/






















































































































































































































































Author: Морачевский А.Г. Кохацкая М.С.
Tags: физика химия термодинамика физическая химия учебное пособие учебник по химии
ISBN: 978-5-7422-2006-0
Year: 2008




Text
a*oHAm
Федеральное агентство по образованию j^lS ЩшЪ
^:%
САНКТ-ПЕТЕРБУРГСКИЙ ГОСУДАРСТВЕННЫЙ \\
ПОЛИТЕХНИЧЕСКИЙ УНИВЕРСИТЕТ
«Образование»
А.Г. Морачевский М.С. Кохацкая
ПРИКЛАДНАЯ ХИМИЧЕСКАЯ
ТЕРМОДИНАМИКА
Учебное пособие
^\
Санкт-Петербург
Издательство Политехнического университета
2008
ББК24.53я73
М79
Морачевский Л. Г. Прикладная химическая термодинамика : учеб.
пособие /А. Г. Морачевский, М.С. Кохацкая. — СПб.: Изд-во Политехи.
ун-та, 2008. - 254 с.
Представляет собой краткое изложение основ химической
термодинамики применительно к описанию металлургических систем и процессов.
Обсуждены общие вопросы термодинамики фаз переменного состава,
гетерогенных равновесий. Рассмотрены основные методы
экспериментального исследования термодинамических свойств преимущественно
металлических систем.
Предназначено для студентов старших курсов и аспирантов
металлургических и химико-технологических специальностей, может быть
использовано при подготовке магистров по направлению "Металлургия"
Табл. 20. Ил. 95. Библиогр.: 47 назв.
Работа выполнена в рамках реализации Инновационной
образовательной программы Санкт-Петербургского государственного
политехнического университета «Развитие политехнической системы
подготовки кадров в инновационной среде науки и высокотехнологичных
производств Северо-Западного региона России».
Печатается по решению редакционно-издательского совета Санкт-
Петербургского государственного политехнического университета.
© Морачевский А.Г., Кохацкая М.С, 2008
© Санкт-Петербургский государственный
ISBN 978-5-7422-2006-0 политехнический университет, 2008
ОГЛАВЛЕНИЕ
Введение 7
Глава 1. Основные понятия и определения 14
Глава 2. Законы термодинамики и их практическое использование .... 19
2.1. Первый закон термодинамики 19
2.2. Энтальпия индивидуальных веществ 21
2.3. Закон Гесса 23
2.4. Энтальпия образования 24
2.5. Зависимость теплового эффекта реакции от температуры. ... 26
2.6. Энтропия. Второй закон термодинамики 28
2.7. Изменение энтропии индивидуальных веществ
в различных процессах 29
2.8. Критерии направления процесса и условия равновесия
в закрытых системах 30
2.9. Характеристические функции.
Уравнение Гиббса—Гельмгольца 32
Глава 3. Основы термодинамики фаз переменного состава 34
3.1. Общие соотношения 34
3.2. Химический потенциал 38
3.3. Относительные термодинамические функции 40
3.4. Идеальный раствор и избыточные
термодинамические величины 41
3.4.1. Термодинамические функции идеального раствора 41
3.4.2. Избыточные термодинамические функции 43
3.5. Активность и коэффициент активности.
Интегрирование уравнения Гиббса—Дюгема 45
3.6. Закон Генри. Термодинамические свойства
разбавленных растворов 49
3
3.7. Выбор стандартного состояния. Переход от одного
стандартного состояния к другому 56
3.8. Приближенные модели растворов 61
3.8.1. Регулярные растворы 61
3.8.2. Атермические растворы 64
3.9. Особенности концентрационной зависимости
термодинамических функций. Функции стабильности
и избыточной стабильности 65
Глава 4. Термодинамика фазовых равновесий 68
4.1. Условия равновесия компонентов в сосуществующих фазах.
Правило фаз Гиббса 68
4.2. Равновесие в однокомпонентных системах.
Уравнение Клапейрона—Клаузиуса 71
4.3. Давление насыщенного пара 74
4.4. Зависимость энергии Гиббса и энергии Гельмгольца
от давления и температуры в однокомпонентных системах 75
4.5. Диаграммы энергия Гиббса — состав
для двухкомпонентных систем 78
4.6. Фазовые диаграммы двухкомпонентных систем
в конденсированном состоянии 82
4.6.1. Общие принципы расчетов фазовых диаграмм 82
4.6.2. Уравнения кривых ликвидуса при кристаллизации
чистого компонента из идеального раствора 83
4.6.3. Уравнения кривых ликвидуса и солидуса в системах
с неограниченной растворимостью в жидком и твердом
состояниях в приближении идеальных растворов 88
4.6.4. Расчет фазовых диаграмм на основании
термодинамических данных 90
4.6.5. Диаграммы состояния и термодинамические функции
систем с образованием интерметаллических соединений 94
4.6.6. Влияние интерметаллических соединений на свойства
жидких сплавов 105
4.7. Извлечение термодинамической информации
из фазовых диаграмм 109
4.8. Двухфазные равновесия жидкость-жидкость
в двухкомпонентных системах. Закон распределения 114
4.8.1. Концентрационная зависимость термодинамических
функций в системах с областью расслаивания 114
4.8.2. Коэффициент распределения 119
4
4.9. Термодинамическое описание равновесия жидкость-пар
в двухкомпонентных системах 123
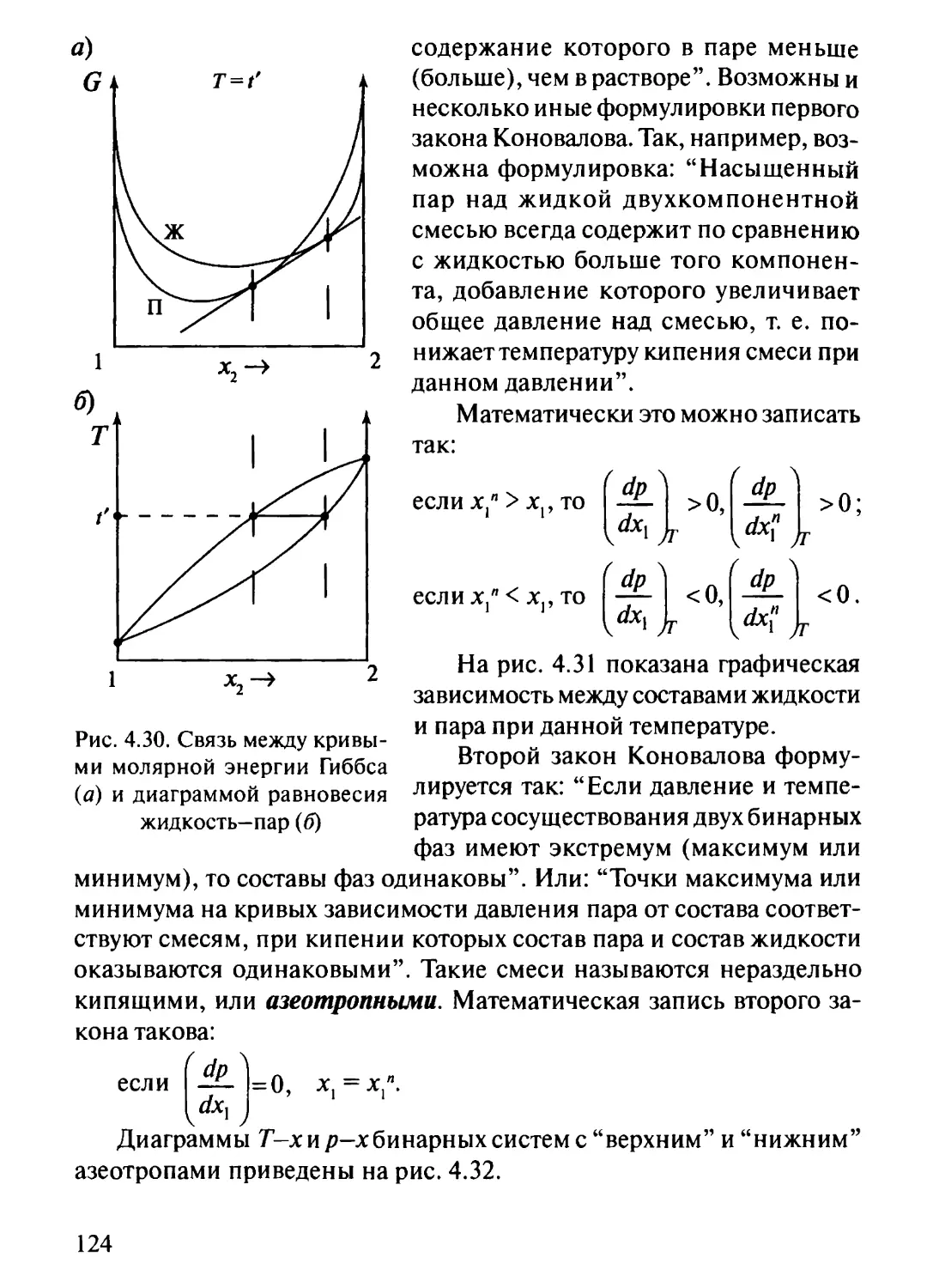
4.9.1. Законы Д.П. Коновалова 123
4.9.2. Законы М.С. Вревского 126
4.9.3. Дистилляционные процессы с участием металлов
и металлоидов 127
4.10. Корректная и некорректная постановка задач
при расчетах фазовых равновесий 133
4.11. Фазовые превращения первого и второго рода 136
Глава 5. Термодинамическое описание химических процессов 138
5.1. Уравнение изотермы химической реакции и константа
равновесия 138
5.2. Стандартная энергия Гиббса химической реакции 142
5.3. Зависимость константы равновесия реакции от температуры
и давления 144
5.4. Абсолютная энтропия. Тепловая теорема Нернста.
Постулат Планка 146
5.5. Расчет стандартной энергии Гиббса и константы равновесия
реакции при заданной температуре с использованием
абсолютных энтропии 150
5.6. Расчет стандартной энергии Гиббса с применением
приведенных функций 153
Глава 6. Трехкомпонентные системы. Термодинамическое описание . . 155
6.1. Методы изображения состава 155
6.2. Термодинамические соотношения для трехкомпонентных
систем. Уравнение Гиббса-Дюгема и его интегрирование 158
6.3. Приближенные методы расчета термодинамических свойств
тройных систем поданным о граничных двойных системах . ... 161
6.3.1. Геометрические модели 162
6.3.2. Полиномиальные методы расчета термодинамических
свойств трехкомпонентных систем 169
6.3.3. Применение правила Здановского к жидким
металлическим системам 173
6.3.4. Метод изопотенциалов 176
6.4. Равновесие между двумя жидкими фазами
в трехкомпонентных системах 177
Глава 7. Основные экспериментальные методы исследования
термодинамических свойств металлургических систем 181
7.1. Общая характеристика методов исследования 181
5
7.2. Метод измерения электродвижущих сил 183
7.2.1. Цепи с расплавленными электролитами 185
7.2.2. Цепи с твердыми электролитами 187
7.3. Методы измерения давления насыщенного пара 198
7.3.1. Статические методы 199
7.3.2. Динамический метод измерения давления насыщенного
пара 204
7.3.3. Кинетические методы измерения давления насыщенного
пара 207
7.4. Метод исследования гетерогенных равновесий
(циркуляционный метод) 214
7.5. Калориметрические методы исследования 218
7.5.1. Общие сведения о классификации калориметров 219
7.5.2. Проведение калориметрических измерений 223
7.5.3. Примеры калориметрических исследований
различных процессов 225
7.6. Обработка результатов термодинамических исследований . . 232
7.6.1. Аналитическое представление концентрационной
зависимости термодинамических функций в однофазных
двухкомпонентных системах 232
7.6.2. Применение модели ассоциированных растворов
к жидким двухкомпонентным металлическим системам 240
7.6.3. Обработка экспериментальных данных на основании
второго и третьего законов термодинамики 246
Рекомендуемая литература 249
Монографии 249
Учебники и учебные пособия 250
Справочники 251
Обзоры, статьи 251
ВВЕДЕНИЕ
Термодинамика является одним из важных разделов теоретической
физики. Применение основных положений термодинамики к
процессам, протекающим в тепловых машинах, двигателях внутреннего
сгорания, составляет предмет технической термодинамики.
Соответственно приложение законов термодинамики к химическим процессам
составляет предмет химической термодинамики.
Возникновение термодинамики как науки обычно связывается
с работами французского инженера Сади Карно (1824 г.). Основное
положение в этих работах заключается в утверждении, что получение
движущей силы в машинах требует разности температур: "Где имеется
разность температур, возможно возникновение движущей силы".
Важным этапом в развитии термодинамики явилось установление
принципа эквивалентности теплоты и работы, который нашел
отражение в законе сохранения энергии. Его открыли независимо друг от
друга Р. Майер (1841 г.), Дж. Джоуль (1843 г.) и Г. Гельмгольц (1847 г.).
Дальнейший крупный шаг был сделан немецким физиком
Р. Клаузиусом (1865 г.), который ввел понятие об энтропии как мере
превращаемости теплоты в работу. Несколько позднее В. Томсон ввел
понятие об абсолютной температуре. Работами Клаузиуса и Томсона
было завершено создание основ классической термодинамики.
Приложение термодинамики к химическим объектам также
связано с рядом фундаментальных исследований. В 1870-1880 гг.
в значительной степени благодаря работам норвежских физикохи-
миков К. Гульдберга и П. Вааге, австрийского физика Л. Пфаундлера
завершается формирование представлений о подвижном равновесии,
т. е. о состоянии, когда одновременно протекают две противоположные
7
химические реакции. Большую роль в развитии этих представлений
сыграло открытие французским ученым А. Сент-Клер Девиллем явления
термической диссоциации, или разложения тел под действием тепла,
при этом "отталкивательная сила теплоты образует равновесие со
сродством" (1857 г.). Закон действия масс в сочетании с молекулярно-
кинетическими представлениями составил основу учения о равновесии
в теоретической химии конца 1870 — начала 1880-х годов.
В 1873 г. немецкий физик А. Горстман на основании работ Клау-
зиуса пришел к выводу, что в изолированных системах критерием
достижения равновесия является максимальное значение энтропии.
В 1882 г. было опубликовано фундаментальное исследование
Гельмгольца "К термодинамике химических реакций", в котором
применительно к химическим процессам вводится представление о
свободной и связанной энергии. Ученый исходил из того, что подобно
тому как теплота может превращаться в работу лишь частично, так и
"в случае химических процессов должно быть принято разделение
между частью сил сродства, способной к свободному превращению
в другие формы работы, и частью, которая может выступать только в
виде тепла". Далее Гельмгольц пишет: "Я позволил себе коротко
обозначить обе эти части энергии как свободную и связанную энергию".
В той же работе указывается, что "при изотермических процессах
работа производится только за счет свободной энергии". Для
изотермических систем условием равновесия является минимальное значение
свободной энергии.
Большим вкладом в описание равновесий химических реакций
послужили исследования Я. Вант-Гоффа, представленные в
"Очерках по химической динамике" (1884 г.) и в цикле статей по теории
разбавленных растворов (1886-1887 гг.). Вант-Гофф подчеркивает
общность природы физических и химических равновесий,
рассматривает их с позиций термодинамики. Он выводит уравнение изохоры
реакции в дифференциальном и интегральном виде, уточняет понятие
"сродство", рассматривая его не как силу, а как работу, которую эта
сила может произвести. Все это позволило объединить термохимию,
электрохимию, исследования равновесий в единое учение о
химическом равновесии и сродстве.
Логическое завершение развития основных положений химической
термодинамики связано с работами американского физика Дж. Гиббса
8
(1876-1878 гг.). Он сформулировал важнейшие понятия современной
химической термодинамики, разработал критерии равновесия, ввел
в употребление весь тот математический аппарат, который позволяет
решать многочисленные задачи научного и прикладного характера.
В работах Гиббса были указаны общие методы и главные направления
для последующих исследований. Большую роль в распространении
идей Гиббса в Европе сыграла голландская школа физикохимиков,
возглавляемая в те годы И. Ван-дер-Ваал ьсом. Уже начиная с 1881 г. он
излагал в своих лекциях по термодинамике идеи Гиббса. Активными
последователями Гиббса были и другие представители голландской
школы — Я. Ван-Лаар, П. Дюгем, Г. Розебом.
Начиная со второй половины восьмидесятых годов XIX века
можно говорить о химической термодинамике как о самостоятельном
большом разделе физической химии. Как отмечается в монографии
А.Я. Кипниса "Развитие химической термодинамики в России"
(1964 г.), последняя четверть XIX века была для химической
термодинамики эпохой наиболее бурного роста, проникновения ее во
все области химии. Начиная с XX века происходит обстоятельная
разработка отдельных направлений и разделов химической
термодинамики, сформировавшихся в предшествующие годы. В течение
двух десятилетий (1890-1910 гг.) в мире было выполнено несколько
тысяч экспериментальных и теоретических исследований в области
химической термодинамики.
К числу наиболее крупных достижений начала двадцатого века
в теоретической термодинамике следует отнести результаты работ
немецких ученых — тепловой закон В. Нернста (1906 г.) и постулат
М. Планка (1911 г.), дающий более общую и более простую
формулировку этого закона. Тепловой закон Нернста в формулировке Планка
позволил вычислить абсолютные величины энтропии, что сделало
возможным проведение полного расчета равновесий на основе
калориметрических данных (теплового эффекта реакции при любой
температуре и теплоемкостей всех участников реакций).
Большую роль в развитии термодинамики неидеальных систем
сыграл предложенный американским физикохимиком Дж. Льюисом
(1907 г.) метод активностей. Метод позволяет сохранить простую форму
уравнений термодинамики идеальных систем при описании систем,
отклоняющихся от идеального поведения.
9
Методы химической термодинамики при наличии современной
справочной базы и расчетных возможностей позволяют наиболее
рациональным путем найти ответ на многие вопросы, связанные с
химическими или фазовыми превращениями. Основная задача химической
термодинамики — установление направления и границ протекания
самопроизвольных процессов. Поскольку эти процессы весьма
разнообразны, то и круг задач становится очень широким. Сюда относятся
многие задачи химической технологии и металлургии, неорганическое
материаловедение, синтез веществ с заданной совокупностью свойств,
включая синтез полупроводниковых соединений, композиций,
характеризующихся высокотемпературной сверхпроводимостью. С
помощью методов химической термодинамики определяются границы
устойчивости различных материалов в агрессивных средах, при
воздействии окружающей среды, при нагревании. В сферу действия законов
термодинамики входит большая совокупность систем преобразования
энергии, в основе которых лежат химические реакции.
Классическими примерами предварительного
термодинамического анализа технологических процессов являются определение области
давлений и температур, в которой возможно получение искусственных
алмазов, синтез аммиака из азота и водорода. Многие важнейшие
редкие металлы (титан, ниобий, тантал, уран, торий и др.) получают
восстановлением их оксидов или галогенидов с помощью различных
восстановителей (Н2, Na, Mg, Ca, A1, С, Si). Методы химической
термодинамики без проведения сложных экспериментов позволяют дать
рекомендации по выбору того или иного восстановителя.
К числу конкретных прикладных задач химической
термодинамики относятся расчет и оптимизация фазовых диаграмм систем
различной сложности, а также решение обратных задач — извлечение
термодинамической информации из фазовых диаграмм. Работы в этом
направлении получили значительное развитие во второй половине
XX века в связи с прогрессом вычислительной техники. С 1977 г.
издается международный журнал CALPHAD, специально посвященный
расчетам фазовых диаграмм.
В связи с развитием новых областей техники важное
значение приобрели термодинамические расчеты процессов,
происходящих при высоких температурах (до 3000-10000 °С) и давлениях
(до ЮОООатм).
10
Интересной областью применения химической термодинамики
остается полупроводниковое материаловедение — кристаллические
фазы переменного состава, полупроводниковые твердые растворы и
соединения. Методы термодинамики растворов могут быть полезны
на любой стадии создания полупроводникового прибора — от очистки
исходного сырья и подбора условий синтеза соединения до
получения {р - л)-перехода и определения температурного режима работы
готового изделия.
Каковы задачи прикладной химической термодинамики в начале
XXI века? Для того чтобы ответить на этот вопрос, следует принять во
внимание такие факторы:
истощение ресурсов ископаемого топлива и других видов сырья
ведет к экономии имеющихся ресурсов и к поиску новых.
Вовлекаются в переработку более бедные руды, большое значение приобретает
утилизация вторичного сырья;
реальная опасность глобального загрязнения окружающей среды
требует существенного изменения целого ряда технологических
процессов прежде всего в металлургической промышленности;
необходимость создания новых материалов, включая материалы
для микроэлектроники, сверхлегкие и сверхпрочные композиционные
материалы и многое другое, стимулирует развитие экспериментальных
и теоретических исследований, в том числе и химической
термодинамики;
дальнейшее развитие вычислительной техники, создание
современных банков данных, повсеместная их доступность ведут к
расширению расчетных возможностей химической термодинамики.
Химическая термодинамика, базирующаяся на фундаментальных
законах природы, ее расчетный аппарат, сформировавшийся более
ста лет назад и постоянно совершенствующийся, экспериментальные
методы исследований всегда будут занимать важное место в системе
наук о материалах.
В предлагаемом вниманию читателей издании авторы предприняли
попытку кратко описать широкий круг вопросов, представляющих
интерес при термодинамическом рассмотрении и экспериментальном
исследовании самых различных металлургических систем и процессов.
При этом мы постоянно стремились сохранять простоту и доступность
изложения. Книга включает семь глав и снабжена списком
рекомендуемой литературы.
11
В небольшой первой главе уточняются основные понятия и
определения, которыми оперирует химическая термодинамика.
Во второй главе очень кратко рассмотрены первый и второй
законы термодинамики, непосредственно связанные с ними расчетные
возможности.
Третья глава посвящена термодинамическому описанию фаз
переменного состава, прежде всего рассмотрены двойные системы.
Специальное внимание уделено активностям компонентов, выбору
стандартного состояния, интегрированию уравнения Гиббса-Дюгема,
разбавленным растворам, концентрационной зависимости
термодинамических функций.
В четвертой главе обсуждаются фазовые равновесия в одно- и
двухкомпонентных системах, зависимость энергии Гиббса от состава
в двухкомпонентных системах, общие принципы расчета фазовых
диаграмм на основе термодинамических данных и решение
обратной задачи — извлечение термодинамической информации из
фазовых диаграмм. Рассмотрены равновесия жидкость—жидкость,
жидкость-пар. Раскрывается понятие корректной и некорректной
постановки задач в термодинамике фазовых равновесий.
Пятая глава содержит краткое термодинамическое описание
химических процессов. Сюда включены уравнение изотермы химической
реакции, зависимость константы равновесия от температуры, понятие
об абсолютной энтропии, различные способы расчета стандартной
энергии Гиббса и константы равновесия химических реакций.
Шестая глава посвящена термодинамическому описанию трех-
компонентных систем. Особое внимание уделено оценке
термодинамических свойств тройных систем на основании данных о граничных
двойных системах с применением геометрических моделей и
полиномиальных методов расчета.
До настоящего времени основным источником данных о
термодинамических свойствах систем различной сложности является
экспериментальное исследование. В связи с этим в заключительной, седьмой
главе описаны методы исследования, наиболее широко применяемые
при изучении термодинамических свойств различных
металлургических систем, преимущественно металлических сплавов.
Авторы посчитали целесообразным привести довольно обширный
список рекомендуемой литературы, в основном металлургической
12
направленности, но содержащий и ряд изданий
общетермодинамического характера.
Главы 3,4,6 и 7 учебного пособия представляют собой полностью
переработанные и значительно дополненные аналогичные разделы
книги: А.Г. Морачевский. Термодинамика расплавленных
металлических и солевых систем. М.: Металлургия, 1987, 240 с, которая
была допущена Министерством высшего и среднего специального
образования СССР в качестве учебного пособия для студентов вузов,
обучающихся по специальности "Физико-химические исследования
металлургических процессов". В то время по этой специальности
велась подготовка инженеров-металлургов в целом ряде вузов СССР.
Данное учебное пособие, охватывающее широкий круг вопросов
термодинамического характера, может быть полезно при подготовке
магистров по направлению "Металлургия", а также для студентов
старших курсов и аспирантов различных металлургических и химико-
технологических специальностей.
Глава 1
ОСНОВНЫЕ ПОНЯТИЯ И ОПРЕДЕЛЕНИЯ
В химической термодинамике применяются те же понятия,
термины и величины, что и в общей термодинамике. Кратко рассмотрим
основные понятия.
Термодинамическая система — тело или группа тел, находящихся
во взаимодействии и выделяемых из окружающей среды мысленно или
с помощью реально существующей поверхности раздела. Система —
это предмет исследования. Поверхность раздела или граничная
поверхность — это неотъемлемый признак системы. Внешняя среда или
окружение — все, что окружает систему, существует за ее границами.
Изолированная система — это система, которая не может
обмениваться с окружающей средой ни веществом, ни энергией. Энергия
и объем системы постоянны.
Закрытая система — может обмениваться с окружающей средой
энергией и не может обмениваться веществом.
Открытая система — может обмениваться с окружающей средой
и энергией и веществом.
Гомогенная система — однородна во всех точках, имеет во всех
частях одинаковые термодинамические свойства (гомогенная
система может быть анизотропной, т. е. иметь свойства, зависящие от
направления).
Гетерогенная система — система, свойства которой в ее пределах
изменяются скачком. Пример: вода + лед. Однородная часть системы,
отделенная поверхностью раздела, носит название фазы. Все
гомогенные системы — однофазные. Гетерогенные системы состоят из двух
или нескольких фаз.
14
Непрерывные системы — системы, в которых отсутствуют
поверхности раздела, но свойства являются непрерывной функцией
координат (например, в вертикальном столбе газа давление будет непрерывно
изменяться с высотой).
Химические вещества, входящие в состав системы,
называются компонентами (более строгое определение этого понятия дано
далее).
Любая система характеризуется совокупностью
термодинамических свойств. Основные термодинамические свойства: масса, объем,
давление, температура, плотность, концентрация, теплоемкость.
По существу к термодинамическим свойствам относятся все свойства,
имеющие количественное выражение и характеризующие систему в
целом или ее макроскопические части, кроме характеристик потоков
массы и энергии (диффузия, вязкость, теплопроводность). Не
относятся к термодинамическим свойствам и кинетические характеристики,
в размерность которых входит время.
Все термодинамические свойства разделяются на интенсивные и
экстенсивные. К числу интенсивных свойств принадлежат давление,
концентрация, температура, другие свойства, имеющие вполне
определенное значение в каждой точке системы. Эти свойства не зависят от
массы системы, они выравниваются при переходе от менее сложной
системы к более сложной.
Экстенсивные свойства — объем, масса, энергия. Они
характеризуют конечную область системы, аддитивно складываются. Не имеет
смысла говорить о значении экстенсивного свойства в данной точке
материального пространства.
Экстенсивная величина, деленная на объем системы, называется
плотностью; деленная на количество вещества в молях — молярным
свойством, или молярной величиной; деленная на массу — удельным
свойством, или удельной величиной.
В международной системе единиц СИ единицей измерения
количества вещества является моль; единицей массы — кг.
Плотности, молярные и удельные свойства являются
интенсивными характеристиками.
Интенсивные свойства отражают физико-химическую
индивидуальность вещества, а экстенсивные — конкретный, представленный в
системе образец вещества.
15
Величины, количественно выражающие термодинамические
свойства, называют термодинамическими переменными. Поскольку
все эти величины связаны между собой, их обычно разделяют на
независимые переменные и функции. Такое деление условно и
определяется прежде всего удобством измерения тех или иных величин или
поддержанием их постоянства. Обычно легко измерить температуру,
давление, химический состав, поэтому эти переменные чаще всего
выступают в качестве независимых переменных, а энтропию, энергию
относят к числу термодинамических функций.
Набор значений независимых переменных определяет
термодинамическое состояние системы. Для полного описания системы
наряду с интенсивными величинами необходимо знать хотя бы одну
экстенсивную величину (массу, объем).
Переменные, которые фиксированы в пределах рассматриваемой
задачи, называют термодинамическими параметрами системы.
Например, температура и давление являются параметрами процесса,
протекающего в изобарно-изотермических условиях. Следует, однако,
иметь в виду, что термин "термодинамические параметры" часто
толкуют расширительно, не делая различия между термодинамическими
параметрами и термодинамическими переменными.
Различают следующие состояния термодинамической системы:
— равновесное, когда все характеристики системы постоянны и
в ней нет потоков вещества или энергии. Дополнительно выделяют:
устойчивое (стабильное) состояние, при котором всякое бесконечно
малое воздействие вызывает только бесконечно малое изменение
состояния, а при устранении этого воздействия система возвращается в
исходное состояние, и метастабильноесостояние, которое отличается
от устойчивого тем, что некоторые внешние воздействия вызывают
конечные изменения состояния, не исчезающие при устранении этого
воздействия;
— неравновесное (неустойчивое, лабильное) состояние, при
котором всякое бесконечно малое воздействие вызывает конечные
изменения состояния системы;
— стационарное, когда независимые переменные постоянны во
времени, но в системе имеются потоки.
Если состояние системы изменяется во времени, то это означает,
что в системе протекает процесс. Термодинамические процессы, их
16
протекание характеризуются не скоростями изменения свойств во
времени, а величинами этих изменений. Процесс в термодинамике —
это последовательность состояний системы, ведущих от начального
набора термодинамических переменных к конечному.
Различают следующие процессы:
самопроизвольные, их протекание не связано с затратами
энергии;
несамопроизвольные, их протекание возможно только при затрате
энергии;
обратимые, когда переход системы из начального состояния в
конечное и обратный переход происходят через последовательность
одних и тех же состояний и после возвращения системы в начальное
состояние в окружающей среде не происходит макроскопических
изменений. Понятие "обратимый процесс" — одно из важнейших в
химической термодинамике;
— квазистатические, или равновесные — протекают при
бесконечно малых воздействиях внешних сил;
— необратимые, или неравновесные— в результате их протекания
систему и окружающую среду невозможно вернуть в первоначальное
состояние.
В последнее время в учебной литературе уделяется внимание
способам аксиоматического построения термодинамики. В
частности, отмечается*, что выводы и соотношения термодинамики можно
сформулировать на основе двух постулатов (исходных положений)
и трех законов (начал). Основной постулат термодинамики: любая
изолированная система с течением времени приходит в равновесное
состояние и самопроизвольно из него не может выйти.
Это положение ограничивает круг систем, к которым можно
применять положения и законы астрономического масштаба, и
макроскопические системы с малым числом частиц.
Самопроизвольный переход системы из неравновесного состояния в
равновесное называют релаксацией. Постулат ничего не говорит о времени
Хм.: Основы физической химии. Теория и задачи: Учебное пособие для
вузов / В.В. Еремин и др. М.: Экзамен, 2005. 480 с. (Серия: "Классический
Университетский учебник").
17
релаксации, так как в классической равновесной термодинамике нет
понятия времени.
Второй постулат иногда называют нулевым законом
термодинамики, он описывает свойства систем, находящихся в состоянии
теплового равновесия: если система А находится в тепловом равновесии
с системой В, а та в свою очередь с системой С, то системы АиС
также находятся в тепловом равновесии.
Системы, находящиеся втепловом равновесии, имеют одинаковую
температуру. Таким образом, этот постулат вводит понятие о
температуре как об интенсивной переменной, характеризующей тепловое
равновесие.
Три закона термодинамики рассмотрены в главах 2 и 5.
Глава 2
ЗАКОНЫ ТЕРМОДИНАМИКИ
И ИХ ПРАКТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ
2.1. Первый закон термодинамики
Первый закон (первое начало) термодинамики по существу
выражает принцип сохранения энергии: в закрытой системе энергия
может изменяться за счет обмена теплотой с окружающей средой и
совершения работы. Записать это можно так:
AU=Q+W, (2.1)
где AU— изменение внутренней энергии системы; Q — количество
тепла, поглощенное системой (Q > 0) или отданное ею в окружающую
среду (Q < 0); W— работа, произведенная над системой (W> 0) или
произведенная самой системой (W< 0). В большинстве
отечественных руководств по физической химии или химической
термодинамике для работы ^используется иная система знаков: работа
считается положительной, если она совершается системой над окружающей
средой. В данном случае использована система знаков,
рекомендованная ИЮПАК (IUPAC).
ВеличинаД^/однозначноопределяется начальным ({/,) и конечным
(U2) состояниями системы, является функцией состояния, ее
изменение не зависит от пути процесса, от способа его проведения. В то же
время величины (?и ^зависят от пути процесса, условий, способа его
реализации. Под термином "путь процесса" понимается изменение
какой-либо величины, характеризующей степень превращения в
рассматриваемом процессе.
19
Если речь идет о бесконечно малых изменениях, то уравнение (2.1)
принимает вид
dU=bQ + bW. (2.2)
Здесь подчеркивается различие между полным дифференциалом
dl/и бесконечно малыми изменениями функций 8(?и b\V.
Иногда для открытых систем первый закон записывают в виде
dU=bQ + bW + YPidni> (2.3)
где последняя сумма характеризует процесс обмена веществом и
условно называется "химическая работа". Подробно о функции ц.,
называемой "химический потенциал", говорится в главе 3 (с. 40).
Если в закрытой системе единственный вид работы —
механическая работа расширения (5 IVmx = -pdV), то уравнение (2.2) принимает
вид
dU=6Q-pdV. (2.4)
Если процесс протекает при постоянном объеме, то
M)=U2-UX = QV. (2.5)
Тепловой эффект химической реакции при постоянном объеме
Qv равен изменению внутренней энергии. Хотя теплота не является
функцией состояния системы, но если задан характер изменения
Q (V= const), то Qv приобретает те же свойства, что и А£/.
Если процесс протекает при постоянном давлении, то можно
записать:
д^е,-/^-^),
Qp = AU+pAV
или
Qp=H2-H,=AH. (2.6)
Функция Н= U + рУност название "энтальпия".
Тепловой эффект химической реакции при постоянном объеме
равен изменению энтальпии.
В системе СИ размерность работы, теплоты, энергии — Дж.
20
2.2. Энтальпия индивидуальных веществ
Под индивидуальными веществами понимаются химические
элементы и их соединения. Как правило, речь идет не об абсолютном
значении энтальпии того или иного вещества, а об изменении функции
в определенном интервале температур.
В химической термодинамике важное место занимает выбор
стандартного состояния. Принято считать, что жидкие и твердые вещества
обладают стандартными свойствами при давлении 1 бар (105 Па). Пока,
однако, в справочных таблицах стандартное состояние соответствует
давлению 1 атм = 1,01325 бар. Для газообразных веществ стандартное
состояние соответствует идеальному газу при давлении 1 бар.
За стандартную температуру принимаются 25 °С (298,15 К), обычно
записывается подстрочный индекс "298".
В справочной литературе часто табулируется величина Щ - Н°1П
с тем или иным шагом (чаще через 100 К). Эта величина рассчитывается
на основании данных о теплоемкости при постоянном давлении (С°)
в интервале температур от 298 К до Т.
т
#f-tf2°98 = \C°pdT. (2.7)
298
Если в интервале от 298 К до Г вещество претерпевает фазовое
превращение, например плавится, то расчет производится с учетом
этого превращения:
Здесь Ств и С ж — соответственно теплоемкости рассматриваемого
вещества при постоянном давлении в твердом и жидком состояниях.
НТ - Я298 = Г C7dT + АН™ + J C?dT- <2-8>
298 7Ьл
Величины Н°т - #298 табулируются в Дж • моль-1 или в кДж • моль-1.
В справочниках, изданных до введения системы единиц СИ,
применяются кал • моль-1 (1 кал = 4,1840 Дж).
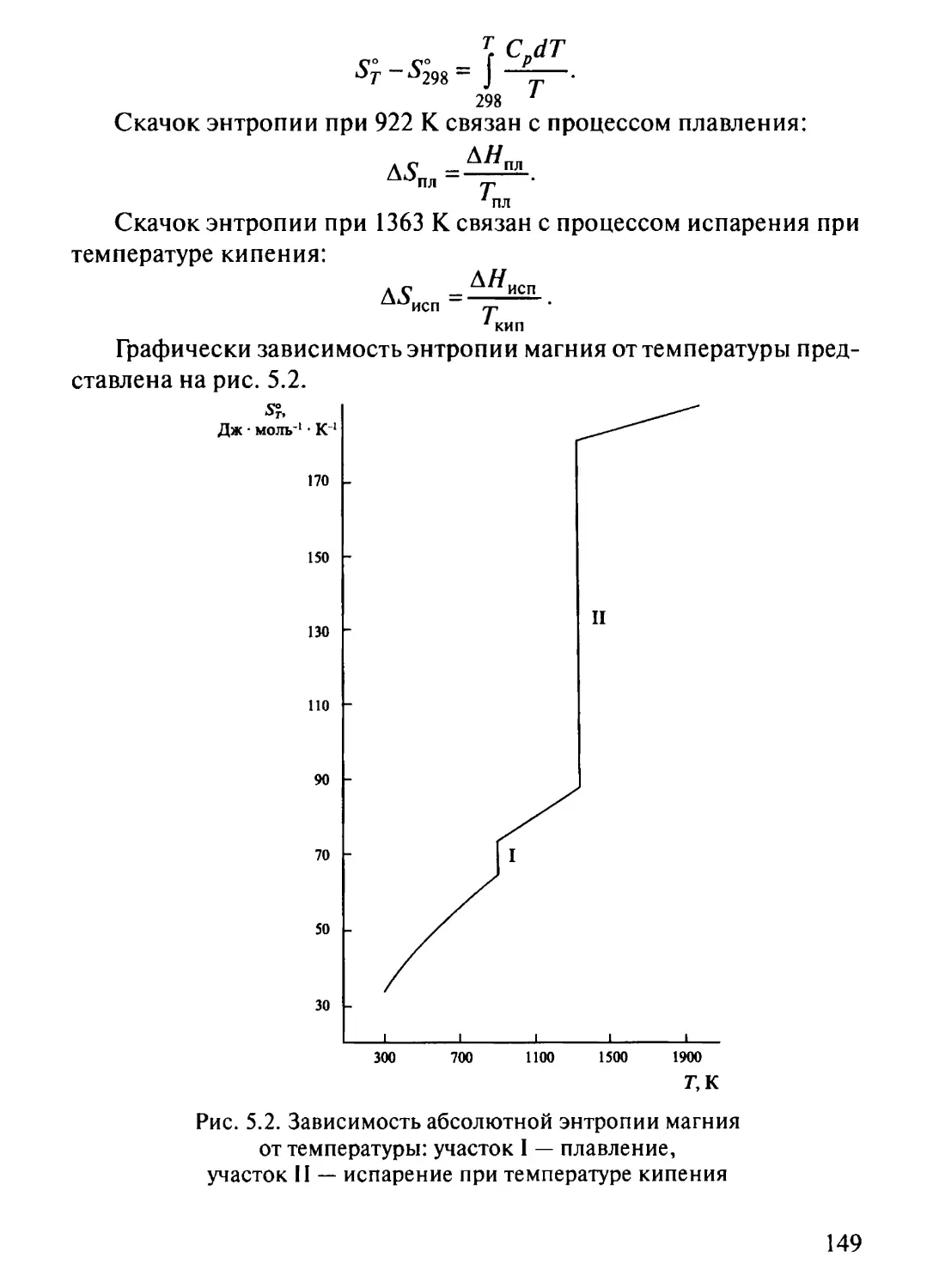
В качестве примера зависимости теплоемкости и функции Н°т - Н°29%
от температуры в табл. 2.1 приведены значения
термодинамических функций для магния, который плавится при температуре 922 К
(649 °С) и имеет температуру кипения при нормальном давлении
1363 К (1090 °С):
21
Таблица 271
Термодинамические функции магния
к к
298
400
600
800
922
922
1000
с\
Дж • моль-1 • К"1
24,89
26,11
28,45
30,79
32,26
32,13
32,97
Нт - #29g,
кДж • моль"1
0
2,60
8,06
13,98
17,83
26,78
29,33
Г, К
1200
1363
1363
1400
1600
1800
2000
Дж • моль-1 • К-1
35,15
36,90
20,79
20,79
20,79
20,79
20,79
Нт - #298,
кДж • моль"1
36,15
42,01
168,57
169,34
173,50
177,66
181,22
Энтальпия плавления равна 8,95 кДж • моль-1, энтальпия испарения
126,57 кДж • моль-1. При плавлении изменение теплоемкости
незначительно, что характерно для металлов и служит одним из указаний
на малые структурные изменения при переходе из твердого состояния
в жидкое.
Функция С ° =/{ 7) может быть представлена в виде полинома:
С^а + вТ+сТ2
или
С° = а + вТ+с'Т-\
р
Предпочтительнее второе выражение. При пользовании первым
из полиномов член сТ2 иногда имеет отрицательный знак, что может
привести к неопределенному максимуму на кривой С° =.Д 7).
Теплоемкость Ср° имеет размерность Дж • моль-1 • К-1.
При наличии экспериментальных данных о теплоемкости
веществ в области низких температур табулируется функция #7°- #0°
или дополнительно к функции #у° - Н°ш указывается величина
и° цо.
298 °' Т 298
Hj - #о = \c;dT, #298 - #о° = J c;dT. (2.9)
о о
Выражения (2.9) предусматривают, что в интервалах температур
от 0 до Гили от 0 до 298 К не происходит фазовых превращений.
22
2.3. Закон Гесса
Соотношения (2.5) и (2.6) служат теоретической базой основного
закона термохимии, установленного экспериментально российским
ученым Г.И. Гессом (1840 г.): Тепловой эффект реакции при
постоянном давлении или при постоянном объеме не зависит от пути (хода)
реакции, т.е. от промежуточных стадий и определяется только
начальным и конечным состоянием веществ при условии, что
единственной работой, совершаемой системой, является механическая
работа. Тепловой эффект относится к постоянной температуре
(Т= const).
В формулировке Гесса закон излагался в следующем виде: "Когда
образуется какое-либо химическое соединение, то при этом всегда
выделяется одно и то же количество тепла независимо от того,
происходит ли образование этого соединения непосредственно или же
косвенным путем и в несколько приемов."
Простейшим примером, иллюстрирующим применение
закона Гесса, может служить образование С02 при сгорании угля
(Т= 1000 К):
С (тв) + 02 (г) = С02 (г), АД = -394,6 кДж; (I)
С (тв) + 0,5О2 (г) = СО (г), АД, = -112,0 кДж; (II)
СО (г) + 0,5О2 (г) = С02 (г), ДД„ = -282,6 кДж. (III)
Поскольку указаны изменения энтальпии в процессе химической
реакции, то применяется оператор химической реакции Ar(rозначает
reaction). Оператор необходимо применять и к другим
термодинамическим функциям, если речь идет об их изменениях при
протекании химической реакции, например, АД АС и т.д.
В соответствии с законом Гесса АД = АД, + ЛД„:
дд
с + о2 ►со2
ДД,\ /ДД,-
со
что подтверждается указанными выше экспериментальными
данными.
23
2.4. Энтальпия образования
В справочной литературе табулированы величины так называемых
энтальпий (теплот) образования Ау#298, которые представляют собой
тепловой эффект реакции образования одного моля соединения из
простых веществ в стандартных состояниях. Энтальпия образования
простого вещества в стандартном состоянии равна нулю при любой
температуре. Оператор Ау указывает, что имеется в виду реакция
образования соединения из простых веществ (^означает formation).
Приведем несколько примеров реакций образования соединений
из простых веществ:
Ti (тв) + 2С12 (г) = TiCl4 (ж)
U (тв) + 02 (г) = U02 (тв)
РЬ (тв) + S (тв) = PbS (тв)
Al(TB) + 0,5N2(r)=AlN(TB)
А/Я298,кДжмоль~
-804,2
-1084,5
-162,6
-316,3
Стандартные энтальпии образования некоторых соединений Ду-Я^
(25 °С = 298,15 К; 1,013 ■ 105 Па) приведены в табл. 2.2.
Таблица 2.2
Стандартные энтальпии образованияДу//^ (кДж * моль1)
некоторых соединений
Соединение
А1203 (тв)
Fe203 (тв)
С02(г)
S02 (г)
Н20 (ж)
HF(r)
НгО (г)
ДуЯ298
-1669,8
-822,0
-393,5
-296,9
-285,9
-269,0
-241,8
Соединение
СО (г)
СН4(г)
NH3(r)
H2S (г)
С2Н4(г)
С2Н2(г)
&/Н°т
-110,5
-74,8
-46,0
-20,2
+52,3
+226,7
Из закона Гесса вытекает важное следствие: тепловой эффект
реакции (изменение энтальпии) равен алгебраической сумме
энтальпий (теплот) образования веществ, участвующих в реакции.
Для реакции
24
аЛ + bB^lL + mM (2.10)
Кнт = 1(&/нж), +т{А/нт)м -
1 м (2.11)
-a(*fHm)A-b(bfH°m)B.
Например, для определения Ar#298 Реакиии
TiCl4 (ж) + 2Mg (тв) = Ti (тв) + 2MgCl2 (тв)
в стандартных условиях при 298 К достаточно в справочных таблицах
найти энтальпии образования MgCl2 (kfH°2n = -644,3 кДж • моль-1)
и TiCl4 (А/#298 = —804,2 кДж ■ моль-1). Тепловой эффект реакции
Дг#2°98= (-644,3 • 2) - (-804,2) = -484,4 кДж • моль-1.
Реакция экзотермическая, сопровождается выделением большого
количества теплоты. В некоторых справочных изданиях* энтальпии
образования приводятся для широкого интервала температур и
различных агрегатных состояний. Например, для GeCl4 (г) значения
С° (Дж • моль-1 • К-1), Н°т- Н°ш и А//7° (кДж • моль1) приведены в
табл. 2.3.
Таблица 2.3
Стандартные термодинамические характеристики
тетрахлорида германия GeCI4 (г)
7, К
298
400
600
800
1000
1200
1400
1600
1800
L 2000
с;
95,22
100,75
104,63
106,10
106,80
107,19
107,42
107,58
107,68
107,76
ПГ "298
0
10,05
30,65
51,74
73,04
94,44
115,91
137,41
158,93
180,48
AfH°T
-500,00
-499,46
-428,24
-497,02
-495,96
-495,18
-531,30
-530,50
-529,73
-528,99 |
Заметный скачок в величине A^#f между температурами 1200 и
1400 К связан с тем, что при 1210,4 К германий плавится. До темпера-
* См. список рекомендуемой литературы.
25
туры плавления энтальпия образования газообразного тетрахлорида
германия соответствует процессу
Ge(TB) + 2Cl2(r) = GeCl4(r).
Выше точки плавления реакция такова:
Ge (ж) + 2С12 (г) = GeCl4 (г).
Энтальпия плавления германия составляет 36,94 кДж • моль-1.
2.5. Зависимость теплового эффекта реакции от температуры
Для реакции общего вида (2.10) изменение теплоемкости при
постоянном давлении АгС равно
\ С, = КС,\ + т(С;)м- а(С;)А - ЦС;),, (2.12)
ИЛИ
&гС,=Ьчс;»- Ъ>мсг. (2.13)
где С ° с соответствующим индексом — теплоемкость при постоянном
давлении того или иного участника реакции (реагента); £vnpC"p и
ХуисхСрСХ — суммы теплоемкостей соответственно продуктов
реакции и исходных веществе учетом стехиометрических коэффициентов
(v и v ).
v пр исх7
Как известно, по определению теплоемкость при постоянном
давлении равна Ср =| -^ | . В соответствии с уравнением Кирхгофа
дТ
Ъэ(]
уравнением , „
)р
зависимость теплового эффекта реакции от температуры выражается
-£L. = LrC,dT. (2.14)
Из уравнения (2.14) следует, что если изменение теплоемкости АгС,
рассчитанное с помощью уравнений (2.12) или в общем виде по (2.13),
для рассматриваемого процесса положительно, то и тепловой эффект
с ростом температуры становится более положительным:
при АС>0 ^^>0, (2.15)
а!
г Р
и наоборот,
26
при ДгС<0 ^?<0. (2.16)
а!
р
Если протекание процесса не сопровождается изменением
теплоемкости, т. е. сумма теплоемкостей исходных веществ с учетом стехио-
метрических коэффициентов равна сумме теплоемкостей продуктов
реакции, то тепловой эффект не зависит от температуры:
при АС=0 ^ = 0.
г ' dT
(2.17)
Графически зависимости, описываемые уравнениями (2.15)-(2.17),
приведены на рис. 2.1.
Изменение теплоемкости при фазовом переходе, например при
плавлении одного моля вещества А: Л(тв) *-> Л(ж), определяют из
уравнения
АС =(С) -(С) .
Д#
ДЯ1
Рис. 2.1. Различные виды зависимости теплоемкости участников реакции
и теплового эффекта реакции от температуры
27
Соответственно зависимость энтальпии плавления от температуры
выражается уравнением
В интегральной форме уравнение (2.14) имеет вид
г
ArHT2=ArHT] + \ArCpdT. (2.18)
Если разность температур Tjn ^относительно невелика, то можно
принять, что АС = const. При большой разности температур
необходимо учитывать зависимость АС =Д7).
2.6. Энтропия. Второй закон термодинамики
Все химические процессы подчиняются первому началу
термодинамики, поскольку оно выражает общий принцип сохранения
энергии, но далеко не все процессы протекают в тех или иных условиях
самопроизвольно. Существует большое число формулировок второго
закона термодинамики, определяющего возможность или
невозможность самопроизвольного протекания процесса. Для решения задач
прикладного характера удобна следующая формулировка: существует
некоторое экстенсивное свойство системы 5, называемое энтропией,
изменение которого связано с поглощаемой системой теплотой Q и
температурой следующим образом:
в самопроизвольном процессе dS > —;
лс 5<3 Т
в равновесном процессе dS = —;
в несамопроизвольном процессе dS < —.
Энтропия — свойство системы, изменение которого не зависит
от пути процесса, а однозначно определяется начальным и
конечным состоянием системы. Если мы имеем дело с изолированной
системой, для которой исключен теплообмен (80 = 0; t/, К, я = const,
п = я,... nN — набор количеств веществ), то приведенные выше
соотношения примут вид
в самопроизвольном процессе dS> 0;
в равновесном процессе dS=0;
в несамопроизвольном процессе dS< 0.
28
Обычно ограничиваются записью
dS>0, (2.19)
которая указывает, что энтропия изолированной системы или
увеличивается, или остается постоянной.
Выражение dS> О является критерием самопроизвольности
протекания процессов в изолированной системе. Энтропия изолированной
системы, в которой протекают самопроизвольные процессы, стремится
к максимуму. При достижении максимума устанавливается состояние
равновесия:
dS=0\d2S<0. (2.20)
Как показано далее, соотношения (2.19) и (2.20) лежат в основе
оценки самопроизвольного протекания процессов и условий
равновесия любых химических процессов. В связи с этим следует остановиться
на методах расчета энтропии и ее изменений в различных процессах.
2.7. Изменение энтропии индивидуальных веществ
в различных процессах
Энтропия имеет ту же размерность, что и теплоемкость: Дж * К-1.
Поскольку энтропия — величина экстенсивная, ее следует относить
к единице массы вещества, т. е. Дж • моль-1 • К-1. Ранее единица
кал • моль-1 • К-1 в научной литературе называлась "энтропийная
единица" (э.е.), с переходом к джоулям этот термин практически не
применяется.
В общем случае при нагревании
AS = S2-Sl=№. (2.21)
1 '
Если процесс протекает при постоянном давлении, то для одного
моля можно записать:
TiC dT
AS= f-2—. (2.22)
Соответственно для п молей
AS = nj
T} CpdT
т.
29
При нагревании индивидуального вещества его энтропия всегда
возрастает. Для изотермических процессов (плавление, испарение)
уравнение (2.21) принимает вид
Д5 = 1/8£ = ^. (2.23)
Если р = const, то Q = Q = АН и
A^-^f"- , Л*„сп = ^- (2-24)
*ПЛ *КИП
Для фазового перехода Н20(тв) -» Н20(ж) при температуре
плавления (273,15 К) АЯ0^ = 6008,2 Дж • моль-1. Плавление одного моля
воды сопровождается следующим изменением энтропии:
ASwi = ^=^^=22,0 Дж моль"1-К'1.
т Т^ 273,15 "*
При кипении воды (Гип = 373,15К, АЯ°исп = 40835,8 Дж • моль"1)
Д#исп 40835,8 1ЛП>|ТТ -1 ~1
ASwcn = ^= —= 109,4 Дж- моль -К.
исп т 37315
*КИП JfJ9LJ
Все фазовые переходы — плавление, испарение, сублимация, в
которых АН > 0, связаны с увеличением энтропии.
2.8. Критерии направления процесса
и условия равновесия в закрытых системах
Применять энтропийный критерий установления направления
процесса и достижения равновесия для закрытых систем, т. е. систем,
обменивающихся энергией с окружающей средой, но не
обменивающихся массой, не всегда удобно. Для изотермических процессов,
протекающих при постоянстве объема или давления, введены функции,
позволяющие решать поставленную задачу:
при постоянстве температуры и объема (Г, V— const)
F= U- TS, (2.25)
при постоянстве температуры и давления (Г, р — const)
G=U+pV- TS. (2.26)
30
В соответствии с существующей номенклатурой первая из функций
носит название энергии Гельмгольца, вторая — энергии Гиббса. Для
решения прикладных задач химической термодинамики наибольший
интерес представляют изобарно-изотермические процессы.
Объединенная формула I и II законов термодинамики может быть записана
так:
dU- TdS + pdV<0. (2.27)
При постоянных Т и р это уравнение преобразуется следующим
образом:
d(U~ TS + pV)<0.
С учетом выражения (2.26) можно записать:
dGTp<0, AGrp<0. (2.28)
Знак неравенства относится к самопроизвольным процессам. Для
конечных самопроизвольных изменений системы ДС7 < 0. Состоянию
устойчивого равновесия отвечает минимальное значение энергии
Гиббса системы:
dGTp = Q,cPGTp>0. (2.29)
Аналогично при изохорно-изотермических процессах для энергии
Гельмгольца
dFrv<^ AFTV<0. (2.30)
Состоянию устойчивого равновесия при Т, V — const отвечают
условия
dFTV=0, cPFTy>0. (2.31)
Таким образом, из неравенств (2.28) и (2.30) следует, что энергия
Гиббса является критерием направления процесса и достижения
равновесия в изобарно-изотермических условиях, а энергия Гельмгольца —
в изохорно-изотермических условиях.
На рис. 2.2 представлено изменение трех критериев направления
процесса и достижения состояния равновесия системы — энтропии,
энергии Гиббса и энергии Гельмгольца. Откладываемая по оси абсцисс
величина должна быть доступна опытному определению. Как видно
на рисунке, при равновесии все три функции — S, Си F— достигают
экстремального значения.
Важно еще раз подчеркнуть, что энтропийный критерий применим
к процессам, протекающим в изолированных системах, а два других
критерия - к закрытым системам.
31
а)
U = const
V = const
В
6)
G
р = const
Т = const
*)
А
В
V = const
Г = const
В
► Путь процесса
Рис. 2.2. Изменение энтропии (д), энергии Гиббса (б), энергии
Гельмгольца (в) при протекании различных процессов:
АВ— необратимый самопроизвольный процесс,
ВС— необратимый и несамопроизвольный процесс,
В — равновесное состояние
2.9. Характеристические функции.
Уравнение Гиббса—Гельмгольца
Характеристической функцией называется термодинамическая
функция, посредством которой или ее произюдных могутбыть выражены
в явном виде термодинамические свойства системы (р, V, Т, S и др.).
Рассмотрим функции G=J{T,p) и F=J{T, V). Выразим полные
дифференциалы этих функций через частные производные:
-(£)-
Гд0 I A
dF-.
dF_
ж
dV.
(2.32)
(2.33)
/г
В то же время дифференцированием выражений (2.25) и (2.26) в
сочетании с объединенной формулой первого и второго законов
термодинамики получаем
dG=-SdT+ Vdp, (2.34)
dF=-SdT-pdV. (2.35)
Сравнивая выражения (2.32) и (2.34), а также (2.33) и (2.35),
приходим к выводу, что
32
dG_
дТ
ЪТ
=-s,
JP
= S, ^
Jy
dp
ЭК
= v;
JT
=-/>•
(2.36)
(2.37)
JT
Таким образом, в соответствии с ранее данным определением
функции G=J{ T,p) и F=J[ T, V) могут быть отнесены к числу
характеристических. Изменение энергии Гиббса можно выразить равенством
AG=AH-TAS, (2.38)
соответственно
дТ
= -AS.
jp
Из этих двух выражений получаем
AG=AH+T
аналогично можно получить
AF=AU+T
дТ
ЭД£
дТ
jp
(2.39)
(2.40)
(2.41)
Ж
Уравнения (2.40) и (2.41) носят название уравнений Гиббса-
Гельмгольца. Величины AG и AF в них имеют смысл максимальной
работы химической реакции, когда она проводится в обратимых
изотермических условиях. Величины АЯи AU' — тепловые эффекты той
же химической реакции, когда она проводится в максимально
необратимых условиях. Вторые слагаемые правых частей уравнений (2.40)
и (2.41) имеют смысл теплоты обратимого процесса.
Глава 3
ОСНОВЫ ТЕРМОДИНАМИКИ ФАЗ
ПЕРЕМЕННОГО СОСТАВА
3.1. Общие соотношения
Фазой переменного состава, или раствором, называется система,
состоящая из двух или большего количества веществ, состав которой
может непрерывно изменяться в определенных пределах. По
агрегатному состоянию растворы могут быть жидкие, твердые или
газообразные. В рамках наших задач наибольший интерес представляют
конденсированные системы, т.е. жидкие или твердые.
Образование любой фазы переменного состава сопровождается
изменением термодинамических функций, описывающих данную
систему. Важнейшие из этих функций — энергия Гиббса ((7), энергия
Гельмгольца (F), энтальпия (Я), энтропия (£), объем (V). Основные
математические соотношения для всех этих функций имеют сходный
вид, поэтому ограничимся рассмотрением уравнений, написанных
только для энергии Гиббса. В общем случае, для любых процессов,
сопровождающихся изменением числа молей компонентов во время
процесса, можно записать:
G=f{T,p, nv nr nv ..., /i.), (3.1)
dG =
'д(Г\
дТ
dT +
УРА
— dp+
Э6-
(3.2)
dnx +
)pJ*j
dG
dn*>+....
}pJ,n,-
34
где nv nv nv ..., л.— массы компонентов, выраженные числом молей.
Полный дифференциал рассматриваемой функции имеет вид
Здесь п. — постоянное количество всех компонентов; п —
постоянное количество всех компонентов, кроме одного, изменение
которого рассматривается.
Обозначим
и давлении
'ас
как G. Тогда при постоянных температуре
JpJsHj
dGrrY.Gdnr
(3.3)
Функция С=Д«,, nv nv ..., п) при постоянных температуре и
давлении обладает важной особенностью: если массы всех веществ
увеличить в к раз, то и величина соответствующего термодинамического
свойства возрастает в то же число раз. Увеличение массы каждого
компонента в определенное число раз идентично увеличению массы
всей системы (фазы) в то же число раз без изменения состава.
Пропорциональность массе внутренней энергии (U), энтропии
(S), объема (V) обусловлена существом этих функций. Поскольку
И= U+ pVw G = U + pV— TS, то величины энтальпии (//) и энергии
Гиббса (G) также оказываются пропорциональными массе системы.
Математически такое свойство системы и функций, ее
характеризующих, можно записать так:
f(knv knv kny..) = kAnv nv ny..), (3.4)
где к — некоторый множитель. Функции нескольких переменных,
подчиняющиеся уравнению вида (3.4), носят название однородных
функций первой степени.
Таким образом, при постоянных температуре и давлении
рассматриваемые термодинамические функции являются однородными
функциями масс компонентов первой степени. Согласно теореме
Эйлера, для однородной функцииДх, у, z) можно записать
Ъ>
К
+ Z
fl=/<w>.
(3.5)
Применительно к функции G = f[nr nv л3, ..., п) по аналогии с
(3.5) получаем
г
G = n,
л
+ /Ь
JpJ,"}
дп2
+ ... + Я:
JPJ^j
[дп,
(3.6)
/Г.Р.Иу
35
С учетом ранее введенного обозначения уравнению (3.6) можно
придать вид
Принимая во внимание уравнение (3.3), очевидно, что
ZndG = Q.
Величины G, а также Fp #, 5, К. носят название парциальные
молярные величины. Они представляют собой частные производные
от экстенсивного свойства по массе компонента, выраженной числом
молей, при постоянных температуре и давлении и массах всех других
компонентов системы.
Рассмотрим прежде всего термодинамические функции для двух-
компонентных систем при постоянных температуре и давлении:
dG=Gldnl + G2dnv (3.8)
G =nlGl + n2Gv (3.9)
nxdGx + n2dG2 = Q. (3.10)
В химической термодинамике основной способ выражения
состава — отношение массы данного компонента к сумме масс всех
компонентов. Если массы выражены числом молей, то получаем
молярные доли (х). Для двухкомпонентной системы
х] — ? х2" •
^+«2 Пх+П2
Тогда уравнения (3.8)-(3.10) примут вид
-^- = (7Л/=хС1+д:2С72, (3.11)
пх+п2
dGM=Gldxl + G2dx2, (3.12)
х, dGx + x2dG2 = 0. (3.13)
В выражениях (3.11)-(3.13): GM — интегральная молярная
энергия Гиббса; Gx и G2 — парциальные молярные энергии Гиббса
компонентов 1 и 2.
Соотношения вида (3.10) и (3.13) носят собирательное
название — уравнение Гиббса-Дюгема, которое может быть записано для
различных термодинамических функций.
36
Величины G} и G2 в уравнениях (3.8)—(3.10) и (3.11)-(3.13) имеют
одно и тоже численное значение. Парциальные молярные величины
как функции состава двойной или более сложной системы можно
найти, если известна зависимость интегрального
термодинамического свойства от состава. Для простоты рассмотрим двойную систему.
Задача состоит в том, чтобы вычислить, например, Gx или Gv если
известна зависимость GM=f[x2). Предположим, что эта зависимость
выражается кривой, показанной на рис. 3.1. Уравнение (3.11) можно
записать следующим образом:
С=<1-*,)(?,+^(?2.
Из уравнений (3.11)-(ЗЛ4) следует
(7)С, Л
= С1-С2+(1-х2) —U
Для двухкомпонентной системы можно записать
С,=Дх2) и С2=Л*2).
Эх7
+ х2
ЭС2Л
V 2 /Г,р
(3.14)
(3.15)
Тогда
dG =
dG]
Эх9
dx2 и dG2 =
Jt.p
С учетом этих выражений и в соответствии с уравнением (3.13)
сумма двух последних членов в уравнении (3.15) равна нулю. Таким
образом, принимая во внимание уравнение (3.14), мы получаем два
уравнения, позволяющие выразить Gx и G2 через значения GM и
{—)
Gl=GM -х
^м
G2=GM+(\-x2)
ъс_Л
дхЛ.Р'
(3.16)
(3.17)
Соотношения (3.16) и (3.17) лежат в основе простого
графического метода определения G{ и Gr Изданных, представленных на
Рис. 3.1 видно, что — = tgcc,a <7W= OB. Соотношения G, = OB- ВС
дх2
и G2 = OB- BA совпадают с уравнениями (3.16) и (3.17). Касательная
37
к кривой 0м =Дх2) в данной точке (точка В) отсекает на осях ординат
отрезки, отвечающие величинам <7, и G2 для заданного на рис. 3.1
состава (точка О). Величины G° и G2° — молярные значения (7м для
чистых компонентов 1 и 2 при постоянных температуре и давлении.
Уравнению (3.16) можно придать вид, сходный с уравнением (3.17):
Рис. 3.1. Схема определения парциальных молярных величин
.Л/
</,=<?"+(!-*,)!
Эх,
(3.18)
)Т,р
Аналогичные зависимости справедливы и для других
перечисленных выше термодинамических функций.
3.2. Химический потенциал
Парциальная молярная энергия Гиббса /-го компонента носит
название также химический потенциал /-го компонента (ц.). Следует
отметить, что понятие химического потенциала можно вывести с
помощью и других термодинамических функций:
fdG\ (bF\ (дн\ (dU^
И,- =
v 3/1/ , ~
V l jyj,nj V ' Jp,Smj
dnt
M,s,nj
38
Химический потенциал
представляет собой частную
производную от какого-либо из
термодинамических потенциалов (Я, {/, G,
F) по числу молей /-го компонента
при постоянстве соответствующих
этому потенциалу параметров
состояния и постоянстве чисел молей U H F G
всех других компонентов, входя-
Рис. 3.2. Схема взаимосвязи тер-
щих в рассматриваемую систему. к
-- модинамических потенциалов
На рис. 3.2 показана взаимосвязь (харакТеристических функций)
термодинамических потенциалов и и их естественных переменных
их естественных переменных.
Для лучшего понимания физического смысла химического
потенциала как частной производной, например, от функций Сил и F,
приведем следующее его определение: если при постоянной температуре
к бесконечно большому количеству фазы переменного состава
(раствору) добавить один моль какого-либо компонента, то химический
потенциал этого компонента будет равен приросту энергии Гиббса в
том случае, если р = const, или приросту энергии Гельмгольца, если
V= const. Понятие "бесконечно большое количество" (иногда просто
"большое количество", "большой объем") означает, что состав системы
практически не должен изменяться при добавлении моля компонента.
Химический потенциал чистого вещества равен энергии Гиббса одного
моля этого вещества, так как при изменении количества чистого
вещества на 1 моль энергия Гиббса изменится на величину, равную энергии
Гиббса одного моля вещества.
В соответствии с уравнением (2.34) при постоянной температуре
dG=Vdp. (3.19)
Для компонента смеси газов, ведущей себя идеально, можно
записать
dGt = d\x. = Vdp. = RTd\upr (3.20)
В интегральной форме выражению (3.20) можно придать вид
\i= »; + RTdLnpl9 (3.21)
где ц° — стандартный химический потенциал; р. — парциальное
Давление компонента в газовой смеси.
39
3.3. Относительные термодинамические функции
При термодинамическом описании систем, особенно когда речь
идет об энергетических характеристиках (G, F, Я, £/), обычно
приходится иметь дело не с полным значением соответствующего
экстенсивного свойства при образовании фазы из чистых компонентов.
В связи с этим вводится понятие об относительных
термодинамических функциях. Разность между парциальной молярной величиной
компонента в растворе, например, G. и молярной величиной для
чистого компонента (7° называется относительной парциальной
молярной величиной: AG. = G. - G* (Ац, = ц, - ц/).
Для чистого компонента в качестве стандартного состояния
преимущественно выбирают термодинамически наиболее устойчивое
состояние при данной температуре. В отдельных случаях за
стандартное состояние принимают переохлажденные жидкости.
Наряду с относительными парциальными молярными
величинами каждого из компонентов вводится понятие об относительных
интегральных молярных величинах:
AG = G"-Xx,.G7=Xx,.AG,.. (3.22)
Величину относительного интегрального молярного свойства для
двухкомпонентной системы можно записать следующим образом:
AG=GM- (xfi{ + x2G2) = JCjAGj + x2AGr (3.23)
Графическое изображение зависимости относительных
интегральных величин от состава всегда будет иметь иной вид, чем
это представлено на рис. 3.3 для полных величин. Парциальные
молярные функции смешения в двойной системе равны нулю для
компонента 1 при х{ = 1, а для компонента 2 при х2 = 1.
Соответственно величина А(7 равна нулю как при л:, = 1 (х2 = 0), так и при
х2 = 1 (х{ = 0). Для связи между относительными парциальными и
интегральными величинами справедливы соотношения того же
вида, что и для полных величин. Так, например, для AG:
AG^AG + il-xA^) , (3.24)
40
AG2=AG + (1-jc2)
ЭАС
Эх
2 )т.р
(3.25)
AG,
Рис. 3.3. Графическое изображение связи между
относительными парциальными и интегральной
величинами энергии Гиббса
Типичная кривая, иллюстрирующая расчеты с помощью
уравнений (3.24) и (3.25), приведена на рис. 3.3. Аналогичного рода
зависимости справедливы и для других относительных функций (АЯ,
At/, А^идр.).
3.4. Идеальный раствор
и избыточные термодинамические величины
3.4. L Термодинамические функции идеального раствора
При анализе термодинамических свойств металлических и
солевых систем, как и любых других фаз переменного состава, большое
значение имеет понятие об идеальном (совершенном) растворе. Под
идеальным обычно понимается раствор, для компонентов которого
пРи всех составах и температурах выполняется закон Рауля:
41
/?. = /?.°х, (3.26)
где р. — давление насыщенного пара компонента / над раствором;
р° — давление насыщенного пара компонента / над чистым
компонентом при той же температуре.
Как известно, закон был установлен экспериментально
французским химиком Ф.М. Раулем (1882 г.). В соответствии с уравнениями
(3.20) и (3.21) для химического потенциала компонента в идеальном
растворе справедливо выражение
jiF-tf+jmn*. (3.27)
Изменение энергии Гиббса, которым сопровождается
образование двухкомпонентного идеального раствора, равно
АСИД = xlRTlnxl + x2RTinx2 = RT(xllnxl + х2\пх2). (3.28)
Из представлений о характеристических функциях (с. 32) следует,
что
№■*- (W1—• <->
Тогда путем дифференцирования уравнения (3.28) по температуре
получаем
А5ИД = -fl^lrix, + х21пл:2). (3.30)
Для относительной парциальной молярной энтропии смешения
компонентов 1 и 2
А51ид = -Л1ах1 и А52ид =-Л1пх2. (3.31)
Величины относительных парциальных и интегральных энергий
Гиббса, энтропии, энтальпии связаны соотношениями
АС = АЯ-7А5; AG=AH-TAS. (3.32)
Из уравнений (3.28)—(3.32) следует, что при образовании
идеального раствора с любым числом компонентов АН. = 0; АН = 0.
На рис. 3.4 представлены кривые, характеризующие образование
идеального двухкомпонентного раствора.
В табл. 3.1 приведены численные значения парциальной
молярной энтропии смешения компонента при образовании идеального
раствора.
42
Рис. 3.4. Кривые парциальных и интегральных молярных
энергий Гиббса (а) и энтропии (б) при образовании
идеальной двухкомпонентной системы
Таблица 3.1
Численные значения парциальной молярной энтропии смешения
1-го компонента в идеальном растворе (в Дж * моль'1 • К1)
1 /
0,000
0,050
0,100
0,150
L 0,200
Г 0,250
[ 0,300
АЛ™
00
24,91
19,14
15,77
13,38 |
11,525
10,010
X
\ i
0,350
0,400
0,450
0,500
0,550
0,600
0,650
"as™
/ 1
8,729
7,618
6,639
5,763
4,971
4,247
3,582
X
\ i
0,700
0,750
0,800
0,850
0,900
0,950
1,000
AS™
2,965
2,392
1,855
1,351
0,8760
0,4264
0,00
3.4.2. Избыточные термодинамические функции
В связи с тем, что реальные системы существенно отклоняются в
своем поведении от идеальных, для характеристики этих отклонений
Целесообразно ввести понятия об избыточных термодинамических
Функциях смешения. Так, для энергии Гиббса можно записать
А<7=АСид + ДСиз6. (3.33)
43
Величина AG1"6 суммарно учитывает все отклонения энергии
Гиббса системы от идеальных значений. Соответственно
AG"36 = AG- АСИД; AG/"6 = AG. - AG™. (3.34)
Для избыточной энергии Гиббса двухкомпонентной системы:
AG"36 = AG - /Щл;,1пх, + х21пх2), (3.35)
AG,"36 = AG, - ЛЛшс,; АС2иэб = AG2 - RT\nxr (3.36)
AG"36 = JCjAG,"36 + x2AG2H36. (3.37)
Для избыточной энтропии двухкомпонентной системы:
А5из6 = AS + /^lnx, + x2lnx2), (3.38)
AS,"36 = AS] + /An*,; А52из6 = А52 + R\nxv (3.39)
AS"36 = xfiS™6 + x2AS2m\ (3.40)
Поскольку АЯИД = 0, применение термина "избыточные функции"
для энтальпии лишено смысла. Для избыточных функций
справедливы все общие соотношения между интегральными и парциальными
величинами.
Величина АСдля равновесных систем всегда отрицательна. Это
следует из общего положения, что при постоянных давлении и
температуре все самопроизвольные процессы сопровождаются убылью
энергии Гиббса. Каких-либо принципиальных офаничений отдельно
для знаков АЯ и AS в изобарно-изотермических условиях нет, однако
надо иметь в виду, что
AG=AH~ 7AS<0. (3.41)
Если АН> 0, то А5> 0 и TAS> АЯ; если АЯ= 0, то AS> 0; если
АЯ< 0, то А5> 0 или AS < 0, но должно выполняться соотношение
(3.41).
Для избыточной энергии Гиббса можно записать:
AGm6 = AH~ TASm6$ 0.
Величина AG"36 определяется характером отклонения системы
от идеального поведения. Типичные кривые зависимости АСиз6 от
состава для ряда систем приведены на рис. 3.5. Для отрицательных
значений АСиз6 ограничений нет. Ограничения для положительных
значений AG"36 рассмотрены в гл. 4 (с. 117).
44
Рис. 3.5. Кривые интегральной молярной избыточной энергии Гиббса
в зависимости от состава для систем: а — с отрицательными
отклонениями от идеального поведения при 1123 К: / — Mg-Bi;
2— Mg—Pb; 3— Pb—Bi; б— с положительными отклонениями
от идеального поведения при 1873 К: 7— Си—Ni; 2— Си—Со.
Стандартное состояние — чистые жидкие металлы,
х2 — для всех систем молярная доля компонента,
указанного вторым. AG*36 — в кДж • моль-1
3.5. Активность и коэффициент активности.
Интегрирование уравнения Гиббса-Дюгема
Как отмечалось выше, для идеальных систем имеется простая
связь между величиной химического потенциала и содержанием
компонента в растворе. Для описания реальных систем вводится
понятие об активности, и выражение для химического потенциала
приобретает вид
ц,= ц; + ДЛ1Ш,. (3.42)
Активностью /-го компонента раствора называется величина,
которую нужно подставить в выражение для химического потенциала
компонента в идеальном растворе, чтобы получить действительное
значение химического потенциала /-го компонента в неидеальном
растворе. Активность af — безразмерная величина.
В общем случае а. =/( Г, />, х,, х2, ..., х), в изобарно-изотермических
Условиях активность компонента зависит только от состава. Связь
45
между активностью компонента и его содержанием в
рассматриваемых фазе или системе представляется в виде зависимости
я =УЛ> (3'43>
где у.— коэффициент активности компонента, суммарно
учитывающий отклонения от идеального поведения. Коэффициент
активности у, — безразмерная величина.
Из уравнения (3.42) следует, что 1пд, =———.
КГ
Таким образом, величина активности определяется через
разность химических потенциалов /-го компонента в данной фазе (ц.) и
в стандартном состоянии (ц.°); ц. = ц.° при а.= 1. Выбор стандартного
состояния — это выбор удобной начальной точки отсчета для
последующего вычисления зависимости химического потенциала от
состава раствора. Во многих случаях за стандартное состояние удобно
принимать чистый компонент: а.— 1 прих.= 1. Более сложные случаи
выбора стандартного состояния, а также переход от одного
стандартного состояния к другому будут рассмотрены далее.
Для двухкомпонентных систем при указанном выше выборе
стандартного состояния можно записать: при х{ = 1 ах = 1 и Yj = 1; при
х2 = 1 а2 = 1 и у2 = 1. Если образуется идеальный раствор, т.е.
выполняется закон Рауля, то при всех составах ах = х{, у1 = 1, а2 = xv у2 = 1.
В общем случае в зависимости от характера взаимодействия между
компонентами могут наблюдаться положительные (а{ > х{, у, > 1),
отрицательные {а{ <xvyx< 1) или знакопеременные (в зависимости
от состава у{ > 1 или у{ < 1) отклонения от идеального поведения.
Типичные изотермы активности для компонента 1 приведены на
рис. 3.6.
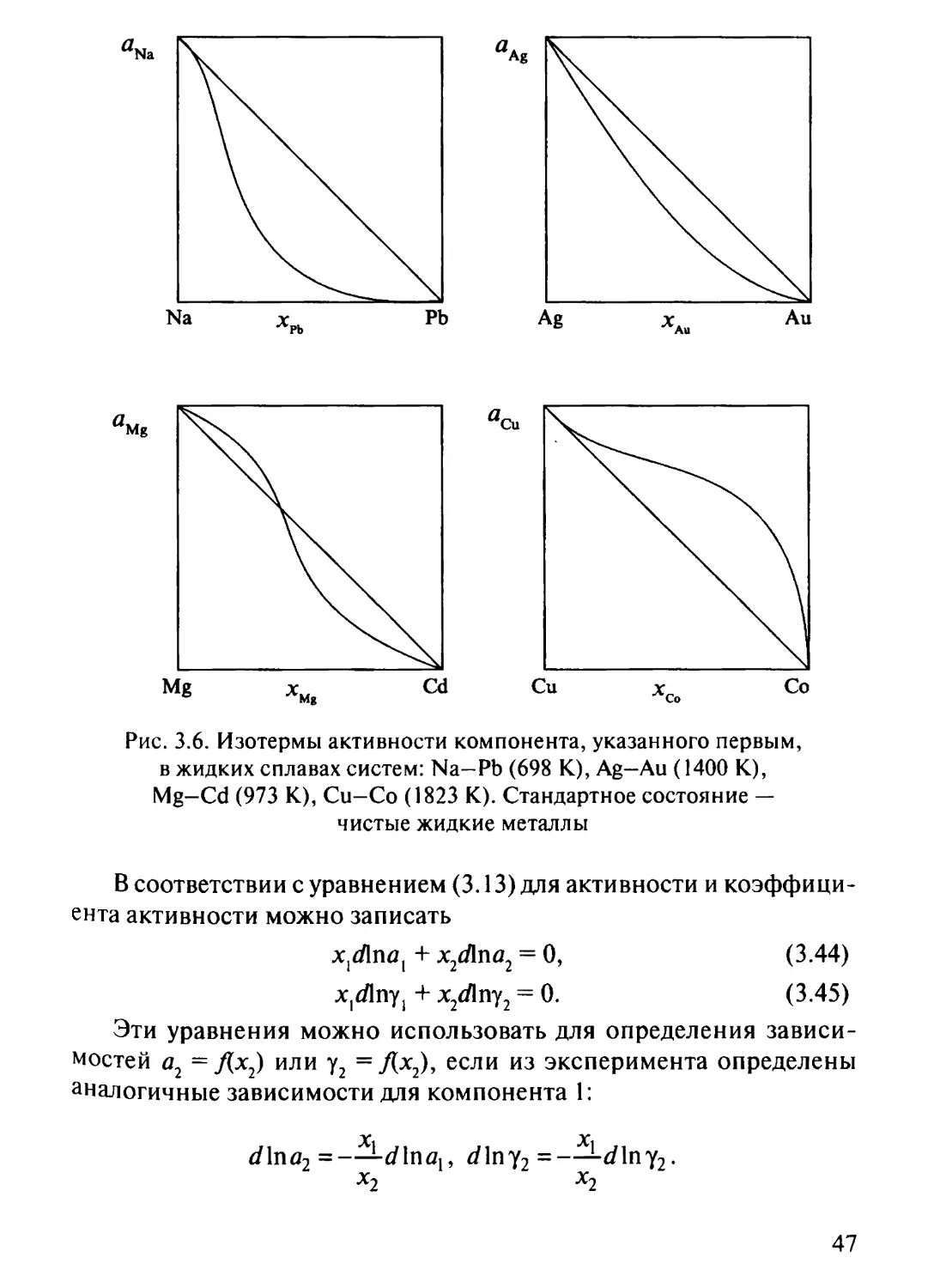
В системе Na— Pb наблюдаются значительные отрицательные
отклонения изотермы активности натрия от идеального поведения, в
системе Ag-Au — умеренные отрицательные отклонения, в системе
Mg-Cd для изотермы активности магния наблюдаются
знакопеременные отклонения, в системе Си-Co — положительные отклонения
от идеального поведения.
Большей частью из эксперимента определяют концентрационную
зависимость активности и соответственно коэффициента активности
только одного из компонентов системы, ддя второго компонента эти
величины определяются расчетным путем.
46
Ag
Na
Pb
Ag
Au
Рис. З.6. Изотермы активности компонента, указанного первым,
в жидких сплавах систем: Na-Pb (698 К), Ag—Au (1400 К),
Mg-Cd (973 К), Cu-Co (1823 К). Стандартное состояние —
чистые жидкие металлы
В соответствии с уравнением (3.13) для активности и
коэффициента активности можно записать
xldlnal + x2dlna2 = 0, (3.44)
xldlr\y] + x2dlny2 = 0. (3.45)
Эти уравнения можно использовать для определения
зависимостей а2 = J{x2) или у2 =Ях2), если из эксперимента определены
аналогичные зависимости для компонента 1:
X X
d\na2=—-d\na{, dlny2=—-d\ny2.
47
Поскольку величина 1пя, при jCj —> 0 стремится к —оо, а lny, при
хх -> 0 имеет конечное значение, предпочтительным является
интегрирование уравнения (3.45). Получаем:
1ПУ!
при
*2
lny2 =- J — dlny^
InYl "
при
(3.46)
Соответствующее графическое построение представлено на
рис. 3.7, а. При х2 = 1 (JCj = 0) величина lnyj имеет конечное значение
и является нижним пределом интегрирования. Заштрихованная
площадь на рис. 3.7,д соответствует подынтегральному
выражению в уравнении (3.46). Для рассматриваемой системы с довольно
сильными отрицательными отклонениями от идеального поведения
(кривые на рис. 3.6 соответствуют жидким сплавам системы Na-Sn
при 850 К) точное определение этой площади не вызывает
затруднений. Однако часто форма графика оказывается значительно менее
удобной для определения подынтегрального выражения.
а)
xjx.
-АщЛХ-х»
0 -0,5 -1,0 -1,5 -2,0
1пу,
1,0 0,8 0,6 0,4 0,2 0
*1
Рис. 3.7. Графическое интегрирование
при расчетах с помощью уравнений: а — (3.46); 6— (3.47)
Даркен (1950 г.) предложил при интегрировании уравнения (3.46)
воспользоваться вспомогательной функцией
48
a^lny/O-x^lny/^2.
Тогда
lir^ = a{/x22, d\nyl = 2alx2dx2 + x22dar
Подставив последнее выражение в уравнение (3.46), получим
х2 х2
dlny2=- \ 2a{xxdx2- \ x{x2da}.
х2-\ х2=\
Дальнейшие преобразования приводят к зависимости
\ny2=-alxlx2+ J axdxx. (3.47)
Уравнение (3.47) для любых двойных систем является удобной
интегральной формой уравнения Гиббса-Дюгема. Величину
интеграла легко определить графически (рис. 3.7,6).
Из уравнения (3.47) можно получить столь же удобные выражения
для интегральных избыточных термодинамических функций двух-
компонентных систем:
AG"*=a-x,) J—i-jdc,, (3.48)
xI=o(l-*l)
Х\ А II
ДЯ=(1-х,) J -^-jdx,, (3.49)
*, доизб
ДГ36 =(!-*,) f -=3-^*fe,. (3.50)
*1=oU-*iJ
3.6. Закон Генри.
Термодинамические свойства разбавленных растворов
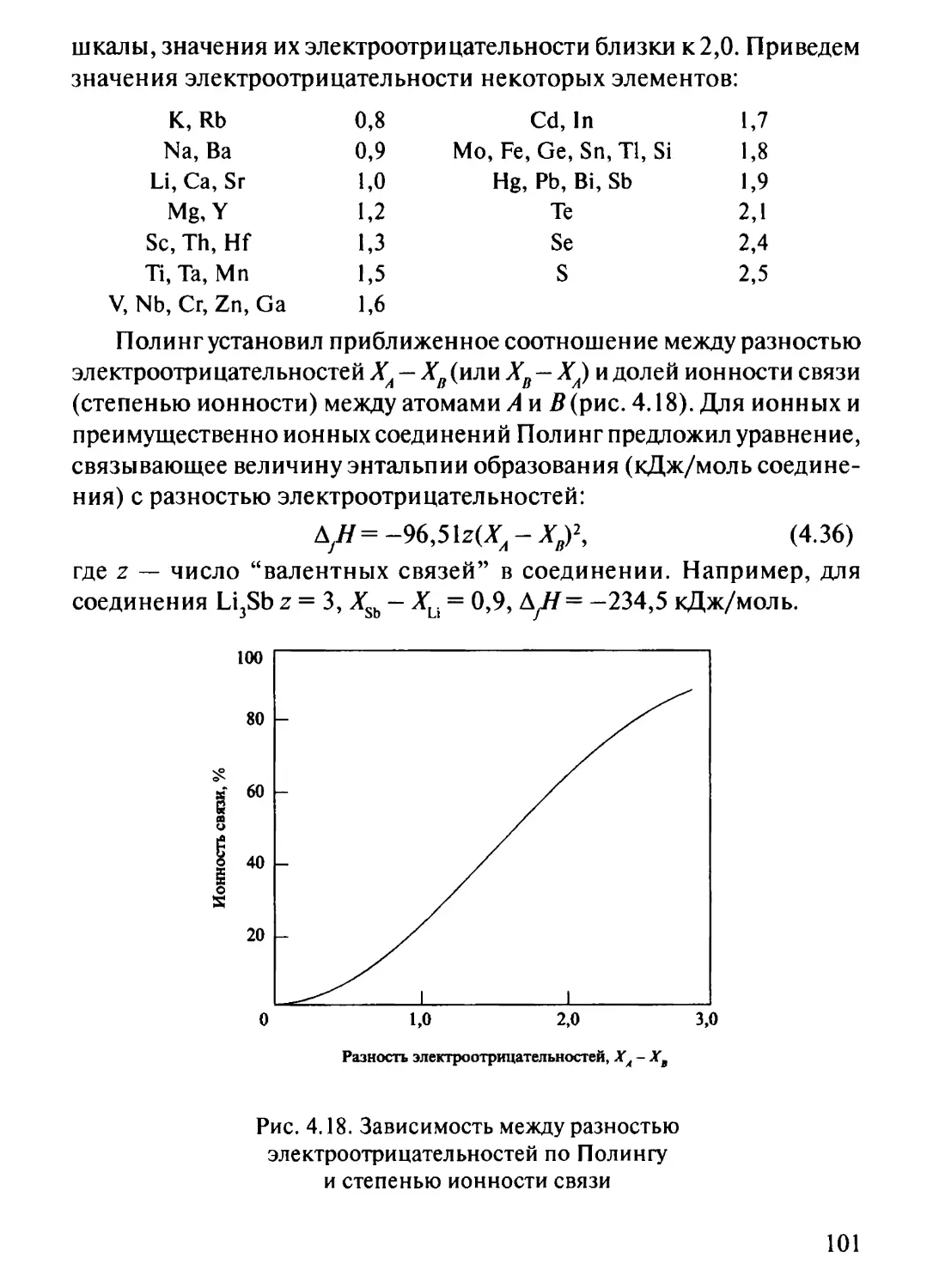
Английский химик У. Генри (1808 г.) при изучении
растворимости газов в жидкостях экспериментально установил, что при малых
содержаниях растворенного вещества его парциальное давление (р2)
пропорционально молярной доле (х2):
Это выражение широко известно под названием закона Генри и
величина Кг носит название константы Генри (постоянной Генри,
49
коэффициента Генри). Следует отметить, что вид уравнения (3.51)
практически не зависит от способа выражения состава, поскольку
при малых содержаниях растворенного вещества его концентрации
различного вида пропорциональны между собой с достаточной
степенью точности. Закон Генри относится к числу предельных, и его
правильнее записывать в форме
lim
Xj-yOl
(Hi
V*2
:*Г.
(3.52)
В уравнении (3.52) предусматривается, что паровая фаза
подчиняется законам идеальных газов, что практически всегда
выполняется. В противном случае давление следует заменить летучестью (f2)
и уравнение (3.52) принимает вид
lim
jc2->0
УХ2
Закон Генри в своей классической форме (3.52) выражает
зависимость давления пара растворенного вещества от состава раствора.
Аналогичная зависимость для растворителя описывается с помощью
закона Рауля. С точки зрения термодинамики различие между
растворителем и растворенным веществом чисто условно. Для идеальных
растворов законы Генри и Рауля тождественны и описываются одним
уравнением (3.26). Из сопоставления уравнений (3.26) и (3.51) видно,
что для идеального раствора константа Генри равна давлению пара
чистого растворенного компонента: Кг = р2°. Для реальных
разбавленных растворов законы Рауля и Генри различны.
Как отмечалось выше, а. = р./р°. Подставив это выражение в
уравнение (3.52), для компонента 2 получаем
а2 =
Гк^
Pi
(3.53)
Для реальных растворов при предельном разбавлении Кг = р2°у2™,
где у2°° — коэффициент активности компонента 2 при бесконечном
разбавлении. Хотя закон Генри и относится к числу предельных,
практически всегда можно выделить область составов, в которой
активность компонента линейно зависит от его молярной доли.
Таким образом, в области малых значений х2 коэффициент
активности у2 постоянен и равен в пределах определенной погрешности
50
предельному значению у2°° при бесконечном разбавлении (подобные
представления могут быть распространены и на системы с
несколькими растворенными компонентами).
Область выполнения закона Генри имеет различную
протяженность, которая, с одной стороны, определяется природой системы,
характером межчастичного взаимодействия, а с другой стороны,
зависит от точности эксперимента. В качестве количественного
критерия применимости закона Генри для жидких металлических
систем рекомендуется принимать в качестве допустимых отклонений
±0,05 логарифмических единиц коэффициента активности
растворенного компонента (±0,051gy.) по аналогии с требованиями,
установленными Международным союзом по чистой и прикладной химии
в качестве допустимого расхождения констант устойчивости
комплексов металлов, используемых для справочной литературы*. При таком
подходе для большого числа жидких двойных металлических систем
с различным характером взаимодействия между компонентами
граница применимости закона Генри лежит в интервале от 0,03 до 0,05
молярных долей растворенного компонента (3-5 мол. %).
Для описания свойств разбавленных растворов может быть
использована целая совокупность термодинамических
функций — предельные парциальные молярные термодинамические
функции смешения. Эти величины имеют четкий физический
смысл, могут быть определены из эксперимента с достаточно
высокой точностью. Особенно важны для технологической практики
и термодинамической оценки взаимодействия между компонентами
значения парциальной молярной избыточной энергии Гиббса
(избыточного химического потенциала) растворенного компонента
при бесконечном разбавлении (АС"36)00 или (Ац изб)ао и связанного с
этой величиной предельного коэффициента активности: (АСизб)°° =
= ЯЛпу.00. В свою очередь величина (АСизб)°° определяется из
выражения (АСизб)°° = А//.00 - ДА5изб)°°, где АН™ и (AS/136)00 представляют
собой также предельные парциальные величины.
* См.: Лебедев В.А. Избирательность жидкометаллических электродов
в расплавленных галогенидах. Челябинск: Металлургия. Челяб. отд-ние,
•993.232 с.
51
Если для двухкомпонентной системы известны значения
интегральной молярной избыточной энергии Гиббса при данной
температуре в широком интервале составов, то величины у,00 и у2°° могут быть
определены следующим образом. Из общего соотношения,
связывающего парциальные и интегральные величины (3.37), следует
AG»
AG"36 AG/136 AGf6
XX
1Л2
*1
Прих
О (л:, -> 1) AG2H36 -► (AG/36)00 и AG,1"6 -> 0. Отсюда
rAG™6] ,. AGf6 ii;_AG2M36
ХгХ>
1Л2
х2-»0
= lim
х7->0
-+lim-
х2 *2->о хх
В свою очередь это можно записать так:
AG
изб
XX
1Л2
-ИЗбЧ00 , ,;_ АС"3
= (AGfT+lim-
х2->0 Xj
Последний член в правой части уравнения представляет собой
неопределенность вида 0:0. По правилу Лопиталя:
hm—-
*2->0 X
изб
8AGI
изб Л
дхч
^=0
к2 у и*2
В соответствии с ранее изложенным (с. 38) уравнение вида (3.13)
для избыточной энергии Гиббса можно записать так:
(
SAG,
изб Л
дХ'у
+ х?
)
SAG
изб Л
дх>>
= 0.
(3.54)
)
Видно, что при х2 = 0, первый член уравнения (3.54) также равен
нулю. Следовательно,
гАстбЛ
v ХЛ j
= (AG2H36)°°=/{riny?.
(3.55)
х2=0
Аналогичным путем можно показать, что
( LQ**\
х\х2
= (AG?*T =RT Iny?
Таким образом, ординаты точек пересечения кривой
AG
изб
XX
(3.56)
- = /(*2>
1Л2
с границами концентрационной области (х, = 0, х1 = 1) дают
предельные значения парциальной молярной избыточной энергии Гиббса
компонентов, что позволяет рассчитать предельные значения ко-
52
эффициентов активности компонентов. Форма кривой зависит от
характера взаимодействия между компонентами (рис. 3.8). Следует
иметь в виду, что использование функции
AG
изб
JCiX
при подобного
1Л2
рода расчетах требует достаточно высокой точности определения
интегральной молярной избыточной энергии Гиббса, особенно при
малых концентрациях какого-либо из компонентов.
Аналогичного рода построения могут быть сделаны и для других
термодинамических функций, в частности для энтальпии смешения.
Построение зависимости = /(х2) позволяет оценить величины
X X
ДЯ,00 и Д#2°° (рис. 3.9).
Все три системы в жидком состоянии характеризуются
положительными значениями энтальпии смешения — очень небольшими в
системе Ni—Co и достаточно значительными в системе Си—Со.
АС?06
^Fe^Ni
-13,0
-11,0 \-
-9,0
-7,0
Fe 0,20 0,40 0,60 0,80 Ni
Рис. 3.8. Зависимость функции AG^/x^x^
от состава для системы Fe—Ni при температуре 1873 К;
ДСиз6 — в кДж • моль-1
53
а) АН
2,0 Ь
1,0
Ni-Co
Ni 0,20 0,40 0,60 0,80 Co
6) AH
16,0
15,0
14,0
*) AH
42,0 U
38,0
34,0
Cu
Ni-Cu
j_
_i_
0,20 0,40
0,60
0,80
Ni 0,20 0,40 0,60 0,80 Ql
Co
Рис. З.9. Зависимость функции A///jc,jc2 от состава
для жидких сплавов систем: Ni-Co (a), Ni-Cu (б), Си-Co (в);
АН — в кДж • моль-1; х2 — молярная доля компонента,
указанного вторым, температура 1873 К
54
Предельные значения энтальпии смешения (кДж • моль-1),
полученные с помощью таких графических построений:
система Ni-Co: A#Ni°° = 1,40, АЯСо°° = 1,95,
система Ni-Cu: АЯ^00 = 14,0, АЯС}° = 16,95,
система Си-Co: АЯСиж = 46,2, АЯСо°° = 40,7.
Предельные значения коэффициентов активности и других
термодинамических функций в двухкомпонентных системах могут
быть вычислены и без графических построений с помощью
полиномиальных зависимостей.
Редлих и Кистер (1946 г.) предложили следующую зависимость
избыточной интегральной молярной энергии Гиббса, точнее —
Q-функции, от состава для двойных систем:
д^»ИЗб
Q=JmRT=XlX2[b*c(<Xl ~Xl)+d(Xl ~*2>2 +"]
или эквивалентно этому
Q = xxx2[b + с(2х, - 1) + d(2xx - х2)2 + ...]. (3.57)
В свою очередь
При постоянных температуре и давлении в соответствии с
известными соотношениями между парциальными и интегральными
величинами в двойных системах получаем
fbQ\
igY,=0+0-*,)
ОХ] уг
1 JT,p
\gy2=Q-X\
dQ
При описании концентрационной зависимости (^-функции, как
правило, достаточно первых трех членов разложения, как это сделано
выше. В таком случае:
lgy, = (1 -х,)2[/> + c(4xt - 1) + d(\2x2- 8x, + 1)], (3.58)
lgy2 = x2[b + c(4xt - 3) + d(\2x2 - 16x, + 5)]. (3.59)
При xt -> 0 и x, -> 1 получаем
\gy™ = b-c + d и lgy2~ = b + с + d. (3.60)
Предельные значения коэффициента активности компонента
V и значения (AG.1136)00 = ДЛпу00 представляют большой интерес
55
для технологической практики, необходимы при решении задач
отделения примесей, разделения веществ, что будет рассмотрено в
соответствующих разделах. На основании данных о температурной
зависимости предельных значений коэффициентов активности
компонентов могут быть вычислены энтальпийные и энтропийные
характеристики разбавленного раствора.
3.7. Выбор стандартного состояния.
Переход от одного стандартного состояния к другому
Как уже отмечалось выше (с. 47), в термодинамике
металлургических систем, как и в термодинамике растворов неэлектролитов,
преимущественно за стандартное состояние принимаются чистые
компоненты в том агрегатном состоянии, которое устойчиво при
рассматриваемой температуре. Такой способ выбора стандартного
состояния обычно называется симметричным, в двухкомпонентной
системе оба компонента раствора равноправны, разделение
компонентов на растворитель и растворенное вещество условно.
Стандартные значения химического потенциала (ц,° и ц2°) равны молярным
энергиям Гиббса чистых компонентов (G° и (72°). Рассмотренное выше
описание свойств растворов с помощью полных и избыточных
функций смешения связано с симметричным способом нормировки.
В разбавленных растворах часто более целесообразно за
стандартное состояние принимать не чистые компоненты, а бесконечно
разбавленный раствор. Такой способ выбора стандартного состояния,
называемый несимметричным, предусматривает, что компоненты
раствора не являются равноправными.
Обозначим в случае несимметричного выбора стандартного
состояния для бинарного раствора 1-2 стандартные значения
химического потенциала ц,°' и ц20', активности компонентов д,' и а2\
коэффициенты активности у/ и у2'. Таким образом, при симметричном
выборе стандартного состояния (компоненты 1 и 2 равноправны):
ц,= ц,* + RT\x\av а{=уххх\ у,-> 1 при*,-» 1, (3.61)
ц2= ц2° + RT\r\av а2 = у2х2; у2-> 1 прих2-> 1. (3.62)
При несимметричном выборе стандартного состояния
(компонент 1 — растворитель; компонент 2 — растворенное вещество):
56
ц^ц.о' + ЛЛпл,', а; = ч;хх\ у^-Ипри^-М, (3.63)
ц2 = ц20' + RT\na2\ а2 = у2' х2\ у2' -> 1 при х2 -> 0. (3.64)
Величины химического потенциала компонентов (ц, и ц2)
определяются объективными факторами — природой раствора,
температурой, давлением — и не зависят от выбора стандартного состояния при
описании концентрационных зависимостей \х. =fix). Из уравнений
(3.62) и (3.64) следует:
ц2° + R Т\щ2 + RT\nx2 = ц20' + ДЛпу2' + ЯЛпх,
или
ц2° + Л Лпу2 = ц20' + ЛЛпу2\
При х2 -> 0 у2 -> у2°° и у2' -> 1. Тогда
ц20' = ц/ + ДЛпу2в. (3.65)
Следовательно:
Я71пу2 = Я71пу2' + Л71пу2°°,
^ = ТГ- (3.66)
Y2
Если при данной молярной доле х2 активность растворенного
компонента при симметричном выборе стандартного состояния (а2)
сравнить с активностью того же компонента при несимметричном
выборе (д2'), получим
% = ^ = Y?. (3.67)
Относительный химический потенциал компонента также
зависит от выбора стандартного состояния:
Ац2 = /Щ1пу2 + 1шс2),
Дц2' = /Щ1пу' + lru2),
A|LL2-A|X2 =Л7'1П
<v
= Л7Чпт£. (3.68)
Как отмечалось выше (с. 51), в соответствии с уравнением (3.52)
Для реальных растворов при предельном разбавлении постоянная
Генри Кг связана с величиной у2°° соотношением Кг = р2ч™. При
несимметричном выборе стандартного состояния у2°° -> 1 при х2^0и
соответственно Кг = р2. Можно записать:
57
<%=-£-, (3.69)
лг
где р2 — парциальное давление пара растворенного компонента.
В отдельных случаях за стандартное состояние выбирают раствор
той или иной концентрации с известной величиной давления пара
растворенного компонента, принимаемой за стандартное значение
(р2ст). В этом случае активность компонента 2 (а") рассчитывается
по выражению
<h =-^- (3.70)
Рг
Выражение для химического потенциала компонента примет
вид
ц2 = ц2°" + RT\nx2 + /mny2". (3.71)
Если компонентом 2 можно насытить раствор при заданной
температуре, то за стандартное состояние целесообразно принять
насыщенный раствор или, что то же самое, состояние чистого
твердого компонента при температуре раствора: рст = рнас = р20ТН. В этом
случае:
<£=-£-. (3.72)
Рг
От выбора в качестве стандартного состояния "твердый металл"
легко перейти к стандартному состоянию "жидкий переохлажденный
металл". Рассмотрим конкретный пример с системой (Ni—Си)ж при
температуре Т(Т< Тк™). Переход от одного стандартного состояния
к другому иллюстрирует схема
ЛГ1п<
Ni (тв) ► (Ni-Cu) (ж)
№(ж)
Здесь RT 1па™{ — относительный химический потенциал никеля
в жидком сплаве Ni-Cu при стандартном состоянии "никель
твердый"; RTlna^i — относительный химический потенциал никеля в
жидком сплаве Ni-Cu при стандартном состоянии "никель жидкий,
переохлажденный". Величина Ац^1 соответствует изменению
химического потенциала никеля при переходе из твердого состояния в
58
жидкое при Т < Т^. Величина Ац^ может быть подсчитана
следующим образом:
АН™1
ДНИ = Д*5? -TAS™ = Atf£? -T—£L.
Из приведенного выше цикла следует:
RT\n <i = Ацй + ДПп а&.
В металлургической практике в отдельных случаях в качестве
стандартного состояния выбирают предельно разбавленный раствор,
причем состав раствора выражается не в молярных долях, а в массовых
процентах (^.). Тогда для растворенного компонента
m
-£- -> 1 при #2 -> 0.
и
Соответственно а2"' = у2"'<?2 для растворов, в которыхр2 имеет
конечное значение. Сравнивая активность компонента 2, отнесенную к
чистому веществу как к стандартному состоянию (д2), с активностью,
отнесенной к бесконечно разбавленному раствору, состав которого
выражен в процентах по массе (а2'"), при одном и том же фактическом
содержании компонента 2 в смеси получаем
££=Т№=7Р.*2.# (3J3)
а2 iiQi Яг
При очень малых содержаниях компонента 2 в растворителе
соотношение между его молярной долей и процентом по массе имеет
вид
fl = _^l_, (3.74)
q2 100 М2
где Л/, и Л/2 — молярные массы (массы одного моля) растворителя
и растворенного вещества. Уравнение (3.73) тогда можно записать
в виде
a = Y~J^l_. (3.75)
я" '2 100 Л/2
В некоторых случаях в системах металлургического характера
за стандартное состояние принимается раствор при концентрации
8 нем растворенного компонента, равной 1 мае. %. В сульфидных
59
расплавах, например в системе Fe—S, за стандартное состояние
принимают состояние серы в моносульфиде железа FeS в жидком
состоянии.
Выбор стандартного состояния особенно при исследованиях
сложных высокотемпературных металлургических систем в
достаточной степени произволен и принципиально ничем не ограничен.
Однако, принимая во внимание, что в общем случае активность
компонента равна отношению его парциального давления над раствором
(/?.) к парциальному давлению в стандартном состоянии (/>.станд), т. е.
а. = р / рстанА для расчета активности из экспериментальных данных
необходимо выбирать такое стандартное состояние, реальное или
гипотетическое, при котором известны давление насыщенного пара
или другая термодинамическая величина, характеризующие
выбранное стандартное состояние.
При переходе от одного стандартного состояния к другому
необходимо всегда иметь в виду, что в уравнениях
ц.= ц/+ЛЛпя, (3.76)
ц.= ц/>' + ЛЛпа/, (3.77)
ц. = ц.°" + RTlna!' и т. д. (3.78)
левая часть при сохранении постоянными р, Т, х. от выбора
стандартного состояния не зависит, как не зависит и от принятой шкалы
выражения состава раствора. Это и служит основой при всех
пересчетах.
Для многокомпонентных систем нежелательно иметь разные
способы выбора стандартного состояния растворенных веществ
(например, для /-го и А>го компонентов). Разные способы выбора
стандартного состояния скажутся на величинах активности
компонентов, затруднят расчет термодинамических характеристик,
константы равновесия и др.
Для многофазных систем допускается выбор для одного и того
же /-го компонента различного стандартного состояния в различных
фазах.
Еще раз подчеркнем, что интегральные функции смешения,
характеризующие систему в целом, имеют четкий физический смысл
только при симметричном выборе стандартного состояния.
60
3.8. Приближенные модели растворов
3.8.1. Регулярные растворы
Термин "регулярные растворы" был предложен американским
ученым Гильдебрандом в 1929 г. в процессе исследований
растворимости йода, серы, газообразных веществ преимущественно в
органических растворителях. Была выявлена группа растворов, в которых, по
мнению исследователей, наблюдалась чисто "физическая" картина
растворения в отличие от "химической", где важную роль играли
процессы сольватации. В дальнейшем группа "правильных" растворов с
отсутствием специфического взаимодействия между компонентами
получила название "регулярные растворы". Считая, что в
регулярном растворе между компонентами отсутствуют ориентационное и
химическое взаимодействия и что расположение частиц в нем столь
же беспорядочно, как в идеальном растворе, Гильдебранд приходит
к заключению, что "...энтропия системы не изменяется при
переносе небольшого количества одного из компонентов из идеального
раствора в регулярный раствор того же состава...". Из этой
формулировки следует наиболее распространенное определение понятия
"регулярный раствор": регулярными растворами называют растворы,
парциальные молярные энтропии компонентов которых равны
парциальным молярным энтропиям компонентов идеальных растворов:
ASf*T = AS™ = -RTlnx., AS**r = А5ИД, (ASm6)^r = 0, (3.79)
а энтальпия смешения при этом не равна нулю:
А#рег ф О, А#Рег ф 0.
Таким образом, получаем, что для регулярного раствора
(дв-ид*- а(д^Г1=адя!1=0
v ' дт ет
Указанные равенства означают, что отклонения свойств
регулярных растворов от свойств идеальных растворов обусловлены
исключительно отличием от нуля энтальпии смешения регулярного
раствора.
Легко показать, что логарифмы коэффициентов активности
компонентов регулярных растворов обратно пропорциональны
абсолютной температуре.
61
Поскольку
(д5изб)рег=_^(АСГб)РеГ=0
1 дТ
а (ДСизб)рег = Л71пу.рег, получаем
5(ДГ1пурег)_0
аг
Самый общий анализ выражений для термодинамических свойств
регулярных растворов показывает внутренние противоречия этой
концепции. Изменение энергии взаимодействия частиц при
образовании раствора, внешним проявлением которого является энтальпия
смешения, взаимосвязано со способом распределения этих частиц в
пространстве, что приводит в свою очередь к отклонению энтропии
смешения реального раствора от величин, характерных для
идеального раствора. Модель регулярного раствора лишь в первом
приближении может быть использована для описания свойств реальных систем
с весьма умеренными отклонениями от идеального поведения.
Другой, более эмпирический подход заключается в том, что
регулярными растворами называют такие растворы, у которых логарифмы
коэффициентов активности пропорциональны квадратам
концентрации. Так, например, для двойного регулярного раствора
Iny^mx*, \ny2 = mxl2, (3.81)
если в качестве стандартного состояния выбраны чистые
компоненты. Равенство коэффициентов пропорциональности (ml = m2 = пг)ъ
указанных выше соотношениях легко выводится с помощью
уравнения Гиббса-Дюгема. Величина/и, входящая в обсуждаемые
уравнения, зависит от температуры и давления. В рассматриваемых нами
конденсированных системах влияние давления можно не принимать
во внимание. Следует отметить, что один и тот же вид зависимости
логарифма коэффициента активности от концентрации может быть
связан с различными значениями энтропии и энтальпии смешения.
Уравнения с коэффициентом m правильнее рассматривать как
эмпирические, хотя они и могут быть обоснованы с помощью некоторых
вариантов "решеточных" моделей растворов. В рамках сделанных
предпосылок можно получить следующие зависимости:
(д G;*)per = RTm( щ^ = к( T)xxxv (3.82)
62
(А5И*Г = -6(AG™T )РеГ=-Ц^2 = а(Т)х,х2, (3.83)
ДЯРег=_T2mG«*/T)=RT2bnXiX2 =Ь{т)хл (3 84)
Как видно из этих уравнений, зависимости интегральных
термодинамических функций регулярного раствора от состава двойной
системы описываются уравнениями парабол, вершины которых
соответствуют концентрации х, = х2 = 0,50. Из уравнений также
следует, что величины (А(?"б)рег, (А*Упб)рег и А№ег пропорциональны друг
другу. В то же время в соответствии с ранее изложенным
термодинамическим подходом к пониманию регулярного раствора величина
(АС*136)^ должна быть равна А№ег, а величина (А^зб)рег должна быть
равна нулю. При этом коэффициенты к (Т) и Ъ (Т) должны быть
равны между собой, а коэффициент а{Т) — равен нулю.
Наряду с понятием "регулярный раствор" в литературе можно
встретить термины "строгорегулярный раствор" и "субрегулярный
раствор". Основные положения модели строгорегулярного раствора
сводятся к следующему:
принимается квазикристаллическая структура чистых жидких
компонентов и раствора (ближний порядок) с одним и тем же
координационным числом z\
взаимодействие учитывается только между соседними
молекулами (атомами), т. е. в основе рассмотрения лежат парные
взаимодействия;
потенциальная энергия разбивается на два слагаемых:
конфигурационную, зависящую от расположения молекул в квазирешетке, и
колебательную, т. е. энергию колебания молекул около центра
равновесия. Принимается, что колебательная составляющая не зависит от
ближайших соседей, молекулы имеют одну и ту же колебательную
энергию в чистой жидкости и растворе;
объемы молекул компонентов близки.
Математическую сторону модели в рамках данного учебного
пособия не будем рассматривать. Другой попыткой расширить области
применения модели регулярного раствора является введение понятия
о субрегулярном растворе (1953 г.). Авторами модели допускается
зависимость энергии взаимодействия компонентов от состава раствора.
Формально в рамках субрегулярной модели зависимость энтальпии
63
смешения от состава описывается с помощью двухпараметрического
уравнения:
AH=Al2xlx22 + A1{x?xv ЛпфЛ2У (3.85)
С помощью такого уравнения можно описать
концентрационную зависимость энтальпии смешения для более широкой группы
систем, чем с применением модели регулярного раствора. Следует
однако иметь в виду, что автор концепции регулярного раствора в
монографии "Regularand Related Solutions" (1970 г.) достаточно резко
возражал против всякого рода "усовершенствования" своей модели,
против попыток заменить простую и ясную в термодинамическом
смысле модель какими-либо другими более сложными моделями.
По мнению Гильдебранда, строгорегулярная модель не может
иметь практического значения, так как очень мало смесей,
построенных из веществ, объемы молекул (атомов) которых столь близки
друг к другу, что позволяют строить квазирешетку.
3.8.2. Атермические растворы
Рассмотрим группу растворов, для которых энтальпия смешения
равна нулю, изменение объема имеет конечное значение. Отсутствие
теплового эффекта при смешении компонентов характерно для
идеальных растворов, но и некоторые реальные системы, не обладая
всем комплексом свойств идеальных растворов, могут иметь очень
малые теплоты смешения. Такие растворы называют атермическими
(атермальными):
АН = 0, Д<Уз6* 0, А№36 = -7Д51*36, (3.86)
т. е. отклонения от идеальности в атермических растворах
обусловлены энтропийными эффектами. Из уравнения (3.86) следует:
ДС-изб
ЯЛпу, = - 7А5.из6, 1пу, = !—. (3.87)
/v
Строго говоря, атермических растворов в действительности не
существует. Модель атермического раствора может быть использована
в качестве первого приближения при анализе термодинамических
свойств таких неидеальных растворов, у которых
|ДЯ| « 7JA5|
и зависимость величин Д<?"6, Д#, А*?136 от температуры и давления
невелика.
64
3.9. Особенности концентрационной зависимости
термодинамических функций.
Функции стабильности и избыточной стабильности
В двухкомпонентных системах при симметричном выборе
стандартного состояния для каждого из компонентов можно выделить
область приближенного выполнения закона Генри. Протяженность
этой области, как это уже обсуждалось (с. 51), зависит от природы
системы и точности эксперимента. С повышением точности
экспериментального метода определения коэффициента активности
область выполнения закона Генри смещается в направлении более
разбавленных растворов.
Если в двухкомпонентной системе для одного из компонентов
справедлив закон Генри, то в этой же области составов для другого
компонента выполняется закон Рауля. Это непосредственно вытекает
из соотношения Гиббса-Дюгема. Таким образом, для каждого из
компонентов раствора можно выделить область составов,
прилегающую к чистому компоненту, где выполняется закон Рауля, и область
предельно разбавленных растворов, где выполняется с той или иной
степенью приближения закон Генри.
Даркен (1967 г.) обратил внимание, что во многих системах
наблюдаемое изменение термодинамического свойства по
существу локализовано в
ограниченной области составов.
Это хорошо видно на примере
системы Mg-Bi (рис. 3.10). В
системе наблюдаются два
линейных участка: в интервалах
от *м =0 до х. = 0,3 и от jcm =
Mg ^ Mg * Mg
- 0,7 до xMg = 1,0. В переходной
области (0,3 < ;cMg < 0,7)
происходит резкое возрастание ум и
отчетливый перегиб кривой lgyMg =
^ЛХщ). Для количественной
характеристики такой зависимости
igy,
0,2
0,3
0,4 0,5 0,6 0,7 1,0
Mg
-2 h
1 1 1 1 1 I i*«\
-8—« o-
J I I L
1,0
Bi
0,8
0,6
0,4
0,2
(1
-V2
Mg
Рис. 3.10. Зависимость логарифма
коэффициента активности магния в
Даркен предложил использовать жидких сплавах системы Mg-Bi от со-
вторую производную энергии става при 973 К (пунктир — в области
Гиббса по составу. В соответствии гетерогенности)
65
с общими условиями равновесия вдоль кривой G = f(x2) устойчивы
растворы с положительной величиной (d2G/dx22)r . Растворы, для
которых (d2G/dx2)Tp < О, неустойчивы (лабильные состояния).
При выполнении конкретных термодинамических расчетов в
отличие от теоретического анализа следует пользоваться
относительными термодинамическими величинами. Функцию (d2G/dx2)T
Даркен предложил назвать функцией стабильности (стабильностью).
В равной степени дифференцирование можно выполнять и по
молярной доле первого компонента. Вторые производные от энергии
Гиббса будут равны между собой. Функция стабильности
характеризует поведение двойной системы в целом. Для идеального раствора
при постоянных температуре и давлении
дх\
\ 1 )
Т,р
Гд2АСГ)
дх}
\ l j
Т,р
RT
xtx2
(3.88)
Соответственно для двойных систем избыточная стабильность
оказывается равной
}2А/зизб^
d'AG*
Va^
/
Т,р
У Ъ*\
ут.р
RT
ххх2
(3.89)
Расчетные выражения для вычисления стабильности и
избыточной стабильности могут быть получены следующим образом. Ранее
(с. 38) было выведено уравнение
AG2=AG + {\-x2) \^-\ .
{ ЭХ2 Ур
Дифференцирование этого уравнения по х2 позволяет
получить:
d2AG
дх2
1
^ эдс2 ^
/г,р
1-х-,
Эх,
= -2
/г,р
ЭД(?л
д(1-х2)2
т,р
Соответственно избыточная стабильность
ч
дх\
= -2!
/
Т,р
dAG?6
5(1-*2Г
(3.90)
(3.91)
AT.p
Функции стабильности и избыточной стабильности могут быть
выражены через активность и коэффициент активности соответству-
66
ющего компонента. Принимая во внимание, что Д(72 = 2,303/?T\gav
а ДС2из6 = 2,303/?rigy2, получаем при постоянных температуре и
давлении
^ = _4,606ЛГ^^2 (3.92)
^^ = -4,606^^1^. (3.93)
Следовательно, избыточную стабильность можно найти,
умножив тангенс угла наклона кривой lgy2 = Д1 - х2)2 на 4,606/?Г
(см. рис. 3.10).
Глава 4
ТЕРМОДИНАМИКА ФАЗОВЫХ РАВНОВЕСИЙ
4.1. Условия равновесия компонентов в сосуществующих фазах.
Правило фаз Гиббса
Рассмотрим соотношение между химическими потенциалами
компонента, входящего в состав нескольких фаз гетерогенной
системы. Переход массы dn. компонента из одной фазы (I) в другую (II)
при постоянных температуре и давлении ведет к изменению энергии
Гиббса d<7системы в целом, которое складывается из изменений d(?
и dOx обеих фаз:
dG=dQ + dGl = jx/dn/ + ц/Ул/1. (4.1)
Так как dn}1 = —dn} в соответствии с постановкой задачи и при
условии сохранения в системе равновесия (dG = 0), получаем
|i,"<//!» + м/Л|/ = \iildn}1 - u/di," = (й/1 - \if)dn}1 = 0
или
И/1 - й/ = 0, ,1" = ,!;. (4.2)
Рассмотрим частный случай, когда система состоит из двух фаз и
двух компонентов. Для перехода массы dnx первого компонента из фазы
I в фазу II на основании уравнений (4.1) и (4.2) можно записать:
rfC=|ilIIrf/iIll-n1irf«il = 0,
Mi" = й,1-
Аналогичным образом можно рассмотреть переход компонента 2
из одной фазы в другую. Общий вывод из изложенного: химические
68
потенциалы данного компонента во всех фазах системы, находящейся
в равновесии, равны между собой.
При отсутствии равновесия в соответствии с уравнением (2.28)
dG=()x}l-\i})dn}l<Q. (4.3)
Отсюда следует, что если \\}1 - ц.1 > 0 (ц.п > |i.'), то dn}1 < О, т. е.
рассматриваемый компонент переходит из фазы II в фазу I. Если
р.11 < ц.!, то компонент будет переходить из фазы I в фазу II.
Таким образом: компонент самопроизвольно переходит из фазы,
в которой его химический потенциал больше, в фазу, в которой его
химический потенциал меньше. Переход будет продолжаться до тех
пор, пока химические потенциалы компонента в обеих фазах не
сравняются.
Соотношения вида (4.1) могут быть распространены на системы
с любым числом фаз и компонентов. Общие закономерности,
которым подчиняются равновесные гетерогенные системы, состоящие
из любого числа фаз и любого числа веществ, устанавливаются
правилом фаз, которое было выведено Гиббсом (1876 г.): число степеней
свободы равновесной термодинамической системы, на которую
влияют только температура и давление, равно числу компонентов
системы минус число фаз плюс два.
Уточним некоторые понятия.
Число степеней свободы характеризует число независимых
переменных (температура, давление, концентрации компонентов),
которые можно изменить в некоторых пределах так, чтобы число и
природа фаз оставались прежними.
Вещество, которое будучи выделено из системы может
существовать самостоятельно, называется составляющим веществом
системы. Составляющие вещества, концентрации которых определяют
состав фаз дан ной равновесной системы, называются независимыми
составляющими веществами, или компонентами системы*. Свойства
системы зависят не оттого, какие составляющие вещества выбраны в
качестве компонентов, а от числа компонентов. Число компонентов
равняется числу составляющих веществ за вычетом числа уравнений,
* Иногда составляющие вещества называют компонентами, а
независимые составляющие вещества — независимыми компонентами. Эта
особенность терминологии должна быть отражена в формулировке правила фаз.
69
связывающих концентрации этих веществ в равновесной системе.
По существу эта же мысль может быть сформулирована иначе: число
компонентов есть наименьшее число составляющих веществ,
достаточное для определения (реализации) состава любой фазы.
Рассмотрим систему, состоящую из/фаз и к компонентов,
которые присутствуют во всех фазах одинаково. Состояние каждой
фазы определяется температурой, давлением, концентрациями всех
компонентов. Для определения состава любой фазы, включающей
к компонентов, достаточно указать содержание (к - 1) компонента.
При записи выражения для химического потенциала нижний индекс,
как обычно, указывает на принадлежность тому или иному
компоненту, верхний индекс соответствует фазе.
Для рассматриваемой системы (к компонентов,/фаз) можно
записать следующую совокупность уравнений:
й/ = И,", И," = йД> М/ = мД ...т. е. {f- 1) уравнение,
ц2' = цД |д2" = цД, й2! = цД»т- е- (/"- О уравнение,
\хк1 = цД \хки = дД, [хк[ = цД..т. е. (/"- 1) уравнение.
Очевидно, что число уравнений в каждой из горизонтальных
строк будет {f- 1), а число этих строк будет соответствовать числу
компонентов к. Ч исло степеней свободы (s) будет равно общему числу
концентрационных переменных f(k — 1), к которым добавляются
температура и давление, за вычетом числа уравнений, связывающих
эти переменные к (f— 1).
5=Л*-1) + 2-*(Л-1),
s = k-f+2. (4.4)
Из уравнения (4.4) следует, что число степеней свободы возрастает
с увеличением числа компонентов системы и уменьшается с ростом
числа фаз. При s = 0 в равновесии находится наибольшее число фаз
для данной системы.
Принято классифицировать системы по числу фаз на однофазные,
двухфазные и т. д., по числу компонентов — на однокомпонентные,
двухкомпонентные (двойные), трехкомпонентные (тройные) и т. д.,
по числу степеней свободы — на нонвариантные (s = 0),
моновариантные (5= 1), бивариантные (s = 2) и т. д.
70
Если на равновесие в системе наряду с температурой и давлением
оказывают влияние другие факторы (магнитное или электрическое
поле, поле тяготения), то уравнение (4.4) может принять вид
s = k-f+n, (4.5)
где п — суммарное число внешних факторов, воздействующих на
систему. В то же время при равновесии конденсированных систем при
обычных условиях часто давление заметно не влияет на равновесие.
В этом случае уравнению (4.4) можно придать вид
*=*-/+!. (4.6)
Следует отметить, что правило фаз Гиббса, выражаемое с
помощью уравнений (4.4)—(4.6), предусматривает беспрепятственный
переход каждого из составляющих веществ из одной фазы в другую.
Оно неприменимо к системам, в которых фазы разделяются какими-
либо полупроницаемыми перегородками.
4.2. Равновесие в однокомпонентных системах.
Уравнение Клапейрона—Клаузиуса
В однокомпонентных системах отдельные фазы представляют
одно и то же вещество в различных агрегатных состояниях или
кристаллических модификациях. Каждую из модификаций следует
рассматривать как самостоятельную фазу.
Для однокомпонентнои системы энергия Гиббса есть функция
только температуры и давления: G = Д71, р). Для равновесия,
например, твердой и жидкой фаз G™ = Сж. Величины энергии Гиббса
относятся к единице массы рассматриваемого вещества. В
координатах G— Т— руравнения G™=f{Typ) или <7ж=/(7",/>) представляют
собой поверхности. Линия пересечения этих поверхностей дает
кривую фазового равновесия, в данном случае — кривую плавления.
Проекции кривых фазового равновесия на плоскость Т — р дают
диаграмму состояния однокомпонентнои системы в координатах
температура-давление (рис. 4.1).
Основные элементы фазовой диаграммы:
моновариантные линии двухфазных равновесий АО, ОВ, ОК.
Составы, лежащие на этих линиях, обладают одной степенью
свободы, 5= 1;
71
а)
Рис. 4.1. Основные типы фазовой диаграммы однокомпонентной системы:
а - dT/dp >0;б- dT/dp < О
нонвариантная точка пересечения линий двухфазных равновесий
О, s = 0.
критическая нонвариантная точка К;
поля существования твердой (тв), жидкой (ж) и парообразной
(п)фаз, к= 1,/= 1,5 = 2.
В соответствии с условиями равновесия сосуществующих фаз
G = G\ dG = dG\ Принимая во внимание уравнение (2.34), можно
записать:
или
(Кп - Vx)dp = (Sn-Sx)dT
dp _ А#ф п
(4.7)
(4.8)
dT TAV У
поскольку изменение энтропии при фазовом переходе в соответствии
с уравнениями (2.23) и (2.24) равно
sn_s\=^[^L (4.9)
Уравнение (4.8) носит название уравнения Клапейрона-Клау-
зиуса.
В уравнениях (4.7)-(4.9) Vx\ V\ Sx\ Sx — молярные объемы и
энтропии равновесных фаз, Д#фп — теплота (энтальпия) фазового
перехода, отнесенная к одному молю вещества.
72
Наклон линий А О, ОВи ОК (см. рис. 4.1), т. е. знак производной
-^-, определяется соотношением молярных объемов равновесных
фаз, так как теплоты фазовых переходов (тв -> п, ж -> п)
положительны.
Линия сублимации (возгонки) АО начинается в точке, где Т= О и
р = О, имеет положительный наклон, — > 0, так как молярный объем
dT
пара значительно превышает молярный объем твердого вещества, A У>
> 0, и заканчивается в точке О.
Линия испарения ОК начинается в нонвариантной точке О и
заканчивается в критической точке К, где парообразная и жидкая
фазы становятся идентичными. Критическая точка соответствует
такой температуре, при которой молярные объемы жидкости и пара
становятся одинаковыми. Критические температура 7*", давление/?^,
объем ^р, отвечающие точке К, являются физическими константами
вещества. При температуре выше критической, Т> Рр, вещество
не может быть сконденсировано ни при каких давлениях. Наклон
линии ОК положителен, AV> О, ЛЯИСП > 0. Молярную теплоту
сублимации можно представить как сумму молярных теплот плавления
и испарения. Изменения объема, А К, при сублимации и испарении
практически одинаковы. Следовательно,
dp]
>
УТусубл
dT
При сопоставимых температурах кривые сублимации идут более
круто, чем кривые испарения. Угловой коэффициент линии
сублимации больше, чем угловой коэффициент линии испарения.
Линия плавления ОД описывает зависимость температуры
плавления от давления. Перевод вещества из твердого состояния в жидкое
требует затраты теплоты, А#пл > 0. Характер зависимости
температуры плавления от давления определяется соотношением молярных
объемов АК= Уж — Vth. Для подавляющего большинства веществ
переход из твердого состояния в жидкое сопровождается увеличением
объема, АК> 0, (dp/dT)nji > 0. С увеличением давления температура
плавления повышается. Для металлов это повышение, как правило,
невелико. Для олова, например, для изменения температуры на
1 градус необходимо изменить давление на 3,04 • 107 Па.
73
В отдельных случаях, как это видно на рис. 4.1, б, молярный объем
жидкой фазы меньше, чем твердой, AV< О, температура плавления
понижается сростом давления. Кчислу таких веществ относятся вода,
Ge, Si, Ga, Bi, ряд полупроводниковых соединений типаЛ,п/?у.
Моновариантные линии двухфазных равновесий АО, ОВ, ОА'пере-
секаются в нонвариантной (тройной) точке, где в равновесии
существуют три фазы. Продолжение моновариантных линий за тройную точку
соответствует метастабильным равновесиям (см. рис. 4.1). Энергия
Гиббса таких состояний превышает равновесные значения, что
приводит к самопроизвольному исчезновению метастабильной фазы.
4.3. Давление насыщенного пара
Для процессов испарения и сублимации уравнение (4.8) удобно
представить в несколько ином виде. Если рассматривать пар при
давлении и температуре, достаточно удаленных от критической точки,
то можно пренебречь молярным объемом конденсированной фазы
по сравнению с молярным объемом пара:
AV= Vn- Кж* V\ (4.10)
Кроме того, можно принять, что пар подчиняется идеальным
законам. Для одного моля
Vn=—. (4.11)
Р
Эти допущения позволяют придать уравнению Клапейрона-
Клаузиуса следующий вид:
d In/? = АЯИСП
dT RT2 '
Поскольку величины в правой части уравнения (4.12) всегда
положительны, то производная d\np/dT также всегда положительна,
т. е. с ростом температуры давление насыщенного пара в равновесии
с конденсированной фазой всегда увеличивается.
В относительно небольшом интервале температур можно
принять, что величина Д//исп не зависит от температуры, сохраняет
постоянное значение. Интефируя уравнение (4.12) в пределах от Г, до
Tv получаем:
]nPL=^ucuT2-Tl
Рх R ТхТг
74
В общем виде при всех сделанных выше допущениях зависимость
p-f(T) можно представить так:
1п^=_Мисп.+Сили lg^=- АЯисп + -^L = - + JB. (4.14)
RT *У 2,303ЛГ 2,303 Т
Таким образом, зависимость давления насыщенного пара от
температуры может быть выражена прямой линией в координатах In p .
В этом случае тангенс наклона прямой равен—-р2- (рис. 4.2).
R
1пр
ЦТ
Рис. 4.2. Зависимость давления
насыщенного пара от температуры
Уравнения (4.13) и (4.14) не охватывают зависимости давления
насыщенного пара от температуры в широком интервале
температур. Одна из основных причин — зависимость А#исп от температуры.
В современной справочной литературе зависимость \%р =Д 7)
представляется в виде полиномов
\gp = AT~l + Яё7+ С7+ D. (4.15)
Для многих веществ коэффициент Сне принимается во внимание.
4.4. Зависимость энергии Гиббса и энергии Гельмгольца
от давления и температуры в однокомпонентных системах
Рассмотрим зависимость энергии Гиббса фаз однокомпонентной
системы от давления и температуры. Как известно,
Ьр
/г
дТ
/р
75
или
отсюда
dG= Vdp-SdT,
dp
= V;
/г
^ | =-S.
ът
Поскольку объем имеет всегда положительное значение, энергия
Гиббса при постоянной температуре должна увеличиваться с ростом
давления.В свою очередь
ЪР2
/г
Ьр
Объем всегда уменьшается с ростом давления, и, следовательно,
(d2G/dp2)r< 0. Таким образом, кривая, выражающая зависимость
G=J{p), обращена вогнутостью к оси абсцисс (рис. 4.3,а).
Энтропия (но не ее изменение) всегда положительна. Поэтому с
возрастанием температуры при постоянном давлении энергия Гиббса
будет уменьшаться. В свою очередь
дТ
При нагревании энтропия системы всегда возрастает.
Следовательно,
дТ2
jp
дТ
<о,
Jp
дТ2
<0.
Jp
а)
G а
6)
Т- const
р - const
Рис. 4.3. Зависимость энергии Гиббса
от характеристических параметров состояния (р и 7)
76
Это говорит о том, что кривая температурной зависимости
энергии Гиббса обращена вогнутостью к оси абсцисс (рис. 4.3,6).
Аналогичным образом, используя выражения
ЭК
дТ
dT
JY
И
dF=-SdT-pdV,
можно построить зависимости F=f{ V) при постоянной температуре
и F=J{T) при постоянном объеме (рис. 4.4, а,6).
а)
б) F
Г-const
К-const
Рис. 4.4. Зависимость энергии Гельмгольца
от характеристических параметров состояния (Ги V)
Для однокомпонентной
системы анализ зависимостей
энергии Гиббса
сосуществующих фаз от температуры при
постоянном давлении дает
отчетливое представление о
температуре фазового перехода и
иллюстрирует равенство
молярных энергий Гиббса этих
фаз при равновесии. На рис. 4.5
представлены зависимости G* =
= Л7) и Ств = Л 7) при
постоянном давлении. В точке
пересечения кривых, соответствующей
G
Рис. 4.5. Относительное
расположение кривых (7= Л 7) твердой и жидкой
фаз для однокомпонентной системы
при постоянном давлении
77
температуре плавления, (7Ж = G™. Ниже Т™ устойчива твердая фаза
(Стн < Сж), выше Т™ устойчива жидкая фаза (<7Ж < GTB).
4.5. Диаграммы энергия Гиббса —
состав для двухкомпонентных систем
В общем случае для двухкомпонентной системы энергия Гиббса
выражается функцией G=/[ Ту />, х), где х — состав в молярных долях.
Когда речь идет о равновесиях в конденсированных системах, т. е.
системах, содержащих только твердые и жидкие фазы, давление
относительно мало сказывается на температуре фазовых переходов. Поэтому
преимущественно двухкомпонентные конденсированные системы
рассматривают при определенном давлении (р= 1 атм = 1,01 • 105 Па).
Проанализируем зависимость энергии Гиббса от состава в
двухкомпонентных конденсированных системах. При общем
теоретическом анализе фазовых диаграмм обычно пользуются функцией G,
а при конкретных термодинамических расчетах — значениями AG.
Если мы имеем двухкомпонентную систему, то всякая однофазная
область будет представлять собой раствор — жидкий или твердый,
всякая двухфазная область — механическую смесь либо чистых
компонентов, л ибо других фаз. Можно легко показать, что энергия Гиббса
механической смеси является аддитивной функцией. Представим
себе, что мы имеем фазу I, характеризующуюся значениями G\ U\
S\ V\ и фазу II с величинами Gn, Un, Sny Vй. Известно, и это легко
представить, что функции аддитивны и для системы в целом:
U=Ul+ U";
S=Sl + Sn;
у= у\ + км.
Поскольку С1 = Ul + pVl- TS\ Gn = Uu +pVu- TSn, получаем
Gx + <7n = Ux + U" + p(Vl + Vй) - T(S{ + S")
или
& + Gu= U + pV- TS=G (4.16)
В соответствии с изложенным для механической смеси чистых
компонентов зависимость энергии Гиббса от состава смеси
выражается прямой линией, описываемой уравнением
G = xlGl° + x2G2° = G; + x2(G2° - G;).
78
Здесь G,° и G2 — энергии Гиббса чистых компонентов, отнесенные
к одному молю вещества. Если же компоненты во всем интервале
составов образуют раствор, твердый или жидкий, то кривая G=f[x2)
должна располагаться ниже аддитивной прямой (рис. 4.6).
Касательная к кривой G=/(x2) будет отсекать на осях ординат отрезки,
соответствующие парциальным молярным энергиям Гиббса (химическим
потенциалам) компонентов при составе хт
И2«ч)
^2
Рис. 4.6. Зависимость энергии Гиббса от состава
в двухкомпонентной системе при постоянных
давлении и температуре
Рассмотрим предельные значения химических потенциалов \хх и
|Д2 при хх = О (х2 = 1) и х2 = 0 (х, = 1). Примем симметричный выбор
стандартного состояния для активности компонентов:
прих, = 1 а, = 1, а2 = О,
при х2 = 1 а{ = 0, а2 = 1.
Тогда
при Xj = 1 |ij = Hj0 + R Tlnal = jij°,
|i2 = |i2° + RT\na2 = -oo;
при x2 = 1 ц, = -oo, ц2 = ц2°.
79
1 JT,p
Таким образом, при х{ = 1 (х2 = 0)
х =0(^=1)
dG_
Эх>
Как известно,
<7=(1 -х2)11{ + х\хг
Учитывая соотношение Гиббса-Дюгема, согласно которому
x{d\xx + x2d\x2 = 0, получаем
fdG^ =112-щ. (4.17)
=—oo-|J,j =-оо , ПРИ
Эх? L
= |Li°-f oo = oo. Кривая G=f(x2) касается как оси
ординат (х2 = 0), так и ординаты х2 = 1. Точки касания соответствуют
величинам ji,° и |д2° (С,° и С2°).
Общие условия равновесия для систем при постоянных
температуре и давлении требуют, чтобы величина энергии Гиббса имела
минимальное значение: dGTp = 0, d2GT > 0. При устойчивом
(стабильном) равновесии системы всякое совместимое с наложенными
условиями (в данном случае — температура и давление постоянные)
бесконечно малое воздействие вызывает только бесконечно малое
изменение состояния системы, причем перемена знака воздействия
вызывает перемену знака изменения состояния системы. Система,
будучи выведена из состояния равновесия, по прекращении
воздействия стремится вновь восстановить прежнее состояние.
На основании указанных выше условий стабильного равновесия
можно сделать заключение о форме кривых G=J{x2). Предположим,
что в бинарной системе зависимость энергии Гиббса от молярной
доли второго компонента выражается кривой, знак кривизны которой
изменяется (рис. 4.7). Двойная касательная определяет наличие в
системе области расслоения раствора на две жидкие фазы с составами
х2 (точка Ь) и х2п (точка/). Точки ewe являются точками перегиба и
отвечают условию (d2G/dx22)r = 0.
На участке кривой между точками сие вторая производная
(d2G/dx22)T меньше нуля. Растворы в интервале составов от точки
с до точки е лабильны, они неизбежно распадаются на растворы,
составы которых соответствуют точкам Ь и/(х2] и х2п). Растворы,
составы которых лежат левее точки с и правее точки е, устойчивы.
Если бы в результате концентрационных флуктуации
образовывались растворы составов с и а, то они самопроизвольно смешались
80
бы, образовав раствор состава Ь. Таким образом, в соответствии с
общими условиями равновесия вдоль кривой G = Дх2) устойчивы
растворы с положительной величиной (d2G/dx22)Tp > 0. Растворы,
для которых (d2G/dx22)Tp < О, неустойчивы (лабильные состояния).
Функция G=f{x2) в области устойчивых состояний имеет
положительную кривизну.
II
Рис. 4.7. Зависимость энергии Гиббса от состава
при наличии области расслаивания (р и Т— постоянные)
Выражение (d2G/dx2)T > О часто называют условием внутреннего
равновесия бинарного раствора. Это условие можно выразить в
несколько ином виде. Дифференцируя уравнение (4.17), получаем
Эх22
(ЭЦ2
/Г.р
Эх
2>„
( Эц, ) ГЭц.
Эх
2>,
Эх,
+
2 /г.р L
Эц,
Э(1-хД
Т.р
Для двухкомпонентной системы уравнение Гиббса-Дюгема
можно записать так:
0-*2)
Тогда
+ Х-,
'Эц^
Эх
2>,
= 0.
Эх-> и,
1 /Г.р
ЭЦ2
Эх-
2>.
1-Х2
Эц,
Эх
2>„
'Э»0
\-х2
дх
2 >>
81
Знаки функций
Э*2 кр
(
И
ЭЛ2 Jr.,
в устойчивых состояниях
совпадают, и условие внутреннего равновесия можно записать так:
1 JT.p
>0. (4.18)
Таким образом, химический потенциал компонента с
увеличением его молярной доли в области устойчивости гомогенного
бинарного раствора должен увеличиваться. Кривая зависимости энергии
Гиббса от молярной доли для устойчивых гомогенных растворов при
постоянных давлении и температуре должна иметь положительную
кривизну. Наличие на кривой участков с отрицательной кривизной
означает, что в этой области должно происходить расслаивание
однородного раствора на две фазы, различающиеся по составу.
4.6. Фазовые диаграммы двухкомпонентных систем
в конденсированном состоянии
4.6.1. Общие принципы расчетов фазовых диаграмм
Диаграммы состояния двухкомпонентных систем следовало
бы рассматривать в трехмерной системе координат: температура-
давление-состав. Однако в конденсированных системах в
большинстве случаев давление не оказывает существенного влияния
на температуру равновесия между твердой и жидкой фазами. Это
позволяет рассматривать диаграммы состояния двухкомпонентных
систем в координатах температура-состав, что существенно облегчает
графические построения. Соответственно в этом случае правило фаз
принимает вид
* = 3-/. (4.19)
В основе термодинамического анализа гетерогенных
равновесий двухкомпонентных систем лежит фундаментальное положение
о равенстве химических потенциалов каждого из компонентов в
сосуществующих фазах. В наиболее простом случае двухфазного
равновесия в двухкомпонентной системе при фиксированных
температуре и давлении
82
ц{Ц1) = ц!,(х||,)| (420)
В скобках указаны составы фаз, при которых наблюдается
равенство химических потенциалов компонентов. В каждой из
сосуществующих фаз сумма молярных долей компонентов равна единице:
х,1 + х21 = 1, х,п + х2и = 1. Решения системы (4.20) при различныхтемпе-
ратурах дают линии равновесия фаз I и II на диаграмме состояния.
Следует, однако, иметь в виду, что в уравнения (4.20) входят
абсолютные значения химического потенциала, величины которых
неизвестны, поскольку экспериментально определяются только
относительные значения основных термодинамических функций.
Выбор абсолютного уровня отсчета этих свойств проблематичен и не
является необходимым для практического использования уравнений
термодинамики, поскольку эти уравнения составляются всегда с
учетом сохранения масс компонентов. Это позволяет из правой и
из левой частей любого равенства вычесть одновременно
соответствующие молярные термодинамические функции компонентов в их
стандартных состояниях и ввести тем самым в термодинамические
соотношения относительные термодинамические функции.
Как уже отмечалось (с. 46), в термодинамике фаз переменного
состава за стандартное состояние обычно принимают соответствующие
чистые вещества при тех же условиях (температура, давление), что и
рассматриваемая система (фаза переменного состава, раствор).
Таким образом, в общем случае для расчета линий равновесия на
фазовых диаграммах нужны сведения о термодинамических свойствах
сосуществующих фаз.
В отдельных частных случаях расчет основных элементов
фазовых диаграмм может быть существенно упрощен. Корректные и
некорректные постановки задач при расчетах фазовых диаграмм
рассмотрены в заключительной части главы 4.
4.6.2. Уравнения кривых ликвидуса при кристаллизации
чистого компонента из идеального раствора
Рассмотрим систему, компоненты которой неограниченно
смешиваются в жидком состоянии и полностью не смешиваются в
твердом состоянии. Примем дополнительно, что в жидком состоянии рас-
83
твор ведет себя как идеальный.
В этом случае из жидкой фазы
кристаллизуется чистый
компонент, например компонент 2 из
любой точки, лежащей на линии
его первичной кристаллизации
(рис. 4.8).
Общее уравнение для линии
первичной кристаллизации
компонента 2:
й2*(х2, 7) = й2тв(*2=1,7), (4.21)
причем Т < Г2ПЛ. Принимая во
внимание, что жидкая фаза
представляет собой идеальный
раствор и кристаллизуется чистый
компонент, получаем
мо,ж + RT\WC2 = Й2°ТВ.
Разность р2°'ж - |д20ДВ представляет собой изменение химического
потенциала (энергии Гиббса) моля чистого компонента 2 при его
переходе из твердого состояния в жидкое:
И2°'ж ~ ^2°,тв = ДМ20,ПЛ = АС2°'ПЛ = АЯ2ПЛ - T&S™.
Величина Дц2опл равна нулю при Г= Г2ПЛ и имеет конечное
значение при температурах ниже точки плавления (Т< Т2пл), А#2ПЛ и
А52пл — соответственно изменения энтальпии и энтропии компонента
2 при плавлении в расчете на один моль. Как известно,
А5Г =^-
х2-> 2(х2 = 1)
Рис. 4.8. Линия первичной
кристаллизации компонента 2 в двухкомпо-
нентной системе
тш
Для простоты вывода принимаем, что в интервале температур
от Гдо Т2™ величина Д#2ПЛ сохраняет постоянное значение. Таким
образом,
W
°>те+дя,пл-г
7тПЛ
2
+ ЛГ1ПХ2=Ц2
1,°»ТВ
отсюда
1пх2 =
ЬИ?{Т-1%*)
RTTm
(4.22)
84
Для линии первичной кристаллизации компонента 1 уравнение
записывается в аналогичном виде:
адг (Г-Г), (4.23)
Уравнения вида (4.22) и (4.23) часто называют уравнениями
Шредера или Ле-Шателье—Шредера. Для практических расчетов удобна
следующая их запись:
=_анг аяГ_ (4 24)
1 2,303*7;™ 2,303ДГ'
lgJC2 =_^Г ^L (4.25)
2,303RT™ 2,303ЛГ
или
АЯ™ _ АЯ™ ,. . _
где Д. = 2 я = *— (/= 1,2).
^ 2,303*77™ 2,303Л х
Угловой коэффициент прямой в координатах lgx,- позволяет
получить сведения об энтальпии плавления соответствующего
компонента.
Проанализируем некоторые особенности кривой первичной
кристаллизации компонента 2, описываемой уравнением (4.18) (для
компонента 1 все аналогично). Из уравнения (4.18) следует
A TTTUlrpTUl
Т = 2 2 . (4.26)
AH™-RT^\nx2
Рассмотрим зависимость температуры ликвидуса Тот состава:
dT = АН?Ч1(Т2™)2
dx2 x2(AH™ - КГ?1 Inx2)2 *
Из уравнения (4.27) следует, что температура первичной
кристаллизации компонента 2 повышается с ростом его содержания в смеси,
поскольку АЯ^ > 0. Знаменатель не может быть равен нулю, 0 < х2 < 1.
Как видно из уравнения (4.26), при х2 = 1 Т= Т2ПЛ.
Рассмотрим вторую производную от температуры по составу:
пл ¥>2/т»пл\3
d2T
АН?Ч1 (Т™)
' АЯГ , Л
2 *—+]пх
ЯТГ
РПЛ - ' 1
(4.28)
dx\ xl(AH^-RT^]nx2)3
85
Знак второй производной определяется выражением, стоящим
в числителе в скобках. При равенстве этого выражения нулю
(знаменатель в нуль не обращается и всегда положителен) на кривой
первичной кристаллизации компонента 2 появляется точка перегиба.
Это происходит при условии
lnx, =-2 +
Afff
Так как \пх2 < 0, это условие можно записать в виде
-2 +
АН™
RT?1
АН?"
<0,
АЯ2ПЛ < 2RT™.
(4.29)
Поскольку AS™ = —, неравенству (4.29) можно придать вид
А52пл < 2&
Таким образом, если выражение, стоящее в скобках в числителе
уравнения (4.28), может быть равно нулю при каком-либо значении
х2, на кривой ликвидуса будет точка перегиба. Обозначим этот
состав х2пер. Если х2 < х2пер, выражение в скобках и вторая
производная отрицательны, кривая обращена вогнутостью к оси абсцисс.
Из изложенного следует, что кривая ликвидуса с точкой перегиба
или даже выпуклая к оси абсцисс может быть и при идеальном
поведении жидкой фазы.
При проведении более точных расчетов может быть принята во
внимание зависимость энтальпии плавления от температуры. Так,
для компонента 2
ГдАН^Л
дТ
С учетом этой зависимости уравнение (4.20) приобретает более
сложный вид:
1 АН™(Т™-Т) С*2-С™2 Т™-Т С*2-С™2 Т^
lg*2 ~ 2^ZRTT^ 2,303г TJ™ R g T
Следует, однако, иметь в виду, что для металлов изменение
величины теплоемкости при переходе из твердого состояния в жидкое
очень невелико. Для ряда металлов в табл. 4.1 сопоставлены значения
теплоемкости в жидком и твердом состояниях.
86
Пересечение кривых Igx, =J{ 7) и lgx2 =f[ 7), описываемых
уравнениями (4.24) и (4.25), позволяет определить положение эвтектической
точки. Рассмотрим следующий пример: кадмий и висмут образуют
простую эвтектическую систему с неограниченной смешиваемостью
между компонентами в жидком состоянии и практически полной
несмешиваемостью в твердом. Жидкие сплавы системы Cd - Bi имеют
относительно небольшие отклонения от закона Рауля, система близка
к идеальной. У кадмия Л#Сс!пл = 6,19 кДж • моль"1, Тс™ = 594,26 К; у
висмута ДЯВ.ПЛ =11,3 кДж • моль"1, Гв.пл = 544,59 К.
Таблица 4.1
Теплоемкость металлов Ср (Дж • моль-1 • К-1) в жидком и твердом
состояниях при различной температуре плавления
Металл
Ag
А1
Cd
Си
Fe
К
Mg
Na
Ni
Sb
Sn
Tl
|_ Zn
Tm, К
1235
934
594
1358
1811
336
922
371
1728
904
505
577
693
31,827
33,723
29,539
33,346
42,551
32,200
32,259
31,246
38,535
30,962
30,731
32,677
29,577
Сж
p
33,472
31,757
29,706
32,635
46,024
32,246
32,133
31,782
38,911
31,380
29,706
29,706
31,380
c*/cr
1,052
0,942
1,006
0,979
1,082
1,001
0,996
1,017
1,010
1,014
0,967
0,909
L,06]
В соответствии с уравнениями (4.20) и (4.21) получаем
323 3 590 2
lgxCd = 0,544-^, lgxBi =1,084-^.
Решение этих уравнений для ряда температур приводит к
следующим результатам:
L Т,К
*Cd
L_*Bi
550
0,904
-
500
0,790
0,801
450
0,669
0,592
400
0,544
0,406
350
0,417
0,250
300
0,293
0,131
87
Графическое построение (рис. 4.9) позволяет заключить, что
температура эвтектики составляют 410 К при jcb. = 0,45. В справочной
литературе указываются температура эвтектики 417 К и содержание
висмута хш = 0,454.
Г, К
500
300 Ь
о'' V
I у
о
Cd
0,2 0,4 0,6 0,8
1,0
Bi
Рис. 4.9. Определение эвтектических
состава и температуры для системы кадмий-висмут
4.6.3. Уравнения кривых ликвидуса и солидуса
в системах с неограниченной растворимостью в жидком
и твердом состояниях в приближении идеальных растворов
В системах, в которых во всем интервале составов образуются
твердые растворы, причем и жидкая и твердая фазы по свойствам близки к
идеальным, уравнения (4.20) можно представить в следующем виде:
li^+jmnjq™ =\1°{ж +RTlnxf,
ц2°тв + RTlnx™ = ц2°ж + RT\nx*.
С помощью ранее описанных преобразований получаем
AHF
х,ж 2,ЗОЗДГ 2,303ЛГ,М'
lg-^-= —
Aff?"
xf 2,303/гГ 2,30ШТ?
(4.30)
(4.31)
88
Рассмотрим систему Ge—Si, в которой образуются близкие
к идеальным жидкий и твердый растворы. У германия Д//Сепл =
= 36,94 кДж • моль"1, ГСепл = 1210,4 К; у кремния A//SinjI =
= 50,55 кДж • • моль"1, Ts™ = 1687 К. В соответствии с уравнениями
(4.30) и (4.31) получаем
iS=i92913_ 1{Д=2640И_ 1565
xGe l *Si *
Результаты совместного решения этих двух уравнений для ряда
температур представлены в табл. 4.2.
Таблица 4.2
Составы твердой и жидкой фаз в системе
Ge-Si при различных температурах
Г, К
1250
1300
1350
1400
1450
1500
1550
L 1600
2й
0,890
0,776
0,684
0,608
0,545
0,492
0,447
0,409
XSi
3,524
2,923
2,458
2,093
1,802
1,567
1,375
1,216
ТВ
0,148
0,304
0,438
0,552
0,652
0,741
0,819
0,890
xSi
0,042
0,104
0,178
0,264
0,362
0,473
0,596
0,732
Расчетные величины вполне удовлетворительно согласуются с
экспериментальными данными, в соответствии с которыми составы
жидкой и твердой фаз при фиксированных температурах
представлены в табл. 4.3.
Таблица 4.3
Г т9 к'
1300
1400
1500
1600
-.ТВ
*Si
0,277
0,508
0,692
0,805
*Si
0,092
0,208
0,404
0,715
89
4.6.4. Расчет фазовых диаграмм
на основании термодинамических данных
Как было отмечено, диаграммы состояния могут быть
рассчитаны, если имеются сведения о термодинамических свойствах всех фаз,
которые образуют данные компоненты. Часто число фаз невелико,
и их термодинамические характеристики определяются с высокой
степенью точности. Тогда диаграммы состояния, рассчитанные из
термодинамических величин, могут лучше отражать существо
явления, чем диаграммы, полученные методом термического анализа.
Выражения для химических потенциалов компонента в
сосуществующих фазах в случае систем с отклонениями от идеального
поведения можно записать так:
для компонента 1
дГ=иГ +
= ц,вт"
Г
+ ЛЛпа™;
(T™-T) + RT\naf;
для компонента 2
ц?=мГ +
u2TB = ц/'™ + Л71па2т";
ГАН^Л
1г
Приравнивая химические потенциалы, получаем
RTlnd^=\
RTlna? =
ГАН™Л
7тПЛ
1
АН?1
1г
(7J"" - 7-)+ RTtoaf;
(T^-T) + RT]naf
i _
о -яг
АЯГ
или
(.
2,№RT
т'
rpTUl
М J
ТВ
(4.32)
lg
(1-х?) AH?
(1
■#)
2,303RT
т ttJ у?
(4.33)
90
Уравнения (4.32) и (4.33) существенно упрощаются, если из
расплава кристаллизуется чистый компонент, как это, например,
наблюдается в системе Ga—Zn (рис. 4.10). В этом случае для ветвей
первичной кристаллизации галлия и цинка можно записать:
lg*& =
А#£
2.303Л
1
7-»пл
Ga
Т
-igy£,;
ig*2,=
АНГп'
2,303*
J \_
7»пл пг
Zn *
-feYzni
АЯ^а
уПЛ
(7Ь™-Г) + ЛЛла£а=0;
(Г^-Г) + ЛЛпа*=0.
Г,'С
400
200
Ga 0,2 0,4 0,6 0,8
Zn
vZn
Рис. 4.10. Диаграмма состояния системы галлий—цинк
(кружки — расчетные значения)
91
Поскольку RTlna. = AC = АН. - TAS., получаем
Т_АН£ + МГЬ
AS£ +
У'ПЛ
п
или Т= ^ |s-
(4.34)
rGa л Zn
в зависимости от того, какой из компонентов кристаллизуется.
Известно, что
АЯ;=Э
дел
л>У
- I и
AS;=~
дТ
)р
Таким образом, для расчета фазовой диаграммы системы Ga-Zn
надо располагать сведениями о зависимостях А(7жСа =/(7) и
AGxZn = Д7) или иметь известные значения A#Ga, ASGa, A#Zn и ASZn.
Необходимо также знать величины теплот и температур плавления
чистых компонентов.
В качестве примера рассчитаем с помощью уравнения (4.34)
температуры начала кристаллизации цинка для ряда составов системы
Ga-Zn. Термодинамические свойства жидких сплавов этой системы
изучались методом измерения ЭДС концентрационных цепей.
Результаты представлены в табл. 4.4.
Энтальпия плавления цинка — 7,322 кДж • моль-1, температура
плавления — 692,73 К.
Таблица 4.4
Термодинамические свойства жидких сплавов системы Ga-Zn при 773 К*
*Zn
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,80
1 0,90
£773К, мВ
62,5
41,4
29,3
21,3
15,7
П,2
7,9
5,3
2,8
dE/dT,
мВ/100 К
11,7
8,0
5,9
4,4
3,2
2,2
1,4
0,9
0,5
А<Ъ
A"zn
Дж • моль-1
-12050
-7990
-5650
-4100
-3035
-2175
-1525
-1025
-545
5395
3950
3140
2470
1715
1110
545
335
210
A*z„>
Дж ■ моль-1 • К-1 |
22,6
15,4
11,4 1
8,49 1
6,15 1
4,22
2,68
1,75
0,96 J
* Герасименко Л.Н., Зайцев В.А., Ложкин Л.Н., Мораневский А.Г. // Изв.
вузов. Цветная металлургия. 1966. № 1. С. 46—48.
92
Линия ликвидуса рассчитывалась с помощью уравнения (4.34),
полученные значения нанесены на рис. 4.10. Эвтектическая точка в
системе Ga-Zn расположена при ;cZn = 0,042 (Рвт = 298,3 К).
Система Ga-Zn характеризуется небольшими положительными
отклонениями от идеального поведения и соответственно малыми
положительными теплотами смешения. Максимальная интегральная
энтальпия смешения не превышает 1650 Дж/моль. Для такого рода
систем при расчетах фазовых диаграмм требуется особенно высокая
точность экспериментальных данных. Действительно, при xZn > 0,6
температурные коэффициенты ЭДС составляют 1-2 мВ на 100 К.
dE
Небольшие погрешности в определении — могут сильно сказаться
на значениях A//Zn и A5Zn.
Расчеты линии ликвидуса, а также других элементов фазовых
диаграмм требуют высокой точности исходных экспериментальных
данных.
Определение линии ликвидуса многих бинарных систем методом
термического анализа не вызывает больших экспериментальных
трудностей, в то время как определение линии солидуса, растворимости
в твердом состоянии значительно сложнее. Если известны
термодинамические свойства жидкой фазы, возможно уточнение фазовых
диаграмм с помощью термодинамических расчетов.
Рассмотрим на примере системы Ga-Sn оценку области твердых
растворов на основе олова (рис. 4.11). Из равенства ц*п =ц^ для
температур ниже точки плавления олова (505,12 К) следует
ЯТЫ& +
ySn )
(Ts™-f)=RTlna£.
Учитывая относительно небольшую протяженность области
твердых растворов на основе олова, можно принять, что flsn =xsn = 1-
Заменив
получаем
RT liu& = AGS* = Д#5* -ГА58ЖП,
A#fn -ГД5£ =-ДЯ8™ +TASg +RTlnx£.
93
г, к
500
400
300
0 0,2 0,4 0,6 0,8 1,0
Ga xSn -» Sn
Рис. 4.11. Диаграмма состояния галлий—олово
(кружки — расчетные значения границы области
твердых растворов)
4.6.5. Диаграммы состояния и термодинамические функции систем
с образованием интерметаллических соединений
Напомним некоторые общие положения, касающиеся
интерметаллических соединений. В 1899 г. была опубликована работа
Н.С. Курнакова (1860-1941) "О взаимных соединениях металлов",
в которой он приходит к выводу о существовании соединений между
металлами (металлических или интерметаллических соединений).
На кривых зависимости свойств от состава (кривые состав-свойство)
этим соединениям соответствуют те или иные характерные точки.
Плавление соединений называется конгруэнтным, если состав
образующейся жидкости тождественен с составом твердой фазы, и
инконгруэнтным, если это условие не выполняется. Если
компоненты А и В образуют соединение АВ(в общем случае АтВп), плавящееся
конгруэнтно, кривая ликвидуса (рис. 4.12) образует максимум в точке
С, когда составы твердой и жидкой фаз совпадают. По обе стороны
от соединения находятся эвтектики Ех и Ег По существу такую
простую систему можно рассматривать как сочетание двух диаграмм
эвтектического типа: А-АВ и АВ-В.
В качестве примера относительно простых диаграмм состояния
систем с образованием соединения можно привести систему индий—
94
сурьма (рис. 4.13). Подобная диаграмма характерна и для ряда других
систем, образованных элементами III и V групп Периодической
системы. Однако диаграммы состояния могут быть и существенно
более сложными (рис. 4.14).
А+АВ
В + ж
АВ + В
В
в
Рис. 4.12. Диаграмма состояния системы
с интерметаллическим соединением,
плавящимся конгруэнтно
г/с
600
400
200
г ж
[■ ^*"—
L / ж + insb
7 155"
Г In +InSb
. . i i i 1_
52
5*
InSb
^J
__^/K + Sb
500ф
InSb + Sb
J 1 1 1 1
In 20 40 60 80
At.% Sb
Sb
Рис. 4.13. Диаграмма состояния системы индий-сурьма
95
г, к
1273
1073
873
673
473
К(
^1
\Al93
-
1403 '
1013 \i
In .,-
KGe .
'1363
■
KGe 1
T1163 J
/848
L.
\
\
\
\
\
\
\
\
337 \
-— -
__i ■ '
Ge 20 40 60 80 К
К, %(мол.)
Рис. 4.14. Диаграмма состояния
системы германий—калий
Рис. 4.15. Различные формы максимума на диаграммах состояния
при образовании интерметаллических соединений. Соединение
при плавлении: а — не диссоциирует; б — диссоциирует
Все приведенные на рис. 4.12-4.14 диаграммы называют
диаграммами с открытым максимумом, точку максимума — дистектикой.
96
В учении Н.С. Курнакова о физико-химическом анализе двух-
компонентных систем существенное значение придается форме
максимума на кривой ликвидуса в области образования соединения.
Типичные кривые приведены на рис. 4.15. Соединение на рис. 4.15,а
обычно называют недиссоциированным при температуре плавления
(относительно более высоких температур диаграмма не дает
указаний). На рис. 4.15,5— аналогичная диаграмма, но уже для случая,
когда соединение хотя бы частично диссоциирует при плавлении.
На первой из диаграмм кривая представляет собой фактически
пересечение двух ветвей ЕХС и Е2С, на второй — плавная кривая. Точка
С на рис. 4.15,а носит название сингулярной точки*.
А. Б. Млодзеевским была установлена количественная связь между
радиусом кривизны в максимуме кривой ликвидуса (р) и некоторыми
термодинамическими характеристиками соединения:
где АЯпл, А5пл и Гпл — энтальпия, изменение энтропии и температура
плавления соединения (Гв К); К— константа диссоциации
соединения на соответствующие компоненты.
В свою очередь величина ^связана со степенью диссоциации
a2 i
соединения а соотношением: К = - -у или ol = JK/(K + \).
(1-а) v
Если К=0 (полное отсутствие диссоциации), то р = 0 и на
кривой плавкости в этом случае имеется сингулярная точка. Кроме р в
уравнение (4.35) входят АЯ^ и 7^. Следовательно, константа
диссоциации, а значит, и степень диссоциации зависят не только от р, но
и от характеристик соединения (АЯ^, 7^). Поэтому нельзя судить о
диссоциации только по одному виду кривой. Степень диссоциации не
следует понимать в обычном смысле этого слова. Вероятно, правильнее
считать, что величина а характеризует степень нарушения ближней
упорядоченности при плавлении кристаллического соединения.
Во многих случаях компоненты А и В образуют соединение АтВп,
плавящееся инконгруэнтно, т. е. с разложением (рис. 4.16). В этом
* Более подробно о сингулярных точках см.: Курнаков Н.С. Избранные
труды. Т. 1.М.:Изд-воАНСССР, 1960. с 565; Аносов В.Я., Озерова М.И.,
Фиал ков Ю.Я. Основы физико-химического анализа. М.: Наука, 1978. 504 с.
97
случае соединение устойчиво только ниже температуры Тс
Температура Гсносит название перитектической температуры, расплав в точке
С— перитектический расплав. При охлаждении расплава состава М
в точке ао, находящейся на линии ликвидуса С В, начнется выделение
кристаллов компонента В. В интервале температур между точками ао
и Ьо система является двухфазной и имеет одну степень свободы (при
фиксированном давлении). В точке Ьо начинается кристаллизация
соединения АтВп, состав которого соответствует точке D. Таким
образом в равновесии оказываются три фазы: расплав, кристаллы АтВп
и В. В этом случае число степеней свободы снижается до нуля, что
указывает на постоянство температуры (соответствует точке С) и
состава (соответствует точке D).
Чтобы состав расплава в процессе кристаллизации соединения
АтВп не изменился, кристаллы Z? должны растворяться, поддерживая
постоянным содержание компонента В в перитектическом расплаве.
Важно отметить следующее: в перитектической точке (С), как и в
эвтектической, находятся в равновесии расплав и две твердые фазы.
Однако процессы при охлаждении этих трехфазных систем существенно
Рис. 4.16. Диаграмма состояния системы
с интерметаллическим соединением,
плавящимся инконгруэнтно
98
различаются. В эвтектической точке одновременно выпадают две
твердые фазы, образующие эвтектическую смесь, а в перитектической
точке одна твердая фаза (соединение) выпадает, а другая —
растворяется. Процесс охлаждения в точке Ьо заканчивается растворением
всех ранее выпавших кристаллов В. Остается двухфазная система,
состоящая из расплава и кристаллов АтВп. Число степеней свободы
становится равным единице (5 = 1). Температура понижается, и из
расплава выпадают кристаллы АтВп. При этом каждой температуре
соответствует определенный состав расплава (кривая СЕ).
Для описания термодинамических свойств интерметаллических
соединений необходим набор следующих величин: изменения
энтальпии, энтропии, энергии Гиббса и теплоемкости при образовании
соединения из чистых компонентов. Для соединений, плавящихся
конгруэнтно, нужны сведения об энтальпии и температуре плавления.
Прежде чем обсуждать эти величины, необходимо отметить, что как
для экспериментального определения термодинамических функций,
так и для выполнения разнообразных расчетов важна протяженность
области гомогенности интерметаллического соединения.
На всех рассмотренных выше диаграммах состояния
образующиеся соединения не имеют различимой области гомогенности,
относятся к числу "линейных". Если соединение не имеет области
гомогенности, то формально получается, что фаза АтВп, имея один и
тот же состав, может находиться в равновесии с любой из двух
других фаз, каждая из которых обладает своим значением химического
потенциала. Такой гипотетический случай представлен на рис. 4.17.
Равновесие такого рода невозможно, и если в системе образуется
соединение, не имеющее области гомогенности, то использовать условия
равновесия в виде равенства химических потенциалов нельзя*.
У большого числа соединений имеются хотя бы очень узкие области
гомогенности. Можно полагать, что они есть и у тех кристаллических
соединений, у которых они вследствие малости пока не найдены.
При наличии даже узкой области гомогенности зависимость
химического потенциала компонента от состава остается непрерывной.
Если промежуточное соединение имеет точно стехиометрический
* Более подробно рассмотрено в книге: Я.И. Герасимов, В.А. Гейдерих.
Термодинамика растворов. М.: Изд-во Моск. ун-та, 1980. 184 с.
99
АВ
В
АЦ«
г
—М
Рис. 4.17. Зависимость химического
потенциала компонента от состава
в системе с интерметаллическим
соединением без видимой области
гомогенности
состав, то целесообразно
рассматривать это соединение
(например, АтВп) как компонент и вместо
системы А - В рассматривать две
самостоятельные бинарные систе-
мы А - А В и А В - В.
т п т п
Энтальпия образования. В
выражении АН = AG + TAS для
процесса образования
соединения из чистых компонентов роль
энтропийного члена, как правило,
невелика; значение AG всегда
отрицательно, а энтальпия
образования в подавляющем числе
случаев отрицательна.
Образование интерметаллических
соединений — процесс экзотермический.
Энтальпия образования непосредственно связана с типом
химической связи в соединении. Переход от выраженной ионной связи к
преимущественно ковалентной ведет к существенному снижению
экзотермического эффекта образования соединения. Еще меньше
энтальпия образования соединений с металлической связью.
Переход от преимущественно ионной к преимущественно
ковалентной связи происходит постепенно, "степень ионности" связи
приближенно может быть оценена с помощью шкалы
электроотрицательности. Чем больше разность электроотрицательностей компонентов
соединения, тем более полярна ковалентная связь. Необходимо иметь
в виду, что электроотрицательность не является строгой физической
величиной, которую можно экспериментально определить. Тем не
менее эта величина, характеризующая способность атома присоединять
электроны, несомненно, полезна при оценке степени ионности связи.
Существует ряд несколько различающихся шкал
электроотрицательности (по Полингу, Горди, Сандерсону, Бацанову и пр.).
По Полингу, шкала электроотрицательности охватывает значения
от 0,7 для цезия до 4,0 для фтора. Фтор — наиболее
электроотрицательный элемент. Водород и типичные неметаллы находятся в центре
100
шкалы, значения их электроотрицательности близки к 2,0. Приведем
значения электроотрицательности некоторых элементов:
К, Rb
Na, Ba
Li, Са, Sr
Mg,Y
Sc, Th, Hf
Ti,Ta, Mn
V, Nb, Cr, Zn, Ga
0,8
0,9
1,0
1,2
1,3
1,5
1,6
Mo
, Fe
Hg,
Cd, In
Ge, Sn, Tl, Si
Pb, Bi, Sb
Те
Se
S
1,7
1,8
1,9
2,1
2,4
2,5
Полинг установил приближенное соотношение между разностью
электроотрицательностей ХА — Хв (или Хв — Хл) и долей ионности связи
(степенью ионности) между атомами А и В (рис. 4.18). Для ионных и
преимущественно ионных соединений Полинг предложил уравнение,
связывающее величину энтальпии образования (кДж/моль
соединения) с разностью электроотрицательностей:
AJH=-96,5lz(XA-Xf, (4.36)
где z — число "валентных связей" в соединении. Например, для
соединения Li3Sb z = 3,XSb — Xu = 0,9, Afl^ —234,5 кДж/моль.
00
80
60
40
20
А
/
/
~~^ i i
1,0 2,0 3,0
Разность электроотрицательностей, Хл - Хв
Рис. 4.18. Зависимость между разностью
электроотрицательностей по Полингу
и степенью ионности связи
101
Если записать соединение Li3Bi в виде Li0 75Bi0 25, расчетная
величина Ду//= -58,6 кДж • моль1; экспериментальное значение Ay#°298 =
= -58,1 ± 4,8 кДж • моль-1. Для соединения Mg0 6Sb0 4 расчетное
значение равно Ду//= -56,7 кДж • моль-1, экспериментальное составляет
-60,0 ± 3,3 кДж • моль'1.
Поскольку электроотрицательность элемента определяется с
точностью до 0,1 отн. ед., энтальпия образования соединений Li3Sb
или Mg3Sb2, вычисляемая по уравнению (4.36), не может иметь
точность более чем ±10 кДж/моль. Если уравнение (4.36) дает хотя бы
приблизительно правильное значение энтальпии образования интер-
металлида, можно предполагать, что связь в нем частично ионная*.
Существует определенная зависимость между энтальпиями
образования бинарных соединений, если входящие в них элементы
образуют несколько соединений. Если известны температуры плавления
соединений, относящихся к данной бинарной системе, то можно
ожидать, что соединение с наиболее высокой температурой
плавления будет характеризоваться наибольшей энтальпией образования,
отнесенной к одному молю сплава.
Энтропия образования. Изменение энтропии при образовании
интерметаллического соединения, как и любого металлического
сплава, условно можно представить в виде суммы различных вкладов:
колебательного, конфигурационного, электронного и магнитного:
AyS= ASkoji + &S + AS3n + А5магн. Более прочная связь в соединении
по сравнению со средней связью в элементах уменьшает
колебательную энтропию. Как правило, ASkojj < 0.
В том случае, если образуется полностью разупорядоченное
интерметаллическое соединение, конфигурационный вклад определяется
выражением
^^ = -R(xA\nX/l + xB\nx„), (4.37)
где хл и хп — молярные доли компонентов при составе соединения.
При образовании полностью упорядоченных соединений А5конф = 0.
Таким образом, в зависимости от степени упорядоченности
конфигурационный вклад может изменяться от 0 до значения, определяемого
уравнением (4.37), А5конф > 0. Электронный и магнитный вклады в
* Об энергетических характеристиках фаз с неионной решеткой (кова-
лентной, металлической) см.: Ормонт Б.Ф. Введение в химию и
кристаллохимию полупроводников. М.: Высш. шк., 1982. 528 с.
102
изменение энтропии для соединений, не содержащих переходные
металлы, незначительны.
При образовании упорядоченных интерметаллидов с
преимущественно ионным типом связи уменьшение колебательной энтропии
особенно существенно и происходит благодаря резко выраженному
возрастанию прочности связи. Величина колебательного вклада в
этих соединениях пропорциональна энтальпии образования. Эта
корреляция справедлива не только для преимущественно ионных,
но и для электронных соединений, когда они упорядочены и можно
пренебречь конфигурационным вкладом. В данном случае речь идет
о корреляции между величинами, характеризующими процесс
образования сплава.
В разупорядоченных фазах переменного состава атомы не
занимают определенных положений в решетке. В этом случае
конфигурационный вклад, определяемый уравнением (4.37), может
быть больше отрицательного колебательного вклада и суммарное
изменение энтропии будет положительно. Для фазы Au025Cd075
при 700 К AS = 1,60 Дж • моль-1 • К-1, для Cu022Zn078 при 773 К
AS =2,51 Дж-моль"1 • К"1.
В качестве примера соединения, для которого надо учитывать
магнитный и электронный вклады в энтропию, можно указать фазу
Mn037Si063. При образовании диамагнитного полупроводникового
силицида практически исчезает атомный магнитный момент
марганца и существенно изменяется электронное слагаемое его
теплоемкости. Электронные и магнитные вклады в энтропию зависят в
каждом конкретном случае от электронной части теплоемкости и
атомного магнитного момента переходного металла. Как известно,
электронную составляющую теплоемкости имеет смысл учитывать
только для переходных металлов, но даже для них при 298 К эта
составляющая очень мала по сравнению с составляющей, связанной с
колебаниями решетки.
Теплоемкость интерметаллических соединений. Согласно
правилу Коппа—Неймана, величина С интерметаллических соединений
равна сумме теплоемкостей составляющих элементов. Однако в
полной мере это правило может быть применено только к
соединениям, в которых связь и кристаллографическое расположение атомов
аналогичны с составляющими элементами.
103
Необходимо учитывать, что точность экспериментального
определения теплоемкости соединений и некоторых чистых
компонентов не всегда достаточна для сравнений. Теплоемкости ионного
соединения Mg3Sb2, соединений с металлической связью CuZn, Ag2Al
хорошо следуют правилу Коппа-Неймана. В целом отклонения от
правила аддитивности составляют от +5 до —8 %. Для соединений с
преимущественно ионной связью более характерны отрицательные
отклонения от аддитивности: С < С Соединения с металличе-
v р,экст\ р,выч ^
ской связью могут иметь небольшие положительные и отрицательные
отклонения, теплоемкости фаз Лавеса имеют отрицательные
отклонения. Не рекомендуется пользоваться правилом Коппа-Неймана
при расчете теплоемкости халькогенидов.
Энтальпии и энтропии плавления. Энтальпия плавления
интерметаллических соединений включает энергию, необходимую для
уничтожения порядка, свойственного твердому состоянию при точке
плавления, и для изменения типа связи, сопровождающего плавление.
Энтропия плавления связана с возрастанием колебательной свободы
при плавлении и с разупорядочением упорядоченного соединения.
Энтропии плавления соединений, существенно разупорядоченных
в твердом состоянии (Bi0 6T10 4, е-фаза системы Au—Zn, е-фаза
системы Ag-Cd), могут быть оценены в первом приближении как сумма
энтропии плавления металлов, входящих в состав соединений. Если
сплав или соединение полностью упорядочено, то к энтропии
плавления, вычисленной по правилу аддитивности, необходимо внести
поправку а, зависящую от состава сплава. Для соединения АхВХх
o = -R[x\nx + (\ — jc)ln(l -x)].
По существу процесс плавления АхВ 1х(тв) = хА(ж) + (1 - х)В(ж)
разбивается на две гипотетические стадии:
АхВ]_х(тъ) = АхВ1_х(ж), (4.38)
АхВх_х{ж) = хА(ж) + (1 - х)В{ж). (4.39)
Реакция (4.38) соответствует гипотетическому процессу
плавления, при котором соединение сохраняет свои структурные
особенности в жидком состоянии. Реакция (4.39) выражает разупорядочение
соединения после плавления, образование полностью
неупорядоченного раствора. Для некоторых упорядоченных в твердом состоянии
фаз (Cd0 5Sb0 5, Mg0 ззз^п0 667) наблюдается довольно хорошее согласие
между а и разностью AS™cn -AS™ . В то же время при наличии су-
104
щественного упорядочения в жидком состоянии величина, которую
нужно прибавить к вычисленной по правилу аддитивности, находится
в пределах 0 - а и может быть оценена лишь приближенно.
Энтропия плавления — одна из важнейших термодинамических
характеристик процесса перехода из твердого состояния вжидкое.
Существенными факторами, влияющими на ее величину, являются изменение
характера химической связи и неидеальность образующейся жидкой
фазы, сохранение структурного упорядочения в жидкой фазе.
Влияние изменения характера химической связи при плавлении
на А^л прослеживается и у простых веществ. Ковалентные кристаллы,
образуемые атомами элементов IV группы Периодической системы
(германий, кремний), отличаются повышенными значениями
энтропии плавления (около 29 Дж • моль-1 • К-1); полуметаллы, к числу
которых относятся галлий, сурьма, висмут, также имеют
относительно высокие значения А5^л (около 21 Дж • моль-1 • К1), и только
для типичных металлов выполняется правило Ричардса и энтропия
плавления составляет около 8,4 Дж • моль-1 • К-1.
4.6.6. Влияние интерметаллических
соединений на свойства жидких сплавов
Многочисленные прямые исследования строения жидких
металлических систем указывают на сохранение в них ближнего
порядка, в основном соответствующего структурам наиболее прочных
высокотемпературных фаз на диаграмме состояния. Выдвинутая
в свое время Я.И. Френкелем идея, что жидкость при небольших
перегревах выше температуры плавления представляет собой как бы
разупорядоченное твердое тело, в котором продолжает существовать
ближний порядок, но нарушается характерный для твердых тел
дальний порядок, сохраняет в определенной степени свое научное
и методологическое значение*. Из многочисленных теорий,
созданных для описания строения реальных жидкостей, наибольший
успех имели те, в основе которых лежат представления о сходстве
ближнего порядка в расположении атомов в жидком и твердом
состояниях.
* Более подробно см.: Френкель Я.И. Кинетическая теория жидкостей.
Л.: Наука, 1975.592 с.
105
Как известно, особенно ясно выраженным стремлением к
образованию интерметаллических соединений обладают щелочные
металлы, как главные носители металлических свойств. Сочетания
щелочных металлов с ртутью, кадмием, оловом, свинцом, висмутом,
другими тяжелыми металлами представляют самую характерную
группу среди известных соединений металлов. Большой интерес
представляют жидкие сплавы магния с висмутом, сурьмой. В данном
учебном пособии ограничимся лишь небольшим числом примеров,
подтверждающих наличие ближнего порядка в жидких сплавах,
диаграммы состояния которых характеризуются образованием
интерметаллических соединений.
В рамках существующих моделей элементарные объемы с
ближним упорядочением рассматриваются какассоциаты (группировки,
кластеры), имеющие вполне определенный стехиометрический
состав; остальные атомы распределяются беспорядочно. Нет
необходимости допускать существование индивидуальных молекул сложного
состава. Время жизни ассоциатов должно превышать на 2—3 порядка
время существования группировок при случайных флуктуациях или
длительность контакта диффундирующих частиц. Для определения
состава ассоциатов могут быть использованы фазовые диаграммы
системы, концентрационная зависимость целого ряда
термодинамических и физико-химических свойств. Полезную информацию
дают характер кривой =/(*■), точки перегиба на кривых пар-
циальных молярных свойств. Весьма чувствительны к образованию
ассоциатов кривые стабильности и избыточной стабильности.
Функция стабильности (с. 68) максимальна при составе, соответствующем
структурным группам в жидкой фазе: о локальной упорядоченности
в жидком сплаве можно судить с помощью структурного фактора
Бхатиа-Торнтона, получаемого из дифракционного эксперимента
и связанного с функцией стабильности:
5„(0) = ЛГ
'Э2Д(Л'
dx} ) „
1 JpJ
(4.40)
Этаже величина, как и некоторые другие
структурно-чувствительные характеристики процесса сплавообразования (изменение
теплоемкости АС, изменение объема AV), может быть непосредственно
вычислена из измерений ЭДС (см. раздел 7.2).
106
Фазовые диаграммы и тер- A5Na
модинамические исследования
дают основания считать, что
в системе Li—Bi
доминирующим ассоциатом является Li3Bi.
15,0
Энтальпия смешения жидких
сплавов при хи = 0,75 имеет
острый экстремум и составляет
-55 кДж • моль-1.
Образование ассоциатов сопровождается
частичным переносом заряда,
электропроводимость при
составе соединения Li3Bi
минимальна.
В системе Na-Hg образуется
целый ряд интерметаллических
соединений, в том числе
плавящееся конгруэнтно соединение
NaHg2. Экстремумы
интегральных кривых Д(?36 и АН лежат
вблизи состава этого соединения.
Характерную форму имеет
кривая зависимости Д£, от состава
Na
10,0 \-
-10,0
-15,0 Ь
Рис. 4.19. Зависимость парциальной
молярной энтропии натрия от состава в
жидких сплавах системы натрий-ртуть
(Д5^а — в Дж • моль-1 • К-1)
(рис. 4.19). Резкий спад
наблюдается в области образования
соединения NaHgr Результаты
прямых структурных
исследований жидких сплавов системы Na—Hg, а также расчет параметров
локального упорядочения на основе термодинамических данных
указывают на предпочтительную координацию разносортных атомов.
В системе Na-Pb также наблюдается образование ряда
интерметаллических соединений, состав которых, однако, достаточно точно не
установлен (наиболее вероятные составы — Na15Pb4, Nal3Pb5, NaPb3).
На кривой избыточной стабильности (рис. 4.20) имеются два
максимума, соответствующие ассоциатам Na4Pb и NaPb. Пик на кривой
избыточной стабильности при эквиатомном составе шире и ниже,
чем другой пик при jcNa = 0,80. Есть все основания считать ассоциат
107
фнзб
200
100
J_
_L
Na
0,4
0,8
Рис. 4.20. Функция избыточной
стабильности в системе
натрий-свинец при 723 К
(Ф — в кДж • моль-1)
Na4Pb доминирующим, причем его
образование связано с частичным
переносом заряда. Удельное
электрическое сопротивление при
составе этого ассоциата максимально,
при этом же составе наблюдается
максимальное сжатие при сплавоо-
бразовании.
Весьма сильны явления
ассоциации в жидких сплавах магния
прежде всего с элементами
четвертой (Ge, Sn, Pb) и пятой (Sb,
Bi) групп Периодической системы.
В первом случае для диаграмм
состояния сплавов характерно
образование соединений типа А2В, во
втором — А3ВГ Температуры
плавления плавящихся конгруэнтно
соединений уменьшаются в ряду
Mg2Ge -> Mg2Sn -> Mg2Pb. В этом же направлении уменьшаются и
отрицательные отклонения жидких сплавов этих систем от идеального
поведения. Резко усиливается взаимодействие между компонентами
при переходе к системам Mg-Bi и Mg-Sb, в которых образуются
плавящиеся конгруэнтно соединения Mg3Bi2 (Тпл = 1094 К) и Mg3Sb2
(Гпл = 1518 К). Энтальпии образования в стандартных условиях в
расчете на состав АхВ]_х равны соответственно -25,4 и -60,0 кДж/моль.
Высокая степень упорядоченности и выраженный перенос заряда при
образовании соединения Mg3Bi2 позволяют отнести его к типичным
жидким полупроводникам. При изменении состава в жидком
состоянии реализуется непрерывный переход от металлического характера
проводимости к полупроводниковому (рис. 4.21).
В заключение отметим, что наиболее четко выражена тенденция к
образованию ассоциатов в жидком состоянии в случаях образования
интерметаллических соединений с преимущественно или частично
ионной связью. Примерами могут служить такие системы, как Mg-
Bi, Li—Pb, Sn-Te и особенно Au-Cs. В последней системе при составе
ассоциата AuCs жидкий сплав состоит из ионов Air и Cs+. Наименее
108
8000
is 6000
i
9 4000
g 2000
20 40 60 80
Me AT.%Bi Bi
Рис. 4.21. Зависимость электрической
проводимости жидких сплавов
магний—висмут (1173 К) от состава
(поданным различных авторов)
выражена тенденция к ассоциации в системах с преимущественно
металлическим характером связи. Явление ассоциации в жидких
сплавах весьма важно для понимания процессов получения аморфных
сплавов.
4.7. Извлечение термодинамической информации
из фазовых диаграмм
Фазовые диаграммы бинарных металлических систем
преимущественно хорошо изучены, и поэтому их можно использовать для оценки
термодинамических свойств, например химических потенциалов или
коэффициентов активности компонентов, интегральных величин.
Расчет термодинамических характеристик из данных фазовой
диаграммы относится к числу так называемых "обратных задач
термодинамики фазовых равновесий". При этом необходимо иметь в
виду естественные ограничения:
\ / \
I \ / \
Г ч / I
109
а. = ух < 1.
Из этой формулы следует
AG = j^xiA[ii=RTJ^xi\nxiyi<0.
i=\ /=1
Рассмотрим ряд конкретных примеров.
Система Sn-Zn относится к числу простых
эвтектических, области образования
первичных твердых растворов пренебрежимо
малы, кристаллизуются практически чистые
компоненты (рис. 4.22). Температуры
плавления олова и цинка равны соответственно
505,2 и 692,6 К, энтальпии плавления — 7,07
и 7,28 кДж • моль-1.
Возьмем какую-либо точку на кривой
ликвидуса, при xZn = 0,5965 Т= 621,3 К.
Равновесие в этом случае можно записать
так:
гп(ж, х£п =0,5965) <->Zn(TB, xJBn =1).
В любой точке вдоль цинковой ветви кривой ликвидуса ц£п = ц^у
Таким образом, для записанного равновесия Д<7= 0 (Г= 621,3 К).
Рассмотрим следующую схему:
650
600
550
500
_.. . 1 1 _J_ . J
0 0,2 0,4 0,6 0,8 1,0
Sn x Zn
Рис. 4.22. Диаграмма
состояния системы
олово—цинк
I
Zn(TB,xZn= 1)
II
-► гп(ж,х£п= 0,5965)
III
гп(ж,*7 = 1)
Изменение энергии Гиббса для процесса I равно нулю.
Процесс II соответствует плавлению чистого цинка при Т < 7^.
В этом случае AG* 0 и
ДС™ = АН% - ГД ^ = 7£Д Jg - TAS% =
79X0
= AS% (Г2™ - Т)=^£^(692,6 - Т) = 10,51(692,6 - Т).
692,6
При температуре Т= 621,3 К AG™ = 749,4 кДж • моль-1. Процесс
111 соответствует переходу цинка из чистого жидкого в сплав соот-
110
ветствующего состава. В этом случае изменение энергии Гиббса
соответствует парциальной молярной энергии цинка в жидком сплаве
AGZn. При этом AG1 = АС11 + АС,П.
Таким образом,
AG^ =RT\naZn = -AG™ =-10,51(692,6-Г).
При температуре Т= 621,3 К A(7Zn =-749,4 кДж-моль-1.
Результаты расчетов для ряда температур представлены в табл. 4.5.
Таблица 4.5
Температуры начала кристаллизации сплавов цинка с оловом
и термодинамические характеристики цинка в жидких сплавах
системы Sn—Zn
*Zn
1,000
0,9819
0,9315
0,8121
0,7220
0,5965
0,4935
0,4062
0,3079
0,2293
0,1913
Г, К
692,6
683,5
666,9
648,5
638,1
621,3
608,7
585,6
554,2
519,8
501,9
A(7Zn, Дж • моль-1
0,0
-95,6
-270,1
-463,5
-572,8
-749,4
-881,8
-1124,6
-1154,6
-1816,1
-2004,3
*Zn
1,0
0,983
0,952
0,918
0,898
0,865
0,840
0,794
0,729
0,657
0,619
Yzn
1,0
1,001
1,022
1,130
1,243
1,450
1,702
1,954
2,369
2,865
3,204
Ветвь кривой ликвидуса, где кристаллизуется олово, как видно
из рис. 4.22, невелика, однако и в этом случае вдоль ветви
термодинамические характеристики олова в жидких сплавах могут быть
рассчитаны аналогичным образом.
Система Cd—Zn (рис. 4.23) также относится к числу эвтектических,
но в этом случае следует учитывать наличие областей твердых растворов.
Так, например, при 619 К жидкий сплав находится в равновесии уже
не с чистым цинком, а с твердым раствором на основе цинка. Составы
равновесных фаз при этой температуре х^ =0,762, х™п =0,984.
Общая схема рассматриваемых процессов несколько
усложняется:
111
Zii(tb, x7n = 1) Л Zn(TB, x7n = 0,984);
III
IV,
II
Zn(x, jcZn = 1) -> 7п(ж, JCZn = 0,762).
г,к
450
Cd
0,2 0,4 0,6 0,8 1,0
jc_ -> Zn
Рис. 4.23. Диафамма состояния
системы кадмий—цинк
Изменение энергии Гиббса процесса I соответствует переходу
цинка из чистого твердого состояния в твердый раствор, т. е.
представляет собой парциальную молярную энергию цинка в твердом
растворе, А (7^ (стандартное состояние — Zn твердый, xZn = 1). В общем
случае AG^ =RT\na™n =RTh\y™nx™n. Поскольку область твердого
раствора невелика, без существенной погрешности можно принять
у5,„=1иЛС7£=ЛГ1пл&.
Изменение энергии Гиббса процесса II равно нулю, так как вдоль
всей ветви ликвидуса \х™п = ц£п. Изменение энергии Гиббса процесса
III соответствует плавлению цинка при Т < Т£* ,и его рассчитывают,
как в предыдущем примере:
AG%=U5%(T%-T) = 10,5l№,6-T).
При температуре 619 К AG^JJ = 773,5 Дж/моль.
Изменение энергии Гиббса процесса IV соответствует
парциальной молярной энергии Гиббса цинка в жидком сплаве AG£n
(стандартное состояние — Zn жидкий, xZn = 1).
112
В рассмотренном цикле AG{ + А<7„ = AG[U + A(7|V, или
AG£n =RThu& = -AG™ + AG™ = -10,51(692,6-Г ) + /гПпл£.
Результаты расчетов представлены в табл. 4.6.
Таблица 4.6
Состав жидкой и твердой фаз и термодинамические характеристики цинка
в сплавах системы Cd-Zn
Г, К
692,6
655,9
639,2
618,3
599,9
586,5
581,4
574,3
560,6
555,0
542,2
xZn
1,0
0,9128
0,8532
0,7586
0,6392
0,5312
0,5010
0,4491
0,3629
0,3388
0,2894
ТВ
XZn
1,0
0,9890
0,9855
0,9844
0,9845
0,9850
0,9852
0,9855
0,9862
0,9865
0,9873
AG^, Дж- моль-1
0,0
-58,6
-75,3
-79,5
-79,5
-75,3
-71,1
-71,1
-66,9
-62,8
-58,6
АС^, Дж • моль '
0,0
-443,5
-636,0
-857,7
-1054,0
-1188
-1238
-1314
-1452
-1506
-1636
Существенный недостаток метода расчета термодинамических
свойств на основании фазовой д и афам мы — невозможность получить
данные для одной температуры, так как все расчеты ведутся вдоль
линии ликвидуса. В связи с этим часто пользуются приближением
регулярного раствора, согласно которому ЛЛпу. = АН. и не зависит
от температуры.
Напомним, что для регулярного раствора AH=x{x2Q., гдеО —
параметр, рассчитываемый на основании экспериментальных данных.
Параметр П может характеризовать либо жидкую Q*, либо твердую
QTB фазу.
В общем случае (/ = 1,2)
АН. = (1 - x)2Q; AG = (1 - x)2Q + Л71шс.;
1пу,= (1--л,)2(П/Д7).
По аналогии с уравнением (4.21) для случая, когда жидкая фаза
представляет собой регулярный раствор и кристаллизуется чистый
компонент, можно записать
ИЗ
АН!
ц°дв + Д//,пл -T^-+RT\nXi+(\-х,уаж = ц°*тв; (4.41)
Пж =
AH™(l-T /Q+flrins,.
(1-*,Г
Более сложным оказывается случай, если в системе наблюдается
неограниченная взаимная растворимость и обе фазы — жидкую и
твердую — рассматривают в приближении регулярных растворов.
Уравнение (4.21) принимает вид
,ДЯ,ПЛ
ц?-тв + АН"Л -T^—+RTlnx* +(\-x?f&K
= n°TB + RT\nxJB+(l-xJB)2nn.
(4.42)
Отсюда получаем
х- 1
RT
AH"
rpWl ПН
rpTL
1/.ж\2|-чж
,тв\2у-\ТВ
+(1-х/жГОж-(1-я/вГО'
4.8. Двухфазные равновесия жидкость-жидкость
в двухкомпонентных системах. Закон распределения
4.8.1. Концентрационная зависимость термодинамических
функций в системах с областью расслаивания
Наиболее распространенными фазовыми диаграммами систем
с двумя жидкими фазами, находящимися в равновесии, в
металлургической практике являются диаграммы состояния систем с моно-
тектическим равновесием. Среди простейших систем с областью
расслаивания можно выделить системы с нормальной и "вырожденной
монотектикой" (рис. 4.24). Последний термин аналогичен термину
"вырожденная эвтектика", когда эвтектическая точка совпадает с
точкой плавления чистого компонента. Иными словами, термин
"вырожденная монотектика" обозначает, что в пределах точности
эксперимента не удается зафиксировать различие между
температурой плавления компонента и температурой монотектики.
Рассмотрим концентрационную зависимость парциальных
термодинамических функций при наличии области гетерогенности в
жидких системах (рис. 4.25). На границах областей гомогенности
М " = ц Y и |д Y = |д р. Соответственно р,а = ц Y = ц.р.
114
б)
т
Tm
А А
/
/
/
I
\
\
\
1
в
Рис. 4.24. Диаграмма состояния системы с "нормальной"
(а) и "вырожденной" (б) монотектикой
Рис. 4.25. Границы фазовых областей
на диаграмме температура-состав
Аналогичный вид имеют зависимости и для компонента 2.
Химические потенциалы компонента равны между собой в
сосуществующих фазах, внутри области расслаивания величина jjjY (Am/,
Д^/) постоянна. Сложнее обстоит вопрос с парциальной молярной
энтропией компонента. Она необязательно одинакова в
сосуществующих фазах для данного компонента. Это, естественно, относится и
к парциальной молярной энтальпии смешения.
115
Величина A|i,Y на границе области гомогенности зависит не только
от температуры, но и от состава: Aji,Y = Дх, 7). Поэтому
дифференцирование этой зависимости следует выполнить как
дифференцирование сложной функции вдоль границы гомогенности фазы а.
Температура является независимой переменной, а граничный состав
фазы х = х в общем случае зависит от температуры*. Тогда
dAfi(x9T)
dT
дТ
дх
Л
dT
(4.43)
УР>х=хч
Частная производная от относительного химического
потенциала компонента 1 по температуре представляет собой парциальную
молярную энтропию компонента 1 в фазе а на границе области
гомогенности, взятую с обратным знаком:
дА\х{
дТ
= -AS?
J
В то же время внутри двухфазной области величина
зависит от состава:
ЭАц[
дТ
(4.44)
не
(
jp
дТ
= -AS7.
Jp
На основании уравнений (4.43)—(4.45) можно записать
AS^AS^-
(
dAtf)
дх
аГ-гф
rfx,
Ф
dT
(4.45)
(4.46)
Таким образом, парциальная молярная энтропия компонента 1
в двухфазной области отличается от парциальной молярной
энтропии того же компонента в фазе а на границе ее гомогенности на
слагаемое
fez
I dT
Это слагаемое представляет собой скачок ASX на фазовой границе.
В двухфазной области AS, сохраняет постоянное значение.
Соответственно, парциальная молярная энтальпия смешения претерпевает
скачок, равный
* Более подробно см.: Воронин Г.Ф. // Современные проблемы
физической химии. Т. 9. М.: Изд-во Моск. ун-та, 1976. С. 29—48.
116
Если
dx,
Эц^
Эх
JpJ=Tm
dx,
гр
dT
гр
dT
'P^-r*
:0, т. е. граница двухфазной области вертикальна,
то скачки парциальных молярных энтропии и энтальпии на фазовой
границе равны нулю: Л5,а = AS{\ AHf = A//,Y.
Все изложенное выше, естественно, справедливо не только для
систем с расслаиванием, но для любых границ раздела между
гомогенной и гетерогенной областями.
Постоянство химического потенциала в двухфазной области
ведет и к постоянству активности компонентов. Изотермы активности
одного из компонентов для различных систем с областью
расслаивания приведены на рис. 4.26.
Система всегда устойчива по отношению к разделению на две
фазы, если она идеальна, и неустойчива в тех случаях, когда
наблюдаются положительные отклонения от идеального поведения (у. > 1).
Простейшим образом связь между характером отклонений от
идеального поведения и величинами энергий межатомных взаимодействий
в системе А-В отражена еще в уравнении Ван-Лаара:
Pa=Pa*a™V
<^>2#
(4.47)
Здесь £— постоянная Больцмана; Ае — "энергия взаимообмена",
учитывающая различие в характере взаимодействия однородных
(связи А-А, В-В) и разнородных (А—В) атомов в пределах первой
координационной сферы. Распространение взаимодействия только
Рис. 4.26. Изотермы активности первого из компонентов в системах
с областью расслаивания: цинк—свинец (926 К), цинк-висмут (693 К),
калий—кадмий (848 К)
117
на ближайших соседей основано на представлении об отсутствии в
жидких фазах дальнего порядка.
Физический смысл Аб поясняет рис. 4.27. На рис. 4.27,я
изображена разнородная конфигурация, когда при условно выбранном
координационном числе 6 каждый из двух разнородных атомов
окружен разнородными атомами. На рис. 4.27,6 представлена
однородная конфигурация.
0 ••• %!•
о°о о0°о
°о° °о°
I конфигурация п конфигурация
(разнородная) (однородная)
Рис. 4.27. Схемы разнородной (а) и однородной (б)
конфигураций в расплаве
Энергии взаимодействия двух изображенных систем различны,
обозначим их 6j (конфигурация а) и е„ (конфигурация б). По Ван-
Лаару, "энергия взаимообмена" составляет половину разности
энергий этих конфигураций:
Де = -(£„-£,).
При Де = 0 силы взаимодействия однородных и разнородных пар
равны, уравнение (4.47) переходит в закон Рауля. При Дг > О
предпочтительна однородная конфигурация, наблюдаются положительные
отклонения от закона Рауля, а при Аг < О — отрицательные.
Соотношение (4.47) не следует рассматривать как
количественную теоретическую закономерность, так как вычислить величину As
только на основании свойств чистых компонентов очень сложно. Тем
не менее это уравнение удобно для анализа и понимания характера
отклонений от закона Рауля. Перепишем уравнение (4.47) в виде
PjPl = ал = *„ехР[( 1 - *|>2РЬ (4-48)
118
где р = Аг/кТ. Вычисление значений
рА/рл° при различных значениях р ведет
к семейству кривых, представленных
на рис. 4.28. При р = 0 раствор следует
рассматривать как идеальный. При
Р > 0 наблюдаются положительные
отклонения, причем при Р = 2
появляется практически горизонтальный
участок, свидетельствующий о
близости области расслоения. Появление
максимума на теоретической кривой
при Р = 3 указывает на область
расслоения, протяженность которой
на рис. 4.28 обозначена пунктирной
линией. Таким образом, нарастание
положительных отклонений от
идеального поведения ведет к области
несмешиваемости в системе. При р < О
наблюдаются отрицательные
отклонения в системе, которые нарастают
по мере увеличения отрицательных
значений р, т. е. с увеличением энергии взаимодействия
разнородных атомов.
4.8.2. Коэффициент распределения
Закон, описывающий распределение какого-либо вещества в двух
несмешивающихся растворителях, был сформулирован Нернстом
(1891 г.)*. "Если вещество распределяется в двух растворителях, в
которых оно образует идеальные растворы, то отношение
соответствующих молярных долей при данных внешних условиях
постоянно". Пусть компоненты А и В образуют систему из двух фаз (фазы I
и II). Если добавим третий компонент (С), который распределится
между фазами, то при равновесии, согласно закону Нернста
* Впервые закон был установлен экспериментально Бертло и Юнгфлей-
шем (1872 г.), термодинамическое обоснование дано Нернстом (1891 г.).
В литературе он чаще называется законом распределения Нернста.
Рис. 4.28. Связь между
характером отклонений от
идеального поведения и параметром р
(цифры у кривых). Пунктир —
область расслаивания
119
4=*- <4-49>
хс
гдехс! ихсп — молярные доли компонента С в фазах I и II; Кх—
коэффициент (константа) распределения. Отступления от закона Нернста
бывают в тех случаях, когда растворенное вещество в одном или в
обоих растворителях претерпевает зависящие от концентрации
химические изменения, подвергается ассоциации или диссоциации.
При равновесии цс,= цсп или
(\хсУ + RTlnacl = (мсв)" + RT\nac". (4.50)
Поскольку, согласно Нернсту, речь идет о достаточно
разбавленных растворах компонента С, то можно принять, что для каждой из
фаз справедлив закон Генри:
рс} = Кг\} ил и .£ = Кг\}9
Здесь/^ и/сЯ — фугитивность (летучесть) компонента С в фазах
I и II. Поскольку при равновесии между жидкими слоями
парциальное давление (или летучесть) компонента С над обеими фазами
одинаково, получаем
х1г *»
^f = -^T = const = Кх(Т,Р).
Область концентраций компонента С, в которой коэффициент
распределения Кх сохраняет постоянное значение при заданной
температуре, связана с областями выполнения закона Генри (с. 51).
При симбатном изменении констант Генри А^,1 и Кг11 с изменением
хс величина Кх может сохранять постоянство в несколько большем
интервале концентраций.
Закон распределения Нернста непосредственно следует из
условий равновесия между фазами (4.46). Если принять симметричную
нормировку активности для компонента С, то {\i°c\ =(nc) и
соответственно ас! = асУ. Отсюда следует
х1 V11
*с Ус
Если концентрации компонента С малы (хс! и хс}1 -» 0), то
120
хс (Ус)
Таким образом, коэффициент распределения связан с
предельными значениями коэффициентов активности растворяемого
компонента Св жидких несмешивающихся растворителях. Уравнение (4.52)
подразумевает, что при рассматриваемых концентрациях компонента
С выполняется закон Генри в обеих фазах и зависимости ас1 =Л*С1) и
асУ =Ахс1) линейны. Из числа металлургических систем примером
может служить распределение серебра между цинком и свинцом.
При содержании серебра до хА = 0,05 для каждой из жидких фаз
выполняется закон Генри.
С помощью закона распределения можно определить активность
компонента в жидком сплаве. Так, например, серебро и железо
практически не смешиваются между собой в расплавленном состоянии.
Экспериментально установлено, что коэффициент распределения
меди между этими металлами при температуре 1873 К равен
jc(Fe)
xCu
Расчет из фазовой диаграммы в приближении регулярного
раствора показал, что при 1873 К коэффициент активности меди в сплавах
с серебром Y£u(Ag) =2,14 (стандартное состояние — чистая жидкая
медь). Отсюда можно рассчитать активность меди в железе из данных
по распределению:
*Cu(Fe) =Ycu,Ag)^ =(2,l4/0,234)xg = 9,12xg.
Аналогичным методом на основании изучения распределения
алюминия между несмешивающимися жидкими фазами серебро-
железо, серебро-никель, серебро-кобальт были определены при
1873 К коэффициенты активности компонентов в жидких сплавах
систем Al-Fe, Al-Ni, Al-Co.
Если для растворенного вещества в качестве стандартного
состояния выбрать бесконечно разбавленный раствор, то(цс) *(м-с) и из
уравнения (4.50) следует
4 = ехр[^)"-^>'1. (4.53)
121
При малых концентрациях растворенного компонента, когда * i
и хс!] стремятся к нулю,
ас _хс _ г°° ,А СЛЧ
ас хс
Понятия "закон распределения" и "коэффициентраспределения"
часто используются для характеристики гетерогенных равновесий
других типов, не относящихся непосредственно к закону Нернста.
Речь идет о распределении /-го компонента (примеси) между
находящимися в равновесии твердой и жидкой фазами основного вещества,
между жидкой и парообразной фазами.
На распределении вещества между двумя несмешивающимися
растворителями основан метод экстракции. Экстракцией называется
извлечение растворенного вещества из раствора при помощи другого
растворителя (экстрагента), практически не смешивающегося с
первым. Для достаточно полного извлечения растворенного вещества
экстракцию проводят несколько раз.
На распределении металлов-примесей между жидкой и твердой
фазами основаны кристаллизационные методы очистки металлов.
В процессе расплавления металла и его последующей кристаллизации
происходит перераспределение металлов-примесей в соответствии с
их коэффициентами распределения К.. Коэффициент распределения
компонента — основной параметр кристаллизации,
представляющий собой отношение его концентраций в твердой и жидкой фазах
{ С л
Сжу
, имеющих общую границу раздела. Различают
равновесный коэффициент распределения К0, определяемый из диаграммы
состояния или из экспериментальных данных в области малых
содержаний компонента-примеси при нулевой скорости
перемещения фронта кристаллизации, и эффективный коэффициент
распределения /Г, значение которого характеризует
распределение примеси в реальной системе и зависит от скорости
перемещения фронта кристаллизации, условий массопереноса и других
факторов*.
* Более подробно см.: Козин Л.Ф., Волков СВ. Химия и технология
высокочистых металлов и металлоидов. Т. 2: Физико-химические и
кристаллизационные методы глубокой очистки. Киев: Наук, думка, 2003. 351 с.
122
4.9. Термодинамическое описание равновесия
жидкость—пар в двухкомпонентных системах
Как уже ранее отмечалось (с. 82),
при постоянных давлении и
температуре зависимость энергии Гиббса
от состава для фазы переменного
состава, образованной компонентами,
смешивающимися между собой во всех
отношениях, выражается непрерывной
кривой, обращенной выпуклостью к
оси х. На рис. 4.29 изображены кривые
(7=Дх) для жидкой и паровой фаз при
температуре, которая выше температур
кипения обоих компонентов и их
растворов при данном давлении. При таких
условиях при всех составах устойчива
паровая фаза, кривая пара (п)
расположена ниже кривой жидкости (ж).
В условиях сосуществования фаз,
при более низких температурах
поверхности энергии Гиббса жидкой и парообразной фаз пересекаются.
На рис. 4.30 построено сечение поверхности G=f(x, 7) при
температуре, промежуточной между температурами кипения компонентов.
Кривые пересекаются, и общая касательная к ним определяет составы
равновесных жидкости и пара при данных условиях. Для наглядности
эти составы спроектированы на х-Г-диаграмму.
Рис. 4.29. Кривые зависимости
молярной энергии Гиббса от
состава для жидкости (ж) и
пара (п) в двухкомпонентной
системе
4.9.1. Законы Д. П. Коновалова
Законы Коновалова (1884 г.) устанавливают связи между
изменениями состава, давления и температуры в двойных системах
жидкость—пар, они лежат в основе теории перегонки и ректификации
бинарных смесей. Первый закон Коновалова формулируется так:
"Давление пара раствора возрастает (уменьшается) при увеличении
концентрации того компонента, содержание которого в паре больше
(меньше), чем в растворе; температура кипения раствора возрастает
(уменьшается) при увеличении концентрации того компонента,
123
содержание которого в паре меньше
(больше), чем в растворе". Возможны и
несколько иные формулировки первого
закона Коновалова. Так, например,
возможна формулировка: "Насыщенный
пар над жидкой двухкомпонентной
смесью всегда содержит по сравнению
с жидкостью больше того
компонента, добавление которого увеличивает
общее давление над смесью, т. е.
понижает температуру кипения смеси при
данном давлении".
Математически это можно записать
так:
еслих^ >хр
еслих1"<х],
то
то
(dp
dx]
l>
>0,
dp
\
dp
dxt
»>
<0,
_dp_
dx[
>0;
<0.
Рис. 4.30. Связь между
кривыми молярной энергии Гиббса
(а) и диаграммой равновесия
жидкость—пар (б)
На рис. 4.31 показана графическая
зависимость между составами жидкости
и пара при данной температуре.
Второй закон Коновалова
формулируется так: "Если давление и
температура сосуществования двух бинарных
фаз имеют экстремум (максимум или
минимум), то составы фаз одинаковы". Или: "Точки максимума или
минимума на кривых зависимости давления пара от состава
соответствуют смесям, при кипении которых состав пара и состав жидкости
оказываются одинаковыми". Такие смеси называются нераздельно
кипящими, или азеотропными. Математическая запись второго за-
а:
dp)
кона такова
если
dx\
= 0,
*i = V
Диаграммы Т—хи /?-хбинарных систем с "верхним" и "нижним"
азеотропами приведены на рис. 4.32.
124
Рис. 4.31. Иллюстрация первого закона Коновалова
Рис. 4.32. Диаграммы температура—состав и давление—состав
в двухкомпонентных системах с образованием "верхнего"
и "нижнего"азеотропов
125
Иногда формулируют и третий закон Коновалова: "В
изотермических (или изобарических) условиях составы раствора и пара изменя-
dx
ются симбатно, т. е. —- > 0. "
dx*
Законы Коновалова могут быть записаны как для изотермических,
так и для изобарических условий. Хотя законы были
сформулированы Д. П. Коноваловым на основании экспериментальных данных,
т. е. получены эмпирически, они имеют строгое термодинамическое
обоснование*.
4.9.2. Законы М.С. Вревского
Если законы Коновалова характеризуют изменение состояния
равновесных двухфазных систем жидкость—пар при изобарных или
изотермических условиях, то законы (правила) Вревского (1911 г.)
определяют закономерности влияния на фазовые равновесия
бинарных систем изменений температуры или давления при наложении
определенных условий на изменение составов равновесных фаз.
Первый закон Вревского формулируется так: "При повышении
температуры раствора заданного состава его пар обогащается тем
компонентом, парциальная молярная теплота испарения которого
больше". Записывается это так:
'л,- ^
>0 если АЯ,ИСП > АЯ2ИСП.
А
dT
Второй закон Вревского определяет смещение состава двойных
азеотропов при изменении внешних условий. Формулируется закон
так: "Если давление (температура) системы раствор-пар имеет
максимум (минимум), то при повышении температуры вазеотропной смеси
возрастает концентрация того компонента, парциальная молярная
теплота испарения которого больше; если давление (температура)
системы раствор-пар имеет минимум (максимум), то при
повышении температуры в азеотропной смеси возрастает концентрация
того компонента, парциальная молярная теплота испарения которого
меньше". Записать это можно так:
* Подробное термодинамическое обоснование законов Коновалова
содержится в монографии: Сторонкин А.В. Термодинамика гетерогенных
систем. Л.: Изд-во Ленингр. ун-та, 1967. Ч. 1—2. 446 с.
126
для азеотропа с максимумом р (минимум 7)
rdxA
-u ^ Л если АЯ.ИСП > А//"сп;
dT
V
>о,
У
аз
>о,
если А//,исп > АЯ2ИСП.
для азеотропа с минимумом р (максимум 7)
К
Законы Вревского, как и законы Коновалова, имеют строгое
термодинамическое обоснование.
Из первого и второго законов Вревского может быть получено
следствие (иногда его называют третьим законом Вревского) такого
содержания: "При изменении температуры (давления) раствора,
кривая давления пара которого имеет максимум, состав пара раствора и
состав азеотропной смеси изменяются в одном и том же направлении;
при изменении температуры (давления) раствора, кривая давления
пара которого имеет минимум, состав пара раствора и состав
азеотропной смеси изменяются в противоположных направлениях".
Еще раз подчеркнем, что в отличие от законов Коновалова законы
Вревского описывают изменения составов фаз систем при условии
постоянства состава одной из сосуществующих фаз.
4.9.3. Дистилляционные процессы
с участием металлов и металлоидов
Дистилляционные методы очистки и разделения веществ пригодны
для довольно широкого круга металлов (щелочные металлы, магний,
кальций, барий, ртуть, кадмий, цинк, свинец, сурьма, индий, галлий) и
металлоидов (сера, селен, теллур, мышьяк). Дистилляционные методы
относятся к числу древнейших методов очистки веществ, они включают
простую перегонку, дробную перегонку, ректификацию, ряд других
вариантов. Эффективность методов зависит от различия между давлением
пара разделяемых компонентов и сопутствующих им примесей. Кратко
рассмотрим некоторые общие положения и конкретные примеры.
Количественной характеристикой однократного акта испарения
в процессе разделения металлов является коэффициент разделения
а, называемый часто относительной летучестью:
ос =
'V
(4.55)
127
где индекс 2 относится к более летучему компоненту. Можно
сделать некоторые общие выводы о влиянии изменений температуры
и давления на величину а. Из приведенного выше уравнения для
бинарной системы следует:
</1п(л£/хГ)
dT
или
rflna
dT
Afff-Aff,"
RT2
АЩСП-АН?СП
RT2
(4.56)
Обычно компонент, имеющий в сравнении с другим меньшую
температуру кипения (большее давление пара), в чистом состоянии
обладает и меньшей теплотой испарения. Если это так, то на
основании уравнения (4.56) можно заключить, что при повышении
температуры (давления) относительная летучесть (А#2ИСП - АЯ,ИСП) < 0.
Эффект разделения компонентов 1 и 2 тем больше, чем больше
величина а. Для идеальных систем в изотермических условиях а не
зависит от состава и оказывается равным
л
ид
аад =-
А
(4.57)
Эту зависимость получаем из следующего простого рассмотрения:
Р{ = P[xv Pi= Ргхгв соответствии с законом Рауля. Общее давление,
согласно закону Дальтона равно сумме парциальных давлений:
Р = Р1+Р2=Р°Х1+Р2ХГ
В то же время для паровой фазы справедливы соотношения
р{=рххпкр2 = рх2п.
Следовательно,
рх2» = р2°х2; (4.58)
рх^рХ (4.59)
Разделив одно уравнение на другое, получаем
xi _ Рг = авд
А
(4.60)
В качестве примера на рис. 4.33 показан ход кривых lg/^e =f(T)
для свинца и сопутствующих ему металлов-примесей: висмута,
сурьмы, таллия, индия и серебра, а также для кадмия, цинка и железа.
128
Видно, что давление паров висмута и сурьмы мало отличается от р°
свинца. Обычно дистилляцию свинца проводят при температурах
1300-1400 К. Для предварительной оценки разделительной
способности дистилляционных процессов ниже приведены значения аид в
соответствии с уравнением (4.57):
Металл
РМе
ат
РЬ
265,3
888,4
—
Bi
354,7
1205,0
1,34
1,35
Sb
465,4
1526,8
1,75
1,72
Tl
1957,8
5945,1
7,38
6,69
In
9,25
32,6
0,035
0,037
Ag
2,16
7,065
0,008
0,008
Примечание. В числителе —данные при 1300 К, в знаменателе — при
1400 К, р°ш -в Па.
Как видно, теоретические значения аид в интервале 1300-1400 К
почти не зависят от температуры. Судя по величинам аид,
эффективность очистки свинца дистилляцией от висмута, сурьмы, таллия
должна быть довольна низка.
Если идеальной является только паровая фаза, а в жидкой фазе
наблюдаются отклонения от
идеального поведения, что характерно
для металлических систем,
уравнения (4.58) и (4.59) принимают вид
рх" = р2°х2у2,
рх.н = р.х.уг
Отсюда
*2/*Г_/>2У2_а<
(4.61)
В случае разбавленных
растворов уравнению (4.61) может быть
придан другой вид. В
термодинамике жидких сплавов
преимущественно распространен так
называемый симметричный способ
нормирования термодинамических
функций, когда за стандартное со-
т,к
Рис. 4.33. Зависимость давления
паров металлов, сопутствующих
свинцу в рудных материалах, от
температуры
129
стояние принимается чистый компонент при определенных давлении и
температуре. При таком способе нормирования компоненты раствора
равноправны, разделить их на растворитель и растворенное вещество
можно лишь условно. В случае сильно разбавленных растворов при
симметричном нормировании коэффициент активности
растворителя уj оказывается близок к единице, а коэффициент активности
растворенного вещества у2 может колебаться в широких пределах,
составляющих несколько порядков.
Как уже отмечалось (с. 57) коэффициенты активности
растворенного вещества при высоком разбавлении (х{ -> 1, х2 -> 0) представляют
собой предельные величины (у2°°). С учетом этого уравнению (4.61)
можно придать вид
«=И-к- <4-б2>
{pi )
Таким образом, значения предельного коэффициента активности
компонента-примеси практически достаточно для расчета
коэффициента разделения (при известных величинах р° и р2° для данной
температуры). При наличии ряда примесей, если они не
взаимодействуют между собой, для каждого примесного компонента можно
считать справедливым уравнение типа (4.62). В табл. 4.7 приведены
значения у^е для ряда металлов, растворенных в жидком свинце. Для
примесей, интенсивно взаимодействующих со свинцом, образующих
с ним системы с интерметаллическими соединениями (калий, натрий,
золото, платина), у^е << 1. Для примесей, образующих со свинцом
системы с расслоением, Уме >> 1.
В табл. 4.8 приведены средние значения результатов анализа
очищенного вакуумной дистилляцией свинца при температуре
1313-1353 К и остаточном давлении 13,3-66,5 Па*.
Для глубокого удаления примесей из свинца необходимо
применять принципы реакционной вакуумной дистилляции — снижение
активности металлов-примесей с помощью введения компонентов,
называемых в вакуумной металлургии "активными добавками".
Другой путь — многостадийные процессы вакуумной дистилляции,
в которых операции испарения и конденсации отдельных фракций
непрерывно повторяются.
* Более подробно см. в монографии: Козин Л.Ф., Морачевский А.Г. Физико-
химия и металлургия высокочистого свинца. М.: Металлургия, 1991. 224 с.
130
Таблица 4.7
Предельные значения коэффициентов активности у"гМе
для ряда металлов-примесей, растворенных в жидком свинце
Примесь
Ag
А1
Г Аи
Bi
Cd
Си
Hg
I_n_ _
Г, К
1273
1200
1200
700
773
1473
600
673
Уме
2,031
22,07
0,314
0,490
3,376
4,872
1,13
1,349
Примесь
К
Mg
Na
Pt
Sb
Sn
Tl
| Zn
г,к
848
973
700
1273
905
1050
773
923
YMe
0,0065
0,120
0,0032
0,043
0,779
6,816
0,796
7,940
Таблица 4.8
Содержание примесей с в исходном и очищенном
вакуумной дистилляцией свинце
Примесь
Ag
Zn
Mg
Си
Fe
Sb
Cd
L_ A1
C- 105,%
(по массе), в свинце
исходном
5-30
5-10
3-10
6-20
5
3-10
4-10
2-5
очищенном
0,01-0,1
0,1-1,0
0,1-0,5
0,02-0,1
1
1
0,01-0,1
0,1-0,2
Примесь
As
Са
In
Hg
Na
Sn
Tl
Bi
C-105,%
(по массе), в свинце
исходном
8-10
8-10
3-10
5-10
8-15
5-10
1-8
4-30
очищенном
1
1-2
0,01-0,1
0,1
1-3
0,1
0,1-1
2-16
Для удаления из металлического индия летучих примесей —
цинка, кадмия и ртути — применяют вакуумную дистилляцию или,
точнее, вакуумный перефев. Расплавленный металл при температуре
873-1073 К выдерживается в течение нескольких часов при давлении
до 1 Па. Дистилляцией можно очистить индий и от менее летучих
131
по сравнению с ним металлов, если производить перегонку индия.
Для этого после отгонки летучих фракций температуру повышают
примерно до 1273 К и при остаточном давлении 10~2— 10~3 Па
перегоняют основную часть металла. Нелетучие примеси — железо, медь,
никель и др. — концентрируются в кубовом остатке.
Одним из самых эффективных методов разделения близких по
свойствам щелочных металлов (например, рубидия и цезия, калия
и рубидия) является дистилляция. Естественно, что при этом
происходит очистка щелочных металлов также от значительно менее
летучих примесей других металлов (железа, никеля, хрома и т. д.).
Для многих бинарных жидких сплавов кривые зависимости
общего давления и температуры кипения от состава фаз имеют сходный вид
с соответствующими кривыми для систем с органическими или
другими низкокипящими компонентами. На рис. 4.34 приведены
кривые фазовых равновесий жидкость-пар для жидких сплавов
системы Mg-Zn.
Mg 0,2 0,4 0,6 0,8 zn
vZn
Mg 0,2 0,4 0,6 0,8 Zn
Рис. 4.34. Зависимости температуры кипения
0? = 40ммрт. ст.) (а) и общего давления пара (Г=960 К)
(б) от состава жидкой и паровой фаз в системе магний—цинк
132
4.10. Корректная и некорректная постановка задач
при расчетах фазовых равновесий
Расчеты равновесий по известным термодинамическим свойствам
фаз — одно из основных практических применений химической
термодинамики. Такие расчеты позволяют, в частности, строить
необходимые для науки и техники диаграммы фазовых состояний
многокомпонентных систем.
Большой интерес представляет и решение обратных задач —
извлечение из фазовых диаграмм любых, даже ограниченных сведений
о неизвестных термодинамических свойствах веществ. В отличие
от расчетов равновесных составов обратные задачи часто не имеют
единственного решения. В настоящее время принято делить все
множество задач расчета равновесий на термодинамически корректные
и термодинамически некорректные. Подробно это рассмотрено в
работах Г.Ф. Воронина (МГУ)*. Задача считается поставленной
корректно, если при имеющихся исходных данных ее решение
существует, является единственным и устойчиво по отношению к исходным
данным. Последнее условие следует понимать так, что небольшое
изменение исходных данных ведет к адекватному изменению
результатов расчета.
Для решения некорректно поставленной задачи необходимо
располагать дополнительной информацией о поведении системы.
Наиболее часто приходится использовать различные модели и вводить
их в исходные условия.
Ранее мы отмечали, что для описания двухфазного равновесия
в конденсированной системе достаточно располагать зависимостями
энергии Гиббса от состава и температуры для каждой из фаз. Это
эквивалентно тому, что мы должны знать для каждой фазы зависимость
энтальпии и энтропии образования фазы от состава. Рассмотрим
двухфазное равновесие в бинарной системе. Обозначим изменения
энтальпии и энтропии при образовании фаз: Л//1, ЛЯ" и Д51, AS11,
Примем, что эти величины не зависят от температуры. Составы
* См. например: Воронин Г.Ф. Некоторые термодинамические расчеты
фазовых равновесий //Термодинамика и материаловедение
полупроводников. М.: Металлургия, 1992. С. 121-152.
133
находящихся в равновесии фаз обозначим х1 и х11. Наиболее часто
приходится решать следующие задачи:
Известно Необходимо найти
Д/Л Atf\ AS1, Д511 xl,x" (1)
Д/Д Д5\ *', х" A/^Atf' (2)
Д/ЛДД11,*1,*" AS1, AS" (3)
Д//\ AS', AS1', x' A/^U" (4)
Д/Л Д//11, Л^\ *' Atf!, jc" (5)
В принципе возможны 15 различных вариантов выбора
рассчитываемых свойств, но указанные пять представляют наибольший
практический интерес. При расчетах предполагается, что известны
во всех случаях энтальпии и температуры плавления чистых
компонентов. Зависимость интегральных термодинамических функций от
состава выражается аналитически, причем параметры
соответствующих математических уравнений определены в пределах, допустимых
экспериментальными погрешностями.
Вариант (1) соответствует расчету диаграммы состояния на
основании сведений о термодинамических свойствах сосуществующих
фаз. В частных случаях это ведет к расчетам с помощью уравнений
(4.32)-(4.34). Вариант (2) позволяет находить неизвестные
термодинамические свойства фазы II по известным свойствам фазы I и
диаграмме состояния. Фазами I и II могут быть жидкий и твердый
растворы или жидкий сплав и фаза постоянного состава. Вариант (3)
характерен тем, что все исходные данные получаются из
калориметрического эксперимента. Зная величины энтальпий образования и
составы сосуществующих фаз, можно получить полное
термодинамическое описание системы. Расчеты по схемам (4) и (5) наиболее
целесообразно использовать для уточнения кривой солидуса.
В основе расчетов для всех вариантов лежат общие
термодинамические соотношения, базирующиеся на равенстве химических
потенциалов компонентов вдоль фазовых границ. Детали расчетов
рассмотрены в специальной литературе (работы Г.Ф. Воронина,
Пелтона, В.М. Глазова и Л.М. Павловой).
Если для интересующего нас варианта известны все указанные
исходные данные, то расчет является корректным. Задачу можно
решить однозначно, не прибегая к каким-либо термодинамическим
134
моделям (идеальный, регулярный растворы и пр.). Это — главный
признак корректной постановки задачи.
При расчете границы твердых растворов в системе Ga—Sn мы
принимали, что твердая фаза представляет собой идеальный раствор.
В наборе исходных данных (вариант (2)) принимали, что А511 = Д5*д.
В этом случае постановка задачи не является корректной.
Необходимо всегда иметь в виду, что свойства чистых
компонентов, свойства фаз в сплаве и координаты границ фазовых равновесий
связаны между собой термодинамическими соотношениями и в то
же время могут быть получены с помощью независимых
экспериментальных измерений. Это открывает широкие возможности для
рационального исследования систем, для анализа результатов с целью
определения согласованности.
При использовании данных о термодинамических свойствах фаз
при расчетах фазовых равновесий важным является вопрос о степени
надежности экспериментального определения тех или иных величин.
Для практики термодинамических расчетов нужны достоверные и
согласованные значения термодинамических функций, и выбор их
для многих бинарных металлических систем представляет сложную
задачу. Процедура оптимизации обычно базируется на статистической
обработке имеющихся результатов исследований и "отбраковке"
выпадающих величин. В последнее время в процессе оптимизации
учитывается фазовая диаграмма системы. Обязательным элементом
процедуры оптимизации должна быть предварительная экспертная
оценка термодинамических функций, в основе которой должны
лежать законы химической термодинамики, зависимости, надежно
установленные для более или менее широких классов систем и фаз,
критическая оценка надежности использованного
экспериментального метода и квалификации исследователей. Такая экспертиза может
быть полезна и при оценке степени достоверности имеющихся для
некоторых систем единичных результатов термодинамических
исследований. Конкретные примеры экспертных оценок результатов
исследований термодинамических свойств бинарных систем на основе
переходных металлов содержатся в работе Г.М. Лукашенко*.
* Лукашенко Г.М. Об экспертной оценке термодинамических данных //
Стабильные и метастабильные фазы в материалах / Ин-т проблем
материаловедения. Киев, 1987. С. 10-29.
135
4.11. Фазовые превращения первого и второго рода
: V. Графически это означает, что каждая фаза
Термодинамическая трактовка фазовых превращений была
впервые предложена Эренфестом (1933 г.). Как уже отмечалось
(раздел 4.2), каждая фаза может быть описана функцией G=flp, 7). При этом
характеризуется своей поверхностью энергии Гиббса. В состоянии
равновесия обеих фаз Gx = Gu поверхности I и II пересекаются по
линии Glu, Согласно Эренфесту, порядок фазового превращения
определяется порядком тех производных энергии Гиббса, которые
испытывают в точке перехода конечное изменение.
Фазовые переходы первого рода характеризуются наличием
скрытой теплоты перехода (плавления, сублимации, полиморфного
превращения), равной TAS, и конечной разностью объемов V11 — V1.
Иными словами, прерывны первые производные энергии Гиббса.
К фазовым превращениям второго рода относятся такие, при
которых G и его первые производные по температуре и давлению
непрерывны, а вторые частные производные претерпевают разрыв.
Это означает, что в точке превращения
В то же время теплоемкость
с,=-т
ьт
d2G
дТ
,T.e.Sl = Su.
ЪТ2
/р
испытывает скачок,
/р
равный Д.С = С" — С. Скрытой теплоты перехода не
наблюдается.
При переходе второго рода молярные объемы фаз равны:
ГдС^
Эр
= Г =
'эсп ^
Л
др
= Vй, а коэффициенты изотермического сжа-
/г
тия р и объемного расширения а прерывны:
р-1
v
^2g^
др
а = —
V
Л
d2G ^
V{dp
л
дрдТ
136
Описанные в предыдущих разделах превращения
кристаллическая фаза — жидкость, кристаллическая фаза — пар, жидкость—пар,
кристаллическая фаза (I) — кристаллическая фаза (II) относятся к
фазовым переходам первого рода. К фазовым переходам второго
рода относятся переходы, связанные с изменением симметрии
кристаллической решетки, превращение ферромагнитных веществ в
парамагнитное состояние, переход в сверхпроводящее состояние в
отсутствие магнитного поля.
Итак: при фазовых переходах первого рода скачкообразно
изменяются первые производные от энергии Гиббса (химического
потенциала): Sи V. При фазовом переходе второго рода первые
производные от энергии Гиббса (химического потенциала) непрерывны,
претерпевают разрыв вторые производные: С, а, р.
Глава 5
ТЕРМОДИНАМИЧЕСКОЕ ОПИСАНИЕ
ХИМИЧЕСКИХ ПРОЦЕССОВ
5.1. Уравнение изотермы химической реакции
и константа равновесия
Если в системе протекает химическая реакция, то в результате
превращения одних веществ в другие происходит изменение всех
термодинамических функций состояния. Для экстенсивной функции
приращение будет равно разности между ее значением в конечном
и в начальном состояниях системы. Приращение энтальпии в ходе
химической реакции рассмотрено в главе 2 (с. 21) при обсуждении
теплового эффекта реакций. В данном разделе рассмотрим изменение
энергии Гиббса по мере протекания химической реакции. Как уже
отмечалось в разделе 2.8 (с. 30), если в закрытой системе
самопроизвольно протекает химическая реакция при постоянных давлении
и температуре, причем не совершается полезной работы, величина
AG< 0; если протекает обратный процесс, то AG> 0; если система
находится в равновесии по отношению к данной реакции, то А (7 = 0.
Рассмотрим изменение количеств реагирующих веществ в
процессе реакции
vyi+Vjfl-^L+v^A/, (5.1)
где А, В, L, М— участвующие в реакции вещества; vA, vB, vrvM —
соответствующие им стехиометрические коэффициенты. Пусть в
начальный момент реакции были только исходные вещества в количествах
138
пл° и пп° молей. В результате реакции образовалось nL и пм молей
продуктов реакции и осталось непрореагировавшими пл и пв молей
компонентов Aw В. Очевидно, что изменение числа молей каждого
из участников реакции пропорционально его стехиометрическому
коэффициенту v.. Обозначим коэффициент пропорциональности £
(этот параметр имеет несколько названий: химическая переменная,
степень полноты реакции, пробег реакции). Величина £ одинакова
для всех участников реакции и всегда положительна. Для бесконечно
малого пробега реакции в общем виде можно записать
d5 = — . (5-2>
В соответствии с уравнением (3.3) при постоянных давлении и
температуре
/'
Подставив в это уравнение значение dn. из зависимости (5.2),
получаем
rf(7=Iv|u^. (5.3)
Величина £, представляющая собой по существу отношение
количества прореагировавшего вещества к его стехиометрическому
коэффициенту, по ходу реакции возрастает от 0 до 1. Проинтегрируем
в этих пределах уравнение (5.3), приняв для простоты постоянными
значения химических потенциалов (ц = const):
g2 i
Jrf^JSv^; AG = Xv/n/, (5.4)
G, 0 /
где G2 и G] — величины энергии Гиббса, соответствующие
конечному и начальному состоянию системы, G2 - С, = АС Необходимо
отметить, что в любых фазах переменного состава (газовых смесях,
растворах, расплавах) в ходе пробега реакции изменяются не только
абсолютные количества, но и соотношение числа молей реагирующих
веществ. Так как концентрация компонентов влияет на химический
потенциал, очевидно, что считать ji. = const можно лишь при
неизменном составе системы. Последнее возможно при постоянных
температуре и давлении лишь в том случае, когда масса системы
настолько велика, что изменение количества веществ практически не
влияет на их соотношение.
139
Раскрывая значение химического потенциала, в соответствии
с уравнениями (3.21) и (3.42) получаем для газовой смеси, ведущей
себя идеально,
^ = Sv/^ + Iv/^ln/7/. (5.5)
Для газовой смеси с отклонениями от идеального поведения
AG = Xviii; + Sv//?71ny;.. (5.6)
Для конденсированных фаз переменного состава
AC = Xv/Ji;+2v^7,lne/. (5.7)
/ i
Следует отметить, что в уравнениях (5.5) и (5.6), записанных для
газообразных систем, значение стандартного химического
потенциала ji.° зависит только от температуры. В уравнении (5.7) в общем
случае величина ц.° является функцией температуры и давления.
По достижении равновесия АС= 0. Следовательно, при
равновесии для реакций, идущих в газовой фазе,
^—ЛПпЩ^Оравн;
(5.8)
]>>X = -/mnn(y;.v<)paBH. (5.9)
Для реакций в конденсированных фазах переменного состава
ZvX— *Т\пЩа?)
равн*
Или, учитывая реакцию (5.1):
П(АУ/)
равн
щур),
равн
p7pV
pI'pIT
p7pvb»
= кп
J равн
= к,
равн
При постоянных температуре и давлении
П
aL аМ
равн
а,' а
м
= К„
JpaBH
(5.10)
(5.11)
(5.12)
(5.13)
Величина К носит название константы равновесия, выраженной
через парциальные давления участников реакции. Соответственно
140
Kfw Kq — константы равновесия, выраженные через фугитивности
(летучести) и активности компонентов. Величины Кр и /^зависят
только от температуры. Величина Ка зависит как от температуры,
так и от давления.
С учетом уравнений (5.5)-(5.7) можно записать
ДС= /тпП(/>/,)исх - ЯШК;, (5.14)
дс= итщ/;днсх- ятк^ (5.15)
AG= ЛЛпП(аЛ)исх- RT\nKQ. (5.16)
Если ПЦу,)исх< Ao,toAG< 0, реакция (5.1) будет самопроизвольно
протекать слева направо. Если П(я//)исх = Ка, то Л (7 = О, реакция
достигла равновесного состояния. Если Ща^)исх > Ка, то необратимый
самопроизвольный процесс будет протекать в обратном направлении,
справа налево. Естественно, что подобные рассуждения справедливы
и для случаев, когда константа равновесия выражается с помощью
парциальных давлений или фугитивностей по уравнениям (5.14) и
(5.15).
Для большей наглядности запишем уравнения (5.14)—(5.16) в
следующем виде:
' " " ч 'pl'pir
AG = RT\n
(р1'р1мЛ
-RT\n
AG = RT\n
Pa Pb Лсх
fvl. fvM
JL JM
Ра'рУ
-фавн
J A J В
\JA
f
-RT\n
AG = RT\n
*м
\ QA aR J
V A b Уисх
-RT\n
JL JM
J A J В
«LaM
равн
равн
(5.17)
(5.18)
(5.19)
Уравнения вида (5.14)—(5.19) носят название уравнений
изотермы химической реакции (реже в учебной литературе для уравнений с
участием активности компонентов применяется термин "уравнения
изотермы-изобары реакции"). Еще раз подчеркнем, что в правой части
уравнений (5.17)—(5.19) первый член соответствует исходному
состоянию участников реакции (5.1), а второй член — равновесному
состоянию. Исходные величины могут быть заданы произвольно, исходя из
состава сырья и других внетермодинамических предпосылок. Конечные
состояния задаются природой системы, температурой, давлением.
141
Уравнение изотермы характеризует изменение энергии Гиббса
в изобарно-изотермических условиях при произвольно заданных
содержаниях всех веществ, участвующих в реакции (как исходных
веществ, так и продуктов реакции) в очень большой системе, в
которой изменение состава не сказывается на начальных содержаниях
веществ (один пробег химической реакции).
Величина константы равновесия, выраженная через парциальные
давления (К), сохраняет постоянное значение лишь до тех пор, пока
газовая фаза подчиняется идеальным законам (при расчетах
технологического характера примерно до 30 атм). Если конденсированная
фаза (или находящиеся в равновесии конденсированные фазы)
подчиняется идеальным законам, то уравнения изотермы реакции могут
иметь вид
AG = Л71пП(хЛ)исх - Д71п*х; (5.20)
А<7 = Л71пП(с/,)исх- RTinKc. (5.21)
Здесь е.— молярная концентрация, т. е. число молей в 1 дм3
раствора.
Если реакция протекает при постоянных температуре и объеме,
то в качестве критерия ее направления и достижения равновесия
следует использовать энергию Гельмгольца. При постоянных
температуре и объеме
Все последующие преобразования и форма записи такие же, как
и для энергии Гиббса. Следует отметить, что уравнения изотермы,
записанные применительно к энергии Гиббса (изотермо-изобары,/?,
7,=const) и к энергии Гельмгольца (изотермо-изохоры, V, 7' = const),
относятся к одному пробегу реакции в бесконечно большой системе
и поэтому величины AG и А^практически равны друг другу.
5.2. Стандартная энергия Гиббса химической реакции
Представляет интерес рассмотреть, как влияет на величину AG
реакции соотношение реагирующих веществ (участников реакции)
в пределах одного пробега реакции. Представим, что в начальный
момент реакции присутствуют только исходные вещества. Тогда в
соответствии с уравнением (5.19) А (7= -оо, т. е. тенденция к протека-
142
нию реакции бесконечно велика. Если одновременно присутствуют
и исходные вещества и продукты реакции и при этом произведение
активностей продуктов реакции меньше произведения активностей
исходных веществ: а\а^в> a\av"M, то, согласно уравнению (5.19),
Д<7< 0 и реакция (5.1) протекает самопроизвольно. Если соотношение
между активностями участвующих в реакции веществ соответствует
равновесному, то AG = 0, в системе наблюдается подвижное
равновесие. В случае преобладания в реакционной смеси конечных веществ
по сравнению с равновесным составом реакция (5.1) идет в сторону
образования веществе и Я(А<7> 0). Если в исходной смеси
находятся только продукты реакции L и Л/, то в соответствии с уравнением
(5.19) AG = +оо, т. е. бесконечно велика тенденция к протеканию
обратной реакции.
Таким образом, при изменении соотношения участвующих в
реакции веществ величина АС, характеризующая химическое сродство,
изменяется в самых широких пределах: от -оо до +оо. Для того чтобы
сравнить между собой по величине сродства различные химические
реакции, введена величина AG°, называемая стандартной энергией
Гиббса (стандартным химическим сродством).
Уравнения (5.17)—(5.19) относятся к одному пробегу реакции,
протекающей в реакционной смеси произвольно заданного состава
при условии, что при этом процессе соотношение компонентов не
изменяется. Поэтому представляется целесообразным сравнивать
изменение энергии Гиббса в разных реакциях при условии, что один
пробег реакции протекает в реакционной смеси, в которой все
составляющие вещества, как исходные, так и конечные, находятся в
стандартных состояниях (|д. = ц.°). Как ранее подробно рассмотрено
(с. 57) выбор стандартного состояния в достаточной мере
произволен. Наиболее целесообразно выбирать такие состояния вещества, в
которых^ или а. всех компонентов равны единице. Тогда из
уравнений (5.15) и (5.16) следует:
AG° = -RT\nKf (5.22)
bG9 = -RT\nKa. (5.23)
Для фугитивности (летучести) за стандартное состояние обычно
принимают состояние газа при давлении, равном 1 атм, т. е. в
условиях, когда практически любой газ ведет себя как идеальный. Тогда
справедливо соотношение, вытекающее из уравнения (5.14):
143
AG° = RT\nKp. (5.24)
Для конденсированных фаз за стандартное состояние
принимаются, как правило, чистые компоненты. Возможен несимметричный
способ задания стандартного состояния, если один из компонентов
условно именуется растворителем.
Так как AG° и все константы равновесия зависят от температуры,
то проводится температурная стандартизация величин AG0. Обычно
за стандартную температуру принимаются 25 °С (298,15 К).
Как уже отмечалось, величина Ка для реакций, протекающих в
конденсированных фазах, зависит также от давления. В
справочных таблицах приводятся величины AG^g wn веществ при давлении
1 бар (105Па) или 1 атм (1,013 • 105 Па; 0,1013 МПа).
В литературе величину AG^g называют стандартной энергией
Гиббса для индивидуальных веществ или для реакций. Следует,
однако, иметь в виду, что термин "стандартная энергия Гиббса" и
обозначение AGf могут применяться при любых температурах, если
выполняются уравнения (5.22)-(5.24)\
Стандартной энергией Гиббса удобно пользоваться для
приближенной оценки направления реакции. Практически с достаточно
большой вероятностью можно считать, что если AGf < —40 кДж, то
реакция термодинамически возможна; если AGf > 40 кДж, то
реакция термодинамически невозможна; если величина AGf находится
между этими двумя значениями, то необходимо делать точный расчет
по уравнениям (5.14)—(5.16).
5.3. Зависимость константы равновесия реакции
от температуры и давления
Уравнения химической термодинамики позволяют определить
зависимость константы равновесия химической реакции от
температуры. Путем дифференцирования уравнения (5.14) по температуре
получаем
* По аналогии с энтальпией реакции и энтальпией образования
индивидуальных веществ из элементов введем дополнительные обозначения:
стандартная энергия Гиббса для реакции при температуре Г— ArG7.\ для
образования соединения из элементов — A G7°..
144
[ST )„
= R\nn(pJ')llcx-R\nK.-RT
(d\r\K,
дТ
(5.25)
При этом считаем, что величина П(оЛ) постоянна и не за-
висит от температуры. Ранее (с. 32) было выведено уравнение
Гиббса-Гельмгольца (2.40), связывающее изменение энергии Гиббса
и тепловой эффект реакции:
AG=AH+T
l^J/
Подставляя в это уравнение выражения (5.14) и (5.25), получаем
(При/7= COnSt)
Э1пАГ^
дТ
jp
АН
RT2
(5.26)
Аналогичным путем с помощью уравнения (2.41) при постоянном
объеме (V= const) можно получить зависимость
Ъ\пК„
AU
(5-27)
ЪТ \ RT2
Выражения (5.26) и (5.27) называются соответственно уравнение
изобары и уравнение изохоры реакции.
Возможен и несколько иной вывод уравнения изобары.
Приведенному выше уравнению Гиббса-Гельмгольца можно придать
иной вид:
дТ
91
т
yJp
Д#°
(5.28)
С помощью уравнений (5.22), (5.23) или (5.24) можно получить
соответствующую форму уравнения изобары, например
Д#°
OlnO
дТ
jp
RT2
(5.29)
Уравнения изобары определяют знак производной константы
равновесия по температуре, а значит, и направление изменения
состава равновесной смеси. Если Л#° > 0, т. е. тепловой эффект реакции
положителен (реакция эндотермическая), то температурный коэф-
л, (д\пК) п
фициент константы равновесия также положителен, >0.
145
Если А//° < 0, то
<0 и равновесие сдвигается в сторону ис-
jp
Это означает, что с ростом температуры константа равновесия
эндотермической реакции всегда увеличивается и равновесие сдвигается вправо.
Э1п£Л
дТ
ходных веществ. Если А/Г = 0, то константа равновесия не зависит
от температуры.
Аналогичные выводы следуют из известного принципа смещения
равновесия Ле-Шателье—Брауна: если к равновесной системе
подводится теплота, то в системе происходят изменения, чтобы ослабить
это воздействие, т. е. процессы с поглощением теплоты.
Интегрирование уравнения изобары наиболее просто
осуществляется, если принять, что в рассматриваемом интервале температур
величина А#° сохраняет постоянное значение. Тогда
In-
КРЛ _ АЯ°
КРЛ R
(5.30)
Представляется, однако, более рациональным рассчитывать
значения ArCf при необходимой температуре и затем переходить к
константе равновесия. Методы расчета ArGf подробно рассмотрены
в следующих разделах этой главы.
5.4. Абсолютная энтропия.
Тепловая теорема Нернста. Постулат Планка
Для расчетов химических равновесий важны значения
абсолютной энтропии химических веществ, участвующих в реакции. Такую
возможность открывает постулат Планка (1911 г.): "Энтропия
правильно сформированного кристалла чистого вещества при
абсолютном нуле равна нулю":
5=0 при Т=0. (5.31)
Объяснение постулата Планка дает статистическая
термодинамика, согласно которой энтропия представляет собой меру
беспорядочности молекулярного состояния системы. Не останавливаясь на
этом подробно, отметим, что в рамках первого и второго законов
термодинамики можно было определить только изменение энтропии,
постулат Планка позволяет найти ее абсолютное значение.
146
Принято считать, что постулат Планка служит дополнением
тепловой теоремы Нернста (1906 г.), называемой также третьим
началом термодинамики.
В теореме Нернста постулируется, что вблизи абсолютного нуля
справедливы следующие равенства:
(5.32)
limAG^limAtf;
Г->0 Г->0
lim
ЭАС
дТ
= lim
г->о
дАН_}
дТ
(5.33)
Первое из равенств говорит о том, что при очень низких
температурах значения AG и АН химической реакции становятся равны;
согласно второму — кривые AG=f{T) и AH=f[7) имеют общую
касательную (рис. 5.1), причем уравнение Гиббса-Гельмгольца(2.40) и
уравнение (5.33) совместимы лишь в том случае, когда
lim
Г->0
ЭАС
дТ
(дАН
т'Щ дТ
= lim
= 0.
(5.34)
Из тепловой теоремы Нернста и постулата Планка следует:
для абсолютной энтропии идеального твердого тела
7 С dT
Sr=l^—, (5.35)
о *
при приближении к абсолютному нулю теплоемкость идеального
твердого тела также стремится к нулю:
lim/C_/-»0.
Г->0
(5.36)
Таким образом, согласно тепловой
теореме Нернста, при температуре вблизи
абсолютного нуля (Г-> 0)
АЯ=АС,
Hmf^UJ—Ц
т^о{ дТ ) т^о{ дТ )
Согласно постулату Планка, при Т=0
S =0; Hm/S/->0.
С учетом постулата Планка можно
записать выражение для энтропии одного
моля какого-либо газообразного вещества
Рис. 5.1. Кривые
ДЯ=ЛЛиАС=Д7)
вблизи абсолютного нуля
температуры
147
при температуре Т, которая будет равна сумме изменений энтропии
при переходе вещества из одного агрегатного состояния в другое и
при нагревании вещества:
s TfCTdT + AH^+TTCldT + AH^+ | &Т
i j j, j, * T T J 71 7
О *пл Тш Л кип Тхип
В интервале температур от 0 до 7^ возможны переходы твердого
вещества из одной кристаллической модификации в другую, что
также должно быть учтено при расчете величины ST. В справочной
литературе преимущественно представляются в виде таблиц значения
теплоемкости, энтропии, других термодинамических функций при
температурах начиная с 298,15 К. В качестве примера в табл. 5.1
приведены соответствующие термодинамические данные для магния.
Таблица 5.1
Термодинамические свойства магния в твердом, жидком и парообразном
состояниях (Ср° и 5Г° в Дж • моль-1 • К_|/7£ -Щ9% в кДж • моль-1)
Г, К
298
400
600
800
922
922
1000
1200
1363
1363
1400
1600
1800
L 2000
р
24,89
26,11
28,45
30,79
32,26
32,13
32,97
35,15
36,90
20,79
20,79
20,79
20,79
20,79
s;
32,68
40,17
51,21
59,71
64,18
73,89
76,53
82,72
87,28
180,10
180,70
183,50
185,90
188,10
Нт - #298
0
2,60
8,06
13,98
17,83
26,78
29,33
36,15
42,01
168,6
169,3
173,5
177,7
181,8 ]
Примечание. 922 К —температура плавления, Л//Пл = 8,95 кДж-моль-1;
1363 К — температура кипения, A//"cn = 126,6 кДж • моль '.
В интервале температур от 298 до 922 К величина Sf -S^ под-
считывается для магния по уравнению
148
о . _ т CpdT
ST-S2g% - J ———.
298
Скачок энтропии при 922 К связан с процессом плавления:
А//„„
ASm =" т
*пл
Скачок энтропии при 1363 К связан с процессом испарения при
температуре кипения:
А С _ А//ИСП
АОисп ~ ~~^ •
'кип
Графически зависимость энтропии магния от температуры
представлена на рис. 5.2.
Дж • моль'1 • К
•к-«
170
150
130
110
90
70
50
30
-
-
у
-
jl
1 1
II
1 1
300
700
1900
Г, К
Рис. 5.2. Зависимость абсолютной энтропии магния
от температуры: участок I — плавление,
участок II — испарение при температуре кипения
149
5.5. Расчет стандартной энергии Гиббса
и константы равновесия реакции при заданной температуре
с использованием абсолютных энтропии
После того как на основании постулата Планка были определены
экспериментально или расчетным путем абсолютные значения
энтропии и составлены сводки стандартных значений этой величины
для большого числа веществ, стал возможен расчет Д/7/ реакций и
соответственно их констант равновесия на основании теплот
образования и стандартных энтропии участников реакции. В основе метода
лежит известное термодинамическое соотношение
a g;=а н:- та s;=-rtii\k. (з.зв)
В данном случае величины АгСу°, Аг#7°, Аг£7°, Ка характеризуют
рассматриваемую реакцию в целом при температуре Т.
Исходными данными для расчета АгС7° (Ка) служат стандартные энтальпии
образования Ау//°298, стандартные энтропии 5°298 и теплоемкости
С =/{ Т) участии ков реакции. Расчет удобнее производить поэтапно.
На первом этапе по уравнению (2.11) (с. 25) определяем тепловой
эффект реакции при 298 К алгебраическим суммированием
энтальпий образования. Аналогичным образом определяем изменение
энтропии реакции A S°m на основании величин S°m. Так, для реакции
(5.1) (с. 142)
АЛ°98 = VL (S29S )L + VA/ (^298 )м ~
~ VA (^298 ) " VB (^298 )B •
В общем виде
(5.39)
\S°m =Zv/(%8W "Zv^^U . (5.40)
/ i
Далее, по уравнениям
т
ArH°T=ArH°m + JAC/Г, (5.41)
298
Ar5f=Ar52°98+!—£— (5-42)
298 l
определяются изменение энтальпии и энтропии при температуре Т.
При этом должны быть учтены все фазовые превращения, которые
150
происходят с участниками реакции (вещества^, Д, L, М) в интервале
температур от 298 до Т. Если в рассматриваемом интервале температур
участники реакции (исходные вещества или продукты) претерпевают
фазовые превращения, то интервал интегрирования в уравнениях
(5.41) и (5.42) разбивается на отдельные участки:
т
Ar#f =АГ#2°98 + J AC\dT±vAHT
298
превр
J ACl]dT, (5.43)
A-«St = ArS
298"
s
298
ACi
dT±v превр
J
■ превр
1
'ьс^
dT. (5.44)
В каждую алгебраическую сумму АС1 и АС" входят теплоемкости
тех фаз, которые устойчивы в данном интервале температур. Теплота
фазового превращения Д#°ревр берется со своим или
противоположным знаком в зависимости от принадлежности к продукту реакции
или исходному веществу.
Как правило, в первом приближении величина Аг(7ув
рассчитывается без учета температурной зависимости величин ArH°m и AriS°298
(полагают, что АС = 0):
ArG°T = ArH°m -TArS°29S. (5.45)
Следует отметить, что это уравнение, выражающее всего лишь
первое приближение в расчете ArGy°, дает вполне удовлетворительные
результаты при термодинамическом анализе высокотемпературных
процессов. Это объясняется тем, что изменение теплоемкости АС
для многих реакций невелико.
Во втором приближении полагают АС = const = AC 298, тогда
Т
ArG°T - Аг #298 - T^rS°m + АС/>,298
Г-298-Лп-
298
(5.46)
Второе приближение энтропийного метода расчета АгСу° в
литературе известно под названием приближения Улиха, который придал
уравнению (5.46) вид
ArG°T -ArH02n-TArS02n +АСрЖТ
In
298
^ 298 ,
Т
/
и, обозначив множитель в квадратных скобках^ 7), рассчитал его
численное значение для широкого интервала температур. В свое время
таблицы, содержащие произведение TJ{ 7) — так называемую функцию
151
Улиха — имели распространение. Таким образом, расчет стандартной
энергии Гиббса химической реакции ведется по формуле
ArG°T = ArH°m-TArS°m +АСРш7П.Т./(Т). (5.47)
В третьем, наиболее точном, приближении учитывается
температурная зависимость термодинамических функций:
т т АС
ArG°T = ArH°m-TArS°m + J АС/Г-Г \-=*-dT. (5.48)
298 298 '
Этому уравнению путем несложных преобразований можно
придать вид
ArCf = ArH°m -TArSfa -T J ^ J АС/Г. (5.49)
298-' 298
Широкое применение компьютерных программ, включающих
банки данных о термодинамических свойствах большого числа
соединений, а также современные справочные издания
значительно упрощают термодинамические расчеты. Приведем в качестве
примера справочник: Thermodynamic Properties of Minerals and
Related Substances at 298,15 К and 1 bar (105 Pascals) Pressure at
Higher Temperatures/ R.A. Robie, B.S. Hemingway// U.S. Geological
Survey Bulletin 2131. Washington: United States Government Printing
Office, 1995. 461 p. В нем в табличном виде для весьма широкого
круга соединений начиная с температуры 298,15 К с интервалом
100 К и до 1800 К представлены данные о стандартных величинах:
о теплоемкости С° (Дж • моль-1 • К-1), абсолютной энтропии 57°
(Дж • моль"1 • К-'1), функциях {Н0Т-Н0Ш)Г\ -(<V - Н\п)Т-{
(Дж • моль-1 • К-1), энтальпии AfH ° и энергии Гиббса AyGy.e
(/—formation; в кДж • моль-1), а также константы равновесия реакции
образования соединения (\gKfI). В примечаниях к таблицам указаны
температуры и тепловые эффекты фазовых превращений. Для веществ в
конденсированном состоянии (твердых или жидких) стандартным
состоянием является устойчивая при рассматриваемой температуре
форма при давлении 1 бар. Для газообразных веществ
стандартным состоянием является идеальный газ при давлении 1 бар. Те или
иные особенности в выборе стандартного состояния всегда
оговариваются.
При составлении справочных таблиц авторы использовали
лучшие из имеющихся в их распоряжении ("best available") величины для
Н°т - #298 или С° = Л 7), S°m и АуЯ°298 или AG°m. На основании этих
152
данных с интервалом 100 К рассчитывались другие указанные выше
функции. Покажем взаимосвязь между табулированными
величинами:
C/r~A/298Jy -\мт~ ит)1 -^т>
AfH°T =A/Я298^-A^Яf -#298],
Af(TT = AfH°m +TA[(G0T -H-m)T~l]9
\gK/T= -AfG;(2,303RT)-1.
Наличие табличных значений функций образования (Ау //7° и
A .G7°) позволяет рассчитать соответствующую термодинамическую
характеристику реакции (Аг#7°и ArG7°) простым алгебраическим
суммированием.
5.6. Расчет стандартной энергии Гиббса
с применением приведенных функций
При расчете равновесий находят применение так называемые
приведенные термодинамические потенциалы (приведенные
функции). Преобразуем уравнение А<77° = —RT\x\Kследующим образом:
Я1п^-^-А(^-Яо>-^ = Дф1-^о.. (5.50)
rri rrt rrt 1 rrt
Здесь А#0° — изменение энтальпии в результате химической
реакции (тепловой эффект) при абсолютном нуле (Т= 0 К). Величина
ф^^Ят—Ш (5.51)
называется первым приведенным потенциалом. Легко видеть, что
Величина Ф7* может быть непосредственно определена из
результатов спектроскопического исследования. Из уравнения (5.50) следует:
ДФ]._АЩ
R RT
Величина Ф7* для рассматриваемой реакции подсчитывается
путем алгебраического суммирования значений Ф7* для всех
участников процесса.
153
Тепловой эффект реакции Д#0° определяется либо из
спектральных данных, либо из термохимических данных для любой
температуры с помощью уравнения Кирхгофа:
т
AH°0=AH°T-JACpdT.
о
Поскольку для большинства веществ известны энтальпии
образования при 298 К, чаще всего исходят из теплового эффекта при этой
же температуре. Следует также иметь в виду, что значения Н° — HQ°
приводятся в различных таблицах и, в частности, могут оцениваться
из спектроскопических данных.
Трудностей в оценке величины #0°, особенно для
конденсированных фаз, можно избежать, если ввести в расчет вместо функции
Ф7Г другую функцию — Ф*\ называемую вторым приведенным
потенциалом:
Фт = -(<Гт-Н*т)Т-х. (5.53)
Функции Ф* и Ф7** связаны между собой соотношением
Фг=Ф*т+(Н°2п-Щ). (5.54)
Второй приведенный потенциал позволяет рассчитать величины
да/ и К:
AG°T =-ТАФ*т +АН°Ш,
1пК-Афт АЯ298
R RT
Глава 6
ТРЕХКОМПОНЕНТНЫЕ СИСТЕМЫ.
ТЕРМОДИНАМИЧЕСКОЕ ОПИСАНИЕ
6.1. Методы изображения состава
При изображении состава систем из трех компонентов (трехком-
понентные, тройные системы) используются методы, предложенные
Гиббсом (1876 г.) и Розебомом (1894 г.). Вершины равностороннего
треугольника соответствуют чистым веществам А, В и С. Стороны
треугольника позволяют описать составы двухкомпонентных систем А+В,
В+С, С+А. Все точки, расположенные внутри треугольника, выражают
составы трехкомпонентных систем. Процентное содержание каждого из
компонентов в системе тем больше,
чем ближе расположена данная точка
к соответствующей вершине. Метод
определения состава по способу
Гиббса (рис. 6.1) основан на том, что
сумма перпендикуляров, опущенных
из любой точки внутри
равностороннего треугольника на каждую из
сторон, равна высоте треугольника,
которую принимают за 100 % (мол.
и мае. %). Состав тройной системы
можно оценить на основании длин
перпендикуляров, опущенных из рис. 6.1. Определение составатрой-
точки р на стороны А В, ВС и СА. ной системы по способу Гиббса
155
Рис. 6.2. Определение состава
тройной системы по способу Розебома
(второй способ Розебома)
Содержание компонента А
пропорционально перпендикуляру
ра и составляет в точке р 27,9 %,
содержание В пропорционально
отрезку рв (24,4 %), содержание С
определяется перпендикуляром рс
(47,7 %).
Графически длина
перпендикуляров оценивается с помощью
сетки, покрывающей
концентрационный треугольник. Сетка
состоит из трех групп прямых линий,
причем прямые проведены
перпендикулярно соответствующей
высоте треугольника и делят эту
высоту на 10 или 100 частей.
По одному из способов Розебома для определения состава системы
используются отрезки трех прямых, параллельных сторонам
треугольника и проходящих от данной точки до пересечения с каждой из сторон
треугольника (рис. 6.2). Сумма построенныхтаким образом отрезков для
любой точки равностороннего треугольника равна длине его стороны:
ра1 + рвх + рс1 = ЛВ — ВС=СА. Отрезок ра1 позволяет определить
содержание компонента А в точке р, отрезок ре1 — компонента В,
отрезок рс1 — компонента С. Как и в способе Гиббса, каждая сторона
концентрационного треугольника делится на 100 частей, одна часть
соответствует одному проценту (мол. и мае. %).
Рассмотрим некоторые свойства концентрационных
треугольников:
все точки, лежащие на прямой, параллельной одной из сторон
треугольной диаграммы, отвечают системам с постоянным содержанием
того компонента, который соответствует противоположной вершине
треугольника (являются изоконцентратами этого компонента);
все точки, лежащие на прямой, проходящей через вершину
треугольника, изображают системы с постоянным отношением
концентраций двух других компонентов, отвечающих остальным вершинам
треугольника.
Эти свойства оказывают определяющее влияние на выбор
составов при экспериментальном исследовании тройных систем или
выполнении термодинамических расчетов. Возможные схемы
расположения составов приведены на рис. 6.3.
156
Рис. 6.3. Рекомендуемые схемы расположения
составов при исследовании трехкомпонентных систем
В некоторых случаях,
особенно при изучении растворимости
двух солей с общим ионом в воде,
целесообразно применять не
равносторонний, а прямоугольный
равнобедренный треугольник,
что, в частности, позволяет
производить построения на простой
миллиметровой бумаге. Пусть
в треугольнике ЛВС (рис. 6.4)
вершины представляют собой
фигуративные точки чистых
компонентов. Если дана система,
содержащая а % компонента А, в %
компонента В и с % компонента С,
то, приняв катет ЛЯ треугольника
за ось абсцисс, другой катет АС —
за ось ординат, а вершину А — за
начало прямоугольной системы
координат, откладывают в как абсциссу, а с как ординату. Таким
образом, при этом способе две концентрации принимаются за
координаты фигуративной точки/? состава рассматриваемой системы.
Наоборот, если дана точка/?, то две концентрации находятся как ее
координаты.
В системах типа вода—соль I—солъ //(соли с общим ионом) по
сторонам треугольника /1С и Л/? (катетам) откладываются концентрации
солей. Начало координат соответствует чистому растворителю.
Рис. 6.4. Прямоугольная система
координат для определения состава
тройной системы (первый способ
Розебома)
157
6.2. Термодинамические соотношения
для трехкомпонентных систем.
Уравнение Гиббса-Дюгема и его интегрирование
Приводимые в начале третьей главы (раздел 3.1) основные
термодинамические соотношения не ограничены каким-либо числом
компонентов. Так, основные термодинамические соотношения для
тройных систем имеют вид
GM = x{G{ + x2G2 + X3G3, (6.1)
dGM = G^dx{ + G2dx2 + G3dxp (6.2)
x^dG{ + x2dG2 + Х3Л/3 = 0. (6.3)
Аналогичным образом можно записать выражения для
относительных и избыточных термодинамических функций тройной
системы. Так, например, для избыточной энергии Гиббса
AG1136 = xfiG™6 + х2А(/2иэб + jc3AG3M36, (6.4)
x^AG,"36 + x2dAG2m6 + x}dAG™6 = 0. (6.5)
Как известно, основные экспериментальные методы
исследования термодинамических свойств трехкомпонентных систем (метод
ЭДС, большая часть методов измерения давления насыщенного
пара, методы, основанные на изучении гетерогенных равновесий)
позволяют определить парциальные молярные термодинамические
характеристики (активность, коэффициент активности) только
одного из компонентов тройной системы. Для вычисления по этим
данным интегральных величин и парциальных молярных
характеристик для других компонентов существует ряд методов, основанных
на использовании уравнений (6.4) и (6.5). Основное условие
рациональной постановки опытов при изучении тройных систем сводится
к строго закономерному расположению исследуемых составов в
концентрационном треугольнике. Возможные схемы изменения состава
приведены на рис. 6.3.
Наибольшее распространение получил метод интегрирования
уравнения (6.5), предложенный Даркеном (1950 г.). За стандартное
состояние принимаются чистые компоненты. Если из эксперимента
определяются парциальные молярные свойства компонента 2, то
при проведении измерений состав следует изменять по разрезам с
постоянным отношением компонентов 1 и 3 (см. рис. 6.3).
Дифференцирование уравнения (6.4) пох2 при постоянном отношении хх/хъ
158
и последующие преобразования с учетом уравнения (6.5), принимая
во внимание, что х{ + х2 + хг = 1, позволяют получить следующее
выражение*:
AG2H36=AGH36+(l-x2)|
SAG
и^
дх-%
(6.6)
Для удобства интефирования разделим все члены уравнения (6.6)
на (1 — х2)2 и представим результат в виде
Гд(7изМ
/5х2
Д#
изб
il (1"*2)2
(6.7)
^3
Интегрирование уравнения (6.7) может быть осуществлено вдоль
линий с постоянным отношением х{: х3 двумя путями: от х2 = О до х2
или от х2 = 1 до х2 (рис. 6.5).
В первом случае получаем
Д(?из0=(1-х2)
АС?^0
♦1
дс2из6 ,
*/=о(1-*2)
(6.8)
чизб
Величина AGX2=0 представляет собой интегральную избыточную
энергию Гиббса граничной двойной системы 1-3 при соотношении
компонентов Xj: xy Аналогичного вида уравнения могут быть
записаны для энтальпии смешения и избыточной энтропии смешения.
Если об интегральных термодинамических функциях системы 1-3
нет сведений, то приходится производить интегрирование уравнения
(6.7) от х, = 1 до х2. В этом случае расчетное уравнение имеет вид
АСизб=(1-х2)
AG
изб
*2=1
'-1<1-*2>
2^2
(6.9)
+ х.
AG,
х,-1
J-0<l-*2)'
rdxj
+ Х,
дс,=0
AG"36 J
—rdxj
U(l-*2)
x2=0
k=o
* Подробные математические преобразования приведены в
монографии К. Вагнера (1957 г.) и в учебном пособии А.Г. Морачевского (1987 г.)
(см. библиогр. список на с. 249). В этих работах рассмотрены и другие
возможности интегрирования уравнения Гиббса—Дюгема для тройных систем.
159
2 (*2=D
Второй и третий члены правой части
уравнения рассчитывают изданных,
относящихся к граничным системам 1—2
и 2-3 соответственно.
Следует отметить, что расчет
интегральных термодинамических функций
тройной системы с помощью уравнений
вида (6.8) существенно более
предпочтителен, чем с помощью уравнений
вида (6.9), так как точность вычисления
s2
Рис. 6.5. Схема двух возможных
путей интегрирования
уравнения (6.7) при постоянном
отношении jc, : х3:
до х2,11 — от х2 — 1 до х2
подынтегральной функции
ACf6
(1-*2)
от х2 = О
хотя эта функция и является конечной
при всех значениях х2, уменьшается
вблизи х2 = 1. В этой области составов
отклонения от идеального поведения,
а следовательно, и величины А(72из6 очень
малы. Очень малы и величины (1 — х2)2. Поэтому для достижения
АС2из6
удовлетворительной точности при вычислении функции —j
(l—Xj)
требуется особенно высокая точность эксперимента. В то же время для
метода ЭДС в целом характерно уменьшение относительной точности
эксперимента в области составов, богатой компонентом, парциальные
молярные величины которого определяются.
Величины парциальных молярных свойств компонентов 1 и 3
могут быть определены с помощью уравнений, аналогичных
уравнению (6.6):
АС"36 :
А(?3из6
АСиз0+(1- Х)
ГдАСизбЛ
дхх
%
АСизо+(1-х3)
'ЗДС«6>*
дх.
(6.10)
(6.11)
Обычно при нахождении величин AG,M36 и АС3из6 с помощью
уравнений (6.10) и (6.11) применяют графический метод: правые
части этих уравнений могут быть получены как отрезки на ординате,
отсекаемые касательными линиями к кривым, выражающим зави-
160
Рис. 6.6. Определение парциальных молярных свойств:
а — компонента 1 (х2: х3 = const); 6— компонента 3 (дс,: х2 = const)
симость AG*436 соответственно от х, и х3 при постоянных отношениях
х2: л:3 и х{: jc2 (рис. 6.6).
Графически термодинамические характеристики трехкомпо-
нентных систем выражают в виде линий, соединяющих при данной
температуре составы сплавов, обладающие равным значением той
или иной термодинамической функции (изолинии интегральных
или парциальных термодинамических свойств, изолинии активности
компонентов).
6.3. Приближенные методы расчета термодинамических свойств
тройных систем по данным о граничных двойных системах
Экспериментальное исследование термодинамических свойств
жидких тройных металлических систем всегда связано с большими
затратами труда. Кроме того, не для всех систем может быть подобран
достаточно надежный метод исследования. В связи с этим большой
практический интерес представляют различные расчетные методы,
позволяющие оценить термодинамические свойства тройной
системы поданным о граничных двойных системах. В основе подхода
лежит представление, что основной вклад в величину интегрального
свойства тройной системы вносят парные взаимодействия, ведущие
к образованию ассоциатов, группировок, включающих разносортные
161
или односортные атомы. Рольтройных взаимодействий, как правило,
не столь существенна.
Методы расчета делятся на две группы:
расчеты с применением геометрических моделей;
расчеты с помощью полиномиальных выражений.
Применению методов способствует относительно простая
геометрическая форма поверхности интеграл ьных молярных энергий Гиббса,
избыточной энергии Гиббса и энтальпии смешения в тройных
системах. В случае геометрических моделей используются разнообразные
геометрические построения, применение полиномов дает возможность
математически описать поверхность интегрального свойства.
6.3.1. Геометрические модели
Геометрические модели делятся на две группы — симметричные и
асимметричные. В первом случае все три граничные двойные системы
являются равноценными, долевой вклад каждой двойной системы
в интегральное свойство определяется только составом тройного
сплава, для которого выполняется расчет. В асимметричных моделях
результат расчета зависит от принятого расположения компонентов
по отношению к лучевому разрезу, вдоль которого ведется расчет.
Для всех геометрических моделей при расчете величин AG, AG136 и АН
алгоритм имеет один и тот же вид. Поэтому в дальнейшем при
описании моделей будем пользоваться обозначением АФ, понимая под
ним любую из трех указанных выше интегральных величин (AG, AG136,
АН). Будут очень кратко описаны только те модели, симметричные
и асимметричные, которые нашли достаточно широкое применение
при термодинамических расчетах. В соответствии со сложившейся в
научной литературе практикой модели обозначаются по фамилиям
предложивших их авторов.
Из числа симметричных моделей при расчетах нашли
применение модели Колера, Колинэ, Муггиану, Шоу. Алгоритмы к моделям
приведены в табл. 6.1, геометрические построения — на рис. 6.7.
Модель Колера. Согласно модели, величина интегрального
молярного свойства тройной системы может быть представлена следующим
образом:
АФ = ф12х,дс2 + ф^х^з + ФзЛ^з-
В этом уравнении ф — функция отношения концентраций х. и
х. Величина функции определяется только отношением молярных
162
долей компонентов соответствующей двойной системы / —j,
сохраняет постоянное значение вдоль всей секущей, добавление третьего
компонента не оказывает влияния на ее величину. В постоянстве
величины ф.. вдоль секущей, проведенной из соответствующей
вершины треугольника, заключается основное допущение метода.
а) 2 б) 2
3 1
Рис. 6.7. Графические построения к симметричным
расчетным моделям: а — модель Колера; б— модель Колинэ;
в — модель Муггиану; г — модель Шоу
Указанное уравнение для двойной системы, например для
системы 1-2, примет вид
АФ12 = ф12х, 'х2\
где л;,' и х2 — молярные доли компонентов в двойной системе 1—2.
Молярные доли компонентов в тройной системе 1-2—3 обозначим
xvx2wxr
Тогда: х{'/х2' = х,'/(1 - *,') = xjx2 или х* = х1/(х] + х2).
Из уравнения для АФ12 следует
АФ12(х!+х2)2
ххх2
Я>12=-
163
Аналогичного вида соотношения можно получить и для других
секущих с постоянным отношением х, : х3 или х2 : хг С учетом
изложенного расчетное уравнение примет вид
ДФ = [ДФ12];1(х1+дс2)2 +
+ [АФ2з]^(х2+х3)2+[АФ3|]х1и1+Хз)2.
Ху X,
В дополнение к рис. 6.7 приведем более детальную расчетную схему
ддя модели Колера (рис. 6.8), например для расчета А(7из6. Эта модель
может быть распространена на четверные и более сложные системы.
Таблица 6.1
Симметричные геометрические модели
Модель
Колера
Колинэ
Муггиану
Шоу
Алгоритм |
АФ = |АФ12]д1(х1+х2)2 +
+ [АФ2зк^2+^з)2+[ЛФ31к^1+Хз)2
*Э *1 |
дф= 1^ Гдф ] +
(2x2+xl)(2xi+xl)1 23J*>
i 4Х|*3 Гдф 1 1
' (2xi+x2)(2xi+x2)L ~и*. '
i 4х'*2 Гдф 1
' (2х1+х2)(2х2+х,)1 '"*i
АФ = -3- ДФ2| (х2) + -3- АФ|3 (*,) + -^-ДФ32 (*з)
1 — Х2 1 — ЛГ| 1 — Ху
Примечание. Соответствующие геометрические построения
представлены на рис. 6.7.
164
Модель Колинэ. Модель была
предложена для расчета энтальпии
смешения тройной системы
поданным о граничных двойных системах,
но она может быть распространена
и на другие интегральные молярные
термодинамические свойства — AG
и АС"36. В общем виде расчетное
уравнение приведено в табл. 6.1.
Особенность модели состоит в том,
что в расчетное уравнение входят
по два значения интегрального
свойства каждой из двойных
систем (рис. 6.9,а). Однако возможно
выполнение расчетов отдельно для
"левой" и "правой" комбинации
(рис. 6.9Дв).
Рис. 6.8. Схема к расчету A<?"6
трехкомпонентнои системы по
модели Колера
Рис. 6.9. Схема к расчету интегральных молярных
термодинамических свойств трехкомпонентнои системы
по модели Колинэ: а — расчет по шести составам двойных систем;
б— расчет по трем составам ("левая" модель);
в — расчет по трем составам ("правая" модель)
Для "левой" комбинации (рис. 6.9,5)
Л4>
^[АфЛ+-*-[Афп1^[Аф1г11.
Для "правой" комбинации (рис. 6.9,<?)
АФП ^[АФД, +^№»\ ^И^ •
165
Приведенное в табл. 6.1 значение АФ представляет собой
полусумму указанных выше величин:
дф=АФл+АФп
2
Модель Муггиану. Расчетное выражение для геометрической
модели Муггиану приведено в табл. 6.1, фафическое построение — на
рис. 6.9,е. В дополнение к табл. 6.1 следует расшифровать значения
x2r, jc/ и х", которые в качестве подстрочных индексов фигурируют
в расчетной формуле:
/ Xi f Х-у 0 X-i
х2=х2+—> х\=х\+— » х\=х\+~г -
Рис. 6.10 иллюстрирует применение модели Муггиану для расчета
энтальпии смешения тройной системы.
Рис. 6.10. Схема к расчету энтальпии смешения
трехкомпонентной системы по модели Муггиану
Модель Шоу. Эта модель является частным случаем модели
Колинэ, "правой" ее комбинацией.
В группе асимметричных моделей для всех трех интегральных
термодинамических характеристик — АС, О136 и АЯ- вид алгоритмов
166
также одинаков, что позволяет и в этом случае использовать
обобщенное обозначение — АФ. Соответствующие алгоритмы приведены
в табл. 6.2, геометрические построения — на рис. 6.11.
Таблица 6.2
Асимметричные геометрические модели
Модель
Бонье
Тупа
Хиллерта-1
Хиллерта-2*
Алгоритм
АФ = -^АФ21(х2) + 7^-АФ23(х2) + (1-х2)[АФ)3К
\-х2 \-хх х 1
АФ-^АФ21(х2) + т^-АФ2зи2) + (1-х2)2[АФ13]х,
АФ =
1
+ —
2
=-^-ДФ21(х2;1 - х2)+-^-ДФ23(1 - х2;х2)
1-Х2 1 — JC|
-^-АФ1з(1-х3;хз)+-^-АФ|з(х,;1-лс|)
1-х, 1-х,
+
АФ = -^АФ21(х2;1-х2) + -^-х
1 — дс2 1 — Xi
хАФ2з(1-х2;х2)+^^-АФ1з(у1з;у31)
VI3V3I
*v.= (1+x + x)/2,/,/=l,2,3:
а)
б)
«)
Рис. 6.11. Графические построения к ассиметричным моделям:
а — модели Бонье и Тупа, б— модель Хиллерта-1,
в — модель Хиллерта-2
167
Дадим краткие пояснения к моделям.
Модель Бонье. Весьма простой геометрический метод был
предложен Бонье для расчета интефальных термодинамических характеристик
тройных систем, в которых одна из фаничных двойных систем близка
к идеальной или регулярной (например, система 1-3, рис. 6.12).
Рассмотрим схему расчета интегральной молярной энергии
Гиббса. Функция АС наиболее удобна для расчета с применением
геометрических моделей, так как во всех трех граничных двойных
системах и в тройной системе всегда отрицательна. Для расчетов
с применением геометрических моделей определенные трудности
вызывают системы, в которых интегральные молярные свойства
фаничных систем имеют разные знаки. Это сильно осложняет форму
поверхности интегрального молярного свойства тройной системы.
а) 2 б)
Рис. 6.12. Схема к расчету интефальной молярной энергии
Гиббса по модели Бонье, пояснения см. в тексте
Для оценки величины АС тройной системы вточке L рассмотрим
такую последовательность процессов: вначале образуются двойные
сплавы в системах 1-2 (точка К), АС12, и 2-3 (точка М), АС23, а затем
происходит их смешение с образованием сплава L. Суммарное
изменение энергии Гиббса АС можно представить в виде
AG^AG^+^AG^ + AC1,
где два первых слагаемых в правой части уравнения характеризуют
вклады (долевое участие) граничных двойных систем 1-2 и 2-3 в
образование тройного сплава по линии с постоянным содержанием
168
компонента 2 (рис. 6.12,5, пунктир), а вел ич и на A G] учитывает участие
в сплавообразовании системы 1—3 (взаимодействие компонентов
1 и 3). Если это взаимодействие невелико, то можно принять, что
величина AG1 линейно изменяется вдоль секущей лучевого разреза
с постоянным отношением хх: х3:
AG] = (l-x2)AGiy
Тогда расчетная формула для АС примет вид
АС =
3_дС12+^_АС23
i-x2 \-x2 Jx2 Хз
+ (1~х2)[АС13]а.
Аналогичного вида уравнения можно записать для АСиз6 или АЯ,
однако желательно, чтобы эти термодинамические характеристики
во всех трех граничных системах были одного знака.
Модель Тупа. Уравнение, предложенное Тупом, внешне мало
отличается от уравнения Бонье:
АСиз6 =
.д^зб+-^-А(72Из36
+ (1-^2)2[А^б]а.
*ч
1 - Х2 1 - *2
Графическое построение для модели Тупа такое же, как и для
модели Бонье (см. рис. 6.12). Однако это уравнение может быть
получено путем интегрирования уравнения Гиббса-Дюгема для
трехкомпонентнои системы по методу Даркена, если предположить, что
жидкий сплав представляет собой регулярный раствор.
Модели Хиллерта (1) и (2). Предлагаемые Хиллертом два
уравнения (см. табл. 6.2) различаются тем, что в первом из них
используются четыре из шести точек уравнения Колинэ (см. рис. 6.11,6),
а во втором — происходит сочетание уравнений Тупа и Муггиану.
Две точки определяются линией, параллельной одной из сторон, а
третья — перпендикуляром, опущенным из точки, соответствующей
составу трехкомпонентнои системы, на неиспользованную сторону
(основание треугольника) (см. рис. 6.11,в).
6.3.2. Полиномиальные методы расчета
термодинамических свойств трехкомпонентных систем
Существует ряд методов расчета термодинамических свойств
тройных систем на основании граничных двойных систем, требующих
полиномиального представления исходных данных.
169
Метод Редлиха—Кистера. Уравнение Редлиха—Кистера (3.56) в
случае тройных систем принимает вид
Q = 2шят = хл1Ьп +Cn(Xl" х'] +
+ dn(x{ '- х2)2 + ...] + х2хъ[Ь2г + с23(х2 - jc3) +
+ d2p2 - х3)2 + ...] + хххг[Ьп + схг{х{ - хъ) +
+ ^^(Xj - х3)2 + ...] + xlx2x3(cl + rfjjc, + rf2x2). (6.12)
Коэффициенты ft.., с , J в уравнении (6.12) относятся к
соответствующим двойным системам; с{, dv d2 определяются по
минимальному числу экспериментальных данных для тройной системы. Опыт
показывает, что для жидких металлических систем вполне достаточно
учитывать три первых слагаемых в уравнении (6.12), коэффициенты
cvdv d2 определять нет необходимости. Таким образом, для расчета
интегральной функции тройной системы оказывается достаточным
только данных о бинарных системах.
Если с помощью уравнения (6.12) определена зависимость
Q-функции от состава, то можно определить коэффициенты активности
каждого из компонентов системы. Для трехкомпонентной системы
Q = ^lgy, + x2lgy2 + x3lgy3. (6.13)
Дифференцирование уравнения (6.13) ведет к выражению
dQ = lgy,rfx, + lgy2dx2 + lgy3</x3 + x,dgy, + x2dgy2 + x3dlgyy (6.14)
В соответствии с соотношением Гиббса-Дюгема сумма трех
последних слагаемых уравнения (6.14) равна нулю. Учитывая, что
dxx + dx2 = -dxv получаем
dQ = \g^-dx{+\g-^dx2i (6.15)
Уз Уз
отсюда следует
Уз
fdQ) ., ,Л2_(Э<Я
вХг L УЗ
1 Л,х2 ••>
дх7
(6.16)
/Г,хх
Для расчета коэффициента активности каждого из компонентов
можно использовать ряд очевидных соотношений:
lgY3=Q-xIte^-x2lg^;
Уз Уз
igyi=ig—+igy3; igY2 = te—+igy3-
170
Уравнение (6.12) без тройных констант было использовано для
расчета Q и соответственно Д(7из6 целого ряда жидких тройных
металлических систем с различным характером взаимодействия между
компонентами. Полученные с помощью метода Редлиха—Кистера
величины хорошо согласуются с результатами интегрирования
уравнения Гиббса-Дюгема на основании экспериментальных данных и с
расчетами с применением тех или иных геометрических моделей.
Метод Вильсона. Уравнение Вильсона имеет малое число
коэффициентов и для двойной системы записывается так:
In Yi =-ln(xj + Л12л:2) + л;2
In y2 =-ln(jc2+A21x1) + x1
42
X\ т J\ 12 Xj
*1+Л12*2
v21
*2+Л21*1
*2+Л21*1
(6.17)
(6.18)
Д<?36 = -R7\ln(xx + Апх2) + ln(x2 + Л21х,)]. (6.19)
Коэффициенты Л12 и Л21 оцениваются по экспериментальным
значениям у, и у2 при нескольких составах, для идеальной системы
Л12 = Л21 = 1. При хх (или х2), стремящихся к нулю, коэффициенты
активности ух (или у2) приближаются к своим предельным
значениям (у* или у2°°). Коэффициенты Л12 и Л21 положительны при любом
характере отклонений двойной системы от идеального поведения.
Принято считать, что уравнение Вильсона наиболее хорошо
описывает концентрационную зависимость термодинамических функций в
системах с умеренными положительными отклонениями от идеального
поведения. Вероятно, это не вполне правильно. В табл. 6.3 приведены
значения Л12 и Л21 для шести двойных систем с различным характером
взаимодействия между компонентами.
Из приведенных в табл. 6.3 систем только для сплавов железа с
кобальтом можно считать положительные отклонения "умеренными",
но концентрационная зависимость термодинамических функций во
всех случаях достаточно хорошо описывается уравнением Вильсона.
Для многокомпонентных систем на основании полинома
Вильсона можно записать:
д^ИЗб П П
——=-5>|in[2>yA,],
К1 /=1 у=1
где AG"36 — интегральная молярная избыточная энергия Гиббса
системы из «-компонентов; Л.. — подгоночные положительные ко-
(6.20)
171
эффициенты. Аналогичного вида уравнение может быть записано и
для энтальпии смешения многокомпонентной системы. На рис. 6.13
приведены в качестве примера результаты расчета методом Вильсона
AGHj6 для одного из разрезов системы Fe-Cu-Co (АСиз6 > 0) и АЯдля
системы Ni-Fe-Co (АЯ< 0).
Таблица 6.3
Значения коэффициентов уравнений (6.17)—(6.19)
и коэффициента корреляции г (температура 1873 К, AG1"6, кДж • моль-1)
Система
Cu-Fe
Cu-Co
Cu-Ni
Fe-Co
Co-Ni
Fe-Ni
A(?"6 при jc2 = 0,5
7,49
7,03
4,73
1,35
-0,63
-2,76
Л12
0,2467
0,3734
0,7196
0,2478
1,0827
0,2930
л21
0,2195
0,1746
0,2790
1,7444
1,0821
3,4138
г2
0,9979
0,9999
0,9984
0,9609
0,9996
0,9850
Рис. 6.13. Зависимость интегральной молярной избыточной энергии
Гиббса, AG136 (кДж • моль1) тройных систем Fe-Cu-Co (xFe: jcCo = 1:3)
(а) и Ni-Fe-Co (jcNi. : хСо = 1:1) (б) от состава при температуре 1873 К.
Рассчитано с помощью: / — геометрических моделей,
2— метода Релиха-Кистера, 3— метода Вильсона
Метод Крупковского—Птака. Согласно Крупковскому,
зависимость коэффициента активности компонента от состава и температуры
в бинарной системе у, =f(xv 7) может быть описана выражением
172
lny^oC^Jc^-wCrKl -x{)'\ (6.21)
где т — постоянная для данной бинарной системы величина.
В соответствии с уравнением Гиббса-Дюгема и принимая во
внимание, что функция со(7) не зависит от состава, получаем
1пу2=ш(7)
_jw w_1+co I (622)
Поскольку при х2 -> 1 1пу2 -> 1, то const = \/(т -1). Уравнение
принимает вид
т
1пу2=со(Г)
i-l 2 т-\\
(6.23)
т-
В первоначальном варианте метода принималась концепция
регулярного раствора, согласно которой величина ЛПпу. не зависит
от температуры, и тогда со(7) = а/Т.
В более общем случае со( 7) = а/7* и уравнения (6.21) и (6.23) для
двухкомпонентной системы принимают вид
1пу1=^-(1-*1Г;
1 а
„/и т „т-1 ,
т-\
m-lj
Основываясь на уравнениях такого вида для бинарных систем,
Крупковский и Птак вывели следующее выражение для расчета
избыточной энергии Гиббсатрехкомпонентной системы:
дсизб=__ЯО|2 х Г1-(1_х )«12-П +
7^-V12-l) 2L ' J
^a23
^-'(^з-!)
x3[l-(l-x2)^-,] +
Да
Г^^Из-!)
12 дс1Г1-(1-дс3ГН1.
» n L J
Величины a , А:.., т.. характеризуют соответствующие граничные
системы.
6.3.3. Применение правила Здановского
к жидким металлическим системам
Около 70 лет назад (1936 г.) А.Б. Здановский на основании
изучения большого числа водных растворов электролитов преимуществен-
173
но с общим анионом установил простую закономерность,
получившую известность как правило Здановского: растворы электролитов,
имеющие одинаковое давление пара воды и химически между собой
не взаимодействующие, образуют раствор с тем же самым давлением
пара воды (изопиестический идеальный раствор). Первоначально
речь шла о системах типа АХ(1)—ВХ(2)—Н20.
Для изображения концентрационных отношений в таких
растворах удобно использовать прямоугольную систему координат (первый
способ Розебомадля изображения состава тройных систем). Если по
осям (рис. 6.14) откладывать моляльность смешиваемых растворов,
то для изопиестических идеальных растворов изменение состава при
смешении будет выражаться прямой линией, соединяющей составы
бинарных смесей.
тл
т,
Рис. 6.14. Иллюстрация к применению
правила Здановского
Обозначим т,°и /и2° моляльность компонентов в исходных
двойных растворах, а через тх и т2 — моляльности тех же компонентов в
образовавшемся смешанном растворе. В случае выполнения правила
Здановского можно записать:
т\ тг
(6.24)
174
AW-)
Mi =А77т
-ГП\
(6.25)
Щ
График зависимости т2 = Д/w,) является прямой линией,
пересекающей оси координат в точках, соответствующих концентрациям
компонентов в бинарных системах с одинаковой активностью воды.
В дальнейшем оказалось, что правило Здановского не
ограничивается системами из солей с общим анионом, а имеет гораздо более
широкие области применения. Правило Здановского может быть
использовано при оценке активности воды и химических потенциалов
компонентов в многокомпонентных растворах электролитов. Если
в многокомпонентной системе обозначить через т° (/ = 1, 2, 3 ...)
моляльность компонентов в исходных смесях, а через т. — моляль-
ностьтехже компонентов в смешанном растворе, то при условии, что
исходные бинарные растворы будут находиться в изопиестическом
равновесии между собой и с образовавшимся многокомпонентным
раствором, математическая запись правила Здановского для системы
из п компонентов примет вид
1^ = 1- (6-26)
1 Щ
Уравнение (6.26) сохраняет свой вид независимо от способа
выражения концентрации, т. е.
5:^=1^=1^=1, (6.2?)
1 /Я/ 1 X/ 1 СО,-
где х. и со — соответствующие
молярные или массовые доли /-го
компонента в двойном исходном
растворе.
Правило Здановского может
быть распространено и на жидко-
металлические системы с
различным характером взаимодействия
между компонентами. На рис. 6.15
построение выполнено для
системы Bi—Sn—Cd, отклонения от
идеального поведения в которой
невелики.
*cd = °>2
Рис. 6.15. Линии постоянной
активности кадмия aCd в системе
Cd-Bi-Sn при 773 К
175
Следует, однако, отметить, что построения, основанные на
правиле Здановского, справедливы и для трехкомпонентных систем, в
которых два компонента мало различаются по химической природе.
К числу таких систем относятся Bi-Pb-Mg, Na-K-Pb, Bi-Sb-Nan
целый ряд других систем подобного типа.
6.3.4. Метод изопотенциалов
Аналогичные предпосылки, лежащие в основе правила
Здановского, относятся и к методу изопотенциалов. Для систем, в которых
два компонента из трех мало различаются по химической природе и
образуемая ими граничная система, например 1—3, близка к
идеальной или регулярной, может быть применен метод изопотенциалов.
В этом случае тройной раствор можно уподобить бинарному по
линиям, характеризующимся постоянством АС2 (линии изопотен-
циала). Линии изопотенциалов компонента 2 можно отождествить с
секущими концентрационного треугольника, соединяющими
составы бинарных систем 1—2 и 2—3, имеющие одинаковые значения AG2.
На примере ряда металлических систем было показано, что
упрощающее предположение о прямолинейности линий изопотенциалов
находится в достаточном согласии с экспериментальными данными
(рис. 6.16). По существу, если учесть возможность интегрирования
уравнения Гиббса—Дюгема по лучевым разрезам, факта
прямолинейности изопотенциалов уже достаточно для определения интегральных
термодинамических характеристик тройной системы по данным о
бинарных. Однако можно избежать интегрирования, полагая, что
абсолютные значения АС компонентов 1 и 3 по линиям изопотенциалов
компонента 2 имеют ту же зависимость от состава, что и в системе 1 -3.
Это не противоречит уравнению Гиббса-Дюгема.
Na Na Na
Sn TI Cd Pb Pb Tl
Рис. 6.16. Линии равных значений AGNa (кДж • моль-1) систем:
Sn-Na-Tl (850 К), Cd-Na-Pb (698 К), Pb-Na-Tl (850 К)
176
Действительно, при AG2 = const
уравнение (6.5) имеет вид xldAGl +
+ Jc3dAG3 = 0, что соответствует
бинарной системе 1-3.
Расчетная формула при исполь-
зовании метода изопотенциалов
такова:
AG=[x2AG2 + Xl(AGlv0
'Lx-) = Q/1A(ivx./x
+
+
(6.28)
+ MlXi = 0) + x,(AG3xr_0
+ ^2 = o)U2^3
Сделаем некоторые пояснения
к уравнению (6.28). AG
рассчитывают для состава тройного сплава,
лежащего на пересечении линии
изопотенциала AG2 и лучевого
разреза х1 : х3 (рис. 6.17). Уравнение
(6.28) полностью соответствует
уравнению (6.4). Здесь
ДЯз-ДСзд.-о + ^-о-
Первые слагаемые правой части
уравнений представляют собой
величины AG, и AG3 соответственно в
точках К и М (см. рис. 6.17). Вторые
слагаемые отвечают изменению
величин AG, и AG3 на участках KL и LM.
Результаты расчета методом
изопотенциалов находятся в
хорошем согласии с
экспериментальными данными (рис. 6.18).
Рис. 6.17. Иллюстрация к
применению метода изопотенциалов
Рис. 6.18. Зависимость AG от
содержания натрия в сплаве Na—
-Tl-Pb (xTI : jcPb = I : 1, 850 К).
Кривая — расчет по
экспериментальным данным методом Дарке-
на, кружки — расчет по данным о
двойных системах методом
изопотенциалов
6.4. Равновесие между двумя жидкими
фазами в трехкомпонентных системах
В дополнение к анализу двухфазного равновесия жидкость-
жидкость в двухкомпонентных системах рассмотрим основные эле-
177
менты диаграммы гетерогенного жидкофазного равновесия тройной
системы А-В-С, приведенной на рис. 6.19. Диаграмма соответствует
простейшей системе, в которой компоненты А и С, г также В и С
неограниченно взаимно растворимы, а компоненты А и В взаимно
ограниченно растворимы. Как видно из диаграммы, добавление
компонента С ведет к увеличению взаимной растворимости
компонентов А и В. Будем считать исходной точку S0 двойной системы
А-В. Смесь состава 50 распадается на два равновесных слоя yQ и у01.
Прибавление все возрастающих количеств компонента С ведет к
перемещению фигуративной точки состава по прямой S0C. При этом
точке состава 5, отвечают равновесные слои у{ и у/, точке S2 — y2 и
у2' и т. д. (в данном случае у — обобщенная характеристика состава,
который описывается молярными долями компонентов: хА, хи и хс).
Ноды ух — у{\ у2 - у2\ у3 — yj, соединяющие равновесные слои, как
правило, не параллельны стороне Л В.
Прибавление к гетерогенной смеси А-В компонента С, который,
как это следует из диаграммы на рис. 6.19, смешивается с
компонентами А и В во всех отношениях, формально ведет к такому же
Рис. 6.19. Изотерма взаимной растворимости
компонентов в системе А—В—С. Пояснения в тексте
178
Sn
Рис. 6.20. Границы области расслоения
в системе Al-Sn-Pb
эффекту, как повышение температуры в двойной системе А—В (если
система Л—В характеризуется верхней критической температурой
расслоения).
Подобного рода системы с областью расслоения достаточно
широко распространены среди жидких тройных металлических систем,
важны для металлургической практики. Типичная диаграмма с
областью расслоения приведена на рис. 6.20.
К числу важных характеристик гетерогенных жидкофазных
равновесий относится расположение нод. Установление каких-либо
корреляций в их расположении позволяет значительно сократить
трудоемкий эксперимент при построении кривой расслоения в
тройной системе. В качестве примера таких корреляций укажем
на правило Д.Н. Тарасенкова (1945 г.), установленное на
основании анализа данных о системах, образованных органическими
соединениями, но применяющееся и в металлургической практике.
Согласно этому правилу, все ноды пересекаются в одной точке,
которая может быть расположена вне пределов
концентрационного треугольника. Рассмотрим правило Тарасенкова на модельной
схеме (рис. 6.21).
179
А Е' F' В D
Рис. 6.21. Иллюстрация к применению правила Тарасенкова
В основе правила лежат чисто геометрические построения.
Примем, что в системе Л—В—С компонент распределяется между фазами
с преимущественным содержанием компонента Л (фаза I) и
компонента В (фаза II); EF— линия, соединяющая составы сопряженных
фаз (нода). Содержание компонента С в точках Ей Fопределяется
отрезками ЕЕ' (сх) и FF' (с2). Соответственно содержание компонента
В определяется отрезками АЕ' (Ь{) и AF' (Ь2). Из подобия
треугольников EDE' и FDFf следует:
FГ _ DF'
ЕЕ'" DE''
где ЕЕ' = с,; FF = c2; DE'= к - b{\ DF' = к- Ь2; к — предполагаемая
константа (абсцисса точки пересечения нод). Тогда получаем
— = или к =-^—1—^. (6.29)
Сл К U\ Су —~ Сл
Глава 7
ОСНОВНЫЕ ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
ИССЛЕДОВАНИЯ ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
МЕТАЛЛУРГИЧЕСКИХ СИСТЕМ
7.1. Общая характеристика методов исследования
Экспериментальные методы исследования термодинамических
свойств сплавов и других металлургических систем можно условно
разделить на прямые и косвенные. Прямыми методами изучаемую
величину определяют непосредственно, а в косвенных методах для
ее нахождения помимо данных эксперимента требуется применить
те или иные термодинамические соотношения. Прямые методы, как
правило, предпочтительнее косвенных, поскольку обычно дают более
точные результаты.
Например, энтальпия образования сплава может быть измерена
в калориметре. Методы гетерогенных равновесий тоже позволяют
найти эту величину, но рассчитав ее на основании температурной
зависимости энергии Гиббса с помощью уравнения Гиббса—Гельмгольца.
При этом в формулу для расчета энтальпии образования входит частная
производная от экспериментально измеряемой функции. Операция
численного дифференцирования сопряжена, как известно, с некоторой
потерей точности, что увеличивает общую погрешность определения
изменения энтальпии и энтропии методами гетерогенных равновесий.
Вместе с тем эти методы являются прямыми по отношению к энергиям
Гиббса и позволяют при их определении избежать накопления ошибок
за счет использования расчетных формул.
181
Условность деления методов на прямые и косвенные проявляется
в том, что в действительности в любом методе напрямую измеряют
не термодинамические функции, а значения таких параметров, как
температура, давление, масса, которые в свою очередь часто
преобразуют в электрические величины.
Методы экспериментальной термодинамики можно
классифицировать и по другим признакам. Принципиально различаются группы
равновесных и калориметрических методов. В каждой из групп
реализован один из двух известных способов обмена энергией между
исследуемой термодинамической системой и окружающей средой.
Равновесные методы измерения термодинамических свойств —
методы гетерогенных равновесий* — основаны на существовании
термодинамического равновесия между исследуемым образцом
вещества и специально выбранной фазой. Эту фазу можно назвать
контрольной, поскольку она выполняет функции контроля за
уровнем химического потенциала компонентов в исследуемой фазе и ее
выбирают исходя из условия, что ее термодинамические свойства
однозначно можно рассчитать по измеренным значениям
термодинамических переменных: температуры, давления, объема, массы,
химического состава. Рациональный выбор такой фазы сравнения
играет важную роль в оценке надежности и в трактовке результатов
термодинамического эксперимента.
Химическое равновесие между исследуемой и контрольной
фазами, как правило, касается не всех компонентов системы, а чаще
всего только одного подвижного компонента. Является компонент
подвижным или нет, зависит от скорости его диффузии в исследуемой
фазе и от свойств межфазной границы.
Таким образом, общим у равновесных методов является наличие,
по крайней мере, двух фаз, имеющих границу раздела, через которую
из одной фазы в другую может переноситься подвижный компонент.
Работа такого переноса при соблюдении определенных условий равна
изменению химического потенциала подвижного компонента. Роль
границы раздела может выполнять третья фаза. Разные варианты
равновесных методов различаются между собой характером и свойствами
* К их числу относятся метод измерения электродвижущих сил и все
методы, где изучается равновесие между конденсированной и газовой фазами.
182
межфазных границ, выбором контрольной фазы (фазы сравнения),
способом определения термодинамических свойств исследуемой
фазы по отношению к фазе сравнения. Чаще всего равновесными
методами изучают химические потенциалы компонентов сплава, но
в ряде случаев можно исследовать и интегральные функции.
В дальнейшем изложении рассмотрим применительно к изучению
термодинамических свойств сплавов различные варианты
равновесных методов: метод измерения электродвижущих сил (ЭДС), метод
измерения давления насыщенного пара и метод изучения
гетерогенных химических равновесий. Будут рассмотрены также
различные калориметрические методы исследования термодинамических
свойств металлических систем.
7.2. Метод измерения электродвижущих сил
Характерной особенностью метода измерения электродвижущих
сил (метода ЭДС)* среди других равновесных методов является
наличие третьей фазы на границе раздела двух фаз, проницаемой только
для определенных, положительных или отрицательных ионов. В этом
случае фазы по обе стороны разделительной фазы (электролита) могут
быть электрически заряженными и иметь различающиеся
электрические потенциалы, что принципиально изменяет условия равновесия
фаз. Рассмотрим это равновесие на примере конкретной цепи:
Электролит,
содержащий
(-) Me,
ионы Me,z+
Ме.,Ме2,Ме3(+) (7.1)
Левым (отрицательным) электродом является чистый металл Мер
правым (положительным) — его сплав. При этом металлы, входящие
в состав сплава (Ме2, Ме3 и т. д.), должны быть более "благородными"
(электроположительными), чем Me,. Химический потенциал чистого
компонента всегда выше, чем химический потенциал этого компонента
в сплаве, и поэтому потенциалобразующим процессом в цепи будет
* Метод ЭДС и другие электрохимические методы определения
термодинамических свойств металлических сплавов детально рассмотрены в
монографии: Электрохимические методы в термодинамике металлических
систем/А. Г. Морачевский, Г.Ф. Воронин, В. А. Гейдерих, И. Б. Куценок. М.:
ИКЦ "Академкнига", 2003. 344 с.
183
изотермический перенос Me, из электрода, состоящего из чистого
металла (левый электрод), в сплав (правый электрод).
На левом электроде: Me, -ze-> Me,z+,
на правом электроде: Ме,2+ + ze -» Me, (в сплаве),
где z — заряд потенциалобразующего иона.
В элементе с замкнутой внешней цепью процесс переноса
происходит самопроизвольно, сопровождаясь разбавлением сплава
компонентом 1, переносимым через электролит. При разомкнутой
внешней цепи переносу Me, будет препятствовать разность
потенциалов, возникающих на границах раздела электрод-электролит.
В разомкнутом элементе каждый электрод находится в равновесии
с электролитом при определенном потенциале электрода,
например,
е{=е;+\-
1п
0+
"Ме.
'Me,
(7.2)
Здесь <7 2, — активность ионов Ме.2+ в электролите; дМе — ак
Ме;
тивность Me, в сплаве (электроде). Поскольку в нашем случае левый
электрод представляет Me,, то для него аш = 1. Электродвижущая
сила цепи (7.1) соответственно равна
(
z7
'Me,
(7.3)
В это выражение уже входит только активность Me, в сплаве.
Естественно, что если оба электрода представляют собой сплавы с
различной активностью металла 1, причем его активность в левом
электроде alMe выше, чем в правом а^ , то выражение для ЭДС цепи
примет вид
zT )
Я|
.и
Me,
Необходимо иметь в виду, что достаточно соединить
электроды внешней цепью, чтобы процесс переноса Me, начал протекать
самопроизвольно и потенциалы электродов сместились бы от
соответствующих равновесных значений. Для измерения разности
потенциалов электродов при равновесии в элементе к электродам
обычно прикладывают внешнее напряжение, равное по величине
и противоположное по знаку измеряемой ЭДС, т. е. пользуются
компенсационным методом. Можно применять и другие способы
184
измерения ЭДС, если при этом ток во внешней цепи будет
достаточно мал.
Из выражения (7.3) следует
RTlnaMC] = AG, = Ац, = -zEJ, (7.4)
где Ац, — изменение химического потенциала компонента 1 при
переносе одного моля его из чистого компонента I (стандартное
состояние) в сплав соответствующего состава.
Измеряя ЭДС при различных температурах, можно получить
температурную зависимость Ац, и соответственно величины
парциальных молярных энтропии и энтальпии для компонента 1:
AHy^AG, +ГА5, =z7
'¥-\-Б
дТ
р
(7.5)
(7.6)
7.2.1. Цепи с расплавленными электролитами
Метод ЭДС применительно к определению
термодинамических свойств металлических сплавов требует в большинстве
случаев проведения экспериментов при повышенных температурах,
чтобы обеспечить скорость диффузии компонентов, достаточную
для выравнивания концентраций в поверхностном слое и объеме
электрода-сплава. По этой причине в качестве электролита широко
применяются расплавленные соли или их смеси. Важным
требованием к эксперименту является обязательное отсутствие в элементе
побочных процессов, например взаимодействия жидких электродов с
материалом контейнера, с атмосферой или с электролитом. Особенно
опасно наличие в приборе-ячейке окислителей, взаимодействие с
которыми может существенно изменить состав поверхностных слоев
изучаемых электродов-сплавов. Поэтому, в частности, в качестве
электролитов малопригодны нитраты, сульфаты и большинство
других солей кислородсодержащих кислот. В электролите должна
отсутствовать электронная проводимость, т. е. должен выполняться
закон Фарадея.
185
Выбор солей при соблюдении указанных выше условий зависит
от температурного интервала исследований и должен учитывать
степень взаимодействия металла и сплава с электролитом. Это
взаимодействие, как правило, не удается полностью исключить, но оно
должно быть минимальным.
Обычно электролитами служат легкоплавкие солевые смеси, к
которым добавляют небольшое количество соли металла,
участвующего в потенциалобразующем процессе. Наиболее удобны смеси
галогенидов, особенно хлоридов. Составы некоторых смесей
приведены в табл. 7.1.
Таблица 7.1
Составы смесей, мае. %, используемых в качестве
электролитов-растворителей в цепях типа (7.1)
Смесь солей
NaCl-55KCl
NaI-44,5KI
CaCl2-38NaCl
UC1-54KC1
LiBr-52KBr
T °C
660
585
500
352
348
Смесь солей
LiCl - 69,7 RbCl
LiBr- 50-55 RbBr
CH3COOK -46 CH3COONa
AICI3 - 30,5 NaCl
T °C
312
270
233
152
При температурах выше 1000-1200 °C целесообразно применять
оксидные расплавленные электролиты. Так, при исследовании
термодинамических свойств систем Pb—Ag, Pb—Au успешно применяли
электролит PbO-SiOr При определении активности никеля в
расплавах системы Ni-S в качестве электролита служил оксидный расплав
(шлак) следующего состава (мае. %): NiO - 2,0, СаО - 20, Na20 - 30,
Si02-33. Температура опытов 1340-1360 °С. При изучении
активности серы в сульфидных расплавах (системы Си—S, Pb-S, Fe-S и др.)
к электролиту Na20-CaO-Si02 (жидкое натрий-кальциевое стекло)
добавлялся сульфид натрия (Na2S).
Большое внимание следует удалять материалу для изготовления
измерительных ячеек. При относительно низких температурах (до
700 °С) ячейки можно изготовлять из тугоплавких сортов стекол,
при более высоких температурах — из кварцевого стекла. Широкое
186
применение при
высокотемпературных измерениях находят
ячейки с использованием сосудов из
огнеупорной керамики (на основе
А1203> ВеО)(рис. 7.1).
При исследовании
низкоплавких сплавов методом ЭДС в
качестве электролита используют
глицериновые растворы солей.
Типичная цепь при таких измерениях
может быть записана следующим
образом:
(-)Cd/0,05MCdBr2, 1,0 М
КВг, глицерин / (Cd, Hg)x (+).
Верхний температурный
предел определяется устойчивостью
глицерина и находится при
температуре около 220 °С.
Электродом сравнения служит твердый
металл.
7.2.2. Цепи с твердыми электролитами
Наряду с расплавленными солями широкое применение
находят твердые электролиты с ионной проводимостью. Электронная
проводимость в таких электролитах не должна превышать 1 % от
общей проводимости. Иными словами, число переноса электронов
в электролите должно быть меньше 0,01. Различают твердые
электролиты с катионной и с анионной проводимостью.
В качестве твердых электролитов с катионной проводимостью
применяют различные стекла, содержащие катионы, свободно
перемещающиеся в структурной сетке стекла (Li+, Na+, K+, Rb+, Cs+,
Ag+, ТГ), а также ряд твердых фаз кристаллического строения,
обладающих аномально высокой проводимостью в соответствующем
Рис. 7.1. Измерительная ячейка с
расплавненным солевым
электролитом: / — вольфрамовые токоот-
воды; 2 — термопара; 3 — крышка;
4 — стакан из корундиза; 5 —
расплавленный электролит; 6— корун-
дизовые тигли для металла и сплавов;
7— чистый металл или сплав
187
(Na, Pb) (ж) (+)
интервале температур (суперионные проводники — преимущественно
твердые электролиты на основе р-глинозема или Р"-глинозема).
При изучении термодинамических свойств сплавов, содержащих
щелочные металлы, применение расплавленных солей практически
исключается из-за их сильного взаимодействия с металлами и
сплавами. В этих случаях хорошие результаты достигаются при работе с
твердыми стеклообразными электролитами.
Известно, что силикатные или боратные стекла, содержащие
оксиды щелочных металлов, являются катионными униполярными
проводниками, т. е. перенос электричества в них осуществляется
только катионами. Числа переноса щелочных металлов в натриевых,
калиевых, рубидиевых и цезиевых стеклах соответственно равны
0,996; 0,998; 0,99; 0,99. Типичную цепь можно записать, например,
так:
Твердый электролит,
(—) Na (ж) содержащий ионы
| натрия
При температурах 280-550 °С электрическая проводимость
стекла вполне достаточна для использования его в качестве твердого
электролита. Применение обычных лабораторных сортов стекол
из-за их размягчения ограничивается температурами 580-600 °С,
специальные сорта тугоплавких стекол могут служить в качестве
твердого электролита при существенно более высоких
температурах.
На примере измерения ЭДС цепей с жидкими сплавами систем
Na-Cd и Na-Pb:
(-) Na / стекло/ Na (xNa = 0,310), Cd (+); (7.7)
(-) Na / стекло/ Na (xNa = 0,484), Pb (+) (7.8)
было показано, что состав стекла, если оно содержит Na20, не
оказывает влияния на величину ЭДС цепи. В табл. 7.2 приведены составы
использованных в качестве твердого электролита стекол
промышленных марок и специально синтезированного борно-натриевого стекла,
обладающего повышенной химической стойкостью по отношению
к жидкому натрию и его сплавам.
188
Таблица 7.2
Содержание основных компонентов в стеклах, мае. %
Марка стекла
j №23
1 №29
№46
ЗС-8
ЗС-5К
B-Na
Si02
68,39
68,60
68,25
66,50
68,00
-
В,!),
2,66
—
16,5
23,0
20,0
60,0
А1203
3,89
3,70
4,00
3,00
3,00
15,00
Na20
9,42
10,00
6,00
3,70
4,00
13,00
СаО
8,51
7,50
—
—
—
12,00
к2о
7,14
3,00
— 1
3,80
5,00
-
ЭДС цепей измеряли в интервале 400—575 °С. Как видно из рис. 7.2,
состав стекла в пределах погрешности измерений не влияет на
величину ЭДС цепей.
Рис. 7.2. Зависимость ЭДС цепей (7.7) (а) и (7.8) (б)
от температуры для стекол различного состава
(составы стекол 1—6 приведены в табл. 7.2)
189
Рис. 7.3. Схемы ячеек для измерения ЭДС цепей
со стеклообразным твердым электролитом:
а — без промежуточного расплавленного электролита;
б— с промежуточным расплавленным электролитом.
/ — вольфрамовые токоотводы; 2 — пробирки—твердый
электролит; 3— сплав натрия или калия;
4— наружная пробирка; 5— натрий или калий;
6 — расплавленный электролит; 7 — тигель
Конструкции измерительных ячеек для цепей со стеклами в
качестве твердого электролита с катионнои проводимостью приведены
на рис. 7.3.
При исследовании термодинамических свойств жидких сплавов
калия с элементами 11—VI групп Периодической системы в качестве
твердого электролита служили стекла, содержащие в достаточном
количестве К20 (3,8-5,0 мас.%)*. Наличие в стеклах оксида натрия
не сказывалось на результатах измерений. При исследовании
термодинамических свойств халькогенидных систем (K-S, K-Se, К-Те)
применялось специально синтезированное стекло с высоким
содержанием оксида калия (мае. %): 56-58 Si02,9-11 В203, 31-34 К20.
* Подробные сведения об исследовании термодинамических свойств
жидких сплавов калия содержатся в монографии: Морачевский А.Г., Бело-
глазов И.Н., Касымбеков Б.А. Калий: Свойства, производство, применение.
М.: Изд. дом "Руда и металлы", 2000. 192 с.
190
Такое стекло обладает высокой электрической проводимостью и
позволяет проводить электрохимические измерения при относительно
низких температурах (150-550 °С).
С конца 60-х годов XX века большое внимание уделяется высо-
копроводящим твердым электролитам на основе полиалюмината
натрия, известным под названием р-глинозема или Р"-глинозема\
Еще в ранних работах русских исследователей (Н.А. Торопов,
М.М. Стукалова, 1939,1940 гг.) была отмечена способность кристаллов
р-глинозема, состав которого приближенно описывается формулой
(Na20)1+jr-11 Al2O3(0,25<x<0,55), кпроцессам обмена ионов,что тесно
связано с подвижностью ионов
натрия в кристаллической решетке.
На высокую электрическую
проводимость этих материалов и
связанные с этим широкие возможности
применения было впервые указано
в 1967 г. Часть фазовой диаграммы
системы NaA102 - А1203 в
несколько упрощенном виде приведена
на рис. 7.4. Рентгеноструктурные
исследования позволяют сделать
вывод, что в р-глиноземе и в
Р -глиноземе имеются
существенные отклонения от стехиометрии.
При отсутствии легирующих
добавок (MgO, Li20) р -фаза
является метастабильной. Выше
температуры 1550 °С Р'-фаза дис-
пропорционирует на р-глинозем
и 5-NaA102 и не возникает вновь
при охлаждении. Увеличение
содержания Р"-фазы в
керамическом электролите резко снижает
величину его удельного сопротив-
t,'C
2000 Ь
1500 h
Расплав
Ь^"*"^
I- Расплав +
L Р-фаза
Г 8-NaA102 + р
U
[Y-NaA102 + р ,
L
Г i
ft
^
1
Расплав + AljOj
7
/
/
/
щ
>%
л L_
Р + А120,
1 1
t t"t
100
I II III AI20„ % (мол.)
Рис. 7.4. Фазовая диафамма систем
Na20—А1203 (заштрихованная область
соответствует области
сосуществования р- и р"-фаз): I — приблизительный
состав электролита 2Na20 • 11A120V
выпускаемого промышленностью;
II —состав, отвечающий
промышленному огнеупору" Монофакс"; III — стехио-
метрический состав 2Na20 • 11А120^
* Подробные сведения о р-глиноземе или Р'-глиноземе содержатся в
монографии: Саудорс Дж., Тилли А. Серно-натриевые аккумуляторы / Пер.
с англ. М.: Мир, 1988.672 с.
191
ления, однако электролит из чистого р'-глинозема подвержен
влиянию влаги, что ухудшает его механические свойства и проводимость,
В качестве электролита преимущественно используют двухфазные
системы р-глинозема и Р"-глинозема, имеющие при 350 °С удельную
электрическую проводимость 0,1-0,2 См ■ см-1.
Благодаря высокой подвижности катионов в слоях проводимости
Р-глинозема возможно их замещение на другие катионы без
существенного изменения остальных фрагментов структуры. Реально при
исследовании термодинамических свойств жидких сплавов
использовали р(Р")-глинозем, содержащий ионы К+ или Rb+.
Наблюдается очень хорошее согласие между величинами Е и
dE/dTцепей с жидкими сплавами натрия, измеренными со стеклом
или с р-глиноземом в качестве электролита. Это служит
дополнительным доказательством, что собственная электронная
проводимость образцов поликристаллического полиалюмината натрия
пренебрежимо мала. Специальные опыты с исследованием жидких
сплавов системы Na-Pb показали, что результаты измерений с
использованием в качестве электролита р-глинозема или р -глинозема
совпадают в пределах погрешности опытов.
В последние 30-40 лет при исследовании термодинамических
свойств металлических систем как в твердом, так и в жидком состоянии
нашли широкое применение твердые электролиты с анионной
проводимостью. Среди довольно многочисленныхтвердых анионпроводящих
электролитов наибольшее распространение при термодинамических
исследованиях получили фазы, обладающие проводимостью по ионам
О2- и F~. Это прежде всего смешанные оксиды на основе ZrOr Th02,
а также CaF2. Твердые растворы диоксидов циркония или тория,
стабилизированных оксидом кальция или оксидами редкоземельных
металлов (0,85 Zr02 + 0,15 СаО; 0,85 Th02 + 0,15 Y203), характеризуются
кристаллической решеткой типа флюорита с высокой концентрацией
вакансий в анионной подрешетке, по которым идет миграция ионов
О2-. Ионная проводимость наиболее широко применяемого твердого
электролита 0,85 Zr02 + 0,15 СаО составляет около 2,4 • 10~2См • см"1
при 1000 °С. Ионное число переноса кислорода (/.) при 1000 °С в
области/^ от 105до 10-10 Па близко к единице (//>0,99). Под величиной
/понимается отношение величины ионной составляющей проводимости
к общей проводимости твердого электролита. Ионная составляющая
проводимости полностью обеспечивается переносом ионов О2-.
192
Для твердого электролита 0,85 Th02 4- 0,15 Y203 число переноса
/. > 0,99 в интервале давлений кислорода от 1 до 10~20 Па (1000 °С).
В общем виде электрохимические элементы с кислородопрово-
дящими твердыми электролитами, применяемые при исследовании
термодинамических свойств твердых или жидких сплавов, можно
записать следующим образом:
А, АО/ О2" - электролит / АО, А-В; (7.9)
D, DOI О2" - электролит / АО, А-В; (7.10)
02 / О2" - электролит /А О, А-В, (7.11)
где А — наименее благородный компонент сплава А-В; АО — оксид
элемента А, находящийся в равновесии со сплавом в исследуемом
интервале температур и давлений; А, АО, D, DO, 02 — электроды
сравнения, представляющие собой равновесные смеси с фиксированным
при данной температуре химическим потенциалом кислорода (типа
металл — оксид металла, газовые смеси типа СО, С02, Н2, Н20;
воздух) или чистый кислород.
Все элементы являются концентрационными относительно
кислорода. Будем обозначать символами Ио2 и Цо2 химические
потенциалы кислорода в левом и правом электродах.
ЭДС (Е) концентрационного элемента типа
О2(ц!0 )/02" - электролит/02(цп0 ) (7.12)
выражается следующим соотношением:
Е=^1^о2- (713)
Как выше отмечалось, ионное число переноса кислорода tj близко
к единице. Тогда ЭДС элемента (7.12) равна
Е = ^о2-^- <7-,4>
Протекающие в элементе (7.9) процессы можно записать так:
на левом электроде А + О ~ =АО+2ё,
на правом электроде АО + 2ё=А(сплав Л -2?) + 02".
Таким образом, суммарная потенциалобразующая реакция
сводится к переносу одного моля компонента А с левого электрода на
правый, где он растворяется в сплаве А-В. Реально процесс выража-
193
ется в переносе одного моля кислорода (02) из правого электрода в
левый. С учетом выражения (7.14) можно записать
-tyA=]L\-tf=tf-\Ll0=2E!F9 (7.15)
гдеИл и №А — химические потенциалы компонента Л в левом и
правом электродах.
Все изложенное позволяет заключить, что, измерив ЭДС
элемента типа (7.9), можно определить термодинамическую активность
компонента А в сплаве А-В (причем сплав может быть и
многокомпонентным: A—B-C—D...).
Если по каким-либо соображениям контакт компонента Л сплава
с оксидом АО реализовать затруднительно (например, образование
фаз переменного состава в системе А—АО при температурах
эксперимента), можно использовать цепь типа (7.10). В качестве примера
рассмотрим цепь
Ni, NiO /Zr02 (CaO)/ Sn-Pd, SnOr
При температуре эксперимента (800-1000 °C) в случае
применения ячейки типа (7.9) пришлось бы иметь в качестве электрода
сравнения жидкую смесь Sn-Sn02. Для элемента типа (7.10) уравнение по-
тенциалобразующего процесса записывается в виде: D+AO = DO +A
(в сплаве А—В). Соответственно
цо« - Ц; = 2Е?= -Ацл +АС;ИО) - АСД/Ю). (7Л6)
В этом случае для нахождения химического потенциала
компонента А в сплаве А—В (точнее — величины Дц^) необходимо знать
величины стандартной энергии Гиббса оксидов АО и DO при
температуре эксперимента. К числу наиболее распространенных электродов
сравнения относится система Ni-NiO, например:
Ni, NiO /Th02 (CaO)/ Ni-V, VO.
ЭДС этой цепи связана с химическим потенциалом ванадия
(менее благородный элемент по отношению к никелю) следующим
уравнением:
-4£J= -Аци + AG(VO) - AG(NiO).
В работе элементов типа (7.11) принимают участие кислород или
воздух. Величина ЭДС цепи при этом может достигать нескольких
сотен милливольт из-за большой разности химических потенциалов
кислорода в газообразной фазе и в оксиде. Это требует особо надеж-
194
ного разделения электродных пространств, чтобы избежать
неконтролируемого переноса кислорода из одного электрода в другой.
Элементы типа (7.9) и (7.10) можно применять и с фторпроводя-
щими твердыми электролитами. В этом случае оксиды АО и DO
заменяют соответствующими фторидами. Из результатов исследования
элементов типа
A, AF/F~ — электролит/^F, А-В (7.17)
по уравнению Ацл = -EJ можно определить химический потенциал
компонента А в сплаве А-В. Чтобы избежать при исследованиях в цепях
типа (7.10) применения при расчетах данных об энергиях Гиббса
образования фторидов AF и DF (аналогично уравнению (7.16)), можно
проводить параллельные исследования ЭДС двух элементов относительно
одного и того же электрода сравнения. Так, при изучении сплавов Со-А1
при 900-1050 К были изучены два электрохимических элемента:
Co-Al, A1F3 /CaF2/ Y, YF3 (E,);
Al,AlF3/CaF2/Y,YF3(E2)
с электродом сравнения Y, YF3. ЭДС первого из элементов равна
RT 1
£. = -wlnaM+—[AG(MF3)-AG(YF3)],
ЭДС второго элемента
E2~[AG(A1F3)-AG(YF3)].
Разность ЭДС этих элементов непосредственно связана с
активностью алюминия в исследуемых сплавах:
RT
Е = Е1-Е2=-—\паА].
Основным условием успешного применения метода ЭДС с
твердыми электролитами является установление равновесий в обоих
электродах и электролите, а также на границах раздела фаз.
Используемые в практических исследованиях методом ЭДС с
твердым анионпроводящим электролитом приборы (ячейки)
отличаются большим многообразием и, как правило, подробно описаны
в оригинальных работах. Все основные требования к конструкциям
ячеек при исследовании твердых или жидких сплавов методом ЭДС
195
с электролитами с проводимостью по ионам О2- или F- обсуждены в
указанной ранее монографии (см. с. 183).
Эксперименты, как правило, ведут при постоянной температуре,
добиваясь постоянства во времени величины ЭДС. Время
достижения равновесной величины ЭДС зависит от многих факторов,
прежде всего от исследуемой системы и температуры проведения
опыта. Обычно оно составляет от нескольких часов до нескольких
суток. После того как ЭДС при данной температуре в течение
нескольких часов не отличается больше чем на I мВ и при отсутствии
монотонного изменения ЭДС от времени, изменяют температуру на
несколько десятков градусов и вновь дожидаются постоянства ЭДС.
В некоторых опытах с жидкими сплавами весь эксперимент ведут при
постоянной температуре, изменяя состав сплава путем добавления в
него взвешенных количеств одного из компонентов. Измерения ЭДС
ведут с помощью высокоомных (входное сопротивление 1О8-1014 Ом)
потенциометров, вольтметров, электрометров. Использование такой
техники особенно важно из-за относительно высокого
электросопротивления многих твердых электролитов. Весь температурный
интервал обычно проходят вверх
и вниз в ходе одного и того же
эксперимента. После опыта часто
проводят дополнительный рентге-
нофазовый анализ исследованных
сплавов.
Для проверки надежности
получаемых экспериментальных
данных используют различные
критерии:
а) проверяют совпадение
результатов, полученных при
прохождении исследуемого
интервала температур вверх и вниз; как
правило, совпадение оказывается
достаточно хорошим (рис. 7.5);
б) проверяют восстановление
равновесной при данной
температуре величины ЭДС после пропу-
Е, мВ о - /
Г, К
Рис. 7.5. Зависимость ЭДС цепи:
W|In(x)|lnCl - (КС1 - NaCl)|
|(In — В1)(ж)|\У от температуры: 1 —
повышение температуры; 2—
снижение температуры. Цифры справа —
молярная для индия в сплаве, х1п
196
екания через ячейку небольшого количества электричества в разных
направлениях;
в) проводят повторные эксперименты с другими образцами
сплавов исследованных ранее составов или различных составов из одной
и той же гетерогенной области;
г) проводят эксперименты относительно других электродов
сравнения или с другими твердыми или расплавленными электролитами
(рис. 7.6).
а) б)
TV 0,2 0,4 0,6 0,8 т г>- 0,2 0,4 0,6 0,8 т„
Bi Y In Bi x In
XIn In
Рис. 7.6. Изотермы активности индия и висмута в системе In—Bi:
а — рассчитанные по измерениям ЭДС с расплавленным электролитом;
б— по измерениям ЭДС цепей с твердым кислородпроводящим электролитом
Кратко рассмотрим погрешности определения
термодинамических величин, которые получаются при измерениях ЭДС с
твердыми анионпроводящими электролитами. Если считать, что средняя
погрешность определения величины ЭДС ±1 мВ, то погрешность
величины Ац.составляет ±96 Дж • моль-1 фтора или ±192 Дж • моль-1
кислорода. Точность определения АН. и AS. существенно ниже,
поскольку для расчета этих величин необходимо знать изменение ЭДС
в зависимости от температуры.
Хотя применение твердых электролитов с анионной
проводимостью при термодинамических исследованиях получило широкое
распространение, необходимо иметь в виду некоторые ограничения.
Основные ограничения связаны с необходимостью проводить измере-
197
ния ЭДС при таких температурах, давлениях и химических потенциалах
компонентов сплавов, при которых твердые электролиты обладают
чисто ионной проводимостью (/. > 0,99). В противном случае при
расчете величины ЭДС необходимо интегрировать уравнение (7.13), зная
зависимость /. =Лц0 )• ^° же относится к твердому электролиту CaFr
Другим важным фактором являются дефекты в керамических
образцах твердых электролитов (пористость, неоднородность
структуры). Для термодинамических исследований подходят лишь изделия
из твердых электролитов, плотность которых составляет не менее
95-98 % от теоретической.
Помимо использования в чисто научных исследованиях твердые
электролиты с проводимостью по ионам кислорода находят широкое
применение в металлургии в качестве датчиков активности
кислорода в расплавленных металлах и сплавах (в том числе в сталях), в
отходящих газах и т. д.
7.3. Методы измерения давления насыщенного пара
Если из эксперимента для какого-либо из компонентов
изучаемой системы получена зависимость/>.=Л 7) и известна аналогичная
зависимость /?.° — J{T) для чистого вещества, то можно вычислить
соответствующие парциальные молярные термодинамические
характеристики этого компонента. Для реальных систем а. = р./р °, тогда
AG, = RT\n
Pi)
= 2,303ЛШ
Pi
(7.18)
Принимая во внимание, что (5AG./97) = -AS. и Д# = Д(? + TASn
получаем
n2plga,
ДЯ,=-2,303ЛГ
дТ
или
ДЯ,=2,303Я
(7.19)
(7.20)
)р
Нахождение производной, входящей в уравнение (7.20), не
представляет трудностей, так как температурные зависимости р( и р° в
198
относительно небольшом интервале температур можно выразить с
помощью уравнений
При изучении термодинамических свойств металлических и
других систем существует большое число различных методов измерения
давления насыщенного пара.
Выбор того или иного метода зависит от величины измеряемого
давления пара, температуры, конкретных особенностей системы.
Можно выделить три основные группы методов: статические,
динамические и кинетические. Каждая из групп имеет свои достоинства
и недостатки.
7.3.1. Статические методы
К их числу относится группа методов, в которых изучаемая
система находится в замкнутом, изолированном от внешней среды
объеме при определенной температуре. Рассмотрим только наиболее
типичные методы исследования, относящиеся к этой группе.
Метод с мембранным нуль-манометром — один из самых
простых статических методов. Исследуемое вещество находится в
замкнутом сосуде, снабженном тонкой стеклянной или кварцевой
мембраной. Мембрана может быть плоской, гофрированной, вогнутой.
За положением мембраны наблюдают с помощью микроскопа либо
применяют фотоэлектрическую систему регистрации отклонения
мембраны от исходного положения. Изменяя внешнее по отношению
к мембране давление газа, возвращают мембрану в нулевое исходное
положение. По величине внешнего компенсирующего давления
судят о давлении насыщенного пара вещества в мембранной камере.
Внешнее давление измеряют с помощью манометров.
Основное достоинство мембранного метода состоит в том, что
весь прибор может быть выполнен из кварцевого или простого стекла.
Это позволяет изучать системы с химически агрессивными
компонентами (сера, фосфор и др.). Мембранный метод, однако, имеет
ограниченную чувствительность, и его целесообразно применять при
давлениях насыщенного пара от 130 Па и выше.
Изотенископный метод. Особенность его заключается в том,
что исследуемое вещество отделяется от внешнего пространства не
199
К A
A W
A u
1 /Z 1^\ /•"
\ H у У п
1 II
i 1 JJJ
И Ik W#~fl
И ^v ^^
и ^
\r f if r/i^.fij
и I
071 J
1 1,1.1' II I nil/ I
I W
( 1Л
I /\ и
H
H
и
K7777
kffl&
//ZZ2.iJ.
И
"""""""""" Pi
rl
7777VI
LL-/
мембраной, а небольшим U-образным
жидкостным нуль-манометром. В
зависимости от свойств исследуемого вещества,
интервала температур манометрической
жидкостью могут служить масло, ртуть,
расплавленные металлы или соли. Так,
например, при исследовании давления
пара над амальгамами применяли в
качестве манометрической жидкости
расплавленную смесь NaN03 — LiN03 — KN03
(Гпя=120вС).
Общая схема изотенископа приведена
на рис. 7.7. Прибор прост по конструкции и
удобен в работе. Главный его недостаток —
обязательный контакт паров исследуемого
вещества с жидкостью нуль-манометра.
Необходимость подбора относительно
инертной манометрической жидкости,
учет ее температуры плавления и
испарения — все это может привести к тому, что
температура исследуемого вещества Т{ и
температура жидкостного манометра Т2 могут быть различны. За
изменением уровня жидкости в нуль-манометре следят через смотровое
стекло в прорези печи. Величину исследуемого давления пара
определяют с помощью компенсационно-измерительного устройства.
Интервал измеряемыхдавлений может быть большим (от 100 Паи
выше), интервал температур в значительной мере зависит от свойств
манометрической жидкости. Наличие двух температурных зон ведет
к дополнительной погрешности, поэтому лучше всего располагать
жидкостной нуль-манометр в той же температурной зоне, что и
исследуемый сплав.
Метод точки росы основан на определении температуры
конденсации, или точки росы. Образец сплава, содержащего летучий
компонент (например, Zn или Cd) помещают в один конец
откачанной кварцевой трубки и выдерживают при постоянной температуре
Тг Температуру другого конца трубки постепенно понижают до тех
пор, пока не начнется конденсация паров металла (температура Т2).
Рис. 7.7. Схема
изотенископа: 1 — исследуемое
вещество; 2 — жидкостной нуль-
манометр
200
Поскольку давление пара во всем
объеме трубки одинаково,
парциальное давление пара металла над
сплавом при температуре Т] равно
давлению пара над чистым
компонентом при температуре Т2: (рш)т =
= (Рме )т \ Тх > Тг Для пользования
этим методом необходимы данные о
зависимости давления пара чистого
летучего компонента от
температуры, т. е. надо знать р°Ме =j{T).
Представляет интерес и
высокотемпературный вариант этого метода,
примененный, в частности, при
исследовании жидких сплавов системы
цинк-марганец. Схема контейнера-
ампулы приведена на рис. 7.8. В
верхней части ампулы, находящейся
при температуре Т{ в
высокотемпературной зоне двухзонной печи,
помещается марганец, в нижнем
конце при температуре Т2 — цинк.
При равновесии давление пара цинка
при низшей температуре Т2 равно его
парциальному давлению над сплавом
системы Mn-Zn при температуре
Ту Опыты проводили при заданных
температурах и различном времени
до достижения постоянного
содержания цинка в сплаве, равновесие с
которым устанавливается в системе.
Температура расплавленного
марганца всегда поддерживалась посто-
Рис. 7.8. Схема
высокотемпературного прибора для определения
давления насыщенного пара
методом точки росы: / — пробирка
из корундиза; 2 — корундизовая
подставка; 3 — легкоплавкий
компонент сплава (цинк); 4 —
пористый наполнитель; 5—тугоплавкий
компонент сплава (марганец);
Тх — высокотемпературная зона;
71, — низкотемпературная зона
янной (1573 К), а цинка в различных
опытах изменялась в пределах от 1050 до 1160 К.
Метод точки росы применим при давлениях от 1,3 до 1300 Па, но
давление пара летучего компонента должно быть, по меньшей мере,
на три порядка выше давления пара другого компонента.
201
Изопиестинеский метод является дальнейшим развитием метода
точки росы. Ряд сплавов различного состава (х,, xv х3 ...), а также
чистый исследуемый компонент находятся при различных
температурах, но имеют общую паровую фазу: px{xr T{) = p2(xv Т2) = ppcv T2) =
= ... = р°Ме( Т0). Если х, > х2 > xv то, естественно, Тх< Т2< Ту
Рассмотрим применение метода на примере системы магний—
олово в жидком состоянии (рис. 7.9). Температуру металлического
магния поддерживали в каждом отдельном опыте постоянной (Г0),
что обеспечивало определенное давление паров магния (р°м£ во всей
камере. Сплавы Mg—Sn были размещены в основном отсеке камеры,
причем температура сплавов была выше, чем температура чистого
магния. Нагревательные элементы обеспечивали наличие в печи
определенного градиента температуры.
V/, У////////Л///////Л
Рис. 7.9. Схема установки для определения давления насыщенного пара
изопиестическим методом: У — печь, создающая градиент температуры;
2— реакционная трубка; 3 — графитовый тигель с чистым металлом (магний);
4— экран; 5— графитовые тигли со сплавами (магний-олово)
Отсек с чистым магнием и труба со сплавами нагревались
раздельно. Опыт длился 48-72 часа, по его окончании создавалась атмосфера
аргона, производились быстрое охлаждение сплавов и их анализ.
Метод сравнения рассмотрим на примере жидких сплавов
кадмия. Главное требование — летучим должен быть только один из
компонентов сплава. В эвакуированную кварцевую ампулу помещают
два сосуда: один — с исследуемым сплавом, другой — со сплавом
202
сравнения, содержащим тот же летучий компонент. После выдержки
при заданной температуре, при которой происходит обмен летучим
компонентом через паровую фазу до установления равновесия,
сплавы быстро охлаждают и определяют их состав. Так как при равновесии
при одинаковом выборе стандартного состояния активности летучего
компонента должны быть равны, а его активность в сплаве
сравнения в зависимости от состава известна, то можно определить
активность летучего компонента в исследуемом сплаве. В частности, при
изучении активности кадмия в жидких сплавах с галлием системой
сравнения служила хорошо исследованная различными методами
система кадмий-олово.
При определении активности свинца в жидких сплавах с
никелем (хрь < 0,10; 1430-1510 °С) в качестве сплава сравнения служила
система Cu-Pb. При равновесии
п =/J . v _ *Pb(Cu-Pb)Ypb(Cu-Pb)
"Pb(Cu-Pb) "Pb(Ni-Pb)' JPb(Ni-Pb) _
*Pb(Ni-Pb)
В этих опытах применялась аппаратура из алунда, графита.
Метод точек кипения не в полной мере является статическим, так
как исследуемая система жидкость-пар не находится в замкнутом
объеме. Тем не менее создаются условия, препятствующие уносу вещества
из зоны с высокой температурой, благодаря вводу в систему инертного
газа и применению специальных диафрагм. Метод имеет целый ряд
вариантов, существенно отличающихся аппаратурным оформлением.
Рассмотрим один из классических вариантов метода. В его основе —
определение момента резкого увеличения скорости
парообразования, когда давление насыщенного пара образца становится равным
давлению, создаваемому за счет введения в систему инертного газа.
Этот эффект может быть достигнут как при повышении температуры
при поддержании постоянного давления (с применением маностата),
так и при понижении давления в условиях постоянства температуры.
В первом случае речь идет об изобарическом варианте метода, во
втором случае — об изотермическом варианте.
Одна из возможных схем прибора приведена на рис. 7.10.
Образец находится в кварцевом, графитовом или алундовом тигле,
подвешенном к кварцевой пружине. В ходе опыта следят за изменением
длины пружины /, которое пропорционально изменению массы
203
образца. График зависимости А/ = J{p) или
А/ = J[T) представляет собой две прямые,
точка пересечения которых отвечает моменту
начала кипения. Как правило, более
отчетливый эффект наблюдается в изотермических
условиях.
7.3.2. Динамический метод измерения
давления насыщенного пара
Метод насыщения инертного газа парами
исследуемого вещества — метод уноса,
переноса, потока — получил самое широкое
распространение при изучении
термодинамических свойств различных металлургических
объектов. Известный объем инертного газа,
медленно пропускаемого над исследуемым
образцом, насыщается летучим
компонентом (или компонентами), который затем
конденсируется в холодильнике. Одна из
типичных конструкций прибора приведена
на рис. 7.11. В ходе опыта измеряют объем
газа и количество конденсата. Если число
летучих компонентов два и более, то
необходим анализ сконденсированного металла.
В общем случае такие экспериментальные
данные позволяют определить плотность
пара вещества, насыщающего при данной
температуре проходящий над ним газ. Для
расчета давления насыщенного пара надо
знать молекулярную массу вещества в
парообразном состоянии.
Расчетная формула для нахождения давления насыщенного пара
имеет вид рНЛС = [п]/(п1 + я2)]/?, гдернгс — давление насыщенного пара
вещества; р — общее давление в системе; п] — число молей
испарившегося вещества при прохождении п2 молей газа-носителя:
л, = Ат/М; п2 = (р- /?нас) V/RT.
Рис. 7.10. Схема прибора
для определения
давления насыщенного пара
методом точек кипения:
/ — шлиф; 2— кварцевая
спираль; 3 — индикатор
изменения длины
пружины; 4 — нагреватель;
5 — тигель со сплавом;
б —термопара
204
^К вакуумной
системе
й 2 3
^~J L*^ К манометру , i
KpeoMGipy Iji / /I
|| )•••••• •/••••• I
_ll ' I H 4
HgQ,i, Д^ ^Q / 4 I I / У
5 » ' IjiT y,Wrl
Очищенный аргон
Рис. 7.11. Схема прибора для определения давления
насыщенного пара динамическим методом (метод уноса,
потока): / — стальная трубка; 2 — лодочка с навеской металла
или сплава; 3 — нагревательная печь; 4 — термопара в чехле;
5— конденсатор с водяным охлаждением
Здесь Am — количество испарившегося (или
сконденсировавшегося) вещества; М — его молекулярная масса; V — объем газа-
носителя.
Если р > ртс, пх < nv то ртс = (Am/MV)RT.
Из эксперимента определяют величины Am и V, отношение
которых и составляет плотность пара.
При использовании этого метода важно учитывать, что перенос
пара из горячей зоны в холодную осуществляется благодаря двум
факторам — собственно переносу и термодиффузии. Роль
диффузии возрастает по мере увеличения давления насыщенного пара и
уменьшения скорости газового потока. Вместе с тем при большой
скорости потока может наблюдаться недонасыщение газа. Вопрос
о действительном насыщении при заданной скорости потока и о
влиянии диффузии необходимо решать экспериментально, так как
оба фактора существенно зависят от конструкции установки. Лучшие
результаты получаются в таком интервале скоростей газа-носителя,
когда давление пара не зависит от скорости. Предпринимаемая
иногда экстраполяция на нулевую скорость газа-носителя, как правило,
приводит к неверным результатам (рис. 7.12).
205
мг • л"1
13 Ь
12
10
J I I I I I I I L
20 40
60
80 100
CM3 • МИ1Г1
Рис. 7.12. Содержание магния в газе-носителе
(аргон) в зависимости от скорости пропускания его
над жидким сплавом системы магний—алюминий
На рисунке четко виден горизонтальный участок ("плато"), в
пределах которого не наблюдается зависимости содержания
испаряющегося компонента в газовой фазе от скорости пропускания газа-
носителя. Этот интервал скоростей предпочтителен при определении
давления насыщенного пара.
Важен также выбор насыщаемого газа. Водород восстанавливает
на металлах оксидную пленку, которая может быть причиной
неполного насыщения газа. Однако из-за большой теплопроводности и
малой вязкости водорода увеличивается термодиффузия. Чаще всего
в качестве газа-носителя применяют аргон высокой чистоты.
Метод прост в экспериментальном отношении и нашел самое
широкое применение при исследованиях сплавов цинка, кадмия,
магния. Его использовали также при изучении равновесий жидкость-пар
в системах Na-Bi, Na-Pb.
Метод уноса особенно эффективен, если оба компонента сплава
летучи (например, Cd-Zn или Mg-Zn), так как в этом случае анализ
конденсата дает отношение числа молей в паровой фазе и,
следовательно, отношение парциальных давлений компонентов.
Полученные независимо одно от другого значения активности
компонентов бинарной системы должны соответствовать уравнению
206
Гиббса-Дюгема. Для проверки согласо- j V
ванности результатов можно использовать
соотношение
*,=!
(7.21)
Выводится это уравнение из общих
термодинамических соотношений очень
просто. Для интегральной избыточной
энергии Гиббсадвухкомпонентной системы
можно записать
AG*36 = RT(xllny] + x2lny2).
В дифференциальной форме это
выражение имеет вид
Yz»
0,2
0,1
0
-0,1
-0,2
-0,3
-0,4
0,2
0,4/
0,6 0,8 1,6
*Mg
Рис. 7.13. Проверка
согласованности
экспериментальных данных по
уравнению (7.21) для
системы магний—цинк
dAOn6 = RT(xldlnyl + x2dlny2 +
+ \nyldxl + lny2rfx2).
В соответствии с уравнением Гиббса-
Дюгема сумма первых двух членов в скобках равна нулю. Учитывая,
что dx2 = -dxv получаем
RT у2 '
Интегрирование следует производить от х{ = 0 до хх = 1.
Величина АО136 при верхнем и нижнем пределах интегрирования
равна нулю. Следовательно,
*1=1лл/?изб *,=!
RT
= Г 1п^х,=0.
J У2
(7.22)
х,=0 х,=0
Если справедливо это уравнение, заштрихованные площади на
рис. 7.13 будут равновеликими.
Динамический метод рекомендуется применять для определения
давления насыщенного пара в интервале от 1,3 • 102 до 1,3 • 104 Па.
7.3.3. Кинетические методы измерения давления насыщенного пара
В группе кинетических методов данные о величине давления
насыщенного пара получаются на основе измерения скорости
испарения вещества с открытой поверхности в глубоком вакууме (метод
207
Лэнгмюра) или скорости истечения пара в вакуум через небольшое
отверстие (метод Кнудсена). И в том и в другом случаях скорость
испарения или истечения может быть связана с величиной
давления пара с помощью уравнения, выводимого на основе простейших
представлений молекулярно-кинетической теории. Равновесие
конденсированная фаза — пар можно рассматривать как
динамическое равновесие, при котором количество молекул, испаряющихся с
данной поверхности в единицу времени, равно количеству
конденсирующихся молекул.
Как известно, масса частиц пара т\ соударяющихся в единицу
времени с единицей поверхности, связана с плотностью пара р и
средней скоростью молекул v°p соотношением
i pvcp
т =-
4
В свою очередь, если паровая фаза подчиняется уравнению
идеального газа, то
р-М
Р- RT ,
где М— молекулярная масса вещества в паровой фазе. Из
молекулярно-кинетической теории следует также, что средняя скорость молекул
равна
vcp=yJSRT/nM.
Совместное решение этих трех уравнений приводит к следующему
выражению для массы /и1:
ml=py]M/2nRT.
В реальных условиях в это уравнение приходится вводить
дополнительный коэффициента — коэффициент испарения. Величина его
может изменяться в очень широких пределах в зависимости от целого
ряда факторов — природы вещества, характера процесса испарения,
рельефа поверхности. Конечное уравнение для расчета давления пара
приобретает вид
mj^Rf 23)
У StocV А/
где m — масса вещества (г), испарившегося за время т (с) с
поверхности S (см2). При таком выборе единиц давление р выражается в
дин -см-2(1 дин -см"2 = 0,1 Па = 7,50- 1(Н мм рт.ст.).
208
Рис. 7.14. Схема эффузион-
ной камеры: / — ввод
термопары; 2 — корпус камеры;
3 — стаканчик с металлом или
сплавом; 4 — диафрагма с эф-
фузионным отверстием
Метод Лэнгмюра обладает
чрезвычайно высокой чувствительностью,
позволяет измерять давление паров до
1,3 • 10~5 Па (10~7 мм рт.ст.). Однако
применение метода существенно
осложняет необходимость оценки величины
коэффициента испарения*.
При использовании метода Кнуд-
сена исследуемый сплав помещают
в сосуд, в крышке которого имеется
маленькое отверстие с точно известной
площадью. Камеры изготавливают из
молибдена, графита, тантала.
Материал должен обладать коррозионной
стойкостью по отношению к парам
исследуемого вещества. Схема камеры
изображена на рис. 7.14.
Пары вещества, вышедшие из отверстия, можно конденсировать
на охлаждаемой поверхности и затем определять количество
конденсата. Можно также непрерывно следить за количеством
испарившегося вещества по изменению массы сосуда. Весь прибор помещают
в печь, термостатируют и откачивают до глубокого вакуума. Убыль
массы вещества А/и связана с истечением (эффузией) его через
отверстие при постоянной разности между давлением насыщенного
пара металлар при температуре опыта и остаточным давлением при
вакуумировании /?ост, она пропорциональна времени т и площади
отверстия 5. Если р » /?ост, то получаем уравнение, аналогичное
уравнению (7.23). Кроме указанных ранее положений молекулярно-
кинетической теории при его выводе для метода Кнудсена
дополнительно сделаны следующие допущения:
1) отсутствуют столкновения молекул между собой как в объеме
эффузионной камеры, так и в области эффузионного отверстия;
2) длина свободного пробега частицы велика по сравнению с
размерами эффузионного отверстия;
* Подробный анализ факторов, влияющих на величину коэффициента
испарения, содержится в монографии: Львов Б.В. Терморазложение твердых
и жидких веществ. СПб.: Изд-во Политехи, ун-та, 2006. 278 с.
209
3) все молекулы, попавшие в эффузионное отверстие, проходят
через него, не изменяя направления движения, так как края отверстия
бесконечно тонки.
Первые два условия обычно выполняются, если давление пара
не превышает 1,3-13,3 Па, а радиус отверстия не более 1 мм.
Влияние конечной толщины краев отверстия и особенности его
формы могут быть учтены с помощью коэффициента Клаузинга к,
который определяется размерами эффузионного отверстия. Основные
требования к геометрическим размерам и форме отверстия таковы:
средняя длина свободного пробега молекул в камере должна,
по крайней мере, на порядок превосходить диаметр эффузионного
отверстия;
канал эффузионного отверстия должен быть минимальной
длины;
площадь испарения вещества должна не менее чем в десять раз
превышать площадь эффузионного отверстия.
Одна из эмпирических формул для оценки коэффициента
Клаузинга при 1/г< 1,5 (/— высота эффузионного отверстия; г— его
радиус): *= 1/(1+0,5//г).
С учетом коэффициента Клаузинга уравнение (7.23) можно
записать так:
^&Шш (7<24)
ЯтакЧ М
Метод Кнудсена позволяет определитьрурМ, для расчета р
необходимы сведения о молекулярной массе вещества в паровой фазе.
В последнее десятилетие при исследованиях
термодинамических свойств металлических систем как в жидком, так и в твердом
состояниях получило широкое распространение сочетание ячейки
Кнудсена с масс-спектральным изучением паровой фазы*. Это
существенно расширяет возможности исследований, повышает их
надежность, дает дополнительную информацию о составе паровой
фазы.
* Детальное рассмотрение проведения масс-спектральных
термодинамических исследований содержится в монографии: Сидоров Л.Н.,
Коробов М.В., Журавлева Л.В. Масс-спектральные термодинамические
исследования. М.: Изд-во Моск. ун-та, 1985. 208 с.
210
Масс-спектрометр — это
устройство, позволяющее разделить пучок
заряженных частиц на фракции с
одинаковым отношением массы к заряду.
Типичная схема эксперимента сводится
к следующему комплексу операций
(рис. 7.15).
Исследуемый сплав помещается в
эффузионную камеру Кнудсена и
нагревается до температуры Т. При этом
давление насыщенного пара сплава должно
лежать в интервале 10~7-10~2 Па.
Молекулярный пучок, истекающий из эффу-
зионного отверстия, проходит систему
коллимирующих щелей и поступает в
область ионизации, где пересекается с
потоком ионизирующих частиц.
Наиболее подходит для выполнения этой
задачи ионизация с помощью электронного
удара. Минимальная энергия
ионизирующих электронов составляет несколько
электрон вольт. Образовавшиеся ионы
вытягиваются из области ионизации,
фокусируются, ускоряются и поступают
в масс-анализатор. После прохождения
масс-анализатора ионы разделяются по величине отношения массы
к заряду, попадают в коллектор, где производится измерение
интенсивности ионных токов для различных массовых чисел.
Масс-спектр является основой определения состава паров.
Не касаясь деталей анализа масс-спектров, укажем на особенности
испарения основных элементов. Пар таких элементов, как В, Al, Ga,
In, Pb, Cr, Mn, Ni, Cu, Ag, Au, Zn, является в основном
одноатомным, но в большинстве случаев содержит небольшие количества
двухатомных молекул. Пары серы и селена содержат сложные
молекулы, которые образуют, по-видимому, непрерывные ряды от S2
до S8 и от Se2 до Se8. В парах Р, As, Sb содержатся только молекулы
Р4, As4, Sb4.
Рис. 7.15. Схема
высокотемпературного масс-спектрального
эксперимента: 7 —эффузион-
ная ячейка; 2— ионизационная
камера; 3 — масс-анализатор;
4 — приемное устройство
211
Уравнение Гиббса-Дюгема для системы А—В в форме
xAd\naA + xBd\naB = 0,
согласно Белтону и Фруэхану, можно преобразовать следующим
образом. Если вычесть из обеих частей равенства величину dlnaB и
принять во внимание, что хл = 1 — хв, получим
dlnaB = -xAdln(aA/aB). (7.25)
Из соотношений хА + хв = 1, dxA + dxB = О и xA(dxJxA) + xB(dxJxB) = О
следует: xAd\r\xA + ^rflnx^ = 0.
Вычитая из обеих частей этого равенства d\nxB и проведя
преобразования, подобные описанным выше, получаем
Лпхв = -xAdln(xA/xB). (7.26)
Вычитание уравнения (7.26) из уравнения (7.25) приводит к
следующему результату:
d\naB -d\nxB =-xA
</ln^L_</lni±
или
d\nyB=-xAd
i аЛ г ХА
In—-In—
Отсюда
*в
|пУя = J xAdln
**=i
(7.27)
Соответственно
1пу^=~ J* xBd\n
aRx
влл
aAx
АЛВ
(7.28)
Для масс-спектрометрического исследования такого рода
уравнениям удобно придать вид
^Уа=~ \ xBd\g
(
*Л=1
I ахв
ч
'gY* = J хА<Н&
(
*u=i
П*в
Здесь // и // -
ионные точки, пропорциональные парциальным
давлениям компонентов в паровой фазе и активностям их в жидкой
фазе. Соответственно для парциальных молярных энтальпий
смешения можно записать
212
А ' J d(l/T)
*™ V Мл
Xg f - ■ ч
ДЯв= 2,303 J xAd
Метод исследования термодинамических свойств жидких
сплавов с использованием масс-спектрометрии наиболее применим
для систем, в которых парциальные давления обоих компонентов
сплава приблизительно равны. Отношения ионных токов в этом
случае будут близки к единице. Этим методом изучены, в частности,
системы Fe-Ni, Fe-Co, Fe-Cu, Co-Ni, многие другие системы как
в твердом, так и в жидком состояниях. Метод высокотемпературной
масс-спектрометрии в сочетании с ячейкой Кнудсена как источником
молекулярного пучка был успешно применен и при исследовании
трехкомпонентных систем Fe-Ni-Co, Fe-Ni-Cr и других.
К числу кинетических методов относятся также торзионный и
торзионно-эффузионный методы. Торзионный метод основан на
измерении силы отдачи, которая возникает при истечении пара из
отверстий эффузионной камеры. Сила отдачи пропорциональна
скорости испарения, массе пара и разности давления по обе стороны
отверстий в камере. Силу отдачи можно измерить, если эффузионную
камеру, имеющую два отверстия (рис. 7.16), направленные в разные
стороны, подвесить на упругую нить.
При истечении пара
возникает закручивающий момент, и угол
поворота служит мерой отдачи:
F=Ocp,
где F — сила, поворачивающая
систему; ф — угол поворота; D —
упругая постоянная нити. В свою
очередь можно записать
F=(qxSx + q2S2)p/2,
где qx и q2 — расстояния от точки
подвеса до центров отверстий; 5,
и S2 — площади отверстий.
эо/п
Рис. 7.16. Схема эффузионной
камеры с двумя отверстиями: / — эффузи-
онные отверстия; 2 — тигель; 3 —
исследуемый сплав; 4 — корпус камеры;
5 — крышка (два эффузионных
отверстия расположены симметрично)
213
Тогда
р = 2/)Ф/иД + <7252)=Сф.
Постоянная С может быть найдена, если известны D, qv qv Sv Sv
или путем градуировки прибора по веществу, давление пара которого
известно. Как правило, при изготовлении эффузионной камеры
принимаются меры, чтобы qx было равно q2 и Sl равно Sr
Торзионно-эффузионный метод представляет собой объединение
в рамках одного прибора двух методов: торзионного и эффузионного.
Технически для этого достаточно закрепить нить подвеса на
коромысле вакуумных весов. Таким образом, одновременно измеряют
силу отдачи и количество испарившегося вещества. Совмещенный
торзионно-эффузионный метод позволяет определить не только
общее давление пара, но и его среднюю молекулярную массу.
7.4. Метод исследования гетерогенных равновесий
(циркуляционный метод)
Циркуляционный метод в некоторых руководствах относится
к динамическим методам*, в некоторых — к статическим**. Метод
применяется, в частности, для изучения процессов диссоциации
труднодиссоциирующих веществ (сульфидов, оксидов), а также для
определения активности одного из компонентов в металлических
системах. Газ циркулирует над навеской исследуемого вещества в
известном ограниченном объеме до достижения равновесия между
конденсированной и газовой фазами в результате протекания
химической реакции. Например:
MeS(TB) + Н2(г) <-> Ме(тв) +
+ H2S(r),^ = pH2S/pH2; (7.29)
1 Рн 'Ps/2
H2S(r)<-> H2(r)+iS2(r), Kn = 2 S2 . (7.30)
2 Ai2s
Парциальное давление паров серы в равновесии с твердой фазой
определяется из равенства
'См., например: Суворов А. В. Термодинамическая химия парообразного
состояния. Л.: Химия, 1970. 208 с.
"См.: Ванюков А.В., Исакова Р.А., Быстров В.П. Термодинамическая
диссоциация сульфидов металлов. Алма-Ата: Наука КазССР, 1978. 272 с.
214
Ph=(KxKn)\ (7.31)
Константа диссоциации сероводорода хорошо изучена для
большого диапазона температур и может быть рассчитана по известным
уравнениям или на основании табличных данных, характеризующих
образование H2S из элементов (табл. 7.3).
Таблица 7.3
Термодинамические характеристики H2S
(стандартное состояние для серы — двухатомный идеальный газ)
Г, К
298
400
600
800
100
1200
1400
1600
1800
I 2000
С0
р
s;
Дж • моль-1 • К-1
34,19
35,58
38,94
42,52
45,79
48,47
50,59
52,26
53,59
54,66
205,65
215,88
230,93
242,62
252,47
261,06
268,70
275,57
281,80
287,51
nT-n2w
ah;
ag;
кДжмоль-1 ]
0,00
3,55
11,00
19,14
27,98
37,42
47,58
57,62
68,21
90,70
-84,87
-85,98
-87,89
-89,26
-90,10
-90,54
-90,73
-90,78
-90,76
-90,70
-73,26
-69,12
-60,26
-50,83
-41,12
-31,27
-21,38
-11,47
-1,55
8,36 '
При правильной постановке эксперимента и прежде всего малом
объеме газового пространства относительно навески вещества, т. е.
когда переход вещества в газовую фазу практически не изменяет
состав твердого сульфида металла (как фазы переменного состава)
или изменение состава может быть уточнено по составу газа и его
количеству, циркуляционный метод может быть использован для
определения давления диссоциации не только для составов, лежащих
на металлическом крае области гомогенности, но и для сульфидов,
лежащих внутри области гомогенности.
Циркуляционный метод нашел самое широкое применение при
определении активности серы в сульфидных расплавах. В этих случаях
наряду с равновесием (7.30) следует рассматривать равновесие
Н2(г) + S(paciwaB) <-> H2S(r), (7.32)
tf = -^L. (7.33)
Рн2Ъ
215
При этом за стандартное состояние для серы принимается S2 (г)
при 1 атм (1 атм = 1,01325 • 105Па). Величина К вычисляется из
значения AGy при соответствующей температуре для равновесия
H2(r)+l/2S2(r)<e>H2S(r).
Типичные кривые, иллюстрирующие время установления
равновесия при определении циркуляционным методом активности серы
в расплавах систем Ni-S и Ni-Fe-S при температуре 1250 °С,
представлены на рис. 7.17.
Путем изучения гетерогенного равновесия с газовой фазой может
быть определена активность одного из компонентов в металлических
сплавах. При этом все другие компоненты не должны принимать
участия в протекающей химической реакции. Поясним принцип
метода на конкретных примерах. Рассмотрим равновесие между
сплавом Cu-Au и газовой фазой Н2+ H2S. Реакцию можно записать
следующим образом:
гн.
i,o h
0,5
/ /°
Г .. 1 ... 1
^"~**0-—Q О ■
д^-0,306
*РС- 0,432
xs- 0,262
^^п—п О п
х,-0,644
xs - 0,356
. 1 _.. i I
О
-О О
Л о
1
12 3 4 5 6 7
Время, ч
Рис. 7.17. Типичные экспериментальные кривые при определении
активности серы в расплавах систем Ni—S и Ni— Fe—S методом
гетерогенного равновесия (1523 К)
216
2Си(сплав) + H2S(r) = Си28(сульфидная фаза) + Н2(г). (7.34)
Если принять, что в условиях опыта Cu2S представляет собой
самостоятельную фазу определенного состава, а газовая фаза следует
законам идеального газа, то при равновесии
*,="
/^H.S^Cu
(7.35)
где рн и рн s — парциальные давления Н2 и H2S в газовой фазе.
Величина Кр в уравнении (7.35) может быть рассчитана на основании
стандартных термодинамических характеристик компонентов
реакции (7.34) с чистой медью (аСи = 1) или определена в этих же условиях
экспериментально:
к -^
Ph2s
Тогда величину активности меди сплава можно определить по
выражению
flCu =
Ри2
Рнл
Pus
V/2
(7.36)
Для определения активности хрома в сплавах с железом может
быть изучено равновесие
2Сг(сплав) + ЗН20(г) = Сг203(тв) + ЗН2(г). (7.37)
Оксид хрома (III) Сг2Оэ представляет собой в условиях опыта
самостоятельную фазу стехиометрического состава, и его активность
принимается равной единице (аСг 0 = 1). Константа равновесия
реакции (7.37) с учетом этого может Ьыть записана следующим
образом:
*,=
р1
Рщоас<
Для реакции с чистым хромом (ас = 1)
Кр =
м
Ко)
Тогда активность хрома в сплаве с железом равна
If .о V/2
uCr
Р\\2о
Рн,
Рн7о
(7.38)
217
Активность углерода, растворенного в железе, может быть
определена методом изучения гетерогенных равновесий. Наиболее удобны
для экспериментального исследования реакции
С (твердый раствор) + 2Н2(г) = СН4(г), (7.39)
С (твердый раствор) + С02(г) = 2СО(г). (7.40)
Константы равновесия этих реакций соответственно равны
Кр=-^-, (7.41)
Рн^с
2
к _Рсо_ (742)
Рсо2ас
При равновесии в реакциях (7.39) и (7.40) с чистым графитом
(ас = 1) выражения (7.41) и (7.42) принимают вид
„ _ Рсн< „ _\Pho)
(РН2) *0,
Каждое из рассматриваемых равновесий позволяет определить
активность углерода:
а =Рсн4(Рн2) = PqoPqo2 (743)
/Ц/>сн4 PCo2(Pcof
7.5. Калориметрические методы исследования
С помощью калориметрии исследуют теплоемкости,
теплосодержания (энтальпии) и тепловые эффекты, сопровождающие химические и
фазовые превращения в веществах. Обычно это интегральные свойства,
но в последнее время есть примеры достаточно удачного
калориметрического определения непосредственно парциальных энтальпий
образования (смешения) жидких сплавов. Методы калориметрии применительно
к металлическим системам очень разнообразны. Например, величину
энтальпии образования сплава можно находить, измеряя теплоту
смешения жидких компонентов непосредственно в калориметре, определяя
тепловые эффекты растворения твердого сплава и отдельно его чистых
компонентов в подходящем растворителе (включая жидкометаллический
растворитель) при различных температурах, измеряя теплоты сгорания
218
сплава и чистых компонентов в кислороде или фторе. В перечисленных
вариантах, кроме первого, энтальпию образования вычисляют из
экспериментальных данных, основываясь на законе Гесса.
Калориметрия в равной мере применима для изучения как
равновесных, так и неравновесных сплавов. Этим она принципиально
отличается от ранее рассмотренных методов. Благодаря успехам в
конструировании калориметров измерения тепловых эффектов могут быть
выполнены с высокой точностью в широком интервале температур.
Современные калориметры в зависимости от их назначения и
конструкции дают возможность проводить исследования при температурах от
0,1 К до тысяч градусов при длительности изучаемых процессов от долей
секунды до нескольких суток и измерять тепловые эффекты от величин
порядка 10~4 Дж до тысяч джоулей. Однако все это далеко не всегда
гарантирует высокое качество конечного результата. Основная причина
ошибок — недостаточная определенность состояния вещества в начале
и особенно в конце калориметрического опыта. Это может быть
связано с присутствием неконтролируемых примесей в исходном веществе
и в продуктах опыта, с неполнотой сгорания взятой навески, с малой
скоростью изучаемых химических реакций или фазового превращения,
с целым рядом других причин, трудно поддающихся контролю.
7.5.1. Общие сведения о классификации калориметров
В монографии Хеммингера и Хене* в основу классификации
калориметров положены три признака: метод калориметрических
измерений, режим измерений и принцип конструкции прибора.
Классификация по этим признакам не является строгой и
исчерпывающей, но дает определенное представление о разнообразии
конструкций калориметров.
Методы калориметрических измерений основываются
наследующих приемах: а) измерение количества превращенного при фазовом
переходе калориметрического вещества, что позволяет оценить
тепловой эффект исследуемого процесса; б) измерение количества
электрической энергии при термоэлектрической компенсации те-
*Хеммингер В., Хене Г. Калориметрия. Теория и практика / Пер. с англ.
М.: Химия, 1990. 176 с. (Пер. с изд.: ФРГ, 1984).
219
плового эффекта реакции; в) измерение изменения температуры
в зависимости от длительности реакции; г) локальное измерение
изменения температуры в пространстве калориметра (измерение
тепловых потоков).
По режиму измерений калориметры можно разделить на
изотермические, изопериболические, адиабатические, сканирующие
(различных типов). Принципиальная схема, иллюстрирующая режим
калориметрических измерений, приведена на рис. 7.18. В калориметре
с изотермическим режимом работы оболочка и калориметрическая
система имеют постоянную и равную температуру, т. е. 7^ = Гзм.
Практически к такого рода калориметрическим системам относятся прежде
всего калориметры фазового перехода (см. далее). В калориметре с
изопериболическимрежимом работы температура оболочки
поддерживается постоянной, Гоб = const, а температура калориметрической
измерительной системы Ттм отличается от температуры оболочки.
Благодаря наличию конечного определенного значения
сопротивления Лу. между калориметрической системой и оболочкой теплообмен
между ними зависит как от Го6, так и от ТИзм, но так как Гоб постоянна,
то тепловой поток является функцией только Гизм. Термин "изопе-
риболический режим" появился в зарубежной научной литературе
в 1962 г. (Кубашевский и Халтгрен), в отечественной литературе ему
эквивалентно название "калориметр с изотермической оболочкой".
При измерениях в адиабатическом режиме теплообмен между
калориметрической системой и оболочкой калориметра полностью
исключается. Этого можно достигнуть тремя способами: 1) реакцию
с участием исследуемого образца проводят так быстро, что за период
измерения теплота не успевает рассеяться; 2) калориметрическую
систему отделяют от оболочки бесконечно большим
термосопротивлением; 3) температуру оболочки в ходе измерений
поддерживают равной температуре калориметрической системы, т. е. Гоб в ходе
опыта постоянно остается равной Гизм. Тепловой поток отсутствует.
На практике третий способ наиболее предпочтителен.
При сканирующем режиме работы калориметра температура
оболочки 7об или калориметрической системы Гизм линейно изменяется во
времени. Существуют различные способы осуществления
сканирующего режима. Один из способов заключается в нагревании оболочки
с постоянной скоростью. При этом температура системы повышается
220
с некоторым опозданием по
времени, так как между системой и
оболочкой существует определенное
сопротивление RT (рис. 7.18). Этот
способ сканирования используется
в калориметрах теплового потока
(сканирования оболочки). Другой
способ создания сканирующего
режима достигается нагреванием
калориметрической системы
таким образом, чтобы ее
температура за время калориметрического
эксперимента оставалось равной
температуре оболочки, которая в
свою очередь повышается линейно
во времени. Этим способом
обеспечивается адиабатический
сканирующий режим.
По принципу конструкций различают калориметры с одной
калориметрической системой и дифференциальные, или двойные
калориметры (калориметры типа Тиана-Кальве).
Таким образом, согласно Хеммингеру и Хене, калориметры с
различными режимами измерения классифицируют на:
изотермические (Т = Т = const), в которых тепловой эффект
00 ИЗМ
процесса преимущественно компенсируется теплотой фазового
перехода;
изопериболические, т. е. калориметры с изотермической
оболочкой (То6 = const; Гизм изменяется во времени), в которых измеряется
временная зависимость разности температур;
адиабатические (Т, = Т ), в которых также измеряется времен-
00 ИЗМ
ная зависимость разности температур, но отсутствует поправка на
теплообмен;
сканирующие калориметры различных типов.
Более простой и распространенный подход к
классификационным признакам калориметров содержится в книге В.П. Колесова*.
* Колесов В.П. Основы термохимии: Учебник. М.: Изд-во Моск. ун-та,
1996.205 с.
Рис. 7.18. Принципиальная схема
калориметра с переменной
температурой: / — оболочка; 2 —
исследуемый образец; 3 — контейнер для
помещения образца; 4 — тепловое
термическое сопротивление
221
Остановимся на этом лишь очень кратко. Автор отмечает, что
создание полной классификации калориметров вряд ли возможно ввиду
многообразия как приборов, так и калориметрических методов.
Следует выделить три принципиально различных метода измерения
количества теплоты: метод смешения, метод ввода теплоты и метод
протока и два типа калориметров: калориметры с постоянной
температурой (калориметры фазового перехода) и калориметры с
переменной температурой.
Калориметры фазового перехода. В них тепловой эффект,
связанный с изучаемым процессом, ведет к фазовым превращениям
вещества, находящегося в калориметре. Такие калориметры в
полной мере являются изотермическими. Примером может служить
ледяной калориметр Бунзена. Процесс протекает при температуре,
соответствующей равновесию вода—лед. При 273,16 К (О °С) 1 г льда
занимает объем 1,09007 см3, а 1 г воды при этой температуре имеет
объем 1,0001 см3. Уменьшение объема системы вода-лед на 1 см3
соответствует плавлению 1/(1,0907- 1,0001)= 11,03гльда. Плавление 1 г
льда связано с поглощением калориметрической системой 333,79 Дж;
отсюда следует, что уменьшение объема системы вода-лед на 1 см3
связано с поглощением 333,79 • 11,03 = 3681,70 Дж. Таким образом,
количество тепла, поглощенное калориметром, можно определить
путем измерения объема системы вода-лед. Калориметр может
быть успешно использован при изучении медленно протекающих
процессов с относительно небольшими тепловыми эффектами. Для
непосредственного определения теплот смешения калориметры
такого типа не применяют.
Калориметры с переменной температурой. Наиболее
распространены при научных исследованиях калориметры переменной
температуры с изотермической оболочкой. Любой калориметр
переменной температуры, как уже отмечалось, состоит из двух
частей — калориметрической системы и оболочки. Калориметрической
системой называют совокупность всех частей калориметра, между
которыми происходит распределение измеряемой теплоты. Оболочка
окружает калориметрическую систему и обеспечивает определенные,
строго фиксированные условия теплообмена калориметрической
системы с окружающей средой.
222
7.5.2. Проведение калориметрических измерений
В калориметрах с переменной температурой тепловой эффект
исследуемого процесса (0) определяют по изменению температуры в
течение реакции (А 7). Теплоемкость калориметра принято выражать
при помощи теплового значения (энергетического эквивалента)
калориметра (W), равного сумме теплоемкостей всех составных частей
калориметра. Расчет величины Сочень прост:
G7.= WAT. (7.44)
Численное значение ^определяют обычно опытным путем,
сообщая калориметру известное количество энергии и измеряя
вызванный этим подъем температуры. Таким образом, и при градуировке
калориметра и при работе с ним измерение величины А Г является
принципиально важным и должно быть сделано с максимальной
точностью. В калориметрии используют ртутно-стеклянные термометры
различной конструкции, платиновые термометры сопротивления,
термопары, термисторы.
Существенным осложнением является то, что величину А Г,
называемую истинным изменением температуры, в калориметрах с
изотермической оболочкой нельзя измерить непосредственно. На эту
величину в большей или меньшей степени, но всегда накладывается
теплообмен с окружающей средой. Возможны и побочные источники
энергии*. Поэтому непосредственно наблюдаемое в опыте изменение
температуры АГ' всегда отличается от истинного. Чтобы получить
истинное изменение температуры, необходимо внести поправку на
теплообмен (8):
АГ=АГ'±5. (7.45)
Все способы оценки величины 8 основаны на наблюдении за
изменением температуры калориметрической системы не только во
время протекания исследуемого процесса, но и в течение
некоторого времени до его начала и после его окончания. В соответствии с
этим в калориметрическом опыте различают начальный, главный и
конечный периоды опыта. В начальном периоде измеряют скорость
изменения температуры калориметрической системы в отсутствие
* Более подробное описание калориметрического эксперимента
содержится в учебнике В.П. Колесова, указанном на с. 221
223
т.+ т,
-т
■Н
I >
1Т I
т>\ !
>т,-тг
АН<0
1, X
2 m
Рис. 7.19. Кривая температура—время
при проведении калориметрического
опыта: 71, — Т2 начальный перио
измеряемой теплоты
("температурный ход" — изменение
температуры, отнесенное к
единице времени). На рис. 7.19
начальный период соответствует
изменению температуры от Г, до
Т2 (время: от т, до т2).
Главный период — часть
опыта, когда происходит быстрое
и неравномерное изменение
температуры калориметра
вследствие протекания изучаемого
процесса. Разность температур
Тг - Т2 (см. рис. 7.19) —
наблюдаемое изменение температуры AT'
за время протекания процесса
(от т2 до х3). Конечный период
следует непосредственно за главным, в нем устанавливается
регулярный тепловой режим (изменение температуры от Тг до Г4, т > т3).
Измеренная разность температур требует внесения поправки 5,
которая оценивается графическим или аналитическим способом.
Простейшее графическое построение для учета поправки на
теплообмен представлено на рис. 7.19. Линия начального хода
экстраполируется до пересечения с координатой, соответствующей времени
тт, ей отвечает значение температуры (Т2 + Г3)/2 = Г . Разность
экстраполированных значений температур начала и конца главного
перехода Гр - Та представляет собой величину А71, учитывающую
поправку на теплообмен по сравнению с Т3— Тг
Теплообмен калориметра с окружающей средой осуществляется
по трем разным механизмам: через теплопроводность, излучение и
конвекцию. Строгий учет тепловых потерь невозможен, поэтому
соблюдают определенные условия, позволяющие при не очень точных
исследованиях ограничиться рассмотренной выше приближенной
поправкой. К числу условий, уменьшающих теплообмен, относятся:
разность температур калориметра и окружающей его оболочки
не должна превышать 2-3 градуса;
внешняя поверхность калориметра и внутренняя поверхность
оболочки должны обладать хорошей отражательной способностью;
224
расстояние между стенками калориметрического сосуда и
оболочки должно составлять не более 10 мм; при этом конвекция
практически исключается, а теплопотери, обусловленные теплопроводностью
воздуха, невелики;
оптимальная величина подъема температуры в ходе
калориметрического опыта лежит в пределах от 1 до 2 °С.
7.5.3. Примеры калориметрических исследований
различных процессов
В калориметрических исследованиях неорганических систем,
особенно при высоких температурах, принято различать
калориметрию нереагирующих систем и реагирующих систем. Для нереаги-
рующих систем методами калориметрии определяют теплоемкость,
энтальпию фазовых превращений, теплосодержание; для
реагирующих систем — тепловые эффекты смешения жидких компонентов,
энтальпии образования твердых фаз. Наиболее точные результаты
при измерении теплоемкости как при низких (ниже комнатной), так
и при высоких температурах могут быть получены с применением
адиабатических калориметров. При этом низкотемпературные
измерения теплоемкости требуют специальных методов и выполняются
в ограниченном числе лабораторий.
Теплосодержание и теплоемкость. Измерение теплосодержания
Н°Т - #298 в Различных интервалах температур позволяет определить
величину средней теплоемкости, отнесенной к единице массы:
С Нт-Нж (746)
" 7-298
Если необходимы сведения об истинной теплоемкости, то следует
аналитически выразить зависимость Hj-H\9% =Д7):
нт ~ нт =аТ+ЬТг + сТ-' + d, (7.47)
соответственно
J(H°T-H°m)= bT т-г (74g)
р дТ
Такого рода расчеты годятся только для фаз вполне определенного
состава. Для фаз переменного состава, например для твердых растворов,
225
интерметаллических соединений с областью гомогенности необходимо
учитывать зависимость Н°т - #298 не только от температуры, но и от
состава. Если принять, что С линейно зависит от состава, то
уравнение (7.47) примет вид
#f -#2°98 = (1 + кх)(аТ + ЬТ2 + сГ1 + d), (7.49)
Поскольку состав фазы (х) не зависит от Г, получаем
С = (1 + кх)(а + 2ЬТ~1 - сГ2).
В более сложном случае, когда С исследуемой фазы зависит от
состава не линейно, уравнение (7.49) записывается следующим
образом:
#f-#2°98 =(\+kx+Lx2)(aT+bT2 + cr{ + d), (7.50)
Ср = (1 + кх + Lx2) (а + 2* 7м - сТ2). (7.51)
При определении теплоемкости достаточно распространен метод
смешения. В этом случае образец запаивается в ампулу из стекла,
кварца или металла, нагревается в термостате до нужной температуры
и сбрасывается в калориметр. При расчете учитываются теплоемкость
и масса ампулы. Этот же метод применим для определения энтальпий
фазовых превращений, но в одном опыте нельзя одновременно
определять и теплоемкость и теплоту фазового превращения. Существуют
и другие методы проведения калориметрического эксперимента при
определении теплоемкости.
Определение энтальпии смешения. Для определения энтальпий
смешения при образовании жидкого сплава из чистых жидких
компонентов применяются высокотемпературные калориметры различных
типов (изопериболические, адиабатические). Из-за ограничений в
выборе конструкционных материалов, устойчивых по отношению
к жидким металлам при высоких температурах, приходится
использовать довольно простые приспособления для смешения
компонентов. Часто в высокотемпературных калориметрах для
определения энтальпий смешения используют принцип количественного
дифференциально-термического анализа (ДТА). Расчет тепловых
эффектов проводят по уравнению /wA# = kS, где т — масса образца;
А# — величина теплового эффекта, отнесенная к единице массы;
S— площадь пика термограммы; к — коэффициент
пропорциональности. Величина к зависит от температуры, от применяемой
аппаратуры. Наиболее рационально определять к по те плотам плавления
чистых металлов в широком интервале температур.
226
Схема калориметра, работающего по принципу ДТА, приведена на
рис. 7.20. Конструкция его очень проста и требует л ишь минимальных
I
!
'Л
1
j
i
i
^ЧУЧЧкУЧЧЧЧУЧЧЛ'^уУЧЧЧ^ЧЧЧЧУЧ.ЧЧУЧУЧЧЧЧЧУ
fg
Рис. 7.20. Схема калориметра для определения энтальпии
смешения жидких сплавов: 1 — запорное устройство для емкости
с одним из компонентов сплава; 2 — спускаемая мешалка; 3 — термопара
227
Рис. 7.21. Типичная
результирующая кривая AT — т при
определении энтальпии
смешения жидкого сплава
f| | пояснений. Если поднять пробку /, то
I fc I один из жидких компонентов
(например В) будет перетекать из верхнего
тигля в нижний (для жидких металлов
горизонтальная штриховка). Благодаря
наличию мешалки 2, спускаемой сверху,
смешение происходит очень быстро,
ток аргона защищает металл от
окисления. Верхний и нижний тигли
расположены в массивных металлических
блоках, имеющих теплоизоляцию, что
обеспечивает постоянство температуры
компонентов перед смешением. Для контроля температуры служат
термопары, расположенные в различных частях блоков, например
J. Изменение температуры в процессе смешения (А7) фиксируется
термобатареей, находящейся под нижним тиглем. Площадь под
результирующей кривой АГ- т (т — время) служит мерой энтальпии
смешения (рис. 7.21).
При оценке различных данных по теплотам смешения жидких
сплавов принято отдавать предпочтение результатам прямых
калориметрических измерений, однако в большинстве случаев величины
АН, рассчитанные на основании
измерений ЭДС, достаточно хорошо
согласуются с калориметрическими
данными (рис. 7.22).
Жидкий сплав можно получить
не только путем смешения двух
жидких компонентов по схеме
тА(ж) + пВ(ж) = АтВп(ж), но и когда
вводимый компонент (В) является
твердым: тА(ж) + пВ(тв) = АтВп{ж).
Обозначим: АН — энтальпию
смеем
шения при образовании жидкого
D 7 лл г. соединения А В из чистых жидких
Рис. 7.22. Энтальпия смешения т п
в системе натрий-свинец (698 К) компонентов в качестве стандарт-
no данным измерений: /-ЭДС; ног° состояния, Л#эксп — наблю-
2 — калориметрии даемый экспериментально тепловой
228
эффект при введении твердого компонента В в жидкий компоненте,
АНипл — энтальпию плавления одного моля компонента В.
Для простоты примем, что исходные компоненты независимо
от агрегатного состояния имеют ту же температуру, что и конечный
сплав. Тогда процесс смешения можно изобразить в виде следующей
схемы:
пАН™
АН \ У АН
эксп ^К 0* см
А В (ж)
Тогда АН =АН -пАН™.
см эксп В
Определение энтальпии образования твердых фаз. Для
решения этой задачи методами калориметрии наиболее часто
растворяют исследуемую твердую фазу и отдельно чистые
компоненты или их механическую смесь в подходящем растворителе.
Разность величин экспериментально наблюдаемых энтальпий
растворения представляет собой энтальпию образования (А#обр)
твердого сплава. Рассмотрим это на конкретном примере.
Для определения энтальпии образования твердой фазы Mg2Al3 из
чистых твердых компонентов в качестве стандартного состояния
подходящим растворителем служит НС1 (концентрация
принципиального значения не имеет):
2Mg + 4HC1 = 2MgCl2 + 2H2, АН{
ЗА1 + 9НС1 = ЗА1С13 + 4,5Н2, Д#2
Mg2Al3 + 13НС1 = 2MgCl2 + ЗА1С13. АЯ3
Величины А//,, Д#2 и Д#3 определяются экспериментально.
Рассмотрим следующую схему:
д"06р
2Mg(TB), ЗА1(тв) ►Mg2AI3(TB)
,,ДЯ2 Ч /М
раствор, Mg2+, Al3+
АЯ, + АЯ2 = ДЯо6р + ДЯ3,
^06p=AHt + AH2-AHy (7.52)
229
Перед реакцией компоненты должны иметь температуру Tr a
калориметр — температуру Тг Результаты измерений не зависят от
температуры растворения, а определяются лишь температурой
образцов. Правильные результаты могут быть получены только тогда,
когда при растворении сплавов и чистых компонентов образуются
одинаковые продукты. Существенный недостаток калориметрии
растворения заключается в накоплении погрешностей, так как
определяется величина (АЯобр), как правило, значительно меньшая
слагаемых правой части уравнения (7.52).
В ранних калориметрических исследованиях, выполненных
рассматриваемым методом, в качестве растворителей применялись
кислоты или растворы типа Вг2 + НВг + Н20 (1:1:5), а также другие
водные растворы, калориметры имели температуру от комнатной
до 100 °С. Однако оказалось, что погрешность не только связана с
малостью величин АЯоб, но и в ряде случаев обусловлена различием
продуктов растворения сплава и чистых компонентов, особенно
металлов с переменной степенью окисления. Вносит погрешность и
выделение газообразных продуктов. Реальный прогресс в
калориметрии растворения был достигнут, когда в качестве растворителя стали
применять жидкие металлы: олово, цинк, алюминий, висмут.
При этом пришлось проводить растворение при температурах
350-850 °С, усложнились конструкции калориметров, но
существенно сблизились величины теплот растворения и энтальпий
образования сплавов, что позволило, несмотря на высокие температуры,
повысить точность метода.
Другой способ оценки энтальпий образования тугоплавких
соединений заключается в определении тепловых эффектов их
взаимодействия с кислородом или фтором. Рассмотрим конкретные реакции:
WC(tb) + 5/202(г) = W03(tb) + С02(г), Д Я,
С(тв) + 02(г) = С02(г), АЯ2
W(tb) + 3/202(г) = W03(tb). АЯ3
Из этого набора реакций, пользуясь законом Гесса, легко
показать, что энтальпия образования WC из чистых компонентов в
твердом состоянии (АЯоб) будет равна
АЯ^АЯ. + АЯз-АЯ,. (7.53)
230
Первоначально метод применялся
исключительно к органическим
соединениям, подавляющее большинство
которых окисляется кислородом с
выделением тепла. Позднее метод был
применен к неорганическим
соединениям. Наряду с кислородом достаточно
широкое использование нашел фтор.
Благодаря его высокой реакционной
способности существенно
расширился круг объектов, которые могли быть
изучены.
Количественный термический
анализ. К числу косвенных
калориметрических методов определения
теплоты смешения относится
количественный термический анализ,
представляющий собой комбинацию
классического термического анализа
с прецизионной калориметрией при
комнатной температуре. Цель
исследования — получение точных сведений
об изменении энтальпии исследуемого
сплава и чистых компонентов. Жидкий
сплав определенного состава с
температурой Т2 вносится в калориметр
простой конструкции (рис. 7.23) и охлаждается в нем до комнатной
температуры Тг Количество выделяющегося тепла, отнесенное
к одному молю сплава, дает разность энтальпий сплава Нт - Нт
между температурами Т2 и Ту Тем же методом определяется разность
энтальпий между температурами Т2 и Г, для чистых металлов Me,
и Ме2. Если компоненты при температуре Т{ не образуют твердых
растворов, то теплоту смешения при температуре Т2 (АНТ) находят
следующим образом:
АН, = (Яг - Я,) - х(Я7 - Я7 )м - хЛНт - Я, )м .
/2 v ?2 /.'сплав Iх 72 7j'Mej 2V /2 /,7Ме2
Рис. 7.23. Простейший
калориметр для
количественного термического анализа:
/ — термометр; 2— термопара;
3— сосуд Дьюара; 4—
пробирка; 5— вода; б— исследуемый
образец; 7 — медная трубка;
8— магнитная машалка
231
7.6. Обработка результатов термодинамических исследований
7.6.1. Аналитическое представление концентрационной
зависимости термодинамических функций в однофазных
двухкомпонентных системах
Использование термодинамических данных о фазах
переменного состава при машинных расчетах любого масштаба требует
аналитического представления концентрационных зависимостей
термодинамических функций, характеризующих процесс
смешения. В настоящее время не существует общепринятого физически
обоснованного способа описания концентрационных зависимостей
термодинамических функций растворов, любых фаз переменного
состава. При создании соответствующего банка данных целесообразно
предусмотреть различные способы описания концентрационной
зависимости термодинамических функций, которые можно было
бы применять по желанию пользователя. При этом следует четко
представлять себе особенности того или иного метода моделирования
термодинамических свойств в зависимости от характера
взаимодействия между компонентами.
Можно выделить две группы математических моделей. К первой
относятся модели, параметрам которых придается тот или иной
физический смысл (модель регулярного раствора и все ее усложнения,
модель идеального или регулярного ассоциированного раствора).
Ко второй группе относятся модели, в основе которых лежит
формальная аппроксимация термодинамических свойств, параметры
моделей не имеют конкретного физического смысла (модели,
реализующие среднеквадратичное приближение и приближение с
помощью сплайн-функций).
Во всех случаях весьма важной является оценка надежности
исходных экспериментальных данных при применении той или иной
модели для их описания. В настоящее время для характеристики
надежности практикой выработан ряд критериев, к числу которых
относятся авторская оценка точности измерений,
воспроизводимость (сходимость) данных и их самосогласование. Объективная
оценка надежности данных при высокотемпературных
исследованиях всегда сложна, должна в каждом конкретном случае учитывать
весь комплекс вероятных ошибок, требует определенного опыта и
232
интуиции. Следует иметь в виду, что применение математических
методов усреднения совокупности имеющихся исследований далеко не
всегда приводит к желаемому результату, так как при такой обработке
могут исчезнуть, сгладиться характерные локальные особенности,
присущие концентрационной зависимости парциальных молярных
функций той или иной системы.
При разработке математических моделей в термодинамике
сплавов ставятся следующие основные цели:
получение точной информации о любой термодинамической
функции в пределах изученных составов и температур;
хранение информации в информационно-справочных системах,
банках данных;
экстраполяция термодинамических свойств в области составов и
температур, для которых нет экспериментальных данных;
выявление и изучение закономерностей образования и структуры
жидких сплавов.
Для достижения этих целей модель должна обеспечивать
качественное описание исходных экспериментальных данных. К
приближающим функциям предъявляются следующие основные требования:
невязки с экспериментальными данными не должны превышать
погрешность измерений, число последовательных невязок с
одинаковыми знаками и число изменений кривизны приближающей функции
должны быть минимальными. По физическому смыслу функции
смешения в области гомогенности должны быть достаточно гладкими
(непрерывными, с непрерывными производными), но могут иметь
особенности. Все это приводит к существенным трудностям при
выборе той или иной математической модели. Если вид зависимости
заранее неизвестен, то обычно прибегают к полиномиальной
интерпретации, используя полиномы возможно более низкой степени.
Однако при небольшом числе коэффициентов в аппроксимирующей
формуле в этом случае может не быть адекватного описания набора
данных. Увеличение числа коэффициентов может привести к
необходимости пользоваться при вычислениях неприемлемо высокой
разрядностью, возможны и другие осложнения. Рассмотрим наиболее
распространенные способы аналитического представления
концентрационной зависимости термодинамических функций.
Все формально-математические модели сначала обеспечивают
аппроксимацию исходных экспериментальных таблично заданных
233
функций, а затем реализуют расчет требуемых термодинамических
свойств. Такие модели различаются между собой методом
аппроксимации и классом используемых при этом функций.
Каждый способ аппроксимации использует свою меру близости
приближаемой и приближающей функций. Наиболее часто
применяются следующие методы: интерполяция, метод наименьших
квадратов, равномерное приближение, сглаживание исходных данных.
Наибольшее распространение получил метод наименьших квадратов,
обеспечивающий минимизацию среднеквадратичного отклонения
функций. В литературе имеются попытки описания
термодинамических свойств различными классами функций: алгебраическими
полиномами, сплайнами, тригонометрическими рядами. Наибольшее
распространение получили алгебраические полиномы.
Алгебраические полиномы. Еще Маргулесом* было показано, что
связь между давлением насыщенного пара каждого из компонентов в
бинарной неидеальной смеси (/>, и р2) и давлением пара над теми же
чистыми компонентами при заданной температуре (р° и /?2°) может
быть представлена уравнениями:
Р\
■ Л *Г° ехр
ах{\-хх) + ?±(\-х{)2 +^L(l-*1)3+...
/>2 = P20-*i)Poexp
ft.
м^^ч^ч...
При бесконечном разбавлении
V
Эх
= Р\
и
А,-и
'д£2
Эх,
= р2.
/*1->0
Отсюда следует, что <х0 = Р0 =1 и oij = Pj = 0. В соответствии с
уравнением Гиббса-Дюгема
р3 = -а2-2а -За - ...,
соответственно
Р\
а{= — = хх ехр
Р\
-а2(\-х{)2 +-а2(\-хх)3 +...
*См.: Вагнер К. Термодинамика сплавов/ Пер. с англ.; Под ред. А.А. Жу-
ховицкого. М.: Металлургиздат, 1957. 179 с.
234
Pi
a2 =—- = x2exp
P\
^p2(l-x2)2+ip3(l-x2)3+...
2I ^ ., у
Для коэффициентов активности эти выражения принимают вид
1 ,1 ,
lnyi=-a2(l-xiy+-a3(l-x]y+...;
(7.54)
(7.55)
1пУ2=^2(1-^)2+^РзО-^)3+"
Если принять, что в уравнениях (7.54) и (7.55) все коэффициенты,
кроме а2 = р2, равны нулю, то выражения для lnyj и 1пу2 примут вид,
характерный для регулярных растворов. Соответствующие кривые
для системы Sn-Tl приведены на рис. 7.24.
Во многих случаях можно ограничиться первыми двумя членами
уравнения (7.54) и записать его в виде
In у! 1 1
О-*,)2 2
-а2+-а3(1-х|).
(7.56)
В этом случае коэффициенты а2 и а3 находят с помощью простого
графического построения. Коэффициенты для у2 равны
Р2 = а2 + а,; р3 = -а3.
£
1
0,4
0,2
л
~™ ft 0° О
О О v
_ _| 1
оо ло
1 1 -
Sn 0,2 0,4 0,6 0,8 Tl Sn 0,2 0,4 0,6 0,8 Т1
Рис. 7.24. Концентрационная зависимость термодинамических функций
в жидких сплавах системы олово—таллий
235
Уравнение, сходное с (7.56), как уже отмечалось (раздел 3.8.1),
следует из концепции "субрегулярного раствора". Согласно этой
концепции, для избыточной энергии Гиббса можно записать
АО1'6 = RTxxx2(ClX2xx + Q2lx2); (7.57)
AG,"36 = Л7х22[(П12 + 2(ft12 +П21)д^]; (7.58)
AGf6 = RTxt[(Q[2 + 2(Q21 -Q12)x2]. (7.59)
При х2 = 1 уравнение (7.58) принимает вид
AG™6/ RT=lny" = Q.2r
При х2 = О из уравнения (7.59) следует
АС2из6/ RT=\ny™ = nir
Если раствор ведет себя как регулярный, то Q12 = Q21 = Q.
Получившее широкое распространение полиномиальное
выражение Реддиха—Кистера ранее подробно рассмотрено нами (раздел
3.6). Отметим только, что применяемая там Q-функция очень
хорошо описывает концентрационную зависимость термодинамических
функций двойных жидких сплавов независимо от характера
взаимодействия между компонентами.
Для интегральной молярной энтальпии смешения был предложен
степенной полином вида
АН = ххх2[А0 + Ах(хх - х2) + А2(хх - х2)2 + Аг(хх -х2П (7.60)
Здесь Л0, Av Av Аъ — коэффициенты, получаемые на основании
экспериментальных данных. Соответственно для парциальных
молярных энтальпий смешения
АН{ = х22[А0 + Ах(Ъхх - х2) + А2(5х{ - х2)(хх - х2) +
+ А3(1х]-х2)(х1-х2)2]\
АН2 = x2[AQ + Ах(хх - Зх2) + А2(хх - 5х,)Ц - х2) +
+ А,(х{-1х2)(хх-х2П
Если кривые AH=f{x) имеют симметричную форму, то А{ = А2 =
= Аъ = 0 и уравнения принимают вид, характерный для регулярных
растворов:
AH = AQx{x2; АНх = А0к22; АН2 = А^с2.
Аналогичные выражения могут быть использованы для других
избыточных величин, в частности для энергии Гиббса и энтропии
смешения.
236
Бонье с сотрудниками (1965 г.) рассмотрели возможность
описания концентрационной зависимости lgyx и ЛЯ, в двухкомпонентных
системах с помощью полиномов различной степени:
Л^аО-лг^ + рО-х,)2; (7.61)
Дх,) = а(1 -*,)4 + Р(1 -х,)3 + у(1 -х,)2; (7.62)
Дх,) = а(1 - х,)5 + р(1 - х,)4 + у(1 - х,)3 + 5(1 - х,)2. (7.63)
Для 38 жидких металлических систем были привлечены данные
различных авторов (обработано методом наименьших квадратов
116 зависимостей Igy, =Лх1) и АЯ, =/(х,)). В 55 случаях для систем с
самым различным характером взаимодействия между компонентами
(In-Sb, Na-Ga, Mg-Sn и т. д.; Igy,: Mg-Pb, Cd-Pb, Na-Tl и т. д.)
полиномы (7.61)—(7.62) в равной мере хорошо описывали
концентрационную зависимость термодинамических функций. В 36 случаях
предпочтительным оказывается полином четвертой степени (7.62).
В трех случаях применение полинома пятой степени (7.63) приводит
к некоторым улучшениям в описании кривой. В целом ряде случаев
не удается добиться удовлетворительного отражения наблюдаемых
зависимостей.
Для большого числа жидких металлических систем применялось
уравнение вида
Дх,) = а0 + аххх + я2х,2 + ар* + я4х,4. (7.64)
Получено хорошее описание экспериментальных данных,
особенно для Igy, = Дх,) (рис. 7.25), так как точность определения этой
зависимости всегда выше, чем зависимости АЯ, =Лх,). Опыт
показывает, что для систем со сложным характером взаимодействия между
компонентами применение простейших степенных полиномов для
Igy, и АЯ, затруднительно только в областях составов, прилегающих
к чистым компонентам.
Применение ортогональных функций — смещенных полиномов
Лежандра или Чебышева — оказывается более эффективным, чем
применение различных степенных рядов, но широкого
распространения не получило*.
'Относительно применения ортогональных функций более подробные
сведения см.: Морачевский А.Г., Сладкое И.Б. Термодинамические расчеты
в металлургии: Справочник. М.: Металлургия, 1993. 304 с.
237
а)
Д# , кДж/моль
1,0 0,8 0,6 0,4 0,2 0
Na v Sn
0
-8
-16
-24
-32
-АО
—х
- \
" \
\
\
1 . 1 1 1
1,0 0,8 0,6 0,4 0,2 О
Na v Sn
Рис. 7.25. Зависимости lgyNa = Л^а) (я) и A#Na = Л*№) (б) для жидких
сплавов системы натрий-олово (850 К): кривая — расчет по уравнению
вида (7.64), кружки — экспериментальные данные
Сплайн-функции. Трудности, возникающие при аналитическом
описании термодинамических функций, в значительной мере
связаны с нелокальным характером описания, они могут быть существенно
уменьшены, если сузить интервал изменения параметров состояния
сплава (уменьшить интервал составов).
При этом можно надеяться, что форма функциональной
зависимости термодинамического свойства сильно упростится и
станет универсальной. Именно исходя из этих предпосылок,
целесообразно использовать сплайны, т.е. функции, составленные
по определенной системе из кусков, каждый из которых является
многочленом невысокой степени. Если на отрезке (д, Ь) задано п
значений аргументах:
а = х. <х, < ... <х = Ь,
то кубическим сплайном называют функцию SF(x), которая
является полиномом степени не выше третьей на каждом из отрезков
х.<х< х.+ 1 (/ = 1, 2,..., /1-1), непрерывна на отрезке [я, Ь] вместе со
своими первой и второй производными и удовлетворяет условию
SIM=yi (/=1,2,..., я).
238
В этом случае кубический сплайн Sp(x) называется
интерполирующим сплайном для функции у = J{x), Кроме интерполирующих
используют также сглаживающие сплайны, которые не проходят
точно через заданные значения уг но наилучшим образом описывают
ход функции у=Лх). При этом максимальная невязка аппроксимации
сплайном не должна превосходить погрешности эксперимента. Таким
образом, для составления таблиц термодинамических функций по
первичным экспериментальным данным целесообразно использовать
сглаживающие сплайны, а для дальнейших расчетов с помощью этих
таблиц — интерполирующие сплайны.
Применение сплайнов для описания концентрационной
зависимости термодинамических функций подробно рассмотрено
Ворониным и Дегтяревым*.
В наших работах на основании ранее полученных
экспериментальных данных с помощью сплайн-функций описана
концентрационная зависимость избыточной парциальной мольной энергии
Гиббса для щелочного металла в жидких сплавах натрия с галлием,
кадмием, индием, ртутью, оловом, свинцом, висмутом, а также калия
с индием, таллием, свинцом. При построении кубических
интерполирующих сплайнов
ЗД = У, + Цх - х) + с (х - х)2 + dt{x - х)3 (7.65)
использовали сетки из десяти равноотстоящих узлов (0,1 < х. < 1,0)
с "шагом", по составу равным 0,1 молярной доли. По найденным
параметрам сплайнов вычисляли значения функций при содержании
щелочного металла в пределах 0,15 <х.< 0,95, "шаг" — 0,1. В качестве
примера на рис. 7.26 приведена концентрационная зависимость
избыточной парциальной молярной энергии Гиббса для калия в системе
К-Т1, построенная по экспериментальным данным и приближаемая
с помощью сплайн-функции.
Описанный прием характерен для вычисления коэффициентов
интерполирующих сплайнов на основании справочных данных —
в справочнике обычно приводятся таблицы сглаженных значений
термодинамических функций бинарных сплавов в равноотстоящих
* Дегтярев С.А., Воронин Г.Ф. // Математические проблемы фазовых
равновесий. Новосибирск: Наука, 1983. С. 53—83.
239
AGK , кДж/моль
-24
Рис. 7.26. Концентрационная
зависимость А(7киз6 в жидких сплавах
системы К-Т1 (680 К): У —
приближение на основании сплайнов (7.65);
2 — экспериментальные данные
точках (узлах), число которых
чаще всего равно 11 ("шаг" по
составу — 0,1 молярной доли) или
21 ("шаг"— 0,05 молярной доли).
Следует, однако, отметить, что
для адекватного описания
экспериментальных данных
сплайнами требуется обычно меньшее
количество равноотстоящих узлов
(5-6). Если же отказаться от
равноотстоящих узлов и перейти к их
рациональному подбору, то число
узлов, достаточное для описания
системы с использованием
интерполирующих сплайнов, может
быть еще меньше.
Сплайны могут описывать
концентрационную зависимость
как интегральных, так и
парциальных термодинамических
функций.
7.6.2. Применение модели ассоциированных растворов к жидким
двухкомпонентным металлическим системам
Как уже отмечалось (раздел 4.6.5) для значительного числа
жидких металлических систем с отрицательными отклонениями от
идеального поведения и прежде всего для систем, диаграммы
состояния которых характеризуются образованием
интерметаллических соединений, может быть применена модель ассоциированных
растворов. В основе модели лежит описание химических равновесий
между образующимися в растворе ассоциатами и исходными
компонентами. Ассоциаты находятся в состоянии стационарного
динамического равновесия с неассоциированными атомами, причем это
равновесие подчиняется закону действующих масс. Предположение
о существовании ассоциатов определенного состава позволяет через
параметры ассоциатов описать концентрационную и
температурную зависимости термодинамических функций рассматриваемой
240
системы. Таким образом, согласно модели, в растворе
устанавливается химическое равновесие между исходными компонентами и
образующимися ассоциатами.
В первом приближении раствор, состоящий из ассоциатов и
исходных компонентов, можно считать идеальным (модель идеального
ассоциированного раствора — ИАР). Можно его рассматривать как
регулярный (модель регулярного ассоциированного раствора — РАР).
При описании некоторых металлургических систем в рамках модели
ассоциированных растворов смесь ассоциатов и находящихся с ними
в равновесии исходных компонентов считали субрегулярной.
Независимо от того, как квалифицировать образующийся
раствор, сильные взаимодействия, ведущие к значительным
отклонениям всей системы от идеального поведения, формально исключаются
из рассмотрения. Энергетические и энтропийные характеристики
образования ассоциатов неявно учитываются через константу
равновесия реакции комплексообразования.
Детальный термодинамический анализ показал, что в рамках
модели ИАР для систем с отрицательными отклонениями от идеального
поведения следует принимать во внимание образование ассоциатов
только из разносортных атомов ЛтВп. В случае положительных или
знакопеременных отклонений от идеального поведения необходимо
учитывать наряду с ассоциатами типа АтВп образование ассоциатов
из атомов одного сорта (Av Ap ...). Еще раз отметим, что
применительно к системам с положительными отклонениями от идеального
поведения модель ИАР в значительной степени утрачивает свой
физический смысл.
При применении модели ИАР к реальным системам с
отрицательными отклонениями от идеального поведения, выбирая тип
ассоциатов, чаще всего все же исходят из состава соединений,
имеющихся в твердой фазе. В некоторых системах соединения состоят из
большого числа атомов, например Al5Bv A9B4, А5В2 и т. д. Очевидно,
что в жидком состоянии такие фуппировки неустойчивы. В расплавах
наиболее вероятно образование ассоциатов, состоящих из 2-4 атомов
(АВ, ABV AB3) и лишь в некоторых случаях имеет смысл предполагать
наличие более сложных ассоциатов (АВ4, АВ5, А2В3).
Многие исследователи справедливо полагают, что в жидких
металлических системах нереально образование большого числа
сортов ассоциатов. Как правило, можно офаничиться двумя-тремя
241
ассоциатами. Только для систем с очень сильными отрицательными
отклонениями от идеального поведения во всем интервале составов
(0,1) можно допустить образование большего числа ассоциатов.
Наибольшее распространение получили способы расчета
параметров модели по предельным значениям коэффициентов активности
и по значениям термодинамических функций в отдельных точках
концентрационного интервала.
Представляется наиболее естественным выбор параметров модели
из условий близости интегральных термодинамических функций во
всем интервале составов (0,1). В качестве критерия близости больше
всего подходит минимум суммы квадратов невязок (5), т.е. параметры
модели следует выбирать так, чтобы
Зс=Ъ*СюЬи)-№£<*и)Ытт9 (7.66)
7=1
S„ -Х[АЯМ0Д(х1у)-АЯэкс(х|у)]2 ^min . (7.67)
Н
Здесь индекс "мод" обозначает величину термодинамического
свойства, рассчитанную по модели ИАР; индекс "экс" —
экспериментально полученную величину; jc,^. —у-е значение молярной доли
первого компонента. Следует иметь в виду, что экспериментальные
погрешности определения интегральных свойств значительно
превышают вычислительные погрешности и погрешности метода расчета.
Поэтому не имеет смысла улучшать описание концентрационной
зависимости термодинамических функций в пределах ошибки их
экспериментального определения путем введения новых ассоциатов.
Разумным следует считать совпадение AG^R(x{) и А#мод(х,) с
соответствующими исходными значениями с точностью (1-5) % в
зависимости от типа и величины отклонений системы от идеального
поведения.
Для каждого фиксированного набора ассоциатов избыточная
энергия Гиббса зависит только от констант равновесия и
температуры, а энтальпия смешения — от К. и А#°, поэтому сначала из условия
(7.66) можно определить константы равновесия, а затем из (7.67) —
энтальпии образования ассоциативных комплексов.
Нахождение оптимальных значений К. усложняется тем, что не
существует формул, непосредственно связывающих АС^ с констан-
242
тами равновесия, т. е. избыточная энергия Гиббса неявно зависит от К..
Задача минимизации суммы квадратов невязок (7.66) в пространстве
параметров К. может быть решена с помощью методов прямого поиска,
субградиентного метода и других специальных методов минимизации
недифференцируемых функций. Для их применения достаточно иметь
алгоритм вычисления функции цели в любой точке из пространства
параметров К*.
Для систем с сильными отрицательными отклонениями (при
эквимольном составе АО136 более отрицательна, чем — 10 кДж/моль)
наиболее вероятными оказались модели с тремя ассоциатами
(в табл. 7.4 указаны система А—В, температура в градусах Кельвина,
оптимальный состав ассоциатов):
Таблица 7.4
Состав и число ассоциатов для систем с сильными
отрицательными отклонениями от идеального поведения
Система
Li-Ga
Li-In
Li-Tl
Li-Cd
Li-Pb
Li-Sn
Na-Hg
Na-Sn
Na-Pb
Na-Bi
Pb-Te
Г,К
1023
1023
1023
833
1073
1073
673
773
1073
1073
1200
Модель
AB+A2B+AB2
AB+A2B+A3B
AB+A2B+A3B
AB+A3B+AB3
AB+A3B+ASB
AB+A3B+A5B
AB+AB2+AB4
AB+A2B+A3B
АВ+А2В+АтВ
АВ+А2В+АЪВ
AB
Система
Mg-Sb
| Mg-Bi
Mg-Sn
Al-Au
Al-Cu
Al-Fe
K-Pb
K-Hg
Rb-Hg
Cs-Hg
Г, К
1123
1123
1073
1400
1373
1873
873
598
573
573
Модель
AB+A3B2+A3B
АВ+АЪВ2+А3В
AB+A2B
AB+A2B+AB2
AB+AB3
AB+A3B+AB3
AB
AB+AB2+AB4
AB+A2B+AB2+ABb
AB+AB3+A3B
Более подробно математическая сторона нахождения констант
равновесия ассоциатов на основании экспериментальных данных изложена в
монографии: Электрохимические методы исследования в термодинамике
металлических систем / А.Г. Морачевский, Г.Ф. Воронин, В.А. Гейдерих,
И.Б. Куценок. М.: ИКЦ "Академкнига", 2003.334 с. Там же приведены ссылки
на оригинальные работы.
243
Попытки учета четвертого и пятого ассоциатов приводили к
улучшению описания концентрационной зависимости термодинамических
функций уже в пределах погрешности определения этих функций.
Исключением явилась система Rb-Hg. Предположение о существовании
двух ассоциатов позволило описать термодинамические свойства
жидких сплавов систем Mg—Sn и Al-Cu. Модель с единственным ас-
социатом оказалась пригодной для двух систем — К-Pb и РЬ-Те.
В подавляющем большинстве случаев для систем с сильными
отрицательными отклонениями от закона Рауля выбор оптимального
числа и состава ассоциатов не представлял трудностей, так как один
из вариантов приводил к результатам, значительно более лучшим,
чем другие варианты.
Для описания термодинамических функций систем с
умеренными и слабыми отклонениями от идеального поведения, как правило,
требуется меньшее число ассоциатов (табл. 7.5).
Таблица 7.5
Состав и число ассоциатов для систем с умеренными и слабыми
отклонениями от идеального поведения
[ Система
Li-Al
Na-In
Na-TI
Mg-Pb
Mg-Ga
Mg-Zn
Al-Ag
Al-Ge
Al-Si
Al-Sb
K-In
K-Tl
Cs-Tl
Pb-Au
Cd-Hg
1 In-Hg
7, К
1023
728
800
923
923
923
1273
1273
1273
1300
733
680
700
1200
573
557
Модель 1
AB+A^B2
AB+AB2
AB+AB2
AB+A2B
AB+A2B+AB2
AB+AB2
AB+AB,+AB,
2 4
AB + AB2
AB
АВ+АЪВ
AB
AB+AB2
АВ+АВЪ
A2B+AB2
AB
AB \
244
Для систем этой группы конкурирующие варианты с ассоциатами
различного состава часто приводят к близким результатам, что не
позволяет однозначно идентифицировать состав ассоциатов.
Следует также отметить, что в системах с небольшими отклонениями от
идеального поведения на результаты моделирования существенное
влияние оказывают погрешности экспериментального определения
приближаемых функций.
Для описания систем с положительными отклонениями от
закона Рауля модель ИАР, допускающая в данном случае образование
ассоциатов только из атомов одного сорта, имеет ограниченное
применение. Теоретический анализ показывает, что в случае сильных
положительных отклонений (АСиз6 > 2,3 Дж • моль-1) не удается даже
формально описать функцию А<?пб. С ростом константы равновесия,
характеризующей ассоциат, АСиз6 медленно возрастает и стремится к
некоторой предельной кривой при К.-><х>.
При описании в рамках модели ИАР систем со
знакопеременными отклонениями от идеального поведения в простейшем случае
необходимо учитывать образование ассоциатов АтВп и А., что
соответствует равновесиям: тА{ + пВх <-» АтВп; iA{ <-* А..
Наряду с моделью ИАР широкое применение имеет модель
регулярного ассоциированного раствора (РАР), позволяющая описать
термодинамические свойства системы, постулируя существование
минимального числа ассоциатов. При этом учитываются
взаимодействия между отдельными свободными атомами, не связанными
в комплекс, и взаимодействие этих атомов с ассоциатами. Обычно
исходной величиной для расчета в этом случае является энтальпия
смешения АЯ, которая разбивается на два слагаемых:
ДЯ=АЯас + АЯре,
Первое слагаемое обусловлено образованием ассоциата,
второе — взаимодействием совокупности беспорядочно расположенных
частиц. В свою очередь величина АЯрег может быть представлена в
виде суммы слагаемых, описывающих взаимодействие мономеров
разных сортов атомов между собой, а также мономеров с каждым из
сортов ассоциатов. Нет указаний на необходимость учитывать
взаимодействие ассоциированных комплексов между собой. Модель РАР
позволяет описать концентрационную и температурную зависимости
термодинамических функций смешения с помощью меньшего числа
245
ассоциатов различных сортов по сравнению с моделью ИАР благодаря
тому, что появляются дополнительные подгоночные параметры в виде
энергий взаимодействия (взаимообмена): Ах — BVA{- А.Вт, В{ - А.Вт
и т. д. Обычно в рамках модели РАР нет необходимости учитывать
более двух сортов ассоциатов.
7.6.3. Обработка экспериментальных данных
на основании второго и третьего законов термодинамики
При определении энтальпии образования металлургических
объектов калориметрическими методами отмечалось, что
получаемые результаты не всегда оказываются достаточно надежными. Так,
например, в случае определения А^Я^для Fe, xS или FeS образцы
этих соединений обычно получаются в неравновесных, закаленных
формах. Это же относится ко многим другим соединениям. Более
надежным путем определения энтальпий образования являются
исследования высокотемпературных равновесий, так как при этом
состав и кристаллическая структура образцов максимально близки
к равновесным.
Калориметрические методы определения термохимических
величин — энтальпии реакции, энтальпии образования соединений,
теплоемкости — основаны только на использовании первого закона
термодинамики и следующих из него зависимостей (закон Гесса,
уравнение Кирхгофа). Измеряемое значение теплового эффекта
реакции Аг#7° позволяет затем вычислить величину энтальпии
образования А //у°. В отличие от калориметрического метода величина
Аг//у°, вычисляемая из определения равновесия реакции, включая
измерения ЭДС, можетбыть найдена двумя независимыми методами,
причем результаты расчетов этими методами могут в общем случае
не совпадать. Иногда расхождения могут быть значительными. Оба
метода основаны на известном соотношении
mnKp=-^ = -^+ArSr. (7.68)
Величины АгС7°, Аг5у°, как и Дг#у' характеризуют изменение
соответствующей термодинамической функции в изучаемой реакции,
К — константа равновесия реакции. Величины Ar//y° и Д^* как
правило, слабо зависят от температуры и в ограниченном интервале
246
температур могут рассматриваться как постоянные величины.
Аппроксимация результатов экспериментальных исследований констант
равновесия или давления пара с помощью уравнения (7.68) позволяет
найти значения АгЯ7°и Аг57°при каком-то промежуточном значении
температуры Г, лежащем внутри исследованного интервала
температур. Определение Дг#7° при Т как коэффициента при Т~х в
уравнении для константы равновесия (7.68) получило в научной литературе
название "расчет по методу второго закона термодинамики"*. Как
видно из изложенного, для нахождения Аг#7° при Т по этому методу
достаточно знать только зависимость константы равновесия реакции
от температуры, а не абсолютные значения К при той или иной
температуре. В отдельных случаях это весьма существенно. Некоторые
экспериментальные методы, например масс-спектрометрический,
позволяют определить только величины, пропорциональные
константам равновесия, а не значения самих констант.
Построив график зависимости \пКр =Д 7м), нетрудно найти оба
параметра уравнения (7.68) — значение Аг#7°по наклону прямой и
значения А^0 — по величине отрезка, отсекаемого прямой на оси
ординат. Если при расчетах используется не константа равновесия, а
величина, ей пропорциональная, то вместо значения Аг57° получаем
некоторую произвольную константу. Все изложенное в полной мере
относится к расчетам с использованием уравнения Клапейрона-
Клаузиуса (4.12), которое после интегрирования в упрощенной форме
принимает вид
RT
Для нахождения величины теплоты испарения достаточно знать
только зависимость давления от температуры, а не абсолютное
значение/?.
Метод второго закона не может быть применен, если измерения
константы равновесия или давления насыщенного пара выполнены
* См. об этом также в справочном издании: Термодинамические
свойства индивидуальных веществ: Справ, изд. /Л.В. Гурвич, И.В. Вейц,
В.А. Медведев и др.; Под ред. В.П. Глушко и др. В 4 т. Т.1, кн. 1. М.: Наука,
1978. 495 сив монографии: Львов Б.В. Терморазложение твердых и жидких
веществ. СПб.: Изд-во Политехи, ун-та, 2006. 278 с.
247
только при одной температуре. В тех случаях, когда измерения
охватывают лишь очень небольшой интервал температур, точность расчета
величины Аг#7°при Т оказывается невысокой. Из выполненного
ряда измерений расчет по методу второго закона термодинамики
позволяет получить только одно значение Аг#у° при Г , которое
очень чувствительно к возможным экспериментальным ошибкам,
особенно на границах температурного интервала. Для вычисления
значений АгЯ7° при Г этим методом не нужны дополнительно какие-
либо термодинамические функции, однако для пересчета к
стандартной температуре (298,15 К) необходимы значения Нт -Н1п для всех
ср
участвующих в реакции веществ.
Значения теплового эффекта реакции Аг#у°могут быть вычислены
и другим путем, называемым "расчет по методу третьего закона
термодинамики". Метод основан на соотношении
АД;=Т[А5;-Л1п^]. (7.69)
В этом случае величина Аг//7°может быть получена для каждого
значения константы равновесия независимо, но для расчета нужно
располагать дополнительно значениями энтропии участников
реакции (или величиной ArS.J) для температур, при которых проводились
измерения.
Таким образом, метод требует более полной исходной
информации (абсолютные значения констант равновесия, термодинамические
функции участников реакции), но и дает более полные данные,
помогающие оценить достоверность экспериментальных измерений.
Расчет по методу третьего закона термодинамики обеспечивает
согласованность между величиной энтальпии данной реакции,
термодинамическими функциями ее компонентов и экспериментально
измеренными значениями констант равновесия. В отличие от метода
второго закона для нахождения энтальпии реакции в принципе
достаточно единственного измерения лишь при одной температуре.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА*
Монографии
Вагнер К. Термодинамика сплавов / Пер. с англ.; Под ред. А.А. Жуховиц-
кого. М.: Металлургия, 1957. 180 с.
ЛамсденДж. Термодинамика сплавов/Пер с англ.; Под ред. Н.Н.Сироты.
М.: Металл ургиздат, 1959. 440 с.
Пригожий И., Дефей Р. Химическая термодинамика / Пер. с англ.; Под
ред. В.А. Михайлова. Новосибирск: Наука, 1966. 510 с.
Hildebrand J.H., Prausnitz J.M., Scott R.L. Regular and Related Solutions.
The Solubility of Gases, Liquids and Solids. N.Y.e.a. Van Nostrand Reinhold
Company, 1970.288 р.
Аносов В.Я., Озерова М.И., Фиалков Ю.Я. Основы физико-химического
анализа. М.: Наука, 1978. 504 с.
История учения о химическом процессе. Всеобщая история химии. М.:
Наука, 1981.448 с.
Кубашевский О., Олкокк СБ. Металлургическая термохимия / Пер. с
англ.; Под ред. Л.А.Шварцмана. М.: Металлургия, 1982. 392 с.
Могутнов Б.М., Томилин И.А., Шварцман Л.А. Термодинамика сплавов
железа. 2-е изд. М.: Металлургия, 1984. 207 с.
Туркдоган Е.Т. Физическая химия высокотемпературных процессов/ Пер.
с англ.; Под ред. В.А. Григоряна. М.: Металлургия, 1985. 344 с.
Деверо О.Ф. Проблемы металлургической термодинамики / Пер. с англ.;
Под ред. В.М. Глазова. М.: Металлургия, 1986. 424 с.
Глазов В.М., Павлова Л.М. Химическая термодинамика и фазовые
равновесия. 2-е изд. М.: Металлургия, 1988. 560 с.
* Рекомендуемая литература располагается в хронологическом порядке.
249
Герасимов Я.И. Избранные труды. Общие вопросы физической химии
и термодинамики. Термодинамические основы материаловедения / Отв. ред.
Г.Ф. Воронин. М.: Наука, 1988. 333 с.
Люпис К. Химическая термодинамика материалов / Пер. с англ.; Под ред.
Н.А. Ватолина и А.Я. Стомахина. М.: Металлургия, 1989. 503 с.
Пригожий И.Р. Молекулярная теория растворов / Пер. с англ.; Под ред.
В.М. Глазова. М.: Металлургия, 1990. 360 с.
Вайсбурд СЕ. Физико-химические свойства и особенности строения
сульфидных расплавов. М.: Металлургия, 1996. 304 с.
Электрохимические методы в термодинамике металлических систем /
А.Г Морачевский, Г.Ф. Воронин, В.А. Гейдерих, И.Б. Куценок. М.: ИКЦ
"Академкнига", 2003. 334 с.
Учебники и учебные пособия
Герасимов Я.И., Гейдерих В.А. Термодинамика растворов. М.: Изд-во
Моск. ун-та, 1980. 184 с.
Глазов В.М. Основы физической химии. М.: Высш. шк., 1981.456 с.
Харьков Е.И., Лысов В.И., Федоров В.Е. Термодинамика металлов. Киев:
Выщашк., 1982.248 с.
Воронин Г.Ф. Основы термодинамики. М.: Изд-во Моск. ун-та, 1987. 192 с.
Морачевский А.Г. Термодинамика расплавленных металлических и
солевых систем. М.: Металлургия, 1987. 240 с.
Физическая химия. Теоретическое и практическое руководство: Учеб.
пособие для вузов/ Под ред. акад. Б.П. Никольского. 2-е изд., перераб. и доп.
Л.: Химия, 1987.880 с.
Термодинамические методы в неорганической химии / Б.К. Касенов,
А.С. Пашинкин, М.К.Алдабергенов, Д.Н.Абишев. Караганда: Изд-во КарГУ,
1989. 133 с.
Дибров И.А. Общая и физическая химия. Ч. 3: Химическая
термодинамика: Учеб. пособие. СПб.: Изд-во СПбГИ, 1996. 171 с.
Глазов В.М., Пашинкин А.С. Аналитический аппарат физической химии:
Учеб. пособие. М.: Изд-во МИЭТ, 2000. 88 с.
Бажин Н.М., Иванченко В.А., Пармон В.Н. Термодинамика для химиков.
М.: Химия, 2000. 408 с.
Дуров В.А., Агеев Е.П. Термодинамическая теория растворов
неэлектролитов: Учеб. пособие. 2-е изд. М.: Едиториал УРСС, 2003. 248 с.
Стромберг А.Г., Семченко Д.П. Физическая химия / Под ред. А. Г. Стром-
берга. 5-е изд. М.: Высш. шк., 2003. 527 с.
Основы физической химии. Теории и задачи / В.В. Еремин, СИ. Каргов,
И.А. Успенская, Н.Е. Кузьменко, В.В. Лунин. М.: Экзамен, 2005. 480 с.
250
Справочники
Pankratz L.B. Thermodynamic Properties of Elements and Oxydes. Bulletin
672. Bureau of Mines. USA. 1982. 509 p.
Pankratz L.B., Stuve J.M., Gokcen N.A. Thermodynamic Data for Mineral
Technology. Bulletin 677. Bureau of Mines. USA. 1984. 355 p.
Pankratz L.B., MahA.D., Watson S.W. Thermodynamic Properties of Sulfides.
Bulletin 689. Bureau of Mines. USA. 1987. 427 p.
Морачевский А.Г., Сладкое И.Б. Термодинамические расчеты в
металлургии: Справочник. 2-е изд. М.: Металлургия, 1993. 304 с.
Thermodynamic Properties of Mineral and Related Substances at 298,15 К and
1 bar(105 Pascals) Pressure at Higher Temperatures / R. A. Robie, B.S. Hemingway//
U.S. Geological Survey. Bulletin 2131. Washington: United States Government
Printing Office, 1995.461 p.
Морачевский А.Г., Сладкое И.Б. Физико-химические свойства
молекулярных неорганических соединений (экспериментальные данные и методы
расчета): Справочник. 2-е изд. СПб.: Химия, 1996. 312 с.
Обзоры, статьи
Герасимов Я.ИМ Воронин Г.Ф. Химическая термодинамика и
некоторые перспективные области ее применения // Вестник АН СССР. 1970. №3.
С. 50-56.
Sharkey R.L., Pool M.Y., Hoch M. Thermodynamic modeling of binary and
ternary metallic solutions// Met. Trans. 1971. Vol.2, November. P. 3039-3049.
Spencer P.J., Hayer F.H., Kubaschewski O. The prediction of the
thermodynamic properties of ternary metallic solutions from binary alloy data //
Revue Chim. Miner. 1972. Vol. 9. N 1. P. 13-29.
Predel В., Arpshofen L, Pool M.J. Calorimetric methods in metallurgy //
Thermochimica Acta. 1978. Vol. 22. P. 211-236.
Velisek J. Termodynamic analysa fazovych diagramu slitin // Chem. Listy. 1980.
Vol. 74. P. 934-950.
Hillert M. Application of thermodynamic models in phase diagram
assessment// Ber. Bunsengesell. Phys. Chem. 1983. Bd.87. S. 762-769.
Морачевский А.Г., Демидов А.И. Термодинамические свойства жидких
сплавов лития //Ж. физич. химии. 1983. Т. 57, № 9. С. 2113-2128.
Ipser H., Komarek K.L. Phase diagrams: new experimental methods //
Z. Metallkunde. 1984. Bd. 75, N 1. S. 11-22.
Воронин Г.Ф. Расчеты фазовых и химических равновесий в сложных
системах // Физическая химия. Современные проблемы. Ежегодник / Под.
ред.Я.М. Колотыркина. М.: Химия, 1984. С. 112-143.
251
Соммер Ф. Модель ассоциатов в теории жидких сплавов // Диаграммы
фаз в сплавах/Ред.: Л. Беннет,Т. Массальски, Б. Гиссен; Пер. с англ. М.: Мир,
1986. С. 128-141.
Sommer F. Modern methods in high temperature calorimetry // J. Thermal
Analysis. 1988. Vol. 33, N 1. P. 15-28.
Predel B. On the thermochemistry of intermetallic compounds//Thermochimica
Acta. 1988. Vol. 129. P. 29-48.
Castanet R. Calorimetric methods in metallurgy // Thermochemistry of alloys
/ Eds.: H. Brodowski, H.-J. Schaller. Kluwer Acad. Publ. 1989. P. 145-168.
Tomiska J. Modern vapor pressure measurement based on the Knudsen-effusi-
on / Eds.: H. Brodowski, H.-J. Schaller. Kluwer Acad. Publ. 1989. P. 247-260.
Okajima A. Touch instant electromotive force method determining activities of
liquid alloys// Met. Fac. Eng., Nagoya University. 1990. Vol. 41, N 2. P. 209-279.
Pratt J.N. Applications of solid electrolytes in thermodynamic studies of
materials: a review// Met.Trans. A. 1990. Vol. 21 A, May. P. 71-79.
Ansara I. Thermodynamic modeling of solution phases and phase diagram
calculations// High Temperature Science. 1990. Vol. 26. P. 215-229; Pure and Appl.
Chem. 1990. Vol. 62, N 1. P. 71-78.
Komarek KX. Experimental techniques in high temperature thermodynamics
// Pure and Appl. Chem. 1992. Vol. 64, N 1. P 93-102.
Морачевский А.Г., Козин Л.Ф. Термодинамика ассоциированных
расплавов // Термодинамика и материаловедение полупроводников / Под ред.
В.М. Глазова. М.: Металлургия, 1992. С. 53—74.
Морачевский А.Г. Физико-химические, структурные и технологические
исследования жидких сплавов калия со свинцом (обзор)//Журн. прикл. химии.
1992. Т. 65, №6. С. 1201-1218.
Морачевский А.Г. Физико-химические и электрохимические
исследования жидких сплавов лития со свинцом (обзор) // Журн. прикл. химии. 1994.
Т67,№ 12. С. 1937-1950.
Морачевский А.Г., Козин Л.Ф. Термодинамические свойства и
структура жидких сплавов рубидия (обзор) // Журн. прикл. химии. 1995. Т. 68, № 5.
С. 705-717.
Морачевский А.Г. Физико-химические свойства и структурные
особенности жидких сплавов цезия (обзор) // Журн. прикл. химии. 1995. Т. 68,
№ 10. С. 1585-1601.
Морачевский А.Г. Сплавы лития с алюминием: термодинамические
свойства и электрохимическое поведение в расплавленных солях (обзор) //
Журн. прикл. химии. 1996. Т. 69, № 4. С. 529-546.
Морачевский А.Г. Физико-химические и электрохимические
исследования системы натрий-сера в расплавленном состоянии (обзор) //Журн. прикл.
химии. 1996. Т. 69, № 9. С. 1409-1426.
252
Морачевский А.Г. Физико-химические свойства, структура и
электрохимическое поведение жидких сплавов системы натрий—свинец (обзор) //
Журн. прикл. химии. 1997. Т. 70, № 7. С. 1057-1071.
Sonuner F. Thermodynamics of liquid alloys // Mater. Science and
Engineering. A. 1997. Vol. A226-228. P. 757-762.
Воронин Г.Ф. Существование и устойчивость химических соединений //
Неорганич. матер. 2000. Т. 36, № 3. С. 342-349.
РусановА.И. Нанотермодинамика: химический подход// Рос. хим. журн.
(Журн. Рос. хим. общ-ва им. Д.И. Менделеева). 2006. Т. 50, № 2. С. 145-151.
Русанов А.И. Д.И. Менделеев и физическая химия: сто лет спустя//Журн.
общей химии. 2007. Т. 77, № 2. С. 193-214.
Морачевский Андрей Георгиевич
Кохацкая Мария Сергеевна
ПРИКЛАДНАЯ ХИМИЧЕСКАЯ
ТЕРМОДИНАМИКА
Учебное пособие
Редактор А. В. Явственная
Технический редктор А#. Колодяжная
Корректор А//. Рогозин
Верстка Н. В. Стасеевои
Директор Издательства Политехнического университета А.В. Иванов
Санкт-Петербургский государственный политехнический университет.
Издательство Политехнического университета,
член Издательстко-полиграфической ассоциации университетов России.
Адрес университета и издательства:
195251, Санкт-Петербург, Политехническая ул., 29.
Лицензия ЛР № 020593 от 07.08.97
Налоговая льгота — Общероссийский классификатор продукции
ОК 005-93, т. 2; 95 3005 — учебная литература
Подписано в печать 26.12.2008. Формат 60x84/16. Печать цифровая.
Усл. печ. л. 16,0. Уч. печ. л. 16,0. Тираж 200. Заказ 4128b.
Отпечатано в Цифровом типографском центре
Издательства Политехнического университета.
195251, Санкт-Петербург, Политехническая ул., 29.
Тел.:(812)550-40-14
Тел./факс: (812) 297-57-76.