/



















































































































































































































































































































Text
Г. Юинг Инструментальные методы химического анализа
Издательство «Мир»
Инструментальные методы химического анализа
Instrumental Methods of Chemical Analysis
Fifth Edition
Galen W. Ewing
Formerly Professor of Analytical Chemistry
Seton Hall University
McGraw-Hill Book Company
New York St. Louts San Francisco Auckland Bogota Hamburg Johannesburg London Madrid Mexico Montreal New Delhi Panama Paris Sao Paulo Singapore Sydney Tokyo Toronto
Г. Юинг Инструментальные методы химического анализа
Перевод с английского канд. хим. наук Е. Н. Дороховой и канд. хим. наук Г. В. Прохоровой
Москва «Мир» 1989
ББК 24.4
Ю22
УДК 543.08
Юинг Г.
Ю22 Инструментальные методы химического анализа: Пер. с англ.— М.: Мир, 1989.—608 с., ил.
ISBN 5-03-000194-8
Это — широко известное в мире учебное пособие, написанное американским автором, вышедшее за рубежом пятым изданием (перевод первого издания опубликован в 1960 г. издательством «Химия»). В нем нашли отражение современные тенденции развития анализа и рассмотрены почти все современные инструментальные методы определения и разделения веществ. Помимо традиционного материала (спектроскопические, электрохимические и хроматографические методы) книга включает еще и совершенно новые для учебной литературы разделы, посвященные применению компьютеров и автоматизации анализа.
Книга предназначена для студентов старших курсов всех специальностей химических вузов и аспирантов.
Ю 1804000000-323 9J_88 j БВК 24 4
041(01)-89
Редакция литературы по химии
Учебное издание
Гален В. Юииг
ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА
Заведующий редакцией академик О. А. Реутов. Зам. зав. редакцией 3. Ф. Ходецкая. Научный редактор Б. М. Комарова. Мл. научный редактор И. И. Землячева. Художник В. А. Медников. Художественный редактор М. Н. Кузьмина. Технический редактор А. Л. Гулииа. Корректор Т. П. Пашковская
ИБ № 6435
Сдано в набор 9.02.88. Подписано к печати 11.10.88. Формат 60X90716. Бумага тип. № 2. Печать высокая. Гарнитура литературная. Объем 19,00 бум. л. Усл. печ. л. 38,00. Усл. кр.-отт. 38,00. Уч.-изд. л. 39,79. Изд. № 3/5526. Тираж 7000 экз. Зак. 537.
Цена 6 р. 50 к.
Издательство «МИР» В/О «Совэкспорткнига» Государственного комитета СССР по делам издательств, полиграфии и книжной торговли, 129820, ГСП, Москва, И-110, 1-й Рижский пер., 2.
Ленинградская типография № 4 ордена Трудового Красного Знамени Ленинградского объединения «Техническая книга» им. Евгении Соколовой Союзполнграфпрома при Государственном комитете СССР по делам издательств, полиграфии и книжной торговли. 191126, Ленинград, Социалистическая ул., 14.
ISBN 5-03-000194-8 (русск.) © 1985, 1975, 1969, 1960
ISBN 0-07-019857-8 (англ.) ' by McGraw-Hill Inc.
All rights reserved
© перевод на русский язык, «Мир», 1989
От переводчиков
Среди учебных изданий по аналитической химии, переведенных в СССР за последние годы, книга Г. Юинга «Инструментальные методы химического анализа» занимает особое место. Первое ее издание на русском языке вышло в свет в 1960 г. в издательстве «Химия». В США книга выдержала несколько переизданий. Перед нами перевод последнего, пятого издания. Сравнивая первое и последнее из них, поражаешься изменениям, которые произошли в этой области аналитической химии за сравнительно короткий период времени. Лишь первые несколько фраз и очень немногие страницы текста перешли из предыдущих изданий без изменений. Введены новые главы и разделы («Автоматические анализаторы», «Компьютеры в аналитических приборах», «Общие вопросы анализа»), описаны новые методы, которые не были известны два десятилетия назад (фотоакустическая и лазерная спектроскопия, ряд ядерных методов и др.). Но дело не только в изменении содержания, изменился подход к изложению материала.
Книга Юинга прежде всего отражает тенденции последних лет в преподавании физических и физико-химических методов анализа и исследовании вещества. Почти любое относительно легко измеряемое физическое свойство элемента или соединения может служить основой метода анализа. Физические свойства очень разнообразны, отсюда и разнообразие методов. Описать все методы в одной монографии или учебнике принципиально невозможно: постоянно рождаются новые методы и открываются новые свойства, которые могут привести к созданию новых методов. Поэтому важно и нужно прежде всего знать фундаментальные свойства и общие закономерности, на которых Основано развитие тех или иных родственных методов. Например, в основе всех спектроскопических методов лежит взаимодействие вещества с электромагнитным излучением. Такая тенденция в преподавании — от общего к частному — находит широкую поддержку среди многих педагогов-аналитиков (см., например, статью Р. Келлнера и Г. Мали<?сы в журнале
6
Предисловие переводчиков
Z. anal. Chem., 1984, v. 319, p. 1), что отражено в современных программах по аналитической химии.
Именно с таких методических позиций построена книга Юинга. Вместо разрозненных глав, описывающих отдельные методы, перед читателем выстраивается здание, в котором каждый метод, как кирпичик, занимает свое место. Главам, посвященным спектроскопическим методам, предпослана большая вводная глава «Введение в оптические методы»; в отдельной главе рассматриваются общие закономерности электрохимических методов.
Итак, настоящее издание не похоже на первое, да и во многом на последующие издания, однако цель, построение и направленность книги остались без изменений. Как и прежде, ее отличают широкий охват материала, сжатость изложения и высокий научный уровень. В сравнительно небольшом объеме Юингу удалось охватить множество методов — давно используемых, новых и, возможно, перспективных. Иногда какой-то модификации метода уделено лишь несколько слов, но при этом обязательно дается ссылка на книгу или обзор, откуда можно почерпнуть более глубокие сведения. Таким образом, книга выполняет одну из главных задач преподавания — пробудить интерес у учащихся к какому-то вопросу, заставить их задуматься и, если интересно, докопаться до истины.
Большая часть их не станет профессионалами-аналитиками, но для них важно уметь выбрать метод, наиболее подходящий в данных обстоятельствах, а не заучивать подробности всех методов— это невозможно, да и не нужно. Из книги Юинга ясно, что не существует метода, пригодного на все случаи жизни. В каждой главе есть раздел «Применение», который может облегчить выбор конкретного метода, а таблицы с сопоставлением характеристик методов будут интересны и полезны специалистам (например, табл. 9-9).
Книга называется «Инструментальные методы химического анализа». Против такого общего названия для физико-химических и физических методов порой выдвигаются возражения, (действительно, основные инструменты . классического анализа— весы и бюретка). Однако в данном случае название вполне оправданно: большая часть книги посвящена описанию аппаратуры, причем излагаются не только принципы действия приборов, но и интересные инженерные решения, нашедшие воплощение в конкретных моделях.
Е. Дорохова Г. Прохорова
Предисловие автора
Цель настоящего издания, как и всех предыдущих изданий этой книги, состоит в том, чтобы дать обзор теории и практики современных инструментальных методов анализа. Особое внимание уделяется возможностям и ограничениям, присущим каждому методу.
Книга рассчитана на студентов старших курсов и аспирантов. К изучению этого курса лучше всего приступить сразу же после ознакомления с курсом классического количественного анализа и годичным курсом физики; его можно также изучать после прохождения курса физической химии или одновременно с ним. В книге нет сложных математических выкладок, и лишь там, где это безусловно необходимо, приводятся простейшие расчеты.
Решить, что исключить и что оставить, всегда было трудной задачей. Не так-то просто дать объективное определение понятий. «аналитический» и «инструментальный». Относительно первого Г. А. Лайтинен писал: «Здесь надо исходить из цели исследования. Если целью исследования является собственно измерение, то его следует отнести к области аналитической химии, тогда как интерпретация результатов измерений относится к другим областям химии» (Anal. Chem., 1966, v. 38, р. 1441). Для того чтобы очертить те области, в которых применим какой-либо метод, я старался включать только достаточно легко интерпретируемый материал.
Что касается охвата конкретного материала,.я стремился прежде всего принести пользу студентам-химикам и старался учитывать уровень их подготовки, считая, что принцип действия аналитических весов уже освоен ими из предыдущих курсов.
В книгу добавлена новая глава, посвященная фотоакусти-ческой спектроскопии, так как практическое применение этого метода не ограничивается какой-либо областью (инфракрасной или ультрафиолетовой — видимой). Расширена глава, в которой описываются электронные схемы; в нее включены сведения
8
Предисловие автора
об аналого-цифровых преобразователях и вопросы стыковки компьютера с прибором. Значительно расширена глава о применении компьютеров в аналитической химии, где особое внимание уделяется серийным приборам с встроенными (автономными) микропроцессорами. Изложенный материал позволяет составить представление о работе этих устройств и их возможностях.
Как и в предыдущих изданиях, глава об электронике дана в конце книги, но сведения из нее по мере необходимости привлекаются по всему тексту со ссылкой на эту главу. По желанию преподаватель может легко перенести этот материал в начало курса.
В книге используется система единиц СИ. Чтобы дать студентам возможность попрактиковаться в переводе одних единиц в другие, в задачах иногда используются единицы, до сих пор еще часто встречающиеся в литературе (при этом дается их определение).
Упоминание об отдельных моделях приборов, выпускаемых теми или иными изготовителями, не означает, что я предпочитаю именно эти модели. Моей целью было описать типичные приборы или модели, имеющие какие-то интересные особенности, а не представлять полный каталог аналитической аппаратуры/
Мне хочется выразить искреннюю благодарность моим коллегам и студентам,чьи советы и замечания помогли устранить отдельные недостатки, и особую признательность профессорам Р. Куку, Ф. Гутрие, А. Хейну, Дж. Иордану и Э. Пипмайеру за внимательное прочтение рукописи и ценную критику. Я в большом долгу перед персоналом компаний и фирм, которых слишком много, чтобы перечислять здесь всех; без сотрудничества с ними книга не могла бы рассчитывать на успех.
Часть работы над настоящим изданием была выполнена во время моего пребывания в Карлтонском колледже, и я хочу поблагодарить сотрудников библиотеки и других подразделений в Карлтоне, которые сделали мое пребывание там очень приятным.
Гален В. Юинг
ГЛАВА
Введение
Аналитическая химия рассматривает принципы и методы определения состава веществ, т. е. входящих в них элементов или соединений. Исторически развитие аналитических методов было тесно связано с внедрением новых измерительных приборов. Первые количественные анализы, проведенные гравиметрическим методом, стали возможны благодаря созданию точных весов. Изобретение спектроскопа в последние десятилетия XIX в. оказало чрезвычайно благотворное влияние на развитие анализа. Сначала спектроскоп применялся для качественного анализа, и единственными методами количественного анализа долгие годы оставались гравиметрия и титриметрия. Постепенно были введены некоторые турбидиметрические и нефелометрические методы. Затем оказалось, что для обнаружения конечной точки титрования можно с успехом использовать электрический сигнал. Быстрое развитие электроники в 30-е годы произвело революцию в инструментальном анализе. Современный химик независимо от того, считает ли он себя специалистом-аналитиком или нет, должен владеть примерно дюжиной методов, которые в сущности не были известны предшествующему поколению.
Практически любое физическое свойство, характерное для отдельного элемента или соединения, может служить основой метода аналитического оцределения. Уже краткий перечень тем, включенных в книгу, указывает на огромное разнообразие аналитических методов. В последующих главах мы прежде всего рассмотрим целый ряд спектроскопических методов, в том числе поглощение и испускание излучения во всех областях электромагнитного спектра. Затем мы проведем обзор электрохимических методов и обратимся к хроматографии газов и жидкостей. Обзор аналитических методов заканчивается главами по термометрическим и ядерным методам.
В последние годы получило развитие совместное использование двух или более методов; Каждый гибридный метод описывается в главе, посвященной одному из входящих в него
10 Глава 1
методов, причем дается ссылка на главу, где описывается второй метод. В основе одного из первых гибридных методов лежит «союз» масс-спектрометрии (МС) и газовой хроматографии (ГХ), который, следовательно, можно обозначить как МС/ГХ; он разбирается в гл. 20.
Аналитические методы имеют много общего. Во избежание ненужных повторений некоторые общие закономерности рассматриваются в гл. 26. Например, в этой главе обсуждается метод добавок, который как один из приемов калибровки используется в разных методах, описанных в предыдущих главах.
Прежде чем перейти к самостоятельным темам, следует сделать несколько общих замечаний о целях инструментализации и результатах, достигаемых с ее помощью.
Основной задачей прибора является перевод химической информации в форму, удобную для непосредственного наблюдения оператором, что осуществляется при помощи преобразователя. В этом узле поступающая информация используется для управления величиной или формой электрического сигнала. Затем с помощью соответствующей электронной схемы ее следует извлечь из электротока, усилить в случае необходимости и передать на считывающее устройство.
Электроника
Аналитические приборы должны обладать максимальной чувствительностью, чтобы точно измерять слабейший сигнал, поступающий с преобразователя. Логические и арифметические действия, например вычитание фонового сигнала, часто осуществляются аппаратно.
Во многих методах необходимо возбуждение (например, поток излучения), которое часто создают, измеряют и регулируют при помощи дополнительных электронных схем. Ввиду того что электронные схемы являются неотъемлемой частью химических приборов, любое обсуждение конструкции прибора невозможно без знания основных принципов электроники. К счастью, современная электроника пришла в своем развитии к блочно-модульному принципу. Создано большое число усилителей и элементов логики в виде недорогих разъемных блоков, которые можно использовать как «кирпичики» при конструировании большинства электронных схем, описанных в этой книге.
Краткий обзор аспектов электроники, имеющих отношение к нашему основному предмету, дан в гл. 27. Этот материал можно изучать отдельно или использовать в качестве вспомогательного при изучении приборов.
Введение 11
Современные микрокомпьютеры
Наиболее значительные успехи в развитии приборостроения последних лет связаны с созданием микропроцессора как электронного устройства общего назначения. Термином «микропроцессор» называют интегральную схему (которую обычно помещают в пластмассовую коробку диаметром 1—5 см) с 40 выводами. Микропроцессор является сердцевиной устройства, состоящего из связанных небольших блоков, называемого микрокомпьютером. Благодаря снижению стоимости этих блоков в конструкцию приборов (за исключением самых примитивных) выгодно включать микрокомпьютерные контролирующие устройства вместо обычных электронных схем, стоящих почти столько же. Приборы нового поколения более универсальны и просты в обращении, чем их предшественники, благодаря автоматическому или полуавтоматическому контролю режимных параметров.
На страницах настоящей книги компьютерам, используемым в разных типах приборов, уделяется не очень много внимания, и не потому, что это неважно, а потому, что они мало отличаются друг от друга.
При решении многих задач, если позволяет время, можно обойтись и без компьютера, но есть области, где без компьютера получить результаты просто невозможно. Например, для изучения быстрых химических процессов нужно получить сотни данных за очень сжатый срок (несколько секунд), и здесь компьютер незаменим. В ряде методов требуется провести математическое преобразование зависимости сигнала от времени в другую зависимость — от частоты (фурье-преобразование). С помощью компьютера это сделать легко, а без него этот ценный для аналитической химии метод был бы неосуществим.
В любом случае компьютерный контроль — большое удобство хотя бы потому, что он освобождает оператора от утомительного однообразия аналитических операций. Однако всегда нужно иметь в виду, что компьютер не в состоянии предотвратить методической ошибки анализа. Легкость, с которой компьютер выдает результаты, может привести неопытного оператора к ошибочным результатам, даже в том простом случае, когда не нужно проверять калибровку и заботиться о других аспектах анализа.
В конце книги дается краткое описание принципов соединения аналитических приборов с компьютерами.
12
Глава 1
Серийные приборы
Обычно новые принципы измерения долгое время разрабатываются в академических лабораториях (и не обязательно химиками). Только после создания опытных образцов прибора и демонстрации его возможностей он поступает на предприятие-изготовитель. В процессе изготовления возникает много инженерных решений, и на основе одного и того же принципа может быть создано несколько моделей;
Для того чтобы научиться пользоваться приборами, необходимо ознакомиться не только с давно выпускаемыми доступными моделями, но и с разрабатываемыми конструкциями. Последнее возможно лишь благодаря Научным публикациям. В этой книге рассматриваются и те и другие модели.
Замечание об единицах
Международное соглашение об единицах и их обозначениях имеет первостепенное значение в науке. Определенный успех в этом отношении был достигнут благодаря проведению ряда международных конференций с участием Национального бюро стандартов США и соответствующих учреждений в других странах. В результате была создана Международная система единиц, сокращенно СИ. Она была опубликована в брошюре, выпущенной Национальным бюро стандартов *.
В нашей книге везде используется система СИ (табл. 1-1). Применение других все еще широко бытующих единиц оговаривается в тексте. В основном это касается таких хорошо известных единиц, как ангстрем, микрон, гаусс и торр (мм. рт. ст.). Приставки для обозначения кратных и дольных единиц приведены в приложении Б.
Литература
Студент, который захочет углубить свои знания по любой теме, упомянутой в настоящей книге, имеет для этого много возможностей. Помимо ссылок, приводимых в конце глав, и общеизвестных изданий, например Chemical Abstracts, выпускается много журналов, посвященных аналитической химии.
Общие вопросы аналитической науки освещаются в журналах «Analytical Chemistry», «Analytica Chimica Acta», «Та-lanta», «The Analist» (включая «Analytical Abstracts»), «The
* В СССР выпущен ее перевод. Номенклатурные правила ИЮПАК. по химии. Т. 1. Неорганическая химия. Физическая химия. Аналитическая химия.— М.: ВИНИТИ, 1979; Аналитическая химия. Т. 4.— М.: ВИНИТИ, 1985.— Прим, перев.
Таблица 1-1. Избранные единицы СИ
Представленные ниже единицы взяты из полной таблицы, опубликованной Национальным бюро стандартов (публикация 330). Сюда включены те единицы, которые, по всей вероятности, пригодятся читателю настоящей книги, и опущены единицы, являющиеся производными приведенных.
Величина Название Символ
Основные единицы
Длина Метр м
Масса Килограмм кг
Время Секунда С
Электрический ток Ампер А
Температура Кельвин К
Количество вещества Моль МОЛЬ
П роизводные единицы
Волновое число Единица на метр м-1
Концентрация Моль на кубический метр моль/м3
Частота Герц 1/с
Сила Ньютон Н = м-кг-с
Давление Паскаль Па = Н/м2
Энергия Джоуль Дж = Н м
Мощность Ватт Вт — Дж/с
Электрический заряд Кулон Кл = А-с
Электрический потенциал Вольт В = Вт/А
Емкость Фарада Ф = Кл/В
Сопротивление Ом й = В/А
Проводимость Сименс См = А/В
Плотность магнитного потока Тесла Тл = В-с/м2
Индуктивность Генри Ги = В-с/А
Необязательные единицы
Объем Литр IO—3 м3
Энергия Электронвольт эВа)
Масса (атомная) Единица атомной а. е. мб)
массы
Единицы, не рекомендуемые к употреблению
Длина Длина Масса Давление Давление Давление Радиоактивность
Энергия Энергия Сила
Сила магнитного поля Объем
Ангстрем Микрон Гамма Бар
Торр (мм. рт. ст.) Атмосфера Кюри Эрг Калория Дина Гаусс Лямбда
А = 10-10 м р. = 10~в м у = 10-9 кг бар == 105 Па (101325/760) Па 760 мм рт. ст.
Ки
эрг = 10~7 Дж 4,184 Дж дииа = 10—5 Н 10-4 Тлв Х= 10~9 м3
а) Экспериментальная единица; 1 эВ = l,6022.10—19 Дж (примерно).
б) Экспериментальная единица; 1 а. е. м. =1,66057-10~27 кг (примерно).
в) Строго говоря, гаусс и тесла не являются мерой одной и той же величины.
14 Глава 1
Fresenius Zeitschrift fur analytische Chemie», «CRC Critical Reviews in Analytical Chemistry», «Analytical Letters» и других. Сведения по более узким вопросам можно найти в таких журналах, как «Applied Spectroscopy», «Analytical Biochemistry», «Journal of Electroanalytical Chemistry», «Journal of Gas Chromatography» и многих других. Что касается специально приборов, то следует обратиться к журналам «Review of Scientific Instruments » и «Journal of Scientific Instruments».
В теоретическом аспекте очень ценными являются оба издания книги «Treatise on Analytical Chemistry», издаваемой И. Кольтгофом и Р. Элвингом (Wiley-Interscience, New York), особенно часть I. Во многих томах серии «Physical Methods of Chemistry», издаваемой А. Вайсбергером и Б. Росситером (Wiley-Interscience), содержится богатая информация об аналитических приборах.
В журнале «Analytical Chemistry», ежегодно в апреле, публикуются критические обзоры («Annual Reviews»), охватывающие все области анализа; в четные годы обзоры посвящены фундаментальным аналитическим проблемам, в нечетные — практическому применению.
Огромное количество полезной информации, в том числе теоретических обзоров, собрано в изданной в 1963 г. под редакцией Л. Мейтиса книге «Handbook of Analytical Chemistry» (McGraw-Hill, New York, 1963), которая, несмотря на дату выпуска, остается одним из наиболее ценных источников информации для химика-аналитика.
ГЛАВА
Введение в оптические методы
Большой класс оптических методов основан на взаимодействии электромагнитного излучения с веществом. В этой главе мы остановимся на некоторых важных свойствах излучения (и вещества), а затем обсудим особенности оптических приборов, применяемых для измерений во всех или нескольких спектральных областях. В последующих главах мы разберем отдельно каждую большую спектральную область (видимую, ультрафиолетовую, инфракрасную, рентгеновскую, микроволновую), останавливаясь на вопросах теории, техники эксперимента и применениях.
Природа излучательной энергии
Изучение электромагнитного излучения привело к дуализму в наших представлениях об его природе. В некоторых отношениях оно проявляет волновые свойства, однако его можно представить состоящим из дискретных сгустков энергии, называемых фотонами. Для строгого описания взаимодействия излучения с веществом почти всегда привлекается концепция фотона, тогда как для ориентировочной оценки поведения большого числа фотонов полезны волновые представления.
Электромагнитное излучение можно охарактеризовать несколькими параметрами. Частота v — число колебаний в единицу времени (используется при описании энергии, как электромагнитной волны); обычно единицей частоты служит герц (1 Гц=1 колебание в секунду). Скорость распространения с в вакууме равна 2,9979-108 м-с-1, в других средах она несколько меньше.
Длина волны К есть расстояние между соседними максимумами. Она равна отношению скорости к частоте. Единицами длины волны служат микрометр (1 мкм=10~в м; прежнее название— микрон, ц) и нанометр (1 нм=10~9 м; прежнее название— миллимикрон, мц). В спектроскопии широко используется ангстрем (1 А= 10~10 м), хотя эта единица не рекоменду
16
Глава 2
ется системой СИ. Еще одной весьма удобной величиной является волновое число v, т. е. число волн, приходящееся на единицу расстояния *. В качестве единицы волнового числа наиболее часто используется обратный сантиметр (см-1), иногда называемый кайзер. И длина волны, и волновое число зависят от показателя преломления среды, через которую проходит излучение, тогда как частота от него не зависит. К показателю преломления мы вернемся в одном из последующих разделов.
Скорость, длина волны и волновое число излучения в вакууме связаны с его частотой соотношением
v = -~ = vc (2-1)
Л
Энергия фотона прямо пропорциональна частоте
E = hv~-~- = hcv (2-2)
где h — постоянная Планка, равная примерно 6,6262 • 10-34 Дж-с. Таким образом, между энергией и длиной волны существует обратная зависимость, а между энергией и частотой или волновым числом — прямая. Поэтому многие спектроскописты предпочитают представлять спектры не в длинах волн, а в волновых числах.
Энергию фотона, особенно в случае ядерного и рентгеновского излучения, удобно выражать в электронвольтах (эВ; 1 эВ = 1,6022-10-19 Дж), что соответствует частоте 2,4180х Х1014 Гц или длине волны (в вакууме) %= 1,2395-10-* м. Часто встречаются также кратные единицы — килоэлектронвольт (кэВ) и мегаэлектронвольт (МэВ).
В обычном спектральном приборе поток излучения переносит энергию от источника через какую-либо среду или ряд сред к приемнику, где она поглощается. На пути к «поглотителю» поток может частично поглотиться средой, через которую проходит, а также изменить направление в результате отражения, преломления или дифракции.
* Выбор символа v для обозначения волнового числа неудачен из-за его сходства с символом v, который используется для обозначения частоты, и возникающей из-за этого путаницы; например, в некоторых разделах физики эти символы имеют как раз обратный смысл. Выражения, подобные «частота 1600 волновых чисел», часто встречающиеся в литературе, строго говоря, неверны. Частота может быть пропорциональна волновому числу, но не эквивалентна ему, поскольку они имеют разную размерность. Более того, волновое число — это не единица, поэтому выражение «1600 волновых чисел» ничуть не лучше выражения «6 расстояний ширины» для обозначения размера этой страницы. Следует сказать: «Волновое число равно 1600 обратных сантиметров»,
Таблица 2-1. Области электромагнитного спектра3)
а) В некоторых случаях числовые множители опущены; точность определения границ области ие позволяет указать большее количество •значащих цифр.
б) Границы областей инфракрасного спектра указаны по рекомендации комиссии по спектроскопии (J. Opt. Soc. Am., 1962, v. 52, p. 476)
18 Глава 2
Часто представляет интерес еще одна величина — мощность потока излучения (т. е. энергия в единицу времени), не совсем точно называемая «интенсивностью». Более правильно определить интенсивность как мощность излучения, испускаемого источником в определенном направлении, на единицу телесного угла. Сигнал фотоэлемента пропорционален скорости фотонов, т. е. мощности падающего излучения. С другой стороны, на фотопластинке суммируется мощность за время экспозиции, и сигнал (количество выделившегося серебра) является функцией общего числа фотонов, поглощенных единицей площади. Чувствительность фотоэлемента и фотопластинки, равно как и человеческого глаза, связана с длиной волны более или менее сложной зависимостью.
Спектральные области
Спектр излучательной энергии удобно разбить на несколько областей (табл. 2-1). Границы областей определяются возможностями методов генерирования и регистрации излучения. Цифрам, приведенным в таблице, не стоит придавать особого значения, их следует рассматривать как ориентировочные.
Для химика деление спектра на области особенно важно, потому что взаимодействие излучения с химической системой в каждой из них протекает по различным механизмам и дает разную информацию. Наиболее важные атомные и молекулярные переходы соответствуют следующей последовательности областей:
Рентгеновская
Дальняя ультрафиолетовая
Ближняя ультрафиолетовая
Видимая
БЛижняя и средняя инфракрасная
Дальняя инфракрасная
Микроволновая
Радиоволновая
К- и L-электроны
Средние электроны
Валентные электроны
Валентные электроны
Молекулярные колебания
Молекулярные вращения и низкочастотные колебания
Молекулярные вращения
Ядерный магнитный резонанс
Для обозначения ультрафиолетового и инфракрасного излучения приняты сокращения УФ и ИК.
Взаимодействие с веществом
Электромагнитное излучение возникает при замедлении движения электрических заряженных частиц, в основном электронов; при обратном процессе оно может поглощаться с передачей энергии ускоряющимся частицам. Поэтому при изучении взаимодействия вещества и излучения необходимо опираться на знание электронного строения атомов и молекул.
Введение в оптические методы 19
Атомные спектры
На рис. 2-1 в соответствии с общепринятыми представлениями показаны некоторые энергетические уровни внешних электронов нейтрального атома натрия. При обычных условиях по существу все атомы в парах натрия находятся в основном состоянии, т. е. их валентные электроны расположены на Зз-уровне. При облучении потоком энергии, включающем длины волн 589,00 и 589,59 нм, внешние электроны многих атомов поглощают фотоны и возбуждаются до Зр-уровней (два очень близких Зр-уровня различаются лишь своими спинами). Возбужденный электрон стремится вернуться в свое нормальное (3s) состояние; при возвращении в нормальное состояние испускается фотон. Испускаемый фотон обладает некоторым количеством энергии, определяемым расположением энергетического уровня. В приведенном примере испускаемое излучение обусловливает знакомый желтый цвет натриевого пламени и натриевой лампы. Излучение, генерируемое в этом простом слу-
Рис. 2-1. Часть схемы энергетических уровней валентных электронов атома натрия. «Символ терма» — это цифровое обозначение разных энергетических уровней. Цифры на прямых указывают соответствующие длины воли в нанометрах.
20 Глава 2
чае, когда внешний электрон переходит в возбужденное состояние, а затем возвращается на свой исходный уровень, называется резонансным излучением, или резонансной флуоресценцией.
Если сообщить электрону более высокую энергию, он, возможно, перейдет на более высокий, чем Зр, уровень, например на 4р- или 5р-уровень. В этом случае он может вернуться на 35-уровень не в результате единичного перехода, а задержаться на промежуточных уровнях, подобно мячику, прыгающему вниз по ступенькам. Эта ситуация мажет быть довольно сложной, и не все мыслимые переходы осуществимы — некоторые «запрещены» правилами отбора квантовой механики.
Под действием источника возбуждения высокой энергии способны возбуждаться в той или иной степени многие электроны (и не только внешние) любого элемента, и в результате испускаемое излучение может состоять из нескольких тысяч дискретных хорошо воспроизводимых длин волн, в основном в УФ- и видимой областях. Это служит основой аналитического метода эмиссионной спектроскопии.
Если же возбуждение осуществляется с еще большей энергией, внутренний электрон может полностью оторваться от своего атома. Освободившееся место может занять электрон с какого-либо более высокого уровня. Такому внутриорбитальному переходу соответствует гораздо более сильное изменение энергии, чем при возбуждении внешних электронов, поэтому испускаемые фотоны должны обладать гораздо большей частотой и соответственно меньшей длиной волны. Так происходит испускание рентгеновских лучей атомами, подвергнутыми бомбардировке потоком быстрых электронов.
Молекулярные спектры
Энергетическое строение молекулы сильно отличается от строения атома; на рис. 2-2 представлены несколько первых энергетических уровней внешних электронов ковалентной молекулы. При нормальных условиях в отсутствие возбуждения такая молекула находится в основном синглетном состоянии, обозначенном на схеме So. На схеме изображены также два более высоких уровня Si и Т\, отвечающие синглетному и триплетному состояниям. (Существует множество других синглетных и триплетных состояний, не указанных на рисунке.) Каждый из этих электронных энергетических уровней имеет ряд подуровней (vi, V2, ...), которые отвечают квантовым колебательным энергетическим состояниям.
Энергия электромагнитного излучения, поглощенная молекулой при возбуждении, вызывает переход на один из более
Введение в оптические методы 21
Рис. 2-2. Диаграмма энергетических уровней органической молекулы, на которой изображены основное синглетное состояние, первое возбужденное синглетное состояние и первое возбужденное триплетное состояние. Сплошными вертикальными линиями показаны поглощение или испускание излучения; штриховыми линиями — безызлучательные переходы. Процесс I: поглощение. Процесс II: колебательная дезактивация. Процесс Ш: колебательная релаксация из синглетного состояния. Процесс IV: флуоресценция. Процесс V: ин-теркомбинационная конверсия в триплетное состояние, вслед за которой могут происходить процессы VI, VII и VIII, аналогичные процессам II, III и IV. Процесс VIII: фосфоресценция.
высоких уровней. Это может произойти только в том случае, если энергия фотонов падающего излучения равна энергии перехода с уровня So на более высокий уровень или подуровень синглетного состояния (этот процесс обозначен на рис. 2-2 цифрой I). Если переход заканчивается на каком-либо колебательном подуровне возбужденного уровня (например, на подуровне уз), то молекула очень быстро переходит на уровень Si; при этом избыток энергии выделяется в виде тепла. На схеме этот процесс обозначен цифрой II.
Молекула в возбужденном состоянии St может расходовать оставшуюся энергию возбуждения по одному из трех механизмов: передать ее другой химической частице при столкновении (процесс III, называемый колебательной релаксацией) или испустить ее в виде излучения (процесс IV, называемый флуоресценцией). При определенных условиях молекула может перейти на какой-либо подуровень триплетного уровня, обладаю
22 Глава 2
щего почти такой же энергией (процесс V, называемый интеркомбинационной конверсией)-, затем возможны процессы VI, VII и VIII, аналогичные процессам II, III и IV. Процесс VIII называется фосфоресценцией.
Молекулярные спектры поглощения
Переходы в молекулах можно изучить, наблюдая избирательное поглощение или излучение или процессы испускания — флуоресценцию и фосфоресценцию. Родственным методом (но который не так-то просто представить наглядно при помощи схемы, подобной приведенной на рис. 2-2) является рамановская спектроскопия (спектроскопия комбинационного рассеяния, спектроскопия КР) • Этот метод разбирается вместе с флуоресценцией и фосфоресценцией в гл. 6.
Переходы между электронными уровнями наблюдаются в УФ- и видимой областях: переходы в пределах одного электронного уровня между колебательными подуровнями лежат в ближней и средней ИК-областях, их можно изучать методом рамановской спектроскопии. Избирательное поглощение в дальней ИК- и микроволновой областях связано с низкоэнергетическими переходами, вызванными вращением молекул или их частей.
Электронные переходы включают «перескоки» электронов на разные колебательные подуровни («туда и обратно»), поэтому поглощение в УФ- и видимой областях складывается из поглощений с близкими частотами. Энергии индивидуальных переходов слишком близки друг к другу, их нельзя разрешить на обычных лабораторных приборах, поэтому электронные спектры состоят из широких полос, хотя часто в них проявляются некоторые особенности строения молекулы. Спектры веществ, находящихся в газообразном состоянии или при низкой температуре (например, при температуре жидкого азота), имеют более тонкую структуру, чем спектры растворов при комнатной температуре.
Абсорбционные спектры в каждой спектральной области легко зарегистрировать: они играют важную роль в аналитических исследованиях, и это будет показано в последующих главах.
Фотоакустическая и фототермическая спектроскопия
За безызлучательными переходами, следующими за возбуждением потоком излучения (процесс II на рис. 2-2), иногда можно наблюдать путем измерения количества тепла, выделяющегося в пробе. Прямые измерения (фототермические) редко
Введение в оптические методы 23
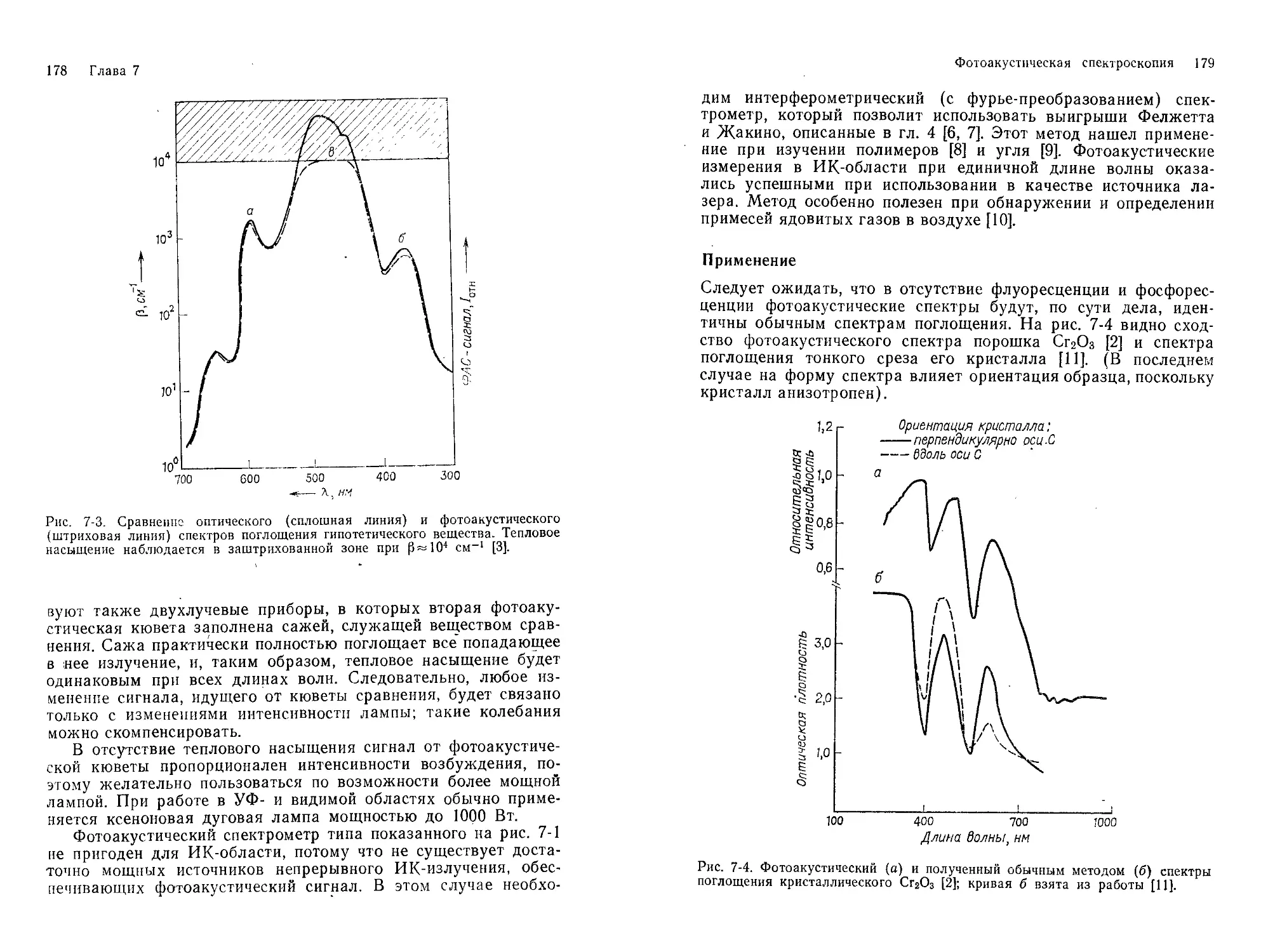
бывают достаточно чувствительными, но косвенные определения, связанные с изменениями давления газа, соприкасающегося с пробой (фотоакустические), лежат в основе важного метода, который применяется для исследований в УФ-, видимой и ИК-областях. Он будет обсуждаться в гл. 7.
Флуоресценция
Процесс испускания, обозначенный на рис. 2-2 цифрой IV, играет важную роль в химии растворов. Из сопоставления длин стрелок на рисунке ясно, что фотоны испускаемого излучения обычно обладают меньшей энергией и, следовательно, большей длиной волны, чем фотоны возбуждающего излучения. (Исключение из этого общего правила представляют резонансное испускание, когда испускаемые и поглощаемые фотоны имеют одинаковую энергию, а также испускание в том случае, когда электрон до поглощения излучения уже находился на возбужденном колебательном уровне.)
Многие органические и некоторые неорганические соединения после возбуждения УФ-излучением флуоресцируют в видимой области. Атомы также способны к флуоресценции в УФ-, ПК- и рентгеновской областях.
Фосфоресценция
В некоторых молекулах безызлучательный переход может происходить с возбужденного синглетного уровня на соответствующий триплетный уровень (процесс V, рис. 2-2). При возвращении молекулы в основное состояние может излучаться избыточная энергия (процессы VI и VIII). Переход из триплетного в основное состояние маловероятен, поскольку сопровождается изменением спина электрона. Поэтому полупериод существования возбужденной молекулы довольно велик, и излучение можно наблюдать в течение измеримого интервала времени после удаления источника возбуждения. Энергетический уровень, переход с которого в основное состояние маловероятен, называется метает обильным, а длительное незатухающее излучение— фосфоресценцией. В отличие от фосфоресценции флуоресценция длится меньше одной микросекунды.
Рамановская спектроскопия (спектроскопия КР)
Эффект Рамана — явление, родственное флуоресценции. Здесь также длина волны испускаемого пробой излучения отличается от длины волны возбуждающего излучения. Однако если для возникновения флуоресценции проба должна поглотить излуче
24 Глава 2
ние, то возникновение эффекта Рамана необязательно связано с поглощением. Сдвиг длины волны в этом эффекте вызван тем, что энергия падающих фотонов расходуется на переведение молекулы в более высокое колебательное состояние. Испускаемые фотоны можно, таким образом, рассматривать как те же самые падающие фотоны, но с меньшей энергией.
Рефракция
Перейдем от процессов, происходящих в атомах и молекулах, к более общим процессам, происходящим при взаимодействии всей массы вещества с излучением.
Важной общей характеристикой оптических свойств среды служит показатель преломления п, который можно определить как отношение скорости излучения определенной частоты в вакууме с к его скорости в какой-либо среде ст-
п = с/ст (2-3)
Изменение показателя преломления вещества при изменении длины волны называется дисперсией. Дисперсия во всех областях электромагнитного спектра тесно связана со степенью поглощения излучения. В областях с высоким пропусканием показатель преломления уменьшается (нелинейно) с ростом длины волны; в областях с высоким поглощением показатель преломления резко возрастает с увеличением длины волны, но здёсь трудно добиться высокой точности измерения.
Показатель преломления — важная характеристика прозрачных материалов. Изменением показателя преломления, связанным с изменением длины волны, обусловлены хроматическая аберрация линз и разложение излучения призмами, которые имеют важное значение при конструировании оптических приборов. Эти и другие вопросы еще будут рассматриваться в настоящей книге.
Поляризация и оптическая активность
Некоторые вещества способны поляризовать излучение или вращать плоскость плоскополяризованного излучения. Неполяри-зованный поток излучения можно представить в виде пучка волн, колебания в которых происходят в плоскостях вдоль линий распространения волны. На‘рис. 2-3, а изображено поперечное сечение луча, который направлен перпендикулярно плоскости бумаги. Если этот поток пропустить через поляризатор, каждая волна в пучке, например распространяющаяся вдоль вектора АОА' (рис. 2-3,6), разлагается на ортогональные составляющие ВОВ' и СОС', направленные вдоль осей х и у
Введение в оптические методы 25
в
а
Рис. 2-3. Векторы колебаний обычного и плоскополяризованного электромагнитного излучения.
поляризатора. Поляризующий материал обладает способностью поглощать одну из составляющих колебаний (например, СОС') и пропускать другую (BOB'). Таким образом, при условии 100 %-ной поляризации в выходящем потоке колебания будут происходить только в одной плоскости; такой поток называют плоскополяризованным. Большинство поляризаторов, применяемых на практике, несовершенны, и поляризация происходит лишь частично.
Второй поляризатор (называемый анализатором), помещенный на пути светового потока, точно так же пропускает только ту составляющую излучения, которая колеблется параллельно его оси. Поскольку поток уже поляризован, все излучение будет проходить через анализатор только при одном угле поворота, а при повороте анализатора на 90° мощность светового потока упадет до нуля (рис. 2-4). Излучение лампы, выходящее из линзы-коллиматора в виде параллельного пучка, проходит через поляризатор А с вертикальной оптической осью. Анализатор Б, также имеющий вертикальную ось, не оказывает влияния на поток, но поляризатор В с горизонтальной осью полностью гасит излучение. При. повороте плоскости В мощность
рующ/ая Поляризатору линза
Рис. 2-4, Схема поляризации излучения.
26 Глава 2
пропускаемого излучения будет меняться с изменением синуса угла поворота. Два последовательно расположенных поляризатора называются скрещенными, если их оси взаимно перпендикулярны. Любой поток излучения можно сделать плоскополя-ризованным в любой степени — от 0 (полная симметрия) до 100 % (полная поляризация).
Важное значение в химии имеет способность некоторых кристаллов и жидкостей вращать плоскость поляризации проходящего через них излучения. Эта способность известна под названием оптическая активность. Более подробные сведения о применении поляризованного излучения, в том числе круговой и эллиптической поляризации, приведены в гл. 10.
Источники излучения
Для многих оптических методов желательно иметь источник монохроматического (т. е. с одной длиной волны) излучения в интересующей области спектра. К сожалению, в настоящее время таких источников нет. Поэтому на практике приходится пользоваться источником в сочетании с монохроматором — устройством, которое можно настроить на пропускание узкого пучка длин волн, приближающегося к монохроматическому.
Нагретые тела в качестве источников. Любое вещество при температуре выше абсолютного нуля испускает излучение вследствие теплового движения электронов. Природа теплового излучения применительно к идеальному источнику, называемому абсолютно черным телом, хорошо изучена. На рис. 2-5 показано, как распределяется энергия излучения абсолютно черного тела по длинам волн при различных температурах [1]. Связь длины волны, соответствующей максимуму энергии Атах, с температурой Т описывается законом смещения Вина
Xmax- Т = 2,8978 -106 (2-4)
(длина волны выражена в нанометрах, температура — в кельвинах). Длина волны и температура связаны обратной зависимостью, поэтому нагретые тела испускают излучение видимой и инфракрасной областей; для получения УФ-излучения источник должен иметь очень высокую температуру, что неудобно в практической работе. Излучение (и форма кривой) абсолютно черного тела описывается законом Планка, согласно, которому общая мощность, испускаемая на единицу площади источника, равна
2лйс2
A5 exp (ftc/AfeT) — 1
Введение в оптические методы 27
Рис. 2-5. Излучение абсолютно черного тела при различных температурах. Вертикальными штриховыми линиями на обоих графиках указаны границы видимой области [1].
где h — постоянная Планка, с— скорость света в вакууме, k — постоянная Больцмана. Излучение реальных веществ при тех же длинах волн значительно слабее, чем это следует из кривой излучения абсолютно черного тела; такие вещества иногда называют серыми телами.
Газоразрядные источники. При использовании в качестве источника газового разряда обычно получается линейчатый спектр. При низком давлении газа каждая линия практически отвечает одной длине волны, но при повышении давления линии уширяются вследствие столкновений и при достаточно высоком давлении сливаются в непрерывный спектр.
Газоразрядные трубки низкого давления в качестве источников излучения используются в ограниченных целях. В атомно-абсорбционной спектроскопии для определения металлов, например хрома, который переведен в парообразное состояние, желательно в качестве источника использовать газоразрядную трубку низкого давления, содержащую хром; резонансное излучение источника избирательно поглощается тем же элементом пробы. В этих целях часто применяется чашеобразный катод, изготовленный из определяемого металла или покрытый им (лампа с полым катодом) (см. гл. 5).
28 Глава 2
Газоразрядные лампы низкого давления служат УФ-детекторами в хроматографах (это еще один пример применения газоразрядных ламп низкого давления). Линия испускания ртути при 253,7 нм, являющаяся самой интенсивной эмиссионной линией в спектре ртутной лампы, поглощается многими органическими соединениями, что можно использовать для обнаружения таких соединений после их вымывания из колонки. Подробно это рассматривается в гл. 20 и 21.
Сплошные спектры можно получить с помощью газоразрядных трубок, заполненных разными газами, например ксеноном при давлении в несколько атмосфер и парами ртути при давлении, превышающем 100 атм. Для получения сплошного спектра широко используется разряд в водороде или дейтерии при давлении 1—2 кПа *, но механизм возникновения спектра в этом случае иной. Молекулы Н2 или D2 переходят в возбужденное состояние (обычно оно обозначается звездочкой), а избыток энергии выделяется в результате процесса D2*->D-f-D + + hv. Поскольку возбужденные молекулы обладают разной энергией, образовавшиеся при диссоциации атомы находятся на разных колебательных уровнях, и тогда возникающее излучение в результате наложения линий тонкой структуры дает сплошной спектр [2].
Рабочие области (в нанометрах) обычной лампы с кварцевым окном составляют: для ксеноновой от 250 до 1000, ртутной высокого давления от 280 до 1400, водородной или дейтериевой от 160 до 365.
Лазеры **
Лазер служит источником монохроматического излучения в УФ-, видимой и ИК-областях. Первый лазер (сообщение о нем появилось в 1960 г.) представлял собой тщательно отполированный стержень из рубина (А12О3 с примесью Сг2О3) с плоскопараллельными торцами. На одном торце помещается зеркало, так что все излучение, идущее изнутри кристалла, отражается обратно. Зеркало на другом торце покрыто тонким слоем серебра, поэтому часть излучения (обычно 80—90 %) отражается, а часть выходит наружу.
Когда на стержень падает интенсивный импульс света, например, от ксеноновой лампы (рис. 2-6), почти все атомы хрома
* 1 кПа (килопаскаль) =7,5 мм рт. ст.
** «Лазер» (Laser) — аббревиатура названия метода усиления света с помощью индуцированного излучения (Light Amplification by Stimulated Emission of Radiation). Симпозиум по лазерам и их применению в химии состоялся в 1981 г. в Нью-Йорке. Материалы его были опубликованы в июне 1982 г. в журнале «Journal of Chemical Education».
Введение в оптические методы 29
Отражатель
Активный элемент ^2
_____Импульсная лампа_______
4т......... ... Ч-
Отражатель
Рис. 2-6. Типичный лазер. Зеркала Afj и М2 ограничивают полость лазера (резонатор); Afj отражает полностью, М2— частично. Активным элементом может быть твердое вещество (например, рубин), газ (например, смесь гелий— неон) и жидкость (например, раствор красителя). Активное вещество заключено между окнами, расположенными под углом Брюстера. Выходящий поток излучения идет направо от полупрозрачного зеркала. Лазер возбуждается под действием ксеноновой импульсной лампы.
возбуждаются и большинство быстро переходит в метастабиль-пое состояние. При переходе первичных электронов из метаста-бильного в основное состояние испускаются фотоны, соответствующие длине волны 694,3 нм. Некоторые фотоны испускаются параллельно оси стержня и многократно отражаются зеркалами, причем часть их при каждом отражении выходит в виде излучения. Действие лазера основано на том, что излучательная энергия определенной частоты вызывает излучение остальных метастабильных атомов хрома, поэтому мощность излучения нарастает очень быстро. Действие лазера настолько эффективно, что мощные импульсы монохроматического излучения испускаются в течение 0,5 мс. Мощность каждого импульса может достигать нескольких мегаватт.
Лазеры можно изготовить на основе многих активных материалов. Из твердых веществ применяются стекла с добавкой нескольких процентов неодима или другого лантанида, а также гранат, содержащий иттрий и алюминий (ИАГ). Многие газы при пропускании через них мощного электрического импульса способны быть активной средой. Стоит отметить лазеры на основе гелия—неона, аргона, азота и диоксида углерода. Газовые лазеры могут давать как непрерывный, так и импульсный потоки излучения.
Все упомянутые здесь лазеры испускают излучение определенной дискретной длины волны, которую можно изменять лишь в очень ограниченном интервале. Существует и другой тип лазера — лазер на красителях с перестраиваемой в широком интервале длиной волны [3, 4]. Активным веществом в них является флуоресцирующий органический краситель, например родамин 6G или флуоресцеин, в водных или спиртовых раство-
30 Глава 2
Рис. 2-7. Лазер на красителях, в котором используется струя раствора красителя. Резонатор заключен между вогнутым полностью отражающим зеркалом Л1, и частично отражающим зеркалом М2. Возбуждение происходит под действием азотного лазера (не показан). Лазер на красителях можно настроить при помощи какого-либо элемента, например интерференционного светофильтра, располагаемого под регулируемым углом. Раствор красителя можно прокачивать при помощи насоса.
рах. Интенсивное излучение какой-либо длины волны внутри полосы поглощения красителя вызывает переход большого числа молекул в возбужденное (Si) состояние, из которого они могут снова перейти в основное состояние по механизму лазерного излучения. Длину волны испускаемого излучения можно перестроить в пределах 40 нм при помощи диспергирующего элемента (см. следующий раздел), который помещают между зеркалами лазера (т. е. в резонатор лазера). Обычно лазеры на красителях получают энергию от другого лазера, например азотного. На рис. 2-7 схематически изображено устройство лазера на красителях. Лазеры такого типа нашли широкое применение в аналитических исследованиях.
Лазерное излучение обладает уникальными свойствами [5] — монохроматичностью и когерентностью. Это значит, что волны, идущие от всех атомов и молекул, находятся в одной и той же фазе (что не характерно для излучения обычных источников). Частично благодаря когерентности коллимированный поток лазерного излучения мало расходится при распространении. Это позволяет концентрировать большое количество энергии на небольшой мишени, находящейся даже на значительном расстоянии. Излучение некоторых лазеров частично или полностью поляризовано.
Важная роль, которую играют лазеры в аналитической химии, связана с высокой монохроматичностью и большой интенсивностью излучения. Среди прочего следует упомянуть об использовании лазера в качестве источника локального нагрева поверхности и источника возбуждения в рамановской и флуоресцентной спектроскопии.
Введение в оптические методы 31
Монохроматизация
При изучении спектров поглощения желательно, чтобы полоса длин волн падающего излучения была как можно уже. Как упоминалось ранее, получить такую полосу можно с помощью устройства, называемого монохроматором. Иногда можно использовать источник линейчатого спектра, но даже в этом случае для выделения единичной линии обычно нужен монохроматор.
Существуют два способа выделения длины волны; во-первых, использование светофильтров и, во-вторых, разложение света с помощью призмы или дифракционной решетки.
Светофильтр — это устройство, способное пропускать излучение некоторых длин волн и поглощать, частично или полностью, остальное. Светофильтрами для видимого излучения обычно служат цветные стекла. Имеется большой выбор таких светофильтров, более или менее равномерно охватывающих видимую область спектра.
Принцип действия светофильтров другого типа основан на явлении интерференции. На рис. 2-8 показано поперечное сечение интерференционного светофильтра. Для его изготовления
на прозрачную пластинку наносят полупрозрачную пленку из отражающего металла, например из серебра. Пленку покрывают очень тонким слоем прозрачного материала, например фторида магния, а затем снова пленкой серебра. Каждая серебряная пленка отражает примерно половину падающего на нее излучения и пропускает остальной его поток. Часть падающего потока повторно отражается слоями серебра, но при каж
дом отражении некоторое количество излучения выходит наружу. Те выходящие лучи, для которых расстояние между серебряными пленками кратно половине длины волны (&Х/2, где X — длина волны,
л=1, 2, 3...), усиливаются. Потоки излучения других длин волн интерферируют в слое MgF2, поэтому их энергия практически не выходит наружу. Тонкие слои в интерференционных светофильтрах, выпускаемых промышленностью, защищены еще одной прозрачной пластинкой. Выделяе-
Рис. 2-8. Схематическое представление интерференционного светофильтра. Светлыми кружками указаны гребни, а темными — впадины волны излучения (*=1).
32 Глава 2
Рис. 2-9. Разложение белого света призмой (а) и дифракционной решеткой на пропускание (б).
мая интерфернционными светофильтрами полоса длин волн значительно уже, а максимальное пропускание гораздо больше, чем у стеклянных светофильтров. Интерференционные светофильтры пропускают потоки излучения многих порядков (в соответствии со значениями k). Излучение нежелательных порядков можно отсечь при помощи подходящего поглощающего слоя. Длины волн второго и более высоких порядков видимого излучения находятся в УФ-области, поэтому их легко устранить при помощи стеклянных пластинок.
Светофильтры того или иного типа или их комбинации позволяют выделить нужную полосу длин волн практически в любой области спектра от рентгеновской до инфракрасной.
Монохроматор
Монохроматор — это прибор, позволяющий выделить очень узкую полосу длин волн из сравнительно широкой части спектра. Его можно настроить автоматически или вручную на любую длину волны.
Монохроматор состоит из диспергирующего элемента (призмы или дифракционной решетки) и двух узких щелей для входа и выхода излучения. Входная щель пропускает узкий пучок, который падает на диспергирующее устройство. Здесь лучи с разными длинами волн отклоняются под разными углами, в результате чего поток расходится в виде веера, как показано на рис. 2-9. Выходную щель можно установить так, чтобы через нее выходил узкий пучок волн в любой области спектра. (В монохроматор входят также линзы и зеркала, которые используются по своему прямому назначению.)
Полихроматоры устроены аналогичным образом, но имеют две или больше выходных щелей, что позволяет одновременно наблюдать несколько полос с разными длинами волн.
Спектрограф — это прибор, сходный с монохроматором, но без выходной щели. На месте щели устанавливают фотопленку
Введение в оптические методы 33
или фотопластинку, чтобы лучи с последовательными длинами волн фокусировались на соседних участках.
Спектрофотометр — это прибор, состоящий из монохроматора, источника излучения и фотоэлектрического детектора. Он служит стандартным прибором для регистрации спектров поглощения и флуоресценции.
Приборы разных типов, но состоящие, по существу, из одних и тех же узлов (исключение составляют источник и детектор), целесообразно рассмотреть вместе.
Разложение излучения призмами
В качестве диспергирующих элементов призмы используются обычно в интервале УФ-область — средняя ИК-область. В принципе призму можно изготовить из любого прозрачного материала с достаточно высокой диспергирующей способностью. Из твердых материалов в УФ-области пригодны только диоксиды кремния и алюминия *. Диоксид кремния используют в виде кварца или в стекловидной форме, иногда называемой «плавленым кварцем». Диоксид алюминия (в виде искусственного сапфира) дороже диоксида кремния и не имеет преимуществ перед кварцем. Интервал пропускания этих материалов простирается от почти 200 нм в УФ-области до почти 4 мкм в ИК-области.
Диспергирующая способность кварца в видимой области ухудшается, поэтому его заменяют оптическим стеклом. Призмы для ИК-области можно изготовить из солей типа NaCl, КВг или CsBr, но их нужно изолировать от атмосферной влаги. Эти и другие прозрачные в ИК-области материалы будут рассматриваться в гл. 4.
Простейший призменный монохроматор состоит из правильной призмы и двух линз (рис. 2-10). Излучение входит через щель Sb собирается в параллельный пучок коллиматорной линзой и падает под косым углом на грань призмы. После разложения излучение фокусируется второй линзой таким образом, чтобы в центр выходной щели S2 попадало излучение нужной длины волны (в форме входной щели).
Этот прибор можно сделать более компактным и экономичным, направив разложенный свет в обратную сторону; в результате получится конструкция Литтрова, показанная на
* Некоторые ионные кристаллы, например NaCl и MgF2, прозрачны для УФ- и видимого излучения, но редко используются в этих областях. Интенсивное УФ-излучение вызывает образование в них окрашенных центров, что снижает прозрачность.
2 Заказ № 537
34
Глава 2
Рис. 2-10. Ход лучей в спектрографе или монохроматоре с правильной призмой (с углами 60°). В случае спектрографа держатель фотопластинки или фотопленки помещают в фокальной плоскости (от Р] до Pi). В монохроматоре имеется выходная щель Si.
Рис. 2-11. Схема монохроматора с призмой Литтрова: а — с полевой линзой и выходной щелью; поток излучения направляется иа нее при помощи зеркала М; б — с вогнутым зеркалом на месте линзы. Диспергирующая призма с углом 30° с зеркальной задней гранью укреплена на столике, который можно вращать для устаиовлеиия длины волны.
рис. 2-11. Правильную призму заменяют призмой с углом 30° (с той же диспергирующей способностью) и зеркальной задней гранью. Теперь одна и та же линза может служить коллиматором для входящего излучения и фокусирующим элементом для разложенного потока.
Во многих случаях желательно заменить линзу сферическим зеркалом (рис. 2-11, б). Достоинство такой схемы заключается в том, что зеркало одинаково пригодно для любого излучения,
Введение в оптические методы 35
способного отражаться от металлической поверхности (т. е. от УФ- до ИК-области). Дело в том, что в этом случае излучение не проходит через поглощающий материал, который может ограничить интервал длин волн; кроме того, зеркало фокусирует излучение всех длин волн в одну точку, а линза не обладает такой способностью.
В особых случаях используются и другие типы призм.
Разложение излучения дифракционными решетками
В современных приборах для разложения излучения чаще всего используется дифракционная решетка. При прохождении потока монохроматического излучения через прозрачную пластинку, на которую нанесено большое число очень тонких параллельных штрихов, наблюдается его расщепление на множество пучков. Один из них проходит прямо, как будто пластинка не разлинована, другие же отклоняются (как это показано на рис. 2-9, б) под разными углами, величина которых зависит от расстояния между штрихами и длины волны излучения. Это явление можно объяснить, предположив, что каждая щель между штрихами при освещении сзади становится как бы источником излучения, распространяющегося во всех направлениях. Однако лучи, исходящие от этих вторичных источников, в большинстве направлений взаимно уничтожаются вследствие интерференции. Только те пучки, которые проходят под одинаковыми геометрически правильными углами, усиливаются. На рис. 2-12 изо
бражен один из отклонившихся пучков: угол отклонения равен 0, разность хода лучей от соседних щелей равна а, расстояние между центрами соседних штрихов (период решетки) составляет d. Как следует из рисунка, C! = d'sin0. Отсюда вытекает фундаментальное соотношение, называемое уравнением дифракционной решетки:
mk—dsinQ (2-6)
где m — любое целое число 0, 1, 2, 3, ..., называемое порядком *>; X—' длина волны.
Рис. 2-12. Дифракция в плоской решетке на пропускание.
*Обычно порядок обозначается буквой п, но в этой книге использована буква m во избежание путаницы с показателем преломления.
2*
36 Глава 2
Из уравнения (2-6) следует, что при прохождении через решетку поток полихроматического излучения разбивается на серию спектров, расположенных симметрично относительно нормали. Спектры, лежащие по обе стороны от нормали, соответствуют каждому из нескольких первых значений т. Из уравнения также вытекает, что для данного угла 0 должно существовать несколько длин волн с одинаковым значением т%. Например, решетка, имеющая 2000 штрихов на сантиметр (период решетки 1/2000 = 5 • 10-4 см), будет отклонять (под углом
0 = 6,89°) излучение с длинами волн, вычисляемыми по уравнению
, dsinO (5-Ю-4) (sin 6,89°) ' (5-10-4) (0,1200)
Л = —-----=-----------------== --------------==
т пг tn
Ниже приведены длины волн, соответствующие последовательным порядкам при этом угле:
Порядок т 12 3 4
Длина волны X, нм 600 300 200 150
Это соотношение представлено схематически на рис. 2-13 (даны длины волн по одну сторону нормали).
Рис. 2-13, Наложение спектров разных порядков при разложении света в решетке на пропускание.
Введение в оптические методы 37
Уравнение (2-6) применимо и к более общему случаю. Если излучение падает на поверхность решетки под некоторым углом ф, то уравнение решетки выглядит так:
tnX = d (sin 0 + sin ф) (2-7)
Перекрывание спектров последовательных порядков кажется на первый взгляд существенным недостатком, однако на практике это не имеет большого значения. Если изучается видимая область спектра, то вопрос вообще не возникает, поскольку в этой области (400—750 нм) разные порядки не перекрываются. Если спектр фотографируют, то степень перекрывания хотя бы частично ограничивается чувствительностью фотопластинки или фотопленки. Перекрывание можно уменьшить или устранить, установив перед решеткой вспомогательную призму с малой дисперсией (называемую призмой для отбора спектра нужного порядка) или используя светофильтры, поглощающие одну область спектра и пропускающие другую.
Описанная выше решетка относится к типу плоской дифракционной решетки на пропускание.
На практике чаще применяют отражательные дифракционные решетки, небольшие по размеру и удобные в обращении. Штрихи наносят на зеркальную поверхность, представляющую собой полированную металлическую пластинку или покрытую пленкой металла стеклянную пластинку.
Отражательную решетку можно разлиновать так, чтобы максимум излучательной энергии приходился на те длины волн, которые дифрагируют при выбранном угле. Это достигается при помощи специального алмаза, укрепляемого под заданным углом. Такая решетка называется эшелеттом; с ее помощью при правильно выбранном угле излучение концентрируется в первом порядке.
На рис. 2-14 представлена часть отражательной решетки типа эшелетта и схематически показан ход лучей. Обозначим угол между более широкой гранью бороздки и плоскостью решетки через ф. Луч, падающий под углом а, отражается от грани бороздки под углом (3; очевидно, что а + ф = |3—ф. Лучи, отражающиеся от соседних бороздок, интерферируют, как описано выше. Благодаря сильному отражению зеркальной металлической поверхностью большая часть энергии при данном значении а будет дифрагировать при угле |3. При углах, близких по величине к (3, энергия будет лишь немного ниже, поэтому решетку можно' успешно использовать в большом интервале длин волн данного порядка. Решетка, дающая максимум дифракции при определенной длине волны в спектре первого порядка, дает вдвое менее интенсивный максимум при той же длине волны в спектре второго порядка, втрое менее интенсив-
38
Глава 2
Рис. 2-14. Ход лучей в концентрирующей отражательной решетке (эшелетте).
ный максимум в спектре третьего порядка и т. д. В направлениях по другую сторону от нормали излучение практически не отражается.
В целом наилучшие результаты получаются в том случае, если период решетки по порядку величины совпадает с длинами волн диспергируемой области. В некоторых случаях более выгодными оказываются другие соотношения между периодом решетки и длиной волны. Например, штрихи в решетке типа эше-летта в сотни раз шире средней длины волны в изучаемой области. Они обладают очень высокой дисперсионной способностью, но требуют использования спектров сотого или более высоких порядков, что создает трудности, связанные с наложением спектров других порядков.
Изготовление хорошей дифракционной решетки — очень скрупулезная работа. Ее выполняют при помощи точного и чувствительного прибора, называемого делительной машиной, которая прочерчивает точечным алмазом тонкие параллельные линии. В спектрографах и спектрофотометрах обычно используют реплики. Их изготавливают, заливая оригинальную решетку пластичным материалом; после затвердевания отливку снимают и укрепляют ее на твердой основе. Искусство изготовления реплик достигло такого совершенства, что эти решетки почти не отличаются от оригинальных.
Введение в оптические методы 39
Рис. 2-15. Положение плоской отражательной решетки в схеме Эберта. Длину волны устанавливают поворотом решетки вокруг вертикальной оси, проходящей через ее центр.
Решетки высшего качества изготавливают при помощи лазера способом голографии. Стеклянную пластинку покрывают фотоэмульсией и освещают одновременно двумя потоками от одного и того же лазера, в результате чего на эмульсии получается интерференционная картина из параллельных полос. После проявления на пластинке появляется ряд параллельных линий, которые составляют прекрасную дифракционную решетку.
Существует несколько способов размещения плоской отражательной решетки в монохроматоре или спектрографе. Один из них — способ Литтрова, о котором уже упоминалось выше. Схематически его можно изобразить так же, как на рис. 2-11, а и б, только вместо призмы на вращающейся подставке укреплена дифракционная решетка (см. рис. 4-5).
Другой удобный способ установки плоской дифракционной решетки был предложен Эбертом в 1889 г., но он почти не использовался, пока в 1952 г. не был пересмотрен и усовершенствован Фастье [6]. Установка (рис. 2-15) состоит из одного большого сферического зеркала, которое служит одновременно для коллимирования и фокусировки, и двух симметрично расположенных щелей. Настройка на нужную длину волны производится вращением решетки. Черни и Тернер [7] предложили заменить дорогое большое зеркало Эберта двумя небольшими симметрично расположенными сферическими зеркалами; большинство выпускаемых приборов сочетают лучшие черты конструкций Черни—Тернера и Эберта. На рис. 2-16 представлены две выпускаемые промышленностью модели.
Модель на основе схемы Литтрова несколько компактнее модели Эберта. В этом приборе установлено всего одно зеркало,
40 ' Глава 2
Коллимирующие
а
Рис. 2-16. Монохроматор Черни — Тернера, а — с углом 90° между входным и выходным потоками излучения; б — двойной монохроматор с промежуточной щелью Sz.
Введение в оптические методы 41
Рис. 2-17. Схема спектрографа с вогнутой дифракционной решеткой, иллюстрирующая правило круга Роуланда.
но обе щели находятся на близком расстоянии друг от друга, что ограничивает его возможности. Благодаря симметричности схемы Эберта излучение здесь меньше подвержено аберрации.
В другом классе приборов используется отражательная решетка, представляющая собой вогнутую сферическую поверхность, на которую нанесены штрихи. В 1882 г. Роуланд сформулировал следующий принцип конструирования таких приборов (носящий его имя): если центр вогнутой дифракционной решетки и входная щель лежат на одной
окружности (круг Роуланда), причем ее диаметр равен радиусу кривизны решетки, то дифрагируемое излучение (в форме входной щели) будет лежать на той же окружности. Этот принцип справедлив для всех длин волн всех порядков дифракции; он иллюстрируется схемой на рис. 2-17. На основе этого принципа разработано несколько механических устройств, используемых как в очень больших спектрографах с высокой разрешающей способностью, так и в малых портативных спектрофотометрах.
Недостатком приборов, в которых используются вогнутые решетки, является астигматизм. Он связан с тем, что изображение входной щели фокусируется в пределах круга очень резко, что позволяет точно измерять длину волны, но высота ее не столь определенна. Это нужно иметь в виду при количественных измерениях, поскольку часть излучения маскируется верхней и нижней частью выходной щели.
Дисперсия
Дисперсию спектрографа определяют как производную d'k/dx, где х— расстояние вдоль фокальной плоскости, например вдоль поверхности проявляемой фотопластинки. Ее называют, обратной линейной дисперсией и выражают в нанометрах на миллиметр. Соответствующая величина для монохроматоров или спектрофотометров называется эффективной полосой пропускания. Это—интервал длин волн, пропускаемых при данной ширине выходной щели. Полоса пропускания равна произведению ширины щели и обратной линейной дисперсии.
42 Глава 2
Длина волны
Рис. 2-18. Полоса длин волн излучения широкополосного источника, выходящего из выходной щели монохроматора.
Входная и выходная щели большинства монохроматоров имеют одинаковую ширину, если же они изменяются, то с помощью простого контролирующего устройства их уравнивают. Полоса излучения, выходящая из щели, включает длины волн по обе стороны от центральной или номинальной длины волны (рис. 2-18). Полоса ДА между точками, находящимися на половине ее высоты, называется эффективной полосой пропускания системы.
На приборах с дифракционными решетками получается нормальный спектр, т. е. излучение диспергируется по длинам волн линейно. Дисперсия и полоса пропускания по существу одинаковы для всего спектра. Призма, наоборот, дает неравномерный спектр: более длинные волны группируются на меньшем расстоянии, чем короткие. Полоса пропускания в призменных приборах изменяется в зависимости от длины волны и типа прибора.
Разрешающая способность
При прохождении излучения через очень узкую щель наблюдается возникновение интерференционной картины (рис. 2-19), состоящей из большого- максимума в центре и ряда менее интенсивных (обычно не принимаемых во внимание) максимумов, симметрично расположенных по обе стороны от основного максимума. Ширина центрального максимума (расстояние между соседними минимумами) равно 2fk/d, где d—ширина щели, f — фокусное расстояние линзы или зеркала, А— длина волны. При широко открытой входной щели пятно света в фокальной плоскости получается размытым и малоинтенсивным, а по мере приближения ширины щели к минимальной пятно сужается; если щель расширять, интенсивность возрастает, поскольку через нее будет проходить больше энергии.
Введение в оптические методы 43
Рис. 2-19. Дифракционная картина от отдельной щели.
Минимальная ширина щели обеспечивает наилучшее разрешение прибора:
с?опт = 2/Ш (2-8)
где w — диаметр линзы или зеркала. В типичном случае, когда сс = 8 cm, f = 40 см, А = 500 нм, оптимальная ширина равна 5мкм.
При оптимальной ширине щели прибор имеет максимальную разрешающую силу (способность к разделению двух соседних длин волн). Этот параметр оценивается критерием, предложенным много лет назад Рэлеем: две длины волны, отличающиеся на ДА, считаются разрешенными, если центральный максимум дифракции одной из них совпадает с первым минимумом второй. В таком случае разрешающая сила равна
Я = А/ДА (2-9)
где А — среднее из двух длин волн. Для дифракционной решетки разрешающая сила выражается произведением Nm, где N — число освещенных штрихов решетки, т — порядок спектра. Для призменных приборов соответствующая величина выражается формулой
Д = &(г1/гШ) (2-10)
где b — ширина основания призмы, п—-показатель преломления.
Если источник дает свет малой интенсивности, то, чтобы получить энергию, достаточную для точного измерения, приходится пользоваться монохроматором с более широкой щелью, чем было рассчитано по уравнениям (2-9) или (2-10); разрешение при этом, конечно, ухудшается.
44 Глава 2
Задачи
2-1. Определите волновое число в обратных сантиметрах и энергию в электронвольтах для каждого из указанных на рис. 2-1 переходов.
2-2. Метр, по официальному определению, принятому в США [8], равен 1 650 763,73 длины волны перехода 2pl0^5d5 чистого изотопа 86Кг в вакууме Рассчитайте для этого излучения волновое число в обратных сантиметрах, длину волны в ангстремах, нанометрах н микрометрах, частоту в герцах и энергию на фотон в электронвольтах. Примите скорость распространения света в вакууме равной (2,99792458± 1,2) • 108 м/с н постоянную Планка равной (6,626176+36) • 10-34 Дж • с, где указанные погрешности представляют собой стандартные отклонения в последних цифрах. Ответ дайте с соответствующей погрешностью. (Если у вас нет калькулятора с достаточным числом цифр, округлите числа н укажите, насколько это снижает достоверность расчетов.)
2-3. Дополните табл. 2-1 еще двумя колонками, представив энергию фотонов в соответствующих областях а) в электронвольтах, б) в калориях на моль.
2-4. Пусть лабораторный источник нзлучення приближается к абсолютно черному телу. Его активная площадь равна 1 см2, а рабочая температура 5500 К. Рассчитайте а) длину волны с максимальной энергией, б) мощность излучения прн этой длине волны.
Литература
1. Jenkins F. A.. White Н. Е., Fundamentals of Optics. McGraw-Hill, New York, 1962.
2. Penner S. S., Quantitative Molecular Spectroscopy and Gas Emissivities. Addison-Wesley, Rading, Mass., 1959; shap. 3.
3. Lengyel B. A., Introduction to Laser Physics. Wiley, New York, 1966. 4. Webb J. P., Anal. Chem., 1972, v. 44, No 6, p. 31A.
5. Green R. B., J. Chem. Educ., 1977, v. 54, p. A365, A407.
6. Fastie W. G., J. Opt. Soc. Am., 1952, v. 42, p. 641.
7. Czerny M„ Turner A. F„ Z. Phys., 1930, v. 61, p. 792.
8 Natl. Bur. Stand. Tech. News Bull., 1963, February and October.
ГЛАВА
Поглощение излучения.
Ультрафиолетовая и видимая области
Если поток белого света пропускать через сосуд (кювету), заполненный жидкостью, то интенсивность выходящего излучения всегда будет меньше интенсивности входящего. Степень уменьшения интенсивности, как правило, зависит от длины волны. Ослабление интенсивности объясняется частично отражением от граней и рассеянием взвешенными в жидкости частицами, но в отсутствие таких частиц в основном поглощением излучения жидкостью.
Если при одних длинах волн в видимой области поглощается больше энергии, чем при других, то выходящий поток оказывается окрашенным. В табл. 3-1 приведены длины волн полос испускания, их цвета и дополнительные цвета. Интервалы длин волн взяты из работ, опубликованых Национальным бюро стандартов [1]; границы их, конечно, в какой-то степени условны. Наблюдаемый цвет раствора является дополнитель-
Таблица 3-1. Цвета видимого излучения [1]
Примерный интервал длин волн, нм Цвет Дополнительный цвет
400—465 Фиолетовый Желто-зеленый
465—482 Голубой Желтый
482—487 Зеленовато-голубой Оранжевый
487—493 Сине-зеленый Оранжево-красный
493—498 Г олубовато-зеленый Красный
498—530 Зеленый Пурпурно-красный
530—559 Желтовато-зеленый Пурпурно-красноватый
559—571 Желто-зеленый Пурпурный
571—576 Зеленовато-желтый Фиолетовый
576—580 Желтый Голубой
580—587 Желтовато-оранжевый Голубой
587—597 Оранжевый Зеленовато-голубой
597—617 Красновато-оранжевый Снне-зел 'ный
617—780 Красный Сине-зеленый
46 Глава 3
ним к поглощенному. Например, раствор, поглощающий в голубой области, кажется желтым, а в зеленой — пурпурным и т. д.
Говоря о цвете, мы имеем в виду видимую область спектра, но большинство рассматриваемых здесь положений приложимо к ультрафиолетовой и инфракрасной областям; то же справедливо и для аналитических методов.
Для химика-аналитика изучение окрашенных растворов очень важно, потому что параметры поглощенного излучения характеризуют поглощающее вещество. Раствор, содержащий аквоионы меди (II), поглощает желтый свет и пропускает синий, поэтому медь можно определить, измерив степень поглощения желтого света при заданных условиях. Таким способом можно количественно определить любое растворимое окрашенное вещество. Кроме того, бесцветное или слабоокрашенное вещество часто можно определить, добавив реагент, который образует с ним интенсивно окрашенное соединение. Например, при добавлении к раствору меди аммиака возникает более интенсивная окраска, чем у аквоиона, что позволяет провести аналитическое определение с большей чувствительностью.
Методы химического анализа, основанные на измерении поглощения излучения, называются абсорбциометрией. Для видимой области применяется термин колориметрия. Раздел абсорбциометрии, в котором измерение проводят на спектрофотометрах, называется спектрофотометрией.
Количественные закономерности
Количественно поглощение излучательной энергии можно описать на основе принципа, получившего название закона Бера. Рассмотрим, что происходит с монохроматическим излучением при его прохождении через стеклянную кювету с плоскими па* раллельными гранями. Пренебрежем отражением на гранях и поглощением стекла. Пусть кювета заполнена поглощающим веществом в непоглощающем растворителе. Мощность излучения уменьшается тем сильнее, чем глубже оно проникает в раствор и чем выше концентрация растворенного вещества. Другими словами, уменьшение мощности пропорционально числу поглощающих молекул на пути потока. Математически это положение выражается законом Бера*: бесконечно малые приращения числа одинаково поглощающих молекул вызывают поглощение одинаковых долей монохроматического излучения, проходящего через раствор.
* Это соотношение называют иногда законом Бера—Ламберта или Бера—Бугера, поскольку Ламберт и Бугер (а также другие ученые) внесли свой вклад в его развитие. Часто при формулировке закона вместо термина «мощность» (Р) пользуются термином «интенсивность» (/).
Поглощение излучения. УФ- и видимая области 47
Рис. 3-1. Иллюстрация к выводу закона Бера.
Число поглощающих молекул в элементарном объеме s2dx (рис. 3-1) равно Ncs^dx, где с — концентрация в молях на литр, а Л/ — число Авогадро. Отсюда закон Бера можно выразить следующим образом:
dP/Ncs2dx = — kP (3-1)
где k — константа, представляющая собой долю мощности потока Р, которая поглощается на пути длиной dx. Число Авогадро можно вынести в постоянный множитель. Для единичного сечения можно записать
dP/P = — kcdx (3-2)
Проинтегрируем это выражение по длине пути Ь: рь ь
\ (dP/P)=r-kc\dx (3-3)
о о
ln(P&/P0) = — kcb (3-4)
Перейдем для удобства к десятичному логарифму, заменим k новой постоянной а, содержащей модуль перехода от натурального логарифма к десятичному, и опустим индекс Ь:
lg (PQ/P) = abc = A (3-5)
Обратите внимание, что в отношении Pq/P числитель и знаменатель переставлены для изменения знака. Величина 1g(Ро/Р), имеющая важное значение и обозначаемая специальным символом А, называется оптической плотностью. Таким образом, самое краткое выражение закона Бера имеет вид A = abc.
Поскольку мощность выходящего потока Р может изменяться от 0 до Ро, логарифм отношения теоретически может меняться от 0 до бесконечности. Однако на практике значения оптической плотности больше 2 или 3 используются редко (из-за рассеяния излучения). Диапазон, в котором обеспе
48
Глава 3
чивается необходимая при аналитических измерениях точность, еще более ограничен; границы допустимых значений определяются в какой-то степени типом используемого измерительного прибора.
Коэффициент поглощения
Константа а в уравнении (3-5) называется коэффициентом поглощения. Он является характеристикой системы, состоящей из растворенного вещества и растворителя, при данной длине волны. Единицы измерения, а также символ, используемый для его обозначения, зависят от выбора единиц измерения b и с (Ь обычно выражают в сантиметрах) (табл. 3-2). Для справки в таблице приведены и другие символы и термины, которые широко применялись в прошлом.
Следует особо подчеркнуть, что коэффициент поглощения является характеристикой вещества (фактор интенсивности), тогда как оптическая плотность—характеристикой отдельной пробы (фактор экстенсивности) и поэтому при прохождении света через кювету оптическая плотность изменяется в зависимости от концентрации и длины пути.
Таблица 3-2. Единицы и символы, рекомендуемые при пользовании законом Бера
Принятый символ Опреде-а) ление ’ Принятое наименование Устаревшие илн альтернативные
символ наименование
т Р/Ро Пропускание (Transmittance) • • • • Прозрачность (Transmission)
А 1g рор Оптическая плотность (Absorbance) D, Е Экстинкция (Optical density, extinction)
а А'Ьс Коэффициент поглощения (Absorptivity) k Коэффициент экстинкции, индекс поглощения (Extinction coefficient, absorbancy index)
е АМ/Ьс Молярный коэффициент поглощения (Molar absorptivity) ам Молярный (молекулярный) коэффициент экстинкции (Molar extinction coefficient, molar absorbancy index)
ь Толщина слоя 1, d
а) Определение Р и PQ дано в тексте. Единицами измерения служат: для с грамм/литр, для b — сантиметр, для М — молекулярная масса. Раньше широко использовался символ который можно определить как А/Ьс', где с'—концентрация
в массовых процентах, a b— I см.
Поглощение излучения. УФ- и видимая области 49
Если прошедшее через раствор излучение представляет больший интерес, чем химическая природа поглощающего вещества, удобнее пользоваться величиной, называемой пропусканием Т, которую обычно выражают в процентах (процент пропускания определяется как 1ООР/Ро)- В процентах пропускания обычно оценивается качество светофильтров, используемых в колориметрии и фотографии. Реально на большинстве спектрофотометров измеряются величины Р и Ро, а оптическую плотность вычисляют по их значениям.
И оптическая плотность А, и коэффициент поглощения а служат для оценки степени поглощения излучения. Коэффициент поглощения а, имеющий размерность л/(г-см), целесообразно использовать в тех случаях, когда природа поглощающего вещества, а следовательно, и его молекулярная масса неизвестны. Если же нужно сравнить поглощение различных веществ с известными молекулярными массами, то следует предпочесть молярный коэффициент поглощения е [с размерностью л/(моль-см)].
Согласно закону Бера, коэффициент поглощения является постоянной величиной, не зависящей от концентрации, длины пути и интенсивности падающего излучения. Закон Бера ничего не говорит о влиянии температуры, природы растворителя или длины волны. Установлено, что на практике температура имеет второстепенное значение, если только она не меняется в очень широких пределах. С изменением температуры немного изменяется концентрация (вследствие изменения объема). Кроме того, если поглощающее вещество находится в растворе в равновесии с другими веществами, следует ожидать влияния температуры в той или иной степени. В то же время некоторые вещества при охлаждении до температуры жидкого азота проявляют совершенно другие абсорбционные свойства. На практике при аналитических измерениях влиянием температуры в большинстве случаев можно пренебречь, особенно если сравнение поглощения неизвестного и стандартного образцов проводится при одной и той же температуре.
Вообще говоря, невозможно предсказать, как повлияет на молярный коэффициент поглощения данного растворенного вещества замена одного растворителя на другой. Аналитик часто ограничен каким-то определенным растворителем или классом растворителей, в которых растворимо данное вещество, так что вопрос о влиянии растворителя может и не возникнуть. При работе в УФ-области, где многие обычно используемые растворители становятся непрозрачными, возникают свои трудности. Вода, спирт, эфир и насыщенные углеводороды пригодны для работы в этой области, но ароматические соединения, хлороформ, тетрахлорид углерода, сероуглерод, ацетон и многие
50
Глава 3
Таблица 3-3. Границы пропускания наиболее употребительных растворителей в ультрафиолетовой области3)
180—195 нм
Серная кислота (96 % -ная) Вода Ацетонитрил 200—210 нм
Циклопентан н-Гексан Глицерин 2,2,4-Триметилпен-тан
Метанол Этанол
210—220 нм н-Бутанол Изопентанол Циклогексан Диэтиловый эфир 245—260 нм
Хлороформ Этилацетат Метилформиат
165—275 нм
Тетрахлорид углерода
Диметилсульфоксид
Диметилформамид
Уксусная кислота 280—290 нм
Бензол
Толуол
л-Ксилол 300 нм
Пиридин
Ацетон
Дисульфид углерода
а) Границами пропускания условно считаются точки, где А = 0,50 при 6 = 10 мм; в пределах каждой группы растворители расположены в порядке возрастания граничного значения длины волны. Данные предоставлены фирмой Matheson Coleman & Bell» Cincinnati, Ohio.
другие растворители использовать нельзя из-за их слишком сильного поглощения (разве что в ближней УФ-области). В табл. 3-3 приведены примерные границы пропускания в УФ-области некоторых распространенных растворителей.
Иногда оказывается, что
Рис. 3-2. Кривые зависимости оптической плотности от концентрации при подчинении закону Бера (1); при положительном отклонении (2); при отрицательном отклонении (3).
даже при постоянной температуре и в определенном растворителе коэффициент поглощения не является, строго говоря, константой. Если построить график зависимости оптической плотности А от концентрации, то, согласно закону Бера, должна получиться прямая, проходящая через начало координат (кривая 1 на рис. 3-2). Однако для некоторых систем наблюдается искривление прямой. Как будет показано в следующем разделе, такие отклонения могут быть скорее кажущимися, чем действительными.
Поглощение излучения. УФ-и видимая области 51
Отклонения называют положительными или отрицательными в зависимости от того, вверх или соответственно вниз искривлена прямая.
Следует иметь в виду, что подчинение закону Бера не является необходимым условием применения поглощающей системы для количественного определения. Любая из кривых рис. 3-2, построенная для какого-либо вещества в определенных условиях, может служить градуировочным графиком. По этой кривой можно найти неизвестную концентрацию вещества, если экспериментально определена оптическая плотность его раствора.
Кажущиеся отклонения от закона Бера
В целом излучение любой данной длины волны должно подчиняться закону Бера, но при изменении длины волны коэффициент поглощения и оптическая плотность будут изменяться. Ширина используемой полосы длин волн, может повлиять на кажущееся значение коэффициента поглощения. На рис. 3-3 представлен спектр поглощения перманганат-иона в водном растворе. Как видно из табл. 3-1, вещество, поглощающее в обла-
Рис. 3-3. Спектр поглощения водного раствора перманганата калия
52 Глава 3
сти 480—570 нм, должно казаться пурпурно-красным, и это, как мы знаем, действительно так. Если измерить поглощение этим раствором излучения, пропущенного через зеленый светофильтр (с границами пропускания, обозначенными А и F), то пики и впадины на кривой поглощения будут сглажены, а значение молярного коэффициента поглощения, рассчитанное при этих условиях, должно составлять примерно 1700—1800. Однако если каким-то образом ограничиться интервалом длин волн между В и Е, то рассчитанное значение, которое, грубо говоря, является средним из истинных значений в этой области, будет лежать около 2300. При сужении полосы до интервала между С и D молярный коэффициент поглощения приблизится к своему истинному максимальному значению при этой длине волны, равному 2500.
То, что при немонохроматическом излучении значения коэффициента поглощения отличаются от истинного, ясно из следующих рассуждений. Пусть какой-либо раствор исследуют при двух длинах волн, Xi .и Хг, и при каждой длине волны он строго подчиняется закону Бера. Оптическая плотность Xi выражается формулой
Ai = lg(P0,i/Pi) = aibc (3-6)
или
P{JP^\Qrbc (3-7)
Аналогичные уравнения можно написать для Л2 при Х2, подставляя индекс 2 вместо 1.
Измерив значения Ро и Р при обеих длинах волн, мы получим кажущуюся оптическую плотность Акаж
Акаж = — lg f Р1+Р2 (3-8)
V. Ро.1 + Ро.2 ) '
Используя уравнение (3-7), получаем
- 1g (Рол + Ро,2)- 1g (Род 10-Я16с+ Po,210~a^) (3-9)
Очевидно, если ах = а2, уравнение превращается в закон Бера, но если они не равны, зависимость между Акаж и с не может быть линейной, и наблюдается отклонение в ту или иную сторону в зависимости от соотношения между щ и а2.
Интересно сравнить монохроматическое излучение, полученное с помощью лазера и монохроматора с узкой щелью. В работе [2] сообщается об измерении оптической плотности краси
Поглощение излучения. УФ- и видимая области 53
теля с четким максимумом при 635 нм прямым методом относительно воды и дифференциальным методом относительно стандартного раствора. Чтобы отношение сигнал/шум было достаточно большим, ширина щели должна быть равна 1,48 мм при использовании лампы накаливания и 0,08 мм при использовании лазера. Кривые зависимости оптической плотности от концентрации, полученные с помощью лазера, оказались линейными во всем интервале измеряемых значений (вплоть до 1,4 единицы оптической плотности), а на кривых, полученных с применением лампы накаливания, наблюдается отрицательное отклонение. Частично оно связано с увеличением ширины полосы пропускания, частично с влиянием рассеянного излучения.
Иногда вид кривой поглощения может меняться при изменении концентрации раствора, что вызывает кажущееся неподчинение закону Бера. Это явление можно объяснить взаимодействием молекул друг с другом или с растворителем.
Ярким примером служит поведение метиленового синего [3]. Его разбавленные растворы обладают интенсивным поглощением при 664 нм, но при увеличении концентрации в 200 раз при этой длине волны остается только плечо, а максимум поглощения наблюдается при 600 нм. Это можно приписать образованию при более высоких концентрациях димеров. На графике зависимости оптической плотности от концентрации следует ожидать отрицательного отклонения от закона Бера при 664 нм и положительного при 600 нм.
Примером кажущегося отклонения, вызванного химическими процессами, служит изменение окраски раствора К2СГ2О7 из оранжевой в желтую при разбавлении водой. Спектры поглощения К2СГ2О7 и КгСгО4 [одинаковой концентрации по Cr (VI)] приведены на рис. 3-4. Соответствующее уравнение реакции можно написать в виде
Сг2О?~ + Н2О ^±2Н+ + 2СгОГ оранжевый желтый
При разбавлении раствора бихромата оптическая плотность постепенно меняется от значений на кривой 2 др значений на кривой 1 (с учетом формы кривых; внешне это выражается в уменьшении действительных значений оптической плотности).
Все «отклонения» от закона Бера, которые рассматривались до сих пор, следует отнести к кажущимся. Они вызваны нарушением условий, для которых сформулирован закон, и не возникают, если учесть действительные, а не рассчитанные по стехиометрии концентрации.
Причина отклонения более фундаментального характера связана с показателем преломления п исследуемого раствора. В самом деле, по теории рассеяния, чтобы получить величину, дей-
54
Глава 3
Длим волны, нм
Рис. 3-4. Спектры поглощения КаСгО4 в 0,05 М КОН (1) и К2СГ2О7 в 1,75 М H2SO4 (2). Концентрация хрома в обоих случаях 0,01071 мг/мл; толщяна слоя 10,0 мм. Для удобства оптическая плотность отложена в логарифмической шкале.
ствительно не зависящую от концентрации, следует умножить коэффициенты поглощения на п/(/г2 + 2)2 [4]. Однако на практической работе аналитика это ограничение не сказывается, поскольку в большинстве случаев измеряется поглощение разбавленных растворов, для которых показатели преломления раствора и растворителя по существу совпадают.
Приборы
Приборы для измерения селективного поглощения излучения растворами называются спектрофотометрами. Простые фотоэлектрические устройства, в которых для выделения длины волны применяется светофильтр, лучше всего рассматривать как упрощенный спектрофотометр, хотя иногда их называют старым неточным названием «колориметр». Для всех типов такого рода приборов изредка встречается более общее название «абсорбциометр».
Фотоэлектроколориметры. На рис. 3-5 представлена схема упрощенного спектрофотометра. Два световых потока от лампы
Поглощение излучения. УФ- и видимая области 55
накаливания пропускают через стеклянный светофильтр и направляют на два фотоэлемента, включенных в одну электрическую цепь, причем один поток проходит через пробу. В обоих фотоэлементах, являющихся фотоэлементами вентильного типа (более подробно описаны в гл. 27), возникают токи, пропорциональные интенсивности падающего на них излучения. Каждый фотоэлемент шунтирован низкоомным резистором, причем резистор фотоэлемента сравнения снабжен скользящим контактом. При работе контакт сначала устанавливают на верхний конец резистора, что соответствует на шкале отметке 100. В кювету помещают растворитель и с помощью подвижной щели устанавливают стрелку гальванометра на нуль, обеспечивая тем самым одинаковую освещенность обоих фотоэлементов. Затем растворитель заменяют анализируемым окрашенным раствором. Поскольку проба поглощает сильнее, чем растворитель, ток в рабочем фотоэлементе уменьшается, а это приводит к уменьшению напряжения на его резисторе. Теперь, чтобы уравнять напряжение от обоих фотоэлементов, нужно передвинуть скользящий контакт (на схеме вниз) и снова привести стрелку гальванометра- к нулю. Со шкалы потенциометра снимают показания в процентах пропускания. Было сконструировано много аналогичных приборов; максимальная точность их составляет 3-4%.
Другой тип упрощенного спектрофотометра — фотоанализатор Du Pont 400 (представлен на рис. 3-6). Он состоит из ряда смодулированных узлов, порядок соединения которых в разных моделях различен (два варианта изображены на рисунке). По схеме а излучение проходит через пробу, а затем расщепляется на два потока. Эти потоки пропускают через разные светофильтры, поэтому индикатор показывает отношение интенсивностей полос излучения при двух длинах волн. Измеряемый поток и поток сравнения выбирают таким образом, чтобы исследуемое вещество их поглощало или соответственно пропускало. По схеме б излучение, прежде чем разделиться на два потока, про-
свете- Фотоэлемент Рабочий
Лампа фильтр „ сравнения фотоэлемент
т Рабочий г---------- --------
фото- _ . элемент Фотоэлемент сравнения Отражатель Подвижная щель
Рис. 3.5. Схема фотоэлектроколорнметра модели Клетт — Саммерсоиа (Klett Manufacturing Company).
56
Глава 3
Полупссеребрснное Фотоэлемент
зеркало светофильтр сроВнения сравнения
Лампа
Проба
Рабочий светофильтр
Рабочий фотоэлемент
। j Логарифмиче-
11 11 ский усилитель
Кизмеритель-
—> ному прибору или самописцу Дифференциаль -
.чая схема.
Кюбета
Рис. 3-6. Два варианта схемы фотоанализатора модели Дюпон 400 (Е. I. Du Pont de Nemours Co).
ходит через один светофильтр. Один поток направляют на раствор сравнения, другой — на анализируемую пробу, так что со шкалы считывается отношение поглощений частицами в исследуемом и стандартном растворах.
Фотометр фирмы Du Pont (модель 400) предназначен в основном для непрерывного контроля в потоках как жидких, так и газообразных проб. В качестве примера можно привести он-
Поглощение излучения. УФ- и видимая области 57
Рис. 3-7. Спектры поглощения SO2 и NO2. Указаны длины волн (280, 436 и 578 нм), используемые для их определения (Е. I. Du Pont de Nemours Co).
ределение оксидов серы и азота в газообразных продуктах горения, которое проводится на приборе, схематически изображенном на рис. 3-6,а (с ртутной лампой). На рис. 3-7 представлены спектры SO2 и NO2 и указаны используемые длины волн ртути. Поток сравнения состоит только из желтого дублета при 578 нм, где анализируемые оксиды не поглощают. Измеряемый поток пропускают через светофильтры, выделяющие лишь серию линий вблизи 280 нм (при определении SO2) и голубую линию при 463 нм (при определении NO2). Предусмотрена возможность окисления NO в NO2 в потоке: измерения через определенное время течения потока при 436 нм дают информацию о содержании обоих оксидов.
Двухлучевые спектрофотометры для УФ- и видимой областей
Нижний предел рабочего интервала длин волн спектрофотометров в УФ- и видимой областях лежит обычно между 160 и 210 нм. Верхний предел находится не ниже чем при 650 нм, но может достигать 1000 нм и даже больше.
Наиболее удобны и общеупотребительны двухлучевые спектрофотометры с автоматической регистрацией. Двухлучевая схема предусматривает два равноценных пути прохождения излучения от одного и того же источника. Один проходит через исследуемую пробу, другой — через такую же кювету, содержащую вещество сравнения. Оба потока измеряют по отдельности или с помощью двух детекторов, или на одном детекторе с модулятором.
58 Глава 3
Рис. 3-8. Соотношение интенсивностей в двухлучевом спектрофотометре. Ро'— интенсивность источника (предполагается, что оиа одинакова для обеих кювет); PR интенсивность потока, прошедшего через кювету сравнения; Ps — интенсивность потока, прошедшего через пробу.
Каким образом закон Бера используется в двухлучевых приборах, будет ясно из следующих рассуждений. Рассмотрим интенсивность потоков, прошедших через одинаковые кюветы (рис. 3-8). Обозначим интенсивность потока от источника (один и тот же для обеих кювет) Р'о. Пусть Ps и Pr— интенсивности потоков, прошедших через растворы пробы и сравнения; величины их, конечно, меньше чем PQ'. Соответствующие оптические плотности по закону Бера выражаются так:
л5=ig (p'qips) и ля=18(р;/ря)
Объединив эти равенства, получаем
= ig (p;/ps)^ig (р;/ря)=ig (Рд/Ps) (з-ю) Если вещество сравнения идентично основе образца, правомочно заменить Рд и Ps на Ро и Р соответственно. После подстановки их в закон Бера получим
X = lg(P0/P) (3-11)
где А — действительное значение оптической плотности с поправкой на светопоглощение вещества сравнения. Заметим, что Ро уже не является интенсивностью падающего потока, скорее это интенсивность потока, ослабленного в результате всевозможных потерь, одинаковых для обеих кювет. Обратите внимание, что Ро' отсутствует в конечном уравнении; изменение (в известных пределах) интенсивности излучения лампы не влияет на измеряемую оптическую плотность. Показатели преломления раствора сравнения и анализируемого раствора, а также содержание примесей в них должны быть по возможности идентичными.
В спектрофотометрах с одним детектором предусмотрено устройство, позволяющее направлять потоки поочередно по двум эквивалентным путям. Это устройство часто изготавливают
Поглощение излучения. УФ- и видимая области 59
Рис. 3-9. Двухлучевая фотометрическая схема с прерывателем.
Рис. 3-10. Изменение сигнала детектора фотометра (рис. 3-9) во времени. Буквами R и S обозначены соответственно потоки от растворов сравнения и пробы.
в форме диска, обычно называемого прерывателем. Одна половина диска имеет зеркальную поверхность, а другая вырезана, как показано на рис. 3-9, а. Диск вращается при помощи электродвигателя с постоянной скоростью. При повороте диска излучение попеременно отражается от зеркальной поверхности и направляется на кювету сравнения (рис. 3-9, б) или проходит через диск и направляется на рабочую кювету. Сигнал, получаемый от детектора, имеет форму, изображенную на рис. 3-10. Высота сегментов, обозначенных буквой R, пропорциональна величине PR, а обозначенных буквой S — величине Ps-Нулевая линия между сегментами S и R соответствует непрозрачным частям диска. Нетрудно сконструировать устройство для разделения этих сигналов и вычисления исправленной оптической плотности. Существует много моделей двухлучевого прибора; две из них показаны на рис. 3-11.
Ограничение закона Бера при высоких концентрациях связано с влиянием рассеянного излучения [5]. Применительно к спектрофотометрам под ним подразумевают любое случайное излучение, попадающее в детектор. Частично оно обусловлено дифракцией более высоких порядков, частично связано с отражением света от поверхностей линз и призм. Его можно уменьшить при помощи непрозрачных экранов, но полностью устра-
60
Глава 3
Рис. 3-11. Два варианта схемы двухлучевого фотометра. В схеме а используются два синхронных прерывателя Dt и D2. В схеме б каретка с двумя небольшими зеркалами передвигается между положениями Afi, Л12 и М/ и М2' (эта схема применяется в спектрофотометрах Бекмана моделей DB и DBG).
нить нельзя. Больше всего влияние рассеянного излучения сказывается на границах областей длин волн монохроматора: чтобы обеспечить достаточную чувствительность, приходится широко открывать щели. Для большинства длин волн остаточное рассеянное излучение хорошего монохроматора не превышает нескольких десятых процента.
Рассеянное излучение можно резко уменьшить при помощи двойного монохроматора. Если один монохроматор пропускает 0,1 % рассеянного излучения, то, направляя поток через другой такой же монохроматор, можно снизить его до 0,1 % от этой величины, т. е. до одной миллионной доли от общего излучения.
В качестве примера рассмотрим схему спектрофотометра для УФ- и видимой областей с двойной монохроматизацией (Varian-Cary, модель 219). В этом приборе используется один монохроматор, но поток направляется на него дважды. На рис. 3-12 показана оптическая схема прибора. Излучение дейтериевой лампы (или для видимой области лампы накаливания, здесь не изображенной) фокусируется системой сферических зеркал AfI( М2 и М3 на входную щель Верхняя часть вогнутого зеркала М4 коллимирует поток, направляя его в плоскость дифракционной решетки. Разложенное излучение фокусируется при помощи нижней части зеркала 7И5 на щель S2. Выделенный узкий пучок проходит через щель и подается с помощью зеркал М6, М7 и М8 на щель S3. Его снова монохроматизируют, используя на этот раз нижнюю часть зеркала М4 и верхнюю часть зеркала М$, и, наконец, выпускают из монохроматора через выходную щель S4.
Далее поток попадает на полукруглое зеркало прерывателя и проходит попеременно то через зеркала М9—Mi7 и кювету сравнения, то через зеркала Alia—Afu и рабочую кювету, а затем попадает на фотоумножитель.
Поглощение излучения. УФ- и видимая области 61
Диск света -'фильтров
Рис. 3-12. Оптическая схема спектрофотометра Вариан — Кери (модель 219). Для устранения излучения других спектральных порядков по мере необходимости поворотом диска с набором светофильтров вводится нужный светофильтр. Щели S| и St расположены над щелями S3 и S2 соответственно. Все щели искривлены, поскольку при такой форме получается наилучшее разрешение для монохроматора Черин — Тернера. Схема несколько упрощена для ясности. (Приборный отдел ассоциации Вариан.)
62
Глава 3
Форма внешнего обода прерывателя позволяет дважды за один поворот пересекать поток между зеркалами и Л17; при этом частота смены потоков излучения от рабочей кюветы и кюветы сравнения удваивается. Электронный усилитель настроен на прием лишь тех сигналов, которые появляются при прерывании потока с удвоенной частотой. Поскольку рассеянное излучение, возникающее при первом прохождении через монохроматор, не подвергается высокочастотному модулированию, это равноценно использованию двух отдельных монохроматоров. Согласно заводским аттестациям, максимальное рассеянное излучение может составлять всего 0,002 %, а для отдельных приборов при длинах волн, больших 220 нм, эта характеристика по крайней мере на порядок меньше.
Однолучевые спектрофотометры
Первый спектрофотометр для УФ-области, разработанный фирмой Beckman (модель DU) [6], появился в США в 1941 г. Это был однолучевой прибор с призмой Литтрова из природного кварца. С его помощью был внесен существенный вклад в наши представления об УФ-области как в теоретическом, так и в практическом аспектах. На протяжении всех этих лет прибор модели DU постоянно совершенствовался, так что последняя из моделей DU-8 внешне мало напоминает своего предшественника. Это по-прежнему однолучевой спектрофотометр, но кварцевая призма заменена голографической решеткой. Выходное устройство снабжено микрокомпьютером, так что благодаря информации, заложенной в память компьютера, в оптическую плотность образца можно вносить поправку на поглощение холостой пробы и проводить сравнение со стандартом.
При проведении серийных анализов часто достаточно измерить оптическую плотность при одной длине волны. Для таких целей использовать дорогой двухлучевой прибор нецелесообразно; более простая однолучевая модель может оказаться вполне приемлемой.
Для работы в видимой области (от 340 до 625 нм, хотя возможно расширение области до 950 нм путем замены фотоэлемента и введения светофильтра) широко используется прибор фирмы Busch & Lomb модели Спектроник-20 (рис. 3-13). На вращающемся столике укреплена плоская отражательная дифракционная решетка (600 штрихов на 1 мм). Схема этого прибора напоминает схему прибора Литтрова, но пучок не проходит через линзы дважды. Первичное излучение собирается в пучок, а не идет параллельным потоком, что приводит к несколько большему уширению полосы излучения, попадающего на выходную щель (устанавливают 20 нм), чем это можно
Поглощение излучения. УФ- и видимая области 63
Рис. 3-13. Оптическая схема спектрофотометра Спектроиик-20 фирмы Bausch & Lomb. Дифракционная решетка связана с ручкой на панели при помощи кулачкового сцепления. Блок-фильтр вводится только при использовании фотоэлемента в инфракрасной области для устранения излучения других порядков.
было ожидать для данной дифракционной решетки; здесь налицо компромисс между точностью и стоимостью.
Детектором служит газонаполненный фотоумножитель в сочетании с линейным усилителем. Шкала на панели прокалибрована в единицах пропускания и оптической плотности. Предусмотрены три ручки управления: установки длины волны и нуля и регулятор чувствительности. Электронный стабилизатор напряжения устраняет любые помехи из сети, которые могут дать ложный сигнал.
Двухволновые спектрофотометры
Прибор можно сконструировать таким образом, чтобы через одну и ту же кювету одновременно проходили два потока излучения с разными длинами волн, и использовать его в нескольких вариантах:
1. Контролировать содержание двух компонентов, регистрируя изменение каждого потока во времени при фиксированных длинах волн.
2. Фиксировать одну длину волны (при которой оптическая плотность очень мала) и сканировать длины волн другого потока; тем самым вносится поправка на колебания интенсивности ламп, как в двухлучевом приборе, но с одной кюветой.
3. Тот же принцип, что и в п. 2, можно использовать для получения истинного спектра поглощения в мутных средах [7]; в этом случае из-за многократного отражения от взвешенных частиц длина пути потока неопределенна, но данный
64
Глава 3
для пробы
Рнс. 3-14. Оптическая схема (упрощенная) двухволнового спектрофотометра Перкин —Элмер модели 356 (Perkin — Elmer Corporation).
Поглощение излучения. УФ- и видимая области 65
принцип обеспечивает одинаковый путь для обоих потоков, каким бы он ни был.
4. Наконец, можно регистрировать спектры потоков одновременно, но со сдвигом в несколько десятков нанометров, так что на получающейся спектрограмме будет отражена зависимость разности АЛ между оптическими плотностями при двух длинах волн от среднего значения длины волны (дифференциальная кривая).
Впервые схема двухволнового спектрофотометра была описана Чейнсом в 1951 г. [8]. На рис. 3-14 представлен ее последний вариант [9]. С помощью экрана с двумя отверстиями («маски») излучение источника делится на два потока, которые проходят через один и тот же монохроматор Черни — Тернера, но разлагаются в спектр разными дифракционными решетками. Благодаря прерывателю оба потока последовательно проходят через пробу и попадают на фотоумножитель. Кюветы с пробой желательно размещать.в одном из двух положений. Первое положение — почти вплотную к фотоумножителю — используется в тех случаях, когда через кювету должны проходить два потока с разными длинами волн; для мутных проб следует помещать кювету как можно ближе к детектору, чтобы полнее собрать рассеянное излучение. Второе положение кюветы с пробой используется в двухлучевых схемах с двумя кюветами.
Те, кто желает подробно ознакомиться с источниками ошибок в двухволновой спектрофотометрии, должны внимательно изучить материалы последней дискуссии по этому вопросу [10].
Производная спектрофотометрия
Дифференциальный спектр можно получить не только на двухволновом спектрофотометре. С помощью подходящей электронной схемы можно легко взять производную любой непрерывной функции; этот принцип вполне применим к спектрам поглощения.
Еще один способ основан на использовании колеблющейся преломляющей пластинки, которую помещают вблизи одной из щелей монохроматора. Как видно из рис. 3-15, поток излучения, проходящий через такую пластинку из кварца, смещается, но остается параллельным первоначальному направлению. При колебании пластинки взад и вперед на несколько градусов длина волны выходящего потока будет колебаться по синусоиде вокруг среднего значения в пределах нескольких нанометров; это пример волновой модуляции [11]. Электронный усилитель настраивают на частоту колебаний и в результате получают дифференциальный спектр.
3 Заказ № 637
66 Глава 3
Рис. 3-15. Колеблющаяся преломляющая пластинка для модуляции длины волны.
Рис. 3-16. Обычный (/) и дифференциальный (по первой производной) (2) спектры поглощения разбавленного раствора, применяемого для никелирования с содержанием сахарина 12 мг/л [12].
Поглощение излучения. УФ- и видимая области 67
Рис. 3-17. Соотношение между обычным (7) и дифференциальным (по первой производной) (2) спектрами [13].
На дифференциальном спектре имеются пики и впадины, соответствующие точкам перегиба на обычном спектре, но степень разрешения гораздо выше. На рис. 3-16 сравниваются обычный и дифференциальный спектры раствора для никелевого гальванопокрытия, содержащего небольшое количество сахарина [12]. (Сахарин, имид о-сульфобензойной кислоты, улучшает физические свойства гальванопокрытия из никеля.) На обычном спектре виднц два небольших отклонения, которые с таким же успехом можно и не заметить, а на производной кривой они превратились в максимумы, легко поддающиеся измерению.
На рис. 3-17 приведены обычный и дифференциальный спектры поглощения двух близких по строению стероидов [13]. Полосы поглощения обоих соединений гораздо легче обнаружить на дифференциальном, а не на обычном спектре.
3*
68 Глава 3
Источники излучения
Лампа накаливания с вольфрамовой нитью остается непревзойденным источником излучения в спектрофотометрии в видимой и ближних УФ- и ИК-областях (примерно от 320 нм до 3,5 мкм). Срок службы этих ламп при высокой температуре можно существенно увеличить введением в баллон небольшого количества паров иода. Такая лампа называется галоген-вольфрамовой (иногда иодно-кварцевой, так как баллон изготовляют часто из кварца, а не из стекла, что позволяет работать при более высокой температуре). Иод реагирует с испарившимися или распыленными атомами вольфрама с образованием летучего соединения, которое при соприкосновении с раскаленной нитью подвергается пиролизу, но при этом атомы металла осаждаются на нити, а не на холодных стенках баллона.
Для УФ-области широко применяются водородные и дейтериевые газоразрядные лампы, используемые при 160—130 нм. Дейтериевая лампа дает несколько более интенсивное излучение, чем водородная, дольше служит, но стоит дороже. Ксеноновая дуговая лампа охватывает большой интервал длин волн, но в абсорбционной спектроскопии редко требуется столь мощное излучение.
Детекторы
Для детектирования в УФ-области наиболее широко используется электронный фотоумножитель (ФЭУ) [14]. Основным элементом этого устройства является светочувствительный катод, на который нанесен тонкий слой полупроводникового материала, содержащего щелочной металл, например Cs3Sb, KzCsSb, Ma2KSb со следами Cs, или слой, полученный последовательным нанесением Ag, О и Cs. Активный слой наносят на металлическую подложку или закрепляют на внутренней поверхности кварцевого или стеклянного баллона. Для изготовления катодов используются материалы с разными спектральными характеристиками. Некоторые фотоэлементы, например широко известный 1Р28, чувствительны в области 200—650 нм. Этот интервал для некоторых специальных фотоэлементов простирается до 150 нм со стороны коротких и до 1,18 мкм со стороны длинных волн. На рис. 3-18 приведены некоторые кривые спектральной чувствительности.
Действие ФЭУ основано на эмиссии вторичных электронов в результате столкновения первичных электронов со светочувствительной поверхностью (рис. 3-19). Энергия излучения, попадающего на фотокатод, высвобождает электроны, которые ускоряются под действием электростатического поля и фокуси-
Поглощение излучения. УФ- и видимая области 69
Рис. 3-18. Кривые спектральной чувствительности фотоумножителей. 1 — катбд изготовлен из Ag—О—Cs, баллон — из боросиликатного стекла; 2— катод состоит из нескольких щелочных металлов, баллон изготовлен из боросиликатного стекла; 3 — катод изготовлен из GaAs(Cs), баллон — из стекла, прозрачного в УФ-области; 4 — катод состоит из двух щелочных металлов, баллон изготовлен из боросиликатного стекла (Hamamatsu Corporation).
руются на первом из серии динодов. Каждое соударение электрона с динодом вызывает испускание п вторичных электронов; п может принимать значения от 3 до 5 в зависимости от приложенного напряжения. Электроны, испускаемые первым динодом, в свою очередь фокусируются на втором и т. д.; в цепи может быть до 10—15 динодов. Усиление равно множителю п, возведенному в степень, равную числу динодов. Так, для 12 динодов при п — 4 должно наблюдаться усиление в 412, или почти в 17-106 раз. Эти 17 миллионов электронов, возникающие из одного первичного электрона, дают импульс, длящийся обычно около 5 нс, что соответствует в среднем силе тока около 0,5 мА.
70 Глава 3.
Путь оптического Электрона от фото- < катода к первому диноду
Электронные умножитель "
Падающее излучение
11±
14
15 1
14
Вакуумная камера
14 .2
-Полупрозрачный фотокатод
13 Анод
14 Фокусирующие электроды
15 Фотокатод
- Типичные траектории электронов
Рис. 3-19. Схема фотоумножителя, на которой показаны траектории нескольких электронов.
14
1-12 Диноды
На рис. 3-20 показана основная электронная схема ФЭУ. Анодный ток попадает в операционный усилитель (на схеме обозначен треугольником). Операционный усилитель — чрезвычайно важный универсальный элемент схемы (он неоднократно упоминается в этой книге и подробно разбирается в гл. 27). А сейчас, чтобы понять его роль в работе фотоумножителя, достаточно усвоить три положения: 1) операционный усилитель
Поглощение излучения. УФ-и видимая области 71
Рис. 3-20. Соединение узлов фотоумножителя. Все сопротивления в цепи делителя напряжения одинаковы, например по 50 кОм каждый. Конденсаторы используют только при измерении флуктуационного или импульсного излучения. Операционный усилитель обсуждается в тексте.
должен быть снафкен контуром обратной связи выхода и входа (обозначен знаком «—»), в данном случае эту связь обеспечивает резистор R-, 2) ни через один из входов в операционный усилитель не должен попадать ток; 3) правильно подобранный операционный усилитель обеспечивает на выходе напряжение, необходимое для поддержания (с помощью петли обратной связи) на обоих выходах практически одинаковых потенциалов. В рассматриваемом случае ток с анода фотоумножителя 1а должен быть равен току (прошедшему через резистор) на выходе усилителя, так как ток не может пройти через усилитель. Значит, вход «—» будет практически заземлен, поскольку заземлен вход « + ». Поэтому напряжение на выходе выражается следующим образом: E^x^IaR- Это эффективный и удобный способ измерения малых токов. Предложено много вариантов схемы, изображенной на рис. 3-20, каждый из которых имеет определенные достоинства. Некоторые из них описаны в работе [15].
Если не требуется высокой чувствительности, то часто используется более простой и экономичный фотоэлемент без умножения. Он состоит из такого же фотокатода, как и ФЭУ, и анода, заключенных в стеклянный и кварцевый баллоны. Как следует из рис. 3-21, операционный усилитель играет здесь точно такую же роль, как и в ФЭУ. Сопротивление обычно со-
72 Глава 3
Рис. 3-21. Схема вакуумного или газонаполненного фотоэлемента.
у ФЭУ, природой
И
ставляет 1—10 МОм, что создает на выходе напряжение от 1 до 10 В при фототоке 1 мкА. Спектральная область этих фотоэлементов, как определяется
материала катода. Фотоэлементы, заполненные аргоном под небольшим давлением, в 10—20 раз чувствительнее вакуумных фотоэлементов, но несколько менее стабильны и надежны при точных фотометрических измерениях.
В видимой и ближяей ИК-областях применяются разнообразные полупроводниковые фотоэлементы — вентильные фотоэлементы, фотодиоды и фототранзисторы. Для ближней ИК-области обычно выбирают фотодиод из PbS, который включают в набор фотоэлементов спектрофотометров, предназначенных для работы в УФ- и видимой областях, для расширения диапазона измерений на этих приборах. Детально полупроводниковые устройства обсуждаются в гл. 27.
. Погрешность фотометрических измерений
Диапазон определяемых фотометрически концентраций ограничен верхним и нижним пределами. При высоких концентрациях поглощающего вещества энергия прошедшего излучения так слаба, что чувствительность фотометра оказывается недостаточной для ее измерения. При низких же концентрациях ошибка в показаниях прибора становится слишком большой по сравнению с измеряемой величиной. Отсюда ясно, что должна быть некоторая промежуточная концентрация, при которой точность максимальна.
Точку, в которой относительная ошибка минимальна, можно найти математически при условии подчинения системы закону Бера. Чувствительность любого детектора ограничена производимыми им шумами. Под «шумами» подразумеваются случайные флуктуации, неизбежные в электронной схеме [16]. Сейчас нас интересуют два типа шумов, которые могут доминировать в том или ином фотодетекторе.
Чувствительность вакуумных фотоэлементов и фотоумножителей ограничена так называемым флуктуационным (дробовым) шумом, вызываемом статистическими флуктуациями скорости, с которой каждый индивидуальный электрон испускается с поверхности катода. Можно показать, что величина флуктуационного шума пропорциональна корню квадратному из мощ
Поглощение излучения. УФ- и видимая области 73
ности падающего излучения. Напротив, чувствительность большинства остальных детекторов ограничена шумами сопротивления (называемыми также тепловыми шумами Джонсона), которые вызваны случайным тепловым движением электронов в элементах схемы, обладающих активным сопротивлением. Этот вид шумов постоянен и не связан с интенсивностью измеряемого излучения.
Для того чтобы достичь максимальной точности измерения оптической плотности, бесконечно малое ее приращение АЛ должно составлять по возможности меньшую часть наблюдаемой оптической плотности А, другими словами, АЛ/А должно быть минимальным. Чтобы найти значение Л, при котором АЛ/А минимально, нужно дважды продифференцировать выражение Л = 1§(Р0/Р) и приравнять вторую производную нулю. Для удобства запишем это выражение в виде
Л = 1ёР0-lg Р , (3-12)
Тогда
dA = 0—(1g е) -j- dP = 0— 0,434 dP (3-13)
Разделив обе части на Л и заменив Р равным ему произведением Ро-10-А, получим
±dA =---^—dP
А АРо10~А
(3-14)
Заменив дифференциалы конечными приращениями, получим выражение
ДХ А
0.434ДР / 1
Ро I А • 10~л
(3-15)
в котором АР — приращение интенсивности излучения, соответствующее АЛ.
При постоянном АР (т. е. при наличии тепловых шумов) второе дифференцирование дает
d(\A!A) 0.434ДР / 10л in 10 10л \
dA ~ Ро \ А ~ А2 )
(3-16)
Величина АЛ/Л минимальна при условии, что правая часть уравнения равна нулю, т. е. множитель в скобках равен нулю:
10л 1п 10/Л = 10л/Л2
(3-17)
Отсюда
Лот= 1/1П 10 = 0,434
(3-18)
74
Глава 3
Рнс. 3-22. Зависимость относительной ошибки фотометрнровання от оптнче ской плотности при тепловых (/) н флуктуационных (2) шумах.
Для детектора, чувствительность которого ограничена флуктуационными шумами, т. е. при ДРосур, аналогичная операция дает
Лопт = 2/1п 10=0,868
(3-19)
Таким образом, оптимальная оптическая плотность для фотоумножителей в два раза выше, чем для фотоэлементов с внут-
Концентрация Мп, мкг/мл
Рис. 3-23. Градуировочные кривые для перманганата. Сплошные кривые получены на спектрофотометре прн длинах волн 526 (7), 480 (2) н 590 нм (3). Штриховая кривая (4) построена по данным, полученным на фотоэлектроколориметре со светофильтром с эффективной длиной волны 430 им (ср. с рнс. 3.3) [18].
ренним фотоэффектом или детекторов других типов. Кривые для обоих этих случаев приведены на рис. 3-22. Минимум для детектора, подверженного флуктуационным шумам, гораздо шире, чем для детектора с тепловыми шумами, поэтому в первом случае можно достаточно точно измерить оптические плотности, значительно превышающие два, тогда как омические детекторы на практике ограничены значением А около 0,8. Нижний предел А в обоих случаях одинаков и составляет примерно 0,25.
Общепринятой формулой градуировочного графика в фотометрическом анализе является прямая линия (рис. 3-2). На ней видна область, в которой выполняется закон Бера, но она не дает никаких указаний об относительной точности при
Поглощение излучения. УФ- и видимая области 75
разных оптических плотностях. Предложен другой способ построения градуировочной кривой [17, 18], позволяющий извлечь некоторую дополнительную информацию. На рис. 3-23 представлены кривые зависимости процента пропускания от логарифма концентрации. Если интервал концентраций достаточно велик, получается S-образная кривая, называемая кривой Рингбома. Если система подчиняется закону Бера, точка перегиба (для детектора с флуктуационными шумами) находится при 37 % пропускания; в противном случае точка перегиба лежит при каком-то другом значении, но форма кривой сохраняется. На кривой, как правило, имеется довольно большой практически линейный отрезок. Границы этого участка указывают оптимальный интервал концентраций.
Часто интервал определяемых с достаточной точностью концентраций оказывается слишком мал. Рабочий интервал можно продлить в область более высоких концентраций, взяв кювету с более короткой длиной оптического пути или изменив длину волны. Как видно из рис. 3-23, рабочий интервал концентраций перманганата составляет 6—60 мкг/мл Мп при 580 нм и 2— 20 мкг/мл при 526 нм.
Счет фотонов
При уменьшении интенсивности падающего на детектор излучения становится все труднее отличить нужный сигнал от случайного шума. В таких случаях сигнал можно существенно усилить, наблюдая за отдельными фотонами. Если конечная ско-
Рнс, 3-24. Серия импульсов, превышающих случайные шумы. Подсчитываются лишь те импульсы, которые расположены над штриховой линией.
76 Глава 3
рость фотонов достаточно низка, чтобы зафиксировать единичные показания на детекторе, можно наблюдать отдельные импульсы от каждого фотона; с помощью электронных устройств эти импульсы можно сосчитать с большей точностью, чем при измерении усредненного постоянного тока на выходе.
Случайные шумы, возникающие в электронной схеме, также дают импульсы, но амплитуда их обычно меньше, чем у импульсов, вызванных фотонами. С помощью электронной схемы, называемой дискриминатором, можно подавить все импульсы, величина которых меньше заданного уровня, и оставить для подсчета только импульсы большей величины (рис. 3-24). При измерении на приборе с регистрацией тока шумы и сигнал складываются, внося каждый свой вклад в общий ток. Если сигнал больше шума, то следует отдать предпочтение этому общеупотребительному способу, но если они сопоставимы по величине, то счет фотонов обеспечивает более правильный результат [19]. Последний способ применяется в абсорбционной спектрофотометрии сильнопоглощающих растворов и имеет очень большое значение в таких областях, как флуориметрия, где приходится иметь дело исключительно с малоинтенсивным излучением.
Эксплуатация спектрофотометра
Каждый спектрофотометр обеспечен целым рядом устройств, необходимых для управления и настройки. Перечисление их поможет лучше понять работу таких приборов.
Спектрофотометры с ручным управлением должны иметь: 1) ручку для настройки на ту или иную длину волны (или волновое число), обычно механически связанную с устройством для вращения дифракционной решетки или призмы, 2) ручку для регулирования ширины щели, 3) ручку для установки прибора на нуль, иногда называемую компенсатором темнового тока, и 4) ручку контроля за усилением. Регистрирующие спектрофотометры, кроме того, снабжены сканирующим и записывающим устройствами и соответствующими ручками управления (развертки и записи).
В более дешевых приборах некоторые из этих устройств отсутствуют, что снижает возможности прибора. Напротив, более сложные модели имеют дополнительные блоки, позволяющие, например, использовать двухлучевой прибор как однолучевой. Большинство современных спектрофотометров управляется с помощью микропроцессорной техники.
Многие спектрофотометры снабжены дополнительными приспособлениями и устройствами, например приспособлениями для получения спектров отражения или флуоресценции или держателями для удлиненных кювет и др.
Поглощение излучения. УФ- и видимая области 77
Способы повышения точности измерений
В спектрофотометрах старых конструкций точность диктовалась точностью считывания со шкалы или записи самописца. Общую точность таких приборов можно повысить путем расширения шкалы. Для этого нужно приготовить один или несколько стандартных растворов с концентрацией, по возможности более близкой к определяемой. Прибор настраивают по самому разбавленному стандартному раствору (но не по растворителю) на максимальное пропускание, а по более концентрированному раствору — на минимальное пропускание. Сейчас такой способ используется редко в основном из-за того, что удобные в обращении цифровые дисплеи обеспечивают существенное снижение ошибки считывания. Точно так же счетчики фотонов повышают точность при измерении высокой оптической плотности. Тем не менее способы расширения шкалы следует иметь в виду как возможную альтернативу; преимущества и ограничения их разобраны в работе Ингла [20].
Практическое применение
Поглощение энергии излучения в УФ- и видимой областях спектра определяется прежде всего числом и расположением электронов в поглощающих молекулах или ионах. Избирательного поглощения неорганическими молекулами следует ожидать в том случае, если незаполненный энергетический уровень экранирован заполненным уровнем, который обычно образован за счет координации с другими атомами.
Рассмотрим в качестве примера медь. В водных растворах не существует простого иона Си2+ (хотя часто его записывают именно так), потому что он легко образует координационные связи с любыми молекулами или ионами, несущими неподелен-ную пару электронов. Такие неподеленные пары имеются в воде, аммиаке, цианид-ионе, хлорид-ионе и многих других частицах. В структуре иона меди(II), координирующейся с любым из этих оснований Льюиса, на третьем (М) основном энергетическом уровне имеется только 17 электронов, тогда как четвертый (N) уровень содержит устойчивый октет. Разные ионы не могут иметь одинаковую окраску, поскольку природа лиганда влияет на энергию электрона.
Избирательное поглощение органическими молекулами тоже связано с дефицитом электронов в молекуле. Полностью насыщенные соединения не проявляют избирательного поглощения ни в видимой, ни в доступной части УФ-области. Соединения, содержащие двойную связь, сильно поглощают в дальней УФ-области (195 нм для этилена). Сопряженные двойные связи вызывают поглощение при больших длинах волн. Чем длиннее
78 Глава 3
сопряженная цепь, тем при большей длине волны наблюдается поглощение. При достаточно большой длине цепи поглощение переходит в видимую область, и в результате наблюдается окрашивание соединения. Так, р-каротин, имеющий II сопряженных двойных связей, интенсивно поглощает в области 420— 480 нм и, следовательно, окрашен в желто-зеленый цвет. В целом вся сопряженная система в соединении называется хромофором.
Длина волны максимума поглощения служит для идентифи-, кации содержащегося в ней хромофора. Под влиянием различных атомных группировок, замещающих атомы водорода в хромофорах, спектры в целом изменяются. При введении заместителей полоса поглощения обычно сдвигается в длинноволновую область и значение оптической плотности изменяется. Заместители, оказывающие такое влияние, называются (несколько произвольно) ауксохромами.
В табл. 3-4 приведен целый ряд органических соединений, содержащих типичные хромофоры, их максимумы поглощения
Таблица 3-4. Типичные хромофоры3
Соединение Хромофор Растворитель А, такс’ им 1g 8
Октен-3 С=С Гексан 185 3,9
230 0,3
Ацетилен СезС (Пары) 173 3,8
Ацетон с=о Гексан 188 2,9
279 1,2
Диазоэтил ацетат N=N Этанол 252 3,9
371 1,1
Бутадиен С=С—с=с Гексан 217 4,3
Кротоновый альдегид с=с—с=о Этанол 217 4,2
321 1,3
Диметилглноксим N=C—C=N Этанол 226 4,2
Октатриенол С=С—С=С--С=С Этанол 265 4,7
Декатетраенол [—С=С—]4 Этанол 300 4,8
Витамин А [—С=С—]в Этанол 328 3,7
Бензол о Гексан 198 255 3,9 2,4
/,4-Бензохннон ^>=0 Гексан 245 5,2
285 2,7
435 1,2
Нафталин 00 Этанол 220 275 5,0 3,7
оо 314 2,5
Дифенил Гексан 246 4,3
а)> Данные заимствованы из разных источников только с целью иллюстрации.
Поглощение излучения. УФ- и видимая области 79
и значения молярных коэффициентов поглощения. В спектрах многих из приведенных соединений, особенно включающих крупные хромофоры, содержится много максимумов с меньшей интенсивностью. По данным этой таблицы нельзя надежно идентифицировать поглощение группы, как это можно сделать, пользуясь соответствующей таблицей ПК-спектров.
Самым простым хромофором ароматических соединений является бензольное кольцо. При наличии двух или более связанных между собой колец, как, например, в нафталине или дифениле, поглощение возрастает, а полоса сдвигается в видимую область. Данные табл. 3-5 иллюстрируют влияние некоторых ауксохромов на поглощение бензола [21].
Хиноидное кольцо более эффективный хромофор, чем бензольное кольцо. Проиллюстрируем это на примере фенолфталеина, который в кислой и щелочной средах имеет соответственно следующее строение:
(6 кислом растворе)
(в щелочном растворе)
Таблица 3-5. Влияние ауксохромов на хромофоры, содержащие бензольные кольца [21]
Соединение Р створнтель Полоса этиленовой группы Полоса бензоидной группы
Члакс’ нм 8макс Ч1акс’ им 8макс
Бензол Гексан 204 7 900 256 200
Анилиний Водный кислый раствор 203 7 500 254 160
Хлорбензол Этанол 210 7 600 265 240
Фенол Вода 210,5 6 200 270 1450
Пирокатехин Вода, pH 3 214 6 300 276 2300
Анизол 2 %-ный метанол 217 6 400 269 1480
Анилин Вода 230 8 600 280 1430
Фенол ят-аннон Водный щелочной раствор 235 9 400 289 2600
Тиофенол Г ексан 236 10 000 269 700
Пирокатехинат Вода, pH 11 236,5 6 800 292 3500
Дифениловый эфир Циклогексан 255 11 000 272 278 2000 1800
80 Глава 3
Рис. 3-25. Спектры поглощения фенолфталеина в кислом (а) н щелочном (б) растворах.
В бесцветной форме сопряжение не выходит за пределы отдельного кольца (за исключением кольца, сопряженного с карбонильной группой). В красной форме, однако, одно кольцо перешло в соответствующий хинон, что привело к удлинению цепи сопряжения за счет включения в нее центрального углеродного атома и благодаря этому двух других колец. Итак, мы должны сделать вывод, что образовавшийся анион включает хромофор, тогда как в кислой среде молекула содержит три отдельные почти идентичные, группы, практически не являющиеся хромофорными. На рис. 3-25 представлены соответствующие спектры поглощения.
Качественный анализ
Спектры поглощения в ультрафиолетовой и видимой областях служат полезным источником дополнительных доказательств при установлении структуры органического соединения. Погло
Поглощение излучения. УФ- и видимая области 81
щение само по себе редко бывает достаточно уникальным, чтобы служить «отпечатком пальцев» какой-либо структуры, но оно может помочь при исключении некоторых альтернатив. Если соединение не поглощает, начиная примерно с 210 нм и вплоть до видимой области, то оно определенно не содержит сопряженных ненасыщенных связей. Если оно остается прозрачным до 180 нм, то следует исключить даже изолированные двойные связи.
Установлен целый ряд эмпирических корреляций между длиной волны в максимуме поглощения и структурой. Одна из них, предложенная Вудвордом [22, 231, относится к а,р-ненасыщен-ным карбонильным соединениям. Основному поглощению можно приписать максимум при 215 нм; дополнительно каждая двойная связь смещает его в длинноволновую область на 30 нм. насыщенный заместитель при С-2 на 10 нм, при С-3 на 12 нм и при С-4 и далее на 18 нм, бромсодержащие заместители при С-2 на 23 нм и т. д. Измеренный максимум поглощения можно сравнить со значениями, рассчитанными для ряда возможных структур.
Изменение спектра поглощения кислотно-основного индикатора в зависимости от pH положено в основу метода определения соответствующего значения рК. На рис. 3-26, а изображены кривые поглощения фенолового красного при разных значениях pH. С ростом pH поглощение при 550 нм увеличивается, а при 425 нм уменьшается. Заметим, что все кривые пересекаются вблизи общей точки при 475 нм, которая называется изобестической точкой. Наличие такой точки указывает на то, что система содержит два хромофора, которые переходят друг в друга, причем общее количество их остается постоянным: коэффициент поглощения обоих хромофоров в изобестической точке одинаков.
Если построить зависимость поглощения в одном из максимумов (в случае фенолового красного при 550 нм) от pH, получится S-образная кривая (рис. 3-26,6). Левый горизонтальный участок соответствует кислой форме индикатора, а верхний правый — почти полному переходу в щелочную форму. Поскольку рК равно значению pH, при котором в каждой форме находится половина индикатора, его значение определяется значением pH в точке, лежащей посередине между двумя горизонтальными участками, например pH 7,0 в приведенном примере.
Подобным образом и даже с большим успехом, чем для видимой области можно часто определить константы диссоциации соединений, поглощающих в УФ-области. В качестве примеров можно привести теобромин [24] (240 нм), теофиллин [24] (240 нм) и бензтриазол [25] (274 нм). Целесообразно пост-
82 Глава 3
Рис. 3-26. Спектры поглощения фенолового красного при различных значениях pH (а) и зависимость оптической ллотности фенолового красного при 550 нм от pH (б) (значение рЛ' индикатора очень близко к 7,0).
Поглощение излучения. УФ- и видимая области 63
Рис. 3-27. Трехмерная диаграмма для бензолазоднфеннламнна. На диаграмме S по вертикальной осн отложена оптическая плотность как функция pH (ось слева направо) и длины волны (ось, перпендикулярная осн pH). Графики F и L — двумерные диаграммы, построенные так же, как на рис. 3-26, а и б соответственно [26].
роить трехмерную диаграмму, как это сделано на рис. 3-27 для бензолазодифениламина [26], где по осям Отложены длина волны, значение pH и оптическая плотность. Изобестическая точка на графике F соответствует прямой линии, параллельной оси pH, на трехмерной диаграмме.
Наличие изобестической точки доказывает существова'ние химического равновесия между двумя поглощающими частицами. Если же при изменении pH, концентрации или другого фактора вместо исчезновения одного максимума поглощения и появления другого (условие изобестической точки) максимум постепенно смещается, то можно предположить, что имеет место физическое взаимодействие поглощающего вещества и его окружения или образуется серия комплексов с близкой устойчивостью.
Определение соотношения лиганд/металл в комплексном соединении
Способность комплексов металлов с органическими лигандами избирательно поглощать излучение в УФ- и видимой областях широко используется для определения их состава и констант
84 Г лава 3
Рис. 3-28. Кривая, построенная по методу йоу — Джонса для комплекса ртути с дифенилкарбазоном (длина волны 520 нм) [29].
устойчивости. Стехиометрию устойчивого комплекса можно установить одним из двух связанных между собой способов: методом молярных отношений Йоу и Джонса [27] и методом изомолярных серий Жоба, усовершенствованным Восбургом и Купером [28].
Метод молярных отношений заключается в измерении оптической плотности серии растворов с разным содержанием од
Рис. 3-29. Кривые, построенные по методу Жоба (метод изомолярных серий) для комплекса ртути с дифенилкарбазоном. По оси ординат отложена разность между оптической плотностью растворов смеси и суммой оптических плотностей растворов реагентов (длина волны 520 нм) [29].
ного компонента и постоянным другого. Затем строят график зависимости оптической плотности от отношения числа молей лиганда к числу молей иона металла. Этот график от исходной точки до точки, где компоненты находятся в эквивалентных количествах, представляет собой прямую линию. Затем он пойдет горизонтально, поскольку один из компонентов полностью израсходован, а добавление второго компонента не может вызывать образования поглощающего комплекса. Если компонент, добавляемый в избытке, обладает собственным поглощением при той же длине волны, график за точкой эквивалентности будет иметь положительный наклон, но меньший, чем до точки эквива
Поглощение излучения. УФ- и видимая области 85
лентности. На рис. 3-28 представлены результаты исследования комплекса дифенилкарбазона с ионами Hg(II) [29].
Метод изомолярных серий заключается в приготовлении ряда растворов с разной концентрацией двух компонентов, но с постоянной суммарной концентрацией обоих. Строят график зависимости разности между измеренной оптической плотностью и рассчитанной в предположении отсутствия взаимодействия между компонентами от мольной доли компонента. При мольной доле, отвечающей составу комплекса, на получившейся кривой будет наблюдаться максимум (или реже минимум) (рис. 3-29).
Следует заметить, что метод Йоу—Джонса, по существу, представляет собой титрование известного количества иона металла раствором лиганда, где конечная точка (пересечение двух линий) дает информацию о стехиометрии комплекса. МакКарти [30] указал, что если оба раствора (иона металла и лиганда) имеют одинаковую молярную концентрацию, то в любой точке титрования суммарная концентрация иона и лиганда обязательно постоянна. Следовательно, кривая Йоу— Джонса содержит всю информацию, необходимую для построения кривой Жоба. Таким образом, построение кривой по методу Жоба представляет собой один из графических способов нахождения конечной точки титрования.
Перегибы на кривых, построенных тем или другим способом, часто оказываются не такими четкими, как хотелось бы. Закругление вблизи точки эквивалентности зависит от значения константы устойчивости комплекса и часто служит для измерения этой константы [31—33].
Количественный анализ
Пользуясь законом Бера, можно непосредственно провести определение поглощающего вещества, если отсутствуют другие поглощающие частицы, мешающие определению, или если эти мешающие частицы можно удалить или внести на них поправку. В качестве примера такого подхода может служить измерение концентрации озона в городском смоге [34]. На крыше какого-либо здания устанавливали ртутную дуговую лампу высокого давления, излучение которой регистрировали с помощью простого призменного спектрофотометра, расположенного на другом здании на расстоянии примерно 100 м. Поскольку двухлучевая схема была непригодна, установку на нуль проводили ночью, когда, как известно, содержание озона в атмосфере падает до пренебрежимо малой величины. Измерению озона мешали другие окислители, например NO2, но их влияние устра
86 Глава 8
няли, определяя соотношение оптических плотностей в максимумах озона при’313 и 265 нм. Было обнаружено, что в полдень содержание озона (по усредненным данным за 50 дней) поднимается примерно до 22 частей на 108 частей.
Вещества, не способные к поглощению, во многих случаях можно определить спектрофотометрически, добавив реагент, образующий поглощающий комплекс, или другой хромофор. Одним из наиболее важных реагентов является дитизон (дифенилтиокарбазон). Это — соединение зеленого цвета, растворимое в хлороформе, реагирует с катионами большинства переходных металлов с образованием красных или фиолетовых комплексов. Селективность реагента можно повысить, регулируя pH. Подробное описание методов с использованием дитизона легко найти в литературе {35, 36], поэтому оно здесь не приводится. Другой пример — образование поглощающего соединения при реакции Hg(II) с красителем 4,4'-бис(диметиламино)дифениламином (зеленый Биндшедлера) в цитратном буфере, что используется для определения ртути [37]. Образующийся комплекс экстрагируют 1,2-дихлорэтаном; получившиеся растворы подчиняются закону Бера в интервале 8-10“7— 4«10~eM Hg(II). Из 21 исследованного металла определению мешает только олово. В монографии [35] приведены сотни аналогичных аналитических методов.
Аддитивность оптических плотностей
При выводе закона Бера мы отметили, что оптическая плотность пропорциональна количеству частиц, способных поглощать излучение с определенной длиной волны. Это положение легко распространить на тот случай, когда в том же растворе находятся и другие поглощающие частицы. Каждый вид частиц поглощает так, как если бы других частиц не было. Мы можем записать, что
<3-20> i I
т. е. оптическая плотность является аддитивным свойством. Это соотношение справедливо, конечно, в отсутствие химических взаимодействий между растворенными веществами.
На принципе аддитивности основан целый ряд приемов и методов, как, например, известный прием введения поправки на холостой опыт путем вычитания оптической плотности, обусловленной растворителем и примесями из реагентов, из измеренной оптической плотности раствора. Пользуясь принципом аддитивности, можно идентифицировать или определить количественно неизвестный хромофор в смеси с известным хро-
Поглощение излучения. УФ- и видимая области 87
Рис. 3-30. Ультрафиолетовые спектры поглощения 4-нитробензоата 7-дегидрохолестерина (а), 4-нитробензоата циклогексила (б) и 7-дегидрохолестерина, полученного вычитанием спектра б из спектра а (растворитель — гексан) [38].
мофором, вычитая оптическую плотность последнего из спектра смеси. Рассмотрим, например, рис. 3-30 138], где нужно по УФ-спектру поглощения точно определить 7-дегидрохолестерин, который трудно получить чистым из-за окисления на воздухе, но эфиры которого очистить легко. Кривая а представляет собой спектр чистого образца 4-нитробензоилового эфира холестерина, из которого вычли спектр циклогексил-4-нитробензоата (кривая б). Полученная кривая в по форме идентична спектру чистого 7-дегидрохолестерина, причем примеси не повлияли на величину молярного коэффициента поглощения.
На аддитивности оптической плотности основан метод анализа многокомпонентных систем — одновременное определение двух или более поглощающих веществ в одном растворе. Условием определения является лишь требование, чтобы кривые поглощения индивидуальных компонентов не были слишком близки друг к другу. Частичное перекрывание допустимо, но чем оно больше, тем меньше точность анализа. Изложенное дегче понять, обратившись к рис. 3-31. Кривые 1 и 2— спектры
88 Глава 3
Рис. 3-31. Гипотетический пример анализа двухкомпоиентиой системы на спектрофотометре.
поглощения чистых компонентов, а кривая 3— спектр их смеси. Предполагается, что другие вещества, поглощающие в этой области, отсутствуют.
Заметим, что максимум кривой 3 слегка смещен по отношению к максимумам кривых 1 и 2. Очевидно, оба вещества вносят свой вклад в поглощение при обеих длинах волн и Пусть Ci и с2 — концентрации компонентов в смеси, а Л1„ представляет собой оптическую плотность вещества Л] при длине
волны Ки и т. д. так, как это показано на рисунке. Тогда оптическую плотность кривой 3 при Kv можно найти по формуле
Л v — л 10 -Т Л 2t> — dlvbc1 ^2^2
а при
Au — A lu r A2u — alu^Cl + a2u^C2
Лг и Au определяют экспериментально для смеси, а значения а — измерением чистых веществ, поэтому приведенные выше уравнения можно решить относительно Ci и с2. Для получения максимальной точности значения alv и а2и должны быть как можно меньше, а ат и a2v — как можно
больше. Совместное решение уравнений легко получить с помощью компьютера или графическим способом [39].
Примером, иллюстрирующим возможности метода, является одновременное определение Mo, Ti и V в виде окрашенных комплексов с Н2О2 [40]. Стандартные спектры приведены на рис. 3.32.
Рис. 3-32. Сравнение спектров продуктов взаимодействия пероксида водорода с Mo, Ti и V, взятых в концентраций 4 мг в 100 мл [40].
Поглощение излучения. УФ- и видимая области 89
Фотометрическое титрование
В классической визуальной титриметрии точку эквивалентности обнаруживают по изменению или появлению окраски одного из реагирующих веществ (например, перманганата). При благоприятных условиях аналитик с нормальным зрением легко достигает точности 0,1 %. Однако трудно или даже невозможно получить хорошие результаты, если окраска изменяется постепенно или неконтрастно.
Этих трудностей можно избежать, если проводить титрование в кювете спектрофотометра или фотометра. Предварительно подбирают оптимальную длину волны (или светофильтр) и проводят установку на нуль. Затем после добавления из бюретки каждой порции титранта проводят фотометрическое измерение. В конструкцию обычных спектрофотометров при этом необходимо внести некоторые изменения: их нужно снабдить сосудом для титрования подходящего размера, в который опускают кончик бюретки, и устройство для перемешивания.
Существуют многочисленные автоматические и полуавтоматические титраторы. Они выполняют все операции титрования при минимальном участии аналитика. В некоторых из них результаты регистрируются на самописце, в других в конечной точке титрования кран бюретки закрывается с помощью электрического устройства. В современных приборах, конечно, предусмотрено компьютерное управление.
Обычно кривые фотометрического титрования строят в координатах объем добавленного реагента — оптическая плотность. Если поглощающие вещества (титрант, титруемое вещество или тот и другой) подчиняются закону Бера, то кривая титрования с поправкой на разбавление должна состоять из двух прямых, пересекающихся в точке эквивалентности. Из-за неполного протекания реакции в точке эквивалентности они несколько искривлены. Как правило, это не имеет значения, так как более удаленные участки кривых почти линейны и их можно экстраполировать до пересечения.
На рис. 3-33 представлена кривая титрования смеси м- и «-нитрофенолов раствором NaOH [41]. Оптическую плотность измеряли при длине волны 545 нм, при которой поглощают анионы обоих изомеров, а у соответствующих кислот поглощение отсутствует. Молярный коэффициент поглощения лъизомера больше, чем у n-изомера. Последний нейтрализуется в первую очередь, поскольку из двух слабых кислот он является более сильной. В этом отдельном титровании ошибка определения по конечным точкам, соответствующим двум точкам пересечения, лишь немного превышает один процент. Определить эти слабые кислоты при их совместном присутствии визуальным
90 Глава 3
Объем О.667М Na ОН
Рис. 3.33. Фотометрическое титрование смеси 50 мл 0,0219 М раствора 4-ни-трофеиола и 50 мл 0,0213 М раствора 3-нитрофеиола 0,667 М раствором NaOH (длина волны 545 им) [41].
методом с индикатором или без него было бы невозможно, поскольку изменение окраски в первой точке эквивалентности происходило бы очень медленно. По аналогичной причине этот анализ нельзя выполнить потенциометрически (на рН-метре).
Удовлетворительные результаты при титровании слабой кислоты можно получить при условии, что произведение молярной концентрации и константы диссоциации кислоты СКа больше чем 10-12 [42]. Такой способ непригоден для титрования сильных кислот, поскольку они всегда находятся в диссоциированной форме. Однако их концентрацию можно контролировать фотометрически по оптической плотности добавленного заранее индикатора. Индикатором, как правило, служит слабая кислота, у которой недиссоциированная и анионная формы поглощают при разных длинах волн. Принцип выбора индикатора здесь иной, нежели при визуальном титровании. При визуальном методе желательно, чтобы рКа индикатора в точке эквивалентности совпадало с pH. При использовании же фотометрической системы, если измерения проводятся в области поглощения анионной формы, следует выбирать индикатор с достаточно.
Поглощение излучения. УФ- и видимая области 91
Рис. 3-34. Кривая фотометрического титрования кислоты основанием с добавлением индикатора.
Рис. 3-35. Фотометрическое титрование смеси висмута и меди 0,1 М раствором ЭДТА [43].
низким значением рКа, чтобы избежать его нейтрализации, пока полностью не прореагирует более сильная кислота. Точку эквивалентности можно затем найти по пересечению двух прямолинейных отрезков (точка а на рис. 3.34). Точка пересечения b соответствует полному оттитровыванию индикатора, следовательно, этот случай можно рассматривать как титрование смеси сильной и слабой кислот.
Одной из самых плодотворных областей применения фотометрического титрования является титрование металлов ЭДТА и другйми комплексонами. На рис. 3-35 приведен пример последовательного определения Bi и Си из одной аликвотной порции [43]. Измерения проводили при 745 нм, когда комплекс Си—ЭДТА интенсивно поглощает, а у комплекса Bi—ЭДТА поглощение отсутствует.
Другая важная область применения фотометрического определения конечной точки — осадительное титрование, но обсуждение этого вопроса мы отложим до гл. 8.
Задачи
3-1. Пропускание раствора окрашеииого вещества, подчиняющегося закону Бера, в 1,00-см кювете равно 80,0%. а) Рассчитайте пропускание (в процентах) раствора вдвое большей концентрации в той же кювете, б) Какой должна быть длина оптического пути в кювете, чтобы пропускание раствора с удвоенной концентрацией осталось прежним (80 %)? в) Рассчитайте пропускание первоначального раствора в 0,5-см кювете, г) Найдите зиачеиие коэффициента поглощения а, если концентрация исходного раствора 0,0050 % (масса/объем),
3-2. При построении градуировочного графика для фотометрического определения были получены следующие данные:
92 Глава 3
Концентрация, мг/л ро Р
0,00 98,0 98,0
1,00 97,0 77,2
2,00 100,0 63,5
3,00 99,5 50,0
4,00 100,0 41,3
5,00 100,0 33,5
6,00 100,0 27,9
7,00 99,0 23,4
8,00 98,2 20,3
9,00 100,0 18,1
10,00 100,0 16,4
а) Рассчитайте оптическую плотность и постройте график зависимости ее от концентрации. Указывают ли эти данные на отклонение от закона Бера и если да, то отрицательное или положительное? б) Постройте по этим данным диаграмму Рингбома и установите приблизительный интервал концентраций, в котором можно получить достаточную точность определения.
3-3. Максимум поглощения витамина D2 (кальциферола, мол. масса 397) лежит при 264 нм, молярный коэффициент поглощения (в спирте) равен 18 200. Закон Бера соблюдается в широком интервале, а) Каково значение коэффициента поглощения? б) Какой интервал концентраций (в граммах на литр) можно использовать для анализа, если желательно, чтобы оптическая плотность лежала в пределах 0,4—0,9? Пусть 6—1,00 см.
3-4. Пользуясь данными рис. 3-4, предскажите знак кажущегося отклонения от закона Бера при построении графика зависимости оптической плотности от общей концентрации Cr(VI) для водных растворов КгСгО4 при длинах волн 350, 370, 445 и 480 нм.
3-5. Для каждого из приведенных ниже случаев предскажите, будет ли отклонение от закона Бера отрицательным, положительным или его практически не будет вовсе, а) Поглощающее вещество является недиссоциированной формой слабой кислоты, б) Поглощающая частица представляет собой катион, находящийся в равновесии со слабой кислотой, в) При определении металла получают окрашенное соединение и измеряют его оптическую плотность на фотоэлектроколориметре с соответствующим стеклянным светофильтром.
3-6. Нужно определить содержание хрома в силикатной горной породе. Пробу истирают до пудры и берут для анализа навеску массой 0,5000 г. Путем соответствующей обработки материал разлагают: хром при этом переходит в Na2CrO4. После фильтрования раствор разбавляют до 50,00 мл 0,1 М раствором H2SO4 и добавляют 2 мл 0,25 %-ного раствора дифенилкарбазида, дающего с хромом (VI) соединение красновато-фиолетового цвета. В качестве стандартного используют раствор чистого К2Сг2О7 с концентрацией 15,00 мг/мл. К аликвотной порции объемом 5,00 мл стандартного раствора добавляют 2 мл раствора дифенилкарбазида и разбавляют до 50,00 мл 0,1 М раствором H2SO4. При измерении оптической плотности обоих растворов на спектрофотометре найдено: Лстанд=0,354; Лнеизв = 0,272. Каково содержание хрома в горной породе (в процентах Сг20з)?
Поглощение излучения. УФ- и видимая области 93
3-7. Используя данные рис. 3-32 и соответствующей таблицы, составьте систему уравнений для определения Mo, Ti и V в смеси. Анализируемый раствор обрабатывали избытком пероксида водорода и хлориой кислоты и разбавляли до 50,00 мл. Были получены следующие значения оптической плотности:
Длина волны, нм 330 410 460
Оптическая плотность А 0,284 0,857 0,718
Рассчитайте количество миллиграммов всех трех элементов в анализируемом растворе. Толщина слоя при всех измерениях была одинакова.
3-8. Оптическая плотность раствора кофеина C8Hio02N4-H20 (мол. масса 212,1) с концентрацией 1,000 мг в 100 мл при 272 нм равна 0,510. Навеску растворимого кофе массой 2,500 растворяли в воде и разбавляли до 500 мл; аликвотную порцию объемом 25 мл переносили в колбу, содержащую 25 мл 0,1 М H2SO4. Пробу осветляли стандартными приемами и разбавляли до 500 мл. Оптическая плотность этого раствора оказалась равной 0,415 при 272 нм. а) Рассчитайте молярный коэффициент поглощения, б) Рассчитайте количество граммов кофеина в фунте (453,6 г) растворимого кофе. Пусть 6 = = 1,00 см.
3-9. По данным Уэттерса и Аглема [44], молярный коэффициент поглощения нитрит-иона при 355 им равен 23,3, а отношение коэффициентов поглощения при 355 и 302 им составляет 2,50. Молярный коэффициент поглощения иитрат-иона при 355 нм пренебрежимо мал, а при 302 нм равен 7,24. Для смеси ионов получены значения: А3о2 = 1,010 и As55=0,730. Рассчитайте молярную концентрацию каждого иона в смеси. Пусть 6=1,00 см.
3-10. В работе [45] приведены следующие данные: a) As и Sb в степени окисления 3+ можно окислить до степени окисления 5-i- бромом, причем As(III) окисляется легче Sb(III). б) В отличие от Sb(V), As(IH) и As(V),Sb(III) образует с хлорид-ионом (в 6 М НС1) комплекс, поглощающий в УФ-области при 326 нм. в) При добавлении к кислому раствору смеси КВгО3 и КВг (вполне устойчивой в водном растворе) количественно выделяется Вг2 по уравнению ВгО3_ + 5 Вг_ + 6 Н+ = 3 Вг2+ЗН2О. г) Свободный бром в избытке Вг~ интенсивно поглощает в УФ-области вблизи 326 нм, хотя максимум поглощения находится в более коротковолновой части спектра. Сам бромид-ион не поглощает в этих условиях. Руководствуясь этими сведениями, покажите, как определить трехвалентные As и Sb при совместном их присутствии в рас-створе НС1, используя смесь КВгО3 и КВг в качестве титранта.
Указание. Оптическую плотность раствора следует измерять после добавления каждой порции титранта. На кривой должны быть два перегиба, соответствующие обоим определяемым элементам.
3-11. Воду определяют спектрофотометрическим титрованием в растворе безводных уксусной и серной кислот [46]. Титрантом служит уксусный ангидрид (АсО)2О. В ходе титрования измеряют поглощение титранта при 257 нм. Проведите схематически кривую титрования и объясните ее форму.
3-12. Описан метод [47] спектрофотометрического определения примеси хлората в перхлорате аммония, используемом в ракетной технике. Он основан на восстановлении хлората до свободного хлора: С1О3_ + 5 С1_+6 Н+ = 3 С12 + +3 Н2О. Хлор затем реагирует с бензидином (I), образуя окрашенный продукт (II) с максимумом поглощения при 438 нм:
+ с1г ----* ^>=/ + 2СГ
(1) 01)
94 Глава 3
Предварительные исследования раствора КС1О3 в описанных выше условиях позволили установить следующую линейную зависимость:
А = (1,17-10s) с —0,186
где с — молярная концентрация КС1О3. а) Объясните, почему продукт II окрашен, а продукт I не имеет окраски, б) Как окрашен раствор соединения II и какого цвета светофильтр подошел бы для его определения? в) Подчиняется ли система закону Бера? г) Член — 0,186 следует приписать наличию примесей восстановителей в реагентах. Объясните, почему он должен иметь отрицательный знак, д) Навеску продажного препарата NH4CIO4 массой 6,000 г обработали по методике, добавили реагент и разбавили до 100,0 мл. Оптическая плотность раствора при 438 нм в 10-мм кювете оказалась равной 0,450. Рассчитайте концентрацию NH4CIO3 в NH4CIO4 (в молярных процентах).
3-13. При добавлении к водному раствору фенола NaOH до щелочной реакции максимум поглощения смещается с 269 до 286 нм. Коэффициент поглощения при этом увеличивается. По данным работы [48], этот сдвиг можно использовать для определения фенола в природных водах с чувствительностью до десятых долей на миллиард, а) Исходя из структуры молекулы, объясните смещение максимума в длинноволновую область и повышение коэффициента поглощения, б) Изучив литературный источник, на который сделана ссылка, объясните, каким образом можно получить отношение оптических плотностей при двух длинах волн, в) Каким образом авторы учитывают различие в спектральных характеристиках разных фенолов?
3-14. Найдено, что в 12-динодном фотоумножителе возникает анодный ток силой в среднем 0,92 мА в виде импульса с периодом 4,2 нс от каждого введенного фотоэлектрона. Рассчитайте фактор умножения динода.
Литература
1. Judd D. В., In Analytical Absorption Spectroscopy (Mellon M. G., ed.), Wiley, New York, 1950, chap. 9.
2. Houle M. I., Grossaint K., Anal. Chem., 1966, v. 38, p. 768.
3. Malkin S„ Cahen D., Anal. Chem., 1981, v. 53, p. 1426.
4. Kortwn G., Seiler M., Angew. Chem., 1939, v. 52, p. 687.
5. Cook R. B., Jankow R., J. Chem. Educ., 1972, v. 49, p. 405.
6. Beckman A. 0., Gallaway W. S., Kaye W., Ulrich W. F., Anal. Chem., 1977, v. 49, p. 280A.
7. Cowles J. C., J. Opt. Soc. Am., 1965, v. 55, p. 690.
8. Chance B., Rev. Sci. Instrum., 1951, v. 22, p. 634; Science, 1954, v. 120, p. 767.
9. Porro T. J., Anal. Chem., 1972, v. 44, No. 4, p. 93A.
10. Ratzlatf K. L., Naiusch D. F. S„ Anal. Chem., 1979, v. 51, p. 1209.
11. O'Haver T. C., Anal. Chem., 1979, v. 51, p. 91A.
12. Fix G. L., Pollack J. D., Anal. Chem., 1980, v. 52, p. 1589.
13. Olson E. C„ Alway C. D., Anal. Chem., 1960, v. 32, p. 370.
14. RCA Photomultiplier Handbook, RCA Corporation, Lancaster, PA., 1980.
15. Ewing G. W., Transducers. In Treatise on Analytical Chemistry, 2d ed., Kolfhoff I; M., Elving P. J. (eds.), Wiley-Interscience, New York, part I, v. 4, chap. 5 (in press).
16. Vassos В. H., Ewing G. W., Analog and Digital Electronics for Scientists, 2d ed., Wiley-Interscience, New York, 1980, pp. 19ff.
17. Ringbom A., Z. anal. Chem., 1939, v. 115, p. 332.
18. Ayres G. H., Anal. Chem., 1949, v. 21, p. 652.
19. Ingle I. D., Crouch S. R., Anal, Chem., 1972, v. 44, p. 785.
Поглощение излучения. УФ- и видимая области 5§
20. Ingle J. D., Anal. Chem., 1973, v. 45, p. 861.
21. Silverstein R. M., Bassler G. C., Morrill T. C., Spectrometric Identification of Organic Compounds, 4th ed., Wiley, New York, 1981, p. 322.
22. Woodward R. B., J. Am. Chem. Soc., 1941, v. 63, p. 1123; 1942, v. 64, p. 72, 76.
23. Физер Л., Физер M. Стероиды.— M.: Мир, 1964.
24. Turner A., Osol A., J. Am. Pharm. Assoc., Sci. Ed., 1949, v. 38, p. 158.
25. Fagel J. E., Ewing G. W., J. Am. Chem. Soc., 1951, v. 73, p. 4360.
26; Archibald R. M., Chem. Eng. News, 1952, v. 30, p. 4474.
27. Yoe I. H., /ones A. L., Ind. Eng. Chem., Anal. Ed., 1944, v. 16, p. 111.
28. Vosburgh W. C„ Cooper G. R., J. Am. Chem Soc., 1941, v. 63, p. 437.
29. Gerlach J. L., Frazier R. G., Anal. Chem., 1958, v. 30, p. 1142.
30. McCarthy P., Anal. Chem., 1978, v. 50, p. 2165.
31. Momoki K., Sekino J., Sato H-, Yamaguchi N., Anal. Chem., 1969, v. 41, p. 1286.
32. Likussar W., Boltz D. F., Anal. Chem., 1971, v. 43, p. 1265, 1273.
33. Lingane P. I., Hugus Z. Z., Inorg. Chem., 1970, v. 9, p. 757.
34. Renzetti N. A., Anal. Chem., 1957, v. 29, p. 869.
35. Сендэл E. Колориметрическое определение следов металлов.— М.: Мир, 1964.
36. Kolthoff I. М„ Sandell Е. В., Meehan Е. J., Bruckenstein S., Quantitative Chemical Analysis, 4th ed., Macmillan, New York, 1969, pp. 351, 1064.
37. Tsubouchi M., Anal. Chem., 1970, v. 42, p. 1087.
38. Huber W„ Ewing G. W., Rriger I., J. Am. Chem. Soc., 1945, v. 67, p. 609.
39. Connors K. A., Eboka С. J., Anal. Chem., 1979, v. 51, p. 1262.
40. Weissler A., Ind. End. Chem., Anal. Ed., 1945, v. 17, p. 695.
41. Goddu R. F., Hume D. N., Anal. Chem., 1954, v. 26, p. 1679, 1740.
42. Underwood A. L., In Advances in Analytical Chemistry and Instrumentation, Reilley C. N. (ed.), Wiley-Interscience, New York, 1964, v. 3, pp. 31ff.
43. Underwood A. L., Anal. Chem., 1954, v. 26, p. 1322.
44. Wetters J. H., Uglam K. L., Anal. Chem., 1970, v. 42, p. 335.
45. Sweetser P. B., Bricker С. E. Anal. Chem., 1952, v. 24, p. 1107.
46. Bruckenstein S., Anal. Chem., 1959, v. 31, p. 1757.
47. Burns E. A., Anal. Chem., 1960, v. 32, p. 1800.
48 Fountaine J. E., Joshipura P. B., Keliher P. N., Johnson J. D., Anal. Chem., 1974, v. 46, p. 62.
ГЛАВА
Поглощение излучения. Инфракрасная область
Если измерение поглощения в ультрафиолетовой и видимой областях целесообразно рассматривать вместе, то измерение поглощения в инфракрасной области лучше выделить в самостоятельный раздел. Это обусловлено двумя важными причинами: во-первых, для измерений в ИК-области требуется совсем иная оптическая техника, поэтому не существует спектрофотометров, которые без изменений были бы пригодны для работы как в ИК-, так и в УФ- и видимой областях; во вторых, механизмы поглощения ИК- и более коротковолнового излучения различны.
В гл. 2 указывалось, что поглощение ИК-излучения связано с увеличением колебательной и вращательной энергий ковалентной связи, если оно приводит к изменению дипольного момента молекулы. Это значит, что почти все молекулы с ковалентными связями в той или иной мере способны к поглощению в ИК-области. Исключение составляют двухатомные молекулы Н2, N2 и О2; только в них не обнаружены колебания или вращения, которые могли бы привести к появлению дипольного момента. Но даже эти простые молекулы при высоком давлении проявляют небольшое поглощение в ИК-области, очевидно, в результате деформации при столкновениях.
Инфракрасные спектры многоатомных ковалентных соединений обычно очень сложны: они состоят из множества узких полос поглощения (см., например, спектр пленки полистирола на рис. 4-1). Они сильно отличаются от обычных УФ- и видимых спектров. Различия вытекают из природы взаимодействий поглощающих молекул и их окружения. Эти взаимодействия (в конденсированных фазах) влияют на электронные переходы в хромофоре, поэтому линии поглощения уширяются и стремятся слиться в широкие полосы поглощения. В ИК-спектре, наоборот, частота и коэффициент поглощения, соответствующие отдельной связи, обычно мало меняются с изменением окружения (в том числе с изменением остальных частей молекулы). Линии расширяются, но не настолько, чтобы слиться в полосу.
Поглощение излучения. ИК-область 97
100
4 Заказ № S3?
98 Глава 4
Рис. 4-2. Инфракрасные спектры стеариновой кислоты в разных физических состояниях: а — в растворах CCU (штриховая кривая) и CS2 (сплошная кривая); б — в пленке при комнатной температуре; в—-то же при —196°C [1].
Из этого правила иногда бывают исключения. Например, для длинноцепочечной молекулы в жидкой фазе можно представить неограниченное число конфигураций, поскольку возможно свободное вращение вокруг множества С—С-связей. Все эти
Поглощение излучения. ИК-область 99
формы имеют близкие, но не вполне идентичные спектры, поэтому будет наблюдаться уширение полос. Следовательно, соединения такого типа лучше исследовать в твердом состоянии. На рис. 4-2 показано, как изменяется характер спектра такого соединения (стеариновой кислоты) в различных условиях. Обратите внимание на то, что в спектре раствора по сравнению со спектром твердого вещества при комнатной температуре произошли уширение максимумов и сглаживание тонкой структуры, а при резком понижении температуры наблюдается противоположный эффект.
Обычно по оси ординат при построении ИК-спектров откладывают пропускание в процентах (как это сделано на рис. 4-1), а не оптическую плотность *. При таком способе построения полосы поглощения выглядят как впадины на кривой, а не как максимумы на УФ-спектрах. Однако такое представление ИК-спектров не является универсальным, и на рис. 4-2 представлены спектры другого вида. По оси абсцисс в ИК-спектрах иногда откладывают длину волны в микрометрах (прежнее название «микроны»), а иногда волновое число в обратных сантиметрах. Многие исследователи предпочитают волновое число, потому что так легче провести корреляцию с колебаниями в молекуле. Напротив, некоторые ученые выбирают для представления спектра линейную шкалу длин волн, потому что выгодно растянуть богатую линиями область (примерно от 5 до 15 мкм), которая служит «отпечатком пальцев», хотя при этом сжимается коротковолновая часть. Оба способа представления спектров сравниваются на рис. 4-1.
Приборы
Большинство спектрофотометров для ИК-области сконструированы так же, как и приборы для более коротковолновой области, но оптическая часть этих приборов изготовлена из других материалов. Кроме того, существует еще один тип приборов (не нашедший широкого применения в УФ- и видимой областях), который основан на интерферометрии.
Оптические материалы. В ИК-спектрофотометрах почти никогда не применяют линзы, их заменяют зеркала с вогнутой поверхностью. Зеркала имеют много преимуществ, поэтому их часто используют не только в ИК-области, но в УФ- и видимой областях спектра. Они не дают хроматической аберрации, т. е.
* Это объясняется инструментальными особенностями первых моделей ИК-спектрофотометров, на которых получены спектры, составляющие банк спектров.
4*
100 Глава 4
Таблица 4-1. Твердые материалы, прозрачные в ИК-области
Материал Интервал длин волна\ мкм Материал Интервал длин волиа\ мкм
Оптическое стекло 0,4—2,6 Ge 1,8—23
Стекловидный NaCl 0,2—25
кварц 0,16—4,0 КВг 0,25—40
LiF 0,12—9,0 CsBr 0,2—55
CaFa 0,13—12,0 КРС-5В 0,55—40
Si6 1,2-15
а) Заимствовано из более подробных таблиц Баркера [2]. Указаны интервалы, в которых пропускание больше 10% при толщине слоя 2 мм.
' При 9 мкм наблюдается минимум пропускания вследствие образования на поверхности связей Si—О.
в) Смешанные кристаллы бромида и иодида таллия.
фокусируют все длины волн в одну точку; их можно изготовить из прочных материалов, например металла или стекла с алюминиевым покрытием, не заботясь о прозрачности; их легче монтировать.
Все материалы, через которые должно проходить излучение, например окна сосудов для проб *, должны быть твердыми и свободно пропускать в нужной полосе длин волн. Некоторые материалы с указанием используемого интервала пропускания приведены в табл. 4-1. Следует заметить, что некоторые из них являются водорастворимыми солями. Это значит, что детали из этих материалов нужно тщательно предохранять от воздействия влаги. Очевидно, что в кюветы со стенками из солей нельзя помещать водные растворы, но на практике это не имеет большого значения, поскольку вода сильно поглощает в ИК-области и не может служить растворителем. Кюветы, сделанные из солей, следует хранить в эксикаторе.
Во многих старых спектрофотометрах дисперсионные призмы изготовлены из NaCl или КВг. В этих приборах монохроматор обычно изолирован от влаги воздуха, а для предотвращения конденсации на его поверхности около призмы помещен нагреватель.
Источники. В ИК-области, представляющей наибольший интерес для аналитической химии, вполне пригодны источники непрерывного излучения. Для ближней ИК-области используется лампа накаливания с вольфрамовой нитью в стеклянном
* В ИК-спектроскопии сосуд для жидких и газообразных проб иногда называют «абсорбционной ячейкой», а не кюветой.
Поглощение излучения. ИК-область 101
или кварцевом баллоне, но при длинах волн выше 3,5 мкм стекло интенсивно поглощает. В интервале от 1 до 30 мкм лучшим источником, пожалуй, является штифт Нернста. Это — стержень или полая трубка около 2 см в длину и 1 мм в диаметре, изготовленные из смеси оксидов таких элементов, как церий, цирконий, торий и иттрий. Штифт устойчив при высокой температуре и не подвержен окислению, поэтому с ним можно работать на воздухе. Он имеет отрицательный температурный коэффициент сопротивления, поэтому, прежде чем пропускать через него ток, его нужно нагреть до собственной температуры. Эта температура лимитирует ток: при очень высокой температуре штифт может перегореть.
Еще один источник, глобар, представляет собой стержень из карбида кремния несколько больших размеров, чем штифт Нернста. Во избежание окисления его следует эксплуатировать при более низких температурах, а концы необходимо охлаждать путем сильной вентиляции *. При длинах волн выше 30 мкм относительная эмиссионная способность глобара больше, чем у штифта Нернста.
Источником ИК-излучения может служить просто спираль из нихромовой проволоки, однако область излучения и интенсивность его в этом случае ограничены. Часто выбор источника определяется стоимостью спектрофотометра.
Детекторы. Детекторы инфракрасного излучения, используемые в абсорбционной спектроскопии [3], можно разбить на две большие группы: 1) так называемые термические детекторы, действие которых основано на измерении тепловых эффектов, возникающих под действием суммарной энергии большого числа падающих фотонов, и 2) фотонные детекторы, полупроводниковые устройства, в которых электрон может поглотить квант ИК-излучения и перейти из валентной зоны в зону проводимости, внося свой вклад в электропроводность. В целом фотонные детекторы обладают быстрой реакцией и более чувствительны, однако интервал длин волн их ограничен, и, кроме того, они действуют при температуре жидкого азота или ниже. Термические детекторы, напротив, применимы в широком, интервале длин волн и не требуют охлаждения, но они инерционны и относительно мало чувствительны. За исключением детектора с внутренним фотоэффектом из PbS, который широко применяется в ближней ИК-области при комнатной температуре, фотонные детекторы редко используются в лабораторных спектрофотометрах и далее не обсуждаются.
* В ряде приборов используется водяное охлаждение.— Прим, перса.
102
Глава 4
Рис. 4-3. Дифференциальная термопара детектора ИК-излучения.
Термические детекторы дают почти одинаковый отклик на любой вид энергии в ближней, УФ-, видимой и ИК-об-ластях (вплоть до миллиметровых длин волн). Их почти не используют в УФ- и видимой областях, поскольку фотоумножители более чувствительны.
Обычно термические детекторы изготавливают из очень маленького кусочка зачерненной золотой фольги, которая и служит поглотителем излучения. В одной из распространенных конструкций (рис. 4-3) два одинаковых кусочка фольги (один из них зачерненный) помещают в разреженное пространство так, чтобы
излучение, которое надо измерить, попадало только на зачерненную поверхность. Таким образом, изменения температуры окружения в равной степени влияют на оба кусочка фольги, но излучение влияет лишь на один из них. Измерительное устройство должно состоять из двух одинаковых приемников, связанных с обоими кусочками фольги и с электрической цепью, регистрирующей разницу
в сигналах.
Одним из наиболее распространенйых термических детекторов ИК-излучения является термопара. На обратную сторону кусочков фольги приваривают стыки проволочек из двух разных металлов (рис. 4-3). Термопары обладают низким сопротивлением и обычно используются в сочетании с трансформатором с большим числом витков (1000:1) и усилителем, реагирующим на низкочастотный переменный ток, возникающий при действии модулированного излучения.
Другим видом термического детектора является болометр — миниатюрный термометр сопротивления с крошечным кусочком платиновой проволоки или термистором в качестве чувствительного элемента. Термистор представляет собой сопротивление, изготовленное путем спекания нескольких оксидов металлов; он обладает в пять раз большим, чем у платины, температурным коэффициентом и быстрее реагирует на модулированное излучение, но его воспроизводимость сигнала несколько меньше. Одинаковые платиновые проволочки или термисторы прикрепляются К задней стороне кусочков золотой фольги, как это описано выше.
Сопротивление болометра можно измерить с помощью подходящей электросхемы, например мостика Уинстона (рис. 4-4). Напряжение на выходе пропорционально отношению сопротив-
Поглощение излучения. ИК-область 103
Рис. 4-4. Схема мостика Уиистоиа, используемая в болометре. R* — облучаемый элемент, Rs — такой же элемент при той же внешней температуре, но защищенный от излучения. Два других сопротивления служат для установки иа нуль.
лений двух одинаковых элементов, и тем самым влияние окружающей температуры, по существу, исключается.
Пироэлектрические детекторы [4] отличаются тем, что реагируют не на саму температуру, а на изменение ее во времени, и поэтому не нуждаются в дублирующей системе, защищенной от' измеряемого излучения. Детектор представляет собой очень маленькую пластинку из кристаллического вещества, молекулы которого обладают постоянным дипольным моментом. Поглощение тепла вызывает изменение кристаллической решетки и, следовательно, дипольного момента, что приводит к изменению электрического заряда, которое фиксируется с помощью электрода из металлической фольги на противоположной грани кристалла [5, 6]. Из кристаллических веществ чаще всего используются сульфат триглицина, титанат бария, цирконат свинца и танталат лития. Скорость отклика у этих кристаллов гораздо больше, чем у термических детекторов, поэтому их можно использовать для регистрации излучения, прерываемого с частотой порядка 105 Гц, тогда как максимальная частота модулирования при использовании термопар или болометров составляет 15—20 Гц.
Спектрофотометры
Можно выделить два основных класса автоматических сканирующих спектрофотометров для средней ИК-области: недорогие модели для серийных работ по идентификации и универсальные приборы для научных исследований. Последние могут быть
104 Глава 4
Рнс. 4-5. Устройство ИК-спектрофотометра: а —оптическая схема; б — деталь зеркала, служащего для обоих потоков (на оптической схеме обозиачеиа С); в — деталь оптического аттенюатора (Д) (Beckman Instruments, Inc.).
снабжены разными приспособлениями, которые обеспечивают более высокую точность, лучшее разрешение и большие возможности для представления спектра. В основе всех этих приборов лежит двухлучевая схема.
Подавляющее большинство ИК-спектрофотометров независимо от стоимости и места изготовления имеют почти идентичную оптическую схему. Они различаются по допускаемой точности многих оптических и механических деталей, что отражается прежде всего на точности настройки на длину волны, точности фотометрических измерений и разрешающей силе. (Более подробное обсуждение разрешающей силы, ее значения и способов оценки содержится в обзоре Стьюарта [7]).
Типичная оптическая система ИК-спектрофотометра представлена на рис. 4-5, а. Излучение источника N отражается двумя наборами зеркал в виде симметричных потоков, один из которых проходит через кювету с пробой, другой — через кювету с холостым раствором или стандартом. Оба потока далее отражаются системой зеркал и фокусируются на щель Sb Приводимый в движение электромотором прерыватель в виде
Поглощение излучения. ИК-область 105
круга С, одна половина которого отражает излучение, а другая вырезана (рис. 4-5,6), попеременно направляет оба потока на щель. Щель Si — это входная щель монохроматора Литтрова, включающего пару приставленных друг к другу дифракционных решеток, охватывающих разные отрезки интервала длин волн. Выделенная полоса выходит через щель S2 и фокусируется на детектор D.
Детектор воспринимает прямоугольный импульс, соответствующий по амплитуде разности интенсивностей двух потоков. Этот переменный сигнал после усиления приводит в движение сервомотор, с помощью которого регулируется положение аттенюатора, имеющего вид гребенки (рис. 4-5,в). Аттенюатор уравнивает интенсивность потоков от рабочей кюветы сравнения, т. е. обеспечивает оптический нуль. Одновременно мотор управляет пером самописца.
Две дифракционные решетки обеспечивают необходимое разрешение во всем интервале длин волн. Первая решетка обычно имеет 300 штрихов на миллиметр и самый яркий максимум дифракции при 3 мкм. При работе она медленно вращается, в то время как спектрофотометр сканирует длины волн в интервале от 2 до 5 мкм. Как только достигается отметка 5 мкм, столик с решеткой резко поворачивается, в действие вводится вторая дифракционная решетка (100 штрихов на миллиметр, дифракционный максимум при 7,5 мкм), и сканирование продолжается в интервале от 5 до 16 мкм. Обе решетки работают в пределах спектра первого порядка. Для устранения излучения более высоких порядков устанавливают четыре светофильтра F, которые вводятся последовательно друг за другом в нужном интервале длин волн с помощью приводного механизма.
Спектры, полученные с помощью призмы или дифракционной решетки, могут быть линейными либо в координатах длины волны, либо в координатах волнового числа в зависимости от конструкции кулачкового устройства, осуществляющего механическую связь между сканирующим мотором и столиком, на котором помещается призма или решетка. Наиболее простым по конструкции является прибор для получения спектра, линейного относительно длины волны, с помощью дифракционной решетки, но, с точки зрения оператора, никакой разницы нет.
Быстросканирующие спектрофотометры
Одним из недостатков обычных ИК-спектрофотометров с термопарой или болометром в качестве детектора является присущая им замедленность сигнала. Для кинетических исследований и непрерывной идентификации компонентов потока, например
106 Глава 4
элюата, выходящего с колонки газового хроматографа, необходимы приборы с высокой скоростью отклика, которая достигается благодаря применению пироэлектрического детектора. Прибор со сканированием длины волны можно сконструировать так, чтобы элюат протекал непосредственно через рабочую кювету, а чистый носитель — через кювету сравнения. Спектр на выходе можно получить за несколько секунд, т. е. за время меньшее, чем обычно нужно для элюирования компонента. Более подробно области применения этих приборов обсуждаются в гл. 20, а сравнительные возможности в жидкостной хроматографии — в гл. 21.
Интерференционные (с фурье-преобразованием) сп ектро фотом етр ы
Излучение представляет собой последовательность электромагнитных волн с разными частотами, поэтому в каждый момент общая картина складывается из наложения множества электрических и магнитных полей, создаваемых излучениями с индивидуальными частотами. Следовательно, существует принципиальная возможность воспроизвести всю заключенную в потоке информацию, если направить его на детектор и фиксировать сигнал во времени. На пути такого прямого подхода имеется трудность, заключающаяся в том, что частота переменных полей (около 1014 Гц) на много порядков выше частоты, которую могут зафиксировать известные детекторы.
Затруднения, связанные со скоростью, можно преодолеть с помощью интерферометра Майкельсона, изображенного на рис. 4-6. Излучение источника собирается линзой 'U в параллельный пучок и затем делится на два одинаковых потока расщепителем излучения. Потоки отражаются обратно плоскими зеркалами М2 к Mit и, наконец, часть излучения потоков попадает на детектор. Если оба зеркала удалены от расщепителя излучения на равные расстояния, детектор воспринимает одинаково изменяющиеся во времени электромагнитные поля так же, как оии воспринимались бы без интерферометра, только с вдвое меньшей интенсивностью. Если теперь передвинуть зеркало М2 вдоль оптической оси вправо, оба потока достигнут детектора в разных фазах, в результате чего будет наблюдаться-интерференция. Сдвиг по фазе при данном увеличении длины пути зависит от длины волны излучения и воспринимается детектором как серия последовательных максимумов и минимумов интенсивности.
Чтобы использовать такую систему в спектрофотометрах, зеркало М2 передвигают с постоянной скоростью на расстояние р несколько миллиметров (что во много раз больше мдксч-
Поглощение излучения. ИК-область 107
Рис. 4-6. Интерферометр Майкельсона, используемый в ИК-спектрометре с фурье-преобразованием.
мально возможной длины волны). Это вызывает флуктуации в сигнале детектора, скорость которых зависит от скорости движения и длины волны излучения. Действие интерферометра эквивалентно действию прерывателя с частотой, равной 2yv, где v — скорость движения зеркала (в см/с), a v —волновое число излучения (в см-1)- В обычных приборах это соответствует модулированию с частотой, непрерывно меняющейся от почти 1250 Гц, при высоких волновых числах спектра (4000 см-1) до 125 Гц при низких значениях (400 см-1). Пироэлектрические детекторы легко улавливают эти частоты.
В идеале, если оптические пути двух потоков не равны, монохроматическое излучение должно иметь форму косинусоиды (представление в форме косинусоиды, а не синусоиды предпочтительнее, потому что здесь при нулевой разнице длин путей амплитуда максимальна). На рис. 4-7,а и б изображены два монохроматических потока с разными частотами. Если оба потока попадают в интерферометр одновременно, получается кривая в. Развивая далее этот подход, можно объяснить сложность спектров (рис. 4-8). Следовательно, любая комбинация частот с соответствующими амплитудами дает единственную интерферограмму, содержащую всю спектральную информацию об исходном излучении.
На рис. 4-9 представлена полная оптическая схема спектрофотометра с фурье-преобразованием. Поток от источника ИК-излучения (вверху слева) собирается вогнутым зеркалом и попадает в интерферометр. Пульсирующий поток из интерферометра направляется на колеблющееся зеркало, расположенное так, чтобы посылать поток попеременно то на кювету с пробой (как показано на схеме), то на кювету сравнения (показано
108 Глава 4
Частота
lilllllllllllllllllllll
в
Частота
Рис. 4-7. Сравнение информации в координатах времени (слева) и частоты (справа): а — косинусоида частот б—косинусоида частот vj; в — обе частоты совмещены.
Рис. 4-8. а — интерферограмма органического соединения и б — спектр того же соединения. Синусоида в верхней части рисунка используется для ка либровки; наклонная линия, обозначенная «развертка», показывает движение зеркала (Spex Industries).
на схеме штриховой линией). С помощью другого зеркала, колеблющегося синхронно с первым, поток фокусируется на детекторе.
Без цифрового компьютера интерферограмму не так легко интерпретировать. С точки зрения математики интерферограмму можно представить интегралом Фурье-, таким образом, в задачу компьютера входит провести обратное преобразование
Поглощение излучения. ИК-область 109
Рис. 4-9. Оптическая схема ИК-спектрофотометра с фурье-преобразоваиием (Digilab Inc.).
Фурье. Вывод и применение соответствующих уравнений можно найти в литературе [8, 9].
Интерференционная, или фурье-спектроскопия, обладает по сравнению с обычно используемыми методами двумя достоинствами. Во-первых, она позволяет использовать все частоты излучения источника одновременно, а не последовательно, как в сканирующих приборах (выигрыш Фелжетта, названный так по имени ученого, впервые описавшего его). Выигрыш Фелжетта обусловлен улучшением соотношения S/N, равным М*'2, где М — число элементов спектра, которые желательно разрешить [8, 9]. Во-вторых, чувствительность фурье-спектроскопии выше, чем у дисперсионных методов, потому что в бесщелевую систему попадает больше излучения; это преимущество называется выигрышем Жакино.
В литературе описаны многочисленные примеры химических исследований, когда преимущества приборов с фурье-преобра-зованием по сравнению с дисперсионными приборами очевидны [8, 10]. В большинстве исследований требовалась высокая чув
110 Глава 4
ствительность: это — исследования, связанные с измерением очень малых потоков излучения, например кинетики быстрых процессов, когда спектр должен быть зарегистрирован быстро, а в противном случае он вообще не нужен. Другим примером использования низкоэнергетической спектрометрии является измерение поглощения в дальней ИД-области (где источники очень слабы) и в эмиссионной ИД-спектроскопии. Например, с помощью ИК-спектроскопии с фурье-преобразованием можно регистрировать испускание горячих газов дымовых труб [И]. Метод также используется в астрономии.
Упрощенные спектрофотометры. Приборы с низким разрешением. Полезные измерения можно выполнить на ручном однолучевом ИК-фотометре, напоминающем фотоэлектроколориметр для видимой области. В целой серии таких приборов (модели Miran *) для выделения длины волны применяются интерференционные светофильтры, которые дают меньшее разрешение, чем дифракционные решетки, но вполне достаточное для многих целей. Эти приборы имеют высокую чувствительность, так как в них используется больший пучок излучения, чем в обычных спектрофотометрах (выигрыш Жакино). Они недороги, надежны и портативны, и их можно использовать для определения соединения или класса соединений, но эти приборы непригодны для идентификации родственных веществ с близкой структурой.
Бездисперсионные фотометры с селективными светофильтрами находят широкое применение для анализа газовых потоков и контроля за загрязнением воздуха. В качестве примера рассмотрим определение СО в присутствии других газов. Поскольку СО поглощает излучение только определенных характеристичных частот, лишь эти частоты и представляют интерес при измерении. Устранить помехи со стороны других частот можно двумя путями (рис. 4-10). На рис. 4-10,а потоки излучения от двух одинаковых источников проходят через кювету с пробой и кювету сравнения и попадают на дифференциальный детектор, который содержит СО (обычно разбавленный аргоном для уменьшения теплоемкости) [12]. Любое различие в интенсивности двух потоков сказывается на разности температур в камерах детектора. Этот сигнал отличается высокой селективностью, потому что вызывать нагревание может только то излучение, которое поглощается СО. Для уменьшения чувствительности прибора к излучению других компонентов газового потока, полосы поглощения которых перекрываются с полосами поглощения СО, служат дополнительные фильтры. Ка-
* Foxboro Analytical, Южный Норфолк, Коннектикут.
Поглощение излучения. ИК-область 111
Источника
Принтеры
Светофильтры
К усилителю
Прерыватель
| j Источник
Сосуд Кювета
с СО с пробой
___К усилителю
Детектор
Рис. 4-10. Безднсперсионные ИК-анализаторы: а — с дифференциальным детектором; б—с источником инфракрасного флуоресцентного излучения.
меры детектора разделены металлизированной гибкой диафрагмой, которая вместе с закрепленной пластинкой составляет конденсатор переменной емкости.
Дифференциальный детектор — это кинетическое устройство, более эффективно реагирующее на быстрые изменения, чем на постепенные, потому что в схеме предусмотрено одновременное прерывание обоих потоков. В высокочастотную электронную цепь, связанную с небольшим сервомотором, включен чувствительный конденсатор, с помощью которого на пути потока сравнения вводят компенсатор для установления оптического нуля. Степень компенсации указывается на шкале или записывается на ленте самописца.
В однолучевом приборе, изображенном на рис. 4-10,6, сосуд, содержащий СО, используется в качестве источника излучения, а не в качестве детектора [13]. Под действием инфракрасного излучения непрерывного источника СО нагревается, а затем испускает излучение с характеристическими частотами, которое пропускают через пробу и направляют в неселективный детектор. Этот прибор менее подвержен влиянию колебаний
112 Глава 4
температуры, чем описанная выше модель. И тот и другой прибор можно сделать чувствительным к любому многоатомному газу.
Спектрофотометры для дальней инфракрасной области. Дисперсионные приборы для области длин волн выше 50 мкм гораздо сложнее, чем для коротковолновой области. Удобным источником дальнего инфракрасного излучения является дуговая ртутная лампа высокого давления в кварцевом баллоне. Кварц непрозрачен при длинах волн ниже 60 мкм, но рабочий интервал тем не менее начинается с более коротких длин волн благодаря излучению, испускаемому раскаленным кварцем. В этой области применяются пироэлектрические детекторы. Трудность работы в дальней ИК-области связана с поглощением компонентов атмосферы, поэтому необходимо эвакуировать спектрофотометр или продуть его сухим азотом.
Чтобы охватить весь интервал длин волн, необходим набор сменных дифракционных решеток; для получения излучения с самой большой длиной волны используется решетка всего лишь с 2—3 штрихами на миллиметр. Чтобы отсечь фоновое излучение и излучение ненужных порядков, необходимы светофильтры. Для дальнего ИК-излучения существуют два типа светофильтров, в которых используется селективное отражение излучения (а не пропускание). Действие одного из них основано на явлении остаточного излучения, т. е. узкие спектральные полосы с высоким отражением в кристаллических веществах соответствуют максимальному показателю преломления, связанному с областями высокого поглощения [14, 15]. Светофильтр другого типа представляет собой рассеивающую пластинку, которой может быть пленка металла, нанесенная на стеклянную подложку, или дифракционная решетка со штрихами, нанесенными не параллельно, а перпендикулярно вертикали щелей. Рассеяние — очень эффективный прием уменьшения числа волн с более высокими частотами, проходящих через оптическую систему, но не затрагивающий более длинные волны.
В дальней ИК-области особенно важно прерывать излучение, прежде чем оно попадет на пробу и монохроматор, потому что тепловое излучение элементов оптической системы может быть отнюдь не слабым. Прерывание в сочетании с правильной настройкой усилителя обеспечивает. получение сигнала детектора только от источника.
В дальней ИК-области спектрометры с фурье-преобразованием играют особенно важную роль. Стоимость их изготовления для работы в этой области ниже, чем при работе в других областях, потому что требования к механической системе менее жесткие,
Поглощение излучения. ИК-область 113
Калибровка и стандартизация
Калибровка ИК-спектрофотометров как по длинам волн, так и по волновому числу со временем может меняться в результате механического износа, потускнения оптических поверхностей или старения деталей, поэтому целесообразны периодические проверки.
Теоретически шкалу длин волн (или волновых чисел) можно откалибровать по геометрии дифракционной решетки с известным периодом, но на практике этот прием не применяется. Наиболее простая проверка заключается в том, что снимают спектр тонкого слоя полистирола в качестве вторичного стандарта. Обычно ИК-приборы снабжены специально для этой цели подготовленными образцами с приложением стандартного спектра для сравнения. Такой спектр (см. рис. 4-1) имеет ярко выраженные полосы поглощения в средней ИК-области.
Другой удобный способ проверки шкалы длин волн состоит в том, что прибор переключают на однолучевую схему работы и регистрируют спектр в отсутствие пробы. При этом должны быть отчетливо видны полосы поглощения атмосферной влаги и диоксида углерода (рис. 4-11), и поскольку их длины волн точно известны, шкалу легко проверить по многим точкам. (Этот спектр делает очевидным одно из важных преимуществ двухлучевого спектрофотометра — исключение поглощения компонентами атмосферы.)
Если спектрофотометр используется для количественных измерений, время от времени следует проверять линейность шкалы. Грубо это можно сделать, регистрируя спектр полистирола, а более точно—-по пропусканию какого-либо растворителя, например бензола, при разной толщине слоя.
Рис. 4-11. Холостая запись, полученная с помощью однолучевой схемы, на которой видны полосы поглощения атмосферной влаги и диоксида углерода [22].
114 Глава 4
Практическое применение
Качественный анализ. Поглощение ИК-излучения обусловлено ковалентными связями,' поэтому ИК-спектры могут служить источником подробной информации о структуре молекулярных соединений.
Благодаря кропотливому изучению огромного числа спектров известных веществ установлены корреляции между положением максимумов поглощения индивидуального колебания и соответствующими атомными группировками. Такие эмпирические корреляции служат мощным средством идентификации ковалентных соединений.
Сформулируем ряд общих положений. Полезно выделить такие виды колебаний, как валентные (периодическое движение атомов вдоль оси связи) и деформационные, при которых происходит искривление или изгиб связи и т. п. Более коротким длинам волн в интервале от 0,7 до 4,0 мкм отвечают валентные колебания между атомами водорода и более тяжелыми атомами, которые составляют ближнюю ИК-область и особенно полезны при идентификации функциональных групп, содержащих водород. Колебания двойных и тройных связей относятся к области от 4,0 до 6,5 мкм. Более длинные волны связаны с деформациями и изгибами связи в скелете молекулы: сюда относится, например, изгиб связи —С—Н.
Поглощение в дальней ИК-области (свыше 25 мкм) соответствует колебаниям связей с участием тяжелых атомов и групп атомов, в том числе связей между углеродом и фосфором, кремнием или тяжелыми металлами, а также связей между тяжелыми металлами и кислородом и т. п. Здесь проявляются также и низкочастотные деформации, например сжатие четырехчленного кольца и торсионные колебания метильной и других групп, и находится большинство чисто вращательных полос.
В табл. 4-2 приведены области, соответствующие наиболее часто встречающимся типам связей. Существуют более обширные и подробные корреляции с учетом внутримолекулярного окружения каждой связи. Они приводятся в удобной форме в таблицах, составленных Годду [16] для области 1,0—3,1 мкм и Колтупом 1[17] для области 2,5—25 мкм. Бентли [18] составил таблицы, охватывающие область до 33 мкм. Все эти таблицы приведены в учебнике Мейтиса «Handbook of Analytical Chemistry». Многие предприятия, выпускающие ИК-спектрофото-метры, издают таблицы, в которых указаны области поглощения и приведены спектры, полученные на изготовленных ими приборах. Подробное обсуждение таких корреляций можно найти в многочисленных учебниках и монографиях, что облег-
Таблица 4-2. Положение полос колебания в инфракрасной области3
Связь Тип колебаний® Относительная интенсивность® Длина волны, мкм Волновое число, см 1
С—Н Валентные с. 3,0—3,7 2700—3300
С—н Валентные (2v) Ср. 1,6—1,8 5600—6300
с—н Валентные (3v) Сл. 1,1—1,2 8300—9000
с—н Валентные (К) Ср. 2,0—2,4 4200—5000
с—н Плоские деформацион- Ср. — с. 6,8—7,7 1300—1500
ные
с—н Неплоскне деформацион- Сл. 12,0—12,5 800—830
ные
с—н Маятниковые Сл. 11,1—16,7 600—900
О—н Валентные С. 2,7—3,3 3000—3700
О—н Валентные (2v) С. 1,4—1,5 6700—7100
О—н Деформационные Ср. — Сл. 6,9—8,3 1200—1500
N-II Валентные Ср. 2,7—3,3 3000—3700
N—Н Валентные (2v) С. 1,4—1,6 6300—7100
N—Н Валентные (3v) Сл. 1,0—1,1 9000—10000
N—Н Валентные (К) Ср. 1,9—2,1 4800—5300
N— Н Деформационные С - Ср. 6,1—6,7 1500—1700
N—Н Маятниковые С. — Ср. 11,1—14,3 700—900
с—е Валентные Ср. — с. 8,3—12,5 800—1200
с-о » Ср. — с. 7,7—11,1 900—1300
С—N » Ср. — с. 7,7—11,1 900—1300
С=С » ср. 5,9—6,3 1600—1700
С=О с. 5,4—6,1 1600—1900
с=о Валентные (2v) ср. 2,8—3,0 3300—3600
с=о Валентные (3v) Сл. 1,9—2,0 5000—5300
C=N Валентные Ср. — с. 5,9—6,3 1600—1700
С==С » Ср. — Сл: 4,2—4,8 2100—2400
teN » Ср. 4,2—4,8 2100—2400
С—F С. 7,4-10 1000—1350
С—С1 С. 13—14 710—770
С—Вг С. 15—20 500—670
С—I С. 17—21 480—600
Карбонаты С. 6,9—7,1 1400—1450
В Ср. 11,4—11,6 860—880
Сульфаты С. 8,-9—9,3 1080—1120
» Ср. 14,7—16,4 610—680
Нитраты с. 7,2—7,4 1350—1390
Ср. 11,9—12,3 820—840
Фосфаты Сл. 9,0—10,0 .1000—1100
Силикаты 9,0—11,1 900—1100
а) Приведены ориентировочные данные; указываются фундаментальные частоты, за некоторыми исключениями, которые оговариваются; данные собраны из разных литературных источников.
(2v) означает вторую гармонику или первый обертон и т. д., К означает составную частоту.
в) С. — сильная, Ср. — средняя, Сл. — слабая.
116 Глава 4
а
Рис. 4-12. Влияние на ИК-спектры переменных параметров прибора: а — полосы поглощения при 861 и 903 см-1 при разной ширине спектральной щели (указана на спектрах); б — спектр, записанный при разных скоростях развертки; эффективное время полной развертки 8 мин (Д), 11 мин (В), 16 мин (В), 3 ч (Г) (Plenum Press [22]).
чает установление структур органических соединений [16, 19—22].
Во многих случаях на поглощение соединением ИК-излучения в большей или меньшей степени влияют условия наблюдения. Такие изменения в положении полос заставляют относиться с большой осторожностью к данным эмпирических таблиц и атласов при установлении структуры неизвестного соединения. Причины отклонений могут быть инструментальными и химическими. На рис. 4-12 [22] показано влияние изменения
Поглощение излучения. ИК-область 117
Таблцца 4-3. Валентные колебания карбонильной группы
Частота
Карбонильная группа
мкм
см '
Кетон
Карбоновая кислота
Дикарбоновая кислота
Сложный эфир
Соль
5,81—5,85
5,67—5,71
5,81—5,85
5,73—5,80
6,21—6,45а
~7,14°
1720—1710
1765—1750
1720—1710
1745—1725
1610—1550а
~1400б
а) Антисимметричные колебания. Симметричные колебания, ширины щели и скорости развертки; большие сдвиги в спектрах могут помешать правильной идентификации.
Примером воздействия другого типа может служить влияние водородной связи на частоту поглощения карбонильной группы. Частота валентной связи С = О в соединении, растворенном в неполярном растворителе, значительно понижается при образовании водородной связи в присутствии гидроксидсодержащих веществ или в гидроксидсодержащем растворителе. Поглощение карбонильной группы меняется также под действием собственного молекулярного окружения: поглощение связи С = О в карбоновых кислотах, способных образовывать внутримолекулярную водородную связь (образование димера), сильно отличается от поглощения связи С=О в сложном эфире, в котором такая связь образоваться не может. В анионе наблюдается резонанс между двумя эквивалентными атомами кислорода, что усиливает ненасыщенный характер карбонильной связи. В табл. 4-3 приведены характерные значения валентных колебаний карбонильной группы в некоторых алифатических соединениях [20]. Рассуждения о поглощении карбонильной группы служат кратким примером структурных рассмотрений, которые могут иметь большое значение как в органической, так и в аналитической химии.
Сложность ИК-спектров объясняется тем, что не существует двух соединений с одинаковыми спектрами. Следовательно, спектр чистого соединения может надежно служить «отпечатком пальцев» при его идентификации, если есть сводка или атлас спектров известных соединений. Опубликовано несколько таких атласов * [23—25].
* Серия стандартных спектров, характеризующихся особо высокой надежностью, собрана обществом Кобленца. Представляют интерес условия получения спектров и необходимые меры предосторожности, рекомендуемые в их трудах [20]. Спектры Кобленца имеются в продаже [25].
118 Глава 4
Применение в неорганической химии. Спектроскопия в инфракрасной области является ценным методом исследования неорганических соединений с ковалентными связями. Сюда относятся также металлорганические соединения и полианионы. В неорганическом анализе ИК-спектроскопия не нашла широкого применения, потому что существует много более удобных методов. Хороший обзор в этой области дан в [20].
Подготовка проб
Анализ газообразных проб на ИК-спектрофотометре не требует предварительной подготовки (за исключением удаления паров воды). Кюветы для анализа газов во многих приборах снабжены зеркалами, что позволяет многократно пропускать излучение через пробу. Длина пути потока в некоторых имеющихся в продаже приборах достигает 40 м.
Затруднения при анализе связаны с выбором растворителя, потому что не существует жидкостей, не обладающих собственным поглощением. В рассматриваемой области спектра в ка-
Длина волны, мкм
Ацетон_________________________
Ацетонитрил___________________-
Бензол-____.___________________
Бромо^рм(стабилиэи/^аннь1йэи^__
Бутанол------------------------
Тетрахлорид углерода -_________
Хлороформ (стйилизированнь >и__
Пиклмксан спиртом>
т, г-Дихлорэтан----------------
Дихлорметан——................—
N, N-Диметилрормамис)___________
Бталобыи эфир__________________
Гептан, ________________-
Гексан ________________________
Метанол _____,_______
Метилциклогексан ____________.—
Нетилрормиат—________________
1-Метил-2~пиррол1Йаюн
Метилсульфоксид________________
Нитрометан—____________________
Изопропанол.___________________
Пиридин ______________________.
Тетрахлорэтилен (стаёилизи^обанныД 2,2,4-Триметилпентан... -им-ом-—
2 4 6 8 10 12 14 16
Н*
»
S
Я! Ж Ж
к к
№ К
*
м
I ж
о,1 од од од 0,05 0,5 ОД од од од 0,03 од од од 0,03 од 0,03 0,03 0,03 од 0,03 од од ОД
5000 ’ 2500 1500 1200 1000 800 700 «25
ЖЖ
Болновое число, см~}
Рис. 4-13. Области пропускания ИК-излучения некоторых растворителей. Темными прямоугольниками обозначены диапазоны пропускания (Eastman Kodac Company).
Поглощение излучения. ИК-область 119
честве растворителя подходит тетрахлорид углерода, но в нем растворяется ограниченный круг веществ. В некоторых узких областях длин волн можно использовать хлороформ, циклогексан и другие жидкости в очень тонких слоях. На рис. 4-13 в виде таблицы представлены спектральные области, в которых можно применять те или иные растворители. Допустимая степень поглощения растворителем зависит от чувствительности спектрофотометра. Ее влияние можно несколько снизить обычным способом, помещая кювету с холостой пробой на пути потока сравнения, но при этом уменьшается количество энергии, попадающей в детектор, поэтому такой прием находит ограниченное применение.
Иногда удается подобрать два растворителя, области поглощения которых дополняют друг друга, так что два спектра полностью охватывают исследуемый диапазон длин волн. Если позволяет растворимость анализируемого вещества, в качестве такой пары широко применяются СС14 (от 4000 до 1335 см-1) и CS2 (от 1350 до 400 см-1) (см., например, рис. 4-2,а).
Для жидких проб в продаже имеется большой набор кювет разнообразных форм — от недорогих доступных до разборных (для очистки). На некоторых кюветах имеется нарезка для регулирования толщины слоя. Удобны кюветы с полостью, которые изготавливают высверливанием отверстия с параллельными стенками в кристалле соли.
На рис. 4-14 изображена разборная кювета. Она состоит из пары пластинок, изготовленных из какой-либо соли, между которыми помещена металлическая или тефлоновая прокладка; получившийся сэндвич скрепляют металлическими зажимами. В металлической рамке и крае одной из пластинок из соли просверлены два отверстия для заполнения кюветы. Обычно такие кюветы заполняют, опорожняют и промывают при помощи шприца (без иглы), острый конец которого вставляют в отверстие в кювете. Кювету можно использовать многократно, црежде чем появляется необходимость в очистке и полировке пластинок из соли.
Высоковязкие жидкости часто помещают в виде слоя между двумя пластинками из соли, поскольку ввести вязкую жидкость в собранную кювету не так просто.
Для подготовки к анализу твердых проб их запрессовывают в пластинки или таблетки из бромида калия (или реже иодида калия и бромида цезия). Навеску истертой в порошок пробы тщательно смешивают в небольшой шаровой мельнице с навеской безводного порошка КВг высокой степени чистоты. Смесь помещают в вакуумированную форму и прикладывают давление 10—20 МПа. При этом получается очень прозрачная пластинка или диск, которые можно закрепить в специальном
120 Глава 4
Рис. 4-14. Разборная кювета для жидких проб в ИК-сПектрофотометре (Beckman Instruments, Inc.).
держателе спектрофотометра. Обычно такой диск имеет около 1 см в диаметре и до 0,5 мм в толщину. Для количественного анализа необходимо точно знать толщину; ее можно определить либо по размерам формы, либо с помощью кронциркуля.
При работе с таблетками из КВг могут возникнуть осложнения. Если размеры частиц не очень малы, возможно появление интенсивного рассеяния. Некоторые чувствительные к нагреванию вещества (например, стероиды) проявляют признаки частичного разрушения, вероятно, под действием тепла, выделяемого при разрушении кристаллов соли. Кристаллический обзор по технике изготовления таблеток дан в статье [27].
Другой распространенный способ состоит в приготовлении суспензии. Порошок пробы смешивают до образования суспензии с небольшим количеством тяжелого парафинового масла (обычно нуйолом медицинской степени чистоты). Масло имеет лишь несколько изолированных полос поглощения при 3,5, 6,9 и 7,2 мкм. Если эти полосы мешают анализу, суспензию можно приготовить на основе фтеролаба (фторуглерода, не поглощающего при длинах волн короче 7,7 мкм). При измерении суспец-Зщо помещают между пластинками из соли,
Поглощение излучения. ИК-область 121
Твердые пробы можно также исследовать в тонком слое, нанесенном на поверхность соляной пластинки сублимацией или путем выпаривания растворителя. Однако этот простой прием может привести к сильному рассеянию излучения.
Количественный анализ
Закон Бера в том виде, в каком он приведен в гл. 3, приложим и к инфракрасной области:
A = lg-^- = abc (4-1)
В ближней ИК-областн, как правило, работают с 10-мм кюветами и с разбавленными растворами, поэтому никаких особых осложнений при использовании соотношения (4-1) не возникает. За пределами этой области ощущается недостаток в прозрачных для ИК-излучения растворителях, поэтому приходится уменьшать длину оптического пути b и увеличивать концентрацию. При больших концентрациях вследствие межмолекулярных взаимодействий возможны отклонения от закона Бера. Кроме того, точное фотометрическое определение часто трудно выполнить из-за наложения полос поглощения; надо также иметь в виду, что полоса пропускания излучения многими ИК-спектрофотометрами шире измеряемых полос поглощения.
Толщину слоя можно измерить несколькими способами. При работе с кюветой с плоскими параллельными стенками на спектрофотометре записывают интерференционные полосы, появляющиеся в результате многократного внутреннего отражения при пропускании излучения через пустую кювету относительно воздуха [20] (рис. 4-15). Значение b затем можно вычислить по уравнению
b =---(4-2)
2 (vi — v2)
где п — число интерференционных полос между волновыми числами V] И V2-
. Не всегда стенки кювет бывают достаточно гладкими, чтобы получилась четкая картина интерференционных полос. Тогда для определения эффективной или средней толщины нужно измерить оптическую плотность бензола при 850 см-1. Экспериментально показано, что 0,1-мм толщина кюветы соответствует 0,22 единицы оптической плотности.
Длину оптического пути в разборных кюветах можно определить измерением толщины прокладки с помощью кронциркуля с микроделениями.
122 Глава 4
Волновое число
1400 1300 1200 1100 1000 эоо
Рнс. 4-15. Интерференционные полосы, используемые для расчета толщины слоя [20].
Для того чтобы избежать ошибок, связанных с отклонениями от закона Бера при высоких концентрациях, необходимо тщательно проградуировать прибор по серии растворов с известной концентрацией. При экспериментальном определении оптической плотности из-за перекрывания полос поглощения часто приходится использовать метод базисной линии. Пусть нужно найти оптическую плотность, соответствующую полосе, изображенной на рис. 4-16, а. Базисную линию проводят через плечи полосы, затем, измерив значения Ро и Р, как это показано на рисунке, можно вычислить значение оптической плотности. Однако, если кривая поглощения выглядит, как на рис. 4-16,6, провести правильную базисную линию не так легко, так как здесь возможно несколько вариантов. В таких часто встре-
Рис. 4-16. Гипотетический ИК-спектр поглощения, на котором проиллюстрированы метод базисной линии (а) и неоднозначность при проведении базисной линии (б).
Поглощение излучения. ИК-область 123
чающихся случаях единственный выход — принять одну из линий за отправную. Правильность полученных с ее помощью результатов следует проверить, измерив другие полосы поглощения того же спектра; объединенные результаты используют далее для доказательства правильности выбора базисной линии.
Спектроскопия внутреннего отражения [28]
Если поток излучения падает на границу раздела двух сред со стороны среды с более высоким показателем преломления, то он полностью отражается при условии, что угол падения больше некоторого критического угла
ac = sin_1— (4-3)
«1
где и п2— показатели преломления, причем ni>n2. Хотя теоретически [28, 29] отражение должно быть полным, на практике часть излучательной энергии проходит через границу раздела и затем возвращается (рис. 4-17). Если менее плотная среда поглощает при длине волны излучения, то отраженный поток будет обладать меньшей энергией, чем падающий, и, измеряя поглощение при разных длинах волн, можно получить спектр поглощения.
В принципе это явление наблюдается в любой спектральной области, но наибольшее применение оно нашло в ИК-спек-троскопии. Расстояние, на которое проникает излучение при внутреннем отражении, зависит от длины волны; для средней ИК-области оно составляет 5 мкм и менее. Такое явление по
лучило название нарушенного полного внутреннего отражения (НПВО).
Для использования НПВО в аналитических целях сконструированы специальные прободержатели, большинство кото-
рых пригодно для работы на обычных спектрофотометрах (рис. 4-18). На рис. 4-18,a изображена конструкция для случая единичного отражения под переменным углом. Грани нижней призмы посеребрены и, следовательно, отражают излучение под любым углом, поэтому при перемещении призмы (рис. 4-18,6) угол падения на пробу может Меняться, но падающий и выходящий потоки остаются на
Рис. 4-17. Схематическое представление полного внутреннего отражения потока излучения с частичным прониканием в субстрат. Коэффициент преломления материала призмы должен быть больше, чем у образца (Foxboro Analytical)
124 Глава 4
Рис. 4-18. Детали аппаратуры для отражательной спектроскопии: а и б — устройство, в котором входящий и выходящий потоки остаются на одной и той же прямой (грани нижней призмы и левая грань верхней должны быть посеребрены; путем перемещения призм в направлении, указанном стрелками, можно менять угол отражения от пробы); в — устройство для многократных отражений; г — подвижный поршень, внутри которого происходят многократные отражения от жидкой пробы (а, б, в,— Foxboro Analytical; г — Harrick Scientific Corporation).
одной прямой. На рис. 4-18, в приведен плоский элемент с НПВО, обеспечивающий многократное отражение [28], который обычно применяется при исследовании твердых образцов. Это устройство представляет собой пластинку, достигающую нескольких сантиметров в длину, из прозрачного твердого материала с высоким показателем преломления. На рис. 4-18,г изображена удлиненная призма с плоскими параллельными гранями, расположенная таким образом, чтобы поток входил и выходил из нее на одном и том же конце [28]. Такую призму можно погрузить в жидкую пробу, как пока
Поглощение излучения. ИК-область 125
зано на рисунке, а можно поместить между кусками твердого образца.
Упоминавшиеся ранее упрощенные спектрофотометры модели Miran снабжены удобными приспособлениями для исследований методом спектроскопии НПВО. В них применяется пластинка из кристаллического селенида цинка (п = 2,89) с горизонтальной гранью площадью в несколько квадратных сантиметров, закрепленная вровень с верхней поверхностью прибора. При спектроскопических измерениях жидкую или твердую пробу помещают непосредственно на эту грань, а затем после удаления пробы грань, если нужно, очищают.
Спектр НПВО не вполне идентичен спектру, полученному обычным путем, но имеет довольно большое сходство с ним. Различие между спектрами возрастает при приближении угла падения к критическому. Однако, чем дальше от критического угла, тем меньше поглощение. Таким образом, чтобы получить удовлетворительную чувствительность, нужно увеличить число отражений. Наибольшее применение спектроскопия НПВО нашла при исследовании непрозрачных твердых материалов, в том числе каучука и других полимеров, адсорбированных на поверхностях пленок, красок и других покрытий.
Поглощение в микроволновой области
Микроволновую область [30, 31] можно рассматривать как продолжение инфракрасной области спектра, поскольку поглощение здесь связано прежде всего с вращательными переходами в молекулах, обладающих постоянным дипольным моментом. При подходящих условиях к поглощению способны также молекулы, обладающие магнитным моментом, например О2, NO, СЮ2, NO2 и свободные радикалы. Особенно ценная для химии спектральная область лежит между 8 и 40 ГГц. Как будет показано в следующей главе, сюда относятся частоты, используемые в методе электронного парамагнитного резонанса (ЭПР), поэтому оба метода имеют много общего, хотя в основе их лежат разные молекулярные механизмы.
В отличие от области низких частот в микроволновой части спектра нет полос поглощения, характерных для специфических типов связи или функциональных групп. Качественный анализ можно провести только путем сравнения с известным спектром. Лишь при идентификации относительно небольших соединений микроволновой спектр дополняет ИК-спектр и служит «отпечатком пальцев». Частично это объясняется очень большим числом возможных частот. Если бы можно было разделить. полосы поглощения со степенью разрешения 200 кГц, чтобы получить приемлемую картину, то на участке 8—40 ГГц
126 Глава 4
наблюдалось бы до 160 000 зон или полос. Для сравнения укажем, что при среднем разрешении, равном 1 см-1, в ИК-обла-сти (от видимой части до 200 см-1, т. е. 50 мкм) наблюдалось бы лишь 10 000 полос. Конечно, многие частоты не проявляются ни в одном из известных спектров, поэтому эти цифры могут до некоторой степени ввести в заблуждение. Тем не менее на микроволновом спектрометре можно в принципе исследовать огромное количество соединений с очень малой вероятностью перекрывания спектров. Изучать можно только газообразные пробы при давлении паров не выше 0,1—10 Па (примерно 10~3—10-1 мм рт. ст.).
Основное ограничение связано с взаимодействиями вращательных уровней, относящихся к различным связям внутри молекулы. При наличии более трех-четырех роторов возникает так много взаимодействий, что спектр представляет собой множество линий, лишь очень немногие из которых достаточно интенсивны для практического использования. Отсюда ясно, что изучать крупные молекулы этим методом нельзя, если только циклические структуры не препятствуют вращению вокруг некоторых связей.
Для математического выражения поглощения в микроволновой области лучше использовать закон Ламберта, связывающий логарифм пропускания с длиной оптического пути в гомогенной среде, а не закон Бера, касающийся концентраций. Закон Ламберта можно представить в виде
Р = Ро-10"“‘ или lgP0/P = a& (4-4)
где Pq и Р — соответственно интенсивность падающего и прошедшего через кювету микроволнового излучения с эффективной длиной Ь. Коэффициент поглощения а соответствует оптической плотности А (величине, которая используется в оптической области) на единицу длины кюветы. В идеале он является функцией только числа поглощающих частиц на единицу длины и, следовательно, должен быть связан с парциальным давлением веществ в смеси газов.
До какой степени это соотношение справедливо, зависит от способа определения коэффициента поглощения и условий его применения. При низких парциальных давлениях и малых интенсивностях, когда влияние насыщения не заметно, давление поглощающих компонентов пропорционально высоте в максимуме поглощения амакс- При более высоких давлениях значение ехмакс становится постоянным, а дальнейшее повышение давления приводит к уширению полосы (ширина измеряется на половине высоты). Можно показать, что начиная с этого
Воглощение излучения. ИК-область 1ST
Рис. 4-19. Изменение полосы микроволнового поглощения с изменением давления. Интенсивность в пике остается постоянной в широком диапазоне давлений [37].
момента парциальное давление почти пропорционально интегральной оптической плотности:
К • р = J a (v) dv
(4-5)
где v — частота, р — парциальное давление, К — коэффициент пропорциональности. На рис. 4-19 представлены кривые поглощения полосы при разных парциальных давлениях.
Несмотря на уширение при изменении давления, перекрывание полос поглощения разных соединений маловероятно, потому что полосы в максимуме достаточно узкие. Следовательно, парциальное давление, определенное по уравнению (4-5), можно рассматривать как аналитический результат, не делая поправок на присутствие примесей. По данным Армстронга [32], при.определении SO2 в пробе воздуха сигнал остается линейным при концентрации от 100 до 2-10-80/о—поистине замечательная рабочая шкала!
В качестве примера прямого аналитического применения можно привести определение следов метанола в вине'[33]. Пробу с помощью шприца вводят непосредственно в рабочую кювету, где она полностью испаряется, и затем измеряют поглощение по полосе при 31,2267 ГГц.
Метод микроволновой спектроскопии позволяет изучать конформации молекул, поскольку транс- и аош-соединения имеют разные спектры и, следовательно, могут быть идентифицированы, а также измерять значения внутримолекулярного вращательного барьера [31]. Изменение дипольного момента соединения при изотопном замещении тоже сказывается на
128 Глава 4
Рис. 4-20. Микроволновый спектр поглощения 4-хлортолуола. Каждый пик представляет собой дублет, соответствующий двум изотопам хлора (Hewlett — Packard Company).
Диапазон частот
Размер пробы
Время развертки
Экспериментальные условия
Источник
Мощность Штарковское поле Постоянная детектора
26,5—40,0 ГГц, или 0,85—1,32 см-1
40 мкм рт. ст. или 2-Ю-8 моль при комнатной температуре
100 с
Устойчивый по фазе
2,5 Вт
2500 В/см
1,5
спектрах: рис. 4-20, на котором иллюстрируется этот эффект, дает представление рб общем характере микроволновой спектрограммы.
Приборы для микроволновой области. Недостаток места не позволяет нам подробно рассмотреть теорию и устройство приборов. По конструкции спектрофотометры напоминают одно- и двухлучевые приборы, используемые в более известных областях спектра. Источником служит либо специальная вакуумная трубка (например, клистрон), либо твердый осциллятор на основе туннельного диода или диода Ганна. Такие осцилляторы, охватывающие значительный диапазон частот, испускают в каждой точке монохроматическое излучение. Вместо прерывателя потока установлена модуляционная система, действующая на основе эффекта Штарка. Детектором служит кристаллический диод. Большинство микроволновых спектрофотометров собирают вручную в лабораториях из модулей.
Поглощение излучения. ИК-область 129
Задачи
4-1. Пробу этилбромида, в которой предполагается наличие следов воды, этанола и бензола, исследовали на ИК-спектрофотометре. Методом базисной линии получены следующие значения относительной оптической плотности: при 2,65 мкм /1 = 0,110, при 2,75 мкм А = 0,220 н при 14,7 мкм А = 0,008. Прибор, кюветы и пр. были в точности такими, как в работах [34, 35]. Рассчитайте количество каждой примеси.
4-2. Сообщалось [36], что гексацианоферраты (II) и (III) при их совместном присутствии можно определить в водных растворах по поглощению в ИК-области. Были получены следующие экспериментальные данные:
Fe(CN)|- Fe(CN)3-
Молярный коэффициент поглоще- 4,23-103 1,18-103
ния е, л/(моль-см)
Интервал линейной зависимости, 0,00031.—0,4 0,0012—0,1
моль/л
Максимальное волновое число, 2040 2115
см-1
В использованных кюветах расстояние между окошками из иртрана-1 (запатентованная форма MgF2) составляло 50 мкм. Из спектрограммы отбеливающего фотографического раствора получены следующие данные: базисная линия горизонтальна при А = 0,043; А = 0,258 при 2040 см-1 и А = 0,515 при 2115 см-1. Найдите концентрации обоих соединений в молях на литр.
4-3. Получены следующие значения кажущейся оптической плотности растворов циклогексанона в циклогексане при 5,83 мкм (толщина кюветы 0,026 мм):
Концентрация, Оптическая Концентрация, Оптическая
г/л плотность г/л плотность
5 0,190 30 0,444
10 0,244 35 0,487
15 0,293 40 0,532
20 0,345 45 0,562
25 0,390 50 0,585
Постройте по этим данным график, а) Определите интервал, в котором выполняется закон Бера и за пределами которого необходимо делать поправку на поглощение фона, б) Рассчитайте значение молярного коэффициента поглощения.
4-4. Газанол состоит из неэтилированного бензина, содержащего 10 % (по объему) этанола. Из других спиртов могут присутствовать метанол, изопропанол, н-бутанол и трет-бутанол. Бензин содержит н-алканы от С4 до С12. Найдите в атласе спектры этих соединений и выберите оптимальное волновое число для ИК-спектроскопического определения этанола в бензине. Составьте методику определения, принимая во внимание возможность наличия примесей в бензине.
4-5. Определите толщину слоя (в миллиметрах), при которой получаются интерференционные полосы, изображенные на рис. 4-15. При каком волновом числе толщина слоя равна десяти длинам волн?
4-6. Высокотемпературный газовый поток при производстве водяного газа состоит в основном из Н2, СО, СО2 и Н2О и, возможно, из небольшого количе
5 Заказ № 537
130 Глава 4
ства (менее 0,5 % по .объему) СН<. Для контроля за содержанием СН< желательно использовать бездисперсионный ИК-аиализатор с максимальной чувствительностью. Перед введением в анализатор газ охлаждают, так что избыток Н2О конденсируется и удаляется. Найдите в атласе подходящие спектры и подробно опишите, что следует поместить в каждое отделение анализатора, изображенного на рис. 4-10, а.
4-7. Рассчитайте примерное число фотонов при длине волны 10 мкм, необходимое для обнаружения с помощью термистора со следующими характеристиками: приемник изготовлен из золота размером 1,0X0,1X0,005 мм; термистор на 10 кОм (при 25 °C), коэффициент сопротивления 500 Ом/К. Минимальное измеряемое изменение сопротивления Д/?= 1 Ом. Теплоемкость золота 0,0308 кал/(г-К), а его удельная масса 19,32 г/см3 (заметим, что из-за отдачи тепла окружающей среде в действительности число фотонов может быть значительно больше, чем предсказывает расчет).
4-8. Спирт имеет слабовыраженный пик поглощения в ближней ИК-области при 0,952 мкм. Это может быть какой-либо обертон (кратный) основного колебания валентной связи С—О или О—Н. Пользуясь данными табл. 4-2, решите, какая из предложенных возможностей наиболее вероятна.
4-9. Оптическая плотность таблетки из КВг диаметром 7,50 мм и толщиной 1,5 мм при длине волны 6,02 мкм равна 0,722. Известно, что коэффициент поглощения вещества, содержащегося в таблетке, при этой длине волны равен 43,21 л/(г-см). Рассчитайте содержание этого вещества в таблетке в миллиграммах.
4-10. Длину оптического пути в разборной кювете определяли двумя способами: а) механически с помощью микрометра измеряли толщину тефлоновой прокладки, которая оказалась равной 22,0; б) кювету заполняли бензолом и измеряли оптическую плотность при 850 см-1, которая оказалась равной 0,125. Насколько согласуются эти измерения? Какой результат вы считаете более достоверным и почему?
4-11. Угол падения потока излучения при каждом отражении равен 45° (рис. 4.18, а). Каким должен быть показатель преломления твердого поршня при измерении проб с показателями преломления более 1,8?
Литература
1. Jones R. N., Sandorfy С., In Chemical Applications of Spectroscopy, West W. (ed.), Wiley-Interscience, New York, 1956, p. 308.
2. Barker J. D., Electro-Opt. Syst. Des., 1970, p. 32.
3. Ewing G. W., J. Chem. Educ., 1971, v. 48, p. A521.
4. Putley E. H., The Pyroelectric Detectop in Semiconductors and Semimetals, Willardson R. K-, Beer A. C. (eds.), v. 5, Academic Press, New York, 1970, chap. 6.
5. Doyle W. M., Electro-Opt. Syst. Des., 1978, p. 12.
6. McLellan E. J., Stotlar S. C., Opt. Spectra, 1981, p. 55.
7. Stewart J. E., Infrared .Spectroscopy, Experimental Methods and Techniques, Dekker, New York, 1970, pp. 276ff.
8. Griffiths P. R. (ed.), Transform Techniques in Chemistry, Plenum Press, New York, 1978.
9. Bell R. J., Introductory Fourier Transform Spectroscopy, Academic Press, New York, 1972.
10. Koenig J. L. Appl. Spectrosc., 1975, v. 29, p. 293.
11. Chenery D. H., Sheppard N., Appl. Spectrosc., 1978, v. 32, p. 79.
12. Walters S. H., In Process and Controls Handbook, Considine D. M. (ed.), McGraw-Hill, New York, 1957, pp. 6—73.
13. Link W. T.. McClatchie E. A.. Watson D. A., Compiler A. B„ AIAA Pa
Поглощение излучения. ИК-область 131
per No. 71-1047, Joint Conference on Sensing of Environmental Pollutants, American Institute of Aeronautics and Astronautics, New York, 1971.
14. Kimmitt M. F., Far-Infrared Techniques, Pion, London, 1970.
15. Stewart J. E., Infrared Spectroscopy. Dekker, New York, 1970, p. 165.
16. Goddu R. F., Delker D. A., Anal. Chem., 1960, v. 32, p. 140.
17. Colthup N. B. J. Opt. Soc. Am., 1950, v. 40, p. 397.
18. Bentley F. F., Wolfarth E. E., Spectrochim. Acta, 1959, v. 15, p. 165.
19. Bellamy L. J., The Infrared Spectra of Complex Molecules, 3d ed., Chapman & Hall, London, 1975.
20. Conley R. T. Infrared Spectroscopy, 2d. ed., Allyn and Bacon, Boston, 1972.
21. Silverstein R. M., Bassler G. C., Morrill T. C. Spectrometric Identification of Organic Compounds, 4th ed., Wiley, New York, 1981.
22. Alpert N. L., Keiser W. E., Szymanski H. A., IR: Theory and Practice of Infrared Spectroscopy, 2d ed., Plenum Press, New York, 1970.
23. American Petroleum Institute Catalog of Infrared Spectrograms, API Research Project 44, Texas & M University, College, Texas.
24. Pouchert C. J., The Aldrich Library of Infrared Spectra, Aldrich Chemical Company, Milwaukee, 1970.
25. Sadtler Research Laboratories. Sadtler Standard Spectra, Philadelphia (непрерывно дополняемое издание).
26. The Coblentz Society, Anal. Chem., 1975, v. 47, p. 945A.
27. Hannah R. W., Instrum. News 1963, v. 14, No. 34, p. 7; (Perkin — Elmer Corporation).
28. Harrick N. J., Internal Reflection Spectroscopy, 2d. ed., Harrick Scientific Corp., Ossining, New York, 1979.
29. Klein M. V. Optics, Wiley, New York, 1970, p. 579.
30. Chantry G. W. (ed.), Modern Aspects of Microwave Spectroscopy, Academic Press, New York, 1979.
31. Varma R., Hrubesh L. W., Chemical Analysis by Microwave Rotational Spectroscopy, Wiley, New York, 1979.
32. Armstrong S., Appl. Spectrosc., 1969, v. 23, p. 575.
33. Kitchin R. W., Willis R. E„ Cook R. L„ Anal. Chem., 1981, v. 53, p. 1190.
34. McCrory G. A., Scheddel R. T., Anal Chem., 1958, v. 30, p. 1162.
35. Williams V. У., Anal. Chem., 1957, v. 29, p. 1551.
36. Drew D. M., Anal. Chem., 1973, v. 45, p. 2423.
37. Gordy W., Smith W. V., Trambarulo R. F., Microwave Spectroscopy, Wiley, New York, 1953 (Dover, New York, 1966).
5*
ГЛАВА
Атомно-абсорбционная спектроскопия
В этой главе мы рассмотрим поглощение атомами, а не молекулярными соединениями. Чтобы наблюдать оптические свойства свободных атомов, необходимо пробу перевести в газообразное состояние, а это в большинстве случаев требует испарения жидкости или твердого вещества и последующей диссоциации молекул на свободные атомы.
Атомная абсорбция (АА) подчиняется тем же основным законам, что и молекулярное поглощение, которое мы рассмотрели в предыдущих главах. Следовательно, атомно-абсорбционные спектрофотометры должны иметь те же узлы, чтобы при необходимости их можно было модифицировать. Наиболее существенное отличие АА заключается в подготовке самой пробы, с этого мы и начнем обсуждение метода.
Атомная флуоресценция (АФ), которую часто рассматривают вместе с АА, будет разбираться в гл. 9 вместе с другими видами атомной эмиссии.
Атомизация
Существует много способов атомизации соединений металлов, осуществляемых в большинстве случаев за счет тепловой энергии электричества или' пламени. Для оптимального перехода в атомный пар необходим строгий контроль за температурой. Слишком высокая температура может быть так же неблагоприятна, как и слишком низкая, потому что часть атомов ионизируется и, следовательно, не поглощает при ожидаемых длинах волн. Но, с другой стороны, высокая температура способствует снижению влияния матрицы, поэтому следует найти компромисс между этими температурами.
Пламенная атомизация. На рис. 5-1 изображена горелка, используемая в пламенной атомно-абсорбционной спектроскопии (ААС). Горючий газ и газ-окислитель подаются в смесительную камеру, где они проходят через ряд перегородок, обес-
Атомно-абсорбциониая спектроскопия 133
Рис. 5-1. Горелка с предварительным смешением газов н безвихревым потоком для ААС.
почивающих их полное смешение, и поступают в верхнюю часть горелки. Отверстие горелки имеет форму длинной узкой щели, что позволяет получить пламя в виде узкой полосы. Анализируемый раствор засасывается в смесительную камеру с помощью небольшой воздушной форсунки. При использовании такого распылителя получаются капельки разного размера, что может быть причиной плохой воспроизводимости. При прохождении через перегородки смесителя более крупные капли задерживаются, так что в пламя попадают более мелкие однородные по размеру капли.
Горелка с предварительным смешением газов не вполне безопасна в работе, потому что, если пламя попадет в смесительную камеру, произойдет сильный взрыв. Для того чтобы свести к минимуму вероятность проскакивания пламени в камеру, щель горелки нужно сделать как можно более узкой (с тем чтобы газы продувались сквозь нее с большой скоростью), а металлический обод вокруг щели как можно массивнее, так чтобы тепло легко отводилось. Но даже в этом случае, если не регулировать газовый поток должным образом, взрыв возможен. В продажных горелках предусмотрены меры безопасности при проскакивании пламени в камеру. При эксплуатации горелки всегда необходимо строго соблюдать правила техники безопасности.
В качестве окислительного и горючего газов в ААС чаще всего выбирают сжатый воздух и ацетилен. Максимально достигаемая температура составляет около 2200 °C. Если нужна более высокая температура, воздух можно заменить оксидом азота (NjO), который разлагается'с образованием смеси азота и кислорода в соотношении 2:1, тогда как для сжатого воздуха это соотношение равно 4:1; максимальная температура, которую можно получить при горении ацетилена, составляет
134 Глава 5
Лоток излучения
Кварцевая трубка
К монохроматору и детектору
Рис. 5-2. Чашечка Дельве пробоотборника. Чашечка укреплена под отверстием в горизонтальной открытой с обоих концов трубке нз кварца, через которую проходит поток излучения.
почти 3000 °C. В горелках с предварительным смешением газов нельзя использовать чистый кислород, поскольку пламя распространяется так стремительно, что проскок в камеру неизбежен.
Пламя — удобный и воспроизводимый источник тепла, но в качестве рабочей кюветы этот источник далек от идеала, потому что два эндотермических процесса (испарение растворителя и последующая атомизация) должны пройти за столь короткий промежуток времени, что каким-то частицам удается пролететь сквозь пламя, не атомизируясь. Кроме того, пламя привносит значительные случайные флуктуации в эффективную длину оптического пути вследствие турбулентности, а это приводит к лишнему шуму при получении сигнала.
Некоторого улучшения можно добиться, используя пламя только как источник тепла. Один из способов заключается в том, что растворенную пробу помещают в небольшую металлическую лодочку или чашечку, называемую чашкой Дельве [1], и высушивают на горячей пластинке. Чашечку затем укрепляют над пламенем для испарения пробы. Образовавшийся пар поступает через центральное отверстие в горизонтальную кварцевую трубку (рис. 5-2), нагреваемую узким пламенем, где собственно и испускается измеряемое излучение. Кварцевая трубка выполняет роль удерживающего сосуда, с помощью которого продлевается время наблюдения за атомами. Этот способ оказался, в частности, полезным при определении следов свинца. Обычно применяются никелевые чашечки, но при анализе кислых проб никель заменяют танталом или другим инертным материалом.
Непламенные атомизаторы. В последнее десятилетие вместо пламени все более широко используется электронагревание. Было предложено несколько типов нагревателей, из которых наиболее удачным оказалось устройство, состоящее из неболь
Атомно-абсорбционная спектроскопия 135
шой графитовой трубки [2], нагреваемой при пропускании через нее тока большой силы (вплоть до 500 А) при низком напряжении. Внутренний диаметр графитовой трубки (модели Ва-риан) составляет несколько миллиметров, а длина около сантиметра. Вверху трубки имеется небольшое отверстие, через которое вводится растворенная проба, как это показано на рис. 5-3. Внутренняя поверхность покрыта устойчивой формой углерода, называемой пиролитическим графитом. Подобные же трубки изготовляются и другими фирмами и различаются деталями. Трубка, часто называемая графитовой кюветой, должна находиться в атмосфере инертного газа, например аргона, для предотвращения окисления пробы и горючего углерода.
При работе с жидкими пробами обычно прибегают к программированному повышению температуры графитовой кю-
Рис. 5-3. Атомизатор с угольными электродами фирмы Varian модели CRA-90. Графитовая кювета представляет собой небольшой цилиндр, закрепленный в поперечном положении .между двумя графитовыми стержнями, являющимися и механическими держателями, и проводниками тока. С помощью шланга слева подводится инертный газ. На фотографии показано введение пробы пипеткой (Varian Techtron).
136 Глава 5
веты в три этапа. Сначала ее поднимают примерно до 300 °C и поддерживают в течение минуты для испарения растворителя. Затем поднимают температуру до 1700 °C и выдерживают еще около минуты; при этом сгорают органические вещества. Только после этого температуру повышают до уровня, необходимого для диссоциации неорганического соединения на атомы; иногда требуется поднять ее до 3000 °C.
Конструкция большинства приборов позволяет оператору изменять интервал времени и температуру в зависимости от характера пробы. В процессе сушки на ленте самописца с разверткой по времени часто обнаруживается посторонний пик, связанный с поглощением паров растворителя. На стадии обугливания при наличии большого количества органических веществ также возможно появление пика. Аналитическую информацию получают, измеряя высоту пика, наблюдаемого при конечной температуре. На высоту пика влияют скорость нагревания и размеры кюветы [3], поэтому при сравнении результатов измерения неизвестной пробы с результатами измерения стандартов следует точно воспроизводить эти и другие изменяющиеся параметры.
Летучие гидриды. Для элементов, образующих летучие гидриды, в частности As, Bi, Ge, Sb, Se и Те [4], существует другой способ подготовки пробы. Гидриды получают реакцией солей этих элементов с щелочным раствором борогидрида натрия. Летучие вещества затем уносятся из раствора с потоком воздуха прямо в атомизатор атомно-абсорбционного спектрофотометра. Часто используется водородно-воздушное пламя; подогреваемая снаружи кварцевая трубка-кювета позволяет спуститься до более низких концентраций вследствие увеличения времени пребывания в ней атомов. Этим способом определяли также олово и свинец, но результаты оказались менее удовлетворительными.
Аналогичным путем можно восстановить ртуть до элементного состояния (если она была в другой степени окисления) и перенести ее с током воздуха в кварцевую трубку-кювету, которая не требует нагревания [4].
Источники излучения
Почти во всех атомно-абсорбционных спектрометрах используются лампы, дающие линейчатые спектры, характерные для отдельных элементов. Эти лампы в сочетании с обычными монохроматорами средней разрешающей силы гораздо эффективнее источников непрерывного света, описанных в гл. 3. Монохроматор служит для выделения нужной линии испускания,
Атомно-абсорбционная спектроскопия 137
Рис. 5-4. Сравнение излучения источника непрерывного спектра и поглощения атомного пара. Кривая а представляет собой полосу длин волн, прошедших через монохроматор; кривая б, которая во много раз уже кривой а,— поглощение атомами в пламени (Unicam Instruments, Ltd.).
а не для сужения полосы поглощения. Нецелесообразность использования источников непрерывного излучения связана с тем, что линии поглощения нейтральных атомов в пламени или кювете чрезвычайно узки. Ширина их составляет порядка 0,001 нм, тогда как полуширина полосы пропускания обычного монохроматора несколько десятых нанометра. Это соотношение иллюстрируется рис. 5-4. Площадь под кривой а, соответствующая пропущенному излучению источника, лишь очень незначительно уменьшается за счет линии атомной абсорбции б.
Наибольшим успехом в качестве источника линейчатого спектра в ААС пользуется лампа с полым катодом. Она представляет собой стеклянный или кварцевый баллон, в котором размещены два электрода. Один из них (катод), имеющий чашеобразную форму, изготовлен из какого-либо определенного элемента (рис. 5-5). Материал анода не имеет значения. Лампа
Рис. 5-5. Лампа с полым катодом.
138 Глава 5
Ni-232,0
Ni
Ni
Рис. 5-6. Испускание лампы с полым катодом, изготовленным из никеля. Атомы никеля в пламени в заметной степени поглощают лишь излучение при 232,0 нм (Westinghouse Electric Corporation).
заполнена благородным газом под низким давлением. При подаче напряжения 100—200 В после непродолжительного разогрева происходит тлеющий разряд с участием большинства частиц, поступающих с полого катода. Положительные ионы инертных газов бомбардируют катод, выбивая атомы металла; этот процесс называется распылением. Атомы поглощают энергию и возбуждаются, испуская характеристическое излучение. Оно состоит из дискретных линий этого металла и линий газа-наполнителя, который выбирают таким образом, чтобы спектральные помехи от него при определении данного металла были минимальными.
На рис. 5-6 представлен спектр испускания лампы с полым катодом. Обратите внимание на то, что чувствительная линия при 232 нм окружена множеством других линий никеля, поглощение которых паром
никеля нежелательно. Нужную линию можно изолировать при помощи монохроматора с узкой полосой пропускания. На рис. 5-7 сравнивается спектр испускания лампы с полым катодом (а) с тем же спектром после прохождения излучения через монохроматор (б) и показано влияние поглощения паром металла (в). Полоса поглощения всегда шире эмиссионной линии, хотя и уже полосы пропускания монохроматора. Очевидно, что уменьшение интенсивности прошедшего в детектор потока излучения непосредственно зависит от числа атомов металлов в зоне нахождения пробы.
Можно изготовить лампы с полым катодом, у которых чаша покрыта смесью нескольких металлов (при условии что они не мешают друг другу при спектральном определении и для их испарения необходима примерно одинаковая энергия). Это делает возможным определение нескольких элементов без смены лампы. В качестве примера можно назвать лампы, катод которых состоит из Са, Mg и Al; Fe, Си и Мп; Си, Zn, Pb и Sn; Сг, Со, Си, Fe, Мп и Ni.
Лампе с полым катодом, как и многим другим источникам, для выхода на постоянный режим работы требуется после включения некоторое время. Особенно это неудобно при опре-
Атомно-абсорбциоииая спектроскопия 139
Рис. 5-7. Атомная абсорбция: а — спектр излучения лампы с цинковым катодом (ширина линии 213,9 им увеличена для наглядности); б —лишние линии циика отсекаются монохроматором, настроенным на 213,9 нм; в — интенсивность линии при 213,9 им резко уменьшается в результате поглощения ато мами циика (Unicam Instruments, Ltd.).
140 Глава 5
делении с помощью одноэлементных ламп нескольких элементов в одной пробе. Один из путей преодоления этой трудности состоит в использовании туррели, на которой укреплено несколько ламп, находящихся в рабочем режиме, так что любую из них можно установить в нужное положение поворотом туррели. Для сокращения задержки при смене ламп используют двухлучевую систему корректировки яркости лампы.
Источником в ААС может служить и безэлектродная разрядная лампа, которая представляет собой запаянную кварцевую трубку, содержащую небольшое количество чистого металла под низким давлением инертного газа. Возбуждение происходит под действием микроволнового поля волнового резонатора, причем испускается, по существу, тот же спектр, что и лампой с полым катодом.
Можно также пользоваться источником непрерывного спектра в сочетании с монохроматором высокого разрешения. О’Хэвер и сотр. [5, 6] показали, что ксеноновая лампа большой мощности с эшелеттом в качестве монохроматора позволяет сузить полосу поглощения, чтобы получить правильные значения оптической плотности. Достоинством такой лампы является то, что при переходе от одного элемента к другому достаточно просто менять длину волны. Авторы описали прибор с 16 фотоумножителями, на котором возможно одновременное определение такого же количества элементов, но с несколько меньшей чувствительностью, чем с помощью лампы с полым катодом. В атомно-абсорбционных спектрофотометрах заводского изготовления непрерывные источники не используются.
Поправка иа поглощение фона
В ААС в отличие от спектрофотометрии растворов нельзя учесть влияние фона с помощью простой двухлучевой схемы. Такой прием потребовал бы дубликатов пламен или графитовых кювет (один рабочий, другой сравнения), а сделать их оптически равноценными было бы чрезвычайно трудно. Тем не менее при количественных измерениях поправка на фон существенна.
Фоновый сигнал частично обусловлен излучением, испускаемым самой нагретой пробой. Этот источник фонового излучения, характерный только для ААС, обусловлен неизбежным возбуждением атомов анализируемого вещества, самопроизвольно испускающих фотоны при тех же длинах волн, при которых исследуется поглощение. Как схематически показано на рис. 5-8, а, если не принять мер предосторожности, наблюдаемая интенсивность света РНабл будет складываться из Р0Т и Ре, где Ро—интенсивность падающего излучения, Т — пропу-
Атомно-абсорбционная спектроскопия 141
а
Лампа с полым катодом
Прерыватель
Атомный пар
Монохроматор и усилитель
Рис. 5-8. Основные узлы АА-спектрофотометра: а — схема без прерывания излучения (испускание нагретой пробы складывается с излучением лампы с полым катодом, что приводит к погрешности); б — схема с прерыванием излучения (детектор фокусирует только излучение лампы).
скание пробы и Ре — та доля интенсивности испускания пробы, которое попадает в детектор. Излучения с интенсивностями Ро и Ре имеют одинаковую длину волны и поэтому при попадании (через монохроматор) в детектор неотличимы друг от друга. Устранить это можно при помощи прерывателя, который прерывает излучение от лампы с полым катодом, как показано на рис. 5-8, б, не влияя на излучение от пробы. Для того чтобы сигнал, обусловленный испусканием пробы, вычитался из общего сигнала, можно поставить электронный усилитель, работающий синхронно с прерывателем. В некоторых приборах тот же результат получают при возбуждении лампы с полым катодом электрическими импульсами; при этом вместо постоянного излучения, прерываемого с помощью диска прерывателя, получается прерывистое излучение.
Даже после введения прерывающего устройства фон остается значительным. Часть его составляет шум, связанный в основном с рассеянием излучения частицами матрицы пробы, а при возбуждении в пламени-—с турбулентностью.
Вклад в фон вносит также поглощение других компонентов пробы. Описано несколько способов снижения такого рода помех. Первый из них заключается в использовании одновременно источника непрерывного излучения, например водородной или дейтериевой лампы, и источника линейчатого спектра [7, 8], как показано на рис. 5-9. Излучение вспомогательной лампы проходит через пробу вместе с резонансным излучением лампы с полым катодом. Электронная система сортирует сигналы от обоих источников и дает их отношение. В одних приборах (фирмы Perkin-Elmer) оба сигнала видоизменяют при помощи
142 Глава 5
Рис. 5-9. АА-спектрофотометр с водородной (или дейтериевой) лампой, с помощью которой вносится поправка иа фои.
вращающегося зеркала, в других (фирмы Instrumentation Laboratories) через оптическую систему проходят сразу оба потока, которые прерываются с разной скоростью. И тот и другой потоки равномерно ослабляются за счет поглощения фоном или рассеяния, но пробой заметно поглощается лишь резонансное излучение.
Второй метод устранения фонового сигнала основан на эффекте Зеемана. Й испускание, и поглощение УФ- и видимого излучения связано со свойствами электронов, вращающихся в атомах, поэтому неудивительно, что эти явления сильно зависят от наличия магнитного поля. Теоретически предсказано [9] и экспериментально подтверждено, что, если источник излучения (лампу с полым катодом) или поглощающую пробу поместить в поперечное магнитное поле, каждая линия испускаемого излучения расщепляется в простейшем случае на три линии, одна из которых имеет несколько большую длину волны, другая несколько меньшую, а третья остается без изменений. Смещенная и несмещенные линии поляризуются перпендикулярно друг другу, следовательно, если на оптическом пути поставить поляризатор, то их можно различить.
На рис. 5-10 изображена оптическая схема атомно-абсорбционного спектрофотометра (фирмы Hitachi), в которой для введения поправки на фон использован эффект Зеемана. Вокруг атомизатора (графитовая кювета, как на рисунке, или пламя) размещают постоянный магнит. Между атомизатором и лампой с полым катодом помещают поляризатор, который можно повернуть так, чтобы плоскость поляризации была параллельна или перпендикулярна направлению магнитного поля. При поляризации излучения под прямым углом к полю оно не поглощается атомным паром, а при параллельной поляризации поглощается, как если бы магнитного поля не было. Однако поглощение, связанное с фоном, не меняется ни в том, ни в дру-
Атомио-абсорбциоииая спектроскопия 143
н: Магнитное поле
и $2; Цели
Lj- £.3'. Линзы
М,- Мц'. Зеркала
G: Дифракционная
решетка
Лампа с полым катодом
Поляризатор
Рис. 5-10. Оптическая схема АА-спектрофотометра фирмы Hitachi (модель 180/70), иа которой показано направление магнитного поля Зеемана вокруг графитового атомизатора (Hitachi, Ltd.)
гом случае, поэтому, вычитая одно (при перпендикулярной поляризации) из другого (при параллельной поляризации), получают спектр поглощения с поправкой на фон. Преимущество этого способа перед описанным выше состоит в том, что здесь используется лишь одна лампа, поэтому не возникает проблем, связанных с выравниванием потоков. В спектрофотометрах с поправкой, вводимой на основе эффекта Зеемана, возможно и другое расположение компонентов [10, 11]. Их относительные достоинства и недостатки обсуждаются в недавно опубликованном обзоре [12].
Третий способ учета фона, который мы разберем, основан на модулировании длины волны, о чем упоминалось в гл. 3. Харнли и О’Хэвер [13] вывели выражение для оптической плотности пробы, для применения которого нужно выделить постоянную и переменную составляющие тока фотоумножителя. Переменную составляющую находят по сдвигу синусоиды длин волн при колебании кварцевой преломляющей пластинки, помещенной посередине выходной щели:
А = lg (Ро/Р) = 1g [(DC+ 0,38AC)/(DC—0,624 С)| (5-1)
где АС и DC — полный размах колебаний переменной и постоянной составляющих соответственно. Авторы показали, что этот способ почти вдвое эффективнее способов с применением дейтериевой лампы или эффекта Зеемана.
Не так давно появилось сообщение еще об одном способе введения поправки на фон [14], достаточно интересном, чтобы рекомендовать его. По этому способу поправку получают с по
144 Глава 5
мощью самой лампы с полым катодом. Сначала, чтобы измерить суммарную оптическую плотность исследуемого элемента и фона, в лампу подают импульс тока малой силы (12 мкА). Затем, чтобы расширить эмиссионную линию лампы, подают короткий импульс тока большой силы (250 мкА). Относительное поглощение элементом пробы резко уменьшается, тогда как фоном поглощается постоянная часть излучения. Разница между обоими сигналами дает истинную аналитическую информацию.
Предел обнаружения. Чувствительность АА-методов связана сложной зависимостью с оптическими свойствами атомного пара, температурой, относительной шириной линий лампы и поглощающих частиц и характером оптической системы (см. рис. 9-11). Наряду со сравнительными данными для некоторых АА-методов, которые будут разбираться в гл. 9, сопоставлены относительные пределы обнаружения многих элементов методом ААС с использованием пламенной или непламенной атомизации. Как будет показано, в целом пределы обнаружения при использовании непламенной ААС в 100 и более раз ниже, чем при использовании пламенной, хотя и имеются некоторые исключения; так, например, пределы обнаружения К, Fe, Sn тем и другим методами почти одинаковы. Табличные значения пределов обнаружения следует принимать с некоторыми оговорками, поскольку другие исследователи на другом оборудовании могут получить совершенно иные значения.
Помехи
В пламенной ААС причиной помех могут быть химические реакции в пламенах. Основные затруднения связаны с неполной диссоциацией или с образованием труднолетучих соединений. Некоторые элементы, например Ti, Al и V, окисляются в пламени, образуя соединения, устойчивые при температуре воз-душно-ацетиленового пламени; большинство таких соединений разлагаются в Ы2О-ацетиленовом пламени. В других случаях интересующий нас элемент образует устойчивые соединения с некоторыми другими компонентами пробы. Например [15], рассмотрим определение Sr в присутствии А1 или Si. На рис. 5-11 показано, насколько сильно они мешают анализу. Эти помехи можно почти полностью устранить добавлением к пробе небольшого количества соли лантана, который связывает А1 или Si и не взаимодействует со Sr.
Помехи возможны и при определении солей Са. Соединения кальция полностью диссоциируют в Ы2О-ацетиленовом пламени, но температура его столь высока, что заметная часть атомов Са ионизируется и, следовательно, не попадает в число
Атомио-абсорбциониая спектроскопия 145
Рис. 5-11. Одни из способов устранения химических помех в ААС. Добавка соли лантана защищает стронций от мещающего действия со стороны алюминия и кремния.
определяемых атомов. Это можно регулировать добавлением более легко ионизирующегося элемента, например Na; при этом атомы Са остаются в нейтральном состоянии.
Другим источником помех является изменение вязкости или других свойств растворов, влияющих на легкость распыления и переноса в пламени. В результате для двух растворов с одинаковой концентрацией металла, но с разным количеством посторонних веществ можно получить разные показания прибора.
Те же проблемы возникают при работе с графитовым атомизатором. Только здесь более вероятно образование труднолетучего соединения с углеродом, а не с кислородом. Карбиды алюминия и кремния при нагревании диссоциируют, но карбиды вольфрама и бора устойчивы при нагревании.
Применение атомной абсорбции. Атомная абсорбция используется для определения большого числа металлов, особенно в следовых количествах. Она широко применяется в таких областях, как анализ воды и фармацевтических препаратов, а также в металлургии.
При любом определении совершенно необходимо скрупулезно соблюдать экспериментальные условия: нужно найти специальные рекомендации и строго следовать им, а если их нет, то провести методические исследования. Выпускаемые промышленностью приборы снабжены обширными руководствами, содержащими методики определения всех известных металлов в различных матрицах. Монография Ван Луна [4] является прекрасным сборником методик и содержит обсуждение принципов АА-анализа.
146 Глава 5
Задачи
5-1. Натрий в количестве 0,5—2 % можно определить методом ААС по нерезонансным линиям излучения цинковой лампы с полым катодом при 330,259 и 330,294 нм. Чувствительность определения при этом почти в пятнадцать раз ниже, чем при использовании дублета резонансных линий излучения натриевой лампы (330, 232 и 330, 299 нм), хотя яркость цинковой лампы на данной длине волны вдвое меньше яркости натриевой лампы. Присутствие цинка в пламени, где происходит поглощение, не мешает определению натрия, а) Объясните такое кажущееся противоречие: почему при яркости, всего лишь вдвое меньшей, чувствительность при использовании цинковой лампы в пятнадцать раз ниже, чем при использовании натриевой лампы? б) Почему цинк не мешает определению?
5-2. Для того чтобы количественно сравнить эффективность источников непрерывного и линейчатого спектров в ААС, были проделаны эксперименты и получены следующие данные. Ксеноновая лампа с обычным монохроматором дает полосу излучения с полушириной 5 нм, а лампа с полым катодом — 0,01 нм. На кривой поглощения соответствующего элемента в пламени имеется полоса с полушириной 0,02 нм (рис. 5-12). В двух опытах, в одном из которых использовалась лампа с полым катодом, а в другом — ксеноновая лампа, была подобрана такая концентрация, при которой высота пика излучения в результате поглощения вдвое уменьшилась по сравнению с высотой при нулевой концентрации. Как сравнить площади под кривыми интенсивность — длина волны для двух источников после прохождения излучения через пламя? Предположите, что полоса имеет форму треугольника и что влиянием рассеянного излучения можно пренебречь.
5-3. Для контроля за наличием паров ртути в воздухе металлургической лаборатории ртутную лампу, дающую основное излучение в виде резонансной линии при 253,7 нм, помещают на расстояние 4 м от соответствующего детектора так, как показано на рис. 5-13. Для построения градуировочной кривой ту же лампу и фотоэлемент размещают близко друг к другу, при этом излучение проходит через кварцевую кювету толщиной 2 см. При температуре кюветы 100 °C капля ртути создает давление паров 36,4 Па (0,273 мм рт. ст.). Каково парциальное давление паров ртути в комнате, если оптическая плотность при измерении в воздухе (на расстоянии 4 м) составляет точно 1,0% от значения, полученного прн использовании кюветы?
Рис. 5-12. Полоса испускания источника непрерывного света с монохроматором (а); испускание лампы с полым катодом (б) и соответствующая линия поглощения (в).
Атомно-абсорбционная спектроскопия 147
Лампа
2м
Зеркало
Фото- а
элемент
Лампа Кювета
C=F—В---------□ Фотоэлемент
Рис. 5-13. Ртутная лампа и фотоэлемент, установленные на расстоянии друг от друга (а) и близко друг к другу для калибровки (б).
5-4. Альдегиды можно определить косвенным АА-методом по реакции окисления ионами Ag+ (реагент Толлена) с образованием 2 моль элементного серебра на каждый моль альдегида. Серебро затем растворяют в азотной кислоте и определяют методом ААС при 328,1 нм. Интервал определяемых концентраций серебра составляет 2—20 мкг/мл. Определите соответствующий ему интервал количеств альдегида (в микромолях) при условии, что объем пробы составляет 10 мл.
5-5. Предположим, что диск прерывателя в АА-спектрофотометре (типа изображенного на рис. 5-8, б) помещен между пробой и монохроматором. Увеличится или же уменьшится при этом ток фотоумножителя? Поясните свой ответ.
5-6. Нужно определить никель в речной воде методом пламенной ААС, Предварительные опыты показали, что для прямого определения концентрация никеля слишком мала, поэтому требуется операция концентрирования. Предложено использовать ионообменную колонку, заполненную катионитом в натриевой форме. При пропускании через колонку порции воды объемом 10 л удерживается весь никель (и другие катионы), замещая при этом ионы натрия. Затем через колонку пропускают НС1О4, вымывая тем самым весь никель. Элюат разбавляют до 50,00 мл. Аликвотные порции этого раствора объемом 10 мл последовательно смешивают с известными количествами стандартного раствора Ni(C104)2, содержащего 0,0700 мкг/мл Ni, и распыляют в пламени. Были получены (при 232,0 нм_) следующие результаты, выраженные в величинах отклонения пера самописца (в мм):
10 мл анализируемого раствора
10 мл того же раствора Н- 5 мл стандарта
10 мл того же раствора + 10 мл стандарта
10 мл того же раствора + 10 мл стандарта
20
48
76
104
Какова концентрация никеля (в частях на 109) в речной воде? (О методе стандартных добавок см. гл. 26.)
Литература
1. Delves И. Т., Analyst (London), 1970, v. 95, р. 431.
2. Woodriff К., Appl. Spectrosc., 1974, v. 28, p. 413.
3. Sturgeon R. E., Anal. Chem., 1977, v. 49, p. 1255A.
4. Van Loon J. C., Analytical Atomic Absorption Spectroscopy: Selected Methods, Academic Press, New York, 1980, pp. 31, 60.
148 Глава 5
5. O’Haver Т. С., Hamly J. М., Zander А. Т., Anal. Chem., 1978, v. 50, p. 1218.
6. Hamly J. M., O'Haver T. C., Golden B., Wolf W. R., Anal. Chem., 1979, v. 5 b, p. 2007.
7. Koirtyohann S. R., Pickett E. E., Anal. Chem., 1965, v. 37, p. 601.
8. Kahn H. L., At. Abs. Newsletter, 1968, v. 7, N 2, p. 40.
9. Jenkins F. A., White H. E., Fundamentals of Optics, 4th ed., McGraw-Hill, New York, 1976, pp. 679—686.
10. Brodie K. G., Liddell P. R., Anal. Chem., 1980, v. 52, p. 1059.
11. Liddell P. R., Brodie K. G., Anal. Chem., 1980, v. 52, p. 1256.
12. Brown S. D., Anal. Chem., 1977, v. 49, p. I269A.
13. Hamly J. M., O’Haver T. C., Anal. Chem., 1977, v. 49, p. 2187.
14. Sotera J. J., Kahn H. L., Am. Lab., 1982, v. 14, No. 11, p. 100.
15. Kahn H. L. J., Chem. Educ., 1966, v. 43, p. A7, A103.
16. Oles P. J., Siggia S., Anal, Chem., 1974, v. 46, p. 911.
ГЛАВА
Молекулярная люминесценция: флуориметрия, фосфориметрия и рамановская спектроскопия
В этой главе мы рассмотрим ряд методов, в которых излучение поглощается молекулярными частицами и снова излучается при другой длине волны. Сюда включена также рамановская спектроскопия (спектроскопия комбинационного рассеяния, КР), хотя механизм поглощения здесь все же иной.
Рис. 6-1 во многом повторяет рис. 2-2, но основное внимание здесь уделяется излучению, испускаемому пробой. Процесс IX на рис. 2-2 вообще не был изображен; он происходит с участием гипотетического нестабильного уровня (который можно обозначить /?) и приводит к возникновению рамановского' спектра.
Целесообразно сравнить интервалы времени между поглощением и испусканием фотона во всех этих процессах. Легче всего увидеть разницу между флуоресценцией и фосфоресценцией. Последняя может иногда длиться несколько секунд после удаления источника возбуждения, тогда как время задержки флуоресценции составляет 10~9—10~7 с. Эти интервалы представляют собой полупериоды существования возбужденных частиц, т. е. промежутки времени, необходимые для того, чтобы половина молекул испустила фотоны и вернулась в исходное состояние. Другими словами, возбужденное состояние флуоресцирующих молекул может продолжаться в среднем 10-8 с, тогда как молекулы фосфоресцирующих веществ, будучи мета-стабильными, находятся в возбужденном состоянии гораздо дольше. Молекулы, активные в рамановской спектроскопии, не найдя подходящего уровня Д, могут сразу же вернуться на основной электронный уровень 50. Очевидно, что эти три процесса протекают по разным механизмам.
Флуориметрия
При возбуждении (процесс I) большинство молекул приобретают колебательную и электронную энергию и переходят на некоторый колебательный подуровень возбужденного электрон-
150 Глава 6
Рис. 6-1. Переходы при флуоресценции и фосфоресценции. Рисунок дает более детальную картину (с последовательной нумерацией) части рис. 2-2. Переход, обозначенный буквой а, соответствует области перекрывания спектров возбуждения и флуоресценции, поскольку его энергия меньше энергии перехода б. Переход IX относится к рамановской спектроскопии.
ного состояния. Большинство таких возбужденных молекул в результате потери энергии при столкновениях стремятся перейти на самый низкий колебательный уровень (процесс II). Этот безызлучательный процесс заканчивается тем, что молекулы оказываются на возбужденном синглетном электронном уровне 51, с которого они могут вернуться в исходное состояние с излучением фотона (флуоресценция, процесс IV). С меньшей вероятностью они могут перед излучением перейти на метастабильный триплетный уровень (фосфоресценция, процессы V, VI и VIII). В любом случае молекула может в конце концов оказаться в каком-либо колебательном состоянии основного уровня. Этим объясняется, почему спектры флуоресценции и фосфоресценции состоят из множества близко расположенных линий, по большей части в видимой области. В присутствии растворителя линии уширяются и сливаются друг с другом, в результате чего получается менее структурированный спектр, внешне напоминающий спектры поглощения в УФ- и видимой областях.
Поскольку молекулы, возбужденные до колебательного подуровня электронного состояния 5Ь перед флуоресценцией пере-
Молекулярная люминесценция 151
а) Возбуждение: щель 10 нм, X 250 нм Испускание: щель Знм
б) Возбуждение: щель 9 нм Испускание: щельЗнм,К397нн
О' -> 0 -переход
250 300 350 400 450 500
Длина волны, нм
Рис. 6-2. Спектры флуоресценции (а) и возбуждения (б) аитацена. Обратите внимание на то, что максимум поглощения при 250 им не имеет зеркальной симметрии [1].
ходят на свой основной уровень большая часть переходов, связанных с флуоресценцией (процесс IV), сопровождается меньшей потерей энергии по сравнению с абсорбционными переходами процесса I. Однако при комнатной температуре часть молекул всегда находится на первом и втором колебательном подуровнях основного состояния? Следовательно, возможен переход (указанный на рисунке буквой а) с такого уровня на уровень 5] с меньшей энергией, чем у перехода, обозначенного-буквой б. Поэтому, хотя основная часть флуоресцентного излучения имеет большую длину волны (энергия фотонов меньше), чем возбуждающее излучение, обычно наблюдается некоторое перекрывание, и небольшая часть испускаемого излучения проявляется в более коротковолновой области, чем самое длинноволновое излучение спектра возбуждения. Расположение колебательных подуровней электронных уровней 50 и почти идентично, поэтому спектр флуоресценции должен быть в общих чертах зеркально симметричным спектру возбуждения.
На рис. 6-2 сравниваются спектры возбуждения и флуоресценции антрацена, из которого очевидно, что они зеркально симметричны [1]. Переход 5q->Si при 378 нм проявляется в обоих спектрах. Интенсивная полоса поглощения при 250 нм обязана своим происхождением переходу 50->-S2; любая молекула, оказавшаяся на электронном уровне S2 (или более высоком), переходит в состояние 5] в результате безызлучательных переходов, поэтому в спектре испускания такой полосы нет..
152 Глава 6
'Рис. 6-3. Конструкции флуориметров. Наблюдение излучения проводится под углом 90° (а), под небольшим углом (б) и под углом 180° (в), Fi и F2— первичный и вторичный светофильтры, один из которых или оба можно заменить монохроматорами.
Флуоресцентное излучение возникает в пробе, поэтому оно распространяется во всех направлениях, и в принципе его можно наблюдать под любым углом. На практике используются три варианта (рис. 6-3). Из конструкционных и других соображений удобнее всего наблюдать излучение под углом 90° (рис. 6-3,а), и обычно этот вариант используется в конструкциях недорогих флуориметров. В очень концентрированных растворах поглощение большей части излучения (и, следова
Молекулярная люминесценция 153'
тельно, большая часть флуоресценции) происходит в непосредственной близости от грани кюветы, на которую попадает излучение (рис. 6-3,6). В данном случае целесообразно наблюдать флуоресценцию под небольшим углом. Эта геометрия предпочтительнее также при изучении растворов, заметно поглощающих при длинах волн флуоресценции, потому что тогда испускаемое излучение проходит через слой раствора минимальной толщины. Если исследование преследует теоретические цели (например, определение квантового выхода), то лучше использовать способ наблюдения на просвет (рис. 6-3, в), поскольку в расчетные уравнения войдет меньше приближенных величин [2].
Математически флуоресценция описывается более сложными зависимостями, чем поглощение, для которого существует сравнительно простой закон Бера. С одной стороны, при прохождении через массу раствора излучение поглощается по экспоненциальному закону. С другой стороны, флуоресцентное излучение всегда, хотя бы в малой степени, поглощается раствором, но толщина слоя, через которую оно проходит; не является постоянной величиной, поскольку излучение возникает во всем объеме раствора, а не в какой-либо одной точке. Полные уравнения имеют слишком громоздкий вид и здесь не рассматриваются, их можно найти в литературе [2, 3].
В простейшем случае в разбавленном растворе находится лишь один вид поглощающих и флуоресцирующих частиц. Основное уравнение для такого случая имеет вид
F = рок(1^-иГА) (6-1)
где F — интенсивность флуоресцентного излучения, попадающего в детектор, Ро — интенсивность падающего УФ-излучения. Множитель К, являющийся постоянной для данной системы и прибора величиной, представляет собой фактор перехода от интенсивности поглощенного излучения к той части непоглощенного флуоресцентного излучения, которая поступает в детектор; А — оптическая плотность раствора при длине волны падающего излучения.
Это выражение можно преобразовать в более удобную форму, применив разложение члена 10-А в ряд Тейлора:
F = Р0К [2,ЗОА -+ J213W _ . . . 1 (6-2)
L 2! 3! J v
В первом приближении для растворов с малой оптической плотностью квадратными членами и членами более высоких степеней можно пренебречь и получить прямую зависимость
154 Глава 6
Рис. 6-4. Зависимость флуоресценции фенола в воде от концентрации. Рисунок наверху является увеличенным изображением начального отрезка основного графика. Возбуждение при 296 нм, флуоресценцию наблюдали при 330 нм, pH 6,5 [5].
между интенсивностью флуоресценции F и оптической плотностью, которая в свою очередь пропорциональна концентрации. Однако включение в расчет члена А2 уменьшает ошибку вычислений примерно в 8 раз [4]. Очевидно, нужно оставлять столько членов, чтобы ошибка при их исключении была меньше, чем ошибка эксперимента.
При проведении серийных анализов ошибку можно уменьшить, построив градуировочный график по растворам с известной концентрацией. На рис. 6-4 представлена зависимость флуоресценции водных растворов фенола от концентрации [5], из которой очевидно, что кривая почти линейна лишь до концентрации 10 мкг/мл, а определение возможно примерно до 70 мкг/мл. За этой точкой интенсивность падает вследствие сильного поглощения испускаемого излучения раствором.
Таким образом, в качестве аналитического метода измерение флуоресценции можно использовать только для очень разбавленных растворов, концентрации которых гораздо ниже, чем при измерении поглощения. При измерении флуоресценции
Молекулярная люминесценция 155
разбавленных растворов фотоэлемент фиксирует слабый свет на темновом фоне, тогда как при измерении оптической плотности того же раствора измеряемая величина представляет собой очень небольшое ослабление падающего излучения. Это равноценно точному определению отношения двух почти одинаковых больших чисел, что является очень неблагодарной задачей. Область определяемых концентраций, указанная на рис. 6-4, как раз попадает в благоприятные условия измерения, поэтому чувствительность аналитического метода очень высока.
Тушение флуоресценции. Так называется явление уменьшения интенсивности флуоресценции (или фосфоресценции) вследствие специфического влияния состава раствора. Тушение, происходящее в результате сильного поглощения раствором падающего или испускаемого излучения, называется концентрационным тушением или эффектом внутреннего фильтра. Если же этот эффект вызван самим флуоресцирующим веществом, он называется самотушением-, примером служит отклонение от прямолинейной зависимости при повышении концентрации фенола (рис. 6-4).
Тушение может быть также вызвано безызлучательными потерями энергии возбужденными молекулами. Реагент, вызывающий тушение, облегчает переход молекул с возбужденного синглетного на триплетный уровень, с которого излучения не происходит. Тушение многих ароматических соединений растворенным кислородом протекает, вероятно, по этому механизму. В некоторых системах тушение флуоресценции может быть вызвано переносом электрона с участием возбужденной молекулы, например флуоресценция метиленового голубого тушится ионами двухвалентного железа.
Уменьшение тушения вследствие изменений в химической природе флуоресцирующего вещества иногда называют химическим тушением. Как правило, этот вид тушения зависит от pH. -Например, в интервале pH 5—13 возбуждение при 290 нм вызывает голубую флуоресценцию анилина. При более низких значениях pH анилин существует в виде катионов анилиния, а в сильнощелочной среде — в виде анионов; причем ни та ни другая частица не флуоресцирует.
Флуоресценция многих соединений зависит от температуры в большей степени, чем этого следовало бы ожидать, основываясь на опыте абсорбционной спектроскопии. В флуориметрии трудности часто связаны с загрязнением пробы флуоресцирующими веществами, которые нельзя обнаружить большинством других методов. Действительно, вода и водные растворы могут извлекать флуоресцирующие вещества из резиновых и даже бакелитовых пробок и пластмассовой посуды [6]. Значительные
156 Глава 6
количества тушителей флуоресценции иногда поступают и из других подобных же источников, таким примером служит загрязнение следами хрома из хромовой смеси, используемой для очистки посуды.
Спектрофлуориметры
Основная особенность конструкции флуориметров заключается в наличии двух оптических систем, одна для возбуждающего, другая для флуоресцентного излучения. Монохроматоры могут использоваться в одной или обеих системах или могут быть заменены светофильтрами.
Приборы с монохроматорами в обеих системах называются спектрофлуориметрами-, в качестве примера можно привести спектрофлуориметр фирмы Perkin-Elmer модели MPF-44B, представленный на рис. 6-5.
Модулированный поток излучения, испускаемый ксеноновой лампой мощностью 150 Вт, проходит через монохроматор Черни—Тернера и направляется на кварцевую кювету, где частично поглощается пробой. Часть флуоресцентного излучения направляется во второй монохроматор. Определенная доля пер-
Рис. 6-5. Схема спектрофлуориметра фирмы Perkin-Elmer (модель MPF-44B) (Perkin-Elmer Corporation).
Молекулярная люминесценция 157
вичного потока отклоняется от основного пути и контролируется с помощью вспомогательного детектора, что позволяет вводить поправку к показаниям основного фотоумножителя на изменение интенсивности ксеноновой лампы при сканировании длин волн.
Работа на спектрофлуориметре обычно состоит из ряда операций. В результате ориентировочных предварительных исследований выбирают подходящую длину волны в спектре испускания и настраивают второй монохроматор на эту длину волны. Затем записывают спектр возбуждения на первом монохроматоре. Аналогично получают спектр испускания сканированием на втором монохроматоре, настроив первый на соответствующую длину волны. Для большинства веществ изменение выбранной длины волны на первом монохроматоре мало влияет на спектр излучения, как бы далеко ни отстояла она от максимума.
Спектрофлуориметры используются для выбора условий определения и изучения влияния мешающих веществ, а также для выполнения серийных анализов. В последнем случае для повышения точности результаты следует сравнивать с результатами, полученными на том же приборе для стандартных растворов. Часто спектры, полученные на разных приборах, отличны друг от друга прежде всего вследствие изменения длины волны излучения источника, абсолютной чувствительности детектора и эффективности монохроматоров. Наблюдаемые спектры представляют некую комбинацию спектральных свойств вещества и ложных сигналов, даваемых прибором. Такие отклонения особенно существенны при измерении квантового выхода флуоресценции.
Спектрофлуориметр можно откалибровать путем строгого сравнения со стандартными источниками и термопарой или предварительной калибровкой фотоумножителя. Полученные таким способом поправки могут быть довольно большими: поправочный коэффициент изменяется от значения менее единицы до значения 50 или выше [7] (рис. 6-6).
Для того чтобы исключить многие неудобства этого метода, для сравнения используют флуоресцирующее стандартное вещество, в качестве которого рекомендован [8] хелат алюминия с азокрасителем понтахромовым черно-синим R. Известно, что родамин 6G обладает характерным откликом на возбуждение в широком интервале длин волн (220—600 нм) и, следовательно, может использоваться как счетчик фотонов, что облегчает калибровку фотоумножителя [9]. Использование способов калибровки, описанных в упомянутых выше работах, в будущем должно привести к уменьшению сообщений о «нескорректированных» спектрах.
158 Глава 6
Рис. 6-6. Скорректированный и нескорректированный спектры возбуждения хинина (0,1 мкг/мл) в 0,05 М H2SO4 [7].
Как показано на рис. 6-5, спектрофлуориметр не является двухлучевым прибором в том смысле, в каком этот термин используется в абсорбционной спектроскопии. Второй фотоумножитель служит только для корректировки изменений режима лампы. Фон, возникающий из-за флуоресценции растворителя и реагентов или из-за рассеяния света частицами пыли и т. п., определяют путем измерения холостой пробы и затем вычитают; эту операцию легко выполнить на современных приборах с микропроцессорной техникой.
Флуориметры
Для аналитических целей часто не требуется разрешения пиков флуоресценции, как, например, при добавлении реагента, селективно реагирующего с определяемым веществом с образованием флуоресцирующего производного. Тогда отпадает надобность в дорогих монохроматорах, их можно заменить светофильтрами. На рис. 6-7 представлен простейший прибор, в ко-
Молекулярная люминесценция 159
тором используются два светофильтра: Fi, пропускающий УФ и задерживающий видимое излучение, и F2, поглощающий любое УФ-излучение, попавшее в детектор, и пропускающий видимое излучение. Этот упрощенный флуориметр может иметь более высокую чувствительность, чем спектрофлуори-метр, так как через светофильтр проходит большее ко-
Ъ КюВета
Рис. 6-7. Простой флуориметр с возбуждением под прямым углом.
личество света, чем через
монохроматор. Мощность питающего лампы и фотоумножитель устройства в обоих типах приборов должна быть строго стабильной.
Применения
Особенностью флуориметрии является ее высокая чувствительность, которая часто в 102—104 раз превышает чувствительность абсорбционных методов. Кроме того, этот метод более селективен, поскольку флуоресцирует меньшее число соединений, чем поглощает.
К флуоресценции в видимой области способны в основном два класса веществ: 1) большое число минералов и неорганические твердые люминофоры и 2) органические и металлоорганические соединения, обладающие интенсивным поглощением в УФ-области. Что касается веществ первого класса, мы упомянем лишь метод определения следов урана в горных породах и природных водах [10]. К растворенной пробе добавляют Са(ЬЮз)2 и затем (медленно) NH4F. Образующийся при этом осадок CaF2 захватывает уран в виде фторида. Осадок отфильтровывают, высушивают, прокаливают при 800 °C, затем измельчают. Получившуюся пудру спрессовывают в таблетки и исследуют на флуориметре. Как показано в цитируемой работе, возбуждение проводилось аргоновым ионным лазером при длине волны 488 нм. По данным авторов, предел обнаружения составляет 0,01 пг/мл.
В неорганической химии флуоресцентный анализ применялся, например, для определения следовых количеств кадмия в виде соединения с кальцеином, являющегося карбоксиме-тильным производным бис (диметиламинометил) флуоресцеина [11]. Кадмий предварительно отделяют от мешающих ионов методами ионного обмена. Флуоресценцию измеряют в 0,5 М растворе КОН при 520 нм с возбуждением при 490 нм; предел обнаружения составляет 2,2 нг/мл.
Глава 6
В органической химии флуоресценция применяется в основном для определения полициклических молекул, в том числе многих веществ, имеющих значение в биохимии и фармакологии. Ниже кратко описаны две такие методики.
Тиамин (витамин Bi) можно количественно определить по голубой флуоресценции его продукта окисления — тиохрома [12]. К пробе добавляют фосфатазу — фермент, вызывающий гидролиз фосфорного эфира тиамина, который часто присутствует в пищевых продуктах. Фосфатазу и другие нерастворимые вещества отфильтровывают, а фильтрат разбавляют до известного объема. Отбирают две аликвотные порции: одну для анализа, другую для приготовления холостой пробы. К первой аликвотной порции добавляют окислитель ^(феррицианид) и к обоим — NaOH и изобутанол. После встряхивания и отделения водного слоя спиртовой раствор исследуют на флуориметре.
Рибофлавин также можно определить флуориметрически [12]. Интенсивность флуоресценции в большой степени зависит от точного соблюдения условий эксперимента, а также от природы и количества присутствующих примесей. Для того чтобы примеси оказывали одинаковое влияние на стандартные и анализируемые растворы, используют метод стандартных добавок. При этом измеряется суммарная флуоресценция определенного количества стандарта и неизвестного количества определяемого компонента. Рибофлавин легко окисляется, превращаясь в нефлуоресцирующее вещество, которое количественно восстанавливается снова в рибофлавин, что используется в данной методике.
Другая область применения спектрофлуориметрии — получение УФ-спектров поглощения очень небольших по размеру проб или очень разбавленных растворов. При этом используется идентичность скорректированных спектров возбуждения флуоресценции и спектра поглощения. Спектр возбуждения имеет менее тонкую структуру, но зато сам метод флуоресценции более селективен, чем методы абсорбционной спектроскопии, потому что многие примеси не флуоресцируют. Если нельзя получить скорректированный спектр флуоресценции, метод нужно использовать с осторожностью, поскольку возможны сильные искажения, связанные с тушением флуоресценции. В этом случае следует снять еще несколько спектров при разных концентрациях; если они идентичны, концентрационное тушение, по всей вероятности, можно исключить. В благоприятных условиях можно получить спектры поглощения растворов, содержащих нанограммовые или микрограммовые количества флуоресцирующих соединений.
Молекулярная люминесценция 161
б
Рис. 6-8. Спектры возбуждения и излучения флуоресценции и синхронный спектр фенантрена (а), антрацена (б) и перилена (в) [13].
6 Заказ 537
162 Глава 6
Рис. 6-9. Обычный (а) и синхронный (б) спектры флуоресценции смеси нафталина, фенантрена, антрацена, перилеиа и тетрацена [13].
Синхронная флуориметрия. При одновременном сканировании длин волн обоих монохроматоров можно получить резко выраженный флуоресцентный сигнал [13, 14]. Пик появляется только в той области, где перекрываются спектры возбуждения и излучения. Часто самый коротковолновый пик в спектре испускания слегка сдвинут и находится в области более длинных волн, чем самый длинноволновый пик спектра возбуждения. Поэтому для получения оптимального результата между двумя монохроматорами следует поддерживать небольшую постоянную разность длин волн (или частот [15]).
На рис. 6-8 показан этот тип сигнала на примере трех ароматических углеводородов. Сдвиг длины волны составляет
Молекулярная люминесценция 163
3 нм. Достоинство этого метода заключается в гораздо лучшем разрешении при исследовании смеси флуоресцирующих соединений (рис. 6-9).
Фосфориметрия
Процесс фосфоресценции отличается от флуоресценции, во-первых, длительностью (для фосфоресценции полупериод составляет от 10-1 до 10 с, а для флуоресценции—10-7—10-9 с) и, во-вторых, сдвигом максимума в более длинноволновую область по сравнению с флуоресценцией. Обе эти особенности можно объяснить, обратившись к рис. 6-10. Переходы с изменением мультиплетности (S->~T или T-+S) запрещены; это значит, что вероятность их мала. Однако возможна интеркомбинационная конверсия с Si на один из колебательных уровней состояния очень близкий по энергии (процесс V на рисунке), затем происходит безызлучательный переход (процесс VI) на основной уровень состояния Т\.
Триплетное состояние Т1 является метастабильным, и находящиеся в нем молекулы обладают избыточной энергией, которая впоследствии должна высвободиться. Потеря энергии может происходить по любому из нескольких конкурирующих механизмов, в том числе в результате фосфоресценции (процесс VIII). Из других возможных путей особенно важны два.
Рис. 6-10. Переходы при фосфоресценции молекул (более подробная картина соответствующей части рис. 6-1).
6»
164 Глава 6
Один из них — кислородное тушение, когда энергия передается молекулам кислорода, в которых осуществляются переходы, поскольку они находятся в основном триплетном состоянии. Поэтому из кюветы фосфориметра нужно тщательно удалять кислород.
Второй путь, по которому может произойти потеря энергии,— столкновения, имеющие для фосфоресценции большее значение, чем для флуоресценции, из-за длительности свечения. Дезактивацию молекул при столкновениях можно уменьшить или устранить любым способом, приводящим к более жесткой структуре (молекулы оказываются иммобилизованными). Этого можно достигнуть повышением вязкости подходящего растворителя при охлаждении до температуры стеклования смеси. В качестве растворителя часто используется смесь этилового эфира, изопентана и этанола (ЕРА) в соотношении 5:5:2 (по объему) при температуре жидкого азота (77 К). Эта смесь не кристаллизуется, что очень важно, поскольку кристаллизация может привести к нежелательному выделению растворенного вещества.
Сравнительно недавно найдены другие методы иммобилизации пробы, позволяющие обойтись без криостата с жидким азотом, неудобного в обращении [16]. Фосфоресценцию при комнатной температуре можно наблюдать в образцах, адсорбированных на фильтровальной бумаге или других твердых подложках. Этот способ прост, но недостатком его является высокий фон, создаваемый носителем.
Другая возможность связана с использованием поверхностно-активных веществ, например лаурилсульфата натрия, образующего мицеллы в водных суспензиях [17, 18]. Мицелла представляет собой крошечный шарик из 40—100 молекул детергента, гидрофильные сульфогруппы которых ориентированы наружу, а гидрофобные углеводородные (лаурильные) остатки обращены внутрь. На рис. 6-11 представлена двумерная схема такой мицеллы [18]. Внутри кластера создаются условия для захвата неполярных молекул, которые запутываются в клубке углеводородных цепей и тем самым оказываются защищенными от столкновений, а также в большой степени от химических тушителей. Это позволяет исследовать спектры фосфоресценции обычными методами на приборах, применяемых в флуори-метрии.
Скорость интеркомбинационной конверсии и Тг->
—>-S0) можно значительно повысить внедрением в пространство, окружающее исследуемый атом, атома с большой атомной массой (эффект тяжелого атома) [19]. Увеличение скорости интеркомбинационной конверсии позволяет получать более ярко выраженный спектр фосфоресценции. С этой целью
Молекуляряая люминесценция 165
wV7Wv’7V/'AVA
ВоЭлая (раза
со
Определяете Вещество
Рис. 6-11. Схема анионной мицеллы в растворе. Молекулы определяемого вещества (нафталина) заключены внутри мицеллы [18J.
Рис. 6-12. Спектры фосфоресценции 6-10“5 М раствора нафталина в мицеллярном водном растворе (а); в мицеллярном растворе, в котором 30 % атомов Na заменены иа атомы Т1 (чувствительность при записи спектра в 10 раз выше, чем в случае рис. а) (6); в этанольном растворе при 77 К (чувствительность записи в 25 раз выше, чем в случае рис. а) (в) [18].
166 Глава 6
в смесь ЕРА можно добавить метилиодид. При получении мицеллы часть (50—60 %) натрия в поверхностно-активном веществе можно заменить на таллий [18]. Типичный пример приведен на рис. 6-12.
Фосфориметры
Фосфоресцирующие соединения обязательно флуоресцируют, поэтому фосфориметр должен различать оба вида свечения. Для этого можно использовать вращающийся прерыватель, так называемый фосфороскоп (рис. 6-13), обеспечивающий определенную задержку между возбуждением и наблюдением свечения. Этот принцип положен в основу конструкции ряда приборов. Теоретические аспекты оптимальной конструкции прерывателя обсуждаются в интересной статье О’Хэвера и Вайнфорднера [20].
Вращающийся прерыватель можно заменить импульсной вспышкой [21], где в качестве источника первичного излучения используется импульсная лампа. С помощью электронной синхронной схемы сначала включается вспышка, а затем через заданный интервал времени — фотоумножитель. Такая система позволяет измерять интервал времени до 10-5 с, что не так просто сделать с помощью механического прерывателя.
Многие органические соединения с сопряженными л-элек-тронными связями фосфоресцируют, что представляет прекрасную возможность для определения их следов. Подробнее эти вопросы обсуждаются Вайнфорднером и Латцем [22]. Ими предложен простой и быстрый способ определения аспирина в сыворотке крови и плазме в присутствии свободной салициловой кислоты.
Дополнительные технические возможности в фосфориметрии дает разрешение во времени [23]. При достаточно большой
Кювета -вращающийся кожух
К монохроматору и фотоумножителю
От монохроматора а лампы
Рис. 6-13. Разрез вращающегося кожуха фосфороскопа. Задержка наблюдения спектра соответствует времени, необходимому для поворота кожуха на 90°.
Молекулярная люминесценция 167
разнице между временами затухания возможен анализ многокомпонентных систем. Разрешения во времени можно легко добиться изменением задержки между возбуждением и наблюдением или (еще успешнее) используя прием логарифмического вычитания [21]. Для двух компонентов А и В интенсивность излучения равна
Рфос = РА + рв = Ро, а ехр (— 1 /тА) + Р0,в ехР (— 1 /тв) (6-3) где т — время затухания, т. е. время, требующееся на уменьшение фосфоресценции вещества на 1/е = 36,8% начального значения. Если регистрировать значения Рф0С в зависимости от времени до прекращения испускания обоими компонентами, то можно определить количество вещества с более длинным периодом затухания по данным, полученным после прекращения испускания другого компонента. Затем можно вычесть его вклад и получить интенсивность излучения для короткоживущего вещества.
В фосфоресценции можно использовать синхронное сканирование [24]. Сдвиг в длине волны между монохроматорами для возбуждающего и испускающего излучений должен соответствовать разности энергий между уровнями Si и Ti, что усложняет данный метод по сравнению с синхронным сканированием в методе измерения флуоресценции, но его преимущество, заключающееся в хорошем разрешении смеси определяемых веществ, сохраняется.
Рамановская спектроскопия [25, 26]
При определенных условиях фотон может временно передать свою энергию какой-либо молекуле независимо от ее энергетического состояния (как это допускается процессом IX на рис. 6-1) и повысить ее энергию до некоторого «фактического» уровня R. Однако в общем случае этот уровень не отвечает устойчивому состоянию, и молекула должна немедленно перейти в свое основное состояние. При возвращении она может остаться в возбужденном колебательном состоянии (переход в на схеме), при этом энергия испускаемого фотона будет меньше энергии, необходимой для возбуждения на определенный колебательный подуровень. С другой стороны (переход д), некоторые молекулы уже могли находиться в возбужденном колебательном состоянии, и тогда испускаемый фотон будет иметь несколько большую энергию. Если же молекула возвращается на тот же уровень энергии, с которого произошел переход (переход г), энергия (или длина волны) не изменяется.
Как будет показано дальше, рамановский спектр и в самом деле состоит из симметричных серий частот (линий) по обе
168 Глава 6
Стоксовы линии
Антистоксовы линии
Рис. 6-14. Рамановский спектр жидкого ССЦ, полученный при сканировании со скоростью 500 см-1/мин 3 мкл пробы при возбуждении гелий-неоновым лазером при 632,8 нм. Сильный сигнал при Av = 0 связан с рэлеевским рассеянием лазерного излучения [34].
стороны от некоторой центральной частоты. Линии в длинноволновой части называются стоксовыми, а в более коротковолновой части — антистоксовыми. Центральная линия, без смещения частоты, соответствует простому процессу рассеяния, который рассматривается в гл. 8. (По этой причине рамановский эффект в чистом виде часто относят к одному из видов рассеяния.)
Возбуждение может произойти под действием излучения любых частот, поэтому при выборе источника перед конструкторами приборов открываются широкие возможности. Необходимо, чтобы излучение было близко к монохроматическому и в то же время достаточно интенсивным. Лучшим источником в рамановской спектроскопии является лазер, который полностью вытеснил ранее используемые ртутные лампы. Для работы в видимой области обычно выбирают гелий-неоновый (632,8 нм) или аргоновый ионный лазер (488,8 или 514,6 нм).
При построении рамановского спектра одной из координат обычно служит рамановское смещение Av, получаемое из соотношения
Av = ± (v4 — Vfl) (6-4)
где vt- и vr — волновые числа падающего и рамановского излучений (рис. 6-14). Стоксовы линии соответствуют положитель
Молекулярная люминесценция 169
ным значениям Av. По шкале ординат откладывают относительную интенсивность излучения, регистрируемую фотоумножителем. Стоксовы линии всегда более интенсивны, чем анти-стоксовы, а линия, обусловленная рассеянным излучением лазера (Av = 0), гораздо интенсивнее, чем те и другие.
Рамановские спектры наблюдаются для тех молекулярных соединений, у которых при колебаниях, соответствующих переходу с волновым числом Av, изменяется поляризуемость. Поляризуемость — способность к наведению диполя под действием электромагнитного поля фотона, попадающего в молекулу. При этом молекула не обязательно должна быть диполем, тогда как для поглощения инфракрасного излучения молекула должна обладать постоянным дипольным моментом, следовательно, частоты, наблюдаемые в ИК-спектре, могут не проявиться в ра-
100
90 80
70 оэ g 60 g 50 “ 40
1 30
20
10 о
2800 2400 2000 1800 1600 1400 1200 1000 800 600
Волновое число, см’’
2800 2400 2000 1800 1600' 1400 1200 1000 800 600 400 200 О
Волновое число, см''1
Рис. 6-15. Сравнение ИК- и рамановского спектров 1,4-диоксана. Спектры, снятые в регистре волновых чисел, во многом сходны. Обратите внимание на полосу при 1220 см-1, очень интенсивную в рамановском спектре и почти невидимую в ИК-спектре. Наоборот, полоса при 620 см-1, наблюдаемая в ИК, отсутствует в рамановском спектре (Courtesy of Cary Instruments Division of Varian Associates).
170 Глава 6
мановском спектре, и наоборот. Некоторые колебательные моды влияют и на поляризуемость, и на дипольный момент, тогда они будут наблюдаться и в том и другом спектрах. Сравнение двух видов спектров. 1,4-диоксана (рис. 6-15) иллюстрирует эти особенности.
Обычно рамановский спектр исследуют в спектральной области, где нет заметного поглощения пробой, потому что иначе детектор не смог бы уловить слабое рамановское излучение. Однако если использовать лазер на красителе с перестраиваемой частотой, близкой по величине, но не совсем совпадающей с частотой максимума поглощения, то чувствительность возрастает во много раз. Это явление, называемое резонансным комбинационным рассеянием [27], еще не нашло широкого аналитического применения, но перспективно в будущем.
Источником помех в рамановской спектроскопии, ограничивающим возможности метода, является флуоресценция. Флу-
Рис. 6-16. Рамановский спектр ССЦ, Симметричные валентные колебания получены при разрешении 0,5 см-1; обратите внимание на то, что здесь разрешен один из пиков, показанных на рис. 6-14.
а — при комнатной температуре; б — при 77 К после нанесения на охлажденный медиый блок. Проявилась сложная структура, отражающая пять возможных комбинаций изотопов хлора. Спектр отчетливо разрешен при низкой температуре, так как полосы стали уже [30].
Молекулярная люминесценция 171
оресцентное излучение обычно гораздо интенсивнее рамановского, поэтому небольшие количества флуоресцирующих примесей (или флуоресценция, вызванная растворителем) могут заглушить наблюдаемый сигнал. Чтобы избежать флуоресценции, нужно подобрать лазер, излучающий свет с длиной волны, находящейся за пределами полосы возбуждения флуоресценции.
Существует несколько, пожалуй, экзотических методов, представляющих собой варианты рамановской спектроскопии, наиболее интересный из которых когерентная антистоксова спектроскопия КР. Этот метод требует одновременного использования двух мощных лазеров; он подробно описан в литературе [27, 28].
Применение рамановской спектроскопии. Как показано на рис. 6-15, рамановский спектр, так же как ПК-спектр, можно использовать для идентификации веществ. Существуют атласы рамановских спектров [29]. Как и при установлении структуры, рамановский и инфракрасный спектры дополняют, а не дублируют друг друга [30] и используются также в количественном анализе.
Подготовка жидких проб включает их тщательное фильтрование для удаления следов суспендированных веществ, которые увеличивают рассеяние. Флуоресцирующие примеси необходимо тщательно удалять. Профильтрованные газы можно исследовать в многоходовой кювете. Твердые вещества или тонкие пленки, нанесенные на твердые подложки, можно' изучать при помощи отраженного от поверхности лазерного луча. Температурный контроль обычно не требуется, но спектры охлажденных проб могут проявлять интересную тонкую структуру (рис. 6-16).
Задачи
6-1. Навеску свинины массой 2 г проанализировали на содержание витамина В! (тиамина) методом, включающим образование тиохрома. Его извлекали НС1, обрабатывали фосфатазой и разбавляли до 100 мл. Аликвотную порцию объемом 15 мл очищали адсорбцией и элюировали, в результате чего объем увеличивался до 25 мл. Из него отобрали две порции объемом 5 мл каждая, к одной из них (А) добавляли КзЕе(СМ)е и обе (А и Б) разбавляли до 10 мл для флуорнметрическпх исследований. Аналогичные операции проделывали со стандартным раствором, содержащим 0,2 мкг/мл тиамина, за исключением того, что порцию, введенную в адсорбционную колонку, разбавляли до исходного объема после элюирования (т. е. не разбавляли). Затем отобрали еще две аликвотные порции по 5 мл, одну из которых (В) окислили, и обе разбавили (В н Г) до конечного объема 10 мл для измерения флуоресценции. Были получены следующие результаты измерений:
172 Глава 6
Раствор Относительная интенсивность флуоресценции
А (станд., окисленный) Б (станд., холостой) В (проба, окисленный) Г (проба, холостой) 62,4 7,0 52,0 8,0
Рассчитайте содержание витамина Bi в свинине в микрограммах на грамм.
6-2. Показано [31], что реакцию Ханца можно использовать для определения альдегидов и первичных аминов, в том числе аммиака, с высокой чувствительностью. В присутствии 2,4-пентандиона (ацетилацетона) при pH 6 образуется циклический аддукт с интенсивной флуоресценцией. Реакция протекает так:
w л,—
Н3С
О=СХ ^н2 R, Кэ
О 1 О
II /с\ II
R—С—С С—С—R
II И
+ ЗН;О
а) Если аммиак находится в избытке, интенсивность флуоресценции после вычитания значения холостой пробы почти линейно зависит от концентрации формальдегида в диапазоне 0,005—1,0 мкг/мл. Каков примерный интервал линейной зависимости при определении следов анилина в присутствии избытка формальдегида? б) В спектре поглощения флуоресцирующего соединения имеется довольно интенсивный максимум при 410 нм (е=8000), слишком большой, чтобы отнести его за счет хромофоров несвязанных 2,3-ненасыщен-ных кетонных групп. Можете ли вы предложить объяснение этому факту?
6-3. При флуориметрнческом определении 7Н-бенз(</е)аитрацен-7-оиа (ВО) (загрязнителя воздуха) возможны значительные помехи со стороны флуоресцирующих полиядерных углеводородов [32]. Флуоресценция последних в сравнении с флуоресценцией БО легче тушится 1,3-динитробензолом (ДНБ), что можно использовать в аналитических целях, а) Добавление к раствору 15 % ДНБ уменьшает интенсивность флуоресценции F 5-10~6 М БО при измерении на флуориметре от 0,7 до 0,4 (относительных единиц). Каким будет выигрыш в чувствительности при использовании тушителя, если указанное количество ДНБ уменьшает флуоресценцию мешающих веществ до 0,005 исходной концентрации? б) Можно ли обойтись без тушителя, если для измерений использовать спектрофлуориметр?
6-4. Спектр поглощения флуоресцирующего вещества, полученный на обычном спектрофотометре, оказался искаженным из-за флуоресценции. Предположим, соединение имеет максимум поглощения (следовательно, и максимум возбуждения) при 290 им, а максимум флуоресценции при 350 нм. При какой длине волны следует ожидать самой большой ошибки? Будет ли наблюдаемая оптическая плотность в этой точке слишком велика или слишком мала? Изменится ли ваш ответ, если измерение проводить на спектрофотометре, в котором излучение лампы сначала проходит через кювету, а затем диспергируется монохроматором? Поясните свой ответ.
Молекулярная люминесценция 173
Время, мс
Рис. 6-17. Зависимость фосфоресценции двухкомпонентной смеси от времени.
6-5. Рамановский спектр чистого соединения был получен с использованием излучения аргонового ионного лазера с длиной волны 488,8 нм. Рамановские линии были найдены при длинах волн 496,6, 498,5, 506,5 и 522,0 нм. а) Рассчитайте для этих линий значения рамановских смещений в волновых числах, б) Какова длина волны антистоксовой линии, соответствующей стоксовой линии при 522,0 нм?
6-6. Рамановскую спектроскопию можно применить для количественного анализа смесей газов. Были изучены смеси трех газов — метана, изобутана и азота. Использованы следующие рамановские смещения (в см-1): при 2917 для метана, два смещения при 800 и 2917 для изобутана и при 2331 для азота. Какие измерения следует провести с чистыми стандартами или смесями известного состава, чтобы разработать методику серийного анализа смесей этих трех газов?
6-7. Имеются сведения, что в благоприятных условиях, используя для освещения гелий-неоновый лазер, можно наблюдать рамановскую линию, смещенную всего на 2 см-1 от возбуждающего излучения. Каковы длина волны и частота соответствующего перехода в ИК-спектре поглощения?
6-8. Объясните качественно форму кривой флуоресценции фенола на рис. 6-4, если она получена на приборе с конструкцией, изображенной на рис. 6-3, а.
6-9. Найдите литературные источники информации об использовании родамина 6G и родственных соединений в качестве «счетчика квантов» и объясните, как оии действуют.
174 Глава 6
6-10. На рис. 6-17 представлены полученные с помощью компьютера данные, описывающие фосфоресценцию раствора, содержащего два активных вещества А и Б. По оси ординат отложен десятичный логарифм интенсивности фосфоресценции Рфос [уравнение (6-3)], фиксируемой фотоумножителем (в таблице на рисунке приведено несколько экспериментальных значений). Рассчитайте постоянные Та и т£ и относительные концентрации А и Б при условии, что чувствительность одинакова для обоих веществ [т. е. константа А в уравнении (6-1) имеет одно и то же значение] и для измерения подходит одна и та же длина полны.
Литература
1. Smith Н. F. Res. and Dev., July, 1968, p. 20.
2. Fletcher M. /7., Anal. Chem., 1963, v. 35, p. 278, 288; Milkey R. G., Fletcher M. HJ. Am. Chem., Soc., 1957, v .79, p. 5425.
3. Holland. J. F., Teets R. E., Kelly P. M., Timnick A., Anal. Chem., 1977, v. 49, p. 706.
4. Lam R. B., Leary J. J., Appl. Spectrosc., 1979, v. 33, p. 17.
5. Williams R. T„ Bridges J. W., J. Clin. Pathol., 1964, v. 17, p. 371.
6. Kordan H. A., Science, 1965, v. 149, p. 1382.
7. Джон IL Флуориметрические методы определения следов элементов. В кн.: Спектроскопические методы определения следов элементов под ред. Вайн-фордпера Дж.— М.: Мир, 1979.
8. Argauer R. /., White С. Е., Anal. Chem., 1964, v. 36, р. 368 (дополнения: р. 1,022).
9. Demas J. N., Crosby G. A., J. Phys. Chem., 1971, v. 75, p. 991.
10. Perry D. L., Riedner S. M„ Bowman H. R., Milanovich F. P., Hirschfeld T., Miller S., Anal. Chem., 1981, v. 53, p. 1048.
11. Hefley A. J., Jaselskis B., Anal. Chem., 1974, v. 46, p. 2036.
12. Association of Vitamin Chemists, Inc., Methods of Vitamin Assay, 3d ed., Wiley-Interscience, New York, 1966.
13. Vo-Dinh T., Anal. Chem., 1978, v. 50, p. 396.
14. Vo-Dinh T., Appl. Spectrosc., 1982, v. 36, p. 576.
15. Inman E. L., Winefordner J. D., Anal. Chem., 1982, v. 54, p. 2018.
16. Miller J. N., Trends in Anal. Chem., 1981, v. 1, p. 31.
17. Cline Love L. J., Skrilec M., Habarta J. G., Anal. Chem., 1980, v. 52, p. 754.
18. Cline Love L. J., Skrilec M., Am. Lab., 1981, v. 13, No. 3, p. 103.
19. McCarthy W. J., Phosphorescence Spectrometry. In Spectrochemical Methods of Analysis. Winefordner J. D. (ed.), Wiley-Interscience, New York, 1971 p. 467.
20. O'Haver T. C., Winefordner J. D„ Anal. Chem., 1966, v. 38, p. 602.
21. Fisher R. P., Winefordner J. D., And Chem., 1972, v. 44, p. 948.
22. Winefordner J. D., Latz H. W., Anal. Chem., 1963, v. 35, p. 1517.
23. Winefordner J. D., Acc. Chem. Res., 1969, v. 2, p. 361.
24. Vo-Dinh T., Gammage R. B., Anal. Chem., 1978, v. 50, p. 2054.
25. Long D. A., Raman Spectroscopy. McGraw-Hill, New York, 1977.
26. Demtroder W., In Analytical Laser Spectroscopy. Omcnetto N. (ed.), Wiley, New York, 1979, pp. 276ff.
27. Melveger A. J. (ed.), Resonance Raman Spectroscopy as an Analytical Tool, Franklin Inst. Press, Philadelphia, 1978.
28. Borman S. A., Anal. Chem., 1982, v. 54, p. 1021A.
29. Sadtler Research Laboratory, Inc., Philadelphia.
30. Sloane H. J., Appl. Spectrosc., 1971, v. 25, p. 430.
31. Belman S„ Anal. Chim. Acta, 1963, v. 29, p. 120.
32. Sawicky E., Johnson H., Morgan M., Mikrochim. Acta, 1967, p. 297.
33. Diller D. E., Chang R. F., Appl. Spectrosc., 1980, v. 34, p. 411.
34. Bulkin B. J., J. Chem. Educ., 1969, v. 46, p. A781, A859.
ГЛАВА
Фотоакустическая спектроскопия
Возбуждение молекулы при -поглощении излучения дает начало целому ряду процессов, в результате которых избыток энергии диссипирует и молекула возвращается в основное состояние. Эти процессы можно разделить на излучательные (флуоресценция и фосфоресценция) и безызлучательные. При изложении материала предыдущих глав мы не обращали особого внимания на безызлучательные процессы, но теперь разберем их подробнее.
Как показано на рис. 2-2, безызлучательные переходы происходят с более высоких колебательных подуровней на основные уровни (So, Si, Ti и т. д.). Если исследуемое вещество представляет собой жидкость или газ, энергия соответствующих переходов расходуется на увеличение теплового движения всех молекул пробы. Если же вещество — твердое тело, энергия сначала идет на усиление колебаний кристаллической решетки (иногда ее выражают в фононах, квантах колебательной энергии решетки), затем она может быть передана любому газу или жидкости, соприкасающимся с пробой. Так или иначе, энергия переходит в тепло, поэтому путем измерения повышения температуры можно получить полезную информацию. Это было осуществлено путем прямого измерения с помощью термистора [1], но более перспективным оказался метод фотоакустической спектроскопии (ФАС), называемый также оптико-акустической спектроскопией.
В ФАС используют прерывающийся (модулированный) поток излучения, в результате чего в пробе возникают тепловые колебания с частотой модуляции излучения. Периодические изменения колебательной энергии распространяются через среду в виде звуковой волны. Любую волну можно охарактеризовать тремя параметрами: амплитудой, скоростью и частотой. В данном случае под частотой имеется в виду частота модуляции излучения, под скоростью — скорость звука в данной среде, а амплитуда соответствует количеству поглощенной и перешедшей в теплоту энергии.
176 Глава 7
Рис. 7-1. Однолучевой фотоакустический спектрометр. Сигналы от фотоаку-стической кюветы и детектора сравнения попадают в разные усилители, синхронизированные с модулятором.
Для измерения акустического сигнала можно использовать два устройства: микрофон и пьезоэлектрический датчик. Если излучение поглощается газом, то звуковую волну можно регистрировать непосредственно микрофоном. Если же проба — твердое вещество, то самый удобный (хотя и не самый чувствительный) способ заключается в том, что пробу вместе с микрофоном помещают в замкнутое пространство, заполненное газом (обычно воздухом) [2, 3]. Звуковая волна, возникшая в пробе, на пути к микрофону проходит через границу раздела твердое тело — газ, где происходит значительная потеря энергии; после прохождения через границу раздела энергию можно усилить с помощью электронного усилителя.
Существует и другой способ, заключающийся в том, что твердую пробу приклеивают каким-либо клеющим материалом [4] к пьезоэлектрическому датчику. При этом энергия преобразуется в акустический сигнал с большей эффективностью, но для серийных анализов такой способ едва ли приемлем из-за трудоемкости операций. Для жидких проб используют и тот и другой приемы.
Приборы, выпускаемые промышленностью, сконструированы на основе газо:микрофонной регистрации, поэтому разберем этот способ более подробно.
На рис. 7-1 представлена блок-схема фотоакустического спектрометра, а на рис. 7-2—обычная кювета. Модулированное излучение проходит через прозрачное окно и попадает на пробу; это может быть газ, которым заполняют кювету, либо твердое вещество или жидкость (показаны на рисунке пункир-ной линией). Чтобы на микрофон не попадало прямое излучение, его обычно помещают сбоку. Чувствительность метода улучшается, если размер кюветы соответствует частоте модулированного излучения, что, правда, требует увеличения размера пробы.
Фотоакустическая спектроскопия 177
Окно
Воздух
Микрофон
Тбердая проба
Рпс. 7-2. Кювета для фотоакустических измерений. Излучение поступает сверху. Микрофон, расположенный на конце бокового отвода, контактирует с воздушной камерой детектора.
Основное ограничение ФАС связано с тепловым насыщением. Причиной его является ограниченная скорость передачи тепла через пробу (от места поглощения фотона до поверхности, где тепло передается окружающему газу). Оба процесса, оптическое поглощение и термодиффузию, можно охарактеризовать константами с размерностью см-1 — коэффициентом поглощения р, определяемым по соотношению
Р/Р0 = е-Р'
(7-1)
где I — длина оптического пути в поглощаемом слое, и коэффициентом термодиффузии а.,, который выражается следующим образом:
где v — частота в герцах, as — коэффициент термодиффузии в см2/с. Если P<as, фотоакустический сигнал пропорционален р, что и требуется в аналитической химии, но если р достигает значения, равного или превышающего а*, сигнал становится постоянным («насыщается»). На рис. 7-3 приведен гипотетический случай, иллюстрирующий этот эффект [3]. Для исследования вещества методом ФАС следует проводить измерения в точках а пли б, но не в точке в. Тепловое насыщение можно свести к минимуму, если перемешать твердую пробу с прозрачным разбавителем, например порошком AI2O3 или КВг [5].
Следует отметить, что спектрометр, изображенный на рис. 7-1, является однолучевым прибором, но он снабжен детектором сравнения, который контролирует поток излучения, выходящий из монохроматора. Чтобы скомпенсировать колебания в излучении лампы, получают отношение сигнала от микрофона и сигнала от детектора (обычно пироэлектрического). Сущест-
178 Глава 7
Рис. 7-3. Сравнение оптического (сплошная линия) и фотоакустического (штриховая линия) спектров поглощения гипотетического вещества. Тепловое насыщение наблюдается в заштрихованной зоне при р»104 см-1 [3].
вуют также двухлучевые приборы, в которых вторая фотоаку-стическая кювета заполнена сажей, служащей веществом сравнения. Сажа практически полностью поглощает все попадающее в нее излучение, и, таким образом, тепловое насыщение будет одинаковым при всех длинах волн. Следовательно, любое изменение сигнала, идущего от кюветы сравнения, будет связано только с изменениями интенсивности лампы; такие колебания можно скомпенсировать.
В отсутствие теплового насыщения сигнал от фотоакустиче-ской кюветы пропорционален интенсивности возбуждения, поэтому желательно пользоваться по возможности более мощной лампой. При работе в УФ- и видимой областях обычно применяется ксеноновая дуговая лампа мощностью до 1000 Вт.
Фотоакустический спектрометр типа показанного на рис. 7-1 не пригоден для ИК-области, потому что не существует достаточно мощных источников непрерывного ИК-излучения, обеспечивающих фотоакустический сигнал. В этом случае необхо
Фотоакустпческая спектроскопия 179
дим интерферометрический (с фурье-преобразованием) спектрометр, который позволит использовать выигрыши Фелжетта и Жакино, описанные в гл. 4 [6, 7]. Этот метод нашел применение при изучении полимеров [8] и угля [9]. Фотоакустические измерения в ИК-области при единичной длине волны оказались успешными при использовании в качестве источника лазера. Метод особенно полезен при обнаружении и определении примесей ядовитых газов в воздухе [10].
Применение
Следует ожидать, что в отсутствие флуоресценции и фосфоресценции фотоакустические спектры будут, по сути дела, идентичны обычным спектрам поглощения. На рис. 7-4 видно сходство фотоакустического спектра порошка Сг20з [2] и спектра поглощения тонкого среза его кристалла [11]. (В последнем случае на форму спектра влияет ориентация образца, поскольку кристалл анизотропен).
ТОО 400 700 1000
Длина волны, нм
Рис. 7-4. Фотоакустический (а) и полученный обычным методом (б) спектры поглощения кристаллического Сг2О3 [2]; кривая б взята из работы [11].
180 Глава 7
1 д Ц/
Красные кровяные
тельца
_________I_________I________I
200 ЧОО 600 800
Длина волны, нм
Рис. 7-5. Фотоакустические спектры цельной крови, красных кровяных телец и гемоглобина, извлеченного из красных кровяных телец [2].
ФАС имеет ряд существенных достоинств по сравнению с обычной абсорбционной спектроскопией. Во-первых, это единственный надежный метод получения спектров непрозрачных твердых веществ; во-вторых, благодаря высокой чувствительности метод ФАС можно использовать для измерения поглощения веществ с очень малой оптической плотностью, и, в-третьих, помехи от рассеянного излучения в данном случае минимальны, так как обнаружимо лишь то излучение, которое действительно поглощается.
Наибольшее применение метод ФАС нашел при исследовании биологических и биохимических систем, в которых часто наблюдается сильное светорассеяние. Рис. 7-5 иллюстрирует возможности метода; фотоакустический спектр цельной крови имеет полосы поглощения, обусловливающие красную окраску; в точности такие же полосы име
ются в спектрах одних только красных телец после удаления плазмы и гемоглобина [2].
ФАС можно применять для контроля за потоками жидкостей, например при детектировании в жидкостной хроматографии. С этой целью Ода и Са-вада [12] сконструировали кювету, использовав пьезоэлектрический датчик, который изолирован от жидкости тонкой платиновой фольгой. Такая кювета должна иметь как можно меньший объем во избежание размывания компонентов потока; описана кювета объемом всего лишь 0,02 мл. Применение в качестве источника аргонового ионного лазера, излучающего при 488 нм, позволило получить хорошее разрешение полос изомеров хлор-4-(диметиламино) азобензола, причем концентрация каждого изомера (3-10~7 М) была гораздо ниже, чем при работе на обычном спектрофотометре.
Фотоакустическая спектроскопия 181
Задача
7-1. Найдите описание ИК-детектора Голэя (см., например, ссылки [3, 7, 14] в гл. 4). Сравните его с фотоакустическим детектором, описанным в этой главе. Укажите их относительные достоинства и недостатки.
Литература
1. Brilmyer G. Н., Fujishima А., Santh.an.um К. S. V., Bard А. /., Anal. Chem., 1977, v. 49, р. 2057.
2. Rosencwaig A., Anal. Chem., 1975, v. 47, p. 592A.
3. Somoano R. B., Angew. Chem., Int. Ed. (English), 1978, v. 17, p. 238.
4. Farrow M. M., Burnham R. К., Auzanneau M., Olsen S. L., Purdie N., Eyring E. M., Appl. Optics, 1978, v. 17, p. 1093.
5. Fuchsman W. FL, Silversmith A. J., Anal. Chem., 1979, v. 51, p. 589.
6. Lloyd L. B., Riseman S. M., Burnham R. K., Eyring E. M., Farrow M. M., Rev. Sci. Instrum., 1980, v. 51, p. 1488.
7. Lloyd L. B., Burnham R. K., Chandler W. L., Eyring E. M., Farrow M. M., Anal Chem., 1980, v. 52, p. 1595.
8. Riseman S. M., Yaniger S. I., Eyring E. M., Macinnes D., Macdiarmid A. G., Heeger A. J., Appl. Spectrosc., 1981, v. 35, p. 557.
9. Rackley M. G., Devlin J. P., Appl. Spectrosc., 1980, v. 34, p. 407.
10. Claspy P. C., in Optoacoustic Spectroscopy and Detection, Y.-H. Pao (ed.), Academic Press, New York, 1977, chap. 6.
11. McClure D. S., J. Chem. Phys., 1963, v. 38, p. 2289.
12. Oda S., Sawada T., Anal. Chem., 1981, v. 53, p. 471.
Литература общего плана
13. Rosencweig A., Photoacoustics and Photoacoustic Spectroscopy, Wiley-In-terscience, New York, 1980.
14. McClelland J. F., Anal. Chem., 1983, v.. 55, p 89A.
Дополнительная литература
Жаров В. П., Летохов В. С. Лазерная оптико-акустическая спектроскопия.— М. Наука, 1984.
ГЛАВА
Рассеяние излучения
Термин рассеяние применительно к взаимодействию излучательной энергии с веществом описывает разнообразные явления. При этом всегда имеется в виду более или менее случайное изменение направления распространения. Рассеяние зависит от длины волны излучения, размера и формы рассеивающих частиц и иногда от их расположения в пространстве.
Электромагнитная теория рассеяния детально разработана в работах Ми [1—4], но она слишком сложна для непосредственного использования. В ограниченных областях можно допустить упрощения: так, удобно различать рэлеевское рассеяние (при котором частицы малы в сравнении с длиной волны) и рассеяние Тиндаля (для крупных частиц). Рамановскую спектроскопию часто рассматривают как рассеяние, при котором происходит сдвиг длины волны.
Рэлеевское рассеяние
В 1871 г. лорд Рэлей показал, что излучение, падающее на небольшую прозрачную частицу, индуцирует в ней электрический диполь, осциллирующий с частотой излучения. Осциллирующий диполь затем сам действует как источник, излучающий энергию во всех направлениях с той же частотой (но с разной интенсивностью).
По теории Рэлея — Ми, рассеяние малыми частицами обратно пропорционально длине волны в четвертой степени; благодаря рассеянию в основном частицами молекулярных размеров мы видим голубой цвет неба и красный цвет заката. Для химических систем показатель степени может меняться от —4 до —2 главным образом из-за наличия более крупных частиц, что указывает на постепенный переход от рэлеевского рассеяния к рассеянию Тиндаля.
Почти все измерения связаны с видимым излучением. Пробу освещают интенсивным ’ потоком Ро (рис. 8-1), а затем, так же как в абсорбционной спектроскопии, измеряют интен-
Рассеяние излучения 183
Рис. 8-1. Картина рассеяния. Ро — интенсивность падающего потока, Pt — интенсивность прошедшего потока; Р45, Р<ю, Pus— интенсивности излучения, рассеянного под разными углами.
снвпость прошедшего излучения Pt или определяют интенсивность излучения, рассеянного под определенным углом (например, 900, 739о). С ростом числа частиц в суспензии отношение РфР0 уменьшается, а отношения вида Р90/Р0 увеличиваются, во всяком случае до умеренных концентраций. Для очень разбавленных суспензий измерение под углом гораздо чувствительнее, чем измерения, когда источник и детектор находятся на одной линии, поскольку при этом можно наблюдать слабый рассеянный свет на темном фоне. Метод, в котором используется линейное измерение, называется турбидиметрией, а метод с измерением под углом 90° (или каким-либо другим)— нефелометрией.
Строгое математическое обоснование этих методов довольно сложная задача, но, к счастью, в практической аналитической работе в нем нет необходимости. При турбидиметрических измерениях величина, называемая мутностью, соответствует оптической плотности и может быть определена из соотношения, аналогичного закону Бера:
S = \g(PjP) = kbN (8-1)
где S — мутность, k — коэффициент пропорциональности, называемый коэффициентом мутности, N—число рассеивающих частиц в миллилитре, b — длина пути. (В литературе иногда встречается функция т, равная 2,303k.) Теоретические рассуждения приводят к следующему соотношению:
k = 0,4343 Г-2- nW* (LY1 (8-2)
L 3 У т2 + 2 7 J
где d — диаметр частицы, X — длина волны, т — отношение показателей преломления частиц и растворителя. (Это уравнение справедливо для разбавленных суспензий с частицами одинаковых размеров, которые меньше длины волны).
Для турбидиметрических измерений можно использовать любой фотометр или спектрофотометр. Если растворитель и
184 Глава 8
рассеивающие частицы бесцветны, максимальная чувствительность достигается при использовании излучения голубой или ближней ультрафиолетовой области. Для окрашенных систем оптимальную длину волны лучше всего подобрать экспериментально.
Используемое в нефелометрии уравнение должно связывать излучение, рассеиваемое под определенным углом наблюдения, концентрацию и другие переменные. В качестве рабочего соотношения лучше всего принять следующее:
Р = КасР0 (8-3)
где К — эмпирическая константа системы, индекс а — угол, под которым проводят измерения, с — концентрация.
Наблюдение под углом 90° не всегда дает наилучший результат. Иногда наблюдается большое увеличение чувствительности при измерении под минимально возможным углом, так что световой поток отклоняется от «прямого» пути всего на несколько градусов. Однако при этом возникают конструкционные трудности, которые не позволяют получить достаточно хорошую точность измерений. При использовании цилиндрической кюветы закругленные поверхности могут действовать как линзы, т. е. деколлимировать падающий свет и направлять в фотоэлемент излучение, возникающее в значйтельном объеме кюветы; при этом рассеяние будет происходить под неопределенным углом и зависеть от показателей преломления растворителя и диспергированных частиц. Напротив, кюветы в виде призмы, параллелепипеда или многогранника (как на рис. 8-1) позволяют наблюдать рассеяние под определенными углами.
Приборы. На рис. 8-2 представлен турбидиметр необычной конструкции (модель 430 фирмы Du Pont). Это двухлучевой прибор, работа которого основана на измерении относительной степени поляризации прошедшего и рассеянного излучений.
Первый поляризатор
Полупосеребренное зеркало Фото-/ элемент 2
Лампа ’ у “ Фильтр
Проба
-""перекрещивающиеся поляризаторы
Фотоэлемент 1
Рис. 8-2. Схема турбидиметра
(модель 430 фирмы Du Pont).
Рассеяние излучения 185
При освещении суспензии плоскополяризованным светом прошедший поток оказывается деполяризованным, причем степень деполяризации пропорциональна концентрации частиц в суспензии. В описываемом приборе поток сначала проходит через поляризатор, а затем через пробу. Прошедшее излучение при помощи полупосеребренного зеркала разделяется на два потока. Один из потоков проходит через другой поляризатор, ось которого параллельна оси первого поляризатора, а другой поток— через такой же поляризатор с осью, перпендикулярной оси первого поляризатора. Сигнал, попадающий на фотоэлемент 1, уменьшается за счет рассеяния, а сигнал, попадающий •на фотоэлемент 2, увеличивается. Электронная схема автоматически определяет отношение обоих сигналов, которое прямо пропорционально концентрации суспендированного вещества. Прибор не реагирует на окраску растворителя и частиц и на флуктуации мощности лампы, но на нем нельзя измерять рассеяние для растворов, содержащих оптически активные компоненты.
В работе [5] описан фотометр для измерения светорассеяния под малым углом с гелий-неоновым лазером в качестве источника света. Лазер дает узкий пучок с очень незначительным расхождением, а детектор может измерять рассеяние под углом всего 2° к первоначальному потоку. Другое преимущество использования лазера состоит в возможности уменьшения размера пробы до 2-Ю-5 мл, тогда как для работы на обычных приборах минимальное количество должно быть порядка 0,5 мл.
Конструкции приборов для нефелометрических и флуори-метрических измерений идентичны, поэтому любой флуориметр можно использовать в качестве нефелометра. Поскольку изменения длины волны при рассеянии не происходит, необходимость во втором монохроматоре или светофильтре отпадает, но если они имеются в приборе, то их следует настроить на длину волны падающего света. Многие серийные флуориметры снабжены специальными приспособлениями для нефелометрических измерений.
Практическое применение
На светорассеяние сильно влияет размер частиц, поэтому при сравнении стандартов и пробы необходимо строгое соблюдение идентичности условий, что позволяет выполнить много аналитических определений с большой чувствительностью и точностью, например, обнаружить 1 часть фосфата в виде малорастворимого соединения с молибденом и стрихнином в 300 млн. частей воды или 1 часть аммиака в 160 млн. частей воды, используя иодидный комплекс ртути (реактив Несслера). Для
186 Глава 8
определения серы в количестве нескольких частей на миллион ее следует превратить в сульфат и осадить в виде сульфата бария в условиях, обеспечивающих получение устойчивой коллоидной суспензии.
Молекулярные массы и размеры частиц. Дебай показал, что метод светорассеяния можно с успехом использовать для определения средневесовой молекулярной массы полимеров в растворах. Для расчетов по уравнению Дебая необходимо знать мутность, концентрацию, показатель преломления, длину волны, производную показателя по концентрации и второй ви-риальный коэффициент, являющийся мерой неидеальности раствора. Использование метода светорассеяния ограничено размерами молекул — они должны быть меньше длины волны; детальное рассмотрение данного метода выходит за рамки настоящей книги [6].
Фотометры для измерения светорассеяния высокомолекулярных полимеров выпускаются рядом фирм. Эти приборы выполняют разнообразные функции, позволяют проводить наблюдения под несколькими углами и снабжены набором кювет и поляризаторами, которые дают возможность получить информацию о форме частиц.
Рассеяние в газах. Дым и туман хорошо видны благодаря светорассеянию, поэтому неудивительно, что приборы для его измерения пригодны для контроля за загрязнением воздуха. Степень уменьшения интенсивности лазерного потока на пути от одного здания к другому пропорциональна количеству частиц, содержащихся в воздухе. Из небольшого лазера и фотоэлемента можно собрать чувствительный детектор, с помощью которого легко уловить несколько микрограммов частиц дыма диаметром от 0,1 до 1 мкм в кубическом метре воздуха.
Турбидиметрическое и нефелометрическое титрование [7]
Еще со времен работ Гей-Люссака по осаждению хлорида серебра (1832 г.) широко используется титрование с турбидиметрическим обнаружением конечной точки. Такое титрование можно выполнить визуально или на абсорбционном фотометре. Следовало бы ожидать, что кривая титрования по реакции А + + В->-С, где С — нерастворимое соединение (по оси абсцисс отложена мутность или интенсивность рассеяния), должна состоять из двух пересекающихся отрезков, так как количество осадка должно возрастать до максимума, а затем оставаться постоянным. В работе [8], однако, показано, что это возможно лишь в том случае, если число частиц до точки эквивалентно-
Рассеяние излучения 187
сти увеличивается линейно и размер их в процессе титрования не меняется. На практике это неосуществимо — более вероятно, что при добавлении реагента одновременно образуется новые частицы и увеличивается размер уже образовавшихся ранее зародышей. В таком случае
Рис. 8-3. Кривые турбидиметрического титрования: 1 — идеальная кривая, кривые 2 и 3 могли получиться при осаждении частиц разного размера, при плохом перемешивании и т. п.
предсказать точную форму кривой титрования невозможно. Если частицы становятся очень крупными, точка эквивалентности плохо выражена или вообще
не видна (кривые 2 н 3 на
рис. 8-3). Кроме того, если суспензия не очень разбавлена, ча
стицы после добавления каждой порции титранта продолжают расти, поэтому титрант следует добавлять довольно медленно (с интервалом в несколько минут).
В работе [9] показано, что добавка перед титрованием кри-
сталлической затравки, равномерно распределяемой по всему объему, заметно повышает точность нефелометрических титрований разбавленных растворов. В серии экспериментов авторы титровали КВг в водном растворе изопропилового спирта раствором AgNO3, добавляя в качестве затравки коллоидную суспензию AgBr в количестве, составляющем примерно 10 % того количества AgBr, который ожидали получить при титровании. Они добились того, что конечную точку удавалось легко обнаружить при концентрации бромида порядка Ю-8 ЛА с относительным стандартным отклонением 1,3%.
Бобтельский и сотр. [10, 11] с успехом провели сотни турбидиметрических титрований при несколько больших концентрациях (обычно 10-3—10~4 М). Они не пытались контролировать размер частиц и титровали с обычной скоростью. Полученные
ими кривые были воспроизводимы и давали ценные результаты, но их нельзя было считать турбидиметрическими, так как нельзя было ожидать, что процесс описывался уравнением 8-1. Поэтому Бобтельский из осторожности назвал свой метод гетерометрия, а свой упрощенный фотометр — гетерометром.
Задачи
8-1. Содержание серы в органических сульфонатах и сульфонамидах можно найти турбидиметрически, используя образование BaSO4 после разложения органического вещества [12]. Навеску препарата моногидрата п-толуолсульфо-кислоты массой 25 мг (СтНуЗОзН-НгО, мол. масса 190,2) подвергли разложению и точно одну десятую часть разбавили до объема 10,00 мл соответствую
188 Глава 8
щим раствором. Затем добавили при контролируемом перемешивании 5,00 мл 1,34 М ВаС12. Мутность, измеренная через 25 мни на приборе Спектроник-20 при 355 нм, оказалась равной 0,295. Мутность стандартного раствора (NH4)2SO4, содержащего 0,200 мг серы, обработанного аналогичным путем, равна 0,322; предварительно показано, что она меняется с разбавлением линейно. Оцените чистоту (в процентах) исходного препарата.
8-2. При увеличении числа частиц в суспензии в конце концов наступает момент, когда проба становится практически непрозрачной и Л и Ры> (рис. 8-1) равны нулю. Означает ли это, что S становится бесконечной? Как это скажется на уравнении (8-3)?
8-3. Иногда пользуются [13] методом, в котором измеряют отношение R рассеянного и прошедшего излучений, т. е. R = Px,/Pt. Выведите уравнение зависимости R от концентрации. Справедливо ли оно для всех концентраций или только для линейного участка? Имеет ли этот метод какие-либо преимущества? Поясните ответ.
8-4. Суспензия AgCl в воде состоит из однородных частиц диаметром 0,040 мкм (плотность 5,56 г/см3 и показатель преломления 2,07). Рассчитайте пропускание в процентах в 10-см кювете, если измерения проводили при 500 нм.
Литература
1. Billmeyer F. W., In Treatise on Analytical Chemistry. Kolthoff I. M., Elving P. J. (eds.), pt. I, v. 5, chap. 56, Wiley-Interscience, New York, 1964.
2. Mie G.. Ann. Physik, 1908, v. 25, p. 377.
3. Van de Hulst H. C., Light Scattering by Small Particles, Wiley, New York, 1957.
4. Gravatt С. C., Jr., Appl. Spectrosc., 1971, v. 25, p. 509.
5. Каце W., Anal. Chem., 1973, v. 45, p. 221A.
6. Vollmert B., Polymer Chemistry, Springer-Verlag, New York, 1973, pp. 348ff.
7. Underwood A. L., in Advances in Analytical Chemistry and Instrumentation. Wiley-Interscience, New York, 1964, v. 3, p. 31.
8. Meehan E. J., Chiu G., Anal. Chem., 1964, v. 36, p. 536.
9. Meehan E. J., Yamaguchi S., Anal. Chem., 1977, v. 49, p. 2268.
10. Bobtelsky M., Anal. Chim. Acta, 1955, v. 13, p. 172.
11. Bobtelsky M., Heterometry, American Elsevier, New York, 1960.
12. Zdybek G., McCann D. S., Boyle A. J., Anal. Chem., 1960, v. 32, p. 558.
13. Vanous R. D., Am. Lab., 1978, v. 10, No. 7, p. 67.
ГЛАВА
Атомная эмиссионная спектроскопия
Еще со времен работ Бунзена и Кирхгофа (1860 г.) было известно, что многие элементы при возбуждении испускают излучение с характерными длинами волн. На этом основаны хорошо известные тесты на присутствие щелочных и щелочноземельных элементов. Если заменить пламя на более мощные электрические источники возбуждения, то таким способом можно обнаруживать металлы и многие неметаллы. Спектры некоторых элементов, например натрия и калия, просты и состоят всего лишь из нескольких длин волн, тогда как в спектрах других элементов, в том числе железа и урана, насчитываются тысячи отчетливо воспроизводимых длин волн.
Количественный анализ на спектрографе основан на эмпирической зависимости между интенсивностью испускаемого излучения при некоторых определенных длинах волн и количеством элемента в пробе. Интенсивность излучения сложным образом зависит от целого ряда факторов, в том числе неконтролируемых. Следовательно, условия определения должны быть строго стандартизированы, а спектры неизвестных веществ следует сравнивать со спектрами стандартных проб, приготовленных в идентичных условиях.
Бурное развитие эмиссионной спектроскопии приходится на период между двумя мировыми войнами, когда были сконструированы громоздкие спектрографы с фотографическим детектированием. С появлением в 50—60-х годах электроники значение приборов с фотодетекторами уменьшилось; во многих областях они был вытеснены аналогичными эмиссионными спектрометрами с фотоумножителями и недавно сконструированными атомно-абсорбционными спектрофотометрами и рентгенофлуоресцентными спектрометрами. Возрождение интереса к атомно-эмиссионным методам произошло в середине 70-х годов в связи с созданием надежных источников плазмы.
190 Глава 9
Возбуждение проб
Как было показано в гл. 2, свободный атом может принимать энергию от внешнего источника и возбуждаться; это означает, что один из его электронов переходит с основного на более высокий энергетический уровень. Возвращаясь в основное состояние, атом испускает фотон с энергией, соответствующей определенной частоте или длине волны.
На практике в эмиссионной спектроскопии существует несколько способов возбуждения, из которых наибольшее значение имеют электрические дуга и искра, пламя и электрогенери-рованная плазма в газе-носителе. Мы разберем каждый из этих способов и одновременно дадим описание соответствующих приборов.
Дуговой разряд. Чтобы получить дугу постоянного тока, пропускают мощный электрический разряд между двумя порциями пробы или между пробой и противоэлектродом, не содержащим искомые элементы. В качестве противоэлектрода лучше всего применять графит — тугоплавкий и труднолетучий материал (не плавится и не возгоняется при температуре дуги), хорошо проводящий электричество и дающий мало собственных спектральных линий. К сожалению, раскаленный углерод медленно реагирует с атмосферным азотом с образованием дициана; последний при возбуждении дает яркие полосы в области 360—420 нм, которые могут помешать наблюдениям. При необходимости этого можно избежать, заключив разряд в кожух, через который пропускается ток инертного газа.
Дуга постоянного тока— высокочувствительный источник возбуждения, особенно при обнаружении металлов. Через дугу пропускают ток силой от 5 до 15 А, регулируемый при помощи реостата, который часто называют балластным сопротивлением (10—40 Ом). Основной недостаток дуги постоянного тока заключается в недостаточно хорошей воспроизводимости. Заряд стремится сосредоточиться в «горячих точках» поверхности электрода, вследствие чего условия анализа в разных участках пробы неодинаковы.
Этот недостаток не столь заметен при использовании дуги переменного тока, когда заряд прерывается 120 раз в секунду. Здесь необходим источник с напряжением 2000—5000 В и силой тока 1—5 А. Балластным сопротивлением может служить реостат или вариометр, включаемые в цепь. Дуга переменного тока удобна при анализе осадков, которые можно нанести на поверхность электрода выпариванием растворов.
Атомная эмиссионная спектроскопия 191
Искра. Искра по сравнению с дугой обладает рядом особенностей: для ее получения используется очень высокое напряжение (15—40 кВ), и вся мощность реализуется в короткой вспышке (в течение миллисекунд). Сам разряд действует как высокочастотный генератор: при каждой вспышке происходит от 10 до 20 осцилляций, с частотами от 104 до 105 Гц. В спектроскопии эти вспышки повторяются 120 раз в секунду. Высокое напряжение получают при помощи повышающего трансформатора или катушки Тесла с конденсатором, который связан с ними через вторичную обмотку.
Электронная схема, в которую входит тиратрон, контролирует электросеть и в оптимальный момент переменного цикла включает пусковое устройство. За время цикла в конденсаторе накапливается энергия, которая в этот момент разряжается в искровом промежутке. Если при анализе нужно точно выдерживать экспозицию, искра как источник лучше, чем дуга; кроме того, она не разрушает пробу, поскольку успевает испариться минимальное ее количество.
Подготовка электродов и проб. Цельнометаллические пробы не требуют специальной подготовки, их просто закрепляют перед собирающей линзой спектрографа. Противоэлектродом служит другой кусок той же пробы или электрод, изготовленный из графита или основного компонента пробы. Например, при обнаружении легирующих добавок в стали противоэлектродом может служить просто не содержащая этих добавок сталь.
Неметаллические пробы наносят на графитовый электрод. По одному из способов пробу переводят в раствор, используя кислоты или, если необходимо, другие реагенты. Несколько капель раствора помещают в углубление,' просверленное в верхней части графитового электрода (рис. 9-1, д), и выпаривают растворитель нагреванием в небольшой печи. Затем электрод закрепляют в нижней части электрододержателя, а угольный противоэлектрод (рис. 9-1, в)—в его верхней части, чтобы между ними могла проскочить искра или загореться дуга. Другой способ заключается в том, что измельченную пробу смешивают с графитовым порошком и набивают в кратер электрода. Графит, обладающий хорошей электропроводностью, повышает стабильность дуги. В литературе приводится множество вариантов подготовки электродов и проб, в которых учтены особенности конкретных проб.
Спектроскопист при использовании дуги в качестве источника должен учитывать различную летучесть компонентов пробы. В некоторых случаях один или несколько компонентов способны быстро улетучиваться и полностью выгорать через полминуты (или около того) после включения дуги, т. е. пре-
192 Глава 9
Рис. 9-1. Типы графитовых электродов. Формы а и б характерны для про-тивоэлектродов. В верхних частях электродов б, г и д имеются углубления для пробы. В центре углублений электродов биг выточены выступы, благодаря которым локализуется дуговой разряд. Узкая шейка вытачивается для уменьшения теплоотдачи верхней частью электрода (Ultra Carbon Corporation).
жде чем другие вещества пробы успевают нагреться, чтобы попасть в дугу. Иногда различия в летучести целесообразно использовать для получения спектров летучих компонентов, что позволяет избежать помех со стороны менее летучих веществ. Примером служит определение примесей оксидов лития, алюминия и других элементов в оксиде урана [1]; спектр урана богат линиями, поэтому определение следовых количеств примесей затруднительно. Согласно описанному в работе [1] методу, уран сначала превращают в нелетучий ПзО8 и добавляют сравнительно летучий оксид СагО3 в количестве 2 % от массы урана. Оксид галлия выполняет роль носителя, захватывающего в дугу очень малые количества примесей, что приводит к высокой чувствительности (вплоть до нескольких частей на миллион) и точности определения.
Приборы
В этом разделе мы опишем некоторые основные спектральные приборы, а затем вернемся к рассмотрению других источников возбуждения. Для наблюдения и измерения эмиссионных спек-
Атомная эмиссионная спектроскопия 193
Рис. 9-2. Оптическая схема спектроскопа Бриланда. Пробу укрепляют над небольшим нагревателем под горизонтально расположенными электродами. Дифракционная решетка и щель расположены вертикально на круге Роуланда (Spectrex Company).
тров существует большой набор приборов, от ручных спектроскопов визуального наблюдения до гигантских фотографических и фотоэлектрических спектрографов с высокими разрешением и чувствительностью.
Из малых спектрометров для качественного и полуколиче-ственного анализа твердых проб применяется спектроскоп Бриланда (Spectrex Company, Palo Alto, California) для видимой области спектра. Он представляет собой настольный прибор с переменнотоковым дуговым источником возбуждения и вогнутой дифракционной решеткой с фокусным расстоянием 42,5 см (600 штрихов на 1 мм). Спектр проецируется на полупрозрачный пластмассовый экран, расположенный на круге Роуланда (рис. 9-2). По обе стороны исследуемого спектра помещают спектры сравнения на позитивных пленках, располагая их так, чтобы можно было сравнивать непосредственно линию исследуемого спектра с соответствующей линией спектра сравнения.
Большая часть классических работ в эмиссионной спектрографии выполнена на больших спектрографах с кварцевыми призмами или вогнутыми отражательными дифракционными решетками и фотографическим детектированием. Многие из этих приборов до сих пор находят применение, хотя выпускаются в малом количестве.
В качестве стандарта для калибровки по длинам волн обычно используется спектр железа, потому что он содержит несколько тысяч линий, довольно равномерно расположенных на протяжении УФ- и видимой областей. Положение линий железа многократно с большой точностью измерено на спек-
7 Заказ № 537
194 Глава 9
трографах с призмами и дифракционными решетками высокого разрешения; этот спектр может служить шкалой при расшифровке других спектров. Идентификацию элементов по спектру на фотопластинке проводят, либо сравнивая его непосредственно со спектром стандартного образца, либо определяя длины волн по спектрам железа с помощью таблиц длин волн.
Количественный анализ
Строгого математического выражения, связывающего количество или концентрацию компонента в пробе и оптическую плотность * осадка серебра (почернение) на фотопластинке, не существует, поэтому градуировку приходится проводить эмпирически. Сравнение стандартного и исследуемого образцов лучше проводить методом внутреннего стандарта.
Этот метод основан на измерении отношения интенсивности данной линии определяемого компонента к интенсивности какой-либо линии другого компонента пробы, который присутствует в известном (или по крайней мере постоянном) количестве. Стандартом может быть элемент, входящий в состав пробы, например железо в стали, или посторонний элемент, добавленный в известном количестве ко всем пробам. Такой прием позволяет устранить ошибки, связанные с различиями в качестве фотопластинок и в условиях их проявления. Длина волны и интенсивность линий, выбранных в качестве стандартных, должны быть как можно ближе к длине волны и интенсивности линий определяемого элемента с тем, чтобы какое-либо нарушение линейной зависимости чувствительности фотоэмульсии от этих параметров не стало серьезным источником ошибки. Две линии, выбранные с этой целью, называются гомологической парой.
Для измерения оптической плотности линий на фотопластинке требуется денситометр — специальной фотометр для измерения пропускания. Он состоит из источника света, линз, фокусирующих световой поток на очень небольшой участок пластинки, и фотоумножителя или другого аналогичного устройства, размещаемого под пластинкой. С помощью моторчика можно медленно перемещать фотопластинку через освещенное пятно. При этом на ленте самописца регистрируется оптическая плотность при разных положениях пластинки. Высота пика над базисной линией практически линейно связана с падающей на пластинку энергией при определенной длине волны.
* Оптическая плотность в данном случае эквивалентна оптической плотности, фигурирующей й законе Бера, и служит для оценки количества серебра иа фотопленке или фотопластинке.
Атомная эмиссионная спектроскопия 195
Чтобы проиллюстрировать метод внутреннего стандарта, разберем некоторые детали типичной аналитической методики. Выберем в качестве примера определение следов магния в растворе с молибденом в качестве внутреннего стандарта. Для более подробного ознакомления с этой и многими другими методиками рекомендуется монография Нахтриба [2].
Найдено, что для такого рода анализов удобно использовать искровой разряд между медными электродами (если не нужно определять саму медь). Электроды представляют собой стержни диаметром примерно 5 мм. Чтобы удалить загрязнения с поверхности и сделать ее однородной, следует заточить на токарном станке верхнюю часть стрежней. После заточки их нужно предохранять от пыли и не трогать руками. Из чистого MgCl2 готовят серию стандартных растворов с концентрацией магния от 0,1 нг до 10 мкг в 1 мл. В каждый раствор добавляют молибдат аммония, так чтобы концентрация Мо была 20 мкг/мл (дистиллированная вода должна быть спектрально чистой относительно Mg). Электроды закрепляют в вертикальном положении над небольшой электроплиткой и с помощью пипетки в верхнюю часть каждого электрода помещают по 50 мкл одного из растворов. Затем осторожно выпаривают растворы досуха, электроды устанавливают на точно отмеренном расстоянии (2,0 мм) друг от друга и пропускают через них искру мощностью 25 кВ. Обычно спектры всех стандартных растворов умещаются на одной фотопластинке.
Рис. 9-3. Градуировочный график для определения магния при помощи искрового разряда между медиыми электродами с молибденом в качестве внутреннего стандарта [3].
7*
196 Глава 9
В спектре присутствуют линии Mg и Мо, а также Си и некоторых других металлов. Почти все имеющиеся в спектре гомологические пары подходят для определения. Одна из таких пар состоит из линии при 279,81 нм (Mg) и 281,62 нм (Мо). На логарифмической бумаге строят график зависимости разницы оптических плотностей двух линий (т. е. отношение интенсивностей испускания при этих двух длинах волн) от количества Mg в пробе (рис. 9-3). Чтобы определить неизвестную концентрацию, пробу обрабатывают по той же методике, включая добавление молибдата аммония. Измеряют оптические плотности двух линий, находят отношение их интенсивностей и по графику определяют количество Mg в аликвотной порции объемом 50 мкл. Этот метод отличается быстротой и удобством в сочетании с высокой чувствительностью. Он позволяет идентифицировать 1 нг Mg в объеме 1 мл с точностью 5—10%, что вполне удовлетворительно, если принять во внимание малое содержание элемента.
Фотопластинка, по существу, представляет собой интегральный детектор, поскольку количество выделившегося серебра пропорционально произведению интенсивности излучения Р и времени экспозиции А/:
Оптическая плотность = kP\t (9-1)
В идеале коэффициент пропорциональности k должен быть константой, но в действительности его постоянное значение сохраняется в ограниченном диапазоне. Поэтому следует подобрать такую гомологическую пару, чтобы обе линии имели близкую оптическую плотность.
Квантометры
Если фотопластинку заменить рядом фотоумножителей, можно обойтись без трудоемкого фотографического процесса. Сканирование длин волн, практикуемое в спектрофотометрии растворов, не так-то легко осуществить при возбуждении дугой или искрой, поскольку они не обеспечивают достаточной стабильности при сканировании, особенно если учесть изменения в летучести, о чем уже упоминалось. Это затруднение можно разрешить, разместив вдоль фокальной плоскости несколько выходных щелей, так чтобы через каждую из них проходила линия, испускаемая определенным элементом. В таком случае, располагая 12 щелями и 12 фотоумножителями, можно одновременно измерять испускание 12 элементов в одной и той же пробе. Каждый усилитель можно снабдить интегратором, который суммирует интенсивности при каждой длине волны за выбранный интервал времени. Один из каналов оста-
Атомная эмиссионная спектроскопия 197
Рис. 9-4. Внутреннее устройство спектрометра фирмы Jarrell-Ash Plasma Atom Comp. Заключенный в кожух источник ИСП находится слева на заднем плане. В металлической коробке справа помещается вогнутая дифракционная решетка. На переднем плане слева можно видеть 15 фотоумножителей, каждый из которых имеет свою выходную щель; все щели расположены в фокальной плоскости.
вляют для внутреннего стандарту, и контролирующий компьютер обеспечивает обработку данных, полученных при сравнении каждого аналитического сигнала с сигналом внутреннего стандарта. Полученная информация может выдаваться в виде распечатки. На рис. 9-4 приведена конструкция такого многощелевого спектрометра.
Следует подчеркнуть, что результаты, полученные на больших и сложных приборах, подобных описанному выше, не отличаются большой правильностью, хотя могут быть очень точными (воспроизводимыми). Правильность часто зависит от матрицы и способа подготовки пробы, а также от правильности приготовления стандартов. Правильность любой аналитической методики, основана ли она на спектроскопических или каких-либо других измерениях, нужно проверять обычно на стандартных образцах, например, выпускаемых в США Национальным бюро стандартов.
198 Глава 9
Возбуждение в плазме
Второй основной механизм возбуждения связан с применением плазмы, возникающей при электрическом разряде в газе, например азоте или аргоне. Плазму можно определить как нейтральный газ, содержащий значительные количества положительных и отрицательных ионов и свободных электронов. Для создания плазмы необходим постоянный подвод энергии, обеспечивающий образование новых ионов, чтобы компенсировать их рекомбинацию с образованием нейтральных атомов. Разновидностью плазмы является пламя, которое питается энергией химической реакции. Благодаря строению энергетических уровней для получения плазмы особенно подходит аргон, обладающий, кроме того, дополнительным преимуществом — химической инертностью.
Существуют два способа получения аргоновой плазмы. В одном из них энергия высокочастотного переменного тока передается газу посредством магнитной индукции (ИСП, индуктивно связанная плазма), в другом возбуждение происходит под действием постоянного тока. ИСП обладает большей чувствительностью, но плазма постоянного тока значительно дешевле.
На рис. 9-5 представлена фотография источника плазмы постоянного тока с тремя электродами. Два нижних являются
Рис. 9-5. Дуговой плазматрон постоянного тока. Катод находится вверху слева, два анода расположены внизу справа, между ними помещена форсунка для введения пробы (Spectra Metrics, Inc.).
Атомная эмиссионная спектроскопия ISO
А
Ток аргона, переносящий Вспомогательный (плазма
аэрозоль (0,8-1,5л/мин) образующий) ток аргона
(до 0,5л/мин)
Рис. 9-6. Схематическое представление факела ИСП [5].
параллельно соединенными анодам.и, а верхний — катодом. Аргон подается по каналам, просверленным в электродах. Температура плазмы в месте стыка трех факелов может достигать 5000 К. Проба впрыскивается с дополнительной струей аргона через форсунку чуть ниже этой горячей точки. Излучение, испускаемое центральной частью столба плазмы, фокусируется на щель спектрографа.
Схема источника ИСП представлена на рис. 9-6. Он состоит из трех концентрических кварцевых трубок, по которым подается ток аргона, и двух- или трехвитковой медной спирали, обвивающей верхнюю часть тфубок. Через медную спираль пропускают переменный ток с частотой 27,14 МГц* мощностью
Эта частота в США запрещена для промышленного использования.
2,00 Глава 9
в несколько киловатт. С помощью вспомогательной схемы создают искру, которая генерирует некоторое количество ионов, и затем посредством магнитной индукции вызывают в ионизированном газе сильный кольцевой ток. Полученная плазма разогревается до нескольких десятков тысяч градусов по Кельвину, что значительно выше температуры, при которой размягчается кварцевое стекло. Очевидно, нужно найти способ защиты источника от саморазрушения, что достигается при помощи тока аргона, выполняющего роль охладителя. Аргон с большой скоростью подается по касательной из внешней трубки (рис. 9-6), при этом образуется вихревой поток (показан на рисунке), и температура понижается. Горячая плазма стремится стабилизироваться на некотором расстоянии от стенок в форме тороида, что также предотвращает перегрев. Проба распыляется в распылителе (не показан на рисунке) и уносится медленным током аргона к центру (к «дырке в пирожке»). Здесь она разогревается за счет теплопроводности и излучения вплоть до 7000 К и полностью атомизируется и возбуждается. Потеря определяемых атомов вследствие ионизации (источник затруднений в плазменной ААС) в спектроскопии ИСП не играет большой роли благодаря наличию более легко ионизирующихся атомов аргона.
Спектрометры с плазменными источниками по существу не отличаются от ранее описанных. В некоторых моделях последовательно сканируют длины волн всех определяемых элементов, в других — на фотопластинке или при помощи фотоумножителей регистрируется испускание сразу нескольких элементов.
Новый в аналитической химии метод эмиссионной спектроскопии плазмы быстро становится одним из самых популярных методов количественного определения металлов, вытесняя во многих случаях атомную абсорбцию.
Возбуждение в пламени
В прошлом газовое пламя как источник возбуждения атомов широко использовалось в методе, называемом фотометрией пламени. Сейчас оно в основном применяется для определения щелочноземельных металлов. Испускание можно измерять на многих атомно-абсорбционных спектрофотометрах, используя то же самое пламя и распылительную систему. В этом случае пламя должно иметь более высокую температуру, чем в ААС, где атомы поглощают резонансное излучение и, следовательно, должны находиться в основном состоянии, тогда как в эмиссионной спектроскопии их нужно перевести в возбужденное состояние.
Интенсивность излучения пламени при характеристической
Атомная эмиссионная спектроскопия 201
длине волны данного элемента почти пропорциональна его концентрации (при условии внесения поправки на фон). Иногда наблюдаются помехи со стороны других металлов (эффект матрицы), увеличивающие или уменьшающие свечение. Эти помехи можно устранить, правда с некоторой потерей в чувствительности, путем добавления большого избытка любого из мешающих элементов. Например [3], при определении Na, К, Са или Mg в природных водах влияние трех других элементов можно предотвратить при помощи спектрохимических буферов. Так, при определении Na к 25 мл пробы добавляют 1 мл раствора, насыщенного относительно хлоридов К, Са и Mg. Любое не слишком большое изменение содержания этих элементов в пробе ничтожно по сравнению с добавленными количествами. При измерении испускания смешанного раствора при длине волны натрия показания фотометра следует сравнивать с градуировочной кривой, построенной с использованием стандартов. Для Na и К легко обнаруживаются различия в концентрациях, составляющие 1—2 мкг/мл, для Са — различия, составляющие 3—4 мкг/мл; менее чувствительно определение Mg.
Другой способ устранения помех заключается в использовании метода стандартных добавок, обсуждаемый в гл. 26. Сначала измеряют испускание пробы, затем ко второй аликвотной порции добавляют известное количество определяемого элемента и снова измеряют испускание. На стандартную добавку влияют те же помехи, что и на компонент пробы, следовательно, возможно определение путем прямого сравнения.
В клинических лабораториях при определении Na, К и Са й крови используются упрощенные пламенно-эмиссионные фотометры с интерференционными светофильтрами, выделяющими линии определяемых элементов. В качестве внутреннего стандарта часто добавляют литий. Такие приборы удобны в работе и высокочувствительны.
Атомная флуоресценция [5]
Другой способ возбуждения атомов связан с облучением УФ-светом той же резонансной частоты, что и в атомной абсорбции, вызывающим излучение этой же или более низкой частоты. Такой метод получил название атомная флуоресценция. Интенсивность излучения зависит от количества флуоресцирующих атомов и, следовательно, от концентрации.
Проба атомизируется в плазме — пламени или индуктивно связанной плазме, флуоресценцию наблюдают под углом (обычно 45 или 90°) к возбуждающему излучению. Так же как и в методе молекулярной флуоресценции, это слабое излучение лучше заметно на темном фоне. Источником возбуждаю-
202 Глава 9
А
Оптический
X А У|
Рис. 9-7. Взаимное расположение источника ИСП, источника излучения и фотоумножителя, контролирующего ИСП (Baird Corporation).
щего излучения обычно служит лампа с полым катодом большей мощности, чем применяемые в ААС, а также ксеноновая дуговая лампа, дающая непрерывный спектр в УФ-области.
Если источником служит лампа с полым катодом, то необходимость в монохроматоре отпадает, и оптическая схема прибора упрощается. На рис. 9-7 представлена комбинация из лампы с полым катодом и фотоумножителя для регистрации одного вещества, возбуждаемого в индуктивно связанной плазме. Чтобы отделить нужную длину волны от остальных (с целью улучшения отношения сигнал/шум), перед фотоумножителем ставят интерференционный светофильтр. В приборе, выпускаемом фирмой Baird Corporation, может одновременно работать десять таких узлов при одном и том же источнике ИСП. Подготовка пробы и работа с ИСП по существу ничем не отличаются от описанных выше.
Недостаточное количество выпускаемых приборов не позволяло раньше широко использовать атомно-флуоресцентную спектрометрию в аналитической практике, но простота и чувствительность метода позволяют надеяться на существенные успехи в будущем.
Атомная эмиссионная спектроскопия 203
Возбуждение лазером. Мощный лазерный поток, сфокусированный на небольшой площади, может превратить в пар заметные количества даже труднолетучих соединений [6]. Иногда для возбуждения пара с последующим испусканием излучения достаточно одной тепловой энергии, а иногда требуется дополнительно использовать электроразряд. С одной стороны, локализация процесса является его достоинством, поскольку позволяет исследовать очень малые поверхности (до 50 мкм в диаметре), но, с другой стороны, она может стать недостатком, потому что анализ крупной пробы оказывается недостаточно представительным. К достоинствам лазерного способа возбуждения следует отнести возможность исследования проб с плохой электропроводностью.
Сравнение плазменных и родственных им методов
Целесообразно сравнить возможности спектроскопических методов, основанных на возбуждении в раскаленных газах и плазмах. Сюда относятся методы с применением ИСП и плазмы постоянного тока, пламенная атомно-абсорбционная спектрометрия и пламенная атомно-эмиссионная спектрометрия, непламенная атомно-абсорбционная спектрометрия, атомно-флуоресцентная спектрометрия и методы с использованием дуги и искры.
Чувствительность. На рис. 9-8 представлены пределы обнаружения многих элементов методами пламенной и непламен-ной ААС, флуоресценции в пламени, пламенной атомно-эмиссионной спектрометрии и ИСП, систематизированные Вайн-форднером и др. [7]. К этому источнику следует обращаться всякий раз, когда возникает необходимость в точных численных значениях пределов обнаружения многих элементов, а также в ссылках на оригинальные работы. Приведенные в нем данные следует рассматривать только как ориентировочные; точные значения зависят от условий анализа, а также от того, что считать «пределом обнаружения», поскольку в определении этого понятия в литературных источниках нет единообразия. Из данных, приведенных на рисунке, следует, что в целом метод ИСП чувствительнее пламенных атомно-абсорбционных и атомно-эмиссионных методов, но уступает в этом отношении непламенным ААС. Как показано в работе [8], флуоресценция с использованием ИСП характеризуется тем же пределом обнаружения, что и пламенная флуоресценция.
На рисунке не указаны сведения- о методах с применением дуги и искры, потому что пределы обнаружения, найденные для твердых проб, трудно сопоставить с пределами обнаружения, выраженными в единицах концентрации растворов. Таб-
204 Глава 9
10~7 Пламенная ААС Непламенная ААС Пламенная Пламенная флуоресценция АЭС ИСП
1Q-6 10"5 - Bezn мд Si Cd -Ag Mn Ca Cs In - Cd Zn Li К Ba Ca Sr Mg Mn
10’4 10~3 10‘2 10'1 10° 101 Мп Na - Ag Cd Li Zn -Be Ca Co Cr К Mg Cu Fe Ni Rb Sr -Pb Au Ba Tl V Asin Mo Sb Bi CsGaPiSn -AlGeSeScSi - Ti H9 -Ir La В NbTa W Zr U - Al Au Ga Rb Sr -Co Cr Pb Ge Li Mo P V Bi Sb Ba AsRh Ni Se - Fe Pl Tl ng К Ti Sc " В Sn - Ag Mg Cu - Mn In Au Ni AL Bi Ca Co Cr FeTl - BeGa Pb Sc Sr~ Se Sb Sn V - AsGeTi Mo Rh St - ca Sr' In Na - BaMn &cr Fe V Rb - CU Ni Tl . Co Rh Ti В Ga Mg - Pb Sn Mo Ir Ge Cs Sb W Cd Sc - Be Au Si Pi Ta U Zr La - As Hq Zn Bi d - Cu Al Mo NaTi V Cr FeLi Be La Ni Zr Ga - HgW Cd Nb Pb Co Rh Sc AgGe - Si In Se Sn Ta U As Au P Bi Pi - SbTl
ю2
Рис. 9-8. Пределы обнаружения некоторых элементов (в мкг/мл) в водных растворах при определении различными спектроскопическими методами [7].
лицы с данными об относительной чувствительности можно найти в специальных источниках, например в монографии Нах-триба [2].
Химические помехи. В целом помехи химического происхождения меньше сказываются'при определении методом ИСП, чем методами с использованием пламени и электротермической атомизации. Это связано частично с отсутствием в ИСП кислорода, но главным образом с гораздо более высокой темпера
Атомная эмиссионная спектроскопия 205
турой и более длительным пребыванием пробы в горячей зоне, где происходит диссоциация. Обратите внимание (рис. 9-8) на значительный выигрыш в чувствительности при определении методом ИСП вольфрама, циркония и урана, образующих в пламени труднолетучие оксиды.
Спектральные помехи. Помехи, связанные с перекрыванием спектральных линий, более вероятны для метода ИСП, чем для методов ААС, так как эмиссионные спектры более богаты линиями. В работе Мак-Ларена и др. [10] этому вопросу уделяется пристальное внимание; при определении следов элементов в морских осадках, содержащих много железа и алюминия, наблюдались сильные помехи со стороны этих компонентов. Авторы разработали методику с компьютерным контролем, в которой для оценки поправки на фон рекомендуется проводить измерения при несколько большей и несколько меньшей длинах волн, чем в максимуме определяемого элемента. Например, при определении меди была выбрана линия при 324,754 нм; показания снимали при 324,719; 324,754 и 324,789 нм, причем первый и третий результаты использовали для вычисления поправки к показанию, полученному для линии самой меди. На интенсивность линии меди сильно влияет линия железа при 324,739 нм; если это влияние очень велико, поправочный коэффициент следует вычислять, используя данные по определению железа при другой длине волны (например, при 259,940 нм).
В атомно-абсорбционных и атомно-флуоресцентных методах редко приходится сталкиваться с проблемами, связанными с перекрыванием линий, так как спектры поглощения имеют очень мало линий, резонирующих с линиями испускания лампы с полым катодом.
Диапазон концентраций. Отклонения от прямолинейной зависимости интенсивности от концентрации, обусловленные са-мопоглощением, столь характерные для методов атомно-эмиссионной спектрометрии и спектрографии с дугой постоянного тока, а также для метода с плазмой постоянного тока, минимальны в методе ИСП, что объясняется физическими особенностями плазменного факела ИСП. Поэтому можно определять методом ИСП очень большой диапазон концентраций, например от 10 нг до 1 мг железа в 1 мл, что позволяет определять на квантометрах одновременно большие, малые и следовые количества компонентов в пробе.
Преимущества. Основное преимущество метода ИСП по сравнению с атомно-абсорбционными методами заключается в возможности определения многих элементов без изменения
206 Глава 9
условий, требуется лишь изменение длины волны и небольшая настройка высоты наблюдения над плазменным факелом. В ААС при переходе от одного элемента к другому необходима смена лампы с полым катодом.
Существенной особенностью ИСП и непламенной ААС является возможность использования только одного газа (аргона), а не двух или более, как в пламенных атомно-абсорбционных и атомно-эмиссионных методах. Стоимость спектрометров ИСП значительно выше аналогичных атомно-абсорбционных приборов.
Задачи
9-1. Требуется проанализировать партию химически чистого металлического алюминия на содержание магния. Навеску массой 1,000 г растворяли в кислоте и добавляли молибдат аммония в таком количестве, чтобы содержание молибдена составляло 2,000 мг. Раствор разбавляли до 100,0 мл; по 50 мкл помещали в кратеры двух медных электродов и выпаривали. Электроды помещали перед щелью спектрографа и пропускали между ними искровой разрид. Оптические плотности линий, полученных на фотопластинке, измеряли иа денситометре; они равны 1,83 при 279,81 нм и 0,732 при 281,62 нм. Найдите процентное содержание Mg в пробе, пользуясь градуировочным графиком, приведенным на рис. 9-4.
9-2. На рис. 9-9 представлено увеличенное изображение участка фотопластинки и миллиметровой шкалы, вмонтированной в объектив увеличителя. В поле зрения находятся участки спектров, полученных при дуговом разриде между железными электродами и электродами, сделанными из неизвестного сплава, предположительно алюминиевого. Линии железа, обозначенные Л, и 7.2, идентифицированы по стандартному спектру железа. Нужно идентифицировать линию х неизвестного сплава. С помощью шкалы сняты следующие показания: 7,1 = 9,990 мм, Х2=8,37 мм и Х3= 10,25 мм. а) Рассчитайте дисперсию
Рис. 9-9. Увеличенный участок фотопластинки со спектром и шкала для сравнения. Длины воли даны в ангстремах.
Атомная эмиссионная спектроскопия 207
спектрографа в этой области в нанометрах на миллиметр, б) Определите длину волны неизвестной линии, в) Пользуясь таблицей длин волн, попытайтесь идентифицировать эту линию, учитывая вероятность присутствия элементов в данном типе сплава. В какой области спектра вы стали бы искать интенсивные линии для подтверждения правильности вашей идентификации?
9-3. Постройте кривую дисперсии для спектрографа, на котором получен спектр, изображенный на рис. 9-3.
9-4. Описан косвенный атомно-эмиссионный метод определения микромоляр-иых количеств первичных амидов [11]. Навеску пробы обрабатывали гипобро-митом бария Ва(ОВг)2, полученным из Ва(ОН)2 и Вг2 по реакции
RCONH2 + 2Ва (ОН)2 + Br2 -> RNH2 + ВаСО3 + ВаВг2 + Н2О
Нерастворимый ВаСОз отфильтровывали, растворяли в HNO3 и анализировали полученный раствор на содержание бария на атомно-эмиссионном спектрометре (пламенном или ИСП). В одном из экспериментов получены приведенные ниже данные. Рассчитайте количество амида в пробе в микромолях на миллилитр.
Проба : 2,00 мл раствора Объем после реакции : 100 мл Анализируемый раствор Показания прибора
0 (холостой) 0,002
5 мкмоль Ва/мл 0,117
10 мкмоль Ва/мл 0,229
15 мкмоль Ва/мл 0,342
Проба 0,157
9-5. При определении следов меди в морских осадках по методу Мак-Лареиа [10] измеряли испускание в ИСП при длине волны меди 324,754 нм, а также, для внесения поправки на фон, испускание при длинах волн 324,719 и 324,789 нм, при которых испускание меди не наблюдается. Кроме того, если в пробе присутствует железо, следует провести измерение црн длине волны испускания железа 324,739 нм.
В одном из экспериментов получены три спектра. Ниже приведены значения токов фотоумножителя (в мкА):
Длина волны, нм
324,719 324,739 324,754 324,789
Сндартиый образец, содержащий Си 23,1 . . . 63,71 8,1
Стандартный образец, содержащий Fe ... 8,75-106 10,5
Проба, анализируемая на содержа- .27,5 9,24-10* 27,49 9,2
низ Си
208 Глава 9
Первый стандартный образец содержал 50,0 мкг/г Си и не содержал Fe. Рассчитайте а) поправку иа фон, б) поправочный коэффициент на перекрывание спектральных линий Fe и Си, в) количество Си в пробе. Результаты выразите в мкг/г.
Литература
1. Scribner В. F., Mullin Н. R., J. Res. Nat. Bur. Standards, 1946, v. 37, p. 379.
2. Nachtrieb N. H., Principles and Practice of Spectrochemical Analysis, McGraw-Hill, New York, 1950.
3. West P. W., Folse P., Montgomery D., Anal. Chem., 1950, v. 22, p. 667.
4. Barnes R. M., In McGraw-Hill Yearbook of Science and Technology, McGraw-Hill, New York, 1978, p. 342.
5. Winefordner J. D., J. Chem. Educ., 1978, v. 55, p. 72.
6. Ishizuka T., Anal. Chem., 1973, v. 45, p. 538.
7. Winefordner J. D.t Fitzgerald J. J., Omenetto N., Appl. Spectrosc., 1975, v. 29, p. 369.
8. Demers D. R., Busch D. A., Allemand C. D., Am. Lab., 1982, v. 14, No. 3, p. 167.
9. Demers D. R., Allemand C. D., Anal. Chem., 1981, v. 53, p. 1915.
10. McLaren J. W„ Berman S. S., Boyko V. J., Russell D. S., Anal. Chem., 1981, v. 53, p. 1802.
11. D’Alonzo R. P., Siggia S., Anal. Chem., 1977, v. 49, p. 262.
12. Van Loon J. C., Anal. Chem., 1981, v. 53, p. 332A.
ГЛАВА
Поляриметрия, оптическая вращательная дисперсия и круговой дихроизм
В предыдущих главах речь шла о лучистой энергии главным образом с точки зрения её поглощения, испускания и распределения по длинам волн. Волновая природа ее представляла интерес только в связи с дифракцией. В этой главе мы рассмотрим явления, связанные с поляризованным излучением.
Многие прозрачные вещества, для которых характерно отсутствие симметрии в молекулярной или кристаллической структуре, способны вращать плоскость поляризованного излучения (краткие сведения о природе плоскополяризованного излучения приведены в гл. 2). Такие вещества называются оптически активными. Вероятно, самые известные из них — это кристаллический кварц и сахара; многие органические и неорганические соединения обладают аналогичным свойством*. Угол поворота плоскости поляризации меняется в широких пределах от одного оптически активного соединения к другому. Вращение называют правым ( + ), если оно происходит по часовой стрелке по отношению к наблюдателю, смотрящему на источник света, и левым (—), если оно происходит против часовой стрелки. Степень вращения зависит от числа молекул на пути излучения или для растворов, от их концентрации и длины сосуда, а также от длины волны излучения и температуры. Удельное вращение [сс]* определяется по формуле
K=-f- (10-1)
где а — угол (измеренный в градусах) поворота плоскости поляризации раствором, содержащим с граммов растворенного вещества в 1 мл в сосуде длиной d дециметров, t — температура и А — длина волнбь Последняя обычно равна 589,3 нм (D-линия натриевой лампы), тогда [сс]* обозначают [а]о'. В-табл. 10-1 приведены значения удельного вращения некоторых веществ.
* Вопросы оптической активности и стереоизомерии подробно изложены в учебниках органической химии и здесь рассматриваться ие будут.
210 Глава 10
Таблица 10-1. Удельное вращение растворов (при 20 °C)
Активное вещество Растворитель . град
Камфора Спирт +43,8
Кальциферол (витамин D2) Хлороформ +52,0
Кальциферол (витамин D2) Ацетон +82,6
Холестерин Хлороформ —39,5
Хинин 0,5 М НС1 —220,0
/-Винная кислота Вода + 14,1
Калий-натрийтартрат (соль Рочелле, сег-нетова соль) Вода +29,8
Сахароза Вода +66,5
ft-d-Глкжоза Вода +52,7
p-d-Фруктозд Вода —92,4
fl-Лактоза Вода +55,4
р-Мальтоза Вода + 130,4
Поляриметры
Для измерения оптической активности обычно используется поляриметр (рис; 10-1). На рис. 10-2 показана схема ручного поляриметра. Монохроматическое излучение от натриевой лампы преобразуется коллиматором в поток параллельных лучей и поляризуется призмой Николя. За поляризатором помещен небольшой вспомогательный николь, который гасит половину потока (объяснение будет дано ниже).. Затем излучение
Рис. 10-1. Прецизионный поляриметр с натриевой лампой (О. С. Rudolph and Sons).
Поляриметрия, ОВД и КД 211
Полутенадой
Коллимирую- НИКОЛЬ щая линза \ о 0 №
Натриебая Николь-
ммпа поляризатор
Защитные / стекла \
□==[
Трубка с пробой
Николь-мализатор
LJ Линзы градуирован- окуляра ныйлимб
Рис. 10-2. Схема поляриметра.
проходит через пробу, находящуюся в стеклянной трубке известной длины, закрытой с обоих концов стеклянными пластинками, и через анализатор попадает в окуляр для визуального наблюдения.
В принципе поляриметр может функционировать и без вспомогательной призмы. В таком случае оба поляризатора должны быть скрещены до попадания потока на пробу, а затем снова уже после пробы. Угол, на который поворачивается анализатор между этими двумя точками, зависит от количества вещества. Однако при таком простом устройстве наблюдатель должен сам_ находить положение, где пропускание излучения равно нулю, что нельзя сделать достаточно точно. Эту трудность можно обойти с помощью дополнительной призмы, через которую проходит половина потока и которая ориентирована таким образом, чтобы ее ось была смещена на несколько градусов относительно оси поляризатора. Можно найти такое положение анализатора, при котором интенсивность излучения обеих половин потока будет одинакова. Этот способ сравнения более удовлетворителен, так как человеческий глаз способен сравнивать с хорошей точностью интенсивности двух потоков.
Фотометрические поляриметры выпускаются многими предприятиями. Это однолучевые устройства, в которых вращение плоскости поляризации до компенсации с вращением пробы осуществляется при помощи сервомеханизма.
Применение. Наиболее широко поляриметрия применяется в сахарной промышленности [I]. В отсутствие других оптически активных веществ при определении сахарозы можно использовать уравнение (10-1), которое при измерении в трубке длиной 2 дм при 20 °C принимает вид
d[a]2D° (2) (66,5) 130
212 Глада 10
В присутствии других активных веществ методика усложняется. Из всех сахаров только сахароза подвергается гидролизу в кислой среде:
С12Н22ОЦ —>- CeHi2Oe -Ь С6Н12О6 сахароза глюкоза фруктоза = + 66,5° + 52,7° —92,4°
Получающуюся смесь глюкозы и фруктозы называют инвертированным сахаром, а реакцию гидролиза — инверсией. В процессе инверсии удельное вращение изменяется от +66,5 до —19,8°; последнее значение соответствует образованию эквимолярной 'смеси продуктов. Измерение вращения до и после инверсии позволяет рассчитать количество растворенной сахарозы. Обычно отбирают 100 мл раствора, измеряют его вращение и добавляют 10 мл концентрированной НС1. Подкисленный раствор следует выдержать при 70 °C не менее 10 мин для полного протекания реакции, затем снова измеряют вращение. Массу сахарозы в пробе рассчитывают по формуле
ws= — 1,17Да—0,00105 [a] (10-3)
где Да —изменение угла вращения; Ца]^°(Х) — удельное вращение других активных веществ, которые могут присутствовать в пробе, wx — масса этих активных примесей. Если почти весь активный материал представляет собой сахарозу, вторым членом можно пренебречь. Наоборот, если нужно определить малое количество сахарозы в большой порции другого активного вещества с известным удельным вращением, можно оценить второй член и рассчитать массу сахарозы по измеренному значению Да.
Рис. 10-3. Ферментное разложение пенициллина при разной концентрации пенициллиназы в фосфатном растворе (pH 7), Цифры обозначают относительную концентрацию фермента [2].
Поляриметрия, ОВД и КД 213
В другом примере анализа, основанного на оптическом вращении, одновременно определяют пенициллин и фермент пенициллиназу [2]. Пенициллин количественно разрушается под действием фермента со скоростью, прямо пропорциональной количеству присутствующего фермента и не зависящей от концентрации пенициллина. График зависимости вращения от времени представляет собой прямую, которая обрывается, когда весь пенициллин израсходован. По наклону прямой можно судить о количестве фермента. На рис. 10-3 приведены такие графики для нескольких концентраций фермента. Как показано на рисунке, кривые закруглены, что объясняется протеканием побочных реакций. Момент исчезновения пенициллина находят пересечением экстраполированных участков прямых. Точность определения пенициллина около ± 1 % и фермента около ±10 %.
Оптическая вращательная дисперсия и круговой дихроизм
Зависимость оптической активности от длины волны (оптическая вращательная дисперсия, ОВД) дает более богатую информацию о структуре асимметрических соединений, чем удельное вращение при единичной длине волны [3]. Это явление тесно связано с явлением, называемым круговым дихроизмом (КД) [4].
В гл. 2 было показано, что обычный световой поток можно разложить на два плоскополяризованных потока. Теперь мы можем сделать следующий шаг в этом направлении. Плоско-поляризованное излучение можно также разложить на два потока, которые поляризованы по кругу в противоположных направлениях (рис. 10-4, а). Показатели преломления оптической среды для лево- и правополяризованных по кругу компонент излучения не обязательно одинаковы и обозначаются соответственно nL и «R. Соответствующие коэффициенты поглощения йь и йн также могут различаться. В изотропной среде, например стекле или воде, «l = «r и aL = aR-t в этом случае говорят, что показатель преломления и коэффициент поглощения не зависят от поляризации. Если же то между двумя компонентами появляется сдвиг по фазе. В этом случае можно сказать, что плоскость поляризации повернулась, т. е. угол а^О (рис. 10-4,6). Если йе^й]{, то одна компонента поглощается сильнее другой, тогда круговая поляризация превращается в эллиптическую с эксцентриситетом 0, который выражается разностью «ь — ак (рис. 10-4, в и г). Значение электрического вектора Е электромагнитной волны пропорционально корню квадратному из интенсивности потока, следовательно, значения .векторов на рис. 10-4, в и г соответственно равны: ^l = ElV^’l и Ек = Екд/^к> где Тъ и Тп— пропускания [5].
214 Глава 10
Рис. 10-4. а — разложение плоскополяризованного излучения на компоненты с левой и правой круговой поляризацией. Два радиуса-вектора EL и Er, вращающиеся в противоположных направлениях, составляют вектор Ео, колеблющийся в вертикальной плоскости. Поток излучения распространяется перпендикулярно плоскости бумаги по центру круга. Векторы являются электрическими составляющими соответствующих потоков излучения; б — соответствующая векторная диаграмма потока излучения, изображенного на рис. а, после прохождения через оптически активный материал. Появляется сдвиг по фазе, соответствующий повороту плоскости поляризации на угол а; в — векторная диаграмма излучения, представленного на рис. а, после прохождения через пробу, которая поглощает компоненту с правой круговой поляризацией сильнее, чем компоненту с левой круговой поляризацией. Результирующий вектор Ео' здесь описывает эллипс, большая ось которого равна El'+Er', а малая ось равна El'—Е8'. Это — гипотетический случай КД без оптического вращения. Эллиптичность равна углу 0; г — векторная диаграмма излучения после прохождения через пробу, которая отражает и оптическую активность, и КД.
Поляриметрия, ОВД и КД 215
Можно показать [3], что угол вращения [уравнение (10-1)], выраженный в радианах, равен
(10-4)
где d — длина оптического пути в пробе.
Эллиптичность, т. е. угол, тангенс которого равен отношению большей оси эллипса к меньшей, выражается (в радианах) следующим образом:
^=~{kL^kR)d (10-5)
4
где k — коэффициент поглощения в законе поглощения, выраженном в форме
р = рое-м (Ю-6)
Уравнение (10-5) можно представить в более удобной форме:
[0] = 0*8О M.^33oo(gL_eR)d (10.7)
я С
где [0] — молекулярная эллиптичность, выражаемая в градХ Хсм2/моль, еь и er — молярные коэффициенты поглощения излучения с левой и правой круговой поляризацией. Зависимости этих величин от длины волны можно видеть на рис. 10-5.
Рис. 10.5. Спектры поглощения в УФ-области, КД- и ОВД-спектры соединения с положительным КД-
216 Глава 10
s
S.-2
^-3 -<15
.§ -4 -
-5-
НО
Пик ОВД S
Максимум КД
сан17
Максимам погмщв-~""~~~ _ ния УФ-излучения
Перегиб
S- z* \ спектра ОВД
2
ед
\ ! Минимум х
Л^06А
250 300 350 400
Л, НМ
7
6
5
4
3
2
Oj
1
Рис. 10-6. Полоса в спектрах ОВД, КД и УФ-поглощения, соответствующая слабому переходу в эписульфидной группе Зр-гидроксихолестаи-5й,6а-эпи-сульфида [6].
Величину (ml—«и) называют оптической активностью, a (kb—&r) — круговым дихроизмом. Обе величины очень малы по сравнению со средними значениями (hl + hr)/2 и (&l+&r)/2 соответственно. Значение а можно измерить непосредственно на соответствующем приборе, а значение 0 можно определить лишь косвенно из данных КД. Наличие в одной среде ненулевой оптической активности и дихроизма получило название эффекта Коттона (по имени открывшего его ученого). Этот эффект проявляется при поглощении хромофорами оптически активных соединений (рис. 10-6). Длины волн в максимуме поглощения КД, в максимуме светопоглощения и в точке перегиба кривой ОВД теоретически должны совпадать, но влияние других хромофоров может вызвать смещение наблюдаемого максимума поглощения (как на рис. 10-6). С помощью спектров КД часто можно обнаружить «скрытый» максимум поглощения, причем более однозначно, чем по спектрам ОВД. С другой стороны, на спектры ОВД довольно сильно влияют более удаленные хромофорные полосы, следовательно, эти кривые более специфичны для отдельных соединений, чем спектры КД. И те и другие спектры могут дать ценную информацию о стереохимии оптически активных веществ. На практике соотношение между
Поляриметрия, ОВД и КД 217
Сканирующий Поляри- Анали- Фотоумножи-
монохроматор затор Проба затор толь
Рис. 10-7. Блок-схема спектрополяриметра.
кривыми для одного и того же вещества может быть более сложным, чем на рис. 10-6. Рамки этой книги не позволяют углубляться в теорию вопроса, поэтому для более детального ознакомления читатель должен обратиться к литературе [4].
Фотометры для измерения ОВД. Сейчас выпускается несколько моделей регистрирующих спектрополяриметров. Как видно из схемы на рис. 10-7, их можно рассматривать как видоизмененные однолучевые спектрофотометры. В этом приборе поток света поступает из обычного монохроматора, проходит последовательно через поляризатор, пробу и анализатор и направляется в фотоумножитель. Поляризованный поток модулируется с частотой 12 Гц при помощи устройства, поворачивающего поляризатор то в одном, то в другом направлении на небольшой угол (порядка 1—2 градусов). Сервоусилитель реагирует только на частоту 12 Гц и заставляет сервомотор постоянно фиксировать анализатор в такой точке, где 12-Гц сигнал расположен симметрично относительно нулевой точки. Сервомотор также управляет пером самописца.
Вместо осциллирующего поляризатора можно пользоваться другими типами модуляторов, например ячейкой Фарадея [7], состоящей из стеклянного или кварцевого стержня, окруженного спиралью, через которую пропускают переменный ток частотой 60 Гц. Это устройство вращает плоскость поляризации на несколько градусов, причем направление вращения изменяется с приложенной частотой.
Приборы для измерения КД. На обычных спектрофотометрах измеряется разница между оптическими плотностями пробы
218 Глава 10
Рис. 10-8. Оптическая схема для измерения КД. Излучение входит слева, отклоняется вниз зеркалами и ЛГ2, плоско поляризуется составной призмой Р и проходит через параллелепипед Френеля R, где подвергается двум внутренним отражениям, что приводит к сдвигу по фазе на четверть длины волны, т. е. к круговой поляризации. С помощью экрана А устраняется нежелательное излучение и пропускается нужное. Всю эту схему целиком помещают в кюветное отделение стандартных спектрофотометров, вторая схема (с противоположной ориентацией) нужна для сравнения. Пробу помещают в точку b при измерении КД или в точку а при изучении пропускания плоскополя-ризованного излучения.
и стандарта, поэтому их легко приспособить к измерению КД, где нужно точно так же измерять разницу между двумя оптическими плотностями.
Круговую .поляризацию проводят в две ступени. Сначала поток излучения нужно сделать плоскополяризованным, а затем поляризованный поток пропустить через устройство, которое разлагает его на компоненты с правой и левой круговой поляризацией. Затем одну из компонент следует сдвинуть по фазе на одну четверть длины волны. Наиболее важное значение имеют три типа устройств для круговой поляризации: параллелепипед Френеля, электрооптический модулятор Покельса и фотоупругий модулятор.
На рис. 10-8 приведено устройство первого типа. Принцип его действия основан на общем внутреннем отражении светового потока от двух поверхностей раздела стекло—воздух; при этом фаза одного из ортогональных плоскополяризованных потоков сдвигается, а другого остается без изменений. При правильном выборе углов падающий поток окажется поляризованным по кругу [7]. Интервал длин волн такого устройства несколько ограничен.
В модуляторе Покельса к пластинке из кристиллического КН2РО4 или подобного материала приложено высокое напряжение (несколько киловатт); под действием электрического поля происходит двойное лучепреломление, так что первоначальное плоскополяризованное излучение поляризуется по кругу [8]. Для того чтобы регулировать сдвиг по фазе, выбирают потенциал и программируют непрерывную корректировку на вы
Поляриметрия, ОВД и КД 219
ходе при изменении длины волны. Интервал длин волн ограничен только областью пропускания кристалла.
В работе фотоупругого модулятора используется принцип двойного лучепреломления под действием механического напряжения [9]. Такие модуляторы изготавливают из плавленого кварца, фторида кальция и сульфида цинка, причем два последних пригодны для работы в УФ-, видимой и ИК-областях.
Подробное обсуждение методов калибровки, необходимой для количественных измерений на спектрофотометрах КД, представлено Шиппером и Деккерсом [10].
Применение кругового дихроизма. Количественный анализ при помощи КД возможен, хотя и не получил широкого распространения. В работе [11] сообщается об определении героина, кокаина и 12 других наркотиков. В принципе возможно определение любого оптически активного соединения в отсутствие других соединений, проявляющих КД в той же области. Например, при подпольном производстве кокаина в качестве разбавителей и добавок используют лактозу, лидокаин и бензокаин. Лактоза оптически активна, но в отличие от кокаина при насыщении не способна к селективному поглощению УФ-излучения (в том же интервале длин волн). Другие упомянутые соединения поглощают в этой же области (благодаря хромофорной бензольной группировке), но оптически неактивны и, следовательно, не мешают анализу.
В большинстве случаев метод измерения КД служит для установления структуры молекул и особенно ее стереометрии и конформации.
Задачи
10-1. Навеску загрязненного препарата соли Рочелле массой 10,0 г растворили в воде, получив 100,0 мл раствора. Сначала кювету поляриметра длиной 20 см заполнили дистиллированной водой (чтобы установить точку отсчета), а затем порцией раствора пробы. Получены следующие данные (при 20 °C):
показание по шкале для воды +2,020°
показание по шкале для раствора пробы +5,750°
Найдите процентное содержание (по массе) соли Рочелле в пробе.
10-2. Рассчитайте ошибку (в процентах) определения сахарозы по уравнению (10-3), в котором опущен второй член, для каждой из следующих проб: а) 30 г сахарозы+2 г лактозы, б) 16 г сахарозы+16 г лактозы и в) 2 г саха-розы+30 г лактозы. Изменением удельного вращения лактозы в интервале 15—20°С можно пренебречь.
10-3. Угол вращения плоскополяризоваиного света водного раствора некоего соединения, измеренный в 1-дм кювете при 500 нм, равен 10,0°. Рассчитайте оптическую активность соединений.
220 Глава 10
Литература
1. Bates F. J. et al., Polarimetry, Saccharimetry, and the Sugars, Nat. Bur. Standards Circ. C440, 1942.
2. Levy G. B., Anal. Chem., 1951, v. 23, p. 1089.
3. Djerassi C„ Optical Rotatory Dispersion: Applications to Organic Chemistry, McGraw-Hill, New York, 1960.
4. Velluz L., Legrand M., Grosjean M., Optical Circular Dichroism, Academic Press, New York, 1965.
5. Duffield J. J., Abu-Shumays A., Robinson A., Chem. Instrum., 1968, v. 1, p. 59.
6. Djerassi C., Wolf FL, Lightner D. A., Bunnenberg E., Takeda K-, Korneno T., Kuriyama K„ Tetrahedron, 1963, v. 19, p. 1547.
7. Bennett J. M„ Bennett N. E„ Polarization in Handbook of Optics, Driscoll W. G., Vaughan W. (eds.), McGraw-Hill, New York, 1978, pp. 10— 120.
8. Hartfield E., Thompson B. J., Optical Modulators in Handbook of Optics, Driscoll W. G., Vaughan W. (eds.), McGraw-Hill, New York, 1978, sec. 17.
9. Koehler M. E., Urbach F. L., Appl. Spectrosc., 1979, v. 33, p. 563.
10. Schippers P. H., Dekkers H. P. J. M., Anal. Chem., 1981, v. 53, p. 778.
11. Bowen J. M., Crone T. A., Kennedy R. K-, Purdie N., Anal. Chem., 1982, v. 54, p. 66.
12. Bowen J. M., Purdie N., Anal. Chem., 1981, v. 53, p. 2237, 2239.
ГЛАВА
Рентгеновские методы
При попадании на мишень пучка ускоренных электронов основная их масса замедляется за счет многократных взаимодействий с электронами мишени. Энергия, теряемая электронами, превращается в энергию рентгеновского излучения с непрерывным спектром, которое называется тормозным излучением. Непрерывный спектр рентгеновского излучения имеет четкую нижнюю границу длин волн Лмин (или максимальную частоту), соответствующую максимальной энергии электронов. Эта граничная длина волны (в • нанометрах) определяется соотношением
Хмин = Лс/Ve = 1240/V (11-1)
где h — постоянная Планка, с — скорость распространения электромагнитного излучения в вакууме, е —заряд электрона, V — ускоряющий потенциал рентгеновской трубки в вольтах. По мере увеличения ускоряющего потенциала энергия достигает величины, достаточной для полного выбивания орбитального электрона из атома мишени. Тогда на вакантный уровень переходит другой электрон и испускается фотон рентгеновского излучения с длиной волны, определяемой разностью энергий соответствующих уровней данного элемента.
Ускоренные электроны более высоких энергий воздействуют главным образом на ближайшие к ядру электроны. Так, например, они могут выбить К электрон, а на его место перейдет электрон L-оболочки. Состояние электронов внутренних оболочек не зависит от химического состояния атомов (за исключением атомов более легких элементов), поэтому характеристики рентгеновского излучения фактически не зависят от химического или физического состояния атома. (В гл. 12 обсуждаются некоторые исключения из этого правила.) Длины волн, соответствующие таким высоким энергиям, малы (порядка 1—1000 пм) *.
* Длины воли рентгеновского излучения обычно выражают в ангстремах (А). С целью распространения системы единиц СИ в этой книге используются нанометры (нм) или пикометры (пм); 1 нм=10 А=1000 пм.]
222 Глава 11
Рис. 11-1. Рентгеновский спектр испускания трубки с мишенью нз молибдена, работающей при 35 кВ. Фон со сплошным спектром обусловлен тормозным излучением; пики соответствуют Ка- и ЛР-переходам [1].
Для исследовательских работ наиболее полезен диапазон длин волн от 70 до 200 пм.
Спектр рентгеновского излучения вещества мишени, облученного электронами высоких энергий, имеет вид, приведенный на рис. 11-1, где на сплошной спектр накладываются отдельные линии. Линии излучения, соответствующие переходам между L-и К-оболочками, обозначены Ка; Kai и К.а.2 соответствуют электронам, выбитым из различных подуровней L-оболочки; рентгеновское излучение, возникающее благодаря переходам между М- и К-оболочками, называется Кр и т. д. Тяжелые элементы дают другие группы линий, соответствующие переходам на L-, М- и более высокие уровни.
Возбуждение может также происходить под действием падающего рентгеновского излучения; в этом случае в спектре появляются только характеристические линии. Такая ситуация благоприятна для рентгеновского эмиссионного анализа, так как существенно увеличивается соотношение сигнал/шум.
Использование рентгеновского излучения дает разнообразную информацию, пригодную для аналитических целей. 1) По-
Рентгеновские методы 223
глощёние рентгеновских лучей дает информацию о поглощающем материале так же, как и в других спектральных диапазонах. 2) Дифракция рентгеновских лучей позволяет идентифицировать кристаллические вещества с высокой степенью избирательности и точности. 3) Измерение длин волн или энергий дает возможность определять различные элементы в возбуждаемом образце. 4) Измерение излучаемой мощности при некоторых длинах волн может быть использовано для количественного определения состава пробы.
Поглощение рентгеновских лучей
Рентгеновские лучи поглощаются веществом точно так же, как и электромагнитное излучение других областей спектра, причем степень поглощения определяется природой и количеством поглощающего вещества. Если наибольший интерес представляет толщина слоя поглощающего вещества, то закон Бера для монохроматического рентгеновского излучения удобно выразить в форме
РХ=Р^ (11-2)
или ln(P0/P) = RX (Н-З)
где Ро — интенсивность падающего излучения и Рх — интенсивность после прохождения через слой толщиной х см. Линейный коэффициент поглощения р, представляет собой часть энергии, поглощенной слоем данного элемента толщиной 1 см. Заметим, что по традиции в уравнении (11-3) используются натуральные, а не десятичные логарифмы. Часто более удобен массовый коэффициент поглощения, определяемый как
Р-м = Р-4> (Н-4)
где р — плотность поглощающего вещества. Если поглощающее вещество состоит только из одного химического элемента, коэффициент связан с длиной волны излучения и атомными характеристиками поглотителя эмпирической формулой
p^-CNZW/A (11-5)
где N—число Авогадро, Z — атомный номер поглощающего элемента, А — его атомный вес, X — длина волны, п — показатель степени, принимающий значения между 2,5 и 3,0, а С — постоянная, имеющая одно и то же значение для всех элементов в пределах ограниченных диапазонов, указанных ниже.
224 Глава 11
Рис. 11-2. Рентгеновское поглощение аргона в диапазоне 50—1000 пм. Обратите внимание на край поглощения при 1g /.=2,59 [2].
Изменение р,м с длиной волны подчиняется степенному закону, поэтому в логарифмических координатах должна получиться прямая с тангенсом угла наклона, равным показателю степени при X. На рис. 11-2 приведена зависимость коэффициента поглощения от длины волны для аргона. Наиболее примечательной особенностью этого графика является разрыв зависимости при lgX = 2,59 (Х=387,1 пм), называемый критическим краем поглощения К. Энергия излучения больших длин волн недостаточна для выбивания /(-электронов аргона, поэтому оно поглощается слабее, чем излучение меньших длин волн. Для более тяжелых, чем у аргона, атомов такие разрывы, соответствующие фотоэлектрическому выбиванию L- и ЛГ-электронов, наблюдаются в более длинноволновой области.
Целесообразно построить зависимость цм от отношения 74/Л для рентгеновских лучей определенной длины волны. На рис. 11-3 показаны значения цм, присущие Да-линии меди (Си), для различных элементов от натрия до осмия. На графике видны критические края поглощения, поскольку по мере увеличения заряда атомного ядра электроны определенной оболочки все сильнее связываются с ним и наступает момент, когда энергии Да рентгеновского излучения меди недостаточно для выбивания этих электронов. «Постоянная» С в уравнении (11-5), в точке разрыва скачком изменяющая свое значение, не является строго постоянной между этими скачками, на что указывает незначительное -искривление зависимости между Д- и Ц-краями поглощения. Это искривление можно приписать изменению показателя степени п. Уравнение (11-5) потребует уточнения по мере более глубокого изучения рассматриваемых явлений. Эмпирические данные по поглощению большинства элементов можно найти в литературе [1, 2].
Рентгеновские методы 225
Рис. 11-3. Зависимость массовых коэффициентов поглощения от атомных постоянных. Излучение Ка линии Си 154,18 пм [2].
8 Заказ № 537
226 Глава ii
Монохроматические источники-рентгеновских лучей
Получить излучение с узкими полосами длин волн в отдельных точках рентгеновского излучения относительно легко, но не так просто создать монохроматор, с помощью которого можно произвольно менять длину волны. Излучение с узким спектром можно получить тремя способами: 1) с помощью фильтров, выделяющих характеристические линии, которые значительно интенсивнее фона; 2) с помощью монохроматора, в котором диф-фракционной решеткой служит кристалл с известными межплоскостными постоянными; 3) с помощью радиоактивных источников.
В некоторых случаях удается получить монохроматический поток с помощью фильтра из какого-либо элемента (или его соединения) с критическим краем поглощения как раз при нужной длине волны. Например, на рис. 11-4 представлен спектр
Рис. 11-4, Наложение кривой поглощения циркония на спектр испускания молибдена при 35 кВ, в результате чего получается монохроматическое излучение /Са-линии Мо [1].
Рентгеновские методы 227
Таблица 11-1. Характеристические длины волн и фильтры для рентгеновских трубок с обычными мишенями
Мишень Длина волны Фильтр Толщина фольги, мкм Поглощенное К₽1, %
Ка,, пм К₽2, пм Элемент К-край, пм
Сг 228,9 208,5 V 226,9 15,3 99,0
Fe 193,6 175,7 Мп 189,6 12,1 98,7
Со 178,9 162,1 Fe 174,3 14,7 98,9
Ni 165,8 150,0 Со 160,8 14,3 98,4
Си 154,1 139,3 Ni 148,8 15,8 97,9
Мо 70,9 63,2 Zr 68,9 63,0 96,3
Ag Wa 55,9 20,9 49,7 18,5 Pd 50,9 41,3 94,6
а) Для излучения вольфрама нет подходящего фильтра.
поглощения циркония (штриховая линия), наложенный на спектр испускания молибдена (ср. с рис. 11-1). /ф-линия испускания Мо и большая часть тормозного излучения поглощаются фильтром из циркония, а Ла-линия Мо, расположенная сбоку от критического края Zr со стороны низких энергий, практически не ослабляется. Выделение этой /Са-линии не будет полным, но тем не менее для многих целей приемлемым. В табл. 11-1 перечислены фильтры, пригодные для выделения /Са-излучения некоторых мишеней.
Кристаллы-анализаторы. Если требуется излучение с большей степенью монохроматичности, чем получаемое с помощью фильтров, необходимо использовать дифракционный монохроматор. Устройство этого прибора будет описано ниже в разделе, посвященном дифракции рентгеновских лучей.
Такой монохроматор может давать узкий участок длин волн в любой области спектра, если есть источник рентгеновских лучей с непрерывным спектром. Однако рентгеновское излучение с непрерывным спектром, получаемое при умеренных напряжениях рентгеновских трубок, не настолько интенсивно, чтобы найти широкое применение, поэтому для практических целей все же ограничиваются источниками с характеристическими длинами волн. Преимущество монохроматора по сравнению с фильтром заключается в существенном снижении фонового излучения, что приводит к улучшению отношения сиг-нал/шум, несмотря на заметное ослабление сигнала.
Радиоактивные источники. Радиоактивные элементы испускают излучение в рентгеновском диапазоне по одному из двух 8*
228 Глава 11
Таблица 11-2. Изотопы, используемые в качестве источников рентгеновских или гамма-лучей а)
Изотоп Период полураспада Излучение^)
55Fe 2.60 года ЭЗ: Мп К, 210,3 пм
5’СО 270 сут ЭЗ: Fe Гамма: 86,1, 10,17 пм
в°Со 5,26 года Гамма: 1,06, 0,931 пм
241 Аги 458 лет ЭЗ: Np L, 88,9 пм, Np L, 69,8 пм, Np L, 59,7 пм Гамма: 20,87, 47,0 пм
а) Данные взяты нз работы [3—5] и при необходимости переведены в соответствующую систему единиц.
ЭЗ — электронный захват
механизмов [3]. Первый из них включает внутриядерные энергетические переходы и приводит к испусканию гамма-излучения, не являющегося истинным рентгеновским излучением. Другой механизм известен под названием электронного захвата, или К-захвата. Электрон, находящийся на К-орбитали, имеет конечную вероятность находиться в течение некоторого времени вблизи ядра, поэтому для многих атомов существует конечная вероятность захвата /(-электрона ядром. Этот процесс понижает атомный номер элемента на одну единицу и образует вакансию в К-оболочке. В результате возникает истинное рентгеновское излучение соседнего, расположенного ниже элемента, которое уже не сопровождается каким-либо значительным излучением с непрерывным спектром. В табл. 11-2 перечислены некоторые изотопы, оказавшиеся полезными в качестве источников гамма-или рентгеновского излучения. Для выделения отдельных линий все же могут потребоваться фильтры.
Преимущество радиоактивных источников состоит в том, что они не нуждаются в сложных высоковольтных источниках напряжения и вакуумных трубках, имеющих ограниченный срок годности. Но, с другой стороны, их нельзя выключить, так что всегда существует потенциальная опасность облучения независимо от того, идет эксперимент или нет.
Детекторы рентгеновского излучения
Впервые рентгеновское излучение было обнаружено по появлению изображения в облученных ими фотоматериалах. В настоящее время фотография не находит широкого применения в количественном анализе, но она удобна для некоторых ди
Рентгеновские методы 229
фракционных методов, где получаются двумерные картины и их геометрию нужно точно зафиксировать. Для увеличения поглощения проникающего рентгеновского излучения фотопленку с обеих сторон покрывают эмульсией. Действие других детекторов основано на способности рентгеновских лучей вызывать в одних веществах вспышки света (сцинтилляции), в других — ионизацию.
Сцинтилляционные детекторы. Некоторые вещества обладают свойством давать очень слабые вспышки света при поглощении рентгеновских фотонов (т. е. флуоресцировать). Примером тому служит кристаллический Nal, в который введено небольшое количество (1 или 2%) ТП. Вспышки света можно регистрировать фотоумножителем, обеспечивающим надежное измерение числа фотонов, падающих на кристалл. Длительность каждой вспышки очень мала (порядка 10~8 с), но возникающий электрический импульс может иметь несколько большую длительность из-за несовершенства электронных схем. Хотя при больших скоростях счета наблюдается наложение импульсов, все же при средних скоростях счета (до 10е имп./с) можно выделить и подсчитать отдельные сцинтилляции. В гл. 3 упоминалось о принципиальных схемах, используемых в сочетании с фотоумножителем.
Газовые ионизационные детекторы. Рентгеновское излучение, проходя через газ, ионизирует его и, следовательно, может быть обнаружено по проводимости газа. Для этого предназначена ионизационная камера, представляющая собой простую металлическую емкость с изолированным центральным электродом, наполненную сухим газом. Электрод находится под напряжением 100 В или выше, а возникающий при ионизации ток измеряют электрометром. Сигналы от отдельных фотонов не разрешаются, и поэтому регистрируемый ток соответствует среднему или равновесному значению.
Сигнал можно значительно усилить, если увеличить напряжение на центральном электроде до значения, при котором электроны, возникающие при первичной ионизации, сильно ускоряются и при столкновении ионизируют другие молекулы. Количество образующихся при этом ионов зависит от энергии фотона рентгеновского излучения, поэтому ток будет пропорционален энергии фотона. Прибор, работающий в таком режиме, называется газовым пропорциональным счетчиком. Он обладает быстродействием, достаточным для счета отдельных фотонов. При дальнейшем увеличении напряжения наступает насыщение, т. е. все импульсы имеют одну и ту же величину независимо от энергии падающего фотона. Детектор, работающий в этом
230 Глава 11
режиме, называется счетчиком Гейгера—Мюллера (или Гейгера). В гл. 24 дано более подробное описание газовых ионизационных детекторов.
Твердотельные ионизационные детекторы. Чистые монокристаллы германия и кремния могут стать чувствительными к рентгеновскому и другим ионизирующим излучениям в присутствии лития. По мере диффузии лития в кристаллическое вещество (технический термин — «дрейф») происходит очистка вещества от примесей. Фотон рентгеновского излучения, проникающий в очищенный кристалл, выбивает электроны из решетки, оставляя вакансии, обычно называемые дырками, которые по своему действию эквивалентны подвижным положительным электрическим зарядам. Число таких актов разделения зарядов непосредственно связано с энергией фотона, поэтому полученный сигнал (увеличение проводимости) также пропорционален этой энергии. Детекторы такого типа должны находиться при температуре жидкого азота (даже при хранении) для предотвращения дальнейшей диффузии лития, которая существенно уменьшает чувствительность и со временем выводит детектор из строя.
Электрический выходной сигнал твердотельного детектора гораздо слабее сигнала газового или сцинтилляционного детектора, поэтому необходим электронный усилитель с большим коэффициентом усиления. На рис. 11-5 приведены сравнительные спектральные характеристики этих детекторов нескольких типов. Отметим, что по оси абсцисс отложены единицы энергии, которые обратно пропорциональны единицам длин волн. Как
0,248 0,124 0,083 0,062 0,050//м
Энергия рентгеновского излучения, кэВ
Рис. 11-5, Сравнение сигналов газового пропорционального, сцинтилляционного н Si (Li)-детекторов.
Рентгеновские методы 231
Fe Ка и кр
6,4 U Ч,05кэВ
энергия рентгеновского излучения, кэВ
Рис. 11-6. Сравнение разрешающей способности сцинтилляционного, газового пропорционального и Si (Li)-детекторов.
показано на рис. 11-6, для строго монохроматического излучения каждый детектор дает несколько уширенный пик. Ка- и Кр-линии железа с длинами волн 193,7 и 175,7 пм (6,40 и 7,05 кэВ) разрешены только с помощью твердотельного детектора на основе кремния Si (Li); ни сцинтилляционный, ни газовый пропорциональный детекторы не дают такого разрешения..
Рентгеновская абсорбционная спектрометрия. Использование рентгеновского поглощения в аналитических целях представляется наиболее полезным в тех случаях, когда в матрице из легких атомов содержится только один определяемый элемент большой атомной массы. К этой категории относятся некоторые аналитические методики, имеющие важное значение в промышленности в контрольно-измерительных целях. Этим способом определяют наличие свинца в бензине [6], хлора в органических соединениях [7], урана в растворах его солей [8]. Датчики на рентгеновском и гамма-излучении используют для контроля толщины пищевой алюминиевой фольги в процессе прокатки.
Аналитические методы, основанные на прямом поглощении рентгеновского излучения, в основном были вытеснены методами рентгеновской флуоресценции (описаны ниже), которые при использовании аналогичной аппаратуры дают как качественную, так и количественную информацию.
Спектрометрия края поглощения. В другом методе, основанном на поглощении рентгеновского излучения, используется понятие о критическом крае поглощения. Поглощение излучения
232 Глава И
Рнс. 11-7. Поглощение на критическом крае [9].
любым элементом образца значительно сильцее при длине волны несколько меньшей длины волны края поглощения, чем при несколько большей длине волны, а так как положение края на шкале длин волн характерно для каждого элемента, то пара измерений при длинах волн, ограничивающих край поглощения, может служить для обнаружения и определения искомого элемента.
Если построить зависимость интенсивности рентгеновского -излучения от длины волны вблизи К-края поглощения элемента, то получится кривая типа приведенной на рис. 11-7. Скачок при длине волны края поглощения Хе не является строго вертикальным, а немного искривлен (как показано на рисунке) из-за того, что щель имеет конечную ширину. Количество определяемого элемента пропорционально расстоянию по вертикали между точками пересечения X и У, которые находят путем экстраполяции. Для того чтобы оценить высоту скачка с хорошей точностью, нужно провести измерения на двух равноудаленных от границы поглощения длинах волн Xi и Хг. Математические детали и обоснование этой методики дано в работе [9], где показано, что для многих элементов вплоть до концентраций 0,1 % относительная погрешность не превышает 1 %.
Аппаратура для абсорбционных измерений. Лабораторный анализ на основе поглощения рентгеновского излучения чаще всего производится при помощи обычных приборов. Промышленная аппаратура, как правило, конструируется для каждой установки. В портативном анализаторе для обнаружения S и РЬ в нефтяных продуктах (Columbia Scientific Industries) используют радиоактивный источник.
Рентгеновские методы 233
Рентгеновские монохроматоры
Рентгеновские лучи, будучи электромагнитными волнами, дифрагируют подобно электромагнитному излучению других спектральных диапазонов (рис. 11-8). Уравнение, приведенное в гл. 2 для дифракции на решетке
mX = dsin9
(И-6)
приложимо к рентгеновским лучам. В данном случае длина волны уменьшается в 1000 и более раз, так что для получения приемлемых значений 0 нужно во столько же раз уменьшить постоянную решетки. Изготовить решетку, удовлетворяющую этому требованию, невозможно, но, к счастью, во многих кри
сталлах расстояние между соседними атомными плоскостями
имеет как раз такой порядок величины. В качестве рентгеновских решеток пригоден целый набор различных кристаллов: к наиболее широко используемым относятся фторид лития, хлорид натрия, кальцит, гипс, топаз, этилен-диамин-й-тартрат (ЭДТТ или ЭДТ) и дигидрофосфат аммония (ДФА). В простейшем устройстве рентгеновский луч отражается от плоскости кристалла (рис. 11-8), и в результате изменения угла падения выделяется определенная длина волны. Волны, отраженные соседними кристаллическими плоскостями, должны дважды проходить пространство между ними, поэтому уравнение (11-6) переходит в уравнение Брэгга
mX = 2dsin0 (Н-7) где d — теперь расстояние между соседними плоскостями в кристалле. Уравнение (11-7) легко вывести с помощью рис. 11-8. В данном случае углы падения и дифракции
Рис. 11-8. Дифракция рентгеновских лучей на последовательных слоях атомов в кристалле. S соответствует падающему плоскому волновому фронту.
Рис. 11-9. Рентгеновский монохроматор, установленный на круге Роуланда. Кристалл деформирован и отшлифован для точной фокусировки.
234 Глава 11
должны быть равны, но это ограничение не распространяется на оптическую дифракцию.
/Ложно использовать фокусирующие решетки Роуланда [10]. Для этой цели кристалл должен иметь искривленную поверхность, удовлетворяющую геометрическим требованиям. Для улучшения фокусировки кристалл предварительно надо изогнуть, чтобы дифрагирующие кристаллические плоскости имели радиус кривизны, вдвое превышающий радиус окружности Роуланда, а затем оптимизировать поверхность кристалла, сделав ее радиус кривизны равным радиусу окружности Роуланда (рис. 11-9). Стоимость фокусирующего монохроматора значительно выше, чём аналогичного прибора с плоским кристаллом, в основном из-за того, что обработка поверхности кристалла требует тонкой ручной работы, но зато этот монохроматор дает в 10 раз большую интенсивность.
Дифракция рентгеновских лучей
Дифракция рентгеновских лучей представляет большой интерес для изучения тех кристаллических веществ, в которых возможна дифракция. Не существует двух химических веществ, которые бы имели кристаллы с совершенно одинаковым расположением плоскостей во всех направлениях, так что полное изучение образца при различной ориентации на пути рентгеновских лучей должно давать однозначный результат для каждого вещества.
Экспериментальная установка для наблюдения рентгеновской дифракции по существу аналогична оптическому дифракционному спектрометру, но, так как для рентгеновских лучей нельзя использовать линзы и ; чается от оптического прибор
жала, внешне она сильно отли-Для получения коллимированного потока от рентгеновского источника используют мишень в виде набора металлических трубок, а если требуется скол-лимировать поток только водной плоскости, то в виде набора параллельных металлических пластин. В некоторых конструкциях испускающая поверхность мишени расположена под скользящим углом к наблюдателю (рис. 11-10), что очень напоминает конструкцию линейного источника с максимальной интен-
Рис. 11-10. Расположение электродов в рентгеновской трубке для получения пучка под скользящим углом.
Рентгеновские методы 235
сивностью. Такую рентгеновскую трубку можно установить вертикально (мишень сверху), а лучи, выводить через несколько каналов в различных горизонтальных направлениях, что позволяет проводить два, три и даже четыре измерения одновременно.
С помощью рентгеновской дифракции можно идентифицировать главным образом кристаллические соединения. Например, каждый из оксидов железа дает свою особую дифракционную картину, и появление ее подтверждает наличие соответствующего соединения. Элементы как таковые могут быть обнаружены, только если они находятся в кристаллическом состоянии. Этим рентгеновская дифрактометрия резко отличается от рентгеновской эмиссии и поглощения, где сигнал зависит от присутствующих элементов независимо от их химического состояния.
Интенсивность дифрагированного луча зависит от содержания соответствующего кристаллического вещества в образце, что позволяет количественно определить состав смеси твердых веществ.
Дифракционные приборы. Дифрагированный пучок рентгеновских лучей можно зарегистрировать фотографически или с помощью одного из описанных выше детекторов. Типичным
Рис. 11-11. Схема порошковой камеры Дебая — Шерера. Излучение входит через фильтр F и коллиматор С и попадает иа образец X. Для предотвра-’ щения засвечивания пленки неотклоиившийся центральный пучок захватывается ловушкой Е (здесь оиа не показана, но ее можно видеть на рис. П-12). Дифрагированные пучки падают на пленку в точках di, di и т. д. [1].
236 Глава 11
Рис, 11-12. Рентгеновская порошковая камера Дебая — Шерера (General Electric Company).
Рис. 11-13. Картины дифракции рентгеновских лучей на пленках, экспонированных в порошковой камере. Использовалось излучение /(-линии Си; перед плевками а—г и верхней частью пленки д помещали никелевый фильтр толщиной 15 мкм. В каждом случае образец наносили на тонкую стеклянную нить, за исключением случая д, когда его помещали в стеклянный капилляр: а — Pb(NO3)2; б — металлический вольфрам; в — NaCl; г — кварц [И].
Рентгеновские методы 237
Рис. 11-14. Гониометр Филипса. Образец закрепляют на игле (слева) и вращают при помощи электродвигателя (справа). Рентгеновский пучок входит через щель, расположенную за образцом (источник рентгеновских лучей не показан). Детектор, заключенный в цилиндрический корпус, расположен наверху и может поворачиваться вокруг образца либо вручную, либо с помощью электромотора.
238 Глава 11
Набор парОЛЛеЛЬ-пых щелей
Линейный току с рент-
/ геноВсКой трубки
/ - Набор параллель-
ных щелей
Образец
Приемная щель
Щель расходимости
Рис. 11-15. Оптическая схема гониометра Филипса.
На детектор Рассеивающая шель<
примером использования фотографической регистрации служит широко известная порошковая камера Дебая—Шерера (рис. 11-11 и 11-12), где на пути рентгеновских лучей помещают достаточно тонкий слой образца, приготовленного в виде тонкодисперсного однородного порошка. Порошок наносят на любой некристаллический материал, например на бумагу с органической текстурой или на клей. Порошкообразный образец содержит множество частичек, ориентированных во всех возможных направлениях относительно рентгеновского луча, что приводит к появлению дифрагированных лучей, соответствующих всем возможным наборам кристаллических плоскостей. Вокруг образца по окружности укрепляют полоску рентгеновской фотопленки, как показано на рисунке. После проявления на фотопленке видны серии дуг, симметрично расположенных по обе стороны от центрального пятна, соответствующего неотклонен-ному потоку. Расстояние от центрального пятна до данной дуги, а также радиус камеры определяют угол дифракции 0. Если длина волны и порядок дифракции известны, расстояние между кристаллическими плоскостями d можно рассчитать по уравнению Брэгга. Несколько образцов фотопленок, полученных с помощью порошковой камеры, приведены на рис. 11-13.
Дифракционные исследования монокристаллов лучше всего проводить с помощью ионизационных или сцинтилляционных детекторов. Детектор удобнее всего закреплять в держателе, который может перемещаться по окружности вокруг образца; такой прибор называется гониометром (рис. 11-14 и 11-15). В рассматриваемой модели мишень рентгеновской трубки представляет собой линейный источник высокой интенсивности с размерами поперечного сечения 0,06X10 мм. На рис. 11-15 расходящимися линиями показана угловая апертура потока, задавав-
Рентгеновские методы 239
мая с помощью экрана с отверстием, который называется щелью расходимости; щель, кроме того, ограничивает падающий пучок размерами образца. Чаще всего работают с апертурой в 1°, используя плоские образцы размером 10X20 мм либо вращая соответствующий образец с помощью небольшого электродвигателя. Выходная щель регулирует ширину отраженного пучка, поступающего на детектор. Набор тонких металлических пластинок из фольги (набор параллельных щелей), расположенных на одинаковом расстоянии друг от друга, ограничивает расходимость пучка в любой плоскости, параллельной линейному источнику. Как показано на рисунке, для достижения высокой разрешающей способности устанавливают два таких набора. Рассеивающая щель предназначена для уменьшения фона, вызываемого побочным излучением. Выходной сигнал детектора усиливается и поступает на самописец. Лента самописца и консоль, несущая детектор, приводятся в движение синхронными электродвигателями; получающийся график представляет собой зависимость интенсивности дифрагированного пучка от угла дифракции, обычно обозначаемого 20.
Рентгеновская флуоресценция
Флуоресцентная эмиссия рентгеновских лучей дает аналитику одно из наиболее мощных средств обнаружения и количественного определения тяжелых элементов почти в любой матрице
90
/
f
Г
кРенпт генод-ская трубка.
Коллиматор (состоит \ из очень тонких параллель- . ных трубочек)
Детектор
45°
. Направление v вращения \ детектора
Центр вращения гониометра
Анализатор (монокристалл, вращающийся вдвое медленней детектора)
Вспомога- \ тельный \ коллиматор^--
0°
Падающее азлучение образец
Рис. 11-16. Принципиальная геометрическая схема рентгенофлуоресцентного спектрометра (Philips Electronic Instruments, Inc.).
240 Глава 11
Рис. 11-17. Кривые дисперсии рентгеновских лучей для некоторых кристаллов.
и в сложных соединениях. Этот метод совершенно неприменим для обнаружения элементов легче натрия и лишь частично применим для обнаружения элементов, стоящих до кальция.
Образец облучают первичным потоком рентгеновского излучения, которое действует так же, как облучение ускоренными электронами, т. е. выбивает электроны с внутренних орбиталей. Первичный поток должен состоять из фотонов, обладающих более высокой энергией, чем наиболее коротковолновое вторичное рентгеновское излучение, испускаемое образцом. Чаще всего для этой цели выбирают коротковолновое излучение рентгеновской трубки с вольфрамовой мишенью. Первичное излучение не обязательно должно быть монохроматическим.
Рентгеновские методы 241
Рнс. 11-18. Спектр рентгеновской флуоресценции сплава серебра и меди с покрытием из никеля н хрома (Philips Electronic Instruments, Inc.).
Каждый тяжелый элемент в образце испускает излучение такой частоты, которую бы он испускал, если бы служил мишенью рентгеновской трубки, но свободное от сплошного спектра тормозного излучения. Излучение образца анализируют с помощью гониометра и детектора, как показано на рис. 11-16. Чтобы охватить разные области, может потребоваться несколько кристаллов, поэтому надо предусмотреть устройство для их замены. Разрешение, достигаемое на каждом кристалле, выража
242 Глава 11
ется его дисперсионной кривой, т. е. зависимостью углового отсчета гониометра 20 от атомного номера Z флуоресцирующего элемента (рис. 11-17).
Рентгенофлуоресцентный метод пригоден для анализа высоколегированных сталей типа Сг—Ni—Со. Определение элементов, присутствующих в низких концентрациях, ограничено влиянием основы, поскольку испускаемое излучение слишком сильно поглощается другими элементами образца. Образцы, основу которых составляют элементы с большими атомными массами, поглощают больший процент излучения, чем более легкие элементы. Например, наличие Ni в сплаве А1 можно обнаружить с большей чувствительностью, чем в стали или свинцовом сплаве, где поглощение Да-линии Ni очень велико. В благоприятных условиях можно достигнуть точности обнаружения порядка 0,5 %. Предел обнаружения может составлять несколько миллионных долей.
На рис. 11-18 приведен спектр флуоресценции хромоникелевого гальванического покрытия, нанесенного на основу из меди и серебра. Отметим, что Да-линии Сг и Ni гораздо сильнее таких же линий Ag и Си, хотя они и возникают от тонкого слоя гальванического покрытия; это обусловлено частичным ослаблением первичного и вторичного излучения при прохождении через слой покрытия.
Рентгеновскую флуоресценцию успешно применили для определения Hf в Zr и Та в Nb [12]. Например, в образце ниобия можно обнаружить 1 % Та с погрешностью ±0,04 % (т. е. ±4 % от количества, находящегося в образце).
Флуоресцентные методы также применяли при анализе следов после предварительного концентрирования. В работе [13] сообщалось об обнаружении различных металлов в коцентра-циях ниже 1 мкг/мл рентгеновским методом после 90 мин электролиза на катоде из пиролитического графита. Пиролитический графит можно расщеплять на очень тонкие пластинки, которые удобно устанавливать в рентгеновскую аппаратуру; кроме того, углерод имеет малый атомный номер и практически не дает фонового излучения.
Рентгеновские спектрометры с энергетической дисперсией [14—16]
Энергия фотона связана с длиной волны X излучения уравнением
EK = hc (11-8)
в котором h и с — фундаментальные постоянные [ср. с уравнением (2-2)]. Эта формулировка подчеркивает связь энергии и
Рентгеновские методы 243
длины волны и подсказывает, что спектральная дисперсия возможна как по длинам волн, так и по энергиям.
Энергетическая дисперсия зависит от наличия детектора с линейным откликом на энергию падающих на него отдельных фотонов. Отклик от отдельного фотона нужно строго отличать от отклика на полную среднюю энергию, или мощность, которая зависит как от энергии фотона, так и от числа фотонов, поглощаемых в единицу времени. Детекторы, которые мы назвали пропорциональными, а именно сцинтилляционные счетчики, газовые счетчики, работающие в промежуточном диапазоне напряжений, и кремниевые или германиевые детекторы, в которые продиффундировал литий, способны измерять энергии фотонов. Эти детекторы позволяют разрешать близкие рентгеновские линии (рис. 11-6). Успехи в практическом использовании твердотельных детекторов сделали возможным создание спектрометров с энергетической дисперсией.
В приборах с волновой дисперсией для дифракции используют кристаллы. Они обладают большей разрешающей силой, чем их аналоги с энергетической дисперсией, но последние в 100 и более раз эффективнее и, следовательно, чувствительнее [17]. Это объясняется тем, что они одновременно детектируют излучение всех частот (еще один пример выигрыша Фелжетта, о котором упоминалось в гл. 4 при обсуждении интерференционных спектрофотометров). Спектры, полученные на обоих спектрометрах, идентичны, они отличаются только разрешением. Поэтому на рис. 11-5 по оси абсцисс нанесены две шкалы. График можно линеаризовать по любой из переменных.
На рис. 11-19 представлена схема спектрометра с энергетической дисперсией и радиоактивным источником [5]. В качестве источника можно было бы использовать рентгеновскую трубку, но это привело бы к дополнительному фону. Независимо от
На усилитель
Рис. 11-19. Рентгеновский анализатор с энергетической дисперсией и радиоактивным источником.
244 Глава 11
выбранного источника излучения фон можно уменьшить, если поместить флуоресцирующую пластинку между источником первичного излучения и образцом. На рис. 11-20 приведены две схемы с радиоактивным источником, в одной из которых используется флуоресцирующая пластинка, а в другой ее нет.
Сигнал твердотельного детектора анализируется набором электронных дискриминаторов энергии, которые позволяют вести расчет импульсных сигналов последовательных энергетических?" диапазонов. Рис. 11-21 дает представление о принципе действия шестиканального анализатора. Импульсы соответствуют фотонам, поступающим на прибор в произвольные мо-
Рис. 11-20. Анализатор рентгеновского излучения с энергетической дисперсией: а — образец облучается непосредственно гамма-лучами радиоактивного источника; б —-на образец попадает только рентгеновское излучение, испускаемое флуоресцирующей мишенью, возбуждаемой первичным гамма-излучением.
Нонер канала И
Рис. 11-21. а — гипотетическое распределение 17 импульсов по шести энергетическим каналам в многоканальном анализаторе; б — результирующий профиль отсчетов в каждом канале.
Рентгеновские методы 245
к л
S
tf сз я
S
246 Глава 11
менты времени. На рисунке выделены два «пика», примерно соответствующие каналам 2 и 5. Ясно, что для серьезного эксперимента необходимо гораздо большее число каналов и экспериментальных точек. Многоканальные анализаторы амплитуды импульсов выпускаются рядом фирм. Чаще всего анализатор имеет 1024 канала. Они нашли широкое применение на практике (помимо описанных здесь областей).
На рис. 11-22 показан спектр, полученный на спектрометре с энергетической дисперсией. В литературе [15, 16] имеется множество примеров применения приборов этого типа. В статье Хенсона [18] (на 25 страницах!) подробно описывается программа для определения элементного состава любого объекта, изготовленного из металла, стекла или керамики, которая включает данные, необходимые для определения 71 элемента. В работе [19] приводятся сравнительные данные по определению in situ содержания Pb в краске для стен, полученные на портативных рентгенофлуоресцентных анализаторах с энергетической дисперсией четырех типов; найдено, что предел обнаружения составляет менее 1 мг РЬ на квадратный сантиметр окрашенной поверхности.
Сурков и др. [20] описали очень интересный .рентгенофлуоресцентный спектрометр, с помощью которого проведен элементный анализ пород на поверхности Венеры. Прибор снабжен двумя радиоактивными источниками, 55Fe и 238Ри. Альфа-линия излучения плутония возбуждает рентгеновское излучение легких элементов, Mg, Al и Si, а при определении тяжелых элементов — К, Са и Ti, предпочтительнее источник на основе изотопа железа. Еще более тяжелые элементы, особенно Мп и Fe, возбуждаются рентгеновскими лучами, испускаемыми плутонием. Спектрометр имеет четыре газовых пропорциональных счетчика и двойной 128-канальный анализатор амплитуды импульсов. В обычную конструкцию приборов были внесены изменения, учитывающие суровые внешние условия на поверхности Венеры (500 °C, 90 атм).
Электронно-зондовый микроанализ [3, 21]
В течение последних десятилетий заметное развитие получили методы фокусировки электронных пучков. С помощью этих методов можно сфокусировать такой узкий пучок, который вызывает рентгеновское излучение в твердом образце размером 1 мкм3 (10~18 м3), Принципиальная схема прибора показана на рис. 11-23. Пучок электронов, выходящий из электронной пушки (вверху диаграммы), фокусируется электромагнитами специальной формы (магнитными линзами) на образец. (Эта часть прибора очень напоминает электронный микроскоп.) Из-
Рентгеновские методы 247
Рнс. 11-23. Схематический вид электронно-зондового мнкроаналнзатора [20].
лучение, возникающее в образце, анализируется рентгеновским спектрометром с волновой или энергетической дисперсией. Прибор должен быть снабжен оптическим микроскопом, чтобы оператор мог точно выделить желаемый участок образца.
Детали этого прибора гораздо сложнее, чем это показано на рисунке. Кроме того, совсем не просто совместить электронную, оптическую и рентгеновскую системы, так чтобы они не мешали друг другу и чтобы каждая из них работала с максимальной эффективностью. Электронный пучок, конечно, должен находиться в высоковакуумной камере, что вносит дополнительные сложности, а вспомогательная электроника должна включать схемы управления электронными линзами, электронной пушкой, рентгеновским детектором и вакуумными насосами и датчиками? Поэтому прибор представляет собой громоздкое, сложное и дорогое устройство. Однако на нем можно получить
248 Глава И
такую богатую информацию, что, несмотря на указанные выше обстоятельства, его выпускают многие фирмы. Им пользуются при исследованиях фаз в металлургической и керамической промышленности, для контроля за процессом диффузии при изготовлении полупроводниковых приборов, установления идентичности примесей и включений и многих других целей.
Задачи
11-1. Какова размерность постоянной С в уравнении (11-5)?
11-2. Металлический образец облучают в рентгеновском спектрометре излучением рентгеновской трубки с вольфрамовой мишенью. В спектрометре установлен кристалл кальцита с межплоскостным расстоянием 302,9 пм. При угле 2 0 = 34°23' наблюдается сильная линия. Рассчитайте длину волны наблюдае-ного рентгеновского излучения и по справочнику найдите, какой элемент присутствует в образце (спектр первого порядка).
11-3. Даны значения цм для кобальта при нескольких длинах волн:
X, пм 50,0 100,0 150,0 200,0 250,0 300,0.
рм 15,5 110,0 345,0 87,0 177,0 270,0
Л'а-нзлученне Ni имеет место при 150,0 пм. Плотность кобальта 8,9 г/см3. Интенсивность Ка-линни Ni примерно в три раза больше интенсивности К|3-ли-ннн Ni, испускаемой мишенью. Постройте зависимость коэффициента поглощения кобальта и определите его значения при длинах волн Ка- и К0-нзлуче-ния Ni. С помощью данных, приведенных выше и в табл. 11-1, определите отношение интенсивностей двух линий излучения никеля после прохождения через фольгу из кобальта толщиной 0,005 мм.
11-4. Длина волны Ка-излучення Ni составляет 165,8 пм. а) Чему равна энергия каждого фотона в килоэлектронвольтах (кэВ)? б) Предположим, что в каком-либо приборе при измерении Да-излучения Ni идеальный детектор (со 100 %-ной эффективностью) дает 100 нмп/с. Подсчитайте мощность (в микроваттах), поступающую на детектор.
11-5. На стальную проволоку, используемую в радиальных шинах, обычно наносят латунное покрытие для улучшения адгезии резины [22]; при этом следует точно контролировать как толщину латунного слоя, так и содержание в нем меди. Придумайте метод, основанный на рентгеновской флуоресценции, для проведения обоих измерений.
11-6. Покажите, откуда появился численный множитель 1240 в уравнении (11-Г). Согласуется лн рис. 11-1 с этим значением?
11-7. Перечислите преимущества и недостатки рентгеновских спектрометров с энергетической и волновой дисперсией.
11-8. а) Точнее всего эффективный,радиус камеры Дебая-Шерера можно рассчитать, зная длину дуги на днфрактограмме кристалла с известным межплоскостным расстоянием и длину волны. В эксперименте, использующем Ка-нз-лученне Мо (Х=70,90 пм), радиус камеры определяли по дифракции рентгеновского луча на кристалле хлорида натрия с межплоскостным расстоянием
Рентгеновские методы 249
Рис. 11-24. Схема камеры Дебая — Шерера.
d, для которого в справочнике находим значение 2,821 А. Расстояние р, измеренное на пленке (рис. 11-24), составляет 1,50 см. Рассчитайте радиус камеры.
Указание. Заметьте, что р относится к длине окружности так же, как угол 2 0 относится к 360°. б) В той же самой камере проводили определение межплоскостного расстояния в металлическом алюминии. Угол 2 0 составлял 20,32°. Рассчитайте d и сравните с табличным значением.
Литература
1. Klug Н. Р., Alexander L. Е., X-Ray Diffraction Procedures, Wiley, New York, 1954.
2. Compton A. H., Allisson S. K-, X-rays in Theory and Experiment, Van Nostrand, New York, 1935.
3. Friedlander G., Kennedy J, W., Marcias E. S., Miller J. M., Nuclear and Radiochemistry, 3d ed., Wiley-Interscience, New York, 1981.
4. Liebhafsky H. A., Pfeiffer H. G„ Winslaw E. H„ Zemany P. D„ X-rays, Electrons and Analytical Chemistry, Wiley-Interscience, New York, 1972.
5. Bowman H. R., Hyde E. K, Thompson S. G„ Jared R. C., Science, 1966, v. 151, p. 562; Hail T., Science, 1960, 153, 320; Bowman H. R., Hyde E. K, Science, 1966, v. 153, p. 321.
6. Vollmar R. C., Petterson E. E., Petruzzelli P. A., Anal. Chem., 1949, v. 21, p. 1491.
7. Griffin L. H„ Anal. Chem., 1962, v. 34, p. 606.
8. Bartlett T. W„ Anal. Chem., 1951, v. 23, p; 705.
9. Dunn H. W., Anal. Chem., 1962, v. 34, p. 116.
10. Rudman R., X-ray Diffraction Analysis. In Topics in Chemical Instrumentation. Ewing G. W. (ed.), Chem. Education Pub. Co., Eeaston, Pa., 1971, p. 75.
11. Parrish W., Science, 1949, v. 110, p. 368.
12. Birks L. S., Brooks E. J., Anal. Chem., 1950, v. 22, p. 1017.
13. Vosses В. H., Hirsch R. F., Letterman H., Anal. Chem., 1973, v. 45, p. 792.
250 Глава 11
14. Porter D. E., Woldseth R., Anal. Chem., 1973, v. 45, p. 604A.
15. Woldseth R., X-Ray Energy Spectrometry, Kevex Corp., Burlingame, Cali!., 1973.
16. Heinrich K. F. J., Newbury D. E„ Muklebust R. L. (eds.), Energy Dispersive X-Ray Spectrometry, NBS Special Publications 604, National Bureau of Standards, Washington D. C., 1981.
17. Heinrich K- F. J., p. 1, of ref. 16.
18. Hanson V. F., Appl. Spectrosc., 1973, v. 27, p. 309.
19. Rasberry S. D., Appl. Spectrosc., 1973, v. 27, p. 102.
20. Surkov Yu. A., Shcheglov О. P., Moskalyeva L. P., Kirichenko V. S., Dudin A. D., Gimadov V. L., Kurochkin S. S., Rasputny V. IV., Anal. Chem., 1982, v. 54, p. 957A.
21. Wittry D. B., In Treatise on Analytical Chemistry. Kolthoff I. M., El-ving P. J. (eds.), pt. I, v. 5, chap. 61; Wiley-Interscience, New York, 1964.
22. Van Lingen R. L. M., Schuurs H. E. C., Veenstra G. Roes J. M. B., Loef E. C. Taianta, 1980, v. 27, p. 641.
ГЛАВА
Электронная и ионная спектроскопия
Бомбардировка вещества фотонами или другими несущими энергию частицами может вызвать несколько явлений: сначала происходит выбивание электронов из атомов мишени с образованием вакансий, затем следует релаксация (т. е. возвращение к нормальной конфигурации), которая может идти по одному из двух путей—испускание характеристического рентгеновского излучения и испускание вторичных оже-электронов. В предыдущей главе рассматривалось значение рентгеновского излучения в анализе, в этой главе будут показаны возможности эмиссии электронов.
Таблица 12-1. Виды электронной и ионной спектроскопии
Источник возбуждения Детектируемые частицы Методы анализа
Рентгеновское излучение Дальнее УФ-излучение Электроны Ионы Электроны Электроны Электроны Рассеянные ионы ОЭС, ЭСХА, ИЭЭ, ФЭСВО РФЭС ФЭС, ФЭСНО, УФ-ФЭС ОЭС, СЭУ, дмэ, смэ СИР, мсви
Сокращения:
ДМЭ дифракция медленных электронов
ИЭЭ индукционная электронная эмиссия
МСВИ масс-спектрометрия вторичных ионов
ОЭС оже-электронная спектроскопия
РФЭС рентгеновская фотоэлектронная спектроскопия
СИР спектроскопия ионного рассеяния
СМЭ спектроскопия медленных электронов
СЭУ спектроскопия электронного удара
УФ-ФЭС ультрафиолетовая фотоэлектронная спектроскопия ФЭСВО фотоэлектронная спектроскопия внутренних оболочек
ФЭСНО фотоэлектронная спектроскопия наружных оболочек
ФЭС фотоэлектронная спектроскопия
ЭСХА электронная спектроскопия для химического анализа
(Среди обозначений есть синонимы; МСВИ рассмотрена в гл. 22.)
252 Глава 12
Рис. 12-1. Схема энергетических уровней электронов в атоме [1]. Врезанный сектор показывает, с каких уровней электроны можно выбить источниками излучения указанных длин волн и энергий:
Атом Длина волны, нм Энергия, эВ
Сг Да А1 Ка YA4g Неи Hej 0,2294 5406,0 0,8342 1487,0 9,34 132,8 30,38 41,0 58,44 21,0
Еще один метод анализа основан на бомбардировке вещества мишени положительными ионами, например Не+ или Аг+. В этом случае по энергии отраженных мишенью ионов (упругое столкновение) можно судить о природе атомов анализируемого вещества. Такого рода методы представлены в табл. 12-1.
На рис. 12-1 дана общая схема расположения электронных уровней в тяжелом атоме [1]. Внешний слой представляет собой непрерывную зону, образованную валентными электронами (принадлежащими молекулярным орбиталям). За этой зоной следует дискретный набор уровней, отвечающих остовным электронам, не участвующим в образовании химических связей.
В правом верхнем углу рисунка указаны различные источники излучения, способные выбивать электроны с последовательных уровней. Дальнее УФ-излучение, испускаемое одно- и двукратно ионизованным гелием, вызывает отрыв только ва
Электронная и ионная спектроскопия 253
лентных электронов с образованием ионов. Мягкое М^-рентге-новское излучение * иттрия способно выбивать из атома внешние остовные электроны. Чтобы затронуть внутренние уровни, необходимо более жесткое излучение, например А1 Ка или Сг Ка.
Рентгеновская фотоэлектронная спектроскопия (РФЭС)
Более употребительное название рентгеновской фотоэлектронной спектроскопии — электронная спектроскопия для химического анализа (ЭСХА). Взаимосвязь энергий выбивающего рентгеновского кванта и выбиваемого фотоэлектрона дается уравнением
E& = hv—(Ек + С) (12-1)
где Есв — энергия связи, т. е. энергия притяжения электрона к атомному ядру. Эту энергию надо затратить, чтобы выбить электрон из атома. Она связана с энергией фотона рентгеновского излучения /iv и кинетической энергией выбиваемого электрона Ек (последнюю и измеряют). С — поправочный член,учитывающий особенности устройства конкретного спектрометра [2]. Энергия связи есть специфическая характеристика электрона данного уровня в атоме данного сорта, так что ее можно использовать для идентификации элементов. На рис. 12-2 представлен график зависимости Есв разных электронов, найденной по данным РФЭС [3], от атомного номера элемента. Для легких элементов (Z<60) наблюдаются два хорошо разрешенных энергетических профиля, отвечающих К- и L-электронам. Для элементов с / выше 70 картина усложняется (N- и М-электроны).
На величину Есв ощутимо влияет химическое состояние атома, например степень его окисления (даже если электроны выбиваются с внутренних оболочек), так как оно влияет на эффективный заряд ядра. Следовательно, Есъ зависит от молекулярного окружения атома (для многих элементов химический сдвиг достигает значений порядка 10 эВ). Так, например, сера (Z=16) может давать фотоэлектроны с энергией связи от 160 до 168 эВ при нормальной величине энергии связи 2/?-электро-нов 165 эВ. Некоторые значения химических сдвигов показаны на рис. 12-3 [4]. Аналогичные корреляционные диаграммы опубликованы для углерода и азота [4, 5].
Два фотоэлектронных спектра на рис. 12-4 иллюстрируют изменение энергии связи электронов, принадлежащих различ-
Иттрин М-дзета.
254 Глава 12
Рис. 12-2. Энергии связи электронов [3].
ным атомам одной и той же молекулы. На оси абсцисс может быть отложена энергия связи сама по себе или энергия, отсчитанная от подходящего пика сравнения (химический сдвиг). Нормированная площадь под последовательно идущими пиками дает примерную оценку числа атомов с одинаковым молекулярным окружением. Поскольку энергии связи 15-электронов метильного и метиленового углеродов (связанного только с атомами водорода и другими атомами углерода) весьма близки [5], пик А на рис. 12-4, а можно отнести к первому, второму и пятому атомам.С в структурной формуле. Остальные два пика отвечают двум другим атомам углерода, связанным с кислородом. Отнести конкретный пик к одному из атомов можно только при сравнении со спектрами других соединений, причем отнесение значительно облегчается при использовании схемы химиче-
Электронная н ионная спектроскопня 255
Рис. 12-3. Корреляции между энергией связи 2р-электронов серы и природой функциональной группы. Отрезки показывают интервалы наблюдаемых энергий; число соединений, использованных при построении схемы, указано в скобках [4].
ских сдвигов углерода. Два пика кислорода, как и следовало ожидать, имеют одинаковую интенсивность. Площади всех четырех пиков на рис. 12-4, б равны.
РФЭС, как и другие виды электронной спектроскопии, являются, по существу, методом анализа поверхности, поскольку выбивание электронов из атомов, отстоящих от поверхности образца более чем на 5 нм, маловероятно. Это позволяет применять метод РФЭС в целом ряде областей, связанных с исследованиями свойств поверхности. Но при исследовании образцов больших размеров метод РФЭС применим только в том случае, если поверхность отвечает составу всего образца. Поверхностный слой многих материалов можно постепенно удалять, бомбардируя образец, например, ионами аргона. Источники таких ионов иногда встраивают в ЭСХА-спектрометр. Это позволяет аналитику исследовать образцы послойно, каждый раз снимая электронный спектр после короткой обработки потоком ионов. Позже мы вернемся к этой возможности.
РФЭС можно применять к решению многих проблем установления структуры или идентификации веществ как в твердой, так и в газовой фазе (рис. 12-5 и 12-6) [6]. Другие примеры можно найти в литературе [2, 4—6].
Нельзя ожидать, чтобы количественный анализ методом РФЭС отличался высокой воспроизводимостью, так как существует слишком много факторов, которые с трудом поддаются контролю. Вагнер опубликовал подробную работу [7], в которой
256 Глава 12
Рис. 12-4. а — фотоэлектронный спектр этилпропионата (обратите внимание на расщепление 1s-лнний кислорода и углерода, вызванное химическим сдвигом); б — спектр ls-электронов углерода в этилтрифторацетате (возбуждение осуществлялось монохроматическим рентгеновским излучением; пикн соответствуют атомам углерода в структурной формуле) [1].
Электронная и ионная спектроскопия 257
Рис. 12-5. РФЭ-спектр элементов образца лунного грунта. Источником возбуждения служит рентгеновское излучение Ка-лииии Mg [6].
139,5 138,1 136,8 £Св.5в
Рис. 12-6. Химические сдвиги в энергии связи 4/-электроиов свинца, возникающие по мере образования поверхностных слоев оксида иа чистом образце. Возбуждение осуществляли рентгеновским излучением Ка-лииии Mg [6].
9 Заказ № 637
258 Глава 12
рассмотрел все потенциальные источники ошибок, сделав вывод, что относительное стандартное отклонение результата анализа порядка 10 % является на сегодня лучшим.
Ультрафиолетовая фотоэлектронная спектроскопия (УФ-ФЭС)
Основное отличие спектроскопии электронов, выбитых дальним УФ-излучением, от РФЭС (см. рис. 12-1) состоит в том, что первая позволяет фиксировать только валентные электроны. Это дает возможность получить прямую информацию о химических связях, степенях окисления и потенциалах ионизации молекул. Однако из-за делокализации электронов на молекулярных орбиталях отнесение спектральных пиков к индивидуальным атомам может оказаться невозможным. УФ-ФЭС может быть специфичным методом идентификации простых молекул. На рис. 12-7 приведен УФ-ФЭС-спектр воздуха, в котором идентифицированы основные компоненты [8]. Аналитические возможности данного метода по сравнению с РФЭС более ограничены.
Спектроскопия электронного удара (СЭУ)
Схема, аналогичная построенной на рис. 12-1 для электромагнитного излучения, показывает, что и при бомбардировке электронами степень воздействия на атомы мишени определяется энергией бомбардирующего потока. Низкоэнергетические электроны обычно приводят к переходу валентных электронов мишени из основного в возбужденные состояния (тогда как вУФ-
Ns • 1-Л'ло/юса
Н2'2-я/»лои
1 г 1 "1 I
ULXZ
_i_I_! I I
16 15 И 13 12
J.
10 эВ
Рис. 12-7. Фотоэлектронный спектр воздуха. Фрагменты показаны при более высокой чувствительности [8].
Электронная и ионная спектроскопия 259
ФЭС сразу образуются ионы). На этом явлении основана спектроскопия электронного удара. Высокоэнергетические электроны вызывают оже-эффект (рассматривается в следующем разделе).
Метод СЭУ основан на измерении уменьшения кинетической энергии бомбардирующих электронов после рассеяния молекулами мишени, т. е. определении энергии, необходимой для про-мотирования (возбуждения) валентных электронов. Существующее оборудование позволяет изучать только газообразные образцы и детектировать только рассеяйные вперед электроны. Полученные данные дают информацию о разности энергий основного и возбужденных состояний, особенно для колебательных и вращательных уровней энергии. Поэтому СЭУ дополняет ИК- и КР-спектроскопию. Метод СЭУ, по крайней мере теоретически, позволяет наблюдать все уровни вплоть до предела диссоциации; здесь не существует никаких правил отбора, запрещающих какие-либо переходы. Отмечаются значительные аналитические возможности метода. Сообщалось [10] об определении в воздухе до 5 • 10~3 % СО, но, несомненно, истинный предел обнаружения значительно ниже.
Оже-электронная спектроскопия (ОЭС)
Если под действием рентгеновского излучения или несущих энергию электронов атом теряет электрон внутренних оболочек, на его место для заполнения вакансии может перейти электрон
Рис. 12-8. Вероятность испускания оже-электронов и рентгеновской флуоресценции в зависимости от атомного номера [4].
9*
260 Глава 12
о 1s
Возбуждение Al Кл
Рис. 12-9. Фотоэлектронный спектр, полученный при возбуждении образца стекла рентгеновским излучением от двух различных источников. Оже-пики легко заметить по изменению их положения на шкале энергии связи [6].
с более высокого энергетического уровня. Высвобождающейся при переходе энергии достаточно для удаления другого электрона с той же оболочки атома. Так, если вначале был выбит /(-электрон, его место может занять L-электрон, и одновременно атом испустит еще один L-электрон. В этом состоит эффект Оже. Вероятность такого процесса, а также вероятность возникновения рентгеновской флуоресценции в зависимости от атомного номера показаны на рис. 12-8 [4]. Из него ясно, что
Электронная и ионная спектроскопия 261
. 1000
Не Ne Аг Кг Хе
J____I__। । ми!_____ill Illi il I I I I I
2 4 6 8 10 20 . 40 60 80100
Атомный номер Z
10
Рис. 12-10. Энергии оже-электронов в зависимости
от атомного номера [11].
оже-электронная спектроскопия наиболее важна при изучении легких элементов, поскольку в этой области рентгеновская флуоресценция снижается.
Кинетическая энергия ЕА выбитого с L-уровня оже-элек-трона выражается формулой
EA = (EK-EL)-EL (12-2)
где Ек и EL— энергии связи электронов на К- и L-оболочках. При переходе электрона с L- на К-уровень выделяется энергия, равная разности Ек—EL- Повторное употребление в уравнении (12-2) величины EL отражает затраты энергии на выбивание оже-электрона с L-уровня. Поскольку все члены уравнения (12-2) служат характеристиками самого атома, энергия оже-электронов не зависит от энергии бомбардирующего пучка [тогда как в соответствии с уравнением (12-1) кинетическая энергия фотоэлектронов меняется при замене источника излучения]. Рис. 12-9 иллюстрирует этот эффект на примере образца, на который действует рентгеновское излучение двух источников с анодами из разных материалов [6]. Для устранения
262 Глава 12
Рис. 12-11. Корреляции между энергией оже-электронов и фотоэлектронной энергией связи, демонстрирующие смысл параметра а' Вагнера и др. Заметьте, как группируются различные соединения, имеющие сходный с атомом железа тип связи. Соединение FeSj проводит электричество и мало отличается спектроскопически от металлического железа [12].
Электронная и ионная спектроскопия 263
влияния фона, обусловленного случайным рассеянием электронов, часто записывают не сам сигнал, а его производную.
Как показано на рис. 12-9, оже-пики наблюдаются и в РФЭС, а не только при электронном возбуждении. Последнее, однако, предпочтительнее, если нужны только оже-спектры. Рис. 12-10 поясняет, как по энергии оже-электронов можно идентифицировать элементы с малой атомной массой [11].
Идентификация веществ на основании химического сдвига в случае ОЭС сильно отличается от описанной для РФЭС. Вагнер и др. [12] указали, что разумное сочетание этих методов существенно повышает их чувствительность к химическому окружению атома. Они ввели параметр а', равный сумме кинетической энергии оже-электрона и энергии связи фотоэлектрона, величина которого не зависит от энергии возбуждающих фотонов. Один из графиков, приведенных в работе [12], воспроизведен на рис. 12-11. Энергия связи отложена по оси абсцисс, энергия оже-электронов — по оси ординат, диагональные линии отве
чают постоянному значению параметра а'. Каждый прямоугольник соответствует отдельному соединению, а по его размерам можно примерно оценить точность измерения.
На рис. 12-12 приведен пример исследования поверхности с помощью оже-спектроскопии [13]. (Заметим, что кривые на рисунке есть производные сигнала.) Исследовалась кремниевая пластинка,, покрытая слоем нихрома. Первоначально на поверхности в наибольшей концентрации присутствовали атомы кислорода. После удаления под действием ионного пучка поверхностного слоя толщиной 10 нм кислород практически исчезал и появлялись хром и никель, а на глубине 20 нм практически исчезали и они, уступая место чистому кремнию. Очевидно, что
До очистки поверх- кость покрыта пленкой трона
Сг
о
Рис. 12-12. Оже-спектры кремния, покрытого 15-нм пленкой нихрома при различной глубине зондирования [13].
Глубина
20 нм
Глубина к, нм
0 200 .400 600 800 1000
Энергия электронов, гв
264 Глава 12
на нихромовой поверхности было адсорбировано некоторое количество СОг-
Известно также о применении оже-электронной спектроскопии для изучения газов [14].
Приборы
Электронный спектрометр должен включать: 1) источник излучения для бомбардировки образца; 2) энергетический анализатор; 3) детектор электронов; 4) систему обеспечения глубокого вакуума. Весь прибор необходимо экранировать от магнитного поля Земли. Некоторые приборы оснащают дополнительными устройствами, например источником ионов (ионной пушкой).
Источники излучения. В качестве источников излучения используют рентгеновские трубки, гелиевые разрядные лампы или электронную пушку. Существенно, что излучение должно быть однородным по энергии. Рентгеновские трубки с алюминиевым или магниевым анодом часто можно применять и без монохроматора, поскольку Ка-излучение этих легких элементов характеризуется высокоинтенсивными и узкими полосами спектра. Алюминиевое окошко в трубке с алюминиевым анодом действует как фильтр, отсекающий Кр-линии и большую часть фонового излучения [15]. В один из серийных приборов (фирмы Hewlett-Packard) вмонтирован монохроматор Роуланда, правда, это привело к снижению интенсивности.
При возбуждении потоком электронов можно использовать монохроматор, а можно обойтись и без него. Электроны, вылетающие с нагретого катода и ускоренные электрическим полем, довольно однородны по энергии. Некоторое уширение полосы энергетического спектра связано с испусканием катодом электронов с разной кинетической энергией. Для получения более однородного пучка следует установить энергетические фильтры (рис. 12-13). В фильтре, изображенном на рис. 12-13, а, электроны между двумя сетками подвергаются воздействию тормозящего поля. Лишь обладая достаточной энергией, они могут преодолеть поле и пройти фильтр. Ясно, что это устройство отсекает низкоэнергетическую часть потока, поскольку оно пропускает только электроны с энергией, превышающей нижнюю границу.
В фильтре, изображенном на рис. 12-13, б, пучок электронов под углом 45° входит в пространство между двумя плоскими проводниками. Через вторую щель могут пройти только электроны, отвечающие некоторой узкой полосе энергии. Размер этой полосы определяется расстоянием между проводниками и приложенным потенциалом.
Электронная и ионная спектроскопия 265
Рнс. 12-13. Энергетические фильтры: а — с использованием тормозящего поля; б — параллельные пластинки; в — цилиндрический; г — сферический.
Если электроды сделать не плоскими, а цилиндрическими (рис. 12-13, в), то электроны, входящие в фильтр расходящимся пучком, будут фокусироваться на выходную щель, а хорошее разрешение по энергии сохранится [9]. Чтобы получить эффект двойной фокусировки [9], электроды должны образовывать круговой сектор с углом 127,28° (т. е. п/^2 радиан). На рис. 12-13, г показан полукруговой (180°), а не цилиндрический (127,28°) сектор. Фокусировка остается хорошей, а фильтр пропускает большую часть электронного пучка.
Специальные разрядные трубки, дающие резонансное излучение Hei и Ней для УФ-ФЭС, описаны в работе [9].
Энергетические анализаторы. От энергетического анализатора (монохроматора), расположенного между образцом и детектором, требуется большая эффективность, чем от ранее рассмотренных (рис. 12-13) фильтров, поскольку интенсивность потока электронов на несколько порядков ниже. Однако при использовании анализаторов с тормозящим полем (рис. 12-13, а) необходимы геометрические ограничители, обеспечивающие перпендикулярность траекторий всех электронов к пластинкам. Анализаторы с магнитным отклонением (рис. 12-14, а) отлича-
266 Глава 12
Рис. 12-14. Магнитный [4] (а) н цилиндрический электростатический (б) анализаторы энергии электронов.
ются эффективностью, но менее удобны, чем электростатические, при конструировании и применении [4]. В них создается неоднородное магнитное поле, и при величине кругового сектора 254,93° (л/^З радиан) достигается двойная фокусировка.
Наиболее распространены монохроматоры с цилиндрической или сферической симметрией электростатического поля. Цилиндрический анализатор сконструирован на основе фильтра, изображенного на рис. 12-13, б (не путать с рис. 12-13, в), как будто плоские электроды свернуты в цилиндр вокруг линии, соединяющей входную и выходную щели (рис. 12-14, б). Расчеты показывают [16], что такой анализатор обеспечивает оптимальную фокусировку, если угол между электронным пучком и осью симметрии близок к 42,3°. На практике в устройствах, принимающих электроны, задний конус имеет несколько больший угол. В сферическом анализаторе (рис. 12-15), представляющем собой почти полностью замкнутую сферу, использована схема, представленная на рис. 12-13, г.
Электронная н ионная спектроскопия 267
Сферический электростатический анализатор
Цилиндрический анализатор повторной фокусировки
Электронный умножитель
"Контроль \
фокусировки Дрдэдебди щель
Область
торможения
Образец
Фотоэлектроны
Рентгеновская трубка
Источник высокого к напряжения >
Камера о образцом
Рентгеновское ""Wi излучение ~ |
Рнс. 12-15. Прибор со сферическим электростатическим анализатором.
Детекторы. Чувствительность и удобство в обращении делают электронный умножитель почти универсальным, хотя и не единственно возможным детектирующим устройством. Разработано несколько видов электронных умножителей, один из которых напоминает фотоумножитель, описанный в гл. 3, с той разницей, что электронный пучок получают непосредственно из образца, а не с фотокатода. Как и в фотоумножителе, система динодов имеет разное устройство. Широко используется также канальный электронный умножитель (рис. 12-16), изготовленный из небольшой изогнутой стеклянной трубки, покрытой изнутри слоем проводящего материала с высоким сопротивлением. Проводящий слой соединен с источником напряжения 2—3 кВ и действует как непрерывный динод и делитель напряжения. Это устройство дает на выходе больше электронов, чем получает их на входе, т. е. действует как усилитель тока. При очень быстром отклике усиление достигает 108 раз, что позволяет детектировать единичные электроны.
Если сконструировать соответствующие электронные схемы, то данные от умножителя можно получать в виде аналогового сигнала или предусмотреть цифровой счет импульсов. В последнем случае можно предусмотреть последовательное сканирование и передачу данных в многоканальный анализатор (см. гл. 11).
Вспомогательные системы. Посторонние магнитные поля, включая магнитное поле Земли, сильно искажают траектории электронов, поэтому электронные спектрометры должны быть
268 Глава 12
К усилителю
+ 3003
Рис. 12-16. а — траектории электронов в канальном умножителе; б — канальный электронный умножитель, электрически связанный со счетчиком импульсов (Galileo Electro-Optics corporation).
магнитно изолированы. Согласно одному из способов, их заключают в кожух из ферромагнитного материала с высокой магнитной проницаемостью. Другой способ состоит в применении катушки Гельмгольца, которая создает магнитное поле, в точности равное, но противоположное по знаку природному полю; эту систему можно сделать автоматической. Вблизи от спектрометра размещают зонд-магнитометр, обеспечивающий протекание по катушке Гельмгольца тока, необходимого для поддержания нулевого суммарного поля. Этот способ делает безопасными даже кратковременные изменения внешних магнитных полей, связанные, например, с работой двигателя подъемника.
Определенные трудности в РФС и УФ-ФЭС вызывает эффект накопления заряда. Когда вещество теряет электроны, оно становится положительно заряженным. Если образец является проводящим материалом и связан с металлическими частями
Электронная и ионная спектроскопия 269
Бор]з
Кислород 1s
Ванадий 2р
—" С омыванием
С омыванием
АВС
Без омывания
Без омывания
550
_1______________i I__________________1_
525 500 230 200
Энергия связи, эВ
_1
170
Рис. 12-17. Электронный спектр диборида ванадия, показывающий влияние омывания образца потоком ннзкоэнергетических электронов [17].
спектрометра, положительный заряд не накапливается. Однако если образец представляет собой изолирующий материал, заряд быстро накапливается, что не позволяет электронам вылетать наружу. В этом случае происходит ослабление наблюдаемого сигнала, а к образцу начинают притягиваться рассеянные электроны (которые всегда присутствуют в том или ином количестве) до наступления равновесия. Эффект накопления заряда может оказаться очень значительным. Для его устранения в прибор встраивают источник низкоэнергетических электронов, нацеленный на образец, что позволяет получить дополнительную полезную информацию. На рис. 12-17 показаны два участка спектра диборида ванадия, подвергнутого действию воздуха [17], на котором видны два пика ls-электронов бора, когда источник омывающего образец потока низкоэнергетических электронов выключен, и только один пик (площадь его равна сумме двух предыдущих) — когда включен. Это означает, что присутствуют атомы бора только одного вида, но две трети этих атомов электрически изолированы от основной массы образца. Пики 2р-электронов ванадия не изменяются при включении и выключении источника. Изменения же ls-пиков кислорода показывают, что пик, помеченный буквой С, относится к атомам, связанным с ванадием, тогда как кислород А- и В-типа связан с бором.
Высокий вакуум обычно создают с помощью масляного диффузионного насоса или сочетания ионного и механического форвакуумного насосов. В некоторых приборах, предназначенных для анализа газообразных проб, необходимы два каскада насосов, что позволяет поддерживать в анализаторе по возможности более низкое давление (обычно 1 мкПа~10-8 мм рт. ст.), а вблизи образца примерно в 100 раз большее.
270 Глава 12
Рнс. 12-18. Схема электронной оптики и электроники в оже-спектрометре (Perkin-Elmer).
Защитные Электронный
кольца} умножитель
Защитные Квадрупольная [кольца /линза
Широкополосный предварительный орильтр
ОкИО Образец
Рис. 12-19. Электронная оптика ЭСХА-спектрометра (Du Pont de Nemours & Co.).
Фильтр низких Тормозящее
энергии поле
Рентгеновское излучение
Фильтр высоких энергий
На рис. 12-15, 12-18 и 12-19 схематически изображены три серийных электронных спектрометра с различной конструкцией энергетических анализаторов.
Электронная и ионная спектроскопия 271
Рис. 12-20. Спектрометр ионного рассеяния [19].
Рис. 12-21. Сравнение спектров рассеяния в обратном направлении 100-кэВ ионов Не+ и Н+ на поверхности кремния, покрытого золотом: Н+ (----------------),
Не* (--------) [19].
Спектроскопия ионного рассеяния (СИР)
При бомбардировке твердого образца пучком положительных ионов происходят упругие столкновения с атомами мишени, в результате которых ионы рассеиваются в случайных направ
272 Глава 12
лениях, а энергия отдачи поглощается массой образца. Если энергия ионов достаточно высока, с поверхности мишени будут вылетать атомы твердого вещества. Это явление, известное как распыление, как уже отмечалось, используется для очистки поверхности в ЭСХА.
Вылетевшие атомы можно собрать и определить их массы с помощью небольшого специального масс-спектрометра. Однако в наших целях интереснее (из-за очевидного сходства с ЭСХА) рассмотреть поведение рассеянных ионов. Энергия рассеянного иона Е связана с энергией Ео, которую он имел до столкновения, уравнением
Е=Е0
М„ — Мв
М п + Л1о
(12-3)
здесь Л4П и Мо — массы атома поверхности и рассеиваемого иона соответственно. Это уравнение верно лишь при Л4О<Л4П. Энергия Е наиболее чувствительна к слабым изменениям МП при условии, что А40 лишь ненамного меньше Л4П. Поэтому важно правильно выбрать подходящие ионы. Чаще всего предпочтение отдают ионам благородных газов, Аг и Не, не вступающим в побочные реакции.
Схема спектрометра ионного рассеяния показана на рис. 12-20. При бомбардировке атомов газа, электронами образуются положительные ионы, которые разгоняются и фокусируются на образце под углом 45°. Ионы рассеиваются по всем направлениям, но электростатический анализатор с круговым сектором 127° отбирает летящие в пределах заданного небольшого угла. Детектор может быть твердотельным (Si), пригоден и канальный электронный анализатор, поскольку несущие энергию ионы вызывают образование электронов в канале. Подробное описание двух приборов можно найти в работах [18 и 19].
Спектр ионного рассеяния показан на рис. 12-21 [19]. Образец представлял собой кристалл кремния, покрытый тонкой пленкой золота. Заметим, что при использовании ионов Н+ несколько пиков смещены в сторону более высоких энергий по сравнению с аналогичными пиками, полученными с использованием Не+ [что можно было предсказать, исходя из уравнения (12-3)]. При бомбардировке ионами такого малого размера ионный пучок при определенной ориентации образца может проникать вглубь на значительное расстояние по каналам в кристаллической структуре. (В действительности энергии, приведенные на рис. 12-21, несколько ниже, чем это следует из уравнения (12-3), поскольку собираемые ионы рассеяны вперед под углом 120, а не 90°.)
Электронная и ионная спектроскопия 273
Задачи
12-1. Из перечисленных на рнс. 12-1 источник Сг Ка обладает наибольшей энергией и в то же время является самым слабым из рентгеновских источников, включенных в табл. 11-1. Можете лн вы объяснить, почему в ЭСХА редко используют рентгеновское излучение с более короткими длинами волн (например, Со Ка или Мо Ка)?
12-2. Объясните, почему сдвиг оже-пиков на рнс. 12-9 не противоречит утверждению о незавнсимостн энергии оже-электронов от энергии возбуждающего излучения. Длины волн А1 Ка лежат прн 832,0 нм, Mg Ка — при 987,0 нм.
12-3. При ЭСХА-анализе соединения кальция электроны выбивали рентгеновским излучением Сг Ка (длина волны 2,294 А). Измеренная кинетическая энергия Ек составила 1,201 кэВ. Константа С в уравнении (12-1) равна 4,5 эВ. Рассчитайте значение энергии связи ЕСв выбиваемых электронов в атоме кальция.
Литература
1. Siegbahn К, Endeavour, 1973, v. 32, р. 51.
2. James Т. L„ J. Chem. Educ., 1971, v. 48, p. 712.
3. Bearden J. A., Burr A. F., In CRC Handbook of Chemistry and Physics, 50th ed., p. E-185, Chemical Rubber Publishing Co., 1967—1970; quoted from Rev. Mod. Phys., 1967, v. 39, p. 125.
4. Hercules D. M„ Anal. Chem., 1970, v. 42, N 1, p. 20A.
5. Swartz IF. E., Anal. Chem., 1973, v. 45, p. 788A.
6. Rendina J. F., Am. Lab., 1972, v. 4, No. 2, p. 17.
7. Wagner C. D„ Anal Chem., 1977, v. 49, p. 1282.
8. Betteridge D., Baker A. D., Anal. Chem., 1970, v. 42, No. 1, p. 43A.
9. Brundle C. R., Appl. Spectrosc., 1971, v. 25, p. 8.
10. Orojean R. E., Rendina J. F., Anal. Chem., 1971, v. 43, p. 162.
11. Harris L. A., J. Appl. Phys., 1968, v. 39, p. 1419.
12. Wagner C. D„ Oale L. H., Raymond R. H., Anal. Chem., 1979, v. 51, p. 466.
13. Palmberg P. W., J. Vac. Sci. Technol., 1972, v. 9,-p. 160.
14. Thompson M., Hewitt P. A., Wooliscroft D. S., Anal. Chem., 1976, v. 48,
p. 1336.
15. Lucchesi С. A-, Lester J. E., J. Chem. Educ., 1973, v. 50, p. A205, A269.
16. Sar-el H. Z., Rev. Sci. Instrum., 1967, v. 38, p. 1210.
17. Kelly M. A., Tyler С. E., Hewlett-Packard J., 1973, v. 24, No. Il, p. 2.
18. Goff R. F., Smith D. P., J. Vac. Sci. Technol., 1970, v. 7, p. 72.
19. Buck T. M„ Wheatley G. H„ Surf. Sci., 1972, v. 33, p. 35.
ГЛАВА
Спектроскопия магнитного резонанса
Совершенно иной тип взаимодействия между веществом и электромагнитным полем можно наблюдать, помещая образец одновременно в два магнитных поля — одно постоянное, а другое радиочастотное. При определенном сочетании полей образен поглощает энергию, что приводит к изменению сигнала на выходе высокочастотного усилителя и детектора.
Поглощение энергии связано с магнитной дипольной природой вращающихся ядер. Квантовая теория характеризует такие ядра спиновым квантовым числом I, которое может принимать положительные значения вида п/2 (в единицах й*), где n = 0, 1, 2... Если /=0, ядро не имеет спинового и магнитного моментов, и, следовательно, его нельзя наблюдать рассматриваемым методом; это относится, в частности, к ядрам 12С, 1вО и 32S. Максимальное разрешение в спектрах достигается для нуклидов с Z=l/2: ’Н, 19Р, I3C, 29Si и др. Спектры на ядрах трех первых изотопов наблюдать легко, поскольку их доля в общем количестве элемента в природе близка к 100 %, а доля других изотопов меньше 5 %.
Вращающиеся ядра ведут себя как микроскопические магниты, поэтому они взаимодействуют с внешним магнитным полем Н. Можно было бы предположить, что все ядра должны быть ориентированы вдоль направления магнитного поля, как стрелки компаса, однако вращательное движение заставляет их прецессировать, подобно гироскопу в гравитационном поле. Согласно квантовой механике, существует 2/+ 1 возможных ориентаций и, следовательно, столько же энергетических уровней. Это значит, что протон, например, имеет два таких уровня. Разность энергий между ними описывается выражением
= (13.1)
где р — магнитный момент вращающегося ядра. Энергия радиочастотного поля с частотой v поглощается системой ядер, если
* й = й/2л, где h — постоянная Планка.
Спектроскопия магнитного резонанса 275
выполняется условие hv = AE. Эта характеристическая частота является частотой прецессии диполей и называется ларморовой частотой. Обычно вводят угловую частоту прецессии со, которая равна 2nv. Объединяя приведенные соотношения, можно получить выражение
7?-—(13-2) п ni ni
Отношение а>/Н есть основная постоянная, характерная для любого ядра с ненулевой величиной I. Она называется гиромагнитным отношением и обозначается буквой у.
При частоте 100 МГц (одна из обычно используемых частот) разность энергий Л£ примерно равна 10~2 кал/моль. Это означает, что радиочастотный генератор может быть не очень мощным, но детектор должен быть высокочувствительным.
Воздействие приложенного радиочастотного поля на ларморовой частоте приводит к тому, что все вращающиеся ядра прецессируют в фазе. Таким образом, мы имеем множество ядер-ных осцилляторов, которые, согласно электромагнитной теории, должны излучать энергию. Поскольку все они находятся в фазе друг с другом, они действуют как когерентный излучатель. Их излучение можно уловить с помощью другой катушки, находящейся вблизи образца, если ось этой катушки взаимно перпендикулярна оси генераторной катушки и направлению постоянного поля.
Существуют два основных типа спектрометров для наблюдения и измерения ядерного магнитного резонанса (ЯМР). В более простом из них измеряется либо поглощение энергии, либо испускание резонансного излучения в момент совпадения частот магнитного и радиочастотного полей (точка резонанса). В приборах второго типа используется фурье-преобразование, что позволяет значительно увеличить отношение сигнал/шум. Мы рассмотрим эти приборы поочередно.
Сканирующие спектрометры ЯМР
Вводный эксперимент. Рассмотрим однокатушечный спектрометр ЯМР (рис. 13-1) с образцом в виде ампулы из боросиликатного стекла, заполненной дистиллированной водой. Радиочастотный (РЧ) генератор настроен на 5 МГц, а магнитное поле изменяется с постоянной скоростью от 0 до 1 Т*; при этом сигнал на выходе детектора непрерывно регистрируется. Самописец регистрирует кривую с резонансными максимумами
*В системе СИ единицей плотности магнитного потока (магнитной индукции) является тесла(Т), которая соответствует 104 Гс.
276 Глава 13
Источник
магнитного поля
Рис. 13-1. Однокатушечный сканирующий спектрометр ЯМР.
6SCu 63cu23hla „
SH ПО
J____И
J______________1___________1 ... .1__________________I____________1____________1______________I____________L
0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9
Магнитное поле, T
Рис. 13-2. Спектр ЯМР пробы воды в ампуле из боросиликатного стекла, полученный на спектрометре низкого разрешения при частоте 5 МГц.
(рис. 13-2), соответствующими каждому из присутствующих в образце изотопов, для которых />0. Значения напряженности магнитного поля, при которых индуцируются сигналы резонанса на частоте 5 МГц, соответствуют гиромагнитным отношениям изотопов. Пики изотопов меди принадлежат материалу проволоки РЧ катушки, другие пики — различным элементам, содержащимся в воде и стекле. Это ясно доказывает возможность проведения эффективного качественного анализа. Воз
Спектроскопия магнитного резонанса 277
можны также количественные измерения путем интегрирования площадей под пиками.
Прибор, пригодный для проведения такого эксперимента, называется спектрометром ЯМР широких линий, или низкого разрешения. В аналитических целях он используется редко отчасти из-за нечувствительности к ядрам, для которых /=0. Спектрометры широких линий нашли применение в исследованиях физического окружения ядер, в том числе для определения параметров кристаллической решетки в твердых телах.
ЯМР высокого разрешения
В аналитической практике ЯМР находит наибольшее применение при изучении тонкой структуры резонанса изолированного ядра; для этого используют спектрометр ЯМР высокого разрешения. Некоторые спектрометры сконструированы только для изучения ядер водорода (протонов), другие позволяют наблюдать также резонанс фтора или фосфора.
В предшествующем обсуждении предполагалось, что на каждое ядро действует магнитное поле, интенсивность которого равна интенсивности приложенного (внешнего) поля. Если бы это точно выполнялось, то наблюдался бы единственный резонансный пик для всех атомов данного изотопа, содержащихся в образце, как в спектрометрах широких линий. Однако это лишь грубое приближение. Наблюдения с помощью спектрометра высокого разрешения показывают, что резонансная линия индивидуального изотопа разрешается в серию близко расположенных пиков. Можно наблюдать два вида тонкой структуры, известных как химический сдвиг и спин-спиновое взаимодействие.
Для удобства мы обсудим вначале протонный резонанс как самый известный и наиболее широко используемый, а затем рассмотрим, какие изменения в теории и приборном оснащении необходимы для изучения других ядер, в особенности 13С.
Химический сдвиг
Каждое ядро в химическом соединении окружено облаком электронов, которые находятся в постоянном движении. Под влиянием приложенного магнитного поля эти электроны начинают вращаться, противодействуя приложенному полю. В результате возникает частичное экранирование ядра от внешнего поля. Следовательно, необходимо слегка изменить либо частоту, либо напряженность поля, чтобы экранированное ядро оказалось в условиях резонанса. В большинстве спектрометров это достигается подстройкой магнитного поля посредством изменения
278 Глава 13
(13-3)
постоянного тока, проходящего через вспомогательную обмотку. Этот вспомогательный ток изменяется линейно, осуществляя развертку поля в узких пределах (менее 100 м. д.). В электронной схеме для удобства воспроизведения спектра на самописце величина приращения поля превращается в ее частотный эквивалент.
Химический сдвиг, обычно обозначаемый символом б, есть отношение изменения силы поля, необходимого для появления резонанса, к силе поля, при которой наблюдается резонанс для эталона:
g ^обр ^эт яэт
Химический сдвиг — безразмерная величина, обычно выражаемая в миллионных долях (м. д.). Величина сдвига, требующегося, чтобы привести данный протон в резонанс, зависит от его ближайшего химического окружения, так как последнее определяет экранирование ядра электронами.
Поскольку мы не можем наблюдать резонанс от протонов, не избежав при этом экранирования электронами, не существует абсолютного эталона для сравнения сдвигов, и мы должны выбрать условный эталон. С этой целью к образцу обычно добавляют небольшое количество тетраметилсилана (ТМС) (СНз)451, служащего внутренним стандартом. Это соединение имеет две особенности, которые объясняют его выбор в качестве эталона: все его 12 водородных атомов находятся в идентичном окружении и экранированы сильнее, чем протоны большинства органических соединений. Положение ТМС на шкале химических сдвигов условно принимают за нуль. Некоторые авторы предпочитают приписывать ТМС величину сдвига 10 и обозначать химический сдвиг символом т, где т= 10—б. Увеличение экранирования соответствует химическому сдвигу «в сильное поле», т. е. поле нужно усилить, чтобы скомпенсировать экранирование. Следовательно, б убывает с увеличением экранирования, в то время как т при этом возрастает.
На рис. 13-3 показаны приблизительные интервалы изменения бит для протонов в различном химическом окружении [1]. Точные величины сдвигов в большей степени зависят от природы заместителя и растворителя, концентрации, наличия водородных связей и т. д., но они воспроизводимы для любого данного набора условий.
Спектры ЯМР обычно снимают на растворах образцов. Набор растворителей ограничен теми, которые не содержат протонов, что может усложнить спектры. Для органических веществ выбирают растворители, по возможности полностью
Спектроскопия магнитного резонанса 279
CH3-Si£
снз-се
$С-СН2-С£
СН2(едале) V/
R-^H __ 7,
CH3-C-0-
CH3-C=Cs
СН^Э 7//
сн,-с-6 11 7/7/,
cHj-nC 7 /////,
^С-СН2-С~ о А
^c~ch2-n< 7, 77,
-с=сн W
сн3-0- ////<
•^с-сн2-о '// V//
JCH-O- '///// 7/.
Ph-SH 77/
R-OH' '// 7/77/ 7////, /////' /////,
Ph-NH2 '//// ///.
-c-ch2-no2 77/.
)с=сн2- 7/.
-сн=сн- 7/777 77/// т////. '7/7
гтн он О N 77/, г//
п п. О^Н N н V///, //////
а " _ 7////Z '7/7.
Ан к К N 7/7,
а '///,
Ph-сн' 77777/, у7/7/Л Y///7 7////г
-с-н II о «Ж.
-с-он’ II 7/М //////' W/7/,
-SO.H 7//М —
8; м.д. 1 t, м.д. 2 2 3 8 2 4 2 6 8 0 10
Рис. 13-3. Диапазоны химических сдвигов протонов в различных молекулярных окружениях.
280 Глава 13
лишенные протонов, такие, как ССЦ или CSa, а также полностью дейтерированные растворители — CDCI3 (дейтерохлороформ), C6D6 (пердейтеробензол) или D2O (тяжелая вода).
Спин-спиновое взаимодействие
Второй тип структуры, часто наблюдаемый в спектрах ЯМР, возникает благодаря взаимодействию спина какого-либо протона со спином другого протона или протонов, присоединенных к соседнему атому углерода. Во взаимодействии участвуют спины электронов всех трех связей (Н—С, С—С и С—Н), однако мы не будем касаться деталей этого механизма. Если протоны находятся в одинаковом окружении, их взаимодействие не проявляется в спектрах, но в противном случае максимумы для каждого химического сдвига расщепляются в узкий мультиплет.
Для примера рассмотрим спектр этилиодида, растворенного в CDCI3 с добавкой ТМС (рис. 13-4). Спектр высокого разрешения показан сплошной линией. Штриховая линия изображает вспомогательную кривую, полученную с помощью встроенного интегратора; высота каждой ступени пропорциональна площади под соответствующей частью основной кривой. Мультиплет при 6 = 3,2 принадлежит двум протонам метиленовой группы, а мультиплет при 6=1,8 — трем протонам метиленовой группы. Площади под кривой (ее интегрируют для каждого полного мультиплета) определяются числом эквивалентных протонов; в данном случае они относятся друг к другу как 2 : 3, как и можно было предвидеть. Пик при 6=0 принадлежит ТМС. Маленький выброс при 6 = 7,25 объясняется остаточной примесью СНС1з в растворителе.
Рис. 13-4. Обычный (------) н интегральный (-------) спектры ЯМР рас-
твора этнлноднда в дентерохлороформе (CDC13) (с разрешения Р. Ф. Хнрша).
Спектроскопия магнитного резонанса 281
Резонансный пик метиленовых протонов расщепляется в квадруплет с примерным отношением интенсивностей 1 : 3 : 3 : 1. Эти цифры связаны с возможными ориентациями протонов метильной группы. Существует равная вероятность ориентации всех трех протонных спинов как по полю (что мы можем обозначить =£ ), так и против поля (4^), в то время как вероятности того, что два протона ориентированы в одну сторону, а один — в противоположную, в три раза больше (
и Таким образом, существуют два возможных
набора ориентаций по полю и два против поля. Это проявляется в том, что возникают четыре пика с отношением амплитуд 1 : 3 : 3 : 1, перекрывающие тот участок спектра, который должен был занимать нерасщепленный пик. Аналогично можно показать, что два метиленовых протона должны вызывать расщепление резонансного пика метильных протонов на пики с отношением амплитуд 1 : 2 : 1. Из рис. 13-4 очевидно, что предсказанные отношения интенсивностей выполняются лишь приблизительно. Связанные между собой мультиплеты часто можно идентифицировать по их несимметричным пикам, крайние пики, находящиеся рядом с другим мультиплетом, более интенсивны, чем удаленные пики (рис. 13-4). Чем меньше отличаются химические сдвиги мультиплетов, тем более заметен этот эффект.
Расстояния между компонентами обоих мультиплетов одинаковы и определяются константой спин-спинового взаимодействия J, измеряемой в единицах частоты. Значения / обычно лежат в интервале 1—20 Гц.
Приборы для ЯМР
К конструкции спектрометров ЯМР высокого разрешения предъявляются очень жесткие требования. Если нужно разрешить пики, отстоящие друг от друга на величину порядка /, однородность магнитного поля должна составлять около 10-8 (для спектрометра на частоте 100 МГц). Чтобы обеспечить достаточную однородность магнитного поля, полюсные наконечники должны иметь гораздо большую площадь, чем площадь, которую фактически занимает ампула с образцом. Даже наиболее тщательно изготовленные магниты не могут удовлетворить этим требованиям, поэтому для компенсации остаточной неоднородности применяют вспомогательные обмотки специальной формы, называемые шиммирующими катушками, которые питаются регулируемым постоянным током. Поле, которое «чувствуют» ядра образца, может также иметь некоторую неоднородность, возникающую внутри самого образца. Влияние этого фактора в значительной степени устраняется вращением ампулы с образцом при помощи маленькой воздушной турбинки.
282 Глава 13
Рнс. 13-5. Спектры протонного ЯМР (ПМР) N-втор-бутилаиилина, полученные на двух частотах: а — 60 МГц (Я=1,4 Т); б — 220 МГц (Я=5,2 Т) [2].
Радиочастотная энергия от генератора с кварцевой стабилизацией частоты передается на катушки датчика и приемник с помощью коаксиальных кабелей.
Достижимое разрешение увеличивается с ростом силы магнитного поля, а следовательно, и частоты. Обычные самые дешевые спектрометры ЯМР высокого разрешения работают на частоте 60 МГц (Н= 1,409 Т для протонов), спектрометры более высокого класса — на 100 МГц (Я=2,350 Т), частота рекордных приборов может достигать 600 МГц (14,4 Т) *. Рис. 13-5 иллюстрирует значительно лучшее разрешение спектрометра на 220 МГц по сравнению с прибором, работающим на 60 МГц. Во многих моделях спектрометров предусмотрена возможность работы на более низких частотах, чтобы величина магнитного поля соответствовала резонансу других ядер. В табл. 13-1 приведены частоты и другие данные, относящиеся к резонансу для ряда ядер с /=’/2-
Частотная стабилизация. Важной частью спектрометра высокого разрешения является автоматическая схема, предназна-
* Катушки магнитов для полей выше 2,5 Т изготавливают из сверхпроводящих материалов и работают при температуре жидкого гелия.
Спектроскопия магнитного резонанса 283
Таблица 13-1. Данные о ЯМР для некоторых изотопов с I = 1/2
Изотоп Распространенность в природе, % рад/(Тс) V, МГц
Н = 1.409Т Н = 2.350Т
99,98 2,68 60,0 100,0
13С 1,11 0,675 15,1 25,2
19J7 100,0 2,520 56,5 94,2
2»Si 4,70 0,531 11,9 19,9
31р 100,0 1,086 24,3 40,5
ченная для поддержания постоянного отношения силы поля к частоте. В ней используется цепь обратной связи (по частоте), которая «захватывает» характерный пик ядерного резонанса и непрерывно настраивает высокочастотный генератор, так чтобы сохранялось максимальное значение опорного сигнала. Без такого автоматического управления характеристики прибора испытывали бы неблагоприятное влияние со стороны посторонних магнитных полей, возникающих, например, при работе электрического оборудования.
В большинстве приборов за опорный принимают сигнал от ядра, содержащегося в самом образце (внутренняя стабилизация), или от ядра, принадлежащего ТМС, который служит эталоном как для химических сдвигов, так и для частотной стабилизации. В некоторых приборах предусматривается внешняя стабилизация, когда опорное вещество помещают в отдельную ампулу, вблизи ампулы с анализируемым образцом. Внешняя стабилизация имеет то преимущество, что управление не прерывается на время смены образца, и тот недостаток, что на опорный и анализируемый образцы действуют не вполне одинаковые магнитные поля; такая схема обеспечивает большее удобство за счет потери в точности. При работе с другими ядрами, отличающимися от !Н, возможен также выбор между гомоядер-ной и гетероядерной стабилизацией. В последнем случае для стабилизации используется ядро другого вида, нежели изучаемое. Например, в ЯМР на ядре 13С можно проводить стабилизацию по ядру дейтерия из CDCh в качестве растворителя. Ядро 2Н со спином /= 1 дает пик без заметного химического сдвига, поэтому оно пригодно как источник опорной частоты.
Двойной резонанс
Спин-спиновое взаимодействие иногда существенно облегчает идентификацию резонансных пиков, однако в относительно сложных молекулах оно может сильно усложнить спектр и сделать идентификацию структуры невозможной. Большую помощь
284 Глава 13
Рис. 13-6. Фрагменты спектров ЯМР циклопентадиенилдикарбонилродия(1), показывающие влияние подавления спин-спинового взаимодействия [3]: а — протонный резонанс с расщеплением вследствие взаимодействия с 103Rh; б — протонный резонанс с подавлением на частоте 103Rh; в — резонанс 103Rh с расщеплением вследствие взаимодействия с протонами циклопеитадиеииль-ного фрагмента; г — резонанс 103Rh с подавлением взаимодействия протона.
в этих случаях оказывает устройство для подавления спин-спи-нового взаимодействия (спин-декаплер). Оно представляет собой вспомогательный генератор, который может работать на избираемой частоте и облучать образец радиочастотным полем значительной интенсивности. Если эту добавочную частоту настроить в резонанс с одной группой взаимодействующих протонов и в то же время наблюдать за сигналом другой группы, то окажется, что мультиплет, возникший в результате спин-спинового взаимодействия, превратится в узкий синглет. Например (рис. 13-4), если вспомогательная частота установлена на 110 Гц ниже по полю от сигнала ТМС (6=1,83), квадруплет при 6 = 3,20 превращается в синглет с тем же положением на шкале сдвигов и той же площадью под пиком. Подобным же образом, если подавляющее поле настроено на 190 Гц (6 = 3,20), триплет при 6=1,83 сожмется в синглет. Еще один пример приведен на рис. 13-6 [3].
Подавление спин-спинового взаимодействия вызвано быстрой переориентацией облучаемых протонов между двумя состояниями, так что наблюдаемые протоны не различают отдельных состояний и их сигнал не расщепляется.
Спектроскопия магнитного резонанса 285
Другой вид двойного резонанса, называемый ядерным эффектом Оверхаузера, связан с нарушением обычных механизмов релаксации [4]. Релаксацией называется процесс, при котором ядро, поглотившее энергию, возвращается в свое обычное состояние. Наиболее важный из таких процессов затрагивает диполь-дипольное взаимодействие между вращающимися ядрами. Это взаимодействие сильно зависит от расстояния между ядрами (оно обратно пропорционально шестой степени расстояния). Если одно из вращающихся ядер с помощью излучения вспомогательного генератора насыщено энергией, другому ядру становится труднее отдавать свою избыточную энергию, и это проявляется в усилении сигнала ЯМР. Если оба ядра — протоны, сигнал может быть усилен в 1,5 раза, в то время как при наблюдении резонанса 13С по мере облучения соседнего протона сигнал может возрасти почти в 3 раза. Этот эффект значительно увеличивает отношение сигнал/шум, но более важно, что он помогает идентифицировать пики ядер, находящихся на близком расстоянии друг от друга. Таким образом, это часто позволяет различить пространственные изомеры. В качестве примера рассмотрим вещество [5]
нч хсоон
И с
\(5>
C=G N-H
(6) / \[4)/ЧП
сн3 с н н
Облучение на частоте протонов метильной группы приводит к усилению сигнала от двух протонов в положении 4, а не в положении 2, показывая тем самым, что метильная группа находится намного ближе к углероду 4, т. е. в транс-положении по отношению к карбоксильной группе.
Применение ПМР
Качественный анализ. Приведенные выше теоретические соображения ясно указывают на большую ценность ЯМР при идентификации индивидуальных соединений. Для ЯМР, так же как и для УФ- и ИК-спектроскопии, составлены атласы спектров (например, издания фирм Aldrich, Sadtler, Varian). ЯМР служит ценным средством и для установления структуры неизвестных соединений. В этой связи полезна информация о химических сдвигах, а также о спин-спиновом взаимодействии.
В первую очередь следует установить, сколько типов атомов водорода (различающихся химическим окружением) имеется
286 Глава 13
в молекуле, и определить при помощи интегратора относительное количество атомов разных типов. Далее, исследуя тонкую структуру (мультиплетность) пиков, можно выяснить, какие типы атомов водорода находятся по соседству друг от друга. На этой стадии очень ценной является информация о подавлении спин-спинового взаимодействия и ядерном эффекте Овер-хаузера.
Количественный анализ. При использовании ЯМР как метода количественного анализа нет необходимости в чистом образце определяемого вещества. (В таких методах, как газовая хроматография и многие виды оптической спектроскопии, образец индивидуального чистого вещества необходим.) ЯМР все же нуждается в каком-либо индивидуальном веществе в качестве внутреннего стандарта, однако им может быть любое соединение с четко идентифицируемым спектром, не перекрывающимся со спектром определяемого вещества. В качестве примера [6] рассмотрим определение степени этерификации пентаэритрита С(СНгОН)4 смесью кислот R—СООН. К аликвотной порции смеси сложных эфиров добавляют известное количество бензилбензоата и регистрируют спектр ЯМР (рис. 13-7). Узкий пик при 6 = 5'43 отвечает СНг-группе бензилбензоата. По его площади осуществляют калибровку спектра, т. е. определяют площадь, приходящуюся на один протон. Три широких пика при 6=1,70, 4,60 и 4,52 соответствуют протонам (выделены курсивом) групп —ОН, —СН2—ОН и СН2—OCOR. Сигнал —ОН неинформативен из-за перекрывания с несколькими узкими пиками, обусловленными протонами R-радикалов; другие же два пика можно использовать для идентификации. Поскольку соотношение между площадью под пиком и числом протонов известно, нетрудно рассчитать концентрации (в мэкв/г) функциональных групп —СН2—ОН и —СН2—OCOR.
Обратите внимание на уширение пиков, используемых для количественного анализа (рис. 13-7), которое обусловлено тем, что общий сигнал от данной функциональной группы складывается из сигналов от разных молекул, в которых эта группа связана с различными радикалами. Следует также отметить, что относительно сильный шум, наблюдаемый в спектре, на интегральной кривой не проявляется, что связано с особенностью операции интегрирования. Шум с равной вероятностью принимает положительные и отрицательные значения (это не всегда верно), и соответствующие вклады в площадь примерно компенсируются. Экспериментальные результаты, полученные в работе [6], характеризуются относительным стандартным отклонением порядка 2—10 %, что следует считать вполне приемлемым.
Спектроскопия магнитного резонанса 287
, м.д
Рис. 13-7. Спектр ПМР продуктов этерификации пентаэритрита. Штриховой линией показана интегральная кривая.
288 Глава 13
Рис. 13-8. Анализ смеси углеводородов методом ЯМР (Varian Associates).
В ряде случаев можно с высокой точностью анализировать смеси веществ. На рис. 13-8 показан спектр ПМР смеси, по составу близкой к некоторым сортам нефти, которая была изготовлена из 1,2,3,4-тетрагидронафталина (тетралина), нафталина и н-гексана. В области а на рисунке наблюдаются перекрывающиеся мультиплеты ароматических атомов водорода. В области Ъ проявляются сигналы протонов атомов углерода, присоединенных к ароматическому кольцу (в а-положении), а в области с — сигналы атомов водорода алифатических цепочек. В тетралине четыре ароматических протона, а в нафталине— восемь. В гексане имеется 14 алифатических протонов, а в тетралине их четыре и еще четыре находятся в а-положении. Для определения мольных долей трех соединений следует решить систему из трех уравнений. Найденные значения приведены на рисунке; они хорошо согласуются с истинным составом приготовленной смеси.
Сдвигающие реагенты в ЯМР
Химический сдвиг сигнала протонов, расположенных вблизи атома, имеющего неподеленную пару электронов (основания Льюиса), может заметно меняться при координации к такому атому какого-либо иона лантанидов (в координации участвует непоДеленная пара). В качестве реагента используют комплекс лантанида с органическим соединением. Структуры некоторых
Спектроскопия магнитного резонанса 289
Обозначение н
THD -C(CH,)j
FOO -С(СН,)з
И
R'
- C(CHj);<
-СЭН,
“ООО соо
\ -----X /
N (Ион Ln координирован с четырьмя
| Ln । атомами N и четырьмя атомами 0)[7]
"ООО COO-
DOTA
Рис. 13-9. Структура некоторых сдвигающих реагентов для ЯМР.
8,м.э.
Рис. 13-10. Спектр ПМР 6-метилхииолина в дейтерохлороформе в диапазоне 6=1—10: а — без сдвигающего реагента; б — в присутствии 0,3 моль Eu(THD)3. Показаны отнесения протонов (Perkin-Elmer Corporation).
из таких реагентов показаны на рис. 13-9. Реагенты, обозначенные THD и FOD, можно применять в неполярных средах, например в CDCh, а реагент, обозначенный DOTA, растворим в воде и, следовательно, может быть использован в виде растворов в D2O. На рис. 13-10 показано улучшение разрешения линий в спектре 6-метилхинолина в CDCI3 в присутствии соединения Eu(THD)3. Под влиянием парамагнитного Eu (III) сигналы ближайших к, азоту протонов сдвигаются в область слабого поля, а для более удаленных протонов эффект менее выражен. Как можно видеть из рисунка, наиболее существен сдвиг сигналов от N-2 и N-8.
10 Заказ № 537
290 Глава 13
Для того чтобы добиться хороших результатов, нужно иметь большой выбор сдвигающих реагентов, поскольку каждый из них имеет свои особенности [7, 8]. Наиболее распространены соединения европия, гольмия, празеодима и тулия.
ЯМР других элементов
Кроме водорода еще 18 элементов имеют изотопы с ядерным спином 1=Чг. Практически же при современном состоянии метода пригодны лишь те из них, для которых обеспечивается достаточная чувствительность:
18F (83,3)
20»Т1 И 205Т| (13,5)
31Р (6,63) и8Хе (0,55)
123Те И 125Те (0,22)
2О7РЬ (0,139)
111Cd и 113Cd (0,134)
U5sn и 118Sn (0,45) 183Pt (0,34)
”Se (0,062) 2»Si (0,037) 13С (0,018)
Цифры в скобках означают относительную чувствительность в процентах от чувствительности протонов при той же силе поля с учетом распространенности изотопов в природе*. Более или менее широко применяют резонанс на ядрах 19F, 31Р и * 13С, суть которого та же, что и в случае протонов. При увеличении атомного номера химический сдвиг обычно возрастает, достигая примерно. 600 м. д. для 19F и 13С, 1000 м. д. для 31Р и до 25000 м. д. (т. е. 2,5 %) для 207РЬ. Следовательно, в этих случаях ценную информацию можно получить на приборе с меньшим разрешением, чем нужно для ПМР [9].
Фтор-19. ЯМР на ядрах 19F не вызывает особых затруднений. При надлежащей установке поля и частоты (см. табл. 13-1) для проведения экспериментов пригодна та же техника, что и для ПМР [10]. Интересна методика приготовления фторсодержащих производных органических соединений, позволяющая обойтись без применения растворителя и устранить другие помехи, мешающие проведению ПМР. Автор работы [11] для получения легко идентифицируемых производных спиртов и аминов использовал гексафторацетон. Он определил химические сдвиги для аддуктов со 125 веществами.
Фосфор-31. За исключением более низкой чувствительности, магнитные свойства 31Р сходны со свойствами *Н и I9F [12]. Известны работы [13, 14] по количественному анализу конденсиро
* Проиллюстрируем вычисление чувствительности на примере ,3С. Собственная чувствительность ядра пропорциональна у3. Если у,н = 2,68, у,зс = = 0,675 и распростраиеиность 13С=1,11 %, то относительная чувствительность
13 С есть (0,675/2,68)3-1,11=0,0177 %.
Спектроскопия магнитного резонанса 291
ванных фосфатов, гидроксиметилфосфинов и тиофосфатов. Данные, полученные этим методом, хорошо согласуются с данными классических методов, а сам метод более экспрессен.
Углерод-13. Чувствительность ядра этого важнейшего изотопа слишком мала, чтобы можно было применять обычный сканирующий спектрометр ЯМР. Некоторые преимущества дает повторное сканирование с сохранением сигналов в электронной памяти и автоматическим усреднением (на многоканальном анализаторе), а также химическое обогащение образца 13С, однако здесь необходим спектрометр с фурье-преобразованием, о котором речь пойдет ниже. Он повышает чувствительность в 100 или более раз по сравнению с обычным прибором, снабженным устройством для усреднения сигнала.
Спектры ЯМР на ядрах 13С характеризуются большей чувствительностью химических сдвигов к деталям структуры, чем протонные. С их помощью легко обнаружить разницу между структурными изомерами и стереоизомерами. Поскольку к атомам углерода в органических соединениях обычно присоединены атомы водорода, важное значение приобретают спин-спи-новые взаимодействия *Н—13С (в спектроскопии ПМР они несущественны из-за низкой распространенности 13С) и, следовательно, подавление этих взаимодействий.
Для подавления пригоден широкополосный генератор, позволяющий насытить все протоны одновременно, чтобы все мультиплеты 13С превратились в синглеты. Другие методы двойного резонанса позволяют получить информацию о спин-спиновом взаимодействии ядра 13С с ближайшими протонами, при этом влияние более удаленных протонов устраняется. В таком варианте (он называется вне резонансным подавлением, или неполной развязкой) четвертичный 13С дает синглет, 13СН — дублет, 13СНг — триплет и 13СНз — квартет. Примеры и подробное описание этого метода можно найти в литературе [15—17].
ЯМР с фурье-преобразованием (ФП) [18—21]
Как и в случае ИКС, эффективность спектроскопии ЯМР можно резко повысить, отказавшись от последовательной развертки спектра в пользу одновременного возбуждения всех возможных резонаторов. В ИКС образец облучают немонохроматическим (белым) светом, изменяя эффективную шкалу времени с помощью интерферометра, а затем преобразуют временную зависимость в частотную. В ЯМР образец также подвергают действию излучения, несущего все частоты из интересующего экспериментатора диапазона, а затем проводят фурье-преобразова-ние зависимости сигнала от времени в зависимость от частоты.
10*
292 Глава 13
Этот метод не требует применения интерферометра, поскольку непосредственная обработка сигналов возможна в шкале времени ЯМР.
Для получения широкого диапазона частот лучше всего использовать короткий радиочастотный всплеск энергии — импульс. Импульс дает диапазон частот, центрированный на генераторной (несущей) частоте; ширина его зависит от длительности импульса: импульс длительностью 1 мкс дает диапазон частот около 105 Гц.
По окончании каждого импульса прецессирующие в магнитном поле возбужденные ядра релаксируют (возвращаются в ос-
а.
Рис. 13-11, Спектр 13С ЯМР метилиодида 13СНз1: а—полученный иа сканирующем спектрометре с многоканальным анализатором; б — спектр СИС (интерферограмма); в — фурье-преобразование интерферограммы.
Спектроскопия магнитного резонанса 293
Рис. 13-12. Спектр СИС 3-этилпиридина [JEOL (USA), Inc.].
новное состояние), давая сигналы соответствующей ларморовой частоты. Зависимость спада намагниченности от времени, регистрируемая по окончании возбуждающего импульса, называется кривой спада свободной индукции (спада индуцированного сигнала, СИС). Спад регистрируется катушками датчика. Каждый импульс дает информацию о полном спектре с интервалом 1—2 с между импульсами. Эти данные с высокой скоростью переводятся в цифровую форму и загружаются в память ЭВМ для последующего усреднения по результатам многих импульсов.
Кривая СИС для одного поглощающего ядра имеет вид экспоненциально затухающей синусоиды с частотой у=|ул—vH|, где л’л — ларморова частота, a vD — несущая частота излучения. В случае мультиплетов суммарная кривая СИС, называемая интерферограммой, приобретает более сложный характер. Рассмотрим рис. 13-11, иллюстрирующий разницу между обычным спектром ЯМР и спектром с фурье-преобразованием. На рис. 13-11, а приведен обычный спектр 13СН31, на котором виден квартет 1 : 3 : 3 : 1, обусловленный расщеплением сигнала 13С на трех протонах, с /=135 Гц (2,35-Т поля). Спектр СИС показан на рис. 13-11,6, где обратная / величина определяет расстояние между четко различающимися группами интерференционных биений. После фурье-преобразования временной зависимости сигнала в частотную получается спектр (рис. 13-11, в), который сходен с обычным спектром, но сигнал в нем существенно выше, а уровень шума ниже.
Как следует из рис. 13-12, даже для такой простой молекулы, как 3-этилпиридин, интерферограмма может быть очень сложной.
Спектрометры с фурье-преобразованием производятся несколькими фирмами, концентрирующими свои усилия в области 13С-ЯМР, однако многие модели пригодны также для протонов и прочих ядер. Такие приборы обязательно оснащены встроенной ЭВМ.
ЯМР в твердых веществах [22, 23]. В последние годы развиваются теория и практика ЯМР в твердых веществах; вероятно, они будут иметь важное значение в анализе. Здесь можно
294 Глава 13
получить больше структурной информации, чем в ЯМР растворов, поскольку существуют факторы, связанные с ориентацией молекул в кристаллической решетке. Так [22], ядро 13С карбонильной группы сильнее экранируется, если магнитное поле перпендикулярно связи С = О, а не параллельно. Это пример анизотропного экранирования. В порошке кристаллического вещества присутствуют все возможные ориентации, так что из множества резонансов образуется наблюдаемый в спектре широкий пик (с тонкой структурой), тогда как в жидкости наблюдается единственная широкая полоса (огибающая).
Химическую анизотропию можно устранить, вращая образец вокруг оси, наклоненной на 54,74° к направлению внешнего поля. Этот «магический угол» обеспечивает превращение широких пиков в узкие полосы.
В спектрах ЯМР твердых образцов влияние прямого диполь-дипольного взаимодействия близлежащих магнитных диполей ядер проявляется сильнее, чем в спектрах жидких образцов, что может затруднять определение химических сдвигов. По этой же причине для подавления спин-спинового взаимодействия в твердых образцах необходимо более мощное радиочастотное излучение, чем в спектроскопии растворов. Все это не позволяет использовать спектрометр ЯМР, предназначенный для жидкостей, при исследовании твердых тел (обратное неверно).
ЯМР в твердых веществах обладает большими потенциальными возможностями для идентификации образцов, для которых не подходят другие средства. С помощью этого метода можно анализировать, например, такие сложные объекты, как биополимеры или ископаемое топливо.
Электронный парамагнитный резонанс (ЭПР)*
Теория магнитного резонанса применима не только к ядрам, но и к электронам, поскольку последние также имеют спин и магнитный момент. Гиромагнитное отношение составляет для электрона 1760 рад/(Т-с) (для протона 2,68). Перепишем уравнение (13-1) для ЭПР:
АЕ =(13-4)
где р — магнетон Бора, универсальная постоянная, равная 9,2732• 1024 Дж/Т, g—безразмерная постоянная (g-фактор). Квантовое число I имеет значение */2, так что для электрона характерны только два энергетических состояния, как и для про
* В англоязычной литературе употребляют и сокращение ESR (от Electron Spin Resonance).
Спектроскопия магнитного резонанса 295
тона. ^-Фактор для свободного электрона равен 2,0023 и на несколько процентов отклоняется от этого значения для свободных радикалов, ионов переходных металлов и других носителей неспаренных электронов. (При спаривании электронов их магнитные моменты взаимоуничтожаются и резонанс не наблюдается.)
Как правило, резонансные полосы неспаренных электронов имеют тонкую структуру, обусловленную взаимодействием со спинами ближайших ядер. В принципе это расщепление сходно с расщеплением, имеющим место в случае протонов, но часто оно является более сложным — вплоть до того, что может оказаться невозможно идентифицировать все компоненты спектра. Так, например, свободный этильный радикал (• С2Н5) дает набор из четырех триплетов. Иногда здесь может оказаться полезным подавление спинового взаимодействия.
Оборудование для ЭПР. Большинство исследований в области ЭПР выполнено при постоянной частоте вблизи 9,5 ГГц и силе поля около 340 мТ. Некоторые приборы работают на частотах до 35 ГГц. Наиболее заметное отличие между приборами для ЭПР и ЯМР в том, что при таких высоких частотах более эффективна передача мощности по жестким волноводам, чем по коаксиальным кабелям. Рабочую кювету помещают внутри волновода в точке, где вектор напряженности магнитного (а не электрического) поля электромагнитной волны достигает максимума. Эффективность подвода энергии к образцу составляет не более 30 %, но все равно метод весьма чувствителен, и с его помощью можно обнаружить от 1010 до 10-8 мол.% свободных радикалов в 1 г образца.
Для-получения лучшего разрешения выгодно записывать спектр ЭПР в дифференциальной форме. Для количественных измерений следует два раза проинтегрировать сигнал по времени.
Применение ЭПР. Как и в ЯМР-спектроскопии, в ЭПР удобно использовать в качестве стандарта вещество с известными свойствами. Часто применяют 1,1-дифенил-2-пикрилгидра-зильный (ДФПГ) свободный радикал, являющийся стабильным соединением с ^-фактором, равным 2,0036. ДФПГ нельзя использовать как внутренний стандарт в присутствии других
296 Глава 13
Рис. 13-13. Спектр ЭПР семихинона, являющегося промежуточным соединением при окислении гидрохинона.
свободных радикалов: поскольку g-фактор меняется слабо, стандарт не удастся отличить от исследуемого вещества. Однако при постоянных условиях эксперимента стандарт и пробу можно вводить друг за другом. В качестве внутреннего стандарта в рабочую кювету обычно вставляют тонкую полоску кристалла рубина. Кристаллическая решетка рубина содержит следы Сг(Ш) и проявляет сильный резонанс при g=l,40.
ЭПР применяют в основном для изучения свободных радикалов в очень низких концентрациях. На рис. 13-13 приведен спектр ЭПР окислительно-восстановительной системы, состоящей из хинона и гидрохинона. Задача исследователя заключалась в доказательстве существования свободного радикала семихинона в качестве промежуточного соединения. Пять линий в спектре обусловлены взаимодействием неспаренного электрона с четырьмя протонами кольца. Из статистических соображений следует, что. отношение интенсивностей должно составлять 1:4:6:4:1. На рис. 13-14 приведены результаты кинетического исследования -образования и распада семихинона.
Пуск потока
Остановка потока
Кривая
! f распаоа
.Деление
соответствует 1с
Кривая образования
Рис. 13-14. Исследование кинетики образования и распада семихшюна, показанного на рис. 13-13 (Varian Associates).
Спектроскопия магнитного резонанса 297
Рис. 13-15. Спектр ЭПР 1-Ю-4 М водного раствора Мп2+ (Perkin-E)mcr Corporation).
Спектрометр ЭПР в течение некоторого времени регистрировал изменение поглощения в максимуме сигнала. Была сконструирована проточная кювета, через которую проходили потоки гидрохинона и кислород со скоростью, превышающей скорость образования радикалов. После остановки потока за их образованием и дальнейшими превращениями легко было следить. Время полупревращения реакции образования радикалов в выбранных условиях составляло 0,15 с.
Сконструированы кюветы, допускающие применение УФ-, гамма- или рентгеновского излучения, а также электролитических окислительно-восстановительных реакций для получения свободных радикалов прямо в образце. Высокая чувствительность метода дает возможность наблюдения переходных состояний, не детектируемых другими методами. Так, например, если этанол облучить рентгеновским излучением, в спектре появляется квинтет этильного радикала.
Еще одна важная область приложения ЭПР — это определение следовых количеств парамагнитных ионов, особенно при биологических исследованиях. На рис. 13-15 показаны шесть пиков спектра иона Мп2+ (водный раствор). Мультиплетность составляет 2/+1; для Мп2+ 1=5/ч, а число пиков равно 6. Этот ион можно определять (т. е. отличать его сигнал от фонового) вплоть до концентрации 10-6 М.
В литературе [24] сообщается и о других применениях ЭПР.
298 Глава 13
Задачи
13-1. С помощью рнс. 13-3 проверьте отнесение пиков на рис. 13-8, а — в. Как можно точнее измерьте высоту трех ступенек интегральной кривой и рассчитайте количество каждого из трех компонентов.
13-2. На рис. 13-16 приведен спектр ЯМР смеси циклогексана, толуола и воды. С помощью рис. 13-3 идентифицируйте четыре пика и установите по интегральной кривой примерный состав смеси.
13-3. Авторы работы [25] использовали ЭПР, чтобы установить, какая из двух формул отражает строение комплекса меди и 8-меркаптохиполииа (C9H7NS): a) Cu^CgHyNS) (C9H6NS_); б) CuII(C9H6NS_)2. Докажите, что эту задачу можно решить с помощью метода ЭПР.
13-4. С помощью ЯМР определяют молекулярную массу неизвестного вещества. Предварительный анализ спектра указывает па наличие пика, отвечающего 6 атомам водорода. Приготовили раствор, содержащий 1,50 г пробы и 1,45 г 1,3,5-тринитробензола (внутренний стандарт). Найденная по интегральной кривой высота, отвечающая пику стандарта, составляет 421 единицу, а отвечающая пику неизвестного вещества — 1024 единицы. Определите молекулярную массу вещества [26].
13-5. а) При какой напряженности магнитного поля можно наблюдать резонанс на ядрах l9F при частоте 220 МГц? б) Рассчитайте значение у для ядра, дающего резонанс при 2,35 Т п 19,7 МГц. Идентифицируйте изотоп по справочникам.
13-6. Перепишите уравнения (13-2) и (13-3) для электронов, используя данные в тексте значения у и g.
13-7. Какова относительная чувствительность детектирования методом ЯМР изотопа 15N, если он имеет следующие характеристики: I=4z, распространенность в природе 0,37 %, резонансная частота при 1 Т равна 4,314 МГц.
Рис. 13-16. Спектр ЯМР среднего разрешения эмульсии циклогексана и толуола в воде (Varian Associates).
Спектроскопия магнитного резонанса 299
Литература
1. Becker Е. D., High Resolution NMR, 2d ed., Academic Press, New York, 1980.
2. Ferguson R. C., Phillips W. D„ Science, 1967, v. 157, p. 257.
3. Dykstra R. W., Harrison A. M., Dombek B. D., Rev. Sci. Instrum., 1981, v. 52, p. 1690.
4. Rennewell P. D., J. Chem. Educ., 1970, v. 47, p. 278.
5. Isono K., Asahi K., Suzuki S., J. Am. Chem. Soc., 1969, v. 91, p. 7490.
6. Ward G. A., Am. Lab., 1970, v. 2, No. 3, p. 12.
7. Rondeau R. E., Sievers R. E., Anal. Chem., 1973, v. 45, p. 2145.
8. Bryden С. C., Reilley C. N., Desreux J. F., Anal. Chem., 1981, v. 53, p. 1418.
9. Sharp R. R., in Applications of the Newer Techniques of Analysis, Simmons I. L., Ewing G. W. (eds.), Plenum Press, New York, 1973, pp. 123ff.
10. Paudler W. W., Nuclear Magnetic Resonance, Allyn and Bacon, Boston, 1971, p. 22.
11. Leader G. R., Anal. Chem., 1973, v. 45, p. 1700.
12. Ссылка 10, p. 24.
13. Colson J. G-, Marr D. IL. Anal. Chem., 1973, v. 45, p. 370.
14. Stanislawski D. A., Van Wazer J. R., Anal. Chem., 1980, v. 52, p. 96.
15. Levy G. C., Nelson G. L., Carbon-13 Nuclear Magnetic Resonance for Organic Chemists, Wiley-Interscience, New York, 1972.
16. Wasson J. R., Anal. Chem., 1982, v. 54, p. 125R.
17. Anet F. A. L., Levy G. C., Science, 1973, v. 180, p. 141.
18. Becker E. D., Appl. Spectrosc., 1972, v. 26, p. 421.
19. Netzel D. A., Appl. Spectrosc., 1972, v. 26, p. 430.
20. Farrar T. C., in Transform Techniques in Chemistry, Griffiths P. R. (ed.), Plenum Press, New York, 1978, chap. 8.
21. Becker E. D., Ferretti J. A., Gambhir P. N., Anal. Chem., 1979, v. 51, p. 1413.
22. Reinhold M., Am. Lab., 1982, v. 14, No. 2, p. 164.
23. Gerstein В. C., Anal. Chem., 1983, v. 55, p. 781A, 899A.
24. Серия из восьми статей в Appl. Spectrosc., 1980, v. 34, pp. 268—310.
25. Corsini A., Fernando Q., Freiser H., Taianta, 1964, v. 11, p. 63.
26. Rahman W. R., Gennaro A. R., Zanger M., Am. Lab., 1981, v. 13, No. 11, p. 42.
ГЛАВА
Введение в электрохимические методы
Ряд важных аналитических методов основан на электрохимических свойствах растворов. Рассмотрим раствор электролита в стеклянном сосуде, в который погружены два металлических проводника. Если такую ячейку присоединить к внешнему источнику с напряжением в несколько вольт, через раствор будет протекать ток, но эта же ячейка может сама по себе действовать как источник электрической энергии и подавать ток во внешнюю цепь.
В принципе подобные электрические эффекты зависят от состава раствора и природы электродов и потому важны для аналитиков. Различия между отдельными электрохимическими методами в основном обусловлены природой электродов и измерительными приборами во внешней цепи. Сначала мы рассмотрим свойства окислительно-восстановительных реакций в водных растворах, позволяющие им реагировать с системой электродов.
Реакция в ячейке
Если через электролитическую ячейку пропускать постоянный ток, в ней протекает окислительно-восстановительная реакция. На электроде, называемом анодом, происходит окисление (потеря электронов), одновременно на катоде идет восстановление (присоединение электронов). Главное назначение внешней цепи — быть проводником электронов от анода к катоду. Электрическая цепь замыкается за счет ионной проводимости раствора.
Запишем уравнение окислительно-восстановительной реакции в общем виде:
/'Лгеа-ЬвВох'Г • • *-^Р^ох4 P^red + • • •
Здесь индексами ох и red обозначены окисленная и восстановленная формы веществ А и В. Для простоты ограничимся рассмотрением случая, когда реакция протекает только с участием А и В (т. е. опустим « + ...»).
Введение в электрохимические методы 301
Выражение для константы равновесия К можно записать следующим образом:
7^ = (Лох)ед (^red)eq (14-1)
(^red)eq (Box)eq
В нем в круглые скобки взяты активности (дальше круглые скобки будем использовать для обозначения активности, квадратные— для обозначения концентрации), а индекс eq указывает, что они относятся к моменту равновесия. Мы можем так же записать выражение для Q:
Q = (Лох)Р (Bred)<? (14-2)
(4red)r (Box)s
в котором активности отвечают фактическим величинам в условиях эксперимента, но не обязательно равновесным величинам. Из термодинамических соображений можно показать, что изменение свободной энергии (максимальная полезная работа при постоянных температуре и давлении) можно выразить уравнением
AG = 7?TlnQ—RTlnK (14-3)
где R— универсальная газовая постоянная [8,316 Дж/(моль• К)]; Т — температура. Для электрохимических реакций свободная энергия связана с электрическими величинами выражением
AG = — nFEa4 (14-4)
в котором Еяч — потенциал ячейки в вольтах, F — константа Фарадея (примерно 96 487 Кл на эквивалент), п — число электронов, переносимое каждым участником реакции. Поэтому
Еяч ... _ '.А __ _ 2LL inQ 'LL )п К (14.5)
nF nF nF
Если подставить выражения для Q и К, то после преобразования получим
. nF (^red)eq nF Ked/ J
(14.6)
L (Sred)e% nF (Bred)* J
Теперь обозначим
£a In (14.7)
nF (Лох)ед
302 Глава 14
и если используем аналогичное выражение для Ев°, то получим
Еяч = Гев—— ln-(-red)l? 1-Г^1—1п 1 (14-8) L nF (B0J J L А nF (4ОХ)Р J v
Таким приемом мы разделили на два слагаемых вклад веществ А и В в потенциал ячейки. Это можно было сделать и на предыдущей стадии, определив, что такое потенциал полуэлемента-.
Е. = Е\- RT In (Ared)r
А А nF (Лох)Р
Е„ = Е°- RT In (^redH
В В nF (W
Тогда из уравнения (14-8) следует, что Еяч = ЕВ-—Ед
(14-9)
(14-10)
(14-11)
Уравнения (14-9) и (14-10) были выведены Вальтером Нерн-стом, и их часто называют уравнениями Нернста.
После того как потенциал ячейки разделили на два слагаемых, можно разделить и уравнение химической реакции в ячейке на две части, называемые полуреакциями:
rAred *^рЛОх4-пе
sBOx+ пе~ ^qBred
в которых символом е~, как обычно, обозначен электрон.
Все полуреакции (особенно для составления справочных таблиц) удобно записывать в одинаковой форме. Согласно современным рекомендациям, мы будем записывать их в форме полуреакций восстановления:
pA<„.+ne-^rAni (14-12)
<?ВОХ + пе-5^ sBred (14-13)
Чтобы получить полное уравнение реакции в ячейке, нужно прежде всего умножить обе полуреакции на подходящий численный фактор для уравнивания в них числа электронов, а затем полуреакцию окисления вычесть из полуреакции восстановления.
Потенциалы Еа и Ев относятся к соответствующим полуреакциям, а относящиеся к этим же полуреакциям Е°Л и Е°в называют стандартными электродными потенциалами. Потенциал ячейки [см. уравнение (14-11) для Еяч], как мы скоро увидим, измерить легко, но доступного способа измерения абсолютной величины потенциала изолированного электрода, или потенциала полуреакции, нет. Отсюда вытекает необходимость выбора какого-то конкретного электрода, которому на шкале по-
Введение в электрохимические методы 303
Рис. 14-1. Электродные потенциалы различных окислительно-восстановительных систем как функции активности ионов (объяснения см. в тексте).
тенциалов для соответствующей полуреакции произвольно приписывается потенциал, равный нулю. С этой целью был выбран стандартный водородный электрод (СВЭ). Для получения СВЭ в водный раствор кислоты с активностью ионов водорода, равной единице, погружают платиновую фольгу и пропускают газо
304 Глава 14
образный водород под давлением в одну атмосферу*. В соответствии с международным соглашением, потенциал СВЭ принимают равным нулю при любой температуре.
Графическое представление уравнения Нернста для ряда окислительно-восстановительных систем дано на рис. 14-1. Для удобства величины потенциалов даны относительно двух электродов: СВЭ и насыщенного каломельного электрода (НКЭ), который будет описан ниже. По абсциссе удобно отложить и величины отрицательных логарифмов концентрации или активности. Если шкала выражена в единицах активности, то зависимости истинных величин потенциалов представлены штриховыми линиями. Если же по оси абсцисс отложены отрицательные логарифмы концентраций, то зависимости величин потенциалов, соответствующих эксперименту, выражаются сплошными линиями. Искривление графиков обусловлено влиянием коэффициентов активности. И действительно, на измерении потенциалов основан один из наиболее употребительных методов их определения. Заметим, что активность и концентрация становятся неразличимыми ниже 10-3 или 10~4 М.
На рис. 14-1 представлены зависимости потенциалов для полуреакций, наиболее полно подчиняющихся уравнению Нернста. Другие из-за комплексообразования или иных причин проявляют заметное отклонение. Так, например, для переходных металлов в первую очередь стандартные потенциалы не представляют особой ценности, потому что для определения активности требуются обычно данные, которые невозможно получить. По этой причине вместо них часто используют величину, называемую формальным потенциалом Е°':
Е°' = Е° — — \и^~ (14-14)
nF Vox
в котором у — коэффициент активности. Перепишем уравнение (14-9), введя коэффициенты активности:
Е. = — In 1Лге^Ул(г^... = Е7 - — In 1ML (14Д5)
А А nF Л nF [40Х]Р
При использовании формальных потенциалов состав раствора, в котором проводят измерения, должен быть точно известен. Значения потенциалов в табл. 14-1, взятой из монографии Мейтиса [1], показывают, насколько сильно состав раствора влияет на формальный потенциал системы. На рис. 14-2 дано
* На платиновую фольгу необходимо нанести платиновую чернь — Прим, перед.
Введение в электрохимические методы 305
Рис. 14-2. Формальные потенциалы различных систем относительно СВЭ. Символом М4|/3 обозначен потенциал, относящийся к равновесию M(1V) + +»5=tM(III). Светлые кружочки относятся к растворам хлоридов, темные — к растворам сульфатов, крестики — к растворам фосфатов. По шкале абсцисс отложена молярная концентрация раствора НС1 пли нормальные концентрации растворов H2SO4 и Н3РО4 [2, 3].
306 Глава 14
графическое изображение зависимости формальных потенциалов ряда окислительно-восстановительных систем от концентрации кислоты. Заметим, что с изменением кислотности окислительно-восстановительные свойства различных систем могут существенно меняться. Так, например, Fe(III) окисляет As (III) в среде 2М НС1, но в 8М НС1 Fe(II) окисляется As(V). Нужно помнить, что потенциалы полуреакций являются термодинамическими величинами и поэтому ничего не говорят о скоростях реакций. Например, хотя потенциал Се(IV) намного выше потенциала As(V), но в отсутствие подходящего катализатора он не будет окислять As (III).
Стандартные и формальные потенциалы полуреакций очень важны, поэтому многие из них точно определены и сведены в таблицы. Стандартные потенциалы некоторых полуреакций даны в приложении А.
Часто удобно или даже необходимо помещать электроды в разные отделения ячейки, электролитически контактирующие друг с другом. Контакт между ними можно установить с помощью перегородки из пористого фарфора или спеченного стекла или соединить оба сосуда солевым мостиком. Такое разделение электродов необходимо, если электролиты в полуэлементах несовместимы или если при их непосредственном контакте протекает окислительно-восстановительная реакция.
На границе между двумя растворами возникает дополнительный потенциал (его называют потенциалом жидкостного соединения) из-за разных скоростей миграции катионов в одном направлении и анионов — в другом. Способа полного устранения этого лишнего потенциала не существует, но его можно уменьшить с помощью мостика, заполненного концентрированным раствором КС1, или NH4NO3, или другой соли с одинаковыми подвижностями ионов.
Таким образом, мы часто случайно используем полуэлементы, не только имеющие свои собственные полуреакции и потенциалы, но также способные к независимому физическому существованию. Всегда, однако, нужно помнить, что одним полуэлементом обойтись нельзя; в любом случае нужна по меньшей мере пара полуэлементов.
Ниже даны примеры нескольких наиболее важных полуреакций.
1. Металлы в равновесии с собственными ионами (электрод I рода).
Zn1 2+ + 2е Zn Е° = — 0,763 В
Cu2+ + 2ei±Cu Е° — +0,337JB
Ag+ + е Ag Е° = + 0,799 В
Введение в электрохимические методы 307
Окисленной формой [Аох в уравнении (14.12)] является катион, восстановленной Area— свободный металл. Величины Е° даны в вольтах относительно СВЭ при 25 °C. Электролитом в этих полуэлементах обычно служит раствор соли металла с анионом сильной минеральной кислоты, выбор которой часто определяется растворимостью соли и комплексообразующей способностью аниона. Как правило, для этих целей пригодны сульфаты, нитраты и перхлораты. Уравнение (14.9) принимает форму
£»=ел+-511п(-4») <14-1б>
поскольку активность чистого твердого вещества всегда принимают за единицу. Величина (Аох) относится к простому (гидратированному) катиону; в присутствии комплексообразующих веществ она меньше (и часто намного) общей концентрации иона металла в растворе.
Простота электродов этого типа может ввести в заблуждение, поскольку на практике полезны всего лишь некоторые из них. Для изготовления электродов нельзя использовать более активные металлы, так как они недостаточно устойчивы в водной среде. Но даже менее активные металлы, например серебро, могут не дать ожидаемого отклика (потенциала) в разбавленных растворах, так как потенциал электрода будет определяться не ионами серебра, а другими веществами. Кроме того, часто важно фактическое состояние поверхности электрода, что трудно контролировать.
2. Металл в равновесии с насыщенным раствором малорастворимой соли (электрод II рода).
AgCl(TB) + е Ag + Cl” Е° = + 0,222 В
(ц=1)
Hg2Cl2(TB) + 2e->2Hg4-2Cl- Е° = 4-0,268 В
(а=1)
Полуэлементы этого типа находят широкое применение в качестве электродов сравнения, которые фактически являются вторичными стандартными электродами и служат для замены неудобного в работе СВЭ. В этом случае активность (или концентрацию) аниона устанавливают на определенном уровне, добавляя раствор хорошо растворимой соли с тем же анионом; для полуэлементов, приведенных выше, обычно применяют насыщенный раствор КС1. Используемые на практике электроды сравнения должны быть просты в изготовлении, иметь воспроизводимый потенциал и низкий температурный коэффициент. Ниже приведено несколько примеров электродов сравнения:
308 Глава 14
а) Насыщенный каломельный электрод (НКЭ)
Hg2Cl2(TB) + 2е 2Hg + 2С1“ Е = + 0,246 В
(нас. КС1)
б) 1 М каломельный электрод (МКЭ)
Hg2Cl2(TB) + 2е 2Hg + 2С1- Е = + 0,280 В
(IM КС1)
в) 1 М хлоридсеребряный электрод
AgCl(TB) + e-> Ag+Cl- £=+0,237 В
(1м ка)
Для электродов II рода уравнение Нернста нужно преобразовать, чтобы ввести в него произведение растворимости малорастворимой соли. Для хлоридсеребряного электрода KSP~ = (Ag+) (CI-) поэтому можно написать, что
Е = Е° + -^- ln(Ag+) = £° + ^ln-^ = nF nF (Cl-)
= E- ^ln(Cl-) + In Ksp (14-17)
Последнее слагаемое при данной температуре является постоянной величиной.
3. Металл в равновесии с двумя малорастворимыми солями с общим анионом (электрод III рода).
Ag2S(TB) —> 2Ag+ + S2
CdS(TB) -> Cd2+ + S2-
С помощью этого полуэлемента можно измерить активность ионов Cd2+. Необходимо, чтобы вторая соль (CdS) была несколько более растворима, чем первая (AgoS). В качестве примера электрода этого рода, широко используемого в практике, можно привести систему, содержащую ЭДТА, ионы Hg2+ и двух-, трех- и четырехвалентных металлов, где мало диссоциированные комплексы играют ту же роль, что и малорастворимые сульфиды в предыдущем примере.
4. Две растворимые частицы электроде.
Се4+ + е->Се3+
2Hg2+ + 2e->Hg2+
Fe3+ + e->Fe2+
в равновесии на инертном
£° = + 1,61 В
£° = + 0,920 В
£° = +0,771 В
Введение в электрохимические методы 309
Инертный электрод выполняет здесь только функцию переноса электронов от восстановленной формы к окисленной. Величины Е° даны для условий, когда активности обеих форм равны, что не обязательно означает равенство общих концентраций окисленной и восстановленной форм. В первую очередь это характерно для случаев, когда присутствуют комплексообразующие вещества, обладающие различным сродством к окисленной и восстановленной формам. Поэтому величины Е°', отвечающие общим концентрациям, а не активностям, могут быть и выше, и ниже табличных значений Е°.
Соглашение о знаках
Мы решили писать полуреакции в форме реакций восстановления, и отсюда вытекает, что величина Е° будет иметь знак минус относительно СВЭ для металлов, являющихся более сильными восстановителями, чем водород, и знак плюс, если они менее сильные восстановители, чем водород:
Сц2+ + 2е Си Е° = + 0,337 В
2Н+ + 2е->Н2 Е° = 0 В
Zn2+ + 2e->Zn Е° = —0,763 В
Эти значения совпадают с экспериментальными данными, полученными для ячеек, состоящих из медного или цинкового электродов, находящихся в контакте с их собственными ионами; медный электрод имеет знак плюс, цинковый — минус. Только для полуреакций, записанных в такой форме, Е° можно называть «стандартным электродным потенциалом». Если же записывать полуреакции в форме реакций окисления, то знак потенциала следует изменить. Существует два противоречивых способа определения знака потенциала электрода. В этой книге в дальнейшем знак потенциала дается в соответствии с рекомендациями Международного союза теоретической и прикладной химии (ИЮПАК), принятыми в Стокгольме в июле 1953 года. Подробное описание соглашений о знаках и рекомендации ИЮПАК можно найти в литературе [4, 5].
Обратимость
Термины обратимый и необратимый используют в разном значении в зависимости от контекста. В чисто химическом смысле реакция необратима, если ее продукты не реагируют друг с другом или если и реагируют, то так, что дают совсем иные вещества, чем исходные. В качестве электрохимического примера рассмотрим ячейку, состоящую из цинкового и хлоридсеребряного электродов, помещенных в разбавленную соляную
310 Глава 14
кислоту. Если цепь замкнуть внешним проводником, то начнут протекать полуреакции
Zn->Zn2+4-2e
2AgCl(TB) 4- 2е 2Ag + 2С1 “
т. е. в ячейке пойдет реакция
Zn + 2AgCl(TB) -> 2Ag 4- 2С1- 4- Zn2+
Если, однако, с помощью внешнего источника напряжения заставить протекать ток в противоположном направлении, то в этом случае пойдут полуреакции
2Н+ + 2е->Нг
2Ag 4- 2С1- 2AgCl(TB) 4- 2е
и полная реакция в ячейке будет иная:
2Ag + 2Н+ + 2С1- 2AgCl(TB) + Н2
Это означает, что хлоридсеребряный электрод является обратимым, а полуэлемент цинк-соляная кислота необратимым.
С термодинамической точки зрения реакция обратима, если даже бесконечно малое изменение движущей силы приведет к изменению ее направления или, иначе говоря, реакция находится в состоянии равновесия. Это означает, что реакция достаточно быстро реагирует на любое малое изменение независимой переменной. Термодинамическая обратимость есть идеальное состояние, лишь приблизительно применимое к реальным системам. Если электрохимическая реакция протекает с большой скоростью даже при достаточно малом отклонении от состояния равновесия, ее можно назвать обратимой. Данную реакцию можно считать обратимой, если наблюдать за ней каким-либо одним способом (например, измерять потенциал в отсутствие тока), но она может проявлять заметное отклонение от обратимости, если ее изучать в других, медленно изменяющихся условиях, как в полярографии, а также стать «полностью» необратимой, если ее подвергать быстрым воздействиям, как в условиях некоторых высокоскоростных методов.
Поляризация
Электрод (и, следовательно, ячейку) называют поляризованным, если его потенциал не равен величине, рассчитанной по уравнению Нернста. Это может наблюдаться, например, если к ячейке приложить какое-то напряжение или если через нее протекает заметный ток. Изменение потенциала, вызванное из
Введение в электрохимические методы 311
менением концентрации ионов на поверхности электрода, иногда называют концентрационной поляризацией. Однако этот термин не рекомендуется употреблять, так как всегда следует рассматривать активности ионов, фактически присутствующих на поверхности электрода.
Перенапряжение
Согласно постулатам термодинамики, полуэлемент ведет себя необратимо, если через него протекает заметный ток. Действительный потенциал электрода в этих условиях рассчитать нельзя, но. он всегда выше потенциала обратимого электрода, рассчитанного по уравнению Нернста, т. е. более отрицателен для катода и более положителен для анода. Разность между равновесным потенциалом и его действительной величиной называют перенапряжением или сверхпотенциалом. Перенапряже
ние можно рассматривать как дополнительную движущую силу, необходимую для протекания реакции с заметной скоростью [6]. Величина перенапряжения зависит от плотности тока (сила
тока, приходящаяся на единицу площади поверхности элек-
трода), температуры, природы окислительно-восстановительной
системы, участвующей в реакции. Особый интерес представляет перенапряжение, необходимое для восстановления Н+ (или
воды) до газообразного водорода. В отсутствие перенапряжения этот процесс протекает при О В (при активности ионов Н+, равной единице, СВЭ). На рис. 14-3 даны характерные величины перенапряжения водорода на некоторых катодах для 1 М раствора НС1 [7].
В некоторых случаях перенапряжение может быть по-настоящему полезным. Например, катионы таких металлов, как железо или цинк, несмотря на то, что их стандартные потенциалы более отрицательны, чем потенциал СВЭ, можно восстановить до металлов на ртутном катоде именно потому, что высокое перенапряжение водорода на ртути препятствует его выделению. На платиновом катоде эти ионы из водного раствора выделить нельзя, так как невозможно превысить потенциал, требуемый для восстановления водорода.
Рис. 14-3. Зависимость величины перенапряжения водорода на различных металлах от плотности тока (А/см2) в 1 М растворе НС[.
312 Глава 14
Электроаналитические методы
Ниже перечислены важнейшие аналитические методы, основанные на электрохимических свойствах. Большинство из них подробно описано в последующих главах.
1. Потенциометрия. Этот метод основан на прямом использовании в аналитических целях уравнения Нернста и включает измерение потенциалов неполяризуемых электродов в отсутствие тока.
2. Хронопотенциометрия. В этом методе через раствор пропускают известный постоянный ток и приобретаемый электродом потенциал изучают как функцию времени. Потенциал остается почти постоянным в течение периода времени, пропорционального концентрации электроактивного вещества. Аналогичный метод, основанный на измерении тока при наложении постоянного потенциала, называют хроноамперометрией.
3. Вольтамперометрия и полярография. Эти методы основаны на изучении состава раствора в электролитической ячейке по кривым зависимости силы тока от потенциала. Обычно потенциал маленького поляризуемого электрода (относительно электрода сравнения) увеличивают в область отрицательных значений до 1 или 2 В и наблюдают за изменением тока. Общее название этих методов вольтамперометрия-, термин полярография относится к методам с использованием капающего ртутного электрода.
4. Кондуктометрия. В этом методе анализа используют два идентичных инертных электрода для измерения проводимости (величины, обратной сопротивлению) изучаемого раствора. При его проведении необходимо как можно полнее устранять влияние электродов.
5. Кулонометрия. Этот метод анализа основан на применении законов электролиза Фарадея, т. е. па эквивалентности между количеством электричества и величиной химического превращения.
6. Разделение при контролируемом потенциале. Часто в процессе электроокисления или электровосстановлеиия при тщательном контроле потенциала электрода можно достичь количественного разделения. Разделенные вещества можно количественно определить гравиметрически или кулонометрически.
Интересные соотношения, существующие между многими из этих электроаналитических методов, можно найти в литературе [8—10], но разобраться в этих работах будет намного проще после детального изучения отдельных методов. Идеи, изложен
Введение в электрохимические методы 313
ные в настоящей и последующих четырех главах, более детально обсуждаются в монографии [11], которая и рекомендуется студентам для дальнейшего изучения.
Задачи
14-1. Выведите уравнение для зависимости потенциала электрода III рода, описанного выше, от произведений растворимости Ag2S и CdS.
14-2. Величины стандартных потенциалов полуреакций Си2+4-2 е^Си и Си+ + + е=^Си даны в приложении А. Исходя из них, вычислите стандартный потенциал полуреакции Cu2++e^Cu+.
Литература
1. Meites L., In Handbook of Analytical Chemistry, Meites L. (ed.), McGraw-Hill, New York, 1963, table 5-1, p. 5—6.
2. Furman N. FL, In Treatise on Analytical Chemistry, Kolthoff J. M. and Elving P. J. (eds.), Wiley-Interscience, New York, 1963, pt. 1, v. 4, p. 2294.
3. )?ao G. G., Rao P. K., Taianta, 1963, v. 10, p. 1251; 1964, v. 11, p. 825.
4. McGlashen M. L., Pure Appl. Chem., 1973, v. 21(1), p. 1.
5. Licht T. S., A. J. deBethune., J. Chem. Educ., 1957, v. 34, p. 433.
6. Bockris J. O’M., J. Chem. Educ., 1971, v. 48, p. 352.
7. Daniels F., Alberti/ R. A., Physical Chemistry (3d ed.), Wiley, New York. 1966, p. 266.
8. Kolthoff J. M., Anal. Chem., 1954, v. 26, p. 1685.
9. Reilley C. N., Cooke W. D., Furman N. H„ Anal. Chem., 1951, v. 23, p. 1226.
10. Reinmuth W. H., Anal. Chem., 1960, v. 32, p. 1509.
11. Vnssos B. FL, Ewing G. WElectroanalytical Chemistry, Wiley-Interscience, New York, 1983.
ГЛАВА
Потенциометрия
В предыдущей главе показано, что уравнение Нернста дает простое соотношение между потенциалом электрода и концентрацией соответствующего иона в растворе. Поэтому на основании измерения потенциала обратимого электрода можно рассчитать активность или концентрацию компонента раствора.
Например, допустим, что мы сконструировали ячейку, состоящую из серебряного электрода, погруженного в раствор нитрата серебра, и соединили ее солевым мостиком с насыщенным каломельным электродом сравнения (НКЭ). Ячейка схематически изображена на рис. 15-1. В левом сосуде находится раствор нитрата серебра, в боковой трубке справа — НКЭ, центральный сосуд служит для соединения раствора КС1 в НКЭ и солевого мостика, погруженного в раствор нитрата серебра. Солевой мостик заполнен агар-агаровым гелем, содержащим для обеспечения электропроводности нитрат аммония. (Это позволяет предотвратить загрязнение НКЭ серебром и загрязнение раствора нитрата серебра КО, а также сводит к пренебрежимо малой величине потенциал жидкостного соединения.)
Если к электродам подключить электронный вольтметр, серебряный электрод будет положительным, а НКЭ — отрицательным. Допустим, что измерительный прибор показывает, что разность потенциалов равна 0,450 В, тогда можно записать, что
Енкэ= +0,246 В
Вычитанием находим, что
Е*ч= ^Ag""^HK3 = ^Ag £'нкэ+ in [Ag+1
Потенциометрия 315
+ nh^nOj с
Агар-агар Насыщенный г—, Нд2С1г
\ КИХ,^ (твердый)
КС I
Ад+М0з NH4+N03
Рис. 15-1. Устройство ячейки с серебряным и насыщенным каломельным электродами.
Решим это уравнение относительно lg[Ag+]* и, учитывая, что для Ag+ п=\, получим (при 25 °C)
t Дй+1 = £ЯЧ~ £Ag + £HK3 _ 0,450 — 0,799 + 0,246 __,
ё1 ё 2,303 (RT/nF) 0,0591
[Ag+] = antilog( — 1,74) = 1,8-10-2 M
Заметим, что при комбинировании двух полуэлемёнтов, как это сделано выше (выражение Еяч=ЕАе—Анкэ)> всегда вычитают более отрицательную величину из более положительной. Здесь действует упоминавшееся выше соглашение о знаках потенциала применительно к соответствующим химическим уравнениям, когда полуреакцию окисления вычитают из полуреакции восстановления. Это гарантирует положительный потенциал ячейки для реакции, записанной в направлении самопроизвольного протекания.
Рассмотрим ячейку с платиновым электродом, погруженным в раствор с 0,1 М концентрацией сульфата железа (II). НКЭ можно погрузить прямо в этот раствор, так как никаких мешающих процессов происходить не может. Наблюдаемая разность потенциалов составляет 0,395В (25°C). Задача заключается в том, чтобы найти процент железа (II), окисленного растворенным кислородом до железа(III). Прежде чем проводить вычис
* Вспомним, что In х=2,303 1g х. Поскольку величина 2,303 (RT/F) встречается очень часто, целесообразно запомнить, что при 25 °C она равна 0,0591. Эти множители даны с гораздо большим числом значащих цифр, чем может понадобиться в действительности, для того чтобы их сразу можно было отличить.
316 Глава 15
ления, запишем выражения для потенциалов полуреакций (rt= 1):
t? । RT' [Fe3+]
£pt ~ fcFe’+/Fe2+ Н--W П ’
г r гс nr [rez+]
Енкэ = + 0,246
^я,= Ept -^НКЭ = £ре3+/Ре2+' [Fe2+1
I1 с J
]tf +1 _____ ^яч ~~ £ре3+/ре2+ ~Ь £НКЭ ___
ь [Fe2+] _ 0,0591 ~
_ 0,395 —0,771 4-0,246 _2 20
~ 0,0591 ~
= antilog (—2,20) = 6,3-10-3 = 0,63 %
Это означает, что окислено 0,63 % железа (II).
Концентрационные цепи
Концентрационными цепями называют две одинаковые ячейки с двумя идентичными электродами, погруженными в растворы, отличающиеся только концентрацией иона, относительно которого электрод обратим. В этом случае разность потенциалов зависит от отношения концентраций иона.
Например, при определении хлорид-иона один хлоридсеребряный электрод можно погрузить в раствор с неизвестной концентрацией хлорида [С1—]ж, а другой — в стандартный раствор с концентрацией хлорида [С1~]ст = 0,1000 М. Как следует из уравнения (14-17), логарифмический член в уравнении Нернста будет отрицательным; постоянные члены, т. е. Е°+ + (RT/nF)\n Ks (равный для Ag/AgCl-электрода при 25 °C 0,222 В), одинаковы для обоих электродов, и поэтому их можно опустить. Поправкой па коэффициенты активности можно пренебречь даже при относительно высоких концентрациях, поскольку растворы идентичны. Потенциал электрода в стандартном растворе равен
£ст= +0,222 — 0,05911g 0,1000= +0,281 В
Если концентрация неизвестного раствора выше, например, 0,1500 М, то
Ех=+0,222—0,0591 IgO,1500=+0,271 В ;
Потенциометрия 317
Потенциал ячейки можно найти как обычно, вычитая более отрицательную величину из более положительной:
Еяч== Ех—Е„ = +0,010 В
Измеренный потенциал (например, +0,010 В) может соответствовать любой из двух неизвестных концентраций, так что концентрационные цепи могут давать неоднозначные для интерпретации результаты. Этого можно избежать, если тщательно соблюдать знаки электродов в стандартном и неизвестном растворах.
Концентрационные цепи служат основой высокочувствительного аналитического метода [1], потому что в методе, основанном на сравнении двух почти идентичных растворов, исключается большинство источников погрешностей, возникающих при измерениях в ячейке. Это означает, что суммарная погрешность метода будет определяться главным образом только погрешностью измерительного устройства. Как и в любом потенциометрическом методе, чувствительность измерения (при 25 °C) составляет 0,0591/п вольт на десятикратное изменение концентрации. Если при измерении используют универсальный электрод сравнения (т. е. не концентрационные цепи), требуется измерительный прибор со шкалой до 2 В, тогда как при работе с концентрационными цепями пригоден прибор для измерения до 20 мВ. Это позволяет получить в 100 раз более точные результаты, чем в первом случае.
Ионоселективные мембранные электроды
Конструкция целого класса очень полезных в практическом отношении электродов, обладающих различной специфичностью и селективностью, содержит мембрану, разделяющую внутренний раствор и электрод и одновременно служащую средством электролитического контакта с внешним (анализируемым) раствором [2]. Несколько таких конструкций показано на рис. 15-2. В каждом из электродов мембрана обладает ионообменными свойствами.
Стеклянный электрод. На рис. 15-2, а изображен хорошо известный стеклянный электрод для измерения pH. Он представляет собой маленький стеклянный шарик из рН-чувстви-тельного стекла, заполненный раствором хлорид-ионов с известным pH, в который погружен внутренний электрод сравнения, обычно хлоридсеребряный или каломельный. Частично гидратированное алюмосиликатное стекло содержит ионы натрия или кальция и часто небольшие количества ионов лантанидов. Оно способно реагировать с ионами водорода, которые могут вайти
318 Глава 15
Стеклянная t-кмбрана
б'
Жидкий-----
ионоабменник
Пористый— диск
в
Кристаллическая мембрана
Внутренний электрод сравнения
Рис. 15-2. Ионоселективные электроды: а — стеклянный; б — с жидкой мембраной; в — с твердой кристаллической мембраной.
в кислородные полости решетки стекла, в то время как другие ионы для поддержания электронейтральности стекла должны уйти с противоположной поверхности. Между ионами водорода во внутреннем и внешнем растворах легко устанавливается равновесие, в результате чего возникает потенциал
Е = К+ — Ы (Н+)внутр (15-1)
F (Н+)внешн
В этом выражении в величину К входит разность потенциалов внутреннего и внешнего электродов сравнения, потенциал жидкостного соединения и небольшой непредсказуемый потенциал (известный как потенциал асимметрии'), обусловленный, вероятно, механическими напряжениями, возникающими в стекле.
После введения определения понятия pH* * уравнение (15-1) (при 25 °C) можно переписать следующим образом:
Е = К' + 0,0591 (pHвнешн1— рНвнутр) (15-2)
* Несмотря на то что существует шкала pH, на которой отложены отрицательные логарифмы активности ионов водорода, найдено целесообразным создать практическую или рабочую шкалу, построенную на измерениях pH серии буферных растворов, приготовленных по стандартной методике. Подробное обсуждение теоретических н практических аспектов концепции pH можно иайти в книге Бейтса [3].
Потенциометрия 319
Для каждого конкретного электрода pH внутреннего раствора постоянен, так что, объединив его с постоянной К, получим
£ = Д/ + 0,0591рН . (15-3)
В практической работе на pH-метре величина К' учитывается на стадии калибровки, когда электроды погружают в стандартный буферный раствор и на шкале регистрирующего устройства устанавливают правильную величину pH. Для стеклянного электрода характеристикой служит «pH нулевого потенциала», т. е. точка, когда внутренний и внешний растворы имеют одинаковый pH.
Многие изготовители выпускают комбинированные зонды, состоящие из стеклянного электрода и электрода сравнения. В большинстве из них контакт с электродом сравнения осуществляется при помощи солевого мостика, находящегося в кольцевом зазоре стеклянного электрода. Раствор КС! служит электростатической защитой для стеклянного электрода, имеющего высокое сопротивление. Комбинированные зонды становятся довольно популярными, потому что они удобны, хотя и стоят несколько дороже, чем обычная пара отдельных электродов.
Применение стеклянного электрода ограничено тем, что в присутствии больших концентраций щелочных металлов он дает сильно завышенные результаты. В этом случае возникает так называемая натриевая ошибка (чаще говорят щелочная ошибка). Ранее выведенное уравнение перестает быть справедливым, и в этом случае пригодно более полное уравнение, известное как уравнение Нернста — Айзенмана
Е = const + In ([Н+] + kr [Na+Ц- k2 [K+] + . . .) (15-4)
Г
в котором k — коэффициенты селективности. Хороший рН-элек-трод должен иметь очень низкие величины k (порядка 10~3 или ниже). Строго говоря, эти коэффициенты не являются константами, и их нельзя использовать для введения поправок в измеренные величины pH, но с их помощью можно оценивать пригодность различных электродов в решении конкретных аналитических задач.
Айзенман [4] и другие исследователи показали, что коэффициенты селективности зависят от констант равновесия ионообменной реакции на поверхности стекла и от подвижностей ионов в фазе стекла. Например, если ограничиться рассмотрением ионов Н+ и Na+, то коэффициент k} определяется выражением k (H+)p(Na+)n0Bl/Na+J
1 (Н+)пов (Na+)pt/H+
в котором в скобках стоят активности ионов в растворе и на по-
320
Глава 15
верхности стекла, a f/Na+ и UH+ — подвижности ионов Na+и Н+ в стекле. Разработаны способы надежного определения влияния Na+ на потенциал стеклянного электрода [5].
Можно изготовить стеклянные электроды, обладающие чувствительностью к ионам Na+ или Ag+ и лишь слабо реагирующие на изменение pH. Электрод такого типа пригоден для определения Na+ или Ag+ в отсутствие других ионов или при по-
Таблица 15-1. Наиболее распространенные иопоселективные электроды [2]
Жндкне мембраны
Определяемый нон Центр обмена3 Основные коэффициенты селективности
Са2+ (RO)2po7 н+107; Zn2+ 3,2; Fe2+ 0,80; Pb2+ 0,63; Cu2+ 0,27; Ni2+ 0,08; Sr2+o,O2; Mg2+0,01; Ba2+0,01; Na+0,0016
N0F nil2+ C1O7 103; 1—20; C1O^2; Br~0,9; S2~0,57; N070.06; Cl-0,006; SO2- 0,0006
сюу FeL2+ OH-1,0; CO,012; NO^ 0,0015;СГ 0,00022; SO2“ 0,00016
ио^+"2) Полиэфир6 h+0,016; Ag+ 0,003; Pb2+0,002 и т. п.
Твердые мембраны
Определяемый ион Мембрана Ионы, мешающие определению
F- La он-
ci- AgCl(Ag2S) Br-, I-, S2-, NHa, CN-
Br- AgBr(Ag2S) I-, S2-, NH3, CN-
I- Agl(Ag2S) S2-, CN-
SCN- AgSCN(Ag2S) Br-, I-, S2-, NH3, CN-
S2-, Ag+ Ag2S Hg2+
CN- AgI(Ag2S) I-, s2-
Cti2+ CuS(Ag2S) Hg2+, Ag+
Pb2+ PbS(Ag2S) Hg2+. Ag+, Cu3+
Cd2+ CdS (Ag.,S) Hg2+, Ag+, Cu2+
Лиганд L замещен 9,10-фенантролиновой группировкой.
б N ,N'-Дигептил- N,N',6,6-тетраметил-4,8-дноксауидекаидиамид.
Потенциометрия 321
стоянной их концентрации в соответствующим образом забуфе-ренном растворе. Имеется также стеклянный электрод, чувствительный к ионам аммония.
Электроды с жидкими мембранами
Электроды, чувствительные к ряду катионов и анионов, можно изготовить на основе мембраны или пленки, содержащей жидкий ионообменник [4]. Конструкция электрода такого типа показана на рис. 15-2,6. Маленький диск из пористого гидрофобного материала разделяет внутренний и внешний растворы электролита. По всему своему периметру диск контактирует с органическим растворителем, не смешивающимся с водой, который находится в кольцевом зазоре. Растворим в этом растворителе соль нужного нам иона с противоионом относительно большой молекулярной массы и со значительно более высокой растворимостью в органической фазе, чем в воде. Под действием капиллярных сил растворитель заполнит поры диска, осуществляя электрический контакт с обоими водными растворами. За счет этого установится равновесие между общими ионами в мембране и растворах. Потенциал внутреннего электрода подчиняется уравнению Нернста практически аналогично стеклянному электроду. Несколько примеров электродов с жидкими мембранами приведено в табл. 15-1.
Можно сконструировать такой ‘же электрод без внутреннего раствора сравнения. Для этого ионообменник вводят в полимер, который затем наносят прямо на. металлическую проволоку [6, 7]. Хотя теория возникновения потенциалов таких электродов не совсем ясна, по-видимому, они электрически эквивалентны другим более разработанным вариантам.
Электроды с двойными мембранами
Сферу применения ионоселективных мембранных электродов иногда можно расширить при помощи второй (дополнительной) мембраны. Для серийных измерений парциального давления СО2 в плазме крови или другой жидкости применяют стеклянный электрод, покрытый тонкой пленкой тефлона или другого газопроницаемого материала. Между пленкой и стеклом находится слой водного раствора NaHCCh, обычно 0,1 М концентрации. В процессе выполнения анализа СО2 диффундирует через пленку полимера в количестве, зависящем от его парциального давления в пробе, и возникающее в результате изменение pH раствора гидрокарбоната фиксируется стеклянным электродом.
11 Заказ № 537
322 Глава 15
Рис. 15-3. Кривые отклика (потенциал — время) электрода для катализируемой уреазой реакции гидролиза мочевины. Концентрация мочевины 0,01 М; 0,1 М трис-буфер с pH 7,0. Единицы активности уреазы: а — 0,04; б—-0,016; в —0,008; г —0,004; д — 0,002.
Иногда для усовершенствования электродов можно использовать специфичность ферментов [8]. Так, например, вторую мембрану можно изготовить из слоя геля, содержащего уреазу, и в результате получить МН4+-чувствительный стеклянный электрод для определения мочевины. Скорость катализируемой ферментом реакции гидролиза мочевины с образованием NH4+-ионов пропорциональна концентрации мочевины [9]. Изменение отклика электрода во времени показано на рис. 15-3. Для установления устойчивого потенциала требуется несколько минут, но правильные результаты можно получить за более короткое время, определив наклон начального участка. О возможностях других ферментных электродов можно прочитать в обзорных статьях [10, 11].
Потенциометрия 323
Электроды с твердыми мембранами
Мембраны для другого важного класса электродов изготавливают из кристаллических материалов в виде пластинок или таблеток. Самым распространенным из таких электродов является фторид-селективный электрод фирмы Orion с мембраной из монокристалла фторида лантана LaF3. Общий вид его показан на рис. 15-2, в. В кристаллической решетке LaF3 фторид-ионы могут довольно легко перемещаться внутри остова из неподвижных ионов лантана. Отклик электрода обусловлен исключительно фторид-ионами, поскольку никакой другой ион не может проникнуть в кристалл. Потенциал электрода подчиняется уравнению Нернста в интервале активности фторид-иона (примерно от 10 до 10-6 М). Отклонение от этой зависимости в чрезвычайно разбавленных растворах обусловлено растворимостью LaF3. Для этого электрода, так же как и хлоридсеребряного, применимо уравнение (14-17), поэтому перед логарифмическими членами стоит знак минус:
Е = К—~ 1п(Х-)1 (15-5)
Фторидселективный электрод стал исключительно ценным средством исследования в аналитической химии, поскольку прежде для определения этого важного элемента располагали лишь трудоемкими и недостаточно точными косвенными методами.
Аналогичные электроды можно изготовить из прессованной поликристаллической таблетки сульфида серебра, чувствительной к ионам Ag+ или S2- до концентраций порядка 10-19 или Ю-20 М. Для того чтобы приготовить хлорид-, бромид-, иодид- и роданид-селективные электроды, нужно спрессовать таблетки из смеси Ag3S и серебряной соли нужного аниона AgX. Так как эти соли более растворимы, чем Ag3S, то в мембране и анализируемом растворе устанавливается равновесие между ионами X ~ и противоионами. Уравнение Нернста принимает вид
В = А—^ln(X-) (15-6)
Г
где в константу К входит логарифм произведения растворимости AgX.
Ряд аналогичных электродов можно приготовить, запрессовывая в таблетку AgaS сульфид другого металла. Если, например, ввести сульфид свинца (п — 2), то получится электрод III рода, для которого применимо уравнение Нернста:
Е = К + ~ In (Pb2+) (15-7)
Примеры электродов с твердыми мембранами приведены в табл. 15-1.
11*
324 Глава 15
Влияние посторонних ионов на потенциал электрода с твердой мембраной отличается от их влияния на электроды с жидкой (и стеклянной) мембраной. В случае электродов с жидкими мембранами этот эффект лучше всего описывается с помощью коэффициентов селективности [уравнение (15-4)]. В случае же электродов с твердой мембраной оно связано главным образом с величиной произведения растворимости. Например [6], электрод на основе AgBr(AgaS) перестает реагировать на бромид-ионы и превращается фактически в роданид-селективный электрод, если отношение активности SCN~-hohob к активности Вг_-ионов равно отношению их произведений растворимости, так как в этом случае AgBr на поверхности электрода превратится в AgCNS. Меньшие же количества SCN- не мешают.
Электроды с твердыми мембранами одни из наиболее доступных и удобных детекторов для обнаружения катионов и анионов в растворе. Можно надеяться, что подобные электроды будут созданы и для многих других ионов [12].
В электродах другого типа, изобретенных Пунгором и выпускаемых в Венгрии, силиконовая мембрана содержит нерастворимую соль типа Agl, введенную в нее в качестве наполнителя. Такую мембрану перед использованием необходимо несколько часов вымачивать в растворе определяемого иона. В США эти электроды не получили широкого распространения.
Электроды сравнения
Требования к электроду сравнения, применяемому при измерениях с помощью ионоселективного электрода, могут быть более жесткими, чем при измерениях pH, что часто диктуется желанием получить более правильные результаты [13]. Обычно pH измеряют с точностью до сотых долей единицы (±0,6 мВ), а при определении других ионов возникает необходимость определения на порядок точнее. Поэтому становятся существенными небольшие отклонения в работе электрода сравнения, которыми при измерении pH можно пренебречь.
В одной из наиболее распространенных конструкций электрода сравнения для измерения pH на участке жидкостного соединения используют асбестовое волокно, вплавленное в стекло. Внутренний электролит (обычно насыщенный раствор КС1) удерживают на несколько более высоком уровне, чем испытуемый раствор, чтобы создать слабый поток КС1, предотвращающий загрязнение электрода сравнения. Однако при работе с таким электродом волокно начинает покрываться крошечными кристалликами КС1, препятствующими оттоку раствора и влияющими на потенциал. В таком случае желательно использовать электрод с жидкостным соединением рукавного типа, обеспечивающий больший поток раствора.
Потенциометрия 325
Потенциометрическое титрование
За протеканием большого числа реакций можно проследить потенциометрически. Для этого необходимо только, чтобы в реакции участвовал какой-либо ион (вводился или выводился), для которого существует подходящий электрод.
Потенциал индикаторного электрода в процессе титрования изменяется в соответствии с уравнением Нернста. Если графически изобразить зависимость потенциала электрода от количества добавленного титранта, то получится кривая, -по которой можно найти конечную точку (или точки) титрования. Эти кривые обсуждаются в учебниках по количественному анализу, поэтому здесь детально не рассматриваются. Типичный пример приведен на рис. 15-4.
Общее изменение потенциала во время титрования составляет лишь небольшую долю от средней разности потенциалов двух электродов. Если это так, то точность можно улучшить при использовании концентрационных цепей. В оба отделения ячейки вводят одинаковые электроды для наблюдения за титруемым ионом, а в одно отделение наливают раствор, по составу идентичный ожидаемому в точке эквивалентности. Как только в процессе титрования анализируемого раствора будет достигнута точка эквивалентности, разность потенциалов станет равной нулю. Повышение точности результатов‘достигается за счет использования измерительного прибора с более высокой чувствительностью.
Рис. 15-4. Кривые потенциометрического титрования кислот различной силы раствором NaOH (значения константы диссоциации указаны на рисунке).
326 Глава 15
Рис. 15-5. Титрование холинэстеразы при постоянном pH (7,40). Одно деление шкалы по ординате соответствует ~ 0,488 мкМ [14].
Титрование при постоянном потенциале. Другой способ потенциометрического титрования основан на измерении количества титранта, необходимого для поддержания постоянного потенциала индикаторного электрода. В этом случае кривую титрования можно построить в координатах объем добавленного стандартного раствора — время. Этот способ титрования чаще применяют в энзимологии.
Например, в присутствии фермента холинэстеразы ацетилхолин разлагается с образованием уксусной кислоты. Фермент чувствителен к изменению pH, и, чтобы создать оптимальные условия протекания реакции, необходимо поддерживать pH вблизи 7,4. Для этого используют pH-метр, присоединенный к сервосистеме, контролирующей движение поршня бюретки, которая подает по мере необходимости раствор NaOH. Такое устройство известно как pH-стат. На рис. 15-5 показана кривая, по которой определяли активность холинэстеразы из образца животной ткани [14]. Навеску ткани массой 0,3 г гомогенизировали в физиологическом солевом растворе (0,9%-ный раствор NaCl). После установления pH 7,4 (точка Р) выжидали 10 мин, регистрируя базовую линию, чтобы убедиться в отсутствии самопроизвольного выделения кислоты. Затем прибавляли избыток иодида ацетилхинолина (Ацх.1), после чего начинался приток NaOH. Активность холинэстеразы, определенная по наклону кривой, полученной в этом эксперименте, оказалась равной 4,96- 10~6 моль/(г-мин).
Этот прием, по-видимому, не нашел распространения, за исключением биологических исследований, но он безусловно заслуживает внимания.
Потенциометрия 327
Аппаратура
Уравнение Нернста справедливо только в отсутствие тока в ячейке, что налагает ограничения на конструктивное решение измерительных устройств. Устройства для измерения потенциала, которое бы гарантировало полное отсутствие тока в цепи, не существует, но можно создать близкие условия, приемлемые для аналитических целей. Классический метод основан на применении потенциометра, в котором потенциал ячейки компенсируют точно измеренной долей напряжения от стабильного внутреннего источника. В современных аналитических лабораториях потенциометры используют редко.
Наиболее распространенным прибором для измерения потенциала ячейки является pH-метр (или иономер). Этот электронный вольтметр имеет усилитель, реагирующий на изменение потенциала. Результат измерения можно считывать со шкалы или цифрового индикатора. Стеклянный электрод может иметь сопротивление до 108 Ом, и если допустима ошибка порядка 0,1 %, то сопротивление на входе усилителя должно быть не менее 1011 Ом. Эта задача легко разрешима с помощью современных полупроводниковых усилителей.
Разработано много моделей pH-метров. Некоторые из них работают от аккумуляторных батарей, другие можно включать в сеть. Условно их можно разделить на три категории: приборы высокой, средней и сравнительно низкой воспроизводимости и правильности (и цены). Приборы первой категории предназначены главным образом для исследовательских работ, приборы второй категории— для работы в лабораториях общего типа, приборы третьей категории применяют там, где небольшие габариты и простота конструкции более важны, чем высокая воспроизводимость.
На панели лабораторного pH-метра- установлены три, а иногда четыре ручки управления: 1) переключатель положений «выключено» и «включено»; 2) регулятор калибровки или стандартизации для установки на нуль, чтобы pH-метр показывал правильную величину, когда электроды погружены в стандартный буферный раствор; 3) температурный компенсатор для регулирования чувствительности согласно нернстов-ской зависимости потенциала от температуры. Некоторые рН-метры имеют также переключатель шкалы, что позволяет прибору давать показания во всем интервале шкалы pH (обычно от 0 до 14) или в ограниченном интервале этой шкалы, возможно в 2 или 3 единицы pH; приборы такого типа называют pH-метрами с растянутыми шкалами.
Иногда забывают, что температурный компенсатор предназначен для регулировки наклона характеристической кривой
328 Глава 15
и, следовательно, его можно использовать для того, чтобы согласовывать показания прибора для двух стандартных буферов. В идеале pH-метр, йастроенный на 7 по буферу с pH 7, должен показать точно 4, если его проверяют по буферу с pH 4, и точно 10, если буфер имеет pH 10. В противном случае плавной регулировкой температурного компенсатора можно установить правильное показание при повторном контроле при pH 7 и вновь при pH 4 или 10. По своей сути эта процедура пригодна для обычных температур.
Автоматические титраторы. В заводских аналитических лабораториях, а также при проведении некоторых исследовательских работ широко применяют автоматические потенциометрические титраторы. Существует два класса титраторов: титраторы, записывающие всю кривую титрования на диаграммной ленте, и титраторы, в которых клапан бюретки закрывается
• . с помощью электрического
Рис. 15-6. Кривая потенциометрического титрования: а — в интегральной форме; б — первая производная; в — вторая производная.
устройства в точке эквивалентности. Титраторы первого класса фактически состоят из pH-метра, присоединенного к самописцу. Титрант нужно добавлять с постоянной скоростью. Поскольку с помощью обычной бюретки это сделать невозможно, необходимо подключать насос с постоянной производительностью или автоматический шприц.
Для обнаружения конечной точки в титраторах второго класса применяют специальные емкостные электронные схемы для двойного дифференцирования изменений потенциала электрода. На рис. 15-6 кривая а представляет потенциал как функцию объема реагента (£=f(V)), кривая б — первую производную dE/dV, а кривая в — вторую производную d^E/dV2. В титраторе используется то преимущество, что левая ветвь положительного пика на кривой в до точки эквивалентно
Потенциометрия 329
сти меняется очень мало. При первом уменьшении второй производной срабатывает реле, закрывающее запорный кран бюретки. Механическая инерция движущихся частей вызывает задержку, которая как раз и компенсирует опережение кривой второй производной, и поток титранта прекращается как бы в нужный момент.
Задачи
15-1. Ячейка состоит из свинцового электрода, погруженного в 0,015 М раствор ацетата свинца, н кадмиевого электрода, погруженного в 0,021 М раствор сульфата кадмия. Растворы соединены солевым мостиком, заполненным нитратом аммония. Каков потенциал ячейки при 25 °C? (Коэффициенты активности ионов в обоих растворах можно считать равными.)
15-2. Каков pH раствора, если потенциал водородного электрода, измеренный при 25 °C относительно СВЭ, равен 0,703 В?
15-3. Ячейка устроена следующим образом: серебряный электрод, используемый раствор, солевой мостик, заполненный насыщенным раствором КС1, НКЭ. а) Назовите индикаторный электрод и электрод сравнения, б) Каково назначение солевого мостика, и каким электролитом следует его заполнить? в) Если измеренный потенциал ячейки равен 0,300 В, а потенциал серебряного электрода более положителен, чем ртутного, то какова концентрация ионов Ag+ в неизвестном растворе?
15-4. Предполагают, что образец свинца содержит примесь серебра. Для выполнения анализа выбирают метод с использованием концентрационной цепи. Навеску анализируемого металла массой 10,00 г растворяют в азотной кислоте, полученный раствор разбавляют до 100,0 мл. Для создания ячейки в два стакана приливают из пипетки по 25,00 мл 0,1000 М раствора нитрата серебра, затем погружают два одинаковых серебряных электрода и растворы соединяют солевым мостиком, заполненным нитратом аммония. Как и ожидалось, разность потенциалов в этом случае равна нулю. Затем в один стакан добавляют 10,00 мл раствора нитрата свинца, а в другой—10,00 мл дистиллированной воды. После этого разность потенциалов серебряных электродов стала равной 0,070 мВ. Рассчитайте процентное содержание примеси серебра в свинце. Определите погрешность анализа, если максимальная погрешность измерительного устройства ±1 мкВ.
15-5. Ячейка состоит из двух проволочных платиновых электродов, погруженных в стаканы, содержащие по 25,00 мл смеси 1 М растворов хлоридов Fe(II) и Fe(III); растворы в стаканах соединены солевым мостиком. Добавление 1,00 мл раствора хлорида олова (II) в один стакан вызывает появление разности потенциалов электродов в 0,260 мВ. Принимая во внимание уравнение реакции SnCl2+2FeC13->-2FeC12+SnC14, рассчитайте концентрацию раствора SnCl2.
15-6. Какова максимально возможная концентрация иона Си2+ в растворе, находящемся в контакте с металлическим цинком?
15-7. Выведите уравнение зависимости потенциала СО2-электрода от парциального давления СО2 в пробе.
15-8. Фторид-селективный электрод можно использовать как электрод II рода для определения ионов La3+. Выведите уравнение зависимости потенциала электрода от активности ионов La3+.
15-9. Водный раствор (pH 5,0) проанализировали на содержание свободных Е~-иоиов. Потенциал фторид-селективного электрода относительно подходящего электрода сравнения, погруженного в 100 мл анализируемого раствора, равен 120 мВ. Если к анализируемому раствору добавить точно 1,00 мл 0,100 М раствора KF и перемешать, потенциал станет равным 108 мВ. Рассчитайте концентрацию Р~-ионов в анализируемом растворе (при 25 °C).
330 Глава 15
15-10. Потенциал стеклянного электрода относительно подходящего электрода сравнения в зависимости от условий принимает следующие значения:
[h+] [Na+] E, В
Ю-w io-• —0,572
io-10 10—7 —0,588
Ю-12 10—4 —0,472
Какова величина коэффициента kt в уравнении (15-4), если других катионов в растворе нет?
Литература
1. Furman N. И., in Trace Analysis, Yoe J. H., Koch H. J. (eds.), Wiley, New York, 1957, ch. 9.
2. Ионоселективные электроды/Под ред. Дарета Р.— М.: Мир, 1972.
3. Бейтс Р. Определение pH. Теория и практика.— Л.: Химия, 1968.
4. Eisenman G., Glass Electrodes tor Hydrogen and Other Cations. Dekker, New York, 1967.
5. Covington A. K., Ferra M. J. A., Anal. Chem., 1977, v. 49, p. 1363.
6. Ross J. W., Jr., гл. 2, ссылка 2.
7. Martin C. R., Freiser H., J. Chem. Educ., 1980, v. 57, p. 512.
8. Guilbault G. G., Smith R. K., Montalvo J. Q„ Jr., Anal. Chem., 1969, v. 41, p. 600.
9. Ansaldi A., Epstein S. J., Anal. Chem., 1973, v. 45, p. 595.
10. Weetail H. H„ Anal. Chem., 1974, v. 46, p. 602 A.
11. Bowers L. D., Carr P. W., Anal. Chem., 1976, v. 48, p. 544A.
12. Senkyr J., Ammann D., Meier P. C., Morf W. E„ Pretsch E., Simon W., Anal. Chem., 1979, v. 51, p. 786.
13. Caton R. D., Jr., J. Chem. Educ., 1973, v. 50, p. A571; 1974, v. 51, p. A7.
14. Jensen-Holm J., Lausen H. H., Milthers K., Moller K. O., Acta Pharmacol. Toxicol., 1959, v. 15, p. 384.
ГЛАВА 16
Вольтамперометрия, полярография и родственные методы
Предыдущая глава посвящена измерению потенциалов электродов в отсутствие тока. Теперь мы приступим к изучению явлений, сопровождающихся протеканием тока, в частности будем иметь дело с изображением кривых зависимости силы тока от потенциала интересующего нас (рабочего, индикаторного) электрода. Общее название такого метода изучения состава раствора — вольтамперометрия. Термин полярография относится только к вольтамперометрии с использованием ртутного капающего электрода.
При прохождении тока возникает необходимость в модификации электрода сравнения или, что лучше, во введении в ячейку третьего электрода. Использование одного и того же электрода в качестве электрода сравнения и проводника тока нежелательно. В этом случае во избежание чрезмерных ошибок электрод должен иметь низкое внутреннее сопротивление: в результате протекания через 1000-Ом сопротивление всего 10-мкА тока возникает ошибка в 10 мВ. Кроме того, граница раздела металл — раствор у электрода сравнения должна иметь гораздо большую площадь, чем поверхность рабочего электрода, чтобы плотность тока и поляризационные эффекты были минимальными. Электроды сравнения, предназначенные для измерений с помощью pH-метра, для этих цепей не пригодны, так как они имеют слишком высокое сопротивление.
Обычно в вольтамперометрии предпочитают трехэлектродные ячейки. Третий (вспомогательный) электрод проводит ток, тогда как электрод сравнения только контролирует потенциал, а ток не проводит. Вспомогательным электродом может служить просто платиновая или серебряная проволока или большой слой ртути. Поскольку электрод сравнения не проводит ток, он может иметь любую удобную форму.
Другое преимущество трехэлектроднрй ячейки состоит в том, что она позволяет проводить измерения не только в обычных водных растворах с низким сопротивлением, но и в растворах с высоким сопротивлением в неводных или сме-
332 Глава 16
Рис. 16-1. Двух- и трехэлектродные ячейки для вольтамперометрии и полярографии (РЭ — рабочий электрод, ВЭ — вспомогательный электрод, ЭС — электрод сравнения).
шанных средах. В этих случаях кончик электрода сравнения (или солевой мостик, ведущий к нему) необходимо помещать как можно ближе к рабочему электроду, так чтобы он не реагировал на возможные изменения падения напряжения в растворе.
На рис. 16-1 сравниваются двух- и трехэлектродные ячейки. Источник поляризующего напряжения и микроамперметр последовательно соединены с токопроводящими электродами. Действительную величину потенциала рабочего электрода измеряют с помощью подходящего электронного вольтметра.
+ 4 Область катодного восстановления на микроэлектроде
-4 - Область анодного окисления на микроэлектроде ________I_____I_____I____I_____1____I_____1 I I
+1,5 +1,0 +0,5 0 -0,5 -1,0 -1,5 -2,0 -2,5 -3,0
Наложенный потенциал (отн. НКЭ), В
Рис. 16-2. Принятая форма изображения вольтамперограмм (символами Hg и Pt у кривых обозначены примерные предельные потенциалы поляризации этих электродов).
Вольтамперометрия, полярография и родственные методы 333
В качестве рабочего электрода в вольтамперометрии используют инертный электрод, способный реагировать на присутствие в растворе любого электроактивного вещества. Выбор электрода определяется главным образом интервалом изучаемых потенциалов. Для потенциалов более положительных, чем потенциал НКЭ, лучшим является платиновый электрод. Ртуть можно использовать при потенциалах более отрицательных, чем примерно +0,25 В относительно НКЭ. Применение платины в области положительных потенциалов ограничивается реакцией окисления воды (2Н2О->О2 + 4Н+ + 4е), протекающей примерно при +0,65 В в зависимости от pH. В области же отрицательных потенциалов платину можно использовать только примерно до —0,45 В, т. е. до потенциала выделения водорода (2Н+ + 2е-э-Нг или 2Н2О + 2е-+Н2 + 2ОН-), тогда как ртуть из-за высокого перенапряжения водорода можно применять до —1,8 В в кислых и до —2,3 В в щелочных средах. На рис. 16-2 изображены граничные потенциалы, а также указаны соглашение о знаках катодного и анодного токов и способ изображения вольтамперных кривых.
Диффузионный ток
В неперемешиваемый раствор, содержащий 0,1 М КС1 и 10-3 М РЬСК, из которого предварительно удален растворенный кислород, поместим инертный рабочий микроэлектрод, а также проволочный серебряный вспомогательный электрод и насыщенный каломельный электрод сравнения. Допустим, что мы замкнем внешнюю цепь и резко подадим на рабочий электрод напряжение —1,0 В относительно НКЭ (рис. 16-1, б). Этот потенциал вполне достаточен для восстановления ионов РЬ2+ до металла, но недостаточен для восстановления воды при pH 7. Для восстановления ионов К+ необходим гораздо более высокий потенциал, и поэтому они не восстанавливаются. На вспомогательном электроде (аноде) серебро в количестве, эквивалентном свинцу, превращается в AgCl, но на рабочий электрод этот процесс не влияет.
Под действием электрического поля ионы РЬ2+ и К+ будут перемещаться к микроэлектроду, а ионы С1" — в противоположном направлении. Ионы К+, не способные восстанавливаться, образуют вокруг электрода слой (толщиной в 1—2 ионных радиуса), и в результате поле будет полностью нейтрализовано. Ионы РЬ2+ не вносят вклада в образование этого слоя, поскольку они восстанавливаются; каждый приближающийся к электроду ион свинца сразу же разряжается и выделяется на его поверхности. Но, поскольку поле нейтрализовано ионами К+, ионы свинца могут приблизиться к электроду только в ре
334 Глава 16
зультате диффузии. (Для выполнения принципа электронейтральности количество ионов С1~, диффундирующих в противоположном направлении, должно уменьшиться до уровня, соответствующего восстановлению ионов свинца.)
Скорость движения любой частицы вследствие диффузии пропорциональна градиенту концентрации в любых двух точках, удаленных друг от друга на расстояние х. Математически это выражается уравнением
dN/di-= —D(dCldx) (16-1)
где dN/dt—число молей, подходящих к электроду в единицу времени, С —концентрация диффундирующей частицы и D — коэффициент пропорциональности, называемый коэффициентом диффузии. (Отрицательный знак указывает на диффузию в область более низких концентраций.) Это выражение называют первым законом Фика [1].
Применение закона Фика к проблемам электролиза приводит к уравнению Коттреля
I = nFA(D/nt)'/2C (16-2)
Здесь 7 —диффузионный ток электролиза (мкА), протекающий за время t (с) с момента начала электролиза; п — число электронов, участвующих в электродной реакции; F — константа Фарадея (96 487 Кл на эквивалент); А — площадь поверхности электрода (см2); D — коэффициент диффузии Фика (см2/с); С — общая концентрация электроактивной частицы (ммоль/л). Предполагается, что концентрация на поверхности электрода равна нулю. Ток принято считать положительным для процесса восстановления на катоде и отрицательным для процесса окисления на аноде. Важно отметить наличие пропорциональности между током и концентрацией, однако для аналитических работ * это уравнение неудобно, поскольку ток уменьшается пропорционально корню квадратному из времени, а не достигает постоянной величины. В следующем разделе будет дано более удобное уравнение.
Капающий ртутный электрод
Наиболее широко используемый ртутный микроэлектрод имеет вид непрерывного потока мельчайших капелек, вытекающих из очень тонкого стеклянного капилляра. Он обладает рядом важ-
* Метод анализа, основанный на изучении зависимостей ток — время при постоянном потенциале и на прямом использовании уравнения Коттреля, называется хроноамперометрией. Он применяется при изучении кинетики переноса электронов в необратимых системах.
Вольтамперометрия, полярография и родственные методы 335
Рис. 16-3. Трехэлектродная полярографическая ячейка. Электродом сравнения может служить электрод, прилагаемый к рН-метру. (РКЭ — капающий ртутиый электрод, ЭС — электрод сравнения, ВЭ — вспомогательный электрод).
электрод сравнения и плати-вспомогательного электрода
ных преимуществ по сравнению с твердыми микроэлектродами, компенсирующих неудобства работы со ртутью,— высоким перенапряжением восстановления водорода на ртутном катоде и непрерывным возобновлением поверхности электрода, что предотвращает ее загрязнение и отравление. Кроме того, увеличение площади поверхности капающего ртутного электрода (РКЭ) за время жизни каждой капли намного превышает уменьшение тока, предсказываемое уравнением Коттреля для электрода с фиксированной поверхностью, что, как мы скоро увидим, более удобно для количественного анализа.
Простейшая трехэлектродная полярографическая ячейка представляет собой просто небольшой сосуд (100—150 мл) с анализируемым раствором. В этот раствор погружают кончик РКЭ, новую проволочку в качестве
(рис. 16-3). РКЭ должен быть укреплен в строго вертикальном положении, чтобы из него вытекали одинаковые капли ртути. Для удаления растворенного кислорода вставляют трубку, через которую поступает инертный газ. Ячейки серийного изготовления обычно снабжены пластмассовыми крышками с отверстиями различных размеров.
Более старые двухэлектродные приборы обычно имеют ячейки Н-образной формы, где НКЭ с низким сопротивлением вмонтирован в одно колено, а РКЭ помещен в другое колено. Соединение между ними заполнено содержащим КС1 агар-ага-ровым гелем, который удерживается стеклянной пористой перегородкой.
Капилляр для капающего электрода изготавливают из стеклянной трубки с внутренним диаметром 0,03—0,05 мм длиной в несколько сантиметров. Он требует очень осторожного обращения, так как при попадании в него водных растворов очистить его практически невозможно. После использования капилляр нужно вынуть из ячейки и промыть дистиллированной водой, не прерывая вытекания из него капель ртути. Затем его
336 Глава 16
Рис. 16-4. Характерные для капающего ртутного электрода кривые ток — время, показывающие, что ток возрастает во времени в степени >/6.
нужно высушить на воздухе, предохраняя от пыли, и уменьшить высоту столбика ртути, чтобы она перестала капать. Не следует оставлять капилляр погруженным в воду.
Чтобы вывести уравнение для РКЭ, соответствующее уравнению Коттреля для стационарного электрода, предположим, что скорость вытекания ртути постоянна и что капля имеет правильную сферическую форму вплоть до момента отрыва от капилляра. На основании этих предположений можно прийти к уравнению Ильковича
Id = 708,2нО1/2Л1/еС (16-3)
где Ц — диффузионный ток, m — скорость вытекания ртути (мг/с). В числовой множитель входят геометрические факторы, константа Фарадея и плотность ртути. Если зависимость тока от времени изобразить графически, то наблюдаются флуктуации тока с периодом в несколько секунд, как показано на рис. 16-4. Период капания можно варьировать, изменяя высоту столбика ртути, или электромеханически, стряхивая капли с кончика капилляра через равномерные промежутки времени. Естественное время жизни капли слабо зависит от величины наложенного потенциала, достигая максимума при потенциале около —0,5 В относительно НКЭ*.
* При этом потенциале, известном как потенциал электрокапиллярного максимума, поверхность раздела ртуть — вода имеет максимальное поверхностное натяжение. Предполагается, что при погружении в воду ртутный электрод принимает потенциал электрокапиллярного максимума.
Вольтамперометрия, полярография и родственные методы 337
Хотя температура в явном виде не входит в уравнение Ильковича, тем не менее это очень важный параметр, так как каждый фактор (кроме п) в этом уравнении в какой-то степени зависит от температуры. Влияние температуры проявляется главным образом через коэффициент диффузии D. При повышении температуры на один градус в интервале комнатных температур диффузионный ток возрастает на 1—2 процента. Поэтому при точных измерениях температуру необходимо контролировать с точностью до нескольких десятых градуса. Для качественных же и полуколичественных работ это не обязательно.
Диффузионный ток зависит также от концентрации фонового электролита; если концентрация последнего в 25 или 30 раз ниже концентрации восстанавливающегося вещества, величина диффузионного тока отличается от его нормальной величины, потому что в этих условиях восстанавливающиеся ионы переносят заметную долю тока и в результате кулоновских взаимодействий ионов с РКЭ возникает миграционный ток. Поэтому при понижении концентрации фонового электролита величина диффузионного тока слегка увеличивается для катионов (кулоновское притяжение), уменьшается для анионов (кулоновское отталкивание) и остается прежней для незаряженных восстанавливающихся частиц. В этой ситуации уравнение Ильковича не выполняется, поскольку диффузия уже не является единственным способом массопереноса восстанавливающихся частиц к электроду.
Полярография с разверткой потенциала
Предыдущий раздел посвящен обсуждению кривых ток — время, полученных при постоянном потенциале. В практической полярографии, однако, более полезную информацию можно получить, регистрируя величину тока как функцию изменяющегося потенциала РКЭ. Такие кривые автоматически записываются на полярографе.
В простейшем полярографе напряжение, подаваемое на ячейку, непрерывно увеличивают обычно в сторону отрицательных потенциалов (т. е. потенциал РКЭ становится отрицательным относительно НКЭ) и регистрируют ток с помощью самописца. Поскольку потенциал во времени меняется линейно, а лента самописца движется с постоянной скоростью, получаемую кривую (полярограмму) можно интерпретировать как истинную зависимость силы тока от потенциала.
Рассмотрим ячейку (рис. 16-3), состоящую из-РКЭ, НКЭ и платинового вспомогательного электрода, погруженных в 0,001 М раствор Cd2+ + 0,l М КС1, из которого удален
338 Глава 16
Рис. 16-5. Сравнение полярограмм 1 • 10~3 М Cd2+ на фоне 1 М KNOs, полученных без размыкания (а) и с размыканием (б) цепи измерения тока. Для наглядности кривая б смещена вверх.
растворенный кислород. Полярограмма, полученная описанным выше способом, имеет вид кривой а на рис. 16-5. Серия глубоких впадин на этой кривой, обусловленная периодическим падением капель ртути, осложняет интерпретацию кривых ток — потенциал. Для их уменьшения разработан ряд инструментальных методов, один из которых основан на использовании электронной стробирующей системы, замыкающей измерительную цепь только на долю секунды незадолго до отрыва каждой капли («стробированная» или «таст»-полярография). Это позволяет получить кривые более удобной формы (кривая б на рис. 16-5).
Для изучения явлений, происходящих в ячейке, удобно использовать идеализированную кривую, изображенную на рис. 16-6. Восходящую часть этой кривой обычно называют «полярографической волной». Саму кривую можно разделить на три участка. На участке А потенциал электрода слишком низок для восстановления одного из присутствующих веществ.
Вольтамперометрия, полярография и родственные методы 339
Рис. 16-6. Идеализированная полярограмма 1 • 10~3 М Cd2+ на фоне 0,1 М КС1.
Небольшой ток, протекающий через ячейку, называется остаточным током. Он складывается из тока восстановления следов примесей (обычно кислорода) и так называемого тока заряжения, обусловленного тем, что поверхность раздела ртуть — раствор с оболочкой из невосстанавливающихся ионов ведет себя как конденсатор. По мере увеличения потенциала электрода в процессе регистрации полярограммы конденсатор приобретает все больший заряд, что и вызывает протекание тока. Остаточный ток невелик и при тщательном удалении кислорода воспроизводим, поэтому несложно ввести поправку на его величину.
По мере приближения к участку В ток начинает возрастать и становится больше остаточного тока. Этот дополнительный ток обусловлен восстановлением ионов Cd2+ из раствора и выделением их в виде металлического кадмия на поверхности электрода. Убыль ионов Cd2+ в приэлектродном слое компенсируется за счет диффузии из глубины раствора.
На участке С ток достигает предельной величины вследствие полного исчезновения ионов Cd2+ вблизи поверхности РКЭ. В результате диффузии все больше ионов Cd2+ непрерывно достигают поверхности электрода, и по достижении устойчивого равновесия начинает протекать постоянный ток. Скорость диффузии определяется исключительно разницей между концентрацией в массе раствора и на поверхности электрода, где она равна нулю. Эта величина предельного тока, за вычетом остаточного тока, называется диффузионным током (Id). Диффузионный ток пропорционален концентрации восстанавливающейся частицы в массе раствора.
340 Глава 16
При потенциалах более отрицательных, чем на участке С, ток медленно возрастает (этот участок параллелен линии остаточного тока) почти до —2В, где он вновь резко увеличивается вследствие восстановления какого-то другого компонента раствора, возможно иона К+ или Н+.
Форма полярографической волны
Уравнение, описывающее зависимость тока от потенциала, для обратимой реакции на РКЭ можно получить на основе уравнения Нернста. Мы сделаем этот вывод для важного специального случая восстановления простого катиона до растворимого в ртути металла. Поскольку полярографически почти всегда изучают очень разбавленные растворы, коэффициенты активности принимают равными единице. Это допущение справедливо и для коэффициентов активности металлов в амальгаме.
Полуреакцию можно записать в таком виде:
Mn++ne-^MKg
где символом Мнй обозначен растворенный в ртути металл. В соответствии с уравнением Нернста потенциал электрода равен
£РКЭ = E°-RTInF In ([М]не/[Мл+]1ч) (16-4)
(верхний индекс s относится к условиям на поверхности ртути). Поскольку величина тока ограничена диффузией *, можно записать, что
I = k (lMn+Jaq-[M"+]Saq) (16-5)
где £=708 nD^rrPW16. На участке предельного тока Л величина [Mn+]aq® становится очень малой, и тогда можно записать, что
Id = k[Mn+]aq (16-6)
и поэтому
/=/<Z-fc[Mn+]Saq (16-7)
Концентрация металла М в амальгаме пропорциональна току, так что
/ = ^[M]Hg (16-8)
* В данном обсуждении предполагается введение поправки на величину остаточного тока холостого опыта.
Вольтамперометрия, полярография и родственные методы 341
Константа k' отличается от k только тем, что здесь вместо коэффициента диффузии D взят коэффициент диффузии металла D' в амальгаме, и поэтому
k/k' = (D/D')1/2 (16-9)
Если величины [М]нй и [Mn+]aqs из уравнений (16-7) и (16-8) и отношение коэффициентов из уравнения (16-9) подставить в уравнение (16-4), то получим
ЕРКЭ = Е° —RT/2nF In (D/D')—RT/nF In [//(Id—1)[ (16-10) f
В точке, где I = /<г/2, последний член равен нулю, и потенциал, называемый потенциалом полуволны, равен
E42 = E°—RT/2nF\n (DID') (16-11)
Поэтому можно записать, что
Еркэ = Е1/2—RT/nF In [1/(1 d—/)] (16-12)
Потенциал полуволны, легко измеримая величина, как показывает уравнение (16-11) связанная простым соотношением со стандартным потенциалом Е°. В отсутствие комплексообразующих веществ различие между D и D' обычно не очень велико, так что Е1/2 мало отличается от Е°.
Уравнение (16-12) описывает форму полярографической волны через параметры Id и Ти.г, что дает удобный способ определения числа электронов п. Наиболее простой способ нахождения п состоит в построении графика зависимости lg[7/(/d—/)] от ЕрКЭ> как показано на рис. 16-7. Согласно уравнению, это будет прямая с наклоном 2,303 RT/nF или 0,0591/п при 25°С. Точка на этой прямой, соответствующая / = 7,^/2, дает правильную величину Е
Рассмотренный вывод относится к восстановлению простого гидратированного иона. В присутствии комплексообразующих веществ £1/2 смещен к более отрицательным потенциалам согласно уравнению
£1/2 = Е°—RT/nF In р—pRT/nF In [X] (16-13)
где р — константа устойчивости комплекса, [X]—концентрация лиганда, р — число молей X, связанных с одним молем металла М. (Это уравнение выведено в предположении, что коэффициенты диффузии простого и комплексного ионов очень близки). В случае восстановления комплексов уравнение (16-12 существенно не меняется, хотя величина Id для данной концентрации может слегка измениться.
Выше обсуждалось катодное восстановление на РКЭ, так
342 Глава 16
Рис. 16-7. График зависимости —£рдэ от lg[//(/d—/)] для восстановления РЬ2+ на фоне: а — 0,1 М NaOH; б—1М К2С2О4 (оксалат) с pH 9,0; в — 0,5 М Na2C4H4O6 (тартрат) с pH 5,5; г—1 М К.С1; д—1 М KNO3. Средний наклон 29,8± 1,7 мВ. Точки пересечения вертикальной нулевой линии с экспериментальными кривыми дают величины соответствующих потенциалов полуволн [2].
как именно оно встречается чаще всего. Но все сказанное справедливо и для процессов окисления на РКЭ.
Для случая восстановления до нерастворимого в ртути металла (например, Fe или Сг) приведенные выше уравнения необходимо модифицировать, но это не столь важно, поскольку все известные случаи представляют собой необратимое восстановление, а к нему уравнение Нернста неприменимо.
Другой важный процесс заключается в восстановлении (или окислении) иона с образованием новой частицы, растворимой в воде, например восстановление Fe3+ до Fe2+. Если в растворе одновременно присутствуют и окисленная (Аох), и восстановленная (Ared) формы полуреакции типа Лох + пе^Лге(ь потенциал РКЭ выражается уравнением
ДРКЭ=Д1/2-ДТ/пЕ1п[(/-/ад)/(/ад-/)] (16-14)
В котором IdW — анодный диффузионный ТОК окисления Ared, ld{k) — катодный диффузионный ток восстановления Аох. Для
Вольтамперометрия, полярография и родственные методы
343
потенциала полуволны можно записать, что
Ец2 = E° — RT/2tlF 1П (Dox/^red) (16-15)
Так как эти коэффициенты диффузии обычно очень близки, стандартный потенциал окислительно-восстановительной системы Е° практически равен ^i/г. Если одна из форм (Аох или Ared) в растворе отсутствует, то соответствующий диффузионный ток равен нулю, но величина Еу2 не изменяется. На рис. 16-8 это показано для системы Fe(III)—Fe(II) [2]. Кривая а соответствует восстановлению Fe(III), кривая б — окислению Fe(II), а кривая в описывает процессы, происходящие в растворе, содержащем почти одинаковые количества Fe(III) и Fe(II). Вертикальной линией отмечены величины fi/2, которые для этой обратимой системы
+0,05 —0Д5
£ркз (отн,НКЭ),В
Рнс. 16-8. Полярограммы: а — 1,4 мМ Fe(III); 6 — 0,7 мМ Fe(II) и 0,7мМ Fe(III); в — 1,4 мМ Fe(II) на фоне насыщенного раствора щавелевой кислоты, содержащего 0,0002 % метилового красного для подавления максимумов [2].
идентичны во всех трех случаях.
Если раствор содержит две или более способные восстанавливаться частицы, то на полярограмме (рис. 16-9) наблюдаются волны для каждой из них. На этом рисунке представлена полярограмма смеси пяти совершенно различных по свойствам катионов: 1) Ag+ восстанавливается очень легко, и его
волну полностью записать невозможно, хотя можно получить четко выраженную площадку предельного тока; 2) Т1+ восстанавливается обратимо, присоединяя один электрон (п=1); 3) Cd2+ также восстанавливается обратимо, но наклон волны указывает на участие двух электронов (ц = 2); 4) Ni2+ образует более пологую волну, чем кадмий, хотя п = 2, что указывает на необратимость процесса восстановления; 5) волна Zn2+ сходна с обратимой волной Cd2+, соответствующей присоединению двух электронов (и = 2).
Максимумы. Иногда после достижения вершины полярографической волны ток вместо того, чтобы оставаться постоянным, несколько снижается, образуя максимум на кривой. Он может
344 Глава 16
Рис. 16-9. .Полярограмма раствора, содержащего по 0,1 М Ag+, Т1+, Cd2+, Ni2+ и Zn2+ на фойе 1 М NH3 и 1 М NH4C1 в присутствии 0,002 % тритона Х-100 (а) и полярограмма фона (б) [2].
иметь вид небольшого горба * или острого пика **, превышающего истинную высоту волны в два или более раз. Это явление обусловлено перемешиванием раствора вблизи растущей капли ртути.
Обычно максимумы можно устранить, добавляя органические поверхностно-активные вещества типа желатины или некоторых красителей. Для подавления максимумов часто используется неионный детергент тритон Х-100 в виде 0,2%-ного раствора, который обычно добавляют в количестве 0,1 мл к 10 мл исследуемого раствора, находящегося в полярографической ячейке. Не следует добавлять слишком много поверхностно-активного вещества, так как в этом случае возможно искажение или даже подавление волны вместе с максимумом и сдвиг потенциала полуволны на несколько милливольт по сравнению с нормальной величиной.
* Максимум 11 рода, образующийся иа площадке предельного тока.— Прим, перед.
** Максимум I рода, образующийся до достижения предельного тока.— Прим, перед.
Вольтамперометрия, полярография и родственные методы 345
Рис. 16-10. Полярограммы 0,1 М КС1 в растворе, насыщенном воздухом, где видны две волны восстановления кислорода (а), и в растворе, из которого растворенный кислород удален частично (б) и полностью (в) [2].
Влияние кислорода. Растворенный кислород восстанавливается на РКЭ во многих средах. Пример влияния кислорода на полярограмму приведен на рис. 16-10. На этом рисунке видны две волны: первая (£1/2 =—0,05 В относительно НКЭ) соответствует восстановлению кислорода из степени окисления 0 до —1 (как в пероксиде водорода), вторая (£1/2=—0,9 В) — восстановлению из степени окисления 0 до —2 (как в воде). В отсутствие поверхностно-активных веществ, а также в разбавленных фоновых электролитах на первой волне часто возникает острый максимум. Эта волна пригодна в аналитических целях для определения растворенного кислорода, но гораздо чаще она мешает определению. Поэтому при проведении большинства полярографических работ необходимо удалять растворенный кислород. В щелочных растворах для этого часто добавляют небольшое количество сульфита калия, количественно восстанавливающего кислород. Чтобы удалить кислород из любого раствора, через него нужно продуть инертный газ, например азот; такой процесс называют разбрызгиванием. Пни использовании стеклянной трубки с оттянутым концом он занимает 20—30 мин; заменив трубку на пористый стеклянный диспергатор, время продувания можно сократить до 2—3 мин. На время регистрации полярограммы пропускание газа необхо-
346 Глава 16
Рис. 16-11. Трехэлектродный полярограф па операционных усилителях.
димо прекратить, так как перемешивание раствора нежелательно. РКЭ не следует вводить в раствор, пока не удален растворенный кислород, так как ртуть может окислиться.
Аппаратура. Трехэлектродный полярограф легко собрать из стандартных электронных узлов в соответствии со схемой на рис. 16-11. Для этого требуется три операционных усилителя (они кратко описаны в гл. 3 в связи с фотодетекторами и более подробно обсуждаются в гл. 27). Линейную развертку сканируемого напряжения, подаваемого на РКЭ, можно получить с помощью одного усилителя (усилитель 1 на рисунке) и конденсатора, присоединенного к его выходу и входу. Если величины Е\, Ri и Ci постоянны, то выходное напряжение Е2 определяют по формуле
-^-ГЛ =------
/?1С1 j) RiCi
(16-16)
Из нее следует, что напряжение Е2 линейно увеличивается во времени, а наклон этой зависимости определяется константами. Поскольку в формуле стоит знак минус, то при отрицательном Ei напряжение Е2 изменяется в направлении положительных величин; это и составляет функцию развертки.
Соответствующую часть напряжения (определяемую величинами R2 и R3) подают на вход В усилителя 2 и таким образом устанавливают на нем потенциал развертки относительно земли. Единственная функция усилителя 2 состоит в пропускании тока через выход к вспомогательному электроду. Через НКЭ этот ток протекать не может, поскольку НКЭ присоединен только к входу усилителя. Поэтому ток должен протекать через РКЭ и резистор Ri. Усилитель 3 вынужден пропускать лишь ток, достаточный для уравнивания тока, протекающего через ячейку, чтобы обеспечить равенство потенциалов на его
Вольтамперометрия, полярография и родственные методы 347
входах, вследствие чего РКЭ по существу все время находится при потенциале земли. По аналогичной причине НКЭ всегда находится при потенциале развертки относительно земли. В результате усилитель 2 пропускает только ток, необходимый для поддержания разности потенциалов НКЭ и РКЭ при заданном линейном увеличении напряжения, причем РКЭ отрицателен относительно НКЭ. Поэтому самописец регистрирует истинную полярограмму. Конденсатор С2 служит для уменьшения в цепи высокочастотного электрического шума. Резистор Ri связан со шкалой самописца. Переключатель S, замыкающий интегрирующий конденсатор, используют для запуска измерения; момент открытия переключателя соответствует нулевому времени. Переключатель диапазонов и сброс пуля на рисунке для ясности опущены.
Измерительные цепи. Как уже упоминалось, одно из слагаемых остаточного тока обусловлено заряжением конденсатора, возникающего на границе раздела электрод—раствор. По мере того как абсолютная величина отрицательного заряда РКЭ в процессе сканирования потенциала увеличивается, все больше невосстанавливающихся катионов втягивается в облако, окружающее электрод. Движение этих ионов и равного числа электронов внутри металла составляет ток переноса, часто называемый током заряжения двойного слоя или емкостным током. Как и для любого конденсатора, он может протекать лишь при изменении потенциала и при раздвижении «обкладок» конденсатора или изменении их поверхности. В случае РКЭ расстояние между обкладками определяется толщиной двойного слоя (ионы в облаке и электроны на поверхности справедливо уравнение
1С = К (dA /dt) + dE/dt (16-17)
где в константу К входит диэлектрическая проницаемость и расстояние между обкладками, А—площадь поверхности электрода. Можно рассмотреть два случая: 1) изменение потенциала dEjdt (развертка) происходит достаточно медленно, чтобы им можно было пренебречь в течение периода жизни капли, и 2) измерение производят вблизи конца жизни капли, когда скорость изменения поверхности (dAjdt} минимальна.
Математически можно показать, что, в то время как фарадеевский ток растет во времени в степени 1/6, начиная с нуля при появлении новой капли, ток заряжения, относительно большой в начале жизни капли, затем уменьшается, согласно уравнению со степенным показателем —*/з- Отсюда следует (рис. 16-12), что в момент, близкий к отрыву капли, когда фарадеевский ток достигает максимума, влияние тока заряжения
348 Глава 16
Рис. 16-12. Рост фарадеевского тока и одновременное снижение емкостного тока (тока заряжения) за время жизни капли ртути. Вертикальными штриховыми линиями отмечен оптимальный интервал времени, необходимый для измерения тока.
уменьшается. Очень эффективный способ увеличения соотношения сигнал/шум основан на использовании стробирующей или синхронизирующей системы, позволяющей измерять ток только в конце жизни капли.
Стробирующую систему можно синхронизировать с РКЭ, либо детектируя отрыв капли с помощью электронного сенсора, либо стряхивая капли в заданный момент времени. Например, в соответствии с рис. 16-12 временную цепь можно сконструировать так, чтобы стряхивать капли через интервалы в три секунды, а самописец подключать только в интервале от 2,4 до 2,9 с. В течение 2,5 с, когда ток не измеряют, самописец будет просто чертить горизонтальный прямолинейный участок. Как видно из рис. 16-5 (кривая б), получающуюся кривую обработать для количественных расчетов гораздо легче, чем немоди-фицированную полярограмму.
Высокоскоростная полярография. Скорость наложения поляризующего напряжения можно сделать достаточно большой, чтобы записать всю полярограмму в конце второй половины жизни капли. От обычной полярограммы полученная кривая будет отличаться характерной пикообразной формой (рис. 16-13). Причина образования волны такой формы заключается в том, что при быстрой развертке потенциала массоперенос восстанавливающегося вещества к электроду за счет диффузии происходит очень медленно и устойчивое состояние никогда не
Вольтамперометрия, полярография и родственные методы 349
достигается. Потенциал максимума Ем связан с величиной Е1/2 соотношением
EM = Ei/2^l,lRT/nF (16-18)
или при 25 °C
Ем = Е1/2—0,028/п вольт (16-19)
Величина тока максимума для обратимой системы описывается уравнением Рэндлса-Шевчика, которое подобно уравнении? Ильковича, но включает скорость развертки потенциала
-Е, мВ
Рис. 16-13. Типичная высокоскоростная полярограмма. Показано соотношение между потенциалом полуволны и потенциалом пика.
/м = W/2m2/3/2/3Z)1/2 (dEldt^C (16-20) .
На рис. 16-14 приведена высокоскоростная полярограмма раствора, содержащего два восстанавливающихся вещества. Второе вещество определить количественно с высокой точ
Рис. 16-14. Высокоскоростная полярограмма смеси двух восстанавливающихся веществ. Остаточный ток при измерении тока второго пика необходимо экстраполировать по линии спада тока первого пика.
ностью невозможно, поскольку не-обходимо экстраполировать начальный участок для вычитания остаточного тока.
Циклическая вольтамперометрия
Это разновидность высокоскоростного метода, в которой направление развертки потенциала после достижения потенциала восстановления изучаемого вещества меняется на обратное. Для этого вместо поляризующего напряжения с линейной разверткой на ячейку подают поляризующее напряжение треугольной формы (рис. 16-15, а). Типичная кривая, получаемая при таком способе поляризации электрода, показана на рис. 16-15, б [3]. Весь процесс происходит во второй половине или даже ближе к концу жизни капли ртути. В начальный момент развертки (вблизи точки
350 Глава 16
Рис. 16-15. Циклическая вольтамперометрия: а — треугольная развертка поляризующего напряжения; б — циклическая вольтамперограмма 10~3 М Zn2+ на фоне 2 М NH3/NH4C1 [3].
А) через ячейку протекает только остаточный ток, пока потенциал не станет достаточно отрицательным, чтобы произошло восстановление какого-либо из присутствующих в растворе веществ. В данном примере в растворе находятся ионы цинка, и поэтому получится точно такая же кривая с максимумом, как показано на рис. 16-13. В точке D направление развертки меняют на обратное, и потенциал электрода начинает уменьшаться с той же самой скоростью, с какой он увеличивался при развертке в прямом направлении. Внезапное падение (от D до Е) вызвано обращением емкостного тока. В интервале потенциалов от Е до F протекает еще катодный ток, т. е. продолжается процесс восстановления ионов цинка из приэлект-родного слоя, как и в интервале потенциалов от точки В до D. Как только потенциал достигнет точки F, наступает устойчивое состояние, контролируемое диффузией, и ток снижается до величины, соответствующей площадке G—Н, по сути дела достигая величины остаточного тока. В интервале потенциалов Н—I—I окисляется металлический цинк, образующийся в предыдущей реакции восстановления, в результате чего протекает контролируемый диффузией анодный ток. В точке / направление развертки потенциала снова меняется на обратное. Ток вновь резко увеличивается от J до К, фактически возвращаясь к исходной величине.
Вольтамперометрия, полярография и родственные методы 351
То, что ток в точке / не равен току в точке С, можно отчасти объяснить неполным удалением металлического цинка из капли ртути и отчасти неравенством коэффициентов диффузии ионов, предположительно [Zn(NHs)4]2+, в водном растворе и атомов цинка в амальгаме. Ясно, что градиенты концентраций, определяющие диффузию этих частиц, также сильно различаются.
Рассмотренная в качестве примера электродная реакция, скорее всего, необратима. На это указывает большая разность (около 0,6 В) потенциалов катодного и анодного пиков; для обратимого процесса при п~2 эта разность при 25 °C должна составлять 0,028 В [см. уравнение (16-19)]. На этом основан другой удобный способ определения обратимости электродного процесса.
Циклическая вольтамперометрия очень ценный способ обнаружения числа присутствующих в растворе электроактивных веществ, одновременно пригодный для установления обратимости электродного процесса. Этот метод применим для изучения кинетики электродных процессов и определения устойчивости присутствующих веществ. В количественном анализе он используется не очень часто.
Дифференциальная импульсная полярография
В этой разновидности полярографии, которая нашла гораздо более широкое распространение, чем классическая постояннотоковая полярография, на линейно увеличивающееся напряжение подают серию кратковременных импульсов напряжения, как показано на рис. 16-16. Продолжительность и амплитуда импульса могут быть разными, но разумно подавать импульсы
Рис. 16-16. Развертка потенциала в методе дифференциальной импульсной полярографии: а — последовательность импульсов, налагаемых на линейно увеличивающееся напряжение; б — детальное представление одного импульса; ta — период капания, 6t — продолжительность импульса.
352 Глава 16
с амплитудой 25 мВ и продолжительностью 50 мс и повторять их на каждую каплю ртути, например каждые 2 с. Протекающий через ячейку ток измеряют, скажем, в течение 15 мс до начала и в конце наложения каждого импульса и разницу фиксируют с помощью самописца. Этот прием позволяет эффективно вычитать емкостный ток и измерять прирост только фарадеевского тока, вызванный изменением потенциала между точками А и В, т. е. прирост тока, приходящийся на ампли
туду импульса дВ (рис. 16-16, б).
Вычитание тока заряжения позволяет заметно улучшить чувствительность. В благоприятных условиях можно с приемлемой точностью определять до 10-8 М
Потенциал (отн, НКЭ), в
Рис. 16-17. Дифференциальные импульсные полирограммы 2-этилаитра-хиноиа на фойе 0,1 М раствора L1C1 в 50 %-ном этаноле. Высота пика почти пропорциональна концентрации (Е. G. & G. Princeton Applied Research Corporation).
Рис. 16-18. Полярограммы 1 мкг/мл Pb2+ и 1 мкг/мл Cd2+ на фоне 0,1 М раствора HNOs, зарегистрированные с помощью различных полярографических методов. Чувствительность регистрирующего устройства при измерении тока везде одна и та же (0,2 мкА на всю шкалу): 1 — классическая полярограмма; 2 — тестированная классическая полярограмма; 3 — обычнаи импульсная полярограмма; 4 — дифференциальная импульсная полярограмма (Е. G. & G. Princeton Applied Research Corporation).
Вольтамперометрия, полярография и родственные методы 353 восстанавливающегося вещества вместо 10-5 М, как в классической полярографии. Как показано на рис. 16-17, дифференциальная импульсная полярограмма имеет форму производной классической полярограммы.
В другой модификации метода — переменнотоковой полярографии— на электрод одновременно с линейно изменяющимся постоянным напряжением подают переменное напряжение малой амплитуды и возникающий переменный ток регистрируют как функцию постоянного напряжения. Этот прием позволяет снизить емкостный ток* и получить полярограмму, напоминающую по форме дифференциальную импульсную полярограмму (рис. 16-17),' с примерно той же чувствительностью, что и для обратимых систем. Необратимйе окислительно-восстановительные системы этим методом изучать нельзя.
На рис. 16-18 показаны классическая и импульсная полярограммы раствора, содержащего 1 мкг/мл свинца и кадмия. Одна из полярограмм зарегистрирована с помощью метода нормальной импульсной полярографии, в котором потенциал электрода в промежутке времени между импульсами всегда возвращается к нулю (относительно НКЭ) **. Это очень удобно в случае, когда какой-то компонент раствора может «отравить» электрод вследствие адсорбции на поверхности.
Более подробно методы импульсной и переменнотоковой полярографии изложены в монографии [1].
Качественный анализ
Поскольку потенциал полуволны служит характеристикой вещества, восстанавливающегося или окисляющегося на электроде, этот параметр полярограммы можно использовать для его идентификации. Для данного конкретного вещества величина Ец2 зависит от природы фонового электролита, главным образом из-за различной способности к комплексообразованию. В табл. 16-1 приведено несколько характерных примеров. Насколько важно правильно выбрать электролит, можно показать на примере свинца и кадмия. Эти катионы имеют практически одинаковые потенциалы полуволны в растворе NaOH, но образуют хорошо разделяющиеся волны в растворах КС1 или Н3РО4 и даже в KCN.
В учебниках и монографиях по полярографии можно найти много данных по величинам потенциалов полуволн [2, 4]. Часто, однако, целесообразнее записывать и непосредственно
* Снизить в случае квадратно-волновой переменнотоковой полярографии и отделить в случае синусоидальной переменнотоковой полярографии.— Прим, перев.
** Чаще потенциал электрода возвращают не к нулю, а к любому заданному начальному потенциалу.— Прим, перев.
12 Заказ № Б37
354 Глава 16
Таблица 16-1. Потенциалы полуволн (в вольтах относительно НКЭ) некоторых катионов в различных фоновых электролитах [2]
Катион Фоновый электролит
Kci (0,1 M) ЫНз (1 М) NH.C1 (1 М) NaOH (1 М) Н3РО, (7,3 М) KCN (1 М)
Cd2+ —0,60 —0,81 —0,78 —0,77 —1,18а
Со2+ —1,20б —1,29б —1,46е —1,20е —1,136 [до Со (I)]
Сг3+ б —1,43б [до Ст (II)] —1,716 [до Ст (0)] * • • • —1,02б [до Ст (II)] —1,38 [до Cr (II)]
Си2+ +0,04 [до Си (I)] —0,22 [до Си (0)] —0,24 [до Си (I)] —0,51 [до Си (0)] —0,416 —0,09 Н. в.г
Fe2+ —— 1,Зб —1,49е
Fe3+ — 1,1б —1,10е —1,12Д [до Fe (II)] — 1,74« [до Fe (0)] +0,06 [до Fe (II)]
Ni2+ —1,18 — 1,36
Pb2+ —0,40 —0,76 —0,53 —0,72
Zn2+ —1,00 —1,35° —1,53 — 1,13б Н. в.
а Из работы Мейтиса. Необратимое восстановление. ® Недостаточная растворимость или отсутствие информации (••). г Н. в. — в этих условиях ион не восстанавливается. д ЗМ раствор КОН +3%-ный маннит.
сравнивать полярограммы известного и изучаемого веществ. Литературные данные при отсутствии точной информации о составе фона и природе электрода сравнения мало пригодны
Рис. 16-19. Графический способ нахождения потенциала полуволны на полярограмме.
для идентификации неизвестного вещества.
Потенциал полуволны можно определить графически непосредственно из полярограммы, как показано на рис. 16-19. Для этого продолжают, как показано на рисунке, отрезки кривой АВ и DF и проводят касательную в точке перегиба кривой С. Отрезок GH делят пополам и проводят линию JK параллельно АВ и DF. Абсцисса точки пересечения линии JK
Вольтамперометрия, полярография и родственные методы 355
с кривой и дает искомую величину £1/2. Это до некоторой степени усложненная методика необходима в часто встречающемся неидеальном случае, когда, как бы этого ни хотелось, DF не параллельна АВ. Согласно описанной методике, небольшие погрешности при проведении линии GH окажут минимальное влияние на конечную величину £1/2. При оценке величины £1/2 по нетастированным полярограммам линии при геометрических построениях следует проводить по верхним зубчикам осцилляций.
Количественный анализ
Зависимость величины диффузионного тока от концентрации восстанавливающегося вещества в методе классической полярографии выражается уравнением Ильковича. В принципе по этому уравнению можно по измеренной величине тока рассчитать неизвестную концентрацию, если остальные факторы в этом уравнении известны или их можно измерить. Практически, однако, это неудобно, и поэтому предпочитают косвенные методы. В упрощенном виде уравнение Ильковича можно записать так:
Id = kC (16-21)
Константу пропорциональности можно оценить по полярограмме стандартного раствора вещества.
Допустим, например, что нужно определить кадмий в образце металлического цинка. Навеску пробы массой 0,1 г растворяют в НС1, добавляют несколько капель тритона Х-100 и разбавляют полученный раствор до известного объема 1,0 М раствором КС1. Часть раствора помещают в полярографическую ячейку, пропускают газообразный азот и записывают классическую или дифференциальную импульсную полярограмму в интервале потенциалов от —0,4 до —0,8 В относительно НКЭ. Наличие волны при потенциале около —0,64 В в этом случае служит качественным указанием на присутствие кадмия. Потенциал восстановления цинка настолько более отрицателен,. что этот процесс не искажает волну кадмия. Если кадмий присутствует, то по виду полярограммы можно уточнить условия выполнения анализа. Если раствор пробы слишком разбавлен, то можно увеличить чувствительность прибора; если же, наоборот, он слишком концентрированный, то его можно дополнительно разбавить раствором КО. Теперь для завершения анализа надо снова записать полярограмму, одновременно приготовить стандартный раствор CdCl2 в 1,0 М растворе КС1 приблизительно с тем же конечным разбавлением и записать его полярограмму. Полученные кривые сходны с при. веденными на рис. 16-20. Измерив графически величины токов 12*
356 Глава 16
Рис. 16-20. Классические полярограммы стандартного (/) и анализируемого (2) растворов, слегка смещенные по вертикали (для наглядности) .
Id для стандартного и анализируемого растворов, по простой пропорции находят искомую концентрацию. Альтернативным методом может быть построение градуировочного графика по серии стандартных растворов с известными концентрациями.
В полярографии выгодно использовать метод стандартной добавки, для чего в анализируемый раствор вводят известное количество стандартного раствора. Из сравнения полярограмм, полученных до и после введения добавки, можно найти всю необходимую для завершения анализа информацию. Преимущество
этого метода состоит в том, что в данном случае сравнивают две полярограммы, полученные в идентичных условиях. Детали метода обсуждаются в гл. 26.
При выполнении любых количественных полярографических измерений необходимо учитывать остаточный ток. Если предварительно зарегистрировать полярограмму фона, то остаточный ток можно приблизительно учесть с помощью экстраполяции, как показано на рис. 16-20. В противном случае придется проводить холостой опыт. Это фактически единственный способ определения высоты первой волны на полярограмме кислорода (рис. 16-10). При определении низких концентраций,
когда влияние остаточного тока сказывается гораздо сильнее, предусмотрены некоторые автоматические способы компенсации тока заряжения. Иногда для этого подключают устройство, которое поставляет чрезвычайно слабые токи в направлении, противоположном току, протекающему через ячейку. Этот добавочный ток, пропорциональный приложенному напряжению, следует тщательно подобрать. Такой способ не позволяет добиться полной компенсации, поскольку остаточный ток меняется не строго по линейному закону, а часто превосходит используемые в полярографии токи примерно в 10 раз.
Другое ограничение точности количественного полярографического определения возникает в случае, когда волны двух восстанавливающихся веществ образуются при слишком близких потенциалах. Если они расположены настолько близко,
что между ними нет разделяющего их горизонтального уча
Вольтамперометрия, полярография и родственные методы 357
стка, то измерить величины индивидуальных токов невозможно. В этом случае нужно заменить фоновый электролит или добавить комплексообразующий реагент. В дифференциальной импульсной полярографии вследствие пикообразной формы кривых такие трудности менее ощутимы, чем в классической.
Полярография органических соединений
Полярография служит удобным методом анализа и установления структуры органических соединений, в принципе не отличающимся от обсужденных выше. Продукты электрохимической реакции с участием органических соединений нерастворимы в ртути, но Почти всегда растворяются в том же растворителе, что и исходное вещество. Для полярографии в принципе подходит любой растворитель, в котором растворим электролит,— различные спирты или кетоны (в чистом виде или в смеси с водой)', диметилформамид, ацетонитрил, этилендиамин и другие. В качестве фоновых электролитов пригодны различные соли четвертичного аммония (например, иодид тет-рабутиламмония), легко растворимые в органических растворителях. В качестве примера на рис. 16-1-7 приведена дифференциальная импульсная полярограмма 2-этилантрахинона на фоне LiCl в 50 %-ном растворе этанола.
На РКЭ восстанавливаются разнообразные органические соединения, среди которых находятся соединения с сопряженными кратными связями, многие карбонильные соединения, галогенсодержащие соединения, хиноны, гидроксиламины; нитро-, нитрозо-, азо-, азоксисоединения; оксиды аминов, соли диазо-ния, многие серусодержащие и гетероциклические соединения; пероксиды и редуцирующие сахара. Более детальные сведения можно найти в литературе [5]. Применительно к органическим соединениям pH и ионная-сила раствора более важны, чем для неорганических соединений, так как ионы Н+ почти всегда участвуют в реакциях органических соединений.
Наглядным примером применения полярографии для органических соединений является определение ароматических карбонильных соединений [5]. На фоне -водно-этанольного раствора LiOH удается получить четкие раздельные волны с потенциалами полуволн (относительно НКЭ) для бензальдегида —1,51 В, для н-пропилфенилкетона —-1,75 В, для изопропилфе-нилкетона —1,82 В и для трет-бутилфенилкетона —1,92 В. Диффузионные токи линейно зависят от концентраций в интервале от 0,2 до 2,5 мМ.
358 Глава 16
Другие амперометрические методы
Здесь удобно обсудить ряд аналитических методов, основанных на измерении тока при выбранном потенциале. Некоторые из них иногда относят к «полярографическим», хотя РКЭ и не используется.
Кислородный электрод. Содержание молекулярного кислорода в растворе можно определить амперометрически с помощью мембранного электрода, называемого кислородным сенсором Кларка. Ячейка для измерений состоит из инертного металлического катода, покрытого тефлоновой или силиконовой газопроницаемой мембраной, и серебряного анода, присоединенных к батарее на 1,5 В (рис. 16-21). Ток протекает только в результате диффузии кислорода из анализируемого раствора через мембрану к катоду, где он восстанавливается до воды. Необходимые для этого ионы водорода потребляются из внутреннего буфера. В результате протекания незначительного тока эквивалентное количество серебра на аноде превращается в AgCL Ток пропорционален ______________-кА концентрации растворенного кислорода в интервале от 1 до 10 мг/л.
1,5 В
Рис. 16-21. Кислородный сенсор Кларка. Микроамперметр можно заменить схемой с операционным усилителем.
Амперометрическое титрование. Титрование можно осуществлять в вольтамперометрической ячейке, регистрируя изменение тока после добавления каждой порции реагента. Поскольку обычно диффузионный ток пропорционален концентрации, кривая титрования состоит из двух прямых, пересекающихся в точке эквивалентности. Можно раз-
Объем в
Рис. 16-22. Типичные кривые амперометрического титрования.
Вольтамперометрия, полярография и родственные методы 359
личить три типа кривых (рис. 16-22). Кривая а получается при титровании электроактивного вещества электронеактивным титрантом. Примером может служить титрование иона свинца в растворе KNO3 оксалат-ионом при —1,0 В (относительно НКЭ) на РКЭ. В начале титрования протекает довольно большой диффузионный ток, который по мере удаления ионов свинца из раствора в результате осаждения оксалатом снижается. Как только будет достигнута точка эквивалентности, ток становится постоянным и не меняется при дальнейшем прибавлении оксалата.
Если провести обратное титрование, т. е. в тех же условиях титровать оксалат раствором свинца, то получим кривую б на рис. 16-22. Пока не осадится весь оксалат, ионов свинца в растворе нет, поэтому ток не протекает.
Кривая в получается при титровании ионов свинца раствором бихромата при —1,0 В, т. е. в условиях, когда и РЬ2+, и Сг2О72- восстанавливаются на РКЭ.
Амперометрическое титрование применимо для многих окислительно-восстановительных реакций, реакций комплексообразования и осаждения. По сути этот метод обладает более высокой точностью, чем соответствующий прямой полярографический метод, так как каждое определение включает ряд отдельных измерений, для которых исключаются случайные ошибки.
Вращающийся платиновый электрод. . При использовании вращающегося платинового' электрода вместо РКЭ чувствительность титриметрических определений увеличивается, поскольку при перемешивании разрушается диффузионный слой. Обычно электрод представляет собой отрезок платиновой проволоки длиной 2 или 3 мм, горизонтально выступающий из вертикальной защитной стеклянной трубки. Трубка вращается со скоростью несколько сотен оборотов в минуту, причем для получения воспроизводимых результатов скорость должна быть постоянной.
Наглядным примером успешного использования вращающегося платинового электрода служит титрование мышьяка (III) раствором ВгОз- в присутствии бромид-ионов [6]. Титрование проводят на фоне 1 М HCl + 0,05 М КВг при потенциале 0,2— 0,3 В относительно НКЭ. В процессе титрования протекает реакция
3AsO7 + ВгОз” -> ЗАэОГ + Вг~
Индикатором служат бромид-ионы, поскольку они окисляются ВгО3~ только после того, как прореагирует весь As(III):
ВгОГ + 5Вг- + 6Н+ -> ЗВг2 + ЗН2О
360
Глава 16
Рис. 16-23. Кривая амперометрического титрования As (III) раствором ВгОз~ [6].
Из всех присутствующих веществ только свободный бром способен восстанавливаться при указанном потенциале электрода. На рис. 16-23 показана кривая, получающаяся при титровании 100 мл 9,18-10-4М раствора мышьяковистой кислоты 0,01000 М раствором КВгО3. При выбранном потенциале кислород не вос-
станавливается, и поэтому нет необходимости продувать инертный газ.
По числу применений
амперометрическое титрование пре-
восходит потенциометрическое титрование, поскольку электроды являются неспецифическими. Известно так много ионов и молекул, способных давать волны либо на ртутном, либо на платиновом электроде, что часто удается найти подходящий
реагент для прямого или косвенного определения почти любого вещества. Метод пригоден для точного определения низких концентраций.
Амперометрическое
методики заключается в нало-
Рис. 16-24. Избранные кривые амперометрического титрования с двумя поляризованными электродами для титрования: 12 раствором NasSjOa (a); Fe(II) раствором Се(IV) (б); V(V) раствором Fe(II) (в); Fe(CN)e4~ раствором Ce(IV) (г) [7].
титрование с двумя индикаторными электродами. Суть упрощенной жении небольшой разности потенциалов на два идентичных инертных электрода. Из оборудования требуется только источник постоянного напряжения на 50—100 мВ и гальванометр. В растворе одновременно должны присутствовать и окисленная, и восстановленная формы обратимой окислительно-восстановительной полуреакции, иначе ток протекать не будет. Например, если присутствуют и Fe2+, и Fe3+, то ток протекает, поскольку Fe2+ может окисляться на одном, a Fe3+ восстанавливаться на другом электроде. Для этих двух процессов необходимы практически равные потенциалы, по-
Вольтамперометрия, полярография и родственные методы 361 этому, чтобы вызвать протекание тока, достаточно даже нескольких милливольт.
Ток может протекать и в случае некоторых необратимых систем. Например, Н2О2 окисляется на аноде с образованием кислорода и одновременно восстанавливается на катоде с выделением ОН~-ионов. Система Мп(VII)/Мп (И) необратима, но тем не менее процесс электролиза возможен, так как Мп2+ окисляется на аноде до МпО2, а МпО4_ восстанавливается на катоде также до МпО2. В качестве примеров некоторых других электроактивных систем назовем пары 12/21-, Вг2/2Вг~, Се4+/Се3+, Fe(CN)63~/Fe(CN)64-, Ti4+/Ti3+, VO3~/VO2+.
Кривые для нескольких титрований по этому способу представлены на рис. 16-24 [7]. Одним из первых было использование титрования иода тиосульфатом (кривая а), которое широко применяется для определения воды по методу Фишера. Форму кривой титрования можно объяснить следующим образом: до точки эквивалентности в растворе присутствуют и иод, и иодид-ионы, и ток может протекать. По мере титрования иод восстанавливается до иодида, в точке эквивалентности иода не остается, и ток протекать не может. За точкой эквивалентностя, когда добавлен избыток тиосульфата, ток также не может протекать, так как система тиосульфат — тетратионат необратима.
При титровании же Fe(II) расвором Ce(IV) (кривая б) ток может протекать и до, и после точки эквивалентности, так как железо и церий образуют обратимые окислительно-восстановительные системы. Наступает момент, когда ток равен нулю (или очень близок к нему), соответствующий полному удалению ионов Fe2+ и отсутствию еще избытка Се4+.
Аналогично можно интерпретировать и другие кривые титрования. Во всех случаях конечная точка титрования выражена четко, так как ток падает практически до нуля.
Задачи
16-1. Навеску металлического цинка массой 1,000 г растворили в 50 мл НС1 и разбавили до 250 мл. В электролизер поместили 25,00 мл полученного раствора, прибавили несколько капель раствора поверхностно-активного вещества, удалили растворенный кислород и зарегистрировали полярограмму в интервале потенциалов от 0 до —1 В относительно Hg-анода. На полярограмме появилась волна с Ец2=—0,65 В высотой 7,6 см. К раствору в электролизере прибавили 5,00 мл 5-10~4 М раствора CdCl2, удалили кислород и вновь записали полярограмму. Потенциал полуволны не изменился, а высота увеличилась до 18,5 см. Рассчитайте процентное содержание примеси кадмия в металлическом цинке. При расчете учтите разбавление раствора. (Обратите внимание, что фоновым электролитом служит смесь НС1 и ZnCl2.)
16-2. Диффузионный ток на полярограмме 5-10-3 М раствора CdCl2 на фоне 0,1 М раствора КС1 при —0,8 В (относительно НКЭ) равен 50,0 мкА. Ртуть вытекает со скоростью 18 капель в минуту. Собрали 10 капель ртути и нашли, что их масса равна 3,82 • 10-2 г. а) Рассчитайте коэффициент диффузии D,
362 Глава 16
Рис. 16-25. Полярограммы купроина (а) и меди (б) на фойе аммиачно-хло-ридного буферного раствора [8].
б) Если капилляр заменить другим с периодом капания 3,0 с (масса 10 капель 4,20-10~2 г), то каким станет диффузионный ток?
16-3. Купроин (а-бензоиноксим) служит осадителем для Си (II) [8]. На фоне 0,1 М NH4Cl+0,05 М NH3 (pH 9) купроин восстанавливается иа РКЭ, образуя волну с Е^2~—1>63 В относительно НКЭ (кривая а иа рис. 16-25). В тех же условиях медь образует две волны (кривая б иа рис. 16-25). Изобразите кривые титрования меди купроином при потенциалах электрода —1,0 и —1,8 В. Какой потенциал наиболее пригоден для титрования меди в отсутствие мешающих веществ? При каком потенциале влияние восстанавливающихся примесей проявляется сильнее?
16-4. Небольшие концентрации нитрат-иона (0,1—10 мкг/мл) можно определить полярографически на фоне 0,1 М раствора цирконилхлорида (ZrOCl2) [9]. Разницу в величинах диффузионных токов до и после восстановления FeSO4 (NH4)2SO4 измеряли на РКЭ при —1,2 В относительно НКЭ. Для двух стандартных и анализируемого раствора получены следующие данные:
Концентрация раствора, мгк/мл Диффузионный ток, мкА
до восстановления п осле восстановления
10,0 87,0 22,0
5,0 48,5 15,2
Анализируемый 59,0 17,0
Рассчитайте концентрацию ннтрат-иона в анализируемом растворе.
16-5. Вычислите величины Еркэ, соответствующие токам 1, 2, 3 мкА (и т. д. до 9 мкА), для трех гипотетических катодных волн с Е^2 =—1,000 В (относительно НКЭ) и Ц= 10 мкА и с п=1, 2 и 3 соответственно (25 °C). Изобразите эти данные на одном графике так, чтобы кривые пересекались при потенциале полуволны. Для этих же данных постройте другой график в координатах lg[//(/a—/)]—Еркэ. Проанализируйте, насколько близко полученные результаты совпадают с соответствующими уравнениями в тексте.
16-6. На фоне NaC104 ион Ag+ восстанавливается на РКЭ при потенциале НКЭ. В тех же условиях С1~ образует анодную волну с Е1/2 =+0,25В (относительно НКЭ). Можно определить, восстанавливается ли комплекс AgCl2_
Вольтамперометрия, полярография и родственные методы 363
Е(отн. НКЭ}, В
Рис. 16-26. Дифференциальные импульсные полярограммы аммиака в 0,4 М ацетатном буферном растворе, содержащем 37 % формальдегида (Е. G. & G. Princeton Applied Research Corporation).
в этих условиях при амперометрическом титровании Ag+ раствором С1_ на РКЭ. Изобразите и объясните возможную форму кривой титрования.
16-7. Полярограммы, изображенные на рис. 16-5, получены с трехэлектродной ячейкой. Предположим, что полярограмму для того же раствора зарегистрировали с двухэлектродной ячейкой, у которой электрод сравнения имеет сопротивление 10 кОм. Какова погрешность (если она будет) измерения а) потенциала полуволны и б) диффузионного тока?
16-8. Уравнение Коттреля [уравнение (16-2)] можно использовать для оценки величины коэффициента диффузии. Допустим, что при следующих параметрах: п=2, объем ртутной капли 0,5 мм3, время капания 2 с, концентрация 2-Ю-4 М — измеренная величина тока равна 0,520 мкА. Рассчитайте величину D.
Формулы для расчетов: А =4 №, о = 4/3 пг3, где г — радиус.
16-9. Емкость двойного слоя у ртутиого капающего электрода может составлять несколько микрофарад. Допустим, что в данном случае она равна 5,0 мкФ при потенциале—1,0 В относительно электрокапиллярного макси
364 Глава 16
мума. а) Каков запас электронов на капле? б) Если капли падают со скоростью 1 капля в 5 с, то какой ток возникает при переносе этого количества электронов?
16-10. В ацетатном буферном растворе аммиак реагирует с формальдегидом, образуя гексаметилентетрамин, согласно уравнению 4NHs+6CH2O-»-->-(CH2)6N44-6H2O. Продукт реакции восстанавливается на РКЭ. На рис. 16-26 приведены дифференциальные импульсные полярограммы, снятые в следующих условиях: фоном служит смесь равных объемов 0,4 М ацетатного буферного раствора с pH 4 и 37 %-ного формальдегида; концентрация стандартного раствора NH4C1 30,2 мг/л деионизованной воды (т. е. 9,6 мкг/мл NH3); при снятии полярограмм к 10,0 мл фона, не содержащего растворенного кислорода, прибавляли 100-мкл порции стандартного раствора; период капания 1 с. а) Постройте градуировочный график в координатах ток пика (с поправкой на холостой опыт) — концентрация добавленного NH3 (расчет сделайте в мкг/л), б) Через порцию фонового электролита в ячейке пропустили 10 л азота из баллона (стандартные температура и давление) и зарегистрировали полярограмму. Величина исправленного тока пика 0,432 мкА. Вычислите содержание аммиака в азоте (млн-1).
16-11. Допустим, что кислородный сенсор Кларка погружен в раствор, содержащий 5 мг/л О2; протекающий в результате ток равен 10 мкА. Рассчитайте, сколько молей О2 фактически диффундирует через мембрану и восстанавливается за минуту. Какую часть это составляет от исходных 5 мг/л О2?
Литература
1. Vassos В. Н., Ewing G. W., Electroanalytical Chemistry, Wiley-Intersci-ence, New York, 1983,- p. 218.
2. Meites L., Polarographic Techniques (2 ed.), Wiley-Interscience, New York, 1965.
3. Schmidt H., von Stackelberg M., Modern Polarographic Methods, Academic Press, New York, 1963.
4. Meites L., in Handbook of Analytical Chemistry, Meites L. (ed.), McGraw-Hill, New York, 1963, p. 5—38.
5. Zaman P., Organic Polarographic Analysis, Macmillan, New York, 1964.
6. Laitinen H. A., Kolthoff I. M., J. Phys. Chem., 1941, v. 45, p. 1079.
7. Stone K. G., Scholten H. G., Anal. Chem., 1952, v. 24, p. 671.
8. Langer A., Ind. Eng. Chem.. Anal. Ed., 1942, v. 14, p, 283.
9. Rand M. C., Heukelekian H., Anal. Chem., 1953, v. 25, p. 878.
ГЛАВА I /
Электровыделение и кулонометрия
Катодное выделение переходных металлов в аналитических целях относится несомненно к старейшим электроаналитиче-ским методам. С середины девятнадцатого столетия металлы, употреблявшиеся для чеканки монет, определяли в сплавах ’и рудах с помощью электровыделения на предварительно взвешенном катоде обычно при плотностях тока порядка нескольких десятков А/см2, которые были слишком велики, чтобы говорить о приблизительно термодинамическом равновесии. В некоторых случаях селективного выделения металлов можно добиться, вводя комплексообразующие агенты. В других случаях в основу селективного разделения можно положить высокое напряжение восстановления водорода на ртутном катоде [11].
Для улучшения методики можно использовать потенциостат. Этот прибор позволяет контролировать потенциал катода в электролитической ячейке относительно электрода сравнения. На рис. 17-1 дана схема потенциостата на операционных усилителях. Как показывает сравнение этого рисунка с рис. 16-11, потенциостат имеет много общего с полярографом. Поскольку в данном случае нужно обеспечить постоянство потенциала, генератор линейно нарастающего потенциала заменяют регулируемым источником напряжения. Если необходим более высокий ток, чем тот, который может давать обычный операционный усилитель, в цепь обратной связи усилителя 1 (управляющего усилителя) вводят дополнительный усилитель мощности. Преобразователь ток — напряжение (усилитель 2) должен быть способен вырабатывать тот же самый ток, и поэтому он также должен иметь усилитель мощности. Если прибор используют для гравиметрического определения выделенного металла, то рабочий электрод можно заземлить, убрав усилитель 2 и самописец.
В процессе электролиза при контролируемом потенциале катода, когда восстанавливается только одно вещество, ток лимитируется диффузией (даже при перемешивании), поэтому он пропорционален концентрации восстанавливающих частиц. Сле-
366 Глава 17
Рис. 17-1. Потенциостат на операционных усилителях. В — усилители мощности, позволяющие оперировать с гораздо более высокими токами, чем это под силу одному операционному усилителю.
довательно, и ток, и концентрация экспоненциально убывают во времени, и можно записать, что
Ct/C0^It/I0=l(Tkt (17-1)
где Ct — концентрация-в момент времени t, Со — концентрация до электролиза, т. е. при t=0, a It и /0— соответствующие токи. Коэффициент k определяется из соотношения DAS/V, где D — коэффициент диффузии, А — площадь поверхности катода, V— -объем раствора, S — величина, пропорциональная скорости перемешивания. Графическая зависимость 1g/ от t выражается прямой с отрицательным наклоном, равным k. Из этой зависимости можно определить время, необходимое для выделения любой доли определяемого вещества. Например, в примере, приведенном Лингейном [2] для выделения меди, найдено, что' k = 0,15 мин-1. Отсюда можно рассчитать, что выделение 99 % меди заканчивается за 13 мин, а 99,9 % —за 20 мин.
Электролиз при контролируемом потенциале находит применение не только в аналитической химии. Контроль потенциала приводит к улучшению результатов в процессах синтеза, основанных на электроокислении или электровосстановлении. Этим способом можно получить Se(II) и Те(II), а также вольфрам в степенях окисления III и V. Сообщалось о получении различных пинаколов, гидроксиламинов и других соединений почти со 100%-ным выходом. Электроосаждение представляет собой ценный метод разделения субмикрограммовых количеств радионуклидов.
Кулонометрия
Даже наиболее совершенный метод измерения количества электричества требует интегрирования. Согласно закону Фарадея, масса вещества, образовавшегося в результате электролиза, пропорциональна количеству прошедшего электричества. Эту
Электровыделение и кулонометрия 367
величину (в кулонах) можно найти путем интегрирования по времени
Q = $Idt (17-2)
На использовании этого соотношения основан метод кулонометрического анализа, позволяющий проводить количественное определение любого вещества, способного участвовать в электрохимической реакций со 100%-ным выходом по току (т. е. в отсутствие побоч/ных реакций).
Возможны два'рарйанта кулонометрических измерений: при постоянной силе тока и при постоянном потенциале. В первом случае количество реагирующего вещества пропорционально затраченному времени, во втором ток экспоненциально уменьшается практически до нуля. Оба варианта находят важное практическое применение.
Величину Q можно непосредственно измерить с помощью ряда интеграторов [3] — химических, электрохимических или электронных. Один из таких интеграторов на операционных усилителях несложно собрать в лаборатории (рис. 17-2). Ток /вх поступает на вход усилителя, но так как он не может пройти в усилитель, он заряжает конденсатор С почти так же, как в генераторе линейно нарастающего напряжения для полярографии. Если бы Q было ограничено величинами порядка 1 мКл (милликулон), то было бы достаточно одного интегрирующего усилителя. Но часто необходимо измерять во много раз большие количества электричества. Один из способов решения этой задачи заключается во введении дополнительных элементов, указанных на схеме. Когда напряжение на конденсаторе достигнет заданного Vm, напряжение на выходе второго усилителя, не имеющего цепи обратной связи (компаратор, см. гл. 27), скачком изменяется от —10 до +10 В. При
Рис. 17-2. Интегратор с цифровым счетчиком, используемый в качестве кулонометра.
368 Глава 17
этом конденсатор разряжается в результате срабатывания управляемой напряжением схемы пропускания (т. е. ключа S). (Этой схемой может быть реле или соответствующее полупроводниковое устройство.) Затем компаратор возвращается в исходное состояние, а конденсатор может снова заряжаться. Каждый раз, когда напряжение на выходе компаратора становится положительным, цифровой счетчик регистрирует импульс, и тем самым регистрируется Q [3, 4].
Кулонометрия при постоянном потенциале. Этот метод пригоден для разделения металлов в соответствии с величинами их формальных потенциалов (см. гл. 14, рис. 14-1 и 14-2). Ток, протекающий через электролитическую ячейку при постоянном потенциале, зависит от концентрации восстанавливающегося вещества и поэтому в соответствии с уравнением (17-1) по мере протекания реакции уменьшается. Принципиальных отличий от классического метода электровыделения здесь нет, за исключением способа получения результата, поскольку измеряют количество электричества, а не массу.
Кулонометрия при контролируемом потенциале широко применяется для определения электроактивных веществ в низких концентрациях. Наглядным примером служит определение плутония (6—12 мг) с относительным стандартным отклонением 0,06 % [5] после ионообменного удаления мешающих примесей. В цитируемой статье дано подробное описание точного метода обработки данных.
Кулонометрия при постоянной силе тока. Этот косвенный кулонометрический метод обычно включает электрогенерацию растворимого реагента, количественно реагирующего с определяемым веществом. Ясно, что он имеет непосредственное отношение к титриметрии, только титрант генерируется in situ, а не добавляется из бюретки, поэтому его и называют кулонометрическим титрованием. Как и в любом титровании, для установления точки эквивалентности необходимо непрерывно наблюдать за изменением какого-то независимого параметра химической реакции.
Лучше всего принцип кулонометрического титрования объяснить на примере. Рассмотрим метод определения Fe(III) в растворе, содержащем НВг, основанный на восстановлении до Fe(II). Это можно сделать в ячейке с платиновым катодом и серебряным анодом. Допустим, что сначала мы попытаемся провести прямое восстановление. На соответствующей вольт-амперограмме (кривая 1 на рис. 17-3) наблюдаются две волны, одна из которых относится к восстановлению иона ЕеВгг+, а другая — к восстановлению иона Н+. Если мы будем пропу
Электровыделеиие и кулонометрия 369
скать через ячейку постоянный ток (на рис. 17-3 его величина обозначена а), начальный потенциал катода будет равен приблизительно +0,40 В (относительно НКЭ), т. е. величине потенциала в точке пересечения кривых 1 и а. В процессе электролиза железо восстанавливается/ и поэтому диффузионный ток РеВгг+ будет постепенно уменьшаться до тех пор, пока потенциал катода внезапно не станет равным примерно —0,3 В (кривая 2), т. е. не достигнет величины, при которой начнется восстановление ионов Н+. С этого момента восстановление Fe (III) и выделение Нг происходят одновременно, и выход по току становится ниже
Рис. 17-3. Кривые ток — потенциал для Fe(III) (7) и Ti (V) (3) в среде НВг, полученные с платиновым катодом и НКЭ. Кривая 2 — то же, что и кривая 1, но при меиьшей концентрации Fe(III).
100 %, что недопустимо в ана-
лизе.
Теперь повторим эксперимент, но введем в раствор избыток титана (IV). В присутствии НВг титан (IV) восстанавливается до титана (III') при несколько более отрицательных потенциалах, чем потенциал НКЭ (кривая 3 на рис. 17-3). Если сила тока снова станет равной а, то железо (III) снова восстанавливается, пока его диффузионный ток не снизится до величины а, но в этот момент потенциал катода будет равен только —0,05 В, а не —0,3 В, как было ранее. С данного момента начнется одновременное восстановление Fe(III) и Ti(IV), но эффективность тока при этом не снижается, поскольку Ti (III) количественно реагирует с Fe(III) и суммарный результат определяется расходом одного электрона на восстановление одного атома железа независимо от того, прямо или косвенно это происходит. В результате, как и требуется, выход по току остается равным 100%. (Если бы сила тока была равна б, а не а, то восстановление титана (IV) и железа (III) с самого начала протекало одновременно, но конечный результат был бы тем же.)
Конечная точка этой реакции соответствует моменту времени, когда количество пропущенного электричества точно эквивалентно количеству Fe(III), первоначально присутствовавшему в анализируемом растворе. Есть много способов идентификации конечной точки. Это можно сделать потенциометри
370 Глава 17
чески с платиновым и насыщенным каломельным электродами, амперометрически при потенциале около +0,25 В, амперометрически с двумя поляризованными электродами, фотометрически с добавлением KSCN или другого хромогенного реагента на Fe(III). Из перечисленных способов, вероятно, самым простым представляется амперометрический с двумя поляризованными электродами.
В описанном выше эксперименте на аноде протекала реакция Ag + Br-->AgBr + e, не вызывавшая осложнений, поскольку образующийся продукт малорастворим. Однако во многих титрованиях на противоэлектроде может образовываться растворимый продукт, который, если не принять мер, может реагировать на генераторном электроде, тем самым уменьшая выход по току. Если бы, например, в рассматриваемом случае вместо серебряного электрода взяли платиновый, то протекала бы анодная реакция 2Вг-->Вг2 + 2е; тогда образующийся бром циркулировал бы в растворе и восстанавливался на катоде или реагировал с Fe(II). В таком случае для предотвращения конвекции противоэлектрод (т. е. анод, если в процессе титрования идет восстановление) можно поместить в стеклянную трубку с пористой диафрагмой на конце. Часто этого достаточно, но иногда таким способом не удается устранить источник погрешностей и приходится прибегать к использованию мостика с агар-агаром или какого-либо другого устройства.
Подобные помехи иногда можно устранить с помощью небольшого слоя ионообменной смолы, заключенного в пористую стеклянную оболочку [6]. Предположим, например, что нужно кулонометрически оттитровать основание электрогенерирован-ными ионами Н+. На аноде, являющемся генераторным электродом, будет протекать известная полуреакция Н2О->2Н++ + '/2О2+2е, а на катоде — комплементарная реакция 2е + + 2Н2О->Н2 + 2ОН-. Конечно, образующиеся на катоде ионы ОН- не должны попадать в титруемый раствор, для чего можно использовать анионит в С1--форме, который обменивает ионы ОН- на С1-. Этот способ, по-видимому, не полуйил должного внимания.
На рис. 17-4 схематически изображена установка для титрования кислоты. Генераторный электрод и противоэлектрод подсоединяют к источнику постоянного тока и электрохронометру; цепь индикации состоит из обычного стеклянного электрода и электрода сравнения, присоединенных к pH-метру. (Источник постоянного тока легко собрать из стандартных электронных элементов, см. гл. 27.)
Электролитически можно генерировать большое число реагентов, например Н+, ОН-, Ag+ и катионы других металлов, окислители Ce(IV), Mn(III), Ag(II), Cl2, Br2, I2, Fe(CN)e3-,
Электровыделение и кулонометрия 371
Цепь индикации
Цепь .генерации
Магнитная мешалка
Рис. 17-4. Установка для кулонометрического титрования при постоянной силе тока с потенциометрической индикацией конечной точки. Сопротивление Л? вводят в цепь источника постоянного тока при отключении ячейки.
восстановители Fe(II), Fe(CN)e4_, Ti(III), CuBr2~ и Sn(II), а также комплексообразующие вещества ЭДТА и CN-. С помощью того или иного из перечисленных реагентов почти все методики классического объемного анализа можно заменить кулонометрическим титрованием. Это дает большие практические преимущества, поскольку отпадает необходимость в приготовлении и хранении стандартных растворов [7]. Роль первичного стандарта в кулонометрическом титровании играет источник постоянного тока в сочетании с электронохронометром, которые пригодны для любых титрований независимо от природы химической реакции. К преимуществам кулонометрического титрования относится также возможность определения меньших количеств вещества (на один или два порядка), чем в обычных способах (как правило, достаточно 0,1—0,001 миллиэквивалента вместо 1—10 миллиэквивалентов, как в классической титриметрии), а также возможность использования таких неустойчивых, т. е. непригодных для титриметрических методов реагентов, как Мп(Ш), Ag(II), CuBr2- и С12.
Кулонометрическое титрование дает сравнимую или более высокую точность по сравнению с классической титриметрией. Экфилд и Шаффер [8] тщательно изучили погрешности кислотно-основного кулонометрического титрования. Им удалось измерить количество электричества с погрешностью ±0,004 %. Один из простых, но эффективных приемов состоял в пропускании слабого потока индифферентного электролита через солевой мостик, что позволило устранить возможные загрязнения
372 Глава 17
Рис. 17-5. Кривая титрования при использовании предварительного кулонометрического титрования. По оси ординат отложен сигнал детектора (фотометрического, амперометрического или др.), линейно зависящий от концентрации.
или потери анализируемого раствора через пористую стеклянную трубку. Эту статью необходимо изучить всем, кто хочет получить точные результаты кулонометрического титрования.
Кулонометрический метод менее подходит для определения высоких концентраций, в чем и состоит его главное ограничение. Это объясняется тем, что при определении больших количеств вещества необходимо работать с гораздо большими токами или в течение более продолжительного времени. В любом случае выход по току, вероятно, уменьшится из-за большой вероятности протекания побочных реакций.
В кулонометрии часто рекомендуется проводить предварительное титрование. После того как установка для титрования собрана, в ячейку вводят небольшую порцию титруемого раствора и проводят электролиз до достижения желаемой конечной точки. Затем вводят измеренный объем титруемого раствора и вновь титруют до достижения конечной точки. Это гарантирует заблаговременное удаление любых примесей, которые могли бы реагировать с генерируемым титрантом, а также исключает возникновение любых погрешностей, связанных с состоянием поверхности электродов (например, образование оксидных пленок). Кривая титрования имеет форму, показанную на рис. 17-5. В процессе предварительного титрования реакция с электрогенерированным титрантом протекает в промежуток времени от t0 до ti, после чего титрант начинает накапливаться (участок А). При введении анализируемого раствора он сразу же вступает во взаимодействие с определяемым веществом, и кривая возвращается к нулю (участок В); затем титрование продолжается до достижения подъема на кривой (участок С). Оба участка кривой экстраполируют на нулевую линию в точ
Электровыделение и кулонометрия 373
ках D и Е и разность t2—ti принимают за время электролиза. Участки А и С будут иметь разные наклоны, если после добавления титруемого раствора произойдет заметное изменение объема.
Кулонометрическое титрование легче поддается автоматизации, чем другие способы титрования, в том смысле, что не нужно вводить такие механические приспособления, как насосы и бюретки. Поскольку титрование проводят при постоянной силе тока, то между количеством добавленного титранта и временем существует линейная зависимость..Для кулонометрической генерации можно легко приспособить полностью автоматический pH-стат, обсужденный в гл. 15.
В продаже имеются различные автоматические и полуавтоматические кулонометрические титраторы, а также ряд анализаторов для непрерывного контроля состава потока жидкости, в которых задается величина тока, необходимого для поддержания постоянной концентрации какого-либо компонента. Их часто называют «кулонометрическими», хотя более подходящим названием могло бы быть «амперометрические» анализаторы.
Предварительное электролитическое концентрирование
При анализе растворов, содержащих следовые количества переходных металлов, часто перед выполнением определения необходимо повысить их концентрацию. Среди пригодных для этого способов одним из лучших является электроосаждение на катоде. Электролиз можно проводить до тех пор, пока не будет накоплено достаточное для последующего определения количество металла. При достаточно отрицательном потенциале катода одновременно выделяются все способные к восстановлению металлы, но при потенциостатическом контроле можно достичь некоторой селективности. Поскольку проведение исчерпывающего выделения или невозможно, или может потребовать очень много времени, чаще выделение проводят в течение только определенного промежутка времени.
Выбор электрода для проведения выделения зависит от метода анализа после выделения. Чаще всего используют электрохимические методы анализа и поэтому обычно предпочитают ртутный электрод (см. следующий раздел). Для проведения фотометрического анализа выделенные металлы необходимо удалить с катода или электролитически, или с помощью кислоты; в этом случае пригоден платиновый электрод. В гл. 11 упоминалось о предварительном выделении на пиролитическом графите с целью последующего рентгенофлуоресцентного определения [9].
374 Глава 17
Инверсионный анализ [10—12]
Это название относится к анализу смеси выделенных металлов методом анодной вольтамперометрии. Согласно обычной методике, электролитическое выделение проводят в порции анализируемого раствора на маленьком ртутном электроде в течение 20—30 мин при потенциале, достаточном для выделения всех присутствующих металлов. Затем потенциал-сканируют от потенциала электролиза в направлении более положительных величин, т. е. растворяют металлы, выделенные'на предварительной стадии. Процесс растворения в условиях вольтамперометрии длится 2—3 мин.
При проведении инверсионного анализа широко используют висящий капельный ртутный электрод. Такая капля образуется на кончике заполненного ртутью капилляра, присоединенного к плунжеру микрометрического устройства. При повороте микрометра на определенный угол выдавливается точно воспроизводимое количество ртути. Менее дорогостоящее устройство
Рис. 17-6. Устройство для получения висящего капельного ртутного электрода, закрепленное в пластиковой крышке сосуда. Каплю ртути при помощи ложечки (7) переносят из капилляра (2) на кончик платинового электрода (3).
для получения ртутной капли, которое можно изготовить самим, показано на рис. 17-6. Под устьем стандартного полярографического капилляра (2) помещают маленькую пластмассовую ложечку или чашечку (1). После того как в чашечку попадает одна или несколько капель ртути, ее поворачивают и поднимают, чтобы ртуть коснулась кончика платиновой проволочки*, впаянной в стеклянную трубку (3), и прилипла к нему.
Применение твердых электродов в методе инверсионной вольтамперометрии не дает удовлетворительных результатов, так как атомы микрокомпонента в выделенном осадке могут находиться под слоем макрокомпонента и поэтому не могут полностью, реокислиться. Если же металлы растворяются в ртути,
Лучше золотой или серебряной.— Прим, перев.
Электровыделение и кулонометрия '375
Потенциал (отн. Ад/АцС1-элвктроЭа),В
Рис. 17-7. Анодная инверсионная вольтамперограмма. Штриховой линией обозначен остаточный ток (подробное описание рисунка см. в тексте) [10].
эта сложность не возникает*. Иногда вместо висящей капли ртути применяют графитовые электроды, предварительно покрытые тонкой пленкой ртути.
На анодной инверсионной вольтамперограмме наблюдается ряд пиков,, соответствующих последовательному окислению металлов. Количество каждого из них можно найти, измерив высоты пиков относительно линии остаточного тока холостого опыта. На рис. 17-7 представлены результаты из работы Эллиса [10]. Анализируемый раствор содержал равные (2-10~7М) количества Сп2+ и Cd2+, предварительное выделение проводили в течение 5 мин при —1,0 В (относительно Ag/AgCl-электрода). Обратите внимание на то, что на вольтамперограмме холостого опыта (штриховая линия) наблюдаются пики Си2+ и РЬ2+, указывающие на их значительное содержание в растворе, хотя анализируемый раствор и не содержал свинца. Это вполне обычно для инверсионного анализа; чувствительность метода настолько высока, что в растворителях и реагентах, считающихся чистыми, часто находят следы каких-то иных металлов.
* Возникает другая сложность — образование интерметаллических соединений.— Прим, перев.
376 Глава 17
Задачи
17-1. Постоянный ток в 15,0 мА пропускали ровно 10 мин через ряд последовательно соединенных ячеек. Во всех ячейках находились платиновые электроды и содержался избыток электролитов, перечисленных ниже. Определите, какие вещества образуются на каждом из электродов, и рассчитайте количество каждого из них в миллиграммах (для твердых или жидких) и в миллилитрах (для газообразных) при стандартных температуре и давлении:
a) Cu(NO3)2 в) K4Fe(CN)e д) РЬ(С1О4)2
б) NaOH г) Hgl2 е) Ag(NH3)2Cl
17-2. Для применения в стандартном элементе Вестона приготовили амальгаму кадмия, содержащую 12 масс. % кадмия. Если взять 20 г ртути, то как долго следует вести электролиз раствора CdCl2 (ток 200 мА), чтобы выделить требуемое количество кадмия?
17-3. Церий(1У) определяли кулонометрическим титрованием электрогенери-рованными ионами Fe2+. Конечную точку титрования определяли потенциометрически, измеряя потенциал платинового электрода относительно НКЭ с помощью pH-метра. В процессе предварительного изучения было установлено, что потенциал платинового проволочного электрода должен быть равен + 0,800 В относительно НКЭ. Подкисленный раствор содержит 0,0005 моль Fe3+ в объеме 250 мл. Мешающие вещества удаляли предварительным титрованием. Для этого добавляли примерно 0,2 мкмоль Ce(IV) и вели электролиз, пока потенциал электрода не достигал +0,800 В. Затем добавляли по 1,00 мл анализируемого раствора и вновь продолжали генерацию титранта до достижения конечной точки титрования. Полученные данные приведены в таблице. Показания кулонометра даны в произвольных единицах, при этом изменение на 1 единицу соответствует 4,79-10-4 микроэквивалента. Рассчитайте концентрацию раствора церия в единицах мкг/мл.
Время, с
Показания кулонометра
Потенциал Pt-электрода (НКЭ), В
Предварительное титрование
20 + 0,830
436,0 120 0,801
472,8 170 0,790
Выполнение анализа
472,8 170 0,893
470 0,861
720 0,820
750 0,814
770 0,810
925,0 790 0,803
939,5 810 0,794
Электровыделение и кулонометрия 377
Рис. 17-8. Кривая ток —время для процесса выделения меди из анализируемого раствора.
17-4. Медь из навески латуни массой 0,400 г выделяли электролизом при контролируемом потенциале. Ячейку последовательно соединяли с регистрирующим. измерительным устройством; полученная кривая изменения тока во времени приведена на рис. 17-8. Чтобы провести интегрирование по времени, нужно подсчитать квадратики под кривой или воспользоваться методом трапеций. Проведите интегрирование и рассчитайте процентное содержание меди в сплаве. Почему эта кривая не экспоненциальная?
17-5. Каким должен быть ток (в миллиамперах) в кулонометрии при постоянном токе, чтобы электрохронометр сразу давал отсчет микроэквивалентов вещества в электродной реакции?
17-6. Антраниловую (о-аминобензойную) кислоту можно с помощью электро-генерированиого брома превратить при pH 4 в триброманилин. Найдено [13], что для определения небольших количеств меди нужно осадить Си(СбН4МН2СО2)2, растворить полученный осадок и оттитровать выделившуюся антраниловую кислоту кулонометрически.
Медь, содержащуюся в 1,000 г биологического образца, после минерализации осаждали избытком антраниловой кислоты. Осадок отфильтровывали, промывали и снова растворяли. Выделившуюся кислоту бромировали при постоянном токе 6,43 мА. На завершение реакции потребовалось 22,40 мин. Рассчитайте содержание меди (в частях иа миллион) в исходном образце.
17-7. Методом инверсионной вольтамперометрии с пленочным ртутным электродом на графитовой подложке в образце озерной воды определяли содержание Си, РЬ и Cd. В одном 'опыте электрод находился в течение 90 мин под напряжением —1,0 В относительно НКЭ (объем анализируемого раствора 200 мл), пока ток не достигал пренебрежимо малой величины; затем регистрировали анодную вольтамперограмму. Интегрированием найдено, что количество электричества при потенциалах пиков Cd, Pb и Си равно 12,5; 41,0 и
378 Глава 17
38,2 мкКл соответственно. Рассчитайте концентрацию каждого металла в анализируемой воде в единицах мкг/л.
17-8. На ртутиой капле проводили предварительное накопление меди из 10~8 М раствора Си2+ для последующего анодного инверсионного определения. Если объем капли 0,0010 см3, а ток электролиза 0,500 мкА, то как долго нужно вести электролиз, чтобы концентрация меди в амальгаме была 10-3М? 17-9. Серебро как главный компонент сплава можно количественно определить кулонометрически при контролируемом потенциале. Фоновым электролитом служит 0,1 М раствор H2SO4; если в этом растворе присутствует азотная кислота, необходимо добавить сульфаминовую кислоту. Для того чтобы избежать загрязнения хлорид-ионами, в качестве электрода сравнения используют Hg/Hg2SO4 (насыщ. K2SO4) электрод с потенциалом — 0,579 В (относительно СВЭ при 25 °C). Выделение серебра проводят при —0,25 В. Для выполнения анализа 1,000 г сплава для припоя растворяли в смеси азотной и серной кислот, кипятили полученный раствор для удаления избытка HNO3 и оксидов азота, раствор переносили в колбу емкостью 250 мл и разбавляли до метки деионизованной водой. Если 0,500 мл полученного раствора ввести в ячейку с фоновым электролитом, то на электролиз потребуется 1,198 Кл. а) Согласуйте с рис. 14-1, правильно ли выбран потенциал электролиза, б) Рассчитайте процентное содержание серебра в сплаве.
Литература
1. Maxwell J. A., Graham R. Р., Chem. Rev. 1950, v. 46, р. 471.
2. Lingane J. J., Electroanalytical Chemistry, 2d ed. Wiley — Interscience, New York, 1958.
3. Ewing G. W., J. Chem. Educ., 1972, y. 49, p. A333.
4. Pareles S. R., Anal. Chem., 1973, v. 45, p. 998.
5. Holland M. K., Weiss J. R„ Pietri С. E., Anal. Chem., 1978, v. 50, p. 236.
6. Eisner U., Rottschafer J. M., Berlandi F. J., Mark H. B., Jr., Anal. Chem., 1967, v. 39, p. 1466.
7. Ewing G. W., Am. Lab. 1981, v. 13(6), p. 16.
8. Eckfeldt E. L., Shaffer E. W. Jr., Anal. Chem., 1965, v. 37, p. 1534.
9. Vassos В. H., Hirsch R. F., Letterman H., Anal. Chem., 1973, v. 45, p. 792.
10. Ellis W. D., J. Chem. Educ., 1973, v. 50, p. A131.
11. Vassos В. H., Ewing G. W., Electroanalytical Chemistry, Wiley, New York, 1983, ch. 10.
12. Shain J., In Treatise on Analytical Chemistry, Kolthoff J. M., Elving P. J. (eds.), Wiley-Interscience, New York, 1963, pt. 1, v. 4, ch. 50.
13. Hargis L. G., Boltz D. F., Taianta, 1964, v. 11, p. 57.
ГЛАВА IO
Кондуктометрия
В предыдущих главах рассмотрены специфические электрохимические реакции на поверхности электрода. Нефарадеевские (не сопровождающиеся химическими превращениями) токи в этих главах трактовались как шумы или нежелательные помехи, искажающие полезную информацию. Теперь же мы рассмотрим несущую полезную информацию нефарадеевскую величину— ионную проводимость.
Ячейку, состоящую из двух платиновых электродов в растворе электролита, можно представить в виде электрического эквивалента из сопротивлений и емкостей, как это показано на рис. 18-1. На этой схеме RLl и Rl2— сопротивления электрических проводников (обычно они пренебрежимо малы), CDli и Cdl2 — емкости двойного слоя на электродах, Ср—межэлектродная емкость, Rso\ — сопротивление раствора между электродами. Компоненты, обозначенные 7?fi и RF2, представляют
Рис. 18-1. Ячейка для измерения электропроводности (а) и эквивалентная схема (б).
380 Глава 18
собой фарадеевские сопротивления на обоих электродах, т. е. электрические эквиваленты какой-либо электродной реакции. Если на эту цепь наложить небольшое постоянное напряжение, не вызывающее протекания электрохимического процесса, то произойдет лишь кратковременный переходный процесс. При увеличении напряжения через компоненты Rf и Rso\ будет протекать ток.
С другой стороны, при наложении переменного напряжения через CDli, Cdl2, /?soi и Ср начнет протекать переменный ток. Каждая CDl представляет настолько легкий путь для переменного тока, что на соответствующих R? потенциал не возникает до начала протекания фарадеевского тока. Поэтому если Ср поддерживать пренебрежимо малой, a Cdli и Сщ.2 — большими, то можно изучить влияние /?Soi. Практически емкость двойного слоя можно увеличить во много раз, если платинировать, т. е. покрыть платиновой чернью, платиновые электроды, так как при этом сильно увеличивается эффективная поверхность. Величина Ср становится значимой только при работе с растворами, обладающими высоким сопротивлением, когда большие электроды приходится располагать близко друг к другу.
Теория
Электропроводностью раствора L, выражаемой в сименсах * (См), называют величину, обратную сопротивлению. Электропроводность связана с отношением площади поверхности электродов к расстоянию между ними и с общей концентрацией ионов выражением
С=10-3(а/б/)£ггСЛг (18-1)
i
Отношение a/d определяется геометрией ячейки, и ее обратную величину называют константой ячейки, обозначая символом 0 (размерность см-1). Величина С; в уравнении (18-1) есть молярная концентрация г-го иона с зарядом г,, а — соответствующая эквивалентная электропроводность. Суммирование проводят по всем ионам обоих знаков.
Величина X дает количественную информацию о вкладе иона в электропроводность раствора. Она до некоторой степени зависит от общей концентрации ионов в растворе, возрастая с увеличением степени разбавления раствора. Величины
* Сименс — единица измерения электропроводности (проводимости) в системе СИ. Она эквивалентна ранее использовавшейся единице «mho». Обычно проводимость обозначают символом G, но для ионной проводимости принято использовать символ L.
Кондуктометрия 381
Таблица 18-1. Эквивалентная электропроводность при бесконечном разбавлении при 25 °C (См-см2/моль) [1]
Катионы % Анноны 4
н+ 349,8 он- 198,6
!/3Со (NH3)3+ 102,3 V4Fe(CN)|- 110,5
к+ 73,5 V3Fe(CN)|~ 101,0
NH+ 73,5 1/3Co(CN)g'~ 98,9
V2Pb2+ 69,5 • v2so2- 80,0
1/3La3+ 69,5 Br~ 78,1
1/3Fe3+ 68,0 I- 76,8
V2Ba2+ 63,6 Cl- 76,4
Ag+ 61,9 Nor 71,4
V2Ca2+ 59,5 VsCO2- 69,3
V2Cu2+ 53,6 v2c2o2- 74,2
V2Fe2+ 54,0 C107 67,3
1/2Mg2+ 53,1 HCO^ 44,5
1/2Zn2+ 52,8 ch3coo- 40,9
Na+ 50,1 HC2O4~ 40,2
Li+ 38,7 C6H5CO^ 32,4
(h-C4H9)4n+ 19,5
Таблица 18-2. Удельная электропроводность растворов КС1 при 25 °C [2]
Концентрация, г/кг раствора, x, См/см
71,1352 0,11134
7,41913 0,012856
0,74526 0,0014088
Хо при концентрации, приближающейся к нулю (предельное разбавление), удобно табулировать. Они отличаются от подвижности ионов на множитель F (константа Фарадея). В табл. 18-1 приведены некоторые характерные величины.
В ряде случаев бывает необходимо знать константу ячейки 0. Однако прямые геометрические измерения обычно не применяются, за исключением ячеек, специально сконструированных
382 Глава 18
для этих целей. Поэтому обычно константу ячейки определяют из данных измерений для растворов с известной электропроводностью (х = Г0). Некоторые величины, используемые при калибровке, приведены в табл. 18-2.
Аппаратура
Для измерения электропроводности служит модифицированный мост Уитстона (рис. 18-2). Мост питается от источника Е с частотой 1 кГц или частотой сети переменного тока. Плечи Ri и /?2 с помощью переключателя можно подобрать так, чтобы соотношение сопротивлений было равно точно 0,1, 1 или 10. Rx — сопротивление кондуктометрической ячейки, шунтированное емкостью Сх (в действительности Сх представляет собой комбинацию нескольких емкостей, показанных на рис. 18-1).
— прецизионное калиброванное переменное сопротивление, шунтированное маленькой переменной емкостью Сз для балансирования емкости ячейки Сх.
Переменное напряжение, возникающее по диагонали моста, усиливают, выпрямляют и измеряют. При проведении измерений подбирают такое положение переключателя и величину калибровочного сопротивления R3, чтобы измерительное уст-
Рис. 18-2. Мост Уитстона для измерения электропроводности. Тройной двухполюсный ключ позволяет выбрать нужное соотношение — 0,1, 1 или 10.
Кондуктометрия 383
ройство показывало нулевое отклонение. В этот момент сопротивление ячейки определяется выражением
а ее электропроводность
(18-2)
(18-3)
Иной подход к измерению электропроводности основан на использовании цепи, контролируемой операционным усилителем (рис. 18-3). Поскольку усилитель может поддерживать одинаковые напряжения на обоих выходах, переменное напряжение Евх появляется непосредственно на концах ячейки, но возникающий при этом в ячейке ток компенсируется равным током с выхода усилителя через подобранное сопротивление Rf. Напряжение на выходе Ео можно представить выражением
Ео = Евх(-^ + 1) = 5вх(^Ах+1) (18-4)
Если R/ имеет достаточно большую величину, то единицей в выражении (18-4) можно пренебречь, и выходное переменное напряжение будет прямо пропорционально электропроводности раствора. Более разработанный вариант описан в работе [3]. Заметим, что этот прибор не требует балансировки, как при использовании моста, а позволяет проводить непрерывное считывание.
Рис. 18-3. Цепь операционного усилителя для измерения электропроводности.
384 Глава 18
Удельное сопро-
тивление, Ом-см 10 ОМ ЮМ 1000 к Юрк
Удельная электро- 0,01 0,1 1 Ю
проводность,мкСм/см | д
Ультра-чистая дода
Юк
100
4
1000 -4— юоо 4
100 ip 1
Юк 100к 1000 К
4 А
Дистиллированная Вода хорошего качества
Неочищенная Вода отличного качества 0,05 % - ный
раствор NaCl
Морская „ Вода 30%-ная
H2SO4'
Рис. 18-4. Удельное сопротивление и удельная электропроводность некоторых материалов.
Применение
В большинстве случаев измеряют электропроводность водных растворов. На рис. 18-4 приведены величины удельной электропроводности и удельного сопротивления ряда материалов. Вода сама по себе очень плохой проводник. Ее удельная электропроводность, обусловленная диссоциацией на ионы Н3О+ и ОН- при 25 °C равна примерно б-Ю-8 См/см. Электропроводность обычной дистиллированной или деминерализованной воды намного ниже. Перегонные аппараты и умягчители воды часто снабжают устройствами для непрерывного контроля электропроводности.
Для растворов сильных электролитов характерна почти линейная зависимость электропроводности от концентрации примерно до величины порядка 10 и 20 масс. %. При более высоких концентрациях электропроводность вновь уменьшается, поскольку межионное притяжение затрудняет движение ионов в растворе. Существует много приборов для измерения электропроводности, специально откалиброванных для конкретных веществ, например снабженных шкалами «0—30 мкг/л NaCl» или «2—25 % H2SO4».
Кондуктометрическое титрование
Для наблюдения за процессом титрования метод измерения электропроводности ; можно применять только при условии, что электропроводность исходного раствора значительно отличается от электропроводности реагента или продуктов реакции. Константу ячейки знать не обязательно, поскольку для нахождения точки эквивалентности достаточно относительных вели-
Кондуктометрия 385
Рис. 18-5. Кривая кондуктометрического титрования НС1 раствором NaOH: а — кривая, записанная прибором; б — та же кривая с указанием вкладов отдельных ионов. Предполагается, что разбавлением раствора при титровании можно пренебречь.
чин. Важно, однако, чтобы расстояние между электродами в процессе титрования не изменялось.
Вклад любого иона в электропроводность раствора пропорционален его концентрации, но при титровании по мере добавления реагента увеличивается количество воды, и поэтому при вычислении электропроводности нужно ввести поправку на разбавление раствора. Отклонение от линейной зависимости может также наблюдаться из-за гидролиза реагирующих веществ или продуктов или частичной растворимости осадка.
Форму кривой титрования легко предсказать. Концентрацию каждого иона в последовательных точках при титровании вычисляют известными методами, основанными на законах стехиометрии, равновесия и разбавления. Умножив концентрации на величину л0 из табл. 18-1, получают относительные вклады всех ионов, суммирование которых дает кривую титрования. Для примера на рис. 18-5 показано титрование НС1 раствором NaOH. Изменение сигнала, пропорционального электропроводности (рис. 18-5, а), можно разложить на составляющие, характеризующие вклады различных ионов (рис. 18-5, б).
Кондуктометрическое титрование сейчас не применяется так широко, как прежде, что отчасти можно объяснить низкой специфичностью метода.
Радиочастотная кондуктометрия (осциллометрия)
Если частоту вибратора, питающего измерительное устройство, повысить от обычной величины в 1 кГц до частот порядка мегагерц, необходимо провести существенное изменение аппаратуры, но получаемая при этом информация мало чем отличается от прежней. За более подробной информацией читателю следует обратиться к литературе [4—6].
13 Заказ № 537
386 Глава 18
Задачи
18-1. Найдено [7], что удельная электропроводность связана с соленостью 5 морской воды при 25° С соотношением
х= 1,82- 10~3S—1,28- 10-BS2+ 1.18- 10-7S3
где 5 выражена в граммах иа килограмм воды. Средняя величина 5 для неразбавленной морской воды равна 35,0.
Удельная электропроводность пробы воды, отобранной вблизи устья реки, равна 1,47-10~2 См/см. Какова ее соленость? Сколько килограммов речной воды смешивается в этом месте с каждым килограммом морской воды? (Электропроводностью речной воды и различием в плотностях можно пренебречь.)
18-2. Изобразите иа одном графике, как вы представляете кривые кондуктометрического титрования 0,001 М AgNO3 0,010 М растворами следующих реагентов: НС1, КС1, NH4C1, СаС12, NaCl и LiCl. В каком случае получатся наиболее точные результаты? Объясните почему.
18-3. Описан метод кондуктометрического титрования органических оснований с помощью ВВгз и наоборот [8]. Для титрования применяли различные апротонные растворители. Например, при титровании ВВг3 в среде нитробензола получится перевернутая V-образная кривая. Напишите реакции, протекающие при титровании, и изобразите графически вклад отдельных частиц в общую проводимость. (В случае необходимости за подробностями обратитесь к первоисточнику.)
18-4. Известно, что Ag2SO4 и ВаС12 хорошо растворимы, a AgCI и BaSOj имеют произведения растворимости порядка 10~10, поэтому Ag2SO4 можно титровать раствором ВаС12 кондуктометрически. Используя уравнение (18-1) и данные табл. 18-1, рассчитайте проводимость в сосуде для титрования (0 = — 0,452 см-1) при титровании 0,1 М раствора Ag2SO4 1,0 М раствором ВаС12 (разбавлением раствора можно пренебречь) в следующие моменты титрования: а) до начала титрования (исходный раствор Ag2SO4); б) после добавления половины эквивалентного количества ВаС12; в) в точке эквивалентности; г) после добавления еще одного эквивалента ВаС12 за точкой эквивалентности. Нарисуйте кривую титрования.
Литература
1. Frankenthal R. Р., In Handbook of Analytical Chemistry, Meites L. (ed.), McGraw-Hill, New York, 1963, p. 5—29.
2. Janes G., Bradshaw В. C., J. Am. Chem. Soc., 1933, v. 55, p. 1780.
3. Mueller T. R., Stelzner R. W., Fisher D. J., Jones H. C., Anal. Chem., 1965, v. 37, p. 13.
4. Reilley C. N., In New Instrumental Methods in Electrochemistry, Delahay P. (ed.), Wiley-Interscience, New York, 1954, ch. 15.
5. Pungor E., Oscillometry and Conductometry, Pergamon, New York, 1965.
6. Svehla G., In Essays on Analytical Chemistry, E. Wanninen (ed.), Pergamon, Oxford, 1977, p. 233.
7. Ruppin E., Z. anorg. Chem., 1908, v. 49, p. 190.
8. IJenry M. C., Hazel J. F., McNabb W. M., Anal. Chim. Acta, 1956, v. 15, p. 187.
ГЛАВА 19
Введение в хроматографию
Анализ множества химических систем необходимо начинать с разделения компонентов смеси. Одним из наиболее распространенных и универсальных методов разделения является хроматография. Хроматография — это динамический процесс, происходящий в системе из двух несмешивающихся фаз, одна из которых подвижная, другая неподвижная. Подвижной фазой может быть либо газ, либо жидкость, а неподвижной — пористое или гранулированное твердое вещество или тонкая пленка жидкости, адсорбированной на твердом теле.
В соответствии с агрегатным состоянием подвижной фазы хроматографию подразделяют на газовую (ГХ) и жидкостную (ЖХ). Исходя из геометрии слоя неподвижной фазы, хроматографию делят на колоночную и плоскослойную. К последней относятся бумажная (БХ) и тонкослойная хроматография (ТСХ). Это также очень полезные методы разделения, но они не относятся к числу инструментальных и поэтому не рассматриваются в этой книге. В данной главе будут обсуждены теоретические вопросы, общие для ГХ и ЖХ, а в последующих главах рассмотрены инструментальные и практические особенности каждого из этих методов.
Хроматографический процесс легко представить на качественной основе. Рассмотрим колонку, заполненную гранулированным твердым веществом (рис. 19-1), через которую протекает поток жидкости. Вещество X, растворенное в жидкости, перемещается вместе с нею, но в то же время имеет тенденцию удерживаться на поверхности твердого вещества за счет адсорбции или другого механизма. Каждая данная молекула X часть времени находится в подвижной фазе и движется вместе с нею, а часть времени удерживается на поверхности неподвижной фазы. Возможность разделения двух таких растворенных веществ X и Y обусловлена различием их относительного сродства к обеим фазам, т. е. одно из них большую часть времени находится в подвижной фазе и поэтому скорее достигает конца колонки.
13*
388 Глава 19
Рис. 19-1. Разрез иасадочиой хроматографической колонки.
При проведении расчетов хроматографисты обычно пользуются коэффициентом емкости или коэффициентом извлечения, обычно обозначаемым k'. Его определяют как
k' njnm
где ns и nm—число молей растворенного вещества X в неподвижной и подвижной фазах соответственно. Этот коэффициент характеризует степень удерживания вещества X в данной хроматографической системе. Степень разделения двух веществ можно выразить через коэффициент разделения а:
а --- k’2/k\ (19-2)
Чем больше молей растворенного вещества в неподвижной фазе, тем больше времени требуется для его элюирования, и поскольку индексы у k в уравнении (19-2) отражают порядок элюирования веществ, то ясно, что а всегда больше единицы.
Чтобы достичь хорошего разделения, пробу нужно вводить в колонку в виде небольшой компактной дозы, чтобы все компоненты начинали перемещаться по колонке в один и тот же момент времени. Помещенный на выходе из колонки детектор регистрирует различные компоненты, а самописец фиксирует сигналы детектора, как показано на рис. 19-2. На этом рисунке приведены пики для четырех присутствующих в пробе веществ, одно из которых удерживается наименее прочно (^),
Разделение смеси на отдельные компоненты происходит в процессе их перемещения по колонке, поэтому длинная ко-
рне. 19-2. Хорошо разрешенная хроматограмма четырехкомпонентной смеси.
Введение в хроматографию 389
Рис. 19-3. Хроматограмма двухкомпоиеитной смеси, иа которой показано, как определяются различные хроматографические параметры. Пик при to принадлежит иеудерживаемому компоненту.
лонка способствует лучшему разрешению пиков. Однако из-за продольной диффузии происходит размывание движущейся зоны, занимаемой каждым компонентом. Степень уширения пиков, обусловленная этими причинами, пропорциональна корню квадратному из времени пребывания вещества в колонке. Чем шире пики, тем менее точно можно определить их положение, поэтому из практических соображений длину колонки ограничивают.
На рис. 19-3 указаны параметры, необходимые для описания хроматографических пиков. Уровень, обозначенный h/2, находится на половине высоты треугольника, образованного касательными, проведенными к кривой через точки перегибов. (Математически можно показать, что он равен 1/]/е действительной высоты пика. Этот коэффициент (0,607) получен из уравнения кривой гауссова распределения и, следовательно, только приблизительно справедлив для реальных пиков.) Ширина каждого пика (ш) является мерой статистического распределения времени удерживания индивидуальных молекул. Стандартное отклонение от среднего, обозначаемое о, равно половине ширины пика на половине его высоты (ПШПМ — половина ширины в половине максимума), и, следовательно, оно равно w/4. Соотношение между шириной пика и временем удерживания выражается уравнением
о). / t. \'/2
= (19-3)
шв Vb /
390 Глава 19
Теория хроматографической подвижности
В основу теории хроматографического разделения положено представление о том, что в процессе перемещения по колонке молекулы растворенного вещества периодически переходят из подвижной фазы в неподвижную и обратно. В результате любая молекула в среднем в течение времени ts находится в неподвижной фазе и в течение времени tm — в подвижной. В течение времени tm она движется по колонке с такой скоростью, с какой перемещается по колонке подвижная фаза и, а в течение времени ts совсем не продвигается вперед. Ее движение, следовательно, является ступенчатым, так как она то переносится в подвижную фазу, то из подвижной фазы. Относительные величины ts и tm определяют, насколько быстро растворенное вещество движется вниз по колонке.
Вклад различных факторов в эффективность разделения можно описать с помощью понятия «высота, эквивалентная теоретической тарелке» (ВЭТТ). Теоретическая тарелка — это чисто абстрактное понятие, не соответствующее ничему реально существующему в колонке. Это очень удобный параметр, что является единственной причиной его существования. ВЭТТ определяют как отношение квадрата стандартного отклонения (дисперсии) к длине колонки, H=a2IL. Это длина участка колонки, на протяжении которого между фазами при определенном наборе условий (скорость потока, температура и др.) может установиться равновесие. Для достижения высокой эффективности разделения желательно, чтобы число (N) теоретических тарелок было достаточно большим. Таким образом, чтобы колонка не была слишком длинной, ВЭТТ должна быть максимально малой. Чем ниже ВЭТТ, тем эффективнее колонка.
Число теоретических тарелок в колонке определяется соотношением 16(х/ш)2. Величины х и w, определение которых дано на рис. 19-3, следует выражать в одних и тех же единицах и обычно удобно измерять по хроматограмме в сантиметрах.
Из соотношения
N — L/H — 16 (х/w)2 или Л^эфф = L/7/эфф = 16(х7ш)2 (19-4)
где ^ФФ и ДЭфф — эффективные параметры, получаем
H = L/N = L/l6(w/x)2 и //Эфф = £Жфф = £/16(ш/х')2 (19-5) Поскольку для данной системы Н— величина постоянная, то из приведенного соотношения следует, что х и w должны изменяться одновременно, поэтому для нескольких пиков на одной хроматограмме чем больше время удерживания (т. е. чем длиннее х), тем больше w и шире пики. Для лучшего разделения
Введение в хроматографию 391
желательно, чтобы w и, следовательно, Н были насколько возможно малыми.
Многими исследователями показано, что ВЭТТ можно выразить соотношением, известным под названием уравнения Ван-Деемтера
Д = Л + В/и + Си (19-6)
где А, В и С для данной системы приблизительно постоянны, а и — линейная скорость движения потока газа-носителя (ГХ) или элюента-носителя (ЖХ), см/с.
Член А отражает тот факт, что не все молекулы растворенного вещества в подвижной фазе при движении по колонке перемещаются на строго одинаковое расстояние. Величина А зависит от степени неоднородности частиц, заполняющих колонку, и определяется соотношением
A = Ku(Dm+ Pdpu) (19-7)
где Dm — коэффициент диффузии растворенного вещества в подвижной фазе, dp — средний диаметр твердых частиц, К и Р — константы, зависящие от нерегулярности упаковочного материала.
Член В, важность которого уменьшается с возрастанием и, определяется выражением
B = 2y(Dm + k'Ds) (19-8)
в котором Ds — коэффициент диффузии растворенного вещества в неподвижной фазе, у — отношение эффективной скорости перемещения молекул растворенного вещества к скорости газа-носителя, a k' — коэффициент емкости. Слагаемое В связано с продольной диффузией растворенного вещества. Величину В/и можно уменьшить, повышая температуру или увеличивая скорость газа-носителя.
В член Си, который оказывает решающее влияние при повышенных скоростях, вносит вклад поперечная диффузия в подвижной фазе, обусловленная различной длиной каналов между твердыми частицами и запаздыванием при установлении равновесия между фазами. Величина С определяется соотношением
в котором q — геометрический фактор, зависящий от размера частиц, d — толщина слоя неподвижной жидкой фазы, нане-
392 Глава 19
Рис. 19-4. Графическое изображение уравнения Ван-Деемтера для газовой хроматографии. На аналогичном графике для жидкостной хроматографии минимум (иопт) был бы настолько левее, что его было бы почти невозможно отличить от вертикальной оси.
сенной на твердые частицы, или диаметр твердых частиц, если жидкая фаза не нанесена, w—параметр, зависящий от упаковки колонки.
На рис. 19-4 изображены качественные зависимости между тремя членами уравнения Ван-Деемтера. Существует оптимальная линейная скорость ц0Пт, при которой И минимальна. Оптимальная скорость не может быть одинаковой для различных компонентов смеси, и ее следует выбирать для таких компонентов, которые труднее всего поддаются разделению. Чтобы сократить длительность анализа, хроматографисты обычно предпочитают проводить разделение при скоростях, превышающих теоретически оптимальные, даже если разрешение при этом несколько ухудшается. Если разделение проводится при «<«0Пг, то результаты его, судя по характеру наклона правой и левой ветвей кривой, могут, по-видимому, оказаться неудовлетворительными.
Уравнение Ван-Деемтера было выведено для газовой хроматографии. В жидкостной хроматографии это соотношение сложнее, чем описываемое уравнением (19-6); в слагаемое С входит несколько дополнительных членов. Минимум на кривой дает величину ц0Пт, приблизительно в 104 раз меньшую, чем в газовой хроматографии, и, следовательно, слишком низкую для практической работы.
Время удерживания и удерживаемый объем. Отрезок времени с момента ввода пробы в колонку до момента элюирова
Введение в хроматографию 393
ния компонента (измеряют время достижения максимума пика) называют временем удерживания tR. Часто более удобно пользоваться исправленным временем удерживания in, скорректированным с учетом времени прохождения через колонку не-удерживаемого компонента. В газовой хроматографии часто за неудерживаемый компонент можно принять воздух: tn=tn— —/возд. В жидкостной хроматографии такого универсального вещества нет, его можно выбрать только применительно к данной конкретной системе.
Результаты измерений можно также выразить через удерживаемый объем VR, который определяется следующим соотношением:
' VR-tRF (19-10)
где F— объемная скорость, измеряемая в см3/с (входящая в уравнение Ван-Деемтера линейная скорость измеряется в см/с). Удерживаемый объем можно скорректировать, вводя поправку на неудерживаемый компонент:
(19-И)
В газовой хроматографии необходимо внести поправку на перепад давления в колонке. Необходима также поправка на перепад температуры. В результате уравнение примет такой вид:
273 Тс
1
ws
(19-12)
в котором /—поправка на перепад давления. В ГХ величина / определяется уравнением
• 3 [(Pf/P0)2— 1]
2 \(Р(!Ро)3-1]
где Pi и Ро — давление на входе и выходе колонки соответственно. Величина / может достигать 0,5 при Л/Л) = 2,75. В ЖХ поправка не нужна, поскольку жидкости несжимаемы), Тс — температура колонки в кельвинах, ws—масса неподвижной фазы в колонке, Vg — объем элюента, необходимый для перемещения половины растворенного вещества через гипотетическую колонку, содержащую 1 г неподвижной фазы, температура которой равна 0 °C, а давление остается постоянным по всей ее длине. Величинами удерживаемых объемов удобно пользоваться для представления данных по удерживанию.
394 Глава 19
Рис. 19-5. Два пика равной высоты с различным разрешением /?, указанным на рисунке.
Разрешение. Разрешение служит мерой степени разделения соседних пиков. Оно определяется соотношением
ц /Я(А)~/Д(В) ffl(A)~*fl(B)
40
(19-13)
°.5(u-(a) + W(B))
На рис. 19-5 показаны два пика равной высоты с различной степенью разделения, т. е. с различными R. Обычно разрешение считают полным при R — 1,5.
Задачи
19-1. а) Взяв в качестве исходного уравнение (19-5), выведите выражение для зависимости от k'. б) При каких условиях Я8фф п II практиче-
ски равны?
Введение в хроматографию 395
19-2. Определите размерность всех величии в уравнениях (19-6) —(19-9).
19-3. Дифференцируя уравнения (19-6), выразите минимальную линейную скорость через А, В и С.
19-4. Полагая, что хроматограмма на рис. 19-3 получена на колонке длиной 0,5 м, определите число теоретических тарелок и соответствующую высоту, эквивалентную теоретической тарелке, для каждого пика. Насколько удовлетворяется уравнение (19-3)? Необходимые данные получите, проведя измерения (в мм) непосредственно по рисунку.
19-5. Решая совместно уравнения (19-4) и (19-13), покажите, что разрешение можно выразить формулой
n А —----------------
f 4
где //=0,5(/а,+ /в') и V=0,5(Va + Vb).
Литература
1. Dal Nogare S., Juvel В. S., Jr., Gas-Liquid Chromatography: Theory and Practice, Wiley-Interscience, New York, 1962.
2. Giddings J. C., Dynamics of Chromatography, pt. 1, v. 1, Dekker, New York, 1965.
3. Heftmann E. fed.), Chromatography, a Laboratory Handbook of Chromatographic and Electrophoretic Methods, Van Nostrand Reinhold, New York, 1975.
4. Snyder L. R., Kirkland J. J., Introduction to Modern Liquid Chromatography, (2d ed..), Wiley-Interscience, New York, 1979.
ГЛАВА
Газовая хроматография
Газовая хроматография — один из наиболее широко используемых в аналитических целях методов разделения. Она позволяет быстро и легко определять число компонентов смеси, включая примеси, а во многих случаях позволяет провести предварительную идентификацию соединения. Главное требование, которому должны отвечать соединения, разделяемые газохроматографически, — это достаточная их устойчивость при температуре, необходимой для поддержания вещества в газообразном состоянии. Газовый хроматограф необходим для химиков, занимающихся синтезом или исследованием соединения со средними молекулярными массами.
На рис. 20-1 схематически изображены основные узлы газового хроматографа. В настоящее время разработано много вариантов основных узлов этого прибора, соответствующих требованиям, предъявляемым к хроматографам, предназначенным для проведения серийных анализов и строгих научных исследований. Сначала рассмотрим физико-химические системы (выбор материалов для неподвижной и подвижной фаз), а затем перейдем к детальному описанию особенностей приборов.
Неподвижная фаза
По природе неподвижной фазы различают два вида ГХ. В одном из них (называемом газо-твердофазной хроматографией, ГТХ) неподвижная фаза представляет собой твердый материал, такой, как гранулированный силикагель, оксид алюминия или уголь. В основе процесса разделения лежит адсорбция на твердой поверхности. Этот вид ГХ находит весьма ограниченное применение главным образом из-за образования хвостов вследствие нелинейности изотерм адсорбции и отчасти из-за слишком сильного удерживания реакционноспособных газов, что снижает доступную площадь поверхности. Определенные ограничения обусловлены также каталитической активностью такой поверхности. Этот метод применяют главным образом
Газовая хроматография 397
Рис. 20-1. Схема типичного газового хроматографа с дифференциальным детектором.
для разделения постоянных газов и низкокипящих углеводородов.
Газо-жидкостная хроматография (ГЖХ) несомненно имеет более важное значение [1]. в этом виде хроматографии неподвижной фазой служит нелетучая жидкость, удерживаемая в виде тонкой пленки на твердом носителе, который в идеале не должен влиять на хроматографический процесс. Наиболее обычный носитель — это диатомовая земля, известная также как кизельгур и доступная в виде различных форм. Получают этот высокопористый кремнийсодержащий материал одним из двух описанных ниже способов. Обработка'щелочью и прокаливание дают белый продукт, обладающий некоторой остаточной щелочностью. Если же вести прокаливание в присутствии связующего вещества, но без щелочного плавня, то получается продукт красного или розового цвета, слегка кислый, известный как измельченный огнеупорный кирпич. Частицы должны быть однородными по размеру и не должны быть слишком мелкими. Типичный диаметр колеблется в интервале 60—80 меш (около 0,25—0,18 мм), 80—100 меш (0,18—0,15 мм) и 100— 120 меш (0,15—0,13 мм). Чем меньше зерна, тем более высокое давление требуется для прохождения газа через колонку.
Диатомовая земля, будучи разновидностью гидратированного силикагеля, содержит на поверхности много свободных групп ОН-, которые могут служить центрами адсорбции молекул растворенного вещества (сорбата). При наличии таких
398 Глава 20
Рис. 20-2. Образование хвостов при хроматографическом разделении .смеси этанола (/), метилэтилкетона (2), метилпропилкетоиа (3) и я-бутапола (4) на хромосорбе Р и устранение их силаннзацией хромосорба [2].
центров замедляется переход сорбата из жидкой пленки в газ-носитель, что приводит к образованию хвостов у пиков — явлению весьма нежелательному. Этот эффект можно снизить, используя полярную жидкую фазу, способную сильно адсорбироваться на носителе. В ряде случаев более эффективна обработка носителя силанизирующим агентом, например гексаме-тилдисилазаном (ГМДС), под действием которого группы Si—О—Н переходят в «безвредные» группы Si—О—Si(CH3)3.
II II
_ Si — О — Si-И [(CH3)3SiloNH —> - Si-О-Si-р NH3
II II-
ОН ОН (ГМДС) о о
I I
НзС — Si-СНз Н,С — Si — СН3
I ' I
СН3 СН3
На рис. 20-2 это иллюстрируется несколькими хроматограммами, полученными в различных условиях [2]. На рис. 20-2, а показана хроматограмма, полученная при использовании неполярной жидкости нуйол, нанесенной на огнеупорный кирпич (хромосорб Р). Полученный в этих условиях пик настолько
Газовая хроматография 399
асимметричен (из-за образования хвоста), что практически бесполезен. Применение более полярного растворителя динонилфталата, ДНФ (рис. 20-2, б), а также обработка силанизирую-щим агентом (рис. 20-2, в) приводят к некоторому улучшению, но пики приемлемой формы можно получить только при использовании обоих этих факторов (рис. 20-2, г).
Капиллярные колонки. В последнее время большое распространение получили капиллярные колонки из стекла или плавленого кварца, в которых носителем неподвижной жидкой фазы служат сами стенки колонки. Типичные размеры капилляра: внутренний диаметр 0,2 мм, длина от 50 до 100 м. Такие колонки могут быть очень эффективными: ВЭТТ составляет примерно 1 мм, а число теоретических тарелок достигает 500000 (вместо 5000 для стандартных'насадочных колонок).
Капиллярные колонки особенно полезны при разделении сложных смесей (см., например, рис. 20-3). Однако работа с ними требует специальных мер предосторожности и методики работы с изучаемым образцом, а также соответствующего выбора детектора, что будет описано позднее.
Неподвижная жидкая фаза
В литературе можно найти описание сотен материалов, рекомендуемых в качестве жидких фаз для специфических разделений. Многие из них при комнатной температуре являются роскоподобными, каучукоподобными или стеклообразными, но становятся жидкими при рабочей температуре ГХ-колонки. Они заметно отличаются по степени полярности и интервалу допустимых рабочих температур. Для решения большинства задач достаточно ограниченного числа жидких фаз. В табл. 20-1 перечислены несколько типичных неподвижных жидких фаз, пригодных для хроматографирования в разнообразных условиях.
В таблице указаны примерные интервалы рабочих температур, но их нельзя считать жесткими, поскольку на величину интервала влияет и ряд других факторов. Так, некоторые детекторы могут выдерживать более высокие парциальные давления летучих неподвижных жидкостей, чем другие. Иногда колонку можно нагревать до минимальной указанной температуры или даже более высокой, правда, на очёнь короткое время. Нижний предел температур определяется такими факторами, как возможность затвердевания или значительное повышение вязкости.
Полярность жидкой фазы характеризуют обычно не величиной диэлектрической проницаемости, а чисто эмпирически по способности разделять подходящие соединения в условиях
400 Глава 20
Время, мин
Рис. 20-3. Сравнение газовых хроматограмм, полученных на стеклянной капиллярной колонке (а) и насадочной колонке (б). Неподвижная жидкая фаза и температура в обоих случаях одинаковы. Размер пробы в случае б в 30 раз больше, а объемная скорость намного выше [3].
Газовая хроматография 401
Таблица 20-1. Некоторые неподвижные жидкие фазы
Неподвижная фаза
Интервал температур, "С
Н еполярные
Апиезон L 50—300
Силикон DC-200 50—350
Силикон SE-30 50—350
Сквалан 20—150
Силикон DC-QF-1
Силикон OV-17
Среднеполярные
0—275
0—280
Полярные
Карбовакс 20 М 60—225
Сукцинат диэтиленгликоля 20—200
Адипинат этиленгликоля 20—190
хроматографирования. Неполярные сорбаты, например, пентан, бутан и пропан, можно легко разделить на такой неполярной жидкой фазе, как сквалан, тогда как при хроматографировании на полярной жидкой фазе их пики располагаются значительно ближе друг к другу. При разделении полярных сорбатов, например спиртов, наблюдается обратная картина.
Количество жидкости, удерживаемой твердым носителем, определяют в массовых процентах. По обычной методике нанесения покрытия для стандартных колонок требуемое количество жидкости растворяют в летучем растворителе. Высушенный носитель тщательно перемешивают с полученным раствором в открытом сосуде, затем растворитель удаляют выпариванием. Носитель с жидким покрытием напоминает свободно текущий песок, и его можно непосредственно загрузить в прямую длинную металлическую трубку, периодически постукивая по ней, чтобы колонка заполнялась равномерно (в этих же целях можно использовать вибрацию). Заполненную трубку, снабженную газонепроницаемыми соединительными устройствами, можно свободно свернуть в спираль.
Нанесение жидкой фазы на стенки капиллярной колонки — процедура более трудная. Многие жидкости склонны образовывать капельки, а не сплошную пленку. Чтобы избежать этого, внутреннюю поверхность капилляра обрабатывают сухим НС1 или другим реагентом, чтобы она стала шероховатой; затем обработанный таким образом капилляр заполняют раствором требуемой жидкости в летучем растворителе и выпаривают
402 Глава 20
растворитель при пониженном давлении и повышенной температуре.
Эффективность капиллярных колонок настолько высока, что часто можно достичь удовлетворительного разделения, даже если неподвижная фаза не идеальна в отношении полярности. Следовательно, для решения большинства проблем достаточно располагать двумя колонками — с полярной и неполярной жидкой фазой соответственно [4].
Существует другой тип насадки для колонок, который можно рассматривать как промежуточный между непокрытым жидкой фазой адсорбентом в ГТХ и покрытым жидкой фазой носителем в ГЖХ. Это пористые гранулы структурированных органических полимеров, например сополимера стирола и дивинилбензола. Максимальная рабочая температура колонки, заполненной таким материалом, может достигать примерно 25'0 °C. Предполагается, что компоненты пробы распределяются между газовой фазой и аморфными гранулами, при этом последние выступают скорее в роли растворителя, чем адсорбента. Разделение на этих материалах может проходить с отличным разрешением. Эти материалы доступны, они поступают в продажу под названиями порапак (фирма Waters), хемипак (Gasukuro Kogyo) и серии хромосорб-100 (Johns Manville).
Большинство насадочных колонок или колонок с нанесенной жидкой неподвижной фазой перед использованием необходимо кондиционировать. Для этого через колонку в течение нескольких часов пропускают сухой газ-носитель при максимально допустимой температуре, чтобы удалить из нее любые летучие вещества, способные быть источниками помех.
Химически связанные фазы. Многие отвечающие всем основным требованиям жидкие фазы, к сожалению, заметно летучи, особенно при высоких температурах. Между тем известно, что это приводит к смещению нулевой линии хроматограммы и, следовательно, мешает наблюдению и измерению микроколичеств компонентов смеси. Эту трудность можно почти полностью устранить, если использовать органические соединения, ковалентно связанные с носителем [5]. Например, диатомовый носитель можно обработать хлорсиланом с длинноцепочечными алифатическими, ароматическими или гликолевыми заместителями. При этом протекающий через колонку газ контактирует только с органическими составляющими насадки, действующими подобно растворителю, в то время как молекулы прочно закреплены и не могут улетучиваться. Химически связанные неподвижные фазы, несмотря на их более высокую стоимость, очень популярны.
Газовая хроматография 403
Газ-носитель
Самый распространенный газ-носитель, несомненно, гелий. Объясняется это главным образом следующим: одним из наиболее важных детекторов в настоящее время является детектор по теплопроводности, измеряющий теплопроводность газов, а у водорода или гелия она значительно выше, чем у любого другого газа. Однако из-за низкой плотности этих газов их приходится пропускать через колонку с гораздо более высокой объемной скоростью, чем тяжелые газы, чтобы уменьшить диффузионные эффекты, вызывающие уширение пиков. У водорода есть два недостатка, препятствующих его применению: во-первых, он огне- и взрывоопасен и, во-вторых, химически реакционноспособен по отношению к ненасыщенным или способным к восстановлению веществам. Однако водород имеет одно специфическое преимущество: его можно генерировать электролитически. Применение этой его особенности описано в статье [6]. Другие газы-носители (азот или аргон), используемые в сочетании с детекторами других типов, будут подробно обсуждены позднее.
Ввод образца
Очень важной особенностью ГХ является ее пригодность для исследования малых проб — от 0,01 до 50 мкл жидкости. Существует два основных способа ввода пробы — при помощи крана и при помощи шприца (последний используется чаще). Применяемый в хроматографии шприц представляет собой по сути дела то же устройство, что и медицинский шприц для подкожных инъекций. При помощи шприцов можно осуществлять дозированный ввод проб объемом от 0,01 мкл и более. Хроматограф снабжен входным отверстием, герметически закрытым заменяемой резиновой мембраной (прокладкой), через которую и можно ввести иглу шприца. Шприцы можно использовать для введения газов или жидкостей с низкой вязкостью. Чтобы введенная проба сразу же испарилась, область ввода иглы необходимо нагреть, но при этом не следует допускать перегревания резиновой прокладки во избежание загрязнения пробы посторонними газами.
Для ввода пробы газа удобно пользоваться краном. На рис. 20-4 показано, как при помощи шестиходового крана-дозатора можно отмерить и ввести в хроматограф газообразную пробу. В положении а отбираемый поток протекает через калиброванную петлю, а через колонку проходит только газ-носитель. При повороте крана на 60° (в положение б) петлю заполняет газ-носитель, и захваченная проба переносится в колонку.
404 Глава 20
Рис. 20-4. Шестиходовой кран для ввода пробы.
При введении жидких образцов в капиллярную колонку требуются специальные меры предосторожности, позволяющие избежать перегрузки колонки. Объем вводимой в колонку пробы ни в коем случае не должен превышать 0,2 мкл, а предпочтительны меньшие объемы (порядка 0,01 мкл). Прецизионный шприц, пригодный для точного измерения таких маленьких объемов, и дорог, и хрупок. Альтернативное решение предусматривает использование делителя потока. В этом устройстве поток вводимого газа делится так, что большая доля его (30— 90 %) выпускается, а остаток поступает в колонку. Интересное описание возникающих при этом проблем и способ их решения, предложенный одним из изготовителей, даны в монографии [7]. В результате постоянных поисков в этой области созданы два новых устройства, описанные в статьях [8, 9].
Твердые образцы
Твердые вещества можно непосредственно исследовать при помощи ГХ только в том случае, если они имеют достаточно высокое давление паров, чтобы их можно было немедленно испарить в нагретом устройстве ввода. Часто их вводят в виде раствора в подходящем летучем растворителе. Многие нелетучие твердые вещества могут при нагревании разлагаться с образованием характерных газообразных продуктов, пригодных для хроматографирования. Этот метод известен как пиролитическая ГХ, или ПГХ [10]. Пробу помещают непосредственно в маленькую петельку из платиновой* проволоки, где ее можно нагреть до нескольких сотен градусов Цельсия за считанные миллисекунды при непрерывном пропускании газа-носителя. Продукты пиролиза поступают сразу в колонку.
Конечную температуру лучше всего контролировать, используя обогрев токами высокой частоты. Пробу помещают на проволоку или полоску из ферромагнитного сплава никеля и железа или кобальта. Каждый такой сплав при определенной температуре, называемой точкой Кюри, теряет ферромаг-
Газовая хроматография 405
Рис. 20-5. Пиролитические газовые хроматограммы трех ионообменных смол при 100°C (неподвижная фаза: полифенпловый эфир на целите) [11].
нитные свойства и становится парамагнитным, т. е. теряет способность поглощать РЧ-энергию. Часто, однако, более важна скорость нагревания, а не конечная температура [10].
Пиролитическая ГХ особенно полезна прн изучении полимерных материалов. Примером тому служит хроматограмма, показанная на рис. 20-5 [11].
Другой способ изучения нелетучих твердых веществ с помощью ГХ заключается в получении нелетучих химических производных, например разделение аминокислот в виде N-ацетил-или «-амиловых эфиров на колонке с карбоваксом [12]. Полезную серию производных можно получить при силанизации — введении ди- и триметилсилильных групп вместо лабильных атомов водорода, например в группах —ОН, —СООН, —SH и —NH2 [13]. Впервые такой подход был применен при хроматографировании сахаров, но он пригоден и для многих других классов соединений. Интересным примером его использования служит отделение бериллия в виде хелата с трифторацетилаце-тоном от микроколичеств других металлов в лунном грунте и осколках метеоритов [14].
Детекторы
В принципе измерение любого свойства, характеризующего различные газы, можно использовать в детекторе для ГХ. Описано несколько дюжин детекторов, которые можно разделить
406 Глава 20
на две большие группы. К первой группе относятся такие детекторы, сигнал которых пропорционален концентрации пробы (выраженной в мольных долях) и не зависит от скорости газа. Сигнал детекторов второй группы зависит от скорости, с которой проба достигает чувствительного элемента, а степень разбавления газом-носителем значения не имеет.
Детекторы первой группы. В эту группу входят детекторы, способные пассивно фиксировать некое физическое свойство газа без химического взаимодействия с ним. Самыми важными детекторами этой группы, безусловно, являются детектор по теплопроводности и детектор электронного захвата. В нее входят и малоиспользуемые детекторы, определяющие поглощение УФ- или ИК-излучения компонентами пробы.
Детекторы по теплопроводности [15]. Этот прибор, иногда называемый катарометром, состоит из металлического блока с двумя (или четырьмя) цилиндрическими полостями, высверленными машинным способом. В центр каждой полости вводят нить из тонкой проволоки (рис. 20-6). Эти сопротивления образуют плечи моста Уитстона (как показано на рис. 20-7), через которые циркулирует газ-носитель. Через мост должен проходить ток такой силы, чтобы температура нитей поддерживалась на несколько градусов выше температуры металлического блока. В исходном положении мост балансируют (регулируя величину /?б) по чистому гелию, проходящему вокруг всех нитей. При прохождении компонента пробы температура нити (рис. 20-7, а) или /?2 и /?з (рис. 20-7, б) повышается, поскольку
Рис, 20-6. Один из вариантов конструкций детектора по теплопроводности. В ячейку сравнения газ поступает в результате диффузии, что обеспечивает большую стабильность, а через измерительную ячейку проходит поток газа, что обеспечивает большую скорость.
Газовая хроматография 407
Рис. 20-7. Электрическая схема детектора по теплопроводности: а — мост с двумя активными плечами; б — мост, в котором активны все четыре плеча. Начальная балансировка осуществляется с помощью сопротивления Т?5. Гальванометр G в приборе заменен электронным усилителем и самописцем. Внешняя цепь относительно активного моста в а и б может быть идентичной.
все анализируемые газы (табл. 20-2) проводят тепло хуже, чем гелий (единственное исключение составляет водород). Температурный коэффициент сопротивления нити вызывает разбаланс моста, что усиливается и регистрируется самописцем.
408 Глава 20
Таблица 20-2. Теплопроводности некоторых газов
Г аз Теплопроводность, кал-с Хсм-^К-1
Н2 Не Ne СН4 О2 N2 со., сн^он Органические газы 44,5 36,0 11,6 8,18 6,35 6,24 3,96 3,68 1—4
В некоторых моделях металлическую нить заменяют термисторами, полупроводниковыми устройствами с высокими отрицательными температурными коэффициентами. Они более чувствительны при комнатных температурах, но несколько менее чувствительны при повышенных.
Детектор электронного захвата. Многие годы в ГХ использовались почти исключительно детекторы по теплопроводности, но с введением капиллярных колонок, работающих с малыми объемами проб, потребовались детекторы с более высокой чувствительностью.
В одном из способов детектирования, который способен обеспечить более высокую чувствительность, применяют модификацию ионизационной камеры, долгое время служившей в качестве радиационного детектора [16]. Поступающий из хроматографической колонки элюент Протекает через такую камеру и облучается в ней постоянным потоком р-электронов из однажды установленного в ней радиоизотопа (рис. 20-8). Наиболее удобный источник излучения — титановая фольга, содержащая адсорбированный тритий, но в этих целях пригоден и 63Ni. Оба источника представляют собой чистые р-излучатели, что позволяет легко создать защиту от опасной радиации.
Через камеру обычно протекает небольшой (наноамперный) ток, переносимый ионами газа. Органические соёдинения, элюируемые из колонки, влияют на ток, переносимый электронами, поскольку ионизируются. Органические ионы, однако, менее подвижны, чем ионы газа-носителя (обычно азота), поэтому ток при их прохождении через камеру уменьшается. Газом-носителем в этом случае может служить аргон, но он имеет тенденцию под действием p-излучения п-ереходить в метаста-бильное состояние, и поэтому приходится добавлять тушащий газ, например метан.
Детектор электронного захвата особенно чувствителен к га-
Газовая хроматография 409
Рис. 20-8. Схема детектора электронного захвата.
логенированным соединениям, и в значительной мере благодаря появлению этого детектора удалось обнаружить повсеместное распространение пестицидов в окружающей среде.
В детекторах электронного захвата величина сигнала пропорциональна xs — мольной доле сорбата, являющейся безразмерной величиной. Площадь пика на хроматограмме равна xsdt. Однако xs = usl (us + uc), где в знаменателе и — сумма скоростей газа-носителя и пробы. Поэтому можно записать, что
A=^xsdt = ^—^~dt (20-1)
Эта площадь пропорциональна массе дающего" пик вещества, т, только при условии, что и поддерживается постоянной, а осуществить это на практике совсем не просто (регуляторы скорости контролируют только ис). Поскольку маленькие пробы незначительно влияют на величину и, то
Л=-—fusd/ = — ^—dt = — (20-2)
и J iu J dt и
Поэтому если измеряется абсолютная величина, найденную площадь необходимо умножить на величину скорости. При измерении относительно стандарта этот фактор можно учесть при общей градуировке.
410 Глава 20
Элюат из колонки
Рис. 20-9. Схема водородного пламенно-ионизационного детектора (ПИД).
Детекторы второй группы. Многие органические соединения легко сгорают в водородно-кислородном пламени с образованием ионов, что в некоторых случаях сопровождается появлением характеристического излучения в УФ- или видимой области. Это их свойство положено в основу ряда высокочувствительных детекторов, предназначенных для ГХ.
Пламенно-ионизационный детектор (ПИД). В этом широко используемом детекторе образующиеся при сгорании пробы ионы собираются на заряженном электроде и возникающий в результате ток измеряют с помощью электрометрического усилителя [1]. На рис. 20-9 приведена схема ПИД. Газ-носитель (обычно азот), выходящий из колонки, смешивается с равным объемом водорода и сгорает в металлической форсунке в атмосфере воздуха. Форсунка (или окружающее ее кольцо) — это отрицательный электрод, а петля или цилиндр из инертного материала, окружающие пламя,— положительный элект
Газовая хроматография 411
род. Чувствительность обнаружения органических соединений примерно пропорциональна числу атомов углерода. Чувствительность ПИД примерно в 1000 раз выше, чем чувствительность детектора по теплопроводности. ПИД — отличный детектор многоцелевого назначения, особенно важную роль, учитывая его высокую чувствительность, он играет в капиллярной хроматографии. Поскольку сигнал этого детектора не зависит от скорости потока, он пригоден для количественных аналитических измерений.
Если окружить пламя блоками из CsBr или Rb2SO4, то можно получить ПИД, чувствительный практически только к фосфороорганическим соединениям, даже если они присутствуют в субмикрограммовых количествах [17]. Как и в немо-дифицированном детекторе, в этом его варианте используют пламя, обогащенное водородом, а не кислородом.
Физические явления, лежащие в основе работы данного детектора, полностью не объяснены. Это лучший детектор для определения микроколичеств фосфорсодержащих пестицидов.
Можно оптимизировать ПИД и для определения кремний-органических соединений [18]. При помощи такого варианта ПИД можно определять силанизированные спирты, амины и другие соединения в присутствии, например, углеводородов, не реагирующих с силанизирующими агентами.
Пламеино-фотометрический детектор. В основе работы этого детектора лежит измерение излучения обогащенного водородом пламени в присутствии серо- или фосфорсодержащих соединений. Эти элементы излучают при 394 и 526 нм соответственно [19]. В детектор (рис. 20-10) этого типа входит водо-
Оптическое волокно к фотоумножителю
Водород
Воздух
-------Элюат из колонки
Рис. 20-10. Схема пламенно-фотометрического детектора. Излучение пламени по линии связи из оптического волокна поступает на фотоумножитель.
412 Глава 20
родно-воздушная горелка, оптически связанная с фотоумножителем. Оптический путь локализован так, что наблюдают только верхнюю часть пламени, так как эта часть в отсутствие серы или фосфора заметного излучения не дает. Взаимозаменяемые оптические фильтры позволяют выделять спектральную линию того или другого элемента.
Поскольку в любом пламенном детекторе компонент пробы расходуется полностью, интегральная площадь под кривой сигнал — время (или сигнал — объем) должна точно соответствовать массе т обнаруживаемого соединения. Высота в любой точке кривой пропорциональна скорости массопереноса пробы us=dm/dt, поэтому площадь, ограниченная наблюдаемой кривой, равна
А = f usdt = { — dt = m (20-3)
J J dt
что позволяет определять m непосредственно.
Продувка детектора. Важной характеристикой любого аналитического хроматографа является мертвый объем систем, включая и детектор. При работе со стандартной насадочной ГХ-колонкой скорость газа-носителя достаточна для продувки детектора. Однако при применении капиллярной колонки эта скорость может быть настолько мала, что в детекторе наблюдается тенденция к застаиванию содержимого, а это приводит к уширению пиков, хорошо разделяемых в обычных условиях. Поэтому очень важно, чтобы компоненты пробы сразу же удалялись из детектора, но здесь существует ряд практических ограничений. Эту проблему можно обычно устранить, подавая дополнительный поток чистого газа-носителя непосредственно
Таблица 20-3. Сравнительные характеристики газохроматографических детекторов [17]
Детектор Предел обнаружения Диапазон линейности
По теплопроводности 10~12 г/мл 105
Пламенно-ионизационный 10~12 г/с 107
(ПИД) Электронного захвата (с три- 10-14 г/мл 10*
тием) Пламенно-ионизационный фос- Ю-16 г/с 103
фор-селективный Пламенно-фотометрический 10~12 г/с —
<фос фор-селективный ИК-спектрофотометр «Выше мкг» —
Масс-спектрометр «Выше мкг» 10°
Газовая хроматография 413
в детектор, чтобы очистить его. Эту процедуру называют продувкой.
В табл. 20-3 [17] приведены величины чувствительности ряда детекторов, которые, однако, следует рассматривать лишь как сравнительные их характеристики, поскольку с изменениями условий хроматографирования пределы чувствительности могут меняться.
Двойное детектирование
Поскольку различные детекторы имеют разную чувствительность для различных классов соединений, часто можно получить дополнительную информацию об изучаемом образце, используя на выходе колонки одновременно два детектора. Их можно разместить последовательно, чтобы поступающий из колонки элюат попадал сначала в один, затем во второй детектор (при этом первый детектор не должен разрушать пробу). Возможен и альтернативный подход: оба детектора подсоединяются к колонке параллельно при помощи делителя потока.
На рис. 20-11 приведен пример хроматограммы, полученной в результате двойного детектирования с помощью ДЭЗ и ПИД [20]. На каждой из хроматограмм можно видеть пики, отсутствующие или слабо выраженные на другой. Величины относительных сигналов этих двух детекторов для ряда соединений
Детектор электронного захвата
Рис. 20-11. Двойное детектирование высококипящих компонентов масла мяты перечной, разделенных методом ГХ. Пик X (а) первоначально был отнесен к метилциннамату, имеющему примерно такое же время удерживания. Результаты, полученные для чистого метилциннамата (б), показали, что детектор электронного захвата дает значительно больший сигнал, чем пламенноионизационный, тогда как X на рис. а наблюдается более отчетливо при обнаружении детектором последнего типа.
414 Глава 20
Таблица 20-4. Приблизительное отношение чувствительности детектора электронного захвата и пламенно-ионизационного детектора [20]
Соединение Отношение чувствительности
Гексан 10-е
Карвон 0,01
Пулегон 0,01
Ментол 0,1
Этилкротонат 0,9
Бензальдегид 1,0
Анисовый альдегид 1,0
Соединение Отношение чувствительности
Метилсалицилат 1,2
Диэтилмалеат 53
Диацетил 53
Этилциннамат 65
Бензилиденацетон 65
Коричный альдегид 200
Тетрахлорид углерода 10°
приведены в табл. 20-4. Вполне понятно, что двойная запись сигнала, подобная изображенной на рис. 20-11, может быть очень полезной при идентификации. Наличие пика на одной хроматограмме и отсутствие его на другой может позволить исключить из рассмотрения некоторые соединения, хотя и не может служить надежным основанием для идентификации.
Авторы статьи [21] разработали такой вариант пламенного детектора, который позволяет осуществлять одновременное наблюдение ионов и фотонов. Этот модифицированный детектор особенно полезен при исследовании металлоорганических соединений, когда спектроскопический детектор можно настроить на соответствующую длину волны с целью наблюдения атомной эмиссии металла. Для детектирования органических составляющих применяют ПИД.
Программирование температуры [22]
При хроматографическом разделении соединений одной группы, значительно различающихся по летучести, при одной и той же постоянной температуре возникает ряд трудностей. Низкокипя-щие компоненты элюируются быстро, и на хроматограмме их пики располагаются рядом друг с другом. Для элюирования же менее летучих соединений требуется намного больше времени, и их пики будут соответственно шире и ниже. Избежать этого можно, повышая температуру всей колонки с постоянной скоростью. Если это осуществить, то изменяются коэффициенты распределения соединений, зависящие от температуры, и в результате пики на хроматограмме будут распределяться намного более равномерно и будут менее отличаться по ширине. Это иллюстрирует хроматограмма (рис. 20-12), взятая из работы [23]. При переходе к высоким температурам (левая часть хроматограммы) нулевая линия повышается, что характерно для
Газовая хроматография 415
Рнс. 20-12. Влияние программирования температуры на разделение спиртов методом газовой хроматографии: хроматограммы, полученные при программировании температуры (а) и в изотермическом режиме (б). Заметьте, что шкала времени дана справа налево [23].
/ —метанол; 2 — этанол; 3— 1-пропанол; 4— 1-бутанол; 5— 1-пентанол; 6— циклогексанол; 7—1-октанол; 8—1-деканол; 9—1-додеканол.
ГХ с программированием температуры и обусловлено увеличением уноса или испарения неподвижной фазы, мало ощутимого при поддержании колонки при постоянной температуре. На рис. 20-12 этот эффект выражен не очень явно, но иногда он бывает настолько заметен, что искажает результаты. Его можно свести к минимуму или даже устранить, если использовать две параллельные колонки с одинаковой набивкой, помещенные в один термостат. Обе эти колонки необходимо подсоединить к идентичным детекторам, или поступающие из колонок элюаты должны проходить через противоположные плечи моста детектора по теплопроводности. Газ-носитель разделяется на два потока перед вводом в колонки, а проба вводится только в один поток. Детекторы включены в одну электриче-
416 Глава 20
скую цепь, и поэтому эффект уноса жидкой фазы, одинаковый в обеих колонках, удается скомпенсировать. Заметьте, что здесь используется тот же принцип, что и в оптических схемах двухлучевых спектрофотометров.
Серийные газовые хроматографы
Конструкции приборов для ГХ, пригодных для точных исследований, в основном предусматривают применение взаимозаменяемых узлов. Пользователь может располагать большим числом взаимозаменяемых колонок и быстро устанавливать любую подходящую (или пару) для решения конкретной задачи. Аналогичным образом можно выбрать подходящий детектор из нескольких имеющихся. Хроматограф снабжен встроенным регулятором программирования температуры. Самописец часто является отдельным, а не встроенным в раму основного прибора блоком, как обычно бывает в спектрофотометрах.
Если хроматограф предназначен для решения какой-то единственной задачи, как это может быть в лаборатории контроля качества, можно ограничиться всего одним детектором и колонкой одного типа. Существует также несколько упрощенных типов хроматографов, снабженных обычно только детектором по теплопроводности и ручным регулятором температуры. (Этого вполне достаточно для выполнения многих серийных анализов.)
Все современные приборы, кроме дешевых моделей, оборудованы встроенными микрокомпьютерами, управляемыми клавиатурой. Они очень помогают при создании условий выполнения конкретного анализа. В некоторых приборах компьютер управляет также графопостроителем и может печатать данные по временам удерживания и площадям пиков для каждого пика на хроматограмме. На рис. 20-13 приведен пример распечатки, которую можно получить при помощи одной из серийных моделей [24].
Качественный анализ
Используя селективность различных жидких фаз колонки и детекторов, можно получить определенную качественную информацию. Однако при этом обычно удается лишь выяснить, к какому классу принадлежит исследуемое соединение. Для получения дополнительной информации следует установить время удерживания — время, прошедшее с момента ввода пробы до момента появления компонента в детекторе. Для данной колонки, заданных скорости газа-носителя и температуры время удерживания конкретного соединения постоянно, но переносить
Газовая хроматография 417
Рис. 20-13. Распечатка с газового хроматографа, управляемого микрокомпьютером. Числа у индивидуальных пиков означают времена удерживания. Обратите внимание на большой динамический интервал измерения (колонка «площадь») главного и второстепенного компонентов. Анализируемый образец— природный газ [24].
Нормализованное ‘к Экспериментальное ‘r Площадь Относительные количества
1,35 1,37 17,590 0,518
1,42 1,47 1905,000 96,247
1,64 1,65 16,620 0,575
1,85 1,85 69,440 2,318
2,55 2,55 503,300 0,208
3,32 3,31 164,400 0,051
3,88 3,88 157,200 0,048
5,66 5,65 84,860 0,021
6,43 6,41 65,870 0,014
эти данные с одного набора условий на другой не представляется возможным (исключение составляют лишь некоторые методики, предусматривающие применение метода внутреннего
14 Заказ № 637
418 Глава 20
стандарта). Относительно того, как все же провести такое сравнение, существует несколько мнений.
Часто пользуются относительным временем удерживания, при определении которого в смесь до подачи ее на колонку вводят стандартное вещество и время удерживания измеряют относительно этого внутреннего стандарта. Таким стандартом часто служит н-пентан, но при хроматографировании на полярной неподвижной фазе при повышенной температуре более подходящими могут быть другие соединения, например метил-пальмитат.
Существует ряд полуэмпирических зависимостей между величинами относительного удерживания и другими параметрами. Как следует из рис. 20-14, график зависимости логарифма времени удерживания на неполярных жидких фазах от числа атомов углерода в гомологических рядах соединений представляет собой семейство параллельных линий. И как следует из наклона этих линий, с увеличением алифатической цепи на одну СНг-группу время удерживания примерно удваивается.
Поскольку от длины цепи зависит и точка кипения, то можно ожидать, что между логарифмом времени удерживания и точкой кипения также будет наблюдаться линейная зависимость. На рис. 20-15 приведена зависимость между логарифмом времени удерживания и точкой кипения для тех же данных, что и на рис. 20-14. Обратите внимание, что несколько параллельных линий (рис. 20-14) теперь слились в одну почти прямую линию.
Решительное улучшение дает введение системы индексов удерживания Ковача, представляющей постоянную шкалу, а не одну фиксированную точку для сравнения [26]. Первоначально индекс удерживания I выражали через объемы, но мы будем пользоваться эквивалентным выражением, в которое входит время удерживания:
/= 100
te^-’g'z
(20-4)
- ^g^Z-J-l ’g^Z
где tx' — исправленное время удерживания соединения х, tz' и t'z+i — исправленное время удерживания нормальных углеводородов с z и z+1 атомами углерода соответственно. Вместо собственно величин времени удерживания принято использовать логарифмы этих величин, поскольку, как мы уже видели, при этом зависимость для гомологического ряда имеет вид прямой. Для такого определения / необходимо, чтобы tx' лежало между t'z и Индекс почти линейно изменяется с изменением температуры, по крайней мере в узком интервале.
Газовая хроматография 419
Логарифм относительного времени удерживания
Рис. 20-14. Относительное время удерживания как функция числа атомов углерода для нескольких гомологических рядов углеводородов [25].
Логарифм относительного времени удерживания
Рис. 20-15. Относительное время удерживания как функция температуры кипения [25].
14*
420 Глава 20
Еще больше информации можно получить, сравнивая индексы Ковача для одного ряда соединений при разделении на полярной и неполярной неподвижных фазах. Наблюдаемое при этом различие в индексах (А/) позволяет составить представление о молекулярной структуре исследуемого соединения. Детали данного метода можно найти в работе [27], в которой он применен для исследования 156 углеводородов, содержащих циклопропановое кольцо.
Имитированная дистилляция. Как выяснилось, ГХ может успешно заменять аналитическую дистилляцию, особенно в нефтехимии. Для завершения высокоточной фракционной дистилляции иногда необходимо порядка 100 ч, поэтому она непригодна для контроля за процессами перегонки. Практически такие же и даже лучшие результаты можно получить всего за 1 ч при помощи двухколоночной ГХ с программированием температуры [28]. Компьютер непрерывно обрабатывает сигналы детектора и выдает распечатку накопленного итога с интервалом в несколько секунд. График зависимости этих данных от температуры по форме идентичен кривой, получаемой при 100-часовой перегонке. Если нужны истинные точки кипения, то необходимо внести исправления в температурную шкалу, так как парциальное давление компонента в газе-носителе не равно 1 атм, как это требуется по определению точки кипения.
Количественный анализ
Количественное газохроматографическое определение известных соединений обычно проводят по хроматограмме, записанной самописцем. Если пики острые и узкие, даже простое измерение высот даст небольшую погрешность. У более широких пиков необходимо измерять площадь. Многие газовые хроматографы снабжены встроенными интеграторами. Если же такого приспособления нет, то проще всего прибегнуть к треугольной аппроксимации гауссовой кривой (упоминавшейся в предыдущей главе), т. е. определить площадь пика, умножая его высоту на половину основания (рис. 19-3).
Другая методика количественного анализа предусматривает отбор фракций пробы при элюировании с колонки или после прохождения через неразрушающий детектор. Каждую полученную фракцию можно затем проанализировать тем или иным подходящим методом. Вариантов устройств для сбора пробы разработано очень много: это может быть простая пробирка, помещенная в бумажный стаканчик со льдом, и сложный автоматизированный коллектор с охлаждением. Если конечной целью является количественное определение, то к таким сложным устройствам не прибегают, они необходимы, лишь когда планируются дальнейшее изучение и идентификация.
Газовая хроматография 421
ГХ в гибридных методах
Газовая хроматография может выполнять полезную роль в сочетании с любым другим инструментальным методом, пригодным для работы с газообразными или летучими жидкими веществами. Наибольшее значение в настоящее время имеют хро-мато-масс-спектрометрия (ГХ/МС) и хромато-ИК-спектрофото-метрия (ГХ/ИК). Эти два метода можно даже использовать одновременно [29].
Особенно плодотворно сочетание ГХ с масс-спектрометрией. Масс-спектрометр дает наилучшие результаты в том случае, если исследуется газообразная проба, содержащая одно вещество, а именно таким требованиям должны отвечать отдельные фракции элюента, поступающего из газового хроматографа в масс-спектрометр. Основная проблема, возникающая при сочетании ГХ и МС, состоит в необходимости удаления газа-носителя, способного затопить насосную систему обычного масс-спектрометра. Этот и ряд других вопросов обсуждаются в гл. 22.
В случае тандема ГХ/ИК существуют две возможности. Максимальной чувствительности можно добиться за счет скорости, задерживая компоненты пробы последовательно один за другим по мере их поступления из газового хроматографа и записывая затем их спектры при помощи обычного ИК-спект-рофотометра. Газопроводящую систему можно сконструировать так, чтобы поток газа попадал прямо в специальную кювету,
Рис. 20-16. Обнаружение диметилфумарата с помощью ИК-детектора. Проба содержит 2 мкг вещества, поток делят так, чтобы одна его половина поступила в ПИД, а другая — к фурве-преобразователю ИК-спсктрофотометра [33].
422 Глава 20
3600-3570
0,200
0,147
3000-3020 0,095
0,042
-0,010
0,200 I-0,147 -0,095
0,042
-0,010
0,200 0,147 0,095 0,042 0,200 - 0,010 0,147
1500-1520 0,095
1760-1000
0,042
-0,010
1170-1155
0,200 0,147 0,095 0,042
- 0,010
5
0 50 100 150 200 250 300 350 Время, С
а
Рис. 20-17. Пять ИК-спектров ГХ-элюата в различных диапазонах длин волн (Nicolet Instrument Corporation).
Состав пробы
Гваякол
Толуол
о-Нитротолуол
Циклогексанон
Изобутилметакрилат
Растворитель метилеихлорид (ввод 6 мкл)
млн—1
10 000
5 000
1 000
500
100
Колонка из нержавеющей стали (6 футХ1/8 дюйм), заполненная OV-17 5% (100/120 г), Не 20 мл/мин.
Система ввода 170 °C
Детектор 100°С
Трубка 106°С
Колонка От 120 до 175 °C со скоростью 80 °С/мин
Аттенюатор 0,5 мВ X 16
где его на время регистрации спектра можно изолировать, закрывая соответствующий кран [30].
Можно также сконструировать ИК-спектрофотометр, способный регистрировать спектры «на лету». Со старыми ИК-детекторами этого сделать было нельзя, но с введением пироэлектрических детекторов столь быстрая регистрация спектра вполне возможна [31, 32]. До сих пор наилучшим вариантом является ИК-спектрофотометрия с фурье-преобразованием ИЗ-За присущей ей высокой скорости,
Газовая хроматография 423
На рис. 20-16 приведен пример использования ГХ/ФП-ИК для идентификации и оценки гомогенности диметилфумарата [33]. Повторяющееся сканирование полосы поглощения карбонильной группы (1650—1850 см-1) со скоростью 0,6 с на всю развертку дает одиночное интенсивное поглощение. Другой пример приведен на рис. 20-17.‘Спектрофотометр программируется так, чтобы можно было контролировать отдельные области, усредняя сигнал в указанном интервале волновых чисел, и записать зависимость получаемых результатов относительно времени удерживания. По полученному графику можно установить, к какому классу соединений относится каждый пик.
Задачи
20-1. Проводя количественный анализ смеси углеводородов, иногда полезно до детектирования окислить полученный элюат. Образовавшиеся пары воды поглощают в ловушке, а СОг пропускают через детектор по теплопроводности. В одном из экспериментов для семи компонентов получены следующие относительные площади пиков:
Пик Соедиие иие Относительная площадь
1 н-Пентаи 2,00
2 н-Гексан 5,72
3 З-Метил гексан 2,21
4 н-Гептан 8,15
5 2,2,4-Триметилпеитан 1,92
6 Толуол 3,16
7 н-Октан 5,05
а) Укажите преимущества и недостатки методики с предварительным окислением. б) Должна ли стадия окисления предшествовать прохождению через колонку или протекать параллельно и почему? в) Исходя из полученных данных рассчитайте состав пробы в мольных процентах от общего количества углеводородов.
20-2. На хроматограмме имеются следующие пики (дано расстояние на ленте самописца от точки ввода):
Соединение
Расстояние, см
Воздух 2,2
п-Гексан 8,5
Циклогексан 14,6
н-Гептаи 15,9
Толуол 18,7
н-Октан 31,5
424 Глава 20
Рассчитайте индексы Ковача для толуола и циклогексана.
20-3. На газожидкостной хроматограмме, записанной на ленте самописца, найдены пики компонентов А и Б, находящиеся соответственно на расстоянии 5,0 и 7,0 см от точки ввода. Пик воздуха удален от точки ввода на 1,0 см. Приведем другие нужные данные: скорость диаграммной ленты 6 см/мин; объемная скорость потока (F) 10 см3/с; давление вверху колонки (Pi) 2 атм, давление внизу колонки (Ро) 1атм; температура колонки (Тс) 100°C; масса неподвижной фазы (w,) 60 г; плотность неподвижной фазы (рв) 2 г/см. Рассчитайте Vg для каждого компонента (см. гл. 19).
20-4. При помощи ГХ можно определить субмикрограммовые количества цианида после превращения, его в CNC1 [34]. Реагентом служит водный раствор хлорамина Т (N-хлор-п-толуолсульфамид-натрий). Образовавшийся CNC1 экстрагируют точно известным количеством гексана и вводят в ГХ-ко-лоику. В качестве газа-носителя используют аргон, содержащий 5 % метана. При помощи детектора электронного захвата установлено, что в описанных условиях время удерживания CNC1 составляет 2,4 мин, а время удерживания гексана 4,8 мин. Пик гексана служит внутренним стандартом. Для графика зависимости отношения площадей пиков CNCl/гексан от концентрации ионов CN- средний наклон равен 2,53 мл/мкг. Пробу 1 мл крови (из следственного управления) обработали описанным выше способом; зарегистрирован пик CNC1 площадью 31,61 ед. и пик гексана площадью 0,2333 ед. Каково содержание (г/мл) цианида в крови?
20-5. Относительную разделяющую способность насадочной (р) и капиллярной (с) колонок можно выразить отношением
F = Л4иакс (С)/Л4макс (Р)> (20-5)
где ЛГмакс — максимальная объемная скорость потока. Для детекторов второй группы отношение Л4Макс к т, общей введенной массе (одинаковой для обеих колонок), описывается выражением
m = 2jMdz, (20-6)
о
где z — координата времени, взятая относительно максимума пика. Решая это уравнение совместно с уравнением для гауссовой кривой, получаем
М — Л4Иаксе«Р
1
2аа
Конечное выражение
F = ар/ас,
эквивалентно выражению
(Ne\l/2 с \ Np)
(20-7)
(20-8)
(20-9)
в котором tn — время удерживания, N — число теоретических тарелок [3].
а) Выведите уравнение (20-8) и подтвердите его адекватность уравнению (20-9). б) Выведите аналогичное уравнение для детекторов первой группы. 20-6. С помощью ГХ проведен анализ очищенного продукта на содержание метилэтилкетона (МЭК) и толуола. В качестве внутреннего стандарта было решено использовать трет-бутилбензол (трет-ББ). Для колонки с 1,2,3-трис-(2-цианокси)пропаном в качестве неподвижной жидкой фазы получены следующие данные:
Газовая хроматография 425
Стандартная смесь Исследуемое вещество
масс. % Высота масс. % Высота пика пика
МЭК 0,050 Толуол 0,050 трет-ББ 0,050 3,20 3,20 4,70 5,21 4,20 0,045 4,11
Каково содержание (масс. %) МЭК и толуола в пробе?
20-7. Согласно сообщению, удельные удерживаемые объемы Vg [уравнение i 19-12)] двух сорбатов составляют 24,0 и 20,0 мл на грамм неподвижной азы. Рассчитайте ожидаемое время элюирования для колонки с той же неподвижной фазой, используя следующие данные: температура колонки 27 °C; давление вверху колонки 2,20 атм, давление внизу колонки 1,00 атм; масса неподвижной фазы 3,50 г; объемная скорость 3,20 мл/с.
Литература
1. McNair И. М., In Chromatography, A Handbook of Chromatographic and Electrophoretic Methods, (3d ed.), E. Heftmann (ed.), Van Nostrand Reinhold, New York, 1975, ch. 9.
2. Dal Nogare S., Juvet R. S., Jr., Gas-Liquid Chromatography, Wiley-In-terscience, New York, 1962.
3. Yang F. J., Cram S. P„ J. High Res. Chromatogr. Chromatogr. Commun. 1979, v. 2, p. 487.
4. Novotny M., Anal. Chem., 1978, v. 50, p. 17A.
5. Grushka E., Ki kt a E. J., Jr., Anal. Chem. 1977, v. 49, p. 1004A.
6 Stevens M. R., Giffin С. E-, Shoemake G. R., Simmonds P. G., Rev. Sci. Instrum., 1972, v. 43, p. 1530.
7. Ettre L. S-, Purcell J. E., In Progress in Analytical Chemistry, v. 8, Simons J. L., Ewing G. W. (eds.), Plenum Press, New York, 1975, p. 119.
8. Wang F. S., Shanfield H., Zlatkis A., Anal. Chem., 1982, v. 54, p. 1886.
9. Peters ,T. L., Nestrick T. J., Lamparski L. L., Anal. Chem., 1982, v. 54, p. 1893.
10. Wolf C. J., Grayson M. A., Fanter D. L., Anal. Chem., 1980, v. 52, p. 348A.
11. Parrish J. R., Anal. Chem., 1973, v. 45, p. 1659.
12. Johnson D. E., Scott S. J., Meister A., Anal. Chem., 1961, v. 33, p. 669.
13. Sweeley С. C., Bentley R., Makita M., Wells ,W. W., J. Am. Chem. Soc., 1963, v. 85, p. 2497.
14. К J. Eisentraut, Grierst D. J., Sievers R. E., Anal. Chem., 1971, v. 43, p. 2003.
15. Johns T„ Stapp A. C., J. Chromatogr. Sci., 1973, v. 11, p. 234.
16. Burgett C. A., Res./Dev., 1974, 25(11), p. 28.
17. Hartmann С. H., Anal. Chem., 1971, v. 43(2), p. 113A.
18. Osman M. A., Hill H. H., Holdren M. W., Westberg H. H., Anal. Chem., 1979, v. 51, p. 1286.
19. Brody S. S„ Chaney J. E„ J. Gas Chromatogr., 1966, v. 4, p. 42.
.20 Hartmann С. H. et al., Aerograph Res. Notes (Varian Aerograph, Walnut Creek, CA), 1963 (fall), 1966 (spring).
21. Aue W. A., Hill H. H., Jr., Anal. Chem., 1973, v. 45, p. 729.
22. Harris W. E., Habgood H., Programmed Temperature Gas Chromatography, Wiley, New York, 1966.
426 Глава 20
23. Dal Nogare S., Bennett С. E., Anal. Chem., 1958, v. 30, p. 1157.
24. Peterson G. V., Poole J. S., Am. Lab., 1974, v. 6(5), p. 70.
25. Ladon A. W., Sandler S., Anal. Chem., 1973, v. 45, p. 921.
26. Ettre L. S„ Anal. Chem., 1964, v. 36(8), p. 31A.
27. Schomburg G., Dielmann G., Anal. Chem., 1973, v. 45, p. 1647.
28. Green L. E., Schmauch L. J., Worman J. C., Anal. Chem., 1964, v. 36, p. 1512.
29. Wilkins C. L., Giss G. N., Brissey G. M., Steiner S-, Anal. Chem., 1981, v. 53, p. 113.
30. Brady R. F., Anal. Chem., 1975, v. 47, p. 1425.
31. Penzias G. J., Anal. Chem., 1973, v. 45, p. 890.
32. Lephardt J. O-, Bulkin B. J., Anal. Chem., 1973, v. 45, p. 706.
33. Wall D. L., Mantz A. W„ Appl. Spectrosc., 1977, v. 31, p. 552.
34. Valentour /. C„ Aggarwal V., Sunshine I., Anal. Chem., 1974, v. 46, p. 924.
Литература общего плана
McNair H. M., Bonelli Е. J., Basic Gas Chromatography (5th ed.), Varian Instrument Division, Palo Alto, Ca, 1969.
ГЛАВА ^1
Жидкостная хроматография
Этот вид хроматографии был первым серьезно изученным хроматографическим аналитическим методом, предназначенным для выделения природных соединений, таких, как растительные пигменты, из растворов. Позднее интерес к жидкостной хроматографии надолго угас из-за стремительного развития газовой хроматографии. Однако в последнее время благодаря применению принципиально новых конструкторских разработок началось ее возрождение [1—3].
В старом варианте ЖХ стеклянную колонку размером, как правило, 1X30 см заполняли гранулированным твердым материалом и пропускали через нее элюент — жидкость-носитель, содержащий пробу. Основное неудобство такого метода разделения — его малая скорость. Если для достижения хорошего разделения колонку заполняли насадкой с зернами малого размера, то под действием только силы тяжести скорость элюирования (прохождения элюата через колонку) могла снизиться до нескольких капель в минуту. Очевидный способ увеличения пропускной способности колонки состоит в использовании нагнетательного насоса или давления газа. Жидкостные хроматографы старой конструкции не способны выдерживать необходимое высокое давление, поэтому для принципиального совершенствования метода необходима была полная реконструкция системы.
Основное преимущество жидкостной хроматографии перед газовой — возможность осуществлять разделение при более низких температурах, интервал которых ограничивается лишь точками кипения и замерзания растворителя. Это означает, что жидкостная хроматография позволяет разделять термически неустойчивые соединения, например белки, которые нельзя испарить без разрушения.
В качестве одного из примеров применения современной ЖХ рассмотрим разделение нуклеозидов [1]. До 1967 г. четкое разделение уридина, гуанозина, аденозина и цитидина на колонке диаметром 6 мм при давлении от 10 до (10—20) • 108 кПа
428 Глава 21
Резервуары
А Б
вакуум-насос
Насос
Кран-смеситель
Обезга-живание (нагреванием)
Змеедик Манометр Ввод пробы
(?) Манометр
Предколонка-
Делитель потока
Термоста-"тируемая камера
Дифференциальный детектор
Аналитическая
На слив или в коллектор фракций
Рис. 21-1. Технологическая схема типичного жидкостного хроматографа с приспособлением для градиентного элюирования.
длилось 1 ч. Два года спустя для аналогичного разделения на колонке диаметром 1 мм при давлении 400•108 кПа требовалось всего 24 мин, а в 1970 г. для такого же разделения на такой же колонке при давлении 5000 • 108 кПа достаточно 1,25 мин. Ясно, что аппаратура, работающая при таких высоких давлениях, должна отвечать особым требованиям. В данной главе мы рассмотрим только современные системы, работающие по принципу 'высокоэффективной жидкостной хроматографии (ВЭЖХ). Краткое превосходное описание состояния ЖХ на 1982 г. можно найти в обзорной статье [4].
На рис. 21-1 приведена технологическая схема многоцелевого жидкостного хроматографа. Два резервуара с растворителями, А и Б, позволяют получать смеси растворителей, используемые в качестве подвижной жидкой фазы. Каждый резервуар связан с камерой обезгаживания, где путем нагревания из растворителей удаляют растворенный воздух, который в противном случае может вызвать появление пузырей в колонке. Специальный кран позволяет подавать на колонку один из растворителей или их смесь в любой пропорции. Краном этим можно управлять так, чтобы соотношение объемов растворителей менялось постепенно по ходу хроматографического раз
Жидкостная хроматография 429
деления. Эта методика известна под названием градиентного элюирования.
Из крана-смесителя жидкость под высоким давлением поступает в собственно хроматограф, помещенный в термостате. Сначала жидкость проходит через змеевик, где она принимает рабочую температуру, а затем через предколонку с такой же* насадкой, что и основная колонка. Использование предколонки гарантирует достижение равновесия между подвижной фазой и насадкой аналитической колонки. Она также выполняет роль фильтра, удаляющего любые оставшиеся примеси.
Пробу с помощью шприца или крана, как в ГХ, вводят между предколонкой и основной колонкой. Элюат из колонки проходит через измерительную ячейку дифференциального детектора к коллектору фракций или сливу. Поток элюента, проходящий через сравнительную ячейку детектора, отделяют от основного потока элюента до ввода в него пробы. В более простых и более дешевых системах контроль температуры не предусмотрен, и соответственно отсутствует змеевик, показанный на схеме. Иногда не используется и предколонка, и не всегда обнаружение осуществляется дифференциальным детектором.
Насосы. Высокое давление, необходимое для продвижения подвижной фазы по колонке с достаточной скоростью, можно получить при помощи управляемого мотором насоса или при помощи сжатого воздуха или азота, поступающего из баллона. В последнем случае требуется мембрана или поршень для предотвращения прямого контакта газа с жидкостью, так как в противном случае огромное количество газа могло бы раствориться и вызвать появление пузырей в колонке или детекторе.
Чаще применяют поршневой или одноходовой насос, т. е. фактически шприц большой емкости, управляемый мотором. Большинство детекторов, применяемых в ЖХ, чувствительны к изменению параметров потока, поэтому насос должен нагнетать жидкость с постоянной скоростью без пульсаций. Для выполнения этого требования применяются различные остроумные методы [2]. Один из них предусматривает использование двух цилиндров и поршней: в то время как один из цилиндров быстро заполняется, другой медленно освобождается, а затем роли их меняются, после чего весь цикл повторяется.
Колонки. Колонки для ЖХ обычно изготавливают из стальных (нержавеющая сталь) трубок внутренним диаметром от 2 до 5 мм и длиной от 10 до 30 см. Ряд фирм продает уже готовые к работе заполненные насадкой колонки, или пользователь сам может подготовить их к работе (при набивке колонок
430 Глава 21
для ЖХ необходимо соблюдать специальные меры предосторожности [3]). Соединения колонок изготавливают из гибких трубок из нержавеющей стали внешним диаметром порядка 1,5 мм. Для устранения возможного уменьшения разрешения необходимо, чтобы мертвый объем всех соединительных устройств был минимальным.
Природа насадки зависит от типа предстоящего разделения и будет описана в следующем параграфе.
Виды жидкостной хроматографии
При рассмотрении ГХ мы говорили о двух различных механизмах удерживания компонентов пробы — взаимодействии с твердой фазой (газо-твердофазная хроматография, ГТХ) или с неподвижной жидкой фазой (газо-жидкостная хроматография, ГЖХ), которая может быть нанесена на твердый носитель (промежуточный вариант). Эти механизмы действуют и в ЖХ, но, помимо того, разделение может быть также обусловлено взаимодействием между растворителем и растворенными компонентами пробы.
Жидко-твердофазная хроматография (ЖТХ)
В данном варианте ЖХ удерживание компонентов пробы обусловлено их адсорбцией на гидроксильных группах субстрата — силикагеля или оксида алюминия. Полярные молекулы удерживаются сильнее, чем неполярные, и обычно наблюдается следующий порядок элюирования [3]: насыщенные углеводороды (небольшие &') <олефины<ароматические углеводороды^ор-ганические галогениды<сульфиды<простые эфиры<нитросое-динения<сложные эфиры ~ альдегиды кетоны < спирты ~амины<сульфоны<сульфоксиды<амиды< карбоновые кислоты (большие k'). (Символом k' обозначен коэффициент емкости, см. гл. 19). Сила адсорбции обычно является характеристикой функциональных групп органических соединений, поэтому этот метод особенно полезен для разделения соединений различных классов. Могут сказываться и стерические эффекты, так что в ряде случаев можно осуществить разделение и геометрических изомеров, например цис и транс.
Основные затруднения, возникающие при количественных разделениях методом ЖТХ, обусловлены нелинейностью изотерм адсорбции, наблюдаемой при повышенных концентрациях компонентов пробы. Желательно, чтобы линейная область (в которой количество адсорбированного вещества пропорционально концентрации) по возможности была более широкой. Экспериментально установлено, что интервал линейности шире для веществ слабоадсорбируемых, чем для сильноадсорбируемых.
Жидкостная хроматография 431
Это говорит о том, что на отдельных адсорбционных центрах возможны дипольные взаимодействия и взаимодействия за счет образования водородных связей, тогда как более слабые ван-дерваальсовы силы могут быть эффективными в более широкой области.
Активность адсорбента можно снизить, вводя в подвижную фазу небольшое количество воды. Молекулы воды селективно адсорбируются на наиболее активных центрах, оставляя какое-то количество менее активных центров незанятыми. В результате линейная область для органических соединений заметно расширяется. Адсорбция воды обратима, поэтому активность адсорбента можно менять, варьируя содержание воды в растворителе.
Жидко-жидкостная хроматография (ЖЖХ)
Эта разновидность хроматографии известна также как распределительная хроматография. Здесь мы рассмотрим системы, в которых гранулированный твердый материал служит только подложкой неподвижной фазы, совсем как в ГЖХ, а подвижной фазой является вторая жидкость. Поскольку эти две жидкости не должны смешиваться, они должны заметно различаться по степени полярности. Фиксировать можно и более полярную, и менее полярную жидкость. Обычно полярный растворитель, например спирт или воду, наносят на пористую подложку из силикагеля, оксида алюминия или силиката магния. Неполярный растворитель может удерживаться на тех же субстратах после их силанизации, после проведения которой они становятся гидрофобными. Такие системы принято называть распределительными хроматографическими системами с обращенными фазами. На рис. 21-2 показаны две хроматограммы, полученные при анализе смеси эфиров фталевой кислоты при помощи хроматографии с нормальными и обращенными фазами [1]. Обратите внимание, что время отложено в противоположных направлениях (рис. 21-2,а и б); сделано это для того, чтобы подчеркнуть изменение порядка элюирования.
ЖЖХ является более эффективным, чем ЖТХ, методом разделения соединений со сходными свойствами, например членов одного гомологического ряда. Важное преимущество ЖЖХ по сравнению с ЖТХ состоит также в том, что неподвижную жидкую фазу можно менять, не прибегая к перенабивке колонки. Однако легкость удаления жидкого покрытия обусловливает и возможность его вымывания. В методе ЖТХ активные центры могут отравляться в результате необратимой адсорбции высокоактивных соединений, и регенерировать колонки обычно Не представляется возможным.
432 Глава 21
Время, мин
а
Рис. 21-2. Хроматограммы пластификаторов на основе эфиров фталевой кислоты дибензилфталата (/), децилбеизилфталата (2) и дидецилфталата (5), полученные методом ЖЖХ с нормальной фазой (а) и обращенной фазой (б) [1]. Обратите внимание на противоположное направление шкалы времени. а: прибор Вариан аэрограф 4000; колонка 50 смХ2,4 мм, 1 % ОДПН иа чешуйчатом силикагеле; подвижная фаза изооктаи; объемная скорость 25,2 мл/ч; УФ-детектор; б: прибор Вариан аэрограф 4100; колонка 50 смХ Х2,1 мм, порасил С с химически привитым октадецилсилаиом; подвижная фаза от 30 % СНзОН в Н2О до 50 % СНзОН в НаО со скоростью 4,2 %/мин; объемная скорость 40 мл/мин; УФ-детектор.
Жидкостная хроматогра'фия 433
Жидкостная хроматография на химически привитых неподвижных фазах. В этом виде хроматографии используют те же типы насадок, что и в соответствующем варианте ГХ. Разделяющая способность колонок данного типа сравнима с аналогичной характеристикой колонок для ЖЖХ. Колонки первого типа стоят дороже, однако они вытесняют последние, поскольку вымывание из них неподвижной фазы практически исключено. На рис. 21-2,6 приведена хроматограмма, полученная при разделении на колонке с химически привитой жидкой фазой.
Ионообменная хроматография
Ионообменные смолы представляют собой высокополимеризо-ванные структурированные органические материалы, содержащие большое число кислотных или основных групп. Хотя смолы нерастворимы в воде, их активные группы гидрофильны и имеют различную степень сродства к ионам в растворе. Четыре
Таблица 21-1. Ионные смолы для хроматографии [2201
Класс «молы Природа смолы Эффективный интервал Область применения
Сильнокислотный ка-тиоиообмеиник Сульфированный полистирол 1 — 14 Разделение катионов, неорганических соединений, лантаноидов, витаминов Б, пептидов, аминокислот
Слабокислотиый ка-тиоиообмеииик Карбоксилирован-ный полиметакрилат 5—14 Разделение катионов, переходных элементов, аминокислот, органических основа- ний, антибиотиков, биохимические разделения
Сильиоосиовиые аииоиообменники Полистирол с группами четвертичного аммония 0—12 Разделение анионов, галогенов, комплексов витамина Б, жирных кислот
Слабоосиовные анионообмеииики Полиаминполисти-рол или фенил-формальдегид 0—9 Разделение анионных комплексов металлов, анионов различных зарядов, аминокислот, витаминов
434 Глава 21
существующих типа смол и некоторые примеры их применения перечислены в табл. 21-1. Важное значение имеет величина рабочего интервала pH. При pH ниже 5 слабокислотные смолы диссоциированы так мало, что обмен катионов практически не происходит. То же справедливо для слабоосновных смол при pH выше 9.
Для иллюстрации многосторонних возможностей ионообменной хроматографии рассмотрим три примера. Вначале обсудим разделение простых катионов на сильнокислотном ионообмен-нике. Относительное сродство однозарядных ионов изменяется в ряду Li+<H+<Na+<NH4+<K+<Rb+<Cs+<;Ag+<Tl+ (т. е. наименее прочно на смоле удерживается Li+). Аналогичный ряд для двухзарядных катионов выглядит так: UO22+<Mg2+< <Zn2+<Co2+<Cu2+<Cd2+<Ni2+<Ca2+<Sr2+<Pb2+<Ba2+.
Рис. 21-3 демонстрирует полное разделение Na+ и К+ [5]. Пробу вводили на колонку и элюировали 0,7 М соляной кислотой. Штриховой линией показаны кривые, рассчитанные исходя из распределения Гаусса. Средняя величина ВЭТТ составляла 0,5 мм.
Следующий пример (рис. 21-4)—разделение ионов ряда переходных металлов на сильноосновном анионообменнике [6]. Смолу предварительно перевели в С1~-форму, обработав 12 М соляной кислотой. Введенную на колонку пробу элюировали последовательно все более разбавленными растворами соляной кислоты. Ионы Ni(II) совсем не удерживаются даже из концентрированной НС1. Разбавление раствора НС1 до 6 М приводит к элюированию ионов Mn(II); ионы Со(П), Cu(II), Fe(III) и Zn (II) элюируются при концентрации НС1 4, 2,5, 0,5 и 0,005 М соответственно. Эта последовательность элюирования отражает относительную устойчивость хлоридных комплексов в реакциях типа СоС142“*^Со2+ + 4С1~, а также различия в сродстве смолы к указанным ионам и хлорид-иону.
Ионообменная хроматография — очень полезный способ разделения смесей биохимически важных соединений. С помощью параллельно установленных колонок с анионо- и катионооб-менниками [7] было осуществлено разделение более чем 100 компонентов мочи.
На рис. 21-5 показаны идентифицированные с помощью фотометрического детектора пики 46 компонентов смеси аминокислот. Несколько фирм выпускают хроматографы, специально предназначенные для анализа аминокислот, так называемые аминокислотные анализаторы.
Следует упомянуть о том, что ионный обмен находит важное применение в аналитической химии помимо хроматографии. Сюда относятся устранение мешающих ионов и предварительное концентрирование микроколичестр ионов. Для дальнейшего
Жидкостная хроматография 435
Объем элюирования, мл
Рис. 21-3. Разделение ионов натрия и калия методом ИОХ на катионите дау-экс-50 при элюировании 0,7 М НС1 [5].
Рис. 21-4. Разделение нескольких переходных металлов методом ИОХ на анионообменной смоле дауэкс-1 при элюировании НС1 с постоянно понижающейся концентрацией [6].
436 Глава 21
ипнпга'(/
unuiorimoj
Hnwy-d
nwog
Hngnujonai/nuja^-e nngntuomi/nujeu-i.
Hngniuonj meny
HniMHdo
(•ионное) HwruJDady Hnenumnodgmoi/wD n -7 ‘a
awoinm/im
-ИПОионпиу-^ -
Duioiror»/ UDgoHomodunngp»Dko»nuy--» -7 HndaoHD+HneoHdoy
яппниу, нпфоштц UnH'DlTOHDWQ
ошотпх mwHiMioenoHmy-i
нптиткнэф—-Hneadnj un'nnai/doy
. undoaif------
HnHoniupiOonhoIiun п-та , нпнопшаи —— (4WHHS2) nnmonfi
(MiUHSl'S) vuioumi UDHUiraDH-H-DHnuy-v (WOHHSL'e) НпМшп'П нпноиу Hn'nnwj Hni/odu ==^Z
ClirOHHSt'S) »шотп>1 гкдонпипеоонпм-' ' , ыпсожЬэ
(4V0HH S‘L) HnHOW^J
. ИПНОдЙ. NwoaunMoc^nj VUIWWM юджп&хЬиэу
Рнс. 21-5. Разделение градуировочной смеси аминокислот методом ионообменной хроматографии. Детектирование проводили фотометрическим детектором при 590 нм по реакции с нингидрином. По вертикали отложена оптическая плотность.
Жидкостная хроматография 437
изучения этого вопроса рекомендуется обратиться к специальной литературе (см., например, [8] и содержащиеся там ссылки).
Ион-парная хроматография (ИПХ)
Этот особый вид ЖЖХ обычно применяется для разделения способных к ионизации соединений, состоящих из катиона С+ и аниона А~, каждый из которых растворим в воде, в то время как ионная пара С+А~ растворима только в неводном растворителе. Колонки для ИПХ могут быть заполнены твердым носителем с нанесенной или химически привитой неподвижной жидкой фазой. Разделение методом ИПХ можно проводить и на нормальных, и на обращенных фазах. ИПХ — очень эффективный метод разделения карбоновых или сульфоновых кислот (при использовании в качестве противоиона иона тетраалкил-аммония), а также аминов (в присутствии перхлората в качестве противоиона). Более подробно этот вопрос освещен в литературе, см., например, [3].
Молекулярные сита и гель-проникающая хроматография [9]
Термином молекулярное сито называют класс высокопористых природных и синтетических кристаллических материалов, включая цеолиты, в которых все поры имеют примерно одинаковые размеры [10]. Синтетические материалы этого класса выпускают в гранулированном виде с диаметром пор от 0,4 до 1,5 нм, т. е. того же порядка, что и размеры органических молекул с малой молекулярной массой. Молекулы, размеры которых меньше, чем диаметр пор, легко диффундируют внутрь гранул и прочно адсорбируются там, а молекулы большего размера в поры вообще не попадают.
Ситовые материалы, используемые в колонках, обеспечивают эффективное удаление маленьких молекул из потока, позволяя более крупным молекулам беспрепятственно проходить через колонку. В табл. 21-2 перечисляется несколько веществ, адсорбирующихся некоторыми типами сит, выпускаемых одной из фирм. Очевидно, что этот тип разделения может иметь большое значение. Поскольку в данном случае разделение основано не на различии в коэффициентах распределения, математические соотношения, выведенные в гл. 19, не применимы, и метод, строго говоря, не является хроматографическим.
Молекулярно-ситовый эффект получил распространение и в хроматографии, в частности в методе, получившем название гель-проникающая или гель-фильтрационная хроматография (ГПХ или ГФХ). Неподвижная фаза состоит из гранул пори-
438 Глава 21
Таблица 21-2. Разделение компонентов иа молекулярных ситах Линде [21]
Адсорбируются на ситах
4А и 5А 5А (4А не адсорбируются) 13X (4А и 5А не адсорби-руютвя)
Этаи, пропан Этилен, ацетилен Метанол, этанол, «-пропанол Вода, аммиак, диоксид углерода, сульфид водорода Пропан и высшие «-парафины Бутен и высшие «-олефины н-Бутанол и высшие н-спирты Циклопропан Фреон-12 Разветвленные парафины Бензол и другие ароматические соединения Разветвленные (вторичные и третичные) спирты Циклогексан Тетрахлорид углерода, гексафторид серы, трифторид бора
Таблица 21-3. Интервал молекулярных масс соединений, удерживаемых гранулами р,-стирагеля Та
Размер пор, нм
Интервал молекулярных масс
10
50 100
103 10* 10ь
700
(0,05— 1) • 104 (0,1—20) • 104
(1— 20)-1О4 (1— 20)-105
(5—>10) • 10е
а С разрешения фирмы Waters Associates.
стого полимерного материала, чаще всего это уже упоминавшийся ранее сополимер стирола и дивинилбензола (ПС-ДВБ) и разнообразные полиакриламидные гели. Эти материалы имеют поры гораздо больших размеров, чем цеолиты, и перед использованием их следует привести в состояние равновесия с растворителем, выбранным в качестве элюента. Растворитель вызывает значительное набухание частиц, что служит характерным признаком геля. Ситовый эффект настолько велик, что его удобно выразить через молекулярную массу неудерживаемых («исключаемых») гелем соединений. В табл. 21-3 приведны такие данные для некоторых сортов ц-стирагеля — сополимера стирола и дивинилбензола, выпускаемого фирмой Waters Associates. Пределы исключения довольно широки, потому что заметное влияние оказывает форма молекулы (глобулярная, линейная, согнутая и т. д.).
На рис. 21-6 приведена типичная хроматограмма, полученная
Жидкостная хроматография 439
Рис. 21-6. Хроматограмма стандартной смеси полистиролов в толуоле, полученная методом ГПХ (Waters Associates). Отмечены молярные массы, соответствующие отдельным пикам. Введено 75 мкл стандартной смеси; 4 колонки 30 смX7,6 мм (внутр, диаметр) с р-стирагелем, 10* А; длительность анализа 10 мин; температура окружающей среды; объем сифона 2 мл.
методом ГПХ. Обратите внимание, что первым элюируется соединение с наибольшей молекулярной массой, поскольку оно не удерживается гелем.
Градиентное элюирование
До сих пор мы рассматривали лишь изократические методы хроматографического разделения, т. е. такие методы, в которых элюирование ведется только одной жидкой фазой. Изократические методы достаточно удобны для разделения подобных веществ, когда можно оптимизировать условия путем соответствующего подбора неподвижной и подвижной фаз. Однако для разделения сложных смесей, компоненты которых могут сильно различаться по степени полярности, эти методики не. вполне пригодны. Времена удерживания различных компонентов (или величины k') слишком разнородны, так что первые их пики, как правило, группируются вблизи стартовой точки, а последние пики настолько уширяются, что их трудно измерить точно.
Эту сложную проблему можно в значительной степени решить при помощи градиентов элюирования, используя смесители, как показано на рис. 21-1. Состав растворителя изменяют постепенно по мере проведения хроматографического разделения, начиная со «слабого» растворителя (дающего большие величины k') и кончая «сильным» растворителем (дающего наименьшие величины k'). В результате разрешение пиков на хромограмме заметно улучшается. На рис. 21-7 показаны хроматограммы одной и той же смеси, полученные при элюировании в изократическом и градиентном режимах [1]. Пример градиентного элюирования в ионообменной хроматографии приведен на рис. 21-4.
440 Глава 21
2-АМР 2-UMP З'-UMP 2'-СМР З'-СМР
Рис. 21-7. Сравнение градиентного и изократического элюирования при разделении нуклеотидов. Наклонная линия на нижнем рисунке характеризует изменение концентрации элюента (шкала справа) во времени. Аббревиатура у пиков — стандартные обозначения различных нуклеотидов [1].
Можно заметить, что градиентное элюирование играет в ЖХ примерно ту же роль, что и программирование температуры в ГХ.
Жидкостная хроматография 441
Детекторы
Фотометрические детекторы. В основе работы одного из наиболее распространенных в ЖХ детекторов лежит измерение поглощения в УФ (или видимой) области. Оптическую систему такого детектора сдедует тщательно сконструировать, чтобы можно было пропустить достаточное количество лучистой энергии через узкие кюветы, диаметр которых во избежание создания мертвого объема составляет около 1 мм. На рис. 21-8 приведена схема типичного фотометрического детектора. Измерительная ячейка и ячейка сравнения представляют собой цилиндрические каналы, высверленные в блоке из нержавеющей стали или тефлона и закрытые кварцевыми окошками. УФ-излучение лампы коллимируется линзой, проходит через каналы и поступает на два фотоэлемента. Обычно источником излучения служит ртутная лампа низкого давления с основной частью излучения при 254 нм. Некоторые модели снабжены флуоресцентным преобразователем, позволяющим получать спектральную полосу с максимумом при 280 нм. Это небольшая кварцевая пластинка, покрытая подходящим люминофором, поглощающим при 254 нм и излучающим при 280 нм. Многие важные классы соединений поглощают при одной из этих длин волн или при обеих.
Для достижения желаемой селективности необходимо иметь спектрофотометр с перестраиваемой длиной волны, хотя при этом будет наблюдаться некоторая потеря чувствительности. Некоторые спектрометры сконструированы так, что позволяют получить узкий интенсивный пучок излучения. Они дают дополнительное повышение селективности, которой не обладают другие детекторы для ЖХ, поскольку разные компоненты поступаю-
Рис. 21-8. Типичный фотометрический детектор для ЖХ.
442 Глава 21
0,500
0,400
0,300
0,200
0,100
0,000
Длина волны, нм
Рис. 21-9. Трехмерная диаграмма элюирования бензола в ацетонитриле с ЖХ-колоики. Детектором служит «гребенка» фотодиодов [11].
щего из колонки элюата обладают различной поглощающей способностью.
В работе [11] описан спектрофотометрический детектор [11] с «гребенкой» из 211 фотодиодов на подложке из одного кремния, допускающий одновременное измерение большого числа полос с узкой длиной волн. Полученную таким способом информацию обрабатывает компьютер (со скоростью 22 500 экспериментальных точек в секунду), он же хранит их в памяти для последующего построения графика. На рис. 21-9 показан пример трехмерной спектрохроматограммы, которую можно получить из этих данных.
Рефрактометрические детекторы. Другим важным детектором в ЖХ является рефрактометрический детектор, позволяющий уловить разницу в величинах показателей преломления всего лишь в одну десятимиллионную часть, что соответствует содержанию органических соединений в воде порядка нескольких микрограммов на миллилитр (эта оценка основана на данных, приведенных в справочнике для мальтозы при 25 °C). Это неселективный способ обнаружения, чувствительность которого зависит только от разности величин показателей преломления растворителя и растворенного вещества. Преимущество рефрактометрического детектора состоит в том, что его можно применять при работе с растворителями, физические свойства которых (например, поглощение в УФ-области) не позволяют использовать другой вариант обнаружения. Недостатком является невозможность применения градиентного элюирования, потому что изменения показателя преломления из-за программирования состава растворителя полностью перекрывают сигналы, обусловленные элюируемыми компонентами.
На рис. 21-10 показаны принципы работы двух рефрактометрических детёкторов. В основе работы модели а лежит из-
Жидкостная хроматография 443
Зеркало
Кювета детектора
Регулировка нуля а
Отраженный (неиспользуемый} луч
. Ячейка „ дь/улЯ
Ввод элюата из колонки
Стеклянная
Фотоэлемент
От источника обета
Рис. 21-10. Рефрактометрические детекторы для-ЖХ фирм Waters Associates (а) и Laboratory Data Control (б).
мерение угла смещения светового потока при прохождении через две призмы, заполненные жидкостью. Если жидкости идентичны, смещения не происходит. Однако изменение величины показателя преломления даже на несколько миллионных долей позволяет определить изменение угла потока. Фотодетектор чувствителен к направлению светового потока, а не к интенсивности его, поэтому выходной сигнал зависит от того, под каким углом входит световой поток. Для установки нуля пригодна простая стеклянная пластинка, которую можно повернуть так, чтобы немного сместить световой поток.
Другая схема детектирования (модель б) основана на измерении относительной интенсивности света, отраженного И про
444 Глава 21
шедшего через границу двух прозрачных сред. Поток элюата пропускают через узкий проход между одной гранью стеклянной призмы с углом 45° и стальной пластинкой. Параллельный пучок света входит в призму, как показано на рисунке; он частично отражается, а частично проходит через межфазную границу и дает световое пятно на задней стенке стальной пластинки. Стальную пластинку наблюдают в телескоп, соединенный с фотоэлементом. Математически можно показать, что если угол падения выбран правильно (немного меньше, чем критический угол полного отражения), то интенсивность наблюдаемого излучения линейно зависит от показателя преломления жидкости. Источник света и фокусирующую систему монтируют на рукоятке, которую можно поворачивать на маленький угол вокруг призмы для начальной регулировки.
Чувствительность этих двух рефрактометрических детекторов примерно одинакова, и они способны обнаружить изменение в показателе преломления в одну десятимиллибнную часть. Детектор отклоняющего типа (рис. 21-10, а) более удобен в работе, но у него наблюдается меньший диапазон линейности, чем у детектора, изображенного на рис. 21-10, б.
Другие детекторы. К УФ-детекторам примыкает детектор, измеряющий флуоресценцию, возбуждаемую излучением ртутной лампы при 254 нм [12]. Этот высокоселективный и чувствительный детектор особенно важен при проведении биохимических исследований, однако при проведении здесь измерений необходимо учитывать возможность тушения флуоресценции другими компонентами раствора или растворенным кислородом.
Еще один способ идентификации и количественного анализа компонентов элюата основан на использовании реакционного детектора. Поток элюата, поступающий из колонки, постоянно смешивается с реагентом, взаимодействующим с определяемым компонентом с образованием соединения с характеристическим излучением или флуоресценцией. Затем объединенный поток проходит через подходящий оптический детектор. Применение этого принципа показано на примере определения белков и аминокислот с использованием флуорескамина (флуорама) [13]. Это многоядерное ароматическое соединение реагирует с первичными аминами с образованием продуктов, сильно флуоресцирующих при 475 нм после возбуждения при 390 нм.
В детекторе по диэлектрической проницаемости [14] узкое пространство между двумя металлическими электродами служит конденсатором. Поступающий из колонки элюат, проходя через этот канал, вызывает изменение емкости в соответствии с величиной своей диэлектрической проницаемости е. Растворители, применяемые в ЖХ, значительно отличаются по величинам е, равным примерно 2 для симметричных молекул типа
Жидкостная хроматография 445
бензола или циклогексана и превышающим 180 для таких высокополярных соединений, как метилформамид. Отклонение, регистрируемое детектором, может быть положительным или отрицательным в зависимости от того, имеет ли компонент по сравнению с растворителем большую или меньшую величину е. Это позволяет создать универсальный детектор с чувствительностью несколько более высокой, чем у рефрактометрического детектора.
Электрохимическое детектирование (амперометрическое или кулонометрическое) также играет важную роль в ЖХ. Оно применимо при хроматографическом разделении на обращенных фазах, если компоненты пробы электроактивны [15]. В самом простом варианте этого метода обнаружения потенциал инертного рабочего электрода поддерживают с помощью потенциостата на уровне плато предельного диффузионного тока детектируемого компонента и записывают изменение тока во времени в процессе элюирования. Этот детектор вполне приемлем для обнаружения легко восстанавливающихся (или окисляющихся) веществ, но если необходимо сообщить электроду значительный потенциал, метод теряет и чувствительность, и селективность.
В одной из модификаций этого метода два рабочих электрода подсоединяют к отдельным потенциостатам. В зависимости от поставленной задачи электроды можно располагать по-разному [16], но лучше всего их поместить последовательно [17, 18]. Вытекающий из колонки элюат проходит через первый электрод, где происходит электрохимическая реакция, продукты которой обнаруживаются вторым электродом. Такая комбинация электродов значительно повышает селективность и чувствительность по сравнению с одностадийным детектированием. На рис. 21-11 показан ряд результатов, которые можно получить описанным способом [16]. В каждой из трех двойных хроматограмм верхняя кривая (m>i) одна и та же, соответствующая анодному току на первом (по движению потока) электроде, где поддерживают потенциал +1,10 В (относительно электрода Ag|AgCl). В примере А второй электрод выдерживали при + 0,95 В; в этом случае пики имеют меньшую высоту из-за частичного истощения на первом электроде. В примере Б второй электрод выдерживали при потенциале +0,35 В. При этом потенциале восстанавливаются соединения 1 и 2, окисленные на первом электроде. В примере В потенциал второго электрода равен 0,0 В; в этом случае восстанавливаются все четыре соединения, окисленные на первом электроде.
В ионообменной хроматографии часто применяют детектор по ионной проводимости. Основная трудность связана с электропроводностью элюента (жидкости-носителя), обязательно
446 Глава 21
I । I i I । I । I । I 0 4 8 12 16 20
Время, MUH
Рис. 21-11. Хроматограммы смесей феиолокислот оксибензойиой (/), ванилиновой (2), кофейной (<?) и метилкатехииа (4), полученные при обнаружении электрохимическим детектором с стеклоуглеродным индикаторным электродом и электродом сравиеиия Ag|AgCl. Аиодиый ток отложен вверх по ординате, катодный — вниз по ординате. Величины потенциалов см. в тексте. Пик, появляющийся примерно через 3 мни, принадлежит иеудерживаемому компоненту раствора [15].
содержащей относительно высокие концентрации электролитов. В работе [19] предложена методика, позволяющая преодолеть трудности. Впоследствии эта методика стала известна под названием ионной хроматографии. В дополнение к обычной ионообменной колонке, на которой ионы разделяются, была введена вторая колонка. Ее функция заключается в удалении фонового электролита без воздействия на изучаемые ионы. В качестве примера было показано отличное разделение ионов щелочных < металлов при элюировании 0,01 М НС1. На второй колонке, заполненной анионитом в ОН_-форме, соляная кислота была нейтрализована
НО + смола • ОН- -> смола • С1_ + Н2О.
Замена НС1 на Н2О позволяет использовать детектор по электропроводности для детектирования катионов щелочных металлов. Авторы указанной работы рассмотрели аналогичные методы детектирования анионов и аминов.
Жидкостная хроматография
447
Задачи
21-1. При разделении нуклеозидов методом колоночной ЖХ с использованием УФ-детектора идентифицированы следующие пики: воздух 4,0 мии, уридин 30 мии, ииозии 43 мии, гуаиозии 57 мин, адеиозии 71 мии, цитидии 96 мии. На колонке других размеров, ио содержащей ту же неподвижную фазу, пик воздуха появляется через 5,0 мин, а пик уридииа—через 53 мии. Через 100 мии элюируется еще один компонент. Идентифицируйте его.
21-2. Смесь нормальных парафинов разделяли методом ГПХ иа р-стирагеле с частицами диаметром 10 им. Получены следующие сигналы детекторов: показатель преломления (ПП) и диэлектрическая проницаемость (ДП):
Соединение пп ДП Время удерживания, мнн
Свн14 45 44 11,8
(-Ю^2Я 1 50 10,5
CiiHjq —29 52 9,4
а) Элюируются ли эти соединения в ожидаемой последовательности? Объясните почему, б) Какова последовательность их элюирования в условиях ЖЖХ с нормальными и обращенными фазами и в условиях ЖТХ? в) Объясните, почему СцНзо дает отрицательную величину при обнаружении рефрактометрическим детектором и положительную величину при обнаружении детектором по диэлектрической проницаемости?
21-3. Предложите принцип работы детектора без потери разрешения, в котором аминокислоты по мере элюирования реагируют с нингидрином, образуя окрашенные продукты, идентифицированные иа рис. 21-5. Подумайте о возможности взаимодействия смеси аминокислот с нингидрином до разделения иа колонке. Схема реакции:
R-CH-COOH
К л X + 1 он nh2 II о нингидрин О 11 ,Н НО |( Л PC + 2NH3 + Л)Н НО II О О II С\ ^-Н R-C-COOH с/ ОН- О II О + NH3 О II II о О О-NH+® И II 0 (ашй) 0
448 Глава 21
21-4. Твердый образец содержит нитраты натрия и калия. Навеску 5,00 г растворили в достаточном количестве воды и разбавили деионизованной водой до 1,00 л. Далее 50,0 мл раствора пропустили через колонку с дауэксом-50 в Н+-форме. Выделившуюся кислоту элюировали водой. После завершения элюирования, что установлено прибором, регистрирующим электропроводность, элюированную кислоту оттитровали 0,1032 М раствором NaOH до достижения конечной точки по метиловому оранжевому. На это потребовалось 27,90 мл титранта. Каков процентный состав пробы?
Литература
1. Johnson Е. J., Stevenson R., Basic Liquid Chromatography Varian Associates, Palo Alto, 1978.
2. Veening H., In Topics in Chemical Instrumentation. Ewing G. W. (ed.), v. 2, American Chemical Society, Washington, 1977, p. 148.
3. Snyder L. R., Kirkland J. J-, Introduction to Modern Liquid Chromatography (2d ed.), Wiley Interscience, New York, 1979.
4. Freeman D„ H., Science, 1982, v. 218, p. 235.
5. Beukenkamp J., Rietnan W., Ill, Anal. Chem., 1950, v. 22, p. 582.
6. Kraus К A., Moore G. E., J. Am. Chem. Soc., 1953, v. 75, p. 1460.
7. Scott C. D., Chilcote D. C., Lee N. E., Anal. Chem., 1972, v. 44, p. 85.
8. Kolthoff /. M., Sandell E. B„ Meehan E. J., Bruckensteln S., Quantitative Chemical Analysis (4th ed.), Macmillan, New York, 1969, p. 291.
9. Yau W. W., Kirkland J. J., Bly D. D., Modern Size-Exclusion Liquid Chromatography, Wiley, New York, 1979.
10. Meier W. M., Uytterhoeven J. B. (eds.) Molecular Sieves, Advances in Chemistry. No 121, American Chemical Society, Washington, 1973.
11. Miller J. C., George S. A., Willis B. G., Science, 1982, v. 218, p. 241.
12. Lyons J. W., Faulkner L. R., Anal. Chem., 1982, v. 54, p. 1960.
13. Udenfriend S-, Stein S., Bohlen P., Dairman W., Leimgruber W„ Weigele M., Science, 1972, v. 178, p. 871.
14. Benningfield L. V., Jr., Mowery R. A., J. Chromatogr. Sci., 1981, v. 19,
p. 115.
15 Kissinger P. T., Jr., Anal. Chem., 1977, v. 49, p. 447A.
16. Roston D. A., Kissinger P. T., Anal. Chem., 1982, v. 54, p. 429.
17. Andrews R. W., Schubert C., Morrison J., Zink E. W., Matson W. R., Am. Lab., 1982, v. 14(10), p. 140.
18. MacCrehan W. A., Durst R. A., Anal. Chem., 1981, v. 53, p. 1700.
19. Small H., Stevens T. S., Bauman W. C„ Anal. Chem., 1975, v. 47, p. 1801.
Глава
Масс-спектрометрия
Масс-спектрометр — это прибор, разделяющий заряженные молекулы (ионы) газообразных веществ по их массам. Хотя реальной связи между масс-спектроскопией и оптической спектроскопией не существует, названия приборов масс-спектрометр и масс-спектрограф были выбраны по аналогии, поскольку ранние приборы регистрировали фотографические записи, похожие на линии оптических спектров.
Аппаратура
Существует несколько различных типов масс-спектрометров, но все они должны содержать узлы, предназначенные для выполнения следующих функций (рис. 22-1): 1) ионизации пробы, 2) ускорения ионов электростатическим полем, 3) распределения ионов согласно отношению их массы к заряду, 4) детектирования ионов по соответствующему электрическому сигналу. Эти функции будут описаны в следующих разделах.
Поскольку в масс-спектрометре используют поток газообразных ионов, дающих четкие траектории вследствие комбинаций электростатического и магнитного полей, то очень важно, чтобы все узлы прибора — от источника ионов до детектора — были вакуумированы. Остаточное давление не должно обычно превышать порядка 10~3 Па (около 10-5 мм рт. ст.). Для этого необходимо эффективное и длительное откачивание с помощью либо ион-геттерного, либо масляного диффузионного насоса с ловушкой, охлаждаемой жидким азотом. Важно иметь чувствительный и точный прибор для измерения давления. На рис. 22-2 показана типичная вакуумная система.
Источники ионов
До начала 70-х годов масс-спектры можно было получить лишь для газов или таких веществ, которые можно перевести в газообразное состояние при пониженном давлении в масс-спектро-
15 Заказ № 537
450 Глава 22
Рис. 22-1. Блок-схема типичного масс-спектрометра.
Рис. 22-2. Типичная вакуумная система для масс-спектрометра. Ионизационный датчик — обычное устройство для измерения остаточного давления.
метре. За прошедшее время разработан ряд способов получения газообразных ионов из твердых веществ и высококипящих жидкостей.
Если образец возможно превратить в низкотемпературный газ, ионы можно получить с помощью пучка электронов из нагретого катода. Этот процесс известен как ионизация с помощью электронного удара (ЭУ). Ионы могут образоваться также в результате вторичного процесса при протекании ион-молекулярной реакции в газовой фазе. Этот способ называют химической ионизацией (ХИ). Разработаны и другие механизмы обеспечения энергии для образования ионов в газах, но они применяются реже.
Масс-спектрометрия 451
Рис. 22-3. Принципиальная схема устройства для ионизации электронным ударом. Катод и анод формируют пучок электронов. Ионы образуются непосредственно над выталкивающими электродами (Л и 5). Положительный заряд выталкивающих электродов и отрицательный заряд фокусирующих электродов (В и Г) заставляют положительные иоиы двигаться вверх (по диаграмме).
Газообразные ионы можно получить из твердых веществ с помощью одного из вариантов методики, известной под названием десорбция (хотя этот термин не всегда описывает происходящие процессы) [1]. Энергию к поверхности образца подводят с помощью пучка ионов или нейтральных атомов. Удар вызывает миниатюрный взрыв, в результате которого небольшая часть молекул отрывается с поверхности и одновременно ионизируется. Десорбцию можно осуществить, воздействуя лучом лазера или электростатическим полем.
Ионизация электронами (электронный удар, ЭУ). На рис. 22-3 показана принципиальная схема устройства для ионизации ЭУ. Образец в виде газа при низком давлении вводят в полость металлического блока, где он подвергается воздействию пучка электронов. Между нагретым вольфрамовым или рениевым катодом и электронной ловушкой устанавливают ускоряющее напряжение 70 В. Этот потенциал сообщает электронам энергию, достаточную для ионизации практически любых межатомных связей.
Положительные ионы * удаляют из полости, в которой они образуются, с помощью электростатического поля между поло
* При бомбардировке молекул высокоэиергетическими электронами положительно заряженных ионов обычно больше, чем отрицательно заряженных, и поэтому в большинстве случаев масс-спектральиый анализ имеет дело только с положительными ионами. Если необходимо изучать и отрицательно заряженные ионы, следует изменить знак потенциала ускорения.
15*
452 Глава 22
жительно заряженным отталкивающим электродом и отрицательно заряженным ускоряющим электродом. Далее их пропускают через узкое отверстие или щель и несколько фокусирующих электродов в масс-анализатор.
Ионы, выходящие из источника, должны обладать по возможности близкими кинетическими энергиями, чтобы их можно было правильно сфокусировать. В процессе ускорения ионы приобретают энергию, равную E = zV, где z— заряд иона, V — наложенный потенциал. После прохождения ионов через щель их кинетическая энергия определяется уравнением
Дкин = zV + До = Vaffio2 (22-1)
где т — масса иона, v — его скорость, Ео — кинетическая энергия, которую ион может иметь в результате ионизирующего удара до ускорения. Энергии выходящих ионов всегда будут различаться из-за колебаний величины Ео и из-за того, что они образуются на различных расстояниях от щели. Тщательная разработка конструкции источника позволяет свести этот разброс к минимуму, так что им можно обычно пренебречь для спектрометров низкого разрешения.
Преобразовав уравнение (22-1) так, чтобы исключить Ео, получаем выражение
o = V2V/(m/z) (22-2)
из которого видно, что ионы движутся со скоростями, зависящими от отношения их массы к заряду, m]z, обычно называемого массовым числом и иногда выражаемого в единицах атомной массы (массовое число очень часто обозначают отношением т/е, а не m/z; е выражено в единицах заряда электрона и численно равно z).
Существует ряд методов введения пробы в источник ионизации. Выбор того или иного метода определяется такими физическими свойствами вещества, как точка плавления или давление паров. Многие спектрометры приспособлены для нескольких способов ввода пробы. Если изучаемый образец является газом или летучей жидкостью, то лучше всего наполнить им заранее откачанный стеклянный или металлический контейнер, сообщающийся с источником через крошечное отверстие, называемое точечной диафрагмой, или молекулярным натекателем (рис. 22-4).
Молекулярный натекатель может состоять из одного и более отверстий, проколотых иглой в тонкой золотой мембране. Отверстия должны быть малы по сравнению со средним свободным пробегом молекулы газа; приемлемым является диаметр порядка 0,01 мм. Это обеспечит условия выполнения закона
Масс-спектрометрия 453
Рис. 22-4. Схема возможной системы ввода пробы в масс-аиализатор. Газы или летучие жидкости вводят при помощи шприца или клапана в большой резервуар, расположенный слева, откуда оии попадают в источник иоиов через молекулярный иатекатель. Твердые вещества вводят с помощью штока, расположенного справа. Диагональный клапан изолирует вакуумную систему от атмосферы во время движения штока.
эффузии Грэхема: газы проходят через отверстие со скоростями, обратно пропорциональными их молекулярным массам. Неиони-зованные молекулы при диффузии от источника ионов подчиняются тому же закону, поэтому относительные парциальные давления различных газов в источнике будут такими же, какими они были во внешнем резервуаре, что является важным условием при проведении количественного анализа. Баллон напуска должен содержать достаточно образца для дублирования большого числа спектров.
Твердые органические вещества, которые нельзя превратить в пар, можно ввести в источник с помощью штока — обычно стержня из нержавеющей стали диаметром 6 мм и длиной 25 см, имеющего на конце крошечную чашечку для удерживания образца. Образец вводят через вакуумный шлюз (рис. 22-4) и нагревают с помощью электрической спирали. Для получения удовлетворительного спектра достаточно миллиграмма или даже меньше образца, но образец откачивается достаточно быстро.
Электронный удар — это довольно жесткий метод получения ионов, разрывающий молекулярную мишень на большое число фрагментов. Он может быть полезен в качестве метода «отпечатка пальцев», позволяет получить много информации об отно
454 Глава 22
сительной силе различных атомных связей, но он может и ввести в заблуждение, особенно при исследовании многокомпонентных образцов. Интерпретация фрагментационной картины будет кратко описана дальше.
Химическая ионизация (ХИ). В этом методе [2] образец до облучения пучком электронов разбавляют большим избытком (порядка 104: 1) газа-«реагента». Вероятность первичных ионизующих столкновений между электронами и молекулами образца после этого настолько мала, что первичные ионы образуются почти исключительно из молекул реагента. В качестве реагентов обычно используют газы с низкой молекулярной массой, например СН4, U30-C4H10, NH3 и инертные газы Не и Аг.
Вторичные ионы образуются в результате переноса атома водорода или электрона. Если реагентом служит, например, метан, реакции протекают в такой последовательности:
CH4+e->CHt+2e
CH4 + e->-CHt+ Н++ 2е
СН^ + СН4->-СН^+СНз CHt+CH4->-C2Ht+H2
R—СНз + CHt R —CHt + СН4
где R—СНз (последняя стадия)—это молекула исследуемого вещества. Образующийся ион R—СН4+ можно обозначить символом (М + Н)+, где М—исходная молекула. Исследования показали, что частицы СН5+ и С2Н5+ в сумме составляют около 90 % ионов, присутствующих в этой системе.
Масс-спектры после химической ионизации заметно отличаются от масс-спектров после ионизации электронным ударом и дают информацию иного рода о процессе ионизации. Масс-спектры, получаемые после химической ионизации, намного проще* содержат меньше пиков, и поэтому их часто легче интерпретировать. На рис. 22-5 показаны два масс-спектра одного соединения, полученные с двумя типами источников ионов.
Камера источника для химической ионизации, так же как и соответствующая вакуумная система, должна быть специально сконструирована так, чтобы можно было поддерживать отношение давлений по обе стороны щели выше 10s.
В качестве интересного примера применения необычного газа-реагента рассмотрим изучение спиртов в присутствии из-
Масс-спектрометрия 455
Рис. 22-5. Масс-спектры днкетопиперазина при ионизации ЭУ и ХИ метаном в качестве газа-реагента. Обратите внимание на высокую интенсивность пика (М+Н)+, обозиачеиного QM+ для «квазимолекулярного» нона. Пик при 225 соответствует продукту присоединения С2Н5+ к молекулярному иону и подтверждает, что молекулярный ион должен быть 196. Это пример спектра, полученного с помощью компьютера (Finnigan Corporation).
бытка оксида азота [3]. Из спиртов разного вида образуются следующие ионы:
|Ион Спирт: первичный вторичный третичный
(М—1)+ X X
(М—2 + NO)+ X X
(М—3)+ (М—ОН)+ X X X
(М + NO)+ X
Наблюдая эти ионы, можно определять и вид спирта, и его молекулярную массу.
Масс-спектрометрия вторичных ионов [4]. В этом методе твердый образец обрабатывают пучком ионов из внешнего источ
456 Глава 22
ника, подобного изображенному на рис. 12-20. Наиболее часто используют ионы Аг+ и О2+. Если изучается непроводящий образец, необходимо предотвратить накопление положительного электростатического заряда на поверхности, мешающее правильной фокусировке. С этой целью образец облучают избытком низкоэнергетических электронов из электронной пушки. Тонко-измельченный образец, помещенный на проводящую подложку, в достаточной мере рассеивает заряд [5]. Органические соединения обычно адсорбируют (или наносят каким-либо другим способом) на подложку из такого металла, как серебро.
Образование ионов любого знака может происходить по различным механизмам [4]. Во-первых, молекула М может присоединиться к заряженному атому металлического субстрата Ag+, образуя металлоорганическую частицу (Ag+M)+. Этот процесс называется катионизацией. Во-вторых, молекула М может ионизироваться в процессе электронного перехода с образованием ион-радикалов М. или М. . В-третьих, ионы, образующиеся из органических солей, могут быть вытолкнуты из образца в результате прямого переноса при столкновении с первичными ионами. На рис. 22-6 приведены системы ионов, образующихся по каждому из этих механизмов. В результате мо-номолекулярных реакций в газовой фазе образуется много ионов с небольшими массами.
Интересным развитием метода является прямое изучение бумажных хроматограмм с помощью масс-спектрометрии вторичных ионов [4].
Бомбардировка быстрыми атомами. Этот метод отличается от масс-спектрометрии вторичных ионов только тем, что облучающий поток состоит из нейтральных атомов, а не из ионов. Атомная пушка по существу та же, что и для получения пучка ионов, но ее геометрия такова, что ионы имеют возможность реагировать с нейтральными атомами или вторичными электронами, так что они теряют свой заряд и сохраняют момент количества движения. Возможные процессы
А+4-А^А4-А+
или
А+ + е^А
(стрелками указаны направления моментов). Пучок атомов, выходящий из пушки, содержит ионы, которые необходимо удалить электростатическим дефлектором [6, 7]. Подходящий пучок образуют атомы аргона, но более высокую чувствительность достигают при использовании более дорогого ксенона [8].
Масс-спектрометрия 457
20
120
60 80 100
m/z
(3
юо
N(ch3)3(c2H5)+ Г/Ад 5кВ,2-Ю~9А (м-|)+
88
-5-25
58
50
72
Ад
20 40 60 80 100
m/z
Рис, 22-6, Спектры, полученные с помощью масс-спектрометрии вторичных ионов, иллюстрирующие три различных процесса ионизации: а) присоединение катиона серебра к молекуле, б) присоединение электрона с образованием отрицательного ион-радикала, в) потерю противоиона (иодид) с образованием иоиа четвертичного аммония. Символ Ag означает, что для ввода в спектрометр органическое вещество нанесено на серебряную подложку, а, в — масс-спектрометрия вторичных положительных ионов; б — масс-спектрометрия вторичных отрицательных ионов. Присутствие Na+ (а) и С1~ (б) обусловлено примесью NaCl [4].
458 Глава 22
Рис. 22-7. Магнитный сектор спектрометра с углом 90° (а) и 180° (б). В каждом случае источник аналогичен показанному на рис. 22-3.
Воздействие быстрых атомов на молекулярные мишени не слишком отличается от того, что происходит при облучении ионами в методе масс-спектрометрии вторичных ионов. Основное преимущество источника облучения быстрыми атомами состоит в его совместимости с магнитным сектором спектрометра, дефокусирующим поступающий пучок ионов [1, 8].
Метод бомбардировки быстрыми атомами весьма успешно применяется при изучении сложных молекул, соединений, участвующих в биохимических процессах, и лекарственных средств. Такие органические соединения можно растворить в капле глицерина, удерживаемой на штоке из нержавеющей стали. Глицерин имеет достаточно низкое давление паров, так что в масс-спектре можно различить лишь следы его [8]. Использование растворителя имеет то преимущество, что доставка изучаемых молекул на поверхность непрерывно возобновляется в результате диффузии [7].
Масс-анализаторы
Разделение ионов по их массовым числам может осуществляться различными способами, и мы ограничимся обсуждением лишь наиболее распространенных из них.
Заряженные частицы движутся в магнитном поле по траекториям, радиусы которых определяются соотношением
r = mvlzB (22-3)
где и — ускоряющий потенциал, В — напряженность магнитного
Масс-спекТрометрия 459
поля. Геометрически легко показать, что гомогенный пучок ионов, выходящих из щели, можно сфокусировать с помощью клинообразного магнитного поля (рис. 22-7). (Необходимо, чтобы обе щели и вершина магнитного сектора лежали на одной прямой, как показано на рисунке. В приборах заводского изготовления используют секторы с углом 60, 90 и 180 °C.) Это основа магнитного сектора масс-спектрометра. Как следует из уравнения (22-1), ионы, выходящие из источника, имеют почти одинаковую кинетическую энергию. Согласно уравнению (22-3), чтобы эти ионы можно было сфокусировать, они должны иметь также равные моменты количества движения (mv). Совместное решение этих двух уравнений позволяет вычислить радиус кривизны траектории
г = — л 2V — (22-4)
в V 2
Из этого уравнения видно, что каждый отдельный ион, характеризуемый конкретной величиной m/z, при данной напряженности магнитного поля движется по собственной траектории. Спектрометр снабжен выходной щелью (рис. 22-7, а), которая выделяет те ионы, для которых длина пробега практически равна радиусу кривизны. Согласно уравнению (22-4'), для отобранных ионов отношение m/z равно k(B2V), где k — постоянная прибора. Отсюда следует, что интервал сканирования масс можно изменять, варьируя либо В, либо V и поддерживая остальные параметры постоянными.
Разрешение секторного масс-анализатора ограничено разбросом кинетических энергий и невозможностью четкого обозначения границ магнитного поля. Улучшить его можно, исправляя любой из этих недостатков. Краевой эффект магнитного поля можно устранить, используя «сектор» в 180°, когда и источник, и детектор находятся в магнитном поле. Такое устройство, однако, неудобно для использования, так как и источник, и коллектор необходимо расположить в ограниченном пространстве между полюсами магнита.
Приборы с двойной фокусировкой
Пучок ионов можно превратить в моноэнергетический с помощью электростатического сектора (анализатора) (рис. 22-8). Пучок движется по круговой траектории, проходя кольцеобразное пространство между двумя концентрическими электродами цилиндрической формы. Радиус г и наложенное поле Е определяют энергию заряженных частиц, которые могут пройти анализатор в соответствии с соотношением
1limoi = zErl2 (22-5)
460 Глава 22
Рис. 22-8, Фильтр энергии в виде цилиндрического электростатического сектора.
Преобразовав это уравнение, получаем m/z=£r/v2 (22-6)
т. е. массу ионов, проходящих через анализатор, можно контролировать, изменяя наложенное напряжение. Этот электростатический сектор (анализатор), иногда называемый энергетическим фильтром, можно использовать в сочетании с магнитным сектором (анализатором) для создания масс-спектрометра с двойной фокусировкой. Такие приборы фокусируют ионы по энергиям и по массам.
Наиболее распространены два типа приборов с двойной фокусировкой. В одном из них, приборе геометрии Нира — Джонсона (рис. 22-9), между двумя 90-градусными секторами (анализаторами) расположена промежуточная щель. В приборе геометрии Маттауха— Герцога, изображенном на рис. 22-10, используют электростатический сектор с углом 31,83°. При такой величине угла все ионы входят в магнитное поле под прямым углом, и поэтому краевой эффект минимален. Магнитный сектор (анализатор) имеет угол 135°, что позволяет фокусировать каждую разновидность ионов на дальней границе поля. Важная особенность этой геометрии состоит в том, что все массы фокусируются одновременно в фокальной плоскости, что позволяет осуществить прямую фотографическую регистрацию. Этого удается достичь только при указанной геометрии масс-спектрометра.
Разрешение. Термин «разрешение», к сожалению, не всегда используют в одном и том же значении. Согласно наиболее широко распространенному определению, разрешение — это отно-
Масс-спектрометрия 461
Рис. 22-9. Масс-анализатор Нира — Джонсона с двойной фокусировкой.
шение М/&М, где АЛ1 —разность масс М и Л4 + АЛ4, пики которых имеют равную высоту, а точка пересечения расположена относительно нулевой линии на расстоянии, составляющем 10 % высоты этих пиков (рис. 22-11). Обычно разрешение считается удовлетворительным, если ДЛ1<1. Во многих типах спектрофотометров разрешение ухудшается по мере увеличения массы. Показателем качества служит «единичное разрешение» — наибольшая величина массы, при которой достигается разделение двух пиков одинаковой высоты. Если этому критерию, например, удовлетворяют массы от 600 до 601, то говорят, что такой спектрометр имеет разрешение 600.
Единичное разрешение магнитного сектора спектрометра может достигать 5000, а для спектрометров с двой
ной фокусировкой — 50 000 или даже больше. Это не означает, что прибор последнего типа позволяет наблюдать массы вплоть до 50 000, но он дает возможность различать частицы, массы которых отличаются менее чем на одну единицу (массы).
Спектрометр с двойной фокусировкой может легко разделить пики ионов с одинаковыми номинальными величинами молекулярных масс, но разным элементным составом. Например, может различить N2 (28,0061), СО (27,9949) и С2Н4 (28,0313) или такую серию, как C5H4N4O (136,0385), СвНбПзО (136,0511), C7H8N2O (136,0637), C8HwNO (136,0762) и С9Н12О (136,0889). Установление эмпирической формулы по данным масс-спектров— задача не простая, но ее можно решить, используя подходящий алгоритм [9].
Квадрупольные масс-анализаторы
Квадрупольный масс-анализатор (известный также как квадру-польный фильтр масс) — это прибор, в котором ионы можно разделить в соответствии с их величинами m/z без применения
462 Глава 22
Рис. 22-11. Иллюстрация способа определения разрешения, используемого в масс-спектрометрии.
Масс-спектрометрия 463
Рис. 22-12. Квадрупольнын масс-спектрометр. Сканирование можно осуществлять, изменяя напряжение постоянного тока и радиочастоты при их постоянном отношении [10].
тяжелого магнита. Он состоит из четырех металлических стержней, абсолютно прямых и расположенных строго параллельно так, чтобы пучок ионов попадал непосредственно в центр (рис. 22-12). В качестве входного отверстия предпочтительно использовать не щель, а отверстие круглого сечения. Стержни, расположенные по диагонали друг против друга, соединены электрически, и обе пары присоединены к противоположным полюсам источника постоянного тока, а также к генератору
радиочастоты.
На продольное движение ионов не оказывает влияния ни поле постоянного тока, ни поле переменного тока, но взаимодействие этих полей вызывает их боковое движение. Это можно рассмотреть [11] в пределах системы координат, показанной на рис. 22-13. Если стержни имеют симметричное гиперболическое поперечное сечение, то потенциал <р в любой точке (х, у) как функция времени определяется уравнением
Ф = (Vdc + ^rf cos ®Z)IU2—У2Уг2\ (22'7) где Vdc — наложенный постоянный потенциал, Vpr —амплитуда напряжения
Рис. 22-13. Расположение стержней в квадруполе. Ионы движутся вдоль осн z перпендикулярной плоскости чертежу
464 Глава 22
переменного тока с частотой со (рад/с) и г— радиальный размер, показанный на рис. 22-13. Это выражение остается почти корректным, даже если стержни с гиперболическим поперечным сечением заменить на менее дорогие цилиндрические.
Силу, вызывающую боковое смещение иона с единичным зарядом z, можно найти, дифференцируя выражение (22-7) по х и по у.
п „ д<р „ (Vdc+ Vrfcos at) х
Г у — 1 Z- —’
дх г2
Р Z дф _(Vdc Н~ cos <о/) у (22-8)
у ду г2
Поскольку ускорение можно выразить как отношение силы к массе, можно записать уравнение движения иона с массой т и зарядом г:
— Ч-------(Vdc + VRF cos at) x = О
dt2 r2 (m/z) v
+ VRF cos at) у = 0 (22-9)
dt2 r2 (m/z)
Из этих уравнений следует, что движение имеет периодическую составляющую с частотой со и зависит от отношения m/z. Проводя дальнейшие математические преобразования, можно найти, что для Уос/Уяр<0,168 существует только узкий диапазон частот, для которых траектории ионов неизменны (т. е. ионы не отклоняются) относительно обоих координат (х и у). За пределами этого диапазона ионы будут сталкиваться с одной или другой парой стержней. Максимальное разрешение достигается при максимальном приближении отношения Уса/Уир к предельной величине 0,168. Если это отношение превышает предельную величину, то частоты, при которой можно найти устойчивую траекторию вне зависимости от отношения mjz, не существует. Разделение по массам может быть достигнуто при одновременном изменении Vac и Vrf при постоянной частоте. Коммерческие квадрупольные спектрометры имеют диапазон массовых чисел вплоть до 1500.
Времяпролетные масс-анализаторы
Рассмотренные выше приборы обеспечивают постоянный пучок ионов при любом заданном наборе регулирующих величин. Однако если применить прерывистый ускоряющий потенциал, то пучок можно разбить на отрезки или импульсы. Это дает возможность сортировать ионы по их скоростям, что равносильно
Масс-спектрометрия 465
Обметь ионизации
Ввод газа
Катод
Управляющая сетка
Фокусирующая сетка / Ускоряющая сетка
Ионы
Отражатель , , ^полевая область
Электронная цаск Область ускорения ионов ловушка Осцилло граф
Рис. 22-14. Схема времяпролетного масс-спектрометра (CVC Products, Inc.).
Катод
'-электрон-
сортировке по массам. Этот принцип положен в основу времяпролетного масс-спектрометра (рис. 22-14) [12, 13]. Пучок электронов ионизирует образец в режиме электронного удара или химической ионизации. К сетке приложен ускоряющий потенциал порядка 2000 В в форме импульсов напряжения длительностью 1 мкс или менее, повторяющихся около 20 000 раз в секунду. Эти импульсы положительного напряжения сообщают ускорение ионам в длинной свободной от полей трубе дрейфа, вдоль которой ионы движутся со своими собственными скоростями. Поскольку все ионы приобрели одинаковую энергию, скорость каждого из них пропорциональна его (т/г)1/2 [уравнение (22-2)]. Пройдя бесполевую область, ионы ударяются о детектор. Время прохождения определяется уравнением
= —(22-10) V2V V Z
в котором L — длина трубы дрейфа, V — ускоряющее напряжение. (Заметим, что при расчетах вместо z необходимо использовать кулоновский заряд электрона е.) Если принять, что А = 1 м, V—2 кВ, т=1,67-10~27 кг и е= 1,60-10"19 Кл, то, согласно уравнению (22-10), время прохождения протона равно 1,58 мкс. Время прохождения ряда других ионов равно: N24 8,37 мкс, Ог+ 8,94 мкс, Хе+ 18,17 мкс.
Детектор, показанный на рис. 22-14, представляет собой специально разработанный электронный умножитель с высоким быстродействием (с малой постоянной времени), необходимым для того, чтобы избежать прерывания следующих друг за другом импульсов. Он состоит из пары стеклянных пластин,
466 Глава 22
покрытых металлической пленкой с высоким электрическим сопротивлением. По длине этих пластин приложен электрический градиент, пересекающий поле, создаваемое небольшими постоянными магнитами. Ионы ударяются об одну из этих пластин, а выбитые вторичные электроны по дугообразной траектории следуют к более отдаленному участку пластины, где процесс повторяется. В конце концов усиленный ток электронов достигает анода, соединенного с регистрирующим устройством. Кроме осциллографа можно использовать электронный преобразователь, посылающий последовательные импульсы на высокоскоростной самописец [13].
Времяпролетные спектрометры позволяют наблюдать ионы с массой до 1500' с единичным разрешением от 500 до 600.
Масс-спектрометрия с фурье-преобразованием (МСФП)
В предыдущих главах рассмотрен значительный вклад в ИК-и ЯМР-спектроскопию, обусловленный применением импульсной техники, основанной на преобразовании Фурье. За последние несколько лет аналогичная картина наблюдается и в масс-спектрометрии и, по мнению некоторых экспертов, может определять будущее масс-спектрометрии [14].
МСФП основана на более старом методе, известном как ион-циклотронный резонанс (ИЦР), см., например, [15]. Этот метод никогда не позволял добиться значительных успехов, исключение составили лишь кинетические, исследования ион-моле-кулярных реакций в газовой фазе. Приемлемый диапазон масс ограничен 280 а. е. м. при единичном разрешении 200. С введением методов, основанных на преобразовании Фурье, как увидим дальше, диапазон масс можно расширить значительно выше 1000, а разрешение при этом может быть таким же, как у масс-спектрометров с двойной фокусировкой.
Сердцем ИЦР-спектрометра (с или без фурье-приставки) является прямоугольная ячейка с длиной ребра в несколько сантиметров, внутри которой молекулы образца ионизуются в постоянном магнитном поле. Ионы с момента образования движутся беспорядочно, но, находясь в магнитном поле, они вынуждены двигаться по циклическим траекториям с так называемой циклотронной частотой, определяемой выражением
сос = Bel^tnlz) (22-11)
где В — напряженность магнитного поля, е — заряд электрона (рис. 22-15, а). Для того чтобы обеспечить возможность разделения ионов, их необходимо задержать в поле на какое-то определенное время (несколько десятых секунды). Это достигается с помощью потенциальной ямы, образованной наложением
Масс-спектрометрия 467
X а
Рис. 22-15. Орбиты иоиов в магнитном поле (в каждом случае магнитное поле перпендикулярно плоскости чертежа): а — множество ионов на малых орбитах, фазы которых относятся друг к другу случайным образом; б — после РЧ-возбуждения все ионы вращаются в фазе на расширяющихся орбитах; в — то же после снятия возбуждения.
Боковая пластина (+W
Коллектор электронов
Торцевая . пластина * (-0,5В)
В
К предусилителю
Балансный конденсатор
Торцевая пластана (-0,56)
Нижняя пластина >
(-0,56)
сетка f Катод
Верхняя пластина (-0,5В) Радио-’ частотный источник
Рис. 22-16. Ячейка ИЦР-масс-спектрометра с фурье-преобразованием объемом около 10 см3 [14].
положительного напряжения ( — 1,0 В) на боковые пластины и отрицательного напряжения (----0,5 В) на верхнюю и ниж-
нюю пластины и две торцевые пластины (рис. 22-16).
Разделение по массам достигается в результате подачи переменного радиочастотного поля с частотой со; через верхнюю и нижнюю пластины. Если подобрать частоту, способную быть в резонансе с циклотронной частотой (mi = coc), то ионы будут поглощать энергию, и их скорость и радиус траектории увеличатся (рис. 22-15, б). Все ионы с данным отношением m/z будут циркулировать в фазе с радиочастотным возбуждением. Если содержащиеся ионы различаются по массе, то условиям резонанса будут соответствовать только такие ионы.
468 Глава 22
C,oH,479Br*
Рис. 22-17. Фрагменты масс-спектрограммы дибромадамантана, показывающие ионы С10Н14Вг+ с изотопами 79Вг и 81Вг при значении массовых чисел 213 и 215. а — временной отрезок ИЦР-интерферограммы; б — спектр, полученный после преобразования фурье-сигнала, изображенного выше (а) [16].
Масс-спектрометрия 469
Рис. 22-18. Спектр высокого разрешения тройной смеси N2, С2Н4 и СО в области массового числа 28 после преобразования Фурье [17].
Существуют два основных подхода к определению условий резонанса. В старых ИЦР-спектрометрах радиочастотное возбуждение обеспечивалось специальной схемой, дающей возможность точно измерить величину энергии, поглощенной ионным резонансом. Эта схема работает только при частотах выше 75 кГц, что ограничивает достижимое массовое число. Усовершенствованный метод требует сканирования в пределах всего интересующего диапазона частот (примерно от 20 кГц до 1 МГц при В= 1,2 Т) за время порядка 1 мс. Это заставляет все ионы в пределах заданного диапазона масс циркулировать в фазе и оставаться в этом состоянии после прекращения возбуждения (рис. 22-15,в). Эти циркулирующие ионы индуцируют ток на верхней и нижней пластинах ячейки, который можно регистрировать электронным усилителем. Полученный в результате сигнал представляет собой совокупность сигналов всех ионов и, следовательно, содержит всю информацию об образце, которую только позволяет получить масс-спектрометр рассматриваемого типа. Переход к традиционному изображению масс-спектра требует преобразования Фурье.
На рис. 22-17 показаны суммарный сигнал и его преобразование [16, 17]. Способность МСФП разделять ионы с одинаковой номинальной массой иллюстрирует рис. 22-18; в данном случае легко просматривается аналогия с масс-спектрометрией высокого разрешения с двойной фокусировкой.
МСФП присуще то же достоинство, о котором упоминалось в связи с обсуждением ИКФП, а именно: все имеющиеся ионы наблюдаются одновременно, а не последовательно [18].
470 Глава 22
Преимуществом этого метода является также отсутствие в спектрометре щели, ограничивающей эффективное количество образца.
В сочетании с МСФП химическая ионизация приобретает несколько иной аспект. Поскольку первичные ионы могут быть задержаны в области активного поля на неопределенное время (вплоть до многих секунд), то вероятность протекания реакции в результате столкновения достаточно велика даже при низком давлении. В отсутствие дополнительного реагента ионы образца могут реагировать с другими фрагментами или нейтральными молекулами самого образца. Это явление известно как самопроизвольная химическая ионизация. В работе [19] подробно обсуждается метод МСФП-ХИ.
Масс-спектрометрические детекторы
Существуют три типа детекторов ионов: простой коллектор, известный как ловушка Фарадея, электронный умножитель и фотопластинка.
Ловушка Фарадея — наименее разработанный и наименее чувствительный из этих трех детекторов. Ее используют во многих спектрометрах, где не нужна чрезвычайная чувствительность, из-за удобства и низкой стоимости. Она состоит из изолированного проводника, присоединенного непосредственно к электролитическому усилителю. Благодаря чашеобразной форме ловушки снижается вероятность утечки вторичных электронов, освобождающихся при столкновении ионов.
Электронный умножитель действует подобно фотоумножителю. Это умножитель традиционного или канального типа, первый катод которого предназначен для детектирования ионов, а не фотонов. Находясь в вакуумированном спектрометре, он не нуждается в стеклянной оболочке, необходимой для обычного фотоумножителя. Чувствительность может быть в 1000 раз выше чувствительности ловушки Фарадея.
Фотографическое детектирование возможно только в приборах с геометрией Маттауха — Герцога. Поскольку фотопластинка интегрирует сигнал иона в течение какого-то периода времени, чувствительность детектирования данным способом выше, чем любым другим детектором. Она позволяет также более эффективно использовать высокое разрешение спектрометров с двойной фокусировкой. Пластинку подвергают обычной фотографической обработке и читают с помощью денситометра.
Картины фрагментации. Серии ионных фрагментов, наблюдаемых для-данного соединения, могут в зависимости от условий эксперимента значительно различаться. Рассмотрим сначала последовательность явлений, происходящих в ионном источнике электронного удара, по мере увеличения энергии
Масс-спектрометрия 471
m/z
Рис. 22-19. Масс-спектр «-бутана (мол. масса 58), снятый на масс-сцектро-метре с магнитным анализатором. Использование гальванометров различной чувствительности обеспечивает одновременную запись с динамическим диапазоном около 30 000. Пик А принадлежит двухзарядному иону, пики Б и В — метастабильным ионам (Е. I. Du Pont de Nemours and Co.).
электронов. Допустим, что в газовой фазе присутствует органическое соединение с несколькими атомами углерода. Первый потенциал ионизации такого соединения лежит в интервале 8— 12 эВ. (Потенциал ионизации соединения или элемента — это энергия (в электронвольтах), необходимая для удаления наименее прочно удерживаемого электрона. В методе ЭУ ее можно принять за ускоряющее напряжение пучка электронов.) При более низких потенциалах положительные ионы не образовываются. Первый ион возникает в результате удаления одного электрона из молекулы, поэтому его называют молекулярным, или исходным, ионом. По мере увеличения напряжения разрываются и другие связи в молекуле, каждая из которых характеризуется потенциалом появления, т. е. потенциалом, при котором это впервые становится заметно. Поэтому масс-спектрометрия позволяет получить данные о величинах потенциалов ионизации и об относительной прочности связей.
Большинство масс-спектров после ионизации ЭУ или ХИ регистрируют, используя пучок электронов с энергией 70 э£,
472 Глава 22
Рис. 22-20. Гистограмма масс-спектра «-бутана.
достаточной для разрыва любых возможных связей. Каждое соединение дает характеристическую серию фрагментов, называемую его фрагментационной картиной, или картиной распада. Такие картины обычно представляют в виде гистограмм, как показано на рис. 22-20, эквивалентном рис. 22-19, изображающем масс-спектр н-бутана. Заметим, что молекулярный ион (т/г=58) дает относительно небольшой пик, а наиболее интенсивным является пик при т/г=43. (Наиболее интенсивный пик в спектре называют базовым пиком-, часто базовый пик не принадлежит молекулярному иону.) Это указывает на то, что фрагмент с массовым числом 43 более устойчив к дальнейшему расщеплению, чем исходная молекула. В этом случае он должен образоваться при потере фрагмента с массой 58—43=15, т. е. метильной группы. Отсюда следует, что базовый пик принадлежит иону С3Н7+. Маленький пик при m/z 59 (=М+1) обусловлен присутствием естественного 13С или 2Н и служит примером сопутствующего пика. Случайный небольшой пик с дробным массовым числом, например 25,5 на рис. 22-19, принадлежит двухзарядному иону (масса 51), а большой пик при m/zVd может принадлежать ионам С4Ню2+ и С2Н5+.
Как уже упоминалось выше, спектры, полученные с источником ХИ, обычно намного проще спектров с ионным источником ЭУ, и в них самый интенсивный пик относится к молекулярному иону.
Масс-спектрометрия 473
Масс-спектры даже лишь при удовлетворительном разрешении могут дать богатую информацию о составе и строении образца. Кроме молекулярной массы часто можно идентифицировать важные фрагменты, включая такие группы, как фенильная (масса 77) или бензильная (91). Серия пиков при m/z, равных 15, 29, 43, 57, 71, 85 и т. д., отделенных друг от друга на 14 единиц, наводит на мысль, что это длинная алифатическая цепь (СН2). Первичные спирты и амины часто идентифицируют по наличию пиков —СН2ОН при 31 и —CH2NH2 при 30 а.е. м. Уникальные возможности открывает использование изотопов. Изотопы 35С1 и 37С1 и содержащие их фрагменты идентифицируют по их характеристическим пикам с отношением интенсивностей примерно 3: 1, разделенным на 2 а. е. м. Аналогично 79Вг и 81Вг также разделены на 2 а. е. м., но их пики имеют почти равную интенсивность. Таким образом, из картин фрагментации часто легко выявить наличие определенных элементов. Полная методика установления элементного состава и молекулярной структуры слишком длинна, чтобы обсуждать ее здесь. Этому вопросу посвящено много книг [11, 20].
Метастабильные ионы. Некоторые ионы, возникающие в ионном источнике, по природе своей неустойчивы, но тем не менее они могут какое-то время существовать. При прохождении через спектрометр эти метастабильные ионы, вероятно, разрушаются. Среди новых фрагментов, образующихся при разрушении таких ионов, должен быть по крайней мере один ион с тем же зарядом. В результате распространения этих ионов по масс-анали-затору в спектре наблюдаются довольно широкие пики, поскольку появляющиеся в различных местах ионы не фокусируются. В масс-спектре на рис. 22-19 заметны два маленьких округлых всплеска, обусловленные возникновением метастабиль-ных ионов (обозначены Б и В). На приведенной на том же рисунке кривой, записанной при максимальной чувствительности (помечено XI), в интервале примерно от 39 до 44 на шкале массовых чисел заметен пологий подъем нулевой линии, обусловленный, вероятно, присутствием метастабильных ионов (ср. с гладкой нулевой линией в интервале 49—59). Наблюдение метастабильных ионов может быть важным при изучении механизмов превращений органических соединений [21].
Отрицательные ионы. До сих пор возможности использования отрицательных ионов в масс-спектрометрии было уделено мало внимания. В газах, ионизованных с помощью ЭУ, преобладают положительные ионы, что отчасти объясняется наибольшей устойчивостью. Отрицательный ион теряет свой избыточный электрон значительно легче, чем положительный ион теряет избыток протонов или присоединяет еще один электрон.
474 Глава 22
Большинство масс-спектрометров пригодны для анализа не только положительных, но и отрицательных ионов, если изменить знаки всех потенциалов и направления всех магнитных полей. В некоторых приборах для этого необходим только перекидной ключ, в других требуется некоторая перестройка.
Наблюдения за отрицательными ионами могут быть, в частности, очень полезными при химической ионизации. Получаемые спектры несложны и в ряде случаев весьма селективны. Таким способом можно измерить субпикограммовые количества хлорированных углеводородов, например ДДТ, применяя в качестве реагента смесь, содержащую 90 % изобутана и 10 % ме-тиленхлорида. В этих же условиях эфир (метилстеарат) не дает ощутимых количеств ионов [22]. В работе [23] сообщается об определении меркаптанов с пределом обнаружения порядка 10 мкг/мл при использовании циклогексана в качестве растворителя и реагента.
Качественный анализ
Для идентификации неизвестного вещества с помощью масс-спектрометрии требуется провести соотнесение пиков на диаграмме с массовыми числами. Масс-спектрометры, снабженные компьютерами, могут идентифицировать пики по мере их появления и соответственно маркировать запись. В противном случае отнесение масс становится более трудным и по мере увеличения масс менее определенным. Для приборов с магнитным отклонением, в которых напряженность магнитного поля В сканируют с постоянной скоростью, из уравнения (22-4) следует, что шкала массовых чисел линейна, так что в принципе по двум известным массам будет проградуирован полный спектр. Часто, однако, необходимо или желательно сканировать напряженность поля экспоненциально или изменяя ускоряющее напряжение. В таких случаях получается нелинейная шкала m/z, и необходима градуировка по известным соединениям. Для идентификации положений масс на нижнем конце шкалы служат пики компонентов воздуха (рис. 22-21). Для проверки правильности установления координат можно использовать пары ртути, дающие характерное распределение пиков изотопных ионов в диапазоне массовых чисел от 198 до 204. Для градуировки в более широком диапазоне часто удобен перфторкеросин (ПФК). В работе [20] дана «неполная» таблица 73 пиков фрагментов из этой смеси фторуглеводородов. Появляются пики, соответствующие массовым числам 69 (CF+3), 93 (СзРз+), 105 (С4р3+), 131 (C3F5+), и пики гомологов с массами, отличающимися от указанных на 50 а. е. м. (CF2). ПФК можно «прогнать» непосредственно до или после неиз-
Масс-спектрометрия 475
Рис. 22-21. Масс-спектр воздуха.
вестного компонента, не меняя настройки, или его можно смешать с неизвестным компонентом. Последний метод, в основном применяемый при работе на приборах высокого разрешения, осуществим потому, что массы фторсодержащих соединений отличаются от масс других соединений и, следовательно, не совсем перекрываются. Как только различным пикам будут приписаны величины m[z, можно идентифицировать соответствующие фрагменты, а следовательно, и исходные молекулы [20].
Количественный анализ
Масс-спектры фрагментов компонентов смеси аддитивны, поэтому смеси можно анализировать, только если имеются спектры различных компонентов, записанные в тех же условиях. Проводимые вычисления предусматривают решение системы из п уравнений с п неизвестными для смеси из п компонентов. Это легкая задача для маленького компьютера. Для компонентов, концентрация которых превышает 10 мол.%, правильность и воспроизводимость результатов анализа могут составить ±0,5 Мол. % (при доверительной вероятности 90 %). Подобные смеси лучше всего анализировать после их разделения на газовом хроматографе, как описано в следующем разделе.
Масс-спектрометр можно применять в методе меченых атомов, используя соединения, обогащенные стабильными изотопами, малораспространенными в природе, например 2Н, 13С, 33S, 40К и многими другими. Некоторых изотопов необходимо избегать из-за возможных помех; так, железо не следует
476 Глава 22
метить 58Fe, так как отличить этот изотоп от 58Ni можно только на приборах высокого разрешения. Достаточно распространенный метод изотопного разбавления детально описан в гл. 24, поскольку его более часто применяют с радиоактивными изотопами.
ГХ/МС и жх/мс
В гл. 20 уже указывалось, что сочетание газовой хроматографии и масс-спектрометрии может быть чрезвычайно плодотворным. Этот симбиоз — интересный пример нового подхода в развитии гибридных инструментальных аналитических методов.
Ранее существовала неоправданная тенденция рассматривать одного участника этого тандема простым довеском другого. Считалось, что или масс-спектрометр — это хорошо разработанный детектор для ГХ, или ГХ — лучшая система обработки пробы для МС. Ни одно из этих представлений не отдает должного объединенному прибору, который представляет собой нечто гораздо большее, чем простая сумма двух его частей. МС не очень хорошо использовать для анализа смесей близких по свойствам веществ из-за обилия фрагментов. В то же время ГХ по природе своей не позволяет идентифицировать компоненты смеси. Объединенный метод не имеет равных в анализе сложных образцов, если они характеризуются достаточным давлением паров и термически устойчивы. ГХ-детектор дает количественную информацию о каждом компоненте, а МС идентифицирует их.
Существуют два важных фактора, которые следует принимать во внимание при разработке приборов для ГХ/МС. Во-первых, газ-носитель нельзя откачать полностью вакуумной системой масс-спектрометра, и, во-вторых, скорость откачки должна соответствовать скорости поступления газа-носителя. Рассмотрим эти моменты несколько подробнее.
Поток газа, выходящий из газового хроматографа, по существу имеет атмосферное давление. В то же время давление газа, входящего в ионный источник масс-спектрометра, не должно превышать 10~3 Па при ионизации электронным ударом или 10 Па при химической ионизации (1 атм~105 Па). Количество компонента пробы, однако, обычно составляет только очень небольшую долю элюата, выходящего из ГХ. Снижение давления приводит к тому, что количество образца оказывается недостаточным, и поэтому необходимо отделение газа-носителя. Этого можно достичь с помощью сепаратора, использующего физические свойства газов. Схема одного из таких сепараторов [24] показана на рис. 22-22. Газовую смесь, в которой носителем служит либо Нг, либо Не, пропускают через пористую
Масс-спектрометрия 477
Вакуумируемая камера
Кольцевой уплотнитель
К Форвакуум- i ному насосу |
К источнику ионов
Вход
Из ГХ
Пористая стеклянная трубка
Рис. 22-22. Обогатительная система, соединяющая масс-спектрометр и газовый хроматограф [24].
трубку, окруженную кожухом, соединенным с вакуумным насосом. Газ-носитель с низкой молекулярной массой легко диффундирует через пористые стенки и отсасывается, а более тяжелые молекулы образца, обладающие большими моментами количества движения, поступают в масс-спектрометр. Этот и другие сепараторы описаны в работе [25].
В принципе почти любой масс-спектрометр можно использовать в сочетании с ГХ, но на практике выбор может быть ограничен рядом параметров. Так, скорость сканирования напряженности магнитного поля может оказаться недостаточной для записи полного спектра фрагментов в процессе элюирования хроматографического пика, хотя напряжение ускорения можно сканировать значительно быстрее. Квадрупольные спектрометры обладают требуемой скоростью, но их разрешение ниже необходимого. ИЦР-спектрометры с преобразованием Фурье идеально отвечают этим требованиям [26].
Тандем ГХ/МС может действовать в двух режимах. Можно настроить предварительно блок МС на конкретное массовое число, соответствующее иону одного типа, образующегося из искомого соединения. В этом случае будет идентифицирован хроматографический пик, соответствующий этому соединению. Квадрупольные спектрометры или спектрометры с преобразованием Фурье можно запрограммировать для последовательной регистрации нескольких специфических массовых чисел, поскольку на это нужно гораздо меньше времени, чем на элюирование пика. В такой ситуации возможна одновременная идентификация нескольких веществ.
В другом варианте режима можно записать полный масс-спектр для каждого компонента, элюируемого из ГХ. Ясно, что для этого необходимо высокоскоростное сканирование наблюдаемых спектров, помещаемых в память компьютера для последующей обработки. Оба эти варианта были использованы при изучении следов органических соединений, найденных в
478 Глава 22
выносах из городских печей для сжигания мусора [27]. В результате проведенных исследований было выявлено около 99 различных соединений; все они, кроме 15, были четко идентифицированы, а все из этих 15, кроме одного, были отнесены к отдельным структурным классам.
Многие масс-спектрометры снабжают вспомогательным электродом, расположенным сразу же за источником ионов. Этот электрод отсекает небольшую часть ионов до того, как они разделятся в масс-анализаторе. Первоначально этот электрод должен был способствовать регулировке источника ионов, но оказался очень полезным в сочетании с ГХ, поскольку его ток можно регистрировать с помощью неселективного ГХ-детектора.
Тандем ЖХ/МС в аппаратурном оформлении более сложен, чем ГХ/МС. Хотя к настоящему времени предложено несколько вариантов приборов [28], внимания заслуживают лишь два из них.
В первом методе перед введением образца в масс-спектрометр растворитель выпаривают. Это можно сделать с помощью движущейся ленты из нержавеющей стали или инертной пластмассы, доставляющей образец из одной точки в другую (рис. 22-23). Поток элюата поступает из жидкостного хроматографа в делитель (точка /), откуда часть потока (до 1 мл/мин) поступает на ленту 2. Инфракрасный нагреватель 3 выпаривает растворитель. Лента проходит через две последовательно расположенные камеры 4, где откачиваются пары растворителя. На участке 5 ленту нагревают, чтобы десорбировать молекулы растворенного вещества, откуда они и диффундируют в ионизационную камеру масс-спектрометра. Нагреватель 6 удаляет все остатки с ленты, чтобы она была пригодна для повторного использования. Движение ленты осуществля-
Рис. 22-23. Схематическое изображение (вид сбоку) поверхности движущей ленты ГХ (обозначения см. в тексте) (Finnigan Corporation).
Масс-спектрометрия 479
ется двумя роликами 7. Эта система очень эффективна для неполярных растворителей. В случае полярных растворителей для получения воспроизводимых результатов необходимы более высокая температура испарения и меньшая загрузка ленты.
Второй способ сочетания ЖХ и МС заключается в прямом введении элюата, содержащего компоненты пробы и растворитель, в масс-спектрометре. Жидкость пропускают через капилляр к точечной диафрагме в стенке ионизационной камеры. Здесь растворитель выполняет роль реагента при получении ионов из растворенного вещества с помощью ХИ. Растворителями в этом случае могут служить вода, метанол или ацетонитрил.
но
он 59“
Л(снзсог)
_1_
60
L___1_
100 120
WZ
(м-н)“
+ 100
л— 1
140 160 181
Рис. 22-24. Отрицательные дочерние ионы, генерированные из карбоксилат-иоиов, наблюдаемые с помощью тандема МС/МС с магнитным анализатором. По этим спектрам можно идентифицировать три кислоты [29].
480 Глава 22
Тандемная масс-спектрометрия
Как следует из предыдущего раздела, роль хроматографа заключается в разделении компонентов смеси таким образом, чтобы в каждый момент в масс-анализаторе присутствовал какой-то один компонент. Аналогичным образом можно использовать один масс-анализатор для выделения иона с определен-
220 240 260 280 300 320 340 360 380 400
m/z
Рис. 22-25. Спектры родительских ионов компонентов мускатного ореха. Две развертки дают профили молекулярных масс всех соединений, образующих дочерние ионы с массовыми числами 193 и 203 в указанных реакциях. Результаты получены на квадрупольном масс-спектрометре, а — спектр всех родительских ионов, дающих дочерние ионы с массой 193+; б — спектр всех родительских ионов, дающих дочерние ионы с массой 203+ [29].
Масс-спектрометрия 481
Рис. 22-26. Схема масс-спектрометра с тремя квадруполями. Большая сфера в задней стенке присоединена к вакуумному насосу. 1, 7, 12 — ионизационные датчики давления; 2 — термопарный датчик давления; 3 — ввод газа для ХИ; 4 — источник ионов (ХИ/ЭУ); 5 — система ввода пробы (твердой или газообразной); 6 — квадруполь 1; 8 — квадруполь 2; 9 — ввод газа для активации столкновений; 10 — квадруполь 3; // — канальный электронный умножитель [31].
ным массовым числом и направить его во второй анализатор для идентификации.
Предположим, например, что смесь родственных органических соединений ионизуют методом химической ионизации так, что преобладают молекулярные ионы. Обычный масс-спектрометр при этом показывает серию пиков, по одному для каждого соединения, но не дает достаточной информации для идентификации индивидуальных соединений. Тандем масс-спектрометров можно отрегулировать так, что через щель, разделяющую первый и второй анализаторы, будут проходить ионы только с одним массовым числом. Затем эти ионы будут подвергнуты дальнейшей ионизации при столкновении с молекулами введенного газа, например гелия, и образуют характерные фрагменты, которые идентифицирует второй анализатор.
Рассмотрим в качестве другого примера идентификацию изомеров, образующих, как показано на рис. 22-24 [29], один и тот же молекулярный ион. Молекулярная масса всех трех соединений равна 182, и все они дают родительский ион (М—Н)_ при 181. Пути вторичной фрагментации идентифицировать легко.
В другом варианте режима работы первичный анализатор может быть сканируемым, а вторичный — настроенным на массовое число, соответствующее важному для диагностики иону. На рис. 22-25 показан такой случай, наблюдаемый при определении компонентов мускатного ореха [29].
Аппаратура для тандемной масс-спектрометрии. Многие ранние работы выполнены с модифицированным спектрометром
16 Заказ № Б37
482 Глава 22
с двойной фокусировкой типа масс-спектрометра Нира—Джонсона, геометрия которого изменена так, что ионы проходили через магнитный сектор раньше, чем через электрический. Если первичный ион с массой тх распадается, образуя дочерний ион с массой т2, то кинетическая энергия последнего связана непосредственно с отношением m2/mi, и поэтому анализатор энергии (электростатический анализатор) может стать масс-анализатором. Однако это приводит к размыванию сигнала из-за разброса величин энергий поступательного движения ионов. Отличные результаты можно получить при использовании двух немодифицированных спектрометров Нира—Джонсона [30], но это слишком дорого для широкого использования.
Более доступным по цене и близким по качеству является тандем спектрометров с квадрупольными элементами [31]. Это показано на рис. 22-26. Фактически прибор состоит из трех квадруполей, из которых квадруполь № 2 управляется только радиочастотным напряжением (постоянная составляющая отсутствует). В этом варианте квадруполь не может обеспечить распознавание масс, но он фокусирует все ионы на выходное отверстие. Этот участок служит камерой для вторичной ионизации.
Задачи
22-1. Ион с массой т— 100 а. е. м. и зарядом z=l подвергли действию ускоряющего напряжения К=1 кВ. Рассчитайте скорость движения нона в единицах м/с.
22-2. Ион, описанный в задаче 22-1, столкнулся с магнитным полем, геометрия которого требует, чтобы радиус кривизны г-, был равен 10 см, если ноны должны достигать выходной щели. Какова должна быть напряженность поля (в теслах)?
22-3. Ион (задачи 22-1 н 22-2) попал в электростатический анализатор с Г2=12 см. Каков должен быть потенциал Е (в кВ), чтобы ион попал в детектор?
22-4. Длина трубы дрейфа времяпролетного масс-спектрометра равна 85,0 см. Рассчитайте разность во времени пролета ионов с массовыми числами 301 н 302 при ускоряющем напряжении 2,00 кВ.
22-5. Опишите каждый главный ион в масс-спектрах, показанных на рнс. 22-19 и 22-21.
22-6. Органическое соединение проанализировали на содержание азота методом изотопного разбавления [32]. Для этого в пробу ввели известное количество соединения, содержащего 15N вместо 14N. После полного превращения азота в N2 был снят масс-спектр, содержащий следующие пики:
т/е 28 29 30
Высота 978,5 360,6 52,5
Рассчитайте процентное содержание 15N.
22-7. В статье [33] описан метод изотопного разбавления для определения С с использованием в качестве метки 13С. Образец смешивают с порцией янтарной кислоты С4Н6О4, содержащей около 30 ат. % 13С. Смесь окисляют до СО2 и Н2О, образующийся СО2 анализируют в масс-спектрометре. Отношение масс
Масс-спектрометрия 483
45/44 (с поправкой на изотопный состав О), принятое за отношение <3С/12С, обозначено г. Следует учесть, что в природном С содержится 1,11 % 13С и 98,9 % |2С. Получена система уравнений:
1зс5=^г
(0,0111)
12С= WT
rs = 13CS/12CS
(0,989)
где 12CS и l3Cs — количество миллиграммов атомов соответствующих изотопов в смешанной пробе, г3 — наблюдаемое отношение, WT и Ws — массы метки и образца в миллиграммах, гт — отношение для чистой метки, окисленной тем же способом, хс — искомая величина, массовая доля С в изучаемом веществе.
а) Объясните приведенные выше уравнения и выведите из них выражения для определения хс через №s, IFt и rs. б) Для выполнения анализа взято 0,156 мг образца и 0,181 мг метки. Найдено, что величина га равна 0,206. Метка содержала 31,41 % углерода в виде 13С. Рассчитайте процентное содержание С в образце.
22-8. В масс-спектре спирта, записанном с использованием NO в качестве реагента при ХИ, содержатся следующие пики:
m/z 17 20 31 120 134 136 155 157
Относительная МСММСБСС высота пика
(М — малая, С — средняя, Б — большая).
Какие выводы об изучаемом образце можно сделать?
22-9. В статье [34] опубликованы исчерпывающие данные по анализу органических материалов на содержание следов металлов с использованием масс-спектрометрии вторичных ионов. Один из проанализированных образцов был стандартный образец Национального бюро стандартов, называемый «Фруктовые листья», в котором средн прочих элементов содержится (2,09±0,030 %)Х Х10~4 % кальция и (1,47±0,03 %) • 10-4 % калия. На основании тщательных измерений, проведенных на приборе для масс-спектрометрии вторичных ионов, авторы статьи разработали таблицу коэффициентов относительной чувствительности (к.о.ч.), выражаемых отношением
К-О-Ч.х, k = (ix/Cxfx)l(iK/CKM
где i—иитенснвность вторичного нона (имп./с), С — концентрация, f — распространенность изотопа. Индексы х и К относятся к определяемому элементу и калию, выбранному в качестве элемента сравнения. Допустим, что в частном случае для образца с матрицей, сопоставимой с «Фруктовыми листьями», найдено, что
fca = 7,2-ю* импульс/с для 40Са и i'k = 1,3-Ю6 импульс/с для 39К
Какова концентрация кальция? (Распространенность изотопов 39К н 40Са 93,1 и 96,9 % соответственно.)
22-10. Молекулярные массы бензофенона и азобензола 182,2214 и 182,2244 соответственно, а) Можно ли ожидать различия в поведении этих индивидуальных соединений при использовании масс-спектрометра с разрешением 1000? б) Можно ли анализировать смеси этих соединений?
16*
484 Глава 22
Литература
1. Busch К. L., Cooks R. G„ Science, 1982, v. 218, p. 247.
2. Munson B., Anal. Chem. 1977, v. 49, p. 772A.
3. Hunt D. F., Harvey T. M-, Brumley W. C., Ryan J. F„ Russel J. W., Anal. Chem., 1982, v. 54, p. 492.
4. Day R. I., Unger S. E., Cooks R. G., Anal. Chem., 1980, v. 52, p. 557A.
5. Christie № H., Eby R. E., Warmack R. J., Landau L., Anal. Chem., 1981, v. 53, p. 13.
6. Rinehart K. L., Science, 1982, v. 218, p. 254.
7. Barber M„ Bordoli R. S., Elliott G. J., Sedgwick R. D., Tyler A. N., Anal. Chem., 1982, v. 54, p. 645A.
8. Martin S. A., Costello С. E., Biemann K„ Anal. Chem., 1982, v. 54, p. 2362.
9. Dromey R. G., Fouster G. T., Anal. Chem., 1980, v. 52, p. 394.
10. Lichtman D., Res. Dev. 1964, v. 15(2), p. 52.
11. Farmer J. B., In /Mass Spectrometry. C. A. McDowell (ed.), McGraw-Hill, New York, 1963, ch. 2.
12. Wiley W. C„ Science, 1956, v. 124, p. 817.
13. McFadden W., Techniques of Combined Gas Chromatography/Mass Spectroscopy. Wiley, New York, 1973, p. 46.
14. Hunter R. L., McIver R. T., Jr., Anal. Chem., 1979, v. 51, p. 699.
15. Baldenschweiler J. D„ Woodgate S. S., Acc. Chem. Res., 1971, v. 4, p. 114.
16. Comisarow M. B., Marshall A. G„ J. Chem. Phys., 1975, v. 62, p. 293.
17. Comisarov M. B., In Transform Techniques in Chemistry, Griffiths P. R. (ed.), Plenum Press, New York, 1978, ch. 10.
18. Marshall A. G., Anal. Chem., 1979, v. 51, p. 1710.
19. Chaderi S., Kulkarni P. S-, Ledford E. B., Jr., Wilkens C. L., Gross M. L„ Anal. Chem., 1981, v. 53, p. 428.
20. McLafferty F. W., Interpretation of Mass Spectra (3d ed.), University Science Books, Mill Valey, CA, 1980.
21. Jennings K. R., In Mass Spectrometry: Techniques and Application. Milne G. W. A. (ed.), Wiley-Interscience, New York, 1971, p. 419.
22. Daugherty R. C., Anal. Chem., 1981, v. 53, p. 625A.
23, Knof H-, Large R„ Albers G„ Anal. Chem., 1976, v. 48, p. 2120.
24. Watson J. T., Biemann K„ Anal. Chem., 1965, v. 37, p. 844.
25. Ryhage R-, Wirkstrom S., In Mass Spectrometry: Techniques and Application. Milne G. W. (ed.), Wiley-Interscience, New York, 1971, p. 91.
26. Wilkins C. L., Giss G. N., White R. L., Brissey G. M., Onyiriuka E. C„ Anal. Chem., 1982, v. 54, p. 2260.
27. Eiceman G. A., Clement R. E., Karasek F. W., Anal. Chem., 1979, v. 51, p. 2343.
28. Arpino R. J., Guiochon G., Anal. Chem., 1979, v. 51, p. 682A.
29, Cooks R. G., Glish G. L., Chem. Eng. News, 1981, v. 59(48), p. 40.
30. McLafferty F. W., Todd P. J., McGilvery D. C., Baldwin M. A., J. Am. Chem. Soc., 1980, v. 102, p. 3360.
31. Yost R. A., Enke C. G., Anal. Chem., 1979, v. 51, p. 1251A.
32. Grosse A. V., Hindin S. G., Kirshenbaum A. D., Anal. Chem., 1949, v. 21, p. 386.
33. Boos R. N, Jones S. L., Trenner N. R., Anal. Chem., 1956', v. 28, p. 390.
34. Ramseyer G. O-, Morrison G. H., Anal. Chem., 1983, 55, 1983.
ГЛАВА 23
Термические методы анализа
Температурные коэффициенты важны для многих ранее обсуждавшихся методов, но аналитики интересовались ими лишь для выбора оптимальных условий проведения анализа. В этой главе рассматривается ряд методов, основанных на измерении какого-либо свойства системы как функции температуры или на измерении количества выделенного или поглощенного в реакции тепла. Эти методы перечислены в табл. 23-1. Некоторые аналогичные, но менее распространенные методы в таблицу не включены (более подробная информация содержится в работах [1, 2]).
Таблица 23-1. Методы термического анализа
Название Регистрируемый параметр Измерительный прибор
Термогравиметрия (ТГА) Термогравиметрия по производной (ТГП) Дифференциальный термический анализ (ДТА) Дифференциальная сканирующая калориметрия (ДСК) Термометрическое титрование Прямая инжекционная эи-тальпиметрия Изменение массы Скорость изменения массы Выделяемое или поглощаемое тепло То же Изменение температуры Выделяемое или поглощаемое тепло Термовесы То же Аппаратура ДТА Дифференциальный сканирующий калориметр Адиабатический калориметр То же
Термогравиметрический анализ (ТГА)
Этот метод основан на наблюдении за изменением массы пробы в течение некоторого периода времени при линейном повышении температуры. Несколько примеров полученных таким способом термограмм представлено на рис. 23-1 [3]. Кривая 1 показывает изменение массы осадка хромата серебра на фильтрующем тигле, Первоначальная потеря массы относится
486 Глава 23
Рис. 23-1. Примеры кривых, полученных при термогравиметрическом анализе [3].
к удалению избытка промывной воды. При температуре чуть выше 92 °C масса осадка достигает постоянной величины и не меняется до температуры примерно 812 °C. Начиная с этой точки и до 945 °C выделяется кислород. Тщательный анализ потери массы показывает, что процесс разложения можно описать уравнением Ag2Cr04->02 +Ag+AgCrO2. Остаток, таким образом, представляет собой смесь металлического серебра и хромита серебра. Из полученных данных следует, что осадок хромата серебра, используемый для гравиметрического определения хрома, можно высушивать в интервале температур примерно от 100 до 800 °C, причем вполне подходящей для этого является температура НО °C (в методиках из старых учебников указывается температура точно 135 °C).
Кривая 2 на рис. 23-1 относится к нагреванию хромата ртути (I). Это соединение устойчиво в интервале температур от 52 до 256 °C, а при дальнейшем повышении температуры разлагается в соответствии с уравнением Hg2CrO4->Hg2O + CrO3. Оксид ртути(1) сублимируется, и при температуре выше 671 °C остается остаток .постоянной массы, содержащий СгО3. Поскольку ртуть имеет большую атомную массу, осадок Hg2CrO4 вследствие низкого гравиметрического фактора удобен для определения хрома. Как и в предыдущем примере, осадок можно высушивать при НО °C.
Термические методы анализа 487
Во многих из опубликованных работ по термогравиметрии ставили задачу нахождения оптимальных условий высушивания или прокаливания осадков, пригодных для гравиметрических определений. Но возможности этого метода значительно шире. Рассмотрим, например, кривые 3 и 4 на рис. 23-1, из которых очевидно, что свойства оксалата кальция существенно отличаются от свойств оксалата магния, что позволяет проводить их одновременное определение. Оксалат кальция разлагается в две стадии: СаС2О4->СаСОз + СО и затем СаСО3->СаО + + СО2. При разложении оксалата магния стадия образования карбоната отсутствует: MgC2O4->MgO + CO-|-CO2. Интервалы температур устойчивости перечисленных соединений таковы (°C):
СаС2О4-Н2О <100
СаС2О4 228—398 MgQC^ 233—397
СаСО3 420—660
СаО >840 MgO >480
Таким образом, при 500 °C устойчивы карбонат кальция и оксид магния, а при 900 °C оба металла существуют в виде оксидов. Сравнив массы осадков при этих двух температурах, можно рассчитать количества обоих элементов в анализируемой пробе.
Граничные температуры различных участков термограмм, как отмечено на рис. 23-1, точно воспроизвести нельзя. Термогравиметрия— динамический метод, и система никогда не достигает равновесия. Поэтому температуры, ограничивающие характерные участки кривых, до некоторой степени будут зависеть от таких факторов, как скорость нагревания и масса осадка.
Термовесы. Регистрирующие аналитические весы с устройством для контролируемого нагревания образца называются термовесами. Наиболее современные из них представляют собой электронные полуавтоматические одночашечные весы. Нагреватель или печь обычно устанавливают под весами, так что держатель образца можно подвесить прямо к коромыслу весов. Поэтому необходимо совершенно устранить конвекционные потоки воздуха от нагревательных приборов, мешающие работе весов. Электронное регистрирующее устройство или чертит график зависимости массы от времени, или с помощью двухкоординатного. самописца прямо дает зависимость массы от температуры. Если записывают зависимость от времени, то желательно одновременно вторым самописцем записывать температуру; примеры программирования температуры показывают кривые 1 и 2 на рис. 23-4.
488 Глава 23
Температура, °C
Рис. 23-2. Соотношение между кривой ТГА и ее производной (ТГП) для пиролиза смеси карбонатов кальция и магния [4].
Дифференциальная термогравиметрия (ТГП). Часто выгодно сравнивать термограмму с ее первой производной, как это сделано на рис. 23-2 [3], где достаточно четко видна площадка при 700 °C, но плечо при 870 °C прорисовывается намного точнее.
Дифференциальный термический анализ (ДТА)
Этот метод позволяет следить за фазовыми переходами или химическими реакциями на основании измерения количества поглощенного или выделенного тепла. Он, в частности, удобен для изучения структурных изменений, происходящих в твердых веществах при повышенных температурах, когда другие методы мало пригодны.
В методе ДТА ведут непрерывную запись разности температур анализируемого образца и инертного материала сравнения (эталона) при повышении температуры окружающей среды. Рис. 23-3 иллюстрирует принцип метода. Два маленьких тигля,
Термические методы анализа 489
содержащих анализируемый образец 1 и эталон 3, помещают в соответствующие углубления металлического блока. В тигли вводят две термопары, соединенные друг с другом так, чтобы сразу получать разность температур между ними. В полость 2 помещают термопару для измерения температуры блока. Затем всю установку нагревают, используя линейный программатор температуры. Эталон должен
Рис. 23-3. Принципиальная схема прибора для ТГА. Ряд точек вокруг металлического блока изображает электронагреватель.
быть термически устойчивым веществом, для которого в интервале изучаемых температур не наблюдается фазовых переходов
или разложения. Для этих це-
лей часто используют оксид алюминия (А12О3).
При постоянном нагревании любое превращение или ини-.
циируемая повышением температуры реакция в исследуемом образце сопровождается появлением пика или впадины на прямой. Для эндотермического процесса температура образца падает ниже температуры эталона, в результате чего возникает потенциал, для экзотермического процесса знак потенциала будет противоположный. Экзотермы принято изображать вверх от базовой линии, а эндотермы — вниз от нее.
Ясно, что если температуру блока поддерживать постоянной, то и исследуемый, и стандартный образцы скоро придут в состояние равновесия, и потенциал станет равным нулю. Очевидно, что это скорее динамический, а не статический процесс. Важно, чтобы скорость нагревания блока была постоянной и хорошо воспроизводимой от одного эксперимента к другому, если предполагается сравнивать их результаты.
Обычная методика работы заключается в подводе тепла к образцам, и поэтому эндотермические эффекты более вероятны, чем экзотермические. Если же проявляется экзотермический эффект, то часто он возникает в результате вторичного процесса. В качестве примера рассмотрим кривые на рис. 23-4 [5]. Кривая 1 практически имеет ту же форму, что и кривая 3 на рис. 23-1; небольшие отличия обусловлены различием условий измерения. Кривая 2 относится к тому же эксперименту, но проводимому в атмосфере СО2, а не воздуха. Как и следовало ожидать, здесь также нет явных различий, но только в атмосфере СО2 разложение СаСО3 происходит при более высокой температуре.
490 Глава 23
Рис. 23-4. Одновременная регистрация диаграмм ДТА — ТГА при разложении СаС2О4 • Н2О на воздухе и в атмосфере СО?.
Кривая .? на дифференциальной термограмме также описывает разложение СаС2О4 в атмосфере СО2. Три точки потери массы соответствуют трем эндотермическим процессам — последовательному отщеплению Н2О, СО и СО2. Наоборот, второй пик на кривой 4, снятой на воздухе, явно экзотермический, обусловленный сгоранием СО на воздухе при температуре печи.
Между дифференциальной и производной термограммами существует некоторое сходство, но они дают различную информацию. Производная термограмма отражает только изменение массы, а дифференциальная термограмма показывает изменение энергии независимо от того, происходит изменение массы или нет. Интересный случай показан на рис. 23-5: явно выраженный эндотермический минимум (при 950 °C) на дифферен-
ТермИчеСкйе методы анализа 491
Температура, °C
Рис. 23-5. Разложение SrCO3 на воздухе, сопровождающееся переходом кристаллической решетки из ромбической в гексагональную, заметным только па кривой ДТА (Mettler Instrument Corporation.)
циальной термограмме не виден ни на интегральной, ни на производной термограмме. .Это, очевидно, связано с переходом кристаллической решетки SrCO3 из ромбической в гексагональную, что не сопровождается какими-либо изменениями массы образца.
Аппаратура для ДТА. На рис. 23-6 схематически изображена основная часть установки для дифференциального термического анализа с устройством, обеспечивающим создание контролируемой атмосферы. Поток газа можно пропускать через слой частиц образца и тем самым очищать его от любых газообразных продуктов разложения. К этому прибору, как и в некоторых моделях термовесов, подключен газовый хроматограф, что позволяет вести непрерывный контроль выделяющихся газов и накапливать полезную информацию об изучаемой системе.
Дифференциальная сканирующая калориметрия. Традиционный метод ДТА позволяет получить качественные данные о температурах и знаках переходов, но этим способом трудно получить количественную информацию об образце или теплоте перехода. Трудности обусловлены тем, что часто неизвестны такие важные факторы, как удельная теплоемкость и теплопровод-
492 Глава 23
Печь
Блок
потока газа
Самописец-регулятор
эталонной термопары
=-> К газсВому хроматографу
Пробода дифференциальной термопары
Камера с контролируемым давлением
Рис. 23-6. Схематическое изображение одного из приборов для ДТА.
Манометр
ность образца до и после перехода. На величину площади под эндотермой или экзотермой влияют также скорость нагревания, расположение термопар и другие экспериментальные параметры.
Для того чтобы получить количественные результаты, нужно превратить отделение для размещения образцов в приборе для ДТА в дифференциальный калориметр [6]. Обычно используют дифференциальный сканирующий калориметр (ДСК) с калориметром изотермического типа [7]. Держатель каждого образца (анализируемого и эталона) снабжен собственным электрическим нагревателем. Когда дифференциальная термопара
Термические методы анализа 493
Рис. 23-7. Разложение C11SO4 • 5Н2О: а — кривая ДТА; б — соответствующая кривая ДСК. Три эндотермы соответствуют потере двух, двух и одной молекул Н2О.
начинает регистрировать напряжение, автоматический контролирующий контур подает к более холодному образцу энергию, достаточную для компенсации температур обоих образцов. Одновременно с помощью второго электронного контролирующего контура температура эталона (т. е. в действительности обоих образцов) линейно увеличивается во времени. Регистрирующее устройство следит за количеством электрической энергии, поданной к тому или другому образцу для поддержания изотермических условий. Получаемая термограмма напоминает обычную кривую ДТА. Но площадь под пиком в этом случае слу
494 Глава 23
жит точной мерой энергии, подведенной к анализируемому образцу для компенсации эндотермических эффектов или к эталону для компенсации энергии, излучаемой образцом при возникновении экзотермических эффектов. Различия в теплопроводности, теплоемкости й другие не имеют значения. Разные фирмы выпускают аналогичные приборы, отличающиеся режимом работы.
. На рис. 23-7 сравниваются кривые ДТА и ДСК для CuSO<X Х5Н2О. На графике ДТА линейный подъем температуры несколько нарушается тепловыми явлениями, происходящими в образце, на кривой же ДСК подобные эффекты невозможны, и пики имеют более правильную форму.
Сканирующий калориметр позволяет проводить точное определение следов примесей в высокочистых органических соединениях по результатам наблюдения за понижением температуры плавления [8, 9]. Детали эксперимента и расчета даны в цитируемой литературе. Сообщается, что стандартное отклонение при определении примесей составляет ±4%.
Термотитриметрия [10]
Поскольку практически все химические реакции сопровождаются тепловым эффектом, за ходом реакции можно проследить, наблюдая за выделением или поглощением тепла при титровании в маленьком адиабатическом калориметре. Подходящий калориметр легко сконструировать из маленьких сосудов Дьюара или изолированных сосудов. Изменение температуры можно контролировать термопарой или термистором. Термометрическое титрование легко сделать автоматическим; такие приборы выпускаются несколькими компаниями.
В большинстве моделей титрант добавляют с постоянной скоростью с помощью автоматического шприца или аналогичного устройства и записывают на самописце разность температур в сосудах, содержащих анализируемый и стандартный образцы.
Были успешно исследованы разнообразные реакции, применяемые в титриметрии. К ним относятся кислотно-основные реакции с участием сильных и слабых кислот и оснований, реакции, сопровождающиеся образованием малорастворимых или комплексных соединений, а также окислительно-восстановительные реакции. Для их проведения можно использовать любые растворители; сообщалось, что наряду с водой применялась уксусная кислота, тетрахлорид углерода, бензол, нитробензол, расплав эвтектической смеси нитратов лития и калия. Стандартное отклонение обычно составляет ±1 %, но иногда удается получить лучшие результаты.
Термические методы анализа 495
Важно, чтобы оба реагирующих раствора мало отличались по составу, так как посторонние вещества могут вызывать заметный тепловой эффект либо при взаимодействии друг с другом, либо при взаимодействии с растворителем (теплота разбавления). Например, при титровании Fe(II) раствором К2СГ2О7 концентрация серной кислоты в пробе и титранте должна быть одинаковой. Большие преимущества дает использование двух идентичных автоматических шприцов для добавления в два калориметрических сосуда равных количеств реагента. Это позволяет устранить влияние теплоты разбавления и любые другие источники помех [11].
Как отмечалось в работе [10], термотитриметрия — это единственный метод титрования, в котором наряду с изменением свободной энергии AG (т. е. константы равновесия) измеряемой величиной является ЛЯ (а не AG), входящая в известное выражение AH=AG + 7’AS. Поэтому результаты термометрического титрования могут оказаться полезными, даже если величина AG равна нулю или положительна. Несколько примеров
иллюстрируют ценность такого подхода.
Борная кислота относится к очень слабым кислотам: константа диссоциации по первой стадии Кх = 5,8-10-10 при 25 °C. Простой термодинамический расчет показывает, что для реакции с ионами ОН-
НзВОз+ОН- Н2ВОГ+Н2О (23-1) величина AG0 составляет —6,5 ккал/моль, что соответствует константе нейтрализации KH = K’i/K’w = = 5,8-104. Это можно
сравнить с реакцией титрования сильной кислоты,для которой Кн=Ю14 и AG°= = —19,2 ккал/моль. Поскольку величина AG0 реакции (23-1) мала, борную
Рис. 23-8. Кривая термометрического титрования 0,01 М водных растворов НС1 (/) и Н3ВО3 (2) раствором NaOH. Титрование начинается в точках А и заканчивается в точках ТЭ [8].
кислоту нельзя оттитровать ни одним из методов, основанных на измерении pH (например, потенциометрическим или фотометриче-
496 Глава 23
Объем титранта
Рис. 23-9. Термометрическое титрование смеси 0,25 ммоль Са2+ и 0,25 ммоль Mg2+ раствором ЭДТА. Титрование начинают в точке В. Конечную точку титрования кальция находят экстраполяцией до точки Сь для магния — до точки С2. Отрезок от точки С2 до точки D соответствует добавлению избытка реагента.
ским), так как активность ионов водорода определяется величиной константы равновесия.
Однако оказалось, что энтропийный фактор T&S° для борной кислоты составляет —3,7 ккал/моль, а для НС1 +5,7. Отсюда можно найти, что энтальпия нейтрализаций составляет —10,2 ккал/моль для борной кислоты и —13,5 ккал/моль для НС1. Эти данные показывают, что обе кислоты одинаково легко оттитровать термометрически (рис. 23-8) [12].
Другим интересным случаем является титрование кальция и магния раствором ЭДТА. Константы устойчивости этих хелатов различаются менее чем на два порядка, и индикаторное титрование невозможно. Однако изменение энтропии в реакции Mg2+ с ЭДТА вдвое превышает изменение энтропии в реакции с Са2+. Это не только дает достаточное различие в величинах АН, но фактически приводит к изменению знака: ДН= = +5,5 ккал/ моль в реакции с ионом магния и —5,7 ккал/моль в реакции с ионом кальция. Кривая титрования смеси этих двух ионов приведена на рис. 23-9 [10].
Термические методы анализа 497
Энтальпиметрия
Этот метод термотитриметрии позволяет получить информацию не только о стехиометрии, но и о величине ДЯ— молярном изменении энтальпии. Методика измерения, называемая прямой инжекционной энтальпиметрией (ПИЭ) [11, 13], основана на том, что измеренный избыток реагента сразу вводят в образец, находящийся в маленьком адиабатическом калориметре.
На рис. 23-10 представлен общий вид кривой, которую можно
Время
Рнс. 23-10. Идеальная энталь-пиметрическая кривая. Реагент вводят в момент времени В.
Рис. 23-11. Инжекционные энтальпиграммы: / — НС1 обрабатывали избытком NaOH; II— Н3ВО3 — избытком NaOH; III — Mg2+ — избытком ЭДТА; IV — Pb2+ — избытком ЭДТА. Содержание образца составляло порядка 0,1 ммоль в 25 мл. В момент времени IT введено 0,300 мл 1М раствора реагента. Д£ — экстраполированный разбалансный потенциал термистора [14].
498 Глава 23
получить с помощью ПИЭ. Отрезок АВ — холостой ход самописца до установления базовой линии. Реагенты вводят в точке В. Вблизи точки С часто наблюдается искривление, а не резкий изгиб, что связано с величиной константы равновесия реакции (и, следовательно, с AG°). От точки С до точки D обычно наблюдается подъем, как это показано на рисунке, соответствующий теплоте разбавления реагента. Но может наблюдаться и спад, указывающий на то, что разбавление представляет собой эндотермический процесс (что бывает очень редко), или на то, что температура реагента ниже, чем содержимого в сосуде для титрования. Изменение температуры (АТ), которое можно прямо связать с изучаемой реакцией, определяют по расстоянию от точки В до точки пересечения экстраполированного отрезка CD с линией, параллельной оси абсцисс. Величина АТ пропорциональна общему количеству выделившегося тепла (Q = —NAH), где N — число молей реагирующего вещества, а АН — теплота, поглощенная молем вещества. Константа пропорциональности k есть теплоемкость сосуда и его содержимого в килокалориях на градус Кельвина, поэтому
T = QJk= — NAHlk (23-2)
Численную величину k можно определить простой калибровкой, например по известному количеству электрической энергии. Затем, если известно АН°, можно найти N (или наоборот), при условии что справедливо предположение, что АН и АН° существенно не отличаются.
На рис. 23-11 приведено несколько примеров инжекционных энтальпиграмм [14]. Этот метод обладает высокой чувствительностью и может найти очень широкое применение. В работе [15] показана возможность определения с помощью ПИЭ 3 нМ нитрит-иона с погрешностью ±5 % по реакции с сульфаминовой кислотой.
Задачи
23-1. На рис. 23-12, а приведены кривые ТГА для безводных AgNO3 (550 мг) и Cu(NO3)2 (285 мг). На рис. 23-12,6 приведены кривые ТГА для бинарной смеси этих солей, а) Путем приблизительной оценки по кривым чистых соединений определите, какие формы устойчивы при 400 и 700 °C. б) Каков состав смеси?
23-2. При нагревании Na(NH4)PO4-4H2O сначала выделяются четыре молекулы НгО, затем еще одна молекула Н2О и молекула NH3 и образуется устойчивый NaPO3. Изобразите форму кривой, которую можно получить при изучении этого соединения а) с помощью термовссов, б) методом ДТА.
23-3, Каков конечный продукт пиролиза SrCO3 (рис. 23-5)?
Термические методы анализа 499
Рис. 23-12. Термограммы для нитратов серебра и меди и их смеси. Перед нагреванием образцы высушивали на воздухе для удаления избытка влаги: а — термограммы индивидуальных стандартных образцов; б — термограммы смеси [3].
23-4. Известно, что соотношение между мольной долей растворенного вещества xs и понижением температуры плавления Д7' дается уравнением
Д7’ =
----xs
bHf
где AHf — теплота плавления (кал/моль); То — температура плавления растворителя (К); R — газовая постоянная, равная 1,987 кал/(К-моль). На кривой, полученной с помощью ДСК [8], для 2,285 мг «чистого» соединения А (молекулярная масса 263,12) есть пик, площадь которого, измеренная планиметром, равна 3302 единицам (единица соответствует 0,0199 мкал) при То— = 308,9 К. Определите ДЯ/ (в кал/моль), а затем содержание примеси А в мольных процентах (мольная доляХЮО); ДТ равно 0,112 К.
23-5. Небольшие количества Ва определяли методом ПИЭ, инжектируя пробу в избыток HSO4- в 0,1 М НС1. Из литературных данных известно, что для реакции HSO4~-|-Ba2+->-BaSO4 (тв.)+Н+ Д//°=—11,1 ккал/моль. (Теплоемкость разбавленного раствора можно принять равной теплоемкости воды, т. е. 1 кал/(г-К), а плотность 1 г/мл.) В одном из экспериментов при инжектировании 2,00 мл пробы в 18,00 мл реагента найдено, что повышение температуры Л.Т= 0,278 К. Какова концентрация [Ва2+] в пробе?
500 Глава 23
Литература
1. Daniels Т., Thermal Analysis, Wiley, New York, 1973.
2. Wendlandt HZ. W., Gallagher P. K-, In Thermal Characterization of Polimer Materials, Turi E. A. (ed.), Academic Press, New York, 1981.
3. Duval C., Inorganic Thermogravimetric Analysis, (2d ed.), American Elsevier, New York, 1963.
4. Paulik F., Paulik J., Edey L., Hung. Sci. Instrum., 1964, v. 1, p. 3.
5. Vaughn H. P„ Wiedemann W. G.; In Vacuum Microbalance Techniques, Waters P. M. (ed.), Plenum Press, New York, 1965.
6. Daniels T„ Thermal Analysis, Wiley, New York, 1973, p. 86, 122.
7. Watson E. S., O’Neill №.. I., Justin J., Brenner N., Anal. Chem., 1964, v. 36, p. 1233.
8. Plato C., Glasgow A. R., Jr., Anal. Chem., 1969, v. 41, p. 330.
9. Plato C., Anal. Chem., 1972, v. 44, p. 1531.
10. Jordan J., In Treatise on Analytical Chemistry, Kolthoff 1. M., Elving P. J. (eds.), Wiley-Interscience, New York, 1968, pt. 1, v. 8, ch. 91.
11. Vassos В. H., Rodriques R. A., Anal. Chem., 1981, v. 53, p. 135.
12. Jordan J., Rec. Chem. Progr., 1958, v. 19, p. 193.
13. Vaughn G. A., Thermodynamic and Enthalpometric Titrimetry, Van Nor-strand-Reinhold, London, 1973.
14, Wasilewski J. C., Pei P. Т.-S., Jordan J., Anal. Chem., 1964, v. 36, p. 2131.
15. Hansen L. D., Richter В. E., Eatough D. J., Anal. Chem., 1977, v. 49, p. 1779.
ГЛАВА 24
Ядерно-физические методы
В предыдущих главах речь шла о массе ядра и спине, представляющих интерес для аналитической химии, не менее существенна также способность некоторых ядер подвергаться характерным превращениям. Эти превращения — спонтанное деление ядер (радиоактивный распад), сопровождающееся испусканием корпускулярного и электромагнитного излучения, а также. реакции присоединения, в которых ядро рекомбинирует с нейтроном или иной частицей. С последним типом превращений связана мёссбауэровская спектроскопия, основанная на резонансном поглощении гамма-квантов некоторыми ядрами.
Р адиоактивность
Вероятно, читатель знает основы теории радиоактивных явлений, однако для удобства изложения их полезно кратко напомнить.
Почти все известные нам элементы имеют несколько изотопов. Многие изотопы не встречаются в природе, однако их можно приготовить искусственно из подходящих изотопов того же или другого элемента. Ядра большинства искусственных и многих природных изотопов нестабильны и склонны к самопроизвольному распаду с выделением различных частиц. Кром.е того, при распаде образуется осколочное ядро, которое несколько легче, чем исходное.
Радиоактивные вещества испускают частицы нескольких типов. Нас будут интересовать (табл. 24-1) электроны, позитроны (позитрон — положительно заряженный аналог электрона), альфа-частицы и нейтроны. Испускание этих частиц обычно (хотя и не всегда) сопровождается испусканием гамма-лучей. К другому типу радиоактивного распада относится спонтанный захват ядром электрона с /(-уровня. Этот процесс, известный как захват электрона или К-захват, вызывает характеристическое для элемента рентгеновское /(-излучение.
502 Глава 24
Таблица 24-1. Частицы, образующиеся при радиоактийнбм распаДё
Частица Обозначение Масса2 Заб ряд6 Проникающая способность Ионизирующая способность
Электрон Позитрон Альфа-частица Нейтрон Фотон (у-квант) !>+ а п V 5,439-10—4 5,439-10—4 3,9948 1,0000 0 — 1 +1 +2 0 0 Средняя То же Низкая Очень высокая То же Средняя То же Высокая Нет Очень низкая
а Относительно нейтрона. 6 В 1,60240-10 *3 Кл.
Частицы и излучение, испускаемые различными ядрами, сильно разнятся по энергии и скорости вылета. Они характеристичны для распадающегося изотопа и, следовательно, могут быть использованы для идентификации.
Частота, с которой происходят атомные распады, характеризуется специфичным для изотопа периодом полураспада. Это время, необходимое для распада половины имеющегося образца. Для известных радиоактивных веществ время полураспада составляет от миллионных долей секунды до миллионов лет. В обоих крайних случаях время полураспада прямо измерить нельзя, но о нем можно судить по косвенным данным. Очень короткоживущие изотопы бесполезны для аналитических целей просто потому, что исчезают слишком быстро. В то же время очень долгоживущие изотопы трудно использовать из-за слишком малой частоты распадов. Для аналитических применений пригодны изотопы с периодом полураспада от нескольких часов до нескольких тысяч лет. Нижний предел может быть уменьшен примерно до 10 мин, если эксперименты с радиоактивными веществами проводить сразу же после получения и в той же лаборатории. В общем длительность эксперимента не должна превышать примерно удесятеренного периода полураспада.
Ряд пригодных в аналитических целях радиоизотопов представлен в табл. 24-2. Радионуклиды могут служить источниками излучения, метками (индикаторами) для контроля за какими-либо реакциями или процессами и определения их полноты. Прежде чем обратиться к этим вопросам, рассмотрим методы детектирования и измерения.
Таблица 24-2. Используемые в анализе радионуклиды’
Изотоп Тип о распада Период полураспада Энергия радиации, МэВ
Частицы у-излучение
3Н 0- 12,26 года 0,0186 Нет
14С p~ 5720 лет 0,155 »
22Na 0+ (90 %) ЭЗ (10%) 2,58 года 0,545 1,27
ззр 0~ 14,3 сут 1,71 Нет
35S 0~ 87 сут 0,167 »
звс| 0~ 3,0-105 лет 0,714 »
«к 0~ (89 %) ЭЗ (11 %) 1,27- 10е лет 1,32 1,46
42К 0- 12,36 ч 3,55 (75 %) 1,98 (25 %) 1,52 (25 %)
45Са 0- 165 сут 0,255 0,32
51СГ ЭЗ 27,8 сут 0,32 (8 %)
66Fe ЭЗ 2,60 года Нет
68Fe 0- 45 сут 0,460 (50 %) 0,27 (50 %) 1,29 1,10
57Со ЭЗ 270 сут 0,122; 0,0144 0,136
“«Со 0- 5,26 года 0,32 1,333; 1,173
63Zn ЭЗ (97,5 %) 0+ (2,5 %) 245 сут 0,33 1,11
85Кг 0~ 10,6 года 0,67 (?)
»»Sr 0~ 29 лет 0,54 Нет
90у 0- 64 ч 2,27 (Y)
95Zr 0“ 65 сут 0,36; 0,40 0,72; 0,76
«5Nb 0~ 35,1 сут 0,16 0,77
noAg 0- 253 сут 0,085 (58 %) 0,44; 2,46
nsm Sn ВП 250 сут 0,065 ; 0,024
1311 0~ 8,06 сут 0,60 (87,2 %) (прочие) 0,364 (80,9 %) (прочие)
187Cs 0~ 30 лет 0,51 (92 %) 1,17 (8%) 0,662 (от дочернего ядра 137тВа)
133Ba ЭЗ 7,2 года 0,360; 0,292 0,081; 0,070
‘«La 0- 40,2 ч 1,34 (70 %) (прочие) 0,49; 0,82 1,60 (прочие)
wpm 0- 2,65 года 0,225 (?)
170Tm 0- 127 сут 0,97 (76 %) 0,88 (24 %) 0,084
203Hg 0- 47 сут 0,21 0,28
19ВА11 0- 2,70 сут 0,96 (прочие) 0,412 (прочие)
204TI 0- (98 %) ЭЗ (2 %) 3,80 года 0,76 Нет
210pb 0~ 22 года 0,015; 0,061 0,046
аДанные отобраны нз таблиц, приведенных в работе [1].
6 ЭЗ — электронный захват, ВП — внутренний переход.
504 Глава 24
Детекторы радиации
Детекторы рентгеновского излучения, описанные в гл. 11, пригодны и для обнаружения радиации. Так, например, гамма-лучи физически неотличимы от рентгеновских. Корпускулярное излучение (кроме нейтронного) имеет меньшую проникающую способность и ослабляется, проходя сквозь стенки или поверхностные слои некоторых типов детекторов, поэтому его нетрудно обнаружить.
Фотографический детектор используют в основном для получения представления о распределении радиоактивного вещества на поверхности твердых образцов с помощью так называемой авторадиографии, или радиоавтографии.
Сцинтилляционные счетчики [3]. Попадая в подходящую флуоресцирующую среду, излучение или частица вызывает мгновенную вспышку видимого света. По числу таких вспышек-сцинтилляций можно судить о количестве частиц или фотонов в потоке радиации. Используемая для этого схема аналогична описанной в гл. 3 схеме счета фотонов в. УФ-спектроскопии.
Сцинтилляторы бывают твердые и жидкие. Из твердых наиболее распространен иодид натрия, активированный таллием. Его можно, как на рис. 24-1, расположить так, чтобы создать вокруг анализируемого образца как бы цилиндрический колодец, что обеспечивает высокую эффективность счетчика. А можно небольшое количество образца поместить между па-
подобие сэндвича), так что будет улавливаться детектором. О такой схеме говорят, что она имеет «4л-гео-метрию». Между сцинтиллятором и фотоумножителем должен быть хороший оптический контакт, а стенки устройства должны хорошо отражать свет, чтобы не было потерь. Счетчик следует поместить в свинцовый «домик» (или в корпус из другого плотного материала), чтобы снизить помехи фонового излучения.
В жидких сцинтилля-
рой сцинтилляторов (получается почти все испускаемое излучение
Рис. 24-1. Схематическое изображе^ ние сцинтилляционного счетчика.
торах активное вещество находится в растворенной
Ядерно-физические методы 505
Рис. 24-2. Жидкостной сцинтилляционный счетчик со схемой проверки совпадений. Образец, фотоумножители и предусилители следует охлаждать для снижения электромагнитных шумов.
форме. Такие счетчики используют преимущественно для количественного измерения потоков низкоэнергетических 0-частиц, испускаемых 14С, 35S и особенно 3Н (тритием). Жидкая среда обеспечивает максимальную эффективность сцинтилляций под действием 0-частиц. Многие органические соединения, растворенные в подходящих растворителях, ведут себя как сцинтилляторы. К ним относятся, например, антрацен, 1,4-дифенилбен-зол, 2,5-дифенилоксазол (ДФО), а-нафтилфенилоксазол (НФО) и фенилбифенилоксадиазол (ФБД). Наиболее эффективен из них ДФО, однако он испускает излучение в УФ-диапазоне, которое поглощается многими растворителями. ДФО удобно смешивать с вторичным осциллятором, который (по флуоресцентному механизму) преобразует УФ-сцинтилляции в видимый свет. Наиболее распространенный вторичный сцинтиллятор 1,4-бис-2-(5-фенилоксазолил) бензол (ФОБ) или его диметиль-ное производное (диметил-ФОБ). В качестве сцинтиллятора можно рекомендовать раствор в толуоле ДФО (5 г/л) щдиме-тил-ФОБ (0,3 г/л).
Чтобы отличить импульсы, вызванные сцинтилляциями, от случайных (возникающих в фотоумножителе вследствие космических ливней или дробового эффекта), следует принимать специальные меры предосторожности. Например, к одному сцинтиллятору можно подключить два одинаковых фотоумножителя и связать их схемой проверки совпадений (рис. 24-2). Последняя включает электронную схему — элемент И, которая пропускает сигнал к счетчику импульсов, только если оба фото
506 Глава 24
умножителя одновременно выдают по импульсу. Беспорядочный флуктуационный (дробовой) шум может случайно вызвать одновременные импульсы. При необходимости, чтобы уменьшить вероятность таких импульсов, можно перейти к тройной схеме проверки совпадений, включающей три фотоумножителя. Возникающий в фотоумножителях шум можно также понизить с помощью охлаждения, ослабляющего тепловое движение электронов.
Сцинтилляционный счетчик является пропорциональным, т. е. энергия каждой вспышки света определяется энергией возбуждающей частицы или фотона. Пропорциональность должна быть обеспечена также конструкцией детектора и умножителя.
Газовые ионизационные детекторы. Радиоактивное излучение (кроме нейтронного) вызывает частичную ионизацию во многих материалах (см. табл. 24-1). Измерение степени ионизации в газах или в полупроводниках служит основой общего метода обнаружения радиоактивного распада.
Рассмотрим явления, происходящие в наполненной газом трубке по мере увеличения разности потенциалов между парой электродов. Один из электродов представляет собой заземленный металлический цилиндр (диаметром около 1 см и длиной 5 см), вторым электродом служит расположенный вдоль оси цилиндра проводник (рис. 24-3). Центральный проводник соединен с инвертирующим входом операционного усилителя (усилитель должен быть рассчитан на работу при высоком напряжении), на неинвертирующий вход которого подается переменный положительный потенциал. Поскольку операционный усилитель уравнивает потенциалы своих входов, в описанной схеме между электродами детектора возникает разность потенциалов, равная напряжению источника питания. Ток, который будет протекать в системе, должен проходить через сопротивление обратной связи На входе операционного усилителя возникает соответствующее напряжение, которое можно при необходимости усилить и вывести сигнал на самописец.
Высокое напряжение
Рис. 24-3. Схема операционного усилителя для газового ионизационного счетчика.
Ядерно-физические методы 507
Рис. 24-4. Число проходящих иоиов в зависимости от напряжения в газовом ионизационном счетчике: а — для а-частиц; б—-для 0-частиц. Обратите внимание на отсутствие масштаба иа оси абсцисс; счетчик, настроенный иа один диапазон напряжений, может плохо работать в других [2].
Теперь предположим, что газ внутри детектора облучают слабоинтенсивным потоком 0-частиц со скоростью около 1000 частиц в секунду. При очень низком потенциале (вблизи начала координат на рис. 24-4) образующиеся ионы будут мгновенно рекомбинировать. При более высоком напряжении (в области Л) все больше ионов будет достигать электродов, вызывая измеримый ток. В области В электрическое поле настолько сильное, что до электродов доходят почти все ионы, и при напряжении около 100 В на графике возникает
плато, поскольку при повышении напряжения количество ионов-носителей тока больше не увеличивается. В этой области детектор работает как ионизационная камера. Сила тока пропорциональна числу образующихся ионов и их энергии, однако она так мала (порядка нескольких наноампер), что для получения полезного сигнала внешние электронные схемы должны давать очень высокое усиление. Если усилитель имеет малое время отклика, можно наблюдать импульсы, обусловленные индивидуальными 0-частицами. Однако сочетать малое время отклика и высокий коэффициент усиления трудно. Поэтому ионизационные камеры обычно работают в статистическом режиме (измеряют силу тока).
При дальнейшем увеличении напряжения (область С) ионы ускоряются полем. Они приобретают энергию, достаточную для дальнейшей ионизации газа (за счет столкновений). Возникающее внутреннее усиление может быть тысячекратным. -Иными словами, величины импульсов от индивидуальных 0-частиц возрастают, но не остаются пропорциональными энергии частиц. Устройство, работающее в таком режиме, называется газовым пропорциональным счетчиком. Здесь (область 1000— 2000 В) необходима точная установка потенциала, поскольку рабочая точка (рис. 24-4) уже не лежит на плато.
Дальнейшее увеличение напряжения (область D) вызывает сильную вторичную ионизацию, и величина импульсов перестает быть пропорциональной энергии частиц ионизирующего
508 Глава 24
излучения. В области Е шириной 100—200 В все импульсы имеют примерно равную величину независимо от энергии ионизирующих частиц. В этой области устройство работает как счетчик Гейгера, очень удобный для подсчета числа частиц независимо от их энергии. Импульсы настолько велики, что усиление (если оно и требуется) должно быть минимальным. Это упрощает конструирование портативных счетчиков. Даже если не удается разделить импульсы по величине, у- и 0-излучение легко различить, используя алюминиевый экран (он Непроницаем для 0-частиц).
При дальнейшем увеличении напряжения до области F в трубке возникает самоподдерживающийся тлеющий разряд.
Рабочие диапазоны для счетчика Гейгера и пропорционального счетчика можно определить, снимая зависимость скорости счета (отсчеты/мин) от напряжения для фиксированного источника радиации. Следует принять меры предосторожности, чтобы ускоренные полем положительные ионы не вызывали вторичную' электронную эмиссию на внешнем (отрицательном) электроде. Дело в том, что вторичные электроны, разгоняясь полем, могут вызвать дополнительную ионизацию газа и непрерывный тлеющий разряд. Во избежание этого в газ, наполняющий трубку, вводят несколько процентов паров органических
Рис. 24-5. Плаишетиое счетное устройство; детектор и образец окружены защитным экраном из тяжелого металла. Общая высота около 40 см, вес 90 кг (Radiation Counters Laboratory, Inc.).
Ядерио-физические методы 509
соединений (метана или низших спиртов) или галогенов. Эти добавки поглощают избыточную энергию положительных ионов.
В другой модификации пропорционального счетчика — без-окошечном проточном счетчике, предназначенном для измерения слабопроникающего излучения, детектор медленно омывается потоком газа (например, смеси аргон—10% метана). При этом органическая добавка расходуется медленно. Кроме того, образец можно помещать прямо внутрь счетчика.
На рис. 24-5 показано устройство торцевого счетчика. Образец, укрепленный на диске из алюминия или нержавеющей стали или помещенный в неглубокий стаканчик (планшет), устанавливают на движущейся платформе внизу счетчика. Расстояние между образцом и счетчиком при работе с образцами разной мощности можно «регулировать. При необходимости в прибор легко ввести алюминиевые экраны.
Полупроводниковые детекторы. Отличные пропорциональные детекторы можно изготовить на основе кристаллов кремния или германия [4]. Существуют два способа, с помощью которых можно подготовить кристаллы этих элементов для использования в качестве детекторов. Оба они основаны на том факте, что под действием радиации электроны в твердом теле смещаются, т. е. возникают электронно-дырочные пары.
В первом способе используется обычный рп-переход (см. гл. 27, где обсуждаются свойства полупроводников). И верхнюю, и нижнюю поверхности кристалла делают проводниками, напыляя тонкую металлическую пленку. Обратное включение (n-зона положительна, p-зона отрицательна) такого диода приводит к тому, что электроны и дырки уходят от перехода, собираясь в п- и p-зонах соответственно. В результате образуется разреженная (лишенная носителей тока) зона толщиной до 1 мм. Толщину зоны можно варьировать, меняя приложен-. ное к кристаллу напряжение.
Во втором методе применяют кристаллы, легированные литием. Ионы лития движутся (дрейфуют) по кристаллу под действием приложенного электрического поля, «очищая» кристалл от естественных носителей заряда. Поэтому наложение электрического поля вызывает формирование глубокого разреженного слоя толщиной до 1 см. Литий-германиевые детекторы следует всегда охлаждать до температуры жидкого азота (77 К), поскольку ионы лития при комнатной температуре достаточно подвижны и могут нарушить геометрическую структуру разреженного слоя.
Для детектирования а- и р-частиц с низкой проникающей способностью пригодны более дешевые детекторы первого типа (с тонким разреженным слоем). Для у-спектроскопии лучше
510 Глава 24
подходят литий-германиевые детекторы, поскольку разреженный слой в них толще. Кроме того, у германия больший коэффициент поглощения, чем у кремния.
Описанные выше детекторы используют так же, как газовые пропорциональные счетчики. Образующиеся в разреженной зоне пары электронов и дырок ускоренно движутся к соответствующим электродам; через кристалл протекает ток, сила которого пропорциональна энергии ионизирующей частицы или излучения. Поскольку сила тока составляет всего несколько микроампер, необходим чувствительный усилитель с низким уровнем шумов.
Фоновая радиация
Поскольку в природе существует радиоактивный фон, любой детектор будет давать некоторый сигнал даже в отсутствие образца. Фон обусловлен естественной радиоактивностью окружающей среды и космическими лучами. Фоновый уровень (до 15—20 отсчетов в минуту) можно существенно снизить, защитив детектор свинцовым экраном («домик») толщиной 5—10 см. Более высокий фон или резкое его возрастание может означать случайное загрязнение рабочего места радиоактивными веществами или поломку счетчика. При проведении количественных измерений или интерпретации данных необходимо ввести поправку на фоновую радиацию.
Пересчетные схемы
Для количественных измерений радиоактивности необходимо устройство, подсчитывающее идущие от детектора импульсы. Для этого обычно используют десятичную пересчетную схему, которая пропускает на свой выход только каждый десятый из поступающих на вход импульсов. Каскад таких схем, соединенный с устройством снятия цифровых отсчетов, обеспечивает достаточную точность представления данных. Обычно пересчетные схемы снабжаются таймером, чтобы счет длился некоторое установленное время. С другой стороны, можно измерять время, необходимое для достижения заранее заданного счета.
Радиоактивный распад — явление статистическое. Законы вероятности определяют, сколько атомов распадается с выделением соответствующих осколков в каждый момент времени. Поэтому наблюдаемые отсчеты (числа импульсов) будут значимы только в том случае, если их накоплено достаточно много, чтобы можно было провести строгий статистический анализ. Это является одним из условий корректного эксперимента [5, 6]. Мы дадим лишь краткое изложение соответствующих сведений.
Ядерно-физические методы 511
Наиболее удобным показателем воспроизводимости последовательных измерений служит стандартное отклонение о. Для измерений радиоактивности, когда период полураспада активного элемента намного больше длительности эксперимента, стандартное отклонение составляет корень квадратный из общего числа импульсов п. Результат эксперимента можно выразить как Теперь ясно, почему лучше измерять время
достижения заданного счета, тогда результаты будут иметь примерно одинаковую воспроизводимость.
Фон тоже имеет статистическую природу. Если а0, оф и Ос —стандартные отклонения измерений счета образца, фона и общего счета, их связывает уравнение
°о = (аф+ас)‘/2 (24-1)
Для уменьшения роли фона его значение можно измерять достаточно долго. Тогда ОфСОс, и первым можно пренебречь. Наилучшая же точность за минимальное время обеспечивается при следующем соотношении периодов счета для образца (вКЛЮЧаЯ фон) tz И ХОЛОСТОГО (ТОЛЬКО фОН) /ф.'
/с//ф=(Яс/Яф)1/2 (24-2)
где Ro и 7?ф — относительные скорости счета, которые следует оценить в предварительных опытах.
Пусть, например, в таких опытах установлено, что активности образца и фона составляют примерно Rc = 1000 имп./мин и 7?ф = 4О имп./мин, а отношение равно (1000/40)1/2 =5. Если желаемая воспроизводимость характеризуется относительным стандартным отклонением 1 %, измеряемый сигнал должен быть на уровне 10 000 импульсов (поскольку 100=1 % от 10 000). Значит, за период измерения (накопления отсчетов) tz следует принять 10 мин. Тогда фоновый счет должен занимать пятую часть времени, т. е. 2 мин. Пусть при этих условиях величина счета для образца составит, например, 10025 импульсов. Тогда активность
D 10 025 ±V10 025 шло к < inni ,
Rc =---------------= 1002,5 ± 10,01 имп./мин
10
Аналогично, если фоновый счет равен 81, п 81 ± V81 . ,
Ь?ф =--------= 4,05 ± 4,5 имп./мин
2
И с учетом уравнения (24-1) исправленная на фон активность образца
Ro = (1002,5 — 40,5) ± [(Ю,01)2+ (4,5)2]1/2 = 962± 11 имп./мин
512 Глава 24
Еще один потенциальный источник ошибок составляют совпадения: два импульса могут идти так близко друг к другу, что счетчик не сможет достаточно быстро обработать первый и откликнуться на второй. Мертвое время т зависит от типа детектора. Учесть его влияние помогает уравнение
R' = R + Rh (24-3)
где R — наблюдаемая, a R' — истинная активность.
Следует также учитывать, что во многих детекторах только часть радиоактивного излучения образца воспринимается счетчиком. Так, геометрическая эффективность простого торцевого счета обычно меньше 50 %. Часто такую, низкую эффективность считают допустимой, поскольку соответствующее оборудование проще, чем в случае полной «4л-геометрии». Важно, чтобы во всех измерениях, результаты которых вы будете сравнивать, была обеспечена одна и та же геометрия. Этого можно достичь при помощи прибора, изображенного на рис. 24-5. Следует также принимать меры предосторожности во избежание частичного поглощения радиации растворителем или фильтровальной бумагой.
Анализ высоты импульсов. В табл. 24-2 показано, насколько сильно различается энергия ионизирующих частиц и излучений, испускаемых разными радионуклидами. Часто по этим энергиям можно идентифицировать индивидуальные радиоактивные частицы. В этих целях применяют стандартные поглощающие экраны: алюминиевый для низкоэнергетических частиц и свинцовый для у-лучей, экран помещают между образцом и детектором. Поскольку толщина слоя металла, ослабляющего активность наполовину, связана с энергией частиц, можно построить и использовать соответствующий градуировочный график.
Более изящный способ состоит в анализе высоты импульсов с помощью какого-либо пропорционального счетчика [6]. Предназначенный для этой йели многоканальный анализатор описан в гл. 11. На рис. 24-6 показан спектр радиоактивного излучения 13Ч, а на рис. 24-7 — спектр смеси у-излучателей, снятые на многоканальном анализаторе.
Для количественного анализа следует измерять площадь под пиками. Часто это сложно сделать из-за отсутствия четко выраженной базовой линии. В работе [8] показано, что по статистическим соображениям для надежного измерения площади пиков пригодна процедура, показанная на рис. 24-8, где каждый вертикальный сегмент соответствует одному каналу анализатора. Линия АВ пересекает контур таким -образом, что слева и справа от вершины пика находится одинаковое число
Ядерно-физические методы 513
Рис. 24-6. Спектр у-излучеиия 131I (Nuclear Chicago Corp.).
Рис. 24-7. Спектр у-излучения, полученный с помощью литий-германиевого детектора и многоканального анализатора. Цифры на рисунке обозначают энергию в килоэлектронвольтах. Пик «генератора импульсов» обеспечивается встроенным генератором и служит внутренним стандартом при определении мертвого времени (Isotopes Inc.).
17 Заказ № 537
514 Глава 24
Рис. 24-8. Количественное определение площади под индивидуальным пиком в спектре у-излучения.
каналов (на рисунке 5). Площадь между кривой и такой линией, измеряемая планиметром, служит мерой интенсивности радиоактивного излучения.
Измерение нейтронных потоков
Поскольку нейтроны не заряжены, они не вызывают ионизации. Их можно обнаружить с помощью счетчика, наполненного газообразным BF3. Нейтроны легко реагируют с ядром 10В, давая 7Li и а-частицы [реакция 10B(n, a)7Li]*. Образующиеся a-частицы регистрируются счетчиком обычным способом. Нейтроны можно регистрировать с помощью сцинтилляционного счетчика, в рабочую среду которого введены соединения бора.
* В такой записи первый символ в скобках обозначает частицу, реагирующую с указанным слева ядром; справа указаны образующиеся в ядерпой реакции ядро и частица, символ которой стоит в скобках вторым.
Ядерно-физические методы 515
Низкоэнергетические нейтроны имеют дебройлевскую длину волны (X = /i/mu) в рентгеновском диапазоне, поэтому, как и для рентгеновских лучей, для них можно наблюдать дифракцию на кристаллической решетке. В другом методе используют развертку по скоростям, когда нейтроны проходят известное расстояние во времяпролетном детекторе. Конструкция этих приборов описана в работе [9].
Селективное поглощение нейтронов различной энергии служит аналитическим средством, но, к сожалению, для его применения требуются очень мощные потоки, которые может обеспечить лишь большой атомный реактор. Менее мощные источники пригодны для обнаружения лишь немногих элементов, ядра которых особенно сильно поглощают нейтроны (имеют высокое сечение захвата): В, Cd, Li, Hg, Ir, In, Au, Ag и некоторых лантаноидов.
Применение радиоактивных источников в анализе. Поглощение а- и 0-частиц можно использовать в аналитических целях. В силу невысокой проникающей способности а-частицы более подходят для анализа в газовой фазе. Описана методика [10], по которой в ионизационную камеру помещают как источник а-частиц (210Ро — «радий-D», 210РЬ), так и образец. При постоянном давлении и потенциале протекающий через камеру ток зависит только от состава газа. В благоприятных случаях так можно анализировать двойные смеси газов (калибровка по смесям известного состава) с точностью ±0,2—0,3 мол. %. Эту методику применяли на практике для определения некоторых токсичных газов, например Ni(CO)4 и РЬ(С2Н5)4 в воздухе в концентрации 1 • 10~7 %. Поглощение 0-частиц, связанное в основном с электрон-электронными взаимодействиями, использовали главным образом для анализа жидких образцов. Элементарные рассуждения показывают, что водород имеет максимальное число электронов на единицу массы, у любого другого элемента эта «плотность» ниже минимум вдвое. Следовательно, данный метод отличается высокой чувствительностью к водороду. Это послужило основой для создания прибора, предназначенного для определения соотношения Н/С в нефтепродуктах [11, 12].
Использование источников у-излучения вместо обычных рентгеновских трубок обсуждалось в гл. И. В гл. 20 рассматривалось применение трития и иных источников 0-излучения в газохроматографических детекторах электронного захвата.
17*
516 Глава 24
Радиоактивные метки
Радиоактивные изотопы легко обнаружить и измерить с высокой точностью даже малые их количества. Поэтому активные изотопы используют во многих способах анализа. Наиболее распространенными являются активационный анализ и изотопное разбавление.
Активационный анализ
Многие элементы приобретают радиоактивность носле облучения потоком нейтронов, протонов, дейтронов или а-частиц. Особенно широко применяют нейтронную активацию; на ней мы остановимся более подробно.
Рассмотрим бомбардировку образца «тепловыми» нейтронами, кинетическая энергия которых меньше 0,2 эВ. Обычно ядро (мишень) захватывает нейтрон, и образуется новое ядро. Его масса на единицу больше, а заряд прежний, т. е. образуется изотоп исходного элемента. Часто новое ядро нестабильно и самопроизвольно распадается с испусканием частицы или у-кванта (или того и другого), иными словами, ядро радиоактивно. Полученные таким путем активные изотопы сильно различаются по периоду полураспада, и благодаря этой и некоторой другой информации, например гамма-спектрам, можно идентифицировать элементы.
При проведении нейтронно-активационного анализа образец помещают в ядерный реактор и облучают его потоком нейтронов, в результате чего все способные к возбуждению элементы пробы становятся радиоактивными. Так называемая кривая распада (потеря активности) отражает зависимость наведенной активности от времени. Эта кривая имеет интегральный характер, т. е. отражает активность всех радиоактивных элементов пробы. Индивидуальные же элементы можно идентифицировать с помощью ЭВМ, разлагая сложный контур на составляющие. Период полураспада самого долгоживущего элемента можно определить по последнему участку кривой, в формировании которого менее стабильные компоненты, вероятно, не участвуют. Активность указанного изотопа можно вычесть из общей кривой («оголить») на участке, отвечающем меньшим временам. Затем так же можно определить следующий долгоживущий элемент, вычесть его вклад и т. д.
Так, в одном из первых сообщений об этом методе был описан анализ алюминиевого сплава [13] (эксперимент выполняли без ЭВМ). Небольшой образец (массой около 25 мг) на 5 мин помещали в ядерный реактор. Затем регистрировали ак-
Ядерно-физические методы 517
Рис. 24-9. Нейтронио-актнвационный анализ алюминиевого сплава [13].
тивность образца с помощью стандартного счетчика в течение 60 ч (рис. 24-9). В природе алюминий существует только в виде изотопа 27А1. После захвата нейтрона образуется 28А1, который подвергается р-распаду с образованием устойчивого 28Si. Поскольку образец в основном состоял из алюминия, излучение 28А1 было очень мощным. Но поскольку период полураспада 28А1 всего 2,3 мин, через полчаса большинство элемента исчезало.
Из рисунка ясно, что после примерно 40-часовой выдержки активность (логарифмическая шкала) образца линейно зависит от времени. Экстраполирование этой прямой линии влево дает для любого момента времени активность, обусловленную наличием элемента с периодом полураспада около 15 ч (значение определяется из графика). По таблице изотопов этот элемент идентифицирован как 24Na с периодом полураспада 15,0 ч. На графике также имеется прямолинейный участок (от 1 до 18 ч), обусловленный присутствием изотопа с периодом полураспада примерно 2,5 ч —56Мп (2,58 ч).
Желаемые результаты можно получить, снимая не всю кривую распада, а регистрируя гамма-спектр образца в определенные моменты времени после облучения. В отчете Агентства по охране окружающей среды .США [14] описано одновременное определение 26 элементов в городском воздухе после концентрирования на фильтрах из стекловолокна. Каждый образец облучали в течение 4 ч, выдерживали 10 дней и измеряли активность с помощью литий-германиевого детектора и 2048-канального анализатора высоты \ импульсов. Полученная экспе-
518 Глава 24
Рис. 24-10. Спектр у-излучення атмосферных частиц (городской воздух), снятый после активации нейтронами.
риментальная зависимость (после обработки на ЭВМ) показана на рис. 24-10.
Чувствительность нейтронно-активационного анализа зависит от мощности потока нейтронов, способности элемента к захвату нейтрона (сечение нейтронного захвата) и периода полураспада образовавшегося радиоактивного элемента. Выполняется соотношение
А = Моср 1 -
— 0,6931 \~|
Т1/2 Л1
(24-4)
где А — наведенная активность к концу периода облучения (имп./с); N — число присутствующих в образце атомов определяемого элемента; а — сечение нейтронного захвата нейтронов (см2); <р — мощность потока [число нейтронов/(см2-с)]; t— время облучения; Ti/2 — период полураспада продукта (Ti/2 и t должны быть выражены в одних единицах). Количественный анализ редко основан на этом уравнении (хотя именно так делали авторы работы [13]), поскольку не хватает надежных данных. Кроме того, поток может быть негомогенным, а <р меняться со временем. На практике одновременно с анализируемыми облучают стандартные образцы и для количественного определения используют сравнение.
Если облучение проводят в большом атомном реакторе, можно обнаружить до 10-10 г элемента. При облучении менее мощными источниками нейтронов активационный анализ применим только к элементам с особыми свойствами ядер. Так,
Ядерно-физические методы 519
источник нейтронов, содержащий 25 мг Ra и 250 мг Be [нейтроны образуются в реакции эВе (а, п) 12С], дает поток мощностью около 100 нейтронов/(см2 • с). С помощью его можно активировать только Rh, Ag, In, 1г и Dy, однако для этих элементов метод удобен и надежен. Источник, состоящий из 124Sb и Be, с мощностью потока 103—104 пригоден для активации 19 элементов. Атомный же реактор дает поток 1014, который в состоянии активировать почти все элементы тяжелее кислорода, но с разной чувствительностью. Для таких элементов, как In, Re, Ir, Sm, Eu, Dy, Ho, Lu, V, As и Sb, чувствительность активационного анализа выше, чем химических методов; для других элементов (Fe, Са, Pb, Bi, Zn, Cd, Na; К) это не так [15].
Активация другими частицами. Активировать образец можно и бомбардировкой заряженными частицами — протонами 'Н+, дейтронами 2Н+, тритонами 3Н+, ядрами гелия 3Не2+ и 4Не2+ [16]. Требуемую энергию (несколько десятков мегаэлектронвольт) дают линейные ускорители, генераторы Ван-де-Граафа или циклотроны. При этом протекает такая же ядерная реакция, как и при облучении нейтронами: ядро-мишень захватывает частицу, образуя ядро с большими массой и зарядом. Новое ядро часто нестабильно и распадается, испуская частицы и излучение, или и то и другое вместе. Этот метод обладает весьма высокой чувствительностью, особенно при обнаружении легких элементов (В, С, N, О), для которых предел обнаружения составляет порядка 1 • 10~7 % [16—18].
Возможна и активация электромагнитным излучением: 9Ве активировали у-излучением 124Sb, а для определения дейтерия в воде использовали облучение излучением 24Na. Эти источники дают практически наиболее мощное излучение (с достаточно большим периодом полураспада), однако и они могут активировать только 9Ве и 2Н соответственно. Более широкое применение должно найти тормозное излучение, генерируемое при поглощении высокоэнергетических электронов мишенью с большой атомной массой. Чувствительность этого метода для некоторых элементов (F, Fe, Pb) выше, чем нейтронно-активационного анализа. Привлекательно и то, что в ряде случаев устраняются матричные эффекты.
Изотопное разбавление
Метод изотопного разбавления применим в том случае, если определяемое вещество можно выделить в чистом виде, но с низким выходом. К пробе добавляют известное количество того же вещества, содержащего активный изотоп, и тщательно
520 Глава 24
перемешивают. Затем из смеси выделяют часть чистого вещества (содержащую и неактивную, и активную формы) и определяют ее активность. Простой расчет позволяет найти концентрацию вещества.
Рассмотрим раствор, содержащий W граммов определяемого вещества, к которому добавляют порцию того же вещества, меченного радиоактивным элементом, весом w граммов с активностью А (имп./мин) и удельной активностью S0=A/w. После смешивания из раствора выделяют g граммов чистого вещества. Пусть измеренная активность чистого вещества В (имп./мин), а удельная активность S = B/g. Общая активность после смешивания не изменяется (распадом пренебрегаем), т. е.
tjySo==(^'+oa)S (24-5)
откуда
W = g—w (24-6)
Если добавка имеет высокую активность, ее вводят в малом количестве, и w по сравнению с W можно пренебречь. Тогда уравнение (24-6) упрощается:
W=g^ (24-7)
В качестве примера рассмотрим определение глицина в смеси аминокислот. Поскольку глицин можно выделить
из смеси, но с низким выходом, целесообразно использовать метод изотопного разбавления. Для начала следует приготовить образец глицина, содержащий примерно один атом В * 10 * * * 14С на миллион молекул, т. е. с удельной активностью (с поправкой на фон) 25 000 имп./мин на 1 г. Смешивают 0,500 г активного вещества с анализируемой пробой, после чего выделяют 0,200 г чистого глицина с удельной активностью 1250 импульсов за
10 мин при фоне 100 импульсов за 5 мин. Подытожим условия:
да = 0,500 г
So = 25 000 имп./мин на 1 г
А = wS0 = 12 500 имп./мин g= 0,200 г
В = 1250/10—100/5= 105 имп./мин
Ядерно-физические методы 521
Отсюда получаем искомое количество глицина в пробе:
UZ = g ——ш = 0,200 12500 —0,500 = 23,2 г В 105
Если пренебречь величиной w, то погрешность составит примерно 2 %, тогда как погрешность счета импульсов составляет прмерно 3,5 %
Еще один пример взят из работы [20], где описывается электрогравиметрическое определение Со в стали. Поскольку Со, выделяемый на аноде в виде Со2О3, образует слабо прилипающий слой, это препятствует обычному гравиметрическому определению. При изотопном же разбавлении потеря некоторых частиц не имеет решающего значения. Для построения градуировочного графика к образцам, содержащим известные количества Со, добавляют одинаковые аликвоты 60Со и проводят электролиз. К анализируемому образцу аликвоту 60Со добавляют сразу же после растворения навески стали. Кобальт выделяют, осадок взвешивают и измеряют активность. Содержание кобальта находят с помощью градуировочного графика. Относительное стандартное отклонение составляет 0,005—0,025 %.
Использование градуировочного графика в таких случаях позволяет устранить некоторые погрешности, так же как при проведении холостого опыта в ряде аналитических методик. Впрочем, в некоторых случаях для количественного определения лучше использовать непосредственно уравнение (24-6).
Мёссбауэровская спектроскопия [1, 21, 22]
Так называют спектроскопию резонансной флуоресценции у-излучения, которая напоминает оптическую резонансную флуоресценцию с той разницей, что вызвана она переходами между ядерными, а не атомными энергетическими уровнями. В соответствующих условиях эксперимента флуоресцентное у-излучение характеризуется предельной резкостью линий. Так, например, резонансное у-излучение 67Zn имеет полуширину всего лишь 4,8- 10~и эВ, что составляет примерно 2-10~13 % энергии у-кванта, равной 93 кэВ. Для сравнения отметим, что для рентгеновского излучения К-линии Zn ее полуширина составляет 4,7-10 8 эВ при энергии фотона 8,6 кэВ (соотношение 2:10й), т. е. линия в 10000 раз шире, чем для у-излучения.
Наиболее часто с помощью мёссбауэровской спектроскопии изучают соединения железа в силу благоприятного расположения ядерных энергетических уровней в изотопе 57Fe. Этот изотоп имеет метастабильный уровнь, лежащий на 14,4 кэВ выше основного, и переход между этими уровнями дает
522 Глава 24
у-излучение, которое легко поглощается ядрами S7Fe, находящимися в основном состоянии. Возбужденные ядра 57Fe * (звездочка справа от символа элемента обозначает возбужденное состояние) получаются в результате распада 57Со (период полураспада 267 сут) по механизму электронного захвата. 57Fe * мгновенно теряет избыток энергии, испуская у-квант (14,4кэВ). Этот квант может резонансно поглощаться невозбужденным ядром 57Fe. Общая последовательность событий такова:
Бвре , 57QO ЭЗ,267 сут , 57ре* — Y , 57ре + 7^, 57ре* а б в г
Процесс а (захват дейтрона и потеря нейтрона) идет в циклотроне при образовании 57Со. Атомы 57Со диффундируют в поверхностный слой металлической фольги, которую применяют как «источник» в мёссбауэровском спектрометре. Отметим, что, так как время существования S7Fe* значительно меньше чем 267 сут, фактически при каждом распаде ядер 57Со испускается у-квант. Поглотителем (образцом) может быть обычное железо в любом химическом состоянии, поскольку распространенность S7Fe в природе составляет около 2,2%, что достаточно для получения приемлемой чувствительности.
Поскольку мёссбауэровские линии очень узки, малейшие изменения энергетического состояния ядра-поглотителя сдвигают частоту резонансного поглощения, причем в такой степени, что энергия облучающего потока перестает соответствовать условию резонанса. Примерно такой порядок величины имеет химический сдвиг, вызываемый влиянием химического окружения атома на ядерные уровни. Для наблюдения и измерения удобно перемещать источник или поглотитель таким образом, чтобы доплеровское смещение точно компенсировало химический сдвиг. Требуемая скорость составляет несколько миллиметров в секунду и легко реализуется на практике.
На рис. 24-11, а приведена блок-схема мёссбауэровского спектрометра. В принципе можно перемещать как источник, так и образец, но на практике образец часто приходится охлаждать для «вымораживания» колебаний решетки, и поэтому удобнее перемещать источник. Для этого используют двигатель, управляемый программируемым генератором сигналов так, чтобы ускорение (сначала в одном направлении, а затем в другом) было постоянным (рис. 24-11,6). В этом случае за один цикл покрывается весь диапазон скоростей-, смещение изменяется в зависимости от времени по квазипараболической кривой.
Сигнал от детектора поступает в одноканальный анализатор (чтобы ограничить отклик резонансом только у-излуче-ния), а оттуда — в многоканальный анализатор, синхронизи-
Ядерно-физические методы 523
anmdwtf чшхдохо эпнэЬюнэ
Рис. 24-11. Мёссбауэровский спектрометр: а — блок-схема; б — последовательность изменения параметров.
524 Глава 24
Рис. 24-12. Мёссбауэровские спектры шести образцов луииой породы и пыли, на которых видно различное соотиошеиие FeTiOa и пироксена (железосодержащего силикатного минерала). Цифры обозначают отдельные образцы [23].
рованный с генератором управляющих двигателем команд. Это обеспечивает соответствие между каналом и узким интервалом скоростей. На встроенном осциллографе отображается зависимость отклика от скорости перемещения источника.
На рис. 24-12 представлены мёссбауэровские спектры нескольких образцов лунных пород [23]. Путем сравнения со спектрами известных пород идентифицированы два минерала железа. Для соединений или сплавов, содержащих элементы, к которым метод чувствителен, мёссбауэровские спектры могут давать информацию о валентном состоянии и кристаллической структуре, но таких немного. Наибольшее количество исследований выполнено с 57Fe, 61Ni и 119Sn. Существует около
Ядерио-физические методы 525
30 элементов, для которых этот эффект наблюдался, и еше примерно 20 элементов, для которых он возможен, однако со многими из них трудно работать, в частности, из-за малой распространенности ответственного за сигнал изотопа. Тем не менее значение в химии Fe, Ni и Sn обусловливает включение мёссбауэровской спектроскопии в список инструментальных методов анализа.
Меры предосторожности
Применяемые в работе с радиоактивными индикаторами небольшие количества активных материалов обычно не представляют такой радиационной опасности, от которой трудно защититься. Изотопы, являющиеся источниками а- и 0-излучения, можно хранить в обычных стеклянных или металлических емкостях, однако при работе с ними следует пользоваться щипцами и надевать резиновые или пластиковые перчатки. Для гамма-излучателей нужна более надежная защита, например слой свинца в несколько сантиметров (толщина слоя зависит от энергии излучения данного изотопа). Набирать жидкость в пипетку, засасывая ртом, вообще нежелательно; при работе с активными растворами это категорически запрещается.
В лаборатории следует иметь дозиметр для контроля за уборкой случайно «разлитой» активности. Вообще «разлитая» активность часто опасна не столько для человека, сколько загрязняет лабораторию (резко возрастает фон). Большинство применяемых в эксперименте радиоактивных индикаторов не опаснее нитрата серебра. Требования техники безопасности не являются очень строгими, за исключением некоторых случаев.
Конечно, при работе с большими, чем следовые, количествами радиоактивных веществ необходимы более серьезные меры предосторожности; с ними можно ознакомиться в литературе [1].
Отметим, что для хранения и работы с радионуклидами требуется разрешение соответствующих органов.
Задачи
24-1. О «совпадениях» говорят как о положительном явлении при обсуждении сцинтилляционных счетчиков и как о нежелательном — в статистике. Объясните, почему этот парадокс кажущийся.
24-2. Ионизационная камера, заполненная воздухом под атмосферным давлением, обеспечивает пробег 8 см fl-частицам, испускаемым источником умеренной мощности (1000 распадов в минуту). Геометрическая эффективность 50 %. Известно, что 0-излучеиие образует 10 пар ионов на 1 мм пробега в воздухе. Рассчитайте силу протекающего тока (заряд электрона равен 1,6-10-1» Кл)
526 Глава 24
24-3. Методика одновременного определения U и Th в минералах [23] включает 1) измерение суммарной радиоактивности U и Th и 2) определение отношения U/Th с помощью рентгеновской эмиссионной спектроскопии. Суммарная активность выражается в урановых эквивалентах, т. е. как количество U в урановой смолке, дающего ту же активность, что и образец. Отношение Th/U определяют по отношению высот соответствующих рентгеновских пиков Thia и ULa. Содержание U и Th определяют из соотношения х + 0,2 ху=урановый эквивалент (в %). Здесь х— массовый процент U, а у — отношение Th/U по массе. Для используемого счетного устройства 1 % уранового эквивалента соответствует счету 2100 имп./мин (фон вычтен). В применении к образцу монацитового песка массой 1,000 г счет, согласно стандартной методике, составил 2780 имп./мин (фон вычтен). Относительная высота пиков — 72,3 для ThLa и 1,58 для Ш.а. Рассчитайте содержание (в % по массе) U и Th в образце.
24-4. Методом изотопного разбавления определяют содержание пенициллина в смеси. К образцу добавляют 10,00 мг радиоактивного пенициллина (активность 4500 имп./мин иа 1 мг). Из смеси выделяют 0,35 мг чистого кристаллического пенициллина с удельной активностью 390 ими./мин на 1 мг (фон вычтен). Каково содержание пенициллина (в граммах) в пробе?
24-5. Согласно методу обратного изотопного разбавления добавляют w грамм неактивного вещества к пробе, содержащей W грамм определяемого активного вещества (удельная активность So). Вещество выделяют в чистом виде, взвешивают и измеряют активность. Покажите математически, что уравнение (24-6) применимо и в этом случае.
24-6. Описана [25] методика определения продуктов окисления пропана с помощью обратного изотопного разбавления. Окисляемый образец был обогащен пропаном, меченным 14С. Среди продуктов найдено значительное количество активного 2-пропанола. К смеси продуктов добавляли отмеренное количество неактивного 2-пропаиола и каким-либо подходящим методом извлекали часть спирта. Получены следующие данные (мкКи— микрокюри, единица измерения активности):
Количество активной о про- 10 ммоль
пана Удельная активность образ- 72,8 мкКи/ммоль
ца Добавка неактивного пропа- 16 ммоль
нола Удельная активность пропа- 5,8 мкКи/моль
нола
Рассчитайте процент превращения пропана в пропанол.
24-7. При изучении растворимости малорастворимых солей для определения оксалата в концентрациях порядка 10~4 % использовали осаждение 45СаС2О4. Рассчитайте содержание оксалата в пробе (в частях на миллион), исходя из следующих данных. Стандартный раствор содержал 0,680 М 45СаС12 с удельной активностью 45Са 20 000 имп./мнн (фон вычтен). К 100 мл анализируемого на оксалат образца добавляли 5,00 мл стандартного раствора. Осадок не выпадал, но появлялась слабая' муть. Добавляли несколько капель FeCl3 и подщелачивали NH3, чтобы осадить Fe(OH)3. Осадок собирали на бумажном фильтре (с отсасыванием), промывали, сушили и измеряли активность. Из предыдущих экспериментов было известно, что эффективность счетного устройства 30 %. Для достижения заданного уровня в 6000 импульсов потребовалось 18,60 мин. Фон составлял 150 имп. за 5 мин. (Учтите, что активность стандартного раствора дана без поправки на эффективность).
Ядерно-физические методы 527
24-8. С помощью счетчика Гейгера измеряли p-излучение. Допустимая погрешность составляла ±1 %. Для образца и фона каждые 5 мин выполнялись такие отсчеты:
Время, мин 0 5 10 15 20 25 30
Фон, имп./мин 0 127 249 377 502 672 793
Образец, имп./мин 0 2155 4297 6451 8602 10 749 12 907
а) Сколько потребуется времени, чтобы погрешность могла снизиться до допустимой? б) Сколько времени следует измерять фон? в) Какова в действительности величина счета (в имп./мин) и ее разброс?
24-9. С помощью нейтронно-активационного анализа определяли следы Ag и Zn в сплавах. По справочникам были найдены такие данные (сюда включены также Cd, In, Си и Au, так как они могут присутствовать в сплаве):
Продукт
Е стествен-
Реакция3 ная распространенность» % Период Радиоактив-
полураспада ность
64Zn (n, y)65Zn 48,9 244 сут ЭЗ
66Zn (n, y)67Zn 27,8 Стабилен
67Zn (/г, y)68Zn 4,1 »
68Zn (n, y)69Zn 18,6 58 мин 1
68Zn (n, y)69mZn 18,6 13,9 ч 1
70Zn (n, y)71Zn 0,62 2,4 мин 1
107Ag (n, y)108Ag 51,8 2,42 мин 93, Р
109Ag (n, y)110Ag 48,2 24,4 с 8
109Ag (n, y)110mAg 48,2 253 сут 8
110Cd (n, p)110mAg 12,4 253 сут В
114Cd (n, a)115Cd 28,9 53,5 ч 3
113In (n, -v)11on>Ag 4,28 253 сут р
113 In (ti, y)116In 95,7 14 с 5
197Au (ti, y)198 Au 100,0 2,69 сут 5
63Cu (n, y)64Cu 69,1 12,9 ч ЭЗ, р
65Cu (ti, y)66Cu 30,9 5,10 мин р
а 68 Zn превращается на 99 % в 69 Zn и на 1 % в метастабильный 69mZn, 109 Ag — на 96 % в 110Ag и на 4% в 110mAg.
После облучения нейтронами образец на 30 сут помещали в автоматическое счетное устройство, которое измеряет общую активность через каждые 2 ч. После этого образец растворяли, добавляли в качестве носителей неактивные перхлораты Ag и Zn. Раствор подвергали электролизу, чтобы выделить серебро и цинк, и определяли их активность, а) Какие изотопы фактически используются для определения Ag и Zn? б) Отметим, что и 110Cd, и 1131п после активации нейтронами могут дать llemAg, что служит потенциальным источником ошибок. Объясните, как можно с помощью кривой распада 115Cd скорректировать данные на присутствие 110Cd. Как устранить мешающее влияние 1131п? в) Что означает т в символах llemAg и 69mZn? г) Как вы считаете, могут ли мешать определению ядерные реакции Си и Au типа (п, р) и (п, а)? Объясните почему, д) 66Zn и 67Zn способны к захвату нейтронов, однако не становятся радиоактивными. Объясните это.
528 Глава 24
Литература
1. Friedlander G., Kennedy J. W., Macias E. S„ Miller J. M., Nuclear and Radiochemistry (3d ed.), Wiley-Interscience, New York, 1981.
2. Hahn R. L., in Guide to Activation Analysis, Lyon W. S. (ed.), Van Nostrand, Princeton, 1964, p. 63.
3. Horrocks D. L., Applications of Liquid Scientillation Counting, Academic Press, New York, 1974.
4. Ouseph P. J., Introduction to Nuclear Radiation Detectors, Plenum Press, New York, 1975.
5. См. [1, ch. 9].
6. Cm. [3. ch. 18].
7. Ross W. A., Multichannel Analyzers. In Instrumentation in Applied Nuclear Chemistry, Krugers J. (ed.), Plenum Press, New York, 1973.
8. Covell D. F., Anal. Chem., 1959, v. 31, p. 1785.
9. Leddicote G. W., Neutron Absorption and Scattering Techniques of Analysis. In Treatise on Analytical Chemistry, Kolthoff I. M., Elving P. J., Sandell E. B. (eds.), pt. 1, vol. 6, chap. 69, Wiley-Interscience, New York, 1965.
10. Deisler P. F„ McHenry K. W., Wilhelm R. H., Anal. Chem., 1955, v. 27, p. 1366.
11. Jacobs R. B., Lewis L. G., Piehl F. J., Anal. Chem., 1956, v. 28, p. 324.
12. Smith V. H., Otvos I. W., Anal. Chem., 1954, v. 26, p. 359.
13. Boyd G. E., Anal. Chem., 1949, v. 21, p. 335.
14. Lambert J. P. F., Wilshire F. W., Anal. Chem., 1979, v. 51, p. 1346.
15. Guinn V. P., In Techniques of Chemistry, Vol. I, Physical Methods of Chemistry, A. Weissberger A., Rossiter B. W. (eds), pt. III-D, Chap. 7, Wiley-Interscience, New York, 1972.
16. Schweikert E. A., Anal. Chem., 1980, v. 52, p. 827A.
17. Sastri C. S., Petri H., Erdtmann G., Anal. Chem., 1977, v. 49, p. 1510.
18. Boulton R. B., Ewan G. T., Anal. Chem., 1977, v. 49, p. 1297.
19. Lutz G. J., Anal. Chem., 1971, v. 43, p. 93.
20. Salyer D., Sweet T. R., Anal. Chem., 1956, v. 28, p. 61; 1957, v. 29, p. 2.
21. Herber R. H., Hazony Y., In Techniques of Chemistry, vol. I, Physical Methods of Chemistry, Weissberger A., Rossiter В. M. (eds.), pt. III-D, chap. 4, Wiley-Interscience, New York, 1972.
22. Stevens J. G., Shemoy G. K-, Mossbauer Spectroscopy and its Chemical Application, Advances in Chemistry Series, no. 194, American Chemical Society, Washington, 1981.
23. Muir A. H., Housley R. M., Grant R. W., Abdegawad M., Blander M., Am. Lab., 1970, v. 2(11), p. 8.
24. Camplell W. J., Carl H. F„ Anal. Chem., 1955, v. 27, p. 1884.
25. Clingman W. H., Hammen H. H., Anal. Chem., 1960, v. 32, p. 323.
ГЛАВА 25
Автоматические анализаторы
Автоматизация и механизация
Многие из приборов, описанных в предыдущих главах этой книги, в высокой степени механизированы: они имеют встроенные электрические и механические устройства, позволяющие оператору не заботиться о многих деталях эксперимента. Механизация позволяет гораздо более полно использовать возможности аналитического прибора, чем при ручном управлении, особенно если аналитический метод требует непрерывного измерения сигнала при сканировании некоторой переменной.
Возьмем, к примеру, ИК-спектрофотометр. Даже если в нем не предусмотрены сервисные электромеханические устройства и вся работа выполняется вручную, можно получить равноценные спектры. Чтобы установить с помощью призмы или дифракционной решетки необходимую длину волны, подобрать ширину щели, измерить пропускание образца и эталона и записать результаты в журнал, опытному оператору нужно не менее 5—10 мин. А чтобы провести измерения в диапазоне 2—20 мкм, описанные действия придется повторить сотни раз. Затем, возможно, понадобится пересчитать данные в каждой точке и вычертить спектр от руки. В результате на получение одного-единственного спектра высокого разрешения может уйти неделя. Ясно, что механизация такого прибора позволяет не только сэкономить время, но и избежать многих ошибок, вызванных невнимательностью.
Конечно, у автоматического регистрирующего спектрофотометра есть свои собственные потенциальные источники ошибок. Несовершенство и сбои механических устройств могут вызвать как систематическую, так и случайную погрешности, которые сложно выявить и устранить. Правда, инструментальные ошибки такого рода обычно удается снизить путем тщательного проектирования и создания оптимальной конструкции прибора; остающуюся вероятность ошибки можно понизить, используя калибровку. Существенно, что через определенные короткие интервалы времени калибровку следует повторять, чтобы обеспечить сравнимость результатов. Такая процедура
530 Глава 25.
в значительной степени компенсирует изменения параметров прибора, обусловленные механическим износом движущихся частей и медленным дрейфом характеристик электронных схем.
Термин автоматизация означает обычно нечто большее, чем просто автоматическое управление работой прибора. Он относится к системе, часто включающей наряду с основным прибором (например, спектрофотометром) другие компоненты и позволяющей последовательно обрабатывать множество образцов и записывать соответствующие результаты. Автоматический анализ особенно важен, если существует необходимость в проведении большой серии однотипных определений. Толчок к развитию в этом направлении дали прежде всего нужды клинического анализа, но автоматизированные приборы широко используются и в лабораториях по контролю качества промышленной продукции (химической, фармацевтической, металлургической и др.).
Вообще говоря, можно сконструировать машину, повторяющую любые действия человека. Такие механические операции, как растворение, отбор раствора, разбавление, машина способна проделывать лучше человека [1, 2]. Менее очевидно, что это относится и к действиям, связанным с принятием решений. Опытный химик или лаборант, наблюдая за ходом реакции, может определить, когда она прошла полностью, и соответственно решить, что пора перейти к следующему этапу. Но и машину можно запрограммировать так, чтобы она проводила измерения и на основе их результатов делала логические выводы. И здесь машина имеет то преимущество, что на правильность ее решений не влияет утомление или однообразие работы.
Однако машина плохо справляется с обработкой неожиданных ситуаций, когда нарушается обычная последовательность событий. Хороший автоматический анализатор должен по крайней мере уметь распознавать неожиданные события и сообщать об этом оператору.
Еще одно требование к автоматической аналитической системе заключается в способности хранить данные. Между последовательностью образцов, вводимых в анализатор, и последовательностью получаемых ответов не должно возникать расхождения, и сохранить соответствие в многостадийном анализе, где одновременно обрабатывается ряд проб, бывает непросто.
Большинство автоматизированных аналитических систем построено на основе одного и того же принципа. Такие операции, как пробоотбор и пробоподготовка, добавление реагентов и т. п., часто реализуются с помощью отдельных модулей. Химизм процесса обычно заимствуют из известных методик не
Автоматические анализаторы 531
автоматизированного анализа, часто делая небольшие изменения, чтобы все операции на данном приборе проходили единообразно (например, в смысле постоянства времени и температуры, необходимых для развития окраски).
Автоматические анализаторы делятся на два класса — проточные и статические. У обоих есть свои преимущества и области применения. В остальной части этой главы будет приведено несколько характерных примеров систем обоих типов, где основное внимание будет уделено химизму и механике процессов. О необходимой электронике (в особенности микропроцессорном управлении) речь пойдет в последующих главах книги.
Проточные системы
Представьте поток раствора, содержащего фотометрический реагент, движущийся по пластмассовым трубкам к спектрофотометру с проточной кюветой. Допустим, что прежде чем поток реагента достигнет места фотометрирования, в него с помощью медицинского шприца вспрыскивается небольшая порция раствора анализируемой пробы (рис. 25-1). При этом образуется окрашенный участок или зона образца, которая движется по трубкам к фотометру. Когда зона проходит через кювету, детектор реагирует на нее, причем интегральный сигнал прямо связан с количеством вещества, определяемого в пробе.
Для того чтобы такая система давала хорошие результаты, необходимо соблюдение нескольких условий: постоянства степени протекания реакции и целостность введенной пробы. Один из приемов выполнения этих условий заключается в разделении потока на сегменты с помощью пузырьков воздуха [3, 4]. Пузырьки создают завихрения в пределах каждого сегмента, чем достигается полнота перемешивания в малом объеме, а также способствуют очистке трубки между отдельными пробами. Недостаток такого подхода состоит в необходимости специального устройства для удаления пузырьков перед входом в детектор.
Другой прием состоит в использовании узких (~0,5 мм) трубок без сегментации воздухом, в которых поток является ламинарным, а перемешивание пробы и реагента обеспечивается в основном диффузией [5, 6], и поэтому воспроизводимо, хотя и менее полно. Этот метод, получивший название проточно-инжекционного анализа (ПИА), более современен, чем метод с сегментацией воздухом, и, пожалуй, предпочтительнее метода сегментации воздухом, так как характеризуется большей скоростью анализа и меньшей стоимостью (хотя, видимо,
532 Глава 25
Рис. 25-1. Примерная схема установки (а) и сигнал детектора (б), иллюстрирующие принципы проточного анализа.
его труднее приспособить к многоэтапным методикам анализа). Оба метода имеют примерно одинаковую воспроизводимость (1—2%) и пригодны для различных определений с фотометрическим или электрохимическим детектированием.
Типичным примером применения проточного анализа служит определение кальция в сыворотке крови (сегментация воздухом). Соответствующая аппаратура показана на рис. 25-2, а диаграмма потоков — на рис. 25-3. На последнем рисунке справа пронумерованными горизонтальными линиями показаны девять каналов стандартного 14-канального перистальтического* насоса; рядом указан внутренний диаметр (в дюймах) соответствующей пластиковой трубки. При работе системы разбавленная НС1 закачивается через канал 4 и смешивается в Т-образном переходнике с воздухом (канал 2). Расходы подобраны так, что поток кислоты разрывается пузырьками воздуха примерно через интервалы в 1 см. Образец сыворотки или плазмы прокачивается через канал 6 и присоединяется к потоку НО. На следующем этапе проводится диализ. Подкисленный образец проходит через длинную спираль с целлофановой диаф-
Автоматические анализаторы 533
Рис. 25-2. Общий вид проточного анализатора Technicon Autoanalyzer. Модули справа налево: карусельный держатель проб, перистальтический насос (в пластиковом корпусе), термостатируемая баня со спиральными смесителями, фотометрический детектор (под самописцем), управляющая микроЭВМ (Technicon Instruments Corp.).
рагмой. По другую сторону диафрагмы движется сегментированный поток НС1 (каналы 8 и 10). Ионные компоненты пробы (Са2+ и Mg2+) диффундируют во второй поток, а белок и другие крупные органические молекулы не проходят перегородку и сбрасываются в сток. В этот момент в систему вводится раствор реагентов крезолфталеина и 8-оксихинолина (для устранения мешающего влияния Mg2+) и сразу же вслед за этим 0,5 М раствор диэтиламина. Проходя спиральный смеситель,, проба хорошо перемешивается с реагентами и движется к фотометрическому детектору. Здесь из жидкости удаляются пузырьки воздуха, и поток, проходя кювету, попадает в сток (канал 14). По каналу 13 закачивается вода на промывку пробоотборника. Описанная схема позволяет проводить до 60 определений в час. Перед вводом каждого образца система промывается для устранения следов предыдущего. Периодически для градуировки вводятся стандартные растворы. Результаты регистрируются самописцем.
Периодические процессы
Системы автоматического титрования (фирмы Mettler Instrument Corporation). Фирма Mettler Instrument Corp, производит автоматизированные аналитические системы, основанные-на титровании. Для них подходит любая реакция титрования^
534 Глава 25
с
к S к <и
<и
Автоматические анализаторы 535
конечную точку которой можно зафиксировать потенциометрически (ионоселективные электроды, pH-метры и редоксимет-рия). В продаже имеются взаимозаменяемые автоматические бюретки различной емкости. Гибкие средства контроля позволяют остановить титрование в конечной точке или записать всю кривую. Данные можно выводить на дисплей или печатающее устройство в цифровом или аналоговом виде или передавать в компьютер.
В- системе расширенной конфигурации электронные весы взвешивают образцы непосредственно в стаканчики (до 44), которые поочередно транспортируются с помощью пневмопривода к головке титрования. Результаты вычисляются автоматически и выводятся на бумажную ленту.
Автоматические анализаторы (фирмы American Monitor Corp.). Эта фирма производит автоматические установки Рго-grammachem для спектрофотометрического (главным образом для клинического) анализа. Спектрометр снабжен дифракционной решеткой, диапазон длин волн 325—800 нм. В прибор можно одновременно загрузить до 89 образцов, скорость анализа 350 проб/ч. Когда очередной жидкий образец перемещается в рабочее положение, в него погружается автоматический пробоотборник, отбирает точную аликвоту и переносит ее в реакционную трубку. Здесь проба разбавляется и к ней добавляется фотометрический реагент. Анализ завершает измерение поглощения при выбранной длине волны. Результаты вычисляются и печатаются на бумажной ленте.
Растворы реагентов для 25 различных методик содержатся в специальном блоке и отбираются при необходимости согласно нанесенным на перфокарту инструкциям. Как и многие другие автоматические анализаторы, этот анализатор сконструирован для выполнения большого числа серийных анализов, но при необходимости может быстро перестраиваться. Например, оператор может выполнить серию определений глюкозы в крови и переключиться на определение холестерина, просто вставив другую программную перфокарту и запустив цикл очистки системы от предыдущих реагентов.
Автоанализаторы для клинического анализа (фирмы Е. I. Du Pont de Nemours & Co.). Автоматические клинические анализаторы этой фирмы довольно необычны. Наборы реагентов для однократных определений изготавливаются производителем и помещаются в специальные пластиковые упаковки, служащие одновременно реакционной камерой и кюветой для спектрофотометра. В некоторых типах анализов упаковки содержат индивидуальные хроматографические колонки для выделения нужных компонентов или молекулярно-массовых фракций.
536 Глава 25
Рис. 25-4. Пластиковая аналитическая упаковка из комплекта анализатора АСА фирмы Du Pont с семью кармашками для реагентов. Цифровой код предназначен для считывания прибором и содержит информацию о необходимом типе анализа. Образец вводят через трубку слева (на рисунке закрыта пробкой) (Е. I. Du Pont de Nemours & Co.).
Для анализа каждого образца используется своя упаковка. На каждой из них нанесены наименование теста (для удобства оператора) и двоичный код — инструкция для анализатора. Для того чтобы задать программу установке, нужная упаковка (или упаковки) вставляется во входной карман под каждый контейнер для образца. Анализатор автоматически впрыскивает порцию пробы последовательно в каждую упаковку, смешивает реагенты, выдерживает установленное время, формирует прецизионную оптическую ячейку внутри упаковки, стенки которой прозрачны, и фотометрически определяет продукт реакции. Все действия контролируются и управляются встроенной ЭВМ, которая также вычисляет концентрации и печатает результаты для каждого образца. В напечатанное для каждого образца сообщение входят результаты всех тестов и информация о пациенте.
На рис. 25-4 показана одна из упаковок. Во внутренних запечатанных кармашках содержится до семи реагентов; кармашки автоматически вскрываются при работе. После впрыскивания образца и последующего разбавления упаковка прикрепляется к движущейся цепи и перемещается по аппарату, показанному на рис. 25-5, от пункта к пункту. После нагре-
Автоматические анализаторы 537
Рис. 25-5. Анализатор АСА фирмы Du Pont (в разрезе) (Е. I. Du Pont de Nemours & Co). 1— приемный лоток для образцов; 2— приемный карман; 3 — челночный привод; 4 — пункт опознания н приема пробы; 5 — пункт заполнения; 6 — нагреватель 1; 7—нагреватель 2; 8 — смеситель 1; 9 —остановочный пункт 1; 10 — остановочный пункт 2; 11 — термостатируемая камера; 12 — остановочный пункт 4; 13 — остановочный пункт 3; 14 — остановочный пункт 5; 15 — транспортная цепь; 16 — смеситель 2; 17 — фотометр; 18— выбрасыватель использованных упаковок; 19 — двигатель транспортной цепи; 20 — печатающее устройство; 21 — карман для бумаги.
вания до 37 °C она попадает в специальные тиски, где вскрываются кармашки с первыми четырьмя из семи реагентов и их содержимое смешивается с пробой. Затем упаковка задерживается в пяти остановочных пунктах, чтобы прошло необходимое для развития окраски время (около 15 мин), затем вскрываются кармашки с тремя оставшимися реагентами. Далее упаковка движется к фотометру, где зажимается между кварцевыми пластинками так, чтобы образовалась кювета толщиной 1 см. Фотометр с интерференционными фильтрами способен работать при длине волны от 340 до 600 нм. Использованная упаковка автоматически выбрасывается.
538 Глава 25
Рис. 25-6. Принцип работы центрифужного анализатора GeMSAEC. а — реагент и образец загружены в полости; б — то же, идет перемещение; в — то же, идет перемещение в положение для проведения оптических измерений, показанное на рис. г; г — вертикальная стрелка показывает путь света [8].
Центрифужный анализатор (фирмы GeMSAEC*). Эта аналитическая система основана на специально сконструированной центрифуге [7]. Все необходимые добавки реагентов, разбавление и даже спектрофотометрические измерения делаются при работающей центрифуге. На рис. 25-6, а [8, 9] видны полости в пластиковом роторе, в которые загружены образец и реагент. Из рис. 25-6,6 ясен результат раскручивания'ротора — центробежная сила перемешивает жидкости, и они стекают в третью полость. Далее смесь перемещается в такое положение, что через нее проходит вертикальный пучок света (25-6, в). При другой форме ротора реагент можно добавлять во все положения во время вращения центрифуги.
Детектор с фотоумножителем регистрирует непрерывную серию импульсов (каждый соответствует одному положению образца в роторе), разделенных периодами темноты. Стандарты и холостые растворы помещены в ротор в 17 или большем числе позиций. ЭВМ исправляет результаты на темновой
* Аббревиатура основана на названиях учреждений-разработчиков: Institute of General Medical Science н U. S. Atomic Energy Comission.
Автоматические анализаторы 539
Рис. 25-7. Лабораторный робот: «рука» н «кисть», манипулирующая образцами под управлением ЭВМ (Zymark Corporation).
ток, вычисляет концентрации и усредняет результаты по многим оборотам центрифуги.
Этот анализатор сейчас изготавливается серийно и находит широкое применение.
Лабораторные роботы (фирмы Zymark Corp). Эта фирма занимается робототехникой — усовершенствованием лабораторных работ при помощи механических устройств для манипуляции образцами под управлением ЭВМ. Такой «робот» показан на рис. 25-7. У него есть «рука» и прикрепленная к ней «кисть», которая может перемещаться вертикально, горизонтально и по кругу. «Пальцы» способны, например, извлечь пробирку из штатива, подставить ее под пипетку (спереди и слева на рисунке) для добавления реагента, встряхнуть и перенести в фотометр (спереди справа), а затем поместить пробирку в штатив для грязной посуды. При этом робот не делает ошибок и не устает. Информация от спектрофотометра и идентификационный код образца закладываются в память ЭВМ для дальнейшего вывода на печать.
Роботизация создает новый вид технологии, который, несомненно, в будущем станет доминирующим.
540 Глава 25
Литература
1. Burns D. A., Anal. Chem.. 1981, v. 53, p. 14O3A.
2. Owens G. D., Eckstein R. J., Anal. Chem., 1982, v. 54, p. 2347.
3. Skeggs L. T., Anal. Chem., 1966, v. 38(6). 31A (см. также публикации Technicon Instrument Corporation).
4. Salpeter J., LaPerch F., Am. Lab., 1981, v. 13, No. 9, p. 78.
5. Betteridge D., Anal. Chem., 1978, v. 50, p. 832A.
6. Ruzicka J., Hansen E. H., Flow Injection Analysis, Wiley-Interscience, New York, 1981.
7. Anderson N. G., Science, 1969, v. 166, p. 317.
8. Scott C. D., Burtis C. A., Anal. Chem., 1973, v. 45, p. 327A.
9. Tiffany T. O., Burtis C. A., Mailen J. C., Thacker L. H., Anal. Chem., 1973, v. 45, p. 1716.
ГЛАВА
Общие вопросы анализа
Итак, обзор наиболее популярных аналитических методов, чаще всего используемых химиками, закончен. Теперь следует обратить внимание на проблему выбора наиболее подходящего метода для решения задачи, которая может возникнуть перед химиком.
Предположим, химику-аналитику нужно разработать методику количественного определения вещества X. Ниже приведен список вопросов, которые следует задать, прежде чем взяться за решение задачи.
1. Зачем нужен этот анализ? Представительна ли проба? Если нет, возможно, нет смысла анализировать ее.
2. Что представляет собой матрица или основа вещества, в котором находится определяемый компонент?
3. Какие примеси могут присутствовать и примерно в каких количествах?
4. Каков ожидаемый интервал концентраций X?
5. Какие требования предъявляются к правильности и воспроизводимости?
6. Какие стандартные образцы можно использовать?
7. Где будет выполняться анализ: в лаборатории, на предприятии или в полевых условиях?
8. Сколько проб в день предполагается анализировать?
9. Имеет ли значение скорость получения ответа, и если да, то как быстро он должен быть дан?
10. Сколько цифр следует принимать во внимание, если операция проводится автоматически, и насколько можно уменьшить их число, чтобы снизить стоимость оборудования.
11. В каком виде желательно представить ответ (письменный отчет, запись на ленте самописца и т. д.)?
12. Какие существуют особые или необычные возможности, которые могут повлиять на выбор метода (например, наличие атомного реактора) ?
542 Глава 26
В ряде случаев нужно идти на компромисс. Например, высокая точность может оказаться несовместимой со скоростью определения. Наконец, при прочих равных условиях личные предпочтения могут оказаться решающими. Так, фотометрический и полярографический методы обеспечивают практически одинаковую точность для растворов близкой концентрации (хотя по селективности они могут различаться), причем время выполнения определения, а также стоимость приборов близки. Аналитик в этом случае волен выбрать метод, с которым он более знаком.
Чувствительность и предел обнаружения
Чувствительность S аналитического метода или прибора можно определить как отношение изменения сигнала R к изменению измеряемого количества вещества или его концентрации С:
S = dR/dC или bR/bC (26-1)
где AR и АС представляют собой малые, но конечные разности. Чувствительность, вероятно, должна зависеть от экспериментальных условий. Наряду с чувствительностью часто используют параметр, называемый пределом обнаружения, и определяемый как наименьшая концентрация, при которой исчезает аналитический сигнал (при С-*О). В большинстве случаев этот сигнал и, следовательно, концентрация должны иметь значительно большую величину при однозначной идентификации и еще большую величину при количественном определении.
Обобщить данные по пределам обнаружения для различных методов трудно, поскольку они сильно отличаются от одного элемента или типа соединений к другому. К’арасек [1] приводит типичные данные для нескольких методов (табл. 26-1). Моррисон [2] проводит очень важное сопоставление имеющихся
Таблица 26-1. Пределы обнаружения и идентификации для некоторых аналитических методов [1|
Метод Предел обнаружения Предел идентификации
Газовая хроматография 10-6—10—12
Спектрофотометрия в ИК-области ю-7 10-6
Спектрофотометрия в УФ-области 10-’ 10-6
ЯМР (с накоплением) 10-’ 10-6
Масс-спектрометрия (с баллоном напуска) 10-6 10—5
Масс-спектрометрия (с прямым вводом) ю-12 ю—11
ГХ—МС ю-11 ю-1»
Общие вопросы анализа 543
в литературе к тому времени данных определения всех элементов несколькими аналитическими методами. Такое же сравнение нескольких спектроскопических методов представлено на рис. 9-9.
Такими сопоставлениями можно руководствоваться при выборе подходящего метода, но точные значения пределов обнаружения могут меняться в широких пределах в зависимости от условий эксперимента и матрицы. Часто пределы обнаружения зависят от предварительных операций.
Максимальный размах концентраций, определяемых с известной надежностью, составляет область определяемых концентраций (динамический интервал). Ее нижняя граница определяется случайными флуктуациями (шумом) в сигнале прибора. Для не очень точного измерения отношение сигнал/шум (S/N) должно быть равно по крайней мере двум, но для точных измерений оно должно быть больше. Верхняя граница определяемых концентраций связана с явлением «насыщения», например с растворимостью анализируемого соединения или возможностями детектора. Область определяемых концентраций для некоторых методов составляет 4—5 порядков измеряемой величины, для некоторых же ограничена одним порядком.
Воспроизводимость и правильность
Воспроизводимость, определяемая как разброс повторных определений, характеризуется стандартным отклонением. Чем меньше его значение, тем лучше воспроизводимость. Воспроизводимость тесно связана с правильностью, т. е. с близостью между полученным результатом и известным «истинным» значением. Результат с высокой воспроизводимостью может оказаться неправильным при наличии источника систематической ошибки, которая одинаково влияет на все параллельные измерения.
Воспроизводимость можно повысить, повторив анализ и проведя соответствующую статистическую обработку данных. В любом методе титрования получают результаты серии измерений, по которым строят кривую титрования для определения положения точки эквивалентности. Проведение плавной кривой через точки улучшает общую воспроизводимость в такой же степени, как и получение того же числа повторных отсчетов без регистрации всей кривой титрования. (Следует, конечно, помнить, что полученные данные могут не совпадать, так как могут относиться к разным равновесным состояниям.)
На воспроизводимость, получаемую при определении разными методами, часто влияют форма кривой, а также возможности детектора прибора. На рис. 26-1 показаны две кривые
544 Глава 26
' титрование Карбонат и избыток бикарбоната
12 3 4 5 6 7 8
0.4
0.3
0.2
О.Т
Объем 1 мнС1,мл
Рис. 26-1. Кривые потенциометрического и фотометрического титрования 3,4 • 10-2 М раствора NajCOs, содержащего 1 М NaHCO3 [3].
титрования, соответствующие одной и той же реакции: титрования карбоната в присутствии большого избытка бикарбоната [3]. Точное определение точки эквивалентности по кривой потенциометрического титрования невозможно, тогда как на кривой фотометрического титрования (при длине волны 235 нм, при которой карбонат поглощает, а бикарбонат нет) видна отчетливая точка эквивалентности, получающаяся при экстраполяции двух прямолинейных участков.
Сравнение со стандартными образцами
Большинство инструментальных аналитических методов основано на сравнении какого-либо физического свойства анализируемого вещества и стандарта или серии стандартов, содержащих то же самое вещество в известных количествах. Такое сравнение можно осуществить с помощью градуировочного графика, который представляет собой зависимость величины, выражающей это физическое свойство, от концентрации определяемого компонента (или некоторой простой функции концентрации, например логарифма или обратной величины). Иногда наклон кривой можно определить теоретически (из закона Бэра, уравнения Ильковича и т. п.), и тогда может быть удобнее рассчитывать концентрацию по уравнению, чем строить градуировочный график. С таким случаем мы сталкиваемся, например, при определении катионов путем измерения потенциала полуэлемента с помощью уравнения Нернста. Это урав нение действительно описывает кривую, которую можно построить и использовать для сравнения анализируемого раствора и стандартного раствора, по которому предварительно оценивается Е° (см. рис. 14-1).
Другим общим приемом является сравнение анализируемого вещества с двумя стандартными образцами, один с несколько меньшим, а другой с несколько большим содержанием определяемого компонента, чем в анализируемом веществе. Преиму
Общие вопросы анализа 545
щество такого приема состоит в том, что и таком небольшом интервале сигнал можно считать линейным. Этот принцип лежит в основе конструкции оптических компараторов, в которых проводится визуальное сравнение окраски.
При всех сравнениях в высшей степени желательно, чтобы стандартные образцы и анализируемые вещества были как можно ближе друг к другу по составу. Это существенно снижает систематические ошибки, которые оказывают одинаковое влияние на все растворы. В некоторых случаях точность можно значительно увеличить, если использовать растянутую шкалу прибора для измерения разности между двумя близкими величинами, а не измерять расстояние по шкале от нуля до каждого значения. Об этом упоминается в связи с фотометрическим анализом в гл. 3, но в принципе этот прием можно использовать шире.
На нем основан тип приборов, в которых сравнение анализируемого и стандартного образцов проводится непосредственно в одной операции. Примерами служат потенциометрическая ячейка, детекторы по теплопроводности в ГХ и фотометры и спектрофотометры, в которых используется уравнивание двух световых потоков, проходящих через две пробы. Следует всегда помнить, что сравнение со стандартами нс может улучшить воспроизводимость анализа; оно повышает его правильность.
Приготовление и хранение очень разбавленных растворов (от микро- до наномолярных) бывает весьма затруднительно. Стенки сосудов, используемых для хранения, могут адсорбировать растворенные вещества, снижая тем самым концентрацию значительно ниже предполагаемого уровня; иногда этого можно избежать, сполоснув сосуд раствором, который будет в нем храниться.
Существенную помощь при стандартизации оказывает обширный набор стандартных образцов, выпускаемых Национальным бюро стандартов США. Каждый образец снабжается паспортом, где указаны концентрации всех компонентов, от основных до таких, концентрации которых составляют лишь несколько тысячных процента. При помощи этих образцов можно проверить правильность множества определений.
Стандартные добавки
Нежелательное влияние основы можно уменьшить или даже устранить полностью с помощью метода стандартных добавок, который заключается в добавлении стандарта непосредственно к пробе. Рассмотрим сначала случай, когда после введения поправки на фон сигнал прибора R прямо пропорционален кон-18 Заказ № 537
546
Глава 26
центрации. Используя подстрочный индекс 0 для исходного раствора, запишем
= (26-2)
Vo
где концентрация представлена в виде отношения числа молей М к объему V. Добавим теперь порцию стандартного раствора, содержащую ЛЬ молей в объеме V). Запишем уравнение (26-2) для получившегося раствора
7?1 = £[(М0 + М1)/(1/0 + О (26-3)
Это выражение упрощается, если Vi будет намного меньше Ио, что легко сделать при использовании достаточно концентрированных стандартных растворов. Тогда
Ri = Аг/Ко (ЛГо 4-Afx) (26-4)
Исключив k/Vo в уравнении (26-4) с помощью уравнения (26-2) и решив его относительно Мо, получим
= R0/(#i~ О (26-5)
Этот прием исключает влияние большинства инструментальных переменных, представленных константой k. Зная объем, можно затем определить М\ и, следовательно, концентрацию.
Можно показать [5], что наибольшая точность достигается, когда добавка немного больше, чем количество определяемого вещества. Если наблюдаются сильные помехи, то требуется более сложная математическая обработка [6]. Важно, чтобы из-
Добадка стандарта
Рис. 26-2. Графическое определение Л10 методом стандартных добавок. График может служить для проверки линейности сигнала при концентрациях, превышающих концентрацию определяемого компонента в пробе.
Общие вопросы анализа 547
Ri-k
(26-6)
мерения с добавкой и без нее были выполнены в присутствии одинаковых количеств всех остальных компонентов. Однако было бы ошибкой считать, что при этом полностью устраняется влияние основы, поэтому следует повторить операцию, вводя дополнительные добавки стандартного раствора, так чтобы получить результаты R2 и /?з- Можно построить график (рис. 26-2) и по нему найти Л40.
В случае экспоненциальной зависимости, когда R=R'+ + Aln (M/V), аналогичная обработка, приводящая к увеличению М, дает
М0 = М1
Применительно к потенциометрии k является множителем RT/nF (где R — газовая постоянная). Аналогичные выражения можно получить для других функциональных зависимостей сигнала от концентрации.
Иногда используется прием, называемый вычитанием стандарта, заключающийся в том, что добавляют известное количество реагента, который эффективно удаляет из пробы соответствующее количество определяемого компонента (в результате осаждения или комплексообразования). Математические выкладки здесь такие же, как и в случае метода стандартных добавок, но с соответствующим изменением знака.
Еще раз обращаем внимание на то, что при любом сравпе нии со стандартом условия должны быть идентичными. Иногда это сделать трудно или даже невозможно, но тем не менее для получения высокой точности необходимо. Например, при использовании метода стандартных добавок ионная сила и концентрация комплексующих реагентов не должны меняться.
Графики
Точность получения данных по графику в большой степени зависит от способа его построения. Целесообразно рассмотреть ряд функций вида /? = /;(С), выражающих соотношение сигнала прибора и концентрационного члена, которые описывают поведение некоторой аналитической системы. На рис. 26-3 построены кривые четырех таких функций: 1) линейной, R = k\C\ 2) квадратной или более высоких степеней, R — k^^RC2 + k2C-, 3) обратной, R = k?i!C\ 4) логарифмической, R = k^C. Построены также производные dRfdC этих функций. Из рисунка видно, что кривые обратных и логарифмических функций круто поднимаются вверх, следовательно, наибольшая чувствительность (по определению, данному выше) будет достигаться при низких концентрациях, а для кривой квадратной функции мак-18*
548 Глава 26
Рис. 26-3. Различные функции концентраций и их производные; кривые отвечают следующим уравнениям:
1) R = krC (l-D) dR/dC k.! ky — 10
2) R = k'2C2 + k2C (2-D) dRIdC = 2k'2C k2 H2 = k2 1
3) R = k3 (1/C) (3-D) dR/dC-- -fe3(l/C2)* k:l - 100
4) R = ki 1g C (4-D) <)R!dC -- 0,434fe4 (1/C) == 100
* Для удобства построения отрицательный знак не принят во внимание.
снмальная чувствительность приходится на высокие концентрации. Как и следовало ожидать, чувствительность для линейной кривой постоянна во всем интервале.
Форма кривых на рис. 26-3, построенных с использованием частных производных, говорит о том, что на чувствительность, вероятно, могут влиять и другие факторы. Например, в спектрофотометрии чувствительность может изменяться при изменении длины волны. Кривая сигнала сохраняет форму, если только ось абсцисс будет сжата или растянута.
Часто целесообразно строить графики в логарифмических координатах (на логарифмической бумаге). Такой прием полезен в двух случаях: 1) если нужно поместить на одной шкале
Общие вопросы анализа 549
величины разных порядков и 2) при определении констант k и п в соотношениях типа y = kxn. С первым из этих случаев мы встречаемся, например, при построении спектра поглощения в координатах 1g Л —длина волны (см. рис. 3-4); теоретического обоснования построения такого графика нет, так как, откладывая A (A — lg(l/T)), а не Т, мы уже используем логарифмическую шкалу.
Применять логарифмические координаты для определения констант следует лишь тогда, когда экспериментальная система, действительно, описывается соотношением y = kxn. Если же откладывать экспериментальные данные в таких координатах в надежде установить неизвестное математическое соотношение, то легко сделать неоправданные корреляции. Возможные «ловушки» и пути их избежания прекрасно описаны в работе [7].
Задачи
26-1. Назовите инструментальные методы, с помощью которых можно получить графики, построенные на рис. 26-3, и приведите уравнения, подтверждающие ваш выбор.
26-2. Закон Бера можно представить в виде соотношения 2?=Й5-10<:. Постройте график этой функции и ее производной в координатах, принятых на рис. 26-3. (Значение й5 выберите так, чтобы 2?=10 при с=4 ед.) Прокомментируйте результаты.
26-3. Как лучше всего количественно определить воду в каждом из следующих случаев (везде, где можно, кратко опишите методику): а) водяные пары в баллоне для сжатых Н2 или Ог; б) примесь воды в «чистом» хлороформе или эфире; в) содержание воды над растворителем в герметически закрытом сосуде; г) воду, собирающуюся на дне большого резервуара для хранения бензин#?
26-4. Предложите инструментальный метод определения тетраэтилсвинца и тетраметилсвинца в бензине при их совместном присутствии.
26-5. Одна из наиболее трудных задач — разделение Zr и Hf. В результате лантанидного сжатия атомы Zr и Hf имеют практически один и тот же размер, и это в сочетании с близким электронным строением делает их химически почти идентичными. Предложите по крайней мере два метода, с помощью которых вы могли бы разделить их. Как вы думаете оценить, успешно ли разделение, проведенное каждым из методов? Какие аналитические методы можно было бы использовать для анализа смеси соединений этих элементов без разделения?
26-6. Марганец п хром в стали можно определить окислением до Mn(VII) п Cr(VI) с последующим фотометрическим определением. Однако, если не принять соответствующих мер предосторожности, окраска Fe(III) может помешать определению. Основную массу железа можно удалить экстракцией эфиром в присутствии НС1 или ионным обменом с последующим окислением Мп и Сг, можно связать железо в комплекс при помощи цитрата, тартрата или другого реагента, чтобы разрушить его окраску, можно оставить железо в растворе и внести поправку па -его светопоглощеиие, проделав измерения до и после окисления Мп и Сг; можно использовать сочетание этих методов. Проведите критическое сравнение этих примеров. Почему нельзя использовать для разделения электролиз на ртутном катоде?
550 Глава 26
26-7. Ранее упоминалось об использовании кристаллов Nal,«активированных» примесью ТП в качестве люминофора в сцинтилляционном счетчике. Предложите недеструктивный метод определения содержания ТП в кристаллах Nal. Оцените ожидаемую точность.
Литература
1. Karasck Г. W., Anal. Chem., 1972, v. 44, No. 4, p. 32A.
2. Morrison G. H., Skogerboe R. K., in Trace Analysis: Physical Methods, Morrison G. H. (ed.), Wiley-Interscience, New York, 1965.
3. Underwood A. L„ Howe L. H., Anal. Chem., 1962, v. 34, p. 692.
4. Vassos В. H., Ewing G. W., Electroanalytical Chemistry, Wiley-Intersci-ence, New York, 1983, pp. 207ff.
5. Ratzlaff K. L„ Anal. Chem., 1979, v. 51, p. 232.
6. Saxberg В. E. H., Kowalski B. R., Anal. Chem., 1979, v. 51, p. 1031.
7. Rowe P. N., Chem. Tech., 1974, v. 4, p. 9.
ГЛАВА 27
Электронные схемы в аналитических приборах
Большинство инструментальных методов, описанных в этой книге, немыслимы без приборов, включающих различные электрические схемы. За исключением простейших случаев, во всех схемах используется электроника. Чтобы лучше понять устройство этих приборов, их возможности и ограничения, необходимы знания основ электроники. Эту и следующую главу следует рассматривать лишь как краткий обзор. Мы не пытались выводить фундаментальные математические уравнения: более подробное обсуждение можно найти в работе [1] и других учебниках. Тем не менее приводимой информации достаточно, чтобы понять суть большинства проблем, связанных с устройством приборов, о которых упоминалось в предыдущих главах.
«Сердцем» любого электронного устройства служит один или несколько элементов, которые воздействуют непосредственно на электроны. К таким элементам относятся разнообразные полупроводниковые устройства, в том числе диоды, транзисторы и фотоэлементы. Эти активные элементы никогда не функционируют сами по себе; для их работы всегда требуются вспомогательные схемы, составленные из большого числа сопротивлений, конденсаторов и индуктивностей. Необходимы также источники питания, которыми служат батареи или выпрямители переменного тока. Кроме того, в схему часто включают и другие элементы из соображений удобств или безопасности— выключатели, предохранители и контрольные лампы.
В настоящее время электронные схемы все в большей степени базируются па интегральных микросхемах (ИМС). Каждая ИМС заменяет большое число индивидуальных транзисторов и диодов, которые связаны в единое целое и расположены на небольшом кристалле («подложке») кремния. ИМС смонтированы в одном корпусе и имеют до 40 соединительных выводов.
Использование ИМС имеет ряд преимуществ в сравнении с ранее применявшимися отдельными транзисторами и дру-
562 Глава 27
гимн элементами. Пользователю не нужно самому конструировать такие сложные устройства, как логические элементы или операционные усилители; они разрабатываются для него изготовителем. Дополнительное достоинство заключается в том, что индивидуальные ИМС недороги и их' можно использовать для замены или создания новых приборов.
Полупроводники
Полупроводником называют твердое вещество, промежуточное по свойствам между металлическими проводниками, с одной стороны, и непроводниками (изоляторами) — с другой. Ои характеризуется относительно высоким отрицательным температурным коэффициентом сопротивления, в то время как для металлов этот коэффициент положителен, что позволяет легко различать два типа проводников. В подавляющем большинстве случаев в качестве полупроводникового материала используется элементарный кремний. Германий используется вместо кремния только в особых случаях.
В чистом кристалле кремния (или германия) каждый атом ковалентно связан с каждым из четырех других атомов (структура алмаза), а так как каждый атом имеет четыре валентных электрона, все валентности при такой структуре насыщены. Чтобы использовать кристалл в электронных устройствах, его следует легировать, т. е. ввести следовые количества примесей. Атомы примеси должны иметь такую природу, чтобы они могли заместить некоторые из атомов полупроводника в кристаллической решетке.
Если примесь представляет собой пятивалентный элемент, например мышьяк или сурьму, каждый атом примеси обладает избыточным валентным электроном, помимо электронов, необходимых для образования ковалентных связей решетки. Избыточный электрон легко отрывается от своего «родительского» атома под действием тепловой энергии, а затем беспорядочно движется по всей решетке. Атом примеси превращается в однозарядный положительный ион, внедренный в решетку.
С другой стороны, если примесь представляет собой трехвалентный галлий, индий или золото, то возникает недостаток одного электрона на атом примеси. Образовавшееся вакантное место называется дыркой. Тепловые колебания электрона нормальной ковалентной связи могут случайно сблизить его с дыркой, так что он полностью уйдет с предыдущего места и рекомбинирует с ней. В результате такого процесса дырка перемещается вдоль решетки с одного места на другое. В легированном таким способом кристалле кремния дырки блуждают по всему кристаллу так же, как и избыточные электроны блуждают в кристалле, легированном мышьяком или сурьмой. Под
Электронные схемы в аналитических приборах 553
вижность дырок, однако, несколько меньше подвижности электронов.
Кремний, в котором примесь служит, донором электронов, называется кремнием n-типа, а легированный электронодефицитными атомами — кремнием р-типа*. Электрическое поле, приложенное к кристаллу легированного кремния, вызывает протекание тока, обусловленного почти исключительно избыточными электронами или дырками, образованными примесью. Таким образом, основными носите
Рнс. 27-1. Схематическое изображение р/г-перехода.
лями в кремнии «-типа служат электроны, а в кремнии р-типа — дырки.
Диоды
Кристаллический диод — это полупроводниковый прибор с двумя выводами, который обладает способностью пропускать ток в одном направлении и не пропускать в обратном. Его изготавливают из кремния или германия частично р- и частично n-типа, как схематически показано на рис. 27-1. Когда источник напряжения соединен отрицательным полюсом с «-областью и положительным с p-областью, основные нреители в обеих зонах движутся к рп-переходу. В области перехода электроны из n-зоны рекомбинируют с дырками p-зоны, и таким образом осуществляется перенос тока. При обратном включении, когда n-зона положительна, а p-зона отрицательна, дырки и электроны уходят от перехода, оставляя промежуточную область без носителей, так что ток протекать не может (исключая короткий переходный период, когда напряжение только что приложено).
Вольтамперные характеристики для кремниевого и германиевого диодов приведены на рис. 27-2. Для положительного, т. с. прямого, потенциала ток экспоненциально зависит от напряжения. Если приложен отрицательный (обратный) потенциал, то ток, называемый током утечки, почти равен пулю и
* Следует отметить, что кремний или германий, легированные даже относительно глубоко, с химической точки зрения все еще остаются высокочи-стымп веществами. Обычно достаточно контролируемой добавки. порядка 1-10-4 %, чтобы придать кристаллу необходимые электрические свойства.
554 Глава 27
Рис. 27-2. а — вольтампсрпые характеристики германиевых и кремниевых диодов; б—обозначение простого диода; в—обозначение диода Зенера (стабилитрона). Шкала токов для нижней части (а) увеличена относительно верхней части.
не изменяется до тех пор, пока (для кремния) не будет достигнута критическая величина Ez, при которой обратный ток возрастает до величины, определяемой только сопротивлением внешней цепи. Эта критическая точка, называемая напряжением Зенера или напряжением пробоя, соответствует напряжению, необходимому для отрыва электронов ковалентных связей в кристалле с созданием положительных и отрицательных носителей, которые движутся к соответствующим полюсам диода. Можно изготовить кремниевые диоды с любым напряжением пробоя в диапазоне от 2 до 200 В. Германиевые диоды имеют более низкое обратное сопротивление, чем кремниевые, и не имеют четко выраженного потенциала пробоя.
Диоды находят применение во многих областях. Диоды большого размера используются как мощные выпрямители для превращения переменного тока в постоянный, от которого питаются другие приборы. Диоды меньших размеров применяются для выпрямления импульсных сигналов; в этом случае они носят название детекторов или демодуляторов. Диоды, рассчитанные на резкое изменение потенциала пробоя (диоды Зенера), широко используются для создания соответствующего напряжения.
Электронные схемы в аналитических приборах 555
Транзисторы
Транзистор представляет собой основной усилительный полупроводниковый прибор. Существует несколько модификаций транзисторов. Наиболее важные для наших целей можно разбить на два класса: 1) биполярные и 2) полевые транзисторы.
Биполярный транзистор (рис. 27-3) состоит из кремниевой пластинки, содержащей две зоны
Рис. 27-3. Схематическое представление дяд-транзистора; /7-область получается сплавлением металла, содержащего примеси p-типа, с пластинкой
p-типа, разделенных тонким слоем кремния «-типа.
материала n-типа; при этом образу-
ются два перехода, каждый из которых обладает свойствами диодов, описанными выше. При работе транзистора переход
между одной p-зоной, называемой эммиттером, и n-слоем, называемым базой, имеет небольшое прямое смещение, тогда как переход база—коллектор (коллектор — это вторая р-зона) имеет обратное смещение. Ток может протекать через переход эмиттер — база. Область базы, будучи тонкой и слаболегированной, бедна электронами. Поэтому большинство дырок с эмиттера диффундируют через нее, так как они имеют большую вероятность быть «собранными» большим коллекторным переходом, чем рекомбинировать с электронами базы. Отношение коллекторного тока к току базы /кДб определяется исключительно геометрией системы. Величины этих токов, однако, определяются напряжением между базой и эмиттером, так как увеличение положительного потенциала базы облегчает проникновение дырок в коллектор. Это и есть источник усилительной
способности транзистора.
Описанный выше транзистор является транзистором рпр-типа, но таким же образом можно изготовить прибор с противоположными характеристиками — лрп-транзистор. Оба типа транзисторов могут использоваться в одних и тех же схемах, только полярности всех напряжений и токов следует поменять на обратные. На рис. 27-4 приведены стандартные обозначения этих двух типов. Стрелка на эмиттере указывает направ-
ление протекания положительного тока для перехода эмиттер — база.
Фактор усиления, обозначаемый |3, есть отношение небольшого изменения коллекторного тока /к к вызывающему его изменению тока базы /Б:
р —d/K/d/5
(27-1)
556 Глава 27
Рис. -27-4. Обозначения для рпр- и црп-транзисторов.
Рнс. 27-5. Усилитель на одном транзисторе. Номиналы всех элементов даны только для иллюстрации. Для рнр-транзистора необходимо изменить знак Уист.
В транзисторах, применяемых для усиления слабых сигналов, значения р обычно лежат между 50 и 200, но мощные транзисторы имеют более низкие значения |3 (от 5 до 10).
Простая схема, демонстрирующая усилитель на одном транзисторе, приведена на рис. 27-5. Цепь питается от батареи или выпрямителя с напряжением УИст относительно земли. Сопротивления R\, Rs и Rs, называемые сопротивлениями смещения, обеспечивают соответствующий постоянный потенциал между базой и эмиттером. При этом потенциале в отсутствие сигнала транзистор пропускает небольшой ток, примерно в середине своего диапазона. Предположим, что переменный сигнал с ам
Электронные схемы в аналитических приборах 557
плитудой в 1 мВ от источника с сопротивлением 7?с= 1 кОм поступает на базу транзистора через связывающий конденсатор Сс. Это дает переменный компонент тока базы, равный отношению Ec/Rc, а именно 10~3 В/10+3 Ом = 10“6 А, или 1 мкА. Умножив на 0 (100), получим, что результирующий переменный компонент коллекторного тока равен 100 мкА. Этот ток, протекающий через нагрузочное сопротивление Rh, дает переменное падение потенциала (100 • 10-6 А) (15 • 103 Ом) = 1,50 В. Итак, 1 мВ на входе дает 1,5 В на выходе. Усиление этой цепи (отношение выхода к входу) можно рассчитать для напряжения, тока или мощности. Получаем следующие значения:
Усиление напряжения = GV = 1,50/(10-3)= 1500
Усиление тока = G; = 0 = 100
Усиление мощности = GP — Gv • G; = 150 000*
Полевой транзистор (ПТ)
Этот прибор работает на совершенно иной основе, чем биполярный транзистор, рассмотренный выше. Обсудим схему на рис. 27-6. Полоска кремния п-типа, называемая каналом, соединена с двух сторон с истоком S и стоком D. Канал находится между слоями р-материала (соединенными вместе), называемого затвором G. В некоторых моделях затвор полностью окружает канал. Существуют ПТ с каналами п- и р-типа.
Если к затвору не приложено напряжение, то ток протекает через канал без помех, причем электроны движутся от истока к стоку. В обычном режиме рп-переход затвор — канал имеет обратное смещение. Это дает эффект обеднения электронами области между затвором и штриховыми линиями. При этом канал сужается, и его сопротивление увеличивается, а ток в направлении исток — сток уменьшается. Усиление напряжения достигается при размещении нагрузочного, сопротивления последовательно со стоком. Изменение потенциала затвора путем изменения тока через нагрузочное сопротивление усиливает изменение напряжения на выходе. Обратное смещение предотвращает также протекание заметного тока в цепи затвора. К затвору может быть приложено напряжение порядка нескольких вольт. Так как ток в этой цепи протекать не мо-
* Мощность — это произведение потенциала на ток, поэтому усиление мощности определяется по формуле
Gp = £вах.-/вых = Gy. Gi
£вх ’ ^вх
558 Глава 27
Рис. 27-6. Схематическое представление ПТ с каналом п-типа.
Рис. 27-7. Стандартные обозначения ПТ: а — ПТ с каналом п-типа; б — ПТ с каналом р-типа; в — МДП-траизистор с каналом Д-типа; г — МДП-транзистор с каналом p-типа; В — обозначает присоединение к основе кремниевого слоя.
пов транзисторов. На рнс. 2' ния для различных типов ПТ.
жет, характеристики затвора даются в вольтах, а не в единицах тока, как для биполярных транзисторов.
Принципиальным преимуществом ПТ является высокий входной импеданс, возникающий благодаря наличию обратного смещения. Этот импеданс может достигать величины десятков или даже сотен мегаом.
ПТ с изолированным затвором, часто называемые МДП-транзисторами (ПТ со структурой металл — диэлектрик — полупроводник), представляют собой модификацию ПТ, в которой тонкая пленка изолирующего материала, обычно диоксида кремния, отделяет затвор от канала. Это устраняет выпрямляющий переход, так что затвору можно придать любую полярность без управляющего тока. Электростатическое поле между затвором и каналом может менять распределение дырок или электронов в канале, определяя таким образом его сопротивление. МДП-транзистор имеет самый высокий входной импеданс среди всех ти-7 даны общепринятые обозначе-
Источники питания
Большинство современных лабораторных приборов питается от сети переменного тока (115 В, 60 Гц в США)*. Постоянный ток получают выпрямлением на кремниевых диодах. Существует несколько типов соединения выпрямительных диодов, каждый из которых имеет свои достоинства в конкретных случаях. Наиболее часто используется соединение четырех диодов в мостиковой схеме (рис. 27-8). Схема работает следующим образом. В каждый полупериод переменного тока два диода прово-
* В СССР 220 В, 50 Гц.— Прим, перев.
Электронные схемы в аналитических приборах 559
Рис. 27-8. Регулируемый 15-В источник питания. Выпрямительный мост помещается в том же корпусе. Для других напряжений замените обозначение в серии регуляторов 7800.
дят ток, в то время как другие два заперты, так что на выходе диодного мостика появляется пульсирующее постоянное напряжение. В следующий полупериод диоды меняются функциями, давая на выходе ту же полярность. Конденсатор большой емкости (Ci) действует в качестве фильтра для сглаживания пульсаций. Элемент, обозначенный 7815,— это регулятор напряжения, который превращает постоянный ток выпрямителя (20—25 В) в стабилизированное напряжение 15 В. Выходной конденсатор (Сг) необязателен, но он увеличивает скорость, с которой регулятор корректирует изменяющееся напряжение. Использование других регуляторов позволяет получать другие напряжения (им соответствуют две последние цифры в маркировке устройства).
Для иопны.х счетчиков и фотоумножителей требуются высокие напряжения при малом токе (несколько киловольт при токе менее чем 1 мА). Эти требования могут быть удовлетворены при использовании генератора (который обсуждается ниже), дающего переменный ток с частотой в несколько сотен килогерц. Напряжение на выходе можно очень сильно увеличить применением трансформатора с воздушным сердечником и выпрямителя. Требования к фильтрам при такой частоте легко удовлетворяются. Сейчас созданы генераторы на двух мощных транзисторах с трансформатором и выпрямителем в одном корпусе.
Источники постоянного тока требуются в кулонометрии и
15-203
Рис. 27-9. Удобный регулятор тока. Ток равен /=12//?.
560 Глава 27
для возбуждения некоторых источников света. Такой источник можно легко собрать, присоединив регулятор, как показано на рис. 27-9. Даваемый им ток /вых = (12//?) +/о, где /0—постоянная для каждого регулятора величина, равная примерно .1 или 2 мА. Таким образом, если нужен ток 25 мА, R должно быть 12/0,025 = 480 Ом (пренебрегая /о). Его следует довести до точной величины при помощи небольшого переменного сопротивления, соединенного последовательно с R.
Операционные усилители
Схемы из отдельных транзисторов, как на рис. 27-5, или расширенные схемы из нескольких транзисторов в настоящее время используются редко благодаря доступности, удобству и компактности ИМС, являющихся операционными усилителями (ОУ). Это название произошло от способности такого усилителя осуществлять математические операции над поступающим на него сигналом. При помощи подходящих внешних соединений можно создать такие условия, что выходное напряжение будет 1) алгебраической суммой двух или более входных
Рис. 27-10. Операционные усилители: а — общее обозиачеиие; б — соединенный как инвертирующий усилитель, ПОЭТОМу Свих — — (Zo.c./Zbx) Свх, ПрН условии что внутренний коэффициент усиления Треугольником обозначен соответствующий источник питания; все потенциалы даются относительно Земли.
напряжений, 2) произведением напряжения на постоянную величину, 3) интегралом входного напряжения по времени, 4) производной входного напряжения по времени. Другие функции — логарифмирование, возведение в квадрат, умножение или деление одной переменной величины на другую можно выполнить при помощи нелинейных компонентов н усилителя. ОУ могут применяться при любой частоте от пуля (т. е. для постоянного токй) до 10 кГц, а специальные модели могут работать в интервале нескольких мегагерц.
ОУ должен иметь следующие признаки: 1) высокий коэффициент усиления напряжения (не менее 104, во многих серийных прибо-
Электронные схемы в аналитических приборах 561
pax 106); 2) высокий входной импеданс* (не менее 105, часто до 1012); 3) давать нулевой выход при нулевом входе; 4) иметь минимальный дрейф (медленное изменение выхода при постоянном входе).
Большинство ОУ имеет две входные клеммы, на одной из которых происходит обращение знака. Клеммы по соглашению помечают знаками + и —, как на рис. 27-10. Это не означает, что выводы должны соединяться только с напряжениями указанного знака, как было бы правильно для подобного обозначения на' вольтметре. Это означает только то, что вывод, обозначенный —, дает инверсию знака, а другой—нет. В случае если неинвертирующий ( + ) вход в данной схеме не нужен, он должен быть соединен с точкой, в которой потенциал равен нулю, называемой точкой заземления.
Основные соединения показаны на рис. 27-10,6. Большинство схем с использованием ОУ, требует отрицательной обратной связи **. Соединение осуществляется через импеданс обратно?! связи Zo. с. от выхода к инвертирующему входу.
Если сигнал, к которому чувствителен- усилитель, представляет собой напряжение, то он должен поступать па вход через входной импеданс ZBX. Так как вход 1 усилителя проводит незначительный ток, то ток, протекающий через ZBX, а именно (евх—e)IZBT должен быть равен току в контуре обратной связи, т. е. (е—еВЫх)/^о. е. Ток обратной связи должен поступать из выхода усилителя. Когда поступает входной сигнал, усилитель саморегулируется, так что ток обратной связи п входной ток в точности равны, или
(евх—e)/ZBX = (е—eBHX)/Zo с. (27-2)
что можно представить в виде
евых К (Zo.c. -I- Zbx) — eBXZo c.]/Zbx (27-3)
Это соотношение можно упростить, если принять во внимание высокий внутренний коэффицпет усиления усилителя (часто называемый усилением разомкнутого контура). Если усиление составляет 106, то тогда 10 В на выходе означает, что потенциал на входе усилителя составляет только 10 мкВ относительно Земли. Это напряжение очень близко к потенциалу Земли, и поэтому суммирующий вход*** обычно считают
* Импеданс в электрической цепи определяется как отношение потенциала вдоль цепи или ее элемента к протекающему току. В цепях постоянного тока импеданс идентичен сопротивлению, но в цепях переменного тока они часто различны.
** Исключение составляет схема компаратора, обсуждаемая в этой главе ниже.
*** Суммирующим входам или суммирующей точкой называют инвертирующий вход ОУ. потому что входной ток и ток обратной связи в сумме дают пуль.
562 Глава 27
Рис. 27-11. Операционный усилитель, соединенный как сумматор. СВ означает суммирующий вход. Для /1>1 еВМх=—^о.сДе^+ег/Аг+ез/Лз).
виртуально (т. е. практически) заземленным (при условие что неинвертирующий вход заземлен). Следовательно, в уравнении (27-3) членом, содержащим е, можно пренебречь:
£вых= --£bxZo.c.A2-BX (27-4)
Уравнение (27-4) является основным рабочим уравнением для подобного включения ОУ. На практике отношение Zo. C./ZBX редко делают больше 100 или меньше 0,01. Если Zo. с. и ZBX— чисто активные сопротивления, их можно заменить соответствующими R.
К суммирующему входу могут быть присоединены одновременно несколько входов, как на рис. 27-11. В этом случае выход становится отрицательной суммой входов, каждый из которых умножен на соответствующее соотношение. На любой из этих параллельных входов можно подать отрицательный сигнал (и осуществить вычитание).
Для осуществления операции интегрирования элементом обратной связи должен быть конденсатор (рис. 27-12). Так как входной ток и ток обратной связи должны быть равны, а ток, протекающий через конденсатор, представляет собой производную потенциала по времени, то из этого следует, что
еВх/Двх=—C[(deK,^/dt) (27-5)
Рис. 27-12. Операционный усилитель, соединенный как интегратор.
Для А 1 Свых= — 1/7?вхС
о
что эквивалентно уравнению
£вых~ —1/RbxC f eBxdt ву—о} (27-6)
При дифференцировании в схеме на рис. 27-12 R и С следует поменять местами, тогда
евь,х=-До.с.С^ (27-7) at
Электронные схемы в аналитических приборах 563
Этот метод дифференцирования на практике используется редко, так как случайные шумы на входе вносят существенные погрешности.
Логарифм переменной может быть получен путем использования экспоненциальной характеристики рп-перехода при прямом включении. На рис. 27-13 показана схема соединения. При выводе уравнения нужно принять равными
Рис. 27-13. ОУ, соединенный для получения логарифмической фуикции. Диод ориентирован на положительный евх.
Для eBMx=~-k lg евх—k', где k п k' — безразмерные константы.
входной ток и ток в цепи обратной связи. Связь ток—напря-
жение для диода приближенно описывается уравнениями
lg7 = kjV или I = /г2 antilog V
(27-8)
из которых следует, что
ввх/^вх = А2 antilog ввых ИЛИ eBbIX = fe3'logeBX—/г4 (27-9)
Эксперимент показывает, что для этой цели больше подходит
кремниевый диод, чем германиевый, но еще лучше кремниевый транзистор с заземленной базой, который дает линейную логарифмическую зависимость по меньшей мере для четырех логарифмических десятичных разрядов.
Для того чтобы найти экспоненциальную (антилогарпфми-ческую) функцию, нужно поменять местами диод и сопротивление, как на рис. 27-14. Умножение или деление переменных можно осуществить логарифмированием, сложением или вычитанием логарифмов, а затем нахождением антилогарифма. Чтобы
обеспечить получение точных результатов, все диоды или экви-
валентные транзисторы должны находиться при одинаковой
температуре (или температурные изменения должны компен-
сироваться) .
Ошибки при работе с операционными усилителями. Наиболее ценное качество ОУ состоит в том, что они строго следуют приведенным выше элементарным математическим уравнениям. Существует, однако, несколько потенциальных источников ошибок.
Рис. 27-14. ОУ, соединенный для получения экспоненциальной (антило-гарифмической) функции, показан для положительных значений евх. Для Л»1 еВых=£ехр(—k'eBX), где k и k'—безразмерные константы.
564 Глава 27
Конечный коэффициент усиления. Все приведенные ранее формулы выведены исходя из предположения, что усиление разомкнутого контура усилителя больше, чем полученное из отношения Zo. c./ZBX, которое может быть названо усилением замкнутого контура. Детальный анализ показывает, что уравнение для работы простого инвертора (рис. 27-10,6) имеет вид евых- -евх-^(—) (27-10)
где А — усиление разомкнутого контура, G — усиление замкнутого контура. Это позволяет оценить ошибку, возникающую, если значение А слишком мало, например если А = 105 и G —103; при этом евь1Х/евх становится равным 1,01 • 103, т. е. ошибка составит примерно 1 %. Если необходима более высокая точность, следует либо поддерживать G ниже 1000 либо использовать лучший усилитель с А> 105.
Напряжение смещения. В идеале усилитель должен давать на выходе нуль при нуле на входе. Существует, однако, небольшая асимметрия во внутренней цепи, так что даже при двух заземленных входах на выходе часто появляется небольшое напряжение, которое можно скорректировать при помощи триммера. Даже после балансировки усилителя вследствие температурных колебаний наблюдается медленный дрейф этого смещения. Этот дрейф может быть причиной, ограничивающей применение усилителей для медленно изменяющихся сигналов. Смещение особенно нежелательно, когда усилитель используется в качестве интегратора, так как эффект в этом случае накапливается.
Ток смещения. Иногда ОУ отклоняется от идеального поведения, так как на его входы проходит значительный ток. Этот ток смещения можно компенсировать, по крайней мере частично, установив между неинвертирующим входом и землей сопротивление в несколько килоом. Падение напряжения на этом сопротивлении будет компенсировать падение напряжения, вызванное токами смещения на входном сопротивлении и сопротивлении обратной связи. Как и в случае напряжения смещения, эта ошибка наиболее существенна в интеграторах.
Применение операционных усилителей в преобразователях
Преобразователь представляет собой устройство для превращения одного вида энергии в другой. В нашем изложении термин относится только к приборам, превращающим информацию о химической системе в электрические сигналы.
Электронные схемы в аналитических приборах 565
Рис. 27-15. Измерение сопротивлений с помощью ОУ. Отношение двух сопротивлений (изображены как фоторезисторы) eBai=—(7?2/-/?1)евх-
Рис. 27-16. Измерение потенциалов с помощью ОУ, схема повторителя напряжения. Если А^>1, то е11ЫХ = еБх.
Преобразователи можно для удобства классифицировать по роду электрической величины, в виде которой представлен сигнал. Рассматриваемые преобразователи могут являться 1) переменным сопротивлением, 2) источником напряжения, 3) источником тока. Класс переменных сопротивлений включает в себя фотопроводящие ячейки, термисторы, металлические термометры сопротивления и электролитические ячейки. В принципе любое из этих устройств может быть использовано как ZBX или Zo. с. в цепи ОУ на рис. 27-10,6. Если переменный евх заменить постоянным потенциалом £вх, тогда измерение евых позволит однозначно определить сопротивление преобразователя. Обратная величина (проводимость) может быть получена непосредственным размещением преобразователя во входной цепи.
Если необходимо получить отношение двух сопротивлений, как, например, в некоторых двухлучевых фотометрах, то вместо двух импедансов в цепи ОУ можно. использовать два одинаковых преобразователя (рис. 27-15).
К преобразователям, являющимся источниками напряже
ния, относятся многие сочетания тродов, фотогальванические ячейки и термопары. В этом случае предпочтительна схема повторителя напряжения (рис. 27-16). Непосредственная обратная связь выхода с суммирующим входом обеспечивает точное воспроизведение входного напряжения на выходе усилителя. Прямое присоединение преобразова
потенциометрических элек-
Рис. 27-17. ОУ для измереиня тока. Если А^>1, то еВых= = —/?о.сТ*вх-
566 Глава 27
теля к входу ОУ не позволяет току протекать через него, поэтому умеренный ток, который может понадобиться для работы последующего оборудования, можно взять с выхода. Схема на рнс. 27-10, б не подходит для потенциометрических измерений, так как требует протекания тока от источника.
К преобразователям, являющимся источниками тока, относятся вакуумные (или газонаполненные) фотоэлементы и фотоумножители, пламенно-ионизационные ГХ-детекторы и электроды для вольтамперометрии и амперометрии. Эти источники тока могут быть соединены прямо с суммирующим входом ОУ, как на рис. 27-17. Для виртуального заземления этого входа через сопротивление обратной связи должен протекать ток равной величины, тем самым повышая потенциал еВых- Примеры такого преобразования ток—напряжение показаны на рис. 3-20 и 3-21 в сочетании с фотоумножителями и фотоэлементами.
Фотоэлементы
Существует несколько типов полупроводниковых фотопреобразователей. Наиболее часто для оптических измерений используется кремниевый фотодиод типа р—i—п. Он представляет собой тонкую пластинку кремния с собственной проводимостью (т. е. состоящую из чистого нелегированного элемента) и с двумя сильно легированными зонами p-типа на поверхности, куда падает излучение, и n-типа сзади. В качестве соответствующих добавок применяют бор и фосфор. Структура р—i—п предпочтительней простой рп-структуры, так как она позволяет приложить более высокое напряжение смещения без пробоя, а это обусловливает широкий динамический диапазон.
Работа фотоячейки этого типа может быть описана семейством характеристических кривых (рис. 27-18). В области справа от начала координат (первый квадрант) ячейка действует как фотопроводящий прибор. В этой области фототок является линейной функцией освещенности. Второй квадрант соответствует работе в качестве фотогальванического прибора, т. е. ячейка действует как генератор электроэнергии, и выход уже не линеен. Прямые линии 1, 2 и 3 соответствуют трем возможным режимам работы: 1) без смещения приложенного напряжения и с сопротивлением измерительной цепи, близким по величине к нулю, что приводит к хорошей линейности и нулевому темновому току; 2) с отрицательным смещением и высоким сопротивлением нагрузки, дающим повышенную скорость отклика, и 3) с нулевым смещением и высоким сопротивлением нагрузки, дающим отклик, пропорциональный логарифму освещенности.
Электронные схемы в аналитических приборах 567
Рис. 27-18. Вольтамперные характеристики кремниевого фотодиода, нагрузочные линии 1, 2 и 3 обсуждаются в тексте (United Detecter Technology).
Селеновые ячейки, иногда называемые ячейками с запирающим слоем, ограничены фотогальваническим режимом. Они впервые были широко использованы в фотометрах пропускания и флуориметрах, но в более современных приборах были заменены кремниевыми фотодиодами. Селеновые ячейки имеют более высокие температурные коэффициенты и подвержены «усталости», т. е. потере чувствительности при длительном нахождении на свету.
Фотопроводящие ячейки, не проявляющие фотогальванических свойств, изготавливают из сульфидов или селенидов свинца или кадмия. CdS- и CdSe-ячейки находят широкое и разнообразное применение для контроля и измерений, не требующих большой чувствительности кремниевых фотодиодов. PbS-ячейки применяются в качестве детекторов преимущественно в ближнем ИК-диапазоне и широко используются в спектрофотометрах для этой области.
Известен ряд комбинированных полупроводниковых приборов, таких, как фототранзисторы или фото-ПТ, в которых управляющая функция транзистора задается излучением. Их характеристики подобны характеристикам фотодиодов с большей чувствительностью благодаря повышенному коэффициенту усиления. Одним из недавних достижений в этой области является изготовление линейного ряда из большого числа фотодиодов, расположенных близко друг к другу на единой подложке’, что позволяет одновременно измерять интенсивность излучения
* Так называемая диодная матрица (diode array), особенно широко используемая как детектор в современных хроматографах.— Прим, персе.
568 Глава 27
Длина волны, МКМ
Рис. 27-19. Спектральная чувствительность фотоэлементов различных типов. CdS-, CdSe- и PbS-элементы являются фотопроводпнками, Se- и Si-элементы фотогальванические.
в последовательных точках фокальной плоскости спектрометра. Его можно использовать для имитации быстрого сканирования спектра или в сочетании с многоканальным анализатором для интегрирования спектра в течение какого-либо периода времени.
На рис. 27-19 представлена спектральная чувствительность нескольких фоточувствнтельных приборов. Вакуумные фотоэлементы и фотоумножители описаны в гл. 3.
Схемы составных усилителей
Схема полярографа на рнс. 16-10 представляет пример сложного прибора, который можно легко собрать из стандартных ОУ. Болес подробно мы рассмотрим другой электрохимический прибор — кулонометр с контролируемым потенциалом, показанный на рис. 27-20. В этом случае измеряются три электрохимические величины: потенциал рабочего (РАБ) электрода относительно электрода сравнения (СРВ); ток, протекающий между рабочим и вспомогательным (ВСП) электродами, и число кулонов (количество электричества), необходимое для протекания химического процесса. Все эти величины можно контролировать или измерять с помощью изображенной схемы.
Отметим вначале, что рабочий электрод непосредственно соединен с точкой виртуального заземления — суммирующим входом усилителя 2. Следовательно, для поддержания необходимого. потенциала между рабочим электродом и электродом сравнения электрод сравнения должен находиться под напря-
Электронные схемы в аналитических приборах 569
Рне. 27-20. Схема установки на ОУ для кулонометрии при контролируемом потенциале. Потенциал электрода сравнения, рабочий ток и количество электричества контролируются одновременно.
жением, отличающимся от потенциала Земли. Это достигается с помощью усилителя 1, который заставляет электрод принимать потенциал Еуст, установленный вручную. Его значение можно считать с панели вольтметра V. Усилитель 1 подает на вспомогательный электрод ток, который необходим для поддержания электрохимической реакции на рабочем электроде. Ток, протекающий через рабочий электрод, должен последовательно проходить через сопротивления R\ и R2, где он заряжает интегрирующий конденсатор С в цепи обратной связи усилителя 3. Выход Eq этого усилителя пропорционален таким образом временному интегралу тока и, следовательно, количеству электричества, прошедшего через раствор. Повторитель напряжения, усилитель 4, обеспечивает измерение электродного тока.
Генераторы синусоидальных импульсол
Иногда в лабораторном приборе необходим источник переменного тока как дополнение к постоянному (линейная частота). Для его получения удобнее всего использовать электронный
570 Глава 27
с
Рнс. 27-21. Схема генератора Хартли.
генератор. В принципе любой усилитель можно превратить в генератор, обеспечив продолжительную обратную связь с соответствующей частотной характеристикой. (Это означает возврат части переменного тока с выхода на вход через ча-. стотно-селективную цепь с таким фазовым соотношением, что сигнал нужной частоты усиливается, в то время как другие ослабляются.)
На рис. 27-21 изображена схема генератора Хартли, являющаяся
одной из многих схем, удовлетворяющих этим условиям. Обратная связь выхода и входа осуществляется через взаимную индуктивность катушки L. Частота генерации в герцах определяется уравнением
2л VLC
(27-11)
где L выражено в генри, а С — в фарадах. Частоту можно легко менять, заменяя конденсатор. Другой путь установки частоты заключается в использовании ДС-цепочки, такой, как заграждающий фильтр (рис. 27-22,а). Этот фильтр пропускает все частоты, кроме одной f0, выражаемой формулой
/о — ~
хьЗТ./\ G
(27-12)
График импеданса как функции частоты имеет очень четкий пик (рис. 27-22,6), резкость которого зависит от того, насколько точно подобраны конденсатор и сопротивление. Если эту цепочку поместить в контур обратной связи ОУ (рис. 27-22, в), коэффициент усиления схемы (G = Z0. с./Хвх) будет низким для всех частот, кроме f0. На неэкранированный суммирующий вход будет поступать достаточно шумов различной частоты (вход показан на рисунке штрихами). Это определяет начало генерации с частотой, задаваемой фильтром. Для поддержания генерации неинвертирующий вход включают в неглубокую неселективную положительную обратную связь через конденсатор Со. с.. Такой генератор удобно использовать при фиксированной частоте, но его трудно сделать перестраиваемым.
Весьма точный (прецизионный) генератор фиксированной частоты лучше всего изготавливать на основе кварцевого кристалла, из которого вырезают небольшую пластинку и прикреп-
Электронные схемы в аналитических приборах 571
ляют к ней с противоположных сторон металлические электроды. Если через эти электроды подвести к пластинке элект роэнергию, то она будет колебаться с характерна стической частотой. Температурный коэффициент расширения кристаллического кварца очень невысок, поэтому частота генератора, изготовленного из надлежащим образом вырезанной пластинки, почти не зависит от температуры. Диапазон частот таких генераторов от 10 кГц до 50 МГц. Кварцевые генераторы обычно изготавливают с одним транзистором (рис. 27-23). ОУ используются редко из-за ограниченных возможностей в области высоких частот.
Сервомеханизмы
Сервосистема прибора
Рис. с = 1/(2л7?С) Гц; б — импеданс фильтра как функция частоты; в — генератор иа ОУ с обратной связью через заграждающий фильтр.
27-22. а — заграждающий фильтр характеристической частотой f0=
в своей обычной форме состоит из небольшого электродвигателя (постоянного тока или двухфазного переменного тока), скорость и направление
вращения которого контролируются усилителем. Система снаб-
жена некоторым типом обратной связи, так что при вращении
электродвигателя возникает напряжение, которое автоматически сравнивается со стандартным. Разница, называемая сигналом ошибки, усиливается по мощности усилителем и передается на электродвигатель с тем, чтобы уменьшить ошибку до
нуля.
Прибор нуждается в сервосистеме, если какой-либо механический эффект должен быть пропорционален переменному сигналу. В качестве примера ее использования можно привести
572 Глава 27
Рис. 27-23. Схема кварцевого генератора. Сопротивления н /?2 устанавливают точное смещение транзистора. Конденсаторы С) н С2 определяют отношение обратной связи. Полученный переменный ток изолирован от источника питания малой индуктивностью («дросселем»).
регистрирующий спектрофотометр, в котором сервосистема контролирует ширину щели. Луч света, проходящий через кювету с раствором сравнения, служит сигналом, поддерживающим постоянство передаваемой энергии во время сканирования спектра. Сервосистема применяется также в самобалансирующемся самописце, описанном в следующем разделе.
Самописцы [2]
Большинство электронных самописцев, используемых в лабораторных приборах, работают по нулевому сервопринципу (рис. 27-24). Потенциал, который нужно зарегистрировать, Ех, включен последовательно (встречно) напряжению Ер; Ер снимают с переменного сопротивления, называемого балансирующим потенциометром (реохордом). При полярностях, показанных на рисунке, если ЕР>ЕХ, то потенциал будет отрицателен, если же ЕР<ЕХ, он будет положителен. Это напряжение управляет вращением небольшого сервомотора постоянного тока с постоянным магнитом. Двигатель выполняет две функции: меняет сопротивление потенциометра, обеспечивая Ер — Ех, и одновременно перемещает перо по диаграммной ленте. Положение пера следует изменениям Ех с точностью до предела, установленного скоростью отклика механической системы. Большинство современных самописцев имеют время отклика не более 1 с, а многие вдвое меньшее.
Электронные схемы в аналитических приборах 573
Рис. 27-24. Сервосистема самописца. Передвигаемая электродвигателем каретка переносит скользящий контакт по делителю напряжения н смещает перо самописца [1].
Цифровая электроника
Обрабатываемые в электронных схемах сигналы, рассматривавшиеся до сих пор, являются по своей природе аналоговыми. Это означает, что они могут принимать любое значение в широком диапазоне, ограниченном только характеристиками обсуждаемых электронных приборов. Компьютер, однако, оперирует цифровыми сигналами, значения которых ограничиваются двумя уровнями «включено» и «выключено» (ON и OFF)*. В системах, которые нас интересуют, эти величины имеют номиналы 0 и +5 В соответственно. Компьютер не способен реагировать иа промежуточный уровень, например 4-2 В; отрицательное напряжение или положительное напряжение, превышающее + 5 В, могут легко его повредить. Следовательно, чтобы подготовиться к изучению компьютеров в аналитических приборах, мы должны рассмотреть некоторые цифровые приборы и преобразователи, которые заполняют пробел между областью цифровых и аналоговых сигналов.
* Логическая 1 и 0, или «истина» и «ложь» соответственно.— Прим, перев.
574 Глава 27
АВС Выход
А в с
ЛВС Выход
0 0 0 0
0 0 10
0 10 0
0 110
iooo 10 10
110 0
1111
0 0 0 0
0 0 11
0 10 1
0 111
10 0 1
10 11
110 1
1111
Рис. 27-25. Логическое устройство И с тремя входами и его таблица истинности.
Рис. 27-26. Логическое устройство ИЛИ с тремя входами и его таблица истинности.
А Выход
1 0
0 1
Рис. 27-27. Логическое устройство НЕ (инвертор) и его таблица истинности.
Логические устройства. Эти устройства управляют прохождением сигнала; те из них, которые мы рассмотрим сейчас, функционируют как модели логических команд. Существует три основных типа устройств, называемых И, ИЛИ и инверторы (НЕ). Их работу удобно описывать с помошью таблиц истинности. На рис. 27-25 изображено устройство И с тремя входами (Л, В и С) и одним выходом. Соответствующая таблица истинности показывает, что для всех входов, равных 1, выход также равен 1, во всех других случаях он равен нулю. Цифры 1 и О часто используются как эквиваленты потенциалов ВЫСОКИЙ (HIGH) и НИЗКИЙ (LOW) соответственно, но в описаниях приборов цифры иногда заменяются буквами Н и L; для уровня сигнала +5 В ставится или Н, или 1.
На рис. 27-26 и 27-27 изображены устройства ИЛИ и НЕ (инвертор). Устройство И и устройство ИЛИ могут иметь в принципе любое число входов, но инвертор имеет лишь один вход.
Э.)ек1ронпые схемы и аналитических приборах 575
л В С
ЛВС
ООО О О 1
О 1 о
О 1 1
1 о о
1 0 1
1 1 о
1 1 1
Выход
1
1
1
1
1
1
1 о
Л--I X Выход
Й=)или-НЕ>О—-
С--1_—'
А Я С | Выход
0 0 0 1
0 0 10
0 10 0
0 110
1 о о о
10 10
110 0
1110
Рис. 27-29. Логическое устройство ИЛИ—НЕ с тремя входами и его таблица истинности.
Рис. 27-28. Логическое устройство И—НЕ с тремя входами и его таблица истинности.
Рис, 27-30. Логическое устройство исключающее ИЛИ (а), построенное из отдельных устройств, его таблица истинности (б), его обозначение (в).
Сочетая инвертор с устройствами И и ИЛИ, можно получить логические функции И—НЕ и ИЛИ—НЕ соответственно (рис. 27-28 и 27-29). Каждое из этих устройств может быть выполнено на основе одного или двух отдельных транзисторов (плюс необходимые вспомогательные элементы), но на практике их оформляют как ИМС, обычно с несколькими устройствами в одном корпусе. Два типа наиболее распространенных устройств носят названия ТТЛ (транзисторно-транзисторная логика) и устройства на МДП-транзисторах. МДП-транзистор-ные элементы используются реже, чем ТТЛ, так как они имеют более медленный отклик из-за относительно высокой входной емкости.
576 Глава 27
Логические элементы можно соединять непосредственно друг с другом н получать более сложные устройства, как, например, на рис. 27-30, которое называется исключающее ИЛИ. Таблица истинности получается при перечислении состояний в промежуточных точках Р и Q, которые при объединении дают состояние выхода. Особенно ценное сочетание представляет собой исключающее ИЛИ, дающее «высокий» выход, если «высок» любой извходов, но не оба входа вместе.
Триггеры, счетчики и регистры. Триггером называют схему, которая в простейшем виде состоит из двух элементов (либо И—НЕ, либо ИЛИ—НЕ), соединенных между собой, как показано на рис. 27-31. Буквы S и R (от англ, set — устанавливать и reset — сбрасывать) обозначают входы, тогда как Q и Q — выходы (обозначение взято из булевой алгебры; Q читается как «Q с чертой» или «не-Q» и означает, что, в каком бы состоянии ни было Q, Q находится в противоположном состоянии).
В нормальном режиме входы R и S находятся в состоянии логической 1. Мгновенный переход входа S в 0 дает Q=1 и Q = 0, тогда как моментальное заземление R дает Q = 0 и Q=l, причем оба состояния стабильны. Отметим, что выход Q может быть установлен (set) до 1 или сброшен (reset) до 0. После установки схемы последующие изменения S несущественны. Это же справедливо для случая сброса. Если R и S оба становятся нулями, тогда Q и Q оба станут единицами; при возвращении обоих входов одновременно к 1 выходы непредсказуемо перейдут в сброшенный или установленный вид, следовательно, таких действий следует избегать.
Рис. 27-31. А\5-триггер, построенный из двух устройств И—НЕ.
Рис. 27-32. D-триггер, построенный из логитсских элементов.
Электронные схемы в аналитических приборах 577
Рис. 27-33. Master/slave /Л-триггер.
Другой вариант триггера, D-триггер, изображен на рис 27-32. Он состоит из 7?5-триггера с добавлением еще двух элементов И — НЕ и инвертора на входе. Вход С присоединен к «часши», которые дают цуг импульсов, представляющих временной сигнал. Анализ схемы показывает, что сигнал (1 или 0), поступающий на вход D («данные»), появится на выходе Q, как только будет получен импульс часов. Затем Q остается неизменным в течение времени, пока часы выдают 1 (HIGH). Эта схема широко используется для задержки, т. е. для хранения в ее памяти одного бита данных, чтобы им могла воспользоваться какая-либо другая схема.
Рассмотрим для примера цифровой вольтметр: он должен почти мгновенно реагировать на изменение входного сигнала, но глазу наблюдателя требуется секунда или более, чтобы считать данные. Устройство задержки данных, дающее один импульс часов за 2 или 3 с, сохранит значение частоты, чтобы его можно было прочесть, прежде чем стереть.
Другой подобной схемой является master/slave (от англ, master — хозяин, т. е. основной, и slave — раб, т. е. вспомогательный) УК-триггер. Это устройство содержит два D-триггера, соединенных между собой (рис. 27-33). Поступающий бит данных (при J или К) проходит на первый, основной триггер (элементы 3 и 4), только если сигнал часов есть 1. Затем он переносится на второй, вспомогательный триггер (элементы 7 и 8) в момент, когда сигнал часов становится нулем. При соединении К и S сигналы также могут прямо идти на вспомогательный элемент. Этот универсальный триггер имеет то преимущество, что благодаря связи master/slave сигналы, временно хранящиеся в основном триггере, не подвергаются воздействию данных, находящихся во вспомогательном триггере, и наоборот.
19 Заказ № 537
578 Глава 27
Рис. 27-34. 4-Битовый двоичный счетчик на основе //(-триггера (а), его таблица истинности (б), его временная диаграмма (в).
Триггеры могут быть соединены в каскады, называемые регистрами, что позволяет им выполнять специальные задачи. Двоичный счетчик, например, может быть сконструирован из последовательно соединенных //(-триггеров, как на рис. 27-34 (/- и /(-входы не используются, но должны быть присоединены к +5 В). Каждый импульс часов вызывает переход выхода Q триггера Т-1, либо 1->0, или 0-И. Этот сигнал приходит на вход Т-2 и так далее вдоль цепи, делящей частоту надвое для каждой строки, как показано на рис. 27-34, в. Значения сигнала на каждом выходе от А до D перечислены в таблице (рис. 27-34, б). Выход А соответствует младшему биту [least significant bit (LSB)], а выход D — старшему биту [most significant bit (MSB)]. Эта таблица показывает, как образуются числа в двоичной системе. Из таблицы видно, что десятичное 6 эквивалентно двоичному ОНО (6ю = 01102). Преобразование двоичного числа в его десятичный эквивалент может быть легко осуществлено разложением на степени числа два, как показано под таблицей. Двоичное число ОНО может быть выражено десятичным как (0х23) + (1 Х22) + (1Х21) + (0x2°) =04-4+2 + 0 = = 6. Для числа больше 15 необходим пятый бит (со значением 24), и в регистр должен быть добавлен еще один триггер.
Электронные схемы в аналитических приборах 579
D С
Импульс
о
1
2
3
4
5
6
7
8
9
(Ю)
о о
о о
о о
о о
О 1
О 1
О 1
О 1
1 о
1 о
о о
б
В ' Л о о О 1 1 о
1 1
о о
О 1 1 о 1 1
о о
О 1 о о
Рис. 27-35. 4-Битовый двоично-десятичный счетчик, построенный как модификация счетчика иа рис. 27-34 (а), и его таблица истинности (б).
Двоичный счетчик можно приспособить для снятия десятичных отсчетов путем замены соединений и (включения дополнительного устройства И, ведущего к /-входу Т-4, как на рис. 27-35. Свойства /-входа таковы, что, когда он заземлен (т. е. при нулевом уровне), он приводит Q-выход к 0 и Q к 1; если же он высок, счет продолжается. Следовательно, показанные на рисунке соединения приведут счет к возврату на 0000 вслед за девятым импульсом. Этот тип счетчика известен как двоично-десятичный [binary coded decimal (BCD)]. Четыре выхода могут быть соединены последовательно с устройствами, которые дают семь параллельных сигналов, питающих соответствующие секции 7-сегментного люминофорного дисплея, имеющегося во многих лабораторных приборах. ,
Счетный регистр можно использовать как таймер для измерения времени, протекающего между двумя событиями, или. чтобы вызвать событие в определенное время. Последнее можно
19*
580 Г лава 27
Вход
Рис. 27-36. Двоичный счетчик, используемый в качестве таймера.
сделать с помощью устройства И (рис. 27-36). Счетчик показывает время (в миллисекундах), в течение которого вход в устройство высок. Для больших интервалов времени были бы удобны часы с меньшей скоростью; часы частотой 1 Гц, например, давали бы отсчет прямо в секундах.
Чтобы установить точно временную последовательность событий, счетный регистр объединяют с серией устройств И, соединенных таким образом, чтобы выбирать различные комбинации выходов Q и Q из последовательных каскадов. Более детальное описание можно найти в работе [1], с. 222—225.
Переходные преобразователи
Теперь мы рассмотрим основные компоненты, которые осуществляют преобразование аналоговых сигналов в цифровой вид и наоборот. Существует множество различных схем таких преобразователей, мы опишем только по одному из каждого типа. Более детальное обсуждение см. в работах [3, 4].
Общий тип цифро-аналогового преобразователя (ЦАП) показан на рис. 27-37. Последовательность электронных переключателей, соответствующих цифрам двоичного числа, соединяет напряжение сравнения с последовательными элементами многозвенной цепи сопротивлений, ведущей к ОУ. Анализ показывает, что напряжение на выходе схемы на рис. 27-37 определяется уравнением
Евых - — ЕсР (Р + C/21 + В/22+ А /2») (27-13)
где буквы от А до О соответствуют положениям переключателей («0», если сопротивление заземлено, и «1», если сопротивление соединено с Еср). Для такого «4-битового» преобразователя в качестве Еср удобно использовать 8,000 В, так как это дает выход, равный десятичному эквиваленту двоичного числа (в вольтах). Таким образом, 01 Юг дает £Вых = 8(0+0,5 + 0,25 + + 0)=6 В. Дополнительные сегменты в звеньях позволяют увеличить точность путем вмещения большего числа битов цифровой информации.
Электронные схемы в аналитических приборах 581
£ср
Рис. 27-37. Цифро-аналоговый преобразователь (ЦАП) с использованием цепи из звеньев сопротивлений R/2R.
Рис. 27-38. Аналого-цифровой преобразователь (АЦП).
На рис. 27-38 показана схема аналого-цифрового преобразователя (АЦП). Он состоит из двоичного счетчика и ЦАП в сочетании с .R/S-триггером и другими управляющими элементами. Мгновенное заземление линии «готов» восстанавливает и триггер, и счетчик. При опускании переключателя «готов» Q становится высоким, позволяя импульсам часов проходить в счетчик. Счетчик увеличивает свой выход до все более высокого счета, пока напряжение Ео. с., даваемое ЦАП, не сравняется с напряжением аналогового входа, что определяется компаратором. Этот последний элемент представляет собой ОУ без отрицательной обратной связи; его выход всегда имеет максимальную величину, положительную или отрицательную. В данном случае выход связан с двумя диодами, которые предохраняют его от поступления положительных импульсов выше + 5 В и отрицательных импульсов. Когда Ео. с,- достаточно велико и превышает входное напряжение, выход компаратора скачком изменяется от 0 до +5 В. Это делает Q низким и ос
582 Глава 27
танавливает часы. Выход счетчика равен двоичному числу, которое ЦАП рассматривает как эквивалентное входному напряжению.
Мы рассмотрели некоторые основные элементы, необходимые для соединения лабораторного прибора с компьютером. Следующая глава предложит некоторые способы осуществления такого соединения.
Задачи
27-1. В схеме на рис. 27-11 положим е^+200 мВ, ег=+700 мВ, ез = —•—500 мВ, Я1 = 2 кОм, Лг=1,4 кОм, 1?з=10 кОм, /?о.с=10 кОм; вычислите значение выходного напряжения евых. Каким будет результат замены /?0.с-па 100 кОм, если все другие величины останутся без изменений?
27-2. Метод амперометрического титрования с двумя электродами, обсуждаемый в гл. 16, требует наложения небольшого постоянного напряжения иа ячейку при контролируемом токе. Сходный метод титрования требует при измерении потенциала протекания небольшого постоянного тока через ячейку. Постройте схему на ОУ, которая бы обеспечивала проведение обоих методов. 27-3. Постройте схему ОУ для решения уравнения
евых = kXYn
где X и У— два переменных напряжения, п. — переменный параметр, k — безразмерная константа. При построении схемы можно исходить из рис. 27-13 и 27-14.
Рис. 27-39. Перестраиваемый усилитель (к зад. 27-4).
Рис. 27-40. Логическая схема к зад. 27-5.
Электронные схемы в аналитических приборах 583
Рис. 27-41. Альтернативная схема для двоично-десятичного счетчика (к зад. 27-6).
27-4. ИК-спектрофотометр имеет вращающийся прерыватель, который модулирует излучение на 17 Гц. Система усиления сигнала включает два заграждающих фильтра, настроенных иа частоты 17 и 60 Гц, расположенных как иа рис. 27-39. В какие положения нужно поместить эти два фильтра? Объясните их функции в схеме.
27-5. Составьте таблицу истинности для схемы из двух элементов иа рис. 27-40.
27-6. Схема на рис. 27-41 может действовать как двоично-десятичный счетчик и как счетчик на рис. 27-35 (хотя последний медленнее). Объясните, как они работают.
27-7. В чем состоит разница в работе триггера, использующего схему рис. 27-31, и такого же триггера, в котором элементы И—НЕ заменены элементами ИЛИ—НЕ?
Литература
1. Vassas В. Н., Ewing G. W., Analog and Digital Electronic for Scientists (2d ed.), Wiley-Interscience, New York, 1980.
2. Ewing G. W., Ashworth H. A., The Laboratory Recorder, Plenum Press, New York, 1974.
3. Zucli E. L. (ed.). Data Acquisition and Conversion Handbook, Datel-In-tersil, Inc. Mansfield, MA, 1980.
4. Sheingold D. H. (ed.). Analog-Digital Conversion Notes, Analog Devices, Inc., Norwood, MA, 1977.
Дополнительная литература
1. Гусев В. Г., Гусев Ю. М. Электроника.— М.: Высшая школа, 1982.
2. Жеребцов И. П. Основы электроники.— Л.: Энергоатомиздат, 19S5.
ГЛАВА х-О
Компьютеры в аналитических приборах
В предыдущих главах мы лишь кратко упоминали об одном из самых важных достижений в приборостроении — речь идет о наделении прибора вычислительными возможностями. Этот предмет слишком сложен для детального обсуждения в этой книге, и самое большее, что можно сделать,— отметить некоторые наиболее существенные моменты.
При работе в аналитической лаборатории, применяющей инструментальные методы анализа, студенту, вероятно придется иметь дело с компьютерами в различных ситуациях. Многие, а возможно, большинство современных аналитических приборов имеют встроенные микропроцессоры и необходимую память. Программы для работы с прибором (программное обеспечение) поставляются изготовителем прибора. Все, что должен сделать пользователь,— это задать значения некоторых параметров, нужных для решения поставленной задачи.
Помимо этого с компьютером приходится встречаться при стыковке приборов с независимой ЭВМ типа «персонального компьютера». Такая ситуация становится все более обычной. Для начала мы рассмотрим общую структуру компьютера.
Архитектура ЭВМ
Большинство цифровых ЭВМ построены на основе общей блок-схемы, изображенной на рис. 28-1. «Сердцем» компьютера является устройство, называемое центральным процессором (ЦП), выполняющее основные логические действия; все остальные блоки ЭВМ присоединены к нему и ему подчиняются. Это относится к большинству устройств памяти и утройствам ввода/вывода (УВВ).
ЭВМ имеет три различных типа памяти. В каждом компьютере есть блок оперативной памяти (ОП) для временного хранения данных и программ *. Термин «случайный» означает, что
* В англоязычной литературе применяют аббревиатуру RAM от Random Access Memory — память, со случайным доступом.— Прим, перев.
Компьютеры в аналитических приборах 585
Рис. 28-1. Архитектура ЭВМ.
запоминаемая информация может храниться в любом свободном месте, откуда ее при необходимости можно извлечь. Второй тип памяти — постоянное запоминающее устройство (ПЗУ) (английская аббревиатура ROM от Read-only Memory—память только для чтения). Информация в ПЗУ заносится при изготовлении ЭВМ, пользователь его содержимое менять не может. В ПЗУ хранятся некоторые простые и часто используемые программы общего назначения, например программа извлечения квадратного корня. ПЗУ содержит также программу инициализации (начальной установки) всех блоков ЭВМ, которая выполняется при включении питания и дает уверенность в том, что все схемы готовы к работе.
Третий тип памяти — это внешняя память на магнитных носителях, дисках или лентах. Соответствующие блоки включают точные и сложные механизмы протяжки ленты или вращения диска, так что чаще их рассматривают как часть периферийного оборудования, а не компьютера как такового. Информация, хранящаяся в ОП, «летуча», т. е. исчезает при отключении питания; для ПЗУ и магнитных носителей это не так*.
В качестве периферийных УВВ обычно используют алфавитно-цифровую клавиатуру для ручного ввода команд л данных,
* Обычно по объему ПЗУ существенно меньше ОП, а ОП — внешней памяти. По быстродействию (времени доступа) соотношение обратное.— Прим, перев.
586 Глава 28
печатающее устройство для представления результатов на бумаге и графопостроитель. Две или все три функции часто выполняет одно устройство. Для удобства программирования и просмотра результатов перед печатью иногда добавляют видеотерминал-дисплей. Особый интерес с точки зрения использования лабораторного оборудования представляют специальные каналы приема данных непосредственно от преобразователей и выдачи команд различным приборам. Этот последний класс УВВ мы сейчас и рассмотрим.
Присоединение ЭВМ к приборам
Из предыдущей главы ясно, что большинство преобразователей дает аналоговый сигнал, функционально связанный с интересующей аналитика химической характеристикой. Этот сигнал можно перевести в цифровую форму с помощью описанных ранее аналого-цифровых преобразователей (АЦП). Обычно в одном приборе несколько преобразователей. Так, например, в УФ-спектрофотометре нужен не только фотоумножитель (или, возможно, два фотоумножителя), но и устройства, сообщающие компьютеру об установленной ширине щели и длине волны. Последние имеют скорее цифровой характер, чем аналоговый, однако обычно имеется несколько аналоговых сигналов, которые следует преобразовать в цифровые. Это вызывает потребность в мультиплексоре — устройстве, которое принимает на входе несколько сигналов и передает их по одному (последовательно) в ЭВМ. Некоторые мультиплексоры работают с аналоговыми сигналами, другие же предназначены для цифровых.
Изготовители интерфейсов предлагают два решения: можно использовать отдельный АЦП для каждого аналогового канала и передавать сигналы с помощью цифрового мультиплексора или, наоборот, передавать через мультиплексор аналоговые сигналы, что требует только одного АЦП. В принципе для обоих способов результаты должны быть правильны. На практике же более экономично применять аналоговый мультиплексор и один АЦП, если все блоки расположены рядом в одном корпусе. Однако если преобразователи находятся далеко от ЭВМ, система будет работать лучше при условии, что .по кабелям, соединяющим прибор и компьютер, идут только цифровые сигналы (поскольку снижается влияние помех). Поэтому иногда лучше пойти на дополнительные расходы и установить отдельные АЦП вблизи соответствующих источников сигнала, передавая через мультиплексор в ЭВМ цифровые данные.
Компьютер и его периферийные устройства обмениваются информацией через линию связи, называемую магистралью, или шиной. Она представляет собой набор проводов, которые могут
Компьютеры в аналитических приборах 587
Память на дисках
ЦП
Индикатор длины волны
Индикатор ширины щели
Электродвигатель управления шириной щели
Самописец
ФЭУ
Рис. 28-2. Магистральная структура системы ЭВМ спектрофотометра.
быть связаны с каждым модулем через буфер — устройство, способное работать в трех режимах: передавать цифровые сигналы в одном из двух направлений или не передавать их вовсе (речь идет о направлениях прибор->ЭВМ или ЭВМ->прибор). Команда ЦП сообщает буферу, в каком состоянии тот должен находиться на каждом этапе работы, Код адреса определяет, какое из многих устройств, подключенных к магистрали, следует активировать.
Спектрофотометр, например, может иметь четыре или больше УВВ, присоединенных к магистрали, не считая ЦП (рис. 28-2). Чтобы установить подходящую чувствительность, ЦП должен получить некоторую информацию от прибора, для чего он поочередно требует от буферов передачи данных. Так, например, ЦП может вначале перевести буфер, подключенный к индикатору длины волны, в состояние «включено» (ON), разрешая ему передачу на вход ЭВМ и одновременно выключая (OFF) буферы щели, ФЭУ и самописца. ЦП получит необходимую информацию об установленной длине волны и сохранит ее в оперативной памяти. После этого процессор изменит установку состояний буферов, чтобы получить информацию от индикатора ширины щели и т. д. В результате обработки полу
588 Глава 28
ченной таким образом информации ЦП может сделать вывод (на основе инструкций, содержащихся в программе) о том, что необходимо изменить ширину щели для приведения сигнала в разумный диапазон. После этого ЦП откроет буфер «мотор управления шириной щели», чтобы мог пройти корректирующий сигнал, и одновременно реверсирует свой буфер, чтобы сигнал мог идти в направлении от ЭВМ к периферии. Следующим шагом может быть считывание выходного сигнала ФЭУ и передача его на самописец.
Очевидно, что все сигналы должны безошибочно кодироваться во избежание неоднозначности. Важно также, чтобы команды выдавались в определенные моменты времени и в определенной последовательности.
Готовые интерфейсы
Пользователь может сам спроектировать и изготовить свой собственный интерфейс. В ряде книг имеются подробные инструкции, как это сделать [1—3]. Пример такого проектирования приведен в работе [4], где описана стыковка с недорогой ЭВМ марки Sinclair ZX81 (или Timex Sinclair 1000).
Целый ряд готовых интерфейсов изготовляется промышленностью. Один из них, «Isaak», выпускаемый фирмой Cyborg Corporation, предназначен для компьютера Apple II. Он может принимать аналоговые данные по 16 каналам и снабжен четырьмя выходными аналоговыми портами для управления режимными параметрами прибора. Более дешевые интерфейсы, пригодные почти для любой ЭВМ, производятся рядом фирм, но не все они позволяют управлять экспериментом.
Программирование
Когда интерфейс, соединяющий компьютер и прибор, сконструирован, необходимо снабдить ЭВМ подробными инструкциями. Это делается посредством программы действий, или программного обеспечения. Поскольку машина воспринимает команды только в цифровом виде, а человек обычно использует только естественный язык сложной структуры, скажем английский, нужен метод, обеспечивающий общение. Для этого служат языки программирования.
Для того чтобы составлять программы, выбирают несколько операторов и обучают их системе нотации, естественной для ЭВМ, так называемому машинному языку. Этот путь позволяет экономно расходовать память компьютера, но программирование на машинном языке — медленное и утомительное занятие. Обычно предпочтительнее перейти к языку ассемблера, использующему рйд трехбуквенных мнемонических сокращений,
Компьютеры в аналитических приборах 589
каждое из которых переводится на машинный язык с помощью внутренней программы ЭВМ, ассемблера. Ассемблер поставляется изготовителем ЭВМ. Такое программирование проще для программиста, хотя и требует большего объема памяти компьютера.
Для пользователя ЭВМ, не являющегося экспертом по программированию, непрактично изучать ни машинный язык, ни язык ассемблера. Ему следует изучить один из языков высокого уровня, таких, как Бейсик, Фортран или Паскаль. Для начала, вероятно, лучше всего выбрать Бейсик. Программы, написанные на этих языках, переводятся в машинные коды с помощью программы-компилятора, для чего требуется большая память, чем в случае языка ассемблера.
Эти языки включают команды, структура которых близка к естественным языкам, поэтому их легко выучить и применять. Научный работник, не знающий хотя бы азов программирования на одном из языков высокого уровня, находится в заведомо невыгодном положении. Существует много прекрасных книг по программированию на конкретных языках, которые могут послужить для него ценным руководством.
Предостережение
МикроЭВМ, несомненно, благо для конструкторов и пользователей приборов, но их использование сопряжено с некоторым риском. Машина работает так превосходно, цифры и напечатанные результаты кажутся такими убедительными, что возникает реальная опасность принимать на веру выходные данные, не обращая серьезного внимания на их «химический подтекст». Например, в большинстве анализов существенно, чтобы образцы сравнения чередовались с пробами, ho компьютер может и не подсказать вам этого. Превосходное обсуждение таких проблем в методе атомной абсорбции содержится в работе [5]; оно приложимо и к другим аналитическим методам.
С другой стороны, компьютер способен провести обработку данных так полно, что аналитик может посвятить освободившееся время решению принципиальных вопросов химии используемого процесса.
Литература
1. Arnold J. Т., Simplified Digital Automation with Microprocessors, Academic Press, New York, 1979.
2 Artwick B. A., Microcomputer Interfacing, Prentice-Hall, Englewood Cliffs, N. Y., 1980.
3. Barnaal D-, Digital and Microprocessor Electronics for Scientific Application, Breton Publishers, North Scituate, Mass., 1982.
4. Webster D. C., Am. Lab., 1983, v. 15, No. 2, p. 48.
5. Koirtyohann S. R., Anal. Chem., 1980, v. 52, p. 736A.
Приложение А
Стандартные потенциалы
Электрод Реакция (oth. НКЭ), В
F2, F- F2 + 2e~ 2F- 4-2,85
Соя+, Со2+, Pt Co3+ -1- e--^Co2+ 4-1,84
Au+, Au Au+ + e~ Au 4-1,68
Се4+, Сс3+, Pt Ce4+ + e~ Ce3+ + 1,46
МпО”, Mn2+, Pt MnO7 + 8H+ 4- 5e“ Mn2+ 4- 4H2O + 1,49
Au3+, Au Au3+ 4 3e~ -► Au + 1,42
Cl2, ci- Cl2 4 2e- 2C1- + 1,360
Cr2O2", Cr3+, Pt Cr2Of“ 4- 14H+4- 6e“ 2Cr3+ 4- 7H2O + 1,33
Tl3+, T1+, Pt Tl3+ + 2e- -> T1+ + 1,25
O2, H2O O2 4- 4H+ 4- 4e~ -> 2H2O +1,229
Pt2+, Pt Pt2+ 4- 2e- Pt + 1,2
Br2, Br- Вг2(ж.) 4~ 2e_ -► 2Br~ +1,065
Hg2+, Hg2+, Pt 2Hg2+ 4- 2e~ -* Hg2+ +0,905
Ag+, Ag Ag+ -1- e- -> Ag +0,800
Hg2+, Hg Hg|+ 4- 2e“ -> 2Hg +0,799
Fe3+, Fe2+, Pt Fe3+ 4- e- Fe2+ +0,770
Ag2SO4, Ag Ag2SO4 + 2e“ -* Ag 4- SO2" +0,653
AgC2H3O2, Ag AgC2H3O2 4- e“ -4- Ag 4- C2H3O^“ +0,64
I2- I" I2 + 2e-->21- + 0,535
Cu+, Cti Cu+ 4- e- Cu +0,522
Ag2CrO4, Ag Ag2CrO4 4- 2e~ 2Ag+ + CrO2~ +0,446
VO2+, V + , Pt VO2+ 4- 2H+ 4- e- -> V3+ 4- H2O +0,337
Fe(CN)3“, Fe(CN)g—,Pt Fe(CN)|— 4- e“ Fe(CN)|~ +0,46
Cu2+, Cu Cu2+ 4- 2e- -> Cu +0,340
Продолжение приложения А
Электрод Реакция (оти. НКЭ), В
UO2+, U4+, Pt ио|+ + 4H+ + 2e~ -4- U4+ + H2O 4-0,334
HggCI2, Hg Hg3Cl3 + 2e- 2Hg + 2CI- 4-0,268
AgCI, Ag AgCI + e~ Ag + Cl- -40,222
HgBr|- Hg HgBr*- 4- 2e~ -> Hg + 4Br- +0,21
Cu2+, Cti+, Pt Cu2+ + e- Cu+ 4-0,158
Sn*+, Sn2+, Pt Sn4+ + 2e~ Sn2+ +0,15
Hg.Br2, Hg Hg2Br2 + 2e- -> 2Hg + 2Br~ +0,140
CuCI, Cu CuCI + e- ->• Cu ci- +0,137
TiO2+, Ti3+, Pt TiO2+ + 2H+ + e~ -> Ti34- + H2O + 0,1
AgBr, Ag AgBr -F e- -► Ag + Br- +0,071
uo|+, uo-^ Pt UO|+ + e“ UO+ -1-0,062
CuBr, Cti CuBr + e_ -> Cu -|- Br- +0,033 .
H + , H2 2H+ 4- 2e~ -4- H3 0,000
Hglf- Hg Hgl2-4- 2e-^Hg+ 41- —0,04
Pb2+, Pb Pb2+ 4- 2e- ^Pb —0,126
Sn2+, Sn Sn2+ 4- 2e~ -► Sn —0,136
Agl, Ag Agl + e- Ag 4- I" —0,152
Cui, Cu Cui 4~ e- -► Cu + I- —0,185
Mo3+, Mo Mo3+ -r 3e~ Mo —0,2
Ni2+, Ni Ni2+ 4 - 2e- -4- Ni —0,23
V3+, V2+. Pt V3+ 4- e- -* V2+ -0,255
PbCl2, Pb PbCI3 4- 2e- Pb 4- 2C1- —0,268
Co2+, Co Co2+ 4- 2e- Co —0,28
PbBr2, Pb PbBr2 4~ 2e- Pb + 2Br- —0,280
T1+, Tl Tl+ 4- e--> Tl —0,336
PbSO4, Pb PbSO.(+ 2e~^Pb 4 SO42~ —0,356
PbJj, Pb Pbl3 4- 2e- -> Pb 4~ 21- - -0,365
Ti3+ Ti2+, Pt Ti3+ (- e- -> Ti2+ —0,37
Cd2+, Cd Cd2+ + 2e- -> Cd —0,403
Cr3+, Cr2+, Pt Cr3+ 4- e- -4- Cr2+ —0,41
Fe2+, Fe Fe2+ 4- 2e~ Fe —0,409
Ga3+, Ga Ga3+ + 3e- Ga ‘ —0,560
T1C1, Tl TIC1 4- e- Tl 4- Cl- —0,557
U4+ , U3+, Pt U44- 4- e- -4- U3+ —0.61
Продолжение приложения А
Электрод Реакция E° (OTH. НКЭ), В
Т1Вг, Т1 TIBr + e-~> T1+ Br- —0,658
Сг3+, Сг Cr3+ + 3e~ Cr —0,74
TH, TI TH + e--^ Tl + i- —0,753
Zn2+, Zn Zna+ + 2e~ -> Zn —0,763
TiO2+, Ti TiO2+ + 2H+ + 4e- -> Ti + H2O —0,89
Mna+, Mn Mn2+ + 2e- ч-Мп — 1,029
V2+, V V2+ + 2e- - + V — 1,12
Ti2+, Ti Ti2 + + 2e- Ti — 1,63
Al3+, Al Al3+ + 3e- Al —1,706
U3+, U U3+ + 3e- U — 1.8
Bea+, Be Be2+ + 2e- Be -1,85
Np3+, Np Np3+ + 3e- Np — 1,9
Th4+, Th Th4+ + 4e- Th — 1,90
Pn3+, Pu Pu3+ 4- 3e- Pu —2,07
AlFg-, Al AlFg- 4' 3e' - Al 4- 6F“ —2,07
Mg2+, Mg Mg2+ 4- 2e~ -*Mg —2,375
Ce3+, Ce Ce3+ A 3e- -^Cc —2,335
La3+, La La3+ 4- 3e~ La —2,37
Na+, Na Na+ 4~ e~ - > Na —2,711
Caa+, Ca Ca2+ + 2e- Ca —2,76
Sra+, Sr Sr2+ + 2e~ -> Sr —2,89
Baa+, Ba Ba2+ + 2e- Ba —2,90
K+, К K+4- e~->- К —2,924
Li+, Li Li+ + e- -> Li —3,045
Приложение Б
Численные префиксы единиц
Фактор Префикс Символ Фактор Префикс Символ
Ю18 экса Е 10-1 деци д
1015 пета П ю-2 санти с
1012 тер a Т ю-3 милли м
10s гига Г 10-6 микро мк
10е мега М 10—9 паио И
103 кило К 10-12 ПИКО п
102 гекто г ю-15 фемто ф
101 дека ~ да 10~18 атто а
Приложения 593
Приложение В
Фундаментальные константы
Скорость света в вакууме Электронвольт Постоянная Планка Константа Больцмана Константа Фарадея Число Авогадро Газовая постоянная Заряд электрона Основание натурального логарифма 1g е - " In 10 2,9979-108 м-с-1 1,6022-Ю-1’ Дж 6,6262- 10-м Дж с 1,3807-10~23 Дж-К-1 9,6485-104 Кл-моль-1 6,0220-1023 моль-1 8,3144 Дж-К~1-моль~1 1,6022-10—19 Кл 2,7183 0,4343 2,3026
Вместо послесловия
Об этой книге и об аналитической химии
Почти любой химический анализ выполняют в наше время с использованием приборов. Разве что титрование с визуальным обнаружением конечной точки можно считать «неинструментальным» методом. Но даже в этом случае при подготовке титранта и часто при отборе пробы используют весы — исторически первый и едва ли не самый распространенный аналитический прибор. Так что анализ теперь практически всегда инструментальный.
Означает ли это, что в книге с названием «Инструментальные методы химического анализа» рассмотрено все, что является содержанием современной аналитической химии? Отнюдь нет. Здесь не представлены классические методы — гравиметрический, титриметрический, приемы традиционного газового анализа. Соответственно не обсуждаются и теоретические основы методов, основанных на химических реакциях,— то, что до сих пор составляет важный раздел учебных курсов. Не так уж много внимания уделено общим вопросам аналитической химии, хотя в книге есть главы, посвященные автоматизации, компьютерам и электронным устройствам для аналитических приборов. Мало места выделено метрологии анализа, и об этом, видимо, стоит пожалеть; практически не делается попыток объективно сравнить разные методы.
И тем не менее книга Г. Юинга — очень хорошая и весьма полезная. Она вполне на уровне своего времени; упомянуты даже лабораторные роботы, а они появились лишь в 1982 году. О книге хорошо написали в своем предисловии переводчики. Можно лишь добавить, что это учебное руководство «обкатывалось» в католическом университете Сетон Холл (шт. Нью-Джерси), где автор руководства был профессором. Маленькая деталь из справочника вузов США 1986 года издания. В университете есть ЭВМ ИБМ 4381 с сорока терминалами, очень много персональных ЭВМ типа Аппл, Макинтош и ИБМ, и все компьютеры предоставлены в распоряжение студентов без ограничения времени счета, с 8 утра до 11 вечера.
В сентябре 1987 года принято постановление Центрального Комитета КПСС и Совета Министров СССР об ускоренном раз
Вместо послесловия 595
витии приоритетных направлений химической науки и технологии. Приоритетных направлений названо в постановлении восемь; одно из них прямо относится к аналитической! химии и созвучно названию этой книги: «Новые методы инструментального химического анализа, химический мониторинг и диагностика химических процессов, свойств материалов и изделий». Академия наук СССР, определяя-основные пути развития химии до конца столетия, также назвала инструментальные методы химического анализа веществ и материалов одним из наиболее важных направлений. Все это дает серьезный импульс развитию аналитической химии в Советском Союзе, значительно поднимает ее престиж и заставляет по-новому взглянуть на преподавание данной науки.
Во всяком случае, сейчас кажется необходимым основательно знакомить всех студентов-химиков с общей методологией анализа и важнейшими современными методами, включая физические и хроматографические. Видимо, пора уже перестать понимать под «физико-химическими методами анализа» только оптические (преимущественно спектрофотометрию в видимой и ультрафиолетовой областях) и электрохимические методы. Каждый студент-химик должен получить представление о газовой хроматографии, атомно-эмиссионном анализе или масс-спектрометрии, и не где-нибудь, а при изучении общего курса аналитической химии. Это нелегкая задача по ряду причин: не хватает приборов, соответствующим образом подготовленных преподавателей; методов много, а время всегда ограничено; считают, что, скажем, ядерно-физические или рентгеновские методы анализа далеки от химии; довлеет груз традиций и т. д. Тем не менее данную задачу нужно решать; альтернативы здесь, кажется, нет.
И новое издание книги Г. Юинга — прекрасное подспорье для такого решения.
’ Академик Ю. А. Золотов
Предметный указатель
Абсолютно черное тело 26 Абсорбциометрия 46 Абсорбция атомная 2, 132 Авторадиография 504 Активационный анализ 516 Амперометрическое титрование 358 кривые 360 с двумя индикаторными электродами 360-
Анализатор; см. также Поляриметр автоматический 529, 535 бездисперсионный ИК НО для клинического анализа 535 масс-спектрометрический 460, 461, 464, 467 многоканальный 244, 246 Техникон 533 центрифужный 538 электронный 265
Аналого-цифровой преобразователь 580
Анод 300
Антистоксовы линии 168
Атомизация 132
Атомно-абсорбционная спектроскопия (ААС) см. Спектроскопия Ауксохромы 78
Базовый пик 472
Базисной линии метод 122
Безокошечный проточный счетчик 509 Безэлектродная разрядная лампа как источник излучения см. Лампа Бера закон 46, 121, 223 отклонения 51
Болометр 102 Буферы кислотно-основные 318 спектрохимические 201
Вакуумные системы 450 Ван-Деемтера уравнение 391 Вина закон 26
Внутреннего отражения спектроскопия см. Спектроскопия
Внутреннего стандарта метод 194, 197
Внутренний фильтр 155
Водородная лампа см. Лампа
Вольтамперометрия 312, 331 инверсионная 374 циклическая 349
Воспроизводимость 543
Время удерживания 392, 418
Вычитания стандарта метод 547
Газовая хроматография 420
на химически связанных фазах 402
пиролитическая 404
с программированием температуры 414
Газовый пропорциональный счетчик 229, 507
Газоразрядная лампа см. Лампа Галоген-вольфрамовая лампа см.
Лампа
Генератор синусоидальных импульсов 569
Гетерометрия 184
Гибридные методы
в ГХ 421
— ЖХ 478
— масс-спектрометрии 471
Гидриды летучие 136
Гиромагнитное отношение 275
Глобар как источник излучения 101
Гомологические пары 194
Гониометр 238, 241
Градиентное элюирование см. Элюирование градиентное
Градуировочная кривая Рингбома 74, 75
Грэхема закон 353
Двойной резонанс 283
Дейтериевая лампа см. Лампа
Дельве чашка 134
Демодуляторы 554
Денситометр 194
Детекторы
в газовой хроматографии 405 пламенно-ионизационные 410 пламенно-фотоэлектрические 410 по теплопроводности 406 электродного захвата 408
— жидкостной хроматографии
по диэлектрической проницаемости 444
по ионной проводимости 445
реакционные 444
рефрактометрические 442
фотометрические 441
электрохимические 445
— инфракрасной спектроскопии 101 пироэлектрические 103 термические 101
Предметный указатель 597
фотонные 101
— масс-спектрометрии 470 фотографические 470
•— УФ- и видимой спектроскопии 68, 71
полупроводниковые 71, 229, 509
радиационные 504, 506, 509
рентгеновского излучения 228
газовые ионизационные 229
сцинтилляционные 229
твердотельные ионизационные' 230
Джансона тепловые шумы 73
Динамический интервал 543
Диоды 553; см. также Зенера дио-
ды, Демодуляторы
Дискриминатор 76
Дисперсия 24, 41
Дистилляция имитированная 420
Дифракция рентгеновских лучей 234
Дифракционная решетка 617
вогнутая 41
голографическая 39
концентрирующая отражательная
38
на пропускание 35
плоская 37
порядок 35
уравнение 35
Черни — Тернера 39
Эберта 39
Дифференциальная сканирующая ка-
лориметрия 491
Дифференциальный термический анализ 488
Диффузии коэффициент 334
Жакино выигрыш 109
/Коба метод см. Изомолярных серий
метод
Зеемана эффект 142
Зенера диоды 554
Излучательная энергия 11
Излучение
рассеянное 59
рентгеновское 221
тормозное 221
Измерительные цепи в полярографии
см. Полярография
Изобестическая точка 81
Изомолярных серий метод 84
Изотопного разбавления метод 519
Инверсионная вольтамперометрия
см. Вольтамперометрия
Индуктивно связанная плазма см.
Плазма
Интегральные микросхемы 551
Интегратор 562 'Интерфейс 588 Интерференционные полосы 121 Интерференционный светофильтр Интерферометр Майкельсона 106
Иодно-кварцевая лампа см. Лампа Ионизационные камеры 229, 507 Ионизация
31
химическая в масс-спектрометрии 454
электронами 451
Иономер 327
Ионоселективные электроды 317
коэффициент селективности 319
Ион-циклотронный резонанс 466
Искра 191
Источники
ионов для масс-спектрометрии 449
нагретые тела 26
питания 558
рентгеновские 227
Иоу и Джонса метод см. Молярных отношений метод 84
Калибровка
ИК-спектрофотометров 113
рН-метров 327
Калориметр дифференциальный сканирующий 492
Капающий ртутный электрод см.
Электрод
Капилляр в полярографии 335
Катарометр 406
Катионизация в масс-спектрометрии см. Масс-спектрометрия
Катод 300
Квантометры 196
Кларка кислородный электрод см.
Электроды
Ковача индекс удерживания 418
Колебания
валентные 114
деформационные 114
Колонка
в газовой хроматографии 399
— жидкостной хроматографии 429 капиллярная 399
Колориметрия 46
Комбинационного рассеяния (КР) спектроскопия см. •Рамановская спектроскопия Компьютеры 584 Кондуктометрия 312, 379 радиочастотная 385
598 Предметный указатель
Концентрационные цепи 316
Концентрирование см. Предварительное электролитическое концентрирование
Коттона эффект 216
Коттреля уравнение 334
Коэффициент
диффузии 334
емкости 388
поглощение 48
разделения 388
Край поглощения см. Спектрометрия Кривая спада свободной индукции 253
Круговой дихроизм 216
Ксеноновая лампа см. Лампа
Кулонометрия 312, 366
при постоянной силе тока 368
— постоянном потенциале 368
схема установки 569
Кулонометрическое титрование 368
Кювета
графитовая 135
разборная 119
Лазер 28
как источник возбуждения 203
на красителях 29
Лампа
безэлектродная разрядная 140
водородная 28
газоразрядная 27
галоген-вольфрамовая 68
дейтериевая 28
иодио-кварцевая 68
ксеноновая 28
накаливания 100
ртутная высокого давления 28
с полым катодом 27, 137
Ларморова частота 275
Ловушка Фарадея 470
Люминесценция молекулярная 149
Магнитный резонанс 274
Масс-спектрометр 406, 460
времяпролетный 464 нон-циклотрониый с фурье-преобразованием 467 квадрупольный 461
с двойной фокусировкой 460
Масс-спектрометрия 449 бомбардировка быстрыми атомами 456 вторичных иоиов 455 катионизация 456 квадрупольная 6, 461 с фурье-преобразованием 466 тандемная 480
Маттауха — Герцога геометрия в масс-'спектрометрах 460, 462 Мембрана 317
Мессбауэровская спектроскопия см. Спектроскопия
Метастабильные ионы в масс-спектрометрии 473
Микрокомпьютер 11
Модулятор 59 электрооптический Покельса см. Покельса модулятор
Модуляция волновая 65
Молекулярные иатекатели см. Точечные диафрагмы
Молекулярные сита 437
Молярных отношений метод 84
Монохроматор 32
двойной 60
рентгеновский 233
Мультиплексор 568
Напряжение пробоя 584
Нарушенного полного внутреннего отражения (НПВО) метод 123 Насосы для хроматографии 429 Нейтронно-активационный анализ 516
Нейтронные потоки 514
Нернста
уравнение 302
штифт как источник излучения 101
Нефелометрическое титрование 186
Нефелометрия 183
Нира — Джонсона геометрия в масс-спектрометрах 460
Обратимость химических реакций 309
Оверхаузера ядерный эффект' 285
Оптическая активность 216
Оптическая вращательная дисперсия 209, 213
Оптическая плотность 47
аддитивность 86
в атомно-эмиссионной спектроскопии 194
Оптические материалы в ИК-области 99 — УФ- и видимой области 33
Осциллометрия 385
Перенапряжение 311
Пересчетиые схемы 510
Период полураспада 502
Пиролитическая газовая хроматография см. Газовая хроматография Пироэлектрический детектор см. Детекторы Плазма
Предметный указатель 599
индуктивно связанная 198
как источник возбуждения 198
постоянного тока 198
Пламена
в ААС 132
как источник возбуждения 200
Планка закон 26
Поглощение
атомное 132
коэффициент 48
критический край 224, 231
микроволновое 125
молекулярное 45
рентгеновское 223
Погрешность фотометрических измерений 72
Показатель преломления 24
Покельса модулятор 218
Полихроматор 32
Полоса пропускания 41, 42
Полупроводники 552
Поляризация
излучения 24
концентрационная 311
электродов 310
Поляризуемость 169
Поляриметр 25, 210
Поляриметрия 209
Полярограф 337
трехэлектродный на операционных усилителях 346
Полярография 312, 331
высокоскоростная 348
дифференциальная импульсная 351, 363
измерительные цепи 347
нормальная импульсная 353
органических соединений 357
переменнотоковая 353
с разверткой потенциала 337
стандартных добавок метод 356
Помехи в ААС 144
Порошковая камера Дебая—Шерера 238
Потенциал
жидкостного соединения 306
полуволны 341
полуэлемента 302
стандартный электродный 302
формальный 304
ячейки 301
Потенциометрическое титрование 325
Потенциометрия 312
Потенциостат 365
Правильность 543
Предварительное электролитическое концентрирование 373
Предел обнаружения 542
в спектроскопии 144, 203
Предколонка 429
Преобразователь 10, 564; см. также Аналого-цифровой преобразователь, Цифро-аналоговый преобразователь Прерыватель 59 Призма Литтрова 33 Программирование' 586 температуры 414
Пропускание 48
Проточно-инжекционный анализ 531
Проточные системы 531
Прямая инжекционная энтальпимет-рия 497
Разрешающая сила 43
Разрешающая способность 42
Разрешение
в масс-спектрометрии 460
— хроматографии 394
Радиоактивность 501
источники рентгеновского излучения 227
Радиоактивные метки 516
Радиочастотная кондуктометрия см.
Осциллометрия
Рамановская спектроскопия 149, 167, 170
Распыление 272
Рассеяние излучения 59, 182
Растворители
в ИК-спектроскопни 118
— УФ-спектроскопии 49
Регистры 576
Рефракция 24
Решетки дифракционные вогнутая 41 голографическая 39 концентрирующая отражательная 38 отражательная 37 плоская 37 порядок 35 уравнение 35
Черни—Тернера 39
Эберта 39
элемент 37
Роботы 539
Роуланда круг 41, 234
Рэлея — Ми теория 182
Рэлея рассеяние 182
Рэндлса — Шевчика уравнение 349
Самописцы 572
Самотушение флуоресценций см.
Флуоресценция
600 Предметный указатель
Сверхиотенциал см. Перенапряжение Светофильтры 31
Сдвигающие реагенты 288
Селеновая ячейка 567 Сервомеханизмы 751 Сечение нейтронного захвата 518, Силанизация 399
Спектр
атомный 19 молекулярный 20 нормальный 42 электромагнитный 17
Спектрограф 32
Спектрометрия
абсорбционная рентгеновская 231 край поглощения 224, 231
Спектрометры
рентгеновские с энергетической дисперсией 242 электронные 264
Спектроскоп Бриланда 193 Спектроскопия
атомно-абсорбционная 132; см. также Помехи в ЛАС атомно-эмиссионная 189 внутреннего отражения 123 ионная 251 искровая 191
когерентная антистоксова КР 171
магнитного резонанса 274
в твердых веществах 293
высокого разрешения 277 иа ядрах углерода-13 191 ----фосфора-31 290 ----фтора-19 290 низкого разрешения 277
с фурье-преобразованнем 291 мессбауэровская 521 оже-электронная 529 оптико-акустическая см. Фотоаку-стическая рамановская (КР) 149, 167 фотоакустическая 175 фотоэлектронная рентгеновская 253
— ультрафиолетовая 258 электронная 251 электронная для химического анализа (ЭСХА) 251, 253 электронного удара 258
Спектрофлуориметры 156 Спектрофотометр 33 быстросканирующий 105 двухволновой 63 двухлучевой 57 для дальней ИК-области 112 однолучевой 62
с фурье-преобразованием 106 Спектрофотометрия 46
видимая 45
производная 65
УФ 45
Спин-декаплер 284
Спин-спиновое взаимодействие 280
Стандартные образцы 544 Стандартный потенциал 302 Стандартных добавок метод 545 Стеклянный электрод 317 потенциал асимметрии 318 щелочная ошибка 319
Стоксовы линии 168
Счет фотонов 75
Счетчик
Гейгера — Мюллера 230, 508 двоичный 579 фотонов 157 электронный 576
Теоретическая тарелка 390
Термистор 102
Термический анализ 485
Термовесы 487
Термогравиметрия 485
по производной 488
Термометрическое титрование 194 Термопара как детектор в газовой хроматографии 102 Термотитриметрия 494 Тиндаля рассеяние 18'2
Титрдторы автоматические 328 Титрование
амперометрическое 358 кондуктометрическое 384 кулонометрическое 368 нефелометрическое 186 потенциометрическое 325 термометрическое 494 турбидиметрическое 186 фотометрическое 89
Ток
диффузионный 339 заряжения 339 остаточный 339 утечки 553
Тормозное излучение 221
Точечные диафрагмы 452 Транзисторы 555 биполярные 555 полевые 557
— с изолированным затвором 558 Триггеры 576
Турбидиметрическое титрование 186 Тушение флуоресценции концентрационное 155
Предметный указатель 601
самотушение 155
химическое 155
Удерживаемый объем 392, 418
Уитстона мост 382
Умножители электронные 267, 470
Уравнение
Ван-Деемтера 391
Ильковича 336
Коттреля 334
Нернста 302
ВэнОлса — Шевчика 349
Усилитель операционный 70, 346, 560
Устройства логические 547
И 547
ИЛИ 547
инверторы 547
Фелжетта выигрыш ПО
Ферментный Электрод 322
Фика закон 334
Фильтры
в рентгеновской спектроскопии 226
заграждающие 570
Флуоресценция
атомная 201
молекулярная 149
рентгеновская 239
самотушение 155, 201
синхронная 162
тушение см. Тушение флуоресценции
Флуориметрия атомная 201 молекулярная 149 синхронная 162
Флуориметры 158
Фон
в атомной абсорбции 140
радиоактивный 510
Фононы 175
Формальный потенциал см. Потенциал Фосфоресценция 145, 163 синхронная 167
Фосфориметрия 163
Фосфорнметры 166
Фосфороскоп 166
Фотодиоды 72
Фотометрическое титрование 89
Фотометрия пламени 200
Фотонов счет 75
Фотопроводящие ячейки 567
Фототранзисторы 567
Фотоумножители, ФЭУ 68, 267, 470
Фотоэлектроколориметры 54
Фотоэлементы 566
вакуумные 72
вентильные см. Селеновая ячейка
газонаполненные 72
Фрагментационные картины 470
Френеля параллелепипед 218
Фурье-преобразоваиие
в ИК-спектроскопип 106
— масс-спектрометрии 486
— ЯМР 291
Химический сдвиг
в мессбауэровской спектроскопии 522
— ЯМР 277
Химически привитые фазы 433
Хроматография 387
бумажная 387
время удерживания 392, 418
газовая 396
гель-проникающая 437
жидкостная 427
ионная 446
ионообменная 433
ион-парная 437
теоретическая тарелка 390
теория подвижности 390
тонкослойная 387
удерживаемый объем 392. 418
Хромофоры 78
Хроноамперометрия 312
Хронопотенциометрия 312
Цеолиты 437
Цепи измерительные в полярографии 247
Циклическая вольтамперометрия см.
Вольтамперометрия
Циклотронная частота 466
Цифро-аналоговый преобразователь 580
Цифровая электроника 573
Чувствительность 542
Шум 72
дробовой, флуктуационный 72 тепловые Джонсона, сопротивления 73
Экранирование анизотропное 299
Электровыделение 365
Электрод
в спектральном анализе 191
второго рода 307
индикаторный 306
ионоселективный мембранный 317
с двойными мембранами 321
— жидкими мембранами 321
602 Предметный указатель
— твердыми мембранами 323 стеклянные 317
капающий ртутный 334
кислородный Кларка 358
первого рода 306
платиновый вращающийся 359
поляризованный 310
сравнения 307, 324
стандартный водородный 303
третьего рода 308
Электроника 551
Электронная спектроскопия 251
Электронно-зондовый микроанализ
246
Электронные схемы 551
Электронный парамагнитный резо-
нанс (ЭПР) 294
Электропроводность 380
Эллиптичность 215
Элюирование градиентное 439
Эмиссия атомная 189
Эмиттер 555
Энергетические анализаторы 256
Энергия связи 253
Энтальпиметрия 497
Эффект
внутреннего фильтра 155
Зеемана 142
Коттона 216
тяжелого атома 164 '
Эффективная полоса пропускания 42
Ядерно-физическне методы 501
Ядерный магнитный резонанс (ЯМР) 273 Ячейка
с запирающим слоем см. Селеновая ячейка
Фарадея оптическая 217
Оглавление
Предисловие переводчиков.......................................... 5
Предисловие автора................................................ 7
Глава 1. Введение................................................. 9
Электроника.....................................................10
Современные микрокомпьютеры.....................................11
Серийные приборы...............................................12
Замечание об единицах ......................................... 12
Литература....................................................12
Глава 2. Введение в оптические методы............................15
Природа излучательной энергии.................................15
Спектральные области..........................................18
Взаимодействие с веществом....................................18
Атомные спектры................................................19
Молекулярные спектры...........................................20
Молекулярные спектры поглощения............................... 22
Фогоакустическая и фототермическая спектроскопия...............22
Флуоресценция..................................................23
Фосфоресценция.................................................23
Рамановская спектроскопия (спектроскопия КР)...................23
Рефракция......................................................24
Поляризация и оптическая активность 24
Источники излучения .......................................... 26
Лазеры.........................................................28
Монохроматизация...............................................31
Монохроматор...................................................32
Разложение излучения призмами..................................33
Разложение излучения дифракционными решетками . . .35
Дисперсия......................................................41
Разрешающая способность........................................42
Задачи.........................................................44
Литература.....................................................44
Глава 3. Поглощение излучения. Ультрафиолетовая и видимая области 45
Количественные закономерности..................................46
Коэффициент поглощения.........................................48
Кажущиеся отклонения от закона Бера............................51
Приборы........................................................54
Двухлучевые спектрофотометры для УФ- и видимой областей ... 57
Однолучевые спектрофотометры..........................., 62
Двухволиовые спектрофотометры..................................63
Производная спектрофотометрия..................................65
Источники излучения ........................................ 68
Детекторы..................................................... 68
604 Оглавление
Погрешность фотометрических измерений..........................72
Счет фотонов...................................................75
Эксплуатация спектрофотометра ................................ 76
Способы повышения точности измерений...........................77
Практическое применение........................................77
Качественный анализ 80
Определение соотношения лиганд/металл в комплексном соединении 83
Количественный анализ..........................................85
Аддитивность оптических плотностей ........................... 86
Фотометрическое титрование.....................................89
Задачи.........................................................91
Литература.................................................... 94
Глава 4. Поглощение излучения. Инфракрасная область...............96
Приборы........................................................99
Спектрофотометры...............................................ЮЗ
Быстросканирующпе спектрофотометры............................105
Интерференционные (с фурье-преобразованнем) спектрофотометры 106
Калибровка н стандартизация....................................ИЗ
Практическое применение.......................................114
Подготовка проб...............................................118
Количественный анализ.........................................121
Спектроскопия внутреннего отражения......................... 123
Поглощение в микроволновой области............................125
Задачи........................................................129
Литература....................................................130
Глава 5, Атомно-абсорбционная спектроскопия......................132
Атомизация.................................................. . 132
Источники излучения ......................................... 136
Поправка на поглощение фона...................................140
Помехи........................................................144
Задачи........................................................146
Литература....................................................147
Глава 6. Молекулярная люминесценция: флуориметрия, фосфориметрия и рамановская спектроскопия...................................149
Флуориметрия..................................................149
Спектрофлуорпметры............................................156
Флуориметры...................................................158
Применения....................................................159
Фосфориметрия.................................................163
Фосфориметры..................................................166
Рамановская спектроскопия ................................... 167
Задачи........................................................171
Литература....................................................174
Глава 7, Фотоакустическая спектроскопия..........................175
Применение....................................................179
Задача........................................................181
Литература....................................................181
Глава 8. Рассеяние излучения.....................................182
Рэлеевское рассеяние ... .....................................182
Практическое применение.......................................185
Турбидиметрическое н нефелометрическое титрование.............186
Оглавление 605
Задачи........................................................187
Литература....................................................188
Глава 9. Атомная эмиссионная спектроскопия.......................189
Возбуждение проб..............................................190
Приборы................:......................................192
Количественный анализ.........................................194
Квантометры...................................................196
Возбуждение в плазме..........................................198
Возбужденно в пламени.........................................200
Атомная флуоресценция..........................................201
Сравнение плазменных и родственных им методов.................203
Задачи........................................................206
Литература..................................................... 208
Глава 10. Поляриметрия, оптическая вращательная дисперсия и круговой дихроизм ................. .......................................
Поляриметры....................................................
Оптическая вращательная дисперсия и круговой дихроизм . . . . Задачи.................................................-. . .
Литература.....................................................
Глава 11. Рентгеновские методы.....................................
Поглощение рентгеновских лучей.................................
Монохроматические источники рентгеновских лучей................
Детекторы рентгеновского излучения.............................
Рентгеновские монохроматоры....................................
Дифракция рентгеновских лучей..................................
Рентгеновская флуоресценция....................................
Рентгеновские спектрометры с энергетической дисперсией .... Электронно-зондовый микроанализ................................
Задачи .....................................................
Литература.....................................................
Глава 12. Электронная и ионная спектроскопия ......................
Рентгеновская фотоэлектронная спектроскопия (РФЭС).............
Ультрафиолетовая фотоэлектронная спектроскопия (УФ-ФЭС) . . Спектроскопия электронного удара (СЭУ).........................
Оже-электронная спектроскопия (ОЭС)............................
Приборы .......................................................
Спектроскопия ионного рассеяния (СИР)..........................
Задачи ........................................................
Литература.....................................................
Глава 13. Спектроскопия магнитного резонанса
Сканирующие спектрометры ЯМР....................................
ЯМР высокого разрешения.........................................
Химический сдвиг . . . . :.................................
Сппн-спнновое взаимодействие............................. ....
Приборы для ЯМР.................................................
Двойной резонанс........................................: :
Применение ПМР .................................................
Сдвигающие реагенты в ЯМР.......................................
ЯМР других элементов....................................'. . :
ЯМР с фурье-преобразованием (ФП) ...............................
Электронный парамагнитный резонанс (ЭПР)........................
Задачи .........................................................
Литература......................................................
209 210
213 219
220
221
223 226
228 233
234 239
242 246
248 249
251
253 258
258
259 264
271
273
273
274
275 277
277
280
281
283
285
288
290
291
294
298
299
606 Оглавление
Глава 14. Введение в электрохимические методы....................
Реакция в ячейке .............................................
Соглашение о знаках . . ..................................
Обратимость...................................................
Поляризация...................................................
Перенапряжение................................................
Электроапалитические методы . ........................ •
Задачи .......................................................
Литература....................................................
Глава 15. Потенциометрия..................................... •
Концентрационные цепи.........................................
Ионоселективиые мембранные электроды..........................
Электроды с жидкими мембранами................................
Электроды с двойными мембранами г.............................
Электроды с твердыми мембранами...............................
Электроды сравнения................................... . . .
Потенциометрическое титрование ...............................
Аппаратура ...................................................
Задачи ............................ ..........................
Литература....................................................
300
300
309
309
310
311
312
313
313
314
316
317
321
321
323
324
325
327
329
330
Глава 16. Вольтамперометрия, полярография и родственные методы . .331
Диффузионный ток............................................ 333
Капающий ртутный электрод.................................... 334
Полярография с разверткой потенциала 337
Форма полярографической волны............................. ... 340
Циклическая вольтамперометрия ................................349
Дифференциальная импульсная полярография.....................,351
Качественный анализ............................... . . . : . 353
Количественный анализ.........................................35о
Полярография органических соединений 357
Другие амперометрические методы........................ ...... 358
Задачи...................................................... 361
Литература................................................... 364
Глава 17. Электровыделение и кулонометрия........................365
Кулонометрия..................................................366
Предварительное электролитическое концентрирование .......... 373
Инверсионный анализ..........................................• 374
Задачи....................................................... 376
Литература....................................................378
Глава 18. Кондуктометрия..................................... . . 379
Теория....................................................... 380
Аппаратура....................................................382
Применение............................................. ..... 384
Кондуктометрическое титрование........................... • . 384
Радиочастотная кондуктометрия (осциллометрия) ............... 385
Задачи....................................................... 386
Литература..................................................- • 386
Глава 19. Введение в хроматографию . '...........................387
Теория хроматографической подвижности . : ............390
Задачи........................................................394
Литература....................................................395
Глава 20. Газовая хроматография
. 396
Оглавление
607
Неподвижная фаза............................................. . 396
Неподвижная жидкая фаза................................. ...... 399
Газ-носитель................................................. 403
Ввод образца................................................. 403
Твердые образцы . ‘......................................... . 404
Детекторы......................................................405
Двойное детектирование . :................................... . 413
Программирование температуры . 414
Серийные газовые хроматографы 416
Качественный анализ .... 416
Количественный анализ 420
ГХ в гибридных методах..........................................421
Задачи.........................................................423
Литература.....................................................425
Глава 21. Жидкостная хроматография................................427
Виды жидкостной хроматографии..................................430
Жидко-твердофазная хроматография (ЖТХ) . . . .'................430
Жидко-жидкостная хроматография (ЖЖХ)...........................431
Ионообменная хроматография.....................................433
Иои-парная хроматография (ИПХ).................................437
Молекулярные сита н гель-проннкающая хроматография (ЭХ) . . 437
Градиентное элюирование........................................439
Детекторы......................................................441
Задачи.........................................................447
Литература.....................................................448
Глава 22. Масс-спектрометрия......................................449
Аппаратура.....................................................449
Источники ионов................................................449
Масс-анализаторы . :......................................... : 158
Приборы с двойной фокусировкой.................................459
Квадрупольные масс-анализаторы.................................461
Времяпролетные масс-анализаторы................................464
Масс-спектрометрия с фурье-преобразованпем (МСПФ)..............466
Масс-спектрометрические детекторы..............................470
Качественный анализ............................................474
Количественный анализ..........................................475
ГХ/МС и ЖХ/МС..................................................476
Тандемная масс-спектрометрия...................................480
Задачи . . . ;...........................................482
Литература.....................................................484
Глава 23. Термические методы анализа..............................485
Термограриметрический анализ (ТГА) - : :......................485
Дифференциальный термический анализ (ДТА)......................488
Термотитриметрия...............................................494
Энтальпиметрия................................................: 497
Задачи...............................•.........................498
Литература.....................................................500
Глава 24. Ядерно-физические методы................................501
Радиоактивность .............................................. 501
Детекторы радиации.............................................504
Фоновая радиация ............................................. 510
Пересчетные схемы..............................................510
Измерение нейтронных потоков...................................514
Радиоактивные метки............................................516
608 Оглавление
Активационный анализ..........................................516
Изотопное разбавление ....................................... 519
Мёссбауэровская спектроскопия.................................521
Меры предосторожности.........................................525
Задачи....................................'...................525
Литература....................................................528
Глава 25. Автоматические анализаторы..............................529
Автоматизация и механизация...................................529
Проточные системы.............................................531
Периодические процессы........................................533
Литература.................................................. 540
Глава 26. Общие вопросы анализа...................................541
Чувствительность и предел обнаружения.........................542
Воспроизводимость и правильность..............................543
Сравнение со стандартными образцами ... 544
Стандартные добавки ........................................ 545
Графики.......................................................547
Задачи........................................................549
Литература....................................................550
Глава 27. Электронные схемы в аналитических приборах.................551
Полупроводники................................................552
Диоды.........................................................553
Транзисторы...................................................555
Полевой транзистор (ПТ).......................................557
Источники питания ......................................... 558
Операционные усилители......................................: 560
Применение операционных усилителей в преобразователях .... 564
Фотоэлементы...................................................566
Схемы составных усилителей.....................................568
Генераторы синусоидальных импульсов............................569
Сервомеханизмы.................................................571
Самописцы......................................................572
Цифровая электроника...........................................573
Переходные преобразователи.....................................580
Задачи.........................................................582
Литература.....................................................583
Глава 28. Компьютеры в аналитических приборах....................584
Архитектура ЭВМ..............................................584
Присоединение ЭВМ к приборам.................................586
Готовые интерфейсы...........................................588
Программирование.............................................588
Предостережение . . . .<.....................................589
Литература ..................................................589
Приложение А.....................................................590
Стандартные потенциалы.......................................590
Приложение Б.....................................................592
Численные префиксы единиц....................................592
Приложение В.....................................................593
Фундаментальные константы....................................593
Вместо послесловия.......................................... 594
Предметный указатель.............................................596