/








































































































































































































































































































































































































































































































































































































Author: Глумчер Ф.С. Трещинский А.И.
Tags: различные формы заболеваний, методы их лечения, применения лекарственных средств фармакология общая терапия токсикология медицина интенсивная терапия
ISBN: 966-642-227-1
Year: 2004




Text
Данный файл представлен исключительно в ознакомительных целях.
Все авторские права на данный файл сохраняются за правообладателем. Любое коммерческое и иное использование кроме предварительного ознакомления запрещено.
Публикация данного документа не преследует никакой коммерческой выгоды. Но такие документы способствуют быстрейшему профессиональному и духовному росту читателей и являются рекламой бумажных изданий таких документов.
Все авторские права сохраняются за правообладателем.
DykoBogcmBo
Гпо интенсивной
терапии
Под редакцией профессоров А. И. Трещинского, Ф. С. Глумчера
Пособие для врачей-анестезиологов, реаниматологов, врачей отделений интенсивной терапии
KI/IIB «В1/1ЩА ШКОЛА» 2004
УДК 616-036.882:615(075.8)
ББК 53.5я73
Р85
Рекомендовано до видання Проблемною KOMicieio «Анестезюлог1я та шпгенсивна тератя» МОЗ та АМН Украши (протокол eid 30 травня 2003 р. № 2)
Авторы: д-р мед. наук, проф. А. В. Беляев, канд. мед. наук, доц. М. В. Бондарь, канд. мед. наук, доц. А. М. Дубов, д-р мед. наук, проф. Ф. С. Глумчер, д-р мед. наук, проф. Е. Н. Клигуненко, канд. мед. наук, доц. И. В. Кузнецова, д-р мед. наук, проф. Л. А. Мальцева, канд. мед. наук Н. Ф. Мосенцев, чл.-кор. АМН Украины, проф. В. Ф. Москаленко, канд. мед. наук К. Н. Олейников, д-р мед. наук, проф. В. В. Суслов, М. А. Трещин-ская, д-р мед. наук, проф. А. И. Трещинский, чл.-кор. НАН и АМН Украины, проф. Л. В. Усенко, канд. мед. наук Л. А. Харченко, д-р мед. наук, проф. А. А. Хижняк, чл.-кор. АМН Украины, проф. В. И. Черний
Рецензе нт и: д-р мед. наук, проф. О. С. Владика (Одеський держав-ний медичний утверситет), д-р мед. наук, проф. Г. I. Белебезьев (Кшвська медична академ!я шслядипломно! осв!ти iM. П. Л. Шу пика)
Викладено сучасш методи штенсивноТ терапй хворих у критичному стан!, способи запоб!гання розвитку критичних сташв при багатьох захворюваннях.
Для л!кар!в-анестез!олопв, реашматолопв, л!кар!в в!дд!лень жтенсивно!’ Tepanii*, а також л!кар!в шших спец!альностей, як! беруть участь у лшуванш тяжкохворих.
Редактор О. Г. Молдованова
Руководство по интенсивной терапии: [Пособие] / А. В. Беляев, Р85 М. В. Бондарь, А. М. Дубов и др.; Под ред. А. И. Трещинского, Ф. С. Глумчера. — К.: Вища шк., 2004. — 582 с.: ил.
ISBN 966-642-227-1
Изложены современные методы интенсивной терапии больных в критическом состоянии, способы предупреждения развития критических состояний при многих заболеваниях.
Для врачей-анестезиологов, реаниматологов, врачей отделений интенсивной терапии, а также врачей других специальностей, принимающих участие в лечении тяжелобольных.
УДК 616-036.882:615(075.8) ББК 53.5я73
© А. В. Беляев, М. В. Бондарь, А. М. Дубов, Ф. С. Глумчер, Е. Н. Клигуненко, И. В. Кузнецова, Л. А. Мальцева, Н. Ф. Мосенцев, В. Ф. Москаленко, К. Н. Олейников, В. В. Суслов, М. А. Трещинская, А. И. Трещинский, Л. В. Усенко, Л. А. Харченко,
ISBN 966-642-227-1 А. А. Хижняк, В. И. Черний, 2004
СПИСОК ПРИНЯТЫХ СОКРАЩЕНИИ И УСЛОВНЫХ ОБОЗНАЧЕНИЙ
ВЕ — сдвиг буферных оснований ЕТ — эндотелиальный пептид GP — гликопротеид
ISS — шкала тяжести повреждений NF-кВ — ядерный фактор-каппа Б
NPY — нейропептид Y
Pg — простагландин
RAF — фактор активации тромбоцитов ТХ — тромбоксан АБ — антибиотик
АД — артериальное давление
АДГ — антидиуретический гормон
АКЛ — антикардиолипиды
АКТГ — адренокортикотропный гормон АЛТ — аланинаминотрансфераза
АНД — антикоагулянты непрямого действия
АПФ — ангиотензинпревращающий фермент
ACT — аспартатаминотрансфераза
АФЕ — антифибринолитические единицы аФЛ — антифосфолипидные антитела АФС — антифосфолипидный синдром АХП — антибактериальная химиопрофилактика
АХТ — антибактериальная химиотерапия АЧТВ — активированное частичное тромбопластиновое время
АШ — анафилактический шок БА — бронхиальная астма
БАВ — биологически активные вещества БОВ — белки острой фазы воспаления
ВА — волчаночный антикоагулянт
ВАП — вентилятор-ассоциированная пневмония
ВБД — внутрибрюшное давление
ВБП — внебольничная пневмония
ВГ — вирусный гепатит
ВИЧ — вирус иммунодефицита человека ВП — внутрибольничная пневмония
ВПГ — вирус простого герпеса
ВПГМ — вторичные повреждения головного мозга
ВХ — внутрипеченочный холестаз ВЧГ — внутричерепная гипертензия ВЧД — внутричерепное давление ГБО — гипербарическая оксигенация ГВК — гипоксическая вазоконстрикция
ГГ — гипоксическая гипоксия
ГД — гемодиализ
ГемГ — гемическая гипоксия
ГЗТ — гиперчувствительность замедленного типа
ГК — глюкокортикоиды
ГР — гипертонический раствор натрия хлорида
ГС — гемосорбция
ГСЗ — гнойно-септические заболевания
ГСП — гемоспленоперфузия
ГФ — гемофильтрация
ГШ — геморрагический шок
ГЭБ — гематоэнцефалический барьер ГЭК — гидроксиэтилированный крахмал ГЭЛ — газовая эмболия легких
ДВС — диссеминированное внутрисосудистое свертывание
ДЗЛА — давление заклинивания в легочной артерии
ДЗЛК — давление заклинивания в легочных капиллярах
ДЛА — давление в легочной артерии ДО — дыхательный объем
4 Список принятых сокращений и условных обозначений
2,3-ДФГ — 2,3-дифосфоглицерат ДЦ — дыхательный центр ЖЕЛ — жизненная емкость легких ЖЭ — жировая эмболия
ИБС — ишемическая болезнь сердца
ИВЛ — искусственная вентиляция легких ИЛ — интерлейкин(ы)
ИОПСС — индекс общего периферического сопротивления сосудов
ИРГТ — интегральная реография тела
ИРНХ — изотонический раствор натрия хлорида
ИСЛС — индекс сопротивления легочных сосудов
ИТП — индекс тяжести поражения
ИТТ — инфузионно-трансфузионная терапия
ИУР — индекс ударной работы
КДД — конечное диастолическое давление КДО — конечный диастолический объем
КЛ — контузия легких
КМ — контузия миокарда
КОД — коллоидно-осмотическое давление КОЕ — колониеобразующие единицы КОС — кислотно-основное состояние
КС — кесарево сечение
КФК — креатинфосфокиназа
ЛДГ — лактатдегидрогеназа
ЛИИ — лейкоцитарный индекс интоксикации
ЛС — легочный сурфактант
ЛУ — лимфатический узел
МА — метаболический ацидоз
МАО — моноаминоксидаза
МАл — метаболический алкалоз
ME — международные единицы
МИК — минимальная ингибирующая концентрация
ММЖ — межклеточная мозговая жидкость МОД — минутный объем дыхания
МОК — минутный объем кровообращения
МОС — минутный объем сердца
МПД — мозговое перфузионное давление
МСМ — молекулы средней массы
МТ — масса тела
НА — норадреналин
НИ — наркозный индекс
НИВ — неинвазивная вентиляция
НМГ — низкомолекулярный гепарин
НПВП — нестероидные противовоспалительные препараты
НФГ — нефракционированный гепарин НШ — нейрогенный шок ОВГ — острый вирусный гепатит ОДП — ожог дыхательной поверхности ОЖГ — острый жировой гепатоз ОИМ — острый инфаркт миокарда
ОИТ — отделение интенсивной терапии ОНД — острая недостаточность дыхания ОПЛ — острое повреждение легких
ОПН — острая почечная недостаточность ОППН — острая печеночно-почечная недостаточность
ОПСС — общее периферическое сопротивление сосудов
ОРДС — острый респираторный дистресс-синдром
ОФВ — объем форсированного выдоха ОФО — острая фаза ответа
ОЦК — объем циркулирующей крови ОЦП — объем циркулирующей плазмы
ПАРС — полиаденозиндифосфатрибозо-син-тетаза
ПВ — протромбиновое время
ПД — перитонеальный диализ
ПДКВ — положительное давление в конце выдоха
ПДФ — продукты деградации фибрина ПДФФ — продукты деградации фибрина и фибриногена
ПИП — парентеральное искусственное питание
ПК — пищеварительный канал ПлС — плазмосорбция
ПНУФ — предсердный натрийуретический фактор
ПОЛ — пероксидное окисление липидов
ПОН — полиорганная недостаточность ППТ — площадь поверхности тела ПСП — плазмоспленоперфузия
ПФ — плазмаферез
ПФОС — перфторанорганические соединения
ПХ — периферические хеморецепторы ПЦР — полимеразная цепная реакция ПЯН — полиморфноядерные нейтрофилы РАл — респираторный алкалоз
РАП — респиратор-ассоциированная пневмония
РАСК — регуляция агрегатного состояния крови
САД — среднее артериальное давление СВ — сердечный выброс
Список принятых сокращений и условных обозначений 5
СДИ — синдром дыхательного истощения СДПД — спонтанное дыхание с положительным давлением
СЗП — свежезамороженная плазма
СИ — сердечный индекс
СКВ — системная красная волчанка
С КФ — скорость клубочковой фильтрации
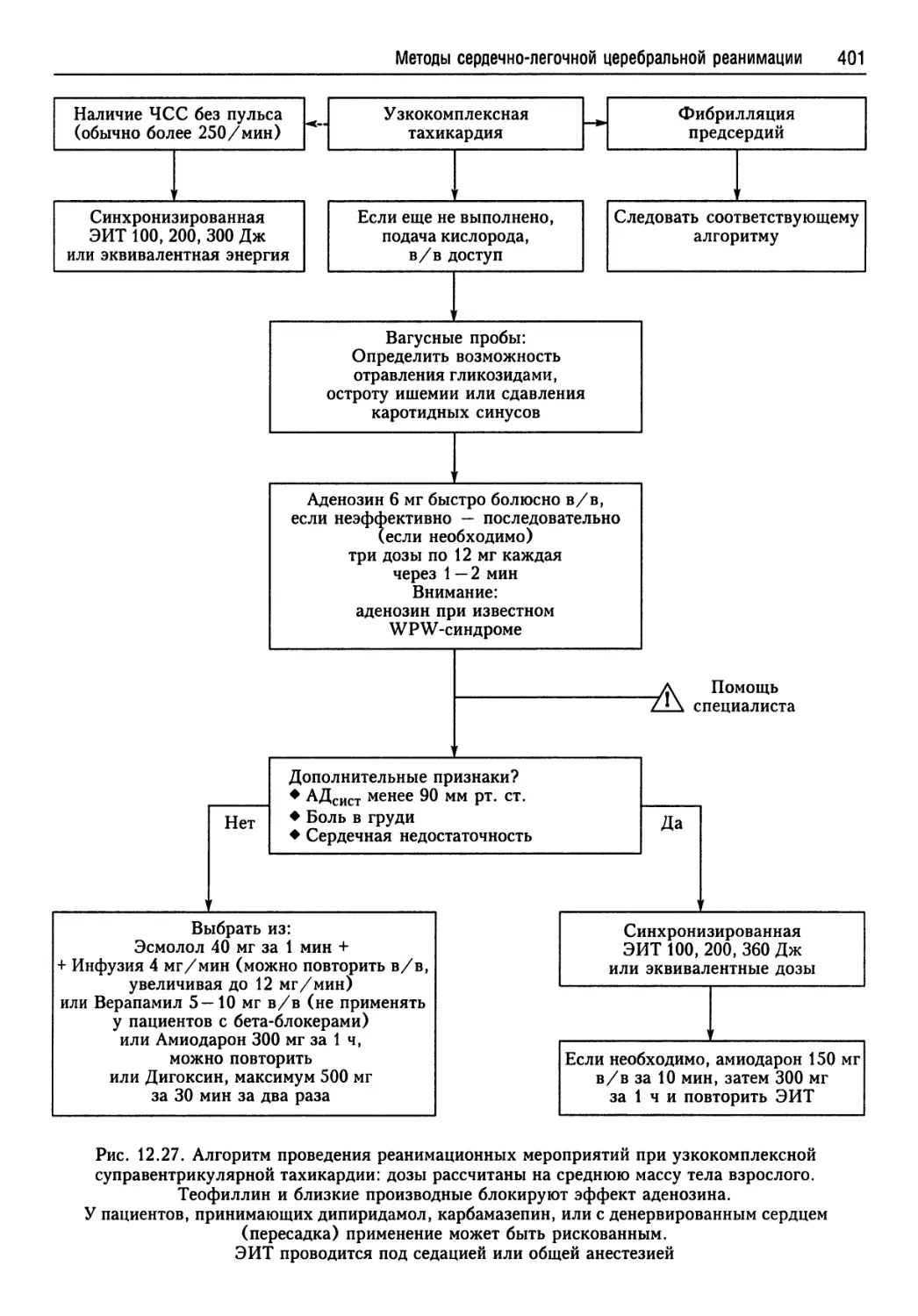
СЛЦР — сердечно-легочная церебральная реанимация
СМФ — система мононуклеарных фагоцитов
СОПЛ — синдром острого повреждения легких
СП — спленоперфузия
СПИ — спинномозговая жидкость
СПОН — синдром полиорганной недостаточности
СРВ — С-реактивный белок
ССВО — синдром системного воспалительного ответа
СШ — септический шок
ТАТ — тромбин-антитромбиновый комплекс
ТГВ — тромбоз глубоких вен
ТД — тканевое дыхание
ТШС — токсический шоковый синдром
ТЭЛА — тромбоэмболия легочной артерии
УЗИ — ультразвуковое исследование
УИ — ударный индекс сердца, мл/м2 поверхности тела больного
У ОС — ударный объем сердца
ФБС — фибробронхоскопия
ФВПМ — факторы вторичного повреждения мозга
ФЕ — фибринолитические единицы
ФЖЕЛ — форсированная жизненная емкость легких
ФНО — фактор некроза опухоли
ФОЕ — функциональная остаточная емкость
ФП — фармакологическое питание ХНЗЛ — хронические неспецифические заболевания легких
ХОЗЛ — хроническое обструкционное заболевание легких
ХПН — хроническая почечная недостаточность
ЦВД — центральное венозное давление
ЦВК — центральный венозный катетер ЦГ — циркуляторная гипоксия
ЦИК — циркулирующие иммунные комплексы
ЦМВ — цитомегаловирус
ЦПД — центральное периферическое давление
ЦХ — центральные хеморецепторы ЧДД — частота дыхательных движений ЧСС — частота сердечных сокращений ЧТВ — частичное тромбопластиновое время
ШКГ — шкала ком Глазго
ЩФ — щелочная фосфатаза
ЭДО — электрохимическая детоксикация организма
ЭКГ — электрокардиограмма
ЭЛОК — эндоваскулярное лазерное облучение крови
ЭнИ — эндогенная интоксикация
ЭОП — эндогенные опиоидные пептиды
ВВЕДЕНИЕ
Термин «интенсивная терапия» (ИТ) за последние 20 — 25 лет стал очень популярным. Почти во всех клиниках и больницах существуют отделения или палаты интенсивной терапии. ИТ используют практически во всех клинических отраслях.
Во многих странах появились циклы постдипломной подготовки врачей по интенсивной терапии в той или иной клинической отрасли. По вопросам интенсивной терапии регулярно проводятся конгрессы, конференции, симпозиумы.
Интенсивная терапия является наиболее молодым, новым разделом медицины, зародившимся на основе реаниматологии, научных исследований и клинического опыта лечения постреанимационных и критических состояний. В ИТ использу
ются достижения целого ряда смежных теоретических и клинических разделов медицины и других отраслей науки: физики, математики, кибернетики, информатики и т. д. ИТ развивается на стыке теоретических разделов медицины с клиническими, что способствует эффективности лечения наиболее тяжелобольных в критическом и терминальном состояниях — пограничных между жизнью и смертью.
Распространение ИТ в клинических отраслях медицины дало основание в ряде случаев отождествлять ее с интенсивной медициной. Образно определяя ИТ, можно сказать, что это терапия, которая позволяет не только удержать больного на краю жизни, но и увести его от «пропасти» смерти.
ИНТЕНСИВНАЯ ТЕРАПИЯ КАК НАУКА
Согласно определению В. А. Неговско-го (1978), реаниматология — это наука об оживлении организма, патогенезе, лечении и профилактике терминальных состояний, а интенсивная терапия является разделом реаниматологии о профилактике реанимации, т. е. о том, что нужно делать, чтобы критическое состояние не перешло в терминальное, а терминальное — не закончилось бы клинической смертью.
Анализ отечественной и зарубежной литературы, а также многолетний собственный клинический опыт позволяют определить ИТ как постоянный контроль за био
химическими показателями и функциональным состоянием жизненно важных органов и систем, своевременное выявление ведущего этиопатогенетического звена болезни и факторов, обуславливающих тяжесть состояния больного, для выбора целенаправленного адекватного лечения. Согласно этому определению, одним из главных компонентов ИТ является интенсивное наблюдение с целью получения информации в динамике о характере и глубине патофизиологических и биохимических нарушений, что способствует выбору лечения. Таким образом, интенсив
Методы интенсивной терапии 7
ная терапия — это комплекс лечебно-диагностических действий, направленных на профилактику клинической смерти.
Уточняя определение В. А. Неговского, необходимо подчеркнуть, что не ИТ составляет раздел реаниматологии, а, наоборот, последняя является разделом ИТ.
В комплексе лечебно-диагностических мероприятий ИТ можно выделить три компонента. Первым и главным из них является интенсивное наблюдение за состоянием больного.
Второй компонент — это собственно терапия, избираемая на основе анализа собранной в динамике информации о состоянии больного.
Третий компонент составляет интенсивный уход, так как больные в критическом состоянии не способны самостоятельно обеспечить все физиологические отправления для поддержания жизни и необходимый санитарно-гигиенический режим.
Основа целенаправленного и адекватного лечения — постоянное получение информации в динамике об особенностях,
характере и глубине функциональных нарушений и метаболических расстройств. Особенностью этого компонента ИТ является необходимость получения большого объема информации постоянно, в динамике, при данной патологии в данный момент и у данного больного, поскольку при одной и той же патологии, но в разные периоды развития и течения заболевания, могут возникать функциональные расстройства и биохимические нарушения, требующие различных подходов и средств для их лечения. Например, при инфекционно-токсическом и гиповолемическом шоке вначале наблюдается уменьшение, а затем — увеличение интерстициального объема.
Информация требуется для получения не отдельного показателя, отражающего то или иное звено процесса, а для представления о характере и степени тяжести процесса в целом, поэтому следующим этапом комплекса интенсивного наблюдения является анализ полученных при наблюдении показателей.
МЕТОДЫ ИНТЕНСИВНОМ ТЕРАПИИ
При критических состояниях все клинические симптомы нарушения физиологических и биохимических процессов можно разделить на четыре группы.
Первая — воздействие этиологического фактора, вызвавшего поражение органа или системы.
Вторая — стресс-реакция организма на воздействие экстремальных факторов, обуславливающих критические состояния. К этой группе относятся однотипные, специфические для стресс-реакции симптомы функциональных и метаболических расстройств.
Третья — нарушения, связанные с адаптационно-компенсаторной реакцией организма. Они имеют определенную специфичность и требуют индивидуального подхода к их оценке.
Поскольку и вторая, и третья группы процессов реализуются через соответствующие звенья регулирующих систем, то
может определяться и четвертая группа симптомов, обусловленная изменениями функционального состояния нейрогумо-ральной системы регуляции.
Для целенаправленной, адекватной терапии необходимо анализировать и оценивать полученную в процессе наблюдения за больным информацию, дифференцировать указанные группы симптомов функциональных и метаболических нарушений, так как одни из них в данной ситуации и в данный момент желательно как можно быстрее устранить или ослабить для спасения жизни или восстановления нарушенных функций, другие, наоборот, сохранить, поддержать.
Оценка состояния по отдельным показателям чревата возможными ошибками, сказывающимися на выборе и эффективности лечения. Поскольку физиологические и биохимические процессы взаимосвязаны, что в ряде случаев может быть вы
8 ВВЕДЕНИЕ
ражено математически, то в определенных ситуациях по одному или нескольким показателям процесса можно рассчитать показатели его сопряженных звеньев (номограмма для определения показателей кислотно-основного состояния (формула Гендерсона — Гассельбаха), методы расчета показателей внешнего дыхания по данным спирометрии и т. д.).
Особое значение для целенаправленного лечения различных вариантов критических состояний имеет информация о показателях водно-электролитного обмена. Основным компонентом внутренней среды является вода, движение которой предполагает ее равновесное распределение в содержащих жидкость пространствах, чей объем может быть установлен с помощью номограммы или расчетных формул.
Стресс является неотъемлемой реакцией организма при критическом состоянии любого происхождения. Косвенное представление о его выраженности можно получить, зная суточную потерю белка (СПБ): СПБ = (суточное количество мочевины х 46,6)/80, где 46,6 — суточное количество азота, г.
Умножая на коэффициент 6,25, соответствующий массовому содержанию (16 %) азота в белке, можно рассчитать количество катаболизированных белков (г).
Большой объем информации, необходимой для выбора направления интенсивной терапии, а также характеристики гемодинамики, обмена воды, характера и глубины нарушений выделительной функции почек, можно получить при рутинном исследовании мочи.
При отсутствии данных о диурезе можно рассчитать экскретируемую (ЭФ) и реабсорбируемую (РФ) фракции воды:
ЭФ = ^креат ; РФ = 10()Г1 ^креат \ ^креат)
где Ркреат — концентрация креатинина в плазме крови; С7креат — концентрация креатинина в моче.
Интегральным показателем концентрационной функции почек может служить концентрационный коэффициент (КК), вычисляемый по формуле
КК = ^>ОСМмочи Росмпл где Росммочи и Росмпл — осмотическое давление соответственно мочи и плазмы крови.
Значение КК ниже 2 свидетельствует о снижении функции почек, а величина, близкая к единице, при олигоанурии указывает на развитие почечной недостаточности.
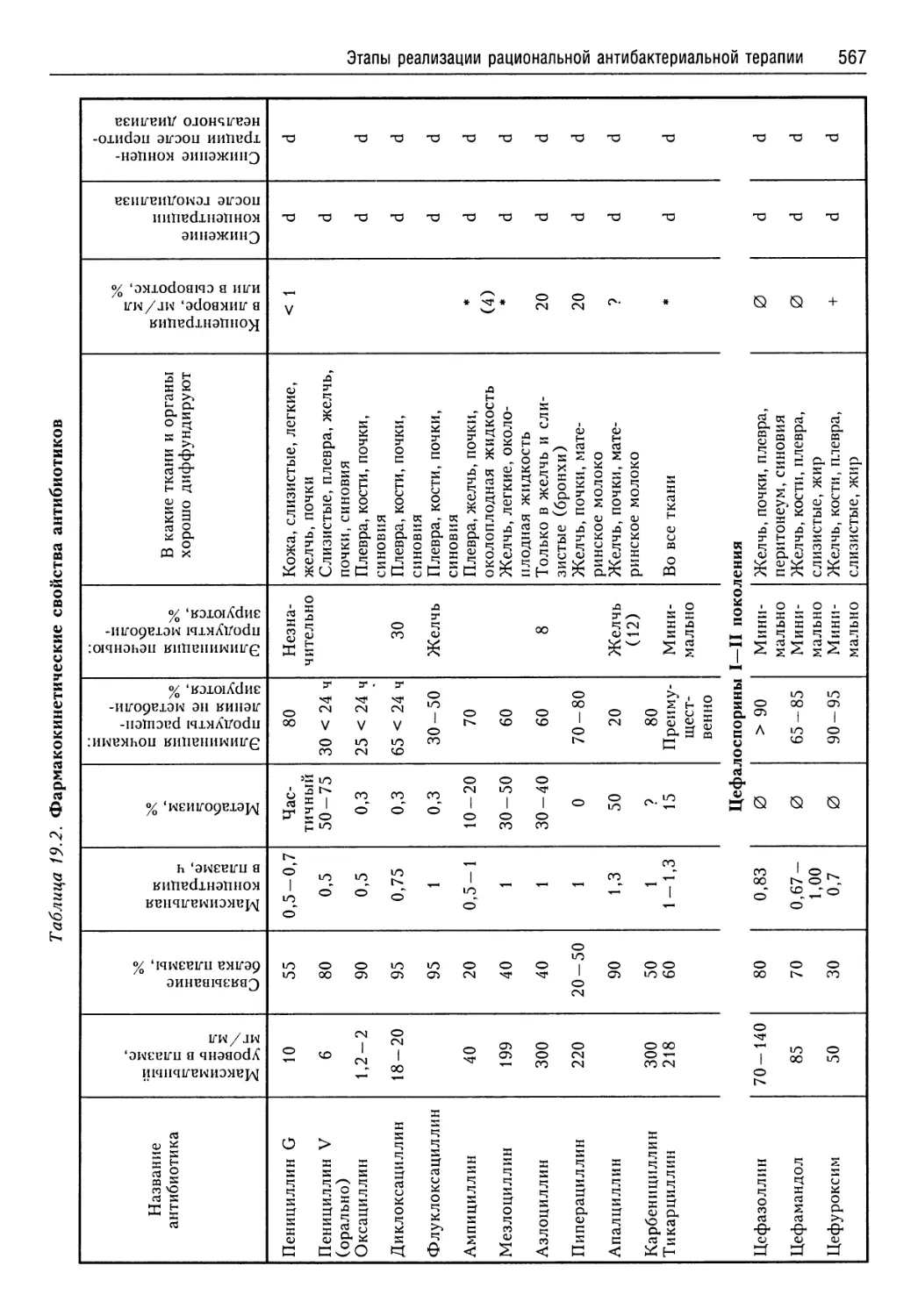
Для выяснения этиопатогенеза критического состояния и выбора целенаправленной терапии требуется огромный объем информации, для получения которой нужны диагностическая аппаратура и реактивы, многие из которых являются зарубежными, а следовательно, дорогими. Поэтому большинство больниц Украины не в состоянии осуществлять мониторинг на необходимом уровне. Вследствие этого расчетные методы сдвигов функциональных и биохимических показателей являются вполне приемлемыми. Естественно, что показатель ошибочности их значительно выше, чем у тех величин, которые могли быть получены при методах прямого определения. С этой точки зрения заслуживает внимания компьютерная программа расчетного получения функциональных и биохимических показателей, разработанная А. В. Малыхиным с соавт. (2002). Используя эту программу, при введении 11 показателей лабораторного анализа крови и сведений о больном можно получить 91 показатель функционального состояния органов и систем, а также метаболизма.
Наряду с информацией, получаемой в процессе непосредственного клинического и инструментального наблюдения, биохимических исследований, по формулам и компьютерным программам, разработанным на основе математического моделирования физиологических и биохимических процессов, можно рассчитать целый ряд дополнительных показателей, которые помогут обеспечить топическую диагностику нарушений кровообращения, газообмена, водно-электролитного обмена, коллоидно-осмотического и кислотно-основного состояний и других звеньев физиологических и метаболических процессов, при помощи которых можно выделить группы па
Направления интенсивной терапии 9
тофизиологических и биохимических сдвигов, обусловленных воздействием этио-патогенетического фактора, стресса, компенсаторно-адаптационными реакциями,
и определить, какие сдвиги подлежат целенаправленному воздействию для устранения критического состояния, поддержания и коррекции функций организма.
НАПРАВЛЕНИЯ ИНТЕНСИВНОМ ТЕРАПИИ
В интенсивной терапии выделяют такие направления:
♦ коррекция функциональных расстройств и биохимических сдвигов;
♦ детоксикация биологических сред организма;
♦ временная замена утраченных функций пораженных органов и систем;
♦ повышение устойчивости к воздействию болезнетворного агента или к последствиям патологического процесса;
♦ управление (поддержание) функции пораженных и интактных органов и систем с целью ослабления последствий патологического процесса и создания условий для восстановления нарушенных функций.
Каждое направление ИТ имеет определенные показания к его использованию и располагает соответствующими методами.
К методам первого направления ИТ относят:
1. Варианты инфузионной терапии с использованием различного состава инфузионных сред для коррекции водносолевого обмена, кислотно-основного состояния, волемических показателей, кислородно-транспортных свойств крови, коллоидно-осмотического давления и т. д.
2. Медикаментозное воздействие непосредственно на органы и системы с нарушенной функцией (допамин, сердечные гликозиды, диуретики, нифедипин, немоди-пин, аминалон и др.).
3. Медикаментозное воздействие на пораженные органы через регулирующие системы (холино- и адреномиметики: хо-линхлорид, эпинефрин, норэпинефрин; холино- и адренолитики (а и р), целенаправленные аналептики и т. д.).
4. Коррекция клеточного метаболизма, нарушение которого запускает патологический процесс (инсулин, глюкокортикоиды, этанол при отравлении этиленглико
лем, метанолом и т. д.). Использование методов коррекционного направления требует соблюдения определенных правил:
1. Коррекции подлежат только те нарушения, при нормализации которых обеспечивается восстановление физиологического или биохимического процесса, а не отдельного показателя. Например, при геморрагическом, гиповолемическом шоке мы наблюдаем тахикардию, снижение среднего артериального, систолического и диастолического давлений, умеренное тахипноэ с гипокапнией, снижение центрального венозного давления (ЦВД). Тахикардию можно уменьшить применением р-адренобло-каторов, а артериальное давление (АД) — повысить р-миметиками, но коррекция этих показателей не восстановит нормальную гемодинамику, перфузию тканей. Она может даже усугубить гемодинамические расстройства. В то же время переливание соответствующих инфузионных сред (плазморасширителей) способствует повышению ЦВД, что приведет к увеличению преднагрузки, диастомического наполнения полостей сердца, силы сокращений желудочков, выбросу крови, повышению АД, перфузии тканей и соответственно рефлекторно уменьшится тахикардия, тахипноэ, нормализуется гемодинамика. Таким образом, выбор коррекции ЦВД (восстановление объема циркулирующей крови) как ведущего симптома в этиопатогенезе процесса является целесообразным.
2. Коррекция должна быть физиологически обоснованной для данного патофизиологического процесса у данного больного.
Например, при кровопотере в результате рефлекторного развивающегося гиперпноэ возникает респираторный алкалоз. Снижение рСО2 артериальной крови повышает сродство кислорода к НЬ и его утилизация в периферических тканях уменьшается,
10 ВВЕДЕНИЕ
при уменьшении количества транспортных средств (эритроцитов) дефицит кислорода в тканях возрастает. Поэтому коррекция гипервентиляции продлевает на несколько часов жизнь больных без компенсации кровопотери. Если же у больного с кровопотерей имеется черепно-мозговая травма с очагом повреждения, то коррекция гипокапнии приведет к расширению сосудов в неповрежденных зонах, уменьшению коллатерального кровообращения и усилит гипоксию в поврежденных зонах. У больных с гиповолемическим шоком коррекция гипервентиляции будет способствовать улучшению гемодинамики и уменьшению кислородного дефицита, а у больных с сочетанием кровопотери с черепно-мозговой травмой — усугубит повреждения структур центральной нервной системы.
3. Коррекция должна быть адекватной, чтобы не вызвать дополнительные сдвиги.
Например, у больной с несколькими свищами на уровне желудка, двенадцатиперстной кишки и верхних отделов тонкой кишки в один из дней при определении электролитов плазмы было обнаружено Na+ — 125 ммоль/л, СГ — 90, К+ — 3 ммоль/л. Для обеспечения адекватности обычно рассчитывают смесь инфузионных растворов, начиная с того электролита, дефицит которого меньше всего выражен. В данной ситуации дефицит калия равен 3 ммоль/л внеклеточной жидкости, хлора — 10, дефицит натрия — 17 ммоль/л. Это электролиты внеклеточного пространства и их соотношение обычно сочетается с умеренной дегидратацией, гиповолемией, снижением АД и респираторно компенсированным ацидозом, что в действительности и определялось у больной. Не существует соединения, введением которого можно было бы одновременно корректировать указанный дефицит всех электролитов, поэтому необходимо подбирать их комбинацию. Обычно первое соединение выбирают, исходя из электролита, дефицит которого минимальный. В данном случае — это К+. Лучшим соединением, содержащим калий, является КС1. Введя на каждый литр внеклеточной жидкости 2 ммоль КС1, мы одновременно с
коррекцией дефицита К+ уменьшим до 8 ммоль/л дефицит СГ. Теперь его дефицит будет минимальным. Для его коррекции следует использовать NaCl. Введя 8 ммоль NaCl на литр внеклеточной жидкости, мы одновременно уменьшим до 9 ммоль/л дефицит натрия.
Чтобы адекватно корректировать натрий, необходимо избрать соединение, содержащее катион натрия и метаболизирующийся анион. Такими соединениями могут быть натрий лактат, натрий ацетат или натрий гидрогенкарбонат. В данной ситуации натрий лактат и натрий ацетат не подходят, поскольку их введение усилит ацидоз и повысит потребность в О2. Более приемлемым будет введение натрий гидро-генкарбоната, ангидрид которого (НСО3) образует с Н+ угольную кислоту, которая под влиянием карбоангидразы диссоциирует до воды и углекислого газа и удаляется из организма при дыхании. Так, дефицит электролитов в данном случае корригируется введением на 1 л внеклеточной жидкости 2 ммоль КС1, 7 ммоль — NaCl и 9 ммоль NaHCO3.
4. Коррекция должна быть оптимальной по времени. При острой или подострой кровопотере для сохранения соответствия между уменьшившимся объемом крови и емкостью сосудистого русла быстро развивается периферический спазм — централизация кровообращения, что способствует притоку крови к сердцу и сохранению кровообращения с пониженными показателями гемодинамики. Прекращение этой компенсаторной реакции происходит медленнее, чем ее развитие. Поэтому при быстром восполнении реально потерянного объема и сохраняющейся централизации кровообращения наступает перегрузка сердца, гипертензия малого круга кровообращения с развитием отека легких. Если инфузию проводить с оптимальной скоростью при контроле гемодинамических показателей, то нормализация кровообращения наступит без осложнений. Это правило необходимо соблюдать и при коррекции гипокалиемии.
5. Коррекция должна осуществляться в определенной последовательности, чтобы обеспечить восстановление не только
Направления интенсивной терапии 11
отдельного показателя, но и физиологического процесса в целом.
Примером могут служить ситуации, когда возникает необходимость в коррекции нескольких показателей. Так, при разлитом или диффузном перитоните развиваются выраженные, требующие коррекции отклонения в виде гипопротеинемии, гипопалие-мии, алкалоза. Последний обычно взаимосвязан с гипокалиемией. Гипокалиплазмия появляется в фазе, предшествующей гипо-калигистии. Поэтому при инфузии растворов калия требуется обеспечить транспорт калия во внутриклеточное пространство, но при гипопротеинемии транскапиллярный объем обычно изменен. В связи с этим следует начать с корректировки гипопротеинемии, а затем коррегировать гипокалиемию во внутриклеточном пространстве. Для ускорения перехода калия во внутриклеточное пространство против концентрационного градиента и для энергетического обеспечения этого процесса желательно вводить соединения калия с растворами глюкозы и инсулина, а также в сочетании с соединениями Mg2+ (на 8 ммоль калия обычно добавляют 1 моль магния и на 4 —5 г глюкозы — 1 ЕД инсулина). По литературным и нашим данным, первоначальное введение калия обычно снижает эффективность коррекции и может привести к серьезным осложнениям.
При детоксикации биологических сред организма обычно используют две группы методов. Первую составляют методы, активирующие или повышающие производительность естественных процессов детоксикации в организме:
1. Активация собственных механизмов детоксикации в печени и других органах и тканях (например, неспецифических окислительных систем с участием цитохрома Р450, процессов конъюгации, подлежащих субстральной индукции). Главное условие при этом — четко определить показания и избежать усиления «летального синтеза».
2. Форсированный диурез. Используя этот метод, следует как можно раньше выявить развитие дегидратации, нарушений реологических свойств крови.
3. Активация и увеличение производительности антиоксидантной системы, а так
же применение средств, связывающих свободные радикалы. Следует помнить, что длительная ингаляция газовых смесей, содержащих более 50 % кислорода, а также длительно проводимая гипербарическая оксигенация могут усилить свободнорадикальное пероксидное окисление липидов.
4. Активация фотохимических реакций детоксикации методом ультрафиолетового облучения крови. Этот метод показан у детей при гемолитической желтухе (Б. С. Шейман).
Вторую группу составляют методы экстракорпоральной детоксикации. В зависимости от частоты использования их можно расположить следующим образом: ♦ гемодиализ, перитонеальный диализ; ♦ плазмаферез;
♦ гемоперфузия (гемосорбция) с использованием сорбентов различных свойств и назначения;
♦ гемо- и ультрафильтрация с использованием молекулярных сит;
♦ лимфодренаж с лимфосорбцией и лим-фоинфузией;
♦ иммуноферез цереброспинальной жидкости с применением специальных фильтров.
Каждый метод экстракорпоральной детоксикации имеет свои особенности применения, показания, о которых будет рассказано в следующих разделах.
Третье направление ИТ — временная замена утраченных функций пораженных органов и систем — также располагает большим количеством сложных методов, многие из которых послужили основанием для создания узкоспециализированных отделений ИТ. Наиболее часто используют метод искусственной вентиляции легких и метод гемодиализа (искусственная почка). Значительно реже — искусственное кровообращение, подключение гетеропечени и гетероселезенки. Применение этих методов имеет свои особенности в зависимости от вида патологического процесса, конструктивных особенностей аппаратуры.
Четвертое направление (протекторное) ИТ располагает значительно меньшим количеством методов, многие из которых в настоящее время используют редко.
12 ВВЕДЕНИЕ
Гипотермия и гипербарическая оксигенация в 60 —70-х годах XX ст. применялись довольно часто, однако из-за отсутствия четкой доказательности положительного влияния на исходы критических состояний эти методы сейчас редко используют.
К фармакологическим средствам протекторного направления можно отнести нейропротекторы ноотропной группы (ноотропил, промистар), антикоагулянты, нестероидные противовоспалительные средства и т. д.
В пятом направлении ИТ в основном используются фармакологические средства повседневной медицинской практики, а также некоторые методы с механическим механизмом действия, например пневмокостюм при синдроме ныряльщика.
Наряду с правилами ИТ, касающимися использования отдельных методов, имеется несколько общих правил ИТ.
1. ИТ следует проводить с учетом возрастных особенностей физиологических и биохимических процессов. Например, у взрослого в процессе обмена воды ежедневно расходуется 1/9—1/12 экстрацеллюлярного объема, а у ребенка — 1/3. У последнего очень слабо выражены неспецифическая окислительная система с цитохромом Р450 и другие дезинтоксика-ционные процессы (конъюгация). Этим можно объяснить высокую резистентность организма ребенка к нагрузкам водой и высокую чувствительность к нагрузкам солевыми растворами из-за еще не сформировавшейся концентрационной функции почек. Вследствие несовершенства процессов детоксикации организм ребенка более чувствителен к таким эндогенным интокси
кациям, как билирубинемия. Все это необходимо учитывать при составлении программы инфузионной терапии.
2. При ИТ следует использовать минимальное количество лекарственных препаратов, чтобы при сочетанном их применении исключить возможное негативное взаимодействие, что является частой причиной осложнений и летальных исходов при полифармации.
Эффективную ИТ можно проводить только в определенных условиях, основными из которых являются следующие:
1. Возможность осуществления интенсивного наблюдения (мониторинга) и ухода (круглосуточное функционирование биологической экспресс-лаборатории, оснащение мониторинговой аппаратурой, соответствующее планирование необходимых площадей под отделения интенсивной терапии).
2. Высокая квалификация, достаточный опыт работы всего персонала, преемственность действий и принципов ИТ, высокая ответственность и добросовестное отношение к своим обязанностям. Особенно важным качеством является честность.
Интенсивная терапия является одним из ведущих направлений медицины, обеспечивающим целенаправленный выбор средств лечения тяжелобольных. Методы интенсивной терапии используются практически в каждой отрасли медицины, поэтому знание принципов интенсивной терапии и применения их на практике необходимо врачу любой специальности, оказывающему помощь критически больным пациентам.
==== Раздел-------
1 физиология автономной нерВной системы
Врач любой специальности не может считаться грамотным профессионалом высокого уровня, если у него отсутствуют элементарные знания физиологии и особенностей анатомии автономной нервной системы (АНС). Особенно это касается анестезиологов и специалистов, занимающихся интенсивной терапией.
АНС является главным компонентом в регуляции функций жизненно важных органов и систем, функциональной их взаимосвязи, поэтому для понимания патогенеза критических состояний, механизмов стрессовых реакций и их последствий, адаптации и компенсации, выбора средств и методов целенаправленной терапии необходимы глубокие знания клинической физиологии АНС. Многие лекарственные
препараты реализуют фармакологический эффект через первичное воздействие на различные виды и уровни структур АНС, поэтому для выбора адекватных средств и целенаправленного воздействия при тех или иных вариантах патологических состояний также важно знание физиологии АНС, что позволит более объективно оценить состояние больного и своевременно диагностировать степень коматозных состояний и смерть мозга.
Знание особенностей медиаторного обмена в АНС и влияния лекарственных препаратов на него дает возможность объективно оценить степень нарушения гомеостаза и избрать наиболее рациональные лекарственные средства для целенаправленной коррекции его нарушений.
1.1. ФУНКЦИОНАЛЬНАЯ АНАТОМИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
В зависимости от функциональной значимости выделяют центральные (надсегментарные) структуры АНС, обеспечивающие регуляцию процесса метаболизма или функции системы жизнеобеспечения с различными направлениями эффекта в зависимости от ситуации, и периферические, активирующие или угнетающие определенный вид функций. Периферические структуры в зависимости от эффекта, проявляющегося при их возбуждении, в свою очередь, объединяют в симпатическую
(СНС) или парасимпатическую нервную систему (ПНС) [1—3]. Вследствие структурных и функциональных особенностей выделяют также отдел энтеральной нервной системы.
Кроме различия эффекта влияния на иннервируемые органы и ткани установлено также различие медиаторов, обеспечивающих передачу возбуждения между нейронами симпатической и парасимпатической НС. В связи с этим нервы традиционно классифицируют по содержа
14 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
нию в них различных медиаторов. Нервы, содержащие ацетилхолин, относят к холинергическим, а содержащие норадреналин или адреналин, — к адренергическим. Кроме того, к холинергическим могут быть отнесены структуры ЦНС и периферические нервы цереброспинального отдела НС, функционирующие благодаря ацетилхолину. Холинореактивные рецепторы содержат протеин в клеточной мембране, реагирующий с ацетилхолином, что вызывает ответ в виде определенного действия (мышечного сокращения, секреции железы).
Агонисты ацетилхолина или лекарственные средства, действующие подобно ацетилхолину на холинорецепторы и вызывающие характерный эффект, относят к холиномиметическим средствам. Холинергические антагонисты — это лекарственные средства, также воздействующие на холинорецепторы, блокирующие доступ ацетилхолина и препятствующие его действию. Их обычно называют холинолити-ками, холиноблокаторами или антихо-линергическими средствами.
Мускарин — химическое вещество, выделенное из грибов, — активирует парасимпатическую нервную систему и считается экзогенным медиатором. Лекарственные средства, действующие подобно мускарину на структуры, иннервируемые ПНС, включая сердце, гладкие мышцы и железы, называются М-холиномиметика-ми, а структуры, реагирующие на них, — М-холинореактивной системой. М-холи-номиметики действуют на ПНС преимущественно как агонисты.
В начале XX ст. было обнаружено влияние никотина на ганглии АНС и нервно-мышечные синапсы скелетных мышц, мембраны чувствительных нервных окончаний. Лекарственные средства, действующие подобно никотину, получили название «Н-холиномиметики», а структуры, реагирующие на эти вещества, — Н-холи-нореактивной системы. Агонисты и антагонисты Н-холинореактивной системы в зависимости от вида анатомических структур (ганглий или нервно-мышечный синапс) разделяют на группу средств, действующих на ганглии (ганглиоблокаторы,
или литики), мышечные релаксанты и миметики (блокаторы холинэстеразы). Агонисты и антагонисты, существующие в каждой категории, оказывают также незначительный перекрестный эффект на ганглии и нервно-мышечные синапсы. Например, пентамин (пентаметоний) и гексамин (гек-саметоний), у которых два азотных четвертичных центра разделены пяти- и шестиуглеродной цепочкой, оказывают влияние преимущественно на Н-холинореак-тивные системы ганглиев АНС, а декамен-тоний, у которого два азотных центра соединены 10-углеродным мостиком, действует преимущественно на нервно-мышечные синапсы как мышечный релаксант [3].
К холинергической системе могут быть отнесены:
♦ все двигательные нервы, иннервирующие скелетные мышцы;
Постганглионарное волокно
Преганглионарное
Симпатическая
нервная система
Преганглионарное
Симпатическая
нервная система
Преганглионарное
Сердце, поперечнополосатые мышцы, железы
Постганглионарное волокно
-----------<АХ
Потовые железы
волокно
Надпочечники нэ, эн
Симпатическая
нервная система
Преганглионарное Постганглионарное
Узел волокно < АХ
Парасимпатическая нервная система
Сердце, поперечнополосатые мышцы, железы
Рис. 1.1. Анатомия нервной системы и нейротрансмиссия ацетилхолина (АХ), норэпинефрина (НЭ), эпинефрина (ЭН)
Функциональная анатомия автономной нервной системы
15
♦ все постганглионарные парасимпатические нейроны;
♦ все преганглионарные парасимпатические и симпатические нейроны;
♦ постганглионарные симпатические нейроны, иннервирующие потовые железы и некоторые сосуды;
♦ преганглионарные симпатические нейроны, входящие в состав большого чревного нерва и иннервирующие мозговое вещество надпочечников;
♦ холинергические нейроны центральной нервной системы (рис. 1.1).
Норадреналин — это медиатор адренергической системы. Лекарственные средства, действующие подобно норадреналину, называют адрено-, или симптоми-метиками. Адреналин, а также норадреналин (в меньшей степени) синтезируются в мозговом веществе надпочечников. Адренергические рецепторы в зависимости от вида эффекта разделяют на а- и Р-рецепторы, которые, в свою очередь, подразделяют на 04-, а2-, Рр, р2. R. Lawhead с соавт. разделяет еще a t-рецепторы на а1л, а1В, а1С, а а2- - на а1Л, а2в, а2С, a2D [4J (Рис- L2).
а-Рецептор активируется агонистами и взаимодействует с протеином G (IIG), в результате чего облегчается открывание рецепторнорегулируемых кальциевых каналов и увеличивается внутриклеточная концентрация Са2+. Механизм работы Р-рецептора более сложный. Этот рецептор активирует фосфолипазу С, которая при взаимодействии с другим IIG катализирует распад 3,4-дифосфата фосфоинозитола в инозитол 1,4,5-трифосфат (ИТФ) и диацилглицерол (ДАГ). ИТФ, в свою очередь, стимулирует освобождение ионов Са2+ из саркоплазматического (эндоплазматического) ретикулума немышечных клеток. Возможно, что ДАГ при участии цАМФ облегчает открытие других управляемых кальциевых каналов, а также увеличивает внутриклеточную концентрацию Са2+. Норэпинефрин может обеспечивать взаимодействие по обоим рецепторным механизмам. Увеличение содержания Са2+ внутри клеток при взаимодействии его с внутриклеточным белком кальмодулином активирует легкую цепочку киназы глад-
__ FIG ДффИ
nG| 1ФЛС1 цАМФ
—»ДАГ,->
ЭР^ИТФ
Протеин-киназа
------4Са%---------i----
^Кальмодулин»
2+ » ।
Са -Кальмодулин |
Г Другие
ГК-киназа киназы
РО43" | Миозин Секреция
Миозин-РО4
| —► Сокращение Актин
Рис. 1.2. Биохимия мембраны а-рецептора:
РРК — рецепторнорегулируемые кальциевые каналы; НЭ — норэпинефрин; ПО — протеин G, ДФФИ — дифосфат фосфоинозотол; ИТФ — инозитол 1,4,5-трифосфат; ЭР — эндоплазматический ретикулум немышечных клеток; ФЛС — фосфолипаза С; УКК — управляемые кальциевые каналы; ДАГ — диацилглицерол; ГК-киназа — киназа гладкомышечных клеток
комышечных клеток, которая фосфорилирует миозин, обеспечивая его соединение с актином и сокращение гладкомышечных клеток. ДАГ также активирует мембранную протеинкиназу С, что обуславливает каскад киназной активации, ответственной за внутриклеточные функции.
а2-Рецепторы локализуются главным образом в пресинаптической мембране а2-синапсов, тогда как cq-рецепторы расположены в гладких мышцах, суживающих сосуды, pj-рецепторы локализуются в сердечной ткани, а р2-рецепторы — в мышцах некоторых органов (бронходилатато-ры и вазодилататоры в поперечнополосатых мышцах) [5].
Адренергические нейроны представлены:
♦ постганглионарными нейронами СНС; ♦ некоторыми вставочными нейронами;
♦ определенными нейронами ЦНС (нейроны восходящей ретикулярной формации).
16 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
1.1.1. Симпатическая нервная система
Центры симпатической нервной системы расположены в боковых рогах серого вещества спинного мозга на уровне всех грудных и двух верхних поясничных сегментов. Тела преганглионарных симпатических нейронов располагаются в области серого вещества между передними и задними рогами, образуя интермедиолате-ральные столбы. Нервные волокна, отходящие от преганглионарных нейронов интермедиолатеральных столбов, выходят из спинного мозга вместе с передними корешками и направляются к нервам, сгруппированным в смешанные спинномозговые нервы и нейроны, которые образуют парные симпатические цепи, состоящие из скоплений нейронов, соединенных между собой дендритами и аксонами пре-и постганглионарных волокон. Нейроны пограничной симпатической цепочки соединены как по горизонтали, так и по вертикали. Это дает возможность импульсам из центров СНС распространяться как гомолатерально, так и гетеро л атерально, краниально и каудально. Нейроны, образующие паравертебральные структуры СНС, можно разделить на три типа: к первому типу, наибольшему, относятся нейроны, сгруппированные в парные симметричные ганглии, образующие паравертебральную симпатическую цепочку, — верхний, средний и нижний шейные ганглии, 22 пары грудных и два (может быть и больше) поясничных ганглия. Второй тип представлен непарными постганглионарными нейронами, расположенными в сплетениях (солнечное верхнее и нижнее брыжеечное, аорто-почечное). Третий тип составляют конечные, коллатеральные ганглии вблизи органов-мишеней (рис. 1.3).
После выхода из спинномозгового канала в составе переднего двигательного корешка аксоны преганглионарных нейронов отходят от корешка в виде миелизи-рованной белой ветви и направляются к нейронам (постганглионарным), образующим паравертебральную симпатическую цепь, на уровне сегмента отхождения, в большинстве случаев они переключаются
Рис. 1.3. Анатомия симпатической нервной системы:
/ — вазо- и пиломоторные волокна и волокна к железам головы; 2 — вазо- и пиломоторные волокна и волокна к железам шеи, верхний шейный узел; 3 — средний шейный узел; 4 — паравертебральные узлы; 5 — волокна к железам, пило-, вазомоторные волокна туловища; 6 — волокна к железам, пило-, вазомоторные волокна нижней части туловища и нижних конечностей; 7 — каротидное сплетение; 8 — глаз; 9 — слезная железа; 10 — сосуды и железы слизистой носа; 11 — околоушная железа; 12 — поднижнечелюстная железа; 13 — сердце; 14 — сосуды бронхов и легких; 15 — печень; 16 — желудок; 17 — поджелудочная железа; 18 — почки; 19 — тонкая кишка; 20 — чревной узел; 21 — надпочечники; 22 — верхний мезентериальный узел; 23 — толстая кишка; 24 — нижний мезентериальный узел; 25 — мочевой пузырь и уретра; 26 — гениталии
на несколько сегментов выше или ниже на постгаглионарные волокна серого цвета, имеющие очень тонкую миелиновую оболочку и образующие соединительную ветвь, которая возвращается к межпозвоночному отверстию, где происходит слияние задней и передней ветвей и образуются смешанные спинномозговые нервы, в составе которых постганглионарные волокна направляются к иннервируемым СНС органам и тканям. Постганглионарные волок
Функциональная анатомия автономной нервной системы 17
на грудного отдела иннервируют потовые железы туловища и конечностей, кровеносные сосуды скелетных мышц и кожи, сердце и легкие. Постганглионарные волокна, иннервирующие голову и шею, отходят от парных ганглиев шейного отдела пограничной симпатической цепочки. Преганглионарные волокна этих шейных структур отходят от преганглионар-ных нейронов верхних грудных сегментов. У 80 % людей нижние шейные симпатические ганглии и один-два грудных с обеих сторон сливаются и образуют один большой ганглий под названием «звездчатый узел».
Непарные околопозвоночные ганглии в виде сплетений расположены в брюшной полости и малом тазу спереди от позвоночного столба. Важнейшими из них являются чревное (солнечное) сплетение, верхнее брыжеечное, аорто-почечное, нижнее брыжеечное.
В образовании мезентериальных (брыжеечных) сплетений принимают участие волокна от Tv—Тх сегментов, подходящие к сплетению в виде большого чревного нерва, и от Тх—Тхп сегментов — в виде малого чревного нерва. В обоих нервах имеется также небольшое количество постганглионарных волокон от нейронов пограничного симпатического столба. Преганглионарные волокна чревных нервов иннервируют мозговое вещесто надпочечников, аналогичное постганглионарным нейронам. Таким образом, надпочечники иннервируются через однонейронный путь. Постганглионарные волокна, идущие от чревного сплетения чаще всего в виде периваскулярных сплетений, иннервируют желудок, печень, почки, селезенку, тонкую кишку и начальные отрезки толстой кишки, а также поджелудочную железу.
Верхнее брыжеечное сплетение осуществляет иннервацию дистальной части толстой кишки, нижнее — иннервацию прямой кишки, мочевого пузыря и половых органов.
Постганглионарные волокна, происходящие из верхних грудных ганглиев, образуют конечные сердечные, пищеводные и почечные сплетения.
Мозговое вещество надпочечников и хромаффинная ткань гомологичны симпатическим ганглиям и имеют общее происхождение. В отличие от симпатических ганглиев мозговое вещество секретирует катехоламины (адреналин и норадреналин).
Симпатические преганглионарные волокна короче постганглионарных. Симпатические ганглии и имеют связь с ЦНС, но расположены далеко от иннервируемых органов, поэтому постганглионарные волокна имеют значительную протяженность.
Распределение симпатических волокон диффузное. Возбуждение распространяется по типу «масляного пятна». Преганглионарные симпатические волокна могут проходить через многочисленные симпатические ганглии, не вступая в контакт с нейронами и только на каком-то отдаленном уровне образуют синапс с нейроном ганглия. Преганглионарные волокна могут контактировать с большим количеством нейронов, образующих постганглионарные волокна. Терминальное волокно преганглионарных ветвей может образовывать синапсы более чем с 20 ганглиями; один нейрон может формировать несколько преганглионарных волокон. Следует помнить, что симпатические преганглионарные нервы связаны не с единственным синапсом в сегментарном ганглии, имеется симпатическая связь с билатерально выше-и нижерасположенными ганглиями. Таким образом, симпатический ответ не ограничен сегментом, он, как правило, генерализован. Эта особенность функциональной анатомии дает представление о сложной ответной реакции всей СНС [3, 6 — 8].
Рефлексы автономной нервной системы сохраняются и после перерезки спинного мозга. Обычно автономные рефлексы ингибируются супраспинальной обратной связью, исчезающей при перерыве спинальных путей. В связи с утратой ингибирования супраспинальными структурами АНС сегментарные рефлексы часто усиливаются. Так, даже незначительная стимуляция (небольшая доза катехоламинов) может вызвать сверхсильный симпатический ответ.
18 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
1.1.2. Парасимпатическая нервная система
Преганглионарные волокна парасимпатической НС отходят от соответствующих нейронов, расположенных в трех зонах ЦНС: среднем и продолговатом мозгу и крестцовом отделе спинного мозга (рис. 1.4). Преганглионарные волокна, отходящие от нейронов Эдингер — Вестфа-левского ядра, идут из среднего мозга в составе глазодвигательного нерва (III пара) к ресничному ганглию, в синапсах нейронов
Рис. 1.4. Анатомия парасимпатической нервной системы:
1 — глазодвигательный нерв; 2 — реснитчатый узел; 3 — глаз; 4 — слезная железа; 5 — крылонебный узел; 6 — большой каменистый нерв; 7 — барабанная струна; 8 — железы и сосуды слизистой носа; 9 — поднижнечелюстная железа; 10 — языкоглоточный нерв; 11 — блуждающий нерв; 12 — сердечная мышца; 13 — мышцы бронхов; 14 — пищевод; 15 — желудок; 16 — тонкая кишка; 17 — толстая кишка; 18 — прямая кишка; 19 — возбуждающий нерв; 20 — уретра; 21 — мочевой пузырь; 22 — гениталии; 23 — средний мозг; 24 — продолговатый мозг
которого переключаются на постганглионарные ветви, иннервирующие гладкие мышцы, сфинктеры радужной оболочки.
В продолговатом мозгу расположены нейроны оливного ядра, преганглионарные волокна которого выходят в составе лицевого нерва, и преганглионарные волокна дорзального ядра, выходящие в составе языкоглоточного и блуждающего нервов. В лицевом нерве преганглионарные волокна образуют барабанную струну и большой поверхностный каменистый нерв, идущие к ганглиям на подчелюстной и подъязычной слюнной железах, а также к крыло-небному ганглию, где переключаются на постганглионарные волокна, иннервирующие слюнные и слезные железы.
Блуждающий нерв является самым большим парасимпатическим нервом. Он обеспечивает 3/4 всей афферентации и эффе-рентации ПНС, иннервирует сердце, тра-хео-бронхиальное дерево, печень, почки, селезенку и весь пищеварительный канал, за исключением дистального отдела толстой кишки (сигма и прямая кишка). Преганглионарные волокна берут начало от нейронов, образующих ядра в продолговатом мозгу. Большая часть волокон блуждающего нерва не образует синапсов, пока не достигнет небольших ганглиев, расположенных вблизи иннервируемых органов в грудной клетке и брюшной полости.
Схема симпатического синапса в стенке органа изображена на рис. 1.5. Норэпинефрин стимулирует постсинаптические рецепторы, вызывая классические cq- и p-эффекты. Стимуляция а2 пресинаптиче-ских рецепторов вызывает ингибирование дальнейшего выделения норэпинефрина.
Преганглионарные волокна образуют синапсы с постганглионарными нейронами в соотношении 1 : 1, а постганглионарные волокна, иннервирующие ауэрбахов-ское сплетение в кишках (энтеральный отдел АНС), могут образовывать синапсы в соотношении 1 : 3 000 [9]. Это объясняет тот факт, что реакции парасимпатической нервной системы более адресные, а симпатической — более генерализованы.
Нейроны, дающие начало преганглио-нарным волокнам, иннервирующим дистальные отделы толстой кишки и моче-
Функциональная анатомия автономной нервной системы 19
Постсинаптическая
клетка
Рис. 1.5. Схема симпатического синапса в стенке органа:
НЭ — норэпинефрин;---------стимуляция;
----- ингибирование
половые органы, расположены в крестцовом отделе спинного мозга [3, 6, 7].
Клинически наблюдаемые эффекты, связанные с анестезией, такие как тошнота, рвота, изменение функций кишок и мочевого пузыря, дали основание предположить наличие третьего отдела автономной нервной системы, получившего название «энтеральная нервная система» (ЭНС). Она состоит из нервов и опорных клеток, расположенных внутри стенки пищеварительного канала, а также нейронов в пределах поджелудочной железы и желчного пузыря. Meissner и Auerbach первыми описали ганглионарные сплетения этой системы.
A. S. Dogiel [10] создал морфологическую классификацию энтеральных нейронов и скорректировал предполагаемые различия формы и функции. W. М. Bayliss и Е. Н. Starling [И] показали существование полярных рефлексов, действующих в кишках независимо от внешних влияний. J. N. Langley [12, 13] установил уникальные особенности ЭНС и создал классификацию «третьей ветви автономной нервной системы». ЭНС содержит много клеток, подобных клеткам спинного мозга. Она происходит из нейробласта и нервного гребешка, которые мигрируют в пищеварительный канал вдоль блуждающего нерва.
Имеется существенное различие между ЭНС и симпатическими и парасимпа
тическими структурами АН С — это необычное анатомическое расположение. Функции пищеварения и перистальтики сохраняются и после перерезки спинного мозга, или эпидуральной анестезии, хотя функция сфинктеров может нарушаться.
Несмотря на функциональную изолированность энтеральной нервной системы, кишки получают как симпатическую, так и парасимпатическую иннервацию. Симпатические преганглионарные волокна от Tv — Lt ингибируют деятельность кишок. Спинальная анестезия на среднегрудном уровне устраняет это ингибирование, восстанавливая и активируя сокращения тонкой кишки, что наряду с глубокой мышечной релаксацией создает хорошие условия для оперативного вмешательства. Сфинктеры расслабляются, восстанавливается перистальтика.
Норадреналин, секретируемый в кишках, является медиатором постганглионарных симпатических нейронов. К примеру, если содержимое верхних отделов пищеварительного канала становится чрезмерно кислым или гипертоническим, то под влиянием адренергических медиаторов желудочно-кишечный рефлекс обеспечивает снижение скорости опорожнения желудка.
В процессе абдоминальных хирургических вмешательств внутренние органы находятся под рефлекторным влиянием адренергических ингибиторов двигательной функции кишок в течение длительного периода. Это адренергическое ингибирование, вероятно, является причиной часто встречающегося состояния, известного как послеоперационный парез.
Утрата парасимпатической нервной регуляции обычно снижает тонус кишечной стенки и замедляет перистальтику, но одновременно на непродолжительное время усиливается активность энтеральной нервной системы. Повреждения спинного мозга могут приводить к утрате парасимпатической крестцовой активности. Но краниальный парасимпатический компонент все еще сохраняет вагусное влияние на иннервируемые им отделы кишок, что приводит к колонодилатации и каловому (фекальному) разобщению, которое может
20 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
активироваться при гипертензии и анатомической дисрефлексии и встречается чаще, чем дисфункция тонкой кишки [8, 14, 15]. Энтеральные нейроны классифицируют на чувствительные (определяют химическую природу содержимого кишок), ассоциативные (функционируют подобно вставочным нейронам) и двигательные (активируют функцию кишок, т. е. вызывают сокращение мышц, дилатацию сосудов, транспорт электролитов). Двигательная функция нейронов энтеральной нервной системы может быть либо возбуждающей, либо ингибирующей.
В кишечной стенке выделяют мезентериальное, или ауэрбаховское, сплетение — сеть нервных волокон и небольших ганглиев, расположенных в слое между наружными продольными и циркуляторными мышцами кишок. В подслизистом слое находится мейснеровское сплетение, состоящее из нервных клеток, включая глиальные клетки и их отростки, но при этом не содержащее соединительной ткани и кровеносных сосудов. Внутри нервных отростков ганглиев имеются пузырьки, которые, возможно, являются депо медиаторов. В отличие от симпатической и парасимпатической нервных систем, где топография размещения клеток соответствует селективности действия, в структурах энтеральной нервной системы кишок это правило не соблюдается. Поэтому наиболее вероятным механизмом деятельности ЭНС является химическое кодирование, что подтверждается наличием около дюжины медиаторов в этой системе. Комбинация медиаторов, аминокислотных остатков, пептидов и энтеральных нейронов обеспечивает кодирование разнообразных функций.
Нервная регуляция пищеварительного канала осуществляется посредством двух типов мотонейронов: возбуждающих и ингибирующих. Эти нейроны действуют согласованно на циркулярный гладкомышечный слой сфинктерной и несфинктерной областей на протяжении всего пищеварительного канала, а также иннервируют мышцы билиарного дерева и слизистых оболочек. В то время как возбуждающие мотонейроны иннервируют одни участки продольных мышц, другие — находятся
под влиянием ингибирующих мотонейронов.
Энтеральные мотонейроны циркулярных мышц активируются местными рефлекторными реакциями, пути которых расположены в стенке кишок.
Растяжение стенки кишок вызывает рефлекторные синхронные оральные сокращения и анальное расслабление, т. е. перистальтику, способствующую перемешиванию и передвижению содержимого кишок. Никотиновые антагонисты угнетают энтеральные рефлексы, возбуждая чувствительные или вставочные нейроны в холинергических путях. Таким образом, в случае холинергической нагрузки, например при отравлении инсектицидами или при чрезмерном введении антагонистов мышечных релаксантов, ингибирующих холинэстеразу, возникает тенденция к гиперактивности кишок, которая в определенной степени может подавляться М-холинолитиками (атропином).
Ацетилхолин является главным возбуждающим триггером несфинктерной части ЭНС, вызывая мышечные сокращения. Холинергические нейроны играют определенную роль в этой системе, способствуя сокращению наружных мышц, активируя двигательные нейроны, увеличивая секрецию воды и электролитов, стимулируя секреторные клетки желудка. Кроме этого, имеется еще много других сложных составляющих, участвующих в регуляции функции кишок. Среди них — SP (субстанция Р), разнообразные опиатные пептиды (эндорфины, энкефалины), VIP (вазоактивный интестинальный полипептид) и растущая популяция пептидных гормонов. Имеются доказательства существования неадренергических и нехолинергических (НАНХ) нейронов в тонкой кишке и полулунных клетках, выделяющих гастрин. В настоящее время доказана роль оксида азота (NO) как соединения, расширяющего сосуды и увеличивающего секреторную функцию эндотелиальных клеток, которое может быть важным компонентом вставочных нейронов и секретироваться совместно с VIP. В сфинктерах SP, опиоидные пептиды и ацетилхолин могут быть возбуждающими медиаторами, хотя в зависимости от сфинктера они могут играть разную роль.
Современные концепции механизма действия медиаторов 21
1.2. СОВРЕМЕННЫЕ КОНЦЕПЦИИ МЕХАНИЗМА ДЕЙСТВИЯ МЕДИАТОРОВ
До недавнего времени в нервной регуляции кровообращения основными медиаторами считались норадреналин и ацетилхолин. В течение многих лет они были единственными признанными медиаторами периваскулярных нервов. В начале 60-х годов XX ст. было открыто множество других веществ, включая моноамины, пурины, аминокислоты, полипептиды, выполняющих функции нейромедиаторов. В дальнейшем G. Burnstock [16, 17] включил неадренергические и нехолинергические медиаторы в состав АНС. Медиаторы, найденные в периваскулярных нервах с помощью гистохимической и иммуногистохимической техники, содержат 5-гидро-кситринтамин (5-НТ, серотонин), 5'-аде-нозинтрифосфат (АТФ), VIP, SP, нейропептид Y, кальцитонин — генрелансирующий полипептид (CGRP).
Иммуноцитохимическое изучение показало, что в одном и том же нерве может содержаться более одного медиатора [18]. Самыми распространенными комбинациями медиаторов в периваскулярных нервах являются: ацетилхолин и VIP в парасимпатических нервах; норадреналин, АТФ и нейропептид Y (NPY) — в симпатических; SP, CGRP и АТФ — в чувствительных и двигательных нервах. Многие из этих предполагаемых медиаторов участвуют в механизме последовательной передачи, что предполагает синтез, хранение и выделение более чем одного передатчика, действующего в нерве.
Первоначально открытие новых медиаторов, выделяющихся в различных комбинациях, казалось случайным. Однако к настоящему времени разработана модель, в которой автономная нервная передача играет роль «химического кода» (индивидуальная нервная комбинация имеет специфическую физиологическую функцию и представлена различным сочетанием медиаторных веществ). Концепция сложной передачи и нейромодуляции является приемлемым механизмом автономной нервной регуляции. Для того чтобы установить, что эти медиаторы в однотипных нервах действуют совместно, необходимо сна
чала выяснить, действует ли каждое секретируемое вещество постсимпатически на свой специфический рецептор, вызывая определенную ответную реакцию. Это доказано для некоторых периваскулярных симпатических нервов: норадреналин и АТФ, секретируемые в однотипных нервах и действующие на а-адренорецепторы и Р2-пуринорецепторы, являются комедиато-рами, вызывая вазоконстрикцию.
Если раньше предполагалось, что АТФ действует только как стабилизатор норадреналина, то в настоящее время считают, что передача через Р2х-рецепторы осуществляется по вольтажзависимым Са2+-ка-налам [8]. Быстрая фаза сокращения реализуется через пуринорецепторы, в то время как норадреналин сам по себе может поддерживать сокращение мышц, воздействуя на а-адренорецепторы через рецепторсвязанные Са2+-каналы. Определенные лекарственные средства могут взаимодействовать с пуринергическим компонентом. Нейромодуляторы изменяют процессы нейропередачи. Такую функцию могут выполнять циркулирующие нейрогормоны, оказывающие местное действие, или нейромодулярные субстанции, выделяемые из однотипных нервов. Нейромодуляторы могут быть либо пресинаптичес-кими (снижающими или увеличивающими суммарное выделение медиатора), либо постсинаптическими, изменяющими степень, время или направление нейропередачи. Пре- и постсинаптическая модуляция, суммируя, ослабляет или усиливает действие медиаторов. В отличие от нервно-мышечного синапса, автономный нейроэф-фекторный синапс существует в динамическом состоянии и подвергается единственной постсинапсальной специализации. Биогенные амины часто должны преодолевать большое расстояние. Установлено, что они имеют короткий период полужизни. Нейромодуляторы обеспечивают биологический механизм усиления или пролонгирования их действия. NPY также действует совместно с норадреналином (НА) и АТФ. Однако в некоторых сосудах (селезенки, скелетных мышц, мозга)
22 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
NPY оказывает незначительное прямое сосудосуживающее действие. Наряду с этим, нейромодулятор, действуя пресинап-тически, ингибирует выделение НА из нерва и/или постсинаптически усиливает действие НА.
В сердце и мозгу имеются местные нейроны, в которых NPY используется как главный медиатор. В селезенке NPY является медиатором, действующим совместно с НА в периваскулярных симпатических нервах. Многократная стимуляция определенных нервных окончаний вызывает освобождение из везикул этих медиаторов. Классические медиаторы, например ацетилхолин, существуют совместно с VIР в парасимпатических нервах многих органов. В этом случае оба медиатора сохраняются
в разных везикулах. Этим объясняется существование различных вариантов их освобождения при разных видах стимуляции в зависимости от их расположения. Например, в слюнных железах могут независимо функционировать ацинарные клетки и железистые кровеносные сосуды. Взаимодействие достигается селективным освобождением ацетилхолина при Н4 (слабой) повторной стимуляции и VIP — при В4 (сильной) повторной стимуляции. На многие биологические состояния, в частности беременность, гипертензию, различные болезни, старение, влияет состояние взаимоотношений между комедиаторами, обуславливающими компенсаторный ответ, который позволяет управлять важнейшими физиологическими функциями [8].
1.3. ФУНКЦИИ автономной нервной системы
Симпатическая нервная система, отвечая на внутренние или внешние воздействия, учащает сердечный ритм, увеличивает артериальное давление и силу сердечных сокращений, расширяет бронхиальное дерево и перераспределяет кровь
из кишок и других внутренних органов, улучшая снабжение произвольных мышц, участвующих в ответной реакции (табл. 1.1). Парасимпатические нервы обеспечивают прежде всего энергетические процессы.
Таблица 1.1. Ответы, выявленные в органах-эффекторах при стимуляции симпатических и парасимпатических нервов
Орган-эффектор Адренергический ответ Вовлекаемый рецептор симпатической НС Холинергический ответ Доминирующие рецепторы А (адренергические) X (холинергические)
Сердце:
ритм сокращений Увеличение ₽1 Снижение X
сила сокращений Кровеносные сосуды: Увеличение ₽1 Снижение X
артерии (большинство) Вазоконстрикция А
скелетных мышц Вазодилатация 02 А
вены Вазоконстрикция а2 А
Бронхиальное дерево Бронходилатация 02 Бронхоконстрикция X
Капсула селезенки Сокращение А
Матка Сокращение Вариабельно А
Капсула предстательной железы Сокращение «1 А
Пищеварительный канал Расслабление а2 Сокращение X
Функции автономной нервной системы 23
Продолжение табл. 1.1
Орган-эффектор Адренергический ответ Вовлекаемый рецептор симпатической НС Холинергический ответ Доминирующие рецепторы А (адренергические) X (холинергические)
Глаз: радиальная мышца Сокращение «1 А
(радужка) (мидриаз)
циркулярная мышца Сокращение X
(радужка) (миоз)
циркулярная мышца Расслабление р Сокращение X
(радужка) Почки Секреция ренина Pl (аккомодация) А
Мочевой пузырь: детрузор Релаксация р Сокращение X
треугольник и сфинктер Сокращение Расслабление А, X
мочеточник Сокращение «1 Расслабление А
Выделение инсулина поджелудочной железой Снижение а2 А
Жировые клетки Липолиз Р1 А
Печень (гликогенолиз) Снижение «1 А
Волосяные фоликулы, Снижение «1 А
гладкие мышцы (пилоэрекция)
Назальная секреция Повышение X
Слюнные железы Повышение Повышение X
секреции секреции
Потовые железы Повышение «1 Повышение X
секреции секреции
Таблица 1.2. Преобладание в иннервации органов-мишеней тонуса определенного отдела автономной нервной системы
Орган Отдел АНС Орган Отдел АНС
Цилиарная мышца глаза ПНС Пищеварительный канал ПНС
Радужка ПНС Матка Мочевой пузырь ПНС ПНС
Атриовентрикулярный узел ПНС Слюнные железы ПНС
Вены СНС Потовые железы СНС (холинергическая иннервация)
Во многих органах выявлена двойная иннервация: симпатическая и парасимпатическая. Часто медиаторы этих систем оказывают противоположный эффект.
Стимуляция одной системы может оказать возбуждающее действие на органы-мишени, в то время как стимуляция второй системы — ингибирующее. Гла
за, сердце, бронхиальное дерево, пищеварительный канал и мочеполовая система имеют специфическую иннервацию. Например, симпатическая стимуляция сердца увеличивает частоту и силу сокращений, ускоряет проводимость через атриовентрикулярный узел, в то время как парасимпатическая стимуляция снижает час
24 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
тоту и сократимость и замедляет проводимость через атриовентрикулярный узел. Одна из двух систем в норме оказывает преобладающее действие на функцию органов. В очень немногих органах (определенные отрезки сосудов в селезенке, мышце, поднимающей волосы) только одна симпатическая система обеспечивает тоническое воздействие (табл. 1.2).
Для того чтобы предсказать фармакологический эффект лекарственного средства, необходимо знать его влияние на АНС в разных органах. Так, блокада симпатического влияния активирует ПНС и наоборот. Например, при атропиновой блокаде мускаринового тонуса преобладающее влияние на сердце оказывает симпатическая нервная система, что является причиной тахикардии.
1.4. ОЦЕНКИ ФУНКЦИОНАЛЬНОГО СОСТОЯНИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ И ЭФФЕКТОВ ЭПИНЕФРИНА
Для оценки характера и глубины патофизиологических нарушений, особенно у больных в критическом состоянии, необходимо определить также характер и глубину нарушений функции регулирующих систем, в частности дисфункциональные признаки АНС. Для тяжелобольных важно знать функциональное состояние симпатической и парасимпатической систем [19, 20].
Наиболее часто первым признаком дисфункции симпатической нервной системы является ортостатическая гипотензия, проявляющаяся головокружением, снижением артериального давления (АД), обмороком и очаговыми неврологическими симптомами. Обычно эти симптомы возникают при переходе больного из горизонтального положения в вертикальное, а при переходе обратно в горизонтальное положение они проходят. Наименее заметным проявлением синдрома является умеренное снижение сердечно-артериального давления без обычного учащения пульса на 10 — 20 ударов в минуту. Отсутствие учащенного пульса в положении стоя и после физической нагрузки предполагает нарушение механизмов учащения сокращений сердца, контролируемых СНС.
Понижение потливости может быть также отнесено к признакам дисфункции СНС. Этот признак считается достоверным, если он сочетается с бледностью, сухостью кожи, мышечной слабостью и фас-цикуляциями.
Реакция зрачка на свет является показателем функции АНС. Небольшой, фик
сированный зрачок или зрачок, не расширяющийся на щипок в области боковых отделов шеи (цилиоспинальный рефлекс), предполагает угнетение функции СНС.
Ночная полиурия как показатель недостаточности функции СНС встречается относительно редко. Она вызвана снижением скорости гломерулярной фильтрации в вертикальном положении и превышает норму в положении лежа на спине.
Слабость, голодные боли, летаргия, зевота и мышечные подергивания могут быть симптомами гипогликемии, обусловленной угнетением функции СНС и снижением метаболизма глюкозы.
Оценивая функцию СНС, необходимо помнить, что многие лекарства влияют на нее. Наиболее часто это влияние выражено у антигипертензивных препаратов. К лекарствам, выражение влияющим на АНС, но применяемым не с гипотензивной целью, относятся фенотиазины, обладающие aj-блокирующей антиадренергической и антихолинергической активностью; амфетамины и некоторые антибиотики, например, изониазид и фуразолидон (ингибиторы моноаминоксидазы (МАО)).
Имеется целый ряд заболеваний, при диагностике которых особое внимание должно быть направлено на определение функционального состояния АНС (табл. 1.3).
Кроме клинических симптомов определенных заболеваний для оценки глубины и характера расстройств АНС могут использоваться физиологические тесты.
Оценки функционального состояния автономной нервной системы и эффектов эпинефрина 25
Таблица 1.3. Заболевания, при которых имеет место недостаточность автономной нервной системы
Невралгические Соматические
Спинная сухотка Сирингомиелия Гематомиелия Повреждение (разрыв) спинного мозга Опухоли дна IV желудочка Паркинсонизм Выраженный церебральный склероз Синдром Ши —Драгера Болезнь Вернике Семейная дизавтономия Сахарный диабет Хроническое заболевание почек Ослабление питания с/без алкоголизма Комбинированные соматические заболевания Ятрогенные симпатэктомия хордотомия Введение лекарств, влияющих на АНС
Наиболее популярным тестом для исследования автономной функции является прием Вальсальва (см.: [20]). Пациенту предлагается сделать выдох при закрытой голосовой щели или в закрытую емкость с манометром (для точности дозирования) в течение 20 —30 с. Это обычно повышает вну-тригрудное давление до 40 мм рт. ст.*, что значительно снижает венозный возврат крови к сердцу. У пациентов с интактной СНС можно наблюдать кратковременное повышение артериального давления, а затем стремительное падение его и пульсового давления, сопровождаемое учащением пульса.
Кратковременный подъем АД в начале пробы обусловлен увеличением венозного притока крови к сердцу в связи с тем, что повышение внутригрудного давления как бы выдавливает кровь из легких и венозных сосудов грудной полости в направлении сердца, что увеличивает выброс. Однако в дальнейшем, в результате сдавливания легких и сосудов, приток крови к сердцу уменьшается из-за повышения резистентности току сдавливанием повышенным внутригрудным давлением сосудов. Когда системное давление падает, прессорные рецепторы в аорте и каротидном синусе инспирируют учащение пульса и периферическую вазоконстрикцию. Последняя, мобилизуя кровь из периферических сосудов, обусловливает повышение ЦВД, преодоление возросшей резистентности внутригрудных сосудов и обеспечивает
♦ 1 мм рт. ст. = 133,322 Па.
венозный возврат крови к сердцу, что предотвращает критические расстройства кровообращения, сохранение АД на оптимальном для жизнеобеспечения уровне. После нормализации внутригрудного давления при прекращении выдоха с сопротивлением возникает кратковременное увеличение систолического, диастолического и пульсового давления, так как венозный возврат восстанавливается и становится даже несколько большим, чем в норме, на время восстановления нормального баланса, что приводит к увеличению ударного и минутного объема сердца. Это вызывает увеличение парасимпатического влияния, что проявляется брадикардией.
Нормальная реакция зависит от функциональной сохранности структуры компонентов, участвующих в пробе барорецепторов, афферентных путей, вазомоторных центров, соответствующих симпатических и парасимпатических путей и центров, органов-эффекторов (артериальные и венозные сосуды, сердце). При дисфункции АНС отмечаются изменения пробы Вальсальва. При патологии СНС отсутствует учащение пульса и более значительное падение АД в фазе положительного внутригрудного давления. При этом не наблюдается повышение систолического и диастолического давления при прекращении выдоха против сопротивления. Это может иметь место при сердечной недостаточности и гиповолемии. Парасимпатическая недостаточность проявляется отсутствием замедления пульса во время повышения АД.
26 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
Холод также может быть использован для оценки функции СНС. У пациентов с нормальной функцией АНС при погружении руки в ледяную воду (+4 °C) на 60 с систолическое давление повышается на 16 —20 мм рт. ст., а диастолическое — на 12 — 15 мм рт. ст. Отсутствие такой реакции указывает на нарушение соответствующей рефлекторной дуги. Такой дефект реакции может иметь место при повреждении соответствующих структур чувствительного нерва, спинномозгово-таламического тракта, СНС или органа-эффектора.
Комбинированное использование приема Вальсальва и холодового теста помогает определить уровень поражения. При ненормальной реакции при пробе Вальсальва и нормальной реакции на холод поражение локализуется, вероятнее всего, на уровне барорецепторов или на уровне афферентного нерва от барорецепторов. Такой эффект обнаруживается у некоторых диабетиков и пациентов со спинной сухоткой.
Изменения объема крови также можно использовать для оценки СНС. Для этого сжимают бедра наложенными на них манжетками. У здоровых людей при раздувании манжеток до 15 — 20 мм рт. ст. в течение 10 мин САД снижается более чем на 10 мм рт. ст. в основном за счет диастолического давления. При угнетении СНС такая «кровопотеря» вследствие флеботомии сопровождается значительным и продолжительным снижением АД.
Инфузия 3000 мл физиологического раствора в час у здоровых людей приводит лишь к незначительным изменениям АД, но у пациентов с нарушением СНС увеличение АД достигает 20 — 35 мм рт. ст. У здоровых людей во время такой инфузии диурез составляет 6 мл/мин, а выделение Na — 15 мэкв/ч; у пациентов с повреждением СНС — соответственно 23 мл/мин и Na — 80 мэкв/ч. Необходимо помнить, что этот тест нельзя применять к пациентам с сердечной недостаточностью из-за риска развития отека легких.
Для оценки СНС может быть также использована реакция на умственную нагрузку, например решение арифметической задачи. У здоровых людей при этом
увеличивается АД и частота пульса, так как умственная нагрузка стимулирует эфферентные пути и не зависит от афферентных. Сочетание этого теста с приемом Вальсальва дает нам информацию об уровне поражения симпатической части рефлекторной дуги. Ненормальная реакция на прием Вальсальва, а также стабильные АД и пульс во время умственной нагрузки предполагают нарушение на уровне вазомоторных центров, эфферентных путей и органа-эффектора.
Фармакологические тесты и биохимические исследования могут дать информацию о состоянии эфферентного отдела АНС.
Фармакологические тесты. Если в конъюнктиву нормального иннервированного глаза закапать 1—2 капли раствора адреналина в разведении 1 : 1000, то эффект будет маловыраженным. Если же глаз денервирован на уровне постганглионарных симпатических волокон, то при закапывании той же дозы зрачок выражено расширится.
У здоровых людей инфузия норадреналина (0,05 мг/кг в минуту) увеличивает систолическое давление менее чем на 23 мм рт. ст., а диастолическое — менее чем на 13 мм рт. ст., в среднем на 8 —9 мм рт. ст. У пациентов с симпатической денервацией (исключая а-блокаторы) такая инфузия норадреналина ведет к значительному повышению АД. Реакция со стороны АД при инфузии норадреналина, меньше ожидаемой, предполагает нарушение чувствительности органа-эффектора, что наблюдается чаще всего у лиц, принимающих лекарства с a-блокирующим эффектом. Важно установить, нет ли а-блокады у пациентов, которым предстоит операция, так как при наличии такой блокады дозы многих прессорных лекарств оказываются недостаточными (левартеренол, метара-минол). В таких случаях более эффективным является ангиотензин, эффект которого не зависит от а-рецепторов.
При недостаточности АНС отмечается усиление реакции на различные вазоактивные вещества. Так, нитроглицерин, принятый под язык, обычно дает незначительное снижение АД, но при нарушении функ
Оценки функционального состояния автономной нервной системы и эффектов эпинефрина 27
ции афферентных путей, вазомоторного центра, симпатических эфферентных путей или органов-эффекторов систолическое давление снижается более чем на 15мм рт. ст., а диастолическое — более чем на 5 мм рт. ст. Таким же достоверным признаком симпатической недостаточности является усиленная гипотензивная реакция на тетраэтиламмония хлорид и окситоцин.
Функция потовых желез, за исключением аксиллярных, контролируется эффекторными симпатическими волокнами, идущими к коже от центра потоотделения в гипоталамусе. Нормальные субъекты потеют, когда температура окружающей их среды настолько высока, что температура в прямой кишке повышается на 0,5 — 1,0 °C.
При внутрикожном введении 0,1 мл раствора ацетилхолина в разведении 1:10 000 или никотина в разведении 1 : 100 000 наблюдается потение кожи вокруг места инъекции. Это свидетельствует об интактности местной иннервации потовых желез. Если ацетилхолин не эффективен, то внутрикожная инъекция пилокарпина в разведении 1 : 1000 может вызвать общее и местное потоотделение, что говорит об интактности потовых желез и их способности отвечать на стимуляцию.
Препараты, блокирующие а-рецепторы, снижают эффект освобождения эндогенных катехоламинов тирамином так же, как и эффекты инфузируемых экзогенных катехоламинов. При низких или обычно используемых дозах блокада а-рецепторов и действие катехоламинов или тирамина уравновешены. Это равновесие может быть нарушено увеличением дозы катехоламинов или тирамина. Дозы последнего, необходимые для получения стандартного прессорного эффекта, прямо пропорциональны степени адренергической блокады. При использовании Р-6локирующих агентов (например, пропранолола) степень блокады сердцеускоряющего эффекта изопротеренола или инфузируемого норадреналина может быть показателем эффективности Р-6локады.
Все вышеизложенное свидетельствует о ведущей роли СНС в регуляции функций
организма, особенно в экстремальных ситуациях и развитии реакций компенсации и адаптации (табл. 1.4). Адренергические нейроны влияют на многие функции организма, наиболее важными из которых являются кровообращение и дыхание, процессы жизнеобеспечения. Симпатическая стимуляция обеспечивает реакции «борьбы-полета». Вентиляция легких возрастает за счет бронходилатации. Производительность работы сердца повышается за счет увеличения сократительной способности сердца и возрастания частоты сокращений. Увеличивается перфузионное давление в жизненно важных органах и возникает вазоконстрикция в других органах. Функция как пищеварения, так и мочеполовой системы, угнетается в результате релаксации мышц этих органов и сокращения их сфинктеров. Желудочно-кишечная секреция активно ингибируется за счет гормонов коры надпочечников. Активация метаболизма вызывает повышение содержания в крови энергетических субстратов: глюкозы и жирных кислот. Оба эндогенных катехоламина, норэпинефрин и эпинефрин, обладают а- и Р-рецепторной агонистической активностью. Однако норэпинефрин имеет минимальную р2-рецеп-торную активность, в то время как эпинефрин стимулирует как Рр, так и р2-ре-цепторы.
Инфузируемые экзогенные и эндогенные катехоламины, освобождаемые нервными окончаниями постганглионарных волокон СНС, отличаются по своему действию. Например, введенный экзогенный норэпинефрин может вызвать снижение частоты сердечных сокращений, в то время как освобождаемый эндогенный норэпинефрин в ответ на стресс всегда вызывает тахикардию. Этот физиологический эффект предопределил использование норэпинефрина для купирования тяжелой пароксизмальной синусовой тахикардии с резким снижением АД.
Физиологические ответы при воздействии медиаторов на а-адренорецепторы могут широко варьировать (табл. 1.5). а-Рецепторы активируются при симпатическом возбуждении, что проявляется сокращением гладких мышц тела, включая
28 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
Таблица 1.4. Эффекты активации симпатической нервной системы
Органы Стимуляция Ингибирование
Сердце Кровеносные сосуды Дыхание Пищеварительный канал Мочеполовая система Метаболизм и эндокринная система Ритм, проводимость, сократимость Вазоконстрикция (кожа, кишки, печень, сердце, почки ) Дыхательный центр Сфинктеры Гликогенолиз (мышцы, печень) Липолиз Гликонеогенез Секреции инсулина (Pi) ренина АДГ Вазодилатация (скелетные мышцы, сердце, мозг) Бронходилатация Гладкие мышцы Мочевые и маточные мышцы Секреция инсулина (a-стимуляция или Р1-антагонизм)
Таблица 1.5. Адренорецепторные различия в органах
Органы Стимуляция Ингибирование
а-Адренорецепторы
Сердце Кровеносные сосуды Пищеварительный канал Мочеполовая система Метаболизм и эндокринная система Вазоконстрикция (кожа, кишки, печень, сердце, почки) Сфинктеры Сфинктеры Секреции инсулина
^-Адренорецепторы
Сердце Кровеносные сосуды Дыхание Пищеварительный канал Мочеполовая система Метаболизм и эндокринная система (Pt) Ритм, проводимость, сократимость (?) Дыхательный центр (р2) Гликогенолиз (мышцы, печень) (pl) Липолиз (р2) Гликонеогенез (Pt) Секреции инсулина (?) ренина (?) АДГ (р2) Вазодилатация (скелетные мышцы, сердце, мозг) (р2) Бронходилатация (р2) Гладкие мышцы (р2) Мочевые и маточные мышцы
Примечание. (0j) — возбуждение в pf-рецепторах; (р2) — возбуждение в р2-рецепторах; ? — спорные данные.
цилиарную мышцу глаз, гладкие мышцы сосудов, бронхов, мочевой системы. Кроме того, желудочно-кишечный и мочеполовой сфинктерные механизмы могут также стимулироваться через а-адренорецепторы. Регуляция выделения инсулина поджелудочной железой осуществляется посредством а-адренореактивного механизма.
В периферических сосудах постганглионарные ар и а2-адренорецепторы находятся как в артериях, так и в венах, и действуют как вазоконстрикторы.
Р-Адренергический механизм является главным в симпатической стимуляции сердца, расслаблении сосудов и гладких мышц бронхов, стимуляции секреции ренина почками, влиянии на метаболические про
Симптомы и признаки дисфункции парасимпатической (холинергической) системы 29
цессы, включая липолиз и гликогенолиз. 0! -Рецепторный механизм включает в себя кардинальные эффекты и реализацию действия кислот и ренина, тогда как р2-ре-цепторный механизм отвечает за релаксацию гладкомышечных структур и гипергликемию. В некоторых случаях р2-рецеп-торы могут также участвовать в регуляции сердечной деятельности. В то время как различные изменения АД или частоты сердечных сокращений могут быть вызваны норэпинефрином или эпинефрином, хроническая гипертензия не возникает при повышении их секреции. В 85 % случаев гипертензия связана с изменениями уровня ренина. Важнейшим действием эпинефрина является усиление микроциркуляции.
Психологическая и физическая стимуляция может вызвать различные компенсаторные ответы. В то время как в публикациях говорится о ведущей роли активации надпочечников, физиологические ответы свидетельствуют о главной роли СНС. Таким образом, ответ на стресс может варьировать в зависимости от интенсивности и вида внешнего воздействия.
Несмотря на то что многие анестезиологические методы могут ослабить ответ на стресс, вопрос об оказании помощи пациенту оставался открытым. Так было до тех пор, пока не появилась методика пролонгированной постоперационной эпидуральной анестезии. В некоторых случаях при операциях на сердце и сосудах использование больших доз опиоидов и другие техники значительно снижают операционный стресс и улучшают результат лечения.
Катехоламины вызывают высвобождение глюкозы в случае системной гипогликемии, что способствует нормализации уровня глюкозы и снабжению клеток энер
гетическим субстратом. Симпатическая нервная стимуляция через Р-рецепторы усиливает гликогенолиз в печени и мышцах и освобождает несвязанные жирные кислоты из жировой ткани, что приводит к повышению уровня глюкозы в крови. У новорожденных эпинефрин участвует в терморегуляционной функции бурого жира (недрожательный термогенез).
а2- и Р2-Рецепторы представлены также в поджелудочной железе. Стимуляция а2-адренорецепторов подавляет секрецию инсулина в островках поджелудочной железы и ингибирует липолиз; блокада этих рецепторов может усилить секрецию инсулина и вызвать значительное снижение уровня глюкозы в крови. Стимуляция Р-рецепторов активирует секрецию глюкагона и инсулина [22].
Кроме того, эпинефрин участвует в регуляции уровня сывороточного калия. P-Адренергическая стимуляция может вызвать преходящую гиперкалиемию вследствие выхода калия из гепатоцитов вместе с глюкозой. В этом случае наблюдается более длительная гиперкалиемия, так как P-стимуляция регулирует перемещение калия в эритроцитах и мышечных клетках. Экзогенно введенный и эндогенно секретируемый эпинефрин стимулирует р2 -рецепторы эритроцитов, активируя аденилатциклазу и Na+-K+-АТФ-азу, транспортируя калий в клетки. Этот эффект приводит к снижению концентрации сывороточного калия и может вызвать сердечные аритмии, инфаркт миокарда и другие стрессовые состояния. Р2 - Адренергическая блокада играет главную роль в ингибировании движения калия. Селективные и неселективные р-бло-каторы используются для защиты сердца в постинфарктном периоде от аритмий.
1.5. СИМПТОМЫ И ПРИЗНАКИ ДИСФУНКЦИИ ПАРАСИМПАТИЧЕСКОЙ (ХОЛИНЕРГИЧЕСКОЙ) СИСТЕМЫ
Симптомы при парасимпатической недостаточности обычно менее заметны, чем при дисфункции симпатической нервной системы. К обычным симптомам парасим
патической дисфункции относятся импотенция и ослабление либидо. Анорексия, недержание мочи и кала, уринарная ретенция, чередование запоров и поносов
30 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
также могут свидетельствовать о нарушении функции ПНС. Кроме этого, может отмечаться атрофия радужки, угнетение реакции зрачка на свет и аккомодации, при повторном раздражении роговицы уменьшается слезотечение, в ответ на раздражение лимоном понижается саливация.
Частый пульс в покое непропорционально увеличивается при ортостатических изменениях. Тонус анального сфинктера и бульбокавернозный рефлекс контролируются сакральными парасимпатическими нервами.
В отличие от генерализованных эффектов СНС в ответ на стресс, ПНС анатомически и функционально более локализована в своих эффектах. Активация ПНС обеспечивает сохранение энергии и поддерживает функциональную активность организма. Генерализованный парасимпатический ответ возникает только в истощенном организме, при этом не контролируются саливация, слезотечение, рвота, мочевыделение, дефекация. В то время как СНС участвует в ответе на стрессовую ситуацию с целью выживания, ПНС играет главную роль в процессе поддержания жизни.
При стимуляции ПНС секретируется ацетилхолин, чье действие почти диаметрально противоположно действию норэпинефрина и эпинефрина. В основном мускариновый эффект ацетилхолина качественно идентичен эффектам при стимуляции блуждающего нерва. Ацетилхолин является единственным эндогенным медиатором, вызывающим одновременно брадикардию и гипотензию. Доза ацетилхолина влияет на его эффекты: небольшая внутривенная доза может вызвать генерализованную вазодилатацию (включая коронарные и легочные сосуды), в то время как большая доза вызывает отрицательный хроно- и дромотропный эффект. В сосудистом русле существуют многочисленные мускариновые рецепторы. Они способствуют секреции ендотелийсвязан-ного релаксирующего фактора (EDRFS), вызывающего вазодилатацию. Однако при повреждении эндотелия стимуляция ацетилхолиновых рецепторов вызывает вазоконстрикцию. Второй механизм сосудис
той релаксации ацетилхолином — это ингибирование норэпинефриновой секреции из адренергических нервных окончаний.
Ацетилхолин замедляет ритм сердечных сокращений, скорость проводимости в синоатриальном и атривентрикулярном узлах, сокращение предсердий, но при этом не происходит увеличения активности СНС. В синоатриальном узле ацетилхолин вызывает гиперполяризацию мембраны, нарушая возобновление порогового потенциала, что способствует задержке следующего потенциала действия. Это замедляет ритм сердца. Несмотря на увеличение продолжительности потенциала действия и эффективного рефрактерного периода, скорость проведения импульса через кардиомиоциты предсердий не изменяется. В атриовентрикулярном узле ацетилхолин снижает скорость проводимости и увеличивает эффективный рефрактерный период. Это снижение узловой проводимости может привести к полной блокаде атриовентрикулярного узла. Такой эффект оказывают холинергические агонисты. В желудочках ацетилхолин угнетает автоматизм волокон Пуркинье, увеличивая порог возбудимости. В сердце мускариновые рецепторы представлены как пре-, так и постсинаптическими составляющими.
Ацетилхолин ингибирует адренергическую стимуляцию сердца, пресинаптически ингибируя секрецию норэпинефрина в симпатических нервных окончаниях и постсинаптически противодействуя эффектам катехоламинов в миокарде.
Парасимпатическая активация по-разному влияет на сердечно-сосудистую систему. Ацетилхолин стимулирует хеморецепторы каротидных и аортальных телец. Холинергическая стимуляция вызывает сокращение гладких мышц, включая бронхиальное дерево. В пищеварительном канале и мочеполовой системе сокращаются гладкие мышцы стенок, но расслабляются сфинктеры.
Ацетилхолин сужает гладкие мышцы радужки, вызывая миоз.
Основными симптомами чрезмерной холинергической стимуляции являются: тошнота, рвота, спазмы кишок, отрыжка, непроизвольное мочеиспускание и дефекация.
Симптомы и признаки дисфункции парасимпатической (холинергической) системы 31
Парасимпатическая стимуляция вызывает усиление секреции слезных, трахеобронхиальных и эндокринных желез.
ПНС принимает также участие в регуляции сосудистого тонуса. Ацетилхолин как медиатор ПНС вызывает фармакологические эффекты. Кроме того, он оказывает дилатирующее действие на сосуды in vivo. В 1980 г. R. F. Furchgott и I. V. Za-wadzki [23] писали, что кровеносные сосуды, имеющие интактный эндотелий, могут дилатироваться под воздействием ацетилхолина, а сосуды с поврежденными эндотелиальными клетками, отвечая на стимуляцию ацетилхолином, продуцируют один или несколько эндотелийсвязанных релаксирующих факторов и имеют немногочисленные нервно-рецепторные агонисты, включая серотонин, аденозин, гистамин и катехоламины.
Радикал NO — первый идентифицируемый EDRFS, продуцируется эндотелиальными клетками в ничтожных количествах, но оказывает расслабляющее действие на гладкие мышцы сосудов и модифицирует эффекты многих вазоконстрикторов. Нитроглицерин снижает содержание NO в миоцитах сосудов. Механизм этого сосудосуживающего действия заключается в активации гуанилатциклазы через каскад фосфорилирования протеинов (в частности, фосфорилирования актомиозином). Таким образом, при поражении эндотелия атеросклеротическим процессом снижается выделение EDRFS и усиливается кон-стрикция. Это объясняет различные эффекты у больных с поврежденными сосудами.
NO, механизм синтеза и выделение которого малопонятны, играет важную роль в нейропередаче и является нейромедиатором в мозжечке и внутренних органах (NMDA вызывает выделение NO в мозжечке). NO продуцируется в цикле превращения аргинина в цитруллин в классе ферментов, известных как NO-синтетазы (NOs). Это семейство энзимов сейчас насчитывает 5 изоформ, большая часть из них была обнаружена в мозжечке, идентифицирована и клонирована. Ткань мозжечка нуждается во флавинах, редуцирующих НАДФ • Н (NADP • Н) и кальмодулин.
Некоторые изоформы возникают при септическом (нетравматическом) шоке или во время химиотерапии. Есть основания предполагать, что NO может вызывать гипотензию. Некоторые изоформы энзимов, например в мозжечке, содержатся в популяции клеток, обладающих тонической активностью. Иногда структура NO-синтетазы весьма сходна с цитохромом Р450. Доклинические исследования с ингибиторами NO-синтетазы позволили предположить, что сосудистая система находится в состоянии активной вазодилатации.
Эндотелиальные клетки осуществляют сложный контроль кровообращения. Кроме продуцирования NO они участвуют также в метаболизме многих вазоактивных аминов, фермента, способствующего превращению ангиотензина I в ангиотензин II, секреции NO, простациклина и вазоактивного эндотелиального пептида-1 (ЕТ-1). NO, ЕТ-1 и простациклин секретируются эндотелиальными клетками под непосредственным влиянием их микроокружения. Простациклин и NO вызывают релаксацию подлежащих гладких мышц на наружной стороне, в то время как на внутренней стороне они могут действовать как разобщенно, так и согласованно, разделенные пластинками из группировок на эндотелии. Наблюдается чистый синергизм между антиагрегатным действием простациклина и подпороговой концентрацией NO. Простациклин обычно стимулирует секрецию NO. При стрессе секреция NO возрастает, активируя расширение сосудов, что является адаптационным механизмом. Имеются доказательства того, что NO функционирует через гуани-латциклазный механизм, а простациклин активирует аденилатциклазу, т. е. они активируют различные вторичные передатчики.
В то время как простациклин и NO являются короткоживущими вазодилататорами, инъекция вазоконстриктора ЕТ-1 вызывает мощный и длительный прессорный эффект. ЕТ-1, состоящий из 21 аминокислоты, соединенных двумя дисульфидными связями, является продуктом генерации фермента из 39 аминокислот — пре-Е-1, или «большого ЕТ-1».
32 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
Для обеспечения высокого уровня интенсивной терапии важно уметь оценить функциональное состояние ПНС, определить причину и уровень ее поражения. При оценке состояния ПНС необходимо иметь сведения о применяемых лекарствах, так как в клинической практике чаще всего приходится сталкиваться с медикаментозно обусловленной дисфункцией.
Из физиологических тестов может быть использован прием Вальсальва. Показателем нормальной функции ПНС после повышения АД является урежение пульса. У большинства здоровых людей массаж каротидных синусов ведет к замедлению пульса. Отсутствие этого эффекта указывает на нарушение функции или целостности парасимпатических путей. На такие же нарушения функции указывает отсутствие брадикардии при прикладывании льда к лицу или при надавливании на глазные яблоки (рефлекс Ашнера). Понижение перистальтики после приема бария также указывает на дисфункцию ПНС.
Фармакологические тесты для оценки ПНС не считают в достаточной степени
информативными. Ацетилхолин является медиатором на всех уровнях ПНС, в синаптических связях с ЦНС, периферических ганглиях, нервных окончаниях и органах-эффекторах. Поэтому имеющиеся фармакологические тесты с использованием атропина и ингибиторов холинэстеразы не дают полной информации для определения уровня поражения ПНС, так как эти препараты действуют на всех уровнях этой системы. При этом следует учитывать, что атропин более эффективен на уровне органных рецепторов, чем на других уровнях ПНС.
Нарушение функции супрасегментар-ных центров АНС является наиболее трудно диагностируемым. Дисфункция центральных отделов ВНС может свидетельствовать о диффузном или очаговом поражении ЦНС. Обычно она сопровождает синдром диэнцефалита или такие заболевания, как болезнь Ши —Драгера, Паркинсона, фамильная дизавтономия, гематомиелия, спинная сухотка, болезнь Вернике, опухоль в области IV желудочка.
1.6. РОЛЬ автономной нервной системы в регуляции метаболизма, ЭНЕРГЕТИЧЕСКОГО ОБМЕНА, УТИЛИЗАЦИИ ЖИРОВ И УГЛЕВОДОВ
Развитие критических и терминальных состояний всегда сопровождается кардинальными нарушениями метаболизма и энергетического обмена. Для оценки тяжести состояния и выбора целенаправленной терапии анестезиолог должен хорошо знать указанные патофизиологии, на которые оказывает выраженное влияние АНС.
Как норадреналин, так и адреналин, являются сильными активаторами мобилизации жира из жировой ткани. Это действие проявляется в быстром повышении плазменного уровня свободных жирных кислот (СЖК) и глицерола-продуктов гидролиза триглицеридов в плазме крови. Концентрация в крови этих быстроизме-няющихся метаболитов является показателем метаболизации жира из жировых депо. Утилизация этих метаболитов из крови происходит одновременно с их мобилизацией. В норме быстрота исчезно
вения из крови СЖК зависит в основном от их уровня в плазме и скорости кровотока в тканях, они потребляются (в основном в зоне спланхникуса и мышцах). В условиях повышенного потребления СЖК увеличенная мобилизация может быть замаскирована. В отличие от глицерола СЖК покидают кровь в основном в печени, даже увеличенный кровоток через мышцы при физической нагрузке выраженно не влияет на скорость снижения глицерола в крови. По этой причине уровень глицерола в крови наиболее точно отражает утилизацию жира.
Жиромобилизирующее действие катехоламинов определяется повышением содержания вторичного мессенджера — аденозинциклофосфата (циклический 3', 5'-АМФ), активирующего гормончувствительную липазу, катализирующую гидролиз триглицеридов на СЖК и глицерол. Этот энзим
Роль автономной нервной системы в регуляции метаболизма, энергетического обмена 33
быстро превращается в неактивную форму, если прекращается активация циклическим 3', 5'-АМФ, который быстро разрушается фосфодиэстеразой. Таким образом, процесс активного липолиза зависит от постоянства синтеза 3', 5'-АМФ, контролируемого системой аденилатциклазы. Катехоламины, поступающие в жировую ткань из крови, или норадреналин, выделившийся в жировую ткань из нервных окончаний при симпатической стимуляции, активируют систему аденилатциклазы, при этом увеличивается скорость образования 3', 5'-АМФ. При быстром круговороте всех компонентов системы возрастает скорость липолиза и соответственно скорость мобилизации жира. Чувствительность системы аденилатциклазы к катехоламинам и другим липолитическим стимулам может быть повышена гормонами щитовидной железы. Гормоны роста совместно с глюкокортикоидами увеличивают синтез липазы, обуславливая более выраженный, но отсроченный липолиз. Метилксанте-лины усиливают липолиз путем торможения фосфодиэстеразы, разрушающей 3', 5'-АМФ.
Инсулин тормозит мобилизацию жира двумя путями: 1) при наличии глюкозы он обеспечивает эстерификацию жирных кислот путем образования а-глицерофос-фата, который представляет собой глицериновую часть молекулы триглицеридов; 2) второй путь, возможно, наиболее важный, заключается в том, что инсулин тормозит образование активной липазы, действуя на систему аденилатциклазы. Это его действие не зависит от метаболизма глюкозы. Таким образом, инсулин противодействует жиромобилизирующему эффекту катехоламинов. Норадреналин и адреналин тормозят секрецию инсулина поджелудочной железы в ответ на различные экстремальные стимулы. Возможно, что катехоламины осуществляют мобилизацию жира путем торможения панкреатической секреции инсулина, но их эффект мобилизации жира хорошо проявляется и у депанкреатизированных пациентов, и у лиц, страдающих ювенильным инсулинодефицитным диабетом. Это дает основания предположить прямое действие ка
2
4-220
техоламинов на гормоночувствительную липазу в жировой ткани. Мобилизация жира катехоламинами оказывает легкоопределяемые эффекты на ткани, в норме потребляющие СЖК в значительных количествах. Повышенное потребление в тканях, которые в норме утилизируют СЖК как главный энергоноситель (печень, почки, легкие, сердце, скелетная мускулатура) сопровождается усилением образования триглицеридов, которые накапливаются в тканях в виде жировых капелек. В большинстве тканей жирные кислоты могут утилизироваться в митохондриях окислительным путем до СО2 и Н2О. В печени они могут окисляться до кетонов, особенно если происходит недостаточная утилизация глюкозы. Повышенная продукция кетонов печенью обычно является следствием повышения потребления СЖК, вызываемым введением катехоламинов или повышенным выделением норадреналина нервными окончаниями при стимуляции СНС. Часть СЖК в печени может после эстерификации в триглицериды ре-транспортироваться в кровь в форме липопротеиновых молекул, которые переносят печеночные липиды в экстрапеченоч-ные ткани для накопления и окисления.
Недавние исследования показали, что гормоночувствительная липаза, активизируемая катехоламинами, также определяется в других тканях, включая печень, сердце и скелетную мускулатуру. Липолитический эффект катехоламинов на другие ткани, кроме жировой, требует дальнейшего изучения.
1.6.1. Влияние автономной нервной системы на метаболизм углеводов
Если в отношении метаболизма жиров эффект больше выражен у норадреналина, чем у адреналина, то в отношении метаболизма углеводов влияние адреналина выражено сильнее. Главными изменениями, развивающимися у животных под влиянием адреналина, являются гипергликемия и гиперлактемия. За эти изменения ответственны сложные физиологические механизмы, до конца еще не изученные. Ги
34 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
пергликемия обычно рассматривается как результат активации печеночного гликогенолиза. Имеются данные, указывающие, что глюкагон, секретируемый поджелудочной железой, является участником или посредником гликогенолиза, активируя аденилатциклазу в печени. Установлено, что адреналин активирует аденилатцик-лазу также во многих других тканях, возможно, его гипергликемическое действие связано с метаболическими эффектами. Например, в скелетных мышцах 3', 5'-АМФ активирует фосфорилазу и ускоряет превращение глюкогенсинтетазы в менее активную £)-форму. В результате этого ускоряется гликогенолиз. Этот и другие эффекты адреналина на метаболизм в мышцах приводят к высвобождению большого количества лактата в кровь. Часть этого лактата превращается в печени в глюкозу (цикл Кори). Этот процесс — глюконеогенез — также ускоряется адреналином. Таким образом, источником повышенного высвобождения глюкозы из печени может служить ускоренный глюконеогенез из лактата. Этот эффект может быть связан с повышенным уровнем 3', 5'-АМФ, так как повышение скорости окисления СЖК (поступающих в печень из жировой ткани или в результате гидролиза триглицеридов в печени) сопровождается изменениями ферментативной активности. В результате упомянутых изменений пируват переводится из окислительного цикла лимонной кислоты «на метаболический путь», приводящий к образованию фосфоено л пиру вата (шунт дикарбоновых кислот), а после — глюкозы.
Жиромобилизирующий эффект адреналина увеличивает окисление СЖК в мышцах. Это сопровождается соответствующим снижением потребления глюкозы, участвующей в развитии гипергликемии. Однако гипергликемический эффект катехоламинов не тормозится агентами, избирательно подавляющими мобилизацию жира. Гипергликемия увеличивается вследствие понижения способности поджелудочной железы реагировать на действие адреналина увеличением продукции инсулина. Но такое действие адреналина является маловероятным, так как уровень
инсулина не падает ниже того, который отмечается до введения адреналина. Увеличение секреции АКТГ, вызванное адреналином, удлиняет гипергликемию.
Меньшая выраженность гипергликемического эффекта норадреналина объясняется его меньшей способностью активизировать систему аденилатциклазы в других тканях, кроме жировой, хотя установлено, что адреналин активирует гликогенолиз и увеличивает выход лактата из мышц.
Этапом, ограничивающим скорость превращения глюкозо-1-фосфата, является фосфорилирование глюкозо-6-фосфата в фруктозо-1,6-дифосфат. Фермент, катализирующий эту реакцию, — фосфофруктокиназа — активируется циклическим 3', 5'-АМФ. Имеются доказательства, что эта реакция усиливается адреналином. Освобождение лактата из мышц также увеличивается при угнетении введения пирувата в цикл лимонной кислоты. Этот эффект объясняется повышенным окислением СЖК в мышцах.
Эфедрин активирует фосфорилазу в сердечной мышце. Это его действие, как считают, лежит в основе инотропного эффекта. Однако недавние исследования четко показали, что активация фосфорилазы не предшествует, а является следствием увеличения сократимости сердца, перфузируемого жидкостью с адреналином в концентрациях, при которых не происходит активации фосфорилазы. Это не исключает возможности участия в обеспечении инотропного эффекта циклического 3', 5'-АМФ, концентрация которого возрастает по мере увеличения инотропного эффекта адреналина.
Как адреналин, так и норадреналин, повышают респираторный газообмен и теплопродукцию, причем эффект адреналина выражен сильнее. Калоригенический эффект адреналина объясняется увеличением транспорта и утилизации лактата в результате повышения потребления О2, вызванного адреналином. Это повышение соответствует наблюдаемому при инфузии лактата. Калоригенический эффект норадреналина связан с увеличением мобилизации и утилизации СЖК. Это подтверждается тем, что специфическое тор
Роль автономной нервной системы в регуляции метаболизма, энергетического обмена 35
можение его жиромобилизирующего действия никотиновой кислотой (которая не влияет на его кардиореспираторное и гипергликемическое действие) снижает наполовину калоригенический эффект норадреналина.
Для реализации различных метаболических эффектов в органах и тканях под влиянием адреналина и норадреналина на клеточных мембранах имеются разнообразные рецепторы.
Симпатическая иннервация жировой ткани четко подтверждается высоким содержанием норадреналина в ней и в нервных окончаниях вокруг артерий этой ткани. Нервные окончания находятся в тесной связи с аденоцитами. Стимуляция симпатических волокон, иннервирующих жировую ткань, сопровождается вазоконстрикцией и снижением кровотока. Но если предварительно введены альфалитики, то стимуляция приводит к вазодилатации, на которую не влияет атропин. Это свидетельствует о том, что дилатация не осуществляется холинергическими нервами. Вазоконстрикция, сопровождающая стимуляцию, приводит к уменьшению освобождения СЖК и глицерола, но этого не наблюдается при интактном артериальном кровотоке жировой ткани. Стимуляция нервов, иннервирующих изолированные участки жировой ткани, приводит к освобождению СЖК, которое можно блокировать предварительной симпатэктомией. Особенно яркая картина симпатического возбуждения наблюдается при электрической стимуляции участков мез-энцефалона и диэнцефалона. При этом обычно повышается плазменный уровень СЖК и глицерола, увеличивается частота сердечных сокращений и повышается АД. Однако при такой стимуляции при наличии эффектов липолиза в жировой ткани практически отсутствуют выраженные эффекты со стороны сердечно-сосудистой системы.
Липолитические эффекты блокировались предварительным введением ганглио-блокаторов и не снимались адреналэктомией. Это дает основание предположить наличие центров, ответственных за симпатическую иннервацию жировой ткани. Они расположены вблизи среднего мозга 2*
и распространяются выше гипофиза и сосочкового тела в таламус (диэнцефалон) и в ограниченных участках колликуляр-ного серого вещества (мезэнцефалон). Кроме того, как свидетельствует ряд авторов, подобные центры обнаруживаются и на уровне спинного мозга (нижних шейных и верхних грудных сегментов).
Адаптация к внешней среде, сопровождающаяся гипертрофией коричневого жира. Коричневый жир отличается от обычного белого тем, что его клетки содержат множество небольших капелек жира вместо одной центральной капли, как в белом жире. Кроме того, в клетках коричневого жира много митохондрий, он больше васкуляризирован, чем белый жир. Норадреналин более выражено увеличивает его перфузию и потребление О2, а также стимулирует липолиз, реэстерифи-кацию и окисление жирных кислот. В ответ на холодовую экспозицию значительно возрастает теплопродукция за счет немышечного термогенеза. В депо коричневого жира мобилизация жирных кислот начинается сразу после рождения, одновременно с «пробуждением» нервной системы. Быстрота реакции, ее торможение гексаметонидином, ее связь с другими признаками «пробуждения» нервной системы — все это свидетельствует о ее адренергической природе.
Коричневый жир играет ведущую роль в поддержании температуры тела у новорожденных, располагаясь в основном в ин-терскапулярном пространстве и средостении. У новорожденных, умерших при гипотермии, количество коричневого жира в местах его запасов значительно уменьшено.
При выходе из состояния гибернации (лечебная гипотермия) первым согревается коричневый жир. Это эффект в значительной степени связан с норадреналином, местно выделяющимся из нервных окончаний. Увеличение потребления О2 при выходе из состояния гибернации в основном связано с возрастанием потребления О2 в ткани коричневого жира.
При кратковременной холодовой экспозиции теплопродукция происходит главным образом за счет дрожания (мышечного термогенеза). Но при длительной
36 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
экспозиции (до нескольких недель) наступает гипертрофия и гиперплазия коричневого жира, и мышечный термогенез сменяется немышечным. В противоположной ситуации, имеющей место у новорожденных и в состоянии гибернации при холодовой экспозиции, увеличение потребления О2 за счет увеличения его в коричневом жире является маловероятным. Однако и в данной ситуации калоригенический эффект был связан с норадреналином, с увеличением степени метаболизма повышалась мобилизация жира, окисление СЖК и глюкозы.
При выполнении физических упражнений мобилизация жира у человека увеличивается в течение 5—10 мин. При более продолжительной нагрузке мобилизация жира может возрасти в несколько раз. Клинические наблюдения свидетельствуют, что в основном это происходит за счет усиления симпатической активности. На это указывают следующие факты: во время упражнений быстро повышается плазменный уровень норадреналина, в то время как уровень инсулина изменяется мало; мобилизация жира при этих условиях уменьшается при введении Р-адренолити-ков. Секреция гормона роста возрастает, но это не вызывает мобилизации жира, так как уровень этого гормона повышается не ранее чем через 20 и более минут после начала упражнений.
Для анестезиологов и специалистов, занимающихся интенсивной терапией, важно знать, как изменяются эти процессы при психоэмоциональном стрессе, травме и хирургических вмешательствах.
Различные ситуационные стрессы вызывают у человека быструю мобилизацию жира, которую могут предотвратить ган-глиоблокаторы. Эта реакция может иметь биологическое значение вследствие возрастания потребности нервной системы в бы-строокисляемых субстратах, когда организм нуждается в повышенной мышечной активности («борьба или бегство»). Эффект более пролонгированного ситуационного стресса, при котором мобилизация жира может оказаться недостаточной, не установлен. Один из потенциально вредных эффектов стресса может возникнуть
в определенных ситуациях с развитием триглицеридемии. Секреция печенью богатых триглицеридами липопротеинов низкой плотности является прямым следствием потребления печенью СЖК. При увеличении мобилизации жиров во время физической нагрузки уровень триглицеридов падает, поскольку происходит потребление СЖК работающими мышцами. При усилении мобилизации жира в состоянии покоя повышенное поступление СЖК в печень увеличивает печеночную продукцию триглицеридов и уровень липопротеинов в плазме возрастает. Повышение уровня липопротеинов всегда сопровождает психоэмоциональный стресс у человека. Тот факт, что плазменный уровень триглицеридов зависит от уровня СЖК, чья продукция стимулируется внутривенной инфузией норадреналина, позволяет предположить, что механизм, посредством которого СНС влияет на плазменный уровень триглицеридов, связан с эффектом норадреналина на уровне СЖК.
Плазменный уровень СЖК увеличивается и после операций. При больших травмах этот признак повышения мобилизации жира указывает на степень повреждения тканей. При этом повышенная экскреция катехоламинов с мочой свидетельствует о возросшей активности СНС. Возможные неблагоприятные эффекты избыточной мобилизации жира в условиях, когда резко ограничена физическая активность, заключаются в жировой инфильтрации сердца, печени и других органов, повышении температуры тела, вызванном ка-лоригеническим эффектом катехоламинов, и тенденции к тромбозам, что объясняется способностью жирных кислот, особенно насыщенных, активировать процессы свертывания. Это служит основанием для использования с целью профилактики антикоагулянтов, особенно у пациентов с ограниченной физической активностью.
При наличии метаболического ацидоза жиромобилизирующий и калоригенический эффект катехоламинов угнетается. Ацидоз является очень важным метаболическим актом, сопровождающим различные виды шока и, возможно, способствует уменьшению количества окисляемых субстратов.
Роль автономной нервной системы в регуляции периферического кровообращения 37
1.6.2. Некоторые патофизиологические проблемы мобилизации жира
Сахарный диабет. У больных с инсулинодефицитным сахарным диабетом торможение мобилизации жира может быть вызвано ганглиоблокаторами. Мобилизация жира у этих больных активирует печеночный патогенез триглицеридемии, а также глюконеогенез и тормозит потребление глюкозы в мышцах. Эти процессы при различных видах стрессов с активацией СНС нарушают компенсацию и контроль инсулинодефицитного диабета.
Феохромоцитома. При этом заболевании обычно наблюдается повышенный уровень СЖК. Гиперметаболические эффекты при этой патологии объясняются вышеизложенными причинами.
Гипертиреоидизм. Эта патология сопровождается повышенной мобилизацией жира, что выражается в умеренном повышении уровня СЖК в плазме (за счет увеличенного сердечного выброса) и в более высоком уровне глицерола. Нет доказательств, что это связано с повышением симпатической активности. При гипертиреоидизме катехоламины, вводимые в организм, значительно усиливают липолиз. Это может свидетельствовать о повышенной чувствительности аденилатциклазы к этим и другим липолитическим стимулам. Субстрат, образовавшийся в процессе липолиза, способен удовлетворить энергетические потребности при гипертиреоидизме. В то же время гиперметаболизм практи
чески не зависит от мобилизации жира. Торможение липолиза (например, никотиновой кислотой) не снижает заметно у человека потребление О2 ни при эутиреоидном, ни при гипертиреоидном поражении щитовидной железы.
Заболевание коронарных сосудов. У людей с выраженным поражением коронарных сосудов отмечается повышение симпатической активности во время бодрствования, как правило, у них превалирует триглицеридемия. Во время проведения психологических тестов у пациентов с триглицеридемией заметно повышается уровень СЖК. Исходя из этого, вполне резонно предположить, что гиперактивность СНС является одной из причин триглицеридемии и что стресс у этих пациентов всегда будет усугублять течение болезни.
Влияние' фармакологических агентов на мобилизацию жира. Метилксантины увеличивают концентрацию 3', 5'-АМФ путем торможения фосфодиэстеразы, гидролизирующей его. Напитки, содержащие кофеин и подобные вещества, могут стимулировать липолиз.
Никотин повышает симпатическую активность, курение, даже нерегулярное, может вызвать мобилизацию жира.
^-Миметики (изопротеренол) являются потенциальными липолитиками. Адреналин и другие адреномиметики увеличивают мобилизацию жира как непосредственно воздействуя на жировую ткань, так и через нервные центры.
1.7. РОЛЬ автономной нервной системы В РЕГУЛЯЦИИ ПЕРИФЕРИЧЕСКОГО КРОВООБРАЩЕНИЯ
Роль АНС в осуществлении реакций компенсации на кровопотерю достаточно детально описана в работах, посвященных шоку. Однако о ее роли в регуляции периферического кровообращения известно мало.
Степень сокращения гладкой мускулатуры стенок артериол и вен в основном определяется активностью нервной системы. При снижении кардиоваскулярной функции производительность сердца уве
личивается за счет регуляции емкости и сопротивления периферического сосудистого ложа вследствие адренергической стимуляции. Роль адренергической регуляции периферической циркуляции возрастает в тех случаях, когда имеет место нарушение равновесия между сердечным выбросом и перфузионными потребностями периферических тканей. Местный контроль гладкой мускулатуры сосудистых стенок сводится главным образом к расширению сосудов
38 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
под влиянием метаболических продуктов, гипоксии и угнетения симпатических сосудосуживающих импульсов (а2). Вазоконстрикция обусловлена усилением симпатической активности. В скелетной мускулатуре рядом авторов обнаружены симпатические постганглионарные волокна, выделяющие ацетилхолин и вызывающие расширение артериол и вен, но их физиологическое значение в регуляции периферического кровообращения, вероятно, невелико.
Наибольшим сосудистым бассейном человеческого организма является бассейн зоны спланхникуса, который получает 25 % сердечного выброса в покое у здоровых людей. Потребление кислорода в этой области составляет четвертую часть общего потребления кислорода всем организмом. В отличие от других зон для данной характерна пропорциональность между потреблением кислорода и кровотоком.
Вторым по величине бассейном циркуляции являются почки, которые получают 20 % сердечного выброса, а насыщение гемоглобина кислородом из почечной вены ненамного ниже насыщения кислородом в артериальной крови.
Мозг получает 12 % сердечного выброса, а потребляет 20 % общего потребления кислорода организмом. Кровоток через сердце составляет лишь 4 % сердечного выброса, но потребление кислорода в этой зоне превышает эту цифру в три раза. Через скелетную мускулатуру протекает 20 % общего кровотока, а потребление кислорода составляет 30%. Она потребляет кислорода больше, чем любой другой орган, и подобно сердцу и мозгу экстрагирует большие количества О2 относительно своего кровотока. Объем кровотока в коже, важной функцией которой является регуляция температуры тела, очень лабилен. Относительная величина кровотока
СИ = 3 л/мин на 1 м2
СИ = 6 л/мин на 1 м2
Рис. 1.6. Региональные изменения кровотока у нормальных субъектов (а, б) и пациентов с сердечной недостаточностью (в, г) в покое (а, в) и при физической нагрузке (б, г):
1 — скелетная мускулатура; 2 — зона спланхникуса; 3 — почки; 4 — головной мозг; 5 — сердце; 6 — кожа; 7 — другие органы; СИ — сердечный индекс
через кожу в 5 раз превышает относительную величину потребления ею О2.
Кроме основной функции венозной системы — обеспечения возврата крови к сердцу — ее сосуды активно участвуют в поддержании нормальной циркулярной функции. Этот процесс регулируется СНС. Под влиянием физиологических стимулов вены могут сокращаться, чтобы предотвратить повышение давления и увеличение венозного возврата. Веноконстрикция возникает в ответ на эмоции, гипервентиляцию, физиологическую нагрузку, переход в вертикальное положение и сопровождается перемещением объема крови в систему центральных вен, лишенных сократительной активности [24] (рис. 1.6).
1.8. АВТОНОМНАЯ НЕРВНАЯ СИСТЕМА И ФИЗИОЛОГИЧЕСКАЯ НАГРУЗКА
Контроль функции сердца во время физической нагрузки включает три важных фактора: частоту сокращений, сократимость и механизм Франка —Старлин
га. Сегодня много известно о роли СНС в увеличении частоты сокращений миокарда. Вагусные влияния действуют на хронотропизм, и их эффект урежения сердеч
Автономная нервная система и физиологическая нагрузка 39
ных сокращений сам по себе обеспечивает хорошую сократимость. При нагрузке на ноги на фоне Р-адренергической блокады пропранололом отмечается ограничение учащения сердечных сокращений и усиление сократимости. Это приводит к уменьшению сердечного выброса. Адренергическая стимуляция сократимости миокарда и тахикардия уменьшают растяжение миокарда в конце диастолы. Кроме того, при введении пропранолола отмечается повышение потребления О2 периферическими тканями при физической нагрузке.
Большое значение в адаптации циркуляции при физиологической нагрузке имеет влияние СНС на артериолы и вены. Повышение симпатической активности уменьшает емкость сосудов и, следовательно, увеличивает венозный возврат во время физической нагрузки. Для преодоления резистентности сосудистого русла СНС увеличивает сердечный выброс в достаточной степени, чтобы удовлетворить повышенную потребность скелетных мышц в О2. На сегодня считается установленным, что повышенный кровоток при физической нагрузке сопровождается не только увеличением сердечного выброса, но и перераспределением кровотока вследствие повышения адренергической активности и действия местных сосудорасширяющих факторов. При физической нагрузке кровоток в скелетной мускулатуре и коронарных сосудах у здоровых пациентов повышается, что сопровождается уменьшением кровотока в почках и бассейнах спланхникуса. При сердечных нагрузках церебральный кровоток сохраняется на прежнем среднем уровне, но при увеличении нагрузки происходит его снижение вследствие уменьшения содержания СО2 в артериальной крови из-за гипервентиляции.
При недостаточности функции сердца как насоса включаются механизмы компенсации. Одним из них является усиление сократимости, обусловленное дилатацией желудочка по принципу Франка — Старлинга, увеличением количества сокращающихся мышечных волокон (гипертрофия желудочков при врожденной сер
дечной недостаточности), усилением адренергической активности СНС, увеличивающей сократимость миокарда. Повышенная экскреция норадреналина с мочой в покое и повышенный его уровень в плазме при физиологической нагрузке у пациентов с врожденной сердечной недостаточностью являются подтверждением повышенной адренергической активности у этих пациентов.
Экспериментальными исследованиями установлено, что при сердечной недостаточности снижается активность тирозин-гидроксилазы — фермента, ограничивающего скорость синтеза норадреналина. Хотя эти данные и объясняют снижение содержания норадреналина в миокарде при его недостаточности, низкая активность фермента скорее связана с уменьшением общего количества адренергических окончаний в сердце, чем со снижением биосинтеза медиатора. Это проливает свет на тот факт, что даже при стимуляции симпатических нервов сердца наблюдается незначительное увеличение частоты и силы его сокращений, что можно объяснить наличием интактных нервов при недостаточной функции сердца. Имеются данные, свидетельствующие, что для поддержания сократительной функции миокарда присутствие норадреналина в миокарде не является необходимым. Недостаточность норадреналина в миокарде не указывает на начальные нарушения биохимизма, ответственные за сердечную недостаточность, но снижение симпатической активности рассматривается как важное расстройство, ограничивающее резервные механизмы для компенсации сердца.
Одной из характеристик сердечной недостаточности у человека является артериальная и венозная констрикция. Эта вазоконстрикция, обусловленная главным образом повышенной симпатической активностью, компенсирует низкую производительность сердца. Увеличение общей периферической резистентности является важным компенсаторным механизмом, поддерживающим АД на необходимом уровне, несмотря на сниженный сердечный выброс. Сердечный выброс, в свою очередь, зависит от достаточного веноз
40 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
ного возврата, обусловленного относительной регидностью сосудистого русла при сердечной недостаточности. Таким образом, роль адренергической иннервации артериол и вен заключается в поддержании адекватного АД. Кроме того, она способствует перераспределению сердечного выброса в организме, влияя на отдельные участки кровообращения. Одинаковое снижение перфузии во всех зонах и органах, обусловленное сердечной недостаточностью, при усилении недостаточности создает условия кислородного голодания в органах с повышенной потребностью в О2. С целью удовлетворения этой потребности происходит перераспределение кровотока. При выраженной сердечной недостаточности перфузия в почках и коже непропорционально снижается по отношению к другим зонам циркуляции. В то же время коронарный кровоток остается нормальным на всех стадиях сердечной недостаточности, если только она не связана с коронарным артериосклерозом. Кровоток в мозгу и скелетной мускулатуре сохраняется на нормальном уровне только при умеренно выраженной недостаточности, а в бассейне спланхникуса снижается пропорционально уменьшению сердечного выброса. Таким образом, за счет снижения кровотока в почках и коже, обусловленного адренергической вазоконстрикцией в этих органах, улучшается кровоснабжение сердца, мозга и скелетной мускулатуры. Следствием такого перераспределения кровотока является снижение почечной функции и теплоотдачи.
У пациентов с сердечной недостаточностью сердечный выброс при физической нагрузке увеличивается мало и не удовлетворяет повышенную потребность тканей в О2. Наибольший кровоток при физической нагрузке отмечается в скелетных мышцах. При уменьшении до минимума кровотока в коже увеличение кровотока в мышцах может достигать уровня здорового человека. Этим можно объяснить, что пациенты с сердечной недостаточностью могут выполнять длительную физическую нагрузку. В то же время кровоток в бассейне спланхникуса в почках и мышцах, находящихся в покое, снижается. Коро
нарный кровоток увеличивается. Перфузия мозга остается неизменной. Таким образом, происходит перераспределение кровотока из органов, находящихся в покое, в работающие органы. При легких нагрузках это перераспределение может быть более выражено, чем у нормальных субъектов. При средней нагрузке перераспределение кровотока у пациента с сердечной недостаточностью становится таким же, как у здоровых людей, выполняющих усиленную нагрузку. Кроме того, при нагрузке у больных имеет место значительное повышение венозного тонуса и ЦВД, обусловленное возросшей симпатической активностью.
Исходя из описанных выше изменений сосудистой резистентности, можно предположить, что повышенный уровень норадреналина в плазме и моче указывает на увеличение симпатического влияния на периферические сосуды. Это соответствует клиническим признакам сердечной недостаточности: генерализованным сосудистым спазмам, тахикардии, олигурии. Имеющиеся данные позволяют исключить участие мозгового слоя надпочечников в повышении симпатической активности, обуславливающем изменение сосудистого тонуса. В недавних исследованиях обнаружены доказательства увеличенной лабильности запасов норадреналина в сосудистом бассейне скелетных мышц у больных с сердечной недостаточностью.
В настоящее время проводится мало исследований, посвященных изучению характера и роли АНС в механизме регуляции объема крови, хотя они имеют важное значение для анестезиологии.
Исследования показали, что можно выделить три вида изменений в механизме регуляции: 1) срочные, проявляющиеся дифференцированной вазоконстрикцией или вазодилатацией; 2) ранние, являющиеся следствием всасывания или потери жидкости через капилляры в зависимости от изменений на пре- или посткапиллярном уровне; 3) отсроченные изменения объема крови, связанные с ретенцией или экскрецией натрия и воды. Роль АНС в последнем типе изменений окончательно не установлена, но имеются данные, позво
Автономная нервная система и физиологическая нагрузка 41
ляющие предположить, что АНС может влиять на канальцевую реабсорбцию Na+ и секрецию ренина (а, следовательно, и на систему ренин —альдостерон) и антиди-уретического гормона (АДГ), т. е. на факторы, изменяющие объем жидкости в организме. Кроме того, секреция АДГ, ренина и альдостерона может изменяться под влиянием рефлексов с барорецепторов низкого и высокого давления, реализуемых АНС.
Срочные изменения проявляются быстрым перераспределением объема крови вследствие вазомоторных рефлексов. Для понимания механизмов, при помощи которых СНС может быстро увеличить приток объема крови, достаточного для наполнения полостей сердца, важно помнить, что артериальная система малорастяжима и поддерживает высокое АД благодаря высокой резистентности кровотока, тогда как венозная система — система низкого давления — легкорастяжима и содержит около 85 % общего объема крови. Внутри-грудные сосуды являются частью системы с низким давлением и лишены сократительной активности. При внутривенном введении крови она может накапливаться во внутригрудном отрезке венозной системы, центральных венах, что обеспечивает объемный резерв для адекватного заполнения полостей сердца и сердечного выброса.
Известно, что вены активно участвуют в поддержании циркуляторной функции под контролем рефлексов СНС. Они могут сужаться в ответ на различные физиологические стимулы, увеличивая венозный возврат крови к сердцу. В ответ на отрицательные эмоции, холод, норадреналин, гипервентиляцию и физическую нагрузку развивается вазоконстрикция, вызывающая перемещение крови в центральные вены. Поскольку периферические вены имеют более выраженную гладкую мускулатуру, чем крупные центральные, то при генерализированном повышении венозного тонуса кровь с периферии перемещается во внутригрудной сосудистый бассейн. Это увеличение эффективного центрального объема крови поддерживает производительность сердца, вследствие чего удовле
творяются метаболические потребности как в покое, так и при физической нагрузке. Локализация эфферентной части веномоторного рефлекса в адренергических нервах не вызывает разногласий, но расположение рецепторных клеток эфферентной части этого рефлекса еще не установлено. Современные данные позволяют предположить, что эти рецепторы локализуются в правом предсердии и, возможно, в других участках системы сосудов низкого давления в грудной клетке.
Консгрикция системного венозного русла развивается в ответ на некоторые физиологические стимулы, исключая обычную стимуляцию рефлекса с барорецепторов высокого давления. Вероятно, рефлекторная вазоконстрикция возникает при значительном снижении АД. При шоке и синкопе имеет место рефлекторное повышение венозного тонуса. Современные исследования, проведенные на людях, показали, что веноконстрикция является компенсаторным рефлексом при вазовагальной синкопе. Однако компенсаторные реакции на небольшие изменения объема крови осуществляются главным образом посредством изменения экскреторной функции почек и сопровождаются небольшими, нерефлекторными изменениями емкости венозных сосудов. Это подтверждается исследованиями, свидетельствующими, что кровопотеря до 500 мл не вызывает вазоконстрикции сосудов руки, а обычно приводит к задержке натрия и воды. Отсутствие вазоконстрикции при средних величинах кровопотери у человека может объясняться тем, что констрикция в зоне спланхникуса выражена сильнее, чем в других сосудистых бассейнах.
Многочисленными исследованиями показано, что при больших кровопотерях развивается вазоконстрикция. Значительное снижение давления внутри полостей сердца стимулируют артериальные объемные рецепторы. Уменьшение сердечного выброса приводит к тяжелой гипотензии и стимуляции барорецепторов высокого давления. Для срочной регуляции кровообращения в ответ на значительное уменьшение объема крови и АД наибольшее значение имеет веномоторный рефлекс.
42 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
Ранние изменения связаны с изменениями транскапиллярной фильтрации в результате активации СНС. В литературе приводятся данные о том, что в ответ на повышение концентрации норадреналина в крови в течение короткого периода времени происходит уменьшение объема плазмы. Механизм этого уменьшения, наиболее вероятно, связан с увеличением транскапиллярного фильтрационного давления между внутрисосудистым и интерстициальным пространствами в связи с повышением гидростатического давления внутри капилляров. Внутрикапиллярное давление определяется отношением между артериальным и центральным венозным давлением и, что особенно важно, посткапиллярной резистентностью. В то же время имеются убедительные доказательства, что адренергическая блокада у здорового человека увеличивает объем плазмы. Таким образом, СНС, благодаря влиянию на венозную резистентность, обуславливает вторичные быстродействующие механизмы регуляции циркулирующего объема крови. Норадреналин уменьшает объем крови, поэтому длительное применение его может усилить неблагоприятные последствия шока. В таких случаях необходимо замещение потери плазмы адекватным количеством плазмозаменителей, не усугубляющих реологические свойства крови.
В основе поздних механизмов лежат рефлекторные процессы, влияющие на почки и баланс натрия и воды. В настоящее время в литературе встречается мало данных, указывающих на влияние АНС на отношение между объемом крови и экскреторной функцией почек. Имеющиеся данные свидетельствуют, что снижение симпатической активности приводит к увеличению экскреции натрия у здоровых людей. Освобождение ренина может быть обусловлено катехоламинами, вводимыми извне, или эндогенными, освобождаемыми при стимуляции симпатического нерва или при метаболизме тирозина. Регуляция объема крови, опосредованная влияниями АНС на экскрецию натрия, включает систему ренин — ангиотензин — альдостерон. Имеются также доказательства автономного контроля секреции альдостерона и
данные о том, что изменение секреции АДГ под влиянием АНС может стимулировать или угнетать диурез [24, 25].
О. Н. Gauer и J. Р. Henry [26] изучали регуляцию объема крови посредством изменения секреции АДГ и объема жидкости тела. У собак с интактным блуждающим нервом при дыхании с отрицательным давлением на выдохе (предсердия растягиваются) или при вливании крови отмечалось повышение диуреза и афферентной импуль-сации блуждающего нерва. При охлаждении нерва до 8 °C этот эффект отсутствовал. С другой стороны, при дыхании с положительным давлением с сопротивлением на выдохе (сдавливание предсердия) или при кровопотере 50 мл (при весе собаки 10 кг) отмечался антидиуретический эффект и уменьшение импульсации блуждающего нерва. На основании этих опытов можно предположить, что стимуляция объемночувствительных рецепторов в стенках левого предсердия вызывает вагальную эфферентацию, которая, в свою очередь, приводит к торможению секреции АДГ и снижению реабсорбции воды в дистальных канальцах. По мнению большинства исследователей, эта рефлекторная регуляция секреции АДГ является наиболее важным компонентом механизма отсроченной регуляции объема крови и изменение секреции альдостерона и адренергической активности фиксируются только в исключительных ситуациях.
Для детализации роли АНС были проведены следующие эксперименты: почки собаки, сохранившие с организмом-хозяином только нервные связи, перфузировались кровью собаки-донора. При инфузии собаке-акцептору физиологического раствора ее почки отвечали на это увеличением экскреции натрия.
Участие механорецепторов левого предсердия в диурезе объясняет полиурию, развивающуюся у пациентов при возникновении пароксизмальной предсердной аритмии. И наоборот, синдром несоответствующей секреции АДГ и олигурия наблюдаются у пациентов, перенесших операцию по поводу тяжелого митрального стеноза, что вызывается выраженным снижением давления и объема левого предсердия.
Автономная нервная система и боль 43
Все вышеизложенное свидетельствует, что существует ряд каналов, через которые реализуется влияние АНС на объем циркулирующей крови. При значительном падении АД объем крови в центральных венах немедленно увеличивается благодаря генерализованной рефлекторной вазоконстрикции, менее тяжелая степень гипотензии компенсируется исключительно рефлекторной артериолоконстрикцией и симпатической стимуляцией частоты и силы сердечных сокращений. При пролонгированной симпатической активации имеет место изменение посткапиллярно-прекапиллярного отношения резистентности. Уменьшение этого отношения приводит к снижению внутрика-пиллярного давления и увеличению внутрисосудистого объема путем реабсорбции жидкости из интерстициального пространства.
До сих пор остаются малопонятными рефлекторные компенсаторные механизмы, включающиеся при уменьшении внутрисосудистого объема. Имеются данные, что небольшое уменьшение объема, воспринимаемое рецепторами растяжения в сосудах с низким давлением, восстанавливается благодаря секреции АДГ без заметного увеличения адренергической стимуляции сосудистой системы. Но при большой, быстрой кровопотере активируются артериальные и венозные рефлексы. Активация барорецепторов высокого давления приводит к активации системы ренин—ангиотензин — альдостерон, но уровень гипотензии, при которой она активируется, трудно точно определить, однако известно, что этот уровень находится в пределах средней и тяжелой геморрагии.
1.9. АВТОНОМНАЯ НЕРВНАЯ СИСТЕМА И БОЛЬ
Термин «АНС» в том смысле, в каком его применил J. N. Langley [27] к системе нейронов, иннервирующих внутренние органы, не включал нервные волокна, ответственные за болевую чувствительность. К автономным нервам были отнесены только те, которые иннервировали внутренние органы, сосуды и ткани и участвовали в рефлекторной регуляции функций и развитии реакций органов и тканей. Афферентные нейроны рассматривались как соматические. Ныне доказано, что в иннервации внутренних органов принимают участие и болевые волокна. Большинство импульсов из внутренних органов участвуют в поддержании «внутреннего устойчивого состояния» (гомеостаза), описанного К. Бернаром в конце XIX в. Часть этих импульсов воспринимается сознанием. Ощущение боли, вероятно, является функцией некоторых интегральных структур, сигнализирующих об опасности. В этом смысле боль можно расценивать как «страж здоровья», защитный механизм.
1.9.1. Висцеральная боль
Между болью висцеральной, исходящей из внутренних органов, и болью, связанной с раздражением соматических чувстви
тельных волокон, имеются значительные различия. Соматическая боль характеризуется более четкой локализацией. Висцеральная — чаще всего плохо локализована. Она вызывается факторами внешней среды (механическими, термическими, электрическими и т. д.) и сигнализирует нам о неблагополучии. Периферический сенсорный аппарат соматической нервной системы высокоразвит, и болевые рецепторы играют в нем важную роль.
Другое существенное различие заключается в том, что многие стимулы, активирующие соматические сенсорные нервы, не вызывают болевой реакции, когда они воздействуют на внутренние органы. Например, человек не ощущает боли при разрезании кишок, но их растяжение сопровождается неприятными ощущениями. Наибольшее значение имеет вид стимула, а не его интенсивность.
При висцеральной боли в результате воздействия стимула (фактора) в каком-либо месте ощущение боли возникает в местах, отдаленных от проекции органа, подвергающегося воздействию стимула. Кроме боли в этих местах — местах иррадиации боли — возникает гипералгезия кожи и иногда кишечные спазмы. Эти наблюдения дали основание К. G. Lennonder
44 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
[28] и J. Mackenzie [29] прийти к выводу, что афферентация из внутренних органов не восходит непосредственно в сенсорные зоны коры головного мозга. J. Mackenzie полагал, что все висцеральные боли отражаются в местах, отдаленных от мест воздействия раздражающих стимулов. Была создана теория о том, что импульсы из внутренних органов перегружают боль-проводящие пути соматической системы в задних рогах спинного мозга, вызывая развитие «очага возбуждения». Из этих очагов боль отражается в соответствующие участки. Стенокардия является наглядной иллюстрацией этой теории. S. Weiss, D. Davis [30] нашли подтверждение концепции отражения боли. Если участок отраженных болей (участок кожной гипер-алгезии) анестезировать прокаином, то ощущение боли исчезает. Однако наблюдения показали, что поверхностная анестезия не эффективна, особенно в случаях болевой стимуляции значительной интенсивности. G. W. Theobald [31] наблюдал при фаррадизации током средней силы шейки матки появление ощущения дискомфорта и отраженной боли в надлобковой зоне, которые исчезали при инфильтрации прокаином кожи, при увеличении интенсивности стимуляции боли всегда «прорывались» в сознание, несмотря на анестезию.
J. Morley [32] отметил, что мышечный спазм участков передней стенки живота, наблюдаемый при заболеваниях органов брюшной полости, возникает рефлекторно вследствие распространения воспалительного процесса на париетальную брюшину. Например, при остром аппендиците сначала появляются боли типа колики вследствие растяжения стенки отростка. Эти боли являются результатом висцеральной афферентации. Позже, когда воспаление переходит на париетальную брюшину, возникает напряжение мышц живота из-за раздражения интеркостальных нервов, иннервирующих нижний правый квадрант живота.
Теория J. Mackenzie не объясняет полной картины висцеральных болей. Сейчас хорошо известно, что афферентные волокна АНС, имеющие одинаковые характеристики с болевыми соматическими, идут из
внутренних органов в задние рога спинного мозга. Предполагают, что висцеральная и кожная болевая афферентации сходятся в клетках вторичных афферентных нейронов в задних рогах и, вполне вероятно, что вторичные спинальные аксоны, идущие в спинноталамические тракты, передают как висцеральные, так и соматические чувствительные импульсы в кору головного мозга. На сегодня доказано наличие автономных болевых волокон. Головные боли при мигрени также связаны с растяжением сосудов.
Таким образом, растяжение является общим фактором для возникновения болевых ощущений, исходящих из сосудов и внутренних органов.
Парасимпатические болевые волокна. Менее известна роль ПНС в передаче болевой чувствительности. Блуждающий нерв является смешанным нервом, он содержит моторные, секреторные и чувствительные волокна. О. Larssell [33] описывает чувствительный компонент блуждающего нерва как: а) общие соматические афферентные волокна от нейронов югулярного ганглия, идущие от кожи уха в составе слухового нерва; б) общие висцеральные афферентные волокна, происходящие от нейронов gan. nodosum, идущие от рецепторов в легких, сердце, пищеварительном канале, слизистой глотки и гортани; в) отдельные висцеральные афферентные волокна от нейронов gan. nodosum, проводящие сенсорные импульсы от вкусовых рецепторов задней трети поверхности языка.
Чувствительная автономная иннервация слизистой и подслизистой отличается от типичной соматической чувствительной иннервации других тканей и гортани. Имеются доказательства, что некоторые анатомические образования (пищевод, бронхи) имеют болевую афферентацию, реализуемую через блуждающий нерв. D. R. Morton с соавт. [34] стимулировал слизистую правого и левого главных бронхов и получали отражение боли на переднюю стенку грудной клетки на стороне раздражения вблизи от грудины или на шее в пределах 2 см от средней линии. При пересечении нерва ниже отхождения от него возвратного гортанного нерва наблюдалось
Автономная нервная система и боль 45
ослабление боли, возникающей при раздражении слизистой бронхов. К. S. Grimson с соавт. [35] обнаружил, что стимуляция блуждающего нерва выше диафрагмы у пациентов в условиях спинномозговой анестезии вызывала жалобы на жжение в области сердца с радиацией в область шеи. Пока нет доказательств, что блуждающий нерв содержит какие-либо болевые волокна, исходящие от органов и тканей, расположенных ниже диафрагмы.
Ряд анастомозов блуждающего нерва с языкоглоточным и подъязычным, так же, как и симпатические волокна, проходящие через нижний шейный симпатический ганглий, затрудняют дифференциацию болевой афферентации. В этот комплекс соматических и автономных нервов при глоссофарингеальной невралгии может включаться и болевая афферентация блуждающего нерва. О. Larssell [33] считает, что висцеральные афферентные нервы глубокой чувствительности в области головы и шеи восходят из клеток коленчатого узла и идут в составе всех ветвей лицевого нерва, включая и chorda tympani. Эти волокна, образуя промежуточный нерв, участвуют в проведении невралгических болей из уха, которые обычно локализуются в наружном слуховом проходе. Хотя анатомия чувствительной афферентации краниальной части ПНС мало изучена, можно предположить, что анатомия болевой афферентации осуществляется в основном этой системой.
Боль и сердечно-сосудистая система. Знание автономной регуляции сердечнососудистой системы является особенно важным для анестезиологов. Все артерии иннервируются вазомоторными нервами; вены и прекапиллярные сфинктеры также находятся под контролем АНС. Имеющиеся сведения свидетельствуют о ведущей роли краниальных и вертебральных симпатических эфферентных волокон в регуляции сосудистого тонуса. Меньше данных о парасимпатическом контроле периферических сосудов конечностей. Исключение составляет сакральный отдел ПНС. Стимуляция симпатических волокон, иннервирующих сосуды конечностей, вызывает их вазоконстрикцию. Сосудистый тонус является результатом увеличения или
уменьшения симпатических вазоконстрикторных импульсов. Однако чувствительная иннервация до сих пор остается окончательно не выясненной. Афферентные волокна, несущие болевую информацию, могут быть соматическими, но их нервные окончания локализуются вблизи артерий и отличаются от чувствительных рецепторов кожи.
J. G. Kramer и T.W. Todd [36] доказали, что сосуды конечностей получают иннервацию по симпатическим волокнам от смешанных нервов, расположенных вблизи сосудов. Когда эти нервы входят в стенку сосудов, они разделяются на мелкие группы волокон, достигающие адвентиции, медии и, возможно, интимы. Исследования лаборатории Н. Н. Woolard [37] показали, что терминальные нервы могут разделяться следующим образом: одна ветвь идет в кожу, другая — в артериолы. Болевая чувствительность из сосудов может косвенно обуславливаться изменением обеспечения кислородом тканей в зависимости от тонуса сосудов. Такой механизм боли имеет место при перемежающейся хромоте и стенокардии. Паравертебральная блокада, устраняя вазоконстрикцию, может надежно улучшить кровоснабжение конечности и предупредить возникновение боли, такой же эффект оказывает блокада периферических нервов и сплетений.
Выяснение природы боли при стенокардии является более сложной проблемой. Иррадиация болей в левое плечо и руку или реже — в правое плечо и шею часто сопровождается прекардиальными болями, типичными для миокардиальной недостаточности. Предполагают, что афферентные волокна из сердца, идущие в дорзальные рога спинного мозга, могут возбуждать соседние соматические нейроны, к которым идут волокна от кожи. В некоторых случаях хронической коронарной недостаточности развивается синдром «плечевой руки». F. Р. Handen [38] составил карту боли в определенных областях верхней половины грудной клетки, имеющих довольно специфическое расположение. Инъекция местного анестетика в соответствующую зону не только улучшала состояние руки, но и уменьшала интен
46 ФИЗИОЛОГИЯ АВТОНОМНОЙ НЕРВНОЙ СИСТЕМЫ
сивность испытываемых пациентом болей в области сердца.
J. С. White с соавт. [39], основываясь на анализе большого количества клинических наблюдений, составил схему проводящих симпатических путей, идущих как к сердцу, так и от него. Все сердечные волокна входят и выходят из него в составе I — IV, иногда V, корешков грудных симпатических нервов. Любая боль сердечного происхождения может быть устранена билатеральной блокадой или перерезкой этих дорзальных корешков. Блокада звездчатого узла может дать временный успех, а его раздражение — вызвать различные варианты проявлений загрудинных болей.
Еще в начале 1960 г. Hertz установил, что боли в животе являются результатом ненормального (чрезмерного) растяжения пищеварительного канала. J. A. Ryle [40] полагал, что противодействие гладкой мускулатуры любого полового органа его пе-рерастяжению является достаточным стимулом для появления висцеральных болей. F. Н. Bentley [41], проводя наблюдения во время операции под местной анестезией по поводу язвы двенадцатиперстной кишки, обнаружил, что непосредственное надавливание на язву вызвало такую же боль, как и при надавливании на брюшную стенку, и что укол иглой в язву также вызывал боль. Она оказывалась менее продолжительной, если была произведена инфильтрация прокаином n. splanchnicus.
Все вышеизложенное служит основанием для утверждения о существовании истинных висцеральных болей и о том, что трансформация спастических сокращений пищеварительного канала в болевое ощущение опосредуется афферентацией собственно АНС. Подобный механизм стимуляции висцеральных и соматических нервов лежит в основе возникающей боли,
исходящей из почек, селезенки и брюшины. Однако автономная афферентация из почек и матки осуществляется по пути симпатических волокон, выходящих на уровне от X грудного до I поясничного сегмента. Растяжение почечной лоханки не вызывает боли, если почечное сплетение блокировано (паранефральная блокада). Желчный пузырь, пузырный и общий желчный протоки имеют болевые волокна, идущие в составе блуждающего нерва и нервов спланхнической зоны.
Абдоминальные боли, возникающие вследствие склеиваний спаечного процесса кишок, менее исследованы. F. Р. Handen [38] наблюдал пациентов, дававших очень точное описание мигрирующих болей в тонкой и толстой кишках, которые могли быть объяснены образованием заворотов и спаечным процессом, что вызывало временное перерастяжение их. Хронические боли этого типа чаще наблюдаются у беспокойных пациентов. Не случайно симптомокомп-лекс этого беспокойства указывает на гиперактивность СНС. J. С. White, R. Н. Smithwick [42] описали успешное облегчение болей этого типа путем спланхнэктомии. S. J. Sarnoff с соавт. [43] предложил дифференциальную спинальную блокаду прокаином как диагностический тест, позволяющий определить эффективность симпатэктомии в облегчении болей, возникающих при спаечной болезни кишок.
Болевая афферентация из толстой кишки осуществляется II, III, IV сакральными нервами так же, как и желчный пузырь и предстательная железа, но пресакральная неврэктомия не обеспечивает облегчения болей из этой локализации. У женщин матка получает афферентные болевые волокна как из торако-люмбальных симпатических, так и из тазовых парасимпатических нервов.
Список использованной литературы
1. Заболевания вегетативной нервной системы / А. М. Вейн, Е. Я. Алимова, Т. Г. Вознесенская и др.; Под ред. А. М. Вейна. — М.: Медицина, 1991. - 624 с.
2. Поленов А. Л., Бондарчук А. В. Хирургия вегетативной нервной системы. — Л.: Мед-гиз, 1947. — 570 с.
3. Хаулике И. Вегетативная нервная система (анатомия, физиология). — Бухарест: Медиз-дательство, 1978. — 350 с.
4. Lawhead R., Blaxall Н., Bylund D. а2А is the predominant а2 adrenergic receptor subtype in human spinal cord // Anesthesiology. — 1992. - 77. - P. 983.
Список использованной литературы 47
5. Ruffolo R. Physiology and biochemistry of the peripheral autonomic nervous system // Human Pharmacology: Molecular-to-Clini-cal. Mosby-Year Book / Ed. L. Wingard, T. Brody, J. Larner, A. Schwartz. — St. Louis, 1991. - P. 77.
6. Ениг В. Вегетативная нервная система // Физиология человека / М. Циммерман, В. Ениг, В. Вутке и др.; Под ред. Р. Шмидта, Г. Тевса. - М.: Мир, 1996. - С. 343-383.
7. Росин Я. А. Физиология вегетативной нервной системы. — М.: Наука, 1965. — 406 с.
8. Moss}., Craigo Р. A. The Autonomic Nervous System. Anesthesia / Ed. R. D. Miller. — 4th ed. - 1994. - P. 523-575.
9. Carpenter F. G. Transmission in Sympathetic Ganglion // Anesthesiology. — 1968. — Vol. 29, N 4. - P. 634-642.
10. DogelA. S. // Arch. Anat. physiol. — 1899. — 53. - P. 130.
11. Bayliss W. M., Starling E. H. The movements and innervation of the small interstine // J. Physiol. - 1900. - 26. - P. 125.
12. Langley J. N. // J. Physiol. (Lond.). — 1898. - 23. - P. 240.
13. Langley J. N. // J. Physiol. (Lond.). — 1901. - 27. - P. 237.
14. Филимонов В. И. Руководство по общей и клинической физиологии. — М.: МИА, 2002. - 958 с.
15. Merin R. G. Pharmacology of the Autonomic Nervous System // Anesthesia. — 2nd ed. — New York; Edinburg; London; Melbourne: Churchill Livingstone. — 1986. — P. 945 — 981.
16. Burnstock G. Local mechanisms of blood flow control by perivascular nerves and endothelium //J. Hypertens Suppl. — 1900. — 8(7). — P. 95.
17. Bumstock G. Overview: purinergic mechanisms // Ann. N. Y. Acad. Sci. - 1900. - 603.
18. Koelle G. B. Functional Anatomy of Synaptic Transmission // Anesthesiology. — 1968. — Vol. 29, N 4. - P. 643-653.
19. Соловьева А. Д., Данилов А. Б. Методы исследования вегетативной нервной системы // Заболевания вегетативной нервной системы. — М.: Медицина, 1991. — С. 39 — 84.
20. Tomson Р. D., Melmon К. L. Clinical Assessment of Autonomic Function // Anesthesiology. - 1968. - Vol. 29, N 4. - P. 724-731.
21. Zaimis E. Vasopressore Drugs and Catecholamines // Anesthesiology. — 1968. — Vol. 29, N 4. - P. 712-732.
22. Havel R. J. The Autonomic Nervous System and Intermediary Carbohydrate and Fat Metabolism // Anesthesiology. — 1968. — Vol. 29, N 4. - P. 702-713.
23. Furchgotf R. F., Zawadzki I. V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine //Nature. - 1980. - 288. - P. 373.
24. Mason D. The Autonomic nervous system and regulation of cardiovascular performance //
Anesthesiology. — 1968. — Vol. 29, N 4. — P. 670-680.
25. Mason D. T., Bartter F. C. Autonomic Regulation of Blood Volume // Anesthesiology. — 1968. - Vol. 29, N 4. - P. 681-692.
26. Gauer О. H., Henry J. P. Circulatory basis of fluid volume control // Physiol. Rev. — 1963. - 43. - P. 423.
27. Langley J. N. // Brain. — 1903. — 26.
28. Lennonder K. G. Observations on the sensitivity of the Abdominal Cavity / Trans, by A. E. Baker. — I. Bale, Sons and Danielson, 1903.
29. Mackenzie J. Symptoms and thier interpretations. — 2nd ed. — London: Shaw and Sons, 1912.
30. Weiss S., Davis D. The significance of the afferent impulses from the skin in the mechanism of visceral pain // Amer. J. Med. Sci. — 1928. - 176. - P. 571.
31. Theobald G, W. The role of the cerebral cortex in the apperception of pain // Lancet. — 1949. - 257, 41. - P. 94.
32. Morley J. Abdominal pain. — Edinburgh: E. and S. Livingstone, 1930.
33. Larssell O. Anatomy of the Nervous System. — New York: D. Appleton-Century Co., 1942.
34. Morton D. R., Klassen К. P., Curtis G. M. The effect of high vagus section upon the clinical physiology of the bronchus // Trans. Amer. Neurol. Assoc. — 1950. — 143.
35. Grimson K. S., Hesser F. H., Kitchin W. W. Early clinical results of transabdominal celiac and superior mesenteric ganglionectomy, vagotomy or transthoracic splanchniectomy in patients with chronic abdominal visceral pain // Surgery. — 1947. — 22.
36. Kramer J. G., Todd T. W. The distribution of nerves to the arteries of the arm, with a discussion of the clinical value of results // Anat. Record. — 1914. — 8.
37. Woolard H. H. The innervation of blood vessels // Heart. — 1926. — 13. — P. 319.
38. Handen F. P. The Autonomic Nervous System and Pain // Anesthesiology. — 1968. — Vol. 29, N 4. - P. 785-792.
39. White J. C., Smithwick R. H., Simeone F. H. The autonomic nervous system: anatomy, physiology and syrgical application. — 3rd. ed. — New York: Macmillan, 1952.
40. Ryle J. A. Visceral pain and referred pain // Lancet. - 1926. - 210. - P. 895.
41. Bentley F. H. Observations on visceral pain. (1) Visceral tenderness // Ann. Surg. — 1948. - 128. - P. 881.
42. White J. C., Smithwick R. H. The autonomic nervous system: anatomy, physiology and syrgical application. — 2nd. ed. — New York: Macmillan, 1941.
43. Sarnoff S. J., Arrowood I. G., Chapman W. P. Differential spinal block // Surg. Gynec. Obstet. - 1948. - 86. - P. 574.
Раздел
Клиническая физиология кровообращения
В 1628 г. Вильям Гарвей сделал величайшее открытие в области медицинской науки: он доказал, что сердце нагнетает кровь в систему сосудов и сформулировал основные принципы деятельности сердечно-сосудистой системы, главной функцией которой он считал обеспечение тканей питанием. В гл. 14 «De Motu Cordis» В. Гарвей охарактеризовал кровь как среду, кото-
2.1. КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ
Масса крови перемещается по замкнутой сосудистой системе, состоящей из большого и малого кругов кровообращения, в строгом соответствии с основными физическими принципами, в том числе с принципом неразрывности потока. Согласно этому принципу разрыв потока при внезапных травмах и ранениях, сопровождающихся нарушением целостности сосудистого русла, приводит к потере как части объема циркулирующей крови, так и большого количества кинетической энергии сердечного сокращения. В нормально функционирующей системе кровообращения согласно принципу неразрывности потока через любое поперечное сечение замкнутой сосудистой системы в единицу времени перемещается один и тот же объем крови.
Дальнейшее изучение функций кровообращения как в эксперименте, так и в клинике, привело к пониманию того, что кровообращение наряду с дыханием относит-
рая «посылается во все тело, проникает в вены и поры ткани и течет обратно через вены в правое предсердие». Он предположил существование капилляров, описанных М. Мальпиги лишь в 1661 г. Таким образом, В. Гарвей заложил фундамент современного понимания феномена кровообращения как непрерывного однонаправленного движения крови по сосудистому руслу.
СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
ся к числу наиболее важных жизнеобеспечивающих систем, или к так называемым «витальным» функциям организма, прекращение функционирования которых приводит к смерти в течение нескольких секунд или минут. Между общим состоянием организма больного и состоянием кровообращения существует прямая зависимость, поэтому состояние гемодинамики является одним из определяющих критериев тяжести заболевания. Развитие любого тяжелого заболевания всегда сопровождается изменениями функции кровообращения, проявляющимися либо в его патологической активации (напряжение), либо в депрессии той или иной степени выраженности (недостаточность, несостоятельность). Первичное поражение циркуляции характерно для шоков различной этиологии.
Оценка и поддержание адекватности гемодинамики являются важнейшим компонентом деятельности врача при прове
Клиническая физиология сердечно-сосудистой системы 49
дении анестезии, интенсивной терапии и реанимации.
Система кровообращения осуществляет транспортную связь между органами и тканями организма. Кровообращение выполняет множество взаимосвязанных функций и обуславливает интенсивность сопряженных процессов, в свою очередь, влияющих на кровообращение. Все реализуемые кровообращением функции характеризуются биологической и физиологической специфичностью и ориентированы на осуществление феномена переноса масс, клеток и молекул, выполняющих защитные, пластические, энергетические и информационные задачи. В наиболее общей форме функции кровообращения сводятся к массопереносу по сосудистой системе и к массообмену с внутренней и внешней средой. Это явление, наиболее четко прослеживаемое на примере газообмена, лежит в основе роста, развития и гибкого обеспечения различных режимов функциональной активности организма, объединяя его в динамическое целое.
К основным функциям кровообращения относятся:
1. Транспорт кислорода из легких к тканям и углекислого газа из тканей к легким.
2. Доставка пластических и энергетических субстратов к местам их потребления.
3. Перенос продуктов метаболизма к органам, где происходит их дальнейшее превращение и экскреция.
4. Осуществление гуморальной взаимосвязи между органами и системами.
Кроме этого, кровь играет роль буфера между внешней и внутренней средой и является наиболее активным звеном в гидрообмене организма.
Система кровообращения образована сердцем и сосудами. Оттекающая от тканей венозная кровь поступает в правое предсердие, а оттуда — в правый желудочек сердца. При сокращении последнего кровь нагнетается в легочную артерию. Протекая через легкие, кровь подвергается полной или частичной эквилибрации с альвеолярным газом, в результате чего она отдает избыток углекислого газа и насыщается кислородом. Система легочных сосудов (легочные артерии, капилляры и
вены) образует малый (легочный) круг кровообращения. Артериализированная кровь из легких по легочным венам поступает в левое предсердие, а оттуда — в левый желудочек. При его сокращении кровь нагнетается в аорту и далее — в артерии, артериолы и капилляры всех органов и тканей, откуда по венулам и венам оттекает в правое предсердие. Система перечисленных сосудов образует большой круг кровообращения. Любой элементарный объем циркулирующей крови последовательно проходит все перечисленные отделы системы кровообращения (за исключением порций крови, подвергающихся физиологическому либо патологическому шунтированию).
Исходя из целей клинической физиологии, кровообращение целесообразно рассматривать как систему, состоящую из следующих функциональных отделов:
1. Сердце (сердечный насос) — главный двигатель циркуляции.
2. Сосуды-буферы, или артерии, выполняющие преимущественно пассивную транспортную функцию между насосом и системой микроциркуляции.
3. Сосуды-емкости, или вены, выполняющие транспортную функцию возврата крови к сердцу. Это более активная, чем артерии, часть системы кровообращения, поскольку вены способны изменять свой объем в 200 раз, активно участвуя в регуляции венозного возврата и циркулирующего объема крови.
4. Сосуды распределения (сопротивления) — артериолы, регулирующие кровоток через капилляры и являющиеся главным физиологическим средством регионарного распределения сердечного выброса, а также венулы.
5. Сосуды обмена — капилляры, интегрирующие систему кровообращения в общее движение жидкости и химических веществ в организме.
6. Сосуды-шунты — артерио-венозные анастомозы, регулирующие периферическое сопротивление при спазме артериол, сокращающем кровоток через капилляры.
Три первых отдела кровообращения (сердце, сосуды-буферы и сосуды-емкости) представляют собой систему макро
50 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
циркуляции, остальные — образуют систему микроциркуляции.
В зависимости от уровня давления крови выделяют следующие анатомо-функциональные фрагменты системы кровообращения:
1. Система высокого давления (от левого желудочка до капилляров большого круга) кровообращения.
2. Система низкого давления (от капилляров большого круга до левого предсердия включительно).
Хотя сердечно-сосудистая система является целостным морфофункциональным образованием, для понимания процессов циркуляции целесообразно рассматривать основные аспекты деятельности сердца, сосудистого аппарата и регуляторных механизмов по отдельности.
2.1.1. Сердце
Этот орган массой около 300 г снабжает кровью «идеального человека» массой 70 кг в течение примерно 70 лет. В покое каждый желудочек сердца взрослого человека выбрасывает 5 —5,5 л крови в минуту; следовательно, за 70 лет производительность обоих желудочков составляет приблизительно 400 млн л, даже если человек находится в состоянии покоя.
Обменные потребности организма зависят от его функционального состояния (покой, физическая активность, тяжелые заболевания, сопровождающиеся гиперметаболическим синдромом). Во время тяжелой нагрузки минутный объем может возрастать до 25 л и более в результате увеличения силы и частоты сердечных сокращений. Некоторые из этих изменений обусловлены нервными и гуморальными воздействиями на миокард и рецепторный аппарат сердца, другие являются физическим следствием воздействия «растягивающей силы» венозного возврата на сократительную силу волокон сердечной мышцы.
Процессы, происходящие в сердце, условно разделяют на электрохимические (автоматия, возбудимость, проводимость) и механические, обеспечивающие сократительную активность миокарда.
Электрохимическая деятельность сердца. Сокращения сердца происходят вследствие периодически возникающих в сердечной мышце процессов возбуждения. Сердечная мышца — миокард — обладает рядом свойств, обеспечивающих его непрерывную ритмическую деятельность, — ав-томатией, возбудимостью, проводимостью и сократимостью.
Возбуждение в сердце возникает периодически под влиянием процессов, протекающих в нем. Это явление получило название автоматии. Способностью к ав-томатии обладают определенные участки сердца, состоящие из особой мышечной ткани. Эта специфическая мускулатура образует в сердце проводящую систему, состоящую из синусового (синусно-предсердного, синоатриального) узла — главного водителя ритма сердца, расположенного в стенке предсердия около устьев полых вен, и предсердно-желудочкового (атриовентрикулярного) узла, находящегося в нижней трети правого предсердия и межжелудочковой перегородки. От атриовентрикулярного узла берет начало предсердно-желудочковый пучок (пучок Гиса), прободающий предсердно-желудочковую перегородку и разделяющийся на левую и правую ножки, следующие в межжелудочковую перегородку. В области верхушки сердца ножки предсердно-желудочкового пучка загибаются вверх и переходят в сеть сердечных проводящих миоцитов (волокна Пуркинье), погруженных в сократительный миокард желудочков. В физиологических условиях клетки миокарда находятся в состоянии ритмической активности (возбуждения), что обеспечивается эффективной работой ионных насосов этих клеток.
Особенностью проводящей системы сердца является способность каждой клетки самостоятельно генерировать возбуждение. В обычных условиях автоматия всех расположенных ниже участков проводящей системы подавляется более частыми импульсами, поступающими из синусно-предсердного узла. В случае поражения этого узла (генерирующего импульсы с частотой 60 — 80 ударов в минуту) водителем ритма может стать предсердно-желудоч
Клиническая физиология сердечно-сосудистой системы 51
ковый узел, обеспечивающий частоту 40 — 50 ударов в минуту, а если оказывается выключенным и этот узел — волокна пучка Гиса (частота 30 — 40 ударов в минуту). При выходе из строя и этого водителя ритма процесс возбуждения может возникнуть в волокнах Пуркинье с очень редким ритмом — примерно 20/мин.
Возникнув в синусовом узле, возбуждение распространяется на предсердие, достигая атриовентрикулярного узла, где благодаря небольшой толщине его мышечных волокон и особому способу их соединения возникает некоторая задержка проведения возбуждения. Вследствие этого возбуждение достигает предсердно-желудочкового пучка и волокон Пуркинье лишь после того, как мускулатура предсердий успевает сократиться и перекачать кровь из предсердий в желудочки. Таким образом, атриовентрикулярная задержка обеспечивает необходимую последовательность сокращений предсердий и желудочков.
Наличие проводящей системы обеспечивает ряд важных физиологических функций сердца: 1) ритмическую генерацию импульсов; 2) необходимую последовательность (координацию) сокращений предсердий и желудочков; 3) синхронное вовлечение в процесс сокращения клеток миокарда желудочков.
Как экстракардиальные влияния, так и факторы, непосредственно поражающие структуры сердца, могут нарушать эти сопряженные процессы и приводить к развитию различных патологий сердечного ритма.
Механическая деятельность сердца. Сердце нагнетает кровь в сосудистую систему благодаря периодическому сокращению мышечных клеток, составляющих миокард предсердий и желудочков. Сокращение миокарда вызывает повышение давления крови и изгнание ее из камер сердца. Вследствие наличия общих слоев миокарда у обоих предсердий и обоих желудочков возбуждение одновременно достигает их клеток и сокращение обоих предсердий, а затем и обоих желудочков осуществляется практически синхронно.
Сокращение предсердий начинается в области устьев полых вен, в результате чего устья сжимаются. Поэтому кровь может двигаться через предсердно-желудочковые клапаны только в одном направлении — в желудочки. В момент диастолы желудочков клапаны раскрываются и пропускают кровь из предсердий в желудочки. В левом желудочке находится двустворчатый, или митральный, клапан, в правом — трехстворчатый клапан. Объем желудочков постепенно возрастает до тех пор, пока давление в них не превысит давление в предсердии и клапан не закроется. В этот момент объем в желудочке представляет собой конечный диастолический объем. В устьях аорты и легочной артерии имеются полулунные клапаны, состоящие из трех лепестков. При сокращении желудочков кровь устремляется в сторону предсердий и створки предсердно-желудочковых клапанов захлопываются, в это время полулунные клапаны тоже пока остаются закрытыми. Начало сокращения желудочка при полностью закрытых клапанах, превращающих желудочек во временно изолированную камеру, соответствует фазе изометрического сокращения.
Повышение давления в желудочках при их изометрическом сокращении происходит до тех пор, пока оно не превысит давление в крупных сосудах. Следствием этого является изгнание крови из правого желудочка в легочную артерию и из левого желудочка в аорту. При систоле желудочков лепестки клапана под давлением крови прижимаются к стенкам сосудов, и она беспрепятственно изгоняется из желудочков. Во время диастолы давление в желудочках становится ниже, чем в крупных сосудах, кровь устремляется из аорты и легочной артерии в направлении желудочков и захлопывает полулунные клапаны. Вследствие падения давления в камерах сердца во время диастолы, давление в венозной (приносящей) системе начинает превышать давление в предсердиях, куда кровь притекает из вен.
Наполнение сердца кровью обусловлено рядом причин. Первая — наличие остатка движущей силы, вызванной сокращением сердца. Среднее давление крови
52 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
в венах большого круга — 7 мм рт. ст., а в полостях сердца во время диастолы стремится к нулю. Таким образом, градиент давления составляет всего около 7 мм рт. ст. Это надо учитывать во время хирургических вмешательств — любое случайное сдавливание полых вен может полностью прекратить доступ крови к сердцу.
Вторая причина притока крови к сердцу — сокращение скелетных мышц и наблюдающееся при этом сдавливание вен конечностей и туловища. В венах имеются клапаны, пропускающие кровь только в одном направлении — к сердцу. Эта так называемая венозная помпа обеспечивает значительное увеличение притока венозной крови к сердцу и сердечного выброса при физической работе.
Третья причина увеличения венозного возврата — присасывающий эффект крови грудной клеткой, которая представляет собой герметически закрытую полость с отрицательным давлением. В момент вдоха эта полость увеличивается, органы, расположенные в ней (в частности, полые вены), растягиваются, и давление в полых венах и предсердиях становится отрицательным. Определенное значение имеет также присасывающая сила расслабляющихся подобно резиновой груше желудочков.
Под сердечным циклом понимают период, состоящий из одного сокращения (систола) и одного расслабления (диастола).
Сокращение сердца начинается с систолы предсердий, длящейся 0,1 с. При этом давление в предсердиях повышается до 5 —8 мм рт. ст. Систола желудочков продолжается около 0,33 с и состоит из нескольких фаз. Фаза асинхронного сокращения миокарда длится от начала сокращения до закрытия атриовентрикулярных клапанов (0,05 с). Фаза изометрического сокращения миокарда начинается с захлопывания атриовентрикулярных клапанов и заканчивается открытием полулунных (0,05 с).
Период изгнания составляет около 0,25 с. За это время часть крови, содержащейся в желудочках, изгоняется в крупные сосуды. Остаточный систолический объем за
висит от величины сопротивления работы сердца и от силы его сокращения.
Во время диастолы давление в желудочках падает, кровь из аорты и легочной артерии устремляется обратно и захлопывает полулунные клапаны, затем кровь притекает в предсердия.
Особенностью кровоснабжения миокарда является то, что кровоток в нем осуществляется в фазу диастолы. В миокарде имеются две системы сосудов. Снабжение левого желудочка происходит по сосудам, отходящим от коронарных артерий под острым углом и проходящим по поверхности миокарда, их ветви снабжают кровью 2/3 наружной поверхности миокарда. Другая система сосудов проходит под тупым углом, прободает всю толщу миокарда и осуществляет кровоснабжение 1/3 внутренней поверхности миокарда, разветвляясь эндокардиально. В период диастолы кровоснабжение этих сосудов зависит от величины внутрисердечного давления и давления извне на сосуды. На субэндокардиальную сеть влияет среднее дифференциальное диастолическое давление. Чем оно выше, тем хуже наполнение сосудов, т. е. нарушается коронарный кровоток. У больных с дилатацией чаще возникают очаги некроза в субэндокардиальном слое, чем интрамурально.
Правый желудочек тоже имеет две системы сосудов: первая проходит через всю толщу миокарда; вторая образует субэндокардиальное сплетение (1/3). Сосуды перекрывают друг друга в субэндокардиальном слое, поэтому инфарктов в области правого желудочка практически не бывает. Дилатированное сердце всегда имеет плохой коронарный кровоток, но потребляет кислорода больше, чем нормальное.
Нейрогуморальная регуляция сердечной деятельности
Нервная экстракардиальная регуляция. Этот вид регуляции осуществляется импульсами, поступающими из ЦНС по блуждающим и симпатическим нервам.
Подобно всем вегетативным нервам сердечные образованы двумя нейронами. Те
Клиническая физиология сердечно-сосудистой системы 53
ла нейронов, отростки которых составляют блуждающие нервы (парасимпатический отдел автономной нервной системы), расположены в продолговатом мозгу. Отростки этих нейронов заканчиваются в интрамуральных ганглиях сердца. Здесь же находятся другие нейроны, отростки которых идут к проводящей системе, миокарду и коронарным сосудам.
Первые нейроны симпатической части АНС, передающие импульсы к сердцу, расположены в боковых рогах верхних сегментов грудного отдела спинного мозга. Отростки этих нейронов заканчиваются в шейных и верхних грудных симпатических узлах, здесь же находятся другие нейроны, отростки которых идут к сердцу. Большая часть симпатических нервных волокон, иннервирующих сердце, отходит от звездчатого узла.
Влияние на сердце блуждающих нервов впервые было доказано в 1845 г. братьями Вебер. Они установили, что раздражение этих нервов тормозит работу сердца вплоть до полной его остановки в период диастолы.
При сильном раздражении блуждающих нервов работа сердца на некоторое время прекращается. В этот период возбудимость мышцы понижена {отрицательный батмотропный эффект). Замедление проведения возбуждения в сердце называется отрицательным дромотроп-ным эффектом. При продолжительном раздражении блуждающего нерва прекратившее сокращения сердце восстанавливает свою работу, несмотря на продолжающееся раздражение. Это явление называется «ускользанием сердца из-под влияния блуждающего нерва».
Влияние на сердце симпатических нервов проявляется в виде учащения сердечной деятельности {положительный хронотропный эффект). При раздражении симпатических нервов ускоряется спонтанная деполяризация клеток — водителей ритма в период диастолы, что ведет к учащению сердечной деятельности. Раздражение сердечных ветвей симпатического нерва усиливает сокращение сердца {положительный инотропный эффект). Этот эффект — усиление сокра
щения сердца без заметного учащения ритма («усиливающий нерв») — обнаружил И. П. Павлов в 1887 г.
Механизм передачи нервных импульсов в сердце, как и в других органах, имеет химический характер. При раздражении периферических отростков блуждающих нервов в их окончаниях выделяется ацетилхолин, а при раздражении симпатических нервов — норадреналин.
Эти вещества являются медиаторами, вызывающими торможение или усиление деятельности сердца. Ацетилхолин оказывает только местное действие, поскольку разрушается холинэстеразой, содержащейся в плазме крови и клетках. У норадреналина этот процесс происходит значительно медленнее и поэтому он действует дольше.
Центры блуждающих и симпатических нервов занимают вторую ступень в иерархии нервных центров, регулирующих работу сердца. Более высокую ступень представляет интегральный центр, который может изменять любые параметры сердечной деятельности и осуществляет интегральную перестройку функций различных систем организма, в том числе и сердечнососудистой, по сигналам из расположенных выше отделов мозга — лимбической системы или коры.
Рефлекторные изменения работы сердца возникают при раздражении рецепторов, возбуждающихся при изменении давления крови в сосудах или при воздействии гуморальных (химических) раздражителей. Участки сосудов, где сосредоточены рецепторы, получили название сосудистых рефлексогенных зон. Наибольшее значение имеют рефлексогенные зоны, расположенные в дуге аорты.
Кроме безусловнорефлекторной существует и условнорефлекторная регуляция деятельности сердца. Кора головного мозга обеспечивает приспособительные реакции организма не только к нынешним, но и к будущим событиям. С помощью механизма условных рефлексов сигналы, предвещающие наступление этих событий или значительную вероятность их возникновения, могут вызвать перестройку функций сердечно-сосудистой системы в той
54 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
мере, в какой это необходимо, чтобы обеспечить предстоящую деятельность организма.
Гуморальная регуляция деятельности сердца. Изменения работы сердца наблюдаются при действии на него биологически активных веществ, циркулирующих в крови. Наиболее важными из них являются катехоламины (адреналин, норадреналин), увеличивающие силу и частоту сердечных сокращений. При физических нагрузках, эмоциональном напряжении и различных патологических состояниях мозговой слой надпочечников выбрасывает в кровь избыточное количество адреналина, что приводит к усилению сердечной деятельности. Указанный эффект возникает в результате стимуляции катехоламинами рецепторов миокардиоцитов, вызывающих активацию внутриклеточного фермента — аденилатциклазы, ускоряющей образование 3', 5'-циклического аденозинмонофосфата (цАМФ). Последний активирует фосфорилазу, вызывающую расщепление гликогена и образование глюкозы (источника энергии для сокращения миокарда). Фосфорилаза необходима также для активации ионов Са2+, обеспечивающих сопряжение процессов возбуждения и сокращения в миокарде. Кроме этого, катехоламины повышают проницаемость клеточных мембран для ионов Са2+, способствуя, с одной стороны, поступлению их из межклеточного пространства в клетку, а с другой — мобилизации Са2+ из внутриклеточных депо. Активация аденилатциклазы в миокарде происходит и при воздействии глюкагона — гормона, выделяемого а-клетками поджелудочной железы.
2.1.2. Сосудистая система
С точки зрения функциональной значимости для системы кровообращения сосуды подразделяют на такие группы:
1. Упругорастяжимые: аорта с крупными артериями в большом круге кровообращения, легочная артерия с ее ветвями — в малом круге, т. е. сосуды эластичного типа.
2. Сосуды сопротивления — артериолы, в том числе и прекапиллярные сфинк
теры, т. е. сосуды с хорошо выраженным мышечным слоем.
3. Обменные (капилляры) — сосуды, обеспечивающие обмен газами и другими веществами между кровью и тканевой жидкостью.
4. Шунтирующие (артерио-венозные анастомозы) — сосуды, способствующие «сбросу» крови из артериальной системы в венозную, минуя капилляры.
5. Емкостные — вены, обладающие высокой растяжимостью, благодаря чему в них содержится 75 — 80 % ОЦК.
Процессы, протекающие в последовательно соединенных сосудах и обеспечивающие циркуляцию крови, называют системной (центральной) гемодинамикой, а процессы, способствующие кровоснабжению органов, — регионарной, или органной, гемодинамикой.
Системная гемодинамика. Левый желудочек нагнетает кровь в системное сосудистое ложе, состоящее из многочисленных регионарных цепей — мозговой, печеночной, почечной, мышечной и т. д., специализированных по строению и соединенных параллельно. Каждая из цепей обеспечивает потребности обмена соответствующей области организма. Этому способствует многократное разветвление артерии.
Во время систолы внутрижелудочковое давление повышается с уровня, близкого к нулю, до 120 в левом желудочке и до 25 мм рт. ст. — в правом. В результате этого систолическое давление в аорте поднимается до 120, а в легочной артерии — до 25 мм рт. ст. По окончании фазы сокращения сердечная мышца расслабляется и внутрижелудочковое давление резко падает почти до нулевого уровня, полулунные клапаны захлопываются, отделяя аорту и легочную артерию от желудочков.
Аорта и крупные артерии (группа упругорастяжимых сосудов) оказывают незначительное сопротивление току крови, но в силу высокой растяжимости смягчают пульсирующий систолический выброс желудочка. После захлопывания полулунных клапанов эластичные сосуды сокращаются, поддерживая этим градиент давления и делая поступление крови на пе
Клиническая физиология сердечно-сосудистой системы 55
риферию более равномерным. Старение эластических элементов артериальной стенки является одной из причин высокого пульсового давления.
Благодаря эластичности больших артерий и сопротивлению току крови в периферических сосудах, артериальное давление колеблется в значительно меньших пределах, чем давление в желудочках, в результате чего диастолическое давление в системном сосудистом ложе составляет примерно 80 мм рт. ст. Поэтому фазовое изменение давления в левом желудочке - от 120 до 0 мм рт. ст. — превращается в артериальное пульсовое давление, равное 120 - 80 = 40 мм рт. ст. Для малого круга эти показатели составляют приблизительно 25 - 10 = 15 мм рт. ст. Функции артериол, прекапиллярных сфинктеров и обменных сосудов, относящихся к системе микроциркуляции, описаны ниже.
Емкостные сосуды, т. е. венозное ложе, играют незначительную роль в создании общего сопротивления сосудов, но они оказывают большое влияние на емкость сосудистого русла, изменяя свою конфигурацию и диаметр просвета. Минутный объем зависит от венозного возврата; в соответствии с этим изменения емкости венозного русла, вызываемые в основном активностью внешних сосудосуживающих симпатических волокон, могут оказывать значительное влияние на «заправку» сердечного насоса.
Микрогемодинамика. Удовлетворительные показатели системной гемодинамики сами по себе не являются гарантией эффективной перфузии органов и тканей. С другой стороны, не всегда системные нарушения влекут за собой снижение адекватности перфузии. Зачастую именно состояние микроциркуляции определяет тяжесть и прогноз заболевания. Измерения регионарного кровотока в покое показали, что кровоснабжение головного мозга составляет 750 мл/мин, печени — 1300, почек — 1200, мышц — 1000, сердца — 250 мл/мин, суммарно — 4,5 л/мин, не считая снабжения кровью кожи, жировой клетчатки и костей. Ввиду того, что снабжение кровью любой регионарной цепи зависит от градиента давления и местно
го сопротивления сосудов и градиент давления практически везде одинаков, ток крови в этих цепях определяется регионарными условиями микроциркуляции.
Микроциркуляция — собирательное понятие. Оно объединяет механизмы кровотока в мелких сосудах и тесно взаимосвязанный с кровотоком обмен жидкостью и растворенными в ней газами и веществами между сосудами и тканевой жидкостью. К системе микроциркуляции кроме обменных сосудов (капилляров) относятся также прекапиллярные сосуды сопротивления (мелкие артерии и артериолы), прекапиллярные сфинктеры и шунтирующие сосуды. На долю сосудов сопротивления приходится большая часть сопротивления кровотоку. Снабжение кровью любого участка, а также гидростатическое давление в капиллярах этого участка определяются главным образом изменениями радиуса этих сосудов. Сосудам сопротивления свойственна высокая степень собственного (миогенного) базального тонуса, постоянно изменяющегося под воздействием множества местных физических и химических факторов. Эти изменения являются почти единственным механизмом адаптации регионарного сопротивления сосудов, снабжающих кровью сердце и головной мозг. В других местах сосуды сопротивления регионарных цепей находятся также под влиянием симпатических нервов.
Прекапиллярные сфинктеры являются частью прекапиллярных сосудов сопротивления. Они в основном определяют площадь обменной поверхности капилляров, влияя на количество капилляров, перфузируемых в каждый определенный момент. Эти сосуды контролируются преимущественно внутренней миогенной активностью, непрерывно изменяющейся под влиянием местных сосудорасширяющих метаболитов.
Шунтовые сосуды осуществляют прямые связи между мелкими артериями и венами в обход капиллярного ложа. Вследствие этого они не выполняют обменной функции, и их роль сводится к регуляции объемного регионарного кровотока и терморегуляции. В патогенезе острых циркуляторных нарушений их значение возраста
56 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
ет (феномен централизации кровообращения).
Капилляры представляют собой тончайшие сосуды диаметром 6 — 8 мкм и длиной 0,5 — 1 мм, расположенные в межклеточных пространствах и тесно соприкасающиеся с клетками органов и тканей организма. Суммарная длина всех капилляров человека составляет около 100 000 км. Физиологическое значение капилляров состоит в том, что через их стенки осуществляется обмен веществ между кровью и органами. Стенки капилляров образованы одним слоем эндотелиальных клеток, покрытых тончайшей соединительнотканной базальной мембраной. Скорость кровотока в капиллярах невысока и составляет 0,5 — 1 мм/с. Кровь течет лишь в «дежурных» капиллярах, содержащих в покое 5 —7 % ОЦК. В условиях патологии емкость капиллярного русла может резко возрасти (вмещает до 90 % ОЦК). Часть капилляров не задействована в кровообращении, а в период интенсивной работы органов (при сокращении мышц или активной секреторной деятельности) обменные процессы усиливаются и количество функционирующих капилляров значительно возрастает. Регуляция капиллярного кровообращения нервной системой и влияние на него физиологически активных веществ — гормонов и метаболитов — осуществляются за счет воздействия на тонус прекапиллярных сфинктеров. Сужение или расширение последних изменяет как количество функционирующих капилляров, распределение крови в капиллярной сети, так и состав крови, движущейся по капиллярам, т. е. соотношение эритроцитов и плазмы.
В некоторых участках тела, например в коже, легких и почках, имеются непосредственные соединения артериол и венул — артерио-венозные анастомозы. Это наиболее короткий путь между артериолами и венулами. Артерио-венозные анастомозы играют роль шунтов, регулирующих капиллярное кровообращение. В обычных условиях они закрыты, и кровь проходит через капиллярную сеть.
Специального рассмотрения заслуживают процессы обмена между кровью и тка
невой жидкостью. Через сосудистую систему за сутки проходит 8000 — 9000 л крови, через стенку сосудов фильтруется около 20 л жидкости и 18 л реабсорбируется в кровь. По лимфатическим сосудам оттекает около 2 л жидкости.
Закономерности, обуславливающие обмен жидкости между капиллярами и тканевыми пространствами, были описаны Э. Старлингом. Гидростатическое давление крови способствует перемещению жидкости из капилляров в ткани. Основной силой, удерживающей жидкость в капиллярном русле, является онкотическое давление плазмы в капилляре. Определенную роль играет также гидростатическое и онкотическое давление тканевой жидкости. На артериальном конце капилляра гидростатическое давление составляет 30 — 35, а на венозном — 15 — 20 мм рт. ст. Онкотическое давление на всем протяжении капилляра постоянное — 25 мм рт. ст. Таким образом, на артериальном конце осуществляются процессы фильтрации жидкости, а на венозном — реабсорбция. Величина онкотического давления тканевой жидкости составляет примерно 4,5 мм рт. ст.
Капилляры различных органов отличаются по ультраструктуре, а следовательно и по способности пропускать в тканевую жидкость белки. Так, 1 л лимфы в печени содержит 60 г белка, в миокарде — 30, в мышцах — 20, в коже — 10 г. Белок, проникший в тканевую жидкость, с лимфой возвращается в кровь.
Нервная регуляция сосудистой активности
Иннервация сосудов. Сужение артерий и артериол (вазоконстрикция) осуществляется преимущественно симпатическими нервами. Сосудодвигательный центр расположен в продолговатом мозгу на дне IV желудочка и состоит из двух отделов: прессорного и депрессорного. Раздражение первого вызывает сужение артерий и подъем АД, раздражение второго — расширение сосудов и падение АД.
Главными сосудосуживающими нервами органов брюшной полости являются
Клиническая физиология сердечно-сосудистой системы 57
симпатические волокна, проходящие в составе внутренностного нерва. Симпатические сосудосуживающие нервы к конечностям идут в составе спинномозговых смешанных нервов, а также по стенкам артериол.
Сосудорасширяющие эффекты (вазодилатация) осуществляются парасимпатическим отделом нервной системы.
Импульсы, идущие от сосудодвигательного центра продолговатого мозга, проходят к нервным центрам симпатической части вегетативной нервной системы, расположенным в боковых рогах грудных сегментов спинного мозга, регулирующих тонус сосудов отдельных участков тела.
Артерии и артериолы постоянно находятся в состоянии сужения, которое в значительной мере определяется базальным тонусом этих сосудов и тонусом сосудодвигательного центра. Последний зависит от афферентных сигналов, приходящих от периферических рецепторов, расположенных в некоторых сосудистых областях и на поверхности тела, а также от влияния гуморальных раздражителей. Рефлекторные изменения тонуса артерий — сосудистые рефлексы — можно разделить на две группы: собственные и сопряженные.
Собственные сосудистые рефлексы вызываются сигналами от рецепторов самих сосудов. При этом важное значение имеют рецепторы, сосредоточенные в дуге аорты и в области разветвления сонной артерии на внутреннюю и наружную. Указанные участки сосудистой системы получили название сосудистых рефлексогенных зон.
Рецепторы, расположенные в дуге аорты, являются окончаниями центростремительных волокон, проходящих в составе аортального нерва. Этот нерв функционально был обозначен как депрессор. Электрическое раздражение центрального конца нерва обуславливает падение АД вследствие рефлекторного повышения тонуса ядер блуждающих нервов и рефлекторного снижения тонуса сосудодвигательного центра. В результате сердечная деятельность тормозится, а сосуды внутренних органов расширяются.
В рефлексогенной зоне сонного (каротидного) синуса расположены рецепторы,
от которых идут центростремительные нервные волокна, образующие синокаротидный нерв, который входит в мозг в составе языкоглоточного нерва. Понижение системного АД обусловлено тем, что растяжение стенки сонной артерии возбуждает рецепторы каротидного синуса, рефлекторно понижает тонус сосудодвигательного центра и повышает тонус ядер блуждающих нервов.
Рецепторы сосудистых рефлексогенных зон возбуждаются при повышении давления крови в сосудах, поэтому их называют прессорецепторами, или барорецепторами.
Снижение АД вследствие гиповолемии, ослабления деятельности сердца или при перераспределении крови и оттоке ее в избыточно расширившиеся сосуды какого-либо крупного органа ведет к тому, что прессорецепторы дуги аорты и сонных артерий раздражаются менее интенсивно, чем при нормальном АД. Влияние аортальных и синокаротидных нервов на нейроны сердечно-сосудистого центра ослабляется, сосуды сужаются, работа сердца усиливается и АД нормализуется.
Рефлекторная регуляция давления крови осуществляется при помощи не только механо-, но и хеморецепторов. Они сосредоточены в каротидных тельцах, чувствительны к СО2, гипоксии, СО, цианидам, никотину. От хеморецепторов возбуждение передается к сосудодвигательному центру и вызывает повышение его тонуса. В результате сосуды сужаются и давление повышается. Одновременно происходит возбуждение дыхательного центра.
Гуморальная регуляция сосудистого тонуса
Сосудосуживающие вещества. К ним относятся гормоны мозгового вещества надпочечников — адреналин и норадреналин, а также задней доли гипофиза — вазопрессин. Адреналин и норадреналин сужают артерии и артериолы кожи, органов брюшной полости и легких, а вазопрессин действует преимущественно на артериолы и капилляры и оказывает влияние на сосуды в очень малых концентрациях.
58 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
К числу сосудосуживающих гуморальных факторов относится серотонин, продуцируемый в слизистой оболочке кишок и в некоторых участках головного мозга. Он образуется также при распаде тромбоцитов. Физиологическое значение серотонина состоит в том, что он сужает сосуды и препятствует кровотечению из пораженного сосуда. Во второй фазе свертывания крови, после образования тромба, серотонин расширяет сосуды.
Еще один сосудосуживающий фактор — ренин — синтезируется в почках, причем чем ниже их кровоснабжение, тем в большем количестве он продуцируется. Ренин представляет собой протеолитический фермент. Сам по себе он не вызывает сужения сосудов, но, поступая в кровь, расщепляет а2-глобулин плазмы (ангиотензиноген) и превращает его в относительно малоактивный ангиотензин I, который под влиянием фермента дипептидкарбоксипептидазы (ангиотензинконвертаза, ангиотен-зинпревращающий фермент) переходит в очень активную сосудосуживающую форму — ангиотензин II. Последний быстро разрушается в капиллярах ангиотензина-зой. В условиях нормального кровоснабжения почек образуется сравнительно небольшое количество ренина.
Открытие ренина и механизма его сосудосуживающего действия объяснило причину высокого АД, сопутствующего некоторым заболеваниям почек.
Сосудорасширяющие вещества. Во многих тканях тела синтезируются сосудорасширяющие вещества, получившие название простагландинов, которые представляют собой производные насыщенных жирных кислот. Из подчелюстной, поджелудочной желез, легких и некоторых других органов выделены сосудорасширяющие пептиды, относящиеся к группе кининов. Наиболее известным из них является брадикинин, вызывающий расслабление гладкой мускулатуры артериол и снижение АД.
К сосудорасширяющим веществам относят также ацетилхолин, который продуцируется в окончаниях парасимпатических нервов, и гистамин, образующийся в слизистой оболочке желудка и кишок, а также в других органах, в частности в коже и скелетной мускулатуре. Эти вещества вызывают расширение артериол и увеличение кровенаполнения капилляров.
В последние годы установлена важная роль эндотелия сосудистой стенки в регуляции кровотока. Эндотелиоциты под влиянием химических раздражителей, приносимых кровью (например, NO), способны выделять вещества, действующие на сосудистый тонус и вызывающие расширение сосудов.
Сосуды ряда органов и тканей обладают специфическими особенностями регуляции, которые определяются структурой и функцией данного органа.
2.2. ПАТОФИЗИОЛОГИЧЕСКИЕ ИЗМЕНЕНИЯ В СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЕ ПРИ КРИТИЧЕСКИХ СОСТОЯНИЯХ
2.2.1. Основные физиологические факторы, определяющие функциональное состояние кровообращения
Как в экспериментальных, так и в клинических исследованиях продемонстрировано, что в патогенезе нарушений циркуляторного гомеостаза при критических состояниях принимают участие разные факторы: гипоксия, токсемия, перераспределение жидкости по секторам при общем
ее уменьшении, водно-электролитный, кислотно-основный и энергетический дисбалансы, нарушения гемореологии и др. Все они вызывают уменьшение венозного подпора, снижение сократимости миокарда и производительности сердца, изменение сосудистого сопротивления, централизации кровообращения, что в конечном итоге приводит к ухудшению перфузии тканей. При всей полиэтиологичности циркуляторных расстройств у больных в критических состояниях существует группа факто
Патофизиологические изменения в сердечно-сосудистой системе при критических состояниях 59
ров, непосредственно определяющих гемодинамический статус пациента, и ряд критериев, позволяющих оценить этот статус.
Главным критерием функционального состояния сердечно-сосудистой системы является величина сердечного выброса. Его адекватность обеспечивают:
а) венозный возврат;
б) сократительная способность миокарда;
в) периферическое сопротивление для правого и левого желудочков;
г) частота сердечных сокращений;
д) состояние клапанного аппарата сердца.
Любые расстройства кровообращения можно увязать с функциональной недостаточностью сердечного насоса, если считать главным показателем его адекватности сердечный выброс:
острая сердечная недостаточность — снижение сердечного выброса при нормальном или повышенном венозном возврате;
острая сосудистая недостаточность — нарушение венозного возврата вследствие увеличения сосудистого русла;
острая недостаточность кровообращения — снижение сердечного выброса независимо от состояния венозного возврата.
Рассмотрим наиболее важные факторы, влияющие на величину сердечного выброса.
Венозный возврат — это объем крови, поступающей по полым венам в правое предсердие. В обычных клинических условиях прямое измерение его практически неосуществимо, поэтому широко используются косвенные методы его оценки, например, исследование центрального венозного давления (ЦВД). Нормальный уровень ЦВД составляет примерно 7 —12 см вод. ст. (686—1177 Па).
Величина венозного возврата зависит от следующих компонентов:
1) объема циркулирующей крови;
2) величины внутригрудного давления;
3) положения тела (при возвышенном положении головного конца венозный возврат уменьшается);
4) изменения тонуса вен (сосудов-емкостей): при действии симпатомиметиков и глюкокортикоидов возникает повыше
ние тонуса вен; ганглиоблокаторы и адре-нолитики снижают венозный возврат;
5) ритмичности изменения тонуса скелетных мышц в сочетании с работой венозных клапанов;
6) адекватности сокращения предсердий и ушек сердца, что обеспечивает 20 — 30 % дополнительного наполнения и растяжения желудочков.
Среди факторов, определяющих состояние венозного возврата, важнейшим является ОЦК. Он состоит из объема форменных элементов, в основном эритроцитов (относительно постоянный объем), и объема плазмы. Последний обратно пропорционален величине гематокрита. Объем крови составляет в среднем 50 — 80 мл на 1 кг массы тела (5 —7 % массы). Наибольшая часть крови (до 75 %) содержится в системе низкого давления (венозная часть сосудистого русла). В артериальном отделе находится около 20 % крови, в капиллярном — около 5 %. В состоянии покоя до половины объема крови может быть представлена пассивной фракцией, депонированной в органах и включающейся в кровообращение в случае необходимости (например, кровопотери или мышечной работы).
Для адекватной функции системы кровообращения важно прежде всего не абсолютное значение ОЦК, а степень его соответствия емкости сосудистого русла. У ослабленных больных и у больных с длительным ограничением подвижности всегда имеется абсолютный дефицит ОЦК, однако он компенсируется венозной вазоконстрикцией. Недооценка этого факта зачастую приводит к осложнениям во время вводной анестезии, когда использование индукторов (например, барбитуратов) снимает вазоконстрикцию. Возникает несоответствие ОЦК емкости сосудистого русла, вследствие чего снижается венозный возврат и сердечный выброс.
В основу современных методов измерения ОЦК положен принцип разведения индикаторов, однако в силу его трудоемкости и необходимости соответствующего аппаратурного обеспечения он не может быть рекомендован для рутинного клинического использования.
60 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
К клиническим признакам снижения О ЦК относятся: бледность кожных покровов и слизистых, снижение кровотока в периферическом венозном русле, тахикардия, артериальная гипотензия, снижение ЦВД. Ни один из этих признаков не имеет самостоятельного значения для оценки дефицита О ЦК и только их комплексное использование позволяет приблизительно оценить его.
В настоящее время сократительная способность миокарда и периферическое сосудистое сопротивление определяются с использованием концепций преднагрузки и постнагрузки.
Преднагрузка эквивалентна силе, растягивающей мышцу перед ее сокращением. Очевидно, что степень растяжения волокон миокарда до диастолической длины определяется величиной венозного возврата. Иными словами, конечный диастолический объем (КДО) эквивалентен пред-нагрузке. Однако в настоящее время не существует рутинных методов, позволяющих в условиях клиники осуществлять прямое измерение КДО. Плавающий (флотационно-баллонный) катетер, проведенный в легочную артерию, дает возможность измерить давление заклинивания в легочных капиллярах (ДЗЛК), которое равно конечному диастолическому давлению (КДД) в левом желудочке. В большинстве случаев это соответствует истине — ЦВД равно КДД в правом желудочке, а ДЗЛК — в левом. Тем не менее КДД эквивалентно КДО только при нормальной растяжимости миокарда. Любые процессы, вызывающие снижение растяжимости (воспаление, склероз, отек, ИВЛ с ПДКВ и др.), приводят к нарушению корреляции между КДД и КДО (для достижения той же величины КДО потребуется большее КДД). Таким образом, КДД позволяет надежно охарактеризовать пред-нагрузку только при неизмененной растяжимости желудочков. Кроме того, ДЗЛК может не соответствовать КДД в левом желудочке при аортальной недостаточности и при выраженной патологии легких.
Постнагрузка определяется как сила, которую необходимо преодолеть желудочку, чтобы выбросить ударный объем кро
ви. Она эквивалентна трансмуральному напряжению, возникающему в стенке желудочка во время систолы и включает в себя следующие компоненты:
— преднагрузку;
— общее периферическое сопротивление сосудов;
— давление в плевральной полости (отрицательное давление в плевральной полости увеличивает постнагрузку, положительное — уменьшает).
Таким образом, постнагрузка создается не только сосудистым сопротивлением, она включает в себя и преднагрузку, поскольку на преодоление последней затрачивается часть систолической работы желудочка, а также компонент, не являющийся частью сердечно-сосудистой системы.
Необходимо различать сократительную способность и сократимость миокарда. Первая является эквивалентом полезной работы, которую может выполнить миокард при оптимальных значениях пред- и постнагрузки, т. е. потенциальной функцией. Сократимость же является функцией актуальной, поскольку определяется работой, выполняемой миокардом при их реальных значениях. Если пред- и постнагрузка неизменны, то систолическое давление зависит от сократимости.
Фундаментальным законом физиологии сердечно-сосудистой системы является закон Франка —Старлинга’, сила сокращения пропорциональна исходной длине миокардиальных волокон, т. е. работа сердца зависит от объема крови в желудочках в конце диастолы. Первые исследования, в результате которых получены эти данные, были проведены в 1885 г. О. Франком и несколько позднее продолжены Э. Старлингом. Физиологический смысл сформулированного ими закона (закона Франка —Старлинга) заключается в том, что большее наполнение полостей сердца кровью автоматически увеличивает силу сокращения и, следовательно, обеспечивает более полное опорожнение.
Как уже упоминалось, величина давления в левом предсердии определяется величиной венозного подпора. Однако сердечный выброс возрастает линейно до определенного потенциала, затем его увеличе
Патофизиологические изменения в сердечно-сосудистой системе при критических состояниях 61
ние происходит более полого. И, наконец, наступает момент, когда повышение конечного диастолического давления не приводит к увеличению сердечного выброса. Взаимосвязь между этими величинами приближается к линейной только на начальном отрезке кривой «давление — объем». В целом, ударный объем увеличивается до тех пор, пока диастолическое растяжение не превысит 2/3 максимального растяжения. Это соответствует уровню конечного диастолического давления, равному приблизительно 60 мм рт. ст. Если диастолическое растяжение (наполнение) превышает 2/3 максимального, то ударный объем перестает увеличиваться. В клинике подобное давление наблюдается редко, однако в случае патологии миокарда ударный объем сердца может снижаться и при более низких цифрах конечного диастолического давления (КДД).
При умеренной сердечной недостаточности способность желудочка реагировать на преднагрузку сохраняется только при давлении наполнения, превышающем норму. Это свидетельствует о том, что сердечный выброс и уровень кровотока на данном этапе еще могут быть сохранены за счет включения компенсаторных механизмов (увеличение венозного подпора), поскольку деятельность желудочка при умеренной недостаточности зависит не столько от постнагрузки, сколько от пред нагрузки. По мере дальнейшего снижения функции сердца деятельность желудочка становится все менее зависимой от предна-грузки. Роль же постнагрузки при тяжелой сердечной недостаточности продолжает возрастать, поскольку вазоконстрикция не только снижает сердечный выброс, но и уменьшает периферический кровоток.
Таким образом, при прогрессировании сердечной недостаточности компенсаторная функция увеличенной преднагрузки постепенно утрачивается и давление венозного подпора не должно превышать критический уровень, чтобы не вызвать перерас-тяжение левого желудочка. По мере увеличения дилатации желудочков пропорционально растет и потребление кислорода. Когда диастолическое растяжение превышает 2/3 максимального, а потребность
в кислороде растет, развивается «кислородная ловушка» — потребление кислорода большое, а сила сокращений не увеличивается. При хронической сердечной недостаточности гипертрофированные и дилатированные участки миокарда начинают потреблять до 27 % всего необходимого организму кислорода (больное сердце работает только на себя).
Физическое напряжение и гиперметаболические состояния приводят к усилению сокращений поперечнополосатых мышц, увеличению частоты сердечных сокращений и минутного объема дыхания. При этом возрастает приток крови по венам, увеличиваются центральное венозное давление, ударный и минутный объемы сердца.
При сокращении желудочков никогда не выбрасывается вся кровь — остается некоторое ее количество — остаточный систолический объем (ОСО). В норме фракция выброса в состоянии покоя составляет около 70 %. При физической нагрузке в норме фракция выброса возрастает, а абсолютное значение ОСО остается прежним вследствие увеличения ударного объема сердца.
Начальное диастолическое давление в желудочках определяется величиной ОСО. В норме при физической нагрузке увеличивается приток крови и потребность в кислороде, а также объем производимой работы. Таким образом, энергетические затраты целесообразны, и коэффициент полезного действия сердца не снижается.
При развитии различных патологических процессов (миокардиты, интоксикация и т. д.) происходит первичное ослабление функции миокарда. Он не в состоянии обеспечить адекватный сердечный выброс, и ОСО увеличивается. При сохраненном ОЦК на ранних этапах (до развития систолической дисфункции) это приведет к увеличению диастолического давления и к усилению сократительной функции миокарда.
В неблагоприятных условиях миокард сохраняет величину ударного объема, но в результате более выраженной дилатации потребность в кислороде увеличивается. Сердце выполняет ту же работу, но с большими энергетическими затратами.
62 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
При гипертонической болезни повышается сопротивление выбросу. Минутный объем сердца (МОС) либо остается неизменным, либо увеличивается. Сократительная функция миокарда на начальных этапах заболевания сохраняется, но сердце гипертрофируется, чтобы преодолеть возросшее сопротивление выбросу. Затем, если гипертрофия прогрессирует, она сменяется дилатацией. Возрастают энергетические затраты, КПД сердца снижается. Часть работы сердца расходуется на сокращение дилатированного миокарда, что приводит к его истощению. Поэтому у гипертоников часто развивается левожелудочковая недостаточность.
Кроме того, сила миокардиального сокращения может увеличиваться в зависимости от пресистолического растяжения в ответ на учащение ритма. Повышение тонуса симпатической нервной системы также увеличивает силу сердечных сокращений. Положительное инотропное действие оказывают симпатические амины, р-ад-реностимуляторы, сердечные гликозиды, эу фи л л ин, ионы Са2+. Эти вещества усиливают сокращение миокарда независимо от его пресистолического наполнения, но при передозировке они могут вызвать электрическую нестабильность миокарда. Сократительная способность сердца угнетается: гипоксией; респираторным и метаболическим ацидозом (pH < 7,3) и алкалозом (pH > 7,5), некрозом, склерозом, воспалительными и дистрофическими изменениями миокарда; повышением или снижением температуры.
Важнейшим фактором сократительной способности миокарда является состояние коронарного кровотока, которое зависит от диастолического давления в аорте, проходимости коронарных сосудов, напряжения газов крови, симпатоадреналовой активности и регулируется только потребностью миокарда в кислороде. Миокард не может «брать кислород в долг», а метаболизм в сердце — происходить в условиях образования кислых продуктов и гипоксии. При прекращении кровотока метаболизм в скелетных мышцах длится еще 1,5 —2 ч, а в миокарде прекращается через 1—3 мин. Сократительная способность
зависит также от внутри- и внеклеточного содержания ионов К+, Na+, Са2+, Mg2+, которые обеспечивают силу мышечного сокращения и электрическую стабильность миокарда.
2.2.2. Мониторинг показателей системной гемодинамики
Рутинная оценка состояния гемодинамики. К сожалению, до настоящего времени в клинике отсутствуют методы простого, быстрого и точного определения гемодинамического статуса. Вследствие этого первым этапом, позволяющим получить ориентировочную информацию о состоянии кровообращения, является физикальное обследование больного. Для косвенной клинической оценки гемодинамического статуса должен приниматься во внимание комплекс различных признаков, каждый из которых сам по себе не имеет точного диагностического значения. К числу наиболее важных из них относятся: уровень сознания, окраска, температура и тургор кожных покровов и слизистых, состояние подкожной сосудистой сети, характер дыхания, наличие периферических отеков, частота и свойства пульса, аускультативные феномены и др. Важнейшим критерием состояния системной гемодинамики является артериальное давление.
К факторам, определяющим величину АД, относятся объемная скорость кровотока и общее периферическое сопротивление сосудов (ОПСС). Объемная скорость кровотока для сосудистой системы большого круга кровообращения определяется минутным объемом крови (МОК), нагнетаемым сердцем в аорту. ОПСС является расчетной величиной, зависящей от тонуса сосудов (в основном артериол), определяющего их радиус, длины сосуда и вязкости протекающей крови.
Определение артериального давления. Во время каждой систолы порция крови поступает в артерии и увеличивает их эластическое растяжение, при этом давление в них повышается. Во время диастолы поступление крови из желудочков в артериальную систему прекращается и происходит отток крови из крупных артерий,
Патофизиологические изменения в сердечно-сосудистой системе при критических состояниях 63
растяжение их стенок уменьшается и давление снижается. Наибольшая величина давления в артериях наблюдается во время прохождения вершины пульсовой волны (систолическое давление), а наименьшая — во время прохождения основания пульсовой волны (диастолическое давление). Разность между систолическим и диастолическим давлением называется пульсовым давлением. При прочих равных условиях оно пропорционально количеству крови, выбрасываемому сердцем при каждой систоле.
Кроме систолического, диастолического и пульсового давления, определяют так называемое среднее артериальное давление (САД) — равнодействующую всех изменений давления в сосудах. При инвазивной регистрации системного АД среднее артериальное давление рассчитывают путем измерения площади, ограниченной кривой АД, и ее деления на длительность кардиоцикла. При расчетном определении используют формулу
САД = АДдиаст + 1/ЗАДПуЛЬС.
Точность измерения САД при помощи автоматической инвазивной регистрации значительно выше, чем при использовании расчетного способа.
У взрослого человека систолическое давление в аорте равно 110 —125 мм рт. ст. По мере прохождения по сосудам оно резко уменьшается и на артериальном конце капилляра составляет 20 — 30 мм рт. ст. С возрастом максимальное давление повышается, у 60-летних оно равно 135 — 140 мм рт. ст. У новорожденных систолическое АД составляет около 50 мм рт. ст., а к концу 1-го месяца возрастает до 80 мм рт. ст. Минимальное диастолическое АД у взрослых людей среднего возраста в среднем равно 60 — 80 мм рт. ст., пульсовое — 35 — 40, среднее — 90 — 95 мм рт. ст.
Особенности измерения и интерпретации АД. В условиях операционной и отделения интенсивной терапии наиболее частым исследованием, влияющим на тактические и стратегические решения, является измерение АД. При этом лишь в редких
случаях врач сомневается в достоверности получаемых результатов. Ниже приведен ряд позиций, которые необходимо учитывать для приближения имеющихся показателей к клинической реальности.
1. Сама процедура измерения АД при помощи манжеты может привести к ошибкам (увеличение объема крови и давления в области плеча). Ложное завышение систолического АД наиболее часто отмечается у пациентов старческого возраста и у страдающих артериальной гипертензией. У больных с ожирением, а также при неплотном наложении манжеты могут завышаться показатели диастолического АД. Занижение АД свойственно чрезмерно плотному наложению манжеты и процедуре, проводимой у астеников и истощенных больных.
2. Ложное занижение систолического и завышение диастолического АД часто происходит при его измерении у больных с брадиаритмиями и при выраженной брадикардии.
3. В связи с тем, что тоны Короткова возникают благодаря кровотоку, у больных с нестабильной гемодинамикой при любом варианте снижения системного кровотока наблюдается занижение показателей АД. Так, у больных с сердечной недостаточностью разница между полученным и истинным значениями АД может превышать 60 мм рт. ст.
4. Систолическое и диастолическое АД в периферических артериях не всегда соответствует таковому в аорте, а САД практически не изменяется. Поэтому динамика САД является наиболее адекватным способом оценки системной гемодинамики при ее нестабильности.
При всей важности АД как критерия состояния системной гемодинамики не следует забывать о том, что давление является не абсолютным показателем состояния сердца и сосудов, а зависимой величиной, которая определяется взаимоотношением между сердечным выбросом и ОПСС. Двойственный характер природы АД не позволяет точно оценивать ни производительность сердца, ни сосудистый тонус. При одной и той же величине АД кровоток может быть различным.
64 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
Инвазивный мониторинг системной гемодинамики. Парадокс использования неинвазивных методов оценки гемодинамики состоит в том, что вероятность и величина погрешности измерений значительно возрастают именно в тех ситуациях, когда точное знание гемодинамических параметров наиболее актуально (критические состояния, нестабильность гемодинамики). Необходимость повышения точности измерений способствовала разработке и внедрению методов инвазивного контроля.
Для инвазивного мониторинга наиболее актуальных гемодинамических показателей необходима и достаточна катетеризация двух артерий: периферической (лучевой или бедренной) — для определения АД и легочной — для определения других параметров гемодинамического статуса.
Хотя наиболее точные результаты при измерении АД достигаются при использовании инвазивного мониторинга, этот способ также не лишен недостатков. Артефакты, обусловленные демпфирующими свойствами измерительных контуров, могут приводить к погрешности измерения порядка 25 — 30 мм рт. ст. Кроме того, вопреки распространенному мнению о снижении АД по мере продвижения крови в сосудистом русле отмечается повышение систолического АД по мере продвижения пульсовой волны дистально от аорты. Диастолическое АД при этом постепенно снижается, САД остается относительно постоянным (речь идет о крупных сосудах; по мере приближения к зоне микроциркуляции все виды АД начинают постепенно снижаться).
С целью полноты оценки функционального состояния сердечно-сосудистой системы помимо катетеризации лучевой или бедренной артерии для регистрации АД в настоящее время наиболее часто исследуют легочную артерию плавающим катетером. Использование этой методики предусматривает прямое измерение: ЦВД, ДЗЛК, сердечного выброса и насыщения кислородом смешанной венозной крови. Ранее указывалось, что ЦВД и ДЗЛК, как правило, равняются КДД в соответствую
щих желудочках, а КДД, в свою очередь, при неизмененной растяжимости миокарда адекватно отражает К ДО.
На основании результатов прямых измерений рассчитывают производные параметры — индексы: сердечный, ударный, ударной работы правого и левого желудочков, ОПСС, сопротивления легочных сосудов, а также наиболее значимые параметры транспорта кислорода (индекс доставки и потребления, коэффициент экстракции).
Принцип данной методики состоит в следующем. Плавающий катетер, предназначенный для проведения в легочную артерию, снабжен у дистального конца раздувающимся баллончиком объемом около 1,5 мл. По стандартной методике катетер вводится в подключичную или внутреннюю яремную вену. После попадания дистального конца катетера в просвет вены баллончик раздувают и катетер медленно продвигают по току крови. Катетер с баллончиком последовательно проходит верхнюю полую вену, правое предсердие, правый желудочек и попадает в легочную артерию. В рентгенологическом контроле нет необходимости. О положении катетера в каждый момент времени судят по характерной форме постоянно регистрируемой кривой давления, специфичной для каждого отдела сердечно-сосудистой системы. Например, кривая давления в верхней полой вене и в предсердии имеет венозный профиль и регистрируемое давление равно ЦВД. После прохождения катетером трехстворчатого клапана и попадания в правый желудочек регистрируется характерная волна систолического давления. За клапаном легочной артерии (при попадании в просвет легочного ствола) на кривой давления появляется диастолическая волна. При дальнейшем продвижении катетера в дистальные отделы легочной артерии наступает момент, когда раздутый баллончик обтурирует просвет сосуда и легочный кровоток прекращается. При этом пропадает систолический компонент пульсовой волны, а регистрируемое в этот момент «конечное» давление получило название давления заклинивания в легочных капиллярах. После регистрации ДЗЛК
Патофизиологические изменения в сердечно-сосудистой системе при критических состояниях 65
баллончик сразу же сдувают до следующего измерения.
Таким образом, последовательное перемещение катетера по сосудам и камерам сердца дает возможность прямо измерять два клинически значимых вида давления: ЦВД и ДЗЛК. Данная методика позволяет исследовать не только давление, но и сократительную активность миокарда. У дистального конца катетера расположен термистор, регистрирующий температуру окружающей крови. Это позволяет непосредственно измерять сердечный выброс методом термодилюции. Двух- или трехпросветный катетер имеет также проксимальное отверстие, расположенное на расстоянии 30 см от дистального конца. В то время как дистальное отверстие катетера попадает в легочную артерию, проксимальное находится в правых отделах сердца. Термоиндикатор (изотонический раствор натрия хлорида или глюкозы комнатной температуры) в объеме 5—10 мл быстро (не более 4 с) вводится в катетер и через проксимальное отверстие поступает в венозную кровь. В правом отделе сердца этот раствор смешивается с кровью и температура последней понижается. Охлажденная кровь выбрасывается в легочную артерию, где термистор регистрирует изменение температуры. Разница температур фиксируется на экране в виде термодилюционной кривой (время —температура), площадь которой обратно пропорциональна объемной скорости кровотока в легочной артерии. При отсутствии внутрисердечных шунтов справа налево объемную скорость кровотока в легочной артерии считают равной сердечному выбросу.
Кроме того, в порции крови, взятой из дистального отверстия катетера, определяют насыщение гемоглобина кислородом для оценки экстракции кислорода тканями как одного из компонентов системного транспорта кислорода.
Ниже приведены нормальные значения величин, получаемых в результате прямых измерений.
1. Группа показателей давления, наиболее важными из которых являются ЦВД и ДЗЛК (мм рт. ст.): правое предсердие (ЦВД) — 0 — 4; правый желудочек —
3 4-220
15 — 30/0 — 4; легочная артерия — 15 — 30/6—12; среднее давление в легочной артерии - 10-18; ДЗЛК - 6-12.
2. Сердечный выброс (ударный объем) — 70 — 80 мл.
3. Насыщение кислородом венозной крови — 68 — 77 %.
Прямая регистрация описанных показателей, дополненная измерением АД, позволяет рассчитать ряд производных параметров, дающих в комплексе детальную информацию о состоянии гемодинамики и кислородного транспорта. Все производные показатели представляют в виде индексов — отношение показателя к площади поверхности тела (ППТ) — для нивелирования индивидуальных антропометрических отличий. Наиболее важные из производных параметров и их нормальные значения приведены ниже.
1. Сердечный индекс (СИ) — отношение сердечного выброса (минутного объема кровообращения, равного произведению У О на частоту сердечных сокращений (ЧСС), определенного методом термодилюции, к ППТ — 2 — 4 л/(мин • м2).
у О
2. Ударный индекс (УИ) = ——— = = (36 — 48) мл/м2.
3. Индекс ударной работы левого желудочка (ИУРЛЖ) характеризует работу желудочка за одно сокращение: ИУРЛЖ = = (САД - ДЗЛК) • УИ • 0,0136 = = (44 — 56) г • м/м2.
4. Индекс ударной работы правого желудочка: ИУРПЖ = (ДЛА - ЦВД) • УИ х х 0,0136 = (7-10) г • м/м2.
5. Индекс общего периферического сопротивления: ИОПСС = (САД -- ЦВД) : СИ • 80 = (1200-2500) дин/(с х х см5 • м2).
6. Индекс сопротивления легочных сосудов: ИСЛС = (ДЛА - ДЗЛЮ/СИ х х 80 = (80 — 240) дин/(с • см5 • м2).
7. Группа показателей системного транспорта кислорода: индекс доставки, индекс потребления, коэффициент утилизации.
Такая подробная информация о функции сердечно-сосудистой системы значительно расширяет как диагностические возможности врача, так и эффективность проводимой терапии. Однако не следует
66 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
абсолютизировать данные, полученные при катетеризации легочной артерии. Это связано как с техническими особенностями самого метода, так и с его интерпретацией.
ДЗЛК само по себе не представляет диагностической ценности, его значение заключается в том, что этот показатель считают равным конечному диастолическому давлению в левом желудочке (аналог ЦВД для правых отделов). Метод измерения ДЗЛК следующий: баллончик на дистальном конце катетера, введенного в легочную артерию, раздувают до тех пор, пока не наступит обструкция кровотока. Это вызывает образование столба крови между баллончиком и левым предсердием, и давление с двух концов столба уравновешивается. При этом давление в конце катетера становится равным давлению в левом предсердии или конечному диастолическому давлению в левом желудочке (КДДЛЖ). В большинстве случаев ДЗЛК действительно соответствует КДДЛЖ, однако эта корреляция может нарушаться при аортальной недостаточности, жесткой стенке желудочка, легочной патологии, ПДКВ — т. е. в ситуациях, не столь уж редких в клинике, что уменьшает диагностическую ценность данного показателя.
Кроме того, ДЗЛК часто применяют в качестве критерия гидростатического давления в легочных капиллярах, что позволяет оценить возможность развития гидростатического отека легких. Однако проблема заключается в том, что ДЗЛК измеряют в условиях полной окклюзии легочной артерии, т. е. в условиях отсутствия кровотока. При сдувании баллончика кровоток восстанавливается, и давление в капиллярах превышает ДЗЛК. Капиллярное давление, в отличие от ДЗЛК, растет при повышении среднего давления в легочной артерии и росте сопротивления легочных вен (например, при остром респираторном дистресс-синдроме) и может превышать ДЗЛК в два раза и более. Если принимать ДЗЛК всегда равным капиллярному гидростатическому давлению, то в некоторых случаях некорректная интерпретация может приводить к серьезным терапевтическим ошибкам.
Тем не менее, учитывая описанные ограничения, результаты, полученные при ка
тетеризации легочной артерии, по праву считают «золотым стандартом» исследования функционального состояния кровообращения. Вместе с тем переоценка значимости инвазивного мониторинга нередко приводит к увеличению частоты осложнений (гемодинамических, септических). Следует помнить, что катетеризация легочной артерии является все же диагностическим, а не терапевтическим мероприятием и далеко не всегда ассоциируется со снижением летальности в соответствующих группах больных.
Таким образом, «эталонная» точность получаемых результатов обеспечивается высокой инвазивностью процедуры, всегда представляющей определенный риск для пациента. В последние годы это побудило даже энтузиастов инвазивного мониторинга — американских специалистов — обратиться к более безопасным альтернативам. Это прежде всего биологическая импедансография (реография) в различных ее вариантах и большой набор версий ультразвукового метода, включая и самую современную — чреспищеводную допплерографию. Выбор метода исследования гемодинамики диктуется не только соответствующим оборудованием и квалификацией персонала, но и такими критериями, как инвазивность, точность, сложность, стоимость, возможность и удобство мониторинга и др. Следует четко представлять, какие гемодинамические параметры обладают наибольшей диагностической значимостью в конкретной ситуации. Так, катетеризация легочной артерии по-прежнему незаменима для точной селективной оценки преднагрузки левого желудочка. В то же время одним из преимуществ использования эхосонографии оказалась возможность исследования локальной кинетики стенки сердца. Необходимо помнить, что при всех своих преимуществах ни один из перечисленных методов не решает конечных диагностических проблем. Это связано с тем, что конечной целью кровообращения является адекватный тканевый кровоток, а возможности использования прямого мониторинга кровоснабжения наиболее важных органов в условиях клиники в настоящее время отсутствуют.
Принципы диагностики и интенсивной терапии некоторых нарушений 67
2.3. ПРИНЦИПЫ ДИАГНОСТИКИ И ИНТЕНСИВНОЙ ТЕРАПИИ НЕКОТОРЫХ НАРУШЕНИЙ ФУНКЦИИ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ ПРИ КРИТИЧЕСКИХ СОСТОЯНИЯХ
2.3.1. Аналитический подход к коррекции гемодинамических нарушений
В сложном комплексе проблем интенсивной терапии особое место занимают нарушения циркуляторного гомеостаза и функционального состояния миокарда. Очевидно, что своевременная диагностика нарушений насосной функции сердца и ухудшения его инотропного состояния, напряжения компенсации и срыва компенсаторных возможностей играет важную роль в определении тактики лечения и прогноза заболевания у больных в критических состояниях. Как уже упоминалось, ведущими факторами гемодинамических расстройств у таких больных являются присущие критическим состояниям гипоксия, перераспределение жидкости по секторам, электролитный и кислотно-основный дисбаланс, токсемия, нарушения гемореологии и метаболизма, симпатоадреналовая активация. Имеют значение также ятрогенные воздействия: использование поливалентной инфузионно-трансфузионной терапии и фармакологических препаратов, ИВЛ с применением различных ее видов и режимов и т. д.
Оценка функционального состояния гемодинамики и дифференциальная диагностика гемодинамических расстройств должны начинаться с оценки ряда клинических признаков, которые, не являясь патогномоничными для циркуляторных нарушений, тем не менее, могут быть с ними связаны. К их числу относятся: состояние кожи (окраска, влажность, тургор), слизистых, подкожной венозной сети, аускультативная картина в легких, размеры печени, наличие периферических отеков и т. д. Определенную косвенную информацию может дать регистрация ЭКГ.
Кроме перечисленных косвенных признаков несложными для определения и гораздо более информативными прямыми критериями оценки состояния гемодина
3*
мики являются: измерение уровня АД, подсчет частоты пульса и пальпаторная оценка его свойств, измерение ЦВД. Важнейшим интегральным критерием состояния системной гемодинамики является АД (об ограничениях метода Короткова и особенностях интерпретации этого показателя у больных с циркуляторными нарушениями упоминалось выше. В частности, нормальное АД при тяжелом состоянии больных отнюдь не всегда является критерием нормализации кровообращения).
К числу несомненных достоинств перечисленных способов оценки гемодинамики относятся простота, быстрота и доступность, позволяющая использовать их в любых условиях. Однако ценой, которую приходится платить за простоту, является низкая информативность. Главным недостатком рутинных способов диагностики гемодинамических расстройств является то, что изменения большинства применяемых критериев зачастую связаны с тяжелыми либо декомпенсированными циркуляторными нарушениями. Это резко ограничивает возможности ранней диагностики и, следовательно, опережающей терапии.
Кроме того, в популяции больных в критическом состоянии изменения сердечно-сосудистой системы, лежащие в основе гемодинамических расстройств, могут существенно отличаться. Так, при массивной кровопотере кроме различных вариантов изменений насосной функции сердца типичным гемодинамическим паттерном является наличие неоднородности изменений в системе малого и большого кругов кровообращения. Для больных с тяжелой изолированной черепно-мозговой травмой к характерным изменениям гемодинамики относятся повышение насосной функции сердца и легочная гипертензия; постреанимационной патологии сопутствует ярко выраженная этапность гемодинамических изменений (гипердинамия — относительная нормализация — гиподинамия) и т. д.
68 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
Ясно, что при использовании только рутинных методов не может быть реализован главный принцип, необходимый для углубленного изучения состояния системы кровообращения, — комплексный анализ системной и легочной гемодинамики, состояния миокарда правых и левых отделов сердца, их систолической и диастолической функции и резервных возможностей, системного и легочного периферического сопротивления. Тем самым значительно сужаются возможности дифференциальной диагностики гемодинамических расстройств, что в конечном итоге обуславливает невозможность проведения доказательной дифференцированной терапии.
У больных, находящихся в отделениях интенсивной терапии, диагностика недостаточности кровообращения должна базироваться не только на клинических данных, но и подтверждаться инструментальными методами и функциональными пробами. Это позволяет диагностировать действительно имеющиеся признаки недостаточности кровообращения и избежать ее гипердиагностики за счет гиперволемии, легочной патологии и др. Кроме того, такой принцип дает возможность выявить слабое звено (правый или левый желудочек, систолическая или диастолическая дисфункция, нарушения сосудистого сопротивления большого или малого круга кровообращения), дать количественную оценку и индивидуализировать терапию. Выбор тех или иных методик (инвазивное или неинвазивное исследование), с помощью которых данный принцип может быть реализован, диктуется клинической ситуацией и оснащенностью отделения.
Наиболее оправданным общим подходом к диагностике и коррекции системных гемодинамических нарушений у больных в критических состояниях является аналитический. В рамках такого подхода выделяется минимальное число критериев, количественный анализ которых позволяет достоверно оценить главные параметры, характеризующие основные аспекты кровообращения: пред нагрузку, производительность сердца и постнагрузку. Наилучшим маркером преднагрузки является ДЗЛК. Сократительная активность
миокарда характеризуется прежде всего ударным индексом, а не МОК, поскольку последний может быть высоким за счет компенсаторной тахикардии. Главными критериями постнагрузки на левый и правый желудочки являются соответственно ИОПСС и ИСЛС. Следует помнить, что низкий УИ может наблюдаться не только при первичной систолической дисфункции желудочков, но и при снижении венозного подпора, т. е. адекватная оценка сердечного выброса возможна лишь после нормализации ДЗЛК.
Данный подход позволяет построить индивидуальный гемодинамический профиль больного (ДЗЛК/УИ/ИОПСС), диагностировать ведущие нарушения гемодинамики и провести дифференцированную терапию выявленного варианта недостаточности кровообращения. Такая возможность обусловлена тем, что каждый из перечисленных параметров играет определяющую роль в генезе различных типов шока: ДЗЛК — гиповолемического, УИ — кардиогенного, ИОПСС — острой сосудистой недостаточности.
Так, гемодинамический профиль, свойственный гиповолемическим типам циркуляторных нарушений, обусловлен первичным уменьшением преднагрузки, т. е. снижением ДЗЛК. Вторичными изменениями являются снижение УИ и компенсаторная (в целях сохранения системного АД) вазоконстрикция, т. е. возрастание ИОПСС. Таким образом, характерный для гиповолемического шока профиль имеет следующий вид: низкое ДЗЛК/низкий УИ/высокий ИОПСС. Соответственно стартовая терапия гиповолемического шока представляет собой индивидуально подобранную по объему, качественному составу и темпу инфузионную программу, главной целью которой является повышение ДЗЛК до максимально возможного уровня, безопасного в отношении отека легких. Таким уровнем считают 18 — 20 мм рт. ст. Следует помнить, что данный уровень ДЗЛК безопасен при условии нормального коллоидно-осмотического давления плазмы. Гипопротеинемия, являющаяся скорее правилом, чем исключением при гиповолемии, снижает порог возникновения гидроста
Принципы диагностики и интенсивной терапии некоторых нарушений 69
тического отека легких. Поэтому на первом этапе лечения целесообразно придерживаться минимального целевого уровня ДЗЛК, который составляет 12 — 14 ммрт. ст. Хотя ДЗЛК и ЦВД не являются взаимозаменяемыми параметрами, поскольку характеризуют преднагрузку на разные желудочки, в ряде клинических ситуаций при отсутствии дисфункции правых отделов сердца показатель ЦВД может быть использован вместо ДЗЛК. Данный этап лечения может включать назначение кортикостероидов.
В ряде случаев оптимизация ДЗЛК является достаточной для коррекции гиповолемических циркуляторных нарушений. В ситуации, когда У И при адекватном ДЗЛК продолжает оставаться низким, необходима инотропная поддержка. Число препаратов для ее осуществления невелико и ограничивается в основном прессорными аминами (дофамин, добутамин, норадреналин, адреналин) и ингибиторами фосфодиэстеразы с кардиотоническим действием (амринон). Выбор препарата для инотропной поддержки обусловлен величиной ИОПСС.
При высоком периферическом сопротивлении предпочтение отдается -агонисту — добутамину, оказывающему выраженное кардиотоническое действие без вазоконстрикторного эффекта, либо сочетанию добутамина с амриноном (последний является препаратом смешанного действия — кардиотонического и вазодилатирующего). При нормальном ИОПСС и низком УИ может использовать дофамин в кардиотонических дозировках. Если при этом выражена артериальная гипотензия, то дозы дофамина должны обеспечивать вазоконстрикторный эффект. Низкий ИОПСС, сопровождающийся артериальной гипотензией, является основанием для использования а-, р-агонистов, иногда — совместно с дофамином, позволяющим сохранить адекватный ренальный и мезентериальный кровоток.
После стабилизации гемодинамических параметров наступает этап контролируемой функции кровообращения, осуществляемой при помощи инфузионной терапии и инотропной поддержки. Заключи
тельный этап коррекции гемодинамических нарушений — отмена инотропных препаратов — должен проводиться после улучшения клинического состояния, восстановления тканевой перфузии, с использованием данных инструментального исследования, с обязательным соблюдением принципа постепенности.
Подробный анализ гиповолемического профиля гемодинамики приведен нами для иллюстрации стандартного синдромного подхода, который в общем виде применим к любым типам гемодинамических расстройств.
Для кардиогенного шока, сопровождающегося первичным поражением сократительной функции миокарда, последующим застоем в малом круге кровообращения и периферической вазоконстрикцией, профиль будет иметь такой вид: высокое ДЗЛК/низкий УИ/высокий ИОПСС. Соответственно основными направлениями коррекции гемодинамики являются: ограничение инфузионной терапии и форсирование диуреза, применение кардиотонических препаратов (добутамин, амринон), назначение периферических вазодилататоров (нитроглицерин, нитропруссид натрия). Достижение нормальных уровней всех параметров профиля будет свидетельствовать об адекватности проводимой коррекции.
Ведущим гемодинамическим нарушением в патогенезе острой сосудистой недостаточности (коллапс, вазогенный шок) является снижение ИОПСС. Другие компоненты профиля могут варьировать в зависимости от этиологии состояния (сепсис, анафилаксия, надпочечниковая недостаточность ит. д.). Однако наиболее часто гемодинамический профиль имеет следующую структуру: низкое ДЗЛК/высокий УИ/низкий ИОПСС. Следовательно, в данном случае коррекция гемодинамики должна включать назначение а-агонистов или дофамина в прессорных дозах и инфузионную терапию.
Рассмотренные ситуации касались в основном нарушений сократительной функции левого желудочка, пред нагрузки на левый желудочек и сосудистого тонуса в большом круге кровообращения. Все ска
70 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
занное в полной мере относится и к правым отделам сердца, если вместо ДЗЛК использовать ЦВД, а вместо ИОПСС — ИСЛС.
Клиническая реальность, разумеется, богаче, нежели приведенные в качестве примеров типичные гемодинамические профили. Нарушения отдельных звеньев кровообращения могут не укладываться в «классический» патогенез и в этом случае нарушается характер профиля. Тем не менее структурный подход к анализу гемодинамических расстройств является универсальным и может быть использован как принципиальная основа коррекции любых вариантов нарушений кровообращения.
Следует отметить, что в большинстве случаев у больных в критических состояниях (при отсутствии первичного поражения циркуляции) увеличиваются сердечный выброс, частота сердечных сокращений, минутный объем кровообращения. Биологическая целесообразность гиперфункции гемодинамики состоит в обеспечении возросших метаболических потребностей организма, и прогноз заболевания в значительной мере зависит от функциональных резервов сердечно-сосудистой системы. Терапевтический подход к гиперфункции гемодинамики в таких ситуациях ограничивается, как правило, обеспечением наиболее благоприятных условий для выполнения усиленной работы сердца: контроль и коррекция (при необходимости) пред- и постнагрузки, кардиоцитопротекция (глюкозо-инсулин-калий-маг-ниевые смеси, неотон, милдронат и др.), оксигенотерапия, тщательное соблюдение базисных направлений интенсивной терапии, способствующих коррекции или поддержанию основных констант гомеостаза (газовый состав крови, водно-электролитный баланс, кислотно-основное состояние и др.).
2.3.2. Диагностика и интенсивная терапия гипертонических кризов
Термином «гипертензивный криз» принято характеризовать значительное, нередко внезапное повышение АД, которое мо
жет сопровождаться развитием тяжелых сосудистых осложнений с поражением жизненно важных органов и требует ур-гентной помощи.
В настоящее время отсутствует однозначная интерпретация уровня АД, позволяющего констатировать гипертонический криз. Наиболее часто в качестве диагностических критериев предлагаются следующие:
повышение диастолического АД до 120—140 мм рт. ст.;
повышение систолического АД на 20 — 100 мм рт. ст., диастолического АД — на 10 — 50 мм рт. ст.;
повышение систолического и/или диастолического АД на 30 % от исходных величин.
Кроме абсолютных значений АД, развитие, прогноз и тактика лечения гипертонических кризов определяются скоростью повышения АД и наличием связанных с артериальной гипертензией сосудистых нарушений и органных поражений.
Общепринятой классификации гипертонических кризов не существует. До недавнего времени в отечественной литературе широко использовалось разделение кризов в зависимости от клинического течения (первый тип — «симпатоадреналовый»; второй — «водно-солевой») и от состояния системной гемодинамики (ги-пер-, гипо- и эукинетический).
Для кризов первого типа, часто отождествлявшихся с гиперкинетическими, свойственно повышение АД за счет возрастания сердечного выброса без значимых изменений ОПСС. Клинически данный тип кризов характеризуется быстрым началом (с развитием головной боли, головокружением, тошнотой, гиперемией лица), преимущественным повышением систолического АД, кратковременным характером (до нескольких часов), относительно редким развитием осложнений.
При кризах второго типа (гипо- и эукинетических) повышение АД обусловлено в основном увеличением ОПСС без значимых изменений сердечного выброса. Характерными клиническими признаками гипертонических кризов этого типа являются: постепенное начало с симптомати
Принципы диагностики и интенсивной терапии некоторых нарушений 71
кой гипертонической энцефалопатии, преимущественное повышение диастолического АД, продолжительный характер, частое развитие осложнений.
В настоящее время широкое распространение получила удобная для практических целей классификация гипертонических кризов, предложенная в 1999 г. рабочей группой Украинского научного общества кардиологов, которая основывается на разделении гипертонических кризов в зависимости от срочности оказания помощи, что определяется наличием нарушений функции органов-мишеней. В соответствии с этой классификацией выделяют:
1. Осложненные гипертонические кризы, характеризующиеся наличием острого или прогрессирующего поражения жизненно важных органов и требующие снижения АД в сроки до одного часа.
К ним относят острую гипертензивную энцефалопатию, внутричерепные геморрагии, ишемический инсульт, острый коронарный синдром, острую расслаивающую аневризму аорты, острую левожелудочковую недостаточность (отек легких), угрожающие жизни аритмии, гипертонические кризы после аортокоронарного шунтирования.
2. Неосложненные гипертонические кризы, при которых отсутствует поражение жизненно важных органов. В этом случае требуется снижение АД в сроки от нескольких часов до 24.
Факторами, предрасполагающими к развитию гипертонических кризов, являются избыточный прием поваренной соли и жидкости, психоэмоциональный стресс и физические нагрузки, метеорологические изменения, неадекватная гипотензивная терапия, интеркуррентные заболевания, травмы, оперативные вмешательства. Эти факторы способствуют активации различных нейроэндокринных, паракринных и аутокринных систем, прежде всего симпатоадреналовой и ренин-ангиотензиновой. В результате развивается быстрое и значительное повышение АД, приводящее к развитию тяжелой перегрузки стенок артериол давлением и механическому повреждению эндотелия артериол. Вследствие структурных и функциональных из
менений стенки артериол и исходно измененной ауторегуляции сосудистого тонуса эти нарушения более выражены у больных, страдающих хронической артериальной гипертензией, чем у исходно нор-мотензивных пациентов. Нарастание этих процессов способствует прогрессированию дисрегуляции, развитию локальной вазодилатации, гиперперфузии органов-мише-нией с их повреждением.
Для дифференциальной диагностики гипертонических кризов в соответствии с приведенной классификацией, а также для определения тактики лечения необходимо установить или исключить наличие острого повреждения жизненно важных органов. Необходимый минимум данных могут предоставить: ЭКГ, офтальмоскопия, целенаправленное неврологическое обследование, определение уровня азотемии, гематокрита, общий анализ мочи, исследование системной гемодинамики.
Общепринятый в настоящее время подход к лечению неосложненных гипертонических кризов состоит в постепенном (в течение 12 — 24 часов) снижении АД и преимущественном использовании оральных антигипертензивных препаратов. Во время лечения необходим тщательный контроль АД.
В результате проведения ряда широкомасштабных рандомизированных клинических исследований изменился подход к фармакотерапии гипертонических кризов. Представлены доказательства того, что применение короткодействующего нифедипина для купирования гипертонических кризов сопряжено с повышенным кардиоваскулярным риском и поэтому должно считаться недопустимым. Наиболее приемлемым оральным препаратом для лечения неосложненных гипертонических кризов является клонидин (клофелин), эффективность и безопасность которого обоснованы в нескольких клинических испытаниях. Существует несколько схем назначения перорального (сублингвального) клофе-лина при гипертоническом кризе (пример высокодозного подхода: 0,075 — 0,15 мг с последующим приемом по 0,075 мг каждые 20 мин до достижения антигипертензивного эффекта или до суммарной дозы
72 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ КРОВООБРАЩЕНИЯ
0,6 —0,8 мг). Для купирования криза возможно парентеральное применение кло-фелина, хотя в настоящее время такой подход не рассматривается как стандартный.
В лечении неосложненных кризов ряд авторов отдает предпочтение лабеталолу (некардиоселективный p-адренолитик с а-адреноблокирующими свойствами), ура-пидилу (периферический блокатор оц-ад-ренорецепторов и агонист центральных рецепторов S-гидрокситриптамина), ингибиторам ангиотензинпревращающего фермента (АПФ) (прежде всего, каптопри-лу).
Общепринятым подходом к лечению больных с осложненным гипертоническим кризом является немедленное начало снижения АД для предотвращения поражения жизненно важных органов. Существенным аспектом лечения является то, что его первичной целью считается не нормализация АД, а уменьшение его значений до таких пределов, при которых могут быть восстановлены ауторегуляторные механизмы. В этой связи в большинстве случаев при осложненных гипертонических кризах целевые уровни АД составляют значения, лишь на 20 — 25 % меньшие, чем исходные, а в некоторых ситуациях снижение АД в течение первых часов должно быть еще более осторожным.
Такой подход является однозначным при лечении гипертонических кризов с неврологическими осложнениями (острая гипертензивная энцефалопатия, внутримозговые и субарахноидальные кровоизлияния, инфаркт мозга). Процесс антигипертензивной терапии в таких ситуациях должен сопровождаться ежечасной оценкой неврологического статуса, и при ухудшении последнего темп снижения АД необходимо снизить или вообще прекратить введение антигипертензивных препаратов. Например, у пациентов с геморрагическими инсультами большинство авторов не рекомендуют снижение систолического АД ниже 180 — 200 мм рт. ст. и диастолического — ниже 110 — 120 мм рт. ст., причем темп снижения АД не должен превышать
5 — 20 % от исходного уровня в течение нескольких часов. Такая тактика мотивирована возрастанием риска ишемии ранее интактных отделов мозга при быстром снижении АД.
Аналогичная тактика (постепенное снижение АД) является определяющей и при купировании кризов, осложненных острым коронарным синдромом, острой левожелудочковой недостаточностью (риск усугубления коронарной недостаточности при резком снижении АД) и другими поражениями органов-мишеней.
Исключение составляют гипертонические кризы, осложненные расслаивающей аневризмой аорты. Общепринятым вариантом интенсивной терапии в этих случаях является активное и быстрое снижение АД с целью уменьшения напряжения стенок аорты и предупреждения ее дальнейшего расслоения и разрыва.
Исходя из необходимости медленного и тщательно контролируемого снижения АД, стандартным подходом к лечению при купировании большинства осложненных гипертонических кризов является внутривенная инфузия гипотензивных препаратов. Данный способ введения позволяет адекватно регулировать антигипертензивный эффект и избежать опасностей, связанных с быстрым неконтролируемым снижением АД. Перевод пациента на оральные антигипертензивные препараты осуществляется после стабилизации АД на целевом уровне.
Число препаратов, применяемых для лечения осложненных гипертонических кризов, довольно велико (нитраты, блокаторы кальциевых каналов, ингибиторы АПФ для внутривенного введения, блокаторы адренергических рецепторов различных классов и др.). Группы препаратов отличаются по механизму действия, скорости и выраженности антигипертензивного эффекта, а также по ассоциированным эффектам в различных популяциях гипертензивных пациентов. В пределах одного и того же класса препараты часто имеют значительные отличия. Выбор того или иного препарата всегда диктуется клинической ситуацией.
Список использованной литературы 73
Список использованной литературы
1. Дядык А. И., Багрий А. Э. Артериальные гипертензии в современной клинической практике. — Донецк: КП «Регион», 2002. — С. 192-233.
2. Зильбер А. П. Клиническая физиология в анестезиологии и реаниматологии. — М.: Медицина, 1984.
3. Иванов Г. Г. Анализ системной гемодинамики и функционального состояния миокарда у больных в критических состояниях // Анестезиология и реаниматология. — 1996. — № 5. - С. 10-13.
4. Марино П. Интенсивная терапия: Пер. с англ. / Под ред. А. И. Мартынова. — М.: ГЭОТАР МЕДИЦИНА, 1998. - С. 17-36, 93-140, 156-169, 232 - 248.
5. Мешалкин Е. Н., Литасова Е. Е., Окунева Г. И., Власов Ю. А, Генеральные нарушения кровообращения и сопряженных процессов у человека // Бюл. Сиб. отд-ния АМН СССР. - 1982. - №2. - С. 17-43.
6. Неговский В. А., Гурвич А. М., Золото-крылина Е. С. Постреанимационная болезнь. — М.: Медицина, 1987. — С. 28 — 43, 368-373.
7. Патологическая физиология: Учеб. / Под ред. А. Д. Адо, М. А. Адо, В. И. Пыцкого и др. — М.: Триада-Х, 2000. — С. 514 — 536.
8. Физиология человека: Учеб. / Под ред. В. М. Поровского, Г. Ф. Коротько. — М.: Медицина, 1998. — 368 с.
Раздел
Клиническая физиология дыхания
Согласно определению Г. Тевса, дыхание — это обмен газами между клетками организма и окружающей средой [1]. При этом кислород попадает в клетку, а углекислый газ выводится из нее. Перенос кислорода из окружающей среды в ткани происходит в несколько этапов:
1. Обмен между воздухом и альвеолярным газом обеспечивается вентиляцией легких.
2. Обмен между альвеолярным газом и кровью легочных капилляров — газообмен в легких.
3. Транспорт кислорода кровью к тканям организма.
4. Потребление кислорода клетками, или тканевое дыхание.
Выведение СО2 происходит в обратном направлении: образование в процессе аэробного гликолиза -> транспорт кровью -> выделение из крови в альвеолы -» -> выведение во внешнюю среду.
Внешнее дыхание включает стадию вентиляции легких и газообмена в них. Таким образом, основной функцией системы внешнего дыхания следует считать оксигенацию крови и элиминацию из нее углекислого газа. Под вентиляцией легких понимают циклический процесс вдоха и выдоха, при котором газ из окружающей среды поступает в альвеолы, а альвеолярный газ — в окружающую среду. Это первый этап доставки кислорода тканям.
3.1. ЦЕНТРАЛЬНАЯ РЕГУЛЯЦИЯ ДЫХАНИЯ
Как известно, процесс дыхания осуществляется при помощи сокращения дыхательных мышц, у которых, в отличие от сердечной мышцы, отсутствует собственный автоматизм.
Сокращениями дыхательной мускулатуры управляет центральный регуляторный дыхательный механизм. В 1885 г. М. Ми-славский открыл дыхательный центр, нейроны которого отвечают за ритмогенез дыхания.
В этом процессе принимают участие три группы нейронов. В латеральном отделе ядра солитарного тракта на дорзальной поверхности продолговатого мозга находятся
нейроны, проявляющие активность главным образом на вдохе, — это инспираторный центр. В вентролатеральной части продолговатого мозга вблизи двойного ядра расположены нейроны выдоха — экспираторный центр. Доказательством того, что импульсы к дыхательным мышцам вдоха и выдоха генерируются именно в этих структурах, служат результаты экспериментов с перерезкой спинного мозга ниже продолговатого на уровне Cj — Сц, вследствие чего спонтанное дыхание у животных полностью прекращалось. Эти выводы подтверждаются клиническими наблюдениями у больных с травмой верхних отделов спинного мозга.
Центральная регуляция дыхания 75
Однако оказалось, что при перерезке ствола мозга на уровне между продолговатым мозгом и варолиевым мостом спонтанное дыхание было возможным, но нарушался ритм дыхания. Marckwald в 1887 г. обнаружил третью группу нейронов, отвечающих за ритмогенез дыхания, — центр пневмотаксиса, функция которого заключается в координации инспираторного и экспираторного центров, т. е. нейроны, находящиеся в ядре Келликера-Фузе в клювовидно-латеральной части варолиевого моста, способствуют цикличности вдоха и выдоха [2].
Перечисленные структуры входят в состав бульбопонтиального дыхательного центра (ДЦ), нейроны которого и обеспечивают ритмогенез дыхания. Потребность организма в О2 и выведении СО2 изменяется в зависимости от условий внутренней и внешней среды. Главной целью регуляции дыхания является приведение в соответствие функций внешнего дыхания метаболическим потребностям организма в конкретный момент времени. Поэтому для адекватной регуляции интенсивности дыхания ДЦ должен постоянно получать информацию о газовом составе крови. Эту функцию выполняют центральные и периферические хеморецепторы.
Центральные {медуллярные) хеморецепторы (ЦХ) расположены непосредственно в продолговатом мозгу (на передне-боковой поверхности около выходов подъязычного и блуждающего нервов) и анализируют химический состав межклеточной мозговой жидкости (ММЖ) (представления о приоритете состава спинномозговой жидкости оказались не совсем точными, хотя при стабильном состоянии химический состав их незначительно отличается). Прежде всего активность ЦХ повышается с увеличением напряжения СО2 и снижением pH ММЖ, вследствие чего повышается активность инспираторных нейронов и подавляется — экспираторных, увеличивается минутная вентиляция легких. Напротив, снижение РДСО2 и повышение pH ММЖ снижает активность ДЦ вплоть до апноэ.
На изменения рСО2 и рО2 артериальной крови реагируют периферические
(ПХ), или артериальные, хеморецепторы. Расположены они в области дуги аорты и в месте деления общей сонной артерии на внутреннюю и наружную (их локализация примерно соответствует локализации барорецепторов кровяного давления). ПХ содержатся в особых тельцах — клубочках, или гломусах, находящихся на поверхности кровеносного сосуда. Они являются высокочувствительными газоанализаторами крови и реагируют даже на небольшие изменения газового состава артериальной крови. Однако для нормального функционирования ПХ необходимо интенсивное кровоснабжение.
Периферические хеморецепторы играют важную роль в адекватном ответе ДЦ на изменение потребностей в О2 и выведении СО2. Они рефлекторно увеличивают легочную вентиляцию в ответ на гипоксемию и гиперкапнию. Даже при нормальном газовом составе артериальной крови они посылают импульсы в ДЦ, им-пульсация прекращается только при дыхании чистым кислородом. ПХ ответственны за быстрый, начальный ответ на повышение напряжения СО2 в артериальной крови. Стимуляция ДЦ происходит при гипокапнических уровнях углекислоты (20 — 30 мм рт. ст.) [3]. В каротидных тельцах клетки первого типа реагируют на гипоксемию двояко [4]: во-первых, ингибируются О2-чувствительные калиевые каналы, что стимулирует вход ионов Са2+ в клетку и образование нейротрансмиттеров, активирующих ДЦ; во-вторых, гипоксемия индуцирует экспрессию тирозин-гидролазы. Этот энзим ответственен за продукцию нейротрансмиттеров.
Центральные и периферические хеморецепторы дополняют друг друга. Особую важность это взаимодействие приобретает в условиях недостаточности дыхания с гиперкапнией, когда ЦХ как бы адаптируются к повышению напряжения углекислоты в интерстициальной жидкости мозга, и основной тонизирующий эффект в ответ на гипоксемию оказывают импульсы из ПХ. В покое основную роль в обеспечении паттерна дыхания играют центральные хеморецепторы, и только 15 % обеспечивается периферическими [5].
76 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
Кора головного мозга И
Афферентация --------- Дыхательный центр -------
t Генератор респираторного
паттерна
Мотонейроны дыхательных мышц
Механо- **__________________________________Сокращения дыхательных
рецепторы мышц
t_________________________________________________ I
----------------------------------------- Вентиляция
Периферические I
и центральные ’
хеморецепторы ----------------------------------- Изменения газов крови
Рис. 3.1. Схема обратной связи центральной регуляции дыхания [5]
Однако данных, получаемых ДЦ от хеморецепторов, недостаточно для эффективной регуляции дыхания. При нормальном дыхании колебания газов артериальной крови незначительны, а человек продолжает дышать. Это происходит благодаря тому, что во ДЦ поступает информация от рецепторов растяжения легких (рефлекс Геринга —Брейера), вследствие чего тормозится глубокий вдох и активируется выдох, и наоборот, раздражение ирритант-ных рецепторов, расположенных в эпителиальном и субэпителиальном слоях дыхательных путей, сокращает выдох, вследствие чего дыхание становится частым и поверхностным. Импульсы от проприоре-цепторов растяжения дыхательных мышц (мышечные веретена и сухожильные ре
цепторы Гольджи) поступают к дыхательным нейронам спинного мозга и корригируют интенсивность и длительность их возбуждения.
У человека регуляторное воздействие на внешнее дыхание оказывает также кора головного мозга.
Получая разностороннюю информацию о состоянии внешнего дыхания и его соответствии потребностям организма, ДЦ посылает импульсы через проводящие пути спинного мозга к мотонейронам дыхательных мышц.
Регуляторная система внешнего дыхания работает по принципу обратной связи, основные принципы которой представлены на рис. 3.1.
3.2. БИОМЕХАНИКА ДЫХАНИЯ
Биомеханика является частью физиологии дыхания, изучающей связи между давлением в разных частях аппарата внешнего дыхания, изменением объема легких и грудной полости, движением воздуха и работой дыхательной мускулатуры. Знание этих процессов необходимо для лучшего понимания патогенеза расстройств дыхания.
Дыхательные движения совершаются в результате сокращения дыхательных мышц. К мышцам вдоха относят диафрагму (основная мышца вдоха) и наружные межреберные, лестничные, грудино-ключично
сосцевидные мышцы. Спокойный нефорсированный вдох обеспечивается за счет сокращений одной лишь диафрагмы. Акт вдоха совершается за счет увеличения объема грудной полости, вызываемого подъемом ребер и опусканием купола диафрагмы. При сокращении купол диафрагмы уплощается и опускается вниз. Сокращение других мышц вдоха, в частности наружных межреберных, увеличивает сечение грудной клетки в передне-заднем направлении. Незначительно увеличивается во время вдоха и поперечный размер грудной клетки. Увеличение объема грудной
Эластическое сопротивление легких 77
клетки вызывает уменьшение давления в плевральной полости на 8 см вод. ст. (784 Па) ниже атмосферного. Таким образом, внутриплевральное давление — это разница между давлением в плевральной полости и атмосферным давлением (по аналогии альвеолярное давление — это разница между давлением в альвеолах и атмосферным давлением). В результате происходит расширение легких с уменьшением давления в альвеолах (до 1 — 2 см вод. ст. (196 Па) ниже атмосферного давления). Разница между внутриплевральным и альвеолярным давлением называется транспульмональным давлением (давление, вызывающее расширение объема легкого). Вследствие разницы давлений между атмосферным газом и альвеолярным давлением происходит движение атмосферного воздуха по дыхательным путям в альвеолы.
Спокойный выдох происходит пассивно за счет эластических свойств легких и грудной стенки. Эти структуры стремятся вернуться к первоначальному объему, что приводит к увеличению давления в альвеолах,
которое становится на 1—2 см вод. ст. (98—196 Па) выше атмосферного (в результате повышается и давление в плевральной полости, которое только на 5 см вод. ст. (490 Па) ниже атмосферного), и газ удаляется из альвеол в окружающую среду. При форсированном выдохе или при увеличенном сопротивлении дыхательных путей выдох становится активным. В его осуществлении участвуют мышцы выдоха, прежде всего мышцы передней брюшной стенки, сокращение которых вызывает увеличение внутрибрюшного давления, пассивный подъем диафрагмы, уменьшение объема грудной клетки и увеличение давления в плевральной полости и в альвеолах.
Таким образом, дыхательная мускулатура способствует движению газа в альвеолы и обратно не прямо, а через изменение давления в плевральной полости и в альвеолах легких. При этом дыхательным мышцам приходится преодолевать эластическое, динамическое и инерционное сопротивление (последнее играет незначительную роль в физиологии дыхания и в дальнейшем рассматриваться не будет).
3.3. ЭЛАСТИЧЕСКОЕ СОПРОТИВЛЕНИЕ ЛЕГКИХ
Эластичность — это мера упругости, в частности легочной паренхимы и грудной стенки. Паренхима легких состоит из эластических коллагеновых волокон, соединяющих бронхиально-сосудистую сеть легких с висцеральной плеврой. Одной из составляющих эластического сопротивления дыханию является сопротивление стромы легких. Но таким же сопротивлением обладает и грудная клетка. Для того чтобы увеличить ее объем на вдохе, также расходуется энергия дыхательных мышц. Для определения состояния эластичности легких и грудной стенки было введено понятие «растяжимость» (compliance). В норме растяжимость (С) обратно пропорциональна эластическому сопротивлению (Рэл):
С другой стороны, растяжимость равна увеличению объема (AV) на единицу уве
личения давления в дыхательных путях (АР) (при условии тотальной релаксации мышц):
г_ AV АР ‘
При этом было определено, что растяжимость легких и грудной стенки одинакова и равна 0,2 л/см вод. ст. (2 л/кПа). Таким образом, общая растяжимость дыхательной системы в два раза меньше и равна 0,1 л/см вод. ст. (1 л/кПа).
При изучении зависимости между увеличением транспульмонального давления и объемом легких оказалась, что она не является прямо пропорциональной, а имеет форму синусоиды (так называемая петля гистерезиса) (рис. 3.2).
При инфляции изотонического раствора NaCl этот эффект нивелируется, а растяжимость значительно увеличивается, кроме того, практически исчезает разница между растяжимостью на вдохе и выдохе [6].
78 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
Давление, см вод. ст.
Рис. 3.2. Эффект силы поверхностного натяжения на растяжимость легких:
Деф — дефляция; Инф — инфляция
Рис. 3.3. Поверхностное натяжение (Т) и давление (Р) пузырька [2]
Рис. 3.4. Закон Лапласа [2]:
Т — поверхностное натяжение; и Р2 — давление соответственно меньшего и большего пузырьков; rt и г2 — радиус кривизны соответственно меньшего и большего пузырьков
Исходя из эластических свойств легочной паренхимы, зависимость должна была бы иметь другой вид. Еще в 1929 г. голландский физиолог К. von Neegard [7] высказал гипотезу, согласно которой часть сопротивления легких, оказываемого дыхательным мышцам на вдохе, зависит от естественной силы поверхностного натяжения молекул жидкости на границе газ — жидкость. Однако в этом случае транспульмональное давление должно было быть значительно выше, поэтому от этой гипотезы отказались. И только спустя 25 лет Р. Е. Pattie и J. A. Clements [8, 9] независимо друг от друга описали вещество, снижающее силу поверхностного натяжения. Наличие такого вещества в альвеолярной жидкости объясняло подобную зависимость между давлением и объемом.
Сила поверхностного натяжения возникает на границе газ — жидкость вследствие большего притяжения между молекулами жидкости по сравнению с притяжением между молекулами внутри жидкости и газа над ней (рис. 3.3 и 3.4).
При одинаковой величине поверхностного натяжения давление в меньшем пузырьке больше, чем в более крупном, поэтому газ будет перемещаться из мелкого пузырька в более крупный [2]. Исходя из закона Лапласа, нерасправленные альвеолы менее стабильны, чем расправленные, происходит их ателектазирование, вследствие чего они не могут принимать участие в дыхании.
Для понимания биомеханики легочного дыхания важным является понятие «сурфактант». Поскольку в специальных руководствах проблеме легочного сурфактанта (ЛС) уделяется сравнительно небольшое внимание, остановимся на этом вопросе подробнее.
Сурфактант — это комплекс липидов и протеинов. Анализ бронхоальвеолярной лаважной жидкости показал, что Л С состоит из 80 % фосфолипидов, 8 % других липидов (холестерол, триацил глицерол, свободные жирные кислоты) и 12 % протеинов. В свою очередь, в состав фосфолипидов Л С входит: 85 % фосфа-тидилхолина, 40 — 55 % которого приходится на дипальмитоил-фосфатидилхолин,
Эластическое сопротивление легких 79
9—12% — на пальмитоилмиристоил-фос-фатидилхолин, 10 % — на пальмитоило-леоил-фосфатидилхолин и 6 % — на паль-митоиллинолеоил-фосфатидилхолин; 9 % фосфатидилглицерола; 3 % фосфатидил-этаноламина и 2 % фосфатидилинозитола [10, И]. Половину белков сурфактанта составляют сурфактант-ассоциированные протеины SP-A (28 — 36 kD), SP-B (8 kD), SP-C (4 kD) и недавно открытый SP-D (43 kD) [12].
Функции Л С представлены в табл. 3.1.
ЛС играет исключительно важную роль в поддержании стабильности дыхательной поверхности, поскольку снижает величину поверхностного натяжения молекул жидкости на границе газ —жидкость в альвеолах, что способствует стабилизации легочного объема, снижению транспульмонального давления и уменьшению работы дыхания.
ЛС способствует стабилизации альвеол разного диаметра, уменьшает риск ателек-тазирования и шунтирования [14].
Компонентом ЛС, наиболее снижающим величину поверхностного натяжения, является дипалметиловый фосфатидилхолин (ДПФХ) [15] — молекула с полярными гидрофильными концевыми группами и неполярной гидрофобной «талией». Таким образом, молекула ДПФХ абсорбируется
на поверхности газ —жидкость, гидрофобной (жировой) частью ориентируясь на газовую фазу, а гидрофильной концевой частью — на жидкую. Гидрофобные апопротеины SP-В и SP-С являются важным фактором повышения абсорбционной способности и стабильности ЛС [16]. SP-А выполняет различные функции, в частности регулирует синтез и секрецию сурфактанта альвеолоцитами типа II [17].
ЛС улучшает мукоцилиарный транспорт, увеличивает клиренс из малых воздухоносных путей, частоту сокращений ресничек и снижает вязкость секрета. Это сопровождается стимулирующим эффектом транспорта ионов хлора через эпителий нижних дыхательных путей.
При повреждении легких наблюдаются продукция кислородных радикалов фагоцитами и снижение функции Л С, который в норме обладает антиоксидантной активностью, поглощая кислородные радикалы, и выполняет протекторную функцию при гипероксическом повреждении легких [18].
Фосфолипиды ЛС формируют очень стабильный липидный слой, который может служить неспецифическим барьером для адгезии и инвазии микроорганизмов в легкие. ЛС обладает бактерицидным и
Таблица 3.1. Классическая и «несурфактантная» функции легочного сурфактанта [13]
Функции
Классическая « Несурфактантная »
Легочная механика Снижение величины поверхностного натяжения газ —жидкость в альвеолах Сохранение легочного объема в условиях низкого транспульмонального давления Предупреждение коллапса и ателектаза альвеол Предупреждение отека легких Участие в газообмене: поддержание дыхательной поверхности легких Неспецифический иммунитет легких Барьер против патогенов Активация мукоцилиарного транспорта Антиоксидантная активность Антибактериальная/антивирусная активность Специфическая иммунологическая: SP-A-и SP-D-опсоническая активность, модуляция хемотаксиса и фагоцитоза SP-A-интеракция с альвеолярными макрофагами посредством специфических рецепторов (Cfq-рецепторы)
80 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
Транспульмональное давление, см вод. ст.
Рис. 3.5. Статистические кривые давление/объем в норме, у больных с фиброзом и эмфиземой легких [6]
антивирусным свойствами, однако не ко всем бактериям (например, Haemophilus influenzae) [19]. В связи с антивирусными свойствами Л С изучается роль апопротеинов, особенно SP-А и SP-D. В частности, SP-А обладает активностью против вирусов гриппа А и простого герпеса.
Важную роль играет сурфактант и при рождении ребенка, препятствуя повторно
му коллапсу альвеол. Наиболее типичной патологией, доказывающей важность Л С для функции дыхания, является респираторный дистресс-синдром новорожденных, при котором в результате резкого увеличения работы дыхания дети не могут самостоятельно дышать. Функция ЛС нарушается также при отеке легких, тяжелой пневмонии и т. д. Нарушение биомеханики дыхания, связанное с повышением эластического сопротивления, может наблюдаться и вследствие поражения легочной паренхимы. На рис. 3.5 показано соотношение между давлением и объемом легких у больных с пневмофиброзом и эмфиземой легких.
При эмфиземе резко повышается растяжимость легких и пациенты стремятся дышать большими объемами. Наоборот, у больных с пневмофиброзом растяжимость резко снижена, легкие становятся неподатливыми, работа дыхания увеличивается, дыхательный объем резко снижается [6].
Все изложенное выше относится к эластическому сопротивлению дыхания в статическом положении. Однако при дыхании наиболее важным является динамический компонент сопротивления сокращению дыхательных мышц.
3.4. ДИНАМИЧЕСКОЕ СОПРОТИВЛЕНИЕ ДЫХАНИЮ
При движении газа по дыхательным путям возникает трение между молекулами газа и между ними и поверхностью дыхательных путей.
Как известно из аэродинамики, существуют три типа потока газа по трубам: ламинарный, турбулентный и переходный (рис. 3.6)
Ламинарный поток представлен слоями движущихся молекул газа, параллельных самим себе и стенкам трубы, преобладает при низких скоростях движения. Турбулентный — характеризуется более хаотичным движением молекул газа вдоль трубки и типичен для высоких объемных скоростей движения и сравнительно не
больших по диаметру труб. Переходному типу присущи завихрения, возникающие в месте разделения трубы.
Общие законы движения газа по трубам подчиняются законам Гагена— Пуазей-ля, согласно которым при ламинарном потоке газа
Р-Л-Г4 8г|/ ’
где V — объемная скорость потока; Р — давление, необходимое для преодоления сопротивления движению; г — радиус трубы; г| — вязкость газа; I — длина трубы.
Динамическое сопротивление дыханию 81
Рис. 3.6. Типы потока воздуха по трубам: ламинарный (а), турбулентный (б), переходный (в) (по J. В. West, 1990)
Таким образом, давление, необходимое для преодоления сопротивления движению, обратно пропорционально радиусу трубы в четвертой степени и прямо пропорционально скорости движения, вязкости газа и длине трубы.
При турбулентном потоке давление прямо пропорционально скорости в квадрате, а вместо вязкости газа необходимо учитывать его плотность.
В дыхательных путях на участках дихотомического деления трахеобронхиального дерева наблюдается переходной тип потока [2], однако в большинстве дыхательных путей поток чаще всего бывает турбулентным.
Большая часть динамического сопротивления в норме приходится на бронхи среднего размера, поскольку их общая поверхность меньше, чем мелких бронхов и бронхиол, где преобладают диффузионные процессы переноса газов.
При уменьшении просвета дыхательных путей значительно возрастает динамическое сопротивление дыханию. При этом основным клиническим признаком нарушения проходимости крупных дыхательных путей (гортани, трахеи, крупных бронхов) будет инспираторная одышка, в тяжелых случаях — стридорозное дыхание. К патологическим состояниям, при которых нарушается проходимость крупных дыхательных путей, относятся: эпиглоттит, острый стенозирующий ларинготрахеит, инородные тела в дыхательных путях, ла-
рингоспазм, стенозирование гортани и трахеи в результате новообразований, специфическое (туберкулез) или неспецифическое воспаление (например, ишемическое при длительной интубации трахеи или трахеостомии), подскладочный отек, поражения хрящей трахеи и бронхов с нарушением их каркасности. По-видимому, основной причиной инспираторного характера одышки является активный вдох с большей, чем на выдохе, скоростью газового потока. Для уменьшения динамического сопротивления в этих случаях кроме увеличения просвета дыхательных путей используют седацию пациентов (чаще всего детского возраста) — для уменьшения скорости потока, гелиевую терапию — для уменьшения плотности вдыхаемой газовой смеси. Однако основным методом является профилактика и своевременная диагностика этой патологии. Последнее особенно важно при злокачественных новообразованиях, когда несвоевременная диагностика (таких больных часто лечат от бронхиальной астмы) угрожает неблагоприятным прогнозом. Для диагностики больных с инспираторной одышкой следует использовать эндоскопические методы диагностики (фибротрахеобронхоско-пию).
В случае обтурации мелких дыхательных путей (мелких бронхов, бронхиол) одышка будет носить экспираторный характер. Это объясняется анатомо-физиологическими особенностями мелких дыхательных путей. Чем меньше размер бронха, тем меньше в составе его стенки хрящей, а в бронхиолах они вообще отсутствуют, т. е. у них отсутствует каркасность. На вдохе эластические волокна, образующие строму легкого, натягиваются между висцеральной плеврой, с одной стороны, и бронхиолами — с другой, увеличивая их диаметр. На выдохе, наоборот, положительное транспульмональное давление способствует уменьшению просвета бронхиол, вследствие чего прежде всего затрудняется выдох. Такие процессы имеют место при обтурации просвета бронхиол содержимым дыхательных путей, воспалением и отеком слизистой бронхиол и мелких бронхов, при повышении тонуса
82 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
гладкой мускулатуры бронхов (бронхиальная астма, стенозирующий бронхит).
Как показали результаты исследований, при малых значениях функциональной остаточной емкости (ФОЕ), например, после форсированного выдоха, внутриплев
ральное давление, особенно в нижних отделах легких, может превышать давление в дыхательных путях, что приводит к коллапсу дыхательных путей малого диаметра, в частности респираторных бронхиол (закрытие дыхательных путей) [17].
3.5. РАБОТА ДЫХАНИЯ
При нарушении биомеханики дыхания, повышении эластического или динамического сопротивления увеличивается работа дыхания. В норме дыхательные мышцы потребляют не более 2 % кислорода, однако при патологии его потребление может значительно возрасти. Благодаря адаптации центральной регуляции дыхания больной дышит в режиме, близком к оптимальному для организма. Например, при увеличении эластического сопротивления дыханию целесообразно дышать
поверхностно и часто, и наоборот, при повышении динамического сопротивления -медленно и глубоко.
При хронической дыхательной недостаточности часто развивается гиперкапния, однако это не свидетельствует о полном истощении резервов внешнего дыхания по элиминации углекислого газа, просто для организма целесообразнее поддерживать сравнительно безопасный повышенный уровень рСО2, чем с большой затратой энергии достигать нормокапнии.
3.6. ВЕНТИЛЯЦИЯ И ГАЗООБМЕН В ЛЕГКИХ
Вентиляция легких способствует поддержанию нормального состава альвеолярного газа. Интенсивность вентиляции определяется величиной дыхательного объема (Vt) и частотой дыхания (f). Частота в покое равна 12 —16 в минуту, минутный объем дыхания (VE) — 6—10 л/мин.
Часть воздуха, попадающего в легкие, расходуется на вентиляцию дыхательных путей, так называемого мертвого пространства (рис. 3.7), где газообмен не происходит (проводящая зона).
Различают анатомическое и функциональное мертвое пространство (VD). В состав последнего кроме анатомического пространства входит еще и объем альвеол, которые вентилируются, но не перфузируются, т. е. в которых также не происходит газообмен. У человека среднего возраста объем анатомического мертвого пространства равен 140 —150 мл, или 1/3 дыхательного объема при спокойном дыхании. В альвеолах к концу спокойного выдоха остается 2500 мл газа (это функциональная остаточная емкость (ФОЕ)). Физиологическая роль ФОЕ состоит в
том, что благодаря ей в альвеолярном пространстве сглаживаются колебания кислорода и углекислого газа на вдохе и выдохе, снижение ФОЕ свидетельствует о нестабильности альвеол (см. биомеханику дыхания). При каждом спокойном вдохе обновляется до 1/7 альвеолярного газа.
Постоянство состава альвеолярного газа обеспечивает та часть минутного объема дыхания, которая непосредственно вентилирует альвеолы, — альвеолярная вентиляция в минуту (Уд). Ее рассчитывают по формуле
Va = v£-(vd7).
Если VE = 6 л/мин, VD = 150 мл, a f= = 12, то VA = 4,2 л/мин.
Из формулы следует, что увеличение мертвого пространства приведет к снижению альвеолярной вентиляции при тех же значениях минутного объема и частоты дыхания. VA также снижается при увеличении частоты дыхания, но при постоянном VE или при снижении VE.
Вентиляция и газообмен в легких 83
Рис. 3.7. Схема воздухоносных путей легких человека [3]
Рассмотрим, какие нарушения функции внешнего дыхания будут возникать при снижении альвеолярной вентиляции (гиповентиляции).
Допустим, что VE снизился до 5 л/мин при частоте дыхания 20 в минуту. В этом случае, VT будет равен 250 мл при неизменном VD = 150 мл. Таким образом, в альвеолы будет поступать: за один вдох — 100 мл воздуха, содержащего 20,91 мл кислорода; за один выдох (в выдыхаемом газе содержится 14 % кислорода) будет выделяться 14 мл кислорода, т. е. за вдох
организм получит 6,93 мл кислорода, а за минуту — 6,91 х 20 = 138,2 мл при минимальном потреблении в минуту 240 мл кислорода. В результате альвеолярной гиповентиляции при дыхании воздухом всегда будет ощущаться дефицит кислорода. Оксигенотерапия даже при выраженной гиповентиляции может полностью компенсировать оксигенацию крови: при тех же самых параметрах дыхания за один вдох при дыхании чистым кислородом организм будет получать: 100 - 18 = 82 мл, а за 1 мин — 1640 мл кислорода.
При тех же параметрах вентиляции подсчитаем баланс СО2: 100 мл выдыхаемого газа содержит 5,6 мл СО2, т. е. за минуту будет выделяться 112 мл СО2 при норме 200 мл СО2. Таким образом, при гиповентиляции всегда будет наблюдаться задержка углекислоты в организме с развитием гиперкапнии. Поскольку нельзя существенно изменить продукцию СО2 в организме, без увеличения объема альвеолярной вентиляции невозможно скорри-гировать гиперкапнию. Напряжение СО2 в артериальной крови четко коррелирует с объемом альвеолярной вентиляции.
Как уже указывалось, целью вентиляции легких является поддержание нормального состава альвеолярного газа. Согласно закону Дальтона, смесь газов в закрытой емкости оказывает на ее стенки общее давление, равное сумме парциальных давлений всех газов смеси, а парциальное давление (напряжение) каждого газа в смеси пропорционально его концентрации в смеси. Так, в атмосфере содержится 20,91 % кислорода, при атмосферном давлении 760 мм рт. ст. у?О2 воздуха будет равно » 150 мм рт. ст. (20 кПа). На больших высотах концентрация кислорода остается практически неизменной, однако снижается общее атмосферное давление, соответственно будет снижаться и рО2 во вдыхаемом газе (РЮ2).
Применяя закон Дальтона к дыхательным газам, необходимо помнить, что в альвеолярном газе имеются водяные пары, определяющие рН2О, а поскольку фракции газов приводятся для «сухой» смеси, то из общего атмосферного давления следует исключить парциальное давление во
84 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
дяных паров, которое в альвеолах зависит только от температуры тела и при 36 °C составляет 47 мм рт. ст. Сумма парциальных давлений О2, СО2, N2 и водяных паров равна атмосферному давлению. Концентрация азота как биологически инертного газа не изменяется и составляет » 79 %. Поэтому на остальные компоненты в альвеолярном газе приходится 21 %. Однако в отличие от атмосферного воздуха в альвеолах содержится и углекислый газ, который диффундирует сюда из венозной крови. В норме РЛСО2 равно 40 мм рт. ст. (5,3 кПа). Для определения парциального давления кислорода в альвеолярном газе используют уравнение
~ ^A^O2 Fi‘O2 +
i-FiO2y
R J’
где FiO2 — фракционная концентрация кислорода во вдыхаемой газовой смеси; PiO2 — парциальное давление кислорода во вдыхаемой газовой смеси; R — дыхательный коэффициент (отношение продукции СО2 к потреблению О2 на уровне клетки).
У здорового человека на уровне моря при РЛСО2, равном 40 мм рт. ст., РЛО2 будет составлять 101 мм рт. ст.
Как следует из уравнения, РаО2 зависит от показателя РЛСО2, т. е. от альвеолярной вентиляции.
Приведенные значения характерны для нормальной вентиляции, однако существуют временные и локальные колебания этого показателя, обусловленные сменой фаз дыхательного цикла и неравномерностью вентиляции в легких.
Ниже приведены типы вентиляции легких [1]:
нормовентиляция — нормальная вентиляция, при которой РЛСО2 = 40 мм рт. ст. (5,3 кПа);
гипервентиляция — усиленная вентиляция, превышающая метаболические потребности организма (РЛСО2 снижается пропорционально увеличению вентиляции);
гиповентиляция — пониженная вентиляция относительно метаболических потребностей организма (РЛСО2 понижает
ся пропорционально уменьшению вентиляции);
повышенная вентиляция — любое увеличение альвеолярной вентиляции по сравнению с уровнем в покое, независимо от уровня РЛСО2;
эупноэ — нормальная вентиляция в покое, сопровождающаяся субъективным чувством комфорта;
гиперпноэ — увеличение глубины дыхания независимо от того, повышена ли при этом частота дыхательных движений или нет;
тахипноэ — увеличение частоты дыхания;
брадипноэ — снижение частоты дыхания;
апноэ — остановка дыхания (обусловлена главным образом нарушением регуляции дыхания вследствие гипокапнии);
диспноэ (одышка) — неприятное субъективное чувство недостаточности или затруднения дыхания;
ортопноэ — выраженная одышка, обусловленная перегрузкой малого круга кровообращения, усугубляется в горизонтальном положении;
асфиксия — остановка или резкое угнетение дыхания, сопровождающееся сильными, опасными для жизни нарушениями газообмена.
3.6.1. Обмен газов между альвеолярным газом и кровью
Этот обмен осуществляется посредством диффузии газов через альвеолярно-капиллярную мембрану. Газ диффундирует из участков с более высоким парциальным давлением в зоны с более низким. Если смесь газов контактирует с какой-то жидкостью, то каждый газ диффундирует в нее в зависимости от парциального давления и растворимости. Таким образом, парциальное давление газа в жидкости -это давление, при котором газовая составляющая уравновешивается с жидкостью и зависит от концентрации молекул этого газа в жидкости [17].
Согласно первому закону диффузии Фика, количество газа, проходящее че
Вентиляция и газообмен в легких 85
рез мембрану за единицу времени, т. е. скорость диффузии прямо пропорциональна разнице парциальных давлений газов по обе стороны мембраны и обратно пропорциональна сопротивлению диффузии, которое увеличивается при большей толщине мембраны и уменьшается с увеличением площади поверхности диффузии
t п
где — — скорость диффузии; А — площадь диффузионной поверхности; h — толщина альвеолярно-капиллярной мембраны; ДР — разница парциальных давлений газа по обе стороны мембраны; К — коэффициент диффузии Крога [20].
Площадь диффузионной поверхности в легких большая, в среднем она равняется 80 м2, толщина мембраны — порядка нескольких микрон, поэтому в легких условия для диффузии очень благоприятные. АР для кислорода равна « 60—70 ммрт. ст., а для СО2 — 6 мм рт. ст.
Вследствие меньшей молекулярной массы СО2 скорость диффузии О2, согласно закону Грэхема, выше, чем СО2, однако, учитывая, что газы при диффузии должны растворяться в альвеолярной и интерстициальной жидкости, а коэффициент растворимости выше у СО2, последний диффундирует примерно в 23 раза быстрее, чем кислород.
Нормальная диффузионная емкость для кислорода равна 25 мл/мин на 1 мм рт. ст. и может увеличиваться в три раза при физической нагрузке за счет увеличения количества активных капилляров [17].
Вследствие благоприятных условий для диффузии нарушения в легких являются очень редкой причиной нарушения газообмена в клинике. Резерв поверхности диффузии настолько велик, что даже при обширных резекциях легких (пневмонэктомия), как правило, не наблюдаются нарушения газообмена. Наиболее частой причиной нарушения газообмена вследствие снижения скорости диффузии является отек легких, когда увеличивается
толщина мембраны, т. е. расстояние диффузии, но даже в этом случае гиперкапния возникает только на терминальных стадиях отека. Нарушение диффузионной способности легких наблюдается при изменении биологических свойств альвеолярно-капиллярной мембраны (пневмосклероз, пневмофиброз, синдром гиалиновых мембран), а также, в определенной степени, при тяжелых пневмониях.
Оксигенотерапия при состояниях, вызывающих расстройства диффузии, будет повышать оксигенацию крови вследствие увеличения АР.
3.6.2. Неравномерность вентиляции в легких
Базальные отделы легких вентилируются лучше благодаря соседству с диафрагмой и большему объему экскурсий по сравнению с верхними отделами легких, где верхние ребра почти неподвижны при обычных дыхательных экскурсиях.
Регионарная вентиляция во многом зависит от положения тела, т. е. от гравитационного фактора. В горизонтальном положении на спине вследствие повышения внутрибрюшного давления значительно уменьшается вентиляция базальных отделов легких, снижается ФОЕ и растяжимость легких.
Еще более выражена регионарная неравномерность вентиляции при нарушении биомеханики дыхания, например при эмфиземе, пневмофиброзе, бронхиальной астме, вследствие различия регионарного сопротивления дыхательных путей и регионарной растяжимости. Резкие различия наблюдаются при ателектазе участков легких, коллабировании их.
3.6.3. Особенности кровотока в малом круге
Малый круг является бассейном низкого давления. Так, давление в легочной артерии равно 20/13 мм рт. ст, а среднее давление в легочной артерии намного меньше, чем в аорте, и составляет 13—15 мм рт. ст. Среднее давление в легочных капиллярах — около 7 мм рт. ст. Разница дав ле
86 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
ний между легочной артерией и левым предсердием — 8 мм рт. ст. В результате значительно возрастает влияние на кровоток гравитационных факторов, т. е. положения тела.
В покое кровь протекает только через половину легочных капилляров, при нагрузке количество функционирующих капилляров значительно возрастает, поэтому несмотря на увеличивающуюся перфузию давление в норме повышается незначительно. С увеличением капиллярной поверхности возрастает и газообмен.
Легочный кровоток отличается выраженной регионарной неравномерностью и степень этой неравномерности так же, как и вентиляции, зависит от положения тела. При вертикальном положении тела нижние отделы легких перфузируются значительно лучше верхушек, так как последние располагаются примерно на 30 см выше оснований легких, вследствие чего образуется перепад гидростатического давления на 23 мм рт. ст. Поэтому артериальное давление в верхних отделах легких ниже альвеолярного. При горизонтальном положении тела кровоток в малом круге более равномерен. В положении на спине задние отделы перфузируются лучше передних.
Важным фактором, влияющим на кровоток в легких, является рефлекс гипоксической вазоконстрикции (ГВК), или рефлекс Euler — Lilijestrand [21]. Суть этого рефлекса заключается в том, что в плохо вентилирующихся отделах легких легочная перфузия снижается в результате повышения сопротивления сосудов в плохо перфузируемых участках. Гладкая мускулатура легочных артерий содержит кислородчувствительные К+-каналы, которые и обуславливают гипоксическую вазоконстрикцию, в отличие от артерий большого круга, содержащих АТФ-зависимые К+-каналы, через которые осуществляется массовый выход ионов калия в случае гипоксии и вазодилатации сосуда [17]. Значение рефлекса Euler —Lilijestrand заключается в том, что местный кровоток адаптируется к местной вентиляции. ГВК усиливается: ацидозом, а- и 0-адренергичес-кой блокадой, низкими дозами серотонина, эпидуральной блокадой, ингибиторами
циклооксигеназы (аспирин, индометацин) и NO-синтетаз. Ингибирование ГВК осуществляется : кальцитонин-гензависимым пептидом, натрийуретическим пептидом, оксидом азота, простациклином, а- и 0-адренергической стимуляцией, ингаляционными анестетиками, увеличением внутри-альвеолярного давления, стимуляцией периферических хеморецепторов [22]. Значение этого рефлекса доказано в клинике у больных с ОРДС. При условии его ингибирования дилтиаземом или простагландином PgEj значительно снижалась оксигенация артериальной крови [23]. У больных с острой недостаточностью дыхания при назначении терапии необходимо учитывать ее возможное влияние на ГВК, а значит, и на газообмен в легких.
3.6.4. Вентиляционно-перфузионные соотношения в легких
Для адекватного газообмена в легких необходимо, чтобы вентиляция соответствовала перфузии, т. е. чтобы были адекватными вентиляционно-перфузионные соотношения в легких (V/Q). У здорового человека в покое альвеолярная вентиляция составляет « 4 л/мин, а перфузия -5 л/мин. Нормальное V/Q равно 0,8 — 1,0. Это соотношение не одинаково в разных отделах легких и зависит от положения тела. В вертикальном положении вследствие увеличения перфузионного градиента снизу вверх оно уменьшается. В этом положении показатель V/Q более высокий в верхних отделах, что увеличивает, в частности, риск развития в них туберкулеза, поскольку сравнительно высокое РЛО2 создает благоприятные условия для роста туберкулезных бактерий [17].
Даже в норме часть легочных альвеол вентилируется, но не перфузируется (V/Q стремится к бесконечности) (рис. 3.8, в), они составляют часть функционального мертвого пространства. При некоторых патологических состояниях, например при микроэмболии малого круга, некоторой сосудистой патологии, гиповолемии, объем этого пространства может значительно возрасти. Это не приведет непосредствен-
Вентиляция и газообмен в легких 87
Отсутствие кровотока в зоне А
Нормальная вентиляция в зонах А и В
в
Альвеолярное мертвое пространство
Рис. 3.8. Три модели соотношения вентиляции к перфузии в легких:
а — норма; 6 — шунт; в — альвеолярное мертвое пространство [6]
но к гипоксемии (снижению рО2 в крови), однако значительно увеличит работу дыхания вследствие бесполезной вентиляции неперфузируемых альвеол и декомпенсации системы внешнего дыхания.
Одним из наиболее частых и опасных нарушений соотношения V/Q является перфузия невентилируемых альвеол, или шунтирование крови (Qs/Qt) из правых отделов сердца в левые (V/Q = 0) (рис. 3.8, б). Это означает, что неоксиге-нированная и не отдавшая СО2 кровь поступает в артериальное русло большого круга кровообращения.
Даже в норме до 10 % минутного объема сердца шунтируется — это так называемый физиологический шунт [24]. К основным причинам шунтирования справа налево относятся:
1) экстрапулъмоналъный шунт — внутрикардиальный кровоток по тебезиевым венам, по которым венозная кровь попадает в левые отделы сердца;
2) внутрипульмональный шунт — кроме выше описанного варианта (рис. 3.8, б), это кровоток по бронхиальным венам, из которых кровь попадает в легочные вены.
В результате шунтирования РаО2 ниже Рл О2 на 5 — 10 мм рт. ст., т. е. альвеолярно-артериальная разница по кислороду £>л-дО2 в норме составляет 10 мм рт. ст. При патологии (некоторые пороки сердечно-сосудистой системы с выраженным сбросом крови справа налево, а также вну-трилегочные нарушения) соотношение Qs/Qt может значительно возрасти, что приведет к тяжелой гипоксемии. Наиболее частой причиной тяжелой гипоксемии в отделениях интенсивной терапии является внутрилегочное шунтирование вследствие множественных ателектазов, пневмонии. Шунтовая гипоксемия характерна для острого респираторного дистресс-синдрома. Внутрилегочный шунт может увеличиваться также вследствие наличия опухолей легких, отток крови из которых происходит в легочные вены.
Влияние фракции шунта на оксигенацию артериальной крови и напряжение в ней углекислого газа представлено на рис. 3.9.
РаО2 прогрессивно снижается по мере увеличения шунта, однако РаСО2 остается практически стаби льным, пока, Qs/Qt не достигнет 40 —50 %. Компенсация элиминации СО2 осуществляется за счет гипервентиляции альвеол, в которых происходит газообмен.
88 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
Рис. 3.9. Влияние величины фракции шунта на РдО2 и РдСО2 (по Р. Marino, 1997)
Рис. 3.10. Влияние величины фракции шунта на показатель РаО2/ГЮ2 (по Р. Marino, 1997).
На кривых указаны величины фракции шунта
Зависимость между РДО2 и FzO2 при различных значениях шунтирования в малом круге представлена на рис. 3.10. Как видно из графиков, повышение FiO2 вызывает увеличение PflO2 только при низких значениях шунта. При Qs/Qt = = 30 % это повышение незначительно, а Qs/Qt > 50 % оксигенотерапия практически неэффективна. Н. D. Covelli с соавт. [25] обнаружили такую корреляцию:
PaO2/FiO2
< 200 мм рт. ст.
>200
Qs/Qt
>20%
<20%
Причин малой эффективности оксиге-нотерапии при увеличении шунтирования несколько, например, кислород не попадает в невентилируемые альвеолы.
Таким образом, оксигенация артериальной крови и элиминация углекислого газа осуществляются благодаря газообмену в легких между альвеолярным газом и капиллярной кровью. Нормальный состав альвеолярного газа поддерживается благодаря вентиляции легких. При гиповентиляции альвеол дыхание воздухом приводит к гипоксемии и гиперкапнии. При
чем гипоксемия, вызванная гиповентиляцией, хорошо корригируется оксигенотера-пией, а гиперкапния практически не корригируется без увеличения вентиляции.
Для ненарушенного газообмена в легких необходимы нормальный процесс диффузии газов через альвеолярно-капиллярную мембрану и адекватные вентиляционно-перфузионные соотношения. При нарушении диффузии и снижении V/Q развивается гипоксемия, которая хорошо корригируется оксигенотерапией, но, в отличие от гиповентиляции, она не сопровождается гиперкапнией. При снижении V/Q до 0, т. е. при шунтировании, оксигенотерапия малоэффективна, хотя гиперкапния развивается только при Qs/Qt > > 40 % минутного объема сердца.
Таким образом, определив газы артериальной крови до и после ингаляции кислорода, можно установить механизм нарушения газообмена в легких.
Основным критерием адекватности функции внешнего дыхания является газовый состав артериальной крови (именно в ней газовый состав отражает оксигенацию и элиминацию СО2 в легких). В норме PflO2 > 90 мм рт. ст., РДСО2 = 40 мм рт. ст.
3.7. ТРАНСПОРТ ГАЗОВ КРОВЬЮ
3.7.1. Транспорт кислорода
После диффузии через альвеолярнокапиллярную мембрану кислород растворяется в плазме капиллярной крови. Со
гласно закону Генри, количество растворенного в жидкости газа пропорционально парциальному давлению газа над жидкостью и коэффициенту растворимости Бунзена, который отражает свойство ра
Транспорт газов кровью 89
створимости и зависит от природы растворенного газа, свойств растворителя и температуры. При t = 37 °C и РаО2 ~ = 100 мм рт. ст. (13,3 кПа) в 1 л крови в растворенном состоянии находится 3,1 мл кислорода. Таким образом, при МОС = 5 л/мин кровь переносила бы 15,5 мл/мин кислорода, что явно недостаточно для жизнедеятельности организма.
Большая часть кислорода переносится с помощью гемоглобина. Количество переносимого гемоглобином кислорода, кроме его концентрации, зависит от состояния самого дыхательного пигмента. У взрослого человека наиболее распространенным видом пигмента является НЬА. Однако в результате катаболизма в норме содержание карбоксигемоглобина (НЬСО) составляет 0,25 — 2,1 %, он не способен присоединять кислород и транспортировать его к тканям. В зрелых эритроцитах содержится также до 2 % метгемоглобина (MetHb), который вместо атома Fe2+ содержит Fe3+ и не может отдавать кислород. Повышение уровня метгемоглобина до 40 % вызывает ишемию тканей, а свыше 70 % — летальный исход [26]. В норме НАД • Н и НАДФ • Н в комплексе с гемоглобинре-дуктазой восстанавливают железо до двухвалентного состояния. В случаях отравления метгемоглобинобразователями введение метиленового синего (2 мг/кг в течение 10 мин) способствует превращению MetHb в нормальный гемоглобин [24].
Степень насыщения гемоглобина кислородом (5йО2) выражают концентрацией НЬО2 в процентах по формуле
5«О2 =
[НЬО2]
[НЬ] + [НЬО2]
•100%.
В соответствии с законом действующих масс, насыщение гемоглобина кислородом зависит прежде всего от напряжения кислорода. В норме при РаО2 = 100 мм рт. ст. 98 % гемоглобина насыщено кислородом. Однако зависимость между РаО2 и 5йО2, как установил в 1907 г. N. Bohr, имеет форму синусоиды, получившей название «кривая диссоциации оксигемоглобина (КДО)» (рис. 3.11).
Благодаря тому что область высокого напряжения кислорода соответствует поч-
/>О2, мм рт. ст.
Рис. 3.11. Кривая диссоциации оксигемоглобина
ти горизонтальному сегменту КДО, при снижении РаО2 до 80 мм рт. ст. не возникает существенного снижения SaO2, одновременно наиболее крутой сегмент КДО соответствует снижению РаО2 примерно с 70 до 40 мм рт. ст., что позволяет гемоглобину легко отдавать кислород тканям. Положение кривой на графике, т. е. сродство НЬ к кислороду, зависит от многих факторов. При РаО2 = 100 мм рт. ст. 5йО2 не всегда будет равно 98 %. Для определения положения кривой на графике предложен показатель Р50, который соответствует такому РДО2, при котором 50 % гемоглобина насыщено кислородом. Нормальное сродство НЬ к кислороду будет соответствовать Р50 — 25,0 — 27,5 мм рт. ст. При снижении этого показателя кривая смещается влево, увеличивается сродство НЬ к кислороду, и наоборот, при повышении Р50 кривая диссоциации смещается на графике вправо и сродство НЬ к кислороду снижается, т. е. НЬ легче отдает кислород.
N. Bohr установил, что повышение температуры тела и РДСО2, а также снижение pH крови увеличивают Р50 и наоборот (рис. 3.12). В 1925 г. Greenvald с соавт. обнаружил в эритроцитах 2,3-дифосфогли-церат (2,3-ДФГ) — фактор, который повышает Р50. В клинической практике установлено, что увеличение Р50 сопровождается повышенной отдачей кислорода тканям и лучше переносится организмом, чем его снижение.
Таким образом, даже при одинаковом РаО2 насыщение НЬ кислородом и снабжение тканей кислородом зависит от Р50.
90 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
100
90
X 60 g 50 | 40 3 30 X 20 10
Сдвиг кривой
О 10 20 30 40 50 6070 8090100
Ра О2, ММ рт. СТ.
Рис. 3.12. Отклонения кривой диссоциации оксигемоглобина, вызванные температурой тела, pH, РдСО2 и 2,3-дифосфоглицератом (2,3-ДФГ)
Сатурация венозной крови (5Ю2) является также очень важным параметром, показывающим, какой процент НЬ остался насыщенным кислородом после экстракции его тканями. Обычно величина 5Ю2 ниже 70 % свидетельствует о низкой доставке и/или высоком потреблении кислорода. На сегодня отсутствует четко обозначенный пороговый уровень 5гЮ2 для каждого больного, однако исследование его, особенно в динамике, позволяет уточнить степень дефицита кислорода, причины гипоксии и методы коррекции [27].
Основная часть кислорода транспортируется кровью в составе НЬ, а общее содержание кислорода в артериальной крови (СйО2) равняется сумме растворенной фракции и связанной с НЬ:
СаО2 = РаО2 • 0,0031 + НЬ • 5йО2 • 1,39.
Один грамм гемоглобина в идеале может связать 1,39 мл кислорода (значение константы Хюфнера в действительности несколько ниже — 1,34 — 1,36).
Очевидно, что наибольшее влияние на СаО2 оказывает концентрация НЬ и SaO2. При содержании НЬ = 150 г/л, SaO2 = = 98 %, СяО2 « 208 мл/л.
Другим важным фактором гомеостаза организма является транспорт, или доставка, кислорода тканям (£Ю2)
DO2 =СаО2 СИ,
где СИ — сердечный индекс.
При обычном показателе сердечного индекса — 2,5 —3,5 л/мин на 1 м2 нормальная величина DO2 = 520 — 720 мл/мин на 1 м2 [24]. В последнее время многие исследователи полагают, что повышение DO2 позволяет снизить летальность у тяжелых хирургических больных [28].
Благодаря гибкости (деформируемости) своей мембраны эритроцит проходит через капилляры с меньшим, чем у него, диаметром. Нарушения микроциркуляции также приводят к нарушению транспорта кислорода к тканям даже при нормальном значении DO2.
Важным показателем для оценки оксигенации тканей является потребление кислорода (VO2), которое можно вычислить по формуле
VO2 =0(СйО2 -сю2), где CvO2 — содержание кислорода в венозной крови.
В норме VO2 равно 110 — 160 мл/мин на 1 м . Поглощение кислорода тканями зависит от метаболических потребностей. Оксигенацию тканей можно считать неадекватной при VO2 < 100 мл/мин на 1 м2 [24].
Показатель экстракции кислорода тканями (О2Ег) рассчитывают по формуле
О2Ег = VO2 /£Ю2 =
= (SaO2-SvO2)/SaO2.
В норме в покое этот показатель равен 0,2 —0,3, или 20 — 30 %, т. е. это та часть доставляемого в капилляры кислорода, которая усваивается тканями. При снижении £)О2, как правило, повышается О2Ег, так как потребление тканями кислорода должно оставаться на прежнем уровне, однако повышение этого показателя при значительном снижении доставки кислорода не может компенсировать его потребление. При некоторых состояниях О2Ег повышается до 0,5 —0,6, а у тренированных атлетов и до 0,8 [29].
Транспорт газов кровью 91
Соотношение между доставкой и потреблением кислорода является важным компонентом мониторинга транспорта кислорода в ОИТ. Его следует использовать для оценки ишемии ткани, выбора терапевтической стратегии [24]. Это соотношение можно увеличить двумя способами: увеличив доставку или уменьшив потребность. Можно повысить £Ю2 несколькими способами: увеличить сердечный выброс за счет коррекции гиповолемии, применения кардиотонических препаратов; повысить содержание кислорода в крови, применяя оксигенотерапию, ИВЛ, увеличивая концентрацию НЬ, однако выбор метода зачастую является сложной задачей для клинициста. Увеличение £)О2 может повысить выживаемость у определенной группы больных, однако этот метод далеко не во всех случаях критических состояний является эффективным, а иногда он может оказаться даже опасным [30, 31]. Нормализация артериального давления (и даже сердечного индекса) и доставки кислорода не отражает в достаточной мере процесс оксигенации и поэтому необходимы дополнительные методы ее мониторинга [32].
Соотношение СИ/О2Ег является комплексным показателем оксигенации тканей [33]. Увеличение потребления кислорода тканями может быть достигнуто как за счет повышения СИ, так и О2Ег, или путем повышения обоих показателей.
Малую эффективность оксигенотерапии при высоком шунтировании крови в малом круге можно объяснить с позиции концепции транспорта кислорода. Так, содержание О2 в крови, протекающей мимо вентилируемой чистым кислородом альвеолы, повысится только на 2 — 3 % за счет увеличения растворимой фракции и повышения 5О2 с 98 до 100 %. Однако в шунтируемой крови содержание кислорода останется на низком уровне, поэтому содержание кислорода в крови после смешения обеих фракций все равно будет низким. Такое неэффективное применение кислорода для повышения оксигенации крови характерно только для шунтирования крови слева направо.
Насыщение кислородом венозной крови. Как указывалось выше, 5гЮ2 — очень
важный показатель оксигенации тканей, который измеряется в смешанной венозной крови, лучше — из легочной артерии, по формуле
SvO2 = 5йО2 - УО2 / (СИ • НЬ).
При отсутствии анемии и гипоксемии 5уО2 может отражать зависимость между VO2 и сердечным индексом. Снижение 5Ю2 свидетельствует о нарушении баланса между потреблением и доставкой кислорода и может служить обобщающим индикатором неадекватной оксигенации, а также важным прогностическим критерием. Однако даже такой существенный показатель не может интерпретироваться независимо от других показателей. Так, у здоровых людей он может снижаться при физической нагрузке, а у больных с нарушенной функцией сердца в некоторых случаях быть нормальным. Если терапия преследует цель только повысить этот показатель до нормальных и супранормальных цифр, то это не всегда будет способствовать выживаемости [32].
Концентрация молочной кислоты (лактата) в крови. Для оценки адекватности оксигенации тканей в клинике используют такой показатель, как уровень лактата в венозной крови. Лактат — конечный продукт анаэробного гликолиза — продуцируется многими тканями, включая эритроциты, скелетные мышцы и кожу. В сутки его образуется 15 — 20 мэкв/кг, а нормальный уровень в крови — 1 ммоль/л. Лактат синтезируется в цитозоле в результате катализируемой лактатдегидрогеназой реакции
Пируват + НАД • Н + +Н+ <=» Лактат + НАД.
Нормальное соотношение лактата к пирувату 10:1. Синтез лактата увеличивается при активации метаболизма (физическая нагрузка), снижении активности пируватдегидрогеназы, а также при гипоксии. В норме осуществляется регенерация НАД • Н в НАД, но при недостатке кислорода количество НАД-Н превышает содержание НАД, как и продукция лактата, уровень которого выше 2 ммоль/л явля
92 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
ется признаком гипоксии тканей, а критический уровень равняется 4 ммоль/л. Превышение этого показателя свидетельствует, что тканевая оксигенация не достаточна и происходит повреждение клетки [24]. Активация провоспалительных клеток, включая лейкоциты, также приводит к повышению уровня лактата в венозной крови. Тест на лактат для определения степени гипоксии тканей не является специфичным, так как недостаточность печени также может привести к гиперлактатемии [34]. Однако в настоящее время общепризнано, что накопление лактата в крови отражает увеличение его продукции, а не сниженный клиренс [24]. При дефиците тиамина, генерализованных судорогах, респираторном алкалозе и даже тяжелой травме уровень лактата может повышаться вне зависимости от ишемии тканей [35]. Необходимо помнить, что и нормальная концентрация лактата в крови не всегда исключает ишемию тканей [24].
Желудочный внутрислизистый (гастроинтестинальный интрамукозный) pH (pHi). Нарушения циркуляции (шок) прежде всего отражаются на перфузии желудка и кишок (см. «Шок»), поэтому степень внутриклеточного ацидоза в слизистой оболочке желудка является важным индикатором гипоксии тканей. pHi определяется методом желудочной тонометрии [36], коррелирует с уровнем лактата и является важным прогностическим фактором. Критический уровень pHi = 7,32, более низкий уровень свидетельствует о гипоперфузии [24].
3.7.2. Транспорт кровью С02
Углекислый газ (углекислота, карбон диоксид, СО2) является конечным продуктом аэробного гликолиза в тканях, откуда он переносится в легкие для удаления во внешнюю среду. Поскольку напряжение СО2 в тканях выше, он диффундирует в плазму капилляров. Продукция СО2 зависит от возраста, пола, температуры тела, состава принимаемой пищи (например, прием углеводов увеличивает его продукцию) и других факторов и в покое составляет « 299 мл/мин.
Так же, как О2, СО2 растворяется в крови. В 1 л крови при рСО2 = 40 мм рт. ст. содержится 24 мл СО2. Подобно кислороду некоторая часть углекислоты переносится гемоглобином, образуя карбами-ногемоглобин:
НЬ—NH2+CO2 -» Hb-NHCOOH' +Н+.
Однако большая часть СО2 транспортируется в составе иона НСО3.
При поступлении в плазму из тканей СО2 диффундирует в эритроцит, где претерпевает ряд преобразований:
СО2 + Н2О —> Н2СО3.
Эта реакция практически не может происходить в плазме, так как для ее осуществления необходим фермент карбоангидраза, который содержится только в эритроцитах. Поскольку Н2СО3 более сильная кислота, она вытесняет НЬ:
КНЬ + Н2СО3 -> КНСО3 + ННЬ.
Часть ионов НСО3 выходит из эритроцита в плазму, в то время как ионы С Г поступают — так называемый сдвиг Хам-бургера (для поддержания электронейтральности). Так, СО2 транспортируется к легким, где ННЬ присоединяет кислород с образованием ННЬО2.
ННЬО2 является более сильной кислотой, чем угольная:
ННЬО2 4- КНСО3 = КНЬО2 + Н2СО3.
Затем в эритроците из угольной кислоты образуется углекислый газ, который диффундирует через альвеолярно-капиллярную мембрану в альвеолярный газ.
В итоге до 12 % СО2 транспортируется в растворенном виде или в составе недис-социированной Н2СО3, И — в составе карбаминовых соединений с гемоглобином, 27 — в составе гидрогенкарбонатных ионов в эритроцитах и около 50 % — в составе гидрогенкарбонатных ионов в плазме крови [1].
Таким образом, для нормального транспорта СО2 необходимо наличие в крови двух форм НЬ, в частности, восстановленной формы. При гипербарической оксигенации более 2,2 атм этот процесс может нарушаться, что сопровождается задержкой СО2 в тканях.
Гипоксия 93
3.8. ГИПОКСИЯ
Адекватное снабжение тканей кислородом является жизненно необходимым для клеточного дыхания, продукции макроэр-гических соединений и поддержания адекватной функции органов и тканей. Гипоксия — это несоответствие между доставкой кислорода тканям и их потребностью.
В ответ на гипоксию в организме возникает О2-регулируемая генная экспрессия [37]. Снижение ГЮ2, кроме активации дыхательного центра (см. «Регуляция дыхания»), вызывает такие реакции организма, как: усиление эритропоэза; пролиферация клеточных фибробластов, клеток поперечнополосатых мышц и эндоте-лиоцитов; ангиогенез и неоваскуляризация в ишемических тканях; усиление эндо-телиозависимой коагуляции, адгезии клеток и проницаемости клеточных мембран; активация вазомоторной деятельности. О2-зависимая генная регуляция играет важную роль в метаболизме и транспорте глюкозы, в метаболизме катехоламинов, апоптозе и гибели клеток.
В результате гипоксии активируется анаэробный гликолиз, увеличивается концентрация лактата, снижается количество оснований в венозной крови, что приводит к лактатацидозу.
В классических работах J. Barcroft (цит. по A. Gorlach, 2002) выделено три вида гипоксии:
1) неадекватное напряжение кислорода в артериальной крови — гипоксическая гипоксия’,
2) неадекватность концентрации циркулирующего гемоглобина — анемическая гипоксия;
3) неадекватный тканевый кровоток — стагнирующая гипоксия.
В современных работах различают следующие виды гипоксии:
1) гипоксическая;
2) циркуляторная;
3) гемическая;
4) тканевая (цитопатическая).
Гипоксическая гипоксия (ГГ) возникает вследствие недостаточности функции оксигенации крови в легких или снижения FiO2. В первом случае РЛО2 может
быть пониженным (при нарушении вентиляции) или нормальным (если в его основе лежат расстройства газообмена в легких), во втором — РдО2 понижено, а соответственно и 5аО2. -Од-дО2 остается в пределах нормы. Компенсация £Ю2 осуществляется за счет увеличения сердечного выброса и смещения КДО вправо. Клинически ГГ проявляется диффузным цианозом кожных покровов, тахикардией, артериальной гипертензией, в терминальных стадиях — брадикардией и артериальной гипотензией. При снижении РДО2 ниже 40 мм рт. ст. нарушается функция ЦНС (даже при хороших компенсаторных возможностях сердечно-сосудистой системы), что может проявляться нарушениями сознания, признаками отека мозга, гипоксической комой. На уровне клеток ГГ вызывает набухание митохондрий и микровакуолизацию. Изменения функции сердечно-сосудистой системы включают независимое от NO снижение сопротивления системных артерий. Кон-стрикция вен вызывает повышение артериального давления, в частности значительно возрастает давление в легочной артерии (гипоксическая вазоконстрикция). Снижается сопротивление церебральных сосудов, что может способствовать развитию отека мозга.
ГГ индуцирует продукцию почками эритропоэтина, что сопровождается активацией эритропоэза и увеличением концентрации гемоглобина [38]. Хроническая гипоксия увеличивает экспрессию тканевых сигнальных протеинов с участием глицеральдегид-3-фосфатазной дегидрогеназы, фактора-1, сосудистого эндотелиального фактора роста и интерлейкина-6 [37, 39]. От баланса этих факторов зависит адаптация организма к ГГ.
Когда компенсаторные механизмы истощаются, ГГ приводит к гибели клеток: растет внутриклеточный ацидоз, развивается тяжелый дефицит АТФ, нарушается функция мембранных насосов, что сопровождается нарушением внутриклеточного баланса электролитов. Повреждение митохондрий, прежде всего нарушение
94 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
транзита через ее мембраны (mitohondrial permeability transition — MPT) [40], усиливает гидролиз АТФ, еще более увеличивая энергодефицит и способствуя смерти клетки.
Циркуляторная гипоксия (ЦГ) возникает вследствие недостаточности кровообращения в тканях (часто ее еще называют ишемической гипоксией). Для этого вида гипоксии характерно увеличение со снижением 5уО2. ЦГ может носить как системный, так и локальный характер.
Местный эффект ЦГ реализуется преимущественно через аденозин. Недавними исследованиями доказано, что аденозин, действуя на Aj-аденозиновые рецепторы, вызывает брадикардию, центральную депрессию дыхания и вазодилатацию мышц. Брадикардия возникает вследствие пресинаптической вагальной стимуляции и ингибирования симпатической системы, непосредственно влияя на синоатриальный узел через Араденозиновые рецепторы; цереброваскулярная дилатация вызывается воздействием на аденозиновые А2А-ре-цепторы [6].
Таким образом, аденозин является первичным медиатором локального эффекта гипоксии в сердечно-сосудистой системе. Вазодилатацию при гипоксии могут также вызвать некоторые простагландины [42].
ЦГ представляет большую опасность для организма, чем ГГ: во-первых, накапливаются продукты метаболизма тканей, чего не наблюдается при ГГ; во-вторых, ведущую роль в патогенезе расстройств функции клетки играет роль синдром ишемия/реперфузия. Именно во время восстановления перфузии происходит повреждение и даже гибель клеток, в которых еще не произошли необратимые изменения во время ишемии (например, свободнорадикальное пероксидное окисление липидов). Кроме того, при ЦГ менее эффективны компенсаторные реакции организма, чем при гипоксической и гемической гипоксии. Доказательством этого могут служить клинические примеры тяжелой ишемии (острый инфаркт миокарда и инсульт).
Гемическая гипоксия (ГемГ) развивается вследствие снижения кислородной емкости крови. Выделяют анемическую гипоксию, связанную со снижением концентрации гемоглобина (вследствие кровопотери или нарушения гемопоэза), и гипоксию, при которой нарушаются свойства гемоглобина (карбоксигемоглобине-мия, метгемоглобинемия). Транспорт кислорода снижен в результате уменьшения кислородной емкости крови. По-видимо-му, ГемГ можно считать самой благоприятной для организма из всех видов гипоксий. Компенсация транспорта кислорода происходит в результате повышения сердечного выброса и, возможно, в значительной мере благодаря снижению вязкости крови и улучшению микроциркуляции [43]. Следует отметить, что компенсация транспорта кислорода при анемической гипоксии (АГ) возможна только при условии коррекции гиповолемии. Есть данные о том, что при нормоволемической гемодилюции снижается свободнорадикальная активность крови и уменьшается активность NO-синтетаз [44], наблюдается повышение перфузии миокарда и мозга — не только в результате увеличения сердечного выброса, но и вследствие коронарной и церебральной вазодилатации, достигающей максимума при Ht = 12,5 % [45], хотя в других органах (печень, почки и др.) это повышение не столь заметно. Еще одним механизмом компенсации при АГ является увеличение экстракции кислорода тканями [37].
При острой анемии при Ht < 15 % повышается содержание 2,3-ДФГ в эритроцитах, что сопровождается увеличением снабжения тканей кислородом, хотя этот механизм компенсации доказан только для хронической анемии [46].
Какой же уровень анемии можно считать критическим, ниже которого обязательно развивается гипоксия тканей в результате снижения транспорта кислорода? Раньше большинство экспертов считало оптимальным уровень Ht = 30 %, однако их рекомендации основывались на теоретических подсчетах максимальной доставки кислорода тканям [37]. Согласно некоторым клиническим исследованиям
Тканевое дыхание 95
[47], независимым фактором риска летальности у хирургических больных является снижение уровня НЬ до 30 г/л и ниже. Большинство экспериментальных и клинических исследований показало, что таким критическим уровнем следует считать « 40 г/л [48]. На этот критический уро
вень анемии влияют многие факторы (см. «Шок»).
Тканевая, или, как ее в последнее время называют, цитопатическая, гипоксия характеризуется снижением продукции АТФ при достаточном поступлении кислорода в ткани [37].
3.9. ТКАНЕВОЕ ДЫХАНИЕ
Тканевым дыханием (ТД) называется обмен дыхательных газов в клетке, происходящий в процессе биологического окисления органических веществ [49]. В результате ТД из питательного субстрата и кислорода образуется углекислый газ и вода с выделением энергии, которая аккумулируется в макроэргических соединениях. Собственно системы внешнего дыхания и кровообращения функционируют для обеспечения этих процессов в клетках и благодаря ТД существуют клетки и организм в целом.
Для осуществления нормального процесса ТД должны поддерживаться на определенном уровне концентрации питательных субстратов (углеводов, жиров, белков) и кислорода. В отсутствие последнего осуществляется анаэробный гликолиз, менее эффективный энергетически, в результате которого образуется лактат. Для получения одного и того же количества энергии в анаэробных условиях в клетке должно потребляться примерно в 15 раз больше глюкозы, чем в аэробных. При аэробном гликолизе при окислении 1 моль глюкозы освобождается 2883 кДж (689 ккал) энергии, при анаэробном — только 208 кДж (50 ккал) [50]. Следует отметить, что несмотря на низкую энергетическую эффективность, анаэробный гликолиз играет важную роль даже при наличии достаточного количества кислорода, например в эритроцитах, поперечнополосатых мышцах во время физической нагрузки, в мозговом веществе почек, сетчатке глаза и др.
Биологическое окисление пищевых субстратов с аккумуляцией энергии происходит в митохондриях клеток. Схема различных путей окислительного метабо
лизма в митохондриях изображена на рис. 3.13.
Пируват, жирные кислоты и аминокислоты переносятся в матрикс митохондрии. После ряда превращений эти вещества распадаются до ацетил-КоА, который поступает в цикл лимонной кислоты. Некоторая часть органических кислот, образовавшихся в матриксе митохондрий при метаболизме аминокислот, может включаться в цикл лимонной кислоты на разных этапах в виде а-кетоглутаровой кислоты, сукцинил-КоА, фумарана и оксало-ацетата. На определенных этапах превращений в цикле Кребса происходит биологическое окисление: отщепление атома водорода из 7?-ОН групп с образованием R =О [17]. В этих реакциях образовавшиеся в цикле лимонной кислоты молекулы никотинамидадениндинуклеотида (НАД) и никотинамидадениндинуклеотид-фосфата (НАДФ) присоединяют водород с образованием восстановленных форм НАД Н и НАДФ Н.
Затем эти субстраты и сукцинат диффундируют из матрикса к внутренней мембране митохондрий, где содержатся ферменты дыхательной цепи: кофермент флавинмононуклеотид (ФМН), флавинаде-ниндинуклеотид (ФАД). ФАД образуется путем фосфорилирования рибофлавина и соединения его с АМФ. Присоединяя водород, он превращается в ФАД • Н и ФАД • Н2. Оба эти флавопротеина передают электроны на убихинон (кофермент Q), который затем окисляется с участием цитохромов.
Из каждой молекулы водорода образуются два протона и два электрона, последние переносятся цитохромом Ь, Fes-бел-ком, цитохромами q и с2 на цитохром-
96 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
Дыхательная цепь
Окислительное
Криста
Рис. 3.13. Схема важнейших этапов биологического окисления в митохондриях [49]
оксидазу (комплекс цитохромов и а2 ), которая и передает их на молекулярный кислород с образованием воды. Согласно теории П. Митчелла (окислительное фосфорилирование), синтез АТФ происходит за счет тока протонов обратно в матрикс митохондрии. При окислении каждой молекулы НАД • Н образуется 3 молекулы АТФ, а при ФАД • Н2 — 2. Интенсивность окислительного фосфорилирования во многом зависит от обеспечения молекулами АДФ, здесь действует система обратной связи: чем больше распадается АТФ с образованием АДФ, тем интенсивнее происходит синтез АТФ [17]. На этот процесс влияет также поступление в ми
тохондрии кислорода и жирных кислот, лактата и метаболитов глюкозы. До 90 % потребляемого кислорода расходуется на синтез АТФ [17].
В покое кислород интенсивно потребляется миокардом, серым веществом головного мозга, печенью и корковым веществом почек. Другие органы и ткани в состоянии покоя расходуют относительно меньшее количество кислорода. При повышении активности какого-либо органа потребление им кислорода увеличивается вследствие усиления метаболизма. Расход О2 также увеличивается при повышении температуры тела. Резерва кислорода в тканях практически нет, только в миогло
Список использованной литературы 97
бине поперечнополосатых мышц имеется ограниченный запас его. Важнейшим показателем, характеризующим адекватность доставки кислорода, является напряжение кислорода в клетке.
К механизмам цитопатической гипоксии относят [37]:
— нарушение транспорта пирувата в митохондриальный ЦТК;
— ингибирование ключевого митохондриального энзима ЦТК — цис-аконитазы или электронного транспорта (НАД • Н-убихинон-оксидоредуктаза, сукцинат-уби-
хинон-оксидоредуктаза), а также активация поли-(АДФ)-рибозо-полимеразы (ПАРС) (см. «Шок»), или коллапс протонового градиента на митохондриальной мембране.
Все это ведет к нарушению окислительного фосфорилирования.
Как показали недавние исследования, цитопатическая гипоксия развивается во всех случаях сколько-нибудь продолжительного течения критического состояния, и именно она является причиной необратимых изменений в клетках и их гибели.
Список использованной литературы
1. Тевс Г. Дыхание // Физиология человека: Пер. с англ. / Под ред. Р. Шмидта, Г. Тевса. - М.: Мир, 1996. - Т. 2, ч. 6. - С. 567 — 604.
2. Гриппы М. А. Патофизиология легких: Пер. с англ. — М.: Бином, 1997. — 344 с.
3. Основы физиологии человека / Под ред. Б. И. Ткаченко. — СПб., 1994. — Т. 1. — 567 с.
4. Acker H.t Hue D. Mechanisms of oxygen sensing in the carotid body in comparison with other O2-sensing cells //News Physiol Sci. — 1995. - 10. - P. 435-444.
5. Hughes J. M. B., Pride N. B. Lung function tests. — W. B. Saunders, 2000. — 314 p.
6. McPhee S. J., Lingappa V. F., Ganog W. F., Lange J. D. Pathophysiology of Disease. — 3rd ed. — McGraw-Hill, 2000. — 662 p.
7. Von Neegard K. Neue Auffas-sungen uber einen Grundbegriff der Atemmeckanik; die Retrak-tionskraft der Lunge, abhangig von der Ober-flachenspannung in den Alveolen // Z. Ges. Exp. Med. - 1929. - 66. - S. 373-394.
8. Pattie R. E. Properties, function and origin of the alveolar lining layer//Nature. — 1955. — 175. - P. 1125-1126.
9. Clements J. A. Surface tension of lung extracts // Proc. Soc. Exp. Biol. Med. — 1957. — 95. - P. 170-172.
10. Kahn M. C., Anderson G. I., Any an W. R., Hall S. B. Phosphatidylcholine molecular species of calf lung surfactant // Am. J. Physiol. - 1995. - 269. - P. L567-573.
11. Bernhard W., Haagsman H. P., Tscheming T. et al. Conductive aireway surfactant: surfacetension function, biochemical composition and possible airway origin // Am. J. Respir. Cell Mol. Biol. - 1997. - 17. - P. 41-50.
12. Persson A., Chang D., Rust K. et al. Purification and biochemical characterization of CP-4 (SP-D): a collagenous surfactant-associated protein // Biochemistry. — 1989. — 27. - P. 6361-6367.
4
4-220
13. Frerking I., Gunther A., Seeger W., Pison U. Pulmonary surfactant: functions, abnormalities and therapeutic options // Int. Care Med. - 2001. - 27. - P. 1699-1717.
14. Hamm H., Kroegel C., Hohlfeld J. Surfactant: a review of its function and relevance in respiratory disorders // Respir. Med. — 1996. — 90. - P. 251-270.
15. Klaus M. H., Clements J. A., Havel R. J. Composition of surface-active material isolated from beef lungs // Proc. Natl. Acad. Sci. — 1992. - 47. - P. 1858-1859.
16. Hamm H., Fabel H., Bartsch W. The surfactant system of the adult lung: physiology and clinical perspectives // Clin. Invest. — 1992. - 70. - P. 637-657.
17. Ганонг В. Ф. Ф1зюлопя людини: Пер. з англ. / За ред. М. Гжегодського, В. Шевчука, О. ЗаячювськоТ. — Л.: БаК, 2002. — 784 с.
18. Ghio A. J., Fracica Р, J., Youang S. L., Piantadosi С. A. Synthetic surfactant scavenges oxidants and protects against hyperoxic lung injury //J. AppL Physiol. — 1994. — 77. - P. 1217-1223.
19. JonssonS., Musher D. M., Goree A., Lawrence E. C. Human alveolar lining material and antibacterial defences // Am. Rev. Respir. Dis. - 1986. - 133. - P. 136-140.
20. Thews G. Der Einfluss von Ventilation, Perfusion, Diffusion und Distribution auf den pul-monalen Gasaustausch // Akadem. Wiss. Lit. — Mainz; Wiesbaden: Steiner, 1979. — 356 S.
21. Von Euler U. S., Liljistrand G. Observations on the pulmonary arterial blood pressure in the cat // Acta Physiol. Scand. — 1946. — 12. - P. 301-312.
22. Naeije R., Brimioulle S. Physiology in medicine: importance of hypoxic pulmonary vasoconstriction in maintaining arterial oxygenation during acute respiratory failure // Review. Crit. Care. — 2001. — 5(2). — P. 67-71.
98 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ДЫХАНИЯ
23. Melot С., Naeije R., Mols Р. et al. Pulmonary vascular tone improves gas exchange in the adult respiratory distress syndrome // Am. Rev. Respir. Dis. — 1987. — 136. — P. 1232-1236.
24. Marino P. L. The ICU Book. — 2nd. — Lippincott Williams& Wilkins, 1997. — 928 p.
25. Covelli H. D., Nessan V. J., Tuttle W. K. Oxygen derived variables in acute respiratory failure //Crit. Care Med. — 1983. — 11. — P. 646-649.
26. Curry S. C., Arnold-Cappell P. Nitroprusside, nitroglycerin, and angiotensin-converting enzyme ingibitors // Toxic effects of drugs used in the ICU / Ed. J. L. Blumer, G. R. Bond. — 1991. - P. 555-582.
27. Schlichtig R. Oxygen delivery and consumption in critically ill patients // Critical Care / Ed. J. M. Civetta, R. W. Taylor, R. R. Kirby. — 3d ed. — Philadelphia, Lippincott-Raven, 1997. - P. 337-342.
28. Jonas M. M., Wolf С. B. Oxygen delivery and the critically ill // Yearbook of Intensive Care and Emergency Medicine / Ed. J. L. Vincent. — Springer, 2002. — P. 603 — 611.
29. Leach R. M., Treacher D. F. The relationship between oxygen delivery and consumption // Dis. Mon. - 1994. - 30. - P. 301-368.
30. Gattioni L., Brazzi L., Pelosi P. et al. A trial of goal-oriented hemodynamic therapy in critically ill patients // N. Engl. J. Med. — 1995. - 333. - P. 1025-1032.
31. Hayes M. A., Timmins A. C., Yau E. H. et al. Elevation of systemic oxygen delivery in the treatment of critically ill patients // N. Engl. J. Med. - 1994. - 330. - P. 1717 — 1722.
32. Vincent J. L. The Available Clinical Tools. — Oxygen-Derived Variables, Lactate, and pHi // W. J. Sibbald, K. Messmer, M. P. Fink. Tissue Oxygenation in Acute Medicine. — Springer, 2002. — P. 193 — 203.
33. Silance P. G., Simon C., Vincent J. L. The ralation between cardiac index and oxygen extraction in acutelly ill patients // Chest. — 1994. - 93. - P. 843-847.
34. Kruse J. A., Zaidi S. A. J., Carlson R. W. Significance of blood lactate levels in critically ill patients with liver disease // Am. J. Med. - 1987. - 83. - P. 77-82.
35. Mizock B. A. Lactic acidosis // Disease-A-Month. - 1989. - 5. - P. 235-300.
36. Fiddian-Green R. G., Pittenger G., Whitehouse W. M. Back-diffusion of CO2 and its influence on the intramural pH in gastric intestines of rats //J. Surg. Res. — 1982. — 33. - P. 39-48.
37. Gorlach A. Oxygen Signaling Cascades in Mammalian Cells // W. J. Sibbald, K. Messmer, M. P. Fink. Tissue Oxygenation in Acute Medicine. — Springer, 2002. — P. 63 — 77.
38. EcKardt K. U., Koury S. T., Tan S. S. et al. Distribution on erythropoietin producing cells in rat kidneys during hypoxyc hypoxia // Kidney Int. - 1993. - 43. - P. 815-823.
39. Graven К. K., Farber H. W. Hypoxia-associated proteins //New Horizons. — 1995. — 3. - P. 208-218.
40. Lemasters J. J., Nieminen A. L., Qian T. et al. The mitohondrial permeability transition in toxic, hypoxic and reperfusion injury // Mol. Cell Biochem. - 1997. - 174. -P. 159-165.
41. Verlato G., Borgdorff P. Endogenous adenosine enhances vagal negative chronotropic effect during hypoxia in the anaesthetised rabbit // Cardiovasc. Res. — 1990. — 24. — P. 532-540.
42. Messina E. J., Sun D., Koller A. et al. Role of endothelium-derived prostaglandins in hypoxia-elicited arteriolar dilatation in rat skeletal muscle//Circ. Res. — 1992. — 71. -P. 790-796.
43. Chapter С. K., Cain S. M. The physiologic reserve in oxygen carrying capacity // Can. J. Physiol. Pharmacol. — 1986. — 64. — P. 7-12.
44. Doss D. N., Estafanous F. G., Ferrario С. M. et al. Mechanism of systemic vasodilation during normovolemic hemodilution // Anesth. Analg. - 1995. - 81. - P. 30-34.
45. Von Restorff W., Hof ling B., Holtz J., Bas-senge E. Effect of increased blood fluidity through hemodilution on coronary circulation at rest and during exersise in dog // Pflugers Arch. - 1975. - 35. - P. 15-24.
46. Rodman T., Close H. P., Purcell M. K. The oxyhemoglobin dissociation curve in anemia // Ann. Intern. Med. — 1960. — 52. -P. 295-301.
47. Spence R. K., Costabile J. R., Yong G. S. et al. Is hemoglobin level alone a reliable predictor of outcome in severely anemic surgical patient? // Am. Surg. — 1992. -58. - P. 92-95.
48. Van Woerkens E., Trouwborst A., van La-schotj. J. B. Profound hemodilution: what is the critical level of hemodilution at which oxygen delivery-dependent oxygen consumption starts in an anesthetized human? // Anesth. Analg. — 1992. — 75. — P. 818 — 821.
49. Гроте Й. Тканевое дыхание // Физиология человека / Под ред. Р. Шмидта, Г. Тевса: Пер. с англ. — М.: Мир, 1996. — Т. 2. — С. 626-641.
50. Burton R., Krebs Н. A. The free-energy changes associated with the individual steps of the tricarboxylic acid cycle, glycolysis and alcohol fermentation and with gydrolysis of the pyrophosphate groups of adenosintripho-sphate // Biochem. J. — 1953. — 54. — P. 94-116.
Раздел
Острая недостаточность дыхания
Недостаточность дыхания — это клинический диагноз, свидетельствующий о том, что система внешнего дыхания не может обеспечить достаточную для метаболизма оксигенацию крови и/или элиминацию углекислого газа.
Острую недостаточность дыхания (ОНД) можно разделить на две большие группы: вентиляционную и легочную, или гипоксемическую/гиперкапническую (тип I), и гипоксемическую (тип II) [1]. Уже из названия понятно, что главной причиной вентиляционной или гипоксемической/ гиперкапнической формы ОНД является недостаточная вентиляция альвеол, а легочной, или гипоксемической — нарушение газообмена в легких при нормальной вентиляции. Основным проявлением ОНД I типа будет снижение РаО2 ниже 80 мм рт. ст. и повышение РаСО2 выше 45 мм рт. ст. при дыхании атмосферным воздухом, и повышение РаО2 вследствие окси-генотерапии. Поскольку наиболее информативным критерием адекватности вентиляции легких является элиминация углекислого газа, то именно уровень повышения РаСО2 будет указывать на степень тяжести этой формы ОНД.
При ОНД II типа на первое место выходит нарушение оксигенации вследствие нарушения диффузии кислорода через альвеолярно-капиллярную мембрану (редкая причина в клинике), вентиляционноперфузионных соотношений в легких, в частности повышения фракции шунтируемой крови из правых отделов сердца в
4*
левые (в последнем случае оксигенотера-пия малоэффективна). При этих видах респираторных расстройств процесс элиминации углекислого газа повреждается мало, и на первый план выходят нарушения оксигенации, основным критерием которых будет уменьшение РаО2 ниже 60 — 70 мм рт. ст. (уменьшение соотношения РаО2 /FiO2 ниже 200 мм рт. ст.).
Основные причины ОНД у больных в ОИТ:
вентиляционная (гипоксемическая/гиперкапническая) форма (тип I):
1. Нарушение центральной регуляции дыхания:
— выраженная гипоперфузия дыхательного центра (артериальная гипотензия, дислокация мозговых структур);
— патологический процесс в ЦНС (менингоэнцефалиты, внутричерепные новообразования);
— черепно-мозговая травма;
— отравление или передозировка препаратов — депрессоров дыхательного центра (опиаты, седативные препараты и др.);
— первичная альвеолярная гиповентиляция (синдром Пиквика и др.).
2. Нарушение проведения импульса к дыхательным мышцам:
— повреждения спинного мозга;
— патологические процессы в спинном мозгу и периферической нервной системе (полирадикулоневриты, инфекционные процессы, полинейропатии).
3. Нейромышечные расстройства:
— мышечные релаксанты;
100 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
— отравление фосфорорганическими соединениями;
— миастения gravis;
— миопатии;
— разрывы или релаксация диафрагмы;
4. Нарушение целостности (каркасно-сти) грудной стенки:
— открытый пневмоторакс;
— флотирующие переломы ребер.
Легочная (гипоксемическая) форма (тип II):
1. Пневмонии.
2. Острый респираторный дистресс-синдром.
3. Легочный фиброз.
4. Ателектазы паренхимы легких.
5. Отек легких.
6. Контузия (травма) легких.
К этой же форме можно отнести эмболизацию ветвей легочной артерии.
Встречаются также патологические процессы, при которых сочетаются обе формы, например тяжелая бронхиальная астма, напряженный пневмогидроторакс и др.
4.1. ВЕНТИЛЯЦИОННАЯ ФОРМА ОНД
Патологические процессы, приводящие к нарушению функции ДЦ. Проявляются нарушением регуляции дыхания: ритм дыхания нарушается вплоть до развития терминальных видов дыхания (гас-пинг, Чейн-Стокса, Биотта), уменьшением частоты дыхания вплоть до апноэ. При инфарктах продолговатого мозга с вовлечением вентрального ядра ДЦ или при повреждении нисходящих проводящих путей (при хирургических вмешательствах, травме) может сохраняться только произвольный (корковый) контроль дыхания — синдром Ондины, когда спонтанное дыхание прекращается во время сна.
Нарушение проводимости импульсов по шейному отделу спинного мозга. Инфаркт (тромбоз питающих сосудов), сдавливание опухолью, гематомой, травматический разрыв приводят к квадриплегии и отсутствию спонтанного дыхания. Вследствие повреждений ниже Сш — Cv (где расположены мотонейроны диафрагмального нерва) развивается паралич или парез межреберных мышц, что в значительной степени уменьшает функциональные резервы внешнего дыхания, а в сочетании со слабостью мышц передней брюшной стенки — снижает эффективность кашля и резко увеличивает риск ателектазов легких, пневмоний. В результате до 80 % таких больных нуждаются в периодической респираторной поддержке [2].
Болезни спинного мозга. Такие заболевания, как полиомиелит, боковой амнио-трофический склероз, приводят к тяже
лой вентиляционной недостаточности дыхания. Наблюдаются случаи, когда подобные явления развиваются через несколько лет после перенесенного полиомиелита. У 50 % таких больных частыми осложнениями являются аспирации, пневмонии. Для большинства больных с нарушением проведения импульса по спинному мозгу необходимо наложение трахеостомии и проведение ИВ Л. В последнее время все чаще используют неинвазивные методы ИВЛ.
Заболевания периферической нервной системы. Острый полирадикулоневрит (синдром Guillain —Barre) — наиболее частая причина периферической нейропатии, вызывающей нарушение вентиляции. В половине случаев дыхательным расстройствам предшествует острая инфекция респираторного тракта или пищеварительного канала, затем наступает восходящий мышечный паралич, в том числе и дыхательных мышц. Паралич может достигать своего максимума на 2 — 4-ю неделю после начала заболевания. До 20 % больных с этой патологией нуждаются в вентиляторной поддержке. Кроме того, эффективными могут оказаться плазмаферез и внутривенное применение иммуноглобулина [3]. Другими причинами периферической нейропатии с ОНД могут быть интермиттирующая порфирия, дифтерия, системная красная волчанка, болезнь Лайма, отравления таллием и триортокрезилфосфатом.
В последнее время синдром полинейропатии все чаще наблюдается преиму
Вентиляционная форма ОНД 101
щественно у пожилых больных после длительного лечения сепсиса и полиорган-ной недостаточности. После ИВЛ их тяжело перевести на спонтанное дыхание [4].
Нарушения нейромышечной проводимости. 10 % пациентов с миастенией gravis нуждаются в респираторной поддержке чаще всего при обострении заболевания, холинергическом кризе или инфекционных заболеваниях. Лечение включает ИВЛ, плазмаферез, применение кортикостероидов, иммуносупрессоров и ингибиторов холинэстеразы. Эффективной может оказаться тимэктомия. К нарушению нейромышечной проводимости может привести также ботулизм, при котором в процесс вовлекаются дыхательные мышцы. Кроме ИВЛ лечение включает элиминацию неабсорбированного токсина из кишок, промывание желудка, применение антитоксина, антибиотикотерапию, хирургическую обработку ран. Иногда больным с ботулизмом ИВЛ требуется в течение нескольких месяцев [3].
К нарушению нейромышечной проводимости приводят также отравления фосфорорганическими инсектицидами, нервнопаралитическими газами. Нередко такие случаи возникают и в анестезиологической практике после применения мышечных релаксантов. Примером может служить применение недеполяризующих миорелаксантов у больных миастенией, сукци-нилхолина — у пациентов с врожденной недостаточностью псевдохолинэстеразы (один случай на 2800), недеполяризующих миорелаксантов (панкурониум, векуро-ниум) при недостаточности почек и др. [5]. В последнее время наблюдаются случаи тяжелой недостаточности дыхательных мышц с их атрофией при длительной ИВЛ с использованием миорелаксантов, аминогликозидов, кортикостероидов, и хотя во многих случаях эти изменения обратимы, они значительно усложняют восстановительный период [6].
Миопатии. Большинство миопатий не вызывают значительного нарушения вентиляции, требующего вентиляционной поддержки. Однако пациенты с мышечной дистрофией часто умирают от недостаточ
ности дыхания через двадцать-тридцать лет заболевания. ИВЛ применяют также при дерматомиозите. В последнее время для этой категории больных пытаются использовать неинвазивные методы ИВЛ. Следует отметить, что продолжительная ИВЛ без периодов тренировки дыхательных мышц почти всегда приводит к мышечной дистрофии.
Пневмоторакс — это наличие воздуха в плевральной полости. Для анестезиолога представляют интерес открытый пневмоторакс и напряженный. Последний сам по себе не приводит к серьезным расстройствам вентиляции и газообмена, однако он должен быть диагностирован у больного до операции, так как применение ИВЛ может достаточно быстро перевести его в напряженный.
Открытый пневмоторакс может быть следствием травмы и носить ятрогенный характер (торакотомия, дренирование плевральной полости без обеспечения герметизации). О его наличии свидетельствует дефект грудной стенки с сообщением между атмосферой и плевральной полостью или присасывающее движение воздуха на вдохе в плевральную полость. Эффективность самостоятельного дыхания при этом осложнении значительно снижается вследствие трех причин: отсутствия вентиляции легкого на стороне пневмоторакса; маятникообразного движения газа со сниженным напряжением кислорода и повышенным — углекислого газа (на вдохе газ в легкое на здоровой стороне поступает как из атмосферы, так и из другого легкого); флотации органов средостения (на вдохе смещение происходит в здоровую сторону в результате снижения давления в плевральной полости, препятствуя нормальному вдоху; на выдохе органы средостения смещаются в больную сторону, ухудшая условия для выдоха). Таким образом, возникает гиповентиляция одного дышащего легкого гипоксической и гиперкапнической газовой смесью. Поэтому если открытый пневмоторакс не связан с торакотомией, его необходимо перевести в закрытый наложением герметичной повязки. Для того чтобы избежать при этом напряженного пневмоторакса, предвари
102 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
тельно следует дренировать плевральную полость. Можно также применить экспираторные методы ИВ Л.
Напряженный пневмоторакс может быть следствием как травмы (в том числе и ятрогенной), так и баротравмы легкого при проведении ИВЛ, результатом спонтанного разрыва легочной паренхимы (буллезная болезнь легких и др.). Особенно часто это осложнение развивается у больных на ИВЛ с повышенным сопротивлением дыханию (как эластическим, так и динамическим), когда для его преодоления необходимо создавать высокое инспираторное давление. Симптомы напряженного пневмоторакса следующие: цианоз кожных покровов, особенно верхней половины тела; артериальная гипотензия, тахикардия, возможны нарушения ритма сердца; отставание при дыхании грудной стенки на стороне пневмоторакса; тимпанический оттенок при перкуссии; смещение верхушечного толчка в противоположную сторону (пальпаторно и аускульта-тивно); внезапное немотивированное повышение РйСО2. При этом осложнении в большей степени страдает кровообращение, чем внешнее дыхание, вследствие смещения органов средостения и перегиба полых вен. В результате может резко снижаться сердечный выброс с развитием обструктивного шока, часто опасного для жизни. Для лечения необходимо экстренно пропунктировать больного во II межреберье по среднеключичной линии с последующей манометрией и, если необходимо, дренировать плевральную полость. Ни в коем случае нельзя до декомпрессии переводить больного на ИВЛ, что может усилить шоковые явления и быстро привести к гибели больного. На ИВЛ больных с напряженным пневмотораксом можно переводить только после декомпрессии плевральной полости.
Общие принципы консервативной (без применения ИВЛ) терапии ОНД. Поскольку одним из компонентов ОНД является недостаточная сатурация крови кислородом, наиболее распространенным методом лечения является оксигеноте-рапия. Показание к ней — гипоксемия, проявляющаяся снижением РаО2 ниже
70 мм рт. ст. или SaO2 — < 90 %. Наиболее часто для ингаляции кислорода используют носовые канюли, носовые катетеры, лицевые маски.
С помощью канюли и катетера, введенного в область носоглотки, при потоке кислорода 5 — 6 л/мин можно достичь FiO2 = = 0,40 —0,44, с помощью простой лицевой маски — FiO2 = 0,50. Имеются специальные маски с частичной рециркуляцией газа (можно достичь FiO2 = 0,70 — 0,80), а также нереверсивные маски (можно добиться ингаляции почти чистого кислорода), а также маски Вентури с подсасыванием воздуха при подаче кислорода, с помощью которой можно поддерживать примерно заданный уровень FiO2.
Ингаляция кислорода не только повышает оксигенацию крови, но и может снижать давление в легочной артерии (рефлекс гипоксической вазоконстрикции). Однако следует помнить, что оксигеноте-рапия часто сопровождается целым рядом осложнений, особенно при неквалифицированном применении.
Гиперкапния. Чаще всего возникает у больных с хронической недостаточностью дыхания — с так называемой гипоксической регуляцией дыхания. У таких больных необходим контроль напряжения углекислого газа в крови.
Токсичность кислорода. Безопасной максимальной концентрацией кислорода при длительной ингаляции является 0,6. При ингаляции кислорода в концентрации более 0,6 свыше 48 ч развивается токсический эффект. Например, дыхание чистым кислородом добровольцами в течение 6 —12 ч вызывало эндобронхит и снижение ЖЕ Л [7], при более длительной ингаляции развивался синдром, напоминающий ОРД С [8].
Поддержание проходимости дыхательных путей. Обтурация нижних дыхательных (н/д) путей является одной из наиболее частых причин развития и прогрессирования ОНД, поэтому поддержание их проходимости — один из наиболее важных компонентов терапии ОНД. Следует отметить, что наиболее эффективным и безопасным методом является фибробронхо-скопия (ФБС) под местной анестезией.
Вентиляционная форма ОНД 103
Этот метод сравнительно хорошо переносится даже тяжелыми больными. С помощью заливки теплого изотонического раствора натрий хлорида и аспирации содержимого дыхательных путей ФБС позволяет под визуальным контролем восстанавливать проходимость вплоть до сегментарных бронхов. Показания к лечебной ФБС: ателектазы, гиповентиляция отделов легкого, стойкие влажные хрипы. При необходимости ФБС следует применять несколько раз в сутки. Часто после ФБС уменьшается чувство недостатка воздуха, другие клинические симптомы гипоксемии, улучшается оксигенация крови. Проведение ФБС позволяет также осуществить дифференциальную диагностику природы тенеобра-зования в легком на рентгенограмме. ФБС можно сочетать с аэрозолетерапией, постуральным дренажем, физиотерапией, применением бронходилататоров.
При невозможности проведения ФБС следует использовать другие методы восстановления проходимости н/д путей: назотрахеальные заливки с аспирацией, чрескожное введение препаратов в трахею с последующим откашливанием, при выраженном трахеобронхите — наложение микротрахеостомии с систематическим введением в трахею растворов антибиотиков. В крайнем случае необходимы поднаркозные аспирации с интубацией трахеи. Следует добиваться восстановления проходимости нижних дыхательных путей, иначе вся последующая терапия ОНД может оказаться неэффективной.
При восстановленной проходимости дыхательных путей для профилактики ате-лектазирования альвеол, увеличения ФОБ, уменьшения шунтирования в легких особенно важным является дыхание с сопротивлением на выдохе. Самый простой метод для его осуществления — выдох больным через трубочку, погруженную в жидкость, но этого бывает недостаточно, больший эффект достигается при раздувании резиновой детской игрушки или шарика. Можно использовать специальные приборы, с помощью которых регулируется сопротивление выдоху. Еще более совершенными являются методы неинвазивной ИВЛ, рассматриваемые в подразделе «ИВЛ».
Бронходилататоры. Для уменьшения сопротивления дыхательных путей применяют агонисты Р2"аДРеноРеЦептоРов — ингаляции сальбутамола, тербуталина, а также внутривенное введение эуфиллина, если нет противопоказаний (тахикардия, нарушения ритма сердца). При этом следует помнить о возможных побочных эффектах действия р2’агонистов и эуфиллина (см. «Бронхиальная астма»).
Муколитическая терапия. С этой целью применяют ацетилцистеин, амброксол внутрь, а также в небольшой дозе инсталлируют в трахею. В аэрозолях ацетил цистеин следует применять с осторожностью, так как он обладает раздражающим действием на гортань, может провоцировать ла-рингоспазм, тошноту и рвоту. С целью улучшения откашливания возможно применение специальных трав, горячего питья.
Относительно использования кортикостероидов путем инстилляции в трахею мы не обнаружили информации о рандомизированных исследованиях, подтверждающую его эффективность. В еще большей степени это касается ингаляционного или интратрахеального введения протеолитических ферментов. Безопасность их применения не доказана, возможен повреждающий эффект на цилиарный аппарат, а также аллергизация больных.
С целью санации трахеобронхиального дерева мы широко используем с хорошим клиническим эффектом внутритрахеаль-ное и внутрибронхиальное введение растворов антибиотиков, учитывая чувствительность к ним высеваемой из нижних дыхательных путей микрофлоры (особенно часто применяем аминогликозиды, фторхинолоны, полимиксин). Необходимы дополнительные контролируемые рандомизированные исследования, доказывающие эффективность этого метода.
Осуществляя профилактику и лечение ОНД, нельзя забывать и о двигательном режиме больных, частой смене положения тела. Особенно большой риск развития ОНД у иммобилизованных (переломы тазовых и бедренных костей) больных пожилого возраста. У пациентов этой категории особое внимание следует уделять профилактике ОНД.
104 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
4.2. ПНЕВМОНИЯ
Несмотря на успехи антибактериальной и интенсивной терапии пневмония остается одной из наиболее распространенных причин госпитализации, часто с неблагоприятным прогнозом лечения. Так, в США она занимает шестое место как причина общей летальности и первое — как причина летальности при инфекционных заболеваниях [9]. Часто с этим заболеванием приходится сталкиваться в ОИТ и врачам-анестезиологам .
4.2.1. Патогенез
Пневмония вызывается микроорганизмами, попадающими в нижние дыхательные пути либо при аспирации, либо инга-ляционно. Гематогенный путь является менее типичным, хотя возможным. Инфекция развивается в случае преодоления механического (мукоцилиарный клиренс) и иммунного (нейтрофилы, макрофаги, гуморальные антитела) барьеров легких. Риск пневмонии более высок у больных в критическом состоянии. Факторами риска развития пневмонии являются [10]: нарушение иммунитета; некроз паренхимы легких; применение стероидных гормонов, цитостатиков, цитотоксических препаратов; нарушение питания; алкогольная интоксикация; сахарный диабет; нарушение секреции эпителия нижних дыхательных путей; ателектазы; курение; хронические неспецифические заболевания легких (ХНЗЛ); неврологические расстройства; ОРДС; контузия легких, вирусная инфекция дыхательных путей. Последний фактор является одним из наиболее частых причин возникновения заболевания.
Согласно современной классификации, пневмонии делятся на внебольничные (ВВП), внутрибольничные (госпитальные, нозокомиальные) (ВП), среди последних выделяют вентилятор-ассоциированные (ВАП).
Внебольничные пневмонии — это те, которые развиваются у амбулаторных, нестационарных больных. В Северном полушарии заболеваемость ВП составляет примерно 12 случаев на 1000 человек в
год, больший риск заболевания отмечается в зимнее время у лиц младенческого и старческого возраста [ 11, 12]. Большинство пациентов с этим заболеванием лечатся амбулаторно, однако при необходимости в госпитализации летальность достигает 25 % [13]. Этиологический фактор ВВП остается невыясненным в 30 — 60 % случаев [14]. Причиной могут быть: эмпирическое назначение антибиотиков перед бактериологическим исследованием, неквалифицированный забор материала для бактериологического исследования, трудности в идентификации небактериальных патогенов.
Диагноз ВВП ставится в основном на основании клинических признаков: острое начало, появление гиповентиляции и специфических хрипов в легких при аускультации, откашливание гнойной мокроты, повышение температуры тела, одышка, появление инфильтратов в легких и др. Подтверждается диагноз ВБП бактериологическими исследованиями секрета нижних дыхательных путей. Наиболее частым (в 25 — 60 % случаев) возбудителем ВБП являются грамположительные кокки (Streptococcus pneumonia, St. aureus) [15], всего известно более 100 возбудителей ВБП [12].
Факторами наибольшего риска развития пневмонии являются: острая вирусная инфекция (особенно гриппозная), пожилой возраст, курение, иммунодефицит, хронические заболевания легких, хроническая недостаточность почек и др. Статистически значимыми факторами, увеличивающими риск летального исхода при ВП, следует считать: принадлежность к мужскому полу, наличие плеврита, переохлаждение, понижение систолического АД, одышку, сахарный диабет, онкологические заболевания, сепсис, лейкопению, множественные инфильтраты в нескольких долях легких [16].
Наиболее часто встречающиеся возбудители внебольиичной пневмонии и соответствующая антибиотикотерапия приведены в табл. 4.1.
Антибиотикотерапию следует выбирать, исходя из чувствительности бактериологического возбудителя. Однако при необходимости эмпирического назначения не
Пневмония 105
Таблица 4.1. Этиология и терапия больных с внебольничной пневмонией [14]
Возбудитель Частота встречаемости, % Антибиотик Альтернативная терапия
Streptococcus pneumonia 25-60 Пенициллин G Эритромицин, первая генерация цефалоспоринов
Staphilococcus aureus 2-10 Нафциллин Первая генерация цефалоспоринов или ванкомицин
Hemophillus influenza 4-15 Цефуроксим Фторхинолоны или тиенам
Klebsiella pneumonia < 1 Вторая-третья генерация цефалоспоринов + аминогликозиды Фторхинолоны
Moraxella catarrhalis < 1 Вторая-третья генерация цефалоспоринов Фторхинолоны
Acinetobacter < 1 Пиперациллин или аминогликозиды Тиенам
Anaerobes 0-25 Клиндомицин Пенициллин G + метронидазол
было выявлено статистически достоверных различий в эффективности амбулаторного применения новых антибиотиков по сравнению с традиционными, такими как пенициллины широкого спектра действия, а также преимущества внутривенного введения по сравнению с приемом вовнутрь, наоборот, в последнем случае увеличивается продолжительность пребывания в стационаре, не найдено убедительных доказательств эффективности применения какой-либо комбинации антибиотиков [16]. Согласно некоторым исследованиям [17], раннее назначение антибиотиков уменьшает летальность у больных с ВБП. Существуют рекомендации [18] по методике перехода с внутривенного введения на прием антибиотика вовнутрь при отсутствии повышенной температуры тела на протяжении 8 ч, уменьшении выраженности одышки, кашля, снижении лейкоцитоза.
Имеются данные об эффективности дыхательной гимнастики и активного двигательного режима у больных с ВБП, а именно — 20 выдохов в день в трубку с водой (столб до 100 мм), изменение положения тела из лежачего в сидячее до 20 раз в день [19].
4.2.2. Внутрибольничная пневмония (ВП)
Это заболевание, развившееся через несколько суток после поступления в стационар. По частоте встречаемости занимает
второе место, а по летальности — первое среди всех внутрибольничных инфекций [20]. Особенно актуальной является проблема лечения ВП для критических больных в ОИТ, где частота развития этого осложнения в 5 — 10 раз выше, чем в других отделениях больниц [21].
Диагноз ВП, по-видимому, более сложный, чем ВБП, так как этот вид пневмонии возникает на фоне другой патологии, часто имеющей признаки воспаления. Клинические признаки пневмонии (повышение температуры тела, одышка, хрипы в легких при аускультации, отхаркивание гнойной мокроты), появление легочных инфильтратов в легких на рентгенограмме являются неспецифическими. Так, трудно отдифференцировать без бронхоскопии ателектаз от пневмонии, участок инфаркта легкого при эмболии ветви легочной артерии. Ложному диагнозу может способствовать отек легких, плевральный экссудат, а также новообразование в легком [10]. Американским Центром по контролю и профилактике заболеваний — The Centers for Disease Control and Prevention (CDC) — предложены следующие критерии для идентификации пневмонии для взрослых [22].
Основным условием установления диагноза ВП является появление на рентгенограмме легких новых или прогрессирование старых инфильтратов, кавитации
106 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
или признаков экссудативного плеврита, а дополнительными — следующие:
— изменение характера мокроты (гнойная);
— выделение патогенов из крови, био-птата легких или бронхиальной лаважной жидкости, взятой с помощью бронхоскопа;
— выделение вируса или вирусного антигена в секрете нижних дыхательных путей;
— выявление титра антител IgM или увеличение в четыре раза титра антител IgG в сыворотке крови;
— гистологические признаки пневмонии.
Среди инфекционных возбудителей ВП преобладают (в 57 % случаев) грамотри-цательные бактерии (Ps. aeruginosa, Klebsiella, Acinetobacter, Enterobacter). Грам-положительные бактерии высеваются в 36 % случаев [23], причем чаще, чем в случаях ВВП, определяются антибиотикорезистентные штаммы.
Факторами риска ВП являются [14]: пожилой возраст, курение в анамнезе, хронические заболевания легких, нарушения сознания (коматозное состояние), аспирация, состояние после операций на органах груди и верхней части живота, применение Н2-блокаторов, нарушение питания.
4.2.3. Вентилятор-ассоциированная пневмония
В структуре ВП выделяют особую форму пневмонии, развивающуюся у пациентов, которым проводят длительную ИВЛ, — вентилятор-ассоциированную пневмонию. Это осложнение является наиболее распространенной формой внутрибольничной инфекции в ОИТ [24]. Пневмония развивается примерно у 35 — 45 % пациентов, которым проводится ИВЛ [25]. Большинство исследователей предлагают считать сроком от начала проведения ИВЛ до начала развития В АП 48 ч и больше [26].
Поскольку основным фактором риска развития В АП считается проведение ИВЛ, то актуальным является вопрос о том, какие же методы ИВЛ наименее опасны (см. «Искусственная вентиляция легких»). Более подробно о В АП читайте в литературе, специально посвященной этому вопросу [27].
4.2.4. Лечение внутрибольничной пневмонии
Основными методами терапии тяжелых форм пневмонии являются: своевременная рациональная антибиотикотерапия, адекватная санация дыхательных путей, включающая аэрозолетерапию, чрескожное внутритрахеальное введение муколитических и антибактериальных препаратов, назотрахеальные заливки и аспирации. Одна из наиболее массовых ошибок при лечении пневмонии — максимальный акцент на антибиотикотерапию. Следует отметить, что даже самая мощная и адекватная по чувствительности микрофлоры антибиотикотерапия будет неэффективной при обтурации бронхов. Наиболее эффективный метод санации и восстановления проходимости нижних дыхательных путей — ФБС под местной анестезией. Показанием к ФБС являются: гиповентиляция и ателектаз (по данным рентгенографии), стойкие хрипы при аускультации, не проходящие после откашливания, кровохарканье. По нашему мнению, ФБС следует проводить всем больным с подозрением на пневмонию. Противопоказанием к ФБС может быть только непереносимость местных анестетиков. Даже больные в тяжелом состоянии переносят эту манипуляцию без тяжелых осложнений и, как правило, с последующим улучшением состояния. Причем чем тяжелее пневмония, тем чаще возникает потребность в ФБС, вплоть до проведения этой процедуры несколько раз в день. Во время ФБС при наличии гнойного бронхита показано промывание бронхиального дерева теплым изотоническим раствором натрий хлорида и неконцентрированным раствором антибиотиков. С этой целью широко используют (при наличии чувствительности к ним микрофлоры) аминогликозиды, фторхинолоны и др.
На фоне систематической ФБС показано периодическое или постоянное дыхание с сопротивлением на выдохе. Часто рутинно применяемый выдох в трубку, погруженную в воду, недостаточен. Более эффективно периодически надувать резиновую игрушку. В случаях тяжелого состояния больного необходимо применение
Бронхиальная астма 107
вспомогательной вентиляции неинвазионными методами. С этой целью можно использовать при отсутствии современной дыхательной аппаратуры ручной дыхательный аппарат (типа «Полинаркон»).
При симптомах гипоксемии показана оксигенотерапия. Поскольку патогенетическими механизмами гипоксемии при пневмонии часто является нарушение вентиляционно-перфузионных соотношений и диффузии в легких, ингаляция килорода может повысить его напряжение в крови.
При развитии таких осложнений, как абсцедирование, пиопневмоторакс с эмпиемой плевры, к лечению необходимо привлекать торакального хирурга. В случаях тяжелого течения, особенно при деструкции легочной ткани, подозрении на анаэробный характер инфекции показаны прямые антикоагулянты (лучше низкомолекулярные гепарины), препараты, улучшающие реологию крови. При этом дозировка антикоагулянтов требует особой осторожности, поскольку нельзя исключить
при деструкции кровохарканья и легочного кровотечения.
Внутривенные инфузии жидкости должны проводиться по показаниям. Следует поддерживать нормальный водно-электролитный баланс, избегать как обезвоживания, так и гипергидратации. И то и другое состояние неблагоприятно для легких. Для восполнения жидкости более предпочтительным является энтеральный ее прием. Важное значение имеет адекватное полноценное питание.
Следует широко использовать постуральный дренаж, физио- и аэрозолетера-пию, прием отхаркивающих средств, при явлениях бронхоспазма — ингаляционные Р-адреномиметики, эуфиллин.
Показания к переводу на ИВЛ у больных с пневмонией не отличаются от общих. Единственной особенностью является то, что ИВЛ будет показана даже при небольшой степени гиперкапнии, свидетельствующей о декомпенсации дыхания [3].
4.3. БРОНХИАЛЬНАЯ АСТМА
Бронхиальная астма (БА) — хроническое воспалительное заболевание дыхательных путей. В США БА болеют 14—15 млн человек, ежегодно умирают более 5 тыс. страдающих этой болезнью [28]. Распространенность данного заболевания постоянно возрастает, почти у 10 % населения земного шара наблюдался хотя бы эдин приступ [29].
Классификация
БА классифицируется по этиопатогене-зу (атопическая, инфекционная, смешанная
форма) и по степени тяжести (табл. 4.2). Анестезиологи чаще всего сталкиваются с тяжелой формой БА у больных.
4.3.1. Патогенез
Фиброоптическая бронхоскопия с лаважем и биопсией позволили прояснить некоторые моменты в патогенезе БА. Исследования показали, что мастоциты, эозинофилы, эпителиальные клетки, макрофа
Таблица 4.2. Классификация бронхиальной астмы по выраженности симптомов [30]
Тяжесть Симптомы
Легкая форма с редкими приступами Появление симптомов реже одного раза в неделю, практически нормальная функция легких
Легкая форма с частыми приступами Появление симптомов чаще одного раза в неделю, но не ежедневно, практически нормальная функция легких
Средней тяжести с частыми приступами Ежедневная симптоматика с легкой или умеренной дисфункцией легких
Тяжелая форма Выраженная ежедневная симптоматика с умеренной или выраженной обструкцией дыхательных путей
108 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
ги и активированные Т-лимфоциты играют ключевую роль в развитии воспалительного процесса при БА [31] (рис. 4.1).
Эти клетки синтезируют медиаторы, непосредственно влияющие на нижние дыхательные пути или опосредованно — через нейрогуморальную регуляцию [32]. Субпопуляция Т-лимфоцитов (ТН2) играет важную роль в регуляции аллергического воспаления дыхательных путей, освобождая селективные цитокины и превращая болезнь в хроническую. Специфические адгезивные протеины, найденные в сосудистой ткани, легочном матриксе, бронхиальном эпителии, могут стать критическим фактором воспалительных изменений [33]. Освобождаемые медиаторы влияют на тонус гладких мышц бронхов, изменяют сосудистую проницаемость, стимулируют бронхиальную секрецию и вызывают характерные структурные изменения дыхательных путей: пролиферацию миофибробластов, накопление интерстициального коллагена в базальной мембране бронхиального эпителия и утолщение ее. Другие изменения включают гипертрофию и гиперплазию гладких мышц бронхов, увеличение подслизистых желез, нарушение соединительнотканного каркаса легких [34]. Быстродействующие медиаторы воспале
ния активируют хемотаксис, способствуют скоплению эозинофилов и нейтрофилов в слизистой оболочке бронхиального эпителия. Воспалительные клетки продуцируют большое количество провоспалительных цитокинов, включая интерлейкины (ИЛ-2, ИЛ-6, ИЛ-8). Усиливается дифференциация В-лимфоцитов в IgE, которые способствуют освобождению длительно действующих медиаторов, в частности провоспа-лительной нейропептид ной субстанции Р. Тормозится образование вазоактивного интестинального пептида, который может служить нейротрансмиттером неадренергической бронходилатации [35]. БА вызывается не каким-то одним фактором, а является, скорее всего, результатом интеракции нескольких воспалительных клеток, медиаторов и ткани дыхательных путей.
Воспаление бронхиального эпителия вызывает гиперчувствительность дыхательных путей, бронхоконстрикцию, отек бронхиального эпителия, образование слизистых пробок. Это, в свою очередь, приводит к нарушению проходимости нижних дыхательных путей и вентиляции. Атопия, генетическая предрасположенность к развитию IgE-зависимого ответа на общие аэроаллергены являются факторами, способствующими заболеванию БА.
Воспаление
Острое ----► Подострое ---► Хроническое
Рис. 4.1. Клеточные механизмы воспаления дыхательных путей [28]
Бронхиальная астма 109
Возникающая гиперреактивность дыхательных путей проявляется бронхиоло-спазмом на различные аллергены, неспецифические раздражители, вирусную инфекцию, холодный воздух, физическую нагрузку, эмоциональный стресс, гастро-эзофагальный рефлюкс, механическую ирритацию нижних дыхательных путей (например, катетером), острые расстройства функции сердца.
Нарушение вентиляции при БА вызывают следующие факторы:
1. Бронхиолоспазм. С увеличением генерации в дыхательных путях возрастает количество гладких мышц. Их иннервация происходит за счет автономной нервной системы. В стенках бронхов и бронхиол расположено большое количество мускариновых холинорецепторов, стимуляция которых повышает тонус гладкой мускулатуры бронхов, и Р2’аДРенеРгических рецепторов, обуславливающих бронходилатацию, в то время как cq- адренорецепторы тормозят этот процесс [36].
Аллерген-индуцируемый острый бронхоспазм возникает в результате высвобождения мастоцитами IgE-зависимых медиаторов: гистамина, триптазы, лейкотриенов и простагландинов [37].
Неаллергический бронхоспазм может развиваться на аспирин вследствие блокирования циклооксигеназного пути метаболизма арахидоновой кислоты и активации липооксигеназного пути с образованием лейкотриенов. Механизм бронхоспазма на ингаляцию холодного воздуха и физическую нагрузку не совсем ясен, но доказано, что он носит воспалительный характер [38]. Возможно, в этих случаях играет роль стрессовая реакция. Острый бронхоспазм является главной причиной нарушения проходимости нижних дыхательных путей только в начальный короткий период времени (от 30 до 60 мин). Затем на первый план выходят отек и воспаление слизистой оболочки нижних дыхательных путей [39].
2. Отек нижних дыхательных путей. Вследствие действия провоспалительных медиаторов увеличивается проницаемость капиллярного эндотелия и развивается отек слизистой оболочки бронхов. Даже
без бронхоконстрикции это может привести к серьезному нарушению просвета дыхательных путей.
3. Образование слизистых пробок. При остром приступе БА в результате увеличения количества и вязкости секрета в просвете бронхиол и мелких бронхов образуются слизистые пробки.
4. Нарушение структуры нижних дыхательных путей. У многих пациентов с БА нарушения проходимости дыхательных путей носят необратимый характер. Причина морфологических нарушений не совсем понятна, возможно, она связана со структурными изменениями матрикса дыхательных путей вследствие воспаления, нарушения обмена коллагена. Это является основной причиной, затрудняющей терапию БА у некоторых больных.
Иммуногистопатологические признаки БА включают:
— денудацию (оголение) бронхиального эпителия;
— нарушение структуры коллагена базальной мембраны бронхиального эпителия;
— отек бронхиального эпителия;
— активацию мастоцитов;
— инфильтрацию воспалительных клеток (нейтрофилов, эозинофилов, лимфоцитов).
4.3.2. Диагностика бронхиальной астмы
Диагностика БА должна основываться на клинических, рентгенологических, бронхоскопических и лабораторных показателях.
Клинически БА проявляется периодически возникающими приступами экспираторной одышки, сменяющимися улучшением состояния. Обязательно должен быть исключен альтернативный диагноз. Симптоматика может обостряться в случаях: физической нагрузки, вирусной инфекции, контакта с животными, плесенью, вдыхания табачного дыма, пыльцы растений, резкого изменения погоды, эмоциональных потрясений, месячных.
Очень зажным диагностическим методом является спирометрия. Типичным для
110 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
обструкции нижних дыхательных путей является снижение соотношения объема форсированного выдоха за 1 с (ОФВ^ к форсированной жизненной емкости легких (ФЖЕЛ) ниже 65 %, в случае нормального показателя ОФВ^ФЖЕЛ и снижения ФЖЕЛ можно предполагать рестриктивный процесс в легких. Для БА характерно повышение O®Bt не менее чем на 12 % после ингаляции быстродействующих бронходилататоров (American Thoracic Society, 1991). Характерным признаком обструкции нижних дыхательных путей является снижение показателя пикового экспираторного потока (PEFR) не менее чем на 20 % от нормальных значений (у взрослых PEFR менее 120—125 л/мин свидетельствует о тяжелой обструкции дыхательных путей [40]).
На начальных этапах обострения БА наблюдается гипоксемия и нормо- или гипокапния. Снижение напряжения кислорода в артериальной крови вызвано интерстициальным отеком легких (вследствие значительного снижения внутриальвеоляр-ного давления на вдохе), уменьшением вентиляционно-перфузионных соотношений и множественным микроателектази-рованием вследствие обтурации мелких бронхов и бронхиол, а, следовательно, и шунтированием. Гиперкапния развивается только при декомпенсации функции внешнего дыхания и свидетельствует об очень тяжелом течении БА.
БА следует дифференцировать от следующих заболеваний: эпиглоттит; нарушение функции голосовых связок; специфическое и неспецифическое стенозирование гортани, трахеи, главных бронхов (постинтубационное, опухоль, острая вирусная инфекция, туберкулез и др.), наличие инородного тела в крупных дыхательных путях, экспираторный стеноз трахеи и главных бронхов. Большинство этих состояний диагностируется с помощью ФБС. Трудности возникают при дифференциальной диагностике БА с тромбоэмболией легочной артерии (ТЭЛА), так как последняя часто также осложняется брон-хиолоспазмом. В этих случаях может помочь длительный анамнез заболевания (хотя нельзя исключить ТЭЛА на фоне
длительного заболевания БА), а в случаях невозможности дифференциального диагноза — применять антикоагулянтную и тромболитическую терапию.
В ОИТ в основном лечат больных с тяжелым обострением БА (астматическое состояние), сопровождаемым гипоксемией и гиперкапнией.
Тяжелое обострение БА клинически проявляется:
— чувством нехватки воздуха у больного;
— цианозом кожных покровов;
— резко выраженной экспираторной одышкой с участием вспомогательной мускулатуры, шумными хрипами при дыхании;
— тахикардией, артериальной гипертензией;
— при аускультации зоны отсутствия дыхания чередуются с большим количеством преимущественно сухих хрипов с удлиненным выдохом.
4.3.3. Лечение обострения бронхиальной астмы
Ингаляция р2-агонистов. Применение р2-агонистов (сальбутамол, альбутерол, метапротеренол) является основой терапии, поскольку, действуя на р2-адреноре-цепторы, они снижают тонус бронхиальной мускулатуры, уменьшают продукцию медиаторов воспаления. При этом наблюдаются побочные эффекты: тахикардия, мышечный тремор, гипокалиемия, гипергликемия, ишемия миокарда (хотя и описана, но встречается редко), возможно некоторое снижение РаО2 вследствие увеличения шунтирования.
Терапию рекомендуется начинать с ингаляции р2-агонистов каждые 20 мин. Если после трехкратной ингаляции эффект не получен — начинают терапию кортикостероидами, а ингаляцию р2-агонистов повторяют через час [41 ]. В тяжелых случаях возможна непрерывная ингаляция альбутерола до 15 мг/ч в течение 2 ч [40].
Подкожное и внутреннее применение р-агонистов менее эффективно, чем ингаляционное. В случаях длительного неку-пирующегося приступа может быть полез
Бронхиальная астма 111
ным подкожное введение адреналина (0,3 —0,5 мг) или тербуталина (0,25 мг). Особенно опасно подкожное применение р2-агонистов больным с сопутствующей патологией сердца и сосудов.
Недостаточно доказательств для рекомендации использования внутривенных р2-агонистов у пациентов с тяжелой острой астмой. Более того, клиническая эффективность их применения кажется сомнительной, в то время как потенциальный клинический риск очевиден. Единственные показания для внутривенного применения р2-агониста существуют у тех пациентов, у которых ингаляционная терапия не выполнима (Travers с соавт., 2002).
Внутривенное введение кортикостероидов. Согласно рекомендации Национальной программы по астме (США, 1991) применяют такие дозы кортикостероидов: метилпреднизолон — 80 —125 мг внутривенно болюсно, затем по 80 мг каждые 6 — 8 ч или гидрокортизон — 2 мг/кг внутривенно болюсно, затем по 2 мг/кг каждые 8 часов.
Нет достоверных данных о преимуществах применения при БА того или иного кортикостероидного препарата, однако, учитывая более выраженный противовоспалительный эффект метилпреднизолона, возможно, следует отдать ему предпочтение. После стабилизации состояния следует перейти на прием метилпреднизолона внутрь (ориентировочная доза — 60 мг в день).
Несмотря на то что применение глюкокортикоидов является наиболее распространенным видом терапии при неэффективности р2'агонистов> некоторые исследователи высказывают сомнения относительно результативности такой терапии [42, 43]. Отмечается много побочных эффектов при длительном (более месяца) применении кортикостероидов, в частности, при использовании больших доз развиваются психотические реакции [44]; миопатии (особенно в сочетании с миорелаксантами); нарушения водно-электролитного обмена и др.
В последнее время появляется все больше данных о благоприятном эффекте лечения ингаляционными кортикостероида
ми. Так, S. F. Lanes с соавт. отмечают, что регулярное использование ингаляционных стероидов уменьшает риск смерти от астмы, а чрезмерное использование р-агони-стов короткого действия — заметно увеличивает риск смерти от астмы.
Ipratropium bromide — ингаляционный антихолинергический препарат, используется в качестве дополнительного средства в сочетании с р2-агонистами, усиливает бронхорасширяющий эффект последних. Доза — 0,5 мг через 2 —6 ч в зависимости от тяжести состояния. Комбинация его с р2-агонистами статистически достоверно повышала ОФВ1 и максимальную скорость выдоха [45].
Аминофиллин (эуфиллин) является наиболее распространенным бронходила-татором в мире. Относится к ингибиторам фосфодиэстеразы — фермента, расщепляющего цАМФ в адренергическом рецепторе. Бронходилатирующий эффект менее выражен, чем у р2-агонистов, а побочные эффекты — более выражены. Кроме того, к отрицательным свойствам этого препарата следует отнести необходимость постоянного мониторинга концентрации препарата в крови, что требует дополнительных расходов и усилий персонала. Начальная доза — 5 — 6 мг/(кг • ч), затем 0,5 —0,8 мг/(кг • ч). Концентрацию препарата в сыворотке крови следует поддерживать на уровне 8—12 мг/л.
В связи с невозможностью обеспечить постоянный мониторинг концентрации препарата в сыворотке крови часто возникают токсические реакции на эуфиллин. По частоте интоксикации среди всех лекарственных отравлений и летальности эуфиллин занимает пятое место в мире [46]. Клинически интоксикация проявляется при концентрации в сыворотке крови 20 мг/л и становится выраженной при концентрации 30 мг/л. Симптоматика включает тахикардию, тремор, тошноту, диарею, нарушение сознания, в более тяжелых случаях — артериальную гипотензию, нарушение ритма сердца, генерализованные судороги, кому, фибрилляцию желудочков сердца [47].
Лечение интоксикации эу фи л лином: — прекращение введения препарата;
112 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
— прием активированного угля внутрь по 20 г каждые 2 часа (лучше на 75 мл 70 % сорбитола [47]);
— в тяжелых случаях применение экстракорпоральных методов очищения;
— симптоматическая терапия: противосудорожные (тиопентал-натрий), селективные рр блокаторы при тяжелых нарушениях ритма сердца; артериальная гипотензия, вызванная передозировкой эуфиллина, может быть рефрактерна к вазоконстрикторам.
Муколитическая терапия. Наиболее часто используют N-ацетилцистеин и ам-броксол. N-Ацетил цистеин не следует вводить непосредственно в трахею, так как в результате своей гипертоничности он может спровоцировать бронхорею [20].
Использования N-ацетилцистеина в аэрозолях лучше избегать, так как, оказывая раздражающий эффект на слизистую нижних дыхательных путей, он может спровоцировать кашель, бронхоспазм, рвоту [20].
Применение антагонистов лейкотриенов. Как указывалось выше, лейкотриенам отводится определенная роль в патогенезе БА. Применение ингибитора лейкотриенов — зафирлукаста (по 20 мг дважды в сутки) уменьшало выраженность симптоматики, увеличивало время ремиссии и уменьшало дозу р2-агонистов [48]. Однако эффективность препаратов этой группы при тяжелом обострении процесса не определена.
Инфузия раствора натрий гидроген-карбоната (соды). Имеются данные о благоприятном эффекте относительно отхождения мокроты и динамики патологического процесса при приеме в дозе 1 — 2 ммоль/кг, в частности у детей [49].
На сегодняшний день не совсем ясна роль ингаляции анестетиков. Ингаляционная анестезия, например, галотаном, может быть полезной при лечении рефрактерных случаев астмы, однако ее применение требует осторожности, так как она может представлять опасность в связи с недостаточной мощностью потока в большинстве респираторов, предназначенных для ингаляционной анестезии [50].
Ингаляция кислорода и гелиево-кис-лородной смеси. Наиболее частой при
чиной смерти у больных с БА является гипоксемия [51], поэтому при тяжелых обострениях БА показана ингаляция кислорода. Ингаляция гелиево-кислородной (1 : 4 и 1 : 3) смеси повышает максимальную скорость выдоха, облегчает общее состояние больного по сравнению с дыханием комнатным воздухом [52].
Применение магния. Есть данные (В. Levin с соавт., 2002), что применение 2 г магний сульфата внутривенно улучшает легочную функцию при использовании в качестве дополнительного средства к стандартной терапии у пациентов с очень тяжелой острой астмой.
Ряд исследований [53] показали эффективность внутривенного и ингаляционного применения раствора магний сульфата у больных с тяжелой обструкцией дыхательных путей.
Перевод на ИВЛ. В настоящее время большинство специалистов (М. Sydow, 2003 и др.) соглашаются, что вспомогательное дыхание должно обеспечиваться прежде всего неинвазивной вентиляцией (НИВ) через лицевую маску. Однако у многих пациентов с тяжелой астмой существуют противопоказания для НИВ или этот метод недостаточен для обеспечения вентиляции. В этом случае необходима эндотра-хеальная интубация и инвазивная ИВЛ. Интубация и вентиляция пациентов с тяжелой астмой или астматическим статусом связаны с высоким риском осложнений по сравнению с пациентами с другой патологией. Поэтому риск инвазивной ИВЛ должен быть тщательно взвешен с оценкой возможности продолжения консервативной терапии и НИВ.
Показания к переводу на ИВЛ при БА мало отличаются от общепринятых при ОНД [54]:
— апноэ;
— прогрессирующая гиперкапния (50 — 60 мм рт. ст.);
— нарушения сознания;
— отсутствие эффекта от интенсивной терапии.
Тяжелая гипоксемия, несмотря на ингаляцию О2 и НИВ, является абсолютным показанием для интубации и ИВЛ. Увеличение РяСО2 с умеренным дыхатель
Бронхиальная астма 113
ным ацидозом само по себе не является показанием для ИВЛ. Однако прогрессирующее повышение РяСО2 или развитие тяжелого метаболического ацидоза после одного часа НИВ — это повод обсудить необходимость перевода больного на ИВЛ. Другие критерии — наличие недостаточности сердца с падением сердечного выброса, развитие аритмии, пневмодиасти-нума или пневмоторакса {должны дренироваться перед переводом на ИВЛ!) (М. Sydow, 2003).
Определение момента перевода больного с БА на ИВЛ связано с большими трудностями для анестезиолога, так как на фоне хронического нарушения функции внешнего дыхания, иногда частых приступов, бывает нелегко определить экстренность вентиляционной поддержки. К. Sykes и J. Young [3] рекомендуют постоянный мониторинг РаО2 и советуют при острой гиперкапнии, а также нарушении функции ЦНС незамедлительно применять вентиляторную поддержку.
Проведение ИВЛ у больных с БА также затруднено вследствие повышенного динамического сопротивления дыханию и высокого давления в дыхательном контуре. Кроме того, в результате затрудненного выдоха и внутреннего сопротивления выдоху развивается гиперинфляция (пере-раздувание) альвеол. Все это может привести к снижению сердечного выброса и артериальной гипотензии. Для уменьшения этих явлений снижают дыхательный объем (Vt= 5 мл/кг) и увеличивают время выдоха (1 : 3). Вследствие высокого инспираторного давления и гиперинфляции альвеол у больных с БА, находящихся на ИВЛ, значительно повышается риск баротравмы легких и развития напряженного пневмоторакса.
Некоторые авторы (G. М. Mutlu с со-авт., 2002) относительно безопасной стратегией респираторной поддержки при бронхиальной астме считают допустимую гиперкапнию. Высокий уровень гиперкапнии и связанный с ней ацидоз хорошо переносятся больными при отсутствии противопоказаний (например, предшествующая внутричерепная гипертензия) [51].
В лечении рефрактерных случаев астмы может быть полезной ингаляционная анестезия, однако ее следует применять с осторожностью, так как в связи с недостаточной мощностью потока в большинстве респираторов, предназначенных для ингаляционной анестезии, она может представлять опасность.
Проведение ИВЛ у больных с БА требует больших физических усилий персонала ОИТ, больного необходимо постоянно поворачивать с боку на бок (рекомендаций о перевороте больного с БА на живот в литературе мы не обнаружили, хотя несколько раз успешно применяли этот прием) для улучшения дренирования нижних дыхательных путей, применять частые регулярные аспирации мокроты с заливкой небольшого количества теплого изотонического раствора натрий хлорида.
Результаты нескольких рандомизированных контролируемых исследований свидетельствуют о снижении летальности у больных с бронхиальной астмой, находящихся на ИВЛ, при использовании методики допустимой гиперкапнии [55, 56].
Иммунотерапия
Показания к иммунотерапии [28]:
1) наличие четкой связи между попаданием в организм аллергена и обострением клинической симптоматики;
2) частые обострения заболевания (на протяжении всего года или большей его части);
3) низкая эффективность фармакотерапии.
Иммунотерапию проводят только после выведения больного из тяжелого состояния.
Факторы риска летального исхода при БА:
♦ частые тяжелые обострения с лечением в ОИТ;
♦ ИВЛ в анамнезе в связи с ОНД;
♦ госпитализация по поводу астмы за последний год более двух раз;
♦ использование р2"агонистов в больших дозах (более двух ингаляторов в месяц);
♦ тяжелые сопутствующие заболевания со стороны сердца, легких;
♦ низкий социальный статус;
♦ непереносимость противоастматических препаратов.
114 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
4.4. СИНДРОМ ОСТРОГО ПОВРЕЖДЕНИЯ ЛЕГКИХ И ОСТРЫЙ РЕСПИРАТОРНЫЙ ДИСТРЕСС-СИНДРОМ
Синдром острой недостаточности дыхания, или острый респираторный дистресс-синдром (ОРДС), — одна из главных причин летальности в О ИТ. Согласно данным разных авторов, частота ОРДС составляет от 1,5 • 105 до (4,8 —8,3) • 105 случаев в год [57, 58]. Исследования, проведенные в странах Северной Европы [59], показали, что частота ОРДС составляет 13,5 случая на 100 тыс. населения и диагностируется у 18 % больных на ИВЛ. Другие авторы [60] обнаружили, что частота ОРДС в ОИТ составляет 6,9 %, а среди всех больных на ИВЛ — 31,3 %.
Многочисленные исследования свидетельствуют о высоком уровне летальности при ОРДС — 50 — 60 % [60, 61]. Смертность, связанная с синдромом острого повреждения легких (СОПЛ) и ОРДС, в Соединенных Штатах Америки сопоставима со смертностью от ВИЧ-инфекции, рака груди и астмы [62]. Однако в последнее время отмечается значительное повышение выживаемости при ОРДС - до 65 % [63].
Синдром острой недостаточности дыхания был описан еще в 60-х годах XX в. под разными названиями, наиболее распространенными из которых были: шоковое легкое, влажное легкое, синдром недостаточности альвеолярно-капиллярной мембраны, постперфузионное легкое и др. Ближе всех к современной трактовке это
го заболевания подошли D. G. Ashbaugh с соавт. [64], описавшие этот синдром в 1967 г. как респираторный дистресс-синдром взрослых (РДСВ). Это название было предложено для того, чтобы отличить его от респираторного дистресс-синдрома новорожденных, главной причиной которого является недостаточность функции сурфактанта. Однако синдром острой недостаточности дыхания наблюдался не только у взрослых. Кроме того, это понятие не включало менее выраженные нарушения дыхания у некоторых больных, поэтому на Американо-Европейской согласительной конференции по ОРДС, состоявшейся в 1998 г., были рекомендованы следующие определения ОРДС и СОПЛ (табл. 4.3).
Главными критериями ОРДС являются тяжелая гипоксемия (РаО2/FiO2 < < 200 мм рт. ст.) при наличии острого начала, этиологического фактора (не обязательно), отсутствие как первопричины кардиогенного отека легких (ДЗЛА > > 18 мм рт. ст.) и наличие характерных рентгенологических изменений в легких. Как правило, при ОРДС снижается растяжимость легких и увеличивается внутри легочное шунтирование.
Термин ОРДС сейчас следует считать общепринятым в мировой литературе, и им следует пользоваться при описании данного синдрома. Поскольку ОРДС пред
Таблица 4.3. Различие и сходство между ОРДС и СОПЛ [65]
Признак СОПЛ ОРДС
Начало заболевания Рентгенологические данные Давление заклинивания в легочной артерии PaO2/FiO2 Острое Двусторонние инфильтр ной рентгенограмме лен < 18 мм рт. ст. или отсу признаков гипертензии . < 300 мм рт. ст. Острое аты в легких на обзор-сих тствие клинических левого предсердия | < 200 мм рт. ст.
Синдром острого повреждения легких и острый респираторный дистресс-синдром 115
полагает тяжелую степень ОНД, был выделен синдром острого повреждения легких, который можно считать ранней стадией ОРДС, но которая совсем не обязательно разовьется в ОРДС [66].
Факторы, способствующие развитию ОРДС:
сепсис;
политравма;
аспирация желудочного содержимого с pH < 2,5;
шок;
внутричерепная гипертензия;
пневмония;
контузия легких;
эмболия малого круга кровообращения;
ожоги;
панкреатит;
массивные гемотрансфузии;
гипероксическое повреждение легких; отравления;
ингаляция токсичных газов;
утопление;
ДВС-синдром;
искусственное кровообращение.
И этим, по-видимому, не исчерпывается список патологических состояний, которые могут осложниться ОРДС. Наиболее частыми причинами ОРДС являются сепсис и пневмония (44 %), шок (15 %), травма (11 %), желудочная аспирация (10 %), другие причины (34 %) [67].
У умерших были значительно выше оценки по шкалам APACHE, SOFA и McCabe. Главные причины смерти: синдром полиорганной недостаточности, сепсис и септический шок; рефрактерная гипоксемия возникала редко. Независимыми факторами смерти были: наличие дисфункции органов на 3-й день, PtzO2/FzO2 на 3-й день и оценка по McCabe [65].
Вероятность развития ОРДС при некоторых состояниях и летальность при ОРДС представлены в табл. 4.4.
На Американо-Европейской конференции было выделено два вида ОРДС: прямой (первичный, легочный) — при непосредственном воздействии повреждающего агента (контузия, аспирация, ингаляция токсичных газов) и непрямой (вторичный, экстрапульмональный) — как
Таблица 4.4. Предрасполагающие факторы и летальность при ОРДС (по L. D. Hudson с соавт., 1995)
Фактор риска Вероят-ность ОРДС, % Летальность, %
при ОРДС без ОРДС
Сепсис 41 69 50
Массивные
трансфузии 36 70 35
Контузия легких 22 49 12
Аспирация 22 48 21
Множественные
переломы И 49 9
Другие причины 26 62 19
результат синдрома системного воспалительного ответа (сепсис, неторакальная травма и др.).
4.4.1. Патогенез
Согласно современным представлениям, ОРДС считают диффузным воспалительным процессом в легких в ответ на действие повреждающего фактора. Очень много исследований посвящено изучению этио-патогенеза ОРДС. Большинство исследователей полагают, что ОРДС — это проявление синдрома системного воспалительного ответа и полиорганной недостаточности в легких. Ведущую роль при этом играют нейтрофилы и легочный эндотелий. Уже в начальной стадии ОРДС в легких обнаруживают секвестрацию нейтрофилов и лимфоцитов. Этот феномен объясняют повышением хемотаксической активности и действием адгезивных молекул. Склеивающие (adherence) рецепторы CD 11/18 нейтрофилов соединяются с адгезивными рецепторами легочного эндотелия ELAM и ICAM. Определенную роль в этом процессе играет L-selectin [68]. Внутриклеточные гранулы нейтрофилов содержат большое количество лизоцима, эластазы, коллагеназы, миелопероксидазы, повреждающих экстрацеллюлярный матрикс и базальную мембрану, что вызывает активацию целого ряда факторов воспаления. Активация нейтрофилов и макрофагов
116 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
приводит к высвобождению большого количества провоспалительных цитокинов (ИЛ-1, ИЛ-6, ИЛ-8, TNF, интерферона-а).
Активированные нейтрофилы, по-видимому, играют главную роль в развитии большинства случаев острого повреждения легких (ОПЛ). В экспериментальных моделях устранение нейтрофилов заметно уменьшало тяжесть ОПЛ [69]. В нейтрофилах, накапливающихся в легких при моделировании ОПЛ, зафиксирована активация киназ Akt и р38, накопление в ядре фактора, регулирующего транскрипцию ядерного фактора каппа В (NF-kB). Характерно также уменьшение апоптоза нейтрофилов в легких. Ингибиция р38, Akt или активация NF-кВ в эксперименте уменьшает тяжесть индуцированного эндотоксином или кровотечением СОПЛ/ ОРДС. Эти механизмы у критических больных пациентов нуждаются в дополнительных исследованиях. Модуляция активации р38, Akt и NF-кВ в нейтрофилах, по-видимому, должна быть терапевтической целью у тяжелобольных пациентов с СОПЛ/ОРДС [70].
Одним из этапов системной воспалительной реакции является активация комплемента, особенно его фракций С3а и С5а, обладающих мембраноповреждающим эффектом. В развитии некардиогенного отека легких важную роль играет интег-рин-индуцированная активация фактора TGFp, что доказывается успешными экспериментами по его ингибированию [70].
При ОРДС нарушаются продукция и метаболизация катехоламинов, серотонина, простагландинов эндотелием. Вследствие нарушения активности ангиотензин-конвертирующего энзима, локализованного на поверхности легочного эндотелия, увеличивается концентрация ангиотензина и брадикинина, что приводит к перфузионным расстройствам в легких [71].
Также нарушается функция сурфактанта, в частности, баланс между альвеолярными макрофагами и сурфактантным протеином А. Если в результате оксидантных повреждений этот белок инактивируется, макрофаги начинают продуцировать супероксидные радикалы, активируется пе-
роксидное окисление липидов при взаимодействии миелопероксидазы нейтрофилов с лактоферином. Это происходит на фоне снижения активности антиоксидантной системы легких (глутатиона, суперок-сиддисмутазы, каталазы), что увеличивает риск свободнорадикального повреждения легких [72].
Некоторые цитокины (ИЛ-1, TNF, ин-терферон-у) индуцируют синтез NO альвеол оцитами II, что нарушает механизм гипоксической вазоконстрикции и увеличивает шунтирование крови в малом круге.
Внутрисосудистый тромбоз, или диссеминированная внутрисосудистая коагуляция, также характерен для острого повреждения легких. Внесосудистое осаждение фибрина способствует дисфункции легкого и острой воспалительной реакции. Кроме того, транзитный фибрин в альвеолярном пространстве подвергается ремоделированию, что способствует ускоренному легочному фиброзу, подобному тому, который происходит при заживлении раны. При СОПЛ/ОРДС осаждение фибрина в альвеолах приводит к последовательным изменениям эндогенной коагуляции и механизмов фибринолиза. Прокоагулянтный эффект усиливается ингибицией фибринолитической активности в альвеолярном пространстве. Инициирование прокоагулянтной реакции происходит в результате локальной суперэкспрессии тканевого фактора, связанного с фактором VII.
Бронхоальвеолярный тканевой фактор вызывает генерацию тромбина и локальную депрессию фибринолиза, индуцируемого активатором урокиназы и плазминогена, путем увеличения активности ингибитора активатора плазминогена.
Эти и другие аналогичные наблюдения привели к имплементации антикоагулянтов или фибринолитических стратегий для профилактики ОПЛ. Успех новых фибринолитических стратегий свидетельствует, что подобный подход можно использовать для предотвращения ускоренного легочного фиброза, развивающегося при СОПЛ/ОРДС [73].
Описанные патологические процессы не объясняют полностью все виды ОРДС. Несмотря на большое количество работ, посвя
Синдром острого повреждения легких и острый респираторный дистресс-синдром 117
щенных этому вопросу, до сих пор не обнаружен ключевой фактор развития ОРДС (если таковой существует). Скопление и секвестрация нейтрофилов в легких, определяемые, как правило, при ОРДС, у больных с лейкопенией могут отсутствовать [73].
В последнее время большое внимание уделяется генетической предрасположенности отдельного индивидуума к развитию СОПЛ/ОРДС [74]. Врачи давно заметили, что пациенты по-разному реагируют на лекарственную терапию и имеют разный риск различных осложнений. Получены некоторые свидетельства того, что клеточные и гуморальные иммунные реакции подчинены полиморфному генетическому контролю, что объясняет известную вариабельность клинических проявлений и результатов лечения у критически больных пациентов с одинаковой патологией. Как обнаружили ученые, нарушение экспрессии некоторых генов в ответ на повреждение легкого и его восстановление и восприимчивость к острому повреждению легких являются наследственными. Недавние исследования показали, что индивидуальный риск и клеточные реакции могут обуславливаться ДНК каждого индивидуума.
4.4.2. Патоморфология
В результате патофизиологических процессов нарушается структура легочной ткани. Структурный ответ на воспаление при ОРДС можно разделить на несколько фаз [75J:
1. Ранняя экссудативная (24 — 96 ч): альвеолярный и интерстициальный отек;
застой в капиллярах;
деструкция альвеолоцитов I типа; формирование гиалиновых мембран.
2. Ранняя пролиферативная (3—10 дней): увеличение количества альвеолоцитов II типа;
инфильтрация межклеточных перегородок;
образование гиалиновых мембран.
3. Поздняя пролиферативная (7 — 10 дней):
пневмосклероз.
Рис. 4.2. Микроскопические изменения в легких на ранней стадии ОРДС:
А — альвеолы; ЭЦ — эритроциты [20]
На фотомикрограмме (рис. 4.2) представлены патогистологические изменения в легких при ранней фазе ОРДС.
Одним из наиболее точных, на наш взгляд, является определение ОРДС К. Weling [76]: ОРДС — легочный ответ на локальную или системную гипоксию тканей, их ишемию и реперфузию — является динамическим патологическим процессом с многофакторной этиологией. Воспалительный процесс в легких связан с многими факторами: активацией полиморфноядерных нейтрофилов, эндотелио-цитов, продукцией свободных кислородных радикалов и др. В патогенезе основную роль играет некорригируемый отек легких вследствие повреждения альвеолярно-капиллярной мембраны.
4.4.3. Клинические проявления
В течение одних-двух суток после начала действия этиологического фактора развивается тяжелая ОНД с прогрессирующими симптомами гипоксемии, одыш
118 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
кой с участием вспомогательной мускулатуры, в легких выслушиваются разнокалиберные хрипы, снижается показатель PflO2/FiO2 и растяжимость легких, увеличивается шунтирование в малом круге. При неэффективности терапии или ее отсутствии развиваются расстройства функции ЦНС, выраженная тахикардия, артериальная гипер-, а затем и гипотензия.
На рентгенограмме легких симптоматика может запаздывать по сравнению с клиникой, но позже появляются застойные явления в сосудах легких, двусторонние инфильтраты, на конечных стадиях ОРДС они носят сливной характер с очагами пневмосклероза. С помощью компьютерной томографии подтверждается наличие диффузных легочных инфильтратов, дис-и микроателектазы, снижение объема функционирующей легочной паренхимы.
На основании только данных компьютерной томографии и рентгенографии легких сложно установить диагноз ОРДС, однако эти исследования очень важны в динамике для оценки течения патологического процесса. Необходим также постоянный мониторинг газового состава крови и кислотно-основного состояния.
4.4.4. Лечение острого респираторного дистресс-синдрома
В лечении ОРДС акцент следует сделать на этиологическом факторе. Без санации септического очага и проведения адекватной антибиотикотерапии гораздо сложнее бороться с ОРДС при сепсисе. Быстрое выведение из шока, устранение гипоксии тканей (в том числе и легочной), ранняя хирургическая стабилизация переломов костей являются профилактикой и лечением возникшего при политравме ОРДС.
Как уже указывалось, отсутствие ключевого фактора патогенеза ОРДС делает проблематичной патогенетическую терапию. Несмотря на предпринимаемые попытки терапевтически прервать неспецифическую воспалительную реакцию организма на действие неблагоприятного фактора, в настоящее время применение моноклональных антител к факторам воспаления и их рецеп
торам и других аналогичных средств оказалось не эффективным в лечении ОРДС [77].
Поскольку мы пока ограничены в методах терапевтического воздействия непосредственно на сам патологический процесс, вызывающий ОРДС, основной целью терапии является поддержание достаточного для жизни газообмена в легких [78, 79].
Вентиляторная стратегия при ОРДС. Механическая вентиляция, необходимая в большинстве случаев ОРДС (см. «ИВЛ»), в свою очередь, вызывает вентилятор-ас-социированное повреждение легких [80]. Недавние исследования на здоровых животных четко демонстрируют, что ИВЛ с высоким инспираторным давлением приводит к баротравме с нарушением механики легких, острому повреждению легочной паренхимы и ее отеку [81]. Однако такие же нарушения наблюдаются и при проведении ИВЛ в режиме низкого инспираторного давления, но с высоким дыхательным объемом [81]. Длительная ингаляция высоких концентраций кислорода также может усиливать повреждение легких при ОРДС [83]. Имеются данные [82], что полиморфноядерные лейкоциты могут быть активированы искусственной вентиляцией легких с освобождением эластазы. Этот процесс коррелировал с тяжестью системного воспалительного ответа и синдрома полиорганной недостаточности. Возможно, эти исследования помогут объяснить снижение смертности у больных с ОРДС при применении протекторной стратегии ИВЛ.
У больных с СОПЛ и ОРДС на ИВЛ с высоким Vt и низким положительным давлением в конце выдоха (ПДКВ) отмечается высокий системный и внутриальвео-лярный уровень цитокинов. Исследования ряда ученых [84, 85] показали, что применение протекторной вентиляторной стратегии, предусматривающей Vt < 6-7 мл/кг и безопасную гиперкапнию, улучшало функцию легких при ОРДС и способствовало снижению летальности при этой патологии. Этими же авторами была высказана гипотеза: поскольку синдром системного воспалительного ответа и поли-органная недостаточность играют ведущую роль в летальности при ОРДС, а протек
Синдром острого повреждения легких и острый респираторный дистресс-синдром 119
торная стратегия уменьшает летальность, то данная стратегия уменьшает и риск развития этих осложнений. В рандомизированных многоцентровых исследованиях [86, 87] сообщается о достоверном снижении летальности у больных с ОРДС, по отношению к которым применялась протекторная стратегия. В. Page с соавт. [82] утверждают, что после внедрения в практику данной стратегии больные с ОРДС умирают преимущественно не от недостаточности легких, а от тяжелых сопутствующих заболеваний.
Таким образом, при проведении ИВЛ у больных с ОРДС следует руководствоваться протекторной стратегией: обеспечивать адекватную оксигенацию при условии минимизации вентилятор-индуци-рованного повреждения легких (ВИПЛ), что предполагает низкий Vt (5 — 6 мл/кг) и ПДКВ = 10—15 см вод. ст. (980,7 — 1470 Па) [86]. Максимальные значения Pplateau следует поддерживать на уровне не более 30 — 35 см вод. ст. (2940 — 3430 Па), как рекомендует Американо-Европейская согласительная конференция по ОРДС 1998 г. [88].
Проспективные рандомизированные исследования подтверждают значительное снижение воспалительной реакции при протекторном режиме ИВЛ у больных с ОРДС [89]. При этом допустима безопасная гиперкапния. В нашей практике не удавалось поддерживать РаСО2 выше 50 мм рт. ст. — повышалось артериальное давление, погрессировала тахикардия, возникала необходимость введения больших доз седативных средств и миорелаксантов. Описано увеличение риска острой недостаточности почек при гиперкапнии у больных с ОРДС [90]. В. Pfeiffer с соавт. [91] сообщает об увеличении шунтирования в легких при гиперкапнии и поддержании РаО2 за счет увеличения сердечного выброса, но при отсутствии декомпенсации функции миокарда. На наш взгляд, применение ИВЛ в режиме гиповентиляции с безопасной гиперкапнией нуждается в дальнейших исследованиях.
Несмотря на менее повреждающий эффект на легкие, ограничение дыхательного объема может привести к распростра
ненному ателектазированию альвеол, снизить ФОЕ, способствовать прогрессированию гипоксемии. Описан риск повреждающего эффекта на легкие ИВЛ с низким дыхательным объемом (так называемое shearing повреждение в результате систематического коллабирования альвеол на выдохе и реинфляции на вдохе). Для профилактики этих осложнений применяют ПДКВ, режим плато, кинетическую терапию, инвертированный режим ИВЛ, а также метод, периодически восстанавливающий инфляцию альвеол (recruitment).
Значение и польза ПДКВ рассматривались выше. Особенно важное значение имеет применение этого режима при ОРДС. Однако при этом серьезной проблемой является определение оптимального уровня ПДКВ при ОРДС. То, что сопротивление на выдохе необходимо у больных с ОРДС не вызывает в настоящее время сомнений. Многочисленные исследования доказали эффективность этого режима ИВЛ (уменьшение шунтирования в легких, улучшение окисигенации), однако «золотой стандарт» уровня ПДКВ до сих пор не найден [92]. У клинических врачей возникают трудности с опре-делениием инспираторной кривой давление/объем, уровня растяжимости легких для оптимизации уровня ПДКВ [93]. Имеются данные [94] о целесообразности выбора уровня ПДКВ по насыщению кислородом венозной крови. Согласно рекомендациям N. S. Ward и М. М. Levy [92], для выбора оптимального уровня ПДКВ следует руководствоваться следующими критериями:
величина PaO2/FiO2; максимальная растяжимость легких; доставка кислорода;
величина шунта.
Дополнительными критериями служат: данные компьютерной томографии; импеданс правого желудочка сердца; соотношение мертвого пространства и дыхательного объема.
Во многих работах подчеркивается, что величина ПДКВ = 10 см вод. ст. (980,7 Па) является достаточной для профилактики ателектазов и улучшения оксигенации [95]. По-видимому, в большинстве случаев эту
120 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
цифру следует считать ориентиром уровня ПДКВ.
Есть данные о целесообразности применения инвертированного режима ИВЛ с увеличенным соотношением времени вдох/выдох (I/Е) в дыхательном цикле. По нашим данным [96], использование режима с соотношением I/E =1:1; 1,2 : 1 улучшало оксигенацию, несколько повышало растяжимость легких, однако применение этого метода требует дальнейших исследований. Так, A. Casetti с со-авт. [69] обнаружил, что увеличение времени вдоха повышает ВИП Л.
Для улучшения оксигенации можно воспользоваться режимом плато (с использованием отечественного респиратора «БРИЗ»). Поскольку во время плато на кривой (длится 0,2 —0,6 с) газ не поступает в легкие и не покидает их, то происходит перераспределение газа из хорошо вентилируемых в плохо вентилируемые альвеолы, что уменьшает шунтирование и улучшает оксигенацию. Не следует сочетать этот режим с инвертированным режимом ИВЛ.
Рекомендуется часто использовать восстанавливающий (recruitment) маневр с кратковременным (на 40 с) повышением давления в дыхательных путях до 40 см вод. ст. (3,3 кПа) [86]. Особенно эффективен этот маневр в положении больного на животе [97]. Есть данные о том, что у больных с первичным (легочная причина) ОРДС этот режим и ПДКВ менее эффективны, чем при вторичных ОРДС (сепсис, неторакальная травма и др.).
Мы использовали recruitment режим у восьми больных, и хотя при этом не у всех пиковое давление достигало 40 см вод. ст. (3920 Па), Vt увеличивалось до 12 — 14 мл/кг. Улучшение оксигенации после этого наблюдалось во всех случаях. Мы применяли этот маневр нерегулярно, так как до сих пор не совсем ясно, с какой частотой его следует использовать.
Как показали недавние исследования [98, 99], периодическое повышение до 35 — 40 см вод. ст. (3430 — 3920 Па) на 40 с с ПДКВ, на 2 см вод. ст. (196 Па) превышающим Pfiex (нижняя точка ин-флексии на кривой давление/объем), и
Vt < 6 мл/кг способствует повышению 28-дневной выживаемости до 62 %.
Хотя в многочисленных работах последнего десятилетия подчеркиваются преимущества использования для ИВЛ у больных с ОРДС респираторов, регулируемых по давлению, недавние контролируемые рандомизированные исследования не выявили таких преимуществ по сравнению с респираторами, регулируемыми по объему при условии Рplateau £ 35 СМ ВОД. СТ. (3430 Па) [100].
В ряде исследований [98, 99] при выборе параметров ИВЛ при ОРДС большое внимание уделено данным компьютерной томографии, с помощью которых можно осуществлять мониторинг регионарной вентиляции, выбирая оптимальный режим ИВЛ для данного больного в данный момент. Если на компьютерных томографических срезах представлены все отделы легкого от верхушек до основания, то можно провести количественный анализ или на целом легком, или на уровне доли. В зависимости от положения тела расположенные ниже доли вентилируются хуже, чем верхние, которые продолжают аэрироваться, несмотря на существенное увеличение в них консолидированной ткани легкого. Объем легких уменьшается, прежде всего, за счет нижних долей, которые сдавливаются снаружи сердцем и внутрибрюшным давлением, когда пациент находится в положении лежа на спине. У пациентов с фокальным компьютерным томографическим ослаблением фронтальная рентгенограмма легких, как правило, показывает двухсторонние затемнения в более нижних квадрантах, но может показаться и нормальной, особенно когда более нижние доли полностью ателектазированы. У пациентов с рассеянными компьютерными томографическими ослаблениями на типичных рентгенограммах наблюдаются так называемые белые легкие. Если эти пациенты лежат на спине, объем легких сохраняется за счет верхних долей и уменьшен в нижних долях, хотя гиповентиляция одинаково распределена между верхними и нижними долями.
Когда положительное внутригрудное давление применяется у пациентов с фо
Синдром острого повреждения легких и острый респираторный дистресс-синдром 121
кальным острым респираторным дистресс-синдромом, в плохо аэрированных и не-аэрированных регионах легких восстанавливается вентиляция вследствие того, что отделы легкого, обычно не вентилируемые при нулевом давлении на выдохе, имеют тенденцию быстро увеличиваться в объеме, повышая риск вентилятор-индуциро-ванного повреждения легких. Поэтому выбор оптимального уровня ПДКВ не должен зависеть только от оптимальной альвеолярной вентиляции. Следует сосредоточиться на ограничении чрезмерного раздувания легкого и сжатия лежащих ниже долей маневрами наподобие изменения положения тела. На животе и в полулежачей позиции облегчается вентиляция отделов легких, частично сдавливаемых сердцем и брюшным давлением, что может улучшать газообмен [101].
К сожалению, как свидетельствует наш опыт, осуществить на практике систематический мониторинг вентиляции с помощью компьютерной томографии затруднительно по нескольким причинам: поскольку аппарат отсутствует в ОИТ, выполнить КТ-исследование больным с тяжелым ОРДС (т. е. тем, кому это показано в наибольшей степени) трудно, так как необходима транспортировка, а, соответственно, и совершенные портативные респираторы; кроме того, нужно иметь в штате квалифицированного специалиста по интерпретации полученных данных. Однако это направление в выборе оптимальных режимов вентиляции следует считать перспективным.
Указанные выше неоднородности регионарной вентиляции в зависимости от положения тела больного обуславливают необходимость и целесообразность так называемой кинетической терапии: перевороты больного на живот, повороты на бок, в вертикальное и полувертикальное положение.
Кинетическая, или позиционная, терапия (ПТ). Первыми об эффективности при ОРДС ИВЛ в положении на животе сообщили М. A. Piehl и R. S. Brown [102]. С тех пор многочисленными исследованиями были подтверждены преимущества этого метода у 60 — 80 % больных с ОРДС. В недавних исследованиях [103] показа
на эффективность 12-часовой позиции на животе у 96 % больных с тяжелым ОРДС. Дело в том, что у больных, находящихся на ИВЛ в положении на спине, развивается коллапс альвеол дорзальных отделов легких, увеличивается шунтирование в легких, величина которого коррелирует со снижением ФОБ [104]. Это объясняется также более низким торакальным комп-лайнсом в положении на спине. В этом положении даже ПДКВ восстанавливает вентиляцию не альвеол задних отделов легких, а только верхних подгрудинных отделов [105]. Эффективность ПТ объясняют тем, что в положении на животе уменьшается шунтирование в легких, увеличивается объем легочных отделов с нормальными вентиляционно-перфузионными соотношениями, улучшаются оксигенация и биомеханические свойства грудной стенки. Исследователи отмечают также уменьшение морфологических изменений в легких при этой позиции пациента [106].
Два проспективных контрольных исследования подтвердили благоприятный эффект позиционной терапии при ОРДС [107—109]. В некоторых исследованиях как альтернатива положению на животе предлагается систематическая ротационная терапия на один и другой бок [110].
Есть данные, что позиционная терапия снижает количество инфекционных осложнений со стороны легких. Так, ВАП при применении ПТ у коматозных больных на ИВЛ снизилась с 38,4 до 20 % (р = 0,148) [111]. Положение на животе способствует ограничению колонизации трахеи и бронхов и оптимизации бронхиального дренажа, что подтверждается данными компьютерной томографии [112].
До сих пор не выработана единая стратегия проведения кинетической терапии, не ясно, как часто и как долго вентилировать пациента в положении на животе [ИЗ], однако не вызывает сомнения, что перевороты больных на живот или повороты на бок следует систематически проводить, начиная с первых суток ИВЛ.
Преимуществом этого метода современной стратегии вентиляторной поддержки является то, что он не требует дополнительных материальных вложений и может
122 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
уже сейчас использоваться практически во всех ОИТ. Для переворотов больного используются специальные кровати (например, Rotoprone). При их отсутствии желательно, чтобы в процедуре переворота больного участвовало не менее трех человек (от 2 до 6) [114].
Осложнения ИВЛ в позиции на животе [103]:
отек лица (проходит обычно через 2 ч после возвращения в положение на спине);
транзиторные расстройства гемодинамики (артериальная гипотензия);
смещение катетера в центральной вене (крайне редко);
кратковременное усиление гипоксемии вследствие быстрого массивного отхождения мокроты в положении на животе;
спонтанная селективная эндобронхиальная интубация;
спонтанная экстубация (крайне редко); нарушение энтерального питания (25 %), рвота (12 %) (отсутствие эффекта от применения средств, улучшающих моторику желудка и кишок);
повышение внутричерепного давления, которое возвращалось к прежним показателям на протяжении нескольких минут после возврата в первоначальное положение;
повышение внутриглазного давления, изъязвление роговицы [115].
Как следует из вышеизложенного, тяжелых осложнений вследствие поворота больных не наблюдалось, а небольшие осложнения быстро устранялись при перевороте на спину.
Противопоказания к позиционной терапии при ОРДС [ИЗ]:
гемодинамическая нестабильность;
повышение внутричерепного давления; наличие абдоминального дренажа;
терапия, замещающая функцию почек; травма лицевого черепа;
отеки;
торакальный дренаж;
беременность.
Абсолютным противопоказанием является повреждение позвоночника. Могут наблюдаться осложнения при наличии силикона в груди у женщин [ИЗ].
Мы согласны не со всеми из этих противопоказаний, в частности, с успехом при
меняем повороты на живот и на бок больных с ОРДС даже при наличии брюшных и торакальных дренажей. В каждом конкретном случае необходимо взвешивать риск и пользу от изменения положения больного и риск гипотетических осложнений.
Несмотря на то что в положении на животе повышается внутричерепное давление (ВЧД) у больных, находящихся в коматозном состоянии, у некоторых из них с тяжелой гипоксемией этот метод может использоваться (при этом повышение ВЧД не должно превышать 30 мм рт. ст.) [107].
Меры, которые следует предпринять перед переводом больного в положение на животе [ИЗ]:
прекратить введение пищи в назогастральный зонд;
более часто аспирировать трахею; изменить параметры ИВЛ;
увеличить FiO2.
Таким образом, положительный эффект от кинетической терапии с переворотами больного на живот у больных с ОРДС можно считать доказанным и рекомендовать этот метод для рутинного применения в этой группе больных. При невозможности переворота на живот или наличии противопоказаний к этому методу следует применять систематическую ротацию больного в положение на бок, которая может служить альтернативой переворотам на живот [110].
Высокочастотная ИВЛ (ВчИВЛ). Предпосылкой являются очень низкие Vt и инспираторное давление при ВчИВЛ и меньшее ВИП Л. На моделях животных [116] и в двух контролируемых исследованиях не было выявлено преимуществ этого метода [117, 118], хотя ультрачастот-ная ИВЛ в нерандомизированных исследованиях улучшала оксигенацию и легочную механику. У нас есть опыт проведения ВЧИВЛ у 46 больных с ОРДС в режиме HFPPV аппаратом «Фаза-8», причем у 21 больного — с трех суток ИВЛ, у остальных — в более поздний период по мере улучшения состояния. После перевода на ВчИВЛ ни один из этих пациентов не умер от ОРДС. Хотя это утверждение не ставит целью пропагандировать
Синдром острого повреждения легких и острый респираторный дистресс-синдром 123
этот метод во всех случаях ОРДС. При очень тяжелой гипоксемии ВчИВЛ не обеспечивает оксигенацию крови и выведение углекислого газа. Применение этого нетрадиционного метода ИВЛ требует дальнейших исследований, однако мы рекомендуем его применение у больных с ОРДС в тех случаях, когда ВчИВЛ обеспечивает газообмен.
Экстракорпоральная мембранная оксигенация (ЭКМО). Этот метод начали применять в 1970-х годах при ОРДС как модификацию кардиолегочного шунта для элиминации СО2. Однако первые исследования не подтвердили благоприятный эффект ЭКМО на выживаемость больных с тяжелым ОРДС [119, 120]. Возможными причинами этого назывались следующие: вено-артериальная перфузия способствует снижению перфузии легких; потеря крови при ЭКМО составляет примерно до 2,5 л/сут; 5-дневная ЭКМО не улучшала функцию легких. Однако в недавних неконтролируемых исследованиях обнаружена более высокая выживаемость при применении вено-венозной ЭКМО [121]. Внедрение в практику ге-паринсвязывающих систем позволило уменьшить количество кровотечений при этом методе [122]. Рандомизированные проспективные исследования последних лет подтверждают эффективность ЭКМО при тяжелой гипоксемии [123], хотя отмечается высокая стоимость такого лечения.
Показания для использования ЭКМО у больных с ОРДС\
РаО2 < 50 мм рт. ст. или SaO2 <85 — 90 % при FiO2 = 1 и ПДКВ > 10 см вод. ст. (981 Па).
Ингаляция NO (iNO) вызывает селективную дилатацию сосудов вентилируемых отделов легких, перераспределяя кровь из неперфузируемых в перфузируемые участки [124]. В четырех проспективных рандомизированных контролируемых клинических исследованиях на 533 пациентах iNO существенно увеличивала оксигенацию на протяжении 24 ч, однако не повышала выживаемость и не снижала продолжительность ИВЛ и пребывание в ОПТ. Неблагоприятных эффектов от такого лечения не отмечено. Подчеркивает
ся эффективность iNO в неонатологии с уменьшением частоты применения ЭКМО [125], хотя эти исследования нельзя механически экстраполировать на взрослых больных. Поэтому сегодня не рекомендуется рутинно использовать iNO для лечения больных с ОРДС (уровень исследования I, рекомендация А). При угрожающей жизни гипоксемии iNO может быть полезным (уровень исследования II, рекомендация С).
Ингаляция простациклина. Простациклин является вазодилататором короткого действия, его эффект при ингаляционном применении подобен эффекту NO. Несколько исследований на небольшом количестве больных подтвердили благоприятный результат этого метода терапии, эквивалентного iNO [126]. Однако пока нет рандомизированных проспективных контролируемых исследований, подтверждающих или опровергающих эффект ингаляции простациклина.
Экзогенный сурфактант способствует снижению величины поверхностного натяжения в альвеолах, предотвращая их ателектазирование, а также выполняет протекторную функцию при ИВЛ и обеспечивает антибактериальную защиту легких [127]. Гомеостаз сурфактанта нарушается при риске развития ОРДС и еще больше — при развитии болезни. Это, в свою очередь, приводит к нарушению легочной механики и воспалительному ответу легочной ткани вследствие ИВЛ [128]. Пилотное исследование ингаляционного применения искусственного сурфактанта (Exosurf) у 725 пациентов с ОРДС не выявило разницы в уровне летальности в группе больных, получающих сурфактант, и контрольной группе, которой было назначено плацебо [129]. Однако исследование бронхоскопического введения бычьего сурфактанта (четыре дозы 100 мг/кг в день) выявило улучшение оксигенации и уменьшение летальности [130]. Следует отметить, что аэрозольное введение сурфактанта не может быть рекомендовано для лечения ОРДС (уровень I). Инстилляции сурфактанта и дозировка требуют дальнейшего изучения с помощью проспективных рандомизированных ис
124 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
следований, целью которых будет определение оптимального времени назначения сурфактанта у пациентов с высоким риском развития ОРДС для предотвращения прогрессирования дисфункции легкого и выбор показаний к лечению сурфактантом конкретного больного. Учитывая роль, которую играет сурфактант в иммунной защите легких, терапия сурфактантом, по мнению некоторых авторов, является перспективной [131].
Парциальная жидкостная вентиляция легких (ПЖВЛ). ПЖВЛ перфлюорокар-боном у пациентов с ОРДС способствует восстановлению вентиляции альвеол, которые спали, увеличивает функциональную остаточную емкость, легочный комплайнс и оксигенацию, снижает впутрилегочный шунт и вентилятор-индуцированное повреждение легких [132]. Проспективное рандомизированное исследование у 90 взрослых пациентов не выявило улучшения оксигенации и уменьшения летальности, хотя у больных моложе 55 лет такая тенденция отмечена [133]. Применение высокого ПДКВ, положения на животе и инвертированного режима вентиляции повышает эффективность ПЖВЛ. На данный момент этот метод является экспериментальным, нуждается в дополнительных исследованиях и не может быть рекомендован для рутинного применения.
Противовоспалительная терапия. При ОРДС ранее рекомендовалось применение глюкокортикоидов (120 мг/кг метилпреднизолона в день), считалось, что такая терапия улучшала результаты лечения.
Кортизол поддерживает сосудистый тонус и эндотелиальную целостность, модулирует большое количество провоспа-лительных цитокинов и подавляет фосфолипазу А2, циклооксигеназу и NOS. Предполагается, что кортизол обладает анти-фиброзным эффектом, включая ингибирование роста фибробластов и осаждение коллагена, а также стимуляцию апоптоза Т-моноцитов. Во время критического состояния нейрогуморальные факторы, цитокины, эндотелии и предсердный натрий-урический пептид могут участвовать в активации воспаления, приводя к повышению плазменной продукции и плазменных
концентраций кортизола. Однако рандомизированные проспективные со слепым контролем исследования не подтвердили эффективность такой терапии [134, 135]. Более широкие (выборка — 304 больных) исследования выявили даже ухудшение результатов лечения (более высокую летальность) при применении метилпреднизолона [136]. Таким образом, можно утверждать, что раннее применение кортикостероидов не показано при ОРДС.
Есть данные о благоприятном результате применения кортикостероидов в низких дозах (2 — 3 мг/кг в день метилпреднизолона) в более поздней пролиферативной стадии ОРДС, такая терапия улучшала функцию легких и снижала летальность [137]. Проспективные рандомизированные исследования у больных с септическим шоком выявили уменьшение продолжительности вазопрессорной поддержки и количества легочных осложнений при применении низких доз гидрокортизона (0,18 мг/кг в час) [138]. Таким образом, можно рекомендовать применение низких доз кортикостероидов в фибропролиферативной стадии ОРДС [74].
Увеличение продукции циклооксигеназы и провоспалительных метаболитов арахидоновой кислоты при ОРДС может быть блокировано применением нестероидных противовоспалительных средств. В рандомизированных проспективных контрольных исследованиях на 455 пациентах ибупрофен снижал остроту сепсиса, но не влиял на частоту развития ОРДС и летальность [139]. Отсутствие эффекта применения ибупрофена в этом исследовании II уровня послужило основанием для рекомендации не применять нестероидные противовоспалительные средства при терапии ОРДС.
В другом рандомизированном исследовании изучалось действие кетоконазола -синтетического имидазола с противовоспалительными свойствами. У 117 пациентов с СОПЛ или ОРДС эффект отсутствовал [140]. В результате этого исследования I уровня даны рекомендации не применять кетоконазол при ОРДС.
Антиоксиданты. Различные антиоксидантные препараты применяются с целью
Синдром острого повреждения легких и острый респираторный дистресс-синдром 125
защиты легких от свободных радикалов. Рандомизированное контролируемое исследование на 66 больных не выявило положительного эффекта внутривенного применения больших доз антиоксидантов (70 мг/ кг N-ацетилцистеина в день) [ 141 ]. В другом рандомизированном контролируемом исследовании у 51 пациента с ОРДС энтеральное применение антиоксидантных витаминов (аскорбиновой кислоты, токоферола, p-каротина, таурина и L-карнитина) улучшало оксигенацию и снижало продолжительность ИВЛ и нахождения в ОИТ. Имеются данные о положительном действии глутатиона при лечении ОРДС [142].
Таким образом, антиоксидантную терапию при ОРДС можно рекомендовать для рутинного применения, хотя требуются дальнейшие исследования для определения комбинации и доз антиоксидантов и методов введения.
Применение диуретиков. Поскольку ОРДС представляет собой отек легких, применение диуретиков кажется логичным терапевтическим методом. Однако отек этот не гидростатического, а воспалительного генеза, и как показали результаты нескольких исследований, применение диуретиков не уменьшало отек легких и не улучшало оксигенацию, поэтому этот метод не может быть рекомендован для рутинного применения при лечении ОРДС [143].
Применение вено-венозной гемофильтрации. В отдельных, большей частью экспериментальных, работах изучалась эффективность гемофильтрации при ОРДС. Она проводится с целью очищения крови от токсинов и, возможно, уменьшения свободной жидкости в легких. Согласно экспериментальным данным R. Ullrich с соавт. [144], высокообъемная вено-венозная гемофильтрация улучшала оксигенацию, способствовала уменьшению отека легких животных, поврежденных бактериальным эндотоксином, однако данных о рандомизированных клинических исследованиях в литературе мы не обнаружили.
Комбинация различных терапевтических подходов. В нескольких исследованиях показаны преимущества комбинированного применения iNO, позиционной
терапии и инфузии вазоконстриктора ал-митрина [145], однако F. Michard с соавт. [146] указывает на неблагоприятный эффект применения алмитрина на функцию правого желудочка и вообще на исход лечения больных с ОРДС. Можно считать практически доказанной пользу от применения комбинации ПДКВ, положения на животе и iNO у больных с тяжелой гипоксемией [98]. В недавних исследованиях выявлены преимущества комбинации iNO с внутривенной инфузией простациклина при тяжелой легочной гипертензии, развивающейся при выраженном ОРДС [147]. Эффективность этих методов нуждается в дополнительных доказательствах, хотя имеются веские основания для применения таких комбинаций на практике.
В табл. 4.5 представлены рекомендации к лечению ОРДС с позиций доказательной медицины.
Учитывая нарушение проницаемости альвеолярно-капиллярной мембраны при ОРДС, объем внутривенных инфузий необходимо максимально ограничивать вплоть до отрицательного баланса жидкости [148]. Введение жидкости энтерально и внутриартериально менее опасно для легких. Однако в литературе мы не обнаружили
Таблица 4.5. Терапия ОРДС с позиций доказательной медицины [79]
Метод Рекомендация Градация
ПДКВ Да С
Протекторная стратегия ИВЛ Да А
Кинетическая терапия ЭКМО (рутинное приме- Да А
нение) ЭКМО (при тяжелой Нет В
гипоксемии) Да Е
Ингаляция NO Ингаляция NO (при тяже- Нет А
лой гипоксемии) Да С
Сурфактант ? ?
ПЖВЛ Кортикостероиды (ранняя ? ?
стадия) Кортикостероиды (поздняя Нет А
стадия) Нестероидные противовос- Да С
палительные препараты Нет С
Антиоксиданты ?
126 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
четких рекомендаций, какой уровень волемии поддерживать. В условиях ПДКВ обычно используемый в клинике такой показатель для оценки волемии, как центральное венозное давление (ЦВД), будет менее информативным — нормальный уровень ЦВД не будет свидетельствовать о нормальном ОЦК. Поэтому в качестве критерия для увеличения объема внутривенных инфузий при нормальном ЦВД предлагается снижение сердечного индекса ниже 3 л/мин на 1 м2 [20]. При повышенном ЦВД и сниженном СИ следует использовать добутамин (15 мкг/(кг х х мин). Применяя допамин, нужно помнить, что он повышает гидростатическое давление в легочной артерии и снижает конечно-диастолическое давление в левом желудочке сердца [20]. При определении количества переливаемой в вену жидкости следует руководствоваться комплексом показателей (центральная сердечная гемодинамика, диурез, тургор кожи, наличие периферических отеков, количество жидкости, вводимой энтерально). Рекомендуется с осторожностью применять вазодилататоры, в частности нитраты, так как они увеличйвают внутрилегочное шунтирование.
Раннее энтеральное питание. Очень важное значение имеет раннее энтеральное питание (РЭП), поскольку введение жидкости в пищеварительный канал позволяет уменьшить объем внутривенных инфузий. Кроме того, РЭП имеет ряд преимуществ по сравнению с тотальным парентеральным питанием. Оно является одним из факторов, позволяющих снизить летальность при ОРДС [149].
Энтеральное питание лучше парентерального, поскольку поддерживает несколько функций кишок: улучшает моторику, барьерную функцию, иммунитет организма [150]. Потенциальная способность энтерального питания предотвращать или значительно снижать интенсивность сепсиса, синдрома системного воспалительного ответа, а следовательно, и ОРДС, является одним из основных преимуществ этого типа питания по сравнению с парентеральным [20]. Для больных с повреждением легких предпочтительнее диета, богатая
жирами и бедная углеводами. Избыточное введение протеинов, аминокислот повышает нагрузку на систему дыхания. Рекомендуют такую диету: 55,2 % — жиры, 16,7 — белки, 28,1 % — углеводы. Особенно важно содержание в ней у-линоле-новой кислоты, со-3-полиненасыщенных жирных кислот и антиоксидантов. Эта диета способствует снижению нейтрофильной инфильтрации легких, продукции про-воспалительных медиаторов, улучшает газообмен, уменьшает продолжительность ИВЛ у больных с ОРДС. Низкое содержание углеводов в пище обуславливает снижение дыхательного коэффициента на 15 — 17 % и соответственно продукции СО2. Избыточное введение протеинов, особенно аминокислот, стимулирует вентиляторный драйв и увеличивает работу дыхания. Поэтому эксперты рекомендуют диету с содержанием белков 15 —20 % общей энергетической ценности, или 1 — 1,5 г/кг, с коррекцией в зависимости от азотистого баланса и концентрации азота в крови [150]. А. V. Casetti [69] обнаружил, что применение энтеральной диеты, содержащей эйкозапентаеновую, у-линоленовую кислоты и большое количество антиоксидантов (ЕРА + GLA; Охера), существенно уменьшало легочное воспаление, увеличивало насыщение крови кислородом и улучшало клинические результаты лечения у больных с ОРДС.
Антибиотикотерапия. На фоне ОРДС могут возникнуть трудности с диагностикой инфекционных процессов в легких вследствие наличия симптомов ОНД, рентгенологических изменений в легких [151]. Это усложняет своевременное и адекватное назначение антибиотиков. Положение о том, что наличие ОРДС само по себе предполагает эмпирическое назначение антибиотиков, сейчас не является неоспоримым. Конечно, патологические изменения в легких при ОРДС, длительное применение ИВЛ, тяжесть состояния больных являются серьезными предпосылками развития пневмонии, однако будет ли способствовать профилактическое применение антибиотиков уменьшению такого риска, пока неясно. При эмпирическом назначении больному с тяжелым ОРДС
Искусственная вентиляция легких 127
антибиотика широкого спектра действия следует руководствоваться общими принципами рациональной антибиотикотера-пии [73].
В заключение следует отметить, что лечение больных с ОРДС требует современ-
4.5. ИСКУССТВЕННАЯ
Искусственная вентиляция легких является одним из наиболее важных и эффективных методов интенсивной терапии больных с недостаточностью дыхания. Большая часть больных в ОИТ на определенный период времени нуждается в проведении ИВЛ, а примерно 6 % больным необходима длительная (более 14 суток) ИВЛ [152].
Первым, кто доказал эффективность для поддержания жизни ритмического вдувания воздуха в легкие у животных с вскрытой грудной стенкой, был Везалий. В 1664 г. его эксперимент повторил в Лондоне R. Нооке.
В 1827 г. Leroy писал о серьезных осложнениях, возникающих при вдувании газа в легкие, в частности о баротравме легких. Эти сведения, а также низкая эффективность применяемых методов ИВЛ привели к отказу от их использования в течение продолжительного времени.
С 30-х годов XIX ст. началась эра инспираторных методов ИВЛ с созданием отрицательного (ниже атмосферного) давления вокруг груди и живота пациента. В 1832 г. шотландский врач J. Dalsiel с помощью этой методики попытался спасти человека с асфиксией, вызванной утоплением. Американец A. Jones в 1864 г. предпринял попытку спасти больных с параличем дыхательных мышц. В 1924 г. Thunberg описал барореспиратор, с помощью которого он пытался проводить ИВЛ больным с полиомиелитом и отравлениями. Через пять лет Р. Drinker из Бостона предложил «железные легкие» — это был практически первый танковый респиратор, который в различных вариантах использовался до 50-х годов XX ст. для обеспечения вентиляции, главным образом у больных с тяжелым полиомиелитом и син-
ных подходов, больших трудозатрат, но в отличие от многих других состояний, нуждающихся в интенсивной терапии, у большинства выздоровевших восстанавливается функция внешнего дыхания и они не становятся инвалидами.
ВЕНТИЛЯЦИЯ ЛЕГКИХ
дромом Guillain —Barre. Несмотря на усовершенствование аппаратов для инспираторной ИВЛ, этот метод оставался ненадежным вследствие невозможности обеспечения проходимости дыхательных путей, трудности синхронизации с дыханием пациента, громоздкости и неудобства в использовании применяемой аппаратуры.
В 1952 г. в Скандинавских странах возникла эпидемия полиомиелита. В Копенгагене анестезиолог Ibsen при содействии инфекциониста Lassen предложил для ИВЛ вдувание газа в легкие с помощью мешка или меха наркозного аппарата с использованием абсорбера для углекислого газа и трахеостомической канюли с раздувной манжетой. Это нововведение позволило уменьшить летальность у больных с тяжелыми бульбоспинальными параличами с 85 до 40% [153].
Совершенствовались методы обеспечения проходимости дыхательных путей. Приоритет в применении трахеостомии для проведения ИВЛ также принадлежит Везалию. В 1858 г. Desault во Франции впервые применил оротрахеальную интубацию для восстановления проходимости дыхательных путей при дифтерийном крупе [154]. Вначале методы интубации трахеи использовались для проведения общей анестезии, однако начиная с 50-х годов прошлого столетия для проведения ИВЛ все большее распространение получает длительная интубация.
Таким образом, экспираторные методы ИВЛ широко применяются для протезирования вентиляции легких более полувека. Сейчас уже невозможно представить себе отделение анестезиологии и интенсивной терапии без аппаратуры для проведения ИВЛ.
128 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
4.5.1. Влияние искусственной вентиляции легких на циркуляцию крови
В 1948 г. Cournand с соавт. обнаружил, что ИВЛ снижает сердечный выброс (СВ). Главной причиной этого является снижение венозного возврата. Исчезает присасывающий эффект грудной стенки вследствие того, что на выдохе при снижении внутригрудного давления уменьшается давление в полых венах и кровь быстрее притекает к правому предсердию. Снижение венозного возврата зависит от повышения среднего внутригрудного давления, которое, в свою очередь, обуславливается пиковым давлением на вдохе, соотношением вдоха к выдоху, формой кривой вентиляции, величиной сопротивления на выдохе.
Другой причиной снижения СВ является повышение сопротивления сосудов легких. Сопротивление интраальвеоляр-ных капилляров в норме составляет 40 % общего сопротивления легочных сосудов. Повышение капиллярного давления в легких увеличивает постнагрузку для правого желудочка, снижает приток крови к левому предсердию, увеличивает объем функционального мертвого пространства. При нормальных легких и оптимальном дыхательном объеме, инспираторном давлении, не превышающем 10—15 см вод. ст. (980—1470 Па), эти явления не выражены. Так, осцилляция АДСИСТ на вдохе и выдохе не превышает 10 мм рт. ст. у пациента с нормоволемией, однако резко возрастает уже на ранних стадиях гиповолемии [155], а также при снижении растяжимости легких и повышении динамического сопротивления дыхательных путей. У больных с хронической патологией легких увеличивается работа правого желудочка, что может приводить к его ишемии. Поскольку венечный кровоток левого желудочка в основном зависит от диастолического давления, а правого — еще и от систолического, то для предотвращения ишемии правого желудочка важно поддерживать эти показатели в норме [3]. Во время ИВЛ снижается растяжимость и ударный выброс левого желудочка сердца.
При некоторых видах патологии сердца и легких (отек легких, приступ бронхиальной астмы) ИВЛ помимо улучшения оксигенации крови улучшает условия для функции сердца.
ИВЛ может влиять на циркуляцию крови за счет изменения ее газового состава. Гипокапния снижает симпатический драйв и способствует уменьшению СВ. Помимо артериальной гипотензии резкое снижение РаС>2 у больных с хронической недостаточностью дыхания может привести к острой ишемии головного мозга. Последствиями гиперкапнии могут быть артериальная гипертензия, нарушения ритма сердца и отек мозга.
Кроме центрального эффекта на гемодинамику ИВЛ может вызывать перераспределение жидкости в секторах организма, приводить к периферическим отекам и снижению ОЦК.
Влияет ИВЛ и на функцию почек. Этот эффект объясняется как снижением почечной перфузии, так и повышением давления в почечных венах. В результате снижается клубочковая фильтрация (хотя подобный эффект больше присущ ПДКВ [156]) и экскреция натрия. Воздействие ИВЛ на диурез может осуществляться посредством изменения активности ренин-ангиотензиновой системы и секреции предсердного натрийурического фактора [157].
ИВЛ, особенно в режиме ПДКВ, снижает печеночный кровоток как за счет снижения СВ, так и за счет выключения массирующего эффекта сокращений диафрагмы и повышения внутрибрюшного давления [158].
ИВЛ нередко осложняется кровотечением из верхних отделов пищеварительного канала у больных в критическом состоянии. Но это объясняется не столько снижением перфузии и повышением венозного давления, сколько стрессовыми факторами, не зависящими от ИВЛ.
Несмотря на описанные побочные эффекты, ИВЛ при тяжелой недостаточности дыхания часто является единственным методом спасения жизни больного; коррекция вентиляции и газообмена в легких с помощью ИВЛ по своему положительному воздействию на организм значительно перевешивает некоторые отрицательные эффекты.
Искусственная вентиляция легких 129
4.5.2. Показания к переводу на искусственную вентиляцию легких
Основанием для показаний к переводу на ИВЛ является появление признаков невозможности поддерживать спонтанным дыханием оксигенацию крови и/или элиминацию углекислого газа на минимальном для организма уровне. Основная сложность заключается в определении этого уровня. ИВЛ необходима больным с остановкой дыхания, терминальными видами дыхания, хотя некоторые больные, например с синдромом Пиквика, у которых по ночам возникают терминальные виды дыхания, не всегда нуждаются в переводе на ИВЛ. Кроме клинических данных в выборе правильного решения могут помочь некоторые лабораторные показатели, данные спирометрии (табл. 4.6).
Наличие одного из этих признаков (кроме апноэ) не всегда является показанием к немедленному переводу на ИВЛ. Следует также учитывать клинические проявления гипоксемии и гиперкапнии, эффективность проводимой консервативной терапии, причину развития недостаточности дыхания. Например, при наличии ателектаза, других признаков обтурационного синдрома своевременное проведение ФБС может улучшить функциональные показатели и газовый состав артериальной крови. С другой стороны, если у больного на фоне гипоксемии развиваются признаки расстройства функции ЦНС (нарушения сознания) и сердечно-сосудистой системы (выраженная тахикардия,
Таблица 4.6. Показания к переводу на ИВЛ
Критерий перевода на ИВЛ Норма
Апноэ, терминальные виды дыхания f (частота дыхания) > 35 мин-1 12-20
ЖЕЛ < 15 — 20 мл/кг 65-75
ОФВ < 10 мл/кг 50-60
Сила вдоха (макс, разрежение на вдохе) < 25 см вод. ст. 75-100
РдО2 / FiO2 < 80 мм рт. ст. > 400
РдСО2 > 55 мм рт. ст. 35-45
стойкая артериальная гипертензия, а затем и гипотензия, нарушения ритма сердца), необходим срочный перевод на ИВЛ.
Шире показания к вентиляторной поддержке при внутричерепной патологии, когда внутричерепная гипертензия вызывает депрессию дыхания, а возникшие гипоксемия и гиперкапния ухудшают течение внутричерепного процесса. В некоторых случаях у таких больных должна применяться ИВЛ в режиме умеренной гипервентиляции с целью уменьшения внутричерепной гипертензии.
Если после некоторого периода наблюдения возникло сомнение, клиницист должен выбрать стратегию перевода на ИВЛ, так как это менее опасно, чем прогрессирование недостаточности дыхания. Следует отметить, что при достаточной квалификации анестезиолога перевод на ИВЛ при отсутствии противопоказаний (напряженный пневмоторакс, прямая ларингоскопия при повреждении шейного отдела позвоночника без профилактических мероприятий) не может ухудшить состояние больного. В ОИТ, где показанием к переводу на ИВЛ является остановка кровообращения, шансы на выживание у тяжелых больных гораздо ниже. Перевод на ИВЛ, улучшение оксигенации крови и элиминации углекислого газа, отсутствие энергетических затрат на дыхание часто позволяют улучшить состояние больных, у которых нарушение функции внешнего дыхания не является доминирующим фактором тяжести состояния.
4.5.3. Выбор параметров искусственной вентиляции легких
С самого начала применения ИВЛ считалось неоспоримым положение, согласно которому дыхательный объем (Vt ) должен составлять 10—15 мл/кг. Кроме того, периодически (6—12 раз в час) рекомендовалось делать глубокие вдохи с Vt = = 15 — 30 мл/кг. Такую методику рассматривали как стандартную для предотвращения коллабирования альвеол [159]. ПДКВ рекомендовалось только в тех случаях, когда не удавалось поддерживать нормальную оксигенацию на безопасном
5 4-220
130 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
уровне ГгО2. Однако D. Dreyfus и G. Sau-mon опубликовали в 1994 г. данные, свидетельствующие о том, что конвенционная ИВЛ вызывает тяжелые повреждающие эффекты в легких, а также имеет системный повреждающий эффект. Внимание было обращено главным образом на отрицательное воздействие большого дыхательного объема и высокого инспираторного давления. В дальнейшем эксперименты на животных подтвердили, что высокий Vt (10—15 мл/кг) вызывает механический стресс в поврежденных легких, увеличение продукции и выброса провоспалитель-ных субстанций. И действительно, клинические и экспериментальные исследования Т. J. Donnelly с соавт. [160] и других ученых показали повышение уровня провос-палительных цитокинов в системном кровотоке во время конвенционной ИВЛ.
L. Tremblay с соавт. [161] изучал содержание в бронхоальвеолярной лаважной жидкости провоспалительных цитокинов при таких моделях ИВЛ:
группа А: неповреждающий (протекторный) режим (Vt < 6 мл/кг, ПДКВ — 5 — 10 см вод. ст. (490 — 980 Па));
группа Б: средний Vt, высокое ПДКВ;
группа В: средний Vt, ZEEP (нулевое ПДКВ);
группа Д: высокий Vt, ZEEP.
Наименьшие значения уровня цитокинов в бронхоальвеолярной жидкости были выявлены у больных в группе А, наибольшие — в группе Д. Авторы обнаружили синергический эффект высокого Vt( 10 — 15 мл/кг) и ZEEP. В группе Д TNFa был выше по сравнению с контролем в 56 раз, в группе Б — в три раза, в группе В — в шесть раз. Сходные результаты были получены и в отношении макрофагального фактора воспаления. При этом оказалось, что увеличение концентрации провоспалительных факторов определяется не только в легких, подобная динамика обнаружена и в системном кровотоке, что, соответственно, увеличивает риск развития синдрома системного воспалительного ответа (ССВО). Два фактора могут быть ответственными за такое повреждение: перерастяжение альвеол и высокое транспульмональное давление.
В последние годы предпринимались попытки объяснить механизм повреждения легких при традиционных режимах ИВЛ. С помощью моделирования циклического перерастяжения в культурах клеток стало возможным исследовать клеточные механизмы активации этих процессов, связанные с механическим напряжением ткани легких [162]. Интегриновые рецепторы, протеины фокальной адгезии клеток и сам цитоскелет участвуют в различных молекулярных взаимодействиях, чувствительных к циклическому стретчу, трансформируют механический сигнал в биологическую реакцию. Активизация нескольких внутриклеточных сигнальных путей приводит к увеличению транскрипции генов, представляющих «стретчзави-симые элементы» и их предшественники. Среди этих сигнальных путей митоген-ак-тивированный протеинкиназный сигнальный каскад, по-видимому, является центральным медиатором перерастяжения и спадания клетки. Другие посттранскрип-циональные механизмы, наподобие мессенджера стабилизации РНК и секреции предшественников медиаторов, также могут принимать участие в секреции провоспалительных медиаторов при циклическом перерастяжении.
Провоспалительные цитокины синтезируются и высвобождаются эндотелиальными клетками и легочными моноцитами. При сравнении таких факторов, как TNFa, ИЛ-lp, ИЛ-6, ИЛ-8 и др., у больных с ОРДС при разных режимах вентиляции также получены аналогичные результаты.
Sadoshim, Vandenberg (1997) показали ведущую роль механического фактора в структурных изменениях в легких. Было обнаружено, что механическое растяжение повреждает молекулы, модулирующие рост и развитие легочной паренхимы, эндотелиальную проницаемость и продукцию сурфактанта. После 15 мин конвенционной ИВЛ увеличивается масса легких. 2/3 легких не вентилируется через 3 суток даже при Vt = 15 мл/кг. К сигнальным механизмам повреждения относят паракринные и аутокринные факторы, влияющие на функцию ионных каналов и непосредственно воздействующие на
Искусственная вентиляция легких 131
содержащиеся в мембранах молекулы, что ведет к активации других мессенджеров. Нарушается регуляция CD 18 и CD64, растет активность се лектина и эластазы. Недавние исследования показали, что механические факторы нарушают структуру и функцию полиморфноядерных нейтрофилов в легких.
Результаты исследований доказывают, что транслокация альвеолярных медиаторов в кровоток играет важную роль в патогенезе вентилятор-ассоциированного ССВО. При повреждающей стратегии ИВЛ происходит также транслокация бактериальных клеток. Многие ученые придерживаются гипотезы, согласно которой большой Vt (10 — 15 мл/кг) и ZEEP нарушают альвеолярно-капиллярный барьер, что приводит к попаданию в системный кровоток из легких цитокинов, эндотоксинов, бактериальных клеток.
Особого повреждающего эффекта на здоровые легкие при непродолжительном применении ИВЛ не выявлено, в частности S. К. Appavu с соавт. [163] не обнаружил увеличения послеоперационных осложнений со стороны легких и летальность при использовании большого дыхательного объема во время операции. Однако у больных с неповрежденными легкими, но с начавшимся системным воспалением, конвенционные методы ИВЛ способствуют прогрессированию этого процесса.
Таким образом, можно сделать вывод, что, по выражению Chiche, механический «инсульт» запускает как многофакторный сигнальный процесс с развитием воспаления в легких, так и системный, с повреждением легочного сурфактанта.
Y. S. Edwards с соавт. [164] продемонстрировал в эксперименте, что механический стретч при конвенционной ИВЛ может запускать механизм альвеолярного апоптоза в легких.
Согласно рекомендациям Р. L. Marino [20], протекторная стратегия ИВЛ предусматривает: Vt = 6 — 7 МЛ/кг; Pplateau< <35 см вод. ст. (3430 Па); ПДКВ = 5 — 15 см вод. ст. (490—1470 Па); допустима «безопасная» (permissive) гиперкапния (не приводящая к повреждению других органов и систем) — pH <7,2.
Результаты недавних исследований позволяют рекомендовать протекторную стратегию при длительной ИВЛ для рутинного применения, поскольку она минимизирует циклическое, связанное с выдохом и вдохом, закрытие дыхательных путей с ателектазированием альвеол и пере-растяжение их на вдохе, вследствие чего ИВЛ в таком режиме оказывает меньший повреждающий эффект на легкие, а также выделяется меньше провоспалительных медиаторов, индуцирующих ССВО и ПОН [165]. Такой режим ИВЛ не вызывает вен-тилятор-индуцированную смерть [166].
Очень важным параметром является конечно-инспираторное пиковое давление (Ppeak)> или пиковое давление в конце вдоха. Ppeak зависит от дыхательного объема, сопротивления дыхательных путей, эластических свойств легких и грудной клетки. Таким образом, при постоянном Vt повышение Ppeak свидетельствует об увеличении сопротивления дыхательных путей и/или снижении эластичности.
Конечное давление плато на выдохе {Рplateau}• На рис. 4.3 изображено давление в проксимальных дыхательных путях на протяжении дыхательного цикла. После вдувания всего дыхательного объема давление в проксимальных дыхательных путях снижается (фаза 1), затем кривая переходит в плато (фаза 2), уровень плато и отражает Рplateau- Поскольку в это время не наблюдается поток газа в дыха-
Фаза 1
Фаза 2
Пиковое давление (Рoeak) Давление плато
(Pplateau)
Вдувание Поддержка Выдох
вдувания
Рис. 4.3. Давление в проксимальных дыхательных путях в течение дыхательного цикла [20]
5*
132 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
тельных путях, исключается влияние сопротивления нижних дыхательных путей, поэтому Рplateau пропорционально эластич-ности легких и грудной стенки, а разница между Ppeak и Ppiateau - сопротивлению нижних дыхательных путей.
Практическое значение учета Ppeak и Ppiateau [20]:
1. Если Ppeak увеличилось, a Poteau не изменилось, это означает, что возросло сопротивление дыхательных путей вследствие нарушения проходимости интубационной трубки, наличия бронхоспазма или обструкции нижних дыхательных путей.
2. Увеличение Ppeak и Poteau скорее всего связано со снижением растяжимости легких и грудной стенки (напряженный пневмоторакс, обширный ателектаз, острый отек легких, тотальная пневмония, ОРДС).
3. Если Ppeak снизилось, следует исключить частичную разгерметизацию дыхательного контура (если причину найти не удалось, необходимо срочно сменить респиратор или перейти на «ручное» дыхание при помощи мешка Амбу). Возможен вариант, когда спонтанный вдох пациента совпадает со вдохом респиратора.
4. Если Ppeak не меняется, изменений в легочной биомеханике не происходит.
Постепенное снижение Ppeak (если оно было повышено) свидетельствует о положительной динамике патологического процесса в легких.
Мы подробно остановились на показателе Ppiateau* поскольку величина этого показателя играет важную роль в развитии ВИПЛ, a Amato (1998) считает повышение Ppiateau СВЫШе 30 — 35 СМ ВОД. СТ. (2940 — 3430 Па) решающим фактором вентилятор-индуцированного повреждения легких. Сейчас в Украине с внедрением в практику нового отечественного респиратора «БРИЗ» появилась возможность осуществлять мониторинг этого показателя.
4.5.4. Специальные режимы ИВЛ
Наиболее часто используемым специальным режимом является ИВЛ с сопротивлением в конце выдоха (ПДКВ). При
обычном режиме ИВЛ давление в конце выдоха падает до нуля, при ПДКВ даже в конце выдоха давление остается положительным. Впервые роль ПДКВ в поддержании стабильности альвеол и улучшении оксигенации была описана в 1963 г. [167]. В 1967 г. D. Ashbaugh с соавт. [64], описывая ОРДС, предложил использовать ПДКВ в качестве эффективного метода улучшения оксигенации крови. Основными эффектами ПДКВ являются: реинфляция ателектазированных альвеол, увеличение ФОЕ, снижение внутрилегочного шунта, увеличение соотношения РдО2/ЕЮ2.
Кроме того, ПДКВ присущи и все негативные эффекты обычной ИВЛ, но в большей степени. Снижение сердечного выброса главным образом зависит не от величины ПДКВ, а от уровня повышения внутригрудного давления. Следует помнить, что повышение внутриальвеоляр-ного давления при ПДКВ не пропорционально уровню ПДКВ, так как, устраняя гипоксическую вазоконстрикцию, развивающуюся при ателектазировании альвеол, и уменьшая шунтирование, ПДКВ снижает периферическое сопротивление легочных сосудов. Режим ПДКВ стал компонентом протекторной стратегии ИВЛ.
Следует отметить, что при локальном повреждении легкого (пневмония, контузия) ПДКВ может отрицательно влиять на газообмен, вызывая перерастяжение здоровых альвеол и перераспределяя кровоток в патологический очаг [20].
Мы поддерживаем мнение L. Gattinoni с соавт. [168], согласно которому режим ИВЛ предполагает соблюдение следующих принципов: предотвращение перерас-тяжения (низкое давление плато в дыхательных путях) и циклического коллапса, а затем — повторного перерастяжения (адекватный ПДКВ и альвеолярная вентиляция). При анализе рандомизированных клинических исследований выяснилось, что транспульмональное давление и стресс альвеол при ИВЛ в большей степени, чем дыхательный объем, влияют на смертность [168].
Вспомогательная ИВЛ (assist-control mechanical ventilation (ACMV)). Этот режим возможен только при наличии спон-
Искусственная вентиляция легких 133
тайного дыхания. Пациент своим дыханием запускает вспомогательное дыхание респиратора. При редком дыхании респиратор, как правило, переходит в обычный режим CMV. Этот метод не получил широкого распространения в клинической практике. Вследствие заданных параметров вентиляции пациент часто испытывает дискомфорт, «борется» с респиратором [3].
Периодическая принудительная вентиляция (intermittent mandatory ventilation (IMV)). Относится к вспомогательным режимам ИВЛ. В отличие от режима ACMV дает возможность пациенту самостоятельно дышать в промежутках между вдохами респиратора. Впервые метод был применен в педиатрической практике в 1973 г., а затем рекомендован к использованию для облегчения отключения больного от респиратора [169]. На фоне спонтанного, но не совсем адекватного дыхания больного респиратор производит периодические принудительные вдохи с заданными анестезиологом параметрами (частотой, дыхательным объемом, временем вдоха, скоростью потока). В дальнейшем методика получила свое продолжение в режиме synchronised (синхронной) IMV (SIMV), при котором респиратор производит вдохи синхронно с вдохами больного. При этом практически во всех респираторах, обеспечивающих этот режим, есть сенсорная триггерная система, позволяющая регулировать силу спонтанного вдоха пациента, при которой включается аппарат. При определенном периоде слабых вдохов респиратор может перейти на обычный режим (CMV).
Главными преимуществами вспомогательного режима SIMV является уменьшение давления в дыхательных путях на высоте вдоха, тем самым обеспечивается более протекторный режим для легких. Кроме того, этот режим делает перевод на спонтанное дыхание более безопасным и управляемым.
Применение SIMV ограничено у больных с ХНЗЛ и низкой растяжимостью легких, когда заданный объем может быть недостаточным для адекватной альвеолярной вентиляции [170].
Инспираторная поддержка давлением (inspiratory pressure support (IPS)). Была внедрена в практику в середине 80-х годов XX в. В отличие от других методик респиратор создает пациенту поддержку на вдохе не потоком, а давлением. Поддержка может осуществляться малым давлением, чтобы только преодолеть сопротивление трубки и дыхательных путей, или большим, полностью замещающим работу внешнего дыхания. Газовый поток подается респиратором при снижении скорости вдоха ниже заданных значений.
Преимуществом этого метода является то, что пациент может сам управлять частотой, МОД, дыхательным объемом, временем вдоха и выдоха, скоростью потока. Этот метод может плохо переноситься больными с повышенным динамическим сопротивлением нижних дыхательных путей [170].
Высокочастотный режим вентиляции легких (ВчИВЛ) (high frequency ventilation (HFV)). Начал применяться в начале 70-х годов XX в. [171]. Отличительным свойством ВчИВЛ является не только высокая частота, но и то, что дыхательный объем меньше объема мертвого пространства (Vt < VD). В отличие от конвенционной методики ИВЛ, газообмен между альвеолярным газом и мертвым пространством осуществляется при ВчИВЛ в основном благодаря диффузии газов.
Существуют три разновидности ВчИВЛ:
1. С перемежающимся положительным давлением (HFPPV):
f (частота) = 60—120 мин-1; Vt = = 3 мл/кг.
2. Струйная (HFJV): f= 100-600 мин’1.
3. Осцилляторная (HFOV): f = 600 — 3600 мин-1.
При этом МОД может составлять 20 — 100 л/мин.
Разновидность HFPPV соответствует конвенционной ИВЛ (кИВЛ), при этом изменены только параметры вентиляции. Для проведения HFPPV в трахею вводится катетер от 3 до 10 мм в диаметре. Выдох пассивный, мимо катетера. За рубежом эта методика не получила широкого распространения и применяется в основ
134 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
ном для обеспечения вентиляции при тра-хеобронхоскопии и операциях на нижних дыхательных путях.
Более эффективным считают режим струйной ВчИВЛ (HFJV), позволяющий использовать более тонкий катетер для инсуфляции (внутренний диаметр 1,7 — 2 мм), чем при HFPPV, однако газовый поток при этом должен быть большим (до 60 — 80 л/мин). Используется для более продолжительной вентиляторной поддержки, особенно показан при наличии бронхоплевральных свищей [172].
Наибольшее распространение за рубежом получил режим осцилляторной ВчИВЛ (HFOV), особенно в детской практике при респираторном дистресс-синдро-ме новорожденных. Метод был разработан Р. Р. Launkenheimer [173].
Используют также комбинацию кИВЛ с ВчИВЛ в одном дыхательном контуре. В отличие от кИВЛ ВчИВЛ позволяет не герметизировать дыхательный контур и поддерживать невысокое инспираторное и среднегрудное давление и соответственно обладает наименее повреждающим эффектом на легкие из всех видов ИВЛ. М. Takata с соавт. [174] сравнил эффект кИВЛ и HFOV на интраальвеолярную экспрессию гена TNFa у кроликов. Наименьшей она оказалась при режиме HFOV. Imai с соавт. (1996) обнаружил через четыре часа кИВЛ активацию в легких фактора активации тромбоцитов, ТгВ2, PgF2, при HFOV эти изменения практически отсутствовали. Кроме того, минимизируется влияние на сердечный выброс, не повышается венозное сопротивление и внутричерепное давление. Очень важным преимуществом ВчИВЛ является возможность сохранять спонтанное дыхание больного.
Главный недостаток этого метода — меньшая, по сравнению с.ПДКВ, эффективность относительно оксигенации крови. Кроме того, возможно нарушение элиминации углекислого газа. Большой газовый поток затрудняет согревание и увлажнение газовой смеси. Возникают сложности с мониторингом эффективности вентиляции. При глубоком смещении катетера возможна гиперинфляция с баротравмой лег
кого, а при поверхностном — неэффективная вентиляция.
Одним из свойств ВчИВЛ является создание внутреннего (intrinsic) ПДКВ. Оно возникает во время окклюзии мелких дыхательных путей в конце выдоха. Внутреннее ПДКВ увеличивает внутриальвеоляр-ное и внутригрудное давление, что способствует снижению сердечного выброса, повышению центрального венозного давления и давления в легочной артерии. При спонтанном дыхании (при бронхиальной астме) этот эффект увеличивает работу дыхания, а при ИВЛ с постоянным потоком внутреннее ПДКВ повышает давление в дыхательных путях. Внутренний ПДКВ характерен и для инвертированного режима.
Мы широко используем режим HFPPV, создаваемый аппаратом «Фаза-8» (другие режимы ВчИВЛ этот аппарат не обеспечивает), в ОИТ у больных в послеоперационном и посттравматическом периоде на фоне сохраненного, но неадекватного спонтанного дыхания. С этой целью применяем катетер с внутренним диаметром 3 мм. Иногда для улучшения оксигенации увеличиваем продолжительность вдоха до 50 — 60 %. По нашим наблюдениям, это очень эффективный режим, облегчающий перевод больного на спонтанное дыхание, хотя у некоторых больных с нетяжелым ОРДС он может играть и самостоятельную роль. Учитывая названные преимущества, методика ВчИВЛ является перспективной для более широкого применения в практике.
Еще одним методом ИВЛ, позволяющим улучшить оксигенацию и уменьшить пиковое инспираторное давление в дыхательных путях, является инвертированная ИВЛ (inverse ratio ventilation (IRV)). Эта методика ИВЛ отличается увеличенным временем вдоха к выдоху в дыхательном цикле — 2 : 1 или 1:1. Впервые была применена в 1971 г. при дистресс-синдроме новорожденных [175]. Более плавный и продолжительный вдох способствует снижению пикового давления на вдохе, уменьшает шунтирование и улучшает оксигенацию [3]. При этом режиме характерно развитие внутреннего ПДКВ вследствие сокращения периода выдоха.
Искусственная вентиляция легких 135
Независимая искусственная вентиляция обоих легких. Поскольку патологический процесс в легких часто носит неравномерный характер с преимущественным поражением одного из них, разной растяжимостью, сопротивлением, то и оптимальные параметры ИВЛ каждого легкого разные. Метод технически сложен. Требует наложения трахеостомии и применения специальных двухпросветных интубационных трубок, устанавливаемых под контролем ФБС, и двух респираторов [3]. При этом методе каждое легкое вентилируется в индивидуальном режиме: для каждого подбирается ПДКВ, соотношение вдоха к выдоху и другие параметры. Широкого применения в практике пока не нашел.
Более подробно об особенностях различных режимов ИВЛ при гипоксемической недостаточности дыхания см. «ОРДС».
4.5.5. Обеспечение проходимости дыхательных путей
при искусственной вентиляции легких
Все рассмотренные выше методы ИВЛ относятся к инвазивным, так как они требуют интубации или трахеостомии. Оротрахеальную интубацию с миорелаксацией начали более широко использовать после внедрения в 1942 г. Н. R. Griffith и
G. Е. Johnson [176] в анестезиологическую практику кураре. Однако длительное время держать в дыхательных путях резиновые интубационные трубки было нельзя, и лишь после внедрения в 50-х годах пластиковых интубационных трубок стала возможна длительная интубация трахеи для проведения ИВЛ. Дальнейшее совершенствование этого метода связано с применением манжеток на интубационных трубках, а затем и манжеток высокого объема с низким давлением. Существуют две методики длительной интубации трахеи: оротрахеальная (ОТИ) и назотрахеальная (НТИ). Показания к ним и осложнения, вызываемые каждой из методик, представлены в табл. 4.7.
Основным преимуществом НТИ перед ОТИ является лучшая переносимость больным, так как оротрахеальную трубку в течение нескольких суток без глубокой седации может переносить только очень ограниченное количество больных. При ОТИ значительно ограничивается подвижность больного, что очень неблагоприятно отражается на легких. При НТИ трубка намного лучше зафиксирована, поэтому гораздо реже встречаются случаи непроизвольной экстубации, практически не бывает эндобронхиального смещения трубки, а также реже наблюдаются стенозы гортани и трахеи. При ОТИ значительно
Таблица 4.7. Показания к применению длительной ИВЛ и осложнения, вызываемые оротрахеальной и назотрахеальной интубациями
Назотрахеальная интубация Оротрахеальная интубация
Показания Осложнения и недостатки Показания Осложнения и недостатки
Повреждение шейного отдела позвоночника Переломы нижней челюсти Обычно применяется у больных, находящихся в сознании Синуситы Некроз хрящей носа Затруднен контроль за проходимостью интубационной трубки Экстренная ситуация Коматозное состояние Переломы костей лицевого черепа Перелом костей основания черепа Непроходимость носовых ходов Плохая переносимость пациентами в ясном сознании Повышение частоты осложнений со стороны гортани и трахеи Эндобронхиальное смещение трубки Затруднена санация полости рта
136 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
затруднена санация полости рта. Все это приводит к тому, что ОТИ очень ограниченно используют для длительной ИВЛ.
К недостаткам назотрахеальной интубации следует отнести некоторую сложность при ее выполнении, затрудненный уход за проходимостью самой трубки и нижних дыхательных путей, особенно при большом количестве гнойной вязкой мокроты или наличии крови в дыхательных путях. Для поддержания проходимости назотрахеальной трубки мы по показаниям, но не реже чем через 4 ч, производим промывание трубки теплым физиологическим раствором с последующим прохождением через трубку катетера максимально широкого диаметра. Таким образом, нам удается поддерживать проходимость интубационной трубки у больных на ИВЛ в течение 4 — 6 суток без переинтубации. Частые переинтубации способствуют риску инфицирования и развития вентиля-тор-ассоциированной пневмонии, введение трубки осуществляется под наркозом с использованием миорелаксантов, что затрудняет перевод на спонтанное дыхание, а иногда сопряжено с более серьезными осложнениями. Поэтому, если необходимо менять трубки чаще, чем через три дня, лучше наложить трахеостому.
К частым осложнением НТИ относят развитие синуситов. Однако в нашей практике (более 200 случаев длительной НТИ) это осложнение встречалось редко (достоверно — в 3 % случаев). Мы наблюдали у трех больных такое осложнение НТИ, описанное в литературе, как некроз хрящей носа (у нас все случаи закончились благополучно после экстубации).
Поздними осложнениями длительной ОТИ и реже — НТИ являются стенозы гортани и верхних отделов трахеи, вызываемые применением интубационных трубок излишне широкого диаметра. Используя трубки размером № 7 — 8 у женщин и №7,5 —8,5 — у мужчин, мы практически не наблюдали этого тяжелого осложнения, требующего длительного лечения.
Трахеостомия. Считается, что наложение трахеостомы показано у больных, нуждающихся в продолжительной ИВЛ. Трахеостомия обеспечивает больший комфорт
пациента по сравнению с интубацией (даже назотрахеальной), позволяет более эффективно санировать дыхательные пути и обеспечивать их проходимость [177]. Оптимальным временем для наложения трахеостомы считают первую неделю проведения ИВЛ, если отсутствуют перспективы скорой экстубации [178]. Кроме того, к преимуществам трахеостомии относятся: возможность проведения ВчИВЛ с сохраненным спонтанным дыханием больного (применяя интубационную трубку использовать этот режим очень затруднительно); большая свобода движений больного; облегченный и более безопасный перевод на самостоятельное дыхание.
Более широкие показания к трахеостомии должны быть у нейрохирургических больных, так как интубация сама по себе повышает внутричерепное давление [179]. С целью санации дыхательных путей наложение трахеостомы показано у пациентов в коме, с хронически нарушенным механизмом кашля.
Однако трахеостомия может сопровождаться целым рядом осложнений и даже летальными исходами в 2 % случаев (для сравнения — при эндотрахеальной интубации один случай на 5000) [180]. Интра-и послеоперационные осложнения включают: пневмоторакс (5 %), кровотечения (5 %), острые нарушения вентиляции в связи со смещением или неправильной постановкой трахеостомической канюли, деканюляцией [177]. Для уменьшения количества осложнений при трахеостомии операция должна выполняться, за редкими исключениями, под интубационным наркозом (даже под местной анестезией безопасней оперировать с интубацией трахеи). Осложнения, связанные с нарушением вентиляции и асфиксией вследствие невозможности реканюлировать больного, могут встречаться в течение всего периода ИВЛ, пока у больного не восстановится адекватное самостоятельное дыхание. Каждая деканюляция пациента, чаще всего связанная со сменой трахеостомической канюли, должна производиться при полной готовности к экстренной интубации. Сейчас все большее распространение получает чрескожная дилатационная трахеосто
Искусственная вентиляция легких 137
мия, не требующая оперативного вмешательства.
К поздним осложнениям трахеостомии относится главным образом стенозирование трахеи, которое наблюдается через некоторое время после деканюляции и сопровождается стридорозным дыханием, а в запущенных случаях — асфиксией. Стенозирование может произойти на уровне: наложения трахеостомы (как правило, это связано с неправильной хирургической методикой, нагноением раны), манжеты трахеостомической канюли и нижнего края канюли. Для уменьшения риска этих осложнений необходимы: квалифицированное наложение трахеостомы, совместный уход за раной анестезиолога и хирурга, систематическая (не реже чем через 4 ч разгерметизация манжеты канюли минимум на 5 мин), применение качественных трахеостом (Portex), периодический фиброскопический контроль за состоянием трахеи.
Через неделю и в более длительные сроки применения ИВЛ может развиться такое грозное осложнение, как позднее кровотечение в дыхательные пути, связанное с эрозией сосуда [20]. Особенно опасно кровотечение из безымянной артерии. Эти кровотечения, как правило, связаны с развивающейся ишемией и некрозом стенки трахеи, вызванными давлением интубационной трубки, трахеостомической канюли или раздувной манжеты.
И интубация трахеи, и трахеостомия часто осложняются инфицированием дыхательных путей с развитием эндобронхита, поэтому борьба с инфекцией является очень важным компонентом терапии у больных на ИВЛ. Основные мероприятия по профилактике и лечению инфекции нижних дыхательных путей сводятся к применению антибактериальных фильтров, соблюдению санитарных мероприятий персоналом ОИТ, борьбе с госпитальной инфекцией, адекватному увлажнению и согреванию вдыхаемой газовой смеси, активной позиционной терапии, своевременной аспирации мокроты из дыхательных путей, рациональной антибиотикотерапии.
Виды осложнений в результате длительной интубации или трахеостомии представлены на рис. 4.4.
Рис. 4.4. Осложнения назо- и оротрахеальной интубации и трахеостомии [20]:
1 — внутричерепная гипертензия; 2 — синуситы; 3 - некротическое кровотечение; 4 — повреждение зубов; 5 — обструкция; 6 — стеноз трахеи; 7 — инфекционные кровотечения; 8 — эрозия безымянной артерии; 9 — фистула пищевода; 10 — повреждение гортани; 11 — аспирация;
12 — геморрагия из задней стенки глотки
Кондиции вдыхаемого газа. Адекватные кондиции вдыхаемого газа являются важным условием успешной ИВЛ. Подаваемая в нижние дыхательные пути газовая смесь должна увлажняться и согреваться. Оптимальными для функции мерцательного эпителия являются следующие параметры: 100 % увлажнение при температуре 32 °C или 75 % увлажнение при 37 °C (100% увлажнение при температуре 37 °C является излишним) [3]. Чрезмерное увлажнение может приводить к повреждению сурфактанта и ателектазированию альвеол. Уровень необходимого увлажнения газовой смеси зависит и от FzO2. Так, при дыхании воздухом абсолютное увлажнение составляет примерно 5,2 мг/л, при вдыхании смеси с 50 % О2 — 4,1, а чистого О2 — 3,6 мг/л. Недостаточное согревание газовой смеси, кроме повреждения мерцательного эпителия нижних дыхательных путей может привести к большим потерям жидкости организмом, особенно у седированных больных. Так же опасно и чрезмерное согревание, поскольку тер
138 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
мальное повреждение эпителия трахеи развивается при температуре газа выше 40 °C [181]. В современных респираторах обеспечивается режим согревания и увлажнения, близкий к оптимальному.
4.5.6. Двигательный режим во время ИВЛ, седация и миорелаксация
Как уже отмечалось в разделе «Клиническая физиология дыхания», регионарная вентиляция и перфузия легких во многом зависит от положения тела, кроме того, при его изменении эффективность бронхиального дренажа меняется. Доказано, что у больных на ИВЛ в положении на спине развивается коллапс альвеол дорзальных отделов легких, увеличивается шунтирование в легких, величина которого обратно пропорциональна величине ФОБ [105]. Применение седативных препаратов и миорелаксантов способствует снижению ФОБ [182] и увеличению шунта [183]. Таким образом, систематическая смена положения больных, находящихся на ИВЛ, является обязательным компонентом терапии. Подробнее о так называемой кинетической, или позиционной, терапии см. «ОРДС».
Длительное систематическое применение седативных препаратов и миорелаксантов, так же, как и иммобилизация, уменьшает вероятность благоприятного прогноза лечения. По нашему мнению, эти средства систематически следует применять только у больных, у которых ИВЛ С Ppeak — < 40 см вод. ст. (3920 Па), а Рplateau^ 30 — 35 см вод. ст. (2940 — 3430 Па) в режиме ПДКВ и с инвертированным вдохом с FiО2 = 0,6 не обеспечивает адекватную оксигенацию (РаО2 80 мм рт. ст.) либо адекватную элиминацию углекислого газа. В остальных случаях синхронизацию респиратора к больному (а не наоборот) следует осуществлять путем подбора соответствующих параметров ИВЛ. Речь идет о систематическом применении этих средств. В случае возникновения экстремальных ситуаций с нарушением вентиляции часто бывает необходимо ввести миорелаксанты до выяснения причины.
Питание у больных на ИВЛ должно быть адекватным по энергетической ценности и составу. Желательно как можно быстрее перейти на раннее энтеральное питание (подробнее см. «ОРДС»).
4.5.7. Осложнения искусственной вентиляции легких
Во время ИВЛ главным образом развиваются осложнения со стороны легких. О вентилятор-индуцированном повреждении речь шла выше. Баротравма легких — это разрывы альвеол с развитием пневмоторакса, пневмомедиастинума (чаще всего при разрыве крупных бронхов), пнев-моперитонеума, подкожная эмфизема мягких тканей шеи, груди и т. д. Особенно опасен напряженный пневмоторакс, так как тяжелые расстройства гемодинамики при этом состоянии развиваются быстрее, чем при спонтанном дыхании. Поэтому при внезапно развившейся артериальной гипотензии, нарушении ритма сердца, отставании при дыхании половины грудной клетки, появлении тимпанического оттенка при перкуссии, повышении РаСО2 следует срочно сделать рентгенографию грудной клетки и исключить напряженный пневмоторакс. При его наличии необходима срочная пункция плевральной полости во II межреберье по среднеключичной линии с последующей манометрией, а в случае невозможности удержать отрицательное давление — произвести дренирование. Однако дренаж не может гарантировать от повторной компрессии. Чаще всего причиной этого является нарушение проходимости дренажной трубки, поэтому ее надо периодически промывать, проводить систематическую манометрию. Особенно высокий риск баротравмы у больных со сниженной растяжимостью легких (ОРДС), повышенным динамическим сопротивлением нижних дыхательных путей (бронхиальная астма).
К осложнениям ИВЛ со стороны легких относят также трахеобронхит (наиболее частое осложнение) и вентилятор-ассоциированную пневмонию.
Методы профилактики ВАП по J. Y. Fa-gon [184] с изменениями следующие:
Искусственная вентиляция легких 139
♦ максимально использовать неинвазивные методы ИВЛ;
♦ избегать длительного применения седативных средств и миорелаксантов;
♦ по возможности быстрее удалять назогастральный зонд;
♦ преимущественно использовать положение пациента с приподнятым на 30° головным концом;
♦ по ограниченным показаниям применять средства, снижающие pH желудочного содержимого;
♦ по возможности быстрее переводить больных на спонтанное дыхание;
♦ раннее полноценное энтеральное питание;
♦ избегать ненужных переинтубаций трахеи;
♦ широко применять кинетическую и физиотерапевтическую терапию;
♦ соблюдать асептику при аспирации трахеи;
♦ тщательно санировать полость рта;
♦ ни в коем случае без стерилизации повторно не использовать респиратор, отсос у другого больного;
♦ контролировать антибиотикотерапию в ОИТ.
Кроме того, одним из профилактических мероприятий является широкое применение ВчИВЛ, при которой риск В АП намного ниже.
Для профилактики и лечения трахеобронхитов очень важно поддерживать адекватные кондиции вдыхаемой газовой смеси. Особое значение имеет своевременная санация дыхательных путей у больных на ИВЛ, прежде всего аспирация их содержимого. Обязательно аспирировать мокроту из обоих главных бронхов (для попадания влево при введении катетера трахею следует смещать вправо). Аспирировать мокроту необходимо в разных положениях больного, часто очень эффективно отхождение содержимого дыхательных путей в положении на животе. При аспирации персоналу ОИТ важно соблюдать санитарно-эпидемиологический режим. При длительной ИВЛ, особенно у больных с трахеобронхитом, необходимы систематический ФБС-контроль и санация, а также рентгенографический контроль.
Нарушение функции пищеварительного канала — довольно частое осложнение у больных на ИВЛ. Чаще всего нарушается моторная функция желудка, поэтому в этих случаях важно проведение назо-еюнального зондового питания.
Кровотечения из верхних отделов пищеварительного канала встречаются у 2 — 10 % больных на ИВЛ [3]. Причиной их чаще всего является ишемия слизистой желудка и двенадцатиперстной кишки вследствие гипоксии и нарушения перфузии. Профилактическими мероприятиями в этом случае будут: раннее энтеральное питание, применение алкализирующих препаратов и Щ-блокаторов внутрь (хотя повышение желудочного pH увеличивает риск пневмонии). При появлении признаков кровотечения необходимо проведение гастроскопии.
Бичом многих ОИТ, особенно у больных на ИВЛ, являются пролежни. Для профилактики этого осложнения следует применять активную позиционную терапию, тщательно систематически обрабатывать кожу в местах наиболее частого образования пролежней. Реже развиваются пролежни у больных в ясном сознании, а наиболее часто — у больных с повреждениями спинного мозга.
Кроме В АП, у больных на ИВЛ повышен риск развития других инфекционных осложнений, связанный с наличием венозных, иногда мочевых катетеров, снижением иммунитета, нарушением питания, неадекватной антибиотикотерапией.
Выбор оптимального режима антибио-тикотерапии при длительной ИВЛ является довольно сложной задачей. Мы отдаем предпочтение местному интратрахеально-му введению раствора антибиотика, к которому чувствительна высеваемая микрофлора (хотя статистически достоверных исследований, подтверждающих это положение, нет). Только в случаях тяжелого острого инфекционного процесса (медиастинит, перитонит, пневмония, сепсис, эмпиема плевры) на период, как правило, не превышающий две-три недели, назначается внутривенное введение антибиотика (ов). При отмене антибиотика часто нормализовалась температура тела при лихорадке неясной этиологии.
140 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
4.5.8. Перевод больного на спонтанное дыхание
Это очень важный и ответственный период для благоприятного исхода у больных после длительной ИВЛ. Для отключения от респиратора необходимы определенные предпосылки: положительная динамика течения основного патологического процесса, вызвавшего недостаточность внешнего дыхания; положительная динамика течения процесса в легких (пневмония, ОРДС); отсутствие тяжелых осложнений со стороны других важных органов и систем (прежде всего сердечно-сосудистой, ЦНС, выделительной) и определенные параметры функции внешнего дыхания.
Как правило, чем длительней период ИВЛ, тем дольше происходит перевод на спонтанное дыхание. Чаще всего он длится 5—7 дней, но может затянуться и на несколько недель. Легочное кровообращение адаптируется к повышенному вну-триальвеолярному давлению, поддерживаемому во время ИВЛ, особенно в режиме ПДКВ. При резком переводе на спонтанное дыхание и снижении внутриальвео-лярного давления, даже если функция системы внешнего дыхания восстановилась, могут развиваться явления интерстициального, а иногда и альвеолярного отека легких.
Для более плавного перехода на самостоятельное дыхание применяют режимы вспомогательной вентиляции, но, по нашему мнению, наиболее целесообразен режим ВчИВЛ через трахеостому с сохраненным спонтанным дыханием. Должен соблюдаться принцип постепенности, а также сохранения периодической ИВЛ с ПДКВ. Со временем периоды самостоятельного дыхания увеличиваются, а ВчИВЛ и подключения ПДКВ уменьшаются.
В период отключения от респиратора очень важен адекватный мониторинг функции внешнего дыхания. Необходимо обращать внимание на жалобы больного (если больной в сознании), нарушение функции ЦНС. После перевода на спонтанное дыхание (или режим вспомогательной ИВЛ) частота дыхания должна увеличиваться не более чем на 10 в минуту, частота пульса — изменяться не более чем на 30 в ми
нуту, диастолическое АД — не более чем на 20 мм рт. ст., а систолическое АД — на 30 мм рт. ст. [10]. Намного сложее отключение от респиратора больных, находящихся в коме. Как правило, это больные после черепно-мозговой травмы, нейрохирургической операции или после остановки кровообращения. По сравнению с трахеостомией затруднен перевод на спонтанное дыхание пациентов с интубацией, тем более, что у них нельзя использовать режим ВчИВЛ.
В литературе приводятся показания, обеспечивающие успешное отключение от респиратора, но они мало чем отличаются от тех критериев, которые являются показанием к переводу на ИВЛ.
4.5.9. Неинвазивные методы искусственной вентиляции легких
Метод неинвазивной ИВЛ (НИВЛ) начал применяться в конце 70-х годов XX в., а в последние годы получил широкое распространение. К неинвазивным относятся методы ИВЛ, не требующие интубации трахеи или наложения трахеостомы. Предпосылками для успешного применения НИВЛ являются [3]:
♦ хороший контакт с пациентом;
♦ возможность для пациента контролировать проходимость дыхательных путей;
♦ адекватный кашлевой рефлекс;
♦ возможность для пациента какое-то время дышать самостоятельно;
♦ стабильная функция сердца и сосудов;
♦ сохраненная функция пищеварительного канала;
♦ отсутствие повреждений лица.
НИВЛ не должна применяться у больных с артериальной гипотензией, опасными для жизни нарушениями ритма сердца, высоким риском аспирации (острые процессы в брюшной полости), выраженными отеками, тяжелой гипоксемией (P«O2/FiO2 < 60 мм рт. ст.).
Так же, как и конвенционные, методы НИВЛ разделяют на экспираторные и инспираторные. Экспираторные методы НИВЛ предполагают использование специальных лицевых масок.
Показаниями к экспираторным методам НИВЛ обычно является хроническая
Искусственная вентиляция легких 141
недостаточность дыхания при ХНЗЛ, бронхиальной астме в период обострений. НИВЛ также может использоваться и при острой недостаточности дыхания, в период отключения от респиратора после длительной ИВЛ. Применение НИВЛ у больных с ХНЗЛ доказало свою эффективность, при этом улучшается вентиляция и снижается частота интубаций трахеи [185].
Риск неэффективности НИВЛ повышен у больных пожилого возраста, с высокой оценкой по шкале ISS при травме, тяжелой гипоксемией [186].
Согласно литературным данным, для экспираторной НИВЛ лучше использовать респираторы, регулируемые по давлению, в режиме поддержки давлением (PSV). При НИВЛ применяют специальные маски (носовые или на все лицо).
Для проведения экспираторной НИВЛ используют следующие методы:
1. Дыхание с постоянным положительным давлением в дыхательных путях (continuous positive airway pressure (СРАР)) обеспечивает положительное давление (выше атмосферного) как во время вдоха, так и выдоха. Увеличивает ФОБ, улучшает оксигенацию крови, однако требует адекватного уровня вентиляции пациента. При гиперкапнии не используется, так как может способствовать задержке углекислого газа.
2. ИВЛ с положительным двухуровневым давлением (biphasic positive airway pressure (BIPАР)) обеспечивает вспомогательный режим вентиляции без интубации трахеи. Чаще всего применяется при легкой и умеренной гипоксемии и гиперкапнии. Для ее проведения используется специальная лицевая маска.
Практически все инспираторные методы ИВЛ относятся к неинвазивным. Для проведения этих методов используют несколько видов респираторов (танковые, кирасные и др.). Подробный анализ аппаратуры и методик представлен в специальных руководствах. Рассмотрим только некоторые из них.
Стимуляция диафрагмы. Электрическая стимуляция диафрагмального нерва для длительного поддержания вентиляции
легких впервые была описана в 1968 г. и у отдельных больных используется уже в течение 10 — 15 лет [187]. Эта методика применима при сохранности нижних отделов мотонейронов и диафрагмальной мышцы. Используется у больных с первичной альвеолярной гиповентиляцией и с повреждением спинного мозга выше Сш [188]. Внешняя батарея генерирует радиочастотный сигнал, который передается петлевой антенной, расположенной вблизи подкожного приемника. От него идут электроды к диафрагмальному нерву. Частоту и дыхательный объем можно регулировать частотой и интенсивностью сигнала.
Высокочастотная осцилляция грудной стенки. Для этой цели применяется осциллятор Hayek с частотой 140 осцилляций в минуту и осцилляторным давлением 10 —26 см вод. ст. (980 — 2548 Па). В последнее десятилетие эту методику начали использовать у больных с хроническими обструктивными заболеваниями легких для улучшения оксигенации и выведения СО2 [189].
По последним данным [190], при ИВЛ продолжительностью более двух недель 68 % пациентов были успешно переведены на спонтанное дыхание. Всего за время пребывания в центре наблюдалось 98 летальных исходов (24,3 %). Факторами благоприятного прогноза были: более молодой возраст, большая продолжительность пребывания в специальном респираторном центре, более низкая оценка тяжести заболевания при поступлении, меньшее количество сопутствующих заболеваний со стороны сердца и сосудов. У 31,5 % был использован неинвазивный метод ИВЛ. Выживаемость в течение 3 месяцев составила 67,6 %, одного года — 49,4 %, 3 лет — 38,1 %. При мультивариант-ном анализе длительность жизни во многом зависела от возраста и основного диагноза (т. е. прогноз при хроническом обструкционном заболевании легких был худшим по сравнению с рестриктивным процессом в легких, нервно-мышечным заболеванием и другими причинами). По нашим данным [98], летальность у больных преимущественно с легочной формой недостаточности дыхания за последние
142 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
5 лет удалось снизить до 38,7 %. Причина смерти у этих больных в 95 % случаев не была связана с нарушением дыхания.
В заключение можно отметить, что ИВЛ стала одним из основных методов интен
сивной терапии, который при квалифицированном применении в большинстве случаев способен протезировать функцию вентиляции легких в течение продолжительного времени.
4.6. ОТЕК ЛЕГКИХ
Отек легких (ОЛ) — это состояние, при котором содержание воды в легочном ин-терстиции превышает нормальный уровень [36]. Характеризуется аккумуляцией вне-сосудистой жидкости в легких вследствие увеличения разницы между гидростатическим и коллоидно-осмотическим давлениями в легочных капиллярах.
Причинами, вызывающими отек легких, являются:
1. Увеличение гидростатического давления в легочных капиллярах:
увеличение давления в левом предсердии (левожелудочковая недостаточность, стеноз митрального отверстия);
легочная гипертензия (ХНЗЛ, состояние после обширных резекций легких);
увеличение объема легочного кровотока (ятрогенная передозировка жидкостью, хроническая недостаточность почек);
2. Снижение давления интерстициальной жидкости в легких (легкое быстро расправляется после ателектаза или коллапса).
3. Снижение коллоидно-осмотического давления в легочных капиллярах (гипо-ал ьбуминемия — нефротический синдром, недостаточность печени, разведение крови).
4. Увеличение проницаемости альвеолярно-капиллярной мембраны (ОРДС).
5. Снижение внутриальвеолярного давления (дыхание через маску со сниженной подачей газовой смеси, нарушение проходимости крупных дыхательных путей — гортани, трахеи, главных бронхов).
6. Снижение лимфатического клиренса (резекции легких с множественным удалением лимфоузлов, обширная лимфангиома легких, трансплантация легких).
7. Неясного генеза: нейрогенный отек; при передозировке наркотиков.
4.6.1. Патофизиология отека легких
Важным механизмом противоотечной защиты легких является резорбция жидкости из альвеол, обусловленная главным образом активным транспортом ионов Na+ из альвеолярного пространства с водой по осмотическому градиенту. Транспорт ионов Na+ регулируется апикальными №+-каналами, базолатеральной Na-, К-АТФ-азой и, возможно, хлоридными каналами. Na-, К-АТФ-аза локализована з альвеолярном эпителии. Результаты исследований свидетельствуют о ее активной роли в развитии отека легкого. Механизмы альвеолярной резорбции жидкости нарушаются при развитии отека [191].
В норме у взрослого человека в интерстициальное пространство легких фильтруется приблизительно 10 —20 мл жидкости в час. В альвеолы эта жидкость не попадает благодаря аэрогематическому барьеру. Весь ультрафильтрат выводится через лимфатическую систему. Объем фильтрующейся жидкости зависит согласно закону Франка —Старлинга от таких факторов: гидростатического давления крови в легочных капиллярах (Ргк) и в интерстициальной жидкости (Рги) коллоидно-осмотического (онкотического) давления крови (Ркк) и интерстициальной жидкости (Рки), проницаемости альвеолярно-капиллярной мембраны [192]:
^=^((рпс-^ги)-^кк-^ки)).
где Vf — скорость фильтрации; Kf — коэффициент фильтрации, отражающий проницаемость мембраны; с — коэффициент отражения альвеолярно-капиллярной мембраны; (Ргк - Рги) — разница гидростатических давлений внутри капилляра
Отек легких 143
и в интерстиции; (Ркк - Рки) — разница коллоидно-осмотических давлений внутри капилляра и в интерстиции.
В норме Ргк составляет 10 мм рт. ст., а Ркк — 25 мм рт. ст. [36], поэтому не происходит фильтрации в альвеолы.
Проницаемость капиллярной мембраны для белков плазмы является важным фактором для обмена жидкостей. Если мембрана становится более проницаемой, белки плазмы оказывают меньшее влияние на фильтрацию жидкости, поскольку при этом уменьшается разница концентраций. Коэффициент отражения (с) принимает значения от 0 до 1.
Ргк не следует путать с давлением заклинивания в легочных капиллярах (ДЗЛК), которое больше соответствует давлению в левом предсердии. Для тока крови Ргк должно быть выше ДЗЛК, хотя в норме градиент между этими показателями небольшой — до 1—2 мм рт. ст. [193]. Определение Ргк, которое в норме приблизительно равно 8 мм рт. ст., сопряжено с некоторыми трудностями.
При застойной сердечной недостаточности давление в левом предсердии возрастает в результате снижения сократительной способности миокарда. Это способствует повышению Ргк. Если его значение велико, жидкость быстро выходит в интер-стиций и возникает отек легких. Описанный механизм отека легких часто называют «кардиогенным». При этом повышается и ДЗЛК. Легочная гипертензия приводит к увеличению легочного венозного сопротивления, при этом Ргк также может возрастать, в то время как ДЗЛК падает. Таким образом, при некоторых состояниях гидростатический отек может развиваться даже на фоне нормального или сниженного ДЗЛК. Кроме того, при некоторых патологических состояниях, таких как сепсис и ОРДС, к отеку легких может привести повышение давления в легочной артерии, даже в тех случаях, когда ДЗЛК остается нормальным или сниженным.
Умеренное увеличение Vf не всегда сопровождается отеком легких, поскольку в легких существуют механизмы защиты. Прежде всего к таким механизмам относится увеличение скорости лимфотока.
Поступающая в интерстиций легких жидкость удаляется лимфатической системой. Увеличение скорости поступления жидкости в интерстиций компенсируется увеличением скорости лимфотока вследствие значительного снижения сопротивления лимфатичесих сосудов и небольшого увеличения тканевого давления. Однако если жидкость проникает в интерстиций быстрее, чем она может быть удалена с помощью лимфатического дренажа, то развивается отек. Нарушение функции лимфо-системы легких также приводит к замедлению эвакуации отечной жидкости и способствует развитию отека. Такая ситуация может возникнуть в результате резекции легких с множественным удалением лимфоузлов, при обширной лимфангиоме легких, после трансплантации легких.
Любой фактор, приводящий к снижению скорости лимфотока, увеличивает вероятность образования отека. Лимфатические сосуды легкого впадают в вены на шее, которые, в свою очередь, впадают в верхнюю полую вену. Таким образом, чем выше уровень ЦВД, тем большее сопротивление приходится преодолевать лимфе при ее дренировании в венозную систему. Поэтому скорость лимфотока при нормальных условиях непосредственно зависит от величины ЦВД. Увеличение ЦВД может значительно снизить скорость лимфотока, что способствует развитию отека. Этот факт имеет большое клиническое значение, поскольку многие терапевтические мероприятия у больных в критическом состоянии, например вентиляция с постоянным положительным давлением, инфузионная терапия и применение вазоактивных препаратов, приводят к повышению ЦВД и, таким образом, увеличивают склонность к развитию отека легких. Определение оптимальной тактики инфузионной терапии как в количественном, так и в качественном аспекте является важным моментом лечения.
Эндотоксемия нарушает функцию лимфатической системы. При сепсисе, интоксикации другой этиологии даже небольшое повышение ЦВД может привести к развитию тяжелого отека легких.
Хотя повышенное ЦВД усугубляет процесс накопления жидкости при отеке лег
144 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
ких, вызванном увеличением давления в левом предсердии или повышенной проницаемостью мембраны, однако мероприятия по снижению ЦВД представляют риск для сердечно-сосудистой системы больных в критическом состоянии. Альтернативой могут стать мероприятия, позволяющие ускорить отток лимфатической жидкости из легких, например, дренирование грудного лимфатического протока.
Повышению разницы между Ргк и Рги способствуют обширные резекции легочной паренхимы (пневмонэктомии, особенно справа, двусторонние резекции). Риск отека легких у таких больных, особенно в раннем послеоперационном периоде, высок.
Из уравнения Э. Старлинга следует, что снижение разницы между Ркк и Рки, наблюдаемое при уменьшении концентрации белков крови, прежде всего альбуминов, также будет способствовать возникновению отека легких.
Отек легких може развиваться при дыхании в условиях резко увеличенного динамического сопротивления дыхательных путей (ларингоспазм, обструкция гортани, трахеи, главных бронхов инородным телом, опухолью, неспецифическим воспалительным процессом, после хирургического сужения их просвета), когда на его преодоление затрачивается сила сокращения дыхательных мышц, при этом значительно снижается внутригрудное и внутриальвео-лярное давление, что приводит к стремительному повышению градиента гидростатического давления, увеличению выхода жидкости из легочных капилляров в ин-терстиций и затем в альвеолы. В таких случаях компенсация кровообращения в легких требует времени и выжидательной тактики, хотя иногда необходимо применить ИВЛ.
Одним из наиболее трудных для коррекции является отек легких, связанный с нарушением проницаемости альвеолярнокапиллярной мембраны, что характерно для ОРДС. Особенности развития отека при этом состоянии рассмотрены в соответствующем разделе.
Этот вид отека легких возникает в некоторых случаях внутричерепной патоло
гии. Патогенез его не совсем ясен. Возможно, этому способствует повышение активности симпатической нервной системы, массивный выброс катехоламинов, особенно норадреналина. Вазоактивные гормоны могут вызвать непродолжительное по времени, но значительное по силе повышение давления в легочных капиллярах. Если такой скачок давления достаточно продолжителен или значителен, происходит выход жидкости из легочных капилляров, несмотря на действие противоотеч-ных факторов. При этом виде отека легких следует как можно быстрее устранить гипоксемию, поэтому показания к использованию ИВЛ в данном случае шире.
Отек легких может возникнуть и при отравлении наркотическими средствами. Причиной могут быть нейрогенные факторы и эмболизация малого круга кровообращения (М. L. Callahan, 1987).
Небольшое избыточное накопление жидкости в легочном интерстиции переносится организмом хорошо, однако при значительном увеличении объема жидкости происходит нарушение газообмена в легких. На ранних этапах накопление избытка жидкости в легочном интерстиции приводит к снижению эластичности легких, и они становятся более жесткими. Исследование функций легких на этом этапе выявляет наличие рестриктивных расстройств. Одышка является ранним признаком увеличения количества жидкости в легких, она особенно типична для пациентов со сниженной эластичностью легких. Скопление жидкости в интерстиции легких снижает их растяжимость (compliance), тем самым увеличивая работу дыхания. Для снижения эластического сопротивления дыханию больной дышит поверхностно.
Основной причиной гипоксемии при ОЛ является снижение скорости диффузии кислорода через альвеолярно-капиллярную мембрану (увеличивается расстояние диффузии), при этом повышается альвеолярно-артериальная разница по кислороду. Усиливает гипоксемию при отеке легких также нарушение вентиляционно-перфузионных соотношений. Заполненные жидкостью альвеолы не могут участвовать
Отек легких 145
в газообмене, что приводит к возникновению в легких участков со сниженным показателем вентиляция/перфузия, увеличению фракции шунтируемой крови. Углекислый газ гораздо быстрее (примерно в 20 раз) диффундирует через альвеолярно-капиллярную мембрану, кроме того, нарушение соотношения вентиляция-перфузия мало отражается на элиминации углекислого газа, поэтому гиперкапния наблюдается только на терминальном этапе ОЛ и является показанием к переводу на ИВЛ.
4.6.2. Клинические проявления отека легких
ОЛ проявляется чувством нехватки воздуха, бледностью и цианозом кожных покровов, одышкой, вынужденным положением больного (сидя) — ортопноэ, пароксизмальной ночной одышкой, появлением III сердечного тона, влажными хрипами в легких, которые часто слышны даже на расстоянии, непродуктивным кашлем, иногда с трудом откашливается мокрота розового цвета. При застойной недостаточности левого желудочка сердца у больного диагностируется сердечная астма. На рентгенограмме легких видны специфические для отека легких изменения: усиленный сосудистый рисунок, нечеткие контуры сосудов, увеличение размеров сердечной тени, инфильтрация в перибронхиальных отделах. Рентгенологические проявления в некоторой степени зависят от этиологии отека и могут отличаться при ОРДС и застойной сердечной недостаточности.
Рентгенографические признаки отека легких (S. J. Allen, 1999):
усиленный сосудистый рисунок, нечеткие контуры сосудов, увеличение размеров сердечной тени, появление линий Керли А (длинные, располагаются в центре легочного поля), появление линий Керли В (короткие, размещены по периферии), инфильтрация в перибронхиальных отделах, появление силуэта «летучей мыши» или «бабочки», выпот в плевральную полость, появление ацинарных теней (участки консолидации, имеющие пятнистый вид).
4.6.3. Лечение отека легких
В случае развития острого отека легких требуется проведение экстренной интенсивной терапии. Тактика лечения во многом зависит от этиопатогенеза отека.
При условии повышенного или нормального артериального давления при гидростатическом отеке кардиогенной этиологии осуществляют следующие мероприятия: больному придают приподнятое полусидячее положение; проводят ингаляцию кислорода; для снижения гидростатического давления в малом круге применяют нитраты, прежде всего нитроглицерин, сначала сублингвально (по 0,8 мг), затем внутривенно капельно (10 — 40 мкг/мин), или натрий нитропруссид (начальная доза 10 мкг/мин) под постоянным контролем АД. М. Я. Руда (1993) считает, что дозу этих препаратов можно повышать до прекращения симптоматики отека легких или до снижения систолического артериального давления у нормотоников до 100 мм рт. ст. Иногда дозу нитроглицерина необходимо увеличить до 100 — 200 мкг/мин. Нитраты снижают гидростатическое давление в легочной артерии, а также улучшают венечное кровообращение.
Рекомендаций по применению ранее широко используемых ганглиоблокаторов (арфонад, пентамин) в современной литературе по лечению отека легких мы не встретили.
В комплексной терапии отека легких применяется морфина гидрохлорид, который вводят внутримышечно (при отсутствии депрессии дыхания — внутривенно) в дозе 5—10 мг. Морфин обладает седативным эффектом, снижает возбудимость дыхательного центра, уменьшает одышку, расширяет вены, уменьшая преднагрузку и постнагрузку для левого и правого желудочков, однако, может вызвать депрессию дыхания (его антагонистом является налоксон).
Для снижения преднагрузки и гидростатического давления в малом круге также применяют быстродействующие мочегонные препараты, в частности фуросемид, который вводят внутривенно по 20 — 40 мг.
146 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
Особенно показано применение этих препаратов, если отек легких развился на фоне передозировки жидкости.
При наличии бронхиолоспазма назначают 2,4 % раствор эуфиллина, который вводят внутривенно по 5 —10 мл. Однако применение этого препарата не всегда безопасно, особенно при тахиаритмиях, острой ишемии миокарда. Также назначают глюкокортикоиды.
В зависимости от вида расстройств деятельности сердца могут применяться другие кардиотропные препараты.
Инфузия раствора альбумина показана при гипоальбуминемии, в противном случае такая терапия может повышать гидростатическое давление и способствовать отеку.
Любая внутривенная инфузионная терапия должна проводиться под контролем ЦВД. При тяжелом, плохо поддающемся лечению отеке легких должна быть катетеризирована центральная вена. Нельзя повышать ЦВД выше нормы, но опасной также является тактика выраженной дегидратации, особенно в течение длительного периода.
При отсутствии эффекта от терапии в случае выраженной гипоксемии и гиперкапнии показано применение искусственной вентиляции легких с сопротивлением на выдохе (ПДКВ) (подробнее см. в «ИВЛ»). Использование ПДКВ повышает внутри-
альвеолярное давление, улучшает диффузию газов через альвеолярно-капиллярную мембрану, уменьшает шунтирование в малом круге, снимает нагрузку, связанную с повышенной работой дыхания.
В тех случаях, когда острый отек легких развивается на фоне артериальной гипотензии, необходимо применение кардиотонических препаратов (допамин, добутамин, ингибиторы фосфодиэстеразы, допекса-мин). Допамин следует использовать в сочетании с инфузией нитратов. В подобных случаях больных часто приходится переводить для купирования отека на ИВЛ. Необходимо помнить, что применение ПДКВ может еще больше снизить сердечный выброс, особенно на фоне гиповолемии, поэтому требуется тщательный мониторинг гемодинамических и респираторных показателей для выбора оптимального режима терапии.
Имеются данные о применении при наличии рефрактерного к терапии отека легких ультрафильтрации крови, однако, если причиной развития отека является недостаточность сократительной способности миокарда, такая терапия даст только кратковременный эффект (М. Я. Руда, 1993).
Отек легких при ОРДС, сужении дыхательных путей требует специальной терапии.
Список использованной литературы
1. Webb A. R. Acute respiratory failure // Critical Care Therapeutics / Ed. R. Elliott. — Pharmceutical Press, 1999. — 340 p.
2. Kelly B. J., Luce J. M. The diagnosis and management of neuromuscular diseases causing respiratoryfailure //Chest. — 1991. — 99. — P. 1485-1494.
3. Sykes K., Young J. D. Respiratory Support in Intensive Care. — BMY Books, 1999. — 305 p.
4. Hund E., Genswurker H., Bohrer H. et al. Predominant involvement of motor fibers in patients with critical illness neuropathy // Br. J. Anaesth. - 1997. - 78. - P. 274-278.
5. Smith C. £., Hunter J. M., Jones R. S. Vecuronium infusions in patients in renal failure in ICU // Anaesthesia. — 1987. — 42. — P. 387-393.
6. Margolis B. D., Khachikian D., Friedman Y. et al. Prolonged reversible quadriparesis in mechanically ventilated patients who received long-term infusions of vecuronium // Chest. — 1991. - 100. - P. 877-878.
7. Lodato R. F. Oxygen toxicity // Crit. Care Clin. - 1990. - 6. - P. 749-765.
8. Winter P. M., Smith G. The toxicity of oxygen // Anesthesiology. — 1972. — 37. -P. 210-212.
9. US Department of Commerce, Bureau of the Census: Statistical Abstract of the United States. — Washington: US Goverment Printing Office, 1993. — 113 p.
10. Marini J. J., Wheeler A. P. Critical Care Medicine. — 2nd ed. — Williams & Wilkins, 1997. - 640 p.
11. Foy H. M., Cooney M. K., Allan L, Kenny G. E. Rates of pneumonia during influenza epidemics in Seattle // JAMA. — 1979. -241. - P. 253-258.
12. Porath A., Shlaeffer F., Lieberman D. The epidemiology of community-acquired pneumonia among hospitalized adults // J. Infect. -1997. - 34. - P. 41-48.
13. Campbell G. D. Overview of community-acquired pneumonia. Prognosis and clinical
Список использованной литературы 147
features // Med. Clin. North Am. — 1994. — 78. - P. 1035-1048.
14. Habibi S., Coursin D. B. Care of the Patient with Pneumonia // Critical Care Medicine / Ed. M. J. Murray,D. B. Coursin,R. G. Pearl, D. S. Prough. — Philadelphia — New York: Lippinctot-Raven, 1997. — P. 415 — 426.
15. Bartlett J. G., Mundy L. M. Community-acquired pneumonia // NEJM. — 1995. — 333. - P. 1618-1624.
16. Мэрри T. Внебольничная пневмония // Доказательная медицина / Пер. с англ. — М.: Медиа Сфера. — С. 82 — 97.
17. Meeban Т. Р., Fine М. J., Krumbolz Н. М. et aL Quality of care, process, and outcomes in elderly patients with pneumonia //JAMA. — 1997. - 278. - P. 2080-2084.
18. Ramirez J. A., Ahkee S. Early switch from intravenous antimicrobials to oral clarithromycin in patients with community aquired pneumonia// Infect. Med. — 1997. — 14. — P. 319-323.
19. Bjorkqvist M., Wiberg B., Bodin L. et al. Bottle-blowing in hospital-treated patients with community-aquired pneumonia // Scand. J. Infect. - 1997. - 29. - P. 77-82.
20. Marino P. L. The ICU Book. — 2nd ed. — Philadelphia: Lippincot Williams & Wilkins, 1997. - 928 p.
21. Fagon J.-Y., Chastre J., Vuagnet A. et al. Nosocomial pneumonia and mortaling among patients in intensive care units // JAMA. — 1996. - 275(11). - P. 866-869.
22. Garner J. S., Jarvis W. R., Emori T. G. et al. CDC definitions for nosocomial infections // Am. J. Infect. Control. — 1988. — 16. — P. 128-140.
23. Smith R. M., Spragg R. G. The lung in the intensive care unit // Ed. R. G. Crystal, J. B. West. The Lung. — New York: Raven Press, 1991. — Vol. 2. - P. 2185-2196.
24. Vincent J. L., Bichari D. J., Suter P. M. et al. The prevalence of nosocomial infection in intensive care units in Europe // JAMA. — 1995. - 274. - P. 639-644.
25. Зубков M. H., Зубков M. M. Госпитальные пневмонии: этиология, патогенез, диагностика, профилактика и лечение // Consilium medicum. — 2000. — 2, № 1. — С. 56 — 64.
26. Torres A., Aznar R., Gatell J. M. et al. Incidence, risk, and prognosis factors of nosocomial pneumonia in mechanically ventilated patients //Amer. Rev. Resp. Dis. — 1990. — 142. — P. 523-528.
27. Глумчер Ф. С., Дубров С. О. Вентилятор-асощйована пневмошя // Bi ль, знеболювання i штенсивна терашя. — 2001. — №4. — С. 58-73.
28. Guidelines for the Diagnosis and Management of Asthma: Expert Panel report 2. — National Institutes of Health, 1997. — N 97. — P. 9.
29. Woolcock A. J., Peat J. K. Evidence for an increase in asthma world-wide // Ciba Found Symp. - 1997. - 206. - P. 122-134.
30. National Heart, Blood and Lung Institute. National Asthma Education and Prevention Program. Expert Panel Report 2. Guidelines for the Diagnosis and Management of Asthma. — NIH Publication N 97 — 4051, 1997. — P. 20.
31. Djucanovic R., Roche W. R., Wilson J. W. et al. Management of Asthma // Am. Rev. Respir. Dis. - 1990. - 142. - P. 434-457.
32. Emanuel M. B., Howarth P. H. Asthma and anaphylaxis: a relevent model for chronic disease? // Clin. Exp. Allergy. — 1995. — 25. - P. 15-26.
33. Albeida S. M. Endothelial and epithelial cell adhesion molecules // Am. J. Respir. Cell. Mol. Biol. - 1991. - 4. - P. 195-203.
34. Roche W. R. Fibroblasts and asthma // Clin. Exp. Allergy. - 1991. - 21. - P. 545-548.
35. Prendergast T. J., Ruose S. J. Pulmonary disease // Pathophysiology of Disease. Ed. S. J. McPhee et al. — 3 ed. — McGraw-Hill, 2000. - P. 185-221.
36. Ганонг В. Ф. ЗИзюлопя людини / Пер. з англ, за ред. М. Гжегодського, В. Шевчука, О. Заячювсько!. — Л.: БаК, 2002. — 784 с.
37. Marshall J. S., Bienenstock J. The role of mast cells in inflammatory reactions of the airways, skin and intestine // Curr. Opin. Immunol. — 1994. — 6. — P. 853 — 859.
38. Busse W. W., Calhoun W. J., Sedgwick J. D. Mechanisms of airway inflamation in asthma // Am. Rev. Respir. Dis. — 1993. — 147. — P. S20-24.
39. Kay A. B. Asthma and inflamation // J. Allergy Clin. Immunol. — 1991. — 87. — P. 893-945.
40. Dellinger R. P. Life-threatening bronchospasm in the asthmatic patient // Critical Care / Ed. J. M. Civetta, R. W. Taylor, R. R. Kirby. — 3d ed. — Philadelphia: Lippincot-Raven, 1997. - P. 1931-1946.
41. Lanes S. F., Garsia Rodriguez L. A., Huerta C. Respiratory medications and risk of asthma death//Thorax. — 2002. — 57(8). — P. 683-686.
42. Morrel F., Orriols R., de Cracia J. et al. Controlled trial intravenous corticosteroids in sever acute asthma // Thorax. — 1992. — 47. - P. 588-591.
43. Eliasson O., Hoffman N., Trueb D. et al. Corticosteroids in COPD // Chest. — 1986. — 89. - P. 484-489.
44. Truwit J. D. Toxic effect of bronchodilatators // Crit. Rare Clin. - 1991. - 7. - P. 639-657.
45. Rodrigo G. J., Rodrigo C. First-line therapy for adult patients with acute asthma receiving multiple dose protocol of ipratroprium bromide plus albuterol in the emergency de
148 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
partment // Am. J. Respir. Crit. Care Med. — 2000. - 161. - P. 1862-1868.
46. Litovitz T. L. Annual Report of the American Association of Poison Control Centers Toxic Exposure Surveillance System // Am. J. Emerg. Med. — 1993. — 11. — P. 494 — 555.
47. Cooling D. S. Theophylline toxicity // J. Emerg. Med. — 1993. — 11. — P. 415 — 425.
48. Fish J. E., Kemp J. P.t Lockey R. F. et al. Zafirlucast for symptomatic mild-to-moderate asthma: A 15-week multicenter study // Clin. Ther. - 1997. - 19. - P. 675-690.
49. Buysse С. M. P., de Hoog M. Trearment of status asthmaticus in children: is there a place for sodium bicarbonate? // Crit. Care. — 2002. - 6 (Suppl. 1). - P. S13.
50. Mutlu G. M.t Factor P., Schwartz D. E., Sznajder J. I. Severe status asthmaticus: management with permissive hypercapnia and inhalation anesthesia // Crit. Care Med. — 2002. - 30(2). - P. 477-480.
51. Molfino N. A., Nannimi A., Martelli A. N., Slutsky A. S. Respiratory arrest in near fatal asthma //N. Engl. J. Med. - 1991. - 324. -P. 285-288.
52. Kass J. E., Torregino C. A. The effect of helios in acute severe asthma: a randomise controle trial // Chest. - 1999. - 116. - P. 296-300.
53. Nannini L. J., Pendino J. C., Corna R. A. et al. Magnesium sulfate as a vehicle for nebulized salbutamol in acute asthma // Am. J. Med. - 2000. - 108. - P. 193-197.
54. Bethel R. A. Asthma // Critical Care Secrets / P. E. Parsons, J. P. Wiener-Kronish. — Philadelphia: Hanley & Belfus, 1998. — P. 103-107.
55. Lum K. N., Mow В. M., Chew L. S. The profile of ICU admissions for acute severe asthma in a general hospital // Singapore Med. J. - 1992. - 33. - P. 460-462.
56. Mansel J. K., Stogner S. W., Petrini M. F., Norman J. R. Mechanical ventilation in patients with acute severe asthma // Am. J. Med. - 1990. - 89. - P. 42-48.
57. Villar J., Slutsky A. S. The incidence of the adult respiratory distress syndrome // Am. Rev. Respir. Dis. - 1989. - 140. - P. 814-816.
58. Thomsen G. E., Morris A. H. Incidence of the adult respiratory distress syndrome in the state of Utah // Am. J. Respir. Crit. Care Med. - 1995. - 152. - P. 965-971.
59. Luchr O. R., Antonsen K., Karlsson M. et al. Incidence and mortality after acute respiratory distress syndrome in Sweden, Denmark, and Iceland. The ARF Study Group // Am. J. Respir. Crit. Care Med. — 1999. — 159. — P. 1849-1861.
60. Roupie E., Lepage E., Wysocki M. et al. Prevalence, etiologies and outcome of the acute respiratory distress syndrome among
hypoxemic ventilated patients // Inten. Care Med. - 1999. - 25. - P. 920-929.
61. Krafft P., Fridrich P., Pernerstorfer T. et al. The acute respiratory distress syndrome: definition, severity and clinical outcome. An analysis of 101 clinical investigations // Inten. Care Med. - 1996. - 22. - P. 519-529.
62. Rubenfeld G. D. Epidemiology of acute lung injury // Crit. Care Med. — 2003. — 31 (4 Suppl.). - P. S276-284.
63. Jardin F., Fellahi J., Beauchet A. et al. Improved prognosis of acute respiratory distress syndrome (ARDS): 1983 — 1993 // JAMA. - 1999. - 273. - P. 306-309.
64. Ashbaugh D. G., Bigelow D. B., Petty T. L., Levine В. E. Acute respiratory distress in adults // Lancet. - 1967. - II. - P. 319-323.
65. Bernard G. R., Artigas A., Brigham K. L. et al. The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination // Am. J. Respir. Crit. Care Med. -1994. - 149. - P. 818-824.
66. Bigatello L. M., Zapol W. M. New approach to acute lung injury // Br. J. Aneasth. -1996. - 77. - P. 99-109.
67. Estenssoro E., Dubin A., Laffaire E. et al. Incidence, clinical course, and outcome in 217 patients with acute respiratory distress syndrome//Crit. Care Med. — 2002. — 30(11). -P. 2450-2456.
68. Donnelly S. C., Haslett C., Dransfield I. etal. Role of selectin in development of acute respiratory distress syndrome // Lancet. — 1994. -344. - P. 215-219.
69. Casetti A. V., Bartlett R. H., Hirschl R. B. Increasing inspiratory time exacerbates ventilator-induced lung injury during high-pressure/high-volume mechanical ventilation // Crit. Care Med. - 2002. - 30(10). -P. 2295-2299.
70. Dhainaut J. F., Charpentier J., ChicheJ. D. Transforming growth factor-beta: a mediator of cell regulation in acute respiratory distress syndrome // Crit. Care Med. — 2003. -31(4 SuppL). - P. S258-264.
71. GropperM. A., Wiener-Kronish P. Acute lung injury and acute respiratory distress syndrome // Critical Care Medicine / Ed. M. J. Murray et al. — Philadelphia: Lippincott-Raver, 1997. - P. 441-451.
72. Halliwell B. Oxidants and human disease: some new concepts // FASEB J. — 1987. -1. - P. 358-364.
73. Трещинский А. И., Беляев А. В., Глумчер Ф. С. Современный взгляд на основные принципы рациональной антибактериальной терапии в интенсивной терапии // 1мль, зне-болювання i штенсивна терашя. — 2002. -№ 2. - С. 54-71.
74. Thompson В. Т. Glucocorticoids and acute lung injury // Crit. Care Med. — 2003. -31(4 Suppl.). - P. S253-257.
Список использованной литературы 149
75. Foner В. J., Norwood S. И., Taylor R. W. Acute respiratory distress syndrome // Critical Care / Ed. J. M. Civetta, R. W. Taylor, R. R. Kirby. — 3rd ed. — Philadelphia: Lippincott-Raver, 1997. — P. 1825 — 1839.
76. Weling K. L. Acute respiratory distress syndrome. Pathogenesis and therapy of acute pulmonary failure //Ugeskr Laeger. — 1996. — 158(25). - P. 3593-3600.
77. YuM., Tomasa G. A double-blind prospective randomized trial of ketanozol, a tromboxane synthetase inhibitor, in the prophylaxis of the adulte respiratory distress syndrome // Crit. Care Med. - 1993. - 26. - P. 183-192.
78. Глумчер Ф. С. Острый респираторный дистресс-синдром // Бш, знеболювання i iH-тенсивна терашя. — 1999. — № 2(7). — С. 27-50.
79. Корр R.f Kuhlen R., Max M.f Rossaint R. Evidence-based medicine in the therapy of the acute respiratory distress syndrome // Inten. Care Med. - 2002. - 28(3). - P. 244-255.
80. International consensus conferences in intensive care medicine: ventilator-associated lung injury in ARDS // Inten. Care Med. — 1999. — 25. - P. 1444-1452.
81. Drey fuss D., Saumon G. Ventilator-induced lung injury: lessons from experimental studies //Am. J. Respir. Crit. Care Med. — 1998. — 157. - P. 294-323.
82. Page B., Vieillard-Baron A., Beauchet A. et al. Low stretch ventilation strategy in acute respiratory distress syndrome: Eight years of clinical experience in a single center // Crit. Care Med. - 2003. - 31(3). - P. 765-769.
83. Capellier G., Maupoil V., Boussat S. et al. Oxygen toxicity and tolerance // Minerva AnesthesioL — 1999. — 65. — P. 388 — 392.
84. Karlstad M., Palombo J., Murray M., DeMichele S. The anti-inflammatory role of gamma-linolenic and eicosapentaenoic acids in acute lung injury // Gamma-linolenic Acid: Metabolism and its Role in Nutrition and Medicine / Ed. Y. S. Huang, D. Mills. — Champagne AOCS Press, 1996. — P. 137 — 167.
85. Mancuso P., Whelan J., DeMichele S. et al. Effects of eicosapentaenoic and gammalinolenic acids on lung permeability and alveolar macrophage eicosanoid syntesis in endotoxic rats // Crit. Care Med. — 1997. — 25. - P. 523-532.
86. Amato M. B., Barbas C. S., Medeiros D. M. et al. Effect of protective-ventilation strategy on mortality in the acute respiratory distress syndrome // N. Engl. J. Med. — 1998. — 338. - P. 347-354.
87. The Acute Respiratory Distress Syndrome Network (2000). Ventilation with lower tidal volumes for acute lung injury and the acute respiratory distress syndrome //
N. Engl. J. Med. - 2000. - 342. - P. 1301 -1308.
88. Ar tig as A., Bernard G. R., Carlet J. et al. The American-European Consensus Conference on ARDS, part 2 // Inten. Care Med. — 1998. - 24. - P. 378-398.
89. Ranieri V. N., Suter P. M., Tortorella C. et al. Effect of mechanical ventilation on inflammatory mediators in patients with ARDS: a randomized controlled trial // JAMA. — 1999. — 282. - P. 54-61.
90. Lapinsky S. E., Aubin M., Mehta S. et al. Safety and efficacy of a sustained inflation for alveolar recruitment in adults with respiratory failure // Inten. Care Med. — 1999. — 25. - P. 1297-1301.
91. Pfeiffer B.f Hachenberg T.t Wendt M., Marshall B. Mechanical ventilation with permissive hypercapnia increases intrapulmonary shunt in septic and nonseptic patients with acute respiratory distress syndrome // Crit. Care Med. - 2002. - 30(2). - P. 285-289.
92. Ward N. S., Levy M. M. Titrating Optimal PEEP at the bedside // Yearbook of Intensive Care and Emergency Medicine / Ed. J.-L. Vincent. — Springer, 2002. — P. 297 — 304.
93. Dos Santos С. C., Slutsky A. S. Advances in ARDS: How do they Impact bedside Management? // Yearbook of Intensive Care and Emergency Medicine / Ed. J.-L. Vincent. — Springer, 2002. — P. 320 — 336.
94. Mamprin F., Pesenti A., Fumagalli R. Extracorporeal Circulation for Acute respiratory Failure //Inten. Care Med. — 2001. — 27. — P. 934-936.
95. Koutsoukou A., Roussos C., Milic-Emili J. Effect of PEEP and Targets during Mechanical Ventilation in ARDS // Yearbook of Intensive Care and Emergency Medicine / Ed. J.-L. Vincent. — Springer, 2002. — P. 305-312.
96. Глумчер Ф. С., Макаров А. В., Суслов Г. Г. и др. Результаты использования современных методов респираторной поддержки // Б1ль, знеболювання i штенсивна терашя. — 2003. - № 2. - С. 8-15.
97. Reutershan J., Schmitt A., Fretchner R. Alveolar recruitment improves arterial oxygenation in responders to prone position // Crit. Care. — 2002. — 6 (Suppl. 1). — P. S9.
98. Valente Barbas C. S. Lung recruitment maneuvers in acute respiratory distress syndrome and facilitating resolution // Crit. Care Med. - 2003. - 31(4 Suppl.). - P. S265-271.
99. Rouby J. J., Puybasset L., Nieszkowska A., Lu Q. Acute respiratory distress syndrome: lessons from computed tomography of the whole lung // Crit. Care Med. — 2003. — 31(4 SuppL). - P. S285-295.
150 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
100. Esteban A.f Alia I., Gordo Е. et al. Prospective randomized trial comparing pressure-controlled ventilation and volume-controlled ventilation in ARDS // Chest. — 2000. — 117. - P. 1690-1696.
Zhang H,, Downey G. P.f Suter P. M. et al. Conventional mechanical ventilation is associated with bronchoalveolar lavage-induced activation of polymorphonuclear leukocytes: a possible mechanism to explain the systemic consequences of ventilator-induced lung injury in patients with ARDS // Anesthesiology. - 2002. - 97(6). - P. 1426-1433.
102. Piehl M. A., Brown R. S. Use of extreme position changes in acute respiratory failure // Crit. Care Med. - 1976. - 4. - P. 13-14.
103. L’Her E., Renault A., Oger E. et al. A prospective survey of early 12-h prone positioning effects in patients with the acute respiratory distress syndrome // Inten. Care Med. - 2002. - 28. - P. 570-575.
104. Cooper K. R., Boswell P. A. Reduced functional residual capacity and abnormal oxygenation in patients with sever head injury // Chest. - 1983. - 84. - P. 29-35.
105. Puybasset L., Cluse I P., Chao N. et al. A computed tomography scan assessment of regional lung volume in acute lung injury. The CT scan ARDS Study Group // Am. J. Respir. Crit. Care Med. — 1998. — 158. — P. 1644-1655.
106. Broccard A. F., Shapiro R. S., Schimitz L. L. et al. Influence of prone position on the extent and distribution of lung injury in a high tidal volume oleic acid model of acute respiratory distress syndrome // Crit. Care Med. — 1997. - 25. - P. 16-27.
107. Beuret P., Carton M. J., Nour dine K. et al. Prone position as prevention of lung injury in comatose patients: a prospective, randomized, controlled study // Inten. Care Med. — 2002. - 28. - P. 564-569.
108. Fridrich P.f Kraft P., Hochleuther H., Mau-ritz W. The effects of long-term prone positioning in patients with trauma-induced ARDS //Anesth Analg. — 1996. — 83. — P. 1206 — 1211.
109. Jolliet P., Bulpa P., Chevrolet J. C. Effects of the prone positioning on gas exchange and hemodynamics in severe ARDS // Crit. Care Med. - 1998. - 26. - P. 1977 — 1985.
110. Staudinger T., KoflerJ., Mullner M. et al. Comparison of prone positioning and continuous rotation of patients with adult respiratory distress syndrome: results of a pilot study // Crit. Care Med. - 2001. - 29(1). -P. 51-56.
111. Beuret P., Carton M. J., Nourdine K. et al. Prone position as prevention of lung injury in comatose patients: a prospective, randomized, controlled study // Inten. Care Med. — 2002. - 28. - P. 564-569.
\\2.Priolet B., Tempelhoff G., Millet J. et al. Ventilation assistee en decubitus ventral: evaluation tomodensi-tometrique de son effica-cite dans le traitement des condensations pulmonaires // Rean Urg. — 1993. — 2. -P. 81-85.
113. Leonet S., Fontaine C., Moraine J. J., Vincent J. L. Prone positioning in acute respiratory failure: survey of Belgian ICU nurses // Inten. Care Med. — 2002. — 28. -P. 576-580.
114. Webster N. R. Ventilation in the prone position // Lancet. — 1997. — 349. — P. 1638— 1639.
115. Stocker R.f Neff T., Steine S. et al. Prone positioning and low-volume pressure-limited ventilation improve survival in patients with severe ARDS // Chest. — 1997. — 111. -P. 1008-1117.
116. Krishnan J. A., Brower R. G. High-frequency ventilation for acute lung injury and ARDS // Chest. - 2000. - 118. - P. 795-807.
117. Carlon G. C., Howland W. S., Ray C. et al. High-frequency jet ventilation. A prospective randomized evaluation // Chest. — 1983. -84. - P. 551-559.
118. Hurst J. M., Branson R. D., Davis K. L. et al. Comparison of conventional mechanical ventilation and high-frequency ventilation // Ann. Surg. - 1990. - 211. - P. 486-491.
119. Zapol W. M.t Snider M. T., Hill J. D. et al. Extracorporeal membrane oxygenation sever acute respiratoryfailure //JAMA. — 1979. -242. - P. 2193-2196.
120. Morris A. H., Wallace C. J., Menlove R. L. et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for ARDS // Am. J. Respir. Crit. Care Med. — 1994. — 149. -P. 295-305.
121. Gattinoni L., Pesenti A., Mascheroni D. etal. Low-frequency positive-pressure ventilation with extracorporeal CO2 removal in sever acute respiratory failure // JAMA. — 1986. -256. - P. 881-886.
122. Bindslev L., Eklund J., Norlander O. et al. Treatment of acute respiratory failure by extracorporeal CO2 elimination performed with a surface htparinized artificial lung // Anesthesiology. — 1987. — 67. — P. 117— 120.
123. Michaels A. J.t Schriener R. J., Kolla S. etal. Extracorporeal life support in pulmonary failure after trauma //J. Trauma. — 1999. -46. - P. 638-645.
124. Rossaint R., Falke K. L., Lopez F. et al. Inhaled nitric oxide for the adult respiratory distress sindrome // N. Engl. J. Med. -1993. - 328. - P. 399-405.
125. The Neonatal Inhaled Nitric Oxide Study Group. Inhaled nitric oxide in full-term and nearly full-term infants with hypoxic respi
Список использованной литературы 151
ratory failure //N. Engl. J. Med. — 1997. — 336. - P. 597-604.
126. Putensen C., Hormann C., Kleinsasser A., Putensen H. G. Cardiopulmonary effects of aerosolized prostaglandin El and nitric oxide inhalation in patients with ARDS // Am. J. Respir. Crit. Care Med. — 1998. — 157. — P. 1743-1747.
127. Griese M. Pulmonary surfactant in health and human lung diseases: State of the art // Eur. Respir. J. - 1999. - 13. - P. 1455-1476.
128. Gregory T. J., Longmore W. J., Moxley M. A. et al. Surfactant chemical composition and biophysical activity in ARDS // J. Clin. Invest. - 1991. - 88. - P. 1976-1981.
\29.Anzuetto A., Baughman R. P., Guntupal-li К. K. et al. Aerosolized surfactant in adults with sepsis-induced ARDS // N. Engl. J. Med. - 1996. - 334. - P. 1417-1421.
130. Gregory T. J., Steinberg К. P., Spragg R. et al. Bovine surfactant therapy for patients with ARDS // Am. J. Respir. Crit. Care Med. - 1997. - 155. - P. 1309-1315.
\3i. Lewis J. F., Brackenbury A. Role of exogenous surfactant in acute lung injury // Crit. Care Med. — 2003. — 31(4 Suppl.). — P. S324-328.
132. Croce M. A., Fabian T. C., Patton J. H. J. et al. Partial liquid ventilation decreases thq inflammatory response in the alveolar environment of trauma patients // J. Trauma. — 1998. - 45. - P. 273-280.
133. Bartlett R. H., Croce M. A., Hirsh H. et al. A phase II randomized, controlled trial of partial liquid ventilation (PLV) in adult patients with acute hypoxemic respiratory failure // Crit. Care Med. — 1997. — 25. — P. A35.
134. Weigelt J. A., Norcross J. F.f Borman K. R., Sayder W. H. Early steroid therapy for respiratory failure // Arch. Surg. — 1985. — 120. - P. 536-540.
\33. Bernard G. R.f Luce J. M., Sprung K. L. et al. High dose corticosteroids in patients with the ARDS // N. Engl. J. Med. - 1987. -317. - P. 1565-1570.
136. Bowe R. C., Fisher C. J. J., Clemmer T. P. et al. Early methylprednisolone treatment for septic syndrome and the adult respiratory distress syndrome//Chest. — 1999. — 92. — P. 1032-1036.
137. Hooper R. G., Kearl R. A. Established adult respiratory distress syndrome successfully treated with corticosteroids // South Med. J. — 1996. - 89. - P. 359-364.
\33. Briegel J., Forst H., Haller M. et al. Stress doses of hydrocortisone reverse hyperdynamic septic shock // Crit. Care Med. — 1997. — 27. - P. 723-732.
139. Bernard G. R.f Wheeler A. P., Russell J. A. et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. The Ibuprofen in Sepsis Study Group //
N. Engl. J. Med. - 1997. - 336. - P. 912 — 918.
140. Acute Respiratory Distress Syndrome Network. Ketokonazole for early treatment of acute respiratory distress syndrome: a randomized controlled trial. The ARDS Network //JAMA. - 2000. - 283. - P. 1995-2002.
141. Jepsen S., Herlevsen P., Knudsen P. et al. Antioxidant treatment with ^acetylcysteine during adult respiratory distress syndrome: a prospective, randomized, placebo-controlled study // Crit. Care Med. — 1992. — 20. — P. 918-923.
142. GadekJ. E., DeMichele S. J., Karlstad M. D. et al. Effect of enteral feeding with icosapen-taenoic acid, gamma-linolenic acid, in patients with acute respiratory distress syndrome // Crit. Care Med. — 1999. — 27. — P. 1409-1420.
143. Suter P. M., Domeninghetti G., Schaller M. D. et al. N-Acetylcysteine enhances recovery from acute lung injury in man: a randomized, double-blind placebo controlled clinical study //Chest. - 1994. - 105. - P. 190-194.
144. Ullrich R., Roeder G., Lorber C. Continuous venovenous hemofiltration improves arterial oxygenation in endotoxin-induced lung injury in pigs//Anesthesiology. — 2001. — 95(2). — P. 428-436.
145. Gillart T.f Bazin J. E., Cosserant B. et al. Combined nitric oxide inhalation, prone positioning and almitrine infusion improve oxygenation in severe ARDS // Can. J. Anaesth. — 1998. - 45. - P. 402-409.
146. Michard F., Wolff M. A., Herman B., Wysocki M. Right ventricular response to high-dose almitrine infusion in patients with severe hypoxemia related to acute respiratory distress syndrome // Crit. Care Med. — 2001. - 29(1). - P. 32-36.
\47.Kuhlen R., Walbert E., Frankel P. et al. Combinftion of inhaled nitric oxide and intravenous prostacyclin for successful treatment of severe pulmonary hypertension in a patients with acute respiratory distress syndrome // Inten. Care Med. — 1999. — 25. — P. 752-754.
148. Глумчер Ф. С., Волъхина И. А., Сергиенко А. В. Профилактика и лечение больных с синдромом острого повреждения легких вследствие травмы // Б1ль, знеболювання i штенсивна терашя. — 2001. — № 3 (16). — С. 2-7.
149. Moore F. A., Moore Е. Е., Kudsk К. L. et al. Clinical benefits of an immune-enhancing diet for early postinjury enteral feeding // J. Trauma. - 1994. - 37(4). - P. 607-615.
150. Clifford D. P. Affecting Clinical Outcomes in Acute Respiratory Distress Syndrome with Enteral Nutrition // Yearbook of Intensive Care and Emergency Medicine / Ed. J.-L. Vincent. — Springer, 2001. — P. 44 — 56.
152 ОСТРАЯ НЕДОСТАТОЧНОСТЬ ДЫХАНИЯ
151. Davey-Quinn A., Gedney J. A., WhiteleyS. М., Bellamy М. С. Extravascular lung water and acute respiratory distress syndrome — oxygenation and outcome // Anaesth. Inten. Care. — 1999. - 27. - P. 357-362.
152. Schonhofer B., Euteneuer S., Nava S. et al. Survival of mechanically ventilated patients admitted to a specialised weaning centre // Inten. Care Med. — 2002. — 28. — P. 908-916.
153. Wackers G. L. Modern anesthesiological principles for bulbar polio; manual IPPR in the 1952 polio epidemic in Copenhagen // Acta Anesthesiol. Scand. — 1994. — 38. — P. 420-431.
154. MacEwen W. Clinical observations on the introduction of tracheal tubes by the mouth instead of performing tracheotomy or laryn-gotomy // BMJ. - 1980. - 2. - P. 122 — 124.
155. Szold A., Pizof R.t Segal E.t Perel A. The effect of tidal volume and intravascular volume state on systolic pressure variation in ventilated dogs // Inten. Care Med. — 1989. — 15. - P. 368-371.
156. Gammanpila S., Bevan D. R., Bhudu R. Effect of positive and negative expiratory pressure on renal function // Br. J. Anaesth. — 1977. - 49. - P. 199-205.
157. Annat G., Viale J. P., Bui Xuan B. Effect of PEEP ventilation on renal function, plasma renin, aldosterone // Anesthesiology. — 1983. - 58. - P. 136-141.
158. Jonson E. E., Hedley-Whyte J. Continuous positive pressure ventilation and portal flow in dogs with pulmonary edema // J. AppL Physiol. - 1972. - 33. - P. 385-389.
159. Bendixen H. H., Egbert L. D., Hedley-White J. Respiratory Care. — St. Louis: P. Mosby, 1965. - P. 137-153.
160. Donnelly T. J., Meade P.f Jagels M. Cytokine, complement and endotoxin profiles associated with the development of the adult respiratory distress syndrome after severe injury // Crit. Care Med. — 1994. — 22. — P. 768-776.
161. Tremblay L., Valenza F.t Ribeiro S. P. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model // J. Clin. Invest. — 1997. - 99. - P. 944-952.
162. Pugin J. Molecular mechanisms of lung cell activation induced by cyclic stretch // Crit. Care Med.— 2003. — 31(4 SuppL). — P. S200-206.
163. Appavu S. K., Haley T. R.t Patel S. R. Does the use of large ventilator tidal volume increase the incidence of postoperative complications? //Crit. Care. — 2002. — 6 (Suppl. 1). — P. S8.
164. Edwards Y. S., Sutherland L. M., Power J. H. et al. Cyclic stretch induces both apoptosis and secretion in rat alveolar type II cells //
FEBS Left. - 1999. - 448(1). - P. 127 — 130.
165. Stuber F., Wrugge H., Schroeder S. Kinetic and reversibility of mechanical ventilation-associated pulmonary and systemic inflammatory response in patients with acute lung injury//Inten. Care Med. — 2002. — 28. -P. 831-834.
166. Pugin J. Is the ventilator responsible for lung and systemic inflamation? // Inten. Care Med. - 2002. - 28. - P. 817-819.
\S7. Bendixen H. H., Hedley-Whyre J., Laver M. B. Improved oxygenation in surgical patients du ring general anesthesia with controlled ventilation//N. Engl. J. Med. — 1963. -263. - P. 991-996.
168. Gattinoni L., Vagginelli F., Chiumello D. Physiologic rationale for ventilator setting in acute lung injury/acute respiratory distress syndrome patients // Crit. Care Med. — 2003. - 31(4 SuppL). - P. S300-304.
169. Downs J. B., Klein E. F., DeSautels D. Intermittent mandatory ventilation: A new approach to weaning patients with from mechanical ventilators // Chest. — 1973. — 64. - P. 331-335.
170. Kirton О. C. Ventilatory support modes // Critical Care / Ed. J. M. Civetta, R. W. Taylor, R. R. Kirby. — 3rd. ed. — Philadelphia: Lippincott-Raver. — 1997. — P. 745 — 755.
171. JonzonA., Oberg P. A., Sedin G., Sjostrand U. High frequency positive pressure ventilation by endotracheal insuflation // Acta Anaes-thesiol. Scand. — 1971. — 43. — P. 1—43.
172. Bishop M. J., Benson M. S., Sato P., Pierson D. J. Comparison of high frequency jet ventilation with conventional mechanical ventilation for bronchopleural fistula // Anesth. Analg. - 1987. - 66. - P. 833-838.
173. Launkenheimer P. P., Rafflenbeul W., Keller H. Application of transtracheal pressure oscillations as a modification of «diffusion respiration» // Br. J. Anaesth. — 1972. -44. - P. 627.
174. Tacata M., Abe J., Tanaka H. Intraalveolar expression of tumor necrosis factor alfa gene during conventional and high-frecquency ventilation // Am. J. Respir. Crit. Care Med. -1997. - 156. - P. 272-279.
175. Reynolds E. O. R. Effects of alterations in mechanical ventilator settings on pulmonary gas exchange in hyaline membrane disease // Arch. Dis. Child. - 1971. - 46. -P. 152-159.
176. Griffith H. R.f Johnson G. E. The use of curare in general anesthesia // Anesthesiology. - 1942. - 3. - P. 418-420.
177. Heffner J. E., Miller S., Sahn S. A. Tracheostomy in the intensive care unit: Pt. 1,2 // Chest. - 1986. - 90. - P. 269-274, 430-436.
178. GallagherT. J. Endotracheal intubation//Crit. Care Clin. - 1992. - 8. - P. 665-676.
Список использованной литературы 153
179. Wolls R. М. Rapid-sequence intubation in head trauma // Ann. Emerg. Med. — 1993. — 22. - P. 1008-1113.
180. Marsh H. M., Gillespie D. J., Baumgartner A. E. Timing of Tracheostomy in the critically ill patient//Chest. — 1989. — 96. — P. 190-192.
181. Tsuda T., Noguchi H., Takumi Y., Aochi O. Optimum humidification of air administered to a tracheosthomy in dogs // Br. J. Anaesth. — 1977. - 49. - P. 965-977.
182. Hedenstierna G., Lofstrom B., Lundh R. Thoracic gas volume and chest-abdomen dimensions during anesthesia and muscle paralysis//Anesthesiology. — 1981. — 55. — P. 499-506.
183. Tokics L., Hedenstierna G., Svensson L. V/Q distribution and correlation to atelectasis in anesthetized paralyzed humans // J. Appl. Physiol. - 1996. - 81. - P. 1822 — 1833.
\M.Fagon J. У. Prevention of ventilator-associated pneumonia // Inten. Care Med. — 2002. - 29. - P. 822-823.
WSNagi H. K.f Habib A. M., Omar S. H., Zakaria A. V. Effect of noninvasive ventilation on pulmonary gas exchange in chronic obstructive pulmonary disease // Crit. Care. - 2002. - 6 (Suppl. 1). - P. S23.
186. Antonelli M., Conti G., Moro M. L. Predictors of failure of noninvasive positive pressure ventilation in patients with acute
hypoxemic respiratory failure: a multi-center study // Inten. Care Med. — 2001. — 27. — P. 1718-1728.
187. Judson J. P., Glenn W. W. L. Radiofrequency electrophrenic respiration. Long-term application to a patient with primary hypoventilation // JAMA. — 1968. — 203. — P. 1033-1037.
188. Moxham J., SchneersonJ. M. Diaphragmatic pacing // Am. Rev. Respir. Dis. — 1993. — 148. - P. 533-536.
189. Spitzer S. A., Fink G., Mittelman M. External high-frequency ventilation in severe obstructive pulmonary disease // Chest. — 1993. - 104. - P. 1698-1701.
190. Schonhofer B., Euteneuer S., Nava S. Survival of mechanically ventilated patients admitted to a specialised weaning centre // Inten. Care Med. - 2002. - 28(7). -P. 908-916.
\§\.Dada L. A., Sznajder L. Mechanisms of pulmonary oedema clearance during acute hypoxemic respiratory failure: role of the Na+-, K+-ATPase // Crit. Care Med. — 2003. — 31(4 Suppl.). - P. S248-S252.
192. Staub N. C. Pathophysiology of pulmonary oedema // Edema / Ed. N. S. Staub, A. E. Taylor. — Raven, 1984. — 159 p.
193. Raijmakers P. G. What is the cause pulmonary oedema after acute miocardial infection? A case study // Inten. Care Med. — 1996. — 22. - P. 591.
Раздел
Клиническая физиология почек
Почки выполняют три важные функции: регуляторную, экскреторную и инкреторную.
Регуляторная функция почек заключается в поддержании постоянства внутренней среды организма (гомеостаза), в частности стабильности концентрации осмотически активных веществ в крови (осморегуляция), регуляции состава внеклеточной жидкости тела (волюморегуля-ция), концентрации каждого из ионов (ионная регуляция) и поддержании кислотно-основного равновесия.
Почки способны обеспечить гомеостаз при весьма значительных колебаниях количества поглощенной жидкости и других веществ, а также продуктов метаболизма. Здоровые люди могут потреблять в сутки 1 — 20 л воды, 5 — 500 ммоль натрия, 20 — 200 ммоль калия. В почках фильтруется большой объем жидкости, при этом необходимые для организма вещества подвергаются обратному всасыванию (реабсорбции), а ненужные — выводятся с мочой. В норме осмолярность крови колеблется в пределах ±1,2 %, концентрация Na+ - ±2,5 %, СГ - ±2 %, pH -±1 %. Именно эти отклонения и являются специфическими раздражителями, включающими в почке тот или иной механизм регуляции, восстанавливающий нарушенное равновесие.
Экскреторная функция почек обеспечивает выведение из организма жидкости, органических веществ, продуктов метаболизма (мочевина, креатинин, мочевая кис
лота) и чужеродных веществ (лекарственные средства, токсины) (табл. 5.1).
При поражении почек ее экскреторная функция сохраняется в течение продолжительного времени и после снижения или прекращения регуляторной деятельности.
Инкреторная функция заключается в выработке почечной тканью таких биологически активных веществ, как простагландины, кинины, ренин, эритропоэтин и др.
1. Простагландины образуются путем циклооксигенации арахидоновой кислоты. Те из них, которые синтезируются в корковом слое почек, принимают участие в регуляции почечного кровотока. Простагландины, продуцируемые мозговым слоем, модулируют реабсорбцию натрия и хлора.
2. Калликреин-кининовая система почек локализуется в дистальных отделах нефрона и является интегральной частью внутрипочечной гормональной системы, контролирующей экскрецию воды и электролитов и принимающей участие в регуляции артериального давления. Каллик-реины представляют собой сериновые протеазы, которые высвобождают кинины из присутствующих в плазме субстратов, называемых кининогенами.
3. Ренин-ангиотензиновая система почек участвует в регуляции внутрипочечного и системного кровообращения. Ренин представляет собой кислую сериновую протеазу, которая вырабатывается в юкстагломерулярном аппарате (ЮГА) почек. Ренин действует на ангиотензино-
Анатомия почки 155
Таблица 5. /. Количественные показатели экскреции различных веществ в сутки почками здорового человека
Вещество Содержание в плазме крови, ммоль/л Фильтруемое количество, ммоль Экскретируемое количество, ммоль Реабсорбция, %
Вода 3,5* 180* 1,5* 99
Натрий 140 24000 100 99
Калий 4 700 50 93
Хлор 105 20 000*** 100 99
Гидрогенкарбонат 27 5100 2 99
Глюкоза 5,55 180** — 100
Аминокислоты 0, 36***** 65** 1** 98
Мочевина 4,99 899 500 45
Креатинин 0,088**** 15,84 15,84 0
* Литров (л).
** Граммов (г).
*** Моль (моль).
**** Миллимоль (ммоль).
***** Граммов на 1 л (г/л).
ген и а-глобулин, образуя неактивный де-капептид ангиотензин I. Последний под действием ангиотензинпревращающего фермента (АПФ) переходит в биологически активный октапептид ангиотензин II. Самым важным физиологическим действием ангиотензина II является сужение гладкой мускулатуры сосудов и стимуляция надпочечников с образованием альдостерона.
4. Эритропоэтин представляет собой гликопротеиновый гормон, участвующий в регуляции эритропоэза. Наиболее важным физиологическим регулятором продукции
эритропоэтина является концентрация кислорода в почечных клетках. Если оксигенация почек уменьшается, продукция эритропоэтина возрастает. Дефицит кислорода приводит к высвобождению ряда простагландинов и простациклина, активации аденилатциклазы, увеличению содержания в почках циклического аденозинмонофосфата и стимуляции биосинтеза эритропоэтина. Регуляция продукции эритропоэтина осуществляется в клетках ЮГА. При почечной недостаточности дефицит эритропоэтина приводит к развитию анемии.
5.1. АНАТОМИЯ ПОЧКИ
Почки расположены в забрюшинном пространстве на уровне XII грудного и III поясничного позвонков. Размеры почки у взрослых составляют примерно 11 х 6 х х 3 см, масса — 120 —170 г. Через ворота почки (медиально вогнутый край) входят почечная артерия и нервы, выходят — почечная вена, лимфатические сосуды и мочеточник. На разрезе почка имеет слоистое строение. Выделяют корковый (наружный) слой толщиной примерно 1 см и медуллярный (внутренний). Последний состоит из 8 —18 пирамид конической фор
мы, вершины которых формируют почечные сосочки.
Почечная артерия отходит от аорты на уровне Ц — Ln. Вблизи ворот почки она разделяется обычно на переднюю и заднюю ветви. Проникая в почечную паренхиму, эти ветви образуют междолевые артерии, которые проходят между почечными пирамидами и, достигнув кортико-медуллярной границы, огибают основания пирамид, образуя дуговые артерии. Последние, разветвляясь, переходят в междольковые артерии. Из них образуются аффе
156 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
рентные артериолы почечных клубочков. Капилляры клубочка переходят в эфферентные артериолы, которые, разветвляясь, образуют перитубулярную капиллярную сеть, сбрасывающую основную массу крови в сосудистую систему пирамид. Это может приводить к ишемии коркового вещества почки и функциональным нарушениям. Анатомической предпосылкой указанного эффекта является строение выносящих артериол клубочков юкстаме-дуллярной зоны: диаметр их больше, чем приносящих артериол, и они образуют множественные артерио-венозные анастомозы с прямыми сосудами почек. Следствием такой сосудистой архитектоники юк-стамедуллярной зоны коркового вещества является большая скорость кровотока через клубочки этой зоны, которая в определенных условиях может в восемь раз превышать кровоток в клубочках коркового вещества. Таким образом, клубочковый аппарат юкстамедуллярной зоны может превращаться в дренажную систему, способствующую шунтированию крови.
Отличительной особенностью кортикального кровообращения является высокая степень ауторегуляции в довольно широких пределах колебаний системного АД (80 — 200 мм рт. ст.). Этот феномен обеспечивается изменениями тонуса гладкой мускулатуры прегломерулярных артериол, благодаря чему поддерживается относительно стабильный уровень клубочковой фильтрации. При снижении систолического АД ниже 80 мм рт. ст. ауторегуляция прекращается.
Давление в сосудах почки на разных этапах почечного кровотока изменяется ступенчато. Недавние исследования показали, что фильтрационное давление в капиллярах клубочков составляет около 45 мм рт. ст., т. е. ниже, чем это было принято считать ранее (60 мм рт. ст.). Относительный тонус и попеременное сужение афферентных и эфферентных артериол обуславливают величину почечного кровотока и скорость клубочковой фильтрации. Нормальная величина почечного кровотока у взрослого человека — в среднем 1100 мл/мин. Корковое вещество составляет 70 —75 % всей массы почки, поэтому
на его долю приходится более 80 % общего кровотока почки, что в пересчете на 100 г ее массы составляет 373 — 538 мл/мин.
5.1.1. Кровообращение в почках
По интенсивности кровоснабжения почки занимают первое место в организме среди других органов. Суммарный кровоток в них составляет 20 — 25 % ударного объема сердца. Кровоток в ткани почки из расчета на 100 г ее массы в 4 раза больше, чем в печени и тренированных мышцах, и в 8 раз больше, чем в мышце сердца. В целом в почках можно выделить два функционально различных круга кровообращения: большой — кортикальный и малый — юкстамедуллярный. В физиологических условиях кровоток в почках распределяется следующим образом: наружное, корковое вещество — 80 %, юкстамедуллярная зона коркового вещества — 15 %, мозговое вещество — 3 %, жировая капсула — 2 %.
В стрессовых ситуациях почечный кровоток может переключаться с большого круга на малый, укороченный юкстамедуллярный путь, который становится своеобразным шунтом (шунт Труэты).
5.1.2. Нефрон
Структурно-функциональной единицей почки является нефрон. Количество нефронов в почках достигает 2 млн. Начальная часть нефрона — почечное тельце, или клубочек, состоит из элементов сосудистой системы и эпителия, обеспечивающих ультрафильтрацию крови. Далее следует проксимальный извитой каналец, который располагается в корковом слое почки -зоне интенсивного кровотока. Здесь происходит реабсорбция большей части фильтрата. Следующим участком является петля Генле, которая входит на различное расстояние в глубь почечной пирамиды, а затем возвращается к тому же клубочку, из которого берет начало этот нефрон. Петля Генле представляет собой эпителиальную трубку, через стенки которой осуществляется осмотическое разведение или концентрирование мочи. Область, гдедис-
Анатомия почки 157
тальный каналец примыкает к клубочку, имеет особую структуру, включающую сосудистый, канальцевый и интерстициальный компоненты. Эта область получила название «юкстагломерулярный аппарат», который имеет важное значение для регуляции функции каждого нефрона. Дистальная часть нефрона, или дистальный извитой каналец, переходит в систему собирательных трубок коркового слоя почки, которые, в свою очередь, сливаются и образуют собирательные трубки мозгового слоя. Длина отдельного канальца равна 3 см, а общая длина всех канальцев (2 млн х 3 см) — 30 км.
Клубочек состоит из капиллярных петель, заполняющих пространство, называемое «боуменовым». Это пространство находится всередине боуменовой капсулы, образованной слоем плоских клеток париетального листка эпителия. Кровь поступает в сосудистый полюс клубочка через афферентную артериолу и покидает его по эфферентной артериоле. Приносящая артериола клубочка по диаметру вдвое больше выносящей, что обуславливает гидростатическое давление в процессе фильтрации.
Стенка капилляра клубочка функционирует по принципу сита, обеспечивая движение воды и низкомолекулярных растворенных веществ и не препятствует прохождению циркулирующих в крови макромолекул (альбумин и др.).
Клубочковая фильтрация. Ультрафильтрация плазмы в клубочках является первым этапом образования мочи. Состав первичной мочи отличается от плазмы крови лишь низким содержанием протеинов. Фильтрационная функция клубочков относится к пассивным процессам, протекающим без затраты энергии. Гидростатическое давление в клубочках создается работой сердца. Скорость клубочковой фильтрации (СКФ) удерживается на относительно постоянном уровне при меняющемся перфузионном давлении. Механизм ауторегуляции СКФ обеспечивается изменением тонуса приносящих и выносящих артериол. В нормальных условиях у взрослого человека СКФ составляет в среднем 120 мл/мин. Таким обра
зом, за сутки образуется 170 —180 л первичной мочи.
Для количественной характеристики процессов мочеобразования используют методы, основанные на принципе очищения. Коэффициент очищения, или клиренс, — это объем плазмы, который полностью очищается от экзогенного или эндогенного вещества за 1 мин. Для определения величины клубочковой фильтрации используют определение клиренса инулина (С) по формуле
Г _UV
С Р ’
где U — концентрация исследуемого вещества в моче, мг%; V — диурез, мл/мин; Р — концентрация исследуемого вещества в плазме, мг%.
Инулин фильтруется в клубочках и обнаруживается в первичной моче в той же концентрации, что и в плазме. Методика заключается в том, что инулин вводят в кровь для обеспечения его постоянного уровня в плазме, затем измеряют количество инулина, экскретируемое за единицу времени, и полученный результат используют для расчета клиренса.
Функциональная роль канальцев. Клетки почечных канальцев высокодиф-ференцированы и выполняют сложные и многообразные функции транспорта веществ, которые должны быть или сохранены для организма, или выделены во внешнюю среду. Процессы реабсорбции и секреции принято обозначать термином «канальцевый транспорт».
В канальцах происходит реабсорбция 99 % ультрафильтрата (первичной мочи) и лишь около 1 % выделяется наружу, т. е. из 170 —180 л первичной мочи обратному всасыванию подвергается 169—179 л.
Проксимальный сегмент извитых канальцев. Клетки проксимального отдела извитых канальцев имеют наиболее сложную организацию по сравнению с другими сегментами нефрона, содержат большее количество митохондрий и широкий спектр ферментов, принимающих участие в реабсорбции веществ. Верхушечная, обращенная в просвет канальца, и базальная плазматическая мембраны различают
158 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
ся по структуре и функции, в частности по степени проницаемости этих мембран для разных компонентов первичной мочи. В проксимальном отделе нефрона всасывается 65 —70 % фильтрата. Этот процесс осуществляется с затратой энергии (расходуется 6 —8 % всего кислорода, поглощенного организмом, или 85 % кислорода, потребляемого почками).
Петля Генле. Структурно петля Генле состоит из трех основных сегментов — толстого нисходящего колена, тонкого сегмента и толстого восходящего колена. Последний сегмент —место, где реабсорбируется около 25 % фильтруемого количества натрия. Жидкость, выходящая из проксимальных канальцев, изотонична плазме крови. В петле Генле, а также в собирательных трубках осуществляется концентрирование или разведение мочи. Предполагается, что моча в основном концентрируется в нисходящем колене петли Генле. В норме в почечной петле реабсорбируется около 10 % профильтровавшейся воды и около 15 % NaCl.
Дистальный каналец. В этом отделе нефрона протекают сложные ионообменные процессы, направленные на поддержание осмолярности плазмы крови и КОС. В начальной части дистального канальца в норме реабсорбируется около 5 —8 % фильтруемого натрия. Этот сегмент извитого канальца, как и восходящее колено петли Генле, непроницаем для воды. Поэтому продолжающийся процесс реабсорб
ции натрий хлорида ведет к дальнейшему разведению мочи.
В дистальных канальцах преобладает ионообменный процесс реабсорбции НСО3 в результате взаимодействия с активно секретируемым ионом Н+. Благодаря указанному ионообменному механизму клетки канальцев почек возвращают в систему циркуляции 99,9 % профильтровавшегося НСО3. С мочой выделяется лишь 1 — 2 ммоль. Дистальные канальцы выполняют также важную роль в экскреции К+. При обычной диете экскреторная функция этого отдела обеспечивает выделение с мочой от 40 до 120 ммоль калия в сутки.
Собирательные трубки. В них происходит дальнейшее качественное изменение мочи. Этот отдел нефрона участвует в поддержании гидропонного гомеостаза, и именно здесь моча достигает окончательной осмотической концентрации.
В отсутствие антидиуретического гормона (АДТ) проницаемость собирательных трубок для воды резко снижается и образуется разведенная моча (минимальная осмолярность мочи составляет 50 — 75 моем/л). При высоком содержании АДГ увеличивается осмолярность мочи (максимальная концентрация достигает 1400 моем/л). В собирательных трубках реабсорбируется около 5 — 7 % профильтровавшегося натрия и секретируются К+ и Н+. Таким образом, в этом сегменте нефрона происходит окончательное формирование мочи.
5.2. ТРАНСПОРТ РАСТВОРЕННЫХ ВЕЩЕСТВ
Транспорт растворенных веществ происходит на протяжении всей длины канальцевой системы нефрона. Различают активный транспорт, облегченную диффузию, пиноцитоз и осмос. Активный транспорт протекает с затратой энергии и заключается в переносе веществ через мембраны против электрохимического и концентрационного градиента. Облегченная, или опосредованная, диффузия осуществляется с участием переносчиков, в частности ферментов (пермеаз), облегчающих диф
фузию ионов через цитоплазматическую мембрану для увеличения транспорта ионов по концентрационному градиенту.
Натрий. Находится в плазме в связанном состоянии, главным образом в форме соединений NaCl, NaHCO3 и фосфатов. Однако вследствие значительной электролитической диссоциации эти соединения в канальцах реабсорбируются как отдельные ионы. Поэтому следует говорить не о транспорте NaCl, а о транспорте ионов Na+ и СГ.
Транспорт растворенных веществ 159
Натрий свободно фильтруется в клубочках, и его концентрация в клубочковом фильтрате идентична таковой в плазме крови. Реабсорбция натрия осуществляется на протяжении всей длины дистального нефрона, однако механизм реабсорбции в каждом из основных сегментов нефрона различен.
Реабсорбция натрия является пороговой. Интенсивность реабсорбции Na+ изменяется в зависимости от его содержания в плазме.
Транспорт натрия в проксимальном канальце. Около 75 % профильтрованного натрия реабсорбируется в проксимальных канальцах. Одновременно реабсорбируются связанные с ним анионы и вода, поэтому жидкость в просвете канальцев остается почти изотоничной относительно плазмы. Реабсорбция натрия в проксимальном канальце носит характер активного транспорта, в основе которого лежит механизм так называемого натриевого насоса (с участием Na+-, К+-АТФ-азы). В люминальной мембране канальцевых клеток локализованы натриевые каналы, а в базально-латеральных мембранах, обращенных к интерстицию, находятся натриевые насосы. Натрий поступает в клетку по электрохимическому градиенту, двигается по цитоплазме и достигает ионных насосов на базально-латеральных мембранах, где транспортируется в межклеточное пространство против электрохимического градиента за счет энергии клеточного метаболизма.
Транспорт натрия в петле Генле. Дальнейшая реабсорбция натрия происходит в толстом восходящем колене петли Генле. В этом сегменте всасывается около 25 % профильтровавшегося натрия. Однако в отличие от проксимального канальца реабсорбция солей в этом сегменте не связана с одновременным всасыванием воды. Эпителий восходящего колена петли Генле непроницаем для воды. Реабсорбция NaCl в сочетании с непроницаемостью мембраны для воды обуславливает гипо-осмотичность люминальной жидкости по отношению к плазме, что, в свою очередь, способствует концентрированию и разведению мочи.
Транспорт натрия в дистальном канальце. В начальной части дистального канальца реабсорбируется около 5 —8 % фильтруемого натрия. Этот сегмент канальца так же, как и восходящее колено петли Генле, непроницаем для воды. Поэтому продолжающаяся реабсорбция NaCl ведет к дальнейшему разведению мочи. Процесс реабсорбции натрия в этом сегменте канальца является активным и заключается в обмене ионов натрия на ионы водорода и калия. Несмотря на то что в количественном отношении реабсорбция натрия в дистальном канальце значительно меньше по сравнению с проксимальным, она имеет решающее значение для сохранения натрия в организме.
Транспорт натрия в собирательных трубках. В собирательных трубках реабсорбируется около 5 % натрия. Так же, как в дистальном канальце, всасывание натрия сопровождается противотранспортом ионов К+и Н+. При нарушении реабсорбции Na+ и воды, а также секреции К+ и Н+ в собирательных трубках расположенных выше сегментов нефрона происходят активные процессы поддержания гидропонного гомеостаза, от их функции зависит окончательная осмотическая концентрация мочи. Следует отметить, что в отличие от двух предыдущих сегментов нефрона этот сегмент обладает свойством изменять проницаемость мембраны для воды в зависимости от концентрации антидиуретичес-кого гормона в крови.
Канальцевый транспорт хлора. Около 70 % всего количества профильтровавшегося хлора реабсорбируется в проксимальном канальце, а остальная часть — в дистальных отделах нефрона. Реабсорбция хлора является в основном пассивным процессом. В проксимальном канальце реабсорбция СГ совершается обычно в том же направлении, что и реабсорбция натрия. Однако оба эти иона переносятся не в виде соединения NaCl, а по отдельности. Нельзя рассматривать СГ простым эквивалентом Na+.
В проксимальном отделе извитого канальца реабсорбция хлора происходит при наличии ферментов, имеющих сульфгидрильные группы, очень чувствительные
160 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
к ртутным диуретикам. При блокировании ртутными диуретиками нормальная реабсорбция хлора увеличивает диурез, поскольку часть воды не реабсорбируется.
Канальцевый транспорт калия. Калий преимущественно находится в клетках и лишь менее 2 % его обнаруживается в биологических жидкостях организма. Почки не сохраняют калий так же эффективно, как натрий. Если потребление калия низкое, то происходит его потеря с мочой. С другой стороны, почки способны адаптироваться к большим его нагрузкам. При потреблении избытка солей калия дистальные извитые канальцы и собирательные трубки интенсивно секретируют калий, и его количество, выделенное с мочой, может превышать фильтруемое в клубочках.
Выведение калия почками осуществляется тремя способами: фильтрацией, реабсорбцией и секрецией. Он свободно фильтруется в клубочках. Около 90 % профильтровавшегося калия подвергается реабсорбции в проксимальном сегменте канальца. Часть ионов К+ реабсорбируется в петле Генле. В дистальном сегменте извитого канальца и собирательных трубках ионы К+ подвергаются секреции в просвет канальца в обмен на ионы Na+. На этот процесс влияют факторы, действующие как с перитубулярной стороны (со стороны кровеносного русла), так и с люминальной стороны эпителия канальцев и собирательных трубок. Наиболее важным из факторов, действующих с перитубулярной стороны, является альдостерон, увеличивающий секрецию калия.
К факторам, влияющим на секрецию калия с люминальной стороны эпителия канальцев, относят разность потенциалов по обе стороны люминальной мембраны, а также скорость движения фильтрата по сегменту нефрона. Секреция калия увеличивается при алкалозе и уменьшается при ацидозе. Некоторые вещества способны влиять на секрецию калия. Например, такой препарат, как амилорид, снижающий трансэпителиальную разность электрических потенциалов, полностью подавляет секрецию калия. Он применяется как ка-
лийсберегающий препарат при лечении диуретиками.
Транспорт ионов Н+. Роль почек в поддержании pH исключительно велика, поскольку они регулируют секрецию ионов Н+ и восполняют буферную емкость крови. Нормальное протекание различных физиологических процессов зависит от строгого поддержания pH крови на среднем уровне 7,4.
Ионы Н+ секретируются в просвет проксимальных и дистальных канальцев. Большая часть ионов Н+ образуется при катализируемой или некатализируемой гидратации СО2:
СО2 ч- Н2О ?=> Н2СО3 <=» Н+ + НСО3 .
Следующим этапом является движение ионов Н+ через люминальную мембрану. В проксимальном канальце ионы Н+ обмениваются на ионы Na+ одной из находящихся в моче натриевых солей. В дистальном сегменте нефрона секреция Н+ обеспечивается главным образом первичной Н+-АТФ-азой, расположенной в апикальной мембране вставочных клеток собирательных трубок коркового и мозгового слоев почки. Попадая в просвет нефрона, Н+ захватывается НСО3. В проксимальном канальце, где концентрация НСО3 относительно высока, эта реакция катализируется карбоангидразой, находящейся в щеточной каемке. Таким образом, секреция Н+ используется для возвращения профильтрованных гидрогенкарбонатов.
Одновременно ионы Н+ участвуют в образовании титруемых кислот:
Н+ + НРО4 2Н+ 4- РО4 ;
Н++А" <=>НА,
где НА является слабой органической кислотой.
Гидроген-ионы фосфатов и неионизиро-ванные слабые кислоты в дальнейшем экскретируются с мочой.
В экскреции ионов Н+ участвует и другая буферная система, обладающая большой емкостью. В проксимальном и дистальном канальцах из глутамина при участии фермента глутаминазы непрерывно образуется аммиак, диффундирующий в
Транспорт растворенных веществ 161
кровь и канальцевую жидкость. В просвете канальцев аммиак (NH3) связывается с Н+, образуя ион аммония (NH4), который экскретируется с мочой.
При нормальном функционировании почек общее количество экскретированных Н+ равно примерно 50—100 мэкв/сут. При гидрогенкарбонатной нагрузке (например, прием с пищей фруктовых соков, содержащих цитроны, инфузия больших количеств гидрогенкарбоната) экскреция Н+ снижается или полностью прекращается, а титруемая кислотность приближается к нулю.
В регуляции экскреции Н+ принимает участие альдостерон, стимулирующий секрецию Н+ и К+ в дистальном отделе нефрона. Избыток альдостерона приводит к развитию метаболического алкалоза, а его дефицит — к метаболическому ацидозу.
Транспорт кальция и магния. Кальций находится в плазме в трех формах: ионизированной, неионизированной (в виде комплексных соединений) и соединения с белком. В норме ионизированный кальций составляет менее половины общего количества (1,136 — 0,126 ммоль/л). Комплексы с ним составляют главным образом три аниона: НСО3, фосфат и цитрат. В почке в сутки фильтруется около 270 ммоль кальция. Суточная его экскреция колеблется от 1,5 до 15 ммоль.
В клубочках фильтруется только ионизированный и неионизированный кальций, поэтому его концентрация в ультрафильтрате составляет около 65 % от общей концентрации в плазме.
Реабсорбция кальция во многих отделах нефрона происходит одновременно с реабсорбцией натрия. Поэтому экскреция кальция с мочой изменяется в зависимости от экскреции натрия.
Около 60 % профильтровавшегося кальция реабсорбируется в проксимальном извитом канальце, 20 —25 % — в петле Генле, 10 % — в дистальном извитом канальце и около 5 % — в собирательных трубках. Терминальный отдел нефрона, где реабсорбируется лишь 5 — 8 % всего профильтровавшегося кальция, является основной зоной окончательной регуляции его экскреции. Реабсорбция кальция в дисталь-6 4-220
ной части осуществляется под воздействием паратиреоидного гормона (паратгормона), который увеличивает реабсорбцию кальция в толстом восходящем колене петли Генле и собирательной трубке.
В регуляции обмена кальция кроме паратгормона принимают также участие активная форма витамина D и кальцитонин. Когда больной получает диету с низким содержанием этого макроэлемента, развитие легкой, преходящей гипокальциемии вызывает высвобождение паратгормона, увеличивающего превращение в корковом веществе почки 25-оксихолекальциферо-ла в 1,25-диоксихолекальциферол (витамин D3). Этот метаболит витамина D повышает всасывание кальция в кишках и увеличивает его реабсорбцию в проксимальных канальцах.
Кальцитонин, секретируемый главным образом клетками щитовидной железы, вызывает натрийурез и увеличивает выведение кальция. Он продуцируется в ответ на увеличение содержания кальция в плазме.
Подобно кальцию часть магния в плазме связана с белком, при этом соотношение фильтруемого магния к общей концентрации Mg2+ в плазме составляет приблизительно 0,75. 20 — 30 % фильтруемого Mg2+ реабсорбируется в проксимальном канальце, большая же его часть (50 — 60 %) — в толстом восходящем колене петли Генле под влиянием паратгормона. В дистальном нефроне реабсорбируется около 2 —10 % фильтруемого магния вне зависимости от действия паратгормона.
Различия в процессах всасывания кальция и магния на всем протяжении нефрона были уточнены при изучении эффекта диуретиков на экскрецию ионов. Так, тиазидовые диуретики снижают экскрецию кальция, но увеличивают экскрецию магния. «Петлевой диуретик» фуросемид увеличивает экскрецию обоих ионов, тогда как действующие на дистальную часть нефрона калийсберегающие диуретики типа спиронолактона, амилорида и три-амтерен снижают экскрецию кальция и магния.
Транспорт фосфатов. Фосфаты принадлежат к наиболее эффективным буферам мочи. В нормальных условиях в моче
162 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
содержатся лишь неорганические фосфатные соединения (Н2РО4, НРО4). За сутки с мочой выделяется около 1 г фосфатов. Неорганические фосфаты свободно фильтруются в клубочках и подвергаются обратному всасыванию в канальцевой системе. Предполагается, что 60 — 70% профильтровавшихся фосфатов реабсорбируется в проксимальном канальце, а также в дистальном.
Паратгормон играет важную роль в регуляции уровня фосфатов в различных сегментах нефрона. Этот пептид связывает рецепторы на контралюминальной стороне проксимального канальца и активизирует систему аденилатциклазы, вследствие этого увеличивается количество циклического АМФ и реабсорбция фосфатов снижается. То же самое происходит и в дистальной части нефрона. Таким образом, при наличии избытка паратиреоидного гормона экскреция фосфатов может повыситься на 20 — 30 %, т. е. развивается значительная фосфатурия. Кальцитонин и глюкокортикоиды уменьшают количество реабсорбированных фосфатов, в то время как гормон роста повышает их реабсорбцию.
Транспорт сульфатов. Концентрация сульфатов в плазме крови колеблется от 0,19 до 2,4 ммоль/л. Неорганические сульфаты плазмы образуются в результате расщепления аминокислот. В клубочках они свободно фильтруются и подвергаются реабсорбции в извитых канальцах. Реабсорбция сульфатов происходит следующим образом: активный сульфат связывается с АТФ (2 АТФ + SO^”) и путем транссульфурирования превращается в «переносчик» с низкой молекулярной массой, от которого в интерстициальном пространстве отделяется сульфат-ион. При нормальной концентрации сульфатов в плазме почти все количество профильтровавшихся ионов подвергается реабсорбции. Повышение концентрации сульфатов вызывает быстро наступающее насыщение канальцевого транспорта и по достижении максимума увеличивается выведение их с мочой. Поэтому сульфаты можно отнести к пороговым веществам.
Ряд гормонов оказывает влияние на реабсорбцию сульфатов. Так, она возрас
тает после введения гормона роста и глюкокортикоидов. Реабсорбция сульфатов ингибируется тиосульфатом, а также глюкозой или аминокислотами в высоких концентрациях.
Транспорт пороговых веществ. К пороговым относятся такие вещества, реабсорбция которых происходит до тех пор, пока их концентрация в плазме крови не превысит определенную пороговую величину, после чего их реабсорбция прекращается и они появляются в моче. К таким веществам относятся глюкоза, хлориды, аминокислоты.
Глюкоза. В обычных условиях в просвет нефрона с ультрафильтратом ежеминутно поступает более 100 мг глюкозы, которая практически полностью реабсорбируется клетками проксимального канальца. Реабсорбция глюкозы является типичным примером выделения веществ путем фильтрации и реабсорбции без секреции. Суточное количество экскретируемой глюкозы не превышает 130 мг. Процесс реабсорбции протекает с затратой энергии АТФ путем энзиматического фосфорилирования. Под влиянием гексокиназы глюкоза превращается в глюкозо-6-фосфат, из которого под действием фосфатазы снова образуется глюкоза, а фосфорный радикал присоединяется к АДФ с образованием АТФ. Этот процесс требует достаточного количества О2.
Максимальная скорость, с которой может происходить реабсорбционный процесс, называется транспортным максимумом (Тт). Он индивидуален для каждого растворенного вещества. Считается, что величину Тт глюкозы (TmG) ограничивает лишь число переносчиков, но не энергоснабжение транспорта. Для количественного определения этого показателя в кровь больного вводят глюкозу («сахарная нагрузка»). Когда количество профильтровавшейся глюкозы превысит ре-абсорбционную способность канальцев, т. е. будут заняты все мембранные переносчики глюкозы, ее избыток в канальцах приведет к глюкозурии. Количество глюкозы, реабсорбируемое при максимальной нагрузке всех ее мембранных переносчиков, является относительно стандартной
Транспорт растворенных веществ 163
величиной и соответствует у мужчин — 380, а у женщин — 310 мг/мин на 1,73 м2 поверхности тела.
Если во всех канальцах количество фильтруемой в них глюкозы соответствует способности клеток к ее реабсорбции, т0 TmG будет одновременно достигнут во всех нефронах. При этом глюкозурия будет увеличиваться пропорционально увеличению гипергликемии. Если же нефроны гетерогенны по способности реабсорбировать глюкозу, то будет наблюдаться расхождение между ними по времени наступления глюкозурии — она будет появляться при разном уровне гипергликемии. TmG в разных нефронах будет достигаться при увеличении концентрации глюкозы в плазме крови от 9,99—11,10 ммоль/л до 22,20 ммоль/л. Это явление носит название расщепления кривой титрования нефронов глюкозой и отражает степень морфологической и функциональной гетерогенности нефронов.
Различают два вида увеличения экскреции сахара с мочой (глюкозурии): почечную и экстраренальную. Первый вариант наблюдается при нормальной концентрации глюкозы в крови, второй — при гипергликемии, например, при сахарном диабете. Глюкозурия почечного происхождения является редким заболеванием, чаще всего она служит одним из симптомов нарушения функции почек при синдроме Фанкони.
Специфическим ингибитором транспорта глюкозы является флоридзин. Последний блокирует процесс фосфорилирования глюкозы и тормозит ее реабсорбцию в проксимальных канальцах.
Аминокислоты. Так же, как и глюкоза, аминокислоты полностью фильтруются в клубочках. Концентрация аминокислот в фильтрате соответствует их концентрации в плазме крови и колеблется от 2,5 до 3,5 ммоль/л.
В обычных условиях более 90 % профильтровавшихся аминокислот подвергается обратному всасыванию главным образом в начальной части проксимального канальца. Механизм трансцеллюлярного транспорта аминокислот подобен таковому при реабсорбции глюкозы. Аминокис
лота связывается в мембране щеточной каемки со специфическим переносчиком, он присоединяет натрий, и весь комплекс перемещается через мембрану в цитоплазму, где переносчик освобождается от аминокислоты и натрия. Системы канальцевого транспорта аминокислот характеризуются насыщением, т. е. имеют ограниченное количество переносчиков, и когда все они связываются с соответствующими аминокислотами, избыток последних из канальцевой жидкости экскретируется с мочой.
Существует, по меньшей мере, четыре специфические активные транспортные системы, специфичные для определенных групп аминокислот и имеющие разное значение Тт. Разные группы аминокислот переносятся отдельными трансмиттерами.
I группа — аланин, гистидин, норвалин, нор лейцин, метионин, а-аминомасляная кислота;
II группа — аргинин, цистеин, лизин, орнитин;
III группа — аспарагиновая кислота, глутаминовая кислота;
IV группа — глицин, оксипролин, пролин.
Перенос аминокислот через мембрану щеточной каемки является стереоспецифическим для их определенных групп, насыщаемым и зависимым от уровня натрия. Процесс их реабсорбции косвенно зависит от потребления кислорода и эффективности энергетического аппарата клетки. Поступившие в клетку аминокислоты перемещаются к латеральной и базальной плазматической мембранам, через которые посредством механизма облегченной диффузии попадают в интерстициальное пространство.
Ряд факторов приводит к увеличению экскреции аминокислот (аминоацидурии), в частности отсутствие или низкая активность в организме ферментов катаболизма некоторых аминокислот; наследственные или приобретенные нарушения одной или нескольких систем транспорта аминокислот в мембране щеточной каемки; дефект трансцеллюлярного механизма переноса аминокислот через клетку проксимального канальца.
6*
164 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
5.3. ЭКСКРЕЦИЯ ПРОДУКТОВ МЕТАБОЛИЗМА
Почки выполняют огромную работу по выведению конечных продуктов метаболизма. Они регулируют экскрецию титруемых кислот, мочевины, мочевой кислоты, креатинина и других продуктов распада.
Экскреция титруемых кислот. В результате разрушения фосфолипидов и белков образуются фосфорные кислоты, представленные главным образом двузамещенным фосфатом Na2HPO4. При нормальном значении pH его содержание в плазме крови составляет 80 %, остальная часть приходится на NaH2PO4. Гидроген-ионы фосфатов и неионизированных слабых кислот экскретируются с мочой. Они определяют титруемую кислотность, измеряемую при титровании мочи до pH плазмы крови. Титруемая кислотность зависит от степени снижения pH, а также от количества фосфатов и органических кислот, доступных для связывания с ионами водорода.
Экскреция мочевой кислоты. Мочевая кислота является слабой кислотой, она транспортируется через проксимальный каналец в двух направлениях (реабсорбция и секреция). Процесс секреции имеет клиническое значение, поскольку повышение концентрации мочевой кислоты в плазме крови может приводить к преципитации кристаллов (подагра).
Суточная секреция мочевой кислоты равна 2,3 —4,5 ммоль, а ее концентрация в крови — 0,3 ммоль/л. Значительная часть мочевой кислоты подвергается реабсорбции в проксимальном извитом канальце. Экскретируемая фракция мочевой кислоты составляет всего 9,8 %.
Повышению мочевой кислоты в крови способствует: высокая скорость ее синтеза, снижение клубочковой фильтрации, усиление канальцевой реабсорбции или снижение секреции мочевой кислоты. Некоторые фармакологические средства нарушают транспорт мочевой кислоты в канальцах. Так, диуретики влияют на процесс транспорта, а также на характер экскреции вследствие изменения объема внеклеточной жидкости. Блокаторами реабсорбции
мочевой кислоты являются салицилаты, кортизон, АКТГ.
Экскреция мочевины. Мочевина является конечным продуктом азотистого обмена и экскретируется почками. Она подвергается ультрафильтрации в клубочках и не расщепляется в моче. Около 40 % профильтровавшейся мочевины реабсорбируется в канальцах. Этот процесс рассматривается как пассивный, так как отсутствуют данные в пользу существования активного транспорта мочевины у человека. Ее реабсорбция осуществляется путем диффузии на протяжении всего канальца. Однако проницаемость мембраны для мочевины не везде одинакова, в проксимальном отделе диффузия более активна, в дистальном — снижена.
Скорость экскреции мочевины фактически определяется фильтрацией и последующей реабсорбцией, последняя, в свою очередь, обусловлена концентрационным градиентом, создаваемым реабсорбцией воды. Поэтому выделение мочевины зависит от диуреза. Максимальная ее экскреция наблюдается при минутном диурезе 2 мл и более, минимальная — при снижении минутного диуреза до 0,35 мл. В последнем случае реабсорбция в канальцах повышается до 70 %. Если же введением какого-либо осмотически активного вещества (глюкозы, сорбитола) вызвать осмотический диурез, то клиренс мочевины может достичь клиренса инулина, т. е. произойдет повышение экскреции мочевины.
Мочевина принимает участие в функционировании противоточно-поворотной множительной системы. Поступая в петлю Генле, она многократно подвергается диффузии из люминального пространства в интерстициальное и обратно и тем самым участвует в разведении и концентрировании мочи. Корковые сегменты дистального отдела нефрона фактически непроницаемы для мочевины, это имеет важное значение для функционирования противоточно-поворотной множительной системы, которая, в свою очередь, способствует тому, что большое количество мочевины экскретируется в малом объеме мочи.
Почечная регуляция кислотно-основного состояния 165
Экскреция креатинина. Креатинин является компонентом плазмы крови. Его продукция варьирует у разных индивидов, в частности, она выше у людей с большой мышечной массой. Креатинин является продуктом метаболизма мышечных клеток, а именно — распада креатинфосфорной кислоты. После отщепления фосфата от креатинфосфорной кислоты образуется креатин. Последний подвергается дегидратации с образованием креатинина. В среднем за сутки почки образуют и экскретируют около 1,8 г (15,8 ммоль) креатинина (концентрация креатинина в крови колеблется от 60 до 100 мкмоль/л). Он полностью фильтруется в клубочках без обратной резорбции, поэтому по клиренсу креатинина судят о СКФ. Клиренс креатинина сходен с клиренсом инулина.
При уменьшении клубочковой фильтрации пропорционально увеличивается концентрация креатинина в плазме крови. Это весьма существенно для клинициста, поскольку по изменению содержания креатинина в крови можно судить о степени ухудшения клубочковой фильтрации.
Выделение аммиака. Аммиак образуется в клетках почечных канальцев путем дезаминирования аминокислот. Основным его источником является глутамин, который, как и некоторые другие аминокислоты, экстрагируется из крови клетками канальцевого эпителия и подвергается дезаминированию под влиянием глутаминазы. Образующийся при этом аммиак (NH3) диффундирует в просвет канальца, так как его парциальное давление в тубулярной жидкости меньше, чем в крови. В просвете канальца аммиак связывается с ионами Н+ и образует аммонийный радикал NH4 (ион аммония). Последний является водорастворимым катионом и не подвергается обратной диффузии через верхушечную мембрану канальцевых клеток. Образовавшийся радикал NH4 обычно связывается в просвете канальца с анионом СГ и в виде аммоний хлорида выводится с мочой. В норме у взрослого человека за сутки выделяется 30 — 50 ммоль аммония. Скорость образования и количество выделяемых солей аммония резко возрастает при ацидозе и может достигать 250 ммоль/сут.
5.4. ПОЧЕЧНАЯ РЕГУЛЯЦИЯ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Поддержание постоянства активной реакции крови является одним из основных условий нормальной жизнедеятельности организма. Кислотно-основное состояние (КОС) зависит от соотношения содержания в плазме кислот и оснований. Показатель pH, характеризующий активную реакцию крови, отражает концентрацию в ней ионов Н+. В норме этот показатель колеблется в пределах 7,35 — 7,45.
Регуляция КОС осуществляется несколькими механизмами: а) буферными системами, которые сводят к минимуму изменения pH; б) вентиляцией легких, быстро реагирующей на колебания /?СО2; в) выделением почками избытка ионов Н+ и титруемых кислот. Последний механизм срабатывает медленнее двух первых. Однако роль его исключительно важна, поскольку почки не только регулируют секрецию ионов Н+, но и восполняют буфер
ную емкость организма. В норме pH мочи колеблется в пределах 4,5 —8,2.
Почечный механизм регуляции КОС включает: а) канальцевую реабсорбцию ги-дрогенкарбонатов; б) канальцевую секрецию ионов Н+ в обмен на ионы Na+; в) образование титруемых кислот в результате связывания ионов Н+ с буферными системами мочи; г) экскрецию аммонийных солей.
Реабсорбция гидрогенкарбоната и секреция ионов Н+ в канальцах связаны с реабсорбцией натрия, являющегося основным внеклеточным катионом. Расширение или уменьшение внеклеточного объема влияет на экскрецию гидрогенкарбонатов. При его возрастании усиливается экскреция гидрогенкарбонатов, а реабсорбция натрия и гидрогенкарбонатов снижается. Противоположный процесс наблюдается при уменьшении внеклеточного объема. Следовательно, концентрация натрия и внекле
166 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
точный объем могут быть отнесены к факторам, влияющим на регуляцию так называемого почечного порога гидрогенкарбо-натов. Реабсорбция НСО3 увеличивается при гипохлоремии. И наоборот, когда запасы хлора в организме возрастают, реабсорбция гидрогенкарбоната уменьшается.
Кислотовыделительная функция почек связана также с содержанием циркулирующих кортикостероидных гормонов. Последние усиливают реабсорбцию натрия и стимулируют выделение кислых продуктов с мочой. При гиперальдостеронизме и синдроме Кушинга концентрация гидрогенкарбоната в плазме увеличивается, в то время как при болезни Аддисона она уменьшается. Это является результатом влияния альдостерона на обмен ионов Na+ и Н+ в дистальных отделах канальцев.
Нарушения кислотовыделительной функции почек преимущественно связаны с канальцевыми поражениями. Это может быть обусловлено уменьшением экскреции ионов Н+ клетками канальцевого эпителия, а также нарушением аммониогенеза. Как правило, такие формы дисфункции почек возникают при хроническом или интермиттирующем пиелонефрите. В результате развивается так называемый почечный ацидоз. При острой почечной недостаточности (ОПН) в олигоанурической стадии и хронической почечной недостаточности (ХПН) в терминальной стадии в результате значительного поражения канальцевой системы возникает декомпенсированный метаболический ацидоз (pH < 7,25).
5.5. КОНЦЕНТРИРОВАНИЕ И РАЗВЕДЕНИЕ МОЧИ
Для поддержания постоянной осмолярности тканевой жидкости и регуляции экскреции воды важное значение имеет способность почек концентрировать и разводить мочу. Это обеспечивается так называемым противоточно-поворотным множительным механизмом, в котором главную роль играет петля Генле.
Разведение мочи достигается путем удаления NaCl из канальцевой жидкости в том участке нефрона, где мембрана канальца непроницаема для воды, т. е. в толстом восходящем колене петли Генле, дистальном канальце и собирательных трубках, причем петля Генле имеет наибольшее значение в образовании разведенной мочи.
Концентрирование мочи — это сложный процесс, зависящий от механизмов активного транспорта в канальцах. Моча в основном концентрируется в нисходящем отделе петли Генле. В собирательных трубках осуществляется конечная концентрация мочи под влиянием АДГ. Последний увеличивает проницаемость клеточных мембран собирательных трубок для воды, которая переходит в интерстиций мозгового вещества, повышая тем самым
осмолярность мочи. Максимальная осмолярность мочи достигает 1400 мосм/л, что соответствует относительной плотности мочи 1040. Наименьшая осмолярность мочи, наблюдаемая при отсутствии АДГ, составляет 290 — 300 мосм/л, что соответствует относительной плотности 1005 — 1008.
Вода, образующаяся вследствие активной реабсорбции осмотически активных веществ через непроницаемую для воды стенку канальца, получила название «осмотически свободная вода». Клиренс осмотически свободной воды (Ch2q) вычисляется по формуле, мл/мин:
^Н2О = ” ^osm ’
где Cosm — осмолярный клиренс, мл/мин; V — диурез, мл/мин.
Величина клиренса свободной воды отражает эффективность водовыделительной функции почек, степень проницаемости стенок канальцев и интенсивность реабсорбции натрия в восходящем колене петли Генле.
Осмолярный клиренс характеризует тот объем плазмы, который за 1 мин очищает
Нейроэндокринная регуляция функции почек 167
ся почкой от осмотически активных веществ. Его рассчитывают по формуле
VU
Г _ v osm ^osm р » *osm
где Unvm и Pnvtn — осмолярная концен-Uu”l Uuill А
трация соответственно мочи и плазмы моем/л; V — диурез, мл/мин.
Осмолярный клиренс отражает интен сивность выделения почками осмотичес ки активных веществ.
5.6. НЕЙРОЭНДОКРИННАЯ регуляция функции почек
Функция почек находится под контролем нейроэндокринной системы организма, который осуществляется как на экстра-, так и на интраренальном уровне. Почки иннервируются волокнами симпатического и парасимпатического отделов вегетативной нервной системы. Волокна симпатических нервов идут от нейронов трех нижних грудных и двух верхних поясничных сегментов спинного мозга, парасимпатические — от блуждающего нерва и тазового сплетения.
Нервные окончания подходят непосредственно к клеткам проксимального и дистального канальцев, будучи отделенными от них только веществом базальной мембраны. Это является морфологическим доказательством возможности прямого влияния нервных импульсов через эфферентные нервы на работу канальцевого эпителия. Однако больше изучено вазомоторное действие эфферентных нервных волокон на почечный кровоток. В стенке артериальных сосудов почек содержатся а- и р-ад-ренорецепторы, количественное соотношение которых до конца не выяснено. В целом, нервные импульсы играют относительно небольшую роль в функционировании почек. Это подтверждается наблюдениями за деятельностью денервированной пересаженной почки. Отсутствие заметных нарушений функции почек отмечено также после симпатэктомии на уровне нижних грудных сегментов. Остается невыясненным, зависят ли от прямых нервных импульсов такие тонкие механизмы деятельности почек, как концентрация и разведение мочи, ионообмен, выделение кислых продуктов и др. Несомненным является факт влияния нервных импульсов на тонус внутрипочечных сосудов. При определенных стрессовых воздействиях может возникнуть
внутрипочечная артериальная вазоконстрикция, которая способна вызвать вторичные функциональные отклонения.
На деятельность почек оказывает прямое действие ряд гормонов (альдостерон, антидиуретический гормон, паратгормон, кальцитонин, катехоламины и др.), для которых почки являются главным органом-мишенью.
5.6.1. Альдостерон
Альдостерон относится к минералокортикоидам коры надпочечников. Он является важнейшим регулятором объема внеклеточной жидкости и главным регулятором гомеостаза калия. Основным объектом действия альдостерона служат собирательные трубки коркового вещества. Его суточная секреция у здоровых людей, потребляющих нормальное количество натрия, колеблется то 50 до 250 мкг, а его концентрация в плазме — от 5 до 15 нг%.
Альдостерон оказывает действие главным образом на реабсорбцию натрия и секрецию калия в почках, подкисление мочи и механизм разведения и концентрации мочи. Он стимулирует реабсорбцию путем обмена ионов натрия на ионы калия и гидрогена в дистальных извитых канальцах и собирательных трубках, вызывает антинатрийурез и калийурез. Калий служит мощным непосредственным стимулятором секреции альдостерона. Поскольку последний, в свою очередь, способствует экскреции калия почками, эта обратная связь является важным фактором регуляции гомеостаза калия.
Альдостерон усиливает секрецию ионов Н+ в просвет канальцев, способствуя образованию аммиака. Это может быть след
168 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
ствием прямого действия гормона на водородный насос или изменения электрического градиента в результате активации транспорта натрия.
Нарушения секреции альдостерона могут обуславливаться первичными дефектами клеток клубочковой зоны надпочечников или возникают вторично вследствие изменения продукции регуляторов альдостероновой секреции.
Гиперальдостеронизм. Альдостеронизм — это синдром, главной особенностью которого является гиперсекреция альдостерона. При первичном альдостеронизме стимулы к повышению продукции альдостерона исходят от самих надпочечников, при вторичном — такие стимулы имеют вненадпочечниковое происхождение, обычно они исходят от ренин-ангио-тензиновой системы.
Первичный альдостеронизм, или синдром Конна, — заболевание, обусловленное опухолью коры надпочечника. Гиперпродукция альдостерона приводит к усилению реабсорбции натрия в обмен на ионы калия. Обеднение организма калием сопровождается мышечной слабостью, парестезиями, приходящими мышечными параличами. Типичными симптомами являются полиурия, никтурия, полидипсия. В результате гипокалиемии внутриклеточный К+ замещается ионами Na+ и Н+, что служит причиной возникновения внутриклеточного ацидоза и внеклеточного алкалоза.
Вторичный альдостеронизм — клинический синдром, обусловленный повышенной инкрецией альдостерона нормальными надпочечниками в ответ на изменения электролитного состава крови и ренин-ангиотензиновой системы.
Диагноз вторичного альдостеронизма подозревается у лиц с гипертензией и гипокалиемией на фоне высокой активности ренина плазмы. При гипертонической болезни или вазоренальной гипертонии на первый план выступает фактор ишемического воздействия на почку. Возникающая ишемия почки ведет к повышению активности ее юкстагломерулярного аппарата, вызывающей усиление продукции ренина и ангиотензина II. Последний стимулирует клубочковую зону коры надпо
чечников, обуславливая усиленную инкре-цию альдостерона.
Вторичный альдостеронизм может наблюдаться и в отсутствие гипертензии. Физиологическим его вариантом является альдостеронизм в условиях сниженного внутрисосудистого объема, например, при приеме диуретиков, ограничении поступления натрия, дегидратации.
Недостаточная продукция альдостерона надпочечниками (например, при болезни Аддисона) приводит к нарушениям в противоточно-поворотном множительном механизме (концентрирование и разведение мочи), вследствие чего снижается выделение осмотически свободной воды.
Гипоальдостеронизм. Общим признаком у всех больных гипоальдостеронизмом является отсутствие адекватного прироста альдостерона в условиях ограниченного приема натрия.
Гипоальдостеронизм может сопровождаться генерализованными деструктивными повреждениями коры надпочечников. При этом снижается продукция кортизола.
В большинстве случаев изолированный гипоальдостеронизм обусловлен недостаточной продукцией ренина (гипоренине-мический гипоальдостеронизм). Этот синдром чаще всего встречается у взрослых с легкой формой почечной недостаточности в сочетании с гиперкалиемией и метаболическим ацидозом. Поэтому наиболее вероятной причиной указанной формы гипоальдостеронизма является недостаточное образование ангиотензина II, наблюдаемое у больных, перенесших двустороннюю нефрэктомию.
5.6.2. Антидиуретический гормон
Антидиуретический гормон (АДГ) синтезируется в супраоптических ядрах гипоталамуса и выделяется из задней доли гипофиза в ответ на осмотический и неосмотический стимулы. Любое повышение осмолярности плазмы активирует осморецепторы, стимулирующие выделение АДГ. Падение осмолярности тормозит высвобождение АДГ, что приводит к водному диурезу и, таким образом, осмолярность
Нейроэндокринная регуляция функции почек 169
плазмы восстанавливается до нормального уровня.
Антидиуретическое действие этого гормона реализуется главным образом на уровне собирательных трубок почек. В норме собирательные трубки непроницаемы для воды. АДГ усиливает проницаемость этих структур для воды и способствует антидиурезу.
Дефицит АДГ связан с невозможностью его синтеза или с патологией задней доли гипофиза, что приводит к развитию несахарного диабета.
Несахарный диабет характеризуется гиперосмолярной гипернатриемией и экскрецией большого количества разведенной мочи с низкой осмолярностью.
Выделяют гипоталамическую и почечную формы болезни. Гипоталамический несахарный диабет — заболевание, обусловленное абсолютным дефицитом АДГ. Эта форма диабета может быть самостоятельной болезнью или симптомом некоторых эндокринных и неэндокринных заболеваний. Заболевание вызывается опухолями гипофиза и гипоталамуса, синдромом Симмондса, акромегалией, гигантизмом, болезнью Кушинга. Несахарный диабет может быть следствием черепно-мозговой травмы. К этиологическим факторам относятся также нейротропные вирусные инфекции (грипп и др.) и токсичность некоторых лекарственных препаратов (литий, демеклоциклин).
Почечный {нефрогенный) несахарный диабет — генетическая патология рецепторов АДГ почечных канальцев (относительный дефицит АДГ). Эта форма диабета встречается редко.
Характерными клиническими признаками являются полиурия и полидипсия (повышенная жажда). Суточный диурез при тяжелых формах заболевания достигает 40 л. Относительная плотность мочи низкая - 1,001-1,005.
5.6.3. Паратиреоидный гормон и кальцитонин
Паратиреоидный гормон (ПТГ) синтезируется в паращитовидных железах. Главным фактором, контролирующим сек
рецию ПТГ, является концентрация ионизированного кальция во внеклеточной жидкости. Этот гормон вызывает множество изменений в почке, но главным его эффектом является снижение реабсорбции фосфатов и повышение реабсорбции кальция. Фосфатурический эффект осуществляется при действии ПТГ на проксимальные канальцы и зависит от реабсорбции натрия. Этот процесс обусловлен изменением активности аденилатциклазы, сопряженной с рецепторами ПТГ в канальцах. ПТГ также снижает клиренс ионов кальция и магния, воздействуя на реабсорбцию кальция в проксимальных канальцах. Фосфатурия, вызываемая повышением секреции ПТГ паращитовидными железами, может способствовать образованию фосфатных камней в почках.
Вторичный гиперпаратиреоз наблюдается при хронической почечной недостаточности. При этом значительно повышается уровень иммунореактивного ПТГ в ответ на задержку фосфатов и потерю кальция при уремии.
Кальцитонин секретируется главным образом клетками щитовидной железы. Он является пептидом, состоящим из 32 аминокислотных остатков. Этот гормон вызывает натрийурез, фосфатурию и увеличивает выведение кальция. Кальцитонин секретируется в ответ на увеличение содержания кальция в плазме.
5.6.4. Катехоламины
Действие катехоламинов на почку осуществляется через а-адренергические рецепторы и Рр и р2-адренорецепторы. Главные, наиболее хорошо изученные эффекты заключаются в вазоконстрикции и стимуляции высвобождения ренина клетками юкстагломерулярного аппарата. Вазоконстрикция в почках обусловлена активацией а-адренорецепторов и ингибируется антагонистами этих рецепторов. Адреналин и норадреналин снижают эффективный почечный плазмоток. Дофамин в небольших дозах увеличивает внутрипочечный кровоток, в больших — угнетает.
170 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
5.7. ВЫВЕДЕНИЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ ПОЧКАМИ
Почки ответственны за выделение не только естественных метаболитов, но и целого ряда лекарственных средств и продуктов их распада. Большинство лекарственных веществ, выделяемых почками, фильтруются в клубочках и затем либо совсем не реабсорбируются, либо реабсорбируются частично. Ряд веществ активно секретируется клетками проксимальных канальцев.
Клубочковой фильтрации подвергаются все вещества, имеющие молекулярную массу не более 5000. Степень клубочковой фильтрации зависит также от того, насколько препарат связан с сывороточными протеинами. Более свободно проникают через клубочковую мембрану препараты, находящиеся в плазме в свободном состоянии.
Большинство лекарственных препаратов являются слабыми электролитами и находятся в организме в неионизирован-ном состоянии. Эти жирорастворимые препараты метаболизируются до водорастворимых дериватов, обладающих менее выраженной фармакологической активностью или вообще неактивных. Некоторые препараты частично растворяются в липидах и находятся в высокоионизированном состоянии при нормальных показателях pH. Они элиминируются главным образом с мочой в неизмененном виде. Длительность их действия в значительной степени зависит от функции почек. К таким препаратам относятся четвертичные аммонийные основания, ганглиоблокаторы, недеполяризующие мышечные релаксанты, многие антибиотики, производные бензодиазепина, сердечные гликозиды и др.
Около 80 % профильтровавшейся в клубочках жидкости реабсорбируется в проксимальных канальцах, что приводит к уменьшению объема жидкости и к повышению концентрации молекул препарата почти в пять раз. Вследствие этого в лежащих ниже сегментах канальцев происходит пассивная реабсорбция путем простой диффузии. Эпителий канальцев является для молекул лекарственных веществ липидным барьером, способствующим быстрой диффузии неионизированных жиро
растворимых лекарств и препятствующим проникновению водорастворимых ионизированных препаратов или их метаболитов. Жирорастворимыми являются лишь неионизированные формы и только они способны реабсорбироваться путем пассивной диффузии.
Увеличение диуреза может повысить почечную элиминацию препаратов, подвергающихся пассивной реабсорбции, поскольку возрастает объем мочи в просвете канальца и соответственно уменьшается концентрационный градиент.
Некоторые лекарственные вещества и органические кислоты активно секретируются специальными транспортными системами проксимальных канальцев. Эти системы для некоторых веществ настолько активны, что даже при низкой концентрации препарата в крови, например, пара-аминогипуровой кислоты (ПАГ), происходит полное очищение плазмы крови за один пассаж через почки. К веществам, выделяемым почками путем канальцевой секреции относятся: кислоты — ПАГ, салициловая, оксаловая, бутадиен, индометацин, диодраст, антибиотики пенициллиновой группы, сульфаниламиды, дихлотиа-зид, метотрексат, глюкурониды; основания — холин, гистамин, гексаметоний, пентамин, новокаин, новокаинамид.
Многие анестезиологические препараты относятся к группе липофильных соединений, распадающихся в организме до гидрофильных метаболитов и лишь частично выделяющихся почками в неизмененном виде. К ним относятся барбитураты короткого действия (тиопентал-натрий), производные бензодиазепина (диазепам, нитразепам, флюнитразепам), кетамин, дро-перидол, опиаты и местные анестетики. Лишь 2,5 % введенной дозы кетамина выделяется почками в неизмененном виде, 19 % экскретируется с мочой в виде неактивных метаболитов, остальная часть метаболитов в конъюгированном виде выделяется печенью. Диазепам выделяется почками в неизмененном виде в незначительном количестве (около 1 % введенной дозы), 30 % препарата экскретируется с мочой в
Выведение лекарственных средств почками 171
виде метаболитов, одним из которых является фармакологический дериват оксазепам, преимущественно выделяемый почками, что при сниженной функции последних может привести к кумуляции оксазепама при повторных введениях диазепама.
Дроперидол и фентанил также относятся к липофильным соединениям, которые вначале подвергаются биотрансформации, а затем выводятся почками из организма. 75 % метаболитов дроперидола выделяется с мочой, 25 % — с желчью. Около 60 % фентанила выводится почками в виде метаболитов, 40 % — выделяется печенью.
Недеполяризующие мышечные релаксанты (павулон, ардуан) в связи с их гидрофильностью, а следовательно — сильной ионизированностью, выделяются почками преимущественно в неизмененном виде (80 %) и лишь незначительная часть (5 %) выводится печенью, 15 % введенной дозы выделяется почками в виде метаболитов.
Многие антибиотики выделяются преимущественно почками, однако степень их экскреции различна. Препараты группы пенициллина экскретируются в количестве 60 — 85 % введенной дозы, причем 40 — 70 % — в активной форме, 30 —50 % — в неактивной. Большая часть препаратов выделяется путем активной канальцевой секреции. Натриевая соль ампициллина выделяется главным образом путем клубочковой фильтрации и активной канальцевой секреции. Период полу выведения препарата в норме составляет 0,4 —0,5 ч.
Препараты группы аминогликозидов, в частности гентамицин, почти полностью выделяются почками, однако эти препараты не секретируются канальцами, а только фильтруются в клубочках. Период полувыведения гентамицина составляет 2,5 ч.
Антибиотики цефалоспоринового ряда в разной степени связываются с протеинами плазмы крови и выделяются из организма преимущественно через почки в неизмененном виде.
Цефуроксим (зинацеф) связывается с белками крови (от 35 % до 50 %) в зависимости от способа введения. Препарат не метаболизируется и в неизмененном виде
выводится почками путем клубочковой фильтрации и канальцевой секреции. Период полувыведения цефуроксима при внутривенном введении составляет 70 мин, 80 % введенной дозы выводится почками в течение первых шести часов. Он не обладает нефротоксическим действием, однако в сочетании с салуретиками (лазикс) или аминогликозидами ухудшает функцию почек. При снижении клиренса креатинина дозы препарата должны быть соответственно уменьшены.
Цефтриаксон (роцефин, лонгацеф) относится к цефалоспоринам третьего поколения. Он активно связывается с белками сыворотки крови, особенно с альбумином (от 80 до 95 % введенной дозы). Время полувыведения цефтриаксона составляет 6 —8 ч, что значительно превышает аналогичный показатель у других цефалоспоринов, поэтому его можно вводить один раз в сутки. Он имеет двойной путь выведения: с мочой выделяется 40 — 60 % неизмененного препарата, остальная часть экскретируется печенью. Особенность фармакокинетики цефтриаксона заключается в том, что при поражении одного из органов (почек или печени) увеличивается клиренс препарата по альтернативному пути. Почечная экскреция способствует высокой концентрации препарата в моче, что позволяет эффективно лечить инфекции мочевого тракта. Печеночная фракция цефтриаксона обеспечивает достаточную концентрацию препарата в желчи, что позволяет проводить терапию инфекций желчных путей.
Тиенам — антибиотик широкого спектра действия из группы карбопенемов — подобно цефалоспоринам выводится почками в неизмененном виде путем клубочковой фильтрации и канальцевой секреции. Дозы и интервалы между внутривенными введениями препарата находятся в корреляционной зависимости от скорости клубочковой фильтрации (клиренса креатинина).
Меронем (меропенем), как и тиенам, относится к группе карбопенемов. Механизм экскреции препарата с мочой аналогичен таковому у тиенама. Период полувыведения меронема почками составляет
172 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
один час. В течение 12 ч происходит полное очищение организма от антибиотика. При снижении клиренса креатинина доза пропорционально уменьшается, а интервал между введениями — увеличивается. При почечной недостаточности возможна кумуляция антибиотиков этой группы.
Препараты из группы фторхинолонов {офлоксацин, норфлоксацин, ципрофлоксацин и др.) выделяются почками путем клубочковой фильтрации и канальцевой секреции. Время полу выведения увеличивается при уменьшении клиренса креатинина, поэтому при снижении функции почек увеличивается интервал между приемами препаратов. При почечной недостаточности возможна кумуляция препаратов из этой группы.
Фосмицин — новый антибиотик широкого спектра действия, после внутривенного введения циркулирует в крови в несвязанном виде (лишь 2,1 % препарата связывается с протеинами) и не подвергается метаболизму. Он почти полностью выводится почками в неизмененном виде, однако механизм экскреции с мочой недостаточно изучен. Период полного очищения организма от антибиотика составляет 10—11 ч после начала внутривенной инфузии 1 —2 г препарата.
5.8. ДИУРЕЗ
Увеличение диуреза обуславливается двумя причинами: уменьшением проницаемости канальцевой стенки для воды или усилением экскреции растворенных веществ и воды, необходимой для их растворения. Хотя теоретически действие диуретиков может осуществляться путем либо увеличения клубочковой фильтрации, либо снижения реабсорбции в канальцах, практически механизм действия всех широко используемых препаратов этой группы реализуется путем воздействия на канальцы. В большинстве случаев диуретики снижают реабсорбцию натрия, поэтому такие препараты называют еще натрий-уретиками.
Одним из путей повышения диуреза является введение веществ, свободно фильт-
Клиренс. В клинической практике для оценки функций почек используют такой показатель, как клиренс. Клиренс вещества эквивалентен объему плазмы, полностью очищенной от этого вещества за единицу времени.
Важное значение имеет клиренс тех веществ, которые выделяются путем клубочковой фильтрации (инулин, эндогенный креатинин) или посредством клубочковой фильтрации и канальцевой секреции (ПАГ, диодраст). В первом случае клиренс является мерой клубочковой фильтрации, во втором — мерой эффективного почечного плазмотока.
В нормальных условиях клиренс инулина составляет 125 мл/мин, эндогенного креатинина — 127 мл/мин (80 — 180 мл/мин), клиренс ПАГ колеблется в пределах 650 ±160 мл/мин.
Измерения клиренса можно проводить для любых веществ, в частности, с целью определения периода полувыведения лекарственных средств. Важно также знать, как этот период изменяется при заболеваниях почек. Клиренс эндогенного креатинина является главным критерием дозирования лекарственных средств при различных функциональных состояниях почек.
И ДИУРЕТИКИ
рующихся в клубочках и нереабсорбиру-ющихся ни в одном из сегментов системы канальцев. Такие вещества известны как осмотические диуретики (маннитол, сор-. битол).
Диуретические препараты могут воздействовать на различные отделы нефрона. По локализации действия на отдельные сегменты канальцев диуретики подразделяют на:
1. Диуретики, действующие на проксимальный отдел канальцев (ацетазоламид, диакарб), ингибиторы карбоангидразы.
2. Диуретики, действующие на толстое восходящее колено петли Генле (петлевые диуретики). К ним относятся фуросемид, буметанид, этакриновая кислота (уре-гит).
Мочекаменная болезнь 173
3. Диуретики, действующие на начальный сегмент дистального канальца (тиазиды). К ним относятся хлортиазид, хлор-талидон, метолазон.
4. Диуретики, действующие на конечную часть дистального канальца и собирательные трубки:
а) блокаторы натриевых каналов (ами-лорид, триамтерен);
б) антагонисты альдостерона (спиронолактон, верошпирон). Эти диуретики известны как калийсберегающие препараты.
В клинических условиях приходится применять два или больше диуретиков различных классов. Этим можно уменьшить нежелательные изменения баланса электролитов (например, амилорид добавляют для снижения потери калия, кислот и магния).
Целесообразность использования комбинации диуретических препаратов определяется необходимостью увеличить на-трийурез в тех случаях, когда развивается устойчивость к диуретикам.
Достичь мощного диуреза можно путем комбинирования «петлевых диуретиков» с тиазидными препаратами, а также с ка-лийсберегающими, действующими на уровне конечной части дистальных сегментов канальца.
Одним из показаний к применению диуретиков является развитие отечного синдрома при таких заболеваниях, как острая сердечная недостаточность с отеком легких, застойная сердечная недостаточность, цирроз печени с асцитом и нефротический синдром. Отеки при хронической почечной недостаточности (ХПН) резистент
ны к тиазидам из-за низкой скорости клубочковой фильтрации. Таким больным требуются высокие дозы «петлевых диуретиков», при этом противопоказано применение калийсберегающих средств. Комбинация салуретиков (лазикс) с осмодиуретиками (маннитол) используется для лечения больных в олигоанурической стадии острой почечной недостаточности (ОПН). Осмодиуретики применяются также при лечении отека мозга.
Диуретики нашли широкое применение для профилактики и лечения артериальной гипертензии. Однако как препараты для монотерапии они утратили свое значение и используются главным образом в качестве одного из компонентов гипотензивной терапии. Известно, что тиазидные диуретики влияют на обменные процесы в клеточных мембранах артериол, уменьшая набухание и приводя к снижению резистентности периферических сосудов. Калийсберегающие тиазидные диуретики целесообразно назначать больным гипертонической болезнью в сочетании с остеопорозом. При тяжелой артериальной гипертензии на фоне почечной недостаточности требуются высокие дозы «петлевых диуретиков» для уменьшения объема внеклеточной жидкости.
При лечении диуретиками следует проводить электролитный мониторинг с целью избежания побочных гидропонных нарушений. Диуретики нельзя применять произвольно или по несоответствующим показаниям, например, с целью уменьшения массы тела или устранения обычного периферического отека.
5.9. МОЧЕКАМЕННАЯ БОЛЕЗНЬ
Мочекаменная болезнь является поли-этиологическим заболеванием. В патогенезе камнеобразования основным элементом являются канальцевые поражения (дистрофия эпителия), вследствие чего происходят изменения в белковом и полисахаридном обменах почки, что при дополнительных условиях приводит к образованию микролитов, каждый из которых может стать ядром (матриксом) для обра
зования мочевого конкремента. Главным условием для образования камня является перенасыщение мочи соединениями, способными к кристаллизации.
Почечные камни различны по составу и структуре. Все камни мочевых путей состоят из двух компонентов: кристалла (или кристаллоподобной структуры) и матрикса. Соотношение между кристаллоидом и матриксом в составе камня широко варьи
174 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
рует от высокоорганизованной переплетенной структуры до беспорядочной массы.
Важную роль в образовании камней играет кислотность мочи. В норме она соответствует pH =6,2 —6,8. Тенденция к камнеобразованию увеличивается главным образом при окислении мочи. Постоянно низкий показатель pH мочи способствует образованию камней, особенно состоящих из оксалата кальция и мочевой кислоты.
Другим важным фактором, способствующим развитию мочекаменной болезни, являются инфекции мочевого тракта, вызванные микроорганизмами, продуцирующими уреазу и изменяющими pH мочи. Первый процесс приводит к увеличению в моче содержания иона аммония, ощелачиванию мочи и образованию камней на основе фосфатов кальция и магния.
Существует несколько классификаций уролитиаза в зависимости от основных компонентов камня. Согласно наиболее упрощенной классификации различают камни:
1) мочевой кислоты — ураты;
2) щавелевой кислоты — оксалаты;
3) фосфорной кислоты — фосфаты;
4) цистиновые.
Эта классификация мочевых камней предполагает их мономинеральность, однако они чаще всего бывают полимине-ральными. Название камню дается цо преобладающему минералу.
5.9.1. Ураты
Ураты — это мочевые камни, состоящие из мочевой кислоты. Образование камней зависит от следующих факторов: 1) суточной экскреции мочевой кислоты; 2) суточного диуреза; 3) pH мочи.
Увеличение суточной экскреции мочевой кислоты может быть обусловлено повышенной эндогенной продукцией уратов или потреблением продуктов питания с высоким содержанием пуринов (мясо, дрожжи и др.). Повышенная экскреция мочевой кислоты наблюдается у больных подагрой. Значительное потребление алкоголя может приводить к расщеплению пуриновых нуклеотидов и повышенной продукции уратов.
Снижение суточного диуреза (меньше 1 л/сут) способствует повышению концентрации уратов в моче, т. е. создает условия для формирования перенасыщенного раствора с последующим кристаллообразованием. Объем мочи зависит от потребления жидкости и ее потери организмом (климатические условия, физическая нагрузка, температура тела, диарея, рвота и другие экстраренальные потери жидкости). При нормальном суточном диурезе (примерно 1,5 л/сут) этот фактор риска уменьшается.
Растворимость уратов в моче в значительной степени зависит от pH мочи. При pH = 7,0 и выше растворимость мочевой кислоты в 10 раз больше, чем при pH = 5,0, т. е. в щелочной среде вероятность образования уратных камней меньше, чем в кислой. На этом основана литолитическая терапия и профилактика уратного нефролитиаза, предусматривающая контроль pH мочи.
5.9.2. Оксалаты
Соли щавелевой кислоты — оксалаты — являются самой частой причиной уролитиаза. По своему химическому составу они относятся к кальциевым солям [СаС2О4 • Н2О].
Большинство оксалатно-кальциевых камней образуются у больных без явного нарушения метаболизма оксалатов или их экскреции, но с нарушением метаболизма кальция (гиперкальциурия).
Оксалаты полностью фильтруются в клубочках и затем подвергаются реабсорбции и секреции в канальцах. В норме экскреция оксалатов с мочой составляет 10 — 50 мг/сут. Вследствие нерастворимости кальциевых солей и высокой ионной силы оксалатов гипероксалурия является причиной оксалатно-кальциевого уролитиаза.
Для больных, страдающих оксалатным уролитиазом, характерна кислая моча (pH = 5,5-6,6).
5.9.3. Фосфаты
Образованию фосфатных камней (чаще всего это соли магния или кальция) способствуют фосфатурия и сдвиг кис
Острая почечная недостаточность 175
лотности мочи в щелочную сторону (pH > >7,0).
Повышенное выделение фосфатов может быть следствием высокого уровня паратиреоидного гормона (ПТГ) в крови. ПТГ способен подавлять канальцевую реабсорбцию фосфатов, оказывая фосфату-рический эффект. Вторым мощным ингибитором канальцевой реабсорбции фосфатов является кальцитонин. Однако сама по себе фосфатурия не может привести к кристаллообразованию, если отсутствует ключевой фактор — щелочная моча. Ощелачивание мочи кроме алиментарных причин вызывает наличие инфекции в полостной системе почки. Микробы, вырабатывающие фермент уреазу, расщепляют аммоний мочи и этим способствуют формированию щелочной среды, что, в свою очередь, приводит к образованию кристаллов из фосфатных солей. Поэтому реци
дивные камни чаще всего бывают фосфатными, поскольку моча таких больных, как правило, инфицирована.
5.9.4. Цистиновые камни
Цистин является продуктом метаболизма метионина. Последний считается наименее растворимым из известных в природе аминокислот. В основе образования этой разновидности камней лежит цистинурия, обусловленная пониженной реабсорбцией как профильтрованного, так и секретированного цистина. При обычных значениях pH мочи в одном ее литре растворено около 300 мг цистина. Факторами риска развития цистиновых камней считаются перенасыщение мочи цистином и наличие кислой концентрированной мочи. Цистиновый уролитиаз наблюдается у 1 — 3 % больных мочекаменной болезнью.
5.10. ОСТРАЯ ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Острая почечная недостаточность (ОПН) представляет собой клинический синдром различной этиологии, характеризующийся значительным и быстрым снижением СКФ, накоплением в крови азотистых шлаков и неспособностью почек поддерживать гомеостаз. СКФ может снижаться с нормальных значений — 100 — 140 мл/мин до 1 —10 мл/мин. При этом наблюдается олигурия (диурез менее 500 мл/сут) или анурия ( диурез менее 100 мл/сут).
Резкое снижение почечных функций приводит к различным клиническим и биохимическим последствиям, тяжесть которых зависит от продолжительности и степени ОПН.
В зависимости от этиологического фактора выделяют три основные формы ОПН: 1) преренальную; 2) ренальную; 3) постренальную. Первая из них обусловлена снижением перфузии почек, вторая — повреждением самой паренхимы почек, третья — обструкцией мочевыводящих путей.
5.10.1. Преренальная острая почечная недостаточность
Выделяют следующие причины прере-нальной ОПН: гиповолемия (кровотечение, ожоги, рвота, диарея), снижение сердечного выброса (кардиогенный шок, инфаркт миокарда, застойная сердечная недостаточность, эмболия легочной артерии), системная гипотензия (сепсис, печеночная недостаточность), повышение внутрипочечного сосудистого сопротивления (большие дозы агонистов а -адренорецепторов) и др.
Общим для всех этих состояний является снижение перфузии почек. При отсутствии изменений почечной паренхимы почки реагируют на снижение перфузии максимальным увеличением реабсорбции натрия и воды, приводящим к уменьшению объема мочи с низким содержанием натрия и высокой осмолярностью. Резкое уменьшение перфузии ведет к снижению СКФ и развитию азотемии. Катехоламины и ангеотензин II яв
176 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
ляются важными местно-выделяемыми гормонами, запускающими данный процесс.
5.10.2. Ренальная острая почечная недостаточность
К причинам ренальной ОПН относятся: острый тубулярный некроз, кортикальный некроз, гломерулонефрит, острый интерстициальный нефрит, васкулит, токсикоз беременных, экзогенные интоксикации и др.
Следует отметить, что нелеченная затянувшаяся преренальная ОПН может трансформироваться в ренальную, т. е. приводить к острому тубулярному некрозу. Последний является наиболее частой причиной ОПН и причинным фактором смерти около 50 % больных, несмотря на прогресс лекарственной и заместительной терапии.
Почечная гипоперфузия относится к наиболее часто диагностируемым пусковым механизмам, приводящим к острому тубулярному некрозу. Гипоперфузия может значительно снизить регионарную доставку кислорода к клеткам почечного эпителия. Последующие после ишемии и гипоксии механизмы клеточного повреждения включают истощение АТФ, приток натрия и кальция в клетку, внутриклеточный ацидоз, повреждение клетки, связанное с накоплением свободных радикалов, а также повреждение цитоскелета. Большинство процессов, повреждающих почечные клетки, характеризуется истощением АТФ из-за нарушения окислительного фосфорилирования в митохондриях.
Излишнее поступление натрия в клетку из внеклеточной жидкости деполяризует клетку и подавляет усиленный транспорт веществ, в норме сопряженный с транспортом натрия (гидрогенкарбонатов, фосфатов, аминокислот и глюкозы). Деполяризация также открывает потенциалзависимые кальциевые каналы, обеспечивая транспорт кальция в клетку. Внутриклеточная концентрация свободного кальция также повышается вследствие его выхода из митохондрий. Это является теоретическим обоснованием применения блокаторов
кальциевых каналов для защиты митохондриального дыхания. Нефротоксическое воздействие занимает второе место по частоте развития острого тубулярного некроза.
Из экзогенных токсинов особый интерес представляют антибиотики, поскольку спектр их применения постоянно расширяется.
Причинами развития нефротоксического острого тубулярного некроза являются:
экзогенные токсины:
нефротоксические антибиотики; рентгенконтрастные препараты;
нефротоксические противоопухолевые препараты;
тяжелые металлы (свинец, ртуть);
органические растворители (этиленгликоль);
фосфорсодержащие анестетики (мето-ксифлюран, галотан);
эндогенные токсины: гемолиз (гемоглобин);
рабдомиолиз (миоглобин);
распад опухоли;
миелома.
Многие цефалоспорины транспортируются в клетки почечного эпителия переносчиком парааминогипурата, расположенным в базолатеральной мембране клеток проксимальных канальцев. Именно здесь локализуется поражение при остром тубулярном некрозе, вызванном цефалоспоринами. Аминогликозиды также накапливаются в этом сегменте нефрона. Основной путь их захвата — это связывание с ними анионных фосфолипидов в мембранах щеточной каемки и последующий их транспорт внутрь клетки. Механизм токсического действия аминогликозидов заключается в повреждении внутриклеточных органелл.
Патогенетический путь развития ОПН — как ишемического, так и нефротоксического происхождения — это повреждение клеток почечных канальцев. Для острого тубулярного некроза характерно наличие зон очагового некроза в клетках канальцев и разрушение базальной мембраны. Очаги повреждения рассеяны в почках диффузно. Наиболее тяжелые поврежде
Острая почечная недостаточность 177
ния обычно обнаруживаются в прямой части проксимального канальца.
Экспериментальными исследованиями на животных выявлено несколько механизмов, снижающих клубочковую фильтрацию при остром тубулярном некрозе. К ним относятся:
1) внутриканальцевая обструкция;
2) канальцевая обратная утечка;
3) вазоконстрикция;
4) снижение проницаемости клубочков.
Эти механизмы не исключают друг друга, чаще всего имеет место их сочетание.
Клиническое течение ренальной ОПН. В клиническом течении острого тубулярного некроза принято различать три основные стадии:
1) начальную;
2) олигоанурическую (стадия выраженных клинических проявлений);
3) восстановления (полиурическая стадия).
Начальная стадия может продолжаться несколько часов или дней и неотличима от обратимой преренальной ОПН. В этой стадии острый тубулярный некроз потенциально предотвратим.
Во второй стадии выраженных клинических проявлений резко снижается СКФ (обычно ниже 5—10 мл/мин). Эта стадия длится 1—2 недели, иногда до 6 недель. В связи с очень низкой скоростью клубочковой фильтрации олигоанурическая стадия характеризуется прогрессивным накоплением в крови азотистых шлаков и калия, а также развитием метаболического ацидоза.
Стадия восстановления начинается с повышения диуреза с последующим переходом в полиурию (диурез до 3 л/сут и более), однако повышение СКФ и нормализация других проявлений уремии обычно наблюдаются через 24 — 48 ч после возобновления диуреза. Канальцевые функции восстанавливаются медленнее, и нарушения концентрационной способности почек и способности к окислению мочи нередко сохраняются месяцами.
Основные нарушения гомеостаза в олигоанурической стадии ОПН. Вследствие снижения почечных функций изменяются состав и объем жидкостей и электро
литов организма, а также экскреция конечных продуктов метаболизма. Это приводит к развитию целого ряда серьезных нарушений гомеостаза.
Гидроионные нарушения. Неспособность почек экскретировать натрий и воду приводит к увеличению объема внеклеточной жидкости и перераспределению натрия между секторами. На фоне гипергидратации и снижения экскреции натрия наблюдается переход натрия во внутриклеточный сектор с развитием гипонатриемии.
Одновременно происходит повышение концентрации ионов калия в плазме крови (гиперкалиемия, К+ > 5 ммоль/л) вследствие снижения его экскреции почками, транспортирования из внутриклеточного во внеклеточное пространство, высвобождения при белковом катаболизме. Наблюдается также гипокальциемия и фосфате-мия.
Основными проявлениями гиперкалиемии являются нарушения сердечного ритма, мышечная вялость, адинамия. При прогрессирующем увеличении уровня калия в сыворотке крови (К+ > 6,0 ммоль/л) на ЭКГ обнаруживается увеличенный зубец Т, расширение комплекса QRS, удлинение интервала PR и в дальнейшем появление сглаженной двухфазной волны QRS —Т. Кроме того, может наблюдаться суправентрикулярная тахикардия, фибрилляция желудочков. Остановка сердца происходит в диастоле.
Метаболический ацидоз. При ОПН выключаются почечные механизмы поддержания КОС. В олигоанурической стадии происходит накопление сульфатов, фосфатов и органических кислот, нарушается выведение Н+, уменьшается количество буферных оснований. Метаболический ацидоз может достигать значительной степени (pH <7,18). Выключение почечного механизма поддержания КОС увеличивает нагрузку на респираторную систему. У больных в этой стадии ОПН гипервентиляция возникает как компенсаторная реакция на метаболический ацидоз. Однако этот механизм компенсации чаще всего оказывается недостаточным.
Азотемия (уремическая интоксикация). Интоксикация организма в олиго
178 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
анурической стадии ОПН обусловлена резким снижением или полной утратой азотовыделительной функции почек, что приводит к накоплению продуктов азотистого обмена (мочевины, креатинина, мочевой кислоты, аммиака). Поскольку ни одно из этих веществ в отдельности не может быть настолько токсичным, чтобы повышением его концентрации в крови можно было бы объяснить картину уремической интоксикации, принято считать уремию результатом совокупности гуморальных расстройств, возникающих при утрате гомеостатической функции почек.
Мочевина крови в этой стадии ОПН нередко достигает очень высокой концентрации (135 — 150 ммоль/л), особенно на фоне острых воспалительных процессов, усиленного протеинового катаболизма. Концентрация креатинина повышается до 0,8 — 1,4 ммоль/л. Скорость нарастания содержания креатинина в крови в сутки колеблется в пределах 0,044 — 0,088 ммоль/л, мочевины — 4,99 —6,66 ммоль/л.
Стадия восстановления диуреза. В этой стадии постепенно увеличивается диурез, достигающий через 3 — 5 дней 2 — 3 л/сут. Наблюдается гипостенурия (относительная плотность мочи 1,005 — 1,012).
Увеличение диуреза еще не является признаком восстановления нормальной функции почек. Повышенное выделение гипотоничной мочи отражает состояние недостаточности канальцев. Клетки канальцевого эпителия еще не в состоянии сохранять воду и регулировать выделение электролитов. В полиурической стадии велика вероятность развития дегидратации при несвоевременной коррекции дефицита жидкости.
В этой стадии ОПН еще не восстановлены процессы активного транспорта натрия в канальцах, что сопровождается потерей этого аниона с мочей и развитием гипонатриемии. Одновременно наблюдается повышенное выделение калия и хлора. Возникает опасность гипокалиемии и гипохлоремии, требующих соответствующей коррекции.
Полиурия в начальный период еще не приводит к ликвидации азотемии. Азотовыделительная функция почек восстанав
ливается на протяжении нескольких недель. Изменения КОС в этой стадии ОПН не закономерны.
5.10.3. Постренальная острая почечная недостаточность
Эта форма ОПН связана с внепочечной обструкцией на различном уровне мочевыводящих путей (лоханка, мочеточник, шейка мочевого пузыря, мочеиспускательный канал). У лиц среднего возраста основной причиной обструкции являются почечные камни и камни мочеточников, у пожилых больных — гиперплазия предстательной железы, различные новообразования.
Выраженность постренальной ОПН зависит как от степени, так и от продолжительности обструкции. Частичная обструкция вызывает медленно прогрессирующее снижение почечных функций с частыми инфекционными осложнениями. Синдром ОПН быстро развивается при полной билатеральной обструкции мочеточников или мочеточника единственной почки. Основным компонентом механизма снижения СКФ является повышение давления в мочеточнике, которое передается на канальцевую систему и на клубочки, что снижает общее гидростатическое фильтрационное давление на уровне клубочков.
Этот механизм функционирует на ранних этапах после обструкции мочеточников, поскольку почечный кровоток сначала возрастает. Начальное повышение почечного кровотока происходит вследствие увеличения синтеза почечного простагландина Е2 (сильного вазодилататора). Позже (спустя 12ч), когда почечный кровоток падает ниже нормы, снижается и клубочковый кровоток вследствие повышения прегломерулярной сосудистой резистентности под действием ангиотензина II или вазоконстрикторного простагландина -тромбоксана А2.
Если устранение обструкции происходит в течение первой недели после ее возникновения, то можно ожидать полное восстановление функции почек. В противном случае наблюдается необратимая потеря функции почек.
Хроническая почечная недостаточность (ХПН) 179
5.11. ХРОНИЧЕСКАЯ ПОЧЕЧНАЯ НЕДОСТАТОЧНОСТЬ (ХПН)
ХПН — это клинический синдром, обусловленный необратимым, как правило, прогрессирующим повреждением почек вследствие различных патологических состояний. ХПН является хроническим процессом, сопровождающимся снижением функциональной способности почек на 50 % и более. В отличие от ОПН, которая считается обратимым состоянием, ХПН необратима вследствие постепенного замещения почечной паренхимы рубцовой тканью.
Независимо от причины патологии снижение почечных функций происходит за счет трех основных механизмов:
1) уменьшения количества функционирующих нефронов (в норме оно составляет один миллион в каждой почке); 2) значительного снижения СКФ в каждом нефроне без уменьшения их количества; 3) сочетания первого и второго механизмов. Следствием действия каждого из этих факторов является снижение СКФ.
Круг заболеваний, способных привести к ХПН, довольно широк. Схематически их можно сгруппировать следующим образом.
I. Первично клубочковые заболевания: 1) хронический гломерулонефрит;
2) подострый злокачественный гломерулонефрит.
II. Первично канальцевые заболевания:
1) интерстициальный нефрит;
2) хронический пиелонефрит;
3) туберкулез почек;
4) радиационный нефрит.
III. Сосудистые заболевания:
1) эссенциальная гипертония;
2) синдром злокачественной гипертонии;
3) стеноз почечной/почечных артерий;
4) тромбоз почечных вен.
IV. Диффузные заболевания соединительной ткани:
1) системная красная волчанка;
2) системная склеродермия;
3) узелковый периартериит.
V. Болезни обмена веществ:
1) диабетический нефросклероз;
2) амилоидоз;
3) подагра.
VI. Обструктивные нефропатии:
1) мочекаменная болезнь;
2) гидронефроз;
3) опухоли мочевых путей, предстательной железы;
4) ретроперитонеальный фиброз.
VII. Врожденные аномалии:
1) поликистоз почек;
2) гипоплазия почек;
3) аномалии мочевых путей.
В подавляющем большинстве случаев основой для развития ХПН являются гло-меруло- или пиелонефрит. Главное проявление этого заболевания — повышение концентрации креатинина и мочевины вследствие падения СКФ. В отличие от ОПН ХПН развивается медленно.
ХПН сопровождается уменьшением количества функционирующих нефронов. При этом интактные нефроны продолжают функционировать на более высоком уровне, чтобы компенсировать потерю части нефронов, т. е. наблюдается гиперфункция оставшихся интактных нефронов. Эта компенсаторная реакция осуществляется путем увеличения размера нефрона, повышения СКФ и усиления экскреции растворенных веществ.
Особенности нарушения экскреторной функции почек
Экскреция воды. Основное нарушение почечных функций при ХПН связано с выделением воды. С развитием заболевания прогрессирует нарушение способности почек концентрировать мочу. В то время как у здоровых людей осмолярность экскретированной мочи может в четыре раза превышать осмолярность плазмы, при прогрессирующей ХПН максимальная осмолярность мочи примерно равна осмолярности плазмы крови. Почки больных ХПН утрачивают способность уменьшать экскрецию воды до уровня, наблюдаемого у здоровых людей. С другой стороны, верхний предел экскреции воды также ограничен вследствие снижения популяции функционирующих нефронов.
Наиболее характерным клиническим проявлением ХПН является полиурия, нередко сопровождающаяся никтурией.
Экскреция натрия. По мере прогрессирования ХПН возрастает фракционная экс
180 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
креция натрия. Если у здорового человека удвоение потребления натрия с 3,5 г/сут до 7 г/сут требует увеличения фракционной экскреции натрия с 0,25 до 0,5 %, то такое же увеличение потребления соли у больного с ХПН делает необходимым повышение фракционной экскреции натрия с 8 до 16 %. Таким образом, при ХПН экскреция натрия может изменяться в очень ограниченных пределах, и эти пределы сужаются по мере снижения СКФ. Нижний предел экскреции натрия, или «порог», определяется неспособностью больных с ХПН максимально удерживать натрий. Больной с ХПН, находящийся на низконатриевой диете, часто не способен уменьшить выведение натрия за нужный период времени настолько, чтобы экскретируемое количество соответствовало поступающему. У него развивается отрицательный баланс натрия и экскретируется соответствующий объем воды.
Экскреция калия. При ХПН длительное время сохраняется нормальный баланс калия в организме. Лишь в терминальной стадии при олигурии наблюдается гиперкалиемия. Поддержание нормальной экскреции калия почками при уменьшении количества функционирующих нефронов зависит от способности дистальных канальцев секретировать калий. При очень низких значениях СКФ скорость секреции калия может приближаться к максимальной для поддержания устойчивого состояния. Остается очень небольшой функциональный резерв для реакции на внезапные изменения потребления калия. Такие факторы, как олигурия, внезапное увеличение потребления калия, развившийся метаболический ацидоз могут привести к гиперкалиемии.
У некоторых пациентов пожилого возраста или больных диабетом наблюдается снижение продукции альдостерона, они предрасположены к развитию гиперкалиемии. Такая же закономерность наблюдается у больных, получающих калийсбере-гающие диуретики, которые блокируют дистальный перенос калия и потенциально опасны при ХПН.
Экскреция Н+. По мере уменьшения СКФ развивается метаболический ацидоз.
Однако он не обнаруживается до тех пор, пока СКФ не упадет ниже 20 мл/мин. Но даже в том случае вариабельность степени ацидоза зависит от природы самого заболевания почек, объема внеклеточной жидкости, баланса калия и эффективной респираторной компенсации.
Нарушение экскреции ионов Н+ при ХПН происходит двумя путями: 1) снижается экскреция аммиака; 2) увеличивается потеря гидрогенкарбоната. Хотя экскреция аммиака в расчете на сохраненный нефрон возрастает, общая его экскреция с мочой снижается. При ХПН падение уровня реабсорбции гидрогенкарбоната может быть частично обусловлена вторичным гиперпаратиреозом. До определенного предела коррекция метаболического ацидоза осуществляется за счет респираторной компенсации. При исчерпании этого механизма компенсации наступает значительная гипервентиляция (аци-дотическое шумное дыхание).
Фосфаты и кальций. Экскреция фосфатов при ХПН не изменяется заметно до определенного уровня СКФ. Только после снижения этого показателя ниже 25 мл/мин происходит накопление фосфатов сыворотки в крови. Гиперфосфатемия постепенно прогрессирует по мере дальнейшего снижения СКФ.
Одновременно наблюдается изменение концентрации сывороточного кальция, вызванное несколькими факторами. Увеличение фосфатов в сыворотке вызывает эквивалентное снижение уровня ионизированного кальция. При СКФ ниже 30 мл/мин уровень 1,25-диоксихолекаль-циферола понижается, что может вызвать снижение всасывания кальция в пищеварительном канале. Способность паратиреоидного гормона мобилизовывать кальций из кости может быть нарушена, и эта резистентность костного скелета к действию гормона также может способствовать развитию гипокальциемии. С другой стороны, персистирующий метаболический ацидоз ведет к постепенному вымыванию кальция из кости и остеопорозу. Таким образом, для ХПН характерными можно считать гиперфосфатемию и гипокальциемию.
Хроническая почечная недостаточность (ХПН) 181
Таблица 5.2. Стадии прогрессирования ХПН
Стадия ХПН Скорость клубочковой фильтрации, мл/мин Содержание креатинина в плазме крови, ммоль/л
1. Начальная стадия >50 0,165
2. Стадия компенсированной ретенции 25-50 0,165-0,66
3. Стадия декомпенсированной ретенции 10-25 0,66-1,05
4. Терминальная стадия < 10 > 1,2
Существуют различные классификации стадий прогрессирования ХПН, однако для практической анестезиологии и интенсивной терапии целесообразно выделение четырех стадий ХПН (табл. 5.2).
В начальной стадии выделительная функция сохраняется, СКФ и концентрационная способность почек снижены. Эта стадия, как правило, протекает незаметно для больного, который может и не подозревать о развивающейся ХПН. Для второй стадии характерны умеренная азотемия, полиурия, гипоизостенурия, нарушение состава электролитов крови. В следующей стадии сохраняются полиурия и изостенурия, нарастает азотемия, появляется метаболический ацидоз, углубляются электролитные сдвиги, в клинической картине отмечаются диспептические расстройства (тошнота, рвота).
Переход второй стадии в третью может произойти под влиянием ряда неблагоприятных факторов: обострение пиелонефрита, присоединение интеркуррентной инфекции, операционная травма, внепочечные потери жидкости и электролитов и др.
Последняя, терминальная, стадия развивается при неблагоприятном исходе консервативной терапии и дальнейшем прогрессировании ХПН. СКФ снижается до 10 мл/мин и ниже. Полиурия сменяется олигурией или анурией, возрастает метаболический ацидоз, появляется гиперкалиемия, развивается клиническая картина уремии.
Благодаря широкому внедрению программного гемодиализа появилась большая группа больных с искусственно пролонгированной ХПН, у которых терми
нальная стадия утратила классические признаки и приобрела специфические особенности.
Наряду с уремическим синдромом при ХПН наблюдается ряд системных поражений. К ним относятся: гематологические нарушения, сердечно-легочные осложнения, гипертензия, нервно-мышечные нарушения, гастроинтестинальные, иммунологические и инфекционные осложнения, эндокринные и метаболические нарушения.
Анемия. Характерный признак ХПН. При падении СКФ ниже 40 мл/мин гематокрит уменьшается пропорционально степени почечной недостаточности (до 20 % в терминальной стадии).
Основным фактором, обуславливающим развитие анемии, является отсутствие эритропоэтина, который в норме продуцируется юкстагломерулярным аппаратом почек. По мере уменьшения количества функционирующих нефронов снижается способность к секреции эритропоэтина, что приводит к уменьшению выработки эритроцитов костным мозгом. Кроме того, развитие анемии обусловлено снижением продолжительности жизни эритроцитов. Развитие анемии при ХПН не связано с недостатком витамина В12 или фолиевой кислоты.
Нарушение свертываемости крови. У больных с ХПН имеется качественный дефект функционирования тромбоцитов, что проявляется в виде удлинения времени кровотечения на фоне нормального протромбинового времени и времени свертывания крови. Время кровотечения удлиняется за счет недостаточного выделения тромбоцитарного фактора III. Агрегация тромбоцитов, их способность к адгезии, потребление протромбина и высвобождение простагландинов — все эти компоненты процесса свертывания крови могут быть подавлены, что будет способствовать появлению геморрагий на коже и слизистых.
Сердечно-легочные осложнения. Вследствие сочетания гипертензии, анемии, перегрузки жидкостью и ацидоза у больных с ХПН развивается застойная сердечная недостаточность. Почти у половины больных, при лечении которых не использова
182 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
ли гемодиализ, наблюдается перикардит. Его патогенез не установлен, однако коррекция уремии с помощью диализа, как правило, вызывает обратное развитие проявлений перикардита.
В поздней стадии ХПН развиваются патологические изменения в легких с характерной рентгенографической картиной, на которой видны застойные явления в сосудах прикорневой зоны. Этот патологический процесс был назван уремическим пневмонитом.
Гипертензия. Более чем у половины больных с терминальной стадией ХПН наблюдается гипертензия. Существует несколько механизмов повышения артериального давления. Почки могут высвобождать повышенное количество ренина, превращающегося в ангиотензин. При этом возрастает общее периферическое сосудистое сопротивление. С другой стороны, пораженные почки частично утрачивают способность вырабатывать некоторые вазодилататоры (простагландины, нейтральные липиды).
Сниженная экскреция натрия приводит к задержке жидкости и повышению ее внеклеточного объема, т. е. возникает гиперволемическая гипертензия.
Нервно-мышечные нарушения. Мышечная слабость и мышечная возбудимость являются характерными признаками поздних стадий ХПН. При уремии снижается трансмембранный потенциал мышечных мембран, во внутриклеточной жидкости мышечной ткани увеличивается содержание натрия и снижается содержание калия.
Наблюдаются также изменения деятельности центральной нервной системы (уремическая энцефалопатия). Они проявляются бессонницей, неспособностью к концентрации, рассеяностью, снижением памяти. Электроэнцефалографические изменения («ритм медленных волн») могут быть связаны с повышением содержания кальция в головном мозгу.
Гастроинтестинальные нарушения. Симптомы нарушений пищеварительного канала появляются при снижении СКФ ниже 10 мл/мин. Они проявляются анорексией, тошнотой, рвотой.
Обычным явлением при ХПН являются геморрагии из пищеварительного канала. Источником чаще всего бывают поверхностные мелкие язвы, которые медленно кровоточат. Патогенез этих язв не установлен, частично он может быть объяснен дефектом тромбоцитов при ХПН.
Иммунологические и инфекционные осложнения. При ХПН снижаются защитные имунные реакции организма. Для этих больных характерна абсолютная лимфопения, у них снижены продукция антител и клеточный ответ на антигены. Ослабление клеточного иммунитета способствует предрасположенности к вирусным и грибковым заболеваниям. Больные с дефектами гуморального иммунитета и фагоцитарной способности значительно более восприимчивы к бактериальным инфекциям.
Инфекции являются наиболее частой причиной смерти как при ОПН, так и в терминальной стадии ХПН. Пневмония, вызванная грамположительными микроорганизмами, служит причиной смерти почти 50 % больных в терминальной стадии ХПН. Значительное количество больных в этой стадии умирает от уросепсиса, вызванного кишечной палочкой.
Почечная остеодистрофия. Снижение экскреции фосфатов при ХПН приводит к гиперфосфатемии, в результате чего уменьшается содержание ионизированного кальция в сыворотке крови. Это стимулирует паращитовидные железы к секреции большего количества паратиреоидного гормона, который, воздействуя на клетки почечных канальцев, снижает реабсорбцию фосфатов. Когда СКФ падает ниже 25 % от нормы, отчетливо проявляются признаки этого вторичного гиперпаратиреоза. Повышение уровня паратгормона в крови приводит к увеличению резорбции кости и уменьшению ее плотности, что обнаруживается при рентгенологическом исследовании (остеопороз). Кроме того, замедляется превращение 25-оксихо-лекальциферола в более активную форму 1,25-диоксихолекальциферол. Это приводит к уменьшению всасывания кальция в пищеварительном канале. Поражению скелета способствует также метаболический ацидоз.
Хроническая почечная недостаточность (ХПН) 183
Гормональные нарушения. В терминальной стадии ХПН меняется количество и действие практически всех гормонов в организме. Это касается как внеклеточных гормонов, так и вырабатываемых непосредственно почками.
Происходит снижение уровня эритропоэтина с последующим развитием анемии. Увеличение активности почечного ренина с повышением уровня ангиотензина в плазме приводит к гипертензии. Уменьшается образование в почках 1,25-диокси-холекальциферола, вследствие чего снижается всасывание кальция в кишках. Наблюдается также повышение уровня паратгормона в сыворотке. Этому способствует снижение почечного катаболизма данного гормона. В почках развивается резистентность к действию антидиурети-ческого гормона и неспособность концентрировать мочу и сохранять содержание воды на нормальном уровне. В терминальной стадии ХПН наблюдается повышение уровня гастрина, глюкагона, гормона роста и базального уровня инсулина. Эти нарушения приводят к отсутствию толерантности к углеводам. У мужчин на этой стадии снижен уровень тестостерона в плазме, а у женщин — прогестерона и эстрогена.
Нарушения метаболизма
Азотистый обмен. В терминальной стадии ХПН резко выражены изменения азотистого баланса. В результате снижения азотовыделительной функции почек в организме накапливаются продукты белкового катаболизма — креатинин, мочевина, мочевая кислота. Однако уремическая интоксикация не может быть объяснена только гиперазотемией. Установлено, что кроме продуктов белкового обмена в синдроме уремической интоксикации принимают участие некоторые ионы, гормоны и неидентифицируемые «средние молекулы». Вопрос о том, имеет ли место при ХПН гиперкатаболизм белков или, наоборот, замедление распада протеинов, оста
ется открытым. Есть данные о том, что продукция креатинина может быть снижена, что сопровождается значительной потерей мышечной массы.
Уремия рассматривается как состояние белковой недостаточности, при которой наблюдаются изменения концентрации специфических аминокислот в плазме. У больных с ХПН снижена активность фенилаланиновой гидроксилазы, что является причиной низкой концентрации тирозина в плазме и низкого соотношения ти-розин/фенилаланин. В то же время повышается внутриклеточная концентрация фенилаланина, глицина и метилированного гистидина, а концентрации валина, тирозина, лейцина и гистидина так же, как и содержание незаменимых аминокислот, остаются низкими.
Обмен углеводов. При уремии нарушается толерантность к углеводам. Это выявляется после пероральной или внутривенной нагрузки глюкозой. Возрастает резистентность периферических тканей к инсулину, чему способствует повышение уровня гормона роста. Происходит увеличение базального уровня инсулина в основном за счет снижения степени его разрушения в почках. Уровень глюкагона также повышается и остается повышенным даже после нагрузки глюкозой.
При ХПН отмечено снижение энергетического обмена. При этом содержание АТФ в эритроцитах повышено, тогда как фосфорилирующая и АТФ-азная активность снижены.
Обмен липидов. Основное нарушение обмена жиров при ХПН — гипертригли-цериемия, сочетающаяся с повышенным содержанием липопротеидов очень низкой плотности. Эти изменения связывают с подавлением активности липазы. Содержание липопротеидов высокой плотности, как правило, снижено.
Гиперхолестеринемия наблюдается реже, за исключением больных с нефротическим синдромом или сопутствующей сосудистой патологией.
184 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
5.12. ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА РАЗЛИЧНЫХ ВИДОВ ПОЧЕЧНОЙ НЕДОСТАТОЧНОСТИ
Важное практическое значение имеет дифференциальная диагностика прере-нальной и ренальной форм ОПН, а также олигоанурической стадии ОПН и терминальной стадии ХПН (табл. 5.3).
При любом виде почечной недостаточности прежде всего следует исключить ее обтурационный генез. На основании УЗИ и рентгенографии следует оценить проходимость мочевых путей. В неясных случаях может быть проведена компьютер-
Таблица 5.3. Дифференциальная диагностика ОПН и ХПН
ОПН (олигоанурическая стадия) ХПН (терминальная стадия)
1. Быстрое, внезапное развитие (почечный анамнез) 2. Снижение диуреза (менее 400 мл/сут) 3. Содержание натрия в моче > 40 ммоль/л 4. Относительная плотность мочи «1,01 5. Выраженная уремическая интоксикация Прирост креатинина крови 0,5 и более ммоль/л в сутки 6. Гиперкалиемия 7. Тяжелый метаболический ацидоз Медленно прогрессирующий процесс (почечный анамнез отсутствует) Диурез более 500 мл/сут Низкое содержание натрия в моче Плотность мочи < 1,01 Адаптация к азотемии Прирост креатинина крови 0,3 —0,5 ммоль/л в месяц Умеренная гиперкалиемия или гипокалиемия Умеренный метаболический ацидоз
ная томография без контрастных веществ, катетеризация мочеточников и ретроградная пиелография. Среди устранимых причин ОПН наиболее распространенными являются острая обструкция (постренальная ОПН) и обратимая преренальная ОПН. Степень обратимости повреждений после обструкции зависит от ее продолжительности. Обратимую преренальную форму ОПН следует дифференцировать от других ренальных причин ОПН, особенно от острого тубулярного некроза, поскольку эта группа фактически имеет ту же этиологию. Ценную информацию могут дать лабораторные исследования. Наиболее типичные тесты при ОПН приведены в табл. 5.4.
У больных с олигурией сохранение показателей канальцевых функций на нормальном уровне является аргументом против внутрипочечного генеза поражения канальцев. Нормальные показатели реабсор-ции натрия (содержание натрия в моче ниже 10 — 20 ммоль/л) и поддержание способности концентрировать мочу (осмолярность мочи выше 500 мосм/л) позволяют предположить, что почечная недостаточность и олигурия являются следствием преренальных причин (артериальная гипотензия, гиповолемия, гипоперфузия почек). Фракционная экскреция натрия (FENa) является наиболее достоверным показателем для диагностики острого тубулярного некроза (FENa > 3 %). Показатель FENa ниже 1 % позволяет предположить преренальную ОПН или потенциально обратимые случаи ренальной ОПН, он характерен также для больных с вы
Таблица 5.4. Лабораторные показатели при различных заболеваниях почек
Показатель Постренальная ОПН Преренальная ОПН Острый тубулярный некроз Гломерулонефрит
1. Осмолярность мочи, мосм/л 2. Осмолярный клиренс, мл/мин 3. Клиренс креатинина, мл/мин 4. Содержание натрия в моче, ммоль/л 5. Фракционная экскреция натрия, % 6. Относительная плотность мочи < 400 1,1 < 20 > 40 > 1 > 500 < 1,3 > 40 < 20 < 1 > 1,018 < 400 < 1,1 < 20 > 40 > 1 « 1,01 > 500 1,3 > 40 < 20 < 1
Лечение острой почечной недостаточности 185
раженной внутрипочечной вазоконстрикцией и задержкой натрия.
Показатель FENa вычисляется по фор-
муле и /Р
FENa= Na^ Na100, UKp ' *кр
где U и Р означают концентрацию натрия и креатинина соответственно в моче и плазме крови. Это уравнение выведено из соотношения клиренса натрия к скорости клубочковой фильтрации по клиренсу креатинина.
5.13. ЛЕЧЕНИЕ ОСТРОЙ ПОЧЕЧНОЙ НЕДОСТАТОЧНОСТИ
На сегодня специфическое лечение ОПН не разработано. Применяется корригирующая и заместительная терапия (гемодиализ, гемофильтрация). Первая направлена на устранение нарушений гомеостаза, вторая — на выведение из организма жидкости, электролитов и продуктов белкового катаболизма. Выбор метода лечения определяется этиологией, формой и стадией ОПН.
Лечение преренальной острой почечной недостаточности. Если ОПН развивается вследствие преренальных причин (артериальная гипотензия, гиповолемия, гипоперфузия почек и др.), то в этих случаях показано быстрое восстановление системного АД и объема циркулирующей крови внутривенным введением кристаллоидных или коллоидных растворов. Все виды инфузионной терапии требуют мониторинга диуреза и центрального венозного давления. Только после стабилизации АД и ликвидации гиповолемии назначаются диуретики: фуросемид (внутривенно 200 — 400 мг каждые три часа) с допамином (3 мкг/кг в минуту) в течение 6 — 24 ч. Осмотические диуретики (10 —20 % раствор маннитола, 40 % раствор сорбита) достаточно эффективны при преренальной ОПН.
Лечение ренальной острой почечной недостаточности (стадия олигоанурии). Лечение ренальной ОПН неспецифично. В то же время существует по крайней мере четыре основных терапевтических мероприятия:
1. Нормализация диуреза.
2. Контроль гидратации, электролитного баланса и КОС.
3. Ранняя диагностика и лечение инфекций.
4. Раннее применение диализа.
Во многих случаях удается увеличить диурез внутривенным введением фуросемида (100 мл/ч) и допамина в низких дозах. Нормализация диуреза позволяет регулировать водный баланс и тем самым снижает потребность в лечении диализом.
Гипергидратация является обычным нарушением при ОПН. Неправильная тактика инфузионной терапии может привести к интерстициальному отеку легких и отеку мозга. Для расчета допустимого объема гидратации следует руководствоваться формулой: суточный диурез + + 500 мл. При повышенной температуре на каждый градус выше 37 °C добавляется около 300 мл жидкости. Прием натрия ограничивается до 40 ммоль/сут.
Гиперкалиемия — характерное электролитное нарушение при ренальной ОПН. Повышение концентрации К+ в плазме крови в олигоанурической стадии достигает высокого уровня (К+ > 8 ммоль/л) и может стать причиной смерти больного. При достижении значения этого показателя 6 ммоль/л проводится следующее лечение:
1. Внутривенное введение 10 % раствора глюконата кальция по 1 — 3 ампулы с интервалом в пять минут до исчезновения изменений на ЭКГ. Суточная доза — до 50 мл. Кальций выступает в роли антагониста калия.
2. Внутривенное введение 20 % раствора глюкозы (500 мл в течение часа) с простым инсулином (30 — 40 ME), что способствует транспортировке калия во внутриклеточный сектор.
186 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
3. Внутривенное введение 50 — 100 мл 8,4 % раствора натрий гидрогенкарбоната в течение 1 — 2 ч. Это способствует транспорту ионов К+ в клетки в обмен на ионы Н+.
4. При выраженном кардиотоксическом эффекте гиперкалиемии — гипертонический раствор натрий хлорида (200 мл 5 % раствора, внутривенно в течение 1 ч).
При повышении уровня К+ > 6,0 ммоль/л показано проведение гемодиализа. Следует отметить, что введение глюкозы с инсулином, глюконата кальция и оксирбути-рата натрия обеспечивает лишь временную защиту от гиперкалиемии, поскольку они не снижают общего содержания калия в организме. Прекращение инфузии приводит к обратному движению калия из внутриклеточного пространства во внеклеточное. Кроме того, каждый из растворов усугубляет проблему перегрузки жидкостью при олигурии, поскольку существует лимит гидратации.
Метаболический ацидоз в олигоанурической стадии ОПН может достичь значительного уровня (pH < 7,18). При декомпрессированном метаболическом ацидозе внутривенно вводятся ощелачивающие растворы по следующим формулам в объеме, рассчитанном:
— для 1,4 % раствора натрий гидрогенкарбоната: BE • Масса тела (кг);
— для 8,4 % раствора натрий гидрогенкарбоната: BE • 0,3 • Масса тела (кг);
— для трисамина: 8,3 мл 0,3 М раствора • Масса тела (кг), где BE — сдвиг буферных оснований.
Лечение ацидоза натрий гидрогенкар-бонатом неизбежно увеличивает поступление в организм натрия и воды, повышая риск перегрузки жидкостью. Подобное лечение должно проводиться с осторожностью, с контролем ЦВД.
Азотемия при ренальном ОПН может прогрессировать даже в тех случаях, когда удается увеличить диурез мочегонными средствами. Скорость нарастания содержания креатинина в крови не зависит от уровня белкового катаболизма, поэтому более достоверно отражает степень поражения функции почек. В неосложненных случаях суточный прирост креатини
на составляет 0,05 — 0,09 ммоль/л, мочевины — 5 — 7 ммоль/л. С целью замедления белкового катаболизма применяются анаболические стероиды (нерабол, ретабо-мил) в повышенных дозах. При достижении концентрации креатенина в крови 0,8—1,0 ммоль/л требуется срочное проведение гемодиализа.
При неэффективном лечении ОПН в течение 2 — 3 суток увеличивается риск осложнений от больших доз фуросемида (ототоксичность) и маннитола (гиперосмолярность, острая сердечная недостаточность). В таких случаях следует переходить к активным диализным методам.
Традиционный гемодиализ остается наиболее эффективным методом внепочечно-го очищения крови при ОПН. В тех случаях, когда его использование затруднено из-за проблем доступа к венозным сосудам, гипотензии или геморрагий, может быть применен перитонеальный диализ, хотя он менее эффективен, чем гемодиализ. При некоторых формах ОПН помогает применение плазмафереза и гемосорбции.
К новым и менее сложным методам, чем гемодиализ, относятся артерио-венозная гемофильтрация и продолжительная веновенозная фильтрация.
Лечение ренальной ОПН (стадия полиурии). Увеличение диуреза в этой стадии еще не является признаком восстановления нормальной функции почек. Наблюдается гипостенурия (относительная плотность мочи 1,005—1,01). Выделение повышенного количества гипотонич-ной мочи отражает состояние канальцевой недостаточности. Происходит потеря натрия, калия и хлора. Полиурия еще не приводит к ликвидации азотемии.
Лечение в этой стадии ОПН направлено на поддержание водно-электролитного баланса, КОС, снижение катаболизма, коррекцию анемии и борьбу с инфекцией.
Лечение постренальной ОПН. Показано срочное деблокирование мочевых путей (эндоскопическим или хирургическим методом). При восстановлении нормального пассажа мочи ОПН в большинстве случаев быстро ликвидируется. Диализные методы применяются в тех случаях, когда, несмотря на восстановление проходимости моче
Лечение острой почечной недостаточности 187
вых путей, сохраняется анурия и нарастает гиперкалиемия. Это наблюдается при апостематозном нефрите, уросепсисе.
5.13.1. Консервативное лечение хронической почечной недостаточности
Малобелковая диета значительно улучшает самочувствие больных с ХПН. Выбор диеты определяется стадией ХПН. При уровне креатинина меньше 0,25 ммоль/л рекомендуется диета с содержанием белка 0,9—1,0 г/кг (60 г/сут) и энергетической ценностью не ниже 35 ккал/кг (146,51 кДж/кг). Используются преимущественно растительные белки.
При уровне креатинина от 0,25 до 0,5 ммоль/л прием белка ограничивается до 0,5 —0,6 г/кг, калия — до 2,7 г/сут, фосфора — до 700 мг/сут. Энергетическая ценность диеты должна составлять 35-40 ккал/кг (146,51 —167,44 кДж/кг).
Диетическая тактика при уровне креатинина более 0,5 ммоль/л заключается в резком ограничении белка до 0,3 —0,4 г/кг (25 г/сут), калия — до 1,6 г/сут, фосфора — до 400 мг/сут. Энергетическая ценность диеты должна быть не ниже 35 ккал/кг (2700 — 2900 ккал/сут, или 11 302,2-12 139,4 кДж/сут).
Энтеросорбция применяется для выведения азотистых шлаков через пищеварительный канал. Используются различные энтеросорбенты (энтеросгель, энтеродез и др.).
Коррекция анемии наиболее эффективна при использовании рекомбинантного эритропоэтина (ЭПО). Лечение ЭПО показано при относительно удовлетворительных показателях гематокрита (29 — 30 %). ЭПО вводится подкожно (20—100 ед/кг) один раз в неделю. Железодефицит, развивающийся на фоне лечения ЭПО, корригируется приемом сульфата железа с витамином С.
Гипокальциемия корригируется метаболитами витамина С (оксидевит, кальци-триол).
Поддержание водно-солевого баланса имеет большое значение, так как при ХПН легко возникает сдвиг как в сторону гипергидратации, так и в сторону дегидратации. При этом могут наблюдаться гипер- или
гипонатриемия, гипер- или гипокалиемия. Водно-солевой баланс поддерживается соответствующим приемом жидкости и солей.
Метаболический ацидоз стимулирует выход калия из клеток и усиливает гиперкалиемию. Он вызывает одышку, слабость и снижает трудоспособность. В начальной стадии ХПН для лечения ацидоза применяют натрий гидрогенкарбонат (3 — 4 г/сут) внутрь или цитраты (80 мэкв/сут). Если снижение уровня гидрогенкарбонатов крови продолжается, дозы гидрогенкарбонатов и цитратов увеличивают.
Декомпенсированный метаболический ацидоз требует внутривенного введения натрий гидрогенкарбоната в виде 8,4 % раствора в количестве, зависящем от дефицита буферных оснований (BE): 8,4 % раствор (мл) = 0,3 BE • Масса тела (кг).
Артериальная гипертония, являющаяся характерным осложнением ХПН, подлежит коррекции, поскольку приводит к прогрессированию ХПН по типу «порочного круга». Гипотензивная терапия проводится длительно и непрерывно. Оптимальный уровень АД, при котором поддерживается достаточный почечный кровоток, находится в пределах 130/80 — 85 мм рт. ст. Лечение гипертонии осуществляется различными гипотензивными препаратами (клофелин, р-блокаторы, блокаторы кальциевых каналов, ингибиторы АПФ).
Препараты и их дозы подбираются индивидуально, начиная с малых доз, и постепенно увеличиваются до терапевтического уровня.
5.13.2. Активные методы лечения ХПН
К активным методам лечения ХПН относят гемодиализ, перитонеальный диализ и трансплантацию почки.
Показания к активному лечению ХПН. Диализное лечение больных ХПН показано на основании клинических и биохимических данных. Оцениваются в динамике показатели креатинина, мочевины, калия и КОС крови. Показанием к началу диализной терапии больных с ХПН является снижение СКФ ниже 10 мл/мин (соответственно повышение креатинина крови до 1,0 ммоль/л), т. е. наступление
188 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПОЧЕК
терминальной стадии ХПН. Также показанием может быть тяжелая гиперкалиемия (К+ > 6,5 мэкв/л), злокачественная артериальная гипертония с признаками застойной сердечной недостаточности, значительная гипергидратация с риском отека легких или мозга, декомпенсированный метаболический ацидоз.
Гемодиализ заключается в обмене веществ между кровью больного и диализирующим раствором, проходящим через полупроницаемую мембрану. В основе гемодиализа лежат механизмы диффузии и ультрафильтрации (конвективный транспорт). При диффузии происходит перенос через мембрану небольших молекул по градиенту концентрации. Мелкие молекулы (мочевина, креатинин, фосфор, калий) диффундируют через мембрану по градиенту концентрации из крови в диализат. Ионы кальция и гидрогенкарбоната переходят по концентрационному градиенту из диализата в кровь. В результате диализа из крови удаляются низкомолекулярные токсины, а плазма крови обогащается молекулами с более низкой концентрацией.
Ультрафильтрация направлена на удаление воды из циркулирующей крови больного. Растворенные вещества вместе с водой переносятся через полупроницаемую мембрану из области с более высоким в область с более низким гидростатическим давлением. За один сеанс гемодиализа путем ультрафильтрации может быть удалено до нескольких литров избыточной воды.
Перитонеальный диализ. Метод вне-почечного очищения крови больного в терминальной стадии ХПН, при котором брюшина выступает в роли полупроницаемой мембраны. Капилляры брюшины доставляют кровь для обмена (около 70 мл/мин) к месту контакта с диализирующим раствором, а диализат подается в брюшную полость через катетер, введенный через переднюю брюшную стенку. Силиконовый
катетер с двумя дакроновыми манжетами устанавливается в брюшной стенке таким образом, что одна манжета располагается сразу над брюшной полостью, а вторая -подкожно. Дакроновые манжеты прорастают соединительной тканью, что препятствует попаданию бактерий с поверхности кожи. Диализирующий раствор вводится в брюшную полость. Он сбалансирован по составу электролитов и содержит глюкозу в высокой концентрации. Уремические токсины диффундируют в брюшную полость через капилляры брюшины, интер-стиций и перитонеальный мезотелиальный слой. Удаление избытка воды из организма осуществляется посредством осмоса. В диализирующий раствор добавляют глюкозу, и избыток воды по осмотическому градиенту переходит из крови в диализат. Концентрация глюкозы в диализате может составлять 1,5%,2,5и4,25%, что позволяет менять скорость удаления жидкости. Количество диализата, вводимого в брюшную полость, колеблется в зависимости от обстоятельств от 0,5 до 3 л.
Удаление диализирующего раствора проводится через промежутки времени от получаса до двух часов. Замена диализата происходит 4 — 5 раз в течение дня. Ночью раствор остается в брюшной полости. Процедура замены диализата на свежий занимает 10 — 20 мин. Перитониальный диализ в определенных ситуациях имеет преимущество по сравнению с гемодиализом. При его применении лучше удаляются высокомолекулярные токсины, отпадает необходимость в гепаринизации крови больного, отсутствуют гемодинамические нарушения, свойственные гемодиализу.
Наиболее вероятным осложнением при перитонеальном диализе является перитонит. Профилактика перитонита основана на обучении больного самостоятельному асептическому и безопасному проведению диализа.
5.14. ТРАНСПЛАНТАЦИЯ ПОЧКИ
Любой пациент с терминальной стадией ХПН считается потенциальным кандидатом на трансплантацию почки. Пересадка почки стала общепринятым методом ле
чения терминальной стадии ХПН и для многих больных представляет наиболее физиологичную и хорошо переносимую форму заместительной терапии.
Список использованной литературы 189
В США ежегодно выполняются 10 — 12 тыс. трансплантаций почек. Органы для пересадки получают от живых доноров (родственники или посторонние лица), а также от трупов людей, умерших внезапной смертью. Чаще всего это лица, пострадавшие от черепно-мозговой травмы, у которых диагностируется «смерть мозга».
Успех операции зависит прежде всего от тканевой совместимости донора и реципиента. У каждого человека индивидуальный «тканевый тип» определяется антигенами на поверхности лейкоцитов человека (HLA), информация о которых локализована на коротком плече 6-й хромосомы. Несоответствие набора HLA-антигенов (их
всего шесть) у донора и реципиента ведет к развитию иммунологических реакций, направленных на отторжение трансплантата. Для предотвращения реакции отторжения проводится иммуносупрессивная терапия, включающая применение кортикостероидов в больших дозах, циклоспорина и азатиоприна. Используются также препараты поликлональных и моноклональных антител против лимфоцитов.
Средняя продолжительность функционирования трансплантата трупной почки на сегодня достигает 8 лет. Удовлетворительная функция трансплантата в течение одного года после операции составляет 80 %.
Список использованной литературы
1. Борисов И. А., Сура В. В., Денисов А. Ю. Почечная недостаточность у лиц пожилого и старческого возраста // Рос. мед. жури. — 1998. - № 6. - С. 54-57.
2. Вандер А. Д. Физиология почки. — СПб.: Питер, 2000. — 252 с.
3. Возианов А. Ф. Колесник Н. А., Самодумо-ва И. М. Энтеросорбция в лечении хронической почечной недостаточности у больных с хирургическими заболеваниями почек // Врач. дело. — 1992. — № 2. -С. 90-93.
4. Ермоленко В. М. Первый опыт применения постоянного амбулаторного перитонеального диализа для лечения больных с терминальной почечной недостаточностью // Терапевт, арх. — 1996. — № 8. - С. 36 — 38.
5. Кокот Ф. Дозировка лекарственных средств у больных почечной недостаточностью // Новости фармации и медицины. — 1997. — № 3. - С. 55-58.
6. Колесник М. О., Дудар I. О. Гостра нир-кова недостатшсть. Нефролопя / За ред. Л. А. Пирога. — К.: Здоров’я, 1995. — С. 233-245.
7. Лопаткин Н. А. Руководство по клинической урологии. — М.: Медицина, 1999. — С. 499-541.
8. Миронов Л. Л. Гомеостаз и особенность клинического течения ренальной острой почечной недостаточности // Нефрология. — 1998. - №4. - С. 25-31.
9. Николаев А. Ю., Милованов Ю. С. Лечение почечной недостаточности. — М.: Медицина, 1999. — 362 с.
10. Руководство по нефрологии / Под ред. Д. А. Витворт, Д. Р. Лоренса. — М.: Медицина, 2000. — 485 с.
И. Серняк П. С., Возианов А. Ф., Коваленко Н. В. Острая почечная недостаточность. — К.: Здоров’я, 1998. — 140 с.
12. Тареева И. Е. Нефрология. — М.: Медицина, 1995. — 496 с.
13. ХрайчикД. Е., Седор Д. Р., ГанцМ, Б. Секреты нефрологии. — М.: Бином, 2001. — 302 с.
14. Шейман Б. С, 1ндивщуал1защя показань терапп у дНей з нирковою недостатшстю в умовах генерал1заци ендотоксикозу // Bi ль, знеболювання i штенсивна терашя. — 2000. — № 1. - С. 123-125.
15. Шейман Д. А. Патофизиология почки. — М.: Бином, 1999. — 205 с.
16. Albright R. С. Acute renal failure: a practical update // Mayo Clin. Proc. — 2001. — Vol. 76, N 1. - P. 67-74
17. Brivet F. G., Kleinkecht D. J., Loirat Ph., Landais P. J. Acute renal failure in intensive care unis — causes, outcome and prognostic factors of hospital mortality: A prospective, multicenter study // Crit. Care Med. — 1996. - Vol. 24, N 2. - P. 192-198.
18. Edward W. L., Humes H. D. Acute renal failure: directed therapy to exchance renal tubular regeneration // Seminars in Nephrology. — 1994. - Vol. 14, N 1. - P. 83-97.
... - Раздел =
6
Клиническая физиология печени
Основными структурными компонентами печени являются сосуды, собственно печеночная клетка, желчные протоки, система мононуклеарных фагоцитов. Нарушение со стороны любого из этих структурных образований может привести к недостаточности печени, например, гипоксия, развивающая
ся в результате уменьшения доставки кислорода из-за нарушения проходимости сосудов или снижения содержания в крови кислорода. Недостаточностью органа может также сопровождаться нарушение проходимости желчных протоков вследствие их механической закупорки камнем.
6.1. КРОВООБРАЩЕНИЕ В ТКАНИ ПЕЧЕНИ
6.1.1. Анатомия сосудов и макроциркуляция в печени
В норме кровоток в печени составляет примерно 1600 мл/мин, или 1 мл/г в 1 мин. Это примерно соответствует 25 % величины сердечного выброса. При этом печень обладает двумя источниками кровоснабжения: 75 % крови поступает по системе воротной вены, а 25 % — из печеночной артерии. Это соотношение может существенно колебаться в различных клинических ситуациях.
В v. portae кровь поступает из верхней мезентериальной вены и из селезеночной вены. Объем кровотока по воротной вене составляет 1200 мл/мин, давление в ней колеблется в пределах 5—10 мм рт. ст. Такой большой объем крови, поступающей к печени по v. portae, обеспечивает доставку веществ от органов пищеварительного канала, многие из которых в последующем подвергаются в печени метаболизму. Кроме этого, в крови v. portae также содержатся гепатотрофические факторы,
в частности глюкагон и инсулин поджелудочной железы.
Воротная вена в области ворот печени делится на две ветви: правая следует к правой доле печени, левая — к левой. Обычно левая ветвь снабжает кровью квадратную и хвостатую доли печени. Кровоток в воротной вене, как правило, носит ламинарный характер. Вследствие этого кровь из разных источников v. portae смешивается мало. В левую ветвь воротной вены преимущественно поступает кровь из селезенки. Здесь определяется повышенное количество непрямого билирубина, железа, продуктов разрушения билирубина. В левую ветвь также поступает кровь от нижней половины толстой кишки, содержащая много продуктов, образующихся при гниении и брожении в кишках. В правую ветвь воротной вены в основном поступает кровь от тонкой кишки, где происходит всасывание продуктов пищеварения.
Печеночная артерия формируется ветвями чревного сплетения. По ней посту-
Кровообращение в ткани печени 191
пает около 400 мл крови в минуту. Давление в этой артерии эквивалентно системному артериальному давлению.
В связи с тем что печень преимущественно получает кровь из v. portae, удовлетворение 60 — 70 % потребности органа в кислороде также происходит за счет кровотока через воротную вену, остальная часть — за счет кровотока по печеночной артерии.
Отток крови от печени осуществляется по печеночным венам, давление в которых составляет примерно 5 мм рт. ст. и менее. В большинстве случаев имеется три печеночные вены. Но их количество может быть и больше. Vv. hepaticae впадают в нижнюю полую вену ниже места ее проникновения через отверстие в сухожильной части диафрагмы.
По внутрипеченочным разветвлениям воротной вены и печеночной артерии кровь поступает в микроциркуляторное русло кровеносной системы печени — в синусоиды, а из них — в отводящие сосуды печени. При этом в местах впадения сосудов в синусоиды, а также последних в печеночную вену расположены гладкомышечные сфинктеры, которых в печени больше, чем в каком-либо другом органе. Те из них, что расположены до синусоидов, регулируют приток крови к последним; сфинктеры, расположенные в выводящих венозных сосудах печени, регулируют отток крови из органа. Вследствие этого с учетом значительного общего объема синусоидов печень обладает мощным запасом циркуляции: в ней может скапливаться до 1,5 л крови. Кроме того, благодаря попеременному действию различных сфинктеров синусоиды содержат преимущественно или артериальную, или венозную кровь. Так, во время пищеварения кровоток в печени за счет портального кровообращения увеличивается и на высоте пищеварения может достигать 100 литров в час.
Давление в воротной вене колеблется в пределах 5—10 мм рт. ст., в печеночной артерии — соответствует системному — 100—120 мм рт. ст., в печеночной вене — 5 мм рт. ст. и менее. Несмотря на большую разницу давлений в воротной вене и печеночной артерии, давление на уровне
внутрипеченочных анастомозов этих сосудов невелико. Как следствие, градиент давления между капиллярной сетью печени и печеночными венами также очень мал. Этим объясняется медленный кровоток через печень, что, в свою очередь, обуславливает возможность неодинакового снабжения кислородом внутрипеченочных структур. Кровь из синусоидов первоначально достигает периферических отделов печеночной дольки. По мере кровотока к центру дольки содержание кислорода в крови уменьшается. При этом следует учитывать, что основная масса крови поступает к печени по воротной вене и имеет более низкое по сравнению с артерией содержание кислорода. Только при нормальном кровоснабжении печени центральные отделы печеночной дольки снабжаются кислородом в достаточной степени. При уменьшении кровотока они сразу начинают страдать от гипоксии.
Печень, как и головной мозг, является органом, чувствительным к гипоксии. Она осуществляет свои функции нормально тогда, когда потребление кислорода ею составляет 20 % общего потребления кислорода организмом. Гипоксия печени ведет к изменениям, которые можно объединить в четыре основные группы: анатомические, метаболические, токсические, инфекционные. Анатомические отклонения характеризуются следующим. Гипоксия вызывает отек, дегенеративные и деструктивные изменения в печеночной клетке, которые первоначально в силу особенностей кровотока в печени максимально выражены в центре печеночной дольки. Отек гепатоцитов ведет к еще большему замедлению кровотока по капиллярам и прогрессированию гипоксии от центра к периферии печеночной дольки. Приведенные механизмы объясняют возникновение патоморфологического симптома гипоксии печени любого происхождения — центрального печеночного некроза.
Чувствительность к гипоксии разных сосудов печени неодинакова. Наиболее чувствительны отводящие сосуды органа. Вследствие этого гипоксия приводит к застою крови в печени и внутренних органах, что еще больше усугубляет гипоксию органа.
192 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
Гепатоцит содержит множество ферментов. Гипоксия прямо или опосредованно изменяет функцию большинства из них, обуславливая метаболические отклонения в организме. Прежде всего нарушается углеводный обмен. Это проявляется активацией гликогенолиза и гипергликемии. При улучшении доставки кислорода гликемия относительно быстро нормализуется. Дополнительным признаком восстановления окислительных процессов в таких случаях может быть гипокалиемия. Механизм активации гликогенолиза при гипоксии печеночной клетки обусловлен опосредованным выбросом адреналина в ответ на недостаток кислорода.
Еще одним примером влияния гипоксии на метаболические процессы в печени является угнетение секреции желчи вплоть до полного прекращения ее образования. Этим можно объяснить обнаружение в крови у таких больных повышенной концентрации прямого и непрямого билирубина. Кроме того, при гипоксии печени могут развиваться различные нарушения обмена жиров и белков.
Токсический аспект воздействия гипоксии на печень также обусловлен влиянием на ферментные системы органа. В результате нарушения действия ферментов могут накапливаться промежуточные продукты обмена веществ, удлиняться метаболизм лекарственных соединений, в том числе и средств для наркоза.
Микробиологические последствия влияния гипоксии на печень обусловлены тем, что у большинства людей в желчных протоках содержится анаэробная микрофлора. Ее вирулентность подавляется определенной концентрацией кислорода. Уменьшение его количества при гипоксии может способствовать проникновению в кровоток как самих бактерий, так и их токсинов.
Основными направлениями ликвидации гипоксии печеночной клетки являются: устранение причины недостатка кислорода, улучшение кровотока в печени и оксигенации притекающей крови. Поскольку в норме во внутрипеченочном кровотоке обычно задействовано 20 — 25 % синусои-дов, для ликвидации гипоксии гепатоцита
можно применять лекарственные средства, увеличивающие кровоток в печени: эуфиллин, никотиновую кислоту. Повысить содержание кислорода в крови, притекающей к печени, можно с помощью методики «интестинального дыхания».
Гемоглобин эритроцитов в системе воротной вены насыщен кислородом примерно на 50 %, в печеночной артерии — на 95 — 96 %, в крови, оттекающей от печени, — на 18 %. Таким образом, степень потребления кислорода печенью значительна. В силу максимальности насыщения гемоглобина кислородом в артериальной крови печеночной артерии в норме увеличение системной оксигенации путем повышения фракционной концентрации кислорода во вдыхаемом воздухе не приведет к существенному росту содержания кислорода в крови печеночной артерии, тогда как степень насыщения гемоглобина кислородом в крови портальной вены останется низкой. Это обуславливает возможность дозированного введения в верхние отделы пищеварительного канала кислорода с целью увеличения насыщения гемоглобина кислородом в крови воротной вены (методика «интестинального дыхания»).
Большая часть крови поступает к печени по воротной вене уже после ее прохождения по системе капилляров органов брюшной полости. В связи с этим концентрация вводимого парентерально препарата в крови, поступающей в печень, может быть существенно ниже той, которая необходима для достижения терапевтической цели. Как свидетельствуют результаты некоторых исследований, концентрация антибиотика в системе воротной вены по сравнению с исходной уменьшается в 3-6 раз. А для эрадикации возбудителя при холангите необходимо, чтобы концентрация антибиотика превышала минимальную ингибирующую концентрацию инфекта в 3 — 8 раз. Это не всегда удается реализовать при парентеральном введении антибиотиков: для большинства из них, даже при использовании максимальных терапевтических доз препарата, не удается обеспечить эффективную терапевтическую концентрацию в очаге гнойно-воспалитель-
Кровообращение в ткани печени 193
кого процесса в печени. Этим объясняются преимущества перорального применения антибиотиков в таких случаях.
6.1.2. Регуляция кровообращения в печени
6.1.2.1. Внутренняя регуляция
Ауторегуляция. Кровоснабжение печени относительно стабильно. Это обеспечивается ауторегуляцией кровотока, а также реципрокностью взаимосвязи кровотока по печеночной артерии и воротной вене. При этом ауторегуляция кровотока (изменение сопротивления сосуда в зависимости от колебаний системного артериального давления) преимущественно характерна для печеночной артерии и в меньшей степени — для воротной вены, это обусловлено тем, что в воротной вене величина давления невелика: основной перепад давлений произошел до поступления крови в V. portae — на уровне сосудистых бассейнов кишок, желудка, поджелудочной железы.
Особенностью ауторегуляции кровотока по печеночной артерии является то, что она преимущественно выражена при функционально активном состоянии органа — после приема пищи, малохарактерна натощак, в том числе и при плановых оперативных вмешательствах, при подготовке к которым для предотвращения аспирации желудочного содержимого пациенту запрещается принимать пищу.
Изменение тонуса печеночной артерии позволяет компенсировать колебания не только системного артериального давления, но и давления на уровне печеночной вены. Так, увеличение венозного давления при сердечной недостаточности сопровождается констрикцией печеночных артериол и уменьшением интенсивности кровотока в них (это не характерно для воротной вены).
Стабильность кровоснабжения печени обеспечивается не только ауторегуляцией кровотока по печеночной артерии, но и реципрокной взаимосвязью кровотока по печеночной артерии и v. portae — при увеличении кровотока в одной системе уменьшается кровоток в другой и наоборот. Но
7 ' 4-220
предел компенсации составляет примерно 20 %. Поэтому при полном прекращении кровоснабжения печени по одному из сосудистых бассейнов полного компенсаторного увеличения доставки кислорода по другой сосудистой системе не происходит и может развиться некроз печени.
Поскольку предел компенсаторного колебания кровотока при нарушении кровообращения по одному из сосудов, обеспечивающих доставку кислорода к печени, составляет 20 %, то выполняемая в отдельных случаях портальной гипертензии окклюзия печеночной артерии для обеспечения реартериализации не эффективна, пока не будут дополнительно наложены анастомозы с диафрагмальными артериями.
Особенностью воротной вены является то, что она связана многочисленными анастомозами с полыми венами (портокавальные анастомозы). Это анастомозы с венами пищевода и венами желудка, прямой кишки, околопупочными венами и венами передней брюшной стенки, а также анастомозы между корнями вен портальной системы (верхней и нижней брыжеечных, селезеночной и др.) и венами забрюшинного пространства (почечными, надпочечными, венами яичка или яичника).
Анастомозы играют важную роль в развитии коллатерального кровообращения при нарушениях оттока в системе воротной вены при портальной гипертензии. Последняя может возникать на различных участках портальной вены и циркуляции крови в печени. Принципиально важно дифференцировать гипертензию в связи с препятствием до синусоидов, а также на их уровне и после них. Пресинусоидаль-ный тип портальной гипертензии может быть обусловлен обструкцией ствола v. portae, а также поражением разветвлений вены в печени (врожденный печеночный фиброз, саркоидоз, миелопролиферативные заболевания, гепатопортальный склероз при идиопатической портальной гипертензии, отравлении некоторыми химическими соединениями (винилхлорид, мышьяк). У таких больных в связи с портальной гипертензией и компенсаторным увеличением кровотока по портокавальным анастомозам течение заболевания осложняется же-
194 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
лудочно-кишечными кровотечениями, но функция печеночной клетки обычно сохраняется. Поэтому в подобных случаях отсутствуют такие типичные проявления поражения печени, как желтуха, асцит и др. В противоположность этому при портальной гипертензии в связи с локализацией процесса на уровне и после синусоидов (цирроз любой этиологии, флебосклеротические процессы при алкогольном гепатите, синдром Бадда —Киарри*) имеет место нарушение функции печеночной клетки. Поэтому прогноз такой группы больных хуже, а возникающее желудочно-кишечное кровотечение вследствие портальной гипертензии существенно усугубляет состояние пациента.
При патологии печени кровь может течь в обход этого органа как по большим естественным портокавальным коллатералям, так и по портопеченочным венозным анастомозам внутри самой печени.
Шунтирование крови, оттекающей от кишечника, при портальной гипертензии является одним из механизмов возникающей при заболевании печени энцефалопатии (портосистемная энцефалопатия). Источником интоксикации при портокавальной энцефалопатии является содержимое кишок, в том числе токсины, вырабатываемые кишечными бактериями. Это служит обоснованием применения у таких больных перорального приема антибиотиков и очищения кишок слабительными, способствующими подавлению микрофлоры кишок.
Метаболическая регуляция. Основными факторами метаболической регуляции печеночного кровотока являются: парциальное напряжение кислорода и углекислого газа в крови, осмолярность крови. Так, системная артериальная гипоксемия с пони
* Синдром Бадда —Киарри характеризуется нарушением оттока крови из печени вследствие тромбоза печеночных вен. Клинические проявления зависят от обширности тромбоза, степени вовлечения нижней полой вены, темпов развития синдрома. Последнее определяет скорость вовлечения коллатерального кровотока. При быстром развитии синдрома возникает гепатомегалия, боль, сердечно-сосудистая недостаточность. В случаях более медленного развития синдрома наблюдаются гепатомегалия, асцит, признаки портокавального анастомозирования, симптомы поражения гепатоцита.
жением парциального напряжения кислорода в артериальной крови до 30 мм рт. ст. и менее повышает сопротивление печеночных артерий практически в два раза и уменьшает печеночный кровоток. Но на тонус печеночных артерий системная ги-пероксия существенного влияния не оказывает. Уменьшение напряжения кислорода в воротной вене, наблюдаемое после принятия пищи, также ведет к увеличению кровотока по печеночной артерии. Повышает кровоток не только по печеночной артерии, но и по V. portae наблюдающееся после приема пищи умеренное увеличение осмолярности крови.
Повышение системного напряжения углекислого газа в крови и сопутствующий этому ацидоз приводят к интенсификации кровотока как по печеночной артерии, так и в системе портальной вены. Уменьшение же системного напряжения углекислого газа при гипокапнии оказывает противоположное воздействие.
6.1.2.2. Внешняя регуляция
Нервная регуляция. Кровоток в печени контролируется главным образом симпатической нервной системой (СНС). Повышение ее активности приводит к уменьшению кровотока в печени. Вследствие этого при шоке и других состояниях, сопровождающихся повышением тонуса СНС, кровь сосудов печени является резервом увеличения объема циркулирующей крови.
Парасимпатическая нервная система на объем кровотока в печени оказывает опосредованное влияние: от ее активности зависит тонус пресинусоидальных сфинктеров и, как следствие, распределение кровотока в печени.
Гуморальная регуляция. Среди факторов гуморальной регуляции кровотока в печени наибольшее значение имеют катехоламины. В печеночной артерии представлены как а-, так и р-адренорецепто-ры. При выбросе адреналина в кровоток тонус артерий печени сперва повышается (активация а-адренорецепторов), затем уменьшается (активация р-адренорецеп-торов). В v. portae представлены только а-адренорецепторы, поэтому при выбро
Метаболические функции печени 195
се адреналина в кровоток тонус воротной вены повышается.
Допамин при физиологических состояниях на кровоток в печени существенного влияния не оказывает. В таких случаях циркуляция в органе преимущественно будет зависеть от адреналина, норадреналина и активности симпатической нервной системы.
6.1.3. Влияние анестезии на кровоток в печени
Нарушение функции печени после наркоза чаще всего обусловлено непосредственным воздействием на гепатоциты, а не на кровоток в органе. А среди факторов интраоперационного воздействия при спектре используемых в настоящее время препаратов для наркоза наибольшее влияние на кровоток оказывает искусственная вентиляция легких (ИВЛ) и само оперативное вмешательство. Так, операционная агрессия при операциях на верхнем отделе брюшной полости в ряде случаев сопровождается уменьшением кровотока в печени на 60 %. Если ИВЛ проводится в режиме
гипервентиляции, то развивающаяся при этом гипокапния также уменьшает кровоток в печени. Дополнительным фактором снижения печеночного кровотока может быть применение положительного давления в конце выдоха, поскольку при этом увеличивается давление в венах печени.
Уменьшение кровотока в печени при использовании средств для наркоза или под влиянием методов анестезии чаще всего опосредованно и обусловлено гипотензией. Например, такой эффект могут вызывать эпидуральная и спинномозговая анестезия. Уменьшение кровотока также может быть вызвано накоплением СО2 и наблюдающимся при этом повышением активности симпатической нервной системы. Непосредственное угнетающее воздействие на печеночный кровоток выявлено у фторотана, энфлурана, нейролептанал-гетиков. Но уменьшение кровотока под влиянием средств для наркоза не означает, что в гепатоците действительно развивается анаэробный метаболизм: анестетики одновременно вызывают снижение интенсивности метаболизма и потребности в кислороде.
6.2. МЕТАБОЛИЧЕСКИЕ ФУНКЦИИ ПЕЧЕНИ
6.2.1. Обмен белков
Печень играет ключевую роль в белковом обмене организма, функционируя как аминостат: несмотря на суточные колебания в поступлении аминокислот и полипептидов в печень из кишок, поглощение и высвобождение в кровоток азотистых соединений периферическими тканями, уровень белков и свободных аминокислот в крови остается строго постоянным. Выполнению функции печени как аминостата способствует, с одной стороны, анатомическое расположение органа, с другой — биохимическая уникальность печени. Анатомическая составляющая обусловлена тем, что печень связана с кишками (посредством воротной вены) и с желчевыводящими путями. После потребления белковой пищи клетки печени принимают на себя «первый удар» потока аминокислот
и других продуктов метаболизма, поступающих из кишок по воротной вене. Органическая связь печени с желчевыводящими путями позволяет выводить некоторые вредные конечные продукты азотистого обмена (в частности желчные пигменты) непосредственно в пищеварительный канал.
Биохимическая уникальность печени обусловлена тем, что в ее клетке содержится полный набор ферментов, участвующих в обмене аминокислот, а также тем, что синтез и распад белков в печени происходит с большой скоростью. При этом скорость обновления белков в печени выше, чем в любом другом органе, кроме поджелудочной железы.
Главными реакциями превращения аминокислот в печени являются, во-первых, взаимопревращение аминокислот, распад углеродного скелета аминокислот с выде
196 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
лением энергии и обеспечением глюконеогенеза реакциями трансаминирования и окислительного дезаминирования; во-вторых, обезвреживание аммиака и других конечных продуктов катаболизма, в том числе мочевины, мочевой кислоты, желчных кислот.
Аминокислоты поступают в обменный фонд печени из трех источников: 1. Экзогенные — по воротной вене из кишок. 2. Эндогенные — это продукты физиологического распада белков в органах и тканях человека. 3. Аминокислоты, образующиеся в процессе обмена веществ из углеводов и жирных кислот.
Реакциями превращения аминокислот являются трансаминирование и окислительное дезаминирование. В реакции трансаминирования аминогруппа (~NH2) одной аминокислоты переносится на а-ке-токислоту с образованием другой аминокислоты в соответствии со схемой:
Амино-а-Аминокислота 1 + трансфераза а-Кетокислота 1 + + а-Кетокислота 2 Пиридоксаль-+ “'Аминокислота 2 фосфат
Эти реакции могут протекать в любом направлении в зависимости от соотношения концентраций реагирующих компонентов и потребности в них. Если концентрация аминокислоты 2 снижена, а аминокислота 1 и кетокислота 2 представлены в изобилии, то реакция переноса аминогрупп при наличии фермента будет идти слева направо, приводя к синтезу аминокислоты 2. Реакция может идти и в противоположном направлении, если аминокислота 2 имеется в ткани печени в избытке. Ферменты этих реакций — аминотрансферазы — при повреждении клеток печени (а также клеток других органов) поступают в кровь, что обуславливает диагностическое значение их определения в кровотоке.
Принцип реакции окислительного дезаминирования иной. В ходе этой реакции аминогруппа освобождается в виде аммиака (NH3), а углеродный скелет аминокислоты окисляется до а-кетоглутарата. Эта реакция требует участия в окислении никотинамидного кофермента (НАД или НАДФ)
и катализируется специфическим ферментом глутаматдегидрогеназой:
Амино-транфе-Аланин + Раза „
+ а-Кетоглутарат------Пируват + Глутамат;
Глутамат + НАД+----► а-Кетоглутарат +
+ NH3 + НАД • Н + Н+.
Суммарная реакция имеет такой вид:
Аланин + НАД+ (или НАДФ+)----►Пируват +
+ NH3 + НАД • Н (или НАДФ • Н) + Н+.
Посредством такой реакции разнообразные аминокислоты, попадающие в печень, могут подвергаться катаболизму с образованием а-кетокислот, восстановленных никотинамидных коферментов, NH3. В дальнейшем кетокислоты могут включаться в цикл трикарбоновых кислот (цикл обмена лимонной кислоты, или цикл Кребса), а восстановленные никотинамидные коферменты — служить источником энергии.
Образующийся в процессе метаболизма азотистых соединений аммиак является токсическим веществом. В печени происходит три процесса фиксации аммиака с образованием органических азотистых соединений: 1. Восстановительное аминирование, обеспечивающее синтез глутамата и других аминокислот при сохранении азота аммиака. 2. Образование амидов, в частности глутамина, способных выполнять функции временного резервуара и транспортной формы аммиака. 3. Образование карбамоилфосфата, который необходим при биосинтезе таких соединений, как пиримидиновые азотистые основания нуклеиновых кислот, или при синтезе мочевины — выводимого из организма конечного продукта азотистого обмена. Среди этих механизмов решающее значение имеет биосинтез мочевины. Процесс образования мочевины целиком протекает в печени в цикле мочевины Кребса. В сутки в печени образуется 20 — 30 г мочевины.
Поражение печени приводит к нарушению обмена аминокислот, что имеет как диагностическое, так и клинико-физиологическое значение. Поскольку процессы синтеза поражаются одними из первых, то нарушение связывания аммиака и увели
Метаболические функции печени 197
чение его концентрации в крови является одним из ранних проявлений заболеваний. Биосинтез мочевины является более «устойчивым» биохимическим процессом. Так, согласно данным экспериментальных исследований, для клинически значимого подавления образования мочевины необходимо удалить по крайней мере 85 % ткани печени. Поэтому диагностически значимое уменьшение концентрации мочевины характерно для терминальных стадий недостаточности печеночной клетки. Фульминантная печеночная недостаточность редко сопровождается снижением концентрации мочевины в крови.
Накопление аммиака в крови оказывает токсическое действие на органы, прежде всего на клетки ЦНС, и является одним из факторов возникновения энцефалопатии у пациентов с печеночной недостаточностью. Токсический эффект аммиака обусловлен прямым действием на мембраны нейронов, а также опосредованным нарушением функции нейронов в результате влияния на глутаматергическую систему. Это объясняется тем, что в головном мозгу цикл мочевины не функционирует, поэтому удаление из него аммиака происходит различными путями. В астроцитах под действием глутаматсинтетазы из глутамата и аммиака синтезируется глутамин. В условиях избытка аммиака запасы глутамата истощаются, а поскольку он является важным возбуждающим медиатором, то следствием уменьшения его содержания будет снижение активности головного мозга.
Нарушение белкового обмена при недостаточности гепатоцита проявляется также уменьшением соотношения в крови различных аминокислот, в частности нарушается баланс между аминокислотами с разветвленной углеродной цепью (лейцин, изолейцин, валин) и ароматическими аминокислотами (тирозин, фенилаланин, триптофан). Обе группы аминокислот проходят через гематоэнцефалический барьер с помощью одного и того же транспортера. Относительное увеличение концентрации ароматических аминокислот ведет к их преимущественному проникновению через гематоэнцефалический барьер в го
ловной мозг. Такие аминокислоты в большом количестве являются предшественниками «ложных медиаторов» (октопамина, р-фенилэтаноламина). Существует точка зрения, что именно они способствуют развитию печеночной комы (теория ложных медиаторов Джеймса). Кроме того, ароматическая аминокислота триптофан является предшественником серотонина — нейротрансмиттера, участвующего в регуляции уровня возбуждения коры головного мозга. Нарушение синтеза серотонина является одним из механизмов развития печеночной энцефалопатии. Этому же способствует и развивающееся при патологии печени нарушение ферментативных систем, отвечающих за обмен серотонина.
Существует и иная трактовка гипотезы ложных медиаторов (рис. 6.1), согласно которой декарбоксилирование некоторых аминокислот в кишках приводит к образованию р-фенилэтиламина, тирамина и октопамина — ложных нейротрансмиттеров. При патологии печени они могут замещать истинные нейротрансмиттеры. Теория ложных медиаторов лежит в основе
Толстая кишка: белки
I
Декарбоксилаза кишечных бактерий
I I
Тирозин Тирозин Фенилаланин
I I
Леводопа Тирамин
I I
Дофамин Октопамин
II"
Норадреналин Ложные 0-Фенил-
| нейромедиаторы этаноламин
Истинные нейромедиаторы
| V
Медиаторы симпатической нервной системы
I
Нарушение церебрального метаболизма
Рис. 6.1. Предполагаемая роль ложных медиаторов симпатической нервной системы в нарушениях церебрального метаболизма у больных с заболеваниями печени
198 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
разработки специальных аминокислотных препаратов для парентерального питания больных с печеночной недостаточностью, содержащих большое количество аминокислот с разветвленной углеродной цепью. Однако при всей теоретической привлекательности клиническая эффективность этих смесей окончательно не доказана.
Из аминокислот в печени синтезируются многочисленные белки: альбумин, аранти-трипсин, а-фетопротеин, а2-макроглобулин, церулоплазмин, компоненты системы комплемента (С3, Сб, СД трансферрин, фибриноген и другие факторы свертывания крови, факторы антикоагулянтной системы, С-реактивный протеин (табл. 6.1). Некоторые из них (фибриноген, гаптоглобин, 04-антитрипсин, С3-компонент системы комплемента, церулоплазмин) являются белками острой фазы. Их концентрация увеличивается при синдроме системного воспалительного ответа у больных с нарушением белковосинтетической функции печени. Это следует учитывать при оценке функции печени по данным биохимических показателей.
Сопоставляя изменение концентрации белков острой фазы с другими признаками воспалительного процесса, можно судить о динамике синдрома системного воспалительного ответа. Некоторые из бел-
Таблица 6.1. Белки плазмы крови, синтезируемые в печени
Белок Нормальная концентрация, г/л
Альбумин арАнтитрипсин* а-Фетопротеин а2-Макроглобулин Церулоплазмин* Компоненты С3, Сб и системы комплемента Фибриноген* Гемопексин Протромбин Трансферрин 40-50 2 — 4 < 10** 2,2-3,8 0,2-0,4 2-4 0,8-1,0 2-3
* Белки острой фазы воспаления.
** Микрограмм/литр (мкг/л).
ков, синтезируемых в печени, имеют особое клинико-физиологическое и диагностическое значение.
Альбумин является основным белком плазмы крови, синтезируемым в печени. Другие плазменные белки — глобулины -синтезируются во всех органах, где есть клетки системы мононуклеарных фагоцитов (печень, костный мозг, легкие).
На долю альбумина приходится 15 % общего синтеза белков печенью. Ежедневно образуется около 120 — 300 мг/кг этого белка. Скорость синтеза альбумина наиболее высока у новорожденных, с возрастом она уменьшается.
Альбумин распределяется во внутрисосудистый и интерстициальный водные секторы. При этом внутрисосудистый пул альбумина составляет около 40 %.
Основное физиологическое значение альбумина заключается в том, что он является ведущим фактором, поддерживающим онкотическое давление крови. В нормальных условиях альбумин крови обеспечивает примерно 75 — 80 % плазменного онкотического давления.
В норме концентрация альбумина в крови колеблется в пределах 40 — 50 г/л. Содержание белка зависит от функции печени, скорости метаболического разрушения альбумина и интенсивности поступления альбумина из интерстициального сектора. Период полуэлиминации альбумина из организма составляет примерно 16 — 21 сут. Поэтому при отсутствии нарушения взаимопревращения альбумина его концентрация в крови является поздним признаком нарушения белковосинтетической функции печени.
В печени синтезируются почти все плазменные факторы свертывания (фибриноген, факторы II, V, VII, IX, X, XI, XII, XIII), за исключением фактора Виллебранда и фактора VIII. При этом факторы II, VII, IX, X являются витамин-К-зависимыми, т. е. содержат на одном конце несколько остатков глутаминовой кислоты, которые в последующем подвергаются превращению при участии карбоксилазы. Для функционирования карбоксилазы необходим витамин К. При отсутствии его поступления в организм витамин-К-зависимые фак
Метаболические функции печени 199
торы свертывания крови остаются структурно незавершенными и не могут выполнять свою функцию. Гипокоагуляционный эффект непрямых антикоагулянтов связан с нарушением образования в печени витамин-К-зависимых факторов свертывания. Наиболее частой причиной недостаточности витамина К является внутри- и внепеченочный холестаз, однако снижение его концентрации может быть также вызвано применением антибиотиков (нарушение микрофлоры кишок), хелаторов желчных кислот (например, холестирамина).
Причиной уменьшения концентрации фибриногена является недостаточность функции гепатоцита. Гипофибриногенемия обычно сопутствует выраженной печеночной недостаточности. Но на ранних этапах нарушения функции органа может наблюдаться нарушение функции фибриногена — дисфибриногенемия. При этом может выявляться фибриноген с низкой молекулярной массой, а также фибриноген, содержащий избыток сиаловых кислот. Дисфибриногенемия сопровождается нарушением полимеризации фибрин-мономеров.
Недостаточность функции печени является также основной причиной уменьшения остальных факторов свертывания крови. При этом, в отличие от альбумина, период полуэлиминации факторов свертывания крови небольшой. Поэтому нарушения гемостаза при нарушении белковосинтетической функции печени проявляются рано. Минимальным периодом полуэлиминации обладает VII фактор свертывания крови (4 —6 ч). Поэтому среди показателей гемостаза часто наиболее чувствительным маркером является протромбиновое время и его производное — протромбиновый индекс.
Кроме факторов свертывания крови в печени синтезируются основные первичные антикоагулянты: антитромбин-Ш, протеины С и 5. При этом синтез протеинов С и 5 зависит от достаточного содержания в организме витамина К. Нарушение белковосинтетической функции печени может сопровождаться уменьшением концентрации в крови первичных антикоагулянтов. Это обуславливает возможность возникновения тромбозов. В клинике недостаточности
функции печени обычно сопутствует геморрагический диатез в связи с отсутствием необходимого количества прокоагулянтов.
Печень также имеет непосредственное отношение к системе фибринолиза. В гепатоцитах синтезируется плазминоген. Его концентрация уменьшается при тяжелом поражении печени. Это наряду с недостатком факторов свертывания является дополнительным фактором возникновения геморрагического диатеза при недостаточности функции печени.
6.2.2. Обмен углеводов
Печень играет важную роль в обмене глюкозы. Она обеспечивает депонирование глюкозы при повышении ее концентрации в крови воротной вены после приема пищи, а также в системном кровотоке. Кроме того, печень обеспечивает поддержание нормальной концентрации глюкозы в крови при голодании. В этом заключается глюкостатическая функция печени. Скорость поглощения глюкозы из крови или высвобождения в кровь пропорциональна степени гипер- или гипогликемии. При этом ведущим механизмом регуляции нормальной концентрации глюкозы в крови является обратимый двусторонний процесс: гликогенез (образование гликогена из поступающих в печень из кишок моносахаридов) и гликогенолиз (образование глюкозы крови из депо гликогена в печени). В течение суток синтезируется и расщепляется приблизительно 65 —70 % гликогена, что свидетельствует о значительной динамичности его состояния.
Первым этапом синтеза гликогена является образование глюкозо-6-фосфата. Глюкоза, из которой синтезируется глю-козо-6-фосфат, поступает в печень, как и в клетку головного мозга, по градиенту концентрации. Инсулин непосредственно на этот процесс влияния не оказывает. Глюкоза как моносахарид свободно диффундирует через мембрану печеночных клеток, поэтому она не может служить резервуаром углеводов в печени. Поскольку фосфатные эфиры глюкозы не так легко проникают через мембраны, фосфорилирование создает «ловушку», изолируя
200 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
реакции в пределах внутриклеточного сектора. Однако фосфаты сахаров не могут накапливаться в больших количествах в печени, поскольку из-за осмоса увеличение запасов подобных растворимых соединений будет сопровождаться накоплением больших количеств воды. Создание в печени запаса недиффундирующей глюкозы без сопутствующего осмотического набухания органа объясняется способностью печени превращать избыток углеводов в нерастворимый полимер с большой молекулярной массой — гликоген.
Инсулин непосредственного влияния на проникновение глюкозы в клетку не оказывает, но дальнейшее превращение глюкозы в глюкозо-6-фосфат уже является ферментативным процессом: его катализирует глюкокиназа. Исходно она обладает более низким сродством к глюкозе по сравнению с аналогичными ферментами в других органах и тканях. Но активность глюкокиназы индуцирует инсулин, секретируемый в ответ на повышение концентрации глюкозы в циркулирующей крови. Таким образом, во время голодания вследствие сниженной секреции инсулина в печеночной клетке, несмотря на свободное поступление через мембрану клетки глюкозы, ее дальнейшее превращение замедлено. Но после приема пищи, повышения концентрации глюкозы и увеличения секреции инсулина печеночная клетка интенсивно превращает глюкозу в гликоген.
Содержание гликогена в печени составляет 2 —8 % (в среднем 5 %) массы этого органа, или 100—125 г. Печень не единственное депо гликогена в организме. Его содержат и другие ткани. Количество гликогена в скелетных мышцах, находящихся в состоянии покоя и получающих достаточное количество питательных веществ, достигает 1 % их общей массы. Поскольку общая масса мышц в организме человека велика, суммарные запасы гликогена в мышцах примерно в пять раз больше, чем в печени, однако его невозможно использовать для ликвидации гипогликемии из-за отсутствия необходимых для этого ферментов. Гликоген в мышцах используется для энергетических потребностей клетки. Депо гликогена в печени, наобо
рот, служит быстромобилизуемым источником глюкозы как для крови, питающей другие ткани, так и для нужд самой печени. В течение первых 12 —24 ч после приема пищи потребности организма в углеводах в основном удовлетворяются за счет распада гликогена в печени.
При исчерпании запасов гликогена (голодание в течение примерно 24 ч) глюкоза синтезируется в печени из углеводных и неуглеводных исходных продуктов (глюконеогенез). Набор ферментов, необходимых для этого процесса, содержат печень, почки и слизистая кишок. Среди них печень играет главную роль. После длительного голодания замедленная реакция гликогенолиза в печени на действие эндокринных факторов приводит к усиленному синтезу ферментов глюконеогенеза.
Исходными субстратами для глюконеогенеза в печени могут быть пируват и его предшественники (аминокислота аланин, жирные кислоты), оксалоацетат и его предшественники (аминокислоты аспартат, глутамат), лактат, образующийся в других органах и тканях и попадающий в печень с током крови. Суммарные реакции образования глюкозы в ходе глюконеогенеза выглядят так:
2 Пируват + 2 НАД • Н + 6 АТФ ---►
----► 1 Глюкоза + 2 НАД+ + 6 АДФ + 6 Фн;
2 Оксалоацетат + 2 НАД • Н + 4 АТФ --►
-----► 1 Глюкоза + 2 СО2 + 2 НАД+ +
+ 3 АДФ + 4 Фн;
2 Лактат + 6 АТФ----►
----► 1 Глюкоза + 6 АДФ + 6 Фн.
Примером роли печени в глюконеогенезе с участием лактата является взаимосвязь печени и скелетных мышц в цикле Кори. Во время напряженной работы скелетных мышц лактат образуется в таком избытке, что митохондрии мышц не справляются с окислением пирувата и НАД х х Н, анаэробный гликолиз преобладает над аэробным катаболизмом. Лактат диффундирует в кровоток и достигает других органов, в частности печени и сердца. Сердечная мышца содержит большое количество митохондрий, поэтому она способна окислять не только пируват, образующий
Метаболические функции печени 201
ся в результате протекающего в ней самой гликолиза, но и лактат, поступивший извне. В печени лактат не окисляется, а используется для получения глюкозы при глюконеогенезе. Образующаяся при этом глюкоза поступает в кровоток, а оттуда — в работающие мышцы, завершая цикл Кори.
Существует предположение, что одной из возможных причин внезапной смерти новорожденного является недостаток ферментов глюконеогенеза. Этим может объясняться неспособность организма ребенка поддерживать нормальный уровень гликемии в промежутках между кормлениями и возможность внезапного летального исхода.
Указанные механизмы помогают поддерживать постоянство уровня глюкозы в крови и стабильность обеспечения глюкозой такого чувствительного к гипо- и гипергликемии органа, как головной мозг. Этому же способствуют и особенности усвоения глюкозы другими органами и тканями. Так, быстрой нормализации гликемии после увеличения концентрации глюкозы в крови способствует поступление глюкозы в жировую клетчатку и в клетки мышц. В противоположность ее поступлению в печень и головной мозг этот процесс активизируется инсулином. Эти явления не наблюдаются при относительной или абсолютной недостаточности инсулина в условиях сахарного диабета. Кроме этого, фермент гексокиназа, содержащийся в клетках жировой ткани и мышц, который, как и глюкокиназа, стимулирует превращение внутриклеточной глюкозы в глюкозо-6-фосфат, имеет более высокое сродство к глюкозе, что обеспечивает в указанных клетках участие глюкозы в реакциях обмена веществ при более низких концентрациях, чем в печени.
Избыток глюкозы печень превращает не только в гликоген, но и в триглицериды жировой ткани, участвуя в создании тканевых энергетических резервов (рис. 6.2). Из глюкозы синтезируются входящие в состав триглицеридов жирные кислоты (синтез глицеринового компонента триглицеридов из глюкозы осуществляется непосредственно в жировой ткани). Образовавшиеся в печени триглицериды в дальнейшем транспортируются в составе липо-
Рис. 6.2. Образование тканевых резервов глюкозы после приема пищи:
Г-6-Ф — глюкозо-6-фосфат; ЛПОНП — липопротеид особо низкой плотности; Триозо-Ф — триозофосфат;
Глицерол-З-Ф — глицерол-3-фосфат
протеидов особо низкой плотности в жировую клетчатку, где они подвергаются гидролизу, катализируемому липопротеин липазой. Высвобождающиеся жирные кислоты (печеночного происхождения) конденсируются с глицерол-3-фосфатом, образующимся из глюкозы, которая поступает в жировую ткань под действием инсулина, и накапливающиеся триглицериды составляют тканевой резерв, в котором сохраняется значительно больше энергии, чем в запасе гликогена. При парентеральном питании и введении избыточного количества растворов глюкозы возможно возникновение жировой дистрофии печени.
Печень также играет главную роль в превращении галактозы и фруктозы в глюкозу. При нарушении ее функции способность организма использовать галактозу и глюкозу уменьшается. На этом основана ранее широко используемая функциональная проба печени с нагрузкой галактозой и фруктозой.
6.2.3. Обмен липидов
Липиды — это соединения, отличительными особенностями которых являются наличие в их молекуле остатков жирных кислот и слабая растворимость в воде. Жирные кислоты содержат длинную не-
202 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
разветвленную цепь углеродных атомов (обычно 16, 18 или 20) и различное число двойных связей (от 0 до 4), а на одном из концов углеродного скелета — карбоксильную группу. Соответствующая формула наиболее распространенной насыщенной жирной кислоты — пальмитиновой — составляет 16:0; ненасыщенной кислоты с одной двойной связью — олеиновой — 18:1; полиненасыщенных жирных кислот 18 : 2 (линолевая), 18 : 3 (линоленовая), 20 : 4 (арахидоновая). При этом организм человека способен синтезировать насыщенные и мононенасыщен-ные жирные кислоты. Полиненасыщенные жирные кислоты являются незаменимыми: они должны поступать с пищей. Значение полиненасыщенных жирных кислот обусловлено их ролью в организме. Во-первых, они являются предшественниками простагландинов, выполняющих различные регуляторные функции. Во-вторых, они поддерживают жидкое состояние липидов клеточных мембран. В-третьих, полиненасыщенные жирные кислоты предотвращают отложение холестерина и других липидов в стенках кровеносных сосудов.
Молекулы жирной кислоты содержат большое количество звеньев ((СН2)„), этим объясняется то, что жирные кислоты являются наиболее эффективным энергетическим субстратом организма. Энергия, запасаемая в ходе биологических окислительных процессов, в основном связана с переносом электронов от атомов водорода по дыхательной цепи и фосфорилированием АДФ до АТФ. Поскольку жирные кислоты построены преимущественно из углерода и водорода, их окисление сопровождается большим выходом энергии, чем, например, углеводов: в состав последних входят и атомы кислорода. Так, при окислении 1 г жиров образуется 9 ккал (37,67 кДж) энергии, 1 г углеводов или белков — по 4 ккал (16,74 кДж). Более половины основной энергетической потребности многих тканей удовлетворяется за счет жиров, особенно в период голодания.
Жирные кислоты редко встречаются в свободном виде. Обычно они входят в состав различных соединений — ацилгли-церидов, фосфоглицеридов, стероидов, жи
рорастворимых витаминов A, D, Е и К. Среди них особое место занимают триацил глицериды. В молекуле глицерина содержатся три гидроксильные группы, которые могут быть этерифицированы жирными кислотами с образованием соответственно моно-, ди- или триацилгли-церидов. Триацилглицериды обычно называют жирами. Их содержание в составе липидов, поступающих с пищей, у человека достигает более 90 %.
Процесс метаболизма липидов пищи в просвете и стенке пищеварительного канала состоит из нескольких основных этапов: 1. Секреция поджелудочной железой гидролитических ферментов, содержащих липазы и буферные системы, разрывающие в жирах сложноэфирные связи. 2. Поступление в просвет ПК в составе желчи желчных кислот, эмульгирующих жиры и продукты их распада. 3. Гидролиз липидов под влиянием секрета поджелудочной железы и желчи до моноглицеридов и свободных жирных кислот. Моноглицериды располагаются между молекулами солей желчных кислот, образуя смешанные мицеллы. 4. Захват слизистой кишок мицелл с последующим превращением в стенке кишок. При этом жирные кислоты и моноглицериды вступают друг с другом в энергозависимые реакции реэтерификации с образованием триацил глицеридов, характерных именно для данного организма. 5. Транспорт липидов к печени как по системе воротной вены, так и по системе печеночной артерии.
По системе воротной вены в печень поступает лишь незначительная часть липидов. К ним относятся соли желчных кислот и жирные кислоты с короткой цепью. Последние переносятся к печени в виде комплексов с альбумином плазмы. Достигая печени, они подвергаются катаболизму в ходе реакций окисления и не играют большой роли в обмене веществ, за исключением детей, в питании которых преобладают жиры молока. Соли желчных кислот, наоборот, захватываются печенью для дальнейшего использования. Они поступают в общий пул из нескольких граммов желчных кислот во внутриклеточном пространстве, в желчных протоках и в концен
Метаболические функции печени 203
трированном виде — в желчном пузыре. Таким образом, совершается энтеросистем-ная рециркуляция желчных кислот. При этом лишь незначительная часть желчных кислот экскретируется с калом.
Основная часть липидов транспортируется к печени по системе печеночной артерии. Ресинтезированный триацил глицерид и холестерин поступают из кишок в лимфатические сосуды, затем через грудной лимфатический проток попадают в кровь. Липиды к печени переносятся преимущественно в виде хиломикронов, хотя пул транспортных форм липидов разнообразен. После приема жирной пищи в крови содержится большое количество хиломикронов, вследствие чего плазма мутнеет и делается похожей на молоко. Это вызывает появление в крови так называемого просветляющего фактора. В его состав входит липаза липопротеидов, расщепляющая триглицериды хиломикронов и липопротеидов. Расщепляющий фактор образуется под влиянием гепарина, содержащегося в печени, легких, мышцах и других органах. На этом основаны рекомендации добавлять небольшие дозы гепарина в растворы жировых эмульсий при проведении парентерального питания.
Еще одним источником липидов, поступающих в печень, являются свободные жирные кислоты, освобождаемые из жировой ткани и поступающие к печени с кровью в виде комплексов с альбумином. Хотя в самой печени запасы жиров незначительны (они составляют менее 1 % общей массы органа), она занимает ключевую позицию в процессах мобилизации, переработки и биосинтеза жиров.
Окисление жирных кислот в печени осуществляется в митохондриях. Процессы связаны с заключительными реакция
ми катаболизма в системе переноса электронов и цикла лимонной кислоты. При этом окисление жирных кислот состоит из трех основных этапов (рис. 6.3). 1. Активация жирных кислот путем энергозависимой реакции образования тиоэфиров ацил-КоА. 2. р-Окисление — замкнутый цикл реакций последовательного отщепления атомов водорода от алкильной цепи и двухуглеродных остатков в виде молекул ацетил-КоА. 3. Окисление в цикле лимонной кислоты образующихся молекул ацетил-КоА до СО2 и дополнительного количества водорода, используемого затем при окислительном фосфорилировании.
Печень играет также ключевую роль в процессах липогенеза — в синтезе жирных кислот в организме. За исключением полиненасыщенных, все другие жирные кислоты, необходимые либо как структурные липиды для построения мембран, либо для создания тканевых депо, синтезируется в организме человека. Любое вещество, конечным продуктом обмена которого является ацетил-КоА, может быть использовано в липогенезе. Но наиболее значимым предшественником являются углеводы (см. «Обмен углеводов»). Во всех случаях, когда потребление углеводов превышает энергетические потребности организма, их избыток превращается в жиры.
Печень также принимает активное участие в обмене холестерина в организме человека. В отличие от триацил глицеридов, играющих роль энергетического резерва, холестерин является компонентом клеточных мембран, предшественником солей желчных кислот, стероидных гормонов, витамина D, а при патологии — узловым патогенетическим звеном развития атеросклероза. Часть холестерина поступает в организм с пищей. Небольшое количество
(У
О (Активация)
Я-С-ОН V*
Жирная /АТФ АМФ+Ф-Ф, кислота/ (ГТФ) (ГДФ+ФН)
KoASH
О
Я-С-S Ко А Алифатический
Ацил-КоА
(?)
(P-Окисление) О
—ЧCH3-C-SK0A
Ацетил-Коа
3'
Жиры Фосфолипиды
(Окислительное
’ фосфорилирование) АТФ
^ЦиклХ трикарбоновых
Чкислот/
2СО2
Рис. 6.3. Три этапа окисления жирной кислоты
204 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
вещества образуют мицеллы с солями желчных кислот и за счет этого проникают в клетки слизистой кишок, в составе хиломикронов попадают в системный кровоток. При этом количество всасывающегося в кишках холестерина ограничено до 0,5 г/сут. Холестерин хиломикронов задерживается в печени, где подвергается метаболизму с образованием производных холестерина в мембранах печеночных клеток и желчных кислот. При этом синтез желчных кислот является важным механизмом элиминации холестерина из организма. Это обусловлено тем, что образование желчных кислот регулируется по принципу обратной связи. При уменьшении количества желчных кислот, всасывающихся в просвете кишок в процессах эн-теросистемной рециркуляции, например, за счет их связи в просвете кишки, в печени увеличивается синтез желчных кислот. За счет этого возможно уменьшение концентрации холестерина в кровотоке. Кроме поступления холестерина с пищей клетки печени и слизистой оболочки кишки могут синтезировать его из ацетил-КоА. Выведение холестерина в составе желчных кислот может вести к его патологическому накоплению в желчных протоках в составе желчных камней.
Печень является местом синтеза кетоновых тел (ацетоуксусная кислота, р-оксимас-ляная кислота и ацетон). В норме их со-
Мозг
Рис. 6.4. Метаболизм углеводов при голодании, избыточное накопление кетоновых тел:
Г-6-Ф — глюкозо-6-фосфат; Триозо-Ф — триозофосфат
держание в плазме не превышает 10 мг/л, но значительно возрастает при декомпенсации сахарного диабета. При этом происходят противоположные описанным при характеристике обмена углеводов процессы превращения триглицеридов жировой клетчатки (рис. 6.4). В условиях голодания, когда экзогенная глюкоза отсутствует, эндогенные триглицериды жировой клетчатки претерпевают обратное превращение путем липолиза в свободные жирные кислоты и глицерин. Эти соединения транспортируются в печень, где глицерин включается в реакции глюконеогенеза. Синтезируемая при этом глюкоза может поступать в кровоток в то время, когда концентрация глюкозы в плазме имела бы тенденцию к снижению, если бы процесс глюконеогенеза не функционировал. Кроме того, свободные жирные кислоты, поступающие из жировой ткани, могут использоваться большинством органов и тканей (за исключением головного мозга) как источник энергии. При превращении свободных жирных кислот в печени образуется ацетил-КоА. Путем ферментативного превращения двух молей ацетил-КоА в печени образуется ацетоуксусная кислота, которая в дальнейшем может быть восстановлена до р-оксимасляной кислоты или декарбоксилирована с образованием ацетона.
Таким образом, взаимосвязь биохимических процессов в печени и жировой ткани способствует поддержанию нормальной концентрации глюкозы в крови при голодании. Благодаря этому, а также за счет поступления и утилизации свободных жирных кислот обеспечивается энергоснабжение органов и тканей в случае отсутствия поступления экзогенной глюкозы. Биохимическим эквивалентом голодания может быть накопление в кровотоке кетоновых тел. При декомпенсации сахарного диабета повышение их концентрации обусловлено не только относительным или абсолютным недостатком инсулина и энергетическим голоданием органов и тканей, поскольку степень накопления кетоновых тел в данном случае зачастую гораздо больше, чем при отсутствии поступления пищи в связи с голоданием или нарушением функции пищеварительного канала,
Метаболические функции печени 205
ЦИТОПЛАЗМА
кттт т
Жирная кислота 1X1X1 1
Ацил-КоА \ *
МИТОХОНДРИЯ
Жирная кислота Ацил-КоА
Аце- г Мало-
тил-КоА к нил-КоА
Пируват
Гликоген
ГЛЮКАГОН
Рис. 6.5. Регуляция кетогенеза в печени: КПТТ I — карнитин-пальмитоилтрансфераза I
но и накоплением контринсулярных гормонов стресса чаще всего вследствие развития сопутствующего заболевания (прежде всего, воспалительных процессов, тромбоэмболических осложнений, инфаркта миокарда). Одним из таких контринсулярных гормонов является глюкагон (рис. 6.5). Кетоновые тела образуются в митохондриях печени. При этом количество соединений жирных кислот, поступающих в митохондрии печени, зависит от активности фермента карнитин-пальмитоилтрансфе-разы, локализованного на внешней стороне мембраны митохондрии. Обычно активность этого фермента снижена за счет повышенной концентрации в цитоплазме соединения малонил-КоА, являющегося первым промежуточным продуктом синтеза жирных кислот, уровень которого регулируется циркулирующим в крови глюкагоном. При накоплении последнего происходит нарушение образования малонил-КоА как вследствие блокирования гликолиза, так и из-за снижения активности малонил-КоА карбоксилазы. В результате увеличивается активность фермента карнитин-пальмитоилтрансферазы. Следствием этих процесов является поступление в митохондрии большого количества жирных кислот с последующим усилением активности кетогенеза. При этом в основном образуются ацетоуксусная кислота и £-гид-роксибутират.
Печень также принимает активное участие в биосинтезе фосфолипидов и липопротеидов.
В норме печень содержит небольшие запасы жиров. В отдельных случаях воз
можно патологическое отложение жиров в органе. Это может быть обусловлено или нарушением их метаболизма при нормальном поступлении жиров в печень, или избыточным поступлением жиров на фоне нормальных процессов метаболизма в печеночной клетке. Типичными, причинами жировой инфильтрации печени являются: 1. Воздействие на печень токсикантов, в частности этанола, хлороформа, четыреххлористого углерода. 2. Инфекционное поражение печени. 3. Поражение печени злокачественным процессом. 4. Избыточное поступление жиров в печень из тканевых депо при голодании, сахарном диабете. 5. Избыточное поступление углеводов в организм при парентеральном питании. 6. Пища, содержащая недостаточное количество белка.
6.2.4. Билирубин и его циркуляция
80 % всей массы билирубина образуется в результате разрушения эритроцитов в системе мононуклеарных фагоцитов, преимущественно селезенки. Высвобождаемый гемоглобин расщепляется до глобина, поступающего в общий фонд белков, и гема, превращающегося в билирубин после потери железа, которое затем используется повторно. К числу других источников билирубина относятся: распад незрелых эритроцитов в тканях костного мозга, а также близких к гемоглобину по составу и свойствам миоглобина и цитохромов.
Образовавшийся в результате распада эритроцитов билирубин называют свободным неконъюгированным билирубином (это билирубин, которому еще не придана водорастворимость в результате конъюгации с глюкуронатом в печени). Он связывается с альбумином плазмы. Вследствие этого билирубин не может проникать ни через почечные клубочки в мочу, ни через биомембраны мозга. Но при низком содержании альбумина в плазме, вытеснении из билирубина связи с альбумином (в том числе и под действием лекарственных соединений), а также при нарушении (незрелости — у новорожденных) механизмов конъюгации в печени в плазме увеличивается фракция свободного неконъюгиро-
206 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
ванного билирубина, который растворим в липидах, проникает через гематоэнцефалический барьер и вызывает токсическое повреждение ткани мозга.
В печени происходит перенос билирубина от альбумина плазмы через свободнопроницаемую мембрану сосудистых синусов в гепатоциты. Этот процесс осуществляется с помощью транспортных белков. Перенос билирубина происходит быстро вследствие наличия в цитозоле связывающих белков, а также благодаря его быстрому последующему метаболизму в гепатоците в реакции глюкуронидизации и выделению в желчь.
В гепатоците происходит активный транспорт поступившего билирубина в гладкий эндоплазматический ретикулум и конъюгация его с глюкуроновой кислотой в ходе процесса, катализируемого уридин-дифосфат-глюкуронилтрансферазой. При печеночно-клеточной желтухе активность этого фермента поддерживается на достаточном уровне, а при холестазе — увеличивается. Однако у новорожденных активность фермента снижена. Дефицит функции конъюгирующих ферментов является основой таких семейных негемолитических гипербилирубинемий, как синдромы Жильбера и Криглера —Найяра. Билирубин-моноглюкуронид поступает к обращенной в сторону желчных канальцев поверхности гепатоцита, где после присоединения второго остатка глюкуро-ната образуется диглюкуронид билирубина, который активно секретируется в желчные канальцы. Последующий транспорт зависит от нормального оттока желчи, а также от состояния транспортных белков. Экскреция билирубина в канальцы происходит с помощью семейства АТФ-зави-симых мультиспецифичных для органических анионов транспортных белков. Желчные кислоты переносятся в желчь также с помощью другого транспортного белка. Наличие разных механизмов транспорта билирубина и желчных кислот обуславливает клиническую картину синдрома Дубина —Джонсона, при котором нарушается экскреция конъюгированного билирубина, но сохраняется нормальная экскреция желчных кислот.
При поражениях печени и повышении давления в желчевыводящих путях наиболее высока вероятность нарушения именно процессов секреции билирубина в желчные протоки, требующих затраты энергии. Поэтому повышение концентрации конъюгированного билирубина в плазме является наиболее ранним проявлением нарушения его экскреции. При печеночной желтухе, как правило, в плазме повышено содержание обеих фракций билирубина, но преобладает конъюгированный, который растворим в воде, менее прочно, чем свободный, связывается с белками и может экскретироваться с мочой.
Конъюгированный билирубин поступает в кишки с желчью. Диглюкуронид билирубина водорастворим (полярная молекула), поэтому в тонкой кишке не всасывается. В толстой кишке конъюгированный билирубин подвергается гидролизу p-глюкуронидазами бактерий до соединений, обозначаемых собирательным термином стеркобилиноген (или уробилиноген кала). При бактериальном холангите часть диглюкуронида билирубина гидролизуется уже в желчных путях с последующей преципитацией билирубина. Этот процесс может играть важную роль в образовании билирубиновых желчных камней.
Небольшая часть стеркобилиногена всасывается, попадает в систему портального кровообращения и преимущественно вновь экскретируется с желчью; очень небольшая его фракция экскретируется с мочой в виде уробилиногена, который, в свою очередь, может быть окислен до уробилина. Уробилин (оген) в отличие от билирубина часто обнаруживается в моче здоровых лиц, особенно в тех случаях, когда экскретируется концентрированная моча. У здорового человека экскреция уробилиногена с мочой настолько изменчива, что клинически значимо только обнаружение очень высокой концентрации уробилиногена в моче во время острых приступов гемолиза.
Неабсорбированный стеркобилиноген окисляется до стеркобилина — пигмента, придающего калу коричневую окраску. Поэтому малоокрашенные фекалии рассматриваются как признак закупорки желчных протоков. Билирубин, уробилин и стер
Желчеобразование и желчеотделение 207
кобилин (желчные пигменты) — окрашены. Уробилиноген и стеркобилиноген — бесцветны.
Конъюгированный и неконъюгирован-ный билирубин иногда обозначают соответственно терминами «прямой» и «непрямой билирубин».
6.2.5. Влияние анестезии на метаболические функции печени
Стресс, оперативное вмешательство сопровождаются повышением концентрации гормонов стресса (катехоламины, глюкагон, кортизол) и одновременным снижением анаболических гормонов (инсулин, тестостерон). В результате катаболизм доминирует над анаболизмом. Это проявляется увеличением интенсивности распада белков, образования мочевины, снижением синтеза белков, повышением концентрации глюкозы. Последнее обусловлено активацией гликогенолиза и глюконеогенеза. Энергетическую ценность образующейся в результате глюконеогенеза глю
козы иллюстрируют следующие реакции:
2 Лактат + 6 АТФ ---► Глюкоза;
Глюкоза----► 2 Лактат + 2 АТФ.
Метаболизм жиров под влиянием оперативного вмешательства изменяется в меньшей степени. Степень этих отклонений зависит от эффективности анестезии: при использовании регионарных методов анестезии, предотвращающих поступление ноцицептивной информации в центральную нервную систему, она меньше.
Непосредственному влиянию средств для наркоза на метаболизм в печени посвящены лишь единичные исследования. Так, оказалось, что фторотан нарушает потребление кислорода в цикле синтеза мочевины, глюконеогенез, усвоение лактата, содержащегося в плазме. При использовании этого анестетика может повыситься концентрация молочной кислоты в крови. В экспериментальных исследованиях на культурах клеток также выявлено, что фторотан и энфлуран могут угнетать синтез белков в печеночной клетке.
6.3. ЖЕЛЧЕОБРАЗОВАНИЕ И ЖЕЛЧЕОТДЕЛЕНИЕ
6.3.1. Состав и функция желчи. Желчные кислоты
Желчь является одновременно и экскреторным, и секреторным продуктом печени. Экскреторный характер желчи подтверждается тем, что в ее состав входят вещества, которые в силу своей баластно-сти и даже токсичности подлежат удалению из организма. Характеристика желчи как секрета обусловлена тем, что она содержит вещества, участвующие в ряде физиологических процессов в кишках, которые способствуют расщеплению и всасыванию пищевых соединений.
Ежесуточно в печени образуется приблизительно 10 мл желчи на 1 кг массы тела, в среднем у взрослого — около 600 мл. В желчевыводящих путях происходит активная реабсорбция электролитов (Na+, СГ, НСО3) и изоосмотическая пассивная реабсорбция воды. В результате образуется пузырная желчь, в 10 раз более концен
трированная, чем печеночная, содержащая в качестве преобладающего катиона натрий, а в качестве доминирующих анионов — соли желчных кислот. Таким образом, желчный пузырь выполняет не только резервуарную функцию, высвобождая после приема пищи необходимую для пищеварения желчь, но и защитную. Высокая осмолярность желчи предотвращает размножение в ней микроорганизмов из просвета кишки. Нарушение концентрирования желчи, дренирование желчных протоков, формирование билиодигестивных анастомозов может создавать условия для инфицирования желчи и развития ангиохолита.
На оценке осмолярности желчи основан метод идентификации протоковой желчи при исследовании больного с подозрением на билиарный сепсис. Согласно данным Украинского центра интенсивной терапии сепсиса, в структуре так называемых «лихорадок неясного происхождения» частота встречаемости би л парно
208 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
го сепсиса составляет 23,4 %. Дифференциальная диагностика и реализация этиотропного обоснования выбора антибиотиков в таких случаях требует получения желчи, выделения из нее возбудителя и оценки его чувствительности к антибиотикам. При этом для разграничения желчи из общего протока, желчного пузыря и внутрипеченочных протоков обычно используется метод Лопеса, в соответствии с которым вид желчи устанавливается на основе внешних признаков. Но такой подход в клинике часто не позволяет четко разграничить источник желчи. Поэтому с целью дифференциальной диагностики более обосновано разграничение вида желчи регистрацией ее осмолярности: осмолярность более 410 моем/л характерна для общего желчного протока (порция «А»), 360 — 400 моем/л — для желчного пузыря (порция «В»), 290 — 360 мосм/л — для внутрипеченочных ходов (порция «С»).
Желчь состоит из желчных кислот, холестерина, фосфолипидов, билирубина, белков, минеральных ионов, воды (табл. 6.2). Из приведенных в таблице данных следует, что основным компонентом желчи являются желчные кислоты.
В организме человека происходит образование четырех желчных кислот. Две из них называются первичными — это холевая и хенодезоксихолевая кислоты. Они синтезируются в печени из холестерина. При этом расходуется около 40 % холестерина, содержащегося в организме. Первичные желчные кислоты с желчью поступают в кишки, где при участии бактерий превращаются во вторичные — дез-
Таблица 6.2. Состав желчи
Вещество Печеночная желчь, г/л Пузырная желчь, г/л
Сухое вещество 23-33 180
Азот 0,8 4,9
Холин 0,4-0,9 5,5
Желчные кислоты 7-14 115
Жирные кислоты 1,6-3,4 24
Лецитин 1,0-5,8 35
Холестерин 0,8-2,1 4,3
Белок 1,4-2,7 4,5
Билирубин 0,3-0,6 1,4
оксихолевую и литохолевую. Часть вторичных желчных кислот подвергается всасыванию в кишках и повторно выделяется печенью. Таким образом, желчь содержит смесь первичных и вторичных желчных кислот. Они представлены натриевыми солями и конъюгированы с аминокислотами глицином и таурином. Конъюгация является важным механизмом, предотвращающим образование в желчных протоках осадка желчных кислот, а в просвете кишки — преждевременную абсорбцию желчных кислот в проксимальном отделе тонкой кишки и таким образом удерживающим желчные кислоты в просвете кишки в концентрациях, достаточных для осуществления переваривания и абсорбции жиров. Это обусловлено тем, что соли конъюгированных жирных кислот выпадают в осадок лишь при pH = 4,3 —5,0, тогда как неконъюгированных — при pH = = 6,5 —7,0. В то же время pH печеночной желчи в норме колеблется в пределах 7,3 —7,7, пузырной — 6 — 7. В физиологических условиях в желчи и в просвете кишки pH = 4,3 —5,0, в пределах которого происходит выпадение в осадок конъюгированных желчных кислот, не наблюдается. Если же в тонкой кишке происходит анормальная пролиферация бактериальной флоры, то может вести к деконъюгации желчных кислот. Вследствие этого они быстро всасываются, что может привести к недостаточной для абсорбции жиров внутрипросветной концентрации желчных кислот и стеаторее.
В норме большая часть желчных кислот не синтезируется вновь, а реабсорбируется из кишок и доставляется в печень. При этом возможны два пути рециркуляции желчных кислот: 1. Портальный путь — вещества, абсорбированные из кишок, попадают в воротную вену и транспортируются в печень. Таким образом реабсорбируется 98 % всосавшихся в кишках желчных кислот. 2. Экстрапортальный путь — всосавшиеся в кишках вещества по лимфатическим путям поступают в лимфатический проток, а затем — в верхнюю полую вену и разносятся током крови по всему организму. Из общего кровотока желчные кислоты захватываются печенью.
Желчеобразование и желчеотделение 209
Недостаточное содержание солей желчных кислот в просвете кишки
I
Нарушение эмульгирования жиров, поступающих с пищей
!
Стеаторея Диарея
Нарушение переваривания Уменьшение содержания белка в плазме
Нарушение поступления витамина А----------- «Куриная слепота»
Гиперкератоз кожи Остеомаляция: кальция ______► деминерализация
и витамина D костной ткани,
переломы витамина К------► Патология системы
гемостаза
витамина Е
Патология метаболизма
Рис. 6.6. Схема клинико-физиологических и патохимических последствий холестаза
В крови желчные кислоты связываются с альбумином. При однократном прохождении через печень извлекается около 90 — 95 % желчных кислот. Благодаря такой эффективности захвата гепатоцитами уровень желчных кислот в периферической крови крайне низок. Почечный клиренс желчных кислот очень мал, поэтому почти все желчные кислоты, попавшие в общий кровоток, возвращаются в печень. Низкий почечный клиренс желчных кислот почками отражает общую закономерность: чем более интенсивная связь вещества с белком, тем менее вероятен почечный клиренс соединения и тем необходимее участие печени в освобождении крови от такого химического вещества.
В просвете кишки под влиянием желчных кислот происходит эмульгирование поступивших с питанием жиров. Желчные кислоты обволакивают жировые капли и, будучи поверхностно-активными веществами, уменьшают их поверхностное натяжение, способствуя тем самым расщеплению капелек на меньшие частицы, а также стабилизируют жировую эмульсию. Поскольку желчные кислоты растворимы в воде, они, соединяясь с липидами, повышают растворимость последних. Наконец,
желчные кислоты активируют липазу поджелудочной железы. Липаза является белком и растворяется в воде, а так как жиры в воде не растворяются, то действие липазы на жиры происходит главным образом на их поверхности, т. е. на границе раздела фаз вода —жир. Таким образом, активируя липазу и создавая условия для ее нормального функционирования, желчные кислоты способствуют перевариванию и абсорбции жиров пищи и жирорастворимых витаминов. Уменьшение поступления желчи в двенадцатиперстную кишку вследствие патологического процесса на каком-либо участке от гепатоцита до фатерова соска называют холестазом. Его клинико-физиологические и патохимичес-кие последствия приведены на рис. 6.6.
6.3.2. Функциональная анатомия желчевыводящих путей
Начальным элементом желчных путей является желчный каналец, контактирующий с гепатоцитом (рис. 6.7). Желчные канальцы впадают в дуктулы, называемые иногда холангиолами, или каналами Геринга. Дуктулы располагаются в основном в портальных зонах и впадают в междольковые желчные протоки, которые первыми из желчных путей сопровождаются
Синусоид
Микро-----|WVUVWW
ворсинки
Гепатоцит —
Септальный желчный “ проток
Междольковый желчный проток
Рис. 6.7. Анатомия желчных протоков
Каналец
Дуктула Портальная триада
210 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
веточками печеночной артерии и воротной вены и обнаруживаются в составе портальных триад. Междольковые протоки, сливаясь, формируют септальные протоки, которые, в свою очередь, образуют два главных печеночных протока, выходящих из правой и левой долей в области ворот печени.
Гепатоцит представляет собой полярную секретирующую эпителиальную клетку. В его структуре выделяют базолатеральную и апикальную мембраны. Базолатеральная мембрана обращена к синусоидам, поэтому ее иногда называют также синусоидальной, апикальная — в сторону желчного канальца, вследствие этого она еще называется канальцевой мембраной.
Образование желчи включает захват гепатоцитом желчных кислот и других органических и неорганических ионов, транспорт их через базолатеральную (синусоидальную) мембрану, синтез ряда соединений в гепатоците, транспорт синтезированных и захваченных веществ через цитоплазму и секрецию в желчный каналец. Процесс переноса соединений через апикальную мембрану сопровождается осмотической фильтрацией воды, содержащейся в гепатоците и парацеллюлярном пространстве. В образовании желчи участвуют такие структурно-функциональные образования гепатоцита:
1. Базолатеральная (синусоидальная) мембрана. Содержит множество транспортных систем для захвата органических анионов.
2. Канальцевая мембрана. Содержит транспортные белки для желчных кислот, билирубина, катионов и анионов, а также микроворсинки, увеличивающие ее поверхность.
3. Внутриклеточные органеллы, представленные аппаратом Гольджи и везикулами. С помощью везикул осуществляется транспорт белков от синусоидальной к канальцевой мембране, доставка сюда же синтезирующихся в клетке транспортных белков для холестерина, фосфолипидов, желчных кислот.
4. Микрофиламенты, представленные актином. Они концентрируются вокруг канальцевой мембраны, определяют сокра
тительную способность и моторику канальцев. Вещества, оказывающие воздействие на микрофиламенты, угнетают моторику канальцев (канальцевый «паралитический илеус») и вызывают холестаз.
5. Промежуточные филаменты, образующие сеть между внутриклеточными структурами. Разрыв промежуточных филаментов приводит к нарушению внутриклеточных транспортных процессов и облитерации просвета канальцев.
Функции вышеназванных структурнофункциональных образований являются энергозависимыми процессами. Выделение таких образований важно, поскольку их поражения — главные звенья формирования внутрипеченочного холестаза (в отличие от внепеченочного холестаза при внутрипеченочном отсутствует обструкция магистральных желчных протоков). Так, повреждение базолатеральной и канальцевой мембран гепатоцита, внутриклеточных органелл развивается при холестатическом варианте вирусного и алкогольного гепатита. Некоторые лекарственные соединения, например аминазин, сульфаниламиды, нарушают активность микрофиламентов, что также обуславливает возникновение внутрипеченочного холестаза.
Контакт гепатоцитов, отделяющий просвет канальцев от кровеносной системы печени, называют плотным контактом, целостность которого обеспечивается специализированными белками. Через плотные контакты между гепатоцитами по осмотическому градиенту могут перемещаться вода и электролиты, но крупные молекулы не проникают. Разрыв плотных контактов сопровождается попаданием в канальцы растворенных крупных молекул, а это приводит к потере осмотического градиента и развитию холестаза. При этом также может наблюдаться регургитация канальцевой желчи в синусоиды.
6.3.3. Желчеобразование
С точки зрения механизмов формирования выделяют три фракции желчи, две из которых связаны с функцией гепатоцита.
Роль печени в гемопоэзе 211
1. Фракция желчи, экскреция которой зависит от желчных кислот. У взрослого человека на ее долю приходится около 225 мл из 600 мл желчи, выделяющейся в сутки. Эта фракция образуется за счет секреции гепатоцитом солей желчных кислот. Осмотически активный транспорт солей желчных кислот сопровождается пассивным транспортом воды.
2. Фракция желчи, секреция которой не зависит от желчных кислот. Ее примерный объем также составляет 225 мл. Гепатоцит выделяет низкомолекулярные растворимые соединения, электролиты. При этом секреция таких веществ, как глутатион и гидрокарбонаты, создает осмотический градиент для перемещения воды.
3. Дуктулярная фракция желчи. Ее образование связано с активностью эпителиальных клеток дистальных протоков, вырабатывающих обогащенный гидрокарбонатами секрет.
В отдельных случаях нарушается выделение желчи и билирубина с желчью, но сохраняется выделение других фракций желчи. У таких пациентов желчь становится бесцветной (белая желчь) и напоминает слизистую желчь.
6.3.4. Желчный пузырь и его моторика.
Влияние анестезии на нарушение моторики у больных в критическом состоянии
Сокращение желчного пузыря находится под холинергическим и гуморальным контролем. Раздражение блуждающего нерва вызывает расслабление сфинктера Одди и сокращение желчного пузыря. Гуморальная регуляция деятельности желчного пузыря связана с холецистокинином, выделяющимся в ответ на поступление в
двенадцатиперстную кишку жира с пищей. Он вызывает сокращение желчного пузыря, усиливает секрецию жидкости и раз-ведение желчи.
Отсутствие поступления пищи в пищеварительный канал у больных в критическом состоянии, а также получающих полное парентеральное питание сопровождается холестазом, а в отдельных случаях может даже приводить к образованию желчных камней.
Лекарственные вещества, которые оказывают влияние на вегетативную нервную систему, могут воздействовать и на моторику желчного пузыря. Так, атропин уменьшает его сократительную способность. К препаратам, вызывающим спазм сфинктера Одди, относятся наркотические анал-гетики. Особенно это характерно для морфина и меперидина. При этом влияние опиатов на сфинктер Одди потенцируется галотаном и энфлураном. Точное значение повышения давления в желчевыводящих путях неясно. Имеющиеся данные позволяют сделать вывод, что это клинически малозначимо. Спазм желчевыводящих путей, возникающий под влиянием наркотических аналгетиков, купируется полным антагонистом опиатных рецепторов налоксоном, частично эффективны глюкагон, нитроглицерин, атропин. Эти данные свидетельствуют, что механизм спазма сфинктера Одди лишь отчасти обусловлен влиянием препаратов на нервную систему. Предполагается также вклад гистаминолибераторного эффекта наркотических аналгетиков. Этим может объясняться то, что спазм сфинктера особенно характерен после применения морфина и меперидина, гистаминолибераторные свойства которых выражены больше, чем у других опиоидов.
6.4. РОЛЬ ПЕЧЕНИ В ГЕМОПОЭЗЕ
6.4.1. Роль печени в эритропоэзе
С 9-й по 24-ю неделю внутриутробного развития гемопоэтическая активность плода связана преимущественно с печенью. Печеночный период кроветворения явля
ется вторым в последовательной смене кроветворных органов плода, сменяя желточный (первые очаги кроветворения появляются на 19-й день развития эмбриона в так называемых кровяных островках, расположенных в стенке желточного мешка).
212 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
После дифференциации костной ткани и костного мозга печеночный период сменяется костномозговым (медуллярным) кроветворением. С этого момента гемопоэтические клетки полностью исчезают из печени. Но при нарушении функции костного мозга или при миелопролиферативных заболеваниях активность гемопоэтических клеток в печени может восстанавливаться.
Роль печени в гемопоэзе также связана с ее участием в обмене витамина В12 и фолиевой кислоты. Давно замечено, что одним из эффективных антианемических средств является сырая печень и препараты, полученные из нее. Были проведены эксперименты по выявлению антиане-мического фактора, изучению его строения и механизма действия. Результатом стало выделение витамина Bt2. Печень является основным депо витамина В12 в организме, куда витамин поступает с пищей животного происхождения (печень, почки, сердце, мышцы и другие органы). Из печени витамин В12 попадает в костный мозг, где используется в процессах кроветворения.
Фолиевая кислота необходима для нормального протекания процессов образования эритроцитов и лейкоцитов. Печень накапливает активную форму расщепленной фолиевой кислоты в организме — тетрагидрофолат, участвующий в различных биохимических процессах.
Заболевание печени часто сопровождается анемией, чье развитие обусловлено следующими причинами: 1. Нарушается необходимый для нормального гемопоэза обмен веществ. 2. Эритроциты распадаются в большем, чем обычно, количестве. Это происходит вследствие часто сопутствующего патологии печени гиперспленизма, а также уменьшения длительности жизни эритроцита, поскольку состав и функции клеточной мембраны нарушаются в результате отклонений от нормы обменных процессов. 3. Развивается кровопотеря в связи с портальной гипертензией на фоне цирроза печени. Это может также обуславливаться сопутствующими язвами желудка и двенадцатиперстной кишки (особенно при алкоголизме). Анемию усугуб
ляют тромбоцитопения и нарушение свертывания крови.
Тяжелому поражению печени часто сопутствует лейкоцитоз. Он обусловлен нарушением функции системы мононуклеар-ных фагоцитов и проникновением микроорганизмов из просвета кишки в системный кровоток. В отдельных случаях этот же механизм является причиной лейкопении.
При патологии печени могут развиваться тромбоцитопения и тромбоцитопатия. Механизмы их следующие. 1. Часто сопутствующий гиперспленизм. 2. Нарушение клеточной мембраны тромбоцитов в связи с патологией обмена веществ, развитием ДВС-синдрома. Это ведет к нарушению агрегации тромбоцитов. 3. Нарушение образования тромбоцитов в костном мозгу вследствие нарушения обмена веществ. 4. У некоторых пациентов с вирусными гепатитами причиной тромбоцитопении может быть выработка антитромбо-цитарных антител.
6.4.2. Роль печени в обмене порфиринов
Гем является составной частью гемоглобина. Он синтезируется в большинстве тканей организма. В костном мозгу происходит его включение в состав гемоглобина. В других клетках гем используется при синтезе компонентов цепи переноса электронов — цитохромов и родственных им соединений. Промежуточными соединениями процесса образования гема являются порфирины (рис. 6.8). Синтез непосредственного предшественника гема -протопорфирина наиболее активно протекает в печени и в эритроцитах.
Группу метаболических заболеваний, при которых имеются дефекты ферментов биосинтеза гема, характеризующихся избыточным накоплением и экскрецией порфиринов и их предшественников, называют порфириями. Клинические их проявления разнообразны и могут варьировать от достаточно ярких до стертых. При этом главными клиническими проявлениями являются неврологические (психоз, вегетативная дисфункция, периферическая нейропатия, боль в животе) и кожные на-
Гуморальная функция печени 213
Глицин + Сукинил-КоА
I
5-Аминолевулиновая кислота
I
Порфобилиноген ........ / .......... 2
У ропорфириноген ..........♦... Копропорфириноген III ..............i 4 Протопорфириноген IX ..............i 5 Протопорфирин IX
6
Гем -> Гемоглобин
Рис. 6.8. Порфирии. Цифры указывают уровень дефекта фермента, обуславливающего: / — острую интермиттирующую порфирию; 2 — врожденную эритропоэтическую порфирию; 3 — позднюю кожную порфирию и гепатоэритропоэтическую порфирию; 4 — наследственную копропорфирию; 5 — вариегатную порфирию; 6 — эритропоэтическую протопорфирию
рушения (чаще всего фотосенсибилизация). Нейропсихические отклонения главным образом характерны для порфирий, в основе которых лежат дефекты ферментов, участвующих в начальных этапах метаболической цепочки синтеза гема. Дефекты ферментов и накопление субстратов на последующих этапах преимущественно ведут к кожным проявлениям. Различные дефекты синтеза гема лежат в основе выделения типов порфирий (табл. 6.3). А с учетом того, что синтез протопорфирина наиболее активно протекает в печени и эритроцитах, порфирии делят на печеночные и эритропоэтические. При этом для острых печеночных порфирий (острая перемежающаяся порфирия, наследственная копропорфирия и вариегатная порфирия)
Таблица 6.3. Классификация порфирий
Острая: нейропорфирия нейрокожная порфирия Острая перемежающаяся порфирия* Наследственная копропорфирия* Вариегатная порфирия*
Неострая (кожная) порфирия Поздняя кожная порфирия* Эритропоэтическая протопорфирия Врожденная эритропоэтическая порфирия
* Печеночные формы порфирий.
характерны обострения с нейропсихичес-кой симптоматикой, рвотой, абдоминальными коликами и периферической нейропатией. Важным моментом является то, что обострение возникает под воздействием лекарств, индуцирующих ферменты печени, включая препараты для наркоза — барбитураты, кетамин, пентазоцин, галотан. Факторами, вызывающими обострение печеночных порфирий, также могут быть алкоголь, пероральные контрацептивы, беременность, предменструальный период. Подобно гемофилии, печеночную порфирию называют «королевской болезнью», так как она передавалась по наследству в королевских домах Ганновера и Пруссии, а также в семье Стюартов (помешательство короля Англии Георга III связывали с этим заболеванием).
Поздняя кожная порфирия, а также эритропоэтические порфирии обычно не сопровождаются острыми неврологическими расстройствами. Препараты для наркоза на них влияния не оказывают. Основным клиническим проявлением этих видов порфирий являются поражения кожи, чаще всего фотосенсибилизация.
6.5. ГУМОРАЛЬНАЯ ФУНКЦИЯ ПЕЧЕНИ
Печень играет ведущую роль в трансформации многих гормонов. Так, она является основным местом разрушения инсулина: примерно 50 % инсулина, секретируемого поджелудочной железой, разрушается в печени после первого же пасса
жа крови. Превращение основного секреторного компонента щитовидной железы — тироксина (Т4) в активную форму — трийодтиронин (Т3) также осуществляется в печени. Здесь же может происходить и инактивация гормонов щитовидной же
214 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
лезы. Таким образом, печень оказывает влияние на суммарную активность и распределение гормонов щитовидной железы между внутри- и внеклеточным водным пространством.
Печень является основным местом инактивации стероидных гормонов — глюкокортикоидов, андрогенов, эстрогенов, альдостерона. Она активно влияет на гомеостатическую регуляцию уровня глюкокортикоидных гормонов; синтезирует специфический транспортный белок крови —
транскортину который связывает гидрокортизон, делая его временно неактивным. При патологии печени нарушается обмен глюкокортикоидных гормонов.
С участием высокоактивной моноами-ноксидазы и гистаминазы в печени инактивируются серотонин и гистамин. Вследствие поражения печени концентрация этих гормонов в крови может увеличиваться.
Таким образом, недостаточность печени сопровождается значительными эндокринными нарушениями.
6.6. ПЕЧЕНЬ И ЕСТЕСТВЕННАЯ ИММУННАЯ РЕАКТИВНОСТЬ
Печень представляет собой самый большой орган системы мононуклеарных фагоцитов. Более 10 % массы органа представлено клетками Купфера. Они являются подвижными макрофагами, образуются из моноцитов крови и имеют ограниченную способность к делению. Купферовские клетки выполняют защитную функцию, поглощая состарившиеся клетки, инородные частицы, опухолевые клетки, бактерии, дрожжи, вирусы и паразиты. Они захватывают и перерабатывают денатурированные белки, накапливающийся при ДВС-синдроме фибрин, а также атерогенные липопротеины низкой плотности. Купферовские клетки активируются при синдроме системного воспалительного ответа. При этом они вырабатывают и/или потенцируют медиаторы воспаления, выделяя фактор некроза опухолей, интерлейкины, коллагеназы, лизосомальные гидролазы и другие соединения. Этому способствует то, что данные клетки содержат специфические мембранные рецепторы, в частности, для фрагмента Fc иммуноглобулина и компонента С3ь комплемента, играющих важную роль в представлении антигена. Выделяющиеся при активации купферовских клеток биологически активные вещества оказывают как системное, так и локальное (печень) действие, хотя сам этиологический фактор (например, эндотоксин грамотрицательных микроорганизмов) может и не оказывать на печень токсического влияния. Эти процессы обуславливают увеличение границ печени, а так
же возможность нарушения функции печени при синдроме системного воспалительного ответа.
Купферовские клетки печени защищают системный кровоток от поступающих в воротную вену из просвета кишок микроорганизмов. Но нарушение функции системы мононуклеарных фагоцитов при печеночной недостаточности может сопровождаться попаданием бактерий в системный кровоток с последующим развитием бактериемии и сепсиса. Относительная недостаточность системы мононуклеарных фагоцитов печени может развиваться при критических состояниях любой этиологии. В таких случаях нарушение состояния слизистой оболочки кишок вследствие уменьшения кровотока и действия других повреждающих факторов сопровождается массивной транслокацией микроорганизмов из просвета кишок в воротную вену, превышающей способность печеночной системы мононуклеарных фагоцитов к их обезвреживанию. Кроме того, развитию сепсиса при критических состояниях способствует целый ряд дополнительных факторов, например, уменьшение функциональной активности полиморфноядерных лейкоцитов, снижение содержания в крови фибронектина, опсонинов, хемоаттрактантов и другие.
Недостаточность системы мононуклеарных фагоцитов может формироваться не только вторично — как следствие печеночной недостаточности или превышения функциональной активности системы в
Клинические и биохимические показатели функции печени 215
результате транслокации микроорганиз- системы мононуклеарных фагоцитов (на-мов из просвета кишок. В отдельных слу- пример, при поступлении в организм не-чаях инфекционные осложнения могут которых токсических веществ).
быть следствием первичного поражения
6.7. КЛИНИЧЕСКИЕ И БИОХИМИЧЕСКИЕ ПОКАЗАТЕЛИ ФУНКЦИИ ПЕЧЕНИ
6.7.1. Клинические симптомы нарушения функции печени
Асцит. Характеризуется накоплением свободной жидкости в брюшной полости. Возникающая при заболеваниях печени портальная гипертензия является не единственным фактором образования асцита. Его формирование сопровождается избыточным накоплением натрия в организме. Гипернатригистия со значительным накоплением натрия в межклеточном пространстве имеет место даже тогда, когда концентрация этого катиона в крови снижена. Несмотря на гипернатриемию функция почек направлена на сохранение натрия и суточная его экскреция с мочой уменьшена.
Задержка натрия при заболеваниях печени, предшествующая накоплению асцитической жидкости, предрасполагает к задержке жидкости в организме и формированию отечного синдрома. Наблюдающееся в последующем преимущественное накопление жидкости в брюшной полости связано с портальной гипертензией. Синусоидальная портальная гипертензия приводит к усилению продукции лимфы в печени. Лимфа из сосудов печени пропотевает в брюшную полость, и устанавливается динамическое равновесие с процессом всасывания ее в кишечные капилляры. Общее количество белка в асцитической жидкости меньше, чем в плазме, но соотношение белковых фракций сохраняется. Содержание белка в асцитической жидкости может увеличиваться при развитии у таких пациентов перитонита. При этом следует учитывать, что асцитическая жидкость может инфицироваться без видимой причины. Таким образом, анализ количества белка и качественного соотношения белковых фракций в асцитической жидкости является диагностическим критерием перитонита у больных с асцитом.
Основной теорией, объясняющей накопление натрия в организме при асците, является теория «недостаточного наполнения сосудистого русла». В соответствии с ней эффективный ОЦК, т. е. та часть ОЦК, которая реально участвует в циркуляции и оказывает регулирующее влияние на функцию волюморецепторов, при формировании асцита уменьшен. Уменьшение эффективного ОЦК обусловлено повышенным венозным давлением в портальной системе, расширением висцеральных и периферических сосудов с открытием артерио-венозных шунтов, гипоальбумине-мией. Этот процесс сопровождается активацией волюморецепторов. В результате активируется ренин-ангиотензин-альдосте-роновая система, наблюдается повышенная активность симпатической нервной системы с увеличением концентрации норадреналина, а также уменьшается образование вазодилататора почечных сосудов — простагландина Е2. Следствием активации симпатической нервной системы и уменьшения образования простагландина Е2 является повышение тонуса сосудов почек, а в результате активации альдостерона происходит усиленная реабсорбция натрия в канальце нефрона.
Боль. Чувство тяжести, давления и ноющие боли в правом подреберье возникают вследствие растяжения фиброзной оболочки печени и преимущественно характерны для вирусного гепатита, застойной сердечной недостаточности, внепеченочного холестаза. Боли в правом подреберье могут быть также связаны с воспалительным поражением капсулы печени, спаечным процессом между фиброзной оболочкой и париетальной брюшиной, раком, абсцессом печени.
Геморрагический диатез. Может быть обусловлен дефицитом витамин-К-зависи-мых факторов свертывания крови вслед
216 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
ствие внутри- и внепеченочного холестаза (холемический геморрагический диатез) и недостаточностью самого гепатоцита. Обычно вначале появляется склонность к образованию петехий, что связано с дефицитом факторов протромбинового комплекса. Позднее появляется склонность к образованию гематом. Это вызвано усугублением дефицита факторов протромбинового комплекса, дефицитом других факторов свертывания крови, а в некоторых случаях — развитием ДВС-синдрома.
Гепаторенальный синдром. Характеризуется уменьшением выделения мочи, азотемией. При этом часто имеет место гипонатриемия, сниженное общее сосудистое сопротивление, иногда с развитием гипотензии. Исследованиями морфологические нарушения в почках обычно не выявляются, что свидетельствует о функциональной природе почечной недостаточности. Это подтверждается также фактами восстановления функции почек после трансплантации печени.
В основе патогенеза лежит ограничение скорости клубочковой фильтрации вследствие вазоконстрикции сосудов. Из-за исходной почечной вазоконстрикции незначительная кровопотеря или перераспределение крови, даже без существенного снижения системного артериального давления (например, кровопотеря при варикозно расширенных венах пищевода, при применении диуретиков, при парацентезе при асците, диарее, рвоте) может приводить к быстрому прогрессированию гепаторенального синдрома.
Механизм почечной вазоконстрикции окончательно не ясен. Он связан с уменьшением эффективного ОЦК так, как это имеет место при асците. Но восполнение ОЦК инфузионными средами с увеличением почечного кровотока дает кратковременный эффект. Поэтому уменьшение эффективного ОЦК является не единственным механизмом гепаторенального синдрома. Его реализация обусловлена также дисбалансом между сосудосуживающим простагландином тромбоксаном А2 и сосудорасширяющим простагландином простациклином Е2, вырабатываемым в почках. В развитии гепаторенального син
дрома участвуют такие сосудосуживающие соединения, как эндотелин-1, эндотелин-2, лейкотриены, NO, а со стороны сосудов выявлено повышение чувствительности к сосудосуживающему действию аденозина. Дисбаланс между сосудосуживающими и сосудорасширяющими соединениями обуславливает наблюдающееся у больных преобладание наряду с почечной вазоконстрикцией расширения артерий во внепо-чечных сосудах и склонность к возникновению гипотензии.
Желтуха. Одним из важнейших симптомов поражения печени является желтуха. Она может быть обнаружена при уровне билирубина в сыворотке крови 34 мкмоль/л и становится явной при билиру бинемии 120 мкмоль/л. Раньше всего она выявляется на склерах и слизистой оболочке мягкого неба. В отдельных случаях желтушное окрашивание бывает парциальным — в области носогубного треугольника, лба, ладоней. При интенсивной желтухе с наличием прямого билирубина цвет кожи со временем становится зеленовато-желтым из-за окисления билирубина в биливердин. Дифференциальная диагностика различных видов желтух приведена ниже.
Желтуха возникает преимущественно вследствие неспособности гепатоцитов метаболизировать билирубин. Поэтому это заболевание в некоторой степени является маркером недостаточности функции гепатоцита. Если же основным механизмом формирования недостаточности печени является развитие портокавального шунта, то желтуха может вообще отсутствовать.
Истощение. Обусловлено нарушением метаболизма в печени, синтеза белка в тканях, а также анорексией и неправильным режимом питания.
Ксантомы. Это внутрикожные желтые бляшки, обычно располагающиеся в ладонных складках, под молочными железами, на шее, груди или спине. Разновидностями ксантом являются ксантелазмы -плоские или слегка возвышающиеся мягкие образования желтого цвета вокруг глаз. На поздних этапах поражения печени возможно появления туберозных ксантом. Они обычно локализуются на раз
Клинические и биохимические показатели функции печени 217
гибательных поверхностях, особенно в области лучезапястных, локтевых, коленных суставов, лодыжек, ягодиц, в местах, подвергающихся давлению, в рубцах. При этом сухожильные влагалища поражаются редко, но могут поражаться кости, периферические нервы. Происхождение ксантом связано с повышением содержания в крови липидов, особенно при длительном холестазе. Ксантомы могут наблюдаться также при других заболеваниях, сопровождающихся гиперлипидемией: атеросклерозе, сахарном диабете, гипотиреозе, эссенциальной гиперлипидемии.
Кожный зуд. Холестаз сопровождается кожным зудом. Долгие месяцы и годы зуд может оставаться единственным симптомом заболевания. Его возникновение связывают с депонированием желчных кислот в печени. Однако в последнее время появились сообщения, опровергающие подобное представление: с помощью точных биохимических тестов не удается выявить корреляцию между степенью зуда и концентрацией эндогенных желчных кислот в сыворотке и коже; в терминальной стадии печеночной недостаточности кожный зуд может исчезать, в то время как концентрация желчных кислот в сыворотке остается повышенной. Предполагается, что возникновение зуда связано с синтезирующимися в печени и экскретирующимися в желчь соединениями, воздействующими на центральные нейротрас-миттерные механизмы, в частности эндогенные опиоидные пептиды.
Лихорадка. Во многих случаях поражения печени развивается лихорадка. Она может вызываться такими факторами: 1. Непосредственное инфекционное поражение печени. Так, температура может повышаться в преджелтушной стадии острого вирусного гепатита. 2. Транслокация микроорганизмов в системный кровоток при холангите. В таких случаях повышение температуры может быть ремитирующим или гектическим с ознобом. 3. Транслокация микроорганизмов в системный кровоток из просвета кишки при тяжелом поражении печени. Причины бактериемии при печеночно-клеточной недостаточности могут быть следующие: нарушение
функции клеток Купфера; проникновение микроорганизмов из воротной вены в системный кровоток через портосистемные коллатерали; часто сопутствующее недостаточности функции печени нарушение функции полиморфноядерных лейкоцитов; снижение содержания в сыворотке защитных факторов — фибронектина, опсонинов и хемоаттрактантов, в том числе компонентов каскада комплемента. 4. Формирование у больных с недостаточностью печени отдаленных очагов инфекции. При этом особенно часто присоединяются инфекции мочевыводящих путей и пневмонии.
Малиновый язык. Этот симптом характеризуется ярко-красной окраской языка, имеющего гладкую, как бы лакированную поверхность. Обусловлен нарушением обмена витаминов.
Печеночная энцефалопатия. Печеночная кома. Печеночная энцефалопатия представляет собой нейропсихический синдром, проявляющийся нарушением сознания и поведения, перепадами настроения, расстройствами интеллекта, а также неврологическими расстройствами, развивающимися вследствие прогрессирования заболеваний печени, портальной гипертензии или искусственного создания портокавальных анастомозов. В качестве клинического описания печеночной энцефалопатии может быть использована клиническая классификация нейропсихических отклонений при патологии печени, в соответствии с которой выделяют следующие стадии: первая стадия проявляется спутанностью сознания, нарушением поведения, перепадами настроения, расстройствами интеллекта. Спутанность сознания на этой стадии характеризуется расстройствами сна, уменьшением числа спонтанных движений, фиксированным взглядом, заторможенностью, апатией, краткостью ответов. Изменения личности проявляются ребячливостью, игривым настроением, эйфорией, раздражительностью. Их связывают с вовлечением в процесс лобных долей головного мозга. Расстройства интеллекта характеризуются нарушением оптико-пространственной деятельности, включая нарушение гностической компоненты (узна
218 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
вание пространственной фигуры или стимула) и конструктивной компоненты (воспроизведение фигуры). При начальной стадии такие отклонения можно обнаружить, применив специальные психометрические тесты, при использовании которых выявляется нарушение начертания букв, неспособность скопировать простой узор и пр.
Вторая стадия характеризуется сонливостью, неадекватным поведением. Сонливость при патологии печени появляется относительно рано. При прогрессировании патологии развивается инверсия нормального ритма сна и бодрствования. Неадекватность поведения может проявляться такими крайними вариантами отклонения, как мочеиспускание и дефекация в неподходящих местах.
Третья стадия характеризуется ступором, дизартрией, выраженной спутанностью сознания.
При прогрессировании заболевания развивается четвертая стадия, главным признаком которой является печеночная кома. Иногда термин «печеночная кома» употребляется в широком смысле, охватывая все клинические проявления церебральной недостаточности при патологии печени. В таком случае выделяют следующие стадии печеночной комы: прекома, развивающаяся кома, ступор, кома.
Печеночная энцефалопатия может быть эпизодической, со спонтанным разрешением или хронической, с неуклонным прогрессированием. Различают печеночную энцефалопатию с острым и постепенным началом. Сложным в диагностике является острое течение, для которого также
характерна высокая вероятность летального исхода. В таких случаях психоневрологический дефицит может развиваться в течение нескольких часов после действия повреждающего фактора.
Механизм возникновения печеночной энцефалопатии окончательно не исследован. Его связывают с нарушением обмена церебральных нейротрансмиттеров. Причиной является расстройство обмена веществ в связи с печеночно-клеточной недостаточностью, нарушение детоксицирующей функции печени по отношению к веществам, поступающим из кишок, а также вследствие шунтирования оттекающей от кишок крови. Нейротрансмиттеры, нарушение функции которых выявлено при печеночной энцефалопатии, приведены в табл. 6.4.
Механизм портокавальной энцефалопатии, роль аммиака и глутамина, ложных нейротрансмиттеров, системы триптофан/ серотонин в развитии нарушения функции головного мозга при патологии печени изложены выше. Развитие печеночной энцефалопатии связывают также с нарушением обмена у-аминомасляной кислоты (ГАМК) в ЦНС. ГАМК представляет собой основной тормозной медиатор в головном мозгу. Она синтезируется в пре-синаптических окончаниях из глутамата при помощи глутаматдегидрогеназы и накапливается в везикулах. ГАМК связывается со специфическим ГАМК-рецептором на постсинаптической мембране. В составе ГАМК-рецептора выделяют собственно ГАМК-субъединицу, субъединицу связывания с барбитуратами и субъединицу связывания с бензодиазепинами. Свя
Таблица 6.4. Нейротрансмиттеры, участвующие в патогенезе печеночной энцефалопатии
Нейро-трасмиттеры Действие в норме Нарушение при печеночной энцефалопатии
Глутамат ГАМК Дофамин Норадреналин Серотонин Возбуждение Угнетение Моторная / когнитивная функция Состояние сознания и цикла сон — бодрствование Уменьшение связывания NH3 Относительное увеличение активности ГАМК Угнетение ложными нейротрансмиттерами (ароматические аминокислоты) Дисфункция ЦНС
Клинические и биохимические показатели функции печени 219
зывание рецептора с любым из лигандов сопровождается открытием хлорных каналов, после поступления в клетку ионов хлора развивается гиперполяризация постсинаптической мембраны и происходит торможение нервных импульсов. Предполагается, что ГАМК, синтезируемая кишечными бактериями, при патологии печени поступает в портальный кровоток, достигает ЦНС и принимает участие в развитии энцефалопатии. Имеются данные, что при печеночной недостаточности могут накапливаться эндогенные бензодиазепины, которые также обуславливают развитие энцефалопатии. Последнее объясняет повышенную чувствительность больных циррозом печени к бензодиазепинам, а также служит обоснованием попыток применения антагониста бензодиазепинов флумазенила для терапии печеночной энцефалопатии. К сожалению, результирующий эффект оказывается временным.
Факторами, потенциально способствующими возникновению печеночной энцефалопатии, являются алкалоз и гипокалиемия. В связи с этим требует осторожности применение салуретиков у пациентов с тяжелым поражением печени.
Возможность развития энцефалопатии и комы вследствие портокавального шунтирования является основанием выделения трех клинико-патогенетических вариантов печеночной комы: 1. Эндогенная печеночно-клеточная (острая дистрофия печени, истинная кома). 2. Экзогенная (портокавальная, шунтовая, обходная, вторичная, ложная кома). 3. Смешанная.
Эндогенная печеночно-клеточная кома обусловлена тяжелыми дистрофическими и некротическими изменениями паренхимы печени. Ее причинами являются вирусный гепатит, отравление гепатотропны-ми ядами (четыреххлористый углерод, те-трахлорэтанол, нитраты толуола, ядовитые грибы, сульфаниламиды, галотан). Экзогенная кома чаще всего развивается вследствие портокавального анастомозирования у больных циррозом печени. При этом обычно разрешающими факторами являются повышенное потребление белка, желудочно-кишечное кровотечение, нерацио
нальное лечение диуретиками, эвакуация асцитической жидкости, наличие острого алкогольного гепатита. Изолированные клинико-патогенетические варианты энцефалопатии встречаются редко. Обычно можно выделить лишь преимущественный механизм развития нейропсихического дефицита.
«Печеночный запах». Имеет сладковатый характер. Такой запах формирует выдыхаемый больным воздух. Этим запахом могут также обладать пот и рвотные массы. Печеночный запах имеет преимущественно кишечное происхождение, поскольку он ослабевает после дефекации и энтерального применения антибиотиков. Его возникновение обусловлено нарушением обмена аминокислот и ароматических соединений, в частности накоплением продукта превращения метионина—метил-меркаптана. Это вещество образуется при подавлении нормального деметилирования поврежденной печенью.
«Печеночные ладони». Пальмарная эритема — симметричное пятнистое покраснение ладоней и подошв, особенно выраженное в области тенара и гипотена-ра, иногда сгибательных поверхностей пальцев. Пятна бледнеют при надавливании и быстро восстанавливают окраску при прекращении давления. Пальмарная эритема чаще всего наблюдается у больных диффузными заболеваниями печени, но также встречается при беременности, септическом эндокардите, тиреотоксикозе. Предполагается, что печеночные ладони (как и сосудистые звездочки) обусловлены артерио-венозными анастомозами вследствие гиперэстрогенемии и/или нарушения соотношения эстрогенов и андрогенов.
Расширение вен на передней стенке живота. Расширенные вены на передней брюшной стенке живота при патологии печени являются анастомозами между системами воротной вены и нижней и верхней полыми венами. Анастомозы вокруг пупка носят название «голова медузы». При портальной гипертензии кровь из воротной вены по коллатералям в брюшной стенке, расположенным выше пупка, поступает в верхнюю полую вену, по коллате
220 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
ралям ниже пупка — в нижнюю полую вену. При затруднении тока крови по нижней полой вене развиваются коллатерали между системами нижней и верхней полых вен, располагающимися в боковых отделах брюшной стенки.
Сосудистые звездочки. Известны также под названием «паучки», «телеангиэктазии», «звездчатые ангиомы». Состоят из пульсирующей центральной части и лучеобразных разветвлений сосудов, напоминающих ножки паука. Центральная артерия звездчатой ангиомы под эпидермисом ампулообразно расширяется, выступает над кожей и распространяется в форме звезды. Сосудистые звездочки располагаются в сосудистом бассейне верхней полой вены и очень редко — ниже линии, соединяющей соски. Чаще всего они выявляются на шее, лице, предплечьях, тыльной стороне кисти. Довольно часто их можно обнаружить на слизистой оболочке верхнего неба, рта, глотки, реже — носа. Размеры сосудистых звездочек колеблются от 1 мм до 1 — 2 см. При достаточно большом размере звездочки можно наблюдать или пальпировать ее пульсацию.
Сосудистые звездочки обнаруживаются преимущественно при активных поражениях печени: остром и хроническом активном гепатите, циррозе печени, циррозе-раке. Иногда единичные сосудистые звездочки возникают у здоровых людей, чаще всего на 2 —5 месяце беременности, а после родов в течение двух месяцев они исчезают. Улучшение функционального состояния печени сопровождается уменьшением количества сосудистых звездочек или их исчезновением. Кроме того, звездочка может исчезать при уменьшении артериального давления вследствие шока или кровотечения. Из звездочки может возникать профузное кровотечение.
Механизм возникновения сосудистых звездочек связывают с повышением количества эстрогенов. Это объясняет возможность появления сосудистых звездочек во время беременности. Эстрогены способствуют увеличению и дилатации спиральных артерий эндометрия. Возможно, аналогичный механизм лежит в основе возникновения и кожных звездочек при пе
ченочной недостаточности: печень инактивирует эстрогены. Существует точка зрения, что механизм формирования звездочек обусловлен не столько увеличением концентрации эстрогенов, сколько нарушением соотношения эстрогенов и андрогенов.
Феминизация. Q повышением активности эстрогенов связана также наблюдающаяся у некоторых больных, преимущественно у пациентов с активным циррозом печени, феминизация. У женщин эти изменения выражены в меньшей степени. Феминизация проявляется гинекомастией, формированием женского типа оволосения. У отдельных больных может развиваться снижение либидо и потенции, гипогонадизм, выпадение волос в местах вторичного оволосения. У женщин может нарушаться овуляция. В пременопаузе исчезают признаки женского телосложения, особенно отложение жира в молочных железах и в области таза. Обычно эти женщины бесплодны, менструации нерегулярные, скудные или отсутствуют, но иногда могут быть обильными.
«Хлопающий» тремор. Наиболее характерным неврологическим признаком печеночной энцефалопатии является «хлопающий» тремор (астерексис). Он связан с нарушением поступления афферентных импульсов от суставов и других частей опорно-двигательной системы в ретикулярную формацию ствола мозга, что приводит к неспособности удерживать позу. «Хлопающий» тремор особенно хорошо выявляется на вытянутых руках с расставленными пальцами или при максимальном разгибании кисти больного с фиксированным предплечьем. При этом иногда наблюдаются быстрые сгибательно-разгибательные движения в пястно-фаланговых и лучезапястных суставах, что сопровождается латеральным движением пальцев. Иногда гиперкинез захватывает всю руку, шею, челюсть, высунутый язык, плотно сомкнутые веки, появляется атаксия при ходьбе. Обычно тремор двусторонний, но не синхронный. Он может быть более выражен на одной стороне тела, чем на другой. Во время комы тремор исчезает.
Клинические и биохимические показатели функции печени 221
Увеличение печени. Наиболее частый симптом поражения печени может быть обусловлен непосредственным поражением гепатоцитов, холестазом, очаговыми поражениями печени при абсцессах, кистах, опухолях, а также развитием регенераторных узлов и фиброза при циррозе печени, застоем крови при сердечной недостаточности, эндофлебите печеночных вен. За край правой доли печени можно принять новообразования желчного пузыря, толстой кишки, правой почки. Имитировать гепатомегалию может также гепато-птоз. Эмфизема легких, экссудативный плеврит, поддиафрагмальный абсцесс приводят к смещению печени книзу. Отличить действительное увеличение печени от этих состояний позволяет пальпация при различных положениях больного, а также инструментальные методы исследования.
В отдельных случаях может наблюдаться сокращение размеров печени в динамике заболевания. Это является следствием не только благоприятного течения заболевания, но и развития массивных некрозов и может свидетельствовать о плохом прогнозе.
Увеличение селезенки при заболеваниях печени связано с портальной гипертензией и системной гиперплазией моно-нуклеарно-фагоцитарной ткани печени и селезенки. Рост селезенки наблюдается при портальной гипертензии; при некоторых формах цирроза она по размеру больше печени; при этих же состояниях обычно имеет место и гиперспленизм. Наоборот, при застое в печени селезенка обычно увеличивается незначительно, и гиперспленизм отсутствует.
В развитии гепатолиенального синдрома болезням печени принадлежит ведущая роль: более чем в 90 % случаев он обусловлен патологией именно печени.
6.7.2. Биохимические показатели нарушения функции печени
При выявлении заболеваний печени не следует переоценивать значение однократного биохимического тестирования. Подтверждением этого является тот факт, что биохимический скрининг практически здо
ровых людей с отсутствием признаков поражения печени в 6 % случаев выявляет нарушение биохимических показателей, отражающих состояние печени, тогда как в целом частота встречаемости заболеваний печени в общей популяции составляет около 1 %. В то же время при подтвержденных заболеваниях печени отклонения со стороны биохимических показателей могут отсутствовать или быть выражены незначительно. Например, у пациентов с вирусным гепатитом С признаки поражения печени могут вообще отсутствовать вплоть до формирования выраженного поражения органа. Кроме того, ряд внепеченочных факторов оказывает влияние на показатели биохимических тестов, обычно используемые для оценки функции печени (табл. 6.5).
Аминотрансферазы. Аспартатаминотрансфераза (АсАТ) и аланинаминотрансфераза (АлАТ) являются ферментами, катализирующими перенос а-аминогрупп аспартата и аланина на а-кетогруппы а-ке-тоглутаровой кислоты. АлАТ преимущественно локализована в печени, тогда как Ас АТ содержится в большинстве тканей, включая сердце, скелетные мышцы, почки, головной мозг. АсАТ расположена в митохондриях и цитозоле гепатоцитов, а АлАТ — главным образом в цитоплазме.
Увеличение активности аминотрансфераз является признаком цитолиза печеночных клеток и выхода ферментов в кровоток. При этом диагностическое значение имеет не только само по себе увеличение активности ферментов в сыворотке, но и степень увеличения их активности (табл. 6.6). Чувствительность лабораторных методов обнаружения увеличения активности аминотрансфераз в сыворотке возрастает при использовании дополнительного разведения биологического субстрата, подвергаемого анализу.
Умеренное увеличение активности АсАТ и АлАТ в сыворотке часто обнаруживается у пациентов при жировой инфильтрации печени, неалкогольной стеаторее, хронических вирусных гепатитах. Более высокие степени увеличения активности ферментов регистрируются при острых вирусных гепатитах, некрозе печени в резуль-
222 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
Таблица 6.5. Внепеченочные причины нарушения функции биохимических показателей, обычно используемых для анализа функции печени
Показатель Внепеченочные причины отклонения от нормы Дифференциальнодиагностический критерий
Уменьшение концентрации альбумина Увеличение активности АсАТ Увеличение активности АлАТ Увеличение концентрации билирубина Увеличение протромбинового времени (уменьшение протромбинового индекса) Потери белка, например, протеинтеряющая энтеропатия Нефротический синдром Сердечная недостаточность Инфаркт миокарда Заболевания скелетной мускулатуры Заболевания костей Беременность Злокачественные новообразования Гемолиз Применение антибиотиков, антикоагулянтов, стеаторея, дефицит поступления витамина К Клиренс агантитрипсина, глобулинов плазмы Анализ мочи Клиническое и инструментальное исследование сердца Креатинкиназа, тропонин, ЭКГ Креатинкиназа Гамма-глутамилтранспептидаза, лейцинаминопептидаза, 59-нуклеотидаза Гамма-глутамилтранспептидаза, 59-нуклеотидаза, хорионический гонадотропин в моче и сыворотке Электрофорез щелочной фосфатазы, ан-тиэритроцитарные антитела Количество ретикулоцитов, лактатдегидрогеназа, гаптоглобин Ответная реакция на введение витамина К
Таблица 6.6. Нормальные величины и степени отклонения активности ферментов печени в сыворотке крови
Показатель Норма, ед/л* Степень увеличения по сравнению с нормой, раз
Умеренная Средняя Выраженная
АсАТ 11-32 < 2-3 2-20 > 20
АлАТ 3-30 < 2-3 2-20 > 20
Щелочная фосфатаза 35-105 < 1,5-2 1,5-5 > 5
Гамма-глутамилтранспептидаза 2-65 < 2-3 2-10 > 10
Нормальная величина зависит от используемого метода регистрации.
тате действия лекарственных препаратов (например, ацетаминофена) или токсинов, ишемии печени вследствие циркуляторного шока. При этом степень увеличения активности аминотрансфераз не всегда коррелирует со степенью некроза, выявляемого при биопсии печени, и поэтому полноценного диагностического значения не имеет. Но быстрое снижение активности в динамике заболевания может отражать массивный некроз печени и является плохим прогностическим признаком.
Диагностическое значение имеет не только увеличение активности аминотрансфераз, но и отношение Ас АТ : Ал АТ. В частности, отношение 2 : 1 чаще встречается у пациентов с алкогольным поражением печени. В то же время увеличение АлАТ свыше 500 ед/л обычно встречается и в отсутствие алкогольного поражения печени, даже при отношении 2:0. Увеличение активности АсАТ без пропорционального роста АлАТ является следствием отличий в восстановлении печеночной АлАТ в связи
Клинические и биохимические показатели функции печени 223
с дефицитом кофактора пиридоксин-5-фос-фата. При вирусном гепатите отношение АсАТ : А л АТ обычно 1 : 0, но в динамике может существенно возрастать при формировании фиброза и цирроза печени.
Щелочная фосфатаза. Термином «сывороточная щелочная фосфатаза» обозначают группу ферментов, катализирующих гидролиз фосфат-эфиров при щелочной реакции pH. Эти ферменты встречаются во многих тканях и органах, в частности в печени, костях, кишках, почках, плаценте. При заболеваниях печени увеличение активности плазменной щелочной фосфатазы обычно отражает поступление фермента в кровоток и является маркером холестаза. Существенное увеличение активности щелочной фосфатазы чаще всего наблюдается при таких заболеваниях печени и желчевыводящих путей, как внепеченочная обструкция желчевыводящих путей, индуцированный приемом лекарственных средств холестаз, первичный склерозирующий холангит, инфильтративные процессы в печени (амилоидоз, поражение печени при гематологических заболеваниях). Вследствие широкого распространения этого фермента его активность может увеличиваться и при других физиологических и патологических состояниях. Так, у детей с активным ростом костного скелета щелочная фосфатаза в норме может быть увеличена до трех раз. При врожденной гипо-фосфатазии, гипотиреоидизме, пернициозной анемии, дефиците цинка активность щелочной фосфатазы может быть уменьшена.
Гамма-глутамилтранспептидаза. Принято считать, что гамма-глутамилтранспептидаза имеет диагностическую ценность в таких случаях:
1. Ее активность может увеличиваться при холестазе. Поэтому определение этого фермента используется для дифференциальной диагностики увеличения активности щелочной фосфатазы и исключения увеличения концентрации щелочной фосфатазы костного происхождения.
2. Изолированное увеличение активности фермента может наблюдаться у людей, злоупотребляющих алкоголем, даже при отсутствии грубых изменений печени. Это, возможно, является следствием индук
ции активности микросомальных ферментов. В таких случаях часто отмечается жировая дистрофия печени. При фиброзе, циррозе и гепатите алкогольной этиологии одновременно с повышением активности гамма-глутамилтранспептидазы в сыворотке возрастает активность и других печеночных ферментов.
Гамма-глутамилтранспептидаза является малоспецифичным маркером заболеваний печени и желчевыводящих путей. Это обусловлено тем, что активность фермента может увеличиваться при достаточно широком спектре заболеваний: при почечной недостаточности, инфаркте миокарда, заболеваниях поджелудочной железы, сахарном диабете. Повышение активности фермента может быть обусловлено также индукцией ферментов печени некоторыми лекарственными препаратами, в частности барбитуратами и фенитоином (ди-лантином).
Гамма-глутамилтранспептидаза также является малоценным диагностическим маркером скрытого алкоголизма. Это обусловлено тем, что период полужизни данного фермента составляет 26 дней. Таким образом, однократный прием алкоголя с увеличением активности фермента может быть обоснованием ложно-положительного диагноза скрытого алкоголизма. В то же время у трети пациентов, злоупотребляющих алкоголем, активность фермента остается неизменной. Выявление повышенной активности гамма-глутамилтранспептидазы нередко служит поводом для проведения необоснованно большого количества исследований у пациентов, не употребляющих алкоголь или употребляющих его лишь изредка и не страдающих алкогольной зависимостью.
5'-Нуклеотидаза в печени взаимосвязана с каналикулярными и синусоидальными плазматическими мембранами. Вследствие этого увеличение активности фермента в крови отражает повреждающее влияние желчных кислот на плазматические мембраны и наблюдается при холестазе. Чаще всего исследование активности 5'-нуклеотидазы используется для верификации причины увеличения активности щелочной фосфатазы. При этом результаты
224 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
исследования более специфичны, чем регистрация активности гамма-глутамил-транспептидазы.
Альбумин и факторы свертывания крови. Большинство циркулирующих в крови белков, в том числе и альбумин, синтезируется в печени. Концентрация альбумина в плазме зависит от интенсивности его синтеза (в организме взрослого этот показатель составляет 12 г/день), скорости его элиминации из кровотока и объема плазмы. Таким образом, уменьшение скорости синтеза альбумина, возрастание интенсивности его элиминации из крови или увеличение объема плазмы может сопровождаться гипоальбуминемией. Уменьшение концентрации альбумина может отмечаться при нарушении белково-синтетической функции печени, при асците, больших потерях белка у пациентов с заболеванием почек, патологией пищеварительного канала и других состояниях. Вследствие длительного периода полувыведения концентрация альбумина при остром поражении печени может оставаться в нормальных пределах.
В отличие от альбумина факторы свертывания крови относительно быстро элиминируются из организма. Поэтому отклонения в показателях системы свертывания крови являются более ранним биохимическим маркером нарушения белково-синтетической функции печени, чем концентрация альбумина. При этом наименьшим периодом циркуляции в кровотоке (4 — 6 часов) обладает VII фактор свертывания крови (проконвертин). Изменения его концентрации сказываются на величине протромбинового времени (протромбинового индекса). Таким образом, увеличение протромбинового времени (уменьшение протромбинового индекса) может быть ранним признаком нарушения синтеза факторов свертывания крови в печени. В некоторых случаях до или одновременно с уменьшением активности VII фактора снижается концентрация IX фактора свертывания крови (фактор Кристмаса). Его дефицит приводит к удлинению активированного частичного тромбопластинового времени (АЧТВ). Этим объясняется необходимость оценки у пациентов с за
болеваниями печени наряду с протромбиновым временем и других показателей системы свертывания крови.
Увеличение протромбинового времени может наблюдаться также и в отсутствие нарушений белково-синтетической функции печени у больных с холестазом вследствие дефицита в организме витамина К. В таких случаях дифференциально диагностическим критерием может быть реакция организма на прием витамина К (10 мг/день на протяжении 3 суток подкожно или внутривенно). Сдвиг показателей минимум на 30 % от исходного в сторону нормализации на протяжении 24 часов является признаком дефицита витамина К.
Сывороточный билирубин. Изменения биохимических показателей при желтухах. Билирубин является эндогенным органическим анионом (см. выше). Образуется преимущественно в результате катаболизма гема эритроцитов, в меньшей степени — вследствие разрушения миоглобина, цитохромов, каталазы, пероксидазы. При образовании связывается с альбумином и транспортируется к печени, где происходит процесс связывания его с глюкуроновой кислотой и экскреция в желчь. У практически здоровых людей концентрация общего билирубина составляет менее 17 мкмоль/л, неконъюгированного (непрямого) — 12 мкмоль/л, конъюгированного (прямого) — 5 мкмоль/л. Патологией является не только увеличение общего билирубина, но и увеличение концентрации конъюгированного билирубина, даже при условии нормальной концентрации общего билирубина. Гипербилирубинемия может быть обусловлена следующими механизмами:
1. Повышенная нагрузка билирубина на гепатоциты.
2. Нарушение захвата и переноса билирубина в гепатоцит.
3. Нарушение процессов конъюгации билирубина в гепатоците.
4. Нарушение экскреции его в желчь.
Гипербилирубинемию с окрашиванием тканей и органов в желтый цвет называют желтухой. В зависимости от клинико-патофизиологической характеристики выделяют три типа желтухи:
Клинические и биохимические показатели функции печени 225
Таблица 6.7. Типичные клинические и лабораторные признаки различных типов желтух
Признак Тип желтухи
Гемолитическая Печеночноклеточная Внутрипеченочный холестаз Внепеченочный холестаз
Часто встречающиеся жалобы Головная боль, боль в суставах Тошнота, рвота, лихорадка, анорексия Желтуха, темная моча, обесцвеченный кал,зуд Желтуха, темная моча, обесцвеченный кал, зуд, печеночная колика
Физикальные данные Спленомегалия Увеличение и болезненность печени,спленомегалия Увеличение и болезненность печени Увеличение печени, симптомы раздражения желчного пузыря
Концентрация общего билирубина, мкмоль/л < 100 Вариабельна Вариабельна (может превышать 500 мкмоль/л) Обычно < 500 мкмоль/л
Прямой билирубин, % общего < 20 > 50 > 50 > 50
АлАТ Норма Увеличение Увеличение Возможно небольшое увеличение
Щелочная фосфатаза Норма Возможно небольшое увеличение Значительное увеличение Значительное увеличение
Протромбиновое время Норма Удлинено Удлинено Удлинено
Коррекция протромбинового времени при назначении витамина К Не применяется Нет Вариабельный ответ Да
Расширение желчных протоков при ультразвуковом исследовании Нет Нет Нет Да
1. Надпеченочная.
2. Печеночно-клеточная.
3. Подпеченочная (холестатическая).
Первый тип вызывается повышенным образованием билирубина при гемолизе и некоторых наследственных нарушениях обмена билирубина. При этом билирубин в основном представлен неконъюгирован-ной фракцией. В моче билирубин не выявляется. Активность сывороточных трансаминаз и щелочной фосфатазы в пределах нормы.
Печеночно-клеточная желтуха возникает вследствие нарушения функции гепа-
Q
° 4-220
тоцита. При этом увеличивается концентрация не только неконъюгированного, но и конъюгированного билирубина. В зависимости от механизма развития желтухи выявляются другие патохимические и клинические признаки печеночно-клеточной недостаточности (увеличение активности трансаминаз, нарушение белково-синтетической функции печени и др.).
Холестатическая желтуха развивается при нарушении поступления желчи в кишки. При этом в сыворотке повышается концентрация конъюгированного билирубина, активность печеночной фракции
226 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПЕЧЕНИ
щелочной фосфатазы, гамма-глутамил-транспептидазы, уровень общего холестерина и конъюгированных желчных кислот. Вследствие стеатореи уменьшается масса тела и нарушается всасывание витаминов A, D, Е, К, а также кальция, что приводит к соответствующим клиническим и пато-химическим нарушениям. Типичные клинические и лабораторные проявления различных типов желтух представлены в табл. 6.7.
В динамике заболевания четкость клинических симптомов, являющихся опорными точками дифференциальной диагностики различных видов желтух, может стираться в связи с формированием новых звеньев патогенеза. Например, исходно жел
туха может быть обтурационной. Но в динамике заболевания может страдать сам гепатоцит. Это ведет к возникновению признаков паренхиматозной желтухи.
Антинуклеарные антитела. Для хронического гепатита характерным является вовлечение иммунных факторов в непрерывное повреждение печеночных клеток. При этом иммунная реакция направлена против компонентов мембраны гепатоцита, выступающих в качестве антигенов. Антинуклеарные антитела обнаруживаются у 75 % пациентов с хроническим активным гепатитом, а антимитохондриальные антитела — практически во всех случаях первичного билиарного цирроза.
Список использованной литературы
1. Бахман А. Л. Искусственное питание: Пер. с англ. — СПб.: Изд-во БИНОМ — Невский диалект, 2001. — 192 с.
2. Гальперин Э. И., Семендеева М. И., Неклюдова Е. А. Недостаточность печени. — М.: Медицина, 1978. — 328 с.
3. Зилва Дж. Ф., Пэннел П. Р. Клиническая химия в диагностике и лечении: Пер. с англ. — М.: Медицина, 1988. — 528 с.
4. Мак-Мюррей У. Обмен веществ у человека: Пер. с англ. — М.: Мир, 1980. — 366 с.
5. Подымова С. Д. Болезни печени. — М.: Медицина, 1984. — 480 с.
6. Шерлок Ш. Ю., Дуй Дж. Заболевания печени и желчных путей: Практ. руководство: Пер. с англ. — М.: Гэотар Медицина, 1999. -864 с.
7. Fluids and electrolytes /Ed. J. P. Kokko, R. L. Tannen. — Philadelphia: W. B. Saunders Company, 1986. — 878 p.
8. Maze M. Hepatic physiology // Anesthesia / Ed. R. D. Miller. — New York: Churchill Livingstone, 1990. — P. 585 — 590.
9. Maze M., Prager M. C. Anesthesia and the liver // Anesthesia / Ed. R. D. Miller. — New York: Churchill Livingstone, 1990. — P. 590 — 600.
— Раздел -----
7 Клиническая физиология пищеварительного канала
Одним из основных симптомов заболевания пищеварительного канала является боль — субъективное ощущение, формируемое центральными структурами периферической импульсации.
Восприятие боли начинается с активации ноцицепторов, содержащих свободные окончания малых А8- и С-афферентных волокон. Сильная механическая стимуляция, высокая и низкая температура могут активировать эти рецепторы. Кроме того, вещества, образующиеся в месте повреждения или воспаления, такие, как брадикинин, гистамин, серотонин и простагландины, либо непосредственно активируют болевые рецепторы, как, например, брадикинин, либо снижают порог их чувствительности к другим стимулам. А8- и С-волокна передают разные типы болевой чувствительности: А8-волокна иннервируют кожу и мышцы и отвечают за передачу интенсивной, локальной боли, например при остром воспалении, С-волокна иннервируют мышцы, надкостницу, париетальную брюшину и внутренние органы и передают импульсы тупой и недостаточно локализованной боли, обычно длительного и непостоянного характера.
Внешняя иннервация пищеварительного канала осуществляется парасимпатическими и симпатическими нервами, по которым происходит передача информации через афферентные (сенсорные) и эфферентные волокна. Сенсорная афферентация от кишок передается по афферентным волокнам блуждающего нерва или
8*
спинномозговым афферентным волокнам. Центральное звено вагусной афферента-ции находится в ядрах солитарного тракта, а эфферентные волокна проходят на периферию в составе блуждающего нерва. Центральное звено спинномозговой аффе-рентации заканчивается в задних рогах спинного мозга, а эфферентные волокна идут на периферию в составе симпатических нервов. Блуждающий нерв не передает боль от кишок, поскольку висцеральная боль передается только по спинномозговым афферентным путям. Эти нервные волокна часто идут в составе симпатических нервов. Клетки задних рогов, передающие боль, получают информацию также от периферических ноцицептивных волокон. Эта двойная иннервация лежит в основе ощущения иррадиирующей боли, которая может сопровождать висцеральные боли.
Высшая нервная деятельность может оказывать сильное угнетающее действие на восприятие боли. Нисходящие волокна, берущие начало в среднем мозгу, пери-вентрикулярном сером веществе и хвостатом ядре, образуют синапсы с различными структурами афферентных путей передачи боли. Эти волокна участвуют в угнетении передачи болевой чувствительности. Нейроны этой системы имеют опиатные рецепторы, а в вышеупомянутых отделах мозга отмечены высокие концентрации эндорфинов. Антагонист морфина, налоксон, снимает угнетение боли, возникающее вследствие активации данной системы.
228 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
Органы брюшной полости обычно не чувствительны ко многим стимулам, которые при действии на кожу провоцируют сильную боль. Так, парез, разрывы или другие повреждения внутренних органов не вызывают болевых ощущений. Основными воздействиями, к которым чувствительны висцеральные болевые волокна, являются растяжения или напряжение стенки кишок: натяжение брюшины (при опухолях), растяжение полого органа (желчная колика), сильное мышечное сокращение (при кишечной непроходимости). Нервные окончания волокон, отвечающих за боль в полых органах (кишки, желчный и мочевой пузыри), локализуются в мышечных слоях. В паренхиматозных органах (печень, почки) нервные окончания нахо
дятся в капсулах и реагируют на растяжение органа при увеличении его объема. Брыжейка, париетальная плевра и перитонеальная брюшина чувствительны к боли, тогда как висцеральная плевра и большой сальник — нет. Для появления боли скорость нарастания напряжения должна быть достаточно большой. Постепенное нарастание напряжения, например при опухолевой обструкции желчных путей, может долго оставаться безболезненным.
Воспаление и ишемия способны вызвать висцеральную боль. Более того, воспаление повышает чувствительность нервных окончаний и снижает порог чувствительности к боли от других стимулов.
7.1. ДИСФУНКЦИЯ ВЕРХНИХ ОТДЕЛОВ ГЛОТКИ
Многие заболевания центральной нервной системы приводят к дисфагии (нарушению глотания). Поскольку нервные структуры, ответственные за иннервацию глоточной части участка прохождения пищевого комка, расположены в стволе мозга, наиболее тяжелые расстройства глотания наблюдаются при инфарктах в этой зоне мозга. Основные наблюдаемые нарушения при поражении мозга включают: выраженное снижение способности к сокращению мышц глотки при глотании, недостаточный контроль со стороны языка за пищевым комком из-за нарушения иннервации мускулатуры языка и потери координированного проталкивания пищи, плохое очищение глотки и аспирация пищевых масс. Результатом дисфагии вследствие цереброваскулярных заболеваний могут быть аспирационная пневмония, дегидратация, потеря массы и даже смерть. Другие заболевания, при которых нарушаются механизмы глотания, включают полиомиелит, способствующий дизартрии и дисфагии вследствие развития слабости мышц глотки, и амиотрофический латеральный склероз с повышенной аспирацией во время и после глотания, возникающий из-за плохого очищения глотки.
Мышечные нарушения, например мышечная дистрофия, могут быть причиной дисфагий за счет вовлечения мышц языка и глотки. Другие состояния, при которых имеются нарушения, клинически похожие на мышечную дистрофию, включают миастению gravis, а также состояния, вызывающие сухость слизистой рта (ксерото-мию).
К основным двигательным расстройствам, поражающим тело пищевода, относятся ахалазия, диффузный спазм пищевода и склеродермия. Ахалазия характеризуется потерей тормозной нервной регуляции гладких мышц тела пищевода. Диффузный спазм пищевода, так же, как и ахалазия, обусловлен потерей тормозного контроля за гладкой мускулатурой пищевода при сохранении нормальной функции нижнего сфинктера.
При склеродермии, относящейся к первичным коллагенозам, поражаются как кожа, так и внутренние органы, наиболее часто — пищевод. Прогрессирующее замещение гладкой мускулатуры пищевода плотной фиброзной тканью приводит к постепенной потере пищеводной перистальтики и заметному снижению давления нижнего пищеводного сфинктера.
Пищевод 229
Основным проявлением дисфункции нижнего пищеводного сфинктера (НПС) является гастроэзофагальный рефлюкс. Снижение давления в этом сфинктере уменьшает эффективность эзофагально-
гастрального барьера, поскольку возникает несоответствующее расслабление НПС, сопровождаемое превышением абдоминального давления над давлением нижнего пищеводного сфинктера.
7.2. ПИЩЕВОД
В процесс продвижения пищи к пищеводу вовлекаются ротовая полость и гортань. Стенки верхних отделов глотки имеют три группы констрикторных мышц — верхнюю, среднюю и нижнюю. Данные констрикторы совместно с мышцами гортани предназначены для поднятия гортани от входа в пищевод, при этом мышцы средней и нижней части глотки сокращаются в задне-переднем направлении. Эти мышцы имеют плотную иннервацию (отношение нервных волокон к мышечным составляет от 1 : 2 до 1 : 6), что обеспечивает исключительно хорошую регуляцию. Центральный контроль и рефлекторная активация нейронов определяют последовательность мышечных сокращений и акт глотания.
Пищевод — полый орган, всегда остающийся пустым, несмотря на прием пищи и рефлюкс, — представляет собой 20 — 22-сантиметровую трубку, стенки которой состоят из гладкой и поперечнополосатой мускулатуры: проксимальный отдел пищевода (5 %) имеет только поперечнополосатую мускулатуру, в то время как средний его участок (35 — 40 %) содержит оба типа мышц, дистальная часть (50 — 60 %, включая нижний пищеводный сфинктер), состоит из гладкой мускулатуры. В отличие от других отделов пищеварительного канала пищевод не имеет серозной оболочки. Внешняя иннервация осуществляется через блуждающий нерв.
Перистальтика пищевода возникает сразу после того, как глоточные сокращения пройдут через верхний пищеводный сфинктер. Средняя скорость перистальтики 2 —4 см/с. Первичная перистальтика стимулируется глотанием, в то время
как вторичная — запускается растяжением пищевода дистальнее области растяжения.
Физиологические механизмы контроля за сокращением поперечнополосатой мускулатуры пищевода осуществляются главным образом за счет возбуждающего действия блуждающего нерва, вызванного сокращением поперечнополосатой мускулатуры пищевода.
Перистальтика гладкой мускулатуры пищевода отличается от перистальтики поперечнополосатой вследствие различий в иннервации для каждой группы мышц.
Внутренний контроль перистальтики гладкой мускулатуры осуществляется разветвленной интрамуральной сетью. Участие этой сети в регуляции перистальтики подтверждается тем, что после перерезки блуждающего нерва перистальтика сохраняется. Возбуждающие нейроны стимулируют сокращение продольных и циркулярных мышечных слоев через холинергические М2-рецепторы. Ингибирующие нейроны преимущественно локализуются в циркулярном мышечном слое и угнетают сокращения через нехолинергические и неадренергические нейротрансмиттеры. Основными тормозными нейротрансмиттерами считают вазоактивный интестинальный пептид и NO. Холинергическая стимуляция возбуждающих нейронов происходит через никотиновые рецепторы, в то время как нехолинергическое и неадренергическое возбуждение может осуществляться через никотиновые и мускариновые рецепторы. Оба типа нейронов иннервируют тело пищевода и нижний эзо-фагальный сфинктер.
230 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
7.3. ДИАГНОСТИКА И ЛЕЧЕНИЕ АБДОМИНАЛЬНОЙ БОЛИ, ЗАБОЛЕВАНИЙ ГЛОТКИ И ПИЩЕВОДА
При абдоминальных болях необходим тщательный сбор анамнеза, внимательное обследование больного и проведение общих лабораторных обследований. Полученная информация поможет установить диагноз или определит дальнейшие диагностические мероприятия.
Острая боль — главный фактор в клинической оценке абдоминальной боли. Ее появление требует от врача принятия быстрого решения о выборе неотложных дополнительных диагностических процедур, т. е. лабораторного и рентгенологического исследования, что поможет определить степень срочности лапаротомии. Если симптомы существуют в течение недель или месяцев, то осуществление диагностического алгоритма может быть не столь срочным.
Пациента необходимо тщательно опросить, обращая внимание на следующие аспекты: локализация боли; характер и интенсивность боли; ее продолжительность и качество; факторы, усиливающие и уменьшающие болевые проявления; дополнительные симптомы и признаки. Клинический анамнез не всегда четко указывает на причину абдоминальной боли. Для установления предварительного и проведения дифференциального диагноза необходимо тщательное обследование пациента.
Нарушения общего характера могут прояснить причину боли. Тахикардия, лихорадка, одышка позволяют предположить наличие сепсиса, перитонита, холангита, пиелонефрита и др. Необходимо определить наличие вздутия живота или асцита. Повышенная перистальтика главным образом характерна для энтерита или ранних стадий странгуляционной непроходимости, ее понижение более характерно для перитонита. Выслушивание сосудистых шумов свидетельствует о наличии сосудистых аневризм.
Ригидность мышц живота — как локальная, так и общая — всегда должна насторожить врача и до выяснения причины ее возникновения больного нельзя оставлять без наблюдения.
У каждого больного с абдоминальной болью необходимо провести обследование половых органов, а также ректальное обследование. При этом нередко выявляется острое воспаление тазовых органов, перекрут кисты яичника или фиброма матки, а при ректальном обследовании — опухоли, абсцессы и скрытая кровь.
Обычные лабораторные исследования кроме клинических анализов крови должны включать исследования биохимических изменений, коагулогического и иммунологического статуса организма. Лейкоцитоз в клиническом анализе крови свидетельствует о воспалении, а анемия позволяет предположить желудочно-кишечное кровотечение. Анализ мочи помогает исключить опухоли или камни в почках, а также инфекции мочевыделительной системы. Повышение уровня амилазы и липазы позволяет диагностировать панкреатит. Повышение уровня щелочной фосфатазы и билирубина предполагает патологию либо поджелудочной железы, либо билиарного тракта, в то время как повышение активности трансаминаз свидетельствует о повреждении гепатоцитов. Остальные тесты должны быть проведены исходя из клинической картины больного.
В последние годы в диагностике заболеваний пищеварительной системы важное место занимают специальные методы диагностики, такие как рентгенография пищеварительного канала, эндоскопическое исследование как верхних его отделов, так и желудка, тонкой и толстой кишок. Пероральная холецистография, ультразвуковое обследование, эндоскопическая ретроградная холецистопанкреография помогают в диагностике заболеваний поджелудочной железы или билиарной системы. Селективная мезентериальная ангиография позволяет выявить стеноз мезентериальных артерий у пациентов, страдающих от болей в кишках. Пищеводная монометрия полезна для дифференциальной диагностики загрудинной боли, а гастродуоденальная — применяется для оценки нарушений двигательной активности соот
Диагностика и лечение абдоминальной боли, заболеваний глотки и пищевода 231
ветствующих отделов пищеварительного канала.
Лечение абдоминальных болей. Зависит от диагноза, тяжести и длительности этого состояния. При диагнозе «острый живот» методом выбора считается лапаротомия.
В лечении абдоминальных болей часто с успехом применяют лекарственные препараты, причем они более эффективны при лечении острых болей, чем при лечении хронического болевого синдрома.
Для нейтрализации болей предложено большое число методов. Это свидетельствует, с одной стороны, об актуальности проблемы, а с другой — о том, что до настоящего времени не найден способ полной ликвидации болевого синдрома. Существующие методы можно разделить на три группы: немедикаментозные, гомеопатические и фитотерапевтические, фармакотера-певтические.
К первой группе относятся: классическая акупунктура и ее разновидности, свето-, термо- и лазеропунктура, ИВЧ-терапия, аутотренинг с биологической обратной связью. Акупунктура как метод аналгезии используется давно, однако в случае невропатического болевого синдрома она недостаточно эффективна. Согласно данным В. Н. Цыбуляка с соавт. [1], отмечается только 59 % эффект от использования акупунктуры.
ИВЧ-терапия — это новый метод физиотерапии, заключающийся в облучении биологически активных точек волнами миллиметрового диапазона нетепловой мощности.
Основным методом лечения болей остается фармакологический. В. В. Чурюка-новым предложена классификация обезболивающих методов в зависимости от локализации и механизма действия [2].
Различают вещества с преимущественно периферическим и преимущественно центральным действием. К первым относятся ингибиторы циклооксигеназы в периферических тканях и ЦНС-нестероид-ные противовоспалительные препараты — ненаркотические аналгетики (ацетилсалициловая кислота, аналгин, кеторолак).
Вещества с преимущественно центральным действием подразделяются на три
группы: наркотические аналгетики (агонисты — антагонисты опиоидных рецепторов — морфин, бупреморфин), неопиоидные средства с аналгетической активностью и вещества со смешанным механизмом действия (трамадол). Во второй группе выделяют такие классы веществ: а2-ад-реномиметики (клофелин), антагонисты возбуждающих аминокислот (кетамин, амантадин), ГАМК-Б-миметики (баклофен), ингибиторы возвратного захвата серотонина (амитриптилин, флуоксетин), блокаторы натриевых канальцев (карбамазепин, мексилетин) [3].
В некоторых случаях при хронических абдоминальных болях может быть полезным использование антидепрессантов. Их роль в лечении хронической нейропатической боли давно известна. Однако механизм их действия выяснен не полностью. Считается, что аналгетический эффект обусловлен преимущественно блокадой повторного захвата серотонина, вследствие чего усиливаются сегментарные и супрасегментарные механизмы контроля боли. Недостаток серотонина приводит не только к возникновению боли, но и к развитию депрессивных явлений, являющихся постоянными спутниками хронической боли. Поэтому использование антидепрессантов при хронических болях преследует две цели: во-первых, обезболивающий эффект, во-вторых, устранение депрессивного состояния. Установлено, что антидепрессант амитриптилин блокирует NMDA-рецепторы, чем можно объяснить аналгетический эффект препарата [4].
Иногда для лечения хронических болей применяется чрескожная электростимуляция нервов. Этот метод предназначен для стимуляции выходящих путей в задних корешках спинного мозга, которые, в свою очередь, активируют интернейроны и таким образом снижают передачу ноцицептивных импульсов.
Лечение больных с хроническими болями может быть разным. Терапия боли осуществляется с учетом прогноза жизни пациента. Больные, находящиеся в терминальном состоянии с неблагоприятным прогнозом, должны получать наркотики и
232 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
антидепрессанты без учета особенностей привыкания к ним.
Терапия доброкачественных заболеваний более сложна. В этих случаях необходимо тщательно следить за назначением лекарственных препаратов, вызывающих привыкание. Основная цель лечения — помочь больному адаптироваться к болезни.
Пациенты с дисфагией и болью в грудной клетке должны быть тщательно обследованы для максимально точного диагноза и лечения.
Наличие в анамнезе кашля, охриплости голоса, регургитации в нос, а, возможно, и серьезных заболеваний центральной нервной системы помогают выявить расстройства на уровне глотки.
Нарушение глотания твердой пищи предполагает наличие механической не
проходимости пищевода, если же нарушается прохождение и жидкой пищи, то это скорее свидетельствует о нарушении перистальтики пищевода. Дисфагия при глотании твердой и жидкой пищи указывает на нарушение двигательной активности пищевода.
Лечебные мероприятия при дисфункциях глоточного сегмента и пищевода в целом направлены на устранение нервных расстройств, явившихся причиной дисфагии или затруднения глотания жидкой или твердой пищи. В случае наличия опухолей — как доброкачественных, так и злокачественных, — главным методом лечения считается оперативный. Из медикаментозных препаратов целесообразно назначение спазмолитиков, при необходимости следует провести антибиотикотера-пию.
7.4. ЯЗВЕННАЯ БОЛЕЗНЬ
Язвенная болезнь представляет собой группу гетерогенных заболеваний, общим проявлением которых является локальный дефект или эрозирование в слизистой оболочке желудка или двенадцатиперстной кишки. Это очень распространенная патология, которая, например, в США встречается у 10 % мужчин и около 5 % женщин. В Украине количество больных язвенной болезнью еще больше и, согласно данным некоторых статистиков, составляет до 20 % страдающих заболеваниями органов пищеварительного канала.
В последние два десятилетия основные усилия ученых были направлены на изучение патогенеза язвенной болезни, что позволило значительно улучшить диагностику и лечение этого распространенного заболевания.
Язвенная болезнь является результатом нарушения соотношений между защитными (секреция слизи, простагландинов, гидрогенкарбонатов, кровообращение, клеточное обновление) и повреждающими (кислота, пепсин, желчные кислоты, панкреатические ферменты, бактерии) факторами. Старое правило Шварца — «нет кислоты — нет язвы» — до сих пор считается вер
ным для большинства случаев язвенной болезни двенадцатиперстной кишки, хотя для язвенной болезни желудка это не обязательно. Причинами формирования язвы также являются: бактериальная инфекция (Helicobacter pylori), употребление некоторых нестероидных лекарственных препаратов (нестероидные противовоспалительные средства), курение, наследственность, нарушение эвакуации пищи из желудка, что в совокупности и приводит к дисбалансу между повреждающими и защитными факторами в желудке и двенадцатиперстной кишке.
Слизистая желудка является основным элементом, который подвергается повреждению и участвует в защитных механизмах при язвенной болезни. В защитном барьере желудка клетки слизистой оболочки являются первой линией защиты от повреждающего фактора, особенно поверхностные клетки, секретирующие слизь и гидрогенкар-бонаты. Этот барьер представляет собой гель, имеющий в норме градиент pH. Гель состоит из неперемешивающихся слоев слизи, гидрогенкарбонатов, фосфолипидов и воды.
Гидрогенкарбонаты необходимы для поддержания pH, близкого к нормально
Язвенная болезнь 233
му, у поверхности эпителия. Важную роль в поддержании базального уровня секреции гидрокарбонатов играют эндогенные простагландины. Механизм снижения секреции гидрокарбонатов до конца не выяснен, хотя недавние исследования показали возможное участие в этом процессе Helicobacter pylori.
В поддержании устойчивости слизистой желудка важную роль играют: способность клеток к репарации, хорошее состояние микроциркуляции и секреция некоторых химических медиаторов защиты, таких как простагландины и факторы роста (эпидермальный фактор роста (ЭФР) и а-транс-формирующий фактор роста (а-ТФР). Простагландины, имеющиеся в слизистой оболочке желудка, могут секретироваться главными, добавочными и париетальными клетками. Простагландины (в частности, простагландин Е2) способствуют защите слизистой посредством угнетения активности париетальных клеток, стимулирования секреции слизи и гидрогенкарбона-тов, увеличения кровотока в слизистой, снижения обратной диффузии ионов Н+ и угнетения клеточного обновления.
Повреждающие факторы слизистой оболочки включают соляную кислоту, пепсин, Helicobacter pylori и другие агенты.
Базальная секреция соляной кислоты является циркадным циклом, с наименьшим уровнем секреции утром и максимальным — ночью. Секреция кислоты в желудке подчиняется холинергической регуляции через блуждающий нерв и гис-таминергической — через локально выделяющийся гистамин. Важнейшим физиологическим стимулятором секреции соляной кислоты является пища.
Процесс пищевой стимуляции секреции кислоты традиционно делится на три фазы: сложнорефлекторную, желудочную и кишечную. Основные стимуляторы секреции кислоты в желудке — гистамин, гастрин и ацетилхолин. Среди множества факторов, угнетающих секрецию кислоты, наиболее важными являются простагландины и соматостатин. Как стимуляторы, так и ингибиторы процесса секреции кислоты в желудке, действуют через специфические рецепторы, находящиеся на обкладочных клетках.
Гистамин, выделяющийся в основном из энтерохромаффинных клеток слизистой желудка, стимулирует секрецию кислоты через Н2-рецепторы, взаимодействующие с цАМФ. Гастрин и ацетилхолин активируют спицифические рецепторы, связанные с системой кальций /протеинкиназа С. После активации соответствующих механизмов происходит стимуляция водород-но-калиевых (Н+/К+) АТФ-азных каналов, приводящая к продукции и выделению ионов Н+.
Базальная секреция кислоты у больных с язвенной болезнью двенадцатиперстной кишки либо нормальная, либо повышенная. В то же время максимально стимулированная секреция кислоты у таких больных значительно повышена.
Главные клетки, преимущественно расположенные в железах слизистой дна желудка, вырабатывают пепсиноген — неактивный предшественник протеолитического фермента пепсина. Патогенетическая роль нарушений выработки пепсиногена в механизме язвенной болезни выяснена не до конца.
В последнее время доказана связь между развитием язвенной болезни и инфи-цированностью желудка Helicobacter pylori — грамотрицательной аэробной палочкой, имеющей жгутик и способной синтезировать уреазу. Ее обнаружили в слизистой оболочке желудка у 95 % больных язвенной болезнью. Этот возбудитель иногда выявляют и у здоровых людей. Лечение гастрита и дуоденальной язвы препаратами, действующими на Н. pylori, например бисмутсодержащими и антибиотиками, приводит к клиническим и морфологическим признакам выздоровления. Исследования показали возможность как прямого, так и непрямого повреждения слизистой желудка. Уреаза, липополисахариды и цитотоксин, вырабатываемые Н. pylori, в свою очередь, могут активировать провос-палительные факторы.
Нестероидные противовоспалительные средства являются важным повреждающим слизистую оболочку фактором, поскольку они угнетают выработку простагландинов, необходимых для защиты слизистой оболочки. Курение также способ
234 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
ствует развитию язвы двенадцатиперстной кишки. Роль в ульцерогенезе других факторов, таких как стресс, нарушение питания и кортикостероиды, не достаточно доказана.
7.4.1. Клиническое обследование
Хотя обычные клинические обследования не дают важной информации при язвенной болезни, необходимо провести клинический анализ крови, а также определить в ней уровень кальция и креатинина для'исключения соответственно кровопотери, гиперкальциемии и почечной недостаточности. Специальные лабораторные исследования следует проводить у тех больных, у которых язвенная болезнь предположительно обусловлена гиперсекреторным синдромом. Эти исследования включают определение уровня гастрина в крови провокационными пробами для стимуляции выброса гастрина (стимуляция секретином, прием пищи), уровня в плазме пепсиногена 1 и анализ кислотности желудочного сока. Учитывая важную роль Helicobacter pylori в патогенезе язвенной болезни, необходимо выявить наличие или отсутствие этого микроорганизма. Методы, используемые для диагностики Н. pylori, разделяют на инвазивные (эндоскопические) и неинвазивные. Инвазивные методы на основе эндоскопической диагностики включают быструю диагностику (окраска биоптата по Граму и тест на уреазу) и отсроченную диагностику (гистологическое исследование, бактериологическое исследование в культуре).
К неинвазивным методам диагностики относят: тест на мочевину в выдыхаемом воздухе, серологическое исследование на антителах к Helicobacter pylori, ферментативную иммуносорбцию (против цельной ослабленной культуры Н. pylori и против высокомолекулярного протеина), иммунодиффузию, связывание комплемента, реакцию пассивной гемагглютинации.
Окончательный диагноз язвенной болезни устанавливается на основании данных рентгенологического или эндоскопического обследования больных.
7.4.2. Терапия
Целью терапевтических мероприятий при неотложной язвенной болезни является: снижение болевого синдрома, стимуляция заживления язвы, профилактика рецидивов язвы и ее осложнений. Наряду с применением противоязвенной терапии больному следует прекратить курение и отказаться от применения нестероидных противовоспалительных препаратов.
В последнее десятилетие резко возросло количество противоязвенных препаратов. Фундаментальные исследования механизмов секреции кислоты привели к созданию препаратов, снижающих кислотность. Изучение защитных механизмов желудка способствовало появлению группы препаратов, повышающих защитные свойства слизистой без влияния на секрецию кислоты в желудке. Кроме того, выявление роли Helicobacter pylori в патогенезе язвенной болезни привело к разработке методов антибиотикотерапии, направленных против этого микроорганизма.
Используемые в лечении язвенной болезни препараты разделяют на такие группы:
1. Ингибиторы кислоты (нейтрализующие препараты).
2. Антациды. Первыми препаратами, ускоряющими процесс заживления язвы, были антациды, снижающие кислотность желудочного сока. Однако они имеют достаточно много недостатков, к которым следует отнести необходимость частого приема, нарушение моторики кишок, проявляющееся диареей, стимулированной магнием, или запорами, вызванными алюминием, связывание фосфатов солями алюминия. В связи с внедрением в клиническую практику Н2-антагонистов антацидные препараты отошли на второй план при лечении язвенной болезни.
3. Н2-Антагонисты. Как упоминалось выше, существуют три основных стимулятора обкладочных клеток: гастрин, ацетилхолин, гистамин. Для каждого из этих активных веществ имеются антагонисты, но наиболее активными оказались антагонисты Н2-гистаминовых рецепторов, снижающие как базальную, так и стимулиро
Язвенная болезнь 235
ванную секрецию кислоты. Обычно используют такие препараты, как циметидин, ранитидин, фамотидин и низатидин. Наиболее важное значение в патогенезе развития язвенной болезни двенадцатиперстной кишки имеет секреция кислоты в ночное время. С учетом этого можно заменить многоразовые приемы препаратов на одноразовый прием перед сном. Через четыре недели после использования ^-антагонистов заживление дуоденальных язв наблюдается приблизительно в 80 % случаев, а при отсутствии лечения — только в 40 %. Применение этих препаратов в соответствующих дозах дает минимальный побочный эффект. Наиболее часто осложнения связаны с передозировкой Н2-анта-гонистов (в основном циметидина) и приводят к нарушениям метаболизма препаратов из-за повреждения цитохрома с в печени. Хронические передозировки могут способствовать развитию гинекомастии, расстройствам сознания: чаще всего подобные нарушения наблюдаются у пожилых пациентов, страдающих печеночной или почечной недостаточностью.
4. Простагландины. Препараты аналогов простагландинов (мизопростол), так же, как и циметидин, эффективны для заживления дуоденальных язв. В некоторых случаях, особенно при воспалении гастродуоденальной слизистой оболочки, вызванном нестероидными противовоспалительными препаратами, простагландины могут быть более эффективны, чем Н2-антаго-нисты.
Основными побочными эффектами являются диарея и сокращение матки, которые при беременности могут быть причиной выкидыша.
5. Ингибиторы Н+-К+-АТФ-азы. Как уже упоминалось, основным ферментом, отвечающим за выработку ионов Н+, является Н+-К+-АТФ-аза обкладочных клеток. Основным препаратом из ингибиторов данного фермента является омепразол (производное бензимидазола), предотвращающий активацию Н+-К+-АТФ-азы ковалентным связыванием с дисульфидными группами. В настоящее время это наиболее сильный ингибитор секреции кислоты в желудке. Выраженная гипохлоргидрия
вследствие применения омепразола часто сопровождается гипергастринемией, клиническая значимость которой при этом не совсем ясна.
Первоначально эти препараты рассматривались как резервные для лечения больных с язвенной болезнью, рефрактерной к Н2-антагонистам, или больных с синдромом Золлингера —Эллисона, но в последнее время их применяют для сокращения сроков лечения дуоденальных язв.
6. Антихолинергические препараты. При использовании самостоятельно неселективные антихолинергические средства имеют второстепенное значение в лечении язвенной болезни, так как слабо угнетают секрецию кислоты. Кроме того, у них имеется достаточно много негативных эффектов.
Кроме вышеперечисленных при лечении язвенной болезни используются препараты, не влияющие на кислотность.
1. Сукральфат — смесь сульфатированных дисахаридов. Препарат стимулирует заживление язвы и по клиническому эффекту сопоставим с Н2-антагонистами. Предполагается, что он усиливает защитный гастродуоденальный барьер.
2. Коллоидный бисмут. Препарат стимулирует заживление язвы и по эффекту не отличается от действия Н2-антагонис-тов. Наблюдаемый эффект препарата способствует иногда более длительной ремиссии течения болезни, чем при использовании Н2-антагонистов.
3. Антибиотики. Эффективное лечение против Helicobacter pylori значительно снижает вероятность рецидива язвенной болезни. Стандартная терапия включает применение комбинации из трех препаратов: метронидазола, амоксициклина или тетрациклина и препаратов бисмута в течение 7 — 10 дней. Такая терапия приводит к положительному эффекту более чем в 85 % случаев. Однако следует учитывать, что подобная комплексная терапия может осложниться побочным эффектом антибиотиков (тошнота, диарея, колит). В последнее время применение этих препаратов стараются сократить. Предложена другая схема лечения, включающая высокие дозы омепразола в сочетании с одним антибиотиком (амоксициклин).
236 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
7.4.3. Хирургическое лечение язвенной болезни
Имеются определенные показания для хирургического лечения язвенной болезни: желудочно-кишечные кровотечения, стеноз пилорического отдела желудка, перфорация или малигнизация язвы. Другими возможными показаниями к оперативному лечению могут быть периодические небольшие кровотечения, пенетри-рующая язва или отсутствие эффекта от консервативной терапии. Хирургические операции включают: резекцию антрального отдела желудка в сочетании с ваготомией; ваготомию в сочетании с пилоро-пластикой; высокую селективную ваготомию.
Цель всех этих операций заключается в уменьшении стимуляции секреции кислоты. Ваготомия снижает холинергическую регуляцию желудка и чувствительность париетальных клеток к гастрину, а резекция антрального отдела приводит к удалению основного источника гастрина. Тип операции выбирается с учетом специфических признаков при оценке локализации язвы.
7.4.4. Кровотечения из пищеварительного канала
Кровотечения из пищеварительного канала — проблема, с которой довольно часто сталкиваются врачи. Степень кровопотери варьирует от небольших медленных кровотечений до угрожающих жизни состояний. Смертность от кровотечений из верхней части пищеварительного канала, например в США, составляет 8 %.
Главную роль в успешном лечении кровотечений играет быстрая оценка тяжести состояния больного и осуществление мероприятий по стабилизации гемодинамики.
Существует множество причин кровотечений из пищеварительного канала, однако преимущественно кровотечение развивается вследствие таких причин:
1. Нарушение целостности слизистой оболочки, приводящее к обнажению глубоких сосудов, их эрозии (например, кровотечение из язвы желудка, из кишок при
инфекционных или идиопатических процессах, из толстой и тонкой кишок при ишемии).
2. Разрыв сосудов при резком повышении в них давления и обширном повреждении стенки кишки (например, кровотечение из дивертикула).
Клиническая картина кровотечения зависит от его остроты и локализации. При хронических кровотечениях выявляется железодефицитная анемия, кровь в стуле. В более тяжелых случаях хронического кровотечения наблюдаются анемия, бледность кожных покровов, слабость, одышка, ангинозные боли.
При остром кровотечении картина очень яркая. Кровотечение из верхних отделов пищеварительного канала проявляется рвотой с кровью, причем при свежем кровотечении рвотные массы окрашены в красный цвет, а при повторных — в цвет «кофейной гущи». Мелена представляет собой черный, кашицеобразный либо твердый зловонный стул. Его темная окраска обусловлена распадом гемоглобина и служит маркером кровотечения из верхних отделов пищеварительного канала либо из правых (восходящих) отделов поперечноободочной кишки. Гематомизия — выделение неизмененной крови из прямой кишки со стулом — служит показателем кровотечения из нижних отделов пищеварительного канала. Если имеет место гематомизия при установлении в верхнем отделе пищеварительного канала источника кровотечения, то это свидетельствует о массивности кровотечения.
При осмотре больного с кровотечением необходимо быстро оценить тяжесть его состояния. Возбуждение, бледность, понижение артериального давления, тахикардия могут указывать на развитие геморрагического шока.
В случае острого кровотечения важно обеспечить внутривенный доступ, а при признаках шока и продолжающемся кровотечении часто появляется необходимость в использовании двух или более вен, одна из которых должна быть центральной. Следует немедленно определить гематокрит, гемоглобин, количество тромбоцитов, факторы свертывания крови, группу кро
Язвенная болезнь 237
ви и сразу начать введение замещающих растворов.
При остром кровотечении уровень гематокрита слабо отражает реальный объем кровопотери, так как гематокрит — это выраженное в процентах соотношение суммарного объема эритроцитов к общему объему крови. Поэтому величина гематокрита снизится лишь после восстановления объема циркулирующей жидкости.
7.4.5. Перфоративные язвы желудка и двенадцатиперстной кишки
Достаточно частым осложнением течения язвенной болезни желудка и двенадцатиперстной кишки следует считать перфорацию. Клиника, диагностика и виды оперативного лечения данного осложнения описаны в специальных руководствах по хирургии.
Важнейшей задачей интенсивной терапии в раннем послеоперационном периоде является коррекция метаболических нарушений, связанных с главным патологическим процессом, а также возникающих в процессе оперативного лечения нарушений основных систем гомеостаза: купирование болевого синдрома, коррекция изменений сердечно-сосудистой и дыхательной систем, нормализация водно-солевого и кислотно-основного состояния, восстановление энергетического баланса, стимуляция иммуногенеза, обеспечение в этот период потребностей организма в питательных факторах.
В связи с увеличением продолжительности жизни людей возросло число лиц пожилого и преклонного возраста, нуждающихся в хирургической помощи. Послеоперационное лечение этой группы больных имеет свои особенности.
Ведение послеоперационного периода у больных, прооперированных в сроки до шести часов после перфорации, не отличается от общепринятого и включает в себя купирование послеоперационных болей. Чаще всего используются наркотические аналгетики (промедол, морфий) по 1 мл 4 — 5 раз в сутки. В последнее время потенцирование аналгетического эффекта обеспечивается дополнительным введени
ем клофелина дважды в сутки, что позволяет снизить прием аналгетиков до 1 — 3 раз в сутки.
Водно-электролитный баланс поддерживается введением жидкости из расчета 40 мл/кг массы тела, 7,5 % растворов КС1 из расчета 0,5 мэкв/кг и инсулина из расчета 1 ед. препарата на 3 — 4 г глюкозы.
В зависимости от исходного состояния больного и сопутствующих заболеваний, назначается терапия, способствующая коррекции данных изменений.
Обычно к 3-м суткам после операции восстанавливается моторно-двигательная функция пищеварительного канала. На 2-й день после операции больному дают воду, чай (до 0,5 л/сут). С 3-го дня добавляют бульон, 1 — 2 стакана фруктового киселя, два яйца и уменьшают количество вводимой парентерально жидкости. На 5-й день разрешают сливочное масло (40 г), протертые супы, манную кашу. Постепенно на 6 —7-е сутки после операции больного переводят на стол № 1. В дальнейшем диету расширяют, учитывая вкус больного, но при этом не нарушают принцип частого кормления небольшими порциями.
Длительный постельный режим приводит к замедлению крово- и лимфотока, атрофии мышц, застойным явлениям и нарушению вентиляции легких. Отсюда понятно стремление к активному ведению больного, предупреждающему легочные осложнения, тромбозы, парезы кишок и т. д. Со второго дня после операции больному разрешают поворачиваться на бок и предписывают обязательное выполнение комплекса упражнений дыхательной гимнастики. Если сроки вставания и активного поведения больного отдаляются, то применяют общий массаж для улучшения кровообращения.
Особенности лечения больных, оперированных спустя 6 ч после перфорации, в послеоперационном периоде во многом определяются выраженностью перитонита, тяжестью нарушения обменных процессов и функции жизненно важных органов, а также наличием сопутствующих заболеваний.
238 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
Все больные с распространенными формами перитонита и некоторые больные с ограниченным перитонитом, но с выраженными нарушениями основных жизненных функций после операции длительное время находятся в отделении реанимации и интенсивной терапии.
Обязательным условием является помещение больного на функциональную кровать, которая дает возможность (если позволяет артериальное давление) поднять головной конец. Это уменьшает ограничение подвижности диафрагмы и улучшает дыхание.
Из-за опасности суммарного эффекта еще неэлиминированного аналгетика дозировку препарата в первые часы после операции уменьшают в два раза, а в некоторых случаях их вообще не вводят, заменяя клофелином.
У подобных больных важным условием обеспечения эффективности корригирующей терапии является создание хорошего венозного доступа. Всем пациентам проводится пункция подключичной или яремной вены. Применение венозного катетера дает возможность увеличить подвижность больного в постели.
Важным условием эффективности и безопасности инфузионной терапии, на наш взгляд, является равномерное распределение в течение суток назначения растворов и медленное их введение под контролем артериального давления.
Всем больным вводится назогастральный зонд, который извлекается только после восстановления перистальтики.
При наличии в послеоперационном периоде олигурии в мочевой пузырь вводят постоянный катетер. Это дает возможность контролировать почасовой диурез. Используется целенаправленная терапия, способствующая его восстановлению.
Для своевременной и правильной коррекции возникающих патологических сдвигов всем больным проводится определенный объем клинических и биохимических исследований. На каждого пациента заполняют индивидуальный листок наблюдения, в котором в течение суток регулярно (через каждые 1—2 ч) регистрируют: частоту дыхания и пульса, сис
толическое и диастолическое давление, центральное венозное давление, диурез, температуру тела, количество и характер отделяемого из желудка по дренажам из брюшной полости. Кроме того, один-два раза в сутки (а при необходимости и чаще) производится клинический анализ крови, мочи. Исследуют электролиты плазмы и мочи, билирубин и глюкозу крови, общий белок и коагулограмму, показатели иммунологии, ЭКГ.
Всем больным в листе назначений фиксируется объем и характер внутривенной терапии, количество выпитой жидкости.
Во время операции и в послеоперационном периоде производится посев экссудата из брюшной полости для выяснения характера микрофлоры и чувствительности ее к антибиотикам.
В уходе за послеоперационными больными особую роль играют профилактика и терапия инфекционных осложнений. Отсутствие приема пищи и процесса жевания приводит к увеличению бактериальной флоры в полости рта, что чревато опасностью развития паротита. Для его профилактики уделяют внимание гигиене полости рта. Тяжелобольным протирают язык, зубы и промывают рот раствором натрия гидрогенкарбоната (0,5 %), натрия хлорида (0,96 %), гидропероксида (0,6 %), калия перманганата (1 : 10 000). Больным после операции проводится ежедневное гигиеническое обтирание.
У пациентов, которые длительное время лежат на спине, могут появиться пролежни. Чаще всего они возникают в области крестца, пяток и лопаток. Профилактика этих осложнений заключается в:
ежедневном перестилании постели и осмотре тела больного;
тщательном контроле за гигиеной кожи;
предупреждении давления на «опасные» участки кожи.
С целью профилактики пролежней применяют резиновые круги, часто меняют положение больного. При появлении пролежней эти места смазывают один-два раза в сутки 5 % или 10 % раствором калия перманганата или облепиховым маслом. Малая подвижность больных в течение продолжительного времени может привес
Язвенная болезнь 239
ти к мышечной атрофии. Для предотвращения этих осложнений, начиная со вторых суток послеоперационного периода, используется специальный комплекс лечебной физкультуры. С помощью физиотерапевтических мероприятий улучшается функция дыхания. С этой целью применяют вибрационный массаж грудной клетки.
Для разжижения мокроты применяют паровые и ультразвуковые ингаляции, у наиболее тяжелых больных с осложнениями в легких выполняется микротрахеостомия с последующим введением в трахеобронхиальное дерево 0,5 % раствора натрия гидрогенкарбоната с трипсином по 2-3 мл через три-четыре часа. Только при выраженной дыхательной недостаточности, связанной с обтурацией дыхательных путей, применяется интубация трахеи с бронхиальным лаважем.
Изменения гемодинамики, наблюдаемые у данной категории больных, занимают центральное место в патогенезе возникающих расстройств гомеостаза. При своевременной и правильной коррекции водно-электролитного баланса и газообмена нарушения гемодинамики встречаются сравнительно редко и возникают преимущественно в терминальной стадии заболевания.
Первичная острая сердечно-сосудистая недостаточность с выраженной артериальной гипертензией, тахикардией, повышением центрального венозного давления, снижением сердечного индекса наблюдается только у 8 — 10% больных. Вторичная острая сердечная недостаточность, обусловленная гиповолемией, отмечается у 15 % больных. Она проявляется снижением величины артериального давления, тахикардией на фоне выраженного уменьшения центрального венозного давления и исчезает после восполнения дефицита жидкости.
Электрокардиографические признаки поражения миокарда отмечаются только у части обследованных больных. Так, из 125 больных с распространенными формами перитонита изменения на ЭКГ наблюдались у 24 больных, что составило 19,2 %, в частности: мерцательная тахикардия — 8 человек (6,5 %), пароксизмаль
ная тахикардия — 7 человек (5,5 %), экстрасистолия — 4 человека (3,2 %), полная или частичная блокада — 5 человек (4,0 %). Эти изменения отмечены на фоне тяжелой диффузной дистрофии миокарда, явлений гипоксии и нарушений коронарного кровообращения.
Первичная острая сердечная слабость, связанная со снижением нагнетательной функции сердца, наблюдается главным образом у лиц пожилого и среднего возраста с выраженной исходной миокардио-строфией, нарушением коронарного кровотока. Чаще всего гемодинамические нарушения обусловлены гиповолемией. ЦВД колеблется в пределах от 4 ± 1,2 (784 ± ± 117,6 Па) до 8 ± 1,23 см вод. ст. (1372 ± ± 120,54 Па).
При анализе динамики развития изменений показателей сердечно-сосудистой системы в послеоперационном периоде установлено, что тахикардия наиболее выражена на 1 —2 сутки после операции, и стабилизация этого показателя намечается (при условии правильной коррекции) только к 4 — 5 суткам послеоперационного периода.
При коррекции гемодинамических нарушений решающее значение имеет восполнение дефицита жидкости в организме, так как у большинства больных наблюдается общая изотоническая дегидратация. Регидратационную терапию осуществляют под постоянным клинико-биохимическим контролем. Показателем ее эффективности является нормализация артериального и венозного давления, исчезновение сухости языка, восстановление кожного тургора, наполнение периферических вен, нормализация уровня гемоглобина и гематокрита, электролитного и белкового состава крови.
При сравнительной оценке эффективности инфузионной терапии установлено, что оптимальной дозой плазмозаменителей у больных с тяжелыми формами перитонита следует считать 60 — 80 мл/кг массы тела. В этих случаях быстрее, чем при меньших дозировках, удается устранить дегидратацию, олигурию, артериальную и венозную гипотензию, восстановить перистальтику тонкой и толстой кишок. У от
240 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
дельных больных повышение ЦВД до субнормальных величин (3 — 5 см вод. ст., или 294 — 490 Па) достигается только после введения 6 —8 л жидкости внутривенно. Даже в пожилом и старческом возрасте для устранения дегидратации приходится вводить значительные объемы жидкости на 3—4 сутки после операции.
Выраженная послеоперационная дегидратация и задержка жидкости в «третьем пространстве» обуславливает необходимость поддержания положительного водного баланса. Такая инфузионная терапия имеет также большое значение для профилактики тромбоэмболических осложнений. Для большинства больных используется введение 400 — 800 мл низкомолекулярных декстранов, что способствует предотвращению этого тяжелого осложнения. У лиц пожилого и старческого возраста со склонностью к гиперкоагуляции применяют значительные дозы гепарина (5—10 тыс. ЕД через 4 ч) для улучшения реологических свойств крови.
Восстановление объема циркулирующей жидкости, как правило, приводит к быстрой нормализации гемодинамики, поэтому нормализация циркуляторного гомеостаза должна быть направлена прежде всего на устранение дефицита жидкости. Меньшее значение имеет «гомеостатическая коррекция», заключающаяся в полной нормализации ОЦК и его компонентов. Это связано с тем, что у большинства больных наблюдается сгущение крови, а не анемия. Поэтому следует отказаться от «рутинного применения крови», особенно эритроцитарной массы, для восполнения ОЦК и стремиться к улучшению реологических свойств крови. Только при наличии выраженной анемии используется гемотрансфузия крови со сроками хранения до трех суток. Переливание крови длительного хранения и эритроцитарной массы приводит к закупорке легочных капилляров белковыми конгломератами, резко ухудшая функцию внешнего дыхания.
Большую роль в послеоперационном периоде играет кардиотонизирующая терапия. Для этого применяется коргликон в суточной дозе 2,5 мл в виде капельного введения в течение 3 — 4 суток, иногда ис
пользуется строфантин по 0,3 мл 0,05 % раствора 3 раза в сутки. Одновременно назначается панангин по 10 мл 2 — 3 раза в сутки. Как правило, всем больным назначают по 100 — 200 мл кокарбо-ксилазы.
У больных с нарушением коронарного кровообращения, обнаруженным при ЭКГ-исследовании, используют курантил и нитраты. При отсутствии выраженной тахикардии всем больным назначают эу-филлин (по 5—10 мл 2,4 % раствора). Обоснованием для его применения является способность этого препарата улучшать легочный, печеночный, почечный и коронарный кровоток. Для устранения тахикардии используют лидокаин (50—100 мг внутривенно капельно).
Улучшение гемодинамики под влиянием вышеописанных мероприятий возможно лишь при полноценной коррекции нарушений белково-азотистого, водно-электролитного и кислотно-основного состояния.
В случае наличия выраженной артериальной гипотензии, когда восполнение дефицита жидкости и весь перечисленный выше комплекс мероприятий не оказывают положительного эффекта, применяют глюкокортикоиды (125 — 250 мг).
У 20 % больных при прогрессивном падении сердечной деятельности и безуспешном использовании других средств применяют симпатомиметики и нитроглицерин (дофамин 5 мкг/кг в минуту внутривенно капельно, нитроглицерин 2 мл на 400 мл физиологического раствора).
Выраженность нарушений гемодинамики находится в прямой зависимости от тяжести клинического течения заболевания, распространенности перитонита.
Применяемая корригирующая терапия, направленная на восполнение объема циркулирующей жидкости, оказывает эффект при вторичной сердечно-сосудистой недостаточности. Худшие результаты получают при первичной сердечно-сосудистой недостаточности, которая, как правило, наблюдается у больных в старческом возрасте.
Одним из наиболее частых и грозных осложнений в послеоперационном периоде является острая дыхательная недостаточность, причиной которой может быть
Язвенная болезнь 241
развитие паренхиматозной недостаточности, что иногда обусловлено влиянием ингаляционных анестетиков и ИВЛ.
Кроме отрицательного воздействия операционной травмы и наркоза, на внешнее дыхание и газообмен оказывает влияние и характер патологического процесса. Нарушение дыхания при перитоните обусловлено стойким ограничением подвижности диафрагмы. Это приводит к развитию ателектазирования.
Решающее значение в определении выраженности дыхательной недостаточности принадлежит исследованию газового состава крови. Гипоксия у больных перитонитом, как правило, является следствием паренхиматозной дыхательной недостаточности.
Профилактике и лечению дыхательной недостаточности после оперативного лечения придается большое значение. Прежде всего используется кислородотерапия в сочетании с улучшением дыхательной функции легких. Улучшение легочной вентиляции достигается при создании возвышенного положения больного, хорошей аналгезии, применении дыхательной гимнастики, ультразвуковой ингаляции.
В некоторых случаях при крайне тяжелом состоянии больного для лечения гипоксии применяется продленная искусственная вентиляция легких. Применение ИВЛ с постоянным положительным давлением в конце вдоха заметно уменьшает гипоксемию.
Для профилактики и лечения легочных осложнений всех больных после выхода из наркоза переводят в положение полусидя, им назначают дыхательную гимнастику, вибрационный массаж, раздувание резиновой камеры. Особое внимание уделяют профилактике и терапии дегидратаций, поскольку они приводят к ухудшению дренажной функции легких. Для ее улучшения используют ультразвуковую ингаляцию и раннее восполнение ОЦК плазмозаменителями. При появлении признаков дыхательной недостаточности больным назначают ингаляции увлажненного кислорода через носовые катетеры. При отсутствии эффекта используется вспомогательное дыхание маской, дыхание под
постоянным повышением давления в дыхательных путях, спонтанное дыхание с выдохом против сопротивления.
Изменение гемодинамики и газообмена приводит к существенным расстройствам кислотно-основного состояния, что сопровождается декомпенсированным метаболическим ацидозом. Его длительное сохранение является плохим прогностическим признаком. Причиной наблюдающегося субкомпенсированного и компенсированного ацидоза следует считать нарушение гемодинамики, микроциркуляции, гипоксии, накопление недоокисленных продуктов, острую почечную недостаточность.
Изменения электролитного состава крови могут приводить к нарушениям кислотно-основного состояния. Наиболее характерным для перитонита является развитие гипохлоремии, которая особенно выражена в первые сутки после операции (С1 — 80,2 ± 1,36 ммоль/л).
В отдельных случаях регистрируется компенсированная гипокалиемия. Постоянная коррекция электролитного баланса предупреждает развитие выраженных нарушений. Следует отметить, что, несмотря на переливание значительных количеств калия (90—120 ммоль/сут) в течение первых 5 — 6 суток, его уровень остается на нижних границах нормы (3,5 ± 0,6 ммоль/л). Это объясняется значительными потерями калия. Так, в первые сутки после операции потери калия с мочой составляют в среднем 60,2 ± 9,6 ммоль/сут.
У одних больных наблюдается гипонатриемия, у других — гипернатриемия. Достоверной разницы в изменении уровня натрия при перитонитах различной этиологии установить не удалось. Нет заметных колебаний в уровне натрия и в различные сроки после операции. Отсутствие выраженных нарушений в уровне натрия в крови связано с активной корригирующей терапией.
Существуют разные мнения о способах терапии нарушений кислотно-основного состояния. В своей работе мы придерживаемся принципов, разработанных IO. Н. Шаниным с соавт. (1978) и основанных на положении о том, что решающим в устранении метаболического ацидоза является
242 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
не вливание щелочей, а лечение гиповолемии, дисгидрии, сниженной производительности сердца. При условии восстановления гемодинамики и электролитного равновесия кислотно-основный баланс восстанавливается автоматически и коррекция буферными растворами не нужна.
Применяемая терапия нарушений электролитного обмена и кислотно-основного состояния имеет патогенетическую направленность. Прежде всего следует нормализовать газообмен и ликвидировать нарушения гемодинамики. При необходимости одновременно проводят коррекцию нарушений электролитного состава крови и кислотно-основного состояния. Коррекцию метаболического ацидоза проводят раствором натрия, лактата или гидрогенкар-боната по формуле
Количество раствора (ммоль/л) = = 0,ЗМасса тела • BE.
Коррекция метаболического алкалоза заключается в восполнении дефицита хлора и калия по следующей формуле
Количество раствора (ммоль/л = = 0,2Масса тела • Дефицит электролита.
Коррекция дефицита калия осуществляется под постоянным контролем его концентрации в плазме крови.
При гиповолемии, сопровождающейся гипохлоремией, коррекция хлора является более трудной. Осуществить ее одними растворами калий хлорида невозможно, поскольку дефицит С1“ обычно больше, чем К+ (в отдельных случаях он достигает 10 — 30 ммоль/л, а К+ — только 2 — 3 ммоль/л). Поэтому используют другие растворы, содержащие хлор, например 10 % растворы калий хлорида и магний хлорида, вводимые вместе с глюкозой.
Одним из частых осложнений тяжелых форм перитонита является нарушение бел-ково-азотистого баланса, что связано с расстройством белковосинтетической функции печени, усиленным распадом белка, длительным голоданием и другими факторами. Показателем этих изменений является гипопротеинемия. Снижение уровня белка плазмы ниже 60 г/л требует коррекции. Одним из показателей нару
шения белково-азотистого обмена является наличие азотемии.
Коррекция нарушений белково-азотистого обмена при перитоните играет большую роль, так как низкое онкотическое давление является одной из причин ухудшения процессов всасывания в тонкой и толстой кишках. Для поддержания белково-азотистого баланса используют растворы аминокислот (аминосол, альвезин), сухую и нативную плазму, альбумин, протеин. Их начинают использовать с 1 —2 суток после восстановления диуреза при отсутствии азотемии. Поскольку у всех больных после операции дренирован желудок через постоянный назогастральный зонд, начиная с первых суток, вводят гидролизат казеина со скоростью 20 кап/мин.
Для предупреждения распада белка вводят углеводы. Как правило, обследованные больные получают в сутки в среднем 150 — 300 г глюкозы. Дозы увеличивают у больных с декомпенсированными нарушениями обменных процессов (до 300 — 400 г/сут).
При длительном парентеральном питании обосновано использование жировых эмульсий. Наиболее широко используют интралипид и липофундин. В среднем переливают по 500 мл интралипида или ли-пофундина через сутки. Активация ферментов белкового, углеводного обмена достигается введением вместе с питательной смесью витаминов, особенно значительных доз витаминов группы В: тиамина, пиридоксина, цианкобаламина, которые при парентеральном введении гидролизатов проявляют выраженный анаболический эффект, нормализуя азотистый баланс.
Кроме коррекции возникающих нарушений белкового и энергетического баланса при перитоните важно снизить резко повышенный катаболизм. Для этих целей используют анаболические гормоны (ре-таболил, нерболил по 1 мл ежедневно, начиная с 2 —3-го дня, в течение 5 — 7 суток после операции).
Большое значение для исхода лечения больных перитонитом имеет борьба с инфекцией. При перитоните в брюшной полости всегда обнаруживают микрофлору. Ведущая роль в инфицировании брю
Язвенная болезнь 243
шины принадлежит кишечной палочке, особенно в ассоциации со стафилококками.
Чаще всего (в 45 % случаев) в перитонеальном экссудате обнаруживают кишечную палочку, на втором месте по частоте высеваемости находится стафилококк, значительно реже — протей, стрептококк. Наиболее эффективными антибактериальными препаратами являются аминогликозиды — мономицин, канамицин, гентамицин.
В лечении больных используют метод повышения эффективности антибактериальной терапии — предупреждение формирования устойчивости микрофлоры к антибиотикам, достигающееся использованием не менее двух препаратов. Наиболее целесообразно применение цефалоспоринов в сочетании с аминогликозидами.
При лечении больного антибиотики вводят в основном внутримышечно, иногда внутрибрюшинно, причем в значительном разведении, так как при этом удлиняется время действия препаратов из-за уменьшения всасывания под действием новокаина.
Показанием к использованию сочетаний препаратов различного спектра действия является тяжелое течение послеоперационного периода, требующее немедленного эффективного лечения до установления бактериологического диагноза. Развитие пневмонии у больных перитонитом также является показанием для сочетанного применения антибиотиков, отличающихся по спектру действия и особенностям фармакокинетики. Наиболее обоснованно применение в течение 2 — 3 суток комбинаций препаратов широкого спектра действия. Внутривенно или внутримышечно вводится ампициллин по 1 г 6 раз в сутки или кефзол 1 г через каждые 4 ч, внутримышечно — канамицин по 1 г 2 раза в сутки или гентамицин по 80 мл 3 раза в сутки. Введение антибиотиков в брюшную полость оказывает бактериостатический и бактерицидный эффект и улучшает течение заболевания.
Антибиотики действуют угнетающе не только на микроорганизмы, но и на иммунную систему организма. В момент по
ступления уровень иммунореактивности у больных перфоративной язвой в среднем значительно отличается от нормы, клеточные реакции у большинства пациентов угнетены, уровень аутоиммунных антител и циркулирующих иммунных комплексов значительно превышает норму. Размеры молекул циркулирующих иммунных комплексов соответствуют уровню значительной патогенности (ЦИКк = 0,72). К третьим послеоперационным суткам уровень иммунореактивности значительно изменяется. В этот период наблюдается тенденция к снижению среднего уровня ЦИК, однако у некоторых больных этот показатель превышает нормальное значение в 3 раза.
К 7 —10 суткам после операции достоверно возрастает число общих и активных многорецепторных розеткообразующих лимфоцитов, составляющих соответственно (26,55 ± 2,74) % и (18,9 ± 2,2) %.
Показатели клеточного и гуморального иммунитета могут служить прогностическими тестами при различных методах комплексного послеоперационного ведения больных. Комплексное иммунологическое обследование позволяет индивидуализировать выбор иммунокорригирующих мероприятий.
В последнее время антибиотики используются совместно с метроджилом по 100 мл три раза в сутки внутривенно. Этот препарат является проводником лекарств, так как быстро проникает через ткани. Кроме того, он оказывает выраженное антимикробное, противовоспалительное, проти-воотечное, десенсибилизирующее действие, изменяет чувствительность микрофлоры.
Особое значение при стафилококковой инфекции придается применению стафилококкового анатоксина. Усиливая специфический антитоксический иммунитет, он значительно повышает эффективность антибиотикотерапии. С этой целью необходимо широко применять гипериммунную антистафилококковую плазму. При использовании антибиотиков широкого спектра действия и их комбинаций хороший эффект дает применение средств, усиливающих защитные силы организма, в частности у-глобулина.
244 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
у-Глобулин назначается со 2-го дня послеоперационного периода, а после окончания пятидневного цикла его введения, особенно в тяжелом состоянии, проводится курс лечения анатоксином. С 3 —4-го дня после восстановления перистальтики обязательно назначаются противогрибковые препараты (нистатин, леворин).
Применение активной антибактериальной и корригирующей терапии при радикальной хирургической коррекции позволяет в большинстве случаев справиться с перитонитом.
Значительное место в патогенезе перитонита занимает парез кишок, вызывающий нарушение процессов всасывания и исключающий возможность полноценного питания больных, что приводит к нарушению всех видов обмена, истощению, обезвоживанию организма. Основным обоснованием комплекса лечебных мероприятий при парезе в настоящее время является представление о ведущей роли нарушений моторной и эвакуаторной функции кишок. Поэтому следует прежде всего применять средства, направленные на восстановление двигательной функции кишок. Для прямой стимуляции сократительной активности мускулатуры кишок используют гипертонические растворы натрий хлорида или глюкозокалиевую смесь.
Немаловажную роль в лечении пареза играет уменьшение перерастяжения петель кишок. Для этого всем больным сразу же
по окончании операции выполняют дренирование желудка и начального отдела тонкой кишки назогастральным зондом.
Большое значение для восстановления двигательной деятельности пищевого канала придается блокаде тормозных нервных влияний. Для этого используют введение раствора новокаина в брыжейку во время операции. Значительным эффектом обладает спинномозговая и перидураль-ная блокада. У больных используется медикаментозная коррекция тормозных нервных сплетений введением симпатолити-ков (аминазин по 0,5 мл 3 раза в сутки), Р-адреноблокатора орнида с последующим введением прозерина. С успехом применяют ганглиоблокаторы (бензогексо-ний, димеколин, церукал).
Терапия пареза кишок является одним из компонентов комплексного лечения перитонита. Она должна быть патогенетической и направленной прежде всего на борьбу с инфекцией и интоксикацией, коррекцию водно-электролитного и белкового обмена.
Таким образом, комплексное лечение больных, оперированных по поводу перфоративной язвы двенадцатиперстной кишки, должно включать четко разработанную схему мероприятий, направленных прежде всего на компенсацию наиболее пострадавших от заболевания систем с постоянным контролем и коррекцией дальнейшего плана лечения.
7.5. ПАНКРЕАТИТЫ
Поджелудочная железа взрослого человека длиной примерно 15 см, массой 90 г расположена за париетальной брюшиной на задней брюшной стенке. Условно железу делят на головку, перешеек, тело и хвост.
Протоки передней и задней частей поджелудочной железы срастаются в 90 % случаев и образуют главный панкреатический проток, через который осуществляется дренаж эндокринного панкреатического секрета.
Поджелудочная железа состоит преимущественно из экзокринной ткани, основ
ным элементом которой являются ацинусы. Вместе с разветвленной сетью протоков они составляют около 80 % массы железы. Ацинарные клетки секретируют ферменты в неактивной форме (в виде проферментов).
Эндокринная часть поджелудочной железы представлена небольшими группами клеток, известных как островки Лангерганса. Они отделены от ацинусов прослойками соединительной ткани.
Кровоснабжение поджелудочной железы осуществляется ветвями чревной, верхней брыжеечной и селезеночной артерий.
Панкреатиты 245
Венозный отток происходит по промежуточным венам в воротную вену печени.
Иннервация осуществляется симпатическими и парасимпатическими нервными системами через чревное сплетение.
В поджелудочной железе выделяют четыре типа эндокринных клеток. В-клетки (р) наиболее многочисленны в островках Лангерганса, они секретируют инсулин и локализуются в центре островков. Другие эндокринные клетки расположены вокруг В-клеток на периферии островков: А-клетки (а) секретируют глюкагон, D-клет-ки — соматостатин, F-клетки (РР) продуцируют панкреатический полипептид.
Экзогенная секреция поджелудочной железы состоит в выделении пищеварительных ферментов и жидкости, богатой электролитами. Гастроинтестинальный гормон секретин стимулирует секрецию эпителием воды, гидрогенкарбонатов, натрия, калия и хлоридов посредством активации аденилатциклазы.
Ферменты поджелудочной железы образуются и хранятся в ацинарных клетках. Каждая гранула содержит в различном соотношении все секретируемые здесь ферменты, которые обычно находятся в «уплотненном» состоянии и после экскреции из клетки растворяются в щелочном секрете поджелудочной железы. Однако растворение ферментов происходит в неактивной форме, а их переход в активную форму осуществляется в двенадцатиперстной кишке. В этом заключается механизм защиты поджелудочной железы от самопереваривания.
Поджелудочная железа секретирует значительное количество пищеварительных ферментов. Большинство из них предназначено для переваривания белков, жиров и углеводов, потребляемых с пищей. Для того чтобы ферменты начали функционировать, они должны быть активированы в двенадцатиперстной кишке. Профермент трипсиноген подвергается ферментативному гидролизу с N-терминального фрагмента благодаря активности пептидазы (энтерокиназы), расположенной на щеточной кайме энтероцитов тонкой кишки. В дополнение к механизмам, обеспечивающим абсорбцию питательных веществ, клетки
щеточного барьера кишки выделяют различные вещества, способствующие пищеварению до момента абсорбции. Энтерокиназа является одним из таких веществ. Активированный трипсин, в свою очередь, катализирует активацию других проферментов, секретируемых поджелудочной железой. Она также секретирует ингибитор трипсина, который является средством защиты поджелудочной железы. Этот пептид инактивируется, соединяясь с трипсином около его каталитического центра.
Различные вещества, ответственные за ингибирование панкреатической секреции, действуют по принципу обратной связи во время и после приема пищи. Панкреатический полипептид представляет собой пептидный гормон, ингибирующий панкреатическую секрецию воды, гидрогенкарбонатов и ферментов. Секреция этого полипептида увеличивается при стимуляции блуждающего нерва, действии холецито-кинина, секретина, вазоинтестинального пептида и, возможно, гастрина и гастрин-рилизинг пептида. Панкреатический полипептид может выступать как антагонист ацетилхолиновых рецепторов и способен ингибировать выделение ацетилхолина из постганглионарных нейронов поджелудочной железы; его конечный эффект проявляется на уровне ацинарных клеток. Пептид YY высвобождается в дистальной части подвздошной кишки и в толстой кишке в ответ на пищу смешанного характера. Этот пептид уменьшает чувствительность поджелудочной железы к действию секретина и холецистокинина, возможно, за счет уменьшения секреции ацетилхолина и норадреналина и ингибирования выделения холецистокинина слизистой оболочкой двенадцатиперстной кишки.
Соматостатин ингибирует секрецию секретина дуоденальной слизистой оболочкой, а также чувствительность к секретину рецепторных полей. Его единственный эффект — снижение секреции ферментов и гидрогенкарбонатов поджелудочной железы. Соматостатин секретируется клетками слизистой оболочки желудка и кишок, а также D-клетками островков Лангерганса. Однако только соматостатин,
246 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
продуцируемый слизистой оболочкой тонкой кишки, оказывает ингибирующее действие на секрецию поджелудочной железы. Выделение соматостатина происходит при участии автономной нервной системы в ответ на поступление жиров и аминокислот с пищей. К ингибиторам, входящим в состав гормонов эндокринных клеток островков Лангерганса, относят панкреатический глюкагон и панкреастатин, а также нейропептиды: кальцитонин — информационный пептид и энкефалины.
Панкреатический глюкагон ингибирует секрецию поджелудочной железы, в частности секрецию гидрогенкарбонатов, воды и ферментов, панкреатин — панкреатическую секрецию, тормозя высвобождение ацетилхолина эфферентными окончаниями блуждающего нерва. Кальцитонин стимулирует выделение соматостатина. Энкефалины и подобные им опиоиды снижают высвобождение секретина слизистой оболочкой двенадцатиперстной кишки и могут также ингибировать высвобождение ацетилхолина.
7.5.1. Клиническая картина острого панкреатита
Почти все пациенты, страдающие острым панкреатитом, жалуются на боли в животе, в том числе при пальпации. Боль обычно локализована в эпигастрии или в верхнем квадранте живота и иногда иррадиирует в спину, может носить опоясывающий характер. Она достигает максимальной интенсивности через несколько часов после начала заболевания и усиливается при наклоне вперед или подтягивании колен к животу. В отличие от больных с перфорацией внутренних органов, стремящихся лежать неподвижно, пациенты с острым панкреатитом беспокойны и все время пытаются найти удобную позу. У большинства больных появляется тошнота, рвота, сопровождаемые незначительным повышением температуры тела.
При обследовании пациента с тяжелым течением острого панкреатита могут наблюдаться проявления артериальной гипотонии, шока, гипоксии, респираторного дистресс-синдрома, нарушение сознания,
отек легких, ригидность мышц живота, а также симптомы забрюшинного кровотечения (симптомы Каллена и Грея —Тернера). Эти симптомокомплексы возникают при проникновении крови из ретроперитонеального пространства в периумби-ликальную область (симптом Каллена) либо в боковые части брюшной полости (симптом Грея —Тернера) и проявляются цианозом этих областей.
Лабораторно выявляется повышение гематокрита в связи с гемоконцентрацией, изредка наблюдается изменение показателей — маркеров поражения печени (АсАТ, АлАТ, билирубин, щелочная фосфатаза), значительное увеличение которых может свидетельствовать о наличии камней в желчном пузыре как причины возникновения панкреатита. Содержание амилазы и липазы в сыворотке крови увеличено более чем в два раза.
У большинства больных панкреатитом легкой и средней степени тяжести улучшение наступает через несколько дней, а полное выздоровление — через неделю после консервативной терапии, включающей восполнение объема циркулирующей жидкости, дефицит которой возникает в связи с воспалением забрюшинного пространства.
7.5.2. Лечение острого панкреатита
В последние годы основным методом лечения острого панкреатита является консервативный. Он используется главным образом при отечных формах панкреатита. Деструктивные формы заболевания являются основанием для оперативного лечения.
Выделяют такие проявления некроза поджелудочной железы:
1) нарастание боли и симптомов раздражения брюшины;
2) отрицательная динамика коллапса и шока;
3) резкое увеличение лейкоцитоза (более 25 • 109 в 1 л крови со сдвигом влево);
4) падение уровня амилазы в крови и моче при ухудшении общего состояния больного вследствие прогрессирующей интоксикации;
Интенсивная терапия панкреонекроза 247
5) повышение температуры до фебрильных цифр;
6) появление признаков паралитической кишечной непроходимости.
Симптомы, требующие неотложного оперативного лечения по жизненным показаниям при остром панкреатите: ♦ клиника перитонита;
♦ клиника локального нагноения;
♦ клиника деструкции поджелудочной железы;
♦ клиника аррозивного кровотечения.
Остальные больные с острым панкреатитом лечатся консервативно, по возможности, в реанимационных отделениях или палатах интенсивной терапии с использованием комплекса дезинтоксикационных средств, инфузионной и антибактериальной терапии, антикоагулянтов.
Сразу после госпитализации в стационар больным назначается лечение голодом: при отечной форме панкреатита до 3 суток, при деструктивных — до 5 суток. На эпигастральную область кладут холодную грелку или используют другие методы локальной гипотермии.
Для купирования болевого синдрома назначаются ненаркотические аналгетики (аналгин, баралгин, спазган). Наркотические аналгетики назначаются в исключительных случаях, так как их использование вызывает спазм сфинктера Одди и гипертензию в протоках, что может стать причиной некроза поджелудочной железы. Используют также нейролептики и спазмолитики (но-шпу, папаверин), холи-нолитики (атропин, метацин). Следует подчеркнуть, что при алкогольном генезе панкреатита атропин нужно вводить с осторожностью, так как препарат может способствовать развитию делирия. Широко
используется новокаин, который вводится внутривенно или в виде блокад — по 100 мл сакроспинально на уровне ThVI на расстоянии 2 см от позвоночника. Для подавления рвоты используют препараты метоклопрамида (церукал, реглан).
С целью обеспечения функционального покоя и прерывания патологических процессов в функциональных клетках поджелудочной железы назначают ингибиторы протеаз — гордокс, трасилол, контрикал, аминокапроновую кислоту. В последнее время для снижения ферментативной активности железы широко используется сандостатин. Для купирования болевого синдрома и снижения активности ферментов назначают цитостатики — 5-фторура-цил или циклофосфан.
Всем больным независимо от формы панкреатита назначается антикоагулянтная терапия: прямые антикоагулянты — гепарин — по 5 тыс. ЕД подкожно через каждые 6 часов или низкомолекулярные антикоагулянты (фраксипарин по 0,3 и 0,6 подкожно один раз в сутки).
Обязательным компонентом лечения острого панкреатита является инфузионная терапия из расчета до 100 мг/кг в сутки с последующим форсированием диуреза; дезинтоксикационная терапия; для улучшения реологических свойств крови назначают реополиглюкин, реомакро-декс.
В случае нарушений гемодинамики в комплекс терапии включают препараты гидроэтиленкрахмала, гормоны, в тяжелых случаях — симпато- и адреномиметики.
На всех этапах лечения проводится коррекция электролитного состава крови и кислотно-основного состояния по принципу баланса.
7.6. ИНТЕНСИВНАЯ ТЕРАПИЯ ПАНКРЕОНЕКРОЗА
Панкреонекроз относится к угрожающим жизни состояниям вследствие развития синдрома системного воспалительного ответа с клинической картиной септического шока и, несмотря на применяемые в настоящее время методы лечения, продолжает сопровождаться высокой смертностью.
При панкреонекрозе в результате гибели большого количества ферментативно активных клеток с последующим развитием некроза ткани поджелудочной железы в брюшной полости образуется очаг, содержащий множество продуктов распада клеток. Последние являются индукторами образования и выброса в системный
248 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
кровоток вазоактивных веществ, медиаторов системного воспалительного ответа — интерлейкинов, прежде всего ИЛ-1, ИЛ-4, ИЛ-6, TNF. Эффект интерлейкинов является в определенной степени «дозозависимым», иными словами, эти вещества вырабатываются и в физиологических условиях, выполняют защитную функцию, вызывая ограниченное воспаление с целью удаления инородных веществ. Основным патологическим эффектом выброса этих веществ в большом количестве в кровь является повышение проницаемости мембран эндотелия сосудов. Это приводит к появлению эффекта просачивания, то есть в норме непроницаемый для белковых молекул эндотелий начинает пропускать из сосудистого русла в интерстициальное пространство крупные белковые молекулы. Кроме того, вазоактивные вещества вызывают расширение артериол на уровне микроциркуляции с замедлением кровотока в капиллярах. Это, в свою очередь, приводит к образованию микротромбов, что еще больше ухудшает кровоток.
В результате замедления кровотока, образования микротромбов снижается доставка кислорода к тканям, метаболизм происходит в условиях дефицита кислорода, накапливаются недоокисленные продукты метаболизма, вследствие чего КОС сдвигается в сторону ацидоза, при этом увеличивается экстракция кислорода тканями. Когда дефицит доставки кислорода превысит возможность компенсаторных сдвигов кривой диссоциации гемоглобина, тонус сосудов в системе микроциркуляции еще больше снижается из-за дефицита энергии в клетках сосудов, что приводит к еще большему застою крови в этом отделе циркуляторного русла. На общем объеме кровообращения этот процесс отразится резким снижением возврата крови к сердцу за счет стремительного увеличения объема сосудистого русла по отношению к объему циркулирующей крови. ОЦК при этом будет снижен по отношению к исходному (для данного пациента в нормальном состоянии) за счет эффекта капиллярного просачивания и перехода в интерстициальное пространство вместе с белками некоторого количества воды.
Процесс развивается достаточно быстро, и больные к моменту поступления в стационар имеют развернутую картину септического, или перераспределительного, шока. Последний термин более точный, поскольку ОЦК значительно ниже объема циркуляторного русла, расстройства микроциркуляции имеют мозаичный характер: в одних участках микроциркуля-торного русла — выраженный парез, в других — вначале развивается компенсаторный спазм (ишемия), при длительном существовании которого и достаточном накоплении продуктов неполного окисления развивается парез всего микроциркулятор-ного русла. В такой ситуации шок часто становится необратимым.
Тактика лечения септического шока является универсальной, но следует помнить, что при панкреонекрозе патологический процесс развивается довольно стремительно и лечение должно быть своевременным и четко аргументированным.
Основной целью интенсивной терапии (ИТ) шока при панкреонекрозе является восстановление адекватной доставки кислорода тканям для прекращения патологического процесса на уровне системы микроциркуляции и ликвидации кислородного долга. Поэтому ИТ на первом этапе должна включать два основных аспекта: восстановление ОЦК, с учетом имеющейся повышенной проницаемости сосудов, *для возобновления доставки кислорода к тканям и восстановления адекватной возросшим потребностям тканей в кислороде оксигенации. Второй аспект -собственно лечение синдрома ишемии-реперфузии.
Следует в обязательном порядке провести адекватное (т. е. в достаточном для проведения лечения объеме) и своевременное лабораторное обследование, которое должно включать информацию о:
а) гемоконцентрационных показателях (гемоглобин, гематокрит, электролиты сыворотки крови, осмолярность; сюда же можно отнести уровень глюкозы и мочевины, поскольку на ранних стадиях процесса они отражают не недостаточность функций органов, а степень стрессовой реакции и метаболических расстройств);
Интенсивная терапия панкреонекроза 249
6) КОС артериальной и венозной крови. Для успешного лечения шока при пан-креонекрозе информация об этих показателях является незаменимой;
в) состоянии свертывающей системы крови, желательно с исследованием факторов свертывания крови; количестве тромбоцитов.
Учитывая, что при шоке всем методам исследования присуща высокая степень погрешности оценки гемодинамики, на эти данные можно опираться только при исследовании в динамике. Однако следует иметь в виду, что клиническая картина иногда может дать более верную информацию. Алгоритм лечения пациента с пан-креонекрозом можно представить следующим образом:
1. При поступлении больного обязательно обеспечить постоянный внутривенный доступ путем катетеризации либо двух центральных, либо одной центральной и одной периферической вен.
2. Забор крови для лабораторных исследований.
3. Кислородотерапия либо через маску с дозатором потока кислорода с FzO2 не менее 0,4, либо при самостоятельном дыхании ИВЛ с исходными параметрами вдыхаемого кислорода FzO2 не менее 0,6 с дальнейшей коррекцией по показателям КОС.
4. Инфузионная терапия: предпочтительной инфузионной средой для восполнения ОЦК являются кристаллоидные растворы. При выраженных расстройствах гемодинамики, клинически проявляющихся резким снижением венозного возврата с нарушением уровня сознания показана стартовая терапия гипертоническим раствором NaCl в концентрации от 3 до 10 % (объем раствора соли в концентрации 10 %, как правило, не превышает 200 мл). Этот метод достаточно оправдан, поскольку дает возможность сократить время для достижения желаемого эффекта увеличения ОЦК и венозного возврата; способствует транспорту воды из интерстициального пространства, куда она переместилась в процессе болезни, во внутрисосудистое, за счет чего также увеличивается ОЦК; является достаточно безопас
ным для перераспределения электролитов между секторами, так как натрий — это естественный компонент человеческого организма; эффект улучшения микроциркуляции наступает вследствие прямого действия натрия на сосуды микроциркуляции и уменьшения степени ишемии.
Введение коллоидов менее оправданно в этой ситуации, поскольку чем тяжелее ситуация, т. е. чем более выражены метаболические расстройства, тем больше проницаемость сосудов и степень проникновения крупных молекул в интерстициальное пространство. Если после восстановления нормального тонуса и проницаемости сосудов электролиты распределяются по секторам в обычном соотношении, то крупные молекулы коллоида обратно в сосудистое русло вернуться уже не могут. Если же расстройства микроциркуляции имеют не настолько тяжелый характер, то можно вводить коллоидные растворы и получать положительный эффект, но следует корректно сравнить группы больных по тяжести метаболических расстройств. То же касается инфузии альбумина.
Инфузия плазмы при низких показателях белка (он частично «провалился» в интерстиций, частично используется для синтеза «острофазовых» белков) обеспечивает описанные выше процессы. При сравнении результатов выживания больных при ее применении с целью коррекции ОЦК не были отмечены какие-либо преимущества, поэтому показанием для инфузии свежезамороженной плазмы остается только уменьшение содержания факторов свертывания крови. При наличии клинических признаков ДВС инфузионную терапию следует начинать именно с инфузии плазмы в объемах не менее 1 л для взрослого человека под контролем параметров свертывающей системы крови.
5. Если в течение 30 мин после начала инфузии нет эффекта от ее проведения (т. е. АД остается на прежнем уровне, сохраняются признаки гипоксии), одновременно с инфузией солевых растворов необходимо проводить инфузию симпатоми-метиков. Препаратом выбора является дофамин из-за его действия на оба типа рецепторов и дозозависимого эффекта. Не
250 КЛИНИЧЕСКАЯ ФИЗИОЛОГИЯ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА
следует стремиться достигать нормальных для больного значений показателей гемодинамики, в оценке адекватности терапии должны быть использованы прежде всего динамика сознания и состояния микроциркуляции, а также темп диуреза и динамика КОС.
6. Поскольку панкреонекроз вызывает развитие клиники септического шока, обусловленного выбросом большого количества цитокинов в кровь, достаточного для поступления в общую систему циркуляции, оправданно проводить антибактериальную терапию аналогично случаям сепсиса любого другого генеза. Однако следует различать клинику сепсиса и септического шока. В последнем случае еще нет септического очага как такового, имеется только очаг некроза ткани, образовавшийся
в результате гибели клеток железы под действием протеолитических ферментов, лишь спустя некоторое время этот очаг превратится в септический. Поэтому в начале развития шока часто наблюдается отсутствие ожидаемого эффекта от введения антибиотиков.
Время начала формирования септического очага имеет существенное значение, поэтому антибиотики назначаются сразу при установлении диагноза панкреонекроза, частично с целью предупреждения образования септического очага в брюшной полости. Однако следует помнить, что антибактериальная терапия при септическом шоке не является первоочередным мероприятием, тогда как при сепсисе она рассматривается в качестве основного метода терапии.
Список использованной литературы
1. Цыбуляк В. Н., Загорулько О. И., Карта-венко С. С. Наша общая хроническая боль (к вопросу о центрах по лечению хронической боли) // Анестезиология и реаниматология. - 1998. - X? 5. - С. 108-112.
2. Чурюканов В. В. Болеутоляющие средства: сравнительная оценка, механизмы действия, перспективы // Экспериментальная и клиническая фармакология. — 1998. — № 4. — С. 23-32.
3. Б1лик В. Д. Каширний С. В. Нейропатич-ний больовий синдром // Б1ль, знеболюван
ня i штенсивна терашя. — 2000. — № 2. -С. 54-58.
4. Book of Abstract II Congress of Europe Federation of J ASP (Chapters «Pain in Europe»). — Barselona, 1997. — P. 509.
5. Klein К. B., Mellinkoff S. M. Approach to the patient with abdominal pain: Textbook of gastroenterology. — 2nd ed. — Philadelphia, 1995. - 660 p.
6. Joseph M., Wend er son M. D. Gastrointestinal pathophysiology. — Moscow; S.-Pet., 1997.
=== Раздел == 8 Физиология и патология водно-электролипшого обмена
В организме человека вода выполняет основополагающие функции — она является средой и непосредственным участником обмена веществ.
Содержание воды в организме значительно превышает содержание всех остальных химических веществ. В большинстве тканей организма, за исключением костной
и жировой, на долю воды приходится от 70 до 85 %. Так, в сером мозговом веществе имеется около 85 % воды, в легочной ткани — 82, почечной — 80, печеночной ткани и поперечнополосатой мускулатуре — 70 % воды. Таким образом, чем интенсивнее обменные процессы в ткани, тем больше эта ткань содержит воды.
8.1. ВОДА И ЖИДКОСТНЫЕ ПРОСТРАНСТВА ОРГАНИЗМА
В костной ткани содержится 22 % воды, в жировой — примерно 10 %. Поскольку последняя особенно бедна водой, то относительное содержание воды в организме в значительной степени зависит от количества жировой ткани — чем ее больше, тем меньше относительное содержание воды в организме. У женщин этой ткани в среднем больше, чем у мужчин, поэтому содержание воды в их организме относительно массы тела примерно на 6 —10 % ниже [1].
Общее содержание воды в организме упрощенно рассчитывают по формулам
Общее содержание воды (л) = = 0,6М (для мужчин);
Общее содержание воды (л) = = 0,5М (для женщин),
где 0,6 и 0,5 — это содержание воды в организме, выраженное в долях единицы; М — масса тела, кг.
У взрослого человека на долю воды приходится 73,2 % безжировой массы тела (т. е. массы всех тканей, кроме жировой, содержание которой может варьировать у каждого индивидуума в значительных пределах). Эта величина не зависит от пола и настолько постоянна, что, исходя из содержания воды, можно вычислить относительное количество жировой ткани в организме по формуле [ 1 ]
Содержание жира (%) =
Общее содержание воды, _ 4 лп % массы тела
0/732 ’
С возрастом содержание воды в организме неуклонно снижается. Больше всего ее у новорожденных — 79 % массы тела. К концу первого года жизни общее количество воды в организме снижается до 60,4 % массы тела. После 60-летнего возраста ее содержание в организме в сред
252 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
нем составляет 56 % массы тела у мужчин и 46 % — у женщин [2].
Часть воды находится внутриклеточно — в так называемом внутриклеточном водном пространстве, или внутриклеточном водном секторе (внутриклеточная вода). Другая часть — внеклеточно, во внеклеточном водном пространстве, или внеклеточном водном секторе (внеклеточная вода).
В норме внутриклеточная вода составляет 35 — 36 % массы тела, а внеклеточная — 22 — 24 %. Более точно объем внеклеточного водного пространства (V, % массы тела) вычисляется по формуле
V = 23,67 - 0,254(ОМТ) +
+ 0,0017(ОМТ)2,
где ОМТ — это отклонение массы тела данного больного от его должных (идеальных) величин, %. При массе тела меньше идеальной ОМТ подставляется в формулу со знаком «минус», а при избыточной массе — со знаком «плюс» [2].
В свою очередь, внеклеточное водное пространство подразделяется на внутрисосудистое водное пространство (содержит жидкость кровеносной и лимфатической сосудистых систем), интерстициальную (межтканевую или межклеточную) воду и трансцел л юлярную жидкость.
В норме жидкость интерстициального пространства составляет 19 %, внутрисосудистая — 5 и трансцеллюлярная жидкость — 2,4 % массы тела. Основные функции внеклеточной жидкости заключаются в обеспечении клеток питательными веществами и удалении из организма продуктов внутриклеточного метаболизма [3].
Внутрисосудистая часть воды соответствует объему плазмы (УПл, л), который вычисляется по формуле [2]
Упл = 0,043М,
где М — масса тела, кг.
После удаления белков из плазмы оставшийся солевой раствор обозначают как воду плазмы. У здорового взрослого человека объем воды плазмы ( Увп, л) вычисляется по формуле [2]
Увп = 0,04М, где М — масса тела, кг.
Из объемов плазмы и форменных элементов крови складывается объем крови, составляющий примерно 7 % массы тела, [4].
Жидкость интерстициального пространства можно рассматривать как ультрафильтрат плазмы. Через поры эндотелия капилляров в норме проходит лишь незначительное количество белков плазмы, поэтому концентрация белка в интерстициальной жидкости относительно низка и равна 20 г/л. Белки, попавшие в интерстициальное пространство, возвращаются в сосудистое русло с лимфой. Большинство электролитов (в основном ионы небольшого размера) свободно проходят через эндотелий капилляров в интерстициальное пространство, чем объясняется почти идентичный электролитный состав плазмы и интерстициальной жидкости (табл. 8.1).
Небольшие различия концентраций электролитов в плазме и интерстициальной жидкости обусловлены тем, что сами белки, являясь электролитами, в пределах pH организма существуют главным образом в виде анионов, а поскольку содержание белков в плазме и интерстициальной жидкости существенно отличается, то происходит некоторый сдвиг катионно-анион-ного равновесия между двумя водными секторами.
Интерстициальная жидкость выполняет в организме роль объемного буфера: при кровопотере из нее мобилизуется жидкость
Таблица 8.1. Электролитный состав секторов внеклеточного водного пространства [3]
Электролиты Внутрисосудистая жидкость (плазма), мэкв/л Интерстициальная жидкость, мэкв/л
Na+ 154 142
К+ 4 4
Са2+ 5 5
Mg2+ 2 2
С1- 105 110
HCOi 24 28
Фосфор 2 2
Белок, г/л 70 20
Вода и жидкостные пространства организма 253
в сосудистое русло, а при передозировке внутривенно введенного раствора в ней депонируется большая его часть.
При восполнении кровопотери сбалансированным кристаллоидным раствором (например, раствором Рингера-лактата) последний быстро распределяется в сосудистом русле и интерстициальном пространстве таким образом: 1/3 введенного количества остается в сосудистом русле, а 2/3 — депонируется в интерстициальном пространстве. Поэтому для получения положительного гемодинамического эффекта, замещение кровопотери кристаллоидами должно осуществляться в следующей пропорции — 3 мл раствора кристаллоидов на 1 мл кровопотери [5].
Следует подчеркнуть, что только 60 % интерстициальной жидкости является лег-кодиффундирующей и, таким образом, функционально активной, а 40 % ее входит в состав протеогликанов и соединительной ткани фасций, хрящей, костей и потому функционально малоактивно.
Гидростатическое давление в интерстициальном пространстве обычно отрицательное и составляет примерно 5 мм рт. ст. При увеличении объема интерстициальной жидкости давление в этом пространстве повышается и, когда оно достигает положительных значений, содержание свободной воды в интерстициальном водном пространстве быстро увеличивается, что клинически проявляется отеками.
В интерстициальную жидкость непрерывно поступают продукты внутриклеточного обмена, поэтому, как правило, ее состав непрерывно меняется, однако постоянство концентрации электролитов и других биологически активных веществ поддерживается благодаря лимфо- и плазмотоку.
Трансцеллюлярная жидкость — это жидкость, содержащаяся в полостях организма (ликвор, внутрисуставная жидкость, желчь, содержимое пищеварительного канала). В норме она составляет 2,4 % массы тела или приблизительно около 2 л. Формирование трансцеллюлярной жидкости связано со специфическими клеточнотранспортными ферментными механизмами, действующими в местах ее образова
ния. Поэтому перечисленные выше секреты, относящиеся к трансцеллюлярной жидкости, существенно отличаются друг от друга по химическому составу: например, биохимический состав цереброспинальной жидкости следующий: [Na+] = = 147 ммоль/л; [К+] = 2,86; [Mg2+] = = 1,12; [Са2+] = 1,14; [СГ] = ИЗ (при туберкулезном менингоэнцефалите концентрация снижена); [НСОз] = 23,3; [неорг. Р] = 1,1 ммоль/л; [белка] = 0,28 г/л; [глюкозы] = 2,8 —4,5 ммоль/л (50 % концентрации в плазме); осмоляльность = = 289 — 300 мосм/кг; pH = 7,307; рСО2 = = 50,5 мм рт. ст. [6].
Понятие «третье водное пространство» используют только при патологических состояниях. Оно включает жидкость, скапливающуюся в серозных полостях при асците, плеврите и перикардите, в слое подбрюшинной клетчатки при перитоните, в глубоких слоях кожи в первые 12 ч после ожога, в замкнутом пространстве кишечных петель при непроходимости кишок, в травмированных тканях. Жидкость, находящаяся в «третьем водном пространстве», не принимает участия в обмене жидкости на микроциркуляторном уровне и по составу подобна плазме или интерстициальной жидкости. Существование «третьего водного пространства» требует дополнительной инфузии жидкости для поддержания нормального объема циркулирующей крови, адекватного сердечного выброса и полноценной перфузии тканей [7].
Некоторые авторы к «третьему водному пространству» относят только скопление жидкости в травмированных тканях, например при ожогах и во время оперативных вмешательств. Установлено, что при проведении открытой холецистэктомии потеря жидкости в травмированные ткани равна примерно 3 мл/кг в час, при резекции кишечника — 6 —8 мл/кг в час, при больших сосудистых операциях (например, резекции аневризмы брюшной аорты) — 10 — 20 мл/кг в час. Потери жидкости с асцитом, экссудативными плевритом и перикардитом, через фистулы пищеварительного канала относят к трансцел-люлярным потерям [4].
254 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
8.2. РОЛЬ ПИЩЕВАРИТЕЛЬНОГО КАНАЛА В ПОДДЕРЖАНИИ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Первый этап обмена жидкости происходит в пищеварительном канале (ПК). Перемещение воды на этом уровне также важно, как и на уровне сердечно-сосудистой системы. В ПК осуществляется обмен жидкости, поступающей с едой и питьем, а также из внутренней среды организма, причем последней поступает значительно больше. Эта вода имеет большое значение. За сутки в организме человека секретируется около 1500 мл слюны с колебаниями ± 500 мл. Нарушение глотания может приводить к потере жидкости со слюной, а также сопровождаться потерей электролитов, так как в слюне содержится, мэкв/л: Na+ - 10-33; К+ - 6-19; СГ - 10,3; НСОз - 10; НРО^" - 10; Са2+ - 3; белка — 0,05.
В норме за сутки в желудке секретируется около 2000 мл желудочного сока с крайними колебаниями от 1000 до 2500 мл. При стимуляции секреция может возрасти до 10 л/сут. Желудочный сок имеет следующий электролитный состав, мэкв/л: Na+ — 60,4 (с колебаниями от 9 при ги-перацидных гастритах до 117 — при ана-цидных); К+ — 9,2 (с колебаниями от 0,5 до 32); С1“ — 89 (с колебаниями от 8 при анацидных гастритах до 155 — при гипер-ацидных).
За сутки печень продуцирует около 500 мл желчи с крайними колебаниями от 50 до 1000 мл. Желчь имеет следующий электролитный состав, мэкв/л: Na+ — 150 (131-164); К+ - 6 (2,6-12); Са2+ -10; СГ - 100 (89-118); НСО3 - 40.
Желчь характеризуется высоким содержанием кальция, поэтому длительно существующие наружные свищи желчевыводящих путей могут вызвать остеопороз и гипокальцийплазмические судороги в результате больших потерь кальция из организма.
За сутки кишки секретируют около 3000 мл кишечного сока с крайними колебаниями от 700 до 3500 мл. Его электролитный состав следующий, мэкв/л: Na+ — 111 (82-150); К+ - 4,6 (2,3-8,0); СГ -104 (43-137); НСО3 - 30; белок - 0,8.
При стимуляции желез кишок объем кишечного сока увеличивается до 6 —9 л. Такая стимуляция, например, происходит под действием токсина холерного вибриона. Секреция преимущественно осуществляется в верхней трети кишок, а всасывание — в нижней части. Поэтому при высокой кишечной непроходимости происходит разобщение между секрецией и всасыванием кишечного сока, отсюда большие потери жидкости с секретом в «третье водное пространство» и выраженные циркуляторные нарушения.
Иногда хирургическая ситуация вынуждает к наложению цекостомы. Электролитный состав цекальной жидкости следующий, мэкв/л: Na+ — 129 (80—143); К+ - 20 (5-40); Са2+ - 6; СГ - 40-60; НСО3 - 30-40.
Суточный объем экзокринной секреции поджелудочной железы равняется примерно 900 мл со следующим электролитным составом, мэкв/л: Na+ — 125; К+ — 5; СГ - 70; НСО3 - 70.
Таким образом, жидкость и электролиты выводятся в больших количествах с пищеварительными секретами в ПК (суммарный объем составляет примерно 8100 мл), но при нормальных условиях большая их часть подвергается реабсорбции. Потери воды с испражнениями составляют в среднем 100 мл. Механизмы резорбции жидкости не нарушаются, если сохраняется нормальная моторика ПК, обеспечивающая продвижение содержимого по пищеварительному каналу. Нормальная моторика (перистальтика) способствует нормальной секреции, всасыванию и осмолярности пищеварительных секретов. Поэтому одной из главных задач интенсивной терапии после операций на органах брюшной полости является восстановление перистальтики и пассажа пищи по ПК. Иногда удается восстановить перистальтику, а пассаж пищи остается нарушенным. Это бывает при стрессах, шоке, некоторых заболеваниях нервной системы. Нарушение эвакуации содержимого из ПК часто сопровождается гиперсекрецией [1,7—9].
Серотонинреактивные структуры организма и водно-электролитный обмен 255
Органы ПК иннервируются симпатическими и парасимпатическими волокнами, которые образуют сплетения в толще стенок. Симпатическое сплетение, располагающееся в подслизистом слое, получило название мейсснеровского, парасимпатическое — находится в межмышечном слое и называется ауэрбаховским. Эти сплетения обладают выраженным автоматизмом. Импульсы, поступающие к ним из высших вегетативных центров, обладают преимущественно сдерживающими свойствами. Поэтому для восстановления перистальтики иногда используют метод фармакологической денервации, применяя дробное введение ганглиоблокаторов (пентамина, димеколина, бензогексония, пирилена). Выявлено, что эта автономная функциональная активность сплетений подавляется перерастяжением (до некоторой степени защитная реакция). С учетом того факта, что перерастянутые кишки всегда отечные, а в таком состоянии их стенка практически не перистальтирует, перистальтику кишок следует стимулировать после снижения давления в них. Поэтому фармакологической денервации должны предшествовать постановка желудочного зон
да, газоотводной трубки или интубация кишок.
Перистальтика зависит от скорости и объема поступления пищи в ПК. Наиболее физиологичным является дробное и в малых количествах питание. После оперативных вмешательств на органах брюшной полости энтеральное питание обычно начинают с теплой кипяченой воды, разведенных соков и разведенного кефира. Высокой всасывательной способностью обладает регидратационный раствор, предложенный учеными Гарвардского университета в 1986 г., содержащий в 1000 мл раствора: 3,62 г натрий хлорида, 20 г глюкозы, 2,49 г натрий гидрогенкарбоната, 1,48 г калий хлорида, до 1000 мл воды дистиллированной [3]. Способ приготовления этого раствора в условиях стационара интенсивной терапии заключается в смешивании следующих количеств растворов, мл: 0,9 % раствор NaCl — 400; 5 % раствор глюкозы — 400; 3 % раствор NaHCO3 - 83; 4 % раствор КС1 - 37; вода дистиллированная — 80.
Общая осмолярность этого раствора равна 330 мосм/л. Рекомендуют принимать его внутрь малыми порциями.
8.3. СЕРОТОНИНРЕАКТИВНЫЕ СТРУКТУРЫ ОРГАНИЗМА И ВОДНО-ЭЛЕКТРОЛИТНЫЙ ОБМЕН
Ранее считалось, что в основе патогенеза функциональной кишечной непроходимости (динамической, паралитической) лежат нарушения функций парасимпатического и симпатического отделов АНС или нарушения их функциональной взаимосвязи. В настоящее время установлено, что наряду с холино- и адреноактивными структурами в регуляции перистальтики ПК принимают участие и серотонинергические структуры.
Дефицит серотонина в организме или блокада серотониновых рецепторов приводят к нарушению сократительной активности гладкой мускулатуры кишок вплоть до полного ее расслабления и паралича. Введение серотонина в организм способствует восстановлению нормальной сократительной активности, при этом эффект
носит дозозависимый характер. Сокращение гладкой мускулатуры под действием серотонина обусловлено его непосредственным взаимодействием с серотонинореактивными структурами (рецепторами) гладкой мускулатуры. Таким образом, он действует, минуя синапсы АНС, что и отличает механизм «серотониновых» сокращений от остальных.
В организме человека постоянно циркулирует около 110 мг серотонина. В норме в организме человека серотонин образуется из L-триптофана, который поступает с пищей. Примерно 1—3 % триптофана превращается в серотонин. Часть его синтезируется и метаболизируется в структурах ЦНС — это так называемый малый серотониновый цикл. Гематоэнцефалический барьер является непроницаемым для
256 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
самого серотонина, но через него проникают предшественники и метаболиты этого вещества. Другая его часть, а именно — от 80 до 95 % общего количества серотонина — синтезируется и сохраняется в виде гранул в энтерохромаффинных клетках ПК. Из этих клеток, расположенных в желудке и кишках, большая часть серотонина адсорбируется тромбоцитами и поступает в кровеносное русло, а остальная часть освобождается в просвет ПК. Серотонин вызывает агрегацию тромбоцитов. Из тромбоцитов он высвобождается в первые 15 — 20 с после начала агрегации и вызывает сильный сосудосуживающий эффект. Содержание серотонина в крови (тромбоцитах и плазме) человека в норме колеблется от 0,0179 до 0,04 мкг/мл. В случаях карциноидного синдрома его содержание возрастает до 5,2 мкг/мл. В плазме крови серотонин соединяется преимущественно с альбумином, причем одна молекула белка максимально может связать две молекулы серотонина.
В головном мозгу серотонин локализован в основном в зонах, осуществляющих регуляцию вегетативных функций. Наибольшее его количество — до 1,96 мкг/мл ткани — содержится в таламусе и гипоталамусе.
Основная часть серотонина распадается в печени, остальная — в плазме крови и других органах. Период полураспада серотонина в тромбоцитах составляет 33 — 48 ч, в желудке — 17, в кишках — И ч, в ткани мозга — от 2 до 20 мин.
В настоящее время выделяют четыре основных типа 5-гидрокситриптаминовых (5-НТ), или серотониновых, рецепторов: 5-HT-l; 5-НТ-2; 5-НТ-З; 5-НТ-4. Кроме того, согласно данным Международного комитета по номенклатуре рецепторов, каждый тип серотониновых рецепторов подразделяют на четыре подтипа, которые обозначают буквами А, В, С, D. Считается, что серотонинергические системы модулируют функцию других синаптических систем, в частности холин- и дофаминергических. 5-НТ-1-рецепторы локализованы в области шва мозга, в базальных ганглиях, на телах дофаминергических нейронов и регулируют состояния агрессивности,
импульсивного поведения, тревоги, депрессии, ухода от борьбы, сна и бодрствования. Стимуляция 5-НТ-1-рецепторов сопровождается вазодилатацией. 5-НТ-2-рецепторы широко представлены в области префронтальной коры, в лимбической системе, тромбоцитах (5-НТ-2А), дне желудка (5-НТ-2В), фибробластах (5-НТ-2С). Стимуляция 5-НТ-2-рецепторов сопровождается развитием вазоконстрикции сосудов легких, почек, сердца, мозга и усилением агрегации тромбоцитов. 5-НТ-З-ре-цепторы преимущественно локализованы на телах нейронов парасимпатической (холинергической) системы. В ЦНС активность этих рецепторов регулирует работу рвотного центра, а также сосудодвигательного солитарного тракта и area post-rema. Этот тип рецепторов отличается от других серотонинергических рецепторов тем, что он связан с лиганд-регулируемым ионным каналом и вызывает быструю деполяризацию каналов транспорта ионов калия и натрия. 5-НТ-4-рецепторы широко представлены в ЦНС в области гипо-кампа, в ПК, где участвуют в регуляции тонуса гладких мышц, перистальтики и секреции желез [10].
Серотониновые рецепторы служат молекулами-мишенями не только для серотонина, но также для многих лекарственных препаратов — антагонистов серотонина, которые взаимодействуют с серото-нин-реактивными структурами гладкой мускулатуры, тромбоцитов, ЦНС, ослабляют или нарушают его действие. Антагонистами серотонина являются все анестетики, многие психотропные препараты, адреналин, аминазин, ампициллин, атропин, верапамил, у-аминомасляная кислота, гентамицин, гидрокортизон, дикаин, димедрол, дипразин, изадрин, кетансерин, кокаин, мек-самин, морфин, новокаин, октадин, перитол, совкаин, тубазид, фентоламин.
Как известно, до, во время и после операции применяются многие из вышеперечисленных лекарственных препаратов -антагонистов серотонина, а их дозировка и количество возрастает в зависимости от тяжести состояния больного. Для снятия их отрицательного действия на гладкую мускулатуру ПК, для восстановления и
Серотонинреактивные структуры организма и водно-электролитный обмен 257
нормализации моторно-эвакуаторной функции последнего в раннем послеоперационном периоде стали применять адипинат серотонина.
Для стимуляции перистальтики доза серотонина для внутривенного введения должна подбираться индивидуально. Существует несколько методик применения серотонина с этой целью. Разовая его доза для стимуляции перистальтики кишок в послеоперационном периоде составляет 10 мг, скорость введения — 2 — 2,5 мг/мин. Для этого 1 мл 1 % раствора адипината серотонина растворяют в 20 мл физиологического раствора натрий хлорида и вводят внутривенно на протяжении 4 — 5 мин. Для предупреждения возможного спазма анального сфинктера предварительно в прямую кишку вставляют газоотводную трубку на глубину 10 —15 см за анальный жом.
Во время введения серотонина отмечается гиперемия кожных покровов на протяжении всего времени введения препарата, кратковременное тахипноэ. Эффект стимуляции перистальтики в виде появления перистальтических шумов, отхождения газов через прямую кишку, появления стула наступает непосредственно во время введения серотонина и продолжается не менее 4 часов. Этот эффект («на кончике иглы») обусловлен тем, что адипинат серотонина действует непосредственно на гладкие мышцы кишок, минуя синапсы АНС.
Стимуляция перистальтики кишок серотонином показана больным с нарушением внутрисердечной проводимости и сердечного ритма, которым противопоказано применение антихолинэстеразных препаратов, а также в тех случаях, когда другие методы стимуляции оказались неэффективными. Стимуляция перистальтики может осуществляться от 1 до 4 раз в сутки в зависимости от длительности полученного эффекта и тяжести состояния больного.
Согласно другой методике на второй день после операции 10 —20 мг адипинат серотонина растворяют в 200 мл 5 % раствора глюкозы или 0,9 % раствора натрий хлорида и вводят инфузоматом в течение 6 — 8—12 час. Обычно к концу 9 7 4-220
суток появляется перистальтика и самостоятельный стул. В случае отсутствия эффекта на следующий день дозу серотонина удваивают. Если парез кишок сопровождается нестабильной гемодинамикой и геморрагическим синдромом, то суточная доза серотонина должна составлять 40 — 60 мг. Осложнения во время стимуляции перистальтики адипинатом серотонина не зафиксированы. Это объясняется небольшой дозой серотонина, который очень быстро инактивируется. 50 % летальная доза серотонина составляет 164 мг/кг массы тела.
Обнаружено, что у больных, получавших серотонин, быстрее стабилизируется гемодинамика. Уже через 15 мин после начала инфузии серотонина отмечается умеренное повышение среднего артериального давления на 20 — 30 мм рт. ст., а через час после начала терапии минутный объем кровообращения увеличивается на 1 л/мин. В случаях дополнения серотонином кардиотонической терапии выявлена следующая закономерность: при увеличении дозы серотонина достоверно уменьшались дозы допамина — вплоть до полной его отмены на фоне стабилизации артериального давления.
Кроме того, в случаях явных анатомических нарушений нервной регуляции ПК (различные виды ваготомий, травматическое повреждение спинного мозга) серотонин восстанавливает нормальную сократительную активность гладкой мускулатуры, нормализуя моторно-эвакуаторную функцию ПК на протяжении первых суток послеоперационного периода.
Кроме симптомокомплекса функциональной кишечной непроходимости серотониновая недостаточность вне ЦНС проявляется:
а) признаками сосудистой недостаточности и нарушением микроциркуляции, в основе которых лежит нарушение сократительной активности гладкой мускулатуры сосудов;
б) появлением в крови свободного гемоглобина и миоглобина;
в) тромбоцитопенией.
Считается, что благодаря серотонину и серотониновым рецепторам поддержива
258 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
ется морфофункциональная целостность тромбоцитов. Ускоренный выход серотонина из тромбоцитов способствует их быстрому разрушению. Свободный гемоглобин и миоглобин взаимодействуют с серотониновыми рецепторами тромбоцитов и вытесняют из них серотонин.
Антагонистическим действием по отношению к серотонину обладают экзо- и эндотоксины. Они нарушают нормальное его взаимодействие с серотониновыми рецепторами нейтрофильных гранулоцитов, в результате чего снижается скорость их перемещения. Экзогенно введенный серотонин нормализует подвижность нейтрофильных гранулоцитов и таким образом улучшает неспецифическую иммунную защиту.
Карциноидный синдром характеризуется повышенным содержанием серотонина в организме. Карциноидные опухоли, возникающие в результате автономной пролиферации энтерохромаффинных клеток, могут локализовываться во всех отделах ПК, реже — в бронхах, желчном пузыре, поджелудочной железе, яичниках. Наиболее часто эти опухоли размещены в аппендиксе, реже — в прямой кишке и желудке. В случаях метастазирования опухоли в печень, когда серотонин и другие биологически активные вещества, минуя портальную систему, через печеночную вену поступают в системное кровообращение, возникают классические клинические симптомы карциноидного синдрома, а именно:
а) диарея;
б) отек лица, приливы крови к лицу; в) похудение;
г) субфебрильная температура тела; д) метастатические повреждения печени; е) повышение концентрации в крови нейроспецифической энолазы, серотонина,
Таблица 8.2. Лабораторная диагностика карциноидного синдрома
Показатель Норма, мкмоль/сут Карциноидный синдром, мкмоль/сут
5-ОУК 10,7-20,4 176,28
Серотонин 0,4-1,2 6,49
Гистамин 0,4-1,2 11,24
гистамина, 5-оксииндолилуксусной кислоты (5-ОУК).
Из внекишечных локализаций особого внимания заслуживают бронхиальные карциноиды, которые также могут метастазировать и формировать карциноидный синдром. Для диагностики используют исследования мочи на гормоны. В суточной моче определяют уровень 5-оксииндолилуксусной кислоты (продукт метаболизма серотонина), серотонина, гистамина (табл. 8.2).
Основной метод лечения карциноидов — хирургический. Концентрация серотонина в плазме может быть снижена под действием сандостатина. Доза сандо-статина для лечения карциноидного синдрома колеблется от 200 до 900 мг/сут.
От 20 до 52 % серотонина разрушается под действием моноаминоксидазы. Поэтому ингибиторы этого фермента (в частности, антидепрессанты) могут также повышать концентрацию серотонина в нервной системе. Кроме того, серотонин инактивируется гемоглобином, цитохромом С, цито-хромоксидазой, церулоплазмином. В эпифизе из серотонина образуется мелатонин. Этанол и резерпин могут изменять метаболизм серотонина в легких и мозгу с образованием буфотенина, вызывающего галлюцинации [И].
8.4. ОБМЕН ВОДОЙ И ЭЛЕКТРОЛИТАМИ МЕЖДУ ВОДНЫМИ СЕКТОРАМИ ОРГАНИЗМА
В организме происходит постоянный обмен воды между различными водными секторами. В течение суток через полости сердца проходит 7 — 8 тыс. л крови. Примерно 4—5 тыс. л воды плазмы за это вре
мя покидает сосудистое русло и возвращается обратно. Объем жидкости, перемещающейся из кровеносного русла в интерстициальное пространство и обратно в течение минуты, превышает величи
Обмен водой и электролитами между водными секторами организма 259
ну сердечного выброса в 45 раз. Вода из поперечнополосатых мышечных клеток обменивается на воду интерстициального пространства за 40 мин. В клетках ЦНС обмен воды происходит в два раза быстрее. В организме человека полный обмен воды между внутриклеточным и внеклеточным водными секторами осуществляется в течение трех часов [12].
Транспорт веществ через биологические мембраны может быть активным или пассивным. Активным транспортом называется процесс, в результате которого поддерживается неравномерное, с точки зрения термодинамики, распределение ионов между цитоплазмой и окружающей средой. Этот вид транспорта происходит только в области клеточных мембран и носит однонаправленный характер. Источником энергии для него служит расщепление макроэргических фосфатов. В процессе активного транспорта вещества могут переноситься против градиента концентрации и благодаря этому накапливаться по одну сторону мембраны в значительных количествах.
Примером активного транспорта является интегральный ферментный механизм, именуемый натрий-калиевым насосом. Сопряженный активный перенос ионов Na+ и К+ через клеточную мембрану связан с деятельностью определенного мембранного фермента. Активность этого фермента зависит от концентрации ионов Na+ в клетке и ионов К+ — во внеклеточной среде. Для переноса ионов используется энергия АТФ. В связи с этим мембранный фермент, отвечающий за деятельность натрий-калиевого насоса, получил название натрий-калий-зависимой АТФ-азы (или сокращенно Ыа+-К+-АТФ-аза). Этот фермент активируется при наличии ионов Mg2+ внутриклеточными ионами натрия, расщепляет АТФ и связывает высвободившийся фосфат. Комплекс фермент — фосфат —натрий обеспечивает перенос ионов Na+ во внеклеточную среду. Затем под воздействием внеклеточного калия происходит дефосфорилирование фермента и перенос ионов К+ в клетку. Таким образом, мембранная Ыа+-К+-АТФ-аза обладает свойствами «двойного фермента»:
9*
она действует как фосфотрансфераза и ацил фосфатаза.
Благодаря активному транспорту поддерживается различная концентрация электролитов во внутри- и внеклеточном водных пространствах: во внеклеточном содержатся главным образом ионы Na+, СГ, НСО3, а во внутриклеточном — К+, Mg2+, фосфаты, сульфаты, белки.
Разность внутриклеточных и внеклеточных концентраций электролитов образует биоэлектрический потенциал мембраны, определяющий функциональную активность (возбудимость) клеток. Мембранный потенциал клеток в норме составляет около 90 мВ. Несмотря на то что клеточная мембрана в покое плохо проницаема для ионов Na+, вследствие действия вышеуказанного активного транспортного механизма, отношение внутриклеточного и внеклеточного натрия остается постоянным и составляет в среднем по организму 1:7. Ионы калия более свободно, чем ионы натрия, проходят через клеточную мембрану, но положительно заряженные ионы калия задерживаются в покоящейся клетке в результате наличия выраженного электроотрицательного внутриклеточного потенциала. Электроотрицательный заряд наряду с большой концентрацией внеклеточных ионов натрия определяют ионы, не проникающие через мембрану, — в основном это белки и нуклеиновые кислоты. Отношение внутри- и внеклеточного калия в среднем по организму составляет примерно 30 : 1. Вода перемещается вслед за электролитами, этот процесс направлен на выравнивание осмоляльности по обе стороны биологической мембраны [1].
Пассивный транспорт воды и растворенных в ней веществ осуществляется под действием трансмембранных градиентов концентрации, осмотического и гидростатического давления. К пассивному транспорту относятся диффузия, осмос и фильтрация.
Диффузией называется процесс перемещения частиц растворенного вещества через мембрану из объема с более высокой их концентрацией в объем с более низкой. Этот процесс прекращается только после выравнивания концентрации ве
260 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
щества по обе стороны мембраны. Диффузия подчиняется таким законам:
1. Закону Фика: скорость диффузионного потока частиц какого-либо вещества прямо пропорциональна разности концентрации этого вещества и площади обменной поверхности и обратно пропорциональна расстоянию, на которое осуществляется перенос.
2. Закону Грехема: скорость диффузии обратно пропорциональна корню квадратному из величины молекулярной массы вещества.
Фильтрация — это перемещение раствора через мембрану под действием градиента гидростатического давления.
Осмосом называют физический процесс перемещения растворителя через полупроницаемую мембрану из более разведенного раствора в более концентрированный. Количественно осмотическое давление оценивается по градиенту гидростатического давления, способному предотвратить перемещение воды через полупроницаемую мембрану. Осмотическое давление раствора зависит от количества осмотически активных частиц, находящихся в определенном объеме раствора, и не зависит от свойств самого растворителя. Такими частицами являются ионы и недиссоцииро-ванные молекулы.
Следует различать два очень близких понятия, отражающих величину осмотического давления раствора, — осмоляльность и осмолярность. Первое отражает количество осмотически активных частиц, содержащихся в 1 кг растворителя, второе — количество осмотически активных частиц, содержащихся в 1 л раствора. На практике разница между этими двумя понятиями невелика. Единицами измерения осмотического давления (осмоляльности, осмолярности) является осмоль и миллиосмоль (1 осм = 1000 моем). Если 1 г/моль недиссоциирующего вещества (например, глюкозы, г/моль которой равен 180,2 г) растворить в 1 кг воды, то этот раствор будет иметь идеальную осмоляльность, равную 1 осм. Если же в 1 кг воды внести 1 г/моль поваренной соли (т. е. 58 г NaCl), то образуется раствор с идеальной осмоляльностью, равной 2 осм, так как при
растворении молекула NaCl диссоциирует на ионы Na+ и СГ, каждый из которых является осмотически активной частицей, вследствие чего в растворе появляется удвоенное число осмотически активных частиц по сравнению с глюкозой. Если при растворении молекула вещества диссоциирует на три осмотически активные частицы, например СаС12 <=* Са2+ + СГ + +С1 , то при растворении 1 грамм-молекулы этого вещества (110 г СаС12) в 1 кг воды образуется раствор с осмоляльностью 3 осм.
Основную часть осмотического давления плазмы создают электролиты, глюкоза, мочевина, белки. Осмолярность плазмы (РОсм> мосм/л) рассчитывают по формуле
Росм = 1,86([Na+] (ммоль/л) + + Глюкоза (ммоль/л) +
+ Мочевина (мммоль/л)) + 5.
1 моем = 17 мм рт. ст. = 2,27 кПа.
Соли натрия обеспечивают 90 — 95 % осмолярности плазмы и интерстициальной жидкости, а внутри клетки осмотическое давление создается в основном солями калия. Осмотическая концентрация внутриклеточной жидкости постоянно подвергается очень точной гомеостатической регуляции. В норме осмоляльности внутриклеточной и внеклеточной жидкости равны и составляют 285 — 295 мосм/кг. Изотоническим раствором называют раствор, физиологически изоосмотичный с внутриклеточной жидкостью.
Та часть осмотического давления плазмы, которую создают белки, называется коллоидно-осмотическим, или онкотическим, давлением. В норме оно равно 1,6 мосм/кг воды, или 28 мм рт. ст. Таким образом, коллоидно-осмотическое давление представляет собой очень небольшую часть всего осмотического давления плазмы (0,55 % общей осмоляльности плазмы). 85 % коллоидно-осмотического (онкотического) давления обеспечивается за счет альбумина. Несмотря на малую долю в общей осмоляльности плазмы, коллоидно-осмотическое давление имеет большое значение, поскольку существенно влияет на распределение воды между плазмой и межткане
Обмен веществ на уровне капилляров 261
вой (интерстициальной) жидкостью. Снижение концентрации альбумина в плазме ведет к снижению способности удерживать воду в сосудистом русле — возникают гипопротеинемические отеки.
Проникновение веществ через клеточную мембрану происходит посредством таких механизмов:
1) путем диффузии через бимолекулярный липидный слой клеточной мембраны;
2) через белковые каналы клеточной мембраны;
3) при помощи обратимого связывания молекулы вещества с белком-переносчиком, способным проникать через клеточную мембрану.
Скорость диффузии вещества через мембрану зависит от:
♦ степени проницаемости мембраны для данного вещества;
♦ разницы концентрации вещества по обе стороны мембраны;
♦ разницы гидростатического давления по обе стороны мембраны, сообщающей мо
лекулам дополнительную кинетическую энергию;
♦ биоэлектрического мембранного потенциала (для частиц, имеющих заряд).
О2, СО2, Н2О и жирорастворимые молекулы проходят через клеточную мембрану без участия переносчиков. Катионы Na+, К+ и Са2+ плохо проникают через клеточную мембрану, поскольку ее наружная поверхность заряжена положительно. Поэтому перенос этих катионов через клеточную мембрану осуществляется только через трансмембранные белковые каналы с участием специфических ферментных систем и затратой энергии. Глюкоза и аминокислоты проникают через клеточную мембрану с помощью белков-переносчиков. Транспорт воды между внутриклеточным и интерстициальным пространством обусловлен осмотическими силами, возникающими в результате разницы концентраций недиффундирующих растворенных осмотически активных частиц [3].
8.5. ОБМЕН ВЕЩЕСТВ НА УРОВНЕ КАПИЛЛЯРОВ
В отличие от транспорта воды через клеточную мембрану, транскапиллярный ее транспорт зависит не только от осмотических сил, но и в значительной степени от разницы гидростатического давления внутри и вне капилляра.
Кровеносные капилляры имеются во всех органах и тканях, они являются продолжением артериол, прекапиллярных артериол (прекапилляров) или чаще всего — боковыми ветвями последних. Отдельные капилляры, объединяясь между собой, переходят в посткапиллярные венулы (посткапилляры). Последние, сливаясь друг с другом, дают начало собирательным венулам, выносящим кровь в более крупные венулы. Исключением из этого правила у человека являются синусоидные (с широким просветом) капилляры печени, расположенные между приносящими и выносящими венозными микрососудами, и клубочковые капилляры почечных телец, локализованные по ходу приносящих и выносящих артериол.
Кровеносным капиллярам принадлежит важная роль в системе кровообращения; они обеспечивают транскапиллярный обмен — транспорт воды плазмы и растворенных веществ из сосудистого русла в ткани и обратно. Неразрывная связь гемодинамической и обменной (метаболической) функций кровеносных капилляров нашла отражение в их строении. Согласно данным микроскопической анатомии, капилляры имеют вид узких трубок, стенки которых пронизаны субмикроскопическими «порами». Капиллярные трубки бывают относительно прямыми, изогнутыми или скрученными в клубочек. Средняя длина капиллярной трубки от прекапиллярной артериолы до посткапиллярной венулы составляет 750 мкм, а площадь поперечного сечения — 30 мкм2. Калибр капилляров в среднем соответствует диаметру эритроцита, однако в разных органах их внутренний диаметр варьирует от 3 — 5 до 30 — 40 мкм. Стенка капилляра имеет толщину 0,5 мкм.
262 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Как показали электронно-микроскопические наблюдения, стенка кровеносного капилляра, часто называемая капиллярной мембраной, состоит из двух оболочек: внутренней — эндотелиальной и наружной — базальной. Эндотелиальная оболочка образована утолщенными клетками — эн-дотелиоцитами. Количество эндотелиоци-тов, ограничивающих просвет капилляра, обычно не превышает двух —четырех. Ширина эндотелиоцита колеблется от 8 до 19 мкм, длина — от 10 до 22 мкм.
В эндотелиоците выделяют три зоны: периферическую, зону органелл, ядросодержащую зону. Размер этих зон и их роль в обменных процессах различны. Половину объема эндотелиоцита занимают ядро и органеллы — пластинчатый комплекс (комплекс Гольджи), митохондрии, зернистая и незернистая сеть, свободные рибосомы и полисомы. Органеллы сконцентрированы вокруг ядра и вместе с ним составляют трофический центр клетки.
Периферическая зона выполняет в основном обменные функции. В цитоплазме этой зоны располагаются многочисленные микропиноцитозные везикулы и фе-нестры. Последние представляют собой субмикроскопические (50 — 65 нм) отверстия, пронизывающие цитоплазму эндоте-лиоцитов, бывают перекрыты истонченной диафрагмой, являющейся дериватом клеточной мембраны. Микропиноцитозные везикулы и фенестры, участвующие в трансэндотелиальном переносе макромолекул из крови в ткани и обратно, в физиологии называют «крупными порами».
Каждый эндотелиоцит покрыт снаружи тончайшим слоем продуцируемых им гликопротеидов, последние играют немаловажную роль в поддержании постоянства микросферы, окружающей клетки эндотелия, и в адсорбции веществ, транспортируемых через них. Соседние клетки объединяются между собой с помощью межклеточных контактов, состоящих из цитолемм смежных эндотелиоцитов и межмембранных промежутков, заполненных гликопротеидами. Эти промежутки в физиологии чаще всего отождествляют с «мелкими порами», через которые прони
кают вода, ионы и низкомолекулярные белки.
Пропускная способность межэндотелиальных промежутков различна, что объясняется особенностями их строения. Так, в зависимости от толщины интерцеллюляр-ной щели различают межэндотелиальные контакты плотного, щелевого и прерывистого типов. В плотных контактах ин-терцеллюлярная щель на значительном протяжении полностью облитерирована благодаря слиянию цитолемм смежных эндотелиоцитов. В щелевых — величина наименьшего расстояния между мембранами соседних клеток колеблется от 4 до 6 нм. В прерывистых — толщина межмембранных промежутков достигает 200 нм и более. Межклеточные контакты последнего типа в медицинской литературе также отождествляют с «крупными порами».
Базальная оболочка стенки кровеносного капилляра состоит из клеточных и неклеточных элементов. Последние представлены базальной мембраной, окружающей эндотелиальную оболочку. Большинство исследователей рассматривает базальную мембрану как своеобразный фильтр толщиной 30 — 50 нм с порами размером 5 нм, в котором сопротивление проникновению частиц возрастает с увеличением диаметра последних.
В толще базальной мембраны расположены клетки-перициты; их называют адвентициальными клетками, клетками Ру же, или интрамуральными перицитами. Перициты имеют вытянутую форму и изогнуты в соответствии с внешним контуром эндотелиальной оболочки; они состоят из тела и многочисленных отростков, оплетающих эндотелиальную оболочку капилляра, которые, проникая через базальную мембрану, вступают в контакт с эндотелиоцитами. Роль этих контактов так же, как и функции перицитов, достоверно не выяснены. Высказано предположение об участии перицитов в регуляции роста эндотелиальных клеток капилляров.
Кровеносные капилляры разных органов и тканей обладают типовыми особенностями строения, что связано со спецификой функции органов и тканей. Принято различать три типа капилляров: со-
Обмен веществ на уровне капилляров 263
магический, висцеральный и синусоидный. Стенка кровеносных капилляров соматического типа характеризуется непрерывностью эндотелиальной и базальной оболочек. Как правило, она малопроницаема для крупных молекул белка, но легко пропускает воду с растворенными в ней кристаллоидами. Капилляры такой структуры обнаружены в коже, скелетной и гладкой мускулатуре, в сердце и коре полушария большого мозга, что соответствует характеру обменных процессов в этих органах и тканях.
В стенке капилляров висцерального типа имеются отверстия-фенестры. Капилляры такого типа характерны для органов, секретирующих и всасывающих большое количество воды и растворенных в ней веществ (пищеварительные железы, кишки, почки) или же участвующих в быстром транспорте макромолекул (эндокринные железы). Капилляры синусоидного типа характеризуются большим просветом (до 40 мкм), прерывистостью эндотелиальной оболочки и частичным отсутствием базальной мембраны. Капилляры этого типа обнаружены в костном мозгу, печени и селезенке. Показано, что через их стенки легко проникают не только макромолекулы (например, в печени, которая продуцирует основную массу белков плазмы крови), но и клетки крови. Последнее характерно для органов, участвующих в процессе кроветворения [13].
Микрокапилляры мозга являются мембранным компонентом гематоэнцефалического барьера и имеют ряд существенных отличий от системных капилляров:
а) наличие плотных контактов между перекрывающими друг друга краями эндотелиальных клеток;
б) отсутствие вследствие этого каких-либо промежутков (фенестр) между эндотелиальными клетками капилляров мозга, присутствующих в микрокапиллярах другой локализации;
в) устойчивость эндотелия капилляров мозга к протеолитическим ферментам и хелаторам кальция;
г) в норме в эндотелии мозговых капилляров крайне ограничены или вообще отсутствуют процессы пиноцитоза;
д) в эндотелии капилляров мозга содержится в пять раз больше митохондрий, чем в системных капиллярах, что указывает на их значительно более высокую метаболическую активность;
е) в эндотелии мозговых капилляров так называемых безбарьерных зон мозга размер пор составляет 70 — 80 нм, что приближает их по строению и функциональным особенностям к системным капиллярам. Биологическое значение безбарьерных зон заключается главным образом в обеспечении адаптивных процессов попадания в кровь физиологически активных веществ, секретируемых определенными структурами мозга (срединным возвышением, нейрогипофизом, шишковидной железой, area postrema, структурами преоптической зоны). По мнению разных авторов, фармакологическое значение безбарьерных зон мозга сравнительно невелико в силу их незначительной протяженности по сравнению с общей поверхностью гематоэнцефалитического барьера [14, 15];
ж) в эндотелии капилляра других зон мозга размер пор гораздо мельче, чем в периферическом, и составляет 7 нм (в периферическом — 65 нм), что делает эндотелий мозговых капилляров непроницаемым для крупных молекул и относительно непроницаемым для небольших полярных молекул (например, ионов Na+ и С1“), но в то же время достаточно легко проницаемым для молекул воды. Вследствие этого ткань мозга чрезвычайно чувствительна к изменению осмоляльности плазмы. Снижение этого показателя (например, при внутривенной инфузии гипотонических растворов кристаллоидов) приводит к увеличению отека мозга, а увеличение — снижает содержание воды в ткани мозга. Поэтому внутривенное введение гипертонических растворов натрий хлорида тормозит развитие отека мозга и подъем внутричерепного давления. Для лечения внутричерепной гипертензии эти растворы не менее эффективны, чем маннитол [5].
Лимфатические капилляры представляют собой систему замкнутых с одного конца эндотелиальных трубок, выполняющих
264 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
дренажную функцию — участвуют во всасывании из тканей фильтрата плазмы и крови (жидкости с растворенными в ней коллоидами и кристаллоидами), некоторых форменных элементов крови (лимфоцитов, эритроцитов), а также в фагоцитозе (захват инородных частиц, бактерий). Лимфатические капилляры отводят лимфу через систему интра- и экстраорган-ных лимфатических сосудов в главные лимфатические коллекторы — грудной и правый лимфатический протоки. Эти капилляры пронизывают ткани всех органов, за исключением головного и спинного мозга, селезенки, хрящей, плаценты, хрусталика и склеры глазного яблока. Диаметр их просвета достигает 20 — 26 мкм, а стенка, в отличие от кровеносных капилляров, представлена лишь резко уплощенными эндотел иоцитами. Последние примерно в четыре раза крупнее, чем эндотелиоциты кровеносных капилляров. Другая особенность лимфатических капилляров заключается в наличии «якорных», или «стройных», филаментов, при помощи которых эндотелий прикрепляется к окружающим капилляры коллагеновым протофибриллам.
В связи с участием в процессах всасывания межэндотелиальные контакты в стенке капилляров имеют различное строение. В период интенсивной резорбции ширина межэндотелиальных щелей увеличивается до 1 мкм [13]. Кислород, углекислый газ, вода и жирорастворимые вещества проходят через мембрану эндотелиальных клеток в обоих направлениях. Через межклеточные щели свободно проникают только низкомолекулярные водорастворимые вещества, например Na+, К+, СГ и глюкоза. Для высокомолекулярных веществ так же, как и для белков плазмы, проникновение через межклеточные щели затруднено (за исключением печени и легких, где щели значительно шире).
В отличие от транспорта воды через клеточную мембрану транскапиллярный транспорт воды зависит не только от осмотических сил, но и в значительной степени от разницы гидростатического давления внутри и вне капилляра. Результирующий эффект осмотических и гидростатических сил на артериальном и веноз
ном конце капилляра разный — на артериальном конце вода транспортируется из сосудистого русла в интерстициальное пространство, а на венозном — в обратном направлении (рис. 8.1). Величина этих сил в различных тканях не одинакова. Гидростатическое давление в артериальном конце определяется тонусом прекапиллярного сфинктера. В капиллярах, где необходимо поддерживать высокое гидростатическое давление (капилляры почечных клубочков), тонус этого сфинктера низкий. В капиллярах скелет-
Артериальный конец т
. т/нниппппн 1 кань
Г идростатическое А давление в капилляре
-5 ммрт. ст.
6 ммрт. ст.
Онкотическое < давление плазмы
13 ммрт. ct.CZ
10 ммрт. ст.I—~~
5 мм рт. ст.
Г идростатическое А давление в v интерстиции
i Онкотическое А давление в
у интерстиции
—I 28 ммрт. ст.
/ Эффективное f фильтрационное А давление (ЭФД)
Гидростатическое к давление в
V капилляре
Г идростатическое давление в интерстиции (-5 ммрт. ст.) Онкотическое > давление в интерстиции
=□ 28 ммрт. ст.
-д—1 7 ммрт. ст.
ЭФД 13 мм рт. ст.
ЭФД 7 мм рт. ст.
6 ммрт. ст. I—
Онкотическое давление плазмы I Эффективное / I фильтрационное i давление
\ Венозный коне]
капилляра
Рис. 8.1. Схема транскапиллярного обмена воды [3]
Обмен водой между организмом человека и окружающей средой 265
ных мышц, где давление низкое, его тонус высокий.
В норме 90 % фильтруемой в интерстициальное пространство воды реабсорбируется в капилляры. Нереабсорбируемая часть воды (объемом около 2 мл/мин) из интерстициального пространства поступает в систему лимфатических капилляров и с лимфой возвращается в кровеносное русло [3].
При острой кровопотере для приведения объема сосудистого русла в соответствие с уменьшенным объемом циркулирующей крови артериолы компенсаторно
спазмируются с повышением тонуса прекапиллярных сфинктеров. При этом уменьшается кровенаполнение капилляров, что сопровождается снижением в них гидростатического давления. В результате этого укорачивается отрезок капилляра, где преимущественно происходит фильтрация воды в интерстициальное пространство, и удлиняется тот его отрезок, где преобладает реабсорция воды по градиенту коллоидно-осмотического (онкотического) давления. Это один из физиологических механизмов компенсации кровопотери.
8.6. ОБМЕН ВОДОЙ МЕЖДУ ОРГАНИЗМОМ ЧЕЛОВЕКА И ОКРУЖАЮЩЕЙ СРЕДОЙ
В норме поступление воды в организм и ее потеря из организма уравновешены. В условиях умеренного климата, при обычном питании и манере одеваться, человек потребляет в среднем 2500 мл воды в сутки. Половина этого количества поступает с питьем, а вторая половина приходится на долю воды в составе пищевых продуктов и так называемой «метаболической воды», образующейся в результате окислительного распада органических веществ. При окислительном расщеплении 1 г углеводов образуется 0,6 мл воды, 1 г жиров — 1,09 мл, 1 г белков — 0,44 мл воды. В среднем за сутки в организме взрослого человека образуется 300 мл метаболической воды [3].
Потребность организма в жидкости тесно связана с уровнем основного обмена. Как показали расчеты, потребность в воде составляет примерно 1 мл на каждую килокалорию продуцируемой энергии, или примерно 2500 мл/сут. Для вычисления часовой потребности организма в воде существует так называемое правило «4-2-1», согласно которому на первые 10 кг массы тела часовая потребность в воде равняется мл/кг (т. е. 40 мл), для следующих 10 кг массы тела — 2 мл/кг (20 мл) и для остальной массы тела — 1 мл/кг. Таким образом, для пациента с массой тела 70 кг часовая потребность в воде будет составлять: 40 мл + 20 мл + (1 мл • 50 кг) =
= 110 мл/ч, а суточная потребность: 110мл/ч • 24 ч = 2640 мл [4].
Выведение воды из организма осуществляется почками, кишками, легкими, кожей и потовыми железами. Суточные потери воды составляют примерно 2500 мл, из них 1500 мл выводится с мочой, 800 мл — испаряется (400 мл — через дыхательные пути с выдыхаемым воздухом и 400 мл — с поверхности кожи), 100 мл — выделяется с калом и 100 мл — теряется с потом. Таким образом, ежесуточный круговорот воды взрослого человека в среднем составляет около 3 — 4 % массы тела, а у новорожденного — около 10 % [1, 3].
Почки играют ведущую роль в регуляции водного баланса — они регулируют выведение воды и электролитов в зависимости от потребностей организма.
Потеря воды через кожу (испарение с поверхности) и через легкие (с выдыхаемым воздухом) получила название «незаметная потеря воды» («perspiratia insensi-bilis»), или перспирационные потери воды. Потеря воды путем испарения играет очень важную роль в терморегуляции организма и в норме обуславливает 20 —25 % теп-лопотерь организма. Величина перспирационных потерь воды определяется уровнем обмена веществ в организме, температурой тела, температурой и влажностью окружающей среды. В наших климатических условиях суточные перспирационные
266 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
потери воды составляют 540 мл в перерасчете на 1 м2 поверхности тела, т. е. 800 — 900 мл в сутки. При повышении температуры тела на один градус выше 37 °C перспирационные потери воды увеличиваются на 50 %, т. е. для температуры тела 38 °C они будут составлять 540 + (540 : 2) = = 810 мл/м2 поверхности тела. При повышении температуры тела до 39 °C перспирационные потери воды увеличиваются на 100 %, а при повышении до 40 °C — на 150 % по сравнению с нормой. Следует подчеркнуть, что перспирационные потери воды из организма осуществляются независимо от функции потовых желез, а теряемая жидкость не содержит электролитов [1,3].
Пот — это жидкость, выделяемая из организма в результате повышенной активности потовых желез. В норме пот выделяется только в складках кожи. Суточные потери воды с потом составляют от 80 до 150 мл (в среднем около 100 мл в день). Образование пота связано с потерей солей. Примерный электролитный состав пота, мэкв/л: Na+ — 46 (15 — 60); К+ - 9,4 (5-21,8); Са2+ - 1,24 (0,18 — 1,29); Mg2+ - 1,3; СГ - 54,5 (19,8-70,2); НРО^“ - 2,0 (1,7-5,9).
Хотя концентрация солей в поте меньше, чем в плазме и интерстициальной жидкости, при обильном потоотделении потери электролитов и прежде всего калия могут быть значительными. У людей, работающих физически, при повышении температуры окружающей среды, потери воды с потом могут достигать 1,6 л/ч.
Минимальные суточные потребности взрослого человека в воде составляют около 1500 мл: 500 мл необходимо для выведения шлаков почками и примерно 900 мл — испаряется. Если минимальные суточные потребности человека в воде не удовлетворяются, то, как правило, развивается гипер-
8.7. ГОРМОНАЛЬНАЯ РЕГУЛЯЦИЯ
Регуляция водно-электролитного обмена в организме осуществляется несколькими гормонами: антидиуретическим гормоном (АДГ), альдостероном, ангиотензи-
тоническая дегидратация. Скорость удаления почками осмотически активных веществ ограничена максимально возможной концентрацией таких веществ в моче, составляющей 1300 — 1400 моем/л. Чем больше таких веществ необходимо вывести, тем выше должен быть суточный объем мочи и тем значительнее потребности в воде.
Осмоляльность (осмотическая концентрация, осмотическое давление) мочи вычисляют по таким формулам:
Осмотическая концентрация мочи (мосм/кг) = 33,ЗД;
Осмотическое давление мочи (кПа) = = 75,59Д,
где Д — две последние цифры плотности мочи [2].
В норме в организме за сутки образуется и удаляется почками около 30 г (500 ммоль) мочевины. Кроме того, ежесуточно с мочой должно выделяться около 12 г (205 ммоль) натрий хлорида, поступающего в организм с пищей. Таким образом, общая осмотическая концентрация мочевины и натрий хлорида, подлежащих экскреции с мочой, составляет: 500 + + (205 • 2) = 910 моем (поскольку молекула натрий хлорида в растворе диссоциирует на две осмотически активные частицы: ионы Na+ и СГ). Для удаления такого количества осмотически активных веществ при максимальной концентрации мочи 1300 моем/л суточный диурез должен достигать 700 мл. Но поскольку с мочой выводятся и некоторые другие осмотически активные вещества, то фактически суточный диурез должен быть выше. В условиях диеты с низким содержанием белка и натрий хлорида осмотическая нагрузка почек может быть уменьшена до 200 мосм/сут. Подобная диета показана больным с олигурией.
ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
ном, ренином, предсердным натрийуретическим пептидом.
АДГ, или вазопрессин, является ведущим гормональным регулятором транспорта
Гормональная регуляция водно-электролитного обмена 267
воды. АДГ и окситоцин вырабатываются в нервных клетках супраоптического и паравентрикулярного ядер гипоталамуса, транспортируются в нейрогипофиз по су-праоптикогипояфизарным трактам, выделяются оттуда по типу медиаторов и сразу же всасываются через сосудистые сплетения в кровяное русло. Естественным стимулом секреции АДГ является возбуждение осморецепторов. Раньше предполагалось, что клетки, секретирующие АДГ, сами содержат осморецепторы. Однако в настоящее время установлено, что в качестве осморецепторов функционируют отростки субэпиндемальных ганглиозных клеток, выступающих в просвет третьего желудочка. Они оказывают влияние на секрецию АДГ посредством синаптических контактов с нейросекреторными клетками супраоптического ядра. Есть данные о наличии осморецепторов в воротной системе печени. Это объясняет тот факт, что диурез быстрее усиливается после перорального приема жидкости, чем после ее внутривенного введения. Изменение секреции АДГ происходит под влиянием рецепторов растяжения в системе сосудов, функционирующих под низким давлением. Информация от этих двух систем рецепторов, вероятно, направляется к нейросекреторным клеткам гипоталамуса по волокнам блуждающего нерва.
АДГ действует на клетки трех типов: клетки почечных канальцев, гладкомышечные клетки сосудов и клетки печени. Действуя на гладкомышечные клетки кровеносных сосудов, АДГ препятствует артериальной гипотензии при кровопотере и участвует в гомеостатическом поддержании АД (суживает сосуды). В печени эффект АДГ сходен с таковым глюкагона (он стимулирует гликогенолиз и глюконеогенез). Различают два вида рецепторов АДГ: У2-тип локализуется в почках; Vj-тип — в сосудах и печени. После взаимодействия АДГ с У2-рецепторами, расположенными на контр люминальной, т. е. обращенной к крови и лимфе поверхности канальца, в клетках стимулируется образование цАМФ. В результате этого резко повышается проницаемость дистальных извитых канальцев для воды. В отсутствие
АДГ люминальные мембраны эпителиальных клеток почечных канальцев практически непроницаемы для воды. Ингибиторы кальмодулина ослабляют биологические эффекты АДГ. Таким образом, в реализации эффекта АДГ важную роль играет комплекс кальций —кальмодулин. АДГ стимулирует синтез простагландинов и других эйкозаноидов в клетках почечных канальцев. PgE2 противодействует анти-диуретическому эффекту АДГ. В то же время вещества, нарушающие синтез простагландинов (например, индометацин), усиливают этот эффект.
АДГ оказывает выраженное влияние на ЦНС и поведение человека. В некоторых отделах мозга обнаружены Vt-рецепторы. АДГ обладает положительным действием как на консолидацию памяти, так и на мобилизацию хранимой информации. Окситоцин оказывает противоположное действие, т. е. является амнестическим пептидом [12].
В идеальном случае под действием АДГ почки экскретируют все вещества, подлежащие удалению из организма с мочой, при значительно меньшем выведении воды. АДГ повышает реабсорбцию воды в дистальных отделах нефрона путем увеличения проницаемости для воды эпителиального слоя дистальных извитых канальцев и собирательных трубочек.
При наличии АДГ реабсорбция воды в дистальных отделах нефрона происходит в два этапа. Сначала первичная моча концентрируется в корковом веществе почек. При этом гипотоническая моча, поступающая из восходящего колена петли Генле в дистальный извитой каналец, становится изотонической. Затем в собирательных трубочках, расположенных в мозговом веществе почек, происходит дальнейшее концентрирование жидкости путем реабсорбции воды, в результате чего образуется гипертоническая моча. Диурез в этих условиях замедляется примерно до 1 мл/мин и менее.
В отсутствие АДГ дистальные отделы нефрона почти непроницаемы для воды и поэтому в них происходит лишь незначительная (по сравнению с нормальным уровнем) реабсорбция воды по градиенту
268 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
осмотического давления. При этом моча гипотоническая, а скорость ее выделения увеличена. Такой тип диуреза называется водным диурезом. Максимальная скорость экскреции мочи в этих условиях может достигать примерно 15 % скорости клубочковой фильтрации, т. е. 25 л/сут, или 18 мл/мин (при максимальном антидиурезе около 0,4 л/сут, или 0,28 мл/мин).
Скорость клубочковой фильтрации — это объем фильтрата (объем первичной мочи), образующийся в почках за единицу времени. У мужчин скорость клубочковой фильтрации составляет около 125 мл/мин, а у женщин — 110 мл/мин. За сутки образуется примерно 180 л фильтрата. Следовательно, общий объем плазмы, составляющий приблизительно 3 л, фильтруется в почках за 25 мин. Иными словами, плазма очищается почками 60 раз в сутки. Аналогично можно вычислить, что вся внеклеточная жидкость организма (14 л) проходит почечный фильтрат примерно 12 раз в сутки. 15 % общей реабсорбции воды в почках регулируется АДГ — это так называемая факультативная реабсорбция воды. Облигатная реабсорбция составляет примерно 84,2 % объема первичного фильтрата и сохраняется даже в отсутствие АДГ.
Больные несахарным диабетом характеризуются абсолютной или относительной недостаточностью АДГ и постоянно пребывают в состоянии водного диуреза. Различают две формы несахарного диабета: центральную и периферическую. Первая вызывается отсутствием или снижением секреции вазопрессина в результате наследственного или вторичного поражения гипоталамо-гипофизарной системы, а именно супраоптических ядер гипоталамуса, супраоптикогипофизарных трактов, задней доли гипофиза.
Периферическая форма несахарного диабета связана с наследственным или вторичным поражением почек с развитием нечувствительности эпителия почечных канальцев к вазопрессину, в связи с этим ее еще называют нефрогенным вазопрес-синрезистентным несахарным диабетом. Причинами развития данной формы несахарного диабета являются: интерстициаль
ный нефрит с фиброзом мозгового слоя почек, поликистоз почек, врожденный гидронефроз. Биохимически нечувствительность к АДГ объясняется недостаточной продукцией цАМФ в эпителиальных клетках канальцев дистального нефрона.
Несахарный диабет может наблюдаться при относительной недостаточности вазопрессина, связанной с повышением инактивации этого гормона в печени. В этих случаях имеет место хронический ауто-имунный гепатит.
При несахарном диабете в результате выведения больших количеств воды из организма вторично развивается полидипсия, предупреждающая развитие дегидратации организма.
Лечение несахарного диабета заключается в применении препаратов вазопрессина. Назначают:
1) адиурекрин — по 0,03 — 0,05 г 2-4 раза в день вдыхать в нос;
2) при невозможности вдыхать адиурекрин его заменяют питуитрином в виде подкожных или внутримышечных инъекций по 0,5 —1,0 мл (5—10 ЕД) через 8-12 ч;
3) применение масляной суспензии ва-зопрессин-танната внутримышечно по 0,2 —0,6 мл (1—3 ЕД) один раз в 2-3 дня. Эффект достигается через 1 — 2 недели, после чего дозу уменьшают.
Умеренный симптоматический эффект при несахарном диабете могут оказывать салу ретики: парадоксальное действие в виде снижения диуреза, механизм которого неясен. Обычно назначают гипотиазид в дозе 50 —100 мг в день.
У некоторых больных с несахарным диабетом эффективно применение хлор-пропамида в дозе 0,125 —0,5 г через день. Полагают, что этот препарат усиливает действие циркулирующих в крови небольших количеств вазопрессина, а также повышает выработку АДГ. Особенно показано лечение хлорпропамидом при сочетании несахарного диабета с сахарным.
Бутадион в дозах 0,4 —0,6 г через день также снижает диурез при несахарном диабете [16—18].
АДГ можно рассматривать как элемент регуляторной системы, обеспечивающей
Гормональная регуляция водно-электролитного обмена 269
поддержание постоянства осмотического давления жидких сред организма. Повышение осмотического давления плазмы крови всего на 5 — 6 % его исходного значения обеспечивает максимальный антидиуретический эффект АДГ, одновременно появляется чувство жажды. Снижение артериального давления вызывает повышение выделения вазопрессина. Афферентные импульсы поступают в гипоталамическую область от барорецепторов каротидного синуса и дуги аорты. При снижении ОЦК импульсы поступают в гипоталамус от объемных рецепторов правого предсердия. Следует помнить, что при значительном уменьшении объема циркулирующей крови осморегуляция утрачивает свое ведущее значение и первостепенной становится задача регуляции и восстановления ОЦК [7].
Увеличение секреции АДГ может быть стимулировано различными состояниями организма: тошнотой, рвотой, болью, физической или эмоциональной нагрузкой, острой гипогликемией, гипоксией, гиперкапнией, а также лекарственными препаратами: изадрином, никотином, изопротеренолом, ацетилхолином, апоморфином, барбитуратами, опиатами, эфиром, калий хлоридом, аминазином, карбамазепином, хлорпропамидом, клофибратом, винкрис-тином, циклофосфамидом. Тормозят секрецию АДГ: норадреналин, дофаминергические антагонисты, опиатные антагонисты, алкоголь, 0-блокаторы, резерпин, дифенин, глюкокортикоиды. На молекулярном уровне освобождению вазопрессина способствуют ионы кальция. При блокаде кальциевых каналов мембран клеток верапамилом выделение вазопрессина угнетается.
Метаболизм АДГ происходит в основном в печени, частично — в почках, некоторое его количество выделяется с мочой в неизмененном виде.
Концентрация АДГ в плазме крови может быть рассчитана на основании величины осмоляльности плазмы по формуле
1g АДГ= 0,0282Р - 8,41,
где 1g АДГ — десятичный логарифм концентрации АДГ, мкЕД/мл; Р — осмоляльность плазмы, мосм/кг Н2О.
Например: при осмоляльности плазмы 300 мосм/кг Н2О концентрация АДГ в плазме равна 1,3 мкЕД/мл [2].
Под влиянием осмотических диуретиков выведение воды с мочой повышается. Осмотический диурез наблюдается в том случае, когда содержание плохо реабсорбируемых веществ в фильтрате значительно повышено по сравнению с нормой (например, после применения гипертонических солевых растворов или маннитола). Скорость диуреза при этом настолько высока, что несмотря на высокую активность АДГ в крови, сберегающий воду эффект этого гормона может полностью нивелироваться. Величина задержки жидкости в организме пропорциональна степени концентрирования мочи. В условиях осмотического диуреза при повышении скорости тока мочи ее концентрация приближается к изотонической.
При изучении внутрипочечных механизмов осмотического диуреза особый интерес представляют два факта:
1) в условиях осмотического диуреза так же, как при водном диурезе и антидиурезе, содержимое проксимальных канальцев изотонично, однако по обе стороны стенки канальца может создаваться градиент концентрации Na+;
2) избыток плохо реабсорбируемых веществ способствует удержанию воды в проксимальных канальцах. В результате этого петля Генле переполняется жидкостью, концентрирующий механизм, расположенный в мозговом слое, перегружается и эффективность его деятельности снижается.
В условиях максимального осмотического диуреза скорость мочеотделения иногда достигает 40 % скорости клубочковой фильтрации. Это может приводить к значительным потерям воды и солей.
Альдостерон, ангиотензин, ренин и предсердный натрийуретический пептид регулируют концентрацию в плазме ионов Na+, К+, Н+, а также их экскрецию с мочой, слюной, кишечным соком и потом. Вследствие этого данные гормоны обладают выраженным влиянием на водно-электролитный обмен.
Альдостерон синтезируется в клубочковой зоне коры надпочечников (кортизол и
270 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
кортикостерон — в пучковой и сетчатой зонах, андрогены — в сетчатой). Является натрийсберегающим гормоном. Увеличивает активный транспорт ионов Na+ через клеточные мембраны. В почке эффект альдостерона заключается в повышении реабсорбции ионов Na+ в канальцах и, вследствие осмотических явлений, реабсорбции воды, а также в облегчении транспорта ионов К+ и Н+ в противоположном направлении (в мочу).
Альдостерон подобным образом влияет на потовые, слюнные и кишечные железы. Содержание натрий хлорида в секрете потовых желез, которое в норме значительно ниже, чем в плазме, при недостаточности альдостерона резко возрастает. В этом случае потоотделение, вызванное повышенной температурой окружающей среды, может быстро привести к опасной потере натрий хлорида из организма. Поэтому одним из факторов адаптации организма к теплу является усиление секреции альдостерона, в результате чего содержание натрий хлорида в поте снижается.
Увеличению секреции альдостерона способствуют:
а) усиленное потребление организмом калия;
б) отрицательный баланс натрия (например, при низком содержании соли в пище или при значительных потерях натрий хлорида с потом);
в) уменьшение объема плазмы (например, при кровопотере или неадекватном потреблении жидкости).
Увеличение скорости секреции альдостерона под влиянием этих факторов осуществляется такими механизмами:
а) секретирующие гормон клетки клубочковой зоны коры надпочечников непосредственно реагируют на изменение концентрации ионов Na+ и К+ в плазме, соответствующим образом изменяя секрецию альдостерона;
б) секреция альдостерона усиливается ангиотензином II, поступающим в кору надпочечников из крови. Его можно рассматривать как «тропный» гормон для секреции альдостерона. Ангиотензин II представляет собой октапептид, образую
щийся из ангиотензина I под действием ангиотензинпревращающего гормона (карбоксикатепсина) при наличии ренина. Ангиотензин II вызывает вазоконстрикцию, стимулирует секрецию катехоламинов, вазопрессина, альдостерона, а при прямом воздействии на нейроны головного мозга — чувство жажды. Согласно современным представлениям, ангиотензин II является одним из основных элементов регуляции ОЦК.
Ренин, в свою очередь, выделяется в результате активации барорецепторов в стенках приносящих сосудов почки и натриевых рецепторов в клетках плотного пятна (это сегмент дистального отдела нефрона, где дистальный каналец контактирует с афферентной артериолой перед тем, как она входит в клубочек). В клетках плотного пятна вырабатывается ренин. Они действуют как хеморецепторы для ионов Na+. Увеличение концентрации натрия в тубулярной жидкости уменьшает высвобождение ренина. Эти два вида рецепторов являются элементами юкстагломерулярной сисгемы;
в) адренокортикотропный гормон также влияет на секрецию альдостерона, однако этот эффект выражен слабее, чем в отношении секреции глюкокортикоидов. Гипофизэктомия не приводит к столь тяжелым нарушениям обмена электролитов и воды, как удаление коры надпочечника [19].
Концентрация натрия в плазме крови и моче может служить количественным отражением уровня циркулирующего в крови альдостерона. Концентрация альдостерона плазмы определяется по формулам:
[Альдостерон] (пг/мл) =
= 1407,53 - 20,55[Na+]njl+
+ 0,080([Na+]njI (ммоль/л))2;
[Альдостерон] (пг/мл) =
= 107,138 + 0,112[Na+]MO4H-
- 0,002([№+]мочи (ммоль/л))2.
В норме концентрация альдостерона в плазме крови равна 65 ± 29 пг/мл. Он
Гормональная регуляция водно-электролитного обмена 271
имеет суточный ритм секреции, хотя и менее выраженный, чем у кортизола. Утром, в положении лежа, уровень альдостерона в плазме составляет 80 — 400 пмоль/л (3 — 14 нг/100 мл), а в вертикальном положении он удваивается [2].
У здоровых людей повышение осмоляльности плазмы на 10 — 15 мосм/кг приводит к появлению сильного чувства жажды, независимо от изменения внеклеточного объема жидкости. В то же время внеклеточная дегидратация вызывает жажду даже в том случае, если объем внутриклеточной жидкости остается на прежнем уровне или имеется гипергидратация клеток. Самым сильным стимулятором жажды является ангиотензин II, который создает крайне высокую питьевую мотивацию через рецепторное воздействие на различные структуры мозга (преоптичес-кая зона и образования вокруг третьего желудочка) [19].
В настоящее время для лечения гипертонической болезни широко используются препараты из группы ингибиторов ангио-тензинпревращающего гормона — каптоприл, капотен, эналаприл, лизиноприл и др. Часто прием этих препаратов вызывает кашель, диспноэ, бронхоспазм, приступ бронхиальной астмы в течение первых двух недель лечения (с интервалом от одного часа до шести месяцев). Возможные причины этих явлений объясняются следующим: карбоксикатепсин (ангиотензинпре-вращающий гормон) представляет собой неспецифическую дипептидную гидрала-зу с высокой активностью расщепления брадикинина и субстанции Р на неактивные метаболиты. Поэтому назначение ингибиторов карбоксикатепсина будет способствовать накоплению в организме брадикинина и субстанции Р. Брадикинин может индуцировать бронхоконстрикцию и предрасполагать к возникновению аллергических реакций.
В 1979 г. в миокарде предсердий были обнаружены гранулы, которые реагировали на изменения электролитного и водного баланса и напоминали гранулы, локализованные в эндокринных железах. В 1981 г. было установлено, что введение экстракта предсердий наркотизированным крысам
вызывает быстрое увеличение диуреза и натрийуреза. Тогда же предположили, что гранулы, содержащиеся в клетках миокарда предсердий, представляют собой гормон, принимающий участие в регуляции объема циркулирующей плазмы. Этот гормон назвали предсердным натрийуретическим фактором (ПНУФ). В литературе встречаются и другие названия ПНУФ — кардионатрин, атриопептин, аурикулин.
ПНУФ представляет собой биологически активный пептид с молекулярной массой от 2500 до 13 300 D. Синтезируется и накапливается в кардиоцитах предсердий, высвобождается в ответ на изменения их объема. Таким образом, синтез ПНУФ и его выброс в кровь происходит вследствие увеличения ОЦП либо под воздействием таких веществ, как адреналин, аргинин, вазопрессин, ацетилхолин. Уменьшение ОЦП, напротив, вызывает уменьшение синтеза и выброса ПНУФ в кровь. ПНУФ вырабатывается и накапливается в виде прогормона, превращающегося в активную форму лишь в плазме крови.
Специфические рецепторы ПНУФ расположены в разных органах и тканях — в корковом и мозговом веществе почек, почечных артериях, эндотелиальных клетках аорты, стенке мезентериальных артерий, ЦНС. После взаимодействия с рецепторами ПНУФ разрушается лизосомальными гидролазами. Это разрушение может тормозиться азидами и динитрофенолом, леупептином и пепстатином (ингибиторами лизосомальных катепсинов); хлорохином, лидокаином, метиламином и дансика-даверином (лизосомотропными веществами).
ПНУФ вызывает релаксацию гладкомышечных клеток, оказывает натрийуретическое и диуретическое действие. Механизм действия ПНУФ на клеточном уровне до конца неясен. ПНУФ не ингибирует мембранную На+-К+-АТФ-азу, не влияет на активность натрий-калиевого насоса в гладкомышечных клетках. Возможно, диуретическое и натрийуретическое действие ПНУФ обусловлено увеличением перфузии почечных артерий вследствие их дилатации. Однако существует мнение, что ПНУФ способен непосредст
272 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
венно воздействовать на процесс реабсорбции воды в почечных канальцах и собирательных трубочках. ПНУФ увеличивает почечную экскрецию ионов Na+ за счет увеличения скорости клубочковой фильтрации и угнетения реабсорбции Na+ дистальным отделом нефрона.
ПНУФ расширяет аорту, почечные и мезентериальные артерии. Однако более мелкие артерии и вены малочувствительны к его действию. Вазодилатирующее действие ПНУФ тормозится норадреналином. Ангиотензин II, потенциальный вазоконстриктор, не влияет на вазодилатирующее действие ПНУФ. По мнению некоторых авторов, ПНУФ является естественным антагонистом ангиотензина. При введении ПНУФ уменьшается сердечный выброс, ингибируется секреция ренина и уменьшается содержание альдостерона за счет угнетения его высвобож-
8.8. ОЦЕНКА ВОДНОГО
Для правильной оценки адекватности гидратации или водного статуса больного необходимо иметь анамнестические данные, оценку клинической ситуации, данные физикального осмотра, оценку функции жизненно важных органов (показатели гемодинамики, мочеотделения, лабораторных методов исследования).
Из анамнеза необходимо установить:
а) время, объем и состав принятой или введенной внутривенно (или другими путями) жидкости;
б) наличие и объем рвоты, диареи и избыточного потоотделения;
в) наличие сахарного или несахарного диабета;
г) злоупотребляет ли больной алкоголем;
д) принимались ли медикаменты, особенно диуретики.
Из клинических ситуаций, сопровождающихся существенными водно-электролитными нарушениями, следует отметить кишечную непроходимость (особенно высокую), перитонит, лихорадки, желудочно-кишечные кровотечения, ожоги, травмы, переломы крупных трубчатых костей и костей таза [4].
дения адренокортикальными клетками. Эти данные свидетельствуют о том, что ПНУФ взаимодействует с ренин-ангиотен-зин-альдостероновой системой и в комплексе с ней регулирует артериальное давление и ОЦП. У больных артериальной гипертензией, с застойной сердечной недостаточностью, пароксизмальной предсердной тахикардией, хронической почечной недостаточностью, первичным альдостеронизмом, уровень ПНУФ повышен. Во всех этих случаях повышение его уровня, по-видимому, является компенсаторным механизмом. ПНУФ можно было бы использовать как диуретик и натрийуретик при застойной сердечной недостаточности, артериальной гипертонии, хронической почечной недостаточности, однако он имеет очень небольшой период полувыведения (около 3 минут), потому его можно вводить лишь внутривенно [20].
СТАТУСА ОРГАНИЗМА
Физикальный осмотр следует производить в полном объеме, акцентируя внимание на существование следующих клинических симптомов нарушения водно-электролитного обмена:
1. Общее состояние — наличие вялости, адинамии, слабости.
2. Наличие жажды, степень ее выраженности и продолжительность.
3. Состояние кожных покровов: их сухость, влажность, тургор, эластичность, температура.
4. Состояние слизистой оболочки полости рта (загубной складки, языка) — сухость или влажность.
5. Тонус глазных яблок.
6. Тонус мышц: миорелаксация или судорожная готовность.
7. Температура тела.
8. Психоневрологический статус: уровень сознания, неадекватность поведения и суждений.
9. Характер дыхания: тахипноэ, бра-дипноэ, поверхностное или глубокое дыхание.
10. Степень наполнения кровью яремных вен и вен кистей.
Нарушения водно-электролитного обмена и их коррекция 273
И. Признаки нарушения микроциркуляции: изменение цвета слизистых, кожных покровов, ногтевых ложе, скорости наполнения капилляров кожи и ногтевых ложе.
12. Мочеотделение: величина диуреза, плотность, или удельный вес, мочи. У взрослых считается адекватным мочеотделение со скоростью 0,5 — 1 мл/кг в час. Скорость мочеотделения меньше этой величины трактуется как олигурия и может свидетельствовать о неадекватном восполнении жидкости или неадекватной гемодинамике. Мочеотделение больше 1 мл/кг в час при отсутствии применения диуретиков может отображать гипергидратацию. Дети в норме имеют более сильный диурез. У новорожденных диурез в норме составляет 2 мл/кг в час.
13. Наличие отеков периферических тканей, конъюнктивального или периорбитального.
14. Наличие отека легких.
15. Наличие асцита или выпота в плевральную полость.
16. Показатели системы кровообращения: характеристика пульса, частота сердечных сокращений (ЧСС), артериальное давление (АД), ортостатические изменения ЧСС и АД, центральное венозное давление (ЦВД), давление в легочной артерии (ДЛА), сердечный выброс и сердечный индекс [4, 9].
Для диагностики водно-электролитных нарушений наибольшую ценность имеют такие лабораторные показатели: концентрация гемоглобина (НЬ), гематокрит (Ht), общий белок плазмы, осмолярность плазмы, электролиты плазмы (прежде всего, концентрация ионов натрия), мочевина, азот мочевины, удельный вес мочи, осмолярность мочи.
8.9. НАРУШЕНИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА И ИХ КОРРЕКЦИЯ
Различают шесть видов нарушений водно-электролитного обмена: три вида дегидратаций — гипотоническую, изотоническую и гипертоническую и три таких же вида гипергидратаций.
При всех этих отклонениях существует несоответствие между скоростью поступления жидкости в организм и скоростью ее выведения из организма. Если потери превышают поступление жидкости, то развивается дегидратация, а если наоборот, то возникает гипергидратация.
В зависимости от степени отклонения от нормы различают:
первую, или легкую, степень дегидратации или гипергидратации, при которой дефицит или избыток жидкости в организме не превышает 5 % массы тела;
вторую, или среднюю, степень дегидратации или гипергидратации — дефицит или избыток жидкости в организме составляет 5 —10 % массы тела;
третью, или тяжелую, степень дегидратации или гипергидратации — дефицит или избыток жидкости в организме превышает 10 % массы тела.
Если в теряемой организмом жидкости концентрация солей больше, чем в жид
ких средах организма (плазме, интерстициальной жидкости), то развивается гипотоническая дегидратация, характеризующаяся пониженной концентрацией натрия плазмы и пониженной осмолярностью плазмы.
Если в теряемой организмом жидкости концентрация солей такая же, как в жидких средах организма, и поступление жидкости в организм отстает от потерь, то развивается изотоническая дегидратация, для которой характерна нормальная осмолярность плазмы.
Если в теряемой организмом жидкости концентрация солей меньше, чем в жидких средах организма, и поступление жидкости в организм не восполняет потери, то развивается гипертоническая дегидратация с присущими ей абсолютным дефицитом жидкости в организме и повышенной осмолярностью плазмы.
8.9.1. Гипотоническая дегидратация
При этом виде дегидратации наряду с потерей воды теряется значительное количество электролитов (прежде всего натрия), обеспечивавших нормальное осмо
274 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
тическое давление плазмы и интерстициальной жидкости. Поэтому при данном виде дегидратации осмоляльность плазмы обычно ниже нормы. В результате возникновения разности осмотического давления между внутриклеточным и внеклеточным водными секторами включается механизм выравнивания осмотического давления и вода перемещается из раствора с меньшей концентрацией осмотически активных частиц (из плазмы и интерстициальной жидкости) в раствор с большей концентрацией (в клетку). Поэтому на ранних стадиях развития этого вида дегидратации клетки не испытывают недостатка в воде.
Причины развития гипотонической дегидратации’.
а) неукротимая рвота у больных с гипер-ацидозными состояниями;
б) обильное потоотделение и неполноценное его возмещение водой, не содержащей электролитов;
в) первичная недостаточность коры надпочечников при аддисоновой болезни, туберкулезе, аутоиммунных заболеваниях, после массивного лечения глюкокортикоидами и последующей резкой их отмене (снижение синтеза альдостерона ведет к снижению реабсорбции ионов Na+ в почечных канальцах и повышенному выделению натрия с мочой и секретом потовых желез, вследствие осмотических явлений увеличивается диурез);
г) заболевания почек, сопровождающиеся нарушением реабсорбции электролитов;
д) форсированный диурез;
е) назначение салуретиков;
ж) диета, бедная натрием;
з) длительное промывание желудка и кишечника водой;
и) длительное лечение слабительными средствами.
Клинические признаки гипотонической дегидратации следующие:
1) отсутствие жажды;
2) кожные покровы не высушенные, так как отсутствует внутриклеточная дегидратация;
3) поскольку во внеклеточном пространстве уменьшается объем воды, в том числе
и во внутрисосудистом секторе, то на первый план выступают клинические признаки гиповолемии:
а) заострение черт лица;
б) спадение вен;
в) снижение АД;
г) снижение минутного объема кровообращения;
д) нитевидный пульс;
е) сухой розовый язык, металлический привкус во рту;
ж) охриплый голос, который после прополаскивания восстанавливается;
з) диурез снижен, но достаточный для выведения всех шлаков;
и) сознание обычно сохранено, и только в последней стадии дегидратации наблюдается астенизация.
Клинические проявления гипотонической дегидратации зависят от степени дефицита натрия в организме. Различают три степени дефицита натрия, ммоль/кг:
первая — дефицит до 9;
вторая — дефицит до 10 — 12; третья — дефицит до 13 — 20.
При последней степени дефицита общее состояние больного крайне тяжелое: он впадает в кому, артериальное давление снижено до 90/40 мм рт. ст.
Для лечения гипотонической дегидратации обычно используют гиперосмолярные растворы натрий хлорида. При умеренно выраженных нарушениях достаточным оказывается вливание 5 % раствора глюкозы с изотоническим раствором натрий хлорида. При значительном дефиците натрия возмещение половины дефицита осуществляется эквилибрированным или молярным (5,8 %) раствором натрий хлорида, а при наличии ацидоза коррекцию дефицита натрия проводят 4,2 % раствором натрий гидрогенкарбоната.
Расчет необходимого для коррекции натрия производят по формуле
Половина дефицита Na+ =
где [Na+]njI — концентрация Na+ в плазме крови больного, ммоль/л; 142 — концентрация Na+ в плазме крови в норме,
Нарушения водно-электролитного обмена и их коррекция 275
ммоль/л; 0,2 — содержание внеклеточной воды, % массы тела; М — масса тела, кг.
Восполнение дефицита жидкости и натрия осуществляют под контролем ЦВД и электролитного состава плазмы [1, 7 — 9].
8.9.2. Изотоническая дегидратация
Возникает при следующих потерях жидкости из пищеварительного канала:
а) аспирация желудочного и кишечного содержимого;
б) повторная рвота;
в) свищи желудка, тонкой и толстой кишок;
г) дренирование желчных путей, кисты поджелудочной железы;
д) кишечная непроходимость (особенно высокая вследствие секвестрации жидкости в просвете кишок);
е) значительное количество жидкости, содержащей электролиты, теряется при диарее, особенно при холере и других видах тонкокишечной диареи.
Другими причинами развития изотонической дегидратации могут быть: кровотечение, асцит, обширные ожоги, обильно сецернирующие раны, чрезмерно энергичная терапия диуретиками, особенно на фоне бессолевой диеты.
Клиника изотонической дегидратации определяется темпом потери изотонической жидкости — при быстрой потере значительных объемов крови и плазмы возможно развитие гиповолемического дегид-ратационного шока. Классическим примером такого шока может быть холерный ал гид. При медленной потере изотонической жидкости происходит равномерное распределение потерь воды из внеклеточного водного пространства и клеток.
В зависимости от выраженности изотонической дегидратации выделяют следующие степени ее тяжести:
I — дефицит до 2 л изотонической жидкости, клинически проявляется повышенной утомляемостью, компенсаторной тахикардией, склонностью к ортостатическим нарушениям кровообращения, хотя в горизонтальном положении регистрируется артериальное давление, близкое к нормальным показателям;
II — дефицит до 4 л изотонической жидкости, клинически проявляется снижением в горизонтальном положении систолического артериального давления ниже 90 мм рт. ст.;
III — дефицит 5 —6 л изотонической жидкости, клинически проявляется помрачением сознания, снижением ЦВД, увеличением сгущения и вязкости крови, нарушением микроциркуляции, олигурией с переходом в анурию.
При изотонической дегидратации выделение с мочой Na+ и СГ снижено вследствие повышенного поступления в кровь вазопрессина и альдостерона в ответ на снижение объема плазмы крови. Нарушения микроциркуляции, возникающие на почве гиповолемии, сопровождаются нереспираторным ацидозом и выходом ионов К+ из клеток, при этом концентрация К+ в плазме крови повышается. Для прогнозирования и правильной оценки нарушений кислотно-основного состояния (КОС) при изотонической дегидратации следует учитывать, что при потере НСО3 с пищеварительными соками из кишок будет развиваться выделительный ацидоз, а при потере ионов Н+ из просвета желудка — выделительный алкалоз. Таким образом, при потерях жидкости через пищеварительный канал в разных случаях характер нарушений КОС может быть диаметрально противоположным.
В зависимости от причины возникновения изотонической дегидратации для ее лечения используют различные изотонические растворы электролитов, коллоидов, декстранов, гидроксиэтил крахмалов.
Если причиной развития изотонической дегидратации является многократная рвота, то основными регидратационными растворами являются раствор Рингера и изотонический раствор натрий хлорида.
При холере тонкокишечное отделяемое, исчисляемое в сутки десятками литров, имеет осмолярность, аналогичную осмолярности плазмы при такой же концентрации Na+, уменьшенной концентрации СГ и в три раза большей концентрации К+. Поэтому для регидратации при холере наиболее подходит раствор Филиппса № 1 («Трисоль»), называемый еще раствором «5-4-1», так как
276 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
он состоит из 5 г натрий хлорида, 4 г натрий гидрогенкарбоната и 1 г калий хлорида на 1 л раствора. Этот раствор является универсальным при лечении холеры.
При диарее показан как можно более быстрый переход к оральной компенсации водно-электролитных потерь. С этой целью могут использоваться растворимые порошки типа элотранса и регидрона. Один пакет элотранса содержит, г: глюкозы — 4,0; натрий хлорида — 0,7; натрий цитрата — 0,59; калий хлорида — 0,3. Его растворяют в 200 мл кипяченой воды. Теоретическая осмолярность этого раствора составляет 323 мосм/л. Принимают до 20 пакетов в сутки (4000 мл).
Один пакет регидрона содержит, г: глюкозы — 10,0; натрий хлорида — 3,5; натрий цитрата — 2,9; калий хлорида — 2,5. Содержимое пакета растворяют в 1 л свежекипяченой воды. Раствор годен к употреблению в течение суток. Показанием к применению регидрона является понос у детей, требующий регидратации. Регидратация ребенка при поносе проводится через рот в течение 6—10 ч. Вводимое количество раствора рассчитывают по предполагаемой потере массы тела. Безвредный для ребенка прием за указанный срок в среднем составляет 60 мл/кг [8, 9].
Для адекватности лечения ожогов требуется в первые 36 ч после ожога восполнить потерю плазмы. Для расчета необходимого для коррекции объема плазмы (Упл, мл) используется формула Muir-Barclay
V = м - ППО
где М — масса тела, кг; ППО — площадь поверхности ожога, %.
При ожоговой болезни внутривенные вливания плазмы дополняются переливаниями изотонических растворов кристаллоидов и гипертонических растворов натрий хлорида.
Тяжесть состояния больных с кровотечениями и выбор инфузионно-трансфузионной терапии определяются объемом и темпом кровопотери. Поэтому немаловажным фактором в лечении кровотечений является определение объема крово
потери. Измерение кровопотери позволяет врачу провести адекватную инфузионно-трансфузионную терапию.
Объем крови у мужчин составляет 70 мл/кг массы тела (при умеренном питании), 60 мл/кг (при ожирении), 2,74 л/м2 поверхности тела. Его можно рассчитать по формуле
VKp = 0,367Р + 0,322М + 0,604, где VKp — объем крови; Р — рост, см; М — масса тела, кг.
У женщин объем крови составляет 60 мл/кг массы тела (при умеренном питании), 50 мл/кг массы тела (при ожирении), 2,37 л/м2 поверхности тела. Для его расчета используют формулу [21, 22]
VKp = 0,356Р + 0,ЗЗМ + 0,183.
Для определения кровопотери в родах и при кесаревом сечение могут быть использованы следующие способы измерения:
1. Способ М. А. Либова (1960). После окончания операции на детских весах взвешиваются салфетки, пропитанные кровью. При ориентировочной кровопотере менее 1000 мл более точный объем кровопотери определяют по формуле
г>
Объем кровопотери = у 15 %,
где В — масса салфеток; 15 % — поправочный коэффициент на околоплодные воды и дезраствор.
При кровопотере свыше 1000 мл ее объем определяют по формуле
ТЭ
Объем кровопотери = у 30 %.
2. Формула Нельсона
Объем кровопотери, мл/кг =
=—^—100.
0,86Ht
Процентное отношение общего объема кровопотери рассчитывают по формуле [23]
гхх о/ 0,36 У ОК
Объем кровопотери, % = • * —,
М • Ht
где У ОК — установленный объем кровопотери, мл/кг; М — масса тела, кг; Ht -гематокрит.
Нарушения водно-электролитного обмена и их коррекция 277
Таблица 8.3. Определение объема кровопотери по плотности крови и гематокриту
Плотность крови, кг/л Гематокрит, л/л Объем кровопотери, мл
1057-1054 0,44-0,40 до 500 мл
1053-1050 0,38-0,32 1000 мл
1049-1044 0,30-0,22 1500 мл
Менее 1044 Менее 0,22 Более 1500 мл
Таблица 8.4. Определение кровопотери по шоковому индексу Альговера
Индекс Альговера Объем кровопотери, % ОЦК
0,8 и выше 10
0,9-1,2 20
1,3-1,4 30
1,5 и выше 40
3. Определение кровопотери по плотности крови и гематокриту (табл. 8.3).
4. Определение кровопотери по шоковому индексу Альговера.
Шоковый индекс Альговера — это отношение частоты сердечных сокращений к систолическому артериальному давлению. В норме индекс Альговера меньше единицы. По величине индекса можно судить о величине кровопотери (табл. 8.4).
Следует отметить, что индекс Альговера не информативен у больных с гипертензивным синдромом.
5. Между величиной дефицита ОЦК и клиническими проявлениями имеется тесная взаимосвязь (М. Вейль, Г. Шубин, 1971) (табл. 8.5).
6. Дефицит объема циркулирующей плазмы может быть рассчитан по формуле
Ht
ОЦП (% исх.) =
100-Ht2 Ht2
где Ht] — исходный (должный) гематокрит; Ht2 — гематокрит у данного больного или в данный момент [2].
Главным направлением интенсивной терапии гиповолемическо-геморрагического шока является как можно более быстрое восстановление и стабилизация показателей центральной и периферической гемодинамики с целью предупреждения развития ДВС-синдрома и связанных с ним осложнений, таких как «шоковая» почка, «шоковое» легкое, «шоковая» кишка и т. д. Это достигается восстановлением дефицита ОЦК, инотропной поддержкой сердца, применением вазоактивных препаратов. Из этих методов в интенсивной терапии первоочередным является полноценное восполнение дефицита ОЦК (более подробно этот вопрос освещается в соответствующих разделах).
Известно, что организм человека способен выжить при потере 85 % функции почек, до 75 % функции печени, до 75 % кровяных телец, но не может пережить некомпенсированной потери 30 % и более общего объема крови [24 — 26]. Поэтому стандартный подход к лечению гиповолемическо-геморрагического шока заключается в проведении массивной инфузионной терапии. Для восстановления ОЦК при его дефиците используются следующие инфузионные среды:
а) сбалансированные изотонические солевые растворы;
б) гиперосмолярные растворы натрий хлорида;
Таблица 8.5. Классификация степеней гиповолемического геморрагического шока
Степень шока Клинические проявления Снижение ОЦК, % Кровопотеря, мл
Не выражена Отсутствуют Не более 10 До 500
Легкая (I) Минимальная тахикардия, снижение АД, признаки периферической вазоконстрикции 15-25 750-1250
Средняя (II) Тахикардия до 120 в минуту, снижение пульсового давления, потливость, бледность, олигурия 25-35 1250-1750
Тяжелая (III) Тахикардия более 120 в минуту, систолическое АД ниже 60 мм рт. ст. (часто не определяется, ступор, резкая бледность, анурия) Свыше 35 Свыше 1750
Таблица 8.6. Содержание электролитов в кристаллоидных растворах, мэкв/л
Раствор Na+ К+ Са2’ Mg2+ ci- НСО- Лактат Ацетат HPOJ- Н2РО; Глюкоза SO2- Осмолярность/ осмоляльность, моем/л
Изотонический (0,86 %) натрий хлорида 154 154 308
Рингера 140 4 6 — 150 — — — — — — 300
Рингера —Локка 156 2,7 3,6 — 160 2,4 — — — — 5,5* — 329
Гартмана ( Рингера-лактата) 136 4 2 2 115 — 27 — — — — — 284
«Лактасол» 140 4 3 2 116 — 30 — — — — — 290
Натрий лактата сложный 132,1 5,4 4,9 — 113,8 — 28,6 — — — — — 284,8
Хартига 45 25 — 5 45 — — 20 — 10 — — 150
Филиппса № 1 («Трисоль») 137,1 13,4 — — 102,9 47,6 — — — — — — 301
Филиппса № 2 («Дисоль») 131,8 — — — 107,4 — — 24,4 — — — — 263,6
«Ацесоль» 109 13 — — 99 — — 23 — — — — 244
Дэрроу 102,7 36,2 — — 138,9 — — — — — — — 278
Элкинтона 72 82 — — 112 — — — — 42 — — 308
Батлера 55 23 — 5 45 — 26 — 12 — 50** — 443
5 % натрий хлорида 855 — — — 855 — — — — — — — 1710
10 % натрий хлорида 1710 — — — 1710 — — — — — — — 3420
7,4 % калий хлорида — 1000 — — 1000 — — — — — — — 2000
10 % кальций хлорида — — 898 — 898 — — — — — — — 1796
25 % магний сульфата — — — 2078 — — — — — — — 2078 4156
4 % натрий гидрогенкарбоната 476 — — — — 476 — — — — — — 952
8,4 % натрий гидрогенкарбоната 1000 — — — — 1000 — — — — — — 2000
11,2 % натрий лактата 1000 — — — — — 1000 — — — — — 2000
Примечание. * Грамм (г); ** грамм/литр (г/л).
ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Нарушения водно-электролитного обмена и их коррекция 279
в) 6—10 % раствор гидроксиэтилкрах-мала;
г) декстраны;
д) свежезамороженную плазму;
е) эритроцитарную массу или отмытые эритроциты;
ж) тромбоцитарную массу.
Преимуществом кристаллоидных растворов является то, что они недороги и всегда имеются в наличии. В связи с этим они рекомендуются для интенсивной терапии кровотечений как препараты первого ряда. Наиболее часто с этой целью используются растворы Рингера, Рингера-лакта-та и изотонический раствор натрий хлорида [5].
В практике интенсивной терапии наиболее часто используются растворы кристаллоидов, приведенные в табл. 8.6.
8.9.3. Гипертоническая дегидратация
Этот вид дегидратации развивается при потерях свободной от электролитов воды (перспирационные потери) или когда потери воды превышают потери электролитов. Причины развития гипертонической дегидратации могут быть следующие:
а) абсолютный недостаток воды в пищевом рационе;
б) нарушение акта глотания при неврологических заболеваниях, ожогах и опухолях пищевода;
в) повышенное потовыделение (пот содержит натрия в 2 —4 раза меньше, чем плазма);
г) диарея с водянистым стулом (дизентерия, сальмонеллез);
д) полиурия при несахарном диабете (из организма в большом объеме выделяется гипотоническая моча вследствие абсолютной или относительной недостаточности АДГ);
е) полиурия при декомпенсации сахарного диабета (за счет высокой концентрации глюкозы в моче развивается осмодиурез);
ж) алиментарная перегрузка организма поваренной солью на фоне снижения поступления воды в организм, особенно в условиях жаркого климата;
з) дыхание через трахеостому (большие потери воды с выдыхаемым воздухом);
и) лихорадки;
к) передозировка осмодиуретиков: маннита, глицерина, мочевины;
л) патология почек, сопровождающаяся гипостену рией;
м) ОПН в полиурической стадии; н) трахеобронхит, пневмония.
При гипертонической дегидратации во внеклеточном водном пространстве организма развивается гиперосмолярность. В результате разности осмолярностей между внутриклеточным и внеклеточным водными пространствами вода перемещается под действием осмотических сил из клеток во внеклеточное пространство, поэтому при этом виде дегидратации потеря воды из организма происходит в основном за счет клеточной воды. Первыми дегидратируются клетки, содержащие большое количество воды, — клетки ЦНС, поэтому в самых начальных стадиях дегидратации появляется церебральная симптоматика: эйфория, заторможенность, делирий, двигательное возбуждение, лихорадка, судороги, кома. Другими клиническими симптомами гипертонической дегидратации являются жажда, снижение диуреза, тургора кожи, сухость кожи и слизистых оболочек, снижение слюноотделения и связанное с этим затруднение глотания, язык покрыт коричневым налетом и несколько уменьшен в объеме. Чувство жажды появляется на ранних стадиях. Лабораторными симптомами гипертонической дегидратации являются: повышение концентрации натрия и хлора плазмы, снижение уровня калия в плазме; симптомы сгущения крови: повышение гематокрита, концентрации белка плазмы, повышение плотности мочи с увеличением содержания натрия мочи (за исключением несахарного диабета).
Истинное содержание воды в организме вычисляют по формуле [9]
[Na+] П71 должная
VB = И • 0,6-—------------,
[Na ]пл истинная
где VB — объем воды, л; И — масса тела, кг; 0,6 — содержание воды в организме, выраженное в долях единицы.
280 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
В зависимости от выраженности клинической картины и объема дефицита жидкости различают три степени гипертонической дегидратации:
I — характеризуется дефицитом 1 — 2 л воды в организме, клинически проявляется жаждой и олигурией;
II — дефицитом 4 —5 л воды в организме, клинически проявляется жаждой, олигурией, сухостью кожи, слизистых оболочек, языка; общей слабостью, беспокойством, двигательным возбуждением — признаками нарастания дегидратации головного мозга;
III — дефицитом 7-8 л воды в организме, клинически проявляется признаками выраженной дегидратации клеток головного мозга (нарушением сознания, гипертермией) и признаками тяжелого де-гидратационного шока (снижение АД до шоковых величин, нарушение микроциркуляции).
В случае наличия гипертонической дегидратации дефицит воды в организме вычисляют по формуле [7]
Дефицит Н2О (л) = [Na+]njI-142 =------ТГ2-----0.6М,
где 0,6 (60 %) — общее содержание воды в организме по отношению к массе тела; И — масса тела, кг.
Лечение гипертонической дегидратации осуществляют возмещением жидкости растворами с низким содержанием электролитов и достаточным количеством свободной воды.
При легкой степени дегидратации для ее коррекции достаточно назначить сладкий чай, если больной может самостоятельно пить. При внутривенном введении жидкости назначают 5 % раствор глюкозы с прибавлением до 1/3 объема изотонического раствора натрий хлорида. Если позволяет состояние больного, то регидратацию проводят в умеренном темпе, поскольку, во-первых, быстрое и обильное введение растворов глюкозы может снизить осмоляльность внеклеточной жидкости и создать условия для стремительного перемещения воды в клетки головного моз
га с развитием его отека-набухания, а, во-вторых, существует опасность усиления диуреза и дополнительной потери жидкости.
При тяжелой дегидратации, явлениях дегидратационного гиповолемического шока, нарушении микроциркуляции и централизации кровообращения необходимо срочное восстановление показателей гемодинамики, которое достигается восполнением объема внутрисосудистого русла не только растворами глюкозы, которые быстро покидают его, но и коллоидными растворами, задерживающими воду в сосудистом русле и снижающими темп поступления жидкости в головной мозг. В таких случаях инфузионную терапию начинают с вливания 5 % раствора глюкозы, прибавляя к нему до 1/3 объема реополи-глюкин, 5 % раствор альбумина и поли-глюкин. Необходимость переливания рео-полиглюкина вызвана тем, что, помимо обеспечения гемодилюции, этот коллоид обладает антиагрегационными свойствами за счет эффекта силиконизации и поэтому предупреждает развитие диссеминированного внутрисосудистого свертывания крови. При тяжелой степени дегидратации показано назначение гепарина по 2,5 тыс. ЕД через 6 ч подкожно в область пупка.
Гипертоническая дегидратация сопровождается выходом ионов из клеток и потерей значительного их количества с мочой. Поэтому при данном виде дегидратации всегда имеет место абсолютный дефицит калия в организме — гипокалий-гистия, которую начинают корригировать с восстановлением диуреза до 30 мл/ч [27-29].
Более упрощенный, без учета массы тела, способ регидратации состоит в вычислении дефицита воды в организме только по избытку натрия. Эмпирически установлено, что на каждые 3 ммоль/л избыточного плазматического натрия в организме необходимо ввести 1 л воды. Разницу между фактической и должной концентрацией плазменного натрия делят на 3 и получают величину дефицита воды в литрах. Следует заметить, что этот расчет коррекции является весьма приблизительным.
Степень дегидратации, а также ориентировочный объем регидратации можно
Нарушения водно-электролитного обмена и их коррекция 281
оценить пробой Мак-Клюра —Олдрича. Она заключается во внутрикожном введении в область сгибательной поверхности предплечья 0,2 мл 0,9 % раствора натрий хлорида и в учете времени рассасывания волдыря. Нормальное время рассасывания волдыря — 50 мин. Если оно составляет 20 мин, то объем дефицита воды у взрослого человека достигает 5 л [9].
Процесс регидратации контролируют по восстановлению диуреза, улучшению общего состояния больного, увлажнению слизистых оболочек, снижению концентрации натрия плазмы крови. Большое значение имеет контроль ионограммы (прежде всего для устранения дефицита калия) и ЦВД, отражающего величину притока крови к сердцу.
8.9.4. Гипотоническая гипергидратация
Возникает в результате введения в организм изотонических растворов метаболизирующих веществ (глюкозы, сорбита, натрия лактата) в избыточном количестве. Гипотоническая гипергидратация сопровождается гипонатрийплазмией, гипохлор-плазмией и наблюдается при следующих состояниях:
1. Избыточном поступлении в организм «свободной» воды в количествах, превышающих возможности выделения из организма, при:
а) промывании водой (без солей) мочевого пузыря и ложа предстательной железы после ее трансуретральной резекции;
б) утоплении в пресной воде;
в) избыточном вливании растворов глюкозы в олигоанурической стадии ОПН.
2. Снижении гломерулярной фильтрации в почках при острой и хронической недостаточности почек.
3. Застойной недостаточности сердца при асците.
4. Дефиците глюкокортикоидов, микседеме.
5. Назначении сильнодействующих диуретиков типа фуросемида и этакриновой кислоты больным с синдромом Барттера (врожденная недостаточность почек — нарушение реабсорбции ионов Na+ и К+ из первичной мочи).
6. Эктопической продукции вазопрессина следующими опухолями: тимомой, овсяно-круглоклеточным раком легкого, аденокарциномой двенадцатиперстной кишки и поджелудочной железы; при туберкулезе и аспергиллезе.
7. Повышенной продукции вазопрессина при поражениях гипоталамической области, менингоэнцефалите, гематоме, врожденных аномалиях и абсцессе головного мозга.
8. Назначении лекарственных средств, повышающих продукцию вазопрессина (вин-кристин, морфин, окситоцин, барбитураты).
Гипотоническая гипергидратация является внутриклеточной и сопровождается церебральной симптоматикой, клиническими признаками отека мозга. Наблюдаются следующие клинические симптомы: общая слабость, разбитость, головная боль, спутанность сознания, судороги, кома, водная лихорадка, мигрирующая очаговая мозговая симптоматика, кишечные спазмы, понос; АД вначале повышено, затем снижается, возможна брадикардия; объем мочи вначале увеличен, она характеризуется незначительной относительной плотностью, позднее — олигурия с переходом в анурию («осмотический нефроз»).
Большинство больных не требует интенсивной терапии и для выравнивания натрийплазмии достаточно ограничения поступления в организм «свободной» воды. Больные, особенно с острым развитием синдрома, выраженными клиническими проявлениями со стороны нервной системы, прежде всего головного мозга, нуждаются в неотложной терапии в связи с угрозой развития отека мозга. В таких случаях рекомендуется осторожное внутривенное введение гиперосмолярных растворов натрий хлорида. Применяются 3 %, 5 %, 5,84 % растворы натрий хлорида. При введении натрия одновременно повышается эффективность диуретиков. При возрастании уровня натрийплазмии выше 130 мэкв/л дальнейшего введения в организм натрия не требуется.
Изложенная выше инфузионная терапия может представлять опасность, поскольку введение гиперосмолярных растворов натрий хлорида увеличивает объем внекле
282 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
точной жидкости в организме и может достигать порога компенсации кровообращения. При возможной декомпенсации сердечной деятельности предпочитают назначать фуросемид с одновременным введением растворов калий хлорида. Методом выбора терапии гипотонической гипергидратации является ультрафильтрация. На фоне гипотиреоза полезно назначение тиреоидина и глюкокортикоидов.
При синдроме неадекватной секреции вазопрессина назначают ингибиторы секреции или действия вазопрессина — этиловый спирт, литий карбонат (0,9 г/сут), димеклоциклин (600—1200 мг/сут). Литий является неконкурентным ингибитором действия вазопрессина на аденилатцикла-зу, а также на следующих за синтезом цАМФ этапах. В результате развивается относительная нечувствительность эпителия почечных канальцев к вазопрессину (АДГ) и нарушается концентрирующая функция почек. Димеклоциклин (диметилхлорте-трациклин) — антибиотик, избирательно отрицательно влияющий на концентрирующую функцию почек и не затрагивающий другие их функции. Этот антибиотик подавляет аденилатциклазную и протеин-киназную активность.
Аналоги вазопрессина, например вазопрессиновая кислота, препятствуют действию АДГ, конкурентно связываясь с рецепторами на плазматической мембране, но при этом не инициируют биологическую активность.
Гипокалиемия и гиперкальциемия снижают чувствительность рецепторов к АДГ, в результате чего уменьшается реабсорбция воды (нарушается концентрирующая способность почек).
При алиментарной дистрофии, хронических истощающих заболеваниях с развитием «синдрома больных клеток» корригирующая инфузионная терапия неэффективна. Основное внимание следует уделить терапии основного заболевания [7].
8.9.5. Изотоническая гипергидратация
Изотоническая гипергидратация возникает при избыточном введении в организм изотонических растворов натрий хлори
да, при развитии отеков, вызванных застойной недостаточностью сердца, циррозом печени, острым гломерулонефритом, хроническим гломерулонефритом с нефротическим синдромом, болезнью Кушинга, терапией кортикостероидами, во время предменструального периода, беременности.
Клиника изотонической гипергидратации зависит от темпа внутривенного введения растворов. При быстром введении развивается внутриклеточная гипергидратация, проявляющаяся интерстициальными отеками, повышением ЦВД и АД, вначале увеличением диуреза, а затем -уменьшением. При хорошей функциональной активности сердечно-сосудистой системы развивается гипердинамический синдром, сопровождающийся повышением АД и учащением пульса.
Лабораторными симптомами изотонической гипергидратации являются: снижение гематокрита ниже 0,4, количества эритроцитов в периферической крови, общей концентрации белка плазмы.
В случаях тяжелой степени изотонической гипергидратации развивается внутриклеточная гипергидратация с клиническими признаками отека мозга.
Построение программы терапии изотонической гипергидратации зависит от основного заболевания, при котором наблюдаются задержки натрия и воды. Наличие отеков при нормальной осмоляльности плазмы свидетельствует о переполнении интерстициального отдела внеклеточного пространства. Главная цель — терапия основного заболевания, вызывающего отеки. Кроме того, для ликвидации изотонической гипергидратации следует прекратить внутривенное введение растворов и применить выжидательную тактику. При этом потеря воды из организма будет происходить с рвотными массами, стулом, мочой. Назначают рисово-фруктовую безсо-левую диету и один из нижеперечисленных диуретиков: маннитол, сорбитол, мочевина в дозе 1 г/кг, этакриновая кислота — 50 мг/сут, диакарб — 250 — 500 мг/сут, альдактон, верошпирон, три-амптерен 1,5 — 2 мг/кг, меркузал, нову-рит — 1 мл внутримышечно. При нали
Обмен электролитов 283
чии дефицита белка (отеки при циррозе печени, нефротические отеки) переливают растворы альбумина. В критических состояниях показано применение ультрафильтрации крови [7].
8.9.6. Гипертоническая гипергидратация
Характеризуется избытком в организме воды и растворенных в ней веществ, по-вышением осмотического давления плазмы, при этом клетки обезвоживаются.
Причины возникновения гипертонической гипергидратации следующие:
энтеральное поступление насыщенных солевых растворов, например питье морской воды;
парентеральное введение изотонических или гипертонических солевых растворов при ограниченной функциональной способности почек, например после операций, при стрессовых состояниях, ОПН, остром гломерулонефрите, особенно при чрезмерном внутривенном введении растворов натрий гидрогенкарбоната (осмолярность 4,2 % раствора натрий гидрогенкарбоната — 1000 мосм/л);
опухоли надпочечников (синдром Конна);
вторичный альдостеронизм при застойной сердечной недостаточности или циррозе печени.
Клиническая симптоматика гипертонической гипергидратации включает признаки внеклеточной гипергидратации и внутриклеточной дегидратации. Клиническими признаками внеклеточной гипергидратации являются: увеличение ОЦК, высокое ЦВД, снижение диуреза вплоть до олигурии, периферические отеки, увеличение концентрации натрия плазмы выше 155 ммоль/л и калия плазмы — выше 5 ммоль/л.
Клиническими симптомами внутриклеточной дегидратации следует считать гиперрефлексию, пирексию, беспокойство, возбуждение, кому вследствие осмотического обезвоживания клеток.
Развитие гипертонической гипергидратации возможно заподозрить в ситуации, когда проводится инфузионная терапия солевыми растворами в большом объеме при одновременном сокращении диуреза.
Лечение гипертонической гипергидратации заключается в назначении безсолевой диеты, инфузии растворов сахаридов наряду со стимуляцией диуреза салуретиками (фуросемид), 5 % раствора альбумина; при тяжелых формах прибегают к гемодиализу.
8.10. ОБМЕН ЭЛЕКТРОЛИТОВ
8.10.1. Обмен натрия
Натрий является одним из основных электролитов человеческого организма. Он содержится в основном во внеклеточном пространстве и считается его главным катионом. Na+ как катион уравновешивается анионами СГ, НСО3, НРО^“, Н2РО/
У человека массой 70 кг содержание ионов Na+ в организме составляет примерно 4200 ммоль, 70 % этого количества, т. е. 3000 ммоль, — это так называемый обменноспособный натрий. Около 55 % общего количества натрия находится в костях, 43 % — во внеклеточной жидкости и 2 % — в клетках. Существует динамическое равновесие между внеклеточным, внутриклеточным натрием и натрием костей.
При избыточном поступлении в организм натрия, он депонируется в костях либо выводится из организма. При недостаточном энтеральном поступлении натрий может быстро мобилизовываться из костей (примерно 10 % общего содержания в костях). При длительно существующей гипонатриемии может использоваться 30 — 40 % натрия костей.
При поступлении натрия в больших количествах он накапливается в костях — это так называемая сухая задержка натрия. Если его поступление продолжается, то развиваются отеки («влажная» задержка натрия). Поскольку в послеоперационном периоде выделительная функция почек снижена, то избыточное введение натрия в это время приводит к его
284 ФИЗИОЛОГИЯ И ПАТОЛО1 ИН видни-JJitM rujii/i i nui и uuivili m
задержке в травмированных тканях, возникновению отека и нарушению проходимости, а, возможно, и несостоятельности анастомозов. Поэтому в послеоперационном периоде следует взвешенно подходить к назначению растворов натрий хлорида.
Натрий постоянно теряется из организма с мочой, потом и стулом. Суточная потребность в нем организма составляет 90мэкв/м2 поверхности тела. В норме концентрация натрия в суточной моче колеблется от 100 до 260 ммоль. Почки регулируют выделение натрия в зависимости от потребностей организма. Выделение натрия с мочой может полностью прекратиться, но он будет теряться со стулом (7 — 8 ммоль/сут) и потом (0,8 ммоль/сут). Если в организм вводить 300 мэкв/м2 натрия в сутки (это примерно 30 г натрий хлорида), то почки еще справляются с выведением избыточного натрия, но при поступлении в большем количестве натрий задерживается в организме. Если же элек-тролитвыделительная функция почек снижена, то и при небольшом введении может наблюдаться задержка натрия в организме. Накопление натрия и воды в местах операционной травмы создает «третье водное пространство».
Натрий всасывается из ПК в неограниченном количестве. Этот процесс зависит от биологической активности слизистой ПК. В течение одной минуты 60 % всосавшегося натрия выходит из сосудистого русла в интерстициальное пространство и примерно такое же его количество возвращается из интерстиция в сосудистое русло. Обмен Na+ между клеткой и межклеточным пространством в норме менее интенсивный, но возрастает в период функциональной активности клетки: в клетку натрий поступает в период деполяризации клеточной мембраны, а из клетки выходит в фазу реполяризации.
Физиологическая роль натрия в организме заключается в следующем:
1. Входит в состав костей.
2. Является основным внеклеточным катионом, определяющим осмолярность внеклеточной среды. Если изменяется концентрация натрия во внеклеточном водном пространстве, то это неизбежно вле
чет изменение осмолярности. За счет других катионов компенсации не происходит.
3. Способствует поддержанию кислотно-основного состояния крови, являясь катионом фосфатной буферной системы: Na2HPO4/NaH2PO4. Снижение концентрации натрия плазмы ведет к уменьшению буферной емкости внеклеточной среды.
4. Один из факторов, способствующих движению и обмену воды. Натрий относится к гидрофильным ионам. В проксимальных извитых канальцах почек происходит активная реабсорбция ионов Na+, вода следует за ним. Потеря натрия из организма сопровождается потерей воды. С потерей 1 мэкв Na+ организм теряет 6 мл воды.
5. Вместе с калием определяет величину мембранного потенциала клетки, а следовательно, и функциональную активность клетки.
6. Регулирует нейромышечную проводимость — повышает ее.
7. Внутриклеточные ионы натрия вызывают конформацию молекул ферментов, необходимую для точной ориентации их каталитических групп.
8. Натрий-кальциевый обмен на уровне клеточной мембраны кардиомиоцита рассматривают как элемент регуляции сократимости миокарда, уровня мембранного потенциала и, возможно, спонтанной электрической активности.
9. Ионы натрия повышают возбудимость симпатических нервных окончаний, а вместе с ионами кальция — сосудистый тонус.
10. Ионы натрия участвуют в регуляции внутриклеточного метаболизма.
Для перехода клетки от стадии роста к делению необходимо затормозить процессы белкового синтеза. Этому способствует снижение концентрации ионов Na+ и увеличение количества ионов К+ в клетке (создаются неблагоприятные условия для функционирования рибосом) при активном участии №+-К+-АТФ-азы. В связи с этим предложено использовать величину активности ферментов для определения стадии клеточного цикла синхронно делящейся культуры клеток. Существует прямая зависимость между активностью Ыа+-К+-АТФ-азы, темпами выделения на
Обмен электролитов 285
трия из клетки и замедлением в ней процессов белкового синтеза.
В основе влияния Ыа+-К+-АТФ-азы на рост клетки лежит действие ионов Na+ на транскрипцию генетической информации. Ионы Na+ — важный фактор дестабилизации структуры ДНК (J. Ling, 1984). Он нарушает водородные связи между нуклеотидами (аденин —тимин, гуанин —цитозин), способствуя развертыванию двойной спирали и увеличению доступности генетической информации в клетке (Р. Hobza, С. Sandorfy, 1984). Таким образом, высокий уровень ионов Na+ в клетке является необходимым условием протекания процессов белкового синтеза.
Однако взаимоотношения между ионным гомеостазом и состоянием генетического аппарата клетки очень сложны. Представление о том, что изменение соотношения Na/К непосредственно управляет считыванием информации с генетического материала, влияя на стабильность ДНК, является слишком упрощенным, а системы, существующие по такому принципу, были бы недостаточно устойчивы.
Если ионы Na+ действительно дестабилизируют структуру двойной спирали, то как же сохраняется ее структура в покоящейся клетке, где содержание натрия постоянно поддерживается на довольно высоком уровне (около 30 ммоль/л в нейронах и 35 — 40 ммоль/л в мышечных клетках)?
Стабилизатором двойной спирали ДНК в клеточном ядре являются молекулы воды. На каждый шаг спирали ДНК приходится 240 — 400 молекул воды. Ионы Na+ в клетке также гидратированы в соответствии с величиной их гидратационного числа. Эта связанная вода предохраняет двойную спираль от дестабилизирующего действия ионов Na+. В период деления, когда ионный состав клетки уравнивается с таковым во внеклеточной среде, происходит повышение содержания ионов Na+ сверх нормального уровня (с одновременным понижением уровня калия). Этот процесс стимулирует белковый синтез и рост клетки, одновременно активируя синтез новых молекул Ыа+-К+-АТФ-азы. В результате активации фермента уровень натрия
в клетке снижается, структура ДНК стабилизируется и рост клетки тормозится до нового деления.
Таким образом, в нормальной клетке этот фермент принимает участие в регуляции длительности клеточного цикла, что недавно было продемонстрировано на синхронизированной культуре фибробластов с помощью специфического ингибитора Ма+-К+-АТФ-азы — уабаина. На стадии интенсивного роста культуры клетки оказались весьма чувствительны к его наличию: ингибирование активного транспорта ионов Na+ и К+ в несколько раз удлиняло эту стадию цикла.
Иная картина наблюдается, когда в клетку попадают гидрофобные индукторы трансляции, вызывающие злокачественное перерождение тканей (например, карболовые эфиры). Они выступают как дополнительный фактор дестабилизации нуклеиновых кислот, что приводит к повышению активности Ка+-К+-АТФ-азы и снижению уровня содержания ионов Na+. Если мощности Ма+-К+-АТФ-азы оказывается недостаточно, то гидрофобная модификация хромосомного материала будет или способствовать развертыванию двойной спирали и экспрессии собственных онкогенов клетки, или облегчать встраивание вирусных онкогенов в состав ДНК.
Согласно данным экспериментальной онкологии, содержание ионов Na+ в злокачественных клетках существенно возрастает, а активности Ыа+-К+-АТФ-азы недостаточно для удаления этого натрия из клетки, поскольку клеточные мембраны становятся высокопроницаемыми для него. Было показано, что активность Ма+-К+-АТФ-азы в злокачественных клетках выше, чем в нормальных тканях или в клетках доброкачественной опухоли. Однако натриевый насос работает в «разобщенном» режиме, гидролизуя АТФ без активного переноса ионов через мембрану. По мнению С. Д. Кузьмина, в злокачественных клетках Ыа+-К+-АТФ-аза не в состоянии выполнять свою функцию, поскольку для них характерен анаэробный гликолиз. Избыточное количество молочной кислоты — основного продукта гликолиза — закисляет внутриклеточную
286 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
среду и заставляет клетку обменивать ионы Н+ на внеклеточный натрий. Вследствие работы Na/H-антипортера в клетку постоянно поступает натрий.
Многие цитостатики, применяемые для торможения роста опухолевых клеток, обладают способностью активировать Na+-, К+-насос. Таков, например, перспективный антибиотик блеомицин.
Другим возможным объяснением того, что активация Ыа+-К+-ЛТФ-азы тормозит белковый синтез, является конкуренция за клеточный фонд АТФ процессов переноса калия и натрия, являющихся весьма энергоемкими.
Стимуляция общего роста организма, осуществляемая при участии гормональных препаратов, например, тиреоидных гормонов, в нормальных условиях всегда сопровождается активацией Na+-K+-АТФ-азы. Между этими процессами наблюдается своего рода динамическое равновесие.
Ма+-К+-АТФ-аза обладает наибольшей ионотранспортной активностью из всех транспортных систем.
Натриевый насос выделяет из организма избыток натрия — по этой причине морские птицы альбатросы не нуждаются в пресной воде, они могут пить морскую воду, опресняя ее. Одновременно натриевый насос участвует в регуляции состояния воды в цитоплазме, что объясняется меньшей гидрофобностью Na+ по сравнению с К+. Гидратное число К+ равно 10,5, в то время как для Na+ оно составляет 16,6 молекулы воды на один ион. Это означает, что в процессе переноса трех ионов Na+ в обмен на два иона К+ (в элементарном акте натриевого насоса) в клетке уменьшается количество связанной воды из-за создания асимметричного распределения ионов, и наоборот, изменение агрегатного состояния воды в клетке может способствовать активизации транспорта ионов.
В норме концентрация ионов Na+ в плазме колеблется в пределах 136 — 145 ммоль/л. Гипернатриемией называется состояние, при котором концентрация натрия в сыворотке крови превышает 145 ммоль/л.
В зависимости от соотношения концентрации ионов Na+ в плазме и абсолютного содержания натрия в организме различают три разновидности гипернатриемии.
1. Гипернатриемия, обусловленная увеличением абсолютного количества натрия в организме.
Причины увеличения абсолютного содержания натрия в организме и соответственно его концентрации во внеклеточной жидкости могут быть различными, но во всех случаях потребление натрия должно превышать его выделение из организма. В хирургической практике это чаще всего связано с дефектами проведения интенсивной терапии: гипернатриемия развивается при избыточном внутривенном вливании растворов натрий хлорида, когда суммарная суточная доза превышает 30 г натрий хлорида. Другими причинами развития этого вида гипернатриемии могут быть: увеличение реабсорбции ионов натрия в почечных канальцах, вызванное повышением уровня альдостерона крови; усиленная секреция глюкокортикоидов, способствующих задержке натрия в организме. Эти явления практически всегда наблюдаются в первые 3 — 5 дней послеоперационного, посттравматического периода, поэтому этот период называется периодом ранней ретенции натрия.
В клинической картине гипернатриемии при выраженном увеличении концентрации натрия в сыворотке крови могут отмечаться: лихорадка, сильная жажда, повышенная возбудимость, утрата сознания, клонико-тонические судороги, остановка дыхания.
Лечение этого вида гипернатриемии заключается во внутривенной инфузии 5 % раствора глюкозы на фоне стимуляции диуреза сосудистыми диуретиками и салу ретиками.
Гипернатриемия в результате первичного альдостеронизма (при опухолях надпочечников) развивается медленно, содержание натрия в плазме повышается до 170 мэкв/л. Характерными для этого вида гипернатриемии являются уменьшение в плазме концентрации калия и хлора, задержка воды во внеклеточном пространстве и образование отеков. Назначение в этой ситуации салуретиков ведет к потере
Обмен электролитов 287
организмом калия. Поэтому предпочтение отдают ингибиторам альдостерона: веро-шпирону, альдактону, амилориту, триамте-рену. К последним двум реже возникает тахифилаксия.
2. Гипернатриемия может развиться при уменьшении общего содержания воды в организме и неизменном абсолютном количестве натрия. Такое состояние соответствует гипертонической дегидратации.
При гипернатриемии с нейротоксическим синдромом в результате патологических процессов в головном мозгу повреждаются центры осморегуляции, расположенные в диэнцефальных отделах мозга (осморецепторы, паравентрикулярные и супраоптические ядра гипоталамуса, гипо-таламо-гипофизарные тракты, задняя доля гипофиза). Это приводит к перестройке осморегуляции на более высокий уровень осмотического давления жидкостных сред организма. Запрограммированное более высокое осмотическое давление плазмы обеспечивается за счет более высокой концентрации в плазме натрия. Назначение салуретиков в этой ситуации с целью устранения гипернатриемии и гиперосмолярности плазмы не дает эффекта, поскольку натрий начинает утилизироваться из костного депо и возникает парадоксальная гипернатриемия на фоне уменьшения абсолютного количества натрия.
При гипонатриемии наблюдается снижение концентрации ионов натрия в сыворотке крови ниже 135 мэкв/л. В основе развития гипонатриемии лежит отрицательный баланс натрия, или положительный баланс воды в организме.
Различают:
а) абсолютную гипонатриемию, когда снижение концентрации натрия в плазме сопровождается уменьшением абсолютного количества натрия в организме;
б) гипонатриемию без уменьшения абсолютного количества натрия в организме;
в) гипонатриемию на фоне увеличения абсолютного количества натрия в организме (парадоксальную).
Одним из основных механизмов развития гипонатриемии при хирургической патологии является потеря организмом жидкостей, содержащих натрий в большей
концентрации, чем внеклеточная жидкость. Это явление наиболее часто встречается при различных заболеваниях пищеварительного канала — свищах, энтеритах, назначении солевых слабительных. В таких случаях отчетливо прослеживается связь между обменом воды и обменом натрия. Гипонатриемия является обязательным симптомом гипотонической дегидратации.
Угнетение синтеза и освобождения АДГ сопровождается большими потерями свободной воды с мочой. Различная патология головного мозга может привести к повышенной секреции АДГ, что сопровождается увеличенной почечной экскрецией натрия (более 20 ммоль/л мочи), несмотря на гипонатриемию и гипоосмоляльность плазмы. При этом осмоляльность мочи по сравнению с осмоляльностью сыворотки крови высокая.
Основой лечения синдрома повышенной секреции АДГ является ограничение инфузии жидкости в организм — обычно не более 1000 мл в день изоосмолярных растворов. Если гипонатриемия тяжелая (менее 110—115 ммоль/л), то следует назначить гипертонические растворы натрия хлорида (3 — 5 %) и фуросемид. Следует опасаться быстрой коррекции гипонатриемии, поскольку это чревато демиелинизацией моста мозга.
Гипонатриемия может развиться при отсутствии нефизиологических потерь натрия из организма. Условием ее развития является нормальное или повышенное выделение натрия из организма физиологическими путями при сниженном его потреблении. Это наблюдается при проведении интенсивной терапии с существенным снижением количества вводимого натрия без должного контроля его баланса.
Нарушение минералокортикоидной функции надпочечников (болезнь Аддисона — Бирмера) также может привести к развитию гипонатриемии за счет снижения реабсорбции натрия в почечных канальцах и его повышенного выделения с мочой, потом, стулом при нормальном его потреблении. Для этого вида гипонатриемии характерно повышение содержания в плазме ионов калия и хлора, а также сдвиг кислотно-основного состояния крови в сторону
288 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
ацидоза. Попытки коррекции концентрации натрия в плазме внутривенной инфузией препаратов натрия или энтеральным приемом солей натрия, как правило, не дают желаемого эффекта. Данную разновидность гипонатриемии корригируют назначением дезоксикортикостерон ацетата (ДОКСА). Другие названия этого препарата — декортон, перкотен. Препарат выпускается в таблетках по 0,005 г для сублингвального приема и в ампулах по 1 мл в виде 0,5 % масляного раствора для внутримышечного введения. Суточные дозы колеблются от 0,005 до 0,01 г, максимальная суточная доза — 0,025 г. Существует еще один препарат подобного действия — дезоксикортикостерон триметилацетат в виде 2,5 % водной суспензии в ампулах по 1 мл.
Гипонатриемия, развивающаяся при избыточном введении в организм воды, называется гипонатриемией разведения. Иногда она классифицируется как водное отравление. Такое состояние можно также характеризовать как гипотоническую гипергидратацию. Тяжесть состояния больных при гипонатриемии разведения определяется скоростью поступления в организм избыточной воды.
Парадоксальная (отечная) гипонатриемия развивается у больных с хронической сердечно-сосудистой недостаточностью, длительно лечившихся с применением диуретиков. Поэтому чувствительность у этих больных к диуретикам либо снижена, либо потеряна. Патогенез развития парадоксальной гипонатриемии сопровождается нарушением осморегуляции с программированием более низкого, чем в норме, осмотического давления плазмы. Определенное количество натрия депонируется внутриклеточно — в клетку поступают ионы Na+ и Н+, а из клетки выводятся два иона К+, в результате чего развивается внутриклеточный ацидоз. Таким образом, на фоне увеличения абсолютного содержания натрия в организме имеет место гипонатриемия.
Существует несколько методов, позволяющих усилить эффект диуретиков у больных с хронической сердечно-сосудистой недостаточностью:
1) предшествующее применению диуретиков внутривенное введение калия в поляризационных смесях (например, глю-козо-калиево-инсулиновая смесь), при этом калий, проникая в клетку, вытесняет натрий во внеклеточное пространство, и применение диуретиков дает эффект;
2) гипербарическая оксигенация;
3) предшествующее применению диуретиков назначение глюкокортикоидов [1,3-5, 7-9, 19].
8.10.2. Обмен калия
Калий является основным внутриклеточным катионом. В организме человека с массой тела 70 кг содержится от 2 до 6 тыс. мэкв (80 — 250 г) калия, в среднем 4 тыс. мэкв (170 г). 98 % калия находится внутриклеточно, 2 % — внеклеточно. Нормальная концентрация калия в плазме колеблется от 4 до 5,5 мэкв/л. Эти колебания зависят от характера пищи. При потреблении пищи, богатой калием (прежде всего, растительной), его содержание в плазме несколько повышается. В среднем с пищей в организм поступает 90 ммоль калия в сутки. За тот же период из организма выделяется с мочой 80 ммоль калия, с калом — 10 ммоль и с потом -1—3 ммоль. У новорожденных нормальная концентрация калия плазмы составляет 3,5 — 7 мэкв.
Клетки различных тканей содержат неодинаковое количество калия — чем функционально активнее ткань, тем больше в ней калия. Мышечные клетки содержат примерно 160 мэкв калия на 1 кг внутриклеточной воды, эритроциты — 87, сердечная мышца — 142, гепатоциты —145, нейроны — 184 мэкв/кг внутриклеточной воды.
Нарушения концентрации калия в плазме проявляются в виде гиперкалиемии (гиперкалийплазмии) (более 5,5 мэкв/л) и гипокалиемии (гипокалийплазмии) (менее 3,8 мэкв/л).
Концентрация калия в плазме снижается при:
уменьшении общего содержания калия в организме — это состояние называют г ипокал иг ист ией;
Обмен электролитов 289
перемещении калия из внеклеточной жидкости в клетки;
увеличении объема внеклеточного водного пространства.
Снижение общего содержания калия в организме (гипокалигистия) возникает при таких состояниях:
а) недостаточном поступлении калия в организм на фоне продолжающейся секреции калия в дистальных канальцах почек с последующим выведением его с мочой;
б) терапии диуретиками, прежде всего петлевыми диуретиками (фуросемидом, этакриновой кислотой, тиазидными производными), и повышенных потерях калия с мочой в полиурической стадии ОПН;
в) избыточных потерях калия через пищеварительный канал: слюнотечение, кишечная непроходимость, стеноз привратника, диарея различного генеза;
г) избыточных потерях калия с обильным потоотделением.
Распределение калия между внутриклеточной и внеклеточной жидкостью зависит от кислотно-основного состояния крови. При алкалозе калий перемещается из внеклеточной жидкости в клетки, а при ацидозе концентрация калия в плазме повышается за счет усиления его выхода из клеток различных тканей.
Транспорту калия в клетки способствует инсулин, причем независимо от поступления в клетки глюкозы. Адреналин вначале повышает выход калия из клеток, а при длительном умеренном воздействии — способствует поступлению калия в клетки печени и мышцы. Транспорт калия в клетку усиливается при интенсивном синтезе белков (в период восстановления после операций, травм, заболеваний; в период интенсивного роста и под действием анаболических гормонов), а также в период клеточной регидратации.
Концентрация калия в плазме снижается при увеличении объема внеклеточной жидкости вследствие повышения в ней концентрации натрия. Это наблюдается при избыточном поступлении натрия в организм и при замедлении его выведения из организма при первичном или вторичном альдостеронизме.
4-220
В условиях отсутствия нарушений кислотно-основного состояния, протеинового обмена и истощения запасов гликогена снижение концентрации калия в плазме до 3 мэкв/л соответствует общему дефициту калия в организме — 100 — 200 мэкв. При гипокалийплазмии ниже 3 мэкв/л каждому снижению концентрации калия в плазме на 1 мэкв/л соответствует увеличение общего дефицита калия в организме на 200 — 400 мэкв. Признаки гипокалийплазмии на ЭКГ появляются при общем дефиците калия в организме, приблизительно равном 500 мэкв, они заключаются в снижении интервала S —Т ниже изолинии, удлинении интервала Р —Т, уплощении зубца Т. Снижение уровня внутриклеточного калия приводит к электрической нестабильности миокардиальных клеток и способствует возникновению аритмий (желудочковых и наджелудочковых экстрасистол, желудочковой тахикардии).
При снижении концентрации калия плазмы до 1,5 мэкв/л может возникнуть предсердно-желудочковая блокада и фибрилляция желудочков сердца.
Повышение концентрации калия в плазме крови наблюдается в результате избыточного его поступления в организм (особенно при наличии почечной недостаточности) при таких ситуациях:
а) пероральный прием препаратов калия; б) употребление пищи, богатой калием; в) употребление калийсодержащих солевых заменителей;
г) внутривенное введение препаратов калия;
д) переливание гемолизированной крови; е) внутрисосудистый гемолиз;
ж) желудочно-кишечное кровотечение;
з) внутривенное вливание калийсодержащих препаратов, например калиевой соли бензилпенициллина;
и) в результате распада клеток после проведения химиотерапии (особенно у больных с лейкемией, лимфомой, миеломой);
к) у больных с рабдомиолизом или краш-синдромом;
л) у больных с тканевым некрозом или с гиперметаболическими состояниями.
290 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Гиперкалиемия может также возникнуть в результате повышенного транспорта калия из клеток во внеклеточный сектор при следующих состояниях:
а) метаболическом и респираторном ацидозе;
б) тяжелых физических нагрузках;
в) на фоне приема р-адреноблокато-ров;
г) дефиците инсулина;
д) дигиталисной интоксикации;
е) гиперкалиемических периодических параличах.
Гиперкалиемия может быть зафиксирована при уменьшении объема внеклеточного водного пространства.
Гиперкалиемия в результате снижения экскреции калия с мочой наблюдается:
а) при почечной недостаточности;
б) на фоне приема следующих лекарственных препаратов: калийсберегающих диуретиков (спиронолактона, верошпиро-на, триамтирена, амилорида), р-блокаторов, нестероидных противовоспалительных препаратов, ингибиторов ангиотензинпрев-ращающего гормона;
в) при недостаточности альдостерона;
г) в результате селективного нарушения почечной экскреции калия при системной красной волчанке, псевдогипоальдостеронизме, обструктивной уропатии, пересадке почки, болезни клеток, врожденной патологии почек.
Ложная гиперкалиемия наблюдается на фоне использования турникета, жгута; в результате гемолиза в пробирке; на фоне лейкоцитоза и тромбоцитоза.
Клинически гиперкалийплазмия проявляется общей слабостью, парестезиями, параличами, рвотой, болью в животе, кишечной непроходимостью. На ЭКГ регистрируется: высокий остроконечный зубец Т, интервал S —Т расположен ниже изолинии, расширение интервала Р — Q, исчезновение зубца Р и деформация комплекса QRS. При гиперкалиемии свыше 7,5 мэкв/л могут возникнуть смертельные нарушения ритма сердца — предсердно-желудочковая блокада и фибрилляция желудочков сердца, а при концентрации калия плазмы 10—12 мэкв/л — диастолическая остановка сердца.
В организме существует функциональная взаимосвязь между обменом натрия и калия. Поэтому могут возникать ситуации, когда концентрация калия в плазме будет нормальной, а внутриклеточная — снижена. Но уменьшение содержания внеклеточного калия всегда сопровождается уменьшением внутриклеточного. Высокая концентрация калия внутри клетки сохраняется до тех пор, пока она нормально функционирует. Введение ионов калия в клетку происходит с затратой энергии, используемой также на удержание калия против градиента концентрации. Обратный переход калия по градиенту концентрации из клетки во внеклеточное пространство сопровождается выделением энергии. Таким образом, поддержание высокой внутриклеточной и внеклеточной концентрации калия сопровождается кумуляцией энергии. Поэтому поддержание электролитного баланса имеет большое значение для энергетического обмена.
Калий оказывает влияние на азотистый обмен. В нормальной клетке на 1 г белкового азота приходится примерно 3 мэкв калия (примерно 100 мг). Таким образом, внутриклеточное соотношение между белковым азотом и калием составляет 10 : 1. Если азотистый баланс отрицательный, то соотношение между белковым азотом и калием уменьшается, а количество теряемого организмом калия увеличивается. Калий из распадающихся белков выводится с мочой. Таким образом, большие потери калия с мочой могут свидетельствовать о происходящем массивном распаде белков, интенсивном катаболизме белков или белковом голодании. Если после успешной сердечно-легочной реанимации в организме нарастает отрицательный баланс калия, то это может свидетельствовать о распаде тканей и является неблагоприятным прогностическим признаком. Калий играет большую роль в процессах синтеза белка. При гипокалиемии утилизация организмом аминокислот снижается, и они выделяются с мочой. Калий имеет большое значение для образования конечных продуктов белкового обмена. При гипокалиемии нарушается мочевинообразовательная функция печени. Крайняя степень пече-
Обмен электролитов 291
ночной недостаточности проявляется гипокалиемией, нарушением синтеза мочевины, аммониемией, энцефалопатией, комой.
Калий является необходимым компонентом углеводного обмена, поскольку входит в структуру гликогена. Снижение концентрации плазменного калия всегда сопровождается снижением синтеза гликогена. У больных с декомпенсированным сахарным диабетом в результате нарушения энергетических процессов из клеток многих тканей во внеклеточное пространство выходит калий. Глюкоза в дистальных извитых канальцах действует как осмодиуретик и вместе с мочой из организма теряется калий. Дефицит внутриклеточного калия является одной из причин инсулинорезистентности при лечении диабетической кетоацидемической комы.
Процесс аэробного гликолиза определяется уровнем внутриклеточного калия. При снижении его внутриклеточной концентрации интенсивность аэробного гликолиза снижается. Гипокалиемия ниже определенного уровня активирует анаэробный гликолиз и в тканях повышается содержание молочной кислоты, одновременно ослабевает синтез АТФ и фосфокреатинина.
Уменьшение содержания калия в тканях ведет к снижению их способности утилизировать кислород.
Калий является основным катионом буферных систем внутриклеточной среды. Он играет важную роль в поддержании электронейтральности этой среды. Если в результате патологического процесса (например, гипоксии, усиления катаболических процессов, снижения запасов углеводов, внутриклеточной дегидратации) клеткой теряется три иона калия, то в клетку поступают два иона натрия и один ион водорода. Этот процесс называется механизмом трансминерализации Дерроу — Жане. Таким образом, истинная гипокалиемия сопровождается внутрпкУкеточпътл ацидозом и внеклеточным алкалозом.
Для нормального функционирования внутриклеточных ферментных систем необходимо восстановить нормальную реакцию внутриклеточной среды. Для этого следует восполнить дефицит внутриклеточного калия. Уровень последнего не вос-10*
станавливается при дефиците в организме магния, поэтому заместительная терапия калием сама по себе, без учета содержания в организме магния, может быть неэффективной. Известно, что при наличии ионов магния активируется Ма+-К+-АТФ-аза. Поэтому препараты калия необходимо вводить в организм вместе с препаратами магния. Примером такого сочетанного препарата является панангин, в 10 мл которого содержится 2,5 ммоль калия и 1,6 ммоль магния. С этой целью в глюко-зо-инсулино-калиевую смесь для внутривенного введения добавляют раствор магния сульфата в такой пропорции, чтобы на 8 ммоль калия приходился 1 ммоль магния.
Калий необходим для нормального функционирования нервной системы. Его ионы обеспечивают определенный уровень потенциала покоя мембран нервных клеток и межнейронную передачу импульсов возбуждения. Если соотношение внутриклеточного и внеклеточного калия меньше, чем 15 : 1, то потенциал действия не возникает и развивается блок синаптической передачи. Наиболее устойчивыми к гипокалиемии являются синапсы диафрагмы, а наименее — синапсы пищеварительного канала. Поэтому дефицит калия в организме сопровождается парезом кишок. При гипокалиемии страдает синаптическая передача и в остальных синапсах ЦНС, поэтому может регистрироваться адинамия, апатия, парестезии. При снижении концентрации калия в плазме крови ниже 3 мэкв/л ослабевают рефлексы, в том числе и кашлевой, повышается бронхиальная секреция и снижается активность мерцательного эпителия. Таким образом, при гипокалиемии снижается дренажная функция нижних дыхательных путей, бронхиальный секрет по составу приближается к плазме, высокое содержание бел-
средой для бактериальной флоры, вследствие чего возникают бронхиты и пневмонии.
Калий принимает участие в синтезе катехоламинов и ацетилхолина. Гипокалиемия потенцирует действие конкурентных миорелаксантов [1, 3 — 5, 7 — 9, 19].
292 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
8.10.3. Обмен кальция
В организме человека со средней массой тела 70 кг содержится 1 — 1,5 кг кальция, или 25 000 — 37 000 ммоль. Основная масса кальция локализована в костях и только 1 % — во внеклеточной жидкости.
Общая концентрация кальция в плазме крови составляет 2,5 ммоль/л, из них 1 ммоль/л образует комплексы с альбумином, 0,17 ммоль/л — с фосфатами, цитратом, гидрогенкарбонатом. Ионизированная, физиологически активная фракция кальция составляет 1,34 ммоль/л [7].
Содержание кальция в плазме регулируют два гормона — паратгормон (паратиреоидин, паратирин) и кальцитонин. Первый вырабатывается паращитовидными железами, а второй — особыми клетками щитовидной железы. Действие парагормона направлено на повышение концентрации кальция в плазме и абсолютного его содержания в организме. Этот гормон представляет собой белок массой 8’500 D. Он стимулирует активность остеокластов путем активизации их адени-латциклазы. Повышенное образование цАМФ приводит к высвобождению ионов кальция и фосфата из минерального вещества, образующего кость, — гидроксиапатита.
Паратгормон усиливает реабсорбцию ионов кальция в почке.
Кальцитонин является антагонистом паратгормона, его действие направлено на снижение концентрации кальция в плазме посредством активации фосфодиэстеразы и депонирования кальция в костную ткань.
В регуляции фосфорно-кальциевого обмена принимает также участие витамин D3 — холекальциферол, который при участии паратгормона превращается в дигидрокальциферол. Последний обеспечивает всасывание кальция в кишках и осуществляет кальцификацию кости. За сутки в организм вместе с пищей поступает около 1 г кальция. Из организма кальций выделяется с мочой — 250 мг/сут и калом — 850 мг/сут.
Секреция паратгормона не регулируется специальным тропным гормоном. Клет
ки паращитовидных желез отвечают на изменения концентраций кальция, изменяя соответственно скорость секреции паратгормона.
Секреция кальцитонина регулируется подобным образом — увеличение уровня кальция в крови вызывает непосредственную стимуляцию секреции гормона С-клетками щитовидной железы.
Таким образом, содержание кальция в плазме определяется равновесием эффектов паратгормона и кальцитонина, уровнем холекальциферола, концентрацией протеинов плазмы, кислотно-основным состоянием [8, 9, 12].
Состояние ионизации кальция зависит от pH крови: при ацидозах содержание ионизированного кальция в плазме возрастает, при алкалозах — снижается.
В нормальной клетке уровень внутриклеточного кальция весьма низок и составляет 10"8-10-4 ммоль/л внутриклеточной воды в зависимости от функционального состояния клетки. В среднем отношение внеклеточного и внутриклеточного кальция равняется 2 ммоль/1 мкмоль (т. е. внутриклеточное содержание кальция в 2000 раз ниже внеклеточного). Ионизированного кальция в клетке содержится еще на два порядка меньше. Такое низкое содержание кальция внутри клетки свидетельствует об очень высокой чувствительности к кальцию ферментных систем, регулируемых им [12].
Поступление ионов кальция в клетку осуществляется через специфические кальциевые каналы, представляющие своего рода поры в клеточной мембране. Различают «медленные» потенциалзависимые кальциевые и потенциалнезависимые рецепторные каналы.
Потенциалзависимые каналы могут находиться в открытом или закрытом состоянии в зависимости от положения двух типов ворот — ворот активации и ворот инактивации. В покое ворота активации закрыты, а ворота инактивации открыты. При деполяризации клеточной мембраны их состояние меняется. В результате этого потенциалзависимые каналы в течение некоторого времени находятся в открытом, или активном, состоянии, и ионы каль
Обмен электролитов 293
ция поступают в клетку. Таким образом, по потенциалзависимым каналам ионы кальция попадают в клетку только в период деполяризации мембраны. При этом концентрация ионизированного кальция возрастает с 10"^ ммоль/л до 10~5 ммоль/л.
На мембранах клеток располагаются особые рецепторы, взаимодействие которых с некоторыми биологически активными веществами открывает в мембране кальциевые каналы, и ионы кальция поступают в клетку. При разрушении этих биологически активных веществ канальные структуры закрываются. Концентрация рецепторов кальциевых каналов на клеточной мембране очень невелика. Так, на 1 мкм2 мембраны мышечных клеток располагаются 300 рецепторов натриевого насоса, два p-адренергических, пять мускариновых и только один рецептор кальциевых каналов [7].
Существуют лекарственные вещества, которые, взаимодействуя с рецепторами кальциевых каналов, закрывают их и прекращают ток ионов кальция в клетку. Это обширная группа производных дигидропиридина: нитрендипин, нифедипин, Д-600, нимодипин, фелодипин, исрадипин (ломир), амлодипин. Последние три препарата являются препаратами 2-го поколения — они более сильные и действуют более продолжительное время. Верапамил блокирует потенциалзависимые «медленные» кальциевые каналы.
К лекарственным препаратам, блокирующим потенциалзависимые кальциевые каналы, относятся: верапамил (фино-птин, изоптин, изоптин SR («sustained release») пролонгированного действия), гал-лопамил, анипамил, фалипамил, RO-5967. Это так называемая фенилалкиламиновая группа лекарственных препаратов.
Из производных бензодиазепина подобным эффектом обладает дилтиазем.
Физиологическая роль кальция (по Эшли и Кемпбеллу, 1979) заключается в следующем:
1) выполнение структурной функции (участие в формировании костной ткани и биологических мембран);
2) кофактор ряда ферментативных реакций как на поверхности клетки, так и внутри нее;
3) проводник тока через клеточную мембрану и ее стабилизатор;
4) внутриклеточный регулятор таких фундаментальных процессов, как секреция, сокращение, регуляция потоков катионов, функция гормонов, оплодотворение, деление и смерть клеток.
Кальций является одним из регуляторов внутриклеточного метаболизма наряду с цАМФ, цГМФ, производными инозитола. Эти вещества называются вторичными посредниками. Внутриклеточный метаболизм изменяется в зависимости от изменения внутриклеточного содержания перечисленных выше веществ. Действие любого биологически активного вещества — эндогенного или введенного извне — например лекарственного вещества, осуществляется таким образом: молекула биологически активного вещества связывается со специфическим рецептором на мембране клетки, после чего внутри клетки происходит повышение концентрации ионов кальция или усиленный синтез цАМФ, цГМФ или производных инозитола. Эти вещества изменяют соответствующим образом внутриклеточный метаболизм. Благодаря вторичным посредникам происходит амплификация (усиление) сигнала, принесенного к клетке биологически активным веществом [16—19].
Для нормального функционирования клетки необходима целостность клеточной мембраны, которая обеспечивается внутриклеточным цитоскелетом и внеклеточным матриксом. Основу цитоскелета, прилегающего к мембране с цитоплазматической стороны, составляют так называемые микрофиламенты (микротрубочки и микрофибриллы), создающие опорную конструкцию клетки, препятствующие резким изменениям ее объема, противодействующие образованию небислойных структур в фосфолипидном матриксе. Кроме этого, целостность клеточной мембраны обеспечивается межклеточными структурами, состоящими из нитей коллагена, насыщенного протеогликанами и гликопротеинами.
294 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Практически все клетки, за исключением клеток крови, обладают внеклеточным матриксом — гликокаликсом. Это вещество углеводной природы, покрывающее мембрану клетки снаружи, отрицательно заряженное, связывающее ионы кальция на своей поверхности, тем самым оно препятствует бесконтрольному поступлению кальция в клетку. Толщина гликокалик-са — 50 нм, снаружи он непосредственно переходит в соединительную ткань межклеточного пространства.
Клеточные мембраны состоят в основном из белков и липидов. В составе мембран обнаружены липиды трех классов: фосфолипиды, гликолипиды, стероиды. Ионы кальция, присоединяя комплексные лиганды и адсорбируясь в определенных местах клеточной мембраны, стабилизируют и упорядочивают расположение молекул фосфолипидов, уменьшая их текучесть и вязкость. Рецепторные свойства клеточных мембран зависят главным образом от микровязкости мембранных липидов, окружающих белок-рецептор.
Как уже упоминалось выше, с внутренней стороны к плазматической мембране примыкают структуры, не относящиеся собственно к мембране, но составляющие скелет клетки. Цитоскелет включает элементы двух типов: микрофибриллы и микротрубочки. Микрофибриллы состоят из актиноподобного белка, его обратимая полимеризация происходит аналогично переходу актина из глобулярного в фибриллярное состояние. Микротрубочки — цилиндрические структуры диаметром 20 — 30 нм, образованные молекулами тубулина. Фосфорилирование тубулина специальными протеинкиназами ускоряет сборку трубочек, а ионизированный кальций вызывает их диссоциацию.
Целостность микрофиламентов в клетке сохраняется лишь при очень низких концентрациях ионов кальция. При повышении концентрации выше определенного уровня цитоскелет разрушается. В нормальной клетке уровень ионизированного кальция измеряется величиной 10"8 при общем внутриклеточном содержании 10“4. Пока клетка функционально активна, она борется с повышением содержания в ней
ионизированного кальция. Избыточное его содержание мобилизуется во внутриклеточные депо (например, в систему моно-нуклеарных фагоцитов митохондрии) или откачивается наружу ионными насосами.
К механизмам удаления кальция из клетки относятся интегральные структуры клеточной мембраны — кальцийзависимая АТФ-аза, или кальциевый насос протоплазматической мембраны, удаляющий кальций из клетки во внеклеточную среду, и кальциевый насос внутриклеточных мембран, переводящий кальций из цитоплазмы во внутриклеточные депо. Работа кальциевых насосов осуществляется с поглощением энергии, с созданием тысячных градиентов концентрации кальция по обе стороны клеточной мембраны. Кроме того, ионы кальция выводятся из клетки в обмен на ионы натрия (так называемый натрий/ кальциевый обменник). Существуют также механизмы, пока малоизученные, обмена ионов кальция на ионы кальция.
Нарушение систем, поддерживающих в ' клетке низкий уровень свободного кальция, означает ее смерть. Механизм гибели клетки представляется таким: повышение концентрации в цитоплазме ионов кальция разрушает цитоскелет клетки, одновременно кальций активирует фосфолипазы бислоя, разрушающие молекулы фосфолипидов мембраны. На фоне повышения проницаемости клеточной мембраны закисляется внутриклеточная среда, активируются лизосомальные ферменты и процессы аутолиза приводят к лизированию клетки.
Важная роль цитоскелета заключается в поддержании жесткой конструкции клетки и противодействии изменениям осмотического объема. Кроме того, цитоскелет «армирован» многими ферментами, ориентированными таким образом, что их последовательность соответствует метаболическим путям, осуществляемым в клетке. Так, большинство ферментов гликолиза расположено на цитоскелете эритроцитарных мембран.
Оказалось, что цитостатические алкалоиды винбластин и винкристин разрушают цитоскелет опухолевых клеток, а также их митотическое веретено вследствие инги
Обмен электролитов 295
бирования ионного кальциевого насоса, при этом уровень внутриклеточного кальция значительно возрастает. Разрушение митотического веретена препятствует делению клетки. К подобному результату приводит любое другое нарушение целостности клеточной мембраны.
Из примеров видно, как важны для жизнедеятельности клетки системы, поддерживающие в ней низкий уровень свободного кальция. Именно поэтому кальций относится к наиболее контролируемым ионам. В клетке условия контроля особенно жесткие: кальций депонируется в самой клеточной мембране, образуя комплексы с АТФ и фосфолипидами, в цитоплазме его ионы буферируются макромолекулами и мелкими молекулами цитоплазмы, кальций накапливается в эндоплазматическом ретикулуме, митохондриях, секреторных органеллах; ионы кальция постоянно откачиваются во внеклеточную среду.
Таким образом, уровень ионизированного кальция в цитоплазме в состоянии метаболического покоя очень низок. Однако этот факт не означает, что он ингибирует метаболические процессы. В клетке содержатся высокочувствительные к кальцию белковые системы, способные реагировать на следовые, микромолярные концентрации этого иона.
В цитоплазматических мембранах и мембранах клеточных органелл существуют белки, влияющие на интенсивность метаболических реакций. Таким белком, оказывающим сильное влияние на обмен кальция, является кальмодулин. Его действие связано с обеспечением и облегчением кальциевой регуляции, в частности он активирует работу кальциевых насосов. Повышение уровня свободного кальция в цитоплазме стимулирует образование комплекса кальмодулин-кальциевый насос и активирует работу последнего.
В клетках, подверженных злокачественной трасформации, обнаруживается повышенный уровень кальмодулина и соответственно более высокая активность Са2+-насоса. Этим объясняется устойчивость таких клеток против внешних воздействий и более низкий, чем в контроле, уровень кальция в их цитоплазме.
Исследование кальмодулиновой регуляции метаболизма показало, что существует множество процессов, и их количество постоянно увеличивается, на которые этот белок оказывает влияние. Содержание кальмодулина в мозгу составляет около 400 мг/кг сырой массы. Менее чем 10 % этого количества достаточно, чтобы с избытком «обслужить» все известные сейчас ферменты мозговой ткани, регулируемые кальмодулином. Зачем нужен этот избыток регулятора, пока неясно. Возможно, что природа сама позаботилась о защите мозга, предусмотрев такое высокое содержание кальмодулина в мозговой ткани. Одной из причин перехода обратимых изменений в необратимые при гипоперфузии и гипоксии мозга является постоянное освобождение ненасыщенных жирных кислот, особенно арахидоновой, в ишемических нейронах из фосфолипидов клеточных мембран под действием фосфолипазы. Активация фосфолипазы на внутренней поверхности клеточной мембраны находится в абсолютной зависимости от уровня ионизированного кальция. Накопление арахидоновой кислоты частично угнетается натрий фенобарбиталом, селективным антагонистом кальция. В процесс реоксигенации вовлекается вазоспастический компонент, обусловленный тромбоксаном А2, одним из продуктов циклооксигеназного пути превращения арахидоновой кислоты. Эта кислота также метаболизируется по липооксигеназному пути, что приводит к образованию кислых свободных радикалов, а также лейкотриенов.
Действие простагландинов в период реоксигенации угнетается преишемичес-ким назначением индометацина. Кроме того, предварительное введение индометацина или простациклина предупреждает развитие гипоперфузионного синдрома в постишемическом мозгу.
В составе мембран саркоплазматического ретикулума миокардиоцитов обнаружен другой регуляторный белок — фос-фоламбан. Он ответственен за проявление положительного инотропного действия норадреналина, осуществляющегося через цепь, которая включает активацию аде-
296 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
нилатциклазы, образование активной формы цАМФ-зависимой протеинкиназы и фосфорилирование ОН-группы серина в молекуле фосфоламбана. В фосфорилированном состоянии фосфоламбан стимулирует кальциевый насос ретикулума миоцитов, ускоряя восстановление кальциевых градиентов в сердечной мышце. Кальмодулин оказывает на этот белок регулирующее действие.
На определенных стадиях клеточного деления внутриклеточным антагонистом ионов кальция является олигоаденилат. Он удлиняет клеточный цикл за счет пролонгирования стадии дифференцировки. Олигоаденилат обладает антивирусным и антипролиферативным действием, образуется под действием цАМФ. Его уровень в клетках возрастает в результате голодания.
Трентал и теофиллин, увеличивающие в клетках уровень циклического аденозинмонофосфата, индуцируют образование олигоаденилата и усиливают иммунный статус организма. Аналогичного эффекта можно достичь с помощью акупунктуры [7, 12, 19].
Возбуждение клеток сопровождается повышением проницаемости клеточной мембраны и трансмембранным перемещением ионов. Так, в миоците потенциал покоя сменяется потенциалом действия, что приводит к поступлению ионов натрия, кальция и выходу из клетки ионов калия. Транспорт ионов в клетку сопровождается передачей информации и команд внутрь клетки, но поскольку натрий плохо соединяется с макромолекулами, его биологическая активность в информационном отношении мала. В аксоне нервной клетки концентрация натрия в 106 раз больше, чем кальция, однако при повышении активности клетки концентрация ионов кальция увеличивается быстрее, чем натрия, в силу того, что концентрация первого в цитоплазме клетки ничтожно мала по сравнению с другими катионами, и небольшие поступления в клетку ионов кальция приводят к значительным изменениям градиентов его концентрации в клетке с соответствующими физиологическими эффектами. Поэтому считается, что ионы
кальция незаменимы при регуляции физиологических процессов внутри клетки. Во время деполяризации клеточной мембраны концентрация ионизированного кальция внутри клетки повышается с 10“8 ммоль/л до 10"5 ммоль/л [7, 12].
В физиологических нормальных условиях кальций обеспечивает также межклеточные связи.
В качестве кофермента кальций способствует переходу протромбина в тромбин во внеклеточном пространстве, активирует аденилатциклазную реакцию и фосфолипазу А2. Совместно с кальцием действуют многие медиаторы: цАМФ, цГМФ, простагландины. Подавляя цАМФ, кальций активирует синтез простагландинов.
Кальций играет решающую роль в процессах проницаемости клеточной мембраны в покое, стабилизируя конформацию рецепторов и регулируя изменение проводимости клеточной мембраны. Этот ион является важным фактором доставки питательных веществ в клетку, положительным модулятором эпителиальной и мезенхимальной пролиферации, регуляции образования ДНК и митотической активности [12].
Роль кальция как регулятора секреции. Для проведения возбуждения в холинергическом нейромышечном синапсе 3 — 4 иона кальция должны освободить один «пакет» гранул ацетилхолина. Аналогичные процессы протекают в головном и спинном мозгу.
Исследования показали, что барбитураты, этанол, транквилизаторы угнетают поток кальция в синаптосому и таким образом блокируют освобождение медиатора из нее. Свое действие барбитураты оказывают, влияя на потенциалзависимые каналы кальция. Возникает вопрос о полезности рекомендаций введения кальций хлорида перед применением барбитуратов с целью вводного наркоза.
Морфин блокирует выделение медиаторов в периферических нервах, конкурируя с ионами кальция. В эксперименте было установлено, что аналгетический эффект морфина может быть частично снят введением в мозг препаратов кальция. В экспериментах на синаптосомах было обна
Обмен электролитов 297
ружено, что морфий снижает поступление ионов кальция в клетки; налоксон блокирует этот эффект морфина. Возникло предположение, что ионы кальция могут быть медиаторами боли. Этилендиаминтетраацетат (ЭДТА), связывающий кальций, вызывает аналгезию. Еще более интересным является наблюдение, что избыток ионов кальция может вызывать толерантность к аналгетикам. Нифедипин достоверно увеличивает аналгетический эффект морфина, не влияя на его метаболизм. Таким образом, ионы кальция играют важную роль в аналгетическом действии опиатов. Блокаторы кальциевых каналов могут быть использованы при лечении боли.
Было проведено лечение послеоперационных фантомных болей и каузалгии внутривенным капельным введением кальцитонина в дозе 100 — 200 МЕД в течение 30 — 60 мин. После 1—2 инфузий боли были купированы у 83 % больных, у 17 % — после пяти инфузий. После двух лет наблюдения рецидив отмечен у 4 из 16 больных. Таким образом, кальцитонин эффективен в лечении фантомных болей в раннем послеоперационном периоде, он обеспечивает продолжительный аналгетический эффект при минимальных побочных эффектах [12, 17].
Эндорфины, подобно морфию, уменьшают поступление кальция в синаптосому, препятствуя тем самым выделению медиатора.
В недавних исследованиях было показано, что как острая, так и хроническая эпилептическая активность сопровождается повышенным поступлением кальция в нейроны. В эксперименте и клинике для предупреждения судорожных припадков оказался эффективным нимодипин.
Кальций поступает в мастоциты и вызывает их дегрануляцию с выделением гистамина. Этот процесс блокируется верапамилом, что нашло практическое применение в терапии некоторых видов бронхиальной астмы.
Ионы кальция активируют подвижность нейтрофилов и реакцию освобождения тромбоцитов.
Согласно популярной гипотезе Репке, сердечные гликозиды угнетают Na+-K+-
АТФ-азу, что ведет к поступлению в кардиомиоцит натрия, а вместе с ним и кальция. Последний, фиксируясь на тропонине, способствует образованию актомиозинового «мостика» и развитию сокращения миоцита. Альтернативная гипотеза предполагает поступление сердечных гликозидов в цитоплазму кардиомиоцита и освобождение кальция из внутриклеточных депо.
Сердечная мышца — единственная мышца, в которую кальций поступает непосредственно из кровотока по мере нарастания его концентрации при внутривенном введении. Верапамил блокирует усиление сокращений, вызванное притоком ионов кальция; возбудимость синусового узла определяется поступлением кальция по потенциалзависимым каналам и подавляется верапамилом; аналогично подавляется и возбудимость атриовентрикулярного узла. Волокна Пуркинье, пучок Гиса, миокард желудочков в связи с поступлением кальция по быстрым рецепторным кальциевым каналам не угнетаются верапамилом, но при появлении эктопического очага в миокарде начинают регулироваться «медленные» каналы, и верапамил подавляет автоматизм эктопических фокусов.
Антагонисты кальция (верапамил, нифедипин и др.) подавляют сократимость, замедляют работу сердца, снижают потребность миокарда в кислороде, расширяют церебральные, кишечные, почечные и бедренные сосуды, снижают АД, действуют как дезагреганты и коронародилататоры [7-9].
Антагонисты кальция не показаны при кардиогенном шоке, но с успехом используются при стенокардии. Поступление в избытке ионов кальция в цитоплазму кардиомиоцита ведет к повышенному потреблению АТФ, при нарушенной функции митохондрий — к дефициту АТФ, нарушению структуры кардиомиоцита и некрозу ткани.
Наиболее распространенная дозировка верапамила внутривенно: однократно — 5 мг — болюсная доза, затем капельно с раствором глюкозы 0,004 мг/кг в минуту (в течение 30 мин — 10 мг) [7].
298 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
Секреция ацинарной клетки поджелудочной железы осуществляется путем эк-зоцитоза и стимулируется холецистоки-нин-панкреозимином (ХЦК-ПЗ), секретином слизистой двенадцатиперстной кишки рефлекторно и уровнем внеклеточного кальция. Еще в 1966 г. было обнаружено, что срезы поджелудочной железы полностью прекращают секреторный ответ на ацетилхолин и ХЦК-ПЗ в среде, не содержащей кальция. Очевидно, существует зависимость экзокринной секреции от внеклеточного кальция, но она нечеткая. В ацинарной клетке поджелудочной железы не обнаружены «быстрые» кальциевые каналы, кальций поступает в клетку вместе с натрием.
Использование верапамила на ранних стадиях острого панкреатита для обезболивания, более благоприятного течения получило клиническое подтверждение.
Терапия при панкреатитах направлена на подавление секреторной активности. Хороший эффект оказывает использование с этой целью цитостатика 5-фторура-цила, но он значительно токсичнее верапамила. Согласно последним сообщениям, определенный эффект в отношении подавления экзокринной секреции создают эндогенные опиоиды — энкефалины. Парентеральное введение их аналога — далар-гина оказалось эффективным при экспериментальном геморрагическом панкреатите. Эндорфины уменьшают поступление в синаптосому кальция.
Обнаружена четкая зависимость в отношении активации кальцием секреции в мозговом веществе надпочечников, нейрогипофизе, слюнных железах, а также секреции инсулина.
В большинстве случаев кальций является антагонистом магния. Отношение магний/кальций в клетке влияет на внутриклеточный метаболизм [7, 8].
При определении концентрации общего кальция в плазме регистрируют гипо-кальциплазмию при снижении его концентрации до 2,25 ммоль/л и ниже и гипер-кальциплазмию при увеличении концентрации кальция выше 2,63 ммоль/л. У взрослых наиболее частая причина ги-
перкальциплазмии — первичный и вторичный гиперпаратиреоз.
При первичном гиперпаратиреозе (аденома паращитовидных желез) в плазме повышается содержание паратирина, кальция, снижается реабсорбция натрия и повышается выделение гидрогенкарбоната почками. В плазме повышается концентрация хлора. Возникает умеренный ацидоз, способствующий ионизации кальция. Первичный гиперпаратиреоз лежит в основе фиброзного остита Реклингаузена и болезни Педжета [16 — 18, 30].
Вторичный гиперпаратиреоз возникает в ответ на гипокальциплазмию при ее длительном существовании. В его основе лежит гиперплазия паращитовидных желез. Затем возникает гиперкальциплазмия. На фоне хронической недостаточности почек количество паратирина может значительно превышать таковое при первичном гиперпаратиреозе.
Под действием паратирина с мочой теряется гидрогенкарбонат, задерживается хлор, в результате развивается ацидоз, последний нарушает комплекс кальция с протеинами, и ионизированная фракция кальция возрастает до 4,25 ммоль/л с возможным развитием гиперкальциеми-ческого криза — острого отравления кальцием. Летальность при кризе достигает 65 % [19]. Криз проявляется острой болью в подложечной области, жаждой, тошнотой, неукротимой рвотой, вначале с полиурией, ведущей к дегидратации, а затем с анурией, гипертермией. При развитии криза реальна угроза острой недостаточности сердца и коллапса. При концентрации кальция в плазме 5 ммоль/л и более развивается терминальное состояние с возможным параличом дыхания и остановкой кровообращения.
Терапия, как симптоматическая, так и специфическая, направлена на снижение концентрации кальция в плазме. Внутривенно вливают: 0,9 % раствор натрий хлорида, растворы глюкозы, реополиглюкин. По показаниям проводят коррекцию ацидоза раствором натрий гидрогенкарбоната.
При аденоме паращитовидной железы показана операция, но до ее проведения
Обмен электролитов 299
необходимо снизить уровень кальциплаз-мии до 2,9 ммоль/л и желательно ниже.
На догоспитальном этапе при гипер-кальциемическом кризе внутривенно начинают переливать изотонический раствор натрий хлорида с преднизолоном (90 мг), который является антагонистом 1,25-ди-оксихолекальциферола.
В стационаре при относительном восстановлении гемодинамики и диуреза, продолжая переливать изотонический раствор натрий хлорида, назначают фуросемид; для блокады синтеза простагландинов (медиаторов деструктивного процесса в костях) назначают индометацин (25 мг каждые 6 ч); для связывания в плазме ионов кальция медленно внутривенно переливают раствор комплексона — динатриевую соль этилендиаминотетрауксусной кислоты (Ма2ЭДТА) - 2,0-4,0 г в 500 мл 5 % раствора глюкозы в течение 3 —4 ч; одновременно внутримышечно назначается кальцитонин по 1 —5 ЕД/кг в сутки. Вся терапия обязательно проводится при постоянном мониторинге сердечной деятельности, повторных контролях концентрации кальция в плазме и при постоянной готовности ввести внутривенно кальций глюконат или СаС12. При неэффективности такой терапии прибегают к проведению гемодиализа [12, 19].
После аденомэктомии возникает опасность развития паратиреоидной недостаточности с транзиторной тетанией, гипо-кальци- и гипомагниплазмией и недостаточностью сердца. Прогностически благоприятными признаками являются: улучшение психического состояния больного и восстановление функции почек. В послеоперационном периоде назначают внутривенно кальций глюконат, гидротахистерол, диету с высоким содержанием кальция и низким — фосфора [19, 47].
Различают следующие формы гипокаль-циплазмии:
1) органическая — при дефиците пара-тирина, хронической недостаточности почек, деструктивном панкреатите, сниженной абсорбции кальция в тонкой кишке, повышенной секреции кальцитонина, поглощении кальция некоторыми остеоблас-тическими опухолями;
2) функциональная — при циррозе печени, нефротическом синдроме, гипопротеинемии, неонатальной тетании, идиопатическом гипопаратиреозе, дефиците витамина D, гипомагниплазмии и алкалозе;
3) гипокальциплазмия экзогенного происхождения — при передозировке Ма2ЭДТА, длительном приеме фенобарбитала, тиазидных диуретиков, слабительных, передозировке кальцитонина, массивном переливании крови на консерванте, содержащем цитрат.
Наиболее частыми причинами гипокаль-циплазмии являются: нарушение кровообращения при травме паращитовидных желез во время операции; субтотальная резекция щитовидной железы; терапия радиоактивным иодом; развитие опухоли, сдавливающей сосуды, питающие паращитовидные железы; случайное удаление паращитовидных желез во время радикальных операций на щитовидной железе и смежных областях шеи.
Псевдогипопаратиреоз обусловлен врожденной нечувствительностью органов-мишеней к паратирину.
Неонатальная гипокальциплазмия у новорожденных развивается при наличии у матери гиперкальциплазмии, подавляющей продукцию паратирина, вследствие чего плод получает его в недостаточном количестве. Поздняя постнатальная гипокальциплазмия связана с прикормом коровьим молоком с высоким содержанием фосфатов.
Различают явную и скрытую формы гипопаратиреоза.
Для клинической картины заболевания характерны повышенная возбудимость нервно-мышечного аппарата с периодическим возникновением тонических судорог при явных формах и наличием парестезий, похолоданием конечностей и чувством «ползания мурашек» — при скрытых. Последние могут переходить в явные в связи с инфекцией, интоксикацией, беременностью, гипервентиляцией, алкалозом.
Судороги при гипопаратиреоидной тетании симметрично захватывают главным образом сгибательные группы мышц. Чаще всего судороги охватывают верхние конечности, затем вовлекаются нижние, в более
300 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
тяжелых случаях — мышцы лица, туловища, диафрагмы, гортани.
При судорогах верхних конечностей плечо приведено к туловищу, предплечье согнуто в локтевом суставе, а кисть — в лучезапястном, пальцы сжаты и слегка согнуты в ладони. Первый палец приведен ко второму и третьему. Такое положение кисти получило название «рука акушера».
При судорогах нижних конечностей ноги вытянуты, прижаты одна к другой, стопы изогнуты вовнутрь, а пальцы — в положении подошвенного сгибания, большой палец накрыт остальными. Судороги мышц лица ведут к появлению «рыбьего рта», или «сардонической улыбки».
Повышенная нервно-мышечная возбудимость в период между приступами при явных формах имеет ряд специфических симптомов:
1. Симптом Хвостека — при постукивании по стволу лицевого нерва у места выхода его возле наружного слухового прохода сокращаются мышцы соответствующей половины лица. Различают: Хвос-тек-1 — сокращение мышц наружного края глазницы, крыла носа и угла рта; Хвостек-2 — сокращение мышц крыла носа и угла рта; Хвостек-3 — сокращение только мышц угла рта.
2. Симптом Труссо — при перетягивании плеча до исчезновения пульса жгутом или манжетой в течение двух-трех минут появляется «рука акушера».
3. Симптом Шлезингера — возникновение судорог в разгибателях бедра с одновременной супинацией стопы при быстром пассивном сгибании выпрямленной в коленном суставе ноги исследуемого.
4. Симптом Эрба — катод-замыкатель-ное сокращение мышц и катод-замыкатель-ный тетанус.
5. Симптом Гофмана — при надавливании на нерв отмечается не только болезненность в месте давления, но и распространение парестезии по ходу данного нерва.
При гипопаратиреозе отмечается повы--шенная возбудимость вегетативной нервной системы, что выражается в тахикардии, потливости, вегетативных реакциях.
Возможны поносы, пилороспазм, гастро-су ккорея, гиперхлоргидрия.
Нередки неврастенические и истерические реакции. Психозы практически не встречаются.
Наблюдаются трофические изменения ногтей (ломкость, исчерченность, расслоение), волос (тусклость, выпадение, раннее поседение), зубов (дефект эмали, выкрашивание, кариес). Нередко развивается катаракта.
Гипокальциплазмия наблюдается при идиопатическом гипопаратиреозе в связи с образованием в паращитовидных железах антител при ХПН, алкалозе, гипогонадизме, неадекватной секреции вазопрессина, сахарном диабете.
Клинические проявления: снижение концентрации плазменного кальция до 1,87 ммоль/л и ниже, а ионизированного — до 1,07 ммоль/л и ниже; повышение нервно-мышечной возбудимости, повышенная готовность к развитию судорожного состояния, типичной тетании.
Приступам судорог предшествуют парестезии, онемение верхней губы, пальцев рук и ног; затем возникают затруднение дыхания, болезненные судороги преимущественно в группах сгибателей; судороги могут сопровождаться ларингоспазмом, развивается цианоз, может наступить асфиксия.
Развивающийся одновременно пилороспазм сопровождается болями, рвотой, одновременно спазмируется кишечник, мочевой пузырь. Кардиотетания проявляется спазмом коронарных сосудов.
Скрытая форма тетании проявляется симптомами Труссо, Хвостека, Эрба; при длительном течении заболевания возможно развитие гипокальциемической катаракты, снижение интеллекта, неврозы.
Терапия гипокальциемии: для купирования приступа внутривенно вводят 20 мл 10 % раствора СаС12 или кальция глюконата;
Внутримышечно вводится паратиреоидин (паратирин) по 2 —4 мл (40 — 80 ЕД); применяется эргокальциферол (витамин D2) — от 10 до 60 капель масляного раствора в сутки (в среднем 1 млн МЕ/сут); назначается диета, богатая кальцием и бедная фосфором (молоко, сыр, капуста, салат, абрикосы, клубника, лимоны) [12, 16, 18, 19, 30].
Обмен электролитов 301
8.10.4. Обмен магния
В организме человека с массой тела 70 кг содержится примерно 2000 мэкв магния (24 г). Половина этого количества приходится на кости, 49 % — на клетки и только 1 % — на внеклеточную жидкость. В норме концентрация магния в плазме колеблется от 1,6 до 2,3 мэкв/л. 65 — 80 % внеклеточного магния находится в ионизированном состоянии. Остальное количество магния связано с белками, в основном с альбумином. Поэтому при гипоаль-буминемии может наблюдаться дефицит магния. Таким образом, магний является преимущественно внутриклеточным катионом. В разных тканях его содержание неодинаковое. Больше всего магния содержат клетки печени и поперечнополосатая мускулатура — примерно 20 мэкв/л внутриклеточной воды.
Обычно при заболеваниях печени гепатоциты теряют магний, поскольку внутриклеточно он удерживается против градиента концентрации с затратой энергии. При хронических интоксикациях у алкоголиков содержание магния в печеночных клетках снижено. При внутривенном введении солей магния происходит активация функций печени, стимулируется желчеобразование. При приеме через рот в силу медленного всасывания магний создает гиперосмолярность в просвете двенадцатиперстной кишки и, таким образом, рефлекторно стимулирует моторику желчевыводящих путей.
По своему физиологическому значению магний занимает второе место после калия. Он является необходимым компонентом каталитических реакций почти 300 ферментных систем и многих обменных процессов, способствует образованию и гидролизу АТФ, креатинфосфата. Магний играет важную роль в процессах гликолиза, включается в различные звенья цитратного цикла, окислительного фосфорилирования, участвует в активировании фосфатаз, нуклеаз, пептидаз. Холинэстераза активируется исключительно магнием, синтез холестерина и его эстерификация (образование растворимых форм) осуществляются при наличии магния. В результа
те такого суммарного воздействия на многие энзимные процессы последний тормозит передачу в нервно-мышечных синапсах, действуя аналогично кураре, вызывает депрессию ЦНС. Поэтому соли магния потенцируют наркоз. При его концентрации в крови 8 мэкв/л возникает поверхностный наркоз, при концентрации 16 мэкв/л — глубокий наркоз, а при концентрации 20 мэкв/л и выше — паралич дыхания и смерть.
По своему влиянию на нервно-мышечную возбудимость ионы разделяются на две группы. Ионы Na+, К+, ОН" повышают нервно-мышечную возбудимость, а ионы Са2+, Mg2+, Н+ снижают ее, поэтому последние называют ионами торможения. Особенно повышается нервно-мышечная возбудимость при одновременном снижении в крови концентрации ионов Са2+ и Mg2+. Если первые влияют на нервно-мышечную возбудимость только поперечнополосатых мышц, то последние еще и гладких мышц. Поэтому магний применяют при лечении гипертонической болезни. Он ослабляет контрактильные свойства гладких мышц и сердечной мышцы, угнетает возбудимость синусного узла и проведение импульса в предсердиях, а в больших дозах может остановить сердце в диастоле.
При внутривенном введении больших доз солей магния дыхание угнетается раньше потери сознания. Это один из недостатков магнезиального наркоза. Поэтому при внутривенном введении раствора магний сульфата под рукой необходимо иметь все приспособления для проведения ИВЛ.
За сутки в организм вместе с пищей поступает в среднем до 30 мэкв магния. Много его содержится в овощах, фруктах, мясе. Почки являются главным органом регуляции содержания магния в организме. При его дефиците они обеспечивают 100 % реабсорбцию магния. В верхней трети кишечника всасывается примерно 25 мэкв магния, а остальная часть (50 — 80 % поступившего с пищей магния) не всасывается и выделяется с калом. Поэтому прием больших количеств магния, особенно магний сульфата, вызывает повышение
302 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
осмолярности кишечного содержимого и перемещение воды из интерстициального пространства в просвет кишок, в результате чего реализуется слабительный эффект.
Дефицит внутриклеточного магния может привести к повышению содержания в клетках натрия и кальция, а также потере ими калия. Определение внутриклеточного содержания магния в мононуклеарах может быть индикатором его содержания в сердечной мышце. Предполагается, что у населения, живущего в регионах с низким содержанием магния в питьевой воде, чаще возникают заболевания сердца, чем у людей, употребляющих воду с высоким его содержанием. У жителей экваториальных областей развивается гипомагниеми-ческий эндомиокардиальный фиброз. Дефицит магния в организме играет важную роль в патогенезе ишемической болезни сердца (ИБС). Нитраты нормализуют уровень магния в сыворотке крови. Есть сведения, что такой же эффект оказывают и р-адреноблокаторы. В экспериментальных исследованиях показано, что прием магния снижает частоту развития атеросклероза.
При сахарном диабете (инсулинозависимом и инсулинонезависимом) имеют место повышенные потери магния с мочой, а также снижение уровня сывороточного и внутриклеточного магния. Назначение препаратов магния внутрь помогает успешно лечить обе разновидности сахарного диабета. Многие ферменты, участвующие в гликолизе, зависимы от магния. У больных с повышенной резистентностью к инсулину в плазме зафиксирован низкий уровень свободного магния. У экспериментальных животных с дефицитом магния в организме обнаруживали повышенную стимуляцию синтеза глюкагона, сниженную секрецию инсулина и сниженный его захват. Инсулин вызывает перемещение магния из внеклеточного сектора в клетку.
Магний нормализует липидный обмен, снижает в плазме уровень холестерина и липопротеидов низкой плотности (ЛПНП) и повышает концентрацию липопротеидов высокой плотности (ЛПВП), уменьшает поступление кальция в клетки. Дефицит
магния в организме ведет к перегрузке гладкомышечных клеток сосудов натрием и кальцием, вследствие чего повышается периферическое сосудистое сопротивление и возникает сосудистый спазм. Но однозначной связи между магнезиальным статусом и артериальной гипертензией пока не установлено. Имеются достоверные наблюдения, что дефицит магния может вызвать спазм коронарных артерий. Его можно рассматривать в качестве естественного блокатора кальциевых каналов биологических мембран. Больных со стенокардией Принцметала удавалось успешно лечить внутривенным введением препаратов магния.
Галогенаты магния подавляют агрегацию тромбоцитов, вызываемую АДФ. В клинических исследованиях отмечено, что под влиянием магния удлиняется время кровотечения у больных, перенесших инфаркт миокарда, снижается свертываемость крови у женщин с преэклампсией, уменьшается повышенная агрегация тромбоцитов у больных сахарным диабетом.
При инфаркте миокарда дефицит магния особенно опасен в постинфарктном периоде. Первичный положительный эффект магния относят за счет снижения частоты возникновения злокачественных аритмий. Инфузия препаратов магния больным с подозрением на наличие инфаркта миокарда может снизить потребность миокарда в кислороде и ограничить размеры инфаркта. После перенесенного инфаркта миокарда имеет место дефицит магния в организме, гипомагниемия может сохраняться в течение полугода. Существует прямая корреляционная связь между гипомагниемией и послеинфарктными желудочковыми аритмиями. Наджелудочковая тахикардия и мерцание предсердий чаще выявляются при гипомагниемии; атриовентрикулярная блокада и наджелудочковая брадикардия — при гипермагниемии. В случаях гипопаратиреоза кардиомиопатии поддаются лечению введением препаратов кальция и магния. При алкогольной кардиомиопатии имеет место дефицит магния в организме. Предотвращение его потерь может предохранить сердце от действия кардиотоксичных агентов.
Обмен электролитов 303
У больных с застойной сердечной недостаточностью часто имеет место дефицит магния в организме из-за потери его с мочой в результате вторичного гиперальдостеронизма. Ситуацию может ухудшить применение диуретиков и дигиталиса, уменьшающих канальцевую реабсорбцию магния, тогда как ингибиторы ангиотензинпревра-щающего фермента, задерживающие магний в организме, снижают летальность больных с застойной сердечной недостаточностью.
Дефицит магния в организме часто сопровождается гипокалиемией, что проявляется на ЭКГ удлинением интервала Q — Т, снижением сегмента S —Т и амплитуды зубца Т.
Повышение уровня магния плазмы выше нормы ведет к брадикардии, увеличению времени проведения возбуждения, снижению автоматизма сердца. Внутривенное введение препаратов магния уменьшает количество желудочковых экстрасистол у больных с дефицитом калия или магния в организме. Даже при нормальном уровне магния в плазме крови в случаях рефрактерности к антиаритмическим препаратам, внутривенное введение препаратов магния может оказать благоприятное действие. В угрожающих жизни ситуациях внутривенно вводят 2 — 3 г магний сульфата в течение одной минуты. Внутривенное введение этого препарата хорошо переносится больными, за исключением пациентов с почечной недостаточностью и блокадой предсердно-желудочкового пучка. Однако вышеуказанная нагрузочная доза магния не должна вводиться быстрее, чем за одну минуту, так как может наступить остановка сердца.
В ряде исследований отмечена эффективность внутривенного введения препаратов магния при специфическом виде аритмии, называемой «пируэт», сопровождающейся выраженным удлинением интервала Q — Т, причем эффективность отмечена даже на фоне рефрактерности к антиаритмическим препаратам. Дозировки могут быть различными: от капельного введения по 50 мг/мин до болюсного введения 1 — 2 г магний сульфата в течение 1 — 2 мин. Магний может оказывать лечебное действие и при наджелудочко
вых тахиаритмиях: предсердной (в том числе политопной) и узловой тахикардии, обусловленной обратным входом волны возбуждения. В этих ситуациях магний сульфат обычно вводят внутривенно в дозе 2 г в течение минуты.
Лечение дигоксином и особенно его передозировка могут сопровождаться потерями магния из организма, в результате чего снижается порог возникновения желудочковых экстрасистолии и фибрилляции. В таких случаях аритмии считают проявлением дигиталисной интоксикации. Введение препаратов магния в подобных ситуациях оказывает лечебное действие, в том числе у больных с гиперкалиемией, угрожающими жизни аритмиями, рефрактерными к лидокаину и дифенину. При дигиталисной интоксикации вводят внутривенно 2 — 3 г магний сульфата в течение минуты, при необходимости продолжают капельное введение до достижения концентрации магния в сыворотке крови на уровне 4 —5 мэкв/л. При этом его концентрацию в сыворотке крови измеряют каждые 2 ч. Инфузию следует немедленно прекратить в случаях возникновения угнетения дыхания или ослабления глубоких сухожильных рефлексов.
Калийсберегающие диуретики задерживают магний в организме. У больных с азотемией очень часто имеет место гипер-магниемия.
У кальция и магния выявлен определенный антагонизм по влиянию на метаболизм. В ряде случаев этот антагонизм не имеет физиологического объяснения. Так, в гладких мышцах, активатором сократительного ответа которых является внеклеточный кальций, повышение уровня магния во внеклеточной среде немедленно индуцирует расслабление, препятствуя действию кальция. В большинстве случаев кальций является антагонистом магния. Соотношение внутриклеточных концентраций магния и кальция управляет клеточным метаболизмом [46].
Повышенное поступление кальция в ишемизированные нейроны осуществляется благодаря избыточному накоплению в мозговой ткани глютамата. Именно избыточная активация специфических N-метил-
304 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
D-аспартатных рецепторов глутамата на поверхности клеточных мембран нейронов приводит к открытию кальциевых каналов, обеспечивающих поступление большого количества кальция через клеточную мембрану внутрь нейронов. Было обнаружено, что магний подавляет избыточную продукцию глутамата и, таким образом, оказывает протекторное действие при ишемии и гипоксии мозга.
Обмен магния находится под контролем паратгормона. Повышение содержания последнего вызывает отрицательный баланс магния. Воздействие паратгормона на обмен магния косвенное. В норме в сутки с мочой теряется до 9 ммоль магния. Суточная потребность в нем для взрослых составляет 12 — 40 ммоль.
Под гипермагниемией подразумевают состояние, когда концентрация магния плазмы превышает 3 мэкв/л (1,5 ммоль/л). Избыток последнего в организме может наблюдаться при снижении выделения магния почками (ОПН, ХПН), избыточном введении его в организм (например, при лечении эклампсии), использовании антацидных средств на фоне ХПН.
Клинические проявления избытка магния в организме прежде всего связаны с торможением выделения ацетилхолина, снижением возбудимости мышц, развитием их пареза и паралича, нарушением возбудимости сердечной мышцы, угнетением дыхания, брадикардией, атриовентрикулярной блокадой, чувствами жажды и жара. При концентрации магния в плазме от 2,5 до 3,5 ммоль/л у больных отмечается сонливость, снижение сухожильных рефлексов, нарушение проводимости импульса по миокарду, при 6 ммоль/л — наступает остановка дыхания, а при 7,5 — 10 ммоль/л — остановка сердца.
Лечение гипермагниемии состоит в промывании желудка, введении солевого слабительного, внутривенном введении 10 —20 мл 10 % раствора кальций хлорида, проведении гемодиализа и ИВЛ по показаниям.
Если больной находится на парентеральном питании и не получает магний, то через полторы-две недели наступает истощение костного депо магния и развивается гипомагниемия.
Гипомагниемией называется состояние, при котором концентрация магния плазмы не превышает 1 мэкв/л. Причины развития гипомагниемии следующие:
1) прием пищи, богатой белками и кальцием, но бедной магнием и витамином В6;
2) голодание и инфузия растворов, не содержащих магний;
3) хронический алкоголизм, цирроз печени;
4) резекция больших участков тонкой кишки или нарушение всасывания в тонкой кишке;
5) потеря пищеварительных секретов через свищи и при диарее;
6) увеличение выведения магния с мочой в полиурической стадии ОПН;
7) первичный гиперпаратиреоидизм;
8) острый панкреатит.
Клиническими признаками общего дефицита магния в организме являются:
а) депрессия;
б) психические расстройства в виде галлюцинаций;
в) повышение рефлекторной активности, тремор, спазм мышц, тетания;
г) атетозоподобные движения;
д) нистагм;
е) парестезии;
ж) гипотензия, тахикардия, желудочковые экстрасистолы;
з) гипокальциемия вследствие нарушения освобождения паратирина из-за дефицита магния в организме.
Подтверждением гипомагниемии является концентрация магния в суточной моче менее 3 мэкв/л или выделение с мочой в течение 16 ч после внутривенной инфузии 3 — 40 мэкв магния менее 70 % введенного количества (этот тест не пригоден, если дефицит магния вызван усиленным его выведением через почки). Лечение гипомагниемии состоит во внутривенном введении препаратов магния. При концентрации магния плазмы менее 0,5 ммоль/л внутривенно в течение 3 ч ввводят 1 л раствора, содержащего 6 г магний сульфата. При парентеральном питании больной должен получать не менее 25 мэкв магния в сутки. Всасывание магния из кишок тормозится кальцием и фосфатами [1, 3-5, 7-9, 19,31].
Обмен электролитов 305
8.10.5. Обмен хлора
Хлор является наиболее распространен-ным анионом в организме — при массе тела 70 кг в нем содержится около 80 г хлора. Биологическая роль ионов СГ состоит в нейтрализации положительно заряженных ионов и создании осмотического давления в жидкостных средах организма. При описании осмотических эффектов натрия в формулы для расчета осмолярности всегда вводится коэффициент 1,86 или удвоенная концентрация натрия плазмы. Это удвоение подразумевает наличие в биологических жидкостях соответствующего количества отрицательно заряженных ионов, значительная часть которых, особенно во внеклеточной жидкости, представлена ионами СГ. Избыток или недостаток хлора в организме всегда сочетается с таковым какого-нибудь катиона. Концентрация хлора во внеклеточном пространстве отличается достаточной стабильностью и составляет 103 — 105 мэкв/л. Концентрация хлора и его суммарное содержание во внутриклеточном пространстве значительно меньше. Так, в скелетной мышце содержание хлора составляет 3 мэкв/л, в миокардиоците — 7, в гепатоците — 12, в нейронах головного мозга — 13, в эритроцитах — 54 мэкв/л внутриклеточной воды.
Большое количество хлора накапливается в воспалительных очагах органов, содержащих соединительную ткань. Поэтому при пневмониях может наблюдаться относительная гипохлоремия. Хлор играет важную роль в поддержании КОС. При ацидозе его концентрация в плазме повышается. Кроме того, он поступает в клетку в обмен на ион НСО3. Хлор играет большую роль в выведении из организма азотистых шлаков. При гипохлоремии нарушается образование в эпителии почечных канальцев NH4C1 из NH3 и НС1. При длительно существующей гипохлоремии аммиак вызывает дегенеративные изменения эпителия почечных канальцев с развитием азотемии и уремии. Гипохлоремия ниже 70 мэкв/л, сохраняющаяся в течение 3 суток, вызывает необратимые изменения в почках. Хлор, соединя
ясь со многими органическими веществами, изменяет их биологические свойства — они как бы становятся индифферентными для организма.
При нормальном содержании остаточных анионов между ионом НСО3 и ионом СГ существуют реципрокные отношения — при увеличении в плазме концентрации ионов НСО3 концентрация хлора снижается, и наоборот.
Под гипохлоремией подразумевают состояния, когда концентрация хлора в сыворотке крови не превышает 95 мэкв/л. Она развивается при многократной рвоте, интенсивных промываниях желудка чистой водой, фистулах желудка, двенадцатиперстной и тонкой кишок. Гиперхлоремией принято считать увеличение концентрации хлора в сыворотке свыше 115 мэкв/л [1,7-9].
8.10.6. Обмен фосфора
Средняя концентрация общего фосфора в плазме взрослого человека колеблется от 3 до 5 ммоль/л (10—15 мг%). Этот элемент в организме находится в двух основных формах: в виде свободного, или неорганического фосфора, представленного ионами фосфорной кислоты, из которых 80 % приходится на двузамещенный фосфат, а 20 % — на однозамещенный, и связанного органического фосфора, содержащегося в различных эстерах фосфорной кислоты. Некоторые авторы выделяют еще липидный фосфор и фосфор нуклеиновых кислот.
В плазме крови концентрация неорганического фосфора в норме составляет 1 — 2 ммоль/л (3 — 6 мг%). Он представлен одно- и двузамещенными натриевыми и калиевыми солями фосфорной кислоты.
Наибольшее количество фосфора находится в костной ткани и внутриклеточной среде. При необходимости он легко мобилизуется из костей. Концентрация неорганического фосфора в клетке и вне ее составляет около 1 ммоль/л.
Органический фосфор входит в состав нуклеиновых кислот, фосфолипидов, сахарофосфатов, нуклеотидов. Одной из основ
306 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ В0ДН0-ЗЛЕКТР0ЛИТН0Г0 ОБМЕНА
ных функций фосфора является его участие в энергетическом обмене. Это обусловлено тем, что при разрыве связей фосфата с такими веществами, как аденин, креатин, аргинин, фосфоэнолпируват, аце-тил-радикал, ацетил-КоА, выделяется большое количество свободной энергии. Не исключено, что интенсивность процессов, сопровождающихся выделением энергии, регулируется концентрацией неорганического (свободного) фосфата. Его концентрация оказывает регуляторное влияние на обмен кальция.
Уровень общего фосфора во внутриклеточной среде зависит от разновидности ткани: в скелетной мышце его содержание составляет 107 ммоль/л, в сердечной мышце — 121, в гепатоците — 176, в нейроне — 184 ммоль/л внутриклеточной воды.
Таким образом, основная роль фосфора в организме заключается в том, что энергия в организме аккумулируется в виде фосфатных макроэргических соединений — АТФ. Фосфор принимает участие в обмене углеводов и белков, поскольку обязательной стадией их утилизации является фосфорилирование. Обмен фосфора связан с обменом кальция и косвенно — с обменом магния. Всасывание фосфора из ПК практически не ограничено.
Из организма фосфор выделяется с мочой почками. Причем этот процесс связан с регуляцией почками КОС. В эпителии почечных канальцев двузамещенный фосфат превращается в однозамещенный и этот радикал выделяется с мочой, а вместе с ним и избыточное количество гидроген-ионов.
Концентрация неорганического фосфора в плазме крови широко варьирует у людей различного возраста и подвержена суточным и сезонным изменениям с максимумом показателя в мае —июне и минимумом в декабре —январе.
Гипофосфатемия наблюдается главным образом во время повышенного анаболизма. При необходимости быстрого поступления фосфора в мышечные клетки осуществляют внутривенную инфузию глюкозы, стимулирующую выделение инсулина, который, в свою очередь, стимулирует анаболизм. Это может привести к недо
статку фосфора в других жизненно важных органах, при этом его резервы в организме остаются незадействованными.
Повышают экскрецию неорганического фосфора с мочой следующие состояния: алкалоз, гипокалиемия, гипомагниемия, сахарный диабет, применение стероидных гормонов, диуретиков.
Выраженная гипофосфатемия наблюдается у пациентов с хронической потерей массы тела, хронических алкоголиков, больных, получающих антацидную и диуретическую терапию. Клинически гипофосфатемия проявляется после снижения концентрации неорганического фосфора в сыворотке крови ниже 0,3 ммоль/л в виде анорексии, тошноты, рвоты, прогрессирующей мышечной слабости, снижения сердечного выброса, рабдомиолиза, тремора, парестезий, атаксии, конвульсий, снижения активности на электроэнцефалограмме (ЭЭГ), нарушения сознания, летаргии, комы, сокращения длительности жизни эритроцитов и тромбоцитов, снижения лейкоцитарного хемотаксиса и фагоцитоза, повышения сродства гемоглобина к кислороду, снижения поступления кислорода в ткани, нарушения минерального обмена (гиперкальциемии, гиперкальциурии, ги-пермагнийурии), в тяжелых случаях — в виде метаболического ацидоза, дыхательной недостаточности, кардиомиопатии (как правило, они заканчиваются сердечно-легочной недостаточностью и летальным исходом).
У хирургических больных достоверное снижение неорганического фосфора в плазме крови проявляется в ответ на операционную травму, особенно при длительности операций более 3 ч. Снижение концентрации плазменного фосфора на 0,2-0,4 ммоль/л наблюдается в ответ на инфузию глюкозы или фруктозы (чаще на инфузию последней). Уменьшение концентрации плазменного фосфора у больных с постоянной 24-часовой инфузией глюкозы обусловлено большей экскрецией фосфатов с мочой. По-видимому, постоянная длительная инфузия глюкозы приводит к нарушению механизма реабсорбции фильтрующихся фосфатов в проксимальных канальцах нефронов.
Обмен электролитов 307
Терапия гипофосфатемии считается трудной и до конца не решенной задачей. В определенной степени коррекцию неорганического фосфора можно осуществить инфузиями жировых эмульсий, например интралипида, содержащего до 18 ммоль/л фосфатов. В послеоперационном периоде гипофосфатемия коррелирует со снижением количества АТФ, 2,3-ДФГ в эритроцитах, повышением ионизированного кальция в плазме крови и эритроцитах. Для коррекции гипофосфатемии может использоваться калий дигидрогенфосфат в виде 6,8 % раствора по 20 — 50 мл в 40 мл 5 — 10% раствора глюкозы — 1 — 2 раза в сутки. При этом увеличение содержания 2,3-ДФГ и АТФ способствует более полной отдаче кислорода гемоглобином, улучшению кислородного и энергетического обеспечения тканей.
Введение препаратов неорганических фосфатов на фоне гиперкальциемии может привести к связыванию кальция и его отложению в виде кальций фосфата, сопровождаться гипокальциемией. Также возможно развитие гиперкалиемии, если для коррекции гипофосфатемии применяется избыточное количество раствора калий дигидрофосфата. Эти ятрогенные осложнения парентерального питания могут приводить к повышению осмолярности плазмы и сопровождаться дегидратацией и гипернатриемией. При олигурии не следует проводить лечение фосфатными препаратами [30].
8.10.7. Обмен микроэлементов
Обмен марганца
Роль этого элемента в организме до настоящего времени полностью не определена, но известно, что марганец, как и магний, является составной частью ферментов, участвующих в синтезе жирных кислот. Марганец активирует 3-фосфоглице-раткиназу, энолазу и некоторые другие ферменты.
Марганец необходим для формирования нормальной структуры костей, нормального пищеварения и усвоения пищи. Этот микроэлемент способствует нормаль
ному функционированию ЦНС и ее регенерации, участвует в синтезе тироксина.
Суточная потребность организма в марганце составляет от 100 до 800 мкг. Однако гипермарганцемия может наблюдаться уже при приеме 500 мкг/сут. Поэтому не следует назначать более 100 мкг марганца в день. Его не прописывают пациентам с заболеваниями печени. Дефицит марганца в организме может проявляться атаксией [12, 30].
Обмен железа
Входит в состав металлопротеидов, одним из основных белковых компонентов которых является гемоглобин. Следует отметить, что в молекуле гемоглобина железо всегда остается двухвалентным. Кроме того, этот микроэлемент входит в состав миоглобина, ферритина, трансферрина. Последний осуществляет в организме транспорт железа.
Ион Fe2+ является активным центром цитохромов, где проявляется его свойство менять свою валентность. В дыхательной цепи с участием цитохромов осуществляется перенос электронов. Железо входит в состав таких ферментов, как сукциноксидаза, каталаза, цитохромоксидаза и активирует альдолазу. При наличии ионов железа повышается продукция гидроксидионов в период реперфузии тканей. Этот элемент входит в состав каталитического центра одного из ключевых ферментов человеческого организма — NO-синтетазы.
Генерализованные бактериальные инфекции сопровождаются резким снижением в сыворотке крови концентрации железа. Измерение концентрации последнего в сыворотке, а также уровней трансферрина и альбумина используется для установления бактериальной природы лихорадки неясного генеза. В норме концентрация железа в сыворотке крови колеблется от 14 до 32 мкмоль/л (8—180 мкг%) [12, 30].
Обмен цинка
Биологическая роль цинка определяется тем, что он является составной частью некоторых белков и ферментов, связую
308 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
щим компонентом между двумя мономерами в молекуле инсулина. Входит в состав карбоангидразы, карбоксипептидазы, алкогольдегидрогеназы, глицерин-3-фос-фатдегидрогеназы. Цинк активирует эно-лазу, лактатдегидрогеназу, альдолазу, 5-нуклеотидазу и некоторые другие ферменты. Наличие ионов цинка способствует образованию кератина, росту костей. При недостатке в организме этого микроэлемента резко замедляется процесс заживления ран. Цинк необходим для синтеза белков и ДНК, он регулирует сократительную способность мышц, участвует в поддержании КОС, способствует поддержанию нормальной функции репродуктивных органов.
Увеличенные потери цинка с мочой наблюдаются на фоне повышенного катаболизма. Увеличение его выделения из организма через ПК зафиксировано при диарее, кишечных фистулах. Восстановление клеточной массы требует большого количества цинка. Потребность организма в этом микроэлементе при проведении парентерального питания может достигать 200 мкмоль/сут. Средняя суточная потребность в нем составляет 12 — 15 мг, или 0,185 — 0,230 ммоль. Чрезмерная потливость может привести к значительным потерям цинка из организма — более 3 мг/день.
Клиническая картина дефицита цинка в организме человека может быть различной. Она сопровождается апатией, депрессией, потерей волос, себорейным дерматитом вокруг носа и рта, в аногенитальной области, на коленях и локтях, пальцах рук и ног, стоматитом, диареей, а в тяжелых случаях — илеусом. Эти проявления обычно возникают в течение 1 — 2 месяцев после начала парентерального питания, не содержащего достаточного количества цинка. Кожные проявления его дефицита исчезают, как правило, через три-четыре дня после назначения 200 — 300 мкмоль цинка в день, а полное заживление поражений кожи происходит в течение 10 дней. Потеря волос прекращается медленнее. Дефицит цинка также проявляется гипертрофией предстательной железы, артериосклерозом, гипогонадиз
мом. В норме концентрация цинка в плазме составляет 12 — 22 мкмоль/л [12, 30].
Обмен молибдена
Молибден принимает участие в регуляции жирового обмена и утилизации железа. Суточная потребность в нем составляет 75 — 250 мкг. Дефицит молибдена встречается редко. В клинической картине его дефицита превалируют неврологические расстройства, головная боль, ночная слепота. Отмечены случаи летаргии и комы. Метаболические изменения связаны с нарушением метаболизма серосодержащих аминокислот и распадом пуринов. Лечение дефицита молибдена осуществляют аммоний молибдатом [12, 30].
Обмен хрома
Хром вместе с инсулином участвует в обмене глюкозы и транспорте протеинов. Точная суточная потребность в нем не-у станов лена, но для взрослых рекомендуемая доза составляет 50 — 200 мкг. С возрастом содержание хрома в организме снижается. О его количестве в организме можно судить по содержанию в волосах.
Хром способствует росту, предупреждает развитие артериальной гипертензии и снижает артериальное давление, препятствует развитию диабета. Дефицит хрома предрасполагает к развитию артериосклероза и диабета, сопровождается диабетоподобными синдромами, периферической нейропатией и дисметаболической энцефалопатией. Много хрома содержится в говяжьей печени, курином мясе, кукурузном масле, пшеничных зернах, пивных дрожжах [12, 30].
Обмен кобальта
Кобальт является составной частью витамина В12, необходимого для синтеза глобина и дозревания эритроцитов. Суточная потребность в нем составляет 8 мкг. Дефицит кобальта проявляется анемией. Много кобальта содержится в мясе, почках, печени, молоке, устрицах [12, 30].
Обмен электролитов 309
Обмен меди
Медь принимает участие в синтезе гемоглобина, тирозина (или активирует эту аминокислоту, являющуюся пигментным фактором для волос и кожи), утилизации витамина С.
Суточная потребность в меди составляет 1,5 — 3 мг. Дефицит этого микроэлемента может быть причиной возникновения гипохромной цитопенической анемии и нейтропении, а также повреждения костей у детей, отеков, дефектов скелета, ревматоидного артрита. Медью богаты сухие бобы, пшеница, чернослив, внутренние органы животных, креветки, большинство морепродуктов. Ее избыток приводит к снижению уровня цинка и является причиной сонливости, выпадения волос, нерегулярных месячных, депрессии. Приготовление и хранение кислых продуктов в медной посуде может увеличить поступление в организм меди с пищей [12, 30].
Обмен фтора
Фтор содержится в питьевой воде в виде натрий фторида и в пищевых продуктах в виде кальций фторида. Снижение в организме содержания фтора сопровождается кариесом, а также обесцвечиванием зубов. Суточная потребность в нем составляет от 1 до 4 мг. Фтор предотвращает кариес зубов и укрепляет кости. Много фтора в морских продуктах, чае. Токсическая доза фтора составляет от 20 до 80 мг/день. Содержание фтора в продуктах резко повышается, если варить их во фторированной воде или в тефлоновой посуде [12, 30].
Обмен ванадия и серы
Ванадий предупреждает развитие атеросклероза, угнетая образование холесте-рола в сосудах [12, 30].
Сера является незаменимым элементом для кожи и ее придатков, участвует в поддержании кислородного баланса мозговой ткани, вместе с витаминами группы В ре
гулирует базисный метаболизм, является составной частью синтезируемых аминокислот, участвует в образовании и секреции желчи [12, 30].
Обмен селена
Селен, как и витамин Е, обладает антиоксидантными свойствами. Мужчины испытывают большую потребность в селене по сравнению с женщинами. Почти половина запасов этого микроэлемента содержится в яичках и простатической жидкости. Много селена теряется со спермой. Суточная потребность в селене для мужчин составляет 70 мкг, для женщин — 50, для беременных —65, для кормящих матерей — 75 мкг. Его можна использовать для профилактики и лечения перхоти. Селен обладает противораковой активностью. Определены роль и значение этого микроэлемента в развитии таких осложнений, как кардиомиопатия и миопатия скелетной мускулатуры [12, 30].
Обмен лития
Литий как химический элемент был открыт в 1817 г. С середины XIX века его используют для лечения почечнокаменной болезни. Эффективность лития при лечении маниакально-депрессивных состояний была обнаружена в 1954 г. Механизм его лечебного действия до конца не изучен. Считается, что антидепрессивное и анти-маниакальное действие этого микроэлемента обусловлено его способностью потенцировать серотонинергическую активность. Литий обладает холинергической активностью, ингибирует биологические системы цАМФ и фосфоинозитола, что проявляется антиманиакальным действием.
Особенностью метаболизма лития является его накопление в тканях с периодом элиминации около месяца. У 60 % больных при лечении препаратами лития возникают побочные реакции, особенно у пожилых лиц, и при сочетанном его применении с нейролептиками и антидепрессантами. Побочные реакции возникают при концентрации лития в крови, равной 0,68 ммоль/л, и заключаются в следующем:
310 ФИЗИОЛОГИЯ И ПАТОЛОГИЯ ВОДНО-ЭЛЕКТРОЛИТНОГО ОБМЕНА
а) общий тремор развивается у 15 % больных (корригируется введением пропранолола);
б) увеличение массы тела наблюдается у 70 % пациентов;
в) диарея появляется у 6 % больных;
г) нарушение памяти без снижения внимания и концентрации;
д) снижение либидо;
е) повышение уровня тиреостимулирующего и снижение уровня тиреотропного гормонов;
ж) развитие гипотиреоза (особенно у женщин), который, в свою очередь, является причиной депрессии (поэтому необ
ходим регулярный контроль функции щитовидной железы);
з) нарушение функции почек вследствие повреждения дистального отдела канальцев, что клинически проявляется несахарным диабетом;
и) при приеме во время беременности у плода развиваются уродства в виде атрезии створок клапанов сердца, коарктации аорты. При беременности препараты лития абсолютно противопоказаны [32].
Таким образом, водно-электролитный обмен играет ключевую роль в поддержании постоянства внутренней среды организма.
Список использованной литературы
1. Хартиг В. Современная инфузионная терапия. Парентеральное питание: Пер. с нем. — М.: Медицина, 1982. — 496 с.
2. Зайцев В. Т., Губский В. И. Номографические методы экспресс-диагностики состояния внеклеточного гомеостаза у больных хирургического профиля: Метод, рекомендации М3 Украины. — Харьков, 1985. — 19 с.
3. Морган Дж. Э. мл., Мэгид С. М. Клиническая анестезиология: Пер. с англ. — М.; СПб.: Изд-во БИНОМ — Невский Диалект, 2000. - Кн. 2. - С. 241-272.
4. Galloway F. М. Fluids and Electrolytes // J. Duke, S. G. Rosenberg. Anesthesia secrets. — Hanley and Belfus Inc., 1996. — P. 18 — 27.
5. Irwin R. S., Cerra F. B., Rippe M. Intensive Care Medicine. — 4th ed. — Vol. 1. — 1999. — P. 911-941.
6. Цветанова E. M. Ликворология: Пер. с болг. — К.: Здоров’я. — 372 с.
7. Жалко-Титаренко В. Ф. Водно-электролитный обмен и кислотно-основное состояние в норме и при патологии. — К.: Здоров’я, 1989. - 200 с.
8. Исаков Ю. Ф., Михельсон В. А., Штатнов М. К. Инфузионная терапия и парентеральное питание в детской хирургии. — М.: Медицина, 1985. — 286 с.
9. Беляевский А. Д., Монченко Г. Д. Интенсивная терапия при сочетанных нарушениях кислотно-щелочного состояния и водно-электролитного обмена. — Ростов-н/Д.: Изд-во Рост. гос. мед. ун-та, 1997. — 136 с.
10. Симоненков А. П. Функциональная кишечная непроходимость, диссеминированное внутрисосудистое свертывание крови и симп-томокомплекс, возникающий при отравлении
психотропными препаратами, как клиническое проявление серотониновой недостаточности: Автореф. дис. ... д-ра мед. наук. -М., 1992. - 50 с.
11. Симоненко В. Б., ЧусевА. Ю., Ивашкин Т. В. Купирование карциноидного синдрома сан-достатином // Рос. журн. гастроэнтерологии, гепатологии. — 1993. — № 1. — С. 54-56.
12. Теппермен Дж., Теппермен X. Физиология обмена веществ и эндокринной системы: Пер. с англ. — М.: Мир, 1989. — 656 с.
13. Большая медицинская энциклопедия. -3-е изд. — М.: Сов. энцикл., 1979. — Т. 10. -С. 97-101.
14. Бредбери М. Концепция гематоэнцефалического барьера. — М.: Медицина, 1988. -С. 479 .
15. Pappas G. Some morphological considerations of the Blood —Brain Barrier // J. Neurol. Sci. - 1970. - Vol. 10, N 2. - P. 241-246.
16. Баранов В. Г. Руководство по клинической эндокринологии. — Л.: Медицина, 1977. -С. 209-221.
17. Фелиг Ф., Бакстер Дж. Д., Бродус А. Е., Фромен Л. А. Эндокринология и метаболизм. — М.: Медицина, 1985. — Т. 1. — 386 с.
18. Жуковский М. А. Детская эндокринология. — 2-е изд. — М.: Медицина, 1982. -С. 389-393.
19. Балаболкин М. И. Гормональная регуляция водно-электролитного обмена // Терапевт, арх. - 1989. - № 10. - С. 83-87.
20. Macchia D. D. Atrial natriuretic factor: a hormone secreted by the heart // Pharm. Wbl. -1987. - Vol. 9, N 6. - P. 305-314.
21. Shoemaker W. Clinical trial of survivors cardiorespiratory patterns as therapeutic goals in
Список использованной литературы 311
critically ill postoperative patients // Crit. Care Med. - 1982. - N 10. - P. 398-403.
22. Buffaloe G. W., Heineken F. G. Plasma volume nomograms for use in therapeutic plasma exchange // Transfusion. — 1983. — N23. - P. 355-357.
23. Кулаков В. И. Профилактика и лечение кровотечений в акушерстве и гинекологии: Метод. рекомендации № 96/120 М3 Российской Федерации. — М., 1997. — 24 с.
24. Марютин П. В. Кровопотеря-гиповолемия, подходы к инфузионно-трансфузионной коррекции // Анестезиология и реаниматология. - 1998. - № 3. - С. 35-41.
25. Баркаган 3. С. Узловые вопросы комплексной терапии острого и подострого ДВС-син-дрома // Вести, интенсивной терапии. — № 1. - С. 11-16.
26. Оборин А. Н., Шичкин В. П. Роль фактора некроза опухолей-альфа при травматическом шоке и острой кровопотере // Журн. АМН Украши. — 1998. — 4, № 2. — С. 253 — 267.
27. Серов В. Н., Абубакирова А. М., Баронов И. И. Современные подходы и новые технологии при профилактике и лечении кровопотери в акушерстве / / Акушерство и гинекология. — 1998. - №4. — С. 44 — 47.
28. KirbyR. R., Taylor R. W., Civettai. М. Hand book of critical care. — 2nd ed. — Lippincott-Raven., 2000. - P. 25-232.
29. Ford-Adams M. E., Edge A. Coma in child with diabetes // Curr. Anaesth. and Crit. Care. - 1999. - Vol. 10, N 5. - P. 257-261.
30. Свиридов С. В., Ломова M. А. Метаболические осложнения парентерального питания // Анестезиология и реаниматология. — 2001. - №2. - С. 64-70.
31. Purvis J. R. Disorders Mg and Cardiovascular Diseases // Clin. Cardiol. — 1992. — Vol. 15, N6. - P. 556-568.
32. Thibaut F. Le lithium ... 30 ans apres // Rev. Prat. (Paris). — 1991. — Vol. 41, N 1. — P. 56-60.
Раздел
Кислошно-осноВное состояние
Кислотно-основное состояние (КОС), или баланс кислот и оснований в организме, является важной составляющей гомеостаза, характеризуется относительным постоянством соотношения гидроген- (водородных) и гидроксид-ионов, оказывает большое влияние на метаболизм и физиологические функции организма.
Для понимания процессов поддержания КОС прежде всего следует рассмотреть характеристики основного биологического растворителя — воды. В незначительной степени вода диссоциирует на ионы Н+ и ОН", которые находятся в равновесии с недиссоциированными молекулами: Н2О <=» Н+ + ОН“. Равновесную концентрацию обычно обозначают с помощью квадратных скобок и выражают в молях ионов в 1 л раствора. Вода, как правило, является нейтральным раствором. Опытом установлено, что в 1 л воды при нейтральной реакции раствора и температуре 22 °C диссоциации подвергаются лишь 10"7 моль и при этом образуется [Н+] = [ОН-] = 10"7. Произведение концентраций ионов Н+ и ОН" в воде называется ионным произведением воды (Кв). При определенной температуре Кв — величина постоянная. Численное значение его при 22 °C равно 10~14. Кв = [Н+] • [ОН"] = 10"14 вне зависимости от реакции раствора: кислой или основной. Из этого следует, что при повышении концентрации гидроген-ионов будет снижаться концентрация гидроксидионов, и наоборот, поэтому достаточно определить концентрацию одного из этих
компонентов. Обычно о реакции раствора судят по концентрации гидроген-ионов при кислой реакции раствора [Н+] > 10"7, при основной — [Н+] < 10“', при нейтральной — [Н+] = 10"7. Концентрация 10"7 соответствует 100 нмоль/л гидроген-ионов. Во внеклеточной жидкости в норме [Н+] = 10"7’4 = 40 нмоль/л. Несмотря на то что эта концентрация очень мала по сравнению с другими основными ионами внутри- и внеклеточной жидкости, поддержание этого показателя в нормальных пределах является очень важным компонентом жизнеспособности организма. Отклонение концентрации гидроген-ионов от нормы существенным образом влияет на каталитическую активность ферментов, функциональное состояние нуклеиновых кислот, мембран клеток и их рецепторов, на ионный состав водных сред организма. Однако манипуляции с цифрами типа 10"7,4 представляют большие трудности, в связи с этим S. Sorensen в 1909 г. предложил для удобства показатель pH, который равняется отрицательному десятичному логарифму концентрации гидроген-ионов в растворе:
pH = -lg[H+].
Другими словами, это величина, обратная концентрации гидроген-ионов, или степень разведения гидроген-ионов. Исходя из этого определения, pH раствора с [Н+] = = 10"7’4 равняется 7,4. При повышении [н+] Рн снижается и в кислой среде (в плазме крови) будет ниже 7,35, в ос-
Буферные системы организма 313
Таблица 9.1. Актуальное pH жидкостных сред организма [1]
Среда организма pH
Артериальная кровь 7,30-7,42
Венозная кровь 7,26-7,36
Спинномозговая жидкость 7,4-7,5
Межклеточная жидкость 7,26-7,38
Слюна 5,8-7,8
Сок поджелудочной железы 7,8-8,4
Желудочный сок 1,4-1,8
Желчь пузырная 6,0-7,0
Секрет тонкой кишки 7,5-8,6
Секрет толстой кишки 8,0-9,0
новной — выше 7,45. Чем ниже pH, тем выше [Н+], и наоборот. Величина pH в различных жидких средах организма не-динакова. Значения pH различных сред организма представлены в табл. 9.1. pH раствора определяется либо с помощью индикаторов, либо электрометрически.
Для оценки кислотно-основного статуса организма необходимо знать, что такое кислота и основание.
Кислота — это вещество, которое при диссоциации в растворе отдает гидроген-ионы (донаторы протона). Кислоты диссоциируют в растворе на гидроген-ион (катион) и анион (А“):
НА -> Н+ + А".
Если реакция идет в обратную сторону (влево), А~ рассматривают как сопряженное основание. В случае сильной кислоты реакция смещена вправо, если же она слабая (например, угольная кислота), реакция диссоциации происходит не полностью и ее степень зависит от константы диссоциации кислоты.
Кислоты образуются в результате метаболических процессов из питательных веществ. Больше всего в организме образуется угольной кислоты, источником которой является углекислый газ:
СО2 + Н2О <=> Н2СО3 <=* Н+ + НСО3.
Угольная кислота является слабой, однако при наличии карбоангидразы хорошо диссоциирует и принимает важное участие в транспорте СО2. Поскольку в легких она является источником выделяемого СО2, эта кислота еще называется летучей.
При окислении в клетках образуются нелетучие органические и неорганические кислоты: из углеводов — пировиноградная и молочная кислоты, из липидов — свободные жирные кислоты, из фосфопротеинов и нуклеопротеинов — фосфорная и мочевая кислоты, из серосодержащих аминокислот — серная кислота.
Основание — это вещество, которое при диссоциации в растворе присоединяет гид-роген-ионы (акцепторы протонов). Основания поступают в организм преимущественно с растительной пищей, но часть из них образуется в организме в процессе секреторной деятельности органов пищеварительного канала (кишок, поджелудочной железы).
Характерная для крови слабоосновная среда поддерживается в очень узких пределах, несмотря на то что в нее поступают в основном кислые продукты. В поддержании относительного постоянства pH принимают участие буферные и так называемые физиологические системы организма (на наш взгляд, название не слишком удачное, так как буферные системы тоже являются физиологическими).
9.1. БУФЕРНЫЕ СИСТЕМЫ ОРГАНИЗМА
Буферные системы организма способны сглаживать (буферировать) колебания pH в условиях кислой либо щелочной агрессии.
Количество ионов Н+ или ОН“, добавленных в раствор, чтобы изменить его активную реакцию на одну единицу pH, на
зывается буферной емкостью — это показатель способности системы создавать буферный эффект.
Буферные системы организма состоят из двух веществ: слабой кислоты и ее соли, образовавшейся в результате реакции этой кислоты с сильным основанием. Как бу-
314 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
ферм могут действовать также амфотерные вещества, которые в зависимости от реакции раствора ведут себя либо как основание, либо как кислота.
К буферным системам относятся: гид-рогенкарбонатная, фосфатная, белковая и гемоглобиновая.
9.1.1. Гидрогенкарбонатная буферная система
Гидрогенкарбонатная система включает в себя угольную кислоту и ион гидро-генкарбоната
Н2СО3
НСО3 ’
Между реакциями ассоциации и диссоциации существует равновесие, подчиняющееся закону действующих масс. Кинетика таких реакций описывается уравнением
тл [Н+][А~]
[НА] ’
где величины, приведенные в квадратных скобках, — это молярные концентрации веществ, участвующих в реакции; К — константа диссоциации.
Для того чтобы определить, чему равно pH, необходимо данное уравнение прологарифмировать:
[Н+].[А-] = К[НА];
[н+] = к[Н41.
[А"]
Отсюда
-lg[H+] = -IgK-или [А ]
pH = pK + lgLJj.
еще
Последнее уравнение называют уравнением Гендерсона — Гассельбаха.
Для угольной кислоты оно будет иметь следующий вид:
pH.pK + lg™
LCU2J
[СО2] = а • рСО2,
где а — коэффициент растворимости СО2 в воде.
Если подставить числовые значения, то уравнение Гендерсона —Гассельбаха примет следующий вид:
, .24 ммоль/л _ , 6,1 + 1g -----'-т- = 7,4.
1,2 ммоль/л
В условиях реакции нейтрализации гид-рогенкарбонатного буфера с кислотами образуется слабая угольная кислота:
НС1 + NaHCO3 = NaCl + Н2СО3, а образовавшийся углекислый газ удаляется легкими.
Избыток оснований, взаимодействуя с гидрогенкарбонатной буферной системой, связывается с угольной кислотой с образованием натрий гидрогенкарбоната, который впоследствии может быть удален почками:
NaOH + Н2СО3 = NaHCO3 + Н2О.
9.1.2. Фосфатная буферная система
Эта система образована одно- и двузамещенной солями фосфорной кислоты
NaH2PO4
Na2HPO4 ’
где однозамещенная соль (NaH2PO4) выступает как слабая кислота, а двузамещенная (Na2HPO4) — как слабое основание.
Если гидрогенкарбонатная буферная система в основном действует в крови, то фосфатная — главным образом участвует в буферировании pH внутри клетки, где фосфаты представлены фосфолипидами и органическими фосфатами. Химические реакции фосфатной буферной системы тесно связаны с физиологическими процессами в почках и гидрогенкарбонатной буферной системой.
9.1.3. Белковая буферная система
Молекулы белка обладают амфотерными свойствами и характер их диссоциации зависит от природы белка и реакции раствора. Важную роль в буферной емкости данной системы играют боковые группы, способные ионизироваться, особенно имидазольное кольцо гистидина. Глобулины обладают более выраженными кислыми свойствами и имеют большое значение
Физиологические системы регуляции pH 315
для нейтрализации щелочей, а белки, содержащие больше диаминовых аминокислот, диссоциируют преимущественно как основания, и принимают участие в нейтрализации кислот. Так же, как и фосфатная, белковая буферная система играет важную роль внутри клетки.
9.1.4. Гемоглобиновая буферная система
Молекула гемоглобина содержит аминокислоту гистидин (до 8,1 %) [1], в состав которой входят как кислые (-СООН), так и основные (-NH2) группы. Гемоглобин играет важную роль в буферировании кро-
9.2. ФИЗИОЛОГИЧЕСКИЕ
9.2.1. Система внешнего дыхания
Как указывалось выше, углекислый газ в значительной степени влияет на кислотно-основной баланс в организме, образуя угольную кислоту. Так, в состоянии покоя образуется до 230 мл СО2 в минуту, или примерно 15 тыс. ммоль/сут. При удалении легкими этого количества СО2 в крови исчезает примерно эквивалентное количество ионов Н+ [2]. Таким образом, удаляя СО2, система внешнего дыхания в значительной степени влияет на КОС.
В поддержании нормальных значений pH большое значение имеет центральная регуляция дыхания (см. «Центральная регуляция дыхания»). При недостаточности функции внешнего дыхания (альвеолярная гиповентиляция) происходит задержка углекислого газа, а вместе с ней накапливаются и гидроген-ионы, снижая pH. При гипервентиляции выводится избыточное количество СО2, что приводит к уменьшению концентрации ионов Н+ и повышению pH.
Респираторными называют такие расстройства КОС, в основе которых лежат нарушения функции внешнего дыхания. В значительной степени влияет на КОС и оксигенация крови, так как ее недостаточность будет способствовать накоплению молочной кислоты и гидроген-ионов, од-
ви благодаря изменению своих кислотных свойств при окислении и восстановлении.
Гемоглобиновую буферную систему можно представить как
ННЬО2 ННЬ '
Это наиболее мощная буферная система цельной крови. При деоксигенации 1 мэкв оксигемоглобина pH повышается на 0,048, что способствует присоединению гидроген-иона, поступающего из кислот тканевой жидкости. Гемоглобиновая буферная система играет важную роль в уменьшении разницы между pH артериальной и венозной крови.
СИСТЕМЫ РЕГУЛЯЦИИ pH
нако эти нарушения уже будут относиться к метаболическим. С помощью механизма центральной регуляции система внешнего дыхания способствует компенсации метаболических расстройств КОС, выводя из организма большее количество углекислого газа при нереспираторном накоплении гидроген-ионов и задерживая элиминацию СО2 при дефиците ионов Н+.
Дыхательная компенсация нарушений КОС происходит через 16—18 ч [1].
9.2.2. Роль почек в поддержании кислотно-основного состояния
Основная роль почек в поддержании КОС сводится к регуляции удаления нелетучих кислот, главным образом сульфатной, а также реабсорбции ионов НСО3. За сутки почки удаляют до 40 — 60 ммоль ионов Н+. В физиологических условиях почки выделяют слабокислую мочу с pH = = 5 — 7, но в зависимости от pH крови почки увеличивают или уменьшают выведение ионов Н+ (pH мочи может колебаться от 4,0 до 8,0). Избыток кислот может выводиться как в свободном состоянии, так и в виде нейтральных солей.
Участие почек в регуляции КОС осуществляется с помощью таких процессов [ 1 ]:
1) секреция гидроген-ионов в мочу клетками канальцевого эпителия;
316 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
2) образование в клетках эпителия канальцев ионов НСО3;
3) образование и диффузия в мочу аммиака, образующего ион NH4 при соединении с ионом Н+;
4) реабсорбция профильтровавшихся в мочу оснований, прежде всего ионов НСО3;
5) фильтрация в первичную мочу кислых или основных соединений;
6) влияние на обмен электролитов, фосфатов, сульфатов.
9.2.3. Секреция гидроген-ионов с мочой
Секреция ионов Н+ клетками эпителия проксимальных и дистальных канальцев в просвет канальцев происходит с затратой энергии. В этом процессе принимают участие только диссоциированные формы угольной кислоты (Н+ +НСО3) ПРИ на‘ личии фермента карбоангидразы. Ион Н+ активно секретируется в просвет канальца через апикальную мембрану клетки эпителия в обмен на ион Na+, обеспечивая равновесие электрических зарядов.
Большая часть (до 65 —75 %) секреции канальцами ионов Н+ происходит путем аммониогенеза. Клетки проксимальных и дистальных почечных канальцев вследствие процессов дезаминирования аминокислот, главным образом глутамина, а также аланина и глицина, образуют ион аммония NH4. В восходящем участке петли Генле происходит его реабсорбция (при гиперкалиемии этот процесс ингибируется), в результате большое количество аммиака накапливается в интерстиции мозгового вещества почек. В дальнейшем аммиак диффундирует (неионная диффузия) в просвет собирательных трубочек и, соединяясь с секретируемым ионом Н+, образует ион аммония (с потреблением энергии), который и выделяется с мочой. Процесс выделения ионов Н+ в процессе аммониогенеза представлен на рис. 9.1.
Кроме выведения секретируемых гидроген-ионов в процессе аммониогенеза, они удаляются из организма в составе титруемых кислот мочи. Основную роль в этом процессе играет фосфатный буфер путем
СО2 + Н2О
\__________/
Петля Медуллярный
Генле интерстиции
NH3 Н
Собирательные канальцы
-nhJ-M?
АДФ
Моча
Рис. 9.1. Процесс аммониогенезного выведения почками ионов Н+ [3]
перевода двузамещенного фосфата в однозамещенный:
Na2HPO4 + Н+ -> NaH2PO4 + Na+.
Освобождающийся ион Na+ реабсорбируется, что способствует сохранению натрия и удалению избытка водорода.
9.2.4. Реабсорбция гидрогенкарбоната в почках
Возвращение в кровь ионов НСО3 происходит двумя способами:
1) продукция НСО3 в проксимальных канальцах при конвертации глутамина в ион NH4, образующийся при этом ион НСО3 поступает в венозную кровь почек;
2) более сложный путь непрямой реабсорбции НСО3 в нижних сегментах нефрона:
из более чем 500_ммоль профильтрованных ионов НСО3 большинство реабсорбируются в петле Генле при секреции ионов Н+ (рис. 9.2).
Вначале происходит фильтрация NaHCO3, затем Na+ реабсорбируется в обмен на секрецию ионов NH4, а НСО3 остается внутри проксимального канальца. NH4 преобразуется в NH3 и Н+ в восходящей петле Генле. Аммиак удаляется через базолатеральную мембрану, секретируется в просвет и образует с находящимся там гидрогенкарбонатом углекислоту и воду. В собирательных трубочках происходит диссоциация Н2СО3 на Н+ и НСО3. Н+ секретируется в мочу в процессе ам-
Основные показатели кислотно-основного состояния 317
Превращение НСО3
Рис. 9.2. Процесс непрямой реабсорбции НСО3 в почках [3]
мониогенеза, а НСО3 вследствие высокой концентрации аммиака в интерстиции мозгового слоя почек реабсорбируется
через базолатеральную мембрану в кровь. Таким образом, основная часть секретируемых гидроген-ионов расходуется на обеспечение реабсорбции гидрогенкарбо-ната.
Кроме ионов Н+ в клетках эпителия дистальных отделов нефрона происходит экскреция ионов К+, причем зависимость между выделением этих ионов конкурентная, например, при гиперкалиемии снижается секреция ионов Н+, и наоборот, при гипокалиемии их выведение увеличивается. Секреция обоих ионов происходит в обмен на экскрецию ионов Na+ и изменяется пропорционально изменению реабсорбции Na+.
Компенсация почками нарушений КОС происходит через двое-трое суток [1].
9.3. РОЛЬ ОРГАНОВ ПИЩЕВАРЕНИЯ В РЕГУЛЯЦИИ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Функция пищеварительного канала играет важную роль в регуляции КОС, влияя как непосредственно на реакцию внутренних сред организма, так и опосредованно, прежде всего через поддержание баланса электролитов.
КОС зависит от характера питания человека. В состав белков животного происхождения входят сера и фосфор, что способствует образованию сильных неорганических кислот и выделению кислой мочи. Растительная пища, наоборот, способствует повышению pH, так как в ней содержится большое количество солей слабых кислот (яблочной, лимонной, щавелевой и др.) и сильных оснований.
Несмотря на то что секрет желудка обладает выраженной кислой реакцией и содержит много ионов Н+ и СГ, а секреты поджелудочной железы и кишок — большое количество ионов НСО3, на КОС при
нормальной функции этих органов это не влияет, поскольку упомянутые ионы впоследствии всасываются назад в кровь. Однако при резком повышении кислотности желудочного сока и/или при выведении его через зонд, гастростому или при частой рвоте (стеноз привратника, тяжелый гастрит) может произойти сдвиг КОС в основную сторону. Наоборот, потери основных валентностей при диарее, свищах кишок и поджелудочной железы могут приводить к избытку кислых валентностей.
Влияние печени на регуляцию КОС. Утилизация молочной кислоты, метаболи-зация кетоновых тел (ацетатная и р-ок-симасляная кислоты), метаболические превращения органических кислот, экскреция желчи, имеющей основную среду, играют важную роль в поддержании КОС. Особенно это проявляется при недостаточности функции печени.
9.4. ОСНОВНЫЕ ПОКАЗАТЕЛИ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Процессы, характеризующие кислотноосновный баланс в организме, можно оценить с помощью таких показателей:
pH раствора — отрицательный десятичный логарифм концентрации гидроген-
ионов. Чаще всего измеряется в крови. Соответствует pH межклеточной жидкости. В норме колеблется от 7,35 до 7,45. Его снижение свидетельствует о сдвиге реакции раствора в кислую сторону, и наоборот.
318 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
рСО2 — парциальное давление, или напряжение углекислого газа в крови. В норме в физиологических условиях при нормальной вентиляции равняется 40 мм рт. ст. (35 — 45 мм рт. ст.). Характеризует респираторный компонент КОС. Гиперкапния понижает pH, гипокапния способствует его повышению;
актуальный гидрогенкарбонат крови (АВ) — истинная концентрация иона НСО3 в КРОВИ» в физиологических условиях нормальный уровень составляет 22 — 25 ммоль/л;
стандартный гидрогенкарбонат крови (SB) — концентрация иона НСО3 в крови в стандартных условиях (при полном насыщении гемоглобина кислородом, рСО2 = 40 мм рт. ст. и температуре 38 °C). Этот показатель важен для диагностики, поскольку на него не влияет респиратор
ный компонент, в норме мало отличается от актуального гидрогенкарбоната;
сумма буферных оснований (ВВ) -общая сумма концентраций всех оснований в крови при полном насыщении гемоглобина кислородом, температуре 38 °C и рСО2 = 40 мм рт. ст. В физиологических условиях NBB, ммоль/л = 41,7 + + 0,043 НЬ, или от 46 до 52 ммоль/л (НЬ — концентрация гемоглобина) [1];
избыток или дефицит оснований (BE) -величина отклонения фактической величины суммы оснований у больного и нормальной величины ВВ:
BE = NBB - ВВ.
Таким образом, если NBB > ВВ, то имеется избыток оснований, если NBB < ВВ -их дефицит. Характеризует метаболический компонент КОС. В норме BE = = ± 2,5 ммоль/л.
9.5. ТЕРМИНОЛОГИЯ, УПОТРЕБЛЯЕМАЯ ПРИ ИЗУЧЕНИИ КИСЛОТНО-ОСНОВНОГО СОСТОЯНИЯ
Снижение pH в крови ниже 7,35 рассматривают как ацидемию, подъем pH выше 7,45 — как алкалемию. Понятия «ацидоз» и «алкалоз» определяются как патофизиологические процессы развития ацидемии или алкалемии. Например, при кетонемии с гипервентиляцией до нормализации pH ни ацидемия, ни алкалемия не определяются, однако, это будет метаболический ацидоз с компенсаторным респираторным алкалозом. Для ацидоза и ал
калоза часто употребляется дополнительное определение, подчеркивающее генез этих процессов, — респираторный или метаболический. Приоритет в определении отдается первичному процессу, хотя иногда это представляет определенные трудности. Для определения первичности одних показателей КОС может быть недостаточно, поэтому необходимо проанализировать патологический процесс и разобраться в генезе расстройств КОС.
9.6. КЛИНИЧЕСКИЕ СОСТОЯНИЯ, СОПРОВОЖДАЮЩИЕСЯ РАССТРОЙСТВАМИ КИСЛОТНО-ОСНОВНОГО состояния
Основные виды расстройств КОС представлены на рис. 9.3.
9.6.1. Метаболический ацидоз
Декомпенсированный метаболический ацидоз (МА) проявляется снижением pH ниже 7,35 и показателей содержания оснований в крови (SB и BE). Определение этиологического фактора МА намного труднее, чем респираторного (РА). Боль
шое значение при этом играет диагностика основного заболевания, однако показатели нарушения КОС в сочетании с биохимическими показателями также могут быть использованы в диагностике основного заболевания. Алгоритм диагностики причин МА представлен на рис. 9.4 [4].
МА можно разделить на две большие группы по этиопатогенезу: с нормальной и повышенной «анионной дырой» (anion gamp, AG).
Клинические состояния, сопровождающиеся расстройствами кислотно-основного состояния 319
[Н+]
।--------------------н----------------------1
Повышена Норма Снижена
I Смешанные расстройства
♦ Снижение рСО2 и НСО3
♦ Повышение /?СО2 и НСО3
♦ Увеличение «анионной дыры»
Ацидемия
Низкая
[НСО3] I Метаболический ацидоз
Высокое
/?СО2 I Респираторный ацидоз
Алкалемия
I----------1---------1
Высокая Низкое
[НСО3] I Метаболический алкалоз
/?СО2 I Респираторный алкалоз
Рис. 9.3. Основные виды расстройств кислотно-основного состояния [3]
Метаболический ацидоз
Нормальная
Концентрация калия
«Анионная дыра»-----1
Повышенная
Реакция на кетоновые тела
Снижена I
Диарея Ингибиторы карбоангидразы Почечный канальцевый ацидоз (тип 1—3)
Повышена
I
♦ Применение NH4CI
♦ Хронический пиелонефрит
♦ Почечный канальцевый ацидоз (тип 4) ।----
Диабетический кетоацидоз
Позитивная Негативная
I I Норма
Осмолярность плазмы Осмолярность------►
плазмы
Повышена Норма Повышена
---- ।----------- Осмолярная +
Т дыра **
Алко- ♦ Отравление |
гольный салицилатами I
кето- ♦ Голодание Креатинин Т
ацидоз ♦ Смешанный I __
респиратор- ♦
ный алкалоз Уремия и метаболичес-
Лактатацидоз Интоксикация: железом фенформином изониазидом
Отравление этиленгликолем, метанолом
кий ацидоз
Рис. 9.4. Алгоритм диагностики метоболического ацидоза
Одним из универсальных принципов существования живого организма является поддержание электронейтральности, это означает, что сумма анионов должна равняться сумме катионов. Основными внеклеточными катионами являются натрий и калий, а анионами — хлор и гидроген-карбонат. Таким образом, в идеале Na* +
+ К* = СГ + НСО3, однако при измерении концентраций этих ионов у здорового человека оказалось, что Na* + К+ > СГ + НСО3. По-видимому, есть группа анионов, которые не определяются обычными исследованиями, эти анионы и составляют AG:
AG = (Na+ + К+) - (СГ + НСО3 ).
320 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
В состав AG входят отрицательно заряженные молекулы белков, прежде всего альбумина (15 мэкв/л), органические кислоты (5 мэкв/л), фосфаты (2 мэкв/л) и сульфаты (1 мэкв/л) — всего 23 мэкв/л. Однако следует учесть и катионы, которые, как правило, остаются вне поля зрения: кальций (5 мэкв/л), магний (1,5 мэкв/л). Поэтому в норме AG = 10—16 мэкв/л и не превышает 20 мэкв/л [5].
Увеличение AG указывает на наличие метаболического ацидоза. Наиболее частой причиной увеличения AG является повышение уровня лактата в крови, а также кетонемия, отравление салицилатами, уремия. Если уровень лактата, креатинина и кетоновых тел нормальный, то для выявления причины повышения AG следует определить «осмолярную дыру», которая вычисляется как разница между расчетной и определяемой осмолярностью плазмы и в норме составляет 10 мосм/л (расчетная осмолярность плазмы равна (2Na+ + азот мочевины)/2,8 + глюкоза/18) [6]. Чаще всего причиной повышения «осмолярной дыры» является интоксикация спиртами (этанол, метанол, этиленгликоль).
Снижение AG наблюдается при гипонатриемии при неизменной концентрации хлоридов и гидрогенкарбоната, при нормальной концентрации натрия, при увели
ченной сумме концентраций хлоридов и гидрогенкарбоната, что часто характерно для гипоальбуминемии; в случае увеличения концентрации других катионов при нормальной осмолярности плазмы, например при множественной миеломе, когда увеличивается концентрация миеломных белков; при интоксикации солями брома. При повышении вязкости крови AG может снижаться вследствие лабораторного артефакта [7].
Наиболее частые причины развития метаболического ацидоза с нормальной и повышенной «анионной дырой» представлены в табл. 9.2 [7].
Метаболический ацидоз с нормальной «анионной дырой». Этот вид ацидоза часто связан с большими потерями гидрогенкарбоната. При этом увеличивается концентрация хлоридов и AG может не изменяться. Наблюдается при нарушении реабсорбции фильтрованного гидрогенкарбоната в проксимальных канальцах. Порог фильтрации снижается и при потерях через пищеварительный канал.
Недостаточность почек обычно сочетается со снижением экскреции гидроген-ионов вследствие уменьшения количества АТФ в клетках почек и снижения окисления глутамина [3]. Однако могут наблюдаться и прямые потери ионов НСО3. Этот процесс могут вызывать нарушение
Таблица 9.2. Наиболее частые причины развития метаболического ацидоза
Нормальная «анионная дыра» Повышенная «анионная дыра»
1. Потеря ионов гидрогенкарбоната: проксимальный канальцевый ацидоз дилюционный ацидоз применение ингибиторов карбоангидразы первичный гиперпаратиреоидизм диарея свищи кишок и поджелудочной железы уретеросигмостома 2. Нарушения регенерации гидрогенкарбоната: дистальный канальцевый ацидоз гипоальдостеронизм применение спиронолактона, триамтерена 3. Избыточное поступление гидроген-ионов: аммоний гидрогенхлорида, лизин гидрогенхлори-да, аргинин гидрогенхлорида парентеральная гипералиментация 1. Снижение экскреции неорганических кислот: недостаточность почек 2. Аккумуляция неорганических кислот: лактатацидоз диабетический кетоацидоз голодание 3. Интоксикации: салицилатами метанолом этиленгликолем
Клинические состояния, сопровождающиеся расстройствами кислотно-основного состояния 321
функции проксимальных канальцев по реабсорбции НСО3, снижение выработки ренин/альдостерона. При нарушении функции дистальных канальцев возникает нарушение регенерации НСО3 вследствие реабсорбции гидроген-ионов с возвращением их в кровь.
При гипоальдостеронизме, часто сочетающемся с интерстициальным поражением почек, наблюдается снижение реабсорбции ионов натрия и секреции дистальными канальцами гидроген-ионов, развивается гиперхлоремический, гиперкалиемический ацидоз. Подобные нарушения могут возникнуть при длительном применении больших доз таких диуретиков, как триамте-рен, спиронолактон и амилорид. Коррекция проксимального канальцевого ацидоза применением растворов гидрогенкар-боната приводит к еще большему увеличению его выделения с мочой.
Дилюционный ацидоз встречается при переливании больших количеств растворов с pH < 7,4. Применение ингибиторов карбоангидразы приводит к снижению реабсорбции НСО3 в почечных канальцах, а первичный гиперпаратиреоидизм снижает гидрогенкарбонатный барьер в проксимальных канальцах и способствует увеличению концентрации хлоридов.
Поскольку в кишках содержится повышенное количество ионов НСО3, понос или кишечные свищи вызывают значительную потерю этих ионов с развитием ацидоза (некоторые авторы называют его выделительным). Понос, не превышающий 1 л в сутки, как правило, не сопровождается тяжелыми нарушениями КОС вследствие почечной компенсации ионов НСО3 (почки могут генерировать их в количестве 200 ммоль/л в сутки).
Пациенты с уретеросигмостомой теряют НСО3 с мочой, которая вымывает его из содержимого кишки.
Причиной гиперхлоремического ацидоза может быть также прием большого количества солей, образованных сильной кислотой и слабым основанием, — аммоний гидрогенхлорида, лизин гидрогенхлорида, аргинин гидрогенхлорида. В процессе их метаболизма образуется соляная кислота (гидрогенхлорид).
П 4-220
Часто гиперхлоремический ацидоз возникает при парентеральном введении большого количества растворов синтетических аминокислот с большим содержанием аргинина, лизина, гистидина.
Метаболический ацидоз с повышенной «анионной дырой» может быть связан со снижением экскреции почками неорганических кислот (серной, фосфорной). Часто наблюдается при хронической недостаточности почек, когда нарушается еще и экскреция аммоний хлорида. Как правило, при этом концентрация НСО3 в крови снижается до 16 ммоль/л и ниже.
Другой и наиболее частой причиной этого вида МА является аккумуляция в организме органических кислот, прежде всего молочной (лактатацидоз), и кетоновых тел. Молочная кислота продуцируется мышцами, эритроцитами и другими тканями в процессе анаэробного гликолиза (см. «Тканевое дыхание»). В норме превращение лактата происходит при участии лактатдегидрогеназы:
Лактат + НАД <=> Пируват + НАД • Н.
При лактатацидозе типа А, который связан со снижением доставки кислорода тканям (шок, респираторная недостаточность, анемия), увеличивается соотношение лактат/пируват и НАД/НАД • Н. При аноксии продукция Н+ составляет 72 ммоль/мин и зависит от степени гипоксии тканей [3]. Чаще всего лактатацидоз наблюдается при шоке, септицемии, выраженной гипоксемии. Лактатацидоз типа В не связан с гипоксией тканей, в его основе лежит нарушение функции печени по конвертации лактата в глюкозу и тканевого дыхания. В мышцах лактатацидоз может развиваться и при нормальном соотношении лактат/пируват, например, при тяжелой мышечной нагрузке. Наблюдается также идиопатический лактатацидоз, например, у дебильных больных.
Еще одной причиной метаболического ацидоза с повышенной «анионной дырой» в результате накопления органических кислот является кетоацидоз при сахарном диабете, голодании, приеме алкоголя, вызываемый активацией липолиза и кетогенеза. После лечения инфузией жидкости
322 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
и применения инсулина этот вид МА часто переходит в ацидоз с нормальной AG, главным образом за счет повышения концентрации хлоридов [8].
Накопление органических кислот с возникновением ацидоза наблюдается при отравлении салицилатами, этиленгликолем, метанолом, паральдегидом.
Клинически МА может проявляться нарушением функции ЦНС вплоть до коматозного состояния. При снижении pH ниже 7,1 развивается артериальная гипотензия вследствие кардиодепрессивного эффекта, повышается давление в легочной артерии, снижается чувствительность к катехоламинам [9].
Механизмы компенсации МА. Компенсация МА происходит прежде всего за счет увеличения вентиляции легких и снижения рСО2. В норме в ответ на снижение концентрации НСО3 на 1 ммоль рСО2 должно снизиться на 1,2 мм рт. ст. Если эта величина меньше, можно предположить наличие дополнительного респираторного ацидоза. Обычно несмотря на тяжесть МА рСО2 не снижается ниже 10 мм рт. ст.
9.6.2. Лечение метаболического ацидоза
Прежде всего следует определить первичный патофизиологический процесс и воздействовать на него. Например, при лактатацидозе необходимо улучшить доставку кислорода тканям, при кетоацидозе — нормализовать жировой обмен. Часто на первый план выходит нормализация водно-электролитного обмена. Однако при критическом снижении pH ниже 7,2 —7,1 необходима срочная коррекция КОС. Обычно с этой целью используют ощелачивающие растворы, прежде всего раствор натрий гидрогенкарбоната. В определении показаний к применению соды при ацидемии нет единого мнения. Основной целью ее назначения в большинстве случаев является нормализация гемодинамики при выраженной артериальной гипотензии за счет увеличения чувствительности адренорецепторов к экзогенным и эндогенным катехоламинам. Однако при этом отмечается отсутствие эффекта и
даже усугубление ацидоза внутри клетки [10]. У больных в критическом состоянии с тяжелым лактатацидозом не было обнаружено разницы в гемодинамическом эффекте при применении эквимолярных объемов соды и изотонического раствора натрий хлорида, использование соды приводило только к снижению концентрации ионизированного кальция [11].
Применение раствора натрий гидрогенкарбоната сопряжено с рядом других неблагоприятных эффектов: гипернатриемией, гиперосмолярностью, усугублением внутриклеточного ацидоза, особенно в нейронах головного мозга (за счет быстрого поступления СО2 в спинномозговую жидкость и внутрь клетки). Передозировка соды создает угрозу перехода в метаболический алкалоз на фоне компенсаторного респираторного алкалоза со смещением кривой диссоциации оксигемоглобина влево и еще большим дефицитом кислорода. Возможно развитие опасной для жизни гипокалиемии [12]. Несмотря на то что некоторые клиницисты продолжают применять раствор натрий гидрогенкарбоната у больных в критическом состоянии, нет сведений, достоверно доказывающих улучшение результатов лечения [13]. Речь идет в основном о больных с лактатацидозом и кетоацидозом.
М. L. Halperin и М. В. Goldstein [3] предложили такие показания к применению соды:
при лактатацидозе: снижение концентрации НСО3 до 8 ммоль/л, сопровождающееся снижением сердечного выброса, артериальной гипотензией или недостаточной респираторной компенсацией;
при кетоацидозе: в случае наличия гиперкалиемии, несмотря на инсулинотера-пию, при выраженном дефиците оснований ([НСО3 ] < 5 ммоль/л), выраженной гипергликемии, не коррегируемой инсулино-терапией.
При таких состояниях, как гемоглобин-урическая недостаточность почек или отравление салицилатами и некоторыми другими токсикантами, применение соды может быть одним из методов выбора. По-видимому, положительный результат от применения раствора натрий гидрогенкар-
Клинические состояния, сопровождающиеся расстройствами кислотно-основного состояния 323
боната следует ожидать при остром ацидозе, связанном с большими потерями гид-рогенкарбоната (понос, кишечный свищ). Мы согласны с мнением, что определение показаний к применению соды является индивидуальным [3], но обязательно следует учитывать положительные и отрицательные последствия такого решения.
Для первичного определения дозы натрий гидрогенкарбоната предложено несколько формул, в частности [13]:
№НСОз (ммоль/л) =
= Масса тела • 0,3 (24 ммоль/л -
- (Актуальный НСО3/2)).
Однако сами авторы отмечают, что выбор этой формулы, как и других, носит преимущественно эмпирический характер. Она пригодна только для начального применения с последующим повторным определением показателей КОС.
В качестве ощелачивающих растворов могут применяться натрий лактат и натрий ацетат. Оба они метаболизируются в печени до гидрогенкарбоната, поэтому по механизму действия мало отличаются от раствора натрий гидрогенкарбоната. При коррекции этими растворами меньше риск развития гипернатриемии, гиперосмолярности плазмы, внутриклеточного ацидоза, повышения уровня углекислого газа, действие их более мягкое и продолжительное, чем раствора натрий гидрогенкарбоната. Их применение противопоказано при недостаточности функции печени, лактатацидозе.
Многие недостатки, присущие раствору натрий гидрогенкарбоната, отсутствуют у трисамина, являющегося органическим акцептором гидроген-ионов как внутри, так и вне клетки. Это щелочной, гиперосмолярный раствор, обладающий осмодиуре-тическим эффектом, вследствие чего он вызывает снижение концентрации ионов калия в плазме крови. При быстром введении развивается депрессия дыхания. Скорость внутривенной инфузии не должна превышать 5 мл/кг в час, при попадании паравазально вызывает некроз клетчатки. Может быть причиной дегидратации, особенно внутриклеточной. Противо
показан при нарушении фильтрационной функции почек.
Доза трисамина определяется по формуле
Объем 3,66 % раствора трисамина (мл) = = BE • Р,
где Р — масса тела, кг.
9.6.3. Респираторный ацидоз
Респираторный ацидоз (РА) возникает в результате альвеолярной гиповентиляции. При декомпенсированном РА наблюдаются снижение pH, повышение рСО2 и нормальный уровень оснований (SB и BE).
Механизм компенсации РА в основном почечный: увеличивается реабсорбция ионов НСО3 и их концентрация в плазме крови. Механизм компенсации реализуется через 24 — 28 ч.
Лечение сводится главным образом к увеличению альвеолярной вентиляции. Ощелачивание крови, повышая pH, не решает проблему, а только может ее усугубить (например, при переливании натрий гидрогенкарбоната увеличивается рСО2).
9.6.4. Зависимость между кислотно-основными балансами крови и спинномозговой жидкости
Как уже указывалось в разделе «Центральная регуляция дыхания», объем вентиляции во многом зависит от рСО2 и pH крови. Гематоэнцефалический барьер (ГЭБ) проницаем для неионизированной молекулы СО2, поэтому изменения pH крови, вызванные гипер- или гиповентиляцией, быстро отразятся на pH спинномозговой жидкости (СМЖ). Однако при метаболических расстройствах изменения pH крови происходят гораздо быстрее, чем изменения pH СМЖ, поскольку ионы Н+ и НСО3 плохо проникают через ГЭБ. Поэтому в острой стадии метаболического ацидоза до начала компенсаторной реакции — увеличения минутного объема крови — проходит несколько часов.
И*
324 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
9.6.5. Метаболический алкалоз
Метаболический алкалоз (МАл) ха-рактеризуется повышением концентрации НСО3 и pH внеклеточной жидкости. Как правило, при МАл увеличивается «анионная дыра» за счет повышения концентрации альбуминов и активации синтеза органических анионов [12]. Повышение концентрации НСО3 может быть следствием двух процессов: увеличения поступления и ретенции НСО3 и снижения объема внеклеточной жидкости.
Увеличение образования НСО3 возможно при снижении концентрации ионов СГ и/или повышенной ретенции катионов (в основном Na+). Потери ионов СГ вместе с ионами Н+ и/или NH4 без потери натрия — одна из наиболее частых причин МАл. Часто при МАл развивается гипокалиемия вследствие увеличения экскреции ионов NH4. Следует отметить, что нормально функционирующие почки выводят избыток гидрогенкарбоната при нормальной концентрации ионов калия, хлора и магния, дефицит этих ионов ингибирует функцию выведения почками НСО3 [12].
При увеличении концентрации натрия во внеклеточной жидкости согласно принципу электронейтральности повышается концентрация НСО3.
При сокращении объема внеклеточной жидкости может повышаться концентрация НСО3 без дополнительного поступления этих ионов, поскольку потери жидкости, как правило, происходят на фоне сниженной потери НСО3 (например, при применении диуретиков, исключая ингибиторы карбоангидразы, обильном потоотделении). Кроме того, при гиповолемии увеличивается секреция проксимальными канальцами гидроген-ионов за счет активации продукции ангиотензина II, а также снижается секреция НСО3 вследствие уменьшения клубочковой фильтрации при увеличении реабсорбции [3].
В клинической практике МАл разделяют на хлорчувствительный и хлоррезис-тентный.
Развитию хлорчувствительного МАл способствуют:
гиповолемия;
потери желудочного содержимого (рвота, гастральный зонд, гастростома);
длительное применение диуретиков (за исключением ингибиторов карбоангидразы);
состояние при компенсации гиперкапнии и компенсаторный метаболический алкалоз.
Хлоррезистептный МАл вызывают: первичный и вторичный альдостеронизм (стеноз почечной артерии, злокачественная артериальная гипертензия, ренинпродуци-рующая опухоль);
увеличение активности минералокортикоидов (за счет увеличения продукции адренокортикотропина или введения их извне);
увеличение активности гликокортикоидов (синдром Кушинга);
состояние гиперкоррекции метаболического ацидоза, в особенности кетоацидоза и лактатацидоза.
Интенсивные потери ионов Н+ и СГ приводят к развитию внутриклеточного метаболического алкалоза с гипокалиемией. При обильной рвоте происходит процесс нарушения кислотно-основного баланса [3]:
НС1 -> Н+ + СГ -> Потери
NaHCO3 -> Na+ + НСО3 -> Потери Н+ + НСО3 СО2 + Н2О
Результат: потери Na+ + СГ + СО2 + Н2О
При потере НС1 ионы НСО3 заменяют ионы СГ во внеклеточной жидкости, возникшая в результате алкалемия ингибирует реабсорбцию в проксимальных канальцах НСО3 и приводит к потерям NaHCO3 с мочой. Комбинированные потери НС1 с желудочным содержимым и NaHCO3 с мочой эквивалентны потерям NaCl, СО2 и Н2О. Возникает гиповолемия с гипонатриемией и гипохлоремией.
При гиперальдостеронизме увеличивается резорбция натрия в обмен на увеличение экскреции водорода и калия. Слабый минералокортикоидный эффект глю
Клинические состояния, сопровождающиеся расстройствами кислотно-основного состояния 325
кокортикоидов может привести к развитию МАл при синдроме Кушинга [12].
Компенсация организмом МАл происходит за счет снижения вентиляции и повышения рСО2. В условиях нормальной компенсации при повышении НСО3 на 1 ммоль рСО2 должно повыситься на 0,6 мм рт. ст. Более низкий уровень свидетельствует о дополнительном респираторном алкалозе. На практике рСО2 редко повышается выше 60 мм рт. ст., этому препятствует возникающая гипоксемия.
9.6.6. Лечение метаболического алкалоза
Основные принципы так же, как и при лечении метаболического ацидоза, заключаются в воздействии на основной патологический процесс. Для лечения МАл применяются восполнение объема циркулирующей плазмы и коррекция дефицита ионов Na+, К+ и СГ. После восполнения жидкостью концентрация НСО3 снижается уже за счет разведения, гидрогенкарбона-турия возникает при перевосполнении ОЦК [3]. Однако инфузия изотонического раствора натрий хлорида, хотя и уменьшает концентрацию НСО3, но без коррекции дефицита калия не корригирует часто сопутствующий внеклеточному алкалозу внутриклеточный ацидоз. Для коррекции дефицита калия используют раствор калий хлорида, однако внутриклеточный дефицит этого иона быстро скор-ригировать трудно, для этого необходимо увеличение внутриклеточной концентрации фосфатов (ДНК, РНК, фосфолипидов).
У пациентов, у которых МАл развился в результате применения диуретиков, при недостаточности миокарда не следует применять для коррекции растворы, содержащие натрий.
В случае толерантности алкалоза к применению растворов натрия, калия и хлора следует исключать избыточную активность минералокортикоидов. У пациентов с гиперальдостеронизмом иногда требуется коррекция дефицита магния, однако одной только заместительной терапией этого достичь бывает трудно. При гиперальдостеронизме полезным может оказаться при
менение ингибиторов альдостерона — спиронолактона, а также амилорида.
В случаях выраженной алкализации крови с повышением pH до 7,7 и отсутствием эффекта на восполнение ОЦК и дефицита калия и хлора требуется дополнительное введение гидроген-ионов в виде соединений НС1 и NH4CL НС1 применяется в виде 0,1—0,2 М раствора в центральную вену со скоростью, не превышающей 0,2 ммоль/кг в час. При расчете дозы следует исходить из значения избытка гидрогенкарбоната, умноженного на половину массы тела [12]. Аммоний хлорид не следует применять у пациентов с недостаточностью почек и печени.
У пациентов, у которых МАл развился в результате нарушений функции почек, его коррекция представляет особую трудность.
У больных с выделением кислого желудочного содержимого через зонд или гастростому для профилактики и лечения МАл можно использовать препараты, повышающие pH желудка (ингибиторы Н2-рецепто-ров), и восполнить дефицит калия и хлора.
9.6.7. Респираторный алкалоз
Причиной респираторного алкалоза (РАл) является альвеолярная гипервентиляция, которая может носить центральный характер (первичная альвеолярная гипервентиляция) и вызываться психогенным фактором (истерия), быть следствием боли, гипоксемии (парциальная форма ОНД), ИВЛ с избыточным МОД, активации легочных рецепторов (пневмония, микроэмболизация легочных сосудов и др.), отравления салицилатами, развиваться после коррекции метаболического ацидоза. Клинически может проявляться одышкой, парестезиями, тетанией, тремором.
Компенсация происходит за счет увеличения экскреции ионов гидрогенкарбоната почками.
9.6.8. Смешанные расстройства КОС
Смешанные расстройства КОС (ацидоз и алкалоз различной этиологии) наблюдаются у больных с нарушением вентиля-
326 КИСЛОТНО-ОСНОВНОЕ СОСТОЯНИЕ
Таблица 9.3. Ожидаемый уровень компенсации при расстройствах КОС [14]
Первичное нарушение КОС Ожидаемые компенсаторные изменения Первичное нарушение КОС Ожидаемые компенсаторные изменения
Метаболический ацидоз Метаболический алкалоз Острый респираторный ацидоз рСО2 = 1,5НСОз + (8 ± 2) рСО2 =0,7НСОз +(21 ±2) ДрН = 0,008(рС02 -40) Хронический респираторный ацидоз Острый респираторный алкалоз Хронический респираторный алкалоз ДрН = 0,003(рС02 — 40) ДрН = 0,008(40-рСО2) ДрН = 0,017(40 -рСО2)
ции, функции почек или возникают вследствие другой патологии, вызывающей метаболические расстройства КОС, у больных на ИВЛ, а также при нарушении компенсаторных механизмов (неполной коррекции первичного нарушения КОС). Уровни ожидаемой компенсации приведены в табл. 9.3.
Например, при pH = 7,4 и рСО2 = = 50 мм рт. ст. компенсация pH выше ожидаемой, можно констатировать метаболический алкалоз с компенсаторным респираторным ацидозом.
Список использованной литературы
1. Основы физиологии человека / Под ред. Б. И. Ткаченко. — СПб., 1994. — Т. 1. — С. 529.
2. Тевс Г. Транспорт газов кровью и кислотнощелочное равновесие // Физиология человека / Под ред. Р. Шмидта, Г. Тевса. — М.: Мир, 1996. - Т. 2. - С. 605-641.
3. Halperin М. L., Goldstein М. В. Fluid. Electrolyte, and Acid-Base Physiology. — 2nd ed. — W. B. Sunders Company, 1994. — 412 p.
4. Gold frank L. Methanol and ethylen glycol // Goldfrank’s toxicologic emergencies. — Nor-wolk: Appleton-Century-Crofts, 1986. — 332 p.
5. Oster J. R.t Perez G. O., Materson B. J. Use of the anion gap in clinical medicine // South Med. J. - 1988. - 81. - P. 229-237.
6. Delaney K., Gold franc L. R. Metabolic acidosis and alkalosis // M. L. Callaham. Current Therapy in Emergency Medicine. — Toronto; Philadelphia: В. C. Becker Inc., 1987. — P. 744-750.
7. Andreoli T. E. Disorders of fluid volume, electrolyte and acid-base balance // Textbook of Medicine / Ed. J. B. Wyngarden, L. H. Smith. — W. B. Saunders Company. — 1988. - P. 528-558.
8. Paulson W. D. Anion gap-bicarbonate relationship in diabetic ketoacidosis // Am. J. Med. - 1986. - 81. - P. 995-1000.
9. Stokke D. B., Andersen P. K., Brinclov M. M. et al. Acid-base interactions with noradrenaline-induced contractile response of the rabbit isolated aorta // Anesthesiology. — 1984. -60. - P. 400-404.
10. Beech J. S.t Nolan К. M.t Iles R. A. et al. The effects of sodium bicarbonate and a mixture of sodium bicarbonate («carbicarb») on skeletal muscle pH and hemodynamic status in rats with hypovolemic shock // Metabolism. -1994. - 43. - P. 518-522.
11. Cooper D. J.t Walley K. R., Wiggs B. R.t Russell J. A. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study // Ann. Intern. Med. — 1990. -112. - P. 492-498.
12. Marini J. J., Wheeler A. P. Critical Care Medicine. — 2nd ed. — Philadelphia: Wil-liams&Wilkins, 1997. — 640 p.
13. Progh D. S.,Mathru M. Acid-Base Disturbances and Lactic Acidosis // M. J. Murray, D. B. Coursin, R. G. Pearl, D. S. Prough. Critical Care Medicine. — Philadelphia; New York: Lippincott-Raven, 1997. — P. 209-217.
14. Marino P. L. The ICU Book. — 2nd ed. -Philadelphia: Lippincott, Williams& Wilkins, 1997. - 928 p.
= Раздел --
10
Оксидантное повреждение тканей
Как уже указывалось, кислород является необходимым субстратом для жизнедеятельности организма. Однако исследованиями, проведенными в последние 20 лет, было доказано, что кислород может быть причиной повреждения клеток у критических больных. Токсический эффект продуктов окисления часто играет решающую роль в неблагоприятном исходе лечения критических состояний.
Кислород вступает в химические реакции с другими соединениями, имеющимися в организме. Поскольку чаще всего кислород взаимодействует в организме с водородом и углеродом, конечными продуктами их окисления являются вода и углекислый газ, при этом происходит разрыв ковалентных связей с выделением энергии в виде теплоты или света.
Сам по себе молекулярный кислород является естественным оксидантом с адекватным окислительным эффектом, однако некоторые его метаболиты с атомарной структурой, обладающие более выраженной окислительной активностью, могут оказывать повреждающий эффект на клетку вплоть до ее гибели [1]. На рис. 10.1 показано превращение молекулярного кислорода в воду при окислении водорода.
В процессе метаболизма кислорода при наличии переходного металла железа образуются промежуточные продукты неполного окисления — свободные радикалы. Это атомы или молекулы, имеющие один или больше непарных электронов на внешней орбите. Некоторые из свободных ра
дикалов являются высокоактивными химическими субстанциями.
Супероксидный радикал. Образуется в процессе реакции: О2 + е -> О2.
Супероксид играет важную роль в процессе повреждения тканей при некоторых патологических состояниях, например при синдроме ишемия/реперфузия [3]. В организме взрослого человека массой 70 кг может образовываться примерно 1,75 кг супероксида [4].
Супероксид водорода (гидроген пероксид). Присоединение одного электрона к супероксиду и двум атомам гидрогена приводит к образованию гидроген пероксида: О2 + е + 2Н4" -> Н2О2.
Гидроген пероксид — очень мобильный и легко проникающий через клеточные мембраны радикал, повреждающий эндотелий. Сам по себе не является свободным радикалом, однако образуемые им радикалы гидроксида обладают активным повреждающим эффектом. Связь между атомами гидрогена и оксигена в гидроген пероксиде очень неустойчивая и легко разрывается с образованием ионов гидроксида, которые уже содержат один непарный электрон. В процессе реакции, катализатором которой является двухвалентное железо, образуется ион гидроксида и гидроксильный радикал (реакция Fenton):
H2O2+Fe(II) -> ОН" + ’ОН + Fe(III).
Железо принимает участие во многих свободнорадикальных реакциях и является опасным прооксидантом.
328 ОКСИДАНТНОЕ ПОВРЕЖДЕНИЕ ТКАНЕЙ
Молекула кислорода
Супероксидный радикал
Электроны внешней орбиты
о,
•02 2Н+
Н2О2
Гидроген
пероксид
Fe(III)*l ЛЛ ль
Гидроксильный^ + 0Н-^ + Ж радикал и ион +
Iе" ® Д!) 2Н.° ® ®
гидроксида Вода
Рис. 10.1. Процесс окисления молекулярным кислородом водорода [2]
Гидроксильный радикал — один из самых активных свободных радикалов. Его активность сдерживает плохая мобильность, и это уменьшает его токсичность. Однако этот радикал остается одним из наиболее опасных, так как может вступать в реакцию практически с любой молекулой в организме человека [2].
Под действием энзима миелопероксидазы в нейтрофилах образуется хлорииат гидроген пероксид
Н2О2 + 2СГ -> 2НОС1.
Гипохлоритная кислота является наиболее мощным оксидантом, активирующим нейтрофильные гранулоциты, особую важность эта реакция приобретает в дыхательных путях, где она способствует повышению проницаемости альвеолярно-капиллярной мембраны и повышает давление в легочной артерии [5]. При активации нейтрофилов превращение кислорода в супероксид возрастает в 20 раз. Этот процесс называют «респираторным взрывом», поскольку резко увеличенное потребление тканями кислорода не сопровождается воз
растанием образования энергетических субстратов. При этом до 40 % гидроген пероксида образует гипохлорит, а оставшееся количество — гидроксильный радикал [6]. Гипохлорит — это сильный бактерицидный агент, который в течение миллисекунд вызывает гибель микроорганизмов [7].
Супероксидный радикал является мобильным, но не обладает высокой токсичностью, а гидроксильный радикал высокотоксичный, но обладает ограниченной мобильностью, поэтому для последнего более характерны локальные повреждения тканей [8].
Конечным этапом метаболических превращений кислорода является присоединение электрона к гидроксильному радикалу с образованием двух молекул воды:
’ОН + ОН” + ё + 2Н+ -» 2Н2О.
В норме концентрация ионов феррума внутри клетки недостаточна для стимуляции этой реакции, однако при остром ацидозе, возникающем при шоке и других критических состояниях, его количество увеличивается. В нормальных условиях до 98 % кислорода подвергается полному превращению с образованием воды, и менее 2 % его метаболитов поступают в клетку [8]. Большую роль в эффективном блокировании образования радикалов играет цитохромоксидаза.
Повреждающий эффект оксидантов часто является результатом реакций со свободными радикалами. Эти реакции можно разделить на два типа [2]: реакция свободного радикала с нерадикалом и реакция между двумя свободными радикалами.
В случае реакции радикала и неради-кала последний теряет электрон и трансформируется в радикал. Таким образом, эта реакция является радикалгенериру-ющей самоподдерживающейся цепной реакцией, которая может привести к тяжелым последствиям [8].
10.1. ПЕРОКСИДНОЕ ОКИСЛЕНИЕ ЛИПИДОВ
Пероксидное окисление липидов (ПОЛ) — это процесс оксидазного повреждения липидов клеточных мембран. Является примером реакции радикала с
нерадикалом. Липофильная внутренняя поверхность клеточной мембраны богата полиненасыщенными жирными кислотами (ПНЖК) (прежде всего арахидоно-
Пероксидное окисление липидов 329
Поли-ненасы-щеные жирные кислоты
R I ’ОН
СН2 V
СН3 н2О
НС* Липидный радикал
R
СН.
2
R
Радикал I пероксида НСОО
СН, Полинена-сыщенные жирные
Липидный пероксид R
НСООН
СН3
Липидный радикал
кислоты
Рис. 10.2. Последовательность реакций перок-сидного окисления полиненасыщенных жирных кислот в клеточной мембране [2]
вой). В результате повреждающего окси-дативного эффекта мембрана теряет свое свойство избирательной проницаемости для различных веществ, что приводит прежде всего к осмотической деструкции клетки [3].
Процесс пероксидного окисления липидов представлен на рис. 10.2.
Начальным агентом пероксидации является гидроксильный радикал, при взаимодействии которого с атомом карбона (углерода) ПНЖК, образуется углеродный радикал (С*), в свою очередь, трансформирующийся в карбонпероксидный радикал (СОО*), инициирующий целую цепь реакций. Заключительной стадией этого процесса является самоподдерживающа-яся цепная реакция, которая продолжается до тех пор, пока не будет подавлена антиоксидантная система.
ПОЛ является компонентом многих патофизиологических процессов в организме (более 100 болезней), однако нет четких доказательств, что именно ПОЛ — основной фактор развития болезни [9].
Реакция радикала с радикалом. Два радикала взаимодействуют с образованием ковалентной связи. При этом они оба могут превращаться в нерадикальные соединения. Это не гарантирует прекращения токсического эффекта, а в отдель
ных случаях токсичность даже возрастает.
Трансформация NO. NO является естественным вазодилататором, нейротрансмиттером и бактерицидным агентом [10], играющим незначительную роль в ПОЛ. Однако при недостатке L-аргинина NO-синтетаза может способствовать образованию супероксида [ 11 ], в присутствии которого NO проявляет токсические свойства. Ион супероксида в комбинации с азотистым центром свободного радикала, например NO*, образует потенциально мощный повреждающий агент — пероксинитрит [12]:
NO* + О2 -> ONOO".
Потенциально пероксинитрит более сильный оксидант, чем супероксид и гидроген пероксид [13]. Он обладает повреждающим эффектом на ткани при непосредственном воздействии, а также вследствие трансформации в гидроксильный радикал и нитроген диоксид:
ONOO" ->*OH + NO2.
Эта реакция является подтверждением непрямого повреждающего эффекта свободных радикалов.
Пероксинитрит крайне нестабилен в водном растворе при физиологическом уровне pH и быстро превращается в лабильную кислоту. Пероксинитрозовая кислота распадается на фрагменты реактивных оксидантов — ион гидроксида или промежуточных веществ, обладающих активностью гидроксильных радикалов. Повреждение интактной клетки экзо- и эндогенными оксидантами (гидроген пероксид, гидроксильный радикал, пероксинитрит) вызывает распад ДНК [14]. In vitro этот процесс наблюдался во многих видах клеток: гладких мышцах сосудов, эндотелио-цитах, макрофагах, миокардиоцитах [14]. Формирование пероксинитрита играет важную роль в пероксидном окислении липидов при ишемии легких [15], повышении проницаемости эпителия в иммуно-стимулированных клетках кишечного эпителия и поврежденных тканей в результате ишемии/реперфузии печени, миокарда, почек.
330 ОКСИДАНТНОЕ ПОВРЕЖДЕНИЕ ТКАНЕЙ
Определенное значение для оксидантного повреждения тканей имеют другие азотсодержащие молекулы: нитрозопероксикарбоксилат (ONOOCOp и нитрозодиоксид (NO2) [16].
Значительную роль в ПОЛ может играть ксантиноксидаза (КО), катализирующая окисление молекулярным кислородом ксантина (или гипоксантина) до мочевой кислоты и О2.
10.2. АНТИОКСИДАНТНАЯ АКТИВНОСТЬ
Как уже упоминалось выше, свободные радикалы образуются в процессе метаболизма кислорода даже в нормальных условиях жизнедеятельности организма. Естественно предположить, что в организме должна быть система, которая бы защищала организм от действия оксидантов. Эта система получила название антиоксидантной, а ее составляющие — антиоксидантов. Эндогенные и экзогенные антиоксиданты и их свойства представлены в табл. 10.1.
Супероксиддисмутаза (СОД). Первый антиоксидант — СОД — был открыт в 1969 г. Роль СОД не до конца выяснена. Возможно, основной эффект СОД заключается в трансформации супероксида в гидроген пероксид (рис. 10.3), однако при недостатке других антиоксидантных энзимов (каталазы, пероксидазы) повышается уровень гидроген пероксида и СОД в этих условиях проявляет прооксидант-ные свойства [17]. Кофактором СОД являются железо, цинк, медь.
Таблица 10.1. Эндогенные и экзогенные антиоксиданты [2]
Антиоксидант Действие Коментарии
Селен Глутатион N-Ацетилцистеин а-Токоферол (витамин Е) Аскорбиновая кислота Церулоплазмин Трансферрин Альбумин Мочевая кислота Аминостероиды (большие дозы метилпреднизолона) Кофактор глутатионпероксидазы Содержит -SH группу Выпускается как муколитический препарат, но может действовать как аналог глутатиона Потенциально блокирует ПОЛ в клеточных мембранах и циркулирующих липопротеинах Повышает активность токоферола (кооперативный эффект). Может оказывать прооксидантную активность, поддерживая железо в двухвалентном состоянии Относятся к антиоксидантам плазмы крови. В основном действуют, соединяясь с железом и медью. Альбумин потенциально связывает НОС1 Ингибирует ПОЛ в дозе 30 мг/кг внутривенно Является естественным ингредиентом пищи, однако часто необходимо дополнительное введение (55 мкг/сут женщинам и 70 мкг/сут мужчинам; максимальная доза 200 мкг/сут) В основном внутриклеточный антиоксидант. Синтезируется в клетках Не проникает через клеточные мембраны Может быть антидотом при некоторых отравлениях Действие наиболее выражено в клеточных мембранах Антиоксидантная активность недостоверна. В основном действует вне клеток, активность выражена в глазах и легких. Прооксидантная активность повышена при наличии переходных металлов Основное действие заключается в удалении из плазмы свободного железа и меди. Церулоплазмин проявляет наибольшую активность в плазме крови Считают эффективными при травмах спинного мозга в течение первых 8 ч
Неэнзимные антиоксиданты 331
2Н2О
Каталаза. Является железосодержащим гемопорфирином, трансформирующим гидроген пероксид в воду. Она содержится в большинстве клеток организма, в мио-кардиоцитах и нейронах — в небольшом количестве. Ее роль также не до конца
Рис. 10.3. Действие трех антиоксидантных ферментов и свободнорадикального антиоксиданта (скавендера):
GSH — редуцированный глутатион; GSSG — оксидазный глутатион как дипептид
выяснена, поскольку даже при снижении активности каталазы токсичность гидроген пероксида не повышается [18].
Глутатионпероксидаза (ГП). ГП также трансформирует гидроген пероксид в воду путем транспортировки электронов с глутатиона на пероксид. Обратное превращение глутатиона происходит под действием редуктазы.
Селен. Активность ГП зависит от наличия селена. Он является естественным компонентом принимаемой пищи. Суточная потребность в нем составляет 70 мкг для мужчин и 55 мкг — для женщин [19]. Нормальный уровень селена в крови — 0,5 —2,5 мг/л. Может применяться внутривенно в виде натрий селенита (максимальная суточная доза 200 мкг — по 50 мкг 4 раза в сутки) [20].
10.3. НЕЭНЗИМНЫЕ АНТИОКСИДАНТЫ
Глутатион. Содержит три аминокислоты: глицин —цистеин - глутамин. Является одним из важнейших антиоксидантов в организме человека. Содержится в молярной концентрации 0,5 — 10 ммоль/л в большинстве клеток организма [21]. Способен связывать гидроксильные радикалы. Глутатион находят в большинстве органов тела, однако больше всего его в легких, печени, эндотелии и слизистой оболочке кишок. Преимущественно это внутриклеточный антиоксидант, в плазме крови его концентрация намного ниже. Сам он непосредственно через клеточные мембраны не может проникнуть, но аминокислоты, из которых он состоит, легко проходят через них. С другой стороны мембраны происходит его формирование. Экзогенный глутатион оказывает небольшое воздействие на внутриклеточном уровне [22], что значительно ограничивает его клиническое применение.
N-Ацетилцистеин. Популярный муколитический препарат. Так же, как и глутатион, содержит сульфгидрильную группу. В отличие от последнего, лучше проникает через клеточные мембраны. Применяют как антидот при некоторых отравлениях (ацетоминофен), однако в последнее время все чаще предпринимаются попытки использовать его в клинике в качестве антиоксиданта. Имеются данные о протекторном эффекте N-ацетилцистеина при ишемии миокарда и реперфузионном повреждении [23], о его положительном действии при лечении ОРДС и воспалительном шоковом синдроме [24, 25].
а-Токоферол (витамин Е). Жирорастворимый витамин. Его функция прежде всего заключается в защите клеток при пероксидном окислении липидов. Локализован на липофильной внутренней стороне мембраны, где расположены также по-линенасыщенные жирные кислоты. При
332 ОКСИДАНТНОЕ ПОВРЕЖДЕНИЕ ТКАНЕЙ
действии пероксидных радикалов а-токо-ферол сам становится радикалом с очень низкой окислительной активностью. Восстановление радикала обратно в а-токо-ферол происходит с участием аскорбиновой кислоты, являющейся донатором электрона в этой реакции [2].
Дефицит а-токоферола часто наблюдается у критических больных. В норме его уровень в крови составляет 10 мг/л, при снижении до 5 мг/л происходит повреждение клеток [26].
Аскорбиновая кислота (витамин С). Протекторная антиоксидантная роль аскорбиновой кислоты заключается в передаче электронов свободным радикалам с переводом их в неактивное состояние. Это водорастворимый антиоксидант, проявляющий свой эффект преимущественно во внеклеточной среде. Аскорбиновая кислота в большом количестве содержится в легких, где играет протекторную роль при поступлении оксидантов через дыхательные пути. Однако существенным ее недостатком является то, что в присутствии переходных металлов (железа и меди) она способствует образованию оксидантов [27, 28]. Аскорбиновая кислота восстанавливает железо до двухвалентной формы:
Аскорбинат + Fe(III) -> Fe(II) + + дегидроаскорбинат;
Fe(II) + Н2О2 -> ОН" + ’ОН + Fe(III).
Витамин С способствует всасыванию железа в кишках, a Fe(II), в свою очередь, принимает участие в образовании гидроксильных радикалов. Таким образом, при увеличении концентрации Fe(II) аскорбиновая кислота может выполнять роль прооксиданта, что часто наблюдается при системном воспалении, гемотрансфузиях, снижении концентрации белков. Это обстоятельство следует учитывать при применении витамина С в качестве экзогенного антиоксиданта при критических состояниях [2].
Антиоксидантная активность плазмы крови в основном обеспечивается белками церулоплазмином и трансферрином (их содержание составляет 4 % всех белков плазмы) [28]. Трансферрин связывает железо в трехвалентном состоянии, а церулоплазмин переводит Fe(II) в Fe (III). Таким образом, оба белка ограничивают концентрацию свободного железа в плазме крови.
Дискуссионной является важность антиоксидантной активности ксантиндегид-рогеназьц которая во время ишемии тканей превращает ксантин (или гипоксантин) в мочевую кислоту, но без образования свободного радикала [29].
10.4. ОКСИДАНТНЫЙ СТРЕСС
Тяжесть оксидативных повреждений тканей детерминирована балансом между активностью оксидантов и антиоксидантов. Состояние, когда активность оксидантов преобладает, называют оксидантным стрессом [30]. Последний играет важную роль в развитии более 100 патологических состояний в клинике, однако при этом он не является основой патологического процесса [2, 9]. К наиболее распространенным из таких состояний относят: повреждение легких (ОРДС, бронхиальная астма, реперфузионный отек легких, аспирационный синдром, гипероксическое повреждение легких); миокарда (острый инфаркт миокарда, реперфузионные повреждения); нервной системы (инсульты,
черепно-мозговая травма, постреанимационные энцефалопатии, травма спинного мозга); органов пищеварения (лекарственное повреждение слизистой оболочки, ишемия желудка и кишок, пептические язвы); почек (ОПН, вызванная применением аминогликозидов, ишемией, миоглобинурией), а также синдром полиорганной недостаточности. Большинство из названных патологических состояний сопровождается или детерминируется прогрессирующей системной воспалительной реакцией, поэтому последнюю можно считать неотъемлемой составляющей оксидантного повреждения. Освобождение свободных радикалов из активированных нейтрофилов и макрофагов подтверждает это.
Антиоксидантная терапия 333
Одним из состояний, при котором активируется продукция супероксида в клеточных митохондриях, является ишемия тканей [31] и гипергликемия [32]. Освободившиеся свободные радикалы взаимодействуют с внутриклеточными белками, ДНК, липидами. Одним из последствий оксидантного стресса может быть инактивация ключевых энзимов клеточного метаболизма, например, глицерол ал ьдегид-3-фос-фатдегидрогеназы, которая ингибируется Н2О2 [33]. Этот же радикал может инактивировать и повреждать внутриклеточный киназный каскад [34]. Н2О2-, *ОН-или ОМОО~-индуцированный оксидантный стресс может привести к повреждению внутриклеточной ДНК, что, в свою очередь, активирует полиАДФ-рибозополиме-разу, которая вызывает снижение НАД в клетке с развитием цитопатической гипо
ксии [35], может повреждать ключевые структурные белки, например, окислять сульфгидрильные группы в актине, нарушая интеракцию между белковыми субъединицами и образование ими внутриклеточных белков [36]. В этот процесс вовлекается и ядерный фактор NF-кВ, регулирующий экспрессию iNO-синтетаз, циклооксигеназы-2 (ЦОГ-2) и многих цитокинов [37]. Этот фактор в норме локализован в цитоплазме и находится в неактивном состоянии благодаря связи с 1кВа [38]. Оксиданты так же, как цитокины, вирусы, обуславливают распад этого комплекса с активацией NF-кВ, который перемещается к клеточному ядру, где взаимодействует с ДНК генов и способствует продукции многочисленных провоспали-тельных трансмиттеров.
10.5. АНТИОКСИДАНТНАЯ ТЕРАПИЯ
С целью повышения антиоксидантной активности применяются препараты — антиоксиданты. Такими препаратами являются субстанции, которые в относительно низкой концентрации могут ингибировать процессы оксидативного повреждения тканей [39]. Естественные антиоксиданты разделяют на энзиматические и неэнзиматические. К первым относятся: супер-оксиддисмутаза (СОД), каталаза, глутатионпероксидаза (ГПО). Для проявления их активности необходимы кофакторы: селен — для ГПО, цинк — для цитозольной СОД, магний — для митохондриальной СОД.
Неэнзиматические антиоксиданты подразделяют на скавендеры и хелаторы металлов. Первые реагируют со свободными радикалами, предотвращая повреждение тканей. Но при этом их молекулы сами активируются, приобретая оксидантную активность. К таким антиоксидантам относятся как водорастворимые (аскорбиновая кислота, глутатион, диметилтиомочевина), так и жирорастворимые (а-токофе-рол, каротиноиды) субстанции. а-Токофе-рол (витамин Е) является антиоксидантом липидной фазы клеточных мембран.
Глутатион в крайне низких миллимоляр-ных концентрациях внутри клетки действует как субстрат для глутатионпероксидазы, как непосредственный скавендер свободных радикалов и их метаболитов. Аскорбиновая кислота проявляет активность преимущественно в плазме крови. Это скавендер супероксида, пероксидных и гидроксильных радикалов. Как указывалось выше, аскорбиновая кислота является ксантиоксидантом а-токоферола, способствуя его регенерации из а-токофе-роксильного радикала. Она также способствует in vitro регенерации уратов, глутатиона и p-каротина из их активных производных [40].
К потенциальным неэндогенным антиоксидантам принадлежат ингибиторы ксантиноксидазы и лазароиды. Первые способствуют образованию супероксида при синдроме реперфузии. Существуют экспериментальные данные об антиксантин-оксидазной активности аллопуринола [41].
Лазароидами являются 21-аминостероиды, ингибирующие железозависимое пе-роксидное окисление. Их значение для клинического применения остается невыясненным [39].
334 ОКСИДАНТНОЕ ПОВРЕЖДЕНИЕ ТКАНЕЙ
В качестве антиоксидантов пытаются использовать кроме перечисленных выше препаратов также ацетил цистеин, амбро-ксол, диметил сульфоксид, маннитол, раствор Рингер-этилпирувата, ингибиторы ПАРС и др. Однако рутинное применение антиоксидантов в клинике нуждается в доказательствах. Полезным является мониторинг концентрации эндогенных антиоксидантов (а-токоферол, аскорбиновая кислота, селен). При снижении их уровня в плазме крови целесообразно экзогенное введение антиоксидантов. Эффективность такой терапии без соответствующего мониторинга не всегда подтверждается рандомизированными исследованиями. Имеются отдельные сообщения об эффективности антиоксидантной терапии при различных состояниях: предотвращение отрицательного воздействия гипергликемии на функцию сердца и сосудов [42], эндоте-лиумзависимой вазодилатации [43]; профилактика системных гемодинамических
расстройств при гипергликемии [44]. Антиоксидантная терапия может снижать концентрацию в плазме крови растворимых адгезивных молекул у женщин с диабетом после менопаузы с высоким кардиальным риском [45].
Однако многие исследователи отрицают положительный эффект антиоксидантной терапии. Некоторые объясняют это тем, что скорость антиоксидантной реакции токоферола и аскорбинатов медленнее, чем реакции NO с супероксидом, поэтому для нейтрализации супероксида антиоксиданты должны применяться в очень большой дозе [46]. Очень важны реци-клические процессы восстановления антиоксидантов, а для этого необходимы аскор-бинаты и глутатион. Оптимальная антиоксидантная терапия требует комбинации различных антиоксидантов. Она может являться дополнительным лечебным фактором у больных в критическом состоянии [39].
Список использованной литературы
1. The oxygen paradox / Ed. К. J. A. Davies, F. Ursini. — Padova: CLEUP University Press, 1995. — 287 p.
2. Marino P. L. The ICU Book. — 2nd ed. — Philadelphia: Lippincott, Williams & Wilkins, 1997. - 928 p.
3. Halliwell B., Gutteridge J. M. Free radicals in biology and medicine. — 2nd ed. — Clarendon: Oxford University Press, 1989. — P. 2-80.
4. Frei B. Reactive oxygen species and antioxidant vitamins: mechanisms of action // Am. J. Med. - 1994. - 97(SuppL ЗА). - P. 5-23.
5. Wahn H., Hammerschmidt S. Influence of cyclooxygenase and lipoxygenase inhibitors on oxidative stress-induced lung injury // Crit. Care Med. - 2001. - 29(4). - P. 802-807.
6. Anderson В. O., Brown J. M., Harken A. Mechanisms of neutrophil-mediated tissue injury // J. Surg. Res. — 1991. — 51. — P. 170-179.
7. Bernovsky C. Nucleotide chloramines and neutrophil-mediated cytotoxicity // FASEB. — 1991. - 5. - P. 295-300.
8. Liochev S. I., Fridovich I. The role of O2 in the production of HO* in vitro and in vivo // Free Radic. Biol. Med. — 1994. — 16. — P. 29-33.
9. Gutteridge J. M. Free radicals in disease processes: a compilation of cause and consequence // Lancet. — 1993. — 344. — P. 721 -724.
10. Anggard E. Nitric oxide: mediator, murderer, and medicine // Lancet. — 1994. — 343. - P. 1199-1206.
11. Xia У., Roman I. J., Masters B. S., Zweier J. L. Inducible nitric-oxide synthase generates superoxide from the reductase domain // J. Biol. Chem. - 1998. - 273. - P. 22635-22639.
12. Salzman A. L. Nitric oxide in the gut // New Horizons. — 1995. — 3. — P. 352 — 364.
13. Freeman B. Free radical chemistry of nitric oxide. Looking at the dark side // Chest. — 1994. - 105(SuppL). - P. 79-84.
14. Szabo C., Zingarelli B., Salzman A. L. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite // Circ. Res. — 1996. - 78. - P. 1051-1063.
15. Ischiropoulos H., al-Mehdi A. B., Fisher A. B. Reactive species in ischemic rat lung injury: contribution of peroxynitrite // Am. J. Physiol. - 1995. - 269. - P. L158-164.
16. Radi R., Peluffo G., Alvares M. N. et al. Unraveling peroxynitrite formation in biological systems// Free Rad. Biol. Med. — 2001. — 30. - P. 463-488.
17. Michiels C., Raes M., Toussant O., Remacle J. Importance of Se-glutathione, peroxidase, catalase, and CU/ZN-SOD for cell survival against oxidative stress // Free Radic. Biol. Med. - 1994. - 17. - P. 235-248.
Список использованной литературы 335
18. Suttorp N., Toepfler W., Roka L. Antioxidant defense mechanisms of endothelial cells: glutathione redox cycle versus catalase // Am. J. Physiol. - 1986. - 251. - P. 671 — 680.
19. National Research Council. Subcommittee on the tenth edition of the RDAs. — Washington, DC: National Academic Press, 1989. — 220 p.
20. World Health Organization. Selenium. Environmental Health Criteria 58. — Geneva, 1987. - 213 p.
21. Cantin A. M., Begin P. Glutathione and inflammatory disorders of the lung // Lung. — 1991. - 169. - P. 123-138.
22. Robinson M., Ahn M. S., Rounds J. D. et al. Parenteral glutathione monoester enchances tissue antioxidant stores // J. Parent. Ent. Nutr. - 1992. - 16. - P. 413-418.
23. Ferrari R., Ceconi C., CurelloS. et al. Oxygen free radicals and myocardial damage: protective role of thiol containing agents // Am. J. Med. - 1991. - 91(SuppL 3C). - P. 95-112.
24. Henderson A., Hayes P. Acetylcysteine as a cytoprotective antioxidant in patients with sever sepsis // Ann. Pharmacother. — 1994. — 28. - P. 1086-1088.
25. Suter P. M., Domeninghetti G., Schaller M. D. et al. N-Acetylcysteine enhances recovery from acute lung injury in man: a randomized, double-blind placebo controlled clinical study //Chest. - 1994. - 105. - P. 190-194.
26. Meydani M. Vitamin E // Lancet. — 1995. — 345. - P. 170-176.
27. Herbert V., Shaw 5., Jayatilleke E. Vitamin C supplements are harmful to lethal for over 10 % of Americans with high iron stores // FASEB. - 1994. - 8. - P. A678.
28. Halliwell B., Gutteridge J. M. Role of free radicals and catalytic metal ions in human disease//Methods EnzymoL — 1990. — 186. — P. 1-85.
29. Jaseschke H.t Smith C. V.t Mitchell J. R. Reactive oxygen species during ischemiareflow injury in isolated perfused rat liver // J. Clin. Invest. — 1988. — 81. — P. 1240 — 1246.
30. Smith С. V. Correlations and apparent contradictions in assessment of oxidant stress in vivo // Free Radic. Biol. Med. — 1991. — 10. - P. 217-224.
31. Dawson T. L.t Gores G. I., Nieminen A. L. et al. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes // Am. J. Physiol. — 1993. — 264. — P. C961-967.
32. Du X. L., Edelstein D., Rossetti L. et al. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Spl glycosilation // Proc. Nat. Acad. Sci. USA. — 2000. - 97. - P. 12222-12226.
33. Janero D. R., Hreniuk D., Sharif H. M. Hydroperoxide-induced oxidative stress impairs heart muscle cell carbohydrate metabolism // Am. J. Physiol. — 1994. — 266. — P. C179-188.
34. Mahadev K., Zilbering A., Zhu I., Goldstein B. J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase lb in vivo and enhances the early insulin action cascade // J. Biol. Chem. — 2001. - 276. - P. 21938-21942.
35. FinkM. P. Cytopathic hypoxia: mitochondrial dysfunction as a mechanism contributing to organ dysfunction in sepsis // Crit. Care Clin. North Am. - 2001. - 17. - P. 219-237.
36. Dallcdonne I., Milzani A., Colombo R. H7O2 -treated actin // Biophys J. — 1995. — 69. — P. 2710-2719.
37. Livolsi A., Bussutil V., Imbert V. et al. Tyrosine phosphorylation-dependent activation of NF-кВ // Eur. J. Biochem. — 2001. — 268. - P. 1508-1515.
38. Ганонг В. Ф. Ф1зюлопя людини / Пер. з англ, за ред. М. Гжегодського, В. Шевчука, О. Заячювсько!. — Л: Бак, 2002. — 784 с.
39. Mullan В. A., McCloskey В. V. Antioxidants and endothelial function: Therapeutic implications // Yearbook of Intensive Care and Emergency Medicine / Ed. J. L. Vincent. — Springer, 2002. - P. 111-120.
40. Halliwell B. Vitamin C: antioxydant or pro-oxidant in vivo // Free Radic. Res. — 1996. - 25. - P. 439-454.
41. Maxwell S. R. J. Prospects for the use of antioxidant therapies // Drugs. — 1995. — 45. - P. 345-361.
42. Cariello A. Acute hyperglicemia and oxidative stress generation // Diabetic Med. — 1997. - 14. - P. S45-49.
43. Beckman J. A., Gold fine A. B., Gordon M. B., Creager M. A. Ascorbite restores endotheliumdependent vasodilation impaired by acute hyperglycemia in humans // Circulation. — 2001. - 103. - P. 1618-1623.
44. Marfella R., Verrazzo G., Acampora R. et al. Glutathione reverses systemic hemodynamic changes induced by acute hyperglycemia in healthy subjects // Am. J. Physiol. — 1995. — 268. - P. El 167— 1173.
45. Goudev A., Kyurkchiev 5., Gergova V. et al. Reduced concentracions of soluble adhesion moleculs after antioxidant supplementation in postmenopausal women with high cardiovascular risk profiles // Cardiology. — 2000. — 94. - P. 227-232.
46. Jackson T. S.f Xu A., Vita J. A., Keaney J. F. Ascorbaite prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations//Circ. Res. — 1998. — 83. - P. 916-922.
----Раздел ==
11
Система гемостаза С норме и при патологии
Система гемостаза обеспечивает нормальную жизнедеятельность организма, его целостность, приспособительные реакции и гомеостаз. Ее основными функциями являются: поддержание жидкого состояния циркулирующей крови; остановка кровотечения при повреждении сосудов. Кроме того, система гемостаза оказывает влияние на реологические свойства крови, микроциркуляцию, проницаемость сосудов, процессы регенерации тканей, иммунологические реакции.
Гемостаз осуществляется при тесном взаимодействии свертывающей, противосвертывающей, фибринолитической и кал-ликреин-кининовой систем организма.
Этапы процесса гемостаза:
1. Первичный, или сосудисто-тромбоцитарный, гемостаз, в котором участвуют сосуды и тромбоциты. Продолжительность фазы — 3 — 5 мин. Конечный продукт — образование тромбоцитарного сгустка.
2. Вторичный гемостаз, или ферментативная коагуляция, в котором принимают участие плазменные факторы свертывания и тромбоцитарный фактор. Эта фаза длится 5 — 10 мин. Конечный продукт — фибриновый тромб.
3. Фибринолиз — процесс, приводящий к растворению тромба. Его длительность — 48-72 ч.
11.1. СОСУДИСТО-ТРОМБОЦИТАРНЫИ ГЕМОСТАЗ
В процессе первичного (сосудисто-тромбоцитарного) гемостаза принимают участие все элементы стенки сосуда (клетки эндотелия, субэндотелиальный слой, гладкомышечные клетки) и клетки крови (тромбоциты, эритроциты).
11.1.1. Роль кровеносных сосудов в первичном гемостазе
Для сохранения крови в жидком состоянии необходима структурная и функциональная целостность сосудистого эндотелия. Нормальный эндотелий представляет собой мощную антикоагулянтную
поверхность, которая не активирует белки свертывания крови и не привлекает к себе клеточные компоненты крови. Несма-чиваемость и интактность обращенного внутрь сосудов эндотелиального пласта обусловлена наличием гликокаликса — пристеночного слоя из гликозаминогликанов, обеспечивающего максимальную текучесть крови [ 1 ].
Клетки эндотелия продуцируют вещества, обладающие диаметрально противоположным эффектом на сосудистый тонус. Так, синтезируемые в эндотелии простациклин и NO расслабляют гладкомышечные клетки сосудов и тормозят адгезию и
Сосудисто-тромбоцитарный гемостаз 337
агрегацию тромбоцитов. Синтез и последующий выброс ангиотензина II вызывает локальный сосудистый спазм. Кроме того, эндотелиальные клетки выделяют аутоки-назу — активатор плазминогена, индуцирующего фибринолиз.
Субэндотелий состоит из базальной мембраны, коллагена, эластиновых волокон, протеогликанов, фибронектина и фактора Виллебранда. После повреждения эндотелия этот слой сосудистой стенки стимулирует адгезию тромбоцитов и активацию каскадной системы свертывания крови.
Первичная нормальная реакция микрососуда на повреждение — немедленный выраженный локальный спазм рефлекторного характера (длительностью менее 1 мин), поддерживаемый освобождением из стенок сосудов и тромбоцитов адреналина, серотонина и других биологически активных веществ. В результате спазма капилляры и венулы временно запустевают и кровотечения из них в течение первых 20-30 с не происходит [2]. Возникающее при этом замедление скорости кровотока улучшает взаимодействие между тромбоцитами, факторами свертывания крови и поврежденным участком сосудистой стенки, способствует сохранению повышенной локальной концентрации активированных факторов в месте образования тромба. Вазоконстрикция как реакция на небольшую травму может остановить кровотечение, а при массивном повреждении лишь снижает кровопотерю.
11.1.2. Тромбоциты
Тромбоциты образуются из мегакариоцитов в костном мозгу и частично в легочных капиллярах. Около 1/3 всей массы тромбоцитов содержится в селезенке (селезеночный пул: при спленомегалии этот пул возрастает, что может приводить к перераспределительной тромбоцитопении). При стимуляции адренорецепторов селезенки (физическая нагрузка, стресс) происходит выброс тромбоцитов в циркуляторное русло, что вызывает временный тромбоцитоз.
Продолжительность жизни тромбоцитов человека составляет 7—10 дней. В крови
здоровых людей содержится 170 — 350 х х 109/л тромбоцитов.
Адекватность реакции сосудистой стенки на повреждение зависит от количественного и качественного состояния тромбоцитов, поскольку одной из их основных функций является ангиотрофическая. Ежедневно около 15 % циркулирующих тромбоцитов «изливают» свое содержимое эндотелиальным клеткам, чтобы поддержать их функциональную полноценность. При недостаточном содержании тромбоцитов в крови и их качественных дефектах наступает дистрофия эндотелия, что позволяет эритроцитам выходить из капилляров диапедезным путем; повышается ломкость микрососудов (легко возникают петехии и экстравазаты).
Структура тромбоцитов и ее функциональное значение. В состоянии покоя тромбоцит представляет собой дискообразную безъядерную клетку с типичной для всех клеток двухслойной фосфолипидной мембраной. Тромбоцитарная фосфолипидная оболочка является местом взаимодействия ингредиентов плазмы и поврежденных эндотелиальных клеток. Мембранные фосфолипиды активируют процесс свертывания крови и в литературе известны под названием «фактор 3 тромбоцитов» (по своим свойствам этот компонент идентичен используемому в лабораторных тестах кефалину). Мембрана тромбоцитов образует множество проникающих вглубь клетки каналов с наружными выходами в виде устьев (так называемая открытая канальцевая система), что увеличивает площадь контакта кровяных пластинок с плазмой.
Тромбоцитарные факторы свертывания крови по международной номенклатуре обозначаются арабскими цифрами и латинской буквой Р (от англ, platelet — тромбоцит).
В цитоплазме тромбоцитов имеются гранулы, содержащие АТФ, АДФ, серотонин, катехоламины, p-тромбоглобулин, ан-тигепариновый фактор (4-й пластиночный фактор), фактор Виллебранда (FW), фибриноген, фактор V свертывания и др. При активации тромбоцитов содержимое этих гранул выходит из клеток (этот процесс
338 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
называется реакцией высвобождения, или дегрануляции). Реакция высвобождения обеспечивает активность процесса агрегации и образования в сосуде гемостатической пробки. Вещества, вызывающие агрегацию, называют индукторами агрегации.
Адгезия и агрегация тромбоцитов как основа первичного гемостаза. Ведущая роль в реализации первичного гемостаза принадлежит адгезивно-агрегационной функции тромбоцитов. В неповрежденном сосуде нестимулированные тромбоциты циркулируют в виде гладких дискообразных клеток с незначительной метаболической активностью. Они не взаимодействуют друг с другом и не адгезируют вследствие электростатического отталкивания от других форменных элементов крови и эндотелия. Физиологическая активация тромбоцитов начинается только тогда, когда поврежден сосудистый эндотелий и обнажен субэндотелиальный слой, т. е. для форменных элементов крови становятся доступными индукторы агрегации (коллаген, FW ). Тромбоциты первыми появляются в месте дефекта. Они прилипают (адгезируют) к поврежденным эндотелиальным клеткам, коллагену базальной мембраны, набухают и образуют отростки. Параллельно адгезии протекает процесс агрегации тромбоцитов, набухание и склеивание их между собой с образованием отростков и наложением агрегатов на участок повреждения сосуда, вследствие чего гемостатическая пробка или тромб быстро растет. В результате реакции дегрануляции из тромбоцитов высвобождаются дополнительные индукторы агрегации, что делает этот процесс необратимым. За этой упрощенной схемой стоят сложные, недостаточно хорошо изученные биохимические процессы, протекающие одновременно с высокой скоростью, так что все попытки их разделения на стадии с целью удобства изучения условны. Выделение стадий и отдельных механизмов целесообразно также с позиций фармакологического воздействия на то или иное звено сосудисто-тромбоцитарного гемостаза. Так, например, одним из ключевых звеньев этапа активации тромбоцитов является мобилизация ионов кальция из внутриклеточных
депо, а также снижение уровня цАМФ. Поэтому фармакологическая коррекция внутриклеточного содержания ионизированного кальция — одно из основных направлений в разработке дезагрегантных средств.
11.1.3. Знамение простагландинов для сосудисто-тромбоцитарного гемостаза
Агрегационная функция тромбоцитов в значительной степени зависит от образования из мебранных фосфолипидов эндотелиальных клеток и тромбоцитов арахидоновой кислоты и циклических производных простагландинов.
Простагландины — это биологически активные вещества из класса кислых липидов, которые синтезируются практически во всех органах и тканях, но не накапливаются в них, а образуются по мере необходимости под действием нейрогенных, физических, химических и других стимулов. Простагландины представляют собой местные гормоны, играющие роль регуляторов клеточного метаболизма и функциональной активности тех клеток, в которых они образуются [3].
При активации тромбоцитов происходит освобождение жирных кислот из фосфолипидных клеточных мембран, в частности предшественницы простагландинов -арахидоновой кислоты. Дальнейшие превращения этой кислоты могут происходить двумя путями: 1) липооксигеназным, в результате которого образуются лейкотриены и ряд гидропероксидов (первые участвуют в иммунном ответе и считаются главной причиной бронхоспазма при бронхиальной астме, физиологическая роль вторых не вполне изучена); 2) циклооксигеназным. Циклооксигеназа тромбоцитов и эндотелиоцитов превращает арахидоновую кислоту в циклические эндопероксиды (PgG2 и PgH2) — очень нестабильные соединения, обладают чрезвычайно высокой биологической активностью, вызывают быструю и необратимую агрегацию в ответ на действие всех известных стимуляторов тромбоцитов, сокращают гладкую мускулатуру кровеносных сосу
Сосудисто-тромбоцитарный гемостаз 339
дов и бронхов. Кроме того, из циклических эндопероксидов в тромбоцитах под воздействием фермента тромбоксан-синте-тазы образуется очень мощный агрегирующий и сосудосуживающий агент тромбоксан Л2 (ТХА2), а в эндотелиальных клетках при участии простациклин-син-тетазы — основной ингибитор агрегации и вазодилататор — простациклин (простагландин I2 — Pgl2).
Тромбоксан А2 вызывает реакцию высвобождения тромбоцитов. Нарушение образования этого агента ведет к выраженному нарушению функции тромбоцитов, способствует развитию кровоточивости. Нарушение синтеза простациклина в сосудистой стенке или снижение его поступления в кровь повышает склонность тромбоцитов к агрегации и создает тромбогенную опасность. При повреждении сосудистой стенки в эндотелиальных клетках также начинает преобладать образование тромбоксана. Дисбаланс между тромбоксаном и простациклином резко усиливает агрегацию и реакцию высвобождения гранул.
Протекающие внутри тромбоцитов реакции превращения метаболитов арахидоновой кислоты в активирующие агрегацию вещества достаточно сложны, но все они являются ответом на воздействие различных стимуляторов. Цикл метаболических превращений арахидоновой кислоты в тромбоцитах и эндотелиоцитах рассматривается как объект фармакологического воздействия для дезагрегатных средств. Для реализации этого процесса необходимы ионы Са2+ и Mg2+. Начинается процесс активизации с взаимодействия индуктора со специфическим рецептором тромбоцитов.
Рецепторы тромбоцитов — это гликопротеиды (GP) с различной молекулярной массой (GP 1а, Па, ПЬ, Ша и т. д.), расположенные не только на поверхности мембраны, но и в открытой канальцевой системе. Одни рецепторы обладают специфичностью, другие — полимодальностью, т. е. способностью реагировать на действие различных индукторов [4].
Рецепторы тромбоцитов можно условно разделить на первичные и вторичные. Первичные непосредственно реагируют на действие индуктора, вторичные становятся
доступными в процессе активизации тромбоцитов. Ключевая роль в агрегации принадлежит тромбоцитарным GP ПЬ/Ша рецепторам, которые могут присоединять фибриноген, FW и другие белки, участвующие в агрегации [ 1 ]. Фибриноген соединяется одновременно с двумя тромбоцитами, способствуя их агрегации. Можно считать, что адгезия и агрегация, вызываемые различными индукторами, в конечном итоге осуществляются за счет экспрессии рецепторов GP ПЬ/Ша к FW и фибриногену, образующим при участии ионов кальция мост между тромбоцитами или между тромбоцитами и поврежденной стенкой. При адгезии тромбоцитов основную роль играет FW, а при их агрегации — фибриноген. Поскольку этот конечный этап агрегации тромбоцитов одинаков при различных стимуляциях кровяных пластинок, то возможность блокировать функцию GP ПЬ/Ша рецепторов считают весьма перспективным направлением анти-тромботической терапии.
11.1.4. Ретракция и консолидация тромба
В осуществлении процесса агрегации и последующей консолидации тромба, а также ретракции кровяного сгустка, важную роль играет сходный с актомиозином сократительный белок кровяных пластинок — тромбостенин, локализующийся в микротрубочках и микрофибриллах этих клеток. При сокращении тромбостенина происходит сближение тромбоцитов в агрегате, который становится плотным и непроницаемым для крови.
В результате взаимодействия тромбоцитарных и плазменных факторов в зоне локального гемостаза образуется тромбин, малые дозы которого усиливают и завершают процесс агрегации. Он способствует также отложению фибрина на тромбоцитарных агрегатах, образующихся в месте сосудистой травмы, и инициирует процесс репарации ткани.
Тромбоциты преимущественно влияют на интенсивность и скорость локального свертывания в зоне микроциркуляции, а не на процесс свертывания вообще.
340 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
Таким образом, сосудисто-тромбоцитарный гемостаз обеспечивается физиологическими (тромбоциты, регуляция тонуса и проницаемости сосудистой стенки) и биофизическими (изменение поверхностного потенциала) механизмами.
11.1.5. Методы исследования сосудисто-тромбоцитарного гемостаза
Тромбоцитарный гемостаз зависит от количества тромбоцитов и их функциональной активности. Методы его диагностики можно разделить на две группы. К первой группе методов, позволяющей заподозрить тромбоцитопатию, относятся: определение времени кровотечения и резистентности стенки микрососудов, микроскопическое исследование тромбоцитов.
1. Определение резистентности капилляров.
Осуществляется с помощью манжеточной и баночной проб, пробы щипка. Резистентность считается сниженной, если число образовавшихся петехий на коже предплечья превышает этот показатель у здоровых людей.
2. Подсчет количества тромбоцитов.
Производится под микроскопом в камере Горяева. В норме количество тромбоцитов составляет: (200,0 — 400,0) • 109/л. Диагноз тромбоцитопения правомочен при уровне кровяных пластинок ниже 150,0 х х 109/л. Уменьшение количества тромбоцитов ниже 80,0 • 109/л способствует появлению кровоточивости, риск которой резко возрастает при уровне ниже 20,0 х х 109/л, а увеличение выше 800,0 • 109/л создает угрозу развития тромбозов. Однако эти цифры условны, поскольку более важным является качественный состав тромбоцитов, наличие в крови ингибиторов их функции, выраженность нарушений в других звеньях системы гемостаза и т. д. [2].
3. Определение длительности кровотечения по Дьюку.
Принцип метода заключается в определении длительности кровотечения из места прокола кожи, нанесенного скарификатором. В норме это время составляет 150 —
300 с. Следует учитывать, что метод Дьюка недостаточно чувствителен и нередко дает ошибочные результаты (из-за раннего склеивания краев ранки и других причин). При многих тромбоцитопатиях его показания не нарушаются [5].
Ко второй группе относятся методы, применение которых имеет наибольшее клиническое значение: определение адгезивно-агрегационной активности тромбоцитов, ретенционные пробы, исследование ретракции кровяного сгустка, определение фактора 3 тромбоцитов.
1. Исследование ретенции (адгезивности) тромбоцитов при контакте со стеклом (по Salzman).
Способность тромбоцитов к адгезии определяют по убыванию их из крови при контакте со стеклом. В норме этот показатель колеблется в пределах от 20 до 50 %.
2. Исследование индуцированной агрегационной функции тромбоцитов фотометрическим методом с графической регистрацией процесса (по Born).
Принцип основан на регистрации изменения оптической плотности богатой тромбоцитами плазмы до и после введения в нее определенного количества различной концентрации АДФ или других агентов, вызывающих агрегацию тромбоцитов. При использовании низких концентраций агонистов определяется тот минимальный их порог, на который реагируют исследуемые тромбоциты. Чем ниже этот порог, тем меньше антиагрегационный потенциал системы и тем значительнее склонность тромбоцитов к тромбообразованию. Повышение агрегационной способности тромбоцитов может служить критерием риска развития тромбозов.
Полное отсутствие или резкое снижение агрегации наблюдается при различных видах качественной неполноценности и дисфункции тромбоцитов.
Важное значение имеет изучение агглютинации тромбоцитов под влиянием антибиотика ристоцетина (ристомицина). Этот процесс происходит только при наличии в плазме кофактора процесса — FW, а в тромбоцитах — рецепторов к нему.
Лекарственные препараты, воздействующие на сосудисто-тромбоцитарный гемостаз 341
11.2. ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, ВОЗДЕЙСТВУЮЩИЕ НА СОСУДИСТО-ТРОМБОЦИТАРНЫЙ ГЕМОСТАЗ
11.2.1. Препараты, восстанавливающие сосудисто-тромбоцитарный гемостаз
Концентрат тромбоцитов (КТ). Трансфузии концентратов тромбоцитов нередко используют в клинической практике как эффективный метод лечения и профилактики геморрагических осложнений, обусловленных снижением числа тромбоцитов в периферической крови (тромбоцитопения) или их дисфункцией (тромбоцитопатия).
При определении показаний к переливанию тромбоцитарных концентратов В. А. Аграненко рекомендует учитывать в динамике абсолютное число тромбоцитов, наличие геморрагий и иммунологический статус пациента [6]. При иммунных тромбоцитопениях трансфузии КТ, как правило, не показаны.
Доза трансфузии концентрата тромбоцитов. Основанием для расчета лечебной дозы переливаемых тромбоцитов является необходимость получения определенного посттрансфузионного их прироста, достаточного для купирования геморрагического синдрома [6]. Полагают, что увеличение количества тромбоцитов на (50,0 — 70,0) • 109/л сопровождается длительным гемостазом.
Через час после трансфузии КТ отмечается увеличение числа тромбоцитов на (6,0—10,0) • 109/л. В повседневной практике рекомендуют использовать простое правило: на 10 кг массы тела переливают один КТ.
Оценка эффективности трансфузий КТ. Основными критериями оценки лечебной эффективности трансфузий тромбоцитов являются клинические признаки прекращения или уменьшения кровоточивости, увеличение числа циркулирующих тромбоцитов через 1 ч и через 18 — 24 ч после переливания. Однако лечебный эффект при переливании тромбоцитов наступает не всегда, поскольку на выживаемость перелитых кровяных пластинок
влияют многие факторы (повышенное потребление тромбоцитов при циркуляции в крови избыточных количеств индукторов агрегации, синтезе антител к тромбоцитам, чрезмерном депонировании их в селезенке и т. д.).
Этамзилат натрия (дицинон). Препарат ингибирует эффекты простациклина, вследствие чего усиливается агрегация тромбоцитов. Стимулирует ферментные реакции тромбоцитов, их новообразование из мегакариоцитов и выход из депо, усиливает «реакцию освобождения тромбоцитов». Умеренно ускоряет образование тканевого тромбопластина. В настоящее время дицинон — один из наиболее эффективных агрегантов. Кроме того, он обладает выраженным эндотелиотропным действием: препятствует десквамации эндотелия, повышает «прочность» эндотелиальной цементирующей субстанции, снижает хрупкость и проницаемость сосудов.
Препарат вводится внутривенно (медленно), внутримышечно или энтерально. При внутривенном введении гемостатический эффект наблюдается через 5—15 мин, а при внутримышечном — через 30 — 40 мин. Терапевтический эффект сохраняется в течение 4 — 6 ч. Назначают 3 — 4 раза в день.
Препараты кальция (кальций хлорид, кальций глюконат). Кальций участвует в агрегации и адгезии тромбоцитов, активирует тромбин и фибрин и, таким образом, стимулирует образование тромбоцитарных и фибриновых тромбов.
Показания к применению: переливание больших количеств цитратной крови, тромбоцитопения.
11.2.2. Препараты, стабилизирующие сосудистую проницаемость (вазопротекторы)
Добезилат-кальций (доксиум). Препарат нормализует микроциркуляцию в стенке сосуда как антагонист брадикинина и
342 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
стабилизатор цементирующей субстанции субэндотелиального слоя. Препятствует контрактильности и десквамации эндотелия артерий, снижает активность кининов, применяется внутрь.
Продектин (пармидин). Является эндотелиальным средством, предупреждает сокращение и десквамацию эндотелия артерий, снижает активность кининов. Препарат применяется внутрь.
Адроксон. Метаболит адреналина, лишенный способности стимулировать адренорецепторы на гладкомышечных клетках сосудов, поэтому не повышает артериальное давление. В результате активации а-адренорецепторов тромбоцитов происходит накопление внутриклеточного несвязанного кальция, что в конечном итоге приводит к реакции высвобождения. Адроксон оказывает гемостатическое действие при капиллярных кровотечениях, обусловленных повышенной проницаемостью стенок капилляров, кишечных кровотечениях. Применяют парентерально и местно, реже внутрь.
Эндотелон (процианидоловые олигомеры). Препарат обладает васкулопротек-торным и венотонизирующим действием, уменьшает проницаемость сосудов. Предохраняет фиброзные протеины, прежде всего коллаген и эластин, от энзиматической деградации и обеспечивает защитное действие в отношении температурной денатурации коллагена. Применяется по 6 таблеток в день в 2 приема курсами длительностью 20 дней.
Сулодексид (Vessel Due-F). Глико-зоаминогликан из группы гепариноидов, оказывает комплексное влияние на стенки кровеносных сосудов: 1) повышает отрицательный заряд эндотелиальных клеток и их резистентность к повреждающему действию экзо- и эндотоксинов, иммунных комплексов, цитокинов, лейкоцитарных протеаз; 2) уплотняет базальную мембрану капилляров, снижает ее повышенную проницаемость, ослабляет формирование рыхлых атеросклеротических бляшек и пролиферацию гладкомышечных клеток; 3) стимулирует выброс простациклина эндотелиальными клетками [2].
11.2.3. Препараты, блокирующие сосудисто-тромбоцитарный гемостаз
Аспирин (ацетилсалициловая кислота). Вызывает необратимую инактивацию циклооксигеназы тромбоцитов. Обладает выраженной антиагрегационной активностью, поскольку снижает синтез тромбоксана в тромбоцитах, тормозит реакцию «освобождения» тромбоцитов. Аспирин, действуя на уровне синтеза простагландинов, не только частично блокирует агрегацию тромбоцитов, но и угнетает продукцию простациклина, который сам по себе является естественным ингибитором агрегации тромбоцитов. Поскольку последние — безъядерные клетки, они лишены способности ресинтезировать белки, в результате чего блокада синтеза тромбоксана при терапии аспирином сохраняется на протяжении всей жизни тромбоцитов — в течение 7 — 10 дней. Это объясняет, каким образом препарат с 20-минутным периодом полужизни в системном кровотоке при однократном приеме в день ингибирует синтез тромбоксана тромбоцитами [7]. Эффективная антиагрегационная доза — 75 мг/сут. В этой дозировке в наименьшей степени угнетается синтез простациклина (естественного дезагреганта).
Индометацин. Вызывает обратимую инактивацию циклооксигеназы тромбоцитов.
Дипиридамол (курантил). Ингибитор фосфодиэстеразы, препятствуя захвату аденозина, образовавшегося из своих предшественников в эритроцитах при микротравмах сосудов, повышает внутриклеточный уровень циклических нуклеотидов -естественных ингибиторов агрегации тромбоцитов.
Производные ксантина (пентоксифиллин, кофеин, теофиллин). Повышают уровень внутриклеточных циклических нуклеотидов. Улучшают микроциркуляцию и реологические свойства крови, усиливают коллатеральное кровообращение, подавляют агрегацию тромбоцитов и эритроцитов.
Реополиглюкин. Уменьшает агрегацию форменных элементов крови и улучшает реологические свойства крови.
Коагуляционный гемостаз (ферментативная коагуляция) 343
Тиклид (тиклопидин гидрогенхлорид). Является мощным ингибитором агрегации тромбоцитов, вызванной АДФ. Тиклид действует на конечный этап агрегации — он ингибирует образование фибриновых мостиков между тромбоцитами [8]. При этом он не затрагивает метаболизма арахидоновой кислоты и, таким образом, не уменьшает производство простациклина стенками кровеносных сосудов, в отличие от аспирина.
Плавике (клопидогрель). Создан как альтернатива другим антитромботическим препаратам, обладает большей эффективностью и лучшей переносимостью. Плавике селективно подавляет связывание аденозиндифосфата (АДФ) с его рецептором на мембране тромбоцита и активацию ком
плекса GP ПЬ/Ша, ингибируя таким образом не только АДФ-индуцированную агрегацию тромбоцитов, но и частично агрегацию, обусловленную коллагеном. Препарат не ингибирует активность фосфо-диэстеразы. В дозе 75 мг в сутки угнетает АДФ-зависимую агрегацию тромбоцитов через 2 ч после перорального приема первой дозы. Стойкий уровень угнетения АДФ-агрегации тромбоцитов (40 — 60 %) достигается на 3 — 7-й день лечения и поддерживается в течение длительного приема препарата. После перорального приема клопидогрель быстро абсорбируется и, пройдя через печень, превращается в активный метаболит. Антитромботическая активность плавикса коррелирует с его антиагрегантным эффектом.
11.3. КОАГУЛЯЦИОННЫМ ГЕМОСТАЗ (ФЕРМЕНТАТИВНАЯ КОАГУЛЯЦИЯ)
Тромбоцитарный гемостаз обеспечивает остановку кровотечения лишь в микрососудах с низким кровяным давлением. В более крупных сосудах, особенно мышечного типа, и в сосудах с достаточно высоким давлением крови, тромбоцитарный тромб уже не в состоянии обеспечить надежной остановки кровотечения. В этих условиях ведущую роль в обеспечении гемостаза играет свертывающая система крови.
Коагуляционный гемостаз, хотя и называется вторичным, включается не строго последовательно за сосудисто-тромбоцитарным (первичным) гемостазом и значительную часть времени функционирует сопряженно с первичным.
Свертывание крови — это сложный многоэтапный {каскадный) ферментактив-ный процесс, в котором последовательно активируются проферменты. Составными частями каскада являются энзимы процессов свертывания, мембранные фосфолипиды. Факторы свертывания в плазме и сыворотке по международной номенклатуре обозначаются римскими цифрами. Если фактор активизируется, то к римской цифре добавляется буква «а» (Па, Vila и т. д.). Факторная часть системы
свертывания представлена девятью гликопротеинами, ионами кальция и тромбопластином, в норме не содержащемся в кровотоке (табл. 11.1).
В норме факторы свертывающей системы циркулируют в неактивном состоянии, а антикоагулянты — в активном, что обеспечивает жидкое состояние крови. Значительный избыток «запаса» факторов коагуляции, с одной стороны, является гарантией надежной остановки кровотечения при травме сосуда, с другой — представляет угрозу массивного тромбоза при снижении активности антикоагулянтного звена и циркуляции высоких концентраций активаторов коагуляции.
Белки свертывания крови можно разделить на три группы:
♦ К-витаминзависимые;
♦ кофакторные (факторы контакта);
♦ чувствительные к тромбину (тромбин-зависимые).
К витамин К-зависимым белкам относятся как прокоагулянты (факторы II, VII, IX, X), так и антикоагулянты (белки С и S). Все эти белки синтезируются в печени при участии витамина К.
Факторы контакта {кофакторные белки) также синтезируются в печени.
344 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
Таблица 11.1. Некоторые физиологические константы факторов свертывания крови
Фактор Название фактора Период полужизни, ч Избыток фактора, раз
I Фибриноген 100 3-6
II Протромбин 72-96 2-3
III Тромбопластин — —
IV Ионы Са2+ — —
V AC-глобулин (проакцелерин) 12-15 8-10
VII Проконвертин 2-6 10
VIII Антигемофильный глобулин А 12-18 3-5
IX Фактор Крйстмасса (антигемофильный фактор В) 20-30 4-5
X Фактор Стюарта — Прауэра 20-40 5
XI Предшественник тромбопластина 10-20 4-5
XII Фактор Хагемана 50-70 Неизвестно
XIII Фибринстабилизирующий фактор 100 10
Они необходимы для активации свертывания при контакте с поверхностью. К ним относятся:
фактор XII (фактор Хагемана);
прекалликреин (фактор Флетчера);
фактор XI;
высокомолекулярный кининоген (фактор Фитцджеральда).
Белками, чувствительными к тромбину, являются фибриноген, фактор XIII, фактор V, комплекс фактора VIII.
Процесс свертывания крови, согласно современным представлениям, протекает в три стадии:
1. Формирование активной протромби-назы.
2. Образование тромбиновой активности.
3. Превращение фибриногена в фибрин: а) процесс полимеризации фибрина;
6) стабилизация фибрина и ретракция сгустка.
Это деление также условно, поскольку фазы нельзя точно разграничить и указать, в какой период заканчивается одна и наступает другая [4].
Первая стадия ферментативной коагуляции наиболее сложная и продолжительная. Формирование протромбиназной активности является результатом многоступенчатого ферментативного процесса.
Протромбиназой называют комплекс факторов Ха, Va, ионов Са2+ и фактора 3 тромбоцитов, активирующего переход протромбина в тромбин.
Имеется два основных независимых механизма активации процессов свертывания — внешний и внутренний. Внешний путь более короткий, что ведет к быстрому тромбинообразованию. В этом механизме участвуют тканевый тромбопластин, фактор VII (проконвертин) и ионы кальция. Образующийся тромбин эффективно усиливает агрегацию тромбоцитов и активирует факторы внутреннего пути, но его небольшого количества недостаточно для образования фибрина.
Внутренний путь активации свертывания определяется как коагуляция, инициируемая компонентами, полностью находящимися в пределах сосудистой системы. Он начинается с образования плазменного тромбопластина в результате активации фактора XII при контакте с поврежденной сосудистой стенкой или инородной поверхностью (игла, катетер и т. д.). Ускоряется эта «контактная фаза» фосфолипидным фактором тромбоцитов (фактором 3). Внутренний путь является поли-каскадным и протекает с последовательной активацией факторов XII, XI, IX + VIII, X + V и II. Сложная система множественных ферментных превращений необходима для многократного усиления инициирующего сигнала, приводящего к эффективному тромбинообразованию.
Существует функциональная взаимосвязь между свертывающей и фибринолитической системами крови, а также связь
Коагуляционный гемостаз (ферментативная коагуляция) 345
этих систем с калликреин-кининовой системой и системой комплемента. Пусковым для всех этих систем является активированный XII фактор (фактор Хагемана). Калликреин (фактор Флетчера) ускоряет взаимодействие между факторами XII и XI, а также активирует фактор VII, функционируя как мост между внутренним и внешним механизмами образования протромбиназной активности.
Как при внешнем, так и при внутреннем механизме свертывания взаимодействие и активация факторов осуществляется на фосфолипидных микромембранах, играющих роль матриц, на которых фиксируются белковые факторы. Роль таких матриц выполняют мембраны оболочек и гранул тромбоцитов (тромбоцитарный фактор 3) и сходные с ними компоненты из оболочек других клеток (эритроцитов и др.).
Протеолитический фермент тромбин отщепляет от молекулы фибриногена по два фибринопептида А и В, в результате чего образуется молекула фибрин-мономера с четырьмя свободными связями. Соединяясь друг с другом, фибрин-мономеры образуют сначала димеры, тетрамеры и более крупные, но растворимые олигомеры. По мере укрупнения фибрин-олигомеры трансформируются в нити фибрина. Неферментативный процесс полимеризации фибрина тормозит гепарин. Ингибирование полимеризации фибрина гепарином получило название «неферментативный фибринолиз». Тромбин активирует также фибрин-стабилизирующий фактор (XIII), который укрепляет фибрин-полимеры дополнительными связями, делая их более прочными и нерастворимыми. В фибриновом сгустке задерживается много форменных элементов крови, вызывающих уплотнение и ретракцию сгустка. Особая роль на этом этапе свертывания принадлежит тромбоцитам.
11.3.1. Методы исследования коагуляционного гемостаза
1. Определение времени свертывания по Ли-Уайту.
В норме этот показатель составляет 420 ± ±8,7 с. Удлинение времени свертывания связано либо с выраженным дефицитом
одного или нескольких факторов, либо с избытком антикоагулянтов в крови.
2. Определение времени рекальцификации плазмы (по Bergerhof и Roka).
В норме этот показатель равняется 112,0 ± 2,6 с. Удлинение времени наблюдается при дефиците всех плазменных факторов, кроме VII и XIII, а также при применении антикоагулянтов и выраженной тромбоцитопении.
Получить представление о состоянии всего гемокоагуляционного каскада и провести ориентировочную дифференциацию между нарушениями внутреннего и внешнего механизмов запуска процесса свертывания можно с помощью таких тестов: аутокоагуляционного (АКТ), протромбинового и тромбинового.
3. Определение протромбинового времени (ПВ).
В норме этот показатель составляет 12 — 15 с. Протромбиновое время характеризует процесс свертывания крови по внешнему механизму (фактор VII) и общему пути (факторы X, V, II и I), что достигается добавлением к исследуемой плазме тканевого тромбопластина.
4. Активированное частичное тромбопластиновое время (АЧТВ).
Процесс свертывания рекальцифицированной плазмы определяется при запуске его по внутреннему механизму при добавлении каолина и кефалина. Его показатели целиком зависят от дефицита плазменных факторов и на них не сказывается дефицит тромбоцитов и их качественная неполноценность.
5. Аутокоагуляционный тест (АКТ) (по Berkarda с соавт.).
АКТ — наиболее чувствителен к нарушениям внутреннего механизма, с его помощью определяется динамика образования и инактивации тромбопластин-тром-биновой активности в исследуемой крови. АКТ дает представление о состоянии как прокоагулянтного, так и антикоагулянтного звеньев свертывающей системы крови, отражая кинетику обоих процессов.
6. Определение тромбинового времени свертывания (ТВ).
В тромбиновом тесте оценивается конечный этап процесса свертывания под
346 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
влиянием стандартного количества тромбина.
Учет показателей этого теста важен для правильной трактовки сдвигов во всех других коагуляционных пробах, поскольку замедление конечного этапа свертывания неизбежно нарушает их показания.
7. Определение рептилазового времени (тест с ядом песчаной эфы).
Тест, в отличие от тромбинового времени, используется для выявления скрытой гиперкоагуляции, в том числе на фоне проводимой гепаринотерапии. Удлинение времени наблюдается при гипофибриногенемии и при накоплении в плазме продуктов деградации фибриногена (ПДФ).
Параллельное исследование рептилазового и тромбинового времени используется в экспресс-диагностике ДВС-синдрома.
8. Определение содержания фибриногена (гравиметрическим, колориметрическим, иммунохимическим методами).
В норме этот показатель составляет (3,4 ± 0,24) г/л (2 — 4 г/л). У здоровых родильниц — (4,5 ± 0,32) г/л.
11.3.2. Препараты, восстанавливающие коагуляционный гемостаз
1. Нативная и свежезамороженная плазма.
2. Антигемофильная сухая плазма.
3. Криопреципитат. Выделяется из плазмы с помощью охлаждения (криоосаждения). Белковый препарат, содержит в значительном количестве фактор VIII, FW, фибриноген и фактор XIII, но мало альбумина и других белков. Низкое содержание альбумина позволяет вводить его в
больших количествах, не опасаясь перегрузки кровообращения и отека легких. Применяются обычно 1—2 дозы. Вводится внутривенно капельно.
4. Витамин К. В клинической практике используют два производных метилнафтохинонов: витамин К] (фитоменадион) и витамин К3 (викасол).
Витамин К принимает участие в синтезе протромбина (фактор II), факторов VII, IX, X и способствует нормальному свертыванию крови. При его отсутствии или недостатке в организме или при приеме антикоагулянтов непрямого действия (кумаринов), вытесняющих нафтохинон из места синтеза коагуляционных факторов, развиваются геморрагические явления.
Фитоменадион (витамин Kt) вводится внутривенно или внутримышечно — 1 мл 1 % раствора или внутрь по 1 таблетке 3 раза в сутки.
Викасол (менадион) — синтетический аналог витамина К3. Применяется: внутривенно или внутримышечно 1 мл 1 % раствора.
5. Кальций хлорид. 10 мл 10 % раствора внутривенно медленно. Ионы кальция необходимы для нормального свертывания крови, способствуют превращению протромбина в тромбин, уменьшают проницаемость сосудов. Применяют также как кровоостанавливающее средство при кровотечениях для повышения свертываемости крови.
6. Гемофобин. Содержит раствор пектинов (3 %) с добавлением кальций хлорида (1 %) и ароматических веществ. Применяется как гемостатическое средство при желудочно-кишечных кровотечениях, внутрь по 2 — 3 чайные ложки 1—3 раза в день.
11.4. АНТИКОАГУЛЯНТНАЯ СИСТЕМА
Активация свертывания крови неизбежно вызывает последующую активацию противосвертывающих механизмов. Биологическая роль антикоагулянтной системы в поддержании гомеостаза очень велика, поскольку она участвует в регуляции системы свертывания крови, сохранении жидкого состояния крови в циркуляции и
препятствует переходу локального тром-бообразования в чрезмерно распространенное или диффузное свертывание.
Все образующиеся в организме антикоагулянты подразделяют на две группы:
1. Первичные (предсуществующие) — самостоятельно синтезируемые в организме.
Антикоагулянтная система 347
2. Вторичные — образующиеся в процессе свертывания крови, фибринолиза и активации других протеолитических систем.
Первичные антикоагулянты:
Гепарин. Образуется во всех органах и тканях. Больше всего его синтезируется в легких и печени. Гепарин является непрямым ингибитором тромбина, поскольку для осуществления антикоагулянтного действия ему необходим кофактор — антитромбин III (AT III). Гепарин тормозит все три фазы ферментативной коагуляции, снижает адгезивность тромбоцитов, способствует дезагрегации эритроцитов, улучшает реологию крови.
Антитромбин III (AT III). Универсальный ингибитор почти всех ферментных факторов свертывания: тромбина, факторов Ха, IXa, Х1а и, возможно, ХПа. На его долю приходится более 75 % всей антикоагулянтной активности плазмы, причем он является основным кофактором гепарина. При уровне AT III ниже 30 % имеется очень выраженная склонность к тромбозам различной локализации (тромбоэмболиям).
Протеин С. Синтезируется гепатоцитами. К-витаминзависимый профермент, активизирующийся тромбином и фактором Ха. Расщепляет и инактивирует основные неферментные факторы VIII и V. Эта активизация идет в комплексе протеина С с фосфолипидом и кальцием.
Дефицит протеина С способствует склонности к рецидивирующим тромбозам. Глубокая вторичная депрессия протеина С наблюдается при острых ДВС-синдромах, респираторном дистресс-син-дроме, тяжелом поражении печени.
Протеин S. К-витаминзависимый кофактор протеина С. При его наличии активированный протеин С расщепляет и инактивирует факторы Va и Villa.
ct-2-Макроглобулин. Белок, обладающий способностью связывать активированные компоненты свертывающей системы и фибринолиза, исключать их из взаимодействия с другими факторами. Дефицит этого белка обычно ведет к развитию тромбозов.
Вторичные антикоагулянты образуются в процессе свертывания крови и фибринолиза в результате дальнейшей ферментной деградации ряда факторов свер
тывания, вследствие чего они после начальной активации утрачивают способность участвовать в гемокоагуляционном процессе и часто приобретают свойства антикоагулянтов. Так, образующийся при свертывании крови фибрин адсорбирует и инактивирует большое количество тромбина, т. е. является как фактором свертывания, так и антикоагулянтом (антитромбин I). Продукты ферментного расщепления фибриногена/фибрина плазмином ингибируют как агрегацию тромбоцитов, так и самосборку фибрин-мономеров в фибрин-полимеры. Продукты фибринолиза обозначаются как антитромбин VI.
Помимо физиологических антикоагулянтов при ряде патологий в плазме могут накапливаться очень мощные ингибиторы свертывания крови, являющиеся специфическими антителами к различным факторам коагуляции. Чаще всего встречаются ингибиторы факторов VIII и IX.
При ряде аутоиммунных процессов и парапротеинемиях в крови могут накапливаться патологические белки, обладающие либо антитромбиновым действием (антитромбин V), либо ингибирующим влиянием на факторы Ха, II или V. Эти парапротеины часто блокируют адгезивно-агрегационную функцию тромбоцитов и одновременно повышают вязкость крови, вызывая выраженные расстройства микроциркуляции.
11 .4.1. Методы исследования антикоагулянтной системы
1. Определение антитромбина III (по Hensen, Loeliger, модификация К. М. Би-шевского).
В норме этот показатель составляет (111 ± ± 0,41) %.
2. Определение содержания гепарина.
В норме количество гепарина в крови равняется (7,4 ± 0,41) мг/л.
3. Определение толерантности плазмы к гепарину.
В норме этот показатель составляет (511,4 ± 4,4) с. Укорочение времени наблюдается при гиперкоагуляции и склонности к тромбозам. Значения этого показателя тесно связаны с количеством антитромбина III.
348 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
11.4.2. Препараты, воздействующие на антикоагулянтную систему
Гепарины. В настоящее время в распоряжении клиницистов имеются два класса гепаринов:
нефракционированные (стандартные), представляющие собой смесь гепаринов с молекулярной массой от 3000 до 30 000 D;
фракционированные (низкомолекулярные) с молекулярной массой от 1000 до 10 000 D.
Различия между гепаринами этих двух видов заключаются в механизме действия, био доступности, скорости элиминации и влиянии на тромбоциты.
Нефракционированный гепарин (НФГ). В основе антикоагулянтного действия НФГ лежит его способность при соединении с кофактором AT III блокировать ферментативную активность факторов свертывания крови Па, Ха и ХПа. Среди этих факторов наиболее чувствительны к инактивации НФГ тромбин (Па) и фактор Ха, причем чувствительность тромбина на порядок выше, чем Ха. Анти-Ха-активностью обладают фрагменты молекул массой меньше 5400 D. Содержание молекул с такой массой в НФГ — менее 5 %, что и обуславливает его невысокую ингибирующую активность по отношению к Ха-фактору.
К недостаткам НФГ следует отнести особенности его сложной и не до конца изученной фармакодинамики. Вследствие связывания гепарина с белками плазмы, эндотелием, макрофагами терапевтический (антитромботический) и побочный (геморрагический) эффекты прогнозируются с трудом, плохо коррелируют с дозой и уровнем гепарина в крови.
Подкожное введение гепарина может быть неэффективным из-за низкой и вариабельной биодоступности, в связи с чем необходимо проводить непрерывные внутривенные инфузии под систематическим контролем АЧТВ для коррекции его дозы. Однако АЧТВ не является совершенным показателем антикоагулянтной активности гепарина, поскольку отражает состояние лишь внутреннего механизма свертывания крови.
Невозможность путем лабораторного мониторинга значительно снизить риск развития геморрагических осложнений применения гепарина обусловлена тем, что его антигемостатический эффект в значительной мере связан с вызываемыми им тромбоцитопатией и повышением сосудистой проницаемости.
Суточная доза гепарина очень вариабельна и колеблется в среднем от 10 000 до 40 000 ЕД. Длительный прием препарата сопряжен с истощением AT III и повышением риска развития тромбоцитопении и остеопороза. Тромбоцитопения развивается у 5 —10 % пациентов при лечении гепарином свыше пяти дней и обусловлена образованием антител к рецепторам кровяных пластинок. В результате может возникать ДВС, часто с образованием артериальных и венозных тромбов, состоящих преимущественно из тромбоцитов и лейкоцитов с низким содержанием фибрина.
В случае передозировки гепарина вводится 5 % раствор протамин сульфата из расчета 1мл препарата на 1000 ЕД гепарина.
Узость терапевтического окна нефрак-ционированного гепарина и ограничения его применения побудили к поиску альтернатив. В результате ферментативной или химической деполимеризации высокомолекулярного гепарина были созданы его низкомолекулярные формы. Они существенно отличаются от нефракциони-рованного гепарина как по фармакодинамике, так и по механизму действия.
Низкомолекулярные гепарины (НМГ) — на др Опарин, эноксапарин, дальтепарин и др. Поскольку тромбин — ключевой фактор в системе гемостаза, его инактивация является основной задачей антикоагулянтной терапии. Однако в работах последних 20 — 30 лет доказано, что 5 ЕД фактора Ха более тромбогенны, чем 100 ЕД тромбина. Это объясняется тем, что при каскадном механизме активации свертывающих механизмов происходит неуклонное и очень интенсивное нарастание количества активированных факторов свертывания. Нейтрализация 32 ЕД фактора Ха равнозначна предупреждению образования 1600 ЕД тромбина, и для ней
Антикоагулянтная система 349
трализации этой дозы тромбина расходуется в 73 раза большая доза антикоагулянта.
Механизм действия НМГ заключается в выраженном ингибировании Ха-факто-ра за счет высокого содержания молекул с массой ниже критической (5400 D) и незначительном подавлении активности Па фактора. Молекулярные цепи массой более 5400 D лучше связываются с белками и теряют часть своей активности. Количество более длинных цепей (> 7500 D) варьирует у разных низкомолекулярных гепаринов, определяя их анти-Ха-активность.
С учетом того факта, что НМГ содержат молекулы, обладающие антикоагулянтным эффектом, составлены рекомендации по их применению: подкожное введение за 2 ч до операции с целью профилактики тромбозов.
В работах последних лет [9] содержатся сведения об открытии нового, ранее неизвестного механизма действия низкомолекулярных гепаринов на систему гемостаза: 70 % противотромботического эффекта препаратов этого класса обусловлено активацией ингибитора внешнего пути свертывания — TFPI-фактора, или липопротеин-ассоциированного ингибитора коагуляции (LACI-фактор), являющегося мощным естественным ингибитором внешнего пути свертывания. Низкомолекулярный гепарин способен повышать его уровень в крови почти на 500 %.
В генезе большинства тромботических явлений ведущая роль принадлежит активации внешнего механизма свертывания. Этот механизм преобладает во время беременности, в перинатальном, послеоперационном периодах, при гнойно-септических осложнениях, антифосфолипидном синдроме, тромбоэмболии легочной артерии (ТЭЛА), остром респираторном дистресс-синдроме (ОРДС), отслойке плаценты, эмболии околоплодными водами и т. д.
Преимущества низкомолекулярного гепарина по сравнению с высокомолекулярным:
♦ предсказуемость действия;
♦ высокая биодоступность (98 — 99 %);
♦ стойкость антитромботического эффекта при подкожном введении, что позво
ляет вводить препарат в фиксированных дозах, определяемых с учетом массы тела (1—2 раза в сутки без лабораторного контроля);
♦ значительно меньшая частота развития тромбоцитопении и остеопороза.
Антикоагулянты непрямого действия (АНД): фенилин, синкумар, неодикума-рин, пелентан, варфарин и др. Применяются перорально. Доза подбирается индивидуально под контролем протромбинового индекса. Механизм действия непрямых антикоагулянтов заключается в блокаде конечного этапа образования К-витаминзависимых факторов свертывания (VII, X, IX, II) в гепатоцитах. Гипокоагуляция развивается при снижении протромбинового индекса до 30 —40 %, более высокие цифры (снижение до 50 —60 %) не гарантируют того, что вторая фаза (переход протромбина в тромбин) не произойдет. Снижение протромбинового индекса ниже 40 — 30 % может осложниться профузным кровотечением. АНД являются препаратами выбора для профилактики и лечения тромбозов, развившихся вследствие дефицита AT III. При передозировке непрямых антикоагулянтов необходимо уменьшение дозы и применение викасола и фитоменадиона.
Противопоказания к назначению АНД: все заболевания, потенциально опасные в плане развития геморрагических осложнений (язвенная болезнь желудка и двенадцатиперстной кишки, цирроз печени, высокая гипертензия, миомы матки и др.). Препараты АНД обладают терратогенным эффектом на плод, поэтому не назначаются беременным женщинам.
В последние годы было выявлено, что АНД не только угнетают синтез К-вита-минзависимых прокоагулянтов, но и снижают активность важнейших физиологических антикоагулянтов — протеинов С и S. Их дефицит приводит к развитию тромбофилии. Если на фоне терапии АНД подавление активности антикоагулянтов более выражено, чем в прокоагулянтном звене, то могут возникнуть «рикошетные» тромбозы, обширные гангренозные пигментные некрозы кожи и другие тромботические осложнения [5].
350 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
11.5. ФИБРИНОЛИТИЧЕСКАЯ СИСТЕМА
Антиподом свертывающей системы крови является фибринолитическая система, осуществляющая асептическое растворение фибрина — фибринолиз. Последний препятствует распространению тромба по сосудистой системе от места его образования и обеспечивает лизис фибрина при его появлении в общей циркуляции, таким образом, поддерживая нормальное кровообращение.
Главным компонентом этой системы является фермент плазмин (фибринолизин), содержащийся в эуглобулиновой фракции плазмы и сыворотки в виде профермента (плазминогена, или профибринолизина). Плазминоген также обнаружен в других тканях организма, особенно высоко его содержание в соединительной ткани, предстательной железе, легких.
Фибринолитическая система, как и свертывающая, имеет два механизма активации — внутренний, осуществляемый ферментными системами самой крови, и внешний, запускаемый тканевыми активаторами.
Внутренняя активация фибринолиза обусловлена образованием комплекса Ха с калликреином и высокомолекулярным кининогеном в ответ на свертывание крови. Благодаря действию внутреннего фибринолитического механизма образующийся фибрин постоянно удаляется из кровотока.
Внешняя активация фибринолиза обусловлена поступлением в кровоток тканевых киназ, в основном из сосудистого эндотелия.
Активаторы фибринолиза содержатся во многих тканях и жидкостях, в том числе в клетках крови — эритроцитах, тромбоцитах, лейкоцитах. Кроме того, гранулы и макрофаги могут секретировать внутриклеточные киназы, которые сами по себе расщепляют фибрин.
Физиологическое значение фибринолиза велико, поскольку в результате не только устраняется фибрин из кровотока, но и образуются высокоактивные антикоагулянты и дезагреганты — продукты деградации фибрина.
Активаторы плазминогена разделяют на:
А. Естественные:
1) тканевые активаторы (тканевые киназы и фибринокиназы). Больше всего их обнаружено в легких и мозговой ткани;
2) активатор плазмы (плазменная киназа) и других биологических жидкостей;
3) урокиназа — активатор плазминогена, содержащийся в моче;
4) трипсин;
5) плазмин — аутокаталитический активатор плазминогена.
Б. Бактериального происхождения:
1) стрептокиназа — мощный активатор фибринолиза, продуцируется гемолитическими стрептококками групп А, С, G;
2) стафилокиназа — продуцируется отдельными штаммами стафилококков.
В зависимости от активатора различают быструю и замедленную активацию плазминогена.
Активированный плазминоген (плазмин) вызывает последовательное асимметричное расщепление фибриногена/фиб-рина на более мелкие фрагменты. Крупномолекулярные продукты фибринолиза (фрагменты X и Y) обозначают как «ранние», а фрагменты D и Е — как «поздние», или «конечные».
Повышенное содержание в крови ПДФ свидетельствует о том, что в ней активирован фибринолиз.
Кроме того, в организме существует система неферментативного фибринолиза, осуществляемого комплексными соединениями гепарина с гормонами (тироксином, адреналином, норадреналином) и со специфическими белками крови — фибриногеном, тромбином, плазминогеном. Неферментативный фибринолиз важен для поддержания жидкого состояния крови и предупреждения тромбообразования при стрессовых ситуациях.
11.5.1. Лабораторная диагностика нарушений фибринолитической системы
1. Определение спонтанной фибринолитической активности.
В норме спонтанный фибринолиз составляет (12,4 ± 0,28) %.
Фибринолитическая система 351
2. Определение уровня плазминогена, плазмина и антиплазмина.
В норме эти показатели соответственно равны: (424,4 ± 19,1)ФЕ; (46,1 ± 4,2)ФЕ; (233,2 ± 26,4)АФЕ.
3. Определение суммарной фибринолитической активности ферментативного и неферментативного фибринолиза.
В норме эти показатели соответственно равны: (18,1 ± 2,6) %; (13,8 ± 1,1) %.
4. Определение естественного лизиса фибринового сгустка.
5. Определение растворимых фибринмономерных комплексов (РФМК, продуктов паракоагуляции): этаноловый, протаминсульфатный, р-нафтоловый тест (фибриноген В).
Этаноловый тест будет положительным при наличии в исследуемой плазме комплексов фибрин-мономера с продуктами фибринолиза и фибриногеном, т. е. этот тест отражает не только тромбинемию, но и активацию фибринолиза.
В период выраженной гипофибриногенемии (менее 0,5 г/л) этаноловый тест может стать отрицательным, при значительной гиперфибриногенемии (свыше 6,0 г/л) - ложноположительным [7]. Хотя в настоящее время эти тесты не считаются достаточно информативными, они сохранены в комплексе методов исследования системы гемостаза в больничных лабораториях.
6. Для определения ранних продуктов деградации фибрина и количества растворимых фибрин-мономерных комплексов'.
в плазме — применяются орто-фенан-тролиновый и латекс-агглютинационный тесты;
в сыворотке — тест склеивания стафилококков.
11.5.2. Препараты, воздействующие на фибринолитическую активность
А. Препараты, активирующие фибринолиз.
1. Активаторы плазминогена:
стрептокиназа, или стрептаза К, — продукт жизнедеятельности бета-гемолитического стрептококка, обладает антиген
ными свойствами, поэтому лечение производится на фоне применения кортикостероидов. Непрямой фибринолитик.
Механизм действия:
1. Стимулирует перевод циркулирующего в крови проактиватора в активатор, который, в свою очередь, трансформирует плазминоген в плазмин.
2. Образует комплекс «стрептокиназа — плазминоген», активирующий остальные молекулы плазминогена в крови, способный проникать внутрь тромба и вызывать его лизис. Вводят внутривенно или внутриартериально. Действие стрептокиназы начинается спустя 30 — 60 мин. Из-за короткого периода полувыведения (около 30 мин) применяется в виде длительной инфузии. Прерывистое введение стрептокиназы с большими (часовыми) интервалами опасно. Как правило, препарат вводят в течение 16 —18 ч со скоростью 20 — 30 капель/мин. Суточная доза — от 500 000 до 1 500 000 ME. Длительность терапии при массивных тромбозах — 4 — 6 сут. Более длительное лечение нецелесообразно из-за выраженного увеличения количества антител и развития резистентности к стрептокиназе. Повторный курс рекомендуется проводить не ранее, чем через 3 — 6 мес. Он более опасен в связи с высокой возможностью развития аллергических реакций и резистентности к препарату;
стрептодеказа — пролонгированный препарат стрептокиназы. Однократное введение средней терапевтической дозы обеспечивает повышение фибринолитической активности крови в течение 48 —72 ч. Для предупреждения ретромбоза стрептодека-зу комбинируют с гепарином, который начинают вводить с конца первых суток после введения лечебной дозы фибринолитика;
урокиназа (укидан) — лишена антигенных свойств, аллергические реакции — большая редкость, в связи с чем возможно повторное введение препарата. Получают из культуры клеток почек человека, а также рекомбинантным путем. Урокиназа — прямой активатор фибринолиза: непосредственно активирует плазминоген и превращает его в плазмин. Препарат активирует фибринолиз как внутри тромба, так и на его поверхности;
352 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
рекомбинантный тканевой активатор плазминогена (актилизе) — гликопротеин (полный аналог эндогенного вещества, вырабатываемого эндотелием), который после системного введения находится в плазме в неактивной форме до момента связывания с фибрином. После активации препарата он способствует переходу плазминогена в плазмин и растворению фибринового сгустка, таким образом, повышая фибринолиз только в ткани тромба. Обладает малым фибриногенолитическим действием и по ряду параметров более эффективен, чем стрептокиназа [10]. Первое же рандомизированное контрольное исследование по использованию актилизе при остром ишемическом инсульте с помощью ангиографии продемонстрировало реперфузию у 21 % больных. Вводится внутривенно (1,1 мг/кг в течение часа). Рекомендуется его применять только в первые 3 — 6 ч после начала развития острого ишемического инсульта.
Лечение вышеперечисленными фибри-нолитиками противопоказано:
1. В течение первых 10 суток послеоперационного и послеродового периодов.
2. При артериальной гипертензии (180/ 100 мм рт. ст. и выше) с ретинопатией.
3. При краниотомии и острых церебральных процессах (кровоизлияния, опухоль).
4. При желудочно-кишечных и урогенитальных кровотечениях.
5. При геморрагических диатезах.
Фибринолизин (плазмин) — получают путем активации трипсином содержащегося в крови человека плазминогена. Вызывает только наружный лизис тромба (преимущественно в венах), так как быстро инактивируется антиплазмином.
Поскольку фибринолизин содержит примесь трипсина, он может вызвать активацию свертывающей системы крови, поэтому его вводят внутривенно капельно с 10 000 ЕД гепарина.
Перечисленные выше фибринолитики — вещества белковой природы — могут вызывать пирогенные и аллергические реакции. Геморрагические осложнения встречаются в 45 — 50 % случаев и частично обусловлены необходимостью сочета
ния фибринолитиков с гепарином. Поэтому при развитии геморрагических осложнений используют как ингибиторы фибринолиза (аминокапроновую кислоту, кон-трикал и др.), так и антагонист гепарина — протамин сульфат.
При лечении тромбоэмболий вероятность рецидива очень велика, поскольку происходит фрагментация тромбов в основном месте их образования;
никотиновая кислота (1—3 мг/кг в сутки), или компламин (5 — 30 мг/кг в сутки). Эти препараты активируют фибринолиз непрямым путем: под влиянием никотиновой кислоты и ее производных активаторы плазминогена освобождаются из стенок сосудов или лейкоцитов.
Б. Препараты, угнетающие фибринолиз:
антифибринолитики животного происхождения (трасилол, инипрол, гордокс, контрикал и др.).
Ингибиторы протеаз природного происхождения (трасилол, контрикал, гордокс и др.) получают либо из поджелудочной и околоушной желез, либо из легких крупного рогатого скота. Действующим началом всех этих препаратов является апро-тинин, который образует неактивные комплексы с протеолитическими ферментами: плазмином, трипсином, химотрипсином, кал-ликреином, кислыми гликопротеидами и липополисахаридами (в том числе и с гепарином). Активность апротинина (соответственно и различных коммерческих препаратов на его основе) измеряется в единицах ингибирования калликреина (ЕИК);
синтетические антифибринолитики (с-аминокапроновая кислота — АКК, парааминометилбензойная кислота, тра-нексамовая кислота и др Л’.
АКК вводится внутривенно капельно в виде 4 —5 % раствора в дозе 100 — 400 мл. Может вводиться перорально. Действие АКК заключается в ингибировании перехода плазминогена в плазмин, она повышает коагуляционную способность крови, увеличивает потребление протромбина, агрегационную способность тромбоцитов и повышает активность фактора VIII.
Транексамовая кислота (экзацил, Тгап-samcha) ингибирует действие активатора
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 353
плазминогена и плазмина, обладает гемостатическим действием при кровотечениях, связанных с повышением фибринолиза, а также противоаллергическим и противовоспалительным действием за счет подавления образования кининов и других активных пептидов, участвующих в аллергических и воспалительных реакциях. Показанием к применению препарата являются кровотечения на фоне усиления фибринолиза (послеродовые кровотечения, ручное отделение последа, заболевания печени, маточные, носовые, желудочно-кишечные кровотечения, гематурия и т. д.).
При генерализованном фибринолизе вводят внутривенно в разовой дозе 15 мг/кг массы тела в изотоническом растворе натрий хлорида или глюкозы. Скорость введения — 1 мл/мин. При профузном маточном кровотечении назначают внутрь по 1 — 1,5 г 3 — 4 раза в день в течение 3 — 4 сут. При носовом кровотечении раствор для инъекций применяют местно путем закапывания или тампонирования носовых ходов марлей, смоченной раствором.
Препарат применяют с осторожностью у больных с тромбогеморрагическими осложнениями.
11.6. ЛОГИКА ОБСЛЕДОВАНИЯ СИСТЕМЫ ГЕМОСТАЗА
Обследование системы гемостаза рекомендуют проводить в два этапа.
Первый представляет собой набор скрининговых, или ориентировочных тестов, позволяющих установить факт наличия нарушений в системе гемостаза и определить дальнейшую тактику обследования пациента. К лабораторным тестам первого этапа относят:
1. Подсчет количества тромбоцитов и определение длительности кровотечения (оценка сосудисто-тромбоцитарного гемостаза).
2. Определение времени свертывания методом рекальцификации (контроль всего каскада свертывания).
3. Определение активированного частичного тромбопластинового времени (оценка внутреннего пути активации системы гемостаза).
4. Определение протромбинового времени (оценка внешнего пути активации системы гемостаза).
5. Определение тромбинового времени или концентрации фибриногена (оценка общего пути с момента активации Х-фактора).
Скрининговые тесты не обладают высокой чувствительностью, однако при наличии клинических проявлений коагулопатии отклонения от нормальных величин в этой группе исследований выявляются практически в 100 % случаев.
Комплекс лабораторных исследований второго этапа (уточняющего) определяется на основании анализа отклонений, выявленных на первом этапе обследования системы гемостаза. Так, например, удлинение АЧТВ и ТВ могут быть связаны с: 1) наличием гепарина; 2) заболеваниями печени; 3) гиперфибринолизом, гипо- и дисфибриногенемией. Удлинение ПВ и АЧТВ при нормальном тромбиновом времени возможно при: 1) дефиците витамина К и заболеваниях печени; 2) приеме антикоагулянтов непрямого действия; 3) дефиците факторов V, X, II, VII [10]. Выбор дальнейших уточняющих тестов при каждом варианте для конкретного пациента должен быть индивидуальным. Подобный подход к обследованию системы гемостаза позволяет экономить время и средства лаборатории.
11.7. НЕКОТОРЫЕ ВИДЫ НАРУШЕНИИ В СИСТЕМЕ ГЕМОСТАЗА В ПРАКТИКЕ ИНТЕНСИВНОЙ ТЕРАПИИ
Приобретенные патологические изменения в системе гемостаза многообразны и касаются всех его звеньев: сосудисто-тромбоцитарного, ферментативного, антикоагу-12 4-220
лянтного и фибринолитического. При этом различают такие варианты клинических проявлений: 1) тромботический; 2) геморрагический; 3) смешанный (тромбо-гемор
354 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
рагический). К наиболее распространенным в клинике интенсивной терапии патологическим синдромам системы гемостаза принадлежат синдром диссеминированного внутрисосудистого свертывания крови (ДВС-синдром) и приобретенные тромбофилии различного генеза.
11.7.1. Синдром диссеминированного внутрисосудистого свертывания крови
ДВС-синдром является неспецифическим общепатологическим процессом, сопровождающим течение практически всех критических и терминальных состояний. Этот сложный патологический процесс в системе свертывания крови развивается при многих заболеваниях, являясь вторичным по отношению к ним. Суть его заключается в развитии рассеянного свертывания крови в сосудистом русле с образованием огромного количества микросгустков и агрегатов клеток крови, что имеет особое значение для системы микроциркуляции, где они блокируют органное и тканевое кровообращение, вызывая глубокие дистрофические изменения. Расходование (потребление) факторов свертывания, тромбоцитов и других клеток в процессе тромбообразования, активация протеолиза и его частного случая — фибринолиза, приводят к образованию продуктов деградации фибриногена и фибрина, блокирующих процесс свертывания, вызывают нарушение процесса гемокоагуляции и патологическую кровоточивость.
В настоящее время существует множество определений ДВС-синдрома, сильно отличающихся друг от друга. В одних из них отражен лишь конечный этап процесса — тромбообразование в зоне микроциркуляции, в других — предпринята попытка перечислить все основные механизмы развития синдрома, из-за чего определения становятся громоздкими и трудновоспроизводимыми. В наиболее часто встречаемых определениях под ДВС подразумевается динамический патологический процесс, обусловленный активацией системы гемокоагуляции с образованием микротромбов, активизацией фибринолитической системы, потреблением и исто
щением факторов свертывания крови и фибринолиза. Таким образом, главной составляющей этого процесса является активизация прокоагулянтного и фибринолитического звеньев. О тесной взаимосвязи этих двух противоположных процессов свидетельствует и общий пусковой механизм.
Для отражения патофизиологической сути происходящего предложено большое количество терминов, которые нельзя признать равнозначными: синдром диссеминированного внутрисосудистого свертывания, тромбогеморрагический синдром, коагулопатия потребления, синдром внутрисосудистого свертывания крови, синдром дефибринации и др. [7].
Течение ДВС-синдрома может быть острым, подострым, молниеносным, затяжным, рецидивирующим, хроническим и латентным. Наиболее неблагоприятные последствия диссеминированного внутрисосудистого свертывания крови отмечаются при его остром течении.
Острое течение ДВС-синдрома чаще всего наблюдается при различных видах шока, критических состояниях в акушерстве, злокачественных новообразованиях, переломах, политравме, травматичных хирургических вмешательствах, ожогах, обморожениях, черепно-мозговой травме, геморрагических инсультах, гнойно-септических заболеваниях, укусах ядовитых змей, остром внутрисосудистом гемолизе и цитолизе, массивных гемотрансфузиях, аллергических реакциях, трансплантации органов и тканей и т. д. [7, 11 — 13].
Причины развития ДВС-синдрома. Пусковым механизмом развития ДВС-синдрома может быть первичная активация любого из звеньев сосудисто-тромбоцитарного или ферментативного гемостаза. Иногда механизм активации носит смешанный характер, вследствие чего процессы запуска обеих систем происходят одновременно, взаимно усиливая интенсивность друг друга.
При активации свертывающей системы происходит вторичная и почти одновременная активация фибринолитической системы. Последняя в данных условиях носит защитный характер и направлена про
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 355
тив диссеминированного микротромбо-образования [3].
В норме система гемокоагуляции состоит из факторов свертывания, находящихся в крови в неактивном виде, а также из противосвертывающих веществ, циркулирующих в активном состоянии, при этом запас прокоагулянтных факторов многократно превышает антикоагулянтный [4]. Возникающие локально незначительные изменения гемостатического потенциала в целом не отражаются на функционировании системы регуляции агрегатного состояния крови (РАСК) и по принципу обратной связи обеспечивают «управляемость» появившегося отклонения [3].
Реализация множества причинных факторов в процессе диссеминированного тромбообразования возможна лишь при наличии особых условий [7]. Главным из них является интенсивное или длительное активирование коагуляционного потенциала, приводящее к истощению и срыву противосвертывающих механизмов, прежде всего антитромбина III и протеина С. Таким образом, развитие ДВС-син-дрома следует рассматривать прежде всего как свидетельство истощения антикоагулянтной системы. Вследствие этого происходит свертывание крови, преимущественно в зоне микроциркуляции, активирование фибринолиза, системы мононуклеарных фагоцитов (СМФ), кал-ликреин-кининовой системы, изменение гемодинамики, pH крови.
Таким образом, установление самого факта активации системы гемостаза позволяет понять, в каком звене этой системы наиболее выражен тромбогенный сдвиг, и даже на ранних этапах разработать возможные пути коррекции выявленных нарушений. Поскольку тромбоопасная ситуация может быть обусловлена первичной активацией как тромбоцитарного, так и плазменного звена гемостаза, следует контролировать маркеры активации обеих систем.
Маркером активации тромбоцитарного гемостаза служит повышение: ♦ спонтанной агрегации тромбоцитов; ♦ индуцированной агонистами агрегации тромбоцитов;
♦ концентрации в плазме компонентов а-гранул тромбоцитов (свидетельство реакции высвобождения) — антиге-паринового фактора (фактора 4) или Р-тромбогл обул ина.
Поскольку активация свертывающей и фибринолитической систем крови в подавляющем большинстве случаев происходит сопряженно, то в плазме крови одновременно возрастает как количество продуктов протеолиза, так и комплексных соединений, свидетельствующих о тромбинемии [5].
Маркерами активации ферментативного гемостаза и фибринолиза являются: ♦ растворимые фибрин-мономерные комплексы (положительные паракоагуляционные тесты подтверждают тромбинемию);
♦ продукты деградации фибриногена и фибрина.
Современная концепция развития дисфункции в системе РАСК предусматривает проникновение в кровоток экзогенных или эндогенных активаторов развития ДВС-синдрома.
Эндогенными активаторами ДВС-синдрома могут быть: тканевой тромбопластин, тромбопластические субстанции, тканевые протеазы, продукты распада клеток и тканей, трипсинемия. К экзогенным факторам относятся: бактериальные эндо- и экзотоксины, обладающие свойствами агрегировать эритроциты, тромбоциты и повреждать эндотелий, яд некоторых змей и насекомых и др.
При различных критических состояниях имеет место сочетанное действие различных этиологических и патогенетических факторов, ответственных за развитие ДВС-синдрома. Реоксигенация и рециркуляция в организме, перенесшем выраженное нарушение системного кровообращения (длительная гипотензия, асистолия), тяжелая гипоксия вызывают ряд новых патофизиологических изменений, среди которых важное место занимает дисфункция системы РАСК.
При гемолитических и цитолитических процессах в кровоток выделяется большое количество внутриклеточного тромбопластина, содержащего фосфолипиды, активирующие внутренний механизм свертывания крови.
12*
356 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
Патогенез ДВС дифференцируется в зависимости от вызвавшей его причины, в связи с чем выделяют такие основные его звенья [3]:
1. Первичное поражение сосудистой стенки, т. е. снижение тромборезистент-ности эндотелия микрососудов. Это звено может быть обусловлено повреждающим действием на эндотелий:
а) инфекционного фактора (вирусов, риккетсий, бактериальных эндотоксинов);
б) неинфекционного агента (активация системы комплемента, образование и фиксация иммунных комплексов, провоспали-тельные цитокины).
Сосудистая стенка играет важную, а иногда и определяющую роль в возникновении и развитии ДВС-синдрома, поскольку она является не только обязательным компонентом гемостаза, но и регулятором процесса свертывания крови и фибринолиза. Однако ее участие в патологическом процессе неоднозначно: генерализованное поражение сосудов может служить пусковым механизмом внутрисосудистой коагуляции или быть вторичным при массивном поступлении тромбопластина и его активаторов, нарушениях реологических свойств крови, повреждении тромбоцитов. Для ангиопатий при синдроме ДВС характерны дистрофические, некротические и пролиферативные изменения. Они представляют собой важный этап в патогенезе внутрисосудистой коагуляции, поддерживая и усугубляя ее [14].
Рассматривая ДВС-синдром как результат дистресс-повреждения системы РАСК, следует остановиться на вопросе взаимосвязи этого синдрома с синдромом системного воспалительного ответа (ССВО).
ССВО — это генерализованная реакция организма на воздействие неспецифического фактора агрессии инфекционной или неинфекционной природы. Первичные модуляторы ССВО — цитокины — гормоноподобные медиаторы, синтезируемые и секретируемые активированными лимфоцитами, моноцитами/макрофагами и другими клетками иммунной системы. Цитокины, активируясь по каскадному механизму, при достаточной силе и продолжи
тельности действия стрессового раздражителя, вызывают быстрый переход локальной реакции защитного характера в деструктивный процесс на уровне целого организма. Характерной особенностью этих иммуномодуляторов является мультифункциональность и избыточность действия [12].
В зависимости от преобладающего эффекта различают провоспалительные и антивоспалительные цитокины. К первым относятся: TNFa, интерлейкины — ИЛ-1, ИЛ-2, ИЛ-8. Ведущая роль в запуске цитокинового каскада принадлежит TNFa и ИЛ-1.
Особое значение для системы РАСК имеет функциональная активность ИЛ-6. Он обладает доминирующим влиянием на стимуляцию синтеза острофазовых белков (С-реактивного белка, комплемента С3, фибриногена). Первоначально ИЛ-6 относили к провоспалительным цитокинам, позже были установлены также его антивоспалительные эффекты. Кроме того, ИЛ-6 стимулирует образование тромбоцитов и развитие реактивного тромбоцитоза [14].
Главным объектом токсических эффектов цитокинов является эндотелий сосудов. В результате их действия активируются циклооксигеназный и липооксигеназ-ный пути метаболизма арахидоновой кислоты, NO-синтетаза. Резкое увеличение продукции оксида азота приводит к периферической вазодилатации и развитию относительной гиповолемии. Активация процессов пероксидного окисления липидов сопровождается накоплением кислородных радикалов и снижением антиоксидантной активности крови. Под влиянием цитокинов и других активированных ими медиаторов воспаления (PAF, эйкозаноидов, NO, гистамина, серотонина и др.) увеличивается проницаемость сосудистой стенки, повреждаются эндотелиоциты. Выход жидкой части крови за пределы сосудистого русла способствует нарушениям реологических свойств и тканевой перфузии, активации воспаления в сосудистой стенке и в окружающих тканях. Развивается цитокин-индуцированный васкулит.
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 357
Генерализованное повреждение эндотелия стимулирует закрытие дефектов тромбоцитами, что приводит к утрате антикоагулянтных свойств внутренней поверхности сосудистой стенки. PAF вызывает быструю агрегацию тромбоцитов, действуя независимо от циклооксигеназного метаболизма арахидоновой кислоты.
В результате «цитокинового взрыва» возникают цитолитические реакции, включая лизис эритроцитов, лейкоцитов и микробных клеток. Этот процесс поставляет в кровоток активный тромбопластин, запускающий первичный гемостаз и ферментативную коагуляцию. На агрегатах форменных элементов оседает фибрин, в просвете сосудов микроциркуляторно-го русла появляются тромбы. Таким образом, на течение ССВО наслаиваются патогенетические механизмы ДВС-синдрома.
Некоторые авторы [15, 16] рассматривают ДВС-синдром как частое осложнение ССВО и предиктор полиорганной недостаточности. Следовательно, коррекцию изменений гомеостаза, обусловленную ССВО, можно считать профилактикой ДВС-синдрома и ассоциированных с ним органных дисфункций.
2. Первичное поступление в кровоток прокоагулянтов. Этот механизм развития ДВС-синдрома является ведущим при попадании в кровоток больших количеств тромбопластина и активации каскада гемокоагуляции по внешнему пути.
Поступление тромбопластина в сосудистое русло может быть связано с массивным разможжением мягких тканей, паренхиматозных органов, травматичными хирургическими вмешательствами, длительным раздавливанием тканей и позиционным сдавлением, обширными некрозами и деструкцией (трипсинемия при панкрео-некрозе), метастазирующим раком, ожогами, эмболией околоплодными водами, укусами некоторых змей и т. п.
Сепсис, аллергические и анафилактические реакции, сопровождающиеся интенсивным выбросом протеаз, активацией каллик-реин-кининовой системы, также обусловливают проникновение тромбопластина в сосудистое русло.
3. Первичное воздействие на тромбоциты. Инициация развития ДВС-синдрома подобным способом возможна при воздействии на тромбоциты сильных индукторов агрегации (высоких доз АДФ, некоторых медикаментов), циркуляции в крови комплексов антиген — антитело при реакции отторжения трансплантата.
4. Формы смешанного патогенеза. Многофакторный механизм развития ДВС-синдрома — наиболее часто встречающийся в клинической практике. К патологическим состояниям, на фоне которых возможно возникновение ДВС смешанного патогенеза, относят шоки, сепсис, острую форму лейкоза, экстракорпоральное кровообращение.
По мнению 3. С. Баркагана [5], в зависимости от патогенеза можно выделить лишь две формы ДВС-синдрома: инфекционно-септическую и первично асептическую, при этом явный приоритет имеет первая форма и частые трансформации второго варианта в первый. Целесообразность подобного деления объясняется автором возможностью рационального подхода к терапии больных, находящихся в критических состояниях: раннее назначение внутривенного введения антибактериальных препаратов позволяет предупредить развитие ДВС или уменьшить интенсивность его проявлений.
Стадии ДВС-синдрома. В настоящее время не существует общепринятой классификации стадий ДВС-синдрома. Большинство специалистов склонны выделять четыре фазы течения, однако интерпретация их различными авторами не всегда совпадает [5, И, 14, 17, 18]. С нашей точки зрения, наиболее удобной для практики является классификация, встречающаяся в работах 3. С. Баркагана [5]:
I стадия — гиперкоагуляция и агрегация тромбоцитов, активация других плазменных ферментных систем (калли-креин-кининовой, комплемента), формирование блокады микроциркуляции в органах;
II стадия — переходная, с нарастающей коагулопатией и тромбоцитопенией, разнонаправленными сдвигами в общих коагуляционных тестах;
358 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
III стадия — глубокой гипокоагуляции (вплоть до полной несвертываемости крови);
IV стадия — восстановительная (при неблагоприятном течении — фаза исходов и осложнений).
Патогенетическая характеристика стадий ДВС-синдрома:
I стадия (гиперкоагуляции).
В результате изложенных выше механизмов независимо от характера пускового фактора в кровотоке появляется активированный фактор X, что приводит к стойкой тромбинемии. Некоторые протеазы (стафилокоагулаза и др.) вызывают тромбинемию, минуя стадию активации тромбокиназной активности [17].
Резкое усиление генерации тромбина сопровождается одновременной активацией противосвертывающих (антикоагулянтных и фибринолитических) механизмов. В этой ситуации уровень тромбинемии является катализатором образования комплекса гепарин — антитромбин III. Однако несовместимая с возможностями этого комплекса нагрузка приводит к истощению антикоагулянтной системы, прежде всего за счет быстрого потребления антитромбина III.
Фибрин начинает откладываться в сосудистом русле только после того, как 25 % его количества образуют фибрин-мономерные комплексы. Возникают условия для формирования микротромбов, вначале пристеночных, затем — обтурирующих. Появление в капиллярном русле микросвертков крови усиливает активацию фибринолитической системы. В результате лизиса и утилизации тромбов в крови накапливаются высокие концентрации патологических антикоагулянтов — ПДФ. В этот период тесты коагулограммы имеют одну гиперкоагуляционную направленность. Продолжающееся потребление факторов приводит к развитию второй стадии.
II стадия (переходная).
Характеризуется истощением механизмов свертывания, накоплением в крови патологических ингибиторов гемокоагуляции и агрегации, выраженным истощением физиологических противосвертывающих механизмов — AT III, который быстро
расходуется на инактивацию тромбина и других факторов свертывания, и трансформировавшегося в плазмин плазминогена. Для коагулогических тестов этого периода характерна патогномоничная разнона-правленность гемостатических потенциалов. Гиперкоагуляционный сдвиг поддерживается циркуляцией в крови высоких концентраций тромбина на фоне истощения депо физиологических антикоагулянтов и при наличии достаточного уровня прокоагулянтов. Потребление прокоагулянтов может быть отсрочено во времени (особенно при подострых и хронических формах ДВС-синдрома) благодаря их изначально значительному количественному превосходству по сравнению с противосвертывающими факторами.
При острой форме течения ДВС-синдрома первые две стадии очень скоротечны и редко диагностируются, поскольку быстро трансформируются в третью стадию.
III стадия (глубокой гипокоагуляции).
Характеризуется качественно новым состоянием системы РАСК, суть которого заключается в доминирующем влиянии на результирующий гемостатический потенциал циркулирующих ПДФ на фоне потребления и функциональной неполноценности как свертывающих, так и противосвертывающих факторов.
Немаловажную роль в усугублении ко-агулопатических расстройств играет ограничение возможности системы мононукле-арных фагоцитов по утилизации ПДФ при резком увеличении нагрузки.
IV стадия (восстановительная или при неблагоприятном течении — фаза исходов и осложнений).
Неблагоприятное течение ДВС-синдрома может быть обусловлено рядом причин:
1. Острая форма синдрома с катастрофически быстро нарастающей коагулопатией вызывает наиболее глубокие нарушения в системе микроциркуляции органов, создавая условия для формирования полиорганной дисфункции и недостаточности.
2. Несвоевременно начатая и нерациональная по составу и объему инфузионнотрансфузионная терапия может не только
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 359
усугублять тяжесть течения ДВС-синдро-ма, но и быть первопричиной его развития при лечении критических состояний различного генеза.
3. Характер основного заболевания, на фоне которого разворачивается ДВС-синдром, может предопределить форму его течения. Например, для эмболии околоплодными водами, преждевременной отслойке плаценты типична острая форма, для тяжелого сепсиса — острая и подострая, для системных коллагенозов — подострая и хроническая формы. При неблагоприятном течении основное заболевание и ДВС-синдром по принципу взаимного отягощения быстро вызывают декомпенсацию друг друга, трансформируя процессы патогенеза в механизмы танатогенеза.
Особого внимания требует рассмотрение условий, в которых функционирует фибринолитическая система. При повышении гемостатического потенциала активация фибринолиза носит вторичный характер и направлена на восстановление проходимости сосудов микроциркуляции. В асептических условиях и при незапредельном для возможностей фибринолитической системы микротромбообразовании активация перехода плазминогена в плазмин не утрачивает своих защитных свойств. Совершенно иные эффекты наблюдаются при наличии инфицированных микротромбов. Их лизис чреват развитием новой волны осложнений для септических больных в виде циркуляторно-метаболических расстройств (шока) и метастазирования гнойных очагов. Свойство фибринолитических агентов повышать сосудистую проницаемость имеет не последнее значение для этих процессов.
Опыт показывает, что данная классификация стадий слишком схематична и ограничена, применима лишь к острой форме ДВС-синдрома, не учитывает многих важных характеристик для правильного ведения больных. Так, хронический ДВС-синдром может протекать с длительной гиперкоагуляцией и рецидивирующими тромбозами вен или волнообразно, с чередованием этих сдвигов и потенциальной возможностью внезапного перехода в тяжелый острый ДВС-синдром.
Поэтому предпочтительнее ориентироваться на форму течения ДВС-синдрома, поскольку именно она обусловливает скорость и характер изменений в системе гемостаза.
Клиническая картина ДВС-синдрома. Складывается из симптомов основного заболевания, послужившего первопричиной его развития, глубины повреждения всех звеньев гемостаза, степени гиповолемии и анемии, дисфункции и дистрофических нарушений в органах, выраженности метаболических сдвигов.
К клиническим симптомам острой и подострой форм ДВС-синдрома можно отнести такие [13]:
1) геморрагические проявления (кожные петехиальные кровоизлияния, гематомы в месте инъекций, массивные кровотечения из полости матки, мочевого пузыря, прямой кишки, полости носа, пищеварительного канала и др.);
2) тромботические проявления (некроз кожи в области кончика носа и мочки уха, внезапная ишемия конечностей, инфаркт-пневмонии, тромбозы мезентериальных сосудов);
3) нарушение функции внешнего дыхания (одышка, акроцианоз, снижение ар-териализации крови и артерио-венозной разницы по кислороду вследствие поражения легких как «первого фильтра» ми-кротромбоэмболами);
4) нарушение функции почек (олигурия и анурия);
5) нарушение функции центральной нервной системы (эйфория, отсутствие критической оценки окружающего, дезориентация, оглушенность, вплоть до глубокого помрачения сознания и комы);
6) внутрисосудистый гемолиз;
7) развитие политопного геморрагического или геморрагически-некротического синдрома с анемизацией и углублением циркуляторных и метаболических нарушений, а также явлений интоксикации продуктами протеолиза, цитолиза и липолиза.
Клинические нарушения, обусловленные хроническим течением ДВС-синдрома, часто остаются нераспознанными ввиду огромной приспособляемости организма и боль
360 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
ших функциональных возможностей жизненно важных органов. При хронической форме течения ДВС-синдрома на первое место выступают нарушения функции отдельных органов. Идентифицировать клинически хронические формы течения ДВС-синдрома без специальных исследований системы гемостаза очень сложно. Для хронически текущего ДВС-синдрома характерны тромбоцитопения и разнонаправлен-ность коагулогических тестов. В этих случаях важное диагностическое значение имеют результаты радиоиндикационных проб с меченными тромбоцитами и фибриногеном, указывающие на ускорение катаболизма фибриногена и укорочение продолжительности жизни тромбоцитов в связи с их хроническим потреблением; появление в плазме промежуточных продуктов превращения фибриногена в фибрин — растворяемых фибрин-мономерных комплексов, а также ПДФ.
Диагностика ДВС-синдрома сложна и трудоемка. Используя общедоступные и общепринятые способы можно поставить диагноз «ДВС» при наличии шока, тромбозов, геморрагий, острой легочной и почечной недостаточности, глубоких нарушений свертываемости крови. Но это будет поздняя и нередко уже бесполезная диагностика.
Помимо клинических симптомов решающее значение в диагностике форм и фаз синдрома ДВС имеет исследование системы гемостаза.
Лабораторная диагностика ДВС-синдрома. Поскольку ДВС-синдром определяется конечным эффектом действия двух ключевых для его патогенеза ферментов — тромбина и плазмина, то процесс диагностики должен включать тесты, отражающие результаты их активности. Лабораторная диагностика состоит из трех групп исследований, исходя из поставленных задач:
1. Скрининговые экспресс-исследования заключаются в определении:
количества тромбоцитов (характерна тромбоцитопения);
АЧТВ;
протромбинового времени; тромбинового времени.
2. Тесты паракоагуляции и ПДФ, подтверждающие действие тромбина и/или плазмина (положительные ортофенантролиновый тест и тест склеивания стафилококков).
3. Дополнительные тесты: определение времени лизиса эуглобулинов, наличия факторов V и VIII, антитромбина III, плазминогена.
В экстренных случаях степень нарушений системы гемостаза определяют с помощью электрокоагулографии или тромбоэластографии .
Существуют более чувствительные, но трудоемкие или дорогостоящие методики, не нашедшие пока широкого применения в клинике. Это определение низкомолекулярных «поздних» фрагментов D и Е, D-D-димера, фибринопептида А, моноклональных антител к отдельным участкам молекул фибриногена и фибрина, маркера клеточных фосфолипидных мембран [5].
В зависимости от степени выраженности изменения одних показателей могут быть значительными, других — незначительными или даже находиться в пределах нормы. Но даже в таких случаях изменения лишь двух-трех показателей не свидетельствует о наличии ДВС-синдрома. Это может быть объяснено выраженностью защитных сил организма, когда они еще не подавлены и способны сопротивляться возникновению патологического процесса. В 15% случаев у больных с острой формой ДВС наблюдается минимальное повышение ПДФ или их нормальное количество.
В 1988 г. в Японии были опубликованы стандарты по диагностике ДВС-синдрома [15]. Протокол включает учет этиологического фактора, клинических симптомов и минимума лабораторных исследований, позволяющих идентифицировать нарушения в системе гемостаза как ДВС-синдром. Эти данные оцениваются по балльной шкале повреждений (maximum = 13, minimum = 0). Если сумма баллов равна 7 и более, выявленные нарушения расцениваются как ДВС-синдром.
К изменениям в системе гемостаза, характеризующим наличие ДВС-синдрома,
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 361
относятся: тромбоцитопения, снижение концентрации компонентов коагуляционного звена, повышение количества ПДФ, снижение уровня предсуществующих антикоагулянтов и истощение депо плазминогена.
Для эффективного лечения больных важна динамичность исследования системы гемостаза, поскольку она выявляет доклиническую фазу процесса, позволяет оценивать динамику его прогрессирования и дает возможность вовремя предупредить внезапное обострение.
Таким образом, проанализировав данные важнейших лабораторных диагностических критериев, полученных у больных с острым, подострым и хроническим ДВС-синдромом, можно сделать вывод о том, что диагноз «ДВС-синдром» должен устанавливаться не на основании результатов отдельных тестов, патогномоничных для синдрома ДВС, а по интегральным показателям нескольких лабораторных тестов.
Лечение ДВС-синдрома. ДВС-синдром является лишь промежуточным звеном в эволюции других патологических процессов. Поэтому и терапия его должна быть комплексной — как этиотропной, так и патогенетической, причем последняя не может ограничиваться только воздействием на свертывающую систему крови, а должна быть направлена на устранение других звеньев его патогенеза — лечение шока, гипоксии, нарушений кислотноосновного состояния, водного и электролитного дисбаланса, восстановление функции «шоковых» органов, купирование цитолиза и протеолиза, восстановление структуры и функции микрососудов, смягчение явлений интоксикации и т. д.
«Классическая» стадийность ДВС-синдрома, обусловленного критическими состояниями и реанимацией, как правило, отсутствует. Поскольку наблюдается различная степень активации, напряжения и истощения звеньев, составляющих систему гемостаза, коррекция нарушений сводится к комплексному «посиндромному» воздействию на отдельные компоненты системы РАСК и восстановление баланса между ними.
Стратегию терапии ДВС-синдрома определяют:
1. Выраженность истощения антикоагулянтной системы крови.
2. Степень потребления прокоагулянтов.
3. Наличие в крови активаторов процесса гемокоагуляции.
4. Выраженность нарушений со стороны сосудисто-тромбоцитарного гемостаза.
5. Соотношение на момент коррекции активности различных составляющих системы гемостаза с учетом характера (геморрагического или тромботического) повреждения органов.
В настоящее время оптимально сбалансированным источником утраченных в процессе диссеминированного свертывания крови про- и антикоагулянтов считается свежезамороженная плазма (СЗП). Существуют различные рекомендации по объему и темпу введения СЗП. Метод проведения заместительной терапии, разработанный в 70-х годах прошлого века 3. С. Баркаганом с соавт., предполагает по возможности более раннее и быстрое внутривенное введение свежезамороженной плазмы (в 2 — 3 приема до 1 —2 л/сут) на фоне капельных инфузий реополиглюки-на как дезагреганта — в дозе 400 мл со скоростью 15 — 25 капель в минуту и в другую вену — гепарина — по 5000 ЕД на каждые 300 — 500 мл криоплазмы. При ДВС-синдромах, протекающих с профузными кровотечениями, доза гепарина должна быть уменьшена до 2500 ЕД на дозу плазмы. В. Г. Лычев [7] рекомендует вводить часть общей дозы гепарина (0,1—0,25 ЕД/мл) непосредственно в переливаемую плазму, осуществляя тем самым немедленную активацию комплекса гепарин —антитромбин III, что позволит снизить общую дозу трансфузии СЗП.
Основная цель применения этого лечебного комплекса состоит не в возмещении объема циркулирующей крови, а в том, чтобы восстановить гемостатический потенциал крови больного путем уравновешивания соотношения факторов свертывания крови и антикоагулянтов, протеаз и антипротеаз, компонентов калликреин-кинино-вой и фибринолитической систем с их
362 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
ингибиторами [3, И]. Свежезамороженная плазма является также источником протеина С — антикоагулянта, защищающего организм от патогенного действия кишечной палочки и бактериального эндотоксина.
Если использование СЗП на любой стадии ДВС-синдрома не вызывает споров, то вопрос применения гепаринов, согласно литературным данным, решается не столь однозначно. Теоретически гепарин должен составлять основу терапии ДВС-синдрома, так как вследствие блокады тромбинемии он предотвращает дальнейшее потребление факторов гемостаза [5, 11, 19]. С этой целью применение гепарина показано в I —III стадиях синдрома, поскольку в I стадии отмечается реальная гиперкоагуляция, а во II и III — потенциальная. Суточная доза гепарина — 10 000 — 20 000 ЕД и более в зависимости от тяжести синдрома. Этот препарат применяется внутривенно капельно в дозе 500—1000 ЕД/ч, а затем — подкожно в дозе, подобранной по результатам коагулограммы. Однако на практике при выраженной гипокоагуляции от введения препаратов гепарина воздерживаются. В частности, В. И. Кулаков с соавт. [17] настаивают на категорическом отказе от гепарина при лечении акушерских кровотечений, развившихся на фоне коагулопатии.
Учитывая опасность развития медикаментозно обусловленных геморрагических осложнений, предпочтительными являются НМГ. Эти препараты обладают преимущественно антитромботичес-ким, а не антикоагулянтным эффектом, следовательно риск кровотечений при их применении ниже, чем у НФГ. У больных с тромбогеморрагическим синдромом, синдромом ДВС, сепсисом, токсикоин-фекционным шоком перспективна замена использования гепарина фраксипари-ном в средней дозе 0,6 мл/сут, затем дозу снижают до 0,3 мл/сут.
Заместительная терапия предусматривает также возмещение утраченных факторов сосудисто-тромбоцитарного гемостаза. При выраженной гипокоагуляции и геморрагиях обязательно проводятся
трансфузии концентратов тромбоцитов, тромбоплазмы или тромбоцитарной массы. При снижении агрегационной функции тромбоцитов применяют натрий этам-зилат (дицинон) — от 4 до 12 мл 12,5 % раствора в сутки.
При проведении заместительной терапии абсолютно противопоказано внутривенное введение фибриногена и препаратов сухой плазмы, поскольку они не сбалансированы с антикоагулянтами и усиливают блокаду микроциркуляции, повышают вязкость крови.
Применение ингибиторов протеолиза. При выраженной гипокоагуляции применяют внутривенные инфузии больших доз антипротеаз широкого спектра действия, предпочтительнее — применение естественных ингибиторов протеолиза (апро-тинина, контрика ла, гордокса, трасилола и др.). Доза вводимого препарата по ап-ротинину от 50 000 до 1 000 000 ЕД в сутки дробно. Препараты апротинина способствуют блокаде не только плазменного, но и лейкоцитарного протеолиза, ослаблению интоксикации, сохранению функции тромбоцитов и купированию неконтролируемых кровотечений. В процессе лечения этими препаратами необходим контроль за состоянием фибринолитической системы.
Применение искусственных ингибиторов протеолиза (фибринолиза) — с-ами-нокапроновой, транексамовой кислот — подавляет преимущественно фибринолиз без блокады других видов протеолиза. Аминокапроновую кислоту (АКК) и ее аналоги следует применять только при выраженной активизации фибринолиза, на фоне геморрагий, угрожающих жизни. Доза 5 % раствора е-АКК составляет 200-400 мл. Терапия АКК позволяет уменьшить смертность от кровотечений, но увеличивает летальность от гемодинамических расстройств, так как они усиливают блокаду микроциркуляции и явления недостаточности почек, легких и других органов, т. е. потенцируют развитие стадии осложнений ДВС-синдрома. Препараты транексамовой кислоты, по литературным данным, подобных осложнений не дают.
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 363
Улучшение микроциркуляции и реологических свойств крови. Результатом тромбо-геморрагического повреждения органов является блокада микроциркуля-торного русла, поэтому вопрос о коррекции этих нарушений рано или поздно (в зависимости от формы и стадии ДВС-синдрома) возникает при лечении каждого пациента. Для улучшения микроциркуляции и реологии назначают трентал, который одновременно с повышением энергетического потенциала эритроцитов и тромбоцитов вызывает сосудорасширяющий эффект, снижает периферическое сосудистое сопротивление, понижает вязкость крови, вызывает дезагрегацию тромбоцитов и повышает эластичность эритроцитов.
При ДВС-синдроме трентал следует вводить с целью восстановления микроциркуляции в период до появления кровотечений или после надежной их остановки. Этот препарат назначают по 5 мл в 250 — 500 мл физиологического раствора. Вводят внутривенно капельно медленно, в течение 90—180 мин.
Реополиглюкин также улучшает реологические свойства крови и дезагрегацию ее форменных элементов. Вводится в дозе 400 мл со скоростью 15 — 25 капель в минуту.
Дипиридамол уменьшает периферическое сопротивление в сосудах, тормозит агрегацию тромбоцитов и препятствует образованию тромбов в сосудах, улучшает коллатеральное кровообращение, стимулирует биосинтез простациклинов и тормозит синтез тромбоксанов. Дипиридамол (2 мл 0,5 % раствора в 250 мл физиологического раствора) следует вводить внутривенно медленно.
Опасным является сочетанное применение аспирина и тромболитической терапии стрептокиназой и ее аналогами с целью дезагрегации и улучшения микроциркуляции, так как это может привести к смертельному кровотечению.
Активация фибринолитической системы крови. Устранить расстройства микроциркуляции с помощью дезагрегантов удается не у всех пациентов, особенно при условии выраженного угнетения фибринолитической системы.
У пациентов с ДВС-синдромом активацию фибринолитической системы крови безопаснее проводить с помощью никотиновой кислоты, эу фи л л ина и компл амина. Никотиновая кислота активирует фибринолиз, не влияя на синтез простациклина, снижает адгезию и агрегацию тромбоцитов, оказывает сосудорасширяющее действие.
Компламин (ксантинол никотинат) расширяет периферические сосуды, улучшает коллатеральное кровообращение, уменьшает агрегацию тромбоцитов, усиливает фибринолитическую активность крови. Применяется в дозе 15 — 30 мг/кг в сутки. Компламин назначают внутримышечно или внутривенно, разводя 10 мл препарата в 500 мл 5 % раствора глюкозы; капельное введение этого количества препарата производят в течение трех-четырех часов.
Некоторые авторы рекомендуют для лечения ДВС-синдрома через 10 —12 ч от начала интенсивной терапии применять тканевой активатор плазминогена (t-PA) [17].
Профилактика и лечение «цитокин-индуцированного» васкулита. Для уменьшения повреждающего действия медиаторов воспаления на эндотелий капилляров уже в первые часы лечения показано:
1. Применение глюкокортикоидов, ингибирующих активность фосфолипазы А2, что уменьшает образование арахидоновой кислоты из фосфолипидов клеточных мембран, поврежденных гипоксией. Дозы глюкокортикоидов — от 120 до 300 мг по преднизолону (в первые сутки).
2. Ингибирование циклооксигеназы, потенцирующей образование гидроген пероксидов липидов, простагландинов, тромбоксана (особенно сильно повреждает эндотелий капилляров тромбоксан А2). Ингибирование достигается систематическим применением нестероидных противовоспалительных препаратов в малых дозах (например, ацетилсалициловой кислоты в дозе 5 — 7 мг/кг массы тела). Показано одновременное применение антагониста тромбоксана А2 — простациклина, который действует так же, как дезагрегант тромбоцитов, увеличивая количество цАМФ в
364 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
тромбоцитах путем потенцирования (увеличения активности) аденилатциклазы. Аналогичный эффект может быть получен при применении дипиридамола, который путем ингибирования фосфодиэстеразы также увеличивает в тромбоцитах содержание цАМФ.
3. Для уменьшения продукции тромбоксана А2 можно использовать ингибирование тромбоксансинтетазы путем применения ибупрофена.
4. Для профилактики отрицательного влияния цитокинов на эндотелий капилляров, моноциты и усиления ДВС крови рекомендуется также применять: а) блокаторы рецепторов токсинов; б) антитела к липополисахаридам грамотрицательных бактерий.
Применение эфферентных методов. Поскольку одним из факторов, в значительной мере определяющих степень выраженности ДВС-синдрома, являются продукты деградации фибриногена, фибрина и протеолиза, то при корригирующей терапии можно использовать гемокарбоперфузию через углеродные сорбенты с целью удаления вышеупомянутых продуктов из кровотока. Это способствует более эффективному восстановлению функции системы мононуклеарных фагоцитов ( ретикулоэндотелиальной системы), снижению явлений интоксикации.
По мнению многих авторов [ 11, 17], наиболее эффективным средством лечения хронического и острого ДВС-синдрома является плазмаферез с переливанием больших количеств (1 — 2 л) свежезамороженной плазмы.
11.7.2. Тромбофилия и гиперкоагуляция
Синдром диссеминированного внутрисосудистого свертывания крови — далеко не единственный приобретенный вид нарушений в системе гемостаза. Широкое использование термина ДВС во всех наблюдениях, спектр изменений которых колеблется от незначительной гиперкоагуляции до выраженного микротромбоза, не только не оправдано, с точки зрения Д. Д. Зербино и Л. Л. Лукасевич [14], но и опасно, так как может повлечь за собой
неверную корригирующую терапию. С целью разрешения сложившейся ситуации в 80-х годах XX ст. Б. И. Кузником [4] было предложено определение: «гипергипокоагуляционный синдром». Это понятие включало возникшие отклонения от нормы в системе гемостаза, которые нельзя было интерпретировать как ДВС-синдром.
В 90-х годах прошлого века в литературе появился новый термин — «тромбофилия», также объединяющий различные патологические состояния, которые не укладываются в рамки существующих представлений о ДВС-синдроме.
К тромбофилиям относят нарушения в различных звеньях системы гемостаза и гемореологии, характеризующиеся повышенной склонностью к развитию тромбозов кровеносных сосудов и ишемией органов. В 1996 г. 3. С. Баркаганом была разработана и предложена классификация основных видов тромбофилий [5]. Дифференциация этих форм патологии принципиально важна, поскольку разные виды тромбофилий, несмотря на подчас очень сходные клинические проявления, требуют применения принципиально отличающихся методов профилактики и лечения. Автор подчеркивает, что термин «тромбофилия» не должен подменяться, как это часто делается, термином «гиперкоагуляционное состояние», поскольку многие виды тромбофилии характеризуются не повышением свертываемости крови, а ее снижением (при дисфибриногенемиях, дефиците фактора XII, антифосфолипид-ном синдроме и др.) либо нарушениями не в гемокоагуляции, а в других звеньях системы гемостаза. В отличие от ДВС-синдрома тромбофилии, вызванные ослаблением всех видов фибринолиза, не имеют предшествующей фазы активации фибринолитической системы.
Гиперкоагуляция может быть определена как состояние, при котором активация свертывания компенсируется системой про-тивосвертывания и отсутствуют клинические признаки тромбозов. Лабораторная диагностика гиперкоагуляции (т. е. определение маркеров активации гемокоагуляции) с установлением конкретного звена нарушения могут существенно повлиять
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 365
на эффективность профилактики тромбозов.
Из всей разновидности тромбофилий подробнее охарактеризуем два вида: анти-фосфолипидный синдром и гиперфибриногенемию.
11.7.3. Антифосфолипидный синдром
Симптомокомплекс, проявляющийся рецидивирующими тромбозами (артериальными или венозными), привычным невынашиванием беременности (более двух случаев потери плода) и тромбоцитопенией при наличии в циркулирующей крови антифосфолипидных антител (аФЛ), получил название антифосфолипидного синдрома (АФС). Полагают, что в основе внутриутробной гибели плода лежит гипоксия, обусловленная недостаточным утероплацентарным кровотоком вследствие тромбоза сосудов плаценты и нарушением имплантации эмбриона.
Антифосфолипидные антитела — это гетерогенная группа антител, различающихся по своей иммунохимической специфичности, что обусловлено существованием нескольких классов мембранных фосфолипидов, различающихся по структуре и иммуногенности.
Наиболее распространены в организме «нейтральные» фосфолипиды:
фосфатиди лэтаноламин;
фосфатидил хол ин.
На втором месте по распространенности — отрицательно заряженные фосфолипиды:
фосфатиди л серин; фосфатидилинозитол.
Отрицательно заряженные фосфолипиды локализуются главным образом на внутренней поверхности биомембран и экспонируются наружу в процессе клеточной активации.
Изучение аФЛ началось задолго до выделения самого АФС. В 1906 г. А. Вассерман описал серологическую реакцию для диагностики сифилиса. В 1941 г. были открыты антитела к кардиолипину, в 1972 г. — так называемый «волчаночный антикоагулянт» у больных с системной красной волчанкой (СКВ). И лишь в 90-х го
дах прошлого века явление усиления синтеза антител к фосфолипидам при наличии определенной клиники получило название «антифосфолипидный синдром».
Термин «волчаночный антикоагулянт» (ВА) является неудачным по ряду причин. Во-первых, В А определяется не только у больных СКВ, во-вторых, он определяется не у всех больных СКВ, в-третьих, ВА — это не какой-то новый специфический антикоагулянт. Под ВА в настоящее время подразумевают спектр антител к нескольким факторам свертывания крови, выявляемых в сыворотках больных АФС, прежде всего антитела к протромбину, фактору V и X. Блокирование антителами этих факторов обеспечивает гипокоагуляционный эффект.
Определение В А основано на том, что гипокоагуляция, обусловленная этими ингибиторами свертывания и выявляемая фосфолипидзависимыми тестами, не корригируется нормальной, бедной тромбоцитами плазмой, однако устраняется при добавлении к исследуемой плазме источника фосфолипидных мембран (разрушенные нормальные тромбоциты, тромбопластин, фосфолипидзависимые змеиные яды и т. д.).
Патогенез тромбоза при АФС. Предполагают, что тромбоз, индуцированный антифосфолипидными антителами, обусловлен несколькими механизмами. После воздействия аФЛ эндотелиальные клетки теряют свою антикоагулянтную активность и могут индуцировать сильные тромбогенные эффекты: снижение синтеза простациклина, экспрессию тромбомодулина, угнетение фибринолиза, повышение уровня PAF. Кроме того, обнаружено, что у больных с АФС падает функциональная активность AT III, протеина С и S в плазме [5].
Причины АФС неизвестны. Увеличение уровня аФЛ (как правило, транзитивное) наблюдается на фоне широкого спектра бактериальных и вирусных инфекций [20].
Клинические признаки АФС различны и зависят от органов, вовлекаемых в процесс. Наличие признаков АФС у больных в отсутствие аутоиммунного заболевания, неопластического или инфекционного про
366 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
цесса послужило основанием для выделения его первичной и вторичной формы. АФС на фоне какого-либо известного заболевания диагностируется как вторичный. При мультиорганном поражении и молниеносном течении АФС называют «катастрофическим».
Особенно большой интерес представляет связь между синтезом аФЛ и апоптозом эндотелиальных клеток. Существуют два типа гибели клеток — некроз и апоптоз. Первый ассоциируется с действием вредоносных агентов и обстоятельств, приводящих к нарушению целостности клеточной мембраны (вследствие повреждения или формирования пор). Второй тип представляет собой активный процесс реализации программы гибели клетки: он может быть вызван действием поступающих извне сигналов, которые сами по себе не являются токсичными или деструктивными.
Слово «апоптоз» в переводе с греческого означает «опадание листьев с деревьев». Этот термин используют для обозначения тех форм гибели клеток, которые являются результатом запрограммированных внутриклеточных процессов и сопровождаются сморщиванием клеток, уменьшением их объема. Перестройка структуры мембран приводит к экспрессии на поверхности клеток определенных детерминант, распознаваемых фагоцитами, что способствует быстрому поглощению ими апоптозных клеток. Нарушения апоптоза лежат в основе некоторых заболеваний: аутоиммунных, лимфопролиферативных и иммунодефицитных.
Явление апоптоза было открыто в 1972 г. Об этом открытии говорят как о «научной революции», раскрывшей ключевые механизмы многих болезней.
Выделяют такие формы апоптоза:
1) гибель клеток вследствие дефицита ростовых факторов;
2) глюкокортикоидиндуцированный апоптоз;
3) «активационный» (включение программы гибели клетки при их активации).
Главная черта клеточного апоптоза — асимметрия фосфолипидной мембраны с экспонированием отрицательно заряженного фосфолипида — фосфатидилсерина (ФС), являющегося мишенью для анти
тел к фосфолипидам. Это служит самым ранним проявлением клеточного апоптоза, предшествующим фрагментации ДНК и нарушению клеточной мембраны. Известно, что при активации тромбоцитов экспонирование ФС ассоциируется с активацией свертывания.
Лечение АФС. Отсутствие достаточных сведений о механизмах клинического полиморфизма и маркерах рецидиви-рования делает крайне сложными лечение и профилактику АФС. Терапия при АФС обычно преследует такие цели:
подавление выработки аФЛ;
механическое удаление аФЛ;
профилактику рецидивов тромбозов и активности процесса.
Подавление выработки антител осуществляют назначением иммуносупрес-сантов:
а) препарат первого ряда — преднизолон (20 мг/сут);
б) пульс-терапия высокими дозами ме-типреда;
в) цитостатики (циклофосфан — 12 г/курс лечения).
Назначение препаратов этих групп показано лишь у пациентов с СКВ.
Механическое удаление антител.
Плазмаферез: отношение к этой процедуре неоднозначное, так как считается, что единичные сеансы приводят к временному снижению концентрации антител. Необходим программный плазмаферез в сочетании с базисной терапией.
Профилактика тромбозов:
а) антималярийные препараты — плак-венил. Ингибирует адгезию и агрегацию тромбоцитов. Суточная дозировка — от 0,4 до 1,0 г;
б) вопрос о назначении антикоагулянтов довольно спорный, поскольку антико-агулянтны (в частности гепарин) могут усугубить имеющуюся тромбоцитопению. Кроме того, характер образования тромбов в венозной и артериальной системе неодинаков и также требует дифференцированного подхода. Из прямых антикоагулянтов предпочтение отдают низкомолекулярным гепаринам. С профилактической целью после прямых назначают непрямые антикоагулянты;
Некоторые виды нарушений в системе гемостаза в практике интенсивной терапии 367
в) пациенты с АФС практически постоянно должны получать дезагреганты. Целесообразно менять группы препаратов (реополиглюкин, дипиридамол, эуфиллин, папаверин, аспирин);
г) переливание свежезамороженной плазмы как источника AT III и плазминогена;
д) назначение ингибиторов протеолиза (трасилол, контрикал).
В литературе имеются сведения об успешном использовании при АФС высоких доз иммуноглобулина, препаратов рыбьего жира.
11.7.4. Гиперфибриногенемия
Воздействие на организм сильного повреждения, обусловленного хирургической операцией, травмой, инфекцией, вызывает взаимосвязанный метаболический, гормональный и гемодинамический ответ. Нервная, эндокринная и иммунная системы — это главные адаптационные системы, взаимное регулирование которых определяет, во-первых, их собственное функциональное состояние, а, во-вторых, характер течения патологических процессов в организме.
Острая фаза ответа (ОФО) является системной реакцией на локализованное повреждение тканей и характеризуется изменением уровня различных белков в плазме, обозначаемых как маркеры острой фазы. ОФО — это прямое следствие повреждающего стресса, который может развиться в ответ на различные стимулы. Наиболее достоверным и хорошо изученным признаком ОФО является изменение уровня белков в плазме. Последние синтезируются главным образом в печени. Выделяют две группы маркеров ОФО:
1) позитивные (оц-антихемотрипсин, комплемент С3, церулоплазмин, фибриноген, гаптоглобин и С-реактивный белок), уровень которых повышается в острую фазу ответа;
2) негативные (альбумин и трансферрин), уровень которых снижается.
Уровень позитивных маркеров ОФО нарастает в течение 24 —48 ч после трав
мы. Характерным является увеличение каждого из них более чем в два раза, при этом степень увеличения соответствует тяжести повреждения. В случае неосложненной травмы или неосложненного восстановительного периода после нетравматичных операций уровень позитивных маркеров снижается в течение 48 ч и возвращается к исходному через 72 —96 ч после воздействия повреждения. Однако при определенных обстоятельствах (сепсис, хронические заболевания) уровень маркеров остается повышенным длительное время и может быть использован как диагностический критерий.
Основными цитокинами, ответственными за стимуляцию системного ответа в острой фазе, являются интерлейкины (ИЛ-1, ИЛ-2, ИЛ-6, а-ТНФ), при этом ИЛ-6 играет доминирующую роль.
ИЛ-6 — плейотропный цитокин, обладающий широким спектром биологического действия, продуцируемый клетками иммунной и неиммунной природы. Обладает свойством повышать синтез фибриногена печенью и увеличивать выработку костным мозгом тромбоцитов.
Таким образом, наблюдаемые у ряда пациентов гиперфибриногенемия и тромбоцитоз обусловлены системным воспалительным ответом и не имеют прямого отношения к активации системы гемостаза. И гиперфибриногенемия, и тромбоцитоз ухудшают реологические свойства крови и создают предпосылки к развитию тром-бофилических состояний. Существующее мнение о том, что гиперфибриногенемия является признаком хронического течения ДВС-синдрома [13], не совсем корректно, поскольку для последнего характерно потребление фибриногена, а не увеличение его концентрации. Кроме того, даже при наличии признаков (например, положительных паракоагуляционных тестов), свидетельствующих об активации свертывающей и фибринолитической систем, следует помнить, что указанные тесты в условиях гиперфибриногенемии могут быть ложноположительными [17]. Однако при определенных обстоятельствах гиперфибриногенемия, как и любое другое тромбофили-ческое состояние, может быть дополнитель
368 СИСТЕМА ГЕМОСТАЗА В НОРМЕ И ПРИ ПАТОЛОГИИ
ным фактором риска для развития ДВС-синдрома.
Лечение гиперфибриногенемии заключается в терапии основного заболевания,
на фоне которого развились эти нарушения, назначении дезагрегантов и прямых антикоагулянтов.
Список использованной литературы
1. Духанин А. С., Губаева Ф. Р. Фармакологическая регуляция активности тромбоцитов // Экспериментальная и клиническая фармакология. — 1998. — № 4. — С. 66 — 77.
2. Баркаган 3. С. Очерки антитромботической фармакопрофилактики и терапии. — М.: Ньюдиамед, 2000. — 148 с.
3. Проблемы и гипотезы в учении о свертывании крови / Под ред. О. К. Гаврилова. — М.: Медицина, 1981. — 288 с.
4. Кузник Б. И., Васильев Н. В., Цыбиков Н. Н. Иммуногенез, гемостаз и неспецифическая резистентность организма. — М.: Медицина, 1989. — 320 с.
5. Баркаган 3. С., Момот А. П. Основы диагностики нарушений гемостаза. — М.: Нью-диамед-АО, 1999. — 224 с.
6. Румянцев А. Г., Аграненко В. А. Клиническая трансфузиология. — М.: ГЭОТАР медицина, 1998. — 575 с.
7. Лычев В, Г. Диагностика и лечение диссеминированного внутрисосудистого свертывания крови. — 2-е изд., перераб. и доп. — Н. Новгород: Изд-во НГМА, 1998. - 191 с.
8. Кириченко А. А. Механизм действия и клиническое применение Тиклида // Клиническая фармакология и терапия. — 1997. — №6. - С. 79-82.
9. Бицадзе В. О., Макацария А. Д. Патогенетическое обоснование и возможности применения низкомолекулярных гепаринов в акушерской практике // Акушерство и гинекология. - 1999. - №2. - С. 37-41.
10. Ангелъсыа С., Якубовсыа 3., ДомМчак М. Г. Юпшчна 6ioxiMin. — Сопот, 1998. — С. 385 — 402.
11. Баркаган 3, С. Геморрагические заболевания и синдромы. — 2-е изд., перераб. и доп. — М.: Медицина, 1988. — 528 с.
12. Возианов А. Ф., Бутенко А. К., Зак К. П. Цитокины. Биологические и противоопухолевые свойства. — К.: Наук, думка, 1998. - 317 с.
13. Серов В. H.t Макацария А. Д. Тромботические и геморрагические осложнения в акушерстве. — М.: Медицина, 1987. — 288 с.
14. Зербино Д. Д., Лукасевич Л. Л. Диссеминированное внутрисосудистое свертывание крови: Факты и концепции. — М.: Медицина, 1989. - 256 с.
15. Satoshi Gando, Takashi Катеие, Satoshi Nandzaki et al. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome // Crit. Care Med. — 1997. — Vol. 25, Nil. - P. 1820-1826.
16. Satoshi Gando, Satoshi Nandzaki, Osamu Kem-motsu. Disseminated intravascular coagulation and sustained systemic inflammatory response syndrome predict organ dysfunctions after trauma //Ann. Surgery. — 1999. — Vol. 229, N 1. - P. 121-127.
17. Кулаков В. И., Серов В. Н., Абубакирова А. М., Баранов И. И. Акушерские кровотечения. — М.: Триада-Х, 1998. — 96 с.
18. Мачабели М. С. Коагулопатические синдромы. — М.: Медицина, 1970. — 304 с.
19. Решетняк Т. М., Алекберова 3. С., Насонов Е. Л., Насонова В. А. Принципы лечения антифосфолипидного синдрома при системной красной волчанке // Терапевт, архив. - 1998. - №5. - С. 83-87.
20. Современные представления о патогенезе антифосфолипидного синдрома / Е. Л. Насонов, А. Г. Кобылянский, Т. В. Кузнецова и др. // Клиническая медицина. — 1998. — № 9. - С. 9-13.
Раздел
12
Сердечно-легочная церебральная реанимация
Наибольшее значение проблема жизни и смерти приобретает для врача, прежде всего реаниматолога, ибо его профессиональным долгом и гуманной общечеловеческой задачей является борьба за сохранение полноценной жизни каждого больного как единственного, уникального и неповторимого индивида на Земле.
Особого внимания заслуживает необоснованная внезапная смерть, наступающая вследствие несчастных случаев (травма, отравление, утопление, поражение электрическим током и др.), сердечных приступов, в основе которых в большинстве случаев лежат потенциально обратимые из
менения при условии своевременного оказания неотложной помощи [1,2].
Изучение умирающего организма позволило установить закономерности умирания и, как отмечает В. А. Неговский, «именно с этого момента, когда было установлено, что смерть есть не только скачок, но и процесс, стало реально говорить об оживлении организма» [3].
Ежегодно в мире регистрируется более 200 тыс. больных, которым проводится сердечно-легочная реанимация в связи с остановкой кровообращения, и около 70 тыс. из них выживают.
12.1. ТЕРМИНАЛЬНЫЕ СОСТОЯНИЯ
Важным этапом в течении любого заболевания является момент превращения патогенеза в танатогенез, когда развиваются процессы, непосредственно ведущие к смерти. О наступлении такого момента в большинстве случаев свидетельствует острое расстройство гомеостаза со снижением функционирования жизненно важных систем организма до критического уровня.
Состояния, непосредственно предшествующие смерти, называются терминальными. Под ними понимается такая степень нарушения функций жизненно важных органов и систем, при которой организм не может справиться с возникшими нарушениями без посторонней помощи. Каж
дое из терминальных состояний имеет свои характерные особенности. Различают: пред-агонию, терминальную паузу, агонию и клиническую смерть. Общим для терминальных состояний является обратимость изменений, происходящих в организме. Центральное звено их патогенеза — гипоксия и ишемия. Нарастание гипоксии в процессе умирания ведет к постепенному выключению функций различных отделов ЦНС, начиная с коры головного мозга [4, 5].
Предагония характеризуется прогрессивным угнетением сознания, снижением рефлексов. Дыхание поверхностное, тахипноэ с переходом в брадипноэ. Возможны различные варианты патологических типов дыхания (Чейн-Стокса, Куссмауля и др.).
370 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Артериальное давление снижается до критических значений, первоначально развивающаяся тахикардия сменяется брадикардией, возможны нарушения сердечного ритма (экстрасистолия, блокады, синусовая аритмия). Расстройства микроциркуляции проявляются мраморностью кожных покровов, акроцианозом. В зависимости от вызвавшей его причины пред-агональное состояние может отсутствовать (поражение электрическим током) или длится от нескольких минут до нескольких часов (кровопотеря).
Терминальная пауза является переходным периодом между предагонией и агонией. Она развивается чаще всего при умирании вследствие кровопотери и характеризуется угасанием рефлекторной деятельности, временным (20 — 90 с) прекращением дыхания и кровообращения.
Сущность этого состояния заключается в дальнейшем углублении торможения коры головного мозга с полным выключением ее из регуляции жизненно важными функциями организма на фоне временного усиления тонуса блуждающего нерва. Длительность терминальной паузы определяется порогом чувствительности дыхательного центра к СО2, содержание которого из-за снижения рефлекторной активности значительно превышает нормальные значения. Вследствие этого прекращается вентиляция и уровень СО2 в крови нарастает. При достижении напряжения СО2 в крови выше порога чувствительности дыхательного центра в нем формируются импульсы, возбуждающие дыхательные мышцы. С возобновлением дыхания начинается стадия агонии.
Агония является этапом, предшествующим смерти и характеризуется последними проявлениями жизнедеятельности организма.
В этом периоде прекращается регуляторная функция высших отделов головного мозга, и регуляция функций жизненно важных органов осуществляется бульбарными и спинальными центрами. При этом само функционирование этих органов требует больших затрат энергии, что ведет к истощению резервов организма. Возобновление дыхания и поступление кислорода
в организм способствует кратковременному подъему АД, появлению синусового ритма, иногда восстановлению проблесков сознания. При этом может наблюдаться патологическое дыхание типа «гаспинг» — короткий максимальный вдох с быстрым полным выдохом. В акте дыхания принимает участие вспомогательная мускулатура.
Но на определенном этапе дальнейшее поддержание жизнедеятельности организма становится невозможным. После некоторого учащения пульса и повышения артериального давления, пульс замедляется до 20 —40 в минуту или ускоряется до пароксизмальной тахикардии, артериальное давление снижается до 20 —10 мм рт. ст. Дыхание становится реже и поверхностнее, наблюдается срыв синхронной работы кардиомиоцитов (фибрилляция) или полное прекращение биоэлектрической активности сердца (асистолия). Наступает остановка кровообращения.
Под остановкой кровообращения понимают состояние, при котором деятельность сердца гемодинамически неэффективна прежде всего по отношению к мозговому и коронарному кровообращению.
Выделяют три вида состояний, развивающихся после остановки кровообращения: клиническая смерть (обратимое состояние), социальная (частично обратимое), биологическая смерть (необратимое состояние).
Клиническая смерть — это последний этап терминального состояния и первый период умирания, начинающийся с момента прекращения основных функций жизнедеятельности организма (кровообращение, дыхание) и продолжающийся вплоть до гибели клеток коры головного мозга. При этом решающим критерием необратимых изменений в ЦНС является временной интервал. Допустимые границы времени остановки кровообращения постоянно пересматриваются: П. Сафар (1997) пределом переносимости полной остановки кровообращения для головного мозга при t = 37 °C считает 10 — 20 мин (в эксперименте), но при обычных условиях окружающей среды клиническая смерть обычно длится 4 — 6 мин, когда возможно пол
Терминальные состояния 371
ноценное возвращение к жизни с полным восстановлением функций мозга.
Продолжительность клинической смерти зависит от многих факторов, но определяющим является запас гликогена в нейронах, поскольку именно гликогенолиз является единственным источником энергии при отсутствии кровообращения. Нейроны не могут содержать большой запас гликогена, так как являются одними из наиболее быстро функционирующих клеток. В обычных условиях его хватает на 4 — 6 мин анаэробного гликолиза. При отсутствии реанимационной помощи или ее неправильном проведении по истечении указанного времени выработка энергии в клетках полностью прекращается, нарушаются все энергозависимые процессы, прежде всего поддержание целостности внутриклеточных и внеклеточных мембран.
Социальная {мозговая, гражданская) смерть — это период, начинающийся с гибели клеток коры головного мозга и продолжающийся до тех пор, пока сохраняется возможность восстановления дыхания и кровообращения. Восстановление функций коры головного мозга не наступает.
Появление этого диагноза связано с развитием реаниматологии. Состояние социальной смерти развивается в тех случаях, когда реанимационная помощь была начата несвоевременно или проводилась с недостаточной эффективностью, в результате чего восстанавливаются функции только тех органов, клетки которых имеют большую толерантность к гипоксии и ишемии. Так, деятельность сердца может быть возобновлена спустя десятки минут, а некоторых других органов — даже через час и позже. Продолжительность социальной смерти колеблется в широких пределах и зависит от ухода и качества интенсивной терапии, выраженности повреждений, возникших во время клинической смерти и проведения реанимационных мероприятий.
Биологическая (истинная, клеточная, панорганная, окончательная) смерть — это последний этап умирания, характеризующийся развитием необратимых изменений во всех органах и тканях организма. Восстановить функции основных жизненно важных систем не удается.
Основными причинами биологической смерти являются неадекватные: легочная вентиляция, транспорт кислорода, работа сердца, а также повреждения ЦНС.
К ранним признакам биологической смерти относятся: высыхание и помутнение роговицы, симптом «кошачьего глаза» (при сдавливании глазного яблока зрачок деформируется и вытягивается в длину).
Поздние признаки биологической смерти: трупные пятна и трупное окоченение.
К жизненно важным системам, выключение функционирования которых приводит к стремительному развитию клинической смерти, относятся дыхание, кровообращение и центральная нервная система. Шансы спасения пострадавшего зависят от того, какая из них прекращает функционирование первой. В случаях нарушения функции дыхания возможности восстановления сердечной деятельности наибольшие, при поражении сердца — ниже, но при этом выше вероятность восстановления функции ЦНС. При первичном поражении центров головного мозга вероятность восстановления сердечной деятельности высокая, а высшей нервной деятельности — самая низкая. В этом случае развитие социальной смерти возможно даже при своевременно начатой и правильно проводимой реанимации.
Остановка кровообращения может быть первичной и вторичной.
Первичная остановка кровообращения происходит при непосредственном поражении сердечной мышцы или проводящей системы сердца вследствие рефлекторных влияний на водитель сердечного ритма.
Основные ее причины: инфаркт миокарда, полная поперечная блокада и другие виды аритмий, ранение сердца, миокардит, поражение электрическим током, раздражение рефлексогенных зон, острое нарушение мозгового кровообращения с поражением бульбарных центров.
При первичной остановке кровообращения через 10—15 с наступает потеря сознания, возможны клонические судороги. Через 30 — 60 с наблюдается апноэ и максимальное расширение зрачков.
Вторичная остановка кровообращения наступает вследствие нарушения деятельности других систем организма.
372 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
К основным причинам, вызывающим ее, относятся: гипоксия, гиповолемия, нарушения электролитного обмена, острые отравления, анафилактический и другие виды шока, травматические повреждения, переохлаждение, перегревание и др.
Факторы, способствующие сокращению периода клинической смерти: предшествующее смерти тяжелое заболевание, физи
ческое и психическое переутомление, эмоциональный стресс, острая кровопотеря, высокая температура тела или нахождение в горячей воде.
Удлинению периода клинической смерти способствуют: предшествующее применение препаратов, замедляющих клеточный метаболизм, низкая температура тела или нахождение в холодной воде.
12.2. ПРИЗНАКИ КЛИНИЧЕСКОЙ СМЕРТИ
Основные признаки клинической смерти: отсутствие пульса на магистральных артериальных сосудах (сонной, бедренной,
6
Рис. 12.1. Определение признаков остановки кровообращения:
а — состояние зрачка; б — пальпация пульса на сонной артерии и определение состояния зрачка
плечевой), самостоятельного дыхания, расширение зрачков.
Дополнительные: отсутствие сознания, бледность или цианоз кожных покровов, атония, арефлексия, адинамия, неестественное положение тела больного.
Диагноз клинической смерти должен быть поставлен в течение 10—15 с. Для успеха реанимационных мероприятий решающее значение имеют фактор времени и технически правильное их выполнение. Ускорение диагностики клинической смерти может достигаться одновременной проверкой наличия пульса и состояния зрачков: одной рукой определяется пульс, другой — приподнимается веко для оценки состояния зрачка (рис. 12.1).
12.3. МЕТОДЫ СЕРДЕЧНО-ЛЕГОЧНОЙ ЦЕРЕБРАЛЬНОЙ РЕАНИМАЦИИ
В настоящее время при остановке кровообращения проводится сердечно-легочная церебральная реанимация (СЛЦР), которая рассматривается как комплекс последовательных мероприятий, позволяющих осуществить окончательную диагностику этапа терминального состояния и незамедлительно начать проведение реанимационной помощи [6 — 8].
Предпосылками для разработки современной программы СЛЦР послужили: ♦ доказательство физиологичности вентиляции «изо рта ко рту» перед ручными приемами по Сильвестру, Шефферу, Хольгеру-Нильссену;
♦ разработка технологии устранения обтурации верхних дыхательных путей — «тройной прием»;
♦ выделение этапов СЛЦР;
♦ проведение первой успешной дефибрилляции сердца через открытую грудную клетку;
♦ проведение первой успешной наружной дефибрилляции;
♦ принятие концепции «сердце слишком хорошее, чтобы умирать» (обратимая неожиданная смерть сердца), выдвинутой в 50-х годах XX в. С. Beck;
♦ принятие концепции «мозг слишком хороший, чтобы умереть», выдвинутой в 70-х годах XX в. П. Сафаром;
♦ доказательство возможности обучения методам СЛЦР парамедиков и населения;
♦ создание учебных пособий, инструкций, программ обучения.
Методы сердечно-легочной церебральной реанимации 373
К настоящему времени программа СЛЦР стандартизирована. Первый стандарт проведения СЛЦР был разработан Американской ассоциацией изучения болезней сердца (ААИБС) в 1980 г. и доработан в 1985 и 1992 гг. В 1998 г. Европейский совет возвращения жизни (European Resuscitation Council for Advance Life Support — ERC ALS) издал «Руководящие принципы по реанимации», созданные на основе документов Международного объединенного комитета (International Liaison Committee on Resuscitation — ILCOR). В 1999 и 2000 гг. были проведены научные согласительные конгрессы представителей ILCOR, ERC ALS, ААИБС и приняты Международные руководящие принципы-2000 для сердечно-легочной реанимации, которые были адаптированы к условиям Европы ERC ALS для взрослых [9, 10].
В настоящее время в большинстве стран мира СЛЦР проводится согласно Руководству Европейского совета по реанимации-2000 и стандарту ААИБС 1992 г. Важным преимуществом последнего является разделение методов СЛЦР на три класса с учетом степени доказанной эффективности каждой рекомендации:
I — определенно рекомендуемые, вмешательства всегда приемлемы и определенно полезные, доказана безопасность;
Па — приемлемые и полезные: рассматриваются большинством экспертов как вмешательство выбора;
ПЬ — приемлемые и полезные', рассматриваются большинством экспертов как факультативные или альтернативные;
III — недопустимые', не существует доказанной пользы, могут быть вредны;
Класс не определен — существующих доказательств недостаточно для принятия решения по классификации.
При изложении мероприятий СЛЦР отнесение их к тому или иному классу указано в скобках.
Целью СЛЦР является поддержание искусственного кровообращения и ИВЛ в пределах жесткого минимума, обеспечивающего профилактику необратимых изменений в жизненно важных органах до момента восстановления самостоятельного кровообращения и дыхания.
Основные задачи СЛЦР:
1. Немедленно приступить к реанимационным мероприятиям.
2. Установить возможную причину и глубину изменений жизненно важных функций организма.
3. Одновременно с проведением основных реанимационных мероприятий оценивать их эффективность, объем дополнительных экстренных мер, характер и масштабы патологических состояний.
Комплекс СЛЦР условно подразделяют на 3 стадии (табл. 12.1).
Первая стадия СЛЦР должна начинаться непосредственно на месте происшествия без промедления любым лицом, зна-
Таблица 12.1. Стадии сердечно-легочной церебральной реанимации (по П. Сафару, Н. Дж. Бичеру, 1997)
I стадия II стадия III стадия
Элементарное поддержание жизни (немедленный период) Цель: экстренная оксигенация A (airway open) — восстановление проходимости дыхательных путей В (breath for victim) — искусственная вентиляция легких С (circulation his blood) — наружный массаж сердца Дальнейшее поддержание жизни (специализированный период) Цель: восстановление самостоятельного кровообращения ABC+D D (drugs) — медикаментозная терапия D (diagnosis) — диагностика вида нарушений сердечной деятельности D (defibrillation) — дефибрилляция Длительное поддержание жизни (постреанимационный период) Цель: восстановление функций мозга, постреанимационная интенсивная терапия 1. Оценка состояния больного, определение возможности восстановления функций мозга. 2. Восстановление нормальной функции мозга. 3. Коррекция нарушений других органов и систем, восстановительная терапия
374 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Таблица 12.2. Потенциально обратимые причины сердечной смерти
Четыре «Г» Четыре «Т»
Гипоксия Гиповолемия Гипер/гипокалие-мия, гипокальциемия, ацидоз Гипотермия Напряженный пневмоторакс (Tension) Тампонада Тромбоэмболическая и механическая обструкция (например, легочная эмболия) Токсические или лекарственные вещества (при передозировке)
комым с элементами сердечно-легочной реанимации.
Показанием к ее проведению является наличие не менее двух основных признаков клинической смерти (потенциально обратимые причины сердечной смерти (табл. 12.2)). При этом на этапе специализированной помощи действия выполняются согласно рекомендациям Европейского совета по реанимации — по универсальному алгоритму.
Противопоказания к проведению сердечно-легочной реанимации', биологическая, социальная или клиническая смерть, наступающая в результате длительно текущих инкурабельных заболеваний, если с момента остановки кровообращения прошло более 25 мин в условиях нормотер-мии, когда больной заранее юридически зафиксировал свой аргументированный отказ от СЛЦР [6, 11, 12].
I стадия — элементарное поддержание жизни
На этом этапе выполняют приемы по замещению жизненно важных функций — дыхания и сердца.
Обеспечение проходимости дыхательных путей. Для восстановления проходимости дыхательных путей пострадавшего укладывают на жесткое основание в положение лежа на спине. Если больной находится на мягкой постели, под грудную клетку следует положить твердый щит. После этого очищают ротовую полость и глотку от инородных тел, слизи, рвотных масс, сгустков крови, сломанных зубов.
Методика туалета верхних дыхательных путей: используется один из трех способов форсированного открывания рта с помощью пальцев (рис. 12.2):
1. Прием с помощью скрещенных пальцев. Используется при умеренно расслабленной нижней челюсти. При этом указательный палец вводят в угол рта пострадавшего и надавливают им на верхние зубы, затем против указательного пальца помещают большой палец по линии нижних зубов и форсированно открывают рот пострадавшего.
2. Прием «палец за зубами» применяется при плотно сжатых челюстях. Указательный палец вводят между щекой и зубами пострадавшего и вклинивают его за последние зубы.
3. Прием «подъем языка и челюсти» используется в случаях очень расслабленной нижней челюсти. Большой палец вводят в рот и глотку пострадавшего и его кончиком поднимают корень языка, а другими пальцами захватывают нижнюю челюсть в области подбородка и выдвигают ее вперед.
При наличии в ротоглотке зубных протезов в случае повреждения их следует удалить, при сохранности — оставить
Рис. 12.2. Три приема открывания рта для туалета верхних дыхательных путей
Методы сердечно-легочной церебральной реанимации 375
в ротовой полости, что позволяет сберечь контур рта и облегчает проведение ИВЛ.
Наиболее распространенной и главной причиной нарушения проходимости дыхательных путей является обтурация гипофарингеальной области корнем языка вследствие расслабления мышц, удерживающих его над задней стенкой глотки.
Оптимальной методикой восстановления проходимости дыхательных путей в этом состоянии (при отсутствии подозрений на травму в шейном отделе позвоночника) является «тройной прием» П. Сафара, состоящий из запрокидывания головы, выдвижения нижней челюсти и открывания рта. Уже только одно запрокидывание головы позволяет восстановить проходимость дыхательных путей у 80 % больных.
Методика выполнения «тройного приема».
Первый способ: оказывающий помощь располагается у головного конца больного справа или слева (рис. 12.3). Одну руку кладут на лоб так, чтобы ребро ладони находилось у начала волосистой части головы (лысых ориентируются на линию, где эта часть головы должна была бы начинаться). Другой — удерживают подбородок (метод выбора) или подкладывают под шею (альтернативный метод). Содружественным движением рук разгибают голову в шейно-затылочном сочленении, запрокидывают ее, выдвигают нижнюю челюсть и открывают рот. В результате этого корень языка поднимается над задней стенкой глотки, а надгортанник — над входом в гортань. Такой вариант тройно
го приема удобен, если возникает необходимость в проведении ИВЛ.
Второй способ: оказывающий помощь располагается у головного конца пострадавшего со стороны затылка, II —V пальцами обеих рук охватывает восходящую ветвь нижней челюсти, выдвигает ее вперед и переразгибает голову в шейном отделе позвоночника (рис. 12.4). Давлением больших пальцев, располагающихся на подбородке, приоткрывает рот. Такой вариант тройного приема П. Сафара в большей степени удобен для восстановления проходимости дыхательных путей у пострадавших с сохраненным самостоятельным дыханием. При подозрении на травму шейного отдела позвоночника голову, шею и грудную клетку пострадавшего укладывают в одной плоскости и выполняют очень умеренное запрокидывание головы, при необходимости выдвигают нижнюю челюсть и открывают рот.
Если пострадавший без сознания, но самостоятельное дыхание и сердечные сокращения сохранены, то для поддержания проходимости дыхательных путей ему следует придать устойчивое положение на боку с запрокинутой головой. Кисть находящейся сверху руки поместить под подбородок для удержания головы пострадавшего в запрокинутом положении. Другая рука пострадавшего, находящаяся за его спиной, не позволит принять ему положение лежа на спине.
В зависимости от места происшествия, клинической ситуации и навыков оказывающего помощь могут использоваться различные приспособления: воздуховоды,
Рис. 12.3. Методика выполнения тройного приема по П. Сафару
376 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.4. Методика поддержания проходимости верхних дыхательных путей
лицевые и ларингеальные маски, пищеводный воздуховод-обтуратор, комбитьюб идр. [13, 14].
Техника их введения проста, легко осваивается, и навыки хорошо сохраняются. Они применяются лишь тогда, когда у больного отсутствует сознание. В противном случае их введение может вызвать ларинго- и бронхоспазм, рвоту.
Имеется несколько разновидностей воздуховодов: S- и Т-образные, орофарингеальные и назофарингеальные, с надувной манжеткой.
Орофарингеальный воздуховод любой разновидности после открывания рта с
помощью методики скрещенных пальцев вводится изгибом от языка, затем вращательным движением его поворачивают на 180° и продвигают вглубь, не заталкивая корень языка назад. Можно использовать другой вариант: корень языка аккуратно отжимают шпателем и вводят воздуховод изгибом к языку без вращения.
Назофарингеальный воздуховод вводится через один из носовых ходов до достижения оптимального воздушного потока. С целью минимальной травмати-зации носовых раковин рекомендуется применять назофарингеальные воздуховоды из мягкой резины или пластика, кото
Методы сердечно-легочной церебральной реанимации 377
рые перед введением смазываются предпочтительнее водным раствором местного анестетика.
Одним из недостатков стандартных воздуховодов является отсутствие герметичности дыхательных путей. Этого недостатка лишены воздуховоды с надувной манжеткой.
Пищеводный воздуховод-обтуратор, объединенный с лицевой маской (класс ПЬ), вводится в пищевод до тех пор, пока лицевая маска герметично не ляжет на лицо (рис. 12.5). После этого раздувается манжетка, предупреждающая заброс желудочного содержимого в пищевод. Поступление воздуха в легкие осуществляется через боковые отверстия трубки, расположенные на уровне ротоглотки.
Комбинированный воздуховод — ком-битьюб (класс Па) позволяет, как и пищеводный воздуховод-обтуратор, предупредить аспирацию желудочного содержимого (рис. 12.6). Особенностью этого воздуховода является наличие двух разобщенных трубок, каждая из которых может использоваться для проведения воздуха. Введение этого воздуховода не требует дополнительных приспособлений и осуществляется так же, как введение оро-гастрального зонда.
Если комбитьюб попадает в пищевод, то после раздувания обеих манжеток воздух проходит по каналу, имеющему перфоративные отверстия между манжетками.
Рис. 12.5. Методика введения воздуховода-обтуратора: а — начальная; б — конечная стадии
Если он попадает в трахею, то после раздувания дистальной манжетки воздух проходит по каналу, имеющему выходное отверстие на конце комбитьюба.
Рис. 12.6. Применение комбинированного воздуховода (комбитьюба)
378 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.7. Ларингеальные маски
Ларингеальная маска (класс Па) является одним из современных видов устройств, поддерживающих проходимость дыхательных путей. Они бывают классические и интубационные (рис. 12.7), различных размеров. Классическую ларин-гомаску вводят в ротоглотку до упора вершины треугольника на конце трубки в мягкие ткани (рис. 12.8). После раздувания манжетки она герметично отграничивает дыхательные пути.
Методика введения интубационной ла-рингомаски не отличается от введения классической. После ее установки эндо-трахеальная трубка проводится через просвет ларингомаски в трахею. При прохождении под элеватором надгортанника эн-дотрахеальная трубка надавливает на него, а тот, в свою очередь, отодвигает надгортанник, открывая путь в трахею.
Оптимальным методом обеспечения проходимости дыхательных путей остается эндотрахеальная интубация (класс I).
Она позволяет надежнее, чем другие способы, изолировать дыхательные пути, поддерживать их проходимость, предотвратить аспирацию, облегчить вентиляцию, оксигенацию и санацию трахеобронхиального дерева.
При СЛЦР интубация трахеи должна осуществляться по возможности в более ранние сроки, но только после преоксигенации и без прерывания массажа сердца более чем на 15 с.
Существует несколько методик введения эндотрахеальной трубки. Наиболее часто применяется оротрахеальная интубация, реже — назотрахеальная.
Оротрахеальная интубация осуществляется при прямой ларингоскопии с помощью ларингоскопа с прямым или изогнутым клинком в классическом (голова находится в той же плоскости, что и туловище) или улучшенном (голова приподнята) положении головы. Следует подготовить три эндотрахеальные трубки: расчетного размера и по одной большего и меньшего размера.
При интубации трахеи правой рукой открывают рот, а левой вводят клинок ларингоскопа в ротовую полость, отодвигая язык вверх и влево. Необходимо следить, чтобы губы больного и кончик языка не попали между клинком и зубами. Клинок ларингоскопа подводят к надгортаннику, поднимают его, захватив край прямым клинком или отдавив основание надгортанника изогнутым клинком. Слиш-
Рис. 12.8. Методика применения ларингеальной маски
Методы сердечно-легочной церебральной реанимации 379
ком глубокое введение изогнутого клинка ларингоскопа может привести к смещению надгортанника вниз, а слишком поверхностное — к выпячиванию корня языка, что затруднит осмотр гортани. Чрезмерно глубокое введение прямого клинка может привести к его введению в пищевод.
Рукоятку ларингоскопа приподнимают под прямым углом к клинку. При этом нельзя использовать верхние зубы в качестве опоры. При необходимости для лучшего обзора просят помощника надавить на щитовидный хрящ с тем, чтобы подать гортань кзади или в одну из сторон. Под контролем зрения эндотрахеальную трубку вводят в просвет голосовой щели. Глубина ее введения определяется по прохождению манжетки. После прохождения через голосовую щель ее проксимального края трубку продвигают в трахею еще на 1 см, после чего удаляют жесткий проводник, если он использовался. Сразу же после введения эндотрахеальной трубки подсоединяют мешок Амбу или другой респиратор, делают пробный вдох. Если трубка находится в дыхательных путях, наблюдается экскурсия грудной клетки, над легкими выслушиваются дыхательные шумы. Раздувают манжетку, фиксируют эндотрахеальную трубку.
Назотрахеальная интубация проводится после предварительной обработки носового хода сосудосуживающими препаратами (капельно или путем орошения) с целью увеличения просвета и профилактики кровотечения в случае травматичного проведения трубки через носовой ход. Дополнительно может быть введен раствор местного анестетика.
Трубка для назотрахеальной интубации должна быть в диаметре на 1 мм меньше, чем для оротрахеальной, достаточно эластичной, но в то же время не слишком мягкой — во избежание перегибов или передавливания. С этой точки зрения оптимальной является армированная эндо-трахеальная трубка.
Для проведения назотрахеальной интубации голову больного укладывают в улучшенное положение и умеренно запрокидывают. Трубку вводят в более проходи
мую ноздрю, для предупреждения повреждения носовых раковин ее скос должен быть направлен к носовой перегородке. Дальнейшее продвижение трубки проводят вслепую или под контролем ларингоскопа.
Слепой метод применяется преимущественно у больных с сохраненным самостоятельным дыханием, у которых по каким-либо причинам нельзя открыть рот. При этом трубку продвигают во время вдохов, определяя наличие потока воздуха через трубку во время выдоха. Допустимы манипуляции трубкой вперед и назад, смещения щитовидного хряща из стороны в сторону, что позволяет определить положение конца трубки. Возможны изменения положения головы — сгибание или разгибание в шейном отделе позвоночника, за исключением пострадавших с подозрением на повреждение шейного отдела позвоночника, у которых эти манипуляции недопустимы.
При использовании ларингоскопа дальнейшее введение трубки осуществляется с помощью щипцов Магилла или большого зажима Келли.
Оротрахеальная интубация по пальцу. Эта методика интубации выполняется без ларингоскопа и применяется у пострадавших с подозрением на повреждение шейного отдела позвоночника, у больных в состоянии глубокой комы.
Для интубации используют хорошо смазанную трубку с проводником. По этой методике в полость рта вводят указательный и средний пальцы так, чтобы указательный палец ложился на язык, отодвигая его книзу, и касался надгортанника. Трубку вводят вдоль пальцев в направлении пальпируемого надгортанника, который отодвигают вперед.
Интубация с помощью светящегося проводника. В последние годы в ряде стран эту методику с успехом используют парамедики под руководством врача даже во внебольничных условиях.
Данная методика также не предполагает использование ларингоскопа.
Светящийся проводник подобно обычному вводят в просвет эндотрахеальной трубки почти до конца и затем сгибают
380 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
под углом более 90°. Язык захватывают через марлевую салфетку и подтягивают вперед. Трубку проводят к гортани и поднимают надгортанник, наблюдают за кожными покровами шеи.
При правильном ее проведении в трахею наблюдается яркое свечение по средней линии. Свечение сбоку свидетельствует о попадании трубки в грушевидную ямку, его исчезновение — о попадании в пищевод. Проводник извлекают после введения трубки в трахею.
Сочетанное использование интубации по пальцу и светящегося проводника довольно эффективно в неотложных случаях трудной интубации.
Интубация с помощью фибробронхо-скопа. При этой методике используется фпбробронхоскоп с наружным диаметром 3 — 4,3 мм, который вводят в просвет эндо-трахеальной трубки, внутренний диаметр последней не должен быть меньше 5 мм. Извлекают изо рта язык, удерживая его марлевыми салфетками, вводят в ротовую полость конец тубуса фибробронхоскопа и под контролем зрения с помощью ларингоскопа проводят его к надгортаннику и между голосовыми связками. Затем по нему как по проводнику эндотрахеаль-ную трубку продвигают в трахею.
При назотрахеальной интубации с помощью фибробронхоскопа эндотрахеальную трубку проводят через носовой ход в ротоглотку и только после этого в ее просвет вводят фибробронхоскоп. Под визуальным контролем тубус бронхоскопа продвигают до надгортанника, проводят через голосовую щель, а затем по нему продвигают в трахею эндотрахеальную трубку. Эта методика может использоваться у больных
отделений интенсивной терапии при сохраненном самостоятельном дыхании, если предполагается трудная интубация. Неприемлема для экстренной интубации.
Интубация с помощью фиброоптического ларингоскопа. Занимает среднее положение между фиброоптической интубацией и интубацией с помощью прямой ларингоскопии. Осуществляется фиброоптическим ларингоскопом «UpsherScope» фирмы «Mercury Medical»,позволяющим интубировать пострадавшего без разгибания шеи и при невозможности достаточно широко открыть рот. Это устройство представляет собой ларингоскоп, у которого угол между рукояткой и клинком значительно больше, чем у стандартного. Вдоль клинка расположены желоб — для направления эндотрахеальной трубки и фиброоптическое волокно — для визуального контроля.
В случаях невозможности выполнения вышеуказанных манипуляций используются хирургические методы восстановления проходимости дыхательных путей: крико-тиреотомия, чрескожная микротрахеостомия, трахеотомия.
Крикотиреотомия — это операция, позволяющая восстановить проходимость дыхательных путей с помощью рассечения связки, соединяющей перстневидный и щитовидный хрящи (рис. 12.9). Она при-
Рис. 12.9. Метод крикотиреотомии
Методы сердечно-легочной церебральной реанимации 381
меняется в случаях, когда просвет голосовой щели вследствие отека или других причин сужен до такой степени, что воздушный поток через нее неадекватен, а интубация трахеи трубкой необходимого диаметра невозможна. Для крикотиреото-мии используются канюли наибольшего диаметра, не вызывающего повреждения гортани. Для взрослых это канюли с наружным диаметром 6 мм, для детей старшего возраста — 3 мм.
С целью предупреждения травматиза-ции хрящей и задней стенки трахеи на лезвие скальпеля предварительно надевают стерильный резиновый ограничитель.
Нащупав промежуток между перстневидным и щитовидным хрящами, производят разрез длиной до 15 мм в направлении, перпендикулярном средней линии тела. После этого в просвет между хрящами вводят зажим, расширяя просвет в вертикальном направлении.
Второй зажим расширяет крикотирео-томное отверстие в горизонтальном направлении. Перед введением канюли проводят расширение с помощью дилататора La Borde. Введенную канюлю фиксируют вокруг шеи.
Чрескожная трахеостомия выполняется при наличии частичной проходимости голосовой щели с последующей инжекционной инсуффляцией кислорода (рис. 12.10). С этой целью производится пункция перстневидно-щитовидной связки иглой большого диаметра, через которую в направлении к легким вводится катетер. После этого последний соединяют с респиратором или другим источником потока кислорода под низким давлением.
При нарушении проходимости дыхательных путей вследствие попадания инородных тел тактика зависит от уровня их нахождения и консистенции. Слизь, сгустки крови, рвотные массы и другое патологическое содержимое ротовой полости можно удалить, обернув пальцы хорошо впитывающей тканью, с помощью марлевого тампона, зажатого в корнцанге, хирургического отсоса или резиновой груши. Твердые инородные тела из ротоглотки можно удалить с помощью двух пальцев, пинцета или корнцанга.
Рис. 12.10. Чрескожная трахеостомия с инжекционной инсуффляцией кислорода
Рис. 12.11. Поддиафрагмальный толчок. Прием Геймлиха
При попадании инородного тела или жидкости за голосовые связки следует создать дренажное положение. Если этого недостаточно, можно использовать методику поддиафрагмального толчка (прием Геймлиха), для чего оказывающий помощь становится на колени с одной стороны пострадавшего или над ним на уровне его бедер. Нижнюю часть ладони одной руки прикладывают к животу по средней линии немного выше пупка и ниже мечевидного отростка (рис. 12.11). Вторую руку кладут поверх первой и 3 — 5 раз надавливают на живот быстрым движением вверх по средней линии. Вместо поддиафрагмального толчка, особенно у бе
382 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
ременных женщин и тучных детей старшего возраста, рекомендуется применять компрессию грудной клетки. Для этого пострадавшего располагают как для выполнения приема Геймлиха, а компрессию грудной клетки производят подобно наружному массажу сердца.
Если же у пострадавшего сохранено сознание и он стоит, может применяться методика, когда оказывающий помощь становится позади пострадавшего, охватывает его руками на уровне груди, прикладывает свой кулак той стороной, где находится большой палец, к середине грудины, избегая надавливания на мечевидный отросток и ребра, и, обхватив кулак кистью другой руки, производит надавливания в направлении назад (рис. 12.12).
Алгоритм обеспечения проходимости дыхательных путей представлен на рис. 12.13.
Искусственная вентиляция легких. После обеспечения проходимости дыхательных путей приступают к проведению искусственной вентиляции легких (ИВЛ). Различают такие методы ИВЛ:
1. Неинвазивные: метод «выдыхаемого воздуха»; «ключ жизни» с клапаном, ли-
Рис. 12.12. Прием освобождения дыхательных путей у пострадавших в сознании
цевая маска с клапаном (Лаердал), мешок Амбу.
2. Полу инвазивные: воздуховоды с искусственным мертвым пространством, пищеводный воздуховод-обтуратор, ларингеальная маска.
3. Инвазивные: интубация трахеи, ко-нико- и трахеотомия.
В настоящее время для вентиляции легких на этапе элементарного поддержания жизни используются экспираторные методы ИВЛ — «изо рта ко рту», «изо рта к носу», «изо рта ко рту и носу» (рис. 12.14).
При проведении ИВЛ методом «изо рта ко рту» оказывающий помощь делает вдох, после чего плотно обхватив губы пострадавшего своими губами и прижав свою щеку к ноздрям пострадавшего для предотвращения утечки воздуха через нос, производит выдох. По окончании выдоха он отстраняется от пострадавшего, чтобы дать возможность воздуху пассивно выйти из легких пострадавшего, а самому сделать очередной вдох. По гигиеническим и этическим соображениям при проведении ИВЛ следует использовать марлевую маску или любую другую неплотную ткань.
С этой же целью можно применить «ключ жизни» с клапаном, представляющим собой брелок, внутри которого находится полиэтиленовая маска со специальным фильтром. Благодаря фильтру во много раз уменьшается риск передачи инфекции от пострадавшего к оказывающему помощь. Полиэтиленовая маска помещается на нижнюю половину лица, а фильтр располагается над ртом.
Показаниями к проведению ИВЛ методом «изо рта к носу» является: судорожное сжатие челюстей пострадавшим; затруднения в обеспечении ИВЛ «изо рта ко рту»; ранения губ, языка, нижней челюсти.
Техника проведения ИВЛ методом «изо рта к носу»: оказывающий помощь делает вдох, после чего обхватывает своими губами нос пострадавшего так, чтобы создать достаточную герметичность, но не вызвать пережатия носовых ходов, и делает выдох, следя за экскурсией грудной клетки. Рот пострадавшего во время выдоха следует открыть, поскольку у трети больных в состоянии комы может быть экспи-
Методы сердечно-легочной церебральной реанимации 383
Рис. 12.13. Алгоритм обеспечения проходимости дыхательных путей
384 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
раторная обструкция носоглотки вследствие клапаноподобного движения мягкого неба.
Метод ИВЛ «изо рта ко рту и носу» применяется у новорожденных и грудных детей ввиду того, что из-за малых размеров рта и носа только такое соотношение позволяет достичь герметичности.
В последнее время изменилось мнение о величине объема воздуха, который необходимо вдохнуть пострадавшему во время ИВЛ экспираторными методами. Согласно стандарту ААИБС 1992 г. он должен соответствовать двойному нормальному дыхательному объему и составлять 800—1200 мл. Однако Европейским со
Рис. 12.14. Методы проведения ИВЛ: «изо рта ко рту» и «изо рта к носу»
ветом по реанимации были приведены данные о том, что объем 400 — 600 мл, особенно при дополнительной подаче кислорода, является достаточным для адекватной вентиляции при СЛЦР взрослых, поскольку образование СО2 во время остановки сердца невелико. По мнению Европейского совета по реанимации при СЛЦР взрослых дыхательный объем должен быть достаточным для обеспечения экскурсии грудной клетки и составлять 400 — 600 мл, что позволяет уменьшить риск раздувания желудка, регургитации и последующей аспирации по сравнению с вдуванием больших объемов. Рекомендуется вначале быстро сделать два пробных вдоха, а затем продолжать вдувания с частотой 10 —12 в минуту с длительностью одного вдоха 1,5 — 2 с. Основным признаком эффективности ИВЛ при этом методе являются экскурсии грудной клетки на вдохе и выдохе.
Несмотря на то, что выдыхаемый воздух содержит всего 16—18 % кислорода, этого достаточно для поддержания минимальной оксигенации крови у пострадав
Методы сердечно-легочной церебральной реанимации 385
шего при проведении ИВЛ методом «изо рта ко рту» или «изо рта к носу». При этом при условии отсутствия выраженных отклонений со стороны системы внешнего дыхания парциальное напряжение кислорода в оттекающей от легких артериальной крови (РаО2) составляет около 75 мм рт. ст. Такому РаО2 соответствует степень насыщения кислородом гемоглобина эритроцитов в артериальной крови (SaO2) — более 90 %. Одновременно эффективно удаляется образующийся в органах и тканях пострадавшего углекислый газ: парциальное давление углекислого газа в артериальной крови (РаО2) пострадавшего колеблется в пределах 30 — 40 мм рт. ст.
Другие методы замещения функции внешнего дыхания (Сильвестра, Хольге-ра-Нильссена, Шеффера) на этапе элементарного поддержания жизни менее эффективны, но могут использоваться в случаях невозможности вентиляции «изо рта ко рту» или «изо рта к носу» (тяжелая травма лица).
При проведении вентиляции легких экспираторными методами могут применяться различные приспособления.
Воздуховоды с искусственным мертвым пространством (S-образная трубка, двойной воздуховод Гведела) позволяют провести вентиляцию «изо рта в воздуховод». Этот метод имеет преимущества перед вентиляцией «изо рта ко рту» с точки зрения эстетичности, поддержания рта пострадавшего открытым и обеспечения проходимости дыхательных путей. Но при использовании воздуховодов у больных в сознании или в состоянии легкого ступора могут возникнуть ларингоспазм, рвота.
Карманная лицевая маска с клапаном Лаэрдал позволяет провести вентиляцию рот—маска. Однако она не увеличивает эффективность вентиляции по сравнению с методом «изо рта ко рту». Единственным ее преимуществом является возможность дополнительной подачи кислорода через имеющийся в ней ниппельный клапан.
Из ручных аппаратов для ИВЛ Европейским советом по реанимации рекомендуется саморасправляющийся мешок Амбу (рис. 12.15) (класс Па). Его можно использовать как с лицевой маской, так и со
13 4-220.
Рис. 12.15. Мешок Амбу
всеми другими приспособлениями для поддержания проходимости дыхательных путей, имеющими стандартный разъем для присоединения респираторов (ларингомас-ка, эндотрахеальная трубка, пищеводный воздуховод-обтуратор и др.). Другой разновидностью ручных респираторов являются устройства с триггерной системой вентиляции, представляющие собой управляемый клапан на шланге подачи кислорода или воздушной смеси. Они позволяют немедленно вручную начать раздувание легких кислородом или воздухом под положительным давлением через маску, пищеводный воздуховод-обтуратор, эндотрахеальную или трахеостомическую трубку. Одна из разновидностей респираторов этого типа с автоматическим переключением фаз вдоха и выдоха по достижении определенного давления в дыхательных путях применяется в службах, оказывающих реанимационную помощь во внегоспитальных условиях (в очагах стихийных бедствий, техногенных катастроф, зонах военных действий). Для проведения длительной искусственной вентиляции легких в условиях стационара рекомендуются автоматические аппараты ИВЛ.
Одним из условий адекватной доставки кислорода к головному мозгу во время проведения реанимационных мероприятий является как можно более ранняя оксигенация (класс I). По возможности, сначала применяют чистый кислород, уменьшая в последующем его процентное содержание во вдыхаемой смеси под контролем напряжения кислорода в артериальной крови.
Ошибки при проведении ИВЛ:
♦ не обеспечена свободная проходимость дыхательных путей;
♦ не обеспечена герметичность при вдувании воздуха;
386 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
♦ недооценка (позднее начало) или переоценка (СЛЦР начата с интубации) значения ИВЛ;
♦ отсутствие контроля экскурсий грудной клетки;
♦ отсутствие контроля попадания воздуха в желудок;
♦ попытка медикаментозной стимуляции дыхания;
♦ вдувание воздуха в момент компрессии грудной клетки.
Искусственное поддержание кровообращения. После обеспечения поступления воздуха в легкие и обогащения крови легочных капилляров кислородом следующей задачей СЛЦР является доставка оксигенированной крови к головному мозгу. Для этого необходимо обеспечить кровообращение в сосудах малого и большого круга кровообращения. С этой целью применяют искусственное воспроизведение работы естественного насоса — сердца.
Существует два вида массажа сердца: наружный и прямой.
Наиболее простым и наиболее приемлемым как на догоспитальном, так и на госпитальном этапах является наружный массаж сердца (класс I), основанный на сжимании сердца при надавливании на грудину (рис. 12.16). Эффект наружного массажа сердца объясняется за счет сердечного (сжатие самого сердца) и грудного насоса (сжатие всех сосудистых емкостей грудной клетки, главным образом легких).
Методика проведения наружного массажа сердца: оказывающий помощь располагается сбоку от пострадавшего, лежащего на спине на твердой поверхности. Прежде, чем приступить к проведению массажа, если с момента остановки сердца прошло
Рис. 12.16. Наружный массаж сердца
не более 30 с, следует нанести одиночный прекордиальный удар, который Европейским советом по реанимации включен в универсальный алгоритм СЛЦР. Он наносится кулаком в область средней части грудины с расстояния 20 — 30 см с размеренной силой так, чтобы не нанести травму.
Прекардиальный удар позволяет восстановить синусовый ритм у отдельных больных с желудочковой тахикардией с отсутствием пульса (эффективность 11-25 %) и фибрилляцией желудочков (эффективность меньше). Ввиду возможности быстрого нанесения удара и отсутствия больших усилий при этом, он рекомендован больным в состоянии клинической смерти, если не представляется возможным немедленно оценить ритм сердца и применить при выявленном нарушении необходимые мероприятия (класс ПЬ). При его неэффективности следует сразу же перейти к проведению общепринятых мероприятий СЛЦР.
Для этого находят месторасположение рук для проведения наружного массажа сердца — на границе нижней и средней трети грудины или в точке, отстоящей на два поперечных пальца выше грудино-мечевидного сочленения (рис. 12.17). В классическом варианте выполнения наружного массажа сердца оказывающий помощь помещает любую из рук на эту точку таким образом, чтобы над местом компрессии располагалось основание ладони, а пальцы были параллельны ребрам. Вторая рука располагается на тыльной стороне нижележащей так, чтобы ее основание также проецировалось на точку компрессии. Пальцы не должны лежать на грудной клетке пострадавшего, поскольку при этом смещается точка компрессии и увеличивается риск перелома ребер и реберных хрящей.
Альтернативным классическому расположению рук является метод скрещенных пальцев (рис. 12.18). При этом пальцы верхней руки пропускаются между пальцами нижней руки и охватывают ладонь. Надавливание производится нижней частью ладони находящейся снизу руки.
Одним из главных условий правильного выполнения наружного массажа сердца
Методы сердечно-легочной церебральной реанимации 387
Рис. 12.17. Классическая методика наружного массажа сердца
Рис. 12.18. Альтернативная методика наружного массажа сердца
является давление не за счет силы пальцев, а за счет массы тела оказывающего помощь. Для этого следует не сгибать руки в локтевых суставах.
Глубина прогиба грудной клетки у взрослых во время наружного массажа должна составлять 3,5 —5 см. У худых она может быть меньше, у тучных, наоборот, больше. Но в любом случае компрессия должна быть достаточной для возникновения пульсовой волны на крупных артериях (признак эффективности наружного массажа) и не вызывать травмы костных элементов грудной клетки.
Частота компрессий у взрослых — в пределах 80—100 в минуту.
На этапе элементарного поддержания жизни чередуют ИВЛ и наружный мас-13*
саж сердца. Если СЛЦР выполняет один человек, соотношение числа вдуваний воздуха в легкие пострадавшего к числу нажатий на грудину должно составлять 2:15 (два вдувания/пятнадцать компрессий), при этом в минуту осуществляется четыре-пять циклов. Если помощь оказывают два человека, соотношение составляет 1 : 5 (одно вдувание/пять компрессий), и таких циклов должно быть не менее 12 в минуту.
Ошибки при проведении наружного массажа сердца'.
♦ пострадавший лежит на мягком основании;
♦ неправильно расположены руки оказывающего помощь;
♦ оказывающий помощь опирается на пальцы и/или сгибает руки в локтевых суставах;
♦ надавливания на грудину проводятся слишком редко;
♦ допускаются перерывы в проведении наружного массажа сердца более чем на 30 с;
♦ нарушается частота массажных движений;
♦ не соблюдается соотношение между вдуваниями воздуха и массажными движениями (1:5 или 2 : 15).
Как варианты наружного массажа сердца Европейским советом по реанимации одобрены и другие методы: вставочная абдоминальная компрессия, активная компрессия — декомпрессия, механическая (поршневая) и жилетная СЛЦР, классифицируемые как класс ПЬ.
Вставочная абдоминальная компрессия используется для усиления притока крови к сердцу. В осуществлении этого метода участвуют три человека, двое из которых выполняют наружный массаж сердца и искусственную вентиляцию легких, а третий надавливает на переднюю брюшную стенку (рис. 12.19). Это увеличивает венозный возврат крови к сердцу за счет сдавления аорты. Надавливание на грудину и переднюю брюшную стенку осуществляется поочередно. Выполняющий абдоминальную компрессию располагает руки в области пупка таким образом, чтобы они находились над позвоночни-
388 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.19. Выполнение наружного массажа сердца со вставочной абдоминальной компрессией
ком. Взаиморасположение рук такое же, как при выполнении наружного массажа сердца.
Метод активной компрессии —декомпрессии в последние годы получил широкое распространение. Идея метода заключается в создании пониженного давления в грудной полости в фазе искусственной диастолы, за счет чего увеличивается приток крови к сердцу. С этой целью используется специальное устройство, напоминающее вантуз, — кардиопамп (дословно — «сердечный насос»). Он представляет собой присоску диаметром около 15 см, к которой присоединен диск со встроенным индикатором силы компрессии-декомпрессии. Присоску располагают на передней поверхности грудной стенки таким образом, чтобы ее центр находился над точкой классического наружного массажа. Оказывающий помощь держит диск двумя руками, которые в процессе выполнения массажа должны быть выпрямлены в локтевых суставах. При первом же нажатии на диск кардиопампа присоска прочно фиксируется на передней поверхности грудной клетки, что позволяет приподнимать грудину относительно ее положения в покое на несколько сантиметров, создавая пониженное давление в органах средостения. При ненарушенной проходимости дыхательных путей в фазе декомпрессии в легкие поступает некоторое количество воздуха, что также увеличивает
эффективность реанимационных мероприятий. Компрессия проводится с такой же частотой и силой, что и при классическом наружном массаже. Давление на грудину у взрослых осуществляется с силой, равной 27 — 41 кг, декомпрессия — с силой, равной 9 кг, что можно оценить по индикатору.
Идея одновременного осуществления массажа сердца и ИВЛ реализована в устройстве фирмы Kendall — кардиовенте, позволяющем проводить реанимационные мероприятия в первой стадии одним лицом с эффективностью, не уступающей, а иногда и превосходящей эффективность оказания помощи двумя реаниматорами.
В отличие от классических ручных респираторов в устройстве устанавливается необходимый дыхательный объем, для чего предусмотрены шесть стандартных положений — 0,2; 0,4; 0,6; 0,9; 1,2; 1,5 л. В кар-диовенте реализовано автоматическое управление соотношением вентиляции и компрессии, возможно добавление кислорода к дыхательной смеси.
Полный цикл реанимации с помощью кардиовента состоит из четырех фаз: 1) вдоха, когда в результате сжатия дыхательного меха осуществляется подача воздуха в легкие пострадавшего; 2) выдоха, осуществляемая пассивно; 3) компрессии на грудину; 4) заполнения дыхательного меха.
Механические устройства для СЛЦР. Большинство из них состоит из поршня, закрепленного на штативе и перемещающегося в вертикальном направлении. Штатив крепится к основанию, которое подкладывается под грудную клетку пострадавшего таким образом, чтобы точка, на которую давит поршень, соответствовала точке приложения сил при классическом наружном массаже. Применение «грудных массажеров» наиболее удобно и оправдано при транспортировке, поскольку независимо от каких-либо внешних факторов обеспечивается постоянство силы и частоты сдавливания сердца. Для обеспечения безопасности необходим постоянный контроль за работой аппарата и наличием пульсовой волны на периферических артериях.
Методы сердечно-легочной церебральной реанимации 389
Кирасная СЛЦР основана на принципе высокочастотной внешней ИВЛ по методике 3. Хайека. Суть этого метода заключается в следующем: грудная клетка больного замыкается в герметичную кирасу, в которой с помощью мощного компрессора попеременно создается повышенное и пониженное давление. Во время первой фазы из-за повышения давления в грудной клетке происходит выдох воздуха из легких пострадавшего и выброс крови из сердца. Во время второй — понижение давления в грудной клетке вызывает поступление воздуха в легкие и наполнение сердца кровью. Для обеспечения адекватного кровотока частота циклов компрессии — декомпрессии должна составлять 80—100 в минуту. Учитывая, что при кирасном методе реанимации воздух в легкие пострадавшего поступает через естественные дыхательные пути, необходимо следить за их свободной проходимостью.
Жилетная СЛЦР. Метод сжатия грудной клетки при этой методике подобен описанному выше. В отличие от кирасы пневматический жилет, надеваемый на грудную клетку, выполняет только фазу компрессии, и поэтому должен сопровождаться проведением ИВЛ.
Как показали исследования, применение методов кирасной и жилетной реанимации обеспечивает достаточный мозговой и коронарный кровоток. Представление о грудном насосе как о главном двигателе крови при методах наружного массажа сердца способствовало появлению особой формы СЛЦР — кашлевой аутореанимации. Она интересна тем, что является единственной формой самопомощи у больных в состоянии клинической смерти. Это особенно важно у пациентов с мерцательной аритмией и другими нарушениями ритма сердца, часто осложняющимися фибрилляцией желудочков.
Этот метод основан на физиологии кашлевого рефлекса, когда выполнение больным в момент прекращения сердечной деятельности (сознание выключается через 10—15 с после этого) судорожных покашливаний позволяет поддержать небольшой кровоток в результате колебаний вну-тригрудного давления.
Естественно, этот метод не может заменить методы классической СЛЦР, но дает возможность больному поддержать свою жизнь до прибытия помощи. В кардиологических отделениях такой «кашель» может быть сигналом медперсоналу.
Поиск новых методов массажа сердца не прекращается. Этому часто способствуют обстоятельства, препятствующие проведению какого-либо из описанных выше методов наружного массажа сердца.
В литературе описаны случаи успешной реанимации при проведении наружного массажа со спины (класс не определен). В этом случае пострадавший находится в положении на животе. Массаж осуществляется так же, как и при классическом наружном массаже, но местом приложения сил является точка на позвоночнике между нижними краями лопаток.
Согласно рекомендации Европейского совета по реанимации возможно использование по показаниям в условиях стационара открытого массажа сердца (класс ПЬ). В настоящее время показания к нему значительно ограничены: остановка сердца во время операций на органах грудной полости и верхнего этажа брюшной полости; ранения сердца и его тампонада; множественные переломы ребер, грудины, позвоночника; напряженный пневмоторакс; массивная тромбоэмболия легочной артерии (если предполагается эмболэктомия); переохлаждение (если предполагается согревание сердца теплым физиологическим раствором). Вскрытие перикарда позволяет устранить тампонаду сердца, производить безопасное интракардиальное введение лекарственных препаратов, накладывать электроды для дефибрилляции на сердце.
Методика прямого массажа сердца'. предполагается, что больной уже интубирован и ему проводится ИВЛ. Разрез кожи делается в пятом межреберном промежутке слева по верхнему краю V ребра во избежание повреждения межреберного нерва и сосудов. Середина разреза должна располагаться примерно на уровне среднеключичной линии. Надсечение плевры производят с помощью скальпеля, а затем продлевают разрез с помощью нож
390 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
ниц. В операционную рану устанавливают ранорасширитель. Сердце пострадавшего захватывают левой рукой так, чтобы большой палец лежал на передней поверхности сердца (над левым желудочком), а остальные пальцы — под сердцем (над правым желудочком).
Сжатие сердца осуществляют с частотой 60 — 80 в минуту или в зависимости от скорости наполнения желудочков сердца.
Во время операций на органах верхнего этажа брюшной полости возможно проведение прямого массажа сердца без вскрытия перикарда и диафрагмы — поддиафрагмальный массаж. В этом случае любой рукой, подведенной под диафрагму, придавливают сердце к внутренней поверхности грудины. При вскрытии диафрагмы проводится чрездиафрагмальный массаж сердца.
Метод баллонной контрпульсации (класс I) является аппаратным методом поддержания кровообращения при его остановке, рекомендуемым Европейским советом по реанимации. Он может быть применен только при наличии ритмичных сокращений сердца, что имеет место при электромеханической диссоциации, когда работающее сердце не дает выброса крови.
Методика баллонной контрпульсации состоит в создании временного препятствия аортальному кровотоку в начале каждой систолы и снятии препятствия с середины систолы до начала диастолы.
К аппаратным методам поддержания кровообращения в жизненно важных органах относятся также метод вспомогательного экстракорпорального кровообращения и экстракорпоральная мембранная оксигенация. Но их применение ограничено условиями специализированных кардиохирургических отделений.
Признаками эффективности проводимых реанимационных мероприятий являются: сужение зрачков, наличие пульсовой волны на центральных артериях, систолическое давление в пределах 70—80 мм рт. ст., изменение окраски кожных покровов с бледной и цианотичной на бледно-розовую.
Признаки оживления пострадавшего'. появление самостоятельных сердечных
сокращений и дыхания, сужение зрачков, восстановление тонуса мышц, уменьшение цианоза кожных покровов и слизистых оболочек. Эти признаки свидетельствуют лишь о начальном периоде оживления.
Возможные осложнения при СЛЦР: ♦ при проведении ИВЛ: регургитация в полость рта содержимого желудка и аспирация его в дыхательные пути; вывих нижней челюсти, разрыв легочной ткани и пневмоторакс;
♦ при проведении наружного массажа сердца: переломы ребер и грудины; повреждения внутренних органов (легких, сердца, печени, селезенки, желудка); пнев-момедиастинум и пневмоперитонеум;
♦ при проведении прямого массажа сердца: повреждение внутренней грудной артерии и кровотечение из нее при восстановлении кровообращения; повреждение сердечной мышцы при сильном сдавливании или кровоизлияние в нее; поворот сердца вокруг оси.
Алгоритм базового поддержания жизни приведен на рис. 12.20.
II стадия — дальнейшее поддержание жизни
Реанимационные мероприятия первой стадии СЛЦР позволяют поддерживать газообмен в тканях на минимальном уровне. Они проводятся до восстановления самостоятельного кровообращения, однако для полного восстановления жизнедеятельности организма методов первой стадии недостаточно. Они дополняются мероприятиями второй стадии СЛЦР, которые проводятся специализированной реанимационной бригадой или в стационаре [15 — 17].
Проведение медикаментозной терапии.
В ее задачи входит: стимуляция сокращений миокардиоцитов, улучшение внутрисердечной проводимости, блокада нежелательных рефлекторных реакций и коррекция грубых метаболических расстройств. Соответственно препаратами первого выбора базисной медикаментозной терапии, применяемыми во время СЛЦР, являются: адреналина гидрогенхлорид, атропина сульфат, лидокаин, амиодарон, натрий гидрогенкарбонат. Бретилиум Европейским советом по реанимации не рекомендуется.
Методы сердечно-легочной церебральной реанимации 391
Определить наличие сознания
Вызвать службу экстремальной медицины
Освободить дыхательные пути
Я
Наличие дыхания
♦ Если пострадавший без сознания — встряхнуть и окликнуть
♦ Позвать на помощь, положить больного на спину
♦ Выполнить тройной прием П. Сафара
♦ При отсутствии явного дыхания — смотри, слушай, ощущай
♦ Поддерживать проходимость дыхательных путей, сделать четыре быстрых вдоха
♦ Убедиться в отсутствии обструкции
Сопротивление вентиляции
Начать искусственную вентиляцию
V Вентиляция проводится V
Восстановление проходимости дыхательных путей
и 1!
Определить наличие пульса Продолжить искусственную вентиляцию г4
J1 Убедиться, что
нет сдавления тела снаружи
Пульс есть Пульса нет
Продолжить . Начать непрямой
ИВЛ массаж сердца
Сопротивление вентиляции
Применить поддиафрагмальный абдоминальный толчок Вручную очистить ротоглотку
Реаниматоры ЧСС/ЧД Частота
1 15:2 80— 100/мин
2 5: 1 80—100/мин
Рис. 12.20. Алгоритм базового поддержания жизни (I стадия СЛЦР)
Адреналина гидрохлорид. Как а-адре-номиметик он способствует централизации кровотока за счет его перераспределения от периферических органов в пользу головного мозга и миокарда, что облегчает восстановление самостоятельных сокращений сердца. Благодаря 0-адреномимети-ческому действию он повышает сократимость миокарда, что способствует возобновлению и усилению собственных сердечных сокращений. При асистолии адреналин помогает восстановить спонтанную сердечную деятельность, при фибрилляции желудочков и пульс-отсутствующей желудочковой тахикардии — благоприятствует восстановлению ритма сердца. Его сочетанное а- и 0-стимулирующее действие приводит к повышению сердечного выброса и артериального давления в на
чале спонтанной реперфузии, что обеспечивает увеличение мозгового кровотока и притока крови к другим жизненно важным органам.
Адреналин вводится внутривенно в дозе 1 мг или эндотрахеально — 2 —3 мг. При отсутствии эффекта повторяют введение адреналина в дозе 1 мг каждые 3 мин до суммарной дозы 5 мг. При введении адреналина через эндотрахеальную трубку его предварительно разводят в 10 мл стерильной воды (разведение 1:10 000).
Центральные вены являются оптимальными путями транспорта лекарственных препаратов в центральное кровообращение. Но могут быть использованы и периферические вены, при этом необходимо предварительно ввести 10 — 20 мл 0,9 % солевого раствора.
392 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Адреналин вводится независимо от вида остановки кровообращения.
Высокие дозы адреналина согласно основным принципам Европейского совета по реанимации не рекомендуются.
Атропина сульфат. Благодаря М-хо-линомиметическому действию, он снимает угнетающее влияние блуждающего нерва на сердце за счет блокады ацетилхолина, тормозящего активность синусового и атриовентрикулярного узлов. В отличие от адреналина атропин применяется в течение всей реанимации один раз в дозе 3 мг внутривенно или 6 мг эндотрахеально. Используется при асистолии и электромеханической диссоциации.
Амиодарон. Препарат первого выбора среди антиаритмических лекарственных средств при фибрилляции желудочков и пульс-отсутствующей желудочковой тахикардии, рефрактерной к электроимпульс-ной терапии (класс ПЬ). Он должен сразу же рассматриваться для применения после адреналина в случае неэффективного третьего импульса дефибрилляции. Это не задерживает последующий импульс. Амиодарон вводится в дозе 300 мг, разведенный в 20 мл 5 % раствора глюкозы болюсно в центральную или периферическую вену. При отсутствии положительного эффекта может вводиться дополнительная доза — 150 мг со скоростью 1 мг/мин в течение 6 ч, а затем — 0,5 мг/мин максимально до двух часов.
Лидокаин и новокаинамид (класс ПЬ). Лидокаин угнетает желудочковые экстрасистолы и повышает порог фибрилляции желудочков. Он рекомендуется в случае отсутствия амиодарона при фибрилляции желудочков и пульс-отсутствующей желудочковой тахикардии, когда дефибрилляция, повторная дефибрилляция или сочетанное применение адреналина и дефибрилляции оказываются неэффективными. Следует помнить, что лидокаин и амиодарон не должны назначаться одновременно. Препарат вводится внутривенно медленно в дозе 1 мг/кг массы тела, затем выполняется дефибрилляция. При необходимости возможна длительная инфузия в дозе 20 — 50 мкг/кг в минуту. Для этого 120 мг лидокаина разводят в 5 % ра
створе глюкозы так, чтобы суммарный объем составлял 100 мл (120 мг/мл) и проводят инфузию со скоростью 1,0 — 2,5 мг/кг в час. Повторные дозы лидокаина (0,5 —1,5 мг/кг) применяются в случаях сохраняющейся фибрилляции желудочков или пульс-отсутствующей тахикардии каждые 5 — 10 мин с последующей дефибрилляцией до суммарной дозы 3 мг/кг.
Новокаинамид используется для лечения фибрилляции желудочков и пульс-отсутствующей желудочковой тахикардии, резистентных к дефибрилляции, введению лидокаина или в случае противопоказаний для применения последнего. Вводится со скоростью 30 мг/мин до суммарной дозы 17 мг/кг.
В отдельных случаях как антиаритми-ческое средство может использоваться магний сульфат: при дефибрилляции желудочков и желудочковой тахикардии, рефрактерных к проводимым мероприятиям СЛЦР (высокая вероятность гипомагниемии), или при подозрении на наличие гипомагниемии. Вводят в дозе 1 —2 г в 100 мл 5 % раствора глюкозы в течение 1 — 2 мин.
Натрий гидрогенкарбонат или аналогичный альтернативный буфер согласно основным принципам Европейского совета по реанимации должны назначаться строго по показаниям:
♦ исходная гиперкалиемия (класс I);
♦ тяжелый метаболический ацидоз (pH < < 7,1);
♦ передозировка трициклических антидепрессантов или фенобарбитала (класс Па);
♦ при отсутствии эффекта СЛЦР в течение 20 — 25 мин после остановки кровообращения, в случае ее неэффективности для восстановления самостоятельной сердечной деятельности (класс ПЬ). Натрий гидрогенкарбонат вводится в исходной дозе 1 ммоль/кг массы тела, в последующем — 1/2 расчетной дозы каждые 10 мин, исходя из газового состава крови и показателей КОС. По рекомендации Европейского совета по реанимации — 50 мл 8,4 % раствора титровано. Для определения количества, необходимо
Методы сердечно-легочной церебральной реанимации 393
го для введения натрий гидрогенкарбоната, учитывают, что 1 мл 8,4 % его раствора содержит 1 ммоль, а 4,2 % — 0,5 ммоль вещества.
В других случаях СЛЦР введение натрий гидрогенкарбоната не предусмотрено в виду его нежелательных эффектов: развитие парадоксального внутриклеточного ацидоза ЦНС; сдвиг кривой диссоциации НЬС>2 и нарушение процесса отдачи кислорода тканям, опасность гиперосмолярности и гипернатриемии, перемещение калия в клетку с развитием гипокалиемии, инактивация одновременно вводимого адреналина, уменьшение эффективности дефибрилляции за счет снижения порога фибрилляции.
Ранее при клинической смерти (асистолия, электромеханическая диссоциация) широко использовали препараты кальция. В настоящее время они не рекомендуются, за исключением подтвержденных гипокальциемии, гипомагниемии, гиперкалиемии или передозировки блокаторов кальциевых каналов (класс Па). В таких случаях применяют 10 % раствор кальций хлорида внутривенно из расчета 0,2 мл/кг массы тела.
Диагностика вида нарушения сердечной деятельности. Ключевым моментом при восстановлении сердечной деятельности является постановка диагноза [18, 19]. Имеется в виду не обнаружение причины терминального состояния, а выявление вида нарушения сердечной деятельности. Ведущими нарушениями ритма сердца при клинической смерти являются:
1. Асистолия. Этот вид остановки кровообращения характеризуется отсутствием электрической активности сердца и механического выброса, регистрируется изоэлектрическая ЭКГ (рис. 12.21, а).
2. Электромеханическая диссоциация (ЭМД). Это состояние представляет собой механическую асистолию при сохраненной электрической активности сердца. В таких случаях кровообращение недостаточно для обеспечения кислородом головного мозга, что приводит к развитию терминального состояния. На ЭКГ регистрируется изоэлектрическая линия, на фоне которой периодически появляются нор-
Рис. 12.21. Виды остановки кровообращения: а — асистолическая; б — электромеханическая диссоциация; в — фибрилляция желудочков
мальные или измененные ЭКГ-комплексы с регулярными или нерегулярными интервалами (рис. 12.21, б). Затянувшаяся ЭМД может привести к асистолии или фибрилляции желудочков.
3. Фибрилляция желудочков — наиболее частая причина внезапной нетравматической сердечной смерти, встречающаяся у взрослых в 80 — 90 % случаев остановки кровообращения. Характеризуется частым (до 400 — 600 в минуту) возбуждением желудочков, что сопровождается хаотической, асинхронной электрической активностью отдельных мышечных волокон или их групп, прекращением систолы желудочков и отсутствием сердечного выброса. На ЭКГ отмечается характерная картина осцилляции без перемежающихся желудочковых комплексов (отсутствие QRS-комплекса и зубца Р) (рис. 12.21, в).
А. Пулъс-отсутствующая желудочковая тахикардия является наиболее опасным предшественником фибрилляции желудочков и представляет собой непрерывно следующие друг за другом преждевременные сокращения. Источник эктопических импульсов находится в проводящей системе желудочков — пучке Гиса, волокнах Пуркинье.
Для регистрации вида нарушения ритма сердца могут быть использованы электрокардиограф , электрокардиомонитор. Удобно производить вначале снятие ЭКГ через электроды дефибриллятора (если это позволяет его конструкция). При этом не требуется тратить время на наложение электродов. Кроме того, в таких дефибрилляторах предусмотрена защита блока
394 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
мониторирования ЭКГ от разряда дефибрилляции, что дает возможность практически сразу же после проведения дефибрилляции регистрировать состояние электрической активности сердца.
Каждый вид остановки кровообращения требует специфических мероприятий, поэтому его определение является обязательным условием патогенетической терапии.
Терапия отдельных видов остановки кровообращения. Асистолия. Этот вид остановки кровообращения является наиболее неблагоприятным с точки зрения прогноза возможности восстановления самостоятельной сердечной деятельности, так как отсутствие какой-либо электрической активности свидетельствует о выраженном истощении резервов организма.
Оказание неотложной помощи при асистолии осуществляется согласно алгоритму базового поддержания жизни — АВС с использованием медикаментозных средств в соответствии с вышеописанными показаниями и методиками их применения.
В результате эффективного лечения асистолии могут возникнуть несинхронные сокращения миокардиоцитов — фибрилляция желудочков и пульс-отсутствующая желудочковая тахикардия.
Электромеханическая диссоциация. Ее лечение так же, как и асистолии, начинается с базового комплекса СЛЦР и проводится с учетом вызвавшей причины, поскольку только ее устранение позволяет добиться восстановления сердечного выброса.
Этиологические факторы, способствующие возникновению ЭМД, можно разделить на три группы: 1) «пустое сердце» — значительное уменьшение притока крови к правым и левым его отделам; 2) «блокада малого круга» — кровь из правых отделов сердца не поступает в левые; 3) «сердечная слабость» — приток крови к сердцу и поступление ее в левые отделы сердца не нарушены, но миокардиоциты не в состоянии выполнять механическую работу. В их основе могут лежать потенциально обратимые причины сердечной смерти (табл. 12.2). Так, при ЭМД, вызванной критической гиповолемией, главным лечебным мероприятием II стадии является восполнение ОЦК путем использования
кристаллоидных и коллоидных растворов. При этом восполнение глобулярного объема отходит на второй план.
При сдавлении сердца вследствие тампонады лечебным мероприятием является пункция полости перикарда с аспирацией содержимого. В случае напряженного пневмоторакса его необходимо разгрузить с помощью постоянной аспирации содержимого плевральной полости или перевести закрытый напряженный пневмоторакс в открытый.
При электромеханической диссоциации, вызванной тромбоэмболией легочной артерии, проводят тромболитическую терапию с использованием препаратов, содержащих стрептокиназу (стрептолиаза), и актилизе. В случае если причиной ЭМД является острая сердечная недостаточность, применяют кардиотонические препараты, прежде всего адреналина гидро-генхлорид, допамин, добутрекс.
Фибрилляция желудочков и пульс-отсутствующая желудочковая тахикардия. Это наиболее благоприятный вид остановки кровообращения, поскольку наличие электрической активности сердца свидетельствует о жизнеспособности структур миокарда и имеющихся резервах. Однако последние быстро истощаются, так как некоординированные сокращения кардиомиоцитов не обеспечивают сердечного выброса, но требуют больших энергетических затрат. Быстрое истощение резервов и возможный благоприятный исход при своевременном оказании неотложной помощи предопределили современный взгляд на место дефибрилляции в комплексе СЛЦР. Американской кардиологической ассоциацией предложено понятие «цепочка выживания», где дефибрилляция рассматривается как одно из первоочередных мероприятий при любом виде остановки кровообращения.
Согласно принципам «цепочки выживания» рекомендуется следующая последовательность оказания первой помощи при клинической смерти:
1) ранний вызов помощи;
2) раннее начало первой стадии СЛЦР;
3) ранняя дефибрилляция;
4) ранняя специализированная терапия.
Методы сердечно-легочной церебральной реанимации 395
Рис. 12.22. Стандартное положение электродов при дефибрилляции: а — два ручных электрода; б — стационарный подлопаточный и ручной электроды
При дефибрилляции (класс I) электрический разряд, проходя через сердце, подавляет эктопические очаги, мешающие восстановлению синусового ритма, что способствует восстановлению синхронной электрической, а вместе с ней и механической активности миокарда.
В современных условиях используются два типа дефибрилляторов. Зарубежные дефибрилляторы генерируют монопо-лярные импульсы тока. В странах СНГ широко используются аппараты, генерирующие биполярные асимметричные квазиси-нусоидальные импульсы, предложенные Н. Л. Гурвичем и названные в его честь. В настоящее время имеются усовершенствованные портативные дефибрилляторы.
При проведении дефибрилляции биполярными импульсами наряду с более высокой эффективностью значительно меньше выражены жизнеопасные нарушения ритма и проводимости сердца, развивающиеся вследствие повреждения миокарда и нарушения его функции.
Электрический разряд опасен как для больного, так и для персонала, поэтому при проведении дефибрилляции требуется тщательное соблюдение правил техники безопасности: при проведении электрического разряда никто из них не должен касаться металлических деталей кровати. Во избежание ожогов кожи и для снижения сопротивления электроды следует плотно прижать к коже. Для уменьшения электрического сопротивления кожи используют специальную токопроводящую пасту, которую наносят на электроды. Вместо пасты допус
тимо применение марлевых салфеток, смоченных физиологическим раствором.
Проведение дефибрилляции начинают как можно раньше, сразу после выявления любого из видов фибрилляции желудочков или пульс-отсутствующей желудочковой тахикардии (мелко-, средне- и крупноволновая). Разряд производится только на выдохе, что уменьшает трансторакальное сопротивление на 15 — 20 %.
В зависимости от конструкции дефибриллятора применяют два стандартных положения электродов (рис. 12.22).
При передне-переднем варианте один электрод помещается в области верхнего правого края грудины под ключицей, другой — над верхушкой сердца по срединно-ключичной линии.
При передне-заднем варианте один электрод устанавливается медиальнее левого соска, другой — под левой лопаткой.
Электрические разряды должны быть достаточной мощности. Первые три разряда производят с минимальными интервалами и нарастающей энергией (табл. 12.3).
У детей энергия 1-го разряда составляет 2 Дж/кг, 2-го — 3 Дж/кг и 3-го — 4 Дж/кг.
Таблица 12.3. Энергия электрических разрядов в зависимости от типа импульса
Разряд Монопо-лярный импульс, Дж Биполярный импульс, Дж Напряжение разряда, кВ
1-й 200 65-90 4
2-й 250-300 140 5,7
3-й 360 190 7
396 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
При неэффективности начальных разрядов все последующие проводятся с максимальной энергией (360 Дж) в сочетании с медикаментозной терапией каждые 30 — 60 с и проведением в промежутках мероприятий I стадии СЛЦР (рис. 12.20). Описана высокая эффективность чрезпи-щеводного способа дефибрилляции, осуществляемой с имплантированного кардиостимулятора, преимущественно для больных, находящихся в кардиологических и кардиохирургических отделениях.
Ошибки при проведении дефибрилляции: ♦ задержка с началом проведения;
♦ плохо смочены прокладки под электродами;
♦ электроды недостаточно плотно прилегают к грудной клетке;
♦ неправильно выбрана энергия разряда; ♦ повторение электрического разряда сразу после введения лекарственных препаратов без предшествующего проведения наружного массажа сердца в течение одной минуты;
♦ использование технически неисправного дефибриллятора;
♦ несоблюдение правил техники безопасности.
Алгоритм проведения реанимационных мероприятий во второй стадии СЛЦР представлен на рис. 12.23.
Универсальный алгоритм реанимации, рекомендуемый Европейским советом по реанимации у взрослых (2000), обобщенно представляет объем и последовательность проводимых мероприятий в I и II стадии СЛЦР (рис. 12.24).
Кроме того, Европейским советом по реанимации в стандарт СЛЦР внесены угрожающие жизни нарушения ритма, такие как брадикардия, предсердная фибрилляция, узко- и ширококомплексная суправентрикулярная тахикардия, введен новый раздел — острые коронарные синдромы.
Брадикардии. Эти формы нарушений ритма легко отличаются от нормальной ЭКГ и определяются как частота сердечных сокращений реже 60 в минуту. Брадикардия независимо от ее происхождения опасна для больного в критическом состоянии, если частота сокращений реже
50 в минуту. Синусовая брадикардия может представлять опасность у пациентов с поражением миокарда, брадикардия желудочкового происхождения обусловлена блокадой проводящей системы сердца, чем представляет угрозу для жизни больного.
Последовательность европейского алгоритма при брадикардии за последние годы изменилась незначительно (рис. 12.25). Не рекомендуется прием изопреналина. Если проведение наружной электростимуляции невозможно, рекомендуются низкие дозы адреналина.
Тахикардии. Европейский совет по реанимации не принял алгоритмы лечения тахикардии, изданные в международных алгоритмах-2000, но несколько изменил существующие и добавил алгоритм оказания помощи при предсердной фибрилляции, ввел новый раздел — острые коронарные синдромы. При этом были сформулированы основные принципы оказания реанимационной помощи:
1. Непосредственное лечение зависит от стабильности состояния больного.
2. При нестабильном состоянии более предпочтительна кардиоверсия.
3. Все антиаритмические препараты обладают проаритмическими свойствами.
4. Нежелательно использование более одного антиаритмического препарата.
5. Если лекарственное средство не действует, должна рассматриваться кардиоверсия, но после антиаритмической терапии.
6. Если у больного нарушена функция миокарда, использование большинства ан-тиаритмических средств приводит к дальнейшему ее ухудшению.
Предсердная фибрилляция. Фибрилляция предсердий иногда может наблюдаться у лиц и без органического поражения сердца, но чаще всего является признаком заболевания сердца в результате нерегулярного, неправильного желудочкового ритма, который отличается характерным типом дизаритмий и может быть немедленно диагностирован даже при пальпации пульса (pulsus irregularis perpetuus — постоянно неритмичный пульС — постоянный патологический ритм).
При этом выделяют три группы риска в зависимости от частоты сердечных
Методы сердечно-легочной церебральной реанимации 397
Рис. 12.23. Алгоритм проведения реанимационных мероприятий (II стадия СЛЦР)
398 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.24. Универсальный алгоритм реанимации:
ФЖ — фибрилляция желудочков; ЖТ — желудочковая тахикардия; СЛЦР — сердечно-легочная церебральная реанимация
сокращений и присутствия дополнительных симптомов и признаков (рис. 12.26). В случае высокого риска предпринимается электрическая кардиоверсия после предварительной гепаринизации. Тактика лечения больных при среднем риске зависит от наличия или отсутствия расстройств гемодинамики или органических нарушений сердца. Лечение начинается тогда, когда известно о наличии предсердной фибрилляции в течение последних 24 ч.
Кардиоверсия может быть предпринята и у больных с низким риском, если известно о наличии .предсердной фибрилляции в течение последних 24 ч. Если продолжительность фибрилляции более 24 ч, кардиоверсия может проводиться лишь после предварительного назначения антикоагулянтов в течение трех-четырех недель.
Суправентрикулярная тахикардия. Различают узкокомплексную и широко-
Методы сердечно-легочной церебральной реанимации 399
Рис. 12.25. Алгоритм реанимации при брадикардии
комплексную суправентрикулярную тахикардию.
При узкокомплексной суправентрикулярной тахикардии частота сердечных сокращений (ЧСС) превышает 250 в минуту. Если при этом отсутствует пульс, следует провести вагусные пробы (проба Вальсальва, каротидный массаж), после чего должна быть предпринята попытка электрической кардиоверсии (рис. 12.27). Средством первого выбора является аденозин (класс Па).
Если у больного обнаруживаются такие дополнительные признаки, как снижение систолического АД ниже 90 мм рт. ст., боль в груди, сердечная недостаточность, следует провести электрическую кардиоверсию, при необходимости — с введением амиодарона. При отсутствии дополнительных признаков используют один из препаратов: эсмолол, верапамил, амиодарон, дигоксин.
При ширококомплексной тахикардии на ЭКГ регистрируется широкий QRS-комп-лекс. В случае отсутствия пульса действуют по алгоритму фибрилляции желудоч
ков. Если у больного имеют место такие признаки, как снижение систолического АД ниже 90 мм рт. ст., ЧСС менее 150 в минуту и боль в груди, вводятся амиодарон и лидокаин, при отсутствии чувствительности к ним проводится электрическая кардиоверсия (рис. 12.28).
Острые коронарные синдромы. Это новый раздел, введенный Европейским советом по реанимации в руководство-2000.
Основные принципы оказания неотложной помощи этой категории больных состоят в следующем:
1. На догоспитальном этапе должна быть доступна ЭКГ с 12 отведениями. Телемедицина или компьютерный анализ ЭКГ могут расширить догоспитальные диагностические возможности.
2. На догоспитальном этапе должны быть доступны средства для непосредственной дефибрилляции и управления аритмиями.
3. При отсутствии противопоказаний всем больным с болью в груди по типу ишемической должны назначаться кислород, опиоиды и нитраты (класс I).
400 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.26. Три группы риска фибрилляции предсердий в зависимости от частоты сердечных сокращений и наличия дополнительных симптомов и признаков: дозы указаны из расчета на среднюю массу тела взрослого
* Не применять у пациентов, получающих 0-блокаторы.
** ЭИТ — электроимпульсная терапия — всегда проводится после седации или под общей анестезией.
Методы сердечно-легочной церебральной реанимации 401
Рис. 12.27. Алгоритм проведения реанимационных мероприятий при узкокомплексной суправентрикулярной тахикардии: дозы рассчитаны на среднюю массу тела взрослого. Теофиллин и близкие производные блокируют эффект аденозина.
У пациентов, принимающих дипиридамол, карбамазепин, или с денервированным сердцем (пересадка) применение может быть рискованным.
ЭИТ проводится под седацией или общей анестезией
402 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.28. Алгоритм проведения реанимационных мероприятий при ширококомплексной суправентрикулярной тахикардии:
дозы рассчитаны на среднюю массу тела взрослого. ЭИТ проводится под седацией или общей анестезией. При пароксизмах мерцания-трепетания используйте магний или электростимуляцию (помощь специалиста)
4. При отсутствии противопоказаний все больные с острым инфарктом миокарда должны получить аспирин и в условиях стационара — p-блокаторы (класс I).
5. Если время «от звонка до иглы в стационаре» превышает 60 мин, на догоспитальном этапе полезно применение фиб-ринолитиков (класс I).
6. В центрах с большим количеством больных и наличием опытного персонала
альтернативой фибринолитической терапии может быть ангиопластика (класс I).
7. Больным в состоянии кардиогенного шока первичная ангиопластика и внутриаортальная баллонизация должны проводиться только в соответствующих специализированных центрах (класс I).
8. Больным с He-Q-инфарктом и высоким риском нестабильной стенокардии может быть предложено антитромботичес-
Методы сердечно-легочной церебральной реанимации 403
кое лечение с применением ингибиторов гликопротеидов ПЬ/Ша. Вместо несанкционированного гепарина могут использоваться гепарины с низкой молекулярной массой (класс не определен).
9. Больные с большим передним инфарктом и/или ухудшением функции левого желудочка при отсутствии противопоказаний должны получать схемы АПФ-ин-гибиторов.
10. Применение смеси «глюкоза —калий—инсулин» может быть полезным у больных с сахарным диабетом и тех, кто подвергся реперфузионной терапии.
Тактические ошибки при проведении СЛЦР:
♦ задержка с началом проведения СЛЦР; ♦ отсутствие единого руководителя;
♦ отсутствие постоянного контроля эффективности проводимых мероприятий;
♦ отсутствие четкого учета лечебных мероприятий и контроля их выполнения;
♦ переоценка нарушений КОС, неконтролируемая инфузия натрий гидрогенкар-боната;
♦ преждевременное прекращение реанимационных мероприятий;
♦ ослабление контроля состояния больного после восстановления кровообращения и дыхания.
III стадия — длительное поддержание жизни
Эта стадия СЛЦР представляет собой долгосрочную интенсивную терапию, направленную на поддержание жизнедеятельности пораженных органов, в основном на восстановление функций головного мозга. Она предусматривает оценку тяжести поражения, критериев раннего прогнозирования, отдаленных результатов, критерии и порядок прекращения реанимационных мероприятий [6, 20].
Оценка состояния больного. Включает определение: 1) причины клинической смерти с целью предупреждения повторных эпизодов остановки кровообращения, каждый из которых ухудшает прогноз полноценного восстановления личности больного; 2) степени тяжести нарушений гомеостаза в целом и прежде всего функций головного мозга для уточнения объема и характера интенсивной терапии.
Исход лечения в смысле выживания и полноценного восстановления всех функций организма (особенно мышления) зависит не только от тяжести и продолжительности ишемического поражения, но и от своевременности и адекватности реанимационных мероприятий, сопутствующих заболеваний, вторичных осложнений, раннего начала высококвалифицированной специализированной интенсивной терапии в постреанимационном периоде, особенно терапии, направленной на восстановление функций мозга, поскольку это определяет качество жизни больных в отдаленном периоде и имеет огромные социально-экономические последствия как для пострадавшего, так и для его семьи и всего общества.
Причина клинической смерти, как правило, выясняется еще во время первых двух стадий СЛЦР, так как без этого часто невозможно восстановить самостоятельное кровообращение. Предотвращению повторных эпизодов остановки кровообращения помогает и оценка тяжести нарушений гомеостаза, поскольку выраженные нарушения со стороны дыхательной и сердечно-сосудистой систем, водно-электролитного обмена, КОС сами по себе могут быть причинами клинической смерти.
Для оценки степени нарушения гомеостаза используется комплекс клиниконеврологических, биохимических, электрофизиологических, рентгенологических исследований, проводится количественная оценка глубины подавления функций ЦНС с помощью шкалы Глазго или Глазго — Питтсбург. Обнаруживаемые при этом изменения отражают стадии функциональных нарушений основных жизненно важных систем организма (табл. 12.4).
Стадии (фазы) восстановления функций центральной нервной системы представлены в табл. 12.5.
В зависимости от полноценности восстановления центральной нервной системы различают [21]:
♦ полное восстановление: быстрое (в течение суток); задержанное (в течение 2 — 3 суток);
♦ прерванное;
♦ восстановление с наличием дефицита, не требующим дополнительного ухода;
404 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Таблица 12.4. Стадии функциональных нарушений жизненно важных систем в постреанимационном периоде
Стадия Время развития Клинические проявления
Сердечно-сосудистая система
I Через 10 — 20 мин после оживления Наблюдается гипердинамический синдром с увеличением сердечного выброса, тахикардией, увеличением коронарного кровообращения и снижением сосудистого сопротивления
II Через 40 — 60 мин Относительная нормализация кровообращения с восстановлением нормального сердечного выброса, периферического сопротивления и других показателей
III Через 90 — 120 мин Развивается гиподинамический синдром с повторным ухудшением функций сердечно-сосудистой системы в виде синдрома «малого выброса» Система дыхания
I Через 10 — 20 мин Нормализация системного дыхания и структуры дыхательного акта. Восстанавливается самостоятельное дыхание
II Через 25 — 60 мин Отмечается гипервентиляция и усиление потребления организмом кислорода
III Через 3 —6 ч Продолжается усиление потребления организмом кислорода
IV Свыше 6 ч При развитии синдрома «малого выброса» продолжается гипервентиляция легких на фоне повторного развития циркуляторной гипоксии
Таблица 12.5. Динамика фаз обратного развития бессознательного состояния, вызванного острым поражением головного мозга (по М. А. Мяги, 1968)
Фаза Нейрофизиологическая характеристика Клинические проявления
I Запредельная кома Полное необратимое прекращение всех функций мозга Полная арефлексия, нарушение дыхания и деятельности сердца, падение АД
t II 4 Острая кома с тяжелыми расстройствами вегетативных функций Тяжелое нарушение всех функций головного мозга Нарушение дыхания, отсутствие кашлевого рефлекса, гипотония мышц, падение АД
t III 4 Кома с некоторой стабилизацией вегетативных функций Восстановление и стабилизация функций понтобульбарных структур Восстановление дыхания, кашлевого рефлекса, повышение тонуса мышц, появление сухожильных рефлексов, симптомов очагового поражения
Т IV 4 Апаллический синдром Восстановление мезентериальной ретикулярной восходящей активирующей системы и улучшение функций нисходящих ретикулярных систем Состояние улучшается, больной спонтанно открывает глаза, но взгляд не фиксирует, контакт невозможен. Периоды бодрствования сменяются периодами сна. Восстанавливаются реакции зрачков на свет
Т V 4 Акинетический му-тизм Восстановление функций таламической системы сетевидного образования и таламо-кортикальной неспецифической проекционной системы, диэнцефальных тормозных систем Больной остается неконтактным, но начинает фиксировать взгляд, следить за движущимися объектами, поворачивать голову в сторону звукового раздражителя. Удлиняются периоды бодрствования
Смерть головного мозга 405
Продолжение табл. 12.5
Фаза Нейрофизиологическая характеристика Клинические проявления
t VI г т VII 1 t VIII Фаза восстановления Фаза глубокой демент-ности и преобладание в клинической картине гипоталамических и лимбических функций Фаза восстановления высших психических и соматических функций Первые признаки восстановления лимбических и неокортикальных функций при недостаточном уровне неспецифической активации Недостаточность восстановления лимбических и неокортикальных функций при достаточной реинтеграции неспецифических активирующих механизмов Восстановление и укрепление связей между кортикальными областями, неокортексом и другими функциональными системами Больной выполняет инструкции, говорит отдельные слова. Дальнейшее удлинение периодов бодрствования Увеличение периода бодрствования, улучшение речи, проявление эмоций, нарушения памяти и речи
♦ частичное восстановление с наличием дефекта, исключающего самообслуживание;
♦ временное частичное улучшение без выхода из комы.
Тяжесть повреждений головного мозга вследствие перенесенной клинической смерти является определяющим фактором прогнозирования исхода и выбора такти
ки интенсивного лечения. Это связано с тем, что ткани головного мозга являются наиболее чувствительными к ишемии и гипоксии, поскольку в норме потребляют до 25 % общего количества поступающего в организм кислорода и именно головной мозг выполняет интегрирующие функции управления жизнедеятельностью организма.
12.4. СМЕРТЬ ГОЛОВНОГО МОЗГА
При выраженном ишемическом и реперфузионном повреждении головного мозга в случае неблагоприятного течения постреанимационного периода развивается смерть головного мозга [22].
Смерть головного мозга — уникальное состояние, являющееся следствием клинической смерти и реанимационных мероприятий. Это ятрогенное заболевание, так как развивается в случае вмешательства медицинского персонала в естественный процесс умирания.
Определение ее наличия или отсутствия у больного является важным не только с точки зрения выбора тактики дальнейшего лечения, но и с позиций клинической трансфузиологии.
Диагностика смерти мозга производится на основании комплекса признаков, включающих результаты клинико-неврологических, биохимических, электрофизиологических, рентгенорадиологических и
морфологических исследований, проведенных в соответствии с установленными принципами.
Клинико-неврологические признаки смерти мозга\
♦ атоническая (запредельная) кома;
♦ расширенные зрачки с отсутствием реакции на свет;
♦ неподвижность глазных яблок, их гипотония, помутнение роговицы;
♦ отсутствие самостоятельного дыхания;
♦ нестабильная гемодинамика (гипотензия, брадикардия);
♦ отсутствие окулоцефалического, роговичного, зрачкового, глоточного и других рефлексов черепно-мозговых нервов и сухожильных рефлексов;
♦ прогрессирующее снижение температуры тела (до 32 °C и ниже);
♦ адинамия;
♦ отрицательная реакция на пробы провокации.
406 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Техника проведения провокационных проб'.
проба с атропином: внутривенно вводят 1 мл 0,1 % раствора атропина сульфата и оценивают изменение ЧСС. Если спустя 2 — 3 мин она увеличивается не более, чем на 5 ударов в минуту, проба считается отрицательной;
холодовая проба: барабанную перепонку орошают водой (около 50 мл) комнатной температуры через предварительно введенный в наружный слуховой ход мягкий катетер. Следят за движением глазных яблок. Отсутствие нистагма свидетельствует об отрицательной пробе;
проба с бемегридом: во время записи ЭЭГ внутривенно вводят 25 — 50 мг бемег-рида и наблюдают за изменениями характера электроэнцефалограммы. Отсутствие электрической активности во всех зонах коры головного мозга после его введения является признаком смерти головного мозга.
Биохимические признаки смерти мозга'.
уменьшение артерио-венозной разницы по кислороду в притекающей к мозгу и оттекающей от него крови ниже 2 об%; доля < 2 %;
уменьшение артерио-венозной разницы по глюкозе в притекающей к мозгу и оттекающей от него крови;
повышение уровня лактата в оттекающей от мозга крови и в ликворе;
значительное повышение активности ферментов в крови и спинно-мозговой жидкости, особенно лизосомальных.
Электрофизиологические признаки смерти мозга:
изоэлектрическая линия на всех отведениях ЭЭГ, регистрируемой непрерывно в течение 30 мин 3 — 4 раза в сутки, лучше использование мониторинга функций мозга;
отсутствие повышения электрической активности на ЭЭГ в ответ на световые, звуковые и болевые раздражители (ЭЭГ с вызванными потенциалами).
Рентгенорадиологические признаки смерти мозга:
наличие участков обеднения сосудистой сети при ангиографии сосудов головного мозга;
наличие участков некроза головного мозга, регистрируемое с помощью компьютерной томографии.
Морфологические признаки смерти мозга — признаки некроза мозговой ткани в биоптатах, взятых через 8 — 10 трепанационных отверстий.
Для исключения ошибочной диагностики смерти головного мозга необходимо исключить воздействие медикаментов, угнетающих церебральный метаболизм, миорелаксантов, интоксикацию нейротоксическими веществами.
Таким образом, при применении методов реанимации в ряде случаев развивается особое состояние организма человека — смерть мозга, характеризующаяся необратимым поражением всего мозга, включая его стволовые отделы и шейные сегменты, при работающем сердце. Кровообращение в этих случаях обеспечивается ИВЛ, введением поддерживающих гемодинамику дорогостоящих лекарственных препаратов и в определенной мере сохранностью регулирующих систем спинного мозга. Поддержание сердечной деятельности и метаболизма требует больших материальных затрат и усилий медицинского персонала в то время, как человек с утратой функций головного мозга перестает существовать как личность. В связи с этим возникла сложнейшая морально-этическая проблема — можно ли признать человека мертвым при тотальном некрозе мозга, но сохранившейся сердечной деятельности и приравнять смерть мозга к смерти человека в целом [23, 24].
В ряде стран смерть мозга признана эквивалентной смерти человека и это положение введено в ранг закона. Это позволяет изъять из такого организма органы, что имеет важное значение для трансплантологии, так как работающее сердце обеспечивает лучшую сохранность их функций. Однако при этом следовало бы принять законы «О трансплантации тканей и органов» и «О смерти мозга», которые бы гарантировали как охрану прав человека, так и создание оптимальных условий для трансплантации органов.
Процессы, происходящие в организме больного во время клинической смерти и
Смерть головного мозга 407
проведения реанимационных мероприятий, вызывают особое состояние организма, которое В. А. Неговским обозначено как постреанимационная болезнь.
Постреанимационная болезнь — это специфическое патологическое состояние, развивающееся в организме больного вследствие ишемии, вызванной тотальными нарушениями кровообращения и реперфузией после успешной реанимации и характеризующееся тяжелыми расстройствами различных звеньев гомеостаза на фоне нарушенной интегративной функции ЦНС. Она может иметь благоприятное и неблагоприятное течение. В зависимости от времени, прошедшего после клинической смерти, выделяют такие стадии течения постреанимационной болезни (по Е. С. Золотокрылиной, 1999) [25]:
I стадия (6 — 8 ч от начала лечения) — стадия нестабильных функций, характерными чертами которой являются максимально выраженное снижение перфузии тканей (в 4 —5 раз) и наличие циркуляторной гипоксии.
II стадия (10—12 ч от начала лечения) — стадия относительной стабилизации основных функций организма. Она характеризуется стабилизацией жизненно важных функций организма, улучшением (часто временным) состояния больных. Сохраняются выраженные нарушения перфузии тканей (их кровенаполнение снижено в 2 —2,5 раза). Сохраняется дефицит ОЦК (даже при отсутствии кровопотери). Отмечается увеличение потерь К+ и ретенция Na+ в организме, сохраняется лактатацидоз, увеличенная сумма органических кислот.
Наблюдается выраженная гиперфер-ментемия: в 4 —7 раз увеличивается уровень креатинфосфокиназы, в 2 — 2,5 раза — щелочной фосфатазы, ЛДГ и других ферментов. Достоверно замедляется фибринолитическая активность плазмы крови. Динамика этой стадии часто определяет исход заболевания.
III стадия (конец 1—2 сутки лечения) — стадия повторного ухудшения состояния больных. У части из них при нормальной температуре тела наблюдается одышка, тахикардия, повышение АД у лиц
молодого и среднего возраста, беспокойство. В результате максимального снижения транспорта кислорода в связи с нарушением свойств гемоглобина и диссоциации оксигемоглобина, сохранением сниженной перфузии тканей, шунтированием кровотока в легких и гиподинамическим состоянием кровообращения формируется гипоксия смешанного генеза.
В этой стадии отмечаются наиболее выраженные нарушения гемостаза и фибринолиза (тромбинемия, гиперкоагуляция, несмотря на активацию антикоагулянтного звена гемостаза, увеличение продуктов деградации фибрина и фибриногена на фоне замедления фибринолитической активности плазмы крови), что создает условия для появления в органах и тканях микротромбов. Именно в 1—2 сутки лечения наблюдаются первые клинические проявления микротромбозов. Почти у половины больных развивается нарушение функции паренхиматозных органов: почек — по типу функциональной олигурии; нарастание ОНД по типу неспецифического поражения легких («шоковое легкое»); реже — печени. Все вышеназванные нарушения носят функциональный характер и при благоприятном течении обратимы.
IV стадия (3 — 4 сутки) может развиваться в двух разных направлениях: стабилизация и последующее улучшение нарушенных функций организма, отсутствие осложнений и выздоровление больных; дальнейшее ухудшение состояния больных в связи с прогрессированием генерализованного воспалительного ответа и нарушений многих функций. При этом отмечается: усиление катаболизма, водноэлектролитные нарушения с появлением и увеличением отека интерстициальной ткани легких, мозга, подкожной клетчатки; углубление гипоксии смешанного типа и гиперкоагуляции, появление гнойно-воспалительных осложнений. На этом фоне развиваются признаки недостаточности функций органов: вторичные кровотечения из верхних отделов пищеварительного канала (эрозии), психозы с галлюцинаторным синдромом, вторичная сердечная недостаточность, углубление ОНД, пан
408 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
креатиты, расстройства функции печени, бескаменные холециститы.
V стадия (5 — 7 сутки). Эта стадия имеет место лишь при неблагоприятном течении постреанимационной болезни. Отмечается дальнейшее прогрессирование воспалительных и гнойных процессов: массивные пневмонии, часто нозокомиальные, абсцедирующие; нагноение ран; перитониты у оперированных больных; абсцессы брюшной полости и мягких тканей; гнойные плевриты. Нередко наступает генерализация инфекции — сепсис, несмотря на раннее применение антибиотиков и антисептиков.
Увеличение антибактериальной резистентности в постреанимационном периоде возникает на фоне выраженных нарушений клеточного и гуморального иммунитета в результате длительной тяжелой гипоксии смешанного типа.
На фоне сепсиса, как правило, начинается новая, вторая волна поражения легких, сердечной мышцы, печени, почек. Органы становятся функционально несостоятельными — развивается полиорган-ная недостаточность.
Постреанимационная болезнь неизбежно сопровождается постгипоксической энцефалопатией, под которой понимают вре
Рис. 12.29. Алгоритм проведения реанимационных мероприятий при постгипоксической энцефалопатии
Интенсивная терапия постреанимационной болезни 409
менное или стойкое нарушение функционирования ЦНС, возникающее вследствие острой тотальной кислородной недостаточности клеток головного мозга [26 — 28]. В течение первых 10 —12 с гипоперфузии и гипоксии в клетках мозга резко падает уровень кислорода, в последующие 3 — 5 мин в нейронах истощаются запасы АТФ и гликогена, в результате чего нарушаются все энергозависимые процессы: транспорт ионов через мембраны, проведение импульсов и целостность мембраны (рис. 12.29). Следствием этого является накапливание в цитозоле ионов кальция. Активируется фосфолипаза Л2, связанная с мембраной, ускоряется распад фосфолипидов мембран с образованием и накоплением в цитозоле арахидоновой кислоты. С устранением кислородной недостаточности и возобновлением поступления кислорода в клетки распад арахидоновой кислоты по липооксигеназному и циклооксигеназному путям приводит к образованию биологически активных веществ — тромбоксанов, лейкотриенов, простагландинов, свободных радикалов кислорода. Эти вещества оказывают двойное действие на нейроны: разрушают мембраны нейронов, вызывая их гибель, и, влияя на мембраны эндотелиоцитов кровеносных сосудов головного мозга, вызывают нарушение свойств сосудистой стенки, в результате чего она из антикоагулянтной становится
прокоагулянтной. Это ведет к тромбо-образованию с мозаичным нарушением кровотока и повторной, более продолжительной ишемией и гипоксией ткани мозга.
Различают две фазы постгипоксической энцефалопатии как основного патогенетического компонента постреанимационной болезни:
I. Острую (катаболическую):
а) активация обменных процессов или их торможение в случае срыва адаптации;
б) развитие в нейронах энергетического дефицита;
в) эндогенная интоксикация интра- и экстрацеребрального генеза.
II. Стабилизации:
а) неустойчивая стабилизация гомеостаза (напряжение и истощение адаптационных процессов);
б) устойчивая стабилизация гомеостаза (анаболическая фаза).
Фаза 1а длится в течение 2 — 3 суток, 16 и 1в — развиваются одновременно, с 3 — 7 до 7 — 10 суток постреанимационного периода. При этом фаза 16 достигает максимального развития на 2 —5, а фаза 1в — на 6 —10 сутки. Фаза Па имеет место при неблагоприятном течении постреанимационного периода, может закончиться смертью или длиться месяцы и даже годы. Фаза Пб развивается в случае благоприятного течения и продолжается от нескольких недель до месяцев.
12.5. ИНТЕНСИВНАЯ ТЕРАПИЯ ПОСТРЕАНИМАЦИОННОИ БОЛЕЗНИ
Основной задачей интенсивной терапии и конечной ее целью в постаноксическом периоде является сохранение нейронов мозга и восстановление его функций. С этой целью проводятся два типа мероприятий:
поддержание функций экстрацеребраль-ных органов и систем, направленное на восстановление адекватного кровообращения и функций других органов, оптимизацию газового состава крови, устранение водно-электролитных нарушений, обеспечение энергетическими и пластическими материалами [20, 27, 29];
специальные мероприятия по оптимизации функций мозга, предотвращающие не
кроз, максимально снижающие действие факторов, инициирующих апоптоз и стимулирующих механизмы саногенеза, а также целенаправленно регулирующие процессы реорганизации межнейронных взаимоотношений путем подавления формирования патологических доминантных систем [17, 20, 27, 30].
Таким образом, проводится комплекс экстра- и интрацеребральных мероприятий с учетом течения постреанимационной болезни [6, 27, 31, 32].
Мероприятия экстрацеребралъ-ного воздействия
1. Коррекция гемодинамических нарушений. Направлена на устранение гипотензии,
410 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
анемии, нарушений реологических свойств крови, восстановление регионарного кровотока, устранение гипоперфузии тканей. Проводится согласно общепринятым принципам с поправкой на необходимость создания умеренной гиперволемической гемодилюции.
2. Нормализация функций дыхания. Предусматривает обеспечение свободной проходимости дыхательных путей как на период комы, так и после восстановления сознания. При отсутствии самостоятельного дыхания или его неадекватности применяется ИВЛ. Учитывая важность предупреждения вторичной гипоксии, приоритет отдается продленной ИВЛ. Согласно рекомендациям Европейского совета по реанимации у больных с ИВЛ величину РаСО2 необходимо поддерживать в пределах нормальных значений (класс Па). Гипервентиляция, приводящая к снижению значений РаСО2 ниже нормальных, может оказаться вредной (класс III).
3. Коррекция нарушений водно-электролитного обмена, КОС. Проводится согласно общепринятым принципам с учетом мероприятий по профилактике отека мозга и проведению умеренной гиперволемической гемодилюции. Водный баланс следует поддерживать на нулевом уровне.
4. Коррекция нарушений системы гемокоагуляции. Состоит из мероприятий по сбалансированию прокоагулянтного и антикоагулянтного звеньев этой системы, а также по устранению нарушений микроциркуляции и восстановлению функциональной активности системы мононуклеарных фагоцитов.
5. Профилактика и лечение острой печеночной и почечной недостаточности. Прежде всего проводятся мероприятия по восстановлению ОЦК, достаточной оксигенации, ускорению выведения из организма или нейтрализации эндотоксинов. При развитии ОПН в случае необходимости проводятся сеансы гемодиализа, ультрафильтрации; при острой печеночной недостаточности — плазмафереза, гемосорбции.
6. Коррекция нарушений иммунной реактивности. Осуществляется частично за счет препаратов, используемых в схеме интенсивной терапии для других целей (аскорбиновая кислота, токоферол, плазма). Специ
фические препараты назначаются в соответствии с иммунологическим диагнозом.
7. Профилактика и лечение гнойно-септических осложнений. Достигается восстановлением микроциркуляции и назначением антибиотиков широкого спектра действия.
У больных с лихорадкой температура тела должна быть снижена и назначены антипиретики (класс Па).
8. Энтеральное и парентеральное питание. До восстановления функции кишок проводится стимуляция перистальтики и применяется парентеральное питание. После восстановления перистальтики — энтеральное питание в соответствии с принципами, изложенными в соответствующем разделе.
9. Детоксикационная терапия. С этой целью применяются методы форсированного диуреза, ультрафиолетового облучения крови, при необходимости — эфферентные методы детоксикации.
Мероприятия интрацеребрального воздействия
Нейрореабилитационное лечение больных, перенесших аноксию головного мозга, проводится в первые сутки постреанимационного периода в зависимости от характера и тяжести выявленных неврологических нарушений.
Г. В. Алексеева (1996) выделяет три группы больных:
первая — с неосложненным восстановлением функций ЦНС, с длительностью нарушения сознания не более 3 —4 ч;
вторая — с осложненным восстановлением функций ЦНС, с длительностью расстройства сознания до 24 ч;
третья — с задержкой восстановления функций ЦНС, с восстановлением сознания позже 24 ч.
Общая схема нейрореабилитационной терапии больных, перенесших терминальные состояния, представлена в табл. 12.6.
Следует отметить, что в первые 3 ч постаноксического периода интенсивная терапия для всех трех групп больных проводится одинаково и включает применение следующих препаратов:
1) снижающих энергетические потребности мозга: диазепам (седуксен, реланиум,
Таблица 12.6. Общая схема нейрореабилитационной терапии больных, перенесших терминальные состояния (по В. В. Семченко, С. С. Степанову, Г. В. Алексеевой, 1999)
Первые три часа Острый период Подострый период Отдаленный период
Стадия угрожающей жизни комы Стадия стабилизации вегетативных функций
В первые три часа постаноксического периода профилактика психоневрологических расстройств у всех больных идентична При продолжении острого периода с клинической картиной среднемозгового синдрома III степени Сократить введение препаратов, снижающих метаболические процессы, и начать введение следующих групп препаратов под контролем электроэнцефалограммы и неврологического статуса Исключить препараты, снижающие энергетические потребности мозга (при отсутствии симптомов раздражения), и продолжить введение следующих групп препаратов Терапия часто проводится амбулаторно таблетированными медикаментозными средствами; один раз в 4 месяца — стационарное лечение под контролем электроэнцефалографии, компьютерной, магниторезонансной томографии
1. Препараты, снижающие энергетические потребности мозга: 2 мл 0,5 % рела-ниума, тиопентал натрия в дозе 200 — 300 мг внутривенно болюсно и далее 7 — 8 мг/кг в час в течение 3 ч, или диприван 2. Антиоксиданты (6 мл 30 % раствора токоферола внутримышечно) 3. Мембраностабилиза-торы (кортикостероиды — предпочтительнее метилпреднизолон в дозе 30 г/кг, болюсное одноразовое введение 1/3 расчетной дозы) 1. Препараты, снижающие энергетические потребности мозга, включая бензодиазепины, опиоиды, энце-фалины, барбитураты, пропофол 2. Антиоксиданты (токоферол) 3. Мембраностабилизато-ры (кортикостероиды, лидокаин, раствор магний сульфата) 4. Антиагреганты и препараты, улучшающие микроциркуляцию (винпоцетин, пентоксифиллин, низкомолекулярный гепарин, низкомолекулярные декстраны) 1. Постепенно уменьшают дозу опиоидов, затем тиопентала натрия (переходят на пероральный прием) и в последнюю очередь — бензодиазепинов* 2. Препараты метаболического действия (вводить в первой половине дня)*: а) прямые антигипоксан-ты (цитохром С, актовегин, солкосерил); б) препараты ноотропно-го действия — не ранее 3 суток (церебролизин, пирацетам, аминалон) 3. Антиоксиданты (токоферол) 1. Препараты, активирующие метаболические процессы в мозгу: а) прямые антигипоксан-ты (актовегин, солкосерил); б) препараты ноотропно-го действия (курсы цере-бролизина, пирацетама, неотона); в) дофаминовые препараты (наком); г) глиатилин внутривенно или внутрь по 400 — 1000 мг 2 раза в день 2. Антиоксиданты (токоферол) 1. Симптоматическая терапия в зависимости от сформировавшегося синдрома 2. Общеукрепляющая терапия (витаминотерапия, АТФ, креатинфосфат, биостимуляторы, адаптоге-ны) 3. Антиоксидантная, антиагрегационная терапия 4. Противосклеротичес-кая терапия (токоферол, липостабил, мисклерон, атероид, ловастатин, окситерм) 5. Препараты ноотропно-го действия (ноотропил,
Интенсивная терапия постреанимационной болезни
Продолжение табл. 12.6
Первые три часа Острый период Подострый период Отдаленный период
Стадия угрожающей жизни комы Стадия стабилизации вегетативных функций
4. Антиагреганты (10 мл 2,4 % р-ра эуфиллина + + 1 мл 1 % никотиновой кислоты в 300 мл физраствора внутривенно капельно, фраксипарин 3500— 4000 ME подкожно 5. Блокаторы кальциевых каналов (нимодипин, нифедипин, верапамил) 5. Препараты группы блокаторов кальциевых каналов (нимодипин, нифедипин, верапамил) 6. р-Адреноблокаторы 7. а-Адреноблокаторы 4. Мембраностабилизато-ры (кортикостероиды, лидокаин, раствор магний сульфата) 5. Антиагреганты и препараты, улучшающие микроциркуляцию (винпоцетин, пентоксифиллин, низкомолекулярный гепарин, низкомолекулярные декстраны) 6. р-Адреноблокаторы 7. Противогрибковая терапия 3. Мембраностабилизато-ры (лидокаин, магний сульфат) 4. Антиагреганты и препараты, улучшающие микроциркуляцию (винпоцетин, пентоксифиллин, низкомолекулярный гепарин) 5. Противосклеротичес-кая терапия — липоста-бил в инъекциях или драже, окситерм 6. Терапия витаминами группы В, С, поливитамины, отонервин 7. Физиотерапия церебролизин, инстенон, энцефабол, ацефен, ре-дергиндегидроэрготок-син мезилат), включая нейропептиды (семакс в разведении по 3 мг капельно в носовой ход за 15 мин до нейропсихологических занятий, гли-атилин) 6. Физиотерапия 7. Эрго- и кинетикотера-пия 8. ЛФК, массаж 9. ГБО 10. Санаторно-курортное лечение 11. Продление больничного листа на долечивание 12. При компенсированной форме энцефалопатии перевод на облегченный труд
Не применяются при пароксизмальной или эпилептиформной активности на ЭЭГ.
СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Интенсивная терапия постреанимационной болезни 413
сибазон) — 10 мг внутривенно струйно; пропофол (диприван) — 15—35 мг внутримышечно; сочетанное применение тиопентала натрия и оксибутерата натрия в субнаркотических дозах, вводимых поочередно: тиопентал натрия (5 % раствор) — 5 мг/кг на одно введение (каждые 3 ч) и оксибутират натрия — 20 мг/кг на одно введение (каждые 2 ч). При уменьшении неврологического дефицита промежутки между введениями увеличиваются, а дозы вводимых препаратов — уменьшаются. Согласно руководству Европейского совета по реанимации целесообразность активной гипотермии после остановки кровообращения не доказана (класс не определен).
2) стабилизаторов клеточных мембран, способствующих уменьшению их проницаемости (глюкокортикоиды, предпочтительно метилпреднизолон в дозе 1 — 4 мг/кг до 30 мг/кг массы тела в сутки); препаратов, направленных на снижение активности калликреин-кининовой системы, уменьшение трипсинемии, ферментемии (контрикал 150 — 800 ЕД/кг массы тела), а также на снижение интенсивности пе-роксидного окисления (а-токоферол до 1800 мг в сутки).
3) препаратов, улучшающих мозговое кровообращение за счет восстановления микроциркуляции в сосудах головного мозга, улучшения венозного оттока из полости черепа, устранения отека мозга, восстановления объемной скорости мозгового кровотока: эуфиллин (до 240 мг) вместе с никотиновой кислотой — 10 мг в 300 мл изотонического раствора натрий хлорида внутривенно, винпоцетин (кавинтон) — 20 мг, для улучшения реологических свойств крови — трентал (100 — 200 мг/сут).
Назначают блокаторы кальциевых каналов — нимодипин (нимотоп) — 1 мг/кг или верапамил (изоптин) — 1 мг/мин.
В дальнейшем комплекс лечебных мероприятий и их длительность зависят от тяжести состояния, характера и глубины поражения нервной системы.
Так, если нарушения психоневрологического статуса отмечаются у 30 % больных первой группы и 50 % — второй и не связаны с соматическими осложнениями, то для больных третьей группы в 70 %
случаев характерны выраженные соматические осложнения, приводящие ко вторичным повреждениям мозга.
Среди новых методов лечения, позволяющих существенно улучшить его результаты, следует отметить использование в комплексе интенсивной терапии кровезаменителя с газотранспортной функцией — перфторана. Последний обладает полифункциональным действием: улучшает кислородно-транспортную функцию крови, увеличивает газообмен и метаболизм на уровне тканей, улучшает реологические свойства крови и микроциркуляцию, имеет мембраностабилизирующий эффект, нейро- и кардиопротекторное действие, дозозависимое иммунопротектор-ное действие, является блокатором медленных кальциевых каналов, оказывает анти-аритмическое действие за счет активации энергетического обмена [33 — 35].
Приведенные выше свойства перфторана позволяют ему воздействовать сразу на несколько звеньев патогенеза при постгипоксической энцефалопатии. При этом благодаря механизму его действия предупреждается не только ранняя, но и отсроченная гибель нейронов, связанная с вторичной гипоксией (рис. 12.30). Нейропротектор-ный эффект перфторана заключается в укорочении длительности коматозного периода, уменьшении глубины комы, ускорении регресса неврологической симптоматики, снижении летальности, улучшении качества жизни больных в отдаленный период.
Препарат вводится в первые 6—12 ч после остановки кровообращения на фоне проведения стандартных реанимационных мероприятий после предварительного размораживания при комнатной температуре и оценки его на пригодность. Перфто-ран вводится внутривенно капельно через отдельную систему в дозе 5 — 7 мл/кг массы тела. В последние годы доказана эффективность малых доз — 2 — 3 мл/кг массы тела. Обязательна биологическая проба. Первоначальная скорость введения — 20 капель в минуту, в последующем — 50 — 60 капель в минуту.
При необходимости купирования смешанной венозной гипоксии и вторичной ишемии головного мозга перфторан мо-
414 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
Рис. 12.30. Механизм действия перфторана при постгипоксической энцефалопатии: действие перфторана: —► — прямое; - - - косвенное
жет назначаться повторно к концу 1 — на 2 сутки в той же дозе.
Для усиления эффекта инфузии перфторана рекомендуется проведение окси-генотерапии (40 — 60 %) в течение 24 ч после начала инфузии.
Не рекомендуется вводить препарат при запредельной коме (III ст.), крайне неблагоприятном течении постреанимационной болезни с клиническими признаками гибели головного мозга, IV —V стадии постреанимационной болезни.
Алгоритм III стадии СЛЦР представлен на рис. 12.31.
Основные ошибки, ведущие к тяжелым неврологическим расстройствам (по Г. В. Алексеевой, 1996):
1. Недооценка ОНД, особенно на ранних этапах постреанимационного периода:
поздний перевод больного на ИВЛ;
раннее прекращение респираторной поддержки, что приводит к вторичной гипоксии, отеку мозга, перераздражению дыхательного центра, судорогам.
2. Недостаточное обезболивание, что вызывает дисциркуляторные изменения, гипоксию, нарушение текучих свойств крови, жировую эмболию, ДВС-синдром, отек
мозга, истощение эндорфинной системы, диэнцефальные кризы.
3. Некорректное и несвоевременное применение препаратов, усиливающих метаболические процессы в головном мозгу, что сопровождается развитием судорог, диэнцефальных и адренергических кризов, отеком мозга.
4. Необоснованное применение дегид-ратационных средств, что обуславливает нарушение соотношения K/Na, развитие гиповолемии, дисциркуляторных нарушений, ДВС-синдрома, полиорганной недостаточности, эндотоксемии, отека мозга, гиперосмолярной комы.
Только 10 % из числа выживших способны возвратиться к прежнему образу жизни, поскольку у остальных после выписки из стационара имеются психоневрологические нарушения различной степени выраженности (А. Н. Коргин, И. Б. Холодов, 1988). Г. В. Алексеева (1998) наблюдала спустя год серьезные неврологические расстройства у 70% выживших после клинической смерти, 50 % из этих больных были инвалидами.
Применение адекватной интенсивной терапии в острой фазе постаноксического
Интенсивная терапия постреанимационной болезни 415
Рис. 12.31. Алгоритм III стадии сердечно-легочной церебральной реанимации
периода и последовательное согласованное применение медикаментозных средств, психотерапевтических методов и интеллектуального тренинга, санаторно-курортного лечения ускоряет регресс неврологической симптоматики уже в остром периоде, увеличивает формирование гибких
адаптационных механизмов с большей сохранностью структур ЦНС [26, 36, 37]. Это способствует снижению инвалидизации, более полному восстановлению личности больных с возвращением их в сферу трудовых ресурсов, к активной жизни в семье и в обществе.
Список использованной литературы
1. Неговский В. А. Очерки по реаниматологии. — И.: Медицина, 1986. — 253 с.
2. Иванов Г. Г., Востриков В. В. Интенсивная терапия и сердечно-легочная реанимация при внезапном прекращении эффективной сердечной деятельности // Анестезиология и реанимация. — 1996. - №5. - С. 70 — 80.
3. Патофизиология и терапия агонии и клинической смерти / Под ред. В. А. Негевского. — М.: Медгиз, 1954. — С. 9.
4. Анестезиология и реаниматология / Под ред. О. А. Долиной. — М.: Медицина, 1998. — 543 с.
5. Белебезьев Г. И., Беляев А. В., Чухрай Т. Г. и др. Сердечно-легочная и церебральная реанимация в детском возрасте: Метод, рекомендации. — К.: Б. и., 2000. — 53 с.
6. Сафар П., Бичер Н. Дж. Сердечно-легочная и церебральная реанимация / Пер. с англ. — 2-е изд. — М.: Медицина, 1997. — 421 с.
416 СЕРДЕЧНО-ЛЕГОЧНАЯ ЦЕРЕБРАЛЬНАЯ РЕАНИМАЦИЯ
7. Сумин С. А. Неотложные состояния. — М.: Медицина, 2000. — 449 с.
8. Зильбер А. П. Медицина критических состояний. Общие проблемы. — Петрозаводск: Изд-во ПГУ, 1995. - 360 с.
9. Latorre F., Nolan J., Robertson C. et al. The ALS working group of the European Resuscitation Council. The European Resuscitation Council Guidelines 2000 for Adult Advanced Life Support Resuscitation. 2000. — 48. — P. 211-212.
10. American Heart Association in collaboration with the International Liaison Committee on Resuscitation (ILCOR). International Guidelines 2000 for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. A Consensus on Science // Resuscitation. — 2000. — 46. - P. 103-252.
И. ЧепкшЛ. П., Усенко Л. В. Гер1атричнаане-стезюлопя та реашматолопя. — К.: Здоров’я, 1994. - 251 с.
12. Экспериментальные, клинические и организационные проблемы общей реаниматологии / Под ред. В. А. Негевского. — М.: Медицина, 1996. — 308 с.
13. Костюченко А. Л. Угрожающие жизни состояния в практике врача первого контакта: Справ. — СПб.: Специальная литература, 1998. - 242 с.
14. Усенко Л. В., КриштафорА. А., ЭстринА. А. Сердечно-легочная и мозговая реанимация. Постреанимационная болезнь: Учеб, пособие. — Днепропетровск: Друкар, 2001. — 78 с.
15. Рябов Г. А. Синдромы критических состояний. — М.: Медицина, 1994. — 368 с.
16. Реанимация на рубеже XXI века. — М.: Медицина, 1996. — 366 с.
17. Мороз В. В. Пути коррекции гипоксии при критических состояниях: Автореф. дис. ... д-ра мед. наук. — М., 1994. — 48 с.
18. Руксин В. В. Неотложная кардиология. — СПб.: Невский диалект, 1997. — 460 с.
19. Неговский В. А., Мороз В. В. Актуальные вопросы реаниматологии // Анестезиология и реанимация. — 1999. — № 1. — С. 6 —9.
20. Неговский В. А., ГурвичА. М., Золотокры-лина Е. С. Постреанимационная болезнь. — М.: Медицина, 1987. — 380 с.
21. Гурвич А. М., Алексеева Г. В., Семченко В. В. Постреанимационная энцефалопатия (патогенез, клиника, профилактика и лечение). — Омск: ИПК Омич, 1996. — 76 с.
22. Уэлш Т. Смерть мозга. Хроническое вегетативное состояние // Неврология / Под ред. М. Самуэльса. — М.: Изд-во Практика, 1997. - С. 187-189.
23. Попова Л. М. Этические проблемы, возникающие при диагностике смерти мозга // Анестезиология и реанимация. — 1992. — № 5-6. - С. 69-72.
24. Биомедицинская этика / Под ред. акад. В. И. Покровского. — М.: Медицина, 1997. - 223 с.
25. Золотокрылина Е. С. Постреанимационная болезнь: этиология, патогенез, клиника, лечение // Реаниматология и интенсивная терапия. - 1999. - № 1. - С. 8-18.
26. Алексеева Г. В. Клиника и терапия постгипоксических энцефалопатий: Метод, рекомендации. — М., 1996. — 36 с.
27. Черний В. И. Постгипоксическая энцефалопатия. — К.: Здоров’я, 1997. — 336 с.
28. Семченко В. В., Степанов С. С., Алексеева Г. В. Постаноксическая энцефалопатия. — Омск: Омск. обл. тип., 1999. — 444 с.
29. Золотокрылина Е. С. Вопросы патогенеза и лечения полиорганной недостаточности у больных с тяжелой сочетанной травмой, массивной кровопотерей в раннем постреанимационном периоде // Анестезиология и реанимация. — 1996. — № 1. — С. 9—13.
30. Рябов Г. А. Логика развития интенсивной терапии критических состояний // Анестезиология и реанимация. — 1999. — № 1. -С. 10-13.
31. Усенко Л. В., Криштафор А. А. Современный взгляд на патогенез и интенсивную терапию постгипоксической энцефалопатии // Укр. мед. часопис. — 1997. — № 1. — С. 17-24.
32. Перфторан в интенсивной терапии критических стояний. Метод, рекомендации / Под общ. ред. Л. В. Усенко, Е. Н. Клигуненко. — Днепропетровск, 1999. — 56 с.
33. Слоан Т. Б. Успехи в решении проблемы защиты мозга // Вестник интенсивной терапии: ИТ в неврологии. — 1993. — № 1. — С. 14-18.
34. Усенко Л. В., Криштафор А. А. Перфторан в профилактике постгипоксической энцефалопатии // Сб. науч. тр. «Перфторорга-нические соединения в биологии и медицине». — Пущино, ОНТИ ПНЦ, 1999. -С. 76-87.
35. Мороз В. В., Веремеенко С. Н., Кичев Т. С., Котляров В. В. Применение перфторана и олифена у больных, перенесших гипоксию // Сб. науч. тр. «Перфторорганические соединения в биологии и медицине». — Пущино, ОНТИ ПНЦ, 2001. - С. 171-178.
36. Усенко Л. В. Стратепя вщновлення особис-TOCTi хворих шеля перенесених критичних сташв // Медичш перспективи. — 2001. -T.VI, №3, ч.2. - С. 73-80.
37. Степанов С. С., Ерениев С. И., СемченкоВ. В. Сопоставление уровня судорожной готовности мозга, долговременной памяти, способности к обучению и их структурные эквиваленты в постреанимационном периоде // Патолог, физиология и эксперим. терапия. — 1999. -№ 3. - С. 15-19.
Раздел
Шок
Шок — одна из наиболее частых причин тяжести состояния и летальности у больных в ОИТ. Несмотря на успехи интенсивной терапии, достигнутые за последние годы, летальность при шоке остается все еще высокой. Так, при септическом шоке она достигает 40 — 60 %, при кардиогенном — 30 — 90 % [1, 2].
Шок — это острое опасное для жизни состояние, характеризующееся неадекватной доставкой и потреблением кислорода тканями вследствие нарушения перфузии.
Определение VO2 является наиболее достоверным диагностическим тестом для выявления шока [3]. Однако при повышенном метаболизме нормальные показатели потребления кислорода не будут адекватно отражать удовлетворение потребности в нем, в этом случае уровень лактата в артериальной крови будет наиболее информативным показателем ишемии.
Шок всегда характеризуется комбинацией артериальной гипотензии и метаболического ацидоза [4]. В результате снижения перфузии тканей и доставки кислорода в недостаточном объеме происходит окислительное фосфорилирование, благодаря которому в организме генерируется до 95 % всей энергии; в клетке образуется недостаточное количество мак-роэргических фосфатов, возникает дефицит энергии, нарушение функции клеток и их гибель. Таким образом, патологические процессы при шоке имеют системный 14 4-220
характер и реализуются на уровне клетки.
Несмотря на полиэтиологичность видов шока, для них характерны общие механизмы развития: неадекватная тканевая перфузия приводит к недостаточной оксигенации клетки, снижается продукция АТФ и цАМФ -> нарушается функция клеточных мембран, в частности ионных насосов -> -> ионы калия выходят из клетки, ионы натрия и вода поступают в нее, повышается концентрация ионов кальция в клетке -> -> увеличивается продукция лактата, усиливается внутриклеточный ацидоз -> повреждаются внутриклеточные органеллы, в частности лизосомы -> происходит самоуничтожение клетки. Даже если на каком-то этапе (кроме самого раннего) удается восстановить перфузию, возникают реперфузионные поражения клетки, прежде всего за счет невозможности быстрого восстановления функции митохондрий.
Одним из основных факторов прогрессирования шока является ишемия кишок, что приводит к утрате барьерной функции кишечным эпителием. В результате сначала в брыжеечный, а затем и в системный кровоток попадают бактерии и их токсины из содержимого просвета кишок. Реакция иммунной системы организма на фоне нарушения циркуляции, гипоксии тканей, гипоэргоза, ацидоза не совсем адекватна и проявляется неконтролируемой системной воспалительной реакцией. Активируются факторы комплемента, особенно С3а и Сза, провоспалительные цито-
418 ШОК
Циркуляторный шок
I
Бактериальный компонент или другая причина провоспалительного ответа
Провоспалительная ПАРС-активация Поли-(АДФ)-рибоза Пероксидное
сигнальная трансдукция Л Протеины повреждение
АР-1, МАР-киназа, ' липидов
активация NF-кВ НАД
Мононуклеарная Провоспалительные Некроз Дисфункция клеточная ** цитокины и хемокины клеток эндотелия инфильтрация |
Активация ------► Воспалительное повреждение клеток
комплемента |
Полиорганная дисфункция Повреждение сосудов Стойкая декомпенсация гемодинамики Смерть
Рис. 13.1. Схема повреждения клетки при шоке и развития полиорганной недостаточности: iNOS — iNO-синтетаза; ONOO" — пероксинитрит; ПАРС — поли-(АДФ)-рибозо-синтетаза;
NF-кВ — ядерный фактор-каппа Б
кины (интерлейкины — 1, 6, 8, фактор некроза опухоли и др.), нарушается метаболизм арахидоновой кислоты с образованием простагландинов, тромбоксанов, лейкотриенов.
Из представленной схемы (рис. 13.1) следует, что после снижения перфузии тканей (циркуляторный шок) как в случае наличия бактериального компонента, так
и без него, но при наличии другого фактора, вызывающего системную воспалительную реакцию организма, ключевым звеном шока является повреждение митохондрий клеток.
Поскольку клетка является центральным объектом повреждения, от обратимости которого часто зависит успех терапии, рассмотрим эту проблему подробнее.
13.1. ПОВРЕЖДЕНИЕ КЛЕТКИ ПРИ ШОКЕ
Свободнорадикальное окисление липидов существенно возрастает при циркуляторном шоке любой этиологии. Оксиданты могут генерироваться в стадии репер
фузии при шоке посредством активации ксантиноксидазы, пуринового метаболического энзима, в малых концентрациях содержащегося и в здоровых тканях, но об
Повреждение клетки при шоке 419
разование которого из предшественника — ксантиндегидрогеназы существенно увеличивается при шоке под воздействием протеолиза. При наличии относительного избытка кислорода ксантиноксидаза образует ион супероксида из ксантина, продукта метаболизма АТФ, формирующегося в ишемических тканях [5]. Активация ксан-тиноксидазы происходит под воздействием стимуляции цитокинов, что имеет большое значение при вирусной и бактериальной инфекции [6]. Супероксид может продуцироваться активированными гранулоцитами, мембранным энзимом НАДФ-окси-дазой. Для увеличения продукции супероксида в процессе ишемии/реперфузии характерна тканевая инфильтрация нейтрофилами [7].
На важную роль пероксидного окисления оксидов (ПОЛ) в развитии полиор-ганной недостаточности при шоке указывают данные, полученные R. Kentner с соавт. [8], об эффективности применения в эксперименте при геморрагическом и септическом шоке нового антиоксидантного препарата темпола, при этом у подопытных животных повышалась выживаемость.
Высокий уровень NO может индуцировать повреждение ДНК. Кроме эффекта свободных радикалов и оксидантного повреждения на активность полиадено-зиндифосфатрибозо-синтетазы (ПАРС) влияют также цитокины. у-Интерферон, освобождающийся при шоке из активированных Т-лимфоцитов и натуральных киллеров, дозозависимо является триггером распада ДНК. Механизм этого процесса неизвестен, возможно, в его основе лежит свободнорадикальное окисление.
Хотя механизм гибели клетки при ишемии и нарушении утилизации кислорода при циркуляторном шоке до сих пор понятен не до конца, в последнее время получено много данных, подтверждающих важную роль ПАРС [9]. Это распространенный ядерный энзим. Его физиологическая роль не совсем понятна. На базальном уровне основными функциями ПАРС являются: восстановление ДНК [10], релаксация хроматина [И], дифференциация клеток [12], репликация ДНК, регу-14*
ляция транскрипции, контроль за клеточным циклом, экспрессией Р53 и апоптозом, трансформация клетки. В условиях патологии, в том числе при радиационном поражении, циркуляторном шоке, местном воспалении, активизация ПАРС может нарушать регуляторные процессы в клетке [13]. Патологическая активация ПАРС ассоциируется со многими базовыми расстройствами при септическом шоке, включая нарушения митохондриального дыхания, снижение контрактильности сосудистой стенки и миокарда [9]. Ингибиция активности ПАРС может оказаться перспективным методом восстановления клеточной оксигенации и дыхания при циркуляторном шоке.
Активация ПАРС индуцирует немедленное образование поли-(АДФ)-рибозы путем каталитического расщепления НАД. Поли-(АДФ)-рибоза быстро деградирует. Поскольку НАД играет основную роль в процессах гликолиза, цикла трикарбоновых кислот, транспорте электронов, его потребление, вызванное ПАРС, обусловливает нарушение процессов окислительного фосфорилирования и образования АТФ даже при достаточном поступлении кислорода [14].
Механизм «суицида» клетки предложен N. A. Berger (1991): под воздействием LPS (липополисахаридов) происходит активация ПАРС -> стимуляция iNOS -> продукция супероксида. Ингибирование активности ПАРС не предотвращает повышение уровня пероксинитрита и повреждения ДНК, однако блокирует снижение продукции энергии в клетке [15].
В повреждении клетки при шоке важную роль играет также NO, являющийся весьма токсичным по отношению к аконитазе (aconitase), критическим энзимом — по отношению к циклу трикарбоновых кислот (циклу Кребса). Даже в условиях аноксии, когда не происходит продукции пероксинитрита, NO ассоциируется с инактивацией не только аконитазы, но и других энзимов, содержащих сульфгидрильные группы, включая электронотранспортный НАД • Н-убихинон-оксидоредуктазу и сукцинат-убихинон-оксидоредуктазу [16]. Несмотря на то что донаторы NO
420 ШОК
редуцируют продукцию АТФ и клеточное дыхание, они мало влияют на обмен НАД. Ингибиторы ПАРС не подавляют ни один из эффектов доноров NO, что подтверждает существование независимого механизма нарушения энергообразования в клетке, вызываемого ПАРС и NO [17].
В отличие от ПАРС-независимого механизма цитотоксичности NO пероксинитрит вызывает истощение в клетках НАД и АТФ, однако этот эффект подавляется ингибиторами ПАРС. Результаты исследований дают основания предположить, что имеется два независимых параллельных механизма энергетического истощения клетки при шоке, вызываемые свободными радикалами и оксидантами [17]. NO ингибирует активность энзимов гликолиза и окислительного фосфорилирования через инактивацию каталитических центров, содержащих серу. Пероксинитрит и другие оксиданты ингибируют интрацеллюлярную энергетику через активацию ПАРС и деградацию ДНК [9]. Это подтверждено экспериментальными исследованиями: при разрушении гена ПАРС мутантные клетки слабо чувствительны к свободным кислородным радикалам, но не изменяют чувствительности к действию NO [18].
Ингибиторы ПАРС блокируют экспрессию внутриклеточных адгезивных молекул (ICAM-1) в иммуностимулированных эндотелиоцитах. В клетках кишечного эпителия ингибиторы ПАРС подавляют вызываемую ИЛ-ip индукцию фактора комплемента С3 и ранний компонент активации С5а, потенциального хемотаксина. Можно сделать вывод, что ПАРС играет определенную роль в сигнальной трансдукции провоспалительной сигнализации.
Сейчас синтезированы фармакологические ингибиторы ПАРС: 3-аминобензамид, никотинамид. Они обладают выраженными побочными эффектами, в частности вызывают артериальную гипотензию, и очень ограниченно могут применяться в клинике как ингибиторы ПАРС, поскольку для достижения эффекта должны использоваться большие дозы этих препаратов. Кроме того, к этой группе веществ можно отнести: 4-амино-1,8-нафталимид (4-amino-l ,8-naphtalimide), 2Н-бенз[с]изо-
квинолин-1-[6(5Н)-фенантридинон]; 1,5-ди-гидроксиизоквинолин, которым также присущи выраженные побочные эффекты. В последнее десятилетие был испытан препарат амино-1,2-бензопирон, проявивший свою эффективность в некоторых исследованиях in vivo [19].
Применение ингибиторов ПАРС показано преимущественно при острых состояниях (шок, сепсис), поскольку при длительном применении проявляются биологические эффекты ингибирования ПАРС. Описаны тяжелые нарушения функции клетки при длительном использовании никотинамида [20].
Имеются сведения об успешном применении ингибиторов ПАРС при эндотокси-ческом шоке. Авторы применяли никотинамид через час после введения LPS, при этом удавалось поддерживать более высокое АД. In vivo ингибирование ПАРС на 50 % восстанавливало вызванное эндотоксином нарушение контрактильности сосудистых мышц [21]. При эндотоксиновом шоке в сосудистой интиме образуется пероксинитрит, нарушающий эндотелиальную регуляцию тонуса сосудов. По-видимому, в этом процессе важную роль играет активация ПАРС. После применения 3-аминобензамида (3-aminobenzamide) при эндотоксиновом шоке значительно снижалась связанная с эндотелием релаксация сосудов [22]. В эксперименте при лечении 3-аминобензамидом наблюдалось значительно меньшее количество летальных случаев у животных при эндотоксиновом шоке [9]. Образование пероксинитрита и активация ПАРС были подтверждены при исследовании сердец и легких септических свиней иммунохимическим методом определения нитротирозина и по-ли-(АДФ)-рибозы. Ингибирование ПАРС с помощью PJ34 нивелировало формирование поли-(АДФ)-рибозы у септических животных. Использование ПАРС-ингиби-тора PJ34 в модели сепсиса у поросят повышало выживаемость и улучшало функцию сердечно-сосудистой системы, а также уменьшало медиаторный компонент воспалительного ответа [23].
Есть данные о потенциальном лечебном эффекте ингибиторов ПАРС при остром
Гиповолемический шок 421
инфаркте миокарда и церебральных расстройствах.
Значительную роль в процессах гибели клетки играет повреждение митохондрий. Это доказывают опыты М. Boulos
с соавт. [24], в которых нарушение функции митохондрий, индуцированное введением сыворотки пациентов с септическим шоком, уменьшалось при использовании ингибиторов NO-синтазы и ПАРС.
13.2. СТАДИИ И КЛИНИКА ШОКА
Большинство исследователей условно выделяют три стадии шока:
1) компенсированная — перфузия жизненно важных органов сохраняется за счет компенсаторных механизмов; как правило, не наблюдается выраженной гипотензии вследствие увеличения общего сосудистого сопротивления;
2) декомпенсированная — компенсаторные механизмы не в состоянии поддерживать достаточную перфузию, запускаются и прогрессируют все патогенетические механизмы развития шока;
3) повреждения носят необратимый характер, развивается массовая гибель клеток и полиорганная недостаточность.
Еще раз отметим условность приведенной стадийности патологического процесса при шоке, а также трудность клинического дифференцирования 2-й и 3-й стадий шока. Большинство авторов подчеркивают, что необратимая стадия шока не
всегда приводит к гибели организма и не является показанием к прекращению терапии, в этом случае представляется не совсем корректным использование названия «необратимая».
Для выбора терапевтической тактики эта классификация по стадиям, на наш взгляд, большого значения не имеет.
Клинически большинство видов шока характеризуется бледностью кожных покровов, тахикардией, одышкой, снижением диуреза, нарушением сознания. Артериальная гипотензия не является ранним признаком шока, который должен быть диагностирован до значительного снижения АД. Степень ишемии органа коррелирует с изменением уровня венозного лактата и системных параметров кислотно-основного состояния — pH, BE [25].
Рассмотрим подробнее отдельные виды шока.
13.3. ГИПОВОЛЕМИЧЕСКИЙ ШОК
Этот вид шока можно разделить на два подвида: геморрагический и негеморрагический (потеря жидкости организмом).
Геморрагический шок (ГШ) — это шок от кровотечения, которое может возникать при травме, патологии пищеварительного канала, во время операции и т. д.
Негеморрагический гиповолемический шок возникает вследствие обезвоживания организма, вызванного:
патологией пищеварительной системы: рвота, диарея, потери через зонд в пищеварительном канале, свищи, панкреатит;
ожогами;
полиурией (несахарный диабет, полиурическая стадия острой недостаточности почек);
недостаточностью коры надпочечников;
потерей жидкости в «третье пространство» (перитонит, непроходимость кишок, асцит).
13.3.1. Геморрагический шок
Это гиповолемический шок, вызванный кровопотерей. Характеризуется снижением ОЦК, ЦВД, ДЗЛА, СИ и повышением ОПС. Но обязательно ли любая кровопотеря сопровождается шоком? При кровопотере до 5 % ОЦК компенсаторные механизмы обеспечивают адекватную перфузию тканей, при кровопотере до 10 % ОЦК (« 500 мл) возможны проявления шока, при кровопотере до 20 % ОЦК (« 1000 мл) — высокая вероятность развития шока, потеря крови свыше этого объема сопровожда
422 ШОК
ется развитием шока, хотя кровопотеря до 30 % ОЦК у пациента в состоянии покоя вначале может не всегда определяться клинически.
Ответные реакции организма на кровопотерю:
1. Активация симпатической АНС (реакция централизации, увеличение ЧСС, увеличение потребности миокарда в О2).
2. Стимуляция системы ренин-ангиотен-зин-альдостерона (спазм сосудов, реабсорбция Na+ в почках — снижение диуреза).
3. Стимуляция продукции вазопрессина (спазм сосудов, реабсорбция воды в почках — снижение диуреза).
4. Стимуляция осморецепторов гипоталамуса (жажда).
5. Стимуляция продукции гликокортикоидов (повышение тонуса адренорецепторов, мембранопротекторное действие (возможно)).
6. Аутогемодилюция.
7. Стимуляция продукции эритропоэтина почками (в более поздней стадии шока).
Первой на потерю крови начинает реагировать симпатическая автономная нервная система. Прежде всего это выражается в веноконстрикции, хотя рецепторы, обуславливающие данную реакцию, пока не обнаружены [26]. Некоторое значение для поддержания волемии имеет выброс крови из органов-депо (селезенка, печень, легкие). Важную роль играет стимуляция адренергических рецепторов. oq-Ад-ренорецепторы расположены в основном в сосудах спланхнической зоны, почек, кожи. Их активация проявляется спазмом сосудов этих областей с развитием ишемии и перераспределением кровотока в органы, где сосуды не содержат aj-адреноре-цепторы или содержат их в небольшом количестве. Таким образом, большая часть крови в большом круге кровообращения направляется в венечные и церебральные сосуды — реакция централизации кровообращения. В результате перфузия мозга и миокарда в течение какого-то периода времени поддерживается за счет ише-мизации других органов, прежде всего кишок и почек. Без симпатической ком
пенсации быстрая потеря 25 % ОЦК (20 мл/кг) привела бы к остановке кровообращения вследствие прекращения венозного возврата к сердцу [27]. Очень существенным фактором пераспределения крови при кровопотере является реакция спланхнического кровотока, которая выражается в спазме артериол, снижении венулярного сопротивления портальной системы и печени, что улучшает отток крови из этого бассейна в системный кровоток [28].
Стимуляция Pj-адренорецепторов вызывает увеличение частоты сердечных сокращений, повышение сократительной способности миокарда, но одновременно увеличение его потребности в кислороде, что у больных с ИБС может усилить ишемию.
Повышенная концентрация катехоламинов в крови может стимулировать ретикулярную формацию ствола мозга, поэтому у больных с кровопотерей довольно быстро могут развиться нарушения сознания [26].
Ишемия почек приводит к активации выработки ренина юкстагломерулярным аппаратом, что способствует образованию из ангиотензиногена ангиотензина I, который с помощью фермента ангиотензинкон-вертазы превращается в легких в мощный вазоконстриктор — ангиотензин II. Этот процесс стимулирует образование в коре надпочечников альдостерона, активирующего реабсорбцию натрия почечными канальцами.
Активируется функция задней доли гипофиза, что проявляется увеличением продукции вазопрессина. Он стимулирует ^’рецепторы в почках, что сопровождается повышенной реабсорбцией воды. Стимуляция -рецепторов в гладких мышцах сосудов приводит к вазоспазму.
Стимуляция осморецепторов гипоталамуса вызывает у человека чувство жажды. Однако даже в случае, когда прием жидкости внутрь не противопоказан (при отсутствии ранения органов живота), этот путь восполнения ОЦК резко ограничен. Вследствие быстро развивающейся ишемии желудка и кишок, последние теряют способность абсорбировать жидкость, при
Гиповолемический шок 423
чем чем больше кровопотеря, тем быстрее теряется эта способность.
Стимуляция функции коркового слоя надпочечников вызывает увеличение продукции гликокортикоидов, имеющих выраженный биологический эффект. Теоретически они повышают тонус адренорецепторов, их реакцию на эндо- и экзогенные катехоламины, а также могут обладать мембранопротекторным эффектом (по-видимому, вследствие этого они являются весьма популярным у анестезиологов средством лечения любого шока).
Аутогемодилюция. Если у больного определить концентрацию НЬ и Ht в крови сразу после большой кровопотери, то она будет близка к исходной. Однако, даже если у больного не производилось восполнение жидкости, через некоторое время (чем больше кровопотеря, тем быстрее) будет определяться прогрессирующее снижение концентрации НЬ и Ht. Это объясняется так называемой аутогемодилюцией. Для понимания ее механизма следует рассмотреть транскапиллярный обмен в большом круге кровообращения (рис. 13.2).
Транскапиллярный обмен жидкости подчиняется закону Франка —Старлинга и зависит от разницы между гидростатическим и коллоидно-осмотическим (онкотическим) давлениями между кровью и интерстициальной средой:
РГ = Ргк - Рги = 30 - (7 - 10)=
= (23 - 20) мм рт. ст.;
рк = Ркк - Рки = (25 - 28) - 15 = = (10 - 13) мм рт. ст.,
где Рги и Ргк — гидростатическое давление соответственно интерстициальной жидкости и крови в капилляре; Рки и Ркк — онкотическое (коллоидно-осмотическое) давление соответственно интерстициальной жидкости и крови.
В нормальных условиях в начальном отрезке капилляра происходит диффузия жидкости, а в конечном — ее реабсорбция в сосудистое русло. При гиповолемии повышается тонус прежде всего прекапил-
Рис. 13.2. Обмен жидкости в капилляре большого круга кровообращения (норма): Рг — гидростатическое давление; Рк — онкотическое (коллоидно-осмотическое) давление
Рис. 13.3. Обмен жидкости в капилляре при шоке:
а — начальная стадия (аутогемодилюция); б — запущенная стадия (декомпенсация периферического кровообращения (pooling): жидкость покидает внутрисосудистое русло); Рк — онкотическое давление; РТ — гидростатическое давление
лярного сфинктера (рис. 13.3, а), что изменяет соотношение между гидростатическим и коллоидно-осмотическим давлениями, поскольку при неизменности последнего гидростатическое давление снижается и в большей части капилляра происходит реабсорбирование интерстициальной, а затем и внутриклеточной жидкости в сосудистое русло. Этот механизм может обеспечить за 10 мин возврат из мышечной ткани, являющейся наибольшим резервуаром воды в организме, до 500 мл интерстициальной жидкости [29].
Все перечисленные выше реакции в той или иной степени помогают организму пережить кровопотерю, хотя при этом прогрессирует ишемия тканей. При умеренной кровопотере объем плазмы может восстановиться в течение 12 —72 ч [26]. Однако уже на ранних стадиях геморрагического шока происходят патологические изменения: прежде всего нарушаются реологические свойства крови. Замедление кро
424 ШОК
вотока, нарушение функции мембран эритроцита вследствие ацидоза, гипоэргоза приводят к потере эритроцитами отрицательного заряда на мембране. Это снижает имеющиеся в норме электростатические силы распора, препятствующие склеиванию эритроцитов, их адгезии. Упомянутые изменения способствуют агрегации эритроцитов, развитию сладж-синдрома — образованию «монетных столбиков», «эритроцитарной грязи», а в дальнейшем — микротромбообразованию. Увеличивается вязкость крови, снижается способность мембран эритроцитов к деформации.
Кроме того, по мере прогрессирования шока под действием накапливающихся метаболитов и ишемии рефлекторное сокращение прекапиллярных сфинктеров сменяется их параличем при сохраненной функции посткапиллярного сфинктера и спазме венул (в англоязычной литературе этот процесс получил название «pooling»). В капилляре повышается гидростатическое давление (рис. 13.3, б) и жидкость выходит из внутрисосудистого сектора, усиливая гиповолемию.
Быстрое восстановление внутрисосудистого объема приводит к восстановлению преднагрузки, однако при нарушенной проницаемости эндотелия вследствие активации системной воспалительной реакции это может спровоцировать потерю жидкости в интерстициальное пространство. В начале гиповолемии и шока просвет капилляров сужается в результате отека гипоксических эндотелиальных клеток и адгезии активированных полиморфноядерных нейтрофилов (ПЯН) с эндотелием в посткапиллярных венулах. Это приводит
к выключению микроциркуляции в некоторых отделах и выраженной гетерогенности перфузии. Окклюзия микрососудов и активация системы гемостаза может привести к тяжелым расстройствам циркуляции крови в жизненно важных органах. Интеракция между венозным эндотелием и ПЯН способствует освобождению вазоактивных медиаторов и токсичных свободных радикалов, еще большему повреждению эндотелия и потере макромолекул. Таким образом, в конечных стадиях шока развивается состояние рефрактерного шока и возникает своего рода «порочный круг», который трудно разорвать [26]. Например, развивающаяся ишемия мозга приводит к угнетению сердечно-сосудистого центра, что способствует вазодилатации, снижению ЧСС, дальнейшему снижению сердечного выброса, вследствие чего ишемия мозга еще больше усиливается. То же самое можно сказать и о связи депрессии миокарда с венечной гипоперфузией. Для достижения терапевтического эффекта при тяжелом шоке следует разорвать этот порочный круг.
Клинические проявления геморрагического шока зависят от объема кровопотери и индивидуальной чувствительности пациента. Например, женщины и доноры лучше переносят кровопотерю, а дети хуже.
Американская коллегия хирургов в зависимости от объема и клинических проявлений выделяет четыре класса кровопотерь (табл. 13.1).
Основными лабораторными показателями при геморрагическом шоке будут такие: анемия, снижение доставки кислоро
Таблица 13.1. Классификация кровопотери [30]
Параметр Класс
I II III IV
Объем кровопотери, % ЧСС, мин-1 АД в горизонтальном положении Диурез, мл/ч Уровень сознания < 15 < 100 Норма > 30 Тревога 15-30 > 100 Норма 20-30 Эйфория, возбуждение 30-40 > 120 Снижено 5-15 Сопор > 40 > 140 Резко снижено < 5 Кома
Кардиогенный шок 425
да, повышение экстракции кислорода тканями больше чем на 50 %, снижение насыщения кислородом венозной крови ниже 50 %, снижение уровня оснований в венозной крови, увеличение уровня венозного лактата свыше 4 ммоль/л, снижение напряжения углекислого газа в выдыхаемом газе (вследствие снижения сердечного выброса и транспорта СО2) [31]. Снижается рНр Уровень дефицита оснований, избытка лактата, снижение интестинального pH, как свидетельствуют данные многих исследований, коррелируют с тяжестью шока. При инфузии жидкости уровень оснований может вначале снижаться, а рСО2 повышаться вследствие вымывания из плохо перфузированных тканей.
Терапия геморрагического шока. Основными принципами лечения ГШ является неотложная остановка кровотечения и своевременная, адекватная по объему инфузионно-трансфузионная терапия. Кровотечение должно быть остановлено в максимально быстрые сроки, при небхо-димости следует применить оперативный гемостаз. Причем чем значительней кровопотеря, чем тяжелее шок, тем быстрее должен быть осуществлен хотя бы первичный гемостаз и начата инфузионная терапия. После остановки кровотечения при низких значениях ДЗЛК и ЦВД на первый план выходит инфузионная терапия. В критических ситуациях важна адекват
ная скорость инфузии, которая должна достигать 500 мл/мин, но без соответствующей аппаратуры такой скорости достичь не удается, поэтому показана струйная инфузия в две-три вены до достижения АД выше критического уровня либо нормализации ДЗЛК (18 — 20 мм рт. ст.) или ЦВД, после чего скорость следует уменьшить. При развитии миокардиальной недостаточности, о чем свидетельствует повышение ДЗЛА или ЦВД в ответ на инфузию при отсутствии эффекта на АД, необходимо применение кардио- и вазотонических препаратов. Более подробная информация о составе инфузионно-трансфузионной терапии и симпатомиметической поддержке содержится в соответствующих разделах.
В результате адекватной инфузионнотрансфузионной терапии ГШ должны быть получены такие лабораторные и гемодинамические показатели: нормализация АД, ЦВД, диуреза, ДЗЛА = 12 мм рт. ст.; СИ > > 3 л/мин на 1 м2, потребление кислорода УО2 > 100 мл/мин на 1 м2, венозный лактат < 4 ммоль/л, BE > 3 ммоль/л. При своевременной (в течение первых часов) терапии исход ГШ, как правило, благоприятный, полиорганная недостаточность возникает редко. Например, по нашим данным, ОРДС развился только у 2,5 %, а ОПН — у 0,1 % больных после массивной кровопотери, превышающей 1500 мл.
13.4. КАРДИОГЕННЫМ ШОК
Низкая перфузия тканей при кардиогенном шоке (КШ) обусловлена снижением сердечного выброса вследствие резкого нарушения насосной функции сердца.
Причинами КШ являются:
♦ резкое снижение сократительной способности миокарда (острый инфаркт миокарда, поражающий до 40—50 % сердечной мышцы, острый миокардит различной этиологии, контузия миокарда, кардиомиопатии в конечной стадии);
♦ повреждение клапанного аппарата сердца, сосочковых мышц;
♦ аневризма желудочков;
♦ фармакологическая/токсическая депрессия миокарда ((J-блокаторы, блокаторы кальциевых каналов, трициклические антидепрессанты).
КШ характеризуется снижением сердечного выброса, сердечного индекса, увеличением периферического сосудистого сопротивления, повышением ДЗЛА и ЦВД. При снижении сердечного выброса АД поддерживается за счет спазма вен и артериол, т. е. увеличения сосудистого сопротивления. Клинически, кроме других признаков шока, быстро прогрессирует отек легких.
При КШ уже на ранних стадиях возникает порочный круг, когда снижение на
426 ШОК
сосной функции сердца сопровождается уменьшением венечной перфузии, что, в свою очередь, еще больше снижает сердечный выброс.
При этом виде шока показан инвазивный мониторинг показателей сердечной и сосудистой функции (АД, ДЗЛА, ОПСС, СИ).
Терапия КШ. Основными методами терапии КШ являются кардиотонические средства и вазодилататоры, применяемые на фоне этиотропной терапии: при остром инфаркте миокарда — тромболитические препараты, механическое расширение просвета венечных артерий; при нарушениях ритма сердца — антиаритмические средства; при механических повреждениях клапанов и папиллярных мышц возможно хирургическое вмешательство. Основная цель терапии — повысить сократи
тельную способность миокарда при уменьшении работы сердца. Для этого применяют добутамин, сочетание допамина с нитроглицерином, а также ингибиторы фосфодиэстеразы — амринон, милренон. В отличие от других видов шока объемная нагрузка жидкостью резко ограничена ситуацией, когда ДЗЛА меньше 12 —15 мм рт. ст., с прекращением инфузий по достижении уровня 18 мм рт. ст. [32]. В связи с быстрым развитием отека легких и гипоксемии необходима оксигенотерапия и часто ИВЛ.
При неэффективности консервативной терапии показаны методы вспомогательного кровообращения — интрааортальная балонная контрпульсация и экстренная операция по улучшению кровоснабжения миокарда.
13.5. ДИСТРИБУТИВНЫМ (ПЕРЕРАСПРЕДЕЛИТЕЛЬНЫЙ) ШОК
В основе этого вида шока лежит перераспределение жидкости в организме, как правило, из внутрисосудистого сектора во внесосудистый. Различают следующие разновидности этого шока:
септический (СШ);
анафилактический / анафилактоидный (АШ);
нейрогенный (подвид — спинальный шок) (НШ).
Элементы дистрибутивного шока встречаются при политравме, ожогах, панкреатите, непроходимости кишок.
13.5.1. Септический шок
Шок в ответ на септицемию и действие бактериальных токсинов. Чаще всего вызывается грамотрицательными бактериями, этиологическим фактором могут быть также грамположительные бактерии, вирусы, грибы, риккетсии.
В ответ на действие бактериальных эндотоксинов, являющихся липополисахаридами, запускается синдром системного воспалительного ответа, сопровождаемый повреждением клеточных мембран, в частности эндотелиоцитов, повышением их проницаемости и перераспределением жидкос
ти из сосудистого русла в интерстициальное пространство и внутрь клетки.
В последнее время при СШ были выявлены нарушения реологических свойств эритроцитов. Реология изменяется под влиянием многих факторов: нарушения обмена внутриклеточного кальция и снижения концентрации АТФ, действия NO, уменьшения концентрации одних компонентов мембраны эритроцита, например сиаловой кислоты, и увеличения других, типа 2,3-дифосфоглицерата, взаимодействия с лейкоцитами и их продуктами (реактивные кислородные субстанции), эффектов колебаний температуры [33].
В процессе развития СШ выделяют гипер- и гиподинамическую фазы. Для первой характерны: лихорадка; теплые, розовые кожные покровы; снижение сосудистого сопротивления; нормальный или повышенный сердечный выброс, благодаря чему удерживается нормальное АД, хотя в отдельных случаях может наблюдаться артериальная гипотензия; умеренная тахикардия; одышка; нормальный диурез. Переход в гиподинамическую фазу характеризуется: нарушением сознания; тяжелыми нарушениями перфузии, бледностью и цианозом кожных покровов; сни
Дистрибутивный (перераспределительный) шок 427
жением сердечного выброса и повышением сосудистого сопротивления; тахикардией, артериальной гипотензией; нарушением функции легких. Снижение сердечного выброса в этой стадии СШ происходит не только за счет гиповолемии вследствие потери жидкости из внутрисосудистого сектора, но и за счет прямого мио-карддепрессивного действия индуцированных эндотоксинами цитокинов (ИЛ-10, TNFa) и NO-синтетаз [34]. Важную роль в патогенезе снижения сердечного выброса и артериальной гипотензии играют нарушения спланхнического кровотока, характеризующиеся выраженной артериолярной констрикцией, повышением венозного давления, секвестрировании крови в спланхнической зоне [35]. Кроме того, бактериальные эндотоксины и цитокины индуцируют активность NO-синтетазы и образование избытка оксида азота [36], что ведет к неконтролируемой вазодилатации, рефрактерной к катехоламинам [37]. Сосудистая гипореактивность к катехоламинам ограничивает успешное лечение гипотензии при септическом шоке. NO реагирует с супероксидом с формированием потенциально токсичных метаболитов, пероксинитрита (ONOO“). Результаты исследования показали, что норэпинефрин окислялся и дезактивировался пероксинитритом. Этим механизмом, возможно, объясняется гипореактивность сосудов к норэпинефрину при септическом шоке [38].
Сепсис, как и политравма, является мощным стимулом гиперкоагуляции, поскольку любые воспалительные стимулы активизируют коагуляцию и провоспали-тельные каскады. При эндотоксемии наблюдается усиление «перекрестной связи» коагуляционных и воспалительных каскадов. Воспалительные медиаторы активизируют коагуляцию. TNFa, ИЛ-1 и ИЛ-6 увеличивают активность тканевого фактора и ингибируют фибринолиз, таким образом активируя внешний путь коагуляции. Кроме того, внутрисосудистая коагуляция стимулирует воспалительный ответ. Коагуляция крови in vitro увеличивает продукцию моноцитами TNFa, ИЛ-1 и ИЛ-8, фактор Ха, a-тромбин и фибрин — синтез ИЛ-6 и ИЛ-8. Генетическая пред
расположенность может увеличивать тенденцию к внутрисосудистой и интрааль-веолярной коагуляции. Полиморфизм генов факторов коагуляции усиливает коагуляцию и ингибирует антикоагуляцию и фибринолиз, что смещает обычно существующее равновесие в пользу коагуляции. Сообщается о роли нуклеотидного полиморфизма в кодировании и продукции a-тромбина, фибриногена, фактора V, протеина С, рецепторов эндотелиального протеина С и активатора ингибитора плаз-могена-1. Нуклеотидный полиморфизм протеина С, фактора V (например, фактора V Leiden) и ингибитора активатора плазмогена-1 связан с повышенным риском тромбоза глубоких вен, легочной эмболии, острого инфаркта миокарда и инсульта. Нуклеотидные полиморфизмы могут обуславливать повышенный риск коагуляции и способствовать прогрессированию у пациентов с сепсисом, травмой и некоторыми другими патологическими состояниями ССВО.
При СШ быстро развивается полиор-ганная недостаточность.
Основные терапевтические мероприятия при СШ:
1. Этиологическая терапия антибиотиками или антивирусными препаратами, к которым чувствительна высеваемая микрофлора, обязательная адекватная санация инфекционного очага.
2. Коррекция гиповолемии внутривенными инфузиями кристаллоидных и коллоидных растворов.
3. Модуляция тонуса сосудов: симпато-миметики; исследуются показания к применению вазопрессина, ингибиторов NO-сите-таз, метиленового синего, нитратов.
4. Заместительная терапия глюкокортикоидами при наличии скрытой или явной недостаточности надпочечников.
5. Симптоматическая терапия возникающих расстройств функций других органов и систем.
6. Для удаления из крови токсинов могут оказаться эффективными экстракорпоральные методы детоксикации (гемофильтрация, ультрафильтрация, плазмаферез).
7. Перспективным считается применение препаратов с антикоагулянтной актив
428 ШОК
ностью (активированный протеин С, антитромбин), так как развивающаяся при СШ гиперкоагуляция является во многих случаях причиной нарушения кровобра-щения и системного воспаления.
Поскольку механизмом, запускающим септический шок, является инфекция, применяется антимикробная терапия и адекватная санация очага. Неадекватная антимикробная терапия приводила к увеличению смертности на 39 % , поэтому антибиотики должны назначаться в соответствии с чувствительностью к ним высеваемой микрофлоры, однако в случаях тяжелой, угрожающей жизни инфекции необходимо раннее эмпирическое назначение антибиотиков.
Одним из патогенетических звеньев СШ является гиповолемия вследствие потери жидкости из внутрисосудистого русла, поэтому назначается инфузионная терапия. В последнее время все больше исследований подтверждают протекторное влияние на эндотелий при сепсисе и эндотоксемии растворов гидроксиэтилирован-ного крахмала (ГЭК) (рефортан), объясняя этот эффект снижением LPS-индуцированной артериолярной и венулярной адгезией лейкоцитов и восстановлением взаимодействия между лейкоцитами и клетками эндотелия [39]. Для плазмозамещающей терапии целесообразно применение гипертонического раствора натрий хлорида/ декстрана, улучшающего деятельность сердечно-сосудистой системы при тяжелом сепсисе без серьезных побочных эффектов [40].
Чаще всего для стабилизации гемодинамики при СШ используют норадреналин или его сочетание с добутамином или допексамином. Менее обосновано при СШ использование адреналина, так как имеются данные [41] о том, что в отличие от норадреналина и допамина этот препарат значительно ухудшает спланхнический кровоток. Наиболее эффективной при СШ считают комбинацию норэпинефрина с допамином или добутамином, при ее применении отмечены наиболее продолжительное выживание и наименее серьезные легочные повреждения [42]. Комбинация добутамина с норэпинефрином сопрово
ждалась улучшением миокардиальной деятельности, большей доставкой и потреблением кислорода, более низкими уровнями лактата крови и рСО2, меньшим анатомическим повреждением. Применение ингибиторов фосфодиэстеразы при СШ нецелесообразно вследствие возможной гипотензии. Перспективным на данный момент представляется использование вазопрессина. Норадреналин-резистентная гипотензия, возникающая при септическом шоке, сопровождается высокой летальностью, снизить которую, возможно, удастся путем инфузии низких доз вазопрессина [39].
Изучается вопрос о применении метиленового синего (3 мг/кг) [43]. Действие этого препарата связывают с ингибированием гуанилатциклазы — фермента, влияющего на вызванные сепсисом дисфункции сосудов и миокарда, гемодинамические переменные, включая объем крови и легочную сосудистую проницаемость во время септического шока. Результаты исследования A. Donati с соавт. [43] подтвердили вазоконстрикторный и положительный инотропный эффекты метиленового синего при СШ. Эти эффекты не были связаны с изменениями объема крови, миокардиальной диастолической функцией или легочной сосудистой проницаемостью.
Очень важным является вопрос о пользе применения кортикостероидов при септическом шоке. Надпочечная недостаточность часто возникает у пациентов с СШ, постановка диагноза в значительной степени зависит от диагностического теста. В настоящее время нет четкого метода определения абсолютной концентрации кортизола в сыворотке крови, которая бы позволяла дифференцировать адекватную и недостаточную реакцию надпочечников. Однако, по мнению некоторых авторов [42], определяемая концентрация кортизола < 25 мкг/дл у пациента в состоянии стресса является полезным диагностическим порогом для определения надпочечниковой недостаточности. 37 % больных оказались стероидчувствительными. Базовая концентрация кортизола в сыворотке составила 14,1 ± 5,2 мкг/дл для стероидчувствительных пациентов против 33,3 ± 18 мкг/дл
Дистрибутивный (перераспределительный) шок 429
для стероиднечувствительных пациентов (р < 0,0001). У 95 % стероидчувствительных пациентов определялась базовая концентрация кортизола < 25 мкг/дл. У больных с септическим шоком и относительной недостаточностью надпочечников низкие дозы гидрокортизона (внутривенно болюсно 50 мг каждые 6 ч) в течение семи дней значительно снижали риск смерти без увеличения риска неблагоприятных осложнений [44]. У больных с септическим шоком без недостаточности надпочечников применение кортикостероидов не оказалось эффективным. Поскольку в клинике часто нет возможности определить уровень кортизола в плазме, следует вводить гормоны в случае рефрактерного к терапии СШ.
Коагулопатия и системное воспаление — почти универсальные факторы патологии у пациентов с тяжелым сепсисом. Часто тромбиновая сигнализация является главным компонентом воспаления во внесосу-дистом пространстве. При низких концентрациях внутрисосудистый тромбин активирует протеиновый С-путь путем преобразования протеина С (связанный с эндотелием рецептор протеина С) в активированную форму, что, в свою очередь, генерирует противовоспалительные сигналы на эндотелиальные клетки. Прямая тромбиновая сигнализация происходит только тогда, когда внутрисосудистые концентрации тромбина превышают порог коагуляции. При системном воспалении, вызываемом бактериальным токсином, ингибирования тромбина недостаточно для ограничения воспаления, однако при остром повреждении легких угнетение тканевого фактора прерывает эскалацию воспаления. Поэтому коррекция гиперкоагуляции и активация фибринолиза, в частности применение активированного протеина С, представляются очень перспективным направлением лечения тяжелого сепсиса и септического шока.
Использование дротрекогина а (активированного), обладающего противовоспалительным, антитромботическим, профи-бринолитическим и другими эффектами, в экспериментах на животных и у людей снижает относительный риск летальных
исходов при грамотрицательной и грам-положительной инфекции, пневмонии, брюшных источниках инфекции, шоке и отсутствии шока. Лечение этим препаратом также уменьшало продолжительность искусственной вентиляции легких и шока, незначительно увеличивало риск кровотечений. Несмотря на столь обнадеживающие результаты, показания и методика применения активаторов фибринолиза нуждаются в дополнительных многоцентровых рандомизированных исследованиях. В Украине, Росии и других странах бывшего СССР при тяжелом сепсисе и СШ используются методы экстракорпоральной детоксикации, в частности плазмаферез. В последнее время в англоязычной литературе появились работы, посвященные этим методам. Так, по мнению R. Busund с соавт., плазмаферез может быть важным и безопасным методом снижения смертности у пациентов с тяжелым сепсисом или септическим шоком, дополняющим традиционное лечение.
Токсический шоковый синдром (ТШС). Шок, вызываемый S. aureus и другими видами кокков, некоторые авторы выделяют как отдельный вид [44]. Этот микроорганизм колонизирует кожу и слизистые оболочки, наиболее часто очагом эндотоксемии является слизистая носа и влагалища. Более чем в 80 % случаев причиной ТШС является тампонирование влагалища в период менструации (тампон повреждает слизистую влагалища и препятствует отхождению содержимого), чаще всего болеют женщины в возрасте 15 — 25 лет. Предрасполагающими факторами являются недавние роды, инфекция в малом тазу, синусит, операции на носовых ходах, грипп, фурункулез.
Симптомами, предшествующими шоку, могут быть лихорадка, головная боль, понос. Часто одним из ранних симптомов является диффузная эритроматозная сыпь. Иногда дескваматозная сыпь появляется на кистях рук и подошвах ног через 1—2 недели после начала ТШС [45]. В течение 24 — 48 ч развивается полиор-ганная недостаточность: ЦНС (дезориентация), пищеварительного канала (рвота, понос), почек, печени и тромбоцитопения.
430 ШОК
В тяжелых случаях ТШС осложняется ОРДС, отеком мозга, миокардиальной недостаточностью. Диагноз ТШС ставится на основании идентификации возбудителя S. aureus и клинических симптомов. Анализ на гемокультуру обычно отрицательный.
Лечение ТШС аналогично лечению СШ. Однако большое значение имеет санация инфекционного очага (извлечение тампона при инфекции во влагалище). Системная антибиотикотерапия менее эффективна, поскольку обычно отсутствует генерализация процесса, однако она необходима для предотвращения рецидива заболевания. Чаще всего эффективны такие антибиотики, как нафциллин, цефалоспорины 1-го поколения (цефалотин), при обнаружении метициллин-резистентных штамов S. aureus (MRSА) показано назначение ванкомицина, тейкопланина. В некоторых исследованиях наблюдали хорошие результаты при применении клиндо-мицина [46].
13.5.2. Нейрогенный шок
Вид дистрибутивного шока, возникающий в результате нарушения вазомоторной функции симпатической автономной нервной системы, что приводит к периферической вазодилатации и перемещению крови в периферические участки.
Наиболее частыми этиологическими факторами являются: травма ствола головного мозга, шейного и верхнегрудного отделов спинного мозга (спинальный шок), отравления или передозировка фармакологических средств с выраженной депрессивной активностью на сосудодвигательный центр (анестетики, антидепрессанты, снотворные и др.), перенесенная острая гипогликемия (чаще всего при передозировке инсулина).
НШ характеризуется: снижением сосудистого сопротивления, сердечного выброса вследствие снижения венозного возврата, ДЗЛА, ЦВД, тахикардией (возможна брадикардия), артериальной гипотензией, в начальных стадиях возможен нормальный диурез.
Основные терапевтические мероприятия при НШ:
1. Коррекция гиповолемии массивными инфузиями жидкости.
2. Применение вазопрессоров.
3. При НШ травматической этиологии (повреждение спинного мозга) — большие дозы кортикостероидов.
4. Симптоматическая терапия для поддержания функции дыхания и других систем.
В случае тяжелого спинального шока для восстановления гемодинамики требуется переливание большого количества жидкости.
13.5.3. Анафилактический шок
Анафилактический шок (АШ) — вид аллергической реакции немедленного типа, возникающей на повторное введение в организм аллергена и сопровождающейся расстройствами функции ЦНС, артериальной гипотензией, повышением проницаемости эндотелия сосудов, спазмом гладких мышц, в частности развитием брон-хиолоспазма. Относится к дистрибутивным видам шока.
Аллергия — иммунная реакция организма, сопровождающаяся повреждением собственных тканей. Аллерген — вещество, вызывающее развитие аллергической реакции, обладает всеми свойствами антигенов (макромолекулярность, преимущественно белковая природа, чужеродность для данного организма) [46].
Аллергическая реакция может вызываться не только аллергенами, но и гаптенами — микромолекулярными соединениями (например, лекарствами), простыми химическими веществами, сложными продуктами небелковой природы (микробные продукты, полисахариды). При попадании в организм сами по себе они не вызывают аллергической реакции, аллергенами они становятся только после соединения с белками организма. При этом образуются конъюгированные или комплексные антигены, сенсибилизирующие организм, которые при повторном введении могут соединяться с антителами или сенсибилизированными лимфоцитами и вызывать реакцию без связи с белками.
Дистрибутивный (перераспределительный) шок 431
Реакции повышенной чувствительности I типа обусловлены образованием го-моцитотропных антител (иммуноглобулины G и Е), Fc-фрагмент которых представляет собой лиганду к Fc-рецептору наружной поверхности мастоцитов и базофилов. В результате гомоцитотропные антитела фиксируются на поверхности этих клеток. Экзогенный антиген, вызывающий образование гомоцитотропных антител, является аллергеном.
Термин «анафилаксия» (от гр. апа — обратный и phylaxis — защита) был предложен Р. Portier и С. Richet еще в 1902 г. для обозначения необычной, иногда смертельной реакции у собак на повторное введение им экстракта из щупалец актиний. Позднее аналогичную реакцию обнаружили у людей.
Причиной анафилактических и анафилактоидных реакций могут быть различные субстанции, но чаще всего они вызываются:
лекарственными препаратами: антибиотиками;
противовоспалительными средствами; аналгетиками;
химиотерапевтическими препаратами; местными анестетиками;
мышечными релаксантами;
рентгеноконтрастными веществами;
пищевыми продуктами:
орехами;
морскими продуктами; цитрусовыми;
химическими субстанциями; инсектицидами;
латексом;
пыльцой растений.
В принципе, любое вещество может вызвать анафилактическую реакцию. Но чаще всего эти реакции возникают на: лекарства — 51 %, определенные виды пищи — 15, укусы насекомых — И %; введение вакцин и сывороток. Среди пищевых продуктов в последнее время участились реакции на арахис, орехи и морские продукты.
В зависимости от патофизиологии и этиологического фактора различают такие виды аллергических реакций (Р. L. Lieberman, 1999):
анафилактические (имму независимые): пищевые продукты (арахис, морские моллюски и др.);
лекарственные препараты (антибиотики, инсулин и др.);
укусы насекомых;
физическая нагрузка (возможно);
анафилактоидные (вследствие освобождения медиаторов из мастоцитов и базофилов):
лекарственные препараты (например, наркотические аналгетики);
вирусные заболевания (простуда); действие солнечного света;
физическая нагрузка;
идиопатия;
препараты, влияющие на метаболизм арахидоновой кислоты (аспирин, другие нестероидные противовоспалительные средства);
плазморасширители: декстраны, альбумин (возможно);
гамма-глобулин;
цитотоксические препараты; трансфузионные реакции;
неантигенная антителозависимая активация комплемента (рентгеноконтрастные вещества, диализные мембраны).
Большинство лекарственных препаратов являются гаптенами, к полноценным антигенам относятся белковые и полипептидные препараты (антитоксические сыворотки, аллогенные гамма-глобул ины, белки плазмы крови, гормоны). Причем доза препарата, к которому произошла сенсибилизация организма, может быть ничтожно малой. Например, описаны случаи развития АШ у больных с аллергией на пенициллин после того, как к ним подходил сотрудник, сделавший перед этим инъекцию пенициллина другому больному. Увеличивается количество случаев АШ вследствие укусов насекомых. Проведение специфической диагностики и гипосенсибилизация у больных с аллергией иногда сопровождается шоком.
Несколько факторов могут повышать риск развития анафилактической реакции, среди них: атопия; путь введения антигена; систематичность попадания антигена; время, прошедшее после последней реакции; возраст и пол. Атопия является важным фактором риска в случаях, когда анти
432 ШОК
ген вступает в контакт со слизистой оболочкой, т. е. при пероральном пути введения. Инцидентность анафилаксии к латексу, физической нагрузке, к приему внутрь рентгеноконтрастных веществ так же, как инцидентность идиопатической анафилаксии, выше у пациентов с атопией.
Причины этой повышенной предрасположенности изучены не полностью, но у пациентов с атопией, включая астму, пищевую аллергию и атопический дерматит, обнаруживается повышенное спонтанное освобождение гистамина из базофилов по сравнению с контрольными группами.
Анафилаксия может возникнуть при любом пути попадания антигена. Однако более часто и более тяжело анафилактические и анафилактоидные реакции протекают при парентеральном пути введения антигена. Кроме того, период начальных проявлений реакции в этих случаях сравнительно короткий.
Постоянство контакта с антигеном — важный фактор риска анафилаксии. Прекращение назначения антигена с последующим его возобновлением может предрасполагать к реакции. Так, анафилаксия на инсулин чаще всего наблюдается тогда, когда применение этого лекарственного средства было прервано и затем возобновлено, например при лечении диабета во время беременности.
Время, прошедшее после последней реакции, также играет важную роль: чем больше времени прошло от последней реакции на лекарственное средство, тем меньше вероятность развития повторной реакции. Причиной этого, возможно, является уменьшение синтеза IgE в результате отсутствия экспозиции с аллергеном.
Вероятность развития анафилактических реакций при контакте с антигеном в определенной степени зависит от пола и возраста. Например, в случаях рентгеноконтрастных веществ, плазмозаменителей, анестезирующих средств риск выше у взрослых, чем у детей; непонятно, является ли эта разница в инцидентности результатом повышенной чувствительности или просто увеличенной экспозиции. Анафилактические реакции на мышечные релаксанты, аспирин и латекс происходят чаще
всего у женщин, а на яд насекомых — у мужчин. Разница, по-видимому, обуславливается преимущественным родом деятельности и соответственно экспозицией с аллергеном. Например, у женщин чаще возникают реакции на внутривенные мышечные релаксанты, вероятно, потому, что эти препараты содержат производные аммония, обнаруженные во многих косметических продуктах.
В некоторых случаях этиологический фактор анафилаксии остается неизвестным. Аллергологи утверждают, что примерно в половине случаев анафилаксии носили идиопатический характер. Диагноз «идиопатическая анафилаксия» ставят в тех случаях, когда другие причины были исключены.
Патофизиология и патогенез. Повышенная чувствительность — это чрезмерная иммунная реакция на антиген. При первичном контакте антиген не вызывает очевидной клинической реакции, но происходит сенсибилизация иммунной системы к определенному антигену. При повторном контакте возникает клиническая реакция. Аллерген реагирует с предшественниками В- и Т-лимфоцитов, активируя формирование Т-хелперов и В-кле-ток. В свою очередь, воссоздается память В-клеток и плазмоцитов, и тогда последние продуцируют специфические IgE антитела — реагины. IgE связываются со специфическими клеточными рецепторами на мастоцитах и базофилах и вызывают продукцию медиаторов анафилаксии, ответственных за локальные и системные реакции. Первичные медиаторы являются молекулами, которые содержатся в гранулах мастоцитов и базофилов, после контакта клетки с антигеном они начинают секретироваться во внеклеточную среду. Эти медиаторы очень быстро появляются в очаге аллергического воспаления, причем сразу в достаточно больших концентрациях. Системное действие этих веществ заключается в повреждении клеточных мембран, повышении проницаемости эндотелия капилляров, интерстициальном отеке, сокращении гладких мышц бронхиол, увеличении секреции клетками слизистых оболочек.
Дистрибутивный (перераспределительный) шок 433
Из мастоцитов и базофилов выделяется много различных медиаторов, количество которых все время растет. Приводим наиболее часто упоминаемые [48]:
гистамин — имидазол-4р-этиламин. Относится к биогенным аминам. Образуется из гистидина путем декарбоксилирования под действием гистидиндекарбоксилазы. Содержится в гранулах мастоцитов и базофильных лейкоцитов. Биологическое действие реализуется через гистаминовые рецепторы двух видов — и Н2, активация которых часто приводит к разновекторному эффекту. Стимуляция Нррецепторов вызывает спазм гладких мышц бронхов, коронарных и легочных сосудов, сокращение эндотелиоцитов, особенно посткапиллярного отдела микроциркуляции, повышение проницаемости сосудов, развитие отека и воспаление. Стимуляция Н2-рецепторов — повышение желудочной секреции и переваривающей способности желудочного сока, подавление пролиферации и цитотоксической активности лимфоцитов;
серотонин — 5-окситриптамин. Также принадлежит к группе биогенных аминов. Его участие в патогенезе аллергических реакций главным образом доказано у животных, несколько меньшее значение он имеет для человека. Однако экспериментальное применение ряда антисеротони-новых препаратов показало их определенную эффективность при некоторых аллергических заболеваниях. В развитии АШ значимая роль его не установлена. Биологические эффекты сходны с гистамином;
эозинофильный хемотаксический фактор анафилаксии — группа тетрапептидов с молекулярной массой 380 — 390. Вызывают хемотаксис эозинофилов и нейтрофильных гранулоцитов, усиливают экспрессию рецепторов эозинофилов для комплемента;
высокомолекулярный нейтрофильный хемотаксический фактор выделен из сыворотки крови человека, болеющего крапивницей, вызывает хемотаксис нейтрофильных гранулоцитов, сопровождающийся снижением их активности;
гепарин — макромолекулярный протеогликан. Активируется после высвобож
дения из мастоцитов, обладает антитромбиновой и антикомплементарной активностью;
фактор, активирующий тромбоциты, — смесь глицеро-3-фосфохолинов. Секретируется базофилами, лимфоцитами, нейтрофилами, тромбоцитами и эндоте-лиоцитами в результате активации фосфолипазой А2. Вызывает агрегацию тромбоцитов, спазм гладких мышц, способствует высвобождению гистамина и серотонина, увеличивает проницаемость эндотелия. Рассматривается как важнейший фактор развития не только анафилаксии, но и других аллергических реакций;
метаболиты арахидоновой кислоты. В результате воздействия на клеточные мембраны фосфолипазы А2 в мастоцитах и других клетках происходит отщепление от фосфолипида полиненасыщённой жирной кислоты, главным образом арахидоновой. В случае осуществления метаболизма по циклооксигеназному пути образуются простагландины и тромбоксаны, по липооксигеназному — лейкотриены. Простагландины и лейкотриены во многих отношениях действуют альтернативно;
лейкотриены — очень сильные медиаторы, оказывают эффект в малых концентрациях. Три лейкотриена — LTC4, LTD4, LTE4 — образуют медленно реагирующую субстанцию анафилаксии. Эти лейкотриены вызывают спазм гладкой мускулатуры (воздействие LTD4 в 100 раз сильнее в отношении гладкой мускулатуры, чем гистамина), могут играть важную роль в развитии медленной фазы анафилаксии, способствуя стойкой бронхокострикции, воспалению, повреждению сердца;
простагландины PGD2, PGE2, PGF2 — могут повышать давление в легочной артерии, снижать тонус периферических сосудов. Накапливаются в очаге воспаления через 4 —24 ч;
растворимая форма CD 23. Этот С-лек-тиновый рецептор присутствует на поверхности 30 % В-лимфоцитов и на небольшом количестве Т-клеток и моноцитов. Стимулирует широкий спектр биологических эфффектов В-клеток и моноцитов.
Анафилаксия лежит в основе таких заболеваний, как аллергическая бронхиаль
434 ШОК
ная астма, аллергические риниты, крапивница, анафилактический шок [49].
Реакции гиперчувствительности могут развиваться с участием как иммунных, так и неиммунных механизмов. Последние называют анафилактоидными. При этом преобладает кожная симптоматика, не всегда сопровождающаяся нарушениями со стороны системы дыхания и кровообращения. Часто такие реакции остаются нераспознанными. Наблюдается гиперемия лица и невыраженная артериальная гипотензия, хотя дальнейшие нарушения могут привести к тяжелым последствиям. По мнению Garret (1978), во время наркоза анафилактоидные реакции проявляются следующими признаками: внезапное покраснение лица, появление на всем теле угреобразной или уртикарной сыпи, артериальная гипотензия, бронхоспазм, отек лица. В его работе приведены данные об анафилактоидных реакциях на введение барбитуратов (тиопентона). Количество сообщений о побочных реакциях на лекарства продолжает расти. Полагают, что ежегодно в каждой европейской стране происходит от 5 до 10 тыс. тяжелых реакций.
При истинной анафилаксии специфическую роль играют антитела IgE и IgG. В других реакциях вазоактивные вещества могут высвобождаться из мастоцитов и базофилов вследствие непосредственного взаимодействия с медикаментами, которые связываются с рецепторами на клеточной поверхности, имитируя активность IgE.
Клинические проявления
Симптомы АШ:
1. Нарушение сознания, вплоть до коматозного состояния.
2. Кожные высыпания (уртикарии, гиперемия, ангиоотек).
3. Бронхоспазм с одышкой, хрипами в легких, участие в дыхании вспомогательной мускулатуры.
4. Артериальная гипотензия с потоотделением, нарастающей тахикардией, возможны нарушения ритма сердца.
5. Возможны спастические боли в животе, рвота, иногда кровянистый стул.
6. Гемоконцентрация вследствие потери жидкости из сосудистого русла.
Наиболее тяжелыми проблемами при анафилаксии являются расстройства дыхания: отек дыхательных путей, одышка и бронхиолоспазм. Асфиксия — одна из наиболее частых причин смерти при АШ.
Как правило, артериальная гипотензия клинически проявляется несколько позже головокружением, обмороком, нарушением сознания.
Симптоматика со стороны пищеварительного канала включает тошноту, рвоту, понос и спастические боли в животе.
Редко возникают симптомы ринита, головная, загрудинная боль и зуд без кожных высыпаний. Повышение температуры тела нехарактерно.
Развивающаяся гипоксемия может вызвать депрессию миокарда, которая сохраняется в течение нескольких дней, что приводит к возникновению артериальной гипотензии. При анафилаксии может наблюдаться спазм коронарных сосудов, иногда развивается инфаркт миокарда. Брадикардия может возникать в результате рефлекса Bezold—Jarisch — рефлекса торможения работы сердца, сенсорная зона которого расположена в нижне-задней стенке левого желудочка.
Симптомы обычно проявляются через 5 — 30 мин после начала контакта антигена в организме. При энтеральном пути введения симптомы развиваются позднее, чем при внутривенном. Симптоматика обычно проявляется в течение первых 2 ч. Отмечается довольно тесная связь между временем, прошедшим от момента попадания антигена до появления симптомов, и тяжестью реакции: т. е. чем более быстрое начало, тем более тяжелая реакция.
Поздняя стадия реакции проявляется возобновлением симптомов после очевидного улучшения состояния. Она сопровождается многочисленными электрокардиографическими изменениями, включая элевацию сегмента ST, уменьшение и инверсию зубца Т, развитие аритмий. Часто определяется повышение кардиальных ферментов. Латентный период с момента введения антигена до возникновения клинических проявлений составляет от нескольких минут до получаса. Чем он короче,
Дистрибутивный (перераспределительный) шок 435
тем обычно тяжелее протекает реакция. Пациенты, у которых развивается поздняя реакция, испытывают двухфазную анафилаксию. Причем такие рецидивы симптомов анафилаксии могут происходить неоднократно после частых улучшений состояния. Это состояние получило название «длительная анафилаксия». Повторные (рецидивирующие) реакции, возможно, обусловлены активацией медиаторов хемотаксиса, высвобождающихся из мас-тоцитов и базофилов. При рецидивирующей анафилаксии смерть больного может наступить в любой момент.
Продромальными симптомами могут быть: беспокойство, зуд, особенно на поверхностях ладоней и стоп, затем развивается АШ.
Тяжесть клинических проявлений от незначительных — до острой остановки кровообращения.
Особенно тяжело протекает АШ у больных с ишемической болезнью сердца, так как анафилактическая реакция часто сопровождается спазмом венечных сосудов и агрегацией тромбоцитов [51].
Диагностика анафилактического шока обычно не составляет труда, основывается на клинической симптоматике. Устанавливается прямая временная связь между введением аллергена и развитием шока. Дифференциальная диагностика проводится с септическим или кардиогенным шоком, астмой, вазовагальным коллапсом, острыми расстройствами функции дыхания и кровообращения другой этиологии.
В случае развития анафилактического шока для установления диагноза достаточно специфической клиники и хронологии развития заболевания, для начала лечения не требуется никаких биомедицинских исследований.
Установлению диагноза анафилаксии могут способствовать некоторые биохимические исследования. Пиковый уровень триптазы в плазме крови достигается через 1 —1,5 ч после начала анафилаксии и может оставаться повышенным до пяти часов. Положительное прогнозирующее значение триптазного теста для диагноза анафилаксии составляет 92,6 %; отрицательное — 54,3 %. Измерение концентрации
гистамина в плазме обычно не столь информативно, как в случае триптазы, поскольку его уровень в плазме повышен не более одного часа после появления симптомов. Однако определение содержания гистамина и его метаболитов в суточной моче может быть полезным, так как для анафилаксии характерно повышение этого показателя. Диагностическое значение анализа на специфический иммуноглобулин Е было подтверждено недавними исследованиями.
Лечение АШ. Прежде всего прекращение введения аллергена. При острой остановке кровообращения применяются мероприятия сердечно-легочной реанимации. При введении аллергена в конечность следует наложить жгут, обколоть место инъекции адреналином. При приеме внутрь — промыть желудок, принять активированный уголь.
При остром АШ проводятся такие мероприятия [52]:
1. Обеспечение проходимости дыхательных путей (при необходимости интубация трахеи либо коникотомия).
2. Оксигенотерапия.
3. Введение адреналина.
4. Инфузия жидкости.
5. Ингаляция р-адреномиметиков.
6. При бронхоспазме — внутривенное введение эуфиллина.
7. Введение Нг и Н2-блокаторов (дифенгидрамин — 50 мг внутривенно и ранитидин в той же дозе).
8. Введение кортикостероидов (хотя не следует ожидать немедленного эффекта).
9. При продолжающейся артериальной гипотензии — инфузия кардиотонических и вазопрессивных препаратов.
Поддержание проходимости дыхательных путей и ингаляция 100 % кислорода. Интубация трахеи применяется не во всех случаях, для более точного определения показаний к интубации трахеи и переводу на ИВЛ необходим анализ газов артериальной крови. В случаях тяжелого отека гортани может понадобиться экстренная коникотомия.
Ингаляция кислорода должна быть налажена немедленно.
Необходимо помнить, что 75 % смерти от анафилаксии происходит вследствие
436 ШОК
асфиксии в результате отека верхних дыхательных путей или тотального бронхио-лоспазма, в большинстве случаев — в течение первого часа после развития клинических симптомов реакции, и только 25 % — от циркуляторного коллапса.
Адреналин (А). Является препаратом выбора при АШ. Стимуляция оц-адрено-рецепторов повышает тонус сосудов, вызывает вазоконстрикцию и увеличивает АД, стимуляция p-рецепторов оказывает бронхо литический эффект, ингибирует освобождение медиаторов, увеличивая внутриклеточное содержание циклического АМФ в мастоцитах и базофилах. Применение 0,75 — 1,5 мкг/кг адреналина в разведении 1 : 100 000 внутривенно или 10 мкг/мл в минуту оказывает сохраняющий жизнь эффект при анафилаксии, ингибирует дальнейшую продукцию масто-цитами медиаторов и помогает избежать передозировки. При отсутствии пульса на периферии необходимо введение реанимационной дозы адреналина — 0,25 —0,5 мг. При невозможности внутривенного введения в случаях тяжелого шока можно вводить А внутритрахеально или в крайнем случае — сублингвально или внутримышечно. Неблагоприятные побочные эффекты при внутривенном введении адреналина чаще наблюдаются при недостаточном разведении, слишком быстром введении и чрезмерных дозах. При внутривенном пути введения необходим мониторинг АД, ЭКГ, пульсоксиметрия. Учитывая неблагоприятные эффекты при внутривенном введении А, при отсутствии критической артериальной гипотензии этот путь введения сейчас не рекомендуется. У пациентов с отеком гортани, но без выраженной гипотензии следует ограничиться внутримышечным или подкожным введением. Рекомендуется раннее внутримышечное введение адреналина в дозе 0,5 мл (1 : 1000) всем взрослым пациентам с клиническими симптомами шока, отека дыхательных путей или затруднения дыхания. Внутримышечно А можно вводить и при шоке, пока не обеспечен венозный доступ или если нет полной уверенности в диагнозе анафилаксии. Этот метод является сравнительно безопасным и эффек
тивным даже в не очень опытных руках. Hughes and Fitzharris (1999) рекомендуют при тяжелом шоке повторение через 5 мин внутримышечного введения А или очень медленное внутривенное введение.
Согласно новым представлениям, дозу адреналина необходимо увеличивать только у пациентов после приема амитриптилина, имапрамина и p-блокаторов, которые, как считают, ингибируют механизм чрез-мембранного поступления норадреналина в адренергические нейроны, у пациентов после применения p-блокаторов преобладает а-адренергический эффект с брадикардией, артериальной гипертензией. Альтернативой увеличения дозы А у этих больных является применение глюкагона, который повышает содержание цАМФ независимо от p-рецепторов. Доза глюкагона: вначале болюсно внутривенно 1 — 5 мг с последующей инфузией со скоростью 5—15 мкг/мин до достижения клинического эффекта. Некоторые авторы рекомендуют ограничить применение у пациентов с повышенным риском анафилактических реакций таких лекарственных препаратов, как р-блокаторы, трициклические антидепрессанты, ингибиторы АПФ, ингибиторы моноаминоксидазы, которые значительно затрудняют лечение артериальной гипотензии.
У пациентов во время АШ преобладает вазодилатация с низким сосудистым сопротивлением, что требует стимуляции а]-адренергических рецепторов. Поэтому в некоторых ситуациях из катехоламинов предпочтение может быть отдано фенилэфрину (мезатону) или норадреналину в дозе 4 — 8 мкг/мин (0,05 — 0,1 мкг/кг в минуту), поскольку применение адреналина у них опасно. Изопротеренол назначается при рефрактерном бронхиолоспазме и легочной гипертензии в стартовой дозе 0,5—1 мкг/мин. Его следует с осторожностью применять у пациентов с гипотензией, так как он обладает р2-адренерги-ческим эффектом и может продуцировать системную вазодилатацию.
При персистирующей артериальной гипотензии используется внутривенная инфузия таких препаратов, как дофамин, добутамин. Выбор препаратов зависит от
Дистрибутивный (перераспределительный) шок 437
клинической ситуации, требующей акцентирования а- или p-адренергического эффекта, т. е. от показателей ЧСС, ритма сердца, системного сосудистого сопротивления давления в легочной артерии.
При развитии тяжелой брадикардии можно дополнительно к адреналину вводить атропин (0,3 —0,5 мг).
Инфузионная терапия. Крайне важной при лечении АШ является адекватная по объему инфузионная терапия. При анафилактическом шоке быстро развивается гиповолемия вследствие потери при тяжелом АШ до 40 % жидкости из внутрисосудистого русла в интерстициальное пространство, что сопровождается гемоконцентрацией. В начале реакции вводят 25 — 50 мл/кг изотонического раствора, в случаях продолжающейся артериальной гипотензии можно добавить раствор коллоидов. Для восполнения объема внутрисосудистой жидкости и коррекции АД в тяжелых случаях следует переливать 5 — 10 мл/кг изотонического раствора натрий хлорида (R. Patterson, М. Schatz, 1975), хотя возможно применение коллоидных растворов (гидроксиэтилкрахмала) и гипертонического 7,5 % раствора натрий хлорида. В особо тяжелых случаях для коррекции гиповолемии необходимо под контролем ЦВД переливать 5 —7 л жидкости.
Как пример необходимости инфузии больших объемов жидкости при АШ можно привести клинический случай, опубликованный J. Dahn с соавт. [53]:
35-летний пациент (ASA I) взят на операцию по поводу параназального синусита; через 5 мин после индукции в наркоз пропофолом (200 мг), фентанилом (0,3 мг), рокурониумом (40 мг) развилась острая артериальная гипотензия со снижением систолического АД ниже 40 мм рт. ст.; тахикардия с частотой сокращений сердца 130 в минуту; выраженная бледность кожных покровов; бронхоспазма не было; были введены норадреналин (1,2 мг титрованно) (адреналин не применялся в связи с выраженной тахикардией), метилпреднизолон (500 мг), Hj- и Н2-блока-торы; концентрация гемоглобина поднялась от 150 г/л (до операции) до 190 г/л (через 25 мин после развития АШ), гематокрит — от 0,45 до 0,59 л/л, pH снизилась до 7,19, BE — до 5 ммоль/л; такая гемоконцентрация соответствует потере 1,5 л жидкости из внутрисосудистого
русла за 25 мин; АД удалось нормализовать в течение 40 мин инфузией 1 л коллоидных растворов и 2 л — кристаллоидных. Как выяснилось в результате проведения аллергических тестов, АШ развился на введение рокурониума.
Применение антигистаминовых препаратов. Поскольку через -рецепторы реализуются многие неблагоприятные эффекты анафилаксии, оправдано внутривенное введение Н]-антагониста (за рубежом с этой целью используют дифенгидрамин в дозе 0,5—1,0 мг/кг ). Применение Ht-антагонистов показано при всех формах анафилаксии. Однако они обладают ан-тидофаминергическим действием и могут усугубить гипотензию при гиповолемии, поэтому вводить их надо медленно, под контролем АД. В последнее время многие исследователи рекомендуют введение Н2-блокаторов при анафилаксии. Ранитидин в дозе 1 мг/кг и циметидин — 4 мг/кг внутривенно следует вводить медленно из-за риска гипотензии.
Применение кортикостероидов. Введение кортикостероидов (КС) не дает немедленного эффекта, однако полезно для предотвращения повторных реакций. Показания к применению КС при АШ четко не определены. В экспериментальных исследованиях было обнаружено, что они снижают метаболизм арахидоновой кислоты путем синтеза ядерных регуляторных протеинов, что способствует стабилизации фосфолипидов клеточных мембран. Кроме того, КС тормозят активацию и миграцию других воспалительных клеток (полиморфноядерных лейкоцитов). КС показаны для купирования рефрактерного бронхиолоспазма или артериальной гипотензии. Точная доза не установлена, рекомендуют 0,25—1 г гидрокортизона или 30 — 35 мг/кг метилпреднизолона. Есть мнение, что применение КС важно для профилактики поздней стадии анафилактической реакции, которая наблюдается через 12 — 24 ч после попадания антигена.
В некоторых случаях длительного тяжелого АШ может понадобиться инфузия раствора гидрогенкарбоната под контролем кислотно-основного баланса крови (при pH < 7,1). Доза зависит от величины pH.
438 ШОК
Ингибиторы фосфо диэстеразы. Аминофиллин — ингибитор фосфодиэстеразы, обладает бронхолитическим эффектом, увеличивает сократимость желудочков сердца и снижает сопротивление сосудов малого круга. Начальная доза — 5 —6 мг/кг в течение 20 мин, с последующей инфузией при продолжающемся бронхиолоспазме 0,5 —0,9 мг/кг в час. Вводится при стабильных показателях гемодинамики.
При рефрактерном бронхиолоспазме может быть полезной ингаляция $-адре-номиметиков.
На догоспитальном этапе пациенты с отягощенным аллергологическим анамнезом, у которых уже были анафилактические реакции, должны сами себе вводить адреналин. За рубежом для обучения таких больных существуют специальные курсы.
Для профилактики развития тяжелых аллергических реакций проводят специальные внутрикожные пробы. В исследованиях in vitro такие тесты, как провокационная проба с лейкоцитами и дегрануляция базофилов, часто являются ненадежными. Внутрикожные пробы с разведенными растворами потенциальных антигенов представляются более информативными. Растворы титруемых препаратов разводят в соотношении 1 : 1000 и 1 : 100, готовят контрольный раствор (например, изотонический раствор натрий хлорида). Затем каплю каждого раствора помещают в зону нанесенной скарификатором царапины (1 мм) в области предплечья. Больной до пробы не должен принимать лекарства, способные повлиять на иммунный ответ, например кортикостероиды или антигистаминные препараты. Результат считается положительным, если появившаяся папула превышает в диаметре 1 см и сохраняется более 30 мин. Подобные тесты выполняются только при наличии полного набора средств первой помощи [54].
Профилактика. Пациенты, у которых случались анафилактические реакции на пищевые продукты или фармакологические препараты, должны быть проинструктированы о необходимости избегать приема этих продуктов и лекарств. Для пре
дотвращения анафилактических реакций необходимо тщательно собирать аллергологический анамнез у пациентов и помнить о риске перекрестной анафилаксии к лекарственным препаратам. Так, пациенты, у которых развились анафилактоидные реакции на аспирин, почти универсально чувствительны к любому лекарственному средству, ингибирующему активность про-стагландинсинтетазы. Наличие р-лактамаз-ного кольца, содержащегося и в пенициллине и цефалоспоринах, способствует перекрестной иммунологической реактивности. Пациенты, у которых повышен риск анафилаксии, должны носить браслет Medic Alert, в котором содержатся сведения о лекарственных средствах, на которые у больного возникала аллергия.
Если у пациента наблюдались системные реакции на введение аллергена, его следует проинструктировать о необходимости постоянно иметь при себе карманный инжектор адреналина в случае последующей реакции. Пациентов также нужно предостеречь относительно потенциальных побочных эффектов введения адреналина, особенно если это люди пожилого возраста или больные с сопутствующими заболеваниями типа ишемической болезни сердца или гипертонии.
В случаях крайней необходимости применения средства, к которому у пациента повышенная чувствительность, иногда проводят десенсибилизацию. В настоящее время не разработаны протоколы «десенсибилизации» для пищевых продуктов и единственным средством профилактики является неупотребление таких продуктов с пищей. Что касается программы десенсибилизации при лекарственной аллергии, то имеются протоколы десенсибилизации, касающиеся пенициллина, аспирина и некоторых других препаратов (например, сульфонамидов).
При частых эпизодах идиопатической анафилаксии (более 4 — 5 раз в год) необходим прием блокаторов гистаминовых рецепторов, преднизолона, а также более тщательное лечение у аллерголога.
Инфузионная терапия 439
13.6. ОБСТРУКТИВНЫМ шок
Возникает вследствие внешнего сдавливания или внутренней обтурации крупного сосуда или сердца:
перегиб магистральных сосудов (напряженный пневмоторакс и др.);
массивные эмболии малого круга кровообращения;
сдавливание магистрального сосуда извне (опухоль, гематома, беременная матка);
тампонада сердца;
закупорка магистрального сосуда (тромбоз);
миксома предсердий.
Гемодинамические показатели и клинические проявления во многом зависят от уровня обструкции. В отличие от других видов шока основным терапевтическим мероприятием при обструктивном шоке является декомпрессия сосудов или сердца.
13.7. ИНФУЗИОННАЯ ТЕРАПИЯ
Одним из революционных событий в медицине можно считать первое успешное вливание «физиологического раствора поваренной соли», выполненное Lan-derer 10 июля 1881 г. С тех пор инфузионная терапия стала неотъемлемой частью не только интенсивной терапии, но и общего стационарного лечения.
Цели инфузионной терапии:
возмещение объема циркулирующей крови при кровопотере; восполнение инт-ра- и экстрацеллюлярного объема при дегидратации, вызванной потерей жидкости при различных патологических процессах (ожоги, нарушение функции пищеварительного канала, травматичные длительные хирургические вмешательства и др.);
улучшение реологических свойств крови и микроциркуляции;
восстановление электролитного, осмотического и кислотно-основного баланса;
компонент дезинтоксикационной терапии.
Основными задачами инфузионной терапии являются [55]: увеличение СИ >4,5 л/мин на 1 м2, £Ю2 > 600 мл О2/мин на 1 м2, VO2 > 160 мл О2 /мин на 1 м2, а также поддержание Ht на уровне 0,30 — 0,32 л/л.
При существующих методах мониторинга, в частности в Украине, трудно проконтролировать эти показатели. Основным, наиболее характерным показателем уровня волемии является ДЗЛА, однако катетеризация легочной артерии дает противоречивые показания, во-первых, вследствие
риска осложнений, а во-вторых, потому что ДЗЛА плохо коррелирует с конечно-диастолическим объемом левого желудочка [56]. В клинике основными критериями скорости инфузионной терапии являются показатели: ЦВД, АД, Hb, Ht, величина диуреза.
Для инфузионной терапии используют плазмозамещающие растворы. Идеальный препарат для замещения плазмы должен обладать такими свойствами [57]:
быстро возмещать потерю объема циркулирующей крови;
восстанавливать гемодинамическое равновесие;
нормализовывать микроциркуляцию;
сохраняться достаточно длительное время в кровеносных сосудах;
улучшать реологию циркулирующей крови;
улучшать доставку кислорода и других компонентов, а также тканевой обмен и функционирование органов;
легко метаболизироваться, не накапливаясь в тканях, легко выводиться и хорошо переноситься;
оказывать минимальное отрицательное воздействие на иммунную систему организма.
В 1861 г. Т. Graham классифицировал внутривенно вводимую жидкость по способности проникать через эндотелиальную мембрану на коллоидные растворы и кристаллоидные: последние свободно проникают через нее, а коллоидные растворы в норме не способны проникать через эндотелий [31 ].
440 ШОК
13.7.1. Кристаллоидные растворы
Основным компонентом кристаллоид-ных растворов является натрий хлорид. Натрий — основной внеклеточный катион — локализован преимущественно во внеклеточном пространстве. 75 —80 % внеклеточной жидкости приходится на интерстициальную. Экзогенно введенный натрий распределяется в такой же пропорции, т. е. аккумулируется в основном в межклеточном пространстве. Таким образом, ресусцитация кристаллоидами предполагает прежде всего увеличение объема внеклеточной жидкости [31].
Изотонический (0,9 %) раствор на* трий хлорида. Распределение изотонического раствора натрий хлорида (ИРНХ) во внутри- и внесосудистом секторах представлено на рис. 13.4.
При переливании 1л ИРНХ 275 мл остается в плазме, а 725 мл переходит в интерстициальное пространство. В действительности, инфузия 1л ИРНХ повышает общий объем внеклеточной жидкости на 1100 мл за счет выхода внутриклеточной, поскольку этот раствор является несколько гипертоническим по отношению к внеклеточной жидкости (ИРНХ содержит более высокую концентрацию натрия и хлора, чем плазма, его pH = 5,7). Существует потенциальный риск метаболическо-
Увеличение
Увеличение интерстициального объема плазмы объема 1000 500 0 500 1000
1 1-----’—I----------Г
D5W (1л) EZZ1
0,9% NaCl (1л) | |
5% Альбумин (1л) I ~ I
7,5% NaCl (250 мл) | |
J I I I
1000 500 0 500 1000
мл
Рис. 13.4. Влияние коллоидных и кристаллоидных растворов на объем внутрисосудистой интерстициальной жидкости
(по A. Imm, R. W. Carlson)
го гиперхлоремического ацидоза, однако на практике гиперхлоремия и ацидоз даже при переливании больших объемов ИРНХ развиваются редко [31]. Поскольку объем плазмы соотносится с объемом интерстициальной жидкости в соотношении 1:4, то для создания такого же объемного эффекта необходимо ввести объем крови, в четыре раза превышающий объем кровопотери [58].
Раствор Рингера был предложен британским исследователем С. Griffith в 1880 г. [59] и содержал кроме натрий хлорида ионы кальция и калия. Hartman в 1930 г. предложил в раствор Рингера добавить лактат с целью коррекции метаболического ацидоза. С тех пор раствор Рингера-лактата известен также под названием «раствор Гартмана». По сравнению с ИРНХ раствор Рингера-лактата содержит меньшее количество ионов натрия и хлора. Наличие лактата предполагает увеличение буферных свойств крови.
Однако нет доказательств преимущества раствора Рингера-лактата по сравнению с ИРНХ, кроме того, нет доказательств, что у первого раствора более выражены свойства буфера. К его недостаткам можно отнести то, что ионы кальция связывают некоторые лекарственные вещества, вследствие чего в донорской крови образуются тромбы, поэтому раствор Рингера-лактата не следует использовать как раствор при гемотрансфузиях [60].
Гипертонический раствор натрий хлорида (ГР). В последнее время получила распространение гипертоническая низкообъемная ресусцитация 7,5 % раствором натрий хлорида. 250 мл такого раствора имеют такой же волемический эффект, как 1 л 5 % раствора альбумина [31]. Первые сообщения об успешном применении ГР при гиповолемии появились в 1980 г.
К преимуществам инфузии ГР относятся:
1) меньший объем;
2) пролонгированный волемический эффект;
3) снижение постнагрузки на левый желудочек;
Инфузионная терапия 441
4) увеличение сердечного выброса;
5) снижение отека тканей;
6) увеличение диуреза.
Механизм этих эффектов не совсем ясен.
Неблагоприятным эффектом является клеточная дегидратация.
Более стойкий волемический эффект оказывает смесь ГР с декстраном или ГЭК в соотношении 1:1. Рекомендуемая доза — 4 —5 мл/кг. В настоящее время эта инфузионная среда дает наилучший гемодинамический эффект.
Растворы глюкозы. Применение растворов глюкозы у критических больных резко ограничено вследствие таких причин:
в результате осмотического эффекта может развиться клеточная дегидратация [31];
повышает продукцию СО2 [61];
повышает продукцию лактата [62];
увеличивает ишемическое повреждение головного мозга [63] и других тканей.
Продукция лактата при инфузии растворов глюкозы возрастает на 5 % у здоровых и на 85 % у критических больных. Это происходит даже при переливании 5 % раствора глюкозы (рис. 13.5).
инфузия
Рис. 13.5. Влияние интраоперационной внутривенной инфузии раствора Рингера и 5 % раствора глюкозы на уровень лактата [62]
При нестабильности кровообращения глюкоза из энергетического субстрата трансформируется в источник токсинов. A. Arieff [64] описал тяжелейшие неврологические расстройства, вызванные гипонатриемией, после плановых операций у 14 здоровых женщин, которым во время общей анестезии проводилась массивная инфузия растворов глюкозы. В настоящее время от рутинного применения глюкозы с целью восполнения ОЦК во время операции и у критических больных полностью отказались [31]. Однако при гипогликемии, гипернатриемии, гипертонической дегидратации показано использование растворов глюкозы.
13.7.2. Коллоидные растворы
Крупные молекулы коллоидов в норме не проникают через эндотелий в интерстиций, поэтому они более эффективно восстанавливают объем плазмы, чем кристаллоиды, повышают коллоидно-осмотическое (онкотическое) давление (КОД) плазмы. Чем выше КОД переливаемого раствора, тем больше увеличивается объем плазмы после его переливания.
Альбумин является транспортным белком, обеспечивающим на 75 % КОД плазмы. Промышленность выпускает 5, 10, 20 и 25 % растворы этого белка. При переливании 5 % раствора, имеющего КОД примерно такое же, как у плазмы, до 50 % его остается в сосудистом русле. Онкотический эффект альбумина длится 12 —18 ч.
25 % раствор альбумина, имеющий КОД = = 70 мм рт. ст., увеличивает объем плазмы в 4 —5 раз. Так, при инфузии 100 мл этого раствора объем плазмы возрастает на 400 — 500 мл за счет поступления в сосудистое русло межклеточной жидкости. Для экстренной ресусцитации гиповолемии раствор альбумина следует применять только в комплексе с другими препаратами, в противном случае это может вызвать гиперосмолярность плазмы и тяжелую дегидратацию клеток [31].
При переливании альбумина отсутствует риск переноса инфекции; редко встречаются аллергические реакции; могут раз
442 ШОК
виться коагулопатии, но они не приводят к тяжелой кровоточивости [65]. Однако есть данные о неблагоприятном эффекте альбумина при септическом шоке, опасно применять его при анафилактическом шоке.
Растворы гидроксиэтилированного крахмала (ГЭК). ГЭК — синтетический коллоидный раствор. Содержит молекулы амилопектина массой от нескольких сотен до миллиона дальтон. Форма выпуска: 6 % раствор в изотоническом растворе натрий хлорида (рефортан). Коллоидный эффект этого раствора незначительно превышает таковой 5 % раствора альбумина [65].
Растворы ГЭК обладают более выраженным волемическим эффектом, чем кристаллоидные растворы, часто повышают уровень амилазы в крови в 2 — 3 раза по сравнению с нормой в течение первых нескольких дней после инфузии. На 5 — 7-й день содержание амилазы понижается до нормы [66]. Выводятся через почки.
Анафилактические реакции на ГЭК встречаются крайне редко (0,0004 %) [66].
Результаты лабораторных гемостатических тестов показывают некоторое увеличение частичного протромбинового времени [61], однако этот эффект не сопровождается увеличением кровоточивости.
Есть данные [66], что в отличие от других коллоидов растворы ГЭКа уменьшают степень повреждения капилляров у больных с тяжелой травмой, улучшают функцию легких и уменьшают выраженность системной воспалительной реакции.
J. Boldt с соавт. [66] отмечает преимущество применения ГЭК по сравнению с альбумином при сепсисе, поскольку ГЭК оказывает благоприятный эффект на эндотелиальнозависимую коагуляцию, хотя при травме эта разница не отмечалась. По сравнению с растворами желатины и декстранами инфузия ГЭК способствует более высокому росту СИ, £Ю2, улучшению кислородного баланса тканей [68]. Растворы ГЭКа не увеличивают, в отличие от альбумина, риск кровотечения, если не превышена терапевтическая доза (10—15 мл/кг) [69]. При сепсисе способствуют нормализации эндокринного статуса [70].
Растворы декстранов. Декстраны являются растворами полимеров глюкозы, продуцируемых бактериями Leuconostoc, которые инкубируют в сахарозе. Предложены к применению в 40-х годах прошлого века. Наиболее распространенными являются 10 % декстран-40 и 6 % декстран-70 в растворе глюкозы или изотоническом растворе натрий хлорида. Оба являются гиперонкотическими по отношению к плазме крови (КОД = 40 мм рт. ст.). Декстран-40 (реополиглюкин) обладает большим волемическим эффектом, но при этом менее продолжительным [65].
Декстраны дают дозозависимый коагу-лопатический эффект, так как ингибируют агрегацию тромбоцитов, снижают активность фактора VIII и активируют фибринолиз. Поэтому максимальная суточная доза не должна превышать 20 мл/кг [66]. Ежедневное введение относительно больших объемов декстрана (500 — 1000 мл/сут) создает угрозу повышения вязкости плазмы крови и агрегации эритроцитов [71].
Анафилактические реакции встречаются чаще, чем при применении ГЭК (примерно в 5 % случаев), однако в последнее десятилетие частота их снизилась до 0,032 % за счет совершенствования технологии приготовления.
Применение декстранов затрудняет определение группы крови пациента.
Предположительно могут служить причиной развития ОПН вследствие гиперонкотического эффекта и снижения клубочкового фильтрационного давления. Однако этот механизм не доказан, и в клинике это осложнение встречается крайне редко [31].
Растворы декстранов со средней молекулярной массой 70 000 D нормализуют главным образом макроциркуляцию, а массой 40 000 D — также и микроциркуляцию [72].
Растворы желатины. Раствор на основе желатины был первым искусственным коллоидом. Был использован J. J. Hogan в 1915 г. В последние годы разработаны новые препараты из желатины (гелофузин, модежель). Желатину производят из коллагеновой ткани крупного рогатого скота методом термической деградации, гидролиза и сукцинирования. У гелофузина пе
Инфузионная терапия 443
риод выведения составляет 9 ч, средняя молекулярная масса — 30 000 D, КОД — 33,3 мм рт. ст., осмолярность — 274 мос-моль/л. Гемодинамические эффекты сравнимы с растворами ГЭК. Даже в больших объемах не оказывает влияния на гемостаз (J. D. Edwards, R. G. Wilkins, 1987).
Анафилактоидные реакции, связанные с переливанием гелофузина, встречаются в пределах от 1 : 6000 до 1 : 13 000 случаев, в основном у предрасположенных к аллергии лиц (J. Ring, К. Messmer, 1977). Этому препарату отдают предпочтение при недостаточности функции почек (А. А. Еременко, 2001).
Искусственные коллоидные растворы представляют собой полидисперсную смесь молекул различной молекулярной массы, от величины которой зависит скорость выведения декстрана и желатины из организма. Для растворов ГЭК существует порог 60 000 — 80 000 D, определяющий предел почечной проницаемости [72].
Одним из недостатков коллоидов является их высокая цена, особенно альбумина. При переливании 250 мл коллоидов и эквивалентного им 1 л кристаллоидов цена первых в три раза выше при использовании ГЭК и в шесть раз выше — альбумина [31].
Сорбилакт и реосорбилакт. Отечественные препараты сорбилакт и реосорбилакт — комплексные инфузионные растворы, основными активными веществами которых являются сорбитол и натрий лактат. Обладают плазморасширяющим эффектом, энергетической ценностью, осмоди-уретическим действием, коррегируют метаболический ацидоз, стимулируют перистальтику кишок. Для подтверждения противошоковой эффективности этих растворов необходимы дополнительные исследования. Есть данные о благоприятном эффекте их применения в клинике неотложной нейрохирургии [73]. К преимуществам сор-билакта и реосорбилакта можно отнести их сравнительно небольшую стоимость.
13.7.3. Выбор раствора для инфузии
Дискуссия о выборе инфузионной среды для восполнения ОЦК ведется уже многие годы. До сих пор нет четких дан
ных о приоритете выбора коллоидных или кристаллоидных растворов. Дефицит интерстициальной жидкости является доминирующим при кровопотере, не превышающей 15 % ОЦК, поэтому в таких случаях достаточно введения кристаллоидов [31]. При кровопотере свыше 15 % ОЦК, приоритетным является восполнение внутрисосудистого объема. Растворы коллоидов эффективнее восстанавливают объем циркулирующей плазмы. Для достижения такого же эффекта кристаллоидами необходимо введение в 3 — 4 раза большего объема. По данным D. McIlroy с соавт. [74], острое увеличение внутрисосудистого объема быстрой инфузией 1 л раствора Рингер-лактата незначительно превышало половину объема, который достигался переливанием 1 л 6 % раствора гидрокси-этилкрахмала. Z. Friedman [75] указывает на то, что жидкостная ресусцитация до достижения среднего АД = 60 мм рт. ст. во время неконтролируемого кровотечения приводила к более высокой доставке кислорода и меньшему уровню системной концентрации лактата при использовании 6 % раствора гидроксиэтилкрахмала по сравнению с ресусцитацией раствором Рингер-лактата до достижения среднего АД = 60 или 80 мм рт. ст. Несмотря на все преимущества, ресусцитация коллоидами у больных с гиповолемическим шоком не сопровождается увеличением выживаемости [65, 76].
Преимущества и недостатки применения коллоидных и кристаллоидных растворов представлены в табл. 13.2.
Вопреки предположительно повышенному риску развития отека тканей, прежде всего легких, при ресусцитации кристаллоидами, на практике риск этого осложнения такой же, как и при переливании коллоидов, при условии отсутствия чрезмерного повышения гидростатического давления в легочных капиллярах [77].
Особые проблемы возникли с применением растворов желатины, так как они могут служить переносчиками губчатой энцефалопатии крупного рогатого скота, и поэтому сейчас в некоторых странах (например, США) введен жесткий контроль за их применением.
444 ШОК
Таблица 13.2. Преимущества и недостатки внутривенного введения коллоидов и кристаллоидов (по D. S. Prough, М. Matru, 1997)
Раствор Преимущества Недостатки
Коллоиды Меньший объем Более длительный волемический эффект Меньше периферические отеки Большая стоимость Коагулопатии (декстран > ГЭК) Больше отек легких (при нарушенной проницаемости капилляров) Снижение клубочковой фильтрации Осмотический диурез
Кристаллоиды Меньшая стоимость Более высокий почечный кровоток Восполнение интерстициального пространства Транзиторный волемический эффект Периферические отеки ( гипоальбу минемия ) Отек легких (гипоальбуминемия + высокое ДЗЛА)
У больных с повышенной проницаемостью эндотелиальной мембраны (сепсис, ОРДС, СПОН и др.) использование коллоидных растворов (в частности, альбумина и декстрана) может оказать неблагоприятный эффект, поскольку альбумин или другие коллоиды могут проникать через поврежденную мембрану в интерстициальное пространство, тем самым способствуя повышению КОД и возникновению отеков [72].
D. Drobin, R. G. Hahn [78], исследуя кинетику различных растворов в сосудистом русле, обнаружили, что наиболее эффективно повышает объем циркулирующей плазмы сочетанное применение 7,5 % раствора натрий хлорида с 6 % раствором декстрана. По-видимому, именно сочетание гипертонического раствора натрия хлорида с коллоидным следует считать близким к оптимальному варианту при восполнении острой, угрожающей жизни гиповолемии.
В 1998 г. G. Schierhout и I. Roberts [79], проанализировав 37 исследований, касающихся преимуществ и недостатков при
менения коллоидов и кристаллоидов у критических больных, пришли к выводу, что пока нет достоверных доказательств приоритета одного метода ресусцитации над другим. W. Hasibeder [80] отмечает, что в настоящее время методологические ограничения исследований не позволяют с позиций доказательной медицины определить, какой из методов является наилуч-шим.
На наш взгляд, до получения новой достоверной информации нет необходимости противопоставлять коллоидные и кристаллоидные растворы. При сравнительно небольшой кровопотере (до 15 % ОЦК) в большинстве случаев можно обойтись без применения коллоидов, учитывая их большую стоимость. При большой кровопотере жидкостная ресусцитация должна включать обе эти среды. При критической кровопотере необходимым элементом инфузионной терапии является гипертонический раствор натрий хлорида в сочетании с раствором коллоида. Из коллоидных растворов приоритет, исходя из имеющихся данных, принадлежит растворам ГЭК и желатины.
13.8. ТРАНСФУЗИОННАЯ ТЕРАПИЯ
Гемотрансфузии. В последнее десятилетие возникла необходимость ограничения использования препаратов донорской крови. Связано это с тем, что обнаружено большое количество неблагоприятных факторов трансфузии аллогенной крови.
Практически в 20 % случаев гемотрансфузий выявляются неблагоприятные побочные эффекты [81]. И это, по-видимому, только верхушка айсберга. Рассмотрим основные осложнения, связанные с гемотрансфузиями .
Трансфузионная терапия 445
Передача инфекции с трансфузирован-ной кровью', в 10 % случаев инфекцию вызывают вирусы (цитомегаловирус, гепатит, ВИЧ), а также бактерии и простейшие (малярия, сифилис, бруцеллез, токсоплазмоз). При переливании одной единицы крови риск заразиться ВИЧ-инфекцией в США составляет от 1 : 200 000 до 1 : 2 000 000, а вирусом гепатита С — от 1 : 30 000 до 1 : 150 000 [82] (по Украине мы таких данных не нашли).
Немедленные гемолитические реакции. Основная причина — переливание крови или ее препаратов, не совместимых по системе АВО. Являются хотя и редким, но опасным для жизни осложнением [83]. Переливание иногруппной крови вызывает внутрисосудистый гемолиз эритроцитов, коагулопатию, шок, ОПН, ОРДС. Первые признаки могут появиться уже после переливания 10 мл крови. Проявляются одышкой, повышением температуры тела (иногда с ознобом), болями в пояснице, костях, выраженной бледностью кожных покровов. Эти симптомы бывают резко сглажены или не проявляются вовсе, если больной под наркозом или находится в бессознательном критическом состоянии. Единственным признаком трансфузии несовместимой крови может быть внезапно развившаяся необъяснимая артериальная гипотензия. Более поздним признаком служит появление гемолитической мочи. ОПН возникает в 5 —10 % случаев и зависит от количества перелитой крови [84].
После диагностики этого осложнения следует немедленно прекратить трансфузию, стабилизировать АД (инфузионная терапия, при необходимости — вазопрессорная (дофамин — 5 мкг/кг в минуту)), провести внутривенную инфузию раствора натрий гидрогенкарбоната (соды) для ощелачивания мочи и профилактики ОПН, с этой же целью используются осмодиуретики, эуфиллин. В случае развития ОПН показан искусственный гемодиализ.
Лихорадочные негемолитические реакции — наиболее частая причина повышения температуры тела при гемотрансфузиях. Возникают в 1 % случаев гемотрансфузий примерно через один час после начала процедуры вследствие наличия анти-
тител у реципиента к поверхностным лейкоцитарным и тромбоцитарным антигенам донора. Антитела вырабатываются у реципиента в ответ на предыдущие трансфузии или беременность в анамнезе. Возможной причиной таких реакций может быть бактериальная контаминация донорской крови, хотя подобные случаи наблюдаются редко [85].
Аллергические реакции. Проявляются различными симптомами (от уртикарной сыпи до анафилактического шока). Возникают у реципиентов с наличием антител IgG к IgA донора. Чаще всего развиваются у больных с наличием дефицита IgA [86]. Лечение такое же, как и при анафилактическом шоке другой этиологии. В дальнейшем показания к гемотрансфузиям у подобных больных должны быть максимально ограничены.
Гиперкалиемия, ацидоз. Внеклеточная концентрация калия в консервированной крови 3-недельного хранения может повышаться до 20 ммоль/л. Однако в клинике при отсутствии ОПН выраженная гиперкалиемия вследствие переливания крови возникает редко. Гемотрансфузии также способствуют развитию метаболического ацидоза, однако необходимости в его коррекции, если он вызван только этим фактором, не возникает.
Гипокальциемия, вызванная переливанием цитрата, может возникнуть при массивных гемотрансфузиях. Специальной терапии препаратами кальция, как правило, не требуется [87].
Пожалуй наиболее существенным осложнением гемотрансфузий у больных в критическом состоянии является выраженный иммуносупрессорный эффект [88, 89]. Еще в конце 70-х годов XX ст. был обнаружен повышенный риск рецидива злокачественных новообразований и послеоперационных инфекционных осложнений, связанный с гемотрансфузией [90]. Е. Fransen с соавт. [91] отмечает увеличение концентрации медиаторов воспаления и количества послеоперационных осложнений после интраоперационных гемотрансфузий. R. W. Taylor с соавт. (2002) доказал, что переливание эритромассы статистически достоверно увеличивает риск
446 ШОК
госпитальной инфекции, летальности и продолжительности нахождения в отделении интенсивной терапии. F. Moore с соавт. [92], проведя проспективное исследование больных с травмой, обнаружил дозозависимый эффект гемотрансфузий на развитие синдрома полиорганной недостаточности.
Гемотрансфузии обладают повреждающим эффектом на сердце и легкие, способствуя развитию конгестивной сердечной недостаточности, отеку легких, ОРДС [93].
Кроме указанных недостатков консервированная кровь обладает низкой кисло-родтранспортной функцией вследствие плохой деформируемости эритроцитов, снижения уровня 2,3-дифосфоглицерата в них, наличия микросгустков, и поэтому может уменьшать оксигенацию тканей [94]. Эти отрицательные качества консервированной крови особенно возрастают после 15-суточного хранения [95].
Проблема показаний к переливанию крови — это проблема анемической гипоксии и адаптационных по отношению к ней возможностей организма.
Основным показанием к гемотрансфузиям являлось снижение концентрации НЬ < 100 г/л и Ht < 0,30 л/л [96]. Однако такие рекомендации основывались на теоретическом расчете максимального показателя транспорта кислорода и не были подтверждены контрольными рандомизированными исследованиями. R. Rawstron [97] сравнил перед операцией две группы пациентов с НЬ больше и меньше 100 г/л и не обнаружил разницы в количестве послеоперационных осложнений. Были опубликованы клинические наблюдения, согласно которым концентрация НЬ = 40 г/л при остром кровотечении не повышала риск неблагоприятного исхода по сравнению с более высоким уровнем НЬ [98]. Клинические наблюдения над больными с острой анемией, по религиозным соображениям не позволявшими переливать себе донорскую кровь, определили уровень НЬ = 30 г/л как критический при остром кровотечении, достоверно увеличивающий летальность [99]. Однако в настоящее время преобладает мнение, что уровни НЬ и Ht не могут рассматриваться как един
ственный критерий для определения показаний к гемотрансфузиям [100].
Знание патофизиологических процессов при анемии, определение клинических факторов, ограничивающих способность организма поддерживать адекватное потребление кислорода в ситуации, когда снижена кислородная емкость крови, позволяют клиницисту принять оптимальное решение о показаниях к гемотрансфузии у конкретного пациента.
Патофизиологические аспекты острой анемической гипоксии были рассмотрены в соответствующем разделе, однако на некоторых вопросах следует остановиться подробнее. Поддержание адекватной оксигенации тканей в условиях острой анемии зависит от двух механизмов: увеличения сердечного выброса (СВ) и экстракции кислорода из крови тканями.
Повышение СВ во многом обусловлено снижением вязкости крови [101]. Например, при Ht = 0,2 л/л абсолютная вязкость равна 1,5, при Ht = 0,3; 0,4; 0,5 л/л соответственно 1,8; 2,3; 2,9 [102]. При анемии повышается ударный объем, снижается общее периферическое сосудистое сопротивление (ОПСС) и постнагрузка при увеличении венозного возврата [103]. Кроме снижения вязкости снижение ОПСС связано с уменьшением способности инактивировать NO [104].
Увеличение сократительной способности миокарда при нормоволемической гемодилюции продемонстрировал О. НаЫег с соавт. [105]. Роль эндогенных катехоламинов в этой компенсаторной реакции остается противоречивой. Следует отметить, что снижение концентрации НЬ ниже 45 г/л не сопровождается повышением СВ [106].
Выяснилось, что при нормоволемической анемии снижение доставки кислорода сопровождается увеличением экстракции кислорода тканями, поэтому УО2 не снижается до достижения уровня гемоглобина 30 г/л, а гематокрита — 0,10 л/л [31].
На системном уровне увеличение экстракции кислорода достигается перераспределением крови, прежде всего в пользу головного мозга и миокарда. Некоторые
Современная стратегия гемотрансфузий 447
исследования обнаружили расширение сосудов мозга и миокарда и непропорционально высокое увеличение перфузии этих органов по сравнению с увеличением СВ при нормоволемической анемии [107, 108]. Причем наибольшая венечная вазодилатация отмечена при уровне гематокрита 12,5 %. Увеличение перфузии мозга и миокарда происходит за счет снижения перфузии других органов, в печени, почках, кишках, селезенке наблюдается вазоконстрикция.
Улучшение экстракции кислорода при острой нормоволемической гемодилюции происходит также на уровне микроциркуляции: увеличивается скорость движения эритроцитов в капиллярах, улучшается диффузия кислорода, увеличивается соотношение микроциркуляторного и системного гематокрита [109]. Кроме того, при Ht < 0,15 л/л кривая диссоциации оксигемоглобина смещается вправо за счет увеличения концентрации 2,3-дифосфо-глицерата, более наглядно этот механизм проявляется при хронической анемии. Имеется еще один терапевтический метод повышения транспорта кислорода при анемии за счет увеличения растворенной в плазме фракции — это увеличение РЮ2. При гемодилюции с Ht = 0,10 л/л DO2 возрастает при дыхании чистым кислородом намного больше, чем при Ht = 0,33 л/л [110]. Появилось много клинических исследований о хорошей переносимости нормоволемической анемии, например больными после резекции легких по поводу злокачественных новообразований [111]. У больных с НЬ = 85 — 100 г/л реже отмечались- послеоперационные осложнения (ателектазы, пневмония, ОРДС), уменьшалось время нахождения в ОИТ и летальность по сравнению с пациентами, у которых уровень НЬ был выше 100 г/л.
Однако при снижении контрактильной способности миокарда и гиповолемии,
а также при наличии другой патологии может резко снижаться переносимость организмом анемии.
Факторы, снижающие переносимость организмом острой анемической гипоксии [89]:
1. Снижение сердечного выброса: гиповолемия;
ИБС;
заболевания сосудов;
заболевания сердца;
применение препаратов с отрицательным инотропным эффектом;
седативные / анестетики.
2. Снижение экстракции кислорода тканями:
ОРДС;
сепсис/ССВО, тяжелая травма; синдром ишемия/реперфузия; применение а-адреноблокаторов; седативные / анестетики;
гипотермия.
3. Повышение потребности тканей в кислороде:
лихорадка, боль, стресс, возбуждение;
сепсис/ССВО;
гипервентиляция.
4. Нарушение газообмена в легких: ОРДС;
ХНЗЛ.
Исходя из вышерассмотренного, больные с патологией сердца и сосудов хуже переносят анемию. A. Nelson с соавт. [112] обнаружил, что при операциях аортокоронарного шунтирования снижение гематокрита ниже 0,28 усиливает ишемию миокарда у больных с ИБС. Особо следует обратить внимание анестезиологов на тот факт, что применение седативных средств и анестетиков также снижает переносимость анемической гипоксии. Этими фактами необходимо руководствоваться при выборе показаний к гемотрансфузиям.
13.9. СОВРЕМЕННАЯ СТРАТЕГИЯ ГЕМОТРАНСФУЗИИ
В 1988 г. на согласительной конференции в Национальном институте здоровья (США) было рекомендовано считать критическим уровнем НЬ у исходно здоровых
людей 70 г/л, а в случаях, когда этот уровень превышает 100 г/л, гемотрансфузии практически не применять при условии нормализации волемии [ИЗ]. В 1992 г.
448 ШОК
была проведена согласительная конференция Американской коллегии врачей, на которой было не рекомендовано во время общей анестезии и операции переливать кровь у пациентов без сопутствующих заболеваний со стороны сердца и сосудов при стабильных показателях гемодинамики, несмотря на уровень НЬ [114].
R. Herbert с соавт. [115] в многоцентровом рандомизированном исследовании сравнил две разные концепции гемотрансфузий'. ограничительную, предлагающую поддерживать НЬ на уровне 70 — 90 г/л, и либеральную, согласно которой НЬ следует поддерживать на уровне 100—120 г/л. Пациенты, при лечении которых руководствовались либеральной концепцией, получили гораздо больше консервированной крови. Госпитальная летальность была ниже в группе с ограничительной тактикой гемотрансфузий. Особенно большая разница в результатах получена у больных с APACHE < 20 и в возрасте менее 55 лет.
. Однако у пожилых пациентов, перенесших инфаркт миокарда,Wen-Chin Wu с соавт. [115] обнаружил, что оптимальным уровнем гематокрита является 0,30 — 0,33 л/л.
Таким образом, у больных при отсутствии активного кровотечения и тяжелых сопутствующих заболеваний со стороны сердечно-сосудистой и дыхательной систем нет оснований для гемотрансфузий при уровне НЬ > 70 — 90 г/л. У пациентов с ИБС, недостаточностью миокарда, мозгового кровообращения уровень НЬ следует поддерживать на уровне 90—100 г/л при условии восполнения объема циркулирующей крови. Как указывалось выше, уровни НЬ и Ht не должны быть единственными факторами при определении показаний к гемотрансфузиям. Нестабильность АД, тахикардия, пожилой возраст, а также тяжелые сопутствующие заболевания расширяют эти показания. Например, у больного со стабильным АД и нормальным ритмом сердца концентрация НЬ = 70 г/л не будет основанием для гемотрансфузий, а при сниженном АД и тахикардии, возможно, стоит принять решение в пользу трансфузии донорской крови, так как, вероятнее всего, это связано с гиповолемией,
а дальнейшее переливание жидкости усугубит анемию. Показания зависят также от фактора наличия крови разных сроков хранения: переливание эритроцитной массы со сроками хранения, превышающими три недели, может допускаться только в экстремальных ситуациях. При массивных профузных кровотечениях, когда нет уверенности в их быстрой хирургической остановке, показания к гемотрансфузиям максимально расширяются.
Р. Marino [31] предлагает такие показания к гемотрансфузиям: ♦ очевидное снижение оксигенации тканей
(снижение VO2 ниже 100 мл/мин на 1 м2 или гиперлактатемия);
♦ O2ER >0,5 при адекватном СВ;
♦ снижение НЬ ниже 70 г/л у больных с нарушениями венечного или церебрального кровотока и с серьезным нарушением функции миокарда.
Таким образом, для определения оптимальных показаний к гемотрансфузии необходим качественный гемодинамический мониторинг, прежде всего определение показателя потребления тканями кислорода или определение уровня лактата в смешанной венозной крови.
Более широкое применение на практике ограничительной стратегии показаний к гемотрансфузиям позволит значительно уменьшить количество осложнений, обусловленных применением этого метода, улучшить результаты лечения и даст экономический эффект.
Свежезамороженная плазма. Одним из наиболее часто употребляемых препаратов крови является свежезамороженная плазма (СЗП), содержащая фибриноген, факторы свертывания крови II, V, VII, VIII, IX, X, XI, XIII и фактор Виллебранда [87]. Основными показаниями для переливания СЗП являются: дилюционная коагулопатия при массивных кровопотере и восполнении; гипокоагуляция вследствие передозировки непрямых антикоагулянтов; врожденные и приобретенные коагулопатии; болезнь Виллебранда [87]. В последнее время обнаружено положительное влияние СЗП на интеракцию между лейкоцитами и клетками эндотелия при системной воспалительной реакции, т. е. про
Инотропные и сосудосуживающие фармакологические препараты 449
текторный эффект СЗП на эндотелий при системном воспалении, сопровождающем сепсис, политравму и другие критические состояния [116].
Возможны аллергические реакции на введение СЗП.
Криопреципитат. Содержит большое количество фибриногена, фактора VIII, фактора Виллебранда. Показания: гипофибриногенемия; гемофилия А; болезнь Виллебранда [87].
13.10. ИНОТРОПНЫЕ И СОСУДОСУЖИВАЮЩИЕ ФАРМАКОЛОГИЧЕСКИЕ ПРЕПАРАТЫ
При лечении шока и других расстройств гемодинамики часто бывает необходимо применить препараты, влияющие на тонус сосудов и сократительную способность миокарда. В настоящее время с этой целью используют дофамин, добутамин, адреналин, фенилэфрин (мезатон), норадреналин, амринон, допексамин. Показанием к применению вазоактивных препаратов чаще всего являются артериальная гипотония (92 %) и олигурия (50 %) [117]. Наиболее часто используют допамин и норадреналин, несколько меньше — добутамин. Частота применения других препаратов этой группы не превышает 6 %. Не-катехоламины в настоящее время редко используются даже у пациентов с недостаточностью сердца.
Поскольку большинство применяемых вазоконстрикторов и кардиотонических препаратов являются агонистами адренорецепторов, в табл. 13.3 приведены локализация и фармакологический эффект активации рецепторов автономной нервной системы.
Дофамин (допамин). Эндогенный катехоламин, предшественник норадреналина, является нейротрансмиттером. Как экзогенный агент вызывает дозозависимую активацию некоторых адренергических и дофаминергических рецепторов [118]. В малых дозах (0,5 — 3 мкг/кг в минуту) допамин активирует дофаминспецифические рецепторы в почечных, брыжеечных и мозговых сосудах; в средних (3—7,5 мкг/кг в минуту) — стимулирует р-адренерги-ческие рецепторы сердца и периферических сосудов, увеличивает сердечный выброс. Инотропный эффект дофамина уступает добутамину [31]. В больших дозах (> 7,5 мкг/кг в минуту) оказывает дозо
зависимый эффект на а-адренергические рецепторы сосудов большого и малого круга кровообращения. Вазоконстрикторный эффект, снижение почечного кровотока, увеличение постнагрузки на сердце и потребление миокардом кислорода ограничивают его применение при сердечной недостаточности и шоке. В средних дозах назначают для лечения шока разной этиологии. Часто применяют в сочетании с вазодилататорами (нитраты). При использовании в высоких дозах допамин теряет преимущества перед адреналином и приводит к выраженной вазоконстрикции, ишемии органов пищеварительного канала и даже к бактериальной транслокации [119].
Из побочных эффектов наиболее часто наблюдаются тахиаритмия, мультифокаль-
Таблица 13.3. Рецепторы автономной нервной системы [117]
Рецептор Локализация Действие
«1 «2 Pl Р2 D1 d2 Миокард Сосуды ЦНС Пресинап-тическая мембрана Миокард Бронхи Периферические сосуды Сосуды почек, спланхникуса Пресинап-тическая мембрана Увеличивает сократимость Вазоконстрикция Вазодилатация, снижает сократимость Уменьшает ЧСС Увеличивает сократимость Увеличивает ЧСС Бронходилатация Вазодилатация Вазодилатация Вазодилатация (ингибиция НА)
4-220
15
450 ШОК
ные эктопические желудочковые экстрасистолы, желудочковая тахикардия. При инфузии в паравенозную клетчатку может вызвать некроз.
Среди исследователей нет однозначного мнения о применении малых доз дофамина, в частности при олигурии. Мы наблюдали стимулирующий эффект низких доз дофамина (2 — 3 мкг/кг в мин) у нескольких пациентов с олигурией, которая плохо поддавалась терапии фуросемидом. Однако последние обзоры по этому вопросу [120] свидетельствуют, что дофамин в низкой дозе не обладает протекторным эффектом на почки. С учетом потенциальных побочных эффектов дофамина этот препарат не следует использовать для лечения и профилактики острой недостаточности почек.
Добутамин (добутрекс) — синтетический катехоламин, часто это инотропный препарат выбора при острой систолической недостаточности сердца [121]. Является преимущественно агонистом рг, в меньшей степени — р2-адренергических рецепторов (бронхо- и вазодилатирующий эффекты). Вызывает дозозависимое увеличение сократительной способности миокарда со снижением системного периферического сосудистого сопротивления, поэтому АД может не меняться. Эффективен при недостаточности желудочков сердца [122].
Инотропный и хронотропный эффект добутамина у больных в критическом состоянии проявляется в разной степени. Частично это зависит от особенностей фармакокинетики, частично — от индивидуальных особенностей организма. Как правило, менее чувствительны к нему пациенты пожилого возраста, но возможна относительная резистентность и у более молодых [123].
Поскольку добутамин не обладает вазопрессорным эффектом, он не показан в качестве монотерапии кардиогенного шока. Основными показаниями к его применению являются септический шок и поли-органная недостаточность с нормальным сердечным выбросом. Повышает метаболизм миокарда и его потребность в кислороде [124]. Часто используется в сочета
нии с вазопрессорами (норадреналин), например при септическом шоке [125]. Начальная доза 5 —15 мкг/кг в минуту, но в некоторых случаях допускается введение до 200 мкг/кг в минуту [126]. Среди побочных эффектов наиболее часто отмечается тахикардия (5—10 %), однако случаи тяжелой, плохо поддающейся терапии тахиаритмии не характерны [127]. У больных с гиповолемией применение добутамина может вызвать артериальную гипотензию [128].
Не показано применение добутамина при диастолических расстройствах функции миокарда и при гипертрофической кардиомиопатии.
Адреналин (эпинефрин). Эндогенный катехоламин, секретируемый мозговым слоем надпочечников. Так же, как дофамин, в очень низких дозах (0,04—0,1 мкг/кг в минуту) преимущественно оказывает p-адреномиметический эффект, но в дозе, превышающей 0,1 мкг/кг в минуту, приводит к выраженному вазоспазму, повышая АД и давление в легочной артерии [129]. В дозе 0,3 мкг/кг в минуту значительно снижает перфузию почек и органов пищеварительного канала. Дозы адреналина, применяемые при сердечно-легочной реанимации и анафилактическом шоке, приведены в соответствующих разделах.
Кроме симпатомиметического действия адреналин обладает выраженным противовоспалительным эффектом, блокируя высвобождение медиаторов воспаления из мастоцитов и базофилов при анафилактической реакции, поэтому рассматривается как первоочередное средство лечения анафилактического шока, но не шока другой этиологии [130].
Адреналин является важнейшим гормоном адаптации у здоровых людей, но может быть крайне опасным у больных в критическом состоянии [127]. Обладает выраженным влиянием на метаболизм, вызывая: гиперметаболизм (выраженный в сердечной мышце), гипергликемию (активируя гликонеогенез и ингибируя освобождение инсулина), увеличивает концентрацию в крови кетокислот (усиливая липолиз), гиперлак-татемию (не связанную с ишемией), снижает концентрацию калия в плазме крови.
Инотропные и сосудосуживающие фармакологические препараты 451
Обладает выраженными побочными эффектами: аритмогенным (особенно на фоне нарушений электролитного баланса и применения галотана); даже в малых дозах вызывает ишемию миокарда, выраженный спазм сосудов почек; способствует развитию тяжелой артериальной гипертензии на фоне применения р-адренобло-каторов [127].
Инфузия адреналина обладает существенными недостатками. Так, по мнению М. М. Jonas и С. В. Wolf [131], адреналин перераспределяет сердечный выброс в пользу поперечнополосатых мышц, уменьшая кровоток в спланхнической зоне и почках.
Учитывая сказанное выше, адреналин следует применять при неэффективности других препаратов этой группы.
Норадреналин (норэпинефрин). Эндогенный катехоламин, предшественник адреналина в процессе биосинтеза. Медиатор симпатической нервной системы. В дозе, превышающей 4 мкг/мин, обладает выраженным cq-адреномиметическим эффектом, вызывая выраженный вазоспазм. Поэтому предпочтительнее примененять его при шоке со сниженным сосудистым сопротивлением (вариант септического шока, неврогенный шок), а также при отравлении трициклическими антидепрессантами и кокаином [132].
Обладает отрицательным хронотропным эффектом вследствие стимуляции барорецепторов. Для уменьшения отрицательного влияния норадреналина на почечный и спланхнический кровоток его применяют в комплексе с небольшими дозами дофамина или добутамина [133]. Рекомендуемая доза норадреналина — 1 — 24 мкг/мин. Вследствие выраженного вазоконстрикторного эффекта может вызвать некроз периферических тканей. Вводить надо строго внутривенно, при попадании в паравазальную клетчатку возникает некроз.
Фенилэфрин (мезатон). Является чистым арадреномиметиком. Обычно используется для повышения АД у пациентов, у которых дальнейшая стимуляция pt-адренорецепторов может быть опасной (выраженная тахиаритмия, ишемия миокарда). 15*
Доза внутривенной инфузии мезатона — 100—180 мкг/мин. По данным М. G. Angelos с соавт. [ 134], в эксперименте на изолированном реперфузируемом сердце применение фенилэфрин вследствие а]-адреномиметического эффекта значительно улучшает постишемическую функцию левого желудочка без ухудшения общего метаболического состояния миокарда и имеет преимущества перед дофамином и адреналином.
Допексамин гидрохлорид. Новый синтетический катехоламин. Обладает р2-ад-реномиметическим эффектом, слабым Р]-адреномиметическим эффектом, не обладает а-адреномиметическим эффектом. Снижает постнагрузку и оказывает положительное инотропное действие вследствие стимуляции барорецепторов. По механизму действия его еще называют иноди-лататором [135], так как он обладает кардиотоническим и вазодилататорным действием. В отличие от адреналина допексамин увеличивает перфузию желудка и кишок и улучшает их функцию у хирургических больных [136], поэтому показан при шоке с ПОН [137]. Предоперационное применение допексамина у больных высокой группы риска со сниженным транспортом кислорода уменьшало летальность [138].
Теоретически можно считать рациональным применение допексамина для увеличения сердечного выброса без увеличения потребления миокардом кислорода при повышении почечного, печеночного и спланх-нического кровотока [131]. D. Boyd с соавт. [138] считает показанной гемодинамическую поддержку допексамином, если £Ю2 < 600 мл/мин на 1 м2, но при достижении этого показателя, при появлении боли за грудиной и депрессии сегмента ST на ЭКГ либо при учащении ЧСС более чем на 20/мин ее следует прекратить.
Начальная доза допексамина — 0,5 мкг/кг в минуту, но через 10—15 мин может быть увеличена до максимальной (6 мкг/кг в минуту) [135]. Дальнейшему расширению применения в клинике препятствует высокая цена препарата.
Препараты изопротеренола гидрохлорида. Относятся к чистым агонистам Р-адренорецепторов. Увеличивают сокра
452 ШОК
тительную способность миокарда и частоту сердечных сокращений. Резко повышают потребность миокарда в кислороде и обладают выраженным аритмогенным эффектом. Поэтому используются в основном при рефрактерной брадикардии до применения искусственного водителя ритма [139], хотя и в этих случаях в настоящее время приоритет имеет адреналин.
Ингибиторы фосфодиэстеразы (амри-нон лактат и милренон лактат). Сравнительно новая группа препаратов, обладающих инотропным и вазодилатирующим эффектом [121]. Адреномиметики, воздействуя на p-адренергический рецептор, активируют аденилатциклазу с образованием из АТФ циклического АМФ (цАМФ). Фосфодиэстераза является ферментом, инактивирующим цАМФ, поэтому ингибиция этого фермента в миокарде и гладких мышцах сосудов, способствуя накоплению цАМФ, улучшает диастолическую релаксацию миокарда, способствует накоплению внутриклеточного кальция и увеличивает сократительную способность миокарда, снижает тонус гладких мышц сосудов и бронхов [129]. Так же, как и допексамин, препараты этой группы могут быть отнесены к иновазодилаторам. Практически не действуют на частоту сокращений сердца.
Представителями этой группы являются амринон, милринон, эноксимон. Амри-нон может использоваться как единственное средство при систолической дисфункции миокарда, однако чаще всего применяется в сочетании с добутамином при рефрактерной недостаточности миокарда [140]. Начальная доза — 0,75 мг/кг, но ее можно увеличить до 1,5 мг/кг [141]. Несмотря на то что амринон не стимулирует p-адренергические рецепторы, по эффективности он сравним с добутамином [140]. При внутривенной кратковременной терапии практически не имеет побочных эффектов. При продолжительном приеме внутрь может развиться тромбоцитопения, однако при внутривенной кратковременной терапии это осложнение наблюдается только в 2 — 3 % случаев [127]. Сообщается также о случаях артериальной гипотензии у больных с гиповолеми
ей [127]. Нельзя применять в растворах глюкозы. Следует хранить в темном месте. Во время инфузии амринона нельзя вводить фуросемид, так как происходит преципитация раствора.
Препараты дигиталиса. Обладают положительным инотропным эффектом вследствие цАМФ-зависимого увеличения концентрации внутриклеточного кальция, воздействуют также на натрий-калиевые и кальциевые каналы мембран миокардио-цитов. Согласно современным представлениям применение дигиталиса при шоке резко ограничено, хотя есть данные о положительной роли его препаратов при грам-отрицательном сепсисе со сниженной контрактильной способностью миокарда [141]. Возможен также синергический эффект при использовании у больных с рефрактерным шоком в сочетании с другими кардиотоническими препаратами [139]. В настоящее время применяют главным образом при хронической недостаточности миокарда, тахиаритмиях.
В табл. 13.4 приведены дозы и методы инфузии основных кардиотонических препаратов.
Предварительное использование инотропных препаратов и вазопрессоров перед восполнением жидкости в большинстве случаев шока не приводит к улучшению перфузии тканей, однако имеются сведения о положительном эффекте допексами-на и ингибиторов фосфодиэстеразы.
Глюкагон. При внутривенном введении обладает положительным инотропным эффектом, улучшает предсердно-желудочковую проводимость, причем этот эффект не обусловлен стимуляцией р-адренерги-ческих рецепторов [142].
Используется для повышения АД при тяжелом ацидозе, передозировке р-адре-ноблокаторов, в случаях неэффективности другой терапии.
Дозировка: вначале вводят болюсно 1 — 5 мг с последующей внутривенной инфузией со скоростью 1 — 20 мг/ч [142].
Возможными побочными эффектами применения глюкагона являются тошнота, рвота, гипергликемия, гипокалиемия.
Вазопрессин (антидиуретический гормон). Увеличивается количество сообще-
Сосудорасширяющие средства 453
Таблица 13.4. Применение инотропных препаратов [117]
Препарат Доза, мкг/кг в минуту Инфузионная среда Концентрация, мг в 50 мл
Адреналин 0,05 0,9 % NaCl 5% глюкоза 2; 4 или 8
Добутамин 2,5-10 0,9 % NaCl 5 % глюкоза 250
Допамин 2,5-5,0 0,9 % NaCl 5 % глюкоза 200
Допексамин 0,5-6,0 0,9 % NaCl 5 % глюкоза 200
Эноксамин 0,5-1* 5-20 0,9 % NaCl Вода для инъекций 100 мг в 40 мл
Изопреналин 1-10** 0,9 % NaCl 5% глюкоза 2-4
Амринон 5-10 0,9 % NaCl 10 мг
Норадреналин Стартовая 0,05 5 % глюкоза 2; 4 или 8 мг
* Миллиграмм на килограмм (мг/кг).
** Микрограмм в минуту (мкг/мин).
ний о положительном эффекте применения низких доз вазопрессина (0,01 — 0,04 ЕД/мин) при катехоламинрезистент-ном шоке, в частности септическом [143]. При шоке декомпенсации гемодинамики способствует снижение продукции вазопрессина, которое наступает после его повышения в начальной стадии шока [143]. АДГ потенцирует эффект норадреналина [144], уменьшает синтез NO-синтетаз и активность протеинкиназы С [145]. Введение терлипрессина (аналог вазопрессина) в дозе 3 мг в сутки увеличивало у больных септическим шоком систолическое АД,
сердечный индекс, ударный объем сердца и позволяло прекратить введение или значительно уменьшить дозу норадреналина, но при этом ухудшало перфузию слизистой оболочки желудка и кишок [146].
Левосимендан. Предлагается в качестве нового средства для лечения острой декомпенсированной недостаточности миокарда. Увеличивает сердечный выброс, не повышая потребность миокарда в кислороде, и обладает сосудорасширяющим эффектом [147]. Этот препарат нуждается в дополнительных исследованиях.
13.11. СОСУДОРАСШИРЯЮЩИЕ СРЕДСТВА
При шоке для уменьшения выраженности спазма периферических сосудов возникает необходимость в применении сосудорасширяющих средств, прежде всего нитратов.
Нитроглицерин. Органический нитрат (глицерил тринитрат), расслабляющий гладкую мускулатуру сосудов и вызывающий генерализованную вазодилатацию. Механизм его действия представлен на рис. 13.6.
Нитроглицерин, взаимодействуя с мембраной эндотелиальных клеток, способствует образованию NO, который, проникая в гладкомышечные клетки сосудов, активи
рует образование цГМФ из ГМФ. Циклический гуанозинмонофосфат является агентом, расслабляющим гладкие мышцы сосудов. До того как NO был идентифицирован, он описывался под названием «эндотелий-зависимый расслабляющий фактор» [148].
Нитроглицерин дозозависимо расслабляет гладкие мышцы артерий и вен как большого, так и малого кругов кровообращения [149]. При постоянной внутривенной инфузии в дозе менее 40 мкг/мин проявляется венорасширяющий эффект, в дозе, превышающей 200 мкг/мин, преобладает артериорасширяющий эффект [118].
454 ШОК
/?ono2 (НГЦ)
2H
-► NO2+H2O
> NO+H2O
Релаксация гладких мышц t ГТФ—►цГМФ
FeNO-CN5^7~~“ гннт Клетка umn/ эндотелия
Клетка гладкой
мускулатуры
Рис. 13.6. Биохимический механизм действия нитроглицерина (НГЦ) и натрий нитропруссида (ННП) [31]:
/?ONO2 — химическая формула нитроглицерина; FeNO-CNs — химическая формула натрий нитропруссида; ГТФ — гуанозинтрифосфат; цГМФ — циклический гу анози н монофосфат
Уже в малых дозах снижает ЦВД и ДЗЛА без выраженного влияния на сердечный выброс. При увеличении дозы сердечный выброс может увеличиваться за счет снижения постнагрузки, однако при использовании в больших дозах может прогрессировать артериальная гипотензия. Действие нитроглицерина управляемое, после прекращения инфузии эффект быстро проходит.
Кроме расслабляющего действия на гладкие мышцы сосудов нитроглицерин обладает антиагрегатным действием, возможно, вследствие этого в значительной степени проявляется его антиангинальный эффект [148].
Нитроглицерин применяют с целью снижения конечно-диастолического давления в желудочках (малые дозы), увеличения сердечного выброса (средние дозы), снижения АД и улучшения перфузии тканей (высокие дозы) [118].
Начинают инфузию с 5 мкг/мин, при необходимости через 5 мин дозу увеличивают. Низкая эффективность введения может объясняться абсорбцией препарата пластическим материалом системы для внутривенного вливания или толерантностью организма пациента к нитратам.
Побочные эффекты нитроглицерина: ♦ значительно повышает мозговой кровоток, что часто сопровождается головной
болью, повышением внутричерепного давления [150], поэтому его не следует применять у больных с внутричерепной гипертензией;
♦ усиление легочного кровотока, снижение эффекта рефлекса гипоксической вазоконстрикции приводит к увеличению перфузии невентилируемых или плохо вентилируемых областей легких и, соответственно, к увеличению внутри-легочного шунтирования, поэтому нитроглицерин противопоказан при ОРДС и другой патологии легких с увеличением внутрилегочного шунта с выраженной гипоксемией [151];
♦ в процессе метаболизма нитроглицерина образуются нитраты и метгемоглобин:
Hb-Fe2+ + NO2 + Н+ -> Hb-Fe3+ + HONO;
♦ при продолжительной внутривенной инфузии нитроглицерина возможно повышение уровня этанола в крови, что проявится нарушением сознания и артериальной гипотензией [152]. Диагностируется это осложнение повышением «осмолярой дыры» (> 10 мосмоль/кг). Снижение чувствительности к нитроглицерину наблюдается редко и только после продолжительной (более 24 ч) инфузии препарата [149].
Натрий нитропруссид. Прямой вазодилататор. Является донатором NO, активирующего цитозольную гуанилатцик-лазу, способствует накоплению цГМФ и расслаблению гладких мышц [153]. Характеризуется быстрым (через 1—2 мин) управляемым эффектом на сосуды, эффективно снижает артериальное давление, но может вызывать «синдром обкрадывания» (усугубляет ишемию мозга, миокарда). Водный раствор разрушается на свету с образованием ионов цианида (лечение отравления заключается в ингаляции кислорода, введении натрий тиосульфата). Часто используется в кардиологической практике при гидростатическом отеке легких.
Список использованной литературы 455
Список использованной литературы
1. Rackow Е. С., Astiz М, Е. Pathophysiology and treatment of septic shock // JAMA. — 1991. - 266. - P. 548-554.
2. CaliffR. M., BengstonJ. R. Cardiogenic shock // N. Engl. J. Med. - 1994. - 330. -P. 1724-1730.
3. Shoemaker W. C. Circulatory mechanisms of shock and their mediators // Crit. Care Med. - 1987. - 15. - P. 787-794.
4. Rosser D. Pathophisiology of acute circulatory failure // Critical Care Therapeutics. /Ed. R. Elliott. — Pharmaceutical Press, 1999. — P. 91-96.
5. Umezawa K.,Akaike T., Fujii S. et al. Induction of nitric oxide synthesis and xanthine oxidase and their roles in the antimicrobial mechanism against Salmonella typh. Infection in mice // Infect Immun. — 1997. — 65. — P. 2932-2940.
6. Akaike T., Ando M., Oda T. et al. Dependence on O2-generation by xanthine oxidase of pathogenesis of influenza virus infection in mice // J. Clin. Invest. — 1990. — 85. — P. 739-745.
7. Salzman A. L. Poly(ADP-ribose) synthetase activation in circulatory shock // W. J. Sib-bald, K. F. W. Messmer, M. P. Fink. Tissue oxygenation in acute medicine. — New York: Springer, 1998. — P. 138—158.
8. Kentner R., Safar P., Behringer W. et al. Early antioxidant therapy with Tempol during hemorrhagic shock increases survival in rats // J. Trauma. - 2002. - 53(5). - P. 968-977.
9. Szabo C., Zingarelli B., Salzman A. L. Role of poly-ADP ribosyltransferase activation in the vascular contractile and energetic failure elicited by exogenous and endogenous nitric oxide and peroxynitrite // Circ. Res. — 1996. — 78. - P. 1051-1063.
10. Satoh M. S., Lindahl T. Role of poly (ADP-ribose) formation in DNA repair // Nature. — 1992. - 356. - P. 356-358.
11. Poirier G. G., de Murcia G., Jongstra-Bilen J. et al. Poly (ADP ribosyl) ation in of poly-nuclesomes causes relaxation of chromatin structure // Proc. Natl. Acad. Sci. USA. — 1982. - 79. - P. 3423-3427.
12. Ohashi У., Ueda K., Hayaiasha O. et al. Induction of murine teratocarcinoma cell differentiation by suppression of poly (ADP-ribose) synthesis // Proc. Natl. Acad. Sci. USA. - 1984. - 81. - P. 7132-7136.
13. Cuzzocrea S., Zingarelli B.t Constantino G. et al. Beneficial effects of 3-aminobenzamide, an inhibitor of poly(ADP-ribose) synthetase in a rat model of splanchnic artery occlusion and reperfusion // Br. J. Pharm. — 1997. — 121. - P. 1065-1074.
14. Berger N. A. Oxidant-induced cytotoxicity: a challenge for metabolic modulation // Am. J. Respir. Cell Mol. Biol. — 1991. — 4. — P. 1-3.
15. Szabo C., Salzman A. L. Endogenous peroxynitrite is involved in the inhibition of cellular respiration in immuno-stimulated J774.2 macrophages // Biochem. Biophys. Res. Commun. - 1995. - 209. - P. 739-743.
16. Stadler J., Curran R. D., Ochoa J. G. et al. Effect of endogenous nitric oxide on mitochondrial respiration of rat hepatocytes in vitro and in vivo //Arch. Surg. — 1994. — 126. — P. 186-191.
17. Kennedy M. S., Denenberg A. G., Szabo C., Salzman A. L. Activation of poly-ADP ribosyl synthetase mediates cytotoxicity induced by peroxynitrite in human intestinal epithelial cells // Gastroenterology (in press).
18. Heller B., Wang Z. O., Wagner J. etal. Inactivation of the poly (ADP-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells // J. Biol. Chem. — 1995. - 270. - P. 11176-11180.
19. Szabo C., Wong H.t Bauer P. I. et al. Regulation of components of the inflammatory response by 5-iodo-6- amino-1,2-benzopyrone, an inhibitor of poly (ADP-ribose) synthetase and pleiotropic modifer of cellular signal pathways // Int. J. Oncol. — 1997. — 10. — P. 1093-1104.
20. Reddy S., Safari-Lak N., Sandler S. Longterm effects of nicotinamide-induced inhibition of poly(ADP-ribose) polymerase activity in rat pancreatic islets exposed to IL-1 beta // Endocrinology. — 1995. — 136. — P. 1907 — 1912.
21. Zingarelli B., Salzman A. L., Szabo C. Protective effects of nicotinamide against nitric oxide mediated vascular failure in endotoxic shock // Shock. — 1996. — 5. — P. 258-264.
22. Szabo C., Cuzzocrea S., Zingarelli B. et al. Endothelial disfunction in endotoxic shock // J. Clin. Invest. — 1997. — 100. — P. 723-735.
23. Goldfarb R. D., Marton A., Szabo E. et al. Protective effect of a novel, potent inhibitor of poly(adenosine 5’-diphosphate-ribose) synthetase in a porcine model of severe bacterial sepsis // Crit. Care Med. — 2002. — 30(5). — P. 974-980.
24. Boulos M., Astiz M. E., Barua R. S., Osman M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated bv inhibition of nitric oxide synthase and polyCADP-ribose) synthetase // Crit. Care Med. - 2003. - 31(2). - P. 353-358.
25. Kvarstein G., Mirtaheri P,, Tonnessen T. I. Detection of organ ischemia during hemor
456 ШОК
rhagic shock // Acta Anaesth. Scand. — 2003. - 47(6). - P. 675-686.
26. Ганонг В. Ф. Ф1з1олопя людини / Пер. з англ, за ред. М. Гжегодського, В. Шевчука, О. Заячювсько!'. — Л.: Бак, 2002. — 784 с.
27. Guyton А. С., Jones С. Е., Coleman Т. G. Determination of cardiac output by equating venous return curves with cardiac output curves // Physiol Rev. — 1955. — 35. — P. 123 — 129.
28. Deschamps A., Magder S. Baroreflex control of regional capacitance and blood flow distribution with or without alpha-adrenergic blo-kade // Am. J. Physiol. 1992. — 263. — P. H1755—1763.
29. Lundvall J., Lanne T. Large capacity in man for effective plasma volume control in hypovolemia via fluid transfer from tissue to blood // Acta Physiol. Scand. — 1989. — 137. — P. 513-520.
30. Committee on Trauma. Advanced trauma life support student manual. — Chicago, American College of Surgeons, 1989. — P. 47 — 59.
31. Marino P. L. The ICU Book. — 2nd ed. — Philadelphia: Lippincott Williams & Wilkins, 1997. - 928 p.
32. Farmer J. A. Cardiogenic shock // Critical Care / J. M. Civetta, R. W. Taylor, R. R. Kirby. — 3d ed. — Philadelphia, Lippincott-Raven, 1997. - P. 389-404.
33. Piagnerelli M., Boudjeltia K. Z., Vanhae-verbeek M., Vincent J. L. Red blood cell rheology in sepsis // Inten. Care Med. — 2003. - 29(7). - P. 1052-1061.
34. Parillo J. E., Parker M. M., Natanson C. et al. Septic shock in humans. Advances in ander-standing the pathogenesis, cardiovascular dysfunction and therapy // Ann. Int. Med. — 1990. - 113. - P. 227-242.
35. Parillo J. E. Pathogenetic mechanisms of septic shock // N. Engl. J. Med. — 1993. — 328. - P. 1471-1477.
36. Moncada S., Higgs A. The L-arginine-nitric oxide pathway//N. Engl. J. Med. — 1993. — 329. - P. 2002-2012.
37. Cobb J. B., Danner R. J. Nitric oxide and septic shock // JAMA. — 1996. — 275. — P. 1192-1196.
38. Takakura K., Xiaohong W., Takeuchi K. et al. Deactivation of norepinephrine by peroxynitrite as a new pathogenesis in the hypotension of septic shock // Anesthesiology. — 2003. - 98(4). - P. 928-934.
39. Hoffmann J. N., VollmarB., Laschke M. W. et al. Hydroxyethyl starch (130 kD), but not crystalloid volume support, improves microcirculation during normotensive endotoxemia // Anesthesiology. — 2002. — 97(2). — P. 460-470.
40. Oliveira R. P., Weingartner R., Ribas E. O. et al. Acute haemodynamic effects of a hypertonic saline/dextran solution in stable
patients with severe sepsis // Inten. Care Med. - 2002. - 28(11). - P. 1574-1581.
41. De Backer D., Creteur J., Silva E,, Vincent J. L. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? // Crit. Care Med. - 2003. - 31(6). - P. 1659-1667.
42. Sun Q., Tu Z., Lobo 5., Dimopoulos G. et al. Optimal adrenergic support in septic shock due to peritonitis // Anesthesiology. — 2003. — 98(4). - P. 888-896.
43. Donati A., Conti G., Loggi S. et al. Does methylene blue administration to septic shock patients affect vascular permeability and blood volume? // Crit. Care Med. — 2002. — 30(10). - P. 2271-2277.
44. O’Brien A., Clapp L., Singer M. Terlipressin for norepinephrine-resistant septic shock // Lancet. - 2002. - 359(9313). - P. 1209 — 1210.
45. Ciesielski C. A., Broome С. V. Toxic shock syndrome: still in the differencial // J. Crit. Illness. - 1986. - 1. - P. 26-40.
46. Bessen M. Toxic Shock Syndrome // Critical Care Secrets / Ed. P. E. Parsons, J. P. Wiener-Kronish. — 2nd ed. — Philadelphia: Hanley & Belfus, 1998. - P. 208-211.
47. Пыцкий В. И., Андрианова Н. В., Артома-сова А. В. Аллергические заболевания. — М.: Медицина, 1991. — 368 с.
48. Реакции немедленного типа при анестезии / Под ред. Дж. Уоткинса, Дж. Леви: Пер. с англ. — М.: Медицина, 1991.— С. 149.
49. Шанин В. Ю. Клиническая патофизиология. — СПб.: Специальная лит., 1998. — 569 с.
50. Клиническая иммунология и аллергология / Под ред. Л. Йегера: Пер. с нем. — М.: Медицина, 1996. — Т. 2. — 560 с.
51. Larsen S. L. Acute myocardial infarction following a wasp sting in a patient with normal coronary vessels // Ugeskr. Laeger. — 2000. — 162(36). - P. 4819-4820.
52. Markovchick V. J. Anaphilaxis // Critical Care Secrets / Ed. P. E. Parsons, J. P. Wiener-Kronish. — 2nd ed. — Philadelphia: Hanley&Belfus, 1998. - P. 455-457.
53. Dahn J., Waschke K., Stuck B., Hormann K. Fluid shifts in anaphylaxis // Europ. J. Anaesth. - 2003. - 20. - P. 331.
54. Крафт T. M., Аптон П. M. Ключевые вопросы и темы в анестезиологии: Пер с англ. — М.: Медицина, 1992. — 132 с.
55. Shoemaker W. С., Appel Р. L., Kram Н. В. et al. Prospective trial of supranormal values of survivors as therapeutic goals in high risk surgical patients // Chest. — 1988. — 94. -P. 1176-1186.
56. Connors A. F., SperoffT., Sawson N. V. et al. The effectiveness of right heart cateterization in the initial care of critically ill patients // JAMA. - 1996. - 276. - P. 889-897.
Список использованной литературы 457
57. Молчанов И. В., Гольдина О. А., Горбачевский Ю. В. Растворы гидроэтилированного крахмала — современные и эффективные плазмозамещающие средства инфузионной терапии. — М.: Изд-во НЦССХ им. А. Н. Бакулева, 1988. — 137 с.
58. Boldt J.t Kling D., Bormann В. V. et al. Praoperative normovolamische Hamodilution in der Herzchirurgie // Anaesthesist. — 1989. - 38. - P. 294-301.
59. Griffith C. A. The family of Ringer’s solution //J. Natl. Intravenous Ther. Assoc. — 1886. — 9. - P. 480-483.
60. American Association of Blood Banks Technical Manual. — 10th ed. — Arlington, VA: American Association of Blood Banks, 1990. — P. 368.
61. TalpersS. 5., Romberger D. J., Bunce S. B., Pingleton S. K. Nutritionally associated increased carbon dioxide production // Chest. — 1992. - 102. - P. 551-555.
62. DeGoute C. S., Ray M. J., Manchon M. et al. Intraoperative glucose infusion and blood lactate: endocrine and metabolic relationships during abdominal aortic surgery // Anesthesiology. — 1989. — 71. — P. 355 — 361.
63. Sieber F. E., Traystman R. J. Special issues: glucose and the brain // Crit. Care Med. — 1992. - 20. - P. 104-114.
64. Arieff A. I. Hyponatremia, convulsions, respiratory arrest, and permanent brain damage after elective surgery in healthy women // NEJM. - 1986. - 314. - P. 1529-1534.
65. Griff el M. L., Kaufman B. S. Pharmacology of colloids and cristalloids // Crit. Care Clin. - 1992. - 8. - P. 235-254.
66. Nearman H. S., Herman M. L. Toxic effects of colloids in the intensive care unit // Crit. Care Clin. - 1991. - 7. - P. 713-723.
67. Boldt J., Heesen M.t Welters I. et al. Does the tipe of volume therapy influence endothelial-related coagulation in the critically ill? //Br. J. Anaesth. — 1995. — 75. — P. 740 — 746.
68. Marx G., Cobas Meyer T., Schuerholz T. et al. Hydroxyethyl starch and modified fluid gelatin maintan plasma volume in a porcine model of septic shock with capillary leakage // Inten. Care Med. — 2002. — 28. — P. 629-635.
69. Falk J., Rackow E., Astiz M. et al. Effects of hetastarch and albumin on coagulation in patients with septic shock // J. Clin. Pharmacol. -1988. - 28. - P. 412-415.
70. Boldt J., Mueller M.t Menges T. et al. Influence of different volume therapy regimens on regulators of the circulation in the critically ill // Br. J. Anaesth. - 1996. - 77(4). -P. 480-487.
71. Гольдина О. А., Горбачевский Ю. В. Гемодилюционная терапия с использованием плазмозамещающих растворов гидроксиэти-лированного крахмала при нарушениях ми
кроциркуляции // Вести, интенсивной терапии. — 1998. — № 3 (инф. трансф. тер.). — С. 25-32.
72. Kapiotis S., Quehenberger Р., Eichler H.-G. et al. Effects of hydroxyethyl starch on the activity of blood coagulation and fibrinolysis in healthy volunteers: comparison with albumin // Crit. Care Med. — 1994. — 22. — P. 606-612.
73. Пол1щук M. Камлнський О. А., Литвиненко А. Л. та in. Застосування rinepocMO-лярного шфузшного препарату сорбмакт в клшщ! невщкладно! Heftpoxipyprii // Укр. Heftpoxip. журн. — 2002. — № 1. — С. 94 — 96.
74. McIlroy D. R., Kharasch Е. D. Acute intravascular volume expansion with rapidly administered crystalloid or colloid in the setting of moderate hypovolemia // Anesth. Analg. - 2003. - 96(6). - P. 1572-1577.
75. Friedman Z., Berkenstadt H., Preisman 5., Perel A. A comparison of lactated ringer’s solution to hydroxyethyl starch 6 % in a model of severe hemorrhagic shock and continuous bleeding in dogs // Anesth. Analg. — 2003. — Jan. - 96(1). - P. 39-45.
76. Imm A., Carlson R. W. Fluid resuscitation in circulatory shock // Crit. Care Clin. — 1993. - 9. - P. 313.
77. Schaeffer R. C., Reeiewicz R. A., Chilton S. W. et al. Effects of colloid or cristalloid solution on edemagenesis in normal and thrombomicro-embolized lungs//Crit. Care Med. — 1987. — 15. - P. 1110-1115.
78. Drobin D., Hahn R. G. Kinetics of isotonic and hypertonic plasma volume expanders // Anesthesiology. — 2002. — Jun. — 96(6). — P. 1371-1380.
79. Schierhout G., Roberts I. Fluid resuscitation with colloid or crystalloid solution in critically ill patients: a systematic review of randomized trials// BMJ. - 1998. - 316. - P. 961 — 964.
80. Hasibeder W. R. Fluid resuscitation during capillary leakage: does the type of fluid make a difference // Inten. Care Med. — 2002. — 28. - P. 532-534.
81. Walker R. H. Transfusion risks // Am. J. Clin. Pathol. - 1987. - 88. - P. 374-378.
82. Goodnough L. T., BrecherM. E., KanterM. H., AuBuchon J. P. Transfusion medicine: Blood transfusion // N. Engl. J. Med. — 1999. — 340. - P. 438-447.
83. Seyfried H.t Walewska I. Immune hemolytic transfusion reactions // World J. Surg. — 1987. - 11. - P. 25-29.
84. Nicholls M. D. Transfusion: morbidity and mortality // Anesth. Inten. Care. — 1993. — 21. - P. 15-19.
85. Gottlieb T. Hazards of bacterial contamination of blood products // Anesth. Inten. Care. — 1993. - 21. - P. 20-23.
458 ШОК
86. Isbister J. P. Adverse -eactions to plasma and plasma components // Anesth. Inten. Care. — 1993. - 21. - P. 31-38.
87. Marini J. J., Wheeler A. P. Critical Care Medicine. — 2nd ed. — Philadelphia: Lippincott Williams&Wilkins, 1997. — 640 p.
88. Spiess B. D. Blood transfusion: the silent epidemic // Ann. Thorac Surg. — 2001. — Nov. - 72(5). - P. S1832 —1837.
89. SurgenorS. D., Hampers M. J., Corwin H. L. Optimizing red blood cell transfusion practice // Yearbook of Intensive Care and Emergency Medicine / Ed. J. L. Vincent. — Berlin; Heilderberg: Springer-Verlag. — 2001. — P. 309-318.
90. Vamvakas E. C. Transfusion associated cancer recurrence and postoperative infection: Metaanalysis of randomized, controlled, clinical trials //Transfusion. - 1996. - 36. - P. 175-186.
91. Fransen E., Maessen J.t Dentener M. et al. Impact of blood transfusion on inflammatory mediator release in patients undergoing cardiac surgery // Chest. — 1999. — 116. — P. 1233-1239.
92. Moore F. A., Moore E. E., Sauaia A. Blood transfusion: An independent risk factor for postinjury multiple organ failure // Arch. Surg. - 1997. - 132. - P. 620-625.
93. Popovsky M. A., Chaplin H. C., Moore S. B. Transfusion related acute lung injury: A neglected, serious complications of hemotherapy // Transfusion. — 1992. — 32. — P. 589 — 592.
94. Marik P. E., Sibbald W. J. Effect of stored-blood transfusion on oxygen delivery in patients with sepsis // JAMA. — 1993. — 269. - P. 3024-3029.
95. Fitzgerald R. D., Martin С. M., Dietz G. E. et al. Transfusing red blood cells stored in citrate phosphate dextrose adenine-1 for 28 days fails to improve tissue oxygenation in rats // Crit. Care Med. - 1997. - 25. - P. 726-732.
96. Allen J. B., Allen F. B. The minimum acceptable level of hemoglobin // Int. Anesthesiol Clin. — 1982. — 20. — P. 1— 22.
97. Rawstron R. E. Anemia and surgery // Aust. NZ. J. Surg. - 1970. - 39. - P. 425-432.
98. Van Woerkens E. C., Trouwborst A., Lan-schotj. J. Profound hemodilution: What is the critical level of hemodilution at which oxygen delivery-dependent oxygen consumption starts in an anesthetized human? // Anasth. Analg. — 1992. — 75. — P. 818 — 821.
99. Spence R. K., Costabile J. P., Young G. S. et al. Is hemoglobin level alone a reliable predictor of outcome in the severely anemic surgical patient? // Am. Surg. — 1992. — 58. - P. 92-95.
100. Janvier G., Annat G. Are there any limits to hemodilution? // Ann. Fr. Anesth. Reanim. — 1995. — 14 (suppl). — P. 9 — 20.
101. Chapter С. K.t Cain S. M. The physiologic reserve in oxygen carrying capacity // Can. J. Physiol. Pharmacol. — 1986. — 39. — P. 453-456.
102. Documenta Scientific Tables. — 7th ed. — Basel: Documenta Geigy, 1966. — P. 557 — 558.
103. Fowler N. O., Holmes J. C. Blood viscosity and cardiac output in acute experimental anemia//J. AppL Physiol. — 1975. — 39. -P. 453-456.
104. Doss D. N., Estafanous F. G., Ferrario С. M. et al. Mechanism of systemic vasodilatation during normovolemic hemodilution // Anesth. Analg. — 1995. — 81. — P. 30-34.
105. HablerO. P.t KleenM. S., Podtschaske A. H. et al. The effect of acute normovolemic hemodilution on miocardial contractility in dog // Anesth. Analg. — 1996. — 83. — P. 451-458.
106. Rosen A. L.t Gould S., Sehgal L. R. et al. Cardiac output response to extreme hemodilution with hemoglobin solutions of various P50 values // Crit. Care Med. — 1979. — 7. - P. 380-382.
107. Van Woerkens E.t Trouwborst A., DunckerJ. et al. Catecholamines and regional hemodynamics during isovolemic hemodilution in pigs // J. Appl. Physiol. — 1992. — 72. -P. 760-769.
108. Crystal J. G., Rooney M. W.f Salem M. R. Regional gemodynamics and oxygen supply during isovolemic hemodilution alone or in combination with adenosine-induced controlled hypotension // Anesth. Analg. — 1988. - 67. - P. 211-218.
109. Lipowski H. H.f FirrellJ. C. Microvascular hemodynamics during systemic hemodilution and hemoconcentracion // Am. J. Physiol. — 1986. - 254. - P. H331-339.
110. Shou H., Perez de Sa V., Larsson A. et al. Nitrous oxyde reduces inspired oxygen fraction but does not compromise circulation and oxygenation during hemodilution in pigs // Acta Anaesth. Scand. — 1997. — 41. — P. 923-930.
111. Dougenis D., Patrinou V., Filos K. S. et al. Blood use in lung resection for carcinoma: perioperative elective anaemia does not compromise the early outcome // Eur. J. Cardiothorac Surg. — 2000. —20. — P. 372-377.
112. Nelson A. H., FleisherL. A., RosenbaumS. H. Relationship between postoperative anemia and cardiac morbidity in high-risk vascular patients in the intensive care unite // Crit. Care Med. - 1993. -21. - P. 860-866.
Список использованной литературы 459
ИЗ. National Institutes of Health Consensus Conference. Perioperative red blood cell transfusion // JAMA. — 1988. — 260. — P. 2700-2703.
114. American College of Physicians. Practice strategies for elective red blood cell transfusion // Ann. Int. Med. — 1992. — 116. — P. 403-406.
115. Wen-Chin Wu, Rathore Saif 5., Wang Yongfei et al. Blood transfusion in elderly patients with acute myocardial infarction // N. Engl. J. Med. - 2001. - 345. -P. 1230-1236.
116. Nohe B., Kiefer R. T., Ploppa A. et al. The effects of fresh frozen plasma on neutrophil-endothelial interactions // Anesth. Analg. — 2003. - 97(1). - P. 216-221.
117. Oldner A., Rossi P., Karason S’., AnemanA. A practice survey on vasopressor and drug therapy in Scandinavian intensive care units // Acta Anaesthesiol. Scand. — 2003. — 47(6). - P. 693-701.
118. Scalea T. M., Donovan R. Amrinone as an inotrope in managing hypermetabolic surgical stress // J. Trauma. — 1992. — 32. — P. 372-379.
119. Thompson В. T., Cockrill B. A. Renal dose dopamin: a siren song // Lancet. — 1994. — 344. - P. 7-8.
120. Marik P. E. Low-dose dopamine: a systematic review // Inten. Care Med. — 2002. — 28(7). - P. 877-883.
121. Zaloga G. P., PrielippR. C., Butterworth J. F.f Royster R. L. Pharmacologic cardiovascular support // Crit. Care Clin. — 1993. — 9. — P. 335-362.
122. Butterworth J. F., Royster R. L.t PrielippR. et al. Amrinone in cardiac surgical patients with left-ventricular dysfunction // Chest. — 1993. - 104. - P. 1660-1667.
123. Klem C., Dasta J. F., Reilley T., Flancbaum L. Variability in dobutamine pharmacokinetics in unstable critically ill surgical patients // Crit. Care Med. - 1994. - 22. - P. 1926 — 1932.
124. Rich M. W.t Imburgia M. Inotropic response to dobutamine in elderly patients with decompensated congestive heart failure // Am. J. Cardiol. - 1990. - 65. - P. 519-521.
125. Colucci W. S., Wright R. F., Braunwald E. New positive inotropic agents in the treatment of congestive heart failure // N. Engl. J. Med. - 1986. - 314. - P. 290-299.
126. Hayes M. A., YauE. H. S’., Timmins A. C. et al. Response of critically ill patients to treatment aimed at achieving supranormal oxygen delivery and consumption // Chest. — 1993. - 103. - P. 886-895.
127. Sundram P., Reddy H. K.t McElroy P. A. et al. Myocardial energetics and efficiency in patients with idiopathic cardiomyopathy
// Am. J. Cardiol. - 1990. - 119. -P. 891-898.
128. Sonnenblick E. H., Frishman W. H., Lejem-tel T. H. Dobutamine: a new synthetic cadrioactive sympathetic amine // N. Engl. J. Med. - 1979. - 300. - P. 17-22.
129. Kulka P. J., Tryba M. Inotropic support of the critically ill patients // Drugs. — 1993. - 45. - P. 654-667.
130. Chiolero R., Flatt J.-P., Revelly J.-P., Jequi-er E. Effects of catecholamines on oxygen consumption and oxygen delivery in critically ill patients // Chest. — 1991. — 100. — P. 1676-1684.
131. Jonas M. M., Wolf С. B. Oxygen delivery and the Critically III // Yearbook of Intensive Care and Emergency Medicine / Ed. by J. L. Vincent. — Springer, 2002. — P. 603-611.
132. Lollgren H.t Drexler H. Use of inotropes in the clinical care setting // Crit. Care Med. — 1990. - 18. - P. S56-60.
133. Shaer G. L., Fink M. P., Parillo J. E. Norepinephrine alone versus norepinephrine plus low-dose dopamine // Crit. Care Med. - 1985. - 13. - P. 492-496.
134. Angelos M. G., Murray H. N., Waite M. D., Gorsline R. T. Postischemic inotropic support of the dysfunctional heart // Crit. Care Med. - 2002. - Feb. - 30(2). - P. 410-416.
135. Badcott S. Inotropic and vasoactive support in acute cardiac failure and shock states // Critical Care Theraputics / Ed. R. Elliott. — Pharmaceutical Press, 1999. — P. 109-119.
136. Smithies M., Yee T. H., Jackson L. et al. Protecting the gut and the liver in the critically ill: effects of dopexamine // Crit. Care Med. - 1994. - 345. - P. 1368 — 1377.
137. Colardyn F. C., Vanderbogaerde J. F., Voglaers P., Verbeke J. H. Use of dopexamine hydrochloride in patients with septic shock // Crit. Care Med. — 1989. — 17. — P. 999-1003.
138. Boyd D., Grounds R. M., Bennett E. D. A randomized clinical trial of the effect of deliberate perioperative increase of oxygen delivery on mortality in high risk surgical patients // JAMA. — 1993. — 270. — P. 2699-2707.
139. Rodrigues R. M., Rosenthal M. H. Etiology and pathophysiology of shock // Critical care Medicine / Ed. M. J. Murray et al. — Philadelphia: Lippincott-Raven, 1997. — P. 185-197.
140. DiBianco B. Acute positive inotropic intervention: thq phosphodiesterase inhibitors //Am. Heart J. - 1991. - 121. - P. 1871 — 1875.
141. Marcus R. H.t Raw K., Patel J. et al. Comparison of intravenous amrinone and do-
460 ШОК
butamine in congestive heart failure due to idiopathic dilated cardiomyopathy // Am. J. Cardiol. - 1990. - 66. - P. 1107-1112.
142. Hall-Boyer K., Zaloga G. P., Chemow B. Glucagon: Hormone of therapeutic agent // Crit. Care Med. - 1984. - 32. -P. 584-588.
143. Landry D. W., Levin H. R.t Gallant E. M. et al.Vasopressin deficiency contributes to the vasodilation of septic shock // Circulation. - 1997. - 95. - P. 1122-1125.
144. Bartelstone H. J., Nasmyth P. A. Vasopressin potentiation of catecholamine actions in dog, rat, cat // Am. J. Physiol. —1965. — 208. — P. 754-762.
145. Umino T., Kusano E.f Muto S. et al. AVP inhibits LPS- and IL-10-stimulated NO and cGMP via Vt-receptor in cultured rat mesangial cells // Am. J. Physiol. — 1999. — 276. - P. F433-441.
146. Auzinger G. M., (У Callaghan P. G., Harry R. A.,Wendon J. A. Terlipressin in the treatment of catecholamine resistant septic shock // Crit. Care. — 2002. — 6(Suppl. 1). - P. S61.
147. Voipio-PulkkiL.-M., Ukkonen H. Myocardial efficiency during levosimendan infusion // Crit. Care. — 2002. — 6 (Suppl. 1). — P. S64.
148. Elkyam U. Nitrates in heart failure // Cardiol. Clin. - 1994. - 12. - P. 73-85.
149. Radermacher P., SantakB., Becker H., Fal-ke K. J. Prostaglandin Ft and nitroglycerin reduce pulmonary capillary pressure but worsen ventilation-perfusion distribution in patients with ARDS // Anesth. — 1989. — 70. - P. 601-606.
150. CurryS. C.'Amold-Cappell P. Nitroprusside, nitroglycerin and angiotensin-converting enzyme ingibitors / Ed. J. L. Blumer, G. R. Bond Toxic effects of drugs used in the ICU // Crit. Care Clin. — 1991. — 7. — P. 555 — 582.
151. Barkers. J., Kemper К. К., Hyatt J. Effects of methemoglobinemia on pulse oximetry and mixed venous oximetry // Anesth. — 70. — P. 112-117.
152. Korn S. H., Comer J. B. Intravenous nitroglycerin and ethanol intoxication // Ann. Int. Med. - 1985. - 102. - P. 274.
Раздел
Кровезаменители с газотранспортной функцией
В последние годы достигнут прогресс в области создания кровезаменителей с газотранспортной функцией. Необходимость их разработки и применения была вызвана возросшей потребностью в донорской крови, возможности заготовки которой с каждым годом существенно уменьшаются (по данным М3 Украины, за несколько лет она снизилась на 39,5 %). Кроме того, она имеет ограниченный срок хранения. Велик также риск передачи больным гемотрансмиссивных болезней.
Искусственные кровезаменители с функцией переноса кислорода имеют ряд преимуществ перед донорской кровью: они универсальны, не требуют изосерологического подбора, практически безопасны в плане переноса инфекций, имеют длительный срок годности, их можно накапливать
14.1. КРОВЕЗАМЕНИТЕЛИ НА ОСНОВЕ
Для получения гемоглобина используют кровь человека, быка, рекомбинантный и трансгенный гемоглобин. Одним из перспективных путей решения вышеуказанной проблемы является создание липосомальной формы гемоглобина в виде искусственных микроэритроцитов.
В настоящее время разработаны и проходят на разных этапах клинические испытания препараты, созданные на основе модифицированного гемоглобина, — Polyheme, HemAssist, Oxyglobin, Hemopure, PHP, PEG-Hb (США), Hemolink (Канада),
в больших количествах и применять немедленно [1,2].
Создание кровезаменителей с газотранспортной функцией ведется в двух альтернативных направлениях:
1. На основе модифицированного гемоглобина;
2. На основе эмульсий перфторуглеродов.
Многие сложные технологические вопросы получения безопасных кровезаменителей с функцией переноса газов, интенсивно разрабатываемые в ряде стран (США, Россия, Канада, Япония, Китай, Нидерланды), уже решены, включая вирусную инактивацию модифицированного гемоглобина и использование безвредных сурфактантов фторуглеродных эмульсий.
МОДИФИЦИРОВАННОГО ГЕМОГЛОБИНА
Геленпол (Россия) и др. К промышленному производству и клиническому применению Минздравом России с 1998 г. разрешен только геленпол.
Этот препарат создан на основе полимеризованного гемоглобина человека и имеет следующие физико-химические свойства: Р50О2 — 26—28 мм рт. ст. при t = 37 °C; рСО2 — 40 мм рт. ст. в 0,05 М трис-буфера; pH — 7,4; средняя молекулярная масса — 120—160 kD; содержание метфор-мы гемоглобина — до 3 %; время полувыве-дения — 12 — 18 ч; срок хранения — 2 года.
462 КРОВЕЗАМЕНИТЕЛИ С ГАЗОТРАНСПОРТНОЙ ФУНКЦИЕЙ
Геленпол вводится внутривенно в дозе 4 — 7 г/кг массы тела. Его применение не сопровождается побочными реакциями и токсическими эффектами [3].
Этот препарат модулирует дыхательную функцию эритроцитов и плазменных белков, обеспечивает временное улучшение транспорта кислорода, оксигенацию и перфузию тканей, повышает уровень гемоглобина в циркулирующей крови, уменьшает степень анемии, улучшает системную гемодинамику и микроциркуляцию, стимулирует гемопоэз. Применение геленпола позволяет на 50 % снизить потребность в
донорской крови и ее компонентах, повысить безопасность глубокой нормоволемической гемодилюции [4].
Показанием к применению геленпола служит пери- и интраоперационная кровопотеря, а также кровопотеря, сопровождающая травмы и другие неотложные состояния.
Однако при использовании растворов модифицированного гемоглобина не исключается опасность развития общей и легочной гипертензии, эзофагального спазма, повышения вязкости крови, временного снижения сердечного выброса.
14.2. КРОВЕЗАМЕНИТЕЛИ НА ОСНОВЕ ЭМУЛЬСИИ ПЕРФТОРУГЛЕРОДОВ
Среди искусственных кровезаменителей с функцией переноса кислорода более перспективными для клинической практики оказались перфторорганические соединения [5 — 8]. На их основе в разных странах был создан ряд препаратов, открывших новую эру в трансфузиологии (Оху-gent, LiquiVent, Oxyfluor — США; Fluosol DA 20 % — Япония; Emulsion И — Китай; Перфторан — Россия).
Перфторан был создан российскими учеными Ф. Ф. Белоярцевым, Г. Р. Иваницким в 1982 г. и является первым и единственным препаратом, введенным ВОЗ в международную анатомо-клинико-химическую классификацию (АТС) в рубрику «Кровезаменители и инфузионные растворы» под № В05АА03.
В настоящее время это единственный препарат, разрешенный к широкому клиническому применению. С 1999 г. он допущен для клинического использования и на территории Украины.
Перфторан представляет собой 10 % субмикронную эмульсию на основе пер-фторорганических соединений (ПФОС), обладающую полифункциональным действием: повышает кислородно-транспортную функцию крови, улучшает газообмен и метаболизм на уровне тканей, восстанавливает центральную и периферическую гемодинамику; улучшает реологические свойства крови и микроциркуляцию, является мембраностабилизатором, обладает кардио-
и нейропротекторным действием, имеет сорбционные и диуретические свойства; оказывает дозозависимое иммунотропное и противоотечное действие; является блокатором медленных входящих кальциевых токов.
Сведения о составе и физико-химических свойствах перфторана представлены в таблице 14.1.
Газотранспортную функцию перфторана обеспечивают перфтордекалин и пер-фторметилциклогексилпиперидин в соотношении 2:1.
Растворимость кислорода в перфтора-не значительно превышает его растворимость в плазме (7,0 об.% против 2,3 об.%), и, хотя она ниже, чем в цельной крови (21 об.%), газообмен в тканях существенно возрастает за счет механизмов, представленных на рис. 14.1.
Субмикронный размер частиц эмульсии перфторана позволяет им проникать в те участки, куда не может проникнуть эритроцит, размеры которого в 70—100 раз больше. Это увеличивает полезную площадь капиллярного массопереноса и обеспечивает снабжение кислородом тканей с недостаточным кровообращением.
Постепенно были обнаружены и другие, указанные выше свойства перфторана, что позволило говорить о его полифункцио-нальном действии и применять не только у больных с кровопотерей, но и при других критических состояниях, сопровожда-
Кровезаменители на основе эмульсий перфторуглеродов 463
Таблица 14.1. Состав и свойства перфторана
Состав, г/100 мл Свойства
Перфтордекалин 13,0 Перфторметилцикло- 6,5 гексилпиперидин Проксанол-268 4,0 Натрий хлорид 0,6 Глюкоза 0,2 Натрий гидрогенкарбонат 0,065 Калий хлорид 0,039 Натрий гидрофосфат 0,02 Магний хлорид 0,019 Вода до 100 мл Содержание ионов фтора < 10~5 М Средний размер частиц 0,07 (0,03 — 0,15) мкм Осмотичность — 280 — 340 моем Вязкость 2,5—3,0 • 10~3 Па • с pH = 7,2-7,8 Растворимость О2 (рОз = 760 мм рт. ст., t = 20 °C) — 7,0 об.% Растворимость СО2 (рСОз = 760 мм рт. ст., t = 20 °C) — 60 об. %
ющихся ишемией или гипоксией различного генеза.
Период полувыведения препарата из кровотока составляет 22 —24 ч. Выводится он в неизменном виде в основном через легкие (93 %).
Перфторан химически инертен, не подвергается в организме человека метаболическим превращениям, не проявляет канцерогенного, мутагенного и эмбриотокси-ческого действия.
Важно подчеркнуть, что введение эмульсий ПФОС не влияет на скорость и качество проведения анализов на групповую и резус-совместимость. Он имеет самую низкую реактогенность среди всех известных препаратов ПФОС, являясь наиболее мелкодисперсным.
Эффекты перфторана при внутривенном введении представлены в табл. 14.2.
Принципиально важным моментом является способность перфторана улучшать газотранспортные свойства собственных эритроцитов больного за счет мембранопротекторного действия, увеличения кислородной емкости гемоглобина, облегчения его диссоциации, предупреждения увеличения дисгемоглобинов [10].
Показания к применению перфторана:
1. Острая и хроническая гиповолемия (травматический, геморрагический, ожоговый и инфекционно-токсический шок).
2. Нарушения микроциркуляции и периферического кровообращения (нарушения тканевого метаболизма и газообмена при внезапной остановке кровообращения и жировой эмболии, черепно-мозговая травма, ишемический отек головного мозга, нарушения мозгового кровообращения, шоковая почка, облитерирующие заболевания сосудов конечностей).
Непосредственный транспорт газов частицами эмульсии ПФОС
'2
Газообмен в легких
Газообмен в тканях Осо2
Каскадная передача молекул газов от одной частицы эмульсии ПФОС к другой, позволяющая осуществлять газообмен даже в участках с отсутствующим кровообращением
Изменение пластичности мембраны аноксичного эритроцита
Рис. 14.1. Механизмы повышения газообмена в тканях при использовании перфторана: ПФОС — перфторорганические соединения
464 КРОВЕЗАМЕНИТЕЛИ С ГАЗОТРАНСПОРТНОЙ ФУНКЦИЕЙ
Таблица 14.2, Эффекты перфторана при внутривенном введении [9]
Показатель Эффект
Кислородная емкость, об.% Вязкость, 10“3 4 5 Па • с Растворимость в жирах Размеры, мкм Первичный гемостаз Вторичный гемостаз Суммарная поверхность газообмена, м2 /100 мл Растворимость кислорода в среде, об.% Осмолярность, моем/л Стабилизация клеточных мембран 7,0 2,5 Растворяется в мембране эритроцитов 1 мл перфторана связывает 10 мг липидов 0,07 (у эритроцитов 7,0) 847 (у эритроцитов 70) До 40 (ПФОС) 280-340 Дополнительная кислородная емкость Уменьшение вязкости, дезагрегация эритроцитов Уменьшение вязкости крови за счет повышения эластичности и деформируемости мембран эритроцитов Предупреждает и устраняет жировую эмболию сосудистого русла Частички перфторана, в отличие от эритроцитов, свободно проходят через спазмированные, склерозированные, частично тромбированные и слад-жированные сосуды Дезагрегация тромбоцитов, что способствует реканализации сосудистого русла Повышение фибринолитической активности, снижение концентрации фибриногена, фактора XIII, что устраняет последствия свертывания крови и способствует реканализации сосудистого русла Возрастает суммарная поверхность газообмена Из частичек ПФОС в кровотоке образуются каналы типа «жемчужных нитей», проводимость кислорода по которым в 20 —25 раз выше, чем по окружающей их плазме. Эстафетный клиренс кислорода в системе «альвеолы/капилляры легких/ эритроциты/ткань» возрастает Осмотический диурез Противоотечный
3. Проведение острой нормоволемической гемоди люции.
4. Противоишемическая защита органов, временно отключенных от кровотока или предназначенных для трансплантации (кардиоплегия при реконструктивных операциях на сердце, предварительная подготовка донора и реципиента к трансплантации).
5. Использование в аппарате искусственного кровообращения при перфузион-
ном сохранении отключаемых от кровотока органов и региональной перфузии.
6. Отсутствие донорской крови и эритроцитарных сред при наличии анемической гипоксии, угрожающей жизни больного.
7. Отказ реципиента от гемотрансфузий по религиозным или иным соображениям, при опасности заражения вирусными инфекциями (СПИД, гепатиты, цитомегаловирус и др.).
14.3. ДОЗИРОВКА И СПОСОБ ВВЕДЕНИЯ ПЕРФТОРАНА
Перфторан вводится внутривенно капельно или струйно после предварительно проведенной биологической пробы. В зависимости от вида и тяжести критического
состояния определены оптимальные, минимальные и предельно допустимые дозы: ♦ минимально эффективная доза — 2 —
3 мг/кг массы тела;
Позировка и способ введения перфторана 465
♦ максимально допустимая доза — 25 — 30 мг/кг массы тела.
Эффективность препарата возрастает при проведении оксигенотерапии (40 — 60 %) во время введения перфторана и после его инфузии в течение суток [11 — 13].
Острая и хроническая гиповолемия. Перфторан вводят внутривенно капельно или струйно в дозе 5 — 30 мл/кг массы тела.
Нарушение микроциркуляции и периферического кровообращения. Препарат вводят внутривенно капельно в дозе 4 — 10 мл/кг массы тела, при необходимости можно вводить 2 — 3 раза с интервалом в 1-4 дня в той же дозе.
Острая нормоволемическая гемодилюция. Замещение предварительно резервированной аутокрови растворами кристаллоидов, коллоидов (препараты ГЭК), пер-фтораном в соотношении 0,4:0,4:0,2.
Нарушение тканевого метаболизма и газообмена (остановка кровообращения). Препарат вводится в первые 6 —12 ч после развившегося эпизода внутривенно капельно в дозе 5 — 7 мл/кг массы тела для предотвращения развития ишемического и реперфузионного синдромов и повторно в той же дозе к концу первых — началу вторых суток для купирования смешанной венозной гипоксии и вторичной ишемии головного мозга.
Противоишемическая защита органов и использование в аппарате искусственного кровообращения (АИК). При кардиоплегии и операциях на отключенных от кровотока органах и конечностях перфторан используется в аппарате искусственного кровообращения как основной или дополнительный дилютант из расчета 10 — 40 мл/кг массы тела. При насыщении перфторана кислородом в АИК с пузырьковым оксигенатором не следует применять пеногасители. Пенообразование необходимо регулировать уменьшением потока газа.
Трансплантация органов. Перфторан вводят донору капельно или струйно в дозе 20 — 30 мл/кг при постоянной оксигенации. Реципиенту перфторан вводится за 2 ч до операции в дозе 10 — 20 мл/кг массы тела [14].
Противопоказания: гемофилия, аллергические и аутоиммунные заболевания. Возможно использование препарата при беременности по жизненным показаниям.
Перфторан совместим в одной линии с донорской кровью, альбумином, изотоническим солевым раствором, глюкозой, антибиотиками. Не совместим с кровезаменителями на основе декстрана, гидрокси-этилкрахмала.
Важно отметить, что ПФОС способны модифицировать фармакокинетику некоторых лекарственных препаратов.
Так, в присутствии ПФОС значительно увеличивается свободная фракция варфа-рина вследствие модулирующего влияния компонентов эмульсии на белковые структуры крови, существенно повышается растворимость наркотических анестетиков, таких как галотан, изофлюран (в чистом перфтордекалине их растворимость в 22 — 35 раз больше, чем в крови).
Установлены модифицирующие эффекты перфторана на фармакокинетику лекарственных средств в зависимости от срока его введения (до, после, одновременно с препаратом) и способа (внутривенно, энтерально).
Так, предварительная инфузия перфторана за 1 ч до введения антибиотика, в частности ампициллина, приводила к достоверно более длительному нахождению свободного препарата в циркулирующей крови при меньшем его содержании в «тканях» и снижению общего удельного клиренса препарата. В то же время при использовании трициклического антидепрессанта амитриптиллина спустя 1 ч после инфузии перфторана выявлено резкое снижение его содержания в сосудистом русле и возрастание общего клиренса при неизменной константе скорости элиминации.
В случаях одновременной инфузии перфторана и введения в желудок сибазона обнаружено более быстрое всасывание препарата из пищеварительного канала в кровяное русло и ускорение его перераспределения в организме при уменьшении общего удельного клиренса препарата; концентрация свободного сибазона в крови была выше в течение всего срока наблюдения.
466 КРОВЕЗАМЕНИТЕЛИ С ГАЗОТРАНСПОРТНОЙ ФУНКЦИЕЙ
Если же перфторан и сибазон вводили одновременно в желудок, то получено достоверное возрастание скорости всасывания и распределения сибазона, но без увеличения его содержания в сосудистом русле и при неизменном общем клиренсе препарата.
При инфузии перфторана через 1 ч после сибазона отмечено повышение мас-сопереноса препарата кровью, но при этом скоростные характеристики всасывания и выведения не изменялись [15].
Перфторан выпускается во флаконах по 100, 200 и 400 мл. Хранится в замороженном состоянии при температуре -18 °C, в размороженном — при температуре +4 °C. Допускается 5-кратная разморозка/заморозка препарата без существенного изменения среднего размера частиц эмульсии.
Рекомендуется самопроизвольная разморозка препарата при комнатной температуре, после чего препарат осторожно встряхивают до полной однородности состава. Перед инфузией согреть до +21 — +23 °C.
Препарат считается непригодным к применению в случаях: расслоения эмульсии (даже после встряхивания), помутнения (до молочного цвета), появления осадка
(прозрачные маслянистые капли на дне флакона). Запрещается хранить препарат при температуре ниже -18 °C и размораживать при температуре выше +30 °C.
Накопленный опыт использования перфторана в комплексе интенсивной терапии различных критических состояний, освещенный в зарубежной и отечественной литературе, и наш собственный опыт свидетельствуют о его высоком клиническом эффекте в остром периоде за счет предотвращения гибели клеток различных органов, определенное время находившихся в условиях неадекватного кровообращения или полной его остановки.
Препарат перспективен для оказания неотложной помощи больным с редкими группами крови, в случаях трудности подбора донорской крови, при отказе от переливания крови по религиозным или иным соображениям, в медицине катастроф.
С каждым годом увеличивается число патологических состояний, при которых доказана эффективность ПФОС, и это связано не с очередным увлечением препаратом, а с его воздействием на два основных патофизиологических процесса -ишемию и гипоксию, сопровождающие многие критические состояния и соматические заболевания.
Список использованной литературы
1. Селиванов Е. А., Софронов Г. А., Хане-вич М. Д. Кровезаменители — переносчики кислорода: настоящее и будущее // Физиологически активные вещества на основе перфторуглеродов в экспериментальной и клинической медицине. — СПб., 1999. — С. Illi.
2. Софронов Г. А., Селиванов Е. А., Хане-вич М. Д. Проблема кровезаменителей — переносчиков кислорода: достижения и перспективы // Физиологически активные вещества на основе перфторуглеродов в экспериментальной и клинической медицине. — СПб., 2001. - С. 73-77.
3. Селиванов Е. А., Быстрова И. М., Ханевич М. Д., Парашин М. Е. Экспериментальное и клиническое изучение кровезаменителя-переносчика кислорода на основе модифицированного гемоглобина // Всеармейская конф. «Физиологически активные вещества на основе перфторуглеродов в военной ме
дицине» (Санкт-Петербург, 1997). — СПб., 1997. - С. 95-96.
4. Софронов Г. А., Селиванов Е. А., Ханевич М. Д. Стратегия поиска искусственных заменителей крови // Физиологически активные вещества на основе перфторуглеродов в экспериментальной и клинической медицине. — СПб., 1999. — С. 78 — 85.
5. Белоярцев Ф. Ф. Перфторированные углероды в биологии и медицине // Перфторированные углероды в биологии и медицине: Сб. науч. тр. / Под ред. Ф. Ф. Белоярцева. - Пущино: ОНТИ НЦБИ АН СССР, 1980. - С. 5-20.
6. Иваницкий Г. Р., Воробьев С. И. Кровезаменитель «Перфторан» // Вести. РАН. -1997. - 67, № 11. - С. 998-1008.
7. Мороз В. В., Крылов Н. Л., Иваницкий Г. Р. и др. Применение перфторана в клинической медицине // Анестезиология и реанимация. - 1995. - № 6. - С. 12-17.
Список использованной литературы 467
8. Перфторан в интенсивной терапии критических состояний: Метод, рекоменд. / Под общ. ред. Л. В. Усенко, Е. Н. Клигуненко. — Днепропетровск, 1999. — 56 с.
9. Перфторан. Кровезаменитель с газотранспортной функцией: Инструкция для врачей. — СПб., 2001. — 22 с.
10. Усенко Л. В., Клиуненко О. М. Перфтор-вуглещ в комплекс! шфузшно-трансфузш-но1 терапп у критичних хворих // Всеукр. симп. «Безкровна xipyprin. Сучасна концеп-щя гемотрансфузшно!терапп». — Л., 2000. — С. 49-54.
11. Брюсов П. Г., Софронов Г. А., Ханевич М. Д. Современное состояние и перспективы использования перфторана в хирургии // Всеармейская конф. «Физиологически активные вещества на основе перфторуглеродов
в военной медицине» (Санкт-Петербург, 1997). - СПб., 1997. - С. 8-9.
12. Усенко Л. В., Шифрин Г. А. Интенсивная терапия при кровопотере. — 2-е изд. — К.: Здоров’я, 1995. — 235 с.
13. Усенко Л. В. Перфторан. «Искусственная кровь» — от мечты к реальности // Л!ку-вання та д!агностика. — 2001. - № 3. -С. 49-54.
14. Spahn D., Brempt R., Theilmeier G. et al. Perflubron emulsion delays blood transfusions in orthopedic surgery // Anesthesiology. — 1999. - Vol. 91, N5. - P. 1195-1208.
15. Применение перфторорганических соединений в экспериментальной и клинической медицине: Сб. метод, рекомендаций / Под общ. ред. акад. РАМН, проф. Г. А. Софронова. — СПб., 2002. - 94 с.
=== Раздел —
15 Полшпрабма
Травма является главной причиной смерти у молодых людей в индустриальных странах [1]. Ежегодно на лечение больных с травмой в США расходуется 150 млрд дол. [2]. От 10 до 15 % таких больных проходят лечение в ОИТ [3]. В последние годы летальность в ОИТ при политравме снизилась до 18 — 24 % [4].
По данным немецких ученых (Regel, 1995), при тяжелой политравме у 85 % больных имеется повреждение конечностей, у 70 % — черепно-мозговая травма, у 35 % — торакальная и у 20 % — абдоминальная. В США среди больных, поступивших в ОИТ, только у 22 % пациентов с тяжелой политравмой отсутствует ЧМТ, а у 19 % — наблюдается изолированная ЧМТ.
15.1. ПАТОФИЗИОЛОГИЧЕСКИЕ ПРОЦЕССЫ ПРИ ТРАВМЕ
Характер патофизиологических расстройств во многом зависит от характера травмы. D. Cuthbertson [5] первым выявил у пациентов с переломами костей отрицательный энергетический и азотистый баланс. Позже снижение энергетических субстратов в клетке при тяжелой травме было подтверждено другими исследователями (см. «Шок»).
D. Cuthbertson выделил в посттравматическом периоде две фазы: катаболизма (Ebb) и анаболизма (Flaw). В острой фазе травмы в печени активируется синтез протеинов стресса: протромбина, фибриногена, церулоплазмина, oq-антитрипсина. Может повыситься температура тела без развития инфекции. Только с улучшением азотистого баланса в период выздоровления катаболизм переходит в анаболизм.
Кровопотеря и шок являются причиной смерти при политравме в 1/3 случаев. У пациентов с политравмой гипотензия и снижение питания тканей продуцируют ло
кальную, клинически не определяемую ишемию, приводят к увеличению продукции макрофагами и полиморфноядерными нейтрофилами провоспалительных цитокинов, комплемента и других медиаторов воспаления, что является ключевым фактором развития синдрома системного воспалительного ответа и полиорганной недостаточности (ПОН) [6].
При проникающих ранениях, не сопровождающихся массивной травмой тканей (характерный пример — ножевые ранения), причиной тяжести состояния в основном является геморрагический шок, а системная воспалительная реакция с развитием ПОН и сепсиса развивается при отсутствии или неэффективности лечения в более поздние по сравнению с политравмой сроки. Это подтверждается данными С. Lucas [7], согласно которым при множественной тупой травме по сравнению с аналогичной по кровопотере проникающей травмой, не сопровождающей
Травма и гемостаз 469
ся массивным повреждением тканей, в посттравматическом периоде требуется гораздо больший объем жидкости для нормализации циркуляции крови. Причиной этого, по мнению С. Lucas, является не
столько вторичная сосудистая гиперемия после шока, сколько повреждение эндотелия сосудов вследствие ССВО, рано развивающегося при политравме, и выход жидкости из внутрисосудистого сектора.
15.2. ТРАВМА И ГЕМОСТАЗ
Нарушения гемостаза в значительной степени обуславливают неэффективность лечения больных с тяжелой травмой.
Гемостаз — защитная реакция организма, в основе которой лежит баланс коагулянтов и антикоагулянтов, достигаемый комплексным взаимодействием различных клеток и факторов коагуляции.
Как уже указывалось, массивное кровотечение наряду с ЧМТ (причина смерти в 52 % случаев) является одним из основных факторов, приводящих к смерти при травмах (38 %), и главной причиной на догоспитальном этапе (Е. J. MacKenzie, С. J. Fowler, 2000) и в раннем (первые 48 ч) посттравматическом периоде [8]. Причиной кровотечения часто является не только повреждение сосудов, но и расстройства гемостаза.
Патогенез коагулопатии при травме имеет комплексный характер. Нарушения гемостаза прежде всего вызываются артериальной гипотензией, гипотермией и ацидозом [9]. При массивных кровотечениях и восполнении жидкостью снижается концентрация факторов гемостаза, агрегация тромбоцитов (при неосложненном течении они восстанавливаются через пять дней). Если у пациентов травма осложнена сепсисом, АДФ- и коллагениндуцированная агрегация тромбоцитов остается значительно сниженной в течение продолжительного периода. Все травмированные со снижением АДФ-агрегации ниже 20 % умирали. У больных с травмой мозга, оцененной по сокращенной шкале травмы (AIS) > > 4 баллов, риск коагулопатии выше, поскольку у них статистически достоверно снижена функция тромбоцитов (R. С. Jacoby, 2002).
Исследователи отмечают четкую корреляцию между продолжительностью артериальной гипотензии и тяжестью коагу
ляционных расстройств. Гипоперфузия сопровождается микроваскулярным кровотечением даже при отсутствии кровотечения из крупного сосуда [10]. При ацидозе, вызванном шоком, длительностью 150 мин наблюдалась значительная пролонгация частичного тромбинового времени (ЧТВ) и активности фактора V [И]. Особенно высок риск коагулопатии при травмах печени [12]. Одними из важнейших причин нарушений гемостаза при тяжелой травме в фазе первичных повреждений являются коагулопатия потребления, дилюционная тромбоцитопения и снижение концентрации фибриногена [13]. Не менее важную роль в развитии коагулопатии может играть гипотермия, часто развивающаяся у больных с тяжелой травмой, которая влияет на весь коагуляционный каскад. Коагуляционные расстройства у больных с продолжающимся кровотечением значительно увеличивают риск неблагоприятного результата лечения.
В случае, если больные выживают, наступает стадия вторичных повреждений, одним из ведущих проявлений которой является гиперкоагуляция — результат увеличения активности прокоагулянтов и снижения антикоагулянтов (R. Jacoby, 2001). К факторам, способствующим этому, относят следующие:
иммобилизация больных (особенно с ЧМТ, переломами костей таза, бедра, голени), статическое положение, кровопотеря и повреждение интимы сосудов, нарушение функции эндотелия;
повышение функциональной активности тромбоцитов;
стимуляция внутреннего и внешнего пути коагуляционного каскада;
активация тканевого фактора коагуляции, коллагена, фактора Виллебранда;
470 ПОЛИТРАВМА
нарушение функции антисвертывающей системы (снижение концентрации антитромбина III, протеинов С и S, нарушение регуляции экспрессии тромбомодулина);
взаимодействие тромбиновых рецепторов с NF-kB;
воспаление;
продукция ингибитора активатора плазминогена (PAI-1), цитокинов, активирующих коагуляцию (TNFa, ИЛ-1, ИЛ-6, ИЛ-12);
подавление фибринолиза: снижение продукции плазмина.
Иммобилизация больных (особенно с ЧМТ, переломами костей таза, бедра, голени), статическое положение нарушают венозный возврат, замедляют и редуцируют кровоток. Стаз способствует выделению тромбопластина, повреждает функцию эндотелия, так как нарушается доставка в эндотелиоциты кислорода и нутриентов. Результаты экспериментальных исследований свидетельствуют, что стагнирующие тромбоциты обладают гораздо большей возможностью активизировать гиперкоагуляцию. Важными факторами, вызывающими гиперкоагуляцию, являются кровопотеря и повреждение интимы сосудов.
В последнее десятилетие многие исследователи обращают внимание на важность функции эндотелия в поддержании баланса коагуляции/антикоагуляции при травме. По мнению М. Knudson [14], она заключается в следующем:
прокоагулянтная активность'.
генерация тканевого фактора коагуляции, активатора ингибитора плазминогена, фактора V, фактора активации тромбоцитов, фактора Виллебранда, активаторов факторов V и XII;
антикоагулянтная активность'.
генерация простациклина, тканевого активатора плазминогена, протеинов S и С, NO.
Интактный эндотелий играет важную антитромботическую и профибринолити-ческую роль, ингибирует адгезию тромбоцитов и лейкоцитов, поддерживает вазодилатацию. В норме эндотелиальные клетки и тромбин не взаимодействуют, поскольку антитромбин III связывает последний с образованием комплекса тромбин/антитромбин.
AT III — гликопротеин, снижающий активность фактора Ха, стимулирующий образование простациклина (что ингибирует освобождение провоспалительных цитокинов), агрегацию тромбоцитов, уменьшает активацию лейкоцитов. Клинически это проявляется улучшением оксигенации тканей, снижением легочной гипертензии, предотвращением гипербилирубинемии в посттравматическом периоде.
Многие исследования подтверждают снижение AT III при травме и шоке. Owings с соавт. (1996) выявил значительное снижение этого фактора у 61 % больных с тяжелой травмой. В норме избыток тромбина активирует образование протеина С, вызывает экспрессию тромбомодулина на поверхности эндотелиальных клеток, при этом связывается избыточный тромбин. При сепсисе и травме нарушается регуляция продукции тромбомодулина, повышается интеракция между тромбином и его эндотелиальными рецепторами, тромбиновые рецепторы начинают взаимодействовать с NF-кВ, что вызывает воспаление и продукцию PAI-1, таким образом, возникает прокоагуляционный профиль эндотелия. Многие цитокины, освобождающиеся при ССВО, в частности TNF, ИЛ-1, ИЛ-6, ИЛ-12, также активируют коагуляцию, изменяя антикоагулянтный фенотип эндотелия в прокоагулянтный.
Снижается продукция протеинов С и S. В норме протеин С инактивирует факторы свертывания Va и Villa и стимулирует фибринолиз, подавляя активность ингибитора активатора плазминогена, поэтому этот протеин является потенциальным антикоагулянтом и профибринолитичес-ким агентом.
Концентрация важнейших антикоагулянтных факторов (антитромбин III, протеины С и S), как и факторов коагуляции, уменьшается при массивном восполнении жидкостью и гемотрансфузиях [15]. Снижение функции естественных антикоагулянтов и стаз крови обуславливают уменьшение активности системы фибринолиза, увеличение концентрации PAI-1, что приводит к снижению продукции плазмина.
Повышается индуцируемая моноцитами и макрофагами продукция тканевого
Терапия тяжелой травмы 471
фактора коагуляции. Эта реакция стимулируется также фактором эндотелия Р-селектином и NF-кВ (Н. ten Cate, 2001). Тканевой фактор на поверхности клеток активирует фактор VII и способствует превращению фактора X в Ха. Последний конвертирует протромбин в тромбин, а тот, в свою очередь, фибриноген в фибрин, что вызывает окклюзию мелких сосудов, синдром ДВС и полиорганной недостаточности, являющейся основной причиной поздней летальности у больных с травмой (50%). Многочисленные исследования выявили строгую корреляцию между нарушением коагуляции и развитием ПОН (J. Marshall, 2001). Был отмечен положительный, иногда спасающий жизнь эффект антикоагулянтной терапии для профилактики выраженной гиперкоагуляции у больных с травмой в стадии вторичных повреждений, что является также профилактикой чрезмерного воспалительного ответа организма на травму и развития синдрома ПОН.
Синдром ДВС, сопровождающийся снижением физиологического антикоагулянтного профиля, резким уменьшением концентрации протеина С и антитромбина, у больных с травмой является фактором высокого риска развития ПОН и неблагоприятного исхода [16].
Однако гиперкоагуляция оказывает и непосредственное влияние на исход тяжелой травмы, вызывая тромбоз глубоких вен (ТГВ) и тромбоэмболию легочной артерии (ТЭЛА) [14].
По данным разных авторов, ТГВ наблюдается при травме в 10 — 90 % случаев, а ТЭЛА — в 1 — 22%. Как отмечает
J. McCartney [17], в 1934 г. в 15 % случаев летальных исходов у больных с травмой и переломом костей голени обнаруживалась ТЭЛА. По данным S. Sevin (1961, 1968), аутопсия 65 % больных, погибших вследствие тяжелых повреждений, показала наличие ТГВ, а 20 % — ТЭЛА, являющуюся основной причиной смерти. Фатальная ТЭЛА без выраженной клинической симптоматики встречается у пациентов с травмой гораздо чаще, чем диагностируется, особенно у больных с переломами костей голени (Geerts с соавт., 1995).
Факторами риска тромбоэмболизма у больных с травмой являются:
1. Необходимость в пролонгированной иммобилизации.
2. Непосредственная травма вен проксимальнее перелома костей нижних конечностей.
3. Пожилой возраст.
4. Переломы костей голени, бедренной кости, таза.
5. Тяжелая травма черепа, спинного мозга.
6. Высокий балл по AIS (сокращенная шкала травмы).
7. Гемотрансфузии, особенно в течение первых суток после травмы.
8. Длительные оперативные вмешательства.
Не рассматриваются как факторы риска: пол, причина травмы, высокий балл по шкале тяжести повреждений ISS, группа крови пациента, количество переливаемой крови (Geerts с соавт., 1995).
Терапевтическая стратегия и тактика при травме должны разрабатываться, исходя из этих положений.
15.3. ТЕРАПИЯ ТЯЖЕЛОЙ ТРАВМЫ
При проникающих ранениях с большой кровопотерей основными принципами терапии должны быть быстрейшая незамедлительная остановка кровотечения и адекватная инфузионно-трансфузионная терапия. Чем дольше длятся артериальная гипотензия и шок, тем выше риск развития осложнений — ССВО и ПОН. В медицинской литературе приобрел популярность термин «золотой час», в течение ко
торого должна быть оказана квалифицированная медицинская помощь, остановлено кровотечение и восполнена гиповолемия. Однако, по нашему мнению, главенствующим должен быть принцип: «чем раньше оказана помощь — тем лучше». ОЦК и АД следует восстанавливать как можно быстрее. В последнее время получены данные, свидетельствующие, что при неостановленном кровотечении, в частное-
472 ПОЛИТРАВМА
ти при проникающих ранениях, интенсивная инфузионная терапия приводит к увеличению объема кровопотери и ухудшению прогноза. Появилась концепция поддержания «безопасной гипотензии», или «гипотензивной ресусцитации», у больных с травмой на догоспитальном этапе до тех пор, пока не остановлено кровотечение [18].
В этом контексте предлагается ограничить внутривенные инфузии до остановки кровотечения на догоспитальном этапе. Справедливость такой концепции подтверждают результаты исследования W. Bickell с соавт. [19]. Интенсивное введение жидкости препятствует образованию тромбов. На примере 598 пациентов с проникающими ранениями груди и живота, часть из которых получали массивную инфузию до остановки кровотечения, а другие — нет, летальность и количество осложнений были достоверно ниже в группе, не получавшей ИТ. По одним данным, при ранней массивной инфузионной терапии до остановки кровотечения летальность составляет 38 %, а при поздней — 30 %, по другим — 31 и 26 % соответственно.
Какое же АД считать оптимальным у больных с неостановленным кровотечением? Т. Kovalenko с соавт. [20] исследовали результаты лечения травмы в трех группах животных, у которых вызывали одинаковую по объему внутрибрюшную кровопотерю: в первой восполнение проводили таким количеством жидкости, чтобы САД поддерживалось на уровне 40 мм рт. ст., во второй — на уровне 80 мм рт. ст., в третьей интенсивную терапию вообще не проводили. Выживание в течение одного часа составило 87,5, 37,5 и 12,5 % соответственно, а кровопотеря — 8,2, 39,9 и 6,7 мл/кг соответственно. Подобные результаты получены и другими авторами [21]. Поэтому по современным представлениям до остановки кровотечения уровень АД достаточно восстановить до критических цифр, хотя это не исключает массивную инфузию и применение кардиотонических средств в ситуациях, когда недовосполнение в ближайшее время угрожает жизни больного.
Пермиссионной артериальной гипотензии следует избегать при ЧМТ, травме
позвоночника с повреждением спинного мозга, исходно нарушенной функции сердца и сосудов.
Не до конца прояснен вопрос о тяжелой тупой политравме, хотя логично было бы предположить целесообразность применения такой же тактики.
После доставки больного с тяжелой кровопотерей в стационар следует как можно быстрее осуществить гемостаз, а в случаях тяжелой гиповолемии и артериальной гипотензии — приступить к восполнению ОЦК. В критических ситуациях скорость внутривенной инфузии должна достигать 500 мл/мин. Конечно, такой объем инфузии можно обеспечить только при помощи специальной аппаратуры, однако и при ее отсутствии можно, используя две-три вены, осуществить инфузию со скоростью не менее 100—150 мл/мин.
При оказании помощи таким больным следует прежде всего решить организационные проблемы [22]: проверить готовность медперсонала к экстренной операции, взаимодействие между специалистами дежурной бригады (анестезиологом, хирургом, рентгенологом, лаборантом), четкое распределение их функций еще до поступления больного и т. д. Все врачи, оказывающие помощь больным с тяжелой травмой, должны руководствоваться едиными принципами терапии. Главная обязанность ведущего хирурга бригады — незамедлительное осуществление гемостаза, а анестезиолога — обеспечение вентиляции легких, венозного доступа, обезболивания для проведения оперативного гемостаза. Порядок выполнения манипуляций может меняться в зависимости от ситуации. Если помощь оказывает один анестезиолог и несколько хирургов, один из них должен помочь в обеспечении венозного доступа (катетеризация центральной вены). Жизнь больного с неконтролируемой кровопотерей может зависеть в буквальном смысле от неэффективно потраченных минут. Если на фоне большой кровопотери произошла остановка кровообращения, результаты сердечно-легочной реанимации часто неудовлетворительные вследствие неэффективности массажа «пустого» сердца.
Терапия расстройств гемостаза при травмах 473
Адекватная жидкостная ресусцитация у больных с проникающими ранениями без массивной травмы тканей в большинстве случаев оказывается эффективной [23]. У больных с политравмой при повышенном ЦВД и низком АД необходимо применить кардиотонические и вазоконстрикторные препараты. В случае шока, когда на фоне низких или нормальных цифр ЦВД обычная объемная ресусцитация жидкостью и симпатомиметическая поддержка не дают эффекта, И. Healey с соавт. [24] предложил восполнение объемом жидкости, в 7 — 12 раз превышающим кровопотерю. В этих случаях другой альтернативы для лечения больных пока не существует. Конечно, такую объемную инфузию лучше проводить под более эффективным гемодинамическим мониторингом (инвазивное определение АД, ДЗЛА и др.). Наряду с инфузией изотонического раствора натрий хлорида применяют инфузию гипертонического раствора (7,5—10 %) натрий хлорида (ГР), быстрее восполняющего кровопотерю и обладающего рядом других преимуществ (см. «Инфузионно-трансфузионная терапия» ).
W. Shumaker и С. Wo [25] обнаружили существенные преимущества ресусцитации при помощи 500 мл 5 % альбумина с 6 % ГЭК по сравнению с использованием 1 л раствора Рингер-лактата у больных с тяжелой травмой. При ресусцитации гипертоническим раствором у больных с ЧМТ отек головного мозга был значительно меньше, чем при применении изотонического. По данным Vassar с соавт. (1994), у более 600 больных с политравмой ГР не вызывал отрицательных эффектов (4 мл/кг 7,5 % раствора). ГР улучшает почечную, мезентериальную и церебральную перфузию, уменьшает повреждение легких и печени при гиповолемическом шоке (Coimbra с соавт., 1997). При его применении меньше отек эндотелия (Mazzoni с соавт., 1992), уменьшается диастолическая дисфункция
миокарда — потенциальная причина гибели при травматическом шоке. ГР может обладать противовоспалительным эффектом, уменьшая выраженность адгезии клеток крови с эндотелием и, тем самым, снижая риск СПОН (Hartl, 1997). Vassar с соавт. (1994) отмечают увеличение выживаемости с 49 до 60 % при ресусцитации ГР у больных с политравмой одинаковой тяжести.
Наряду с показателями АД, ЦВД, ДЗЛА, диуреза важными критериями адекватности инфузионной терапии являются 5О2, pH, BE, лактат смешанной венозной крови.
С целью выведения медиаторов воспаления, что может способствовать лечению тяжелого шока, предлагается использовать вено-венозную гемофильтрацию, улучшающую экстракцию кислорода тканями. Однако наблюдающиеся побочные эффекты, в частности развитие тромбоцитопении, ограничивают применение этого метода при политравме [6].
Профилактику и лечение осложнений: ОРДС, ОПН, сепсиса, пневмоний, являющихся основной причиной поздней летальности, рассматривают как важный элемент оказания помощи больным с политравмой. В этом плане большое значение имеет поддержание адекватного жидкостного баланса в послешоковом периоде, раннее энтеральное питание, современная вентиляторная поддержка, рациональная антибиоти-котерапия.
Одним из эффективных методов профилактики осложнений у больных с политравмой является раннее оперативное вмешательство по поводу стабилизации переломов костей. Так, W. Carash с соавт. [26] отмечает снижение летальности, продолжительности ИВЛ и количества легочных осложнений при интрамедуллярном остеосинтезе переломов костей в первые сутки после травмы по сравнению с отсроченными операциями. Особенно это касалось больных с контузией легких (Р = 0,002).
15.4. ТЕРАПИЯ РАССТРОЙСТВ ГЕМОСТАЗА ПРИ ТРАВМАХ
В фазу первичных повреждений при продолжающемся кровотечении, когда наблюдаются явления гипокоагуляции, для восстановления гемостаза следует поддер
живать нормальную температуру тела (согреть больного), проводить коррекцию артериальной гипотензии, ацидоза, метаболических расстройств. При необходимое-
474 ПОЛИТРАВМА
ти применить препараты, содержащие факторы гемостаза (свежезамороженная плазма, криопреципитат). В последние годы появились сообщения о высокой эффективности рекомбинантного активированного фактора Vila (новосевен) [27]. Механизм его действия заключается в активации тканевого фактора, факторов свертывания XI и X, образовании тромбина и фибриновых сгустков [13].
В фазе вторичных повреждений на фоне остановившегося кровотечения на первый план выходят осложнения, связанные с гиперкоагуляцией.
Методы профилактики ТГВ и ТЭЛА при травме\
1. Пневматическая компрессия (эффективность статистически не доказана).
2. Артерио-венозная импульсная терапия (Foot Pump) (снижает риск ТГВ при операциях на тазобедренном суставе с 40 до 5 %) [27].
3. Установка фильтров в нижнюю полую вену (у больных с высокой степенью риска — наличие предшествующего тромбофлебита и др.).
4. Применение низкомолекулярных гепаринов.
5. Ограничительная стратегия гемотрансфузий, особенно в первые сутки после травмы.
Для профилактики тромбообразования в хирургии, в частности при травме, сейчас широко используют антикоагулянты. Однако наиболее распространенное подкожное введение нефракционированного гепарина является малоэффективным для профилактики ТГВ и ТЭЛА у больных с травмой, так же, как и при ортопедических операциях [28]. Сходные результаты были
получены М. Knudson с соавт. [14] в исследовании, проведенном на 8 тыс. больных с травмой.
При предоперационном (за 2 ч) введении подкожно 5000 ЕД гепарина и повторном его введении через 12 ч риск ТГВ и ТЭЛА снизился на 60 —70 %, однако при ортопедических операциях и травме эта тактика менее эффективна.
Низкомолекулярные гепарины (НМГ) намного более эффективны в профилактике ТГВ и ТЭЛА у больных с травмой (Geerts с соавт., 1995).
Факторами риска при применении антикоагулянтов у больных с травмой являются: тяжелая травма черепа и позвоночника; множественная травма с внутренним кровотечением, не поддающимся оперативному гемостазу;
тяжелая контузия легких, миокарда; выраженная гематома в области шеи; тромбоцитопения (< 50 000 в 1 мм3); коагулопатии.
По-видимому, использование НМГ должно стать рутинной практикой в лечении больных с травмой после остановки кровотечения. Однако остается невыясненным: когда следует начинать такую терапию, кому она противопоказана, точные дозировки. Особенно трудным оказывается решение этих вопросов у больных с множественной тупой травмой (перелом костей таза, множественные гематомы мягких тканей). От правильности выбора момента начала проведения антикоагулянтной терапии может зависеть исход лечения.
При тяжелой травме очень важна адекватная респираторная поддержка. Для ее проведения необходим постоянный мониторинг газового состава крови и КОС.
15.5. ЧЕРЕПНО-МОЗГОВАЯ ТРАВМА
Из-за частоты распространения черепномозговой травмы (ЧМТ) ее назвали «тихой эпидемией» [29]. В 1990 г. в мире зафиксировано 5 563 тыс. случаев ЧМТ, причем в 20 % случаев она была тяжелой [30]. В ФРГ ежегодно диагностируется до 10 тыс. случаев ЧМТ в год [31]. В 44 % случаев именно она является причиной всех леталь
ных исходов при травме, при тяжелой ЧМТ летальность достигает 30 — 50 % [32]. Совершенствование методов лечения этой разновидности травмы является актуальнейшей задачей не только интенсивной травматологии, но и интенсивной терапии вообще.
В зависимости от характера ранения различают проникающую и закрытую ЧМТ.
Черепно-мозговая травма 475
Первая чаще всего связана с огнестрельными ранениями, а вторая — с автотравмой, падением с высоты.
15.5.1. Патогенез
Повреждения головного мозга при ЧМТ делят на первичные и вторичные.
Первичными повреждениями головного мозга при прямом действии травматического фактора являются: аксональные повреждения, контузия, кровоизлияния, повреждение гематоэнцефалического барьера. В течение нескольких минут могут развиться вторичные повреждения головного мозга: диффузное аксональное повреждение, гиперемия и отек, тяжелые метаболические расстройства. Повышается внутричерепное давление (ВЧД), снижается мозговое перфузионное давление (МПД) и мозговой кровоток. Эти изменения, в свою очередь, приводят к ишемии ткани головного мозга, активации пероксидного окисления липидов, увеличению концентрации ионов кальция внутри нейронов, а в наиболее тяжелых случаях — к смерти мозга [33].
Хотя повреждения, непосредственно вызванные травмой, могут быть очень тяжелыми и даже стать причиной летального исхода, более опасными являются вторичные повреждения головного мозга (ВПГМ) [34]. К факторам, вызывающим ВПГМ, относятся [30]:
артериальная гипотензия (систолическое АД < 95 мм рт. ст.);
гипоксия ( РаО2 < 60 мм рт. ст. и/или SaO2 < 90 %);
глобальная ишемия (СИ < 2 л/мин на 1 м2, МПД < 50 мм рт. ст.);
региональная ишемия (вазоспазм);
анемия или сгущение крови (идеальный Ht = 0,33 л/л, НЬ = 100-110 г/л);
гипертермия (увеличение потребности головного мозга в кислороде);
гиперкапния (РаСО2 < 40 мм рт. ст.) или гипокапния (РаСО2 < 30 мм рт. ст.).
Основными внутричерепными причинами вторичных повреждений мозга являются [35]: гематомы, отек, гидроцефалия, инфекция.
К внечерепным факторам вторичных повреждений мозга относятся [36]:
гипоксемия (причина: гиповентиляция, торакальная травма, аспирация, ингаляция токсичных газов);
артериальная гипотензия (гиповолемия, миокардиальная недостаточность, сепсис, повреждение ствола мозга и мозжечка);
анемия (внутреннее или наружное кровотечение);
артериальная гипертензия (боль, расстройства функции автономной нервной системы, недостаточное обезболивание и седация);
гиперкапния (депрессия дыхания);
гипокапния (гипервентиляция в ответ на стресс, инфекцию);
гипертермия (гиперметаболизм, стресс, инфекция, воспаление);
гипергликемия (ответ на стресс, гипотермия, переливание растворов глюкозы);
гипогликемия (недостаточное питание, передозировка инсулина);
гипонатриемия (недовосполнение, переливание растворов глюкозы).
15.5.2. Клинические проявления черепно-мозговой травмы и ее диагностика
Начальными диагностическими показателями наличия ЧМТ у больных с травмой являются нарушения сознания и посттравматическая амнезия. Основными критериями для оценки уровня амнезии в настоящее время являются ориентация на тесты на амнезию (GOAT) [37], а оценка степени нарушения функции ЦНС производится по шкале ком Глазго (ШКГ) [38]. Оценка по этим шкалам тесно коррелирует с тяжестью ЧМТ, исходом и продолжительностью лечения.
Для диагностики важное значение имеет неврологическое обследование больного с ЧМТ, в частности исследование некоторых рефлексов: фотореакции зрачков на свет (при ее наличии прогнозируется летальность ниже 10 %, а при отсутствии реакции — 50 — 75 % [39]), а также фронтоорбитального , окулоцефалического.
Основными диагностическими методами при ЧМТ являются компьютерная томография (КТ), магнитно-резонансная терапия, ультразвуковое исследование. Для
476 ПОЛИТРАВМА
своевременной диагностики и выбора тактики лечения больных с ЧМТ необходим адекватный мониторинг. A. Valentin с соавт. [40] обнаружил достоверное увеличение откорректированного показателя госпитальной смертности в группе больных с тяжелой ЧМТ и кровоизлияниями без мониторинга ВЧД по сравнению с пациентами, которым такой мониторинг проводился. В то же время С. Thees с соавт. [41] в экспериментальном исследовании показал, что использование для диагностики только уровня мозгового перфузионного давления не отражает адекватность перфузии мозга.
W. Mauritz [30] выделил три уровня мониторинга при ЧМТ:
первый: ЭКГ; газы артериальной крови; САД; температура тела; диурез; электролиты, глюкоза, лактат крови; оценка по ШКГ, транскраниальная допплерография, ЭЭГ.
Показания: больные с ЧМТ с оценкой по ШКГ > 8 баллов, минимальными изменениями, зафиксированными с помощью КТ, не требующими седации и ИВЛ;
второй: показатели первого уровня плюс определение ВЧД; МПД (= САД - ВЧД); ^еСО2-
Показания: больные с ЧМТ с оценкой по ШКГ < 8 баллов, более серьезными изменениями на КТ, требующие седации и ИВЛ;
третий: показатели второго уровня плюс измерение ДЗЛА, 5/02-
Показания: больные с ЧМТ с оценкой по ШКГ < 8 баллов, тяжелыми изменениями на КТ, повышением ВЧД выше 20 мм рт. ст., несмотря на дренирование желудочков и применение маннитола.
Таблица 15.1. Клиническая интерпретация показателя SjO2
SjO2 Мозговой кровоток Клиническое значение
> 90 Отсутствует Смерть мозга
90-75 Гиперперфузия Возможна гипервентиляция
75-60 Норма —
60-40 Гипоперфузия Увеличение СИ, РаСО2 ,
< 40 Ишемия — « —
У больных с тяжелой ЧМТ ДЗЛА следует поддерживать в интервале 8 —12 мм рт. ст., СИ — в пределах 3,5 —5,0 л/мин на 1 м2.
Кроме постоянного мониторинга ВЧД у больных с тяжелой ЧМТ необходимо контролировать насыщение кислородом крови из внутренней яремной вены (S/O2), отражающее перфузию и метаболизм головного мозга [42]. Клиническое значение S/O2 представлено в табл. 15.1.
Повышение 5;О2 выше 75 % является показателем избыточной доставки кислорода, что часто наблюдается при гиперемии. ВЧГ (внутричерепная гипертензия), вызванная гиперемией, требует лечения гипокапнией. ВЧГ, сопровождающаяся снижением S/O2 ниже 55 %, предполагает повышение АД (САД > 110 мм рт. ст.) и применение маннитола, тиопентала. Снижение SjО2 ниже 55 % является показателем того, что церебральный кровоток ниже необходимого для мозга и сопровождается увеличением экстракции О2.
15.5.3. Лечение черепно-мозговой травмы
Основной целью терапии ЧМТ является минимизация вторичных повреждений головного мозга, первичные же повреждения не поддаются лечению.
В 40 % случаев ЧМТ диагностируют внутричерепные гематомы. Ранняя хирургическая декомпрессия является императивным методом лечения. При значительной внутричерепной геморрагии, выявляемой при ультразвуковом или КТ исследовании, задержка с операцией в течение 4 ч увеличивала летальность до 90 % [43]. Увеличение ВЧД выше 30 мм рт. ст., снижение ЦПД ниже 45 мм рт. ст. также могут служить показаниями для декомпрессионной краниотомии.
Обеспечение вентиляции и газообмена в легких является очень важным компонентом лечения ЧМТ. Гипоксемия и гиперкапния значительно ухудшают прогноз у больных с ЧМТ [44]. Часто у больных с ЧМТ с нарушенным сознанием возникает рвота и аспирация содержимого желудка, нарушается проходимость верхних
Черепно-мозговая травма 477
дыхательных путей, поэтому прежде всего необходим контроль за проходимостью дыхательных путей. С этой целью используют ФБС, интубацию трахеи и трахеостомию. Показания к ИВЛ при ЧМТ более широкие (см. «ИВЛ»), незамедлительно следует переводить на ИВЛ больных с оценкой по ШКГ < 8 баллов и тяжелой политравмой [30].
ИВЛ следует проводить в режиме умеренной гипервентиляции с интервалом РаСО2 = 30 — 35 мм рт. ст. Кратковременная более глубокая гипервентиляция с РаСО2 < 30 мм рт. ст. может быть показана при мозговой гиперперфузии (5/О2 > > 75 %) со стойкой внутричерепной гипертензией (ВЧД > 25 мм рт. ст.), не реагирующей на введение маннитола и тиопен-тал-натрия, а также при нормальном уровне 5;О2, но с ВЧД > 40 мм рт. ст. [45].
Необходимо обеспечить хорошую оксигенацию крови (РаО2> 100 мм рт. ст.) при минимальных уровнях ПДКВ (< 8 см вод. ст., или 784 Па) и инспираторного давления [30]. Применение режима ВчИВЛ нуждается в дальнейших исследованиях (риск гиперкапнии).
Отдельно следует остановиться на обеспечении газообмена у больных с ЧМТ и ОРДС. Некоторые режимы ИВЛ, предназначенные для улучшения оксигенации при ОРДС такие, как ПДКВ, положение на животе, способствуют повышению ВЧД. Однако положительное влияние ПДКВ, переворота на живот на насыщение кислородом ткани мозга, увеличение насыщения кислородом артериальной крови, по-видимому, перевешивает неблагоприятный эффект этих режимов на мозговое перфузионное давление у пациентов с ОРДС [46, 47].
15.5.4. Поддержание оптимальной гемодинамики
Как уже указывалось выше, следует поддерживать нормальную волемию с ДЗЛА = 8—12 мм рт. ст. (ЦВД = 10 — 12 см вод. ст., или 980—1176 Па). При ДЗЛА < 10 мм рт. ст. показана инфузия жидкости, при ДЗЛА > 14 мм рт. ст. — отрицательный водный баланс, при необ
ходимости — применение симпатомиме-тиков. САД должно быть не меньше суммы: ВЧД + 60 мм рт. ст. Если САД снижено при нормальном ДЗЛА (ЦВД), то показана инфузия вазопрессоров; если САД снижено при низком ДЗЛА (ЦВД) — инфузия жидкости; если САД значительно повышено — отрицательный баланс жидкости, антигипертензивные препараты. Внутричерепная гипертензия, сопровождающаяся снижением SjO2 ниже 55 %, предполагает улучшение мозгового кровотока при помощи повышения АД (САД > > 110 мм рт. ст.) и применения маннитола, тиопентал-натрия. Снижение САД ниже 90 мм рт. ст. ассоциируется с ухудшением неврологического прогноза. Его следует удерживать на уровне 120 мм рт. ст., а МПД — выше 70 мм рт. ст.
Для восполнения ОЦК предпочтительнее переливание изотонического и гипертонического растворов натрий хлорида (3 — 4 %), растворов ГЭК с осмолярностью выше 300 мосмоль/кг. Гипоосмоляр-ных растворов следует избегать [48]. Переливание ощелачивающих растворов, прежде всего натрий гидрогенкарбоната (соды), должно быть исключено у больных с ЧМТ и шоком даже в случае выраженного лактатацидоза. Было доказано, что эти растворы усиливают внутриклеточный ацидоз и провоцируют снижение pH спинномозговой жидкости [39].
Для поддержания АД на необходимом уровне при нормальном или повышенном ЦВД используют допамин (10 — 15 мкг/кг в минуту) и норадреналин (0,5 — 2 мкг/кг в минуту).
Необходимо поддерживать оптимальный гематокрит (0,30 — 0,33 л/л), уровень гемоглобина (100—110 г/л) и нормальную волемию. Дегидратация с развитием гиповолемии и снижением перфузионного давления мозга не должна использоваться у больных с ЧМТ (G. L. Clifton). Превышение порога ВЧД, САД, ЦПД и снижение баланса жидкости могут иметь пагубное влияние на исход при тяжелой ЧМТ. Баланс жидкости ниже 600 мл (отрицательный баланс) ассоциируется с отрицательным влиянием на исход, независимым от уровня ВЧД, САД или ЦПД [49].
478 ПОЛИТРАВМА
Еще одним важным показателем экст-рацеребрального гомеостаза, который необходимо поддерживать при ЧМТ, является концентрация ионов натрия в плазме крови (оптимальный уровень 145 — 150 ммоль/л). Следует избегать гипонатриемии и выраженной гипернатриемии.
Большое внимание уделяется поддержанию нормогликемии, только при гипогликемии у больных с ЧМТ можно вводить гипертонические растворы глюкозы. Уровень глюкозы не должен продолжительное время превышать 10 ммоль/л [39].
15.5.5. Обезболивание
Боль повышает потребность мозговой ткани в кислороде. Наиболее распространенным обезболивающим средством является фентанил, в тяжелых случаях применяется постоянная внутривенная инфузия со скоростью 10 — 20 мкг/кг в час. Опиаты вводят, как правило, только у больных на ИВЛ.
Седация имеет противоречивое значение. Показана у пациентов с оценкой по ШКГ < 8 баллов на ИВЛ или при возбуждении.
Пропофол снижает потребность мозга в кислороде и вызывает констрикцию церебральных сосудов, снижает внутричерепную гипертензию [39]. Требует осторожного применения при нестабильной гемодинамике. Начальная доза —1—3 мг/кг в час, поддерживающая — 2—4 мг/кг в час.
Тиопентал-натрий снижает мозговой кровоток и потребность мозга в кислороде, но влияет на гемодинамику; его накопление в жировой ткани может способствовать развитию ятрогенной комы; большие дозы вызывают артериальную гипотензию, иммуносупрессию и увеличивают риск инфекции. Профилактическое применение больших доз барбитуратов увеличивает летальность. Показан только в случаях, если не удается снизить ВЧД другими методами [50].
Этомидат снижает ВЧД, но не снижает АД. При длительном применении угнетает функцию надпочечников, поэтому применяется в течение короткого периода
времени у больных с нестабильным АД. Начальная доза — 0,3 —0,4 мг/кг, последующая — 0,1 мг/кг в час.
Согласно современным представлениям кетамин также может применяться у больных с ЧМТ. Поддерживает стабильное АД в сочетании с мидазоламом, пропофолом, при однократном введении предпочтительнее, чем фентанил [51]. Начальная доза — 1—3 мг/кг.
Применяют также небольшие дозы бензодиазепинов, предпочтение отдается мидазоламу (1—20 мг/ч) [52]. Болюсное введение бензодиазепинов может оказывать неблагоприятный эффект (снижение САД и повышение ВЧД) [53].
С целью лечения судорог используют: фенобарбитал — 200 мг/сут и вальпрои-новую кислоту — 30 мг/кг в сутки. При развитии судорожного припадка важно как можно быстрее его купировать для предупреждения повышения ВЧД.
Мышечные релаксанты не рекомендуется применять постоянно, только ситуационно перед манипуляциями, которые могут повысить ВЧД. С этой целью чаще всего используют векурониум (6—10 мг болюсно) [30]. J. К. Hsiang с соавт. [54] сообщают об отрицательных эффектах длительной кураризации: увеличивалось количество инфекционных осложнений (сепсиса, пневмоний), время госпитализации, ухудшался неврологический статус.
15.5.6. Лечение внутричерепной гипертензии
Повышение ВЧД до 20 — 25 мм рт. ст. требует терапевтического вмешательства. С этой целью используется прежде всего маннитол. Этот препарат снижает ВЧД посредством таких механизмов: уменьшает вязкость крови, оказывает вазоконстрикторный эффект, снижает объем интерстициальной жидкости в мозгу [55]. Существует гипотеза, что он обладает антиоксидантным эффектом [56]. Однако продолжительное применение маннитола снижает его эффективность, вызывает гиперосмолярность как плазмы, так и цереброспинальной жидкости, осмотический нефроз, требует компенсации электролитов [57].
Черепно-мозговая травма 479
Последствиями его использования могут быть гиповолемия и усиление ишемии мозга. Длительное применение этого препарата приводит к его накоплению в поврежденной зоне и усилению ее отека. Поэтому маннитол вводят болюсно за 25 мин в дозе 0,25 — 1,0 мг/кг. Осмолярность плазмы не должна превышать 320 мосмоль/кг.
Фуросемид менее эффективно снижает ВЧД, однако при совместном использовании с манниолом может оказать синергический эффект. При гиповолемии его применение противопоказано.
В качестве осмотерапевтического средства в последнее время рекомендуют применять гипертонический раствор натрий хлорида, который, по данным некоторых авторов [58], в дозе 2 мл/кг 7,5 % раствора (приблизительно 480 мосм/70 кг массы тела) оказался эффективным и безопасным методом лечения внутричерепной гипертензии у пациентов с черепномозговой травмой и был даже более эффективным, чем 2 мл/кг 20 % раствора маннитола, в тех случаях, когда показана осмотерапия.
Профилактическая антибиотикотерапия показана только при открытых ЧМТ.
Позиция головы: при нестабильной гемодинамике — горизонтальная, при стабильной — голова приподнята на 20 — 30°.
Начинать питание следует в первые 24 ч после травмы.
Целью нейропротекторной терапии является ограничение вторичных повреждений мозга медиаторами, пероксидным окислением липидов, связанным с накоплением внутри клетки ионов Са2+, что ведет к деструкции нейронов и глии. Потенциально эффект могут оказать блокаторы кальциевых каналов, антиоксиданты, антагонисты глутаматовых рецепторов, блокаторы возбуждающих кислот, ноотроп-ные препараты, хотя с позиций доказательной медицины до сих пор не подтверждена эффективность в клинике ни одного из этих препаратов.
Лечебная гипотермия. Гипотермия уменьшает потребность головного мозга в кислороде и глюкозе. Снижение температуры тела до 32 — 33 °C считается благоприятным у больных с ишемией мозга [59].
Во многих работах подчеркивается важность ранней (в первые сутки) хирургической фиксации переломов костей (интрамедуллярный остеосинтез), прежде всего бедренной и тазовых, при ЧМТ и торакальной травме. После операций, выполненных на 2 — 5-е сутки после травмы, значительно увеличивалось количество осложнений со стороны легких, а также головного мозга [60]. Однако до устранения гипоксемии и артериальной гипотензии ортопедических операций следует избегать [61].
При рефрактерной к обычно применяемой терапии ВЧГ показана так называемая Lund-концепция, предложенная сотрудниками Ландского университетского госпиталя [62]. Она предусматривает снижение прекапиллярного давления вазоконстриктором — дигидроэрготамином и АД с помощью p-блокатора и а2-агониста (кло-нидина), но с поддержанием МПД выше 50 мм рт. ст.
В заключение следует отметить, что главной задачей интенсивной терапии при тяжелой ЧМТ является эффективная профилактика вторичных повреждений мозга. Е. Je-remitsky с соавт. [63] отмечает, что у пациентов с ЧМТ в раннем периоде после травмы необходимо любой ценой предотвращать и коррегировать факторы, способствующие вторичным повреждениям мозга. Особое внимание следует уделять предотвращению артериальной гипотензии, гипоксемии, гипергликемии и гипотермии, которые являются независимыми факторами, увеличивающими смертность. Поскольку эти факторы вторичного повреждения мозга потенциально предотвратимы, необходимы протоколы лечения, способствующие уменьшению их частоты и более грамотной их коррекции.
480 ПОЛИТРАВМА
15.6. ТОРАКАЛЬНАЯ ТРАВМА
Травма груди является одной из наиболее частых травм и сопровождается высокой летальностью. В США ежегодно регистрируется до 500 тыс. случаев торакальной травмы. Вследствие травмы груди большинство пострадавших умирает на месте происшествия. Если пациента удается доставить в стационар, летальность минимальная [64].
Тяжесть состояния при травме груди может быть обусловлена разными факторами, в том числе: открытым и напряженным пневмотораксом, множественными флотирующими переломами ребер, контузией сердца и легких.
Наиболее частым видом торакальной травмы является перелом ребер (чаще 5 — 9-х). Повреждения верхних ребер встречаются реже вследствие анатомических особенностей, однако они чаще сопровождаются повреждением дыхательных путей и крупных сосудов. Переломы 10—11-х ребер происходят не часто, но сопровождаются повреждениями внутренних органов живота.
15.6.1. Переломы ребер по одной линии без флотации
Наиболее часто осложняются повреждением межреберных сосудов, внутренней грудной артерии и легких с развитием пневмогемоторакса. К более поздним осложнениям относится развитие пневмонии и недостаточности дыхания.
Диагностика основывается главным образом на данных рентгенографии грудной клетки, хотя при этом выявляются не все переломы.
Основными патогенетическими факторами легочной недостаточности при переломах ребер без флотации фрагментов и тяжелой контузии легких являются: боль, поверхностное дыхание, нарушение механизма кашля, обтурация дыхательных путей, ате-лектазирование участков легких, пневмония.
При отсутствии внутриплеврального кровотечения и напряженного пневмоторакса переломы ребер, даже множественные, не вызывают тяжелый шок.
Обезболивание. Осуществляется блокадой местным анестетиком мест переломов, паравертебральной блокадой, интраплевральным введением местных анестетиков (кроме новокаина), эпидуральной анестезией. Нельзя для обезболивания использовать наложение сдавливающих ребра повязок, которые, хотя и имеют обезболивающий эффект, однако значительно увеличивают риск развития пневмонии, так как участки легких, лежащие под повязкой, недостаточно вентилируются, часто ате лектазиру ются.
На фоне адекватного обезболивания показаны: ранняя активизация больных (если нет противопоказаний, связанных с другой травмой), эффективная санация дыхательных путей, дыхание с сопротивлением на выдохе. Если отсутствует контузия легких, недостаточность дыхания при переломах ребер без флотации фрагментов развивается вследствие неэффективной или несвоевременно оказанной терапии.
15.6.2. Множественные переломы ребер с флотацией фрагмента
Переломы ребер по двум или нескольким линиям приводят к образованию фрагмента, реберной створки. Поскольку фрагмент не фиксирован к позвоночнику, он флотирует при дыхании: на вдохе западает внутрь, на выдохе выпячивается наружу. В результате может значительно нарушаться вентиляция легкого, причем как участков, непосредственно предлежащих к флотирующему фрагменту, так и всего легкого, а при двусторонних переломах — обоих легких. При флотации обширного переднего фрагмента грудной стенки возможно развитие недостаточности миокарда вследствие его тампонады.
Лечение. По нашим данным, до 8 —10 % больных с флотацией фрагмента нуждаются в проведении ИВЛ даже при адекватной терапии. Последняя означает использование таких же мер, как и при нефлотирующих переломах ребер, однако применять их следует более интенсивно. Часто,
Торакальная травма 481
иногда несколько раз в день, необходимо проведение санационной ФБС с последующими неинвазивными методами вспомогательной ИВЛ. Кроме того, может понадобиться хирургическая стабилизация грудной стенки. Хирургический остеосинтез ребер позволяет стабилизировать реберный каркас, уменьшить риск развития ОНД, а при необходимости проведения ИВЛ — сокращает время ее использования. Хирургическая стабилизация ребер и грудины значительно улучшает функцию внешнего дыхания у таких больных в посттравматическом периоде. I. Dimo-poulou с соавт. [65] обнаружил, что факторами риска длительной ИВЛ у пациентов с травмой груди, госпитализированных в ОИТ, являются: наличие двусторонних повреждений груди, пожилой возраст и тяжесть нейротравмы. Эти сведения могут помочь в определении перспектив долговременного лечения таких пациентов.
Показания к профилактическому применению антибиотиков при переломах ребер без контузии легких остаются противоречивыми.
15.6.3. Контузия легких (КЛ)
Возникает в результате повреждения легких в момент удара при травме. Перед столкновением при автокатастрофе или перед падением с высоты человек рефлекторно задерживает дыхание, при этом смыкаются голосовые связки и в момент удара резко повышается внутриальвеолярное давление. Это приводит к множественному разрыву альвеолярно-капиллярных мембран, кровоизлияниям, интерстициальной эмфиземе, микроателектазам. В легких увеличивается шунтирование, нарастает гипоксемия. Одним из возможных симптомов КЛ является кровохарканье, в некоторых случаях — легочное кровотечение. При КЛ в 90 % случаев имеются другие повреждения грудной клетки и органов груди.
Лечение контузии легких. Специфического лечения КЛ, к сожалению, нет. По нашим данным [66], высокое ПДКВ, инвертированный режим ИВЛ и восстанавливающий маневр представляют опасность при тяжелой контузии легких.
КЛ является ведущей причиной летальности при торакальной травме, достигающей 25 % [67, 68]. К факторам, увеличивающим риск летального исхода при КЛ, относятся: наличие шока с артериальной гипотензией, требующего проведения массивной инфузионно-трансфузионной терапии, ISS > 25, сочетание с ЧМТ, избыточный вес, предшествующие заболевания легких и сердца. КЛ может привести к ОРДС и смерти в результате тяжелой недостаточности легких. По нашему опыту, особенно неблагоприятен прогноз при сочетании контузии легких с тяжелой травмой спинного мозга (спинальный шок), требующей массивных инфузий жидкости. Показания к внутривенной инфузии жидкости у больных с контузией легких следует максимально ограничить, при необходимости скорость инфузии должна быть невысокой. В первые двое-трое суток опасно применение антикоагулянтов, увеличивающих зону контузии и степень легочной недостаточности.
В редких случаях КЛ осложняется легочным кровотечением. Интенсивная терапия и мониторинг, при необходимости с эндотрахеальной интубацией, являются важными мероприятиями при массивном кровохарканьи. Для определения локализации кровоточащего участка следует применить бронхоскопию. Эмболизация является первоочередным методом лечения при легочном кровотечении, оставляя в резерве хирургические методы по неотложным показаниям для случаев, когда упомянутые выше меры оказались недостаточными для остановки кровотечения [69].
15.6.4. Повреждения сердца
Контузия миокарда (КМ) часто встречается при тупой травме груди, однако редко диагностируется. По литературным данным [70], КМ встречается в 8 —71 % случаев закрытой травмы груди (ЗТГ). Как показала ЭХО-кардиография, из 105 больных с ЗТГ контузия миокарда была выявлена у 31 пациента, из них у 17 (55 %) — зафиксирована контузия обоих желудочков сердца, у 12 (39 %) — только правого, у 2 (6 %) — только левого [71].
16
4-220
482 ПОЛИТРАВМА
КМ возникает вследствие прямого повреждающего воздействия травмы на миокард, сопровождающегося повреждением и некрозом кардиомиоцитов, отеком и интерстициальными кровоизлияниями. Различают диффузные трансмуральные повреждения и локальные — субперикардиальные и субэндокардиальные.
В диагностике КМ нет «золотого стандарта» . Обычная и чрезпищеводная ЭКГ не могут выявить четкие критерии диагноза КМ. Для диагностики КМ может иметь значение определение креатинкина-зы, однако этот тест нельзя считать специфическим [72]. Одним из наиболее информативных критериев может быть повышение уровня тропонина 1 — белка, специфического для миокарда [73]. Однако М. Ferjani с соавт. [74] считают этот тест более специфичным для ишемии миокарда, чем для КМ.
Клинически КМ, как правило, не проявляется. Боль в груди, часто сопровождающую КМ, относят преимущественно за счет других повреждений. Классическая ишемическая загрудинная боль наблюдается редко. Выраженная упорная тахикардия и нестабильность АД могут являться клиническими признаками КМ, однако эти симптомы неспецифические. Наличие эктопических экстрасистол при нормальном электролитном балансе и отсутствии гипоксемии может быть признаком КМ. Шок вследствие КМ развивается редко и является признаком тампонады сердца или более серьезных осложнений, требующих хирургического вмешательства.
Терапия контузии миокарда. Если КМ не проявляется тяжелой клинической симптоматикой, нет необходимости в специальной терапии. Лечение в основном симптоматическое: стандартная терапия аритмии, недостаточности сократительной способности миокарда [75].
Осложнения КМ: разрыв предсердий или желудочков сердца, перфорация межжелудочковой перегородки, развитие желудочковых аневризм и псевдоаневризм. Повреждение венечных артерий может приводить к инфаркту миокарда. Возможно развитие острой и хронической недостаточности миокарда.
При разрыве миокарда желудочков летальность достигает 75 %, несмотря на быструю хирургическую операцию. Диагноз обычно устанавливается на торакотомии после перикардиотомии. Важна интенсивная объемная поддержка жидкостью, часто необходимо применение кардиотонических средств, внутриаортальная балонная контрпульсация, в некоторых случаях — осторожное применение вазодилататоров (нитратов) [72].
15.6.5. Тампонада перикарда
При травме груди вследствие повреждения миокарда, венечных и перикардиальных сосудов может возникнуть внут-риперикардиальное кровотечение с тампонадой сердца. Наиболее тяжелой бывает тампонада при ранении левого желудочка.
Симптомы: наполнение шейных вен, тахикардия, артериальная гипотензия, обструктивный шок, высокое ЦВД, глухость сердечных тонов. Уточнить диагноз можно с помощью рентгенографии и эхокардиографии.
Лечение заключается в декомпрессии перикарда (пункция, дренирование, торакотомия с перикардиотомией), остановке кровотечения. Несмотря на высокое ЦВД, нельзя вводить вазодилататоры, диуретики, так как это еще больше снизит сердечный выброс и усугубит состояние.
15.7. БРЮШНОЙ компартмент-синдром
Увеличение внутрибрюшного давления нарушает регионарную циркуляцию кишок, печени, почек. При отсутствии адекватного лечения брюшной компартмент-синдром (БКС) может приводить к летальному ис
ходу. Абдоминальная травма и расслаивающаяся аневризма брюшной аорты — наиболее частые причины БКС. Клинически преобладают, как правило, симптомы ОНД, олигурия, нарушения гемодинамики.
Список использованной литературы 483
Пальпация живота не является эффективным приемом для определения повышения внутрибрюшного давления. Наиболее распространенный метод диагностирования внутрибрюшного давления — определение давления в мочевом пузыре.
J. М. Doty с соавт. [76] сообщают, что у больных после геморрагического шока с БКС развивается более выраженный внутриклеточный ацидоз и гипоперфузия эпителия слизистых желудка и кишок, однако без увеличения бактериальной транслокации. J. В. Rezende-Neto с соавт. [77] обнаружили, что внутрибрюшная гипертензия провоцирует продукцию провоспали-
15.8. ТРАВМЫ
Тяжелые травмы таза сопровождаются массивной кровопотерей, летальность в таких случаях достигает 35 %. В случае кровотечения из костей таза необходима срочная ангиография и эмболизация сосудов. Причиной тяжести состояния кроме кровопотери является быстрое развитие синд-
тельных цитокинов, которые могут служить вторичным фактором развития полиорганной недостаточности.
Абдоминальная декомпрессия является методом выбора лечения БКС. Она должна выполняться как можно раньше, если давление в брюшной полости (ВБД) превышает 25 мм рт. ст. [78]. После декомпрессии могут развиться осложнения, связанные с реперфузионными расстройствами органов брюшной полости. Перед декомпрессией важно скорригировать гиповолемию [78]. Сообщается, что ВБД было выше у умерших больных с травмой живота по сравнению с выжившими [79].
КОСТЕЙ ТАЗА
рома системного воспалительного ответа и полиорганной недостаточности. В определенной степени это касается и переломов бедренной кости, которые, как и переломы костей таза, часто осложняются жировой эмболией. При сочетанной травме показана ранняя хирургическая стабилизация.
Список использованной литературы
1. Rion В., Laudais Р., Vivien В. et al. Distribution of the probability of survival is a strategic issue for randomized trials in critically ill patients // Anesth. — 2001. — 95. - P. 56-63.
2. Elliot D. C., Rodriguer A. Coet effectiveness in trauma care // Surg. Clin. North Am. — 1996. - 76. - P. 47-62.
3. National Safety Council. Accident Facts. — 1994 Edition. — Itasca, II: National Safety Council, 1994.
4. Fleming A., Bishop M., Shoemaker W. et al. Prospective trial of supranormal values as goals of resuscitation in severe trauma //Arch. Surg. - 1992. - 214. - P. 289-297.
5. Cuthbertson D. P. Surgical metabolism: historical and evolutionary aspects // Metabolism and the Response to Injury / Ed. A. W. Wilkinson, D. P. Cuthbertson. — Chicago: Year Book, 1976. — P. 1—34.
6. Bauer M.t Marzi I., Ziegenfub T.f Riegel W. Prophylactic hemofiltration in severely traumatized patients: effects on post-traumatic organ dysfunction syndrome // Inten. Care Med. - 2001. - 27. - P. 376-383.
7. Lucas С. E. Resuscitation of the injured patient: the three phases of treatment // Surg. Clin. North. Am. - 1977. - 57. - P. 3-15.
8. Sauaia A., Moore F. A., Moore E. E. et al. Epidemiology of trauma death: a reassessment // J. Trauma. — 1995. — 38. — P. 185 — 193.
9. Rotondo M. F., Zonies D. H. The damage control sequence and underlying logic // Surg. Clin. N. Am. - 1997. - 77. - P. 761-777.
10. Reed R. L,, Ciavarella D., Heimbach D. M. et al. Prophylactic platelet administration during massive transfusion // Ann. Surg. — 1986. - 203. - P. 40-48.
11. Harke H., Rahman S. Haemostatic disorders in massive transfusion // Bibl. Haematol. — 1980. - 46. - P. 179-188.
12. Rotondo M. F., Reilly P. M. Bleeding and coagulation complication // Trauma / Ed. K. L. Mattox, D. V. Feliciano, E. E. Moore. — 4th ed. — New York: McGraw-Hill, 2000. — P. 1274-1275.
13. Lynn M., Jeroukhimov I., Klein Y. Coagulopathy in Trauma Patients // Yearbook of Intensive Care and Emergency Medicine / Ed. J.-L.Vincent. — Springer, 2002. — P. 661-669.
14. Knudson M. Use of low molecular weight heparin in preventing thromboembolism in trauma patients // J. Trauma. — 1996. — 41(3). - P. 446-459.
16*
484 ПОЛИТРАВМА
15. Hippala S. T., Myllyla G. L., Vahtera E. M. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates // Anesth. Analg. — 1995. — 81. — P. 360-365.
16. Gando 5., Kameue T., Morimoto У. et al. Marked reduction of protein C and antithrombin in post-trauma DIC have close relation with MODS and poor outcome // Crit. Care. — 2002. - 6 (Suppl. 1). - P. S55.
17. McCartney J. S. Pulmonary embolism following trauma // Am. J. Pathol. — 1934. — 10. - P. 709.
18. KirpatrickA. W., Dulchavsky C. A., Builan-ger B. R. et al. Extraterrestial resuscitation of hemorrhagic shock: Fluids //J. Trauma. — 2001. - 50. - P. 162-168.
19. Bickell W. Wall M. J., Pepe P. E. et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries // N. Engl. J. Med. — 1994. — 331. - P. 1105-1109.
20. Kovalenko T., Stern S., Dronen 5., Wang X. Improved outcome with hypotensive resuscitation of uncontrolled hemorrhagic shock in the swine model // J. Trauma. — 1992. — 33. - P. 349-353.
21. Capone A. C., Safar P., Stezoski W. et al. Improved outcome with fluid restriction in treatment of uncontrolled hemorrhagic shock // J. Am. Coll. Surg. - 1995. - 180. -P. 49-56.
22. Глумчер Ф. С., Макаров А. В., Скубрий В. M. Организация специализированной помощи при лечении тяжелой сочетанной травмы // Матер1али 2-го Нацюнального конгресу анестезюлопв Украши. — К.: Вища шк., 1996. - С. 23.
23. Глумчер Ф. С., Макаров А. В., Суслов Г. Г., Дубров С. А. Шок при травматических повреждениях // Пол1травма — сучасна кон-цепщя надання медично! допомоги: Тез. I Всеукр. наук.-практ. конф. (Кшв 16—17 травня, 2002). - К., 2002. - С. 13.
24. Healey М. A., Samphire J., Hoyt D. В. et al. Irreversible shock not irreversible: a new model of massive hemorrhage and resuscitation //Trauma. - 2001. - 50(5). - P. 826-834.
25. Shumaker W. C., Wo C. Invasive and non-invasive monitored effects of resuscitation fluids in the emergency department and operating room//Int. J. Inten. Care. — 1998. — 5(2). — P. 42-46.
26. Carash W. E., Fabian T. C., Croce M. A. Delayed surgical fixation of femur fractures is a risk factor for pulmonary failure independent of thoracic trauma // J. Trauma. — 1994. - 37(4). - P. 667-672.
27. Martinowitz U.t Kenet G., Segal E. et al. Recombinant activated factor VII for adjunctive hemorrhage control in trauma // J. Trauma. - 2001. - 51. - P. 431-439.
28. Ruiz A. J., Hill S. L.f Berry R. E. Heparin, deep venous thrombosis, and trauma patients //Am. J. Surg. - 1991. - 162. - P. 159.
29. Goldstein M. Traumatic Brain Injury: A silent epidemic // Ann. Neurol. — 1990. — 27. — P. 327.
30. Mauritz W. Management of Brain Trauma //Trauma Care. — 1998. — 8(2). — P. 50 — 55.
31. Lehr D.t Baethmann A., Reulen H. J. et al. Management of patients with sever head injury in preclinical phase//J. Trauma. — 1997. — 42(Suppl. 5). - P. S71-75.
32. Andrews В. T. The intensive care management of patients with head injury // Neurosurgical Intensive Care / Ed. В. T. Andrews. — New York: McGraw-Hill, 1993. - P. 227-242.
33. Egeler-Peerdeman S. M. Pathophysiology of head trauma // Manual of Neurosurgery / Ed. J. D. Palmer. — New-York: Churchill Livingstone, 1996. — P. 500 — 510.
34. Chesnut R. M. Secondary brain insults after head injury: clinical perspectives // New Horizons. — 1995. — 3. — P. 366 — 375.
35. Gentleman D., Dearden M., Midgley 5., Maclean D. Guidelines for resuscitation and transfer of patients with serious head injury // Br. Med. J. - 1993. - 307. - P. 547-552.
36. Jones P., Andrews P.t Midgley S. et al. Measuring the burden of secondary insults in head injured patients during intensive care // J. Neurosurg. Anesth. — 1994. — 6. — P. 4-14.
37. Levin H. 5., O’Donell V. M., Grossman R. G. The Galveston orientation and amnesia test. A practical scale to assess cognition after head injury // J. Nerv. Ment. Dis. — 1979. -167. - P. 675-684.
38. Teasdeale G., Jennet B. Assessment of coma and impaired consciousness. A practical scale //Lancet. - 1974. - 2. - P. 81-84.
39. Albanese J., Leone M., Martin C. Sever Head Injury in Patients with multiple Trauma // Yearbook of Intensive Care and Emerjency Medicine / Ed. J.-L. Vincent. — Heildelberg: Springer-Verlag, 2001. — P. 353 — 373.
40. Valentin A., Lang T., Kamik R., Ammerer H. P. et al. Intracranial pressure monitoring and case mix-adjusted mortality in intracranial hemorrhage // Crit. Care Med. — 2003. -31(5). - P. 1539-1542.
41. Thees C., Scheufler К. M., Nadstawek J. et al. Monitoring of cerebral perfusion pressure during intracranial hypertension: a sufficient
Список использованной литературы 485
parameter of adequate cerebral perfusion and oxygenation? // Inten. Care Med. — 2003. — 29(3). - P. 386-390.
42. Robertson C. S., CormioM. Cerebral metabolic management//New Horizons. — 1995. — 3. — P. 410-422.
43. The Brain trauma Foundation: Guidelines for the management of sever head injury // J. Neurotrauma. — 1996. — 13. — P. 639— 734.
44. Miller J. D., Sweet R. C., Narayan R., Becker D. P. Early insults to the injured brain //JAMA. - 1978. - 240. - P. 439-442.
45. Muizelaar J. P., MarmarouA., Ward J. D. et al. Adverse effects of prolonged hyperventilation in patients with sever head injuriy // J. Neurosurg. — 1991. — 75. — P. 731—739.
46. Reinprecht A., Greher M., Wolfsberger S. et al. Prone position in subarachnoid hemorrhage patients with acute respiratory distress syndrome: effects on cerebral tissue oxygenation and intracranial pressure // Crit. Care Med. - 2003. - 31(6). - P. 1831-1838.
47. Пилипенко M. M.t Шлапак I. П., }Псянсь-кий M. С. Сучасн! аспекти ресшраторноТ шд-тримки при тяжкш черепно-мозковш травм! // Б!ль, знеболювання i штенсивна тера-шя. - 2003. - № 2. - С. 31-43.
48. Hemmer М. Fluid administration in sever head injury // Yearbook of Intensive Care and Emerjency Medicine / Ed. J.-L. Vincent. — Heildelberg: Springer-Verlag, 1993. — P. 353-373.
49. Clifton G. L., Miller E. R., Choi S. C., Levin H. S. Fluid thresholds and outcome from severe brain injury // Crit. Care Med. — 2002. - 30(4). - P. 739-745.
50. Eisenberg H. M.t Frankowski R. F., Con-tant C. F. et al. High-dose barbiturate control of elevated intracranial pressure in patients with sever head injuries // J. Neurosurg. — 1988. - 69. - P. 15-23.
51. Albanese J., Arnaud S., Rey M. Ketamine decreases intracranial pressure and electroencephalogram activity in traumatic brain injury patients during propofol sedation // Anesth. - 1997. - 87. - P. 1328-1334.
52. Prielipp R. C., Coursin D. B. Sedative and neuromuscular blocking drug use in critically ill patients with head injuries // New Horizons. - 1995. - 3. - P. 456-468.
53. Papasian L., Albanese J., Thirion X. Effects of bolus doses of midazolam on intracranial pressure and cerebral perfusion pressure in patients with sever head injuries // Br. J. Anaesth. — 1993. - 71. - P. 267-271.
54. Hsiang J. K., Chesnut R. M., Crisp С. B. et al. Early, routine paralysis for intracaranial pressure control in sever head injuries: is it ne
cessary? //Crit. Care Med. — 1994. — 22. — P. 1471-1476.
55. Donato T., Shapira У., Artra A. et al. Effect of mannitol on cerebrospinal fluid dynamics and brain tissue edema // Anesth. Analg. — 1994. - 78. - P. 58-66.
56. Bullok R. Mannitol and other diuretics in sever neurotrauma // New Horizons. — 1995. — 3. - P. 448-452.
57. Polderman К. H., van de Kraats G., Dixon J. M. et al. Increases in spinal fluid osmolarity induced by mannitol // Crit. Care Med. — 2003. - 31(2). - P. 584-590.
58. Vialet R.t Albanese J., Thomachot L. et al. Isovolume hypertonic solutes (sodium chloride or mannitol) in the treatment of refractory posttraumatic intracranial hypertension: 2 mL/kg 7.5 % saline is more effective than 2 mL/kg 20 % mannitol // Crit. Care Med. — 2003. - 31(6). - P. 1683-1687.
59. Marion D. W., Penrol L. E.t Kesley S. F. et al. Treatment of traumatic brain injury with moderate hypotermia // N. Engl. J. Med. — 1996. - 336. - P. 540-546.
60. Brundage S. McGhan R., Jurkovich G. J. et al. Timing of femur fracture fixation: effect on outcome in patients with thoracic and head injuries // J. Trauma. — 2002. — 52(2). — P. 299-307.
61. JaicksR. R., CohnS. M., Moller B. A. Early fracture fixation may be deleterious after head injury // J. Trauma. — 1997. — 42. — P. 1-5.
62. Asgeirsson B.t Grande P. O., Nordstrom С. H. A new therapy of post-trauma brain oedema based on haemodynamic principles for brain volume regulation // Inten. Care Med. — 1994. - 20. - P. 260-267.
63. J eremitsky E.f Omert L., Dunham С. M. et al. Harbingers of poor outcome the day after severe brain injury: hypothermia, hypoxia and hypoperfusion // J. Trauma. — 2003. — 54(2). - P. 312- 319.
64. Marini J. J., Wheeler A. P. Critical Care Medicine. — Philadelphia, Willias&Wilkins, 1997. - P. 640.
65. Dimopoulou I., Anthi A., Lignos M. et al. Prediction of prolonged ventilatory support in blunt thoracic trauma patients // Inten. Care Med. - 2003. - 29(7). -P. 1101 — 1105.
66. Глумчер Ф. С., Вольхина И. А., Сергиенко А. В. Профилактика и лечение больных с синдромом острого повреждения легких вследствие травмы // Б!ль, знеболювання i штенсивна терашя. — 2001. - № 3 (16). - С. 2-7.
67. Richter М., Krettek С., Otte D. et al. Correlation between crash severity, injury severe-
486 ПОЛИТРАВМА
ty, and clinical course in car occupants with thoracic trauma: a technical and medical study //J. Trauma. - 2001. - 50(1). - P. 10-16.
68. Moore F. A., Haenel J. B. Flail chest and pulmonary contusion // Critical Care Secrets / Ed. P. E. Parsons, J. P. Wiener-Kronish. — Philadelphia: Hanley&Belfus, 1998. — P. 396 — 405.
69. Ong T. H., Eng P. Massive hemoptysis requiring intensive care // Inten. Care Med. — 2003. - 29(2). - P. 317-320.
70. Messenger J. C., Wolfel E, E. Myocardial contusion // Critical Care Secrets / Ed. P. E. Parsons, J. P. Wiener-Kronish. — Philadelphia: Hanley&Belfus, 1998. — P. 405 — 409.
71. Karalis D. G., Victor M. F., Davis G. A. et al. The role of echocardigraphy in blunt chest trauma // J. Trauma. — 1994. — 36. — P. 53-58.
72. Duke J. C. Blunt Cardiac Trauma // Trauma Care. - 2001. - 11(2). - P. 69-72.
73. Adams J. E., Davila-Roman V. G.t BesseyP. Q. et al. Improved detection of cardiac contusion with cardiac troponin // I. Am. Heart J. — 1996. - 131. - P. 308-312.
74. Ferjani M., Droc G., Dreux 5. et al. Circulating cardiac troponin T in myocardial contusion//Chest. - 1997. - 111. - P. 427-433.
75. Wisner D. H., Reed W. H., Riddick R. S. Suspected myocardial contusion: triage and indication for monitoring // Ann. Surg. — 1990. - 212. - P. 82-86.
76. Doty J. M., Oda J., Ivatury R. R. et al. The effects of hemodynamic shock and increased intra-abdominal pressure on bacterial translocation//J. Trauma. — 2002. — 52(1). — P. 13-17.
77. Rezende-Neto J. B.t Moore E. E.t Melo de Andrade M. V. et al. Systemic inflammatory response secondary to abdominal compartment syndrome: stage for multiple organ failure // J. Trauma. - 2002. - 53(6). - P. 1121 — 1128.
78. PottecherT., Segura P., Launoy A. Abdominal compartment syndrome // Ann. Chir. — 2001. - 126(3). - P. 192-200.
79. Pouliart N., Huyghens L. An observational study on intraabdominal pressure in 125 critically ill patients // Crit. Care. — 2002. — 6 (Suppl. 1). - P. S3.
= Раздел == 16 Интенсивная терапия оЖоговой болезни
Лечение ожоговой болезни имеет давнюю историю, однако интерес к нему не ослабевает. Это связано с увеличением частоты получения ожогов в быту и на производстве, в условиях катастроф мирного времени и региональных военных конфликтов, со сложностью патогенеза и лечения ожоговой болезни, а главное — с высокой летальностью при ожоговой болезни. Лечение ожоговых больных значительно улучшилось после окончания второй мировой войны. Наиболее важные нововведения заключались в агрессивном восполнении жидкости, ранних некрэктомиях и применении трансплантатов в лечении ожоговых ран, использовании более эффективных антибиотиков, достижений энтеральной нутритивной поддержки и в появлении многодисциплинарных ожоговых центров. При условии своевременного лечения шансы на выживание у большинства пациентов с площадью ожогов 80 % поверхности тела и более повышаются. К факторам, увеличивающим риск летальности у больных с термическими повреждениями, относятся: возраст свыше 60 лет; ожоги площадью 40 % поверхности тела и более; сопутствующее ин
галяционное поражение дыхательных путей.
Под ожоговой болезнью подразумевают комплекс патологических изменений, возникающих в организме в ответ на воздействие термического агента.
В ожоговой болезни выделяют четыре периода:
I — ожоговый шок (от нескольких часов до двух-трех суток);
II — токсемия (с 3-х до 10 — 15-х суток после ожога) — наблюдаются явления выраженной интоксикации;
III — септикотоксемия (с 10—15-х суток после ожога до восстановления кожных покровов) — характеризуется развитием инфекционных осложнений;
IV — реконвалесценция (наступает после восстановления целостности кожных покровов).
Такое деление ожоговой болезни на периоды является условным, тем более что внедрение новых технологий лечения позволяет существенно ослабить проявления ожоговых токсемии и септикотоксемии. Поэтому в последнее время ожоговый шок и токсемию объединяют в единое понятие «острый период ожоговой болезни».
16.1. ЭТИ0ПАТ0ГЕНЕЗ
Ожоговое повреждение вызывает немедленное, тотальное или частичное повреждение кожи и расположенных под ней тканей.
Нагревание кожи и развитие термических ожогов зависят от источника тепла. Перенос тепла осуществляется двумя путями:
488 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
конвекцией (воздействие горячего пара или газа);
проведением (прямой контакт с горячим предметом или жидкостью).
Изменения в тканях зависят от уровня их нагревания. Если воздействующая температура не превышает 60 °C, возникает влажный, или колликвационный, некроз. При более интенсивном прогревании ткани высыхают и развивается сухой, или коагуляционный, некроз. Поскольку интенсивность прогревания тканей на разных участках неодинакова, то эти разновидности некроза комбинируются в ожоговой ране в различных сочетаниях. Таким образом, по механизму возникновения ожоги можно разделить на шесть категорий (У. Эймс, 2000): ♦ контакт — прямой контакт с горячей поверхностью;
♦ ошпаривание — воздействие горячей жидкости/газа (обычно вызывает поверхностный ожог);
♦ вспышка — быстрый ожог (обычно на всю глубину тканей);
♦ пламя — обычно на всю глубину;
♦ химический;
♦ электрический.
Ожоги разрушают кожу как непосредственно при воздействии источника тепла, так и косвенно (вторично) — в результате ишемических нарушений в тканях. Гибель клеток в ожоговой ране обусловлена прогрессирующей денатурацией клеточного протеина, приобретающей необратимый характер при температуре свыше 45 °C. Одновременно происходит инактивация клеточных энзимов респираторной цепи. Клетка погибает в случае, если более 50 % их инактивируется. К гибели клеток приводит также разрыв клеточных мембран вследствие чрезмерного увеличения внутриклеточного объема жидкости. Таким образом, пусковым механизмом патологических изменений при ожоговой болезни являются разнообразные морфологические и функциональные изменения в области ожоговой раны (местная реакция).
Ожоговая рана имеет три зоны, описанные Джексоном в 1953 г.:
♦ некроза и коагуляции;
♦ ишемии и стаза;
♦ гиперемии.
Зона коагуляционного некроза — это участок, где высокая температура разрушила морфоструктуру и кровообращение в тканях. Зона ишемии и стаза примыкает к зоне коагуляции и некроза. Кровообращение в ней замедлено до стаза. В этой зоне ткани еще сохраняют свою жизнедеятельность, но сосудистая сеть уже повреждена, и клетки эндотелия экспрессируют поверхностные рецепторы молекул адгезии. Вследствие этого активируются нейтрофилы, запускающие при контакте с тромбином процесс необратимой коагуляции. Эти процессы с неизбежностью приводят к дальнейшему нарушению микроциркуляции, дополнительному повреждению эндотелия и тромбозу (рис. 16.1), усиливающему тканевую ишемию. Полное прекращение кровотока (стаз) сопровождается тромбозом и увеличением зоны первичного некроза.
Зона стаза и ишемии окружена зоной гиперемии, в которой клетки и сосуды повреждены обратимо, усилен кровоток, эффективна микроциркуляция. Практически сразу же после воздействия термического фактора в обожженных участках кожи происходят существенные изменения: содержание в них воды увеличивается на 75 %, натрия — на 100 %, белков в интерстициальной жидкости — на 350 %.
Все поврежденные в той или иной степени ткани вырабатывают или высвобож-
Инфекция или травма
Гуморальный ответ -------► Ответ клетки
Сладжи
Кружащиеся
Увеличение нейтрофилы проницаемости Агрегаты
Повреждение ^^°леКул^*Им1™ энДотелиальныхЮх^г13"и_Х^ клеток*
Увеличение NO п ♦
Дилатация артериолТ^ууу^ув
Рис. 16.1. Схема предполагаемых нарушений микроциркуляции в различных органах при системном воспалительном ответе
Этиопатогенез 489
дают различные химические медиаторы воспаления (кинины, гистамин, тромбоксан, простагландины, лейкотриены, свободные кислородные радикалы, липоперекиси). Среди медиаторов воспаления важную роль играют производные арахидоновой кислоты, которая входит в состав всех клеточных мембран и освобождается из них под воздействием фосфолипазы А2, появляющейся в больших количествах при термическом повреждении тканей. Каскад превращений арахидоновой кислоты осуществляется двумя путями: циклооксигеназным и липооксигеназным.
При циклооксигеназном пути окисления арахидоновой кислоты образуются короткоживущие эндопероксидазы: PGG2 и PGH2, метаболизирующиеся в тромбоксан (ТхА2), простациклин (PGI2) или простагландины (PGD2, PGE2, PGF2a). При липооксигеназном пути окисления свободной арахидоновой кислоты образуются эндопероксидазы (НРЕТЕ), в дальнейшем превращающиеся в аналоги алкоголя либо в лейкотриены.
Метаболиты арахидоновой кислоты активно влияют на микроциркуляцию. Так, тромбоксан А2 вызывает спазм микрососудов и стимулирует агрегацию тромбоцитов. Простациклин, способствующий активному расширению сосудов, в то же время является сильным ингибитором агрегации тромбоцитов. Простагландин Е2 — сильный вазодилататор, а простагландин F2a — вазоконстриктор. Лейкотриены LTC4, Д4, Е4 в 1000 — 5000 раз превышают сосудопроницаемое действие гистамина. Нарушения проницаемости сосудистой стенки в виде патологических пор в эндотелии способствуют активному выходу внутрисосудистой жидкости в межклеточное пространство, а затем — ее поступлению в клетку, что вызывает отек. Гипергидратация клеток ведет к разрыву клеточных мембран и гибели клеток, что увеличивает зону первичного некроза.
Повышение в крови уровня таких главных воспалительных цитокинов, как интерлейкины — 1,6,8 (ИЛ-1, ИЛ-6, ИЛ-8), свидетельствует о переходе местной воспалительной реакции в общую, т. е. в синдром системного воспалительного ответа.
Последний при неблагоприятном течении ожоговой болезни приводит к полиорган-ной недостаточности.
Ожоговое поражение сопровождается болевым синдромом и психоэмоциональным стрессом, запускающими нейроэндокринный ответ в организме пострадавшего. Клинически это проявляется спазмом сосудов, повышением общего периферического сосудистого сопротивления и централизацией кровообращения, что ухудшает системную тканевую перфузию, приводит к тканевой гипоксии и ацидозу, которые увеличиваются с ростом гиповолемии и гемоконцентрации .
Гиповолемия формируется различными механизмами:
1. Повышенная проницаемость сосудистой стенки обуславливает уменьшение ОЦК вследствие перехода жидкой части крови (плазмы) из русла в интерстициальное пространство как обожженных, так и неповрежденных тканей.
2. Увеличение содержания белка и повышение онкотического давления в интерстициальном пространстве способствуют активному поступлению в него жидкости из сосудов, что приводит к снижению ОЦК.
3. Повышенное осмотическое давление в обожженных тканях вызывает усиление притока жидкости в пораженную зону и увеличение ее отека, вследствие чего снижается ОЦК.
4. Нарушение функции клеточных мембран необожженных тканей ведет к пропотеванию интерстициальной жидкости в клетки.
Прогрессирующая гиповолемия способствует развитию ожогового шока, под которым понимают острую сердечно-сосудистую недостаточность, обусловленную преимущественно потерей плазмы. Различают четыре степени тяжести ожогового шока.
Клиническая картина. Ведущими симптомами ожогового шока являются олигоанурия, низкая температура тела и, в последнюю очередь, падение артериального давления. Основные клинические признаки ожогового шока представлены в табл. 16.1.
Первая и вторая степени ожогового шока чаще наблюдаются у пострадавших
490 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
Таблица 16.1. Клинические признаки ожогового шока
Признак Степень тяжести ожогового шока
I II III IV
Симптом «белого 1 — 2 2-3 > 3 > 3
пятна»,с Температура тела Субфебрильная Нормальная < 36-35 °C < 35 °C
Пульс, уд/мин или нормальная До 100 90-110 > 120 Брадикардия
АД систолическое, 140-120 110-90 < 90 < 70
мм рт. ст. ЦВД, см вод. ст. 8-4 8-4 4-0,0 Отрицательное
Диурез, мл/ч Кратковременная 30 < 30 Анурия
Парез кишок задержка до начала инфузии Отсутствует Имеется Имеется Имеется
молодого и среднего возраста с неотяго-щенным анамнезом, при ожогах 15 —20 % поверхности тела. При неглубоких поражениях кожи больные испытывают сильную боль, поэтому они могут быть возбуждены. Гемодинамические показатели не выходят за нижние границы нормы, поча-совый диурез не снижен. Если инфузионная терапия запаздывает на 6 — 8 ч, развиваются гемоконцентрация и олигурия.
Третья степень ожогового шока (тяжелый ожоговый шок) возникает при поражении 21 —60 % всей поверхности тела. Для нее характерны быстро развивающиеся заторможенность и адинамия при сохраненном сознании. Гемодинамика поддерживается инфузионной терапией с использованием вазопрессоров. Больные жалуются на жажду, диспепсию. Может быть парез кишечника, острое расширение желудка. Выражены олигурия, поддающаяся медикаментозной коррекции, гемоконцентрация (гематокрит достигает 65 %), умеренный метаболический ацидоз с респираторной компенсацией.
Четвертая степень ожогового шока (крайне тяжелый ожоговый шок) развивается при поражении более 60 % полной поверхности тела. Сознание нарушено до сопора. Пульс нитевидный. Гемодинамика нестабильная, с тенденцией к стойкой гипотонии, не поддающейся медикаментозной коррекции. Дыхание поверхностное,
частое. Как правило, наблюдаются рвота, парез кишечника. Первые порции мочи с признаками гематурии, быстро наступает анурия. Гемоконцентрация с гематокритом свыше 70 %. Нарастают гиперкалиемия и некомпенсированный смешанный ацидоз.
Диагностика. Основана на индексе тяжести поражения (ИТП), клинических данных и лабораторных показателях. ИТП рассчитывают, исходя из глубины и площади поражения.
Глубину ожогового поражения определяют в зависимости от степени повреждения кожных покровов:
I — эритема (покраснение и отек кожи);
II — фликтена (гиперемия и отек кожи с отслойкой эпидермиса и образованием пузырей);
II 1а — дермальный ожог или частичный некроз кожи (ростковый слой кожи сохранен);
III6 — тотальный некроз кожи (ростковый слой утрачен полностью)
IV — омертвление не только кожи, но и глубже расположенных слоев.
Площадь ожоговой поверхности оценивают согласно введенному Wallace «правилу девяток», устанавливающему, какой процент от площади полной поверхности тела приходится на передние и задние поверхности головы, конечностей и туловища: голова и шея — 9 %, руки — по 9,
Этиопатогенез 491
Таблица 16.2. Токсичные компоненты дыма
Материал Токсичный продукт Воздействие на организм
Дерево, бумага, хлопок Акролеин, этаналь, метаналь, уксусная кислота Раздражение верхних дыхательных путей, бронхоспазм. Некроз слизистых оболочек
Пластик Фосген, хлор, хлористый водород Раздражение верхних дыхательных путей. Острое поражение легких
Синтетические материалы (нейлон, искусственный шелк) Гидроген цианид, окислы нитрогена Отравление цианидами, тканевая гипоксия. Отек легких
Все вышеперечисленные Угарный газ Тканевая гипоксия
передняя и задняя поверхность туловища — по 18, ноги — по 18, промежность — 1 %. Для учета частично обожженных областей можно использовать площадь ладони потерпевшего, которая приравнивается к 1 % от общей поверхности тела.
Ожог считается обширным, если его площадь составляет 15 — 20 % и более от полной поверхности тела (ППТ) для взрослых и 5—10 % — для новорожденных и детей младшего возраста. Наличие ожогов такой площади требует госпитализации в ОИТ.
Критерии госпитализации для взрослых пострадавших (по Дж. Марини, 2002):
ожог II степени, охватывающий более 20 % ППТ;
ожог III степени, охватывающий свыше 5 % ППТ;
любой ожог II или III степени у больных старше 60 лет;
ингаляционные поражения;
ожоги по окружности туловища или конечностей;
ожоги руки, лица, ног или промежности.
Площадь ожога также можно определить путем нанесения изображения ран на схему Долинина, в которой каждый отдельный фрагмент соответствует 1 % поверхности тела, или с помощью схемы Лунда— Браудера, учитывающей возрастные изменения соотношения частей тела.
Сопутствующее поражение органов дыхания пагубно отражается на состоянии пострадавшего и ухудшает прогноз. В зависимости от тяжести поражения
ожоги дыхательных путей (ОДП) можна разделить на три степени:
♦ легкая — респираторные расстройства в первые сутки не отмечаются;
♦ средняя — респираторные расстройства наблюдаются в первые 6—12 ч после ожога;
♦ тяжелая — дыхательная недостаточность выражена с момента получения ожога.
В зависимости от вида горящих материалов выделяют токсичные вещества, поражающие дыхательные пути или вызывающие системное отравление организма (табл. 16.2).
Следует помнить, что угарный газ (СО) является первопричиной смерти жертв пожаров в 75 % случаев. Клинические проявления при остром отравлении СО такие же, как и при гипоксии тканей, и хорошо коррелируют с уровнем карбоксигемоглобина в крови (табл. 16.3.)
Определив глубину и площадь поражения, устанавливают тяжесть ожогового
Таблица 16.3. Корреляция между уровнем НЬСО и признаками отравления СО
Уровень карбоксигемоглобина, % Симптомы отравления
Менее 15 Обычно отсутствуют
15-20 Головная боль, спутанность сознания
21-40 Дезориентация,ослабление зрения, рвота
41-60 Галлюцинации, кома, шок
Более 60 Смерть
492 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
шока, используя индекс тяжести поражения (ИТП). При этом:
1 % ожога I и II степени принимают за 1 ед. ИТП;
1 % ожога Ша степени — за 2 ед. ИТП;
1 % ожога Шб степени — за 3 ед. ИТП;
1 % ожога IV степени — за 4 ед. ИТП.
При наличии поражения дыхательных путей к полученному результату добавляют:
при ОДП легкой степени — 15 ед. ИТП;
при ОДП средней степени — 30 ед. ИТП;
при ОДП тяжелой степени — 45 ед. ИТП.
За каждый год после 60 лет прибавляют 1 ед. ИТП.
С учетом полученного результата ожоговый шок по степени тяжести подразделяют на:
♦ легкий — ИТП до 30 ед.;
♦ средней тяжести — ИТП 31—60 ед.;
♦ тяжелый — ИТП 61—90 ед.;
♦ крайне тяжелый — ИТП свыше 90 ед.
16.2. ЛЕЧЕНИЕ ОСТРОГО ПЕРИОДА ОЖОГОВОЙ БОЛЕЗНИ
Оказание помощи при тяжелых ожогах можно разделить на три этапа:
первый — догоспитальный, или первоочередные мероприятия, — оказание помощи на месте происшествия и транспортировка в ближайшее лечебно-профилактическое учреждение;
второй — госпитальный, или очередные мероприятия, — купирование острых проявлений шока, обработка раны и пере-транспортировка в специализированный ожоговый центр;
третий — специализированный, или отсроченные мероприятия, — лечение острого периода ожоговой болезни в специализированном ожоговом центре.
Таким образом, алгоритм лечения острого периода ожоговой болезни включает:
1. Первоочередные мероприятия (догоспитальный этап)
Цель: первая помощь и восстановление водных секторов организма.
1.1. Устранение воздействия повреждающего фактора
Первая помощь должна быть оказана немедленно на месте происшествия. Прежде всего необходимо прекратить действие термического фактора. Поскольку повреждающее термическое действие продолжается и после прекращения непосредственного влияния открытого пламени, то рекомендуется как можно быстрее остудить обожженную поверхность прохладной проточной водой (15 °C) в течение 10 — 15 мин, аппликацией охлажденных предметов (криопакетов, грелок с холодной во
дой и др.) или орошением веществами с низкой температурой кипения. Но при обширных ожогах следует помнить об опасности развития гипотермии, особенно у детей и пожилых больных.
Необходимо как можно быстрее удалить с пострадавшего тлеющую одежду и одежду, пропитанную химикатами. Если сгоревшая одежда прилипла к телу, то удалять ее нецелесообразно. У пациентов, подвергшихся химическому ожогу, следует в течение не менее 30 мин обильно промывать пострадавшую кожу прохладной водой.
Охлаждение ожоговой поверхности патогенетически обосновано в первые два часа после ожога, поскольку оно приводит к резкому уменьшению разрушающего действия тепла на ткани, спазму сосудов и уменьшению отека, инактивирует выделение гистамина, кининов, простагландина Е2, уменьшает образование молочной кислоты. Охлаждение проводят сразу же после получения травмы как элемент первой помощи или при поступлении в отделение, если до этого такая процедура не была произведена. Сама процедура заключается в орошении пораженных поверхностей охлажденными растворами антисептиков или в применении пеленок, смоченных холодной водой, до того момента, когда они перестанут быстро нагреваться до температуры тела. После этого зону ожога укрывают чистыми простынями или пеленками, сухими марлевыми или контурными повязками, в крайнем случае чистой сухой одеждой.
Лечение острого периода ожоговой болезни 493
Наложение любых мазевых повязок противопоказано!
Пострадавшего необходимо согреть. Можно дать горячий чай. При сильной жажде дать выпить раствор регидрона, минеральную воду или щелочно-солевой раствор (1,5 — 2 г питьевой соды и 3 — 4 г поваренной соли на 1 л воды).
Бригада скорой медицинской помощи, прибывшая на место происшествия, должна быстро оценить ситуацию и очередность проводимых мероприятий.
1.2. Обезболивание
Для снятия болевого синдрома больным с сохраненным спонтанным дыханием показаны агонисты, агонисты —антагонисты морфиновой группы, купирующие боль, не угнетая при этом дыхания: нальбуфин (иубаин) — 1 % или 2 % — 1,0 мл; норфин (аифин) — 0,2 % — 1,0 мл; бупренорфин (бупремен, нопен, торгезик) — 0,2 % — 1,0 мл; стадол — 0,2 % — 1,0 мл каждые 3 — 4 часа. Возможно использование трамадола, анальгина в сочетании с антигистаминными препаратами.
Тревога и беспокойство, характерные для ожоговых больных, могут быть устранены небольшими дозами бензодиазепинов (си-базон, реланиум, седуксен, диазепам) или нейролептиков (дроперидол, галоперидол), вводимыми в дополнение к анальгетикам. В случае недостаточной эффективности проводимой терапии показан медикаментозный сон, достигаемый внутривенным введением оксибутирата натрия (50—100 мг/кг массы тела). Использование наркотических анальгетиков возможно только при тяжелых и крайне тяжелых поражениях.
1.3. Обеспечение адекватного газообмена
Необходимо проверить и обеспечить проходимость дыхательных путей и дыхание кислородом (4 — 8 л/мин) через маску. В случае любого клинического проявления обструкции дыхательных путей интубация трахеи и ИВЛ показаны больным с тяжелыми ожогами лица и шеи II или III степени, при глубоких ожогах, захватывающих более 60 % поверхности тела, при обнаружении признаков ожога дыхательных путей (сажа в носовых ходах, охриплость голоса, стридорозное дыхание).
1.4. Начало инфузионной терапии
К инфузионной терапии приступают на месте происшествия. Она заключается во внутривенном струйном введении солевых сбалансированных растворов через катетеры диаметром 1,2 — 1,4 мм, установленные в одну или две периферические вены. Объем и темп инфузии определяются тяжестью состояния больного и суточным объемом жидкости, необходимым для возмещения дефицита ОЦК. При оказании помощи на догоспитальном этапе используют исключительно кристаллоиды, в которых основным ионом является натрий. Кристаллоиды могут использоваться в виде гипертонического, изотонического или гипотонического раствора.
Для расчета необходимого объема инфузии (Уинф) на догоспитальном этапе используют модифицированную формулу Паркланда:
Уинф (мл) = 2 (мл) • Масса тела больного (кг) • Площадь ожога (%).
Рассчитанный объем вводят в течение первых 6 —8 ч после ожога.
1.5. Транспортировка пострадавшего
Показана всем больным. Своевременная и хорошо организованная эвакуация пострадавших является важным фактором дальнейшего лечения. Транспортировку обожженных рекомендуется проводить врачебно-фельдшерской бригадой машины скорой медицинской помощи после выполнения пунктов 1.1 —1.4 настоящих рекомендаций.
2. Очередные мероприятия {госпитальный этап)
Цель: лечение и профилактика органной дисфункции и недостаточности.
2.1. Обеспечение адекватного газообмена
При поступлении пострадавшего в стационар продолжаются оксигенотерапия и ИВЛ, проводится санация трахео-бронхи-ального дерева, а при подозрении на ожоги дыхательных путей — диагностические ларингоскопия или бронхоскопия. Наличие отека надгортанника является показанием к обязательной интубации. Гипоксемия, диффузные инфильтраты на рентгенограмме — прогностические при
494 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
знаки, указывающие на необходимость ранней интубации и ИВЛ.
2.2. Продолжение инфузионной терапии
Поскольку ожоги вызывают гиповолемию вследствие массивного перемещения жидкости из внутрисосудистого пространства во внесосудистое и экссудации ее через поврежденную кожу, то на госпитальном этапе проведения инфузионной терапии руководствуются следующими правилами:
1. Темп инфузии и компонентность растворов определяют временем, прошедшим от момента поражения.
2. Первые 24 ч ожогового шока:
а) суточный объем инфузии рассчитывают по формуле Паркланда
Уинф (мл) = 4 (мл) • Масса тела больного (кг) • Площадь ожога (%).
При сопутствующем ожоге дыхательных путей жидкости может потребоваться на 50 % больше (до 6 мл/кг на 1 % поверхности ожога от ППТ)\
б) скорость инфузии:
50 % расчетного объема вводят в первые 8 ч;
25 % — в последующие 8 ч;
25 % — в третьи 8 ч.
в) компонентность растворов: в первые 24 ч используют кристаллоиды (раствор Рингера-лактата или нормотонический солевой раствор).
Быстрое восполнение объема сосудистого русла солевыми растворами ликвидирует спазм сосудов, уменьшает вязкость крови, обеспечивает производительность миокарда, снижает степень ацидотических сдвигов.
Такие растворы являются наполнителями внеклеточного сектора и обеспечивают оптимальное перераспределение электролитов между клеточными и внеклеточными секторами — основу коррекции нарушений при ожоговом шоке. Электролитные растворы интенсифицируют обмен тканевой жидкости за счет увеличения лимфообразования и лимфодренажа в тканях с редуцированной микроциркуляцией. Введение больших количеств электролитных растворов в первые часы после травмы обеспечивает быстрое наполне
ние венозного отдела сосудистого русла, создает так называемый венозный подпор, что способствует адекватному сердечному выбросу. Все методы возмещения потери жидкости сопровождаются развитием отека тканей, однако при вливании гипертонических солевых растворов (250 ммоль/л) они выражены в большей степени, что имеет отрицательные последствия.
Отрицательные последствия формирования отеков (К. К. Nagy с соавт.):
сдавление капилляров;
снижение капиллярного тока крови;
нарушение транспорта кислорода и питательных веществ;
увеличение массы тела;
увеличение срока пребывания на ИВЛ; ухудшение заживления ран;
увеличение срока пребывания в отделении интенсивной терапии.
Однозначного мнения относительно использования коллоидов нет. Высокомолекулярные коллоиды, в частности поли-глюкин, в первые часы после травмы применять нецелесообразно из-за их высокой вязкости, агрегатного действия, низкой скорости выведения из организма, образования коллоидной губки в микроциркуля-торных зонах, что резко ухудшает ранее нарушенный лимфодренаж.
Из низкомолекулярных коллоидов вводят препараты на основе ГЭК (до 20 мл/кг), например рефортан, инфекол, HESS — 6 %, которые обеспечивают:
изоволемическое (6 %) или гиперволемическое (10 % раствор) и объемозамещающее действие в течение 4 — 6 ч после введения;
восстановление гемодинамики (увеличение сердечного индекса, повышение давления заполнения сосудов, а также увеличение доставки и потребления кислорода);
предотвращение развития синдрома повышенной проницаемости капилляров путем закрытия пор в их стенках («латание» стенки капилляров);
предотвращение активизации эндотелиальных клеток и блокирование развития вторичных поражений тканей;
защиту моноцитов крови, играющих важную роль в системе клеточного и гуморального иммунитета;
Лечение острого периода ожоговой болезни 495
отсутствие активации системы комплемента;
нормальные проявления реакций иммунитета вследствие отсутствия воздействия на экспрессию поверхностных антигенов крови.
П. Гуньо [2] рекомендует в первые 24 ч после ожога следующую инфузионную тактику:
первые 6ч — введение раствора Рингера (1 мл/кг на 1 % площади ожога от ППТ);
последующие 18 ч — сочетанное применение кристаллоидов (1 мл/кг на 1 % площади ожога от ППТ) и коллоидов (1 мл/кг на 1 % площади ожога от ППТ).
В первые 24 ч после ожоговой травмы общий объем инфузий не должен превышать 4 мл/кг на 1 % площади ожога от ППТ!
Растворы глюкозы в первые сутки после получения тяжелой ожоговой травмы не используют, так как вследствие нарушения функции капиллярной мембраны они проникают в межклеточное пространство и способствуют образованию отеков, нарушению питания тканей и углублению уже имеющихся повреждений. Через 2 — 4 часа от начала инфузионной терапии электролитными растворами целесообразно применение плазмозаменителя, обладающего функцией переноса кислорода, — перфторана в дозе 2,5 —3,5 мл/кг на одно введение.
Однократное введение перфторана позволяет снизить общую летальность при ожоговом шоке средней степени тяжести на 20,2 %, двукратное — способствует дополнительному снижению общей летальности на 12,6 %.
2.3. Коррекция агрегатного состояния крови
Предполагает применение низкомолекулярных гепаринов (НМГ): фраксипари-на, клексана, фрагмина или нефракциони-рованного гепарина (НФГ) уже в первые часы после ожоговой травмы. НМГ используют в умеренных дозах только внутривенно, что обусловлено нарушенной тканевой перфузией. В частности, фраксипа-рин вводят внутривенно в дозе 0,3 мл один или два раза в сутки. Первоначальная
болюсная доза гепарина составляет 5 — 10 тыс. ЕД (100 ЕД/кг) с последующей внутривенной инфузией из расчета 1 — 2 тыс. ЕД/ч. В случае невозможности постоянной инфузии препарат вводят по 5 тыс. ЕД. каждые 4 —6 ч. Критерием эффективности проводимой терапии являются активированное частичное тромбопластическое время (АЧТВ) и содержание тромбоцитов.
Уменьшение агрегации форменных элементов крови достигается внутривенным введением таких дезагрегантов, как:
трентал (пентоксифиллин) — по 200 — 400 мг внутривенно капельно на 400 мл физиологического раствора 1—2 раза в сутки;
ксантинола никотинат — по 2 мл 15 % раствора внутривенно 1—3 раза в сутки;
аклотин — по одной таблетке (250 мг) 2 раза в сутки во время еды.
2.4. Лечение и профилактика полиор-ганной дисфункции
Предполагает проведение комплекса мероприятий, направленных на поддержание таких жизненно важных органов, как сердце, почки, печень и др.:
а) инотропная поддержка миокарда.
Для устранения синдрома низкого сердечного выброса миокардиального происхождения, наблюдаемого при тяжелом термическом поражении, возможно применение препаратов инотропного действия. С этой целью лучше использовать препараты, обладающие преимущественно кардиальным адренергическим эффектом (дофамин, добутреке, добутамин).
Из вазоактивных средств препаратом «первого ряда» для поддержания деятельности сердца и почек является дофамин (400 мг в 250 мл изотонического раствора). Скорость инфузии препарата выбирают в зависимости от желаемого эффекта:
2 — 5 мкг/кг в минуту («почечная» доза): расширяет мезентериальные и почечные сосуды без увеличения частоты сердечных сокращений и артериального давления;
5—10 мкг/кг в минуту: выраженный инотропный эффект, мягкая вазодилатация вследствие стимуляции р2’аДРе’
496 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
норецепторов либо умеренная тахикардия;
10 — 20 мкг/кг в минуту: дальнейшее усиление инотропного эффекта, выраженная тахикардия;
свыше 20 мкг/кг в минуту: резкая тахикардия с угрозой тахиаритмии, сужение вен и артерий вследствие стимуляции at-адренорецепторов, ухудшение инфузии тканей.
Добутамин (400 мг в 250 мл изотонического раствора натрий хлорида). Скорость инфузии — 2 — 20 мкг/кг в минуту. Показан при низком сердечном выбросе и высоком ОПСС на фоне нормального давления наполнения желудочков сердца. Препарат обеспечивает сильный инотропный эффект, значительное расширение сосудов большого и малого кругов кровообращения, дает незначительную тахикардию, не влияет на почечный кровоток.
Адреналин (4 —8 мг в 250 мл изотонического раствора натрий хлорида). Скорость инфузии — 0,02 — 0,2 мкг/кг в минуту. Используют при тяжелой сердечной недостаточности, поскольку обеспечивает сильный инотропный эффект (введение в высоких дозах чревато выраженной тахикардией), несильное сосудосуживающее действие.
Норадреналин (8 —16 мг в 250 мл изотонического раствора натрий хлорида). Скорость инфузии — 0,02 — 0,5 мкг/кг в минуту. Обеспечивает мощный инотропный эффект и незначительную тахикардию, возможна сильная вазоконстрикция. Для уменьшения последней рекомендуется вводить его в сочетании с а-адренобло-каторами (8 мг норадреналина и 10 мг фентоламина в 250 мл изотонического раствора натрий хлорида, начальная скорость введения — 0,17 мл/мин, увеличивается до наступления эффекта).
Амринон — ингибитор фосфодиэстеразы. Вводят 0,75 мг/кг внутривенно струйно в течение 2 — 3 мин, затем переходят на инфузию со скоростью 5 — 10 мкг/кг в минуту. Повышает сердечный выброс без увеличения энергетических потребностей миокарда, снижает пред- и постнагрузку, не вызывает тахикардии, вазоконстрикции.
Высокие дозы у-адренергических агонистов (например, норадреналина) уменьшают питающий кровоток в уже поврежденной коже\
б) профилактика и лечение острой почечной недостаточности.
Предполагает адекватное восполнение дефицита ОЦК, контроль за почасовым диурезом, уровнем калия, креатинина в моче и плазме. При почечной недостаточности уровень креатинина в крови превышает 200 — 250 ммоль/л, скорость клубочковой фильтрации ниже 30 мл/мин. Стимуляцию диуреза проводят на фоне восстановления дефицита ОЦК (ЦВД выше 1 —1,2 см вод. ст., или 980— 1176 Па), удовлетворительного сердечного выброса, инфузии дофамина в «почечной» дозе, введения 2,4 % раствора эуфиллина по 5 мл каждые 4 —6 ч. Преимущество отдается петлевым диуретикам, в частности фуросемиду, первоначальная доза которого (40 мг внутривенно) при необходимости увеличивается до 160 — 240 мг. Отменяют нефротические препараты. Не назначают сосудосуживающие средства.
в) коррекция КОС.
Натрий гидрогенкарбонат вводят при тяжелом метаболическом ацидозе (pH < < 7,25), рассчитав его количество по формуле
Количество натрий гидрогенкарбоната (мэкв) = Масса тела (кг) х х 0,3(25 - [НСОз]пл).
44-48 мэкв (50-100 мл 7,5 % NHCO3) можно ввести сразу, остальное количество — в течение следующих 24 —36 ч. При невозможности определения КОС коррекцию ацидотических сдвигов проводят вслепую, доводя pH мочи до 7,5.
Излишнее введение натрий гидрогенкарбоната создает предпосылки для развития метаболического алкалоза, гипокалиемии, аритмий!
г) стабилизация клеточных мембран.
В первые часы после получения ожоговой травмы возможно применение глюкокортикоидов из расчета по дексаметазону 0,5 мг/кг в сутки или по преднизолону — 3 мг/кг в сутки. Снижая активность комплемента, они оказывают противовоспали
Лечение острого периода ожоговой болезни 497
тельное действие за счет уменьшения пристеночного стояния лейкоцитов и снижения их адгезивности к сосудистой стенке.
Длительное применение глюкокортикоидов ускоряет развитие инфекционных осложнений в ране!
2.5. Профилактика и лечение раневой инфекции
Лечение ожоговых ран при обширном поражении не является первоочередным мероприятием. Исключение составляют циркулярные, захватывающие всю толщу кожи ожоги, требующие немедленного рассечения ожогового струпа для улучшения кровообращения в дистальных отделах конечностей, включая пальцы, или для обеспечения адекватного дыхания при поражениях грудной клетки.
Декомпрессионная некрэктомия выполняется в первые 6—12 ч после травмы. Она уменьшает вероятность инфицирования и ускоряет заживление ран, а также обеспечивает поддержание направления вектора жидкости из кровеносного русла в раны и удаление токсического экссудата вместе с повязками, что способствует снижению эндотоксикоза. Глубокий ожог должен быть иссечен в течение 48 — 72 ч после травмы. Перевязку ожоговой раны следует производить дважды в день после промывания ее йодистым поливидоном или хлоргексидином с использованием местных антибиотиков (сульфадиазин серебра). Периоперативное ведение ожоговых больных связано со многими факторами риска.
Факторы, затрудняющие периопера-ционное ведение ожоговых больных (по Е. К. Sherwood, I. С. Woodson, 2002):
скомпрометированные дыхательные пути;
легочная недостаточность;
нарушения в психическом статусе; сопутствующие повреждения;
ограниченный сосудистый доступ; быстрая кровопотеря;
нарушенная перфузия тканей вследствие:
гиповолемии;
снижение сократительной функции миокарда;
анемии;
сниженное коллоидно-осмотическое давление;
отеки;
аритмия;
нарушения терморегуляции;
измененная реакция на медикаменты; почечная недостаточность;
иммуносупрессия;
инфекция / сепсис.
Точная оценка объема кровопотери имеет решающее значение в планировании периоперационного ведения ожоговых больных. При выполнении хирургической обработки ожоговой раны возможна быстрая и массивная кровопотеря. Адекватная подготовка (мониторинг, сосудистый доступ, наличие крови и ее препаратов) имеет огромное значение. Хирургическая кровопотеря зависит от площади иссекаемых тканей (см2), времени с момента получения травмы и наличия инфекции (табл. 16.4).
Наличие критериев эффективности терапии ожогового шока является основанием для решения вопроса о межбольничной перетранспортировке обожженного в специализированное ожоговое отделение или центр.
Критерии эффективности терапии ожогового шока (по Е. К. Sherwood, I. С. Woodson, 2002):
восстановление адекватного сознания; нормализация артериального давления; диурез (1—2 мл/кг в час);
Таблица 16.4. Расчет предполагаемой кровопотери во время иссечения и пластики у ожоговых больных (по Е. К. Sherwood, I. С. Woodson, 2002)
Хирургическое вмешательство Предполагаемая кровопотеря, мл/см2 площади ожога
Менее 24 ч с момента ожога 0,45
1—3 сут с момента ожога 0,65
2 — 16 сут с момента ожога 0,75
Свыше 16 сут с момента ожога 0,5
Наличие инфицированной раны Более 1
498 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
лактат сыворотки (< 2 моль/л); дефицит оснований (< “5);
pH внутри слизистой желудка (< 7,32); центральное венозное давление (7 — 12 см вод. ст.);
сердечный индекс (4,5 л/мин на 1 м2);
индекс доставки кислорода (600 мл/мин на 1 м2).
2.6. Перетранспортировка пострадавшего в специализированный центр
Своевременная и хорошо организованная эвакуация пострадавших является важным фактором дальнейшего лечения. Транспортировку обожженных рекомендуется проводить бригадой в составе врачей комбустиолога и анестезиолога. Выполнение алгоритма транспортировки пострадавшего в состоянии шока (В. К. Гусак с соавт., 2001) позволяет повысить объективность оценки состояния пациента путем замены субъективного произвольного толкования оценкой объективных, доступных для воспроизведения в экстремальных условиях показателей стандарта. Этим алгоритмом предусмотрено проведение ряда последовательных этапов для установления целесообразности, показаний и сроков межбольничного перевода пострадавшего в специализированный ожоговый центр.
Алгоритм подготовки больного с ожоговым шоком к перетранспортировке в специализированный ожоговый центр (по В. К. Гусаку с соавт., 2001):
1-й этап — медицинская экспертиза состояния пострадавшего (оценка общего состояния пациента, степени шока и эффективности проведенной терапии).
2-й этап — определение показаний для межбольничного перевода в специализированный центр (наличие показаний к специализированному хирургическому лечению или необходимость проведения ИТ с применением специальных технологий).
3-й этап — определение противопоказаний для межбольничного перевода (наличие травмы, несовместимой с жизнью, нестабильной гемодинамики, отека мозга, нарушений мозгового кровообращения).
4-й этап — определение степени риска транспортировки.
5-й этап — получение добровольного сознательного согласия на транспортировку.
6-й этап — определение объема трансфузионной терапии при транспортировке.
7-й этап — транспортировка больного в специализированный центр.
Лечение больных с площадью глубоких ожогов более 10—15 % ППТ, электроожогами, ожогами дыхательных путей и ожогами, чреватыми грубыми косметическими или функциональными деформациями (ожоги лица, рук, суставов), должно проводиться в специализированном ожоговом центре!
Перед транспортировкой пострадавшего необходимо выполнить «правило трех катетеров»:
♦ обеспечить катетеризацию центральной вены, желательно, вне ожоговой поверхности (лучше бедренной вены);
♦ обеспечить катетеризацию мочевого пузыря для учета диуреза;
♦ установить назогастральный зонд для декомпрессии и срочной диагностики возможного желудочно-кишечного кровотечения.
Температура воздуха в машине скорой помощи должна быть в пределах 25 — 33 °C. Инфузионная и дыхательная терапия не должны прерываться. Во время пе-ретранспортировки следует обеспечить седацию больного и минимальный мониторинг (контроль АД, пульса, частоты дыхания, пульсоксиметрия или кардиоскопия).
При отсутствии подтверждения недавней иммунизации, пострадавшему до перетранспортировки необходимо ввести анатоксин столбняка!
3. Отсроченные мероприятия (специализированный этап)
Цель: лечение и профилактика поли-органной недостаточности.
3.1. Полное выведение пострадавшего из шока
При поступлении в специализированное ожоговое отделение или центр больных помещают в бокс на кровать-сетку, под источник инфракрасного излучения, создавая таким образом управляемую воздушную среду с температурой 34 — 36 °C, и продолжают осуществлять интенсивную
Лечение острого периода ожоговой болезни 499
инфузионно-трансфузионную терапию шока.
Приступая к терапии ожогового шока, на этом этапе следует помнить, что сосудистая мембрана и целостность эндотелия начинают восстанавливаться в течение вторых суток после ожога. Это требует повышенных количеств коллоидов, с одной стороны, и уменьшения общего объема инфузий — с другой. Поэтому в последующие 24 ч ожогового шока инфузионно-трансфузионную терапию проводят согласно таким правилам:
1. Общий объем инфузии, представляющий сумму минимальных потребностей и потерь с раневой поверхности, уменьшают на 1/3—1/2 объема первых суток;
2. Минимальные потребности в воде составляют 1500 мл на 1 м2 поверхности тела в сутки. Площадь поверхности тела (5тела, и2) вычисляют по формуле
= /Р-М тела v 3600 ’
где Р — рост человека, см; М — масса тела, кг.
3. Потери воды за счет испарения через раневую поверхность составляют (мл/ч) для взрослых
(25 + ПО) • ППТ (м2), где ПО — площадь обожженной поверхности тела, %;
для детей
(35 + ПО) • ППТ (м2).
4. Для восполнения дефицита ОЦК используют растворы 0,9 % натрий хлорида в сочетании с 5 % раствором глюкозы. На каждый литр вводимых растворов добавляют 20 мэкв калий хлорида.
5. Коллоидные растворы (декстраны, ГЭК, альбумин, плазму) вводят в течение четвертых 8 ч после ожоговой травмы.
Поскольку в результате глубокого повреждения мышц освобождается миоглобин, а термическое воздействие на эритроциты способствует их гемолизу, увеличиваются как гемохромогенная нагрузка на почки, так и содержание калия в сыворотке крови. Поэтому для профилактики ос
трого некроза почечных канальцев показаны мочегонные препараты и ощелачивание. Некоторые коллоиды (декстраны, альбумин) способствуют развитию интерстициального отека легких как признака ОРДС и второй фазы ПОН.
3.2. Лечение полиорганной дисфункции и профилактика полиорганной несостоятельности
3.2.1. Профилактика и лечение раневой и системной инфекции
Уже через 36 ч после ожога инфекция представляет наибольшую угрозу для жизни пострадавшего. К факторам, способствующим генерализации инфекции, относят: утрату большой поверхности кожного покрова, нарушение обменных функций организма, угнетение факторов антиинфек-ционной защиты. В настоящее время ожоговую болезнь рассматривают как иммунодефицитное заболевание, протекающее на фоне острейшего дефицита энергетических и пластических ресурсов. Увеличенную восприимчивость к инвазивной инфекции при ожогах связывают с комбинацией недостаточной местной резистентности и разрушения кожи вследствие термического воздействия.
Кожа и слизистые, будучи первым компонентом иммунитета, выступают в роли механического препятствия, защищающего организм от проникновения бактерий и других агрессивных факторов, а клетки Лангерганса, расположенные в глубине кожи, выполняют специфическую иммунную функцию.
Неспецифическая система, выступающая вторым компонентом иммунитета и представленная клетками тканей, макрофагами, нейтрофилами, активизируется в момент разрушения механического барьера. Вследствие этого и вторжения микроорганизмов развивается местная воспалительная реакция, характеризующаяся экссудацией и притоком клеточных элементов в поврежденную область. Экссудат содержит коагулированный белок, фибриноген, белки системы комплемента, иммуноглобулины. Покрывая бактерию извне подобно смазке, они способствуют быстрому выявлению ее макрофагами и нейтрофилами с целью последующего уничто
500 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
жения или опсонизации. Основными клеточными элементами воспаления на данном этапе выступают нейтрофилы, которые преимущественно функционируют в зонах с уменьшенным кровотоком или в зонах полного стаза. Вместе с тем они не влияют на бактерии, циркулирующие в крови. Последние уничтожаются системой мононуклеарных фагоцитов (СМФ), представленной определенным, фиксированным числом макрофагов, локализованных в почках, селезенке, лимфоузлах.
На уничтожение бактерий, циркулирующих в крови, направлено также третье, специфическое звено иммунитета. В его состав кроме макрофагов входят лимфоциты и плазматические клетки. Последние представляют собой активированные лим-фокином р-лимфоциты, синтезирующие специфические антитела. Синтез антител всегда специфичен, и поэтому антитела выступают в роли специфического опсонина, стимулирующего активность нейтрофилов, с одной стороны, и классический путь активации комплемента — с другой.
Нарушая барьерную функцию кожи, ожоги денатурируют клеточные белки. Жидкость из ожоговых волдырей ухудшает местный иммунитет, поскольку под ее влиянием снижается опсонизация синегнойной палочки, подавляются пролиферация лимфоцитов, хемотаксис и перемещение нейтрофилов. При этом снижается активность клеток СМФ, что обусловлено нарастающим уменьшением уровня фиб-ринектина как вследствие повышенного потребления его, так и вследствие дополнительного инвазивного инфекционного поражения пострадавших.
По мере снижения активности клеток СМФ функция уничтожения бактерий, циркулирующих в крови, переходит к макрофагам легочной ткани, а продукты агрегации нейтрофилов обуславливают клинические проявления ОРДС, приводящего зачастую к летальному исходу.
Поверхностные ожоги заживают самостоятельно в течение 2 — 4 недель, поэтому при поражениях II степени до 20 %, Ша степени — до 10 % поверхности тела применяют только медикаментозное, консервативное лечение. При обширных по
площади поражениях II 1а степени с развитием ожоговой болезни показана ранняя (в 1 —5-е сутки) секвенциальная не-крэктомия с одномоментным закрытием раневого дефекта после иссечения лиофилизированными ксенодермотрансплантата-ми, другими биологическими или современными композиционными синтетическими материалами.
Глубокие ожоги Шб —IV степени, при которых гибнут все слои дермы, только при ограниченных, точечных (1,5 — 2,0 см) поражениях кожи могут зажить спонтанно путем генерации эпителия с краев раны. Остальные раны претерпевают изменения всех фаз раневого процесса и заживают с образованием рубца.
Глубокие ожоги приводят к развитию ожоговой болезни, как правило, при поражении 10 % и более поверхности тела, а иногда и при поражении 5 —7 % поверхности тела (у детей, лиц пожилого возраста, ослабленных пациентов). Местное лечение таких ожогов обязательно включает консервативный и оперативный этапы. Оно направлено на быстрейшее восстановление утраченного кожного покрова путем дерматомной пластики собственной кожи пострадавшего (аутодермопластики). Как было указано выше, при этом большое значение имеет раннее (до 5-х суток) иссечение некротических тканей после травмы и осуществление одномоментной или отсроченной аутодермопластики в первые две недели.
Проведение радикального иссечения позволяет полностью абортировать или по крайней мере существенно уменьшить манифестацию интоксикации и ожоговой болезни в целом. Однако для осуществления подобных процедур необходимы адекватная интенсивная инфузионно-трансфузионная терапия, изоляция и антибактериальная защита пациента, наличие заменителей кожи для одномоментного закрытия ран и дополнительное медикаментозное обеспечение.
При химических повреждениях наложение каких-либо жировых повязок или мазей на жировой основе категорически противопоказано, поскольку агрессивное вещество часто оказывается жирораство
Лечение острого периода ожоговой болезни 501
римым, что ускоряет всасывание и может вызвать тяжелое отравление пострадавшего с поражением печени, почек и т. д.
После туалета все раны, за исключением поверхностных ожогов лица и промежности, ведут закрытым способом. Такие ожоги ежедневно обрабатывают антисептиками и используют аэрозоли, антисептическую пленку или кремы (гели). Раневую поверхность на лице можно вести открытым способом, пока нет обильной экссудации или нагноения, затем также используется повязочный способ лечения. Прежде всего назначаются местные антибактериальные препараты на гидрольной основе.
Повязки, пропитанные гидрольными кремами, создают щадящие условия для ран, обладают анестезирующим и антисептическим эффектами, выраженными сорбционно-дренажными свойствами. Антибактериальная эффективность их определена клинически (in vivo) и лабораторно (in vitro) для различных штаммов патогенной и условнопатогенной микрофлоры, в отличие от влажно-высыхающих и других типов повязок. Использование гид-рольных кремов позволяет сократить сроки эпителизации ожогов II — Ша степени на 6 суток, Шб степени — на 10 суток.
Если есть угроза развития влажного (колликвационного) некроза, то применение закрытого метода ведения ран дополняется использованием источников инфракрасного облучения: электронагревателей, ламп накаливания, фенов и других приспособлений. В таких случаях не потеряли своей актуальности тканевые или порошкообразные сорбционные материалы. Использование последних не представляет сложностей для специалистов, не имеющих достаточного опыта ведения больных с обширными раневыми поверхностями, поскольку при использовании аппликационных сорбентов проще контролировать развитие влажного некроза и раневого сепсиса.
Подозрение на развитие системной инфекции (сепсиса) у ожоговых больных требует посевов культуры с обожженной кожи, подкожной ткани и смежной нормальной кожи. Рост микроорганизмов свыше 105 КОЕ подтверждает предположение о тяжести инфекции.
Острое повышение температуры, лейкоцитоз и нейтрофилез являются показанием для системной антибиотикоте-рапии!
В зависимости от тяжести термической травмы назначают либо один антибиотик (монотерапия), либо два-три препарата (тройной стандарт). Применяются антибиотики спектра. Как правило, назначается комплексная терапия, включающая два или три препарата в дозировке, обеспечивающей адекватную концентрацию действующего вещества в тканях.
Гиперметаболизм и перемещение жидкости из одного сектора в другой для достижения терапевтического эффекта требуют значительного увеличения дозы лекарственных препаратов!
3.2.2. Коррекция энергетического обмена
Обширные ожоги вызывают выраженную метаболическую нагрузку. Резкое увеличение уровня метаболизма сохраняется от момента получения ожоговой травмы до момента полного заживления ожоговой раны.
В основе метаболических расстройств лежат:
повышенная теплопродукция;
повышенная температура тела;
повышенное расщепление тканевых белков;
глубокое катаболическое состояние.
Увеличение уровня обмена веществ у ожоговых больных зависит от площади ожога (табл. 16.5).
При обширных ожогах повышается температура тела в покое. Необходимость поддержания «температуры комфорта» на
Таблица 16.5. Влияние площади ожога на повышение уровня обмена веществ (по Wilmore, 1998)
Площадь ожога, % Увеличение уровня обмена веществ, %
10 26
20 50
30 71
40 87
50 98
60 107
70 ИЗ
502 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
более высоком уровне влечет за собой увеличение энергозатрат для поддержания постоянного градиента между температурой тела пострадавшего и температурой окружающей среды. Поэтому температура воздуха, окружающего ожоговых больных, должна поддерживаться на более высоком уровне, что уменьшает затраты тепла организмом, устраняет дрожь и стресс.
Следствием глубокого катаболического состояния, во время которого все основные ткани организма используются для поддержания удвоенного уровня обмена, является постоянная и значительная (до 1 кг в сутки) потеря веса больным. Поэтому все больные с обширными ожогами тела после стабилизации жидкостного баланса, т. е. через 24 —36 ч после травмы, требуют усиленного питания, которое может быть парентеральным, энтеральным или смешанным.
До 80 % всей поступающей энергии должно быть обеспечено небелковыми продуктами. Обычно глюкоза обеспечивает от 1/2 до 3/4 общего количества энергии (2 — 4 г/кг в сутки). Однако если скорость ее введения превышает 7 мг/кг в минуту, возникают осложнения в виде перегрузки объемом, гипергликемии, увеличения содержания СО2 и жира в печени. Остальная энергия (до 40 %) обеспечивается поступлением жиров. Увеличение распада липидов, наблюдаемое у пострадавших с ожогами, требует повышения доли энергии, вводимой с жиром. Точные пропорции глюкозы и жиров не имеют принципиального значения. Вместе с тем для предотвращения распада собственных аминокислот должно быть введено 100 —
16.3. КОМПОНЕНТЫ РАСТВОРОВ
Глюкоза. Один из наиболее распространенных ингредиентов парентерального питания (ПП). Из общего количества вводимой внутривенно глюкозы 65 % циркулирует в крови и распределяется по организму, 35 % задерживается в печени, превращаясь в гликоген или жир. Помимо выполнения функции источника энергии глюкоза усиливает окислительно-восста-
150 небелковых калорий на 1 г азота (104,67 — 146,54 Дж/г белка).
Без исследования азотистого баланса потребность в белке можно установить только приблизительно, исходя из того, что: здоровому человеку необходимо 0,6 — 0,8 г/кг белка в сутки;
больному на стационарном лечении — 0,8— 1,0 г/кг белка в сутки;
для купирования белкового дефицита требуется 1,1 —1,5 г/кг белка в сутки;
только при тяжелых ожогах необходимо 2 г/кг белка в сутки.
Избыток белка, вводимого с питанием, не ускоряет репарацию тканей!
Таким образом, показаниями к парентеральному и энтеральному питанию являются:
1. Шокогенная травма.
2. Ожоги лица и шеи, затрудняющие обычный прием пищи.
3. Нарушения питания в стадии токсемии и септикотоксемии.
4. Инфекционные осложнения.
5. Дисфункция пищеварительного канала, обусловленная ожоговой болезнью.
При этом следует учитывать необходимость проведения питания на уровне ги-пералиментизации, использования питательных смесей с высоким энергетическим обеспечением и непрерывное проведение питания в течение суток.
Парентеральное питание противопоказано: больным с нестабильной гемодинамикой (гиповолемия, кардиогенный или септический шок), при тяжелом отеке легких или перегрузке жидкостью, анурии (без диализа), выраженных метаболических и электролитных расстройствах.
ДЛЯ ПАРЕНТЕРАЛЬНОГО ПИТАНИЯ
новительные процессы, улучшает антитоксическую функцию печени, стимулирует сократительную способность миокарда, секрецию инсулина.
Глюкоза является единственным углеводом, необходимым для нормальной функции мозга. При гипогликемии возникают различные формы энцефалопатий: психические расстройства, эпилептические при-
Компоненты растворов для парентерального питания 503
падки, делирий и комы. Глюкоза необходима для предотвращения избыточных потерь воды, некоторых микроэлементов.
Суточная потребность в глюкозе зависит от общеэнергетической потребности организма, но не должна быть меньше 100 —150 г, иначе начинается синтез глюкозы из аминокислот.
Экономически целесообразнее использовать стандартные растворы, исходя из того, что 1 г глюкозы (декстрозы) обеспечивает 17,17 кДж.
При инфузии в периферическую вену концентрация глюкозы не должна превышать 10 % (при концентрации аминокислот не более 3,5 %). Максимальная осмоляльность растворов для периферического введения не должна быть выше 900 мосмоль/кг. Поэтому у крупных людей практически невозможно обеспечить полную потребность в энергии периферическим путем. При инфузии в центральную вену максимальная концентрация глюкозы составляет 35 %.
Минимальная потребность в глюкозе — 200 г в сутки (это количество преимущественно обеспечивает потребности головного мозга).
Максимальная скорость окисления глюкозы — 7 мг/кг в минуту, или 0,5 г/кг в час.
Белки (аминокислоты). Важнейшей составной частью организма человека являются белки, которые, помимо выполнения функции структурного элемента, регулируют многие метаболические и ферментативные процессы, участвуют в иммунных процессах и других многочисленных жизнеобеспечивающих реакциях. При недостаточном поступлении белковых субстанций возникают глубокие изменения адаптивной и репаративной регуляции.
Посредством внутривенных инфузий цельной крови, эритроцитов, плазмы и альбумина нельзя обеспечить организм человека белками. Несмотря на то что в 500 мл цельной крови содержится 90 г белка, использовать кровь как источник аминного азота для парентерального питания не представляется возможным, поскольку средняя продолжительность жизни эритроцитов составляет 120 дней, после чего
белки эритроцитов расщепляются до аминокислот и могут быть задействованы в процессах синтеза. Аналогично обстоит дело с инфузиями альбумина, период полураспада которого составляет до 20 дней.
Основными источниками аминного азота при парентеральном питании являются белковые гидролизаты и растворы кристаллических аминокислот. Главное требование, предъявляемое к данному классу инфузионных сред, — обязательное содержание всех незаменимых аминокислот, которые не синтезируются в организме человека: изолейцина, фенилаланина, лейцина, треонина, лизина, триптофана, метионина, валина. Шесть аминокислот синтезируются в организме из углеводов: аланин, глицин, серин, пролин, глутаминовая и аспарагиновая кислоты. Аргинин, гистидин, тирозин и цистин могут быть синтезированы в достаточном количестве, в связи с чем их относят к полузаменимым аминокислотам.
Аминокислоты должны поступать в организм человека в строго определенном количестве и пропорциях. Например, соотношение незаменимых аминокислот (НА) и общего азота (ОА) при проведении парентерального питания у детей и истощенных больных должно быть равно трем. Если же парентеральное питание проводится для поддержания малонарушенно-го азотистого баланса, величина НА/О А может быть более низкой — 1,4 —1,8.
Международным комитетом по питанию за стандарт наиболее полноценного белка для питания человека принят яичный белок. В настоящее время все препараты белка сравнивают с этим стандартом. Огромный практический опыт, накопленный ведущими клиниками мира, показывает, что несбалансированность аминокислотного состава в используемых средах для парентерального питания может нанести существенный вред организму человека.
Аминокислотные смеси имеют стандартную концентрацию от 8,5 до 15 %. При определенных показаниях можно приготовить растворы и другой концентрации. Стандартный раствор аминокислот разводится в соответствующем количестве глюкозы до желаемой концентрации (обычно
504 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
это 3,5 —5,0 % растворы). При почечной недостаточности без олигурии и/или печеночной недостаточности (без диализа) конечная концентрация растворов должна быть менее 3,5 %. При показаниях к ограничению жидкости — более 4,5 %.
Разные концентрации растворов глюкозы и аминокислот имеют различное соотношение небелковых калорий и азота.
Смеси для парентерального питания содержат разный белковый состав (табл. 16.6).
В качестве консерванта в некоторые аминокислотные смеси добавляют натрий дисульфит. Эти смеси нельзя назначать больным с повышенной чувствительностью к соединениям серы.
Конечное соотношение небелковой энергии и азота должно рассчитываться с учетом энергии, полученной при парентеральном введении жировой эмульсии:
(Энергетическая ценность глюкозы + + Энергетическая ценность жиров) х
х 6,25 - Кол-во аминокислот (г).
Жиры. Жировые эмульсии находят широкое применение в качестве энергетического обеспечения при парентеральном питании. Высокая калорийность жира в малом количестве вводимой жидкости позволяет обеспечить 30 — 40 % и более небелковых энергетических потребностей организма. Сырьем для производства жировых эмульсий являются растительные масла: соевое, хлопковое, сафлоровое. Для эмульгирования масел до хиломикронов размером до 1 мкм используют либо яичный лецитин, либо соевые фосфолипиды. Изотоничность с кровью достигается путем добавления глицерина.
Жировые эмульсии являются изотоничными растворами и могут вводиться в периферическую вену. Это наиболее ценный источник энергии при периферическом парентеральном питании. Применяются в качестве источника энергии (обеспечивают 20 — 30 % общей энергетической потребности и предотвращают дефицит незаменимых жирных кислот).
10 % эмульсия содержит 4,61 кДж/мл, а 20 % — 8,37 кДж/мл (для сравнения: 25 % глюкоза содержит 3,56 кДж/мл, а 35 % декстроза — 4,61 кДж/мл).
Тест-дозой для введения 20 % жировой эмульсии является 0,5 мл/мин в течение 15 — 30 мин, а для 10 % жировой эмульсии — 1 мл/мин. Тест рекомендуется для проверки стабильности состояния больного и выявления гиперчувствительности к жировой эмульсии.
Чтобы не вызвать перегрузки системы мононуклеарных фагоцитов, максимальная скорость инфузии 10 % жировой эмульсии не должна превышать 100 мл/ч, а 20 % — 50 мл/ч. Во избежание «синдрома жировой перегрузки» (характеризуется гепатомегалией, желтухой, тромбоцитопенией и может привести к смерти) максимальное количество вводимых внутривенно липидов составляет 2,5 г/кг в сутки.
Инфузия любого флакона с жировой эмульсией не должна продолжаться более 12 ч. Доказано, что растворы «три в одном» можно переливать более длительное время.
Жировую эмульсию не следует переливать больным с аллергией на яйца (фосфатиды яиц используются в качестве эмульгаторов). Описаны редкие случаи анафи
Таблица 16.6. Состав различных аминокислотных смесей
Препарат Условный белок, г/л Общий азот, г/л Аминокислоты, г/л Калий, моль/л Натрий, ммоль/л pH Осмолярность, мосм/л
Аминон 76,0 12,0 100,0 2,5 45,0 5,6 790
Аминосол 80,0 12,7 67,0 0,5 160,0 6,1 800
Полиамин 70,6 11,3 80,7 — — 6,8 1190
Мориамин 78,1 12,5 91,2 — — 6,8 1210
Альвезин 33,3 5,3 40,0 25,0 35,0 — 1140
Вамин 58,8 9,4 70,3 20,0 50,0 5,3 340
Аминостерил КЕ 70,0 11,2 66,0 25,0 30,0 4,8 1410
Компоненты растворов для парентерального питания 505
лактического шока. Аллергия к соевым бобам не является противопоказанием к применению жировых эмульсий, поскольку в них не содержится соевый белок.
Редко встречаются такие побочные эффекты, как лихорадка, головная боль, боль в спине, одышка, озноб, тошнота, боль в груди, масляный привкус во рту. Способность жировых эмульсий вызывать преждевременные роды у человека не доказана.
Жировые эмульсии могут применяться при остром панкреатите без повышения уровня триглицеридов в крови. Во время инфузии жировых эмульсий у таких пациентов, как и у всех других больных, должен проводиться мониторинг концентрации триглицеридов в сыворотке крови.
Чтобы убедиться в адекватности жирового клиренса, следует контролировать уровень триглицеридов в крови спустя 4 — 6 ч после прекращения инфузии.
Жировые эмульсии могут вводиться самостоятельно или одновременно с растворами для парентерального питания через инъекционный порт «piggyback» (подсоединение ко второй инфузионной линии через разветвитель). Они также входят в состав смеси «3 в 1» (глюкоза, жиры, аминокислоты).
Жировые эмульсии противопоказаны при нарушении жирового обмена, тяжелых геморрагических диатезах, нестабильном диабетическом обмене веществ, беременности в первом триместре, эмболии, остром инфаркте миокарда, коме неясной этиологии. Как и другие растворы для парентерального питания, жировые эмульсии не следует применять при острых и угрожающих жизни состояниях (коллапс, шок, тяжелая дегидратация, гипергидратация, гипогликемия, дефицит калия).
Многочисленные преимущества энтерального питания перед парентеральным обуславливают необходимость максимально раннего перехода к нему. Зондовое питание дешевле, препятствует образованию язв в пищеварительном канале, сохраняет целостность слизистой оболочки и функцию тонкой кишки лучше, чем парентеральное питание, а высокобелковые энтераль
ные диеты с соотношением калорий и азота, равным 100 : 1, позволяют увеличить выживаемость обожженных.
Преимущества питания через тонкую кишку:
поддерживает слизистую оболочку кишки;
улучшает секрецию гормонов в тонкой кишке;
устраняет необходимость в центральном катетере, уменьшая риск сепсиса и осложнений;
нейтрализует кислоту желудочного сока в кишке;
снижает вероятность гипергликемии;
обеспечивает введение уникальных и сложных питательных веществ без необходимого тотального парентерального питания: глутамина, клетчатки, низко- и среднемолекулярных жирных кислот;
резко уменьшает стоимость питания.
Введение смесей энтерально позволяет нормализовать гипергликемию, сохранить нормальные желудочно-кишечные гормональные реакции, обеспечить освобождение желчного пузыря и секрецию поджелудочной железы. Таким образом, ни умеренное вздутие живота, ни уменьшенные кишечные шумы не должны быть причиной отказа от попытки зондового питания, которое проводят через тонкие полиэтиленовые зонды, вводимые в желудок, двенадцатиперстную кишку и более дистальные отделы кишечника.
Начинать введение питательных смесей через зонд можно только при гарантии надлежащего размещения его!
Введение зондовой смеси лучше производить непрерывно, так как это способствует увеличению pH желудочного содержимого, уменьшает риск аспирационных пневмоний, вызывает меньшее вздутие и дает меньший остаточный объем в желудке. Если в последнем остается свыше 200 мл содержимого или если при ежечасном измерении желудочный остаток увеличивается точно на объем вводимого через зонд питания, то его лучше прервать. Наиболее частым осложнением энтерального питания, встречающимся у 30 — 40 % пациентов, является диарея. По мнению Дж. Марини [5], в большинстве случаев
506 ИНТЕНСИВНАЯ ТЕРАПИЯ ОЖОГОВОЙ БОЛЕЗНИ
ее вызывает не питание, а другие причины:
медикаментозная терапия: антибиотики;
антациды (особенно содержащие магний);
блокаторы гистамина;
средства, поддерживающие перистальтику (метоклопрамид, цизаприд);
холинергические средства (физостигмин);
пероральные препараты, содержащие сорбитол (KCI, метоклопрамид, эликсир теофиллина);
хинидин;
альфа-метилдопа;
кишечные инфекции:
Clostridium difficile;
тонкокишечная патогенная флора (ток-сигенная Escherichia coli, Salmonella, Shigella, Campylobacter);
гипоал ьбу минемия;
ущемление, относящееся к ободочной и толстой кишке;
непереносимость лактозы; основное заболевание (гипертиреоз). Если невозможно установить причину диареи, то следует уменьшить вводимый объем, его осмолярность или содержание углеводов в нем. В ряде случаев к питательному раствору добавляют пектин.
Методом выбора может быть применение нового варианта метаболической поддержки — фармакологического питания (ФП). Под ФП понимают использование нутритивной поддержки с применением известных нутриентов в дозах, в 2 —7 раз превышающих нормальную потребность, с одновременным снижением сопутствующих компонентов для предотвращения гипералиментации. Основными компонентами ФП являются аминокислоты и их производные (аргинин, глутамин, орнитин-альфа-кетоглутарат), липиды (3-омега по-линенасыщенные жирные кислоты, короткоцепочечные триглицериды) и антиоксиданты (а-токоферол, глутатион и витамин С). Одним из наиболее распространенных вариантов фармакологического питания является так называемое иммунное питание. В состав используемых смесей входят в различных комбинациях
L-глутамин, L-аргинин, 3-омега-жирные кислоты, антиоксиданты, нуклеотиды и пищевые волокна в дозах, в 2 — 7 раз превышающих нормальную физиологическую потребность. Все компоненты иммунного питания вводят в составе безлактозных изокалорических изонитрогенных энтеральных смесей (нутризон).
3.2.3. Профилактика и лечение желудочно-кишечных кровотечений
Тяжелая термическая травма сопровождается, как правило, развитием синдрома полиорганной дисфункции. Кишечная трубка в условиях ожогового шока является типичным органом-мишенью. Рано реагируя на ишемию, кишки, являясь естественным резервуаром условно-патогенной микрофлоры и эндотоксина, служат дополнительными «входными воротами» для инфекции помимо обширной ожоговой поверхности. Поэтому у больных с ожогами с начала 2-х суток начинают проводить комплексную органопротекторную терапию, направленную на профилактику возникновения язв Курлинга и кровотечений из пищеварительного канала. Для этого используют блокаторы гистаминовых рецепторов (квамател по 20 мг два раза в сутки, рантак). Местное лечение включает в себя чередование антацидных (маа-локс, альмагель) и обволакивающих (смекта) препаратов. Со вторых суток к терапии подключают очистительные клизмы, которые проводятся 2 — 3 раза в сутки до восстановления эвакуаторной функции кишок. Для восстановления нормальной микрофлоры кишок и профилактики дисбактериозов используются эубиотики (А-бактерин, бифидум-бактерин, биоспо-рин, бификол, лактобактерии и др.).
Антациды менее эффективно поддерживают pH желудочного содержимого выше 4,0!
3.2.4. Ранняя интоксикация
В развитии ранней интоксикации участвуют несколько факторов, наиболее важными из которых являются продукты термической денатурации белка, представленные ожоговыми токсинами и среднемолекулярными пептидными комплексами. Кроме того, обусловленная развитием ожоговой болезни активация ферментных сис
Список использованной литературы 507
тем приводит к массивной ферментемии. Одновременно при нарушении целостности кожного покрова и снижении иммунной защиты в крови начинает циркулировать большое количество бактериальных токсинов. Все это происходит на фоне значительно сниженной антитоксической резистентности. В условиях ожоговой токсемии естественные процессы детоксикации резко снижены, что указывает на необходимость проведения детоксикационной терапии с первых дней ожоговой болезни.
Простейшим детоксикационным методом, который может проводиться в лечебном учреждении, где оказывают первую помощь ожоговым больным, и не требует дополнительного оборудования, является форсированный диурез. Метод можно применять после выведения больного из ожогового шока.
В настоящее время в период ожоговой токсемии наиболее часто применяют метод плазмафереза — метод экстракорпоральной гемокоррекции, основанный на замене плазмы больного препаратами крови или кровезаменителями. В силу больших возможностей варьирования методик (скорость, объем перфузии, объем и качество плазмозамещения) он может иметь де
токсикационную , иммунокорригирующую и реокорригирующую направленность.
Исходя из патогенеза ожогового шока, можно предположить развитие острой почечной недостаточности, сопровождающейся гипергидратацией и азотемией. В этом случае возможно использование гемодиализа — метода детоксикации, основанного на принципе диффузного и фильтрационного переноса через полупроницаемую мембрану низкомолекулярных токсических веществ и внутрисосудистой жидкости из циркулирующей экстракорпорально крови в диализирующий раствор. Одной из основных задач гемодиализа является удаление жидкости из организма с использованием ультрафильтрации. Последняя обусловлена трансмембранным давлением, т. е. суммой онкотического и гидростатического давления. Во время гемодиализа и ультрафильтрации создается управляемое трансмембранное давление за счет создания разницы давления между кровью и диализатом.
Несмотря на успехи, достигнутые в лечении больных с тяжелыми ожогами, лечение ожоговой болезни все еще представляет серьезную проблему для врачей интенсивной терапии.
Список использованной литературы
1. Алексеев А. А., Лавров В. А., Дутиков В. Н. Ожоговый шок: патогенез, клиника, лечение // Вести, интенсивной терапии. — 1995. — № 2. - С. 31-36.
2. Грунъо П. Первичное реанимационное пособие при обширных ожогах. — Архангельск, 1998. - С. 196-201.
3. Perioperative management of the sevely burned patient // Yearbook of intensive Care and Emergency Medicine / Ed. Z. Vinsert. — Springe, 2002. — P. 853 — 863.
4. Ожоговая травма / С. В. Слесаренко, Г. П. Ко-зинец, Е. Н. Клигуненко и др. — Днепропетровск, 2002. — 63 с.
5. Марини Дж., Уиллер А. П. Медицина критических состояний. — М.: Медицина, 2002. - С. 961-973.
6. Клиническая хирургия / Под ред. Р. Кон-дена, Л. Найхуса. — М.: Практика, 1998. — С. 304-318.
— Раздел —
17
Эмболизм
Эмболизм — это миграция с кровотоком в легочную артерию и сосуды большого круга тромбоэмболов, жировых, газовых эмболов, эмболов амниотической жидкости, септических эмболов, паразитарных (шистоматоз), фрагментов опухолей (ренальноклеточная карцинома с инвазией в нижнюю полую вену). С легочной эмболией неизбежно сталкиваются представители как хирургических, так и терапевти
ческих специальностей, поскольку она может возникнуть при различных заболеваниях. По данным Американской медицинской ассоциации, ежегодно в Соединенных Штатах отмечается до 650 тыс. случаев ТЭЛА, из них 35 тыс. заканчиваются смертью. Вместе с тем даже массивное эмболическое поражение легочных артерий прижизненно не диагностируется у 40—70 % пациентов [1].
17.1. ТРОМБОЭМБОЛИЯ ЛЕГОЧНОЙ АРТЕРИИ (ТЭЛА)
ТЭЛА — наиболее часто встречающийся и клинически значимый вид эмболий. На аутопсии у 25 — 50 % умерших госпитализированных пациентов находят ТЭЛА, и во многих случаях именно это осложнение является причиной смерти. Однако прижизненно ТЭЛА диагностируют только в 10 — 20 % случаев [2].
17.1.1. Этиология
Обычно ТЭЛА возникает при флотирующих (плавающих) тромбах, которые свободно располагаются в просвете сосуда и имеют единственную точку фиксации в своем дистальном отделе. ТЭЛА часто является осложнением другого заболевания: тромбоза венозных сосудов. Более чем в 95 % случаев источником ТЭЛА являются тромбы из вен нижних конечностей (подколенных, бедренных, тазовых). Тромбы в поверхностных венах голени редко вызы
вают ТЭЛА. При бластоматозном поражении почек возможен тромбоз почечных вен, распространяющийся на нижнюю полую вену. Встречается и тромботическое поражение печеночных вен. Иногда источником тромбоэмболии может стать тромб, находящийся в правом предсердии, который развивается на фоне мерцательной аритмии и дилатационной кардиомиопатии. Эмболизация легочного сосудистого русла возможна также при эндокардите трехстворчатого клапана и эндокардиальной элекгрокардиостимуляции, осложненной тромбозом правых отделов сердца [1]. Крайне редко легочной эмболией осложняется тромбоз в бассейне верхней полой вены. ТЭЛА стала настоящим «бичом» послеоперационного и послеродового периодов, являющейся доминирующей причиной смерти, выделяющейся на фоне снижения общей летальности от инфекционных и других осложнений.
Тромбоэмболия легочной артерии (ТЭЛА) 509
Таблица 17.1. Категории риска развития венозного тромбоза и ТЭЛА [4]
Категория риска Факторы риска Вероятность проксимального ТГВ, % Вероятность фатальной ТЭЛА, %
Низкий Малая хирургия (< 30 мин) Большая хирургия (> 30 мин) в возрасте менее 40 лет < 1 < 0,1
Средний Большая хирургия (> 30 мин) в возрасте свыше 40 лет Малая хирургия + ТГВ или ТЭЛА в анамнезе Тяжелая травма или ожог ОИМ или инсульт 1-10 0,1-1
Высокий Большая хирургия или тяжелая травма + ТГВ или ТЭЛА в анамнезе Онкологические операции на органах живота и таза Операции на коленном суставе и костях таза Паралич ног 10-30 1-10
Выделяют три категории ТЭЛА [3]:
1) массивная окклюзия легочной артерии (до 50 % всех случаев);
2) инфаркт легкого — эмболизация приводящей ветви легочной артерии с некрозом участка легочной паренхимы (в 10 % случаев);
3) ТЭЛА без инфаркта легкого (представляет трудность для диагностики, так как не имеет специфических симптомов).
Еще в 1856 г. немецкий патолог Р. Вирхов выделил три основных фактора риска тромбообразования: венозный стаз, повреждение сосудистой интимы, активация свертывающей системы. Сейчас этот перечень расширен. Факторами риска венозного тромбоза являются ([2] с дополнениями): венозный стаз: постельный режим;
иммобилизация после ортопедических операций;
синдром низкого сердечного выброса; беременность;
повышение вязкости крови; большие хирургические операции; пожилой и старческий возраст;
гиперкоагуляция;
повреждение тканей (операции, травмы, ОИМ);
онкологические заболевания; нефротический синдром;
применение оральных контрацептивов, в частности производных эстрогена;
наследственные коагулопатии (дефицит антитромбина III, плазминогена, протеинов С и S, дисфункция фибриногена);
повреждения сосудов: травма.
Консенсусным решением для послеоперационных больных, не получавших антикоагулянты, выделены группы риска венозного тромбоза и ТЭЛА (табл. 17.1).
Одним из наиболее важных факторов риска считают иммобилизацию больного, особенно после проведенного ортопедического оперативного вмешательства. В течение 5 — 7 дней после травматичных операций повышен риск тромбоза глубоких вен (ТГВ) голени и ТЭЛА вследствие таких факторов: иммобилизации, повреждения сосудов, гиперкоагуляции (в это время повышено образование тромбопластина и снижено — антитромбина III). Вероятность ТГВ голени после протезирования коленного сустава без профилактики составляет 84 %, после операций на тазе и простатэктомии — более 50, риск фатальной ТЭЛА доходит до 5 % [2]. К факторам высокого риска также относят онкологические операции и генетические расстройства, вызывающие гиперкоагуляцию.
17.1.2. Патофизиология тромбоэмболии легочной артерии
В случае попадания тромбоэмбола в легочную артерию он вызывает ряд патологических сдвигов (табл. 17.2).
Нарушения гемодинамики. Основным гемодинамическим последствием ТЭЛА является развитие гипертензии малого круга кровообращения. Непосредственной
510 ЭМБОЛИЗМ
Таблица 17.2. Патологические изменения при легочном эмболизме (модифицировано и адаптировано по С. G. Elliot [5])
Вид нарушений Эффект ТЭЛА Механизм
Гемодинамика Газообмен Вентиляция Работа дыхания Увеличение легочного сосудистого сопротивления Увеличение альвеолярного мертвого пространства Гипоксемия Гипервентиляция Увеличение сопротивления дыхательных путей Снижение растяжимости легких Обструкция сосудов Вазоконстрикция серотонином, тромбоксаном А2 Обструкция сосудов Увеличение соотношения V/Q Увеличение соотношения V/Q Увеличение Qs/Qt Снижение сердечного выброса со снижением РоО2 Рефлекторный Рефлекторная бронхоконстрикция Нарушение функции легочного сурфактанта
причиной ее возникновения является увеличение сопротивления сосудов легких, т. е. возрастание постнагрузки для правого желудочка, что препятствует полноценному его опорожнению во время систолы и требует повышения сократимости. У больных с неизмененным преэмболи-ческим статусом кровообращения пороговым уровнем, превышение которого инициирует развитие легочной гипертензии, является окклюзия 50 % легочной циркуляции. Дальнейшее увеличение распространенности эмболической обструкции ведет к повышению давления в легочном стволе и правых отделах сердца, снижению сердечного выброса и напряжения кислорода в артериальной крови. У пациентов с предшествующей патологией со стороны сердца и сосудов легочная гипертензия и циркуляторные расстройства развиваются при меньшей степени обструкции.
Максимальная величина систолического давления в малом круге кровообращения в острой стадии ТЭЛА у пациентов без исходных нарушений со стороны сердечно-сосудистой системы никогда не превышает 70 мм рт. ст. Негипертрофированный правый желудочек из-за ограниченных резервных возможностей не может преодолеть более выраженной гипертензии. Превышение этого уровня указывает на длительный характер эмболической окклюзии или наличие сопутствующей
сердечно-легочной патологии. Снижение легочного кровотока более чем на 75 % является критическим уровнем эмболической обструкции, поскольку приводит к депрессии сердечной деятельности. Прогрессирующее падение сердечного выброса, несмотря на централизацию кровообращения, вызывает системную гипотензию, шок и асистолию. Таким образом, механическая обструкция легочного артериального русла является ведущим фактором в генезе гемодинамических расстройств и развитии депрессии сердечной деятельности при ТЭЛА. При попадании массивного эмбола в легочную артерию сердечный выброс может внезапно снизиться до нуля и произойдет остановка кровообращения. Это встречается в 5 % всех случаев ТЭЛА.
Для легочной эмболии характерно двустороннее поражение легких за счет фрагментации тромбов, которая происходит в правых отделах сердца. При этом даже полная эмболическая обструкция легочных артерий не всегда приводит к развитию инфаркта легкого. В условиях кровоснабжения легкого из двух кругов кровообращения тромбоэмболия вызывает инфаркт легочной паренхимы только у одного из десяти больных.
Нарушения вентиляционно/перфу-зионных соотношений в легких. Эмбо-
лизация приводит к редуцированию кровотока дистальней эмбола, что является
Тромбоэмболия легочной артерии (ТЭЛА) 511
причиной увеличения соотношения V/Q в большинстве отделов. Происходит рост функционального мертвого пространства за счет альвеол, которые вентилируются, но не перфузируются. Кроме этого, в плохо перфузируемых отделах легких возникает ишемия легочной паренхимы. Это приводит прежде всего к нарушению продукции сурфактанта, как высокоэнергозатратного процесса, снижению стабильности альвеол, что вызывает их ателекта-зирование и увеличение шунтирования в легких. Все эти процессы приводят к увеличению DA_aO2 и развитию тяжелой гипоксемии при сохранении элиминации СО2. Увеличивается работа дыхания. Плохо перфузируемые участки легких представляют большой риск инфицирования и развития пневмонии.
17.1.3. Диагностика тромбоэмболии легочной артерии
Клинические проявления ТЭЛА не специфичны, тем не менее, именно они позволяют заподозрить это заболевание и ориентировочно судить об объеме эмболического поражения. Классическая триада симптомов при ТЭЛА — внезапная одышка, плевральная боль в груди и кровохарканье — возникают только в 20 % случаев ТЭЛА, по одному эти симптомы выражены в 85, 75 и 30 % случаев соответственно [2]. Одышка является следствием рефлекторной бронхоконстрикции, снижения растяжимости легких, нарастающей гипоксемии. Плевральная боль характерна для развития инфаркта легочной паренхимы, однако это не всегда подтверждается на рентгенограммах грудной клетки. Кровохарканье также чаще возникает на фоне инфарктирования легочной паренхимы и является следствием высокой легочной гипертензии, повышения проницаемости альвеолярно-капиллярной мембраны и пропотевания эритроцитов в альвеолы. Окклюзия крупных легочных артерий сопровождается признаками острой недостаточности сердца и легких. «Классический» синдром массивного поражения легочного русла включает: внезапный коллапс, появление болей за грудиной, одышку,
цианоз лица и верхней половины туловища, набухание и пульсацию шейных вен. Между тем гораздо чаще (в 60 % случаев) при массивной эмболии наблюдается бледность кожных покровов, являющаяся следствием спазма периферических сосудов в ответ на внезапное снижение сердечного выброса. Помимо тахикардии и тахипноэ для тромбоэмболии главных стволов легочной артерии характерен акцент II тона на легочной артерии, который начинает регистрироваться при систолическом давлении в малом круге выше 50 мм рт. ст. Аускультативно в легких выслушиваются разнокалиберные хрипы, связанные как с бронхоконстрикцией, так и с отечным участком легочной паренхимы. Возможными симптомами при ТЭЛА являются лихорадка и лейкоцитоз.
При исследовании газов крови обнаруживается снижение РаО2 и РаСО2 и увеличение £)л_йО2, однако в 30 % случаев ТЭЛА гипоксемия отсутствует. Важным фактором, увеличивающим вероятность диагноза ТЭЛА, является наличие тромбоза глубоких вен.
Электрокардиография в большинстве случаев помогает заподозрить массивную ТЭЛА по появлению признаков острого легочного сердца', синдром Me Ginn-White (S1Q3 ТЗ), смещение переходной зоны (глубокий SV5-6 в сочетании с отрицательными TV1-4). Изменения на ЭКГ не являются специфическим и обязательным признаком ТЭЛА, в 25 % случаев их не выявляют. Наиболее часто ТЭЛА проявляется синусовой тахикардией, инверсией зубца Т, возможны изменения интервала S —Т. Типичные для легочной гипертензии глубокий зубец S в I отведении, зубец Q и инвертируемый Т в III отведении встречаются только у И % больных с ТЭЛА [2].
Отсутствие ЭКГ-проявлений не исключает наличие ТЭЛА. Трудности трактовки ЭКГ-изменений возникают у пациентов пожилого возраста с органическими поражениями коронарных артерий. Порой даже опытные кардиологи остро возникшие ЭКГ-симптомы, обусловленные ТЭЛА, связывают с проявлениями инфаркта задней стенки левого желудочка.
512 ЭМБОЛИЗМ
Хотя рентгенография грудной клетки должна быть обязательным компонентом диагностики ТЭЛА, почти в 40 % случаев она не выявляет патологии, однако помогает исключить другую патологию легких [6]. Типичным для ТЭЛА является: появление признаков инфаркта легкого, инфильтрата в легком, элевация купола диафрагмы на стороне ТЭЛА.
Дилатация правых отделов сердца, сопровождающаяся расширением вен, высокое стояние диафрагмы на стороне окклюзии и обеднение легочного сосудистого рисунка указывают на массивный характер эмболического поражения. К сожалению, почти у трети больных рентгенографические признаки эмболии вообще отсутствуют. «Классическая» треугольная тень легочного инфаркта выявляется крайне редко (в менее 2 % случаев). Можно сделать вывод, что на основании клинических, лабораторных и рентгенологических данных диагностировать или исключить ТЭЛА не представляется возможным [7].
Эхокардиография позволяет обнаружить возникновение острого легочного сердца, исключить патологию клапанного аппарата и миокарда левого желудочка. С ее помощью можно определить выраженность гипертензии малого круга кровообращения, оценить структурное и функциональное состояние правого желудочка, обнаружить тромбоэмболы В полостях сердца и в главных легочных артериях, визуализировать открытое овальное окно, которое может влиять на выраженность гемодинамических расстройств и являться причиной парадоксальной эмболии. Вместе с тем отрицательный результат эхокардиографии ни в коей мере не исключает диагноза легочной эмболии.
Наиболее информативными критериями диагноза ТЭЛА являются: радиоизотопное сканирование легких, компрессионная ультрасонография, импедансная плетизмография, ангиопульмонография, спиральная компьютерная томография. Радиоизотопное сканирование легких позволяет выявить ТЭЛА в 87 % случаев, а при нормальной сканограмме легких этот диагноз можно исключить [3]. Высоковероятным критерием эмболии является сег
ментарное отсутствие кровотока в легких, не сопровождающееся изменениями на обзорной рентгенограмме грудной клетки. Если строгая сегментарность и множественность перфузионных дефектов отсутствуют на сцинтиграммах, диагноз ТЭЛА маловероятен (нарушения могут быть обусловлены бактериальной пневмонией, ателектазом, опухолью, туберкулезом и другими причинами), но не исключен и требует ангиографической верификации.
Ангиография легочной артерии применяется для диагностики ТЭЛА по строгим показаниям, например при нестабильном состоянии с целью быстрейшего выявления эмбола и возможной эмболэктомии. Этот метод абсолютно показан во всех случаях, когда не исключается массивное эмболическое поражение сосудов легких (в том числе при сомнительных данных сканирования) и решается вопрос о выборе метода лечения. Применение этого метода ограничено его высокой стоимостью и риском осложнений [8].
17.1.4. Методы профилактики ТГВ и ТЭЛА у хирургических больных
Общими методами профилактики являются ранняя активизация больного после операции, систематические движения в голеностопном суставе. Однако у больных группы риска этих мер недостаточно и необходимы специальные методы профилактики ТЭЛА.
Компрессия нижних конечностей (перед операцией рекомендуется сдавливающая повязка с внешней компрессией голеностопного сустава и лодыжек 18 мм рт. ст. и бедра — 8 мм рт. ст. [9]. Этот метод малоэффективен при среднем и высоком риске ТЭЛА [10]. Более результативен метод периодической пневматической компрессии давлением 25 мм рт. ст. — лодыжек и 20 мм рт. ст. — бедра [10].
Артерио-венозная импульсная терапии (Foot Pump) — эффективный, но не рас пространенный в Украине метод профи лактики ТЭЛА. Является довольно резуль тативным, например, при операциях н; тазобедренном суставе, снижает риск ТП с 40 до 5 % [И].
Тромбоэмболия легочной артерии (ТЭЛА) 513
Установка фильтров в полую вену. В США наибольшее распространение получил фильтр Greenfield. Устанавливается чрескожно через внутреннюю яремную или бедренную вену.
Оптимальным методом является непрямая трансвенозная имплантация кава-фильтров различной конструкции непосредственно ниже устьев почечных вен. После постановки этого фильтра ТЭЛА возникает у больных с высоким риском только в 2 —5 % случаев, а летальные исходы вследствие ТЭЛА практически не встречаются [12]. Показан у больных с высокой степенью риска: тромбоз проксимальных вен бедра или ортопедическая операция с высоким риском в сочетании с одним из перечисленных ниже факторов [13]:
противопоказания к введению антикоагулянтов;
развитие ТЭЛА, несмотря на введение антикоагулянтов;
легочная гипертензия;
тяжелые заболевания легких.
При установке фильтра частота развития ТЭЛА не превышает 3 % [13].
Применение антикоагулянтов. Наиболее распространенным методом профилактики является применение антикоагулянтов. С этой целью чаще используется не-фракционированный гепарин, который в настоящее время рекомендуют применять в малых дозах. Обычная профилактическая методика для хирургических больных заключается в введении за 2 ч до операции 5000 ЕД гепарина, с последующим введением подкожно в той же дозе через 8 — 12 ч в послеоперационном периоде. Такая методика является весьма эффективной, однако не оптимальной у больных высокой группы риска и при ортопедических операциях [7].
Нефракционированный гепарин содержит смесь гетерогенных полисахаридных молекул, из которых только 30 % обладают антикоагулянтной активностью [14]. В последнее время для профилактики ТЭЛА все чаще стали использовать низкомолекулярные гепарины — эноксапарин натрий, дальтепарин натрий, надропарин натрий.
Преимущества низкомолекулярных гепаринов:
1. Более низкая молекулярная масса (4000 — 5000) по сравнению с нефракцио-нированным гепарином (12 000—16 000). Проявляют более высокие антикоагулянтные свойства в более низких дозах (J. Hirsh, M.N. Levine, 1992).
2. Выше биодоступность и продолжительность действия, на 25 — 30 % более эффективен для профилактики ТГВ и ТЭЛА (М. Т. Nurmohamed с соавт., 1992).
3. Меньше риск кровотечений (ниже способность ингибировать тромбин, более выраженный аффинитет к антитромбину III). (Hull с соавт., 1992)
4. В меньшей степени индуцируют тромбоцитопению в результате образования аутоантител IgG (Т. Warrentin с соавт., 1995).
5. Нет необходимости в постоянном контроле свертываемости крови, возможно применение в амбулаторных условиях.
НМГ применяют для профилактики в следующих дозах: клексан — по 20 — 40 мг; фраксипарин — по 0,3 дважды в сутки подкожно. В большинстве исследований НМГ назначались за 2 — 12 ч до операции, а затем в послеоперационном периоде один раз в сутки в течение минимум 7 дней.
Противопоказанием для введения НМГ так же, как и для нефракционированного гепарина, являются: активное кровотечение, тромбоцитопения.
Для больных высокой группы риска, кроме дооперационного введения антикоагулянтов, во время операции показано применение растворов декстрана-40 или гидроксиэтилкрахмала (рефортан). Прием аспирина считается малоэффективным для профилактики ТГВ и ТЭЛА.
17.1.5. Терапия тромбоэмболии легочной артерии
Развитие клинически значимой ТЭЛА является опасным для жизни осложнением и требует немедленной терапии, иногда с проведением мероприятий сердечно-легочной реанимации, часто — перевода
514 ЭМБОЛИЗМ
больного на ИВЛ. Одновременно следует приступить к антитромботической терапии.
Тромболитическая терапия (ТлТ). Именно с нее необходимо начинать терапию очевидной массивной ТЭЛА, сопровождающейся нестабильной гемодинамикой и шоком. Она более эффективна, чем терапия антикоагулянтами и дает лучшие и более быстрые результаты [15]. Особенно показана ТлТ при развитии недостаточности правого жедудочка вследствие ТЭЛА. Однако по многим причинам от применения ТлТ часто отказываются, прежде всего из-за высокого риска кровотечения. В одном исследовании [16] указывается, что у 93 % пациентов с ТГВ были противопоказания к ТлТ. Кроме того, начинать ее следует в первые семь дней с начала тромбообразования, иначе эффективность ее значительно снижается [15]. Однако, как правило, начало ТГВ протекает клинически незаметно и своевременно не диагностируется [7].
Современные тромболитические средства могут вводиться в общий кровоток как через центральные, так и через периферические вены. Из таких препаратов в настоящее время используют стрептокиназу, урокиназу и тканевой активатор плазминогена (t-PA). Начальная доза стрептокиназы — 250 000 ЕД в течение 30 мин с последующей инфузией 100 000 ЕД/ч, или 4400 ЕД/кг в час [15].
Применение тромболитиков высокоэффективно (полный и частичный лизис наблюдается у 90 % больных), но небезопасно, поскольку приводит к выраженной кровоточивости и чревато геморрагическими осложнениями.
Абсолютными противопоказаниями к применению ТлТ являются [17]:
активное внутреннее кровотечение;
АД > 200/120 мм рт. ст.; расслаивающаяся аневризма аорты; аллергическая реакция на тромболитические препараты;
геморрагический инсульт.
К относительным противопоказаниям относятся:
новообразование — внутричерепное или спинного мозга;
геморрагическая ретинопатия; беременность.
Не рекомендуется применять тромбо-литики в течение двух недель после тяжелой травмы, восьми недель — после нейрохирургических операций и острой черепно-мозговой травмы.
В качестве контроля ТлТ следует использовать тромбиновое время, которое должно увеличиться в два раза, если этого не удалось достигнуть при применении базовой дозы, следует повторно ввести двойную дозу [18]. При неэффективности двойной дозы необходимо сменить тромболитическое средство. Продолжительность лечебного тромболизиса обычно составляет двое-трое суток.
Терапия антикоагулянтами. Большинству больных с эмболией долевых и сегментарных ветвей достаточно адекватной антикоагулянтной терапии. Малый круг кровообращения обладает большими компенсаторными возможностями, велика также вероятность спонтанного лизиса небольших тромбоэмболов в результате активизации собственных фибринолитических механизмов. Антикоагулянты в адекватных дозах позволяют предотвратить вторичное тромбообразование в легочном сосудистом русле и прогрессирование венозного тромбоза — источника эмболии. При диагнозе ТЭЛА необходимо сразу же внутривенно ввести 80 ЕД/кг гепарина (при отсутствии противопоказаний). В дальнейшем, при отсутствии необходимости в тромболитической терапии внутривенно капельно вводят 18 ЕД/кг в час гепарина (20 000 ЕД гепарина разводят в 500 мл раствора — 40 ЕД/мл). Для выбора дозы ориентируются на актуальную массу тела и частичное тромбопластиновое время (ЧТВ). Основным критерием при выборе дозы должно быть увеличение ЧТВ в 1,5 —2,5 раза (а не время свертывания крови на стекле) [19]. Рекомендуемые дозы гепарина представлены в табл. 17.3. После достижения необходимой дозы контроль ЧТВ осуществлять через каждые 6 ч.
Продолжительность гепаринотерапии по стандартам составляет 10—14 дней (хотя в последнее время считается достаточным применение в течение пяти дней [19]).
Тромбоэмболия легочной артерии (ТЭЛА) 515
Таблица 17.3. Рекомендуемые дозы гепарина при ТЭЛА [20]
ЧТВ, с ЧТВ (соотношение) Дозировка
Болюс, ЕД/кг Капельная инфузия, ЕД/кг в час
< 35 < 1,2 80 Увеличить до 4
35-45 1,2-1,5 40 Увеличить до 2
46-70 1,5-2,3 — Уменьшить до 2
71-90 2,3-3,0 — Уменьшить до 2
> 90 > 3 — Прекратить инфузию на 1 час с последующим контролем ЧТВ
Существует мнение, что антикоагулянтную терапию следует продолжать до полного восстановления двигательной активности больного. Перед снижением дозы гепарина назначают непрямые антикоагулянты, которые после подбора адекватной дозы больной должен принимать не менее шести месяцев для предотвращения рецидива флеботромбоза и ТЭЛА.
К осложнениям при гепаринотерапии относятся: тромбоцитопения (обязателен контроль уровня тромбоцитов), повышение уровня трансаминаз (не связано с нарушением функции печени), гиперкалиемия [21].
Как указывалось раньше, низкомолекулярные гепарины имеют ряд преимуществ перед нефракционированным гепарином. Это проявляется и при лечении ТЭЛА. В лечебных целях НМГ вводят дважды в сутки под кожу живота. Их использование не требует частого лабораторного контроля состояния системы гемостаза. Рекомендуемые дозы НМГ для лечения ТЭЛА: фраксипарин — 0,1 мл/10 кг дважды в сутки, фраксипарин форте/фраксоди — 0,1 мл/10 кг один раз в сутки; клексан — 1 мг/кг дважды в сутки или 1,5 мг/кг один раз в сутки (A. Lensing с соавт., 1995). Методика применения низкомолекулярных гепаринов для лечения ТЭЛА перспективна, но нуждается в дальнейшем изучении.
Прогрессирующее ухудшение состояния больных с массивной ТЭЛА может потре
бовать экстренного хирургического вмешательства. Эмболэктомия показана больным с тромбоэмболией легочного ствола или обеих главных его ветвей при крайне тяжелой степени нарушения перфузии легких, сопровождающейся резко выраженными гемодинамическими расстройствами: стойкой системной артериальной гипотензией, рефрактерной к введению вазопрессоров, повышением уровня систолического давления в правом желудочке выше 60 мм рт. ст. при высоких цифрах конечного диастолического давления. В таких условиях у пациента очень мало шансов выжить, даже при проведении тромболитической терапии. Риск операции оправдан прежде всего у лиц молодого возраста. Однако, поданным К. Е. Wood [22], наиболее эффективным методом терапии ТЭЛА следует считать тромболитическую терапию, а хирургическое лечение применять только в случаях наличия противопоказаний к ТлТ.
Большинство врачей уверено в фатальности всех случаев ТЭЛА. Однако, согласно данным А. И. Кириенко с соавт. [1], лечение более 3000 больных с ТЭЛА оказалось успешным, 580 из них была проведена тромболитическая терапия, 80 — эмболэктомия из легочных артерий. Результаты этих авторов свидетельствуют о том, что в настоящее время существуют реальные возможности предотвращения и лечения этого распространенного и опасного заболевания.
516 ЭМБОЛИЗМ
17.2. ГАЗОВАЯ ЭМБОЛИЯ ЛЕГКИХ
Причинами газовой эмболизации легочной артерии и церебральных сосудов (ГЭЛ) являются ятрогенные осложнения во время выполнения следующих манипуляций:
пункция и катетеризация центральных вен (основная причина ГЭЛ);
нейрохирургические вмешательства, особенно в положении с приподнятым головным концом;
проведение гемодиализа и других экстракорпоральных методов;
искусственное кровообращение; трансплантация печени.
Реже ГЭЛ возникает при кесаревом сечении, лапароскопических операциях, операциях на венах.
При градиенте между атмосферным и венозным давлением 4 мм рт. ст. скорость проникновения воздуха в сосуд через катетер 14g составляет 90 мл/с, т. е. экспозиции в доли секунды достаточно для фатальной ГЭЛ [23]. Симптоматика газовой эмболии легочной артерии сходна с ТЭЛА. Если эмболизация происходит в лежачем положении, эмбол попадает в правый желудочек и легочную артерию со снижением сердечного выброса, шоком, отеком легких, симптомами острой гипоксемии. Попадание массивного эмбола (более 50 мл) в легочную артерию может быстро привести к остановке кровообращения и необходимости в проведении мероприятий сердечно-легочной реанимации [24].
17.2.1. Диагностика газовой эмболии легких
Диагноз может основываться на остром развитии симптоматики после проведения приведенных выше манипуляций. При аускультации часто над сердцем выслушивается симптом «кошачьего мурлыканья», «мельничного колеса». При мониторинге определяется повышенное ЦВД при нормальном или сниженном ДЗЛА, при капнографическом исследовании выявляется снижение РЕСО2-
Профилактика ГЭЛ. Основным методом профилактики является повышение венозного давления выше атмосферного (задержка пациентом дыхания на вдохе, положение Тренделенбурга), аккуратные манипуляции с катетером в центральной вене.
17.2.2. Терапия газовой эмболии легких
1. Немедленно положить пациента на левый бок в положение Тренделенбурга.
2. Попытаться аспирировать газ через катетер или с помощью пункции правого желудочка.
3. Ингаляция чистого кислорода.
4. Как можно быстрее провести сеанс гипербарической оксигенации (ГБО), уменьшающей объем газового эмбола и улучшающей оксигенацию тканей. Применять ГБО следует даже спустя некоторое время после эмболии.
17.3. ГАЗОВАЯ ЭМБОЛИЯ ВЕН ГОЛОВНОГО МОЗГА
Является ятрогенным осложнением инвазивных вмешательств во время общей анестезии и интенсивной терапии, наиболее часто, так же, как и ГЭЛ, возникает при катетеризации центральных вен [25]. Если эмболизация происходит в приподнятом положении пациента газовый эмбол
попадает в яремные и мозговые сосуды. Больной при этом испытывает чувство жара, затем могут развиться судороги, потеря сознания, возможен летальный исход. Срочные терапевтические мероприятия уменьшают риск неврологического дефицита.
17.4. ВОЗДУШНАЯ ЭМБОЛИЯ АРТЕРИИ БОЛЬШОГО КРУГА КРОВООБРАЩЕНИЯ
Может вызываться: пункцией плевральной полости с попаданием иглы в сосуды легкого; баротравмой легких при ИВЛ;
тяжелой травмой органов грудной клетки и проникающими ранениями груди; искусственным кровообращением; выполнением
Жировая эмболия 517
коронарной балонной ангиопластики и других инвазивных вмешательств на артериях, а также через венозное русло при незаращенном овальном отверстии.
В зависимости от локализации эмболии развивается ишемия органов и тканей: ОИМ, ишемический инсульт и др. Соответствуют локализации и клинические симптомы. Диагноз ставится на основании данных: клиники, ультразвукового исследования и компьютерной томографии. В комплекс терапевтических мероприятий входит оксигенотерапия, ГБО, часто необходима ИВЛ.
Первичной целью терапии должно быть поддержание жизненных функций организма.
У больных с газовой эмболией сосудов мозга показан немедленный перевод в положение Тренделенбурга для более быстрой эвакуации газового эмбола в другие сегменты тела, хотя это может способствовать развитию отека головного мозга [26].
Больным в коматозном состоянии, как правило, необходима ИВЛ чистым кислородом [27]. Судороги, часто возникающие при церебральной газовой эмболии, рефрактерны к лечению бензодиазепинами, поэтому показано применение барбитура
тов [28]. При условии адекватной респираторной поддержки ГБО чистым кислородом под давлением 202,6 кПа способствует улучшению транспорта кислорода и более быстрому устранению эмбола, вымывая из него азот [29].
Для поддержания волемии, снижения вязкости крови и улучшения микроциркуляции применяют инфузионную терапию [30].
В случае необходимости используют кардиотоническую и вазоконстрикторную поддержку.
Показания к антикоагулянтной терапии при газовой эмболии артерий головного мозга являются противоречивыми вследствие повышенного риска кровоизлияний в зону инфарктирования.
Поскольку кортикостероиды могут усиливать ишемию головного мозга при окклюзии церебральных сосудов, их применение не рекомендуется [31, 32].
В нескольких исследованиях отмечен благоприятный эффект использования лидокаина [33 — 36]. Рекомендуют внутривенное болюсное введение 1,5 мг/кг с последующей поддерживающей инфузией. Однако следует помнить, что передозировка лидокаина может привести к депрессии функции ЦНС, судорогам, брадиаритмиям.
17.5. ЖИРОВАЯ ЭМБОЛИЯ
Характеризуется появлением свободного жира и жирных кислот в крови, легких, головном мозгу, почках и других органах. От преобладающей локализации эмболов зависит клиническая картина. Классическая триада ЖЭ: острое нарушение дыхания, глобальная неврологическая дисфункция и петехиальная сыпь встречается в 0,5 —2 % случаев переломов длинных трубчатых костей и в 5 —10 % случаев в сочетании с повреждениями костей таза [37].
17.5.1. Патогенез
Не существует общепринятой теории развития жировой эмболии (ЖЭ). Полагают, что жировые эмболы из места перелома попадают в сосуды легких и концентрируются там; активируют свертывающий
4-220 +'/н
каскад, факторы V и VIII, увеличивают активность тромбоцитов и фибринолитическую активность, вызывают катехоламинозависимую мобилизацию свободных жирных кислот. Последние повреждают сосудистый эндотелий и увеличивают его проницаемость. Сообщается о развитии при ЖЭ синдрома ДВС, однако без выраженной кровоточивости. Одним из проявлений тяжелой ЖЭ является ОРДС.
По другой гипотезе, жировые эмболы являются только очагами, на которых агглютинируют хиломикроны плазмы крови с вовлечением в процесс некоторых белков крови, в частности С-реактивного протеина [38].
ЖЭ могут вызывать: переломы костей (свыше 90 % случаев); ортопедические операции, не связанные с травмой; ожоги;
518 ЭМБОЛИЗМ
остеомиелит; коллагеновые сосудистые заболевания; панкреатит; терапия стероидными гормонами; сахарный диабет; внутривенное введение жировых эмульсий; трансплантация костного мозга; липосакция; эпилепсия; искусственное кровообращение.
17.5.2. Клиническая картина
Обычно латентный период длится 12 — 72 ч. У 30 % больных с ЖЭ развивается легочная форма с явлениями ОРДС различной выраженности: от случаев легкой гипоксемии до асфиксии. Церебральные расстройства возникают в 60 % случаев и развиваются чаще всего после легочных. Выраженность их также разная: от незначительных расстройств сознания до локального неврологического дефицита, комы, судорог. Петехиальная сыпь на конъюнктиве, шее, туловище встречается примерно в 50 % случаев (по нашим данным, значительно реже). Сыпь, как правило, быстро проходит, примерно в течение шести часов.
S. A. Schnofeld с соавт. [39] предложил шкалу для диагностики ЖЭ (табл. 17.4).
Диагноз ЖЭ следует считать положительным при сумме баллов > 5.
Однако более надежными для установления диагноза ЖЭ являются специфические тесты. Не существует отдельного теста для поставки подобного диагноза, только совокупность биохимических нарушений может значительно увеличить ве-
Таблица 17.4. Шкала диагностических критериев ЖЭ
Симптом Баллы
Петехии 5
Диффузные альвеолярные инфильтраты 4
Гипоксемия 3
Спутанность сознания 1
Лихорадка (> 38 °C) 1
ЧСС > 120/мин"1 1
Частота дыханий > 30/мин-1 1
роятность правильного диагноза. Для ЖЭ характерно повышение концентрации продуктов деградации фибрина, протромбинового времени, СОЭ, С5а-фракции комплемента и снижение гематокрита, количества тромбоцитов и концентрации ионов Са2+ в крови. По данным S. A. Deppe с соавт. [40], наличие жировых включений в моче, мокроте, спинномозговой жидкости, повышение уровня липазы крови являются ненадежными и неопределенными методами диагностики ЖЭ, однако наличие их в бронхоальвеолярной жидкости является более специфичным и достоверным тестом [37].
17.5.3. Лечение
Не доказана эффективность обычно применяемого лечения ЖЭ гепарином, растворами низкомолекулярных декстранов, этанола, гипертоническими растворами глюкозы [40]. Имеются данные о пользе профилактического применения больших доз кортикостероидов — 7,5 мг/кг метилпреднизолона внутривенно через 6 ч, всего 12 доз [39] или 30 мг/кг через 4 ч, всего 2 дозы [41]. Авторы обосновывают эффективность применения стероидов их мембраностабилизирующим эффектом, ограничением образования свободных жирных кислот в крови, ингибированием комплемент-индуцированной агрегации лейкоцитов. Эффективность этой методики лечения ЖЭ нуждается в дополнительных исследованиях. S. A. Deppe с соавт. [40] ее отрицают. При необходимости должна проводиться поддерживающая терапия (ИВЛ, коррекция гиповолемии, назначение катехоламинов), от эффективности которой часто зависит исход лечения. Кроме того, важным профилактическим мероприятием является ранняя хирургическая стабилизация переломов костей (в первые сутки после травмы).
Летальность при неосложненных формах ЖЭ сравнительно ниже, чем при других видах ОРДС, и достигает 10 %, однако при церебральной форме ЖЭ часто остается неврологический дефицит [37].
Список использованной литературы 519
Список использованной литературы
1. Кириенко А. И., Матюшенко А. А., Андри-яшкин В. В., Чуриков Д. А. Тромбоэмболия легочных артерий: диагностика, лечение и профилактика // Consilium medicum. — 2001. - Т. 3, № 6. - С. 5-8.
2. Prendergast Т. J., Ruoss S. J. Pulmonary Disease // Pathophisiology of Disease / Ed. S. J. McPhee, V. R. Lingappa, W. F. Ganong, J. D. Lange. — 3rded. — McGraw-Hill, 1999. — P. 184-221.
3. Moss M. Venous Thromboembolism // Critical Care secrets / P. E. Parsons, J. P. Wie-ner-Kronish. — 2nd ed. — Philadelphia: Hanley&Belfuws, 1998. — P. 133 — 138.
4. Thromboembolic Risk Factors Consensus Group // BMJ. - 1992. - 305. - P. 567.
5. Elliot C. G. Pulmonary physiology during pulmonary embolism // Chest. — 1992. — 101 (4 Suppl.). - P. 1635.
6. Mehra M. R., Bode F. R. Venous thrombosis and pulmonary embolism // J. M. Civetta, R. W. Taylor, R. R. Kirby. Critical Care. — 3rd ed. —Philadelphia: Lippincott-Raven, 1997. - P. 1887-1903.
7. Marino P. The ICU Book. — 2nd ed. — Philadelphia: Lippincott Williams & Wilkins, 1997. - 928 p.
8. Hull R. D., Hirsh J., Carter C. L. et al. Pulmonary angiography, ventilation lung scanning, and venography for clinically suspected pulmonary embolism with abnormal perfusion scans // Ann. Int. Med. — 1983. — 98. — P. 891-899.
9. Goldhaber S. Z., Marpurgo M. For the WHO/ISPC Task force on Pulmonary Embolism. Diagnosis, treatment and prevention of pulmonary embolism // JAMA. —1992. — 268. - P. 1727-1733.
10. Clagget G. P., Anderson F. A., Levin M. N. et al. Prevention of venous thromboembolism. The Third ACCP Consensus conference on Antithrombotic Therapy//Chest. — 1992. — 102 (SuppL). - P. S391-407.
11. Fordyce M., Ling R. A venous Foot Pump reduces thrombosis after total hip replacement //J. Bone Joint Surg. Br. — 1992. — 71. — P. 45.
12. Becker D. M., Philbrick A. Inferior vena cava filters. Indication, safety, effectiveness//Arch. Int. Med. - 1992. - 152. - P. 1985-1994.
13. Kempezinski R. F. Surgical prophylaxis of pulmonary embolism // Chest. — 1986. — 89. - P. S384-388.
14. Hirsh J., Dalen J. E.f Deykin D.t Poller L. Heparin: mechanism of action, pharmacokinetics, dosing considerations, monitoring, efficasy and safety // Chest. — 1992. — 102(SuppL). - P. S337-351.
15. Hyers T. M., Hull R. D., Weg J. G. Antithrombotic therapy for venous thromboembolic
disease//Chest. — 1992. — 102 (SuppL). — P. S408-425.
16. Market A., Manzo R. A., Strandess E. Jr. The potential role of thrombolytic therapy in venous thrombosis//Arch. Int. Med. — 1992. — 152. - P. 1265-1267.
17. National Heart Attak Alert Program Coordinating Committee, 60 Minutes to treatment Working Group // Ann. Emerg. Med. — 1999. - 23. - P. 311-329.
18. Rogers L. Q., Lutcher C. L. Streptokinase therapy for deep vein thrombosis // Am. J. Med. - 1992. - 152. - P. 1265-1267.
19. Hull R. D., Raskob G. E., Rosenbloom D. et al. Optimal therapeutic level of heparin therapy in patients with venous thrombosis // Arch. Int. Med. - 1992. - 152. - P. 1589 — 1595.
20. Raschke R. A. et al. The weight-based heparin dosing nomogram compared with the «standard care» nomogram // Ann. Int. Med. — 1993. - 119. - P. 874.
21. Oster J. R., Singer I., Fishman L. M. Heparin-induced aldosterone suppression and hyperkaliemia // Am. J. Med. — 1995. — 98. — P. 575-586.
22. Wood К. E. Major pulmonary embolism: review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism // Chest. — 2002. — Mar. - 121(3). - P. 877-905.
23. Sladen A. Complication of invasive hemodynamic monitoring in the intensive care unit // Curr. Probl. Surg. — 1988. — 25. — P. 69-145.
24. PalmonS. C., Moore L. E., Lundberg J., Toung T. Venous air embolism: a review // J. Clin. Anesth. - 1997. - 9. - P. 251-257.
25. Oberbaugh S. L. Venous air embolism: clinical and experimental consideretions // Crit. Care Med. - 1992. - 20. - P. 1169-1177.
26. Moon R. E.t Dear G. L., Stolp B. W. Treatment of decompression illness and iatrogenic gas embolism // Respir. Care Clin. N. Am. — 1999. - 5. - P. 93-135.
27. Annane D., Troche G., Delisle F. et al. Effects of mechanical ventilation with normobaric oxygen therapy on the rate of air removal from cerebral arteries // Crit. Care Med. — 1994. - 22. - P. 851-857.
28. Tovar E. A., Del Campo C., Borsari A., WebbR. P., Dell J. R., Weinstein P. B. Postoperative management of cerebral air embolism: gas physiology for surgeons // Ann. Thorac. Surg. — 1995. — 60. — P. 1138 — 1142.
29. Workshop Panel. Final summary of recommendations: diving accident workshop 1990 // Diving accident management / Ed. P. Bennett, R. E. Moon. — Bethesda, Md.: Undersea
17*
520 ЭМБОЛИЗМ
and Hyperbaric Medical Society, 1990. — P. 366-369.
30. Smith R. M., Van Hoesen К. B., Neuman T. S. Arterial gas embolism and hemoconcentration // J. Emerg. Med. — 1994. — 12. — P. 147-153.
31. Sapolsky R. M., Pulsinelli W. A. Glucocorticoids potentiate ischemic injury to neurons: therapeutic implications // Science. — 1985. - 229. - P. 1397-1400.
32. Dutka A. J., Mink R. B., Pearson R. R., Hallenbeck J. M. Effects of treatment with dexamethasone on recovery from experimental cerebral arterial gas embolism // Undersea Biomed. Res. - 1992. - 19. - P. 131-141.
33. Evans D. E., Kobrine A. I., Le Grys D. C., Bradley M. E. Protective effect of lidocaine in acute cerebral ischemia induced by air embolism // J. Neurosurg. — 1984. — 60. — P. 257-263.
34. Evans D. E., Catron P. W., McDermott J. J. et al. Effect of lidocaine after experimental cerebral ischemia induced by air embolism // J. Neurosurg. — 1989. — 70. — P. 97 — 102.
35. Dutka A. J., Mink R.t McDermott J. J.t Clark J. B., Hallenbeck J. M. Effect of lidocaine on somatosensory evoked response and cerebral blood flow after canine cerebral air
embolism // Stroke. — 1992. — 23. — P. 1515-1520.
36. McDermott J. J., Dutka A. J.t Evans D. E.t Flynn E. T. Treatment of experimental cerebral air embolism with lidocaine and hyperbaric oxygen//Undersea Biomed. Res. — 1990. — 17. - P. 525-534.
37. Geraci M. W. Fat Embolism sindrome // Critical Care secrets / Ed. P. E. Parsons, J. P. Wiener-Kronish. — 2nd ed. — Philadelphia: Hanley&Belfuws, 1998. — P. 402 — 404.
38. Gosling H. R., Pellegrini V. D. Fat Embolism syndrome//Clin. Orthop. — 1982. — 165. — P. 68-82.
39. Schnofeld S. A., Ploysongsang Y., Di Lisio R. et al. Fat Embolism prophylaxis with corticosteroids: A prospective study in high-risk patients//Ann. Int. Med. — 1983. — 99. — P. 438-443.
40. Deppe S. A., Thompson D. R., Burette R. Other embolic syndrome// J. M. Civetta, R. W. Taylor, R. R. Kirby. Critical Care. — 3d ed. — Philadelphia: Lippincott-Raven, 1997. — P. 1905 — 1916.
41. Lindeque B. G. P., Schoeman H. S., Dommis-se G. F. et al. Fat embolism and the fat embolism syndrome // J. Bone Joint Surg. — 1987. - 69B. - P. 128-131.
Раздел
Сепсис
В настоящее время сепсис рассматривается как инфекционно-индуцированный синдром, характеризующийся многими клиническими симптомами и признаками, включая лихорадку (гипотермию), лейкоцитоз (лейкопению), тахикардию, тахипноэ.
Попытки поиска специфической терапии для снижения заболеваемости и летальности при сепсисе не увенчались успехом, но эти неудачи заставили по-новому взглянуть на патофизиологию сепсиса и понять, что сепсис — это нечто большее, чем просто воспалительный процесс.
В патогенетической терапии сепсиса, с позиции медицины доказательств, эффек
тивными признаны: ранняя диагностика инфекции, хирургическая санация септических очагов, рациональная антибактериальная терапия и поддерживающая интенсивная терапия, направленная главным образом на обеспечение адекватных тканевой перфузии и оксигенации, клеточного метаболизма.
Концепция эмпирической реоксигенации путем достижения супранормальных значений доставки и потребления кислорода [1, 2] в последние годы признана неэффективной, а в ряде случаев даже вредной [3].
18.1. ЭПИДЕМИОЛОГИЯ СЕПСИСА
Сепсис с острой органной дисфункцией представляет серьезную проблему для современного здравоохранения. Так, только в США ежегодно регистрируется более чем 750 тыс. случаев тяжелого сепсиса, являющегося основной причиной летальности в отделениях интенсивной терапии некардиологического профиля. Несмотря на достижения медицины критических состояний, тяжелый сепсис продолжает ассоциироваться с уровнем летальности от 28 до 50 % [4].
По оценкам экспертов государств — членов Организации экономического сотрудничества и развития (OECD), ежегодно в мире выявляется более чем 1,5 млн случаев тяжелого сепсиса, а экономичес
кие затраты на лечение пациентов составляют 16,7 блн дол. По прогнозам, в ближайшие десять лет ожидается резкое увеличение числа больных с риском развития сепсиса в связи с развитием инвазивных медицинских технологий, бесконтрольным применением антибиотиков широкого спектра действия, увеличением количества микробов, устойчивых к антибиотикам и антисептикам [5].
За последние 30 лет отмечено достоверное изменение локализации инфекции у септических больных [6]. Брюшная полость и мочевыводящие пути наиболее часто манифестируют инфекцию (27 и 21 % соответственно). Инфекция легких является доминирующей формой (36 % от
522 СЕПСИС
всех инфекций). Первичная инфекция крови (без других источников инфекции) была зарегистрирована у 20 % больных.
Локализация инфекций была идентифицирована в девяти исследованиях у 2803 больных (92 %) [7], в остальных случаях первичный источник инфекции не удалось установить.
R. A. Balk (2002) приводит данные четырех когортных исследований, демонстрирующие распространенность случаев тяжелого сепсиса у госпитализированных пациентов и летальных исходов вследствие этого (табл. 18.1).
Участниками Европейской септической группы (ЕСГ) Европейского консорциума по интенсивной терапии были представлены результаты международного проспективного мультицентрового когортного исследования, проведенного в течение года в 28 отделениях интенсивной терапии шести стран Европы, Канады и Израиля. В исследование было включено 14 364 пациента, из которых 6011 находились в ОИТ до 24 ч, а 8353 — более чем 24 ч [7]. При поступлении пациентов в ОИТ были установлены 3946 эпизодов инфекции, их характеристика представлена в табл. 18.2.
Особый интерес представляют микробиологические данные, характеризующие
этиологию сепсиса в последние годы. В отличие от характеристик, представленных экспертами многоцентрового европейского исследования EPIC в 1992 г., спектр возбудителей сепсиса в последние годы изменился: увеличилась доля грамполо-жительных кокков (до 37 %), при доминировании грамотрицательных бацилл (45 %), Candida spp. и грибы составляют до 10 % всех изолятов.
В табл. 18.3 представлены микробиологические характеристики возбудителей сепсиса (по данным ЕСГ).
В другом проспективном когортном исследовании, проведенном D. Р. Raymond с соавт. [5] в университете Вирджинии с использованием системы VITEK, установлено, что 21,96 % грамотрицательных бактерий устойчивы ко всем аминогликозидам, цефалоспоринам, карбапенемам и фторхинолонам, что приводит к более тяжелому течению сепсиса, развитию пневмоний или катетерных инфекций, суперинфекции резистентными штаммами грам-положительных кокков и грибами, высокой летальности (27,1 %). В то же время значимых различий в летальности между заболеваниями, вызванными резистентными и чувствительными штаммами Pseudomonas aeruginosa не выявлено [8].
Таблица 18.1. Наблюдательные когортные эпидемиологические исследования при сепсисе (R. A. Balk, 2002)
Дизайн исследования Количество наблюдений Длительность исследования Тяжелый сепсис
Исследование Частота Летальность, %
Rangel-Frausto с соавт. (США, Айова, 1995) Проспективное 3708 пациентов (2527 с критериями ССВО) 28 дней, последующие 9 месяцев 13 % в трех ОИТ 20
Brun-Buisson с соавт. (Франция, 1995) Проспективное, в 170 ОИТ И 828 пациентов 2 месяца 9 % поступивших в ОИТ 56 (до 28 дня)
Sands с соавт. (8 центров, США, 1997) Проспективное, в 8 академических центрах 12 759 пациентов 15 месяцев 10 % всех пациентов ОИТ 34 (до 28 дня) 45 (до 5 мес.)
Angus с соавт. (семь штатов США, 2001) Ретроспективное, в 847 госпиталях 6 621 559 пациентов 12 месяцев 0,3 % всей популяции США, 2 % всех госпитализированных 28,6
Эпидемиология сепсиса 523
Таблица 18.2. Характеристика эпизодов инфекции, установленных при поступлении пациентов в ОИТ
Характеристика Общее количество случаев (п = 3946) Инфекции без ССВО (п = 707, 17,9 %) Сепсис (п = = 1115,28,3%) Тяжелый сепсис (и = 944, 23,9 %) Септический шок (и = 1180, 29,9 %)
абс. абс. % абс. % абс. % абс. %
Внегоспитальная инфекция 1432 287 20,0 453 31,6 301 21,0 391 27,3
Приобретенная в госпитале 1114 177 15,9 291 26,1 272 24,4 374 33,6
Приобретенная в ОИТ 1400 243 17,4 371 26,5 371 26,5 415 29,6
По локализации: респираторные 2438 478 19,6 702 28,8 621 25,5 637 26,1
желудочно-кишечные 533 57 10,7 143 26,8 88 16,5 245 46,0
первичная бактериемия 489 57 11,7 114 23,3 131 26,8 187 38,2
мочевыводящий тракт 433 87 20,1 107 24,7 108 24,9 131 30,3
множественные очаги 531 69 13,0 125 23,5 157 29,6 180 33,9
Микробиологически документированы 2804 456 16,3 785 26,2 728 26,0 885 31,6
Клинически документированы 1142 251 22,0 380 33,3 216 18,9 295 25,8
Инфекции кровотока 774 66 8,5 173 22,4 216 27,9 319 41,2
Некровяные инфекции 3172 641 20,2 942 29,7 728 23,0 861 27,1
Грамположительные 1525 232 15,2 367 24,1 428 28,1 498 32,7
Грамотрицательные 1890 304 16,1 539 28,5 460 24,3 587 31,1
Candida, fungi 411 78 19,0 84 20,4 89 21,7 160 38,9
Полимикробные 968 156 16,1 249 25,7 249 25,7 314 32,4
Таблица 18.3. Микробиологические характеристики возбудителей сепсиса (по данным ЕСГ)
Характеристика инфекции Внебольничные инфекции (и = 1504) Госпитальные инфекции (и = 1192) Инфекции ОИТ (п = 1581)
абс. % абс. % абс. %
Микробиологически документированные инфекции 824 54,8 841 70,6 1356 85,8
Инфекции кровотока 241 29,2 280 33,3 309 22,8
Полимикробные инфекции 236 28,6 349 41,5 459 33,8
Общее количество микроорганизмов 11 147 1- 147 1S >85
S. aureus 134 11,7 185 13,7 295 14,9
Другие грамположительные кокки 315 27,5 296 22,0 447 22,5
Общее количество кокков 449 39,1 481 35,7 742 37,4
524 СЕПСИС
Продолжение табл. 18.3
Характеристика инфекции Внебольничные инфекции (и = 1504) Госпитальные инфекции (и = 1192) Инфекции ОИТ (п = 1581)
абс. % абс. % абс. %
Haemophillus, Moraxella 71 6,2 39 2,9 67 3,4
Е. coli, Proteus 154 13,4 150 11,1 210 10,6
Enterobacteriaceae 80 7,0 178 13,2 263 13,2
Аэробные грамотрицательные бациллы 81 7,1 251 18,6 381 19,2
Другие грамотрицательные бациллы 19 1,7 27 2,0 56 2,8
Общее количество грамотри-цательных бацилл 405 35,3 645 47,9 977 49,2
Анаэробы 37 3,2 35 2,6 31 1,6
Микобактерии, вирусы, паразиты 104 9,1 25 1,9 И 0,6
Candida spp., fungi 103 9,0 140 10,4 192 9,7
Внутриклеточные микробы 25 2,2 8 (0,6) 2 (0,1)
Другие 24 2,1 13 1,0 30 1,5
В результате исследования антимикробной резистентности в ОИТ десяти медицинских центров Гонконга, проведенном в 2003 г. на 1697 пациентах, выявлено 12,1 % метициллин-резистентных штаммов S. aureus, 14 % цефтазидимрезистентных грам-отрицательных бацилл, 13 % ванкомицин-резистентных энтерококков, что сопровождалось удлинением сроков пребывания пациентов в ОИТ, применением большого числа антибиотиков, а также значительным риском трансмиссии этих резистентных штаммов другим пациентам [8].
Установлено, что сепсис повышает риск смерти в течение пяти лет после перенесенного септического эпизода, что, возможно, объясняется персистирующей органной дисфункцией вследствие отрицательных эффектов используемых методов поддерживающей терапии, в частности ИВЛ [9].
Тот факт, что более 500 тыс. больных ежегодно умирает от тяжелого сепсиса (свыше 1400 человек ежедневно), показывает, что до сих пор влияние этой патологии на всю систему здравоохранения недооценивается [10].
18.2. ТЕРМИНОЛОГИЯ
В 1991 г. Американская коллегия торакальных хирургов и общество медицины критических состояний провели согласительную конференцию, целью которой была разработка концептуальных и практических границ определения системной воспалительной реакции на инфекцию, сопровождающуюся прогрессирующим повреждением органов. Реакция определялась общим термином «сепсис» и включала сепсис-ассоциированную органную дисфункцию [11].
На конференции был рассмотрен широкий спектр определений сепсиса с целью поиска наиболее адекватных, которые можно было бы использовать для диагностики, мониторирования и лечения сепсиса, а также для разработки стандартизированного исследовательского протокола.
В 1992 г. Консенсусная конференция ACCP/SCCM ввела понятие SIRS (systemic inflammatory response syndrome — синдром системного воспалительного от
Терминология 525
вета — ССВО), вызываемого локальной или генерализованной инфекцией, травмой, ожогами или стерильными воспалительными процессами. Были разработаны и внедрены в клиническую и исследовательскую работу критерии ССВО.
Однако спустя несколько лет результаты многочисленных клинических испытаний потребовали пересмотра определений сепсиса, принятых в 1992 г. В 2001 г. в Вашингтоне была проведена конференция по выработке консенсуса относительно нового определения сепсис-синдрома, организованная Обществом медицины критических состояний (SECM), Европейским обществом интенсивной терапии (АССР), Американским торакальным обществом (ATS) и Обществом хирургической инфекции (SIS).
Определения
1. ССВО
Концепция ССВО рассматривает его как системную воспалительную реакцию, запускаемую различными инфекционными и неинфекционными факторами. Согласно критериям, выработанным на согласительной конференции 1992 г., ССВО характеризовался наличием двух или более клинических признаков:
♦ температура тела > 38 °C или < 36 °C; ♦ ЧСС > 100 уд. в 1 мин.;
♦ Ч Д Д > 20 дыханий в минуту или РаСО2 < < 32 мм рт. ст.;
♦ лейкоциты > 12 • 109 л или < 4 • 109 л; ♦ юных форм лейкоцитов > 10 %.
При наличии двух признаков синдром оценивали как умеренной (легкой) степени тяжести, трех — как средней. При трехчетырех признаках ССВО риск прогрессирования возрастал.
Предложенные критерии были слишком широкими и недостаточно специфичными для того, чтобы можно было их использовать для диагностики причин, вызвавших развитие синдрома или идентификации индивидуального паттерна реакции у каждого больного [12—14].
В отличие от клинических проявлений системного воспаления, которые многообразны, биохимические маркеры являются более информативными. Было выявлено
повышение уровня циркулирующих ИЛ-6 [15], адреномодул л ина [16], растворимых (S)CD 14, ELAM-1, MIP-lcc [14], внеклеточной фосфолипазы А2 [17] и С-реактив-ного белка [18] у больных с проявлениями ССВО, согласно критериям 1992 г.
В будущем на основе эпидемиологических исследований станет возможным использование исключительно биохимических критериев в идентификации воспалительной реакции.
Выделяют следующие стадии ССВО [19]:
Стадия А: нормальный ответ на стресс, характеризующийся умеренным уменьшением системного сосудистого сопротивления и соразмерным увеличением сердечного выброса, физиологической артериовенозной разницей по кислороду, увеличением сердечного индекса, повышением потребления кислорода, нормальной концентрацией лактата.
Стадия В: чрезмерный ответ на стресс, характеризующийся потерей системного сосудистого сопротивления. При адекватно поддерживаемой преднагрузке и нормальном физиологическом резерве левого желудочка, сердечный выброс увеличивается для удовлетворения потребностей, возникших в результате значительного уменьшения постнагрузки из-за системной вазодилатации. Уменьшается артерио-венозная разница по кислороду. Несмотря на адекватность системного АД наблюдается развитие синдрома полиорганной дисфункции (MODS).
Стадия С: декомпенсированный ответ на стресс, характеризующийся потерей системного сосудистого сопротивления. Сердечный выброс находится в пределах нормы или слегка повышен. Снижение постнагрузки приводит к тому, что физиологические резервы левого желудочка не способны поддерживать АД. Низкое АД сохраняется даже при адекватной преднагрузке. Снижение АД и нарушение периферической утилизации О2 вызывают тяжелый лактат-ацидоз. Это состояние гипотензии традиционно приписывается септическому шоку или шоку, возникающему при «естественном» развитии сепсиса.
Стадия D: претерминальная стадия, характеризующаяся выраженным ССВО,
526 СЕПСИС
сопровождающимся сердечной недостаточностью, гиподинамической циркуляцией с низким сердечным выбросом. Системное сосудистое сопротивление резко повышено. Общее потребление кислорода снижено вследствие нарушения утилизации на периферии. Концентрация лактата значительно повышена. Летальный исход в этой стадии вероятен у большинства больных.
2. Сепсис
В отличие от ССВО критерии сепсиса должны быть пересмотрены. На конференции 1992 г. сепсис был определен как клинический синдром, характеризующийся наличием как инфекции, так и системной воспалительной реакции. Относительно того, какие диагностические критерии инфекции или системной воспалительной реакции должны быть пересмотрены, М. М. Levy с соавт. [20] предлагают исходить из следующих принципов:
♦ возможность широкого использования критериев как в клинической практике, так и для проведения клинических испытаний с целью познания природы сепсиса и оптимальной тактики его интенсивной терапии;
♦ критерии должны быть достаточно чувствительны для идентификации у большинства больных с синдромом;
♦ критерии должны быть удобными для запоминания и использования клиницистами;
♦ лабораторно-зависимые критерии должны учитывать методы лабораторного анализа как используемые в настоящее время, так и те, которые будут разработаны в недалеком будущем;
♦ критерии должны быть применимы как для взрослых, так и для детей и новорожденных.
2.1. Инфекция
Ранее инфекцию определяли как патологический процесс, вызванный инвазией в нормальные стерильные ткани, биологические жидкости или полости тела патогенных или потенциально патогенных микроорганизмов. Однако это определение не лишено ряда недостатков. Так, колит возникает в результате чрезмерного размножения Clostridium difficile в толстой кишке, не являющейся стерильным, т. е. кли
ническая манифестация колита обусловлена не бактериальной инвазией нормальных стерильных тканей, а секрецией экзотоксина в организме. Часто встречаются случаи с подозрением на инфекцию без микробиологического подтверждения [21].
2.2. Системное воспаление как реакция на инфекцию
Поскольку вокруг определения ССВО до сих пор возникают дискуссии, М. М. Levy с соавт. [20] составил перечень возможных признаков системного воспалительного ответа на инфекцию. Указанные признаки неспецифичны. Так, высокий сердечный выброс наблюдается во время проведения большинства хирургических вмешательств или политравме; гипотензия может быть следствием других состояний и т. д.
Диагностические критерии сепсиса
Инфекция, документированная или подозреваемая; наличие следующих признаков:
1. Общие переменные:
1.1. Лихорадка (температура тела >38,3 °C).
1.2. Гипотермия (температура тела <36 °C).
1.3. ЧСС > 90 уд./мин или в два раза выше нормального возрастного уровня.
1.4. Тахипноэ.
1.5. Нарушение сознания.
1.6. Достоверные отеки или положительный водный баланс (> 20 мл/кг более 24 ч.).
1.7. Гипергликемия >120 мг/дл или 7,7 ммоль/л в отсутствие диабета.
2. Переменные воспаления:
2.1. Лейкоцитоз > 12 • 109/л.
2.2. Лейкопения < 4 • 109/л.
2.3. Нормальный уровень лейкоцитов с содержанием > 10 % юных форм
2.4. Содержание С-реактивного белка плазмы в два раза выше нормы.
2.5. Количество прокальцитонина в два раза выше нормы.
3. Гемодинамические переменные'.
3.1. Артериальная гипотензия (АДСИСТ < <90 мм рт. ст.; САД < 70 мм рт. ст.; А Деист снижается до 40 мм рт. ст. и ниже исходного уровня у взрослых или в два
Терминология 527
раза меньше нормального возрастного уровня.
3.2. 5^О2 > 70 % (однако поскольку это соответствует норме у детей (75 — 80 %), данный показатель не должен использоваться как признак сепсиса у детей и новорожденных).
3.3. СИ > 3,5 л/(мин • м2) (это также является нормой у детей и потому не может быть использовано у них как признак сепсиса).
4. Признаки органной дисфункции:
4.1. Артериальная гипоксемия (РаО2/ FiO2 < 300).
4.2. Острая олигурия (диурез менее 0,5 мл/кг в час).
4.3. Повышение уровня креатинина > 0,5 мг/дл.
4.4. АЧТВ > 60 с.
4.5. Парез кишок.
4.6. Тромбоцитопения (< 100 • 109/л).
4.7. Гипербилирубинемия (общий билирубин > 4 мг/дл, или 70 мкмоль/л).
5. Признаки тканевой перфузии:
5.1. Гиперлактатемия > 1 ммоль/л.
5.2. Снижение капиллярного наполнения (симптом белого пятна).
Примечание. Диагностические критерии сепсиса в педиатрии: признаки и симптомы воспаления плюс инфекция с гипер-/гипотермией (ректальная температура > 38,5 °C или < 35 °C), тахикардия (может отсутствовать у пациентов с гипотермией) и наличие как минимум одного из следующих признаков органной дисфункции: нарушение сознания;
гипоксемия;
повышение уровня лактата в крови.
3. Тяжелый сепсис
Тяжелый сепсис определяется как сепсис с органной дисфункцией. Наиболее частая причина смерти в некардиологических ОИТ: ежегодно от него умирают 150 тыс. пациентов в Европе и более 200 тыс. — в США [22]. Органная дисфункция определяется с использованием критериев шкалы SOFA [23], в педиатрии — с помощью критериев, предложенных J. D. Wilkinson с соавт. [24], F. Proulx с соавт. [25] и L. A. Doughty с соавт. [26], или с использованием шкал PEMOD и PELOD [27].
4. Септический шок
Определяется как циркуляторная недостаточность с персистирующей артериальной гипотензией: АДСИСТ < 90 мм рт. ст. (у детей в два раза ниже нормального возрастного уровня), САД < 40 мм рт. ст. от исходного, несмотря на адекватное волемическое восполнение дефицита ОЦК в отсутствии других причин гипотензии.
У детей и новорожденных сосудистый тонус более высокий, чем у взрослых, поэтому септический шок будет иметь место до развития гипотензии. В связи с этим септический шок в педиатрии определяется тахикардией с признаками снижения перфузии, включая:
♦ снижение периферического пульса по сравнению с центральным;
♦ нарушение капиллярного наполнения > 2 с;
♦ снижение диуреза;
♦ холодные конечности.
Гипотензия является поздним признаком декомпенсированного шока у детей.
Следовательно, ССВО, сепсис, тяжелый сепсис и септический шок определяют различные градации в процессе болезни, проявляющиеся комбинацией изменений витальных функций, лабораторных параметров, гипоперфузией или органной дисфункцией. Течение сепсиса, тяжелого сепсиса и септического шока коррелирует с возрастающей органной дисфункцией и летальностью.
Наряду с ССВО существует понятие CARS (compensatory antiinflammatory response syndrome) — синдром компенсаторного противовоспалительного ответа. При этом последовательность возникающих звеньев сепсиса можно представить в виде следующей схемы, удачно соединяющей фазы воспалительного, противовоспалительного ответов, ранней и поздней органной дисфункции и нозокомиальной (госпитальной) инфекции (рис. 18.1).
5. Синдром полиорганной недостаточности
Этот синдром характеризуется наличием острого повреждения функций органов и систем, организм не в состоянии стабилизировать гомеостаз.
Синдром полиорганной недостаточности (ПОН) — это универсальное поражение
528 СЕПСИС
СПОН поздняя стадия
ССВО СПОН ранняя стадия
СКПО
TNF, ИЛ-1
ИЛ-6, ИЛ-8
Нозокомиальная инфекция
ИЛ-10, TGFP, INFy, ИЛ-13
Рис. 18.1. Последовательность развития сепсиса (по R. С. Bone с соавт., 1996):
СПОН — синдром полиорганной недостаточности; ССВО — синдром системного воспалительного ответа; СКПО — синдром компенсаторного противовоспалительного ответа
всех органов и тканей агрессивными медиаторами критического состояния с временным преобладанием симптомов той или иной органной недостаточности. Различают:
♦ ПОН, возникающую в связи с усугублением определенной патологии;
♦ ятрогенную ПОН, обусловленную медицинскими действиями — профилактическими, диагностическими, лечебными.
♦ ПОН развивается сравнительно недолго: несвоевременная или неадекватная интенсивная терапия быстро превращают патогенез в танатогенез.
Выделяют несколько физиологических механизмов развития ПОН [28]:
♦ медиаторный, при этом ПОН возникает в результате аутоиммунного поражения;
♦ микроциркуляторный и связанные с ним реперфузионные механизмы;
♦ инфекционно-септический (гипотеза «кишки как недренированный абсцесс»);
♦ феномен «двойного удара» и другие механизмы.
Из методологических соображений перечисленные механизмы представлены раздельно. Однако в клинической практике отмечено их совместное действие, хотя каждый из них может преобладать на разных этапах развития ПОН.
1. Медиаторный механизм
В 1987 г. на I Международном симпозиуме по эндотелиальной биологии эндотелий было предложено рассматривать как
орган, обладающий специфическими анатомическими и функциональными особенностями в зависимости от вида ткани, где он располагается, но несмотря на это выполняющий одинаковые функции в организме.
К функциям эндотелиальной системы относятся:
♦ активное изменение проницаемости сосудистой стенки, обеспечение пассажа жидкости с содержащимися в ней веществами из кровотока в ткани и обратно;
♦ регуляция просвета сосуда, который она выстилает, вследствие продуцирования расширяющих или суживающих веществ;
♦ участие в свертывающей и фибринолитической системах крови, атерогенезе;
♦ участие в адгезии, агрегации и трансформации клеток крови;
♦ участие в аутоиммунных реакциях организма.
Цитокиновая система. Цитокины — это низкомолекулярные белковые медиаторы, вырабатываемые различными клетками (эндотелием, лейкоцитами, фибробластами и др.) в ничтожных количествах и принимающие участие в различных функциях клеток. Биологическая активность цитокинов реализуется через специфические рецепторы, имеющиеся на поверхности клеток.
В состав цитокиновой системы входят пять классов веществ, объединенных по их доминирующему действию на другие клетки:
1) интерлейкины;
2) фактор некроза опухоли a (TNFa);
3) факторы роста и дифференцировки лимфоцитов;
4) факторы, стимулирующие рост колоний макрофагов и гранулоцитов;
5) факторы, вызывающие рост мезенхимальных клеток.
Основные функции цитокинов:
♦ участие в воспалительных реакциях;
♦ участие в регуляции роста и дифференцировки отдельных клеток;
♦ воздействие на опухолевый рост;
♦ обеспечение иммунной защиты;
♦ участие в регенерации поврежденной ткани.
Терминология 529
Основную роль в регуляции острого воспаления посредством активации цитокинов играют молекулы белка NF-kB [29].
Продукция цитокинов зависит от состояния организма. В физиологическом состоянии их секреция ничтожно мала, они участвуют во взаимодействии между продуцирующими их клетками и другими медиаторами воспаления. Когда медиаторы иммунных реакций образуются в избытке, в том числе и с участием побочных механизмов их продукции, происходит: ♦ не просто увеличение энергопродукции, а самосжигание организма;
♦ капиллярная утечка жидкости с интерстициальными отеками;
♦ деструкция тканей.
Такое, лишенное защитного значения, воспаление приобретает патогенный характер и служит одной из причин СПОН.
2. Микроциркуляторный и реперфузионный механизмы
Медиаторный механизм запускается возбуждением эндотелиальной клетки с последующей адгезией к эндотелию различных клеток и структур, подлежащих уничтожению, а также адгезией и агрегацией тромбоцитов по типу сосудисто-тромбоцитарного гемостаза.
Развитию этих реакций противодействуют биологически активные вещества, обладающие вазодилататорным эффектом. В результате происходит дальнейшее замедление кровотока, нарушение реологии с явлениями агрегации, секвестрации крови и капиллярной утечки — неизбежно возникает гиповолемический порочный круг.
Если на фоне адекватной интенсивной терапии ишемия успешно ликвидируется и кровоток в тканях восстанавливается, то запускается реперфузионный механизм. Происходит дальнейшее ухудшение состояния тканей, возникают три парадокса.
Кислородный парадокс. При реперфузии в ткани с ферментными системами биологического окисления, поврежденными ишемией, содержание кислорода неадекватно увеличивается. Происходит пероксидное окисление липидов, повреждаются мембраны клеток и органеллы протоплаз
мы. Пероксидное окисление белков приводит к инактивации многочисленных ферментов, а оксид углеводов — к деполимеризации полисахаридов и повреждению состоящего из них межклеточного вещества матрикса.
Кальциевый парадокс. При ишемии нарушается структура внутриклеточных рибосом. При восстановлении кровотока кальций входит в клетку и оказывает разрушающее действие на рибосомы, при этом нарушается продукция белка и АТФ. Кальций способствует возникновению вазоспазма, активирует образование медиаторов, что приводит к еще большим расстройствам микроциркуляции, нарушает проницаемость мембран.
Ионный парадокс. В условиях ишемии растет осмолярность тканей, в среднем на 40 — 50 моем (1 моем эквивалентен 19 мм рт. ст.). При восстановлении кровотока интерстициальный сектор активно притягивает воду (осмолярность достигает 760 — 950 мм рт. ст.), в результате возникает отек тканей.
3. Инфекционно-септический механизм
Пищеварительный канал на всем своем протяжении заселен микроорганизмами (табл. 18.4).
Нормальная микрофлора пищеварительного канала принимает участие в процессе пищеварения, важна для формирования иммунной реактивности организма, препятствует развитию патогенной микрофлоры кишок, оказывает влияние на скорость обновления энтероцитов, кишечнопеченочную циркуляцию компонентов желчи, принимает определенное участие в инактивации биологически активных веществ. Кроме того, анаэробная часть кишечной микрофлоры обеспечивает колонизационную резистентность и подавляет рост потенциально патогенной аэробной бактериальной фракции.
В организме существует система барьеров, отделяющих его внутреннюю среду от содержимого кишок [30]:
1) гликокаликс энтероцитов и контакты между ними;
2) IgA, высвобождаемый в просвет пищеварительного канала в составе слюны,
530 СЕПСИС
Таблица 18.4. Распределение микроорганизмов в пищеварительном канале (по А. Л. Костюченко и др., 1996)
Отдел пищеварительного канала Норма Патология
Желудок Двенадцатиперстная кишка Тощая кишка Проксимальная часть подвздошной кишки Дистальная часть подвздошной кишки Ободочная кишка 0 — 102 в 1 мл; грибы, стрептококки, молочно-кислые бактерии 102 в 1 мл 104 в 1 мл; стрептококки, энтерококки, бактероиды 105 в 1 мл 105—106 в 1 мл; фекальный тип флоры 1012 в 1 мл; различные бактерии До 109 в 1 мл; преимущественно грамотрицательные бактерии и кокки До 109 в 1 мл; фекальный тип флоры До 109 в 1 мл; фекальный тип флоры До 109 в 1 мл; фекальный тип флоры
желчи и кишечного секрета и предотвращающий фиксацию бактерий на энтероцитах;
3) мукоциты, секретирующие муцин, содержащий IgA и покрывающий слизистую оболочку;
4) лимфоциты и резистентные макрофаги подслизистого слоя стенки кишки;
5) локальные лимфатические образования, главным компонентом которых являются М-клетки, покрывающие пейеровы бляшки;
6) система резидентных макрофагов печени и биотрансформационная активность гепатоцитов, которые могут участвовать в дезинтоксикации иммуночужеродных субстанций, поступающих с кровью из кишки;
7) цитолиз бактерий под влиянием протеолитической активности желудочного сока и кишечного секрета, который считают главным бактерицидным механизмом;
8) неспецифические антибактериальные механизмы (лизоцим, лактоферрин и др.);
9) желчные кислоты, во многом обуславливающие биотрансформацию микробных эндотоксинов в просвете кишок;
10) дефекация, периодически снижающая внутрипросветное давление, повышение которого может способствовать миграции кишечных бактерий через эпителиальный покров кишки.
Состояние биологического комфорта в пищеварительном канале нарушается вследствие неумеренного приема антацидных препаратов, нерациональной антибакте
риальной химиотерапии, нарушения иммунной реактивности, длительного парентерального питания, а также гиперосмолярного энтерального пареза кишок, застоя кишечного содержимого [31].
Это способствует возникновению условий для развития транслокации — феномена, приводящего к повышению активности системы мононуклеарных фагоцитов (СМФ) и особенно клеток Купфера в печени. Как результат нарушений слизистого барьера кишок и СМФ развивается системная эндотоксемия. Последняя ингибирует функции клеток Купфера, вызывает нарушения в работе различных органов, повреждает слизистый барьер. Транслокация эндотоксинов осуществляется через лимфатическую систему мезентериальных лимфоузлов, грудной лимфатический проток, достигает легких. Поскольку легкие расположены между кишками и печенью, с одной стороны, и системой циркуляции — с другой, они принимают первый удар на себя, являясь первичным фильтром.
Таким образом, транслокация может быть первичным или вторичным механизмом запуска синдрома системного воспалительного ответа — важнейшего звена сепсиса.
4. Феномен двойного удара
В свете современных представлений о системной воспалительной реакции выделяют два основных пути развития ПОН.
Первичная ПОН непосредственно возникает под воздействием определенного
Терминология 531
Таблица 18.5. Шкала APACHE II
А. Экстренная оценка физиологических функций
Показатель Баллы
4 3 2 1 0 1 2 3 4
Температура, °C САД, мм рт. ст. ЧСС в 1 мин ЧД в 1 мин Дл.иО2, мм рт. ст. (если FiO2 > 50 %) Р«О2, мм рт.ст. (если FiO2 < 50%) pH артериальной крови Содержание НСО3 в сыворотке крови, ммоль/л (учитывается при отсутствии данных газового состава артериальной крови) Содержание Na+ в сыворотке крови, ммоль/л Содержание К+ в сыворотке крови, ммоль/л Содержание креатинина в сыворотке крови, мг% Показатель гематокрита, % Общее число лейкоцитов, х 109/л Сумма баллов по шкале ком Глазго >41 > 160 > 180 >50 >500 >7,7 >52 > 180 >7 >3,5 >60 >40 39 — 40,9 130 — 159 140 — 179 35-49 350 — 499 7,6- 7,69 41,0 — 51,9 160 — 179 6,0-6,9 2,0-3,4 ИО-129 ПО-139 200 — 349 155 — 159 1,5-1,9 50,0 — 59,9 20,0 — 39,9 38,5 — 38,9 25-34 7,5- 7,59 32,0 — 40,9 150 — 154 5,5-5,9 46,0 — 49,9 15,0- 19,9 36,0-38,4 70-109 70-109 12-24 < 200 > 70 7,33 — 7,49 23,0 — 31,9 130 — 149 3,5-5,4 0,6-1,4 30,0-45,9 3,0- 14,9 13-15 34,0 — 35,9 10-11 61-70 3,0-3,4 10-12 32,0 — 33,9 55-69 55-69 6-9 7,25 — 7,32 18,0 — 21,9 120 — 129 2,5-2,9 < 0,6 20,0 — 29,9 1,0-2,9 7-9 30,0 — 31,9 40-54 55-60 7,15- 7,24 15,0 — 17,9 111 — 119 4-6 < 29,9 < 49 < 39 < 5 < 55 < 7,15 < 15 < 110 < 2,5 < 20 < 1 3
Сумма баллов:
Б. Оценка возраста В. Влияние сопутствующих заболеваний
Возраст, годы < 44 45-54 55-64 65-74 > 75 Баллы 0 2 3 5 6 Баллы добавляют в следующих случаях: 1) цирроз печени, подтвержденный биопсией; 2) ишемическая болезнь сердца. Стенокардия напряжения, IV функциональный класс; 3) тяжелая хроническая обструктивная болезнь легких; 4) хронический диализ; 5) иммунодефицит. При наличии какого-либо заболевания добавляют 2 балла терапевтическим или плановым хирургическим больным. 5 баллов добавляют в случае экстренного хирургического вмешательства
Сумма баллов Б > I В:
532 СЕПСИС
Таблица 18.6. Интерпретация (по W. A. Knaus с соавт., 1985)
Сумма баллов по APACHE II Летальность, %
0-9 до 7,5
10-14 ДО 11
15-19 до 24
20-24 до 30
25-29 до 51
30-34 до 71
35 и более 85 и выше
повреждающего фактора любой этиологии, при этом признаки органной дисфункции проявляются рано. Примером этого вида ПОН может служить полиорганная дисфункция при политравме, тяжелых ожогах.
Вторичная ПОН развивается после латентной фазы и является результатом генерализованного системного ответа организма на повреждающий фактор. Септический вариант ПОН можно рассматривать как классическую вторичную органную недостаточность, проявление крайне тяжелого системного ответа на инфекционную инвазию.
Согласование понятий течения сепсиса требует использования систем оценки тяжести состояния больных, что позволит
субъективно оценивать системы жизнеобеспечения, эффективность проводимой терапии, прогнозировать уровень летальности. Наибольшее распространение в мире получили системы оценки APACHE (Acute Physiology Age Chronic Health Evaluation), впервые предложенные W. A. Knaus с соавт. в 1981 г. (табл. 18.5 и 18.6).
Оценка по системе APACHE II включает:
1. Экстренную оценку физиологических функций на основании 12 показателей, полученных в первые 24 ч нахождения больного в ОИТ. Наибольшие отклонения от нормы каждого показателя, выраженные в баллах, суммируют для получения балльной оценки тяжести состояния пациента. Единственным субъективным показателем является оценка по шкале ком Глазго.
2. Значение возраста оценивают по шестибалльной шкале.
3. Значение сопутствующих заболеваний оценивают по пяти дополнительным критериям в зависимости от степени вовлечения основных систем организма.
Окончательный результат тестирования по APACHE II определяется суммой баллов трех составных ее частей: А + Б + В = = 0...71 в зависимости от тяжести состояния больного.
Таблица 18.7. Шкала SOFA
Показатель Оценка, баллы
1 2 3 4
Оксигенация РйО2/ГгО2, мм рт. ст. < 400 < 300 < 200 < 100
Коагуляция Тромбоциты, х 103/мм3 < 150 < 100 < 50 < 20
Печень Билирубин, мг/дл (мкмоль/л) 1,2-1,9 (20-32) 2,0-5,9 (33-101) 6,0-11,9 (102-204) > 12,0 (> 204)
Сердечно- Гипотензия САД < Допамин < 5 > 5 или адре- > 15
сосудистая система или степень инотропной поддержки < 70 мм рт. ст. или добутамин налин <0,1, или норадреналин <0,1 V V о о
ЦНС Показатель по шкале ком Глазго 13-14 10-12 6-9 < 6
Почки Креатинин, мг/дл (мкмоль/л) или олигурия 1,2-1,9 (110-170) 2,0-3,4 (171-299) 3,5-4,9 (300 — 440) или < 500 мл/сут > 5 (> 440) или < 200 мл/сут
Современный взгляд на патогенез и диагностику сепсиса 533
Таблица 18.8. Интерпретация: летальность в зависимости от количества пораженных систем (по J. L. Vincent с соавт., 1998)
Число систем Балл SOFA Летальность
0 0-2 До 9 %
1 3-4 До 22 %
2 6-8 До 38 %
3 9-12 До 69 %
4 и более 13 и более 83 % и более
Для оценки органной недостаточности, связанной с сепсисом, была предложена объективная, простая и доступная шкала SOFA (Sequential (Sepsis-related) Organ Failure Assessment), принятая Европейским обществом интенсивной медицины (European Society of Intensive Care Medicine — ESICM) по согласованию с рабочей группой ESICM по проблемам сепсиса (табл. 18.7, 18.8).
Дисфункция каждого органа (системы) оценивается отдельно, в динамике, ежедневно на фоне проводимой интенсивной терапии.
Разработчиками шкалы SOFA выведена зависимость количества пораженных органов, недостаточность которых оценивалась 3 — 4 баллами, от наличия в организме инфекции. Так, у 17 % обследованных с идентифицированным возбудителем вообще отсутствовали выраженные нарушения каких-либо органов; у 31 % больных имелась моноорганная недостаточность; недостаточность двух органов отмечена у 47 %; трех — у 55; четырех и более органов — у 74 % пациентов.
Неврологический статус в приведенных системах оценивался при помощи шкалы
Таблица 18.9. Шкала оценки комы Глазго
Признак Характер ответа Оценка в баллах
Открывание глаз Спонтанное 4
В ответ на просьбу На болевой раз- 3
дражитель 2
Не отмечается 1
Спонтанная дви- Выполнение
гательная команды 6
активность Отталкивание
раздражителя Отдергивание 5
конечности 4
Патологическое сгибание Патологическое 3
разгибание 2
Нет реакции 1
Вербальные Ориентированные 5
реакции Спутанная речь Бессмысленные 4
слова 3
Неразборчивые звуки 2
Нет реакции 1
Ин?перпретация\ 15 баллов — ясное сознание; 13 — умеренное оглушение; 11 — 12 — глубокое оглушение; 8—10 — сопор; 6 — 7 — умеренная кома; 4 —5 — глубокая кома; 3 балла — смерть мозга.
ком Глазго (Glasgow), основанной на оценке трех критериев (открывание глаз, двигательный ответ и речевая реакция) и касался только комы при травме черепа (табл. 18.9). В дальнейшем эта шкала была модифицирована для комы любой этиологии и получила название «Глазго-Питтс-бургская шкала».
18.3. СОВРЕМЕННЫЙ взгляд на патогенез и диагностику сепсиса
Новая парадигма, радикально меняющая взгляд на патофизиологию сепсиса, рассматривает нарушения гомеостаза как неконтролируемый каскад изменений в системах коагуляции, фибринолиза и воспаления, происходящих одновременно, с последующим повреждением сосудистого эндотелия, микрососудистой дисфункцией, ишемией, органной недостаточностью и вероятностью летального исхода, представлена на рис. 18.2 [32 — 35].
18.3.1. Активация воспаления при сепсисе
Воспаление является нормальным ответом организма на инфекцию. Инициальный ответ организма индуцирует провос-палительное состояние с выделением таких провоспалительных медиаторов, как TNFa, интерлейкины (ИЛ-1, ИЛ-6), фактор активации тромбоцитов (PAF), простагландины. Одновременно вырабатыва-
534 СЕПСИС
Рис. 18.2. Схема патогенеза сепсиса в свете новой парадигмы
ются компенсаторные противовоспалительные медиаторы ИЛ-4 и ИЛ-10, нормализующие (ограничивающие) провос-палительные ответы организма.
При сепсисе регуляция раннего ответа организма на инфекцию утрачивается. В итоге развивается массивная системная реакция, являющаяся избыточной. TNFa и ИЛ-1 вызывают неконтролируемую реакцию органов, приводящую к тканевому повреждению и заключающуюся в развитии диффузного капиллярного повреждения, органной дисфункции [36].
18.3.2. Активация коагуляции при сепсисе
Процессы воспаления и коагуляции тесно взаимосвязаны: многие провоспа-лительные цитокины индуцируют выделение тканевого фактора из эндотелиаль
ных клеток и моноцитов, инициируя коагуляцию.
Тканевой фактор (TF) представляет собой связующее звено между иммунной системой и коагуляцией, одновременно являясь основным активатором коагуляции.
TF взаимодействует с фактором Vila, который превращает фактор IX в фактор 1Ха, а фактор X — в фактор Ха. Последний способствует образованию из фактора II (протромбина) фактора Па (тромбин). Каждая из этих реакций происходит на активированной поверхности клеток. Генерализованный фактор Па инициирует образование фибрина из плазменного фибриногена.
Реакция превращения тромбомодулин — протеин С — протеин S ингибирует факторы Va и Villa. AT III инактивирует факторы Xia, 1Ха, Ха и Па в реак
Современный взгляд на патогенез и диагностику сепсиса 535
ции, ускоряемой в присутствии гепарин сульфата. В ходе фибринолиза тканевой тип активатора плазминогена (t-PA) и урокиназный тип плазминоген-активато-ра (u-РА) способствуют превращению плазминогена в плазмин. Последний катализирует гидролиз фибрина [37].
В норме каскад прокоагуляции уравновешен механизмами антикоагуляции.
При сепсисе антикоагулянтные системы истощаются или потребляются и их сниженная активность прогрессирует по мере развития тяжелого сепсиса и септического шока. При сепсисе активация TF наряду со снижением активности многих естественных антикоагулянтных механизмов, смещает гемостатический баланс в сторону коагуляции.
18.3.3. Внешний путь модели коагуляции
В клинических исследованиях особое значение придается ведущей роли внешней коагуляции при сепсисе. В настоящее время этот путь известен как критический медиатор сепсисиндуцированной активации коагуляции.
Сепсис проявляется индукцией цитокинов, которые прямо или опосредованно активируют тканевой фактор Vila в периваскулярных клетках, на поверхности моноцитов и, возможно, эндотелиальных клеток. Исследования С. Т. Esmon с соавт. [38] показывают, что повреждение эндотелиальных клеток в стенках сосудов может подавлять выделение антикоагулянтных комплексов (тромбомодулин, протеин S, фактор Va-инактивирующую формацию), что может привести к развитию гиперкоагуляционного состояния. В последующем TF активирует внешний путь коагуляции и регулирует образование тромбина. TF выделяется периваскулярными клетками и вызывает тканевое повреждение. Его появление в моноцитах и эндотелиальных клетках может индуцироваться такими провоспалительными цитокинами, как TNFa и ИЛ-1, являющихся центральными медиаторами сепсиса.
При сепсисе многие из естественных антикоагулянтных механизмов или сис
тем, включая гепарин — антитромбин— протеин С — тромбомодулин, угнетены, нарушены или повреждены.
Гемостатический фенотип — кровотечение или тромбоз — определяется нарушением баланса между прокоагулянтами и антикоагулянтами. Механизм этого дисбаланса может быть клиническим парадоксом, вытекающим из системной недостаточности функции с локальным тромботическим исходом. Итог может быть предопределен, когда системное повреждение гемостатического механизма связано не с общим тромбообразованием, а с отдельными, локальными повреждениями, включая сосудистое дерево.
Разгадка парадокса «системный ста-тус/локальный эффект» заключается в эндотелиальной системе. Эндотелий нельзя рассматривать как инертный физический барьер, отделяющий кровь от подлежащей ткани. Недавними исследованиями установлено, что он является высокоактивной метаболической системой, принимающей участие во многих гомеостатических процессах, включая поддержание жидкого состояния крови, контроль вазомоторного тонуса и перенос нутриентов и клеток между кровью и подлежащими тканями. Антикоагулянтные возможности эндотелия обусловлены выделением гепарин сульфата, простациклина, тромбомодулина, тканевого активатора плазминогена, ингибитора тканевого фактора и эндотелиальной NOs, что обеспечивает нетромбогенность клеточной мембраны. Кроме того, эндотелиальные клетки обладают следующими прокоагулянтными качествами: выделяют фактор Виллебранда, ингибитор активатора плазминогена тип 1, активируют рецепторы клеточной поверхности для TF и тромбина, открывают соединительные участки для комплексов коагуляционных факторов, привлекают тромбоциты и моноциты к участкам активации.
В норме эндотелиальная система устанавливает баланс между указанными механизмами. Эти механизмы способны интегрировать многие сигналы для генерации ответа таким образом, что эндотелий способен поддерживать гемостатический
536 СЕПСИС
баланс при нарушении функции определенных сегментов сосудистого русла. В то же время эти свойства эндотелия делают его уязвимым к локальной дисфункции и патофизиологическим нарушениям. Про-и антикоагулянтные факторы дифференцированно распределены в сосудистом русле. Например, фактор Виллебранда имеет высокую экспрессию в сердце и легких; рецепторы тромбомодулина обнаруживают высокую активность в пределах эндотелия легких, но едва чувствительны в пределах гематоэнцефалического барьера.
В физиологическом состоянии печень, костный мозг и другие органы продуцируют различные необходимые прокоагулянты, антикоагуляторные факторы и протеины. Этот постоянный выброс интегрирован в каждой сосудистой области и обеспечивает весь гемостаз. При сепсисе имеет место чередование или расстройство выброса факторов и протеинов. Например, при тяжелом сепсисе коагуляционные печеночные факторы не синтезируются в нормальном количестве, уровень моноцитов повышается и они могут индуцировать TF на клеточных поверхностях. Эти системные изменения в протеинах печеночного, а, возможно, и костномозгового происхождения по-разному проявляются в каждом специфическом сосудистом ложе. В результате может быть нарушен гемостатический баланс. В дальнейшем течение сепсиса осложняется «втягиванием» в процесс эндотелиальной системы.
Повреждение эндотелиальных клеток вызывает васкулиты и часто индуцирует цитокиновый каскад, что усугубляет повреждение функции эндотелия, изменяет локальный баланс вазомоторов, клеточную трофику и гемостаз. В итоге при сепсисе нарушение функций эндотелиальной системы может сочетаться с системными изменениями и способствовать генерации сосудистого специфического тромбина и образованию фибрина.
Во многих клинических исследованиях обнаружена активация коагуляции при сепсисе. Например, М. М. Levi с соавт. [39] выяснил, что значительная активация тромбин-антитромбинового комплекса
(ТАТ) является параметром, коррелирующим с продукцией тромбина, наблюдаемой у здоровых лиц вследствие применения эндотоксина и TNFa. Наличие D-димеров (продуктов деградации фибрина) в плазме крови указывает на то, что одновременно с активацией свертывающей системы происходит активация фибринолиза, во время которого фибрин гидролизуется плазмином [40].
18.3.4. Угнетение фибринолиза при сепсисе
Фибринолитическая система принимает непосредственно участие в септическом процессе. У многих больных сепсисом фибринолиз как нормальный ответ организма на избыточное тромбообразование угнетается, в то время, как активация коагуляции продолжается.
Плазмин, являющийся принципиальным эффектором фибринолиза, образуется тогда, когда тканевый плазминоген-активиру-ющий фактор (t-PA) индуцирует преобразование плазминогена в плазмин, последний разрушает нити фибрина. Происходит деградация фибриногена и факторов коагуляции V и VIII.
Угнетает фибринолиз ингибитор активатора плазминогена-1 (PAI-1) и тромбин-активирующий ингибитор фибринолиза (TAF-1). PAI-1, продуцируемый эндотелиальными клетками и тромбоцитами, является важным быстродействующим ингибитором t-PA. Эндотоксин грамотрицательных патогенов повышает активность PAI-1.
У пациентов с сепсисом наблюдаются следующие фибринолитические нарушения: повышение активности PAI-1, снижение активности t-PA, уровней протеина С и плазминогена. Супрессия фибринолиза в сочетании с активацией коагуляции способствует процессу коагулопатии.
Результаты экспериментальных исследований, в которых добровольцам назначали эндотоксин или TNFa, демонстрируют угнетение фибринолитических процессов [41]. Отмеченные изменения являются ведущими в возникновении органной дисфункции и прогрессировании сепсиса [42, 43].
Новые агенты при сепсисе 537
18.4. НОВЫЕ АГЕНТЫ ПРИ СЕПСИСЕ
Существуют агенты, прерывающие каскад коагуляции или предположительно способные принимать участие в коррекции микроваскулярных нарушений, имеющихся у больных сепсисом. К ним относятся AT III, активированный протеин С, ингибитор TF, каждый из которых можно использовать для прерывания на разных стадиях процесса тромбообразования.
Антитромбин III является плазменным ингибитором протеазы. Подавляет активацию тромбина так же эффективно, как многие компоненты внутреннего, внешнего и общего каскадов свертывания. Он может ограничивать агрегацию тромбоцитов, уменьшать продукцию провоспали-тельных цитокинов эндотелиальными клетками. При сепсисе уровень AT III снижен, причем чем ниже его содержание, тем вероятнее летальный исход заболевания. Для проверки гипотезы о том, что AT III предупреждает септический шок и ДВС, F. Fourrier с соавт. [43] провели рандомизированное двойное слепое, плацебо-кон-тролируемое исследование у пациентов с документированным септическим шоком и ДВС. Больные получали плацебо или АТ III (90— 120 МЕ/нг в секунду) в течение 4 дней. Свежезамороженная плазма, тромбоциты и концентрат фибриногена применялись только у пациентов с кровотечениями, выраженным удлинением протромбинового времени, снижением уровня тромбоцитов и фибриногена. Установлено, что высокие дозы AT III существенно улучшают течение сепсисиндуцированного ДВС при септическом шоке, снижая летальность до 44 %. У больных с оценкой тяжести состояния по APACHE II — 20 и более баллов, концентрацией ИЛ-6, превышающей 1000 пг/л при применении ингибитора TF уровень летальности снижался с 62 % до 41 % [44]. В исследованиях S. Opal с соавт. и М. J. Kemme с соавт. [45, 46] не отмечено позитивных эффектов при применении этого агента.
Особого внимания заслуживает протеин С, изменение которого ассоциируется с различными критическими состояниями, включая сепсис. Так, его недостаточность
зафиксирована у более чем 85 % больных сепсисом. Замечено, что недостаточность протеина С коррелирует с негативными клиническими исходами при таких состояниях, как тяжелый сепсис, септический шок, ДВС и СПОН, заместительная терапия с активированным протеином С может улучшать исход у пациентов с сепсисом.
При физиологических условиях многочисленные регуляторные механизмы включают натуральные ингибиторы коагулопатии, активаторы фибринолиза и противовоспалительные медиаторы, поддерживающие состояние баланса в токе крови, принимают участие в эндотелиальном ответе на воспаление или травму [47]. Эти гомеостатические механизмы включают протеин С-путь, систему антитромбин III — гепарин —сульфат и ингибитор TF-пути. Данные системы действуют по принципу негативных и позитивных обратных связей и предупреждают наступление генерализации [42].
Эндогенный активированный протеин С является наиболее значимым ингибитором протеолиза факторов коагуляции Va и Villa, вовлекаемых в ограниченные в пределах нормы шаги коагуляции. Когда тромбин переходит в тромбомодулин как эндотелиальный поверхностно-клеточный и растворимый протеин, их активность переключается с формирования фибрина и активации тромбоцитов на активацию протеина С.
Активированный протеин С (АПС) связывает тромбин с тромбомодулином. Усиление этого процесса в сосудистом эндо-телиуме (первоначально микрососудис-том), где имеются высокие концентрации тромбомодулина, является рецепцией эндотелиального протеина (EPCR). АПС связывается с EPCR, рецептор взаимодействует с протеином С, инактивирующим тканевой фактор Va. В результате происходит саморегулирование системой балансирования коагуляции.
Протеин С способен также усиливать фибринолиз. Во время коагуляционного каскада активатор тканевого плазмина резко угнетается ингибитором плазменной
538 СЕПСИС
протеиназы, называемой ингибитором активатора плазминогена (PAI-1), который инактивирует t-PA, препятствуя тем самым лизису тромбов. АП С взаимодействует с PAI-1, в результате PAI удаляется из циркуляции, что приводит к усилению фибринолиза.
W. W. Hancock с соавт. [48] отмечают, что АПС блокирует кальциевый ток и СД-14-снижение регуляции на моноцитах в ответ на эндотоксин. Согласно исследованиям О. Р. Smith с соавт. [49], связывание АПС с протеиновыми рецепторами на мононуклеарных фагоцитах блокирует TNF-продукцию этими клетками. АПС тормозит выделение TF в EPCR-зависи-мых формированиях. EPCR может подвергаться транслокации из плазменной мембраны к клеточному ядру и модулировать ген экспрессии, что имеет важное значение для способности АПС корригировать воспалительный ответ в эндотелии. Эндогенно активированный протеин С (ЭАПС) может принимать участие в прерывании цикла- коагуляции и воспаления [50], так как, во-первых, ЭАПС обладает потенциально ингибиторными эффектами относительно образования тромбина; во-
вторых, он оказывает непосредственное противовоспалительное действие путем снижения продукции цитокинов и ингибиции сродства лейкоцитов к эндотелию. ЭАПС способен также восстанавливать баланс между процессами, происходящими при сепсисе: коагуляцией, угнетением фибринолиза и воспалением.
К эндогенным модуляторам гомеостаза относятся:
♦ антикоагулянтные (антитромби-ческие) — активированный протеин С (АПС), антитромбин III — гепарин сульфат, тканевой фактор ингибирования пути (TFPT). Основная их функция — предупреждение генерализации процесса коагуляции;
♦ фибринолитические — тканевой активатор плазминогена (t-PA), АПС. Ингибируют образование PAI-1 и TAFIa, удаляют сформировавшиеся микротромбы и сохраняют текучесть крови;
♦ противовоспалительные — противовоспалительные цитокины, АПС. Ингибируют тромбин-опосредованную воспалительную активность; адгезию лейкоцитов к эндотелию, тормозят воспалительный ответ.
18.5. КОАГУЛОПАТИЯ ПРИ СЕПСИСЕ — ГЛАВНЫЙ ПУТЬ К ОРГАННОЙ ДИСФУНКЦИИ И ЛЕТАЛЬНОМУ ИСХОДУ
При прогрессировании сепсиса нарастает коагулопатия. Во время развития септического шока коагулопатия проявляется следующими лабораторными изменениями: значительным дефицитом протеина С, удлинением АЧТВ и ПВ, увеличением количества фибриновых мономеров, снижением уровня фибриногена и возрастанием уровня D-димеров (рис. 18.3).
В большинстве экстремальных случаев дисбаланс между процессами воспаления, коагуляции и фибринолиза переходит в распространенную коагуляцию, микросо-судистый тромбоз, СПОН. Лабораторные маркеры, характеризующие наличие коагулопатии при сепсисе (ранний, средний и тяжелый варианты течения), представлены на рис. 18.3 [34].
Коагулопатия, именуемая ДВС-синдро-мом, — наиболее частое осложнение сепсиса. ДВС-синдром представляет собой спектр патофизиологических явлений — от повышения фибринолиза до сверхактивной коагуляции. Клинически ДВС-синдром может проявляться как диффузное кровотечение (фибринолиз-доминантный ДВС) или диффузная гиперкоагулопатия (доминантный ДВС) [34].
Следовательно, ПОН является обобщенным клиническим лицом ДВС с преобладанием коагуляции и предвещает быстрый неблагоприятный исход [33].
Снижение уровня протеина С является одним из значимых и широко распространенных нарушений, наблюдаемых при ДВС, причем нарушения коагуляции, включая
Основные направления патогенетической терапии сепсиса 539
Инфекция/ травма
Воспаление Микротромбы Органная
недостаточность
* л*
Нарушение Микрососудистая
коагуляции дисфункция
Ранние признаки
протеина С
Tf,+2
? D-димеров
>1 TAT
Признаки средней тяжести
протеина С
tF1+2
АЧТВ/ПВ
Т фибриногена
? D-димеров
?ТАТ
тромбоцитов
Признаки тяжелой тяжести
ФФФ протеина С
Tf1+2
? АЧТВ/ПВ фибриногена
Т D-димеров
?ТАТ
4< тромбоцитов
Рис. 18.3. Лабораторные маркеры коагулопатии при сепсисе:
F1+2 — фибриновые мономеры; ТАТ — тромбин-антитромбиновый комплекс; АЧТВ/ПВ — активированное частичное тромбопластиновое время/протромбиновое время
Рис. 18.4. Порочный круг воспаления и коагуляции при сепсисе
дефицит протеина С, развиваются значительно раньше, чем главные клинические симптомы: за 2 ч до начала лихорадки, за 12 ч до развития септической формы ОПН и за 16 ч — до развития клинических признаков тяжелого сепсиса и септического шока.
Диагностически значимым является увеличение фибриновых мономеров (F1+2) (от 20 до 50 %) и D-димеров (от 80 до 250 %) [34,40].
Согласно концепции продленной коагулопатии, при сепсисе вследствие инфекции или травмы нарастание нарушений микрососудистого эндотелия формирует порочный круг воспаления и коагуляции (рис. 18.4).
18.6. ОСНОВНЫЕ НАПРАВЛЕНИЯ ПАТОГЕНЕТИЧЕСКОМ ТЕРАПИИ СЕПСИСА
Основные направления патогенетической терапии сепсиса представлены на основе методологии доказательной медицины, разработанной согласно рекомендациям руководства Управляющего комитета интернационального септического форума (ISF) для включения в соответствующие протоколы лечения [35].
Преобразование результатов исследований в ясные, четкие рекомендации для вра
чей составляет методологию доказательной медицины (evidence-based medicine) [51]. Подготовка таких рекомендаций обеспечивается путем включения в состав групп по их разработке специалистов по всем аспектам проблемы. Для выработки соответствующих рекомендаций по лечению сепсиса и септического шока создана международная неправительственная ассоциация, члены которой являются профес
540 СЕПСИС
сионалами в области медицины критических состояний и инфекционных болезней [52]. Члены Управляющего комитета акцентируют свои усилия на достижении международного консенсуса с целью понимания научных и клинических результатов исследований по проблемам сепсиса в мире. Лечебные рекомендации базируются на доказательной методологии с категоризацией по предварительному описанию в соответствии с правилами рейтинговой системы.
Рейтинговая система для сообщений:
уровень I — большие рандомизированные исследования, соответствующие четким критериям и характеризующиеся низким риском ложноположительных (а) и ложноотрицательных (0) ошибок;
уровень II — малые рандомизированные исследования с сомнительными результатами, умеренным риском ложноположительных (а) и ложноотрицательных (0) ошибок;
уровень III — нерандомизированные сравнительные исследования;
уровень IV — нерандомизированные исследования с историческим контролем и экспертной оценкой;
уровень V — серии случаев, неконтролируемые исследования с экспертной оценкой.
Рейтинговая система для рекомендаций:
степень А — поддержанные, по крайней мере, двумя наблюдениями I уровня;
степень В — поддержанные хотя бы одним наблюдением I уровня;
степень С — поддержанные лишь наблюдениями II уровня;
степень D — поддержанные, по крайней мере, одним наблюдением III уровня;
степень Е — поддержанные только наблюдениями IV или V уровня.
Методология использует содержание базы данных Medline за десятилетний период публикаций рукописей и релевантных изданий. Первое обсуждение с участием членов ISF состоялось в ноябре 1999 г. Согласительные рекомендации, выработанные в процессе совещания, предложены для включения в существующие лечебные протоколы [35].
Начало инфекции и диагноз сепсиса должны быть идентифицированы как можно раньше, когда хирургическое дренирование и антибактериальная химиотерапия наиболее эффективны для предупреждения прогрессирования болезни.
1. Определения сепсиса [52]:
1.1. Употребляемые в прошлом различные определения сепсиса, используемые как взаимозаменяемые, приводят к путанице;
1.2. Сепсис — это системный воспалительный ответ на инфекцию;
1.3. Не существует простых физиологических или лабораторных параметров, универсально определяющих сепсис;
1.4. Сепсис, тяжелый сепсис и септический шок определяют различные градации в процессе болезни, проявляющиеся комбинацией изменений витальных признаков, лабораторных параметров, гипоперфузией или органной дисфункцией;
1.5. Течение сепсиса, тяжелого сепсиа и септического шока коррелирует с возрастающей органной дисфункцией и летальностью;
1.6. Начало инфекции и диагноз сепсиса должны быть идентифицированы как можно раньше, когда хирургическое дренирование и антибактериальная химиотерапия наиболее эффективны для предупреждения прогрессирования болезни.
2. Диагностика инфекции при сепсисе [33]:
2.1. Бактериемия:
2.1.1. Лихорадка, озноб, гипотермия, лейкоцитоз, сдвиг влево нейтрофилов, нейтропения и, в случае подозрения на наличие инфекционного процесса, гипоальбу-минемия, развитие ренальной дисфункции или признаков гемодинамической нестабильности являются специфическими показателями для забора крови на гемокультуру;
2.1.2. Культура крови должна быть взята как можно раньше от начала лихорадки или озноба;
2.1.3. Культуру крови следует брать из новой вены;
2.1.4. Участки, ассоциированные с кожной контаминацией или повреждением кожи, должны быть исключены;
Основные направления патогенетической терапии сепсиса 541
2.1.5. Перед венопункцией кожа должна быть дважды обработана 70° спиртом или йодсодержащим раствором; культура крови забирается в чистую пробирку;
2.1.6. Игла для венопункции перед введением крови в культуру флакона должна быть заменена;
2.1.7. Объем крови должен быть адекватным (20 — 60 мл) — по 10 — 30 мл на флакон питательной смеси;
2.1.8. Для каждого эпизода подозреваемой бактериемии целесообразен забор минимум двух, максимум — трех проб крови;
2.1.9. У больных, находящихся в критическом состоянии, интервал между забором кровяной культуры и началом интенсивной терапии не является необходимым.
2.2. Инфекции, связанные с центральным венозным катетером (ЦБК) [33]:
2.2.1. Если ЦБК подозревается как источник инфекции, то диагноз подтверждается с помощью гемокультуры;
2.2.2. ЦБК следует удалить, если исключены другие источники бактериемии и позволяет состояние больного;
2.2.3. В случае нагноения места пункции отделяемое отправляется на бактериологическое исследование;
2.2.4. Наличие нагноения в области катетера требует его перестановки независимо от результатов бактериологического посева.
2.3. Вентилятор-ассоциированная пневмония (ВАП) [33]:
2.3.1. ВАП должна рассматриваться как источник сепсиса у части вентилируемых более 48 ч пациентов, после интубации, аспирации, применения назогастрального зонда для питания, использования антацидов;
2.3.2. При подозрении на ВАП обязательны двукратная гемокультура и рентгенография грудной клетки;
2.3.3. Плевральный выпот более 10 мл должен быть аспирирован и направлен на бактериологическое и биохимическое исследование (белок, ЛДГ, глюкоза) с одновременным биохимическим исследованием крови;
2.3.4. Аспират из эндотрахеальной трубки направляется на бактериологическое
исследование с экспресс-оценкой по Граму и на грибки;
2.3.5. При отсутствии противопоказаний выполняется бронхоскопия;
2.3.6. Инвазивные методы диагностики не имеют преимуществ перед указанными.
2.4. Хирургические инфекции и интра-абдоминальный сепсис [34]:
2.4.1. При подозрении на хирургическую или абдоминальную инфекцию культура крови обязательна;
2.4.2. Наличие гноя в ране или целлюлита — показание для ревизии раны;
2.4.3. При раневой инфекции подразумевается анаэробная коинфекция;
2.4.4. Первые признаки инфекции могут быть выявлены на УЗИ, при отрицательных результатах показана АКТ;
2.4.5. При выявлении скопления жидкости необходимо дренирование под УЗИ-или рентгенконтролем и бактериальное исследование аспирата.
2.5. Острый бескаменный холецистит (ОБХ) [32]:
2.5.1. ОБХ вероятен у септических больных, если имеются признаки напряжения в правом верхнем квадранте живота или повышены маркеры холестатического синдрома;
2.5.2. При подозрении на ОБХ должно быть ургентно выполнено УЗИ;
2.5.3. Если УЗИ неинформативно, показано АКТ;
2.5.4. Если АКТ невозможно, следует через 24 ч повторить УЗИ.
2.6. Синуситы [32, 33]:
2.6.1. Подозрение на острый синусит возникает: у больных с назогастральным зондом или назогастральной интубацией; при повреждении головы;
2.6.2. При определении наличия жидкости в придаточных полостях необходима рентгенография;
2.6.3. При неинформативности рентгенографии показана АКТ;
2.6.4. При выявлении жидкости обязательна пункция синусов до начала анти-биотикотерапии.
2.7. Инвазивные грибковые инфекции [33,34]:
2.7.1. Отсутствуют данные в пользу рутинного скрининга Candida-колонизации,
542 СЕПСИС
однако у септических больных грибковые инвазии более часты у пациентов, колонизированных до болезни;
2.7.2. Если сепсис развился у больных, колонизированных Candida в двух или более участках организма (кожа, слизистые, мочевой тракт), культура крови направляется для септических тестов;
2.7.3. Мазки из подозрительных участков направляются на сепситивные тесты.
3. Антибактериальная химиотерапия [33,35]:
3.1. Антибиотики при сепсисе:
3.1.1. Раннее применение адекватных антибиотиков снижает летальность у пациентов с грамотрицательными бактериями (градация А);
3.1.2. При кандидемии рекомендована противогрибковая терапия;
3.1.3. Противогрибковые агенты, например флюконазол, не должны применяться рутинно как эмпирическая терапия;
3.1.4. Для эмпирического лечения больных сепсисом без нейтропении монотерапия карбапенемами или цефалоспоринами III и IV генерации так же эффективна, как и комбинированная терапия 0 -лактамами и аминогликозидами;
3.1.5. Карбоксипенициллины или уре-идопенициллины расширенного спектра в комбинации с ингибиторами 0-лактамаз эффективны для лечения инфекций с лихорадкой, нейтропенией у больных с перитонитами или нозокомиальной пневмонией;
3.1.6. Монотерапия азтреонамом эффективна у пациентов с документированным грам [отрицательным] сепсисом;
3.1.7. Фторхинолоны показали высокую эффективность для лечения документированной грам [отрицательной] инфекции и удовлетворительную эффективность против грам [положительных] бактерий;
3.1.8. Неконтролируемое применение гликопептидов (ванкомицин или тейко-планин) для предполагаемой грам [положительной] инфекции у больных с тяжелым сепсисом или септическим шоком должно быть исключено; показано только у больных с тяжелым сепсисом, где доминирует метицилинрезистентный S. aureus.
3.2. Принципы деэскалационной антибактериальной терапии [36]:
3.2.1. Изначальная эффективная антибактериальная терапия является жизнеобеспечивающей;
3.2.2. Стартовый режим антибактериальной терапии определяет исход борьбы с патогенными возбудителями;
3.2.3. Правильным подходом к выбору антибактериальной терапии следует считать деэскалационную терапию;
3.2.4. Деэскалационная терапия — это эмпирическое антиинфекционное лечение, характеризующееся широким стартовым спектром с высокой вероятностью охвата возможных возбудителей и последующим (от 48 до 72 ч) переходом на терапию суженного спектра на основании микробиологических данных.
3.3. Клиническая эффективность антибактериальной терапии:
3.3.1. Выздоровление — полное исчезновение всех исходных симптомов и признаков заболевания;
3.3.2. Улучшение состояния, но без полного исчезновения признаков и симптомов заболевания при отсутствии необходимости дополнительной антибактериальной терапии;
3.3.3. Отсутствие эффекта — отсутствие динамики на фоне терапии, потребность в назначении дополнительной или другой антибактериальной терапии;
3.3.4. Рецидив — выздоровление или улучшение состояния к концу лечения с последующим ухудшением или повторным появлением инфекции;
3.3.5. Невозможно оценить — при прекращении лечения по любой причине через менее чем 48 ч от начала лечения или при прогрессировании другого патологического процесса;
3.3.6. Положительная клиническая эффективность включает случаи выздоровления и улучшения; другие результаты терапии — свидетельство неэффективности терапии.
3.4. Бактериологическая эффективность антибактериальной терапии:
3.4.1. Эрадикация возбудителя — исчезновение первоначальных возбудителей;
3.4.2. Предполагаемая эрадикация — невозможность получения материала для
Основные направления патогенетической терапии сепсиса 543
бактериологического исследования (в связи с зашиванием раны, исчезновением раневого отделяемого, мокроты или по другой причине) при условии положительного клинического эффекта;
3.4.3. Эрадикация с суперинфекцией — выделение новых микроорганизмов при повторном микробиологическом исследовании из места первичной локализации инфекционного процесса при появлении или усугублении клинических признаков инфекции;
3.4.4. Персистирование — сохранение первичного возбудителя к концу лечения;
3.4.5. Рецидив — эрадикация с последующим появлением первичного возбудителя во время лечения;
3.4.6. Случаи эрадикации и предполагаемой эрадикации расцениваются как положительный бактериологический эффект.
3.5. Характеристика неадекватной антибактериальной терапии:
3.5.1. Ранняя быстрая прогрессия ассоциирована с несвоевременным началом адекватной антибактериальной терапии;
3.5.2. Персистирующая инфекция также предполагает неадекватность антибиотического спектра и должна вести к смене режима терапии; если бактериологические данные указывают на адекватность спектра, следует увеличить дозу или заменить антибиотик на другой, лучше проникающий в ткани;
3.5.3. Начальное улучшение, за которым следует ухудшение, вызвано индукцией Р-лактамаз; следует предположить нецелесообразность выбора цефалоспоринов, суперинфекцию резистентным штаммом возбудителя, осложнения местного характера;
3.5.4. Медленное улучшение требует продолжения антибактериальной терапии.
4. Гемодинамическая поддержка.
Гемодинамическая поддержка включает жидкостную ресусцитацию, вазопрессорную терапию и инотропную поддержку [37, 38,43,44].
4.1. Жидкостная ресусцитация.
Септический шок характеризуется снижением эффективной капиллярной перфузии вследствие общих и распределительных нарушений системного и микроцир-
куляторного кровотока. Ведущим фактором, вызывающим нарушение тканевой перфузии, является гиповолемия. Начальные фазы экспериментального и клинического септического шока проявляются как синдром низкого сердечного выброса с низким АД, развитием гипердинамического соотношения только после восстановления объема. Недооценка степени гиповолемии может обусловить низкий сердечный выброс. У больных с септическим шоком наблюдается значительный дефицит жидкости. Во время начальной ресусцитации требуются кристаллоидные (6—10 л) и коллоидные растворы. Несмотря на сеп-сис-индуцированную миокардиальную депрессию, при ресусцитации жидкостью сердечный индекс может возрастать с 25 до 70 %. Приблизительно у половины септических больных с начальной гипотензией благодаря жидкостной ресусцитации удается восстановить гемодинамическую стабильность.
Внутрисосудистый объем можно пополнить переливанием эритроцитарной массы, кристаллоидов и коллоидов. Инфузионно-трансфузионную терапию лучше начинать с предварительной болюсной титрации по таким клиническим признакам, как ЧСС, диурез и АД. У больных, не реагирующих на начальную инфузию болюсов или имеющих низкие физиологические резервы, следует коррегировать терапию под гемодинамическим мониторингом. Повышение АД на 12 — 15 мм рт. ст. оптимизирует сердечный выброс. Более высокий подъем АД не улучшает конечно-диастолический или ударный объем, но повышает риск отека легких.
Ресусцитация должна титроваться по конечным признакам кислородного метаболизма и органным функциям; должен наблюдаться параллелизм между увеличением процента выживаемости, оптимизацией уровня системной £>О2, обратным развитием лактат-ацидоза и повышением рНР
Трансфузионная терапия. Оптимальные уровни НЬ и Ht для септических больных с септическим шоком не выявлены. Концентрация НЬ обычно колеблется в пределах 8—10 г/дл. Снижение концентрации
544 СЕПСИС
НЬ вызвано многими факторами, включая нарушения эритропоэза и гемодилюцию. Снижение этого показателя на 1 —3 г/дл отмечено во время ресусцитации с кристаллоидами и коллоидами. Эта степень анемии обычно хорошо переносится больными, так как ассоциируется со снижением вязкости крови, постнагрузки и повышением венозного возврата, в результате возрастают ударный объем и сердечный выброс. Снижение вязкости крови улучшает ее реологические свойства и облегчает микроциркуляцию. Кардиальная дисфункция может ограничивать повышение сердечного выброса в ответ на снижение вязкости крови, что приводит к неадекватному уровню DO2. При значительном гиперметаболическом состоянии повышения сердечного выброса не всегда достаточно для компенсации вследствие снижения артериального содержания кислорода, потенциально контролирующего системный окислительный метаболизм. Неспособность к экстракции кислорода вследствие анатомической несостоятельности (при коронарной болезни) или физиологических причин (при сепсисе), может обуславливать значительную зависимость между содержанием О2 и поддержанием окислительного метаболизма.
Ряд исследователей, изучив эффект трансфузионной терапии у критических больных с концентрацией НЬ от 8 до 10 г/дл, не выявили существенного улучшения тканевой перфузии. Другими авторами установлено, что повышение содержания О2 после трансфузионной терапии не столь эффективно для восстановления спланхнической перфузии, как повышение сердечного выброса. Трансфузия эритроцитарной массы с большим сроком хранения сопровождается снижением гастральной pH и усугубляет реологические нарушения, наблюдаемые при сепсисе. Более того, рандомизированные исследования критических больных с трансфузионными порогами 7 — 10 г/дл не выявили различий в клинически значимом исходе. Выраженная тахикардия, тяжелая десатурация 5г?О2, сердечная дисфункция вследствие коронарной болезни, лактат-ацидоз или низкие значения гастральной pH являются пока
занием для повышения уровня кислорода.
Эффективность жидкостной ресусцитации. Пациенты с септическим шоком могут быть успешно ресусцитированы как кристаллоидами, так и коллоидами. Повышение сердечного выброса и системного £)О2 пропорционально увеличению внутрисосудистого объема. Если кристаллоиды и коллоиды титруются по уровню подъема АД, они одинаково эффективны для восстановления тканевой перфузии.
Растворы кристаллоидов могут требовать в 2 — 4 раза большие объемы и больший период времени для достижения необходимых гемодинамических показателей, чем растворы коллоидов. Последние значительно дороже, чем растворы кристаллоидов. 5 % альбумин и 6 % ГЭК эквивалентны в количествах, требующих ресусцитации.
Осложнения жидкостной ресусцитации. Главными осложнениями при жидкостной ресусцитации являются легочный и системный отеки. Эти осложнения обусловлены тремя принципиальными факторами:
1) повышенным гидростатическим дав-. лением;
2) сниженным коллоидно-осмотическим давлением;
3) повышенной микрососудистой проницаемостью, наблюдаемой при септическом шоке.
Кристаллоиды и коллоиды по-разному поддерживают коллоидно-осмотическое давление. Большие объемы введенных кристаллоидов приводят к его снижению, в то время как инфузия коллоидов способствует его поддержанию на определенном уровне. В эксперименте снижение в плазме коллоидно-осмотического давления повышает внесосудистое содержание жидкости в легких. Снижение уровня гидростатического давления приводит к накоплению воды в легких. В ряде клинических исследований наблюдали корреляцию между снижением коллоидно-осмотического давления в легочной артерии, легочным окклюзионным градиентом давления и наличием легочного отека. В других работах отмечено отсутствие различий меж
Основные направления патогенетической терапии сепсиса 545
ду растворами и частотой повышения легочного отека при применении кристаллоидов. Эксперименты на моделях сепсиса не показали повышения внесосудистой жидкости в легких при поддержании гидростатического давления на низком уровне. Следовательно, если поддерживается низкий уровень давления, нет существенных различий между кристаллоидами и коллоидами в формировании легочного отека. Если для оптимизации сердечной деятельности у больных с сердечной дисфункцией требуется высокий подъем АД, то коллоиды могут уменьшать внесосудис-тое накопление жидкости. При повышенной сосудистой проницаемости коллоиды частично могут мигрировать в интерсти-ций, задерживать жидкость в легких, увеличивать легочный отек. Экспериментальные исследования, включая различные модели повышения микрососудистой проницаемости, не показали повышения содержания воды в легких или негативного влияния на легочные функции при их применении.
Системный отек является частым осложнением жидкостной ресусцитации. Роль увеличенной сосудистой проницаемости, повышенного гидростатического давления и сниженного коллоидно-осмотического давления в плазме крови в развитии этого осложнения при сепсисе не ясна. Отек тканей может снижать тканевое /?О2 вследствие увеличения расстояния для диффузии кислорода в клетки. При экспериментальных перитонитах терапия кристаллоидами в сочетании с коллоидами приводит к повышению набухания клеток эндотелия и снижению функциональных возможностей системных капилляров. Другие исследователи, применяя большие объемы кристаллоидов, не обнаружили их влияния на скелетные мышцы, интестинальный кислородный метаболизм, не выявили повреждения окислительного метаболизма, несмотря на формирование значительного тканевого отека. Снижение коллоидно-осмотического давления и развитие тканевого отека после ресусцитации кристаллоидами не отразились на целостности гастроинтестинальной мукозы как барьера для бактериальной транслокации. Причем главным для бактериальной транслокации
является не выбор жидкости (кристаллоиды, коллоиды), а эффективность ресусцитации.
Алгоритм жидкостной ресусцитации
Цель: восстановление тканевой перфузии и нормализация окислительного метаболизма.
Рекомендация 1 — степень С: инфузия жидкости должна быть начальным этапом гемодинамической поддержки.
Рекомендация 2 — степень С:
начальная жидкостная ресусцитация должна быть титрована по клиническим критериям: АД, ЧСС, диурез, перфузия почек, ментальный статус, концентрация лактата в крови; ЛО2; изотонические кристаллоиды или изоонкотические коллоиды одинаково эффективны.
Рекомендация 3 — степень D: инвазивный гемодинамический мониторинг следует применять у тех пациентов, которые не отреагировали на инициальную ресусцитацию; инфузия жидкости должна быть титрованной до уровня подъема давления, ассоциированного с наибольшим подъемом сердечного выброса и ударного объема; у многих пациентов этот уровень может определяться, когда ДЗЛК достигнет 12 — 15 мм рт. ст.
Рекомендация 4 — степень D:
концентрация гемоглобина должна удерживаться на уровне 80— 100 г/л; эта степень анемии ассоциируется со снижением вязкости крови, постнагрузки и повышением венозного возврата, ударного объема и сердечного выброса, улучшением реологических свойств крови и микроциркуляции; у больных с низким сердечным выбросом, десатурацией смешанной венозной! крови, лактат-ацидозом, увеличенным гастро-артериальным градиентом рСО2 или коронарной болезнью может быть рекомендована трансфузия до более высокого уровня гемоглобина.
4.2. Вазопрессорная терапия.
Если применение жидкостной ресусцитации не приводит к восстановлению адекватного АД и тканевой перфузии, то следует начать вазопрессорную терапию. К вазопрессорным агентам относятся допамин, норэпинефрин, эпинефрин и фенилэфрин.
546 СЕПСИС
Вазопрессорная терапия при наличии жизнеугрожающей гипотензии может быть начата даже тогда, когда сердечный компонент давления еще недостаточен для поддержания адекватной перфузии. Поскольку применение этих препаратов может привести к снижению органного кровотока путем артериальной вазоконстрикции, их конечные эффекты зависят от суммы прямых эффектов и повышения в органах перфузионного давления. В ситуациях, когда органная ауторегуляция потеряна, кровоток в органах зависит от давления. Нормальное перфузионное давление поддерживается, если кровоток в нем оптимальный. У пациентов с низким сердечным выбросом клиническое значение приобретают не только вазопрессорные, но и инотропные эффекты. Если начата инфузия вазопрессора, доза должна титроваться до восстановления САД без ухудшения ударного объема. Если ударный объем снижается, то соответственно должна снижаться доза вазопрессора или следует рассмотреть вопрос о применении добутами-на. При дисфункции правого желудочка во время инфузии вазопрессоров легочное сосудистое сопротивление следует удерживать на самом низком уровне на фоне восстановления адекватной системной гемодинамики. Особое внимание должно уделяться ренальной и гепатоспланхничес-кой циркуляции. Установлено, что после восстановления САД на фоне терапии вазопрессорами повышается почечное перфузионное давление, улучшаются ренальные функции, в частности, повышаются диурез и клиренс креатинина. У некоторых больных механизмы почечной ауторегуляции могут изменяться в зависимости от повышения перфузионного давления для увеличения почечного кровотока. Среднее кровяное давление зависит от преморбидного кровяного давления, но должно быть не меньше 75 мм рт. ст., т. е. индивидуальные ответы не должны превышать уровень, необходимый для восстановления диуреза. У части больных восстановление диуреза может достигаться при САД от 60 до 65 мм рт. ст.
Пищеварительный канал, в частности спланхническая перфузия и целостность
слизистой кишок, играет ключевую роль в патогенезе ПОН при сепсисе. Различные вазопрессорные агенты оказывают специфические эффекты на спланхническую циркуляцию, что следует учитывать при их выборе для применения у больных.
Осложнения при терапии вазопрессорами. Все катехоламиновые вазопрессоры могут вызывать значительную тахикардию, особенно у больных с неадекватной жидкостной ресусцитацией. У пациентов с сопутствующей коронарной болезнью, ва-зопрессориндуцированное повышение потребления кислорода миокардом может способствовать миокардиальной ишемии и инфаркту. При наличии миокардиальной дисфункции избыточная вазоконстрикция может снижать У О, сердечный выброс и £>О2. При инфузии вазопрессоров дозы должны быть осторожно титрованы для восстановления САД, чтобы не повредить У О. Если наблюдается нарушение У О, дозы вазопрессоров следует снизить или рассмотреть вариант применения добута-мина. При наличии дисфункции правого желудочка при септическом шоке, повышение преднагрузки в правом предсердии может ухудшить функцию правого желудочка. С целью профилактики подобной клинической ситуации при инфузии вазопрессоров следует удерживать легочное сопротивление на низком уровне, одновременно восстанавливая адекватную системную гемодинамику. Такие мощные вазопрессоры, как НА, снижают кровоток, вазопрессорная терапия должна обеспечивать адекватный уровень СИ для оптимизации почечного кровотока.
Применение вазопрессоров может приводить к повреждению кровотока в спланх-нической системе, это проявляется стресс-язвами, илеусом и мальабсорбцией. Проницаемость мукозного слоя кишок играет важную роль в патогенезе ПОН, поэтому снижение спланхнической микроциркуляции делает кишки более уязвимыми для £Ю2, чем другие органы. Следует избегать эпизодов интрамукозного ацидоза, которые могут выявляться снижением гастрального pH или повышением гастрального мукозного рСО2, поскольку исследования выявили снижение летальности у
Основные направления патогенетической терапии сепсиса 547
больных с септическим шоком при наличии физиологических величин pH или гастрального рСО2.
Алгоритм вазопрессорной терапии
Если соответственная жидкостная ресусцитация не приводит к восстановлению адекватного АД и органной перфузии, должна быть начата терапия с вазопрессорными агентами.
Рекомендация 1 — степень Е: допамин является агентом первой линии для повышения АД; катетеризация легочной артерии применяется для определения давления в легочной артерии, измерения сердечного выброса и 5гЮ2.
Рекомендация 2 — степень С:
допамин и НА эффективны для повышения АД при условии адекватной жидкостной ресусцитации; допамин повышает сердечный выброс больше, чем НА, но его применение может быть лимитировано тахикардией; НА обладает выраженным вазопрессорным эффектом.
Рекомендация 3 — степень D: адреналин должен назначаться при рефрактерной гипотензии.
Рекомендация 4 — степень Е:
рутинное применение низких доз допамина для поддержания почечной функции не рекомендуется, но они могут повышать почечный кровоток у некоторых больных, когда используются дополнительно к НА.
4.3. Инотропная поддержка.
Сепсис характеризуется гипердинамическим состоянием с нормальным или сниженным АД, нормальным или повышенным СИ, низким ОПСС. Поскольку у септических больных сердечный выброс обычно поддерживается объемной ресус-цитацией, многие исследования показали у них нарушение сердечной деятельности. Миокардиальная дистрофия характеризуется сниженными фракциями выброса, желудочковой дилатацией, снижением сократительного ответа на повышенный объем, низким пиковым давлением в конце систолы. Механизмы кардиальной дисфункции не известны. Миокардиальная ишемия не выражена, если коронарный кровоток адекватный и не наблюдается продукции лактата в области, окружающей
коронарные сосуды. Исследования у животных с эндотоксемией или бактериальной инфекцией показали, что миокардиальный отек, повреждение сарколемм или нарушение внутриклеточного кальциевого гомеостаза, снижение или прерывание р -адренорецепторной передачи могут вызывать кардиальную сократительную дисфункцию. Динамика воспалительных медиаторов, включая простагландины, PAF, TNFa, ИЛ-1, ИЛ-2, NO, также указывает на наличие миокардиальной депрессии. Исследования конца 80-х — начала 90-х годов XX ст. показали, что у септических больных обычно наблюдается линейная зависимость между DO2 и УО2 («патологически обусловленная зависимость»); нормальные величины DO2 были недостаточными для метаболических потребностей. Эти выводы легли в основу гипотезы о том, что ресусцитация выше нормальных значений СИ, DO2, VO2 («гипер-ресусцитация») может улучшать исход сепсиса. Недавние наблюдения внесли изменения в концепцию патологически обусловленной зависимости и гиперресусцита-ции. Поскольку величины СИ и £>О2 коррелируют с исходом сепсиса, неясно, что является причиной возрастания выживаемости: повышение этих показателей или наличие физиологических резервов у больных. Неопределенность существует во взглядах и на другие конечные результаты инотропной терапии. Дефицит £)О2 может вызывать лактат-ацидоз, в то же время повышение концентрации лактата не определяется дефицитом £>О2. У септических больных сатурация смешанной венозной крови обычно высокая, но значение этого показателя практически не коррелирует с сердечным выбросом. Мониторинг pHj является достоверным для определения адекватности кишечной перфузии. Таким образом, инотропные агенты следует использовать для поддержания адекватных СИ, САД, Sz?O2, pHj и диуреза.
Осложнения инотропной поддержки
У септических больных с неадекватной жидкостной ресусцитацией все инотропные агенты вызывают значительную тахикардию. При сопутствующей ишемичес
548 СЕПСИС
кой болезни изменение потребления кислорода миокардом может обусловить миокардиальную ишемию и инфаркты. Избыточные дозы катехоламинов могут вызывать миокардиальные некрозы независимо от наличия коронарной болезни. Применение инотропных агентов, имеющих вазодилататорную активность, приводит к снижению АД, которое сохраняется в течение длительного времени. Использование инотропных агентов, имеющих прессорную активность, вызывает нарушение кровотока в гепатоспланхническом регионе.
Алгоритм инотропной терапии
Цель: приведение сердечного выброса к физиологическим величинам; конечные результаты инотропной терапии: контроль инвазивного мониторинга сердечного выброса, концентрации лактата крови, гастроинтестинальной интрамукозной pH.
Рекомендация 1 — степень Е:
добутамин является агентом первого выбора для пациентов с низким СИ (< 2,5 л/мин на 1 м2) после адекватной ресусцитации жидкостью и достижения допустимого САД.
Рекомендация 2 — степень D:
у пациентов с доказанной тканевой гипоперфузией применение добутамина может быть полезным для повышения сердечного выброса и улучшения органной перфузии.
Рекомендация 3 — степень D:
вазопрессор норадреналин и инотропный агент допамин могут быть тщательно титрованы для поддержания САД и сердечного выброса.
Рекомендация 4 — степень С:
эпинефрин и допамин могут использоваться для повышения сердечного выброса, но при применении эпинефрина может снижаться мезентериальная перфузия, при применении допамина — гастральная мукозная.
5. Общие принципы хирургического лечения сепсиса'.
5.1. У больных с некрозом мягких тканей вследствие инфекции хирургическое вмешательство должно быть выполнено немедленно после гемодинамической ста
билизации (степень Е, рекомендуемый уровень IV и V).
5.2. При инфицированном панкреонекрозе для адекватной демаркации тканей хирургическое вмешательство должно быть выполнено у стабильных больных (степень С).
5.3. Диагноз интраабдоминальной инфекции следует устанавливать на основании данных УЗИ и КТ; УЗИ предпочтительнее, так как обладает аппаратной мобильностью, дешевле; КТ особенно показана при затеках в ретроперитонеум.
5.4. Выявление интраабдоминального абсцесса требует чрескожного дренирования.
5.5. Лапаротомия показана, если нечетко определена локализация, невозможно транскутанное дренирование или его выполнение недостаточно эффективно.
5.6. Хирургическое вмешательство необходимо для перитонеальной контаминации; риск недостаточности возрастает, если откладываются показания для чрескожного дренирования и только после повторного КТ вырабатываются показания для этой процедуры.
5.7. Данные из практики свидетельствуют в пользу концепции релапаротомии «оп demand» (по требованию), показанной в ряде клинических ситуаций, когда отсутствует эффект или прогрессирует органная дисфункция.
5.8. Плановая ре лапаротомия показана у больных с ишемией кишок («second look»), некротическими панкреатитами, при нечеткой демаркации некроза тканей, развитии кровотечений.
5.9. Диагностическая операция показана, если тканевой некроз определяется по характерным рентгенологическим признакам или если некроз относится к угрожающим жизни состояниям (например, интестинальная ишемия).
5.10. Инфицированный центральный венозный катетер должен быть удален (степень В, уровень II); однако нет доказательств, что перестановка катетера снижает риск катетерзависимой бактериемии; ЦВК должен быть заменен только если инфекция очевидна: имеются признаки воспаления, нагноения в месте пункции или
Основные направления патогенетической терапии сепсиса 549
если катетер не функционирует (степень С рекомендации для ЦВК, поддержанных уровнем II наблюдений и степень Е рекомендаций для периферических катетеров, поддержанных уровнем V наблюдений).
5.11. В случаях гастроинтестинальной перфорации резекция повышает риск потенциальных осложнений (степень D рекомендаций, основанных на уровне III наблюдений).
5.12. Первичный анастомоз или коло-ностомия одинаково эффективны; выбор плана оперативного вмешательства должен диктоваться такими факторами, как состояние больного, наличие сопутствующих хронических заболеваний, степень или длительность перитонеальной контаминации, опыт хирурга; при наличии факторов риска предпочтительнее колоностомия (степень D рекомендаций, основанных на уровне III наблюдений).
5.13. Интраабдоминальные инфекционные осложнения требуют контроля причин инфекции, что предусматривает применение современной диагностической техники; рентген-исследование не всегда обеспечивает диагностическое решение соответствующей хирургической проблемы.
6. Дыхательные пути и легкие при сепсисе'.
6.1. Обеспечение адекватной дополнительной оксигенации до достижения SaO2 > > 90 % путем применения простых систем доставки кислорода (назальная канюля или лицевая маска); у пациентов с эндо-трахеальной интубацией положительное давление в конце выдоха, приводящее к повышению среднего давления в дыхательных путях, может использоваться для снижения концентрации вдыхаемого О2 ниже потенциально токсичного (НО2 < 0,6).
6.2. Избегать применения неинвазивной вентиляции с позитивным давлением у пациентов с сепсисзависимым острым повреждением легких.
6.3. У септических больных с СОПЛ/ ОРДС показаны ранняя интубация и ИВЛ при следующей симптоматике: тахипноэ более 40 в минуту с участием дополнительной дыхательной мускулатуры, нарушении ментального статуса, тяжелой гипоксии, несмотря на использование О2.
6.4. У больных с ОРДС с низким легочным комплайнсом и/или значительным риском баро- или волютравмы возможна ИВЛ в режиме допустимой гиперкапнии путем снижения дыхательного объема вентиляции.
6.5. ИВЛ у больных с ОРДС должно проводиться малыми объемами (примерно 6 мл/кг идеальной массы тела) для поддержания конечно-экспираторного плато на уровне < 30 см вод. ст.
6.6. Позиция на животе должна применяться у пациентов, зависимых от высокого уровня вдыхаемого О2 (Fz’O2> 0,6), которым не противопоказаны позиционные изменения.
6.7. Применение NO показано у больных с жизнеугрожающей гипоксемией, не поддающейся традиционной стратегии ИВЛ (степень Е).
6.8. Умеренное применение кристаллоидов при установленном ОРДС; при ги-поонкотическом состоянии кристаллоиды сочетаются с коллоидными растворами; ограничение объема вводимой жидкости не улучшает прогноз (степень В).
6.9. Не показано рутинное применение кортикостероидов; рассматривается возможность применения пульс-терапии метилпреднизолоном при рефрактерном ОРДС после активного лечения инфекции (степень Е).
6.10. Постепенное отлучение от респиратора стабильных пациентов.
7. Иммунотерапия при сепсисе'.
7.1. Кортикостероиды не должны использоваться при тяжелом сепсисе или септическом шоке в высоких дозах (30 мг/кг) и коротким курсом (один-два дня); при рефрактерном шоке может применяться гидрокортизон (100 мг трижды в сутки в течение 5 — 10 дней со снижением дозы в соответствии с гемодинамическим статусом).
7.2. Ибупрофен не должен применяться при тяжелом сепсисе и септическом шоке; возможен положительный эффект ибупрофена у больных с гипотермией.
7.3. Простагландины не должны применяться при ОРДС во время сепсиса.
7.4. Пентоксифиллин не следует применять у взрослых с тяжелым сепсисом
550 СЕПСИС
до тех пор, пока не будет подтверждена его эффективность.
7.5. N-Ацетил цистеин не следует применять при тяжелом сепсисе.
7.6. Дополнительные данные не дают оснований для подтверждения первоначальных положительных рекомендаций относительно применения селениума.
7.7. Антитромбин III не следует применять при тяжелом сепсисе; страны, допускающие свободное его применение, должны пересмотреть свои позиции.
7.8. Иммуноглобулины не следует применять у взрослых и новорожденных с сепсисом; страны, допускающие широкое применение иммуноглобулинов, должны пересмотреть свою позицию.
7.9. Гранулоцитколониестимулирующий фактор не должен применяться при сепсисе.
7.10. Гормон роста (СТГ) не должен применяться при сепсисе, поскольку он повышает летальность.
7.11. Гемофильтрация не должна применяться у больных с тяжелым сепсисом и септическим шоком, пока дополнительные независимые исследования не подтвердят ее эффективность.
8. Другие виды поддерживающей терапии при сепсисе'.
8.1. Профилактика тромбозов глубоких вен снижает летальность у септических больных; особенно это касается больных с тяжелым сепсисом, септическим шоком и ПОН, имеющих низкий кардиопульмональный резерв и эпизоды малых тромбоэмболических ситуаций (степень А).
8.1.1. При отсутствии противопоказаний к гепарину, больные с сепсисом должны получать профилактику или низкими дозами гепарина (5000 ЕД два-три раза в сутки), или низкомолекулярными гепаринами в рекомендуемых дозах (степень А).
8.1.2. При наличии противопоказаний (тромбоцитопения, тяжелая коагулопатия, активное кровотечение, подозрение на внутричерепную гематому) применяются варианты механической профилактики (степень Е).
8.2. Исходя из того, что у септических больных развивается гиперметаболическое состояние, приводящее к белково-энерге
тической недостаточности и плохим прогнозам, должна обязательно проводиться нутритивная поддержка; виды и сроки этой поддержки у септических больных требуют уточнения.
8.2.1. Энтеральное питание является предпочтительным (степень С).
8.2.2. При наличии противопоказаний (мезентериальная ишемия, кишечная непроходимость) показано парентеральное питание (степень Е).
8.2.3. Специфические рекомендации для септических больных: ежедневная потребность в калориях составляет 104,67 — 125,7 кДж/кг, в протеинах — 1,3 —2,0 г/кг; в глюкозе — 30 — 70 % общих небелковых калорий с поддержанием уровня глюкозы примерно И ммоль/л, в липидах — 15 —30 % общих небелковых калорий, в эссенциальных жирных кислотах — 1 г/кг ежедневно.
8.2.4. Нет специфических рекомендаций по применению триглицеридов со средними по размеру цепями, аминокислот с короткими цепями или специальных добавок микроэлементов к питательным формулам; использование этих стратегий, отстаиваемых в ряде концепций, не имеет достаточных доказательств относительно улучшения клинического исхода у септических больных.
8.3. Больные с сепсисом предрасположены к риску возникновения стресс-язв и гастроинтестинальных кровотечений.
8.3.1. Профилактика стресс-язв рекомендована в подгруппе септических больных с гипотензией, шоком, коагулопатией, продленной ИВЛ (степень А).
8.3.2. Применение антацидов, сукраль-фата, Н2-блокаторов одинаково эффективно.
8.3.3. У септических больных с факторами риска раннее энтеральное питание является предпочтительным для предупреждения кровотечения из стресс-язв (степень В).
8.4. Выбор конкретных препаратов для профилактики тромбоэмболических осложнений и стресс-язв определяется индивидуально.
9. Экспериментальные направления терапии сепсиса.
Основные направления патогенетической’терапии сепсиса 551
В различных экспериментальных стратегиях лечения сепсиса с целью улучшения параметров гемодинамики применяются допексамин, ингибиторы NO, экстракорпоральная циркуляция для выведения токсических медиаторов.
Допексамин является синтетическим катехоламином, имеющим универсальное сочетание мощных 0 -адренергических и допаминергических эффектов. Этот препарат может стимулировать р2-, ДАр, Д2-рецепторы, восстанавливать органный кровоток вследствие своего вазодилатирующего действия. Теоретически препарат может повышать гепатоспланхническую перфузию самостоятельно или в сочетании с другими вазоактивными агентами, потенциально ее усиливать и предотвращать некоторые негативные эффекты сепсиса и септического шока. Допексамин улучшал тканевую оксигенацию в кишках, печени, скелетных мышцах у собак с эндотоксемией. A. Petros с соавт. [53], сравнивая почечные эффекты допексамина, допамина и добутамина в экспериментальных моделях сепсиса, установил, что у крыс после инфузии Escherichia coli почечная экскреторная функция наиболее эффективно улучшалась при приеме допексамина. Этот препарат лучше поддерживает АД, чем добутамин; по влиянию на САД разницы в указанных препаратах не отмечено.
Допексамин был исследован и в клинике. Colardyn с соавт. наблюдали кратковременное повышение СИ и ЧСС и дозозависимое снижение ОПСС. При длительном приеме может развиваться толерантность, и эти эффекты угасают. Отмечено повышение печеночного кровотока, значительное улучшение показателя гастральной pH. Улучшение спланхнической оксигенации не зависит от воздействия препарата на системную гемодинамику. Применение допексамина у больных с сепсисом и септическим шоком может ограничиваться значительным возрастанием ЧСС и возможным развитием гипотензии. Допексамин не может быть включен в режим стандартной терапии для лечения гемодинамической недостаточности на основе терапевтических доказательств у больных сепсисом.
Ингибиторы NO. Цитокины и другие медиаторы воспаления стимулируют макрофаги, моноциты, гладкие мышечные клетки и сосудистые эндотелиальные клетки к продукции NO — потенциального эндогенного вазодилататора. У больных с септическим шоком было отмечено повышение продукции NO, о чем свидетельствует увеличение его концентрации. Вазодилатация, стимулированная NO, играет важную роль в инициации и поддержании гипотензии. NO является также ведущим медиатором сепсисиндуцированной рефрактерности к вазопрессорным эффектам катехоламинов. Концентрация нитрата у септических больных коррелирует с системной сосудистой резистентностью. В настоящее время проводятся исследования с целью раскрытия путей активации NO-синтетаз, поскольку ингибиция этих механизмов, возможно, снизит частоту геморрагических нарушений при септическом шоке. NO синтезируется из эндогенного L-аргинина при участии энзима NO-син-тетазы (NOs), который может ингибироваться аналогом L-аргинина. Ингибирование NOs улучшает АД, снижает сердечный выброс и повышает ОПСС в эксперименте.
A. Petros с соавт. [53] использовали ингибитор NOs-NG-methyl-L-arginine у больных с септическим шоком, требующих прессорной терапии, и выявили повышение САД, снижение сердечного выброса и повышение легочного сосудистого сопротивления.
Продукция NO с помощью конституированной NOs в эндотелиальных клетках является важным модулятором сосудистой проницаемости, лейкоцитарной адгезии, ингибирования адгезии тромбоцитов и их агрегации. NO является важным компонентом в поддержании микрососудис-того кровотока, защищает печень от эндо-токсининдуцированного повреждения. Однако дальнейшие исследования ингибиторов NOs, в частности NG-methyl-L-arginine, были приостановлены вследствие повышения летальности в группе больных, получавших эти препараты.
NO расслабляет гладкую мускулатуру сосудистой стенки, активирует раствори
552 СЕПСИС
мую гуанилатциклазу и повышает концентрацию внутриклеточного цГМФ. Метиленовый синий подавляет эффекты NO на гуанилатциклазу. J. С. Preiser с соавт. [54] вводил 2 мг/кг метиленового синего бо-люсно больным с тяжелым септическим шоком, требующим адренергической терапии и доказали, что препарат повышал САД без снижения сердечного выброса или VO2, однако эффекты носили преходящий характер. Противоречивость полученных результатов как в эксперименте, так и в клинике, требует дальнейших исследований.
Экстракорпоральная мембранная оксигенация. С помощью экстракорпоральной циркуляции в виде продленной веновенозной и артерио-венозной гемофильтрации и экстракорпоральной мембранной оксигенации можно выводить медиаторы септического каскада, участвующие в гемодинамической декомпенсации. J. Boldt с соавт. [55] провели ретроспективный анализ у детей, получавших экстракорпоральную мембранную оксигенацию при рефрактерном септическом шоке, и обнаружили, что пациенты длительное время оставались живы, сокращался период продленной ИВЛ, что доказывает целесообразность применения указанного метода. Экстракорпоральная мембранная оксигенация облегчает гемодинамическую поддержку у педиатрических пациентов с менингоэнцефалитом, резистентных к традиционной терапии. У взрослых длительная гемофильтрация повышает клиренс TNF.
К сожалению, исследования были выполнены лишь в ограниченных ситуациях
и не могут быть экстраполированы на популяцию септических больных; данные об исходах заболевания весьма разноречивы. Если эндогенная продукция и клиренс медиаторов у септических больных является столь значимым, то экстракорпоральный клиренс не ясен. Данные о кинетике воспалительных медиаторов являются спорными. Выведение цитокинов и медиаторов приводит к осложнениям септического шока и не способствует улучшению клинических исходов, снижению летальности, улучшению гемодинамического статуса и восстановлению функций органа.
Указанные виды терапии нуждаются в дополнительной оценке после рандомизированных и контролируемых исследований, прежде чем быть рекомендованными для практического применения.
Не могут быть рекомендованы для рутинного клинического применения в силу отсутствия аргументированных экспериментальных и клинических доказательств эффективности [43] следующие методы:
гемосорбция;
лимфосорбция;
дискретный плазмаферез;
ультрафиолетовое и лазерное облучение крови;
электрохимическое окисление крови, плазмы и лимфы;
инфузия ксеноперфузата;
инфузия озонированных растворов кристаллоидов;
эндолимфатическая антибиотикотера-пия;
иммуноглобулины для внутримышечного применения.
Список использованной литературы
1. Shoemaker W. С. Relation of oxygen transport patterns to the pathophysiology and therapy of shock states // Inten. Care Med. — 1987. — N 13. - P. 230-243.
2. Shoemaker W. C. Early invasive and non-invasive monitoring of high risk surgical patients improves outcome // Тез. докл. и co-общ. 6-го Всероссийского съезда анестезиологов и реаниматологов (Москва, 7 — 10 октября 1998 г.). — М., 1998. — С. 44 — 45.
3. Wheeler А. Р., Bernard G. R. Treating patients with severe sepsis // N. Engl. J. Med. — 1999. - Vol. 340, N 3. - P. 207-214.
4. Advances in the diagnosis and management of the patients with sever sepsis / Ed. R. A. Balk. — The Trinity Worcester Press, 2002. - 118 p.
5. Raymond D. P., Pelletier S. I., Crabtree T. D. et al. Impact of antibiotic-resistant Gramnegative bacilli infections on outcome in hospitalized patients // Crit. Care Med. — 2003. - Vol. 31, N 4. - P. 1035-1041.
6. Linde-Zwabee W. T., Angus D. C.t CarcilloJ. et al. Age-specific incidence and outcome of sepsis in the US // Crit. Care Med. — 1999. — N27. - P. 33.
Список использованной литературы 553
7. Alberti С., Brun-Buisson Ch.t Burchardi Н. et al. Epidemiology of sepsis and infection in ICU patients from an international multicentre cohort study // Inten. Care Med. — 2002. - Vol. 28, N 2. - P. 108-121.
8. Ho P. L. Carriage of methicillin-resistant Staphylococcus aureus, ceftazidim-resistant Gram-negative bacilli, and vancomycin-resistant enterococci before and after intensive care unit admission // Crit. Care Med. — 2003. - Vol. 31, N 4. - P. 1175-1182.
9. Opal S. M.t Cohen J. Critical gram-positive sepsis: does it fundamentally differ from gramnegative bacterial sepsis? // Crit. Care Med. - 1999. - N 27. - P. 1608-1616.
10. Friedman G., Silva E., Vincent J. -L. Has the mortality of septic shock changed with time? // Crit. Care Med. — 1998. — N 26. — P. 2078-2086.
11. Members of the American College of Chest Physicians Society of Crit. Care Med. Consensus Conference Committee: American College of Chest Physicians / Society of Crit. Care Med. Consensus Conference: Difinitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis // Crit. Care Med. - 1992. - N20. - P. 864-874.
12. Vincent J.-L.'. Dear SIRS, I’m sorry to say that I don’t like you // Crit. Care Med. — 1997. - N25. - P. 372-374.
13. Ramsay G., Gerlach H., Levy M. M. et al. An international sepsis survey: A study of doctors’ knowledge and perception about sepsis // Crit. Care Med. - 2003. - N 31. - P. 300-305.
14. Stoiser B., Knapp S., Thalhammer F. et al. Time course of immunological markers in patients with the systemic inflammatory response syndrome: Evaluation of sCD14, sV-CAM-1, sELAM-l,MIP-l alpha and TGF-beta2 // Eur. J. Clin. Invest. - 1998. - N 28. - P. 672-678.
15. Taniguchi T.t Koido Y., Aiboshi J. et al. Chang in the ratio of interleukin-6 to interleukin-10 predicts a poor outcome in patients with the systemic inflammatory response syndrome // Crit. Care Med. - 1999. - N 27. -P. 1262-1264.
16. Ueda S., Nishio K.f Minamino N. et al. Increased plasma levels of adrenomedullin in with the systemic inflammatory response syndrome // Am. J. Respir. Crit. Care Med. — 1999. - Vol. 160. - P. 132-136.
17. Hietaranta A., Kemppainen E.t Puolakkainen P. et al. Extracellular phospholipases A2 in relation to systemic inflammatory response syndrome (SIRS) and systemic complication in severe acute pancreatitis // Pancreas. — 2002. - Vol. 18. - P. 385-391.
18. Bernard G. R., Vincent J-L., Laterre P. F. et al. Efficacy and safety of recombinant human activated protein C for severe sepsis //
"Lso
N. Engl. J. Med. - 2001. - Vol. 344. -P. 699-709.
19. Marshall J. C: SIRS and MODS: what is their relevance to the science and practice of intensive care? // Shock. — 2000. — N 14. — P. 586-589.
20. Levy M. M., Marshall J. C., Fink M. P. et al. 2001 SCCM/ ESICM/ АССР/ ATS/ SIS International Sepsis Definitions Conference //Crit. Care Med. - 2003. - Vol. 31, N4. -P. 1250-1256.
21. Angus D. C., Linde-Zwirble W. T., LidicerJ. et al. Epidemiology of sever sepsis in the United States: Analysis of incidence, outcome and associated cost of care // Crit. Care Med. - 2001. - Vol. 34. - P. 1303-1310.
22. Zeni F., Freeman B., Natanson C. Antiinflammatory therapies to treat sepsis and septic shock. A reassessment // Crit. Care Med. - 1997. - N 26. - P. 1095-1100.
23. Ferreira F. L., Bota D. P.t Bross A. et al. Serial evaluation of the SOFA score to predict outcome in critically ill patients // JAMA. — 2002. - Vol. 286. - P. 1754-1758.
24. Wilkinson J. D.y Pollack M. M.y Ruttiman A. et al. Outcome of pediatric patient with multiple organ system failure // Crit. Care Med. - 1986. - N3. - P. 271-274.
25. Proulx F., Fagan M.y Farrell C, A. et al. Epidemiology of sepsis and multiple organ dysfunction syndrome in children // Chest. — 1996. - N 109. - P. 1033-1037.
26. Doughty L. A., Carcillo J. A., Kaplan S, S. et al. Plasma nitrite and nitrate concentration and multiple organ failure in pediatric sepsis // Crit. Care Med. - 1996. - N 109. -P. 1033-1037.
27. Carcillo J. A. Task Forse Members, Fields Al: Clinical Practice Parameters for Hemodynamic Support of Pediatric and Neonatal Patients in Septic Shock // Crit. Care Med. — 2002. — N30. - P. 1-13.
28. Cook R.,Cook D. J ..Tilley J. et al. Multiple organ dysfunction: Baseline and serial component scores // Crit. Care Med. — 2001. — Vol. 29. - P. 2046-2050.
29. Christmann J. W.y Lancaster L. M.y Black-well T. S. Nuclear factor kappa B: a pivotal role in the systemic inflammatory response syndrome and new target for therapy // Inten. Care Med. - 1998. - Vol. 24 (11). -P. 1131-1138.
30. Сепсис и нозокомиальная инфекция / В. Ф. Саенко, В. И. Десятерик, Т. А. Перцева, Ю. М. Кривицкий, В. В. Шаповалюк. — Кривой Рог: Минерал, 2002. — 226 с.
31. Костюченко А. Л.. Костин Э. Д.у Куры-гин А. А. Энтеральное искусственное питание в интенсивной медицине. — СПб.: Спе-циал. лит-ра, 1996. — 330 с.
32. Angus D. С., Birmingham М. С., Balk R. А. et al. Е5 murine monoclonal antiendotoxin antibody in Gram-negative sepsis: a rando
554 СЕПСИС
mized controlled trial. E5 study Investigators //JAMA. - 2000. - N 283. - P. 1723 — 1730.
33. Levi M., ten Cate H. Disseminated intravascular coagulation // N. Eng. J. Med. — 1999. - N341. - P. 586-592.
34. Mesters R. M.y Helterbrand J., Utterback B. Q. et al. Prognostic value of protein C levels in neutropenic patients at high risk of severe septic complications // Crit. Care Med. — 2000. - Vol. 28, N 7. - P. 2209-2216.
35. Sprung C. L.y Bernard G. R.y Dellinger R. P. Introduction // Inten. Care Med. — 2001. — N27. - P. 1-2.
36. Astir M. E.y Rackow E. C. Septic shock // Lancet. - 1998. - N361. - P. 1501-1505.
37. Rosenberg R. D.y Aird W. C. Vascular-bed-specific hemostasis and hypercoagulate states // N. Engl. J. Med. - 1999. - N 340. -P. 1555-1564.
38. Esmon С. T. Inflammation and thrombosis. Mutual regulation by Protein C // Immunologist. - 1998. - Vol. 6, N 2. - P. 84-89.
39. Levi M.y ten Cate H.y Van der Poll T. et al. Pathogenesis of disseminated intravascular coagulation in sepsis // JAMA. — 1993. — N 270. - P. 975-979.
40. Mammen E. F. The haematological manifestations of sepsis // J. Antimicrob. Chemo-terapie. — 1998. — N 41. — P. 17 — 24.
41. Lorente J. A., Garsia-Frade L. J.y Landin L. et al. Time course of hemostatic abnormalities in sepsis and its relation to outcome // Chest. — 1993. - N 103. - P. 1536-1542.
42. Vervloet M. G., Thijs L. G., Hack С. E. Derangements of coagulation and fibrinolysis in critically ill patients with sepsis and septic shock. // Semin. Thromb. Hemost. — 1998. — N24. - P. 33-44.
43. Fourrier F.y Chopin C., Gondermand J. et al. Septic shock, multi pie organ failure,and disseminated intravascular coagulation. Compared patterns of antithrombin III, Protein C, and Protein S deficiencies // Chest. — 1992. — N 101. - P. 816-823.
44. Gando 5., Nanzaki S.y Sasaki S. et al. Activation of the externic pathway in patients with severe sepsis and septic shock // Crit. Care Med. - 1998. - N 26. - P. 2005-2009.
45. Opal S'., Thijs L.y Cavaillon J. M. et al. Roundtable I: relationships between coagulation and inflammatory processes // Crit. Care Med. - 2000. - N 28. - P. 34-37.
46. Kemme M. J., Burggraaf J., Schoemaker R. et al. The influence of reduced liver blood flow on the pharmacokinetics and pharmacodynamics of recombinant tissue factor pathway inhibitor // Clin. Pharmacol. Ther. — 2000. — N67. - P. 504-511.
47. Kidokoro A., Iba T.y Fukunaga M. et al. Alteration in coagulation and fibrinolysis during sepsis // Shock. — 1996. — N 5. — P. 223-228.
48. Hancock W. W.y Bach F. H. Immunobiology and therapeutic applications of Protein C (Protein S) thrombomodulin in human and experimental allotransplantation and xenotransplantation//Trends Cardiovasc. Med. — 1997. - N 7. - P. 174-183.
49. Smith О. P., White B.y Vanghan D. et al. Use of protein-C concentrate, heparin, and haemofiltration in meningococcus-induced purpura fulminans // Lancet. — 1997. — N 350. - P. 1590-1593.
50. Esmon С. T. The protein C pathway // Crit. Care Med. - 2000. - N 28. - P. 44-48.
51. Флетчер 3., Флетчер С., Вагнер Э. Клиническая эпидемиология. Основы доказательной медицины: Пер. с англ. — М.: Медиа Сфера, 1998. — 352 с.
52. Matof J.y Sprung С. L. Definition of sepsis // Summary of recommendations // Inten. Care Med. - 2001. - N 27. - P. 128.
53. Petros A., Zamb G., Zeone A. et al. Effect of a nitric oxide synthase inhibitor in humans with septic shock // Cardiovasc. Res. — 1995. — N28. - P. 34-39.
54. Preiser J. C.y Leyeune P.y Roman A. et al. Methylene blue administration in septic shock. A clinical trial // Crit. Care Med. — 1995. — N23. - P. 259-264.
55. Boldt J., Muller M., Helsen M. et al. Influence of different volume therapies and pentoxi-filline infusion on circulatory soluble adhesion molecules in critically ill patients // Crit. Care Med. - 1996. - N 24. - P. 358-391.
==^= Раздел------
19
Принципы рациональной антибактериальной терапии
Основными принципами рациональной антибиотикотерапии являются:
1. Оценка необходимости назначения антибиотика (АБ).
2. Обоснование выбора АБ.
3. Анализ степени проникновения АБ в пораженный инфекционным процессом орган и возможность обеспечения концентрации АБ, необходимой для эрадикации возбудителя.
4. Оценка необходимости комбинированного применения АБ.
5. Анализ безопасности применения выбранного препарата у конкретного пациента.
6. Выбор пути, дозы, кратности введения АБ.
7. Предполагаемая длительность терапии.
8. Выбор методов контроля за эффективностью и безопасностью применения АБ.
19.1. ЭТАПЫ РЕАЛИЗАЦИИ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
19.1.1. Оценка необходимости назначения антибиотиков
АБ показаны для терапии инфекционного процесса бактериальной этиологии. Правомочность назначения АБ обосновывается данными клиники, инструментальных и лабораторных методов исследования. При этом достоверными признаками бактериальной этиологии воспаления являются гнойно-воспалительные очаги, выявленные в ходе исследований или во время операций, наличие однотипной положительной гемокультуры при двух-трех бактериальных исследованиях в течение суток. К современным лабораторным методам верификации бактериальной природы воспалительного процесса относится регистрация прокальцитонина, в меньшей степени — С-реактивного протеина. Перспективный лабораторный метод — 18*
выявление увеличенной экспрессии относительно нормы активационных молекул HLA-DR+ на Т-лимфоцитах. Одновременный дополнительный анализ других маркеров системного воспалительного ответа позволяет детализировать характеристику воспаления. Так, зафиксированное на фоне увеличения экспрессии HLA-DR+ на Т-лимфоцитах уменьшение циркулирующего пула натуральных киллерных клеток (NK-клеток) может свидетельствовать об активном воспалительном процессе, тогда как нормальное или увеличенное количество NK-клеток — о хроническом процессе или длительном течении заболевания. Превышение активности противовоспалительных цитокинов над провоспа-лительными, выявленное в ходе лабораторного исследования, позволяет прогнозировать неблагоприятный исход процесса в связи с неэффективностью иммунно
556 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
го ответа. Последнее может быть обусловлено врожденным нарушением механизмов формирования иммунного ответа или неэффективностью иммунной реакции, которая может развиться при длительном течении патологического процесса или неправильной терапевтической тактике.
19.1.2. Эмпирическое и этиотропное обоснование выбора антибиотика
Ведущим принципом рациональной АБ терапии является обоснование выбора конкретного препарата. При этом возможны два методологических подхода — этиотропный и эмпирический. При первом возбудитель и его чувствительность к АБ идентифицируются дополнительными исследованиями. При эмпирическом подходе возбудитель предполагается. Предположение базируется на клиническом диагнозе, при этом учитывается локализация инфекционного процесса и механизм инфицирования — вне- или внутрибольничный. После формирования гипотезы о вероятном возбудителе выбирается АБ для его эрадикации на основе данных о природной резистентности и эпидемиологических сведений о приобретенной резистентности предполагаемого возбудителя к АБ в регионе и стационаре, результатов контролируемых клинических исследований эффективности отдельных препаратов. При этом для многих инфекционных процессов различной локализации эмпирический выбор АБ в настоящее время алгоритмизирован.
Клинический диагноз позволяет также вне зависимости от выбранного методологического подхода к микробиологическому обоснованию выбора АБ прогнозировать степень проникновения АБ в пораженный орган и возможность обеспечения концентрации препарата, необходимой для эрадикации возбудителя, выбрать путь, дозу и кратность введения АБ.
О значении клинического диагноза для эмпирического обоснования выбора АБ может свидетельствовать такой пример. При перфорации язвы двенадцатиперстной кишки кишечное и желудочное содержимое, попадающее в брюшную полость,
минимально инфицировано (ниже инфицирующей концентрации), поэтому у больных, профилактически не получавших АБ терапию, послеоперационные инфекционные осложнения встречаются не чаще чем в 12 % случаев, а бактериальная флора чаще всего бывает условнопатогенной и чувствительной к большинству АБ 0-лак-тамной группы. При перфорации язвы желудка бактериальное загрязнение желудочного содержимого почти в 30 тыс. раз выше, в большинстве случаев высевается патогенная флора, а частота послеоперационных инфекционных осложнений в среднем достигает 38 %. При наличии злокачественного новообразования желудка и перфорации его стенки бактериальная загрязненность еще выше, и частота послеоперационных гнойно-воспалительных осложнений даже при профилактическом назначении нерационально подобранных АБ средств достигает 58 % и выше. В желудочном содержимом, наряду с аэробной грамотрицательной флорой, как правило, высеваются неклостридиальные анаэробные возбудители. Располагая такой информацией, врач в большинстве случаев эмпирически может выбрать эффективные АБ средства.
Кроме того, по некоторым клиническим признакам инфекционного процесса с определенной степенью вероятности может быть установлен и микробиологический диагноз. Имеется целый ряд клинических признаков анаэробной неклостридиальной инфекции. По отдельным признакам можно даже предположить семейство, род и вид возбудителя. Например, серый, телесный или темно-коричневый цвет пораженных тканей или отделяемого из бактериального очага чаще всего свидетельствует о наличии В. melaninogenicus, а инфекция, сопровождающаяся резким снижением уже в первые дни уровня альбуминов в крови, гаптоглобина, гемопектина и трансферрина, как правило, обусловлена темно-пигментированными видами бактероидов. В. gingivalis вызывает разрушение всех четырех видов белка. При развитии абсцедирования в брюшной полости часто обнаруживают ассоциацию анаэробных и аэробных инфекционных возбудителей. Весь
Этапы реализации рациональной антибактериальной терапии 557
род бактероидов устойчив к аминогликозидам, а при наличии некоторых видов возбудителей применение АБ этого класса даже способствует прогрессированию инфекционного процесса. Рост культуры Fusobacterium сопровождается гнилостным запахом, который ранее отождествлялся с кишечной палочкой, поэтому так называемый колибациллярный запах свидетельствует о наличии фузобактериаль-ной флоры.
Эмпирический подход позволяет лишь предположить возбудитель инфекционного процесса, в ряде случаев такое предположение оказывается ошибочным. Так, в соответствии с данными, приведенными на 21-м Международном симпозиуме по интенсивной терапии и неотложной медицине (2001), неэффективность эмпирической АБ терапии в отделениях интенсивной терапии на примере анализа вентилятор-ассоциированной пневмонии (ВАП) составляет 22 — 73 % случаев. Это ведет к увеличению в два-три раза вероятности летального исхода по сравнению со стартовой терапией, к которой патогенные возбудители чувствительны. Таким образом, при микробиологическом обосновании выбора АБ предпочтение должно быть отдано этиотропному подходу. Возрастание роли этиотропного выбора АБ на современном этапе развития клинической микробиологии объясняется расширением круга возбудителей, вовлечением в спектр инфектов внебольничных процессов внут-ригоспитальных микроорганизмов, дальнейшим эволюционным совершенствованием механизмов АБ-резистентности. Следствием этого является наблюдаемая в ряде случаев неэффективность алгоритмов эмпирической АБ терапии и необходимость их постоянного совершенствования. Реализация этиотропного подхода обеспечивает идентификацию наиболее частых возбудителей болезни и чувствительности инфектов в каждом конкретном регионе и стационаре, что позволяет адаптировать алгоритмы эмпирического выбора АБ, повысить эффективность терапии и в конечном итоге уменьшить затраты на лечение. Кроме того, этот подход к обоснованию выбора АБ способствует эради-
кации причинного инфекта при оптимальном соотношении эффективность/стои-мость терапии и предотвращает селекцию резистентных микроорганизмов.
Этапы реализации этиотропного обоснования выбора АБ включают: выделение из исследуемого материала инфекта, доказательство его участия в развитии воспалительного процесса (исключение роли возбудителя как контамината), определение чувствительности причинного инфекта к АБ.
Успех выделения возбудителя и доказательство его участия в развитии воспалительного процесса при реализации этиотропного подхода зависят от многих факторов: правильного выбора вида и метода забора исследуемого материала, условий транспортировки, обработки, подбора методов выделения микроорганизма, определения чувствительности к АБ препаратам и интерпретации полученных данных. При этом при определении вида и метода забора изолята необходимо учитывать, что обсеменение (контаминация) слизистых оболочек, поверхности кожи микроорганизмами не обязательно означает, что именно они вызывают воспалительный процесс. Так, синегнойная палочка является наиболее частым возбудителем нозокомиальных процессов. Прекрасно сохраняясь во влажной среде во внутригоспитальных условиях, она в течение 2 — 3 суток контаминирует слизистые оболочки и кожу пациента. Это особенно характерно для больных в отделениях интенсивной терапии. Поэтому, например, у пациента с пневмонией, наиболее часто встречающимся инфекционным процессом, вероятность того, что возбудитель, выделенный с поверхности слизистой ротоглотки и трахеи, действительно является причиной пневмонии составляет всего 20 %. У таких больных специфичность результатов идентификации причинного инфекта можно увеличить до 80 % при условии выделения микроорганизма из глубоких отделов дыхательных путей, особенно с использованием бронхоскопии методом защищенной бронхоскопии или бронхоальвеолярного лаважа. Потребность в постоянном микробиологическом контроле слизистых обо
558 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
лочек верхних дыхательных путей у пациентов в отделениях интенсивной терапии обусловлена не обоснованием выбора терапии у конкретного пациента, а эпидемиологическими соображениями. Такой мониторинг с созданием базы данных частоты встречаемости возбудителей и их чувствительности к АБ позволил бы определить микробиологический паспорт отделения и обосновать выбор стартовых препаратов для терапии нозокомиальных инфекций. Этот подход к решению вопросов лечебной тактики обладает несомненным преимуществом перед контролем за микробиологическим состоянием отделения, который заключается в периодической регистрации микроорганизмов, контаминирующих поверхность стен и предметов.
В случае необходимости микробиологического исследования крови идеальным вариантом является ее забор до назначения АБ. Если больной уже получает АБ, по возможности их следует отменить как минимум на 24 ч, после чего осуществить забор крови. В тех случаях, когда отмена АБ невозможна, кровь следует забирать непосредственно перед очередным введением препарата. При этом стремление забора материала на «высоте температуры», практикующееся во многих отделениях, не обосновано вследствие закономерностей развития лихорадочной реакции.
Кровь для исследования забирают из периферической вены. При условии бактериальной природы воспалительного процесса не выявлено преимуществ забора крови из артерии. Имеются данные о предпочтительности анализа артериальной крови при грибковой этиологии воспаления, так как периферические органы и ткани выполняют роль своеобразного сита, задерживающего грибки. Кровь из катетера можно забирать только в случае подозрения на катетерсвязанный сепсис, при этом необходим микробиологический анализ материала, полученного из периферической вены и катетера. Если в обоих образцах обнаружен один и тот же возбудитель, высока вероятность роли катетера как источника сепсиса. При любом подозрении на связь сепсиса с катетером необхо
димо его извлечь. Уже только за счет этого возможно купирование сепсиса.
При заборе крови образец может быть контаминирован кожными сапрофитами. Поэтому для подтверждения истинной бактериемии необходимы две положительные гемокультуры. Для этого следует как минимум два раза забрать кровь из разных венозных доступов с интервалом 30 — 60 мин. Существенно повышает выявление возбудителя увеличение количества отбираемых проб крови до трех. Но дальнейшее их увеличение не имеет преимуществ перед трехкратным забором.
Исследованиями доказано, что объем отбираемой для микробиологического исследования крови более важен для результата, чем выбор среды или обеспечение условий для роста бактерий. В большинстве случаев в среду необходимо ввести 2 — 10 мл крови. В соответствии с наблюдениями увеличение объема крови с 2 до 20 мл повышает вероятность положительного результата на 30 —50 %. Увеличение объема более 30 мл существенно не повышает чувствительность культурального исследования, но при этом возрастает риск развития приобретенной анемии. Большой объем крови не может быть отобран у детей. Рекомендуется у новорожденных забирать для посева 1 мл крови, у детей до 10 лет — 1 мл на один год жизни. Соотношение объема крови к объему среды должно составлять 1 : 5 — 1 : 10. В таком случае соотношение между объемом крови и ее бактерицидным влиянием на рост микроорганизмов наиболее оптимально.
Предпочтительнее использовать стандартные коммерческие флаконы с готовыми питательными средами, а не флаконы со средами, приготовленными в местных лабораториях. Это обусловлено тем, что коммерческие флаконы не требуется вскрывать, таким образом предотвращается контаминация питательной среды микрофлорой окружающей среды. Кроме того, коммерческие среды стандартизированы. Кровь не должна забираться в емкости с антикоагулянтами (гепарин, цитрат, оксалат и др.) из-за их губительного действия на некоторые микроорганизмы.
Этапы реализации рациональной антибактериальной терапии 559
Материал из патологического очага одновременно с выполнением посева должен быть окрашен по Граму. Информация, полученная при этом, позволяет выяснить, относятся возбудители к палочкам или коккам, к грамположительным или грам-отрицательным бактериям, имеет место ассоциация микробов или монофлора. По окраске, форме, особенностям расположения при микроскопировании можно заподозрить анаэробную неклостридиальную инфекцию.
В соответствии со многими алгоритмами АБ терапии в настоящее время при идентификации возбудителя инфекционного процесса и исключения выделенного возбудителя как контамината рекомендуется определение колониеобразующих единиц (КОЕ). КОЕ — это количество жизнеспособных микроорганизмов в единице объема (1 мл, 1 г твердого материала). Проиллюстрировать значение определения КОЕ можно на примере постановки диагноза ВАП.
ВАП определяется как пневмония, развивающаяся через 48 ч и более после перевода больного на искусственную вентиляцию легких. При этом в большинстве клиник для постановки диагноза ВАП применяются лишь клинические критерии: подтвержденное рентгенологическим исследованием возникновение новых или прогрессирование имеющихся инфильтратов в легких, лихорадка, воспалительные изменения со стороны белой крови, гнойное отделяемое из трахеобронхиального дерева. Но такие критерии малоспецифичны. Постановка диагноза ВАП и соответственно определение тактики АБ терапии особенно затруднены при развитии у пациента респираторного дистресс-синдро-ма взрослых. Для установления диагноза ВАП, доказательства этиологической роли выделенного микроорганизма и исключения того, что идентифицированный инфект просто контаминирует слизистую оболочку дыхательных путей, обоснован и введен диагностический порог 106 КОЕ/мл. Это позволяет обосновать выбор АБ и повысить эффективность АБ терапии. Результаты проведенных исследований свидетельствуют о том, что указанный по
рог сохраняет свое диагностическое значение даже при условии введения больному АБ на протяжении 72 ч. Если же смена АБ произведена в течение последних 24 ч перед забором материала, для повышения чувствительности метода исследования целесообразно уменьшение порога до 102 КОЕ/мл. Применение приведенного критерия для выяснения роли выделенного возбудителя в генезе ВАП обосновано при условии использования для забора мокроты инвазивных методов: фибробронхоскопии с миниальвеолярным лаважем или защищенной биопсии слизистой оболочки.
Для определения у выделенной культуры микроорганизмов чувствительности к АБ применяют либо диффузионные методы (с использованием дисков с АБ или с помощью Е-теста), либо методы разведения (в жидкой питательной среде или в агаре). При этом наиболее простыми являются диффузионные методы определения чувствительности. Из них более совершенным является Е-тест, позволяющий определить значение минимальной ингибирующей концентрации (МИК) АБ для данного возбудителя. Необходимость более широкого внедрения в клиническую практику Е-теста объясняется тем, что при его использовании возбудитель может быть классифицирован не только как чувствительный или резистентный, но и как обладающий промежуточной чувствительностью. Такая градация обусловлена четкими количественными порогами: она определяется путем сопоставления зафиксированного МИК с общепринятым МИК для данного возбудителя. При этом в большинстве стран используются критерии чувствительности, разработанные Национальным комитетом по клиническим лабораторным стандартам США (National Committee for Clinical Laboratory Standards — NCCLS). Установив чувствительность или резистентность выделенного возбудителя к АБ, можно по данным исследования in vitro прогнозировать эффективность АБ in vivo. При промежуточной чувствительности возбудителя прогнозировать эффективность АБ in vivo более сложно. В таких случаях назначение тестируемого АБ пра
560 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
вомерно при условии использования препарата в максимальных дозах и/или локализации инфекции в органе, где АБ накапливается в больших концентрациях. Кроме того, анализ распространения в стационаре и на территории региона чувствительных, резистентных и с промежуточной чувствительностью штаммов возбудителя способствует повышению эффективности эмпирической АБ терапии.
Традиционные методы определения чувствительности к АБ in vitro в определенных случаях дают ошибочные результаты. Одной из причин этого является применение указанных методов к бактериям, продуцирующим 0-лактамазы расширенного спектра действия (extended-spectrum P-lactamases — ESBL). Отличие ESBL от других 0-лактамаз заключается в том, что они обладают способностью инактивировать АБ, содержащие оксииминовую цепочку: цефалоспорины III поколения (цефтазидим, цефтриаксон, цефотаксим, цефподоксим), оксииминовый монобактам азтреонам. Цефалоспорины III поколения во многих случаях рассматриваются как препараты стартовой АБ терапии преимущественно тяжелых инфекционных процессов, развивающихся по внебольнично-му механизму. Способность ряда микроорганизмов вырабатывать ESBL часто сопровождается устойчивостью к другим АБ — цефамицинам, комбинированным 0-лактамным препаратам, иногда к фтор-хинолонам и аминогликозидам. Кроме того, ESBL-продуценты могут быстро распространять гены АБ резистентности (опосредованно через плазмиды) между видами. Таким образом, идентификация ESBL-продуцентов как первопричины воспалительных процессов способствует обоснованию ряда эпидемиологических мероприятий в отделении и стационаре.
С диагностической точки зрения актуальность идентификации ESBL-продуцентов обусловлена тем, что традиционные микробиологические методы определения чувствительности возбудителя к цефалоспоринам III поколения во многих случаях могут давать ложноположительный результат: микроорганизм расценивается как чувствительный к этой группе препаратов,
в действительности он к ним резистентен. Поэтому, учитывая прогрессирующую способность ряда бактерий продуцировать ESBL, традиционные этапы этиотропного обоснования выбора АБ в настоящее время следует дополнить этапом исключения способности микроорганизма вырабатывать ESBL (апроксимирующий тест двойных дисков, специализированный Е-тест диагностики способности вырабатывать ESBL).
Наиболее частыми продуцентами ESBL являются нозокомиальные штаммы Klebsiella spp., в меньшей степени — Escherichia coli и Proteus mirabilis. Реже продукция ESBL отмечается у других представителей семейства Enterobacteriaceae, а также у некоторых неферментирующих грамот-рицательных палочек, включая Pseudomonas aeruginosa. Эти эпидемиологические данные являются обоснованием необходимости идентификации способности микроорганизма вырабатывать ESBL у всех нозокомиальных штаммов Klebsiella spp. и Е. coli, а также представителей Enterobacteriaceae, которые по данным предварительного тестирования проявляют пониженную чувствительность к одному из цефалоспоринов III поколения. В случае же выявленной способности синтезировать ESBL назначение цефалоспоринов III не показано даже при зафиксированной чувствительности микроорганизма к отдельным представителям этого класса препаратов; нецелесообразным также является назначение цефалоспоринов IV поколения. Возможно применение аминогликозидов и фтор-хинолонов при подтвержденной чувствительности in vitro; препаратами выбора являются карбапенемы (имипенем и меро-пенем).
Традиционные культуральные методы имеют низкую диагностическую ценность для идентификации так называемых атипичных микроорганизмов (хламидий, микоплазм, легионелл); микобактерий; вообще некультивируемых бактерий; некоторых микроорганизмов, весьма чувствительных к условиям забора исследуемого материала, транспортировки и культивирования, наличию специальных факторов роста (пневмококки, гемофиллы, нейссерии,
Этапы реализации рациональной антибактериальной терапии 561
микоплазмы, облигатные анаэробы и другие). В таких случаях используют либо серологические методы обнаружения (реакция связывания комплемента (РСК); реакция торможения гемагглютинации (РТГА); реакция иммунофлюоресценции; реакции пассивной и обратной пассивной гемагглютинации (РИГА, РОПГА); различные варианты иммуноферментного анализа), либо прямые методы идентификации микроорганизмов в исследуемом материале (реакция иммунофлюоресценции, имму-ноферментный анализ и другие). Примером является внедрение в клиническую практику методов диагностики микроорганизмов, вызывающих атипичную пневмонию: микоплазмы (М. pneumoniae), хламидии (С. pneumoniae), легионеллы (L. pneumophila). По результатам отдельных наблюдений, до 20 % выявленных пневмоний имеют смешанную этиологию с участием атипичных возбудителей. Для выявления роли атипичных инфектов в генезе пневмоний используются реакции иммунофлюоресценции, позволяющие идентифицировать антигены микроорганизма в мокроте. Для диагностики легионелл перспективным является метод обнаружения антигена возбудителя в моче. Хотя эта экспресс-методика специфична только для одного серотипа возбудителя, она позволяет документировать роль легионелл в 75 — 95 % случаев. Приведенные методы диагностики атипичных возбудителей являются относительно дорогостоящими и не всегда доступными. Поэтому в большинстве случаев, когда высока вероятность участия в воспалительном процессе таких микроорганизмов, эмпирически применяют АБ, эффективные в отношении атипичных микроорганизмов.
Доказательство участия выделенного инфекта как причинного в развитии воспалительного процесса основывается на сопоставлении вида микроорганизма и зоны локализации воспаления. Например, идентификация Е. coli в спинномозговой жидкости у больного с менингитом свидетельствует о том, что возбудитель является причинным, тогда как однократное выделение из ликвора какого-либо из эпидермальных микроорганизмов объясняется скорее
всего загрязнением образца. Кроме того, для доказательства роли выделенного инфекта в воспалительном процессе используется вышеприведенный анализ КОЕ. Теоретически «идеальными» для разграничения контамината и причинного инфекта являются серологические методы диагностики. Они позволяют зафиксировать ответную реакцию организма на бактериальный агент. Таким образом, в отличие от предыдущих методов, не просто идентифицируется возбудитель, а подтверждается его роль в развитии заболевания. Наиболее чувствительным методом является обнаружение специфических IgM и IgG при помощи иммуноферментного метода ELISA. Увеличение концентрации антител класса IgM свидетельствует об острой фазе инфекционного процесса.
Перспективным методом идентификации бактериальных агентов является полимеразная цепная реакция (ПЦР) — осуществляемый in vitro искусственный процесс амплификации (многократного копирования) специфической последовательности ДНК из мономерных нуклеотидных звеньев с помощью фермента ДНК-полимеразы. Таким образом, ПЦР является своеобразной заменой биологической амплификации (роста на искусственных средах) на ферментативное копирование нуклеиновых кислот in vitro. При этом реакция полимеризации инициируется специфическими короткими фрагментами ДНК — праймерами. За счет способности праймеров связываться со строго определенными участками ДНК согласно принципу молекулярной комплементарное™ достигается диагностическая специфичность ПЦР. Амплифицированные ДНК улавливаются специальными диагностическими системами, чаще всего методом гель-фильтрации.
ПЦР высокоспецифична и высокочувствительна, позволяет получить результат в течение короткого промежутка времени. Практическому внедрению ПЦР в клинику с целью идентификации бактериальных агентов препятствуют следующие моменты:
1. ПЦР узконаправлена: она не позволяет выявить сразу несколько микроорга
562 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
низмов как при посеве на среды. А во многих случаях, особенно при хирургической патологии, развитие инфекционного процесса обусловлено микробными ассоциациями.
2. ПЦР обладает чрезвычайной чувствительностью, что существенно увеличивает вероятность ложноположительного результата исследования. Так, реакция может быть положительной даже при незначительном количестве генетического материала в исследуемом образце, причиной появления которого может быть контаминация в клинических или даже лабораторных условиях без участия выявленного микроорганизма в развитии заболевания.
3. Возможность ложноотрицательного результата вследствие ингибирования реакции компонентами биологических образцов, например при использовании антикоагулянтов.
4. Высокая стоимость реакции.
Возможно, в ближайшее время практическая ценность ПЦР для клинической бактериологии возрастет вследствие использования реакции для идентификации в выделенном возбудителе генов, ответственных за антибактериальную резистентность. Так, разработан метод идентификации у стафилококка с помощью ПЦР генов, ответственных за появление метициллиновой резистентности. Это существенно ускоряет определение чувствительности возбудителя к АБ.
19.1.3. Методика деэскалации как вариант эмпирической антибактериальной терапии тяжелых инфекций
В большинстве случаев назначение АБ не может откладываться до идентификации возбудителя. Как показали результаты проведенных исследований, в таких случаях уровень смертности соответствует наблюдаемому при неадекватной АБ терапии. Поэтому эмпирическое назначение АБ у большинства больных является стартовым, а также доминирует в случаях неудачного выделения инфекта, что может иметь место даже в хорошо оборудованной микробиологической лаборатории.
Ведущим методологическим подходом к эмпирическому назначению АБ являет
ся разработанная для лечения пневмоний методика деэскалации (от препарата широкого спектра действия к препарату с узким спектром), или Таррагонская стратегия (название дано в честь города Тар-рагон в Испании, где она была разработана). Основные принципы деэскалацион-ной терапии инфекционных процессов от тяжелых до жизнеугрожающих: в качестве стартового назначается АБ широкого спектра в большой, индивидуально подобранной дозе, что позволяет захватить вероятные возбудители инфекционного процесса. Чаще всего при воспалении, развивающемся по внебольничному механизму, стартовыми являются цефалоспорины III поколения, защищенные аминопенициллины; при тяжелом инфекционном процессе внутрибольничного происхождения — карбапенемы (имипенем, меропенем). В течение последующих 48 — 72 ч осуществляется переход к АБ более узкого спектра, оказывающим действие на идентифицированный причинный инфект. Этот этап является обязательной составной частью деэскалации. Таррагонская стратегия позволяет обеспечить наилучший исход лечения при небольшой вероятности селекции микроорганизмов, резистентных к АБ.
19.1.4. Анализ степени проникновения антибиотика в пораженный орган
Не все обоснованные эмпирическим или этиотропным методом АБ могут быть назначены пациентам, поскольку они неодинаково распределяются между различными органами, тканями и средами организма. Поэтому на основе клинического диагноза следует выбрать препарат, обладающий способностью накапливаться в пораженных органах и тканях в концентрациях, превышающих МИК для предполагаемого или идентифицированного возбудителя (табл. 19.1 и табл. 19.2).
19.1.5. Выбор кратности введения антибиотика
Обычно прием лекарственных средств назначают 3 — 4 раза в день. Такой подход не всегда допустим при применении АБ.
Таблица 19.1. Антимикробный спектр важнейших антибиотиков
Микроорганизм Минимальная ингибирующая концентрация, мкг/мл
бензилпенициллина, ЕД/мл оксациллина диклоксациллина метициллина ампициллина карбенициллина
Staphylococcus aureus: чувствительные к бензилпенициллину 0,001-0,5 0,1-0,3 0,05-0,1 1,0-2,5 0,06-0,1 0,4-0,8
устойчивые, пенициллиназоположительные 7,5-100 0,3-0,6 0,1-0,5 1,2-3,2 12,5- > 100 12,5- > 50
Streptococcus pyogenes 0,001-0,25 0,01-0,4 0,1 0,15-0,2 0,02 0,25
Streptococcus viridans 0,001-0,5 0,5-0,9 0,1-1 0,8-3,1 0,06-1,6 0,2-0,6
Streptococcus faecalis 1,0-10 6,2-50 0,5-25 5-50-100 0,4-6 6,2-25
Streptococcus pneumoniae 0,02-0,04 0,02-0,1 0,1-1 0,1-0,2 0,01-0,15 0,5
Neisseria gonorrhoeae 0,001-0,02 0,1-4,8 0,1-0,3 2-12,5 0,01-0,6 —
Neisseria meningitidis 0,015-0,3 0,12-3,1 0,1-1 0,12-1 0,02-0,25 —
Escherichia coli 10- > 100 У У У 3,1-12,5 1,6- > 5,0
Enterobacter aerogenes У У У У У 0,5- > 5,0
Brucella spp. 0,15-6 — — — — —
Haemophilus influenzae 0,1-1 — 0,5-25 — 0,1-0,5 0,5
Proteus mirabilis 1- > 10 — — — 0,5 2,5- > 50
Proteus spp. (индол +) 2,5- > 100 — — — 1,56- > 50 1,6-5,0
Pseudomonas aeruginosa У У У У У 12,5- > 50
Klebsiella pneumoniae У У У У У > 100
Salmonella typhi 1,25-7,5 — — — 0,4-1,5 12,5
Salmonella spp. 3-16 — — — 0,7-8 6,2- > 25
Shigella spp. У — — — 0,6-8 5,0
Corynebacterium diphtheriae 0,002-0,5 — — — — —
Mycobacterium tuberculosis 5-100 — — — — —
Listeria monocytogenes 0,01-0,5 — 0,1-0,5 0,1 2,5 —
Actinomyces israeli 0,01-0,5 — — — —
Nocardiae asteroides 50-100 — — — — —
Bacillus anthracis 0,15-1 — — — 1,0 0,3-1,0
Clostridia spp. 0,3-0,5 — 0,01-0,1 0,025 0,25 0,5-1,6
Vibrio cholerae У — — 5,0 2,5 —
Yersinia spp. У У У У У У
сл
Этапы реализации рациональной антибактериальной терапии
Продолжение табл. 19.1
Микроорганизм Минимальная ингибирующая концентрация, мкг/мл
сизомицина тобрамицина амикацина рифампицина полимиксина эритромицина линкомицина фузидина
Staphylococcus aureus: чувствительные 0,03-0,5 0,05-1,6 1-5 0,001-0,1 — 0,3-2 0,4-1,6 0,1-5
к бензилпенициллину устойчивые, пенициллиназоположительные Streptococcus pyogenes 0,1-0,6 3-10 0,2-8 0,005-0,1 0,05-0,75 0,8- > 25 4-10
Streptococcus viridans — 10-25 0,5-10 — — 0,1-0,5 0,04-2 1-2
Streptococcus faecalis 1-32 3-25 5-25 0,001-1(5) — 0,1-2 0,02- > 5 1-5
Streptococcus pneumoniae 0,06-0,2 3-25 3-10 0,005-0,1 — 0,05-0,3 12,5- > 100 2— > 10
Neisseria gonorrhoeae — — — 0,001-0,1 20-100 0,05-1 0,005-0,4 0,1-1,0
Neisseria meningitidis — — — 0,001-1 20- > 100 0,1-1 6,25-25 0,1-0,75
Escherichia coli 0,1-4 0,3-5 0,5-8 1- > 10 0,1-5 50 > 25 100
Enterobacter aerogenes 0,01-1 0,3-6 0,5-4 5-20 0,5-5 50 > 100 —
Brucella spp. . — — — 1-10 0,5-5 10 > 100 —
Haemophilus influenzae — 0,5-5 0,5-2 0,1-5 0,1-3 1,5 6,25- >100 —
Proteus mirabilis Proteus spp. (индол +) 0,1-4 0,8-5 6-12 1-10 100 > 100 > 100 —
Pseudomonas aeruginosa 0,1-1 0,2-0,6 0,5-5 1-20 0,1-5 > 100 > 100 —
Klebsiella pneumoniae 0,02-1 0,2-5 0,5-4 1-10 0,3-5 5 > 100 —
Salmonella typhi 0,1-2 0,3-10 0,8-5 1-10 0,5-25 40 > 100 —
Salmonella spp. 0,2-5 1-10 1-5 5-20 0,2-2,5 100-500 > 100 —
Shigella spp. 0,1-5 0,5-2,5 1,6-6,2 1-10 0,1-3 25 > 100 —
Corynebacterium — — 0,5-5 1-5 — 0,1-2 2-16 —
diphtheriae Mycobacterium — — — 0,005-0,5 40-50 — — —
tuberculosis Listeria monocytogenes (синт. среда) 0,5-10 (яичная среда) 0,1-0,5 0,3-3 0,75-3
Actinomyces israeli — — — 1 0,1-1,0 0,3-0,5 1-4 —
Nocardiae asteroides — — 0,8-10 100 50-100 — 0,1-5 —
Bacillus anthracis — 1,6-10 — 0,1-1 0,1-1 0,5-2,5 0,75-5 4-12
Clostridia spp. — — — 0,01-0,1 0,5-5 1-5 7,5-25 0,01-15
Vibrio cholerae — 1,6-12,5 3,2-12,5 0,1-5 25 2,5- > 10 > 50 —
Yersinia spp. 0,5-3,2 1,6-10 1,6-6,2 0,8-3,1 У У — —
ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
Продолжение табл. 19.1
Микроорганизм Минимальная ингибирующая концентрация, мкг/мл
цефалотина цефалоридина цефалексина левомицетина тетрациклина стрептомицина канамицина гентамицина
Staphylococcus aureus: чувствительные к бензилпенициллину 0,05-0,2 0,01-0,1 0,5-6,2 0,5-7 0,1-7 0,5-10 0,5-10 0,03-0,4
устойчивые, пенициллиназоположительные 0,1-6,2 0,01-12,5 1,6-6,25 0,5-5 0,1-5 2-10 10- > 50 0,5-6
Streptococcus pyogenes 0,001-0,05 0,001-0,02 0,03-0,2 0,5-5 0,1-3 0,5-10 25-100 6-12
Streptococcus viridans 0,05-0,8 0,02-0,8 0,4-3,1 0,5-10 0,3-5 10-50 15- > 50 1,5- > 10
Streptococcus faecalis 25-50 6,2-25 10-50 0,2-5 0,1-5 2-10 1-50 0,1-6
Streptococcus pneumoniae 0,01-0,05 0,003-0,1 0,8-3,1 0,5-3 0,1-1,5 2-10 2,5-12,5 0,8-6,2
Neisseria gonorrhoeae — 0,1-0,8 0,3-0,2 0,5-3 0,15-1,5 2-10 2-12,5 6,2-3,1
Neisseria meningitidis — 0,8-6,2 0,6-1,6 0,5-12 0,5-5 0,5-10 5-5 0,2-3,1
Escherichia coli 3,25 1,6-25 6,1-25 1-15 0,75-15 3-10 2,5-15 0,8- > 10
Enterobacter aerogenes 1,5-25 100 100 1-5 0,1-2 1-5 2,5-5 0,08-1,6
Brucella spp. — — — 0,5-5 0,1-5 1-10 2,5-10 1,5
Haemophilus influenzae — 12,5- > 100 3-5 2,5-10 1,0- > 100 1,5 — —
Proteus mirabilis 1,5-25 1,5-25 3,1-25 2,5-15 7,5-100 1-15 10-15 0,5-4
Proteus spp. (индол +) 100 100 100 10- > 100 3- > 100 2— > 10 25-100 0,1-2
Pseudomonas aeruginosa > 100 > 100 100 2,5- > 10 0,5- > 100 1- > 100 1-10 0,1-4
Klebsiella pneumoniae 5- > 100 2,5- > 100 3,2- > 50 1,5-10 0,5-10 0,5-10 0,1-3,2 —
Salmonella typhi 1,5-6,0 1,6-6,2 3,2-12,5 2,5-12,5 1-5 0,5-10 1-10 0,25-2
Salmonella spp. 3,2-12 3,2-12,5 6,2- > 25 2,5-12,5 0,3-5-10 1,0-5,0 1-10 0,5-6
Shigella spp. 1,5- > 10 1,0-8,0 6,2-12,5 0,5-5 0,5-5 1-6 0,5-5 0,1-0,2
Corynebacterium diphtheriae — 1,0 — 1-10 0,5-5 5-15 0,1-0,5 —
Mycobacterium tuberculosis — 0,5-5 — 0,3-1 1-2,5 1-5 — —
Listeria monocytogenes — 1-5 — 0,5-5 10-15 0,5-5 — —
Actinomyces israeli — 4-10 — 1,5-10 5 5-25 — —
Nocardiae asteroides — 4-10 — 0,5-5 0,5-5 0,5 — —
Bacillus anthracis 0,1-1,0 1-62 — 2-10 0,1-5 1-10 100 —
Clostridia spp. 0,1-1,0 0,1-1 0,5-5 0,01-25 0,5-50 0,2-20 1,6-12,5 —
Vibrio cholerae — 0,5-10 — 2,5-10 0,2-6,2 1,5-3,0 — —
Yersinia spp. — — 12,5-25 — — — — —
СП сл
Этапы реализации рациональной антибактериальной терапии
Продолжение табл. 19.1
Микроорганизм Минимальная ингибирующая концентрация, мкг/мл
имипенема (тиенама) меропенема (меронема) левофлоксацина (таваника)
Staphylococcus aureus: чувствительные к бензилпенициллину устойчивые, пенициллиназоположительные Streptococcus pyogenes Streptococcus viridans Streptococcus faecalis Streptococcus pneumoniae Neisseria gonorrhoeae Neisseria meningitidis Escherichia coli Enterobacter aerogenes Brucella spp. Haemophilus influenzae Proteus mirabilis Proteus spp. (индол +) Pseudomonas aeruginosa Klebsiella pneumoniae Salmonella typhi Salmonella spp. Shigella spp. Corynebacterium diphtheriae Mycobacterium tuberculosis Listeria monocytogenes Actinomyces israeli Nocardiae asteroids Bacillus anthracis Clostridia spp. Vibrio cholerae Yersinia spp. 0,008-4,0 0,03-32 0,004-0,06 0,004-0,16 0,5-8,0 0,008-0,5 0,03-1,0 0,016-0,03 0,03-0,5 0,12-2,0 0,016-2,0 0,12-8,0 0,25-8,0 0,25-8,0 0,12-0,5 0,06-1,0 0,03-2,0 0,004-4 0,12-0,5 < 0,008-4 0,008- > 128 < 0,008-4 < 0,008-0,25 < 0,008-64 < 0,008- > 16 < 0,008- > 16 < 0,008- > 128 < 0,008-32 < 0,008-8 0,25-2 0,5-2 0,5-1 0,004-0,008 0,008-0,015 0,015- > 64 0,015-8 0,008-0,06 0,03-32 0,03-0,5 0,06-32 0,06-1,0 0,015-0,06 0,015-8 0,12-0,25 0,25-1 1-2 0,25-32 0,5-64 0,25 0,002-0,06
Примечание. У — устойчивые.
ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
сл СП СП
Таблица 19.2. Фармакокинетические свойства антибиотиков
Название антибиотика Максимальный уровень в плазме, мг/мл Связывание белка плазмы, % Максимальная концентрация в плазме, ч Метаболизм, % Элиминация почками: продукты расщепления не метаболизируются, % Элиминация печенью: продукты метаболизируются, % В какие ткани и органы хорошо диффундируют Концентрация в ликворе, мг/мл или в сыворотке, % Снижение концентрации после гемодиализа Снижение концентрации после перитонеального диализа
Пенициллин G Пенициллин V (орально) Оксациллин Диклоксациллин Флуклоксациллин Ампициллин Мезлоциллин Азлоциллин Пиперациллин Апалциллин Карбенициллин Тикарциллин Цефазоллин Цефамандол Цефуроксим 10 6 1,2-2 18-20 40 199 300 220 300 218 70-140 85 50 55 80 90 95 95 20 40 40 20-50 90 50 60 80 70 30 0,5-0,7 0,5 0,5 0,75 1 0,5-1 1 1 1 1,3 1 1-1,3 0,83 0,67 — 1,00 0,7 Частичный 50-75 0,3 0,3 0,3 10-20 30-50 30-40 0 50 ? 15 Цефа^ 0 0 0 80 30 < 24 ч 25 < 24 ч 65 < 24 ч 30-50 70 60 60 70-80 20 80 Преимущественно носпорины > 90 65-85 90-95 Незначительно 30 Желчь 8 Желчь (12) Минимально I—II поко. Минимально Минимально Минимально Кожа, слизистые, легкие, желчь, почки Слизистые, плевра, желчь, почки,синовия Плевра, кости, почки, синовия Плевра, кости, почки, синовия Плевра, кости, почки, синовия Плевра, желчь, почки, околоплодная жидкость Желчь, легкие, околоплодная жидкость Только в желчь и слизистые (бронхи) Желчь, почки, материнское молоко Желчь, почки, материнское молоко Во все ткани нения Желчь, почки, плевра, перитонеум, синовия Желчь, кости, плевра, слизистые, жир Желчь, кости, плевра, слизистые, жир < 1 (4) * 20 20 ? ♦ 0 0 d d d d d d d d d d d d d d d d d d d d d d d d d d d
сл со
Этапы реализации рациональной антибактериальной терапии
сл CD со
Продолжение табл. 19.2
Название антибиотика Максимальный уровень в плазме, мг/мл Связывание белка плазмы, % Максимальная концентрация в плазме, ч Метаболизм, % Элиминация почками: продукты расщепления не метаболизируются, % Элиминация печенью: продукты метаболизируются, % В какие ткани и органы хорошо диффундируют Концентрация в ликворе, мг/мл или в сыворотке, % Снижение концентрации после гемодиализа Снижение концентрации после перитонеального диализа
Цефокситин Цефотетан Цефотаксим Цефоперазон Цефтазидим Цефтриаксон Цефалоксим Гентамицин Тобрамицин Сизомицин Нетилмицин Амикацин Имипенем Азтреонам 75 185 20-80 244 40-200 120-260 30 5-8 4-8 4-8 6-8 20-25 | 20-40 | 48 50 ? 40 90 ? 90 50 0-5 0-5 0,50 0,50 4-10 1 15 I 60 0,6 3,5 1,25 2 1,7-2,1 6-9 1,7-9 2 2 2 2 2,3 | 1 | 1,7-2 < 5 0 Цеф; 30-50 75 < 5 Незначит. < 5 0 0 0 0 0 | 30 I 20-30 95 80 1ЛОСПОрИН1 55 25-30 90 40-60 85 Аминогл 90 < 24 ч 90 < 24 ч 85 < 24 ч 80 < 24 ч 95 < 24 ч Карбо] | 65-80 Моноб |70 < 24 ч Минимально Желчь 4 III ПОКОЛ< Желчь Желчь (70) Умеренно Желчь (35-40) Умеренно икозиды Минимально Минимально Минимально Минимально Минимально пенемы актамы Хорошо Кости, почки, желчь, жир, гной Почки, желчь, перитонеум ения Кости, почки, желчь, жир, кожа Во все ткани Кости, желчь Кости, почки, желчь, жир, кожа Кости, почки, желчь, жир Почки,синовия, умеренно в плевру, слизистые Почки,синовия, умеренно в плевру, слизистые, жир; плохо в желчь, кости Почки,синовия, умеренно в плевру, слизистые Почки,синовия, умеренно в плевру, слизистые Почки, синовия; умеренно в слизистые, плевральную полость, плаценту Почки, желчь, плевра, 1 кости, синовия | Желчь, почки 0 0 10-20 Вариабельно <15 мг/мл * * * *** *** 10-20 d d d d d d d d d d d d d d d d d d d d
ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
Доксициклин Норфлоксацин Ципрофлоксацин Офлоксацин Эритромицин Метронидазол Клиндамицин Ванкомицин Фосфомицин Левомицетин Тейкопланин Триметоприм Сульфаметоксазол Амфотерицин В Флюконазол Миконазол Кетоконазол 1,8-2,9 1-2 1-25 3-3,5 2 — 4 13-15 2,5-3 25-35 40-60 6-12 71-74 1,5-3 50-60 2-3 10-50 5-7 1-20 90 15 30 10 20-50 (90) 20 90 10 0 30-50 90 15-60 70 10 10 92 98 15-17 3-4 3-4 3-10 1,5 7-8 2,5 4-8 2 3 44-70 9-12 9-11 0,33-0,40 3-6 20 (2 фазы) 2-8 (2 фазы) 50 20 20 5-10 40-50 40 Высоко Незначительно 0 80-90 0 10-15 20-30 ? 0 Печень Печень Тетрац! 35 Другие ан 30-50 30-60 80-95 10-15 Хорошо 15 80-90 Высоко 90 43-67 ? 80-90 < < 24 ч Противог 2,3-13 < < 24 ч 40 < 1 не- деля 80 15-20 5 (метаболиты) иклины Желчь тибиотики Хорошо (30) Желчь 20-30 Желчь Желчь (плохо) Минимально Умеренно Минимально рибковые ? Желчь Желчь Плевра, синовия, почки, желчь, кости, плод, материнское молоко Желчь, почки, легкие, мышцы, простата, матка Желчь, почки, слизистые, мышцы, простата Желчь, почки, слизистые, мышцы, простата Плевра, синовия, почки, желчь, кости, плод, материнское молоко, простата Желчь, легкие, кости, жир, кожа, матка, перитонеум, плод, материнское молоко, полости Почки, плевра, кости, желчь, абсцессы, материнское молоко Плевра, синовия, абсцессы, почки Раневой секрет, кости, мышцы, кожа, плевра, плод Во все органы Кожа, кости, легкие, почки, желчь Слизистые, почки, кости, простата, легкие Плевра Почки, плевра, синовия, асцитическая жидкость Легкие, почки, синовия, слизистые, асцит Во все ткани Синовия; умеренно в почки, материнское молоко; плохо в желчь и кости ♦ * ♦ * * * * * ♦ ♦♦* (ликвор и мозг) * * 10-20 % * 50 % * ♦ 0,1-0,5 60-90 % ♦ * * d d d d d d d d d d d d d d d d d d d d d d d d d d d d
Примечание. 0 - не действует; ? - неизвестно; d — диализируется; * — умеренно проникающий АБ; ** — плохо проникающий АБ; * — незначительно проникающий АБ; **** — высокопроникающий АБ.
Этапы реализации рациональной антибактериальной терапии
570 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
Выбор дозы и кратности введения АБ является одним из главных принципов рациональной АБ терапии. 50 лет назад Eagle с соавт. в эксперименте на животных доказали возможность уменьшения суммарной дозы пенициллина G, необходимой для терапии сифилиса и стрептококковой инфекции, если препарат вводить в виде постоянной инфузии. Авторы также установили, что максимальной активностью пенициллины обладают при превышении МИК для данного микроорганизма в три раза. Эти результаты были подтверждены последующими исследованиями. Они послужили основанием для обоснования выбора наиболее оптимального способа введения АБ, при котором наблюдается увеличение эффективности препаратов и уменьшение токсичности.
С математической точки зрения активность АБ может быть описана как производное концентрации препарата и длительности контакта АБ с микроорганизмом (фармакокинетический параметр — площадь кривой концентрация: время, concentration-time curve, AUC). При таком подходе можно выделить две группы АБ. Препараты первой группы называют «время-зависимыми». К ним относят все р-лак-тамные АБ, гликопептидные препараты, некоторые макролидные АБ (эритромицин), рифампицин, клиндамицин. Механизм действия таких АБ связан с влиянием на клеточную стенку бактерий. Время-зависимые АБ обладают максимальной активностью тогда, когда их концентрация постоянно превышает МИК для данного возбудителя в 2-4 раза. При дозировании время-зависимых АБ следует обратить внимание на величину однократной дозы и периодичность введения. При условии превышения МИК в 2 —4 раза эффективность время-зависимых АБ максимально выражена в случае частого введения препаратов с минимальными интервалами между инъекциями.
Препараты второй группы называют «концентрационнозависимыми» АБ. К ним относят аминогликозиды, фторхинолоны, метронидазол, амфотерицин В, некоторые макролиды (кларитромицин, азитромицин). Механизм действия концентрацион
нозависимых АБ связан главным образом с угнетением синтеза белков и нуклеиновых кислот в микроорганизме. Препараты этой группы обладают максимальной активностью при условии существенного превышения МИК. Так, активность аминогликозидов и фторхинолонов максимальна при превышении МИК в 10—12 раз. Вследствие этого дозирование таких АБ преследует цель достичь максимально возможную концентрацию в крови и пораженной ткани. Это способствует гибели бактерий, а постантибиотическое действие (задержка пролиферации микроорганизма после удаления АБ из инкубационной среды) предотвращает повторный бактериальный рост. Таким образом, увеличение интервала между очередными инъекциями препаратов из группы концентрационнозависимых АБ способствует усилению их активности. Однократное суточное введение аминогликозидов вызывает не только увеличение активности лекарственного вещества, но и уменьшает вероятность возникновения побочных эффектов.
19.1.6. Комбинированная антибиотикотерапия
При этиотропном подходе после идентификации возбудителя предпочтительнее монотерапия с использованием препаратов узкого спектра действия, хотя не исключена возможность применения комбинированной терапии. Напротив, при эмпирическом подходе преимущественно назначается комбинированное лечение.
Основные принципы рациональной комбинированной АБ терапии:
1. Использование препаратов с разным механизмом действия. Примером может служить сочетание р-лактамных АБ с аминогликозидами. Первые взаимодействуют с ферментативными системами транс- и карбоксипептидазы микроорганизма, ответственными за синтез пептидогликана мембраны бактерий. Аминогликозиды подавляют синтез белка посредством необратимого связывания с бактериальными рибосомами. Комбинация АБ с разным механизмом действия повышает эффективность терапии, тормозит развитие резистентности
Этапы реализации рациональной антибактериальной терапии 571
возбудителя, позволяет при необходимости проводить длительные курсы терапии без смены препаратов.
2. В случае эмпирической терапии до получения результатов микробиологического исследования возможно сочетание препаратов широкого спектра действия, но при этом повышается риск вторичной инфекции, вызванной устойчивыми возбудителями. При этиотропной терапии предпочтительнее сочетать препараты узкого спектра.
3. Из-за опасности суммации побочных эффектов следует использовать препараты с различным спектром побочных действий. Например, опасна комбинация двух ото- или нефротоксичных препаратов.
4. Целесообразна комбинация препаратов с синергическим эффектом (повышение активности комбинации по сравнению с суммарным действием составных лекарственных средств). Следует избегать назначения АБ с антагонистическим действием. Под последним понимают уменьшение активности комбинации лекарственных препаратов по сравнению с действием каждого препарата в отдельности. Это явление встречается нечасто. Кроме того, антагонизм обладает видо- и штаммоспецифичностью относительно возбудителя. Отмечено снижение активности каждого из препаратов при: комбинации рифампицина с кетоконазолом, тетрациклина, доксициклина, левомицетина — с рифампицином, клиндамицина — с эритромицином. Пенициллин не сочетают с тетрациклинами и левомицетином, аминогликозиды — с полимиксином и метициллином, левомицетин — с цефалоспоринами (С. И. Бидненко).
19.1.7. Выбор пути введения антибиотика
Учитывая клинический диагноз, а также особенности кровообращения и микроциркуляции в очаге инфекции, можно выбрать рациональный путь введения АБ препаратов, который в одних случаях помогает предотвратить генерализацию инфекции из гнойного очага или ограничить транслокацию инфекции из кишечника, а в других — способствует созданию более высокой концентрации АБ в органах и
тканях, пораженных инфекционным процессом. Например, при локализации инфекции в печени преимущество имеет энтеральный способ приема АБ.
Данные о клиническом течении инфекционного процесса способствуют выбору дополнительных (вспомогательных) средств и методов для борьбы с инфекцией. Так, создание необходимой концентрации АБ на всех уровнях желчевыводящих путей без использования таких средств практически невозможно вследствие физиологических особенностей перфузии печени. В связи с этим необходимо использовать в определенной последовательности целый комплекс мероприятий: вначале — применение средств, усиливающих всасывание АБ из кишок, затем — средств, усиливающих перфузию печени и желчеобразования, в конце цикла необходимо применение препаратов, замедляющих желчеотделение, и, наконец, на 3 —4-й день к лечению присоединяют через определенные интервалы времени желчегонные средства. В других клинических ситуациях, например при послеродовом сепсисе, когда в большинстве случаев очаги инфекции локализуются в тромбированных околоматочных сосудах, трудно достигнуть положительного результата без сопутствующего применения антикоагулянтной и тромболитической терапии.
19.1.8. Длительность введения антибиотика
АБ применяют до выздоровления пациента. В случае эффективности нет необходимости менять АБ каждые 7 — 8 дней. Критериями, требующими смены препарата, являются: возникновение побочных эффектов; значительная токсичность препарата, ограничивающая суммарную длительность его применения; клиническая неэффективность. Последняя при остром воспалительном процессе устанавливается в течение 48 —72 ч от начала терапии. Этот интервал может быть продлен у больных с нарушением отдельных звеньев иммунной системы (почечная или печеночная недостаточность, онкологическое заболевание, использование иммуносупрессивных веществ, сахарный диабет и др.).
572 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
19.2. ЗНАЧЕНИЕ ИММУНОЛОГИЧЕСКОГО, ПАТОФИЗИОЛОГИЧЕСКОГО И ПАТОБИОХИМИЧЕСКОГО ДИАГНОЗОВ ДЛЯ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
Для эффективной терапии инфекционного процесса клинического и микробиологического диагнозов недостаточно, необходимы также иммунологический, патофизиологический и патобиохимический диагнозы. Так, без сохранности или восстановления собственных механизмов иммунитета больного никакими методами и средствами справиться с инфекцией невозможно, что наглядно видно на примере больных СПИДом, пациентов с иммуносупрессией при трансплантации костного мозга, других органов. В результате действия бактериальных токсинов и ферментов, применения медикаментов (особенно АБ) и гормонов, возникают разные варианты нарушения иммунитета, наиболее частыми из которых являются: нарушение процессов кооперации при индукции иммунного ответа; расстройство функции макрофагов при первичных инфекциях и функций других иммунокомпетентных клеток вследствие влияния продуктов секреции инфекционных агентов — токсинов; разрушение антител бактериальными протеазами; возникновение гиперергии, перерастающей в аутоиммунный процесс (медикаментозно спровоцированный синдром красной волчанки и др.). Поэтому для правильной коррекции иммунологических расстройств и целенаправленного воздействия на звенья иммунной системы необходимы соответствующие исследования с постановкой иммунологического диагноза, отражающего характер и глубину расстройств этой системы организма.
Немаловажное значение для эффективного лечения заболевания, обоснования выбора методов и средств интенсивной терапии имеют также патофизиологический и патобиохимический диагнозы. При любом гнойно-воспалительном процессе возникают патофизиологические и пато-биохимические расстройства различного характера и степени выраженности. Первые можно классифицировать на следующие синдромы: почечный, печеночный, различные варианты сердечно-сосудистой и дыхательной недостаточности, шок, расстройства функции пищеварительного канала с явлениями транслокации бактериальной флоры в лимфатическую систему, а затем и в системный кровоток с развитием синдрома полиорганной недостаточности. Патобиохимические расстройства проявляются нарушениями водно-электролитного и кислотно-основного балансов и др. Каждый из этих синдромов требует своего подхода, индивидуального применения определенных методов и средств, которые охватывают фактически все разделы интенсивной терапии. Без коррекции патофизиологических и патобиохимичес-ких сдвигов лечение не даст ожидаемого эффекта. Поэтому нередко при терапии гнойно-септических заболеваний приходится прежде всего применять методы и средства поддерживающей терапии и только затем подключать специфическое лечение инфекции.
19.3. ПРИНЦИПЫ ПРОФИЛАКТИЧЕСКОГО ИСПОЛЬЗОВАНИЯ АНТИБИОТИКОВ В АНЕСТЕЗИОЛОГИИ
Анестезиологу ежедневно приходится сталкиваться с необходимостью профилактики послеоперационных инфекционных осложнений. Актуальность этой проблемы подтверждают данные отдельных исследований, в соответствии с которыми развитие раневой инфекции способствует
удлинению сроков пребывания больного в стационаре в среднем на одну неделю, что сопровождается увеличением стоимости терапии на 10 — 20 %.
Современный методологический подход к профилактике послеоперационных инфекционных осложнений был заложен
Принципы профилактического использования антибиотиков в анестезиологии 573
крупномасштабным анализом примерно 14 800 операций еще в 1964 г. Этот анализ был выполнен Национальным исследовательским советом, учрежденным Национальной академией наук США. В результате исследования были выделены четыре типа операций:
1. Чистые оперативные вмешательства. Основные критерии определения чистых операций: операции без контакта с органами пищеварительного канала, респираторным и мочеполовым трактами; на момент выполнения операции отсутствуют симптомы острого воспаления или инфицирования; нетравматичные; выполняются без нарушения правил асептики.
2. Условно контаминированные. Их признаками являются: проникновение во время операции в просвет органов пищеварительного канала или респираторного тракта без существенной контаминации окружающих тканей; проникновение в билиарный тракт при условии отсутствия инфицирования желчи; операции на ротоглотке, влагалище, мочеполовом тракте при условии отсутствия инфицирования мочи; незначительные нарушения асептики в ходе операции.
3. Контаминированные операции. Критериями их выделения служат: значительная контаминация при операциях на органах пищеварительного канала или респираторном тракте; операции на мочеполовых или желчевыводящих путях на фоне острого инфекционного процесса; первичная хирургическая обработка травмированной ткани; значительные нарушения асептики во время оперативного вмешательства.
4. Инфицированные операции. Их признаками являются: острое бактериальное воспаление или наличие гноя в месте оперативного вмешательства; поврежденные ткани с наличием нежизнеспособных тканей, чужеродных материалов, каловой контаминации и/или задержкой в хирургическом лечении.
Был также определен риск развития послеоперационных инфекционных осложнений: для первой группы он составил 3,3 %, для второй — 10,5, для третьей — 16,3, для четвертой — 28,6 %.
В последующем были проведены еще два крупномасштабных исследования эпидемиологии и профилактики послеоперационных инфекционных осложнений: Study Efficacy of Nosocomial Infection Control (SENIC) проект (1985), National Nosocomial Infection Surveillance (NNIS) System (1991). В результате этих исследований, а также целой серии менее масштабных уточнена вероятность послеоперационных инфекционных осложнений, выявлены дополнительные факторы риска.
На современном этапе общепризнанным является выделение таких операций:
1. Чистые', риск послеоперационного нагноения составляет 5 % (нетравматические плановые операции без признаков воспаления, не затрагивающие ротоглотку, дыхательные пути, пищеварительный канал или мочеполовую систему; ортопедические операции; мастэктомия; струм-эктомия; грыжесечение; флебэктомия у больных без трофических нарушений; протезирование суставов; артропластика; операции на аорте и артериях конечностей; операции на сердце; пластические операции).
2. Условно чистые', риск послеоперационного нагноения составляет 7 —10 % (чистые операции с риском инфекционных осложнений, плановые операции на ротоглотке, пищеварительном канале (например, холецистэктомия), женских половых органов, урологические (например, трансуретральная простатэктомия) и пульмонологические операции без признаков сопутствующей инфекции; флебэктомия у больных с трофическими нарушениями, но без трофических язв; повторное вмешательство через «чистую» рану в течение семи дней; погружной остеосинтез при закрытых переломах; ургентные неотложные операции по другим критериям, входящие в группу «чистые»; тупые травмы без разрыва полых органов).
3. Загрязненные', риск послеоперационного нагноения составляет 12 —20 % (оперативные вмешательства на желчных и мочеполовых путях при наличии инфекции; оперативные вмешательства на пищеварительном канале при высокой степени его контаминации; операции при наруше
574 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
нии асептики или при наличии воспалительного процесса (но не гнойного воспаления); операции при травматических повреждениях, проникающих ранениях, обработанных в течение 4 ч).
4. Грязные', риск послеоперационного нагноения составляет более 20 % (оперативные вмешательства на заведомо инфицированных органах и тканях при наличии сопутствующей или предшествующей инфекции; раны или перфорация пищеварительного канала (например, перфоративный дивертикул); прокто-гинекологичес-кие операции; проникающие ранения и травматические раны, обработанные по истечении 4 ч; флебэктомия у больных с трофическими нарушениями и язвами; операции при гнойном воспалении на инфицированных тканях).
Вне зависимости от типа выделяемых оперативных вмешательств микробное обсеменение раны регистрируется в большинстве случаев к концу операции. Но приведенная классификация обоснована с позиций необходимости профилактики раневой инфекции применением АБ, поскольку целью их применения является не полная эрадикация микробов, а уменьшение количества попадающих в рану микроорганизмов ниже некоторого порогового уровня, когда защитные силы организма будут способны самостоятельно справиться с инфектом. В соответствии с таким подходом, подтвержденным опытом работы многих клиник, профилактическое применение АБ не показано при чистых операциях, за исключением случаев, когда развитие инфекционного осложнения представляет серьезную угрозу для жизни больного (примерами могут служить эндопротезирование тазобедренного сустава, аортокоронарное шунтирование) или защитные силы организма снижены (сахарный диабет, заболевания сердца, почечная недостаточность, опухоли, применение иммуносупрессивных средств, ВИЧ-инфици-рование). Профилактика АБ показана при всех условно чистых и загрязненных операциях. При грязных операциях АБ терапия проводится в полном объеме независимо от того, проводилась ли профилактика АБ или нет.
19.3.1. Выбор временного интервала и пути введения антибиотика
Условием эффективного использования приведенного методологического подхода в практической деятельности является введение АБ до оперативного вмешательства (т. е. до контаминации зоны операции или развития раневой инфекции). Эффективность назначения АБ с профилактической целью после операции существенно ниже. Это подтверждается тем, что при введении препарата до операции необходимая длительность антибиотикопрофи-лактики составляет 24 ч. Дальнейшее назначение АБ уже следует рассматривать не как профилактику, а как АБ терапию. Показанием для нее является документированная бактериальная инфекция.
В настоящее время определен наиболее оптимальный временной интервал начала АБ профилактики. Результаты исследований свидетельствуют, что минимальная вероятность инфекционных послеоперационных осложнений наблюдается при внутривенном введении препарата за 30 — 60 мин до разреза. Если длительность операции превышает 2 — 3 периода полу жизни АБ, то необходимо повторное введение препарата. При отдельных, небольших по объему операциях (например, трансуретральная резекция предстательной железы, ударноволновая литотрипсия) возможно применение АБ внутрь.
Дооперационное введение АБ при условно чистых и загрязненных операциях позволяет предотвратить инфицирование зоны оперативного вмешательства. С другой стороны, длительное введение АБ в послеоперационном периоде не предотвращает гематогенное инфицирование, не предупреждает колонизацию ротоглотки и кожных покровов микроорганизмами и развитие инфекционных осложнений вне раны. Более того, уничтожение чувствительных микроорганизмов создает экологическую нишу для контаминации прооперированного больного внутригоспитальны-ми микроорганизмами и создает условия для развития инфекционных осложнений и вне зоны оперативного вмешательства. Так, согласно Руководству по предотвра-
Принципы профилактического использования антибиотиков в анестезиологии 575
щению нозокомиальной пневмонии, изданному Центром по контролю и предотвращению заболеваний США в 1997 г., не поддерживается тактика рутинного ежедневного использования АБ для профилактики нозокомиальной пневмонии, в том числе в послеоперационном периоде, а также у больных на ИВЛ. При этом такая рекомендация в соответствии с изложенными в Руководстве критериями доказательной медицины отнесена к классу IA: однозначная рекомендация для всех стационаров. Безусловно, такая рекомендация не может быть внедрена в практику работы отечественных отделений интенсивной терапии изолированно от других методов профилактики нозокомиальной пневмонии, используемых за рубежом.
19.3.2. Применение других методов профилактики инфекционных осложнений
Современный методологический подход к АБ профилактике послеоперационных инфекционных осложнений предполагает выделение собственно АБ профилактики и АБ терапии раневой инфекции, АБ профилактики раневой инфекции и АБ терапии инфекционных осложнений другой локализации. При этом АБ профилактика является составной частью профилактики инфекционных осложнений, включающей обработку операционного поля, удаление волос, обеспечение правильного ухода за катетерами и др. Даже самая продуманная АБ профилактика не в состоянии компенсировать грубую хирургическую технику: наличие в ране большого количества крови, желчи или других сред, развитие гипоксии уменьшают активность лейкоцитов и других защитных элементов.
19.3.3. Выбор антибиотика для профилактики послеоперационных инфекционных осложнений
Выбор препарата для периоперативной АБ профилактики в каждом конкретном случае должен быть микробиологически обоснован. Одним из источников инфицирования во время операции являются
экзогенные бактерии операционной, в том числе на поверхности рук хирурга. При этом чаще всего встречаются Staphylococcus aureus и S. epidermalis. Контаминацию такими бактериями можно относительно легко уменьшить соблюдением обычных правил асептики, применяемых в операционной. Кроме того, такие микроорганизмы обычно относительно легко подвергаются эрадикации АБ, применяемыми для профилактики. Это объясняет факт отсутствия в рандомизированных исследованиях последних лет зависимости возникновения послеоперационных инфекционных осложнений от случайного нарушения целостности перчаток у оперирующих хирургов или от наличия масок у персонала операционной. Для возникновения послеоперационных инфекционных осложнений при используемых в настоящее время в операционной правил асептики более значимо обсеменение эндогенными микроорганизмами во время оперативного вмешательства. При этом при операциях на поверхностных тканях, костях, суставах ведущая роль принадлежит микроорганизмам поверхности кожи — стафилококкам и стрептококкам. При операциях на органах брюшной полости к характерным возбудителям послеоперационных осложнений относятся типичные микроорганизмы просвета кишки — грамотрицательные энтеробактерии, энтерококки, анаэробные бактероиды, пепто-стрептококки, клостридии.
Разработаны частные алгоритмы выбора АБ для профилактики послеоперационных инфекционных осложнений в зависимости от выбора метода оперативного вмешательства и состояния больного. В большинстве случаев такими препаратами являются цефалоспорины II поколения, фторхинолоны II поколения, защищенные аминопенициллины. Применение цефалоспоринов III —IV поколения, кар-бапенемов, ванкомицина для АБ профилактики возможно лишь у ослабленных больных с длительным пребыванием в стационаре: в таких случаях естественная эндогенная флора может замещаться нозокомиальными инфектами. Бесконтрольное применение АБ широкого спектра дейст
576 ПРИНЦИПЫ РАЦИОНАЛЬНОЙ АНТИБАКТЕРИАЛЬНОЙ ТЕРАПИИ
вия с профилактической целью в конечном итоге увеличивает степень антибиотического пресса и способствует селекции антибиотикорезистентных штаммов.
19.3.4. Экономическая целесообразность применения антибиотиков
Приведенный методологический подход медленно внедряется не только в отечественных, но и в зарубежных клиниках. Так, например, в главном госпитале Ванкувера — высокоспециализированном медицинском учреждение на 1100 коек, являющемся базовой клиникой университета, ежегодно выполняется около 12,5 тыс. оперативных вмешательств. При анализе обнаружилось, что в нем отсутствовали единые для всего лечебного учреждения правила, способные гарантировать оптимальные сроки предоперационного введения АБ препаратов. Это послужило толчком к применению двухэтапного междисциплинарного вмешательства. Были установлены правила, в соответствии с которыми предоперационная парентеральная антимикробная профилактика проводилась медицинскими сестрами или анестезиологами только в предоперационном помещении или операционной. Во внутреннем информационном бюллетене была опубликована статья, содержащая информацию об общих принципах антимикробной профилактики в хирургии, обоснование инициативы и одобрение руководством. В ней приводилось описание правил, касающихся предоперационного введения антимикробных препаратов; они предшествовали «Стандартизированным распоряжениям по антимикробной профилактике». На второй стадии вмешательства фармацевты были наделены полномочиями, в случае необходимости изменять рецептурные прописи, противоречащие «Стандартизи
рованным распоряжениям». В каждом случае профилактического применения АБ время введения препарата оценивалось по отношению ко времени разреза кожи. Предлагалось выделять досрочное введение (более чем за 2 ч до разреза кожи), своевременное (за 0 —2 ч), запоздалое (2 —3 ч после разреза) и позднее (после разреза кожи прошло более 3 ч). Демографическая характеристика больных, использованные антимикробные препараты, оперативные вмешательства были схожими в обеих фазах. В фазе, предшествующей организационному вмешательству, в 68 % случаев профилактика была проведена своевременно, в 22 % — преждевременно, частота случаев запоздалой и поздней профилактики была одинаковой. В фазе «после организационного вмешательства» частота своевременной профилактики возросла до 97 %. Внедрение этой стратегии позволило сократить ежегодное число случаев послеоперационной раневой инфекции. При этом удалось уменьшить суммарное стационарное лечение на 153 стационарных дня и сохранить 99 807 канадских долларов. Из них 9100 канадских долларов было затрачено на внедрение и поддержание «Стандартизированных правил». Поэтому действительная экономия составила 90 707 канадских долларов (цит. по данным Международного союза за рациональное использование антибиотиков с разрешения Украинской ассоциации за рациональное использование антибиотиков).
Рациональное применение АБ, преимущественное использование этиотропного подхода, внедрение современных методологических подходов к профилактике инфекционных осложнений в послеоперационном периоде позволяют повысить эффективность использования АБ, уменьшить вероятность летального исхода и осложнений, сократить затраты на лечение.
Список использованной литературы 577
Список использованной литературы
1. Куценко О. В. Маркери ендотоксемп при гншно-запальних процесах: Автореф. дис. ... канд. мед. наук. — К., 2002. — 18 с.
2. Alvarex-Lerma F. Modification of empiric antibiotic treatment in patients with pneumonia acquired in the intensive care unit // Inten. Care Med. — 1996. — Vol. 22. — P. 387-394.
3. BodiM.,Ardanuy C., OlonaM., Castander D., Diaz E., Rello J. Therapy of ventilator-associated pneumonia: the Tarragona strategy // Clin. Microbial. Infect. — 2001. — Vol. 7, N 1. - P. 32-33.
4. Hey land D. K,, Cook D. J., Griffith L. et al. The attributable morbidity and mortality of ventilator-associated pneumonia in the critically ill patient. The Canadian Critical Trials Group // Am. J. Respir. Care Med. — 1999. - Vol. 159. - P. 1246-1256.
5. Kleff M. H., Ward S. The influence of mini-BAL cultures on patient outcomes: implications for the antibiotic management of ventilator-associated pneumonia // Chest. — 1998. - Vol. 113. - P. 412-420.
6. Luna С. M.f Vujacich P., Niederman M. S. et al. Impact of BAL data on the therapy and outcome of ventilator-associated pneumonia // Chest. - 1997. - Vol. 111. - P. 676-685.
7. Rello J., Diaz E. Optimal use of antibiotics for intubation-associated pneumonia // Inten. Care Med. - 2001. - Vol. 27. - P. 237-239.
8. Rello J., Gallego M., Mariscal D. et al. The value of routine microbial investigation in the ventilator-associated pneumonia // Am. J. Respir. Care Med. - 1997. - Vol. 156. -P. 196-200.
9. Tablan О. C., Anderson L. J.t Arden N. H. et al. Guidelines for Prevention of Nosocomial Pneumonia Centers for Disease Control and Prevention // MMWR. Morbidity and Mortality Weekly Report. — 1997. — Vol. 46, N RR-1. - P. 1-79.
10. Украинская ассоциация за рациональное использование антибиотиков (http:/uad а.boom.ru)
ОГЛАВЛЕНИЕ
Список принятых сокращений и условных обозначений 3
Введение
(д-р мед. наук А. И. Трещинский) 6
Раздел 1. Физиология автономной нервной системы (д-р мед. наук А. И. Трещинский,
М. А. Трещинская) 13
1.1. Функциональная анатомия автономной нервной системы 13
1.1.1. Симпатическая нервная система 16
1.1.2. Парасимпатическая нервная система 18
1.2. Современные концепции
механизма действия медиаторов 21
1.3. Функции автономной нервной системы 22
1.4. Оценки функционального состояния автономной нервной системы и эффектов эпинефрина 24
1.5. Симптомы и признаки дисфункции парасимпатической (холинергической) системы 29
1.6. Роль автономной нервной системы в регуляции метаболизма, энергетического обмена, утилизации жиров
и углеводов 32
1.6.1. Влияние автономной нервной системы на метаболизм углеводов 33
1.6.2. Некоторые патофизиологические проблемы мобилизации жира 37
1.7. Роль автономной нервной системы в регуляции периферического кровообращения 37
1.8. Автономная нервная система
и физиологическая нагрузка 38
1.9. Автономная нервная система и боль 43
1.9.1. Висцеральная боль 43
Список использованной литературы 46
Раздел 2. Клиническая физиология кровообращения
(чл.-кор. АМН Украины В. И. Черний, канд. мед. наук К. Н. Олейников) 48
2.1. Клиническая физиология сердечно-сосудистой системы 48
2.1.1. Сердце 50
2.1.2. Сосудистая система 54
2.2. Патофизиологические изменения в сердечно-сосудистой системе
при критических состояниях 58
2.2.1. Основные физиологические факторы, определяющие функциональное состояние кровообращения 58
2.2.2. Мониторинг показателей системной гемодинамики 62
2.3. Принципы диагностики и интенсивной терапии некоторых нарушений функции сердечно-сосудистой системы
при критических состояниях 67
2.3.1. Аналитический подход к коррекции гемодинамических нарушений 67
2.3.2. Диагностика и интенсивная терапия гипертонических кризов 70
Список использованной литературы 73
Раздел 3. Клиническая физиология
дыхания (д-р мед. наук Ф. С. Глумчер) 74
3.1. Центральная регуляция дыхания 74
3.2. Биомеханика дыхания 76
3.3. Эластическое сопротивление легких 77
3.4. Динамическое сопротивление дыханию 80
3.5. Работа дыхания 82
3.6. Вентиляция и газообмен в легких 82
3.6.1. Обмен газов
между альвеолярным газом и кровью 84
3.6.2. Неравномерность вентиляции в легких 85
Оглавление 579
3.6.3. Особенности кровотока в малом круге 85
3.6.4. Вентиляционно-перфузионные соотношения в легких 86
3.7. Транспорт газов кровью 88
3.7.1. Транспорт кислорода 88
3.7.2. Транспорт кровью СО2 92
3.8. Гипоксия 93
3.9. Тканевое дыхание 95
Список использованной литературы 97
Раздел 4. Острая недостаточность
дыхания (д-р мед. наук Ф. С. Глумчер) 99
4.1. Вентиляционная форма ОНД 100
4.2. Пневмония 104
4.2.1. Патогенез 104
4.2.2. Внутрибольничная пневмония 105
4.2.3. Вентилятор-ассоциированная пневмония 106
4.2.4. Лечение внутрибольничной пневмонии 106
4.3. Бронхиальная астма 107
4.3.1. Патогенез 107
4.3.2. Диагностика бронхиальной астмы 109
4.3.3. Лечение обострения
бронхиальной астмы ПО
4.4. Синдром острого повреждения легких и острый респираторный дистресс-синдром 114
4.4.1. Патогенез 115
4.4.2. Патоморфология 117
4.4.3. Клинические проявления 117
4.4.4. Лечение острого респираторного дистресс-синдрома 118
4.5. Искусственная вентиляция легких 127
4.5.1. Влияние искусственной вентиляции легких на циркуляцию крови 128
4.5.2. Показания к переводу
на искусственную вентиляцию легких 129
4.5.3. Выбор параметров
искусственной вентиляции легких 129
4.5.4. Специальные режимы ИВЛ 132
4.5.5. Обеспечение проходимости дыхательных путей при искусственной вентиляции легких 135
4.5.6. Двигательный режим во время ИВЛ, седация и миорелаксация 138
4.5.7. Осложнения искусственной вентиляции легких 138
4.5.8. Перевод больного
на спонтанное дыхание 140
4.5.9. Неинвазивные методы искусственной вентиляции легких 140
4.6. Отек легких 142
4.6.1. Патофизиология отека легких 142
4.6.2. Клинические проявления
отека легких 145
4.6.3. Лечение отека легких 145
Список использованной литературы 146
Раздел 5. Клиническая физиология почек
(д-р мед. наук В. В. Суслов) 154
5.1. Анатомия почки 155
5.1.1. Кровообращение в почках 156
5.1.2. Нефрон 156
5.2. Транспорт растворенных веществ 158
5.3. Экскреция продуктов метаболизма 164
5.4. Почечная регуляция кислотно-основного состояния 165
5.5. Концентрирование и разведение мочи 166
5.6. Нейроэндокринная регуляция функции почек 167
5.6.1. Альдостерон 167
5.6.2. Антидиуретический гормон 168
5.6.3. Паратиреоидный гормон и кальцитонин 169
5.6.4. Катехоламины 169
5.7. Выведение лекарственных средств почками 170
5.8. Диурез и диуретики 172
5.9. Мочекаменная болезнь 173
5.9.1. Ураты 174
5.9.2. Оксалаты 174
5.9.3. Фосфаты 174
5.9.4. Цистиновые камни 175
5.10. Острая почечная недостаточность 175
5.10.1. Преренальная
острая почечная недостаточность 175
5.10.2. Ренальная
острая почечная недостаточность 176
5.10.3. Постренальная
острая почечная недостаточность 178
5.11. Хроническая почечная недостаточность (ХПН) 179
5.12. Дифференциальная диагностика различных видов почечной
недостаточности 184
5.13. Лечение острой почечной недостаточности 185
5.13.1. Консервативное лечение хронической почечной недостаточности 187
5.13.2. Активные методы лечения ХПН 187
5.14. Трансплантация почки 188
Список использованной литературы 189
Раздел 6. Клиническая физиология печени
(д-р мед. наук А. И. Трещинский,
д-р мед. наук А. В. Беляев) 190
6.1. Кровообращение в ткани печени 190
6.1.1. Анатомия сосудов и макроциркуляция в печени 190
580 Оглавление
6.1.2. Регуляция кровообращения в печени
6.1.2.1. Внутренняя регуляция
6.1.2.2. Внешняя регуляция
6.1.3. Влияние анестезии
на кровоток в печени
6.2. Метаболические функции печени
6.2.1. Обмен белков
6.2.2. Обмен углеводов
6.2.3. Обмен липидов
6.2.4. Билирубин и его циркуляция
6.2.5. Влияние анестезии
на метаболические функции печени
6.3. Желчеобразование и желчеотделение
6.3.1. Состав и функция желчи.
Желчные кислоты
6.3.2. Функциональная анатомия желчевыводящих путей
6.3.3. Желчеобразование
6.3.4. Желчный пузырь и его моторика.
Влияние анестезии на нарушение моторики у больных в критическом состоянии
6.4. Роль печени в гемопоэзе
6.4.1. Роль печени в эритропоэзе
6.4.2. Роль печени в обмене порфиринов
6.5. Гуморальная функция печени
6.6. Печень и естественная
иммунная реактивность
6.7. Клинические и биохимические показатели функции печени
6.7.1. Клинические симптомы нарушения функции печени
6.7.2. Биохимические показатели нарушения функции печени
Список использованной литературы
Раздел 7. Клиническая физиология пищеварительного канала
(д-р мед. наук А. А. Хижняк)
7.1. Дисфункция верхних отделов глотки
7.2. Пищевод
7.3. Диагностика и лечение абдоминальной боли, заболеваний глотки и пищевода
7.4. Язвенная болезнь
7.4.1. Клиническое обследование
7.4.2. Терапия
7.4.3. Хирургическое лечение язвенной болезни
7.4.4. Кровотечения из пищеварительного канала
7.4.5. Перфоративные язвы желудка и двенадцатиперстной кишки
7.5. Панкреатиты
7.5.1. Клиническая картина
193 острого панкреатита 246
193 7.5.2. Лечение острого панкреатита 246
194 195 195 7.6. Интенсивная терапия панкреонекроза 247
Список использованной литературы 250
195 Раздел 8. Физиология и патология
199 водно-электролитного обмена
201 (канд. мед. наук М. В. Бондарь) 251
205 8.1. Вода и жидкостные пространства
207 207 организма 8.2. Роль пищеварительного канала 251
в поддержании водно-электролитного обмена 254
207
8.3. Серотонинреактивные структуры организма и водно-электролитный обмен 8.4. Обмен водой и электролитами 255
209 210
между водными секторами организма 8.5. Обмен веществ на уровне 258
211 211 капилляров 261
8.6. Обмен водой между организмом человека и окружающей средой 8.7. Гормональная регуляция 265
211 212 213
водно-электролитного обмена 8.8. Оценка водного статуса организма 266 272
214 8.9. Нарушения водно-электролитного обмена и их коррекция 273
215 8.9.1. Гипотоническая дегидратация 273
8.9.2. Изотоническая дегидратация 275
215 8.9.3. Гипертоническая дегидратация 279
8.9.4. Гипотоническая гипергидратация 281
221 8.9.5. Изотоническая гипергидратация 282
8.9.6. Гипертоническая гипергидратация 283
226 8.10. Обмен электролитов 283
8.10.1. Обмен натрия 283
8.10.2. Обмен калия 288
8.10.3. Обмен кальция 292
227 8.10.4. Обмен магния 301
8.10.5. Обмен хлора 305
228 8.10.6. Обмен фосфора 305
229 8.10.7. Обмен микроэлементов 307
Список использованной литературы 310
230 Раздел 9. Кислотно-основное состояние
232 (д-р мед. наук Ф. С. Глумчер) 312
234 9.1. Буферные системы организма 313
234 236 9.1.1. Гидрогенкарбонатная буферная система 314
9.1.2. Фосфатная буферная система 314
9.1.3. Белковая буферная система 314
236 9.1.4. Гемоглобиновая буферная система 9.2. Физиологические системы 315
237 регуляции pH 315
244 9.2.1. Система внешнего дыхания 315
Оглавление 581
9.2.2. Роль почек в поддержании кислотно-основного состояния 315
9.2.3. Секреция гидроген-ионов с мочой 316
9.2.4. Реабсорбция гидрогенкарбоната в почках 316
9.3. Роль органов пищеварения в регуляции кислотно-основного состояния 317
9.4. Основные показатели кислотно-основного состояния 317
9.5. Терминология, употребляемая при изучении кислотно-основного состояния 318
9.6. Клинические состояния, сопровождающиеся расстройствами кислотно-основного состояния 318
9.6.1. Метаболический ацидоз 318
9.6.2. Лечение метаболического ацидоза 322
9.6.3. Респираторный ацидоз 323
9.6.4. Зависимость
между кислотно-основным балансом
крови и спинномозговой жидкостью 323
9.6.5. Метаболический алкалоз 324
9.6.6. Лечение метаболического алкалоза 325
9.6.7. Респираторный алкалоз 325
9.6.8. Смешанные расстройства КОС 325
Список использованной литературы 326
Раздел 10. Оксидантное повреждение тканей
(д-р мед. наук Ф. С. Глумчер) 327
10.1. Пероксидное окисление липидов 328
10.2. Антиоксидантная активность 330
10.3. Неэнзимные антиоксиданты 331
10.4. Оксидантный стресс 332
10.5. Антиоксидантная терапия 333
Список использованной литературы 334
Раздел 11. Система гемостаза в норме и при патологии
(чл.-кор. АМН Украины В. И. Черний, канд. мед. наук И. В. Кузнецова) 336
11.1. Сосудисто-тромбоцитарный гемостаз 336
11.1.1. Роль кровеносных сосудов
в первичном гемостазе 336
11.1.2. Тромбоциты 337
11.1.3. Значение простагландинов
для сосудисто-тромбоцитарного гемостаза 338
11.1.4. Ретракция и консолидация тромба 339
11.1.5. Методы исследования сосудисто-тромбоцитарного гемостаза 340 11.2. Лекарственные препараты, воздействующие на сосудистотромбоцитарный гемостаз 341
11.2.1 . Препараты, восстанавливающие
сосудисто-тромбоцитарный гемостаз 341
11.2.2 . Препараты, стабилизирующие сосудистую проницаемость
(вазопротекторы) 341
11.2.3 . Препараты, блокирующие сосудисто-тромбоцитарный гемостаз 342
11.3. Коагуляционный гемостаз (ферментативная коагуляция) 343
11.3.1. Методы исследования коагуляционного гемостаза 345
11.3.2. Препараты, восстанавливающие коагуляционный гемостаз 346
11.4. Антикоагулянтная система 346
11.4.1. Методы исследования антикоагулянтной системы 347
11.4.2. Препараты, воздействующие
на антикоагулянтную систему 348
11.5. Фибринолитическая система 350
11.5.1. Лабораторная диагностика
нарушений фибринолитической
системы 350
11.5.2. Препараты, воздействующие
на фибринолитическую активность 351
11.6. Логика обследования системы гемостаза 353
11.7. Некоторые виды нарушений в системе гемостаза
в практике интенсивной терапии 353
11.7.1. Синдром диссеминированного
внутрисосудистого свертывания крови 354
11.7.2. Тромбофилия и гиперкоагуляция 364
11.7.3. Антифосфолипидный синдром 365
11.7.4. Гиперфибриногенемия 367
Список использованной литературы 368
Раздел 12. Сердечно-легочная церебральная реанимация
(чл.-кор. НАН и АМН Украины
Л. В. Усенко) 369
12.1. Терминальные состояния 369
12.2. Признаки клинической смерти 372
12.3. Методы сердечно-легочной церебральной реанимации 372
12.4. Смерть головного мозга 405
12.5. Интенсивная терапия постреанимационной болезни 409
Список использованной литературы 415
Раздел 13. Шок
(д-р мед. наук Ф. С. Глумчер,
чл.-кор. АМН Украины
В. Ф. Москаленко) 417
13.1. Повреждение клетки при шоке 418
13.2. Стадии и клиника шока 421
13.3. Гиповолемический шок 421
13.3.1. Геморрагический шок 421
13.4. Кардиогенный шок 425
582 Оглавление
13.5. Дистрибутивный (перераспределительный) шок 426
13.5.1. Септический шок 426
13.5.2. Нейрогенный шок 430
13.5.3. Анафилактический шок 430
13.6. Обструктивный шок 439
13.7. Инфузионная терапия 439
13.7.1. Кристаллоидные растворы 440
13.7.2. Коллоидные растворы 441
13.7.3. Выбор раствора для инфузии 443
13.8. Трансфузионная терапия 444
13.9. Современная стратегия гемотрансфузий 447
13.10. Инотропные и сосудосуживающие фармакологические препараты 449
13.11. Сосудорасширяющие средства 453
Список использованной литературы 455
Раздел 14. Кровезаменители с газотранспортной функцией
(чл.-кор. НАН и АМН Украины
Л. В. Усенко) 461
14.1. Кровезаменители на основе модифицированного гемоглобина 461
14.2. Кровезаменители на основе
эмульсий перфторуглеродов 462
14.3. Дозировка и способ введения перфторана 464
Список использованной литературы 466
Раздел 15. Политравма
(д-р мед. наук Ф. С. Глумчер, канд. мед. наук А. М. Дубов) 468
15.1. Патофизиологические процессы
при травме 468
15.2. Травма и гемостаз 469
15.3. Терапия тяжелой травмы 471
15.4. Терапия расстройств гемостаза при травмах 473
15.5. Черепно-мозговая травма 474
15.5.1. Патогенез 475
15.5.2. Клинические проявления черепно-мозговой травмы и ее диагностика 475
15.5.3. Лечение
черепно-мозговой травмы 476
15.5.4. Поддержание оптимальной гемодинамики 477
15.5.5. Обезболивание 478
15.5.6. Лечение внутричерепной гипертензии 478
15.6. Торакальная травма 480
15.6.1. Переломы ребер по одной линии без флотации 480
15.6.2. Множественные переломы ребер
с флотацией фрагмента 480
15.6.3. Контузия легких (КЛ) 481
15.6.4. Повреждения сердца 481
15.6.5. Тампонада перикарда 482
15.7. Брюшной компартмент-синдром 482
15.8. Травмы костей таза 483
Список использованной литературы 483
Раздел 16. Интенсивная терапия ожоговой болезни
(д-р мед. наук Е. Н. Клигуненко) 487
16.1. Этиопатогенез 487
16.2. Лечение острого периода ожоговой болезни 492
16.3. Компоненты растворов для парентерального питания 502
Список использованной литературы 507
Раздел 17. Эмболизм
(д-р мед. наук Ф. С. Глумчер, чл.-кор. АМН Украины
В. Ф. Москаленко) 508
17.1. Тромбоэмболия легочной артерии (ТЭЛА) 508
17.1.1. Этиология 508
17.1.2. Патофизиология тромбоэмболии легочной артерии 509
17.1.3. Диагностика тромбоэмболии легочной артерии 511
17.1.4. Методы профилактики ТГВ
и ТЭЛА у хирургических больных 512
17.1.5. Терапия тромбоэмболии
легочной артерии 513
17.2. Газовая эмболия легких 516
17.2.1. Диагностика газовой эмболии легких 516
17.2.2. Терапия газовой эмболии легких 516
17.3. Газовая эмболия вен головного мозга 516
17.4. Воздушная эмболия артерий
большого круга кровообращения 516
17.5. Жировая эмболия 517
17.5.1. Патогенез 517
17.5.2. Клиническая картина 518
17.5.3. Лечение 518
Список использованной литературы 519
Раздел 18. Сепсис (чл.-кор. НАН и АМН Украины Л. В. Усенко, д-р мед. наук Л, А. Мальцева,
канд. мед. наук Н. Ф. Мосенцев) 521
18.1. Эпидемиология сепсиса 521
18.2. Терминология 524
18.3. Современный взгляд на патогенез и диагностику сепсиса 533
18.3.1. Активация воспаления при сепсисе 533
18.3.2. Активация коагуляции при сепсисе 534
Оглавление 583
18.3.3. Внешний путь модели коагуляции 535
18.3.4. Угнетение фибринолиза при сепсисе 536
18.4. Новые агенты при сепсисе 537
18.5. Коагулопатия при сепсисе — главный путь к органной дисфункции
и летальному исходу 538
18.6. Основные направления патогенетической терапии сепсиса 539
Список использованной литературы 552
Раздел 19. Принципы рациональной антибактериальной терапии
(д-р мед. наук А. В. Беляев, канд. мед. наук Л. А. Харченко) 555
19.1. Этапы реализации рациональной антибактериальной терапии 555
19.1.1. Оценка необходимости назначения антибиотиков 555
19.1.2. Эмпирическое и этиотропное обоснование выбора антибиотика 556
19.1.3. Методика деэскалации как вариант эмпирической антибактериальной терапии тяжелых инфекций 562
19.1.4. Анализ степени проникновения антибиотика в пораженный орган 562
19.1.5. Выбор кратности введения антибиотика 562
19.1.6. Комбинированная антибиотикотерапия 570
19.1.7. Выбор пути введения антибиотика 571
19.1.8. Длительность введения антибиотика 571
19.2. Значение иммунологического, патофизиологического
и патобиохимического диагнозов
для рациональной антибактериальной
терапии 572
19.3. Принципы профилактического использования антибиотиков
в анестезиологии 572
19.3.1. Выбор временного интервала
и пути введения антибиотика 574
19.3.2. Применение других методов профилактики инфекционных
осложнений 575
19.3.3. Выбор антибиотика
для профилактики послеоперационных инфекционных осложнений 575
19.3.4. Экономическая
целесообразность применения антибиотиков 576
Список использованной литературы 577
Науково-практичне видання
Беляев Андрей Викторович, Бондарь Михаил Владимирович, Дубов Андрей Михайлович и др.
DykoBogcmBo 1 по интенсивной терапии
Под редакцией профессоров Трещинского Анатолия Ивановича, Глумчера Феликса Семеновича
ПОС1БНИК 3 1НТЕНСИВНОТ ТЕРАПП
За редакц1сю професор!в Трещинського Анатол1я 1вановича, Глумчера Фелмсса Семеновича (рос. новою)
Оправа i титул художника I. С. Бринюк Художнш редактор Г. С. Муратова Техшчний редактор А. I. Омоховсъка Коректори: Л. М. Байбородша, Н. М. Мельник, Р. Б. Попович, Н. Г. Поташна Комп’ютерна верстка Н. П. Довлетукаевог
Шди. до друку 02.09.2004. Формат 70 х 100/16. Пашр офс. № 1. Гарштура Peterburg. Офс. друк. Ум. друк. арк. 47,45 + 0,81 вкл. Обл.-вид. арк. 58,36 + 1,3 вкл. Тираж 2000 пр.
Вид. № 10531. Зам. № 4-220
Видавництво «Вища школа», вул. Гогол1вська, 7г, м. Кшв, 01054
Свщоцтво про внесения до Держреестру в!д 04.12.2000 cepia ДК № 268
Надруковано з пл!вок, виготовлених у видавництв! «Вища школа», у АТЗТ «Книга», вул. Артема, 25, м. КиГв, 04053 Свщоцтво про внесения до Державного реестру виготтвниюв Cepia ДК №1911