамовой проволоке. Соответствующие данные для тем-
ператур 1200 и 1450° С приведены на рис. 55. По этим данным терми-
ческая диссоциация нитрида титана представляет собой реакцию вто-
рого порядка, константа скорости которой подчиняется уравнению
К = —-----—~(ммрт. ст.-1 -мин,-1],
т а (а — х) '
где а — концентрация газа при предельном давлении р0, х — концент-
рация газа при давлении р, т — время. Ниже приведены значения кон-
станты скорости диссоциации нитрида титана от времени при темпе-
ратуре 1200° С (ро=0,59 лш рт. ст.):
Т, мин. 5 10 20 30 40 50 60 70 80 90
К, мм pm. ст^1-мин-1 0,19 0,23 0,23 0,22 0,22 0,23 0,22 0,22 0,23 0,23
Последующие исследования поведения нитрида титана в вакууме
выполнены в работе [203], в которой испарение TiN определяли эффу-
зионным методом Кнудсена при температурах 1987—2241° К. Установ-
137
лено, что нитрид испаряется с образованием газообразного титана и
азота
TiNTB — Т1газ 1/z N2.
Константа равновесия этой реакции Кр=ртгРмг72,
Из данных работы получено
1g Кр - — 0,60-10“4Т 4- 12,245,
Рис. 55. Давление диссоциации TiN:
/ — TiN на вольфрамовой проволоке при
1200° С; 2 — то же при 1450° С; 3 — воль-
фрамовая проволока при 1200° С (эталон
без нитрида титана).
Таблица 54
Скорость испарения нитрида титана
и давление диссоциации титана и
азота над нитридом
Температу- ра, °к Скорость испарения G-10*, г/см*-сек Давление пара титана PTi10*» am Давление азота "N/IO*, am
1987 1,510 1,697 0,650
2017 1,972 2,234 0,854
2050 3,129 3,573 1,366
2155 15,963 18,685 7,146
2212 33,254 39,435 15,08
2241 70,555 84,221 32,2
а парциальные давления титана и азота
над нитридом
lgpTi = _ 2^. _ 0,40 • 10-4T -Ь 8,263,
1g_ о,40 • 10~4Г + 7,963.
Теплота реакции диссоциации Д№ была установлена равной
191,2 ккал/моль, а стандартная теплота образования нитрида титана
Л^298==—79,4 ккал/моль. (По [1076] ДЯ®98=76,76 ккал/моль) .
Эти данные проверяли В. В. Фесенко, А. С. Болгар и Э. А. Рыклис,
которые установили, что состав нитрида титана при испарении заметно
изменяется, а скорость испарения из ячеек Кнудсена резко зависит от
площади истечения. Поэтому данные работы [203] являются прибли-
женными.
Зависимость составов конгруэнтного испарения от температуры
(1993—2243° К) выражается уравнением: х=1,46—2,8-10-4 Т, где
x=;N/Ti; коэффициент испарения нитрида титана а=1,3-10~2.
138
Исследование микротвердости нитрида титана в области его гомо-
генности выполнено в работе [147]. Показано (см. рис. 4), что концент-
рационная зависимость микротвердости является линейной и подобна
в этом отношении зависимости микротвердости карбидов титана, цир-
кония [147] и тантала [148] от содержания связанного углерода. Одна-
ко, несмотря на то, что в последних трех случаях экстраполяция линии
микротвердости на содержание углерода, равное нулю, дает приблизи-
тельное значение микротвердости соответствующих металлов, для нит-
рида титана это не наблюдается. Если в карбидах характер распреде-
ления электронной концентрации мало отличается от такового для ме-
таллов, в нитриде он изменяется в результате появления значительной
доли ионной связи. Как видно из рис. 4, доля ионной связи возрастает
с понижением содержания азота в решетке нитрида. Последнее может
быть связано с образованием связей Me—Me, меньшим возмущением
вследствие этого валентных электронов атомов азота и соответствую-
щим увеличением энергетического разрыва между d- и s-состояниями
титана и p-состоянием азота.
Сжимаемость нитрида титана, по данным Бриджмена [149], выра-
жается следующими соотношениями:
ду/у0 = 3,32• 10-7р — 2,13- 10~12р2 при 30 С,
АК/К0 = 3,51 • 10~7р — 2,13- 10-12р2 при 75°С,
где Ко — объем при р = 0, р — давление, к.г/см2-.
Модуль упругости нитрида титана, полученный на образцах с низ-
кой плотностью (3,03 г/сл«3), равен 8060 к.г!мм2 [125, 139], это занижен-
ное значение. Затем модуль упругости нитрида титана был определен
равным 25600 кг/мм2 [166]. Модуль упругости TiN в пределах темпе-
ратур до 600° С изменяется мало [195].
По данным [1031], модуль упругости беспористого нитрида титана
равен 61600+2000 кг/мм2 (чему соответствует характеристическая тем-
пература 796+15° К), а по [1077] --44000 кг/мм2 (0=847° К).
Механические свойства нитрида титана почти не изучены; имеются
сведения только о пределе его прочности на сжатие при 20° С, кото-
рый составляет 100—130 кг)мм2 [146].
Для нитрида титана характерно низкое удельное электросопротив-
ление, равное около 25 мком-см, что приблизительно в два раза ниже
электросопротивления металлического титана [144]. Исследование
электрических свойств нитрида титана в области гомогенности, прове-
денное нами совместно с Т. С. Верхоглядовой [145], показало, что с
уменьшением содержания азота увеличивается доля ионной связи в
решетке с соответственным изменением не только уровня, но и харак-
тера проводимости. Изменение электросопротивления нитрида титана
139
в области гомогенности (см. рис. 5) нелинейно и отличается от хода
сопротивления карбида титана. Появление энергетической щели в ре-
шетке нитрида титана с неполным содержанием азота, предположен-
ное для нитрида титана в работах [151, 152], подтверждается измере-
ниями электросопротивления при высоких температурах (см. рис. 6).
Показано, что нитрид титана с содержанием азота, близким к стехио-
метрическому (49,8 ат. %N), обнаруживает линейный рост электро-
Рис. 56. Температурная зависимость электросопротивления пленок TiN,
нанесенных на подкладки:
/ — SiO2 (толщина пленки 3 мк); 2— SiO2 (толщина пленки 9 мк); 3 — А12О3 (тол-
щина пленки 7 мк); 4 — вольфрамовая проволока.
Рис. 57. Зависимость толщины пленки на образце из TiN от времени
окисления при различных температурах.
сопротивления с температурой. При уменьшении содержания азота до
48,4 ат. % появляется максимум сопротивления при 1800° С. При даль-
нейшем снижении содержания азота сопротивление возрастает с одно-
временным смещением максимума в область более низких температур:
750° С для нитрида с 43,4 ат.% азота; 550° С для 38,6 ат.%. Ширина
энергетической щели с уменьшением содержания азота увеличивается.
Таким образом, снижение содержания азота в нитриде титана в пре-
делах области его гомогенности приводит к увеличению доли ионной
связи.
Теперь понятна чувствительность электрических свойств нитрида
титана к примесям, особенно таким, которые могут содействовать по-
вышению степени поляризации атомов азота. Так, уже в ранней работе
140
Мюнстера и Загеля [172] было показано, что электропроводность пле-
нок нитрида титана, нанесенных осаждением из газовой фазы на раз-
личные подкладки, существенно зависит от состава последней и воз-
можности перехода ее компонентов в слой нитрида. Пленки нитрида,
нанесенные на подкладки из металлического вольфрама, а также из
окиси алюминия обнаруживают металлический характер проводимости.
Пленки нитрида, нанесенные на подкладку из SiO2, имеют отрицатель-
ный температурный коэффициент электросопротивления (рис. 56). Это
может быть связано с относительно легкой восстановимостью SiO2 и
переходом в процессе осаждения нитрида определенной части кисло-
рода SiO2 в слой TiN, по-видимому, с образованием твердого раствора
TiN — TiO. Последующая работа [174] подтвердила эти соображения.
Данные об электрических свойствах пленок из нитрида титана приведе-
ны в табл. 55.
Таблица 55
Электрические свойства пленок из нитрида титана, полученных оса-
ждением из газовой фазы на различных подкладках
Материал подкладки Толщи- на слоя TiN, мк Удельное электросопротивле- ние, ом-см Температурный коэффициент сопротивления в интервале 700—800°С Константа Хол ла, см3/к
20°С | 800°С
Вольфрам — 1,65-10—5 5,82-10—5 +3.2-10-2
A12O3 7 4.6-10-5 1,2. ИГ"* + 2-10-2 1,2-Ю-4
SiO2 3 1,4-Ю-2 1,8-Ю-4 —2-10—2 —
(кварц) 9 3,9-10—4 1,6-10-4 —4-10“4 —1,3-10—4
Эти данные подтверждаются исследованиями электрических свойств
системы TiN—TiO, выполненными в [174]. Показано, что добавление
кислорода к нитриду титана вызывает полупроводниковый характер
его проводимости.
В работах [1057, 1078] исследована зависимость электросопротив-
ления нитрида титана состава TiNo.es от пористости образцов.
С увеличением давления электросопротивление нитрида титана
понижается, как и для большинства металлов; средний барический
коэффициент электрического сопротивления при давлениях до
12 000 кг/см- составляет 9,35 ПО-7 при 30° С и 9,97-10~7 кг/см?- при
75° С [149].
Электропроводность нитрида титана при низких температурах ис-
следовали неоднократно, причем точку перехода к сверхпроводимости
принимали в широких пределах — от 1,1 до 5,6° К [168, 169]. Наиболее
достоверным значением Тк считается 4,85° К [1001].
141
Термоэмиссионные свойства нитрида титана изучены мало [170],
работа выхода электронов <р = 2,92 эв, постоянная Ричардсона А =
— 120 а/см2-град2 [140]; по [252] <р=3,70 эв.
Нитрид титана слабо парамагнитен с молярной магнитной воспри-
имчивостью 48- 1 О-6 [171] *.
Химические свойства. Первое систематическое исследование стой-
кости нитрида титана к окислению выполнено в работе [154]. На
рис. 57, по данным этой работы, показана зависимость толщины пленки
на компактном, полученном горячим прессованием образце TiN от вре-
мени окисления при разных температурах. На основе химического и
электронографического исследования окисных пленок установлено, что
уже при низких температурах на нитриде образуется плотный тонкий
слой твердого раствора TiN—TiO, подобный псевдоморфным слоям,
наблюдающимся при окислении металлов. При достижении определен-
ной критической толщины этот слой во внешней своей части начинает
превращаться в Т1гО3 и ТЮг. Первой стадии — образованию слоя твер-
дого раствора TiN—TiO — отвечает начальный участок изотерм (кри-
волинейный), а второй стадии — прямолинейный участок. Диффузия
ионов кислорода через слой твердого раствора TiN—TiO связана со
значительной кажущейся энергией активации, составляющей, по дан-
ным работы [154], 54,94 ккал!моль.
В работе [238] проведено изучение окисления дисперсного порошка
(удельная поверхность 1500 г!см2) нитрида титана в сухом и влажном
воздухе при 600—750° С. Подтверждены основные выводы работы [154],
показано, что кривые окисления состоят из двух участков — началь-
ного прямолинейного, соответствующего химическому взаимодействию
нитрида титана с кислородом, и последующего — нелинейного, соответ-
ствующего диффузии кислорода к нитриду титана через окисную плен-
ку. Установлено, что кривые окисления нитрида во влажном воздухе
(до 6 об. % водяных паров) практически совпадают с кривыми окисле-
ния в сухом воздухе, что указывает на принципиально одинаковый ме-
ханизм окисления. Энергия активации для первого этапа окисления
(химического взаимодействия нитрида с кислородом) составляет
44,9 ккал/моль, для второго этапа (диффузия через окисную пленку) —
54,60 ккал!моль, что хорошо согласуется с данными [154] — 54,94 и
[239] — 54,30 ккал!моль.
Технически заметное окисление нитрида титана начинается при
температурах выше 700—800° С. По данным [238], порошок нитрида
титана при 850° С полностью окисляется менее чем за 10 мин.
Дальнейшее подробное исследование кинетики и механизма окисле-
ния нитрида титана было проведено Мюнстером и Шлампом [155], ко-
* По данным [978], магнитная восприимчивость 37 • 10~в.
142
торые также проанализировали данные соответствующих работ других
исследователей. Авторы [155] исследовали скорости окисления нитрида
титана в интервале температур 625—1075°С в чистом кислороде при
нормальном его давлении на образцах нитрида титана, нанесенного в
форме слоев на алундовые пластинки (по реакции TiCl4 + 2H2 + 1/2N2->
->TiN+4HCl, содержание азота в слое составляло 22,2—22,9%). Фор-
мы изотерм, полученных в этой работе, аналогичны изотермам рис. 57,
т. е. состоят также из двух участков — параболического и линейного,
которым отвечают энергии активации соответственно 46 400 и
24 500 кал/моль. В принятых пределах температур изотермы удовлетво-
рительно описываются уравнением t—ay2-\-by, выше 900° С при
больших продолжительностях окисления наступают некоторые от-
клонения.
Мюнстер и Рупперт [157] обнаружили, что поверхностный слой из
нитрида титана толщиной меньше 1 мк полностью разрушается на воз-
духе за несколько часов при 700° С. Поллард и Вудворт [158] изучали
взаимодействие нитрида титана с кислородом при 1200° С по измене-
нию давления в системе. В первые 15 мин давление резко падает (при-
близительно в два раза), затем начинается медленный рост последнего.
Анализ остаточного газа показал, что атомное отношение поглощенного
кислорода к выделившемуся азоту равно 1,7, в то время как для ре-
акции
TiN ]- О2 = TiO2 -[- V2 N2
ожидалось отношение, равное 2. Падение давления в начальный период
объяснено медленным удалением азота из решетки, а несоответствие
количеств поглощенного кислорода и выделившегося азота — раство-
рением кислорода решеткой нитрида. В основном эти наблюдения со-
гласуются с данными работы [154] по образованию при окислении ни-
трида титана в начальный период слоя твердого раствора TiN—TiO.
В работе [159] приводится завышенная граница стойкости компактного
нитрида титана против окисления, соответствующая 1100—1400° С.
Порошкообразный нитрид титана сгорает в токе кислорода уже
при 700—800° С.
По отношению к водороду стехиометрический нитрид титана инер-
тен. Так, по данным [НО], спеченный нитрид до точки плавления сохра-
няет в среде водорода полное содержание азота, авторы некоторых
ранних работ наблюдали только небольшое поглощение водорода при
нагревании нестехиометрического нитрида при 1000° С, а также очень
незначительное выделение аммиака в подобных условиях [139]. Смеси
азота и водорода не взаимодействуют с нитридом титана при
1200° С [158].
Нитрид титана стехиометрического состава устойчив по отношению
к действию СО, нитрид с недостатком азота легко разлагается СО и
143
СО2. Окисление стехиометрического TiN углекислым газом идет мед-
ленно по реакции
2TiN + 4СО2 -> 2TiO2 + 4СО + N2,
константа скорости которой выражается уравнением для реакций вто-
рого порядка [158]
к 4АР
рт (р — 4Др) ’
где р — начальное давление СО2, Др — изменение давления за время т
реакции (табл. 56).
Водяные пары на нитрид титана практически не действуют; по-
пытка в начале 900-х годов использовать нитрид титана для получения
аммиака действием на него сме-
Таблицабб сями пара Н2О и кислорода при
Константы скорости окисления нитрида ти- пониженном давлении не дали по-
тана углекислым газом ложительных результатов [139].
т, мин 1 р. мм рт. ст. к, мм рт. ст. 1 •MUH,~~l т, мин р, мм рт. ст. К, мм рт. ст. ' мин~1 При низких температурах (до 270° С) на нитрид титана хлор не Действует, а при темпера- турах от 300 до 400° С TiN хлори- руется газообразным хлором [160]. При нагревании втокеНС! при 600—650° С нитрид титана
0 2,75 0,021 50 3,18 0,013
10 3,00 0,021 60 3,22 0,013 количественно переходит в TiCh
-20 3,08 0,013 70 3,24 0,013 [161]*.
30 40 3,11 3,14 0,012 0,012 80 90 9,26 3,28 0,013 0,014 Нитрид титана разлагается
НС1 (при температурах выше 1200—1300° С) с образованием
смеси газообразного TiCl4 с водородом и азотом. Наряду с TiCU образу-
ются также низшие хлориды титана Т1С1з и TiCl2. Эта смесь способна
взаимодействовать на металлических поверхностях (например, молиб-
дена) с образованием вновь нитрида титана [237].
Нитрид титана энергично взаимодействует с NO и NO2, однако ко-
личественное исследование еще не проведено [158].
При нагревании нитрида титана в токе водорода с введенными
в последний парами серы при температре 700—800° С нитрид превра-
щается в сульфид титана [139].
Отношение нитрида титана к кислотам и щелочам наиболее де-
тально исследовано в последние годы [162]. TiN в течение 24 ч
При комнатной температуре практически не растворяется в соляной,
серной, хлорной, фосфорной кислотах, смесях хлорной и соляной, ща-
* Несколько иные данные о температурах хлорирования нитрида титана приводят-
ся в работе [230].
144
целевой и серной кислот. Слабо действуют на него в течение 2 ч кипя-
щие соляная, серная и хлорная кислоты, малоустойчив на холоду про-
тив действия растворов едкого натра. Обзор ранних работ по этому
вопросу приведен в [139].
Огнеупорные свойства [163, 164]. Нитрид титана стоек против дейст-
вия расплавленных олова, висмута, свинца, кадмия и цинка (табл. 57).
Жидкие чугун и основной ваграночный
шлак при температуре 1500° С смачивают
нитрид титана, но не реагируют с ним. Чех
и Сейбал в Институте порошковой метал-
лургии ЧССР исследовали смачиваемость
нитрида титана в вакууме при 1550° С. Най-
дено, что нитрид титана не смачивает желе-
зо, никель смачивает хорошо, сплав никеля
и хрома (1:1) разъедает нитрид. При дей-
ствии расплавленного алюминия наблюдает-
ся небольшая поверхностная коррозия ни-
трида титана (за 30 мин при 1000° С).
Тигель из нитрида титана (20,75% N),
спеченный при 1800° С в вакууме при давле-
нии 1,5 мм рт. ст. в течение 15—30 мин,
не разъедался и не разрушался в расплав-
ленном электролитическом железе (при
1700° С), сыром железе (1600° С), никеле,
кобальте, хроме (1600° С), марганце
(1400° С), меди (1200° С), алюминии
(1000° С) и фосфористом железе (1200° С)
рид титана в вакууме также не взаимодег
При сильном нагревании нитрид не взаимодействует с порошком
вольфрама, добавка до 10% TiN к вольфраму почти не изменяет тем-
пературы плавления последнего, при дальнейшем увеличении содер-
жания нитрида точка плавления снижается, однако металлографиче-
ски наблюдать взаимодействие не удается. Таким образом, по данным
работы [ПО], нитрид титана не взаимодействует с вольфрамом в широ-
кой области температур и составов. Отмечено, что вольфрамовая нить
не реагирует с нитридом титана и при получении последнего методом
наращивания из газовой фазы.
Тигли из нитрида титана при высокой температуре сильно разъе-
даются окислами Fe2O3, MnO, SiO2 и стеклом [164]. Порошок нитрида
титана окисляется при нагревании с легковосстановимыми окислами
типа CuO, PbO, HgO, причем окисление часто сопровождается сильным
искрением и самовозгоранием.
Авторы [165] исследовали электрохимическое поведение анодов из
нитрида титана в хлоридном расплаве LiCl-|-КС1 и показали, что даже
Поведение нитрида титана в
контакте с расплавленными
металлами
Металл Темпе- ратура, °C Время контак- та, ч Содержание Ti в метал- ле после переплавки, %
Sn 350 10 Не обна-
ружен
350 40 0,26
Bi 380 10 Следы
380 40 0,07
Pb 450 10 0,04
450 40 0,20
Gd 450 10 0,20
450 40 0,07
Zn 520 10 0.06
[139]. Очень чистый нит-
твует с железом.
10—272
145
при низких плотностях тока (Da = 0,004—0,035 а/см2) длительный
электролиз сопровождается выделением на них азота и переходом в
электролит четырехвалентного титана. Ниже 1,5* 10—3 al см2 анодный
потенциал мало меняется с плотностью тока, значительная поляриза-
ция нитридных анодов наблюдается в интервале 0,002—0,2 а]см2, где
потенциал возрастает на 0,6—0,7 в. Выше 0,2 а)см2 анодный потенциал
становится практически постоянным.
Результаты изучения поведения нитрида титана в контакте с нио-
бием, танталом, молибденом и вольфрамом в твердом состоянии при
температурах 1600—2100° С, проведенного
нами совместно с Г. А. Ясинской и
Э. А. Пугач по методике, описанной в ра-
боте [163], показывают, что наиболее стоек
нитрид титана в контакте с молибденом
(табл. 58).
Взаимодействие нитрида титана с угле-
родом исследовали в [1'57], найдено, что
Рис. 58. Изменение свободной
энергии при науглероживании
нитрида титана:
/ — по реакции TlN+C^TiC + 72N2;
2 — то же, но с учетом образования
твердых растворов TiC—TiN (XTIC =
=0,7).
реакция
TiN + C^ TiC 4-72N2
сопровождается убылью свободной энергии,
составляющей в зависимости от темпера-
туры
&р°= 39650 — 20,157’’ (рис. 58, прямая /).
Таким образом, при повышении температуры устойчивость нитрида
по отношению к углероду снижается, очевидно, в связи с его наклон-
ностью к диссоциации, и частично образуется карбид. Это уравнение
для AF0 выведено без учета образования твердых растворов TiN—TiC;
если учесть последнее, то изменение свободной энергии выразится урав-
нением
^iN-Tic И pN = RT In + RT In ,
2 *TiN "TiN
где x — молярная концентрация, f — коэффициент активности. Прямая
2 рис. 58 показывает изменение свободной энергии при образовании
твердых растворов, хтю=0,7, причем принято [цс=[™=1. Как видно,
образование твердого раствора TiN—TiC мало влияет на взаимодейст-
вие нитрида титана с углеродом.
В [231, 232, 1079] вновь исследовали реакцию взаимодействия нит-
146
рида титана с углеродом (TiN + C^TiC + '/2^2) в пределах температур
1480—2480° С при атмосферном давлении азота. Найдено выражение
для температурной зависимости константы равновесия
5600
Т
2,78,
а также показано, что лимитирующим звеном при установлении равно-
весного состояния являются процессы диффузии — подвода углерода
к поверхности частиц нитрида титана, а также диффузии внутри частиц.
Методы получения нитрида титана. Основными ме-
тодами получения нитрида титана являются:
1) азотирование металлического
титана или его гидрида;
2) взаимодействие в газовой фазе
между TiCl4 и аммиаком или смесями
азота и водорода;
3) разложение аминохлоридов ти-
тана и других подобных соединений,
содержащих титан и азот;
4) восстановление TiO2 углем или
металлами в среде азота.
Азотирование металлического ти-
тана впервые проведено Либихом в
1831 г. В работах [175, 141, 143] нит-
рид титана получали азотированием
Таблиця 58
Поведение нитрида титана в контак-
те с тугоплавкими металлами при
высоких температурах
Темпе- ратура, °C I Nb Та Мо W
2ч 5ч 2ч 5 ч 2ч1 5 ч 2 ч 5 ч
1600 Н н Н —
1800 — сл — н н И Н н
2000 В в в — сл сл сл в
2100 с с — с — в в •—•
титанового порошка В молибденовых П р и м е ч а н и е : в—взаимодействуют, с —
_________________________________ пап сильно взаимодействуют, сл — слабо взаимо-
ЛОДОЧКАХ при температуре 1 100 действуют, н—не взаимодействуют.
1200° С в азоте или токе Нг+Nz в фар-
форовой трубке в течение 1—2 ч. Але-
ксандер [176] предложил для получения нитрида титана азотировать
его гидрид, что было использовано Дувецом и Оделлем [177], ко-
торые азотировали гидрид при 1800—1900° С чистым азотом. Анало-
гичный метод разработан Фостером [196], который азотировал гидрид
титана нагреванием в токе NH3 в течение почти 100 ч при 1000° С.
Эрлих [135] получал нитрид титана азотированием 99,9 %-ной стружки
титана чистым азотом в течение 2—3 ч при 1200° С (в корундовой
трубке). При этом после однократного азотирования образуется нит-
рид состава TiN0>95- После измельчения и повторного азотирования этого
продукта содержание азота возрастает до TiNi.oo, т. е. до полагающихся
50 ат.%. Нагревая смеси TiN+Ti в вольфрамовой лодочке, Эрлих [135]
ю*
147
получил сплавы титана с азотом в широких пределах содержания по-
следнего с условными формулами от TiN0,i до TiNi.o-
Гвоздов и Журенкова [204], также исследовавшие взаимодействие
титана с азотом, установили, что до 500° С азот не действует на титан,
поглощение начинается при 500° С и при 800° С развивается энер-
гично с образованием нитрида титана. Отмечено, что поглощение азота
титаном резко снижает последующее поглощение водорода, причем
это снижение значительнее, чем в случае предварительного окисления
титана.
Процесс азотирования порошкообразного титана в условиях, прак-
тически исключающих одновременное окисление, подробно исследован
в [178].
Для азотирования использовали порошок титана чистотой 99,98%
(периоды решетки а = 2,96; с = 5,64 А), температура азотирования
варьировалась от 500 до 1200° С, а выдержки при каждой температу-
ре— от 15 до 240 мин. Температурная зависимость константы ско-
рости азотирования приведена в табл. 28. Результаты азотирования
чистого порошка титана (рис. 59) показывают, что зависимость содер-
жания азота при температурах 700—900° С подчиняется линейному, а
выше — параболическому закону насыщения, чего и следовало ожи-
дать, имея в виду широкую область гомогенности нитрида титана и ус-
ложнение процесса диффузии азота в кристаллическую решетку по
мере заполнения пор между металлическими атомами и приближения
к верхней границе области гомогенности. Основное насыщение титана
азотом происходит в первые 10—15 мин, а при дальнейшей выдержке
содержание азота растет незначительно. Полное завершение реакцион-
ной диффузии с образованием нитрида предельного состава достига-
ется при выдержке в течение 2—4 ч при 1200° С. Опыты азотирования
гидрированого титана (с 95,2% Ti), проведенные в той же работе, по-
казали, что водород, внедренный в решетку титана, сильно замедляет
диффузию азота при относительно низких температурах порядка 500—
800° С, когда гидрид титана достаточно устойчив. При прочих равных
условиях продукты азотирования содержат в два раза меньше азота.
Начиная с 1000° С, когда гидрид титана заметно диссоциирует, насы-
щение азотом проходит более интенсивно, и максимальное насыщение
азотом достигается при 1200° С в течение 2—4 ч, т. е. при тех же ус-
ловиях, что и для чистого исходного титанового порошка. При азотиро-
вании порошка титана аммиаком (рис. 60) диффузия азота затрудня-
ется.
Азотирование гидрида титана (Т1Ы194) в пределах температур
700—1300° С показало, что стехиометрический TiN может быть получен
в течение 5 ч при 1300° С [1000].
Исследователи [226] готовили нитрид титана азотированием по-
рошка титана (99,5%) полученного восстановлением TiO2 гидридом
148
кальция. Азотирование проводили в печи ТВВ-2 хорошо очищенным
азотом по режиму: продувка печи водородом 20 мин, продувка печи
азотом 20 мин, нагрев в течение 1 ч до 1200° С, выдержка в токе азота
при 1200—1250° С в течение 2,5 ч, охлаждение с печью. Полученный
по этому режиму нитрид титана содержал 77,35% Ti и 22,39% N, т. е.
был близок к стехиометрическому составу.
Рис. 59. Зависимость содержания азота в порошке титана, азотированного при
500—1200° С от времени выдержки:
/ — теоретическое содержание; 2 — 1200; 3 — 1100; 4— 1000; 5 — 900; 6 — 800; 7 — 700 , 8 —
600; J —500° С.
Рис, 60. Температурная зависимость содержания азота в титане при обработке
его аммиаком.
Гульбранзен и Эндрю [179], изучавшие скорость азотирования титана
при давлении 769 мм рт. ст. с образованием нитрида титана по упро-
щенной реакции* Ti + 1/2N2 = TiN, установили характер зависимости
константы скорости от температуры (рис. 61) и времени (рис. 62) азо-
тирования при 500—900° С. Для температурной зависимости константы
скорости реакции азотирования получено:
Температура, °C К, г/см3-сек Температура, °C К, г/см9-сек
550 5,28-Ю-15 775 1,08-10~12
600 2,97-10~и 800 1,46-Ю-12
700 2,78 -10-13 880 3,2-Ю-12
* Не учитывающей образования фаз переменного состава и различной толщины.
149
Рассчитанная из этих данных теплота активации при диффузии
азота в титан с образованием нитрида титана оказалась равной
36300 кал/моль. Временная зависимость привеса при азотировании вы-
ражается эмпирическим уравнением
W2 = Кт,
где W — привес, мг/см2 поверхности листового титана (99%), К — кон-
станта, т — время азотирования. Азотирование иодидного титана высо-
кой чистоты идет гораздо медленнее.
Изменение давления в пределах
76—760 мм рт. ст. мало влияет на
скорость азотирования, что объясняет-
Рис. 61. Температурная зависимость константы скорости реакции Ti+'/jNs
от температуры.
Рис. 62. Зависимость привеса при азотировании титана от времени:
/ — 500; 2 — 600; 3 — 700, / — 800, 5 — 900° С.
ся образованием пленки нитрида на титане, скорость растворения кото-
рой в металле определяет скорость реакции азотирования.
Ричардсон и Грант [180] исследовали взаимодействие азота с ти-
таном в области более высоких температур — от 700 до 1050° С. Ис-
пользовали технический титан, который азотировали при начальных
давлениях азота в 0,2—0,5 ат. Скорость азотирования определяли по
уменьшению давления в системе, вызванному поглощением азота ме-
таллом. Было установлено, что скорость азотирования приблизительно
в 50 раз меньше скорости окисления титана. В поверхностных пленках
на азотированном титане установлено только наличие нитрида TiN.
Добавка к порошку титана 5°/о KHF2 в качестве катализатора су-
щественно ускоряет процесс азотирования [246].
Одним из технологических приемов получения нитрида титана яв-
150
ляется плавка титана в среде азота, впервые предложенная для этой
•цели В. П. Реминым [181]. Впоследствии режимы этого приема были
разработаны в работе [132]: расплавление титана проводят током 15О.а,
затем силу тока увеличивают до 300 а; при обоих значениях силы тока
материал выдерживают в течение 2—3 мин. Недостатком этого приема
является значительная ликвация азота, что затрудняет получение од-
нородного продукта.
Авторы [306] обращают внимание на то, что образующиеся при азо-
тировании тугоплавких металлов, в частности титана, фазы являются
достаточно тугоплавкими, что затрудняет диффузию через них азота,
замедляет, а в некоторых случаях делает практически невозможным,
технологическое проведение азотирования. Поэтому рекомендуется по
мере насыщения образца азотом повышать температуру азотирования
для увеличения подвижности азота в поверхностном слое азотируемого
металла.
Взаимодействие в газовой фазе между TiCl4 и аммиаком или сме-
сями азота и водорода. Метод получения нитрида титана взаимодействи-
ем между TiCl4 и аммиаком — один из наиболее старых способов полу-
чения этого нитрида, предлагавшийся многими исследователями, начи-
ная с Велера и Сент-Клер Девиля (1858 г. [139]. В основе способа ле-
жит реакция
2 TiCl4 + 2 NH3 = 2 TiN + 6 НС1 + Cl2,
которая проводится при температурах выше 1000° С, т. е. практически
восстанавливают тетрахлорид титана смесью водорода с азотом. Тех-
нологическая разработка этого метода подробно дана в патенте [182] для
получения тонкодисперсного и однородного по химическому составу
нитрида титана. Сущность технологии заключается в подаче предва-
рительно нагретых до температуры 700—900° С аммиака и тетрахлори-
да титана в кварцевую реакционную камеру через форсунку, по внут-
реннему каналу которой подается тетрахлорид, а по внешнему кольце-
вому пространству — аммиак. Реакционная температура в камере
поддерживается в пределах 750—950° С, на 1 моль тетрахлорида ти-
тана в газовой фазе приходится 2—4 моля аммиака, скорость пропус-
кания газов в реакционную камеру — от 2 до 20 м[сек. Показано, что
лучшие результаты получают при ламинарных потоках газов, для чего
впуск последних в реакционную камеру осуществляют через большое
число форсунок с диаметром внутренней трубки 0,5—8 мм. Температу-
ра реакции непосредственно у выхода из форсунки должна составлять
100—1200° С, что обеспечивает достаточно высокую полноту реакции:
Температура, °C 800 1000 1200
Степень превра-
щения в нит-
рид, % 75,6 95,7 99,6
151
Продукт, выходящий из реакционной камеры, состоит из очень
тонкой взвеси нитрида в газообразной среде из хлора, паров НС1, не-
прореагировавших реагентов, примесей прочих газов, которые могли
проникнуть при подаче реагентов в камеру.
Эта смесь сначала охлаждается в трубчатых теплообменниках, а
затем нитрид титана отделяется в циклоне электростатическим осажде-
нием или фильтрацией на керамическом фильтре.
Получаемый описанным методом нитрид титана представляет по-
рошок размером частиц 0,1—0,6 мк с содержанием азота 22,2—22,6%.
Согласно [185], нитрид титана можно получить также взаимодейст-
вием тетрахлорида титана с аммиаком. Установлено, что ниже 1400° С
образуется нитрид, в решетке которого 15—20% пустых титановых уз-
лов, что не согласуется с вышеприведенными данными.
Вариант этого метода заключается в наращивании из газовой фазы
нитрида титана на металлической нити обычно по реакции
2 TiCl4 + N2 + 4 Н2 = 2 TiN % 8 НС1.
Ван-Аркель [183] получал этим методом нитрид титана на вольф-
рамовой нити, нагретой до 1400—2000° С, причем осадок состоял из
крупных монокристаллических образований. Аналогичные работы вы-
полнены Моерсом [184], а также Кемпбеллом с сотрудниками [159], ко-
торыми подробно исследовано влияние температур, давлений, тока на
процесс наращивания нитрида из газовой фазы. В работе [158] указы-
вается, что нитрид титана, точно отвечающий формуле TIN, образуется
методом осаждения из газовой фазы на проволоке, нагретой до
~ 1450° С, при общем давлении в реакционной камере 300—400 мм рт. ст.,
парциальном давлении TiCU 17 мм рт. ст. и отношении содержаний
N2:H2=1 : 1.
В недавно выполненной работе [1050] кристаллический нитрид ти-
тана получен аналогичным способом: осаждением на вольфрамовом
прутке. При температуре 1400° С и соотношении Ti : N в газовой фазе,
равном 1:4 создаются условия роста кристаллов размером 3X3 ММ'„
содержание азота в них составляет 21,0—21,3% (TIN 0,э).
Разложение аминохлоридов титана. Наиболее по-
дробное исследование этого старейшего метода получения нитрида
титана выполнены А. X. Брегером. В работе [186] Брегер получал нит-
рид титана реакцией в газовой фазе между TiCU и ЫНз при 800° С над
инертными веществами с последующим нагреванием при 300—350° С
полученного промежуточного продукта Ti/NH2/Cl2 • 2NH4CI до амино-
хлорида TiCU - nNH3 (где п=4, 6, 8) при высокой температуре с разло-
жением по реакции типа
TiCU -4 NH3 -> TiN 4- НС1 % NH3.
152
Разложение TiCl4 • 4NH3 при низких температурах порядка 400—
800° С приводит к образованию соединений титана с азотом, содержа-
щих недостаток титана (и соответственный избыток азота против фор-
мулы TiN—TixN, где х=0,85—0,95). При повышении температуры про-
каливания аминохлоридов состав все более приближается к формуле
TiN, достигая ее при 1400—1600° С.
При использовании для получения нитрида титана аминохлорида
TiCU • 6NH3 значительная часть последнего испаряется, и выход ни-
трида титана составляет не более 8—10% после 6 ч прокаливания при
1500° С [187].
Предполагается, что аминохлориды разлагаются не сразу до нит-
рида титана, а с образованием промежуточного соединения TiNCl (со-
единение черного цвета с тетрагональной структурой, образующиеся
при 350—400° С. При нагревании этого промежуточного продукта в
течение 6—8 ч при 400° С получается кубический продукт с решеткой
TiN, но еще содержащий атомы хлора, для полного удаления которых
из решетки необходим нагрев до 1000° С [188]. Таким образом, получе-
ние нитрида титана через аминохлориды может быть в целом представ-
лено следующей схемой:
300 — 350°С
TiCl4 + NH3 - Ti (NH2)C12'2 NH4CI--------->-
C < 1000°
TiCU • n NH3----> TiCIN-------> TiN
Восстановление ТЮ2 углем или металлами в сре-
де азота. Получение нитрида титана восстановлением TiO2 углем в
среде азота основано на реакции
TiO2 + 2C + 1/2N2 = TiN + 2CO
и заключается в восстановлении TiO2 углем до титана или его низших
окислов с одновременным их азотированием до нитрида или оксинитри-
дов. В работе Фридериха и Зиттиг [НО] TiO2 или рутил нагрева-
ли в смеси с сосновой сажей (1% золы) в вольфрамовой, молиб-
деновой или угольной лодочке в токе азота в продолжение нескольких
часов при 1250°С. Образования оксинитридов в этом исследовании не
наблюдалось и продукт реакции содержал 98—99,5% нитрида титана
(содержание азота порядка 21,9%) [ПО, 187].
Руфф и Айснер [187] также приготовляли нитрид титана прокали-
ванием смеси двуокиси титана с сажей в токе азота при 1100—1200° С
и указали, что продукт отвечает по составу формуле TiN.
В дальнейшем Ремин [181] подвергал азотированию смесь дву-
окиси титана с активированным углем, составленную в весовом отно-
153-
I
шении 2:1 (т. e. с количеством углерода меньшим, чем полагается по
вышеприведенной реакции). Азотирование осуществлялось тщательно
очищенным азотом, аммиаком и смесью азота с водородом, причем
шихту помещали в железные, никелевые или фарфоровые лодочки.
В результате было установлено, что при трехкратном азотировании
аммиаком (каждый раз в течение 3 ч при 1250° С) содержание азота
в полученном продукте составляет 17,61%, при такой же обработке
азотом 17,47%, а при азотировании смесью N2 + H2 в результате 6-ча-
совой операции (подъем температуры до 1250° С около 3 ч и выдержка
на этой температуре также 3 ч) содержание азота достигает всего
16,4%. В этой работе дополнительным азотированием не удалось уве-
личить содержание азота в нитриде.
При указанном методе образование нитрида титана, очевидно,
возможно только через стадию образования низших окислов титана,
в первую очередь закиси титана TiO, которая образует непрерывный
ряд твердых растворов с образующимся побочно карбидом титана по
схеме:
ПО2 + С = TiO 4- СО,
TiO2 -+- 3 С = TiC Т 2 СО,
TiO + TiC = TiO — TiC.
При азотировании твердого раствора образуется сначала тройной
твердый раствор TiO—TiC—TiN, из которого далее получается нитрид
титана
TiO — TiC — TiN 4- N2 = 3 TiN + CO.
Эта схема реакции подтверждается данными работы Беляковой,
Комара и Михайлова [191], согласно которой получающийся при азо-
тировании смеси TiO2 с углем при температуре 1100—1500° С нитрид,
а также карбид титана растворяются в TiO, так что получаются твер-
дые растворы TIN—TiC—TiO; при проведении реакции при 1900° С в те-
чение 3 ч закись титана полностью восстанавливается и образуется
твердый раствор TiN—TiC, концентрация которого определяется соот-
ношениями, установленными в работе Зеликмана и Горовиц (см. ниже).
В той же работе [191] при попытке непосредственного азотирования
TiO2 смесью N2+H2 установлено, что при температурах 1000—1500° С
происходит образование промежуточных окислов титана, а при 1700° С
образуется твердый раствор TiN—TiO.
Возможны и другие схемы реакций, приведенные, например, в мо-
нографии [146], однако важнейшей из побочных реакций при всех об-
стоятельствах является образование карбида титана, дающего устой-
154
чивые твердые растворы с нитридом титана. При спекании металло-
керамических твердых сплавов азот, находящийся в их составе в форме
твердого раствора TiN—TiC, практически не выделяется из сплава, ос-
таваясь в карбидном твердом растворе [189]. При этом до температуры
1200° С проявляется преимущественно окисляющее действие подса-
сывающего в печь воздуха, т. е. имеет место образование оксинитридов,
а в диапазоне температур 1200—1800° С преобладает азотирующее
действие.
Поэтому важное значение имеет для получения нитрида титана
восстановлением двуокиси титана углем в среде азота работа Зелик-
мана и Горовиц [190] по изучению равновесия TiC—TiN при действии
•азота на карбид титана
хПС 4- z//2N2 £ TiNx[(%- i/)C, z/N] ±yC.
Константу этой реакции [190] определяли как: Лр=р№-
(TiC)
(TiN)
и
нашли, что при давлении азота 0,12 ат (90 мм рт. ст.) в зависимости от
температуры составляет: при 1300° С — 0,14, при 1500° С — 0,22 и при
1800° С—0,36. На основании этих данных для pN2 = 0,12 am выведено
уравнение изохоры реакции
IgKP = -
2700
Т
-4- 0,85.
В работе [231], однако, показано, что в написании уравнения кон-
станты реакции авторами [190] была допущена ошибка и его следует
записывать в виде
ir 1- (TiC)
Ар - И (TiN) .
что приводит к хорошему согласованию с экспериментальными данны-
ми работы [190].
Отсюда следует, что насыщение азотом при данном давлении по-
нижается с повышением температуры. Таким образом, для получения
безуглеродистого нитрида титана при обычном давлении азота в печи,
равном 1 ат, необходимо проводить азотирование при температурах
не выше 1000—1300° С, а при более высоких температурах для полного
азотирования TiC давление азота должно превышать 1 ат.
Эти данные подтверждены экспериментальной работой, проведен-
ной нами совместно с В. С. Нешпором по азотированию смеси TiO2 с са-
жей в токе азота в графитотрубчатой печи [146]. Найдено, что азоти-
рование начинается при 1000° С, при одночасовой выдержке выход
нитрида всего 6—7%, при 1300° С выход повышается до 15—20%,
при 1400° С выход резко возрастает до 65—70% и при 1600° С дости-
гает 93—94%. При дальнейшем увеличении температуры до 1700° С
155
выход падает до 77—78% с одновременным увеличением в продукте
азотирования карбида титана в связи со сдвигом равновесия в сторону
образования TiC, что хорошо согласуется с данными [190]. Наибольшее
содержание азота в продукте азотирования (при 1600° С) составляет
21—22%. Увеличение времени выдержки при этой температуре не при-
водит к сколько-нибудь заметному изменению содержания азота.
Положительные результаты при проведении процесса азотирова-
ния при относительно низких температурах (порядка 1200° С) дает
многократное азотирование с протирками промежуточных продуктов.
Так, в нашей работе с В. С. Нешпором содержание азота после первого
азотирования составляло 12,5% при 1200° С после протирки (для раз-
рушения пленок нитрида на частицах окислов) и повторного азотирова-
ния— 18,5% и после очередной протирки и третьего азотирования —
20,06%. Крупность зерен нитрида при этом остается практически неиз-
менной и равной 2—3 мкм.
Одним из вариантов технологии получения нитрида титана восста-
новлением его кислородных соединений углем с одновременным азоти-
рованием является предложенный в работе [194] метод образования
порошка нитрида, заключающийся в азотировании смеси TiP2O7 (по-
лучаемого действием Н4Р2О7 на Ti(OH)4, взятую в стадии гидролиза
в процессе производства TiO2 и имеющую размер частиц 0,05—0,2 мкм)
с углем в стехиометрическом соотношении, которое производится при
1500—1600°С в среде азота. В результате получается нитрид титана в
виде порошка с размером частиц 1—50 мкм.
Резюмируя изложенное, следует отметить, что метод азотирования
смесей TiO2 с углем непригоден для получения чистого нитрида, но
является весьма удобным и технологичным для приготовления крупных
количеств нитрида титана технического качества для использования
его, например, в составе огнеупоров и других изделий, не требующих
высокой чистоты нитрида.
Александер [192] предложил аналогичный способ получения нитри-
да титана с той лишь разницей, что восстановителем двуокиси является
не углерод, а гидрид кальция СаН2. Смесь тонкого порошка TiO2 с не-
большим избытком гидрида против расчетного количества по уравне-
нию реакции
TiOa +- СаН2 = Ti + Н2О + СаО
помещали в печи и нагревали для образования титана в атмосфере
инертного газа, после чего в печь вводили азот или аммиак для обра-
зования нитрида титана
Ti ]-N2(NH3)^TiN + (H2).
После отмывки продукта азотирования от окиси кальция этим ме-
тодом получается весьма чистый нитрид титана.
156
По мнению Александера, использование гидрида кальция для вос-
становления окиси титана обеспечивает весьма активное восстановление
в результате диссоциации гидрида с образованием атомарного водо-
рода. Однако это мнение было опровергнуто опытами Меерсона и Кол-
чина [193], показавшими, что время рекомбинации атомарного водорода
в На настолько мало, что он не успевает участвовать в атомарном со-
стоянии в процессе восстановления, которое осуществляется кальцием.
Выделяющийся при диссоциации гидрида водород служит в основном
защитной средой, предотвращающей окисление титана.
Аналогичные опыты проведены нами совместно с Г. 3. Ланкиной
с использованием магния в качестве восстановителя TiO2
2 TiO2 + 4 Mg + N2 = 2 TiN + 4 MgO,
однако даже при температурах восстановления 1400—1600° С не дали
содержания азота в продуктах азотирования выше 7—8% при содер-
жании титана 72—73%, т. е. продукты азотирования представляют
оксинитриды титана.
При действии аммиака на TiO2 при температуре 900° С образуются
смеси нитрида титана (я = 4,242 А) с низшими окислами и оксинитри-
дами [252].
Спекание нитрида титана. Для получения плотных изделий из нит-
рида титана наиболее эффективно использование метода горячего прес-
сования его порошка, которое проводится при 1900—2100° С под
давлением 200—300 кг/смг [232]. Минимальная остаточная пористость
спеченных горячим прессованием образцов составляет 2—4%. При спе-
кании порошка нитрида титана происходит резкое укрупнение частиц
(от 2—3 до 16—18 мкм) и сильное охрупчивание, не свойственное дру-
гим нитридам (например, при спекании нитрида тантала крупность
частиц возрастает от 4 до 6—7 мкм). На рис. 63 показана микрострук-
тура спеченного нитрида титана.
При спекании брикетов, спрессованных из порошка нитрида ти-
тана в вакууме, пористость изделий, спеченных при температуре 2200° С
в течение 4 ч, составляет 22% [232].
Оригинальным методом приготовления изделий из нитрида титана
является реакционное спекание, заключающееся в одновременном об-
разовании нитрида титана из металла и спекании, и осуществляющееся
спеканием заготовок, спрессованных из порошка титана в среде азота.
Этот способ был предложен одновременно в 1963 г. Самсоновым и
Верхоглядовой [233, 225], Гудингом и Пэрратом [234]. Так как при
образовании нитрида титана из металла происходит увеличение удель-
ного объема (на 7,5%), то при реакционном спекании это увеличение
реализуется в снижении общей пористости заготовки из титана,
что позволяет, по данным работы [235], получать изделия из нитрида
титана пористостью до 13% (при этом использовались образцы из гид-
157
рида титана, спрессованные под давлением 1,1—1,3 т!см2, обработан-
ные для удаления водорода в вакууме при 400—750° С и спеченные
затем при 950—1000° С в течение 1—2 ч. Азотирование проводили на-
чиная с 1100° С с последующим подъемом до 1400° С и спеканием при
этой температуре 10—20 ч.
С уменьшением содержания азота в нитриде титана в области его
гомогенности пористость спеченных изделий уменьшается и, например,
Рис. 63. Микроструктура спеченного нитрида титана.
для нитрида с 12% N после 4 ч спекания в вакууме при 2200° С состав-
ляет всего 4%, после горячего прессования в аргоне при 2100°С—
5% [232].
Нитриды циркония. Диаграмма состояния системы цирконий —
азот в исследованной области составов, построенная Домагалой, Мак-
Ферсоном и Хансеном [198], показана на рис. 64. В качестве исходного
материала использовали иодидный цирконий. Для получения сплавов
цирконий азотировали при температуре 800—1200° С в течение 4—12 ч.
Полученные образцы переплавляли в дуговой печи с нерасходуемым
электродом. Гомогенизацию проводили при 1200° С в течение 48 ч с
последующим отжигом под закалку.
^-Твердый раствор образуется при охлаждении по перитектической
158
Рис. 64, Диаграмма состояния цир*
конин—азот.
реакции: жидкая фаза — 0,7 вес.% (4,4 ат.%) N+a-фаза — 3,0 вес.%,
(16,8 ат.%) N-^p-фаза — 0,8 вес.% (5,0 ат.%) N при 1800+10° С.
'Добавление азота стабилизирует a-модификацию циркония, по-
вышая температуру а-^р-превращения. а-Твердый раствор образуется
при 1985+15° С по перитектической реакции: жидкая фаза — 2,4 вес. %
(13,5 ат. %) N-J-ZrN-xx-фаза — 4,8 вес. % (25 ат. %) N.
Максимальная растворимость азота в 0-Zr составляет 0,8 вес. %
(5,0 ат.%) при температуре перитектической реакции и снижается до
нуля при температуре а->[3-превращения,
равной 862° С.
Растворимость азота в a-Zr состав-
ляет при температуре образования
a-твердого раствора 4,8 вес. % и понижа-
ется с уменьшением температуры, дости-
гая 4 вес. % при 600° С.
Нижняя граница области гомогенности
ZrN лежит при 11,5 вес.% (~ 46 ат.%) N
при температуре 1985° С. Следует отме-
тить, что в работе [198] не удалось полу-
чить нитрида циркония стехиометрическо-
го состава (13,3 вес.% N) и исследовать
область гомогенности этого соединения.
Температура плавления ZrN бвгла приня-
та равной 2980±50°С.
Данные по изучению системы Zr—N
[198] хорошо согласуются с более ранними
результатами других работ; так, повыше-
ние температуры а~-*-|3-превращения при
добавлении азота отмечалось в работах
, 200], растворимость азота в a-Zr,
равная около 20 ат. %, также хорошо
совпадает с данными работы [198].
Таким образом, в системе обнаружено только одно стабильное
соединение — нитрид ZrN, хотя ранее были сообщения о существова-
нии еще одного нитрида с 13 ат. % N [201].
Однако, как отмечалось в [248, 249], действием аммиака на тетра-
галогениды циркония получены нитридные фазы, содержащие азота
больше, чем в нитриде ZrN. Согласно [248], действием аммиака
на Zr при ~750° С, а также на ZrCl4 и ZrBr4 при более высоких
температурах был получен нитрид ZrxN темно-голубого цвета с
0,940>х>0,812. Определение плотности (6,1—7,4 г/см3), изменения
параметра кристаллической решетки, электропроводности (около
3-10~2 ом~1 • см-1) и магнитных свойств (препараты диамагнитны)
дает возможность полагать, что нитрид содержит четырехвалентные
159
ионы циркония, а также трехвалентный азот (Zr3N4). Этот нитрид
кристаллизуется в дефектной решетке типа NaCl с вакантными мес-
тами в подрешетке циркония и параметром решетки, изменяющимся в
указанных пределах составов от 4,566 до 4,440 А. В работе [249] анало-
гичным путем по схеме
750°C ZrxN (голубой) I000°C
+ ->ZrN
250°С 500°С
ZrJ4 (Zrl4 • nN3) ZrNI
700°С
1000°C
Zr3N4 (коричневый) -> ZrN
был получен нитрид циркония коричневого цвета, состава, соответст-
вующего формуле Zr3N4 (плотность 5,9 г/см3, магнитная восприимчи-
вость при комнатной температуре yg = 0,03 • 10 ~6). Оба нитрида при
температуре 1000° С теряют избыток азота и переходят в стабильный
мононитрид ZrN.
Причины образования нитридов такого состава могут быть следую-
щими. Образование нитрида Zr3N4 отвечает ионной связи между ато-
мами циркония и азота, возникающей при передаче атомом циркония
всех валентных электронов (4d25s2) атомам азота, которые в этом слу-
чае должны приобретать £2р6-конфигурацйю Однако для азота (элект-
ронная конфигурация изолированного атома s2p3) возможно как обра-
зование £2р6-конфигурации за счет приобретения электронов партнера по
соединению, так и передача одного электрона партнеру с образованием
устойчивой s/Аконфигурации. Как правило, у атомов азота реализу-
ется вторая возможность (например, в нитридах BN, TiN, ZrN), а реа-
лизация первой возможности, т. е. полного привлечения валентных
электронов партнера, становится вероятной по наследственным причи-
нам (в данном случае — четырехвалентность циркония в галогениде,
образование азотом 52р6-конфигурации в аммиаке). Таким образом, в
ряду соединений ZrN ... Zr3N4 повышается статистический вес $2р6-кон-
фигураций атомов циркония. Этому переходу от набора d°- и ^-кон-
фигураций для атомов циркония и s/Аконфигураций атомов азота со
значительной долей нелокализованных электронов к высокому стати-
стическому весу d°- и 52р6-конфигураций с сильной локализацией элек-
тронов в этих конфигурациях отвечает изменение характера проводи-
мости от металлического для ZrN до полупроводникового для Zr3N4, а
также снижение парамагнетизма до полного его исчезновения у Zr3N4.
В работе [998] исследована структура одного из препаратов с из-
бытком азота (недостатком циркония). Состав препарата был
(Zr, 2% Hf) о,95±о,о3 Nlro, плотность его 6,884+0,003 а/щи3 (плотность
стехиометрического ZrN по этим данным 7,284 г/см?). Ячейка содержит
160
3,913 молей с 3,5% пустых мест (8 на ячейку) катионов и 1,1% анио-
нов. Избыток азота составляет 0,19 атомов на ячейку.
Ширина области гомогенности фазы ZrN, по данным [202], состав-
ляет от 40 до 50 ат. % (9,5—13,3 вес. % N). С увеличением содержания
азота в области гомогенности фазы ZrN, по данным [202], повышает-
ся абсолютное значение термо-э. д. с., уменьшаются коэффициенты
Холла и удельное электросопротивление и возрастает твердость
(табл. 59).
Таблица 59
Изменение физических свойств нитрида циркония в области
гомогенности
Содержание N Термо-э. Д. с.. мкв/град Удельное электро- Коэффици- ент Микротвер-
формула вес. % ат. % (по отно- шению к сопротив- ление, Холла Я-104, см3/к дость, кг/мм*
меди) мком-см
ZrN0.675 9,41 40,3 —0,98 190 4-5,5 -—
Zr^0,76 10,5 43,3 —4,0 75 + 1,2 ——
Zr^0,818 И.1 45,0 —5,4 58 —1,4 850 ±42
ZrN0,87 11,93 46,8 —7,7 37 —1,45 1060 ±53
ZrN n Zr№Q93 ZrN0’56 0,99 12,52 48,2 —7,5 30 —1,4 1260±76
12,83 48,9 —7,4 28,5 —1,3 1390±110
13,2 49,8 —7,3 28 —1,3 1520 ±85
Электросопротивление при этом изменяется нелинейно, что харак-
терно для нитридов с большей долей ионной связи (см. рис. 5), и зна-
чительно круче, чем для нитрида титана, что указывает на боль-
шую скорость нарастания в ZrN доли ионной связи с уменьшением
содержания в нем азота. Эффект Холла вблизи нижней границы
области гомогенности нитрида циркония положителен, что указывает
на преимущественно дырочную проводимость сплавов этого со-
става.
Следует отметить, что, по данным [978], электросопротивление нит-
рида циркония в области от 45 до 50 ат. % азота изменяется линейно
и достигает при 45 ат. % азота больших значений, чем указано в
табл. 59 (~200 мком-см по сравнению с 58 мком-см). Име-
ется также некоторое различие в ходе изменения термо-э. д. с. в
области гомогенности. По [978], термо-э. д. с. очень мала и практически
постоянна в пределах 35—40 ат. % азота, а затем строго линейно воз-
растает до 50 ат. % азота, достигая для этого предельного состава по-
рядка 8 мкв/град, что близко к данным табл. 59.
По данным [978], в пределах области гомогенности магнитная вос-
11—272
161
1
приимчивость нитрида циркония строго линейно изменяется от ~40
(при 35 ат. % N) до 22- 10~6 (при 50 ат. % N).
На рис. 65 показана температурно-концентрационная зависимость
электросопротивления нитрида циркония от содержания азота в преде-
лах области его гомогенности. Видно, что для сплавов с содержанием
азота, близким к 50 ат.%, зависимость электросопротивления от тем-
пературы практически линейна и типична для металлической проводи-
мости. С понижением содержания азота изменение электросопротивле-
ния проходит затем через максимум при температурах, уменьшающихся
при понижении содержания азота. Затем сопротивление уменьшается,
как для полупроводников, что указывает на расширение энергетической
щели между sd-состояниями циркония и sp-состояниями азота, отвеча-
ющее росту доли ионной связи. Более быстрый рост доли ионной связи
с уменьшением содержания азота в нитриде циркония в сравнении с
нитридом титана хорошо виден на рис. 4, на котором пунктирными
линиями показан характер изменения микротвердости составов, типич-
ный для металлического состояния.
Результаты изучения влияния хемосорбции азота на эффект Холла
и электросопротивление тонких пленок металлического циркония [250]
показали, что электросопротивление при этом возрастает, что связы-
вается с уменьшением эффективной толщины металлического слоя. На
самом деле эффект обусловлен передачей атомами азота части электро-
нов (при перестройке конфигурации s2p3 в sp3) в нелокализованное со-
стояние— в общий коллектив с нелокализованными электронами цир-
кония и возрастанием при этом доли электросопротивления, обуслов-
ленной взаимодействием между нелокализованными электронами.
В работе [252] исследованы термоэмиссионные свойства нитрида
циркония на образцах, приготовленных реакционным спеканием брике-
тов из порошка циркония в среде азота при температуре 1300° С в те-
чение 4 ч. Работа выхода ф=3,78 эв.
Коэффициент излучения нитрида по этим данным возрастает в
пределах температуры 800—-2000° С от 0,73 до 0,76 (при А,=655 ммк).
Коэффициент катодного распыления нитрида циркония при бом-
бардировке ионами аргона с энергией 50—200 эв равен 0,026 мол/ион,
что существенно меньше катодного распыления металлов (например,
для вольфрама коэффициент катодного распыления равен 0,23 ат]ион).
Нитрид циркония ZrN предельного состава — порошок желто-ко-
ричневого цвета с золотистым оттенком; в компактном состоянии имеет
лимонно-желтый цвет с металлическим блеском.
В отличие от нитрида титана, разлагающегося при нагревании в
вакууме на пары титана и азот, нитрид циркония испаряется с разло-
жением на твердый цирконий и азот [203]
ZrN Тв ZrTB ф-
162
Константа равновесия К-р=р'!/, равна, по [203]:
^2
1g кр = — + 1,48 • 10~4Т + 4,467,
давление азота в зависимости от температуры определяется выраже-
нием
1g + 2,96 • 10“4Т + 8,934.
Таблица 60
Скорость испарения нитрида
циркония и давление азота
над нитридом
Температу- ра, °C Скорость испарения G-10\ г/см2- сек Давление азота PN/10’, ат
2236 0,534 0,915
2259 1,299 2,397
2318 1,979 5,583
2333 2,714 4,477
2344 3,020 5,536
2451 14,981 28,96
2466 16,127 31,04
В табл. 60 приведены данные
по скорости испарения и дав-
лению паров циркония и азота в
зависимости от температуры.
Определенная по этим дан-
ным теплота образования ZrN
—ДЯ02Э8=72,53+0,43 ккал/моль,
что близко к данным, получен-
ным ранее в работе [117], со-
гласно которой:
Рис. 65. Температурно-концентраци-
онная зависимость электросопротив-
ления ZrN в области гомогенности:
/ — 40,3 ат.% N; 2 — 34,3 ат.% N; 3 — 48,8
ат.% N; 4 — 49,4 ат.% N.
/, °к
298,16 500 1000 1500 2000
—AZZ, ккал!моль 82,2 82,1 81,7 81,6 80,9
А/7°298 is для ZrN равно — 87,3±0,4 и A770298 i5 = —80,5 ккал!моль
[213].
В противоположность данным [203], в работе [210], где исследова-
лось равновесие в системе цирконий — азот при высоких температу-
рах, установлено, что при температурах от 2200 до 2800° К и давлениях
от 0,1 до 300 мм рт. ст. нитрид циркония (в области его гомогенности)
11*
16»
диссоциирует с выделением азота и образованием не металлического
циркония, а более бедных азотом по сравнению с исходными фаз
ZrN, = ZrNx-д + 4 Na‘
На рис. 66 показано изменение х в формуле ZrNx в зависимости от
температуры при различных давлениях. Авторы работы [210] допуска-
паров циркония над
ют возможность наличия заметной упругости
Рис. 66. Зависимость состава нит-
рида циркония от температуры
азотирования:
2— 10—100; 2— /4^0,1 мм рт. ст.
2600—2800° К.
Зависимость теплоемко-
сти ZrN от температуры
представляется выражени-
ем [206]
Таблица 61
Изменение теплот образования и свободной
энергии в области гомогенности нитрида цир-
кония [207]
Формула нит- рида Теплота образо- вания, ккал/моль Изменение энтропии д5°298 Свободная энергия об- разования Д-^298 Для бескисло- родных нит- ридов
ZrI%,56 О0,02 57,5±0,7 —12,7 —52,3
Zr^0,69 О0,00 68,7±1,8 —15,4 —64,1
Zr^O,74 *“*0,00 72,2±0,9 —16,5 -67,3
ZrN08g О0 03 84,5±0,5 —20,4 —76,3
ZrNi Oq,04 90,7±0,2 —22,9 —81,1
Ср = 11,10 + 1,68- 103Т— 1,72-10 5Т 2 [кал/град-моль].
В работе [207] определены теплота и свободная энергия образова-
ния нитрида циркония в области гомогенности. Показано, что с ростом
содержания азота в нитриде циркония обе эти величины возрастают
(табл. 61). Отмечается, что эффект изменения теплот образования и
убыли свободной энергии существенно больше (в ~2 раза), чем для
карбидных систем. Это объясняется резким изменением характера хи-
мической связи при изменении содержания азота в пределах области
гомогенности.
Диффузия азота в компактный и порошкообразный цирконий изу-
чалась во многих работах. В работе [208] исследована кинетика азоти-
рования циркония в интервале температур 862—1043°С при давлениях
азота от 10 до 300 мм рт. ст., по полученным данным рассчитана энер-
гия активации, оказавшаяся равной 52 ккал/моль. В работе [209] изу-
чена диффузия азота в p-Zr при температурах 920—1640° С и давлении
1 ат. Азотируемый цирконий содержит 97,8—98,2% Zr, 1,8—2,2% Hf и
164
следы О, N, Н, Fe, Si. Константа азотирования описывается выраже-
нием
К = 5,0-103-ехр(48000//?Т) [(мл/см2)2сек],
температурная зависимость коэффициента диффузии
D = 1,5 • 10~2ехр(- 30700/КТ)[см?/сек\.
Рис. 67. Кинетические кривые азотирования циркония.
Рис. 68. Температурная зависимость константы скорости реакции Zr-t-’/aNa
(£--39,2 ккал/моль).
Растворимость азота в p-Zr линейно возрастает с температурой и
при 1600° С достигает 0,6 вес.%. В интервале 920—1640° С раствори-
мость может быть определена из уравнения [199, 204]
С = — 2810 Т-1 + 1,42 [вес.%].
По данным Гулбранзена и Эндрю [179], энергия активации азоти-
рования a-Zr составляет 39,2 ккал/моль (рис. 67, 68).
Результаты исследования кинетики азотирования порошка цирко-
ния [178] показали, что диффузия азота в a-Zr с образованием твердого
раствора проходит с энергией активации 20,0 ккал/моль (при 500—
600°С), в a-Zr с образованием нитрида циркония — с энергией актива-
ции 5 ккал/моль (при 600—1000° С) и в p-Zr— с энергией активации
2,8 ккал/моль. Малые значения энергии активации связаны с использо-
ванием очень тонкого порошка циркония, в то время как в указанных
выше работах использовали компактный цирконий либо стружку или
грубые порошки.
165
Авторы [222] исследовали азотирование при 700—1200° С компакт-
ного магниетермического циркония, выплавленного в тигле. Показано,
что нитрид циркония, образующийся на поверхности металла при тем-
пературе до 1000° С, не проникает вглубь металла, а дает тонкую
пленку на его поверхности. При длительном нагревании (до 24 ч) при
более высоких температурах (1100—1200° С) глубина азотирования
достигает около 1,5 мм. Рентгенографически показано, что нитрид с
кубической решеткой образуется только в
Рис. 69. Температурная зависи-
мость теплопроводности TiN (I) и
ZnN (Щ.
тонком поверхностном слое, под ним рас-
положен слой твердого раствора в a-Zr и
уже под ним основа из ^-твердого раст-
вора. 1
При увеличении дисперсности порош-
ка азотируемого циркония резко сокра-
щаются время азотирования и кажущие-
ся энергии активации, входящие в урав-
нение константы реакции азотирования.
Это сокращение при увеличении степени
дисперсности усиливается вследствие вы-
сокой теплоты образования нитрида цир-
кония, что также приводит к уменьшению
кажущейся энергии активации. Послед-
нее влияние сказывается в тем большей
степени, чем выше отношение веса обра-
зовавшегося нитрида к весу азотируемых
частиц, возрастающее с ростом дисперс-
ности. Это необходимо иметь в виду и в
случаях образования нитридов других ме-
таллов, имеющих высокие значения те-
плосодержания.
Добавка 5% KHF2 к цирконию каталитически ускоряет процесс
азотирования [246].
В работе [253] проведено исследование диффузии азота в цирконии
методом внутреннего трения, однако определенных выводов о механиз-
ме элементарного акта диффузии не получено.
Теплопроводность нитрида циркония исследовали при температу-
рах до 1100° С [211]. Теплопроводность нитрида циркония несколько
меньше, чем нитрида титана, и изменяется умереннее с температурой,
чем TiN (рис. 69).
Термическое расширение нитрида циркония довольно подробно
изучал Бейдлер [212] на нитриде, полученном нагреванием циркония в
течение 4 ч при температуре 1250° С в азоте и содержавшем 52,7% Zr,
47,2% N и 0,05% Mg. Параметр решетки а при 17, 445 и 680° С оказал-
ся равным соответственно 4,5745, 4,5861 и 4,5965 А. Коэффициент тер-
166
мического расширения а составляет в интервале 17—445° С
(6,0+0,5) • 10 6, а в интервале 17—680° С сс= (7,0+0,5) • 10~6.
ZrN — сверхпроводник [140, 219] с критической температурой, воз-
растающей от 3° К для ZrNo,932 до 9,5° К для ZrN0,984 [1069]. Термоэмис-
сионные характеристики ZrN не представляют технического интереса
[170, 221], ток насыщения при 2000° К составляет всего 0,05 а (см. так-
же стр. 162).
По данным [1031], модуль упругости беспористого нитрида цирко-
ния равен 51000+1500 кг/мм1- (характеристическая температура
580+10° К). Согласно [1077], £=40 000 кг/лш2, 0 = 645° К.
Химические свойства. Нитрид циркония в порошкообразном состоя-
нии на холоду довольно стоек против действия многих минеральных
кислот, особенно соляной, азотной, хлорной, а также смесей кислот с
окислителями [214]. При нагревании растворимость в кислотах и их
смесях резко увеличивается, за 2—3 ч нитрид циркония полностью раст-
воряется в концентрированной H2SO$, смеси HNO3 + HF, H2SO4+K2SO4.
В растворах щелочей (с концентрацией до 10%) нитрид циркония не
растворяется даже при нагревании, в более концентрированных ще-
лочах и смесях щелочей с окислителями типа пергидроля раство-
рение проходит незначительно. Кислородом воздуха нитрид циркония
не окисляется до 900—1000° С. ZrN — один из наиболее устой-
чивых в этом отношении нитридов (несколько уступает нитриду ти-
тана) [9].
Огнеупорные свойства. В работе [215] проведено исследование ха-
рактера взаимодействия нитрида циркония с тугоплавкими металлами
при нагревании до высоких температур в вакууме. Нитрид циркония
устойчив в этих условиях, намного устойчивее нитрида титана. До
2100° С ZrN не взаимодействует в течение 5 ч с танталом, вольфрамом;
с молибденом слабое взаимодействие начинается при нагревании при
2100° С в течение 5 ч, с ниобием значительно реагирует уже при 2000° С,
ниже этой температуры в контакте с ниобием цирконий совершенно
устойчив.
Методы получения нитрида циркония. Основным методом приготов-
ления нитрида циркония является непосредственное азотирование по-
рошка циркония [178]. Оптимальной температурой азотирования по-
рошка циркония с азотом является 1200° С. Медленнее происходит про-
цесс насыщения циркония азотом в диссоциированном аммиаке: необ-
ходима температура не ниже 1400° С. Гидрированный цирконий обра-
зует нитрид при взаимодействии с азотом при высоких температурах
порядка 1300—1400° С. Метод непосредственного азотирования цирко-
ния с образованием нитрида использовался в работе [213], где доволь-
но грубая циркониевая стружка (0,002—0,003 дюйма) азотировалась
в течение 5 ч при 1300—1400° С. В работе [203] при азотировании гид-
рида циркония ZrH2 в токе азота при 1050° С в течение 21 ч был полу-
чен продукт, состав которого описывается формулой ZrN0,9Z3-
Оригинальный метод получения технического циркония азотирова-
нием металлического циркония предложен в работе [216]. Он заклю-
чается в азотировании влажного порошка циркония с использованием
для разогрева порошка до температур, необходимых для активного азо-
тирования, теплот окисления циркония и образования нитрида цирко-
ния. По этим данным, при нагревании влажного порошка циркония в
токе азота при 450—600° С в течение 15 мин получаются продукты, со-
держащие 9,5—10,2% азота, т. е. оксинитриды циркония или техниче-
ский нитрид, который в ряде случаев может быть использован вместо
чистого ZrN.
Другой метод получения нитрида циркония, впервые применен-
ный в работе Фридериха и Зиттиг [ПО], заключался в восстановлении
углем двуокиси циркония в среде азота
ZrO24-2C -p/2N2 - ZrN + 2CO
при температурах до 1300° С. Полученный продукт содержит, кро-
ме нитрида циркония (которому они впервые приписали правильную
формулу ZrN), 7—10% примеси, нерастворимой в серной кислоте и
состоящей из ZrO2 и примесей. Воспроизведение этого метода пока-
зало невозможность получения чистого нитрида циркония. Очевидно,
это связано с тем, что нитрид циркония, согласно термодинамическим
расчетам, устойчив в смеси с углем до 1000° С, а с повышением темпе-
ратуры реакция
ZrN + С £ ZrC 4- V2 N2
должна сдвигаться вправо с образованием более обогащенного угле-
родом твердого раствора ZrC—ZrN. Температурная зависимость кон-
станты равновесия реакции взаимодействия нитрида циркония с угле-
родом выражается уравнением
р = +2,76.
которое количественно выражает низкую устойчивость нитрида в кон-
такте с углеродом при повышенных температурах [232, 1079].
Нитрид циркония получают методом наращивания из газовой фазы
на вольфрамовой или циркониевой проволоке из смеси ZrCl4 и NH3
или N2+H2 при температуре нити 2000—2400° С, а из смеси ZrCl4+
+ N2 —при 3000° С [45, 159, 184, 217, 218].
Авторы [251] разработали метод получения нитрида циркония вос-
становлением двуокиси циркония магнием в присутствии азота
ZrO2 + 2 Mg + % N2 = ZrN + 2 MgO.
Приведенная реакция имеет формальный смысл, так как восстанов-
ление ZrO2 магнием по термодинамическим условиям должно прохо-
168
дить не до металла, а до твердого раствора кислорода в цирконии
(9,3 ат.% кислорода при 1000° С), который далее восстанавливается
азотом за счет дополнительного высвобождения свободной энергии в
результате образования нитрида циркония.
Технологически при восстановлении ZrCb магнием в среде азота
при температуре 1100° С образуется нитрид циркония, содержащий
13,3% азота, обладающий кубической гранецентрированной решеткой
с параметром а = 4,575 А, т. е. полученный нитрид близок
по составу к ZrN. Недостатком этого метода является
необходимость отмывки продукта восстановления от оки-
си магния, что удлиняет и усложняет технологический
процесс.
В последние годы разработаны и довольно широко
применяются непрерывные методы получения ZrN
[1047]. Схема установки для непрерывного получения
нитрида циркония показана на рис. 70. Порошок цирко-
ния загружается в бункер 1 под действием вибрации,
сообщаемой бункеру вибратором, 2, просыпается через
сетку 3 и в распыленном состоянии под действием соб-
ственного веса опускается вниз, входит в зону нагрева
реактора, где вступает во взаимодействие с азотом. По-
лученный нитрид циркония собирается в приемнике 5, по-
Рис. 70. Схема
установки для непрерывного получения нитрида цир-
кония.
мещенном в камере 6. Обогрев реактора осуществляется с помощью
электропечи 7. Температура в реакторе 1200° С, расход порошка цирко-
ния 0,5 кг/ч, выход нитрида 0,55 кг/ч, расход электроэнергии 16 квч/кг
нитрида.
Нитриды гафния. Диаграмма состояния системы гафний — азот
не построена, известно лишь, что гафний образует с азотом один нит-
рид HfN, впервые полученный и идентифицированный Ван-Аркелем
[217] и Моерсом [184]. Это соединение металлического характера с
весьма высокой точкой плавления и кубической структурой.
Азотирование металлического гафния при температурах 876—
1034° С и давлениях 38—402 мм рт. ст. исследовано в работе [223]. Азо-
тированию подвергался листовой гафний (толщиной 0,2 мм), тщатель-
но очищенный после прокатки и отожженный в вакууме 10—4 мм рт. ст.
при 970° С в течение 1 ч. Показано, что кинетика азотирования отвечает
простому параболическому закону скорости насыщения
V2 = Kpt + С,
169
я
где W — вес азота на единицу площади поверхности листового гаф-
ния, t — время азотирования, — константа скорости реакции, С —
постоянная, представляющая ошибку в первоначальный момент наблю-
дения. Рассчитанная из полученных данных энергия активации реак-
ции азотирования гафния оказалась равной 57±3 ккал!моль. Рентге-
новский анализ прочных золотисто-желтых пленок, образующихся на
поверхности образцов при азотировании, показывает наличие наряду
с линиями металла также линии кубического нитрида гафния. Нитрид
гафния получают при азотировании гафния в течение 1 ч при темпера-
туре 1400° С и давлении азота,
Таблица 62 равном 1 ат. Параметр ре-
Сравнение энергий активации и скоростей реак- ций азотирования титана, циркония и гафния [223] щетки такого продукта состав- лял 4,50 А (расчетная плот- ность 14,0 г/см3). Из сравне-
Металл Энергия актива- ции, ккал/ио ль Константа ско- рости реакции, (г/см2)г/мин Температу- ра, °C Литера- тура ния энергий активации и кон- стант скорости реакции азоти- рования титана, циркония и гафния видно, что максималь- ной энергией активации и наи- меньшей скоростью азотиро- вания обладает гафний (табл. 62). В работе [224] нитрид гафния получали азотировани- ем при 1400—1500°С в те- , содержащего 2,4% Zr, а Zn, Al, Si, Си и следы Мп, нитрида были использованы
a-Ti a-Zr ₽-Zr a-Hf чение также Mo, 36 39 52 57 10 ч приме ЭЪ, Mg 1,9-10~9 7,8-10“10 3,8 -КЩ10 1,8 • 1О~10 ТОНКОЙ си Fe, С, Sn. Пол 550—850 400—825 862—1043 876—1034 стружки Ti, Сг, ученные [179] [179] [208] [223] гафния W, Ni, образцы
для термохимических исследований. Найдены значения теплоты обра-
зования ДЯ°298,16=—88,24 ±0,34 ккал!моль и убыли свободной энергии
при образовании нитрида гафния AF°298,16 = —81,4 ±0,5 ккал/моль. Эн-
тропия нитрида гафния, определенная в этой же работе, S°298,i6 =
13,1 кал/град моль.
Авторы [252] предполагают, что работа выхода нитрида гафния при
термоэлектронной эмиссии должна составлять при 1700° К 3,85—3,90 эв.
В работах [232, 255] исследовано взаимодействие между нитридом
гафния и углеродом по реакции
HfN -l-C^HfC + ^Na.
Показано, что лимитирующим звеном в процессе достижения равнове-
сия является диффузия в твердой фазе. Температурная зависимость
константы равновесия в области 1700—2300° С описывается уравнением
1g Кр = -~- 4-2,00,
we
из которого следует, что при низких температурах более устойчив нит-
рид, а при высоких — карбид гафния. Уже при 1200—1300° С около
50% нитрида гафния переходит в твердый раствор с карбидом гафния.
Азотирование порошка гафния азотом исследовали в работе [225].
Установлено, что нитрид гафния стехиометрического состава образуется
при 1200° С в течение 1 ч азотирования. Температурная зависимость
константы скорости азотирования изменяется при 700—800 и 1000—
1100°С.
Ksoo—?оос° = 1,51 . 10~4ехр (—910/Т) (образование твердого раствора в a-Hf)
Квоо-юоо’С = 2J5• 10~4 ехр(— 2940/Т) (образование HfN)
Кпоо-ноо’с = 2,75-10~4ехр(—4000/Т) (образование HfN).
По методу Ван-Аркеля [217], нитрид гафния получают осаждением
из газовой фазы, состоящей из смеси хлорида гафния, водорода и
бромистого азота на вольфрамовой проволоке, нагреваемой до 1100—
2700° С [159]. Указывается на возможность получения нитрида гафния
восстановлением окиси гафния углем в токе азота [159].
Нитрид гафния — желто-коричневый порошок с оливковым оттен-
ком, обладает металлической проводимостью с очень невысоким удель-
ным электросопротивлением. Нитрид гафния более теплопроводен, чем
нитриды титана и циркония, обладает наименьшим среди этих нитри-
дов термическим расширением, а также умеренной твердостью. Темпе-
ратура плавления, по данным Руффа [29], равна 3310° С, а по более
точным данным [254] — около 3000° С.
Химическая стойкость нитрида гафния почти не изучена; предпола-
гается, что она должна быть весьма высокой [9].
Как сообщается в [1034], при растворении нитрида гафния в 6 %-ной
HF происходит выделение водорода в результате прохождения реакций
HfN + 3HF = HfF3 + NHS,
NH3 + Hf = NH4F,
HfF3 + Hf = HfF4 +1/2H2.
Таким образом, гафний в нитриде гафния имеет валентность 34-,
а азот валентность 3—, иначе говоря, атомы азота образуют за счет
присоединения трех электронов атомов гафния №р6-конфигурацию, а
атомы гафния — преимущественно конфигурации d° и d5, что опреде-
ляет известную долю ионной связи в нитриде гафния наряду с метал-
лической и ковалентной.
Для стехиометрического нитрида с периодом решетки а = 4,5118 А
и рассчитанной отсюда плотностью 13,386 г]см3 установлена пикномет-
171
рическая плотность, равная 11,696 г/см3, что указывает на наличие де-
фектности в обеих подрешетках нитрида. Эта дефектность может быть
оценена в 12,63%- Следует отметить, что данные были получены не для
чистого нитрида гафния, а для нитрида гафния, содержащего 4,5%
нитрида циркония.
3. НИТРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ V ГРУППЫ
Нитриды ванадия. Система ванадий — азот изучена мало и можно
лишь считать установленным существование в ней двух нитридных
фаз — V3N и VN [256]. Наиболее полно система исследована Ханом
Рис. 71. Зависимость периодов решеток фаз в
системе V—N от содержания азота.
Таблица 63
Атомный состав кислородсодержаще-
го нитрида ванадия при различных
температурах
Температура, Формульный состав
600
800
1000
1200
1300
°.9 N0,94 °0,06
0,94 ^{о,94 °0.06
У1.0 ^0,98 °0,02
1,03 ^0,96 °0,04
V N Г)
1.14 0,98 0,02
[257] на основе данных рентгенографического анализа. Азот ничтожно
мало растворим в ванадии. Сплавы, содержащие до 9,3% азота
(VNo,37), двухфазны (a-твердый раствор и р-фаза).
р-Фаза V3N гомогенна в области от VN0i37 до VN0,43 (V3N — V2N),
т. е. от 9,3 до 10,5% азота в ванадии. С увеличением содержания азота
периоды решетки гексагональной p-фазы возрастают (рис. 71). В об-
ласти от 10,5 до 16,4% N сплавы двухфазны (.р- и у-фазы), с 16,4 до
21,5% N(VNo,7i—VNi.oo) простирается область гомогенности у-фазы,
отвечающей формуле VN. Период решетки этой кубической фазы так-
же резко изменяется с повышением содержания азота (рис. 71).
Нитрид ванадия VNX имеет структуру вычитания, причем в интер-
вале 600—1200° С вакантна часть узлов ванадия, а выше 1200° С—
часть узлов азота. При низких температурах могут образовываться ок-
синитриды VxNyOs-y. Атомы азота при этом замещаются атомами кис-
лорода, т. е. на структуру вычитания накладывается структура заме-
щения. Период решетки (табл. 63, 64) имеет максимум при соотно-
шении Vi,o-Ni,o, этому же составу отвечает и максимальное зна-
172
Таблица 64
Зависимость состава и свойств кислородсодержащего нитрида ванадия от
температуры реакции V2O2+2H2+N2=2VN+2H2O
Темпе- ратура, •С Время, ч Химический состав, % Формула* Период решетки а, кХ Плот- ность, г/см* Удельное электро- сопротив- ление. мком см Шлифо- вальная способ- ность, условные единицы
V N О
600 16 76,6 21,8 1,0 VNo.96 4,113 5,12 5,8 0,020
800 2 77,2 21,28 1,52 VNo,98 4,126 5,4 3,8 0,033
1000 2 78,4 21,1 0,5 vnX 4,13 5,83 3,2 0,064
1200 2 78,9 20,8 0,3 VN1>U 4,128 5,86 6,6 0,066
1400 2 80,1 18,9 0,5 vn1>17 4,12 5,78 5,6 0,056
* Без учета содержания кислорода.
Таблица 65
чение шлифовальной способности. Период решет-
ки VN, определенный прецизионно, составляет
4,129кХ [259].
Как видно из табл. 65, по данным [257], раз-
личие пикнометрических и рентгеновских значе-
ний плотности невелико, что свидетельствует об
ограниченном числе пустых мест в решетке, во
всяком случае, гораздо меньшем, чем в нитриде
титана [257].
Физические свойства. Порошкообразный
нитрид ванадия — серо-коричневого цвета с фио-
летовым оттенком, препараты с максимальным
содержанием азота — бронзового цвета. Нитрид
Зависимость удельного
веса нитрида ванадия от
содержания азота
Удельный вес
Состав пикно- метричес- кий расчетный
VN1>00 6,040 6,102
VNq,87 5,988 6,089
VNfl,72 5,972 6,066
VNo,38 5,967 5,987
ванадия термодинамически довольно прочен, убыль свободной энер-
гии при образовании из металла и азота выражается уравнением
Л7 = — 60000 — 1,737 lg Т + 26,387 (± 10% в интервале 298 — 2000 °К).
Теплосодержание изменяется по уравнению [260]
Нт — ЯЯ8,|6 = 10,947 + 1,05 • 10-372 + 2,21 • lOV"1 —4096 ккал/моль,
а теплоемкость
Ср = 10,94 4- 2,10-10 37— 2,21-1057 2 [ккал/арад-моль] (от 400 до 161 ГК).
Энтропия VN равна, по [261], 8,91 ±0,04 ккал/град • моль.
173
Рентгеноспектральное исследование К-спектров поглощения вана-
дия в нитриде VN сравнительно с гидридами, карбидами, боридами это-
го металла, в также с пятиокисью ванадия показало, что тонкая струк-
тура основного К-края поглощения ванадия в пятиокиси, карбиде и
нитриде качественно близки [267]. Однако по изменению относительной
интенсивности длинноволновой белой линии поглощения при переходе
от окиси к нитриду можно сделать вывод об изменении ковалентности
связи.
Следует полагать, что при образовании нитридных фаз ванадия
происходит некоторое повышение статистического веса ^-конфигура-
ций атомов ванадия, имеющих в изолированном состоянии 3d34s2-KOH-
фигурацию как за счет привлечения части s-электронов ванадия на
d-оболочку, так и за счет электронов азота, освобождающихся при пре-
образовании конфигурации s2p3 в sp3. Образование высоких статисти-
ческих весов достаточно сильно обособленных в энергетическом отно-
шении d5- и 5р3-конфигураций вызывает некоторые особенности физи-
ческих свойств нитридов ванадия, в частности плавление VN при
2050° С с разложением. С этим же связана способность VN переходить
в сверхпроводящее состояние при сравнительно высокой температуре
(7,5° К) [275]. Замечательным свойством нитрида VN является очень
низкий термический коэффициент электросопротивления, составляю-
щий от 100 до 1200° С всего 0,01 %/град. Основные физические свойства
нитридов ванадия приведены в табл. 66.
Примесь кислорода в нитриде ванадия (около 3% О) вызывает
аномальный ход температурной зависимости электросопротивления: до
450° С сопротивление такого нитрида заметно растет с температурой,
затем спадает, снова возрастает [955].
Химические свойства. Нитрид ванадия VN нерастворим в соляной
и серной кислотах, разлагается при кипячении в азотной кислоте, при
нагревании с едким натром и кипячении в растворе едкого кали [ПО].
При комнатной температуре водные растворы щелочей практически не
разлагают нитрид ванадия VN [276]. При длительном кипячении в кон-
центрированной серной кислоте медленно разлагается с выделением ам-
миака. При нагревании на воздухе нитрид ванадия VN довольно устой-
чив, окисляясь только при 500—800° С. Нитрид ванадия V3N гораздо
менеее устойчив в химическом отношении, чем VN.
Методы получения. Действием азота на порошок ванадия можно
получать нитрид ванадия точного состава VN [257, 177]. Для приготов-
ления сплавов с более низким содержанием азота Хан [257] прессовал
брикеты из смесей VN и порошка ванадия с последующим нагревом их
в закрытой кварцевой трубке при температуре 1000—1100° С в течение
24 ч. При нагревании ванадиевой проволоки в азоте образуется поро-
шок нитрида ванадия бронзового цвета [273]. Для приготовления ни-
трида ванадия одновременным восстановлением окисла углем и азоти-
174
Таблица 66
Свойства нитридов ванадия
Характеристика P-V.N T-VN
Область гомогенности vn0>37-vnM3 vn0>7. -VN11OO
Содержание азота, вес. % 9,22—10,6 16,3—21,5
Кристаллическая структура Периоды решетки, д: Гексагональная , Кубическая
а с с/а 2,835 [257] 4,541 1,60 4,12, [259]
Плотность, г/СМ3 5,987 [257] 6,102 [257]
Температура плавления, °C Теплоемкость (при 298° К, — 2050° С разлагается
ккал/моль- град) Теплота образования, — 11,57 [260]
ккал/моль — 60,0±2,0 [47]
Энергия (298° К)> кал/град — 21,0±0,1 [261]
Удельное электросопротивле- 123—10 [289] 85,4 [289]
ние> ом-см Т ермический коэффициент электросопротивления, (VNo,338) (vn0i93)
%/град — 0,01*
Коэффициент термо-э. д. с., —5,3±0,2 [289] —4,6±0,8 [289]
мкв • град (VN0,138 ) (VN0-93)
Коэффициент ХоллаХ! О4, —0,9±0,1 [289] —0,45±0,16 [289]
см3/к Т еплопроводность, (VNo,33e) (VNo,93) 0,0270±0,007 [289]
кал/см-сек-град Точка перехода в сверхпро- — (VN0,93) 7,50—8,20 [304 , 275,
водящее состояние, °К Микротвердость, кг/ леи2 Коэффициент излучения (Х=655 мкм) при 800— — 1001]
1900±102 [289] (VN0,338) 1520±115 [289 (VNo>74)
2000° С Работа выхода при термо- — 0,77 [252]
эмиссии (1700° К), эв 3 252
* Определено С. Н. Львовым и В.Ф. Немченко.
рованием обычно используется V2O3, так как V2O5 очень летуч. Однако
и при этом получить нитрид трудно, ибо при 1200° С нитрид в присут-
ствии углерода превращается в карбид. Впервые довольно чистый нит-
рид получали таким способом Фридерих и Зиттиг [ПО]. Они нагревали
смесь УгОз+ЗС в молибденовой лодочке в трубчатой печи с фарфоро-
вой трубкой при 1200° С в среде азота (нитрид содержал 21,1% N
175
вместо 21,55%, полагающихся для VN). Прокаливая ванадий в токе
азота при 1200°С, получали нитрид, содержащий 21,69% N.
Нитрид ванадия приготовляли также нагреванием VOClj или
УгО3 в токе аммиака [256].
Эпельбаум и Ормонт [258] получили нитрид ванадия разложением
ванадата аммония в токе аммиака при 1000—1100° С (при более высо-
ких температурах образуются смеси VN с окислами ванадия). Позднее
этот способ был с успехом применен Ханом [257] для приготовления
очень чистого нитрида ванадия. Эпельбаум и Ормонт [258] получали
нитрид ванадия одновременным восстановлением и азотированием низ-
шего окисла ванадия V2O2 при 500—600°С по реакции
V2Oa 4- 2Н2 4- N2 — 2 VN 4- 2НаО 4- 16 ккал.
Систематическое исследование условий образования нитрида ва-
надия азотированием порошка металлического ванадия проведено в
работе [178]. Азотирование проводили при 500—1500° С. Насыщение
азотом металлического ванадия при 500—800° С подчиняется линей-
ному закону, при более высоких температурах — параболическому за-
кону с некоторыми отступлениями в начальный период. Повышение
температуры гораздо сильнее сказывается на росте содержания азота,
чем увеличение времени выдержки. При температурах порядка 1400—
1500° С максимальное содержание азота достигается за 10—15 мин.
Насыщение азотом ванадия в среде аммиака происходит более мед-
ленно, чем в среде азота. Из данных рентгеновского анализа следует,
что при 800—900° С образуется фаза V3N, а при повышении темпера-
туры азотирования — фаза VN. Параметр кристаллической решетки
ванадия с повышением содержания азота от 0 до 3,5% увеличивается
от 3,03 до 3,09 А. При содержании азота 2,5—3,0% рентгенографически
обнаруживается фаза V3N, которая сохраняется до содержания 11,3%
азота. После 12,8 вес. % N обнаруживается фаза с параметром решетки
а=4,13—4,14 А. Константа скорости реакции азотирования уменьша-
ется от 1,7 • 10~5 при 500° С до 3,71 • 10~4 г/см? при 1200° С. Энергия
активации азотирования порошка ванадия невелика и составляет в
области 500—800°С (образование V3N) около 4640, а в области 900—
1200° С (образование VN) — около 4400 кал/моль.
Энергия активации при диффузии в компактном ванадии выше,
например, в твердом растворе азота в ванадии она составляет 36—
37 ккал/молъ [305]. Оптимальный режим получения фазы V3N состоит в
нагревании порошка при 900° С в течение 1 ч, a VN — при 1200—
1300° С в течение 4 ч.
Можно приготовить нитрид ванадия методом осаждения на вольф-
рамовой проволоке из смесей VC14 с эквимолярным газом N24-H2. Оп-
тимальные условия: температура 1540—1570° С, общее давление 50—
60 мм. рт. ст., упругость пара VC14— 8 мм рт. ст., диаметр вольфрамо-
176
вой проволоки — 0,20 мм. Получаемый продукт близок по составу к
VN [301].
Чистый нитрид ванадия VN образуется при разложении различ-
ных солей, содержащих ванадий и азот. Хорошие результаты дает раз-
ложение при 600°С соли (NH4)3VF6 или при 800°С— (VH4)3VO2F4 в
течение 1 ч [274].
Приготавливают изделия из нитрида ванадия либо горячим прес-
сованием порошка в среде аргона (оптимальные условия: для V3N —
температура спекания 1850° С, давление 100 кг[см2, время 3—5 мин, ос-
таточная пористость 6%; для VN — температура 1800° С, остаточная
пористость 4%), либо реакционным спеканием в среде азота предва-
рительно спрессованных заготовок из порошка ванадия [232]. В послед-
нем случае, хотя удельные объемы нитридных фаз ванадия на 28—
29% больше удельного объема металла, увеличение объема не реали-
зуется за счет сокращения пористости и последняя составляет 46—49%.
Нитриды ниобия. Система ниобий — азот исследовалась неодно-
кратно. По ранним данным, обобщенных в [138], в системе имеется два
химических соединения — Nb2N и NbN. Достаточно полное исследова-
ние системы Nb—N проведено Брауэром и Яндером [277]. Растворимость
азота в ниобии менее 4,8 ат.%, так как уже сплав, отвечающий формуле
NbNo.osi содержит фазу нитрида ниобия. а-Твердый раствор имеет ку-
бическую объемноцентрированную решетку с параметром, очень мало
отличающимся от параметра решетки чистого ниобия (3,2948 кХ).
P-Фаза iNbaN гомогенна в области NbNo,4o—NbNo.so, имеет гексагональ-
ную плотноупакованную решетку со статистическим распределением
атомов азота в крупных порах решетки. Параметр решетки а=3,046 кХ
в области гомогенности не изменяется, а значение параметра с при
уменьшении содержания азота снижается от 4,986 (NbNo,5o) до 4,947 кХ
на границе с a-фазой (NbNo,4o)- у-Фаза (NbNo,8o—NbNo,go) изоморфна
6-фазе в системе Та—N и имеет структуру типа NiAs. Атомы азота рас-
положены в центрах двух тригональных призм из атомов ниобия. В ин-
тервале концентраций NbNojs—NbNo,94 имеются две фазы: NbN0,75—
NbNo,79 с тетрагональной структурой типа деформированной NaCl и
NbN0,87—NbNo,94 со структурой NaCl. Однако последующей работой
Шенберга [278] было показано, что наличие двух фаз следует отнести
за счет присутствия кислорода в образцах, т. е. образования фаз си-
стемы Nb—N—О.
Гексагональная 6-фаза (NbN—л-фаза Брауэра), в предельном слу-
чае соответствующая формуле NbN0,95, имеет решетку с антиникельар-
сенидной конфигурацией атомов. Атомы азота расположены в центрах
октаэдров, построенных из атомов металла. е-Фаза NbNi.oo имеет гек-
сагональную решетку, пространственная группа £)64—F36; аналогичную
структуру имеют МоС (у-фаза), а также некоторые фосфиды — TIP,
p-ZrP [279]. Брауэр и Яндер [277] обращают внимание на несоответствие
12-272
177
F
между приводимыми данными о фазах систем Nb—N и данными Уман-
ского [280], который для образца NbN0,65 получил гексагональную ре-
шетку, параметры и расположение атомов которой соответствуют 6-фазе
(NbN—л-фазе Брауэра и Яндера). Это возможно только при условии,
что нитрид Уманского содержал кислород, который, внедряясь в ре-
шетку ниобия вместе с атомами азота, доводил общий состав фазы до
NbNo,6sOo,3- Причиной несоответствия могло быть неполное азотиро-
вание, что вызвало образование наряду с 6-фазой металлического нио-
бия или a-фазы. Изложенные
результаты Брауэра были под-
тверждены и уточнены в [278,
281, 282, 294].
Исследователями [283]
предложен предварительный
вариант диаграммы состояния
системы Nb—N. Система весь-
ма сложна, в настоящее вре-
мя принимается существова-
ние твердого раствора азота в
ниобии и пяти нитридных фаз
(рис. 72). Область существо-
вания твердого раствора от
Nb до NbN0,o25. Далее следуют
Рис. 72. Фазовая диаграмма состояния системы
ниобий—азот.
гексагональная р-фаза NbzN
(NbN0,4—NbN0,s), тетрагональ-
ная у-фаза Nb4N3 (NbNo.zs—
NbNo,8s), гексагональная 6-фа-
за NbN (NbNo,97—NbNoj98) с антиникельарсенидной конфигурацией,
кубическая 6-фаза NbN (NbNo.ee—NbNo.gs) co структурой NaCI и
гексагональная е-фаза NbN (предельного состава NbNi,Oi). Как
следует из фазовой диаграммы (рис. 72), выше 1400° С стабильны
фазы а, р, у и 6, а ниже 1230° С существуют фазы а, р, у и е.
Схема взаимного превращения фаз в области температур от 1200 до
1400° С и условия этих превращений показаны на рис. 73 [281, 282].
На рис. 74 изображена диаграмма состояния системы Nb—N, по-
строенная по результатам исследования [283]. Подтверждено существо-
вание ,р-, у-, 6- и е-фаз, найденных Брауэром, б'-фаза, найденная Брау-
эром и Шенбергом, не была обнаружена в работе [283].
Область диаграммы от 0,1 до 30 ат. % N в интервале температур
1600—2200° С исследована кинетическими методами в работе [284]
Показано, что в этих условиях однозначно существуют две твер-
* Кинетика потери азота твердыми растворами азота в ниобии исследовалась так-
же в работе [981].
178
дые фазы. При низких концентрациях азота обнаружена область a-твер-
дого раствора, концентрация которого с температурой и давлением
меняется по уравнению
CNO =Ур • 7,73 -1(Г4- ехр (21400/7'),
где Cn,<z — концентрация N, ат. %, р— давление N, тор; Т — темпера-
тура, °К- Растворимость азота в ниобии зависит от температуры и
-N2 >1230 °C
" .. — 6-фозо
+ N2>1230°C 21230 ° С
I
I
I
i
II
-N2 Г .
Г <розо ——«------------ <5 -фозо
||______________-%< 1230 °C
(^<1230 ° С
Рис. 73. Превращения нитридных фаз ниобия.
Рис. 74. Диаграмма состояния системы ниобий—азот (по [283]).
возрастает от 3,7 ат. % при 1600° С до ~ 18 ат. % при 2300° С. Внутри
этой области растворимость изменяется по зависимости
CN,U = 1,48-103-ехр(— 11300/Т).
макс
При выходе за пределы твердого раствора образуется р-фаза (МЬгМ).
При достаточно длительной обработке азотом a-фаза переходит в р-фа-
зу с энтальпией перехода АЯ— —22,6 ккал/г-атом. и с изменением пар-
циальной молярной стандартной энтропии AS°=—8,1 кал/град • г-атом..
Растворимость азота в ниобии в интервале температур 300—
2300° С изучена также в [297]. При температуре выше 1150° С раство-
римость С—720-ехр (—20 000/7??), где С — ат.% N.
В результате тщательно проведенного исследования растворимости:
азота в ниобии [1049] установлены несколько иные, по сравнению с дан-г
ными предыдущих работ, параметры уравнения растворения:
А-10—4 ДДК.1О’ Ли™рРа’
6,2 —46,2 [297]
7,73 —42,4 [284]
7.6 —46,2 [1049]
12* 17W
Согласно [284], минимальное содержание азота в p-фазе составляет
~25 ат.%, т. е. меньше, чем предельное минимальное содержание по
Брауэру, равное около 28 ат.%. Между 700 и 2100° С граничная кон-
центрация или не зависит от температуры, или несколько возрастает
с повышением температуры. Таким образом, p-фазе следует приписать
область гомогенности не NbNo,4—NbN0,s, как у Брауэра, а более ши-
рокую — NbNo,33—NbN0,5.
Существование в системе ниобий — азот а-, р-, у-, 6- и е-фаз было
подтверждено в [285] на основе данных рентгено- и металлографиче-
ского исследований. Показано, что нижняя граница области гомоген-
ности p-фазы лежит при ~26 ат. % N, а не при 28 ат. % N, как опреде-
лено Брауэром. Данные верхней границы области гомогенности прак-
тически совпадают с данными Брауэра и Шенберга (50 ат. % N). Ав-
торы [285] по изменению электропроводности и эффекта Холла в области
б-фазы при комнатной температуре делают предварительное заключе-
ние о неправильности вывода Брауэра о существовании этой фазы в
стабильном состоянии только при температурах выше 1230° С.
Брауэр и Кирнер [295], уточнив состав б- и е-фаз, показали, что
предельным составом кубической 6-фазы является NbNi.oo, а гексаго-
нальной е-фазы — Nb0,94N.
Сравнивая значения плотности и периодов решетки, авторы при-
шли к выводу, что фазу с избытком азота NbNi+д правильнее рас-
сматривать не как фазу внедрения дополнительных атомов азота в
решетку NbN, а как фазу вычитания ниобия из решетки NbN, т. е.
фазу типа Nbi-AN.
Суммируя все имеющиеся данные, можно считать достаточно до-
стоверно установленным в системе ниобий — азот существование нит-
ридных фаз, указанных в табл. 67.
Физические свойства. Основные физические свойства нитридов нио-
бия наиболее изучены в [285, 289], результаты которых приведены в
табл. 67. Выше дана краткая характеристика структур нитридных фаз
ниобия, показаны элементарные ячейки кристаллических решеток этих
фаз (рис. 75).
Как показывают положительные значения коэффициента Холла
(рис. 76, [285]), электропроводность твердого раствора азота в ниобии
и смеси 6- и б'-фаз, так же, как и самого ниобия, преимущественно
дырочного характера. Дырочный характер электропроводности ниобия
подтверждается рентгеноспектральными исследованиями его Лр2-поло-
сы, которые показывают заполнение большей части первой зоны Брил-
люэна [286]. В области твердого раствора сохраняется преобладающее
влияние дырочного характера электропроводности металлического нио-
бия. По мере обогащения сплавов азотом дырочный вклад в электро-
проводность снижается и при содержании 27,3 ат. % 'N электропро-
водность становится преимущественно электронной и только для об-
iso
Таблица 67
Свойства нитридов ниобия
Характеристика ₽-Nb2N V-Nb3N4 б-NbN e-NbN
Область гомогенности NbN0,38 NbN0 75 NbN088— №N1i00-
NbN0,50 —NbN085 -NbN, >00 -NbNli06
Содержание азота, вес.% 4,72—7,02 10,'—11,26 11,61—13,1 13,1—13,8
Кристаллическая структу- Гексагональ- Кубическая Кубическая Гексагональна»
ра ная плотно- упакованная типа NiAs типа NaCl типа у-МоС
Температурная область стабильного существо-
вания, °C Периоды решетки, д: До 2400 До 1500 1230 До ~2300
а 3,057—3,050 [283] — 4,386—4,394 [283] 2,953 [283]
с 4,957—5,005 — 11,234—11,257
с! а 1,622—1,641 — 3,804—3,813
Плотность, г/см3 8,31—8,33 8,32 8,30 7,98
[277] [277] [277] [295]
Микротвердость, кг/мм2 1720± 100 1780 1525+136 1396+26
Удельное электросопротив- [289] [285] [285] [285, 289]
142±6 90±8 85 ±2 78±4
ление, мком’СМ Коэффициент термо-э. д. с., [285, 289] [285] [285, 289] [285, 289]
4,6±0,7 1,6±0,1 —2,24
мкв-град [285, 289] — [285, 289] [285, 289]
Коэффициент ХоллаХ 104, + 1,9±0,4 —0,7±0,1 +0,5±0,2 +0,5±0,2
см3/к [285, 289] [285] [285, 289] [285, 289]
Теплопроводность, 0,0200 ±0,008 0,0191 ±0.004 0,009±0,002
кал/см-сек-град Теплота образования [285,289 ] 67,0 [297 ] [285] [285],
AH°9qR 1SoK, ккал/моль 61,1 ± 1Д303] — 56,8 [213] —
Стандартная энтропия,
кал/г-град-моль Теплоемкость (298° К), 22,3[297 — 10,5±02 [47] —
ккал/моль-град — — 10,41 [302] —
Коэффициент теплового расширениях 10е,
град~х 3,26[298] — 10,1 [298] —
Теплота испарения,
ккал/моль — — 91,5 [298]
Температура перехода в
сверхпроводящее со- 6,8—8,9 13,1—15,6 <1,94 [291]
стояние, °К <1,94[291] [290, 291] [290,291,275]
Коэффициент излучения
при 800—2000° С — — 0,83 [252] —
Работа выхода при термо-
эмиссии, (1700°К,) эв — — ~3,90 [252] —
Молярная магнитная вое-
приимчивость х 10б — .— +30 [978] —
Модуль упругости, кг/мм2- — — 49300 ±2000 —
18)
ласти б-фб'-фаз снова наблюдается дырочная электропроводность.
Электронный характер электропроводности при относительно высоких
содержания азота также согласуется с данными рентгеноспектральных
исследований Ьр2-полосы ниобия в его соединениях с азотом [286], сог-
ласно которым уже при содержании 6,32 вес. % N происходит значи-
тельное уменьшение степени заполнения первой зоны Бриллюэна.
Результаты рентгеноспектрального исследования [287] показывают,
что для NbN, NbC и NbBa по уменьшению интенсивности полос и
Рис. 75. Кристаллическая структура нитридных фаз ниобия.
Lv ниобия при образовании указанных соединений можно сделать од-
нозначный вывод о понижении плотности d-состояний в обобщенной
полосе. Это свидетельствует о том, что при образовании некоторых
соединений, в частности ниобия с азотом (NbN), не происходит пере-
ход валентных электронов неметаллических атомов в d-полосу метал-
лического атома. Так как, согласно вышеприведенным данным
(стр. 13), при образовании металлического кристалла из изолирован-
ных атомов ниобия 3,7—3,9 валентных электрона переходят в локали-
зованное состояние (обеспечивающее ковалентную связь), а остальные
1,1—1,3 электрона коллективизируются, то можно предположить, что
при образовании нитрида ниобия NbN происходит переход части нело-
кализованных электронов к атомам азота с образованием ими ста-
бильных «2ре-конфигураций. Этот процесс не должен привести к замет-
ному изменению плотности d-состояний атомов ниобия по сравнению с
таковой в металле. Однако при этом должен относительно повыситься
статистический вес d5-кoнфигypaций, образуемых локализованной
частью валентных электронов, при одновременном уменьшении концент-
рации свободных электронов и росте статистического веса атомов
азота, имеющих стабильные х2р6-конфигурации. Это вызывает, в част-
182
пости, некубическую симметрию е-фазы нитрида ниобия NbN (если бы
азот отдавал часть электронов по схеме s2pa-^spi-^-sp5+р ниобию в
нитриде NbN, то последний имел бы кубическую структуру), а также
его высокую электропроводность.
При уменьшении содержания азота в нитриде ниобия NbN (пере-
ходе от е- к d-фазе), по-видимому, создаются условия превращения
атома азота по указанной схеме с образованием хр3-конфигураций и лег-
коподвижного электрона,
могущего частично перехо-
дить к атомам ниобия, а ча-
стично пополнять коллектив
нелокализованных электро-
нов. Последнее приводит к
некоторому росту электро-
сопротивления (см. табл.
67). Кроме того, появление
^реконфигураций вызывает
кубическую симметрию
б-NbN и повышение его твер-
дости по сравнению с е-фа-
зой. Далее это прогрессиру-
ет при переходе к у-фазе.
Предложенная выше
картина электронного строе-
ния мононитрида ниобия б-
фазы NbN хорошо подтвер-
Рис. 76. Концентрационная зависимость элек-
тросопротивления и коэффициента Холла спла-
вов ниобия с азотом.
ждается данными по теплопроводности как малым ее значением, так и
аномально высоким отношением расчетной электронной теплопроводно-
сти к измеренной общей теплопроводности для этого нитрида [289].
Так же хорошо она интерпретируется по данным исследования
сверхпроводимости в широком интервале содержаний азота в ниобии
от NbNo.oz до NbNi.oo [290, 291, 1069]. Гексагональные е-фаза
NbN и p-фаза NbzN не обнаруживают сверхпроводимости вплоть до
1,94° К. Для у-фазы NbsN4 точка перехода равна по этим данным
6,8—8,9° К, а кубическая б-фаза переходит к сверхпроводимости при
10,62° К (по другим данным этих же авторов — при 13,1° К, а по обще-
принятым сейчас данным—15,6° К). Образцы с небольшой примесью
кислорода дают совпадающие кривые нагревания и охлаждения в об-
ласти перехода, а образцы без кислорода обнаруживают сильный гисте-
резис. В [292] показано, что в полях 0,5—4 эрст и при токе до 20 а на-
блюдается увеличение магнитного потока (на 63%) в области перехода
б-NbN из нормального в сверхпроводящее состояние (парамагнитный
эффект), характерное для чистых металлов. Точка перехода NbN с 11,1
и 12,8% лежат в интервале 16,2° К (начало) и 13,5° К (конец) [293].
183
Сверхпроводимость фазы б-NbN отмечается на ходе температурной
зависимости теплоемкости [288]. Критическая величина магнитного
поля, соответствующая переходу в сверхпроводящее состояние, равна
257 гс/град. Таким образом, сверхпроводимостью обладают только ку-
бические б- и у-фазы, в которых образуется высокий статистический вес
атомов ниобия со стабильными ^-конфигурациями и атомов азота со
стабильными эр3-конфигурациями локализованной части валентных
электронов. При этом образуется своеобразный «канал с инертными
стенками» из стабильных электронных конфигураций, по которому без
заметного рассеяния могут перемещаться носители тока при тех темпе-
ратурах, когда эти носители объединяются в куперовские х2-пары и
мало термическое возбуждение стабильных конфигураций, образующих
«стенки» канала.
При образовании 0-фазы можно предположить повышение стати-
стического веса ^-конфигураций атомов ниобия за счет обмена элек-
тронами между ними с освобождением при этом части электронов
атомов ниобия, бывших локализоваными в металлическом кристалле,
и переходом их частично в нелокализованное состояние, а частично к
атомам азота с увеличением статистического веса з2р6-конфигураций
этих атомов. Появляется значительное число носителей тока, что уси-
ливает электрон-электронное взаимодействие и приводит к несверхпро-
водимости Nb2N [275]. По тем же причинам этот нитрид имеет относи-
тельно высокое электросопротивление, а также гексагональную, а не
кубическую структуру.
В работе [284] исследована термическая устойчивость фазы Nb2N
в равновесных условиях и получено следующее выражение температур-
ной зависимости превращения р-фаза->а-фаза + П2:
Р₽->а = 5,28-1012 ехр (— 66000/Т),
где р — давление азота, тор.
В работе [298] по методу Лэнгмюра измерена температурная зави-
кт , г , 20000
симость давления паров нитрида NbN : lgp = 5,l---?—
Химические свойства. Нитрид ниобия NbN (по-видимому, б-фаза,.
либо получаемая смесь б- и е-фаз) весьма устойчив на холоду и при
нагреве в концентрированных соляной, хлорной, серной кислотах, не-
сколько менее устойчив в разбавленных соляной, азотной, серной кис-
лотах, а также смесях кислот (смесью азотной и плавиковой кислот
быстро и нацело разлагается). Еще более сильно действуют на него
перекись водорода, смеси щелочей с перекисью водорода. Водные раст-
воры щелочей при нагревании разлагают нитрид ниобия не интенсив-
но, степень действия щелочи на нитрид возрастает с повышением кон-
центрации щелочи в растворе. Для аналитических целей рекомендуются
184
Ри.с77. Кинетические кривые азотирования нио-
бия.
компактного ниобия или же нагревание
в качестве растворителей нитрида ниобия концентрированная H2SO4 и
смесь NaOH с пергидролем.
Nb2N устойчив против действия кислот, а при нагревании в раст-
ворах сильных щелочей или при сплавлении с ними выделяется не ам-
миак, а азот [296].
Окисление нитрида ниобия на воздухе происходит при 500—800° С
с образованием Nb2Os.
Методы получения. Фридерих и Зиттиг [ПО] получали нитрид NbN
путем азотирования смеси окисла (Nb2O3) с углем, причем этот окисел
приготовляли предваритель-
ным восстановлением пяти-
окиси ниобия водородом при
температуре 1100—1200° С.
Содержание азота в полу-
ченном нитриде составляло
12,8% (вместо 13,1% по рас-
чету). В работе [280] иссле-
довали нитрид ниобия, при-
готовленный М. М. Бабичем
трехчасовым прокаливанием
смеси ниобия с его гидри-
дом в токе азота при 1100 и
1300° С.
Основным методом по-
лучения низших нитридов
ниобия является азотирова-
ние порошкообразного или
смесей высшего нитрида ниобия с ниобием.
Кинетику азотирования ниобия изучали Гульбранзен и Эндрью
[179]. Найденная ими зависимость скорости азотирования от времени
при 400—800° С показана на рис. 77. Энергия активации при диффузии
азота в ниобий составляет около 25,4 ккал:моль. В работе [284] была
подробно исследована растворимость азота в ниобии (см. стр. 178).
Установлено, что растворение подчиняется закону Сивертса (Уд). Вы-
числено, что теплота растворения равна 21,4 ккал!моль. Подобные дан-
ные были получены и в [297], где для равновесной концентрации азота
в растворе найдено следующее уравнение
С = |/ pN; • 6,2 • 1(Г4 • ехр (— 46000Ж).
Растворы азота в ниобии близки к идеальным.
Исследование азотирования порошка ниобия с азотом в интервале
температур 500—1200° С с выдержками 15—120 мин при каждой тем-
пературе [225, 298] показало, что до 500° С за короткое время азотиро-
185-
вания (15—30 мин) образуется твердый раствор азота в ниобии (а-фа-
за), а при температурах от 600 до 1200° С — смеси нитридных фаз —
р, у, 6 и е. При коротких выдержках (30—60 мин) при 900° С образу-
ется ф-фаза (Nb2N), а при 1200° С — е-фаза (NbN) без примесей
других фаз. Анализ кривых азотирования показывает, что насыщение
ниобия азотом с образованием a-фазы и однофазного нитрида Nb2N
подчиняется закону, близкому к линейному, а фазы NbN — параболи-
ческому. Энергия активации при азотировании порошка резко снижа-
ется по сравнению с энергией азотирования компактного ниобия и со-
ставляет от ДО ’А от указанных выше значений при азотировании
компактного ниобия.
Брауэр, Яндер [277] и Иов [299] приготавливали нитриды ниобия
азотированием порошка или стружки ниобия в токе азота или аммиака.
Подобное азотирование ниобиевой стружки в течение 4—5 ч при тем-
пературе 1300° С позволяет получить высший нитрид ниобия NbN, а при
1450° С — NbNo.ss—NbNo,94- Нитриды, более бедные азотом (от NbNo.ss
до NbN0j7i), получают частичным удалением азота при нагревании нит-
ридов NbN в высоком вакууме при 1300—1400° С в течение 3—6 ч. На-
конец, при нагревании смесей нитридов, богатых азотом, с ниобиевой
стружкой при 1450° С в течение 3 ч в среде аргона образуются сплавы,
бедные азотом (вплоть до NbNo.os). Аналогичным путем приготовлял
нитрид ниобия Шенберг [278], который азотировал порошок ниобия и
гидрида ниобия при температурах 700—1000° С в течение от 1 ч до
2 недель.
Брауэр и Кирнер [295] проводили азотирование порошка ниобия
в среде азота при 1200—1500° С под давлением до 160 ат. При темпе-
ратурах 1400—1500° С, давлениях от 50 до 150 ат и временах выдержки
от 2 до 24 ч образуется чистая 6-фаза, а при более низких температу-
рах 1200—1300° С и высоких давлениях 150—160 ат — смеси 5- и
е-фаз.
При азотировании ниобиевой проволоки аммиаком в течение 1 ч
при 1400—1800° С на проволоке образуется наружный слой из NbN и
внутренний из Nb2N [300].
Кристаллизованные осадки нитрида получаются при реакции па-
ров NbCl5, образуемых испарением NbCl5 при 175° С, со смесью экви-
молекулярных объемов N2+H2 при 1340—1350° С и общем давлении
газа в 600 мм рт. ст. на вольфрамовой проволоке [301].
Исследователи [274] нитрид NbN приготавливали пиролитическим
разложением (NH^NbF? в токе аммиака при 700° С.
Изделия из нитрида ниобия получали спеканием предварительно
спрессованных брикетов из порошков Nb2N и NbN при 1900° С в вакууме
с относительно небольшой потерей азота [232]. Полученные изделия
имеют пористость 0,5—1%. Другим методом приготовления является
горячее прессование при 1800—1850° С под давлением 100 кг/см2 (оста-
186
точная пористость 0,2%, загрязнение углеродом до 0,3% при спекании
в графитовых прессформах). Изделия из нитридов ниобия можно по-
лучать реакционным спеканием спрессованных образцов из ниобиевого
порошка в азоте при 900—1200° С в течение 4—8 ч, однако пористость
изделий при этом оказывается не ниже 20%.
Нитриды тантала. Систематическое изучение нитрида тантала,
открытого в 1876 г. Жоли, который приписал ему формулу TaN [138],
началось лишь в 1924 г., когда Ван-Аркель получил в результате газо-
фазной реакции препарат достаточно высокой чистоты, определил его
химический состав и кристаллическую структуру [183]. Его работа была
подвергнута критическому рассмотрению Хэггом [307].
Подробное рентгенографическое исследование системы Та—N было
проведено в 1954 г. Шенбергом [308], а также Брауэром и Цаппом [309].
Шенберг показал наличие в системе Та—N следующих фаз: твердого
раствора азота в тантале с условной формулой TaN-o.os (p-фаза), нит-
ридов TaN~o,4o—TaN~o,45 (у-фаза), TaN~o,8o—TaN~o,9o (6-фаза) и TaN
(е-фаза). Кубическая решетка p-фазы незначительно отличается от ку-
бической объемноцентрированной решетки чистого тантала (а-фазы).
Эта сверхструктурная фаза имеет период элементарной ячейки а =
= 3,369 кХ (период решетки тантала а = 3,311 кХ).
Гомогенная в пределах TaN~o,4o—TaN~o,45 (у-фаза), близка по со-
ставу к нитриду Ta2N. Это классический пример нитрида, образован-
ного по типу фаз внедрения с расположением атомов металла в плот-
ноупакованной гексагональной решетке и атомов азота в двух октаэдри-
ческих порах, в центрах элементарной ячейки. 6-Фаза, с областью го-
могенности от Ta>N~o,8o до TaNo.go, имеет простую гексагональную ре-
шетку с расположением металлических атомов ААА... Атомы азота рас-
положены в одном из центров двух тригональных призм, образуемых
атомами металла.
Гексагональная е-фаза, соответствующая нитриду TaN в элемен-
тарной ячейке TaN содержит три формульные единицы нитрида.
Брауэр и Цапп [309] обнаружили только две нитридные фазы Ta2N
и TaN (табл. 68). При определении структуры фазы TaN получились
результаты, полностью совпадающие с результатами Шенберга
(рис. 78). Кратчайшие расстояния между атомами тантала составляют
2,90; 2,99; 3,33 кХ, расстояние Та—N 2,08 или 2,59 кХ. Область гомо-
генности этой фазы очень мала, так как сплав TaNo.gs уже содержит
низший нитрид Ta2N. Последний имеет область гомогенности от TaNo,4i
до TaNo.so (вместо TaNo,4o—TaN0,45 по Шенбергу). Атомы тантала об-
разуют плотнейшую гексагональную упаковку.
По данным [312], область гомогенности фазы Ta2N при комнатной
температуре составляет от 28 до 33 ат. % N, что близко к данным Брау-
эра и Цаппа. 6-Фаза, установленная Шенбергом, Брауэром и Цаппом,
в этой работе не была обнаружена.
187
Таблица 68
Свойства нитридов тантала
Характеристика TaaN TaN
Область гомогенности ^а^0,41“^а^0,50 TaN
Содержание азота, вес. % 3,1—3,86 7,19
Кристаллическая структура Периоды решетки, кХ: Гексагона льная
а 3,042—3,0415 [ 309] 5,1808 [309]
с 4,905—4,9088 [309] 2,9049 [309]
с/а Плотность, г/сл43: 1,62—1,61 0,562
расчетная 15,78—15,86 [309] 14,36 [309]
пикнометрическая 15,42—15,46 [309] 13,80 [309]
Температура плавления, °C — 3087 + 50 [211]
Теплота образования— 64,7±3 [303] 60,0+0,6 [213]
ДН°98, ккал/моль Удельное электросопро- тивление Q (20° С), 50,0 1311 (для 24 ат. % N и 1600—2380° С) 65,2±5 [314]
мком • см Коэффициент Холла 220±15 [318] 180±10 [318]
ЙХЮ1, см3/кул Коэффициент термо-э. д. с. —1,4±0,3[318] —0,6±0,09 [318]
а, мкв/град Работа выхода при термо- —2,17±0,4 [289] —1,6+0,3 [289]
эмиссии (1700° К), эв Теплопроводность, — 4,20 [252]
кал/см -сек -град Теплоемкость (298° К), 0,0240 ± 0,005 [289] 0,0205+0,009 [289]
кал /моль град Энтропия (298° К), — 13,10 [47]
град • моль Температура перехода в сверхпроводящее со- —W 12,4±1,5 [47]
стояние, °К Микротвердость, кг/мм3 Коэффициент излучения 9,5 [31 <1,20 [304]
1220 + 120 [318] 1060±75 [318]
при 800—2000° С Молярная магнитная вое- — 0,79 [252]
приимчивость X- 10s — <25 [978]
Модуль упругости, кг/мм2 —- 58700 +2000
Однако Брауэр и Лессер [310] полностью подтвердили свои преж-
ние результаты, а в обзоре [256] предполагается, что фаза TaNo.so—
TaNo.go Шенберга является кислородсодержащим нитридом тантала, а
не соединением системы тантал—азот.
188
Растворимость азота в тантале мала. В [311] показано, что в интер-
вале температур 1600—2000° С и давлении КН ат тантал в твердой
фазе растворяет не более 7 ат.% N. Согласно данным [312], тантал
при комнатной температуре растворяет 2 ат. °/о N, а при 2300° С —
13 ат. % N. В области температур
1600—2380° С связь между равновес-
ным давлением, концентрацией азота
и температурой выражается соотноше-
нием
С = р2’4^- 104-ехр(- ^29),
где С — концентрация азота, ат. %,
р — давление, ат.
Согласно исследованию кинетики
потери азота твердыми растворами
азота в тантале в области температур
1670—2170° С [984], теплота растворе-
ния азота в тантале равна
82 ккал/моль Na. Диффузия азота в
тантал изучена методом внутреннего
трения в [313].
Некоторые данные о взаимодей-
ствии тантала с азотом и воздухом
приведены в [972].
П. Чиотти [43] показал, что TaN
Рис. 78. Кристаллическая структура
TaN (по Брауэру и Цаппу):
при ВЫСОКИХ температурах, теряя азот, а — элементарная ячейка; б — ее верти-
гг хт тг г кальный разрез по АВ.
переходит в 1а21М. Как отмечалось в
[312], Ta2N в свою очередь распадается
с выделением азота. Это в интервале температур 1600—2380° С описы-
вается уравнением
р = 4,5.10» 'ехр (— [ат].
Таким образом, можно считать установленным образование в си-
стеме Та—N твердого раствора азота в тантале и двух нитридных фаз
Ta2N и TaN.
Физические свойства. Нитриды тантала обладают относительно
высоким удельным электросопротивлением, что свидетельствует об
энергетическом обособлении атомов тантала и азота в решетках нит-
ридов (табл. 68). Из рис. 79 видно, что резкие экстремумы отвечают
нитриду Ta2N.
Электропроводность до 25 вес. % N носит преимущественно дыроч-
ный характер, а при более высоких содержаниях азота — электронный,
о чем, в частности, свидетельствует отрицательный знак термо-э. д. с.
189
О сильной локализации валентных электронов у остовов атомов тан-
тала и азота говорят невысокие значения твердости, высокая темпера-
тура плавления TaN и низкие значения термо-э. д. с. По данным [289],
теплопроводность нитрида тантала решеточная, так же как и у полу-
проводникового нитрида хрома CrN, т. е. нитрид тантала TaN занимает
промежуточное положение между металлическими и полупроводнико-
выми соединениями, причем, по-видимому, он ближе к полупроводни-
кам, чем к металлам. Нитрид тантала TaN не обладает сверхпроводи-
мостью до температуры 1,20° К; нитрид Ta2N является сверхпровод-
ником при 9,5° К-
Научный и практический интерес представляют электрические ха-
рактеристики тонких пленок из нитрида тантала TaN.
Порошкообразный нитрид TaN имеет окраску от голубовато-серого-
до темно-серого.
Химические свойства. Нитрид тантала TaN весьма устойчив по от-
ношению к большей части минеральных кислот любых концентраций
как на холоду, так и при нагревании [276, 315]. Неустойчив он в разбав-
ленной H2SO4, а также в концентрированной при кипячении, легко раст-
воряется с гидролизом. Быстро растворяется в HNO3 + HF и H2SO4 +
+ Кг8О4. Неустойчив в водороде [256]. В работе [316] приведены данные
по устойчивости нитрида тантала против действия технических газов..
В целом химические свойства нитридов тантала близки к химическим
свойствам тантала, за исключением окисления, которое для тантала
начинается при 500—600° С, а для нитридов тантала — лишь слабо при
800° С.
Методы получения. Нитриды тантала получают нагреванием по-
рошка тантала в токе азота или аммиака. Хорн и Циглер [317] приго-
товляли очень чистые сплавы тантала с азотом обработкой порошка
тантала азотом при температуре 1100—1200° С, Шенберг [308] — азоти-
рованием тантала и гидрида тантала при 700—1100° С в течение от 1 ч
до 2 недель. Брауэр и Цапп [309] получали богатые азотом сплавы си-
стемы Та—N обработкой порошка тантала азотом в течение 6 ч при
1400° С. Кинетику процесса азотирования тантала изучал Гульбранзен
[179], данные которого приведены на рис. 80. Значение константы ско-
рости реакции изменяется в пределах 500—850° С от 9,6 • 10~15 до
2,84 • 10-12 г!смг-сек, энергия активации азотирования равна
39 ккал/моль. При более высоких температурах кинетика азотирования
исследовалась в [311, 312].
Оптимальными режимами получения нитридов тантала обработкой
порошка тантала азотом являются: для Ta2N температура 800—900° С,
для TaN— 1200° С при времени азотирования 1—2 ч [178]. Для полу-
чения нитрида TaN при 1100° С необходимо 5—6 ч азотирования.
Чиотти [43] исследовал условия приготовления нитрида тантала
азотированием металла аммиаком, при температуре 800—900° С и вы-
190
держке от 5 до 18 ч содержание азота в сплаве достигало всего 6,12%
и на рентгенограмме были обнаружены только слабые линии, относя-
щиеся к TaN. Сплав при нагревании в вакууме при 2000° С терял часть
азота, переходя в нитрид Ta2N. При дальнейшем нагревании в вакууме
на рентгенограммах появлялись линии тантала, что указывает на даль-
нейший распад нитрида.
Фридерих и Зиттиг [ПО] приготавливали нитрид тантала с содер-
жанием 6,9% N нагреванием гидрида тантала в азоте.
При нагревании смеси Та2О3 и
углерода в токе N2 и NH3 получить
чистый нитрид не удается, содержа-
ние нитрида в лучшем случае до-
стигает 10—15%, остальное — кар-
Рис. 79. Концентрационная зависимость электросопротивления и коэффициента Холла
сплавов тантала с азотом.
Рис. 80. Кинетические кривые азотирования тантала.
ты получаются при обработке порошка тантала азотом, содержащим
следы соединений углерода.
Сплавы тантала с азотом можно получить осаждением из газовой
фазы смеси TaCi5 + N2 + H2, однако вместе с TaN выделяется тантал,
восстанавливаемый из Та2О3 водородом. Поэтому лучше проводить оса-
ждение смеси TaCls + N2, но при высоких температурах порядка 2500—
2800° С.
Исследователи [274] указывают, что при разложении при 700—
800° С соединения (NH4)2TaF7 образуются препараты, состав которых
может быть выражен формулой Ta3N5(TaNli67), имеющие красновато-
коричневый цвет и растворимые в кипящей концентрированной H2SO4
и плавиковой кислоте.
Существование этого соединения недавно было подтверждено в.
19Г.
[980], где Ta3Ns получен при действии аммиака на пятиокись тан-
тала при 800—900° С
ЗТа2О5 -F 10NH3 = 2Ta3N6 + 15Н2О.
В этой же работе получены дополнительные характеристики
свойств соединения. Найдено, что оно окисляется на воздухе при 200° С,
в вакууме 10-3 мм рт. ст. при температурах выше 800° С не полностью
теряет азот, превращаясь в некоторую новую нитридную фазу черного
цвета. При дальнейшем нагреве выше 1100° С эта фаза переходит в
смесь нитридов TaN и TaaN. Плотность Ta^Ns равна 9,85 г/см?, электро-
проводность спрессованного порошка — ниже 104 ом-см-1, он является
диамагнитным. Кристаллическая структура пока не исследована доста-
точно однозначно, по предварительным данным TasN5 имеет тетраго-
нальную решетку с периодами а= 10,264; с = 3,893 А.
Образование подобного нитрида не согласуется с данными по си-
стеме тантал — азот, однако его существование вполне возможно так
как А. X. Брегеру удавалось получать аналогичным способом нитрид
титана TiNi,i6 (см. стр. 152). Такое соединение, очевидно, должно об-
ладать высокой долей ионной связи и атомы азота иметь высокий ста-
тистический вес стабильных ^//-конфигураций.
Для получения ультратонкого порошка нитрида тантала исполь-
зовано действие струи водорода при температуре 3000° С на хлористый
тантал [1023]. Содержание азота в таком нитриде составляет 6,5% (по
сравнению с расчетным содержанием 7,19% для TaN), примесь кисло-
рода около 1%, размер частиц — несколько сотых долей микрона.
Приготавливают изделия из нитридов тантала горячим прессова-
нием порошков нитридов при температуре 1800° С и давлении
100 кг) см1 в среде аргона. Полученные продукты имеют остаточную по-
ристость 1,5—2%, загрязнение углеродом графитовой прессформы — до
0,1%. Изделия из нитридов тантала приготавливают спеканием в ваку-
уме предварительно спрессованных заготовок из нитридов; реакцион-
ным спеканием брикетов, спрессованных из порошка тантала в токе
азота при 900° С в течение 8 ч для Ta2N и при 1200° С в течение 4 ч
для TaN. Полученные изделия имеют остаточную пористость 9—14%.
4. НИТРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ VI ГРУППЫ
Нитрид хрома. Растворимость азота в твердом хроме мала [321, 334].
В работе [323] показано, что растворимость азота в хроме составляет:
при 1100° С —0,04 вес.%; при 1200° С —0,09 вес.%, при 1300° С —
0,14 вес.% и при 1400° С — 0,26 вес.%.
Данные по растворимости азота в хроме в интервале температур
700—1320° С, определенные в работе [943], близки к данным, получен-
ным в [323].
192
Предельное поглощение азота жидким хромом составляет при
р = 1 ат около 4 вес. % [322, 323]. Температурная зависимость кон-
N2
станты растворения азота в хроме в интервале температур 1600—
1750° С имеет вид [322]:
1g К = _ 0,2782.*
Понижение общего состояния азота в хроме с повышением темпера-
туры связано с экзотермичностью обратимых реакций образования нит-
ридов хрома из жидкого хрома и азота [324]. Это отчетливо доказано
в [322], где установлено, что содержание азота в хроме снижается от
4,0842 вес. % при 1600° С до 3,5413 вес. % при 1750° С.
По данным [320, 325], в системе хром — азот образуются два нит-
рида CrNx и Cr2Nx. Нитрид Cr2N (p-фаза) имеет область гомогенности,
находящуюся по старым данным в пределах, от 11,3 до 11,9 вес. % N,
а по данным Эриксона [325] —от 9,3 до 11,9 вес. % N. Его структура
характеризуется неупорядоченным расположением атомов азота в гек-
сагональной плотнейшей упаковке решетки из атомов хрома, т. е. это
типичная фаза внедрения азота в решетку хрома.
Фаза CrN (у-фаза) имеет простую кубическую решетку типа NaCl.
В работах [343, 350] приведены результаты электронографического ис-
следования нитрида CrN.
Физические свойства. Нитрид хрома в порошкообразном состоянии
черного, a Cr2N — светло-серого цвета. Физические свойства этих нит-
ридов подробно исследованы в [326—328], где отмечено, что CrN —
полупроводник с шириной запрещенной зоны около 0,24 эв, с вы-
сокими электросопротивлением, коэффициентом Холла и термо-э. д. с.
(табл. 69). Температурная зависимость термо-э. д. с. и удельного элек-
тросопротивления CrN показана на рис. 81. В работах [326, 328, 329]
изучены магнитные свойства нитридов хрома, установлено, что они яв-
ляются парамагнетиками с восприимчивостью, несколько большей, чем
чистого хрома. В [329] отмечается, что наличие парамагнетизма у нит-
ридов хрома является указанием на присутствие у них резонирующих
ковалентных связей (по Полингу), так как при обычных ковалентных
связях они являлись бы диамагнетиками или слабомагнитными вещест-
вами. В работе [330] проведено рентгеноспектральное исследование
нитридов хрома в сопоставлении с другими соединениями хрома. На-
личие закономерного изменения интенсивности начального поглощения
в ряду Сг20з, CrN, СгВ2, а также уменьшение магнитной восприим-
чивости в этом ряду свидетельствует о возрастании доли ковалентной
связи наряду с наличием ионной связи, а также об уменьшении числа
* По данным [939], хром при 1600° С и давлении N2=l ат растворяет 6,5 вес.%
азота (в состоянии переохлажденной жидкости).
13—272
193
Таблица 69
Свойства нитридов хрома
Характеристика Cr2N CrN
Содержание азота, вес. % Область гомогенности, 11,87 21,7
ат. % 32—33,3 [401] Очень узкая
Кристаллическая структу- Гексагональная плотно- Кубическая гране-
ра Периоды решетки, кХ: упакованная центрированная
а 4,806—4,760 [401] 4,148 [401]
с 4,479—4,438 —
с/а Плотность, г/см3-. 0,928 —
расчетная 6,51 6,18
пикнометрическая — 5,8—6,1 [1]
Температура плавления, °C Теплота образования — При 1500°С разлагается
—А^298’ Ккал/МОЛЬ Температуры (°C), при ко- торых упругость при диссоциации состав- ляет: 7,6 [322] 23,5 [953]
1 мм рт. ст. — 719 [74]
10—3 мм рт. ст. Удельное электросопро- тивление (20° С), — 533 [74]
МКОМ-СМ Термический коэффициент электросопротивления, 79±3 [ 326 , 327] 640+40 [ 326,327]
град~1 +1,78 [326, 327] 3,75 [ 326 , 327]
Термо-э. д. с., мкв/град Постоянная Холла (20° С), —2,2±0,1 [326, 327] —92+6 [ 326 , 327]
RX 104, см3! к Эффективная концентра- ция носителей тока —0,7±0,1 [326, 327] —260±13 [326(327]
«ХЮ-23, см~3 Подвижность носителей 0,89 [326, 327] 0,002 [326,327]
тока, и, см3! сек Магнитная восприимчи- 0,83 [326, 327] 41,0 [326, 327]
5,7 [329] 18,0 [329]
вость XX 10s Теплопроводность, 5,6 [326, 327] 12,1 [326, 327]
кал!см-сек-град 0,519 [289] 0,0300** [289]
Микротвердость, кг/мм3 Коэффициент излучения (Х=655 ммк) при температуре 800— 1571±49 [289] 1093+93 [ 289]
1300°С 0,69 [340] 0,60—0,55 [340]
Модуль упругости, кг!мм3 32000 [1077] 33000 [1077]
• Магнитная восприимчивость хрома, по данным [329], равна 3,410
•• Для CrN0j926.
194
нескомпенсированных спинэлектронов на Sd-оболочке атомов хрома.
В соединениях Cr3C2, CrB, Cr7C3, Cr2N наблюдается большая, чем в вы-
шеуказанном ряду, начальная область К-спектров поглощения и суще-
ственно меньшая магнитная восприимчивость, что позволяет предполо-
жить наличие в них резонирующей ковалентной связи. В гибридизации
принимают участие почти все электронные Sd-орбиты с уменьшением
при этом числа свободных со-
стояний и со значительным
увеличением числа d-состоя-
ний с р-характером. В целом
у CrN — ионно-ковалентный
характер, а у Cr2N — кова-
лентно-металлический [329]. К
такому же общему выводу
можно прийти и с позиций
представлений об образова-
нии атомами в соединении ста-
бильных электронных конфи-
гураций с минимумом свобод-
ной энергии. При образовании
нитрида хрома можно пред-
положить приобретение ато-
мом хрома стабильной d5s2-
конфигурации в результате
приема одного электрона ато-
ма азота, имеющего при этом
конфигурацию локализован-
ных «р3-электронов. Подобная
комбинация конфигураций вы-
зывает кубическую структуру
Рис. 81. Температурная зависимость тер-
мо-э. д. с. и удельного электросопротив-
ления нитридов хрома:
;-“GrN; 2—₽GrN; s~₽CrN (15,5% N): 4~₽Cr2N.
5-“Сг2 N;’
этого нитрида, а также появление энергетического разрыва между
состояниями атомов хрома и азота со следующими отсюда полупровод-
никовыми свойствами. Образование неспаренных электронов на d-ор-
бите обусловливает также повышенный парамагнетизм этого нитрида.
В нитриде Cr2N следует полагать возможным образование высокого
статистического веса ^'^-конфигураций атомов хрома (такие конфи-
гурации в предельном случае должны обусловливать антиферромагне-
тизм *) с образованием атомами азота «реконфигурации и электрона,
который преимущественно находится в нелокализованном состоянии и
обусловливает металлические свойства этого нитрида. Естественно, что,
при этом понижается парамагнетизм нитрида по сравнению с CrN и
значение его приближается, как и следовало ожидать, к парамагне-
* Об антиферромагнетизме нитридов хрома см. также [331].
13*
195
тизму хрома. Большая локализация валентных электронов в стабиль-
ные конфигурации в CrN вызывает большую теплоту его образования
по сравнению с Cr2N, а энергетическая разобщенность атомов — лег-
кую диссоциацию на элементы (при 1500° С). На высокую локализацию
валентных электронов в CrN указывают также большое значение эф-
фекта Холла и малая концентрация эффективных носителей тока при
их высокой подвижности за счет слабого рассеивания на стабилизи-
рованных остовах атомов.
Химические свойства. Подробное исследование химической стой-
кости нитридов хрома проведено в [276, 315, 341], из которых следует,
что нитрид CrN значительно устойчивее, чем Cr2N при действии мине-
ральных кислот. Водными растворами щелочей оба нитрида почти не
разлагаются. Следует отметить [276], что нитрид CrN является наряду
с нитридом тантала TaN одним из самых устойчивых среди нитридов
переходных металлов, а нитрид Cr2N — одним из наименее устойчивых
наряду с нитридом циркония ZrN. Оба нитрида хрома наиболее полно
разлагаются хлорной кислотой, a Cr2N, кроме того,— разбавленной и
концентрированной НС1, а также разбавленной H2SO4 (1:4). Нитрид
хрома CrN не реагирует с водородом, сгорает в кислороде до Сг2О3.
Методы получения. Сплавы хрома с азотом приготавливают азоти-
рованием порошка хрома аммиаком при температуре 800—1400° С
[332]. Нитрид хрома CrN получают также обработкой азотом при 600—
900° С тонкого пирофорного порошка хрома, приготовленного разложе-
нием амальгамы [55]. Можно получать нитриды и непосредственной
возгонкой амальгамы в азоте.
Авторы [953] исследовали равновесие в системе хром—азот в зави-
симости от давления и температуры при азотировании чешуйчатого
электролитического хрома чистотой 99,40% (основная примесь кисло-
род 0,588%). Температуру варьировали в пределах 1100—1400° С, дав-
ление азота 0,5—45 мм рт. ст. Во всех случаях были получены двух-
фазные образцы, состоящие из нитрида хрома Cr2N и твердого раст-
вора азота в хроме. Измерение давления азота над двухфазным об-
разцом приблизительно эквимолярного состава показало, что давление
при 1000° С составляет 0,5; при 1100° С—1,5; при 1200° С — 6,6; при
1300° С — 19; при 1400° С — 43,5 мм рт. ст.
Систематическое исследование азотирования порошка хрома для
получения нитридов было проведено в работе [178]. Показано, что мак-
симальное поглощение азота хромом происходит в интервале темпера-
тур 800—1000° С. Температура 900—950° С является оптимальной для
Получения фазы CrN. В этой температурной области реакция между
порошкообразным хромом и азотом происходит настолько активно, что
скорость подачи азота в печь приходится увеличивать в 8—10 раз.
Выше 1000° С фаза CrN неустойчива, превращается в Cr2N. Для полу-
чения однофазного нитрида Cr2N необходимо после 2—4 ч выдержки
196
при 1200—1300° С резко охлаждать (закаливать) полученный продукт
вместе с реактором, так как при медленном охлаждении происходит
значительное поглощение азота с образованием смеси фаз CrN и Cr2N.
Чем медленнее происходит охлаждение в температурном интервале
1000—800° С, тем больше CrN образуется в полученном продукте. Ре-
акцию между компактным хромом и азотом изучали в [334] в области
температур 1000—1300° С. Установлено, что кинетические кривые по-
глощения азота носят параболический характер. Энергия активации
диффузии определена 23,3 ккал!моль, что хорошо совпадает с данными
[335] и [336], в которых найдены значения энергии активации диффузии
соответственно 25,7 и 27—28 ккал/моль. Максимальная скорость погло-
щения азота наблюдается при 1100° С, константа реакции равна
2,4- 10“9 (г/сыРу/сек.
Реакционная диффузия азота в хром начинается при температуре
700° С, возрастает сначала медленно, а при 1030° С — весьма быстро.
Это, вероятно, связано с образованием выше 1030° С только фазы Cr2N
(ниже образуется Cr2N и снаружи образцов — CrN), также отмечается,
что обратной диффузии хрома не происходит.
Кисслинг и Лиу [320] приготовляли нитриды хрома обработкой бо-
рида хрома аммиаком: при 735° С образуется CrN, между 800 и
1100° С — смесь CrN и Cr2N, а при 1180° С — Cr2N.
Ольсон с сотрудниками [337] получал пленки нитрида хрома, про-
пуская аммиак над хромом, обезгаженным в вакууме (10-4 мм рт. ст.).
Исследование пленок, полученных при десятиминутном азотировании
при 950—1000° С, показало, что они состоят преимущественно из CrN,
при более высоких температурах — из смеси CrN и Cr2N и при самых
высоких температурах — из Cr2N. Эти данные хорошо совпадают с ре-
зультатами работ [178, 320]. Хром в тонкодисперсном состоянии погло-
щает азот при 800° С [338], при повышении давления азота поглощение
несколько увеличивается [339].
Известны [332] способы получения нитридов хрома нагреванием
СгС1з или СгО2С12 с аммиаком или с нитридами магния, лития и др.,
однако все эти препараты вряд ли могут считаться чистыми.
Исследователи [274] указывают на возможность получения нитрида
хрома CrN разложением (NH4)3CrF6, которое начинается при 300° С
и проходит до конца при 600° С.
Приготовление изделий из нитридов хрома наиболее эффективно
проводить реакционным спеканием заготовок, спрессованных из порош-
ка хрома в токе азота [232]. При этом оптимальной температурой спе-
кания заготовки, спрессованной под давлением 1,8 т]см2 с образованием
изделия из Cr2N, является 950° С. Полученные изделия имеют остаточ-
ную пористость 11%. Сокращение пористости спеченного изделия по
сравнению со спрессованной заготовкой происходит в результате уве-
личения удельного объема при образовании нитридов из хрома, это
197
увеличение составляет 40—50% Аналогичный способ получения изде-
лий из Cr2N описан в работе [234]. Рекомендуется сначала спекать за-
готовки из порошка хрома в аргоне при 1300—1400° С в течение 16 ч,
затем азотировать их при 1300° С. Однако результаты получаются ху-
же, остаточная пористость выше — 30—35%, вследствие инактивации
хрома при длительном спекании в аргоне.
Нитриды молибдена. Впервые нитриды молибдена получил Ур-
лауб в 1857 г. и приписал им формулы M05N3 и M05N4 [345]. Розенгайн
молибдена при реакциях МоОз, M0CI3
или M0CI5 с аммиаком и приписали
формулу Mo3N2.
Систематическое исследование ни-
тридов молибдена методом рентгенов-
ского анализа выполнено Хэггом [347],
который установил в системе Мо—N
наличие трех нитридных фаз: |3-фазы
M05N2 (МоЫоло), которой раньше при-
писывали формулу M03N, с тетраго-
нальной искаженной решеткой метал-
лических атомов, у-фазы Mo2N с куби-
ческой, гранецентрированной решеткой
и 6-фазы MoN с простой гексагональ-
ной решеткой, аналогичной решетке
никельарсенида. [1-Фаза устойчива при
температурах выше 600° С. Область
гомогенности фазы Mo2N при вы-
в направлении повышенных содержа-
ний молибдена (рис. 82). Отсюда следует, что продукты, описанные
Урлаубом, Розенгайном и Брауном, представляли собой либо смеси
нитридов молибдена, либо дефектные нитридные фазы.
Шенберг [348] подтвердили данные Хэгга, несколько подробно изу-
чив структуру 6-фазы (MoN).
Дальнейшее исследование структуры и составов нитридов молиб-
дена было проведено в работах 3. Г. Пинскера с сотрудниками
[349—352].
Электронографическое изучение структуры у-фазы (Mo2N), имею-
щей кубическую решетку, показало, что фаза склонна к образованию
структур вычитания с дефектом атомов молибдена. При исследовании
гексагонального нитрида б-MoN было обнаружено, что на самом деле
имеются два нитрида молибдена этого состава со слегка отличными пе-
риодами решетки (б'- и б^-фазы), б'-фаза отличается некоторым смеще-
нием атомов молибдена из частных положений [349, 351]. При недо-
статке азота образуется пластинчатая текстура MoN (б'-фаза), а при
большем содержании азота — б"-фаза. Внедрение большего количества
!9в
и Браун [346] получали нитрид
400
600
О 10 20 30 40 Nfom%
6(MoN)
!(МогЫ)
маМФ
MoN(-6)
10 н,0ес.7о
Рис. 82. Диаграмма состояния систе-
мы молибден—азот.
соких температурах смещается
о
азота не увеличивает, а уменьшает периоды решетки (для б'-фазы а =
= 5,72; с = 5,60, для б"-фазы а = 5,665; с = 5,520 А), что объясняется
следующими обстоятельствами. Решетка б-фазы состоит из пакетов,
каждый из которых образован тремя атомными слоями (MoNMo), т. е.
эта решетка слоистого типа. Внедрение добавочного азота в призмы
между пакетами приводит к уплотнению упаковки, т. е. к «стягиванию»
атомов молибдена. 0-Фаза является тетрагональной высокотемператур-
ной модификацией кубической у-фазы с точкой превращения 650° С
[355].
Растворимость азота в молибдене очень мала, область a-фазы узка,
можно считать, что твердый молибден практически не растворяет азота
[353, 354]. Уже при содержании в плавленом молибдене 0,0008% азота
наблюдается выделение фазы Mo2N [356].
Результаты исследования адсорбции аргона, кислорода, азота и
окиси углерода порошкообразным молибденом (чистота 99,9%, удель-
ная поверхность ~0,5 мг/г) при —195 и —183° С показали, что адсорб-
ция азота мала и близка к адсорбции аргона (близкие значения ем-
кости мономолекулярных слоев) [357].
Физические свойства. Основные физические свойства нитридов мо-
либдена приведены в табл. 70. Фаза Mo2N имеет, по-видимому, метал-
лический характер [289]. В отличие от нитридов других переходных
металлов Mo2N имеет положительный знак коэффициента термо-э. д. с.,
что свидетельствует о дырочном характере проводимости. На это же
указывает положительный знак постоянной Холла. Весьма низкая мик-
ротвердость этого нитрида (630 кг/жж2) является следствием сильной
асимметрии распределения электронной плотности либо дефектной
структуры, на которую указывалось выше. Нитрид Mo2N термически
неустойчив и легко переходит в низший нитрид M03N.
Физические свойства остальных фаз практически не исследованы.
Нитриды Mo2N и MoN являются сверхпроводниками с высокими зна-
чениями точек перехода [275, 304]. Температура перехода в сверхпро-
водимое состояние для Mo2N ниже, чем для MoN (5,0 и 12,0° К соот-
ветственно). Можно предположить, что при образовании MoN часть не-
локализованных электронов атомов молибдена передается атомам
азота с образованием у последних повышенного статистического веса
стабильных з2р6-конфигураций, что обеспечивает создание «энергети-
ческого канала» из стабильных конфигураций атомов молибдена (<75)
и азота, малое рассеяние электронов в котором приводит к высокому
значению Тк. В случае же нитрида Mo2N происходит принципиально
то же, однако высокая концентрация при этом нелокализованных элек-
тронов от двух атомов молибдена не может быть существенно умень-
шена за счет захвата электронов атомами азота, что приводит к умень-
шению значения Тк. Возможно также, что в этом случае, судя по куби-
ческой симметрии Mo2N, атомы азота образуют ^-конфигурации с ос-
199
Таблица 70
Свойства нитридов молибдена
Характеристика Mo3N Mo2N MoN
Содержание азота, вес. % 5,4 6,75 12,73
Область гомогенности, ат. % Узкая [348] ~32—33 [348] Узкая [348]
Кристаллическая структура Периоды решетки, кХ: Гранецентриро- ванная тетра- гональная Кубическая гра- нецентриро- ванная Гексагональная
а 4,180 [347] 4,155—1,160 [347] 5,725 J348]
с 4,016 — 5,608
с/а Плотность, г! см?; 0,962 — 0,980
расчетная — — 9,18 [956]
пикнометрическая —- 8,04 [347] 8,60 [347]
Температура плавления, °C Разлагается Разлагается при 895° С Разлагается
Теплоемкость (298° К), кал)моль-град — 8,98 [360] —‘
Теплота образования (—ЬН(]^&),к>мл; моль Температуры (°C), при которых упру- гость диссоциации составляет: — 16,6±0,5 [47] —
1 мм рт. ст. — 329 [74] —
10~3 мм рт. ст. Удельное электросопротивление, — 209 —
34КОЛ1 • СМ Коэффициент термо-э. д. с., мкв!град — 19,8±7 [289] 2,18±0,5 [289] —
Коэффициент Холла, см3/к — 2,83±1,2 [289] —
Теплопроводность, кал]см • сек-град — 0,0427± 0,007 —
Микротвердость, кг]мм2 Температура прихода к сверхпроводи- — 630 ±86 [289] —
мости, °К — 5,0 [275 , 304] 12,0 [275, 304]
вобождением одного электрона на атом, что также должно приводить
к понижению Тк для Mo2N по сравнению с MoN.
Химические свойства. При нагревании на воздухе нитриды молиб-
дена окисляются до МоО3. Имеются указания об их самопроизвольном
возгорании на воздухе [1].
При нагревании в аргоне p-нитрид не изменяется до 1000° С; при
нагревании в вакууме теряет азот, перехода в a-фазу, т. е. практи-
чески чистый молибден [359]. MoN в вакууме разлагается медленнее,
чем WN.
Методы получения. При пропускании аммиака над молибденом
при температуре 850° С в сплав с азотом переходит лишь небольшая
доля металла [358]. Хэгг [347] приготовлял сплавы молибдена с азотом
200
пропусканием аммиака над свежевосстановленным молибденовым по-
рошком при 400—720° С в течение 4—24 ч. При этом образовывались
сплавы, содержащие от 0,77 до 7,15% N. Выше 725° С эти сплавы дис-
социируют. Для получения более высоких содержаний азота до 11,95%
необходимо длительное азотирование (до 120 ч) при 400° С. Шенберг
[348] приготавливал нитриды молибдена обработкой порошка молиб-
дена азотом при температуре 800° С в течение нескольких часов. Мак-
симальное содержание азота составляло при этом 49,6 ат.%.
Урлауб [345] получал нитриды молибдена пропусканием аммиака
над МоС15 или МоО3. Аналогичным путем получали нитриды молибдена
Розенгайн и Браун [346].
В работе [359] подробно исследовано образование нитридов молиб-
дена одновременным восстановлением и азотированием трехокиси мо-
либдена и молибдата аммония смесью 70 об.% N2 и 30 об.% Н2. В ре-
зультате при температуре 750—800° С получается p-нитрид. Показано,
что образующаяся на частицах молибдена пленка нитрида, плохо про-
ницаемая для азота, гидрируется с образованием различных соеди-
нений типа аминов, что вызывает разрыхление пленки и обеспечи-
вает возможность продолжения процесса азотирования. При азотиро-
вании порошка молибдена аммиаком образуются две нитридные фа-
зы р и у.
По данным [ 1781 ,.дпм-абработке порошка молибдена азотом полу-
чение нитридов затруднено. Нитрид M.o2N образуется при обра-
ботке порошкообразного молибдена аммиаком при 700° С в течение 4 ч,
причем роль водорода сводится, кроме указанной выше, к восстанов-
лению молибдена, что активизирует дальнейшую реакцию с азотом.
Нитрид молибдена Mo2N может быть также получен путем прокали-
вания молибдата аммония в среде аммиака при температуре 1100° С
в течение 1 ч.
Изделия из нитрида молибдена Mo2N приготавливают реакцион-
ным спеканием — обработкой предварительно спрессованных под давле-
нием 1,8 т/сж2 заготовок из порошка молибдена азотом при температуре
700° С в течение 8 ч [232]. Пористость полученных таким образом из-
делий составляет около 20%.
Нитриды вольфрама. Система вольфрам—азот впервые была
исследована Хэггом [347], который обнаружил наличие а-фазы — прак-
тически чистого вольфрама, и P-фазы, которая имеет кубическую гра-
нецентрированную решетку и отвечает по составу формуле W2N. Не-
растворимость азота в вольфраме была обнаружена еще И. И. Жуко-
вым [338]. р-Фаза аналогична у-фазе системы Мо—N. Кисслинг и Лиу
[320] обнаружили вторую модификацию фазы W2N, которая образуется
при азотировании вольфрама при температуре 825—875° С и назвали
ее не совсем удачно у-фазой. Эта фаза имеет плотную кубическую гра-
нецентрированную решетку. По-видимому, у-фаза идентична р-фазе.
201
Шенберг [348] обнаружил фазу WN (6-фазу) с содержанием около
50 ат. % азота, имеющую гексагональную решетку. Кисслинг и Лиу
подтвердили существование W2N, однако кубическая у-фаза WN ими
не была обнаружена. Кисслинг и Петерсон [361] показали, что у-фаза
является на самом деле оксинитридом, образующимся при действии на
вольфрам аммиака, содержащего примеси кислорода, и паров воды.
К такому же выводу пришли и авторы [362].
Авторы [988] обнаружили кубический нитрид вольфрама состава
W3N4 (y'-WsN.}) с периодом решетки а=4,122 А, подробно рассмотрена
его структура. Однако возможно, что эта фаза является оксинитридной,
так как в ней содержится кислород (состав W3[No,950o,o5]4, т. е. соотно-
шение между атомами азота и кислорода 19 : 1).
Дальнейшее исследование фазового состава системы W—N прово-
дилось электронографическим методом 3. Г. Пинскером с сотрудни-
ками [350, 363—368]. Ими была получена в тонких пленках р-фаза
(W2N), подтверждена ее кубическая гранецентрированная структура.
По данным [347], период решетки этой фазы а=4,118 А при содержа-
нии ~18,2 ат. % N, что является нижней границей области гомогенности,
верхняя граница соответствует стехиометрическому составу W2N
(33,3 aT.%N). В решетке кубического нитрида вольфрама (р-фазы)
атомы вольфрама образуют не дефектную гранецентрированную ре-
шетку, а атомы азота статистически распределены в центре куба и на
серединах ребер [364, 366]. Дальнейшими электронографическими ис-
следованиями [366—368] установлено существование в системе W—N
большой серии гексагональных и ромбоэдрических 6-фаз, схема рас-
пределения которых на концентрационной диаграмме приведена на
рис. 83. Особенностью структуры 6-фаз (табл. 71) является образова-
ние атомами вольфрама одно-, двух- или трехэтажных пакетов, кото-
рые образуют гексагональную или ромбоэдрическую последователь-
ности. Атомы азота располагаются либо внутри пакетов, либо между
ними. Расстояния между атомами вольфрама невелики, а расстояния
между атомами вольфрама и азота по величине могут быть подразде-
лены на два типа, что указывает на разницу в природе сил взаимодей-
ствия между атомами компонентов [366]. Типичный элемент структуры
одной из 6-фаз показан на рис. 84. Имеется сходство между структу-
рами 6-нитридов вольфрама и боридами переходных металлов как по
расположению атомов, так и по величине межатомных расстояний. Так,
атомы азота в нитриде 6VR образуют слегка гофрированные слои по-
добно тем, какие наблюдаются в боридах со структурами типа W2B5.
Это сходство представляет большой интерес с точки зрения особеннос-
тей электронного строения атомов бора и азота и его изменения при
образовании соответствующих фаз. Для атома азота возможна пере-
стройка конфигурации валентных электронов по схеме: s2p3-+sp4-+sp3 +
202
, Таблица 71
Структурные характеристики б-фаз системы W—N
Фаза* Приб- лижен- ная формула Простран- ственная группа Периоды решетки, A Межатомные расстояния, A Расчетная плотность, г/см%
a c c/a W—N N-N
Р63- —mmc— 2,885 15,30—15,46 5,3—5,35 Wj—N 2,16 2,60—2,62 1,36—15,7
-wI>35N — — -— Wji—N 2,26 — —
w2n РЗ—С'^ 2,89 22,85 7,9 Wj—N 2,88 W„—N 3,09 WnI-N 2,91 2,89 12,0
ый °н Wo,64N PQ:t!mmc— ~Dlh 2,87 11,00 3,81 Wj-N 2,14 W—N 2,16 Wj—N 2,16 2,75 и,о
AIV Д'/ W0,6N 2,89 10,8 3,73 1,67 10,63
ftv w0,5n 2,89 16,4 5,67 W—N 2,915 3,03 3,04 1,71 9,0
wM7n D3d 2,89 23,35 8,07 Wj—N 2,13 Wn-N 2,12 3,12 13,6
—гексагональная решетка, К—ромбоэдрическая решетка.
4-р с освобождением одного слабосвязанного электрона. Для атома
бора по энергетическим соображениям за счет 8->р-перехода происхо-
дит преобразование конфигурации валентных 52р-электронов в энерге-
тически более устойчивую sp2, которая, в свою очередь, стремится к
еще большей энергетической стабилизации до sp3 по принципиальной
схеме: 2 sp2-^sp3 + 2p. Таким образом, превращения конфигураций ва-
лентных электронов в обоих случаях аналогичны с той лишь разницей,
что в случае бора образуется большое число легкоподвижных элек-
тронов.
203
Рис. 83. Фазовая диаграмма состояния системы воль-
фрам—азот.
[НО]
Рис. 84. Элемент структуры б-фазы системы W—N.
Физические и химиче-
ские свойства. Свойства
нитридов вольфрама
представлены в табл. 72.
Высший нитрид вольф-
рама WN — порошок
коричневого цвета,
W2N — черного цвета.
При 600° С WN разлага-
ется в вакууме с образо-
ванием 3-фазы по реак-
ции: 2WN=W2N + 1,2N2.
Разложение WN проис-
ходит гораздо быстрее,
чем MoN. Нитриды
вольфрама устойчивы
против действия азотной,
разбавленной серной кис-
лоты и раствора соды
[369, 370]. Горячая кон-
центрированная серная
кислота разлагает нит-
риды с образованием
WO3 и выделением амми-
ака. Так же действует
царская водка. При
сплавлении с Na2CO3 об-
разуется вольфрамат
натрия и аммиак. При
нагревании на воздухе
нитриды легко окисляют-
ся до WO3. Нитрид W2N
не обнаруживает сверх-
проводимости.
Методы получения.
Получить соединения
вольфрама с азотом при
обработке вольфрама ам-
миаком весьма трудно, а
по мнению некоторых
исследователей [358, 371]
вообще невозможно. Од-
нако Лаффитом и Гран-
дадармом [372] было ус-
204
Свойства нитридов вольфрама
Таблица 72
Характеристика WjN WN
Содержание азота, вес. % 3,67 7,09
Цвет Черный Коричневый
Кристаллическая структура Периоды решетки, д: Кубическая гране- центрированная Гексагональная
а 4,118 [347]* 2,893 [348]
с — 2,826
с/а Плотность, г/смя: — 0,977
расчетная — 10,52
пикнометрическая 12,2 12,08—12,12 [348]
Температура плавления, °C Упругость диссоциации в ва- кууме, мм рт. ст.: Разлагается при 600 °C
344°С 1 [74] —
296°С 10~1 —
256°С 10~2 —
221°С Теплота образования—ДЯ°298, 10-3 —
ккал/моль Т чка перехода к сверхпрово- 17,2±3,0 [47] —
димости, °К <1,28 [275, 304] —
Коэффициент преломления — 5±0,5 [370]**
* Для кубической у-фразы <з=4,122—4,133 А
** Прозрачен в тонких слоях.
тановлено, что аммиак реагирует с вольфрамом при температуре уже
140° С, но при 200° С сплав снова разлагается на азот и вольфрам.
При низких температурах (от 20 до 80° С) и сравнительно боль-
ших величинах давлений (от К4 до 300 мм) превалирует адсорбция
аммиака порошком вольфрама, причем адсорбционный слой приблизи-
тельно мономолекулярен. При высоких температурах между 250 и 750° С
происходит сначала образование имида (от 250 до 360° С), а затем
нитрида W2N.
Механизм реакции порошка вольфрама с аммиаком изучали Мит-
тиас и Франкенбергер [374], которые установили, что при 250—360° С
образуется в качестве промежуточного соединения имид вольфрама
(WsNeHg). Хэггу [347] действием аммиака на вольфрам при 700—
800° С в течение 48 ч удалось получить продукты с содержанием только
1,67% N. Однако Шенберг [348] сообщает о приготовлении им нитридов
вольфрама с содержанием до 41,7 ат.% N путем обработки вольфра-
205
мового порошка сухим аммиаком в течение нескольких часов при
800° С.
По данным, приведенным в справочнике Гмелина, нитрид вольфра-
ма можно получить действием азота на вольфрам уже при обычном
давлении, при 550—600° С, но в присутствии катализаторов. Без катали-
заторов азот слабо действует на вольфрам практически до 900° С. Ни-
трид образуется только на активных местах поверхности, покрывая
около 20% ее площади мономолекулярным слоем. Нитрид вольфрама,
которому приписывали в работах [332, 373] формулу WN2 с 13,2%
азота, образуется при очень высоких температурах порядка 2500° С.
3. Г. Пинскер с сотрудниками для электронографического иссле-
дования приготовлял пленки нитридов вольфрама азотированием
вольфрамовых пленок (нанесенных испарением на скол кристалла ка-
менной соли) с помощью диссоциированного аммиака с достаточно
высокой скоростью последнего. При медленном прохождении аммиака
наблюдается неполное азотирование, а также образование неупорядо-
ченных структур. Температура азотирования в этих работах составляла
700° С и выше [364].
Исследование азотирования вольфрамового порошка проведено
также в [362]. Показано, что нитриды образуются только при исполь-
зовании очень тонкого порошка вольфрама и предварительного пропус-
кания водорода для восстановления вольфрама с последующим азоти-
рованием в токе аммиака. Содержание азота быстро повышается при
температурах от 350 до 470° С, достигая состава нитрида WN. При тем-
пературе 400° С образуется нитрид W2N. При обработке вольфрамата
аммония аммиаком образуется при 560° С нитрид W2N [362]. Восстанов-
ление вольфрамового ангидрида аммиаком приводит к образованию
оксинитрида.
Напротив, по данным, полученным в нашей лаборатории Т. С. Вер-
хоглядовой, восстановление WO3 аммиаком приводит к образованию
при 300° С нитрида W2N, а при 600° С — нитрида WN, при 400° С об-
разуются смеси этих фаз. Обработка аммиаком вольфрамового порошка
нагревом до 500° С и при выдержке при этой температуре в 30—60 мин
приводит к образованию фазы W2N, а при нагреве до 1100° С — к обра-
зованию смеси фаз W2N и WN.
5. НИТРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ VII группы
Нитриды марганца. Первые работы по получению и исследованию нит-
ридов марганца были проведены Прелингером [385] и Жуковым [338,
386]. Система Мп—N рентгенографически впервые исследована Хэггом
[387].
Растворимость азота в а-Мп составляет всего 0,156%, период ре-
шетки при этом увеличивается с 8,894 до 8,897 кХ. Растворимость азота
206
в р-Mn несколько выше, период решетки (а) увеличен с 6,289 до
6,305 кХ. Растворимость азота в жидком марганце при р = 1 ат вы-
^2
ражается уравнением [388]
lgK = lgCN(%) = -^-1,457,
с изменением при этом свободной энергии AF°= —13780-]-6,65 Т.
По данным Хэгга, в системе существует пять нитридных фаз. 6-Фа-
за содержит около 2% N, существует только при высоких темпера-
турах, между 400 и 600° С распадается на a-твердый раствор и
е-фазу.
е-Фаза при низких температурах (до 500° С) имеет, по данным
Хэгга, узкую область гомогенности — от 6 до 6,5% N, при высоких
температурах эта область расширяется в сторону сплавов, бедных
азотом от 3—4 до 6,5% N. е-Фазу можно рассматривать как Mn4N
(5,99% N) с кубической гранецентрированной решеткой атомов Мп и
упорядоченным расположением (аналогична у'-фазе в системе Fe—
N(Fe4N).
g-Фаза (MnaN—MngNa) имеет область гомогенности 9,2—11,9%,
причем атомы марганца образуют гексагональную решетку с плотной
упаковкой, а атомы азота распределены статистически в междоузлиях
этой решетки. При увеличении содержания азота внутри области гомо-
генности происходит рост периодов решетки.
т]-Фаза (Mn3N2), содержащая минимально 13,5%N, а максималь-
но— 14—15% N, имеет тетрагональную гранецентрированную решетку.
Атомы азота расположены в ней упорядоченно.
В дальнейшей работе [389] были уточнены температурные области
существования фаз и их области гомогенности. Было показано, что в
поле е-фазы необходимо выделить область новой фазы е', лежащей
между 9,8 и 15,0 ат. % N, обладающей парамагнитными свойствами. По
этим же данным 6-фазу следует рассматривать как стабилизированную
азотом тетрагональную гранецентрированную фазу у-Мп, а g-фазу как
стабилизированную азотом кубическую гранецентрированную фазу
у-Mn. На рис. 85 показана схематическая диаграмма состояния системы
марганец — азот, построенная Хэггом, с внесением в нее уточнений по
данным работы [389]. Исследователи [390] подтвердили существование
фйз Mn4N (е-фаза), MnsN2 (g-фаза), Mn3N2 (ц-фаза). Обнаружена но-
вая фаза Mn6Ns (О’-фаза), наиболее богатая азотом и кристаллизиру-
ющаяся в тетрагональной гранецентрированной решетке, т. е. такой же,
как и Mn3N2, отличаясь от последней беспорядочным расположением
атомов в решетке. •О-Фаза стабильна до температуры 580° С включи-
тельно. Расположение фаз на диаграмме Мп—N может быть пред-
ставлено схемой рис. 85, а. Некоторые данные о сплавах системы Мп—N
приведены также в работах [391, 392].
207
Физические и химические свойства. Свойства нитридов марганца
исследованы мало; имеющиеся данные представлены в табл. 73.
Наиболее полно изучены магнитные свойства нитридов марганца
Рис. 85. Система марганец—азот:
а — схема фазовых полей, б —диаграмма состояния.
g-фазы (MnsNa—Ma2N). При изменении содержания азота в Mn4N, т. е.
при увеличении числа дефектов в подрешетке азота, происходит умень-
шение магнитного насыщения, магнитного момента и рост точки Кюри
(табл. 74).
В работе [392] отмечается, что магнитные свойства нитридов мар-
ганца зависят только от состава, а не от особенностей условий полу-
чения.
Нитриды марганца в химическом отношении не являются стой-
кими, при нагревании легко отдают азот. При нагревании нитридов
Mn2N и Mn3N2 с водородом образуется аммиак. Аммиак выделяется
также при взаимодействии Mn3N2 с водными растворами NH4OH [6].
208
Таблица 73
Свойства нитридов марганца
Характерис- тика б-фаза 8-фаза (Mn4N) 8'-фаза Е-фаза Mn6N2— —Mn2N Ч-фаза (MnsNs) 0-фаза (MneN,)
Содержание азота, ат. % 400°С 19,5—20,0 28,4—34,7 45,7—47,9
600°С [385] 16,7—20,7 [389] 27,5—34,7 [390] 46,5 (500°С)
800°С 5,3—9,1 15,6—20,7 — 24,4—? —
1000°С [389] 3,8—9,8 15,0—20,7 9,8—15,0 24,4—? 38,0—44,6 —
Кристалла- Тетраго- Кубическая Кубическая Гексагональ- [389] Тетрагональ- Тетрагональ-
ческая нальная гранецент- гранецент- мая плот- кая гране- ная гране-
структура гране- рированная рированная ноупако- центриро- центриро-
Периоды ре- шетки, кХ: а центри- рован- ная 3,764 3,860 [389] 3,800 [389] ванная 2,773—2,825 ванная 4,20 [387] ванная 4,214 А
С [389] 3,729 . [387] 4,520—4,528 4,03 [390] 4,148 А
с] а 0,99 — — 1,630—1,601 0,97 0,984
Плотность, — 6,76 — 6,2—6,6 6,6
г/см3 Теплота об- 30,3±0,4 48,2±0,5 45,8
разования ~Д#298,5’ ккал/моль Энтроп ИЯ, кал/градмоль Точка Кюри, [394] [394] 45,9 [47] [112] 35,8 [112]
485—513 475 [389]
°C Теплота его- [389] 411,4—0,2 . 504,0—0,4
рання, ккал! моль Эффективный [394] 1,2 [140] [394] 3,94 [140]
магнитный момент, магнето- нов Бора
14-272
209
г
Т а б л и ц а 74 Методы получения.
Магнитные свойства дефектной по азоту фазы Сплавы марганца с азотом
Mn4M I393J легко получают непосред-
С5 ственным азотированием
©S5 s’ o' S S3 “8 ®3 Я П марганца азотом или ам-
X Я £ £ а «5. т # ч . 2 О . д а> t; ь Э миаком при температуре
л ст о*4 । О S3 S3 X я § к д В 4) 15 3- Йбо sgs 800—1200° С [332, 338, 386,
5g U я Й иЙ - & о с а г4 о <2 s 390, 392, 395] или азотиро-
ванием в струе аммиака
15,23 0,2819 3,810 505 2,45 0,101 амальгамы марганца, обра-
16,20 17,40 18,61 0,2266 0,1568 0,1500 3,820 3,830 3,834 513 510 509 6,58 13,50 14,26 0,272 0,560 0,592 зуемой на ртутном катоде электролизом МпС1г, при
19,25 0,0852 3,844 500 19,95 0,828 накаливании амальгамы в
19,87 0,0099 3,856 494 24,73 1,032 среде сухого азота получа-
20,00* 0,0000 3,860 483 28,55 1,192 ется нитрид M113N2 [6, 385].
Авторы [390] нитриды
* Экстраполированные значения. марганца также приготов-
ляли обработкой амальгамы
MnaHgs (которая получалась электролизом на ртутном катоде сульфа-
та марганца) аммиаком, азотом и смесями аммиака и водорода.
В табл. 75 приведены оптимальные режимы такого процесса.
Таблица 75
Оптимальные режимы получения нитридов марганца азотированием
амальгамы
Фаза Состав газовой среда, об. % Темпера- тура, °C Время, ч
NH3 N, Н2
е (Mn4N) 0 100 0 400 10
£ (Mn5N2-Mn2N) 0—2 0—100 98—0 400—500 10—15
т) (Mn3N2) 30—50 0 50—70 400 3—6
& (MneN5) 100 0 0 400—550 1—3
Нитриды технеция. В работе [384] нитрид технеция был получен
нагреванием NH4TcO4 в токе аммиака при температуре 700—1100° С.
При 900—1100° С обнаружена новая фаза, имеющая кубическую гра-
нецентрированную решетку с периодом а = 3,980—3,985 А. Максималь-
ное содержание азота соответствует составу TcNo,76- Нитрид предель-
ного состава отвечает формуле TcN.
Нитриды рения. В широкой области температур до 2200° С не уда-
лось обнаружить взаимодействия рения с азотом [375, 376]. Также не
обнаружено образования нитридов рения при термическом разложении
210
хлораммиакатов трехвалентного рения при 350° С [377]. Предполага-
лось, что образование нитридов рения возможно при взаимодействии
окиси рения с аммиаком [378]. Впервые нитриды рения были получены
исследователями [382], которые сначала пытались получать нитриды
действием аммиака на рений при 200—1000° С, что не дало результа-
тов, а затем приготовили их нагреванием NH4ReO4 при 270—450° или
ReCl3 при 350—390° С в токе аммиака. Образуемые при этом продукты
имели состав между Re2N (3,63% N) и Re3N (2,45% N). Аналогичным об-
разом был приготовлен нитрид рения Re3N (ReN0>34) [380]. Авторы [381]
нитрид рения получали обработкой перрената аммония аммиаком или
азотом при 300—350° С в течение 20—25 ч.
Согласно [383], нагревание порошка рения в токе азота при тем-
пературах 300—900° С в течение 4 ч не приводит к образованию ни-
тридов рения. В то же время при взаимодействии рения с аммиаком
уже при 250° С образуются продукты, содержание азота в которых воз-
растает с повышением температуры, достигая при одночасовой вы-
держке при 600° С значения 3,5%, что отвечает приблизительному
составу нитрида Re2N. Практически при всех температурах азотирова-
ния порошка рения содержание азота наибольшее при наименьших
выдержках и понижается с увеличением времени азотирования, что
объясняется образованием при коротких выдержках метастабиль-
ных продуктов, постепенно переходящих в форму устойчивых ни-
тридов.
Аналогичные результаты получены при исследовании взаимодей-
ствия перрената аммония с аммиаком, образование нитридов наблю-
дается при температуре 300° С.
Таким образом, можно предварительно считать установленным
наличие в системе рений—азот двух нитридных фаз — Re3N и Re2N.
Физические и химические свойства. Нитриды рения представляют
собой порошки черного цвета, по внешнему виду не отличающиеся от
порошка рения, полученного восстановлением перрената водородом,
В химическом отношении поведение нитридов рения аналогично пове-
дению рения, но нитридные препараты несколько более инертны. При
длительном пребывании на влажном воздухе нитриды рения окисля-
ются до HReO4. Вода на нитриды не действует. Легко растворяются в
разбавленной HNO3 и особенно бурно — в концентрированной HNOa-
Нагретая концентрированная H2SO4 медленно растворяет нитриды ре-
ния, HF и НС1 на них не действуют. По отношению к кислороду и
хлору нитриды рения ведут себя подобно металлическому рению, в
водных растворах щелочей растворяются только при наличии кисло-
рода с образованием перренатов. Термически неустойчивы, при мед-
ленном нагревании в вакууме разложение начинается уже при 280° С,
Нитрид ReNo,43 (~Re2N) имеет кубическую гранецентрированную ре-
шетку с а=3,92±0,02 А [382, 383]. Область гомогенности этой фазы не
14*
21t
менее 2% азота. Теплота образования от —1 до —2 ккал!моль. Уста-
новлено; что нитрид ReNo,34 («Re3N) является сверхпроводником с
точкой перехода между 4 и 5° К [380].
6. НИТРИДЫ МЕТАЛЛОВ СЕМЕЙСТВА ЖЕЛЕЗА
Нитриды железа. Диаграмма состояния системы железо—азот приведена
на рис. 86 [29, 396]. Растворимость азота в a-железе при эвтектоидной
температуре составляет ~0,10% *, при снижении температуры до
200°С растворимость уменьшается до ~ 0,004 %. Решетка а-железа
при растворении в ней азота незначительно расширяется. Максимальная
растворимость азота в у-железе при температуре 600° С составляет
2,8%. При 585—590° С у-фаза претерпевает эвтектоидный распад (у->
->-у' + а) с образованием у'-фазы.
у'-Фаза (FetN) имеет, как и у-фаза, гранецентрированную кубиче-
скую решетку с узкими пределами гомогенности от 5,7 до 6,1 вес. % N,
пространственная группа .
Область гомогенности у'-фазы при температуре 400° С составляет
5,64—6,14% N, при 500° С—5,60—6,01 % N, при 600° С—5,51—5,90% N,
при 680° С—5,70—5,81 % N [940].
е-Фаза, содержащая от 5,7 до 11,0 вес.% N, имеет гексагональную
плотноупакованную решетку с упорядоченным расположением атомов
азота. При 650° С она претерпевает эвтектоидный распад на у- и
у'-фазы.
g-Фаза (Fe^N) обладает ромбической структурой [387, 398] с упо-
рядоченным расположением атомов азота и узкой областью гомоген-
ности от 11,0 до 11,35 вес.% N. Стехиометрический состав g-фазы соот-
ветствует 11,14% N.
Установлено, что ранее считавшаяся однофазной е-фаза представ-
ляет собой ряд нитридных фаз с компактной гексагональной решеткой
атомов железа и с различным расположением атомов азота в октаэд-
рических промежутках [396—398]. Электронографические исследования
тонких пленок азотированного аммиаком железа на сколе кристалла
каменной соли подтвердили эти предположения [397, 947]. Установлено
наличие четырех гексагональных е-фаз, одна из них отвечает составу
Fe3N и три — Fe2N. Структуры е-фаз могут быть описаны, если принять
за основу построенную из атомов железа компактную гексагональную
решетку с периодами ат и ст [146]. Нитрид Fe3N имеет гексагональную
решетку с периодами а=ат У3=2,76 УЗ и с=ст=4,42 А. В элементар-
* По данным [940], растворимость азота в a-железе составляет при эвтектоидной
температуре 0,115%, по [941] — 0,095%, по [942] — 0,108%. Растворимость азота в рас-
плавленном железе при 1600°С и давлении Nj=l ат составляет 0,0438±0,0007 вес.%
[939].
212
ной ячейке имеется 6 атомов железа и 2 атома азота, пространствен-
ная группа £>з'- На рис. 87 приведена проекция кристаллической ре-
шетки нитрида Fe3N на плоскость (001). Б пределах ячейки атомы
железа расположены в плоскости проекции и в плоскости, проходящей
на половине высоты. Один из атомов азота (1) лежит в плоскости,,
проходящей на !/ч высоты ячейки,
другой (2) на 3Д высоты ячейки..
Структура фазы Fe2N (е'-фа-
за) отличается от структуры фазы
Fe3N только тем, что в одной из
плоскостей (либо на ’Д, либо на
3/4 высоты ячейки) вместо одного-
атома имеются два атома азота.
Кроме нитрида Fe2N с боль-
шой элементарной ячейкой уста-
новлено наличие двух нитридов,
того же состава с малой ячейкой
и с~ст) [397]. Один из
имеет упорядочен-
(а—ат
них (е"-фаза)
Олр •»
системы железо—азот.
о *
Рис. 86. Диаграмма состояния
Рис. 87. Проекция кристаллической решетки нитрида Fe3N на плоскость
(001) (а) и элементарная ячейка Fe2N—е (6).
ное распределение атомов азота, отвечающее слоистой решетке CdJ2,
пространственная группа D3d (рис. 87, а), во второй (е/х/-фаза) атомы
азота размещены статистически неупорядоченно в октаэдрических пусто-
тах, пространственная группа £>♦ , тип iNiAs. Степень стабильности
е-фаз пока не установлена, возможно, что некоторые из них являются
метастабильными соединениями, образованными в качестве переходных
ступеней при перераспределении атомов азота.
Пинскер исследовал электронографически кубический нитрид
Fe4N [399, 400] и упорядоченность в нитридных фазах железа [401].
213
Физические свойства. Нитриды железа, особенно Fe4N, заметно дис-
социируют при температуре 500° С [402], процесс диссоциации заканчи-
вается при 1270—1527° С [403, 404]. Давление диссоциации Fe4N в за-
висимости от температуры изучено в [405], а кинетика удаления азота
из нитридов железа — в [406]. Показано, что энергия активации при
удалении азота из у'-фазы составляет 42100+1400 ккал]молъ при 350—
500° С. Скорость удаления атомов азота выражается экспоненциальной
зависимостью от температуры. Приближенный расчет показывает, что
в секунду удаляется около 1,8 • 10-3 атомов азота на 100 атомов же-
леза. При изотермическом разложении в вакууме Fe2N необратимо
отдает азот, переходя в Fe3N и Fe4N (этот переход начинается при
330° С) [407].
Нитрид Fe4N при закалке не изменяется, е-фаза при содержании
выше 7,5% N закаливается без изменений, а при содержании азота
ниже 7,5% неустойчива.
Нитрид Fe4N обладает сильными магнитными свойствами (средний
магнитный момент на молекулу 8,88). Предполагается, что железо на-
ходится в нитриде не в ионной форме, а в форме «нейтральных атомов»,
соединенных ковалентными связями [408]. Это предположение позволяет
объяснить природу магнитных свойств нитрида. Подобные взгляды раз-
виваются Юм-Розери, который считает, что введение в железо азота
приводит к акцептированию части электронов железа атомами азота и
освобождению соответствующих З^-состояний для связи [409]. При этом
возникает гексагональная плотноупакованная структура сплавов же-
леза с азотом.
Внедрение азота в железо вызывает замедление движения дисло-
каций [410]. Твердость нитридов железа Hv колеблется в широких пре-
делах от 224 до 687 кг)мм1 [413]. Наибольшей твердостью, по-видимому,
обладает е-фаза на границе с е+у'— полем диаграммы. Физические
свойства нитридов железа изучены в [411—415]. В работе [985] прове-
дено рентгеноспектральное исследование фаз Fe4N и Fe2N.
Физические свойства нитридов железа приведены в табл. 76.
Химические свойства. Нитрид Fe2N растворяется в разбавленной
НС1, разлагается хлором и бромом, но не поддается действию йода.
Нитриды разлагаются водой [112]. При действии углерода на нитрид
железа при 400° С образуется Fe5C2 [454].
Методы получения. Азот действует на железо крайне медленно,
Поэтому нитриды обычно получают обработкой железного порошка ам-
миаком. Так, Fe2N образуется при действии аммиака на порошок кар-
бонильного железа при температуре 600—700° С. Исследователи [63]
для получения Fe2N обработкой железа аммиаком применяли более
низкую температуру — 350° С, в [398] — 450° С. Нитриды железа полу-
.чают также действием аммиака на тонкие пленки напыленного железа
при 250—600° С [391]. Процессы реакционной диффузии азота в железо
214
Таблица 76
Свойства нитридов железа
Характеристика Y'-Фаза Fe4N е-Фаза (Fe3N, г', е", е'" — Fe,N) £-Фаза (Fe2N)
'Содержание азота, вес. % 5,7—6,1 5,7—11,0 11,0—11,35
Кристаллическая структура .Периоды решетки, А: Кубическая гране- центрированная Гексагональная плотноупакованная Ромбическая
а 3,791—3,801 [396]* 2,660—2,764 [398]** 2,764 [398]***
в — —— 4,829
с — 4,343—4,420 4,425
с/а Плотность, г/см3: — 1,683—1,599 —
расчетная 7,21 — 7,08
пикнометрическая 6,57 — 6,35
Теплота образования, ккал) моль 2,6±2 [112, 47] — 0,9±2 [47]
Эффективный магнитный момент на 1 атом же- леза, магнетонов Бора 2,22 [451] —-
Почка Кюри, °К 761 [452] 548 [453] —
* В работе [399] приводятся несколько большие значения периода (3,81—3,97 А), в работе
[940] 0=3,797 А. ** По данным [940], при 10,99% N для е-фазы о0=4,787, с0=4,418 А. *** По дан-
ным [940], для £-фазы при 11,07% N а0=5,525, /><?=4,827, с0=4,422 А.
с образованием нитридов изучены в [414, 415]. Особенности поведения
сплавов системы железо—азот при высоких давлениях рассмотрены
в [416].
При выборе оптимальных условий получения нитридов железа не-
обходимо иметь в виду близость температур прохождения двух процес-
сов— образования нитридов (2xFe + 2NH3->2FexN + 3H2) и их диссо-
циации (2FexN->2xFe + N2) [414].
Низшие нитриды железа образуются при нагревании высших ни-
тридов в вакууме, например, Fe2N при нагревании до 440—600° С,
переходит в FetN.
Авторы [274] получали нитрид железа Fe3N термическим разло-
жением (NH^sFeFg в токе аммиака при температуре 600° С.
В связи с высокими свойствами азотированных слоев на железе,
сталях и чугунах, а также ролью азота в сталях в многочисленных
работах исследованы вопросы азотирования железа и его сплавов как
в научном, так и в производственном аспектах [402, 447].
Нитриды кобальта. Растворимость азота в а-кобальте при темпе-
ратуре 600° С составляет около 0,6 вес. %. В системе кобальт—азот об-
наружено существование двух нитридов — Co3N и C02N [29, 455, 458].
215
Область гомогенности нитрида C03N лежит в пределах от 7,7 до-
8,0 вес. % N. C03N имеет гексагональную решетку. Нитрид C02N изо-
морфен Со2С [456], имеет ромбическую искаженную гексагональную
упаковку металлических атомов. Расположение внедренных атомов,
азота у Co2N такое же, как Fe2N.
Физические и химические свойства. Оба нитрида имеют цвет от
серого до черного, плохо поддаются действию слабых кислот на холоду,
концентрированной H2SO4 растворяются медленно, в концентрирован-
ной соляной и азотной — быстро. При нагревании оба нитрида быстра
растворяются в разбавленных минеральных кислотах. Физические свой-
ства нитридов кобальта
Таблица 77 Свойства нитридов кобальта представлены в табл. 77. Методы получения. Азот медленно действует на ко- бальт, поэтому азотирование ведут аммиаком с образо-
Характеристика CosN Co2N
Содержание азота, вес. % 7,34 10,71 ванием при температуре 250—300° С нитрида C03N,
Кристаллическая струк- Гексагональ- Ромбическая а при 380—500° С — Co2N.
тура Периоды решетки, д: а ная 2,658 [455] 2,842 [455] Нитриды кобальта получают также нагреванием при 2000° С цианамида кобальта
в — 4,627 Co(CN)2 и его смеси с окис-
с с/а Плотность, г/см3 4,531 1,71 7,1 4,330 лом кобальта в среде азота
6,3 [6]. В работе [457] нитриды
Теплота образования, 2 [460] — кобальта получались терми-
ккал/моль ческим разложением в ва- кууме амида кобальта
Co(NH2)3 с удалением при этом аммиака. В зависимости от температуры
разложения образуются либо нитриды при 50—70° С, либо малоконцен-
трированные твердые растворы азота в кобальте при 220—250° С.
При обработке окиси Со3О4 аммиаком при 330—390° С в течение
2—3 ч происходит образование не нитридов, а оксинитридов ко-
бальта [455].
Нитриды кобальта могут быть получены азотированием CoF2 при
360° С. C03N обычно образуется при термическом разложении Co2N
при 276° С [455].
Нитриды никеля. По данным [459], растворимость азота в никеле
не превышает 0,32% (при одновременном растворении 0,17% водорода,
выделяющегося при азотировании аммиаком *. По более надежными
* Растворимость азота в жидком Ni при 1600° С и давлении N2=l ат равна
0,001 ±0,001 вес.% [939].
216
данным [458] при 450° С в никеле растворяется только 0,07% N. Рент-
генографически обнаружено существование в системе никель—азот
только одного нитрида никеля — NisN, имеющего гексагональную ре-
шетку из металлических атомов с размещением в междоузлиях атомов
азота. Нитрид NisN разлагается выше 360° С, при 440° С процесс раз-
ложения протекает очень быстро [458].
Физические и химические свойства. Нитрид никеля NisN — вещество
темно-серого цвета. Периоды решетки NisN (7,37 вес.% N) а = 2,670,
с=4,307 А, с/а= 1,613 [458], удельный вес — 7,66 г/см?, теплота образо-
вания нитрида никеля NisN из элементов составляет — 0,2±
0,1 ккал/моль [460]. Нитрид NisN устойчив в едком натре, медленно раз-
лагается 2-н. соляной, серной и фосфорной кислотами на холоду, кон-
центрированной H2SO4 не разлагается, концентрированными НС1 и
HNO3 на холоду разлагается бурно. При нагревании энергично (на хо-
лоду) и со взрывом (при нагревании) взаимодействует с концентриро-
ванными и разбавленными минеральными кислотами, серная кислота
действует на него медленнее и спокойнее, чем другие. При растворении
в разбавленных, не содержащих кислорода кислотах выделяется во-
дород, что связано с переходом никеля в двухвалентное состояние. Кон-
центрированная H2SO4 при нагревании восстанавливается нитридом до
SO2, а разбавленная — до H2S [458]. На воздухе нитрид при темпера-
туре 800° С сгорает до NiO.
Методы получения. Азот в широкой области температур, по край-
ней мере до 900—1400° С, не действует на никель, поэтому приготовле-
ние нитридов никеля осуществляется нагреванием порошкообразного
никеля в среде аммиака при температуре 440—450° С, либо нагреванием
в токе аммиака комплексных галогенидов — NiFa^NFBF при 390—
410° С. Нитриды никеля получают также нагреванием в токе аммиака
NiBf2, из которого при действии аммиака сначала образуется аммиа-
кат, который при 450° С разлагается с получением NisN [458]. В работе
[461] подробно исследовалось взаимодействие тонкого никелевого по-
рошка (полученного восстановлением оксалата при 600° С) с аммиаком.
Показана зависимость содержания азота в никеле от скорости тока ам-
миака, обнаружено сильное каталитическое действие добавки 1 % Fe
на процесс азотирования никеля. Оптимальная температура реакции —
300° С, скорость тока аммиака — 40 мл/мин, время реакции—10—
20 ч, получающийся продукт содержит 7,3—7,4% N.
Можно также приготовлять NisN нагреванием Ni(CN)2 в среде
азота при температуре 2000° С [6].
В работе [462] описана методика получения сплавов нитридов ни-
келя и кобальта и приведены данные по магнитным свойствам и твер-
дости этих сплавов.
217
1
7. НИТРИДЫ ПЛАТИНОИДОВ
Практически никаких сведений о соединениях платиноидов с азотом
нет [463, 464, 6, 29]. Известно только, что при исследовании взаимодей-
ствия палладия с азотом, растворения азота в Pd не обнаружено до
1400° С [29]. Однако при вакуумном анализе некоторых платиноидов
найдены ничтожные примеси азота (в Rh— 0,0003%; в Ru — от<0,002
до 0,0118% [465]. В обзоре [Г56] указывается, что небольшое количе-
ство нитрида палладия образуется в дуге между палладиевыми электро-
дами в среде азота.
8. НИТРИДЫ АКТИНОИДОВ
Нитриды тория. Ранее считали [121, 483, 493], что торий образует один
нитрид Th3N4, существующий в двух модификациях, отличающихся по
структуре, свойствам и цвету (одна — желтого, другая — коричневого
цвета). Предполагали, что коричневый нитрид представляет собой ин-
дивидуальное соединение состава Th2N3 [493].
В настоящее время однозначно установлено, что торий образует
три нитрида — ThN, Th2N3 и Th3N4 [43, 495—497].
Растворимость азота в тории, по [498], возрастает от 0,05 вес.% N
(0,8 ат.% N) при 845°С до 0,35 вес.% N (5,2 ат.% N) при 1490°С.
В этой области растворимость может быть представлена уравнением
lg С = —+ 0,9115.
где С — концентрация азота, вес.%.
Установлены температурные зависимости константы реакции азо-
тирования и коэффициента диффузии
k = 5,9 ехр (— 24300/RT) [мл/см?-сек],
£>=2,1 • 10~3-ехр(— 22500/7??) [см2/сек].
Нитрид ThN имеет кубическую решетку типа NaCl, a Th2N3—
гексагональную.
Физические и химические свойства. Нитрид тория ThN имеет вы-
сокую температуру плавления, стабилен при высоких температурах в
вакууме. Нитрид тория Th3N4 в вакууме при 1500° неустойчив, в среде
азота устойчив при 1730° С. Разлагается Th2N3 в вакууме, образуя
ThN [496].
Свойства фазы Th3N4 изучены мало [996], исследована в основном
ее кристаллическая структура [1022]. Согласно этим данным, Th3N4
имеет ромбоэдрическую решетку с а=9,398 А, а=23,78° (периоды соот-
218
ветственной гексагональной решетки: а=3,871, с—27,385 А); расчетная
плотность 10,55 г/сж3.
ThN — желтого цвета, устойчив по отношению к кислотам. Корич-
невый Th2Ns легко гидролизуется под действием воды или влаги воз-
духа [493, 623]. Оба нитрида при комнатной температуре медленно реа-
гируют с кислородом, образуя ThO2 [497]. Свойства нитридов тория
приведены в табл. 78.
Таблица 78
Свойства нитридов тория
Характеристика ThN Th3Na
Содержание азота, вес. % Кристаллическая струк- тура 5,69 Кубическая гранецент- рированная типа NaCl 8,30 Гексагональная ти- па La2O3
Периоды решетки, д: а с с/а Плотность, з/сЛ43 Температура плавления, °C Теплота образования, ккал/моль Энтропия, кал/град-моль Давление диссоциации Точка перехода в сверх- проводящее состоя- ние, °к 5,20 [494]—5,144 [93] 11,50 2790±30 [1019] 90,6 [996] 23,5 [996] IgP (am) = —8,086—33,24/T-i + +0,958-10"'7 [1019] 3,875 [495] 6,175 1,595 10,51 2100 [499]* 309,5 ±4,0 [47]* 42,7±2,5 [47]* 88 мм pm. cm. при 2230°C [496] <1,2 [ 275 , 304]*
* Для Th3N4.
Методы получения. Нитриды тория получают обычно обработкой
тория аммиаком либо действием аммиака на гидрид тория [493]. В по-
следнем случае при температуре 1000° С образуется ТЬ2Ыз- Нитриды
тория образуются при непосредственном азотировании металла сухим,
очищенным азотом при 800° С в течение 3 ч [24].
Нитрид тория можно получать методом наращивания из газовой
фазы,например на вольфрамовой нити, нагреваемой до 1000° С в смеси
паров ТЬСЦ с N2 + H2 [24].
Имеются сведения о возможности образования нитридов тория
взаимодействием карбида тория с аммиаком и аммиака с хлоридом
тория, а также термическим разложением амидов тория Th(NH2)4 и
Th(NH)2[493],
219
Нитриды протактиния. Нитриду протактиния приписывается состав
PaN2 [493]. PaN2 — светло-желтое твердое соединение, образующееся
при действии аммиака на Pads или РаСЦ при температуре 800° С;
изоструктурен UN2.
Нитриды урана. Система уран—азот неоднократно исследовалась,
особенно за последние 20 лет [500]. Установлено существование трех
соединений урана с азотом — мононитрида UN, полуторного нитрида
U2N3 и динитрида UN2. Область между UN и U2N3 двухфазна, U2N3
с увеличением содержания азота переходит в нитрид UN2, т. е. область
между этими двумя соединениями представляет область однородного
твердого раствора. Исследование системы продолжено в работе [501],
где даются технологические рекомендации получения отдельных нитри-
дов, особенно в плавленом состоянии. В этой работе построены
диаграммы состояния системы U—N для нескольких давлений азота
(рис. 88).
Наиболее полное исследование системы выполнено в работе [502],
результаты которой приведены в виде диаграммы состояния системы
U—N для давления азота в 5 ат (рис. 89).
Растворимость азота в уране мала [497].
Мононитрид урана имеет кубическую гранецентрированную ре-
шетку типа NaCl, плавится конгруэнтно при температуре 2835±30°С
(отношение N : U = 0,96±0,03). Полуторному нитриду урана U2N3, по*
данным [500, 503], отвечает кубическая объемноцентрированная ре-
шетка типа Мп20з (О5з), по данным [43] — гексагональная решетка
типа Th2S3. Теперь доказано [502], что те и другие данные правильны,
так как нитрид претерпевает при 1315° С полиморфное превращение,
переходя от кубической объемноцентрированной решетки к гексаго-
нальной. Динитрид урана UN2 имеет кубическую гранецентрированную*
решетку типа CaF2. Диаграмма состояния системы U—N построена
также в [516, 1004].
Физические свойства. Мононитрид урана — тугоплавкое соединение-
с температурой плавления 2835° С (по старым данным Чиотти [43] —
2650° С, по данным [504]—2850° С при давлении азота более 2,5 ат).
При более низких давлениях происходит инконгруэнтное плавление с
разложением на жидкий уран, насыщенный азотом, и газообразный
азот. Давление диссоциации выражается уравнением [504]
lgp[am] = 8,193 —(29,54- 103)T-' + (5,57-10-18) Т5.
Нитрид урана UN, загрязненный кислородом, разлагается при
более высоких температурах, чем чистый нитрид. Кубическая струк-
тура этого нитрида подтверждена нейтронографическим методом [505].
При облучении нейтронами из мононитрида урана установлено, что
влияние превращения азота в водород и углерод ничтожно мало [506].
Основное влияние оказывает деление урана, входящего в нитрид.
220
Полученные данные при облучении нейтронами мононитрида урана
представлены в табл. 79.
В работе [964] дуговой плавкой приготовлены монокристаллы моно-
нитрида урана с плотностью 99—100,1% от расчетной. Их твердость,
по Кнупу, оказалась равной 555, по Виккерсу — 500. Температура кон-
^°СО 20 40 бОКот.%
3000 (гв503>Ь.______
2000-
(JN+NZ
((52(f)
и(ж)*- -
'°00' U(T5t*UN
груэнтного плавления под давлением азота
2,5 ат равна 2850±50°С.
По данным [1012], где измерялась теп-
лопроводность мононитрида урана при тем-
пературах 900—1650° С, выше 1200° С теп-
лопроводность не зависит от температуры
и составляет при 1650° С около 0,26 вт/смХ
Хсек. В этом интервале температур пре-
5o<n.Nz
(3820) <<
3000-
nap(U*Nf)
U(>*<[* nop
1/ЛкГ'>.(2800 °>
2000
UN*NZ
(1345°)
i000’u(r6H//^}
fam. N2
3000-
(2230)
2000-
Пар(О*Нг)
UM* nag
1000 -
~2030s
UN*N2
.. UN*U,Nj
Ю*атКг г
0 3 6 до.Вес.%
if
(ЦЦ+ОЩ)
(U0%<>)
(l/fl'Mil
Рис. 88. Диаграмма состояния системы уран—азот для разных давлений
азота.
Рис. 89. Диаграмма состояния системы уран—азот (pNj =5 ат).
валирует электронная составляющая теплопроводности, например, при
1300° С она составляет до 80%.
Электрофизические свойства нитридов урана исследованы в работе
[507], в частности — температурная зависимость электропроводности
(рис. 90) и термо-э. д. с. (рис. 91) мононитрида, то же — для дини-
трида (рис. 92) и термо-э. д. с. для нитридов с различным отношением
221
Влияние облучения да Таблица 79 нейтронами мононитри- урана * содержаний азота и урана (рис. 93). Высокие значения тер- мо-э. д. с. могут быть использо-
Обога- щение урана, % Выгорание урана, % Максимальная температура на поверхности, °C Уменьшение плотности, % ваны в известных условиях для термоэлектрических генераторов, тока. Мононитрид имеет р-тип проводимости. Магнитная вое-
0,22 0,72 5 * Исс; щей стали оболочки изменений о,1 0,4 2—3,5 1едовали обр марки 304 разрушаютс 65—70 ЮО—105 275—580 азцы с оболочке или без оболочк я, а в оболочк 0,73—1,6 1,1—2,1 2,3—6,4 й из нержавею- и. Образцы без е остаются без приимчивость мононитрида, рав- ная, по этим данным, 3050 • 10-6, может быть объяснена наличием двух неспаренных бй-электронов (теоретическое значение для двух неспаренных 6б?-электро- нов — 3333- 10-6) и участием од-
ного 6с?-электрона на атом урана
в связи Me—Me. Отрицательный температурный коэффициент электро-
проводности обусловлен рассеянием носителей с температурой. При низ-
ких температурах эти носители могут обеспечиваться связью Me—Me, а
при высоких должно происходить возбуждение носителей в пределах по-
лос, относящихся к связям U—N. Автор [507] полагает, что относитель-
но большая разница электроотрицательностей компонентов UN, составля-
ющая 1,5, может вызывать наличие большой энергетической щели, что
обусловливает поставку некоторого числа носителей тока при низких
температурах. Для нитридных фаз UN2-x установлена проводимость
n-типа; низкие значения проводимости при высокой концентрации но-
сителей, что соответствует х = 0,41, могут быть приписаны низкой под-
вижности носителей. Можно ожидать, что UN2, будучи менее ионным
соединением, чем изоморфная ему двуокись урана (разность электро-
отрицательностей 2,0), имеет более высокую подвижность носителей,
чем 10 см2-в-сек, установленную для UO2.
При некоторой величине х, большей 0,41, происходит фазовое пре-
вращение UN2 из структурного типа CaF2 в структурный тип La2O3.
Полуторный нитрид урана имеет по сравнению с высокопроводящим
мононитридом низкую электропроводность и одновременно малое зна-
чение термо-э. д. с. Предполагается, что U2N3 может иметь только сте-
хиометрический состав.
Электропроводность и термо-э. д. с. мононитрида урана при низких
и средних температурах были измерены в работе [831]. На рис. 94 по-
казана зависимость от температуры электросопротивления, а на рис. 95—
термо-э. д. с. (по отношению к меди).
Более подробное исследование магнитных свойств нитридов урана
выполнено в работах Тжебятовского с сотрудниками [508, 509]. Показа-
но, что в мононитриде урана высокая отрицательная константа Вейсса
(—310) указывает на строго антиферромагнитное сочетание спинов.
222
электронов. Это совпадает с наиболее коротким межатомным расстоя-
нием U—U в этом соединении с азотом, равным 3,449 А. Фаза p-U2N3
(гексагональная модификация) характеризуется ферромагнитными
свойствами при низких температурах в метастабильном, закаленном с
высокой температуры состоянии; точка Кюри равна +186° К- Фаза
(Z-U2N3 (кубическая модификация) имеет магнитный момент 1,92 маг-
нетонов Бора; для объяснения этого
значения можно предположить, что
часть атомов урана находится в со-
стоянии ионов U4+ (p,U4+=2,83 маг-
нетонов Бора), а к ним подмешана
часть диамагнитных ионов U6+:
: U4+ U6+ N3" , либо имеется
смесь четырех- и пятивалентных ио-
нов урана: U4+ 1,0%-и5+ 1,0 U3~ 3.
Рис. 90. Температурная зависимость электропроводности нитрида
UN:
7 — 1,08% С; 2 — 0,2% С; 3 — 0,06% С.
Рис. 91. Температурная зависимость термо-э. д. с. UN.
Нитрид U2N3 при температурах 700—800° С в вакууме отдает азот
до 1300° С, при этой температуре и низких давлениях разлагается на
мононитрид и азот, а при высоких — на моно- и динитрид [500]. В по-
следнем случае объем сплава уменьшается на 13%. Теплота декомпо-
зиции U2N3->-UN + N2 равна 57—58 ккал!моль N2 [963].
В работе [1055] приводятся данные по измерению равновесного дав-
ления над нитридными, карбонитридными фазами урана в зависимости
от давления и температуры и вычислены энтальпии и энтропии этих
фаз, а также уточнены параметры гексагональной решетки фазы
₽-U2N3 (а=3,698, с=5,839 А).
Физические свойства нитридов урана приведены в табл. 80.
Химические свойства. Важным свойством нитридов урана является
их способность активно поглощать газы, что используется для очистки
азота от кислорода, влаги и позволяет в некоторых случаях применять
нитриды урана в качестве геттеров. При окислении спеченного моно-
нитрида урана в кислороде при температурах 350—480° С и давлениях
223
до 1 ат образуется ряд промежуточных окислов между изО8 и UO3.
Энергия активации окисления в указанном интервале температур
24,5 ккал/моль [510, 969].
В работе [962] показано, что окисление мононитрида урана кисло-
родом, СОг, воздухом, насыщенным влагой, выражается линейным ки-
Рис. 92. Температурная зависимость электропроводности и
термо-э. д. с. UN2.
Рис. 93. Зависимость термо-э. д. с. от отношения N/U.
Рис. 94. Температурная зависимость электросопротивления UN.
Рис. 95. Температурная зависимость термо-э. д. с. UN.
нетическим законом с энергией активации от 10,4 до 15,4 ккал/моль.
Продукт окисления при 300° С состоит преимущественно из U3O8. Кор-
розия водой при 100° С и натрийкалиевым сплавом при 820° С проходит
очень медленно. В целом, нитриды урана легко окисляются на воздухе,
224
Свойства нитридов урана
Таблица 80
Характеристика UN a-U2N3 ₽-u2N, un2
Содержание азота, вес. % 5,55 8,22 8,22 10,5
Кристаллическая структу- Кубическая гра- Кубическая объ- Г ексагопальная Кубическая
ра нецентрирован- емноцентриро- гранецентри-
ная ванная рованная
Периоды решетки, А:
а 4,880 [500] 10,678 [500] 3,69 [511] 5,31 [500]
с — — 5,83 —
с/а — — 1,58
Плотность, г/см3 14,32 [500] 11,24 [500] 12,44 [508] 11,73 [514]
Температура плавления, °C 2850 ±50 [964] —
Теплота образования из —
элементов, ккал/моль 68,5 [47] 256 [9] —
Энтропия, кал! град моль 12,5 [47] —
Область устойчивого су- —
ществования, °C: •—
при давлении N2 5 ат — 1520 [501] .—
» » » 1 ат —— 1345
» » » 10~4ат — 780 —
Т еплопроводность,
кал/сек -см -град 0,0325 [507] .—
Электропроводность,
ом—1 • см—1 3200 [507] —
Термо-э. д. с., мкв/град 57 [507] —
Магнитная восприимчи-
вость 3050-ИГ6 [507] См. [508, 509] —
Температура перехода в
сверхпроводящее состо-
яние, °К <1,20 [275,304] —
Коэффициент термическо-
го расширения, а-10е
(при 20—1600 °C) 10,1 [976] —
Модуль упругости, кг/см? 2,2—2,5-106[97б] —
Модуль сдвига, кг/см2 0,8—1 -106(976] —
Коэффициент Пуассона 0,1—0,26 [976] —
Твердость (по, Кнупу),
кг/мм2 600—700 [976] —
труднорастворимы в кислотах и растворах щелочей [514], но легко раз-
лагаются расплавленными щелочами. Высшие нитриды урана восстанав-
ливаются водородом.
В работе [43] показана высокая стойкость нитрида урана к дейст-
вию нитридов тантала и тория при 2000° С. Высокая температура
плавления мононитрида позволяет использовать его для огнеупорных
изделий, эксплуатирующихся в азоте или инертных газовых средах.
15—272
225
Методы получения. Нитриды урана обычно приготовляют азотиро-
ванием урана или гидрида урана.
Изучение реакций азотирования урана проведено в [511, 512]. При
температурах азотирования 650—900° С в диффузионных слоях обна-
ружены три фазы — мононитрида, динитрида, а также гексагональной
модификации полуторного нитрида. В слоях, полученных [512] азоти-
рованием урана при 550—750° С также обнаружен преимущественно
нитрид UN2 с малыми примесями U2N3. При температурах азотирова-
ния между 775 и 900° С, как и в [511], обнаружены все три нитридные
фазы. Скорость азотирования урана в обеих указанных областях тем-
ператур подчиняется параболическому закону, однако скорость диффу-
зии и энергии активации различны
7Э = 202-ехр(—25500/7??) при 550 — 750°С,
£> = 3,95-ехр (—15100/7??) при 775 —990°С.
Изменение параметров диффузии связывается с ру-превращени-
ем металлического урана (по новым данным [502], превращение проис-
ходит при 771—778°С).
Установлены энергии активации процесса азотирования, равные
при 630° С 16 ккал/моль, а при более высоких температурах —
7 ккал/моль (температуре [3у-превращения урана), указывается на
далеко идущую аналогию между механизмом взаимодействия урана с
азотом и кислородом [515].
В работе [522] исследовано азотирование порошка урана, получен-
ного кальцийтермическим восстановлением в области температур 300—
700° С под давлением азота 50—700 мм рт. ст. Частицы порошка урана
имели сферическую форму с размерами 2—200 мк. На первых стадиях
реакции получается UN, а при 600° С за 30 мин реакция заканчивается
с образованием продукта, близкого по составу U2N3. Энергия активации
процесса при 500—700° С равна 27±2 ккал/моль, что хорошо согласу-
ется с величиной, определенной в [512]. В процессе азотирования части-
цы урана измельчаются, что приводит к обнажению новых поверхностей
и ускорению процесса азотирования. До наступления процесса разру-
шения зерен реакция протекает не по параболическому закону, а по за-
кону типа \m=ktn, где п близко к 7з-
Минимальное давление азота, требующееся для получения литого
UN, должно составлять 2,5 ат. При более низких давлениях невозмож-
но добиться насыщения азотом урановой жидкой фазы до получения со-
става UN. Возможно получение UN путем разложения U2N3 при низ-
ких температурах в откачиваемой системе, в которой поддерживается
необходимый вакуум. Однако поскольку скорость процесса при низких
температурах мала, минимальной температурой такого способа получе-
226
ния нитрида урана является 1000° С. Более высокие температуры ухуд-
шают условия вакуума, необходимые для разложения.
При азотировании урана под очень большим давлением порядка
120—130 ат образуется смесь моно- и динитрида урана с общим содер-
жанием азота в сплавах до 55—66 ат.%.
Чиотти [43] приготовлял мононитрид урана пропусканием азота, ам-
миака над стружкой урана при температурах 400—940° С. Азот тща-
тельно очищали пропусканием также над стружкой урана при 500° С.
При выбранных Чиотти режимах получались смеси нитридных фаз ура-
на, перевод которых в термически устойчивый мононитрид производил-
ся нагреванием продуктов азотирования в вакууме при температуре
около 1500° С.
Азотирование металлического урана аммиаком было вновь иссле-
довано в [987]. В интервале температур 470—1000° С (при давлении в
1 ат) получены продукты составов соответственно от UNi,74 aoUNi,55-
Все нитридные фазы UNX с х^= 1,50 имели кубическую ОЦК-структуру
типа МП2О3 с постепенным переходом при увеличении значения х в
КГЦ-структуру типа CaF2, характерную для UN2.
Исследователи [500] получали нитриды урана взаимодействием
гидрида урана с аммиаком при температуре 200° С либо с азотом при
350° С в условиях тщательной изоляции от попадания в реакционное
пространство кислорода. В работе [485] рекомендуется метод получения
нитрида урана с промежуточной стадией образования гидрида урана.
Куски урана предварительно гидрируются при температуре 200° С, затем
азотируются при 350—400° С и некотором избыточном давлении азота
(2—3 кг/см2). При этом с большой скоростью реакции образуются про-
дукты, имеющие состав UNX, с х=1,77—1,78, т. е. состоящие из смеси
U2N3—UN2. При нагревании этого продукта в течение нескольких часов
в вакууме при 800° С получают мононитрид урана.
Наиболее чистый мононитрид урана получают дуговой плавкой
урана в среде азота при высоких температурах [513].
Нитрид урана, которому приписывается состав U3N4 (смесь фаз),
образуется при взаимодействии UF4 с аммиаком при 800° С [274], или
UCI4 с аммиаком [514].
Изделия из мононитрида урана приготавливают обычно методами
порошковой металлургии. Например, в [43] изделия из порошка круп-
ностью частиц —100 меш получали прессованием заготовок под давле-
нием 3,1—3,7 т/см2 с последующим спеканием при температуре 2000—
2100° С. Такие изделия имеют пористость 16%, линейная усадка при
спекании составляет около 3%. Пористость изделий крупных размеров
колеблется в пределах 23—30%. Анализ материала изделий после спе-
кания показывает на близость его состава к мононитриду урана с неко-
торым дефицитом содержания азота. При попытке изготовления тиглей
из смесей U2N3 с металлическим ураном, составленных в расчете на об-
15*
227
разование мононитрида, заготовки при спекании коробились и треска-
лись. Спекание в обоих случаях производилось в индукционной печи.
Условия приготовления изделий из мононитрида урана изучены в
[513]. Изделия получали методом горячего прессования в графитовых
прессформах при температурах 1370—1678° С, давлениях 200—
400 кг/см2 в течение 30 мин. Получаемые за 18 мин спекания изделия
имеют невысокую плотность (около 80%), которая может быть повы-
шена еще на ~10% последующим спеканием при 1700° С в течение 2 ч,
а также утончением частиц мононитрида. Загрязнение изделий угле-
родом в результате контакта с графитовой прессформой невелико и со-
ставляет максимально 0,44 вес.%. Содержание кислорода существенно
уменьшается по сравнению с исходным порошком и составляет в изде-
лии 0,06 вес.% (см. также [1080]).
Согласно [507], изделия UN также приготавливают методом горя-
чего прессования при температуре 1800°С в течение 15 мин; их плот-
ность составляла 10,3 г/см3, примесь углерода 0,06%, кислорода—0,13%.
В связи с перспективой использования мононитрида урана в каче-
стве ядерного топлива его получению, свойствам и методам приготов-
ления из него изделий посвящена обширная литература, сводки которой
даны в подробном обзоре [976], а также в библиографии [977].
Нитриды нептуния. Нитрид нептуния NpN имеет кубическую струк-
туру типа NaCl с периодом решетки «=4,8987 А [495, 1005]. Получается
взаимодействием гидрида нептуния с аммиаком при температуре 750—
800° С [517, 496]. При нагревании тетрахлорида нептуния с аммиаком
в пределах 350—1000° С нитрид нептуния не образуется.
Исследование реакции разложения NpN(TB) -> Np(ra3)+0,5И2(газ)
при 2210—2830° показало [1005], что давление паров подчиняется урав-
нению
lg р (ат) = 8,193 — 29’54' — + 7,87 • 10"18Г.
NpN плавится конгруэнтно при 2830±30°С под давлением азота
до 10 ат.
NpN не растворим в воде, растворим в соляной кислоте [517].
Нитриды плутония [1028]. Плутоний образует один нитрид PuN
[518]. По данным Цахариазена [495, 519], PuN имеет кубическую гране-
Центрированную решетку с периодом (для состава PuNo.go?) « = 4,9069 А,
линейно изменяющимся с температурой: а=4,9069 (14-12,29-10-6 t) А,
где 20</<900°С.
Плотность нитрида 14,23 г/см?, свободная энергия образования
PuN при 700° К равна —60± 1 ккал/моль, при 298° К она более отри-
цательна, чем —70 ккал/моль [997]; энтропия образования из элементов
ASC 298 = —22+1 кал/град • моль. Температура плавления мононитри-
да плутония равна 2750±75° С. Начинает интенсивно испаряться
228
при 1600° С с диспропорционированием по реакции PuNi_r->Pu+
+ (1—x)PuN. Давление диссоциации по реакции PuN (тв) —Ри (ж) 4-
+ 0,5Ы2(газ) в области 2290—2770° С определяется уравнением
lgp(am)= —11,28-10-1V +8,193.
Коэффициент термического расширения PuN по результатам ди-
латометрических измерений равен 9,30 • 10~6 град~!, а по данным рент-
генографического метода 12,29—13,80- 10-6 град~\
Рис. 96. Температурная зависимость электросопротивления PuN.
Рис. 97. Температурная зависимость термо-э. д. с. PuN.
Цахариазен приписывает нитриду плутония полуметаллические
свойства. В работе [831] были определены электросопротивление и тер-
мо-э. д. с. PuN при низких и средних температурах (рис. 96, 97), кото-
рые в общем подтверждают это предположение.
Мононитрид плутония представляет собой плотное и хрупкое ве-
щество черного цвета, неустойчивое во влажном воздухе и кислороде.
Под действием влажного воздуха гидролизуется по реакции [496]:
PuN + 2Н.2О -> PuO2 + NH3 + V2 Н2.
В холодной воде PuN гидролизуется медленно, а в горячей — очень
быстро с образованием Ри(ОН)4.
В соляной и фосфорной кислотах нитрид растворяется быстро,
несколько медленнее — в азотной кислоте, еще медленнее — в плавико-
вой и азотной. При растворении в соляной и серной кислотах на холоду
образуются соли трехвалентного плутония.
Окисление кислородом происходит интенсивно начиная с 500 и осо-
бенно быстро — при 1000° С.
229
Азот очень медленно действует на плутоний как при низких темпе-
ратурах, так и при 800—1000° С [518]. Поэтому нитрид плутония обыч-
но получают действием аммиака как на плутоний, так и на его соеди-
нения: действием аммиака на плутоний при 1000° С; взаимодействием
трихлорида плутония с аммиаком при 800—900° С; действием аммиака
при давлении 250 мм на гидрид плутония при 600° С с последующим
медленным охлаждением до комнатной температуры [493, 520]. Видо-
изменением последнего способа является описанное в [824] получение
PuN нагреванием гидрида плутония в среде азота при температурах
выше 230° С.
Авторы [485] нитрид плутония приготавливали гидрированием по-
верхности кусков плутония при температуре 200° С с последующей об-
работкой азотом при 250° С. Реакция азотирования проходит с огром-
ной скоростью. Период решетки а нитрида, полученного таким обра-
зом и затем отожженного при температуре 1000—1500° С, равен
4,912 А. Указывается, что с увеличением температуры отжига период
решетки несколько возрастает, что свидетельствует о частичной поте-
ре азота.
При попытке получить PuN методом дуговой плавки [521] образу-
ются смеси PuN, PuO2 и свободного плутония. Продукт испаряется при
температуре 2650° С в гелии без плавления.
Подробные данные о методах получения и свойствах нитридов ура-
на и плутония приведены также в отчете [1041].
ГЛАВА VI
НИТРИДЫ
ЭЛЕМЕНТОВ
ПОДГРУППЫ
БОРА
1. НИТРИДЫ БОРА
Нитрид бора BN, открытый более ста лет назад
Бальменом [625], известен в трех модификаци-
ях— a-BN (гексагональный), [3-BN (кубиче-
ский) и y-BN (гексагональный плотноупако-
ванный).
a-BN имеет гексагональную кристалличе-
скую структуру, аналогичную структуре графита.
Она состоит из графитоподобных сеток, располо-
женных в отличие от структуры графита точно
друг под другом с чередованием атомов бора и
азота по оси z (рис. 98, а). Пространственная
группа C6m2 \ , Z—2 [36, 626]. Из-за бли-
зости структуры и некоторых физических свойств
графита и нитрида бора последний часто назы-
вают «белой сажей» или «белым графитом» (в
зарубежных источниках эту модификацию часто
обозначают как s-BN). 0-BN имеет кубическую
кристаллическую решетку, аналогичную решетке
алмаза (рис. 99), т. е. кристаллизуется в струк-
туре цинковой обманки ZnS. y-BN имеет гексаго-
нальную плотноупакованную или тетрагональ-
ную [628] решетку (часто обозначают как Д-BN).
По наиболее достоверным данным y-BN кристал-
лизуется в ромбоэдрической структуре, подобной
структуре (3-графита [733] с равным смещением
между шестиугольниками в последовательных
слоях [627]. Эта структура показана на рис. 98, б.
Периоды решетки: а=2,504; с= 10,01 А.
Имеются указания о существовании в сис-
теме бор — азот нитрида состава B3N [629],
однако его существование весьма проблема-
тично.
Согласно [1084], гексагональный нитрид бо-
ра может иметь неупорядоченную структуру, ко-
торая превращается в нормальную при нагрева-
нии до температуры выше 2000° С. Дефекты
упаковки гексагонального BN подробно рассмот-
рены в [1013, 1085—1087].
Физические свойства. Наиболее полно изу-
чены свойства гексагонального нитрида бора
231
a-BN. Природа химической связи в нитриде бора изучалась во многих
работах [631—633].
Распределение электронной плотности в a-BN аналогично таково-
му в графите (рис. 100), причем между слоями (сетками) находится
15—16% всех электронов, что соответствует двум электронам от каж-
Рис. 99. Структура кубической модификации нитрида бора (боразона):
ф — атомы бора; О — атомы азота.
о-N
Рис. 98. Структура гексагональных модификаций нитрида бора:
а — графитоподобный; б — ромбоэдрический.
Рис. 100. Распределение электронной плотности в кристалле;
а — нитрида бора; б — графита.
дой пары атомов В—N в нитриде (в графите — одному электрону от
каждого атома). Брегер и Жданов [631] приходят к выводу о наличии
ионной связи между атомами бора и азота в сетках (приблизительное
валентное строение B+N") и отсутствии металлической связи между
сетками в отличие от графита, где металлическая связь обеспечивается
одним электроном. Наличие у нитрида бора определенной доли ионной
232
связи следует также из его положения в изоэлектронном ряду LiF—
ВеО—BN—СС, где он расположен между окисью бериллия, имеющей
четкий ионный характер, и графитом с большой долей ковалентного и
металлического типа связи.
Полингом [1005] структура и свойства нитрида бора рассмотрены
с позиций теории резонанса (предполагается резонанс между / В—N<^
и —В” = N+<^).
Рассматривая нитрид бора с позиций представлений о стремлении
атомов при образовании соединений к наиболее стабильным электрон-
ным конфигурациям, можно полагать, что при образовании гексаго-
нального нитрида бора атомы бора преимущественно передают валент-
ные з2р-электроны атомам азота, в результате чего атомы бора приоб-
ретают устойчивую конфигурацию s2, а атомы азота — s2p6. Наличие
определенного (причем высокого) статистического веса з2/76-конфигура-
ций атомов азота обусловливает, как и обычно, ионную долю связи, а
энергетическая обособленность этих конфигураций — наличие широ-
кой энергетической щели со следующими отсюда диэлектрическими
свойствами нитрида бора (интервал между заполненной и пустой л-под-
зонами в BN составляет приблизительно 4,6 эв [674]).
Дворкин с сотрудниками [634] на основании определенной ими теп-
лоты образования нитрида приходит к выводу о наличии двойных свя-
зей между бором и азотом в звеньях слоев структуры.
По данным [635], различие в энергии решеток BN и графита со-
ставляет только 0,077 ккал/моль при принятии, как это делают Брегер
и Жданов, одного электрона на атом.
Исследование ИК-спектра поглощения нитрида бора, проведенное
в [636, 637], показало наличие двух полос интенсивного поглощения при
длинах волн 7,28 и 12,3 мк (рис. 101), отвечающих, по-видимому, двум
основным кристаллографическим направлениям с резко разнящимся
характером связей.
Расстояние между сетками в решетке нитрида бора, равное 3,34 А,
меньше, чем у графита (3,40 А), что свидетельствует о более прочной
связи между сетками в структуре нитрида бора по сравнению с гра-
фитом.
Удельное электросопротивление q горячепрессованного образца
нитрида бора снижается с температурой следующим образом [638, 628]:
Температура, °C 25 500 1000 1500 2000*
Удельное электросопротив-
ление, ом-см 1,7 • 1013 2,3-Ю10 3,1-Ю4 2-103 1.10а
По данным [639], сопротивление BN при 2000° С составляет 2- 103 ом-см.
233
С повышением плотности изделий из BN их сопротивление быстро
-уменьшается, составляя для пористости 80% — 1012, для 50%—7 • 1010
и для 10% — 5 • 109 ом • см.
Эти цифры относятся к сухим образцам нитрида, при увлажнении
электросопротивление быстро снижается, например:
Относительная влажность, % 20 50 90
Удельное электросопротивление
при 25° С, мком-см 7-Ю12 7-1012 5-Ю9
Рис. 101. Инфракрасный спектр поглощения нитрида бора.
Ширина запрещенной зоны по данным, приведенным в [640], со-
ставляет 4,6 эв, что характерно для изоляторов. Новые измерения по-
зволяют считать ширину запрещенной зоны более узкой — от 3,6 до
3,8 эв.
Электросопротивление нитрида бора без структурных дефектов
составляет, как указывалось, 1012—1013 ом • см, с понижением содер-
жания азота уменьшается, особенно резко при 38—40 ат. % азота (вме-
сто положенных по формуле BN 50 ат.%), когда начинается разруше-
ние основы структуры — плоских сеток из атомов бора и азота.
При длительном нагреве в вакууме при высоких температурах
электросопротивление технических образцов нитрида бора стабилизи-
руется, как это видно из данных [641], приведенных в табл. 81. Образцы
нагревались в течение длительного времени при температуре 500° С с
последующим нагревом при 1000, 1100 и 1400° С в течение 30 мин, пос-
ле чего измерялось их электросопротивление вновь при 500° С.
В работе [1038] измерено электросопротивление образцов из нит-
рида бора, а также нитрида бора с 2,5; 5 и 10% в области температур
'20—2000° С. Установлено несколько более высокое электросопротивле-
ние нитрида бора при комнатной температуре — 1014 ом см по срав-
нению с ранее приводившимися данными (1013 ом-см), при 2000° С
электросопротивление составляет 2• 102 ом • см. С увеличением содер-
234
жания борного ангидрида в нитриде бора сопротивление сначала нес-
колько увеличивается (до 5- 1014 при 2,5% В20з), а затем (при 5—10%
В2О3)—падает (до 1012—1013 ом • см). В работе принимается преиму-
щественно вакансионный механизм проводимости, указано, что энер-
гия активации собственной проводимости нитрида бора состав-
ляет 5,2 эв.
Диэлектрическая постоянная и коэффициент рассеяния (tg б) про-
каленных сухих образцов из нитрида бора, измеренные [638] в электри-
ческом поле, параллельно давлению, прилагавшемуся при горячем
прессовании образцов (за исключением частоты 1010 гц, когда поле бы-
ло перпендикулярно давлению горячего прессования), приведены в
табл. 82. Диэлектрические потери сильно зависят от влажности [641].
Диэлектрическая прочность (напряжение пробоя) составляет 1,97—
3,94 кв • мм [628].
Теплопроводность и коэффициент термического расширения нитри-
да бора снижаются с температурой (табл. 83 и 84 [638]).
Таблица 82
Частотная и температурная зависимость
диэлектрической постоянной и коэффициен-
та рассеяния нитрида бора
Таблица 81
Электросопротивление нитрида бора при
температуре 500° С после предваритель-
ного нагрева в вакууме
Температу- ра термо- обработки в вакууме, °C Сопротивление (ом см) после тер- мообработки в вакууме при 500 °C Ча- сто- та, гц Диэлектрическая постоянная Коэффициент рассеяния
0 ч 10 ч 40 ч 10° с 330° с 500° С 10° С 330° с 470° С
1000 1100 1400 1,75-10’ 3,5-Ю’о 2,0-Ю11 4,2-Ю10 2,0-Ю11 4,9-Ю10 2,0-Ю11 102 ю4 10е 4,15 4,15 4,15 4,4 9,0 4,5 4,2 0,00103 0,00042 0,00020 0,32 0,0043 0,0012 1,000 0,100 0,0056
Ю8 4,15 — 0,00009 — —
1010 — — — 0,0003 0,004 0,0005
В среднем коэффициент тер-
мического расширения составляет
около 2 • 10~6 град-1. Тем не менее относительно высокая теплопровод-
ность (перпендикулярно С-оси она самая высокая среди всех известных
диэлектриков) и особенности строения обусловливают высокую стой-
кость изделий из BN против теплового удара [638]. Это относится толь-
ко к сухим изделиям, в то время как влажные образцы разрушаются
уже при теплосмене 20—600° С вследствие механического воздействия
испаряющейся влаги [641]. В порошкообразном состоянии нитрид бора,
особенно полученной взаимодействием в газовой фазе между ВС13 и NH3
с насыпным весом приблизительно 0,1 г/см3 является, как и сажа, от-
личным теплоизолятором.
235
Таблица 83'
Температурная зависимость
теплопроводности нитрида
бора
Теплопроводность, кал /см- сек- град
Температу- ра, °C Параллель- но направ- лению горя- чего прес- сования Перпенди- кулярно на- правлению горячего прессова- ния
300 0,036 0,069
500 0,034 0,067
700 0,032 0,065
900 0,030 0,063
1000 0,029 0,064
Таблица 84
Температурная зависимость
коэффициента термического
расширения нитрида бора:
Температу- Коэффициент термиче- ского расширения, ХЮ-”®’
ра, °C Параллельно направле- нию горя- чего прессо- вания Перпенди- кулярно на- правлению горячего прессования
25—350 10,15 0,59
25—700 8,06 0,89
25—1000 7,51 0,77
* Коэффициент термического рас-
ширения измерен для образцов с по-
ристостью 4—5%
Из калориметрических измерений [642] вытекает следующая зави-
симость молярной теплоемкости чистого BN от температуры в интер-
вале от 400 до 900° С
Ср = 5,030 + 12,6192 • 10~3(/— 22) — 6,5770 10“6 (/ —22)~2.
Келли для молярной теплоемкости в интервале от 0 до 900° С (точ-
ность ±5%) предлагает иную зависимость
Ср =1,61 г 4,00-ИГ Т.
Выполненное в [638] измерение теплоемкости нитрида бора при
низких температурах показало, что в пределах температуры 20—65° К
теплоемкость подчиняется закону квадрата абсолютной температуры
(а не закону куба, как отвечало бы дебаевскому закону)
Су = 14,4-а№/0',
где Cv — молярная теплоемкость.
Это объясняется слоистым строением нитрида, наличием у него
квазидвухразмерной решетки.
В табл. 85 приведены значения энтальпий и теплоемкости BN по
[655], которые подтверждены в работе [775].
Характеристическая температура нитрида бора 0 = 598° К.
Наиболее полно термодинамические свойства и поведение нитри-
да бора при высоких температурах исследованы в работе [643]. Полу-
ченные при этом данные представлены в табл. 86—89.
236
Таблица 85
Энтальпии и теплоемкость нитрида бора
[655] *
т/к Яр—«298 СР Т, °к СР
300 7,45 4,69 1000 5998,4 10,6
400 559,2 6,28 1100 6975,2 10,9
500 1252,2 7,50 1200 8084,4 11,2
600 2051,4 8,42 1300 9218,6 11,4
700 2929,4 9,09 1400 10371 11,6
800 3868,5 9,66 1500 11537 11,7
900 4860,5 10,2 1600 12708 11,7
* Для состава В—4?,81. N—56,85±0,4%.
Таблица 86
Изобарный потенциал, константа
равновесия и давление азота реак-
ции BN т в — В т в Т V2N2
т, °к AZ°, ккал -1g К «N2. am
1500 29,4 4,29 2,62-10“9
1600 27,2 3,72 2,66-10“8
1700 25,1 4,88 1,06 10~7
1800 23,1 2,81 2,44-10“6
1900 21,0 2,42 1,48-10“5
2000 19,2 2,11 6,17.10“5
2100 16,8 1,75 3,11-10“4
2200 14,7 1,47 1,16- io—3
2300 12,7 1,21 3,76 10—3
Таблица 87
Изобарный потенциал, константа равновесия и давление
азота реакции ВМтв = ВГаз + 1/2М2 *
Т, °к AZ', ккал -1g Д', am AZ", ккал -1g «N2. am
2500 23,2 2,03 2,80-Ю“2 63,2 5,54
2600 17,9 1,51 6,25 -10-2 57,9 4,88 —
2700 12,5 1,01 1,34-IO-1 52,5 4,25 —
2800 7,2 0,57 2,67.10“' 47,2 3,69 2,22-10“3
2900 1,8 0,14 5,12.10“' 41,8 3,16 4,97.10“3
3000 —3,4 — >1 36,6 2,67 1,05.10“2
* AZ', и —соответствуют теплоте реакции 163 ккал;
AZ", J(2 и ^N2~теплоте реакции 203 ккал.
Температурная зависимость упругости диссоциации нитрида бора
выражается уравнением
1 z \ л л 6450
1g р (мм рт. ст.) = 4,0----.
Теплота сгорания нитрида бора BNTB + ^Ог = V2B2O3 аморфн+'/г^
равна 90,2 ккал/моль. При теплоте образования В2О3 301,8±
±1,4 ккал/моль для теплоты образования BN из элементов получено
237
Таблица 88
Изобарный потенциал и
давление сублимации ре-
акции BNTB = BNraa
Таблица 89
Изобарный потенциал, константа равновесия
н давление нитрида бора реакции
Вгаз + V2N2 ~ BNra3
т, °к Д2°, ккал PBN, am т, °к AZ, ккал К"-102 pNe am PBN- am
2500 44,4 1,14-10—4 2500 24,4 1,35 2,80-10“2 1 ,3-10“4
2600 40,0 4,25-10~4 2600 22,2 1,32 2,65-IQ—2 4,2-10“4
2700 35,6 1,21 - IO-3 2700 23,1 1,29 1,34-Ю-1 1,3-10“3
2800 31,3 3,15-10“3 2800 24,3 1,26 2,67-IO-1 3,5-10“3
2900 26,9 9,12-10—3 2900 25,3 1,23 5,12-10“’ 8,8-10“3
3000 22,6 2,10-10—2 3000 26,3 1,20 —
значение 60,7 ккал/моль [644], что резко отличается от старых данных
Рота (33,5 ккал/моль) [645].
Близкое значение теплоты образования получено калориметрически,,
и в [1020] оно равно 59,97±0,37 ккал/моль.
Нитрид бора плавится под давлением азота (для подавления дис-
социации) при температуре 3000° С [646].
Энергия диссоциации BN, по данным [611], находится в пределах
93—203 ккал/моль.
Спектр паров нитрида бора дает две группы линий: одну от 4371,2
до 3772,7 А, другую — от 3496,0 до 2180,0 А [642].
Коэффициент излучения нитрида бора (монохроматический, при
Z = 655 ммк) уменьшается в пределах температур 800—1700° С от 0,64
до 0,62 [340].
Термоэмиссионные свойства нитрида бора изучали при нанесении
порошка нитрида на биндере на вольфрамовый керн [170]. Ток насыще-
ния при 2000° К равен 0,04 а.
Важным свойством нитрида бора является способность к люмине-
сценции, которая была замечена уже Бальменом. Эта способность объ-
яснялась многими авторами влиянием примесей борного ангидрида,
угля и наличием в «аморфном» нитриде кристаллической его формы.
Ремеле [647] в 1911 г. наблюдал эффекты действия на фотопленку,
ионизацию воздуха, образование озона, вызов флюоресценции экрана,
покрытого цианоплатинатом бария. Отмечалось, что такими свойствами
обладал только нитрид бора, приготовленный из буры и борного анги-
дрида [648]. Аналогичные работы проводил Тиде с сотрудниками [649—
652]. Люминофорными свойствами обладает кристаллизованный нитрид
бора (перекристаллизованный через расплав борного ангидрида, буры,,
хлорида, сульфата и фосфата натрия) [633]. В работе сделаны следую-
щие выводы:
238
1) активация нитрида бора металлами в отличие от обычных кри--
сталлолюминофоров не вызывает люминесценции;
2) наилучшим активатором служит углерод, который при низких
концентрациях вызывает голубое, а при высоких — желтое свечение
фосфора;
3) увеличение содержания борного ангидрида в BN-фосфбре ме-
няет цвет свечения от голубого к бледно-зеленому;
4) нитрид хорошо активируется как люминофор при возбуждении
светом, ультрафиолетовыми, рентгеновскими лучами, а-частицами и
электронами (при возбуждении светом обнаруживается очень сильное
послесвечение в течение 5 мин, при катодном возбуждении послесвече-
ние очень слабое);
5) свечение стойко удерживается с повышением температуры до
700—800° С.
Подробное исследование люминесцентных свойств нитрида бора,
полученного различными способами и с различными активаторами, вы-
полнено в [1088]. В работе [1089] исследована электролюминесценция
весьма чистого нитрида бора и показано, что зависимость интегральной
яркости от напряжения может быть представлена выражением
В = В0 ехр (-&/]/V).
Нитрид бора, полученный при температуре 1200° С взаимодейст-
вием ВС1з и NH3 в газовой фазе, активируется остающимся в нем хло-
ром [547]. Он флюоресцирует в ультрафиолете светло-голубым цветом
и обнаруживает сильную желто-зеленую фосфоресценцию.
Как отмечалось в [654], разбухание, а также растрескивание и раз-
рушение изделий из борсодержащих материалов при нейтронном облу-
чении и связанное с выгоранием изотопа В10 вызывается сначала на-
коплением, а затем выделением гелия, образующегося при этой ядер-
ной реакции. В каркасных кристаллических решетках атомы гелия
накапливаются в значительной степени, что в конечном счете вызывает
нарушение решетки, а в случае слоистых решеток могут проходить
между слоями-сетками и эффект разбухания может быть в значитель-
ной степени снижен. Это относится в первую очередь к нитриду бора,,
действие на который облучения нейтронами должно быть незначи-
тельным.
Механические свойства изделий, спеченных из гексагонального нит-
рида бора горячим прессованием, подробно изучены в работе [638].
Свойства сильно зависят от направления измерений — параллельно
(||) и перпендикулярно (±) направлению горячего прессования свой-
ства существенно различаются. Предел прочности при сжатии для об-
разца с плотностью 2,12 г) см3 (4—5% пор) составляет 32 и 24 кг]мм2.
Модуль упругости и предел прочности при разрыве резко уменьшаются
с температурой, особенно между 400 и 700° С (табл. 90 и рис. 102). Од-
239
нако при дальнейшем повышении температуры, как это было обнару-
жено в исследовании [1090] прочность нитрида бора вновь повышается,
достигая при 1500° С максимума (рис. 103). Горячепрессованный при
низких температурах (900° С) нитрид бора, содержащий до 14% борно-
ТаблицаЭО
Температурная зависимость проч-
ности и модуля упругости нитрида
бора
Предел проч- Модуль упру- гости, /сг/жи2
ности, кг/жи2
Темпе- О । к о , я
ратура. °C : Параллельн растяжение О S 03 Я щ о. Й. х £ к £ С Параллельн растяжение S Я D ф о 2 g к Я О. о, Я с н ф С
25 11,12 5,10 8650 3440
350 10,60 4,90 6150 2430
700 2,70 1,33 1080 360
1000 1,53 0,76 1160 —
го ангидрида, после нагревания в ваку-
уме при 1400° С обнаруживает вначале
некоторое повышение прочности, а затем
прочность во времени уменьшается [641].
По тем же данным испарение борного
ангидрида из нитрида бора в аргоне или
азоте при 1800° С вызывает снижение
прочности на 70%.
Прочность нитрида бора на изгиб
практически не изменяется до 1900° К
(при 1500° К наблюдается незначитель-
ное повышение прочности) [970].
Относительно высокая механическая
прочность изделий из нитрида бора поз-
воляет обрабатывать их резанием [638].
При возрастании в них содержания бор-
ного ангидрида обрабатываемость их
значительно ухудшается [641]. Механиче-
можно изготовлять из нитрида бора сложные изде-
ской обработкой можно изготовлять из нитрида бора сложные изде-
лия— нарезные болты, гайки и т. и. (рис. 104, по [641]).
Твердость нитрида бора равна 1—2 по минералогической шкале
[656], однако она существенно изменяется при наличии борного ангид-
рида и других примесей. Удельная поверхность порошка нитрида бора
составляет 20 000—40 000 см2!г. Исключительная тонкость частиц по-
рошка нитрида бора наряду с особенностями его строения и низкой
твердостью обусловливает его смазочные свойства, более высокие, чем
у графита и дисульфида молибдена.
Коэффициент трения свежеотрезанной поверхности нитрида бора
0,03—0,07, при повторных износах он возрастает от 0,11—0,23. По дан-
ным [657], коэффициент трения возрастает при температуре 150° С до
0,4, а при 600° С уменьшается до 0,1, что объясняется плавлением при-
меси В2О3 и появлением жидкой смазки. При 900° С величина ко-
эффициента трения значительно возрастает благодаря интенсивному
юкислению (см. также [1091]).
Изделия из нитрида бора стойки к абразивному износу. Например,
стойкость к действию песка эквивалентна стойкости к действию стекла.
Крупность частиц нитрида бора существенно зависит от способа
получения. В работе [658] рентгенографическим методом были измере-
ны размеры кристаллов: ширина графитоподобного слоя La и толщина
пакета из параллельных слоев Lc нитрида бора, полученного различ-
240
ними методами. Измерено также межслоевое расстояние d(002). Резуль-
таты измерений приведены в табл. 91 и показаны на рис. 105. Для ни-
трида бора BN, полученного путем газофазных реакций, Д002) больше,
Рис. 102. Температурная зависимость предела прочности нитрида бора:
/ — параллельно направлению прессования образца; 2 — перпендикулярно направлению
врессования.
Рис. 103. Температурная зависимость предела прочности при изгибе:
1— нитрид бора; 2 — материал системы В—Al—N; 3 — Si3N(.
Рис. 404. Изделия из нитрида бора.
a La и Lc меньше, чем для BN, полученного путем твердо-газофазных
или твердо-твердофазных реакций. Кристалличность BN более заметна
с повышением температуры реакции и температуры отжига образован-
ного нитрида, причем для BN, полученного по твердо-твердофазной ре-
241
акции, наблюдается в большей мере, чем для BN, полученного по газо-
фазной реакции.
Порошок нитрида бора обычно состоит из агломерированных ча-
стиц. Такие агломераты имеют форму либо круга с диаметром
мк, либо форму вытянутых
ло 0,5 и длиной до 20 мк.
пластинок с поперечным размером око-
Рис. 105. Рост кристаллов ни-
трида бора при термообработ-
ке (отжиг в среде азота при
200 мм рт. ст.):
Таблица 91
Зависимость размеров кристаллитов нит-
рида бора от способа и температуры по-
лучения [658]
Метод получе- ния Темпе- ратура реак- ции, °C rf(002), А Сг, А LC, А
BC13+NH3 900 3,619 45 12
1050 3,591 49 13
Разложение 800 3,624 56 14
BC13-4NH3 1000 3,594 68 14
800 3,389 71 45
1000 3,339 290 66
Na2B4O,4-NH3 300 3.349 265 88
1000 3,339 1160 282
B2O3-j-NaNH2 800 3,389 243 115
1 — после синтеза; 2 — отжиг при 1400° С; 3 — отжиг при 1600° С; 4— отжиг при 1800° С; Т от
жиг при 2000° С; О —' BN, полученный взаимодействием ВС13 и NH3; ф — BN, полученный
взаимодействием В,О3 н NH3.
Рентгенографическое исследование линейной сжимаемости нитрида
бора при давлениях до 160 000 кГ/см2 [659] дало следующую зависи-
мость:
~ - 34 1 (Г7р — 54 • 10~12р2.
Сжимаемость гексагонального нитрида бора при более высоких
давлениях (до 300 кбар) исследована в работе [992]. Установлено, что
сжимаемость нитрида бора при всех давлениях выше, чем у графита.
При умеренных давлениях она намного больше, чем у алмаза, а при
высоких — значительно меньше, чем у алмаза.
Кубический нитрид бора |3-BN. Под высоким давлением гексаго-
нальный нитрид бора переходит в кубическую модификацию аналогич-
но переходу гексагонального графита в кубический алмаз. Указания о
кубической модификации нитрида бора имеются давно [660, 110]. Куби-
ческий нитрид бора p-BN был получен Р. Уэнторфом в лаборатории
фирмы «Дженераль электрик», США [661, 662, 663] и назван «боразо-
ном». Кристаллы кубического нитрида бора были получены при высо-
ких давлениях (45—75 кбар) и температурах (1200—2000° С) из сме-
242
сей гексагонального нитрида и нитридов Li или Mg (в качестве катали-
заторов) с добавками некоторых примесей [664]. Добавка 0,01—1% Be
в форме металла или соли вызывает образование кристаллов кубиче-
ского нитрида p-типа, голубого цвета, с электросопротивлением
103 ом • см (в некоторых случаях получаются образцы с q = 200 ом-см).
Энергия активации проводимости, определенная из измерений электро-
сопротивления в интервале температур 25—400° С, составляет 0,19—
0,23 эв. Тип проводимости был установлен по знаку термо-э. д. с. Пред-
полагается, что бериллий замещает бор или азот в решетке кубическо-
го BN. Добавка вместо бериллия элементарной серы приводит к обра-
зованию боразона n-типа, бледно-желтого цвета, с электросопротивле-
нием 103—104 ом • см. Сера замещает азот в боразоне, на что указывает
существование соединения BS с кубической решеткой, образующегося
при высоких температуре и давлении. Энергия активации проводимости
боразона n-типа в интервале температур 25—250° С составляет 0,05 эв.
При добавке к исходной смеси соединений, содержащих азот и углерод,,
образуются кристаллы p-BN с @=105—107 ом • см и энергией активации
проводимости 0,28—0,41 эв. Иногда ₽-BN с q= 106—109 ом • см обра-
зуется непосредственно из исходной смеси нитридов бора и лития либо
магния без всяких присадок, возможно, вследствие влияния примеси
кислорода. Контакт p-BN с n-BN и p-BN с алмазом (допированным А1
или В) обнаруживает выпрямляющее действие. Измерения проводи-
лись на постоянном токе 10-6 а и напряжении 5 в.
Авторы [1014] получили p-тип BN (из смеси 1 вес. ч. Be, 4 вес. ч.
нитрида лития, 150 вес. ч. гексагонального нитрида бора под давлением
58 кбар при 2000° С в течение 15 мин). Его сопротивление оказалось
равным 1 • 10-6—5 • Ю“6 ом • см.
Согласно недавно опубликованным результатам [1067], удельная по-
верхностная энергия кубического нитрида бора равна 4720 эрг/см\
что уступает только величине поверхностной энергии алмаза
(5378 эрг!см2). По тем же данным, ширина запрещенной зоны боразона
равна около 10 эв, т. е. чистый боразон является диэлектриком.
В работе [665] методом самосогласованного поля вычислены энер-
гетические зоны кубического BN, показано, что энергия щели между
связями, которая может быть принятой за ширину запрещенной зоны,
равна около 3 эв. Зонная структура боразона исследована также в
[1093].
Геллер [666] рассмотрел связь В—N в гексагональном и кубиче-
ском нитриде бора. Расстояние В—N в кубическом BN (1,57 А) больше,
чем в гексагональном (1,45 А). Сокращение длины связи ведет к боль-
шему изменению энергии, чем для С—С-связи. Энергия связи в куби-
ческом BN — 4 ккал!моль, в гексагональном — 2—3 ккал/моль да
0,01 А. Энергия связи в кубическом BN (35 ккал/моль) ниже, чем в ал-
мазе (84,9 ккал/моль). ц-.;и
16* 243
Электронная схема образования боразона может быть представле-
на следующим образом. Атомы бора, имеющие в изолированном состо-
янии конфигурацию валентных электронов s2p, в результате «—>-р-пере-
хода приобретают «/^-конфигурацию, затем вследствие привлечения
легкоподвижного электрона атома азота — «р3-конфигурацию. Валент-
ные электроны атома азота соответственно совершают следующую
трансформацию: «2р3—>-«р4«/?3-|-р и, передавая р-электрон бору, при-
обретают «реконфигурацию. Таким образом, в боразоне создается вы-
сокий статистический вес атомов, обладающих «реконфигурациями ло-
кализованных электронов, что обеспечивает кубическую алмазоподоб-
ную структуру этого соединения. Однако статистическая подвижность
р-электрона атома азота вызывает некоторое понижение статистическо-
го веса «реконфигураций атомов бора и азота, что, в свою очередь,
обусловливает меньшую энергию связи, менее высокое электросо-
противление, меньшую ширину запрещенной зоны и меньшую твердость
боразона по сравнению с алмазом. С другой стороны, меньшая доля
жестких, направленных связей в решетке боразона вызывает несколько
большие степени свободы при термическом возбуждении, в частности,
более высокую температуру полиморфного превращения в гексагональ-
ный нитрид, которая по некоторым данным составляет даже 2000° С [662],
в то время как для алмаза она лежит в районе 900° С. По этой же
причине боразон обладает большей теплостойкостью, чем алмаз, и более
значительным сопротивлением удару [667] в результате приобретения
известной «пластичности», вызванной появлением некоторой статисти-
ческой доли нелокализованных электронов.
Плотность боразона 3,45 г/см3, период решетки а = 3,615 А.
Физические свойства ромбоэдрической модификации нитрида бора
y-BN практически не изучены. В табл. 92 приведены основные свойства
нитрида бора трех модификаций (см. также обзор [1094]).
Химические свойства. В химическом отношении нитрид бора устой-
чив в нейтральной и восстановительных газовых средах. Водород и йод
на нитрид не действуют, с хлором он реагирует при красном калении,
образуя треххлористый бор. Хлорирование горячепрессованных образ-
цов из нитрида бора идет очень медленно при температуре 700° С и
быстро — при 1000° С [638] (табл. 93). Сухим кислородом и СО2 BN
окисляется быстро при температуре 700—800° С с образованием
B2O3+N2. При действии влажного воздуха, кипящей воды или разбав-
ленных кислот гидролизуется с образованием аммиака и борной кисло-
ты. Активное окисление порошка нитрида бора влажным воздухом про-
исходит при температуре 800—900° С, а при двухчасовом прокаливании
на воздухе — при 1000—1100° С степень его окисления составляет 96—
97% [668]. Окисляемость порошка нитрида существенно зависит от тем-
пературы его предварительного прокаливания («стабилизации»). Так,
при повышении температуры предварительного прокаливания в азотсо-
244
Основные свойства модификаций нитрида бора
Таблица 92
Характеристика a-BN P-BN V-BN
Содержание бора, вес. % 43,6 43,6 43,6
Кристаллическая структура Гексагональная Кубическая типа Ромбоэдр и ческая
Периоды решетки, Д: типа графита алмаза типа Р-графита
а 2,504 [626, 719] 3,615 [661—664] 2,504 [627]
с 6,661 10,01
с/а Плотность, г/см.3: 2,662 —' 4,0
рентгеновская 2,29±0,0 [626] 3,45 [661—664] ~1,80 [627]
пикнометрическая 2,20±2,35 [ 648] —. —
Насыпной вес, г/см3 0,1—0,7 [638] — —
Удельная поверхность, см2/г Теплота образования из эле- 20000—40000 [638] — —
меитов, ккал)моль 60,7 [644] —• ——-
Теплота сгорания, ккал/моль Температура плавления, °C 90,2 [644] — —
(под давлением азота) ~3000 [646] .— —
Энтропия, кал/моль .град Удельная теплоемкость, 20,77 [634] •— —
кал/моль • град Т еплопроводность, 4,65 [655] — —
кал/см-сек-град Удельное зяектросопротивле- 0,036—0,069 [638] — —
ние, ом-см Твердость по Моосу Предел упругости при сжатии, ~1013 [638] Юз—104 [664] —
1—2 [648] ~10 [661—663] —
кг/мм?' Предел упругости при растя- 24—32 [638] — —
жении, кг/мм2 5,11—11,12 [638] —•
Модуль упругости, кг/мм2 Коэффициент термического 3440—8650 [ 638] —. —
расширения X 10~6, град-1 0,5—1,7-10-6 [628] — —
Показатель преломления 1,74 [638] 2,22 [664] —
Двойное лучепреломление ~0,3 — —
держащей среде с 1000 до 1600° С скорость его окисления уменьшается
в четыре раза [639].
Особенно резко повышается стойкость нитрида бора против окис-
ления при предварительном его прокаливании в азоте при температу-
рах 2200—2400° С; при этих температурах сильно сокращается удельная
поверхность порошка нитрида и его стойкость против окисления при-
ближается к стойкости компактного нитрида бора, полученного горя-
чим прессованием [1030]. Окисление порошка нитрида начинается при
770° С и проходит тем медленнее, чем более порошок нитрида загрязнен
245
борным ангидридом. При более высоких температурах преобладает уле-
тучивание В2О3 и порошок окисляется с большой скоростью. Горяче-
прессованные, достаточно плотные образцы нитрида бора (пористость
4—5%) окисляются воздухом слабо. Убыль веса образцов при окисле-
нии, по данным (638], приведена в табл. 94.
Таблица 93
Убыль веса при хлорирова-
нии нитрида бора, мг/см2
Время, ч 700° С 1000° с
3 . - 2,7
20 0,25 17,0
40 0,55 —
Таблица 94
Убыль веса при окислении
нитрида бора на воздухе,
мг! см2
Время, ч 700° С 1000° с
2 0,014 0,35
10 0,062 0,85
30 0,138 4,8
60 0,235 10,0
С углеродом нитрид бора реагирует при температурах выше 2000° С
•с образованием карбида бора и азота [669], а при взаимодействии с ту-
гоплавкими металлами или их карбидами при высоких температурах об-
разуются соответствующие бориды [670, 671]. Стойкость нитрида бора в
форме горячепрессованных образцов в различных коррозионных сре-
дах при комнатной температуре в
Таблица 95
Коррозионная стойкость нитрида бора
Коррозионная среда Убыль ве- са, мг/см? Уменьшение предела проч- ности образ- цов* прн рас- тяжении, %
HjSOa (конц.) Нет Нет
HjSOa (20 % -ная) 10,7 60
Н3РО4 (конц.) 1,3 23
HF (конц.) 17,5 55
НЬЮ3 (конц.) 8,9 70
NaOH (20 %-ный) 8,9 82
СС14 1,3 20
Газолин 1,6 Нет
Бензин 0,4 Нет
Спирт этиловый
(95 %-ный) 14,6 48
Ацетон 13,0 32
• Исходное значение о составляло
5.18 кг/мм2.
продолжение семи дней исследована
в [638], полученные результаты при-
ведены в табл. 95. Образцы стаби-
лизированного нитрида бора доста-
точно устойчивы при температуре
190—300° С в НС1, H2SO4, Н3РО4
(чистых и с добавками окислите-
лей — КМпО4, К2СГ2О7, КС1О4).
Наиболее быстро разлагается 5%-
ной H2SO4, а лучшим активатором
разложения является КС1О4.
При химическом анализе нитри-
да бора на содержание азота (по
Кьельдалю) его обычно разлагают
кипящей концентрированной H2SO4
с добавкой K2SO4. Методики хими-
ческих анализов нитрида бора пред-
ставлены в [276, 673].
Сильно действует на нитрид
246
бора плавиковая кислота. Горячие щелочи разлагают его с выделением
аммиака [574]
3BN + ЗН2О + ЗОН--* (ВО2)3 + 3NH3.
Нитрид бора не смачивае тся расплавленным стеклом при 750° С на
воздухе; свинцовая глазурь, которая плавится при 850—900° С, смачи-
вает нитрид на воздухе, но не смачивает в инертной газовой среде; фос-
фатом бора при 1400° С в азоте нитрид смачивается. Кремний, алюми-
ний, бронзы не смачивают нитрид [641]. Последний почти не взаимо-
действует также с расплавом криолита.
Гексагональный нитрид бора адсорбирует аргон, как и графит,
причем различие в теплотах адсорбции аргона этими веществами неве-
лико (2,2 ккал!моль для нитрида; 2,3 ккал/моль для графита), при этом
адсорбированная пленка аргона нелокализована [675].
При взаимодействии с газовыми потоками, имеющими высокие ско-
рости, при температурах 6000—7000° К и атмосферном давлении [676]
нитрид бора быстро эродирует в воздухе (0,117 ф/сек • дюйм2) и более
медленно в ракетном выхлопном газе (0,061), а также в азоте (0,026).
Анализ полученных данных показывает, что повышенная эрозия в по-
токах воздуха только в небольшой степени может быть отнесена за
счет окисления, а в основном вызывается процессами массо- и тепло-
переноса, на долю которых относится 76% эрозионного удаления
нитрида (см. также [1092]).
Нитрид бора весьма устойчив в парах лития при температуре
2000—2500° С и давлении 0,1—1 мм рт. ст., существенно превосходя в
этом отношении все прочие огнеупорные материалы, особенно окиси
алюминия и циркония, а также циркон [677].
Высокую стойкость обнаруживает нитрид бора по отношению к
действию шлаков, образующихся при выплавке ферромарганца, мар-
ганца и силикомарганца, не разрушаясь при температурах 1600—
2000° С в течение 10—15 ч [678].
Методы получения нитрида бора. Известно много способов приго-
товления гексагонального нитрида бора, которые могут быть классифи-
цированы следующим образом.
1. Нагревание борного ангидрида, борной кис-
лоты или буры с цианистым натрием, калием или
кальцием [625,679] либо с амидами [717]
В2О3 г 2NaCN = 2BN г Na2O % 2СО.
Реакция осуществлялась с использованием цианистого кальция при
температуре 1200° С [630], цианистого натрия при 2000° С [680],. циани-
стого водорода с бором при 750° С [681], действием мочевины на борный
ангидрид [682]. Все эти способы сложны, требуют высоких давлений и
температур и дают в результате загрязненный нитрид.
247
Несколько лучше описанный в патенте [717] метод получения нит-
рида бора путем взаимодействия борного ангидрида, либо борной кис-
лоты или ее щелочных солей с амидом щелочного металла в интервале
температур 210—1000° С (в большинстве случаев — при 300—500° С).
Из амидов наиболее целесообразно использовать амид натрия NaNH2r
а из борсодержащих соединений — буру или борный ангидрид
В2О3 + 3NaNH2 = 2BN 4- NH3 -4- 3NaOH,
N?2B4O7 4- 5NaNH2 = 4BN + NH3 + 7NaOH.
Процесс осуществляется в расплаве, в среде аммиака, при избыт-
ке амида. Продукт реакции обрабатывают водой для удаления приме-
сей, а затем стабилизируют при нагревании при температуре 1800—
2200° С в течение 2 ч. При этом нитрид становится устойчивым против
действия кислот и щелочей. Выход нитрида бора по этому методу 60—
70%, что значительно превышает выход при использовании реакции с
цианистым калием, где он составляет 22%.
2. Обработка борного ангидрида, борной кис-
лоты или ее солей хлористым аммонием [652, 683—
685]
В2О3 + 2NH4C1 = 2BN % 2НС1 4 ЗН2О,
а также хлористым аммонием с добавками магния [686]. В последнем
случае две части борного ангидрида нагревают в смеси с одной частью
магния и тремя частями NH4C1 при температуре 300° С. Тиде и Тома-
шеком [652] этот способ осуществлялся с использованием буры, смесь
которой с NH4CI в молекулярном отношении 1 : 2 прокаливается
2NH4C1 + Na2B4O7 -> 2NaCl 4- Н2О 4- 2NH3 4- 2В3О3,
В2О3 4- 2NHS -> 2BN -у ЗН2О.
Полученный продукт промывается водой и сушится.
3. По Майеру и Цаппнеру [687], нитрид бора получается пропу-
сканием тока хлорида бора в смеси с водородом с
избытком аммиака через кварцевую трубку, нагретую до 600° С. Затем
температура постепенно поднимается примерно до температуры 1000° С,,
при которой происходит реакция
4NH3 + ВС13 = BN -Ь 3NH4C1.
Этот продукт прокаливают при 1000° С в токе азота. Содержание
азота в полученном таким образом продукте составляет 55,68% (по-
сравнению с 56,02% по расчету для BN), содержание BN в продук-
те 99,44%.
248
Позднее этот метод был запатентован [705] в следующей форме.
Карбид бора подвергается хлорированию с образованием треххлори-
стого бора: В4С+6С12=4ВС134-С. Затем ВС1з обрабатывается аммиа-
ком BC13 + 4NH3 = BN + 3NH4C1. Первая стадия осуществляется пропус-
канием хлора над нагретым до 500° С карбидом бора, вторая — при на-
гревании смеси ВС1з с аммиаком сначала при 500—1000° С, а затем до
1600—2200° С (оптимальная температура 1800° С). Готовый продукт
стабилизируют нагреванием при 1000—2000° С.
Аналогичный способ образования нитрида бора использован в [574],
где BN получали пропусканием смеси N2 + BCI3 через внутреннее сопло
кварцевой «форсунки» и аммиака — через внешнее. Реакция протекала
при температуре 800—1500° С, слой нитрида осаждался на графито-
вой пластинке, расположенной над форсункой. Указывается, что вместо
аммиака можно использовать нитрилхлорид фосфора (NPC12)X и дру-
гие подобные соединения.
В патенте [706] описывается метод приготовления нитрида бора
взаимодействием в газовой фазе между этилборатом (или метилбора-
том) и аммиаком при температуре 850—900° С с последующей обработ-
кой полученного продукта аммиаком при более высоких температурах
(950—1100° С). Выход нитрида бора при этом составляет до 96%, со-
держание бора до 42,9 и азота — до 54,4%.
Второй вариант способов, основанных на использовании галогени-
дов бора, состоит в реакции бромистого бора с жидким аммиаком [689],
в результате чего образуется смесь бромистого аммиака, Вг(МН2)з и
Вг(МН)з. После выпаривания избытка аммиака смесь нагревают при
750° С в токе аммиака, получая в сухом остатке нитрид бора. Близок к
этому способу также метод Штока и Бликса [691], состоящий в разло-
жении B2NH3, которое при температуре 125° С количественно распада-
ется на BN и аммиак. Выход при этом мал, но нитрид получается очень
чистый.
4. Обработка борного ангидрида аммиаком [692,
693], азотом либо аммиаком в присутствии угля в
качестве восстановителя [639, 694—698]
В2О3 + 2NHS = 2BN 4- ЗН2О.
При прокаливании борной кислоты в смеси с углем в токе азота
при температуре 1000—1100° С выход нитрида бора не превышает 3—
4% даже при больших давлениях азота до 7 ат [696], что связано с
образованием на поверхности частиц борного ангидрида в токе аммиа-
ка очень тонкой и плотной пленки нитрида, препятствующей течению
реакции [685]. Толщина этой пленки, по данным Подсцуса [639], при
нагревании в течение 100 ч борного ангидрида в токе аммиака при тем-
пературе 1100°С составляет 10—20 мк.
249’
Авторы [694] для получения нитрида бора нагревали смесь из
12 вес. ч. борного ангидрида с 11 вес. ч. угля в токе азота. При атмос-
ферном давлении и температуре около 1700° С максимальный выход
составляет 26,6%. При проведении реакции под давлением азота 70 ат
и температуре 1600° С выход повышается до 85,8%.
Для увеличения газопроницаемости шихты и реакционной поверх-
ности помимо измельчения порошка исходного борного ангидрида пред-
ложено наносить борный ангидрид на подкладки, служащие для уве-
личения поверхности раздела фаз, а также вводить в шихту вещества,
выделяющие при нагревании летучие составляющие. Так, в работе [696]
сообщается, что при предварительном прокаливании 1 вес. ч. борной
кислоты с 2 вес. ч. фосфорнокислого кальция и последующим нагрева-
нием смеси в токе аммиака выход нитрида бора достигает 80—90%.
Хорошие результаты дает также нагревание в токе азота в графито-
трубчатой печи смеси из 2,5 вес. ч. бората кальция и 1 вес. ч. угля при
1400° С.
Известны два варианта такого способа приготовления нитрида бо-
ра [697]. Один из них рассчитан на создание губки борного ангидрида
с очень пористой и тонкой структурой и состоит в нагревании до 1200° С
смеси 1 вес. ч. борной кислоты и 3 вес. ч. NH4C1 с пропусканием аммиа-
ка со скоростью 1 л/ч • см2.
Другой вариант предполагает использование шихт с подкладками
таких составов (по весу):
32 % Н3ВО3 ф 30 % Са3 (РО4)2 ф 38 % NH4C1,
40 % Н3ВО3 ф 20 % СаСОз % 40 % NH4C1,
45%Н3ВО3 % 15%MgCO3 + 40%NH4Cl.
Процесс азотирования ведется, как и в первом случае, в течение
24—40 ч с последующей отмывкой порошка нитрида кислотами.
В патентах [699, 707] рекомендуется использовать в качестве под-
кладки фосфорнокислый кальций, смешивая его с борным ангидридом
в эквимолярном соотношении, с нагреванием смеси в среде аммиака
при 800—1000° С (оптимальная температура 900° С).
Такие же рекомендации приведены в патенте [708], однако в каче-
стве подкладки используют не один фосфорнокислый кальций, а его
смесь с нитридом бора. Продукт азотирования смеси такой подкладки
с борным ангидридом при 900° С обрабатывают сначала кислотами для
отмывки примесей и борного ангидрида, а затем стабилизируют нагре-
вом при температурах порядка 1200—1500° С (длительность при 1200° С
составляет 40—60 ч, при 1300° С — 5—16 ч, при 1400° С — 1—5 ч, при
1500° С — 0,5—1 ч). Содержание BN в таком продукте 95%, плотность
1,9 г/см3, максимальный размер частиц стабилизированного нитрида бо-
ра 1,5 мк, а средний размер — около 0,3 мк.
250
Подобного рода технологические процессы рекомендованы для
производства [641].
Подсцус [639] в качестве подкладки, составляющей до 70% веса
шихты, использовал нитрид бора, азотирование осуществлялось в печи,
показанной на рис. 106, при температуре 1000° С. Два угольных элек-
трода 1 (8X8 см) с медными шинами 2 обеспечивают подвод тока к
реакционной смеси <3. Между электродами помещен тонкий угольный
стержень 9, играющий роль нагревателя в начале процесса. Для тепло-
изоляции печь футеруется нитридом бора 4, цирконом 5 и шамотным
кирпичом 6, далее следует железный кожух 7 с соответствующей арма-
турой. Аммиак подается по трубе 8. Кроме того, им была разработана
печь для непрерывного процесса получения нитрида, основанная на
встречном движении через трубу печи борного ангидрида и тока аммиа-
ка. Производительность такой печи диаметром 60 мм составляет 0,6 кг
нитрида в 1 ч.
Рентабельность использования нитрида бора в качестве подкладки
подтверждается также Москвиным [653].
Нитрид бора можно получить из «губки» (В2О3 + С), образуемой
дегидратацией борной кислоты в смеси с углем [638]. Губка помещается
в лодочки из нитрида бора и обрабатывается аммиаком. Установлено,
что наиболее удобно получать нитрид бора путем обработки шихты
B2O3 + NH4CI аммиаком при температуре 1100—1200° С [700, 701]. Ших-
та приготавливается на основе спека В2О3 + СаО, полученного путем на-
гревания смеси борной кислоты с мелом, во время которого происходит
обезвоживание борной кислоты и разложение СаСОз с образованием
весьма тонкого распределения борного ангидрида по частицам СаО и
частичным образованием бората кальция (рис. 107). При соотношении
компонентов в смеси (в молях) Н3ВО3: NH4C1 :CaSO3= 1:1:0,25 по-
лучается нитрид бора, почти точно соответствующий формуле BN, с
суммой содержаний бора и азота, близкой к 100%, а выход достигает
‘92—93% вместо 70—80% по ранее предлагавшимся аналогичным спо-
собам. Необходимо отметить, что таким способом образуется наиболее
тонкозернистый порошок нитрида бора.
К этой же группе способов получения нитридов бора относится на-
гревание в токе азота борнокислого кальция в смеси с углем [648]
СаВ4О7 + 8С + 3N2 = 4BN + Ca(CN)2 У 7СО.
Для приготовления нитрида бора для люминофоров использовали
также прокаливание буры с хлористым аммонием [702].
Авторы [703] детально исследовали процесс азотирования В2О3 в
смеси с углем, описываемого суммарной реакцией
В2О3 + ЗС 4- N2 = 2BN + ЗСО.
251
При этом, как и в [638], использовали «губку», полученную дегид-
ратацией смеси борной кислоты с сажей.
Для азотирования спек порциями по 200—300 г загружали в гра-
фитовые лодочки, которые помещали в электропечь сопротивления с
графитовым трубчатым нагревателем (печь Таммана), в которую пода-
вался ток азота, очищенного от кислорода и влаги.
Результаты предварительного исследования показали, что из ших-
ты стехиометрического состава 71 вес.% Н3ВО3 и 29 вес.% сажи полу-
Смешение
О | Шихта! [ СО2
НН. О.
Прокаливание-
I Спек I
—
Измельчение
ПросеВ
NH3 | Шихта /7~|
Азотирование
HCL \BN+CoO\
Отмывка,
I BN | V CaClt
Сушка Троп
| Порошок нитрида 6ооа\
Рис. 106. Печь для получения нитрида бора (по Подсцусу).
Рис. 107. Технологическая схема получения нитрида бора.
чается продукт, сильно загрязненный углеродом, поэтому дальнейшие
опыты проводили с шихтами, содержащими меньшее количество са-
жи— 25, 20, 15 и 10 вес.% против 29% по стехиометрии. Предполага-
лось, что будет достигнут баланс между оставшимся в шихте после ча-
стичного улетучивания борным ангидридом и уменьшенным содержа-
нием сажи в шихте, а отчасти недостаток углерода в шихтах будет
восполняться за счет графита лодочки и трубы печи путем переноса
через газовую фазу, содержащую окись углерода. Смешение и дегидра-
тацию проводили так же, как указано выше, однако время размола опе-
ка с уменьшением содержания сажи в шихте увеличивалось и для спека,
полученного из шихты с 10% сажи, составляло 8—10 ч. Как следует их
252
рис. 108 и 109, начиная с температуры азотирования 1600° С в большин-
стве случаев сумма содержаний бора, углерода и азота в продуктах
азотирования близка к 100%, что указывает на окончание процесса
восстановления борного ангидрида.
Максимальное содержание азота в продуктах азотирования дости-
гается при 10% сажи в шихте, равномерно снижаясь при 15 и 20% и
резко при 25% сажи в исходной шихте. Выход нитрида бора, напротив,
увеличивается с повышением содержания сажи в шихте. Получаемые
Рис. >108. Зависимость содержания азота в продуктах азотирования опеков борного ан-
гидрида с сажей от температуры и содержания сажи в исходной шихте:
1 — 10; 2—15; 3 — 20; 4 — 25 вес.% сажи.
Рис. 109. Зависимость выхода нитрида бора от содержания сажи в исходной шихте и
температуры азотирования:
1 — 10; 2—15; 3 — 20; 4 — 25 вес.% сажн.
при больших содержаниях сажи продукты состоят из смеси карбида и
нитрида бора, что свидетельствует о преимущественном образовании
карбида бора.
Механизм восстановления и азотирования на основе полученных
данных может быть описан следующим образом. Вышеприведенная ре-
акция процесса является суммарной и состоит из реакций восстановле-
ния борного ангидрида углем до бора и науглероживания или азотиро-
вания последнего. При малых содержаниях углерода в исходной шихте
весь он расходуется на восстановление борного ангидрида, образую-
щийся бор азотируется до нитрида, а избыток борного ангидрида уле-
тучивается, что обусловливает малый выход. С повышением содержания
сажи в шихте количество восстановленного борного ангидрида увели-
чивается, степень азотирования его при прочих равных условиях сни-
253
жается, а выход возрастает. При 25 %-ном содержании сажи в шихте-
образующийся восстановленный бор связывается с углем в карбид, на-
ряду с этим часть бора азотируется с образованием нитрида. Из
рис. 109 видно, что максимальный выход нитрида бора во всех случаях
достигается при 1600—1700° С, когда высокая летучесть борного ангид-
рида подавляется реакциями восстановления и азотирования. При бо-
лее высоких температурах преимущественное значение имеет высокая
летучесть борного ангидрида, то же наблюдается и при более низких
температурах, недостаточных для развития реакций восстановления и
азотирования, как было показано в [714].
Оптимальными условиями получения нитрида бора являются, та-
ким образом, использование шихт, при азотировании которых наиболее
полно проходит процесс восстановления борного ангидрида, но не оста-
ется избытка угля, могущего связаться с бором в карбид; температуры,
при которых превалируют процессы восстановления и азотирования над
процессом испарения борного ангидрида; время азотирования, достаточ-
ное для азотирования образовавшегося бора. Исходя из полученных ре-
зультатов, для трехчасового азотирования, оптимальным режимом
является температура 1700° С, а наиболее благоприятным содержанием
сажи в исходной шихте—15%. Установлено, что выход нитрида и со-
держание азота в нем могут быть еще повышены при использовании
двухстадийного азотирования при температурах 1500 и 1700° С. Химиче-
ский состав такого нитрида: 43,1—43,4% В, 55,2—55,9% N и до 0,1% С.
Следовательно, неудовлетворительные результаты опытов Штелера
и Эльберта [694] следует отнести за счет использования ими не спеков,
а смесей борного ангидрида с углем, обладающих малой реакционной
поверхностью. Для подтверждения этого в работе проведены опыты с
шихтами, полученными механическим смешением. Обнаружено, что вы-
ход нитрида бора не превышает 3—4%, а продукты азотирования силь-
но загрязнены углеродом.
Таким образом, показано, что содержание сажи в шихте, а также
температуру восстановления (азотирования) нужно выбирать в расче-
те на максимальное подавление высокой летучести борного ангидрида.
При 15 вес.% сажи в шихте Н3ВО3 + С и температуре азотирования
1700° С достигается состав нитрида бора, близкий к стехиометрическо-
му, при выходе 60—70% по сравнению с выходом в 26% и низким ка-
чеством нитрида, получавшимися в предыдущих работах азотированием
механических смесей борного ангидрида с углем.
Подобный способ эксплуатируется в СССР в крупных масштабах
промышленного производства нитрида бора для последующего изготов-
ления огнеупорных изделий.
5. Непосредственное азотирование бора. Еще Му-
ассан [709] предлагал получать нитрид бора действием на бор закиси
азота, в работе [710] описано непосредственное азотирование аморфно-
254
го бора в струе азота. Продукт, получавшийся при температуре 1600° С,
содержал 94,3% BN, повышение температуры до 2000° С увеличивает
выход до 99,5 %.
Температурная зависимость константы азотирования порошка бора
(рис. НО) резко изменяется при переходе от 1200 к 1300° С >[704]. Так
как рентгенографическое исследование не обнаруживает при этом ка-
а— изотермы азотирования; б — температурная зависимость лога-
рифма константы азотирования.
ких-либо структурных изменений и появления новых фаз, то следует
полагать, что замедление диффузии при температурах выше 1200° С и
одновременное уменьшение энергии активации происходит из-за исправ-
ления решетки, сводящегося к достройке уже образовавшихся плоских
слоев из атомов бора, не все свободные узлы которых заполнены ато-
мами азота. Таким образом, происходит заполнение узлов уже постро-
енной основы решетки, на что требуется энергия, меньшая, чем для
организации самой решетки, однако процесс заполнения идет относи-
тельно медленно вследствие «поиска» атомами азота вакантных мест.
Из уравнений температурной зависимости коэффициента реакционной
диффузии азота в бор
£>боо-12оо°с = 30100 ехр (— 30650/7),
^1зоо—15оо°с = 20,3• 10 5ехр(—2000/Т)
видно, что энергия активации при переходе от температуры 1200 к
1300° С уменьшается с 61,3 до 4 ккал/моль. Последняя величина близка
255
к энергии сцепления между слоями атомов в решетке графита, равной
4,36 ккал/моль, т. е. миграция атомов азота происходит не в плоских
сетках дефектной структуры нитрида, а между ними. Величина
4 ккал/моль близка к величине энергии связи между плоскими сетками
в структуре нитрида.
Метод получения нитрида бора непосредственным азотированием
бора дорог и не может использоваться в промышленности [641].
Выше было упомянуто об образовании кубической модификации
нитрида бора — боразона, которое происходит аналогично образованию
искусственного алмаза — под высокими давлениями и при высоких
температурах с использованием в качестве катализаторов добавок ще-
лочных и щелочноземельных металлов (по [663] — под давлением
62 000 ат и при температуре выше 1350° С).
Без катализаторов кубические кристаллы нитрида бора не образу-
ются д<1же под давлениями в 100 000 ат и температурах более 2000° С.
Уэнторфф [664] показал, что добавки переходных металлов не содейст-
вуют образованию боразона. Положительные результаты дают добавки
щелочных, щелочноземельных металлов, сурьма, олово и свинец. Заме-
чено, что давления и температуры, необходимые для аллотропического
превращения, возрастают с увеличением атомного веса добавки. Можно
полагать, что «каталитическое» действие указанных добавок заключа-
ется в передаче электронов атомам бора с повышением статистического
веса «реконфигураций, необходимого, в свою очередь, для образования
боразона. Именно поэтому хорошими катализаторами являются щелоч-
ные и щелочноземельные металлы, легко отдающие свои валентные
электроны на стабилизацию «/^-конфигураций, а также олово и свинец,
которые, кроме способности достаточно легко передавать нелокализо-
ванные электроны, имеют сами по себе известный статистический вес
зр3-конфигураций, образованных локализованной долей валентных
электронов. Так же и сурьма является источником «^-конфигураций и
легкоподвижных электронов, образующихся по схеме «2/?3^>-«/24->-«/23-J-p.
Следующий за ней висмут не должен обладать такими свойствами, так
как часть его валентных электронов переходит на вакантную /-оболоч-
ку. По этим же соображениям эффективность «катализаторов» умень-
шается с повышением атомного веса добавки. Для щелочных металлов,
а также и щелочноземельных при формальном увеличении атомного ве-
са происходят переходы валентных электронов на вакантные d-состоя-
ния с увеличивающейся при росте главного квантового числа стабиль-
ностью конфигураций электронов на этих состояниях. Последнее при-
водит к уменьшению возможности передачи валентных электронов для
повышения статистического веса «/^-конфигураций атомов бора в нит-
риде бора. С этими же обстоятельствами связано «каталитическое» дей-
ствие нитридов указанных выше металлов. При образовании нитридов,
например, щелочных металлов, повышается вероятность перехода ва-
256
лентных электронов атомов этих металлов к атому азота, что предотв-
ращает переходы электронов на d-состояния и образование ими трудно-
нарушаемых стабильных конфигураций. С другой стороны, лишь часть
этих электронов образует с азотом э^-конфигурации, они слабо привя-
заны как к атомам металла, так и азота, и могут принимать участие в
образовании зр3-конфигураций атомами бора. Роль этих нитридов как
сред, через которые происходит перекристаллизация гексагонального
нитрида в кубический либо сомнительна, либо играет второстепенную
роль [614]. О механизме действия катализаторов см. также [1095—
1098].
Оригинальный метод получения боразона описан Виккери [718], со-
стоящий в замещении фосфора азотом в фосфиде бора ВР, имеющем
алмазоподобную структуру
ВР н- NH3 - BN <РН3.
Авторы [817] предлагают получать кубический нитрид бора взаимо-
действием боранов или хлоридов бора с аммиаком, азотом или смесью
азота с другими газами, например, водородом, с введением в зону ре-
акции затравок, имеющих кубическую структуру (a-Fe, цинковая об-
манка) в форме тончайших частиц аэрозоля.
Ромбоэдрическую модификацию нитрида бора получают взаимо-
действием NaBCh с NH4C1 при температуре 1000° С, буры с цианистым
калием, а также разложением трихлорборазола при 900° С [627]. В чи-
стом виде ромбоэдрический нитрид не получается, а образуется в сме-
сях с гексагональным.
Приготовление изделий и материалов из нитрида бора. Изделия из
нитрида бора приготовляют либо спеканием предварительно спрессо-
ванных различными способами заготовок, либо горячим прессованием.
В первом случае используют гидростатическое прессование в эластич-
ных формах под давлением 7 т)см2 с последующим спеканием в среде
азота или сухого аммиака [669]. Отмечается благоприятное влияние на
процесс спекания добавок к порошку нитрида летучих примесей, осо-
бенно В2О3. Согласно [641], заготовки из порошка нитрида бора прес-
суют с использованием в качестве связки воды, добавляемой в неболь-
ших количествах. После сушки при температуре 110° С заготовки спе-
кают в среде аммиака при 1800° С или среде азота при более высоких
температурах. Одновременно со спеканием происходит очистка нитрида
бора от летучих примесей.
Для прессования заготовок в металлических прессформах рекомен-
дуются давления 0,7—2 т!см2 с последующим спеканием при темпера-
туре ~ 1400° С. Плотность изделий при этом способе мала и составляет
1,1—1,2 г/смг, усадка в процессе спекания не наблюдается.
Для получения плотных изделий из нитрида бора применяется го-
рячее прессование. Так, в [638] горячим прессованием образуются об-
17—272
257
разцы нитрида бора с плотностью до 2,1 г!см3, т. е. с пористостью 5—
7%. Рекомендуется проводить горячее прессование в графитовых прес-
формах при 1500—1900° С под давлением не менее 16 кг/см2 [699, 707],
или при 1700—1900° С под давлением 140 кг!см2 [708].
Существенно влияет на плотность горячепрессованных изделий из
нитрида бора давление прессования. Так, изделия из нитрида бора с
относительно высокой плотностью (94%) получаются горячим прессо-
ванием под давлением 600 кг/см2 при температуре 1900° С [974].
Исследование горячего прессования BN, выполненное в работах
[975—1099], показало, что наиболее активно спекается при горячем
прессовании порошок нитрида бора, синтезированный при низких тем-
пературах (из шихты НзВОз + Са3(РО4)2 в токе аммиака при 900—
1000° С). Высокая дисперсность этого порошка и несовершенства струк-
туры обусловливает его рекристаллизацию и активное спекание. По-
этам данным оптимальными условиями горячего прессования являются:
температура 1800° С и давление 300 кг/см2. Полученные при этих ре-
жимах образцы имеют плотность 2,06—2,08 г/см3, т. е. пористость по-
рядка 0,1 % При спекании частицы нитрида бора резко укрупняются
от 0,06—0,2 мк в порошке низкотемпературного нитрида до 60—80 мк
в горячепрессованном образце.
Авторы [641] изучали влияние добавок борного ангидрида к нитри-
ду бора при горячем прессовании при 1700—1900° С и давлениях 100—
200 кг!см2. Прибавление борного ангидрида позволяет увеличить плот-
ность изделий до 2,6 г/сл*3, но химическая стойкость получаемых изде-
лий сильно снижается. Для приготовления плотных технических изделий
на основе нитрида бора в этой работе использовали добавки алюмоси-
ликатов кальция (5—10%), фосфата бора ВРО4 (10%), а также алю-
миний (до 5%) и нитрид алюминия (1—5%).
Новым видом технических материалов из нитрида бора является
пиронитрид бора, представляющий кристаллоориентированные образо-
вания, аналогичные пирографиту. Структура и свойства пиролитическо-
го нитрида бора описаны в [711—714]. Получение пиролитического нит-
рида аналогично получению пирографита [715—716] и заключается во
взаимодействии летучих соединений бора и азота при определенных
технологических условиях.
Пиронитрид бора осаждается на горячей подложке в форме плотно-
го мелкозернистого вещества, прозрачного в мелких кусочках и полупро-
зрачного в слоях толщиной около 0,5 мм. Структура материала пред-
ставляет столбчатые конусы, характерные для пирографита. Однако,
как отмечается в [712], предпочтительная ориентация пиронитрида бора
выражена гораздо слабее, чем пирографита.
Свойства пиролитического нитрида бора существенно зависят от
направления — параллельного (а) или перпендикулярного (с) поверх-
ности осаждения
258
В табл. 96 приведены некоторые свойства пиролитического нитри-
да бора.
В сравнении со свойствами обычного гексагонального нитрида бо-
ра (см. табл. 92), пиролитический нитрид обладает О’ень высокой теп-
лопроводностью в «с»-направлении, значительно более высокими проч-
ностными свойствами и отрицательным коэффициентом термического
расширения при температурах 25—700° С (в «н»-направлении).
Таблица 96
Свойства пиролитического нитрида бора
Характеристика Параллельно к поверхности осаждения (а—направле- ние) Перпендику- лярно к поверх- ности осажде- ния (с—направ- ление)
Кристаллическая структура Гексагональная типа
грае жта
Коэффициент термического расширения,
град—1: 20° С —2,9-Ю-6
700° С 0 40,5- 1О'~с:
Кажущаяся плотность, г!см? 2,2 —
Относительная плотность, % 97 —
Теплопроводность, кал/см-сек-град 0,496 0,0019
Модуль упругости, кг/см2 * 31-Ю4 * —
Прочность при изгибе, кг/мм? Прочность при растяжении, кг: мм1-. 19,25 —
25°С 4,12 —
1650°С 5,60
2000° С 7,63 —
2200° С 8,22 —
По скорости окисления пиролитический нитрид бора более стоек
по сравнению с пирографитом, а также с пиролитическим карбидом бо-
ра (рис. 111) [714].
2. НИТРИДЫ АЛЮМИНИЯ
Первая опубликованная работа по соединениям алюминия с азотом от-
носится к 1862 г. [571]. Дальнейшие исследования условий получения и
свойств этих соединений описаны Меллором [572].
По существу во всех этих работах было обнаружено только одно
соединение — A1N, которому иногда приписывали формулу AI2N2.
Нитрид алюминия A1N кристаллизуется в гексагональной решетке
типа вюртцита. По различным данным значения периодов решетки ко-
17*
259”
леблются в пределах а = 3,10—3,13; с = 4,93—4,98 А в зависимости от
степени чистоты препаратов [573].
Азот практически не растворяется в алюминии [602, 603, 604].
Физические свойства. Порошкообразный нитрид алюминия обычно
белого цвета, монокристаллы — водянисто-белого (прозрачны), при за-
грязнении примесями оксикарбида
алюминия АЬОС нитрид приобре-
тает голубую или голубоватую
окраску (в этом случае содержание
оксикарбида достигает 4—7%).
Данные по температуре плавле-
ния A1N весьма разноречивы (от
Рис. 111. Скорость окисления различных пиролитических материалов
./ — пирографит; 2 — пиролитический В4С; 3— пиролитический BN; 4 — пироли-
тический сплав BN—С.
Рис. 112. Температурная зависимость электросопротивления AIN.
2000 до 2500° С), что понятно, так как нитрид алюминия разлагается до
достижения температуры плавления на компоненты, температура нача-
ла разложения определяется особенностями условий проведения опре-
деления «температуры плавления» [573, 574]. Твердость по минералоги-
ческой шкале Мооса определена также в широком интервале (от 5 до
9—10 единиц), твердость по Кнупу (микротвердость при нагрузке 100г)
в среднем составляет около 1200 кг/мм2-. Примесь оксикарбида не из-
меняет этого среднего значения [573]. Физические свойства нитрида
алюминия наиболее полно исследованы в [575, 1100]; образцы содер-
жали до 4% С, по-видимому, в форме оксикарбида. Температурная за-
висимость электросопротивления типична для полупроводников и ди-
электриков (рис. 112), рассчитанная ширина запрещенной зоны A1N
равна A£=4,26 эв. На рис. 113 показана температурная зависимость
коэффициента теплопроводности. По величине электросопротивления и
260
теоретическому значению числа Лоренца для полупроводников рассчи-
тана электронная составляющая теплопроводности, оказавшаяся равной
для 673° К 4,6- 10'15, а при 1473° К —2,5- 10“7 вт[м-град. На рис. 114
показаны частотные зависимости диэлектрической проницаемости и
угла диэлектрических потерь.
Отмечается аналогия электрических свойств A1N и AI2O3 [573]. Ди-
электрическая константа при комнатной температуре составляет 8,5,
Рис. 113. Температурная зависимость коэффициента теплопроводности A1N.
Рис. 114. Частотная зависимость диэлектрической проницаемости и угла ди-
электрических потерь A1N.
что близко к значению диэлектрической константы А120з, для которой
ее значение находится между 9 и 10. Диэлектрическая константа A1N
быстро возрастает с температурой при низких частотах и медленнее —
при высоких, так что при частоте 8,5 • 109 сек-1 изменение диэлектри-
ческой константы с температурой весьма незначительно. На рис. 115 и
116 показано изменение диэлектрической константы и фактора диэлек-
трических потерь с частотой и температурой. Отличие этих значений от
значений диэлектрической константы, показанной на рис. 114, по дан-
ным [575], связано с загрязненностью в последнем случае нитрида алю-
миния карбонитридом. При низких частотах диэлектрические потери
быстро возрастают с температурой, однако при частоте 8,5 • 109 сек'1
изменение с температурой вплоть до 500° С происходит медленно. При
комнатной температуре коэффициент потерь находится в пределах
0,01—0,001, в то время как для А120з— в пределах от 0,001 до 0,0001.
При высоких температурах и низких частотах коэффициенты потерь A1N
и А12О3 сравнимы, в то время как при высоких температурах и часто-
тах рассеяние у А12О3 гораздо ниже.
Физические свойства нитрида алюминия показывают, что A1N яв-
ляется типичным диэлектриком с большой шириной запрещенной зоны,
261
высоким электросопротивлением, доходящим, по данным [574], до
Ю20 ом • см. Расчет электронной составляющей теплопроводности пока-
зывает, что теплопроводность осуществляется только колебаниями ре-
шетки. Так как для неполярных твердых диэлектриков справедливо со-
отношение е = п2, где е — диэлектрическая проницаемость, п — коэффи-
циент преломления (для A1N : 8=8,5, п = 2,13, т. е. это соотношение не
выполняется), то поляризация нитрида не может быть объяснена одной
Рис. 116. Температурная зависимость угла диэлектрических потерь A1N.
Рис. 115. Температурная зависимость диэлектрической проницаемости A1N.
только электронной составляющей и она, по-видимому, носит ионный ха-
рактер. В работе [575] даются следующие представления о механизме
образования связи в нитриде алюминия. Один из электронов азота пе-
реходит к атому алюминия с образованием таких электронных конфи-
гураций: АК 3s23p2; N+ 2s22p2. В соединениях со структурой типа
вюрцита каждый атом металла окружен четырьмя атомами неметалла,
расположенными на равных расстояниях от него по вершинам правиль-
ного тетраэдра (в A1N это несколько искажено). Таким образом, на каж-
дый атом приходится четыре равноценные связи, каждая из которых
осуществляется s-р-электронами, поступающими по одному от каждого
атома. Такие связи ковалентного типа, упрочненные наложенными на
них ионными связями, приводят к высокой жесткости решетки, что оп-
ределяет малое значение коэффициента термического расширения, вы-
сокие значения модуля нормальной упругости, характеристической тем-
пературы и фононной составляющей теплопроводности.
Такая схема представляется в общем правильной, однако с учетом
s-+p-переходов необходимо полагать, что конфигурация части атомов
алюминия и азота имеет вид sp3. Кроме того, наряду с переходом элек-
262
трона от атома азота к атому алюминия происходит переход валент-
ных электронов от части атомов алюминия к атомам азота с образова-
нием при этом конфигураций Al 2s22pe и N 2s22p6, что и обусловливает
наличие доли ионной связи в нитриде алюминия.
В работе [576] для нитрида алюминия в отсутствие в спектральной
области 2,5—5 эв края поглощения найдено большее значение ширины
запрещенной зоны (А£>5 эв), с другой стороны, в [577], где исследо-
ваны оптические и электрические свойства A1N, показано, что АЕл; 3,8 эв.
В этой же работе установлено, что основная полоса поглощения лежит
в ультрафиолетовой области с границей при 3200 А. В той же области
наблюдается слабая фотопроводимость и образование фото-э. д. с.
Ширина запрещенной зоны A1N близка к зонам SiC и ZnO, имею-
щим сходную структуру и близкие значения показателей преломления
(атомы Si могут замещать А1, а атомы С или О — атомы азота).
Обнаружены две полосы поглощения в области 0,60—0,65 мкм и в
близкой к УФ-части спектра, что объясняет окраску кристаллов и ука-
зывает на существование двух групп примесных уровней со средними
энергиями активации ~2 и ~3,2 эв. Третья полоса обнаруживается в
УФ-области, что соответствует третьей группе уровней с энергией ак-
тивации -~0,6 эв. При действии ультрафиолетовых лучей нитрид алю-
миния дает кратковременную люминесценцию (желтую или желто-зе-
леную) в интервале %=0,45—0,65 мкм. Фотоэлектрический эффект на-
блюдается лишь при очень интенсивном облучении светом электриче-
ской дуги.
В работе [1016] исследована фотопроводимость в монокристаллах
A1N, наведенная лазером.
Тщательное исследование структуры A1N выполнено в [578]. Пока-
зано, что структура A1N отличается от идеальной структуры вюртцита,
так как отношение da равно 1,600 вместо 1,633 и параметр U, опреде-
ляющий расстояние А1—N вдоль тригональной оси, равен 0,385 вместо
0,375. Сжатие тетраэдра вдоль оси С в структуре A1N приводит к не-
которому искажению правильного тетраэдрического расположения свя-
зей А1—N. Увеличение значения параметра U указывает на то, что
центр электронной плотности в каждом атоме не совпадает с центром
тетраэдра, образованного его ближайшими соседями, что атом смещен
вдоль оси С к основанию тетраэдра на 0,05 А. Следовательно, углы
между связями А1—N колеблются от 107,7 до 110,5°, а расстояния А1—N
от 1,885 до 1,917 А.
При исследовании спиралей роста и политипии кристаллов A1N, по-
лучаемых из газовой фазы [579], показано наличие спиралей, хорошо из-
вестных для SiC (рис. 117), причем они варьируют от полигональных
до почти круглых, а высота ступенек слоев роста составляет несколько
сотен А. В противоположность изоструктурным SiC и ZnS для нитрида
алюминия рентгенографически не были установлены политипные струк-
263
туры, „поэтому спирали роста не могут объясняться на основе полити-
пии, как это делается, например, в случае карбида кремния.
Термодинамические свойства нитрида алюминия наиболее подробно
изучены в [580], результаты исследования приведены в табл. 97. Изме-
нение теплосодержания в области 298—1800° К Ят—Н№& = 10,98Г +
Рис. 117. Спирали роста
на плоскости (0001) AIN. Х160.
+ 0,4 • 10 3Т2 +3,58 Ю5?В * * 11—4,51, температурная зависимость теплоем-
кости ср= 10,98 + 0,80 • 10-3Т—3,58 • 105Т“2. Изменение изобарного по-
тенциала реакции A1+1/2N2 = A1N с температурой следующее:
Т, °К
— &F°, ккил
298 1000 1100 1400 1500 1700 1900 2000
68,15 50,20 47,40 39,00 35,25 30,70 25,20 22,44
В работе [584] проведено масс-спектрометрические исследования
испарения A1N, определено приблизительное теплосодержание
—63 ккал)моль, которое значительно ниже калориметрически опре-
деленного.
Прочность нитрида алюминия при высоких температурах (порядка
1400° С) может быть сопоставлена с прочностью окисной керамики, при
264
Свойства нитрида алюминия
Таблица 97
Характеристика A1N
Содержание, вес. % 34,9
Кристаллическая структура Гексагональная типа вюртцита
Периоды решетки, д: 3,104 [581]
а
с 4,965
da 1,600
Плотность, г/см2:
рентгеновская 3,27 [583]
пикнометрическая 3,12
Температура плавления, °C 2400 [578] разлагается
Теплота образования, ккал/моль 76,47±0,20 [582], см. также [580]
Энтропия, кал/град -моль 4,8±0,02 [580]
Теплопроводность, кал/см-сек-град: 0,072 [578], см. также [575]
200° С
400° С 0,060
600° С 0,053
800° С 0,048
Коэффициент термического расширения, 10е-грлд—1: 4,03 [578], см. также [585]
25—200° С
25—600° С 4,84
25—1000° С 5,64
25—1350° С 6,09
Удельное электросопротивление, ом-см >1013 [575]
293° К
673° К 2,25-10й
773° К 109
873° К 8-Ю7
1073° К 4-10°
1173° К 7-105
1273° К 105
1373° К 4-Ю4
1473° К 9-Ю3
Ширина запрещенной зоны, эв 3,8 [577] до >5 [576]
Диэлектрическая константа 8,5 [573]
Коэффициент преломления 2,13 [583]—2,20 [1003]
Коэффициент излучения (Х=655 мк): 0,80 [340]
800—2000° С (в среде аргона)
800—1400° С (в вакууме) 0,85 [340]
Твердость по Моосу ~9 [573]
Твердость по Кнупу, кг/мм2 1230 [573]
Модуль упругости, кг/мм2: 35050 [573]
20° С
1000° С 32300
1400°С 28100
Предел прочности при разрыве, кг/мм2: 27 [573]
25° С
1000° С ~19
1400° С 12,7
265
Продолжение таблицы 97
Характеристика
Давление паров, мм рт. ст.:
1000° К
1200° К
1400° К
1600° К
1760° К
2173° К
Энергия диссоциации, ккал/моль
AIN
5-10—12 [610]
2-10-8 [610]
1-10-5 [6Ю]
0,9-10—4 [610]
1 [150, 1101]
14 [45]
82—137 [611]
обычной температуре несколько уступает окислам. Теплопроводность
технического A1N на порядок меньше теплопроводности плотного кар-
борунда и в 2—3 раза выше теплопроводности керамики из А12О3 [573].
Термическое расширение при средних температурах немного превышает
тепловое расширение карборунда. Сопротивление тепловому удару вы-
сокое, так при теплосменах 22'00°—20° С изделия из BN не разрушаются
в течение нескольких циклов. После 30 циклов нагрева в течение 2,5 мин
до 1400° С и быстрого охлаждения до комнатной температуры на воз-
духе потеря прочности составляет 12% [585].
Химические свойства. Изделия из нитрида алюминия медленно рас-
творяются в горячих минеральных кислотах (табл. 98, [578]).
На порошкообразный нитрид алюминия соляная, азотная, серная
кислоты, царская водка на холоду действуют очень слабо [610]. При
нагревании в H2SO4 (1 : 1) нитрид алюминия полностью разлагается за
четыре дня (2A1N + H2SO4+6H2O = 2A1(ON)3+ (NH4)2SO4). Холодная
плавиковая кислота на A1N также не действует. Концентрированные
горячие растворы щелочей разлагают нитрид алюминия с выделением
аммиака:
A1N + ЗН2О + ОН~ -> А1 (ОН)Г + NH3.
Компактный нитрид алюминия значительно устойчивее к действию
кислот и щелочей, чем порошкообразный. Так, компактные образцы
A1N совершенно устойчивые при кипячении в концентрированной серной
кислоте, трудно поддаются воздействию кипящих азотной и соляной
кислот. Устойчивость возрастает в разбавленных кислотах и растворах
щелочей [612].
Сухие галогены медленно действуют на нитрид, хлор начинает раз-
лагать при температуре 760° С с образованием А1С13 [610], сухой хло-
ристый водород практически не действует. При нагревании с серой и
парообразным сероуглеродом нитрид алюминия частично разлагается,
парами хлорида серы A1N разлагается быстро, фосфор частично его
266
разлагает, РС13 не действует. Реакция с углеродом начинается при тем-
пературе 1200° С. Перекись натрия разлагает нитрид алюминия с обра-
зованием нитратов, полностью и быстро A1N разлагается бихроматом
свинца [572] *.
Окисление порошка на воздухе начинается при температуре 1200° С
[574], а по данным [612] — при 900° С. Теплота реакции окисления:
A1NTb+3/4O2= V2AI2O3+ V2N2 составляет —АН°298 = 124,6+0,0370 ккал
[580].
Скорость коррозии при действии горячих газов зависит от плот-
ности изделий из нитрида. Показано [578, 585], что при окислении спе-
ченного нитрида при 1200° С в течение 1 ч превращается в окись алю-
миния 11 % нитрида, при окислении горячепрессованного образца при
1400° С в течение 30 ч в окись превращается только 1% нитрида.
В табл. 99 приведены данные по коррозии горячепрессованного, плотно-
го нитрида алюминия в различных газовых средах.
Таблица 98
Коррозия горячепрессованного ни-
трида алюминия в воде и мине-
ральных кислотах (время испыта-
Таблица99
Коррозия горячепрессованиого нитри-
да алюминия в газовых средах
тания — 72 ч) Среда t, °C Время, ч Переход нитри- да в окись или другие соеди- нения, %
Среда t, °C Скорость коррозии, мм/год
н2о 100 14 Воздух 1000 30 0,3
НС1 (конц.) 72 320 Воздух 1400 30 1,3
НС1 (1:1) 100 570 Воздух 1700 4 10,6
H2SO4 (коиц.) 305 180 Кислород 1400 30 0,9
H2SO4 (1:1) 145 550 Сухой пар 1000 30 0,3
HNO3 (конц.) 120 150 Хлор 500 30 0,1
HNO3 (1:1) 111 200 Хлор 700 30 19,2
HF (конц.)+ 57 160 Водород 1700 4 Не взаимо-
4-HNO3 (конц.) HP (1:1) 57 215 действует
HF (1:1) 57 170
(монокрис- таллы A1N' На нитрид алюминия не дейст- вуют расплавленный алюминий (до
2000° С), галлий (ДО 1300°С), борный ангидрид (до 1400° С) [585].
расплавленного криолита и алюминия в течение
A1N устойчив в смеси
66 ч при 1200° С [578], расплавленный борный ангидрид при температу-
ре 1000° С вызывает за 4 ч потерю в весе только в 0,02%.
Нитрид алюминия активно взаимодействует с базальтом (1400° С;
0,5 ч), с медным штейном (1300° С; 5 ч), медленнее — с никелевым
* Подробное изучение стойкости A1N в минеральных кислотах и растворах щело.
чей проведено в работе [612].
267
штейном (1250° С; 5 ч), слабо — с медным шлаком (1300° С; 5 ч), не
взаимодействует с расплавом NaCl+BaCh [541].
Высокую стойкость обнаруживает нитрид алюминия по отношению
ко многим полупроводниковым соединениям, в частности, к полупровод-
никам типа AniBv. В работе [610] показано, что он исключительно стоек
по отношению к расплавленному GaAs, что позволяет использовать кон-
тейнеры из нитрида алюминия для очистки и получения монокристаллов
из этого полупроводника.
Устойчив также в контакте с графитом до высоких температур, с
вольфрамом и молибденом — до 1800° С [610].
Методы получения. Разработаны многочисленные методы получе-
ния нитрида алюминия, использующиеся как в лабораторных, так и в
промышленных масштабах.
1. Непосредственное азотирование алюминия.
Нитрид алюминия был получен впервые в 1862 г. действием азота на
алюминий при температуре 700° С [571].
Кинетику азотирования алюминия исследовали в области темпера-
тур 530—625° С [75]. При низких температурах до 580° С азотирование
подчиняется линейному закону, при более высоких температурах — па-
раболическому.
В линейной области температур константа скорости азотирования
выражается 7(=58 ехр (—17 900//??') мг/смР-ч, в параболической —
А=4,2-1010 ехр (—23 700/7??') мг/слр-ч.
Константа окисления алюминия в линейной области температур
А=2,34-1011 ехр (—47 700/7??'), в параболической — А=1,35-103 ехр
(—22 800/7??'), т. е. в обоих случаях скорость окисления намного выше
скорости азотирования.
Особенностью азотирования порошка алюминия является образо-
вание на поверхности частиц окисных пленок (у-А^Оз), которые удаля-
ются, по-видимому, в результате образования летучих низших окислов
алюминия: А12О3-|-4А1=ЗА12О. Нарушение пленки при перестройке А1гО3
в А12О позволяет азоту проникнуть к ювенильной поверхности алюми-
ния и провести азотирование.
При этом образуется тонкая пленка A1N, плохо проницаемая для
азота, что и вызывает переход от линейного закона азотирования к па-
раболическому при более высоких температурах, когда пленка уже
начинает образовываться.
Таким образом, для ускорения реакции образования нитрида алю-
миния необходимо использование наиболее тонких алюминиевых по-
рошков, которые, хотя и являются более окисленными, но тем не менее
позволяют более полно провести реакцию азотирования с образованием
нитрида. При азотировании одновременно происходит спекание алюми-
ниевого порошка, которое сокращает реакционную поверхность. Для
предотвращения спекания используются ступенчатые режимы азотиро-
268
вания, рассчитанные на то, что уже при низких температурах наблюда-
ется покрытие частиц пленками нитрида, что задержит спекание по-
рошка.
Нитрид алюминия может быть приготовлен азотированием алюми-
ниевой пудры (ПАК-4) [586]. Для получения нитрида алюминия стехио-
метрического состава обрабатывают алюминиевую пудру азотом при
800° С в течение 1 ч (скорость подъема температуры до 800° С
10 град/мин) с перемешиванием образованного продукта и повторным
азотированием при 1200° С в течение 0,5—1 ч (со скоростью подъема
температуры до 1200° С в 40 град/мин). Технический нитрид алюминия
(с содержанием 33% N) получают путем однократного азотирования
при 1200° С со скоростью подъема температуры 10 град/мин.
Медленный подъем температуры, а также ступенчатый режим азо-
тирования с промежуточными перетирками позволяет избежать спека-
ния алюминиевого порошка до азотирования.
В работе [587] проведено азотирование алюминиевой пудры (марки
ПП-1) и алюминиевого порошка с «подкладкой» из нитрида алюминия,
имеющего благодаря тонкости частиц сильно развитую поверхность и
предотвращающего спекание частиц алюминия друг с другом. Оказа-
лось, что введение 30% A1N в шихту с алюминиевым порошком или пуд-
рой достаточно для получения при одностадийном азотировании каче-
ственного нитрида алюминия (при 1200° С в продолжении 1 ч). Однако
при этом образуються рыхлые, легкоизмельчаемые спеки; производи-
тельность получения нитрида снижается из-за оборота 30% нитрида
алюминия, вводимого в свежие шихты.
Авторы [558] исследовали более подробно азотирование пудры и по-
рошка алюминия. Показано, что образование нитрида алюминия при
азотировании пудры ПАК-4 начинается уже при температуре 400° С;
при 500° С в нитрид переходит до 15% пудры, а при 600° С — до 20%
(при четырехчасовой выдержке). Особенно интенсивное образование
нитрида происходит при 720—730° С, что требует подачи дополнитель-
ных количеств азота. Температура в реакторе за 1 —1,5 мин возрастает
до 1400—1500° С и через 15—20 мин реакция практически заканчи-
вается.
Образование нитрида алюминия при азотировании алюминиевого
порошка ПА-4 начинается при 600° С, но количество получаемого нит-
рида при этом весьма незначительно и до 800° С составляет не более
1%. При повышении температуры выше 800° С часто наблюдается спе-
кание порошка и максимальный выход нитрида удается получить рав-
ным только 50—60%. Дальнейшему азотированию препятствует сплав-
ление алюминия и резкое сокращение реакционной поверхности.
С уменьшением крупности частиц алюминиевого порошка от 160—
250 до 70—100 мк и далее до 45—60 мк количество образующегося нит-
рида довольно резко увеличивается.
269
Так как с повышением температуры убыль свободной энергии реак-
ции образования нитрида уменьшается, то следовало бы ожидать более
полного прохождения реакции с понижением температуры. Результаты
исследований показывают, что, наоборот, с ростом температуры реак-
ция идет полнее.
Такое несоответствие объясняется тем, что скорость реакции обра-
зования A1N существенно зависит от скорости диффузии азота к алю-
минию. Плотная пленка окиси алюминия на частицах порошка является
существенным препятствием диффузии азота. Это подтверждается и
опытами Смиттелса и Ранслея [589], показавшими, что при удалении
окисной пленки с поверхности алюминия скорость диффузии водорода
увеличивается в десять раз. Скорость диффузии азота как через окис-
ную пленку, так и через нитридную резко возрастает с температурой,
это приводит к большей скорости азотирования, перекрывая уменьше-
ние скорости реакции алюминия с азотом, вытекающее из чисто термо-
динамических соображений. Так как реакция образования нитрида алю-
миния сопровождается высоким тепловым эффектом, то первые же
порции азота, проникшие через пленку к алюминию, вызывают сильное
повышение температуры, которое в свою очередь вызывает увеличение
скорости диффузии азота, т. е. реакция имеет своеобразный «цепной»
характер. Если в начале этого процесса прекратить внешний обогрев,
то реакция продолжается и «на холоду», проходя при достаточной до-
ставке азота до конца. Реакция образования нитрида из пудры ПАК-4
приобретает «цепной» характер при температуре 720—730° С, а из по-
рошка алюминия — при температуре выше 800° С.
Поэтому в [588] рекомендовано при получении нитрида алюминия в
юке азота из пудры ПАК-4 температуру поднимать до начала самопро-
извольной реакции, которая проходит до конца в течение 30—40 мин.
При азотировании же порошка алюминия самопроизвольная реакция
не может быть доведена до конца и обрывается при образовании 50—
70% нитрида.
В работе [590] изучали влияние давления азота на процесс азоти-
рования алюминиевого порошка ПА-4 и алюминиевой пудры ПАК-4.
При азотировании пудры увеличение давления азота способствует обра-
зованию нитрида (рис. 118). С ростом температуры положительный эф-
фект увеличения давления на скорость образования нитрида уменьша-
ется и при 665° С для времени выдержки Г20 мин, а при 690° С для
времени выдержки 60 мин становится отрицательным. При прочих рав-
ных условиях реакция образования нитрида алюминия ускоряется с по-
нижением давления.
При азотировании порошка ПА-4 увеличение давления азота уско-
ряет реакцию образования A1N (рис. 119), см. также [1102].
Такое неожиданное влияние давления на процесс азотирования алю-
миниевой пудры можно предварительно объяснить следующим образом.
270
Прежде чем вступить в реакцию с алюминием, азот должен продиффун-
дировать через окисную пленку у-А^Оз, имеющуюся на каждой частице
алюминия, а также через пленку образовавшегося нитрида алюминия.
Количество газа, диффундирующего через единицу площади на единицу
длины в единицу времени через вещество, определяется формулой Сми-
телса [591]:
где п — константа,
энергия активации
d — толщина слоя вещества, р — давление газа, Ев—
(теплота диффузии). Из данного выражения видно,
что скорость диффузии пропорцио-
Рис. 118. Температурная зависимость
вании пудры ПАК-4:
/ — давление азота 3 ат, время выдержки
держки 60 мин; 3 — давление азота 2 ат,
нальна ]р и с повышением темпера-
туры должна резко возрастать. Как
уже указано, скорость реакции об-
разования A1N лимитируется ско-
ростью диффузии азота через окис-
выхода нитрида алюминия при азотиро-
120 мин; 2 — давление азота 3 ат, время вы-
время выдержки 120 мин; 4 — давление азота
2 ат, время выдержки 60 мин.
Рис. 119. Зависимость выхода нитрида алюминия при азотировании порошка
ПА-4 от температуры и давления азота.
нонитридную пленку к алюминию. До температуры плавления алюминия
закон у р соблюдается. При расплавлении поверхность алюминия стре-
мится сократиться за счет сил поверхностного натяжения, а частицы
алюминия, имевшие до того форму лепестков (ПАК-4), капель, дендри-
тов, лепешек (ПА-4), стремятся принять шарообразную форму, причем
этому процессу препятствует окиснонитридная пленка. С ростом темпе-
ратуры прочность пленки снижается, при определенной температуре по-
верхностное натяжение начинает превалировать над прочностью плен-
271
ки, последняя разрывается и расплавленный алюминий выливается из
частиц. В момент выхода капли алюминия из окиснонитридной оболоч-
ки происходит почти мгновенное азотирование алюминия, так как вели-
чина этих капель в случае тонкой пудры ПАК-4 очень мала.
Чем тоньше пленка, тем меньшие силы необходимы для ее разру-
шения, а следовательно, и меньшие температуры процесса. Поскольку
до расплавления алюминия увеличение давления азота способствует об-
разованию нитрида алюминия, то во время процесса азотирования при
этих температурах образуется более толстая и более прочная пленка.
Для разрушения такой пленки требуются более высокие температуры,
при которых прочность пленки становится меньше сил поверхностного
натяжения капли алюминия.
При азотировании порошка ПА-4 механизм процесса азотирования
в общем остается таким же, как и при азотировании пудры ПАК-4, но
капли алюминия, получаемые после разрушения пленки, здесь значи-
тельно крупнее. Поэтому капли успевают проазотироваться только ча-
стично с поверхности и имеют тенденцию к слиянию (экспериментально
наблюдаются крупные частицы размером до 2—3 мм). Вследствие слия-
ния капель резко сокращается и до того гораздо меньшая по сравнению
с пудрой ПАК-4 поверхность и соответственно уменьшается скорость
азотирования. Дальнейшее азотирование происходит в результате диф-
фузии азота через пленку и подчиняется закону у р (рис. 119).
Для ускорения процесса получения нитрида алюминия непосредст-
венным азотированием металла в патенте [592] предложено вводить в
порошок алюминия 5% KF в качестве катализатора. Смесь чистого по-
рошкообразного алюминия (НО—220 меш) и катализатора KF нагре-
вается в среде азота при температуре ниже точки плавления алюминия
до начала реакции азотирования, затем температура повышается до
точки плавления алюминия до окончания образования A1N. В получен-
ном продукте содержится 90—95% A1N, выход составляет 95—100%.
Значительно ускоряет процесс азотирования добавление к алюминию
5% KHF2 [246] или 1% NaF [619].
В работе [585] предложен метод получения нитрида алюминия сжи-
ганием алюминия в кислороде с последующим замещением кислорода
азотом при высоких температурах. Нитрид высокой чистоты получают
распылением алюминия с помощью электрический дуги в среде азота
[585]. Оригинальный метод получения нитрида алюминия плавкой алю-
миния во взвешенном состоянии (в высокочастотном поле) в азоте опи-
сан в [1103]. Кристаллы нитрида алюминия длиной до 30 и до 0,5 мм
в поперечнике образуются при испарении алюминия в азоте при тем-
пературе 1800—2000° С [573].
Для получения монокристаллов нитрида используют также метод
сублимации из газовой фазы при 1900° С [593—595], аналогичный мето-
272
ду получения монокристаллов карбида кремния. Образуются кристаллы
зеленовато-голубого цвета размерами до 2 мм.
Монокристаллы нитрида алюминия приготавливают пропусканием
в течение 24 ч смеси азота и аргона над расплавленным при 1500°С
алюминием, помещенным в корундовой лодочке, находящейся в корун-
довой трубе [601, 614]. На стенках последних, имеющих температуру око-
ло 1450° С, вырастают монокристаллы A1N в форме пластинок и игл
(размерами соответственно 1X1 и 4X0,1 Х0,1 мм). Указывается, что по-
лучение более крупных монокристаллов A1N возможно с помощью газо-
транспортных реакций [614].
Очень чистый (до 99,999%) порошок A1N образуется [619] при на-
греве порошка алюминия (99,999%) в тиглях из A1N в среде тщательно
очищенного азота. Реакция проходит при температуре 1600° С и давле-
нии 100 ат.
Иногда для удаления примесей проводится дополнительная обра-
ботка полученного нитрида алюминия хлором при 600° С.
При действии аммиака на алюминий также образуется нитрид алю.
миния: 2Al + 2NH3->2AlN + 3H2+2,57 ккал [6].
2. Восстановление — азотирование окиси алюми-
ния. В работе [596] указывается на возможность получения нитрида
алюминия действием аммиака при температуре 1600° С на тонкоизмель-
ченную окись алюминия, образованную при разложении при 650° С аце-
тата алюминия. Несколько более подробное исследование этого процес-
са выполнено в [597], где показано, что аммиак реагирует очень медлен-
но с окисью алюминия при 1000° С с образованием нитрида алюминия.
При более высоких температурах (около 2000° С) при плавлении обна-
руживается появление двух фаз: у-фазы и б-фазы. Область гомогенно-
сти у-фазы при температуре 1700° С простирается от 68 до 84% А12О3
(32 или 16% A1N), имеет структуру шпинели, близкую к структуре
А120з. б-фаза менее богата азотом (приблизительный состав 93% А12О3,
7% A1N), структура близка к структуре окиси алюминия, у- и б-Фазы
представляют оксинитриды алюминия (в диаграмме системы А12О3—
A1N) и получаются при реакции окиси алюминия и нитрида алюминия
в твердом состоянии.
3. Получение нитрида алюминия из газовой фа-
зы (методы термического разложения). Впервые в
1938 г. в [598] упомянуто, что можно получать нитрид алюминия раз-
ложением А1С13 • 6NH3 через стадию образования промежуточного про-
дукта AICI3 • NH3. Реннер [574] также получил нитрид алюминия терми-
ческим разложением А1С13 • NHs->AlN+ЗНС1 в установке, показанной
на рис. 120. В длинной кварцевой трубке на тонкой кварцевой трубке
подвешена графитовая шайба, обогреваемая током высокой частоты с
помощью водоохлаждаемой медной спирали. Основную кварцевую
трубку заполняют в нижней части определенным количеством
18—272
273
AlCh-NHs, который нагревают надвигаемой на трубку печью до опре-
деленного давления паров АЮз-МНз. При достаточно высоких темпе-
ратурах образуются при разложении кристаллы, обладающие четко
выраженными гранями (рис. 121). Этот метод был вновь исследован в
работах [1102, 1104, 1105].
Исследователи [596] осуществляли такой метод получения A1N оса-
ждением нитрида алюминия на вольфрамовой нити разложением сме-
Рис. 120. Установка для получения нитрида алюминия из газовой фазы:
J — кварцевая трубка; 2— графитовый диск; 3 — медный индуктор; 4—печь.
Рис. 121. Кристаллы A1N, полученные из газовой фазы.
сей парообразных AICI3 с аргоном и аммиаком. При температурах 800—
1100° С осадок нитрида представляет сплошной слой небольшой толщи-
ны, прочно соединенный с нитью. При температуре нити выше 1200° С
осадок получается рыхлым, неравномерно покрывает нить и слабо с ней
связан; его структура типично дендритная.
274
Можно приготовлять нитрид алюминия термическим разложением
А1(СНз)-ЬГН3 при 200—300° С [599]. Получаемый при этом нитрид
очень неустойчив, легко гидролизуется на влажном воздухе с выделе-
нием аммиака, полный гидролиз завершается за 48 ч [596].
При термическом разложении (МН4)зА1Р6 в токе аммиака уже при
температуре 500° С образуется чистый нитрид алюминия [274]. Окраска
и химическое поведение его сильно зависят от температуры приготовле-
ния (при 500—600° С цвет A1N меняется от коричневого до оливково-
зеленого, при 700—800° С — черно-зеленый с фиолетовым блеском). По-
лученный таким методом нитрид алюминия менее активен в химическом
отношении, чем A1N, образуемый азотированием алюминия.
4. Восстановление окиси алюминия с одновре-
менным азотированием. Для промышленного производства
технического нитрида алюминия использовался так называемый метод
Серпека [6, НО, 600, 596], основанный на реакции
А12О3 + ЗС 4- N3 = 2A1N + ЗСО.
Реакция эндотермична, идет с поглощением большого количества
тепла, температура восстановления-азотирования порядка 1600—
1800° С. В присутствии железа, действующего как катализатор, темпе-
ратура процесса снижается до 1400° С. В промышленности данный ме-
тод (предназначенный для последующего разложения нитрида алюми-
ния водяным паром с образованием гидроокиси алюминия либо содой
с образованием алюмината натрия) осуществляется во вращающихся
печах длиной 60 м и диаметром 3—4 м.
5. Прочие методы получения нитрида алюминия.
Нитрид алюминия получается нагреванием тонкого порошка фос-
фида алюминия в медленном токе сухого аммиака в течение 1—2 ч при
температуре 1000—1100° С [599]
А1Р + NH3 -> AIN + V4P4 + 3/2Н2.
При этом образуется аморфный порошок A1N, только после много-
часового прокаливания при 1100° С рентгенографически обнаружива-
ются линии, характерные для гексагонального нитрида алюминия.
Получение изделий из нитрида алюминия. Изделия из нитрида алю-
миния получают различными методами: 1) спеканием предварительно
спрессованных заготовок из порошка нитрида алюминия; 2) реакцион-
ным спеканием заготовок из порошка алюминия в азоте или аммиаке;
3) горячим прессованием порошка нитрида алюминия.
Первый из этих методов может осуществляться по технологии, опи-
санной подробно в [605]. Порошок нитрида в смеси с порошком алюминия
или пудрой в количестве 10% прессуют с пластификатором (5—6% до-
бавкой 5%-ного раствора синтетического каучука в бензине), заготов-
ки просушивают и спекают по определенному режиму с окончательной
18* 275
температурой спекания 1900° С в среде азота. При спекании алюмини-
евая пудра переходит в нитрид [609], образуются изделия из нитрида
алюминия пористостью 12—16% [586], а по данным [609] — даже по-
ристостью 2—6%. Аналогичный метод описан в патенте [606], где поро-
шок нитрида алюминия смешивают с небольшим количеством алюми-
ния и связкой, в качестве которой используют церезин, озокерит или
полигликоль, а также с пластификатором — трихлорэтиленом, прессуют
и спекают в среде азота при температуре выше 1400° С. Возможно по-
лучить изделия из A1N предварительным активированием прессовок на-
гревом при температуре 450° С в окислительной среде с последующим
спеканием в азотсодержащей среде при ~ 1400° С. На этом же прин-
ципе спекания смесей порошков нитрида и алюминия основаны реко-
мендации патентов [607, 608].
Заготовки из нитрида алюминия могут приготовляться шликерным
литьем [585]. Так как нитрид алюминия гидрофобен, то в качестве сус-
пендирующей среды может применяться диоксан.
Хорошие результаты дает гидростатическое прессование порошка
нитрида алюминия с последующим спеканием в среде аргона при
1950—2050° С [585].
Вторым принципиальным методом приготовления изделий из нитри-
да алюминия является реакционное спекание заготовок из порошка
алюминия в среде азота. Этот метод, основанный на азотировании за-
готовок из порошка алюминия, спрессованных под давлением 8 т/см2,
не позволяет получать изделия плотностью выше 50—60% от теорети-
ческой [234].
Горячее прессование осуществляется под давлением 300 кг/см2 при
температурах порядка 2000—2100° С с получением изделий, имеющих
практически нулевую пористость [605] или порядка 1,5% [609].
3. НИТРИДЫ ГАЛЛИЯ
В системе галлий — азот обнаружено одно соединение — нитрид галлия
GaN. Его структура впервые исследована в работе [613], где показано,
что GaN обладает гексагональной структурой типа вюртцита.
Физические свойства. Нитрид галлия относится к полупроводнико-
вым соединениям типа AniBv, отличается от прочих соединений галлия
такого типа (фосфида, арсенида, антимонида, стибнида, висмутида)
более высокими температурами плавления и сублимации, а также стой-
костью к окислению и действию химических реагентов. Ширина запре-
щенной зоны нитрида галлия по разным измерениям составляет 3,25—
3,6 эв [614], чему соответствует и высокое электросопротивление нитри-
да, которое, по данным Реннера [574], составляет 10—-100 ом • см, а по
данным [33], полученным на более чистом препарате,— 4,0 • 103 * * * * 8 ом • см.
Температура плавления GaN составляет ~ 1500° С [574], упругость
276
паров при температуре 1130° С достигает 4* 10-11 ат [617], что подтвер-
ждается измерениями, проведенными в [574], причем предполагалось, что
нитрид сублимирует моно- или полимолекулярно, без диссоциации. Од-
нако в [613] утверждается, что нитрид галлия термически неустойчив и
диссоциирует уже начиная с температур ниже 600° С, а при 1000° С дис-
социирует полностью на галлий и азот. В то же время по значениям
свободной энергии и теплоты образования [617] можно вычислить рав-
новесное давление пара GaNra3, которое при 1500° С оказывается рав-
ным 4 • 10“4 мм рт. ст., что значительно ниже давления пара чистого
галлия (2- 10-1 мм рт. ст.). Из этих данных вытекает, что нитрид гал-
лия не только не диссоциирует, но испаряется не в виде двухатомных
молекул, а образует в парах сложные полимеры. Поэтому авторы [616]
исследовали состав пара над нитридом галлия масс-спектрометриче-
ским методом. Показано, что нитрид галлия испаряется в основном в
форме димеров, а не диссоциирует (диссоциация в парах возможна
только при дополнительных возбуждениях, например, при соударениях
полимеров с электронами).
Нитрид галлия обладает люминесцентными свойствами, в частно-
сти, при исследовании катодолюминесценции на порошках GaN обна-
ружены два максимума интенсивности при 3200 и 5200 А [615, 614].
Нитрид галлия — сверх-
проводник сточкой перехода к Таблица 100
сверхпроводимости 5,85° К, Т. е. Свойства нитрида галлия
является пока довольно ред-
ким сочетанием сверхпровод-
ника и полупроводника [618].
Сведения о физических свой-
ствах GaN приведены в табл.
100.
Химические свойства. Ни-
трид галлия устойчив к дей-
ствию различных химических
реагентов [612]. На порошкооб-
разный нитрид не действуют
при кипячении серная и азот-
ная кислоты, в то же время
растворами щелочей быстро
и полностью разлагается, но
медленнее, чем A1N щелоча-
ми [574].
На воздухе начинает окис-
ляться (в форме порошка) при
температуре 800° С, окисление
полностью завершается при
Характеристика GaN
Содержание азота, вес. %
Кристаллическая структура
Периоды решетки, д:
а
с
с! а
Плотность, г/см3
Температура плавления, °C
Теплота образования,
ккал/моль
Энергия диссоциации,
ккал/моль
Удельное электросопротив-
ление, ом-см
Ширина запрещенной зо-
ны, эв
Температура перехода к
сверхпроводимости, °К
Давление пара при 1100°С ат
16,72
Гексагональная, ти-
па вюрцита
3,180 [31]
5,166
1,625
6,1
~1500 [614] (раз-
лагается)
24,9 [112]
72—101 [611]
4.0-108 [33]
3,25—3,60 [33, 614]
5,85 [618]
4-10-" [617]
1200° С с образованием Ga2O3 [612]. Водород на GaN не действует [614].
Методы получения. Нитрид галлия получают нагреванием металли-
ческого галлия в токе аммиака при температуре 1200° С [613].
По данным Хана и Юца [31], а также Джонсона с сотрудниками
[62], нитрид галлия может быть получен двухкратным нагреванием ме-
таллического галлия в корундовой лодочке в быстром токе аммиака в
течение 2 ч при 1100° С с измельчением образующегося после первого
азотирования промежуточного продукта. Можно приготовить нитрид
галлия разложением фторгаллата аммония (NH4)3GaF6 в токе аммиака
при 900° С [102]. По Реннеру [574], нитрид галлия получается разложе-
нием соли GaCl3 • NH3 при 900—1000° С.
В работе [599] нитрид галлия получали прокаливанием тонкого по-
рошка GaP или GaAs в токе сухого аммиака. Превращение GaP в GaN
начинается при температуре 900° С, полное превращение в нитрид про-
исходит при нагреве в течение 2 ч при 1000—1100° С. GaAs с аммиаком
начинает реагировать при 700° С, количественный переход арсенида в
нитрид галлия наблюдается в течение 1—2 ч при 1000° С. Охлаждение
продуктов реакции производится в среде аммиака. При использовании
фосфида получается нитрид галлия желтоватого цвета, при использова-
нии арсенида — белого.
Многочисленные методы приготовления нитрида галлия приведены
в [619].
Подробное исследование условий образования нитрида галлия вы-
полнено в [620]. Трудностью азотирования галлия является образование
легкоплавким галлием (т. пл. 29,7° С) зеркала расплава, что вызывает
резкое сокращение реакционной поверхности. Поэтому необходимо мно-
гократное азотирование с получением нитрида стехиометрического со-
става. Авторы [620] использовали разрыхлители, которые, разлагаясь во
время нагрева, выделяют газы, перемешивающие жидкий галлий, и об-
легчают доступ к нему азотирующего агента. Наиболее удачным раз-
рыхлителем является углекислый аммоний, при разложении которого
выделяется аммиак, участвующий и в разрыхлении и в процессе азоти-
рования, а также СО2, сильно разрыхляющая расплав. Результаты ис-
следования приведены на рис. 122. Такой способ позволяет получать
при соотношении Ga : (NH4)2CO3=1 : 1, при использовании в качестве
азотирующего агента аммиака при температуре 1050—1200° С и вы-
держке 0,5—2 ч нитрид галлия точного стехиометрического состава,
85—95%-ным выходом галлия в продукт азотирования.
Разработан метод получения нитрида галлия восстановлением оки-
си галлия аммиаком с одновременным азотированием [620]
Ga2O3 + 2NH3 = 2GaN + ЗН2О.
При использовании в этом случае в качестве разрыхлителя угле-
кислого аммония (1:1) образуется нитрид расчетного состава при дли-
278
тельной выдержке при 1050° С либо при выдержке в течении 1—2 ч при
1100—1200° С (рис. 123). GaN также получается восстановлением окиси
галлия аммиаком при 600—1100° С [615].
Оба эти метода удобны для приготовления нитрида галлия в круп-
ных количествах.
Рабенау [619] указывает на возможность получения монокристал-
лических образцов GaN путем возгонки его при температуре 1000—
ЮОО 1100 1200
. Температура озотиробонир, °C
Рис. 122. Зависимость содержания азота и выхода галлия в продукте азотирования
металлического галлия в смеси с углекислым аммонием от температуры и времени
азотирования.
Время азотирования: 1 — 1 ч; 2 — 4 ч; 3 — расчетное содержание азота в GaN; выход галлия в
продукт азотирования при азотировании в продолжение: 4 — 1 ч, 5 — 4 ч.
Рис. 123. Зависимость содержания азота и выхода галлия в продукте азотирования
окиси галлия в смеси с углекислым аммонием от температуры и времени азотирова-
ния (обозначения те же, что и на рис. 122).
1100° С. Более перспективным методом является выращивание монокри-
сталлов GaN из растворов, например, нитрида в галлии [614]. Однако
так как растворимость мала (не превышает 1—2%), получение более
или менее крупных монокристаллов и этим методом маловероятно.
4. НИТРИДЫ ИНДИЯ
Как алюминий и галлий, индий образует один нитрид — InN, имеющий
структуру вюртцита (а = 3,533; с = 5,693; с/а — 1,611 [33]).
Физические свойства. Нитрид индия — полупроводник, но свойства
его в этом отношении совершенно не изучены. В работе [33] отмечается,
что InN имеет металлический характер с сопротивлением порядка
4 • 10~3 ом-см и температурным коэффициентом сопротивления
279
Рис. 124. Кинетика окисления InN
на воздухе.
+ 0,00037 град-1. Авторы [574] также указывают на его хорошую про-
водимость и сопротивление, значительно меньшее 1 ом-см. Из этих
данных следует, что InN представляет собой полуметаллическое соеди-
нение. Теоретические оценки показывают, что ширина запрещенной зо-
ны нитрида индия, если он является полупроводником, должна быть
порядка А£ = 2,4 эв.
Твердость InN меньше, чем A1N и GaN, плотность 6,88 г/см3, теп-
лота образования из элементов 4,6 ккал/моль [33]. Энергия диссоциа-
ции равна 70—116 ккал [611].
Химические свойства. По данным
[612], нитрид индия весьма неустойчив
в химическом отношении. Он быстро раз-
лагается серной, азотной и соляной кис-
лотами, водными растворами щелочей
при кипячении (в работе [574] нитриду
индия приписывается более высокая хи-
мическая устойчивость). На воздухе ус-
тойчив до температуры 300° С, а при
350° С почти полностью и скачкообразно
окисляется до 1п3О3, окисление полностью
заканчивается при 600°С (рис. 124).
Методы получения. Нитрид индия
обычно получается разложением гекса-
фториндата аммония (NH4InF3) при
600° С [6, 31, 33, 102]. Позднее Реннером [574] был описан метод полу-
чения InN разложением полиаммиакатов хлористого индия InCL -xNH3
при температурах порядка 400° С. В той части реакционной трубки, ко-
торая имееет температуру 600° С, осаждается в течение нескольких ча-
сов черный слой, при очень медленном процессе и соответствующем
температурном режиме нитрид индия образуется в форме коричневых
прозрачных кристаллов.
В работе [622] нитрид индия получали взаимодействием окиси ин-
дия с аммиаком
In2O3 + 2NH3 = 2InN + 3H2O,
с использованием в качестве разрыхлителя (для увеличения реакцион-
ной поверхности) углекислого аммония. За 4 ч при температуре 610° С
при соотношении In: (NH4)2CO3=1 :3 образуется нитрид индия прак-
тически стехиометрического состава.
В обзоре [614] обсуждаются принципиальные возможности получе-
ния монокристаллов нитрида индия.
280
5. НИТРИДЫ ТАЛЛИЯ
В некоторых источниках [1, 6] указывается на существование нитрида
TIN, образуемого при температуре 620° С взаимодействием паров тал-
лия с аммиаком [623]. При нагревании в водороде восстанавливается до
металлического таллия. Энергия диссоциации равна 52—92 к.кал1моль
[611].
Получают нитрид таллия состава TI3N по реакции ЗТ1ЫОз +
+ ЗКПНз = Т1зЫ + ЗКПОз+2ЫН3, которая приводит к выпадению нитри-
да таллия в форме черного осадка [624]. TI3N взрывается при соприкос-
новении с водой или разбавленными кислотами, при нагревании или
ударе. Растворим в аммиачных растворах TINO3 и KNH2.
При взаимодействии NaN3 с раствором сернокислого или азотно-
кислого таллия образуется азид T1N3, хорошо растворимый в горячей
воде. Следует отметить, что при попытке воспроизвести эти данные не
были получены положительные результаты; итак, существование нитри-
дов таллия весьма проблематично.
281
ГЛАВА VII
НИТРИДЫ
ЭЛЕМЕНТОВ
ПОДГРУППЫ
УГЛЕРОДА
1. НИТРИДЫ КРЕМНИЯ
Исследование условий получения и свойств сое-
динений кремния с азотом, начатое еще в
1844 г., показало наличие в этой системе нес-
кольких химических соединений, которым при-
писывались составы SiN, Si2N2, Si3N4. В настоя-
щее время однозначно доказано существова-
ние в системе кремний—азот только одного
химического соединения — нитрида кремния
Si3N4.
Одно из первых исследований структуры
этого соединения было выполнено в [525, 1025].
Установлена ромбическая структура с периода-
ми решетки: а= 13,38, 6 = 8,60, с = 7,74 А (см. так-
же [529, 532]). Затем было сообщено о гексаго-
нальном нитриде кремния [526]. Туркдоган,
Биллс и Типпет [527], а также авторы [526] об-
наружили, что имеются две модификации нитри-
да а- и |3-Si3N4, рентгенографически исследован-
ные в [528, 559]. Обе модификации оказались
гексагональными.
Обе структуры построены из тетраэдров
SiN4 и различие состоит в способе сочленения
этих тетраэдров (тетраэдры SiN4 почти правиль-
ные, расстояния Si—N 2,72—1,75 А).
Структура (3-Si3N4 (пространственная груп-
па Р6з/т) может быть выведена из структуры
фенакита Вег5Ю4 замещением атомов кислоро-
да и бериллия соответственно азотом и кремни-
ем. Каждый атом кремния находится в центре
слегка неправильного тетраэдрона из атомов
азота. Ячейки Si3N4 соединяются общими угла-
ми таким образом, что каждый атом азота яв-
ляется общим для всех тетраэдров. Иначе струк-
тура может быть представлена в форме восьми-
членных колец Si3N4, соединенных между собой
соответствующим образом.
Структура a-Si3N4 (пространственная груп-
па Р31с) иная, чем ^-модификация: в послед-
ней плоскости соединены вдоль направления
(001) в последовательности АВАВАВ..., в то
время как в решетке а-модификации — в по-
следовательности ABCDABCDABCD...
282
Число формульных единиц в элементарной ячейке а-модификации
равно четырем, а в р — двум.
Получены следующие данные для периодов решеток [530, 531]:
Модификация a,k С-А с/а Литера- тура
a-SigNj 7,748 5,617 0,725 [528]
7,76 5.62 0,726 [530]
7,76 5,64 0,727 [531]
P-Si3N4 7,608 2,9107 0,3826 [528]
7,59 2,90 0,383 [530]
7,59 2,92 0,335 [531]
Растворимость азота в твердом кремнии достаточно определенно
не установлена, растворимость в жидком кремнии порядка
1019 атомов/см3.
Физические свойства. Атомы кремния и азота в SisNi соединены
ковалентными, насыщенными связями, наличие которых следует из то-
го, что каждая четвертая гиб-
ридная 5р3-орбита атома пере-
крыта с гибридной 5Д2-орбитой
атома азота [528]. Оставшие-
ся, полностью занятые р-орби-
ты каждого атома азота, пер-
пендикулярны к трем плос-
костям хр2-орбит и не могут
принимать участия в связи.
О ковалентности связей свиде-
тельствует, в частности, высо-
кое электросопротивление ни-
трида, составляющее более
1012 ом-см [529]. Подробное
исследование электросопротив-
ления нитрида кремния выпол-
нено в [534], где показано, что
электросопротивление SiaN.*
при комнатной температуре со-
ставляет 1013—1014 ом -см, с по-
Рис. 125. Зависимость электропроводности чи-
стого нитрида кремния и сплавов нитрида кре-
мния с титаном и углеродом от температуры.
вышением температуры быстро
падает, доходя при 300° С до 2-108 ом-см (рис. 125). Излом на кривой
электропроводности при 700° С отвечает модификационному превраще-
нию, установленному также дилатометрически (рис. 126). Фазовое пре-
вращение смещает примесный уровень в энергетическом спектре SisN^
Значение энергии активации примесей Ео= 1,13 эв при 300—650° С и
3,91 эв при 650—1200° С. Во втором случае, по-видимому, наблюдается
283
преимущественно собственная проводимость и величина Ео приблизи-
тельно характеризует ширину запрещенной зоны нитрида кремния. До-
бавление к нитриду углерода уменьшает, а титана — увеличивает элек-
тросопротивление нитрида (см. рис. 125).
В соответствии с сильными ковалентными связями между атома-
ми, нитрид кремния обладает высокой твердостью, малым коэффициен-
том термического расширения. Энергетическая обособленность входя-
щих в его состав групп атомов
вызывает его диссоциацию до
плавления, как и всех других
ковалентных нитридов, обла-
дающих неметаллическими
свойствами.
Давление диссоциации ни-
трида кремния выражается
уравнением [74]
1g Р =----f---1-8,54.
Давление диссоциации, равное
Рис. 126. Дилатометрическая кривая нитрида 1 ат, достигается при темпе-
кремния. ратуре 1977° С (см. также
[1106—1108]).
Ниже (табл. 101) приведены основные данные о свойствах нитрида
кремния.
Химические свойства. Одним из важнейших свойств нитрида крем-
ния является его исключительно высокая химическая стойкость [530,
535, 542]. Химические свойства представлены в табл. 102.
Нитрид кремния устойчив к действию кислорода. По данным [530],
при действии кислорода на порошок Si3N4 при 1000° С в течение 3 ч
окисляется 14,7%; при 1300° С — 23,6% и при 1400° С — 49,5% этого
порошка. Еще более устойчив нитрид кремния к окислению воздуха.
(По данным [527], при нагревании в токе воздуха при 1450° С в течение
20 ч увеличение веса составляет всего 11,3%.) Компактные и плотные
изделия могут использоваться на воздухе до температуры 1200° С в те-
чение достаточно длительного времени [536]. В работе [547] исследована
стойкость нитрида кремния в форме порошка при 600—1400° С на воз-
духе и при 1200° С в парах воды. Показано, что окисление на воздухе
начинается при 1000° С со степенью превращения нитрида в окислы
0,11%, далее степень разложения возрастает на каждые 100° С на 0,2—
0,3% до 1400° С, когда скорость разложения резко возрастает, и дости-
гает 1,41%. В парах воды за 1 ч при 1200° С степень разложения состав-
ляет более 2%. Увеличение веса образцов Si3N4 за 80 ч при 1200° С
составляет всего 5 мг/см2, в то время как образцов из TiB3— 10, а из
TiC — 42,5 мг/см2. Еще большую устойчивость к окислению на воздухе
284
Свойства нитрида кремния
Таблица 101
Харак теристик а a-Si3N4 ₽-Si3N4
Содержание Si, вес. % 60,2 60,2
Кристаллическая структура Гексагональная Гексагональная
Периоды решетки, д: а 7,76 [531] 7,59 [531]
с 5,64 2,92
с/а 0,727 0,335
Плотность, г/см3: рентгеновская 3,184 [527[ 3,187 [527]
пикнометрическая 3,19 [527( 3,21 [530]
Температура плавления, °C 1900 535]
Теплота образования, ккал/моль 179,5 [551, 1118]
Теплоемкость (298—900°К), кал! моль град Теплопроводность, кал/с'м-град-сек 16,83+23,6 .Ю-ЗГ И?]
0,011 [542], см. также [551]
Коэффициент термического расши- 2,75 [535]
рения (20—1000 °C), ХЮ6 град~~1 Энтропия (S02g8), кал/град-моль 23,0±2,5 [47]
Удельное электросопротивление 1013—1 0i« [534]
(20°С), ом-см Диэлектрическая константа (при 28°С) 9,4 [530]
Микротвердость, кг/мм2 3337+120 [544]
Твердость по Роквеллу (Рд ) 99 [535]
Модуль упругости, кг/мм2 11600—14500 [535
Предел прочности при изгибе, кг/мм2: 20°С 16,0 [5421
1200°С 14,7 [542]
Коэффициент излучения (/.--655 ммк) 800—1600°С 0,77 [340]
имеет при этой температуре карбид кремния на связке из нитрида крем-
ния (привес 2,5 мг/см2) .
По данным [991], нитрид кремния на воздухе при 1100—1500° С
окисляется в основном с образованием кремнезема; добавка NaF спо-
собствует окислению нитрида кремния до оксинитрида. В коксовой за-
сыпке при 1450—1550° С нитрид кремния разлагается с образованием
оксинитрида и кубического карбида кремния; добавки CaF2 и MgO
способствуют переходу нитрида кремния в среде окиси углерода при
1550° С в оксинитрид кремния.
Хлор слабо действует на нитрид кремния при средних температу-
рах. Так, при 350—420° С за 2 ч потеря веса при обработке порошка
нитрида хлором составляет 0,77—0,95% [530]*.
* О химических свойствах и методах анализа нитрида кремния см. также [НО.
530, 532].
285
Таблица 102
Реагенты, против которых устойчив нитрид кремния
Реагент Состояние Стойкость
20 % НС1 65 % HNO3 HNO3 10 % H2SO4 77 % H2SO4 85 % H2SO4 HPO3 H4P3O7 20 % NaOH Cl8 H2s H2SO4+CuSO4+ +KHSO4 NaNO3+NaNO2 50 % NaOH NaOH 48% HF* 3% HF+10% HNO3 NaCl+HCl NaB (SiO2)2 -|-V2OS NaF+ZrF4 Кипящая » Дымящая При 70°С » 20°С То же » » » » » » При 30—900°С » 1000°С Концентрированный кипящий Расплав при темпера- туре 350°С Расплав при темпера- туре 790°С кипящий Расплав при темпера- туре 450°С При 70°С » 70°С Расплав при 900°С При 1100°С » 800°С (Не разлагается в течение ^>500 ч) То же » » » » » » » » » » » » » » » » » » » » » » Устойчив в течение 115 ч Устойчив в течение 5 ч Устойчив в течение 3 ч » » 116 ч » » 144 ч » » 4 ч » » 100 ч
* См. также [547].
Нитрид кремния отличается весьма высокой стойкостью по отно-
шению к расплавленным металлам. В табл. 103 приведены данные, по-
лученные при испытаниях тиглей из нитрида кремния, приготовленных
шликерным литьем [535]. Указывается, что особенно нитрид кремния
стоек к расплавленному алюминию, устойчивость в котором при 982° С
доходит до 3000 ч без какого-либо заметного разрушения [537]. Эти же
данные подтверждают высокую стойкость нитрида кремния к действию
расплавленного свинца, олова, цинка. Несколько менее устойчив он к
действию расплавленного магния при 750° С. Высокая стойкость нитри-
да кремния к действию алюминия и магния отмечена также в работе
[547]. Магний действует в основном на окись кремния, содержащуюся
в нитриде, с образованием окиси и силицида магния: SiO2+4Mg=
= 2MgO+Mg2Si. По данным работы [538], стойкость нитрида кремния
в расплавах хлоридов в процессе электролиза не высока.
286
Таблица 103
Стойкость нитрида кремния против действия расплавов
Расплав Темпера- тура, °C Время контакта, ч Результат действия на нитрид кремния Литера- тура
Алюминий 800 950 Не действует [535]
Алюминий 1000 3000 В в |537|
Свинец 400 144 в » [535]
Олово 300 144 в в [535]
Цинк 550 500 в в [535]
Магний 750 20 Слабо действует [535]
Медь 1150 7 Сильно действует [535]
Медный штейн 1300 5 Действует 15411
Медистый шлак 1300 5 Не действует 15411
Никелевый штейн 1250 5 В в |Ь41|
Никелевый шлак 1250 5 в в 1541|
Базальт 1400 2 Слабо действует 15411
NaCl+CaCl2 700 50 Не действует 15411
NaCl+BaCla 1100 3 Слабо действует [Ь41]
На рис. 127, по данным Г. А. Ясинской [539], показаны результаты
сравнительного исследования стойкости изделий из нитрида кремния и
других огнеупорных материалов при кратковременном контактирова-
нии. Видно, что сильнее действуют на нитрид кремния переходные ме-
таллы и почти не действуют непереходные металлы и неметаллы. Это
довольно хорошо согласуется с величинами межфазных энергий при
смачивании расплавами нитрида кремния [540]. По данным [541], при-
веденным в табл. 103, нитрид кремния практически не взаимодействует
со штейнами и шлаками никелевых плавок, медистыми шлаками, рас-
плавами хлористых Натрия и кальция, медленно взаимодействует с мед-
ными штейнами и очень слабо с расплавленными базальтом и смесями
хлоридов натрия и бария.
Методы получения. Для получения нитрида кремния известно мно-
го способов.
1. Непосредственное азотирование кремния. Впер-
вые этот метод описан Бальменом [543], который нагреванием кремния
в среде азота, выделявшегося при разложении цианистого калия, полу-
чал нитрид кремния неопределенного состава в форме пористого хруп-
кого спека.
В [535] для получения нитрида использовали нагревание кремния в
среде азота при температуре 1300° С. Для азотирования наиболее бла-
гоприятны размеры частиц порошка — 150 меш, при использовании ко-
торого получается порошок нитрида с крупностью частиц 1—10 мкм.
287
0
МеХ
Рис. 127. Характер взаимодействия тугоплавких соединений с расплавленными металлами: I
1 — не взаимодействуют (1-й балл); 2 — слабо взаимодействуют (2-й и 3-й балл); 3 — взаимодействуют (4-й балл);
5 •— температура опыта, 0 К; 6 — продолжительность опыта, мин; 7 — продолжительность опыта, ч.
288
Кинетику азотирования порошка кремния (чистотой 99,2% и макси-
мальной крупностью частиц 40 мкм) исследовали в [544]. Полученные
при этом кинетические кривые насыщения азотом кремния показаны на
рис. 128. Образование нитрида кремния начинается при температуре
970° С. Константа реакции азотирования составляет при этой темпера-
туре 7,49 • 10-7 г/см2 • сек, при 1490° С — 2,00 • 1СГ4 г! см2 • сек. При даль-
нейшем повышении температуры до 1600° С константа скорости азоти-
рования остается постоянной, при более высоких температурах — резко
уменьшается.
Таким образом, азотирование при температурах выше 1600° С вы-
зывает уменьшение привеса по сравнению с привесом при температуре
1600° С, а при температурах выше 1600° С скорость диссоциации пре-
валирует над скоростью образования нитрида. По данным этой работы,
энергия активации при реакционной диффузии азота в кремний состав-
ляет 33 800+7190 кал/моль.
Следует отметить, что улетучивание нитрида кремния (подавляе-
мое высокой скоростью образования) обнаружено уже в интервале тем-
ператур 1200—1400° С [545]. Предполагается, что нитрид кремния Si3N<
диссоциирует при этих температурах не на кремний и азот, а, частично
теряя азот, переходит в низшие нитриды кремния, например Si2Ns. Од-
нако в этой же работе отмечается натянутость такого объяснения, так
как низшие нитриды кремния в системе кремний — азот не обнару-
жены.
Несколько отличные данные по непосредственному азотированию
кремния получены в [530]. Установлено, что азотирование, регистрируе-
мое по привесу, начинается при температуре 1230° С, что же касается
оптимальной температуры образования нитрида, то указываемая в этой
работе температура в 1450° С хорошо согласуется с установленной в
[544] температурой в 1400—1490° С.
Так же хорошо согласуются и данные о времени, необходимом для
предельного насыщения кремния азотом при температуре 1400° С (при
более низких температурах в [530] насыщение до предела не произво-
дилось) .
Авторы [526, 547] нитрид кремния получали азотированием при тем-
пературах 1200—1600° С. Наиболее обстоятельное исследование азоти-
рования кремния проведено в [532], где показано, что характер кинети-
ческих кривых насыщения кремния азотом несколько отличен в интер-
валах 1240—1315° С; 1315—1385° С; 1385—1415° С и выше 1415° С
(рис. 129). Предполагается, что первый из указанных интервалов соот-
ветствует простому растворению азота в кремнии (чисто диффузионный
процесс). Ускорение реакции, наблюдаемое в интервале 1315—1385° С,
может быть объяснено различием в коэффициентах термического рас-
ширения кремния и нитрида кремния, которое вызывает напряжения в
пленке нитрида, появление в ней разрывов, пустот, трещин с соответст-
19-272
289
вующим обнажением поверхности кремния и ускорением азотирования.
Определенную роль играет и давление кристаллизации образующегося
нитрида кремния. При температуре выше 1385° С и до 1415° С реакция
протекает до полного азотирования кремния; ее высокая скорость так-
же, как и на предыдущем этапе, объясняется растрескиванием слоя нит-
Рис. 128. Кинетические кривые насыщения кремния азотом.
Рис. 129. Кинетические кривые азотирования кремния [532].
рида, превращая ее в микропористое тело, через которое легко проходит
азот.
В работе [527] показано, что реакция образования нитрида крем-
ния при азотировании порошка кремния резко ускоряется при добав-
лении в последний 1 °/о CaF2, являющегося катализатором этой
реакции.
Авторы [1046] исследовали влияние добавок различных металлов
(в количестве 2 вес.%) на степень превращения кремния в нитрид при
азотировании в течение 5 ч при 1300° С в токе азота. Оказалось, что наи-
более активными катализаторами процесса служат марганец, железо,
кобальт, палладий, никель, а также медь; гораздо более слабое влияние
оказывают титан, ванадий, хром, молибден, золото, а серебро и цинк
даже препятствуют реакции азотирования кремния. Так как азотирова-
ние ведется молекулярным азотом, то можно предположить, что дейст-
вие катализатора заключается в нарушении связи между атомами в
молекуле азота, что возможно, если катализатор является акцептором
электронов. Как раз все металлы, обнаруживающие каталитическую ак-
тивность при азотировании кремния, являются сильными акцепторами,
стремящимися к локализации валентных электронов с образованием
290
максимального статистического веса атомов со стабильными электрон-
ными конфигурациями. К числу металлов-катализаторов относится, на-
пример, никель, акцепторные свойства которого настолько велики, что
он способен акцептировать валентные электроны даже вольфрама; ана-
логичными сильными акцепторами являются также платина, палладий,
медь, марганец.
Промышленная технология получения нитрида кремния описана в
[548]. Кремний дробят, измельчают, тщательно отмывают от железа, пос-
ле чего азотируют по двухступенчатому режиму: при 1300—4350° и при
1500—1550° С по 3 ч на каждой ступени. Расходные коэффициенты (на
1 кг нитрида): 1 кг кремния, 6 м3 азота, 70 кеч электроэнергии; получа-
емые технические продукты содержат 58—59 % Бйбщ, ДО 20 SiCBo6 и 35—
38% азота.
Непосредственное азотирование кремния можно также проводить
аммиаком [532]. В [531] этим же методом нитрид кремния приготовляли
действием на порошкообразный кремний аммиака при 1200—1500° С, а
в [559] a-Si3N4 получали при 1350—1450° С; a p-SisN4 при 1500° С в те-
чение 3 дней.
2. Нагревание смеси кремнезема и угля в сре-
де азота. Реакция 3SiO2+6C + 2N2 = Si3N4 + 6CO, как показали ре-
зультаты исследования [544], при температуре 1000—1800° С проходит
не в направлении образования нитрида кремния, а карбида кремния,
содержание которого в продуктах реакции повышается от 15% при тем-
пературе 1400° С до 85% при 1800° С; содержание нитрида при всех
температурах не превышает 2—4%.
Поэтому Вейсс и Энгельгардт [549] исследовали влияние добавок
в шихте, направляющих реакцию в сторону образования нитрида и уско-
ряющих ее. Они обнаружили, что при добавлении к шихте 10% окиси
железа реакция при температуре 1250—1300° С проходит с преимуще-
ственным образованием нитрида кремния, который отмывается от сое-
динений железа соляной кислотой.
3. Прочие методы получения нитрида кремния.
Шутценбергер и Кольсон [550] получали нитрид кремния, которому они
приписывали состав Si2N3, действием азота на карбазот кремния
C2Si2N при высоких температурах. Указывается на образование нитри-
да кремния состава SiN2 нагреванием при температуре 400°С триимидо-
дисилана [551]. Описано получение нитрида кремния азотированием
CaSi2 с последующей отмывкой продукта реакции соляной кислотой
[552].
Авторы [530] нитрид кремния a-модификации получали нагрева-
нием при 1350° С соединения Si(NH)2; аналогичный способ был пред-
ложен ранее Бликсом и Бирбельбауэром [553] и в [554].
Билли [532] исследовал реакции взаимодействия тетрахлорида и тет-
рабромида кремния с аммиаком, показал, что при этом образуется не
19*
291
нитрид кремния, а, например, имид кремния SiN2H2 либо бромаммиа-
каты аммония (МЩВг • xNH3).
В работе [527] нитриды кремния получали азотированием железо-
кремниевых сплавов, содержащих от 2,83% Si с последующим выделе-
нием нитрида обработкой бромметиловым эфиром уксусной кислоты.
Рентгеновское исследование показало наличие только a-Si3N4. После
этого было проведено азотирование различных сплавов железа, никеля,
марганца с кремнием и показано, что в составе сплава нитрид кремния
имеет структуру, отличную и от а- и от p-Si3N4, но, будучи выделен из
сплава химическим путем, всегда превращается в a-Si3N4. Предполага-
ется, что это вызвано образованием в сплавах сложных нитридов
MexSiyNj, которые при химической обработке разлагаются с выделе-
нием Si3N4. Напротив, в [560], где также было замечено отличие струк-
туры нитрида кремния в железокремнистых сплавах от структуры Si3N4,
было предположено, что в сплавах образуются низшие нитриды кремния,
что, вообще говоря, маловероятно, хотя о их существовании указыва-
ется в ранних работах. В присутствии железа нитрид кремния разла-
гается с выделением азота [530].
Вопрос о поведении азота в кремнистых сплавах железа, в част-
ности в трансформаторных сталях, обсуждается также в [561—563].
Таким образом, как указано выше, единственным реальным мето-
дом получения нитрида кремния является непосредственное азотирова-
ние кремния азотом или аммиаком.
Приготовление изделий из нитрида кремния. Наиболее технологич-
ным методом приготовления изделий из нитрида кремния является так
называемое реакционное спекание, заключающееся в азотировании
спрессованных из кремния заготовок, при котором совмещаются процес-
сы образования нитрида кремния и его спекания. Впервые этот процесс
был изучен в работе [535, 558], где заготовки из порошка кремния, сфор-
мированные методом шликерного литья, подвергались обработке азо-
том. Образующийся при азотировании каждой частицы нитрид запол-
няет объем пор, так как удельный объем нитрида существенно превы-
шает удельный объем исходного кремния; практически в этом случае
могут быть получены малопористые или вообще беспористые изделия.
По этим же причинам, т. е. вследствие того, что увеличение объема при
азотировании реализуется в результате заполнения пор, изменение
внешних размеров заготовок при реакционном спекании невелико; на-
пример, по данным [535], оно не превышает +0,005 мм/мм; в [542] так-
же не наблюдалось какого-либо изменения размеров.
Реакционное спекание нитрида кремния исследовано также в [557,
234]. Указывается, что для приближения к получению изделий теорети-
-ческой плотности необходимо использовать заготовки из кремния, имею-
щие наибольшую возможную плотность (80—85%), что однако практи-
чески затруднительно в связи с плохой прессуемостью порошка кремния.
292
Технологическое оформление этого способа приготовления изделий
подробно описано в [548].
Для получения крупногабаритных изделий из нитрида кремния
обычно удобнее применять способ прессования заготовок из порошка
нитрида с последующим спеканием в среде азота [548, 555]. Хотя поро-
шок нитрида прессуется лучше, чем порошок кремния, для прессования
заготовок применяют различные пластификаторы-клеи, в частности
5%-ный раствор каучука в бензине, 5%-ный водный раствор поливини-
лового спирта, либо крахмальный клейстер (преимущественно первые
два, так как при выгорании крахмального клейстера остается много зо-
лы). Прессование ведут под давлением 1—2 т/см2 (остаточная порис-
тость заготовок около 30%), спрессованные заготовки сушатся, спека-
ются в среде азота, сначала при 500—600° С для удаления (выгорания)
пластификатора, а затем при 1550—1600° С в течение 2—3 ч. При ис-
пользовании порошка нитрида кремния с высоким содержанием свобод-
ного кремния вводится еще одна температурная ступень — 1350° С для
азотирования кремния. При спекании усадка изделий отсутствует и по-
ристость не изменяется, т. е. остается практически такой же, как и в
спрессованных заготовках.
Исходные заготовки, особенно для приготовления сложных фасон-
ных изделий, удобно получать шликерным литьем [555, 556] в гипсовых
формах с использованием шликеров, приготовленных на 1—1,5%-ном
растворе поливинилового спирта (Ж:Т=40:60, рН = 5, электролит —
НС1).
В работе [564] предложено получать плотные изделия из нитрида
кремния методом горячего прессования с так называемыми флюсующими
добавками, из которых лучший результат дают MgO и Mg3N2, при вве-
дении их в порошок кремния в количестве от 0,1 до 10% (обычно —
5%). Горячее прессование осуществляют в графитовых прессформах
под давлением 210 кг/сл2 и температуре 1600—1900° С (обычно —
1850° С). Открытая пористость полученных изделий не превышает 20%,
в основном достигает до 0,5%, прочность при изгибе 35—84 кг[мм2 при
20° С и 14—42 кг!мм2 при 1200° С. Потеря в весе при нагревании на воз-
духе при 1100° С составляет менее 1 мг!см2.
Таким образом, при горячем прессовании требуется вводить в со-
став нитрида добавки, а чистый нитрид кремния спекается горячим
прессованием плохо. В работе [542] для получения плотных компактных
изделий из нитрида предлагается горячее прессование его смесей с
кремнием ( с содержанием кремния в смеси более 20%), что при тем-
пературе 1400° С и давлении в 800 кг/см2 приводит к получению после
дополнительного азотирования изделий с довольно высокой плотностью
(этот процесс аналогичен получению самосвязанного карбида кремния).
Изучение в этой работе влияния температуры азотирования на проч-
293
ность нитрида кремния показало, что наиболее высокой прочностью
при изгибе обладают образцы, полученные азотированием при
1600° С (Оизг= 16 кг 1мм2).
Без горячего прессования раздельным прессованием и спеканием
могут быть получены относительно плотные изделия из нитрида крем-
ния с окисью магния либо с окисью алюминия, стойкие против окисле-
ния при 1200° С и обладающие высокими огнеупорными свойствами, осо-
бенно в расплавленных буре, цинке и т. п.
Рис. 130. Внешний вид изделий из нитрида кремния.
В настоящее время сделаны попытки приготовления волокон из
нитрида кремния [557].
Исследованы также условия образования и некоторые свойства пи-
ролитического нитрида кремния [1006] в форме тонких пленок. Плотность
пиронитрида равна 3,02—3,21 г]см3, коэффициент преломления 2,0—
2,06, диэлектрическая прочность 107 в/см, его химическая стойкость в
различных реагентах очень высока. Так, скорость коррозии в 48 %-ном
HF составляет всего 75—100 А/мин.
Изделия из нитрида кремния хорошо поддаются действию обыч-
ных методов механической обработки (они подобны в этом отношении
слоновой кости), очень легко обрабатываются ультразвуком [776].
Внешний вид изделий из нитрида кремния, в основном огнеупорно-
го назначения, показан на рис. 130.
294
2. НИТРИДЫ ГЕРМАНИЯ
В системе Ge—N различными исследованиями найдены соединения
Ge3N2 и Ge3N4 [6, 525, 565—567]. Однако результаты недавно выполнен-
ной работы [568] показали, что нитрид Ge3N2, по-видимому, не сущест-
вует. Следовательно, германий образует с азотом один нитрид — Ge3N4.
Структура Ge3N4 была исследована Юца и Ханом [102], которые
обнаружили, что это соединение изоморфно фенакиту BeaSiO^ имеет
гексагональную решетку с параметрами а—13,84; с=9,25 A, c/a=Q,668.
В дальнейшем структура этого соединения исследовалась неоднократно,
так, в [525] для него была указана ромбическая решетка с параметрами
а= 13,38; 6=8,60; с—7,74 А; в [529] обнаружены две модификации —
<x-Ge3N4 с ромбической решеткой (а=4,10; 6 = 7,10; с=5,94 А) и |3-Ge3N4
с ромбоэдрической (а=8,62; а=108°). При этом было установлено, что
a-Ge3N4 образуется при обработке германия аммиаком, a [+Ge3N4 —
при взаимодействии GeOs с аммиаком.
Следует полагать, что обе кристаллические модификации нитрида
германия — гексагональные, изоструктурные соответствующим модифи-
кациям нитрида кремния.
Физические и химические свойства. Нитрид Ge3N4 — светло-корич-
невое вещество с красноватым оттенком, обладает высоким электросо-
противлением (порядка 108 ом-см [33]), плотность 5,29—5,31 г/см^ [33,
102]. При температуре более 850° С нитрид германия полностью разла-
гается на элементы (до 850° С в среде азота устойчив). Теплота обра-
зования 15,6+1,7 ккал/моль [569].
Однако в [1060] это значение существенно скорректировано и со-
ставляет от 90+8 до 94+3 ккал/моль. Исследованное в той же работе
испарение нитрида германия в области температур 923—963° К эффу-
зионным методом Кнудсена показало, что при испарении этот нитрид
диссоциирует на твердый германий и азот. Давление азота над нитри-
дом германия составляет при 933° К 2,40 • 10~2 ат, возрастая при 963° К
до 4,27 • 10~2 ат.
Нитрид германия начинает окисляться на воздухе при 750—800° С,
окисление полностью заканчивается при 950° С (с обращением GeO3).
Методы получения. Нитрид германия Ge3N4 можно получить раз-
личными методами [568], из которых наиболее распространенным явля-
ется обработка германия аммиаком. Как видно из рис. 131 (кривая 2),
азотирование начинается при 700° С и особенно активно проходит при
750° С. Однако и при 800° С нитрид германия стехиометрического со-
става не образуется, дальнейшее повышение температуры вызывает раз-
ложение уже образовавшегося продукта. Для развития поверхности азо-
тирования в работе [568] было предложено смешивать порошок герма-
ния с разрыхлителем, в качестве которого был выбран углекислый ам-
моний. При использовании шихты из порошка германия и углекислого
295
аммония в соотношении 1 :2 азотирование при 750° С в продолжение
1 ч позволяет получить нитрид германия практически точного стехио-
метрического состава (рис. 131, кривая 3).
Можно также получать нитрид германия взаимодействием между
двуокисью германия и аммиаком в присутствии углекислого аммония и
без него в токе аммиака. Применение углекислого аммония в качестве
разрыхлителя позволяет снизить температуру азотирования до 750° С.
Рис. 131. Результаты азотирования гер-
мания:
I — расчетный состав Ge3N4*, 2 — обработка
германия аммиаком; 3 — то же в присутствии
разрыхлителя (NH4)2)CO3.
Без разрыхлителя реакция завершается за 4 ч при 800° С, при 850° С
время азотирования сокращается до 1 ч, но технологически эта темпе-
ратура опасна, так как небольшое ее превышение приводит к разложе-
нию нитрида.
В работе [569] a-Ge3N4 получался действием аммиака на порошок
германия, а p-Ge3N4 — на GeO3 при 750° С.
Азот на германий не действует.
3. НИТРИДЫ ОЛОВА И СВИНЦА
Сообщается о получении нитрида олова с вероятной формулой Sn3N4
путем испарения олова в среде азота [570, 29]. Растворимости азота в
свинце до 600° С не обнаружено. Сведений об образовании им нитридов
нет [29].
ГЛАВА VIII
НИТРИДЫ
ЭЛЕМЕНТОВ
ПОДГРУППЫ
АЗОТА
И ПРОЧИХ
ЭЛЕМЕНТОВ
1. СОЕДИНЕНИЯ ФОСФОРА С АЗОТОМ (НИТ-
РИДЫ ФОСФОРА)
В системе Р—N установлено существование со-
единений P2N5, P3N5 и PN. Наиболее распро-
страненным способом приготовления фосфида
P3N5 является реакция взаимодействия сульфи-
да P4N10 с газообразным аммиаком при нагре-
вании [720]. Термическим разложением P3N5-
•7NH3 в токе водорода [721] или нагреванием
PNCh в токе аммиака до 825° С [722] трудно
приготовить фосфид P3N5 точного состава.
Фосфид PN получают термическим разло-
жением P3N5 в интервале 750—810° С.
Авторы [723] исследовали равновесие меж-
ду паром PN и эквимолярной смесью фюсфора и
азота при 900° С, скорость разложения пара PN
и кинетику синтеза нитридов фосфора из элемен-
тов (на накаленной вольфрамовой нити). Рас-
сматривались равновесия: (PN)x,TB=?a:^PNra3;
2РМГаз^Р2,газ+Н2,газ С учетом рЭВНОВеСИЯ
Р 4,газ:*:±:2Р 2,газ-
Установлено, что равновесная концентрация
фосфора при 900° С составляет 1—3 об. % при
0,7—1 ат, что приводит к значению энергии дис-
социации D для PN равному 7,1+0,05- эв. Для
константы равновесия синтеза PN из элементов
получены значения от 1 • 10_2 при 900° С до
2-10“1 при 2000° С. Среднее значение теплоты
образования PN из элементов по реакции
Рг.газ+Ыг ,газ — 2PNras составляет 14,2 ккал/моль.
Энергия активации данной реакции равна
31 ккал/моль, эта величина показывает, что ре-
акция синтеза PN проходит не через стадию сво-
бодных атомов Р и N. Скорость образования
твердого PN из Р2 и N2 на вольфрамовой нити
при 1720—2127° С пропорциональна корню квад-
ратному из концентрации фосфора. Энергия ак-
тивации при температуре выше 1800“ С равна
59 ккал)моль, что близко к энергии диссоциации
фосфора на атомы. Отношение N:P s твердых
продуктах конденсации изменялось ок 1,03 до
1,19 (в исходной газовой смеси это отношение
было 1). Это отклонение объясняется юбразова-
297
нием на поверхности вольфрама комплексов из атомов Р и молекул
N2 типа PNN, реагирующих затем с атомами Р или N с образованием
полимеров вплоть до состава (P3N5)X.
Фосфид PN может быть также получен при термическом разложе-
нии P3N5 в интервале 750—810° С.
При пропускании тока азота с парами фосфора через электриче-
скую дугу получаются аморфные нитриды фосфора с отношением
N:P в интервале 1—1,5. При нагревании их до 800—900° С в токе ам-
миака это отношение возрастает до 1,7, т. е. образуется нитрид P3N5,
имеющий кристаллическое строение [724] *.
При комнатной температуре нитриды фосфора совершенно инертны,
холодная вода на них не действует, а горячая реагирует медленно, не
разлагаются при нагревании в концентрированной и разбавленной НС1,
разбавленной HNO3, растворах щелочей. Незначительно разлагаются
при длительном кипячении в концентрированной HNO3 и разбавленной
H2SO4. Хлор на них не действует [726].
При нагревании до 500—700° С нитриды фосфора начинают дейст-
вовать как сильные восстановители, вследствие разложения на азот и
элементарный фосфор [724].
Плотность P3N5 равна 2,57—2,58 г/см3 [720], теплоемкость до 300° С
выражается как Ср = 5,20+102 • 10-3 Т [84].
Практически в большинстве способов получения образуются нит-
риды фосфора непостоянного состава и в аморфной форме; перевод их
в соединения определенного состава производится при нагревании в оп-
ределенных условиях.
2. СОЕДИНЕНИЯ МЫШЬЯКА, СУРЬМЫ И ВИСМУТА С АЗОТОМ
О непосредственных соединениях As, Sb и Bi с азотом достоверных све-
дений нет.
3. СОЕДИНЕНИЯ ЭЛЕМЕНТОВ ПОДГРУППЫ КИСЛОРОДА С АЗОТОМ
Соединения кислорода с азотом. Азот с кислородом образует окислы:
закись N2O, окись (моноокись) азота NO, трехокись азота N2O3, дву-
окись азота NO2, четырехокись азота N2©4 и пятиокись азота N2Os, ме-
тоды получения которых и физико-химические свойства подробно опи-
саны в курсах неорганической химии [727]. Все эти соединения имеют
ковалентный характер связи.
О рентгеновском анализе нитридов фосфора см. [725].
298
Закись азота N20 — бесцветный газ, температура кипения—89,5° С,
температура плавления — 102,4° С, критическая температура — 36,5° С*
критическое давление 71,1 ат. Структура твердой закиси азота куби-
ческая (изоморфна СО2), с плотнейшей кубической упаковкой молекул
N2O, причем линейные молекулы СО2 направлены по четырем непересе-
кающимся осям. Период решетки а—5,656 А. Лэнгмюр предложил для
N2O следующее электронное строение: N : : N : : О, которое было затем
дополнено резонансными формами: N_ : : N+ : : О и : N+ : : N : О- :
В соответствии с этим строением молекула N2O имеет очень малый
дипольный момент, равный 0,14 • 10-18 эл. ст. ед. С точки зрения обра-
зования состояний с минимумом свободной энергии можно представить
молекулу N2O как результат преобразования конфигурации атомов
азота «2р3 в sp^ и передачи легкоподвижных электронов атому кисло-
рода с образованием конфигурации «2р6. Таким образом, в молекуле
N2O следует ожидать высокого статистического веса «реконфигураций
атомов азота и «^-конфигураций атомов кислорода, связанных между
собой обменом через часть электронов, оставшуюся нелокализованной в
этих конфигурациях. Такое строение удовлетворительно объясняет то,
что, несмотря на метастабильность, закись азота начинает разлагаться
при относительно высоких температурах.
Моноокись азота NO — бесцветный газ, температура кипения
—151,8° С, температура плавления —163° С. В соответствии с представ-
лениями Паулинга в NO осуществляется типичная трехэлектронная
связь: : N : : б : с соответствующими резонансными формами [727]. На-
ложение трехэлектронной связи на обычную гомеополярную, образо-
ванную электронными парами, несколько усиливает связь азота с кис-
лородом. Это усиление невелико, так как димеризация молекул NO в
N2O2 со спариванием неспаренных электронов имеет место только в
жидком, но не в газообразном состоянии, где существуют мономолеку-
лы NO. Наличие трехэлектронной связи обусловливает парамагнитность
моноокиси азота. Дипольный момент NO равен 0,6 • 10“18 эл. ст. ед.
В жидком состоянии полностью полимеризована, что вызывает высокую
ее теплоемкость, сильно зависящую от температуры, и высокую энтро-
пию испарения.
Как и в предыдущем случае, можно предположить, что в окиси азо-
та атомы азота и кислорода образуют «р3- и «2р6-конфигурации, причем
образуют их попеременно; эти конфигурации связаны электрона-
ми, находящимися в состоянии обмена между ними. Такой тип связи
условно можно назвать бегущим.
Трехокись азота N2O3, температура плавления 102° С, легко распа-
дается на NO и NO2, либо на NO и N2O4- Теплота образования N2Os
из NO и NO2 только 9,6 ккал/моль, а свободная энергия образования
равна 0,44 ккал/моль.
299
Двуокись азота NO2 и четырехокись азота N2O4 • NO2 — бурый газ,
температура кипения — 22,4° С, температура плавления—10,2° С. При
охлаждении NO2 димеризуется, переходя в бесцветную N2O4 • NO2, па-
рамагнитна, N2O4—-диамагнитна. Диамагнетизм последней объясняет
стремление NO2 к димеризации со спариванием неспаренных электро-
нов. По Полингу, в молекуле NO2 имеет место наложение трехэлектрон-
ной связи на обычную, парноэлектронную, с мезомерией двух состояний
,.,О
ХО
О
жидкой N2O4 при 17° С равна
Удельная электропроводность
1,3 • 10-12 ом~1 • см,—*, диэлектрическая проницаемость 2,42, показатель
преломления 1,420, при 20° С молярная рефракция 15,2 сл<3, молярная
поляризация 26,5 сл«3. В твердом состоянии N2O4 имеет кубическую ре-
шетку с а=7,77 А.
Пятиокись азота N2O5 образует бесцветные, твердые кристаллы
гексагональной сингонии, а = 5,410; с=6,570 (при 80° С), удельный вес
1,63 г/см3, температура плавления 30° С, температура кипения 45—
50° С, в твердом состоянии имеет солеобразную структуру, построен-
ную из треугольных ионов (NO3)~ и линейных ионов (NO2)+.
Имеются данные о существовании еще более богатого кислородом
окисла азота NO3 — весьма неустойчивого соединения, образующегося
при реакции NO2 или N2Os с озоном.
Соединения серы с азотом. Сера образует с азотом два соедине-
ния — [727, 728] — нитрид серы S4N4 и четырехсернистый азот S4N2.
Нитрид серы получают взаимодействием серы с жидким аммиаком
10S + I6NH3 -> 6(NH4)2S + S4N4.
Структура этого соединения, по данным [729], показана на рис. 132
(см. также 730). На рис. 133 показаны возможные резонансные формы
электронного строения нитрида S4N4 [720]. Представляет собой золо-
тисто-желтые кристаллы с температурой плавления 178° С, соединение
сильно эндотермично (при образовании из элементов поглощается
128 ккал/моль), поэтому при нагреве или детонации распадается со
взрывом. Несколько растворим в органических растворителях, гидро-
лизуется водой при продолжительном кипячении с образованием аммиа-
ка и кислородных кислот, при сублимации в вакууме деполимеризуется
с образованием S2N2 — кристаллического вещества, устойчивого только
при низких температурах, а при комнатной температуре медленно пре-
вращающегося в смесь S4N4 и высокополимерного соединения (SN)X.
Последнее соединение устойчивее, чем S2N2 и S4N4, и обладает полуме-
таллической проводимостью.
300
Четырехсернистый азот — кристаллическое вещество темно-красно-
го цвета, температура плавления 23° С, плотность 1,71 г/см3, получается
действием сероуглерода на нитрид серы при 100° С под давлением либо
при разложении нитрида серы. Разлагается при обычной температуре и
вспыхивает при нагревании, растворяется многими органическими раст-
ворителями, водой постепенно разлагается с выделением серы и аммиа-
ка. Предполагается, что это соединение имеет циклическую структуру
Рис. 132. Схема структуры соединения S4N4.
Рис. 133. Возможные резонансные формы строения S4N4.
с содержанием в молекуле S4N2 ионов S4+, S2+, двух нейтральных ато-
мов S при отрицательных ионах азота [727].
Соединения селена с азотом. Селен образует с азотом соединение
Se4N4 — оранжево-красное вещество с триклинной решеткой и парамет-
рами: а=6,47; 6 = 6,85; с = 6,85 А, а = 99,5°, (3 = 100,4°, у= 100,4° [731].
По последним данным [965], это соединение относится к пространст-
венной группе С 2]с с периодами решетки а=9,65; 6 = 9,73; с = 6,47 А,
3=104,9°.
Его плотность 4,2 г/см3, теплота образования из элементов
42,6 ккал!моль. Легко разлагается при ударе или нагревании до 200—
230° С с выделением селена. Нерастворим в холодной воде, медленно
разлагается водой при кипячении с образованием Н28еОз, селена и ам-
миака. Получается нитрид селена по реакции
12SeCU + 64NH3 = 3N4Se4 + 48NH4C1 + 2N2,
а также разложением Se(NH)2, действием сухого аммиака на раствор
Se2C12 в сероуглероде, взаимодействием бензола с сухим аммиаком в
присутствии SeO(OC2H5)2-
301
Известен также полимер (NSe)x с моноклинной решеткой с пара-
метрами: а = 9,65; 6 = 9,73; с=6,47 А, (3=104,9°.
Предполагалось, что (NSe)x состоит из четырех молекул Se4N4. Од-
нако в [732] было показано, что наиболее удовлетворительное соответ-
ствие рентгеновской плотности (4,22 г/см3), пикнометрической (4,2 г!смл)
получается при принятии димеров, т. е. (NSe)8.
Соединения теллура с азотом [1027]. Известны два нитрида теллура:
TeN (9,89 вес.% N) и Te3N4 (12,76 вес. % N). Они получаются взаимо-
действием тетрабромида теллура с аммиаком
ЗТеВг4 4- 16NH3 = Te3N4 4- 12NH4Br.
4. СОЕДИНЕНИЯ ГАЛОГЕНОВ С АЗОТОМ [727]
Галогены с азотом образуют два класса соединений — общей формулы
XN3 (производные азотистоводородной кислоты) и NX3 (продукты за-
мещения водорода аммиака на галоген). Соединения первого из этих
классов носят название азидов. Известны азиды фтора (FN3 — желто-
зеленый газ, температура плавления —154° С, температура кипения
—82° С, в жидком состоянии взрывчат), азиды хлора (CIN3 — бесцвет-
ный, сильно взрывающийся газ), азиды брома (BrN3 — жидкость), йода
(JN3 — желтовато-белое твердое взрывчатое вещество).
Соединения второго класса — фторид азота NF3 (бесцветный газ,
температура кипения —129° С, температура плавления —208,5° С, плот-
ность при температуре кипения 1,885 г]см\ стойкое экзотермическое
соединение), хлорид азота (NCI3 — темно-желтая летучая маслянистая
жидкость, удельный вес 1,65 г/см3, сильно эндотермичен — теплота об-
разования —54,7 ккал/моль, взрывается при нагревании до 93° С или
при соприкосновении с веществами, способными хлорироваться), йодид
азота (NJ3 — при обычной температуре черное твердое вещество, сильно
экзотермичное).
ГЛАВА IX
СЛОЖНЫЕ
НИТРИДЫ
В настоящее время накоплен значительный экс-
периментальный материал по многокомпонент-
ным системам, содержащим азот, в частности по
системам, в которых образуются тройные, чет-
верные нитридные фазы. Не имея возможности
подробно изложить и систематизировать эти ма-
териалы в настоящей монографии, а также учи-
тывая, что они в известной мере изложены в [29,
933], приводятся данные только об основных и
типичных системах, в которых образуются слож-
ные карбидные фазы, представляющие научный
и практический интерес. Эти данные сгруппи-
рованы по системам, которые образованы раз-
личными металлами с азотом (Mei—Мег—N),
металлами с бором и азотом (борнитриды Me—
В—N), с углеродом и азотом (карбонитриды
Me—С—N), с кислородом и азотом (оксинит-
риды Me—О—N), с галогенами (галогенонитри-
ды Me—F—N, где F — галоген), с кремнием и
азотом (силиконитриды Me—Si—N), а также
неметаллами с азотом (Xi—Хг—N, где X — не-
металл). Подобная классификация естественно
не имеет особых научных оснований и предназ-
начена лишь для упорядочения имеющихся в
литературе многочисленных и разрозненных све-
дений о сложных нитридах и системах, в кото-
рых они образуются.
1. СИСТЕМЫ МЕТАЛЛОВ С АЗОТОМ
Литий—алюминий (галлий) — азот [426]. IJ3AIN2
получают нагреванием лития с алюминием в
токе азота при температуре 730° С либо нагрева-
нием Li3Al в токе азота, либо нагреванием сме-
сей нитридов лития и алюминия. Белый до свет-
ло-серого, устойчив до 1000° С, в среде азота,
плотность 2,33 г]см?. Легко гидролизуется, может
присоединить два моля аммиака. Li3GaN2 полу-
чается из Li3Ga и азота при 600° С, светло-серый,
термически устойчив до ~800°С, плотность
3,35 г!см\ Легко гидролизуется с образованием
аммиака и азота. В токе NH3 и Нг разлагается
при ~400°С. Рентгенографически для обоих со-
303
единений обнаружена сверхструктура с решеткой CaF2, пространствен-
ная группа Т7 , 16 формульных единиц в элементарной ячейке. Построе-
ны по структурному принципу: плотная упаковка атомов азота с атома-
ми металла, расположенными в тетраэдрических пустотах этой упаковки.
Обладают значительно меньшими объемами элементарных ячеек по
сравнению с Li3N вследствие замены однозарядного катиона лития на
многозарядный. Аналогичные фазы образуют литий с цирконием и
торием.
Литий — магний (цинк) —азот [911]. В этих системах обнаружены
соединения LiMgN и LiZnN. Нитрид LiMgN получают совместным на-
греванием нитридов лития и магния в токе азота при температуре
1050° С. Порошок красно-коричневого цвета, плотность 2,41 г]см3, легко
гидролизуется, может присоединять 1 моль аммиака. Решетка CaF2 с
азотом в положениях атомов кальция и статистическим распределением
лития и магния в положениях атомов фтора; а=4,970 А. Нитрид LiZnN
готовится спеканием смеси нитридов лития и цинка при 400° С в токе
аммиака. Черный, плотность 4,61 г/сд<3, легко гидролизуется. Структура
выводится из структуры CaF2, литий и цинк статистически распределены
в положениях фтора; п = 4,877А.
Литий — ванадий (ниобий, тантал) — азот [917]. При нагревании
Li3N и VN в среде азота при температуре 680° С образуется соединение
Li7VN4, которое термически более устойчиво, чем оба исходных нитрида.
Кристаллизуется в сверхструктуре плавикового шпата с а=9,60 А. Ана-
логичные соединения образуют ниобий и тантал: Li7NbN4 и Li7TaN4—
они химически и термически более стабильны, чем тройной нитрид ва-
надия.
Литий — ванадий (марганец) — азот [436]. В этих системах обна-
ружены тройные нитриды Li7VN4 и Li7MnN4, кристаллизующиеся в
сверхструктуре плавикового шпата с удвоенным периодом решетки (про-
странственная группа Тр. Ионы металла в решетке CaF2 занимают по-
ложения фтора. Указанные фазы имеют соответственно периоды решет-
ки: а = 9,604 и а=9,57[ А и расчетные значения плотности 2,33 и
2,42 г/см3.
Литий — хром (молибден, вольфрам) — азот [912]. Нагреванием при
температуре 616—856° С смесей нитрида лития с хромом, молибденом
или вольфрамом в среде азота были получены тройные нитриды LigCrNs,
LigMoNs и LigWNs. Тройной нитрид хрома темно-коричневый, на возду-
хе гидролизуется с выделением аммиака, а при длительном пребывании
на воздухе желтеет вследствие образования хромата. Нитрид имеет пре-
имущественно солеобразный характер. Так как нитрид LigCrNs образует
непрерывный ряд твердых растворов с Li2O, то можно считать, что он
кристаллизуется в сверхструктуре плавикового шпата.
304
Тройной нитрид молибдена коричневый, но более светлого оттенка,
чем тройной нитрид хрома, а тройной нитрид вольфрама — еще светлее,
почти серый. Оба нитрида разлагаются с выделением аммиака, раство-
ряются в разбавленных кислотах, тройной нитрид вольфрама наиболее
термически устойчив из всех нитридов (при 870° С давление над ним
азота всего 3 мм рт. ст.), а наиболее неустойчив — нитрид хрома. Кри-
сталлическая структура тройных нитридов молибдена и вольфрама не-
ясна, предполагается, что они имеют по две модификации.
Литий — кобальт (никель, медь) — азот [918]. В этих системах нит-
риды компонентов образуют твердые растворы (Li, Co)3N, (Li,Ni)3N,
(Li,Cu)3N.
Магний — германий — азот [1061]. При действии аммиака на герма-
нид магния образуется соединение MgGeN2 : 3Mg2Ge + 8NH3 =
=3MgGeN2+Mg3N2+ 12Н2.
Это соединение может быть получено также действием аммиака на
смесь нитрида магния и германия: MggN2+3Ge+4NH3=3MgGeN2-[-6H2,
либо взаимодействием нитридов магния и германия при 850—950° С:
Mg3N2+Ge3N4=3MgGeN2.
Соединение представляет порошок серого цвета, стабильный в при-
сутствии воды или растворов щелочей; действует на него HCL Кристал-
лизуется в ромбической системе (протранственная группа C92V ), пара-
метры решетки: <2 = 5,504; 6 = 6,660; с=5,172 А. При нагревании в арго-
не при 750° С разлагается на германид магния, германий и азот, а при
950° С полностью диссоциирует на элементы. Окисление начинается при
575° С и полностью заканчивается при 950° С. Продуктами окисления
являются MgO и MgGeO3.
Стронций (барий) — рений (осмий) — азот [994]. При нагревании
нитридов бария и стронция с рением в азоте образуются тройные нит-
ридные фазы Sr9Re3Nio, Ba9Re3iNio, Ba9Os3Nio, имеющие ромбиче-
скую структуру, металлическую проводимость; измерение магнитных
свойств показало сильное взаимодействие атомов переходных металлов
в этих фазах. Существует также фаза S^rReg^e — кубическая типа
NaCl.
Алюминий — галлий — азот. Исходя из подобия кристаллической
структуры и размеров решетки, AIN и GaN должны образовывать не-
прерывный ряд твердых растворов, однако попытки приготовить
твердые растворы (Ga, A1)N не дали положительных результа-
тов [599].
Нагревание смесей этих нитридов при температуре 900° С в течение
недели в среде азота не привело к образованию твердого раство-
ра, а при 1100° С смеси взрывались из-за высокого давления
паров GaN.
Ванадий (титан) — хром — азот. При действии смеси аммиака и во-
дорода при температурах 800—1000° С на осадки гидратов хрома и ти-
2U—272
305
тана образуются твердые растворы нитридов ванадия и хрома либо ти-
тана и хрома [1024], аналогичным образом получаются тройные твердые
растворы нитридов титана — ванадия — хрома. Все указанные твердые
растворы имеют кубическую гранецентрированную решетку. Термоди-
намическая устойчивость CrN и VN повышается при образовании ими
твердых растворов друг в друге либо каждым из них в нитриде тита-
на [1044].
Титан — алюминий — азот. В работе [418] обнаружено соединение
состава Ti2AlN (получается горячим прессованием смеси TiN, металли-
ческого титана и алюминия). Оно изоморфно СггА1С и Ti2AlC и имеет
периоды решетки: а = 2,994; с= 13,61 А; с/а=4,544. Рентгеновская плот-
ность 4,30 г!см3.
Титан — хром — азот. Установлено, что при азотировании хромоти-
тановых сплавов образуются пленки, содержащие двойной нитрид Сг—
Ti—N [337]. Кинетика процесса азотирования сплавов титана с 5% Сг
исследована в [925].
Титан — молибден — азот [439, 226]. Сплавы системы TiN—Мо по-
лучены спеканием смесей порошков компонентов горячим прессованием
при температуре 1750—1800° С. Сплавы состоят из двух фаз — молиб-
дена и нитрида титана, причем их взаимной растворимости не замечено.
Сплавы, содержащие 10—30% Мо (остальное — TiN), имеют плотность
5,59—6,59 г/см3, оизг=28—38 кг/мм3. Твердость сплавов с 5—30 об. % Мо
составляет 1400—1500 кг/мм2, при 400° С снижается до 764—913, при
800° С — до 540—608 кг/мм2. Наиболее окалиностойкими являются спла-
вы (керметы) с наименьшим содержанием молибдена, в данном случае
при 5% Мо.
Титан — кобальт (никель) — азот, молибден — кобальт (никель) —
— азот. В этих системах обнаружены и исследованы тройные нитриды:
Tio,zCoo,3N; Ti0,7Ni0,3N; Moo,aCoo,2N0,2 и Moo,8Ni0,2No,9. Все они имеют
структура типа WC [916].
Переходный металл — Al, Ga, In, TI, Ge—азот [419]. В этих систе-
мах обнаружены четыре группы тройных соединений: Me2XN, MesCe3Nx,
Me3X2N, Me2XNx. Соединения первой из этих групп имеют структуру
типа Сг2А1С: Zr2TllN, Hf2InN, V2GaN, V2GeN. Ко второй группе относятся
соединения: V5Ge3Nx, Nb5Ga3Nx, Ta5Ga3Nx, Ta5Ge3Nx, Ta5Al3Nx (стаби-
лизованное кремнием), которые, за исключением последнего, имеют
структуру типа Mn3Si3; только Ta5Al3Nx имеет структуру типа Сг5В3 с
вакансиями на местах атомов азота. Соединения V3Zn2N, V3Ga2N и
Nb3Al2N имеют решетку типа fJ-Mn, а соединения Ti2ZrNx, Zr2ZnNx-,
Hf2ZnNx относятся к типу Ti2Ni.
Авторы [420] получали соединения Ti2GaN, Ti2lnN, Zr2InN путем на-
гревания смесей мононитридов титана или циркония с галлием или ин-
дием в кварцевой ампуле при 850° С в течение 500—850 ч. Все они име-
ют структуру Сг2А1С с периодами решеток и плотностью:
306
Фаза а, А с, А с/а Рентгенов- ская плот- ность, г/см3
1 i2GaN 3,004 13,30 4,428 5,73
Ti2InN 3,074 13,975 4,54, 6,52
Zr2InN 3,27, 14,84 4,52в 7,49
Еще одну группу составляют соединения со структурой перовскита
и общей формулой Me3XN [421]:
Рентгеновская
Фаза а, А плотность, г/см3
Ti3InN 4,190 6,15
Ti3TlN 4,191 8,16
В работах [422, 423] дан подробный обзор структуры фаз типов
Me3X2N, MesXaNi-x, Me2XN, Me3XNi_x.
Ниобий — (гафний, молибден, вольфрам) — азот. В работе [1049]
исследована растворимость азота в богатых ниобием сплавах Nb—Hf,
Nb—Мо и Nb—W в функции давления и температуры. Установлено, что
гафний, молибден и вольф-
рам снижают раствори-
мость азота в ниобии. Так,
если при 2000° С и давлении
азота 2,8-10-1 мм рт. ст.
растворимость азота в нио-
бии составляет 9,4 ат.%, то
в сплаве ниобия с 10% Hf
или Мо она уменьшается
до 1,0 ат.%, а в сплаве нио-
бия с 10 ат. % W — до
Рис. 134. Температурная зависимость электросо-
противления нитрида NbN и нитрида с добавками:
/ — NbN; 2 —NbN + 0,1% Fe; 3 — NbN+0,05% Ti+0,1% Fe;
4 —NbN + 0,5% Fe; 5 —NbN+l%Fe.
6,8 ат. % N.
Ниобий—железо — азот
[431]. При исследовании
растворимости NbN в y-Fe в
интервале температур 1191 —
1336° С (по содержанию азота в сплавах Fe—:Nb с 0—0,92 вес. % Nb,
находившихся в равновесии с газовой смесью из азота с 1 % Н2) уста-
новлено, что температурная зависимость произведения растворимости
NbH в y-Fe описывается формулой
(%Nb)(%N) = — 10,230/7" -г 4,04.
Молярная теплота растворения нитрида ниобия в y-Fe равна 46,8 ккал.
При введении в нитрид ниобия небольшого количества железа воз-
растает как его электросопротивление, так и ТКС. При увеличении со-
держания железа ТКС уменьшается, для сплава NbH с 1 % Fe ТКС в
широком интервале температур практически равен нулю (рис. 134). Од-
20*
307
новременно происходит некоторое снижение коэффициента термо-э. д. с.
нитрида ниобия
Тантал — хром (марганец, железо, кобальт, никель) — азот [428].
Сплавы этих систем приготовляли азотированием смесей порошков ме-
таллов сухим аммиаком при температуре 650—950° С в течение от двух
дней до двух недель. Ре-
зультаты рентгенографиче-
ского анализа представле-
ны в табл. 104.
Таблица 104
Фазы, наблюдающиеся в двойных сплавах нитрида
тантала с нитридами Cr, Мп, Fe, Со, Ni
Система Исходный сплав Фазы, образующиеся при азотировании
Та—Cr—N Fa0,75 ^Г0,25 Та0,50 (-'Г0,50 Та0,25 (-'г0,75 X-J-TaN X + следы неизвестной фазы Cr(Ta)N-|- следы Х-фа- зы
Та—Мп—N Та0,90 Мп0.10 Та07,5 Mn0j25 Та0,50 Мп0.5о Та0,25 МпО,75 Ta(Mn)N0 8+ следы Та Ta3MnN4 X + Ta3MnN4 X + нитриды Мп
Та—Fe—N Та0,80 Fe0,20 Fa0,67 Fe0,33 Fa0,50 Fe0,50 Ta2FeN2+Ta Ta2FeN2 Ta2FeN2 + нитриды Fe
Та—Со—N Fa0,80 C°o,2O Та0,67 Co0,33 Ta0,50 C°o 50 Ta2CoN3 + TaN Ta2CoN3 Ta2CoN3 -p a-Co
Та—Ni—N Ta0,80 Ni0,20 Fa0,67 NiOj33 Ta0,50 Ni0,50 Ta2NiN3 + TaN Ta2NiN3 Ta2NiN3 + Ni
В системе Та—Сг—N
обнаружена тройная фаза
(фаза X), рентгенограмма
ее имеет сложный характер.
Нитрид тантала раство-
ряется в нитриде хрома,
при этом период решетки
CrN изменяется от а =
— 4,149 кХ (CrNi,oo) до а=
— 4,239 кХ. Последнее зна-
чение соответствует макси-
мальной растворимости TaN
в CrN (около 25 мол.% Та).
В системе Та—Мп—N
найдены две тройные фазы,
одна из которых изострук-
турна фазе X системы Та—
—Сг—N, а другая имеет
состав, выражаемый форму-
лой Ta3MnN4. Тантал не
растворяется в MnNo,6i-o,63
(ц-фазе), а марганец в
s-TaN.
Хром—марганец—азот.
В системе Сг—Мп—N
обнаружено [438] трой-
ное соединение состава
CrMnaN3/4Qi/4 (где □—вакансии). Образование этой фазы обусловли-
вает повышение на два порядка магнитной восприимчивости сплавов
хрома с марганцем при их азотировании. Юм-Розери [441] указывает
на существование тройного нитрида Cr56>1MnI8,3N25,6.
Молибден — кобальт — азот [958, 956]. Смешение Mo3N с 20% Со
с последующим прессованием и спеканием в течение 30 мин в среде ам-
миака при температуре 1500° С приводит к образованию сплава, имею-
щего металлический характер излома с твердостью 86,5 Rc. Несколько
308
более низкой твердостью обладает сплав из 20% MoN, остальное М02С.
Молибден — цирконий — азот [982]. В системе Мо—Zr—N были
приготовлены сплавы с 10, 30, 50,70 и 90 ат. % молибдена, превращенные
в нитриды азотированием аммиаком при 740° С. От чистого циркония до
соотношения Zr0,5Mo0,5 образуется смешанная нитридная фаза (Zr,
Mo)N, имеющая кубическую гранецентрированную структуру, анало-
гичную структуре нитрида ZrN. Сплав Zro,iMoo,9 и чистый молибден об-
разуют при азотировании кубическую гранецентрированную фазу — у-
(Мо, Zr)2 N. Вблизи соотношения металлов Zro,3Moo,? образуются обе
нитридные фазы, не смешивающиеся друг с другом.
Марганец — медь (цинк)—азот [393]. Установлено образование
твердых растворов, которым можно условно приписать составы
Мп4-жСижМ1_ж/4Пх/4Мп4-жСижЫ1_ж/4Пж/4 (где □—вакансии). Проведено
исследование их структуры и магнитных свойств.
Марганец — медь (серебро, галлий) — азот, хром — галлий — азот
[429]. В указанных системах образуются тройные нитриды со структурой
перовскита, из которых Mn3CuN, Mn3AgN и MnaGaN являются продук-
тами замещения одного атома марганца в нитриде Mn4N на атом дру-
гого металла, a Cr3GaN — производным гипотетического нитрида хрома
Cr4N. Периоды решетки этих фаз: Mn3CuN а — 3,906; Mn3AgN tz=4,019g;
Mn3GaN a=3,898; Cr3GaN а=3,8755 А.
Марганец — галлий (германий, индий) — азот; железо — галлий
(индий, олово)—азот; кобальт — галлий (германий, индий, олово) —
азот; никель — галлий (германий, индий, олово) — азот [437]. Из этих
систем тройные фазы образуются в системах
Со — Ga — N (CoeGa3N), Со — Ge — N (Co59,iGe20,5N2o,4);
Co — In — N (C061,5! 1120,2^8,3); Co — Sn — N; (Co63Sn2iNi6);
Ni — In — N (Nies,7^21,9^2,4)-
Все эти фазы имеют кубическую структуру типа L72 с периодами, ука-
занными в табл. 105. Тройной нитрид образуется также в системе Fe—
Ge—N с тетрагональной решеткой (см. табл. 105). Показано также, что
Таблица 105
Периоды решетки тройных нитридов фаз, А [437]
Фаза Кристаллическая решетка а С с/а
Co6Ga3N Кубическая 3,56—3,68 — —
СО59,1Се20,5^20,4 » 3,57—3,61 — —
С 061,5^20,2^18,3 » 3,85—3,86 — —
СО63^П21^]6 » 3,82 — —
N165,7In21,9^12,4 » 3,84 — —
Fe Ge Ы Л у л Тетрагональная 5,318 7,733 1,454
30®
Ga, Ge, In и Sn существенно расширяют области гомогенности бинар-
його нитрида железа (у'-фазы системы Fe—N).
Обнаружено тройное соединение в системе Мп—Ga—N [937]. Оно
имеет состав Mn3GaN и идентично соответствующим фазам в системах
Сг—Ga—N, Мп—Си—N и Мп—Ag—N (стр. 309). Это соединение при
298° К претерпевает антиферромагнитное превращение первого по-
рядка.
Марганец — золото (ртуть, олово, платина, палладий, родий) —
азот. Установлено существование тройных соединений состава Mn3XN со
структурой перовскита и следующими периодами решетки [1033]:
Соединение а д Соединение
а, А.
Mn3AuN 4,0235 Mn3PtN 3,9685
Mn3HgN 4,O72o Mn3PdN 3,9796
Mn3SnN 4,0586 Mn3RhN 3,9280
• Cr,iec.%
Рис. 135. Растворимость азота в расплавах Fe—
Сг—Ni при 1600° С ио =1 ат.
n2
Исследованы магнитная восприимчивость и магнитные превраще-
ния этих химических соединений.
Железо — хром — азот [930]. Определен состав нитридов, теплота
растворения азота в сплавах железа с хромом. Показано, что хром уве-
личивает растворимость
азота в а- и у-Fe. При
600° С образуется CrN с
периодом решетки а =
=4,140 А. Азот стабили-
зирует область у-фазы в
сплавах железо—хром.
Железо — хром — ни-
кель—азот. Исследована
растворимость азота в
жидких сплавах Fe—
—Сг—Ni при температу-
ре 1600° С и давлении
азота 1 ат (рис. 135),
определены теплоты рас-
творения [939].
Неодим — алюминий
(галлий, индий, таллий,
олово, свинец) — азот
[1063]. В этих системах
образуются тройные нитридные фазы состава Nd3MeN (где Me—Al, Ga,
In, TI, Sn, Pb) co структурой перовскита и периодами решетки соот-
ветственно: 4,910; 5,063; 4,94э; 4,957; 5,057; 5,067 А.
Уран — плутоний — азот [1028, 1041]. В системе U—Ри—N образует-
ся непрерывный ряд твердых растворов, которые получаются спеканием
310
смесей мононитридов урана и плутония. Азотирование сплава плутония
с ураном проходит с большим трудом, сплавлением эти твердые раство-
ры получить нельзя вследствие диссоциации и удаления части азота и
металлов.
2. СИСТЕМЫ МЕТАЛЛ — БОР — АЗОТ
Литий (кальций, барий) — бор — азот [913]. При нагревании смесей нит-
рида лития, нитрида кальция или амида бария с нитридом бора при
температуре 700—1000° С образуются тройные нитриды LisBNz, СазВ2П4
и Ba3B2N4. Структура их не исследована, изучение ПК-спектров указы-
вает на наличие в них иона N = B = N3~. Борнитрид лития плавится при
870° С, плотность 1,755 г]см\ имеет черно-фиолетовую окраску, очень
тверд. Борнитрид кальция — вещество белого цвета с желтоватым от-
тенком, полностью растворяется в разбавленной НС1, гидролизуется в
воде с выделением аммиака (значительно устойчивее, чем борнитрид
лития).
Титан (цирконий, гафний, ниобий, хром, вольфрам, железо)—бор—
азот. В работе [901] при совместном борировании и азотировании нио-
бия не было обнаружено тройных фаз.
Система Ti—В—N исследована Г. В. Самсоновым и Е. В. Петраш
[468] по разрезу TiB2—TiN в пределах от 3 до 97 мол. % каждого компо-
нента. Образцы сплавов получали горячим прессованием с последую-
щим отжигом. Измерение температур плавления, проведенное визуально,
позволило построить кривую плавкости для этой системы. Микро-
скопическое исследование сплавов показало, что все сплавы до допол-
нительного спекания и отжига двухфазны. После отжига двухфазные
сплавы, содержащие 3—8 мол. % TiB2, становятся однофазными, что мо-
жет связано с растворимостью борида в нитриде титана.
Микротвердость белой (на основе TiB2) и желтой (на основе TiN)
фаз практически не зависит от состава сплавов и близка к твердости
соответственно TiB2 и TiN (рис. 136). Такие же результаты дает и рент-
геновское исследование, малая точность которого не могла дать возмож-
ности обнаружить растворимость компонентов. Последняя выявляется
на кривой электросопротивление — состав (рис. 137). Термическое рас-
ширение в системе TiB2—TiN представлено на рис. 138.
Наиболее стоек к окислению сплав, содержащий 40—50% TiN
(рис. 139). Вероятно, этому составу отвечает наиболее стойкая пленка
бората титана, защищающего образцы. В сплавах, содержащих большие
количества борида, образующийся при окислении борный ангидрид вы-
зывает пузырение пленки. В сплавах, богатых TiN, пленка бората не
сплошная из-за интенсивного выделения окислов азота при окислении
TiN кислородом воздуха.
311
Обнаружены признаки растворимости гексагонального TiB2 в ку-
бическом TiN и практическое отсутствие обратной растворимости TiN
в TiB2, что напоминает такое же явление при исследовании раствори-
Рис. 136. Микротвердость сплавов системы TiB2—TiN:
I — фаза на основе TiB2; 2 — фаза на основе TiN.
Рис. 137. Электросопротивление сплавов системы TiBj—TiN.
Рис. 138. Коэффициент термического расширения сплавов системы TiB2—TiN.
в химической прочности TiB2 и TiN меньше, чем между WC и TiC, по-
этому область растворимости TiB2 и TiN значительно меньше, чем WC
в TiC.
Исследование системы Ti—В—N продолжено в работе [903], где
подтверждено, что в системе не существует тройных соединений.
В исследованной аналогичным образом системе Zr—В—N [904] так-
же не были обнаружены тройные соединения, но показано образование
твердых растворов Zr—N—В состава Zr(N, В)1_ж, где около 37 ат.°/о N
может быть заменено бором. Аналогичная фаза твердых растворов су-
ществует и в системе Hf—В—N [904], причем границы ее однофазности
сильно зависят от температуры.
В работе [416] исследованы следующие области и разрезы системы
Zr—В—N: Zr—ZrB2—ZrN, ZrB2—В—BN, ZrN—ZrB2—BN, ZrB2—ZrN,
312
Рис. 139. Стойкость к окис-
лению сплавов системы
TiB2—TiN.
ZrB2—BN. Для приготовления образцов смеси гидрида циркония, нит-
рида циркония, нитрида бора и бора подвергались горячему прессова-
нию с последующим гомогенизирующим отжигом в течение 4—6 ч при
температуре 1400—1900° С (в зависимости от состава) в среде аргона.
В результате металло- и рентгенографического исследования установ-
лено, что в этой системе тройных фаз не образуется. В дибориде цирко-
ния, имеющем ничтожно малую область го-
могенности, не растворяются ни углерод, ни
азот. При малых содержаниях циркония в
системе образуются двухфазные области
ZrB2—BN и твердый раствор (ZrN—ZrB) —
—iBN при температурах ниже 1600° С. При
содержаниях в средней области, т. е.
~50 ат.% Zr, 25 ат.% В и ~25 ат.% N, об-
наруживаются три фазы: a-твердый раствор
на основе циркония, ZrB2 и твердый раствор
(ZrN—ZrB). В плавленых образцах в области
ZrB2—BN—В обнаруживается фаза ZrBi2. На
разрезе ZrN—BN наблюдается преимущест-
венно ZrB2, который образуется по реакции:
ZrN + 2BN = ZrB2 ф- 3/2N2,
что согласуется с прежними наблюдениями
Брауэра и Гаральдсена [417]. Исследование
твердых растворов ZrN—ZrB показывает, что
преимущественно происходит растворение
ZrB в ZrN.
Кисслинг и Лиу [320], обрабатывая амми-
аком при высоких температурах различные
бориды хрома, железа и вольфрама, обнару-
жили, что при этом происходит разложение
боридов и образуются нитриды металлов и
бора. Устойчивость исходных боридов сильно
для боридов железа и хрома увеличивается с
бора в фазе.
В системе Сг—В—N азотированию аммиаком подвергались все бо-
риды хрома, обнаруженные в свое время Кисслингом: б(~Сг2В),
c(Cr3B2), g(CrB), т](Сг3В4) и 0(СгВ2).
Фазы, полученные азотированием в течение 10 ч, приведены в
табл. 106 (фазы, содержащие хром, перечислены в порядке убывания
концентрации хрома, нитрид бора указан последним).
Таким образом, в порядке возрастающей устойчивости против дей-
ствия азота фазы системы Сг—В могут быть расположены в следующий
ряд: Сг2В (735° С), Сг3В2 (800° С), СгВ (1000° С), Сг3В4 (900—1000° С)
313
зависит от их состава и
повышением содержания
Таблица 106
Результаты азотирования боридов хрома [320]
Темпе- ратура, °C 6(~Сг2В) e(Cr3B2) 5(CrB) T](Cr3B4) 9(CrB3)
552 СгаВ Cr3B2 CrB СГ3В4 CrBe
735 Cr2B+CrN+BN Cr3B2 CrB cr3BJ —
800 Cr2B+CrN-hBN Cr3B2+CrN CrB cr3B4 CrB2
900 — CrN+Cr2N+BN CrB+CrN Cr3B4 —
954 CrN+Cr2N+BN — — — CrB2
1000 — CrN-]-Cr2N+BN CrB+CrN+BN — CrB2
1050 CrN+Cr2N+BN — CrN+CrN+BN Cr2N+Cr3B4 —
1100 1 — — Cr2N+BN CrB24-CrN
1180 CrgN+bJN Cr2N+BN Cr2N+BN Cr2N+BN Cr2N+BN
и СгВ2 (~ 1100° С). Стойкость к азоту возрастает с усложнением эле-
ментов структуры из атомов бора.
О системе Fe—В—N достаточно точных данных не получено, но
большей устойчивостью против действия азота, по-видимому, обладает
борид железа FeB (табл. 107), в структуре которого имеются борные
Таблица 107
Результаты азотирования боридов железа[320]
Темпе ратура, °C Fe2B FeB
352 Fe3B FeB
400 ^-фаза+BN Fe2B+g+BN(?)
448 e-f-BN —
505 e+y'+BN —
550 y'+e+BN y'+e+BN
602 a-Fe-f-y'-j- Y'+e+a+
+e+BN +Fe+BN
702 a-Fe+BN —
768 a-Fe-]-BN a-Fe+BN
цепочки, в то время как в струк-
туре Fe2B атомы бора изолиро-
ваны друг от друга.
В работе {902] исследована
растворимость азота в расплавах
железо—бор. Обнаружено обра-
зование при этом нитрида бора.
В системе W—В—N азоти-
рованию подвергались все три
борида — W2B, WB и W2Bs
(табл. 108). Заметного увеличе-
ния устойчивости боридов с воз-
растанием числа связей В—В в
этой системе не обнаружено, все
бориды начинают взаимодейст-
вовать с азотом при температуре
800—900° С с образованием нитридов W2N (|3-фаза Кисслинга), BN, а
также кубической нитридной у-фазы системы W—N, открытой в той же
работе Кисслингом (о системе W—В—N см .также [1109, 1110]).
3. СИСТЕМЫ МЕТАЛЛ — УГЛЕРОД — АЗОТ
Кальций — углерод — азот. Исследование взаимодействия между карби-
дом кальция СаС2 с азотом [240] при температурах 850—1100° С и в
присутствии различных ускоряющих добавок (CaCI2, CaF2, BaF2 и др.)
314
Таблица 108
Результаты азотирования боридов вольфрама [320]
Темпе- ратура, °C W2B WB W2B5
700 W2B WB WB2
750 W2B WB WB2
800 W2B WB+BN —
850 W2B+a-W+(Y+BN) a-W-t-BN-Hy+BN) a-W-(WB2)+(y+BN)
900 a-W+(W2B)+BN a-W+(WB)+BN a-W+(WB2)+BN
1000 a-W+(W2B)4-BN a-W-f-BN —
1100 a-W+BN a-W+BN a.W+BN
показало, что реакция взаимодействия линейна и имеет первый поря-
док по давлению. Это позволяет сделать вывод о том, что азотирование
карбида кальция является типичной топохимической реакцией. В резуль-
тате образуются смеси CaCN2 + C.
Алюминий — углерод — азот. В этой системе достоверно установле-
но существование тройных карбонитридов А16СзН2, AI7C3N3, AhCgN^, а
также предполагается существование соединений AI9C3N5 и А1юСзЫ6
[908], т. е. наличие гомологического ряда соединений общей формулы
(AlN)n • AI4C3. Характеристики основных свойств этих соединений в
сравнении со свойствами нитрида и карбида алюминия приведены, по
данным [908, 909], в табл. 109.
Таблица 109
Структура и плотность карбонитридов алюминия
Соединение Число атомов Простран- Периоды решетки, А Плотность, г/см?
ментар- ной ячейке ственная группа а С с! а расчет- ная пикно- метри- ческая
AIN 2 Р63тс 3,111 4,978 1,600 3,262 3,20
ai4c3 3 R3m 3,330 24,89 7,474 3,000 2,93
ai5c3n 2 Р63тс 3,281 21,67 6,005 3,039 2,99
Al^CgNg 3 R3rn 3,248 40,03 12,33 3,076 3,04
A'7C3N3 2 PG3mc 3,226 31,70 9,826 3,102 3,046
A18C3Ns 3 R3m 3,211 55,08 17,15 3,133 3,05
ai9c3n5* 2 Р63тс 3,197 41,69 13,04 9,141 3,06
A110C3N6* 3 R3m 3,186 70,00 21,97 3,157 3,07
•Предполагаемые соединения.
315
Карбонитридные системы переходных металлов. Большая часть
карбидов и нитридов переходных металлов IV и V групп образуют не-
прерывные ряды твердых растворов, за исключением сплавов с TaN,
имеющих гексагональную структуру и некоторых сплавов с VN и VC с
периодами решеток, существенно отличных от периодов решеток других
карбидов и нитридов. Данные по взаимной растворимости нитридов и
карбидов IV и V групп сведены, по
Таблица 110 Кифферу [333], в табл. ПО.
Взаимная растворимость карбидов и Результаты экспериментального ис-
нитридов переходных металлов IV и следования смешанных карбидони-
V групп * тридных систем приведены по дан-
Соединение TiC ZrC HfC VC NbC TaC
TiN A (A) (A) A A (A)
ZrN A A (A) Б Б (A)
HfN (A) (A) (A) (A) (A) (A)
VN A (B) (A) A A (A)
NbN A (A) (A) A A (A)
TaN (B) (B) (B) (A) (B) В
* А—полная взаимная растворимость; Б—
отсутствие или очень малая растворимость;
В—область твердого раствора на стороне ку-
бической фазы. В скобках даны вероятные ха-
рактеристики неисследованных систем.
ным [177, 244], на рис. 140—142. При
этом в системах TiN—VC, TiN—NbC,
ZrN—TiC, VN—VC, VN—NbC, VN—
—TiC, NbN—TiC, NbN—VC, NbN—
—NbC установлена непрерывная, a
в системах ZrN—VC и ZrN—NbC —
ограниченная и малая растворимость.
Наиболее подробные данные по
ряду систем этого типа приведены
в работе [978], где были исследованы
условия приготовления, структура и
физические свойства систем TiC—
—ZrN, TiC—TaN, ZrC—NbN, ZrC-
TaN, ZrN—NbC, NbN—TaC. Bee
они представляют системы с непрерывными рядами твердых растворов,
за исключением систем TiC—TaN и ZrC—TaN, в которых твердые раст-
воры образуются в области до содержаний TaN соответственно в 80
и 85 мол.%. В системах TiC—TaN, TiC—ZrN, ZrC—NbN изменение мо-
лярной магнитной восприимчивости однотипно — она плавно возрастает
от отрицательных значений для соответствующих карбидов до положи-
тельных значений для нитридов; изменение это во всех случаях нели-
нейное с пересечением кривой восприимчивости оси составов при
~20 мол.°/о нитридного компонента.
Исследование систем из мононитридов с монокарбидами было про-
ведено в [905]. Подтверждено образование непрерывного ряда твердых
растворов в системах TiC—ZrN, TiC—NbN, ZrC—ZrN, ZrC—NbN, ZrN—
NbC, NbC—NbN, NbN—TaC. Гексагональный TaN растворяется в TiC
до 70 мол.% и в ZrC до 80 мол.%. Исследование магнитной восприим-
чивости этих сплавов показало, что влияние углерода в нитриде в об-
щем значительно менее выражено, чем азота в карбиде.
Приведено аналогичное исследование ряда систем монокарбидов и
мононитридов с участием гафния и показано, что в системах HfN с TiN,
ZrN, TiC, ZrC, HfC, NbC, TaC, а также в системах HfC c TiN, ZrN.
316
NbN образуются непрерывные ряды твердых растворов [906]. Нитриды
VN и TaN значительно растворяются в HfN, также значительно и раст-
ворение TaN в HfC.
В работах [232, 255] исследована кинетика замещения азота в нит-
ридах титана, циркония и гафния углеродом.
Титан — углерод — азот. Карбид титана TiC образует с нитридом
титана TiN непрерывный ряд твердых растворов [175]. Результаты рент-
Период решетки, их
Рис. 140. Изменение периода решетки в системах TiN (ZrN)—TiC (VC; NbC).
Рис. il41. Изменение периода решетки в системах NbN—TiC (VC; NbC).
генографического исследования сплавов TiN—TiC, приготовленных на-
греванием смесей TiN+TiC в течение 4 ч при температуре 2425° С [177],
позволяют считать, что период решетки изменяется с составом практи-
чески по правилу Вегарда (см. рис. 140). Однофазность сплава, отвеча-
ющего эмпирической формуле TiioN8C2 и имеющего решетку типа NaCl
с периодом 4,243 кХ, была устновлена ранее. Кристаллы карбонитри-
дов титана, извлеченные из стали, содержащей 0,1—1 % С и до 6% Ti,
имели составы в широких пределах от почти чистого TiN до почти чи-
стого TiC [243]. В шлаках доменных печей при плавке высокоти-
танистых шихт присутствуют карбонитриды титана [191]. Вследствие
высокой температуры плавления они сильно понижают жидкотекучесть
шлаков.
Свойства сплавов TiN—TiC подробно исследованы в работах [126,
170, 830]. При переходе от TiN к TiC значение коэффициента тер mo-
в. д. с. возрастает, проходя в области содержания TiC порядка 25 мол.%
через небольшой относительный максимум. Ход удельного электросопро-
тивления также обнаруживает довольно резкий максимум электросо-
противления при 20—300/о TiC. Коэффициент Холла, напротив, возра-
стает монотонно. Термический коэффициент электросопротивления при
317
переходе 70—80 мол. % TiC обнаруживает максимум, а затем несколько
убывает к чистому TiC (рис. 143). Магнитная восприимчивость резко
уменьшается при растворении TiC в TiN (см. рис. 16 [830]). В работе
[798] проведено исследование температурной зависимости постоянной
Холла карбонитридов титана с низкими содержаниями азота
(TiCo.gesNo.oog; TiCo.geiNo.oas! TiCo,g48No,o47’, TiCo,930X0,075; TiCg,952X0,042) - По-
казано, что с повышением темпера-
туры в области до 300° К расхожде-
ние в значениях коэффициента Хол-
ла уменьшается до практически пол-
ного исчезновения этого различия
при ~ 300° К-
Рис. 142. Изменение периода решетки в системах VN—TiC (VC; NbC).
Рис. 143. Физические свойства сплавов в системе TiN—TiC:
1~ удельное электросопротивление; 2 — коэффициент Холла; 3 — коэффициент термо-э. д. с.;
4 — термический коэффициент электросопротивления.
В работе [232] исследована кинетика замещения азота углеродом
в системе Ti—N—С.
Цирконий (гафний) — углерод — азот. Исследования некоторых
электрофизических свойств сплавов системы ZrC—ZrN [789] показало
металлический характер их проводимости по крайней мере до темпера-
туры 300° К (с низким термическим коэффициентом электросопротивле-
ния), а также слабую температурную зависимость постоянной Холла в
этой области. Аналогичное исследование проведено в той же работе для
некоторых сплавов системы HfC—HfN.
По данным [978], молярная магнитная восприимчивость в системе
ZrC—ZrN монотонно и нелинейно возрастает от отрицательного значе-
318
ния для ZrC (—20,3-10 6)
(+22-10-6).
Ванадий—углерод—азот.
1100—1400° С обнаружены
[433] три различные фазы:
металлическая a-фаза, два
карбонитрида — гексаго-
нальная р-фаза (тип Ь'з)
V2(C, N) и кубическая 6-фа-
за (тип Bi) V (С, N). Две
области существования этих
фаз показаны на рис. 144
[1082].
Ниобий—углерод—азот.
В работах [920, 923] пост-
роена фазовая диаграмма
системы Nb—NbC—NbN,
изображенная на рис. 145
(изотермическое сечение
при 1250—1450° С). Как
следует из этих данных, в
системе имеются две облас-
ти карбонитридных фаз.
Область существования ку-
бической 6-фазы типа NaCI
имеет форму широкой поло-
сы между бинарными фаза-
ми того же типа. Карбонит-
ридная р-фаза, имеющая
гексагональную плотней-
шую упаковку металла, так-
же распространяется через
всю тройную область и свя-
зывает оба бинарных сое-
динения одинаковой струк-
туры — Nb2C и Nb2N. Изоб-
раженные на рис. 145 поло-
жения фазовых границ со-
ответствуют азотированию,
до положительного значения для ZrN
Рис. 144. Фазы тройной системы V—VC—VN
в области температур 1100—1400°С:
О — однофазные; ф — двухфазные.
проведенному при темпера- Рис. 145. Фазы в тройной системе Nb—NbC-
туре 1250—1300° С и давле- NbN при 1250—1450° С.
нии азота 1 ат. При азоти-
ровании под давлением азота 4,5—5 ат, температуре 1450° С и вы-
держке 1,5—2 ч, смеси ниобия и карбида ниобия либо гомогениза-
319
цией при указанных условиях смесей карбида и нитрида ниобия в ра-
боте [966] были получены твердые растворы NbC—NbN (карбонитриды)
в широкой области составов, а также показано, что составу NbCo.sNbNo,?
соответствует максимальная критическая температура перехода к сверх-
проводимости, равная 17,9° К- Этому же составу соответствует электрон-
ная концентрация около 4,85 валентных электронов на атом. Наличие
максимума связывается
с наличием максимума плот-
ности электронных энергети-
ческих состояний у поверх-
ности Ферми при указанной
электронной концентрации.
О сверхпроводимости
кубического карбонитрида
ниобия см. также в работе
[1018].
Молярная магнитная
восприимчивость сплавов в
системе NbC—NbN доволь-
но круто возрастает от
18 • 10~6 для NbC до
~30- 10 6 при содержании
NbN в 40 ат.%, после чего
с дальнейшим увеличением
Рис. 146. Фазы в тройной системе Та—TaC—TaN содержания нитрида нио-
при 1250—1450°С: бия (вплоть до 100 мол.%)
о — однофазные; ф — двухфазные; А — трехфазные. ПраКТИЧбСКИ Не ИЗМеНЯбТ -
ся [978].
Карбидонитрид ниобия (6-фаза) образуется в жаропрочном сплаве
на кобальтовой основе, содержащем 1% Nb и 0,20% С.
Тантал—углерод — азот. Авторами [526] обнаружен непрерывный
ряд твердых растворов Та2С—Ta2N. Растворимость TaN в TaNo.so-o.go
(6-фазе) порядка 5 ат.%, а растворимость ТаС в TaN очень мала, ра-
створимость TaN в ТаС не обнаружена. Тройные нитридные фазы также
не обнаружены.
В работе [923] исследована тройная система Та—ТаС—TaN. Най-
дены следующие фазы: металлическая a-фаза с кубической объемно-
центрированной структурой (растворяющая до 5 ат.% углерода и азо-
та), p-фаза с гексагональной плотной упаковкой (существующая в двух
кристаллических формах), g-фаза с неизвестной структурой и гексаго-
нальная ц-фаза, соответствующая ТаСо,б4 или TaNi;oo,‘ 6-фаза типа NaCl,
соответствующая структуре ТаС (рис. 146).
Хром — углерод — азот. В работе [434] показано, что при карбиди-
зации хромовых покрытий в среде, содержащей азот, на хроме образу-
320
ется слой нитрида хрома Cr2N, затем следует слой карбида хрома Сг7С3
и, наконец, внешний слой карбида Сг3С2. В связи с наличием гексаго-
нальной структуры у нитрида Cr2N и карбида Сг7С3, они ограниченно
растворяются друг в друге. Исследователи [435] спекали смеси порош-
ков Сг3С2 и Cr2N методом горячего прессования при 1500° С. При рент-
генографическом исследовании обнаружено существование карбонитри-
да хрома состава Сг2Мо,зСо,б, являющегося производным от нитрида хрп-
ма Cr2N (замена части атомов азота атомами углерода). Другой кар-
бонитрид хрома представляет производное карбида хрома Сг7С3 и имеет
состав, описываемый формулой Cr7C2;7N0,5. Еще один карбонитрид
Сг2Мо,бСо,3 является упорядоченным насыщенным твердым раствором
азота и углерода в хроме и представляет тот раствор, который образу-
ется при обработке хрома азотом и углеродом, обнаруженный в работе
[434] на границе слоев Cr2N и Сг7С3.
Более подробное изучение системы Сг—С—N было выполнено Этт-
майером [1021], который исследовал рентгенографически два изотерми-
ческих разреза при температуре 1100 и 1400° С, при давлениях от 1 до
30 am. При этом установлено, что карбиды хрома практически не раст-
воряют азот, а мононитрид хрома не растворяет углерода; в то же вре-
мя нитрид растворяет значительные количества карбида хрома. При вы-
соких давлениях азота обнаружено тройное соединение примерного со-
става Сго.вгСо 35N0 оз, имеющее ромбическую структуру (а—6,952; Ь =
= 9,255; с = 2,845 А).
Согласно [993], в системе хром — углерод — азот имеется соедине-
ние Сг3(С, N)2 (пространственная группа D^h структура которого род-
ственна структуре Сг3С2 (содержит октаэдрические и тригонально-
призматические структурные элементы).
Вновь проведенное исследование фаз, образующихся в системе Сг—
С—N [1056] показало наличие карбонитридных фаз Cr2CN (гексагональ-
ная типа e-Fe2N; (а = 4,83 до 4,85; с = 4,48 до 4,45 А; плотность 6,44 г) см?,
существует до 15% С в сплавах), Cr2CN (ромбическая типа g-Fe3N; а=
= 4,85; 6 = 5,60; с=4,44 А; плотность 4,44 г/см3) и Cr3CN2 (ромбическая,
структурный тип с пространственной группой D™ ; а=6,95; 6 = 9,25; с =
= 2,84 А; плотность 6,50 г/сж3, плавится инконгруэнтно при 1600° С, твер-
дость по Виккерсу 1600 кг/мм2).
Молибден — углерод — азот [427]. При взаимодействии молибдена
с СО и азотом при 450—850° С образуются только смеси [1- и б-нитрида
молибдена; образования карбонитридов не установлено (в отходящих
газах содержатся циан и дициан).
Вольфрам — углерод — азот. В работе [838] исследована реакцион-
ная диффузия в системе W—С—N2. Показано, что при этом образуются
только бинарные фазы, причем в присутствии азота процесс диффузии
азота в вольфрам замедляется.
121—272
321
Марганец — углерод — азот. При взаимодействии нитрида с карби-
дом марганца и металлическим марганцем при 1000° С (от 8 до 28 «)•
образуется карбонитрид марганца MruNi-xCx с х=0,102—0,798 и повы-
шением в этой области составов точки Кюри от 515 до 626°С [393].
Железо — углерод — азот. Система исследована Бриделлем и Ми-
шелем [432]. Сплавы готовили азотированием карбида железа FesC с
аммиаком при температуре 400—500° С в течение 1—70 ч, науглерожи-
ванием нитрида железа (у'-фазы) при 450° С смесью Н2 + СО, а также
одновременно науглероживанием и азотированием Ре20з при 450—
550° С.
Во всех случаях обнаружено образование тройного соединения
FexNyCz с гексагональной решеткой, периоды которой составляют: а =
2,749; с=4,400 кХ. Точный состав этой фазы остается неопределенным;
по-видимому, она имеет довольно широкую область гомогенности как по
содержанию азота, так и углерода. Джек [924] указывает, что эта об-
ласть гомогенности простирается от FesN-j до Ре8СзМ. Последний изо-
морфен Fe2N. В патенте [430] обсуждены методы получения карбонитри-
да железа нагреванием при 750° С смесей чистого железного порошка с
цианистыми солями щелочных металлов.
Схема основных реакций в системе Fe—С—N приведена в рабо-
те [936].
Уран (торий) — углерод — азот. Проведено рентгенографическое
исследование смесей порошков UC—UN и ThC—ThN, нагретых в среде
аргона при 1600° С в течение 4 ч [935]. В системе ThC—ThN образуется
непрерывный ряд твердых растворов с практически точным соответстви-
ем изменения периода решетки правилу Вегарда. В аналогичной системе
UC—UN наблюдаются две фазы, одна из которых представляет твердый,
раствор UC—UN.
Напротив, как отмечалось в [1041], при правильном выборе режи-
мов гомогенизации монокарбид и мононитрид урана образуют непре-
рывный ряд твердых растворов без появления каких-либо дополнитель-
ных фаз.
Авторы [1055] приводят дополнительные данные по системе U—С—
N и свойствам карбонитридов урана как перспективным материалам
дисперсного ядерного горючего.
Уран — плутоний — углерод —азот [1041]. В системе UC—PuN об-
разуется непрерывный ряд твердых растворов. Изменения параметров
решетки незначительно отличаются от правила Вегарда.
4. СИСТЕМЫ МЕТАЛЛ — КИСЛОРОД — АЗОТ
Литий—титан (германий, кремний) — кислород — азот [425]. При
сплавлении U3N с ТЮ2 и TiN (или Li2O) образуются оксинитриды об-
щей формулы: Li5TiN3-% Li2O (х=0,5; 0,9; 1,6; 2,0; 5,2), имеющие струк-
322
туру типа флюорита с атомами лития и титана в положениях фтора, и
атомами азота и кислорода в положениях кальция. Аналогично образу-
ются оксинитриды LigSiNs-LiaO и LigGeXs^LiaO. Легко растворимы в
разбавленных кислотах, схожи по свойствам с тройными нитридами
Li5XX3 (где X = Ti, Ge, Si).
Алюминий (галлий, германий) — кислород — азот. Оксинитрид
алюминия А18+жП1_ж О4_хХх образуется замещением атомами кисло-
Т 'Т
рода и содержит значительную долю дефектов □ [538]. Этот оксинитрид
обозначается как у-фаза системы А1—О—N. Кроме того, обнаружен еще
один оксинитрид: б-фаза, аналогичный по структуре 6-AI2O3 с периодами
решетки а = 7,943; с= 11,72 А. При уменьшении содержания азота ре-
шетка превращается в ромбическую с периодами а=7,943; /> = 7,962; с =
= 11,71 А.
В системе Ga—О—N оксинитридных фаз не обнаружено, а в систе-
ме Ge—О—N установлено существование оксинитрида состава Ge6O9N2
с большим числом дефектов решетки. Его структура аналогична струк-
туре рутила; периоды решетки а=4,395; с = 2,864 А.
Кальций — кислород — азот. Образцы карбонитрида кальция со
структурой СаО и составом, изменяющимся от СаО0,944X0,024 («=
= 4,8192 А, пикнометрическая плотность 3,283, рентгеновская плотность
3,294 г/смъ} до СаОо,932X0,029 (а=4,8216 А, пикнометрическая плотность
3,287, рентгеновская — 3,283 г/с.и3), получены в работе [967] нагревани-
ем СаО в парах Са и Х2, образующихся в результате диссоциации
Са3Х2 при 1100° С в герметичном железном контейнере. Эти продукты
рассматриваются как твердый раствор нитрида Са2Х (или, возможно,
CagX2) в СаО; он имеет высокое электросопротивление. Подобные же
фазы обнаружены в системах Sr(Ba)—О—X и Са—-S—X.
Титан — кислород — азот. Стон и Марголин [921] исследовали фа-
зовые диаграммы на образцах, полученных обработкой в дуговой печи
расплавленного иодидного титана с помощью ХО. Сплавы гомогенизи-
ровали, отжигали с последующей закалкой в воде. При температуре
1770° С наблюдается тройная перитектическая реакция, система Ti—X—
О характеризуется областями а-, р- и а + Р-фаз, расширяющимися с по-
вышением температуры. Установлено, что добавление к сплавам Ti—X
кислорода понижает действие азота как стабилизатора а-фазы.
В работе [77] определена зависимость периода решетки от состава
в системе TiO—TiX, которая сопоставлена с ходом кривой
плотности образцов, отожженных при 1700° С. В области от 0 до
60 мол. % TiX период решетки возрастает, а затем, достигнув значения
«tin, остается постоянным. Плотность при изменении состава от 0 до
30 мол.% TiX падает, при 30—60 мол.% TiX линейно возрастает, а
далее остается постоянной. Эти данные объяснены тем, что вплоть до
состава TiOo,7X0,3 происходит образование твердого раствора, а до
21*
328
TiOo,4No,6 заполняются дефектные места (в TiO, по данным Эрлиха
[135], около 16% дефектов). В области выше 60 мол.% TiN образуется
гетерогенная смесь TiOo,4No,6 и TiN.
Подробное исследование физических свойств сплавов этой системы
выполнено в работе [174] (табл. 111). На кривых концентрационной за-
висимости электросопротивления, коэффициента термо-э. д. с., коэффи-
циента Холла обнаруживаются мини-
Та блица 111 мумы, отвечающие приблизительно
Состав и результаты измерений элек- 20—30 мол. % TiO в сплавах.
трических свойств сплавов TiN—ТЮ Как следует из данных по измене-
Содержание, мол. % Удельное электро- сопротив- ление, МКОМ.-см Коэффи- циент Холла R-104, см3/к Коэффи- циент термо- Э.Д.С.. мкв град
TiN TiO
100 0 26 —0,67 —9,3
90 10 17,9 —0,4 —7,1
79,6 20,4 13,1 —0,17 —6,4
73,1 26,9 11,3 —0,48 —6,75
65,0 35,0 12,1 —0,82 —
62,7 37,3 12,7 — 1,36 —8,6
47,6 52,4 14,2 —1,64 —
46,7 53,3 — —1,70 —13,4
46,0 54,0 14,3 —2,02
41,8 58,2 27,0 —2,64 —
нию электросопротивления на участке
системы TiN—TiO, добавление TiO к
TiN вначале вызывает довольно резкое
уменьшение электросопротивления до
содержания TiO в 25—30 мол.%, после
чего сопротивление вновь медленно
возрастает до 50—55 мол.% TiO, ко-
гда это возрастание становится резким.
Такой ход изменения свойства обычно
характерен для случаев взаимного
влияния двух конкурирующих факто-
ров, которыми в данном случае, оче-
видно, являются атомный радиус и
ионизационный потенциал кислорода.
При малых добавках кислорода основ-
ную роль играет меньший его иониза-
ционный потенциал (13,57 эв) по сравнению с атомом азота (14,51 эв),
что облегчает переход электронов за счет атомов кислорода в зону про-
водимости и уменьшает суммарную отрицательную поляризацию энер-
гетических комплексов решетки, состоящих из атомов азота и кислоро-
да. Однако одновременно нарастает влияние геометрического фактора—
большего радиуса атомов кислорода по сравнению с атомами азота, что
вызывает постепенное уменьшение перекрытия энергетических зон, со-
путствующего увеличению периода решетки твердого раствора. Начиная
с 30 мол. % TiO этот фактор сказывается во все большей степени, что
вызывает постепенный рост сопротивления, и в области 50 мол. % TiO
происходит резкое повышение сопротивления за счет превалирования
атомов кислорода в решетке твердого раствора и своеобразного «обра-
щения фаз», когда закись титана становится растворителем, а не раство-
ренным компонентом. При этом важную роль играет, по-видимому, спо-
собность атома кислорода передавать на связь только два электрона по
сравнению с тремя электронами для атома азота.
Минимум э. д. с. для содержания ТЮ в 20 мол. % и его совпадение
с минимумом на кривой электросопротивление — состав хорошо согла-
324
суются с представлениями, заключающимися в том, что термо-э. д. с.
уменьшается по абсолютной величине с увеличением степени перекры-
тия (размытия) энергетических зон.
По данным концентрационного изменения коэффициента Холла до
20 мол.% ТЮ, можно предположить относительное возрастание дыроч-
ной доли в процессе электропровод-
ности, что согласуется с уменьшением
абсолютной величины отрицательного
значения термо-э. д. с. Напротив, пос-
ле 20 мол. % ТЮ дырочный вклад в
электропроводность уменьшается. Из
рис. 147 следует, что все исследован-
ные сплавы по достижении определен-
ной температуры изменяют знак
температурного коэффициента элек-
тросопротивления на отрицательный,
причем эта температура в общем воз-
растает с повышением содержания ни-
трида в твердом растворе с одновре-
менным уменьшением абсолютного
значения максимума электросопро-
тивления. Более ранний температур-
ный переход через максимум сплавов,
богатых ТЮ, связан, очевидно, с
ролью геометрического фактора —
большего размера ионного радиуса
кислорода, приводящего к снижению
размытия энергетических зон. Обра-
щает внимание различный характер
максимумов на кривых — вероятно,
острые максимумы отвечают преобла-
дающему влиянию одного из неме-
таллов (кислорода или азота,) а от-
Рис. 147. Температурно-концентраци-
онная зависимость электросопротив-
ления сплавов системы TIN—TiO:
/ — 79,6% TiN + 20,4% TiO; 2 — 73,1% T1N+
+26,9% ТЮ; 3 — 62,7% TiN+37,3% TiO; 4 —
65% TiN+35% TiO: 5 — 46% TiN+54% TiO.
носительно плоские — взаимному их
влиянию, близкому по окончательному результату одно к другому,
В работе [1066] определена энтальпия образования оксинитридов
титана TiNxOy с суммой индексов при азоте и кислороде, равной 1; 0,8;
0,7. Показано, что энтальпия образования оксинитридов аддитивно скла-
дывается из энтальпий образования TiN и TiO.
Цирконий — кислород — азот. В системе обнаружены оксинитриды
общего состава ZrO2-2XNx+y [440, 538].
Результаты исследования взаимодействия между ХгОг и аммиаком
[440] показали, что при 950° С наолюдается образование фазы белого
цвета (по обозначениям [440] — p-фазы) при более длительном взаимо-
325
действии — образование у-фазы желтого цвета, а при нагревании дву-
окиси циркония в токе аммиака при более высоких температурах, на-
пример при 1100° С, у-фаза разлагается с образованием (3-фазы и нит-
рида ZrN. Наконец, при очень высоких температурах (выше 2000° С)
р-фаза разлагается и образуется нитрид ZrN, а также р'-фаза, структу-
ра которой напоминает структуру p-фазы. Фазы р, р', у стабильны при
температурах их образования лишь в среде азота, а при нагревании в
инертной среде (например, аргона) они разлагаются с образованием
нитрида циркония и моноклинной ZrO2. При этом наибольшее количе-
ство нитрида образуется при разложении у-фазы (75%), меньшее р-фа-
зы (40%) и наименьшее — при разложении р'-фазы (20%). Рентгено-
графическое исследование показало, что все эти фазы являются оксинит-
ридами циркония различных составов. Так, у-фаза имеет состав Zr2ON2
и кристаллизуется в кубической объемноцентрированной решетке с пе-
риодом а= 10,135 А; р-фаза — ромбоэдрическая с а=6,246 А, а—99°35',
ее состав приблизительно отвечает формуле Zr7O8N4; р'-фаза — также
ромбоэдрическая и отвечает составу Zr70nN2.
Ванадий — кислород — азот [445]. При термическом разложении ме-
таванадата аммония в среде азота или аммиака, разбавленного водоро-
дом, при температурах 500—1000° С образуется оксинитрид ванадия. Со-
ставы оксинитридов согласуются с данными [67], где показано, что при
разложении метаванадата образуется соединение VxNyOz с 0,9<х< 1,14;
0,94 < г/<0,98; 0,02<z<0,04.
Ниобий — кислород — азот. В работе [443] исследована часть систе-
мы Nb—О—N с отношениями (N, O)Nb<Jl. При этом установлены две
тройные оксинитридные фазы: NbN~o,90~o,i со структурой NaCI (пе-
риод решетки а = 4,373—4,390 А), а также NbNo.gOo.s с тетраэдрически
деформированной решеткой типа NaCI. Период решетки изменяется от
4,370; с=4,295 А; с/а = 0,983 до а=4,386; с=4,325 А, с/а=0,986.
Других тройных фаз не обнаружено.
Попытка получить оксинитриды ниобия взаимодействием окислов
ниобия с нитридами ниобия или с другими азотсодержащими соедине-
ниями ниобия не дала положительных результатов: во всех случаях об-
разовывалась фаза б-NbN с очень малым содержанием кислорода [910].
Оксинитриды ниобия получены обработкой Nb2Os аммиаком при темпе-
ратуре 750—800° С. Все они имеют структуру либо кубического б-NbN,
либо гексагонального е-NbN. Оксинитриды характеризуются невысокой
устойчивостью, разлагаются при нагревании до 1000° С (табл. 112, [920]).
Оксинитриды ниобия имеют сильную дефектность в подрешетке ниобия.
В [1062, 1081] исследовали растворимость смесей азота и кислорода
в ниобии. При растворении возрастает электросопротивление ниобия (на
4,1 мком/см на каждый ат.% растворенного газа), уменьшается удель-
ная магнитная восприимчивость, возрастает микротвердость (с 64 кг/мм2
для чистого ниобия до 525 кг/мм2 при содержании азота и кислорода
326
4,5 ат.%), причем влияние
растворенного азота на твер-
дость ниобия больше, чем
Таблица 112
Условия получения и свойства оксинитридов
ниобия
кислорода. Тантал — кислород — азот [546]. В системе обнаружены четыре оксинитрида составов: Состав Темпе- ратура, °C Время реак- ции, ч Струк- турный тип Пикно- метри- ческая плот- ность, г/см8
TaNo,9oOo,io (структура сход- на с е-TaN, а = 10,34; с — ^^0,76^0,82^0,19 750 482 б-NbN 6,86
= 5,802 А; с/а=0,561), Nb0>7oN0i7600_24 800 12 б-NbN 6,48
TaN-о,750-0,25 (имеет структу- Nbfl,69^0,75^0,25 800 20 б-NbN 6,42
ру б-фазы системы Та—N, пе- риоды решетки: а=5,988; с= Nbfl, 82^0,81°0,19 800 625 e-NbN 7,27
= 2,879 А; с/а=0,481),
TaN-о,650~о,з5 (сверхструкту-
ра типа таковой для фазы б-TaN, периоды решетки: а— 10,34; с = 2,864 А,
с/а—0,277) и TaN~o,5oO~o,5o (гексагональная решетка с периодами:
а=5,939; с = 2,866А; с/а=0,483). Цвет оксинитридов изменяется от
черного для TaN~o,9oO~o,io до красноватого для TaN~o,5oO~o,so. Межатомные
расстояния сравнительно мало отличаются от межатомных расстояний
в TaN.
Буслаев [979] из нитрилхлорида тантала Ta2N3Cl получил оксинит-
рид тантала TaON, индивидуальность которого подтверждена исследо-
ванием ИК-спектра. Это вещество устойчиво по отношению к неоргани-
ческим кислотам и щелочам, разлагается смесью плавиковой и серной
кислот. В инертной среде по крайней мере до 900°С устойчив; предпо-
лагается, что при 840° С происходит его полиморфное превращение с
уплотнением кристаллической решетки и изменением цвета с красного
на зеленый. ТаОН в обеих модификациях диамагнитен.
Существование этого соединения подтверждено работами [980,986],
где оно получалось действием аммиака на пятиокись тантала при тем-
пературе около 800° С. Это соединение является промежуточным продук-
том азотирования пятиокиси тантала с получением в конце концов нит-
рида тантала Ta3Ns. По предварительным данным кристаллическая
структура TaON аналогична структуре бадделеита (моноклинной моди-
фикации 2гОг) с периодами решетки: а=5,145; 5 = 5,207; с = 5,311;
0 = 99,23°.
Хром — кислород — азот [444]. При действии тока аммиака и водо-
рода на гидроокись хрома или оксалатный комплекс хрома
(NH4)3 | Сг(СгО4) | ЗН2О при 600—1000° С образуется оксинитрид хро-
ма CrNxOy, где 0,8<х<0,9; 0,1 <у<0,2.
Вольфрам — кислород — азот [919]. Вольфрам образует оксинитри-
,ды при взаимодействии WO3 с аммиаком при 700° С, состав которых мо-
жет быть описан общей формулой WxNyOi_y с х~у«0,62.
327
Цирконий — тантал — кислород — азот [900]. Установлено сущест-
вование четверного нитрида состава ZrTaON, имеющего гексагональную
структуру с периодами: а = 3,645; с=3,881 А, с/а— 1,065. Расчетная плот-
ность 11,24 г!см\
Торий — кислород — азот [968]. Указывается на существование в
этой системе соединения Th2N2O, получаемого нагреванием спрессован-
ных смесей 3ThN + ThO2 под давлением азота 2 ат при температуре
1700° С. Это соединение кристаллизируется в ромбоэдрической решетке
с параметрами: а = 9,398; с=23,78 А (параметры соответственной гек-
сагональной решетки: а = 3,871; с=27,385А), плотность 10,55 г/см\
Уран — углерод — кислород — азот [949]. При восстановлении ПО2
углеродом в среде азота вследствие растворимости кислорода в карбо-
нитриде образуется оксикарбонитрид урана UNi_x_yCxOy, являющийся
более близким к химическому индивиду, чем к твердому раствору. Ра-
створимость О2 в карбонитриде зависит от концентрации углерода и
парциальных давлений азота и окиси углерода. Термодинамическим рас-
четом установлены границы области гомогенности оксикарбонитрида и
предельное содержание примеси кислорода; результаты расчета прове-
рены экспериментально. Например, при нагревании карбонитрида с х=
=0,43 в среде аргона при Р.Т =10-5 и Рс.о = 3,5-10 6 ат концентрация
примеси кислорода соответствует у=0,006. Об оксинитридах урана
см. [1083].
5. СИСТЕМЫ МЕТАЛЛ — ГАЛОГЕН — АЗОТ
Барий — галоген — азот [442]. Установлено существование тройных со-
единений в системах ВазМ2—ВаС12, ВазМ2—ВаВг2, ВазМ2—BaJ2, имею-
щих составы Ba2NCl, Ba2NBr, BaeNJg. Их структуры однозначно не ус-
тановлены; теплоты образования соответственно равны: 44,4; 46,3;
53,5 ккал/моль. Эти соединения плавятся конгруэнтно при соответствен-
ных температурах 965, 918 и 810° С.
Титан (цирконий) — иод (хлор, бром) — азот. В системе Ti—J—N
установлено [914] соединение TiJN, которое легко гидролизуется и тер-
мически неустойчиво (наименее устойчиво из всех рассматриваемых
соединений). Структура типа EOs, пространственная группа Z)^3(FeOCl)
подробнее описаны ниже.
В системе цирконий — иод — азот образуется соединение ZrNJ,
имеющее вид вытянутых оранжевого цвета листочков длиной около
3 • 10~3 см и шириной 0,5 • 10-3 см. Кристаллическая решетка ромбиче-
ская типа ЕО5 (тип FeOCl) с периодами: а=4,114; Ь = 3,724; с = 9,431 А,
число формульных единиц в ячейке 2, рентгеновская плотность.
5,34 г/см?. Устойчив в отсутствие влаги [424]. Структура типичная слой-
чатая с порядком слоев вдоль оси z : NZrJ/JZrNNZr/JJZrN. Расстояние
328
между обоими слоями J относительно большое (3,12 А), а между слоя-
ми N — малое (0,43 А). Принимается, что азот занимает промежуточное
положение между, с одной стороны, анионом слойчатой структуры, с
другой — составными частями группировки (ZrNNZr)2^.
Установлено также существование соединений ZrNCl, ZrNBr, TiNCl
и TiNBr [914], которые образуют по две модификации, причем все «-мо-
дификации имеют описанный выше структурный тип EOs, большая часть
этих соединений имеет малую химическую и термическую устойчивость,
кроме р-ZrNCl и 0-ZrNBr, отличающихся сравнительно высокой стой-
костью в химическом отношении и при нагревании. Исследование струк-
туры этих двух соединений показало [915], что они имеют гексагональ-
f ную слойчатую структуру (слои xZrNNZrx) с периодами: р-ZrNCl а=
=2,08; с=9,23; р-ZrNBr а=2,10; с = 9,75А.
II
J 6. СИСТЕМЫ МЕТАЛЛ — КРЕМНИЙ — АЗОТ
] Литий — кремний (германий, титан)—азот [425]. Нагреванием смесей
двойных нитридов при температуре 850—1300° С в металлической бом-
бе в условиях, исключающих доступ влаги и кислорода (в среде азота
или аммиака, ускоряющего реакцию образования тройных нитридов),
были получены нитриды LisSiNg, LisTiNs и LisGeNs. Все эти нитрида
имеют кубическую структуру с периодами решетки соответственно 9,44;.
9,70; 9,61 А (пространственная группа La3). Нитриды имеют светлую
окраску, легко гидролизуются, растворяются разбавленными кислотами,
термически устойчивы (LisSiNe плавится выше 1300° С).
Бериллий — кремний — азот [907]. При спекании в среде аммиака
смесей Be3N2 и SisN^ взятых в стехиометрическом соотношении, при
; 1750—1800° С в тиглях из нитрида бора образуется соединение состава
BeSiN2 напоминающее нитрид алюминия. Гексагональная структура с
периодами решетки: а—2,87; с=4,67А; с/а= 1,62, рентгеновская плот-
ность 3,24, пикнометрическая — 3,12 г!смъ.
Дальнейшее исследование условий получения и свойств соединения’
BeSiN2 было выполнено в [1035], где подтверждены режимы получения,
установленные Рабенау и Эккерлингом в [907], указанные выше пе-
риоды решетки. Показано, что образцы из BeSiN2 устойчивы при нагре-
вании на воздухе до 900° С; при 1000° С окисление усиливается, причем
скорость окисления убывает во времени. Конечным продуктом окисления
является силикат Be2SiC>4 (фенакит). Соединение термически устойчиво
при нагревании до 2000° С, при более высоких температурах разлагается
(в среде азотно-водородной смеси); предполагается также, что при
1950° С оно сублимирует без разложения.
Магний—кремний—азот [1043]. В этой системе образуется соедине-
ние MgSiN2, получающееся при совместном нагревании нитридов магния
329
и кремния при 1200° С в среде азота. Тройной нитрид негигроскопичен,
он нерастворим в щелочах на холоду, а также в разбавленных и кон-
центрированных минеральных кислотах; медленно реагирует лишь с
40%-ной HF и нагретыми растворами щелочей. В присутствии кислоро-
да при 1250° С полностью окисляется, превращаясь в метасиликат
магния.
Кальций — кремний — азот [938]. В этой системе обнаружено суще
ствование трех соединений: CaSiN2 (фаза I), Ca5Si2N6 (фаза II) и
Ca4SiN4 (фаза III), получающихся взаимодействием Ca3'N2 и Si3N4, либо
сложного нитрида (фазы II) с нитридами кальция или кремния
5Ca3N2 + 2Si3N4 = 3Ca6Si2Ne (при~600°С),
Ca5Si2N6 + Si3N4 = 5CaSiN2 (при~ 800°C),
Ca5Si2Ne + Ca3N2 = 2Ca4SiN4 (при ~ 800°C).
Фаза II может образовываться также взаимодействием фаз I и III
при температуре 1180° С в течение 24 ч
CaSiN2 -ф Ca4SiN4 = Ca5Si2N6,
т. е. является наиболее устойчивой.
В работе [983] построена ориентировочная фазовая диаграмма со-
стояния системы Ca3N2—Si3N4, показывающая области расположения
указанных трех фаз, а также твердого раствора Si3N4 в Ca3N2. Раство-
римость нитрида кремния в нитриде кальция при температуре 1050° С
составляет 1,12 мол.%.
Переходный металл — кремний — азот [314, 928, 929]. При взаимо-
действии образца, содержащего 62% Ti и 35% Si, с азотом образуется
TiN. Никаких других фаз в системе Ti—Si—N не обнаружено. В систе-
ме Zr—Si—N при нагревании циркония с Si3N4 в тигле из ZrO2 в при-
сутствии азота при температуре 1800° С появляется наряду с элементар-
ным кремнием гексагональная фаза неопределенного состава с перио-
дами: а=7,603; с —2,906 А, которая, очевидно, представляет тройное
соединение циркония с кремнием и азотом. При исследовании системы
Та—Si—N образцы готовились спеканием смесей Si3N4 с Ta2N или TaSi2.
Нагревание таких образцов до 1327°С показало, что TaSi2 неустойчив
в присутствии азота и взаимодействует с ним, образуя тройную фазу
Tas-j-Sh-^Nz. В равновесии с этой тройной фазой находится Ta2N и, ве-
роятно, TaN. По результатам рентгеновского анализа была построена
предварительная диаграмма системы (рис. 148). Система Nb—Si—N
построена аналогично; в ней образуется тройное соединение
Nbs-xSis-^Nz. Обе фазы имеют широкие области гомогенности.
В системе церий — кремний — азот тройных соединений не обнару-
жено.
330
Железо — кремнии — азот. Влияние малых количеств кремния на
растворимость азота в а- и у-железе может быть выражено уравнением
Ig5x/So=—Ах, где Sx и So— растворимость соответственно в сплаве
железо—кремний и в чистом железе, х — атомное содержание кремния.
Величина А уменьшается с повышением температуры, причем больше
для a-фазы, чем для у-фазы [944]. В
легировании сплавов системы Fe—Si
SisNi. Кремний понижает раствори-
мость азота как в а-, так и в у-же-
лезе.
7. Системы неметалл — неметалл —
азот.
Одной из наиболее полно изучен-
ных является система кремний—уг-
лерод—азот [873, 874, 876, 931,
1036], в которой, несмотря на мно-
гочисленные поиски, не удалось об-
наружить тройных нитридов: спла-
вы данной системы состоят из нит-
рида и карбида кремния.
В системе алюминий—бор—
работе [945] установлено, что при
азотом образуется нитрид кремния
Рис. 148. Фазы тройной системы Та—
Si—N.
азот предполагается существова-
ние тройного соединения [884, 932].
Исследование сплавов, полученных азотированием смесей A1N4-B и
BN+A1 (при температурах соответственно 1600—2000° С и 1200—1400° С
показало, что наряду с фазами, являющимися исходными компонентами
или продуктами их азотирования, появляется фаза высокой твердости
(порядка 5000 кг/мм2), а сами материалы — продукты азотирования —
имеют высокое электросопротивление.
В работе [898] описан огнеупорный материал системы бор — угле-
род— азот, состоящий из 80% нитрида бора и 20% карбида бора и по-
лучаемый горячим прессованием смесей этих компонентов. Далее, в ра-
боте [771] предложено получать аналогичные материалы азотированием
карбида бора, причем установлено, что они принципиально отличаются
по свойствам от механических смесей нитрида и карбида бора. В связи
с этим сделано предположение, что в этой системе образуется тройное
соединение, близкое по структуре к нитриду гексагонального бора, но в
отличие от него имеющее повышенный статистический вес «реконфигу-
раций атомов бора, углерода и азота, что придает ему повышенные свой-
ства (высокая стойкость в вакууме, диэлектрические свойства при высо-
ких температурах, химическая устойчивость). Предполагается, что при
высоких давлениях и повышенных температурах это соединение должно
331
приобретать алмазоподобную структуру, аналогично нитриду бора и
графиту.
В работе [974], посвященной исследованию условий приготовления
изделий из карбида бора, в частности, исследовано смазывающее дейст-
вие добавок нитрида бора к порошку карбида бора при горячем прессо-
вании. Показано, что наибольшая плотность получается при горячем
прессовании эквимолярных смесей (B4C:BN-=1: 1) и достигает 97%.
Можно предположить, что это связано с образованием при данном со-
ставе в процессе горячего прессования значительного количества указан-
ного выше тройного соединения.
Сера — фтор — азот [926]. В системе обнаружено соединение
S4F4N4 с температурой плавления 153° С (разлагается при плавлении),
плотностью 2,326 г/см\ диэлектрическая постоянная е = 2,257, дипольный
момент 111 = 0. Кристаллизуется в гексагональной системе, пространствен-
ная группа D4 , периоды решетки: а = 9,2; с = 4,3 А. Гидролизуется в
теплом разбавленном растворе NaOH. Известно также газообразное со-
единение SF^N2 с температурой плавления 108° С, температурой кипе-
ния — 11° С, плотностью 1,57 г[см?.
Сера — кислород — азот [927]. При реакции S4N4 со смесью SO2 и
SOCI2 образуется твердая фаза желтого цвета S3N2O2.
ГЛАВА X
НИТРИДНЫЕ
ПОКРЫТИЯ
Многие нитриды, одновременно обладающие вы-
сокой твердостью и износостойкостью, находят
широкое применение для создания защитных по-
крытий на металлах, сплавах, неметаллических
веществах, графите.
Особенно широко развито азотирование ста-
лей, как одно из основных направлений химико-
термической обработки [741, 742]. Азотированные
стали обладают высокой поверхностной твердо-
стью, повышенным пределом выносливости, кор-
розионной и кавитационной стойкостью, а также
высокой износостойкостью. Еще большее распро-
странение получило одновременное насыщение
поверхности стали углеродом и азотом из газо-
вой среды (нитроцементация) и из расплавлен-
ных цианистых ванн (цианирование).
Этими повышенными свойствами азотиро-
ванные и азотированно-цементированные стали
обладают благодаря наличию в поверхностном
слое азота в форме нитридов или карбонитридов.
Важно также повышение стойкости сталей при
сварке в азоте против межкристаллитной корро-
зии [761], повышение их усталостной и коррози-
онно-усталостной прочности [955].
Вопросу азотирования, цианирования, нит-
роцементирования сталей посвящена обширная
литература, обзор которой дан в указанных
выше и других монографиях по химико-терми-
ческой обработке сталей (например, [743, 744,
1032]).
Азотированию железа с соответствующим
исследованием механизма и кинетики образова-
ния диффузионного слоя и фазовых превращений
посвящена большая серия работ, выполненных
под общим руководством В. Г. Пермякова, на-
пример, важная работа [973].
В настоящей работе остановимся только на
вопросах создания покрытий нитридного типа на
тугоплавких металлах и сплавах [745, 747].
Возможно образование при азотировании
простых или сложных твердых растворов азота
либо химических соединений — нитридов, обычно
и обеспечивающих наиболее высокие свойства
покрытий.
333
Азотирование титана азотом и аммиаком приводит при использо-
вании аммиака к большим глубинам диффузионных слоев и к большей
их твердости, однако одновременно происходящая диффузия водорода
вызывает повышенную хрупкость [748], очевидно, обусловленную увели-
чением жесткости связей при внедрении водорода в пустоты решетки
нитрида и образовании при этом гидридно-нитридных фаз, о которых
было упомянуто в главе II.
Хотя водород может быть в значительной степени удален вакуум-
ной обработкой, все же метод азотирования с помощью аммиака оказы-
вается технологически и экономически нецелесообразным.
Толщина и свойства слоев на титане после азотирования в среде
азота приведены, по данным [748], в табл. 113.
Таблица ИЗ
Влияние параметров диффузионной обработки титана азотом на свойства
азотированных образцов
Темпе- ратура, °C Время, ч Глуби- на слоя, мк Твер- дость на по- верхно- сти Hv, кг/мм2 Твер- дость на глу- бине 100 мк кг/мм2 Предел пропор- цио- наль- ности, кг/мм2 Поедел прочно- сти при растя- жении, кг/мм2 Удли- нение, % Удар- ная вяз- кость. кгм/см2
8 25 1132 250 29,0 37,8 36,5 22,8
16 42 1206 360 26,0 36,4 45 27,6
/ UU 24 33 1332 285 23,5 37,3 45 26,0
48 35 1246 445 27,0 36,6 48,5 25,4
8 13 1003 205 26,0 36,7 43,0 25,1
ОЛП 16 40 1333 325 26,4 36,4 45,0 21,1
oUU 24 25 1520 280 24,5 39,5 45,0 26,8
48 44 1426 360 26,0 37,4 39,5 23,5
8 53 1500 375 36,0 41,7 36,5 17,1
900 16 61 1500 470 22,5 46,7 25,0 н,о
24 76 1500 500 39,0 47,7 30,0 10,6
48 31 1500 390 43,5 48,1 43,5 9,0
При азотировании аммиака в той же области температур удлинение
падает до 2%, а ударная вязкость до 0,3—0,5 см2. Как видно из приве-
денных данных, глубина слоя не подчиняется параболическому закону
роста от времени азотирования. Аналогичные результаты получены
А. Н. Минкевичем с сотрудниками [749], которые азотировали чистый и
легированный хромом и вольфрамом титан в аммиаке при температу-
рах 850—1050° С. Обнаружено, что при малых расходах аммиака слой
в некоторых случаях глубже, чем при больших, причем с повышением
температуры увеличивается оптимальный расход аммиака, необходимый
334
для получения слоя максимальной глубины. Глубина слоя изменялась
не по кривой, близкой к параболе, а по кривой с максимумом. Рентге-
нографическое исследование слоя показало, что при температуре азоти-
рования 1050° С слой представляет твердый раствор нитрида и гидрида
титана (и TiO — при использовании недостаточно очищенного от кисло-
рода аммиака). При температуре 950° С обнаружен только твердый ра-
створ азота и водорода в титане. Азотирование титана в аммиаке в де-
сятки раз превышает его износостойкость, однако снижает прочность и
пластичность, особенно если процесс ведется при высоких температурах
и длительных выдержках.
Легирование титана вольфрамом приводит к резкому уменьшению
глубины азотированного слоя. Сплав титана с 5% Сг после азотирова-
ния обнаруживает повышение износостойкости в 40—170 раз (в зависи-
мости от режима насыщения) по сравнению с неазотированным про-
мышленным титаном. Поскольку азотирование в аммиаке неизбежно
приводит к значительному охрупчиванию изделий, были проведены ра-
боты по азотированию титана в смеси аргона с азотом [750—752]. При
разрежении диффузия азота в титан ускоряется, что относится за счет
более медленного образования при этом поверхностного тонкого слоя из
TiN, задерживающего дальнейшее азотирование [752]. Азотирование в
среде с дозированным количеством азота связано с трудностью введе-
ния малого количества азота, который следует добавить к аргону. Опти-
мальное содержание азота в аргоне при 800° С составляет 1% [752].
Азотирование титана в смеси аргона и азота, прошедших предваритель-
ную тщательную очистку, описано в работе [751]. Рентгенографическое
исследование азотированных образцов, выполненное в этой работе, по-
казывает, что поверхностный слой при содержании азота в смеси с ар-
гоном более 0,5% состоит из TiN и TiNx, где х<Л.
Состоящая из нитридов корочка препятствует диффузии азота
вглубь металла, и тем самым уменьшается толщина общего насыщенно-
го азотом поверхностного слоя. Уменьшение концентрации азота ДО'
0,5% и менее приводит к образованию на поверхности твердого раство-
ра азота в Ti с включениями TiN и постепенному переходу к чистому
твердому раствору азота в Ti. При парциальном давлении азота 0,005—
0,04 ат и при азотировании в течение 20 ч при температуре 850° С мож-
но получить поверхностный слой без образования нитрида титана. При
больших временах выдержки, температурах и парциальных давлениях
азота в смеси с аргоном на титане всегда образуется тонкий слой нит-
рида титана. Легирование алюминием ускоряет диффузию азота в ти-
тан, увеличивая глубину диффузионного слоя, и марганец, олово и осо-
бенно кремний действуют в противоположном направлении (табл. 114).
Процесс азотирования титана в струе чистого азота при температу-
ре 700—1050° С длительностью до 10 ч изучался в [753]. Обнаружено,
что заметное приращение веса начиналось при 800° С и следовало пара-
335
Таблица 114
Влияние легирования титана на глубину диффузии азота, при-
вес образцов и распределение микротвердости при 850° С
Легирую- щий эле- мент Содержа- ние легирующе- го элемента % Наличие нитридного слоя (по микротвер- дости и цве ту) Микротвер- Дость, кг/мм2, на глубине 0,01 мм при нагруз- ке 100 г Глуби- на слоя, мк Привес образ- цов, мг]см2 поверх- ности Вы- держка, ч
1,74 Нет 824 50 0,92 4
3,00 » 1000 56 0,95 4
Алюминий 3,75 » 1000 50 0,85 4
5,00 » 1100 48 0,73 4
5,00 Есть 1200 120 — 24
1,35 Нет 824 27 0,72 4
Марганец 2,45 3,76 » » 1000 1100 28 0,71 0,70 4 4
6,54 Есть 1200 25 0,70 4
6,54 — 1500 68 — 24
0,66 Нет 1100 20 0,52 4
Кремний 1,62 » 1100 18 0,46 4
1,62 Есть 1200 65 — 24
0,86 Нет 824 28 0,53 4
Олово 1,58 » 824 24 0,74 4
1,58 Есть 1100 83 — 24
Титан йо- — Нет 824 25 0,82 4
дидный — Есть 1200 90 1,05 24
•болическому закону до 1000° С. Электронографически установлено, что
после азотирования при 800° С и выше в поверхностном слое образуется
нитрид TiN. Повышение твердости мало заметно при 800° С, однако при
температуре более 900° С толщина и твердость слоя прогрессивно возра-
стали со временем азотирования.
Мнения о пользе азотирования при малых парциальных давлениях
азота и азотировании чистым азотом расходятся, что вызвано различ-
ными задачами азотирования. В некоторых случаях необходимо обеспе-
чить большую глубину азотированного слоя, не доводя его до состояния
нитрида, в других — сохранить свойства основной массы титановой де-
тали, повысив лишь ее поверхностную износо-, коррозиоустойчивость и
твердость. Последние случаи, как правило, преобладают [754—756].
Авторы [754] азотировали титан при 850° С в течение 16—80 ч с
целью повышения износостойкости, коррозионной устойчивости и меха-
нических свойств. Азотированный титан удовлетворительно работал без
смазки в паре с чугунным литьем, твердыми* хромистыми покрытиями и
неазотированным титаном. При работе со смазкой получались хорошие
336
I
результаты в паре с бронзой, углеродистой и низколегированной сталью,
бакелитом, хромированными поверхностями. Высокую коррозионную
стойкость показал азотированный титан в нагретых соляной, серной,
фосфорной, азотной, плавиковой и других кислотах. Азотирование не
влияет существенно на механические свойства титана.
В работе [755] образцы технического титана и жаропрочного тита-
нового сплава с 6% А1 и 4% V подвергали азотированию в очищенном
азоте при 980° С в течение 4—168 ч. На техническом титане азотирован-
ный слой был значительно толще, чем на сплаве титана с алюминием и
ванадием, а твердость его достигала 1650 Hv. Отмечено, что значения
ударной вязкости азотированного и неазотированного титана почти не
отличались, в то время как у сплава они были значительно ниже. Это
связывалось с наличием в азотированном сплаве азотированных зерен,
расположенных под углом 45° к поверхности и распространенных на
большую глубину. Являясь, по-видимому, концентратами напряжений,
они вызывали значительное падение ударной вязкости. Изменение тол-
щины азотированного слоя на техническом титане происходило со вре-
менем по параболе, толщина составляла после 168 ч азотирования
0,2 мм.
Исследование процесса азотирования титана и его сплавов в чистом
азоте было проведено также в работе [756]. Оптимальным режимом азо-
\ тирования было насыщение при температуре 950° С в течение 24—30 ч
\ и скорости подачи азота 0,12—0,15 л)мин. При более низких температу-
i рах диффузия азота проходит медленно, при более высоких сильно воз-
! растает хрупкость слоя и самого металла. При указанных оптимальных
! условиях азотирования на образцах кованого титана ВТ1 получен слой
' глубиной 80 мк с поверхностной твердостью Яу = 780—850 кг/.w.w2. Азо-
j тированный слой состоял из нескольких зон. На поверхности образовы-
валась золотистая пленка TiN толщиной в десятые доли микрона, над
ней располагалась зона толщиной 8—10 мк с содержанием азота 10—
12%, которая состояла, по-видимому, из нитрида титана в нижней об-
ласти его гомогенности. За нитридной зоной следовала зона самая
большая по глубине (60—80 мк) с постепенно убывающей микротвер-
достью (от 1300 до 700 кг/мм2) с содержанием от 4 до 1,5% N. Эта зона
представляет a-Ti с увеличенным параметром решетки. Исследование
механических свойств азотированных образцов, прошедших различную
дополнительную обработку, показало (табл. 115), что отжиг в ва-
[ кууме при 800° С уменьшает хрупкость слоя и повышает характеристи-
j ки пластичности на 10—15%. В работе [756] принимается, что главной
I причиной уменьшения (на 25—30%) пластичности титана после азоти-
i рования является рост зерна в результате длительного высокотемпера-
турного нагрева. Другой причиной снижения пластичности является
глубокое проникновение азота в титан (с увеличением глубины снятого
слоя пластичность титана повышается).
22—272
337
Таблица 115
Влияние азотирования и дополнительной обработки после азо-
тирования на механические свойства образцов из сплава ВТ1
Обработка
кг/ммг
ak-
\кгм/сна
Исходный сплав, отожженный в
вакууме при 800°С, 2 ч
Азотирование, 950°С, 30 ч
(азотированный слой не снят)
То же (слой снят на глубину
0,5 леи)
То же (слой снят на глубину 1 мм)
То же (слой снят на глубину
1,5 мм)
Азотирование при 950°С, 3 ч +
-|-отжиг в вакууме 800°С, 2 ч,
давление 3-10—4 леи рт.ст.
(азотированный слой не снят)
45,1 33,1
55,5 15,1
51,0 20,1
48,2 23,2
54,2 19,4
65,2
39,2
53,9
54,2
14,8
5,3
7,6
9.7
12,0
35,0 10,6
Исследование влияния легирования титана различными элементами
показало, что 2—2,5% А1, до 2% Si и 1,5% Мп вызывают увеличение
глубины азотированного слоя. При большем содержании этих элементов
глубина слоя уменьшается. Хром и железо снижают глубину азотиро-
ванного слоя независимо от их содержания в сплаве. В этой же работе
опробовано азотирование некотоорых тройных сплавов титана, установ-
лено, что сплавы марок ВТ2, ВТЗ, ВТ5 и ВТ6 азотируются хуже, чем
сплав ВТ1.
По данным [1064], при азотировании титана, легированного А1, Си и
Sn, в азотированном слое обнаруживаются два нитрида — внешний слой
состоит из мелкозернистого нитрида TiN, а следующий — из низшего
нитрида состава Ti2il2_2,36N, образующего крупнозернистую структуру.
Образование таких двух слоев обнаружено, в частности, при азотирова-
нии сплава ВТ5 в течение 30 ч при 960° С в токе очищенного азота.
Преимущества азотирования титана с применением нагрева изде-
лий токами высокой частоты показаны в [757]. При азотировании ВТ4
и ВТ6 таким путем (при давлении азота в камере 40—50 мм рт. ст.),
температуре 1100° С за 20 мин получали слой толщиной 30 мк с микро-
твердостью 2000 кг!мм2. Испытания на износ в паре с закаленной
сталью У12А в режиме сухого трения практически не показали измене-
ния (износа) азотированных образцов. Жаростойкость азотированных
образцов на воздухе при 1000° С оказалась в четыре раза выше жаро-
прочности неазотированных.
Преимущества индукционногоо нагрева токами ВЧ при азотирова-
нии заключаются в ускорении образования диффузионного слоя и воз-
338
можности повышения температуры только поверхностного слоя, не за-
трагивая основной массы металла, оптимальные структуры и свойства
которой заданы предварительной обработкой [760].
Азотирование титана, по-видимому, наиболее существенно упроч-
няет поверхностные свойства изделий в техническом отношении, имея
ряд преимуществ перед карбидизацией различными способами [758].
В работе [237] описан метод нанесения нитрида титана, в частности,
на поверхность молибдена газотранспортным методом, основанным на
разложении нитрида титана парами НС1
TiN -Ь 4НС1 = TiCl4(ras) ф 0,5 N2,
с последующим переносом хлорида титана на молибден и его превра-
щением в нитрид за счет реакции с азото-водородной смесью и осажде-
нием на молибдене. Тонкие слои нитрида прочно удерживаются на мо-
либдене, толстые — растрескиваются.
Наряду с методом газотранспортных реакций для азотирования на-
чинают использовать различные электрометоды, из которых следует упо-
мянуть ионное азотирование, заключающееся в возбуждении тлеющего
разряда в разреженном пространстве, содержащем атомы и ионы азота
между катодом (деталью) и анодом. Тогда к поверхности детали на-
правляется поток ионов азота, которые осуществляют быстрое азотиро-
вание, кроме того, разогревая поверхность в результате ударов о ка-
тод [759].
Существенно интенсифицируется азотирование и при использовании
ультразвука [762].
Исследованию реакционной диффузии в системах металл — слож-
ный газ посвящены работы школы В. И. Архарова [763—765]. В. И. Ко-
нев [763] исследовал структуру и свойства карбонитридных покрытий на
хроме. Образцы после хромирования обрабатывали в смеси бензина с
азотом при температуре 700—1100° С. Рентгенографический анализ и
металлографическое исследование покрытия показали, что его внешний
слой состоит из Сг3С2, средний — из Сг7С3 и внутренний, прилегающий
к металлической основе,— из Cr2N. Если хромовый слой имел текстуро-
ванное строение, то и слой, состоящий из нитрида хрома, также обладал
текстурой, подчиняясь принципу ориентационного структурно-размерно-
го соответствия. Как следует из рис. 149, реакционная диффузия в си-
стемах Сг—N, Сг—С, Сг—С—N подчиняется параболическому закону,
причем диффузионная подвижность атомов азота гораздо выше, чем уг-
лерода [1111].
Испытания на кислото- и износостойкость показали, что карбони-
трированные покрытия на хроме не уступают по свойствам карбидизи-
рованным. Одинаковые результаты дало и испытание этих покрытий на
стойкость против окисления, что объясняется тем, что в обоих случаях
верхний слой покрытия состоит из карбидов хрома. Карбонитрирован-
22*
339
ный слой образуется в результате диффузии атомов азота и углерода
через соответствующие слои карбидов и нитрида хрома, заметной
встречной диффузии атомов хрома не наблюдалось. Фронт реакции азо-
та с хромом находится на межфазовой границе Сг—Cr2N, а углерода
с хромом — на границе Cr2N—Сг7С3. Четкая граница между Cr2N и
Сг7С3 указывает на ограниченную растворимость этих фаз.
Рис. 149. Зависимость привеса образцов хрома при
1100° С от продолжительности процессов карбидизации
(/), карбонитрирования (2) и азотирования (3).
Рис. 150. Зависимость привеса образцов Мо в среде ам-
миака от времени при температурах:
1 — 700; 2 — 750; 3 — 820; 4 - • 880; 5 — 900; 6 — 940; 7 - 1000; 8 —
1050; 9 — 1120° С.
Исследовались азотирование, карбидизация и карбонитрирование
молибдена [764]. Насыщение азотом производилось в среде аммиака и
молекулярного азота при температурах соответственно 700—1150° и
800—1200° С, причем во втором случае взаимодействие молибдена с азо-
том при атмосферном давлении не было обнаружено. В среде аммиака
насыщение молибдена азотом подчиняется параболическому закону во
всем температурном интервале (рис. 150). До температуры 940° С на
поверхности молибдена образуются два слоя: внешний MoN и внутрен-
340
ний Mo2N. При температуре выше 940° С устойчива только фаза Mo2N„
при температуре выше 1150° С исчезает и она. Совместное насыщение
поверхности молибдена азотом и углеродом производилось так же, как
хрома в [763], только азот был заменен аммиаком. Образование диффу-
зионного слоя, состоящего из карбонитрида молибдена с решеткой Мо2С,
наблюдалось при температуре 1000—1200° С и подчинялось параболиче-
скому закону (рис. 151). Исследование кинетики образования диффу-
зионных слоев в системах молибден — азот, молибден — азот — углерод
и молибден — углерод показывает, что рост их идет за счет преимуще-
ственной диффузии азота и углерода через продукты реакции к метал-
лу, причем скорость азотирования молибдена значительно выше скоро-
сти его карбидизации, а скорость совместного насыщения азотом и уг-
леродом имеет промежуточное значение. Результаты рентгеновского и
металлографического исследования поверхностных диффузионных слоев
в указанных системах приведены в табл. 116.
Таблица 116
Результаты азотирования, карбидизации и карбонитрирования мо-
либдена
Система Область тем- ператур, °C Число сло- ев, выявля- емых ме- таллогра- фически Результаты фазово- го рентгеновского ана- лиза Наличие тек- стуры
Мо—N 700—900 2 Внешний слой—MoN Внутренний — Mo2N Нет
940—1150 1 Mo2N »
1150—1250 — Мо »
Мо—С 800—1200 1 Фаза с решеткой Мо2С » (текстура при 1200°С)
Мо—С—N 900—1200 1 Фаза с решеткой Мо2С Нет
В работах [956, 957] сообщается об удвоении твердости сплава мо-
либдена с 1% Ti при обработке в азоте при температурах 1100—1500° С
в течение 27 ч. Это увеличение твердости относится за счет образования
нитрида титана, дисперсионно распределенного в молибденовой матри-
це. Указывается также [956], что при азотировании сплава молибдена с
0,5% Ti твердость возрастает от 290 до 660 Hv, а при азотировании
сплава молибдена с 0,5% Ti и 0,08% Zr твердость возрастает до 835—
858 Hv.
Исследование азотирования молибдена (молибденового сплава
ЦМ-2А 0,003% С; 0,09% Ti; 0,14% Zr) проводили в [1051] различными
341
средами (аммиак, диссоциированный аммиак, смесь азота и 1 % аммиа-
ка смесь аргона и 1% аммиака, разреженный до 10~* мм рт. ст. аммиак)
при 700—1600° С в течение от 0,25 до 10 ч. В результате установлено,
что при азотировании сначала происходит растворение азота в молиб-
дене, затем образуется наиболее бедная азотом 0-фаза (Mo3N), затем
более богатые азотом фазы Mo2N и MoN (до температуры 1000° С) либо
один нитрид MoN (при температуре азотирования выше 1000° С), что
Время, ч
Рис. 151. Зависимость привеса образцов Мо в среде паров бензина и аммиака от вре-
мени при температурах:
/ — 1000; 2 — 1050; 3 — 1100; 4—1150; 5—1200’С.
Рис. 152. Зависимость квадрата привеса образцов вольфрама от времени:
А — система W—С; О — система W—С—N.
противоречит данным [764], по которым более устойчива фаза M02N, а
при температурах выше 1150° С не образуется ни одна из этих фаз.
Полученные в [1051] азотированные слои на молибдене обладают
высокой твердостью (порядка 2000 кг/льи2) и высокой хрупкостью (балл
3—4 по шкале микрохрупкости), причем объемные изменения, происхо-
дящие при азотировании, вызывают высокие внутренние напряжения,
снятие которых приводит к появлению микротрещин. Жаропрочность
(измеряемая сопротивлением пластической деформации при испытании
на ползучесть) повышается при поверхностном азотировании молибдена
на 60—90% при 1000—1400° С. Установлено, что это повышение дости-
гается при оптимальном режиме азотирования листового молибдена
(толщиной 1—1,5 мм) при 900—1000° С в течение 1 ч.
Раздельная и совместная диффузии углерода и азота в вольфрам
изучены в [765] (табл. 117).
Из сравнения хода кинетических кривых (рис. 152) можно заклю-
чить, что присутствие азота замедляет диффузию углерода в вольфрам
342
Таблица 117
Результаты азотирования, карбидизации, карбонитрирования воль-
фрама
Система Область темпе- ратур, °C Число сло- ев, выяв- ляемых ме- таллогра- фически Результаты фазового рент- геновского анализа слоев Наличие текстуры
W—N 1000—1100 2 Внешний—WN, внутренний—W2N Нет
W—с 1000—1200 2 Внутренний—W2C, внешний—WC
W—C-N 1000—1200 2 Внешний—с решеткой WC, внутренний—с решеткой W2C »
(для хрома и молибдена наблюдалось обратное явление). Поскольку и
при карбидизации и при карбонитрировании парциальное давление уг-
лерода в реакционном пространстве оставалось одинаковым, авторы ра-
боты [765] предполагают, что образующиеся в диффузионной зоне кар-
биды вольфрама содержат растворенный азот, который затрудняет диф-
фузию атомов углерода через карбидные фазы дальше вглубь металла.
Наблюдающийся во всех системах параболический закон роста толщи-
ны поверхностного слоя свидетельствует о преимущественной диффузии
азота и углерода через кристаллические решетки образующихся фаз.
Авторами [766] описан метод нанесения нитридных покрытий на
титан, цирконий и ниобий путем взаимодействия галоидных соединений
этих металлов с аммиаком. При этом образуются комплексные соедине-
ния различных составов, разлагающиеся при высокой температуре, с
выделением активных атомов металлов и азота, диффундирующих в на-
сыщаемую поверхность. Для нанесения покрытия из нитрида титана
реакцию взаимодействия TiCl4 с NH3 рекомендуется осуществлять при
температурах 900—1200° С. Во избежание выделения частиц нитрида в
свободном состоянии TiCU и NH3 вводят в реакционную зону раздель-
но, причем хлорид титана в струе водорода.
Теории нанесения защитных покрытий из нитридов через газовую
фазу посвящена работа [1065].
Как сообщалось в [271], при нанесении нитридного покрытия на
магний (его сплав с 0,6% циркония и другие под давлением 30 ат)
образуются относительно стойкие слои, предохраняющие магний от суб-
лимации при повышенных температурах.
Большой интерес представляют неметаллические нитридные покры-
тия на металлах и неметаллах, в частности жаро- и коррозионностойкие
покрытия из A1N [767], покрытия из пиронитрида бора и пирокарбонит-
343
рида бора [746], а также покрытия из металлоподобных нитридов на не-
металлах, например, из нитрида титана на графите [768].
Согласно [842], покрытия из нитрида алюминия ifa графите наноси-
лись путем плазменного напыления на графит алюминия с последую-
щим азотированием. Сцепление слоя алюминия с графитом осуществля-
ется только в результате механического закрепления при последующем
азотировании при 1300° С в течение 0,5 ч, слой алюминия полностью
превращается в нитрид алюминия и слой плотно прилегает к графито-
вой подложке, прочно на ней удерживаясь. Особенно качественно про-
ходит азотирование при засыпке покрытой алюминием графитовой де-
тали алюминиевой пудрой, которая взаимодействует с окисными плен-
ками на алюминиевом слое с удалением моноокиси алюминия AI2O3 +
+ А1-ЗА1О.
ГЛАВА XI
ОБЛАСТИ
ПРИМЕНЕНИЯ
НИТРИДОВ
Изменение в широких пределах характера хими-
ческой связи и физико-химических свойств нитри-
дов обеспечивает им весьма разнообразные об-
ласти применения. Однако наиболее развито в
настоящее время использование огнеупорных и
электроизоляционных свойств некоторых нитри-
дов — бора, кремния, алюминия, а также их
сложных соединений и различных материалов на
их основе.
Нитрид бора. Высокие электроизоляционные
свойства, устойчивость по отношению к тепловым
ударам, химическая стойкость, прочность при
высоких температурах, хорошая теплопровод-
ность, устойчивость в вакууме, большое сечение
захвата нейтронов в сочетании с хорошей обраба-
тываемостью, а также высокими огнеупорными
свойствами обеспечивают использование нитрида
бора и изделий из него в различных отраслях тех-
ники. Следует отметить особо использование ни-
трида бора для изготовления защитной изоляци-
онной соломки для термопар, печной фурнитуры,
труб муфельных печей, креплений транзисторов,
цоколей электронных ламп, тиглей, трубок и кла-
панов, устройств ядерной техники [628]. Большое
значение имеет применение нитрида бора для
изготовления деталей металлопроводов, электро-
магнитных насосов для перекачки расплавлен-
ных металлов [896], футеровки каналов МГД
генераторов, некоторых устройств ракетной тех-
ники. Перспективно использование люминесцент-
ных свойств нитрида бора, легированного различ-
ными примесями. Реальное применение нашел
нитрид бора в качестве термоизолятора в высоко-
частотных индукционных печах, так как он не
прогревается индукционными токами даже при
высоких температурах [769]. Из него изготовля-
ются огнеупорные обмазки форм и тиглей, туго-
плавких держателей электродов при автоматиче-
ской сварке, предполагалось его применение для
изготовления сливных носков миксеров и конвер-
торов, однако была обнаружена не очень высо-
кая стойкость изделий из нитрида бора по отно-
шению к расплавленной стали при температуре
выше 1590° С [783]. Перспективно также исполь-
345
зование нитрида бора в качестве высокотемпературного смазочного ма-
териала [770], для изготовления легконагруженных подшипников сколь-
жения. Введение его в медные электроды для электроэрозионной обра-
ботки металлов резко повышает их относительную стойкость. Сравни-
тельно малая величина диэлектрических потерь во всем интервале
вплоть до ультравысоких частот позволяет использовать нитрид бора в
составе диэлектриков [773].
В химическом машиностроении может быть использована высокая
стойкость нитрида бора против действия многих химических реагентов,
стойкость к абразивному износу—для изготовления сопел различных
распыляющих аппаратов.
Пиролитический нитрид бора используется в ракетной технике [713]
в качестве экранирующего материала.
Материал, состоящий из 80% BN и 20% В4С [898], стоек к резким и
частым теплосменам, химически инертен, прочен и тверд. Из него
изготовляют плазменные горелки, облицовку для сопел эксгаустеров,
ракетных камер сгорания. Добавлением порообразователей получа-
ют пористый материал для фильтров в качестве носителей ката-
лизаторов и т. п.
Высокими свойствами обладает также карбонитрид бора (B^N^Cz)
[771], имеющий высокую устойчивость против испарения в вакууме, близ-
кую к таковой карбидов и боридов тугоплавких металлов, стойкость
против действия многих расплавленных металлов, солей и шлаков, вы-
сокие электроизоляционные свойства, плазмоустойчивость.
Карбонитрид бора имеет удельное электросопротивление при 20° С
порядка 1014—1015 ом • см, снижающееся при 1400° С до 5-Ю5 ом • см,
а при 2000° С — до 2 104 ом • см; он обладает высокой теплопровод-
ностью, хорошо обрабатывается механически [771]. Характеризуется вы-
сокой устойчивостью против действия расплавленной буры при 1000° С.
Тигли из карбонитрида бора устойчивы в продолжение 10—15 ч в угле-
родистом ферромарганце, металлическом марганце, силикомарганце при
температурах до 2000° С [678], в расплавах железо-титанового концент-
рата и продуктов его восстановления при 1600°С [772], литиевой плазме
при 2000—2500° С [677], медных штейнах и шлаках, никелевом штейне
при 1250—1300° С [541].
Кубический нитрид бора — боразон, обладающий высокой твер-
достью, используется для изготовления различных абразивных инстру-
ментов этого же типа и назначения, что абразивные инструменты, при-
готовленные из алмаза [774], а в легированном примесями виде
может найти применение в качестве полупроводника [664], могущего
эксплуатироваться при повышенных температурах без сильной потери
свойств.
Перспективно использование люминофорных свойств нитрида бора,
легированного различными примесями [653], особенно углеродом.
346
Не исключена возможность использования нитрида бора в качест-
ве наполнителя резин, особенно микропористой резины для промыш-
ленного и бытового использования.
Нитрид алюминия. Известные огнеупорные свойства нитрида алю-
миния — стойкость его в расплавленных алюминии, олове, галлии, бор-
ном ангидриде до температур в 1300—2000° С [586] — обеспечивают перс-
пективы его использования в качестве материала футеровок ванн, элек-
тролизеров, резервуаров для высокотемпературных галлиевых термо-
метров. Электроизоляционные свойства нитрида алюминия позволяют
применять его для изготовления соломки для защиты металлических
термопар, а стойкость в кислотах — в химическом машиностроении. Осо-
бенно перспективно изготовление покрытий из нитрида алюминия на
сталях, графите, различных металлических и неметаллических материа-
лах. В работе [866] описан огнеупорный материал, состоящий из нитри-
да алюминия с 10—20% углерода и плавиковым шпатом, который может
использоваться до 2300° С для футеровки стеклоплавильных ванн, ванн
электрохимических производств.
Особенно интересна и важна роль нитрида алюминия в сталях и же-
лезе [563, 572, 867, 870—872]. Установлено, что одной из причин хрупко-
го разрушения литой стали являются выделения нитрида алюминия по
границам аустенитного зерна [867],что в присутствии нитрида алюминия
существенно снижаются магнитные характеристики трансформаторной
стали [563].
Однако в последнее время показано, что нитрид алюминия, который
принято обычно считать вредным компонентом сталей, обеспечивает в
определенных условиях и содержаниях (до 0,08% азота в форме дис-
персного нитрида) одновременно повышение прочности, пластичности и
улучшение свариваемости стали [871]. Нитрид алюминия вводится в
сталь в форме тонкодисперсного порошка (~1 мк), при затвердевании
слитка распределяется равномерно и предотвращает рост зерна при на-
греве. Размер зерен при этом весьма мал — от 8000 до 31 000 зерен на
1 мм2; при содержании 0,07% нитрида алюминия рост зерен не наблю-
дается даже при длительной выдержке при 1100° С. Для малоуглеро-
дистых сталей лучшее сочетание прочности свойств и ударной вязкости
при низких температурах дает введение 0,06—0,08% A1N, а для мало-
легированных— 0,04—0,06%. Околошовная зона сварного соединения,
возникающая при сварке и являющаяся причиной охрупчивания, в этой
стали почти не возникает.
Показано, что введение нитрида алюминия в алюминий и его ме-
таллокерамические сплавы резко повышает модуль упругости и тепло-
устойчивость. При этом образуются сплавы типа САП, в которых окись
алюминия заменена нитридом алюминия [868]. Известные перспективы
имеет использование полупроводниковых свойств легированного нитри-
да алюминия [614].
347
Высокими электро-, термоизоляционными и огнеупорными свойст-
вами обладают материалы, состоящие из нитридов алюминия и бора, а
также из нитрида алюминия с карбидом кремния. Например, в [884]
описаны свойства сплавов системы В—А1—N, которые обладают высо-
ким электросопротивлением, более медленно снижающимся с темпера-
турой, чем у нитридов бора и алюминия в отдельности [1112, 1113].
Нитрид кремния. Тугоплавкость, высокая стойкость против тепло-
вых ударов, хорошая жаростойкость до 1200—1300° С обусловливают
применение нитрида кремния в составе различных жаропрочных матери-
алов, особенно важными в практическом отношении свойствами обла-
дают материалы SiC—SiglSU, в которых нитрид кремния является связ-
кой, соединяющей между собой зерна карборунда [873—877]. По дан-
ным [873], пористость таких материалов 15—20%, предел прочности при
сжатии при 20° С около 20—24 кг/мм2, при разрыве при 1000° С— 1,9—
2 кг/мм2, теплопроводность при 850° С равна 8,3 ккал/м • ч • град, коэф-
фициент термического расширения (20—1200° С) 3,8 • 10-6 град~1, тер-
мостойкость от 1600° С — 7 водных теплосмен. Такие же материалы, но
изготовленные по технологии [876], имеют <Тразр = 2—4 кг!мм2, <тИзг=4—
7 кг/мм2, сгсж = 22—38 (20° С); 22—40 (600° С); 27—40 (800° С) кг!мм2.
Такие материалы могут использоваться для изготовления холодильни-
ков скрубберов, работающих с горячими газами, насадок сопел для раз-
брызгивания химически активных жидкостей, мешалок, стойких против
коррозии, а также против одновременного действия твердых составля-
ющих суспензий и пульп, для облицовки химической аппаратуры, камер
электросварки под флюсом. В последнем случае футеровка камер выдер-
живает 2 ч под флюсом без каких-либо измерений. Особенно перспек-
тивно использование карбида кремния на связке из нитрида кремния
для изготовления деталей насосов для перекачки расплавленного алю-
миния [877], изготовления футеровки алюминиевых электролизеров [878].
Такие материалы обнаруживают высокую стойкость против действия
медных и никелевых штейнов и шлаков при 1250—1300° С, слабо вза-
имодействуют с расплавленным базальтом (1400° С), расплавами хло-
ридов (700—1100° С) [541]. Материал SiC—Si3N4 устойчив против дей-
ствия цинка и его паров [897].
В работе [879] показана перспективность использования сложных
материалов SiC + Si3N4 + MoSi2 в качестве излучателей в инфракрасной
области спектра.
Чехлы из Si3N4 и материалов SiC-|-Si3N4 используются для защиты
металлических термопар при измерении температуры фторидных рас-
плавов в алюминиевых электролизерах (стойкость 100 ч при 940—
970° С) [880].
Высокое электросопротивление Si3iN4 делает перспективным его ис-
пользование в составе объемных сопротивлений, термисторов и в составе
нагревательных электродов электропечей сопротивления [881].
348
Рекомендовано использование Si3N4 в составе жаропрочных спла-
вов [882, 883].
Высокая твердость нитрида кремния позволяет использовать его в
•составе абразивных материалов. Так, по патенту [946], порошки кремния
или феррокремния с добавкой до 50% SiC или без нее прессуются на
связке из глюкозы в виде абразивных кругов, которые затем обжигают-
ся в среде азота или аммиака при 1100—1600° С в течение 24—108 ч, и
получают абразивные изделия, состоящие из SisN-t: SiC = 2 : 1. Можно
также использовать пасты из подобных материалов. Твердость абразива
близка к твердости SiC, по шлифующим свойствам (при шлифовании
чугуна, сплавов кобальта с хромом и вольфрамом, а также рубина) он
превосходит карборунд и электрокорунд.
Большой практический интерес представляют материалы, состоящие
из нитрида кремния с окислами магния и алюминия [1040]. Материалы
MgO—Si3N4 [1114] обладают пределом прочности на изгиб 12—
14 кг/мм2, на сжатие 30—35 кг/мм2; привес при окислении при 1200° С
3—6 мг!см2, электросопротивление 1010—1013 ом- см, термостойкость при
1500—20° С — 20—30 теплосмен; они устойчивы против действия рас-
плавов буры и цинка. Материалы ;412О3—Si3N4 [1115] имеют пре-
дел прочности на изгиб 6—7 кг/мм2, на сжатие 16—18 кг/мм2, привес
при окислении при 1200° С 2—5 жг/сж2, электросопротивление 109—
1011 ом • см, термостойкость 30—35 водяных теплосмен (1500—20° С).
В [1058, 1116, 1117] исследованы физико-технические свойства еще
одной группы важных сплавов на основе нитридов бора и кремния,
которые представляют механические смеси BN и SisN4 и принад-
лежат к классу неоднородных диэлектриков. Удельное электросопро-
тивление материала, содержащего 80% BN и 20% Si3N4 при 20° С со-
ставляет 4,5 • 1013 ом • см, снижаясь при 1400° до 8 • 104 ом • см; макси-
мальным удельным электросопротивлением (1014 ом-см при 20° С; 2,5X
ХЮ5 ом • см при 1400° С) обладает материал, содержащий 40% BN и
60% Si3N4. Величина диэлектрической проницаемости имеет тот же по-
рядок, что и для исходных компонентов (Si3N4 е—4,4; BN — е = 4,25—
3,2). Такого рода материалы являются хорошими высокотемпературны-
ми изоляторами.
Нитриды галлия и индия являются перспективными полупроводни-
ками типа AnIBv [614]; особенно сложные нитридные сплавы с дру-
гими изоморфными соединениями этого типа.
Нитриды переходных металлов. Основными свойствами нитридов
переходных металлов, обеспечивающими их применение в технике, яв-
ляются высокие температуры плавления, довольно хорошая химиче-
ская стойкость, а также некоторые специфические свойства, как-то: спо-
собность переходить в сверхпроводящее состояние, относительно высо-
кая электропроводность металлического типа либо полупроводниковые
свойства. Известное значение имеют и высокие твердость и износостой-
349
кость некоторых нитридов. Основной областью использования нитридов
в настоящее время является, по-видимому, их применение в качестве-
огнеупорных материалов, обладающих высокими температурами плав-
ления, удовлетворительной стойкостью против действия расплавленных
металлов и против окисления (особенно нитридов титана, циркония,
тантала, урана и тория [885]). Так, в [43] разработан способ производ-
ства тиглей из нитрида титана, в [886] — способ изготовления тиглей из
нитридов титана и циркония. Сплавы MgO—TiN—Ni [197], в которых
TiN является своеобразной связкой между MgO и никелем (TiN обра-
зует непрерывный ряд твердых растворов с MgO и растворяется в Ni),
хорошо сопротивляются тепловым ударам; их прочность при 1090° С на
30% выше, чем при 20° С. Иногда используются сплавы, в которых Ni
заменен на NiO, восстанавливаемый в процессе спекания [887].
Лодочки, изготовленные из сплавов системы Ti—В—N успешно
используются в качестве контейнеров для испарения алюминия при ва-
куумной металлизации. Такие лодочки весьма стойки против корроди-
рующего действия расплавленного алюминия и могут использоваться в
течение нескольких дней, что дает возможность организовать непрерыв-
ный процесс металлизации стали, пластмасс, бумаги [1048].
Известны антикоррозионные, износостойкие и твердые покрытия из
нитридов [45], свойства которых описаны в главе X.
В качестве примера можно указать на высокие эксплуатационные
свойства фильер для текстильной промышленности (протяжки синтети-
ческих волокон), производимых лионской фирмой Ж- Босюз (Франция).
Эти фильеры получаются карбонированием молибдена; поверхность
имеет твердость 1400 Hv, а по эксплуатационным свойствам они пре-
восходят стальные хромированные фильеры [956].
По данным [1052], хорошим материалом для изготовления тиглей
для испарения алюминия является композиция из 50% TiB2 и 50% BN,
которая достаточно электропроводка, чтобы ее можно было нагревать
токами высокой частоты. Преимущество такого нагрева заключается в
возможности испарения тонких слоев (пленок) расплавленного алюми-
ния, который стремится мигрировать из тигля во время работы. Для
предотвращения миграции применяются тигли, толщина стенок которых
меньше, чем толщина скин-слоя в высокочастотном поле, которая в
свою очередь зависит от удельного электросопротивления материала
тигля и частоты. Так как сопротивление композиции TiB2 + BN при
1100—1200° С (температуре, необходимой для испарения алюминия)
равно 2 • 10-3 ом • см, то при частоте 200 кгц толщина скин-слоя поряд-
ка 5 мм, что и определяет выбор толщины стенки тигля с учетом также
таких факторов, как предотвращение турбулентности расплава в резуль-
тате его экранирования от высокочастотного поля.
Нитрид титана успешно используют для припыливания поверхности
изложниц с целью получения чистой поверхности отливок.
350
Высокая твердость некоторых нитридов позволяет их применять для
правки шлифовальных кругов обкаткой кружками, например, из литого
нитрида титана [181].
Нитриды титана и циркония использовались в качестве проводяще-
го материала ториевых катодов [888], в производстве зажигателей к вы-
прямителям применялась композиция, содержащая 25% TiN + 75% ВеО
[888]. Сложный нитрид титана и хрома рекомендуется в качестве высо-
коомного сопротивления [889]. Нитрид ниобия используется в качестве
детектора для радиоаппаратуры, работающей при 170° С [890, 891], в
конструкции трубок для передачи изображений [892], а также для сверх-
проводящих болометров [893, 894, 954]. Во всех этих случаях использу-
ется способность нитрида ниобия переходить в сверхпроводящее состо-
яние. Добавлением к NbN карбида ниобия удается повысить точку пе-
ревода в сверхпроводящее состояние до 17—18° К [895], что позволяет
расширить использование сверхпроводящих свойств нитрида ниобия.
Нитрид хрома CrN является перспективным полупроводником, в
частности, для использования его в термоэлектрических генераторах
[327], для этих же целей перспективен нитрид скандия [466].
Некоторые нитриды, в частности нитриды хрома, являются перспек-
тивными катализаторами [899].
По данным патента [1053], нитриды переходных металлов в форме
покрытий на самих переходных металлах являются перспективными ма-
териалами для изготовления электродов солионов — электрохимических
приборов, предназначенных для измерений весьма малых акустических
давлений, преобразования энергии последних в электрическую и осно-
ванных на транспорте малых количеств вещества в ионных растворах
[1054]. Электроды солионов из переходных металлов (с нитридными по-
крытиями) обладают хорошей электропроводностью и устойчивостью к
химическим и электрохимическим воздействиям, т. е. практически инерт-
ны в растворе электролита солиона.
Образование нитридов редкоземельных металлов (например, лан-
тана, церия) в сталях, содержащих азот, является одним из средств
улучшения таких свойств конструкционной стали, как хладостойкость и
склонность к старению [934].
Мононитрид урана является перспективным ядерным топливом, так
как в отличие от карбида и двуокиси урана не взаимодействует с боль-
шинством элементов, которые могут быть использованы в контакте с
ним, имеет более высокую теплопроводность при температурах выше
800° С, чем монокарбид урана, менее склонен к возгоранию и гидроли-
зу, чем карбид, имеет высокое содержание урана, а по радиационной
стойкости не уступает карбиду и двуокиси урана. Его можно использо-
вать в тепловыделяющих элементах дисперсионного типа, в реакторах
различных типов [976].
ЛИТЕРАТУРА
1. Славинский М. П.— Физико-химические свой-
ства элементов. Металлургиздат, М., 1952.
2. Ария С. М„ Прокофьева Е. А.— В цц.: Сбор-
ник статей по общей химии, 1. Изд-во АН СССР, М., 1953.
3. Б о л ь ш а к о в К. А., Федоров П. И., Сте-
пина Л. А. — Изв. вузов. Цветная металлургия,
1959, 4, 52.
4. Dafert R., Miklauz R.— Mh. Chemie, 1910, 31,
981.
5. Ш а м p а й Ф. И. Литий и его сплавы. Изд-во АН
СССР, М., 1952.
6. Б у с е в А.— Уч. зап. Ленингр. гос. пед. ин-та, 1940,
29 303
7. Deslandres Н.— С. г., 1895, 121, 886.
8. Ouvrard L.— С. г., 1892, 114, 120.
9. В о л А. Е. Строение и свойства двойных металли-
ческих систем, 1. Физматгиз, М., 1959.
10. Frankenburger W.—Z. Elektrochem., 1926,
32, 48.
11. Клинаев В. М.— В кн.: Редкие щелочные эле-
менты. Изд-во СО АН СССР, Новосибирск, 1960.
112. Беляев А. И., Фирсанова Л. А., Поме-
ранцев И. Н.— В кн.: Научные труды Моск, ин-та цвет-
ных металлов и золота им. М. И. Калинина. Металлург-
издат, М., 1955, 25, 172.
13. Остр оу ш ко Ю. И., Бучихин П. И. и др.
Литий, его химия и технология. Атомиздат, М., 1960.
14. М а с d u р u 1.— Ann. Chim., 1959, 2, 527.
15. Алабышев А. Ф. и др. Натрий и калий. Гос-
химиздат, М., 1959.
16. Gmelins Handbuch der anorg. Chemie, 21, Nat-
rium, 1928.
17. Browne A., Houlehan A.— J. Amer. Chem.
Soc., 1911, 33, 1749.
18. Franklin E.— J. Amer. Chem. Soc., 1924, 46,
2142.
19. Ho th W., Pyl G.— Z. angew. Chem., 1929, 42,
890.
20. Matter O.— Герм. пат. 302561, 1920.
21. Davy H.— Phyl. Trans., 1809, 99, 450.
22. D г e у f u s R., Levy P.— Proc. Roy. Soc., 1958
A246, 233.
23. Перельман Ф. M. Рубидий и цезий. Изд-во
АН СССР, М„ 1960.
24. Руководство по препаративной неорганической хи-
мии. Под ред. Г. Брауэра. ИЛ, М., 1956.
25. Suhrmann R., Clusius К.— Z. anorg. Chem.,
1926, 152, 57.
26. Sieverts A., Krumbhaar W.—Z. phys. Chem.,
1910, 74, 280.
27. R 6 n t g e n P., Moller F.— Z. Metallwirtschaft,
1934, 13, 97.
28. Laffitte P., Grandadam G.— C. r., 1935,
200, 1039.
29. Хансен M., А н дер ко К. Структуры двойных
сплавов. Металлургиздат, М., 1962.
352
30. W i 1 s d о r f H.— Acta cryst., 1948, 1, 115.
31. Juza R., Hahn H.— Z. anorg. Chem., 1938, 239, 282.
32. Juz a R.—Z. anorg. Chem., 1941, 248, 118.
33. J u z a R., Rabenau A.— Z. anorg. Chem., 1956, 285, 212.
34. D e n i g e s G.—- C. r., 1942, 214, 651.
35. Suzuki S.— J. Chem. Soc. Japan, Pure Chem. Sect., 1953, 74, 269.
36. Б о к и й Г. Б. Введение в кристаллохимию. Изд-во МГУ, М., 1954.
37. Straumanis М., Cirulis А.— Z. anorg. Chem., 1943, 251, 315.
38. К е 11 е у К., Department U. S. of the Interior Bureau of Mines. Bull., 476,
Washington, 1949.
39. H a h n H., G i 1 b e r t E.— Z. anorg. Chem., 1949, 258, 77.
40. Wohler L., Martin F/—Z. Ges. Schiess. Sprengstoffwesen, 1918, 12, 1.
41. Gulbransen E., Andrew K.— J. Electrochem. Soc., 1950, 97, 383.
42. W h i t e D., Burke I. The Metal Beryllum. Amer. Soc. Metals, Cleveland,
Ohio, 1955.
43. C h i о 11 i P.— J. Amer. Cer. Soc., 1952, 35, 123.
44. Kaufmann A., Gordon P., Lillie D.— Trans. ASM, 1950, 42, 785.
45. Техника высоких температур. Под ред. И. Кемпбелла. ИЛ, М., 1959.
46. Stackelberg М., Paulus R.— Z. phys. Chem., 1933, В22, 305.
47. К у б а ш е в с к и й О., Эванс Э. Термохимия в металлургии. ИЛ, М., 1954.
48. Deville Н. St., Caron Н.— Ann. Chim. Phys., 1863, 67 (3), 349.
49. BriglebF., Gluther A.— Lib. Ann., 1862, 123, 236.
50. Hartung H. Beitrage zur Kenntnis der Metallnitride. Hannover, 1912.
51. M e r z W.— Герм. пат. 243940, 1891.
52. Kaiser К.—Герм. пат. 181657, 1904; 346112, 1920; 336437, 1920.
53. Smits А.— Rec. Trav. Chim. Bays., 1896, 15, 135.
54. Paschkowetsky A.— J. Prakt. Chem., 1893, 47 (2), 89.
55. Neumann B., Kroger C., Habler H.— Z. anorg. Chem., 1931, 196, 61.
56. Kirchner W.— Chem. Ztg., 1901, 25, 395.
57. E i d m a n n W., Moser L.— Ber., 1901, 34, 393.
58. Eidmann W.— J. Prakt. Chem., 1899, 59 (2), 1.
59. Szarwasy E — Pharm. Post., 1896, 29, 53; Ber., 1897, 30, 806.
60. M e h n e r H.— Герм. пат. 88999, 1895.
61. Neuberger W.— Z. angew. Chem., 1905, 18, 1761.
62. Vournasos A.— Bull. Soc. Chim., 1911, 9 (4), 506; 1917, 21 (4), 282.
63. Жуков И. И.— Изв. физико-хим. анализа, 1926, 3, 1.
64. Neumann В., Kroger С., Kun Н.— Z. anorg. Chem., 1932, 207, 138.
65. Р о г t е г J.— Пат. США 2487472, 1949.
66. Davis L.— Пат. США 2488054, 1949.
67. Эпельбаум В. А., Ормонт Б. Ф.— ЖФХ, 1947, 32, 319.
68. Mitchell D.— Ind. Eng. Chem., 1949, 41, 2027.
69. H e к p а с о в Б. В. Курс общей химии. Госхимиздат, М., 1952.
70. Whitehouse N.— J. Soc. Chem. Ind., 1907, 26, 728.
71. Mellor J. A Comprehensive treatise on inorganic and theoretical chemistry,
VIII, 1931, 104—106.
72. Fichte F„ Schollig C.— Helv. Chem. Acta, 1920, 3, 298.
73. Агеев H. В., За мотор ин M. И., Шойхет Д. М.— Металлургия, 1936,
4, 27.
74. Д э ш м а н С. Научные основы вакуумной техники. ИЛ, М., 1950.
75. Staphitanonda Р., Margrave L.— J. Phys. Chem., 1956, 60, 1628.
76. Кубашевский О., Гопкинс Б. Окисление металлов и сплавов. ИЛ,
М., 1955.
77. A n t г о р о f f A., Falk F.— Z. anorg. Chem., 1930, 187, 405.
78. W 6 h 1 e г L., M a r t i n F.— Z. angew. Chem., 1917, 30, 33.
79. Hartmann H., Frohlich H.— Z. anorg. Chem., 1934, 218, 190.
80. Paulus R.— Z. phys. Chem., 1933, B22, 305.
23—272
353
81. А р и я С. М., П р о к о ф ь е в а Е. А., М а т в е е в а И. И. ЖОХ, 1955, 2S 634.
82. Erlich Р., Hein Н.— Z. Elektrochem., 1953, 57, 710.
83. Ария С. М., Ерофеева М. С., М о ч а л о в Г. П— ЖОХ, 1957, 27, 1740.
84. Термические константы неорганических веществ. Изд-во АН СССР, М., 1948.
85. А р и я С. М., Прокофьева Е. А.— ЖОХ, 1955, 25, 849.
86. Rem y-G 1 u n e t e P — Ann. Chim., 1933, 19, 289.
87. F i g о u r H.— Bull. Soc. Chim. France, 1947, 1096.
88. Giinter P., Porger J., Rosband P — Z. Phys. Chem., 1929, 6, 459.
89. Sieverts A., KrumbhaarW.— Ber., 1910, 43, 894.
90. H о f f m a n n W., M a a t s c h J.— Z. Metallkunde, 1956, 47, 89.
91. I w a s e K.— Sci. Rep. Tohoku Univ., 1926, 15, 531.
92. Juz a R., Neuber A., Hahn H.—Z. anorg. Chem., 1938, 239, 273.
93. HiittigG.—Z. anorg. Chem., 1920, 114, 161.
94. H e r z e r H. Beitrage zur Kenntnis der Nitride. Hannover, 1927.
95. White A., Kirschbaum L.— J. Amer. Chem. Soc., 1906, 28, 1344.
96. Frankland E.— Phyl. Mag., 1858, 15, 149.
97. В e n 11 у W., Sterne P.— Science, 1921, 53, 143.
98. Rossel A.—C. r„ 1895, 121, 941.
99. G г о w e W.— Phil. Mag., 1841, 19, 98.
100. Fischer F., S chroter F.— Ber., 1910, 43, 1465.
101. R a v n e r O.— Chem. Met. Engn., 1921, 24, 886.
102. Juz a R., Hahn H.— Z. anorg. Chem., 1940, 244, 125.
103. Sato S.— Sci. Pap. Inst. Tokyo, 1939, 35, 182.
104. Vournasos A.— Z. anorg. Chem., 1927, 164, 263.
105. Rossini F. et al.— Cirvul. Bureau Stand., 1952, 500, 181.
106. В о h a r d G.— J. Phys. Chem., 1915, 19, 137.
107. H a h n H., J u z a I.— Z. anorg. Chem., 1940, 244, 111.
108. Franklin E.— J. Amer. Chem. Soc., 1907, 29, 35.
109. Stettbache r.— J. Chem. Soc., 1921, 119—120, 48.
110. F r i e d e r i c h E., S i 11 i g L.— Z. anorg. Chem., 1925, 143, 293.
111. Becker K-, Ebert F.— Z. Phys., 1925, 31, 268.
112. Brewer L. Chemistry and Metallurgy of Misellanous, Materials, Thermodyna-
mics, E. Quill, National Nuclear Energy Series. N. Y., 1950.
113. Cempter K-, К r i с о r i a n N., Me. Gurre J.—J. Phys. Chem., 1957,
61, 1237.
114. Ковальченко M. С., Нешпор В. С., Самсонов Г. В.—ЖПХ, 1958,
31, 1427.
115. Young R., Ziegler W.— J. Amer. Chem. Soc., 1952, 47, 5251.
116. Muthmann W., Kratt F.— Ann. Chim., 1902, 325, 262.
117. Neumann B., Kroeger C., Kunz N.— Z. anorg. Chem., 1932, 207, 133.
118. I an dell i A., Botti E.— Atti acad. nazl. Lincei, Classe Sci. fis. mat. e nat.,
1937 25 129.
119. Klemm W., Winkelmann G.— Z. anorg. Chem., 1956, 288, 87.
120. Foster L. Atomic U. S. Energy Comission Publ., 1950, (AECD 2941);
J. Inst. Metals, Metall. Abstracts, 1952, 20, 242.
121. M a t i g n о n C.— C. r., 1900, 131, 837.
122. Muthmann W., Kraft F.— Lieb. Ann., 1919, 1, 156.
123. T a m m a n G.— Z. anorg. Chem., 1922, 124, 25.
124. DafertF., Miklauz R.— Mh. Chemie, 1912, 33, 911.
125. Fichter F., Scholl у C.— Helv. Chem. Acta, 1920, 3, 164.
126. Ziegler W., Young R.— Phys. Rev., 1953, 90, 115.
127. Иверонова В. И., Тарасова В. П., Уманский М. М.— Вести. МГУ.
сер. физ.-мат. и ест. наук, 1951, 5, 8, 37.
128. Eick Н., Baenziger N., Eyring L.—J. Amer. Chem. Soc., 1956,
78, 2987.
129. I a n d e 11 i A.— Z. anorg. Chem., 1956, 288, 81.
354
130. E n d t е г F.— Z. anorg. Chem., 1948, 257, 127.
131. Liebigs Ann., 1902, 325, 262.
132. Palty A., Margolin H., Nielsen I.— Trans. Amer. Soc. Metals 1954
46, 312.
133. Корнилов И. И.— Изв. АН СССР, ОХН, 1954, 3, 392.
134. Самсонов Г. В., Нешпор В. С., Ланге Л. В.— Металловедение и об-
работка металлов, 1956, 1, 51.
135. Erlich А.— Z. anorg. Chem., 1949, 259, 1.
136. Б р е г е р А. X.— ЖФХ, 1939, 13, 272; 1939, 13, 1483.
137. Munster A., Sagel К.— Z. Elektrochem., 1953, 57, 571.
138. Уманский Я. С. Карбиды твердых сплавов. Металлургиздат, М., 1947.
139. GmelinsHandbuch, 41 «Titan», 1951, S. 278.
140. Самсонов Г. В. Тугоплавкие соединения. Металлургиздат, М., 1963.
141. Naylor В.— J. Amer. Chem. Soc., 1946, 68, 370.
142. Sato S.— Sci. Pap. Inst., Tokyo, 1938, 34, 888.
143. S h о m a t e C.— J. Amer. Chem. Soc., 1941, 68, 310.
144. Львов С. H., Немченко В. Ф., Самсонов Г. В.— ДАН СССР, 1960,
135, 577.
145. Самсонов Г. В., Верхоглядова Т. С.— ДАН СССР, 1961, 138, 342.
146. Самсонов Г. В., Уманский Я. С. Твердые соединения тугоплавких ме-
таллов. Металлургиздат, М., 1957.
147. Самсонов Г. В., Верхоглядова Т. С.— Журн. структ. хим., 1961,
2, 617.
148. Самсонов Г. В., Рукина В. Б.— ДАН УССР, 1957, 3, 247.
149. Bridgman Р.— Proc. Amer. Acad., 1932, 66, 255, 259, 268.
150. Kelley К.— Bur. Mines Bull., 1937, 407.
151. Фогель Я — ЖЭТФ, 1945, 15, 545.
152. Самсонов Г. В.— Журн. структ. хим., 1960, 1, 447.
153. W i 1 k i n s о n М. et al.— J. Appl. Phys., 1960, 31, 358.
154. Самсонов Г. В., Голубева Н. К.— ЖФХ, 1956, 30, 1258.
155. Munster A., Schlamp G.— Z. phys. Chem., 1957, 13, 1/м.
156. Савицкий Е. М., Полякова В. П., Т ы л к и н а М. А. Сплавы палла-
дия. «Наука», М., 1967.
157. Munster A., Ruppert W.— Z. Elektrochem., 1953, 57, 558.
158. Pollard F., Woodwart P.— Trans. Farad. Soc., 1950, 46, 190.
159. Campbell J. et al.— J. Electrochem. Soc., 1949, 96, 318.
160. ЛевеН. Ф., Ю p e в и ч А. Б.— Зав. лаб., 1940, 9, 957.
161. Montemartini С., Los ana L.— Gior. Chim. ind. Appl., 1924, 323.
162. Самсонов Г. В., Пилипенко А. Т., Назарчук Т. Н. Анал1з твердих
тугоплавких сполук. Вид-во АН УРСР, К., 1961.
163. Самсонов Г. В., Ясинская Г. А., Шиллер Э. А—Огнеупоры, 1961,
7, 335.
164. М а у е г О.— Вег. Deutsch. Keram. Ges., 1930, 11, 358.
165. Смирнов М. В., Краснов Ю. Н.— ЖНХ, 1958, 3, 1876.
166. Самсонов Г. В., Нешпор В. С. Вопросы порошковой металлургии и
прочности материалов. Изд-во АН УССР, К., 1958, 5, 3.
167. Сирота Н. Н., Самсонов Г. В., Стрельникова Н. С.— В кн.: Фи-
зико-химический анализ. МИЦМИЗ. Металлургиздат, М., 1957, 30, 4, 368.
168. Самсонов Г. В., Нешпор В. С.— В кн.: Технология цветных металлов.
Тр. МИЦМИЗ. Металлургиздат, М., 1958, 29, 361.
169. Н а г d у G., Н u 1 m J.— Phys. Rev., 1954, 93, 1004.
170. Goldwater D., Haddad R.— J. Appl. Phys., 1951, 22, 70.
171. Klemm W., S c h fl t h W.— Z. anorg. Chem., 1931, 201, 24.
172. MflnsterA., Sagel K.— Z. Phys., 1936, 144, 139.
173. Sagel K., Munster A., Schlamp G.— Nature, 1954, 174, 1154.
174. Самсонов Г. В. и др.— ДАН СССР, 1962, 142, 862.
355
23*
175. AgteC., Moers К.— Z. anorg. Chem., 1931, 198, 233.
176. Alexander P.— Metals and Alloys, 1938, 9, 179.
177. DuwezP., Odell F.— J. Electrochem. Soc., 1950, 97, 299.
178. Верхоглядова T. С., Дубовик T. В., Самсонов Г. В.— Порошко-
вая металлургия, 1961, 1, 9.
179. Gulbransen Е., Andrew К.— J. Metals, 1949, 187, 741; 1950, 188, 568.
180. Richardson L., Grant N.— J. Metals, 1954, 6, 1, 69.
181. Ремин В. П.— Вестник металлопромышленности, 1938, 18, 5763.
182 Hughes W., Harris В.— Англ. пат. 787516, 1917.
183. van А г k е 1 А. Е.— Physica, 1924, 4, 286, 293.
184. Moers К.— Z. anorg. Chem., 1931, 198, 243, 165, 259.
185. О р м о н т Б. Ф.— ЖФХ, 1947, дополнит, том, 72.
186. Б регер А. X,—ЖФХ, 1939, 11, 617.
187. R u f f О., Е i s n е г F.— Вег., 1908, 41, 2254.
188. Б р е г е р А. X,— ЖФХ, 1939, 11, 630.
189. 3 е л и к м а н А. Н., Лосева С. С., Ц е й т и н а Н. Я.— Цветные металлы,
1947, 20, 41.
190. Зеликман А. Н., Горовиц Н. Н.—-ЖПХ, 1950, 23, 689.
191. Белякова Е., Комар А., Михайлов В.— Металлургиздат, 1940, 15, 5.
192. Alexander Р.— Chem. Abstr., 1949, 43, 5162.
193. Меерсон Г. А., Колчин О. П.— В кн.: Научные труды Моск, ин-та цвет-
ных металлов и золота им. М. И. Калинина. Металлургиздат, М., 1955, 25, 195.
194. Огорд Л., Эспеншид Г.— Пат. США 2822246, 1958; Металлургия, 1959, 10.
195. Koster W., R a u s с h е г.— Z. Metallkunde, 1948.
196. Foster L., Atomic Energy Comission, Report AECD, 1950, цит. no [197].
197. Schwarzkopf P., Kieffer R.— Refractory Hard Metals, 1953.
198. Domagala R., Ph er son D. Me., Hansen M.— J. Metals, 1956, 8, 98.
199. M a 11 e 11 M. et al.— J. Electrochem. Soc., 1953, 100, 103.
200. В о e r J. De, Fast J.— Rec. trav. chim., 1936, 55, 459.
201. Guladner W., Wooten L.— Trans. Electrochem. Soc., 1948, 93, 223.
202. Самсонов Г. В., Верхоглядова Г. С.— ДАН УССР, 1962, 1, 48.
203. Hoch М., Dingledy D., Johnston Н.— J. Amer. Chem. Soc., 1954, 77,
304.
204. Гвоздов С. П., Журенкова А. А.— Научные доклады Высшей школы,
сер. Металлургия, 1958, 3, 32.
205. Агеев Н. В, Мод ель М. С.— ЖНХ, 1958, 3, 1439.
206. Kubaschewski О., Evans Е. Metallurgical Thermochemistry, Pergamon
Press. London — N. Y., 1958.
207. Смагина E. И., Куцев В. С., Ормонт Б. Ф.— ДАН СССР, 1957, 115,
354.
208. Dravnieks А.— J. Amer. Chem. Soc., 1950, 72, 3518.
209. Mallet M., Belle J., Cleland B.— J. Electrochem. Soc., 1955, 101, 1.
210. Смагина E. И., Куцев В. С., Ормонт Б. Ф.— ЖФХ, 1960, 34, 2328.
211. V a silos Т., Kinger у W.— J. Amer. Cer. Soc., 1954, 37, 409.
212. В a k е г Т. W.— Acta Cryst., 1958, 11, 300.
213. М a h А.— J. Amer. Chem. Soc., 1956, 78, 3261.
214. Попова О. И., Кабанник Г. Т.— В кн.: Труды семинара по жаростой-
ким материалам. Изд-во АН УССР, К., 1961, 6, 64.
215. Самсонов Г. В., Страшинская Л. В., Шиллер Э. А.—Изв. АН
СССР, Металлургия и топливо, 1962, 5, 167.
216. Портной К. И., Левинский Ю. В.— ЖПХ, 1961, 34, 1413.
217. Van Arkel А. Е., Boer J.— Z. anorg. Chem., 1925, 148, 347.
218. Boer J., Fast J.— Z. anorg. Chem., 1926, 153, 1.
219. Самсонов Г. В., Нешпор В. С —В кн.: Технология цветных металлов.
Научные труды Моск, ин-та цветных металлов и золота им. М. И. Калинина. Метал-
лургиздат, М., 1958, 29, 361.
356
220. Немченко В. Ф., Львов С. Н., Самсонов Г. В.— Укр. физ. ж., 1962
7, 117.
221. Фоменко В. С. Эмиссионные свойства элементов и химических соедине-
ний. «Наукова думка», К., 1964.
222. Hayes Е., Roberson А.— Trans. Electrochem. Soc., 1949, 96, 142.
223. Edwards R., Malloy G.— J. Phys. Chem., 1958, 62, 45.
224. Humphrey G.— J. Amer. Chem. Soc., 1953, 75, 2806.
225. Верхоглядова T. С. Технология почучения и некоторые физические
свойства нитридов переходных металлов. Автореферат канд. дисс., КПЙ, К., 1962.
226. Брохин И. С., Фридман А. А.— В кн.: Твердые сплавы. «Металлургия»,
М., 1965, 7, 92.
227. Вайнштейн Э. Е., Васильев Ю. Н.— ДАН СССР, 1957, 114, 53.
228. Жураковский Е. А., Вайнштейн Э. Е.— ДАН СССР, 1959, 129, 2669.
229. Жураковский Е. А., Дзегановский В. П.—Порошковая металлур-
гия, 1964, 5, 57.
230. Остроумов Э. А.— Зав. лаб., 1935, 5, 506.
231. Портной К. И., Левинский Ю. В.— ЖФХ, 1963, 37, 2627.
232. Портной К. И., Левинский Ю. В.— В кн.: Исследование сплавов
цветных металлов. ИМЕТ им. А. А. Байкова, Изд-во АН СССР, М., 1963, 4, 279.
233. Samsonov G. V. Technologia a vlasnosti spekanych ziaruvzdornych materi-
alov. Prob^emy praskovej metalurgie, Slovenska Akademia Vied., Bratislava, 1963, 30.
234. Gooding R. W., Parratt N. J.— Powder Metallurgy, 11, 42, 1963.
235. Holmberg B.— Acta Chem. Scand., 1962, 16, 1253.
236. Гришин В. К. и др. Свойства лития, Металлургиздат, М., 1963.
237. Schafer Н., Fuhr W.— Z. anorg. Chem., 1962, 319, 52.
238. Федосеев И. В., Немко ва О. Г.— ЖНХ, 1962, 7, 980.
239. Munster А.— Angew. Chem., 1957, 69, 281.
240. Голдберг Н. А., Знеменский Ю. Д.—ЖФХ, 1962, 36, 2748.
241. Glemser О., Fild М., К 1 е i n e-W е i s с h е d е К.— Z. anorg. Chem., 1964,
332 257
242. Дубовик Т. 'В., Полищук В. С., Самсонов Г. В.— ЖПХ, 1964, 37,
1828.
243. Hum e-R о t h е г у W., Raynor G., Lillie А.—J. Iron and Steel Inst.,
1942.
244. Murgulescu J. G., Cismaru D.— Rev. Chim. Acad. Rep. Pop. Roumanie,
1960, 251.
245. S i f f e г 1 e n R.— C. r., 1964, 259, 1520.
246. Khan J. A., В h a t T. R.— J. Less-Common Metals, 1965, 9, 388.
247. M а к о л к и н И. А. и др.— ЖПХ, 1960, 4, 824.
248. J u z a R. et al.— Z. anorg. Chem., 1964, 329, 136.
249. Juza R„ Rabenau A., Nitschke J.— Z. anorg. Chem., 1964, 332, 1.
250. Hansen N.— Z. anorg. Chem., 1964, 329, 68.
251. Меерсон Г. А., Рой C.— Порошковая металлургия, 1963, 6, 71.
252. Самсонов Г. В., Фоменко В. С., Верхоглядова Т. С.— В кн.: Хи-
мия и физика нитридов. «Наукова думка», К-, 1968, 162.
253 Bungardt К-, Preisendanz Н., Horn Е.— Z. Metallkunde, 1962, 53,
495.
254. Nowotny Н., Braun Н., Benesovsky F.— Radex-Rundschau, 1962,
53, 495.
255. Портной К. И., Левинский Ю. В.— ЖФХ, 1963, 37, 2467.
256. Kieffer R., Benesovsky F. Hartstoffe. Wien, Springer-Verlag, 1963.
257. Hahn H.— Z. anorg. Chem., 1949, 258, 58.
258. Эпельбаум В. А., Ормонт Б. Ф.— ЖФХ, 1947, 21, 3.
259. Брегер А. X., Эпельбаум В. А.— ЖФХ, 14, 1602, 1940.
260. King Е.— J. Amer. Chem. Soc., 1949, 71, 1.
357
261. Shomate C., Kelley К.— J. Amer. Chem. Soc., 1949, 71, 1.
262. Жураковский E. А., Вайнштейн Э. E.— ДАН СССР, 1959, 127, 534.
263. Hartmann, Frohlic h.— Z. anorg. Chem., 1934, 218, 190.
264. Moissan h.— C. r., 1898, 127, 497.
265. Moissan H.— Ann. Chim. Phys., 1899, 18, 318.
266. D u t о i t, S c h п о г f — C. r„ 1929, 187, 300.
267. Schmitz-Dumont O., Steinberg U.— Naturwis., 1954, 41, 117.
268. Franek, Bredig, Hoffman n.— Naturwis., 21, 1933, 330.
269. J u z a R.— Chemie, 1945, 58, 25.
270. Laurend Y., David L, Lang I.— C. r., 1964, 259, 1132.
271. S i 11 e r 1 e n R.— C. r„ 1964, 259, 1520.
272. Belin P.— C. r„ 1964, 258, 4715.
273. Ж У ков И. И,—ЖРФХО, 1910, 42, 40.
274. Funk Н., Bohl and Н.— Z. anorg. Chem., 1964, 334, 155.
275. Самсонов Г. В.— В кн.: Металловедение и металлофизика сверхпровод-
ников. «Наука», М., 1965, 65.
276. Самсонов Г. В. и др. Анализ тугоплавких соединений. Металлургиздат,
М., 1962.
277. Brauer G., Jander J.— Z. anorg. Chem., 1952, 270, 160.
278. Schonberg N.— Acta Chem. Scand., 1954, 8, 202.
279. Schonberg N.— Acta Chem. Scand., 1954, 8, 226.
280. Уманский Я. С.— ЖФХ, 1940, 14, 332.
281. Brauer G.— J. Less-Common Metals, 1960, 2, 131.
282. Brauer G. Conference on Niobium, Tantalum, Molybdenum and Tungsten,
Department of Metallurgy University of Scheffild, 1960.
283. Elliott R. P., К о m j a t h у S. Columbium Symposium Met. Soc. Conf., vol.
10, Intersci-Publ, N. Y„ 1961, 367.
284. Gebhardt E., Fromm E., Jakob D.— Z. Metallkunde, 1964, 55, 423.
285. Верхог лядова T. С., Львов С. H., Немченко В. Ф.— Укр. хим. ж.,
1964, 30, 667.
286. Корсунский М. И., Генкин Я. Е., Верхоглядова Т. С.— Порошко-
вая металлургия, 1962, 4, 35.
287. Корсунский М. И., Генкин Я. Е.— Изв. АН СССР, сер. физ., 1960,
24, 461.
288. Armstrong G.— J. Amer. Chem. Soc., 1949, 71, 3583.
289. Самсонов Г. В., Верхоглядова Т. С.— ДАН СССР, 1962, 142, 608.
290. Lautz С., Schroder Е.— Z. Naturforschung, 1956, 11, 517.
291. Schroder Е.— Z. Naturforschung, 1957, 12, 247.
292. S е 11 m a i е г А.— Z. Phys., 1955, 141, 550.
293. Cook D. В., Zemansky М. W., Boorse H. A.— Phys. Rev., 1950,
79, .1021.
294. Brauer G., Jander J., Rogener H.— Z. Phys., 1953, 134, 432.
295. Brauer G., Kirner H.— Z. anorg. Chem., 1964, 328, 34.
296. Kieffer R., Schwarzkopf P. Hartstoffe und Hartmetalle, Wien, 1953.
297. Cost J. R„ Wert C. A.—Acta Metallurgica, 1963, 11,231.
298. Самсонов Г. В., Верхоглядова Т. С.— ЖНХ, 1961, 6, 2732.
299. Love А,—С. г., 1902, 134, 1577.
300. Septier A., Ganzeit М., N ar u ch Р.— С. г., 1952, 234, 105.
301. Pollard F., Fowles G.— J. Chem. Soc., 1952, 2444.
302. Krings W., Kemp ken s J.— Z. anorg. Chem., 1929, 183, 225.
303. M a h A. D.— J. Amer. Chem. Soc., 1958, 80, 3872.
304. Roberts B. W. Superconductive Materials and some ef their Properties, Ge-
neral Electric Research Laboratory, Schenectady, N. Y., 1963.
305. Tompson B. W., Carlson O. N.— J. Less-Common Metals, 1964, 7, 321.
358
306. Портной К. И., Левинский Ю. В., Салибеков С. Е.— Порошковая,
металлургия, 1965, 12, 36.
307. Н a gg G.—Z. Phys. Chem., 1931, 13 (), 33.
308. Schonberg N.— Acta Chem. Scand., 1954, 8, 199.
309. Brauer G., Zapp K.— Z. anorg. Chem., 1954, 277, 129.
310. Brauer G., Lesser R.— Z. Metallkunde, 1959, 50, 512.
311. Gebhardt E., Seghezzi H. D., Diirrschnabel W.— Z. Metallkunde
1958, 49, 577.
312. Gebhardt E., Seghezzi H., Fromm E.— Z. Metallkunde, 1961, 52
464.
313. T i n y-S u i-K e.— Phys. Rev., 1948, 74, 914.
314. Brewer L., Krikorian O.— J. Electrochem. Soc., 1956, 103, 38.
315. Попова О. И., Кабаник Г. М,— ЖНХ, 1960, 5, 930.
316. В о о s s H. J.— Metall, 1956, 10, 130.
317. Horn F, Ziegler.— J. Amer. Chem. Soc., 1947, 69, 2762.
318. Верхоглядова T. С. и др.— Изв. вузов, сер. физ., 1967, 8, 31.
319. Hulm Т., Matthias Н.— Phys. Rev., 1952, 82, 273.
320. К i е s s 1 i и g R„ L i u Y — J. Metals, 1951, 3, 639.
321. В 1 i x R.—Z. Phys. Chem, 1929, B3, 229.
322. Мозговой В. С, Самарин А. М.— Изв. АН СССР, ОТН, 1950, 10, 1529.
323. S е у b о 11 A. U, О г i а и i R. А.— J. Metals, 1956, 8, 556.
324. В г i с R, С г i w i D — Met. Techn, 1940, 7, 2.
325. Eriksson S, Jerkon t.— Ann, 1934, 118, 530.
326. Верхоглядова T. С. и др.— ФММ, 1961, 12, 622.
327. Л ь в о в С. H. и др — УФЖ, 1963, 8, 1372.
328. Самсонов Г. В, Львов С. Н, Немченко В. Ф.— ДАН УССР, 1962,
7, 936.
329. М е н ь ш и к о в А. 3, Рудом а но в П. Г.— Тр. Ин-та физики металлов
АН СССР, 1959, 22, 69.
330. Немнонов С. А, Меньшиков А. 3.—-Изв. АН СССР, сер. физ, 1959,,
23, 578.
331. Corliss L, Elliot N, Hastings J.— Phys. Rev, 1961, 117, 929.
332. Хансен M. Структуры бинарных сплавов. Металлургиздат, М, 1941.
333. Kieffer R. Pulvermetallurgie, 1. Planseeseminar «De re metallica», 22—26
Juni, Reutte Tirol, 1953, S. 268.
334. Buck R. H, Waterhouse R. B.— J. Less-Common Metals, 1964, 6, 36.
335. Weaver C.W.— Acta Metallurgica, 1962, 10, 1151.
336. Bungardt K, Preisedanz H.— Arch. Eisenhiittenwesen, 1958, 29, 241.
337. Olson E, Layer E, Middleton A.—J. Electrochem. Soc, 1955, 102, 73.
338. Жуков И. И.— Изв. сектора физ.-хим. анализа АН СССР, 1926, 3, 14.
339. Dupare L, Wenger Р, Schussele W.— Helv. Chem. Acta, 1930, 13,
917.
340. Самсонов Г. В, Фоменко В. С, Падерно Ю. Б.— Огнеупоры,
1962, 1, 40.
341. Попова О. И.—УХЖ, 1961, 27, 22.
342. Архаров В. И, Конев В. Н, Меньшиков А. 3.— ФММ, 1959, 7, 64.
343. Пинскер 3. Г, Абросимова Л. Н.— Кристаллография, 1958, 3, 281.
344. DuwezP, Odell F.— Powder Metall. Bull, 1950, 5, 43.
345. U r 1 a u b E.— Pogg. Ann, 1857, 101, 605.
346. Rosenhein A, Braun H.— Z. anorg. Chem, 1905, 46, 311.
347. H a g g G.— Z. Phys. Chem, 1930, B7, 339.
348. Schonberg N.— Acta Chem.' Scand, 1954, 8, 204.
349. Пинскер 3. Г, Каверин С. В, Троицкая Н. В.—Кристаллография,
1957, 2, 179.
350. Пинскер 3. Г, Каверин С. В.— Кристаллография, 1957, 2, 386.
359
351. Троицкая Н. В., Пинскер 3. Г.— Кристаллография, 1961, 6, 43.
352. Троицкая Н. В., Пинскер 3. Г.— Кристаллография, 1959, 4, 38.
353. Norton F. J., Marshall A. L.— Trans. Amer. Inst. Min. Eng., 1944,
156, 351.
354. S p a c i 1 H. S., Wulff J. The Metal Molybdenum. Amer. Soc. Metals, Cleve-
land, 1958.
355. Ev ans D. A., J ack К. H.— Acta Cryst., 1957, 10, 833.
356. Olds L. E., Rengstorff G. W. P.— J. Metals, 1956, 8, 150.
357. Hearley F. H., Chessiek J. J., Zettlemayer A. C.— J. Phys. Chem.,
1953 57 178
358. Henderson G., Galletly J.— J. Soc. Chem. Ind., 1908, 27, 387.
359. Hegediis A. J., S as v ar i K., Neugebauer J.— Z. anorg. Chem., 1957,
293, 56.
360. Pease R., Cook R.— J. Amer. Chem. Soc., 1926.
361. Kiessling R., Peterson L.— Acta Metallurgica, 1954, 2, 675.
362. Neugebauer J., Hegediis A. J., Millner T.— Z. anorg. Chem., 1959,
303, 50.
363. Хитрова В. И., Пинскер 3. Г.— Кристаллография, 1958, 3, 545.
364. Хитрова В. И., Пинскер 3. Г.— Кристаллография, 1959, 4, 545.
365. X и т р о в а В. И., Пинскер 3. Г.— Кристаллография, 1960, 5, 711.
366. Хитрова В. И., Пинскер 3. Г.— Кристаллография, 1961, 6, 882.
367. Хитрова В. И.— Кристаллография, 1961, 6, 549.
368. X и т р о в а В. И.— Кристаллография, 1963, 8, 39.
369. Крестовников А. Н., Шахов А. С. Физико-химические и термодина-
мические свойства редких металлов. Металлургиздат, М., 1939.
370. Gmelins Handbuch «Wolfram», Berlin, 1933, 74.
371. Sieverts A., Bergner E.— Ber. deutsch. Chem. Ges., 1911, 44, 2394.
372. LaffiteP., Grandadarm P.— C. r., 1935, 200, 1039.
373. Smithells C., Rooksby H.— J. Chem. Soc., 1927, 1882.
374. Matthias A., Frankenberger W. F.— Z. Elektrochem., 1929, 35, 920.
375. HeyneG., Moers K.— Z. anorg. Chem., 1931, 196, 156.
376. Trzebiatowski W.— Z. anorg. Chem., 1937, 233, 377.
377. Klemm W., Frischmuth G.— Z. anorg. Chem., 1937, 230, 210.
378. Gillmann W., Wrigge F.— Z. anorg. Chem., 1931, 199, 70.
379. Hagg G.— Z. Phys. Chem., 1933, B12, 33.
380. Matthias A., Zachariasen W. H.— Phys. Chem. Solids, 1958, 7, 98.
381. Дубовик T. В. Получение нитрида рения Re3N. Информписьмо Ин-та ме-
таллокерамики и специальных сплавов АН УССР, № 251. 1960.
382. Hahn Н., Konrad А.— Z. anorg. Chem., 1951, 264, 174.
383. Самсонов Г. В., Верхоглядова Т. С.— УХЖ, 1964, 30, 143.
384. Т г z е b i a t о w s k i W., Rudz inski J.—J. Less-Common Metals, 1964,
6, 244.
385. P г e 1 i n g e r O.— Mh. Chem., 1894, 15, 315.
386. Жуков И. И,—ЖРФХО, 1894, 40, 315.
387. Hagg G.— Z. Phys. Chem., 1929, B4, 346; B6, 229.
388. Бараташвили И. Б. и др.— ДАН СССР, 1961, 139, 1354.
389. Juza R., Puff Н., Wagneknecht F.— Z. Elektrochem., 1957, 61, 84.
390. Lihl F., Ettinger P., KutzelniggA.— Z. Metallkunde, 1962, 53, 715.
391. Z w i c k e r U.— Z. Metallkunde, 1951, 42, 274.
392. Guilland C., Wyart J.— Rev. Metallurgie, 1948, 45, 271.
393. Juza R., Puff H.— Z. Elektrochem., 1957, 61, 810.
394. M a h A. D.— J. Amer. Chem. Soc., 1958, 80, 2954.
395. Guilland C., Wyart J.— C. r., 1946, 222, 71.
396. Paran jpe V., Cohen M., В e v e г M., Floe C.— J. Metals, 1950, 188, 261.
397. Пинскер 3. Г., Каверин С. В —ДАН СССР, 1954, 96, 519.
360
398. Jack К.— Proc. Roy. Soc., 1948, 195, 35; 1951, 208, 400; Acta Cryst., 1950
3, 392; 1952, 5, 404.
399. Пинскер 3. Г., Каверин С. В.— ДАН СССР, 1954, 95, 797.
400. Дворник и на Г. Г., Пинскер 3. Г.— Кристаллография, 1958, 3, 438.
401. Дворянкина Г. Г., Пинскер 3. Г.— Кристаллография, 1960, 5, 153.
402. Королев М. Л. Азот как легирующий элемент стали. Металлургиздат,
М., 1961.
403. Fry А.— J. Iron Steel Inst., 1932, 3, 191.
404. Satos S.— Sci. Papers Inst. Phys. Chem. Res. Tokio, 1938, 28, 135.
405. E m u r e t P. et al.— J. Amer. Chem. Soc., 1930, 52, 1956.
406. GoodeveC., J ack К. H.— Trans. Farad. Soc., 1948, 4, 82.
407. Chretien A., Mathis M.— C. r., 1949, 228, 91.
408. Дорфман Я. Г. Магнитные свойства и строение вещества. ГИТТЛ, М.,.
1955 332
409. Hum e-R о t h е г у W.— Phil. Mag., 1962, 7, 1955.
410. Rawlings R., Robinson P. M.— Acta Metallurgica, 1959, 7, 659.
411. Конторович И. E., Совалова A. A.— Изв. АН СССР, OTH, 1949, 11,
2675.
412. Конторович И. E., Совалова A. A.— Tp. MATH, Трансжелдориздат,
4, 1948.
413. Конто p ов и ч И. Е., Совалова А. А.— ЖТФ, 1950, 20, 53.
414. Кричевский И. Р., Хазанова Н. Е.— ДАН СССР, 1950, 71, 481; 1950,
71, 677.
415. Кричевский И. Р., Хазанова Н. Е.— ЖФХ, 1945, 19, 676.
416. Nowotny Н., Rudy Е., Benesovsky F.— Mh. Chemie, 1960, 91, 963.
417. Brewer L., Haraldsen H.— J. Electrochem. Soc., 1953, 102, 399.
418. J e i t s c h k о W., Nowotny H., Benesovsky F.—Mh. Chemie, 1963,
94, 1198.
419. J e i t s c h k о W., Nowotny H., Benesovsky F.— Mh. Chemie, 1964,
95, 156.
420. J e i t s c h k о W., Nowotny H., Benesovsky F.— Mh. Chemie, 1964,
95, 178.
421. J e i t s c h k о W., Nowotny H., Benesovsky F.— Mh. Chemie, 1964,
95, 438.
422. Jeitschko W., Nowotny H., Benesovsky F.— J. Less-Common Me-
tals, 1964, 7, 133.
423. Nowotny H., Jeitschko W., Benesovsky F.— Planseeberichte file
Pulvermetallurgie, 1964, 12, 31.
424. Juza R., Klose W.— Angew. Chem., 1959, 17, 161.
425. Juza R.— Z. anorg. Chem., 1953, 273, 48.
426. Juza R., Hund F.— Z. anorg. Chem., 1948, 257, 13.
427. Ghosh S. P.— J. Indian Chem. Soc., 1953, 30, 98.
428. Schonberg N.— Acta Chem. Scand., 1954, 8, 213.
429. Samson C., Bouchand К- P-, Fruchart R.— C. r., 1964, 259, 392.
430. Kloepfer H., Muller J., Sperr F.— Пат. ФРГ 1178219, 1965.
431. Smith R — Trans. Met. Soc. AIME, 1962, 224, 190.
432. BridelleR., Michel A.— Rev. Metallurgie, 1954, 51, 278.
433. Brauer G., Schnell W. D.— J. Less-Common Metals, 1964, 7, 23.
434. Конев В. H.— В кн.: Исследования по жаростойким сплавам, 3. Изд-во
АН СССР, М„ 1958, 415.
435. Архаров В. И., Катанов Л. М., Конев В. Н., Самсонов Г. В.—
Уч. зап. Уральского гос. ин-та им. А. М. Горького, Свердловск, 1966, 79.
436. Juza R., Anschutz Е., Puff Н.— Angew. Chem., 1959, 71, 161.
437. Stadelmaier H. H., Fraker А. С.— Z. Metallkunde, 1962, 53, 48.
438. Wachtel Е., Bartelt С.— Z. Metallkunde, 1964, 55, 88.
36»
439. Бр ох ин И. С., Фридман А. А.— Порошковая металлургия, 1965, 7, 34.
440. Gilles Р.— Corrosion et anticorrosion, 1964, 12, 15.
441. Carlill S. J., Hume-Rothery W.— J. Inst. Met., 1949, 76, 195.
442. E r 1 i c h P. et al.—Z. anorg. Chem., 1964, 328, 243.
443. Schonberg N.— Acta Chem. Scand., 1954, 8, 208.
444. R о u b i n M. P., P a r i s J. M.— C. r., 1965, 260, 3981.
445. R о u b i n M. P., P a r i s J. M.— C. r., 1965, 260, 3088.
446. Кричевский И. P., Хазанова H. E.— ЖФХ, 1947, 21, 719.
447. M и н к e в и ч А. Н.— Химико-термическая обработка металлов и сплавов.
Машиностроение, М., 1965.
448. Юргенсон А. А. Азотирование в энергомашиностроении. Машгиз, Сверд-
ловск—Москва, 1962.
449. Юргенсон А. А.— ФММ, 1959, 7, 110.
450. М а г a t г а у.— Docum. Metallurgique, 1953, 16, 127.
451. Brauer G., Lesser R.— Z. Metallkunde, 1959, 50, 487.
452. Wiener G., Berger J.— J. Metals, 1955, 7, 2, 360.
453. Займовский А. С., Чудновская Л. А., Магнитные материалы. Гос-
энергоиздат, М., 1957.
454. Senateur J., Fruchart R., Michev A.— C. r., 1962, 255, 1615.
455. Juza R., Sachsze W.— Z. anorg. Chem., 1945, 253, 95.
456. Clarke J., Jack K-— Che. Ind., 1951, 17, 1004.
457. Schmitz-Dumont O., Krin N.— Angew. Chem. 1955, 67, 231.
458. Juza R., Sachzse W.— Z. anorg. Chem., 1943, 251, 201.
459. Hagg G.— Nova Acta Reg. Soc. Sci. Upsaliensis, 1929, 7 (4), 22.
460. Hahn H., Konrad A.— Z. anorg. Chem., 1951, 264, 181.
461. Reinacker G„ Hohl К. H.—Z. anorg. Chem., 1964, 333, 291.
462. N u г у G.— C. r„ 1953, 237, 654.
463. Гинзбург С. И. и др. Руководство по химическому анализу платиновых
металлов и золота. «Наука», М., 1965.
464. Звягинцев и др. Химия рутения. «Наука», М., 1965.
465. Д ж а ф ф и Р., Мейкат Д. Дж., Дуглас Р. У. Рений и тугоплавкие
.металлы платиновой группы. ИЛ, М., 1963, 49.
466. Самсонов Г. В., Лютая М. Д., Нешпор В. С.— ЖПХ, 1963, 36, 2389.
467. Самсонов Г. В., Косолапова Т. Я., Лютая М. Д., Макарен-
ко Г. Н.—В кн.: Редкоземельные элементы. Изд-во АН СССР, М., 1963, 8.
468. Самсонов Г. В., Петраш Е. В.— Металловедение и обработка метал-
лов, 19S5, 4,119.
469. КоломоецН. В. и др.— ЖТФ, 1958, 23, 238.
470. Нешпор В. С., Самсонов Г. В.— ДАН УССР, 1964, 9, 1175.
471. Ж у Р а к о в с к и й Е. А., Дзегановский В. П.— ДАН СССР, 1963,
150, 1260.
472. Beattie Н., Snyda F.— Trans. ASM, 1953, 35, 397.
473. Самсонов Г. В., Лютая М. Д.— УХЖ, 1963, 29, 251.
474. Михеева В. И. Гидриды переходных металлов. Изд-во АН СССР,
М„ 1960.
475. Gschneidner К. Rare Earth Alloys, Toronto. London—N. Y., 1961.
476. Самсонов Г. В., Лютая M. Д.— ЖПХ, 1962, 35, 2359.
477. М и х е е в а В. И„ К о с т М. Е — ЖНХ, 1958, 3, 260.
478. Essen U., Klemm W.— Z. anorg. Chem., 1962, 317, 25.
479. V e у s s i e J. J., В г о c h i e r D., N emoz A., Blanc J.— Phys. Letters, 1965,
14, 261.
480. Veyssie J. J., Chaussy J., Berton A.— Phys. Letters, 1964, 13, 29.
481. Veyssie J. J., Haen P., Chaussy J., Anselin F.— C. r., 1965,
260, 4980.
482. S h i 1 d H. R. et al.— Phys. Rev., 1963, 131, 922.
•362
483. Kohlschutter V,— Ann., 1901, 317, 158.
484. Shulman R., Wyluda B.— J. Phys. Chem. Solids, 1962, 23, 166.
485. A n s e 1 i n F.—C. r., 1963, 256, 2616.
486. Самсонов Г. В. Тугоплавкие соединения редкоземельных металлов с не-
металлами. Металлургия, М., 1964.
487. D а о u J.— С. г., 1960, 250, 3635.
488. Didchenko R., Gorstema F.— J. Phys. Chem., 1963, 24, 863.
489. Радзиковская С. В., Марченко В. И. Сульфиды редкоземельных
элементов и актиноидов. «Наукова думка», К., 1966.
490. McClure J.— J. Phys. Chem. Solids, 1963. 24, 871.
491. Scl ar N.— J. Appl. Phys., 1964, 35, 1534.
492. Пермяков Л. H. и др.— Изв. АН СССР. Металлургия и топливо,
1964, 4, 68.
493. Актиниды. Под ред. Г. Сиборга, Дж. Каца. ИЛ, М., 1955.
494. R u n d 1 е Р. S.— Acta Cryst., 1948, 1, 180.
495. ZachariasenW. Н.— Acta Cryst., 1949, 2, 388.
496. С и б о p г Г. Т., Кац Дж. Химия актинидных элементов. Атомиздат,
М., 1962.
497. Кутайцев В. И. Сплавы тория, урана и плутония. Госатомиздат,
М„ 1962.
498. Gerds A. F., Mallett М. W.— J. Electrochem. Soc., 1954, 101, 175.
499. N о г t о и Р. Refractories, 3 Ed., N. Y., Me Graw Hill, 1950.
500. R u d 1 e R. et al.— J. Amer. Chem. Soc., 1948, 70, 99.
501. Nucleonics, 1964, 22, 64.
502. Benz R., Bowman M. G.— J. Amer. Chem. Soc., 1966, 88, 264.
503. Karlowitz C., Urban A.— Bohrtechniker Ztg., 1937, 9, 265.
504. Olson W., Mulford R.— J. Phys. Chem., 1963, 67, 952.
505. M u 11 e r M. H., К и о 11 H. W.— Acta Cryst., 1958, 11, 751.
506. Chubb W.— Reactor Materials, 1962, 5, 35.
507. Warren J. H.— Canad. Mining and Metallurgical, 1963, LXVI, 147.
508. Trzebiatowski W., Troe R., Leciejewicz J.—Bull. Academie Polo-
naise, Ser. Chimiques., 1962, 10, 395.
509. Trzebiatowski W., Troe R.— Bull. Acad. Pol., Ser. Chim., 1964, 12, 681.
510. Besson J., Moreau C., Philippot J —Bull. Soc. Chem. France,
1964, 1069.
511. Vaughan D.~ J. Metals, Sect. 2, 1956, 8, 78.
512. Mallett H., Gerds A.— J. Electrochem. Soc., 1955, 102, 292.
513. Thiimmler F., Ondraceck G., Dalal K.— Z. Metallkunde, 1965,
56, 535.
514. Вдовенко В. M. Химия урана и трансурановых элементов. Изд-во АН
СССР, М, 1960, 80.
515. Бессонов А. Ф., Власов В. Г.— ФММ, 1962, 14, 478,
516. Busi J., В а и е г А.— J. Amer. Cer. Soc., 1964, 47, 425.
517. S h е f t J., F r i e d S.— J. Amer. Chem. Soc., 1953, 75, 1236.
518. Бэгли К. Плутоний и его сплавы. Атомгиз, М., 1958.
519. Zachariasen W. Н.— Natl. Nuclear Energy, Ser. Div. IV, 148, Transura-
nium Elements, 1949, 11, 1448.
520. Abraham B., Davidson N., Westrum E.—Natl. Nuclear Energie, Ser.
Div. IV, 148; Transuranium Elements, 1949, 11, 945.
521. Porude W.— Reactor Materials, 1962, 5, 24.
522. More a u С., P hi 1 ippot J.— C. r„ 1961, 251, 1100.
523. MatignonC. A., Delepine M.— C. r., 1901, 132, 36.
524. Brown F., Ockenden H. M., Welch G. A.— J. Chem. Soc., 1955, 4196.
525. Leslie W., Caroll K., Fisher R.— J. Metals, 1952, 4, 204.
526. W a s s i 1 о n B„ W i 1 d e F. G — Nature, 1957, 179, 435.
363
527. Т и г к d о g a n E. T., Bills P. M., Tippet V.—-J. Appl. Chem., 1950, 8, 296.
528. H ar die D„ Ja ck К. H.— Nature, 1957, 180, 332. ।
529. Popper P., Ruddlesden S.— Nature, 1957, 179, 1129. I
530. Glemser O., Beltz K-, Naumann P.—Z. anorg. Chem., 1957, 291, 51.
531. Narita K., Mori K.— Bull. Chem. Soc. Japan, 1959, 32, 417. I
532. Billy M.— Ann. Chimie, 1959, 795.
533. Kaiser W., Thurmond C.— J. Electrochem. Soc., 1958, 105, 251. <
534. Самсонов Г. В., Цебуля Г. Г.— УФЖ, 1960, 5, 35. (
535. Collins J., Gerby R.— J. Metals, Sect., 1955, 7, 612.
536. Самсонов Г. В. Силициды и их использование в технике. Изд-во АН
УССР, К-, 1959.
537. Промышленно-экономич. газета, 72 (372), 15.VI 1958. »
538. G i 11 е s J. Р.— Rev. Hautes Temper, et Refract., 1965, 2, 237. I
539. Ясинская Г. А. Исследование взаимодействия расплавленных металлов I
с тугоплавкими соединениями. Автореф. дисс. ИПМ АН УССР, 1964. I
540. Allen В. С., К i n g е г у D.— Trans. Metal. Soc. AIME, 1959, 215, 30.
541. Бурыкина А. Л., Страшинская Л. В.— Физ.-хим. механика мате-
риалов, 1965, 5, 557.
542. Б р о х и н И. С., Ф у н к е В. Ф.— Огнеупоры, 1957, 22, 562.
543. Balmain W.— Ann. Chim., 1844, 52, 324.
544. Функе В. Ф., Самсонов Г. В.— ЖОХ, 1957, 28, 267.
545. Evans J. W., Chattergi S. Н.— J. Phys. Chem., 1958, 62, 1064.
546. Schonberg A.— Acta Chem. Scand., 1954, 8, 620.
547. К у л e ш о в И. М,— ЖНХ, 1959, 4, 488.
548. Самсонов Г. В., Ковальченко М. С., Добровольский А. Г.
Изготовление изделий методом порошковой металлургии, вып. 8. Опыт производства
порошка нитрида кремния и изделий из него. Изд-во ЦИТЭИН, М., 1960.
549. Weiss L., Engelhardt Т.— Z. anorg. Chem., 1910, 65, 38.
550. SchutzenbergerA., К о 1 s о n H.—C. r., 1882, 92, 326.
551. Евстропьев К. С., Торопов Н. А. Химия кремния и физическая химия j
силикатов. Госстройиздат, 1950, 223.
552. Wohler J.— Z. Elektrochem., 1926, 420.
553. Blix A., Birbelbaue г.—Berichte, 1903, 36, 4220.
554. Glemser О., Naumann P.— Z. anorg. Chem., 1959, 298, 134.
555. Самсонов Г. В., Добровольский А. Г.—Огнеупоры, 1966, 6, 55.
556. Добровольский А. Г. Шликерное литье. «Металлургия», М., 1967.
557. Popper Р., Ruddlesden S.— Trans. Brit. Ceramic Soc., 1961, 60, 9, 603.
558. Фр. пат. 1278723, 1960.
559. Ruddlesden S., Popper P.— Acta Cryst., 1958, 11, 465.
560. Turkdogan E. T., Ignatovicz S.—J. Iron Steel Inst., 1957, 185, 2, 200.
561. Beltz K.— Deutsche Nationalbibliographie, 1954, 17, 1422.
562. Johnson E., Krohn C.— Nature, 1961, 190, 523.
563. Аверин В. В., Г a p н ы к Г. А., Самарин А. М. — Изв. АН СССР, 01Н,
Металлургия и горное дело, 1963, 2, 40.
564. Англ. пат. 9700639, 1964. X
565. Шварц Р.— Успехи химии, 1936, 5, 1448. V
566. Johnson W.— J. Amer. Chem. Soc., 1930, 52, 5160. 1
567. Johnson W., R i g 1 e у G.— J. Amer. Chem. Soc., 1934, 56, 2395. i
568. Самсонов Г. В., Лютая М. Д., X о р п я к о в О. Т.— ЖНХ, 1964, 9, Ч
1529. ’
569. Джонсон О.— Успехи химии, 1956, 25, 1. j
570. Berraz G.— Anal. Soc. Sci. Argentina, Seccion Santa Fe, 1935, 7. 6; Chem.
Abstr., 1937, 31, 949.
571. Briegleb F., Geuther A.— Libigs Ann., 1962, 123, 238.
364
572. Mellor I. W. A Comprehensive treatise on inorganic and theretical chemistry
1928,8,111.
573. Taylor К. M., L e n i e C.-—J. Electrochem. Soc., 1960, 107, 308.
574. Renner T.— Z. anorg. Chem., 1959, 298, 22.
575. Андреева T. В. и др.— Теплофизика высоких температур, 1964, 2, 829.
576. Kauer Е., Rabenau А.— Z. Naturforschung, 1957, 12а, 943.
577. Lagrenaudie J.— J. Chim. Phys, et Phys. Chim. Biol., 1956, 54, 222.
578. G e 1 f г e у G. A., P a r r i G. S.— J. Chem. Phys., 1955, 22, 201.
579. Kleber W., Witzke H. D.— Naturwissensch., 1963, 50, 372.
580. M a h M. D. et al.— Thermodynamic Properties of the Aluminium Nitride,
Report 5716, Bureau of Mines, U. S. Dep. of the Inform., В. M., 1961.
581. О 11 H — Z. Phys. 1924, 22, 201.
582. N e u g e b a u e r C. A., Margrave J. L.—Z. anorg. Chem., 1957, 290, 82.
583. Беркс Дж. Б., Шульман Дж. Г. Прогресс в области диэлектриков, I.
Госэнергоиздат, М., 1962.
584. S с h i s s e 1 P. O., Williams W. S.— Bull. Amer. Phys. Ser. 2, 1959, 4, 139.
585. Long G., Foster L.— J. Amer. Ceram. Soc., 1959, 42, 53.
586. Самсонов Г. В., Дубовик Т. В.— Цветные металлы, 1962, 3, 56.
587. Полищук В. С,— ЖПХ, 1963, 36, 1142.
588. Репкин Ю. Д., Самсонов Г. В.— Порошковая металлургия, 1963, 5, 68.
589. Smithells С., Ransley С.— Proc. Roy. Soc., 1935, 152, 706.
590. Самсонов Г. В., Репкин Ю. Д.— Порошковая металлургия, 1965, 2, 1.
591. Смиттельс К. Газы и металлы. Металлургиздат, М., 1940, 81.
592. Charlton I., Evans С. Пат. США 784126, 1957.
593. Kleber W„ Witzke Н,— Z. Krist., 1961, 116, 126.
594. Witzke Н.— Phys. Stat. Sol., 1962, 2, 1109.
595. Witzke H — Z. Phys. Chem., 1962, 221, 259.
596. L e j u s A. M. et al.— C. r., 1963, 257, 157.
597. Gilles J. C., L e j u s A. M., Collongues R.— Corrosion et Anticorrosion,
1964, 12, 99.
598. Tiede E., Thimann M., S e n s s e K.— Ber. deutsch. Chem. Ges., 1938,
61, 1568.
599. Ad damiano A.— J. Electrochem. Soc., 1961, 108, 72.
600. Беляев А. И., Раппопорт M. Б., Хазанов E. И. Алюминий. Цветмет-
издат, M., 1932, 78.
601. Matsumura Т., Tanabe Y.— J. Phys. Soc. Japan, 1960, 15, 203.
602. Колобнев И. Ф., Альтман M. Б. Газы в алюминиевых сплавах. Изд-во
АН СССР, М., 1948.
603. Воронов С. М. Газы в алюминиевых сплавах и методы дегазации распла-
вов. ОНТИ, М„ 1938, .10.
604. К л ючко Ю. А,—ЖПХ, 1941, 14, 84.
605. Самсонов Г. В., Дубовик Т. В.— Порошковая металлургия, 1964,
2, 99.
606. Пат. ФРГ 1057005, 1959.
607. Фр. пат. 1149539, 1957.
608. Пат. США 2929126, 1960.
609. Р е п к и н Ю. Д.— Огнеупоры, 1965, 2, 41.
610. Long G., Foster L. М.— J. Electrochem. Soc., 1962, 109, 1176.
611. Margrave I. L., Staphitanonda P.—J. Phys. Chem., 1955, 59, 1231.
612. Лютая M. Д., БуханевичВ. Ф.— ЖНХ, 1962, 7, 2487.
613. Жданов Г. С., Мирман Г. В,—ЖЭТФ, 1936, 6, 10.
614. Шмарцев Ю. В., Валов Ю. А., Борщевский А. С. Тугоплавкие
алмазоподобные полупроводники. «Металлургия», М., 1964.
615. Lorenz ЛА. R„ Binkowski В. В.— J. Electrochem. Soc., 1962, 109, 26.
24—272
365
616. Гордиенко С. П., Самсонов Г. В., Фесенко В. В.—ЖФХ. 1'Кй,
38 2974.
617. Sime R., Margrave I.— J. Phys. Chem., 1956, 60, 810.
618. Алексеевский H. E., Самсонов Г. В., Шулишова О. И.—ЖЭТФ,
1963, 44, 1413.
619. Rabenau A. Compound Semiconductor, 1, N. Y.— London, 1962, 174.
620. Самсонов Г. В., Лютая M. Д.— ЖПХ, 1962, 35, 1680.
621. Johnson W., Parsons I., Grew M.— J. Phys. Chem., 1952, 36, 2651
622. Самсонов Г. В., Лютая М. Д.— ЖПХ, 1963, 36, 1416.
623. Juza R.— Angew. Chem., 1938, 13, 189.
624. F г а и k 1 i и Е. С.— J. Phys. Chem., 1912, 16, 683.
625. Balmain W.—J. Pract. Chim., 1842, 27, 422; Phil. Mag., 1842, 21, 170.
626. P e a s e R. S.— Nature, 1950, 165, 722.
627. Herold A., Mar luff B., Perio P.—C. r., 1958, 246, 1866.
628. Catalog Section H-8745 EC, National Trade Mark, Boron Nitride, Union Carbi-
de Corporation, 1963.
629. Kroll W.— Z. anorg. Chem., 1918, 102, 17.
630. Wiberg E., Michand H.— Z. Naturforsch., 1954, 96, 497.
631. Б регер A. X., Жда нов Г. С.— ДАН СССР, 1940, 28, 630.
632. Brill R., Neumann C., Peters C.— Naturwiss., 1941, 29, 784.
633. Coulson C., Taylor E.— Proc. Phys. Soc., 1952, A65, 825, 834.
634. Dworkin A., Sasmor D., Van Artsdalen E.— J. Chem. Phys., 1954,
22, 837.
635. Lorn er W., Morton K.— Proc. Phys. Soc., 1953, 66A, 772.
636. Brame E., Matgrave J., Meloche V.—J. Inorg. Nucl. Chemistry, 1957,
5, 48.
637. M i 11 e r F., W i 1 k i n s C.— Anal. Chem., 1955, 24, 1253.
638. Taylor K.—Ind. Eng. Chem., 1955, 47, 2506.
639. P о d s z u s E.— Z. anorg. Chem., 1917, 30, 156.
640. Иоффе А. Ф. Физика полупроводников. Изд-во АН СССР, М., 1957.
641. Ingles Т. A., Popper Р. Special Ceramics. London, 1961, 144.
642. Gmelins Handbuch der anorg. Chemie, Bor, Erganzunsband. Verlag Chemie,
Wien, 1954, 160.
643. Фесенко В. В.— Порошковая металлургия, 1961, 4, 80.
644. Гальченко Г. Л., Корнилов А. Н., Скуратов С. М.—ЖНХ, I960,
5, 1651.
645. Roth W.— Z. Naturforschung, 1946, 1, 574.
646. Campbell J. et al.— J. Electrochem. Soc., 1949, 96, 318.
647. R e m e 1 e.— Verh. Phys. Ges., 1911, 13, 777; Phys. Z., 1911, 12, 971.
648. Pascal P.— Traite de chimic minerale, 1933, 4, 608.
649. T i e d e E., В u s c h e r F.— Berichte, 1920, 53, 2203.
650. T i e d e E., S c h 1 u d e A.— Naturwiss., 1923, 11, 745.
651. TiedeE., Tomaschek R.— Z. anorg. Chem., 1923, 29, 303.
652. Tiede E., Tomaschek R—Z. anorg. Chem., 1925, 147, 111.
653. Москвин А. В. Катодолюминесценция, 2. ГИТТЛ, M., 1949, 515.
654. Самсонов Г. В.— Атомная энергия, 1963, 14, 588.
655. McDonald К. A., Stull D. R.— J. Phys. Chem., 1961, 65, 1918.
656. Goldschmidt V.— Norsk. Geol. Tidskr., 1926, 9, 258.
657. Deacon R. F., Goadman I. F.— Proc. Poy. Soc., 1957, 243, 464.
658. Hiroaki T., Kikou J.— Bull. Chem. Soc. Japan, 1962, 35, 1425.
659. Кабалкина С. С., Верещагин Л. Ф.—ДАН СССР, 1960, 134, 330.
660. Jager, Westenbrink.— Proc. Acad. Amsterdam, 1926, 29, 1218.
661. W e n t о r f f R.— J. Chem. Phys., 1957, 26, 956.
662. Wentorff R.— Geochem. News, 1957, 5.
663. Neuhaus A., Meyer H.— Angew. Chemie, 1957, 69, 551.
366
664. W e n t о r f f R.— J. Chem. Phys., 1962, 36, 1990.
665. Kleinman L., Phillips J. C.— Phys. Rev., 1960, 117, 460.
666. Geller S.— Phys. Chem. Solids, 1959, 10, 340.
667. M e c h a n.— Marquett, 1957, 2969.
668. Загя некий И. Л., Самсонов Г. В.— ЖПХ, 1952, 25, 557.
669. Finlay G., Fetterley G.— Amer. Cer. Soc. Bull., 1952, 31, 141.
670. Schwarzkopf P., Glaser F.— Z. Metallkunde, 1953, 44, 353.
671. Brewer L., Haraldsen H.— J. Electrochem. Soc., 1955, 102, 399.
672. Serichi Nakamura, Azumo N., Aray Z.— Nagoya Kogyo Gijntsu
Shkensha. Kokoku, 1960, 9, 59.
673. Ш а ф p а н И. Г,— ЖПХ, 1940, 13, 1885.
674. Убеллоде A. P., Льюис Ф. А. Графит и его кристаллические соедине-
ния. «Мир», 1965, 136.
675. Pierotti R. A., Petricciani J. С.—J. Phys., 1960, 64, 1596.
676. Broi Р., Steinberg S.— J. Amer, rocket. Soc., 1961, 31, 4460.
677. Полубояринов Д. H. и др.— Огнеупоры 1964, 2, 82.
678. Чепеленко Ю. В. и др.— Огнеупоры, ,1964, 6, 253.
679. Weintraub Е,— Пат. США 1135232; Chem. Abstr., 1915, 9, 1535.
680. Weintraub F.— J. Soc. Chem. Ind., 1914, 33, 421.
681. Vournasos A.— Bull. Soc. Chem., 1911, 9, 508.
682. Dannstadt A.— Bull. Soc. Chem., 1969, 12, 348.
683. Wohler P.— Liebigs Ann., 1850, 74, 71.
684. Rose H.— Poggend. Ann., 1950, 80, 255.
685. H a у d e r R — J. Soc. Chem. Ind., 1913, 32, 600.
686. H a у d e r R.— Chem. Ztg., 1914, 38, 29.
687. Mayer F., Zappner R.— Berichte, 1902, 35, 535.
688. Laubengayer A., Condicke G.— J. Amer. Chem. Soc., 1948, 70, 2274
689. Stock A., Hoile W.— Berichte, 1908, 41, 2095.
690. Пат. США 2824787, 1958.
691. S t о с k А., В 1 i x A.— Berichte, 1901, 34, 3044.
692. PodszusE. Герм. пат. 282748, 1913.
693. Clark H., Hoard J.— J. Amer. Chem. Soc., 1943, 65, 2115.
694. Stabler A., Elbert J.— Berichte, 1913, 46, 2075.
695. H e m p e 1.— Berichte, 1890, 23, 3388.
696. Moser L., Eidmann W.— Berichte, 1907, 35, 535.
697. Шафран И. Г., Ормонт Б. Ф.— Авт. свид. СССР 50556, 1936.
698. Mayer F., Zappner R.— Berichte, 1921, 54, 560.
699. Англ. пат. 742236, 1955.
700. Меерсон Г. А. и др.— Огнеупоры, 1955, 20, 72.
701. Самсонов Г. В,— ВКник АН УРСР, 1958, 5, 66.
702. Ж и р о в Н. Ф. Люминофоры. ОНТИ, М., 1940, 351.
703. Слепцов В.М., Самсонов Г. В.— ЖПХ, 1961, 34, 501.
704. Слепцов В. М., Самсонов Г. В.— В кн.: Кинетика и катализ. Изд-во
АН СССР, М, 1960, 129.
705. Пат. США 2801903, 1957.
706. Пат. США 2824787, 1958.
707. Пат. США 2808314, 1957.
708. Англ. пат. 777000, 1957.
709. Moissan Н.—С. г., 1897, 115, 206; Ann. Chim. Phys., 1895, 6, 312.
710. Stein G.— Z. anorg. Chem., 1907, 55, 166; 1912, 75, 218.
711. Belforti D., Blum S., Vavarnik B.—Nature, 1961, 190, 907.
712. GuetertO., Mozzi R.— Nature, 1962, 193, 570.
713. Schiff D., Mater. Sci. Technol. Advan. Appl., 1962, 236.
714. Pentecost J. High Temperature inorganic Goatings Ed. by J. Humenik,
N. Y„ 1963, 51.
367
li
2
715. Графит как высокотемпературный материал. «Мир», М., 1964.
716. Конструкционные углеграфитовые материалы. «Металлургия», М., 1964.
717. Пат. США 2865715, 1958.
718. Vickery R.—• Nature, 1959, 184, 268.
719. Pease Р.— Acta Crysts., 1952, 5, 356.
720. Плотников В. Ф., Кузьмин М. Л.— ЖПХ, 1935, 8, 429.
721. Stock A., Hoffmann В.— Berichte, 1903, 36, 317.
722. Moser Н., Fiequelmont А.— С. г., 1934, 198, 1417.
723. Huff mann Е. О. et al.— J. Amer. Chem. Soc., 1954, 76, 6239.
724. Ван Везер. Фосфор и его соединения. ИЛ, М., 1962.
725. Herzog А. Н., Nielsen М. L.— Anal. Chem., 1958, 30, 1490.
726. Самсонов Г. В., Верейкин Л. Л.— Фосфиды. Изд-во АН УССР,
К-, 1961.
727. Р е м и Г. Курс неорганической химии. ИЛ, М., 1963.
728. Вознесенский С. А.— Успехи химии, 1955, 24, 440.
729. Sharma В. D., Donohue J.— Acta Cryst., 1963, 16, 891.
730. Lundquist J.— J. Inorg. Nucl. Chem., 1958, 6, 159.
731. Чижиков Д. M., Счастливый В. П. Селен и селениды. «Наука»,
М„ 1964.
732. Barnighausen Н., Volkmann Т., Junder J.—Acta Crysta., 1953,
14, 1079.
733. Ko c h ano vs ka A.— Czechosl. J. Phys., 1953, 3, 193.
734. Farber M.— J. Chem. Phys., 1953, 21, 172.
735. Самсонов Г. В,— УХЖ, 1965, 31, 433.
736. Б р е г е р А. X,— ЖФХ, 1939, 10, 593.
737. Эпельбаум В. А., Брегер А. X,—ЖПХ, 1940, 13, 595.
738. Рауб Э., Плате В.— Проблемы современной металлургии, 1952, 4, 102.
739. Верхоглядова Т. С.— В кн.: Редкие и редкоземельные элементы в тех-
нике. «Наукова думка», К-, 1964, 104.
740. Samsonov G. V. Problemy praskovei metalurgie, Vyd. Slov. Akademie
Vied., Bratislava, 1964, 27.
741. МинкевичА. H. Химико-термическая обработка металлов и сплавов. «Ма-
шиностроение», М, 1965.
742. Юргенсон А. А. Азотирование в электромашиностроении. Машгиз, Сверд-
ловск—Москва, 1962.
743. Лахтин Ю. М. Физические основы процесса азотирования. Машгиз, М.,
1948.
744. Попов А. А. Термические основы химико-термической обработки стали.
«Металлургиздат», Свердловск, 1962.
745. Самсонов Г. В., Эпик А. П. Покрытия из тугоплавких соединений.
«Металлургия», М., 1964.
746. High-Temperature inorganic Coatings. Ed. by J. Humeniks, Reinhold Publ.
N. Y.—London, 1963.
747. Samsonov G. V. et al.— Coating of High-Temperature Materials, Plenum
Press, N. Y., 1966.
748. Bungardt K., Rudinger K.— Z. Metallkunde, 1956, 47, 577.
749. M и н к e в и ч A. H., Таймер А. Д., 3 о т ь e в Ю. А.— Металловедение и
обработка металлов, 1956, 7, 39.
750. Смирнов А. В., Начинков А. Д.— Металловедение и термическая об-
работка металлов, 1960, 3, 22.
751. Смирнов А. В., Начинков А. Д.— Металловедение и термическая об-
работка металлов, 1960, 7, 42.
752. Gulbransen Е., Andrews - К-— Trans. Amer. Inst. Min. Met. Eng.,
1949, 185.
753. Takeuchi Y. et al.— J. Japan, Inst. Met., 1959, 23, 7.
368
754. N а к а п о К- et al.— J. Japan. Inst. Met., 1960, 24, 8.
755. Metal Ind., 1960, 97, 87.
756. Новикова E. H.— В кн.: Титан и его сплавы, 3, Изд-во АН СССР
М„ 1960.
757. Грдина Ю. В., Г о р д я е в а А. Г., Тимошина Л. Г.— Изв. вузов, Чер-
ная металлургия, 1962,’ 6, 128. В кн.: Диффузионные покрытия на металлах. «Наукова
думка», К., 1965, 109.
758. Wyatt J. L., G г a n t N. J.— Iron Age, 1954, 173, 122.
759. Буренко О. И.— Металловедение и термическая обработка металлов, 1965
10, 57.
760. Калашникова М. И., Зотьева А. С.— Металловедение и термическая
обработка металлов, 1964, 4, 46.
761. Акулов А. И.— Автоматическая сварка, 1961, 9, 3.
762. Земсков Г. В. и др.— Металловедение и термическая обработка метал-
лов, 1964, 1, 52.
763. Конев В. Н. Исследования по жаропрочным сплавам, 3, Изд-во АН СССР,
М., 1958.
764. Архаров В. И., Конев В. Н., Герасимов А. Ф.— ФММ, 1960, 9, 659.
765. Герасимов А. Ф., Конев В. Н., Тимофеева Н. Ф.— ФММ, 1961,
11, 596.
766. Ruppert W. Пат. ФРГ 1089240, 1961.
767. Financial Times, 13.7, 62, 15.
768. Metal Progress, 1961, 80, 143.
769. Foster L. et al.— J. Amer. Cer. Soc., 1950, 33.
770. Summer G.— Meeh. Engr., Jah., 1948.
771. Самсонов Г. В., Семенов Ю. Н., Бородулин П. Я.—Огнеупоры,
1962, 7, 332.
772. Дегтярев В. С. и др.— Огнеупоры, 1966, 5, 52.
773. Larsen Р. А.— Insulation (USA), 1963, 9, 27.
774. С ем ко И. Ф. и др. Алмазные инструменты и их применение в машино-
строении. «Машиностроение», Харьков, 1965.
775. Mathien J. С., Durand F., Bonnier Е.— Thermodynamics, 1966,
1, 75.
776. Мукасеев А. А. и др.— Вестник машиностроения, 1961, 3, 63.
777. Уманский Я- С., Финкельштейн Б. Н., Б л антер М. Е. Физиче-
ские основы металловедения. Металлургиздат, М., 1949.
778. Хаган М. Кратные соединения включения. «Мир», М., 1966.
779. Б р е г е р А. X.— ЖФХ, 1941, 15, 927.
780. Уманский Я. С.— В кн.: Металловедение и термическая обработка. Тр.
Моск, ин-та стали, 20, Металлургиздат, М., 1940, 3.
781. Беликов А. М.— Автореф. канд. дисс., Моск, ин-т стали, М., 1958.
782. С а м с о н о в Г. В,— ДАН СССР, 1953, 93, 689.
783. Самсонов Г. В.— Изв. сектора физ.-хим. анализа ИОНХ АН СССР,
1956, 27, 97.
784. С а м с о н о в Г. В., Латышева В. П.— ФММ, 1956, 2, 309.
785. Самсонов Г. В.— ЖТФ, 1956, 26, 716.
786. Самсонов Г. В., Нешпор В. С.— ЖЭТФ, 1956, 30, 1143.
787. Самсонов Г. В., Нешпор В. С., Кудинцева Г. А.— Радиотехника и
электроника, 1957, 2, 631.
788. Самсонов Г. В., Нешпор В. С.— Инж. физ. журн., 1958, 1, 30.
789. Самсонов Г. В,— ЖФХО, 1960, 5, 515.
790. Самсонов Г. В., Падерно В. Н.— Изв. вузов, сер. физ., 1966, 2, 64.
791. Part he Е.— Powd. Met. Bull., 1956, 1, 138.
792. R u n d 1 e R. E.— Acta Cryst., 1948, 1, 180.
793. Engel N.— Powd. met. Bull., 1954, 7, 1.
369
794. Palmer W. G., Valency, Classical and modern, Me Millan Co, N. Y., 1944.
795. К i e s s 1 i n g R.— Powd. Metallurgy, 1959, 3, 177.
796. R о b i n s D. A.— Powd. Metallurgy, 1958, 1—2, 172.
797. В i 11 z H.— Z. Phys., 1958, 153, 338.
798. Piper J. Compunds of Interest in Nuclear Reactor Technology, Ed. by J. Wa-
ter and P. Chiotti, AIME, Michigan, 1964, 29.
799. С о s t a P., С о n t e R. R. Там же, p. 3.
800. D e n к e r S. P. T а м ж e, p. 51.
801. Goretzki H. Untersuchung der magnetischen, elektrischen und thermo-
elektrischen Eigenschaften der Karbide und Nitride der 4a- und 5a-Ubergansmetalle,
Dissertation, Universitat Wien, 1963.
802. Dempsey E.— Phyl. Mag., 1963, 8, 285.
803. Самсонов Г. В.— УХЖ, 1965, 31, 1233.
804. Берсукер И. Б., Аблов А. В. Химическая связь в комплексных соедине-
ниях. Изд-во АН Молд.ССР, Кишинев, 1962.
805. О pre л Л. Введение в химию переходных металлов. «Мир», М., 1964.
806. Боресков Г. К. Катализ гетерогенный. Краткая хим. энциклопедия, 2, М.,
1963. 468.
807. Самсонов Г. В.— Порошковая металлургия, 1966, 12, 49.
808. Пр яд ко И. Ф.— Порошковая металлургия, 1966, 12, 61.
809. Козлова И. Р., Гурин В. Н., Обухов А. П.— Порошковая металлур-
гия, 1966, 12, 68.
810. Pauling L. The Nature of the Chemical Bond, Third Edition, Cornell Univer-
sity, Press, 1960.
811. Д аркен Л. С., Гурри P. В. Физическая химия металлов. Металлургиздат,
М., 1960, 77.
812. Корсунский М. И., Генкин Я- Е.— ДАН СССР, 1962, 142, 1276.
813. Корсунский М. И., Генкин Я. Е.— Изв. АН СССР, сер. физ., 1964,
28, 832.
814. Корсунский М. И., Генкин Я. Е.— Изв. АН СССР, сер. неорг. мате-
риалы, 1965, 1, 1701.
815. М е н ь ш и к о в А. 3., Н е м н о н о в С. А.— ФММ, 1965, 19, 57.
816. Львов С. Н., Немченко В. Ф., Самсонов Г. В.— ДАН СССР, 1960,
135, 577.
817. Романов В. Д., Самсонов Г. В., Никитин Д. И. Авт. свид. СССР,
120509, 1959.
818. Львов С. Н., Немченко В. Ф., Самсонов Г. В.— Укр. ф!з. ж., 1963,
8, 1372.
819. Ковальский А. Е., Макаренко Т. Г.— ЖТФ, 23, 265, 1953.
820. Ковальский А. Е., Макаренко Т. Г.— В кн.: Микротвердость. Изд-во
АН СССР, М„ 1951, 187.
821. Самсонов Г. В., Розинова Н. С.— Изв. сектора физ-хим. анализа
ИОНХ АН СССР, 1956, 27, 126.
822. Фогель Я — ЖЭТФ, 1945, 15, 545.
823. Самсонов Г. В.— Журн. структ. хим., 1960, 1, 447.
824. Woddington G.— Trans. Farad. Soc., 1957, 53, 901.
825. Львов С. Н., Немченко В. Ф.— В кн.: Высокотемпературные неоргани-
ческие соединения. «Наукова думка», К., 1965, 100.
826. Mott N., Jones Н. The Theorie of Properties of Metals and Alloys, Claren-
don Press, Oxford, 1936.
827. Нешпор В. С., Самсонов Г. В,—ДАН СССР, 1964, 157, 834.
828. Ш у л и ш о в а О. И. Сверхпроводимость карбидов, нитридов переходных
металлов и их твердых растворов со структурой NaCl. Автореф. дисс. ИМФ АН УССР,
К., 1966.
829. Klemm W., Schutt W.— Z. anorg. Chem., 1931, 201, 24.
370
830. Самсонов Г. В., Нешпор В. С., Стрельникова Н. С,—ДАН УССР
1958, 8, 838.
831. Costa Р. et al. Compounds of Interest on Nuclear Reactor Technology Ed.
by J. Waber and Chiotii P. AIME, Michigan, 1964.
832. Л ь в о в С. Н. та ш.— Укр. ф!з. ж., 1963, 8, 1372.
833. Шмелев Б. А.— Зав. лаб., 1951, 6, 671.
834. Fukke, Morl е.— Stahl und Eisen, 1943, 46, 845.
835. Фесенко В. В., Болгар А. С. Испарение тугоплавких соединений. «Ме-
' таллургия», М., 1966.
836. Жураковс кий Е. А., Вайнштейн Э. Е.— ДАН СССР, 1959, 127, 534
837. Вайнштейн Э. Е„ Чирков В. И.—ДАН СССР, 1962, 145, 1031.
838. Герасимов А. Ф., Конев В. Н., Тимофеева Н. Ф.— ФММ, 1961
И, 596.
839. Корсунский М. И., Генкии Я. Е.— В кн.: Высокотемпературные ме-
1 таллокерамические материалы. Изд-во АН УССР, К-, 1962, 36.
840. Вайнштейн Э. Е. и др.— ФММ, 1961, 12, 360.
841. Нешпор В. С.— В кн.: Высокотемпературные металлокерамические мате-
риалы. Изд-во АН УССР, К-, 1962, 46.
842. Бурыкина А. Л. и др.— Теплофизика высоких температур, 1965, 6, 940.
843. Полищук В. С.— Порошковая металлургия, 1965, 7, 100.
844. Самсонов Г.' В.— В кн,: Редкие щелочные металлы. Изд-во СО АН СССР,
Новосибирск, 1967.
845. Самсонов Г. В., Падерно Ю. Б., Фоменко В. С.— ЖТФ, 1966, 36,
1435.
846. Самсонов Г. В., Фоменко В. С., Падерно Ю. Б.— Укр. ф!з. ж., 1963,
8, 700.
847. Fomenko V. S., Handbook of Thermoionic Properties Ed. by Samsonov G. V.
Plenum Press, N. Y., 1966.
848. Rentschler H., Henry D.— Trans. Electrochem. Soc., 1945, 87, 289.
jl 849. Haas G., Jensen J.— J. Appl. Phys., 1963, 34, 3451.
850. Самсонов Г. В. и др. Физико-химические свойства элементов. «Наукова
думка», К., 1966.
i 851. Самсонов Г. В.— В кн.: Методы испытания на микротвердость. «Наука»,
М„ 1965, 59.
Ь 852. Nowotny Н., Vitovec F. Plansee Seminar «De re metallica», 1953, 46.
e 853. Acta Metallurgica, 1955, 3, 104.
К 854. Беликов A. M., Уманский Я. С.— Научные доклады высшей школы,
I 1958, 1, 192.
855. Самсонов Г. В., Нешпор В. С.— ДАН СССР, 1955, 104, 405.
* 856. Самсонов Г. В., Нешпор В. С., Хренова Л. М.— ФММ, 1957, 4, 181.
* 857. Самсонов Г. В., Иванько Г. А., Баженова Л. Н.— Изв. АН СССР,
Неорг. материалы, 1966, 2, 1194.
858. О р м о н т Б. Ф. Тезисы постоянного межинститутского коллоквиума по
; твердым фазам переменного состава, 3, ФХИ им. Л. Я. Карпова, М., 1956.
j 859. Самсонов Г. В. Нитриды. Краткая хим. энциклопедия, 3, М., 1964, 490.
* 860. Самсонов Г. В.— В кн.: Химия и физика нитридов. «Наукова думка»,
|К.. 1968, 9.
861. Самсонов Г. В.— В кн.: Высокотемпературные металлокерамические ма-
териалы. Изд-во АН УССР, К., 1962, 5.
862. Верятин У. Д. и др. Термодинамические свойства неорганических ве-
ществ. Атомиздат, М., 1965.
863. Бродский А. И. Физическая химия, 1. Госхимиздат, М., 1948.
864. В о с k г i s J. О. М., W h i t е J. A., Mackenzie I. D. Physicochemical Mea-
surements at High Temperatures, 1959, 353.
865. Раковский А. В. Введение в физическую химию. ГОНТИ, М., 1938.
24*
371
866. Johnson А,— Пат. США 2480473, 1949.
867. Бугаков Д. К-— Сталь, 1958, 5, 457.
868. Репкин Ю. Д. Металлокерамические жаропрочные сплавы типа САП.
Изд-во АН УССР, К., 1964.
869. J. Iron and Steel Inst. Japan, 40, 279, 1954.
870. Nature, 1958, 182, 4630, 255.
871. Korbin C. L.— Iron Age Metal Work International, 1964, 3, 20.
872. Nature, 1927, 181, 4622.
873. Кайнарский И. С., Дегтярева Э. В., Кухтенко В. А.—Огнеупоры,
1960, 4, 175
874. Кайнарский И. С., Дегтярева Э. В. Карборундовые огнеупоры.
Металлургиздат, М., 1963.
875. Самсонов Г. В.— В кн.: Огнеупорное производство. Металлургиздат, М.,
1965, 482—512.
876. Дуглас Д., Фа ликов Л.— В кн.: Сверхпроводимость. «Наука», М., 1967.
877. Engineering, 1960, 139, 4908, 572.
878. Brown R. W., Landback C. R.— Ceramic Bulletin, 1959, 7, 352.
879. Самсонов Г. В., Пеньковский В. В.— Оптика и спектроскопия, 1961,
11, 410.
880. Л а х В. И. и др.— Цветные металлы, 1961, 8, 38.
881. Самсонов Г. В., Кислый П. С. Авт. свид. СССР 126203, 1959.
882. F i и 1 а у G. Пат. США 2529333, 1950.
883. A b b е у А. Англ. пат. 716836, 1954.
884. Приходько Л. И.— Порошковая металлургия, 1966, 1, 17.
885. Finlay G.— J. Electrochem. Soc., 1952, 99, 58.
886. Foster L.— ASM Review of Metal, Literature, 1952, 9, 737.
887. Chem. Eng., 1953, 60, 186.
888. Bush O., Vandergriff R,, Hanley T.— J. Appl. Phys., 1949, 20, 295.
889. Olson E., Layer E., Middleton A.— J. Electrochem. Soc., 1955, 102, 73.
890. Chem. and Eng. News., 1946, 24, 3361.
891. Chem. Abs, 1949, 43, 4957.
892. Chem. Abs., 1950, 44, 10524.
893. Fuson N.— J. Appl. Phys., 1949, 20, 59.
894. M i 11 о n R.— Chem. Rev., 1946, 39, 149.
895. Matthias B.— Phys. Rev., 1953, 92, 874.
896. Самсонов Г. В.— В кн.: Техническая магнитная гидродинамика. «Метал-
лургия», М., 1965, 39.
897. Ясинская Г. А., Коган С. Ф.— Огнеупоры, 1963, 10, 472.
898. Пат. ФРГ 1075839, 1953.
899. Самсонов Г. В.— Кинетика и катализ, 1964, 6, 424.
900. Schonberg N.— Acta Chem. Scand., 1954, 8, 627.
901. Асанова M. П., Герасимов А. Ф., Конев В. Н.— ФММ, 1960, 9, 689.
902. Исаев В. Ф., Морозов А. Н.— Изв. АН СССР, металлургия и горное
дело, 1964, 2, 13.
903. NowotnyH. eta 1.— Mh. Chem., 1961, 92, 403.
904. RudyE., Benesovsky F.— Mh. Chem., 1961, 92, 415.
905. В i 11 n e r H. et a ].— Mh. Chem., 1963, 94, 518.
906. Nowotny H., Benesovsky F., Rudy E.— Mh. Chem., 1960, 91, 348.
907. RabenauA., Eckerlin P.— Naturwiss., 1959, 46, 106.
908. J e f f г e у G. A., W u V. Y — Acta Cryst, 1963, 16, 559.
909. J e f f г e у G. A., W u V. Y.— Acta Cryst., 1966, 20, 538.
910. Brener G., Esselborn R.— Z. anorg. Chem., 1961, 308, 52.
911. J u z a R„ H u n d F.— Z. anorg. Chem., 1961, 257, 276.
912. Juza R., Haug J.— Z. anorg. Chem., 1961, 309, 276.
913. Goubeau J., Anselment W.— Z. anorg. Chem., 1961, 310, 248.
372
914. JuzaR., Heners J.— Z. anorg. Chem., 1964, 332, 159.
915. Juza R-, Friedrichsen H.— Z. anorg. Chem., 1964, 332, 173.
916. Schonberg N.— Acta Metallurgica, 1954, 2, 427.
917. Juza R., Gieren W., Haug J.— Z. anorg. Chem., 1959, 300, 61.
918. Sachse W., Juza R.— Z. anorg. Chem., 1949, 259, 278.
919. Kiessling R., Peterson L.— Acta Metallurgica, 1954, 2, 675.
920. Brauer G.— J. Less-Common Metals, 1960, 2, 131.
921. Stone L., Margolin H.— J. Metals, 1953, 5, 1498.
922. Еременко В. H. Многокомпонентные сплавы титана. Изд-во АН УССР
К-, 1962.
923. Brauer G., Lesser R.— Z. Metallkunde, 1959, 50, 512.
924. J a с k К. H.— Proc. Roy. Soc., 1948, A195, 41.
925. SpernerF., Meixner J.— Z. Metallkunde, 1959, 50, 691.
926. GlemserO. eta 1.— Naturwiss., 1955, 42, 44.
927. G e r i n g.— Z. anorg. Chem., 1955, 278, 53.
928. Kieffer R., Benesovsky F., Lux B.— Planseeberichte, 1956, 4, 30.
929. Nowotny H„ Lux B., Kudielka H.— Mh. Chem., 1956, 87, 447.
930. Turkdogan E. T., Ignatovicz S.— J. Iron Steel Inst, 1958, 188, 242.
931. Казаков В. K-, Городецкий С. С.— Порошковая металлургия, 1966,
4, 65.
932. Приходько Л. И.— В кн.: Вестник Киевского политехи, ин-та, сер. ме-
ханико-технологическая. Изд-во КГУ, К-, 1965, 59.
933. Диаграммы состояния металлических систем, I—VIII. Под ред. Н. В. Агеева.
ВИНИТИ, М, 1959—1964.
934. Пермяков Л. Н. и д р.— Изв. АН СССР, Металлургия и горное дело,
1964, 4, 68.
935. Street R., Waters Т.— J. Less-Common Metals, 1963, 5, 295.
936. Scheil E., Mayr W., Mil Iler J.—Arch. Eisenhuttenwesen, 1962, 33, 385.
937. В о u c h a г d J. P. et al.— C. r., 1966, 262, 640.
938. Laurent Y., Lang J.— C. r., 1966, 262, 103.
939. Умбер Д., Эллиотт Д.— Проблемы современной металлургии, 1961, 1,
(55), 2.
940. В u г d е s е А.— Metallurgia ital., 1955, 47, 357.
941. F a s t J. D., V е г г i j р М. В.— Acta Metall., 1955, 3, 203.
942. Rawlings R., Tambini D.— J. Honstal Inst. Nov., 1956, 302.
943. Ductile Cromium and its alloys, PubL by the American Soc. for Metals, Cleve-
lend, Ohio, 1957.
944. Rawlings R.— J. Iron and Steel Inst., 1957, 185, 441.
945. Turkdogan E. T., Ignatowic S.— J. Iron and Steel Inst., 1956, 185, 200.
946. Пат. США 2750268, 1956; РЖХ, 1958, 9, 325.
947. Пинскер 3. Г., Каверин С. В.— Тр. Ин-та кристаллографии АН СССР,
1956, 12, 3.
948. Toman К.— Czechosl. Journ. Phys., 1956, 6, 5.
949. Stocker H., Niekel H-, Imota S.—Ber. Deutsch. Keram. Ges., 1966,
43 130
950. Stokes C. S., К n i p e W. E.— Eng. Chem., 1960, 52, 287.
951. Meyer H.— Ber. Deutsch. Keram. Ges., 1962, 39, 115.
952. Levinstein M. A.— Metal Finishing Journ., 1960, 6, 72.
953. Seybolt A. H., Orians R. A.— J. Metals Sect., 1956, 11, 8, 556.
954. Конозенко И. Д.— Успехи физ. наук, 1955, 283, 1.
955. Д а л и с о в В. Б., Замиховский В. С., Похмурский В. И.— Физ.
хим. механика материалов, 1966, 2, 173.
956. MabzoneM. G., Briggs J. Z.— J. Less-Common Alloys, of Molybdenum,
Climax Molybdenum Company, 1962, 93.
957. Mikherriee A. K-, Martin J. W.— J. Less-Common Metals, 1960, 2, 392.
373
958. Meyer О., Eilender W.— Arch. Eisenhuttenwesen, 1938, 11, 545.
959. A n s e 1 i n F. et a 1.— C. r., 1965, 19, 174.
960. Ж у p а к о в с к и й Е. А., Дзетановский В. П.—ДАН СССР, 1963, 150,
1260.
961. Но enig С. L., Searcy A. W. Vaper Pressure and Evaporation Coefficient
Studies of the Berillium Nitride (Be3N2), Technical Conference on Compounds of Interest
in Nuclear Reactor Technology, University of Colorado, 1964.
962. В u g 1 J., Bauer A. A. Corrosion and Oxidation Characteristics of Uranium
Mononitride (там же).
963. La pet P. E., Holden E. B. Thermodynamics of the Uranium-Nitrogen
System (там же).
964. Ende Brock R. W., Foster E. L., Keller D. L. Characterization and
Properties of Arc-Melted Uranium Moninitride (там же).
965. Barnighausen H., Volkmann T.— Acta Cryst., 1966, 21, 571.
966. Шулишова О. И.— Изв. АН СССР, Неорг. материалы, 1966, 2, 1434.
967. Bevan D., Lincoln F., Richardson F.— Austral. J. Chem., 1966,
19, 725.
968. Benz R., Zach ariasen W. H.— Acta Cryst., 1966, 21, 838.
969. Morean C., Philippot J.— C. r., 1963, 257, 5366.
970. Самсонов Г. В., Харченко В. К-, С тру к Л. И.— Порошковая метал-
лургия, 1967, 12.
971. ЖураковскийЕ. А., Горский В. В.—ДАН УССР, 1966, 11, 1428.
972. Гаврилюк М. И., Ершова В. Т., Константинов В. А.—Металло-
ведение и термическая обработка металлов, 1966, 12, 37.
973. Белоцкий А. В., Мохорт А. В., Пермяков В. Г.—Изв. вузов, Черная
металлургия, 1966, 5, 147.
974. Janes S., Nixdorf J.— Ber. Deutsch. Keram. Ges., 1966, 43, 136.
975. Кузнецова И. Г., Полубояринов Д. Н.— Огнеупоры, 1967, 3, 48.
976. Jakesova L., Jakes D.— Atomic Energy Rev., 1963, 1, 3.
977. Uranium Carbides, nitrides and silicides, Bibliographicak Series, 14, Internatio-
nal Atomic Energy Agency, Vienna, 1965.
978. Goretzki H. Untersuchung der magnetischen elektrischen und thermo-
elektrischen Eigenschaften der Karbide und Nitride der 4a und 5a-Ubergangsmetalle.
Dissertation, Universitat Wien, 1963.
979. Буслаев Ю. А. и др.— Изв. АН СССР, Неорг. материалы, 1966, 2, 2120.
980. Brauer G., Weidlein J., Strahle J.— Z. anorg. Chem., 1966, 348, 298.
981. HorzG., Gebhardt E.— Z. Metallkunde, 1966, 57, 737.
982. Brauer G., Leibbrandt F.— J. Less-Common Metals, 1967, 12, 57.
983. Laurent Y., Lang J.— C. r., 1966, 263, 340.
984. Horz G., Gebhardt E.— Z. Metallkunde, 1966, 57, 812.
985. ЖураковскийЕ. А,— ДАН УССР, 1967, 3, 240.
986. Brauer G., Weidlein J. R.— Angew. Chem., 1965, 77, 913.
987. Berthold H. J., Delliehausen C.—Angew. Chem., 1966, 78, 750.
988. Гюнтер Ф., Шнайдер X. Г.— Кристаллография, 1966, 11, 683.
989. Tsuchida Т., Wallace W. E.— J. Chem. Phys., 1965, 43, 2885.
990. Стр у к Л. И., Дубовик T. В., Казаков В. К-—Огнеупоры, 1967, 2, 59.
991. Красоткина Н. И.— Огнеупоры, 1967, 6, 33.
992. Lynch R., Drikamer Н.— J. Chem. Phys., 1966, 44, 181.
993. Ettmayer P., V in ere G., Rassaerts H.—Mh. Chemie, 1966, 97, 1258.
994. Petterson F., Ward R.— Inorg. Chem., 1966, 5, 1312.
995. Flasser L, Hog G.— J. Phys. Chem., 1966, 70, 281.
996. Aronson S., Auskern A.— J. Phys. Chem., 1966, 70, 3937.
997. Campbell J., Leary J.— J. Phys. Chem., 1966, 70, 2703.
998. Straumanis M., Faunle C., James W.—Inorg. Crem., 1966,
5, 2024.
374
999. OkomotoU., Goswami L.— Inorg. Chem., 1966, 5, 1281.
1000. Shinziro Wakao, Kiyoshi Yoshimura, Tsuwemi Natehabe,
Ceizo Matsuda, Proc. Facult. of Science of Tokey University, 1966, 1, 83.
1001. Toth L. E., Wang С. P., Yen С. M.— Acta Metallurgica, 1966, 14, 1403.
1002. Krikorian E., Sheed R.— J. Appl. Phys., 1966, 37, 3674.
1003. Pastrnak J., Roskovcova J.— Phys. Stat. Sol., 1966, 14, K5.
1004. Benz R., Bowmen M. G.— J. Amer. Chem. Soc., 1966, 88, 2932.
1005. Olson W., Mulford R.— J. Phys. Chem., 1966, 70, 2832.
.1006 . Doo V. Y., Nishols D. R., Silvey G. A.— J. Electrochem. Soc., 1966
113, 1279.
1007. W h a n D. В. M c.— J. Chem. Phys., 1966, 44, 3528.
1008. JunodP., Levy F.— Phys. Letters, 1966, 23, 11.
1009. В u s c h G. et a 1,— Phys. Letters, 1965, 14, 264.
1010. Junod P., Menth A., Vogt O.— Phys. Letters, 1966, 23, 626.
1011. Fed der R., Cooper B., Schumacher P.— Bull. Amer. Phys. Soc.,
1966, 11, 15.
1012. Atomic U. S. Energy Comiss., PWAC.—481, 1965; C. A. 1966. 65, 1413c.
1013. Sato R.— J. Phys. Soc. Japan, 1966, 21, 1623.
1014. Brit. Patent 1005425, 1965.
1015. Pauling L.— Proc. Nat. Accad. Sci. America, 56, 1646, 1966.
1016. Kazuo Kawabe, Katsumi Yoshino, Yoshio Jnnischi.—J. Phys.
Soc. Japan, 1966, 21, 1604.
1017. Moody L., Thomas J.— Chem. Education, 1966, 43, 205.
1018. W i 11 i a m s M., Balls K., Pickus M.— J. Phys. Chem. Solids, 1967,
28, 333.
1019. Olson W., Mulford R.— J. Phys. Chem., 1965, 69, 1223.
1020. W i s e S. et a L— J. Phys. Chem., 1966, 70, 7.
1021. Ettmayer P.— Mh. Chemie, 1966, 97, 1248.
1022. Benz R., Zachariasen W. H.— Acte Cryst., 1966, 21, 838.
1023. Neuenschwander E.— J. Less-Common, Metals, 1966, 11, 365.
1024. R о u b i n M. P., P a r i s J. M.— C. r„ 1966, 263, 1381.
1025. Bradley R. S., Munro D. C., Whitfield M.— J. inorg. nucl. Chem.,
1966, 28, 1803.
1026. Чижиков Д. M. Кадмий. «Наука», M., 1967.
1027. Чижиков Д. М., Счастливый В. П. Теллур и теллуриды. «Наука»,
М., 1966.
1028. Вольский А. Н., Стерлин Я. М. Металлургия плутония. «Наука»,
М„ 1967.
1029. Тарасов В. В., Демиденко А. Ф., Мальцев А. К-—Неорг. мате-
риалы, 1967, 3, 957.
1030. Печентковская Л. Е., Дубовик Т. В., Назарчук Т. Н.— Неорг.
материалы, 1967, 3, 963.
1031. Портной К- И. и др.— Порошковая металлургия, 1968, 3, 32; 5, 87.
1032. Юргенсон А. А.— Металловедение и термин, обработка металлов,
1967, 7, 2.
1033. М a d а г R. et а L— С. г., 1967, 264, 308.
1034. Straumanis М. Е., Faun се С. А.— Z. anorg. Chem., 1967, 353, 329.
1035. Е с k е г 1 i и F., Rabenan A., Nortmann Н.— Z. anorg. Chem., 1967,
353, 113.
1036. Billy М., Colombeau F — С. г., 1967, 264, 392.
1037. Самсонов Г. В., Падерно Ю. Б., Рудь Б. М.— Изв. вузов, Физика,
1967, 9, 129.
1038. Полубояринов Д. Н., Шишков Н. В., Кузнецова И. Г.— Изв.
АН СССР, Неорг. материалы, 1967, 3, 1828.
1039. Гельд П. В. и др.— Изв. АН СССР, Неорг. материалы, 1967, 3, 1835.
1040. Казаков В. К-—Изв. АН СССР, Неорг. материалы, 1967, 3, 1661.
375
1041. Ansel in F., Preparation et Etude des nitrures et carbonitrunes d'uranium
et de plutonium, commiseriat a 1’energic atomique France, centre d’etudes nucleaires de
Fontenay-aux-Roses, Rapport CEA-R, 988, Juin, 1966.
1042. Kotsch H., Putzky G., Bericht fiber die III. International! Pulvermetal-
lurgische Tagung in Eisenach, vom 13—45 Mai 1965, Akademie-Verlag, Berlin, 1966, 249.
1043. D a v i d J., L a n d J.— C. r„ 1965, 261, 1005.
1044. Roub M.P., ParisJ. М,— C. r., 1966, 262, 1151.
1045. RebouillatJ. P., Veysiie J.— 1964, 259, 4239.
1046. Muller K„ Holl er er H. Konferencja metalurgii proszkow, 20—23. C6.
рефератов 11.IX 1967, часть II, 691, Краков, 1967.
1047. Полищук В. С. Разработка технологии получения нитридов некоторых
металлов. Автореферат дисс., ИПМ АН УССР, К., 1967.
1048. J. Industrial Heating, 1962, 29, 2004.
1049. Taylor A., Doyle N. J.— J. Less-Common Metals, 1967, 13, 299.
1050. Айвазов M. И., Мелехин В. Ф.— Неорг. материалы, 1967, 3, 2109.
1051. Лахтин Ю. М., Коган Я. Д-—Металловедение и термическая обработ-
ка металлов, 1968, 1, 24.
4052. Ames I., Kaplan J. Н., Roland Р. А.— The Review of Scientific Instru-
ments, 1966, 37, 100.
1053. Фр. пат. 1300785; пат. США 34969, 1960.
,1054. Hurd, Lane.— J. Electrochem. Soc., 1957, 104, 12.
1055. N aoumi dis A.— Ber. Kernforschungsanlage Julich, 1967 , 472.
1056. Kieffer R., Ettmayer P., Dubsky T.— Z. Metallkunde, 1967, 58, 560.
1057. Айвазов M. И., Голубев И. И., Дмитриев В. П.— Изв. АН СССР,
Неорг. материалы, 1968, 4, 152.
1058. А н д р е е в а Т. В., Казаков В. К-, Рогоз и иска я А. А.—Изв. АН
СССР, Неорг. материалы, 1968, 4, 54.
1059. Кутолин С. А., Вулих А. И.— В кн.: Методы получения химических
реактивов и препаратов, 16, Изд-во ИРЕА, М., 1967, 52.
1060. Рык л ис Э. А., Болгар А. С., Лютая М. Д., Фесенко В. В.— По-
рошковая металлургия, 1968, 2, 64.
1061. David J., Lang J.— С. г., 1967, 265, 581.
1062. Gebhardt Е., Rothenbach R., Kverens I.— Z. Metallkunde, 1967,
58, 703.
1063. Haschke H., Nowotny H., Benesovsky F.—Mh. Chem. 1967, 98,
2157.
1064. Лашко H. Ф., Глазова А. И., Гуськова E. И.— Изв. АН СССР,
Металлы, 1968, 2, 224.
1065. Клевцур С. А., Седельников Т. X.— Изв. АН СССР, Неорганические
материалы, 1968, 4, 604.
1066. М о р о з о в а М. П., Гальбрайх Э. И., Смирнова В. А.—Изв. АН
СССР, Неорганические материалы, 1968, 4, 523.
1067. О р монт Б. Ф.— Изв. АН СССР, Неорганические материалы, 1968, 4, 611.
'1068. Стасовская В. В.— Автореф. канд. дисс., ИПМ АН УССР, К., 1967.
1069. Ш у л и ш о в а О. И.— В кн.: Химия и физика нитридов. «Наукова думка»,
К., 1968, 157.
.1070 . Самсонов Г. В.— Физ.-хим. механика материалов, 1968, 5, 502.
1071. Кутолин С. А., Вулих А. И.— В кн.: Промышленность химических реак-
тивов и особо чистых веществ. Изд. ИРЕА, М., 1968, 13. (19), 26.
1072. Laurent Y., Lang J., Le Bihan M. Th.— Acta Cryst., 1968, B24, 494.
1073. Лютая M. Д., Вдовенко C. A.— В кн.: Химия и физика нитридов.
«Наукова думка», К-, 1968, 101.
1074. Seybold D., Denicke К.— Z. anng. Chem., 1968, 361, 277.
1075. Лютая М. Д., Гончарук А. Б.— В кн.: Химия и физика нитридов.
«Наукова думка», К-, 1968, 62.
376
1076. Ария С. М., Морозова М. П., Хернбург М. М.— В кн.: Химия и
физика нитридов. «Наукова думка», К-, 1968, 130.
1077. Францевич И. Н., Лященко А. Б.—ДАН УРСР, 1968, сер. А, 11, 1051.
1078. Айвазов М. И., Домашней И. А.— Порошковая -металлургия 1968
9, 51.
1079. Курганов Г. В., Левинский Ю. В. и др.— В кн.: Химия и физика
нитридов. «Наукова думка», К-, 1968, 47.
1080. Ondracek G., Petrow G.— J. Nuclear Materials, 1968, 25, 132.
1081. Gebhardt E., Rothenbacher R., Kverens I.— Z. Metallkunde, 1967
58, 790.
1082. Roubin M., Paris I. M.— J. Less-Common Metals, 1968, 15.
1083. Blum P. L., L a n g i e r J., M a r t i n J. M., M о r 1 e v a t J. P.— C. r., 1968
266, 1456.
1084. T h о m a s I., Weston J., O’Connor T.— J. Am. Chem. Soc,, 1963
84. 4619.
1085. Хусидман M. Б., Шару пин Б. H.— Радиохимия, 1967, 9, 279.
1086. Хусидман М. Б., Нешпор В. С.— физ. твердого тела, 4968, 10, 1229.
1087. Хусидман М. Б., Нешпор В. С.— Теор. и эксп. химия, 1967, 2, 270.
1088. Файн ер И. С. Автореф. канд. дисс., ИПМ АН УССР, К-, 1969.
1089. Джузеев У. Д., Рамазанов П. Е.— Изв. вузов, физика, 1968, 7, 74.
4090. Самсонов Г. В., Харченко В. К., С тру к Л. И.— Порошковая ме-
таллургия, 1968, 3, 59.
4091. Афанасьев В. Ф., Карп иное Д. М.— Порошковая металлургия,
1966, 7, 49.
4092. Душин Ю. А., Дмитриев А. В.— Изв. АН СССР, Неорг. матер., 1968,
4, 1596.
4093. S t a k е г D.— Proc. Roy. Soc., 1962, 270, 397.
1094. Ниденцу К-, Даусон Дж. Химия боразотных соединений, «Мир», М.,
1968, 227.
1095. RodewaldH. I.— Chimia, 1960, 14,162.
1096. RodewaldH. I.— Chimia, 1961, 15, 251.
1097. Филоненко H. E. и др.— Труды ВНИИАШ, 1966, 2, 5.
1098. Никитина Г. П., Филоненко Н. Е.— Труды ВНИИАШ, 1967, 5, 16.
1099. Кузнецова И. Г., Полубояринов Д. Н.— В кн.: Химия и физика
нитридов. «Наукова думка», К-, 1968, 105.
1100. Падерно Ю. Б., Андреева Т. В. и др.— В кн.: Химия и физика
нитридов. «Наукова думка», К, 1968, 168.
1101. Болгар А. С. и др.— В кн.: Химия и физика нитридов. «Наукова дум-
ка», К., 1968, 151.
1102. Миронов И. А., Строева И. А.— В кн.: Химия и технология люмино-
форов. «Химия», Л., 1968, 71.
ПОЗ. Купер К- Ф- и др.— В кн.: Специальная керамика. «Металлургия», М.,
1968 39.
’ 1104. Плетюшкин А. А., Славина Н. Г.— Изв. АН СССР, Неорг. матер.,
1968, 4, 893.
1105. Славина Н. Г., Плетюшкин А. А.— В кн.: Химия и физика нитридов.
«Наукова думка», К-, 1968, 90.
1106. Фесенко В. В., Болгар А. С. Испарение тугоплавких соединений.
«Металлургия», М., 1966.
1407. Pehlke, Elliot J.— Trans. Met. Soc. AIME, 1959, 215, 781.
1108. Wolff E., Alcock C.—-Trans. Brit. Ceram. Soc., 1962, 61, 667.
1109. Артамонов А. Я. и др.— Порошковая металлургия, 4967, 9, 58.
1110. А р т а м о н о в А. Я., Джафаров Я. А.— Порошковая металлургия,
1967, 12, 78.
1111. Катанов Л. М., Конев В. Н.—-В кн.: Химия и физика нитридов.
«Наукова думка», К-> 1968, 172.
377
1112. П р 11 х о д ь к о Л. И.— В кн.: Химия и физика нитридов. «Наукова дум-
ка», К., 1968, 84.
1113. Приходько Л. И., Соболевский Л. И.— Порошковая металлургия
1968, 12, 22.
1114. Андреева Т. В., Казаков В. К., Рогозинская А. А.— Теплофиз.
выс. темп., 1967, 5, 612.
1115. А и д р е е в а Т. В., Казаков В. К., Рогозинская А. А.— Изв. АН
СССР, Неорг. матер., 1967, 3, 1418.
1116. Самсонов Г. В., Казаков В. К.— Огнеупоры, 1965, 7, 29.
1117. Городецкий С. С., Казаков В. К.— Изв. АН СССР, Неорг. матер.,
1968, 4, 1067.
1118. Рык лис Э. А., Болгар А. С., Фесенко В. В.— Порошковая метал-
лургия, 1969, 1, 95.
1119. Л е в и н с к и й Ю. В. и др.— Изв. АН СССР, Неорг. матер., 1968, 4, 2068.
ОГЛАВЛЕНИЕ Предисловие............................................. 3
Введение.................................... 5
ГЛАВА I. СТРУКТУРА И СВОЙСТВА НИТРИДОВ
1. Электронная и кристаллическая структуры ни-
тридов ..................................... 7
2. Физические свойства нитридов ..... 18
3. Термодинамика нитридов *..................42
4. Классификация нитридов...................67
ГЛАВА II. СПОСОБЫ ПРИГОТОВЛЕНИЯ НИТРИДОВ (69)
ГЛАВА III. НИТРИДЫ МЕТАЛЛОВ I ГРУППЫ ПЕРИО-
ДИЧЕСКОЙ СИСТЕМЫ
1. Нитриды щелочных металлов.................81
2. Нитриды металлов подгруппы меди .... 88
ГЛАВА IV. НИТРИДЫ МЕТАЛЛОВ II ГРУППЫ ПЕРИО-
ДИЧЕСКОЙ СИСТЕМЫ
1. Нитриды бериллия, магния и щелочноземельных
металлов............................... 92
2. Нитриды металлов подгруппы цинка .... 106
ГЛАВА V. НИТРИДЫ ПЕРЕХОДНЫХ МЕТАЛЛОВ
1. Нитриды редкоземельных металлов (скандия,
иттрия и лантаноидов)...................ПО
2. Нитриды переходных металлов IV группы . . 133
3. Нитриды переходных металлов V группы . .172
4. Нитриды переходных металлов VI группы . .192
5. Нитриды переходных металлов VII группы . . 206
6. Нитриды металлов семейства железа .... 212
7. Нитриды платиноидов......................218
8. Нитриды актиноидов.....................218
ГЛАВА VI. НИТРИДЫ ЭЛЕМЕНТОВ ПОДГРУППЫ
БОРА
1. Нитриды бора.............................231
2. ( Нитриды алюминия.......................259
3. Нитриды галлия.........................276
4. Нитриды индия............................279
5. Нитриды таллия...........................281
ГЛАВА VII. НИТРИДЫ ЭЛЕМЕНТОВ ПОДГРУППЫ УГ-
ЛЕРОДА
1. Нитриды кремния..........................282
2. Нитриды германия.......................295
3. Нитриды олова и свинца...................296
ГЛАВА VIII. НИТРИДЫ ЭЛЕМЕНТОВ ПОДГРУППЫ
АЗОТА И ПРОЧИХ ЭЛЕМЕНТОВ
1. Соединения фосфора с азотом (нитриды фос-
фора) .....................................297
2. Соединения мышьяка, сурьмы и висмута с азо-
том .......................................298
3. Соединения элементов подгруппы кислорода с
азотом.....................................298
4. Соединения галогенов с азотом [727] . . . 302
ГЛАВА IX. СЛОЖНЫЕ НИТРИДЫ
1. Системы металлов с азотом...............303
2, Системы металл—бор—азот.................314
3. Системы металл—углерод—азот.............314
4. Системы металл—кислород—азот............322
5. Системы металл—галоген—азот.............328
6. Системы металл—кремний—азот.............329
ГЛАВА X. НИТРИДНЫЕ ПОКРЫТИЯ (333)
ГЛАВА XI. ОБЛАСТИ ПРИМЕНЕНИЯ НИТРИДОВ (345)
Литература.................................352