/






























































































































































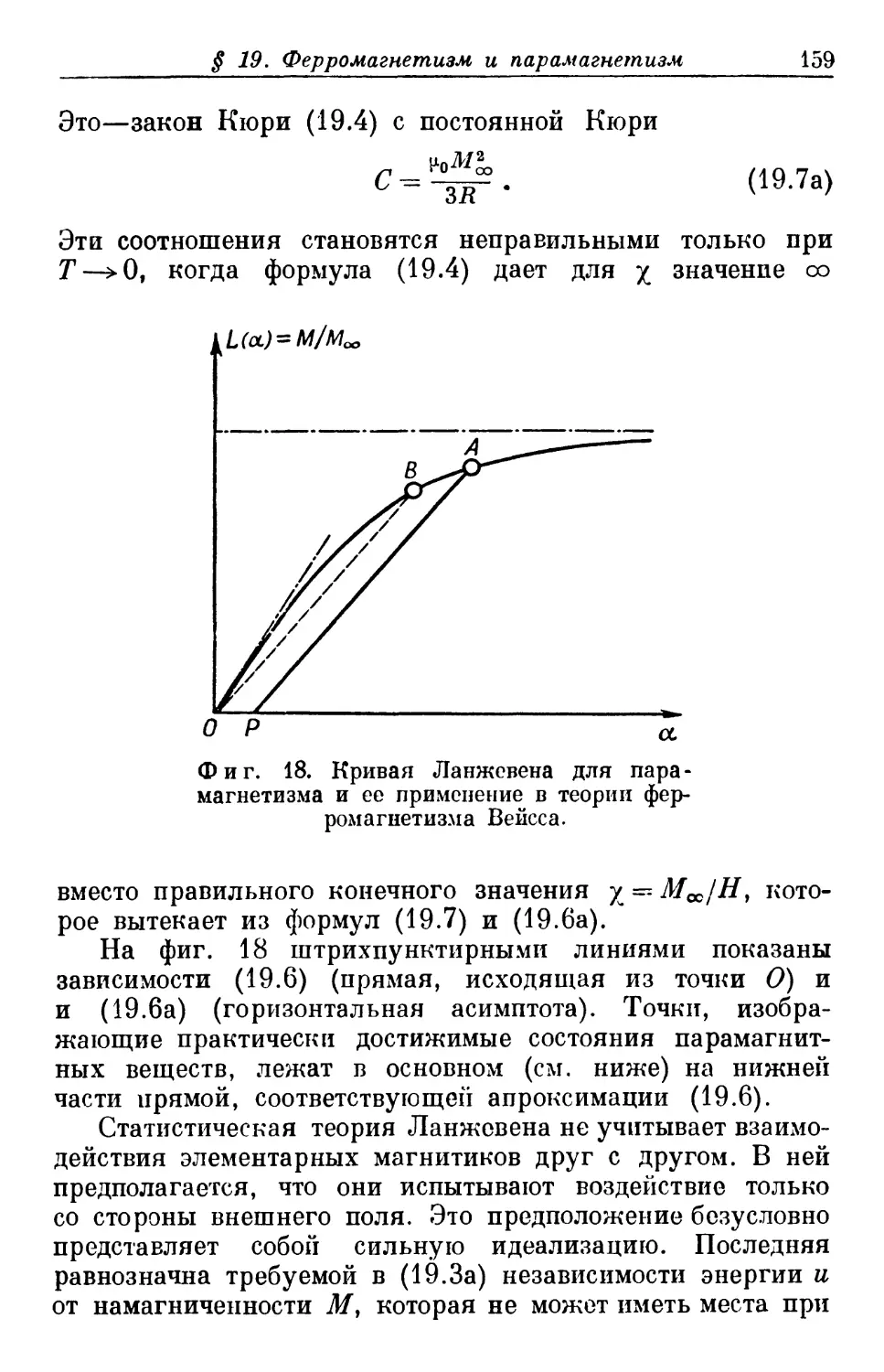



































































































































































































































































































































Text
А. ЗОММЕРФЕЛЬД
ТЕРМОДИНАМИКА
И СТАТИСТИЧЕСКАЯ
ФИЗИКА
Перевод с немецкого
В. Л. БОНЧ-БРУЕВИЧА
и В. Б. САНДОМИРСКОГО
И * Л
ИЗДАТЕЛЬСТВО
ИНОСТРАННОЙ ЛИТЕРАТУРЫ
Москва, 19 5 5
VORLESUNGEN
UBER THEORETISCHE PHYSIK
BAND V
THERMODYNAMIK
UND STATISTIK
von
ARNOLD SOMMERFELD
WIESBADEN
1962
от РЕДАКЦИИ
Предлагаемая вниманию читателя книга А. Зоммер-
фельда посвящена изложению основ термодинамики п
статистической физики. Она является пятым,
хронологически последним, томом серии «Лекции по теоретической
физике» ^) п вышла уже после смерти автора. Зоммерфельд
не успел закончить подготовку книги к печати, и это
было выполнено Бонном и Мейкснером.
По сравнению с другими томами, которые, как правило,
содержали изложение избранных вопросов данной
дисциплины, в настоящей книге круг рассматриваемых проблем
отличается значительно большей полнотой.
Как обычно для классических курсов термодинамики,
автор ведет изложение аксиоматическим путем, начиная
с формулировки «начал»; статистическое обоснование
дается лишь значительно позднее. Для данной книги,
как и для других томов этого курса, характерно
стремление к максимальной конкретизации получаемых
результатов. Расчеты там, где это возможно, доводятся до числа
и до непосредственного сравнения с опытом; в ряде случаев
рассматриваются задачи, имеющие не только теоретический
(или иллюстративный), но и непосредственный практический
интерес. Так, например, подробно рассматривается
индикаторная диаграмма паровой машины с водяным паром
в качестве рабочего тела, а не с идеальным газом, как это
делается в обычных курсах термодинамики; в одной из
задач разбирается вопрос об отоплении помещений, ко-
^) Из этой серии уже выпущены: т. I, Механика A947); т. II,
Механика деформируемых сред A954); т. IV, Оптика A953); т. VI,
Дифференциальные уравнения в частных производных физики A950).
От редакции
торый оказывается далеко не тривиальным, и т. д. Однако
некоторые важные вопросы термодинамики изложены
недостаточно полно. В особенности это относится к теории
фазовых переходов, в которой совершенно не освещена
важная и актуальная проблема фазовых переходов
второго рода.
Статистическая физика излагается в три этапа:
элементарная кинетическая теория газов, общие методы
статистики (метод Больцмана и метод ансамблей Гиббса),
ироблемы кинетики. В этой последней части выводится
кинетическое уравнение для идеального газа и
рассматривается его приближенное решение методом моментов.
Следует отметить, что в курсах и монографиях, имеющихся
на русском языке, этот круг задач еще не освещен с
достаточной полнотой.
Такое построение курса с методической стороны
представляется удачным. Вместе с том «статистическая» часть
книги страдает наибольшей неровностью изложения.
Возможно, это связано с тем, что она подвергалась наибольшей
доработке без участия самого автора. К сожалению, весьма
неполно рассмотрена вся теория флуктуаций. Нет даже
упоминания о новых методах решения равновесных и
кинетических задач, связанных с введением «цепочек»
функций распределения (работы Н. Н. Боголюбова, Борна и
Грина и др.).
Как и в остальных томах своей серии, Зоммерфельд
не проявляет должной объективности в исторических
ссылках, упоминая в основном лишь о работах немецких
ученых. Достижения же ученых других стран, как правило,
остаются в тени. Несмотря на отмеченные недостатки,
книга представляет несомненный интерес как оригинальное
и глубокое изложение термодинамики и статистической
физики, написанное крупным немецким ученым и
талантливым педагогом.
ПРЕДИСЛОВИЕ АВТОРА
Термодинамика представляет классический пример
аксиоматически построенной науки. В противоположность
классической механике революция, произведенная квантовой
теорией, не поколебала основных положений термодинамики.
За сто лет ее существования в термодинамику было
сделано лишь несколько значительных вкладов. Это—тепловая
теорема Нернста(§ 12), теория разбавленных растворов (§ 15),
применение второго начала термодинамики к теории
электричества и магнетизма (§ 18 и 19). Весьма перспективным
обобщением классической «равновесной» термодинамики
представляется нам термодинамика необратимых процессов
(§ 21), основанная на соотношениях взаимности Онзагера
и охватывающая реальные процессы, текущие с конечной
скоростью. По свидетельству Планка, имеющемуся в его
воспоминаниях, даже Кирхгоф хотел ограничить понятие
энтропии обратимыми процессами. Твердая уверенность
в общности этого понятия, которую Планк выразил уже
в своей диссертации, привела его в 1900 г. к его закону
излучения и к теории квантов.
Мы не будем проводить аксиоматический метод
настолько последовательно, чтобы пытаться построить изложение
на основе возможно меньшего числа аксиом. Это выполнил
Каратеодори в своем доказательстве второго начала
термодинамики. В настоящей книге будет дано понятие о работе
Каратеодори, однако мы не можем предпочесть ее
рассуждению Карно—Клаузиуса. В последнем содержится так много
поучительных идей, что мы считаем его, особенно в
начальной стадии изложения, совершенно незаменимым. В том,
что это рассуждение оперирует с техническими
представлениями, мы видим скорее пpеимyщecтвo, чем недостаток.
Ведь и термодинамика первоначально развилась в связи
с задачами конструирования паровых машин.
Предисловие автора
Существует известное соперничество между методом
циклических процессов и методом термодинамических
потенциалов. Первому методу вследствие его наглядности
оказывают особое предпочтение в технике. Однако мы
будем почти везде пользоваться последним методом. Он
допускает значительно более сжатое математическое
рассмотрение, свободное от всякого произвола, который
содержится в искусственно придумываемых циклических
процессах. Мы будем рассматривать четыре потенциала Гиббса
(§ 7) как равноправные, хотя свободная энтальпия (часто
называемая просто термодинамическим потенциалом) имеет
наиболее важные применения.
Привлеченный нами экспериментальный материал очень
неполон. В случае реальных газов мы ограничиваемся
уравнением Ван-дер-Ваальса, которое, несмотря на его
простую форму—только с двумя эмпирическими
постоянными,—удовлетворительно отражает основные черты
парообразного и жидкого состояний. Такую же роль по
отношению к ферромагнетизму играет теория Вейса с ее
единственной константой внутреннего поля в
ферромагнетике. Критику этих упрощенных теорий следует отнести
в специальные труды.
В первоначальных университетских лекциях я отвел
больше места статистике, чем чистой термодинамике,
поскольку первая благодаря своей связи с квантовой
теорией меня лично больше привлекала. Фактически в
современном изложении квантовую теорию следует в принципе
исключить, лишь изредка привлекая ее в дополнение к
статистике Больцмана. Поэтому в настоящей книге гл. I и
П, в которых излагается термодинамика, количественно
преобладают над гл. III—V, посвященными статистике.
Статистика Ферми излагается лишь в небольшой части,
касающейся электронов в металле.
В гл. III дано предварительное введение в статистический
метод, насколько это можно сделать элементарным путем.
Этому служат приводимые здесь примеры (постоянные
Ван-дер-Ваальса, теория парамагнетизма Ланжевена),
несколько восполняющие пробелы, допущенные в
термодинамике. Как важнейший пример статистических флуктуа-
циояных явлений рассматривается броуновское движение
о связи с теорией крутильных весов. Проблема длины
Предисловие автора
свободного пробега представлена лишь весьма схематично,
так как она принадлежит к труднейшим задачам статистики.
Материалы, изложенные в гл. IV, можно считать
вершиной наших статистических исследований. Я думаю, что метод
ячеек Больцмана, хотя он и ограничен (по крайней мере
в настоящее время) только стационарными состояниями,
превосходит по плодотворности и смелости своего соперника—
динамический метод кинетического уравнения Больцмана.
Поэтому в первых параграфах этой главы излагается метод
ячеек в первоначальной больцмановской форме, в которой
молекулы газа рассматриваются как физическая реальность.
В § 32—35 мы освобождаемся от недостатков этого метода,
вводя дискретные квантовые уровни энергии. Однако при
этом еще не возникает собственно квантовая статистика.
В последней метод Больцмана в своей первоначальной
форме—распределение частиц по состояниям—становится
несостоятельным, так как с квантовой точки зрения
молекулы газа не различимы. Напротив, в квантовой теории
состояния суть первично заданные объекты. Объектами
новой статистики являются наборы чисел, характеризующие
•заполнение различных состояний неиндивидуализируемы-
ми частицами. Это излагается в § 36 и 37.
Соответствующие примеры приведены в § 38 (газ световых квантов)
и в § 39 (электроны в металле).
Может быть нуждается в оправдании тот факт, что мы
не основываемся на этой собственно квантовой статистике
состояний с самого начала, а начинаем с явно устаревшей
больцмановской статистики частиц. Это объясняется
дидактическими соображениями. Первоначальная больцманов-
ская статистика сделала уже так много и является столь
наглядной, что, повидимому, она и впредь будет
необходимой основой для понимания новой статистики состояний.
Гл. V по сравнению с гл. IV сделана возможно более
короткой. Необходимые здесь модельные представления
являются гораздо более cпeциaльными, а окончательные
вычисления—гораздо более трудными, чем в методе ячеек.
Правда, в этой области имеется созданная Гильбертом
пocлeдoвaтeльнaя теория необратимых процессов (таких,
как внутреннее трение, теплопроводность и т. д.), над
которой неоднократно работали Максвелл и Больцман,
однако без полного успеха. Кроме того, математически
Предисловие автора
разработан метод Энскога и Чэпмена и результаты его
доведены до сопоставления с данными наблюдений. Однако
эти применения далеко выходят за рамки настоящей
книги. Они отчетливо показывают, как трудна при точном
математическом рассмотрении проблема длины свободного
пробега, лишь обрисованная в гл. III. Наше изложение
ограничено освещением основной проблемы, поставленной
Больцманом в его статистических работах: объяснить
противоречие между механикой обратимых процессов и
вторым началом термодинамики.
Арнольд Зоммерфельд.
из ПРЕДИСЛОВИЯ ИЗДАТЕЛЕЙ
Зоммерфельду не суждено было полностью закончить
свой курс теоретической физики. Во время работы над т. V,
посвященным термодинамике и статистике, который
мыслился автором как последний, он умер в результате
несчастного случая. По его желанию нижеподписавшимся
было поручено дополнение и издание этого тома.
Раздел, посвященный термодинамике, был в основном
закончен. Однако автор уже не успел прочесть § 21,
который был вчерне написан одним из нас. Вновь переработан
§ 8, который имелся в двух редакциях.
Разделы, посвященные кинетической теории и
статистической механике (вплоть до § 35), были готовы, было
также довольно подробно разработано содержание § 37.
Однако из многочисленных бесед можно заключить, что
автор не был вполне удовлетворен этим параграфом. Мы
пытались исправить это, включив § 36, посвященный
методам Гиббса, но сознавали, что автор, возможно,
предпочел бы другой путь. Рубрикация и содержание § 38—40
были обсуждены с автором, однако они не были
сформулированы письменно.
О гл. V, кроме замечаний в предисловии, никаких
записей не было. Автор еще не приступил к написанию
этой главы и иногда говорил, что предполагает включить
ее в следующее издание. Что касается электронной теории
металлов в § 45, то мы в основном следовали первой
части известной книги Зоммерфельда и Бете^).
1) Г. Бете, А. Зоммерфельд, Электронная теория
металлов, ОНТИ, 1938.—Прим. перев.
10 Иэ предисловия издателей
Задачи частично заимствованы из сборника,
составленного автором. Они дополнены, согласно его неоднократно
высказанному желанию. Некоторые новые задачи были
просмотрены автором. Однако он не мог уже указать
их место.
Ф. Бопп, Ж. Мейкснер.
Глава I
ТЕРМОДИНАМИКА. ОБЩИЕ ПРИНЦИПЫ
§ 1. ТЕМПЕРАТУРА КАК ФУНКЦИЯ СОСТОЯНИЯ
В термодинамике появляется новое
понятие—температура. Оно чуждо классической механике, равно как и
электродинамике и атомной физике (исключения:
омическое сопротивление, интенсивность спектральных линии
как результат действия множества отдельных объектов).
Качественной мерой температуры является наше
ощущение тепла; количественным измерителем, правда еще в
некоторой степени произвольным,—любой термометр.
Тело, находящееся в тепловом равновесии, имеет во всех
точках одинаковую температуру. То же самое справедливо
для двух тел, которые достаточно долго имели тепловой
контакт. Равенство температур во всех точках системы
является необходимым условием термодинамического равновесия.
Температура является функцией состояния. Она не
зависит от предистории тела и полностью определяется
его состоянием в данный момент времени. Качественно
мы определяем температуру тела по его состоянию в
данный момент, точное значение температуры—по показаниям
термометра в данный момент.
Термодинамика, как мы уже говорили в
предисловии,—аксиоматическая наука. В соответствии с этим
введем температуру при помощи следующей аксиомы:
Сугцествует функция состояния—температура.
Равенство температур во всех точках есть условие теплового
равновесия двух систем или двух частей одной и той ^нсе системы.
Эта аксиома сцециально сформулирована аналогично
приводимым ниже формулировкам первого и второго
начал термодинамики: это позволяет нам назвать ее,
согласно предложению Фаулера, «нулевым началом» ^).
^) Это название было предложено в связи с обсуждением
термодинамики известным индусским астрофизиком Саха и его
сотрудником Зривартава (М. Saha, В. Srivartava, Allahabad,
1931, 1935).
12 Гл. /. Термодинамика, Общие принципы
Чтобы раз навсегда установить математический смысл
понятия «функция состояния», рассмотрим ее
дифференциал. Мы напишем его для двух независимых
переменных X и у, которыми, конечно, должны быть самые
характерные и легко измеримые свойства системы
(например, давление и объем):
dT = Xdx-iYdy; X = g; 7 = 0. A.1)
огда, очевидно, справедливо соотношение
дХ^дУ
ду дх
A.2)
Это необходимое и достаточное условие того, чтобы
выражение Xdx t- Ydy являлось полным дифференциалом.
Оно равносильно утверждению, что Т есть функция
состояния.
То же самое условие мы можем записать в
интегральной форме
^dT^O A.3)
§
для любого замкнутого контура в плоскости х, г/.
Обозначим двухмерный вектор с компонентами X, Y через Z
и для доказательства соотношения A.3) используем
специализированную в применении к двухмерному вектору
теорему Стокса
$Zrfs= { TotZdxdy. A.4)
Так как rotZ, согласно соотношению A.2), исчезает, то,
действительно, условие A.3) тождественно утверждению,
что Т есть функция состояния.
При п независимых переменных условие того, что
данное выражение является полным дифференциалом,
заключается в равенстве нулю w-мерного ротора и
изображается л (л—1)/2 уравнениями вида A.2). Обобщенное
таким образом выражение A.1) называют
«дифференциалом Пфаффа». В случае двух независимых переменных
Xj у выражение Xdx + Ydy, даже когда оно не является
полным дифференциалом, т. е. когда не выполняется
условие A.2), всегда можно превратить в полный диф-
§ 1, Температура как функция состояния 13
ференциал, введя вспомогательную функцию N{Xy у)
(«интегрирующий делитель»).
При трех переменных х, у, z это, вообще говоря,
не всегда возможно. В этом случае требование
интегрируемости накладывает на компоненты X, F, Z
трехмерного вектора Z ограничение, которое мы исследовали
в т. II ^): вектор Z должен быть перпендикулярен к своему
ротору, т. е. должно выполняться равенство
(ZrotZ) = 0. A.4а)
На примере картины силовых линий и потенциала было
показано, что это требование не определяет однозначно
интегрирующий делитель (или, как мы там говорили,
множитель), т. е. таким интегрирующим делителем
(множителем) могут быть различные функции.
Это предварительное замечание будет полезно для
понимания второго начала термодинамики (см. § 6).
Подобно тому как в электродинамике появившемуся
там новому понятию заряда была приписана новая
размерность (наряду с механическими размерностями длины,
массы и времени), мы припишем особую (четвертую)
размерность и новому, введенному в термодинамике,
понятию температуры (при рассмотрении
электрохимических вопросов в качестве пятой единицы будет
присоединен еще заряд). Однако размерность температуры
будет обозначаться не особым символом, а, как это
общепринято, словом градус (град).
Вт. 1^) мы понимали под «механической системой»
совокупность материальных точек или твердых тел,
поведение которой определяется силами или геометрически
заданными связями; здесь мы будем говорить о
«термодинамической системе», когда для описания системы
требуется, помимо всего прочего, знание температур
ее составных частей и условий теплообмена между ними.
Простейшей термодинамической системой является
однородная жидкость (в это понятие мы включаем и спе-
1) А. Sommerfeld, Mechanik der deformiei:baren Median
(Bd. II), Leipzig, 1949, Aufgabe 1.7. (См. перевод: A. Зоммер-
фельд, Механика деформируемых сред, ИЛ, 1954, задача 1.7.)
2) А. Sommerfeld, Mechanik (Bd. I), 1944. (См. перевод:
А. Зоммерфельд, Механика, ИЛ, 1947.)
14 Гл. /. Термодинамика. Общие принципы
циальные случаи газа или пара). Жидкость имеет только
одну механическую степень свободы — объем и одну
термическую степень свободы — температуру. Объему V
(экстенсивной величине) сопоставляется в качестве
канонически сопряженной переменной ^) интенсивная
величина — давление р (соответствующая величина с обратным
знаком называется напряжением). Температуру Т следует
рассматривать как термическую интенсивную величину.
В § 5, п. 4, мы выясним, какая экстенсивная величина
является ей сопряженной. Давление, вообще говоря,
является функцией. Г и V, Соотношение/? = /(Г, V)
называют уравнением состояния.
Взаимная зависимость между величинами F, р и Г
выражается коэффициентом теплового расширения а
и термическим коэффициентом давления р, отнесенными
соответственно к данным значениям V или р:
Нижние индексы означают, что при дифференцировании
по Т величины р и V соответственно остаются
постоянными. Оба выражения имеют размерность 1/град. О
значениях а и р, в частности для газов, пойдет речь
несколько ниже. Зависимость между /?, Г и F
характеризуется также (изотермической) с^нсимаемостъю х:
Между величинами а, р и х имеется замечательное
соотношение (см. задачу 1.1).
Переходы, при которых Г, р или V остаются
неизменными, будем называть соответственно изотермическими,
изобарическими и изохорическими.
*) Термин взят из гампльтоновой механики [ср. т. I
(Механика), § 41]. Там мы назвали координату д (экстенсивную
величину) и импульс р (интенсивную величину) канонически
сопряженными, перенося затем эту терминологию и на случай более общего
выбора пар величин Q, Р. Этого указания достаточно, чтобы
объяснить соответствующие названия в тексте. Подробнее см. § 7 и 14
настоящей книги.
§ 2. Работа и количество тепла 15
§ 2. РАБОТА И КОЛИЧЕСТВО ТЕПЛА
Пусть жидкость находится в цилиндрическом сосуде
с площадью поперечного сечения F. К внутренней
боковой поверхности сосуда плотно прилегает поршень. На него
действует со стороны жидкости сила pF, При
перемещении поршня на расстояние dh жидкость совершает работу
dW = pFdh = pdV. B.1)
Это выражение справедливо не только при
положительных значениях dV (поднятие поршня), но и при
отрицательных значениях (опускание поршня). Равным
образом оно справедливо не только для цилиндрической
поверхности, но и для граничной поверхности
произвольной формы и для любого ее изменения. В последнем
случае надо лишь алгебраически просуммировать
изменения объема по всей граничной поверхности.
Дифференциал dW определен выражением B.1).
Соответствует ли ему функция состояния W? Конечно нет.
Чтобы выражение B.1) было полным дифференциалом,
согласно A.3), должно выполняться условие
§
dlV = 0. B.1а)
Имеется в виду, что жидкость совершает циклический
процесс, т. е. из некоторого начального состояния
возвращается по произвольному пути в то же начальное
состояние. Такие процессы для нашей системы с двумя
степенями свободы можно изображать графически в плоскости
двух независимых переменных (выбранных в зависимости
от обстоятельств), например V и Т (одна механическая
и одна термическая переменная) или р ж V (две
механические переменные). Последний график представляет
собой хорошо известную индикаторную диаграмму
(фиг. 1), предложенную еще 150 лет назад Джемсом
Уаттом. Такую диаграмму обычно йолучают для каждой
паровой машины путем автоматической записи.
Верхняя горизонтальная прямая р = Pi изображает
процесс, при котором цилиндр паровой машины соединен
с котлом высокого давления; нижняя прямая р^р^ —
процесс, при котором цилиндр соединен с атмосферой
16
Гл, /. Термодинамика, Общие принципы
ИЛИ котлом низкого давления. Правая и левая кривые
соответствуют расширению и сжатию^). Абсцисса
пропорциональна соответствующему перемещению поршня h и,
Pi
V-P,
Стотие
Расширение
Р-Р,
Фиг. 1. Индикаторная диаграмма паровой
машины.
следовательно, также объему цилиндра, наполненному
паром. Площадь, ограниченная циклом, дается
выражением
^pdF=^
dW,
B.2)
которое, очевидно, отлично от нуля. Согласно B.1а),
отсюда следует, что дифференциалу dW не соответствует
функция состояния W.
В паровой машине результирующая работа B.2)
совершается за счет подводимого тепла. Обратный переход
1) При рассмотрении этого циклического процесса могут
возникнуть недоуменные вопросы в связи с тем, что в ходе его
изменяется масса системы. Действительно, при открывании клапана,
соединяющего цилиндр с котлом высокого пли низкого давления,
количество пара в цилиндре меняется; оно остается постоянным
лишь во время расширения или сжатия. В противоположность
этому в циклах, рассматриваемых ниже, масса системы обычно
будет оставаться неизменной. Однако и в данном случае можно
добиться фактически или мысленно постоянства массы путем
конденсации выпущенного пара и возвращения его обратно в
нагревательный котел. Как бы то ни было, индикаторная диаграмма
может служить классическим примером наглядного изображения
циклического процесса в плоскости р, V.
§ 2, Работа и количество тепла 17
работы в тепло происходит в любом процессе, связанном
с трением. Решающим и исторически важнейшим
доказательством этого был опыт, произведенный Румфордом
в 1798 г. в Мюнхене: сверление пушечного ствола
привело к кипению воды.
Обозначим соответствующее приращение количества
тепла через dQ. Что касается его измерения, то оно, как
известно из определения единицы количества тепла,
сводится к измерению температуры: количество тепла,
которое необходимо для нагревания 1 кг воды при
атмосферном давлении от 14,5 до 15,5°С называется
(большой) калорией (ккал] часто в литературе употребляют
сокращенное название Кал). Напомним далее об
определении удельной теплоемкости, которую мы также должны
отнести к единице массы 1 «г. Количество тепла,
сообщаемое единице массы, будем обозначать посредством dq:
dq==c^dT, B.3а)
где с^—удельнаятеплоемкссть при постоянном объеме, или
dq^Cj^dT, B.36)
где Ср—удельная теплоемкость при постоянном давлении.
Между значениями с^ и с^ для газов имеется
существенное различие. Для капельных жидкостей это различие
в большинстве случаев пренебрежимо мало. Из B.36),
положив dq = l ккал/кг и dT = 1 граду получаем для воды
при 15°С
Ср = 1 ккал/град-кг, B.4)
Это выражение, очевидно, эквивалентно нашему
определению калории.
Во всех процессах, связанных с трением,
затрачиваемая работа dW находится в постоянном, не зависящем
от условий опыта, отношении к выделяющемуся
количеству тепла dQ. Джоуль доказал это различными опытами
(хотя и не очень точными количественно). В частности,
укажем на измерение количества тепла, выделяющегося
при прохождении электрического тока (джоулево тепло).
Еще несколько ранее Роберт Майер убедился качественно
18 Гл. I. Термодинамика. Общие принципы
В ТОМ, что при интенсивном перемешивании вода
нагревается^). Положим
dW = JdQ B.5)
и назовем величину J механическим эквивалентом тепла.
Измеряя dQ в калориях, а dW в технических единицах
работы, кгм, получим численное значение /:
/ = 427 кгм/ккал. B.6)
Здесь кг означает, как известно, килограмм-силу,
в связи с чем его называют в настоящее время «кило-
пондом» {кп)у сохраняя килограмм массы (кг) в качестве
наименования единицы массы в системе MKS. Мы имеем
тогда
1 «n = g.l хг«9,81 MKS-2 = 9,81 Дин.
Дина в этой системе единиц есть единица силы, равная
10^ дин. Мы назовем ее вместе с Р. В. Полем «большой
диной». Таким образом,
/ = 4,19.103M2KS-V««a^ = 4,19 Эрг/кал. B.7)
Единица энергии в системе MKS Эрг («большой эрг»)
равна 10' эрг=1 д:нсоуль = \ вт-сек. Единица тепла
малая калория {кал) находится в таком же отношении
к 1 г воды, как ккал к \ кг воды.
К экспериментальному доказательству соотношения
B.7) мы скоро возвратимся. Впрочем, в дальнейшем можно
будет отказаться от специальных тепловых единиц
{ккал или кал)у полагая в связи с B.6) или B.7)
1 ккал — АП кгм или 1 «a./i = 4,19 Эрг, т. е. /=1.
Здесь же прежде всего констатируем в связи с
уравнением B.5), что dQy так же как и dW, не является
полным дифференциалом. Не существует не зависящей
от предистории тела функции состояния— количества
^) В одном письме (в сентябре 1841 г.) Майер писал, что он
часто с неизхменным успехом производил этот опыт (см. Ostwald,
GroBe Manner, Leipzig, 1909). В этой связи couiwieMcn также
на утверждение Альбрехта фон Галлерса A708—1777), что теплота
животных должна возникать за счет трения крови в кровеносных
сосудах. Это представление сохранилось в неприкосновенности
вплоть до XIX в.
§ 3. Идеальный газ 19
тепла в теле в данном его состоянии. Тем самым
исключается старая теория теплорода.
Необходимо специально отметить, что данное нами выше
определение калории дает способ измерения количества
тепла dQ (или Q, если это конечная величина),
определенным образом подведенного к системе, а не общего
количества тепла Qy содержащегося в системе. То, что
эта величина зависит от способа подведения тепла,
очевидно уже из уравнений B.3а) и B.36).
В литературе стараются избегать обозначений dQ, dW
и заменяют их различными символами, например 8^, 8Ж,
чтобы не путать их с полными дифференциалами. Мы не
находим это необходимым. Напротив, мы считаем
существование функций состояния и связанных с ними
дифференциалов совершенно особым случаем, который, как
в § 1, каждый раз будет особо подчеркиваться.
§ 3. ИДЕАЛЬНЫЙ ГАЗ
Газ тем ближе к идеальному газу, чем труднее его
сжижитьпри нормальном давлении 760л«жрт. ст. = IWmopp
и чем ниже лежит его точка кипения. Насколько близки
к идеальному газу различные газы, показывает следующая
таблица точек кипения в градусах Цельсия при давлении
760 торр\
Не На Na 0^ СО2 Н2О
— 269 —259 —210 —218 —78,5 +100
Стоящий на последнем месте в этой таблице водяной пар,
конечно, не является идеальным газом. Идеальный газ—
это пределгное состояние реальных газов при бесконечном
разрежении их. Следующие законы относятся к этому
идеальному состоянию газа.
1. Закон Бойля — Мариотта
/?F = const. C.1)
Это уравнение справедливо при постоянной темпера-^
туре. Давление р обычно измеряется в атмосферах. Под
одной атмосферой понимают либо давление, равное
давлению воздуха, когда барометр показывает 760 мм
,20 Гл. I, Термодинамика. Общие принципы
рт. ст. = 760 торр (физическая атмосфера = 1 атм), либо
давление, производимое срлой в 1 «л на площадь в 1 см^
(техническая атмосфера = 1 am). Техническая атмосфера
С достаточной точностью равна весу столба воды высотой
10 ж и поперечным сечением 1 см^. Соответственно
техническая атмосфера выражается следующим образом:
1 am = 981 см/сек'^ЛООО zfcM^.
Здесь первым множителем является ускорение силы
тяжести g, вторым —масса столба воды; деление на см^
соответствует переходу от веса к давлению. Имеем, следо-
Штельно,
1 am = 0,981 • 10» дин/см^ = 0,981 Бар. C.2)
Соответственно
1атл«= 1,013 Бар. C.2а)
Введенная здесь единица Бар имеет значение
1 Бар= 10® дин/см^.
В метеорологии обычно употребляется единица давления
миллибар, равная 0,001 Бар. Выражая Бар в нашей
системе MKS, получаем
1 Бар = 10 Дин/см^ = 10^ Дин/м^. C.26)
Если в уравнении C.1) ввести вместо V плотность р,
равную массе, деленной на объем F, то получим
/? = р-const. C.3)
2. Закон ГеЁ-Люссака
pV = CT. C.4)
Здесь С —временно введенная постоянная, которая вскоре
будет выражена через универсальную газовую постоян-
)иую R. Прежде всего следует заняться определяемой
Посредством уравнения C.4) температурной шкалой,
которая, как показывает опыт, при соответствующем выборе
С явлется одной и той же для всех (идеальных) газов.
Будем исходить из выражения для коэффициента
теплового расширения A.5). Если использовать темпера-
§ 3, Идеальный еаа 21
турную шкалу, определяемую уравнением C.4), то
выражение A.5) примет вид
1 /дУЛ 1 С 1 ,о / V
Отсюда следует, что а не зависит от природы газа и
определяется только температурой. То же самое
справедливо и для термического коэффициента давления
Q 1 / дР\ 1 С 1 .г. ,^.
Сравнив 100-градусную шкалу Цельсия с
температурной шкалой, определяемой уравнением C.4), получим,
что температуре таяния льда (при давлении 760 торр)
соответствует значение
Го = 273,15°. C.5)
Вообще
T==T, + t, C.5а)
где ^ — температура в градусах Цельсия. Коэффициент
теплового расширения и термический коэффициент
давления при 0°С равны
а = р «^ 1/273 г/?аа = 0,00366 град'\ C.6)
Определяемая выражениями C.4), C.5) и C.5а)
температура называется температурой газового термометра.
Как видно из выражения C.5), нулевая точка последнего
сдвинута относительно нулевой точки термометра Цельсия
на 273 (точнее 273,15) град.
Воздушный термометр (еще лучше был бы водородный
или гелиевый) можно использовать для измерения
температуры как при постоянном давлении, так и при
постоянном объеме. Последнее является более
употребительным. Согласно уравнению C.4), при этом температура Т
пропорциональна давлению газа р. В соответствии с этим
подлежит измерению (барометром) разность давлений
р — Ро, определяемая сравнением положений столба ртути
при температурах газа Г и Г^. Для большинства
практических целей определение температуры при помощи воз-
Душного термометра является достаточно точным. Этот
22 Гл, I. Термодинамика, Общие принципы
способ неприменим лишь при низких температурах, когда
воздух уже не ведет себя как идеальный газ. Как
определяется температура (температура Кельвина) в этом
случае, мы увидим позже. Для реального газа (или для
идеального газа при низких температурах) уравнение Гей-
Люссака C.4) заменяется уже упоминавшимся выше общим
уравнением состояния жидкой или газообразной системы
T = F{p, V). C.6а)
3. Закон Авогадро и универсальная газовая
постоянная. Закон кратных отношений Дальтона,
справедливый для всех химических соединений, для газов
дополняется законом простых объемных отногиений Гей-Люссака.
Например: 1 л водорода-f 1 л хлора = 2 л хлористого
водорода в соответствии с химической формулой
H2 4Cl2 = 2HCl.
Другой пример: 2 л водорода +\ л кислорода = 2 л
водяного пара, или в химической записи
2Н2 4-02 = 2Н20.
Законы Дальтона и Гей-Люссака, очевидно, объединяются
правилом Авогадро A811 г.): все газы при одинаковом
давлении и температуре в равных объемах codepofcam
равное число молекул (Авогадро вместо молекул говорил
о корпускулах). Это правило, не получавшее признания
в течение десятилетий, примерно с 1860 г. является
основой для всех определений молекулярных весов. Нернст
подчеркнул его значение, дав своему большому учебнику
название: «Теоретическая химия на основе правила
Авогадро и термодинамики».
Атомистика чужда термодинамике. Поэтому в
последней вместо молекул, по примеру Оствальда, говорят о
молях. Под молем понимают, как известно (см. также
т. И, § 7, примечание 1), массу, измеряемую числом
граммов или килограммов (пишут грамм-моль = леоль или килог-
рамм-моль = «жо^ь), равным сумме атомных весов
элементов, образующих данное вещество. Следовательно, грамм-
моль Og равен 32 г, килограмм-моль Hg — приблизительно
2 кг, грамм-моль HCI —примерно A + 35,5) г. Заметим
в связи с этим, что наличие в молекуле воды именно
§ 3, Идеальный газ 23
двух атомов водорода было впервые установлено на
основании правила Авогадро. Еще в 1850 г. формулу воды
писали в виде НО.
Если в качестве естественной единицы измерения
пространства ввести молярный объем, определив его как
объем, который при данных давлении и температуре
содержит один моль газа, то правило Авогадро можно
выразить в следующей простой форме: при одинаковых
внешних условиях молярные объемы всех газов одинаковы.
Конечно, это утверждение, равно как и предыдущие
законы, строго говоря, относится только к идеальным газам,
а на реальные газы, или, лучше сказать, пары, оно
распространяется лишь с определенными оговорками.
Найдем теперь молярный объем идеального газа при
давлении 1 атм и температуре 1°С. За исходный пункт
можно взять плотность водорода, которая при этих
условиях [см., например, т. IV, уравнение A7.14)] с
достаточной точностью равна
9,00.10-2 хг/л«з = 9,00.10-2 г/./г.
Отсюда следует, что 2 г Hg занимают объем
g-^j ./1 = 22,2 ./г.
Точное значение молярного объема получим, если примем
во внимание, что атомный вес водорода несколько больше
единицы,
^моль = 22,4 л/моль ^ 22^А м^/кмолъ при 7Q0 торр и 0°С.
C.7)
Согласно правилу Авогадро, это значение молярного
объема характерно не только для Hg и является универсаль-
ным для всех идеальных газов.
Уравнение состояния газа также принимает
универсальную форму, если отнести его к молярному объему.
Запишем его в обычном виде
pVuonb=^IiT C.8)
и назовем R универсальной газовой постоянной. Вычислим
ее, подставив в уравнение C.8) ^моль из C.7) и соответ-
24 Гл. I. Термодинамика. Общие принципы
ствующие значения р и Т:
р = 760 торр = 1,03323 am = 9,81 -1,03323 Дин/см^
[см. C.2) и C.2а)],
Г = Го = 273,15 град [см. C.6)],
^моль = 22,4 л/л<оль = 22,4 м^/кмолъ.
Получаем
„ 9,81.1,03323.224 jj , ^ q ол г. , г,
Н = - 27315 Дин »м/град* моль = 8,31 Эрг/град* моль.
C.9)
Здесь Эрг —уже употреблявшаяся в B.7) единица работы
в системе MKS.
Из уравнения C.8) следует, очевидно, что для объема
F, содержащего п молей газа,
pV = nRT. C.10)
Отсюда находим значение постоянной С, фигурирующей
в уравнении C.4):
С = пВ C.10а)
Обозначим через v так называемый удельный объем, т. е.
объем единицы массы. Таким образом, имеем
^моль = [Аг;. C.11)
Здесь J1 —молярный вес (т. е. масса одного моля)
соответствующего газа; например, для 0^ ji = 32 кг/кмолъ.
Обычно вместо термина «молярный вес» употребляют
менее правильный термин «молекулярный вес». Подставив
C.11) в уравнение C.8), получим
pv=^^T, C.11а)
§ 4. ПЕРВОЕ НАЧАЛО ТЕРМОДИНАМИКИ.
ЭНЕРГИЯ И ЭНТАЛЬПИЯ КАК ФУНКЦИИ СОСТОЯНИЯ
Поело того как теория теплорода была признана
неудовлетворительной, ее место заняла так называемая
«механическая теория тепла». Название показывает, что
теплоту понимали как хаотическое движение
материальных частиц, связывая ее с «живой силой» последних.
§ 4» Первое начало термодинамики. Энергия и энтальпия 25
В ЭТОМ смысле Гельмгольц дал своей работе 1847 г.
название «О сохранении силы». Эта работа основывается на
гипотезе, что вся физика может быть сведена к механике
и что взаимодействие между частицами обусловлено
центральными силами.
Название «механическая теория тепла» безусловно
является неполным. Солнечное излучение бесспорно входит
в тепловой баланс Земли и равным образом несомненно,
что оно не является механическим процессом. Поэтому
в наше время предпочигают менее красочное название
«термодинамика». Также и двусмысленное название
«живая сила» заменяется удачно предложенным
Вильямом Томсоном термином «кинетическая энергия». Слово
«энергия», встречающееся еще у Аристотеля, было
введено Ранкином в технику в 1853 г.; у него встречается уже
и слово «энергетика». При желании перевести слово
«энергия» (это, впрочем, отнюдь не следует рекомендовать)
можно употребить термин «запас работы» (Ф. Нейманн).
Смелые идеи Роберта Майера («ОЬег die Krafte der unbe-
lebten Natur», 1842) вышли далеко за границы
классической механики и вполне соответствуют современному
общему пониманию закона сохранения энергии, хотя
самим Майером они и не были математически оформлены
с той степенью законченности, с какой это позже сделал
Гельмгольц. Особая 3acviyra Майера состояла также в том,
что он подчеркивал важность процессов высвобождения
(на первый взгляд противоречащих закону сохранения
энергии), которые являются основными для весьма важной
в настоящее время техники катализаторов.
Мы введем понятие энергии аксиоматически, не
ссылаясь на механику, и вместе с тем сформулируем первое
начало термодинамики:
Калсдая термодинамическая система обладает
характеристической функцией состояния -энергией. Эта
функция состояния возрастает на величину сообщенного
системе количества тепла dQ и уменьшается на величину
совершенной системой внешней работы dW. Для
замкнутой системы справедлив закон сохранения энергии.
1. Эквивалентность тепла и работы. Если
использовать введенное Клаузиусом обозначение U для сохра-
26 Гл. /. Термодинамика. Общие принципы
няющейся энергии системы, то математическая
формулировка первого начала гласит:
dU^dQ-dW. D.1)
Здесь dU в противоположность dQ и dlV является
полным дифференциалом. Поэтому для каждого циклического
процесса справедливо соотношение
§
dU = 0. D.1а)
Количество тепла dQ в правой части уравнения D.1)
измеряется, конечно, не в калориях, а считается
выраженным в механических единицах работы, согласно
равенствам B.6) или B.7),
Специализируем уравнение D.1) для простейшей
термодинамической системы - гомогенной жидкости,
рассматривая при этом единичную массу ее. Энергию в этом
случае назовем удельной энергией и обозначим ее малой
буквой м, подобно тому как удельный объем в уравнении
C.11а) и удельное количество сообщенного системе тепла
в соотношениях B.3а) и B.36) мы обозначали малыми
буквами V и dq. Следовательно, имеем
du^dq— pdv. D.2)
Прежде всего используем это уравнение для того, чтобы
вычислить механический эквивалент тепла У, и тем самым
подтвердим равенство B.7). Рассмотрим для этой цели два
процесса. Пусть первый из них происходит при
постоянном объеме (система из состояния v^ Т переходит в
состояние V, T-rdT), а второй—при постоянном давлении,
причем система переходит из состояния v, Т в состояние
V -\- dv, Т -Y dT при тех ^нсе значениях Т и dT, что и в
первом случае. Тогда, принимая во внимание определения
B.3а) и B.36), имеем
du^ = c^dT, D.3)
du^ = c^dT---pdv. D.3а)
Пусть рассматриваемая система представляет собой
идеальный газ. Тогда для второго процесса, согласно
уравнению C.11а), имеем
pdv^--dT.
g 4. Первое начало термодинамики. Энергия и энтальпия 27
Следовательно, уравнение D.3а) принимает вид
du,= (^c^-^^dT. D.36)
Дополним наше прежнее определение идеального газа еще
одним «калорическим» условием: будем считать, что
удельная энергия и (м, конечно, такж:е полная энергия U)
есть функция только темпер ату ры Т. Следовательно,
при данной температуре она не зависит ни от объема, ни
от давления газа. Тогда, согласно уравнению D.3), с^
также является функцией только Т [с^ — du/dT — и' (Т)].
Поэтому уравнение D.3) можно записать в интегральной
форме
u{T)=^c„iT)dT. D.4)
Поскольку и есть функция состояния, то величина этого
.^тнтеграла не зависит от характера процесса (в частности,
от того, происходит ли он при постоянном или
изменяющемся объеме).
Здесь мы не касаемся экспериментального п
теоретического обоснования дополнительно принятого
«калорического» условия, отсылая читателя к § 5, п. 3, или
к §7.
Так как, по предположению, Т и dT в обоих
процессах одинаковы, мы получаем
du^=:du2 = u'{T)dT. D.4а)
Из уравнений D.3) и D.36) на основании этого следует,
что
или
KS-0 = ^- (^-ба)
Слева стоит разность обеих молярных теплоемкостей,
которая для всех идеальных газов приблизительно равна
2 кал/град-моль. Следовательно,
(с — с^моль = 2 кал/град'моль = 2 ккал/град-кмоль. D.56)
28 Гл. /. Термодинамика, Общие принципы
Подставляя в соотношение D.5а) это значение и значение
R из C.9), находим
1 «ал = 4,16Э/?г. D.6)
Это согласуется с точностью до 1% с ранее полученным
значением [см. B.7)], причем неточность обусловлена тем,
что принятое в D.56) значение с^ — с^ является
приближенным. Рассмотрим этот пример еще раз, выразив единицы
работы через калории. Соотношение D.5а) дает в силу D.56)
Л = 2 кал1 град-моль, (^-7)
и уравнение состояния C.8) приобретает своеобразный вид
Р^иопъ = 27* кал/град*моль. D.8)
При этом давление выражается в калориях на единицу
объема.
2. Энтал1:пия как функция состояния. Наряду с
энергией рассмотрим еще одну функцию состояния, особенно
ваянную для техники. Мы назовем ее энтальпией Н и
определим при помощи соотношения
H=:U + pV. D.9)
Слово энтальпия означает «тепловая функция», обозначение
Н (первоначально имелось намерение воспользоваться
греческой буквой «эта») взято из американского учебника
термодинамики Льюиса и Рендала. Различные обозначения
термодинамических функций будут приведены в § 7, где
также определение D.9) будет выведено на основании
общих математических принципов. Соотношение D.9) вместе
с уравнением dU=-dQ — pdV дает
dH = dQ + Vdp. D.10)
Следовательно, при постоянном давлении {dp = 0) dH равно
количеству тепла, сообщенному системе извне, чем и
объясняется название «тепловая функция».
Обозначая через h энтальпию, приходящуюся на 1 моль
(или в данном случае также на единицу массы), имеем
h = u-\-pVy D.9а)
dh = dq + vdp. D.10а)
§ 4. Первое начало терлюдинамики. Энергия и энтальпия 29
Отсюда ^) для молярной теплоемкости с^ находим
(—') =^\ =с,. D.11)
\дТ J-P dT |p=const Р ^ '
Это равенство аналогично уравнению D.4а), которое, будучи
записано подробно, имеет вид
(^а=
dq
It
= с^, D.11а)
v=const " ^ '
Для идеального газа в силу равенства pv = RT /г
наряду с и зависит только от температуры. В этом случае
индексы р и V в левых частях D.11) и D.11а) можно
опустить; тогда, вычитая D.11а) из D.11), получаем
d(h — u)
Это согласуется с D.5а), если принять во внимание
равенство h — u==pv = RT.
Для техники понятие «энтальпия» особенно важно
потому, что для стационарного рабочего процесса оно дает
непосредственное представление о потоке энергии.
Представим себе паровую турбину. В каждый момент времени
в нее входит определенное количество сжатого перегретого
пара, который расширяется, охлаждается и выходит из
турбины. Нас интересует общий энергетический баланс
произвольной машины, совершающей стационарный рабочий
процесс. Отнесем все термические величины, так же как
и всю механическую работу, к единице массы газа (или
вообще рабочего вещества), наполняющего машину.
Рассмотрим поперечное сечение 1 (плоскость Fj)
подводящей трубы машины. Пусть через сечение Fj^ протекла
как раз единица массы газа. Тогда через сечение Р^^ была
перенесена внутренняя энергия Wj (индекс 1 относится
к состоянию газа в поперечном сечении 1 трубы).
Протекающий газ под давлением р^ перемещается на
расстояние vJFj^j так как единица массы газа в сечении 1
занимает объем v^. Следовательно, внешнее давление (например.
^) Мы избегаем в D.11) и D.11а), так же как и в последующих
уравнениях, выражений типа (dq/dT)p и (dq/dT)^, так как q не
является функцией состояния*
30 Гл. I. Термодинамика, Общие принципы
обусловленное паровым котлом) совершило работу (равную
произведению силы на путь) {PiF-^{v^lF^ = p^v-^. Если
пренебречь кинетической энергией, то поток энергии через
сечение F^ равен
Те же самые соображения относятся к течению газа через
поперечное сечение 2 выхлопной трубы. Пусть машина
производит внешнюю (в технике говорят «полезную») работу
I (рассчитанную на единицу массы газа). Ради общности
предположим, кроме того, что к машине было подведено
количество тепла q, (Конечно, в частном случае q может
быть равно 0.) Из закона сохранения энергии следует
простое уравнение
h^^q=.l-th^. D.12)
Преимущество этой формы записи энергетического баланса
состоит в том, что здесь не фигурируют явно конкретные
закономерности процессов, происходящих внутри машины.
Мы возвратимся к этому в § 5, п. 3, при рассмотрении
одного- физически особенно важного примера.
3. Соотношение между удельными теплоемкостями
Ср и с^. Здесь мы вставим одно замечание, которое выходит
за пределы термодинамики. Термодинамика дает только
соотношения ме:нсду материальными константами, такие
как, например, соотношение D.5а), но не может дать никаких
абсолютных значений материальных констант. Для
получения последних необходимо привлечь модельные
представления типа используемых в кинетической теории газов. Тогда
для молярных тсплоемкостей газов или паров, согласно
закону одинакового распределения энергии по стененям
свободы, получим формулу [см. § 31, п. 2, перед
уравнением C1.9)]
с,= ^//?. D.13)
Здесь / — число степеней свободы. Оно имеет следующие
значения:
/ = 3 для одноатомных молекул (принимается во
внимание только трансляционное движение, вращение
одноатомной молекулы не учитывается);
§ 4. Первое начало термодинамики. Энергия и энтальпия 31
/ = 5 ДЛЯ двухатомных молекул (пм приписывается
симметрия «гантели»; тогда они имеют 2 вращательные и 3
трансляционные степени свободы, вращение же вокруг
прямой, соединяющей атомы, не учитывается; равным
образом не принимается во внимание возможность
колебаний-атомов относительно цоложения равновесия, так как
эти колебания влияют на удельные теплоемкости лишь при
высоких температурах);
f ^6 для молекул произвольного вида C вращательные
н 3 трансляционные степени свободы; возможность
внутренних движений также не уч11тывается).
Формула D.13) показывает, что с^ является константой,
характерной для каждого газа, и, следовательно, не зависит
не только от v, но также и от Т. Соответствующее
значение теплоемкости при постоянном давлении (рассчитанное,
как и значение теплоемкости при постоянном объеме,
на 1 моль), согласно D.5), равно
Из D.13) и D.13а) следует, что
Величина f (она, очевидно, одна и та же как для
удельных, так и для молярных теплоемкостей) принимает
следующие значения:
1 + 1 = 1,66 1 + |- = 1,40 1 + 1=1,33
Примеры газов, для которых / = 3: пары Hg, инертные
газы Не, Ne, Аг. Примеры газов, для которых /=5: Hg,
Ng, Og, воздух. Примеры газов, для которых / = 6: все
многоатомные газы.
Однако в то время как термодинамическое
соотношение D.5) является совершенно точным и революция,
произведенная квантовой теорией, оставляет его в неприкосно-
32 Гл. I. Термодинамика. Общие принципы
венности, значения величин в формулах D.13) и D.14),
полученные из модельных представлений, являются лишь
более или менее хорошими приближениями и требуют
уточнения с точки зрения квантовой теории. В частности,
значение 7 = 1»33 есть лишь некоторое среднее значение,
около которого группируются эмпирическрш значения для
многоатомных молекул. С другой стороны, весьма
замечательно, что случай / = 4, ^=1,50, которому не
соответствует никакая геометрическая модель и никакая
молекулярная симметрия, действительно в природе не
реализуется.
Рассмотрением этого вопроса мы хотели
продемонстрировать сильные и слабые стороны термодинамики, с одной
стороны, и кинетической статистики—с другой.
§ 5. ОБРАТИМЫЙ И НЕОБРАТИМЫЙ АДИАБАТИЧЕСКИЕ
ПРОЦЕССЫ
Начнем с различия между обратимым и необратимым
процессами. Обратимые процессы — это, собственно говоря,
не процессы, а цепи следующих друг за другом
равновесных состояний. То, что мы всегда наблюдаем, — это
необратимые процессы, процессы, приводящие к
восстановлению нарушенного равновесия. Вместо слов «обратимый
процесс» можно было бы сказать: бесконечно медленный,
квазистатический процесс, при котором используется вся
работа системы и никакая энергия не пропадает даром.
Несмотря на нереальный характер обратимых процессов,
они играют основную роль в теоретической термодинамике,
так как только они приводят к некоторым равенствам;
необратимые процессы в термодинамике равновесных
состояний можно описывать только посредством неравенств.
Основной критерий обратимости состоит в том, что при
протекании процесса в прямом, а затем в обратном
направлении в окружающей среде не должно остаться никаких
изменений.
1. Адиабатический обратимый процесс.
Адиабатическим называется процесс, происходящий в системе,
находящейся в условиях, при которых она не поглощает
и не отдает тепла. Примером адиабатической оболочки
§ 5. Обратимый и необратимый адиабатические процессы 33
может служить термос, изобретенный Дюаром.
Изотермический процесс является противоположностью
адиабатическому, так как для поддержания неизменной температуры
необходим теплообмен между системой и окружающей
средой (как это имеет место, например, в водяной бане,
в которой находится исследуемый газ).
Пусть единица массы идеального газа совершает
адиабатический процесс. Принимая во внимание равенство D.3),
положим в уравнении D.2)
dq = Oj du^c^dT.
Тогда получим
c^dT^—pdv. E.1)-
Чтобы найти отсюда соотношение между р и у, необходимо
воспользоваться уравнением состояния C.11а). В
результате вместо уравнения E.1) получаем
-^ с„ {pdv -Ь vdp) + pdv = О,
(с« + у ) pdv + c^vdp = О,
или, согласно D.5),
c^pdv + c^vdp^Q,
Отсюда находим [см. D.14)]
^ + т4^ = 0- E-2)
Будем считать ^ численной константой (см. конец § 4),
т. е. примем условие, несколько более сильное по
сравнению с «калорическим» условием, согласно которому
функции м, а также с^, Ср и ^ зависят только от температуры.
Уравнение E.2) при этом сразу же интегрируется и дает
1п/? + 7 In?^ = const.
Получаем уравнение адиабатического процесса, впервые
выведенное Пуассоном,
^9с;1^ = const. E.3)
Оно играет особенно важную роль в метеорологии.
Напомним также, что при помощи, уравнения Пуассона вычи-
34
Гл, I. Термодинамика, Общие принципы
слялась скорость звука в т. II [уравнение A3.17а)]; там
оно называлось уравнением политропы с показателем л = 7»
Используя общее уравнение состояния C.11а), уравнение
Пуассона можно переписать в переменных Г, v или Г, р\
Гут-1 = const, Тр -^ = const. E.3а)
Константы, входящие в уравнения E.3) и E.3а),
следующим образом выражаются через параметры исходного
состояния системы:
i-Y
const = jOqI^J, const = rorJ-*, const = Го/?о ^ •
В TO время как изотерма в плоскости /?, V
изображается, согласно уравнению Бойля — Мариотта,
равносторонней гиперболой, адиабата, описываемая уравнением
П
Фиг. 2. Адиабата А и изотерма /
идеального газа в плоскости /?, К.
Пуассона E.3), идет вниз более круто (фиг. 2). В
плоскости Г, V адиабата благодаря показателю степени
7 — 1 в уравнении E.3а), естественно, имеет менее крутой
ход (фиг. 2а).
Чтобы наглядно представить себе обратимый процесс,
рассмотрим газ в цилиндрическом сосуде с
теплонепроницаемыми стенками и площадью поперечного сечения Fi
л 5. Обратимый и необратимый адиабатические процессы 35
сверху газ закрыт невесомым поршнем, плотно
прилегающим к внутренней боковой поверхности цилиндра.
Поршень находится в равновесии, так как на нем лежит
груз, вес которого Р равен силе pFy действующей на
поршень со стороны газа. Пусть груз состоит из очень
большого числа маленьких грузиков весом 8Р, которые мы
будем по одному снимать с поршня. При этом поршень
Фиг. 2а. Адиабата А и изотерма
/ идеального газа в плоскости Г, К.
медленно поднимается и давление газа уменьшается.
Каждый грузик оР снаружи цилиндра оставляется на той же
высоте, с которой его сняли, никакой работы для этого
совершать не надо. Давление уменьшается от некоторого
начального значения р (например, 2 am) до конечного р^
(скажем, 1 am), а объем увеличивается от начальной
величины V (например, 1 л) до конечной F^ (в нашем
примере 2^^^ л). Центр тяжести всех грузиков ЬР поднялся
по сравнению с его первоначальным положением. Эта
работа по переменхению центра тяжести равна работе газа
по поднятию поршня. Последняя не пропала даром, но
пошла на увеличение потенциальной энергии поднятых
грузиков ЬР. Если их снова один за другим' перенсстЕ<
на поршень, не затрачивая при этом работы, то газ
сожмется, нагреется и вернется в свое начальное состояние..
Таким образом, процесс обратим цри условии, что ои
состоит из последовательности бесконечно малых измене-
36 Гл, /. Термодинамика, Общие принципы
нийу следовательно, происходит достаточно медленно^),
т. е. при разбиении груза Р на достаточно малые доли ЬР.
2. Адиабатический необратимый процесс. Если
поршень (вместе с грузом Р) мгновенно поднять, то сначала
газ расширяется в вакуум, не совершая при этом
никакой внешней работы. Возникающие при этом
турбулентные течения постепенно прекращаются. Каково конечное
состояние газа? Нагревается ли он из-за внутреннего
трения или охлаждается в связи с расширением? Ни то, ни
другое. Что касается конечного состояния, то с тем
приближением, с которым газ можно рассматривать как
идеальный, процесс протекает не только адиабатически, но
также изотермически.
Этот опыт впервые был произведен в 1807 г. Гей-
Люссаком (расширение газа в пустоту) и с несколько
большей точностью повторен Джоулем. Вместо цилиндра
были использованы две стеклянные колбы, соединенные
друг с другом стеклянной трубочкор! с краном. Одна колба
откачивалась, в другой находился изучаемый газ.
Измерения температуры после того, как крап был открыт и
система достигла конечного состояния, показали
(особенно четкие результаты наблюдались, когда колба была
наполнена воздухом или водородом), что температуры
в начале и конце опыта одинаковы.
Прежде чем пытаться объяснить этот результат,
рассмотрим циклический процесс^ приведший Роберта Майера^)
к вычислению механического эквивалента тепла и открытию
первого начала термодинамики. Пусть в начальном
состоянии 1 (фиг. 3) газ находится под атмосферным
давлением Pj^ и занимает объем V^, Нагреем его при постоян-
^) Чтобы процесс был обратимым, ои должен происходить
бесконечно медленно. Обратное утверждение, однако, неправильно;
бесконечно медленный процесс не всегда обратим. Примером
служит разрядка конденсатора через очень большое сопротивление.
2) В бумагах, оставшихся после рано умершего Сади Карно
A796—1832), сына известного геометра и генерала Лазаря Карно
(см. т. I (Механика), вблизи уравнения C.286)], найден тот же
самый расчет, в основу которого положены удельные
теплоемкости. Следовательно, Сади Карно можно считать пионером не только
в отношении второго, но, отчасти, и в отношении первого начала
термодинамики.
§ 5. Обратимый и необратимый адиабатические процессы 37
НОМ объеме Fj и доведем давление в нем до р^у это
состояние газа изображается на фиг. 3 точкой 2. При
расширении в пустоту объем газа увеличивается до
значения Fg, причем процесс совершается по изотерме
(равносторонняя гипербола), проходящей через точку 2
(считаем, что турбулентных потоков нет). Эта часть пути,
Фиг. 3. Цикл для определения
механического эквивалента тепла.
будучи сама по себе неопределенной, изображена на
фигуре пунктиром в отличие от гипербол, лежапшх за
точками 2 и 3. Если объем Fg выбран таким образом, что
соответствующее давление равно р^у то, затратив работу,
газ можно сжать при постоянном давлении р^ до
первоначального объема.
Изменения энергии (рассчитанные на единицу массы
газа) на трех отрезках пути 1 2у 2 3 и 3 1 равны
соответственно
\c^dT; 0; [c^dT-p, {v, - v,). E.4a)
Ti To
Их сумма, согласно уравнению D.1 а), должна быть равна
нулю. Отсюда, если воспользоваться уравнением состоя-
88
Гл, /• Термодинамика. Общие принципы
ния идеа^тьного газа и допустить, что разность
температур Т^^Т^ может быть сколь угодно мала, вытекает
уравнение D.5а) и полученное в D.6) значение
механического эквивалента тепла. Тот факт, что рассмотрение
данного цикла приводит к тем же результатам, к каким
приводит дифференциальный метод, является
самоочевидным. Действительно, оба метода основаны на одном и
том же предположении, а именно, что энергия идеального
газа является функцией только температуры.
3. Процесс Джоуля —Кельвина. Для увеличения
точности опыта по расширению газа в пустоту Вильям Том-
сон предложил «метод пробки из ваты» и осуществил его
Фиг. 4, Процесс Джоуля—Кельвина.
экспериментально вместе с Джоулем. Опыт заключался
в том, что медленный стационарный поток газа протекал
через пробку из ваты по направлению от большего
давления с одной стороны пробки к меньшему давлению
с другой стороны. В пробке, которая помещалась в трубке,
сделанной из почти теплонепроницаемого самшита, газ
тормозился. Когда устанавливается стационарное давление,
температура пробки и газа слева и справа от нее также
становится стационарной (каким бы сложным ни было
распределение температуры внутри пробки).
Рассмотрим порцию газа (фиг. 4), которая находится
между произвольным поперечным сечением А трубы и
правым концом пробки В и, проходя через пробку,
занимает новое положение А'В' (при этом частицы газа,
вышедшие из сечения А, входят в левый конец пробки).
На всем этом пути слева действует сила pF (F —
поперечное сечение трубы), справа — противодействующая сила
p'F» Перемещение газа слева от пробки равно V/F^
§ 6. Обратимый и необратимый адиабатические процессы 39
справа от пробки — F'/^f*, поэтому вся совершенная
работа равна
^dW = pV-p'V\ E.5)
С другой стороны, так как тепло не подводится для
поддержания одинаковой температуры в газе слева и справа
от пробки и не уходит через самшитовую оболочку, имеет
место равенство
\dQ = 0. E.5а)
Поэтому из закона сохранения энергии следует, что
U-U'=-pV + p'V\ E.6)
В таком виде наше рассмотрение справедливо для
любого газа. Общий закон, характеризующий процесс
Джоуля—Кельвина, запишем в форме
U + pV = U'+p'V\ или Н = Н\ E.7)
Или, в словесной формулировке: процесс Дджоуля
—Кельвина характеризуется тем, что при протекании газа
энтальпия остается постоянной. Напомним в связи с этим
замечание, сделанное в конце § 4, п. 2, о потоке энергии
через подводяшую и выхлопную трубы паровой машины.
Очевидно, уравнение E.7) (при соответствующем выборе
единицы времени) как раз представляет имевшийся там
в виду поток энергии. Таким образом, полученный
результат может служить обоснованием приведенного там
специального примера.
Для случая идеального газа правая часть уравнения
E.6) имеет вид
где Л/ —масса газа, занимающего на фиг. 4 объем АВ.
Опыт показал, что разность Т' — Т для воздуха очень
мала, а для водорода - едва измерима. Отсюда с гораздо
большей точностью, чем из опыта Гей-Люссака, следует,
что в идеальном предельном случае справедливо
утверждение
U'= и и не зависит от F. E.8)
40 Гл, I. Термодинамика. Общие принципы
Этим впервые дано убедительное экспериментальное
обоснование нашему дополнительному «калорическому»
условию, использованному в уравнении D.4).
4. Одно очень важное следствие. Будем исходить из
первого начала термодинамики, примененного к
обратимому изменению состояния идеального газа (например,
к единице массы последнего). Согласно теперь уже
твердо установленным соотношениям и = и{Т), с^ = с^{Т)у
du = c^{T)dT и уравнению состояния идеального газа
имеем
dq=:du + pdv = с„ (Т) dT + -^ dv. E.9)
Разделив обе части этого уравнения на Т, получим
p-^c,{T)'f + f'^. E.9а)
Хотя, как мы знаем, dq не является полным
дифференциалом, уравнение E.9а) можно проинтегрировать.
Сделав подстановку ds=-dq/T, получим
\ds=^s-s, = c„ln^ + ^lnl. E.10)
То, V0
Здесь сделано удобное, но не необходимое пpeдпOw^oжeниe
о том, что с„=const. Величина s является функцией
состояния, значение которой не зависит от пути ме:нсду
начальным и конечным состояниями и при произвольном
задании первого определяется только текущими
параметрами второго. Вместе с Клаузиусом назовем эту функцию
состояния энтропией. Это слово означает «способность
к превращению».
Чтобы уже здесь разъяснить, по крайней мере
формальный, смысл энтропии, напишем, учитывая, что dq=Tds,
закон сохранения энергии E.9) в виде
du = Tds-pdv, E.11>
Отсюда заключаем, что 5 —величина, сопряженная Г,
подобно тому как V—величина, сопряженная р\ s — та
§ в. Второе начало термодинамики 41
самая экстенсивная величина, сопряженная интенсивной
величине Г, о которой упоминалось в § 1.
Очевидно, определение энтропии E.10) относится не
только к единице массы газа, но и к одному молю и
вообще к произвольной массе М (в последнем случае вместо
малых употребляют большие буквы S, F).
Полученную в п. 1 адиабату можно было бы назвать
также «изэнтропой», т.е. кривой постоянной энтропии, так
как для нее dq=0. В самом деле, легко убедиться, что
ее уравнение E.3а) в плоскости у, Т идентично
уравнению 5 = const, вытекающему из E.10).
§ 6. ВТОРОЕ НАЧАЛО ТЕРМОДИНАМИКИ
Чтобы прийти к основному пункту термодинамики, мы
будем следовать классическому пути, начало которому
было положено Сади Карно в 1824 г. и по которому шли
затем Рудольф Клаузиус A850 г.) и Вильям Томсон A851 г.).
Заглавие работы Карно «Размышления о движущей силе
огня и средствах, потребных для ее получения»,
указывает на историческую связь термодинамики с развитием
паровых машин.
В этой работе Карно проводит следующую
гидравлическую аналогию: подобно тому как вода при падении
с более высокого на более низкий уровень может
совершать работу, теплород также должен быть способен
производить работу при переходе от более высокой к более
низкой температуре. Эта аналогия, конечно,
несостоятельна, так как никакого неразрушимого теплорода не
существует. Тем не менее идеи Карно имеют непреходящее
значение. Они сыграли важную роль в открытии второго
начала термодинамики, последовавшем через 25 лет.
Мы сформулируем второе начало термодинамики
аксиоматически, подобно тому как мы сформулировали первое
начало в § 4 (и «нулевое» в § 1):
Ка^исдая термодинамическая система обладает
функцией состояния^ называемой энтропией. Энтропия
вычисляется следующим образом. Система переводится из
произвольно выбранного начального состояния в
соответствующее конечное состояние через последовательность
состояний равновесия; вычисляются все подводимые при
42 Гл. I. Термодинамика. Общие принципы
этом К системе порции тепла dQ, делятся калиевая на
соответствующую ей абсолютную температуру Т и все
полученные таким образом значения суммируются,
(Первая часть второго начала термодинамики.)
При реальных (не идеальных) процессах энтропия
замкнутой системы возрастает. (Вторая часть второго начала
термодинамики.)
Когда мы познакомимся с приводимыми ниже
«доказательствами» этого утверждения, то станет ясно, что все
они лишь сводят его к более простым, кажущимся
самоочевидными, однако по существу не доказуемым
предположениям. Простейшим из них представляется следующее:
тепло не мож'ет само по себе перейти от системы с
меньшей температурой к системе с большей температурой
(Клаузиус). Однако при этом надо уточнить смысл слов
«само по себе». Они означают, что в среде, окружающей
тела, которые участвуют в теплообмене, не должно
произойти каких-либо изменений, связанных с этим процессом.
Эквивалентным постулату Клаузиуса является постулат
Кельвина: иевозмоэ^сно непрерывно получать работу^
только охла^нсдая отдельное тело ниже температуры самой
холодной части окружакщей среды. В противном случав
можно было бы полученную при этом работу превратить
(например, трением) в тепло и таким образом передать
тепло от более холодного тела к более теплому. Оствальд
выразил этот принцип в общеупотребительной в настоящее
время формулировке: neeosMOOiCHO построить «вечный
двигатель второго рода»у т. е. периодически работающую
машину, которая производила бы только подъем груза
за счет охлаждения теплового резервуара^). (Первое начало
термодинамики, как известно, запрещает осуществление
«вечного двигателя первого рода».)
1. Цикл Карно и его коэффициент полезного действия.
Используем произвольное, но гомогенное рабочее вещество.
Слово «гомогенное» означает, что состояние этого вещества
всегда возможно задать двумя механическими переменными
F, /?, значения которых определяют величину термической
^) Формулировка взята из книги: Planck, Thermodynamik,
S Aufl., § 116.
§ 6» Второе начало термодинамики
43
переменной О при помощи какого-либо общего уравнения
состояния. Обозначение О вместо Т показывает, что для
измерения температуры используется произвольно
калиброванный термометр (в частности, это может быть термоэлемент
или какое-либо подобное устройство).
dQ=0
в=в,
Фиг. 5. Цикл Карно.
Цикл Карпо (фиг. 5) состоит из двух изотерм 1 2 и 3 4
и двух адиабат 2 3 и 4 1. Ur отрезке пути 1 2 от
«нагревателя» (теплового резервуара при температуре 9^)
подводится определенное количество тепла Q^] на отрезке пути
3 4 некоторое количество тепла Q2 отводится в
холодильник (тепловой резервуар при температуре Ь^). Полное
количество тепла, полученное системой, равно
§dQ^Q,-Q,
Совершенная рабочим веществом работа, как и в случае
индикаторной диаграммы в § 2, равна
^dW = i^pdV = W.
Так как энергия системы U возвращается к своему
исходному значению в точке i, то, согласно первому началу
44 Гл, /. Термодинамика, Общие принципы
термодинамики, справедливо уравнение
W=Q^-Q,- F.1)
Коэффициент полезного действия, как и для паровой
машины, определяется формулой
Произведенная работа __ 1^ _« i ^2 /А 9\
'"" Подведенное количество тепла ~~ Qi" Qi * \ * ^
Карно рассматривал машину Л/, которая совершает
процесс 12 3 4 бесконечно медленно (без потерь на
трение и излучение), так что рабочее вещество в каждый
момент времени находится в состоянии теплового
равновесия. (Поэтому адиабаты должны иметь качественно тот
же характер, что в § 5, п. 1 для идеального газа.) Такая
машина обратима. Она может проходить через
последовательность равновесных состояний в направлении 14 3 2
так же хорошо, как и в направлении 12 3 4, причем
в первом случае она работает не как силовая, а как
холодильная машина {W < О, ^g > ^1» ^ ^'^^^ случае
необходимо подводить работу | И' |, чтобы еще более понизить
температуру холодильника).
Карно доказывает, что коэффициент полезного
действия такой машины не зависит от вида рабочего
вещества. Для этой цели он рассматривает две обратимые
машины М и М\ работающие с различными рабочими
веществами, но между двумя одинаковыми тепловыми
резервуарами при температурах б^, в^ и совершающие равные
работы W. Количество тепла, подведенное к машине il/'
и отведенное от нее, равно Q[ и Q'^ соответственно.
Предположим, что
V>^. F.3)
Соединим машины М я М' так, чтобы М работала как
холодильная машина, т. е. в направлении 14 3 2, и
приводилась в действие от М\ Из F.2) и F.3) вытекает, что
\W \ \W\
^-q7^ > -т^ и, следовательно, Q^ > Q[.
Таким образом, горячий тепловой резервуар будет
получать больше тепла от машины il/, чем отдавать машине М',
Разность количеств тепла ^Q ='Qi — Q[ будет благодаря
§ 6, Второе начало термодинамики 45
связи машин М и М' заимствоваться из холодного
теплового резервуара. Общий эффект будет следующий:
количество тепла ^Q переходит с более низкого уровня Ь^
на более высокий уровень б^ без совершения работы и
без того, чтобы в машинах М и М' или окружающей их
среде произошли какие-либо изменения. Но это, согласно
предшествующему постулату, невозможно. Следовательно,
предположение F.3) недопустимо.
Так же обстоит дело с предположением if] > т^' — стоит
лишь поменять ролями машины М и М\ чтобы опять
получить противоречие с нашим постулатом. Следовательно,
7i = V. F.4)
Все обратимые машины ^ имеющие тепловой контакт
с окруон:ающей средой только при двух температурах
б^ ц 02» обладают одинаковыми коэффициентами
полезного действия. Согласно формуле F.2), вместо равенства
F.4) можно написать
% = f^h'K)> F.5)
где / — универсальная функция, не зависящая от вида
рабочего вещества и конструкции тепловой машины.
2. Первая часть второго начала термодинамики.
Чтобы функцию двух переменных /F,, Ь^ представить как
две функции одной переменной, свяжем оба уровня б^, 6^
посредством обратимого процесса Карно с тепловым
резервуаром, имеющим произвольно выбранную, но
фиксированную промежуточную температуру 6^. При этом
тепловой резервуар 6^ для одного процесса может служить
холодильником, отнимая количество тепла Q^^ а для
другого—нагревателем, сообщая то же самое количество
тепла. Тогда резервуар 6^ исключается из теплового
баланса и простая машина (в^, Sg) работает с теми же
количествами тепла, как и сложная машина (б^, б^) -Ь (б^, 62).
Поэтому наряду с уравнением F.5) справедливы (и
притом с теми же самыми значениями Q^ и Q^ также и
уравнения
46 Гл'. I, Термодинамика, Общие принципы
Перемножая их, получаем
^l=f{K^)-f{KO,). F.6а)
Из сравнения уравнений F.6а) и F.5) следует, что
/(^.o,)=/(9i,eo)-/@o. У- F.66)
Подставляя сюда значение 0^ = Og, получаем
так как, согласно уравнению F.5), /@^, 9^) = 1. В связи
с этим уравнение F.66) м.ожно записать также в виде
Так как величина 6^ исключается, то, согласно F.5) и
(б.бв), имеем
Теперь произвольной температурной шкале 6 можно
сопоставить абсолютную так, чтобы каждому показанию 9
на первой из них соответствовало показание
Г = ср(9) F.7а)
на второй. В § 10 мы увидим, каким образом это
осуществляется практически. Здесь же заметим только^
что эта абсолютная температура Т совпадает при
соответствующем выборе содержащегося в ср@) произвольного
постоянного множителя с температурой газового
термометра внутри той области температур, в пределах которой
термометрическое вещество ведет себя подобно идеальному
газу. Это будет доказано в задаче 1.3.
Объединяя вместе уравнения F.7) и F.7а), получаем
пропорцию Карно
Яг-Яг = Тг-Т^. F.8)
Отсюда, с одной стороны, вытекает формула д^ля
коэффициента полезного действия
¦П = ^^. F.8а)
§ в, Bmofoe начало термодинамики
47
С другой стороны, специализируя пропорцию F.8) на
случай бесконечно тонких диаграмм Карно (когда разность
температур конечна, но количества подведенного и
отведенного тепла dQi и dQ^ бесконечно малы), имеем
i^^=^. F.86)
Перейдем теперь к рассмотрению произвольного
обратимого кругового процесса. В плоск-ости /?, V (фиг. 6) он
изображается непрерывной замкнутой кривой, на которой
dQ,
Я,
Фиг. 6. Представление произвольного^про
цесса в виде суммы бесконечно малых
циклов Карно.
взяты две произвольные точки А ia В, Представим весь
процесс в виде совокупности бесконечно тонких циклов
Карно. При этом непрерывная замкнутая кривая
превращается в кривую с зубцами (сторонами которых
являются попеременно адиабаты и изотермы), однако после
интегрирования это различие исчезает. Если количества
отводимого тепла dQ^ в уравнении F.86) считать
отрицательными (что вполне последовательно), то, согласно
уравнению F.86), при интегрировании по всему циклу получаем
''%^=0. F.9)
Ф-
Индекс при dQ указывает на обратимый характер
рассматриваемого кругового процесса. Равенство @.9),
48 Гл. I. Термодинамика, Общие принципы
согласно § 1, есть необходимое и достаточное условие
того, чтобы выражение
ds='^-^^ F.10)
являлось полным дифференциалом. Оно имеет место в том
случае, если количество тепла dQ подведено к системе
обратимым путем (т. е. при использовании всей
доступной работы). Эта обратимость будет гарантирована, если
в F.10) подставить значение dQ из первого начала
термодинамики: dQ=-dU-\-dW, Тогда для рассматриваемого
простого рабочего вещества вместо выражения F.10) имеем
^g^dU+pdV^ F.10а)
Таким образом, абсолютная температура, определенная
уравнением F.7а), является интегрирующим делителем
для неполного дифференциала, стоящего в числителе
выражения F.10а).
Равенство F.9) описывает процесс, идущий по
произвольному пути; однако оно доказано пока лишь для
специальной термодинамической системы (гомогенной
жидкости). Фактически оно справедливо для любой системы,
состоящей из различных веществ в любых фазах и с
произвольными степенями свободы (в частности, также
электрическими или магнитными), при условии лишь, что
в системе не происходит необратимых процессов
{подобных трению, выделению д:нсоулева тепла и т. д.).
Рассмотрим прежде всего отдельную, скажем i-ю,
гомогенную составную часть системы (подсистему) с двумя
степенями свободы. Согласно F.10), выражение
A3, = Щ F.106)
является полным дифференциалом. Здесь Т^ — абсолютная
температура i-й составной части, dQ-^ - полное количество
тепла, сообщенное ей обратимым путем извне или от
внутренних составных частей системы^).
1) Если число степеней свободы больше двух, то можно,
фиксируя в различных комбинациях все степени свободы, кроме двух,
применять выражение F.106) для каждого частного процесса.
§ б. второе начало термодинамики 49
Составим сумму
i i
которая также является полным дифференциалом,
независимо от того, при помощи каких параметров системы
мы будем описывать процесс. Это выражение для суммы
проще, чем сумма выражений F.106), для отдельных
частей системы, так как в (б.Юв) члены, учитывающие
теплообмен между составными частями системы,
сократятся. Действительно, поскольку эти процессы
теплообмена, по предположению, являются обратимыми,
температуры подсистем должны быть одинаковыми
(обыкновенная теплопроводность исключена). Следовательно, в
случае двух подсистем, приписывая им индексы / и Г, имеем:
Ti=Ti' и dQ^= —dQif (количество тепла, подведенное
к i-ii подсистеме, есть количество тепла, отведенное от
Г-й подсистемы). Следовательно, вклады таких
процессов теплообмена в сумму (б.Юв) взаимно уничтожаются.
То же самое справедливо для всех процессов теплообмена
при равновесии фаз, которое, как подчеркивается в § 8,
п. 2, предполагает равенство температур любых двух фаз.
Таким образом, мы имеем право считать, что dQ^ есть
количество тепла, подведенное извне к i-u составной части
системы.
Если i-я и Г-я подсистемы отделены друг от друга
адиабатической перегородкой, то неравенство температур
!Г| п 7*1/ не исключено. Однако в этом случае была бы
необходима довольно искусственная конструкция системы
из отдельных компонент. Обычно имеют дело со случаем,
когда одинаковы не только температуры всех частей
каждой отдельной компоненты (Г^ постоянна внутри
/-компоненты), но и вся система в целом характеризуется
некоторой постоянной температурой {Т^ = Т), Тогда сумма
(б.Юв) принимает вид
i
^10 тождественно выражению F.Ю). Здесь величина dQ,
так же как и в выражении (б. 10), имеет смысл колк-
50 Ул. t. Термодинамика. Общие принципы
честна тепла, подведенного ко всей системе извне
обратимым путем. Этого достаточно, чтобы стала понятной
справедливость выражения F.10) для любой
термодинамической системы. Благодаря выражениям F.10), F.10а),
а также несколько более общему выражению F.10в)
теперь непосредственно установлено существование
энтропии как функции состояния и тем самым доказана первая
часть второго начала термодинамики»
Вычислим разность энтропии двух произвольных
состояний системы А и В при помощи уравнения
^ dO
Sb-Sj,^\^. F.11)
Подчеркнем, что выбор пути интегрирования в
уравнении F.11) не имеет ничего общего с тем, каким-образом
в действительности система переходит из состояния А
в состояние В. Реальные процессы всегда, по крайней
мере частично, необратимы. Уравнение F.11), однако,
требует, чтобы выбранный переход был вполне обратимым.
Выбор пути процесса не имеет значения, так как
энтропия S является функцией состояния, и, следовательно,
интеграл в уравнении F.11) не зависит от пути
интегрирования,
В качестве простейшего примера такого расчета
рассмотрим расширение газа в пустоту, о котором шла речь
в § 5 (см. фиг. 3). Точки 2 и 3 на фиг. .3 соответствуют
состояниям Л и S в уравнении F.11). Поскольку
расширение газа в пустоту происходит адиабатически {dQ = 0),
имеем для реального перехода
независимо от тех отклонений от (вычерченной
пунктиром) изотермы, которые могут быть обус^ловлены
турбулентностью. Напротив, для мысленно выбранного
обратимого процесса, если избрать путь вдоль изотермы,
имеем dU = О, с?^обр. ^dU -\- pdV = pdV, Вычислим теперь
изменение энтропии одного моля газа, расширяющегося
§ 6. Второе начало термодинамики 51
В пустоту. Согласно уравнению F.11), получаем
^ 2 } Т \ V Vo
2 2
Конечно, то же самое значение Аб* получится, если вместо
изотермы выбрать путь 2 3-^^31 (см. фиг. 3). (Это можно
проверить и непосредственным расчетом.) Отметим также,
что при предварительном вычислении энтропии в
уравнении E.10) расчет был произведен для случая обратимого
подвода тепла [в смысле, определяемом выражением F.10а)].
То же замечание относится и к расчету в § 9, п. 2, для
случая газа Ван-дер-Ваальса.
Разобранный пример ясно показывает, что
существование и значение энтропии в конечном состоянии зависят
только от самого этого состояния, каким бы путем
(обратимым или необратимым) оно ни достигалось. Значение
энтропии (в нашем примере S^) определено с точностью до
постоянной величины F*2).
Заметим еще, что переход от выражения F.106) к
выражению (б.Юв) предполагает аддитивность энтропии
отдельных составных частей системы. Это положение лежит
в основе классической термодинамики. Однако оно не
является обязательным с точки зрения статистики (см.
§31, п. 1).
Назовем систему замкнутой, если она не подвергается
внешним воздействиям, т. е. если ей не сообщается
никакого количества тепла и она не совершает работы.
Энергия такой системы имеет постоянное значение, так как
dW^zQ и dQ = 0. Согласно уравнению F.14), энтропия
такой системы также постоянна:
Sb = Sa^ F.12)
Это обстоятельство является парадоксальным и на
первый взгляд противоречит второй части второго начала
термодинамики. Причина этого парадокса лежит в том,
что в выражении F.106) и в последующем изложении мы
сузили понятие «термодинамической системы», исключив
все необратимые взаимодействия между ее составными
частями и предположив, что имеет место термодинамическое
равновесие. Действительно, для вычисления разности
52 Гл. I. Термодинамика, Общие принципы
энтропии в уравнении F.11) это было необходимо. При
этом, и только при эгом, условии справедливо
утверждение, содержащееся в равенстве F.12): энтропия
замкнутой системы постояшш, если система находится в
тепловом равновесии,
3. Вторая часть второго начала термодинамики.
Предположим теперь, что из двух машин М и М\
рассмотренных в п. 1, одна, например М\ является
необратимой. Тогда можно осуществить соединение машин,
о котором говорилось после неравенства F.3) (il/работает
в качестве холодильной машины между теми же
тепловыми резервуарами, что и М\ и приводится в действие
машиной ЛГ). Рассмотрение работы этого агрегата
позволяет доказать невозможность неравенства tq' > tq. Обратное
соединение машин в данном случае осуществить нельзя.
Поэтому вместо равенства F.4) теперь имеем
7|>7|', F.13)
причем равенство ir) = tq' также исключается в связи с
необратимостью машины АГ, Обратимый процесс Карно
имеет больший коэффициент полезного действия, чем
любой необратимый, происходящий между теми же
тепловыми резервуарами и дающий ту же работу. Таким
образом, необратимый процесс менее экономичен; он требует
затраты большего количества горючего материала для
выполнения той же работы: ^( > ^i-
Из неравенства 1 — тг] < 1 — tq' при сохранении данного
в уравнении F.8) определения абсолютной температуры
и согласно формулам F.8а) и F.2) вытекает: TjTi =
= Q2/Q1 < QilQv Отсюда следует, что
Для бесконечно малых циклов Карно вместо равенства
F.86) получаем
Точно так же для произвольного кругового процесса,
полностью или частично необратимого, если считать отводи-
§ 6. Второе начало tnepMoдинамики 53
мые количества тепла dQ'^ отрицательными, получаем (при
помощи разделения всего процесса на совокупность
бесконечно малых процессов)
J)-^-<o. F.14)
Представим весь процесс в виде двух переходов Л-^В
^ В—>А и предположим, что переход В—^ А состоит
только из бескснсчно малых обратимых процессов, а
переход А—>В включает и необратимые процессы. Найдем,
согласно уравнению F.11), разность энтропии для
перехода В—^А л перепишем неравенство F.14) в виде
в
А
В
Sb-Sj,>\^. F.15)
А
Это неравенство справедливо для любой сложной системы,
причем теперь естественно отменить наложенное на систему
при рассмотрении выражения (б.Юв) требование,
состоявшее в том, что внутри системы не должны иметь место
необратимые процессы теплообмена. Тогда количество
тепла dQ' состоит из dQfj^ (количество тепла, подведенное
к системе извне) и dQ^ (количество тепла, подведенное
путем внутреннего необратимого теплообмена). Поэтому
для термически замкнутой системы {dQ^ = 0) имеем
в
Sb-Sa>^\-P. F.15а)
А
Для каждого отдельного процесса dQi интеграл в неравенстве
F.15а) положителен (примерами такого рода процессов
являются расширение в пустоту и теплопроводность при
конечной разности температур). Отсюда вытекает
Sb > Sa. F.16)
Энтропия адиабатически изолированной системы мо^нсет
талька.возрастать. Таким, образом, второе начало термо-
54 Гл. I* Термодинамика. Общие принципы
динамики устанавливает определенное направление
течения процессов в природе; в механической картине мира
это избранное направление отсутствует.
Чтобы разъяснить противоположный смысл
соотношений F.16) и F.12), введем понятие о заторможенном
равновесии. Пусть состояние А как в случае неравенства
F.16), так и в случае равенства F.12) является
равновесным. При этом, однако, система состоит из различных
компонент, которым мешают реагировать друг с другом
искусственные приспособления. Представим себе,
например, что теплонепроницаемая перегородка разделяет две
газовые фазы и делает невозможным их смешивание.
Если удалить такую перегородку, что можно сделать,
затратив сколь угодно малую работу (например,
открытием крана или замыканием электрического контакта), то
начнется необратимый процесс, который прекратится при
достижении нового равновесного состояния В. Или можно
представить себе, что два вещества в условиях А не
вступают в химическую реакцию друг с другом, но начинают
реагировать при добавлении катализатора. Последний не
участвует в обмене энергии, но делает возможным
необратимый химический процесс и переход в новое равновесное
состояние В. О деталях этого процесса второе начало
термодинамики ничего не говорит, но оно позволяет
вычислить изменение энтропии при переходе из равновесного
состояния А в равновесное состояние В. Для этого надо
придумать произвольный обратимый процесс,
протекающий от состояния А к состоянию В, и вычислить интеграл
-s,= \
который не зависит от выбора пути между состояниями А
и В. Таким образом, ввиду неравенства F.16) в
равновесном состоянии В условия оказываются иными, чем в
состоянии А. Это значит, что существует заторможенный
необратимый процесс, при котором система переходит из
состояния А в равновесное состояние В {Sb > S^).
4. Простейший численный пример. Коэффициент
полезного действия идеальной тепловой машины, совершаю-
§ 6. Второе начало термодинамики 55
щей цикл Карно, согласно формуле F.8а), равен
7] = ^^^^ = ^ = 22%. F.17)
Здесь принято, что 7'i=100''C, Г2=20°С. Правда, реальный
процесс, происходящий в паровой машине, не является
циклом Карно (ср. фиг. 1 и 5). Все же верхняя кривая
на фиг. 1 примерно соответствует изотерме кипящей воды,
а нижняя—изотерме с температурой воздуха. Указанное
предельное 'Значение коэффициента полезного действия,
равное 22%, приближенно достигается в современных
конструкциях паровых машин, но не может быть
превзойдено.
Если в формуле F.17) фиксировать температуру T^f
то коэффициент полезного действия будет расти с
увеличением температуры Т^,
Перегретый водяной пар, используемый в паровозах,
эффективнее, чем водяной пар при обычной температуре
кипения. Однако давление водяного пара нельзя
увеличивать беспредельно. Поэтому в США уже в течение ряда
лет работают над созданием ртутных паровых турбин.
Путем соединения ртутной паровой турбины и турбины
с водяным паром может быть создан агрегат, работающий
между температурами 535 и 35°С. Коэффициент полезного
действия соответствующей идеальной паровой машины
равен
7] = 62%.
Мотор дизеля (температура воспламенения 400°С)
работает с большим перепадом температур, чем паровые
машины, и соответственно имеет значительно больший
идеальный коэффицент полезного действия. Заметим при
этом, что коэффициент полезного действия процесса
в двигателе Дизеля нельзя получить непосредственно из
схемы Карно, так как индикаторная диаграмма этого
процесса очень сильно отличается от цикла Карно.
Вообще можно сказать, что тепло от источника с более
высокой температурой ценнее, чем тепло от источника
с более низкой температурой. Работу можно считать
эквивалентной теплу, получаемому от источника с
бесконечно высокой температурой.
56 Гл. I. Термодинамика. Общие принципы
Если бы можно было понизить температуру
холодильника тепловой машины до абсолютного нуля, то коэффициент
полезного действия составил бы т] = 100%. Почему такой
коэффициент полезного действия не может быть достигнут,
объясняется в § 12.
Сделаем в связи с этим одно попутное замечание
исторического характера. Коэффициент полезного действия,
будучи записан при помощи нашей первоначальной 9-шка-
лы для случая бесконечно малой разности температур
^1 = 6» Ь2 = 0 — М, согласно формулам F.2) и F.7), дается
выражением
^^-'-'-^ = 'vW<^'-(^i')d^>¦ F-17а)
Функция C{^) — (f'{0)/ip{0) в старой литературе называется
«функцией Карно». Так же называется в Г-шкало функция
С{Т) = -У F.176)
В задаче I. 4 содержится несколько своеобразное
применение второго начала термодинамики к выводу
алгебраических неравенств.
Применение второго начала термодинамики ко всей
вселенной, которое впервые было сделано Клаузиусом и
привело к предсказанию им «тепловой смерти вселенной»
(так как с возрастанием энтропии все разности температур
нивелируются и тепло как источник получения работы
обесценивается), здесь не будет обсуждаться. К тому же
мы считаем, что структура вселенной—замкнутая или не
замкнутая, расширяющаяся (возможно пульсирующая) или
статическая—является еще проблематичной.
Планк^) возражал (конечно правильно) против мнения
энергетиков, что сущность второго начала термодинамики
заключается в тенденции энергии к «обесцениванию».
Правда, во многих случаях увеличение энтропии означает
уменьшение имеющегося температурного перепада и,
следовательно, также уменьшение работы, которую можно
1) Planck, Theorie der Warme, der Einfuhrung in die theo*
retische Physik, Bd. V, § 36, Thermodyuamik, § 108. (См. перевод:
М. Планк, Введение в теоретическую физпку, часть 5, Теория
теплоты, § 30, ОНТИ, 1935.)
§ 6, Второе начало термодинамики 57
получить за счет передачи тепла. Однако Планк приводит
сам собой напрашивающийся пример полного
превращения тепла в работу, а именно изотермическое расширение
идеального газа с подведением тепла от источника с
высокой температурой при полном использовании
давления газа для совершения работы. В этом процессе
энергия не будет «обесцениваться», а, наоборот, будет
становиться «ценнее» (тепло полностью превращается в работу).
Для нас, так же как и для Планка, сущность второго
начала термодинамики состоит в существовании энтропии
и в том, что при некоторых, вполне определенных условиях
она не может уменьшаться.
5. Некоторые исторические замечания. Приведеннсе
выше доказательство второго начала термодинамики
принадлежит Клаузиусу^). Доказательство, данное Планком,
возможно, прощен бесспорно точнее, но оно более
абстрактно и, пожалуй, менее поучительно, чем приведенное
выше. Еще абстрактнее доказательство Каратеодори^);
оно также и проще, если о простоте доказательства судить
по малости предположений, необходимых для его
проведения. В самом деле, Каратеодори для системы из двух
жидкостей, которые по мере надобности разделяются
теплопроницаемой или теплонепроницаемой перегородкой,
делает единственное предположение, а именно, что в
окрестности любого адиабатически дости;нсимого состояния
имеются другие состояния, которых нельзя достичь
адиабатическим и обратимым путем, т. е, либо недости:нсимые
вообще, либо такие, в которые система мо^неет попасть
лишь в результате необратимого процесса.
Этого чрезвычайно экономичного постулата достаточно,
чтобы математически доказать существование функции
состояния—энтропии.
Приведем сначала точку зрения, высказанную по
поводу этой теории самим Каратеодори^): «Можно поставить
^) R. СI а U S i U S, Mechanische Warmetheorie, 1876.
2) С. Gar а theodory, Math. Ann., 67 A909) und PreuB.Akad.,
Januar, '1925. См. также работу М. Born, Natural Philosophy
of Cause and Chance, Oxford, 1949, в которой, согласно
собственному мнению Каратеодори, его метод показан особенно ясно.
») Sitzungsher. PreuB, Akad,, 3. Juli A919), Nr. XXXIII.
58 Гл, I. Термодинамика. Общие принципы
вопрос, каким образом должна быть построена
феноменологическая термодинамика, чтобы при расчетах
использовать лишь непосредственно измеримые величины, т. е.
объем, давление и химический состав тела? Теория,
которая при этом возникает, логически неоспорима и
математически совершенна, так как она, исходя из реально
наблюдаемых фактов, обходится минимальным количеством
гипотез. Однако именно это преимущество делает ее мало
пригодной с точки зрения исследователя, не только потому,
что в ней температура появляется в качестве производной
величины, но прежде всего потому, что гладкие стены
искусственно воздвигнутых в этой теории зданий не
позволяют установить какой-либо связи между миром
видимой и осязаемой материи и миром атома».
В этой связи Планк^) отмечает, что для десяти молекул,
находящихся в замкнутом объеме, первое начало
термодинамики, правда, справедливо, однако с такой системой
нельзя сконструировать никакой тепловой машины
вследствие слишком больших флуктуационных явлений;
поэтому второе начало термодинамики для подобной системы
просто бессмысленно. Однако доказательство Каратеодори
не исключает с самого начала такую систему. Чтобы не
вступить в противоречие с действительностью, к этому
доказательству следует добавить еще некоторые
ограничивающие условия.
Чтобы хотя бы дать понятие о методе Каратеодори,
рассмотрим уже упоминавшиеся две жидкие системы
Lj и Eg, состояния которых будем описывать давлением,
объемом и некоторым параметром 6. Взаимная
зависимость этих переменных для каждой системы выражается
уравнением состояния. Эти уравнения состояния можно
представить в виде
Приведем обе системы в тепловой контакт, что можно
обозначить равенством 6j = 62. Тогда для такой сложной
системы E = Ei-f-E2 имеем уравнение
') Sitzungsber. Preufi. Akad., 453 A921).
§ 6, Второе начало термодинамики 59
Т. е. ИЗ четырех переменных только три являются
независимыми. Обозначим их через х, у и z. Тогда первое
начало термодинамики гласит, что количество тепла,
подведенное к системе S обратимым путем, выражается
дифференциалом Пфаффа
dQ = Xdx + Ydy-]'Zdz, F.18)
где X, F, Z—функции ж, у, z, В общем случае
выражение F.18) не является полным дифференциалом, как это
следует из A.4а). Соответствующие двучленные
дифференциалы Пфаффа для компонент S^ и Sg можно всегда
сделать полными дифференциалами при помощи
интегрирующего делителя, что уже отмечалось в § 1. Отсюда
при учете постулата Каратеодори следует, что
выражение F.18) также обладает интегрирующим делителем
(собственно говоря, множеством последних). Это означает
существование абсолютной температуры и энтропии как
функций состояния.
6. К вопросу о взаимоотношении энергии и энтропии.
Приведем здесь одну заметку^) Роберта Эмдена, который
показал глубокое понимание термодинамики в своих
фундаментальных работах по астрофизике (газообразное
ядро!) и метеорологии (серая атмосфера): «На вопрос,
почему мы топим зимой, неспециалист ответит: чтобы
сделать комнату теплее; знаток термодинамики выразится,
возможно, таким образом: чтобы подвести недостающую
энергию. В таком случае правым окажется профан, а не
ученый».
«В соответствии с фактическим положением вещей
предположим, что давление воздуха в комнате всегда равно
атмосферному. Энергия единицы массы воздуха в комнате
равна
^и*
и, следовательно, энергия единицы объема равна
иг^с.рТ, F.19)
1) R. Е m d е п, Why do we have Winter Heating? Nature,
141, 908 A938).
60 г л, I, Термодинамика, Общие принципы
или, при учете уравнения состояния,
и^ = ^. F.20)
Следовательно, количество энергии в комнате не зависит
от температуры, полностью определяясь
барометрическим давлением. Вся энергия, которую мы вводим в
комнату при отоплении, уходит через поры в стенах наружу».
«Я приношу бутылку красного вина из холодного
погреба, и она принимает температуру теплой комнаты.
Она становится теплее, однако внутренняя энергия ее
увеличивается за счет наружного, а не ]юмнатного воздуха».
«Почему же всё-таки мы топим? —По той же самой
причине, по которой жизнь на Земле была бы невозможна
без солнечного излучения. При этом дело заключается
не в падающей энергии. Последняя будет снова излучена
вплоть до пренебрежимо малой доли, подобно тому как
человек не меняет своего веса, несмотря на принятие
пищи. Условия нашего существования требуют известной
температуры тела и чтобы ее поддерживать, используется
не увеличение энергии, а понижение энтропии».
«Будучи студентом, я с пользой прочел небольшую
книгу Ф. Вальда «Царица мира и ее тень». Имелась
в виду энергия и энтропия. Достигнув более глубокого
понимания, я пришел к выводу, что их надо поменять
местами. В гигантской фабрике естественных процессов
принцип энтропии занимает место директора, который
предписывает вид и течение всех сделок. Закон сохране-
нения энергии играет лишь роль бухгалтера, который
приводит в равновесие дебет и кредит».
Вычисления и критические замечания к этому пункту
даны в задаче 1.2.
§ 7. ТЕРМОДПНАМПЧЕСКПЕ
ПОТЕНЦИАЛЫ П СООТНОШЕНИЯ ВЗАИМНОСТИ
Для описания простой гомогенной системы (с одной
механической и одной термической степенями свободы,
например газа, пара или жидкости) мы располагаем двумя
парами переменных
р, V и Т, S.
§ 7. Термодинамич. потенциалы и соотношдниА езаи^^ности 61
Первое начало термодинамики, записанное в этих
переменных для одного моля или для единицы массы вещества,
принимает вид [см. § 5, п. 4, уравнение E.11)]
du^Tds-pdv. G.1)
Здесь независимыми переменными являются две
экстенсивные величины S ж V, зависимыми — сопряженные им
интенсивные величины Тир. Таким образом, энергию и
следует рассматривать как функцию переменных s ш v:
u = u{s, v).
Две другие переменные, согласно уравнению G.1), заданы
производными
Однако независимые переменные можно выбирать
самыми различными способами. Так, возможны четыре
варианта, в которых независимыми являются одна
термическая и одна механическая переменные:
5, v; 5, р; Т, V] Г, р. G.3)
Здесь уместно вспомнить о преобразования Лежандра,
важное значение которого для математического анализа,
механики и термодинамики подчеркивалось в т. I, § 42.
Это преобразование говорит о следующем: чтобы в
дифференциале Пфаффа G.1) od?iy независимую переменную
{например^ s) заменить ей сопряженной, надо из
зависимой переменной {в нашем случае и) вычесть произведение
двух независимых сопрялсенных переменных {в нагием
случае Ts).
Аналогичное утверждение справедливо также в случае,
когда надо заменить две первоначальные переменные или
когда число переменных превышает два. Вследствие этого
четыре возможные варианта G.3) соответствуют
следующим четырем выражениям:
и E, v)\ hE, р)=и-\'pv\ f{T,v)=-u- Ts\
Энергия Энтальпия Свободная энергия /п А\
g{T, p)==u-Ts + pv.
Свободная энтальпия
(термодинамичесний потенциал)
62 Гл. I. Термодинамика. Общие принципы
Функция, обозначенная через Л, соответствует уже
введенной при помощи соотношения D.9) функции Я,
рассчитанной на один моль или единицу массы.
Общеупотребительные названия и обозначения термодинамических
функций представлены в таблице.
Выясним сначала чисто формально целесообразность
введения этих функций, составив соответствующие
дифференциалы и подставив в них du из уравнения G.1):
dh = du + р dv '\-vdp=^T ds + vdp,
df = du-Tds-'SdT= -pdv.-sdT, G.5)
dg — du — 'rds — sdT-\-pdv + vdp= —sdT -{-vdp.
Правые части этих уравнений показывают, что
дифференциалы rf/г, df и dg, будучи выражены через
соответствующие независимые переменные, приобретают такой же
простой вид, как и дифференциал du в переменных 5 и г;.
Функции G.4) называют термодинамическими
потенциалами, так как, зная их, можно получить сопряженные
переменные дифференцированием этих функций по
соответствующим аргументам, подобно тому как получают
компоненты силы из потенциала последней. Энергия также
заслуживает этого названия, как видно из формул G.2).
Соответствующие выражения для потенциалов Л, / и g
приведены в таблице. Специально отметим, что с выбором
потенциала устанавливаются также и соответствующие
независимые переменные. Например, свободная энергия /
имеет свойства потенциала только по отношению к
переменным V, Т я теряет эти свойства при другом выборе
переменных.
Из формул G.2) и соответствующих им формул в
таблице теперь непосредственно вытекают крайне важные
и глубокие термодинамические соотношения, которые
сведены в предпоследнем столбце нашей таблицы.
Сначала рассмотрим третье из этих соотношений
(^).-A)г- <'•«)
Полагая здесь ds = dq/T, имеем
\ дТ Jv Т dv\ T=const. ¦
G.7)
Таблица
Потенциалы
и, и
Я, h
h=zU'\- pv
g=h--Ts =
= f + pv =
= u—Ts + pv
Независимые
переменные
V, s
du — Tds — pdv
Pf s
dhz=Tds-\-vdp
V, T
df^^—sdV—pdv
dg= -'SdT-\'Vdp
Сопряженные
переменные
~V ds )p
/ dh\
\dTjr,
Термодинамические
соотношения
(^ дТ \ _ f др\ _ д^и
\ dv Js \ ds Jv~dvds
\др /8~~\д8 Jp'^dpds
\dv jT'^KdT Jv'^ дидТ
vWt"" \dTjp дрдт
Обозначения
и названия
Энергия и (Клау-
зиус) е (Гиббс)
Энтальпия Я
(Льюис и Рендал)
ЛГ (Гиббс) J
(теплотехника)
Свободная энергия
F (Гельмгольц) Ц/
(Гиббс)
Свободная
энтальпия С (Гиббс);
называют также просто
термодинамическим потенциалом
64 Гл. I. Термодинамика. Общие принципы
В этом соотношении слева стои<г (с точностью до
знаменателя р) термический коэффициент давления Р [см.
выражение A.5)]. Второй множитель справа есть «теплота
изотермического расширения», т. е. то количество тепла ^),
которое необходимо сообщить системе для расширения при
постоянной температуре.
Замечательно, что соотношение G.7), которое было
обосновано только для гомогенной системы, дает важное
указание относительно перехода между двумя
гомогенными системами, а именно, двумя агрегатными
состояниями одного и того же вещества. Особый интерес
представляет равновесие между родой и водяным паром. Если
интерпретировать р как давление пара при температуре Т
и заменить {dq/dv)^ через Дд/Ду, где Дд обозначает
теплоту испарения 1 моля (или 1 г), а Дг; — изменение объема
при испарении 1 моля (или 1 г), то уравнение G.7)
становится идентичным известному уравнению Клапейрона,
сыгравшему очень важную роль в теории паровых
машин (см. § 16).
Теперь применим соотношения таблицы для вывода
замечательной я совершенно общей формулы для разности
Ср—с^. Из первого начала термодинамики, записанного
в форме G.1), получаем при фиксированной температуре
Отсюда в силу G.6) следует
С другой стороны, если написать первое начало
термодинамики в форме
dg = du + pdv=[(^^^\ + p]dv + (^-l;-\dT,
^) В прежней литературе эту величину обозначали через М
и измеряли в калориях. Если 1/Т обозначить через функцию Карно
С(Т) [см. формулу F.176)], а механический эквивалент тепла—
через /, то правая часть соотношения G.7), примет вид JCM.
С этим связан мнемонический прием, используемый для запомина-
вия формулы Клапейрона: ее произносят как James Clerk Maxwell
dp/dt .(первые буквы образуют произведение JCM).
g 7 * Термодинамич, потенциалы и соотношения взаимности 65
то при постоянном объеме v получаем
а при постоянном давлении
',=-'^U.-[C-*-)r+^] (wKwl- ('¦«)
Разность выражений G.86) и G.8а) дает
Подставляя сюда G.8), находим
Два последних множителя в правой части представляют
собой термический коэффициент давления р и
коэффициент теплового расширения а, если в определяющих их
выражениях A.5) отбросить знаменатели р и v. В связи
с этим формулу G.9) можно записать в виде
Так как для идеального газа а = р = l/T, то для этого
частного случая формула G.9а) принимает вид
пг, i R для одного моля,
с—с= —=1 G.96)
р » Т I R/^ для единицы массы, ^ * ^
как это и должно быть согласно § 3.
Отметим здесь, что в этом выводе формулы G.96) для
идеального газа не было использовано дополнительное
калорическое условие (см. § 4, п. 1), согласно которому
внутренняя энергия идеального газа зависит только от
температуры. Это связано с тем, что в действительности
калорическое условие не является независимым, а
представляет собой одно из следствий второго начала
термодинамики, примененного к идеальному газу. Чтобы
убедиться в этом, выразим правую часть соотношения G.8)
через давление при помощи уравнения состояния
идеального газа: {др/дТ)^-^р/Т. Подставляя это в соотноше-
66 Гл. I. Термодинамика, Общие принципы
ние G.8), получаем (9гг/9?;)т = 0, т е. внутренняя
энергия не зависит от объема, а определяется только
температурой.
Рассмотрим также несколько подробнее последнее
термодинамическое соотношение нашей таблицы. Подставляя,
как и раньше, ds = dq/Ty получаем
Саг jp= '"Тдр\т=.сотх' ^'^•^^^
Слева здесь стоит (умноженный на v) коэффициент
теплового расширения а. Последний множитель справа назовем
«теплотой изотермического сжатия». Эта величина,
вообще говоря, отрицательна, т. е., для того чтобы тело
находилось в условиях повышающегося давления при
постоянной температуре, необходимо отводить тепло (в
противном случае при сжатии происходит нагревание
тела). Этому отвечает тот факт, что а, вообще говоря,
положительно [два отрицательных знака в правой части
соотношения G.10) компенсируются]. Однако имеются
исключения. Наиболее известным из них является вода
между О и 4°С. Согласно формуле G.10), теплота
изотермического сжатия в этом температурном интервале будет
положительна: чтобы не охладить воду при увеличении
давления, необходимо подводить к ней тепло (см. также
задачу 1.6). То же самое в определенных температурных
интервалах имеет место для каучука и йодистого
серебра. Рентген высказал предположение (позднее
подтвержденное другими исследователями), что аномалия в
поведении воды обусловлена ее склонностью к полимеризации
вблизи точки замерзания; поэтому кристаллизация,
имеющая место при 0°, фактически частично происходит еще
раньше.
Все четыре термодинамических соотношения были
выведены Максвеллом в его «Теории тепла» (Лондон, 1883 г.)
на основании элементарных геометрических соображений;
Максвеллом же были даны и соответствующие словесные
формулировки. Повидимому, он чувствовал сам, что в
этом случае дифференциальный метод все же много
проще, чем произведенное в его книге элементарное
рассмотрение. Поэтому в примечании к гл. IX он добавил
содержащиеся в нашей таблице аналитические формулировки.
ff 8. Термодинамические равновесия 67
Интуитивное толкование смысла знаков в этих
соотношениях взаимности приводит к «принципу Ле-Шателье—
Брауна» (аналогичному правилу Ленца в
электродинамике); однако последний менее точен, чем утверждения,
содержащиеся в нашей таблице ^).
Ясной систематичностью нашей таблицы мы обязаны
великому термодинамику и статистику Вилларду Гиббсу.
Его первые работы, затерявшиеся в Трудах
Коннектикутской академии от 1876 и 1878 гг., стали общеизвестны
лишь после того, как в 1902 г. Оствальд издал их на
немецком языке под названием «Термодинамические этюды».
С точки зрения Гиббса четыре потенциала, м. Л, /, g, или
и, Я, F, G, рассматриваются как равноправные. Выбор
между ними связан только с выбором независимых
переменных [см. G.3)]. В уравнении E.7) уже
подчеркивалось, что простейшая формулировка процесса Джоуля —
Кельвина состоит в условии равенства энтальпий: Н' = Л".
Соответствующее равенство свободных энтальпий G
определяет равновесие фаз. В физической химии и
электрохимии основную роль играет свободная энергия F, которая
также дает меру химического сродства, Планк
обыкновенно предпочитал «потенциальную функцию»
Ф - g J u+pv
^*^ rp *-* fp 1
которая действительно очень удобна в статистических
вопросах, но несколько выпадает из прекрасной
систематики Гиббса.
§ 8. ТЕРМОДИНАМИЧЕСКИЕ РАВНОВЕСИЯ
1. Незаторможенное термодинамическое равновесие
и максимум энтропии. В § 6, п. 3, мы показали, что
энтропия замкнутой системы не может уменьшаться. Усло-
1) См. Р. Ehrenfest, Zs. Phys. Chem., 77 A911); Planck,
Ann. d. Phys., 19 A934), с добавлением там же, 20 A935); далее
Т. Ehrenfest, de И аа s-Loren t z, Physica, 2 A935), с
возражением Планка там же. Главной проблемой в этой дискуссии было
выяснение различия в понятиях интенсивных и экстенсивных
величин, имеющего решающее значение для четкой формулировки
принципа Ле-Шателье—Брауна.
68 Гл. I, Термодинамика. Общие принципы
вия замкнутости состояли в том, что система не получает
тепла и не совершает работы. Их можно представить
также, как условия постоянства внутренней энергии
системы и и ее объема V{dU = Oy dV = 0). Если в
замкнутой системе устранить все связи, то она будет стремиться
к конечному состоянию, в котором энтропия имеет
максимальное значение. Назовем это состояние
«незаторможенным термодинамическим равновесием».
Самопроизвольное изменение незаторможенного
равновесия невозможно. Действительно, это означало бы
повторное увеличение энтропии, что находилось бы в противоречии
с нашим предположением о том, что энтропия уже достигла
максимума. Мы можем, однако, рассматривать виртуальные
изменения состояния, 8, совместимые с условиями dU = 0^
с?К = 0, которые, конечно, не могут происходить
самопроизвольно. (Например, в сосуде с газом, температура
и давление в каждой точке которого одинаковы,
виртуальное изменение состояния может состоять в повышении
на 87* температуры одной его половины и таком же
понижении температуры другой половины.) Для таких
виртуальных отклонений от незаторможенного
термодинамического равновесия справедливо неравенство
86" < О, когда 8C/ = 0, 8F = 0, (8.1)
или, иначе,
*5' = *5'макс.> когда ^7 = const, F = const. (8.1а)
Если бы можно было указать такое виртуальное
изменение состояния (при ьи-О, 8К = 0), для которого 86*>О,
то исходное состояние не описывало бы незаторможенного
равновесия. Это означало бы, что существуют еще какие-
то связи, по устранении которых энтропия продолжает
возрастать.
Соотношение (8.1) или (8.1а) является одним из двух
условий равновесия, установленных Гиббсом. Другое
условие, менее важное для нас, гласит:
Ьи > О, когда IS = 0, 8F = О, (8.2)
или
U = Uuim.y когда 15 = const, F = const. (8.2а)
§ 8» Термодинамические равновесия
Внутренняя энергия в состоянии равновесия имеет
минимум. Это утверждение напоминает критерий равновесия
(минимум потенциальной энергии) в общей механике^).
2. Система в незаторможенном термодинамическом
равновесии является изотермической и изобарической.
Из определения незаторможенного термодинамического
равновесия (8.1) следует, что давление и температура не
зависят от положения точки внутри системы.
Действительно, предположим, что это не так. Рассмотрим тогда два
элемента пространства, температуры и давления в
которых равны Tj, /?1 и 7*2, Р2» ^ произведем такое
виртуальное изменение состояния, чтобы внутренняя энергия
первого элемента изменилась на BC/j, а его объем —на bV^.
Тогда для второго элемента пространства в силу (8.1)
изменения соответствующих величин будут равны 5^2 =
= —Ьи^, 8^2= — 8Fj. Изменения концентраций или
количеств вещества, имеющихся в отдельных фазах, при
этом не должны приниматься во внимание. Таким
образом, согласно условию (8.1), получаем
-(^"-A)»^.+(t-ft)^''.-
Виртуальные изменения bU^ и oFj произвольны и не
зависят друг от друга. Поэтому эти виртуальные изменения
можно выбрать так, что при Т^ФТ^, р^ ф р^ данное
неравенство не будет выполняться. Следовательно, наше
предположение неверно.
3. Дополнительные степени свободы в
заторможенном равновесии. Состояние системы, которая находится
в незаторможенном термодинамическом равновесии, часто
определяют заданием внутренней энергии U, объема V
и числа независимых компонент (см. § 14). Рассмотрим
^) См., например, А. Sommerfeld, Partielle Differential-
gleichurengen der Pbysik (Bd. VI) Leipzig, 1948, § 25. (См. перевод:
A. Зоммерфельд, Дифференциальные уравнения в частных
производных физики, ИЛ, 1950, § 25.)
70 Гл. I, Термодинамика, Общие принципы
теперь систему Е, которая еще не находится в
равновесии. Ее состояние будет определено, если, кроме U, V
и числа независимых компонент, задать еще другие
величины х^. Они могут описывать, например, распределение
компонент по отдельным фазам или концентрации
отдельных компонент, между которыми возможны химические
реакции. Далее, эти величины могут характеризовать
локальные различия, если всю систему разбить на
достаточно малые элементы объема и пронумеровать все х^
в соответствии с нумерацией элементов объема. Мы
рассматриваем только такие неравновесные состояния,
которые при фиксированных ж^ можно считать состояниями
заторможенного равновесия (это предположение
существенно также для § 21). Таким образом, энтропию системы
мо^нсно вычислить как сумму энтропии всех элементов
объема для этого затормо^нсенного равновесия.
Ограничимся здесь рассмотрением изотермических и изобарических
систем. Изменение энтропии при переходе от
заторможенного равновесия (С/, F, х^) к заторможенному равновесию
{U + dU, V + dVf Xi + dx^) найдем из следующего
уравнения, являющегося обобщением уравнения G.1):
TdS = dU + pdV + J^ X^dXi. (8.3)
i
Характер перехода (обратимый или необратимый) при
этом не существенен, так как dS означает разность
энтропии конечного и исходного состояний, которые оба
являются бесконечно мало отличающимися друг от друга
состояниями заторможенного равновесия. Назовем
коэффициенты Х^ обобщенными силами, соответствующими
дополнительным степеням свободы ж^.
4. Экстремальные свойства термодинамических
потенциалов. Присоединим к системе S «внешнюю среду»,
которую можно представить себе в виде очень большого
теплового резервуара Е^. Все величины, относящиеся к
Sq, снабдим индексом 0. Пусть полная система,
составленная из S и Eq, является термически изолированной.
В этих предположениях энтропия всей системы не может
уменьшаться:
dS + dSQ>0, (8.4)
§ 8. Термодинамические равновесия 71
Знак равенства мы исключаем, предполагая, что имеющие
место в системе S изменения состояния, как и все
реальные процессы, необратимы. Однако, как уже упоминалось,
система Е всегда должна быть изобарической й
изотермической, т. е. она должна находиться в механическом и
термическом равновесии (но не обязательно в химическом или
фазовом равновесии). Теплообмен между системами Е и
Eq допускается только тогда, когда Е находится при
температуре теплового резервуара Tq. Будем предполагать,
что система Е^ настолько велика, что температура ез не
меняется сколько-нибудь заметно при теплообмене с
системой Е. Тогда
rf^o = ^"- (8.5)
•*¦ О
Изменения объема системы Е всегда должны быть
таковы, чтобы давление р в ней равнялось внешнему
давлению. Первое начало термодинамики, примененное к
системе Е и подведенному к ней от Е^ количеству тепла
dQ= - dQ^y дает
dU + pdV^^dQ^. (8.6)
Тогда справедливо неравенство
dS>-^(dU-VpdV). (8.7)
Так как имеет место теплообмен, то, по предположению,
Г = 7*0 и неравенство (8.7) вытекает из выражений (8.4) —
(8.6). Если же Г =7^ Г^, то никакого теплообмена нет; при
этом dU -\-pdV исчезает, согласно первому началу, и (8.7)
означает просто, что dS > О, как уже было выведено
в F.16) для теплоизолированной системы. Следовательно,
неравенство (8.7) справедливо при любом обмене теплом
п работой между нашими системами.
Из соотношений (8.3) п (8.7) далее следует, что для
каждого изменения состояния dU, dV, dx^y которое может
протекать самопроизвольно,
J]X^dXi>0. (8.8)
Для обратимых изменений состояния всей системы
Е + Sq, а, следовательно, также и для одной системы S
72 Гл. /, Термодинамика. Общие принципы
В (8.4) следует поставить знак равенства. Соответственно
знак равенства появится и в (8.8). Таким образом,
справедлива теорема: обратимое изменение состояния
изотермической и изобарической системы ^ имеет место тогда^
и только тогда, когда: а) теплообмен с окру писающей
средой происходит обратимым путем {т. е. при условии
равенства температур системы Е и окру^исающей
среды), б) внутреннее давление р равно внешнему давлению
и в) в процессе изменения состояния соблюдается
равенство
2^Ai = 0. (8.9)
i
Последнее условие выполняется, например, когда все
Xi имеют постоянные значения. Равным образом оно
выполняется, если х^ фО, а соответствующая величина Х^
обращается в нуль. Каждый переход из состояния 1, с
параметрами U^, Fj, ж^^, в состояние 2 с параметрами C/g»
Fg, ^i2 можно (и притом различными способами)
произвести так, что равенство (8.9) выполняется в течение
всего перехода. Один такой пример рассмотрен в п. 5.
Рассмотрим теперь изотермическую систему с
фиксированными температурой и объемом (близкие к этому
условия можно создать, помещая систему в водяную баню
с постоянной температурой Г). Дифференциал свободной
энергии равен dF = dU —TdS. Отсюда, учитывая
неравенство (8.7) и условие dV — O, получаем для
самопроизвольного процесса
rfF<0, (8.10)
т. е. свободная энергия уменьшается^). Существует
состояние, в котором свободная энергия имеет минимум;
по достижении его дальнейшие процессы становятся
невозможными. Следовательно, условие равновесия
рассматриваемой системы гласит
F = Fmiih.t когда 7" = const, F = const. (8.11)
^) Условие постоянства давления во всех точках
рассматриваемой системы (см. п. 3) не является необходимым для
справедливости этого нслснчения. Действительно, в случае различия
давлений в разных частях системы доказательство неравенства
(8.7) не изменяется, так как dV^O.
S 8» Термодинамические равновесия 73
Соответственно при любом виртуальном изменении
состояния
8F>0, когда 8Г = 0, 8F = 0. (8.11а)
В случае изотермически-изобарической системы, т. е.
системы с фиксированными температурой и давлением (эти
условия можно осуществить, поместив систему в водяную
баню с температурой Т и давлением р и обеспечив
возможность непрерывного выравнивания температуры и
давления), будем исходить из свободной энтальпии G = U —
— TS + pV. Ее дифференциал в данном случае равен
dG = dU-TdS + pdV, так как dT = 0, dp = 0. Отсюда,
пользуясь неравенством (8.7), получаем для изменения
С, С/, 6", F в самопроизвольном процессе
dG<0, (8.12)
т. е. свободная энтальпия уменьшается.
В состоянии, которому соответствует минимум свободной
энтальпии у система никаких дальнейших изменений не
претерпевает. Условие равновесия, следовательно, имеет вид
С = Смин., когда Г = const, /? = const, (8.13)
или, в эквивалентной форме,
5G>0, когда 8Г = 0, 8/? = 0. (8.13а)
В теории фазовых превращений эта последняя
формулировка играет особенно важную роль. Вообще в
дальнейшем свободная энтальпия будет занимать первое место
среди четырех потенциалов, введенных в § 7.
Предоставляем читателю самому доказать соотношения
(8.2) и (8.2а) и вывести аналогичное экстремальное
свойство энтальпии В для изобарической системы при
условиях 66' = О, 8/? = 0.
5. Теорема о максимальной работе. Вычислим теперь
работу, которую может отдать система Е окружающей ее
внешней среде, когда она перейдет обратимым путем из
состояния 1 с определенной температурой TqB другое
состояние 2 с той же самой температурой. Будем считать, что
переход не обязательно является изотермическим, но
система Е может обмениваться теплом с окружающей
74 Гл. I, Термодинамика, Общие принципы
средой только при температуре Т^\ иначе говоря,
изменения состояния системы при Т Ф Tq должны быть
адиабатически обратимыми.
Согласно определению свободной энергии F = U —TS,
для бесконечно малого изменения состояния системы
имеем в силу (8.3)
dF = dU-TdS--SdT= ^SdT^pdV-^X^dXi.iSAi)
i
Условие обратимости означает, что во время полного
перехода 1 —> 2 сумма 2 ^i ^^i = 0. Поэтому для работы,
i
совершенной системой, получаем выражение
2 2
\^pdV=:F,-F^^^SdT.
1 1
Докажем теперь, что
2
^pdV=.F,^F,, (8.15)
1
т. е. что \ S dT при наших предположениях исчезает.
Для этой цели заметим, что переход 1—>2 может
содержать изотермические участки при температуре Т = Т^,
На этих участках dT = 0, Остальная часть пути
представляет собой один или несколько адиабатических
процессов, каждый из которых характеризуется постоянной
энтропией и происходит между одинаковыми начальной
и конечной температурами Tq. Следовательно, для них
То То
^ SdT=^S^dr = 0.
То То
Если процесс проводить необратимо, то работа,
совершаемая системой, меньше, чем F^ —F^. В противном
случае можно было бы обратимым образом перевести
систему в исходное состояние, совершив при этом
неотрицательную работу за счет эквивалентного количества
тепла, заимствованного из окружающей среды, находящейся
при температуре Т^. Это, однако, противоречит второму
§ 8. Термодинамические равновесия 75
началу термодинамики. Следовательно, изменение
свободной энергии в некотором процессе определяет также
максимальную работу, которую система Е может
совершить при этом над внешней средой при условии, что
начальная и конечная температуры системы совпадают
с температурой среды Tq и теплообмен со средой
происходит лишь при этой температуре.
Во многих изложениях термодинамики эта теорема
о максимальной работе доказывается при специальном
предположении об изотермическом характере перехода
1-^2. Наша формулировка, однако, шире, так как для
некоторых систем невозможно найти обратимый
изотермический переход 1-^2, но том не менее переход,
соответствующий максимуму работы, все же возможен.
Свойство (8.15) дало Гельмгольцу основание назвать
потенциал F «свободной энергией». Разность ее значений
для двух состояний 1 и 2, имеющих равные температуры
Гд, выражает ту часть изменения энергии U^ — U^,
которая при обратимом процессе 1 —> 2 может пойти на
совершение внешней работы или (в обратном процессе) должна
покрываться за счет работы, производимой над телом.
При этом предполагается, что теплообмен с окружающей
средой происходит обратимым образом и только при
температуре Tq. Поэтому функцию и — F =TS можно
назвать «связанной энергией».
Покажем теперь на одном простом, но характерном
для общего случая примере, каким образом можно провести
процесс 1 —> 2 так, чтобы при этом постоянно
выполнялось равенство 2 ^i ^^i = О- Д^^ этого рассмотрим дис-
i
социирующий газ, скорость диссоциации которого можно
произвольным образом уменьшать. Пусть а; — степень
диссоциации. В состоянии незаторможенного равновесия она
имеет значение х(Т, V). Вместо (8.3) теперь мы имеем
TdS = dU + pdV f Xdx. Пусть начальное состояние
характеризуется величинами Г^, Fj, х^, а конечное — Tq, Fg,
х^. По крайней мере одно из значений степени диссоциации,
т. е. Xi или ^2, соответствует отклонению от равновесного
значения. (В противном случае последующие рассуждения были
бы тривиальны.) В первом процессе величины,
характеризующие систему, обратимо адиабатически изменяются
76 Гл, I, Термодинамика. Общие принципы
при неизменном х-^ от значений Г^, F^, х^ до таких
значений Т', V\ чтобы Xi = x{T\V'). Мы находим,
следовательно, такие значения температуры и объема, для
которых заданное значение х^ соответствует степени
диссоциации в состоянии незаторможенного равновесия. Тогда
можно устранить связи, оставляющие Xj^ неизменным,
причем дальнейших изменений в системе происходить
не будет.
Затем совершим адиабатический обратимый процесс,
при котором величина Xj^ переходит в Х2 = х{Т'', F"),
где Г", F" —новые значения температуры и объема.
Система проходит исключительно через состояния
незаторможенного равновесия, так что, согласно равенству (8.9),
величина Xdx всегда исчезает (очевидно, что при с?ж =?^ О
условие равновесия диссоциации дает X = 0). Теперь
фиксируем значение iCg, а Г" и V" изменяем обратимо
адиабатически до достижения неходкой температуры Т^
и объема F'". Наконец, при фиксированном х^ обратимо
изотермически изменяем объем системы, пока он не
достигнет заданного конечного значения Fg.
В этом примере можно было бы и весь процесс
провести изотермически, так как диссоциационное равновесие
зависит от давления. Для этого следовало бы вести
изотермический обратимый процесс при фиксированном ж^
(начиная от давления р^) вплоть до наступления
равенства Xj^ — x{Tq, /?'), т. е. до тех пор, пока заданная
степень диссоциации не окажется равновесной. Далее надо
было бы опять устранить связи и изменить давление от р'
до /?", где /?" выбрано так, что Х2 = х{Т^у р"). (В течение
этого процесса Х = 0.) Наконец, при фиксированном х^
следовало бы изменять давление до тех пор, пока не был
бы достигнут заданный конечный объем.
В заключение отметим еще следующее. Максималгная
работа, которую можно получить при бесконечно малом
изменении состояния с одинаковыми начальной и
конечной температурами Г^, равна pdV-\-^X^dx^, Поэтому
i
данное выражение следует назвать обобщенным
дифференциалом работы. Это полный дифференциал и,
следовательно, ему соответствует некоторая функция состояния,
§ 9, Уравнение Ван-дер-Ваальса 77
характеризуюш;ая совершенную работу (при фиксированной
температуре Г^, так же как и свободная энергия F),
§ 9. УРАВНЕНИЕ ВАН-ДЕР-ВААЛЬСА
Перейдем теперь от идеального газа к реальным газам,
следуя при этом диссертации Ван-дер-Ваальса:
«Непрерывность газообразных и жидких состояний» (Лейден, 1873 г.).
Действительно, Ван-дер-Ваальсу удалось
количественно исправить уравнение состояния идеального газа и
установить уравнение состояния, которое качественно
описывает сжижение газов. Больцман ^) назвал Ван-дер-Ваальса
«Ньютоном реальных газов».
Уравнение Ван-дер-Ваальса, написанное для одного
моля газа, имеет вид
-"¦» а //-V л V
P-UiTb-r^- (9-1)
Введенная здесь константа b описывает собственный объем
молекул; константа а характеризует силы сцепления между
молекулами газа и связана с капиллярными явлениями.
(Атомистический смысл констант а и b рассмотрен в § 26.)
При а = Ь = 0 или, что то же самое, при достаточно
больших значениях v уравнение (9.1) переходит, как это и
должно быть, в уравнение состояния идеального газа.
Вместо (9.1) можно также написать
{p + Pa){v-b) = RT, ;7„ = ^, (9.1а)
где /?д — «внутреннее давление», добавляющееся к
«кинетическому давлению» р,
В качестве прямого следствия уравнения (9.1)
вычислим коэффициент теплового расширения. Дифференцируя
(9.1) и полагая dp = 0у получаем
^"v-b \{v-bf Rv^J^^'
откуда
1) Cm. Encykl. der Ma them. Wiss., Bd. V, 1, S. 550.
78 Гл. I, Термодинамика, Общие принципы
Это выражение обобщает формулу для коэффициента
теплового расширения идеального газа а^1/Ту
получающуюся из (9.2) при a = fc = 0.
Вычислим еще разность
а — 7^ =
2а / у-Ьу
RT\ V J
-^(^у ¦
(9.2а)
Сохраняя здесь лишь первые степени а и Ьи считая, что
V ^ by RTv ^ а, получаем
Для большинства газов, например для кислорода
и азота, правая часть выражения (9.26) при обычных
температурах положительна. Следовательно, коэффициент
теплового расширения для этих газов больше, чем для
идеального газа. Только водород и инертные газы
являются исключениями. Для них силы сцепления, мерой
которых является константа а, настолько малы, что при
обычных температурах правая часть выражения (9.2а)
оказывается отрицательной. Поэтому прежде водород часто
называли «более чем идеальным» газом.
1. Ход изотерм. Картина вандерваальсовых изотерм
представлена на фиг. 7. Асимптотами изотерм служат ось
объемов и прямая v = b, параллельная оси давлений.
В области V <С Ь уравнение (9.1) не имеет физического
смысла.
Согласно уравнению (9.1), точка пересечения
изобары (р = const) с изотермой {Т = const) определяется
кубическим уравнением. Последнее имеет один или три
вещественных корня. Границу между двумя этими
случаями образует критическая изотерма T = Ti^^,, на
которой три точки пересечения сливаются в одну точку
перегиба {критическая точка г = Гкр., Р=Ркр.)у
касательная в которой горизонтальна.
§ 9, Уравнение Ван-дер-Ваальса
79
Чтобы определить v^^, и Гкр., вычислим dpjdv и
4- d'^p/dv^ и приравняем их нулю; получим
RT _2fl
гK »
ВТ
За
Отсюда находим
DT» ТУТ ^
(9.3)
Соответствующее значение давления р находим из
уравнения (9.1):
Фиг. 7. Изотермы в плоскости p^v согласно
уравнению Ван-дер-Ваальса.
Выражая при помощи этих соотношений константы а и b
через критические параметры /?кр., Vk^, и Гкр.» можно
переписать уравнение (9.1) таким образом, чтобы в него
80 Гл. I. Термодинамика. Общие принципы
входили лишь относительные величиньх
V р Т
"кр. ^кр. ^ кр.
Тогда получим
(^+^)C?-1) = 8х. (9.5)
Это уравнение выражает вандерваальсов закон
соответственных состояний—всегда справедливый закон подобия.
[Естественно, он выполняется лишь с той точностью, с какой
справедливо уравнение (9.1).] Впрочем, соответствующий
закон подобия существует для каждого уравнения
состояния, которое содержит только три индивидуальные
константы^). Чтобы получить его, следует лишь
исключить эти три константы, введя новые (безразмерные)
2. Энтропия и калорические свонства газа Ван-
дер-Ваальса. В то время как для идеального газа мы
вывели существование энтропии при дополнительном
калорическом условии (см. § 5, п. 4), в случае вандер-
ваальсова газа, наоборот, из факта существования
энтропии будут выведены определенные заключения о
калорических свойствах газа. В то время как калорические
свойства идеального газа выражались уравнением E.8)
du/dv = Oy теперь мы получим более общее соотношение
Физически это понятно: внутренняя энергия газа
складывается теперь не только из кинетической энергии
молекул, но и из потенциальной энергии их сил сцепления,
которые связаны с константой а. Эта потенциальная
энергия, как и для системы гравитационно
взаимодействующих материальных точек, отрицательна и
приближается к нулю при расширении газа. Следовательно,
энергия газа и должна расти с увеличением объема v,
как это и предсказывается соотношением (9.6).
^) См. J. de В о е г и сотрудники, Physika, 14, 139, 149, 320
A948).
§ 9. Уравнение Ван-дер-Ваальса 81
Ван-дер-Ваальс при установлении своего уравнения
состояния уже располагал основными законами
термодинамики. Он мог, следовательно, приспособить форму
своего уравнения состояния к принципу энтропии. Мы
покажем, что и уравнение (9.6) можно вывести из этого
основного принципа.
Положим, согласно определению F.10а),
ds^'Ji+f^. (9.7)
Подставив сюда величину р из уравнения (9.1) и
рассматривая и как функцию Т и v, получим
-Tp^+iVi+eb-^)""- (9-8)
Необходимое и достаточное условие того, чтобы это
выражение являлось полным дифференциалом, имеет вид
1 дЪ
Т дидТ
^±A^J^.^ ^Л (99)
дТ\Т dv^ v—b v^T J ' ^^'^^
При выполнении дифференцирования в правой части
средний член исчезает, а последний дает a/v^T^, Из
первого члена возникают два выражения, первое из которых
взаимно уничтожается с левой частью, в то время как
второе равно — j^ g^ • Таким образом, уравнение (9.9)
принимает вид
О— S. — A^-L± /о о \
Отсюда сразу получаем доказываемое соотношение (9.6).
Дифференцируя соотношение (9.6) по Г, получаем
^ = -^^=—- = 0 Г9 10)
Следовательно, теплоемкость с^, как и в случае
идеального газа, зависит только от температуры.
Вычислим также разность молярных теплоемкостей
Ср —с^, которая для идеального газа была равна газовой
82 I\i. I. Термодинамика, Обгиис принципы
постоянной R. Будем исходить из общего соотношения G.8в)
Производные {dv/дТ)^ и {du/dv)T даются выражениями
(9.2) и (9.6). Таким образом, для газа Ван-дер-Ваальса
имеем
RT v^
Постоянную а здесь можно считать малой. Пренебрегая
произведением малых величин аЬ, получаем вместо (9.12)
'^р-^» = -4г • (9.13)
RTv
Вернемся теперь к выражению для энтропии (9.8).
Принимая во внимание (9.6) и (9.10), его можно записать
в более простом виде:
ds = '^ + R^^. (9.14)
В интегральной форме, если, как и для идеального газа,
считать молярную теплоемкость с^ не зависящей от
температуры, это уравнение имеет вид
T,v
5 ds = s-s, = Onf^+R\n-^. (9.15)
То, го
§ 10. СЖИЖЕНИЕ ГАЗА ПО ВАН-ДЕР-ВААЛЬСУ
1. Интегральный и дифференциальный процессы
Джоуля —Кельвина. Условие постоянства энтальпии Н
справедливо не только для идеального, но и для любого
газа. Это уже подчеркивалось в связи с уравнением E.7).
При помощи таблицы, приведенной в § 7, получаем,
заменяя фигурирующие там дифференциалы ds, dp малыми
конечными приращениями Д5, Д/?,
dih = TAs-j'V^p. A0.1)
§ 10. CotcwHcenue газа no Ван-дер-Ваальсу 83
Переходя от перемевных s, р к переменным Г, /?, получим
Но, согласно определению с^^,
С другой стороны, из таблицы, помещенной в § 7, видно,
что
KdpJT^ \^дт)р
Поэтому уравнение, A0.1) принимает вид
Ah = c^AT+[v-T(^%\]^p. A0.2)
Таким образом, из условия постоянства энтропии
следует, что
где а — коэффициент теплового расширения. Подставив
сюда его значение из (9.2), придем к специальному
случаю газа Ван-дер-Ваальса. При учете уравнения (9.26)
соотношение A0.3) принимает вид
АТ^ВТ_
Ар Ср ^ '
Отсюда следует, что при расширении газа {Ар < 0) он
охлаждается (АГ < 0), если
Ij > fc. A0.4а)
Это неравенство выполняется для воздуха и для
большинства других газов. Путем достаточно длительного
расширения воздух моофсно произвольно охладить и^
наконец^ перевести в (нсидкое состояние — сконденсировать.
При промышлейном получении жидкого воздуха (и его
разделении на Ne, Аг и т. д.) с помощью машины Линде,
естественно, необязательно осуществлять в точности про-
84 Гл, /. Термодинамика, Общие принципы
цесс Джоуля — Кельвина. В этих машинах вместо пробок
из ваты используют понижающие вентили и увеличивают
коэффициент полезного действия путем применения метода
обратного потока, предложенного Линде.
Соотношение A0.3) еще встретится нам в § И. Оно
описывает конечный процесс Джоуля —Кельвина.
Последний получается экстраполяцией начального, точно
определенного дифференциального процесса, для которого
в силу A0.3) справедливо равенство
2. Кривая инверсии и ее техническое применение.
Исследуем теперь в общем виде, в какой части плоскости
переменных /?, Т расширение газа (А/? < 0) связано, как
и в случае A0.4а), с понижением его температуры
(АГ < 0). Иначе говоря, надо определить значения р тТу
при которых {dT/dp)f^ > 0. Ихменяо эта область значений
р и Т представляет интерес. Назовем ее пололсительной.
Она ограничена кривой инверсии, на которой (97'/9/?);^ = 0,
т. е., согласно A0.5),
а{р,Т) = ^. A0.5а)
Кривая инверсии отделяет положительную область от
нежелательной в технике отрицательной области. Как уже
говорилось, воздух и большинство других газов при
обычных условиях (давлении и температуре) всегда
находятся в положительной области. Это подтверждает фиг. 8.
Беря за основу точное уравнение Ван-дер-Ваальса,
находим в силу (9.2а) следующее уравнение кривой
инверсии:
|j(^y = 6. A0.56)
Здесь надо еще выразить v через р и Т, Переходя
сначала к приведенным координатам тс и т, получаем после
некоторых преобразований
IT = 24 УЗх - 12х - 27. A0.5в)
На фиг. 8 наряду с кривой инверсии, построенной по
уравнению A0.5в), изображена также экспериментальная
^ 10. Союьинсение газа по Ван-дер-Ваальсу
85
кривая инверсии для водорода (В. Мейсснер). В подписи
к фиг. 8 приведены коэффициенты для пересчета
приведенных значений в обычные для водорода и воздуха.
На фиг. 8 видно, что для воздуха производная {дТ/др)^^
при комнатных температурах и давлениях вплоть до
давления 450 am положительна; напротив, для водорода при
комнатных температурах она всегда отрицательна. Это
обстоятельство очень резко проявляется при катастрофах,
происходящих в тех случаях, когда сильно сжатый водород
Фиг. 8. Кривая инверсии для
дифференциального эффекта Джоуля—Кельвина в
приведенных переменных.
А—экспериментальная кривая для Иг, согласно Мейс-
снеру, В—теоретическая кривая для газа Ван-дер-
Ваальса, согласно уравнению A0.5в). Для Иг
Т=33,2':*' К, р=:13,2тс am. Для воздуха Т=132,5т;** К,
p=:34,57t am.
самопроизвольно воспламеняется при истечении из
поврежденных труб. Вообще при внезапном расширении водород
может охлаждаться лишь при температурах ниже —80° С.
Возвращаясь к охлаждению, происходящему в
результате процесса Джоуля—Кельвина, определим технически
важный интегралгный эффект
Р2
Pi
A0.5г)
В технических устройствах давление р^ после
разрежения в большинстве случаев приблизительно равно
атмосферному. Температура Т^ определяется в основное)
8в Гл. I. Термодинамика. Общие принципы
выбором условий предварительного охлаждения (при
сжижении воздуха пользуются холодной водой, при сжижении
водорода — жидким азотом). Произвольным (в известных
границах) остается только начальное давление р^. Чтобы
найти значение р-^, при котором охлаждение максимально,
продифференцируем интеграл в A0.5г) по нижнему
пределу и приравняем получающееся выражение нулю; получим
Однако это как раз условие того, что точка /?j, Т^ лежит
на кривой инверсии дифференциального эффекта Джоуля —
Кельвина. В соответствии с этим и конструируют
холодильные машины. Так, например, для сжижения
водорода наиболее благоприятная температура предварительного
охлаждения равна 64,5° К (ее получают при кипении азота
под пониженным давлением). Из кривой инверсии
находят, что соответствующее наиболее благоприятное
давление равно 160 am. Практически часто работают при
температуре 72° К и давлении 140 am. Для сжижения гелия
пользуются предварительным охлаждением до 14° К при
давлении /?i = 29 am. Температура 14° К достигается или
при кипении жидкого водорода под пониженным
давлением (Камерлинг-Оянес), или при охлаждении
газообразного гелия при помощи адиабатически обратимого
расширения (при этом газ совершает внешнюю работу).
Последний способ применяли Капица и с более полным
учетом термодинамических условий Мейсснер.
3. Границы области сосуществования жидкой и
газообразной фаз в плоскости р^ v. Слово фаза имеет
много значений. В общем смысле оно означает «форму
явления» (например, фаза световых колебаний, фазы луны
различные фазы политического развития). Ниже, в
главах, посвященных статистической физике, мы будем
говорить о многомерном «фазовом пространстве». В
термодинамике под «фазами» понимают различные агрегатные
состояния отдельного вещества, причем в твердом
состоянии различные структуры (кристаллические структуры,
аморфное состояние) считают различными фазами; при
наличии нескольких веществ в понятие «фаза» включаются
§ 10. Смсшисение газа по Ван-дер-Ваальсу^
87
различные химические группировки, которые они
способны образовывать.
В теории Ван-дер-Ваальса речь идет о равновесии
между газообразной (i) и жидкой {2) фазами, т. е. о
насыщенном паре и жидкости. Механическим условием этого
равновесия является равенство давлений, термическим —
равенство температур в обеих фазах. Среди четырех
Pi
^Пересыщение
В
Фиг. 9. Определение прямой Максвелла.
потенциалов таблицы, приведенной в § 7, наиболее
подходящим для случая одинаковых давления р и
температуры Т является свободная энтальпия G, Равенство
значений р и Т в силу (8.13а) влечет за собой равенство
потенциалов g обеих фаз: gi = g2- Пусть т^ и /Wg
—количество вещества соответственно в фазах 1 и 2; тогда,
согласно (8.13а),
8G = 8 (m^gj^ + m^g^) = (g^ - ^2) ^Щ = О,
так как Sm^ = — Ьт^: полное количество вещества т^ + /Wg
остается при варьировании постоянным.
Выберем на фиг. 7 одну из изотерм Ван-дер-Ваальса
при Т < Гкр. и проведем пересекающую ее изобару р < /?кр..
Из трех точек пересечения (фиг. 9) обозначим две внеш-
88 Гл. I. Термодинамика, Общие принципы
ние через А ii В\ соответствующие значения потенциала
g пусть будут gA ^ 8в- Они равны друг другу, так как
соответствуют одинаковым значениям р и Т. Поскольку
g==u — Ts'\-pv, из равенства gA и gB вытекает соотношение
ub-Ua-T {sb -Sa)-tP {vb - Va) = 0. A0.6)
Разность sb—Sa дается уравнением (9.15). Учитывая
равенства температур в точках А и В, получаем
sj,-SA = Rln^^^. A0.7)
Vj^ — O
Разность энергий ив — Ua можно найти, проинтегрировав
du вдоль изобары р = const (любой другой путь
интегрирования привел бы к тому же результату):
А А
Пользуясь соотношениями (9.6), (9.10), получаем
в в
UB-UA=^\^dv+^c,(T)dT.
А А
Второй интеграл в правой части равен нулю, так как
Та = Тв и, следовательно,
«B-UA=-f+ f . A0.8)
^В ^А
Подставляя A0.7) и A0.8) в A0.6), имеем
^±^ETln{vB-b) + ;^ + RTln{vA-b) + p(vB-VA]=^0.
^В ^А
A0.9)
Вычислим теперь площадь в плоскости р^ у,
заключенную между изотермой Т = const, осью абсцисс и двумя
прямыми v = Va и v==VB' Согласно уравнению Ван-дер-
Ваальса, она дается выражением
АЛА "^
A0.10)
§ 10. CotciuHcenue газа no Ван-дер-Ваальсу
89
Правая часть этого равенства с точностью до знака равна
первым четырем членам уравнения A0.9). Пользуясь этим,
получаем
в
^pdv==p{vB-VA)^ A0.11)
А
Выражение p{vb — va) представляет площадь
прямоугольника ABCD на фиг. 9. Согласно A0.11), этот
прямоугольник равновелик вычисленной в A0.10) части площади
Pi
[
/\
\
\
\
Лииил \
шидности 1
шидиостьШ
\^ир.
bJ=
I газ
X
Рнр.
= R-
4 :
Е жидкость •
\
\
\
1
/ \
У!цнил
Газ
пара
)—
Фиг. 10. Границы области сосуществования фаз
газ+жидкость в плоскости р, v.
между изотермой и осью абсцисс. Следовательно, две
заштрихованные области на фиг. 9 равновелики, но имеют
«противоположные знаки». Это дает удобный метод
графического построения граничных точек фазового
равновесия Л и S. На это обстоятельство впервые указал
Максвелл (Nature, 1875), в соответствии с чем прямая АВ
называется «прямой Максвелла».
Произведя аналогичное построение для каждой
изотермы ниже критической точки, получим (фиг. 10) границу
той области в плоскости р, у, в которой обе фазы —
W г л. I. Термодинамика. Общие принципы
хнсидкая и газообразная— я^хо]щтся в равновесии. Справа
ют нее веш;ество может суш;ествовать только в
газообразном состоянии, слева — только в жидком. Высшая точка
4)той области является критической. В ней сливаются две
ветви: линия пара — совокупность точек В — и линия
оФсидкости — совокупность точек А, Если идти от чисто
газообразной фазы, то на линии пара появляются первые
капли жидкости. Если же идти от жидкой фазы, то на
линии жидкости наблюдаются первые пузырьки газа.
Вернемся еще раз к фиг. 9 и рассмотрим, что
означают физически различные точки отрезка АВ. Эти точки
•соответствуют различным объемам, но одинаковым
давлениям и температурам. В этих различных объемах
присутствуют различные доли жидкой и газообразной фаз.
Точка В описывает чистый насыщенный пар с молярным
объемом VBy точка А — чистую жидкость с молярным
объемом Уд. Если обозначить относительное количество
ткидкости через гг, а относительное количество пара через
1~л;, то в точке В х = 0, в точке А x^i. В
произвольной точке Р отрезка АВ имеем
Vp = xva + {^ — x)vb.
Отсюда
Следовательно, как видно из фиг. 9, величины х и I — х
равны отношениям длины соответствующих частей
отрезка АВ к его полной длине.
Далее следует объяснить еще смысл надписей «пере-
-сыщение» и «перегревание» на фиг. 9. Эти названия
показывают, что в соответствующих точках существует
нестабильное равновесие в отличие от точек прямой
Максвелла. При благоприятных обстоятельствах (например,
в случае водяного пара в атмосфере, свободной от пыли)
пар можно перегреть, причем при одинаковой
температуре он будет иметь более высокое давление, чем в точке
линии пара В. Равным образом в сосуде, тщательно
предохраненном от сотрясений, можно получить жидкость
при температуре, несколько превышающей точку кипения,
причем давление будет меньше, чем в точке А (при той
§ 10. С^исьинсение газа по Ван-дер-Ваальсу 91
же температуре). Однако эти нестабильные пограничные
состояния реализуются лишь в малой части. Состояния,
соответствующие точкам между А' vl В\ недостижимы,
так как в них увеличение давления было бы связано
с расширением. Тот факт, что теория Ван-дер-Ваальса
может объяснить, по крайней мере качественно, и
нестабильные состояния (ветви А А' и ВВ'), является особенно
замечательным.
Возвратимся теперь к фиг. 10. Рассмотрение ее
немедленно позволяет прийти к следующим выводам:
1. При температурах Г > Гкр.» сколь бы велико ни
было давление, газ невозможно сконденсировать в жидкость.
2. Чтобы сконденсировать газ, недостаточно понизить
температуру до значений Г < Г^. Как видно из A0.56)
и (9.3), температура инверсии Т^ примерно в 7 раз
больше, чем Гкр.. Однако по достижении температуры Т < Т^
можно при помощи процесса Джоуля — Кельвина, как в
случае воздуха, добиться частичного, а затем и полного
сжижения газа.
3. Произведя обход над критической точкой, можно
перейти из области разреженной газовой фазы (на фиг. 10
справа внизу) в область жидкой фазы (слева внизу),
минуя заштрихованную область сосуществования двух фаз.
Состояния, через которые при этом проходит система,
образуют, согласно уравнению Ван-дер-Ваальса,
непрерывную последовательность устойчивых* состояний. Выше
критической изотермы также не происходит никакого
разрыва ни при низких давлениях (/?</?кр.)» ни при
высоких (/?>/?кр.). В этом заключается смысл заглавия
работы Ван-дер-Ваальса «Непрерывность газообразных и
жидких состояний».
4. В частности, рассмотрим переход от конца В
прямой Максвелла к другому концу А по пути, показанному
на фиг. 10 пунктирной линией. Аналитическое
выражение для gB при этом непрерывно переходит в gA, что в
сущности уже предполагалось в соотношении A0.6).
Поскольку до сих пор нулевые значения энергии и
энтропии еще не были определены, в это соотношение можно было
бы ввести еще линейную функцию температуры. Однако
в силу непрерывности газообразного и жидкого
состояний эту функцию фактически следует приравнять нулю.
Я2 Гл. I. Термодинамика. Общие принципы
До сих пор МЫ имели дело с понятиями критической
точки и прямой Максвелла только для идеализированной
модели газа Ван-дер-Ваальса. Действительные изотермы
реальных газов качественно согласуются с изотермами
Ван-дер-Ваальса. В частности, всегда существует
критическая точка, определяемая условиями (см. § 9)
Г^^Л =0- Г^^ =0
K^dvjr ^' К^ди^ Jt
и уравнением состояния р — р(р, Т).
Как легко показать, соотношение A0.11), определяющее
прямую Максвелла, остается в силе для общего случая
любого аналитически заданного уравнения состояния.
Рассмотрим изотерму, лежащую ниже критической (см. фиг. 9).
Поскольку g-=f-\-pv, из равенства свободных энтальпий
в точках А и В следует, что
/a + PVa^/b + PVb. A0.12)
Но
/л^/в=-^(Й),й.. A0.13)
А
Однако, согласно формулам таблицы § 7, {df/dv)T= —р-
Таким образом, из A0.12) и A0.13) получаем, что
р (Рв - ^а) = 5 pdo,
где путь интегрирования совпадает с экстраполированной
изотермой АА'В'В, Отсюда следует, что соотношение
A0.11) справедливо для любого уравнения состояния.
§ 11. ШКАЛА КЕЛЬВИНА
В § 6 было определено понятие абсолютной
температуры как такой функции ср(б) какой-нибудь измеряемой
обычным способом температуры 6, которая удовлетворяет
пропорции Карно F.7) и F.8)
Qi_^lM^Il /11 \\
§ 11. Шкала Кельвина 93
Это определение было предложено В. Томсоном уже в
1848 г. Кроме того, мы подчеркивали, что температура
газового термометра подчиняется условию A1.1),
поскольку в рассматриваемой температурной области
термометрическое вещество ведет себя подобно идеальному газу.
Теперь надлежит выяснить, каким образом можно
температуру 6, отсчитываемую по реальному газовому
термометру, свести к абсолютной температуре Г.
Очевидно, что уравнение A1.1) мало подходит для
этой цели, так как ка^чориметрические измерения
количества тепла недостаточно точны. Можно, однако,
воспользоваться вместо него любым следствием второго
начала термодинамики, содержащим абсолютную
температуру. Томсон указал, что для этого особенно удобным
является соотношение, описывающее процесс Джоуля —
Кельвина (см. § 10). Другим практически удобным
исходным пунктом может служить уравнение Клапейрона
(см. § 16).
Согласно соотношению A0.3),
Стоящие справа величины а и Ср (теоретически
определяемые дифференцированием по Т) можно выразить через
производные по 6, так как Г = срF) является функцией
только 6; получаем
dT '
S - \^дТ Л=соп8г "~ V^0/P=con8t dT ~ ^Р rfT •
Введенные здесь величины а' и Ср рассматриваются как
известные из экспериментальных данных функции б.
Тогда правая часть соотношения A1.2), если числитель и
знаменатель поделить на dS/dT, принимает вид
t/Г/ , i dT\ /44 о \
94 Гл. I, Термодинамика. Общие принципы
Левую часть соотношения A1.2) можно написать в виде
причем Ав/^р также считается известной из опыта
функцией 6 (ее можно получить, например, при исследовании
процесса Джоуля-Кельвина). Приравнивая A1.2а) и
A1.26), получаем
откуда
или
, Д0 1 dT_ f , 1 dT\
т db '' ГТ^в
^Ap
^+<^PTZ
A1.3)
^-^= -ТЖ^«- fii-4)
^о^ + 'РГр
Фигурирующий здесь интеграл, который содержит лишь
эмпирически заданные функции в (к ним надо добавить
также объем у), в общем случае берется, конечно, только
численно. Если температуру 6 измерять в 100-градусной
шкале и затем перейти к абсолютной шкале Г, то
получим соотношения
Г = Го при 6 = 0,
Г = Го+100^ при e = loo^
Для определения температуры Tq на основании A1.4)
получаем уравнение
Установленная таким образом температурная шкала
называется шкалой Кельвина. Пишут . Г= .. .°К. Разница
между температурами Гиб для большинства
термометрических веществ внутри области О —100° С очень
маЛа. Согласно данным имперского физико-технического
§ и. Шкала Кельвина 95-
комитета, максимальная величина этой разности
составляет — 0,0026°, если 6 определяется по воздушному
термометру (и, следовательно, процесс Джоуля —Кельвина
проводится с воздухом). Она еще меньше для водорода —
газа, более близкого к идеальному газу (а именно, она
равна 0,0007°).
С приближением к точке конденсации используемого
термометрического вещества эта разница увеличивается.
Ниже этой точки процесс Джоуля — Кельвина нельзя
провести. Эффективным средством получения самых низких
температур, очень близких к абсолютному нулю (который
не может быть достигнут), оказывается магнитокалори-
ческий эффект [открытый Дебаем A926 г.) и Джиоко\1
A927 г.)]. В качестве наиболее подходящего вещества
была предложена (независимо обоими авторами)
парамагнитная соль —сульфат гадолиния. Поступают
следующим образом; сначала вещество находится в тепловом
контакте с гелиевой баней, имеющей очень низкую
температуру ('^ 1,3° К); затем включают сильное внешнее
магнитное поле и дожидаются установления термического
равновесия. При достаточно сильном поле все
элементарные магниты ориентированы практически одинаково. В этом
упорядоченном состоянии энтропия меньше, чем в
неупорядоченном, когда магнитное поле отсутствует (см. § 19).
Следовательно, в итоге достигается уменьшение энтропии.
Затем парамагнитную соль термически изолируют и поло
выключают. При этом полная энтропия должна оставаться
постоянной. Однако теперь —в отсутс1Вие внешнего поля —
устанавливается определенное неупорядоченное состояние
элементарных магнитов, т. е. магнитная часть энтропии
увеличивается на величину, существенно отличную от
нуля. Так как полная энтропия при адиабатическом
процессе остается постоянной, то температура должна
понизиться. Благодаря малости удельной теплоемкости с^ при
этих низких температурах магнитная часть энтропии
значительна. При помощи такого адиабатического
размагничивания де-Гааз понизил температуру до 0,001° К. Однако
в § 12 будет показано, почему таким путем нельзя
достигнуть абсолютного нуля. Здесь мы лишь констатируем,
что с созданием термодинамической теории этого
адиабатического размагничивания (см. § 1,9) определение шкалы
ti6 Гл. I. Термодинамика, Общие принципы
Кельвина становится возможным вплоть до ближайшей
окрестности абсолютного нуля.
Скептическая точка зрения, иногда высказывавшаяся
и идущая от Маха^), согласно которой нуль абсолютной
температуры имеет смысл только для специального случая
газа, была в корне опровергнута с созданием шкалы
Кельвина. Без этой (существующей, но недостижимой) нижней
гранищл температуры разрушилось бы все здание
термодинамики.
§ 12. ТЕПЛОВАЯ ТЕОРЕМА НЕРНСТА
Тепловая теорема Нернста с полным правом называется
третьим началом термодинамики. Хотя она и йе вводит
в термодинамику новой функции состояния, как первое
и второе начала, однако именно она делает функции
состояния Sy F, G, ... численно определенными и
практически полезными.
Энтропия определяется через ее дифференциал, dS.
Поэтому сама велична S определена лишь с точностью
до постоянной интегрирования Sq. Так как обычно имеют
дело с дифференциалом энтропии, то сама по себе эта
неопределенность не повела бы ни к чему плохому. Однако,
поскольку в потенциалы входит произведение TS,
в них остается неопределенной уже линейная функция
температуры вида 8^Т-\-соп^1, вследствие чего их
практическая полезность для состояний с различными
температурами (а также при установлении условий химического
равновесия) становится иллюзорной.
Таким образом, возникает вопрос об абсолютном
значении энтропии. Как всегда при постановке
фундаментальных вопросов, природа дает математически наиболее
удовлетворительный и самый простой ответ: при абсолют-
ном нуле температуры энтропия принимает значение
Sq, не зависящее от давления, агрегатного состояния и
других характеристик вещества. Эту величину можно
положить равной нулю, получив таким образом абсолютную
нормировку энтропии любого вещества. Если теперь
1) МасЬ, Prinzipien der Warmelehre, Leipzig, 1896, S. 341.
В этой книге в качестве дополнения напечатана, правда трудно
иоспринимаемая, работа Гей-Люссака о его опыте по расширению
газа в пустоту.
§ 12. Тепловая теорема Нернста 97
считать, что в интегральном представлении энтропии
[см. уравнение F,11)] нижний предел совпадает с Г = 0, то
тем самым устраняется также и неопределенность в
потенциалах F и G.
Такую окончательную реакцию тепловой теореме
Нернста придал Планк. Посмотрим, однако, как
первоначально сформулировал ее Нернст.
Нернст, который, как и другие исследователи, с
успехом применял второе начало термодинамики, особенно
в своих электрохимических работах, странным образом
не любил понятие «энтропия», предпочитая ему понятие
«максимальная работа», служившее мерой химического
сродства. Если ввести обозначение Нернста Aj то
соотношение (8.15), в которое входит эта величина, примет вид
A = ^F, где AF = F^-F^, A2.1)
Из уравнения F = U ^ TS и соотношения
(см. таблицу в § 7) получаем
и, следовательно, согласно A2.1),
А-Ш = Т(^^^^, где AU==U,^U,, A2.2)
Нернст рассматривает A2.2) как фундаментальное
уравнение^), объединяющее первое и второе начала
термодинамики, и именно на нем основывает все дальнейшие выводы.
Величина ДС/, согласно Вертело и Нернсту, имеет смысл
теплоты процесса при постоянном объеме ^). Согласно
Вертело, левая часть уравнения A2.2) должна равняться
нулю. Однако фактически это не имеет места. Вот что
говорит об этом Нернст: «Тот факт, что на опыте разность
^) Nernst, Theoretische Chemie, 1926, S. 30, 795. Вместо
принятого у Нернста обозначения U мы использовали обозначение AZ7
п приписали у производной индекс F, опущенный у Нернста.
2) В большинстве случаев целесообразнее рассматривать
теплоту процесса при постоянном давлении, которая определяется как
разность энтальпий АЯ.
98 Гл. I. Термодинамика. Общие принципы
между А и АС/ часто очень мала, навел меня на мысль,
что здесь мы имеем дело с некоторым предельным
законом, согласно которому величины А и Af/ не только равны
между собой при абсолютном нуле, но и асимптотически
стремятся друг к другу при Г—>0. Следовательно,
lim^ = lim^ = 0 при Г = 0». A2.3)
Приведенная цитата дает представление о происхождении
гениальнейшего расширения классической термодинамики,
достигнутого в нашем столетии.
Возвращаясь от ^ к
получаем непосредственно из A2.3)
,. / d^u .о rrd^s\ т d^u
А^-^0. т. е. ^2->^1 при Г->0. A2.4)
Тем самым формулировка Планка S—^S^ и возможность
считать Sq равной нулю для каждого вещества выведены
из формулировки Нернста. Очевидно, справедливо также
и обратное: уравнение A2.3) вытекает из A2.4).
Первоначально Нернст ограничил свою теорему
конденсированными веществами (твердые тела или жидкости),
исключив, следовательно, газообразное состояние. Однако
в более поздней публикации ^) он подробно дискутировал
носившиеся уже тогда в воздухе идеи о вырождении газа,
которые вскоре после этого приняли явную форму в
статистиках Бозе —Эйнштейна и Ферми — Дирака. Обе
статистики показывают, что при очень низких температурах
газы становятся вырожденными. В частности, автор
в своем очерке теории металлов A927 г.) показал, что
свойства газа свободных электронов соответствуют теореме
Нернста. Тот факт, что выражения E.10) и (9.15) для
энтропии идеального газа и газа Ван-дер-Ваальса не
подчиняются условию 6* —> О при Г —> О, не является удивитель-
откуда
^) Nernst, Die theoretischen und experiment alien Giundlagen
des neuen Warmetheorems, Halle, 1918.
§ 12. Тепловая тедрема Нернста 9Й
ным, так как соответствующие уравнения состояния
неприменимы в этой температурной области. Кажущееся
противоречие с теоремой Нернста, наблюдающееся в
соединениях дейтерия, было объяснено Клузиусом и
другими авторамп при помощи представления о
замороженном (т. е. метастабильном) равновесии.
О вероятной величине времени жизни такого особого
состояния термодинамика ничего сказать не может. Этот
вопрос должна решить квантовомеханическая
кинетическая теория. Во всяком случае, мы не имеем оснований
считать, что теорема Нернста ограничивается
конденсированными веществами, и полагаем ее справедливой для
вещества в любом состоянии.
Сделаем теперь ряд простейших выводов из теоремы
Нернста для гомогенного вещества. Так как при этом
рассматривается единица массы (или 1 моль), будем
использовать вместо употреблявшихся до сих пор больших букв
малые буквы 5, v.
1. Коэффициент теплового расширения при Г—>0
обращается в нуль. Согласно таблице в § 7, имеем
\др)т \dTjj,'
Левая часть этого равенства при Г—>0 обращается в нуль,
так как предельное значение энтропии Sq не зависит от
давления. Следовательно, и правая сторона также
обращается в нуль. То же самое справедливо для
коэффициента теплового расширения а.
2. Термический квэффициент давления при Г—»О
обращается в нуль. Согласно таблице в § 7, имеем
\dvjT \с
Левая часть обращается в нуль, так как предельное
значение энтропии Sq не зависит от объема v. То же самое
относится и к правой части равенства, т. е. к
термическому коэффициенту давления р при р фО,
3. Удельные {или молярные) теплоемкости с^ и с^ при
Т — 0 обращаются в нуль. Из определений
КЮ.'^'-КЮ,
loo Гл. /. Термодинамика. Общие принципы
следует
т т
siv,T)=\^^dTMs{p,T)=Y-^dT. A2.5)
О о
Если здесь ввести постоянные интегрирования, то в первом
случае соответствующая постоянная была бы некоторой
функцией объема у, а во втором — функцией давления р.
Наличие таких функций при достаточно малой температуре
Т привело бы к противоречию с теоремой Нернста, которая
требует, чтобы предельное значение энтропии Sq
«асимптотически» не зависело как от у, так и от р. Поэтому в обоих
случаях следует считать Sq равной нулю, в соответствии
с чем и были приравнены нулю нижние пределы
интегралов. Отсюда получаем, что при Т = 0 теплоемкости с^
и Ср должны исчезать, так как в противном случае интегралы
оказались бы расходящимися на нижнем пределе.
Под руководством Нернста были проведены обширные
измерения, которые полностью подтвердили, в частности,
заключение об уменьшении с^ при приближении к
абсолютному нулю температуры. Скорость убывания теплоем-
костей с температурой формулами A2.5) еще не
определяется. Для упругих тел, согласно Дебаю^), она
пропорциональна Г^, для электронного газа — пропорциональна
Т (см. § 39). Эти теоретические результаты были также
подтверждены обширным экспериментальным материалом.
4. Абсолютного нуля температуры нельзя достигнуть
ни в каком конечном процессе; к нему можно лишь
асимптотически приближаться. Охлаждение реальных газов
производят при помощи адиабатического расширения (см.
§ 5). Для достижения очень низких температур его место
занимает эффективный метод адиабатического
размагничивания (см. § И).
Изобразим в плоскости /5, Т кривую S (Т) для
намагничиваемой соли в заданном магнитном поле Я^), которое
считаем постоянным, Я = const (фиг. И). Давление при
этом также считаем постоянным (например, /? = О, что
соответствует вакууму; зависимость от объема для твердой
^) См. т. IT, Механика деформируемых сред, § 44.
*) Здесь и в § 19 и 25 буква Н употребляется для обозначения
не энтальпии, а напряженности магнитного поля.
§ 12. Тепловая теорема Нернста
101
СОЛИ отсутствует). Эта кривая проходит через начало
координат, и наклон ее с повышением температуры
увеличивается, так как с ростом Т все более и более нарушается
обусловленная магнитным полем упорядоченная
ориентация элементарных магнитов. Далее проведем кривую
S {Т) для ненамасниченной соли, т. е. при Я = 0. Она идет
выше, чем при /Г = const, так как при И = 0 полностью
И=const
Sab
Фиг. и. К вопросу о достижении
абсолютного нуля температуры при помощи
адиабатического размагничивания.
отсутствует магнитное упорядочение i). Однако в этом
случае в соответствии с теоремой Нернста при Т = 0
энтропия равна нулю. Если теперь адиабатически (т. е.
по горизонтали S = const) перейти от намагниченного
состояния (при температуре Т^) к ненамагниченному, то
температура последнего jg окажется заметно меньше Т^.
Однако, как видно из фиг. И, ни в коем случае не удастся
получить значения Г = 0.
С другой стороны, если бы теорема Нернста не была
справедлива, то предельное значение энтродии на кривой
1) Своеобразная извилистая, форма этой кривой обусловлена
тем, что парамагнитные вещества при очень низких'температурах
обнаруживают некоторое спонтанное намагничивание (одинаковая
ориентация магнитных моментов) подобно ферромагнетикам.
102 Гл, I. Термодинамика. Общие принципы
Н = 0 при Г = О должно было бы равняться Sq-Ф О (см.
пунктирную кривую на фиг. И). В этом случае при
нашем специальном выборе исходной температуры Т^
на кривой Н = const можно было бы сразу достичь
температуры 7" = О (в точке S = Sq при Я = 0).
Казалось бы, если вещество при температуре 7*2
намагнитить, а затем вторично размагнитить, то можно
еще ближе подойти к абсолютному нулю температуры.
Однако этому мешает ряд практических трудностей.
Следующим эффективным шагом явится только
размагничивание ядра. Однако это дело будущего.
Во всяком случае, из третьего начала термодинамики
непосредственно следует, что абсолютный нуль
температуры достижим только асимптотически. Именно поэтому
нельзя осуществить цикл Карно с температурой
холодильника Г = О и соответствующим коэффициентом полезного
действия 7] = 1 (см. § 6).
Глава П
ПРИМЕНЕНИЕ ТЕРМОДИНАМИКИ К КОНКРЕТНЫМ
СИСТЕМАМ
§ 13. СМЕСЬ ГАЗОВ; ПАРАДОКС ГИББСА.
ЗАКОН ГУЛЬДБЕРГА И ВААГЕ
Окружающий нас воздух представляет собой смесь
следующих газов:
78% N2, 21% Og и почти 1%Аг.
Присутствие удивительно большой доли аргона, который
благодаря своей химической инертности ускользал от
прежних наблюдателей, было установлено лишь в конце
прошлого столетия. Релеем и Рамзеем. Приведенные выше
числа суть молярные проценты, следовательно, они
пропорциональны количествам соответствующих молекул
в смеси газов. Чтобы получить весовое процентное
содержание газов, следует эти числа умножить на
соответствующие молекулярные веса 28, 32 и 40.
Согласно нашему представлению об идеальных газах,
которое в данном случае допустимо при обычных
температурах (молекулы — материальные точки, силы сцепления
отсутствуют, вандерваальсовы постоянные 6 = 0, а = 0),
каждый газ, входящий в смесь, занимает объем V так,
как если бы он был один. Поэтому можно говорить
о парциальных давлениях р^, из которых аддитивно
складывается полное давление газа /?,
P = IiPi A3.1)
i
{закон Дальтона), Так как для каждого отдельного газа
в смеси справедлив закон Гей-Люссака в форме C.10).
Vp, = n,RT, A3.1а)
то из соотношения A3.1) получаем для смеси газов
Vp = nRT, где ^i=2^i. A3.2)
104 Гл. II. Применение термодинамики к конкретным системам
Энергия смеси газов также аддитивно складывается
из молярных энергий составных часте^'г:
C/ = 2^iWi, где Ui = c^,J, A3.3)
i
Для теплоемкостей С^, С^ смеси газов отсюда вытекает
C.= 2«Ai. Cp=S«iV- C^~C, = nR. A3.3а)
i
Чтобы пояснить смысл уравнения A3.3), представим
себе, что сначала все отдельные составные части были
Фиг. 12. Обратимое разделение двух газов.
изотермически сжаты до меньших объемов
Vi=V^ A3.36)
и заключены в ящики объема V^ и одинакового
поперечного сечения F (фиг. 12). Все ящики расположены рядом
и разделены перегородками, причем в сумме их объемы
составляют V = ^V^. Если перегородки устранить, то г-й
г
газ растечется по большему объему F, причем его
температура Т и молярная энергия и^ (а следовательно, и
полная энергия n^Ui) остаются неизменными. В соответствии
с нашим представлением о пространственном
распределении прочие компоненты смеси при этом можно не
принимать во внимание. Физическая природа этого явления
гораздо лучше передается термином диффузионный про-
цесс, чем прежним названием «процесс растекания».
§ 13. Смесь газов; парадокс Гиббса. Закон Гульдперга и Вааге 105
1. Обратимое разделение газов. В дальнейшем
ограничимся двумя газами i и 2. Для разделения смеси
воспользуемся полупроницаемыми мембранами. Такие
мембраны в готовом виде встречаются в природе в виде
стенок клеток и реализуются с известным приближением
химическим путем (мембрана из ферроцианида меди).
В практику и теорию они были введены ботаником Пфеф-
фером (см. § 15). На фиг. 12 показаны два цилиндра
(/ и //) равных объемов F, вдвигающиеся без трения
один в другой. Крышки цилиндров и их боковые
поверхности (G^ —боковая поверхность цилиндра/, G2 —
Цилиндра //) должны быть непроницаемы для обоих газов.
Напротив, мембрана Н^ цилиндра // должна быть
непроницаемой только для газа 2, а мембрана Н^ цилиндра
/ — только для газа 1.
Первоначально оба цилиндра были вдвинуты один
в другой, следовательно, газы i и 2 были смешаны.
Выдвинем цилиндр // бесконечно медленно на отрезок dx.
На участок GJI^ может проникать только газ 1 (для него
мембрана Н^ как бы отсутствует). Следовательно, на
мембрану //g действует только давление р^, направленное
против смещения dx. Работа, совершаемая при этом
перемещении, равна
dW= ^Fp^dx. A3.4)
Мембрана Я^, оставаясь на месте, не входит в
рассмотрение при вычислении работы, так же как стенка G^.
Стенка G^, смещается на dx и поэтому должна
рассматриваться при вычислении полной работы. Так как на участке
Hfi^ находится только газ 2 под давлением; равным
давлению на участке HJI^ (Н^ для газа 2 как бы
отсутствует), то работа, совершаемая им при перемещении
стенки Gg, равна
dW= +Fp2dx. A3.4а)
Это выражение равно правой части равенства A3.4),
взятой с обратным знаком. Разделение смеси происходит
без затраты работы и, согласно нашему предположению,
без подвода тепла. Но тогда энергия и, следовательно,
температура обеих составных частей смеси остается нсиз-
менцой. Конечный результат таков: оба газа 1 и 2 рас-
106 Гл. II. Применение термодинамики к конкретным системам
положены рядом в объемах V при давлениях
соответственно pi и Р2* Пусть их энтропии равны S^ и S^. Так
как разделение смеси оказалось возможным провести
обратимым путем — без затраты работы и без сообщения тепла,
то для энтропии смеси S имеем
S = S, + S,. A3.5)
Очевидно, что процесс, изображенный на фиг. 12,
допускает обращение, т. е. оба цилиндра можно
постепенно вдвигать друг в друга. Смешение газов,
проведенное таким образом, также будет обратимым.
При помощи уравнения A3.5) можно определить
энтропию смеси. В подробной записи это уравнение имеет вид
S (Г, V) = S, (Г, V) + S, (Г, V), A3,5а)
или (если рассматривать S как функцию Тир)
S{T,p) = S,{T,p,) + S,{T,p,), A3.56)
В словесной форме этот вывод гласит: чтобы вычислить
энтропию смеси газов, следует просто сложить энтропии
ее компонент. При этом надо считать, что каждая
компонента имеет объем, равный объему смеси У, и
соответствующее парциальное давление.
Обобщение этого результата на случай более двух
компонент очевидно:
S {Т, р, п„п^,...) = ^ n,Si {Т, р,). A3.6)
i
Здесь Аг^ —число молей, а 5^ —энтропия одного моля 1-й
составной части смеси.
2. Увеличение энтропии при диффузии и парадокс
Гиббса. В начале рассмотренного процесса диффузии
расположенные рядом составные части занимали объемы V^,
не равные друг другу и меньшие чем объем V. Пусть
в результате изотермического сжатия давление каждой
составной части доведено до суммарного давления р,
и пусть Sq — сумма энтропии составных частей перед
диффузией. Аналогично A3.6) имеем
S,=^^nMT,p). A3.6а)
§ J3. Смесь газов; парадокс Гиббса. Закон Гульдберга и Вааге 107
Из формул A3.6) И A3.6а) получаем изменение энтропии
при диффузии
5 - 5„ = 2 «i [Si (Т, рд - S, {Т, р)]. A3.66)
г
Пользуясь первоначальным определением энтропии
идеального газа E.10), находим, считая температуру
неизменной^) и относя все к одному молю,
s^(T,p,)-s^{T,p)=^Rln-^.
В силу A3.36) правую часть этого равенства можно
написать также в виде Н1п{р1р^). Тогда уравнение A3.66)
принимает вид
S-S, = R2nan^. A3.7)
г
Таким образом, диффузия является необратимым
процессом подобно теплопроводности. Определяемое
уравнением A3.7) увеличение энтропии можно предотвратить
лишь при затрате работы.
В силу уравнени!! состояния A3.2) и A3.1а) имеем
plPi = nln^. Таким образом, увеличение энтропии будет
функцией только чисел молей п^ и их суммы дг = ^ w^:
i
^-^S„ = /?2«iln-^ = i? [nlnn-^niln/ii] . A3.8)
i i
Назовем правую часть формулы A3.8) энтропией
смешения. Последняя зависит только от числа молекул, но не
зависит от их свойств. Отсюда вытекает парадокс Гиббса:
если перейти к предельному случаю смеси тождественных
молекул, то формула A3.8) не меняется. Это нелепо, так
как при удалении перегородки между газами, состоящими
из совершенно одинаковых молекул, не может быть и речи
ни о каком процессе диффузии. Следовательно, предель-
^) Клузиус и Вальдманн показали, правда, что при диффузии
появляются заметные разности температур. Однако нас здесь
интересует только конечное состояние, которое наступает после
выравнивания этих разностей температур.
108 Гл. II. Применение термодинамики к конкретным системам
ный переход здесь недопустим. Он противоречит атомизму
вещества и тому факту, что между различными видами
атомов (например, атомами Н и Не) нет никакого
непрерывного перехода.
Чтобы выразить это яснее, рассмотрим случай очень
похожих молекул, например изотопов какого-нибудь
благородного газа. Они отличаются физическими свойствами,
но неразличимы химически. Для них формула A3.8)
справедлива в приведенной форме. То же самое относится
к смеси орто- и параводорода, компоненты которой
отличаются только спинами, или к смеси молекул в
основном состоянии и тех же молекул в возбужденном
состоянии. Напротив, при полной тождественности
составных частей формула A3.8) неприменима.
3. Закон действующих масс. До сих пор
предполагалось, что газы в смеси не реагируют между собой.
Если допустить возможность химических превращений
между ними, то возникает вопрос о том, какое
равновесие установится в ходе реакции между компонентами
смеси. При этом будем предполагать, что реакция
протекает при постоянном (например, атмосферном) давлении р
и при постоянной температуре Т. В большинстве случаев
этого можно достигнуть путем соответствующего
проведения опыта. Условие равновесия для этого случая,
согласно § 8, имеет вид
8G = 0, G = U-TS+pV = H-TS, A3.9)
В качестве простейшего примера рассмотрим
диссоциацию водяного пара на водород и кислород, при которой
возможно возникновение гремучего газа. (Тот факт, что
водяной пар при обычных температурах не является
идеальным газом, здесь нет необходимости учитывать,
так как заметная степень диссоциации наступает лишь
при очень высоких температурах.) Уравнение химической
реакции
2Н,0^2Н2 + 0,
показывает, что с исчезновением 2 молей Н2О должны
появиться 2 моля Hg и 1 моль Og. Обозначим числа молей
Нз, Og и HgO, присутствующие в реакционном сосуде,
§^13. Смесь газов; парадокс Гиббса. Закон Гульдберга и Вааге 109
через ^1, ^2 и щ. Соответствующие числа молей в
уравнении реакции суть целые числа Vj, Vg и Vg (они
считаются положительными для веществ, стоящих в правой
части уравнения, и отрицательными для веществ, стоящих
в левой части). В нашем случае имеем
о А о A3.9а)
Изменения чисел молей при реакции (как виртуальные,
так и действительные) составляют пропорцию
8^1: 8^2: Ьп^ = v^: V2: Vg. A3.10)
Выразим теперь условие равновесия A3.9) через
переменные р. Г, w^ и параметр v^. Энергия одного моля, как
известно, является функцией только температуры Г.
Следовательно,
и=^п,и,{Т).
i
То же самое справедливо в силу уравнений A3.1) и A3.1а)
для энтальпии Н и молярных энтальпий, которые мы
будем обозначать через А^. Таким образом,
я=2«Л(^)- A3.11)
i
Иначе обстоит дело с энтропией S, Здесь к молярным
энтропиям s^ {Ту р) добавляются отдельные компоненты
энтропии смешения A3.8)
6' = 2«i[«i(?',/') + ^lii^] . A3.12)
i
Из формул A3.9), A3.11) и A3.12) получаем
G = ^n,[h,{T)-Ts,{T,p)-RT\n^^] .
i
Это выражение можно также записать в виде
G = ^n,[g,{r,p)-RTlnf] , A3.13)
110 Гл. fl. Применение термодинамики к конкретным системам
где под g^ понимается молярная свободная энтальпия
отдельных компонент.
Из выражения A3.13) при постоянных р и Т получаем
ЬО
=^2^n,[g,{T,p)-RTln^]-Rr'2^nfi\n^,
Благодаря соотношению дг = 2 ^i последний член здесь
обращается в нуль. Следовательно, условие равновесия
8G = О при учете соответствующим образом обобщенной
пропорции A3.10) приводит к соотношению
^^i{T,p)-RT\n^]=^0. A3.14)
i
Производя в этом соотношении потенцирование,
непосредственно получаем закон действующих масс
Ц(^-^У = К, InK^-^-^'iSiiP'^)- A3-15)
i i
Этот закон был установлен в 1867 г. норвежцами Гульд-
бергом и Вааге, которые исходили из статистических
соображений (связанных с вероятностью соударения). Вскоре
после этого Гиббс не только термодинамически доказал
этот закон для идеальных газов, но и дополнил его,
вычислив постоянную К. С известными ограничениями
этот закон оказался справедливым и для паров ^) и явился
фундаментом расцветавшей в то время физической химии.
Переходя к молярным концентрациям, т. е. к
относительным числам молей с- = п^/п^ получаем из
уравнения A3.15)
Цс^'^К. A3.15а)
^) Процессы полимеризации и диссоциации, которые могут
происходить в смеси паров, также можно включить в
вышеупомянутую схему, описывая их дополнительным и уравнениями реакций.
В этом случае устанавливают условия равновесия для большого
числа одновременно протекающих реакций.
§ 13. Смесь газов; парадокс Гиббса. Закон Гульдберза и Вааге 111
Назовем К константой закона действующих масс.
В нашем примере гремучего газа уравнение A3.15а)
принимает вид
^'- = К. A3.16)
Чтобы определить по отдельности три неизвестные с^,
Сз и Сд, воспользуемся, кроме уравнения A3.16), еще
соотношением (вытекающим из условия ^ri^ = n)
i
Ci + C2 + C3 = l A3.16а)
и отношением концентраций водорода и кислорода в смеси
^Н _2с1 + 2сз
Nq 2С2 + Сд
A3.166)
(Это отношение считается либо известным заранее, либо
доступным измерению.) В общем случае, когда в реакции
участвует больше трех веществ и больше двух видов
атомов, для определения всех величин с^ также
необходимо соответствующее число уравнений.
Если в уравнении A3.15а) концентрации с^ заменить
парциальными давлениями р^, то оно примет вид
П/^> = ^^р, где К^=^р' \. A3.17)
i
Эта форма уравнения полезна в том отношении, что
константа К^ не зависит от давления р и, следовательно,
является функцией только температуры Т, Действительно,
из выражения для \пК A3.15) следует, что
д\пК_ 1 ^ \ ^^-dgiip^T)
dp
= -i^3["^^]- ('"8)
Из соотношений таблицы, приведенной в § 7, получаем
""^-v.ip. П Ф = -s,{p^ ту A3.18а)
др ^x\t^^ * h ST
Величина Vi — молярный объем г-й компоненты при
давлении р, следовательно, v^^RTfp. В силу этого уравнение
112 Гл. II. Применение термодинамики к конкретным системам
A3.18) принимает вид
др р др ^
Интегрируя по /?, получаем отсюда
К = Ср ' \ A3.186)
где С не зависит от р. Но, согласно второй формуле
A3.17), эта величина С идентична К^. Таким образом,
наше утверждение относительно константы К^ доказано.
Аналогично можно определить и зависимость
константы К от температуры Т. Дифференцируя выражение для
\пК A3.15) по 1\ находим
дт ^ ~Ш^ 2j ^i^* " 7^ 2j ^i дт ' A3.19)
i i
Правая часть этого равенства в силу A3.18а) равна
i i
Поскольку зависимость hi К от р и Т теперь нам
известна, константа К задана с точностью до постоянного
множителя, зависящего только от природы реагирующих
веществ в смеси. В § 14, п. 3, будет показано, каким
образом обычно определяют этот множитель.
Мы не можем здесь останавливаться на всех тех
бесчисленных приложениях, которые находит закон
действующих масс в химии и технике. Укажем лишь на
некоторые следствия общего характера, которые
получаются из выведенных уравнений. Для этого запишем
важнейшие из этих уравнений в несколько дополненном виде:
A3.15а)
A3.18) и A3.18а)
A3.19) и A3.19а)
din к
др -
д\пК
дТ
^c'A^Kip,!),
i ^V
RT ~ RT*
2 '^'^ л.
г АЛ
~RT^ ~ RT^ '
§ 13. Смесь еазов; парадокс Гиббса. Закон Гульдберга и Вааге ИЗ
Здесь Ду изменение полного молярного объема, А/г
—изменение полной молярной энтропии при однократном
протекании процесса согласно уравнению реакции слева
направо; при этом предполагается, что реакция
происходит при постоянных температуре и давлении.
Первое из этих уравнений показывает, что увеличение
константы К влечет за собой рост молекулярных концен-
традий С| тех веществ, для которых коэффициенты v^
положительны (т. е. которые стоят в правой части
уравнения реакции).
Второе из приведенных уравнений показывает, что
увеличение давления при постоянной температуре сдвигает
равновесие в пользу той стороны уравнения реакции,
которой соответствуют вещества с меньшим объемом. Так
как, по предположению, вещества, участвующие в
реакции, представляют собой идеальные газы, то, согласно
закону простых объемных отношений Гей-Люссака (см. §3,
п. 3), изменение объема iv получается непосредственно
из уравнения реакции. Так, в нашем примере с
.гремучим газом A3.9а) водяной пар имеет меньший молярный
объем B), чем сумма молярных объемов B-|-1 = 3)
продуктов распада 2Н2 и Og. Поэтому повышение давления
вызовет увеличение концентрации водяного пара.
При реакции образования газообразного хлористого
водорода
H2 + CI2 ^ 2НС1
изменение объема Аг; = 0, т.е. положение равновесия
зависит только от температуры.
Из последнего уравнения следует, что повышение
температуры при постоянном давлении смещает равновесие
в пользу той части уравнения реакции, вещества которой
обладают более высокой энтальпией. При низких
температурах вещества, стоящие в той части уравнения реакции,
которой соответствует более высокая энтальпия, почти
отсутствуют. В нашем примере с гремучим газом это
соответствует тому факту, что водяной пар еще не
распадается при температуре кипения A00° С).
При этом, конечно, следует отметить, что
термодинамика равновесных состояний имеет дело только с
конечными стабильными состояниями и ничего не может ска-
114 Гл. It. Применение термодинамики к конкретным системам
зать о скоростях реакций, с которыми устанавливается
равновесие. Эти скорости могут быть настолько малы, что
оказываются возможными «метастабильные» состояния,
которые не учитываются классической термодинамикой.
Поэтому, например, смесь из двух частей Hg и одной
части Оз при комнатной температуре может существовать
сколь угодно долго, хотя она и не находится в
термодинамическом равновесии. Такими же метастабильными
состояниями можно считать определенные в § 6, п. 3,
«состояния заторможенного равновесия».
В технике очень большое значение имеет процесс
синтеза аммиака из водорода и азота воздуха. Согласно
уравнению реакции
N2 + 3H2^2NH3,
молярный объем при реакции уменьшается с 4 до 2.
Следовательно, согласно второму из приведенных выше
уравнений, высокое давление способствует увеличению
концентрации аммиака. Полный анализ условий
термодинамического равновесия (Габер), преодоление технических
трудностей, связанных с применением высокого давления
(Бош), и выбор соответствующего катализатора для
ускорения реакции (Митташ) были необходимыми
предварительными условиями для успешного проведения
этого синтеза.
§ 14. ХИМИЧЕСКИЕ ПОТЕНЦИАЛЫ И ХИМИЧЕСКИЕ
ПОСТОЯННЫЕ
В предыдущем параграфе наряду с переменными /?, Т
или V, Т и т. д. были введены параметры п^ (числа молей
отдельных компонент). Они являются функциями
состояния и типичными экстенсивными величинами.
Уже в уравнении (8.3) мы допускали возможность
существования (коль угодно большого (но всегда
конечного) чпсла функций состояния. На базе этого уравнения
объединим первое и второе начала термодинамики в
соотношении
TdS = dU ^pdV+s^i^^i' A^-^)
§ 14. Химические потенциалы и химические постоянные 115
Пусть входящие сюда величины х^ совпадают с п^. Если
обозначить вместе с Гиббсом интенсивные величины^
канонически сопряженные п^, через —ji^, то уравнение A4.1)
примет вид
TdS = dU + pdV^^li^dni, A4.2)
i
dU = TdS^pdV+^ li^drii, A4.2a)
i
Вместо уравнения A4.2a) можно написать также
уравнения
dH = dU-\d ipV) ^TdS + Vdp + '^iiidn^, A4.26)
i
dF = dU-d(TS)= -SdT -pdV+^ [lidrii, A4.2b)
i
dG = dH-d {TS) = -SdT+Vdp+'2iV'idni. A4.2r)
i
1. Химические потенциалы ji|. Прежде всего
очевидно, что если считать новые переменные W|
фиксированными, то в таблице, приведенной в § 7, в которой мы
первоначально ограничились лишь двумя переменными,
ничего не изменится. Однако если принять во внимание
возможность изменения величин л^, то к уже имеющимся
дифференциальным соотношениям добавляются новые ^),
которые можно получить, используя уравнения A4.2) —
A4.2Г),
1) Величина лу, написанная в виде индекса, показывает, что
все г?;, по которым не ведется дифференцирование, являются
постоянными.
116 Гл. tl. Применение термодинамики к конкретным системам
Пользуясь тем же способом, что и раньше, можно,
конечно, вывести соотношения, аналогичные
соотношениям Максвелла в § 7. Например, из уравнения A4.26)
получаем
(слева производится дифференцирование при постоянных
Sy р и п^ Ф п^, справа —при постоянных S, п^).
Важнейшим из уравнений A4.3а) —A4.Зг) является последнее.
Чтобы изучить его подробнее, представим себе нашу
систему увеличенной. Для этого умножим все экстенсивные
величины (количества вещества w^, объем F, энтропию S)
на одно и то же число, например у, оставив неизменными
все интенсивные величины (давление, температуру,
потенциалы р.^). Из уравнения A4.Зг) заключаем, что
потенциал G, так же как и п^, увеличится в ^ раз. Отсюда
следует, что потенциал G является однородной линейной
функцией величин д^, т. е.
G{p, Т, '(щ) = ^С{р, Т, щ). A4.5)
То же самое можно утверждать и относительно U, Н и F,
если рассматривать их как функции Г, /?, п^. Но,
например, для свободной энергии, представленной как функция
переменных Г, F, п^, для которых она является
потенциалом (см. § 7), справедливо соотношение
F{T, -iV, ^п,) = -^Р{Т, V, п,).
Пользуясь теоремой Эйлера об однородных функциях
(ее можно вывести, дифференцируя по f и полагая т = 1),
находим
i •'
откуда в силу A4.Зг)
i
Величины р,. зависят от р, Г и п^, причем относительно
последних величин они являются однородными функциями
нулевой степени, т. е. \i^ зависят только от отношений
чисел молей, т. е. от «концентраций».
§ 14» Химические потенциалы и химические постоянные 117
Из уравнения A4.Зг) вытекает также, что условие
равновесия 8G = О можно записать в виде
ЬС
= 2(f^X,,..K = 2i^iK = 0, A4.8)
8^9 = 0, 8Г = 0.
К этому добавляются еще условия, обеспечивающие
сохранение числа атомов. В случае одного уравнения
реакции соответствующее условие имеет вид [см.
формулу A3.10) в связи с законом действующих масс]
8п1: 8^2: 8^3 : ... = Vi: Vg: Vg : ... A4.9)
Используя его в уравнении A4.8), получаем
2t^iVi = 0. A4.10)
i
При этом не использовалось предположение о том, что
реагирующие вещества являются идеальными газами, как
это было при выводе закона действующих масс.
Следовательно, уравнение A4.10) можно рассматривать также
как наиболее общую формулировку закона действующих
масс.
Специфические трудности возникают при определении
величин [X.. Эта задача занимает центральное место в
физической химии и может быть решена только
экспериментальным путем. К счастью, объем необходимых
измерений ограничивается благодаря наличию некоторых
тождеств, которые мы сейчас выведем. Из уравнения A4.7)
прежде всего вытекает совершенно общее соотношение
dG = ^n,dii, + ^li,dn,. A4.11)
i i
Однако, поскольку G есть функция состояния,
Согласно уравнению A4.2г), множители, стоящие здесь
при dp и dT, равны соответственно F и iS; в то же время
из соотношения A4.Зг) следует, что множители при dn^
118 Гл. II. Применение термодинамики к конкретным системам
как раз равны ji^. Таким образом, сравнивая A4.11) и
A4.11а), получаем
^nidiii = Vdp-SdT. A4.116)
i
Это уравнение имеет всеобщую применимость.
Специализируем его, считая, что из независимых переменных
Pi Т, щ, фигурирующих в выражении A4.11а), р иТ
фиксированы и только Hi изменяются. Тогда уравнение
A4.116) принимает вид (с учетом того, что [х. суть
функции состояния)
2KJ^X^_„dn, = 0. A4.11b)
i k ^
Так как rif^ теперь являются независимыми переменными
[дополнительные условия, которые учитывались,
например, в законе действующих масс, не играют здесь
никакой роли, так как уравнение A4.116) справедливо не
только при химическом равновесии, но и при изменении
числа участвующих в реакции атомов], уравнение
A4.Ив) должно выполняться для каждого drij^^ в
отдельности. Следовательно,
2«*Шг.р.„. = 0 (Л = 1.2,3 Z). A4.12)
Так как, кроме того, величины ji^ зависят только от
отношений w^j, т. е. от концентраций с^^, то систему
уравнений A4.12) для случая К компонент и, следовательно,
К — \ концентраций можно записать в виде
2'^*(Ют.р.с. = 0 (* = 1.2.3,....if-l),A4.12a)
г ^
где jJLi понимаются как функции К -—i переменных
Ci, С2, ..., сх-1. Уравнения A4.12а) восходят еще к Гиббсу,
однако в литературе они обычно называются условиями
Дюгема—Маргуле.
2. Связь величин {i^ и g^ для смесей идеальных газов.
В § 13 выражение A3.13) представляет свободную
энтальпию для специального случая идеальных газов в
зависимости от всех переменных /?, Г, л^. Но это выражение
§ 14. Химические потенциалы и химические постоянные 119
как раз имеет вид A4.7). Следовательно, сравнивая A4.7)
и A3.13), можно немедленно написать выражение для
химического потенциала
v^, = gdT,p)-RT\n^., /i = 2"i- A^-13)
i
Как уже подчеркивалось в § 13, g^ обозначает молярную
свободную энтальпию чистой i-\\ компоненты. Как видно
из выражения A4.13), химический потенциал не
совпадает с этой величиной. Дополнительный член ЛПп (w/w^)
связан с увеличением энтропии при смешении (см. § 13,
п. 2). Назовем смеси, для которых справедливо уравнение
A4.13), идеальными (также и в том случае, когда мы
имеем дело не с идеальными газами). Легко показать,
что химические потенциалы, заданные выражением A4.13),
удовлетворяют условиям Дюгема—Маргуле (см. задачу
II. 1). В случае неидеальных смесей благодаря
взаимодействию между компонентами могут происходить
термические процессы, изменения объема и другие явления.
3. Химические постоянные идеальных газов.
Возвратимся еще раз к поднятому в конце § 13 вопросу о
том, в какой мере можно заранее определить постоянную
К{р, Т) в законе действующих масс. Для этого должны
быть полностью известны величины g^, которые теперь мы
будем записывать в виде
g^ = h,-Ts,. A4.14)
Известно, что для случая одного идеального газа при не
слишком низких температурах
h, = c^,T + h,,, A4.15)
s, = c^,\nT--R\iip + s,,, A4.16)
Здесь h^Q и s^Q—постоянные интегрирования, значения
которых нельзя найти при помощи одной лишь
термодинамики. Третье начало термодинамики непосредственно
здесь также не помогает, так как экстраполяция
уравнения состояния идеального газа на случай Г—^О
недопустима. С другой стороны, уже здесь следует
подчеркнуть, что квантовая статистртка для достаточно высоких
температур также приводит к уравнению A4.16) и дает
120 Гл. II. Применение термодинамики к конкретным системам
при этом определенное значение s^. Равным образом из
квантовой статистики получается точное значение с^, так
что величину
i,=^^ A4.17)
МОЖНО вычислить для каждой /-й компоненты. Величины
i. называются химическими постоянными. Их смысл
станет ясным, если подставить A4.14)—A4 16) в
уравнение A3.15). Тогда мы получим
У
где теперь последний член в скобках совпадает с ij. Таким
образом, закон действуюш;их масс принимает вид
l[cyi = p i Ti ei . A4.18)
i
Единственная величина, которую здесь еще надо
определить из измерений и которую можно формально назвать
«теплотой реакции при абсолютном нуле температуры», есть
'•o = SviV A4.19)
i
Это и есть та самая постоянная, о которой уже
упоминалось при выводе уравнения A3.19а).
§ 15. РАЗБАВЛЕННЫЕ РАСТВОРЫ
Раствор называется разбавленным, если количество
растворителя (например, воды) во много раз превышает
количество растворенного вещества (например, сахара).
Разбавленные растворы отличаются от
концентрированных той же простотой, которой идеальные газы отличаются
от реальных (если при этом не рассматривать так
называемые сильные электролиты).
1. Общие принципы и история. Благодаря процессу
диффузии растворенное вещество из произвольного началь-
1 ого состояния равномерно распределяется в среде рас-
§ 15. Разбавленные растворы 121
творителя, подобно тому как газ распределяется по всему
доступному ему объему. Это объясняется, как и в случае
газа, действием давления, под которым находится
растворенное вещество. Назовем это давление осмотическим
давлением и обозначим буквой Р, Измеряют его при
помощи мембраны (см. § 13), проницаемой для растворителя
и непроницаемой для растворенного вещества. На эту
мембрану действует только осмотическое давление Р,
давления растворителя такая мембрана «не чувствует».
Если такая подвижная мембрана помещается между
раствором и растворителем, то при отклонении ее в сторону
раствора должна совершаться работа; при этом
растворитель беспрепятственно проходит сквозь мембрану
и раствор становится более концентрированным. Наоборот,
работа выигрывается, если мембрана перемещается в
сторону растворителя; при этом растворитель входит
в раствор и последний разбавляется. Следовательно,
имеется тенденция к разбавлению^ которая проявляется в
возможности совершения этой положительной работы. Можно
сказать, что мембрана оказывает на растворитель
всасывающее действие, которое способствует стремлению
растворенного вещества к расширению и пропорционально
его осмотическому давлению.
На этом основаноустройство,котороеПфеффер^)впервые
применил для измерения осмотического давления. В стакан
с водой частично погружается трубка, нижний конец
которой закрыт полупроницаемой мембраной (сделанной из фер-
роцианида меди, см. § 13, п. 1). Поскольку для воды эта
мембрана проницаема, жидкость в трубке
устанавливается сначала на том же самом уровне, что и в стакане.
Однако если к воде, находящейся в трубке, прибавлять
сахар, то уровень жидкости внутри трубки поднимается
тем выше, чем больше добавлено сахара. Равновесие
установится тогда, когда гидростатическое давление на
нижний конец трубки будет равно осмотическому
давлению раствора Р. Высота поднятия для
концентрированных растворов измеряется несколькими метрами,
вследствие чего Пфеффер вынужден был перейти от открытой
трубки к закрытому ртутному манометру.
1) Р f е f f е г, Osmotische Untersuchungen, Leipzig, 1877,
122 Гл. II. Применение термодинамики к конкретным системам
В природе осмотическое давление и полупроницаемые
пленки имеют очень большое значение. Только
осмотическое давление решает загадку о том, каким образом
древесный сок проникает вплоть до самого конца высокого
дерева. Стенки клеток в растительных и жиеотных
организмах всегда полупроницаемы. Протоплазма внутри
клеток непременно должна иметь то же самое
осмотическое давление, что и внешняя жидкость, в которой клетка
находится: оба раствора должны быть «изотоническими»
(изоосмотическими). Если внешнее осмотическое давление
больше внутреннего, то клетка сморш;ивается, если
внутреннее больше внешнего, то она лопается. Также и в
медицине (например, при болезни кровяных шариков)
обе возможности играют заметную роль.
2. Уравнение состояния разбавленных растворов
Вант-Гоффа. Чтобы перейти от этих обш;их рассуждений
к количественному исследованию, необходимо
ограничиться рассмотрением разбавленных растворов и
обратимых процессов. Именно на этом пути Вант-Гофф в 1885 г.
открыл известное сходство между разбавленными
растворами и идеальными газами.
Обычно для доказательства этой аналогии используют
циклический процесс с подвижным поршнем (который при
движении в одном направлении предполагается
полупроницаемым, а при движении в обратном направлении —
непроницаемым) и затем сравнивают полученную и
затраченную работы. Мы достигнем цели гораздо быстрее,
исходя из общих условий равновесия.
Рассматриваемая система состоит из двух частей —
чистого растворителя и раствора, отделенного от него
полупроницаемой пленкой. Все величины, относящиеся к
растворителю, снабдим индексом 1, все величины,
относящиеся к растворенному веществу, — индексом 2.
Подсистему «раствор» на одной стороне полупроницаемой пленки
будем отмечать верхним индексом 1, подсистему «чистый
растворитель» на другой стороне пленки — верхним
индексом 2. Соответственно этому различаем числа молей п\, п\
и п\. Так как в подсистеме 2 никакого растворенного
вещества нет, то
п\^0. A5.1)
§ 15. Разбавленные растворы 123
Будем считать, что полупроницаемая пленка не
препятствует теплообмену, так что температура во всей газовой
системе предполагается всегда одинаковой. С другой
стороны, следует допустить, что на разные стороны пленки
могут действовать различные давления р^ и р'^.
Уже при выводе условий равновесия в § 8 можно было
учесть случай Т = const, р^ = const Ф р'^ — const. Однако
легко сообразить, что ничего нового при этом не
получается, т. е. старое условие
8G = 0 A5.2)
остается справедливым также и при р^ Ф р^.
Дополнительные условия имеют вид
8wl + 8wJ = 0, A5.3)
1п\^Ъп\ = ^. A5.4)
Уравнение A5.3) выражает сохранение вещества
растворителя, а уравнение A5.4) есть математическое
выражение полупроницаемости пленки. Вводя в A5.2)
химические потенциалы из § 14, п. 1, получаем, принимая во
внимание уравнение A5.4),
fiJ8nl +{1^18^2 = 0. A5.5)
В силу A5.3) отсюда следует
t^?(^'^Л = l^}(л^. ф. A5.6)
При этом для ясности указаны переменные, от которых
зависят химические потенциалы. Тот факт, что величина
[X зависит только от отношений чисел молей
(концентраций), был установлен уже в § 14. То, что в \х\ должен
быть опущен аргумент п\\п\, очень важно, так как
означает, что здесь мы имеем дело с гомогенной
подсистемой, в которой концентрация п\\п\, согласно A5.1), равна
нулю. Теперь совершенно очевидно, что при п\ф^ ^
необходимостью должно быть р'^ — р^ф^О, В противном
случае условие равновесия A5.6) не могло бы быть выцол-
124 Гл. II. Применение термодинамики к конкретным системам
нено. Причина этого лежит в характерном для
равновесия соотношении 8nJ + S/ij = О, выражающем закон
сохранения вещества. Разность
Р = р1-р2 A5.7)
называется осмотическим давлением.
Уравнение A5.6) представляет собой точное уравнение
состояния произвольного раствора. Чтобы воспользоваться
им, необходимо знать химические потенциалы как
функции их аргументов. Так ках в общем случае это
неизвестно, то, как и при исследовании реальных газов,
пользуются результатами измерений и полуэмпирическими
формулами. Подобно тому как для газов идеальный газ
является предельным случаем, так и здесь допустимо чисто
теоретическое рассмотрение предельного случая сильно
разбавленного раствора. Пусть
Единственный результат введения в растворитель
растворенного вещества состоит в увеличении молекулярного
беспорядка, т. е. в появлении энтропии смешения. Выводы,
вытекающие из этого представления, подтверждаются
большим экспериментальным материалом. Впрочем, его можно
обосновать также термодинамически или статистическим
путем.
Если обозначить молярную свободную энтальпию
растворителя через giy то в соответствии с § 14, п. 2, имеем
\^\ = 8i{p\ T)-RTln'^^^ ,
\^\ = 8Лр\Т).
В силу предположения п\ < п\, 1п{1+ п11п\) ^ пЦп].
В связи с этим из равенств A5.6) и A5.8) следует, что
«1
gAp',T) = g,(p\T)-RT^,. A5.9)
Разложим левую часть в ряд Тейлора, ограничиваясь
первым членом,
$ 15. Разбавленные раствори 125
Из таблицы § 7 следует, что
'^ = v„ A5.11)
где ^1 —молярный объем чистого растворителя. Внося
выражения A5.10), A5.11) и A5.7) в уравнение A5.9),
получаем
Pv^^RT^,, A5.12)
«J
Так как парциальный объем растворенного вещества мал
по сравнению с объемом раствора V, то с той же
точностью, как и все прежние формулы, справедливо
соотношение
Тогда, обозначая число молей растворенного вещества п1
через л, получаем
PV = nRT. A5.13)
Это и есть уравнение Вант-Гоффа. Таким образом, осмо-
тическое давление разбавленного раствора, содержащего п
молей растворенного вещества, равно тому давлению
п молей идеального газа, которое он имел бы, будучи
помещен в сосуд, занимаемый данным раствором,
В случае нескольких растворенных веществ,
характеризуемых числами молей п^, П2, ..., вполне
аналогичным путем получаем
РУ = {п^^-щ+...)НТ, A5.14)
Т. е. уравнение состояния для смеси идеальных газов.
Поэтому парциальное давление Р^ отдельной составной
части раствора можно определить уравнением
P^V^n^RT. A5.14а)
Следовательно, как и для смеси газов, мы имеем
Неожиданная форма уравнения A5.13), разумеется,
столкнулась сначала с рядом выражений. Они, однако, были
опровергнуты не только колоссальным экспериментальным
материалом, но также и кинетическим рассмотрением
(между прочим, при участии Г. Лорентца).
126 Гл, II. Применение термодинамики к конкретным системам
§ 16. РАЗЛИЧНЫЕ АГРЕГАТНЫЕ СОСТОЯНИЯ ВОДЫ.
К ТЕОРИИ ПАРОВОЙ МАШИНЫ
В настоящем параграфе будет специально рассмотрено
равновесие между различными фазами воды: тем самым
мы коснемся происхождения термодинамики, а именно, ее
связи с развитием паровых машин (см. о Карно в начале § 6).
1. Кривая давления пара и уравнение Клапейрона.
Рассмотрим следующую хорошо известную систему: пусть
нижняя часть цилиндрического сосуда наполнена чистой
водой; подвижный поршень, плотно (герметически)
прилегающий к внутренней боковой поверхности цилиндра,
вначале лежит на поверхности воды. Если поршень поднять
при постоянной температуре, то при этом будет
испаряться как раз такое количество воды, которое
соответствует некоторому, не зависящему от объема, а
определяемому только температурой, давлению пара в
пространстве между поршнем и поверхностью воды. В этом случае
образуется насыщенный пар. Если поршень опускать, то
пар почти не будет сжиматься, но при этом
сконденсируется такое количество воды, что пар останется
насыщенным. Следовательно, существует не зависящее от
объема уравнение
9iP,T) = 0, A6.1)
связывающее данные температуру Т и давление р
насыщенного пара. Уравнение A6.1), будучи представлено
графически в плоскости /?, Г, дает кривую давления пара,
показанную на фиг. 13. Приведем некоторые численные
значения
t, °с
/7, am
0
6-10-3
50
0,126
100
1,03
120
2,02
200
15,9
Эта независимость от объема согласуется с вандерва-
альсовой моделью процесса сжижения. Представим себе,
что на фиг. 10 в качестве третьей координаты введена
температура, ось которой расположена перпендикулярно
§ 16, Различные агрегатные состояния воды
127
К плоскости Ру F, и рассмотрим возникающую при этом
«поверхность состояний». Если проектировать ее на
плоскость Ру Ту то каждая прямая Максвелла АВ на
плоскости Ру Т изобразится точкой. Тот факт, что на каждой
из этих прямых объем изменяется согласно соотношению
между количествами жидкости и пара, заданному
уравнением A0.12), никак не отражается на кривой давления
пара. В частности, две кривые —линия пара и линия
Вода или
пересыщенный
пар
Перегретый
пар
t^T-273
—^
150 200
Фиг. 13. Равновесие фаз между водой и
водяным паром.
жидкости, различающиеся в плоскости/?, F, при
проектировании на плоскость /?, Т совпадают и дают гладкую
кривую давления пара A6.1). Она заканчивается в той
самой точке, которая на поверхности состояний
соответствует критической точке. Следовательно, уравнение Внн-
дер-Ваальса действительно передает сущность явлений,
описываемых уравнением A6.1), которое справедливо не
только для водяного пара, но и для любого
конденсирующегося газа.
Исследуем уравнение A6.1). Так как оно записано
в переменных р, Ту то будем исходить, согласно § 8, из
128 Гл. II, Применение термодинамики к конкретным системам
свободной энтальпии G и условий равновесия 8G = 0. Для
нашей двухфазной системы имеем
G = ng,^{N~n)g,. A6.2)
Индекс 2 обозначает «более высокую» фазу (пар), индекс
1 — «более низкую» (жидкость). Одна фаза называется ка-
лорически более высокой, чем другая, если переход от
второй к первой требует подвода тепла. В силу этого
твердой фазе будет соответствовать индекс 0. В уравнении
A6.2) g2 и gi~~ малярные свободные энтальпии
соответственно пара и жидкости, w —число молей пара, N —
число молей пара и жидкости вместе. Величины g^ и gg
зависят только от р и Т,
Условие ЬС = 0 приводит, согласно A6.2), при
постоянных р, Т и N к соотношению (gg — gj Ьп = 0 и,
следовательно, к равенству
g2=gx- A6.3)
Это и есть искомая аналитическая формулировка
уравнения A6.1). Прежде чем его использовать, выведем отсюда
следующее дифференциальное уравнение.
Рассмотрим две соседние точки Р и Р' (см. фиг. 13)
с координатами /?, Т и p-\-dp, T + dT. Пользуясь
соотношениями таблицы, приведенной в § 7, получаем
= g{p,T) + dp^v^dT^s. A6.4)
Составим теперь разность величин g^ и gg, вводя при этом
обозначения
Ау = У2-^1, A5 = 52-5i, Ag=---g2-"g'i;
получаем из A6.4)
^g{P + dp, T-\-dr) = Ag{p,T)-\^dp^v-dTAs. A6.4а)
Левая часть и первый член npaBoii части этого
уравнения исчезают, так как равенство A6.3) относится к обеим
точкам Р и Р\ Поэтому A6.4а) дает
$-1^- (".5)
§ 16, Различные агрегатные состояния воды 129
Иыразим разность As через энтальпию Л. Поскольку
g=h-Ts (см. § 7), Ag = О и АГ = О, мы имеем
А/г = ГА5. A6.5а)
Величина А/г = /г2 —^i имеет смысл количества тепла,
которое надо сообщить одному молю вещества для
осуществления фазового перехода 1—^2 при постоянном давлении.
Ее можно назвать «теплотой испарения» или «скрытой
теплотой испарения». Мы будем обозначать ее буквой г.
Подставляя A6.5а) в уравнение A6.5), получаем
% = Ш- A6.6)
Это — знаменитое уравнение Клапейрона, которое впервые
было доказано термодинамически Клаузиусом. Если под
величиной Ау понимать разность не молярных, а удельных
объемов, то соответственно меняется смысл величины г
(удельная теплота испарения вместо молярной).
Сравнение уравнения A6.5) с уравнением G.6) пока-
:швает, что вместо частных производных {др/дТ)^ и {ds/dv)T
(для однофазной системы) для рассматриваемой системы
фаз появляются полная производная dpjdT и отношение
конечных разностей As/Av.
Если величины г и Ау измерены, то уравнениег
Клапейрона дает возможность построить кривую давления пара
но касательным к ней в каждой точке. Вместо того чтобы
производить это постепенное интегрирование, вернемся
к равенству A6.3), которое как раз и представляет собой
точный результат такого интехрирования. При этом будем
считать давление настолько низким, что пар можно
принять за идеальный газ. Тогда, согласно уравнениям
A4.14)-A4.17), имеем
g,^/?r[ln/.-^4nr-f^|,«i,] . A6.7)
^1тобы иметь возможность задаться также определенным
выражением для gi, пренебрежем, кроме того,
изменениями объема для жидких и твердых тел. В этом случае
исчезает различие между Ср и с^ и можно пользоваться
130 Гл. II, Применение термодинамики и конкретным системам
ОДНОЙ удельной теплоемкостью с^идк.- Отсюда для
энтальпии и энтропии получаем
т
^=Л1,+ 5^жидк.йгГ, A6.8)
s, = s,,+ Y-^dT^ A6.9)
Те
где Aj^ и 5^5 —значения энтальпии и энтропии при
произвольной начальной температуре Г^, в качестве которой
обыкновенно выбирают температуру плавления. В
последнем случае постоянные Л^^ и ^^g можно определить при
помощи тепловой теоремы Нернста и калорических свойств
твердой фазы.
Таким образом, свободная энтальпия жидкости равна
т т
^1= $Сж„дк.с/Г-г5^ + А,,-Гб-,., A6.10)
Те т,
и условие равновесия A6.3) вместе с A6.7) и A6.10) дает
т
Те
т
'_j^^T_]4^^_^l^_!^ A6.11)
Те
Принципиально все величины в этой формуле, вклю-
-н
рений в твердой фазе. Если значение i^, даваемое
квантовой теорией, вызывает сомнения, то его можно
проконтролировать, измеряя давления пара. На фиг. 13
показана полученная таким образом кривая давления пара.
Она, как уже можно было заключить из уравнения
Клапейрона, только поднимается вверх. В самом деле, г всегда
положительно (это равносильно нашему прежнему
определению «более высокая фаза») и, кроме того, конечно,
^v> О (так как v^ > v^.
Самая высокая температура, которую приходится
рассматривать для водяного пара (см. выше), — это его кри-
§ 16. Различные агрегатные состояния воды 131
тическая температура. Здесь кривая заканчивается (на
фиг. 13 эта точка не изображена, так как она лежит
очень высоко). Кривая имеет также начальную точку^.
положение которой определяется физическими свойствами
системы. Для водяного пара она лежит в
непосредственной близости от начала координат на фиг. 13 (см. § 17^
п. 1) и может служить для прямого контроля значения i^^
2. Фазовое равновесие между водой и льдом. Лед
относительно воды является более низкой фазой, так>
например, для плавления 1 г льда к рассматриваемой
системе необходимо подвести теплоту плавления г (скрытая
теплота конденсации). Твердая фаза обозначается
индексом 0. Под величиной At; теперь следует понимать
разность объемов Увода и Улед; ТОТ же смысл имеет символ'А,
примененный к g, h и s. Из уравнений A6.4) и A6.5)
снова получаем формально не изменившееся уравнение
Клапейрона A6.6), однако с той разницей, 'что Ау те-^
перъ отрицательно:
Ai; = ?;1-Уо = A,00-1,091) си*з/2= - 0,091 сж^/а.
Это обстоятельство имеет фундаментальное значение для
жизни на Земле. Лед плавает на воде. В
противоположном случае все рыбы зимой замерзали бы, и в наших
широтах не могла бы возникнуть никакая жизнь.
(Сухопутные животные также, как известно, происходят от
животных, живших в воде.) Вода расширяется при
замерзании. Этим в значительной степени обусловлена эрозия
гор (взрывное действие замерзшей воды в расщелинах гор^
ных пород), которая приводит к образованию плодородных
почв в долинах.
Из уравнения Клапейрона следует, что кривая
плавления с ростом температуры падает вниз в
противоположность кривой давления пара. Для области температур
вблизи 0°С имеем
/• = 80 кал1г = 80 • 42,7 am • см^/г,
[с учетом формул D.6), C.2) и C.2а)]. Отсюда для Т ^ 273*"
п t — T—273° (^ — температура по шкале Цельсия) имеем
^^ " 273.М91 «'^/^/^«^= — 138ат/г/?аа. A6.12)
132 Гл. II, Применение термодинамики к конкретным системам
Поэтому кривая плавления ср (/?, ^) = О идет следующим
образом: выходя из точки /? ^^ О, / ^ О, она проходит во
втором квадранте на фиг. 13 в виде очень крутой и почти
прямой линии. Необходимо повысить давление до 138 am,
чтобы снизить температуру плавления до — 1°С. Это
понижение точки замерзания существенно при всей его
малости для передвижения ледников. Под давлением
вышележащих слоев льда более глубокие части ледника
становятся подвижными, однако они снова замерзают, если давление
уменьшается (режеляция). Этим же обусловлена большая
подвижность конькобежца: под давлением коньков лед
расплавляется и образовавшаяся вода действует как смазка.
3. Удельная теплоемкость насыщенного водяного
пара. До сих пор говорилось лишь об удельных теплоем-
костях Ср и с„. Можно, однако, определить удельную
теплоемкость для любого процесса, т. е. для любого контура
в плоскости /?, V, Очевидно, что для адиабатического
процесса {dq = 0) Сз = 0. С другой стороны, можно утверждать,
что ст ^ оо, так как в изотермическом процессе при любом
dq изменение температуры пренебрежимо мало. Так же
как и для контуров в плоскости р, v, можно говорить
об удельной теплоемкости для любого контура в
плоскости р, Т.
Особый интерес для наших целей представляет
удельная теплоемкость пара при продвижении вдоль ь-ривой
давления пара ср(/?, Г) = 0. Обозначим ее через с^. Будем
исходить из определения теплоты испарения A6.5а) г = АЛ.
Принимая во внимание, что h-=u-\-pv, имеем
dr dAu , dAv , dp . /ла ло\
Согласно первому началу термодинамики, для любого
контура
%'% + Р%' ('"За)
следовательно, для специального случая пара на кривой
давления пара имеем
/ 16. Различные агрегатные состояния еоды 133
Соответствующее выражение для жидкой фазы имеет вид
сжи„к. = ^ + р^, A6.13b)
так как для жидкости, как уже отмечалось перед
выражением B.4), разница между с^^ и с^ исчезает, и можно
положить Ср «^ с^ г^ Сжидк.. Из выражсния A6.136) следуст,
что
_ dAu j^ dAv
С^ — С'жидк. --df^P'df'
Перенося это в уравнение A6.13), получаем
g, = С; - Сжидк. +^^v. A6.13г)
Если учесть еще уравнение Клапейрона A6,6), то мы
получим
с^ = ^ +Сжидк.--^ . A6.14)
Согласно очень точным измерениям ^) (проведенным с
техническими целями), при Т ^ 273 °
^= —Оу^Акал/г-град. A6.14а)
Следовательно (поскольку г =539 кал/г для Г = 373^,
Сжидк. = 1 кал/г' град),
с,^ = A—О, 6А—1М)кал/г-град=-1,07кал/г»град. A6.146)
При изменении состояния вдоль кривой насыщения насы-
ценный водяной пар не только не требует никакого
подвода тепла, но в состоянии давнее отдавать тепло. Если,
с другой стороны, заставлять его расширяться без подвода
тепла, то пар попадет (см. фиг. 13) в область слева от
кривой давления насыщенного пара (на фиг. 13 эта
область носит название «вода или пересыщенный пар»).
Здесь пар склонен к конденсации.
Приведем два примера—один тривиальный, а другой
фундаментальный для современной физики. В бутылках
с минеральной водой между поверхностью жидкости и проб-
^) См. также задачу II. 2.
134 Гл. //. При^ненение термодинамики к конкретньин системам
кой образуется атмосфера насыщенного водяного пара.
Если бутылку открыть, то происходит мгновенное
адиабатическое расширение и водяной пар конденсируется в
капельки. Этот процесс находит важное применение в
камере Вильсона A912 г.). Камера, содержащая пересыщенный
водяной пар, подвергается быстрому расширению. Если
незадолго перед этим через камеру прошли ионизирующие
частицы, то возникающие ионы действуют как центры конденсации
и путь частиц становится видимым. Известно, какое большое
.4'
Адиабатицесиое
расширение
-^ ^-
V
Фиг. 14. Индикаторная диаграмма,
исправленная для отрицательной удельной
теплоемкости вдоль кривой давления пара.
значение имел этот метод для целого ряда исследований
(космические лучи, открытие позитронов, мезонов и т. д).
Тот факт, что с5 отрицательно, имеет также большое
значение для теплотехники. При расширении адиабата
водяного пара идет менее круто, чем адиабата
идеального газа /?у^ = const. Вследствие этого индикаторная
диаграмма для водяного пара имеет по сравнению с
диаграммой для идеального газа дополнительную площадь
(заштрихованную на фиг. 14). Это обстоятельство очень выгодно
<• точки зрения применения воды в паровых машинах.
§ 17. ОБЩИЕ ПРИНЦИПЫ ТЕОРИИ РАВНОВЕСИЯ ФАЗ
Различные агрегатные состояния воды представляют
собой только случайно выхваченный пример гораздо более
общей проблемы сосуществования произвольных фаз
вещества произвольного состава. Уже в специальном случае
§ 17. Общие прмнципы пкориы равновесия фаз 135
)юды только ЧТО нарисованная картина является неполной.
Б твердом состоянии существует не только обычный
гексагональный лед, структура которого лучше всего видна
на снимках снежинок, сделанных через микроскоп, но
также, согласно Тамману, и многие другие
кристаллические модификации, существующие в других областях
плоскости Ру г. Кроме того, полное описание свойств воды
должно включать и ее диссоциацию на кислород и
водород (см. § 13), причем в этом случае мы имеем дело не с
гомогенной (HgO), а, вообще говоря, с гетерогенной
системой, включающей также Hg и Og.
1. Тройная точка воды. Прежде всего вернемся к
диаграмме состояний воды в плоскости /?, Г. В отличие от
фиг. 13 нам будет удобно теперь откладывать давление р
1Ю горизонтальной, а температуру t (измеренную по
шкале Цельсия) по вертикальной оси; для обеих величин мы
выберем весьма большие единицы измерения. Кривая
давления пара теперь будет выпуклой вверх, а кривая
плавления в соответствии с § 16, п. 2, будет почти прямой
линией, составляющей весьма малый (^/lae) угол с осью
абсцисс.
Выясним, как разграничены в плоскости /?, t области
сосуществования льда и пара. Известно, что лед
может переходить в пар не только через промежуточное жид-
1юе состояние, но и непосредственно. Этот процесс
называется сублимацией (название, собственно говоря,
происходит не от воды, а от ртути). Сублимация наблюдается
весной при небольшом морозе и сильном солнце, когда
снег исчезает в больших количествах без таяния; в
действительности он улетучивается в воздух в виде водяного
пара. Условие такого перехода дается также уравнением
'f (/?, Т) = О, определяющим кривую сублимации в плоскости
/?, Л Покажем, что эта кривая проходит через точку
пересечения кривых плавления и давления пара. Для этой цели
])ассмотрим аналитическое представление трех
перечисленных кривых.
Кривая плавления: cpoi = 0, g^^g^.
Кривая давления пара: cpi2 = 0, g^ = g^, A7.1)
Кривая сублимации: cp^g^O, go = g^*
136 Гл, II. Применение термодинамики к конкретным системам
Отсюда следует, что равенство go^gz выполняется, если
одновременно справедливы равенства go~?i ^ Si^So-
Точка пересечения всех трех кривых называется тройной
точкой. Она задается соотношениями
«^0 = ^1 = 82
A7.1а)
Скрытые теплоты, соответствующие этим трем
переходам, в дальнейшем будем обозначать через г^^, r^g, г^^:
t/c
Фиг. 15. Окрестность тройной точки в
плоскости р, г,
величина г^^ называется теплотой сублимации. Согласно
первому началу термодинамики, Tq^, r^g и Tqj связаны
соотношением
^•01 +''12+''20 = 0.
A7.2)
(Оно получается, если рассмотреть циклический процесс,
в котором совершается обход вокруг тройной точки;
при этом энергия возвращается к своему
первоначальному значению.) Из соотношения A7.2) вытекает, что
'-02=-''го = ''01 +''и- A7.2а)
Пользуясь полученным в § 16, п. 2 значением,
§ 17. Общие принципы теории равновесия фаз 13/
Го1 = 80 кал/г-град и принимая во внимание, что г^2=^
= 603 кал/З'град^), получаем
Го2 = 683 кал/г. A7.20)
Так как аналитические выражения для энтальпий g^
и g^ неизвестны (можно получить лишь приближенное
выражение для g^ из уравнения состояния идеального
газа) и, следовательно, соотношения A7.1) использовать
нельзя, координаты тройной точки приходится определять
из опыта. Они равны (фиг. 15)
^ = 0,0074° С, /? = 0,0061 атм. A7.3)
Вдоль кривой сублимации также справедливо
уравнение Клапейрона
W-it' A7.4)
где теперь
Ду = Ау^о = ^2 - ^0 == ^2 - ^1 + ^1 - ^0 = ^^'21 + ^^10- A7.4а)
На фиг. 15 эти три кривые (изображающие
последовательности некоторых состояний равновесия) продолжены
:^а тройную точку пунктиром. Последнее показывает, что
в этих частях плоскости /?, t они соответствуют
состояниям неустойчивого равновесия.
Резюмируя, можно утверждать, что сосуществование
трех рассматриваемых фаз воды возможно только в одной
точке плоскости р, t; сосуществование любых двух фаз
имеет место вдоль определенной кривой, каждая фаза
в отдельности существует в определенной части
плоскости Ру t.
На фиг. 16 показана сделанная из картона
пространственная модель /?, у, Г-диаграммы для воды.
Температура Т откладывается по вертикальной оси, объем у — пого-
ризонтальной (назад), давление /?-слева направо. Фазе
пара, соответствует круто поднимающаяся поверхность
гиперболоида T = pv/R с пересекающимися
прямолинейными изобарами и изотермами. Основание гиперболоида
*) Это значение получено экстраполяцией па основании
знамений г = 539 кал/г при i = 100°C и производной dr/dt=^
==—0,64кал/г.грод [cNf. A6.14а)].
138 Г.1. И. Применение термодинамики к конкретным системам
ограничено линией пара. Твердая фаза (лед) появляется
в виде валика слева внизу вдоль кривой плавления и за-
1чанчивается у :нсидкой фазы. Последняя ограничена сверху
линией жидкости. Между ней и линией пара расположена
в виде правильной поверхности область вода-^ пар. Если
смотреть на нее слева вдоль оси v, т. е. проектировать ее
Фиг. 16. Модель трех фаз воды.
i—пар, 2—вода, 5—лед, 4—вода+пар, 5—лед+пар, б—линия пара, 7~трой-
ной край, 5—линия жидкости. Для более удобного сравнения с фиг. 15
нужно представить в последней ось р направленной налево.
на ПЛОСКОСТЬ /?, Г, то она представляется в виде кривой
давления пара, как это уже указывалось выше, в связи
€ уравнением A6.1). Выпуклость внизу справа изображае!
область сосуществования лед + пар (она тоже изображается
правильной поверхностью). Вдоль тройного края она
переходит в область вода-{-пар. Передняя точка тройного
края и есть тройная точка, Кривая сублимации
получается, если эту часть модели спроектировать на
плоскость Pj Т,
g 17. Общие принципы теории равновесия фаз 1'^9
2. Правило фаз Гиббеа. До сих пор мы ограничива-
jmcb одним веществом (HgO); рассмотрим теперь систему
из произвольного числа компонент, снабдив их номерами
1,2, ...А, ...if.
Далее, до сих пор принималась во внимание
возможность сосуществования только двух или трех фаз; теперь
допустим, что данные К компонент могут находиться
в произвольном числе раз^чичных физических состояний
и химических соединений; все их мы будем попрежнему
называть фазами и снабдим номерами
1, 2, ...i, .../.
Очевидно, / обозначает максимальное число
сосуществующих фаз, тогда как К есть полное число реагирующих
веществ.
Пусть число молей Л-го вещества в i-й фазе есть п^^^,
Свободная энтальпия G аддитивно слагается из вкладов
отдельных фаз и веществ. Из условия 8G = 0 получается
система уравнений
S S?iftS«ife = 0. A7.5)
{=1 k=i
Если бы все вариации 8/г^^ были независимы, то система
уравнений A7.5) состояла бы из JK уравнений. Однако
фактически величины 8w^^ связаны между собой условиями
постоянства полных (распределенных по всем фазам) чисел
молей всех компонент. Эти условия имеют вид
J
2 8л^;, = 0 для А = 1, 2, ...л:. A7.5а)
Поэтому число независимых уравнений составляет не JK,
а только
JK-K. A7.6)
Найдем число неизвестных, которые надлежит
определить из этих уравнений. Это, во-первых, числа молей л^^,
полное число которых есть JK, Однако так как G является
однородной функцией л^^^ (см. § 14, п. 1), то в условия
равновесия A7.5), A7.5а) для каждой фазы входят только
140 Гл. II, Применение термодинамики к конкретным системам
отношения величин л^^^, число которых для каждой фазы
равно К—\\ суммируя по всем фазам, получаем
/(Z-l) = /Z-/.
К этому добавляются еще две переменные р л Т^
определяющие точку в плоскости р, Т. Поэтому полное число
независимых переменных равно
JK^J+2, A7.7)
Если это число меньше числа уравнений A7.6), то
решение вообще невозможно. Следовательно, должно
выполняться условие
JK^J + 2>JK^K,
откуда следует
J<K + 2. A7.8)
Это и есть знаменитое правило фаз Вилларда Гиббса.
Можно также написать
J + F = K + 2, A7.8а)
где F обозначает число степеней свободы, которыми
обладает система с К веществами и / сосуществующими фазами.
Если F = Oj то состояние системы называется
инвариантным. Этот случай соответствует воде {К = I) в
тройной точке. Здесь 7 = 3 и одновременно сосуш.ествуют лед,
вода и пар.
Если F=l, то система называется моновариантной.
Для специального случая К=^ I при этом / = 2, что
соответствует сосуществованию двух фаз на кривых давления,
плавления или сублимации.
Если F=2, то система называется дивариантной.
Для ЛГ=1 будем иметь /=1, т. е. имеется чистая фаза
(она может существовать в некоторой области на
плоскости р, Т).
Если принять во внимание, что кроме обыкновенного
(гексагонального) льда могут существовать еще и другие
модификации (Тамман), то из правила фаз следует, что
две твердые модификации могут сосуществовать еще только
либо с жидкой, либо с газообразной фазой. «Четверной»
точки при jK'=l не существует. Такая точка может по-
§ 17, Общие принципы теории равновесия фаз 141
явиться впервые в «бинарной» системе (содержащей две
компоненты): при К = 2 и F = 0 / = 4.
Применение правила фаз к системам с тремя или более
степенями свободы приводит к построению многомерных
поверхностей фазовых равновесий. Однако изучение их
относится уже к области физической химии.
3. Закон Рауля для разбавленных растворов. Особенл-^
простым случаем равновесия фаз является испарешл
разбавленного раствора, при котором растворенное
вещество не улетучивается. При этом можно воспользоваться
выведенным в § 15, п. 2 уравнением Вант-Гоффа, если,
как и раньше, исключить из рассмотрения важный случае!
электролитов. Относительно обозначений заметим
следующее: если до сих пор фазы различали нижними
индексами, то теперь мы будем отмечать жидкую и
парообразную фазы растворителя верхними индексами 1 и 2,
которые в § 15 примеБШЛись для обозцачения двух подсистем —
«раствора» и «чистого растворителя». Нижние индексы 1 и 2
понадобятся, как ив § 14, для обозначения
«растворителя» и «растворенного вещества». Таким образом, роль
«чистого растворителя» теперь играет парообразная фаза
(число молей п1); «полупроницаемой пленкой» является
не проницаемая для растворенного вещества поверхность
жидкости. Числа молей разбавленного раствора
обозначаются здесь, как и в § 15, через п\ и п1.
Различие состоит только в том, что теперь давление
одинаково в обеих фазах. Прежнее условие равновесия A5.5)
f.J8nt + [x?5n? = 0 A7.9)
и дополнительное условие A5.3)
Ьп1 + Ьп1 = 0 A7.10)
не изменяются, так что в качестве уравнения,
определяющего теперь «кривую давления пара», получается прежнее
уравнение A5.6), однако с учетом того, что /?^ = /?2 = /?,
т. е. уравнение
\^Цр>Т) = А(^Р,Т,^'). A7.11)
142 Гл. П. Применение термодинамики к конкретным системам-
Аналогично прежним выражениям A5.8) следует положить
,l=^g\{p,T)-RTlnJ!^,
\^\ = 8\{Р,П
где в отличие от § 15 g^ обозначает сюбодную энтальпию
пара, а gj —свободную энтальпию чистого растворителя.
Тогда условие равновесия имеет вид
gl{p, Т) = g\{p,T)-RTIn 1^^. A7.13)
Так как для достаточно разбавленного раствора л^ < nj,
то вместо логарифма можно написать п1/п\ или просто
ng/Wi (опустив верхние индексы, которые теперь
становятся излишними). Тогда уравнение A7.13) принимает вид
gl(P,T) = g\{p,T)-RT^. A7.14)
Это уравнение изображает в плоскости /?, Т кривую
давления пара над разбавленным раствором.
Для сравнения приведем кривую давления пара над
ЧИС1ЫМ растворителем (для которого в отличие от р, Т
будем писать /?*, Т*):
g\iP*,T*) = gl{p*,T*). A7.14а)
Вычитая A7.14) из A7.14а), получаем
gi{P*,T*)-g\{p,T) = g\{p*,T*)-g\{p,T) + RT-^^.
A7.15)
Разлагая левую и правую части A7.15) в ряд Тейлора
и ограничиваясь первыми членами, получаем
-(р*-/')^ + (Г-п[-#]+-»г^^- A7.16)
Подставляя сюда знечения производных из таблицы,
приведенной в § 7, имеем
(P*-p)[tA{p,T)-v[{p,T)] +
+ (T-T*)[sUp, T)-s\{p, T)] = RT^. A7.17)
Рассмотрим это уравнение с двух точек зрения.
§ 77. Общие принципы теории равновесия фаз 143
а) Представляет интерес выяснить, как изменяется
давление пара при постоянной температуре вследствие
добавления ^2 молей вещества 2 к растюрителю 1. Тогда,
чтобы определить пониэФсение давления Ар = р* — р^ в
уравнении A7.17) следует положить Т = Т*; получаем
^Р=1^^^- A7.18)
Эта формула справедлива для любого разбавленного
раствора, т. е. во всех случаях, когда п,2<кп^- В
частности, если пар можно рассматривать как идеальный газ^
т. е. положить
2_ RT
и пренебречь молярным объемом жидкости по сравнению
с объемом пара, то формула A7.18) упрощается и при-
нил1ает вид
-^=-^. A7.19)
Р Пу ^ ^
Это и есть закон относительного пони^неения давления пара.
Последнее не зависит от природы растворителя и
растворенного вещества, определяясь только отношением чисел
молей этих веществ. Этот удивительно простой закон был
установлен эмпирически Раулем в 1886 г. Вскоре после
этого он был выведен на основании термодинамических
соображений Вант-Гоффом.
б) Исследуем, как меняется температура кипения при
постоянном давлении вследствие добавления вещества 2
к растворителю 1, т. е. вычислим разность Т^Т*=АТ.
Для этого положим в уравнении A7.17) р=^р* я примем
во внимание, что
{sl~sl)T = n
где г —теплота испарения. Из уравнения A7.17) тогда
получаем
ДГ = ^^. A7.20)
Таким образом, к закону понилсения давления пара
присоединяется закон повышения точки кипения. О послед-
144 Гл. II. Прьименрние термодинамики к конкретным системам
нем законе и его историческом происхождении можно
«сказать то же, что было сказано выше о законе
понижения давления пара.
Повышению точки кипения соответствует в случае
фазового перехода 1 —> О (замерзание) пониэ^сение точки
замерзания. Действительно, тогда в формуле A7.20) (которая
остается справедливо!!) вместо г = г^^ появляется теплота
реакции г^^ = — г^^, равная отрицательной теплоте
плавления.
До сих пор предполагалось, что масса молекулы остается
яеизменной; тем самым процессы полимеризации и
диссоциации вьшадали из рассмотрения. Однако их можно
учесть, если, например, в правую часть уравнения A7.11)
ввести соответствующее отношение масс т^/т^. Значение
этого обстоятельства для определения молекулярных весов
растворителей и пара было подчеркнуто Плавком ^).
4. Закон абсорбции Генри A803 г.). Условие
равновесия 8G = О находит таьже простое применение при
изучении растворимости газа в жидкости, давление пара
которой исчезаюпю мало по сравнению с давлением газа.
В этом случае имеют значение только химические
потенциалы растворяющегося газа в газовой фазе ji^ и в
растворе [i^ (нижние индексы, обозначающие, что речь идет
именно о газе, здесь и в дальнейшем можно опустить).
Дополнительное условие
дает, как и в случае A7.11),
[xi = (i2. A7.21)
^1,ля газовой фазы, которую мы опять будем рассматривать
как идеальный газ, имеем
^^ = g^ = BTlnp + ^{T). {П.22)
Функция фG'), в которую входит химическая постоянная
газа, в явном виде нам не потребуется. Для раствора
можно написать выражение, аналогичное первому из выра-
1) Р1 а п с к, Thermodynamik, § 269, 270.
§ 18. HanpnofcPHue галъваничегкого элемента 145
жений A7.12),
[xi = gi(p, Г) + ЛГ1пс, A7.23)
где с обозначает молярную концентрацию растворенного
газа [в выражении A7.12) она обозначается через п\/п,
где п = п\-{-п1]\ в определенных границах свободная
энтальпия жидкости неизменного состава практически не зависит
от давления, так что выражение A7.23) можно записать
в виде
Из выражений A7.21) —A7.23) получаем
И, следовательно,
c^pf{T), причем 1п/(Г)=1^^^-^. A7.24)
Это и есть известный закон Генри: количество газа,
поглощенного мсидкостью, пропорционально парциальному
давлению газа, оставшегося над ^нсидкостгю. Множитель
пропорциональности зависит только от температуры (общей
для газа и жидкости) и не зависит от количества газа
в объеме над жидкостью.
§ 18. НАПРЯЖЕНИЕ ГАЛЬВАНИЧЕСКОГО ЭЛЕМЕНТА
До сих пор мы рассматривали термодинамические
системы, состоящие только из электрически нейтральных частиц
(атомов и молекул). Исследуем теперь, что изменяется,
если в состав системы входят также заряженные частицы
(электроны или ионы). Сюда относятся такие проблемы,
как термические и калорические уравнения состояния
электролитов, электронов в металле, ионизированных газов
и т. д. Ограничиваясь пока термодинамикой равновесных
состояний, мы с самого начала исключим все
неравновесные задачи, как, например, прохождение тока через металлы
или электролиты.
Из всего множества остающихся проблем будет
рассмотрен вопрос о напряжепии между электродами
разомкнутого гальванического элемента, так как в этом случае
можно без особого труда получить определенные утвержде-
440 Гл. II. Применение термодинамики к конкретным системам
ния общего характера. Мы добавляем здесь слово
«разомкнутый» к названию, указанному в заглавии параграфа,
чтобы подчеркнуть, что нас интересует только статический
случай. Равновесие должно быть уже достигнуто, и оно
не должно больше нарушаться необратимыми процессами,
например выделением джоулева тепла при прохождении
тока. Кроме того, следует считать, что напряжение
элемента измерено при помощи электростатического вольтметра.
Подобно тому как два раствора различных
концентраций, разделенные полупроницаемой перегородкой, могут
находиться в термодинамическом равновесии только в том
случае, если между ними постоянно поддерживается
осмотическая разность давлений, между двумя фазами,
способными обмениваться заряженными частицами, при
равновесии устанавливается определенная электрическая разность
потенциалов.
1. ^Элeктpoxимичecкиe потенциалы. Рассмотрим, как
и в § 8, термодинамическую систему S и окружающую
ее среду I'. Пусть система Е характеризуется функциями
состояния F, Т п п^. Согласно соотношению A4.1), для
системы S имеем
TdS = dU ^- pdV ^^^^йщ, A8.1)
i
Представим себе мысленно бесконечно малое изменение
состояния
dV = 0, dT = О, dn^ = О при i Ф /, dn^ Ф 0.
Тогда, как и раньше, величина (Лус/пу представляет работу,
которую следует совершить над системой -, чтобы обрати-
мым путем изменить число молей п^ на duj.
Пусть теперь /-я компонента не является, как прежде,
электрически нейтральной, а имеет положительный заряд
2, т. е. состоит из молекул или атомов, потерявших z
электронов. Следовательно, один моль /-й компоненты несет
заряд zF, где F - число Фарадея, равное 96 494 кулонам
(элементарный заряд, умноженный на число атомов в одцом
моле). Тогда система И обладает относительно системы S'
электрическим потенциалом Ф. Не уменьшая общности
рассуждений, можно считать систему S' заземленной и
§ 18. Напрямсение гальванического элемента 147
принять ее потенциал за нуль. Так как в данном случае
рассматривается мысленный эксперимент, то нас не
интересует, каким образом будет поддерживаться напряжение
Ф. Если теперь к системе S подвести drij молей
заряженных частиц, то к совершаемой «химической» работе jiyrfn^.
добавится еще «электрическая» работа
Uyz.Fdrij.
Следовательно, полная работа, выполненная над системой
S, равна
В силу условия обратимости процесса перенос зарядов
должен происходить бесконечно медленно и, следовательно,
джоулсво тепло не может выделяться. Таким образом,
вместо A8.1) следует написать
TdS = dU + pdV^^7l^dni, A8.2)
7|i = jx, + 2,m A8.3)
В отличие от химических потенциалов ji^ величины Тц
называются элекарохимическими потенциалами. Для
отрицательно заряженных частиц естественно считать z^
отрицательными.
В общем случае гальванический элемент
характеризуется не одним потенциалом. Напротив, поскольку элемент
составлен из «цепочки» веществ в различных фазах, каждая
фаза находится в равновесии только с двумя соседними
и имеет свой собственный электрический потенциал. Можно
представить, по крайней мере в пределах нашего
мысленного эксперимента, что различные фазы отделены друг
от друга полупроницаемыми пленками, через которые
проходят только определенные ионы. Поясним это на примере
элемента Даниэля.
2. Элемент Даниэля A836 г.). Элемент Даниэля
схематически изображен па фиг. 17. Отдельные фазы обозначены
римскими цифрами. Через перегородку М (из глины) могут
проходить только ионы SO4. Представим себе, что цин1чсь
вый электрод IV заканчивается медным отростком V. Тогда
напряжение Ф/ — Фу разомкнутого элемента будет изме-
148 Гл. II, Применение термодинамики к конкретным системам
ряться между одинаковыми металлами и при измерении
напряжения электростатическим способом не появится
никакой контактной разности потенциалов. В дальнейшем при
написании электрохимического потенциала т] и числа молей п
будем слева наверху указывать соответствующую фазу,
а справа внизу —сорт частиц, например ^^iqcu++- Для
обозначения электронов используем символ 9. Если произвести
Фиг. 17. Схема элемента Даниэля:
Zn I ZnS04+H.O I CUSO4+H2O I Си
Твердая фаза Жидкая фаза М Жидкая фаза Твердая фаза
варьирование при постоянных р и Т и химические
потенциалы заменить на электрохимические, то условие
равновесия между фазами / и // SG = О и дополнительное
условие (сохранение числа молей), согласно уравнениям
A7.9) и A7.10), будут иметь вид
Из условий A8.4) и A8.5) получаем
A8.4)
A8.5)
A8.6)
Согласно A8.3), это означает (при 2—4-2)
Фазы / и //: ^р.си+*4-2^Ф/ = ^^р.си+++2/^Ф//. A8.7)
§ 18. Напряжение гальванического элемента 149
Аналогичным путем получаем условия равновесия между
другими парами фаз:
Фазы // и ///: 1%^--- 2РФи = ^^^sor " 2^Ф///,
A8.8)
Фазы /// и IV: ^^Vzn+++ 2^Фт = ^^^1гп--+2^Ф/у,
A8.9)
Фазы IV и V: ^ V - ^Ф/у = % - ^Фу. A8.10)
Кроме того, следует описать реакции, происходящие
на медном и цинковом электродах:
Cu^Cu++ + 29, Zn;:^Zn++ + 2e.
Это—нейтрализация ионов Си++ при осаждении их на
медном электроде и двукратная ионизация атомов Zn при
прохождении их через граничную поверхность Zn —ZnS04.^
Этим процессам соответствуют дополнительные условия
Vcu-2%-Vcu-, A8.11)
^>zn-2^Ve = ^Vj,^n-. A8.12)
Наконец, благодаря тождественности материалов ив
которых состоят фазы V и I (Си), получаем еще одно
уравнение
V = Ve- A8.13)
Приведенная система уравнений полностью и во всех
подробностях описывает реакции, происходящие между
рассматриваемыми фазами в состоянии равновесия.
3. Сведение отдельных реакций к упрощенной брут-
то-реакции. Из уравнений A8.7) — A8.10) можно
последовательно исключить потенциалы Ф//, Фщ и Ф/у. Поступая
таким образом, находим
2F {Фг ~ Фу) = (^V - V)cu- + (^V - ^^V)zn- -h
+ (^V-^^Ho7-+ 2(^'^t^- '^i^)e- A8.14)
Первый член справа в силу A8.11) можно преобразовать
к виду
^Vcu--^Fbcu + 2Ve. A8.15)
150 Гл, II. Прьиченение термодинамики к конкретным системам
Равным образом второй член справа, согласно уравнению
A8.12), записывается в виде
^Vn-^^Vzn- + 2^VH. A8.16)
Благодаря условию A8.13) четвертый член в A8.14) и два
предыдущих члена с (хв взаимно уничтожаются. Поэтому
из уравнения A8.14) получаем
Ф/ — Фу =
2^[^^[xzn-^^Vzo- + ^Vcu- - Vcu + (^V-^^V)soГ^
A8.17)
Разность Ф/ — Фу представляет собой напряжение между
электродами неразомкнутого элемента, которое обычно
называют электродвижущей силой (э. д. с.) и обозначают
через Е,
Можно положить ^Vso'^^^^^P'so""' ^^^ равносильно
равенству Ф// = Ф///. Выполнения этого равенства можно
добиться при помощи некоторого искусственного приема.
Тогда вместо A8.17) имеем
E.2F = ^Vn^-^Vcu--^^Vzn- -^[Jbcu A8.17а)
Физико-химический смысл этого уравнения состоит в
следующем. Представим себе, что во внешнем пространстве
происходит обратимый (электростатический) перенос заряда
2F от положительного полюса к отрицательному, а внутри
элемента Даниэля перенос такого же количества заряда
от отрицательного полюса к положительному. Последний
процесс осуществляется благодаря реакции (в которой
участвует один моль каждого вещества)
Zn->Zn++
Си++--» Си A8.176)
Zn + Cu++->Zn+++Cu ,
текущей при постоянных давлении и температуре. При
этом величина 2FE составляет (электрическую) работу,
совершенную элементом над внешней средой. Теперь
правую часть уравнения A8.17а) можно толковать как
приращение свободной энтальпии ^G в процессе A8.176). Это
§ 18. Напря:нсение гальванического элемента 151
следует из выражения
ДС = 2(.Д-Дп^), A8.18)
i
которое получается, например, из A4.17), при ^/2zn= - 1,
^'?zii++ = +1, Д^си = +1> ^'^си++ = — 1 и постоянных
химических потенциалах. Впрочем, строго говоря, для того
чтобы соблюдалось последнее из перечисленных условий,
в приведенной реакции должна участвовать бесконечно
малая часть моля.
Реакция A8.176) есть брутто-реакция в элементе.
Она представляет co6oii результат совокупности реакций
A8.7)-A8.13). Уравнения A8.7)-A8.13) вытекают из
виртуальных перемещений, а уравнение A8.18)
соответствует реальному перемещению заряда. Однако из
уравнений A8*. 17а) и A8.176) видно, что брутто-реакция совсем
не зависит от частных реакций. Последние служат лишь
для разъяснения того, что э. д. с. возникает благодаря
установлению равновесия ме:нсду отдельными частями
системы.
При перемещении заряда 2F, т. е. в результате брутто-
реакции одного моля вещества, выделяется определенная
теплота реакции, которую можно непосредственно измерить.
Выведем теперь соотношение между этой теплотой
реакции и э. д. с.
4. Уравнение Гиббса—Гельмгольца. Прежде всего
отвлечемся от специального случая элемента Даниэля и
перейдем к произвольному гальваническому элементу.
Очевидно, что уравнения A8.17а) и A8.18) должны быть
справедливы и в общем случае, т. е.
^ = i-AG, A8.19)
zF
где AG — приращение свободной энтальпии при
превращении одного моля в результате реакции, текущей при
постоянных р и Г, а 2 — валентность аниона или катиона
или общее наименьшее кратное этих валентностей.
При помощи таблицы, приведенной в § 7, например,
из соотношений G = H — TS, S = — [dGjdT)^, образуя соот-
152 Гл. II. Применение термодинамики к конкретным системам
ветствующие конечные разности, находим
ДС = Г(^у^)^ = А//. A8.20)
Если допустить, что реакция протекает при F = const,
7" = const, то вместо уравнений A8.19) и A8.20) мы
получим уравнения ^)
Е = ±А^, ¦ A8.21)
Д^-Г(^-^)^ = АС/, A8.22)
где А^^, АЯ и АС/— приращения ^^, Н и U при
превращении одного моля в брутто-реакции. Величины А// и MJ
имеют также смысл теплот реакции соответственно при
/? = const и F = const. Из уравнений A8.19) — A8.22)
немедленно следует
^-Г A^)^ = 4^- (Гельмгольц). A8.24)
Эти уравнения сыграли решающую роль при
установлении Нернстом третьего начала термодинамики.
Результаты измерения показали, что уже при не слишком низких
температурах оказывается правильной упрощенная формула
Е^^, или^~-^. A8.25)
Отсюда Нернст заключил, что кривые, изображающие
функции и ж ^, при абсолютном нуле температуры не
только пересекаются, но и соприкасаются (см. § 12).
5. Численный расчет. Вернемся к рассмотрению
элемента Даниэля. Измерение э. д. с. дает:
? = 1,0999 в при температуре таяния льда,
-Угр = —4,3-10"^ Gjepad при температуре таяния льда.
1) В этом параграфе свободная энергпя обозначается буквой ^
для отличия от числа Фаралея F, Б остальных случаях свободная
энергия обозначается буквой F.
§ 18. Напряозсение гальванического элемента 153-
Так как вследствие конструкции элемента Даниэля нельзя
ожидать (и не наблюдается) никакого различия между
{dE/dT)j^ и {дЕ/дТ)^, то не получается также никакого
различия и между теплотами реакции Д// и АС/,
рассчитанными по формулам A8.23) и A8.24). Обозначая их
одной буквой д, получаем из формулы A8.23) или A8.24)
g = 2.96494 кулон • A,0999 -f 273• 4,3• 10-^)б =
= 192988 «?/ло«.A,0999+-0,1173)^ = 2,35.105 кулон-в.
Единицей измерения является здесь кулон-в = дмсоуль=^
^Эрг. Так как 1 5/?г = 0,239 кал, то, следовательно,
д = 56100 кал.
Измерения дают значение 7 = 55200 кал.
Рассчитывая по формуле A8.25) и принимая для q
экспериментально найденное значение, получаем
^ = "^У64У4.0,239 ^ = 1»195б
вместо значения 1,0999 в, полученного из опыта.
Следовательно, примитивная формула A8.25) здесь еще
достаточно хороша, что понятно, так как величина TdEldT^
= 0,1173 мала по сравнению с величиной ?' = 1,0999.
Другой результат получается в случае элемента
Hg I HgCI, + КС11 кон + HgJ Hg.
Здесь
-?' = 0,1483 в при температуре таяния льда,
—- = 8,37-10"* в 1 град при температуре таяния льда.
Наблюдаемая на опыте теплота реакции равна — 3280 кал,
В этом случае при использовании формулы A8.25)
получилось бы отрицательное значение э. д. с. В самом деле,
здесь TdEjdT = 0,221 в при О^С, т. е. больше Е, что
находится в соответствии с отрицательной теплотой
реакции.
В литературе иногда указывается, что э. д. с.
элемента (в частности, элемента Даниэля) можно вывести из
первого начала термодинамики и что второе начало вообще
дает только поправку. Из последнего примера видно, что
154 Гл, II. Применение термодинамики к конкретным системам
ЭТО утверждение неправильно. Таким образом,
принципиально всегда следует исходить из уравнений Гиббса —
Гельмгольца, выведенных при помощи первого и второго
начал термодинамики.
6. Интегрирование уравнения Гиббса—Гельмгольца.
Благодаря тождеству
^_rif=-7'2^(f) A8.26)
уравнение A8.23) можно записать в виде
Разделим А^ на не зависящую и зависящую от
температуры части MIq и Д//т. Последняя очень быстро
убывает при Г —» 0. Действительно,
Так как, согласно третьему началу термодинамики, при
Т —> О ^Ср обращается в нуль, то ДЯт убывает бьгст/?ее,
чем Т (в большинстве случаев, как Т*; наличие в ^Нт
постоянной части, выпадающей при дифференцировании
в A8.28), не играет роли в данном случае, так как она
включается в ДЯд)- Интегрируя теперь уравнение A8.27),
немедленно получаем
О
Положив нижний предел в этом интеграле равным
нулю, мы правильно определили постоянную
интегрирования, а именно так, что она соответствует прежнему
определению Е A8.19). В самом деле, из третьего начала
термодинамики следует, что при Г —» О, поскольку
TS -> О,
^Gq = M{q и, следовательно, ?' = —^.
При абсюлютном нуле температуры, согласно A8.29),
указанное предельное условие будет вьшолняться. Этим
и обосновывается выбор предела интегрирования.
§ 19. Ферромагнетизм и парамагнетизм 155
Уравнение A8.29) дает возможность заранее вычислить
э.д.с. и ее температурную зависимость, если известна
теплота реакции как функция температуры. Таким
способом были установлены э.д.с. многих элементов,
оказавшиеся в прекрасном согласии с опытом. Это
обстоятельство означает по существу не что иное, как новое
подтверждение справедливости второго и третьего начал
термодинамики.
§ 19. ФЕРРОМАГНЕТИЗМ И ПАРАМАГНЕТИЗМ
В то время как диамагнетизм от температуры не
зависит, парамагнетизм и ферромагнетизм зависят от
температуры решающим образом, возрастая с ее понижением.
Выше определенной температуры (точка Кюри)
ферромагнетизм переходит в парамагнетизм. Эти процессы.
допускают термодинамическое рассмотрение. Правда, как
и в других случаях, термодинамика дает лишь общие
рамки, в пределах которых разыгрываются эти явления.
Детали должны быть выяснены статистической физикой
и дополнены данными атомной физики (магнитные моменты
электронов, см. т. III ^)). Диамагнетизм полностью относится
к области атомной физики.
I. Работа намагничивания и магнитное уравнение
состояния. Дифференциал плотности магнитной энергии
задается выражением (Н, rfB)^), где В = (Хо(Н-}-М),
причем М обозначает намагниченность (магнитный момент
единицы объема), а (Iq — некоторую константу, введенную
из соображений размерности. Часть этого дифференциала
(jlq(H, б/Н) нас не интересует, так как она имеется также
и в отсутствие магнетика. Та часть плотности энергии,
которая для нас единственно важна, если отвлечься от
векторного характера процесса намагничивания (что
недопустимо в случае монокристаллов), будет выражаться
в виде (см. примечание 2 на стр. 100)
^^HdM. A9.1)
1)А. Sommerfeld, Elektrodynaraik (Bd. HI), Leipzig, 1949,
§14B.
2) Cm. a. Sommerf eld, Elektrodynamik (Bd. Ill), Leipzig,
1949, формулы E.6b) и A2.2).
156 Гл. IT. Применение термодинамики к конкретным системам
В дальнейшем под величиной М будет удобно понимать
намагниченность одного моля (вместо единицы объема).
Тогда выражение A9.1) представляет работу, которую
совершает внешнее поле при увеличении
намагниченности одного моля на с/Л/, т. е. энергию, подводимую при
этом к телу.
Наряду с термическими переменными Г и 5 мы имеем
здесь две магнитные переменные^) Н и М. От двух
механических переменных р и V можно отвлечься (они
играют роль только в вопросах магнитострикции). Первое
и второе начала вместе дают
du^Tds + ^^HdM, A9.2)
или
ds = ^-^dM. A9.2а)
В своей первой работе в этой области Пауль Ланже-
вен A905 г.) сделал предположение, что оба члена в
выражении для ds являются полными дифференциалами-
Тогда величина i^qH/Т не должна зависеть от Г и должна
определяться только значением М, Вместо этого можно
также сказать, что М должно быть функцией только
от Н/Т:
М
= /(cf). A9.3)
Равным образом энергия м, согласно уравнению A9.2а),
должна быть функцией только от Т и, следовательно,
(w)t = 0. A9.3а)
Будем считать, что в материальную константу С,
присутствующую в аргументе функции / [см. A9.3)],
включена также константа [л^ из уравнения A9.2а). Вместе
с тем ее следует выбрать так, чтобы аргумент функции /
1) Последовательнее было бы выбрать в качестве первой
магнитной переменной \з^Н вместо Я, так как эта переменная,
являясь интенсивной величиной, соответствует интенсивным
величинам р и Т двух других пар переменных. Однако, чтобы как
можно меньше перегружать текст не интересующей нас константой
jjto, остановим свой выбор на переменной Н.
§ 19. Ферромагнетизм и парамагнетизм 157
был безразмерным. В связи с равенством A9.3а) заметим
еще, что в данном случае а следует считать функцией
Г и il/, а не функцией 5 и Л/, как это сделано в
уравнении A9.2) (помимо зависимости от р и у, которую здесь
и в дальнейшем также можно не принимать во
внимание).
Написанные уравнения напоминают уравнения для
идеального газа, если заменить в них магнитные
величины М, Я, С на механические 1/у, /?, 1//?. Тогда A9.3а)
переходит в основное калорическое уравнение идеального
газа {du/dv)T = 0, а уравнение A9.3) при специальном
выборе функции f (х) = х превращается в уравнение
состояния идеального газа
1 Р
V КГ '
Этот параллелизм между идеальным газом и
рассмотренным типом магнетиков заставляет рассматривать
уравнение A9.3; в качестве «уравнения состояния» идеального
магнетика. Правда, это сравнение оправдывается лишь
для парамагнитных веществ. Если опять положить / (х) = х.
то получим
М = С^, 4 = Х.=т- A9.4)
Это закон Кюри для парамагнетизма^). Здесь С
—постоянная Кюри, у^ —магнитная восприимчивость одного моля.
Из соотношения A9.4) ясно, что С имеет размерность
температуры.
Уравнение состояния диамагнитных веществ не
подпадает под схему A9.3), так как в этом случае М
пропорционально 7/ и не зависит от температуры. Конечно,
уравнение A9.2) для диамагнитных веществ также
справедливо. Интересующее нас главным образом уравнение
состояния ферромагнетиков также отклоняется от схемы
A9.3). Поэтому для ферромагнетиков несправедливо также
равенство A9.3а), как это подробнее показано в п. 4
этого параграфа.
1) См. А. Sommerfeld, Elekrodynamik (Bd. Ill), Leipzig,
1949, уравнение A3.10).
158 Гл. II. Применение тс р.м о динамики к конкретньич системам
2. Функция Лапжевена для парамагнетиков. Если
элементарный магнитик с моментом т, который может
вращаться, поместить во внешнее магнитное поле, то
он ориентируется по направлению последнего. Когда все
имеющиеся в одном моле п элементарных магнитиков
ориентируются таким образом, намагниченность достигает
насыщения
Моо=тп. A9.5>
Этому противодействует тепловое движение. Поэтому каждой
температуре соответствует некоторое промежуточное
состояние между полным насыщением М — Моо и полной
неупорядоченностью Л/ ^ 0. Ланжевен пришел к этому выводу
сравнительно простым путем при помощи статистики Больцмана
(см. § 25). Его результат, как будет показано в § 25, имеет вид
_ = ctha—J-, а = -^^— . A9.5а>
Здесь а —безразмерная величина, так как и числитель и
знаменатель в выражении, определяющем а, имеют
размерность плотности энергии.
Функция
L(a) = ctha--i A9.56)
называется функцией Ланжевена. На фиг. 18 она
изображена монотонно поднимающейся кривой ОБА. Приа—>О
и а —> со функция Ланжевена приближенно
представляется в виде
'О, L(a):
а—^ сх), L (а) =
1-Ьа24+ ...
1
а
1-
1 -4
+4
а
¦>|-,A9.6)
{19.6а>
Из выражений A9.6) и A9.5) следует, что почти для всех
практически достижимых значений а мы имеем
^-^ Н " //.3 ~"злг • ^^^-'^
§ 19. Ферромагнетизм и парамагнетизм
159
Это—закон Кюри A9.4) с постоянной Кюри
С-
ък
A9.7а)
Эти соотношения становятся неправильными только при
Г—>0, когда формула A9.4) дает для ^ значение со
kL(oL) = MM
Фиг. 18. Кривая Ланжсвена для
парамагнетизма и ее применение в теории
ферромагнетизма Вейсса.
вместо правильного конечного значения у^ =-- Мос/Н,
которое вытекает из формул A9.7) и A9.6а).
На фиг. 18 штрихпунктирными линиями показаны
зависимости A9.6) (прямая, исходящая из точки О) и
и A9.6а) (горизонтальная асимптота). Точки,
изображающие практически достижимые состояния
парамагнитных веществ, лежат в основном (см. ниже) на нижней
части прямой, соответствующей апроксимации A9.6).
Статистическая теория Ланжевена не учитывает
взаимодействия элементарных магнитиков друг с другом. В ней
предполагается, что они испытывают воздействие только
со стороны внешнего поля. Это предположение безусловно
представляет собой сильную идеализацию. Последняя
равнозначна требуемой в A9.3а) независимости энергии и
от намагниченности Л/, которая не может иметь места при
160 Гл. II, Применение термодинамики к конкретным системам
наличии взаимодействия между элементарными
магнитиками. Отсюда ясно также, что ланжевеновское уравнение
состояния A9.5а) подчиняется общей зависимости A9.3),
которая терлюдинампчески связана с равенством A9.3а).
Оправданием столь далеко идущей идеализации считается
тот факт, что для парамагнитных веществ насыщение
в обычных условиях не наблюдается и ожидается лишь
при очень низких температурах. (Это подтверждается
наблюдениями .над сулгфатом гадолиния, проведенными
в Лейденсгюй криогенной лаборатории Вольтьером и Ка-
мерлинг-Оннесом, при температурах вплоть до 1,3° К,
см. п. 5 этого параграфа.) Дебай^) показал, что при
таких температурах функция Ланжевена A9.56) уже не
может быть правильной, так как при Г —> О она
противоречит теореме Нернста.
3. Теория ферромагнетизма Вейсса. Пьер Вейсс
выдвинул весьма плодотворную гипотезу о том, что фер-
рома1нитные тела состоят из небольших областей
(доменов), в которых все элементарные магнитики
ориентированы одинаково. Это обусловлено существованием
внутреннего молекулярного поля /^,,,, в десят1чи раз
превосходящего наблюдаемые внешние поля 2). Вейсс предположил,
что поле, создаваемое каждой областью, пропорционально
ее намагниченности М с очень большим коэффициентом
пропорциональности Л^, зависящим от материала
ферромагнетика,
H^ = NM. A9.8)
Внутри каждой области элементарные магнитики
ориентированы одинаково, но при переходе от области к области
направление внутреннего молекулярного поля Н^ меняется.
Поэтому в отсутствие внешнего поля Н вещество в целом
оказывается ненамагниченным. При наличии этих областей
намагниченности результат действия внешнего поля Н
оказывается совершенно другим, чем в случае отдельных
1) Debye, Ann. d. Phys., 81, 1154 A926). Необходимое
усовершенствование функции Ланжевена дается квантовой статистикой
(см. п. 4 этого параграфа).
2) См. А. Sommerfeld, Elektrodynamik (Bd. П1), Leipzig,
1949, § 14 А.
§ 19. Ферромагнетизм и парамагнетизм 161
элементарных магнитиков, а именно, поле ориентирует
все области по своему направлению и тем самым вызывает
появление большой намагниченности, характерной ^^ля
ферромагнетиков.
Процесс намагничивания (по крайней мере в слабых
полях) происходит не путем вращения целых областей,
а посредством необратимой переориентации элементарных
магнитиков, находящихся на поверхности наиболее выгодно
ориентированных областей, в результате которой
происходит смещение границ последних ^). Поведение всей системы
областей в целом нас здесь не интересует; мы ограничимся
лишь исследованием зависимости намагниченности
отдельной области от внешнего поля.
Для проведения количественного расчета Вейсс
поступает следующим образом. В формуле Ланжевена для а
к внешнему полю Н добавляется внутреннее поле Н^
а = !^ (Я + Я„,) = ^ (Я + NM). A9.9)
Поскольку Н^, > Я, а теперь будет иметь гораздо большее
значение, чем в случае парамагнитных веществ. Кроме
того, при подстановке а из A9.9) в уравнение A9.5а)
последнее превращается в трудно разрешимое уравнение
относительно Л/, так как теперь эта величина входит
не только в левую часть уравнения A9.5а), но также
и в правую.
На фиг. 18 представлено графическое решение этого
уравнения. Точка, определяемая двумя неизвестными М/Моо
и а, должна лежать как на кривой Ланжевена, так и на
прямой линии, соответствующей уравнению A9.9). Иначе
говоря, в этой точке удовлетворяются следующие два
уравнения:
^=L(a), A9.9а)
°^ = ^ + Рл7^, где S = —йг"-' ^^^•^т^- A996)
Прямая A9.96) пересекает ось абсцисс в точке а = ад
(на фиг. 18 обозначена буквой Р). Последняя, согласно
^) См. А. Sommerfeld, Eleklrodynamik (Bd. Ill), Leipzig.
1949, § 14 С.
162 Гл. II. Применение термодинамики к конкретным системам
A9.5а), как раз характеризует значение абсциссы для
парамагнитного случая и лежит очень близко от начала
координат. Тангенс угла между, этой прямой и осью
абсцисс, согласно A9.96), равен
Таким образом, угол наклона зависит от температуры
и он тем меньше, чем ниже температура. Точка
пересечения А прямой A9.96) с кривой Ланжевена при
понижении температуры перемещается направо. При этом
намагниченность М все более приближается к
намагниченности насыщения Л/с», которая соответствует одинаковой
ориентации всех элементарных магнитиков.
Положение точки пересечения лишь незначительно
изменится, если выключить внешнее поле,*т. е. положить
Н = 0. Прямая РА при этом перемещается параллельно
самой себе до точки О (так как теперь а(,==0), и точка
пересечения переходит в точку В. Поле отдельной области
остается при этом почти неизменным. Таким образом,
существование постоянных магнитов объясняется тем,
что области могут оставаться в состоянии
самопроизвольной намагниченности. Итак, уравнение A9.9)
действительно передает существенные свойства ферромагнитных
веществ при низких температурах. Из него вытекает
возможность существования самопроизвольной
намагниченности, увеличивающейся с уменьшением температуры.
Тот факт, что эта самопроизвольная намагниченность
не всегда наблюдается, обусловлен взаимной компенсацией
магнитных моментов различных областей, так как
направление намагниченности может изменяться при переходе
от одной области к другой.
До сих пор предполагалось, что прямая РА проходит
менее круто, чем, например, касательная к кривой
Ланжевена в точке О, Если же предположить обратное, то при
достаточно слабых внешних полях эти линии пересекутся
вблизи точки О. Тогда для функции L(a) справедливо
приближение A9.6) и^ согласно A9.9а) и A9.96), мы имеем
М 1 \^оМ^
§ 19. Ферромагнетизм и парамагг^тизм 163
Отсюда находим
М[Т-^^)=^'^Н. A9.10)
Согласно выражению A9.7а), в правой части множителем
перед Н является постоянная Кюри С. В левой части
стоит член NC, который мы обозначим через в:
Величина в называется температурой Кюри.
Так как множитель ^/д в предыдущем выражении
происходит от первого члена в функции Ланжевена L(a)
и пoэтo^Iy идентичен L'@), то вместо A9.11) можно
также написать
0 = ^^L'(O)^. A9.11а)
Это выражение предпочтительнее, чем A9.11), так как
оно делает некоторые приводимые ниже расчеты не
зависящими от специального выбора функции Ланжевена
L (а) и более удобно для квантовотеоретического обобщения.
При использовании обозначения в уравнение A9.10),
согласно A9.11) или A9.11а), принимает вид
^^ = 7^- A9-12)
Выше температуры Кюри вещество становится
парамагнитным и следует закону Кюри—Вейсса A9.12).
Последнее уравнение изобразится прямой линией, если нанести
на график зависимость отношения НjM от температуры Т.
Строго говоря, опыт показывает, что надо различать две
несколько отличающиеся друг от друга точки Кюри
в зависимости от того, определяют ли в графически на
основе закона A9.12), или по исчезновению
самопроизвольной намагниченности. Однако мы здесь не можем входить
в детальное рассмотрение этого обстоятельства, равно как
и многочисленных других опытов в области
ферромагнетизма.
Следует, однако, подробнее рассмотреть процессы
в окрестности точки Кюри. Очевидно, что при Г—^в
164 Гл, II, Применение термодинамики к конкретным системам
уравнение A9.12) справедливо только тогда, когда
одновременно (и притом достаточно быстро) стремится к нулю
напряженность магнитного поля Н. Следовательно, для
случая Н = 0 уравнение A9.12) можно применять и при
температурах выше точки Кюри. Тогда получим, что
М = Оу т. е. самопроизвольная намагниченность
отсутствует.
Рассмотрим теперь намагниченность при Я = О, т. е.
самопроизвольную намагниченность при температурах
немного ниже точки Кюри. Соответствующее значение а
обозначим через адр. Чтобы охватить и точку
пересечения В на фиг. 18, следует взять функцию Ланжевена
не в виде прямой линии (несмотря на то, что Я = 0),
а учесть более высокие производные в разложении Z/(a).
Так как L (а) — нечетная функция, то при а = О все четные
производные исчезают. Пренебрегая 5-й, 7-й и следующими
производными, получаем, согласно A9.9),
м '
-^ = a,pL'@)M-^L"'@). A9.13)
00
с другой стороны, формула A9.9) при Я = 0 с учетом
A9.11а) дает
М„
-^^;;Ш1,--^^'(^Ы- A9-1^)
Сравнивая A9.13) и A9.14), находим уравнение для agp,
решение которого отлично от нуля и равно
«sp = /=?PV)(l-|0- A9.15)
Производная L'" @), как можно вычислить из A9.5а),
равна — ^/i5» Подставляя A9.15) в A9.14), получаем
после небольших преобразований следующее выражение
для самопроизвольной намагниченности непосредственно
нижнее точки Кюри:
Л/зр = Мсо/-^4?^|-]/"A-|-). A9.16)
На фиг. 19 показан резкий скачок il/gp в точке Кюри
дри уменьшении температуры в соответствии с выраже-
§ 19. Ферромагнетизм и парамагнетизм
165
нием A9.16). Это увеличение il/gp продолжается до Г = 0^
где Afsp = Afc». Однако фиг. 19 имеет лишь качественное
значение. В действительности благодаря квантовым
эффектам (пространственное квантование спинового момента)
Фиг. 19. Самопроизвольная
намагниченность ниже точки Кюри согласно теории
Вейсса (цсправлепная в соответствии с
квантовой теорией).
кривая имеет другой вид (она подходит к точке М^р = Afooi
имея горизонтальную касательную, а не так, как показано
на фиг. 19).
Само собой разумеется, что фиг. 19 относится только
к отдельной ферромагнитной области. Вопрос о величине
постоянной намагниченности макроскопического агрегата
из таких областей выходит за рамки данной теории,
и решение его зависит от структуры материала.
4. Удельные теплоемкости Сп и Гм» Магнитное
уравнение состояния благодаря дополнительному члену NM
в A9.9) уже не принадлежит к простому типу A9.3).
Поэтому калорическое условие A9.3а) не будет
выполняться. Оно заменяется другим условием, которое можно
найти из выражения для энтропии A9.2а), записывая его
166 Гл. II. Применение термодинамики к конкретным системам
В независимых переменных Т и М:
Беря частные производные от множителя при dT по Л/ и от
множителя при dM по Г, получаем соответственно
1 д^и
Т дМдТ
И
М Т^
Так как обе производные должны быть равны друг другу,
имеем
i^>^.[«-K^X]. (.9.18,
Введение сюда индексов Т п М необходимо для
дальнейшего. В отношении предыдущей производной отметим, что
она в точности аналогична вандерваальсовои производной
du/dv в уравнении (9.6). Подставляя A9.18) в A9.17),
находим количество тепла, подведенного обратимым путем,
^^^ = (*)П^-^'^о(#Х^М- A9.19)
Используем теперь в качестве независимых переменных Т
и Н вместо Т и М. Для этого необходимо лишь
произвести замену:
Тогда из формулы A9.15) получаем
Совершенно аналогично молярным теплоемкостям с^
и с определим теперь молярные теплоемкости при
постоянной намагниченности см и при постоянной напряженности
поля Ся. Они вычисляются из уравнений A9.19) и A9.20).
§ 19. Ферромагнетизм и парамагнетизм 167
Получаем
Отсюда находим разность сн — см'-
с,-с..-Т,.(§.Х(^Х. A9.22,
Это опять точная копия хорошо известного общего
выражения для с — с„ [выражение G.9) при замене —/?, у на
и,Я, Л/].
Обе производные в правой части выражения A9.22)
определяются из параметрического представления A9.9а),
A9.96). Дифференцируя по Т при постоянном М или Я,
получаем соответственно
»=^'(»)[=^Aг).-т]. ("-^з,
(»), = A,^.w[^;?-(#)„-f].(i9.24)
Из A9,23) непосредственно вытекает, что
Пользуясь A9.24) и принимая во внимание
определение в A9.11а), находим
/дЛ?\ M^L'{0)L'(a)a
Таким образом, разность удельных теплоемкостей^
сог-часно A9.22), равна
сн-см^ \ — • A9.27)
L'@)-^L'(«)
Обсудим этот результат для специального случая Н = 0
(внешнее поле выключено), когда il/ = il/sp, т. е. имеется
лишь самопроизвольная намагниченность (фиг. 20). При
Г > в, как известно, il/gp = О (вещество представляет собой
168 Гл. И. Применение термодинамики к конкретным системам
парамагнетик). Поэтому, согласно A9.9), из равенства
Н = 0 вытекает, что и а = 0. Следовательно,
выражение A9.27) дает
ся=^см для Г>е и Я = 0. A9.28)
В области Г<в прежде всего интересен случай Г < в,
который, согласно A9 9), соответствует а > 1. Тогда
|^//"^iH
в
Ф и г. 20. Качественное изображение
максимума cjj—Cj^ в точке Кюри.
Поведение при Т=0 вопреки уравнению
A9. 29) изображено так, как это ожидается,
согласно квантовой теории. Для Т>в се—сзг=^0.
ИЗ A9.6а) следует, что
L'(a)^^.
Поэтому из A9.27) получаем
ся — См = —
1
BL' @)
^•<»)-ii
A9.29)
Формула A9.9) при Я = 0 и M = Msp = Moo (см. фпг. 17)
дает
а= ¦
RT
причем, согласно A9.11а), можно наппсать
е/г
а =
V @) •
§ 19. Ферромагнетизм и парамагнетизм 101>
Подставляя это выражение в A9.29), получаем
сн-см^ -^ ^ Л. A9.29а>
Поскольку рассматривается окрестность точки Кюри,
положим
в-Г< е. A9.296)
Дальнейшие вычисления протекают так же, как и в
случае уравнения A9.13), а именно, в знаменателе
выражения A9.27) оставляем лишь первую и третью
производные функции Ланжевена, а в числителе заменяем L' (а)
на L'@), что возможно, поскольку a = asp<Cl. Тогда
получаем
,,_,,=. ^-^^^^1Щ ^. A9.30)
1''@)-|-[^Ч0)+4-^'"@)]
Подставляя сюда значение а из формулы A9.15) и
сокращая числитель и знаменатель на общий множитель в — Г,
получаем
Сн-См = Ък4^Щ^ -J. A9.31)
Поскольку при 7* > в сн = см, мы видим, что при Г- 9^
происходит скачок удельных тешюемкостей. Согласно»
приведенным выше значениям, L' @) = Va ^ L'"@) = — ^f^^.
Поэтому этот скачок равен
ся-См = !-/?. A9.31а>
В действительности благодаря размазанности точки
Кюри (см. выше) этот резкий скачок превращается в
максимум, круто падающий со стороны Г > в и медленно
понижающийся в области Г < в. Такой максимум остается
также и в случае Н ^ О, причем он обусловлен не только
самопроизвольной намагниченностью, но и
намагниченностью, зависящей от поля.
В п. 3 этого параграфа подчеркивалось, что
полученный там результат справедлив только для отдельной
области самопроизвольной намагниченности и не может
170 Гл. II. Применение термодинамики к конкретным система.ч
считаться правильным для макроскопического образца,
когда играет роль структура материала. В
противоположность этому для удельных теплоемкостей это ограничение
отпадает. Удельные теплоемкости в отличие от полей
суть скалярные, а не векторные величины. Поэтому
полученные здесь формулы справедливы в неизменном
виде и для макросьопического образца.
Конечно, все результаты, полученные в этом пункте,
должны быть исправлены в соответствии с квантовой
теорией. Это следует уже из того, что уравнение A9.29а)
при Г—>0 дает ch — cm-^R, тогда как из тепловой
теоремы Нернста следуе!*, что сн — см —> О (см. замечание 3
в конце § 12).
Квантовая теория дает вместо значения 5Д/2 в
уравнении A9.31а) заметно меньшую величину, например
ЗЛ/2, в соответствии с типом пространственного
квантования, положенным в основу при выборе функции Лач-
жевена L.
Исчерпывающее -и критическое изложение теории
ферромагнетизма содержится в книге Беккера и Дёринга ^),
на которую мы уже ссылались в т. П1^). В то время
как мы просто считали модель Вейсса достаточно точным
отражением действительности, в книге Беккера и Дёринга
эта модель обсуждается с точки зрения всего
относящегося к данному вопросу опытного материала.
5. Магнитокалорический эффект. Адиабатическое
размагничивание в случае ферромагнитных и
парамагнитных веществ сопровождается понижением температуры,
которое легко вычислить из уравнений A9.20) и A9.21).
Находим
Это явление называется магнитокалорическим эффектом.
(Наоборот, быстрое и, следовательно, адиабатическое
1) R. Becker, W. Do ring Ferromagnetismus, Berlin, 1930.
В этой книге данная проблема рассмотрена также с точки
зрения атомной физики.
2) А. Sommerfeld, Elektrodynamik (Bd. Ill), Leipzig, 1949,
5 14D.
§ 19. Ферромагнетизм и парамагнетизм 171
намагничивание вызывает соответствующее повышение
температуры.) В конце § И и в § 12 этот эффект был
наглядно истолкован как результат нарушения
упорядоченности ориентации элементарных магнитиков при
размагничивании. Здесь этот эффект будет подвергнут
количественному исследованию.
Ограничимся особенно интересным случаем
парамагнитных веществ (к их числу относится, например,
сульфат гадолиния) и предположим, что они вплоть до самых
низких температур следуют закону Кюри. Тогда имеем
м-^.// /"^Л ^^ /^^^ -?.
и, следовательно, согласно A9.32),
^ fdT\ гр М С С „
Отсюда следует, что течение рассматриваемого
адиабатического процесса описывается дифференциальным
уравнением
TdT = ^-^HdH. A9.33)
Пусть при Я—* о, T—^Tq\ тогда, считая множители С
и сн постоянными и интегрируя уравнение A9.32),
получаем
у2 Т2 __ f^P^
е
с
|Я^ T, = tYi-^^. A9.34)
Следовательно, в самом деле происходит понижение
температуры, причем тем более значительное, чем сильнее
первоначальное поле i7 и чем ниже исходная температура Т.
Однако этот расчет носит несколько оценочный
характер, так как предполагалась справедливость закона
Кюри вплоть до ближайшей окрестности абсолютного
нуля температуры и, следовательно, пренебрегалось
взаимодействием элементарных магнитиков, что в данном
случае недопустимо. Тем не менее формула A9.34) дает
представление о том эффективном способе, при помощи
которого Дебаю, Джиоку и Камерлинг-Оннесу удалось
получить температуры, очень близкие к абсолютному нулю.
172 Гл. II. Применение термодинамики к конкретным системам
§ 20. ИЗЛУЧЕНИЕ В ПОЛОСТИ
Нагретое тело испускает электромагнитное излучение.
С увеличением температуры происходит переход от
красного каления к желтому и затем к белому. Однако и
при обычных и низких температурах тела также испускают
лучи, которые лежат в инфракрасной области спектра.
Все тепловое излучение имеет волновой характер. Однако
при рассмотрении можно воспользоваться правилами
геометрической оптики, т. е. представлять излучение
в виде пучков лучей. Представим себе эвакуированную
полость, стенки которой имеют постоянную, одинаковую
во всех точках температуру. Имеющееся внутри полости
излучение находится в тепловом равновесии со стенками.
Поэтому ему следует приписать ту же температуру,
которую имеют стенки. Это справедливо для каждой
единицы объема полости и обусловливает изотропное,
однородное во всей полости излучение. Полость
представляет собой термодинамическую систему универсального
характера, не зависящую от специальных физических
и химических процессов испускания и поглощения,
происходящих на стенках (доказательство этого
утверждения будет дано в п. 1 этого параграфа).
Если в эвакуированной полости сделать небольшое
отверстие, через которое излучение изнутри сможет
выходить наружу и наблюдаться, то это почти не нарушает
равновесия внутри полости. Излучение, падающее
снаружи на отверстие полости, не отражается, а, пройдя сквозь
отверстие, благодаря многократным отражениям и
поглощениям на стенках полости остается внутри. Так как
абсолютно поглощающая поверхность называется черной,
то выходящее из полости и находящееся внутри нее
излучение также назовем «черным» излучением.
Если в полость внести «угольную пылинку», т. е.
абсолютно поглощающее тело с очень малой теплоемкостью,
то равновесное состояние черного излучения не изменится.
Однако если внутренняя поверхность стенок полости
является идеально отражающей и поэтому не может
влиять на характер падающих лучей, то внутри полости
может существовать неравномерное излучение. При
внесении угольной пылинки оно будет превращаться в чер-
§ 20, Излучение в полости 173
ное (пылинка играет в данном случае роль
катализатора).
Картина, представляющаяся наблюдателю, находящемуся
внутри полости, не слишком интересна: в каждой точке
и в любом направлении яркость излучения одинакова.
Наблюдатель ничего не может сказать ни о форме полости,
ни о расстоянии до стенок в том или ином направлении.
При помощи призмы Николя можно установить, что
излучение повсюду не поляризовано. Только
интенсивность и цвет воспринимаемого излучения меняются с
изменением температуры.
1. Закон Кирхгофа. Из электродинамики известно,
что излучение обладает энергией и импульсом^).
Плотность энергии обозначается буквой W, Для случая
монохроматического поля излучения усредненные по времени
энергия и импульс зависят от координат данной точки,
частоты V и амплитуды или ее квадрата (т. е.
интенсивности). Рассмотрим теперь не монохроматическое
излучение, а излучение, заключающее малую область спектра cfv.
Плотность энергии, соответствующую этой области,
обозначим через K^cfv, а плотность энергии, соответствующую
всему спектру, — через м. Тогда
оо
и{Т)=\^ u^{^,T)d^, B0.1)
о
Добавление аргумента Т в функцию и показывает, что
амплитуда (или интенсивность) излучения в полости при
равновесии всегда определяется температурой, которая
одинакова во всех точках полости. Обозначение и
отклоняется от общей системы Термодинамических обозначений,
поскольку и не является энергией единицы массы (или
одного моля), а имеет смысл энергии единицы объема:
[к] = 9/?г/си*з. B0.1а)
Отсюда, согласно B0.1), находим размерность и^\
[иу,]=^эрг'сек1см^. B0.16)
Кирхгоф A859 г.) доказал, что и^ не зависит от
свойства полости у а зависит лишь от v и Г. Это утвержде-
*)А. Sommerfel d,Elektro(iynamik (Bd. Ill), Leipzig. 1949. §31
174 Гл. II. Прьиченение термодинамики к конкретными системам
ние называется законом Кирхгофа. Чтобы дать
представление о его доказательстве, рассмотрим две полости А
и В с различными свойствами стенок. Пусть значение
функции Kv для определенной спектральной области
(v, dv) в полости А больше соответствующего значения
в полости В. Соединим эти полости небольшой трубкой,
прозрачной только для излучения частоты v (светофильтр).
Тогда от А к В будет переходить больше теплового
излучения, чем от В к А. Тепловое равновесие будет
нарушено: полость В будет нагреваться, а полость А
охлаждаться до тех пор, пока не сравняются соответствующие
функции. Таким образом, «сама собой» (без затраты
работы) образуется разность температур, что противоречит
второму началу. Поэтому и^ долблена быть универсальной
функцией ^ и Т. Согласно B0.1), и является тогда
универсальной функцией Т,
Перерщем от плотности энергии к потоку энергии^
который в электродинамике обозначается через S. Вектор S
имеет смысл энергии, проходящей через единицу
поверхности в единицу времени. Направление вектора S
соответствует нормали к рассматриваемой единице
поверхности. Так как излучение в полости изотропно (по всем
направлениям одинаково), то векторный характер потока
энергии не играет роли, и можно говорить о скалярной
интенсивности излучения. Ее следует, однако, связывать
не с дискретным направлением (для каждого отдельного
направления поток энергии был бы равен нулю), а с
узким конусом лучей dQ. Если этот конус описан около
нормали к рассматриваемой поверхности, то энергию,
излучаемую в конусе с?2, обозначают через KdQ. Для
конуса, ось которого составляет с нормалью к прверх*
ности (острый) угол 6, излучаемая энергия равна
Kcos^dQ, где dQ^smbdbd^jf. B0,2)
Поэтому излучение, проходящее через «лицевую» или
«оборотную» сторону произвольно расположенного элемента
поверхности df за время dt, дается выражением
7t/2 2ic
Kdfdt \ cosBsinedB { d<f=:TzKdfdt, B0.2a)
§ 20, Излучение в полости 175
Производя спектральное разложение величины К и
различая два направления поляризации штрихом, можно
написать
00 оо
К{Т)=\ [К, (V, т) + а:; (v, т)] d>^ = 2^K, (v, т) d^. B0.3)
о о
Последнее выражение получено при условии, что
излучение не поляризовано. Величина К имеет размерность
потока энергии S, а размерность величины К^, можно
определить из выражения B0.3):
[К]^9рг1см^^сек, [К^]=:эрг/см^. B0.3а)
Между величинами и и К имеет место соотношение
м = ^. B0.4)
Мы не будем здесь останавливаться на его
доказательстве, так как оно основано на простейших положениях
геометрической оптики. При этом предполагается, что
в полости—вакуум. В противном случае вместо скорости
света с появилась бы величина с/п (где п — показатель
преломления). Из формулы B0.4) в силу B0.3) и B0.1)
следует, что
и^ = ^. B0.4а)
Если, наконец, распространить постулат о равновесии
излучения и на стенки полости, то придем к обобщению
сформулированного выше закона Кирхгофа.
Обозначим через А поглощательную способность
элемента поверхности стенки cf/, т. е. ту часть падающего
излучения jK'v(v, Г), которая при проникновении в стенки
полости будет превращаться в тепло. Энергия, теряемая
при этом равновесным излучением (рассчитанная на
единицы поверхности, времени и телесного угла), равна
АК^(у,Т). B0.5)
Эта энергия должна возместиться благодаря
излунательной способности Е того же элемента поверхности
i76 Гл. II. Применение термодинамики к конкретным системам
стенки. Для черной поверхности (Л = 1) имеем
Е=^К^{^,Т). B0.5а)
В случае идеально отражающей (абсолютно белой)
поверхности Л = О и Е = 0, При этом, как уже отмечалось выше,
стенки полости никак не влияют на установление
термодинамического равновесия. В общем случае должно
осуществляться равновесие между энергией, поглощенной
и излученной стенками. Следовательно, должно быть
§ = Z,(v,r). B0.6)
Для чисто теплового излучения отношение излуча-
телъной и поглощателъной способностей представляет
собой универсальную функцию частоты излучения и
температуры.
Закон Кирхгофа и это его обобщение имеют
фундаментальное значение не только для теории теплового
излучения, но и. для всех вопросов светотехники. В
особенности важен этот закон для спектрального анализа,
одновременно открытого Кирхгофом и Бунзеном.
2. Закон Стефана—Больцмана. Как было сказано
в начале п. 1, излучение обладает не только энергией,
но и импульсом. Именно это обстоятельство
обусловливает предсказанное Максвеллом давление света. Давление
света, падающего под углом 6 на элемент поверхности rf/,
равно ксоз^б^). Отсюда давление света в случае
излучения, падающего на одну сторону элемента поверхности
под любым углом, составляет
/? = tt \ cos2esine6fe=|-. B0.7)
о
Данная формула справедлива как для частично
отражающей, так и для черной поверхности. Это обусловлено
тем, что импульс отдачи отраженного излучения
действует так же, как импульс отдачи испущенного света.
1) См. А. Sommerfeld, Elektrodynamik (Bd. Ill), Leipzig,
t949, § 31 (последняя формула).
S 20, Излучение в полости 177
Представим себе эвакуированный цилиндрический
сосуд с подвижным поршнем, внутри которого содержится
черное излучение при температуре Т, Объем V можно
изменять (бесконечно медленно) на произвольную
величину, перемещая поршень. Таким образом, мы имеем
термодинамическую систему, описываемую двумя
переменными F и Г. Ее энергия равна
U^Vu{T).
Работа по перемещению поршня, согласно B0.7),
составляет
dW = pdV = ju{T)dV,
а изменение энтропии дается выражением
as^du+m^v^ ^ат^^ ^dv. B0.8)
Так как dS является полным дифференциалом, то
должно выполняться условие
Т dT~^ Ъ dT\T )'
Отсюда без труда получаем
— = 4-^, 1пм = 41п7' + const,
и т
или
к = аГ*. B0.9)
Чтобы определить введенную здесь постоянную
интегрирования а, перейдем от w к К,, На основании B0.4)
имеем
К=^^Т^. B0.10)
Согласно B0.2а), левая часть уравнения B0.10) имеет
смысл полного излучения, испущенного в единицу
времени единицей поверхности (например, отверстия в стенке
эвакуированной полости). Поэтому константу в правой
части са/4тс (обычно обозначаемую через о) можно
определить экспериментально. Уравнение B0.10) выражает
178 Гл, II. Применение термодинамики к конкретным системам
закон излучения, эмпирически найденный Стефаном.
Приведенный выше термодинамический вывод этого закона
был дан Больцманом в 1884 г. Г. Лорентц назвал его
в своей речи, посвященной памяти Больцмана,
«истинным перлом теоретической физики»^).
Подставляя выражение B0.9) в B0.8), получаем
dS=^Aa(^VT^T + jT^V^=^ad{TW). B0.11)
При интегрировании этого уравнения никакой постоянной
интегрирования не появляется, так как, согласно
тепловой теореме Нернста, при 7* = О 6' = 0. Поэтому
S = ^aT^V, B0.12)
Отсюда получаем уравнение изэнтропы в плоскости Г, V:
Г37 = const. B0.12а)
Это уравнение показывает, как изменяется температура
и, следовательно, согласно B0.9), энергия и при
адиабатически обратимом изменении объема. Уравнение B0.12а)
совпадает с уравнением изэнтропы идеального газа, если
отношение удельных теплоемкостей положить равным
3. Закон Вина. Вину принадлежит замечательная
идея, позволившая выяснить характер частотной и
температурной зависимости излучения в полости. Эта идея
состоит в исследовании изменения спектрального состава
излучения при отражении от движущегося зеркала. Из
т. IV, § 13^) известно, что, когда зеркало движется по
направлению, перпендикулярному к его поверхности,
частота отраженного света отличается от частоты падающего
света. То же самое справедливо и для интенсивностей
отраженного и падающего света. Требуя, чтобы при
подходящем процессе превращения излучение с таким измененным
спектром оставалось равновесным, можно, в частности,
1) Verb. d. Deutsch. Phys. Gesellschaft A907).
2) A. Sommerfeld, Optik (Bd. IV), Wiesbaden. 1950.
(Cm. перевод: A. Зоммерфельд, Оптика, ИЛ, 1954.)
§ 20. Излучение в полости 179
определить смещение, которое испытывает максимум
интенсивности (и, следовательно, цвет) при изменении
температуры.
Мы откажемся здесь от доказательства закона Вина,
основанного на определенной модели^), и прежде всего
займемся часто дискутируемым вопросом о возможности
вывести этот закон только из соображений размерности,
т. е. сделать его понятным, пользуясь только теорией
подобия^). Как обычно, у нас имеется четыре единицы.
Одной из них является единица температуры (символ 6),
остальные три — механические. При этом для дальнейшего
удобно выразить единицу массы через эрг (символ е)^).
Длину и время обозначим соответственно через I и t.
Величина Mv, согласно B0.1а), имеет размерносль
etH^. Выразим и^ через v и Г (размерности их равны
соответственно Г^ и 9) и некоторые универсальные
константы, в качестве которых в нашем распоряжении
находятся скорость света с (размерность /Г^) и газовая
постоянная /?. Размерность последней есть еб"^, так как RT^
согласно уравнению газового состояния, имеет размерность
;)нергии. Постоянная R относится к одному молю какого-
либо вещества. Для дальнейшего, однако, удобнее ввести
газовую постоянную, отнесенную к одной молекуле, что
достигается делением постоянной R на число молекул
в одном моле. Эту новую константу, называемую
постоянной Больцмана, обозначим через А:. Она, так же как
и /?, имеет размерность е^^.
Сведем названные пять величин и их размерности
в следующую таблицу (к последнему ее столбцу мы вскоре
вернемся):
^) Простейшее доказательство такого вида дано Лауэ
[Laue, Ann. d. Phys., E) 43, 220 A943)]. Моделью служил
отдельный монохроматический пучок лучей. Доказательство
основывалось на инвариантности последнего относительно
преобразования Лорентца. Наше рассмотрение предполагает инвариантность
только относительно преобразования масштабов.
*) См. также замечание В. Глазера (W. Glaser, Sitzungsbe-
richt der Akademie in Wien, 156, 87), к которому мы отчасти при
соединяемся.
3) Предполагается, что четвертая единица нашей электродина
мической системы единиц—единица количества электричестпа
Q—здесь не входит в рассмотрение.
"v
el-H
V
ri
T
8
с
It-^
к
eG-i
h = ka
et
180 Гд. //. Применение термодинамики к конкретным системам
B0.13)
Постараемся теперь найти такое произведение этих
пяти величин (в положительных или отрицательных
степенях), которое имело бы нулевую размерность по отно-
шению ко всем четырем единицам
ej,t и е. B0.14)
Если показатель степени одной из названных величин
выбрать произвольно (например, положить равным единице,
что не имеет какого-либо особого смысла), то степени
остальных четырех величин будут однозначно
определяться четырьмя линейными уравнениями, которые
получаются приравниванием нулю суммы степеней для каждой
единицы из B0.14). Следовательно, имеется только одно
такое произведение. Полагая степень величины и^ равной
единице, непосредственно из таблицы B0.13) видим, что
и^с^ имеет размерность еГ^] такой же размерностью
обладает и v^Ar. Следовательно, искомое произведение есть
П=--^
B0.15)
Величина П представляет собой неизвестное
универсальное число. Отсюда для спектральной функции
распределения получаем
Mv =
П.
B0.16)
Таков (с точностью до неопределенного численного
множителя И) однозначный ответ, который может дать
классическая физика на вопрос о спектре излучения в
полости. Эпитет «классическая» подразумевает при этом
ограничение двумя издавна укоренившимися в физике
универсальными постоянными с и А.
Формула B0.16) была получена из классической
статистики Релеем в 1900 г., причем множитель Показался
равным 8ir. Эта же формула была вторично получена
§ 20. Излучение в полости 181
Джинсом (отсюда название — формула излучения Релея —
Джинса). Однако эта формула абсурдна в области
больших частот, так как при v —» оо из нее получается
бесконечно большое значение и^ и при переходе к полному
излучению интеграл к = \ u^d^ расходится.
Чтобы прийти к согласию с опытом, необходимо при
выводе закона излучения отказаться от ограничения двумя
универсальными постоянными. Следует ввести третью
постоянную, так как из закона Кирхгофа следует, что
никаких других переменных, кроме Kv, v и Г, в искомой
формуле быть не может.
Эта новая постоянная дает новое, не связанное с B0.15),
безразмерное произведение П', относительно которого без
ограничения общности можно предположить, что оно не
зависит от Kv и содержит частоту v в первой степени ^).
Следовательно, имеем
П'=avГ^ B0.17)
В константу а включены величины с, А и новая
универсальная постоянная. Отсюда имеем
П = /(П'), B0.17а)
или
Wv (V, Г) = -^ АГ./(avr**). B0.176)
Степень п следует выбрать таким образом, чтобы инте^
грал по всему спектру давал выражение B0.9). Из
равенства
^) Если бы это не получилось сразу же, то необходимо было бы
лип1ь умножить П' на соответствующую степень П, чтобы
исключить и^. Затем полученное при этом значение П' нужно было бы
возвести в такую степень, чтобы v оказалось в первой степени.
Последнее возможно, так как из опыта известно, что П' saBHcnt
от V.
182 Гл, II, Применение термодинамики к конкретным системам
полагая av7'" = a:, находим
оо
^ f{x)x
^dx.
Правая часть пропорциональна Т^ только при условии,
если положить п= — 1. В этом случае соотношение B0.176)
дает закон Вина
«v(v,r)=-^/(-^). B0.18)
Таким образом, неизвестная функция двух переменных
Wv (v, Т) сводится к неизвестной функции f одной
переменной а^/Т, В этом и заключается основное достижение
закона Вина.
Если в аргумент функции / ввести постоянную Больц-
мана к у положив Ла = /г, то получим формулу Вина в
обычном ее виде:
«v(v,T) = ^/(-^). B0.18а)
Здесь постоянная h имеет размерность et^ т. е.
размерность действия. Она дополняет нашу таблицу B0.13). Для
предварительной ориентации читателя укажем, что мы
имеем здесь дело с планковским квантом действия Л,
который считается теперь одной из фундаментальных
констант физики. Закон Вина предсказывает размерность
этой величины. Вводя в дробь перед функцией / в
формуле B0.18а) произведение ^v, получим
«v(v,r) = -^/-^ = -^/,(x), где х = -^. B0.186)
Отсюда находим постоянную Стефана — Больцмана а
(см. B0.9)]
ОО
а = кС^Ур, где F=[x%{x)dx. B0.19)
о
В заключение приведем объяснение названия «закон
смещения Вина». Найдем частоту v, соответствующую
(при заданной температуре Т) максимуму интенсивности Wv,
§ 20. Излучение в полости 183
Т. е. частоту, для которой 3kv/9v = 0. Из формулы
B0.18а) находим для этой частоты
2/(ж) + х/'(:с) = 0, где х^^. B0.20)
Пусть а; = х^ — вещественный положительный корень этого
уравнения, а v = v^ —соответствующее ему значение
частоты. Очевидно, что
v„ = ^. B0.20а)
Следовательно, с увеличением Т положение максимума
интенсивности смещается в сторону больших частот. Так
1^ак положение этого максимума определяет окраску,
воспринимаемую при наблюдении всего спектра, то формула
B0#20а) объясняет переход от красного каления к белому,
имеющий место при повышении температуры.
Обычно наше ощущение цвета характеризуют не
частотой V, а длиной волны X. Из соотношений
v = y, |dv| = -^|cfX|, u^\d^\^Ux\dX\
получаем для распределения интенсивности по длинам
волн Ux выражение
кТ . Г ас \\ d^ \ кТ ./ ас \ /оп о4ч
Введем новую переменную у и новую функцию g{y)
соотношениями
у = ^. g{y)=yf(^j').
Тогда из B0:21) получим
акс , V
откуда находим
S=-"^t5^B/)-yg'(y)]-
B0.21а)
B0.22)
B0.22а)
184 Гя. II, Пргиченение термодинамики к конкретным системам
Таким образом, положение максимума интенсивности по
шкале X определяется уравнением
5g{y)-yg'iy)^0. B0.23)
Находя положительный вещественный корень этого
уравнения г/^, получаем, согласно B0.21а),
КТ^^су^- B0.23а)
Вследствие того, что величины у и х имеют разный смысл,
корень этого уравнения у^ отличается от корня
уравнения B0.20) х^^. Однако качественный вывод об изменении
окраски, естественно, остается в силе. При увеличении Т
максимум интенсивности смещается в сторону меньших
длин волн (больших v).
4. Закон излучения Планка. Планк рассматривает
помещенный в поле излучения линейный осциллятор,
являющийся до некоторой степени реактивом на поле.
Осциллятор представляет собой диполь Герца с определенной
собственной частотой (Oq и размерами, малыми по сравнению
с рассматриваемыми длинами волн. Если такой
осциллятор колеблется, будучи предоставлен самому себе, то он
испытывает затухание благодаря собственному
электромагнитному излучению и при малом затухании остро
реагирует на падающее излучение с частотами о), близкими
к частоте (о^. Если колебания падающего излучения и
вынужденные колебания осциллятора описываются
выражениями
Csino)^ и Z) sin (A)^ + 8), B0.24)
то, согласно т. I [Механика, формула A9.10)], получаем
2) = -^[(аJ-аJJ-|-4р2аJ]-1/2. ^ B0.24а)
При этом уравнение колебаний имеет вид
т (х + 2pi -f- (о^ж) = еЕ^ B0.25)
в соответствии с уравнением A9.9) т.1. Величина Е^
представляет собой ж-компоненту напряженности
электрического поля падающего излучения (ось х совпадает с
§ 20, Излучение в полости 185
направлением колебаний диполя), е и m — соответственно
заряд и масса колеблющегося электрона. Сила
торможения, согласно B0.25), равна
Л= -2pmi. B0.26)
Сравним ее с силой торможения излучением [т. III ^)г
уравнение C6.4)]
р е^ "•
^¦" бтгеосз ^'
которую можно также записать в виде [поскольку
Из формул B0.26) и B0.26а) получаем
p=i-;^«>''- B0.266)
Найдем теперь энергию осциллятора. Кинетическая энергия
его равна
^i2 = -JZ)X'cos2((o^ + 8),
а потенциальная энергия, согласно B0.25), равна
Сумма обоих выражений [при учете B0.24а)] дает
усредненное по времени значение полной энергии
Энергия и и амплитуды С л D снабжены индексом ю,
так как в данном случае они относятся к отдельному
монохроматическому колебанию. Однако в поле излучения
осциллятор будет возбуждаться непрерывным спектром
тесно располо:нсенных некогерентных колебаний С^. Им
соответствуют также некогерентжые (и с близкими
частотами) колебания осциллятора. 9to требование некогерент-
*) А. S о m m е г f е 1 d, Elektrodynamik (Bd. Ill), Leipzig, 1949.
186 Гл. II. Применение термодинамики к конкретным системам
ности необходимо для рассматриваемого здесь «черного»
излучения в той же степени, что и для естественного
«белого» света (т. IV, Оптика, § 49).
Из него вытекает, что в данном случае (в отличие от
^когерентного излучения) аддитивными являются не сами
41мплитуды, а квадраты их (т. е. интенсивности). Поэтому
И8 формулы B0.27) получаем
U=\u^d. = ^\ci (,3_:',+;V^, d.. B0.28)
Подинтегральное выражение в правой части является
быстро меняющейся функцией ш, которая вблизи (o = u)q
обладает острым максимумом (вследствие малости члена
•с p^u)^), в то время как функция С2> изменяется медленно
и может быть заменена своим значением при а) = (о^.
Поэтому вместо формулы B0.28) мы имеем
О
Этот интеграл допускает дальнейшие упрощения, так как
числитель и член 4р2аJ также медленно меняются. Поэтому
их можно заменить соответственно выражениями
2u)^ и 4 @0JJ (oj, B0.286)
где, согласно B0.266),
^=-kj^' B0.28b)
Далее можно написать
(COJ-0JJ = (о,-@^J 4<
Тогда рассматриваемый интеграл принимает вид
о —1/3
и, так как ао)^ < 1, получаем
-Wo
-2^arct.g$|^^ = ^.
§ 20. Излучение в полости 487
Формула B0.28а) принимает вид
>^-8i^(^o- B0.29)
Теперь следует еще выразить величину Cq через
плотность энергии черного излучения к^ Плотность энергии
равна удвоенной электрической энергии, следовательно,
•она равна
{Е, В)^г,Е\
Усредняя это выражение по времени для изотропного
черного излучения, находим
So?2 = 3so^ = K„,. B0.30)
Последний член в этом равенстве показывает, что в
данном случае плотность энергии измеряется в шкале
круговых частот (О, а не просто частот v. Согласно B0.24) и
<20.25), С равно амплитуде еЕ^.. Отсюда при усреднении
110 времени вблизи ш^ш^ получаем
А Г2 = е'^'Е^
Подставляя значение Е% из B0.30), получаем
С? = -!-««>. .B0.30а)
Величина и^ имеет смысл плотности энергии, отнесенной
jx интервалу частоты dim вблизи а) = а)^. Таким образом,
имеем соотношение
1
U^^ do) = Wv rfv, Ku) = 2" ^
«'V
Поэтому вместо выражения B0.30а) имеем
^«=5S:i^«v. B0.306)
Подставляя это выражение в B0.29) и учитывая B0.28в),
исключаем связанные со специальной моделью
осциллятора величины е и m (постоянная Sq, очевидно, также
выпадает, чего, впрочем, можно было ожидать уже из
188 Гл. II, Применение термодинамики к конкретным системам
соображений размерности). Окончательно получим
^ = es»v = gSi»v. B0.31)
Таким образом, энергия осциллятора оказывается
такой же универсальной функцией, как и плотность
энергии теплового излучения. Поэтому в дальнейшем
исследовании Планка вместо теплового излучения
рассматривается осциллятор. Планк приписал осциллятору не только
температуру Г, но и энтропию S. Последняя при
постоянном объеме излучения {dV = 0) дается выражением
dS = ^. B0.32)
Планк в своем нобелевском докладе 1920 г. дал
классический пример объективного изложения истории. Его
окончательный закон излучения появляется там прежде
всего как «счастливо угаданная интерполяционная
формула» .
Казалось, что измерения, произведенные до 1900 г.
для малых длин волн (Пашен, Луммер, Прингсгейм),
подтверждают гипотетический закон, предложенный
Вином. Этот закон можно получить из формулы B0.186),
если положить (в принятых нами обозначениях)
/i (х) = ^е-*, ж = ^ .
Следовательно, получаем
Mv(v, r) = ^v3e-«WT. B0.33)
Согласно B0.31), этому закону соответствует
и=А^е--^1Т, ^1 = 1^ v^. B0.33а)
Определяя отсюда 1/Т и принимая во внимание B0.32),
находим
m-~KV) <2о.ззб)
И
Ж^=-^- B0.33В)
§ 20. Излучение в полости 189
Напротив, более поздние измерения Рубенса и Курль-
баума, произведенные в области длинных волн
(инфракрасная область остаточных лучей), привели к
совершенно другим соотношениям, подтверждающим, казалось бы,
скорее закон Релея —Джинса B0.16). Этому закону
соответствует в силу B0.31) [если использовать для
численного множителя П в B0.16) полученное Ре леем значение
8т:] следующее значение энергии осциллятора:
и = кТ. B0.34)
Отсюда в силу определения B0.32)
§ = 1 = А, B0.34а)
Ж^=-т' B0.346)
Область промежуточных частот [между областями,
описываемыми соответственно уравнениями B0.33в) и B0.346)]
Планк описывает следующей интерполяционной формулой:
"^'^ ^ B0.35)
Если правую часть написать в виде
то B0.35) можно проинтегрировать. Появляющиеся при
этом постоянные интегрирования определяются из условия,
что при и = оо также и Г= оо и, следовательно, dS/dU =
= 0. Тогда получаем
W=-^J-i^- B0-36)
Выражая здесь diS/cf?/, согласно B0.32), через 1/7*, находим
^ТТт=-'г' ^ = ?^Г B0-36а)
ИЛИ, подставляя значение р из B0.35а), имеем
190 Гл* II, Применение термодинамики к конкретным система.\е-
Величина а/г, согласно таблице B0.13), имеет размерность
энергии, умноженной на время, т. е. размерность
действия. Здесь, следовательно, появляется уже упоминав-
шаяся выше новая универсальная постоянная — квант
Оействия
h = ak. B0.37)
Энергия осциллятора записывается теперь в виде
и из формулы B0.31) вытекает закон излучения Планка
«v = ^;nwSzT- B0.39)
Гораздо глубже, чем этот несколько затянувшийся вывод,
идет статистическое понимание уравнения B0.38), в
котором впервые выступает в должном свете революционный
характер константы h (см. § 33). Выше мы набросали
первоначальный ход мыслей Планка; мы это сделали не
только потому, что он имеет исключительную историческую
важность, но также для того, чтобы показать, что
решающую роль сыграло перенесение на осциллятор понятия
энтропии.
На фиг. 21 представлена пространственная модель
поверхности, описываемой формулой Планка B0.39). В
вертикальном направлении откладывается Kv, в
горизонтальном—направо V и от читателя Г. Эта модель состоит из
шести последовательно расположенных плоских картонных
профилей, которые представляют зависимость KvC"^) для
Т = 100, 200, ..., 600° К. Эти профили удерживаются
плоским вертикально установленным профилем,
проходящим через точки v^ [уравнение B0.20а)]. Благодаря
линейности последнего уравнения данный профиль также
вырезается из плоского картона.
Покажем теперь, каким образом из формулы B0.39)
вытекают предельные случаи законов Релея —Джинса и
Вина. Для малых v при заданном Т знаменатель в формуле
B0.39) можно разложить в ряд. Это дает
и^^^^^Т. B0.40)
§ 20. Излучение в полости 191
Для больших V при фиксированном Г, пренебреггя в
знаменателе B0.39) единицей, имеем
u^^^^^e-^^i^T^ B0.40а>
Формула B0.40) совпадает с B0.16), в частности, и в
отношении уже отмечавшегося там выбора численного
Фиг. 21. Модель поверхности, описываемой законом излучения
Планка и^ = f (v, Т).
Ось V направлена направо, ось Т—назад. Светлая и темная окраска pacuojo-
женных уступами профилей вызвана освещением. Температуре бОО^'К
соответствует максимум при частоте Vin==4»10i»ceK.""i, температуре 200°К—очень
низкий максимум при vm=12-10i2 сек.. Температуре 100*'К
соответствует профиль, который едва виден на фигуре, благодаря своему малому
возвышению.
множителя п. Формула B0.40а) переходит в B0.33), если
постоянную А в последней формуле также приравнять
8ic. Наконец, для постоянной а в законе Стефана — Больц-
мана также получается определенное теоретическое выра-
192 Гл. II. Применение термодинамики к конкретнъин системам
/кение. Сравнение формулы B0.186) с формулой Планка
<20.39) дает для фигурирующей в B0.186) функции Д
выражение
Следовательно, интеграл F в B0.19) принимает вид
оо
^ = 8u5^c?x. B0.41)
О
Перепишем его, принимая во внимание, что для всех
л: > О функция е-^ есть правильная дробь:
оо оо
о о
B0.41а)
Заменяя во втором, третьем и т. д. членах ряда 2х,
Зх, ... череа f, преобразуем B0.41а) к виду
со
(l+^ + ^-f ...M5'e-=rft B0.416)
О
Здесь интеграл равен ГD) = 3!. Значение ряда,
стоящего в скобках перед интегралом, приведено в т. IV
[Оптика, формула B.18)]. Оно равно ic*/90. Поэтому все
выражение B0.416) равно 11^/15 и интеграл в B0.41а) есть
/'=^^'.. B0.41b)
Подставив это выражение в B0.19), найдем теоретическое
значение постоянной а в законе Стефана — Больцмана:
«=?(-^3. B0.42)
Так как а и постоянная а = h/k в законе смещения
Вина B0.23а) известны из опыта, то соотношения B0.42),
B0.23а) и B0.37) представляют собой уравнения для
определения постоянных Л и А. В настоящее время
наиболее точными значениями последних считают следующие:
А = 6,624.10-27 эрг.сек, Л = 1,380.10-1» эрг/град.. B0.43)
§ 21, Необратимые процессы 193
§ 21. НЕОБРАТИМЫЕ ПРОЦЕССЫ. ТЕРМОДИНАМИКА
НЕРАВНОВЕСНЫХ СОСТОЯНИЙ
1. Теплопроводность и появление локальной
энтропии. До сих пор мы рассматривали в основном только
состояния термодинамического равновесия, а в отношении
необратимых процессов смогли лишь установить, что в
адиабатически изолированной системе их протекание
связано с возрастанием энтропии. Теперь установим более
точно, где именно имеет место возрастание энтропии и
как оно зависит от соответствующих параметров системы.
Прежде всего рассмотрим особенно простой пример,
а именно процесс теплопроводности в твердом однородном
изотропном теле, отвлекаясь при этом от теплового
расширения последнего. Если температура меняется от точки
к точке, то внутренняя энергия единицы массы к (ж, у, z, t)
зависит, вообще говоря, от координат точки и от
времени. Последнее справедливо также для потока тепла W.
Тогда из закона сохранения энергии получаем [см. т. VI,
Дифференциальные уравнения в частных производных
физики, уравнение G.11); там через и обозначена
температура]
pg + divW = 0, B1.1)
где р — плотность вещества. Для полного описания
теплопроводности необходимо еще знать зависимость
внутренней энергии и от температуры Т. Она дается
дифференциальным соотношением
du^cdT, B1.2)
где с— удельная теплоемкость. Далее, надо связать поток
тепла с градиентом температуры. Эта связь определяется
законом Фурье [см. т. VI, § 44, 45, уравнение G.12)]:
W--xgradr, B1.3)
где X — коэффициент теплопроводности.
Отвлечемся на время от соотношений B1.2) и B1.3)
и сосредоточим свое внимание на уравнении B1.1).
Поскольку изменения объема можно не учитывать, между
194 Гл. II. Применение термодинамики к конкретным системам
удельной энергией и удельной энтропией существует
соотношение
du=Tds. B1.4)
В силу B1.1) это дает
p?=-T'*^^W. B1.5)
или после некоторых преобразований
pg4 div -^ = —y^(W, gradT). B1.6)
Уравнение B1.1) представляет собой уравнение
непрерывности, т. е. выражает некоторый закон сохранения
(закон сохранения энергии). Уравнение B1.6) было бы
уравнением непрерывности, если бы его правая часть
обратилась в нуль. Как известно, для энтропии не существует
какого-либо закона сохранения. Напротив, в ходе
необратимого процесса (в качестве которого здесь выступает
процесс теплопроводности), в замкнутой системе энтропия
возрастает. Это возрастание мы должны будем связать с правой
частью уравнения B1.6). Интегрируя для этого B1.6) по
объему проводящего тепло тела и применяя теорему f аусса
[см. т. II, Механика деформируемых сред, формула C.1)],
получаем
P^5sflfF + ^^"d/'=-5^(W,gradr)dF. B1.7)
Прежде всего представим себе, что поверхность
проводящего тепло тела адиабатически изолирована.
Вследствие этого нормальная составляющая потока тепла на
поверхности тела обращается в нуль. Тогда слева в
уравнении B1.7) стоит изменение энтропии тела в единицу
времени. Правая часть выражает его через температуру,
1'радиент температуры и поток тепла. Так как, согласно
второму началу термодинамики (в формулировке Клаузиу-
са), тепло не перетекает само, по себе от тела с более
низкой температурой, то скалярное произведение
(W, gradT) должно быть отрицательным, если W ^ О и
grad Т фО. Поэтому в соответствии со вторым началом
термодинамики правая часть уравнения B1.7) положительна.
Таким образом, изменение энтропии в единицу време-
§ 21, Необратимые процессы 195
ни изображается объемным интегралом. Это приводит
к мысли определить его подинтсгральное выражение как
энтропию, возникающую в единицу времени в единице
объема. Назовем это выражение локальным приращением
энтропии
e=-^(W,gradr). B1.8)
Величина 6 зависит только от состояния тела в
соответствующем объеме в данный момент времени. При этом
следует, конечно, расширить понятие состояния по срав-
вению с тем, что мы имели до сих пор. Положим в
определении B1.8) температуру постоянной. Для
характеристики состояния следует также задать градиент
температуры и, следовательно [см. B1.3)], поток тепла.
Откажемся теперь от адиабатической изоляции
поверхности проводящего тепло тела и представим себе, что
каждая точка поверхности связана с соответствующим
термостатом. Тогда величина W^ представляет
количество тепла, отдаваемое в единицу времени единицей
поверхности тела находящемуся рядом с ней термостату; WJT
есть отдаваемая энтропия. Естественно в связи с этим
назвать величину
S=^ B1.9>
потоком энтропии. При этом уравнение B1.6) принимает
вид
p|j + divS = e B1.10)
и интегрирование по произвольной части объема тела дает
р~ С sdV + ^SJF== \ edV. B1.11)
Если читать это уравнение справа налево, то оно
получает следующее ясное толкование: часть энтропии,
возникающей в рассматриваемом объеме, вытекает наружу,
а другая ее часть увеличивает содержание энтропии в
данном объеме (это последнее изменение энтропии может быть,
конечно, я отрицательным).
Таким образом, для случая теплопроводности мы
установили источники энтропии и их мощность; кроме того,
196 Гл. II. Применение термодинамики к конкретным системам
В соответствии со вторым началом было показано, что
эта мощность не может быть отрицательной. Уравнение
B1.10) вместе с условием 0>О можно назвать
дифференциальной формулировкой второго начала термодинамики.
Вместо интегрального утверждения о том, что энтропия
замкнутой системы не убывает, появляется
дифференциальное, согласно которому локальное приращение
энтропии не отрицательно независимо от того, является ли
система замкнутой или незамкнутой и обратимы или
необратимы протекающие в ней процессы.
Сравним теперь закон Фурье B1.3) и определение
локального приращения энтропии B1.8). Мы найдем, что
последнее содержит в качестве множителей как раз те
величины W и Г, зависимость между которыми
описывается законом Фурье. Этот факт, как мы увидим ниже,
имеет весьма общее значение. Из соотношения
e = ^(gradrJ>0
непосредственно следует, что
2. Теплопроводность в анизотропных телах и
соотношения взаимности Онзагера. Рассмотрим теперь
общий случай теплопроводности в анизотропных телах с
произвольной кристаллической структурой. В наших
предыдущих рассуждениях при этом ничего не изменяется,
кроме уравнения B1.3). Последнее теперь заменяется
тензорным соотношением между компонентами градиентов
температуры и потока тепла (координаты обозначаются
через Xj^, х^, х^)
W --Х ^-х ^-х ^^ B112)
*^з- bi^ ^32^0:2 ^33^^^*
Это соотношение приводит к выводу, что в произвольном
кристалле градиент температуры и поток тепла, вообще
говоря, не параллельны (точнее, не антипараллельны)
друг Друху.
§ 21, Необраттимые процессы 197
Сравним теперь соотношение B1.12) (мы будем
называть это соотношение или соответствующие соотношения при
других необратимых процессах феноменологическими) с
выражением для локального приращения энтропии B1.8),
предварительно переписав последнее_в виде
^=-^(^if + ^^^f+^^3g). B1.13)
При этом отметим, что феноменологическое соотношение
B1.12) линейно и однородно связывает первые
множители W-^, W^ и PFg в B1.13) с соответствующими вторыми
множителями дТ/дх^у дТ/дх^ и дТ/дх^. Конечно,
феноменологическое соотношение B1.12) не является произвольным.
Во-первых, оно должно удовлетворять условию 9>0 для
любых температурных градиентов, т. е. должно иметь
место неравенство
SS''i.SS>0. B1.14)
i k
И, следовательно, компоненты тензора х^,^ не могут быть
отрицательными. Во-вторых, доля^ны выполнятся
соотношения
^ik = 4i (/,Л = 1,2,3), B1.15)
т. е. тензор теплопроводности должен быть симметричен.
Последние соотношения подтверждены опытом^), они
вытекают из кинетической теории теплопроводности и,
наконец, представляют собой частный случай очень общих
соотношений симметрии, открытых Онзагером^).
Относительно их общей формулировки еще будет говориться ниже.
С точки зрения общей термодинамической теории в
случае только что рассмотренного примера следует
говорить о наложении трех элементарных необратимых
процессов, каждому из которых соответствует одна
пространственная координата. При этом из соотношений B1.12) я
B1.13) явствует, что при наличии нескольких необратимых
1) W. Voigt, Nachr. Ges. Wiss. Gottingen, Math. Phys.
Klasse, S. 87 A903); Ch. Sore t, Arch, de Geneve, 29, 355 A893); 32,
611 A894).
«) L. 0nsagar, Phys. Rev., 37, 405 A931); 38. 2265 A931).
198 Гл. II. Применение термодинамики к конкретньим системам
процессов локальное приращение энтропии склады-
вается, повидимому, из трех частей, каждая из которых
относится только к одному из названных процессов;
однако феноменологические соотношения могут привести
к связи этих необратимых процессов в том смысле, что
перепад температуры в направлении х^ может обусловить
наличие компонент теплового потока также и в
направлениях х^ и х^.
Это явление имеет очень общий характер. Оно может
иметь место также при одновременном протекании весьма
различных необратимых процессов, как, например,
теплопроводности и диффузии или теплопроводности и
электропроводности. В этих случаях связь необратимых
процессов, описываемая феноменологическими соотношениями,
приводит к существованию явления термодиффузии и
диффузного термоэффекта или к термоэлектрическим
явлениям.
3. Термоэлектрические явления. В
термоэлектрических явлениях мы имеем дело, с одной стороны, с
переносом энергии и заряда и, с другой стороны, с
причинами, их порождающими,—перепадом температуры и
напряженностью электрического поля. Рассмотрим металл,
в котором течет электрический ток и суп1;ествует перепад
температуры. Напишем для этого случая закон
сохранения энергии и из него выведем уравнение для энтропии,
соответствующее равенству B1.6). Предположим, что
удельная внутренняя энергия и и удельная энтропия s не
зависят от плотности тока J. Это предположение такого же
сорта, как и неявно подразумевавшаяся в п. 1 гипотеза
о том, что внутренняя энергия зависит от температуры,
ко не зависит от потока тепла и перепада температуры.
Более детально оно может быть обосновано в электрон-
1ЮЙ теории металлов (см. также § 45).
Без ограничения общности положим, что подвижными
1юсителями тока являются отрицательно заряженные
частицы с зарядом — е(е>0). Это представление лежит
в основе электронной теории металлов.
При формулировке закона сохранения энергии следует
принять во внимание два обстоятельства: во-первых, к
каждой единице :>бъема в единицу времени подводится
§ 21. Необратимые процессы 199
электрическая энергия (J, Е); во-вторых, поскольку заряд
единицы объема в единицу времени возрастает на
величину — divj, возникает дополнительная потенциальная
энергия — OdivJ. (Здесь Ф — электрический потенциал,
Е =—grad Ф — напряженность электрического поля.) При
этом предполагается, что сила тока изменяется
достаточно медленно. Следовательно, закон сохранения энергии
в данном случае принимает вид [ср. с уравнением B1.1)]
t) J= -divW+(J, E)-ФdivJ. B1.16)
Подставляя сюда выражение для дифференциала
энтропии S из A8.2) и A8.3) (т. е. пренебрегая, как и выше,
изменениями объема), имеем, поскольку 2= —1,
Tds^du - (ji- РФ) dn. B1.17)
Здесь Lw —число носителей заряда в 1 г металла (L —
число Лошмидта), [х — химический и [х - ^Ф —
электрохимический потенциалы носителей заряда. Принимая во
внимание, что F = Le, а величина — pLwe есть заряд единицы
объема, получаем
7-т 5л д (— Lne) J. т
Таким образом.
Здесь \i/F = С/е, т. е. С = [x/L. Величину С в электронной
теории металлов называют химическим потенциалом,
рассчитанным на один электрон. Исключая du/dt из B1.16)
и B1.17), находим
p5- + div-^(w + f J)=4r(w. -^grad7') +
+ ^(j,E + rgrad^). B1.176)
Это уравнение подобно уравнению B1.6). Соответственно
величину "тг ( W -]— J 1 следует истолковать как поток
энтропии, в то время как правая часть B1.176) представ-
200 Гл, II. Применение термодинамики к конкретным системам
ляет собой локальное приращение энтропии 9.
Руководствуясь результатами п. 1 и 2, можно и в данной задаче
из выражения для локального приращения энтропии
получить законы тепло- и электропроводности. Прежде всего
из уравнения B1.176) следует, что 9 является линейной
функцией потоков W и J. Последние в свою очередь
представляются линейными функциями стоящих при них коэф-
1 С
фициентов — -jr grad Г и Е + Г grad — *
а
W=-4gradr + p(Efrgrad^)
J= -Xgradr + 8 (E + Tgrad-^) .
B1.18)
Разрешая эти уравнения относительно Е и W, получаем
в обычных обозначениях
W= -xgradr-(^n + -^)j, B1.18а)
E = -^J-egradr- grad-^-. B1.186)
Смысл коэффициентов х, И, е, 1/а подлежит исследованию.
Для дальнейшего целесообразно отметить их связь с^иу,
Р=-а(п + |), 7=-а(еГ + |). B1.18в)
Из соотношения B1.186) вытекает, что на граничной
поверхности между двумя металлами вследствие скачка
величины С возникает скачок потенциала. Интегрируя
B1.186) вдоль очень малого отрезка пути, проходящего
через граничную поверхность между двумя металлами /
и //, получаем (принимая во внимание, что вклад первых
двух членов в правой части сколь угодно мал)
Фи-Ф/=у{С//-С/). B1.18Г)
Величина Ф// —Ф/ есть не что иное, как контактная
разность потенциалов между двумя металлами. При
равновесии мы имеем J = 0, W = 0, grad 7* = О, и поэтому
Е = — grad С/е. Следовательно, в равновесных условиях
напряженность электрического поля, согласно B1.186)
§21. Необратимые процессы 201
отлична от нуля только там, где (при постоянной
температуре) изменяется величина С, т. е. там, где материал
неоднороден (в частности, следовательно, в переходном слое
между двумя различными однородными металлами).
Если электрический ток отсутствует, то уравнение
B1.18а) представляет собой закон теплопроводности Фурье
и X, как показывает сравнение с B1.3), обозначает
коэффициент теплопроводности. В этом случае, согласно
уравнению B1.186), всегда существует напряженность
электрического поля Е= — sgradT* —fijTadC/e. Коэффициент е
называется абсолютной термодлектродеи:нсущей силой, [См.
в связи с этим формулу D5.25), которая дает также и
точное выражение для е.]
Если температура везде одинакова и материал однороден,
т. е. gradC = 0, то уравнение B1.186) есть не что иное,
как закон Ома с электропроводностью а. В общем случае,
т. е. когда температура неодинакова во всех точках системы,
равенство B1.185) можно записать также в виде
J = a(E4^E^). B1.19)
Величину, для краткости обозначенную через Е^, иногда
называют напряженностью стороннего электрического поля.
Наряду с этим равенство B1.18а) показывает, что поток
энергии существует также и в отсутствие какой-либо
разности температур, если только J =7^ 0. Следовательно,
перенос энергии связан также с переносом заряда. Это
обстоятельство обусловлено тем, что как перенос заряда, так
и перенос энергии имеют общую причину — движение
электронов проводимости в металле. Электронная теория
металлов во всех отношениях подтверждает это утверждение.
Энергия, переносимая одним кулоном, в наших
обозначениях равна (П + С/е).
Перейдем теперь к исследованию термоэлектрических
явлений. Начнем с эффекта Томсона. Он состоит в том,
что при протекании тока через проводник, в котором
имеется градиент температуры, помимо джоулева тепла
выделяется еще так называемое тепло Томсона, Последнее,
будучи рассчитано на единицу объема и единицу времени,
равно |х (J, gradT*). Величина [х [не следует путать с [л
из уравнения B1.17)] ndi^uBdieicn коэффициентом Томсона.
Это количество тепла может быть как положительным,
202 Гл. II, Применение термодинамики к конкретньим системам
так и отрицательным, в зависимости от взаимной
ориентации J и grad Т, Иногда этот эффект называют обратимым,
так как он меняет знак при изменении направления J
или grad Г на противоположное. Все же такое название
вводит в заблуждение, так как этот эффект представляет
лишь часть всего процесса и, кроме того, неразрывно
связан с теплопроводностью и выделением джоулева тепла,
т. е. с типичными необратимыми процессами.
Эффект Томсона содержится в наших соотношениях.
Чтобы показать это, рассмотрим уравнение для количества
тепла, выделяющегося в единице объема в единицу времени
р {du/dt) = (J, Е) — div W — Ф div J, и подставим в него
выражения B1.18а) и B1.186) для W и Е. Тогда, поскольку
div J = О (это равенство точно выполняется для постоянного
тока и с достаточным приближением —для тока, медленно
меняющегося), получаем
p|f = div(xgradr) + (^^-e)(J,gradr)-i-ij^.
Первый член в правой части соответствует чистой
теплопроводности, последний — джоулеву теплу, а второй имеет
структуру, предполагаемую для тепла Томсона. Из
определения коэффициента Томсона вытекает первое
соотношение Томсона
tx = |^-e. B1.20)
Впервые это соотношение было выведено Томсоном. Точнее,
им было указано, что в общем случае ( -^jT" ^ ) ^ О» и
отсюда в силу закона сохранения энергии сделан вывод
о существовании эффекта Томсона.
Величина П называется коэффициентом Пелътъе. Он
связан с эффектом Пелътъе, состоящим в выделении
положительного или отрицательного количества тепла
в месте контакта двух различных металлов. Рассмотрим
систему, изображенную на фиг. 22, и предположим, что
температура во всех точках одинакова. Тогда в металле
/ слева направо течет энергия Г И/ Н— С/ ) /, а в металле
// соответственно f П/j + — ^iijJ (если площадь попереч-
§ 21. Необратимые процессы 203
КОГО сечения проводников в месте соприкосновения равна
единице).
Таким образом, выделяется энергия
[п,^-11, + 1(С,,-С,)]/. B1.21)
Однако часть ее, равная A/е) (С// —С/)/, затрачивается
на перемещение носителей тока через граничный слой
от потенциала Ф/к потенциалу Фц [см. B1.18г)]; поэтому
выделяется в виде тепла лишь энергия (И// —П/)/.
Металл I
1 ъ
J ^
Металл II
J ^
Фиг. 22. К эффекту Пельтье.
Наконец, вычислим еще э.д.с, возникающую в
замкнутом контуре, составленном из двух различных
металлов. Она дается выражением
^ (E^ rfr) = $ S (grad Г, dv) + ^ Tgrad |-, tfr) = $ s йГ.
Разлагая интеграл на два слагаемых, каждое из которых
относится только к одному металлу, получаем (Г^ и 7*2 —
температуры спаев, е/ и e/j — абсолютные
термоэлектродвижущие силы обоих металлов)
Т2 Ti То
^ (E^ rfr)= ^ ^iidT-V YidT=\^ ^eu-^j)dT. B1.22)
Ti Го Ti
Отсюда следует, что э.д.с. такого контура зависит только
от температур спаев. Далее, отсюда вытекает, что термо-
э.д.с. исчезает, если оба металла одинаковы (так как
тогда е/ = е//), и, наконец, при достаточно малой разности
температур э.д.с. оказывается равной {вц — 8/) {Т^ — Т^),
Заметим также, что из измерений эффекта Пельтье, как
и из измерений термо-э.д.с, можно установить только
разность коэффициентов Пельтье и абсолютных термо-э.д.с.
204 Гл. II. Применение термодинамики к конкретньич системам
для двух металлов. Поэтому соотношения B1.20) и B1.23)
можно проверить только в применении к разностям
соответствующих величин для двух металлов. Однако в
электронной теории металлов определяют II и е также и для
одного металла.
Уравнение B1.20) указывает на связь между тремя
термоэлектрическими эффектами. Другое важное равенство
дают соотношения взаимности Онзагера, если применить
их к рассматриваемому примеру. Согласно этим
соотношениям, матрица, составленная из коэффициентов уравнений
B1.18), должна быть симметричной. Отсюда вытекает так
называемое второе соотношение Томсона Р = 7» или,
согласно B1.18в),
П = Гв. B1.23)
Томсон вывел это соотношение другим путем. Именно,
в его рассуждениях термоэлектрический эффект был
мысленно отделен от сопровождающих его процессов
теплопроводности и выделения джоулева тепла и термоэлемент
рассматривался как стационарно работающая машина Карно.
Нагревание осуществляется тогда только за счет тепла
Пельтье, выделяющегося на подогретом спае. Поэтому
если разность температур спаев составляет dT и
возникающая электрическая энергия равна J сБ (Е, dr), то
коэффициент полезного действия имеет вид
т п^^-п^ •
Вместе с тем это выражение является следствием
соотношения B1.23).
Это разделение на так называемые обратимые и
необратимые эффекты мало убедительно^), несмотря на то,
^) Томсон в своем письме «О термодинамической теории тепла»
(Ostwalds Klassiker dor exakten VVissenschaften, Nr. 193)
указывал на то, что здесь мы имеем дело только с гипотезой, для
доказательства которой необходима экспериментальная проверка:
«Дело не только в том, что условия, предписываемые вторым
началом динамической теории тепла, выполняются здесь неполностью,
но и в том, что доля процессов, которые этим условиям
удовлетворяют, во всех известных термоэлектрических явлениях чрез-
§ 21, Необратимые процессы 205
что оно приводит к экспериментально подтверждаемому
выводу. Поэтому весьма отрадно, что электронная теория
металлов смогла подтвердить второе соотношенпе Томсона
без помощи такого рода дополнительных гипотез^). Тем
самым был дан один из первых примеров
непосредственного вывода соотношений Онзагера и оказалось возможным
понять существо этого принципа, в дальнейшем столь
остроумно обобщенного Онзагером, как принципа
микроскопической обратимости (т. е. инвариантности
элементарных законов относительно изменения знака времени).
Укажем в связи с этим на обобщения, возникающие
в связи с термоэлектрическими явлениями в анизотропных
телах и в присутствии внешнего магнитного поля В.
Специально следует отметить видоизменение соотношений
Онзагера, происходящее при наличии магнитного поля.
В простейшем случае для тензора теплопроводности вместо
B1.15) мы получаем
>Ci,(B)==x,,(-B).
Это связано с тем, что в данном случае принцип
микроскопической обратимости справедлив только при
одновременном обращении направления магнитного поля.
Действительно, уравнение движения электрона в магнитном поле
mv= --e[v-B] остается неизменным при замене t, В
на —/, —В.
Так же как термоэлектрические эффекты, можно
рассмотреть и термодиффузию и открытый Дюфуром^)
и особенно убедительно продемонстрированный Клузиусом
и Вальдманом^) диффузионный термоэффект. Внешнее
усложнение проявляется только в том, что в этих примерах
учитываются также изменения объема и явления переноса.
При этом применение соотношений взаимности Онзагера
также дает связь между обоими эффектами, которая экспе-
вычайно мала по сравнению с неразделимо связанными с ними
процессами, нарушающими эти условия в существенных пунктах».
Больцман очень тщательным анализом показал несостоятельность
этого метода и тем самым полностью подтвердил сомнения Томсона
(Sitzber. d. Akad. d. Wiss. Wien. Math. Naturwiss. Klasse, II Abt.,
96, 1258 A888)].
1) A. Sommerfeld, Zs. f. Phys., 47, 1, 43 A928).
2) Dufour, Ann. de Phys., 28, 490 A873).
8) Kl. Glusius, L. Waldmann, Naturwiss., 30, 711 A942).
206 Гл, II. Применение термодинамики к конкретным системам
риментально подтверждена и в настоящее время дала
возможность весьма точного определения
термодиффузионных факторов из исследования диффузионного
термоэффекта вместо того, чтобы использовать измерения
термодиффузии ^).
4. Внутренние превращения. Если до сих пор мы
имели дело с необратимыми явлениями переноса (перенос
энергии и электрического заряда), то теперь рассмотрим
необратимые процессы в гомогенной среде, так называемые
внутренние превращения, или релаксационные явления,
не сопровождающиеся явлениями переноса. (В случае,
если внутренние превращения представляют собой
химические реакции в обычном смысле, они называются также
гомогенными реакциями.) Кроме того, должен оставаться
неизменным объем и по условию исключается подвод тепла;
таким образом, cfF = 0, dU = 0. Рассмотрим 1 г вещества,
состоящего из трех компонент А^, А-^ и А^у между которыми
могут протекать две реакции: А^^А^^, А^Т^А^. Все другие
мыслимые возможности, произвольное число компонент
и любые типы химических реакций, можно принципиально
рассмотреть по той же схеме, почему и достаточно
ограничиться этим простейшим примером. Тогда, согласно A4.2),
мы имеем (поскольку dV = dU = 0)
Tds = — [Xq drif^ — |Xj dn-^ — [Xg dric^.
Здесь p.^ — химические потенциалы отдельных компонент,
рассчитанные не на один моль, а на 1 г, в то время как
'^о» ^1» ^2 — количества отдельных компонент в граммах
на 1 г вещества. Следовательно, nQ + /?i-}-n2= 1. Таким
образом, независимыми переменными являются только щ
и AZg, и предыдущее уравнение принимает вид
Tds = ((х, - (х,) dn, + A^0 - Н^2) dn^. B1.24)
Изменение энтропии со временем (которое в силу
отсутствия явлений переноса совпадает в данном случае с
локальным приращением энтропии) определяется выражением
pS=f(.o-..)f + f(.o-.Уl^ B1.25)
1) L. Waldmann, Zs. f. Phys., 124, 30 A944).
§ 21, Необратимые процессы 207
Это выражение также является линейной функцией dnjdt
и dnjdt, а коэффициенты при последних величинах
характеризуют отклонения от равновесия. В состоянии
равновесия для каждого виртуального изменения dn^ и dn^,
согласно (8.1), ds< 0. Это означает, что [J^o ~ F^i = F^o "~ F^2 = ^•
При незначительных отклонениях от термодинамического
равновесия в данном случае допустимо представить dnjdt
и dnjdt в B1.25) как линейные функции их
коэффициентов, т.» е. в виде
B1.26)
- = «21 (н-о - M-i) + «22 ^и - Р'г)»
где а|^ = 01^G*, v) и, кроме того, справедливо
соотношение взаимности Онзагера a^^ = a2i. ^ связи его с
принципом детального равновесия в химии будет сказано при
решении задачи П.7.
Сделаем еще одно замечание. Условие с?С/* = 0 было
необходимым, так как мы исключили из рассмотрения
явления переноса. Изменение внутренней энергии может
происходить только в результате совершения работы
(т. е. при изменении объема) или вследствие подвода
тепла. С другой стороны, предположение о том, что
протекают лишь две реакции {А^^^А^ и А^'^А^), не было
необходимым. В наших рассуждениях ничего не
изменится, если возможна также реакция Aj^'^A^. Важно только,
чтобы реакции были независимыми. Их число определяет
число независимых величин л^. Однако в нашем случае
реакция А^^^А^ была бы зависимой реакцией и
получалась бы при помощи операций вычитания из реакций
Aq^^Ai и AqT^Ao. Конечно, в качестве независимых
можно взять любые две из этих трех реакций.
Необязательно также, чтобы записанные уравнения реакций
отражали их действительный молекулярный механизм, т. е.
представляли собой так называемые элементарные реакции.
Вместо независимых элементарных реакций можно
исходить из произвольных независимых линейных
комбинаций этих элементарных реакций с вещественными
численными коэффициентами; при этом никаких изменений в
наших рассуждениях не произойдет. В этом находит отра-
208 Гл, II, Применение терлюпинамики к конкретным системам
жение тот факт, что термодинамическая теория
необратимых процессов иcпoльзvp.т только феноменологические
законы, не* вдаваясь в детали молекулярного механизма
явлений.
Напомним здесь еще раз наше утверждение (8.9), что
условием обратимости процесса является равенство
2-X^iCfiCi = 0. Из выражения B1.25) следует, что
величина р 2 -^i dx^ представляет локальное приращение энтро-
i
ПИИ 9 за время dt (за которое происходят изменения dx^),
следовательно, для необратимого процесса действительно
всегда 2 -^i ^^i > 0.
i
5. Общие закономерности. Мы не можем здесь
входить в рассмотрение необратимых процессов общего типа,
при которых внутренние превращения связаны с
одновременным протеканием явлений переноса. Заметим лишь,
что в каждом случае изменение энтропии произвольно
выбранной единицы массы можно получить из законов
сохранения и соотношения Гиббса A4.2) и записать
в виде^)
pg-bdivS = e. B1.27)
Величина S есть поток энтропии у который линейно
зависит от потока энергии, присутствующих иногда
диффузионных потоков или от плотности электрического тока.
В него не входит поток энтропии, обусловленный
конвекцией, т. е. энтропии, переносимой потоками вещества,
так как уравнение B1.27) относится к наблюдателю,
движущемуся вместе с веществом. Величина 6 представляет
€обой локальное приращение энтропии и всегда имеет вид
0 = l2^i-^i' B1-28)
i
где переменные Х^ суть обобщенные потоки. Они могут
обозначать поток энергии (три его компоненты), диффузи-
^) Это впервые совершенно ясно было высказано, повидимому,
Г. Яуманном.
§ 21. Необратимые процессы 209
онный поток (соответственво, три его компоненты), поток
импульса, обусловленный внутренним трением, т. е.
компоненты тензора р^^ [см. т. II, уравнение A0.18)],
величину p{dnjdt) из предыдущего пункта и т. п. Стоящие
при них коэффициенты К^, которые называются термо-
динамическими силами, равны — ^grad Г, Т grad -^ ,
j-^, химическим потенциалам или их разностям [см.
уравнение B1.25)] и т. д. Таким образом, величина ТО,
характеризующая диссипацию энергии, всегда является
суммой произведений потоков на силы.
При малых отклонениях от термодинамического
равновесия потоки Xi линейно зависят от сил К^:
^i=S«ift^ft- B1.29)
/1=1
Теперь можно сформулировать соотношения взаимности
Онзагера. Однако они не всегда выражаются в виде
Hk" "'hi^
B1.30)
как можно было бы ожидать согласно предыдупщм
разделам. Казимире) первый указал, что эти соотношения
правильны только тогда, когда обе силы А''^ и /iT^,
соответствующие паре индексов i, к, являются либо четными,
либо нечетными функциями скоростей потоков или
скоростей молекул. Если же одна из этих сил дается четной,
а другая нечетной функцией, то для этой пары индексов,
как указал Казимир, справедливо соотношение
flift =-«;»• B1.31
Мы не можем здесь входить в детали общего
доказательства соотношений B1.30) и B1.31). Его можно было
бы провести при помощи теории возмущений на основе
принципа макроскопической обратимости, как это сделали
Онзагер и Казимир, или рассматривая специальные
случаи, например пользуясь кинетической теорией газов в
1) Н. В. G. Casimir, Rev, Mod. Phys., 17, 343 A945).
210 Гл. II. Применение термодинамики ^ конкретным системам
применении к газовым потокам. Однако в этом примере,
чтобы получить нетривиальное соотношение Онзагера,
следует рассматривать по меньшей мере смеси двух газов.
Действительно, для однородного газа диссипация энергии
имеет вид (см. также гл. V)
^S = yS2/'.(S + tLW' -r^-dr) B1.32)
[в отношении второго члена см. п. 2, в отношении
первого - т. II, уравнение A0.18)]. Следовательно, если
применить феноменологические соотношения B1.29), то
компоненты . W^, W , W^ будут представлены в виде
V „ i дТ
линейных функции величин —^д~ ^ шести величин
Г ^ + ^ j . В силу изотропности газа эти три
феноменологических соотношения должны быть инвариантны
относительно вращений системы координат, т. е. в новой
системе координат они должны сохранять прежний вид
с теми же коэффициентами. Отсюда вытекает, что
коэффициенты при ( л~^ •+ 5-^} исчезают (если среда
изотропна, то вектор не может линейно зависеть от тензора).
Кроме того, из изотропии среды следует, что тензор
теплопроводности сводится к тензору, кратному
единичному тензору Ь^^.
Аналогичные соображения относительно
феноменологических соотношений для р^^ показывают, что последние
не выражаются через температурные градиенты.
Следовательно, почти все смешанные феноменологические
коэффициенты выпадают; остается только несколько смешанных
коэффициентов, связывающих р^^ и (л~ + л-^ ;• Именно,
как показано в т. II [уравнение A0.21)] и как будет
выведено в гл. V этого тома,
^..-<?+||)+о-^)(ё7+ё+&)'».B1.зз)
где тг] и С —первый и второй коэффициенты вязкости.
Сопоставляя коэффициенты при dvjdx^ в р^^ и при dvildx^
§ 21, Необратимые процессы 211
В /?22» найдем, что они равны. Отсюда видно, что все
соотношения взаимности Онзагера в этом примере
вытекают из соображений симметрии. Однако для смеси двух
газов имеется уже одно нетривиальное соотношение
взаимности, которое устанавливает связь между
термодиффузионным и диффузионнотермическим эффектами.
6. Область применимости термодинамической теории
необратимых процессов. Все до сих пор изложенное
относилось лишь к случаю малых отклонений от
термодинамического равновесия. Теперь следует уточнить смысл
этого условия, т. е. установить, какие отклонения можно
считать малыми. Естественно, требуется, чтобы имели
смысл термодинамические понятия температуры и
термодинамических функций. Однако фактически определить
пределы применимости данной теории необратимых
процессов можно, лишь владея теорией более общего
характера, из которой данная теория получалась бы как
частный случай.
В случае газов такой более общей теорией была бы
кинетическая теория газов. Как мы увидим в гл. V, из
нее действительно получаются законы сохранения в форме,
которую мы используем в гидродинамике и
аэродинамике; далее, она дает выражение B1.32) для диссипации
энергии и приводит к нашим феноменологическим
соотношениям для потока тепла и вязкого тензора
напряжений.
Для определения области применимости этих законов
необходимо перейти к следующим членам разложений,
выписанных в гл. V. Это было проделано, например, Энскогом^).
Тогда можно получить весьма наглядное выражение для
условий, при которых этими следующими членами можно
пренебречь. Именно, оказывается необходимым, чтобы
изменения температуры на расстоянии, равном длине
свободного пробега, были малы по сравнению с абсолютной
температурой, а изменения скоростей должны быть малы
по сравнению со скоростью звука или со средней
скоростью хаотического теплового движения молекул.
Принимая во внимание, что при нормальных условиях длина
1) D. Enskog. Zs. f. Phys., 54, 498 A929).
212 Г л, II, Применение термодинамики к конкретным системам
свободного пробега имеет величину порядка 10"* см,
видим, что пределы применимости теории ^ще очень
широки и могут быть нарушены разве только в случае
ударных волн. Если для многих других типов
необратимых процессов в настоящее время еще невозможно простым
способом количественно определить область применимости
их термодинамической теории, то все же результаты,
полученные для газов, вселяют уверенность, что и в других
случаях существует достаточная для ряда задач область
применимости.
Глава III
ЭЛЕМЕНТАРНАЯ КИНЕТИЧЕСКАЯ ТЕОРИЯ ГАЗОВ
Зарождение кинетической теории газов восходит к
Даниилу Бернулли. В его книге «Гидродинамика» (С'ррас-
бург, 1738 г. ^) уже выведено давление газа из
изменения импульса молекул газа, сталкивающихся со стенкой
сосуда. Однако дальнейшее развитие относится лишь к
середине XIX века 2) и связано с именами Кронига
A856 г.), Клаузиуса A857 г.). Максвелла A860 г.). Высшей
точкой являются работы Людвига Больцмана, который
придал закону Максвелла о распределении скоростей
самую общую форму.
§ 22. УРАВНЕНИЕ СОСТОЯНИЯ ИДЕАЛЬНОГО ГАЗА
Если представить себе толчки, которые испытывает
твердая, плоская (или с непрерывной кривизной), гладкая
(и поэтому не испытывающая сил трения) стенка сосуда,
содержащего газ, вследствие ударов молекул газа, то
соответствующая картина для каждого элемента
поверхности do благодаря громадному числу этих толчков
изобразится в виде кривой f{t) с огромным количеством
«зубцов». Давление (сила, действующая на единицу
поверхности) определяют как усредненное по времени
значение этой функции. Вклад отдельного толчка равен
изменению импульса молекулы при ударе и последующем
отражении. Если скорость сталкивающейся со стенкой
молекулы равна с, а угол между направлением ее дви-
^) См. также А. Зоммерфельд, т. II, Механика деформируемых
сред, §11.
*) В качественной форме ряд руководящих идей кинетической
теории был высказан в aVIII веке М. В. Ломоносовым. — Прим,
перев.
214 Гл, III. Элементарная кинетическая теория газов
жения и нормалью к стенке равен б,
пульса дается выражением
2тс cos 0.
то изменение им-
B2.1)
ТГ7777ТП7777777777777777т
Фиг. 23. К вычислению кинети-^
ческого давления.
Множитель 2 появляется здесь вследствие отдачи,
которую испытывает стенка при отражении (угол отражения
равен углу падения, абсолютное значение скорости | v | = с
сохраняется благодаря абсолютно упругому характеру
столкновения). Если разложить вектор v на декартовы
составляющие f, т], Си счи-
тать, что положительная
полуось направлена
перпендикулярно к стенке
наружу, то вместо
выражения B2.1) можно также
написать
2mf, S>0. B2.1а)
Чтобы сложить все толчки
"такого рода, построим над
рассматриваемым
элементом поверхности do косой
цилиндр (фиг. 23), образующая которого наклонена под
углом О к нормали и имеет длину с. Внутри этого цилиндра
находятся все те молекулы, которые имеют скорости между
с и c-J-^c и направления движения в интервале углов
от 6 до 6 + cf0 и от :р до cp-f cf,p, где 9 и ср —углы
относительно нормали к поверхности. Концы векторов,
изображающих скорости всех этих молекул, достигают
данного элемента поверхности cfa. Следовательно, в единицу
времени с ним сталкиваются эти, и только эти, молекулы.
Объем цилиндра равен произведению площади на
высоту, т. е. dB\. Если обозначит» через п число молекул
в единице объема, то в нашем цилиндре будет
nldfs B2.2)
молекул. Отберем отсюда только сталкивающиеся со
стенкой молску^лы, которые одновременно находятся в
определенной области cfu) пространства скоростей. До сих пор
эта область характеризовалась дифференциалами rfc, db и
dcp. Таким образом, пространство скоростей описывалось
§ 22, Уравнение состояния идеального гааа 215
В полярных координатах, что с точки зрения
координатного пространства представлялось наиболее естественным.
Однако можно также (и для дальнейшего это будет
удобнее) отнести с?а) к прямоугольным координатам, т. е.
положить cfo) = rfS с?тг] cfC.
Во всяком случае независимо от выбора системы
координат число сталкивающихся со стенкой молекул дается
выражением
dv = vUat/a), B2.2а)
где V в отличие от п означает плотность молекул на
единицу объема и единицу пространства скоростей с/о).
Очевидно, что
»ч
vt/o).
Умножая B2.2а) на B2.1а), получаем выражение для
(Соответствующего изменения импульса
2vm?2dac/a). B2.26)
Поэтому вклад этой группы столкновений в давление
составляет [надо разделить B2.26) на do]
dp = 2vm?2^a), J>0. B2.2г)
Отсюда, произведя аналогичное построение для всех
возможных областей пространства скоростей, получим для
полного давления
p^nmW, ^2 = -^^ v^2^a), S>0. B2.3)
Величина t^ возникает в результате усреднения по
полупространству скоростей S > 0. Однако, так как каждой
молекуле, падающей на стенку (S > 0), соответствует
с равной вероятностью молекула, летящая от стенки
(? < 0), то интегрирование в B2.3) можно распространить
на все пространство скоростей, если вместо B2.3) написать
р = птТ^ ^2"= ~ [ v?2 с/о), S ^ 0. B2,3а)
Здесь ?2 - среднее значение 1^ в некоторой определенной
точке. Важное не зависящее от выбора системы коорди-
216 Г л. III. Элементарная кинетическая теория вааов
лат соотношение можно получить, приняв во внимание
эквивалентность всех направлений (изотропию
пространства скоростей). Именно,
^=^ = ^2"=--? B2.36)
(последнее равенство здесь обусловлено тем, что ?;2 = 52-|-
+ 7J-f С* = с*). Таким образом, из B2.3) получаем
Р^-'^-^-^пЕ,,, :Etr=y^, B2.4)
тде Ех,т — кинетическая энергия поступательного движения.
Энергия, связанная с вращением или с другими
возможными внутренними движениями при вычислении
давления не играет роли.
Уравнение B2.4) содержит не только кинетическое
объяснение давления, но также и кинетическое
определение температуры. Чтобы убедиться в этом, положим
л=-^, B2.4а)
где N — полное число молекул в объеме V. Тогда из
B2.4) получаем
pV^^NE,,. B2.46)
Если применить уравнение B2.46) к специальному
случаю одного моля, то F = ^мол. и N = L равно числу
молекул в одном моле (числу Авогадро). Равенство B2.46)
при этом принимает йид
pV,,o^=jLEu. B2.4в)
Если теперь принять во внимание уравнение состояния
идеального газа в форме C.11) и C.11а), то правая часть
B2.46) должна быть равна ЯТ. Следовательно, получаем
Str = |-5^ = |-*r, B2.5)
где к обозначает введенную уже в § 20 постоянную Больц-
мана, т. е. отнесенную к одной молекуле газовую
постоянную R/L. Поступательному движению б трехмерном
пространстве соответствугот три степени свободы. Поэтому
§ 22, Уравнение состояния идеального газа 217
содержание уравнения B2.5) (при замене нижнего индекса
tr на fr) можно также выразить следующим образом:
средняя кинетическая энергия на одну степень свободы равна
Etr:={kT. B2.6)
Это утверждение и составляет (предварительное)
кинетическое определение температуры.
Далее, в уравнении B2.5) уже заключена теория
удельных теплоемкостей одноатомного газа. Так как
энергия последнего зависит только от температуры, то
уравнение B2.5) для величин и и с^., рассчитанных на один
моль, дает
и = LEi^^-^RT, c„ = ^ = y/?<^3 ккал/кмоль^). B2.6а)
В силу равенства c^ — c^ = R отсюда можно найти и с^,
а также отношение
с
Р __
2
= 14 4= 1,66. B2.66)
Тем самым мы подтвердили то, что уже говорилось в § 4,
п. 3: кинетическая теория в состоянии заполнить общие
рамки термодинамики конкретными численными
значениями, согласующимися с опытом. К приведенным там
сведениям относительно многоатомных газов мы вернемся
в гл. IV.
Выше были выведены средние квадратичные величины
?^, с^. Среднее значение f, конечно, равно нулю, так как
в силу изотропии пространства скоростей положительцая
полуось ? статистически не отличается от отрицательнбй
или какой-либо другой полуоси. О среднем значении с,
которое, конечно, не равно нулю, будет идти речь
в § 23, п. 2. Непосредственно из опыта определяется
средняя квадратичная скорость
{?f^'\ B2.7)
Вычислим ее, например, для газов Н^, Не, О2, Ng. Пока
газ остается идеальным, она зависит только от темпера-
^) 1 клюль = 1000 моль=1 килограммолекулярный вес.
218 Гл, III. Элементарная кинетическая теория еазов
туры и не зависит от давления. Из соотношения B2.5)
вытекает, если обозначить через т массу молекулы, что
или, умножая на число молекул в граммолекуле L,
Величина Lm представляет собой вес одного моля р., так
что средний квадрат скорости молекулы с^ = 3RT/\i
вычисляется исключительно из макроскопических данных. Для
юдорода, например, р. = 2 кг/кмолъ. При 7* = 273°К и
значении R из C.9) получаем
F2 = -|.8,31.273.103 л«2.се«-2,
откуда
1/Р=1,85 кМ'Сек'К B2.8)
Для гелия, представляющего собой одноатомный газ
с атомным весом 4 (т. е. вдвое больше молекулярного
веса Hg), соответственно получаем
Ус^ = —-— км.сек'^ = 1,30 км-сек'^.
/2
Молекулярный вес кислорода в 16 раз больше
молекулярного веса водорода. Поэтому скорость B2.8) следует
разделить на |/16 = 4; аналогично для азота ее надо
разделить на |/14 = 3,74.
Эти скорости уже при 0°С удивительно высоки. Они
еще несколько возрастают с увеличением температуры,
согласно закону A -\- //273)^/2^ где t - температура Цельсия.
Известное объяснение такая высокая скорость все же
находит в том, что скорость звука, которая не может
быть больше, чем скорость молекул, участвующих в
распространении звука, имеет тот же самый порядок вели-
ЧИНЫ, а именно с^ = — [ср. т. П, уравнение A3.17а)].
/ г*
Аналогично обстоит дело при истечении сжатого газа
из сопла.
§ 23. Распределение Максвелла 219
В атмосфере средние скорости молекул газов Og, Ng
и т. д. различны у так же как и в любой другой смеси
газов. При тепловом равновесии, согласно нашему
определению температуры, средние энергии поступательного
дви:нсения для всех газов одинаковы. Полная энергия
поступательного движения распределяется в среднем
равными долями по различным составным частям смеси
газов в соответствии с их относительными количествами.
Это простейший пример общего закона о равномерном
распределении энергии.
Здесь говорилось только о давлении на стенку. Однако
все результаты переносятся также на давление внутри
газа, если представить себе, что в какую-либо его точку
внесена небольшая мембрана, являющаяся измерителем
давления. При этом давление внутри газа (так же как
и давление на стенки) оказывается не зависящим от
координат (см., однако, в связи с этим § 26). Это результат
того, что мы не учитывали внешних сил (например, силы
тяжести). Каким образом они входят в статистическую
теорию газов, будет показано в § 23, п. 3.
§ 23. РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА
Выше говорилось о координатном пространстве и
пространстве скоростей. Мы считали, например, в равенстве
B2.4а), что координатное пространство равномерно
заполнено молекулами (это, повидимому, подтверждается
макроскопическими наблюдениями), причем неявно
предполагалось, что на молекулы не действуют никаг^ие
внешние силы. Обратимся теперь к пространству скоростей,
в котором до сих пор нам были известны только средние
значения i?, .. ., с^.
1. Распределение Максвелла в случае одноатомного
газа. Вывод 1860 г. Компоненты скорости какой-либо
(произвольно выбранной) молекулы газа имеют некоторые
значения ?, т], С. Можно ввести представление .о
вероятности того, что первая компонента лежит в области между
? и ? -h с?$ (т. е. в плоскопараллельном слое пространства
скоростей, перпендикулярном к оси ?). Эта вероятность
обозначается через
220 Г л III, Элементарная кинетическая теория газов
То же можно написать и для компонент т) и С, причем
в силу изотропии пространства «функция вероятности»
для них будет той же самой. Однако отнюдь не очевидно,
что математическое ожидание, например, величины т] не
зависит от значения f, коль скоро последнее уже найдено.
В лотерее, как известно, это имеет место: если я в этом
году получил главный выигрыш, то имеется вероятность,
что я получу его и в следующем году, и притом точно
такая же, как в предыдущем году. Максвелл сначала
просто предположил, что такая независимость вероятностей
имеет мбсто и в теории газов; в дальнейшем же ему
удалось доказать это утверждение (ср. п. 3 и гл. V).
Скорость V, составленная из ?, т], С, принадлежит
элементу объема cfJcf^rfC пространства скоростей (этот
элемент образуется пересечением трех
плоскопараллельных слоев, перпендикулярных к осям 5, if], С);
соответственно вероятность того, что койец вектора v находится
в этом элементе, благодаря предположению о назависи-
мости вероятностей равна
f{^)f{^)fi(,)dkdrid(.. B3.1)
Так как в силу изотропии все направления скорости
равновероятны, то для вероятности B3.1) можно ввести
новую неизвестную функцию F, зависящую только от
абсолютной величины скорости, а именно:
F{c)d^, d(u = did'ndL B3.1а)
Сравнение B3.1) и B3.1а) дает
F(/F+?TC^)-=/(S)/(t))/(C). B3.2)
Чтобы из этого функционального уравнения
определить функции F и f, поступаем (довольно формально)
следующим образом:
а) Возьмем логарифмическую производную от B3.2)
по S
с F(c) - /(e) • V^^-^>
б) 1^ведем обозначения
л./ \ 1 ^' (с) п\ 1 /' E) /оо о ч
S 23. Распределение Максвелла 221
тогда B3.3) примет вид
Ф(с) = срE). B3.36)
в) Дифференцируя B3.36) по т] или С, получаем
Ф' (с) = О, Ф (с) = const. B3.3в)
Если положить ^) const = —2-1', то, согласно B3.36),
ср($)==-2т.
Поэтому в силу B3.3а)
^^)=--2т5. ln/(S) = a--fS2 B3.3Г)
И, следовательно, обозначая е""" через а, имеем
f{l)^ae-r^\ B3.4)
Замечательно и достойно внимания, что здесь появляется
нормальная форма всех статистических распределений —
кривая ошибок Гаусса. Наиболее вероятным значением
составляющей скорости I является значение ? = 0. Вокруг
этого значения симметрично с обеих сторон группируются
отклонения в виде колоколообразной кривой.
Определим значение постоянной а в,B3.4) из условия,
что ? с достоверностью (т. е. с вероятностью 1) должна
иметь какое-нибудь значение между — оо и + оо:
+ 00
5/(S)rfl==l. B3.5)
—оо
Отсюда, пользуясь известным выражением для интеграла
Лапласа, получаем
a'\e-r^-d\=^a(^^y' = i, а={^у\ Bа.5а)
— 00
с другой стороны, чтобы определить т» вычислим среднюю
кинетическую энергию, приходящуюся на одну степень
1) Знак минус необходим хотя бы для того, чтобы
выполнялось равенство B3.5) Множитель 2 присоединен для удобства. .
22i Гл. Ill, Элементарная кинетическая теория еазоё
свободы (на компоненту $):
т="'-т(^)"'Т'*'-"«--1Ш"'|Т^-'"«-
—с© —оо
_ т / t \^l2 d / п '\Ч2 ___ т
Если, согласно B2.6), приравнять ее А Г/2, то получим
Т = ^. B3.6)
На основании B3.5а) и B3.6) выражение B3.4)
принимает следующий окончательный вид:
/(^) = B5гТ'^"^'""'' ^1 = Т^'- B3-7)
Величина -Б^ обозначает здесь кинетическую энергию,
соответствующую составляющей 5. Такие же выражения
справедливы и для компонент т] и С Отсюда на
основании уравнения B3.2) сразу получаем функцию
распределения
где Е обозначает теперь кинетическую энергию
поступательного движения, направление которого может быть
любым (оно определяется значениями компонент 5, 7]иС).
2. Вычисления и эксперимент. Будем интересоваться
сейчас не самой скоростью v, а только распределением
по абсолютным значениям скоростей (они, как обычно,
обозначаются буквой с). Рассмотрим сферу, описанную
около начала координат, с внутренним и внешним
радиусами с и c + dc соответственно. В этой сфере содержится
некоторое конечное число частиц, пропорциональное dc.
Математическое ожидание их обозначим через
(f{c)dc.
Оно равно объему сферы 4тсс^б?с, умноженному на
функцию F{c) B3.8). Таким образом, получим
§ 23. Распределение Мйксвеллй
223
Отсюда следует, что закон распределения уже не будет
изображаться симметрично!! колоколообразной кривой.
Ф и г. 24. Распределение Максвелла.
Действительно, кривая, показанная на фиг. 24, не
симметрична относительно максимума вероятности в
противоположность гауссовой кривой (фиг. 24а). Как и эта
последняя, кривая B3.9) экспоненциально стремится к нулю
Фиг. 24а. Распределение Гаусса.
при больших с; однако при малых с (т. е. при с—>0)оиа
спадает лишь параболически (при с < О функция ср,
естественно, не определена).
224 Гл. Ill, Элементарная кинетическая теория газов
В точке, где ср' (с) = 0, кривая B3.9) имеет максимум.
Соответствующее значение с = с^^^ представляет собой
наиболее вероятную скорбеть. Для нее мы получаем
выражение
Она отличается как от средней квадратичной скорости
так и от среднего значения
У7\
с= \c<f{c)dc. B3.10а)
о
Как будет показано в задаче И 1.2, отношения этих трех
величин равны
с^: с :'К^= 1:1,13:1,22. B3.11)
Качественным подтверждением распределения
Максвелла является наблюдаемое у испускающих свет газов
увеличение ширины спектральных линий с повышением
температуры. Оно обусловлено эффектом Допплера. Пусть
\ — собственная частота излучающей частицы (атома или
молекулы) и Xo = cl/vo (где cl —скорость света); тогда
наблюдатель, производящий измерения в направлении
оси Ху воспримет свет с длиной волны Х^ от тех частиц,
у которых составляющая скорости S почти равна нулю;
в общем же случае длина волны будет равна X^-f^^^-
Здесь, согласно т. IV (Оптика) §11 (отвлекаясь от
релятивистских поправок), M/\q = ^/cl и, следовательно,
АХ = 1. B3.12)
Все частицы с одинаковыми значениями S дают вклад
в плотность интенсивности на спектрограмме в точках
с положительными или отрицательными смещениями ДХ
относительно ее середины. Число их, следовательно,
определяется функцией / (S) [выражение B3.4)]. Так как можно
предположить, что все частицы возбуждены в одинаковой
степени и так как в силу некогерентности излучения
складываются интенсивности, а не амплитуды, то наблю-
§ 23. Распределение Максвелла 225
даемая интенсивность- будет также прямо
пропорциональна / (S), где ?, согласно B3.12), следует приравнять \^\.
Поэтому из B3.4) получаем
/ = /^e-Y(VoAAJ. B3.13)
Здесь Jq — плотность интенсивности в середине
спектрограммы, а 7, согласно B3.6), обратно пропорциональна
температуре Т и определяет ширину спектральной линии.
Полуширина линии характеризуется значением аргумента ^
при котором J=rJ^l2, и, следовательно, равна
Контур спектральной линии, соответствующий
выражению B3.13), непосредственно изображает кривую ошибок
Гаусса, а следовательно, и закон распределения Максвелл?.
Первые измерения формы и полуширины линий
испускания в зависимости от температуры и атомного веса т
были выполнены Майкельсоном^). Для астрофизики
фундаментальное значение имеет форма фраунгоферовых линий
поглощения. Наряду с эффектом Допплера здесь
учитывается также уширение, обусловленное давлением
(ударное гашение), в то время как естественная ширина линии
(см. т. П12), § 36) отходит на задний план.
Прямую проверку распределения Максвелла впервые
удалось произвести Отто Штерну ^) при помощи его метода
атомных пучков.
3. Общие соображения о распределении по энергиям.
•Множитель Больцмана. До сих пор мы основывались на
первоначальном несколько примитивном доказательстве
Максвелла. В гл. IV предположение о независимости
вероятностей будет доказано для объектов классической
механики при помощи более общего и в основе своей
более простого «метода ячеек», который для одгоатомного
газа, находящегося вне силового поля, приводит также
к распределению Максвелла. Ограничение случаем
одноатомного газа нашло свое выражение в заглавии п. 1 этого
1) Michel son, Phil. Mag., 34, 280 A892).
*) A. Sommerfeld, Elektrodynamik, Leipzig, 1949,
3) 0. Stern, Zs. f. Phys., 3, 417 A920).
226 r.t. ///. Элементарная кинетическая теория газов
параграфа. Условие, что газ находится вне силового поля,
содержится, например, в уравнении B3.4а), где
предполагается, что плотность частиц одинакова во всем
координатном пространстве. Очевидно, что это условие не
выполняется, если газ находится, например, в поле силы тяжести.
Дадим теперь, предворяя результаты гл. TV,
обобщение для случая многоатомного газа, который, кроме того,
может находиться во внешнем силовом поле с потенциалом Ф.
рудем исходить из формулы B3.8), в которой, однако,
энергию поступательного движения Е следует заменить
полной энергией частиц
e = ?tr + ^rot-h...+*. B3.14)
Следовательно, к энергии поступательного движения
(единственной, имеющейся в случае одноатомного газа)
добавляется еще вращательная энергия -Erot и обозначенная
многоточием энергия возможного внутреннего движения
частиц (колебательная энергия и т. п.), а также
потенциальная энергия Ф в силовом поле. Далее, сделаем еще
одно обобщение, введя вместо интервала пространства
скоростей cfo) = cfS с?7] cfC интервал «фазового пространства»
(определяемого ниже) с?2. Тогда вместо B3.8) получим
FdQ^AQ-^l^T^dQ. B3Л5)
Введенная здесь постоянная А определяется, как и
постоянная а ь формуле B3.4), из условия нормировки
^FdQ^i. B3.15а)
Относительно значения dQ заметим только, что в случае
одноатомного газа cfQ = б?х. m^cfo). Здесь dx = dxdydz^
элемент объема в координатном пространстве; множитель
т^ обусловлен тем, что в фазовом пространстве вместо
скоростей ?, т], С фигурируют импульсы т?, /пт], тС. Нас
сейчас интересует содержащийся в уравнении B3.15)
множитель
е-*/^^ B3.16)
(множитель Больцмана). Так как предполагалось, что от
координат X, у у z зависит; только Ф, то множитель B3.16)
дает верЬятность того, что частица газа окажется в еди-
§ 2d. Броуновское двион:ение 227
нице объема с координатами х, г/, z. Следовательно» он
позволяет вычислить пространственную плотность частиц р.
Если ро — плотность для нулевого уровня потенциала,
то в общем случае
А = 6--*/'^^. B3.17)
В простейшем случае поля тяжести Ф = mgz и B3.17)
переходит в барометрическую формулу [ср. т. II,
Механика деформируемых сред, § 7, уравнения G.15а), G.15в)].
Модельные опыты Перрена (см. там же) можно
рассматривать как макроскопическую проверку правильности
формулы B3.17).
§ 24. БРОУНОВСКОЕ ДВИЖЕНИЕ
Ботаником Робертом Броуном в 1826 г. было описано
явление, состоящее в том, что мельчайшие частицы
(частицы пыли, коллоидные частицы и т. д.), взвешенные в
жидкости или газе и рассматриваемые под микроскопом,
обнаруживают беспорядочное движение. Это долго
оставалось загадкой. Окончательно.е объяснение было дано
Эйнштейном в 1905 г. Однако в 1906 г. Рентген, критически
относившийся к этпм опытам, пытался рядом контрольных
экспериментов выяснить, не вызывается ли это движение
энергией света, сообщаемой частицам при освещении их под
микроскопом.
Броуновское движение принадлежит к общему кругу
флуктуационных явлений, т. е.,отклонений от
термодинамического равновесия, В соответствии с элементарным
характером этой части книги мы ограничимся здесь лишь
частным рассмотрением вопроса, принадлежащим Ланже-
вену^), из которого весьма наглядно получается результат
Эйнштейна.
В т. IV (Оптика) § 33 была доказана следующая
теорема: если на плоскости образовать сумму большого числа
N единичных векторов, ориентированных вполне
беспорядочным образом, то результирующий отрезок будет равен
УП, в данном случае мы имеем дело с ударами, которые
испытывают коллоидные частицы вследствие теплового
1) Langevin, Compt. rend., 530 A908).
228 Гл. У//. Элементарная кинетическая теория Зазде
движения в окружающей среде. Полная равновероятность
их направлений является статистически гарантированной.
Число их пропорционально времени наблюдения t. Пути,
проходимые частицами между двумя ударами
(подразумеваются проектирующиеся на поле зрения микроскопа
отдельные отрезки пути г, из которых составляется полный
зигзагообразный путь), не являются, конечно, единичными
векторами, а представляют собой небольшие отрезки, длины
которых колеблются около некоторого среднего значения
и зависят от свойств окружающей среды и сталкивающихся
частиц. Результирующее смещение частицы при движении
ее по зигзагообразной траектории определяется формулой
C3.4) т. IV (Оптика). Заменяя принятое там обозначение
iS на г и обозначая единичный вектор через г^, получаем
r^=^^7i = '?iN = Pt. B4.1)
Чтобы найти введенный здесь коэффициент
пропорциональности Р, надо воспользоваться уравнением движения
частицы
Д/*г = К@-Сг, B4.2)
где Л/—масса коллоидной частицы, г — радиус-вектор ее
центра тяжести относительно некоторой фиксированной
начальной точки, R (/) — случайная (по величине и
направлению) сила, характеризующая удары, испытываемые
частицей; последний член в правой части представляет собой
силу трения, которая, согласно Стоксу, принимается
пропорциональной скорости г (таким образом, окружающая
среда рассматривается как континуум). Очевидно, это
допустимо только в том случае, если частица весьма
велика по сравнению с молекулярной структурой
окружающей среды (это предположение законно). . Принимая
частицу за шарик радиуса а и обозначая коэффициент
вязкости окружающей среды через т], получаем на
основании формулы C5.20) т. II (Механика деформируемых
сред)
С:=&ща. B4.2а)
Умножая уравнение B4.2) скалярно на г, получаем
M(r;r) = (r,R)^C(r,f), B4.3)
^ 24, Броуновское двьинсение 229
где
Величина (r,R) в механике точки называется «вириалом
силы R». Полезность его для кинетической теории газов
была известна уже Клаузиусу. Совершим обычное для
теоремы вириала элементарное преобразование
•• d • * * i d^
при этом уравнение B4.3) принимает вид
Проинтегрируем это уравнение по времени от О до /
и затем поделим почленно на /. В течение времени t
величина (г,Н), т. е. проекция быстро изменяющейся силы
удара на направление г, очень часто меняет знак. Можно
ожидать, что для больших значений t [больших по
сравнению с интервалом, в котором происходит изменение
знака (r,R)], мы получим
t
|^(r,R)c/i = 0. B4.4а)
о
Тогда проинтегрированное уравнение B4.4) при Го = 0
(начальное положение частицы) имеет вид
М dr^
2t
Х-+1'^=И^^'^'- B4.5)
В правой части уравнения B4.5) стоит усредненная по
времени удвоенная кинетическая энергия частицы.
Согласно закону равномерного распределения по степеням
свободы, среднее значение кинетической энергии, взятое
по большому количеству частиц, равно кТ (соответственно
двум степеням свободы плоского движения; для
одномерного движении следовало бы написать кТ 12), При этом
можно сослаться на наш результат, полученный при
рассмотрении смеси газов (§ 23, п. 2): средние - скорости
230 Гл, III. Элементарная кинетическая теория газов
коллоидных частиц вследствие их большой массы много
меньше, чем скорости молекул окружающей среды, однако
средние кинетические энергии и тех и других одинаковы
и поэтому выражаются указанным образом через
абсолютную температуру окружающей среды. Обозначая «среднее
по частицам» горизонтальной чертой сверху (т. е. переходя
от рассмотрения одной отдельной частицы к совокупности
их), получаем
t
'^-V^t = kT. B4.5а)
о
1 Г il/ -
Как мы убедимся ниже, первый член слева в уравнении
B4.5) экспоненциально убывает со временем i, поэтому
при предварительном рассмотрении мы опустим его. Тогда
из B4.5), переходя в левой части также к среднему по
частицам и учитывая уравнения B4.2а) и B4.5а), находим
•^=§^J- B4-6)
В смысле зависимости от времени эта формула содержит
прежнее уравненпе B4.1), причем здесь явно определен
фигурирующий там коэффициент пропорциональности Р.
Если наблюдаются лишь смещения в одном измерении,
например в направлении оси х, то вместо формулы B4.6),
согласно закону равномерного распределения по степеням
свободы, получаем формулу
J^^4^t. B4.6а)
Это формула Эйнштейна^ которая была подтверждена
разнообразнейшими экспериментами и использовалась,
например, для определения постоянной Больцмана к или
числа Авогадро L^Rjk,
Дополним теперь наш вывод строгим интегрированием
уравнения B4.5), в котором уже до интегрирования
перейдем к среднему по частицам. Интегрируя B4.4) один раз,
находим Aюлагая г^ = м)
§ 24. Броуновское двиоюение 231
Решение соответствующего однородного уравнения имеет вид
Ui = Ae-^^l^^, B4.7а)
а в качестве частного решения неоднородного уравнения
легко находим
«^ = ^(^-^)- B4.76)
Но Л/ = D7:/3) ра^, где р — плотность частиц; поэтому,
согласно B4.2а),
С "¦ 9 Y) •
Полагая а = 10^ еле (предел видимости), у]^ 10"^ г/см» сек
(вода) и р «^ 1 г-слГ^ (частица плавает в воде), получаем
^=^.10-6 сек. B4.7в)
Следовательно, член М/С в скобках в выражении B4.76)
дает лишь неизмеримо малое смещение нуля шкалы
времени, а множитель С/М в экспоненте B4.7а) приводит
к чрезвычайно быстрому уменьшению любого имевшегося
вначале возмущения А. Поэтому наше решение м = м^ + Mg
упрощается и принимает вид
к j^ Kg ^ ~7^ ^>
что согласуется с формулой B4.6).
Конечно, наблюдения над отдельными частицами
приводят к значительным отклонениям от средних значений
B4.6) или B4.6а); как можно показать, эти отклонения
распределены по закону ошибок Гаусса. Следовательно,
для экспериментальной проверки формул для средних
значений необходимо произвести много отдельных
наблюдений.
Чрезвычайно поучительный вариант броуновского
движения представляет поведение крутильных микровесов.
Их исследование, начатое еще Смолуховским, Капплер^)
1) Е. Kappler, Ann. d. Phys., 11, 233A931), ср. Natttrwws.,
27, 649, 666 A939).
232 Гл, III, Элементарная кинетическая теория газов
усовершенствовал до такой степенц, что смог, определить
число Лошмидта с точностью до 1%.
В то время как при броуновском движении имеют дело
со средним значением квадрата пути (г^ или ж^), в случае
крутильных весов определяют среднее значение квадрата
угла отклонения ср^.
Условия опыта обычно следующие. На кварцевой нити
длиной в несколько сантиметров и толщиной в несколько
десятых микрона висит тонкое зеркальце площадью около
1 мм^. Повороты зеркальца, обусловленные действием
ударяющихся о него молекул воздуха, благодаря
отражению света регистрируются на фотографической пленке.
Для успеха опыта необходимо отсутствие сотрясений
и постоянство температуры. Коэффициент упругости
кварцевой нити D вычисляется известным способом путем
исследования свободных колебаний дополнительно
нагруженной системы. Во избежание радиометрического эффекта
давление воздуха рекомендуется поддерживать или очень
низким (например, 0,01 мм рт. ст.), или достаточно
высоким (например, 1 атм). Продолжительность регистрации
составляла для одной пленки примерно 10 часов.
Для теоретической обработки результатов исследования
этого флуктуационного явления следует принять во
внимание, что зеркало обладает не только кинетической энергией
уср2 (/ —момент инерции 1)), B4.8а)
но и потенциальной энергией
-^-ср^ (D — коэффициент упругости). B4.86)
Так как в среднем по времени кинетическая и
потенциальная энергии равны друг другу, то каждая из них вносит
в статистическое среднее энергию кТ/2 на одну степень
свободы. Поэтому, усредняя B4.8а) и B4.86), получаем
?^ = ^, B4.9а)
^ = ^. B4.96)
^) Ср. § 31, замечание к уравнению C1.7).
§ 24, Броуновское двиинсение 233
Статистические отклонения от этих средних значений
регистрируются по схеме Капплера. Их распределение имеет
•
характер строго гауссовых кривых. Распределение ср^ около
среднего значения B4.9а) можно назвать «распределением
Максвелла» для угловой скорости.
Так как равенство R = Lk позволяет при известном к
вычислить число Авогадро, то метод Капплера дает
элементарный и достаточно точный способ определения L.
При броуновском движении свободных взвешенных
частиц соотношение, аналогичное B4.96), естественно
отпадает, так как для этих частиц нет какого-либо
специального положения равновесия. Соотношение B4.9а)
заменяется прежним соотношением B4.5а). Далее, как
было показано еще Перреном, для среднего угла
поворота броуновских частиц (измеряемого относительно
некоторой неподвижной оси) также справедливо соотношение,
подобное формуле Эйнштейна B4.6а); при этом лишь
соответственно видоизменяется выражение для
коэффициента вязкости по сравнению со стоксовым значением
B4.2а) [см. т. II, Механика деформируемых сред, формула
C5.21)].
Для изучения динамической стороны поведения
крутильных весов изберем опять метод Ланжевена.
Уравнение движения в этом случае аналогично B4.2) и имеет
вид
h = mt)-C^-Db B4.10)
где 9Л (О ~ закручивающий момент, обусловленный
столкновениями молекул с зеркалом. Сер —момент, вызванный
стоксовой силой трения окружающего (возможно,
разреженного) воздуха, /)ср — закручивающий момент силы
упругости кварцевой нити [единственный член, не имеющий
себе аналога в уравнений B4.2)]. Умножая B4.10) на ср
и пользуясь теоремой вириала, получаем вместо B4.4)
(тЙ + Т-^)?'-^ + ^?^ = ?5Ш@- B4.11)
После интегрирования. по t и деления на t правая часть
этого уравнения исчезает. Средние значения по частицам
от двух последних членов слева, согласно B4.9а), B4.96),
234 Гл, III. Элементарная кинетическая теория газов
взаимно уничтожаются. Таким образом, уравнение B4.11)
принимает вид
]^^Си = 0, где и = ^^
(постоянная интегрирования равна нулю, так как при
усреднении моменты исчезают). Второе интегрирование
дает
гг = Мое-С'/Л B4.11а)
Уменьшение начального угла отклонения (или угловой
скорости), описываемое уравнением B4.11а), было изучено
Капплером. Уменьшая давление воздуха, можно сделать
константу трения С сколь угодно малой и, следовательно,
увеличить время затухания I/C до значений порядка
нескольких секунд. Именно поэтому в случае
крутильных весов можно экспериментально проверить также
и соотношение B4.9а).
Невозможность такой проверки в случае свободных
взвешенных частиц [соответствующем уравнению B4.5а)]
обусловлена вычисленным в B4.7в) порядком величины
М/Су одновременно характеризующей время, в течение
которого движение можно считать практически
прямолинейным (без значительных изменений направления).
Вследствие малости М/С непосредственно наблюдаемое
движение броуновских частиц фактически представляет
собой результат сильного усреднения.
Именно поэтому в очень тщaтew^ьныx измерениях
Франца Экснера A900 г.) были получены значения
скоростей, заниженные на несколько десятков процентов по
сравнению с B4.5а). Вероятно, по этой же причине
объяснение броуновского движения как результата
движения молекул смогло победить лишь после 1905 г., когда
Эйнштейн теоретически вычислил величину, доступную
эксперименту; таковой оказалось среднее значение квад-
рата смещения.
Возвращаясь опять к крутильным весам, сделаем
в заключение одно замечание общего характера: факт
существования теплового дви^нсения в случае крутильных
весов, допускающий количественное исследование и проверку у
ставит непреодолимую границу чувствительности всех
§ 25. Статистика парамагнитных веществ 235
измерительных инструментов (впервые это было
обнаружено Изингом в 1926 г. для высокочувствительного
гальванометра).
§ 25. СТАТИСТИКА ПАРАМАГНИТНЫХ ВЕЩЕСТВ
Распределение Больцмана B3.15) позволяет дать
простой статистический вывод введенной в § 19 функции
Ланжевена.
PaccMOTppiM парамагнетик, составленный из отдельных
элементарных магнитиков. Будем считать, что они
движутся вполне хаотично, независимо друг от друга и
способны свободно вращаться. Магнитные моменты их равны
m =: pin {р — магнитный заряд полюса, / — расстояние
между полюсами, п —единичный вектор). Такая модель
удовлетворительно описывает не только парамагнитные
газы (Og, N0 и т. д.) и жидкости, но также и твердые
соли, и притом не только в классическом (см. п. 1),
но и в квантовом (см. п. 2) случае. Согласно
классическим представлениям, все направления магнитного момента
априори считаются возможными и равновероятными.
В квантовом случае допустимы лишь некоторые
дискретные ориентации магнитного момента относительно
внешнего магнитного поля. В обоих случаях вероятность той
или иной отдельной ориентации определяется двумя
противодействующими факторами — магнитным полем и
тепловым возбуждением (т. е. температурой системы).
1. Классическая функция Ланжевена. Обозначим
через 6 угол между осью элементарного магнита* и
направлением внешнего поля; индукцию последнего
обозначим через В (в § 19 эта величина обозначалась через i^qH).
Вращательный момент, действующий на каждый магнит
со стороны поля, равен
D = mBsme.
Он вызывает уменьшение угла 6 и, следовательно, стремится
повернуть т параллельно В. Работа, которую надо
совершить для увеличения 6 против сил поля, составляет
t)dB = mB^indde^ -mBdco^O.
236 Гл, III. Элементарная кинетическая теория газов
На такую же величину изменяется потенциальная
энергия Ф элементарного магнитика при повороте его на угол
сЮ. Имеем, следовательно,
с1Ф = --mBd созв, Ф = -~тВ созв. B5.1)
С увеличением угла 6 величина Ф возрастает, подобно
тому как растет потенциальная энергия в поле силы
тяжести Ф = mgz при увеличении высоты подъема над
поверхностью земли z, В данном случав значение Ф
нормировано так, что его минимум Ф= —тВ соответствует
стабильному положению элементарного магнитика 6 = 0,
а максимум — нестабильному положению 6 = ir.
С классической точки зрения вероятности тех или иных
ориентации dW нашего элементарного магнитика априори
одинаковы для равных интервалов телесного угла cfo) =
= sin О dO cfcp. Согласно распределению Больцмана, они
оказываются различными —в той или иной степени —
в зависимости от температуры. Именно,
dPy = ^e-*/^^6fa). B5.2)
Здесь ясно видно противоположное влияние температуры
и поля. При высоких температурах все ориентации
приблизительно равновероятны, как это было ясно априори,
так как в этом случае е-*/^^ «^ 1. Для низ^шх температур
над всеми прочими возможностями преобладают
ориентации вблизи стабильного положения 9 = 0, Ф = Фмин.-
Фигурирующий в формуле B5.2) множитель А удобнее
всего определить из того условия, что элементарный
магнитик с достоверностью имеет какую-либо ориентацию,
т. е. вероятность этого события равна единице. Это
означает, что
2л It
^dW=l^A[d^f\ е-*/'^^8швй9.
и, следовательно,
те
-i- = 2ic с е-*/''Т sin б d0. B5.2а)
$ 2S. Статистики парамагниУпных вегцедшв 23?
Подстановка в B5.2) дает после интегрирования dW по ср
^^=;Т1т"''' ' B5.3)
Вычислим теперь среднее значение компоненты m
в направлении поля. Обозначив ее через т, получим
т
= \ mco^bdW. B5.4)
[Сюда надо подставить значение dW 1лг B5.3).]
В случае большого числа элементарных л1агнитиков
важна только эта компонента и ее вероятное значение,
так как компоненты, перпендикулярные к полю, имеют
всевозможные азимуты ср и поэтому взаимно
компенсируются. Умножая среднее значение т на число
элементарных магнитиков в одном моле л, находим величину Л/,
которая в § 19 была названа намагниченностью,
М = пт\
с другой стороны, величина Л/о© = пт обозначает
намагниченность насыщения (в достаточно сильных полях все
элементарные магнитики ориентируются одинаково). Вводя
обозначения
ж = cose, а = ^^-, B5.5)
в силу сказанного получаем из B5.4)
+ 1
~ J e'^dx
-1
Интегрирование в знаменателе дает
i-(e«- e-«) = -|-sha.
B5.6)
238 Гл. III. Элементарная кинетическая тедрия газов
Числитель является частной производной знаменателя по
а и, следовательно, равен
2 2
— cha ;-sha.
Поэтому из B5.6) получаем
М _ cha
B5.7)
Правая часть представляет собой функцию Ланжевена
из уравнения A9.56). Наше определение а в B5.5)
совпадает, как это непосредственно видно, с прежним
определением в A9.5а). Вывод уравнения B5.7) восполняет
допущенный в § 19, п. 2 пробел, и классическая теория
парамагнетизма (равно как и связанная с ней теория
ферромагнетизма Вейсса) становится законченной.
2. Функция Ланжевена, видоизмененная согласно
квантовой теории. С точки зрения квантовой теории
возможные ориентации элементарного магнитика не образуют
континуума — 1 < cos 9 < + 1, а ограничиваются
определенными дискретными значениями cos 9 (<шростран-
ственное квантование»). Их число определяется из
характера спектра атома (или молекулы) в основном
состоянии. Оно равно 2, 3, 4, ... в зависимости от того,
принадлежит ли основное состояние к дублетной, триплет-
ной, квадрупольной и т. д. системам. (Если основное
состояние представляет собой сингулет, то магнитный
момент в нем отсутствует] в этом случае атом не
является элементарным магнитиком.)
Подробное рассмотрение показывает, что
пространственное квантование приводит к следующему правилу,
которое здесь, конечно, не может быть обосновано. Пусть
г —число состояний, соответствующих терму системы (г = 2
для дублета, г = 3 для триплета и т. д.); полагают
г = 2/+1 (/ называется «внутренним квантовым числом»)
и cos6 = 5// (следовательно, |5|</, так как |cos6|<l).
Тогда допустимы только такие значения 5, которые
отличаются друг от друга на единицу и, таким образом,
§ 25. Статистика парамагнитных вещесШв 239
даются следующей таблицей:
г 2 3 4 5
S ± V2 ± 1; о ± Vz; ± V2 ±2; ± i; о
Следовательно, для дублетной системы возможны лишь
две ориентации (cos6= ± 1)—параллельно и антипарал-
лельно полю. В случае триплетной системы к этим двум
добавляется еще ориентация перпендикулярно к полю,
cos 9 = 0. При квадрупольной конфигурации существует
четыре ориентации: cos9=± 1, cos9= ± g- и т. д. Эти
ориентации априори являются равновероятными. При
вычислении среднего значения т компоненты магнитного
момента m в направлении поля следует опять учесть
множитель Больцмана. Вместо интеграла в уравнении B5.4)
теперь появляется сумма, вообще говоря, г членов. В
обозначениях B5.5) имеем, в частности, при г = 2
= tha, B5.8)
при г = 3
при г = 4
jn _ б''—g"*' _ 2shg
т "~ g« ^ 1 ^ е~* "" 1+ 2 oh а *
B5.8а)
B5.86)
Таким образом, функцию Ланжевена в уравнении B5.7)
следует в соответствии со спектроскопической структурой
состояния атома заменить правой частью одного из
предшествующих равенств. Классическая функция Ланжевена
получается только в пределе при г —» оо. В дальнейшем
будем обозначать ее через Loo (а), а указанные выше
видоизмененные формы функции Ланжевена соответственно
через L^{o), L^{(x) и т. д. Функция L^ была введена уже
240 Гл. III. Элементарна,ч кинетическая теория газов
В 1920 г. В. Ленцем^). Сравним ее с классической функ*
цией Ланжевена:
Кривые для Lg, L4, ... располагаются по порядку между
двумя границами, описываемыми уравнениями B5.9).
Это сказывается, в частности, на угле наклона
касательной в нулевой точке. Согласно B5.9), имеем [ср.
также A9.6)]
l;@) = i, l;,@) = |,
в то время как из B5.8а), B5.86) получаем
^;@)=|-. l;@)=|....
Следовательно, указанный наклон является наибольшим
для Z^2 ^ постепенно уменьшается до ^з ^ случае Loc.
Так как температура Кюри 9, согласно A9.11а),
зависит от L' @), то ее квантовотеоретическое значение
также изменяется по сравнению с классической
величиной. То же самое справедливо И71;ля постоянной Кюри С,
классическое значение которой дано в A9.7а). Например,
в случае дублета выражение A9.7а) заменяется следующим:
Что касается других изменений, вносимых квантовой
теорией в классические результаты, то они указаны в конце
§ 19, п. 3 и на фиг. 19. Не входя в детали, заметим
только, что теорий пара- и ферромагнетизма в
соответствии с их статистическим происхождением часто
обнаруживают различие между классическими и квантовыми
результатами.
§ 26. СТАТИСТИЧЕСКИЙ СМЫСЛ ПОСТОЯННЫХ
В УРАВНЕНИИ ВАН-ДЕР-ВААЛЬС А
Постоянные Ван-дер-Ваальса а и fc были введены в § 9
феноменологическим путем; мы ограничились лишь беглым
указанием на их физический смысл. Сам Ван-дер-Ваальс
i) W. Lenz, Phys-. Zs., 21, 613 A920).
§ 26, Статистический смысл постоянных в у р. Ван-дер-Ваальса 241
В своей диссертации дал им подробное статистическое
обоснование, а Больцман ^) смог это обоснование
упростить. Мы следуем здесь (в п. 1) изложению Ф. Зау-
тера 2), представляющему собой новое оформление в
сущности того же метода Больцмана. Подчеркнутое Заутером
обстоятельство, что при этом необходимы лишь
простейшие сведения из элементарной теории газов, дает
возможность уже здесь восполнить пробел, допущенный в § 9,
не обращаясь к общим методам гл. IV.
1. Собственный объем молекул и постоянная &.
В случае идеального газа собственный объем молекул
считался равным нулю, что оправдывается при известных
условиях (не слишком низкие температуры и достаточно
высокая степень разрежения). В общем случае для
реальных газов следует предположить, что наличие у
молекул (очень небольшого) собственного объема v^
препятствует проникновению в их сферу действия других
молекул с таким же собственным объемом. Достаточно
рассматривать молекулы как твердые шарики объема г^^.
Тогда сферу молекулярного действия можно определить
как объем шара, поверхность которого простирается до
центров соседних молекул (этим обеспечивается отсутствие
«перекрывания» двух собственных объемов). Очевидно, что
радиус этой сферы действия вдвое превосходит радиус
молекулы и, следовательно, объем ее равен Sy^.
Рассмотрим некоторую единицу объема внутри газа.
Пусть /i| — плотность молекул, содержащихся в этом
объеме. Тогда часть единицы объема, недоступная для
центров других молекул, составляет Ъп^и^, и,
следовательно, доля свободного пространства равна
| = l-8n,tv B6.1)
Здесь 17^ обозначает единичный объем, помещенный в
знаменателе только из соображений размерности.
Представим себе теперь мысленно некоторый элемент
поверхности do внутри газа. Для него также справедливо
1) L. В о 11 Z m а п 11, Vorlesungen йЬег Gastheorie, 1898 (см.
перевод: Л. Б о л ь ц м а н, Лекщ1и по теории газов, М.—Л., 1953).
2) F. Sauter, Ann. d. Physik, F), 6, 59 A949).
242 Г.I. J и. Элементарная кинепсическая теория газов
соотношение B6.1). Однако если cfa представляет собой
элемент стенки, непроницаемый для молекул, то следует
учитывать лишь половину их, а именно те, которые
находятся со стороны газа. Таким образом, надо различать
н^ и п^ (плотность молекул вблизи стенки). Доля
свободного для других молекул пространства в последнем
случае больше, чем в B6.1), а именно,
,^=1-4пл. B6.2)
V
Отношение v^JVi одновременно представляет собой
отношение вероятности пребывания других молекул вблизи стенки
к вероятности их нахождения внутри газа (т. е.
отношение чисел способов, которыми можно разместить
молекулы в этих двух случаях). Следовательно, оно равно
/г,у/л^. Поэтому, согласно B5.2) и B6.1), имеем
^ = ^^ = ^^ —"^^ta^o /26 3)
Vi П[ 1—SniVQ * v • /
Отсюда, разрешая это уравнение относительно л^., находим
B6.3а)
[ — AriiVo
Число частиц п м, следовательно, плотность газа вблизи
стенки несколько больше, чем внутри. Это является
истинной причиной возникновения вандерваальсовой
поправки Ъ, как подчеркивает Заутер, ссылаясь на Больцмана.
Чтобы доказать это утверждение, возвратимся к
выводу давления газа в § 22. Там мы рассматривали
цилиндр с малым основанием rfa и конечной высотой ?.
Можно, однако, сколь угодно уменьшить значение ? и,
следовательно, ограничить весь цилиндр непосредственной
близостью стенки, если одновременно соответственно
увеличить основание. Поэтому уравнение B2.4) остается
в силе, но только теперь следует заменить п на л^.. Тогда,
принимая во внимание определение температуры B2.5) и
соотношение B6.3а), получаем
^' = ".^-^^ = Т^- B6.4)
Чтобы привести это уравнение к форме уравнения Ван-
дер-Ваальса в § 9, специализируем его для случая одного
§ 26, Статистический смысл постоянных в ур. Ван-дер-Ваальса 243
МОЛЯ газа. Тогда число всех молекул равно числу Аво«
гадро L, а число молекул в единице объема п-^ равно
Ljv, где теперь v обозначает молярный объем. При этом
из B6.4) вытекает
LhTjv ВТ ,ос / \
— ' — B6.4а)
^ 1—^Lvq/v V — 4Lro
Сравнение с уравнением (9.1), в котором положено а = О,
так как мы пока еще не учитываем силы сцепления, дает
b = ^iLv^. B6.5)
Постоянная b равна учетверенному собственному объему
Vq всех молекул, содер^нсагцихся в одном моле газа. Таков
(вскрытый уже Ван-дер-Ваальсом) физический смысл
постоянной Ь,
2. Вандерваальсовы силы сцепления и постоянная а.
Пренебрежем теперь вновь собственным объемом, но зато
учтем наличие сил сцепления, действующих между
соседними молекулами. Эти силы характеризуются малым
радиусом действия; например, в случае благородных газов
они убывают с увеличением расстояния между
молекулами г по закону г~'. Тогда можно положить п^ = п,
как если бы никаких сил сцепления не cyщecтвoвaw^o^
так как внутри газа они действуют на каждую молекулу
равномерно со всех сторон и потому взаимно
компенсируются. Иначе обстоит дело в граничном слое, где силы
сцепления дают результирующую, направленную внутрь
газа, так как по другую сторону стенки молекул газа
нет. Мы имеем здесь нечто вроде седиментации по
направлению «вверх», т. е. внутрь от стенки. В результате
внутри газа устанавливается нормальная плотность п,
а вблизи стенки (в граничном слое) несколько
пониженная плотность Пу}<П,
Таким образом, качественно влияние сил сцепления
(как и собственного объема молекул) сводится к
отклонению плотности газа у стенок от нормального значения.
Количественное рассмотрение проще всего провести,
основываясь на барометрической формуле B3.17). В этой
формуле фигурировал гравитационный потенциал, соот-
244 Гл. III. Элементарная кинетическая теория газов
ветствовавший не зависящей от координат и
направленной вниз силе тяжести:
<S> = mgz, или —-x- = —mg. B6.6)
В противоположность этому в нашем случае сила не
постоянна. Вне граничного слоя она равна нулю
(благодаря взаимной компенсации сил сцепления); внутри же
этого слоя она направлена «вверх». Будем рассматривать
сферу действия сил сцепления как поверхность шара,
описанную около точки наблюдения, и обозначим через
7 (z) площадь сегмента, вырезаемого из нее стенкой
сосуда. Равнодействующая сил сцепления, направленная
«вверх», пропорциональна этой (геометрически
определенной) величине 7 B). Кроме того, в данном случае мы имеем
дело не с молекулой в заданном силовом поле, как в
случае силы тяжести, а с взаимодействием между
некоторой определенной молекулой и ее соседями. Это
взаимодействие зависит от той степени заполнения соседними
молекулами сегмента ^(z), которая имела бы место в
отсутствие стенки, т. е. оно пропорционально числу
молекул п. При этом, поскольку речь идет о поправочном
члене, можно пренебречь зависимостью п от положения
точки. Таким образом, включая множитель
пропорциональности в 7, следует положить в противоположность
B6.6)
оо
-g = n7B), Ф = «5т-(г)^2. B6.7)
Z
Здесь величина Ф нормирована противоположно тому,
как это было сделано для поля тяжести, а именно, здесь
Ф = 0 при 2=00 (а там Ф = 0 при 2 = 0). Тогда имеем
у стенки
•о
Ф@) = п7, 7= 5 ТB)Й2.
о
Прежняя барометрическая формула
По
§ 26. Статистический смысл постоянных в ур. Ван-дер-Ваальса 245
переходит, в частности при z = 0, в
!Ье = е~ф@)/^^ = e-«^/fe^. B6.8)
Поэтому для давления из уравнения B6.4) (или из схемы
Бернулли) получаем
р = nJiT = пкТе-^'^1^'^ B6.9)
Разлагая показательную функцию для больших кТ
в ряд и пренебрегая высшими членами, получаем
р^ пкТ-п^^. B6.9а)
Опять специализируем уравнение для случая одного моля,
полагая в связи с этим n = L/Vy где v = VJлoл.^ Таким
образом, получаем ^)
Р = ^-Цг- B6.10)
Первый член здесь имеет такой же вид, как и для
идеального газа, что естественно, так как мы пренебрегали
собственным объемом молекул. Однако второй член при
сравнении с уравнением Ван-дер-Ваальса (9.1) позволяет
понять статистический смысл постоянной а\
a = L^'^. B6.11)
^) Естественно спросить, не дает ли уравнение B6.9) более
точную поправку к давлению, чем уравнение Ван-дер-Ваальса. Если
исходить из него, а не из приближения B6.9а), то вместо
уравнения Ван-дер-Ваальса получается уравнение Дитеричи
v — b
Отсюда для критических значений находим (см. § 9, п. 1):
а для критической постоянной имеем
^ = ^ = 4-^' = 3,69
РкЩ 2
(вместо jK' = V3 = 2,67, согласно Вап-дер-Ваальсу).
Экспериментальные значения для Не, Аг и Хе соответственно равны 3,31, 3,42
и 3,57.
246 Гл. III. Элементарная кинетическая теория газов
Вандерваалгсова постоянная а равна увеличенному в L^ раз
действию сил сцепления на границе газа; внутри газа это
действие исчезает. Не лишне, однако, заметить, что если
внутри газа поместить манометр, то при этом мы создадим
стенку и измерим около нее «когезионное давление» p^ = a/v^.
Заутер в цитированной выше работе выводит
когезионное давление из статистического поведения совокупности
молекул, а не из поведения отдельной произвольно взятой
молекулы в поле всех остальных. Метод Заутера
теоретически более удовлетворителен, чем наш, однако он несколько
сложнее. Заутер также показал, чго свойства стенки и
возможные дальнодействующие силы адгезии между стенкой
и газом не оказывают никакого влияния на измеряемое
давление.
§ 27. ПРОБЛЕМА ДЛИНЫ СВОБОДНОГО ПРОБЕГА
Понятие длины свободного пробега было введено уже
Клаузиусом в 1858 г. Под этой величиной понимают
путь /, проходимый некоторой молекулой (произвольно
выделенной из совокупности всех остальных) от некоторого
произвольного начального положения до первого
столкновения с какой-либо другой молекулой. В случае идеального
газа, молекулы которого предполагаются точечными, значение /
было бы бесконечно большим. Следовательно, как и в §26,
необходимо учесть собственный объем молекул i^q. В то время
как там он выражался вандерваальсовой постоянной 6,
равной учетверенному собственному объему молекул, здесь
будет фигурировать учетверенная площадь диаметрального
сечения шара с объемом Vq. Мы ограничимся, как и
раньше, моделью молекул как твердых шариков.
Вычислим I для предельного случая, когда скорость
одной молекулы значительно превышает скорость другой.
Тот же порядок величины имеет и «средняя длина
свободного пробегав, вычисленная еще Максвеллом в
предположении, что скорости молекул распределены по максвел-
ловскому закону (отличие состоит лишь в множителе |/2
в знаменателе). Наша специализация проблемы, повидимому,
обоснована, так как в строгой теории (см. § 44) знание
длины свободного пробега излишне; эта величина
заменяется там некоторым параметром разложения, значение
§ 27> Проблема длины свободного пробега 247
которого особо определяется для каждой отдельной
проблемы.
Заметим, что задачи, рассматриваемые в п. 2 п 3,
касаются необратимых процессов и, следовательно, строго
говоря, выходят за рамки обычной термодинамики и
статистики (см. § 21 и гл. V).
1. Вычисление длины свободного пробега в одном
частном случае. Если одна из частиц («ударяющаи»)
движется значительно быстрее другой («ударяемой»), то
последнюю можно считать просто покоящейся. Как и в § 26,
п. 1, припишем собственные объемы обеих молекул одно1'[
из них («ударяюпхей»), определив таким образом их
«эффективное сечение». Обозначим его радиус через s
(он равен сумме радиусов г^ и г^ обеих молекул; в нашем
случае r^=r^ = r, следовательно, 5 = 2г). Часть
пространства, охватываемая сферой действия в единицу времени,
представляет собой круговой цилиндр с основанием тгб-
и высотой с. Если единица объема содержит п частиц,
то внутри этого кругового цилиндра в среднем находится
ударяемых частиц, которые теперь, после того как мы их
лишили собственных объемов, следует считать точечными.
Вместе с тем эта величина равна числу столкновений
в единицу времени v. Так как число столкновений
соответствует всему пути, пройденному в единицу времени,
то длина свободного пробега, определяемая как путь, прой-
денныр! до первого столкновения, будет равна
/ = ~ = -V- B7.1)
В случае жестких шарообразных молекул с
эффективным сечением ти^^ длина свободного пробега / равна
толщине слоя, эффективные сечения молекул которого, не перс-
крываясь друг с другом, покрывают всю его площадь.
Установим порядок величины /. Для этого введем
эталонную длину /j, равную ребру куба, в котором в
среднем помещается одна молекула. Toi^a w = l//J и,
согласно B7.1),
248 Гл. III. Элементарная кинетическая теория газов
Численное значение радиуса атома водорода (радиуса
первой боровскои орбиты) составляет приблизительно
0,5'10'® см. Если для радиуса молекулы водорода
приближенно взять значение, равное удвоенному радиусу
атома, то величина 5, представляющая радиус сферы
молекулярного действия, окажется равной 2-10'® см. Величина п
есть число частиц в единице объема; она вычисляется
из числа Авогадро и составляет при температуре 0°С
и давлении 1 am 2,8-10^^ в 1 см^. Поэтому
1
1\ = У0Гсм^ А = 4:__ = | B7.2а)
И, согласно B7.2),
i = ?-100. B7.26)
Следовательно, в силу B7.2а), B7.26) имеем пропорцию
/:/i 15= 104:102:6. B7.3)
Длина свободного пути 1 при указанных условиях в 100 раз
больше^ чем среднее расстояние между молекулами Z^,
а последнее при атмосферном давлении значительно больше
радиуса сферы молекулярного действия s {т. е. диаметра
молекулы).
Так как длины 1^ для всех идеальных газов, согласно
Авогадро, одинаковы и размеры молекул также мало
отличаются друг от друга, то та же самая пропорция B7.3)
справедлива для Og и No с тем же приближением, что
и для Hg.
Отсюда следует, что в идеальном газе молекула
испытывает бесчисленное множество столкновений, прежде чем
она удалится на несколько сантиметров от своего
начального положения. Вследствие этого поразительно большие
значения, которые мы вывели в § 22 для Кс^, имеют
место на чрезвычайно малых расстояниях. С другой
стороны, из уравнения B7.1) видно, что, поскольку / обратно
пропорционально давлению, при очень сильном разрежении
понятие длины свободного пути становится иллюзорным —
молекулы могут достичь стенок сосуда, не сталкиваясь
друг с другом. Этот предельный случай кинетической
§ 27» Проблема длины свободного пробега
249
теории газов был экспериментально и теоретически
исследован в обстоятельной работе М. Кнудсена. В другом
предельном случае (сжижение, соприкосновение молекул)
величина / становится почти равной s и тогда понятие
длины свободного пробега также теряет смысл.
2. Вязкость. Допустим теперь, что наряду с
молекулярной молекулы газа обладают и молярной скоростью.
Пусть она направлена по оси ж и по абсолютной
величине линейно возрастает вдоль оси у. В
феноменологической гидродинамике доказывается
(см. т. II, Механика деформируе- ц(у)
мых сред, § 10), что на сечение,
расположенное перпендикулярно У
к оси у, действует напряжение
сдвига в направлении оси х
'ух
= Tir,
ди
B7.4)
Фиг. 25. Средняя
скорость течения Куэтта.
которое замедляет движение
верхнего (более быстрого)
полупространства и ускоряет движение
нижнего (более медленного). Мы
должны это доказать с точки зрения
кинетической теории газов и
вычислить коэффициент вязкости
(или внутреннего трения) т]. Вывод основан на
исследовании переноса импульса.
Обозначим через т массу молекулы, через иш\ —
соответственно компоненты молярной и молекулярной
скоростей в направлении оси х. Тогда полная составляющая
импульса молекулы в направлении оси х будет равна
g^ = m(« + 5). B7.5)
Скорость и зависит от у (фиг. 25); распределение
вероятности значений 5 от у не зависит, если, как мы и
предполагаем, температура одинакова во всех точках
пространства. Через плоскость г/ = 0 переходят из нижнего
полупространства в верхнее более медленные молекулы
и из верхнего полупространства в нижнее — более быстрые.
Точное расположение мест, откуда приходят медленные
250
Гл. ///. Элементарная кинетическая теория газов
И быстрые молекулы, зависит от того, где именно они
испытали последнее столкновение, в нижнем или
соответственно верхнем полупространстве. Чтобы определить
вероятные места столкновений, рассмотрим траектории
молекул и отложим на них от плоскости ?/ = О
(противоположно направлению движения) отрезки, равные длине
^•^Icosd
Фиг. 26. Частицы,
испытывающие столкновения в
верхней полуплоскости (/—радиус
круга).
\/=-lcos
Фиг. 26а. Частицы,
испытывающие столкновения в
нижней полуплоскости.
свободного пробега /. Если 6 —угол между направлением
траектории и осью г/, то ординаты мест столкновений
равны —/cos9 для молекул, приходящих снизу, и +lcosO
для молекул, приходящих сверху (фиг. 26 и 26а).
Вычислим теперь молярный импульс g = mUy
переносимый в верхнее полупространство (?/ > 0) молекулами,
пересекающими единицу площади у1чазанного выше
сечения (при у = 0). Молекулярный импульс т$ при этом
не следует учитывать, так как величина ? не зависит от у,
и импульсы, переносимые вверх и вниз, взаимно
компенсируются.
Рассмотрим прежде всего группу молекул, летящих
вверх, скорости которых лежат между с и с-f cfc и
траектории которых составляют с положительным направлением
оси у углы между Ь и в-\-Aв, В соответствии с их
происхождением из нижнего полупространства (фиг. 26а)
§ 27. Проблема длины свободного пробега 251
ИХ вклад составляет
gr| = ;y2M(-/cose) = mM@)-m/cose(-~-) . B7.5а)
[В силу линейности функции и {у) более высокие члены
разложения и в ряд Тейлора здесь не появляются.] Для
молекул, летящих вниз (угол О для них отсчитывается
относительно отрицательной полуоси у) соответственно
получаем
g 1 = ти ( +1 cosB) ^ти{0) +ml cose f-^^ . B7.56)
Разность этих выражений равна
gt-gl=-2m/cose(|^)^. B7.6)
Мы должны ее умножить на (одинаковое для обеих групп)
число dv молекул, которые в единицу времени
пересекают единицу площади сечения у = 0. Согласно
формулам B2.2а) и B3.9), число молекул, подлетающих в
единицу времени из показанного на фиг. 23 объема к единице
поверхности, дается выражением (предполагается, чго
нормаль к поверхности направлена по оси С, которая
является также полярной осью)
dv = -^ср {с) с cos б sin е de d^ с2 dc. B7.7)
Полный импульс, переносимый этими молекулами, мы
получим, если умножим это выражение на величину B7.6).
После интегрирования по азимутальному углу ср и
соответствующей группировки сомножителей это дает
— птк^ {с) dc cos- О sin ^ ^6 ( "т- ) •
Заметим, что g\ и gl были умножены на одну и ту же
величину, так как распределение Гаусса является
симметричным. Весь перенесенный импульс получится отсюда
путем интегрирования по всем скоростям с и по всем
направлениям траекторий 6. При этом последнее
интегрирование выполняется лишь в пределах от О до 7:/2.
252 Гл. III. Элементарная кинетическая теория гааов
Таким образом, для полного изменения импульса имеем
'g-=\{g\-g\)d-^ =
со и/2
-'^(М)М'^^''^"'- B7-8)
В такой форме записи учитывается, что величина /
сама зависит от с. Эту зависимость можно установить,
даже если она не совсем проста, при условии
справедливости распределения Максвелла. Однако для
рассматриваемой проблемы характерно то, что она не относится
к какому-либо равновесию и, строго говоря, распределение
Максвелла для нее несправедливо (ср. п. 4). Поэтому
мы не будем производить более подробное исследование
оставшегося в B7.8) интеграла, а просто заменим /
некоторым средним значением, например (см._^ начало этого
параграфа) значением /, деленным на ]/ 2, и вынесем
эту величину за знак интеграла. Тогда из выражения
B7.8) получим
g=_^(_-)^^c<p(c)dc. B7.8а)
тп1 / ди
О
Воспользовавшись теперь формулой B3.10а), можно
заменить интеграл в B7.8а) через с. Таким образом, B7.8а)
принимает вид
Такой же формулой (но с положительным знаком)
определяется изменение импульса для отрицательного
полупространства
- mnlc / ди \ ,гO п \
§ 27. Проблема длины свободного пробега 253
Сравнивая B7.9) и B7.9а) с B7.4), получаем для
коэффициента внутреннего трения
7] = ^. B7.10)
Заметим, что произведение тп представляет собой массу
единицы объема, т. е. плотность газа р. Формула B7.10)
была выведена еще Максвеллом в 1860 г. Зависимость
от давления в ней лишь кажущаяся. Действительно,
составляя произведение величин
Р^^'"" ^ ^"'Tlbi' B7.11)
видим, что число частиц п в числителе и знаменателе
сокращается и остаются лишь величины, зависящие от массы
и размеров частиц. Этот казавшийся сначала
парадоксальным вывод о независимости у\ от давления был
подтвержден в 1875 г. Кундтом и Варбургом вплоть до
давления 1/60 am. Однако при весьма больших давлениях этот
вывод становится уже несправедливым, так как тогда газ
приближается к жидкому состоянию; кроме того, для таких,
а также для очень низких давлений понятие длины
свободного пробега теряет смысл.
Представляет интерес также температурная
зависимость 7] в B7.10), определяемая множителем с.
Действительно, с [как и (с^) ^^] растет пропорционально
квадратному корню из температуры. В то время как вязкость
жидкости (например, масла) с температурой сильно падает,
вязкость газа возрастает пропорционально УТ,
3. Теплопроводность. Предыдущие рассуждения можно
применить к переносу не только импульса gy но и любой
другой величины G, транспортируемой молекулами,
летящими вверх и вниз. Выберем в качестве такой величины
энергию Е, т. е. рассмотрим газ, не находящийся в
тепловом равновесии. Предположим, что температура газа
линейно возрастает в направлении оси у. Для пояснения
опять могут служить фиг. 26 и 26а.
Величина Е дается выражением B3.14), в котором
для нашего случая потенциал Ф можно зачеркнуть. Обо-
254 i\?. ///. Элементарная кинетическая теория газов
значим число степеней свободы через / (сюда входят
поступательное движение, вращение и внутренние колебания)
и воспользуемся законом равномерного распределения
энергии по степеням свободы. Тогда на каждую степень
свободы приходится энергия кТ/2у так что для полной
энергии мы получаем
^- 2 '
или; если явно указать зависимость от у,
^4[no,+.(f).]-
Значение перенесенной энергии G мы найдем, подставив
эту величину вместо ти в формулу B7.6). Получим
Gt-Gi = -2f/cosO(|)^.
Вместо B7.8а) имеем
«=-l=f(f).W')^--Tf(|).-
о
Сравним теперь это выражение с феноменологическим
законом Фурье для теплопроводности при соответствующем
температурном перепаде
Здесь ^ — переносимое количество тепла, х — коэффициент
теплопроводности. Тогда для газокинетического значения х
находим
x = y^-J'- B7.12)
Величина х, так же как и т], не зависит от давления
(так как в нее входит лишь произведение п1) п зависит
от температуры (благодаря множителю с). Независимость
от давления (при не слишком низких температурах) была
подтверждена Кундтом и Варбургом одновременно с
исследованием 7].
Представляет также интерес простое значение, которое
получается для отношения х/т], так как из него выпадают
§ 27. Проблема длины свободного пробега 2Ъо
проблематичные величины с и /. В силу B7.10) и B7.12)
оно равно
Y) ~ 2т
Если умножить числитель и знаменатель этого отношения
на число Авогадро L, то получим
% ^ /Д/2
(поскольку kL = R — универсальная газовая постоянная
и mL/a — молярный вес). В числителе здесь стоит, согласно
выражению D.13), молярная теплоемкость газа при
постоянном объеме с^.. Следовательно, в противоположность
величинам си/, первоначально присутствовавшим в
выражениях для ХИТ], здесь фигурируют лишь непосредственно
измеримые величины. Получающееся таким образом
соотношение
~ = ^ , B7.13)
где Су/р. — удельная теплоемкость, содержит при более
точном расчете в правой части еще числовой множитель,
который для твердых шарообразных молекул равен 2,52
(Энског) и несколько меньше для многоатомных молекул.
Соотношение B7.13) до некоторой степени напоминает
закон Видемана — Франца в теории металлов (см. § 45):
Т=?(тУ^: B7.13а)
здесь о — электропроводность, е - заряд электрона.
Аналогия состоит в том, что и из последнего соотношения
выпадает проблематичная величина / (равно как и число
свободных элегтронов п). То обстоятельство, что в
соотношении B7.13) выпадает также температурная зависимость,
отличает B7.13) от B7.13а).
4. Общие замечания к проблеме длины свободного
пробега. Уже неоднократно подчеркивалось, что
использование распределения Максвелла при изучении
неравновесных состояний представляет собой грубое приближение.
Собственно говоря, в таких задачах проблема состоит
256 Гл. Ill» Элементарная кинетическая теория газов
в том, чтобы найти распределение скоростей в
зависимости от координат и времени. Необходимость такого
рассмотрения была осознана еще Больцманом;
математическая формулировка ее принадлежит Гильберту. При ее
решении сама собой появляется некоторая длина /,
которую истолковывают как длину свободного пробега,
но определить которую можно лишь вместе с
распределением скоростей. При этом отнюдь не очевидно, что она,
как это было принято выше, окажется одной и той же
как для внутреннего трения, так и для теплопроводности.
Мы еще вернемся к этому вопросу в § 44.
Особые трудности возникают в связи с задачей о
диффузии, которую мы здесь не будем рассматривать. Однако
следует разобраться в одном осложнении, которое
содержится в самых основах исчисления вероятностей и
становится особенно ощутимым в проблеме длины свободного
пробега.
Поясним его суть на примере игральной кости.
Спросим себя, при котором бросании вероятно первое
выпадание какого-нибудь заданного числа (например, единицы)?
Этот довольно неясно поставленный вопрос уже давно был
уточнен при помощи понятия математического ожидания
i= 2 ^^«- B7.14)
л:=1
Здесь о;^ — вероятность того, что данное число впервые
выпадет при х-м бросании. В случае игры в кости
значение w^ хорошо известно и потому сумму в B7.14) можно
немедленно вычислить. Это проделано в задаче П1. 4;
результат оказывается равным шести. Это число
представляет собой, следовательно, «длину свободного пробега
единицы» в серии бросаний.
Однако рассматриваемую последовательность событий
можно обратить, поставив вопрос о том, когда в
последний раз до данного бросания кости было вероятно
выпадание единицы. Математическое ожидание этого события
опять дается формулой B7.14) и попрежнему равно шести.
Таким образом, «расстояние» между последним
выпаданием единицы в прошлом и первым выпаданием ее
в будущем оказывается равным двенадцати, что на первый
§ 27. Проблема длины свободного пробега 257
взгляд противоречит найденному ранее значению «длины
свободного пробега». Однако это противоречие целиком
обусловлено полцой беспорядочностью рассматриваемых
событий и связано с произвольностью выбора начального
момента в цепи бросаний.
В кинетической теории газов этому «начальному
моменту» соответствует произвольное сечение, проведенное
на фиг. 26 и 26а при 2/ = 0. Выпаданию единицы в буду-
jueM или в прошлом соответствует в данном случае факт
столкновения данной молекулы с какой-нибудь другой
в верхнем или нижнем полупространстве. Не удивительно
поэтому, что расстояние, пройденное молекулой между
этими двумя столкновениями, оказывается равным
удвоенной длине свободного пробега, которая определяется
формулой B7.14) как математическое ожидание одного
отдельного столкновения и приближенно выражается
соотношением B7.1).
Глава IV
ОБЩИЕ ПРИНЦИПЫ СТАТИСТИКИ. МЕТОД ЯЧЕЕК
В гл. Ill было показано, что статистика равновесных
состояний приводит к таким простым общим
закономерностям, как закон распределения Максвелла—Больцмана,
уравнение состояния идеальных газов и формулы для
их удельных теплоемкостей. Напротив, в конце гл. III
мы установили, что необратимые процессы и все вопросы,
связанные с понятием длины свободного пробега, приводят
к необходимости использовать довольно произвольные
апроксимации и требуют для своего решения весьма
обстоятельных рассуждений. Поэтому естественно
предположить, что при рассмотрении с более общей точки
зрения можно получить простую обобщающую трактовку
равновесных состояний; напротив, для удовлетворительного
решения проблем, связанных с необратимыми процессами,
требуются более сложные методы. Последние мы
рассмотрим в гл. V.
Настоящая гл. IV овеяна духом Людвига Больцмана
A847 — 1906) и удивительно ясна с идейной стороны.
Действительно, о простоте следует судить но с точки
зрения элементарной, тривиальной наглядности, но с точки
зрения высшей математической ясности. В этой главе
впервые появляется понятие энтропии. Это основное
понятие термодинамики, которое вообще не фигурировало
в элементарной гл. III, только здесь занимает подобающее
место и выступает в принципе Больцмана как чисто
арифметическое следствие принятого метода расчета.
§ 28. ТЕОРЕМА ЛИУВИЛЛЯ. Г-ПРОСТРАНСТВО
И {1-ПРОСТРАНСТВО
Прежде чем применять теорию вероятности,
необходимо ввести понятие равновероятных событий. Например»
§ 28, Теорема Лиувилля. Т-пространство и [^-пространство 259
ври игре в кости однородность материала и
геометрическая форма куба гарантируют равновероятное появление
чисел от одного до шести. При игре в карты тщательная
тасовка колоды уравнивает шансы всех игроков.
1. Многомерное Г-пространство. В теории газов
естественно ожидать, что в силу колоссальности числа частиц
и хаотичности их движения закономерности, возможно,
существовавшие в пространстге скоростей и координат
в некоторый начальный момент, с течением времени
нивелируются и устанавливается состояние
молекулярного беспорядка («молекулярного хаоса»), для описания
которого можно применять законы теории
вероятностей.
Будем рассматривать газ как механическую систему с F
степенями свободы. Если N - число молекул,
рассматриваемых как тождественные, а / — число степеней свободы
каждой отдельной молекулы, то F = fN, Будем описывать
эту систему при помощи канонических уравнений
Гамильтона [см. т. I, Механика, уравнение D1.4)]
P^^'^dfu' ^^^'^Ук' ^ ^^ ^^''' '-^Р^^^^' --.^Ы. B8.1)
где q^ и ^?^J —cooTBeiCTBCHHo обобщенные координаты
и импульсы. Величины q и р пронумерованы здесь в
соответствии с т. I, § 12, так что к одной молекуле
относятся несколько значений индекса к. Функция
Гамильтона Н есть полная энергия системы, представленная
как функция р л q» Сюда включается, в частности,
энергия взаимодействия молекул при столкновениях и
отталкивающее взаимодействие их со стенками. Функция
Гамильтона выводится из функции Лагранжа
L = L{q^, • • •» 9f» 9i» • . -^Ы
ири помощи соотношений [см. т. I, уравнение D1.1)]
^=^Pkk-L, Pk = ^ .
В 2F-MepH0M (/?, д)-пространстве, которое мы назовем
260 Гл. IV, Общие принципы статистики. Метод ячеек
Г-пространством^), соответствующее состояние системы
задается определенной точкой, которая с течением времени
описывает некоторую кривую. Мы рассмотрим не просто
отдельную Г-точку, а множество таких точек, в частности
множество точек, .заключенных в малом элементе
Г-пространства
А2 = Ар Ад, где А/? = A/?i ... Арр, Aq = Aq^... Aqp, B8.2)
II мысленно выделим описываемые ими траектории. Такие
множества точек в фазовом пространстве соответствуют
газам, состоящим из одинаковых молекул, которые лишь
с точки зрения микросостояний несколько отличаются
друг от друга. Макроскопически эти системы неотличимы
друг от друга, если они имеют одинаковые или
приближенно одинаковые энергии 2).
Форма элемента объема AQ изменяется с течением
времени самыми разнообразными способами. Однако
величина его при этом остается неизменной.
2. Теорема Лиувилля. Доказательство последнего
утверждения проще всего проводится при помощи обычных
кинематических соотношений (см. т. II, Механика
деформируемых сред, § 1). Как было показано в т. II,
относительное изменение объема дается формулой
e=g + g + i. B8-3)
где X, г/, 2 —координаты произвольной точки тела в
неподвижной декартовой системе координат, а ?, тг], С
—смещения точки тела по осям гг, у и z соответственно.
Если распространить эту формулу на многомерный
случай и считать, например, что хну соответствуют
1) Термин Г-пространство был пред-южен П. Эренфестом
и Т. Эренфест в их работе Mathem. Enzykl., Bd. IV, 4, Artikel 32,
заложившей основы теории газов.
2) При одинаковых энергиях число степеней свободы 2F
Г-пространства превращается в 2F — i благодаря уравнению
Н (/?, д) = const. При приближенно одинаковых энергиях Hi< Н < Н,^
в рассмотрение входит только часть 27^-мерного Г-пространства^
Последнее имеет место, например, в том случае, если система*
находится в тепловом равновесии с окружающей средой.
(
§ 28. Теорема Лиувилля. Т-пространство и [^.-пространство 261
/?1 и д^, ТО $ И 7] в Г-иространстве будут обозначать
смещения p^dt и q^dt. Тогда 5^ + ^ переходит в
и, используя систему уравнений Гамильтона B8.1).,
получаем
-^г^г + ^.ё]Г.Г' = ^- B8.4)
Точно так же обращаются в нуль и два следующих
члена суммы, соответствующей в нашем случае
формуле B8.3). Правда, эти частные суммы не имеют
никакого самостоятельного физического смысла, так как они
представляют собой проекции уравнения в многомерном
пространстве на произвольно расположенную
координатную плоскость. Однако сумма этих проекций 9
инвариантна относительно поворота осей координат в Г-про-
странстве. Геометрический смысл ее, очевидно, следующий:
^ dAQ/dt
Таким образом, окончательно получаем
^ = 0, А2 = const. B8.5)
Это и есть теорема Лиувилля.
Этот результат можно интерпретировать следующим
образом: если точки фазового пространства,
изображающие различные микросостояния, распределить по фазовому
пространству с одинаковой плотностью, то они будут
двигаться как однородная нес^нсимаемая снсидкостъ.
Рассматривая эту плотность как меру вероятности
обнаружить некоторую фазовую точку в элементе объема А2,
можно высказать следующее предположение: если в какой-
либо момент времени вероятности нахождения точки
фазового пространства в равных элементах объема А2
одинаковы, то это будет верно и для любого другого момента
времени. В силу этого становится возможным вместо
начальных условий, употребляемых в механике, принять
^статистическое предположение о равновероятности состоят
262 Гл. IV. Общие принципы статистики. Метод ячеек
НИИ, изображаемых фазовыми элементами равного объема.
Эта гипотеза аналогична условию равновероятности
выпадения каждой стороны игральной кости. Выполняется ли
это предположение фактически, может установить только
опыт. Выше было показано, что основная гипотеза
статистической механики совместима с уравнениями движения.
Такие статистические условия всегда заменяют собой
начальные условия механики системы точек, коль скоро
остаются неизменными уравнения движения.
Резюмируя, подчеркнем еще раз, что рассуждения о
Г-пространстве относятся не к одному какому-то объему
газа, а ко множеству частиц исследуемого газа (или
любых других систем); имеется в виду, что мы
одновременно следим за поведением всей эгой совокупности систем,
микросостояния которых различны, но макросостояния
одинаковы.
3. Равновероятность состояний для идеального газа.
Перейдем теперь к рассмотрению простого случая, когда
фазовое пространство описывает поведение только одной
молекулы газа. Согласно терминологии, предложенной
П. Эренфестом и Т. Эренфест, это случай так называемого
fx-простраиства. Число измерений ^.-пространства равно 2/,
а не 2УУ/, как для Г-пространства. Таким образом, для
одноатомного газа ji-пространство шестимерно (/ = 3), для
двухатомного газа, если считать, что атомы в молекуле
жестко закреплены, — десятимерно (/=5), так как
добавляются две вращательные степени свободы, и т. д.
Переход от Г-пространства к [х-пространству возможен
только тогда, когда механическая система, определяемая
функцией Гамильтона Я (р,д), обладает весьма
специальными свойствами. Если до сих пор рассматривался самый
общий случай и учитывались любые взаимодействия между
молекулами (например, силы отталкивания при
столкновениях или дальнодействующие силы сцепления), то теперь
мы будем рассматривать систему молекул как идеальный
газ (объем молекулы Vq = 0, следовательно, никаких
столкновений нет и длина свободного пробега бесконечно
велика). Тогда молекулы движутся независимо друг от друга
и их фазовые пространства идентичны и друг с другом
не связаны. Функция Гамильтона всей системы представ-
§ 28. Теорема Лиувилля, Т-простраиство и \l-пространство 263
ляет собой сумму функций Гамильтона для отдельных
молекул. Очевидно, в ней нет членов, включающих
координаты разных молекул.
Таким образом, элементы фазового пространства,
описывающие отдельные молекулы, при движении остаются
отделенными друг от друга. Это обстоятельство в
сочетании с теоремой Лиувилля и статистической гипотезой
(постоянство Д2г и равновероятность состояний с равными
1'-объемами) приводит к выводу о равновероятности
состояний с равныхми значениями AQp^. Этот факт лежит в
основе метода Больцмана, который мы рассмотрим ниже.
В этом методе делается предположение об априорной
равновероятности фазовых объемов
Д2 = А/?Ад, где А/? = Api ». . А/?^, ^q = Aq^ . . . A^'y. B8.6)
Следует отметить, что эта формула проще, чем B8.2), так
как здесь индексы 1, 2, ..., / относятся к одной
молекуле, в то время как в формуле B8.2) индексами 1,2, ..., F
были пронумерованы все молекулы системы.
При выводе формулы B8.2) мы говорили о малости
элементов объема AQ Г-пространства и в выражении B8.4)
рассматривали приращения А/? и Ад как дифференциалы.
Однако мы не решали вопроса о том, насколько малы
должны быть элементы объема AQ. Больцман при
изложении своего метода неоднократно подчеркивал, что нужно
брать конечные значения А/?, Ад, так чтобы в данном
фазовом элементе содержалось еще большое число молекул.
Однако в конце вычислений он проводил предельный
переход А/? —> О, Ад —» 0. Только квантовая теория смогла
полностью ответить на вопрос о том, насколько малы
должны быть элементы объема.
Забегая вперед, заметим по этому поводу, что
произведение Apk^qk имеет размерность действия. Для случая
декартовых координат {[q] = см [р] = [mg] =г-см-сек'^)
это ясно непосредственно; но, как видно из приведенной
выше формулы р = dL/dq, это верно и для любых
обобщенных координат д. Действительно, L имеет размерность
энергии {эрг); поэтому [р^д^] = э/?г-се« (размерность
действия).
264 Гл, IV, Общие принципы статистики. Метод ячеек
Согласно квантовой теории, действие является
дискретной суммой элементарных квантов. В дальнейшем будет
показано, что наименьшая величина произведения каждой
координатной пары Pf^ q^ равна кванту действия Планка
^Pk^qk = h. B8.7)
Этим соотношением мы воспользуемся уже здесь. Для
молекул с / степенями свободы на основании формулы
B8.6), получаем, что величина элементарной ячейки
фазового пространства равна
А2 = /гЛ B8.8)
Два состояния молекулы, для которых р п д относятся
к одной и той же ячейке фазового пространства,
статистически неразличимы между собой. Определенные таким
образом ячейки AQ являются теми элементами, с
которыми оперируют в методе Больцмана, и соответствуют,
согласно нашей аналогии, шести равновероятным
возможностям при игре в кости.
Мы должны выяснить еще один довольно трудный
вопрос: достаточно ли велики эти элементарные ячейки,
чтобы заключать в себе, как того требует Больцман,
большое число молекул? Обычно это условие не
выполняется. Раньше считали фазовые ячейки достаточно
большими для того, чтобы не возникали трудности, связанные
со слишком малым числом молекул. Однако, после того
как при помощи квантовой механики были определены
размеры элементарной ячейки, подобное рассмотрение
стало невозможным. Но можно показать, что простое
преобразование позволяет объединить большое число ячеек
фазового пространства в один достаточно большой элемент
(см. § 29, п. 3). Тем самым создаются условия,
существенно необходимые для применения метода Больцмана.
Поэтому малые размеры элементарных ячеек фазового
пространства не представляют серьезного препятствия.
Дарвин и Фаулер предложили другой метод
вычисления средних значений, который годится, даже если числа
заполнения элементарных ячеек малы. Их результаты
совпадают с теми, которые дает метод Больцмана, но
область применимости их метода шире, так как объеди-
§ 29. Принцип Болъцмана 265
нение слишком многих ячеек в один элемент все же
невозможно вследствие изменения энергии от ячейки к ячейке.
Мы не будем излагать здесь метод Дарвина—Фаулера^),
но ниже по другому поводу с успехом применим его
(см. § 37).
Последующие рассуждения обычно будут относиться
к [х-пространству; однако вытекающая из квантовой
механики невозможность различить тождественные частицы
вынудит нас в конце концов вернуться к Г-простран-
ству.
Для Г-пространства справедливы формулы, очень
похожие на те, что будут выведены для (л-пространства (их можно
получить вполне аналогичным путем). Все же в § 36 эти
формулы будут строго обоснованы методами,
разработанными В. Гиббсом, который наряду с Л. Больцманом яп-
ляется основателем статистической механики. Будет
показано, что метод Гиббса применим совершенно независимо
от того, насколько малы элементарные ячейки.
§ 29. ПРИНЦИП БОЛЬЦМАНА
Под принципом Больцмана мы понимаем сведение
понятия энтропии к вероятности состояния системы.
Математически принцип Больцмана выражается краткой
формулой
S = k\nW. B9.1)
Высеченная на памятнике Больцману на Венском
кладбище эта формула парит на фоне облаков, плывущих
над могилой великого Больцмана. Неважно, что сам Больц-
ман никогда не писал этой формулы. Это сделал Планк
в первом издании лекций по теории теплового излучения
A906 г.). Планку же принадлежит введение постоянной к.
Сам Больцман говорил только о пропорциональности
между энтропией и логарифмом вероятности состояния.
Термин «принцип Больцмана» был введен Эйнштейном; он
^) Для более подробного ознакомления можно использовать
книгу R. Н. Fowler, Statistical Mechanics, Cambridge. 1929,
или небольшую, но очень ценную книгу М. Born, Natural
Philosophy of Cause and Chance, Oxford, 1949.
266 Гл. IV, Общие принципы статистики. Метод ячеек
же использовал обращенную форму этого принципа:
PF = eS/ft. B9.1а)
Здесь энтропия S считается известной из эксперимента,
а вероятность состояния W подлежит определению.
Поэтому вторая часть второго начала термодинамики означает,
как это уже было известно Гельмгольцу, переход от
искусственно созданного порядка к более вероятному
беспорядку .
В правую часть формулы B9.1) следует ввести
(особенно в случае неоднородных систем) постоянное
слагаемое, т. е. величину, которая зависит лишь от числа
молей в составных частях системы и не зависит от
переменных, характеризующих ее состояние. Таким образом,
в общем случае формула B9.1) имеет вид
6' = A:lnP1^-f-const. B9.16)
В 1877 г. Больцман сделал одно довольно
неопределенное замечание по поводу (не написанной им) формулы
B9.16)^): «Можно было бы даже из отношения числа
различных распределений, характеризующих состояние,
вычислить вероятность. Это, возможно, привело бы к
интересному рассмотрению теплового равновесия». Вскоре после
этого он добавляет 2):
«Я, однако, не думаю, что мы вправе без дальнейших
рассуждений представить этот результат как нечто само
собой разумеющееся, по крайней мере пока мы не
определим совершенно точно, что именно следует
понимать под наиболее вероятным распределением».
Там же в качестве условия, ограничивающего
возможность выбора характеризующих систему величин, была
указана теорема Лиувилля.
1. Статистический вес как мера вероятности
состояния. Рассмотрим идеальный газ. Предположим, что он
состоит из Л'' молекул, заключенных в заданный объем F,
1) L. Boltzmann. Die gesammelten Werke, Nr. 39, S. 121,
Wien.
2) L. В 011 z m a n n, Die gesammelten Werke, Nr. 42, S. 193,
Wien.
§ 29. Принцип Больцмана 267
п имеет полную энергию U. Если каждая молекула
имеет / степеней свободы, то число измерений фазового
пространства равно 2/. Разделим фазовое пространство на М
ячеек:
1, 2, ..., I, ..., il/.
Поскольку заданные значения (/ и F, так же как и
размеры ячеек [см. формулу B8.8)], конечны, М
представляет собой также конечное, хотя и очень большое число.
Пусть молекулы совершенно произвольно распределены
по ячейкам. Обозначим числа заполнения ячеек черев
п^, п^, ..., ^i, ..., wm. B9.2)
При этом, естественно,
^n^^N, B9.2а)
Каждое такое распределение чисел п^ характеризует
определенное микросостояние газа.
Найдем, сколько различных распределений из N
молекул по М ячейкам соответствуют одному и тому же
микросостоянию газа. Назовем это число распределений
термодинамической вероятностью состояния W^)\
величина W определяется статистическим весом 2) данного
распределения
ЦТ —. ^^ B9 3)
П^\П2\ ... П^\ ' \ • /
Для пояснения рассмотрим сначала случай малых
значений М и N. Пусть Л/= 2, N = 2; тогда возможны сле-
^) Существенно, что термодинамическая вероятность, будучи
целым числом, не нормируется к единице и в силу этого зависит
от величины элементарной ячейки.
*) Автор пользуется введенным Больцманом термином Permuta-
bilitat, считая его более содержательным, чем часто употребляемый
термин «число комплексии». Поскольку в русской литературе этот
термин не употребляется, мы пользуемся принятым у нас
термином «статистический вес».—Прим. перев.
268 Гл. IV. Общие принципы статистики. Метод ячеек
дующие варианты:
а) ^1 = 1, Л2 = 1, PF:-j^=2,
б) П1 = 2, п, = 0, W = ^ = l,
в) П1 = 0, П2 = 2, vy = g^=l.
В варианте «а» первая молекула может находиться либо
в первой, либо во второй ячейке. Положение второй
молекулы при этом однозначно определено; таким образом,
W = 2» В вариантах «б» и «в» нет никакой свободы
выбора и поэтому W = 1.
Хотя формула B9.3) хорошо известна из
элементарной комбинаторики как выражение для коэффициентов
полинома, мы, полноты ради, выведем ее методом полной
математической индукции. Предположим, что формула
B9.3) верна для случая, когда число молекул равно N — I.
Чтобы из известного выражения для Wn-i получить
значение H'iv, следует исходить из одного из следующих
распределений:
^1-
Wj,
-1, Л2,
Л2— 1,
Щ, . . . ,
. . . , Пму
. . . , Пму
пм —if
j^B) ___GV-|)!__
цгт_ (^-1I
_ (iV-l)!n,
_ (ЛГ-1)!па
ni\ ... njj! '
(ЛГ-1)!пд^
Если к каждому такому распределению
соответствующим образом добавить iV-ю молекулу, то получится
исходное распределение B9.2). Таким образом, если
просуммировать все правые части VFiv-i, то в силу уравнения
B9.2а) получим результат, совпадающий (с 29.3):
§ 29, Принцип Больцмана 269
Чтобы упростить полученное для W выражение,
можно воспользоваться формулой Стирлинга. При большом
целом N достаточно хорошую апроксимацию дает формула
Л^!=(у/. B9.4)
Более точная оценка дается формулой
N\ = {2i:NY/2(^^y, B9.4а)
Формула Стирлинга выводится при помощи
следующего соотношения:
lniV-! = lnl + ln2-b...+lnyV^ \lnxdx,
1
(Здесь ступенчатая кривая приближенно заменяется
логарифмической.) Далее
N
\ lnxdx = [x (In X — l)]x=iv — [х (In X — i)]x=i =
1
= 7V(lnA^-l) + l ^iVln^.
Потенцируя это равенство, получим формулу Стирлинга
B9.4).
Формула B9.4) будет применяться не только к Л^,
но и к числам л^, которые, таким образом, также
считаются достаточно большими. Это предположение не
вызывает возражений, пока в ряду п^ вообще попадаются
большие числа, потому что тогда факториалы малых
чисел можно заменить единицами. Однако этот метод не
годится, когда все числа п^ малы. Это, к сожалению, как
раз тот случай, когда можно применять метод Больцмана
(если мы пользуемся фазовым элементом AQ = Л^).
Можно показать, например, что в идеальном газе при
нормальных условиях примерно из 30000 квантовых ячеек
максимум в одной содержится одна молекула, прочие же
ячейки не заполнены (см. § 37). Попадание двух молекул
в одну ячейку в этом случае совершенно невероятно.
К счастью, как уже было упомянуто в конце § 28, это
270 Гл. IV, Общие принципы статистики. Метод ячеек
не представляет серьезного препятствия. Поэтому мы
пренебрегаем указанным затруднением и ведем расчет так,
как если бы среди п^ имелись большие числа.
Исследуем в заключение, что изменится в нашем
рассуждении, если соединить много элементарных ячеек
в один большой элемент, так что среди щ действительно
появятся большие числа.
Для rill возьмем, следуя Больцману, приближенные
значения
И, подставив в формулу B9.4), получим
м
In Ж = const- ^п^1пщ. B9.5)
Под символом const понимаются все члены, которые не
зависят от /г^, т. е.
м
const = TV (In TV-1)+2 n^-iVlniV. B9.5a)
2. Максимум вероятности как мера энтропии.
Выясним теперь, какое распределение молекул «встречается
чаще всего», т. е. при каких значениях п^ W имеет
максимум. Для этого мы будем варьировать л^,
учитывая, что в силу B9.2а) вариации связаны условием
28л^ = 0. B9.6)
Из формулы B9.5) получим
blnW= -S4(lnni-bl).
В силу B9.6) единицу под знаком суммы можно
вычеркнуть и окончательно условие наиболее вероятного
распределения [когда числа п^ подчиняются условию B9.6)}
принимает вид
8 In Vr = - S S^i In Wi = 0. B9.7)
Это означает, в согласии со статистической гипотезой, что
числа п^ равнораспределены по всем ячейкам, так как
уравнения B9.6) и B9.7) совместны лишь при /г^ = Пд = ...
§ 29, Принцип Больцмана 271
Однако числа п^ должны удовлетворять еще одному
условию. Именно, энергия системы U является заданной
(см. начало этого пункта). Представим U в виде суммы
по всем фазовым ячейкам
?/-1«^е^. B9.8)
Здесь е^ — полная энергия молекулы, принадлежащей
ячейке фазового пространства (с координатами /?^, q^).
Значение е^ изменяется от ячейки к ячейке, но внутри
данной ячейки оно считается постоянным в соответствии с
квантовой теорией. Из уравнения B9.8), варьируя п-^ при
постоянных С/ и 8^, получаем
2ei4 = 0. B9.8а)
Чтобы одновременно учесть условия B9.6) и B9.8а), лучше
всего воспользоваться методом множителей Лагранжа
[см. т. I, Механика, уравнение A2.5)]. Таким образом,
уравнение B9.7) заменяется следующим:
8 In Ж = - 2 SAii (In Wi -f а + P^i) = О- B9.9)
Благодаря наличию множителей а и ^ с числами л^
теперь можно оперировать так, как если бы они были
независимыми. Тогда из уравнения B9.9) следует
\пп^= -а~ре^, Wi = e-«.e-P«^ B9.10)
Если это значение \пп^ подставить в формулу B9.5), то
для максимума \iiW получим
In Жмакс. = const + а ]l AZi + Р 2 п^^^. B9.11)
Величины аир характеризуют состояние всей системы,
а не отдельной ячейки, и потому их можно вынести из под
знака суммы.
Использовав B9.2а) и B9.8), а также значение
постоянной B9.5а), получим просто
lnWMaKc.=A^lniV-fa.V-fpC/. B9.12)
Полученное здесь максимальное значение И^ как мы
увидим ниже, очень велико по сравнению с состояниями,
соответствующим]? несколько измененным значениям п^.
Поэтому состояние, осуществляемое с максимальной веро-
272 Гл. IV. Общие принципы статистики. Метод ячеек
ятностью, можно отождествить с «реальным состоянием»,
которое мы должны обнаружить при наблюдении газовой
системы. Но тогда, согласно принципу Больцмана,
формула B9.12) дает также величину S/k, Поэтому из
формулы B9.11) следует (для таких процессов, при
которых N остается постоянным, а энергия U изменяется под
влиянием внешнего воздействия)
-^^NdoL + Ud^ + ^dU. B9.13)
При этом изменения величин а и р ограничены следующим
условием:
iV = 2 ^i = е-* 2 е-^"'' = const. B9.14)
Сумма в формуле B9.14) представляет так называемую
сумму состояний («функцию состояний» по английской
терминологии):
Zo = ^e-^'i, B9.15)
i
Точнее говоря, речь идет здесь о сумме состояний в (i-npo-
странстве. Значение ее основано на том, что, зная Zq,
можно вычислить все термодинамические величины [см.
также уравнение C3.14)].
Из формулы B9.14) следует, что 0L = ln{ZJN). Таким
образом, уравнение B9.12) окончательно принимает вид
1 ^ = In РГмакс. = N\nZ, + p^7, B9.12а)
а для чисел заполнения п^ и энергии U в силу B9.8)
и B9.10) получаем
3- _ N dlnZo
п^ = е-
Р дн
U=^e'-^z,e-^4=^N^^
B9.16)
3. Выводы относительно метода элементарных ячеек.
Вне всякого сомнения, приближение, использованное
в п. 2, недопустимо. Однако результаты, полученные таким
путем, правильны. Легко видеть, что это имеет место,
по крайней мере, в тех случаях, когда значения энергии
§ 29, Принцип Больцмана 273
мало изменяются при переходе от одной элементарной
ячейки к другой. Тогда можно считать энергию
постоянной в довольно большой области фазового пространства
и потому объединить большое число ячеек х в более
крупную единицу, так называемую большую ячейку.
Если /Vj, iVg, ..., iV^ —числа заполнения больших
ячеек, то по аналогии с формулами B9.2а) и B9.8)
справедливы соотношения
2iV^. = iV, ^N~z.==U, B9.17)
у у
где суммирование проводится по всем большим ячейкам,
причем е^. — среднее значение энергии в /-й ячейке.
Термодинамическую вероятность распределения по большим
ячейкам можно получить, суммируя элементарные
вероятности, которые соответствуют данному распределению по
большим ячейкам, т. е.
^'=N^''\^^ /'' , Л' ,•••B9.18)
Математически это означает, что суммирование
производится по всем разложениям N-^, N^, ..., начиная
с Л^1, Л^2 ° "^^ Д. В этом случае сумма распадается на
произведение сумм
которые можно вычислить при помощи теоремы о
коэффициентах полинома. При этом получаются соответственно
величины х^1, х^2, . . .^ так что
Эта формула отличается от формулы B9.3) только
множителем х^. К W можно применить метод Больцмана,
так как числа х можно выбрать настолько большими, что
среди чисел заполнения N^ безусловно окажутся большие
числа.
Аналогично равенству B9.10) имеем
/V, = e-""^V. B9.20)
274 Гл. IV. Общие принципы статистики. Метод ячеек
Соответственно вместо формулы B9.12) получаем
In l^iiaKc. = iV In xiV + aN + ^U B9.21)
и вместо формулы B9.14) получаем
iV = e-«2e-Pv. B9.22)
Наконец, сумму состояний в fi-пространстве B9.15) можно
написать в виде
Zo = x2e~4 B9.23)
у
так что из формулы B9.22) следует, что
е«=|^, a=lnZo-lnxiV.
Соответственно из формулы B9.21) снова получаем
формулу, аналогичную B9.12а):
In Т^^акс. = iV In Zo + р?^ - In Жмакс B9.21а)
Таким образом, значение W не зависит от х. То же самое
относится и к другим величинам; [следовательно, мы
получаем такие же результаты, как и при х=1, но теперь
они строго обоснованы.
§ 30. СРАВНЕНИЕ С ТЕРМОДИНАМИКОЙ
1. Изохорический процесс. Так как объем V
постоянен, то разделение фазового пространства на ячейки
остается прежним. Взяв логарифмическую производную
от обеих частей уравнения B9.14), получим
0= -d<i-d^^^= -^da-d^^L . C0.1)
Таким образом, равенство B9.13) принимает вид
dS=^k^dU. C0.2)
Но из второго начала термодинамики в применении к изо-
хорическому процессу следует (см. § 6)
^sJ^^^. C0.2а)
§ 30. Сравнение с термодинамикой ^75
Сравнивая формулы C0.2) и C0.2а), получаем
C0.3)
Ниже мы увидим, что это фундаментальное
соотношение соблюдается и в случаях, рассматриваемых
в п. 2 и 3.
2. Общий случай процесса в газе в отсутствие
внешнего поля. В общем случае изменяется не только
энергия, но и объем. При этом к старым фазовым
ячейкам от 1 до Л/ прибавляются новые от М-у I до Л/' (при
положительных значениях изменения объема dV; при
отрицательных dV пропадают фазовые ячейки от Л/' н- 1
до Л/). Связанное с этим относительное изменение суммы
м
2 обозначим через
^2= J , или с/2=- 2 . C0.4)
i=M+l г=Л1'+1
Величины аир сохраняют здесь те же значения, что
и раньше, поскольку они характеризуют всю систему
в целом, а не отдельные элементы фазового прострацства.
Вычислив логарифмическую производную от обеих
частей равенства B9.14), получаем вместо C0.1)
^da-d^-^ + ^=^0. C0.4а)
Если внешние силы отсутствуют, то, как мы покажем
ниже, имеет место соотношение
^- V ¦
Подставив C0.4а) и C0.46) в B9.13), получим
C0.46)
dS = k^dU-\-kN^.
Сравнивая это со вторым началом термодинамики
^с ''^обр. dU + pdV
27^ Гл, IV. Общие принципы статистики. Метод ячеек
получаем наряду с C0.3) уравнение состояния идеального
газа
^ = ^, или pv = RT. C0.5)
Последняя формула верна, если число молекул равно
числу Авогадро, т. е. когда V выбрано равным объему
одного моля.
Для доказательства формулы C0.46) рассмотрим
внимательнее структуру элемента фазового пространства AQ.
Если пространственные координаты молекулы (например,
координаты се центра тяжести) суть ж, у, z, то
д2 = АхД2', ^x = ^x^y^z. (зо.ба)
Здесь ДЙ' —элемент объема, включающий все импульсные
координаты молекулы, а также пространственные
координаты, соответствующие внутренним степеням свободы.
К сделанному в § 28 утверждению о том, что все элементы
объема Д2 равны между собой (а именно, равны А^),
можно добавить, что и значения Дт также могут быть
выбраны одинаковыми. В самом деле
дх ду dl _ д^Н , __/^^, ^_п
дх"^ ду~^ dz "дхдрх '•••"" К^дрх'^ ... j—yj,
так как мы предполагаем, что внешняя сила К = 0.
В этом случае значение энергии е^ не зависит от
Ху у у Z. При суммировании по i в уравнении B9.14) для
каждош Д2' появляется столько равных членов, сколько
имеется пространственных ячеек Дх. Это число равно
V/Az. Поэтому вместо B9.14) можно написать
Ar=2«i = «-''-tS«"^ C0.6)
i >
причем индекс / относится к суммированию по фазовым
элементам Д2у.
После логарифмического дифференцирования
равенства C0.6) при переменных F, а и р условие
постоянства Л^ дает
-da-d?-^+^^^ = 0, C0,6а)
§ 30. Сравнение с термодинамикой 277,
Появляющийся здесь добавочный член dV/V соответствует
слагаемому с?1/11 в уравнении C0.4а). Тем самым
соотношение C0.46) доказано.
3. Газ в силовом поле. Формула Больцмава.
В уравнении C0.5) давление во всем газе считается
одинаковым. Однако это правильно только в том случае,
если энергия е не зависит от координат. Если в системе
действуют внешние силы [мы предполагаем, что они имеют
однозначный потенциал Ф (ж, у, 2), поскольку иначе вообще
невозможно состояние равновесия (ср. т. II, Механика
деформируемых сред, конец § 7)], то можно положить
е = Ф(д:, г/,2) + е', C0.7)
где е' — не зависящая от а:, г/, z часть энергии молекулы
(в е' входит энергия вращения и т. п.). Теперь существенно,
где именно производится изменение объема dV (т. е. где
вводятся новые ячейки в координатном пространстве. А*:)
Ограничимся, так же как в п. 1, рассмотрением изо-
хорического процесса. Так как изменение состояния
системы не влияет на внешние силы, то вследствие того,
что F = const, мы получаем, подобно C0.1)—C0.3),
Р = 4г- C0.S)
кТ
Здесь А—константа системы, т. е. величина, не зависящая
от координат, хотя последние и фигурируют в Ф.
Следовательно у Т такоке не зависит от координат. Этот
вывод, в частности, справедлив и для земной атмосферы,
находящейся в поле тяжести, если только в ней имеет
место состояние теплового равновесия. (Метеорологи раньше
иногда протестовали против этого утверждения.)
Напротив, давление и плотность зависят от
координат, что непосредственно вытекает из уравнения B9.10).
Если из формулы C0.7) исключить г- и провести
суммирование по фазовым элементам Д2', то для числа частип
в пространственной ячейке Ат получим
п = е-»-*/'^^ 2 e"V^^. C0.9)
Умножая п на массу молекулы т и деля на Дт, н ходим
плотность газа р. Сравнивая ее с плотностью Ро при Ф = 0,
,278 Гл. IV. Общие принципы статистики. Метод ячеек
получаем
А = е-ф/лт. C0.10)
Это и есть уже многократно применявшаяся нами формула
Вольцмана [см. B3.16) и B3.17)].
4. 1'аспределение скоростей по Максвеллу—Больц-
ману. Так же просто выводится максвелловское
распределение скоростей для одноатомного газа, свободного от
внешних воздействий. Рассмотрим определенную ячейку
пространства импульсов Ао)^. и некоторую (в отсутствие
внешнего поля произвольную) ячейку координатного
пространства Ах. Первая характеризуется координатами
где под ?, 7], С мы понимаем, как и раньще,
составляющие вектора скорости.
В случае одноатомного газа, молекулы которого не
имеют внутренних степеней свободы, ячейке в пространстве
имцульсов соответствует энергия
е = -|E^ + 11'' + С^).
Поскольку всегда ^ = 1/^7*, из выражения B9.10)
следует, что
«, = e-.exp[-f<i^bMil]. C0.11)
Просуммировав по всем пространственным элементам,
получим
i
Положим
^ = ^,Дш,.. C0.116)
Величина Fj^^j представляет собой вероятность того,
что, выбрав один атом из общего числа N, мы обнаружим
его в импульсной ячейке Аш^ (т. е. вероятность того, что
1хомпоненты его скорости суть S, тг], С). Так как iV =^ 2 ^j^
§ 30. Сравнение с термодинамикой 279
то ИЗ C0.11а) и C0.116) вытекает, что
е.р[_-(!1±й±С!)]
Z^^pL-T кт—J/"*'
Здесь множитель До)^., который в формуле C0.116)
находится при Fjy перенесен в знаменатель и приписан
к отдельным членам суммы. [Вследствие равенства фазовых
ячеек Д2. и предположенного в C0.5а) равенства
элементов Дх импульсные ячейки Дш^ также равны между
собой.]
Для вычисления знаменателя в формуле C0.12),
который, как мы видим, тесно связан с суммой состояний Z^
B9.15), совершим предельный переход при До)^.—> 0. Тогда
знаменатель принимает вид
+ 00 +00 +00
с е-^"^'/2ЛТй$ С e-.m,2/2ftT^^ ^ e-rnc«/2ftT^(; C0.12а)
т**
и после интегрирования [см. B3.5а)] оказывается равным
гп^ Q^J^y^ =. {2^ткТУ'2. C0.126)
Следовательно, из формулы C0.12) получаем
F = Bum*r)-^/2exp [ -^(^1±^±^] . C0.13)
Это то же самое выражение, что и B3.8), с той лишь
формальной разницей, что там F относится к элементу
пространства скоростей, а здесь —к тому же элементу
пространства импульсов. Именно по этой причине
множитель т®/2 находится здесь в числителе, тогда как
в выражении B3.8) он был в знаменателе.
Путь, которым мы шли при выводе максвелловского
распределения скоростей, непосредственно приводит к больц-
мановскому обобщению на случай многоатомных молекул —
обобщению, которое было указано уже в конце § 23.
В самом деле, все наши рассуждения можно без
изменения применить и к многоатомному газу с внутренними
степенями свободы, а также к газу, находящемуся в поло
280 Гл, IV. Общие принципы статистики. Метод ячеек
потенциальных сил, так как появляюп1иеся в числителе
и знаменателе дополнительные множители в формуле
C0.12) сокращаются.
5. Смесь газов. Мы можем ограничиться случаем
смеси двух газов, которые находятся в объеме V и имеют
энергию и. Массы молекул обозначим через т^ и т^,
числа частиц —через iVj и Л^. Для каждого из двух газов
введем отдельное фазовое пространство. Числа заполнения
фазовых ячеек равны п^^ и п^^-
Статистические веса, согласно B9.3), равны
W -^ -^ W - ^^•
Вследствие независимости обоих распределений
термодинамическая вероятность состояния для смеси будет равна
Применяя формулу Стирлинга, вместо B9.5) получим
In W = const - B п.^ In Wii + 2 ^2 In М- C0.14)
i У
Максимум W следует искать при трех дополнительных
условиях:
S «il = N„ 2 «У2 = Л'2- 2 «il^il + 2 И;2^-2 = U. C0.15)
Чтобы удовлетворить им, надо ввести три множителя
Лагранжа: а^, ag, р. Из формулы C0.14) находим
b\iiW^--y Ь?г,,(In п,, + а,+ ^в,,)-
i
— 2 Ц2 (In 'г^-з + S + PS-2)»
следовательно,
In Mil = -cci-psii, 1плу2= -a2-Pe^2.
Подставив в формулу C0.14), получим
In Тумаке. = const—а^ 2 ^ii—^2 2 ^-з—Р B Щг^гЛ 2 ^/«^^г)-
У i У
Таким образом, в силу условий C0.15)
In ^^макс. = const - a^iVi - S^2 - Р^- C0-16)
§ 31. Энергия еааа из абсолютно твердых молекул 281
Согласно принципу Больцмана, эта величина представляет
собой значение энтропии для смеси. Так же как и выше,
р = ^. C0.17)
Итак, смесь газов характеризуется одной
температурой; уравнение состояния смеси определяется формулами
P = Pi + Pv Pi = -y-» P2 = -#- » C0.18)
где Pi и /?2 —парциальные давления.
Каждый газ можно рассматривать независимо от
другого, считая, что он занимает весь объем V. Точно так
же как парциальные давления, накладываются друг на
друга и распределения скоростей, причем каждое имеет
максвелловскую форму.
S 31. УДЕЛЬНАЯ ТЕПЛОЕМКОСТЬ И ЭНЕРГИЯ ГАЗА
ИЗ АБСОЛЮТНО ТВЕРДЫХ МОЛЕКУЛ
Представление об абсолютно твердых молекулах, так же
как и представление об абсолютно твердых телах
механики, без сомнения, является «не физическим». Тем не
менее тщательное изучение термических cbojictb таких
молекул представляет интерес особенно потому, что оно
указывает на пределы применимости классической
статистики.
Трудности, встречающиеся при изучении этого вопроса,
Кельвин ^) назвал «тучами, сгущающимися над
динамической теорией теплоты XIX века». Эти трудности
заставили его сделать в высшей степени революционный для
того времени вывод о необходимости отказа от теоремы
о равномерном распределении энергии по степеням
свободы. В § 33 — 35 мы увидим, что физика XX века, а
именно квантовая теория, проливает свет на все до сих
пор темные области статистики.
1. Одноатомный газ. Поскольку одноатомная молекула
не имеет структуры, предположение об ее абсолютной
^) Kelvin, Baltimore Lectures, 1884, Appendix В.
282 Гл. IV. Общие принципы статистики. Метод ячеек
твердости не вызывает еще трудностей. В § 22 мы нашли
для энергий и теплоемкости одного моля одноатомного
газа выражения [см. B2.6а) и B2.66)]
« = |/гг. с„=|/?. ^=|. C1.1)
Здесь нужно еще показать, как при помощи статистики
выражается уже известная нам из термодинамики
энтропия газа.
Согласно принципу Больцмана, из уравнения B9.12)
следует
S^kNlnN-^akN-^ ЦП. C1.2)
Здесь последний член справа есть константа, равная
3 3
— kN (так как ^к=1/Т и U = '^NkT)\ следовательно,
его можно объединить с первым членом, после чего
формула C1.2) примет вид
.S = ^iV (|-f а + In iVy C1.2а)
Значение а для одноатомного газа можно получить из
вычислений, произведенных в § 30, п. 4. Исходим из
формулы
Величина в квадратных скобках здесь та же, что и в
формуле C0.12). Суммирование по / распространяется здесь,
так же как и там, только на ячейки импульсного
пространства. Множитель F/At обозначает число
пространственных ячеек, содержащихся в объеме F, т. е. кратность,
с которой надо брать каждое слагаемое в сумме по /,
чтобы вернуться к исходной сумме по i. Умножим обе
части равенства C1.3) на А2 = Ат-До). Величину Ао) можно
ввести под знак суммы по / [см. замечание к формуле
C0.12)]. Получаем
1
§ 31. Энергия газа из абсолютно твердых молекул 283
Сумма в этой формуле полностью совпадает со
знаменателем в формуле C0.12); поэтому, согласно C0.12а) и
<30.12б), она равна {2жткТу/2,
Таким образом, находим
a = \nV + ^lnT + ^lnB7zmk)-ln{N^Q). C1.4а)
При этом значении а формула C1.2а) принимает вид
S = kN(^lnV + ^lnT^ + C, C1.5)
Ото не что иное, как хорошо известное нам [§ 5,
формула E.10)] термодинамическое выражение для энтропии
в том случае, когда она относится не к 1 г, а к iV
частицам одноатомного газа. Однако наш результат
значительно содержательнее термодинамического, так как
константа С в формуле C1.5) имеет вполне определенное
численное значение. Именно, она равна сумме констант,
фигурирующих в уравнениях C1.2а) и C1.4а), причем
последние надо еще умножить на kN:
С = у AiV [ 1 + In BirmA) - |- In AQ ] . C1.5a)
Если у Больцмана значение элемента объема Д9
оставалось неопределенным, то, согласно квантовой механике,
^Q=lh^ (в частности, в случае одноатомпого газа Д2 = /г^).
Таким образом, формула C1.5а) рает
C = kN]n (^-'^'^1^'^''1 , C1.56)
Подставив это выражение в формулу C1.5), получим
(IчОнчательно для энтропии
^transl = kN\nV ^ f^ . C1.5в)
Для одного моля
5transl = ^?lnt;-^ f^ . C1.6)
Индекс у S указывает, что эта формула для энтропии
годится не только для одноатомного газа, но дает и ту
284 Гл. IV. Общие принципы статистики. Метод ячеек
часть энтропии многоатомных газов, которая связана
с перемещением центра тяжести частиц.
Формулу C1.6) нельзя, конечно, экстраполировать на
случай Г = О, так как при Г = О вещество уже не является
идеальным газом. Таким образом, между ней и теоремой
Нернста нет никакого противоречия.
Формула C1.6) была выведена Саккуром^). Почти
одновременно и независимо от него Тетроде 2) получил
соотношение, лишь формально незначительно
отличающееся от C1.6). Мы вернемся к знаменитой формуле
Саккура — Тетроде в § 37, п. 1.
Здесь мы должны указать еще на два обстоятельства.
Во-первых, в постоянную энтропии С входит конечный
объем фазовой ячейки. Это означает, что мы не можем
произвольно распоряжаться величиной фазового элемента,
как это делал Больцман. Напротив, объем фазовой ячейки
определяется постоянной энтропии. К сожалению, этот
факт вместе с тем означает, что наше предположение о
возможности больших чисел заполнения для отдельных
ячеек не соответствует действительности из-за малости
кванта действия (см. § 29, п. 3).
Во-вторых, энтропия должна быть пропорциональна
числу частиц Л^. Однако из C1.5в) следует, что это имеет
место, только если из правой части C1.5в) вычесть
величину Л^1пЛ^. Действительно, в противном случае энтропия
будет расти быстрее, так как фигурирующий под знаком
логарифма объем V при постоянной температуре
пропорционален числу частиц. Эта трудность устраняется только
в квантовой теории. Вывод формулы Саккура — Тетроде
будет обсуждаться еще в § 37, п. 1.
2. Газ двухатомных молекул. Будем рассматривать
двухатомную абсолютно твердую молекулу как гантель.
Оба атома, рассматриваемые как материальные точки,
как бы соединены невесомым стержнем длиной 1^).
Помимо масс атомов т^ и гпс, эта модель характеризуется
двумя моментами инерции относительно осей, перпенди-
1H. Sack иг, Ann. d. Phys., 36, 958A911); 40, 67A913).
2) Н. Tetrode, Ann. d. Phys., 38, 434 A912).
*) Возможность пренебречь смещениями атомных ядер и элек-
тропов обусловлена существованием кванта действия.
§ 31. Энергия газа из абсолютно твердых молекул 285
кулярных К линии, соединяющей атомы т^, /Wg. Эти два
момента инерции равны друг другу; момент инерции
относительно «линии центров» (линии, соединяющей
т^ и /Wg) равен нулю. То же самое справедливо и для
любого расположения атомов на одной прямой (например,
для молекулы COg).
Эта модель обладает пятью степенями свободы,
соответственно координатам центра тяжести ж, г/, z и двум
угловым координатам б и ф, определяющим по известным
формулам положение линии центров относительно
некоторой произвольно выбранной начальной оси. Третий
угол ср, характеризующий поворот относительно линии
центров, не учитывается, так как ему соответствует
нулевой момент ко^чичества движения.
Кинетическая энергия вращения, согласно т. I
[Механика, формула C5.12)], равна (в данном случае А=1,
С = 0)
erot=4(^' + '^^"^'^')- (^^•'^)
Здесь /—момент инерции ротатора, который в т. I
обозначался символом е. Перемена обозначения вызвана тем,
что букву в мы будем использовать, начиная с § 33,
для обозначения характеристической температуры ротатора.
Импульсы, соответствующие углам 6 и ф в силу C1.7)
определяются соотношениями
Ре = ^* = /е, рф = ^=/8ш2е.ф. C1.7а)
дЬ 4
Фазовое пространство в данном случае десятимерно;
фазовый элемент имеет вид
AQ = ^x^y^z•m^^l^riM^^вц^p^^p^ C1.76)
Для дальнейшего, однако, гораздо удобнее
представить Erot в виде квадратичной формы с постоянными
коэффициентами. Как известно, это можно сделать, вводя
угловые скорости относительно двух взаимно
перпендикулярных (и перпендикулярных к линии центров осей:
О), = а = —-- , A)„ = sm
i-v- ^ , -g-^^'-Y-Asin
+ = rio- C1-7^)
286 Гл, IV. Общие принципы статистики. Метод ячеек
Величины (Oj, AJ представляют собой «неголономные»
скорости [см. т. I, Механика, § 35, формула C5.4)].
Величины /(Oj и /(Og суть, по выражению Больцмана, «момен-
тоиды» или, как мы будем говорить, «импульсоиды».
Фазовый элемент пространства импульсоидов /(Oj, lo)^
получается из элемента объема в пространстве /?в, /?ф.
умножением на якобиан
d(/cOi)» (9(/Ш2)
др^ ^Рф
О
sin(
= sine. C1.7г>
дAьУ^)' 5G@2)
Соответственно выражение C1.76) в результате
преобразования C1.7в) переходит в
Д2 = АжАуА2-тЗД?А7].ЛС-8твА9Дф.Аа)^АаJ. C1.8)
Одновременно формула C1.7) принимает обычный в
механике вид
erot=^^ {< + <)' C1.8а)
Вполне общие результаты § 29 (такие, как равенство
Р = -г^, вид уравнения состояния, множитель
Больцмана и т. д.), естественно, не изменяются при увеличении
числа измерений фазового пространства с 6 до 10, Однако
на добавочные степени свободы, появляющиеся благодаря
вращению, приходится определенная доля энергии —по
RT/2 на каждую (по крайней мере, если придерживаться
классического метода расчета). Следовательно, вместо
выражений C1.1) мы получаем
« = |/?Г, с„=|.Д, ^ = 1+1 = 1
сь
C1.9)
Для доказательства этих выражений запишем энергию
одного моля, согласно всегда применимой формуле B9.16),
в виде
1=-^^1п2е-'\ C1.10)
Перейдем от суммирования по i, которое проводится и по
координатному и по импульсному пространствам, к сум-
§ 31, Энергия газа из абсолютно твердых молекул 287
мированию по /. Последнее, очевидно, охватывает только
импульсы, причем каждое слагаемое, как и в формуле
C1.3), надо умножить на число ячеек в координатном
пространстве. Это число равно теперь
где Ат = Дгг А?/A'z, Да = 8ш6А6Дф, 4тс = \ sin ЙАбАф
—поверхность шара единичного радиуса. Умножим числитель
и знаменатель этой дроби на пятимерный элемент
импульсного пространства Аю. При этом в знаменателе мы
получим AQ = Ах Аа До) = Л^. В числителе внесем Aa) = Au)^
под знак суммы по /. Тогда для суммы состояний,
фигурирующей в формуле C1.10), найдем
^0=S^ 2 «-'•'• Ч- C1-11)
В сумме состояний можно выделить множитель,
соответствующий вращению, так как энергии, соответствующие
разным степеням свободы, аддитивны. Полагая
^0 ^^ ^transl • ^voti
имеем
Zrot = ^^e^p[-j{^\ + 0] Aa).(rot) =
У
Этот интеграл представляет собой произведение двух
интегралов Пуассона, каждый из которых равен ]Л2тс/р/.
Таким образом,
2rot=-^. C1.12)
Согласно B9.16), это дает следующий вклад в энергию:
U.ot=-N'-^ = -^ = NkT. C1.13)
Учитывая трансляционные члены, находим (для одного
моля)
U = ^RT, c, = ^R, C1.13а)
288 Гл, IV» Общие принципы статистики. Метод ячееш
Соответственно изменяется и энтропия двухатомного газа.
Для вращательной части се, согласно B9.12а), получаем
(на один моль)
s,ot-=-R\nT + Rln^^^^. C1.14)
Для полной энтропии одного моля, в согласии с
термодинамическим уравнением E.10), имеем
5 = /?lnt; + |-/?lnr + const. C1.15)
3. Газ многоатомных молекул и трудности его
рассмотрения, отмеченные Кельвином. В общем случае
абсолютно твердой молекулы, в которой атомы
расположены не на одной прямой, добавляется еще одна степень
свободы. Эта система описывается тремя угловыми
координатами 6, ф, ср и тремя угловыми скоростями (о^, (Og, Шд
относительно главных осей эллипсоида инерции.
Соответствующие главные моменты инерции обозначим /j, /g, /3.
Вычисления, аналогичные тем, которые привели к
формуле C1.12), дают теперь для суммы состояний
\^?т J UA У K^hJ V ?/з J •
Вместо C1.9) мы получаем
u = -kRT, с =3/?^6 кал/град*моль, -^ = —. C1.16)
?d Су О
Весьма удовлетворительно, что таким путем можно
понять простое правило § 4: с^^ = 5Я/2 или с^, = ЗЯ. Однако
при более пристальном рассмотрении это правило
оказывается слишком простым. Вспомним, например, о
треугольной модели молекулы воды. Валентные штрихи,
соединяющие атом О с атомами Н, образуют угол ^ > ir/2
(согласно спектроскопическим данным, полученным из
анализа полосатого спектра водяных паров). Три момента
инерции не равны друг другу и отличны от нуля. Если
рассматривать водяной пар как почти идеальный газ,
то теплоемкость с^ — независимо от температуры и
моментов инерции — должна быть равна 6 кал/град * моль. То же
самое справедливо и для молекул с еще более тупыми
^ 32, Теплоемкости упругих молекул и твердых тел 289
углами. Только при 7 = ^» когда расположение атомов
становится линейным, с^ скачкообразно принимает
значение, характерное для двухатомного газа E/?/2«^
*^5 кал/град * моль).
Дискретность значений с^, C, 5 и 6 кал/град-моль),
отражающая наличие трех, пяти и шести степеней свободы
у одно-, двух- и многоатомных молекул, составляет одну
пз трудностей, стоящих перед кинетической теорией газов.
Однако имеется еще более серьезная трудность.
Эйкен, побуждаемый представлениями Нернста о
вырождении газов, измерял молярную теплоемкость Hg при
понижающейся температуре. Он нашел, что теплоемкость
неуклонно уменьшается от значения 5 кал jград * моль
и при 80° К составляет 3 кал/град-моль. Степени свободы,
связанные с вращением, отпадают, или, как часто
говорят, вымерзают. Молекулярный водород ведет себя как
одноатомный газ.
Перефразируя Шиллера, автор указывал в 1911 г. на
собрании естествоиспытателей в Карлсруе, что «степени
свободы следует взвешивать, а не считать». Как это
делать, показывает квантовая теория.
§ 32. ТЕПЛОЕМКОСТИ УПРУГИХ МОЛЕКУЛ И ТВЕРДЫХ ТЕЛ
В этом параграфе мы отбросим «нефизическую» модель
молекулы с жестко закрепленными атомами и учтем, что
атомы в молекуле могут совершать малые колебания
около положений равновесия. В этом случае атомы
обладают не только кинетической, но и потенциальной
энергией. Все сказанное относится также к атомам,
образующим огромную молекулу — твердое тело.
1. Двухатомная молекула. Сила взаимодействия
между атомами направлена по линии центров независимо
от того, имеет ли она электростатическую или гомеопо-
лярную природу. Пусть Tj и Tg —смещения атомов из
положений равновесия, причем положительными считаются
значения Tj и Tg, соответствующие удалению атомов друг от
друга. В силу третьего закона Ньютона оба смещения
связаны друг с другом. Для простоты мы проведем расчет,
считая связь квазиупругой, т. е. для гармонических колебаний.
290 Гл. IV. Общие принципы статистики. Метод ячеек
Положив r = rj + ''2» легко найти потенциальную и
кинетическую энергию системы:
^пот. = у''"» ^кин. = -2~^^- C2.1)
Здесь С —константа упругой связи, а «приведенная масса»
М^ ^^^^ C2.1а)
т^^ + т2 ^ ^
(см., например, т. IV, Оптика, задача III.1). На
колебания вдоль линии центров накладывается еще
вращательное движение. Строго говоря, колебательное и
вращательное движения нельзя рассматривать совершенно
независимо друг от друга, так как момент инерции зависит
от расстояния между атомами. Однако этим эффектом
в первом приближении можно пренебречь, так как
смещения атомов из положений равновесия малы по
сравнению с междуатомным расстоянием (при Т = 2000° для
молекулы Hg смещение составляет около 10%
междуатомного расстояния).
Число измерений фазового пространства колеблющейся
двухатомной молекулы увеличивается с 10 до 12. В
фазовый элемент ^Q из C1.8) надо включить еще множи-
тели ^r и il/Ar. Однако благодаря тому, что
добавляющаяся энергия квадратично зависит от г и г, это не
приводит к трудностям. Надо лишь умножить прежний
интеграл C1.12) на множитель
С e-(P/2)Cr2^^_|/|j^ C2.2а)
+ °° Г-.
jj e-i^i2)MT2dr=Y -щ C2.26)
(изменения момента инерции, мы не учитываем). Поскольку
нас интересует лишь зависимость от р, дополненное таким
§ 32. Теплоемкости упругих молекул и твердых тел 291
путем выражение C1.13) непосредственно дает
7 7
u~-K-RT, с^ = -^Rt:^ 7 кал/град-молъу
-^ = 1+^=1,29. C2.3)
Из вышеизложенного вытекает важное замечание и еще
более важный вопрос.
1. Потенциальная энергия в C2.2а) выступает на
равных правах с кинетической энергией в C2.26). Доля
обеих в распределении энергии составляет в силу C2.3)
RT12 на один моль.
2. Почему для воздуха и других двухатомных газов
при обычных условиях не возбуждается колебательная
степень свободы? В противном случае вместо известного
значения Ср/Су= 1,4 наблюдалось бы меньшее значение
Ср/с„= 1,29.
2. Многоатомный газ. В общей теории колебаний
хорошо известна следующая теорема (см., например,
т. VI, Дифференциальные уравнения в частных производи
ных физики, § 25). Для любой механической системы
число свободных колебаний около положения устойчивого
равновесия равно числу степеней свободы. Для каждого
из этих колебаний справедливы все утверждения,
сделанные выше для линейного колебания молекулы
двухатомного газа. В частности, каждое из этих колебаний
увеличивает значение с^ на RT, Следовательно, с
усложнением структуры молекулы значение с„ должно было бы
неограниченно возрастать, а отношение c^jc^
приближаться к единице. Возникает вопрос: почему же тогда
для сложных органических молекул среднее значение
Ср/Су составляет 1,33 (ср. § 4, п. 3), что соответствует
наличию только трансляционных и вращательных
степеней свободы?
3. Твердые тела и закон Дюлонга и Пти.
Исследование показывает, что все кристаллы состоят из атомов,
образующих решетчатую структуру и благодаря наличию
сил связи колеблющихся около положений равновесия
совершенно независимо от перемещения и вращения всего
292 Гл. IV. Общие принципы статистики. Метод ячеек
тела как целого. Поскольку каждый атом имеет 3, а
решетка из N атомов —3iV степеней свободы, то, вычитая
из 3iV 6 степенец свободы твердого тела, мы находим,
что на колебательное движение приходится 3iV — 6
степеней свободы. Столько же имеется и независимых
осцилляторов, так как движение системы связанных
осцилляторов всегда можно представить как совокупность такого
же числа независимых нормальных колебаний^) (т. VI,
Дифференциальные уравнения в частных производных
физики, § 25, п. 3). Потенциальную энергию их надо
учитывать так же, как в п. 1.
Таким образом, в состоянии термодинамического
равновесия каждое колебание обладает средней энергией кТ.
Поэтому молярная теплоемкость твердого тела (каждое
настоящее твердое тело является кристаллическим)
составляет с^ = ЗЛ ^ 6 кал!град-моль независимо от
температуры. Это и есть знаменитый закон Дюлонга и Пти.
Однако этот закон противоречит теореме Нернста, согласно
которой Су—»0 при Г—>0 (см. § 12, п. 3), а также
экспериментальным данным, полученным при низких
температурах. Полезно добавить также, что для очень твердых
веществ (алмаз, карборунд) уменьшение с^ (и cj
наблюдается уже при комнатных температурах.
§ 33. КВАНТОВАНИЕ ЭНЕРГИИ КОЛЕБАНИЙ
Открытие Планком кванта действия h побудило нас
с самого начала считать элемент фазового пространства
Д2 конечным. Естественно, что при этом значения
энергии е^, соответствующие фазовым элементам, образуют
дискретный ряд. Оказалось, что это обстоятельство
существенно для определения константы в выражении для
энтропии. Этот ряд значений можно рассматривать как
непрерывный только в том случае, когда разность
энергий кТ соседних ячеек много меньше энергии,
приходящейся на одну степень свободы:
Ч^г - Si <С кТ. C3.1)
1) Нормальные колебания отличаются тем, что атомы движутся
в известном смысле упорядоченно. Достаточно вспомнить, например,
синфазное (или противофазное) движение двух связанных
маятников или собственные колебания струны.
§ 33. Квантование энергии колебаний 213
Только в этом случае закономерен переход от суммы
состояний
2о = 2е-''""' C3.2)
к статистическому интегралу [см., например, выражение
C0.12а)]
^^е-Ч'^га^^ C3.2а)
1. Линейный осциллятор. В дальнейшем вместо
индекса i будем употреблять индекс /г, а собственную
частоту осциллятора обозначим через v. Тогда, согласно
первоначальной гипотезе Планка A900 г.),
е^ = пЬ, C3.3)
или, согласно его предположению, сделанному в 1911 г.
и полностью совпадающему с результатами квантовой
механики,
вп = (п + 4)^^- C3.3а)
В обоих случаях условие C3.1) сводится к неравенству
Ь < кТ. C3.4)
Введем характеристическую температуру
в = ^^. C3.4а)
Тогда неравенство C3.4) принимает вид
в < Г. C3.5)
Только в этом случае законен наш прежний расчет,
в котором сумма состояний заменяется интегралом. Мы
покажем, что в другом предельном случае, когда в > Г,
теплоемкость стремится к нулю. Значение v, а
следовательно, и в можно получить, изучая полосатые спектры
молекул (инфракрасные вращательные спектры).
Например, для НС1 (в этом случае инфракрасный спектр
изучен особенно хорошо) мы имеем в ^ 4000°.
294 Гл. IV. Обгцие принципы статистики» Метод ячеек
Произведем теперь расчет в форме, пригодной для
всех температур (Г ^ 9). При этом сначала мы
используем формулу C3.3).
В силу B9.16) для энергии одного моля мы имеем
оо
«--'f'lE^-^"'''- C3-6)
п=0
Фигурирующий здесь ряд представляет собой обычную
геометрическую прогрессию и сходится при всех
значениях Р > 0. Сумма его равна
1
Таким образом, формула C3.6) дает
« = -f'bj—^3ph^. C3.6а)
Подставляя сюда значения р = iJkT и в = h^jk, находим
"= в^/?-1 ' ^33.7)
В двух указанных выше предельных случаях мы имеем
T>Q
c.=--R
Т^в
а = Лве-в/Г-^0 C3.9)
В первом предельном случае колебательные степени
свободы полностью участвуют в распределении энергии (так
же, как и в § 32, п. 1). Во втором случае роль
колебательных степеней свободы совершенно ничтожна.
Промежуточный случай представлен на фиг. 27 и 27а.
Например, при Г = в мы имеем, согласно C3.7) и C3.8),
гг = J?5- = 0,58Лв, с, = —?— = 0,34i?.
§ 33, Квантование энергии колебаний
295
Кривые на фиг. 27 и 27а дают ответ на вопрос, постав-
пленный в конце § 32, п. 1. При таком подходе к задаче
колебательные степени свободы не просто «считаются»
(как в классической теории), а «взвешиваются», т. е.
имеют различный статистический вес б зависимости от
температуры.
Фиг. 27. Молярная энергия квантового
осциллятора как функция температуры. (Единицы
масштаба Лв, 0.)
Теперь ясно, что при характеристической
температуре в порядка нескольких тысяч градусов колебательные
степени свободы в обычных условиях опыта не
участвуют в распределении энергии.
К сказанному следует добавить еще два замечания.
1) Если вместо старой формулы C3.3) взять
правильное значение энергии осциллятора C3.3а), то, заменяя
А
В C3.6) п на ^ + "» легко получим вместо C3.7)
«=^i?e.
i?e
е«/Г_1
C3.10)
296 Гл. IV. Общие принципы статистики. Метод ячеек
К значению энергии, определяемому формулой C3.7),
добавляется, таким образом, еще «нулевая энергия» /?в/2,
реальность которой доказана целым рядом исследований,
проведенных при низких температурах. Кривая и{Т) на
фиг. 27 будет в силу этого несколько сдвинута вверх
(на отрезок, одинаковый для всех температур). Кривая
cJT остается неизменной. Поэтому все предыдуш;ие
выводы, относящиеся к теплоемкости, остаются в силе.
Ф н г. 27 а. Молярная теплоемкость
квантового осциллятора как функция температуры.
(Единицы масштаба Л, 0.)
2) Естественно, что нас особенно интересуют
симметричные молекулы (например, Hg, Ng, Og и т. п.). К
сожалению, для них предыдущие формулы не вполне
верны. Однако серьезное рассмотрение этого вопроса здесь
не может быть проведено, так как это завело бы нас
слишком далеко в тонгости квантовой механики. Мы
удовлетворимся констатацией того, что при в > Г и в этом
случае имеет место «вымерзание» колебательных степеней
свободы. Для молекул с большим числом колебательных
степеней свободы следует, естественно, взять целый ряд
собственных частот v^, Vg, ... и соответствующих им
характеристических температур в^, во, ... ; при этом
вклады различных колебаний в энергию просто суммируются.
Вместо кривой на фиг. 27а мы получим тогда для с^
ступенчатую кривую, причем высота каждой ступеньки
будет равна -R. Для обычных температур играет роль,
очевидно, только первая ступенька.
§ 33. Квантование энергии колебаний 297
2. Твердое тело. Противоречие с теоремой Нернста,
установленное нами в § 32, п. 3, исчезает, если
рассматривать каждый атом твердого тела как «квантованный»
независимый осциллятор. Тогда при Г—>0, так же как и
в правом столбце C3.9), теплоемкость экспоненциально
стремится к нулю.
Эта идея была высказана уже в 1907 г. Эйнштейном i).
Однако произведенные Нернстом опыты показали, что
теплоемкость стремится к нулю не экспоненциально, а
медленнее. Причину этого легко понять — фактически
атомы колеблются не независимо друг от друга, а более
или менее крупными группами, в зависимости от
температуры (см. § 32, п. 3, а также § 35).
3. Обобщение на любые квантовые состояния. В об-
nicM виде молекулярная сумма состояний определяется
как
2o=Sg„e-'/"''^. C3.11)
n=0
От прежнего определения C3.2) формула C3.11)
отличается весовым множителем g^. Последний позволяет
объединить различные квантовые состояния с равной
энергией в одно слагаемое. Для колебаний двух неравных
атомов §^^=1, но уже в случае двух одинаковых атомов
значения g^ различны в разных членах суммы (ср. с
замечанием 2 в п. 1).
Зная Zq, можно при помощи формулы C3.6) найти
общую энергию U. При этом мы будем рассматривать
систему, состоящую не из одного моля, а из любого
числа частиц Л\ Кроме того, удобно будет
дифференцирование по р заменить дифференцированием по Т. Тогда
d dT d jL7^2 ^
~"Щ^~ '^~^df~~ df
И формула C3.6) принимает вид
U^NkT^^^. C3.12)
1) А. Einstein, Ann. d. Phys., 22.
298 Гл. IV. Обгцис принципы статистики. Метод ячеек
Для определения энтропии мы воспользуемся уравнением
B9.12а), из которого получим
S==Nk\nZ^+^ . C3.13)
Чтобы доказать это термодинамическим путем, про-
варьируем Г, считая объем V и другие параметры
постоянными. Тогда из C3.13) получим
dS = Nk^-^--^dT + ^-^. C3.13а)
На основании C3.12) второй член в правой части A3.13а)
принимает вид
-Nk^lnZ.dT^ -Nk^
И взаимно уничтожается с первым членом.
Следовательно, как это и должно быть при изохорическом процессе.
Тем самым формула C3.13) доказана с точностью до
аддитивной постоянной, значение которой определяется
теоремой Нернста. Комбинируя C3.12) и C3.13), получаем
S^Nk^{T\nZ,), C3.136)
Особенно просто выражается свободная энергия F = 17 —
— TS, Из формулы C3.13) непосредственно получаем
F= -^NkT In Z^, C3.14)
§ 34. КВАНТОВАНИЕ ВРАЩАТЕЛЬНОЙ ЭНЕРГИИ
Значения вращательной энергии, допускаемые
квантовой механикой, также образуют дискретную
последовательность. Однако формула Планка для линейного
осциллятора C3.3) в этом случае не имеет места. В
простейшей модели ротатора (двухатомная молекула) возможны
следующие значения энергии:
s„ = n(n + l)|J, п = 0, 1,2, .... Й = |^. C4.1)
§ 34, Квантование вращательной энергии 299
Квантоюмеханическое доказательство формулы C4.1)
опирается на теорию шаровых функций, но здесь его
придется опустить.
Для вычисления средней энергии найдем сумму
состояний C3.11). Фигурирующий там весовой множитель
g^ в данном случае равен
gn = 2n + l. C4.2)
Это следует из теории шаровых функций, согласно
которой кроме зональных шаровых функций, зависящих
только от 9, имеется еще 2л присоединенных
поверхностных шаровых функций, зависящих от 9 и ср. Эти
геометрически различные состояния соответствуют одной и той
же энергии C4.1). Поэтому каждое значение энергии в
последовательности C4.1) является, как принято говорить,
Bл4-1)-кратно вырожденным. Весовой множитель g^
соответствует, следовательно, суммированию по
координатному пространству, которое должно быть проведено
наряду с суммированием по пространству импульсов.
Поэтому, в согласии с C3.11), сумма состояний имеет вид
Zrot= S Bп+1) 6-^(^+1)9 ='1 + Зе'2Ч5е-б«-Ь..., C4.3)
п=0
где
Вычислим в для особенно интересного случая
молекулы Hg. Из спектроскопических данных, полученных
при изучении линейчатого спектра, следует ^)
/ = 0,46.10-40 8'Cm\
Вспомнив значения ^ = 1,06.10"^^ эрг-сек и А: = 1,38-10"^*
эрг/град (см. § 20), получаем для в, согласно C4.3а),
в ^80° К. C4.4)
^) Для расстояния а между атомами водорода из формулы
/ = 2mjj(a/2J при mjj = 1,67-10-24g и / = 0,46.10-*о г^см^ получаем
значение а = 0,74-10-8 еж = 0,74 А. Эта величина того же порядка,
что и диаметр атома водорода. Отсюда видно, что принятое нами
значение момента инерции / является вполне разумным.
300 Гл. IV. Общие принципы статистики. Метод ячеек
Отсюда следует, что при Т < 80° К уже второй член
ряда C4.3) экспоненциально мал но сравнению с первым.
Таким образом,
Zfot ^ 1, InZrot ^ 0.
Согласно C3.12), это означает, что вращательные
степени свободы ничего не вносят в значения U и с^,.
Теплоемкость сводится к чисто трансляционной теплоемкости
с„ = ^. C4.5)
Водород оказывается, так сказать, «одноатомным». Это
совпадает с результатами Эйкена, которые в конце § 31
были охарактеризованы как самая серьезная трудность,
стоящая перед классической статистикой. Теперь же эти
результаты являются блестящим подтверждением
квантовой статистики^).
Рассмотрим также другой предельный случай, когда
Г > в, т. е. д< 1. Ряд в формуле C4.3) сходится тогда
очень плохо, так как его члены стремятся к нулю лишь
при очень больших значениях п. По своему
аналитическому характеру этот ряд принадлежит к группе 6-функ-
ций, которые применяются не только в теории
эллиптических функций, но и в теории теплопроводности (ср.
т. VI, Дифференциальные уравнения в частных
производных физики, § 15). По аналогии с «формулой
преобразования б-функций» [т. VI, формула A5.8)] можно
было бы и в нашем случае преобразовать ряд к виду,
допускающему удобное и точное численное суммирование.
Однако здесь достаточно будет приближенной оценки для
предельного случая Г > в (более точный расчет приведен
в задаче IV.6).
В силу малости q величину
р„ = п(п-Ы)? C4.6)
1) По тем же самым причинам не возбуждается вращение
электронной оболочки и атомного ядра. Характеристическая
температура в исключительно велика в первом случае из-за малости
массы электюона, а во втором—из-за малости радиуса ядра
(соответственно 6 = 105 и 0 = 10^ град).
,§ 34. Квантование вращательной энергии 301
можно рассматривать как непрерывную переменную,
изменяющуюся вместе с л от О до оо. Разность между р^
и /?„_! равна
A/? = /?^-/?„^i = w(n+l)g-w(w-l)g = 2Aig C4.6а)
и для всех конечных значений п, при которых
соответствующие члены в формуле C4.3) имеют еще заметную
величину, является сколь угодно малой. В дальнейшем мы
обозначим ее через dp. Для весового множителя,
фигурирующего в C4.3), без большой погрешности ^) можно
написать 2п^- \—dplq. Таким образом, мы получаем
оо
1
^rot =
. = \\or^dp = \ = ^. (МЛ)
о
Из уравнения C3.12) находим теперь
U.= NkT^^lR^==NkT, % = Nk. C4.7а)
Пз последней формулы видно, что вклад вращательного
движения в молярцую теплоемкость при Г > в составляет
как раз R.
Температурный ход средней энергии и теплоемкости,
приходящихся на вращательные степени свободы,
изображен на фиг. 28 и 28а. Так же как и в случае
колебаний, здесь ясно видно постепенное вымерзание
вращательных степеней свободы при Г < Н. Полный вклад в
теплоемкость, равный R = 2 кал/град-моль, они вносят только
при Т>в.
Чтобы установить прямую связь с работами Эйкена,
мы говорили все время о водороде. На самом деле под
«водородом» следовало бы понимать «полутяжелый»
водород HD (D — дейтерий), так как для обычного водорода Hg
(как, впрочем, и для дейтерия Dg) возникают
осложнения, указанные в § 33, п. 1. Однако описанный здесь
качественный ход теплоемкости существенно не изменится и для
газов, молекулы которых состоят из двух одинаковых
атомов.
^) Оценку погрешности можно получить, воспользовавшись
формулой суммирования Эйлера.
302 Гл» IV, Общие принципы статистики. Метод ячеек
На квантовом рассмотрении вращения сложных
многоатомных молекул мы не будем останавливаться. Уже
фе
Фиг. 28. Молярная энергия квантового
ротатора как функция температуры. (Единицы
масштаба ЯО, 9.)
в частном случае симметрично построенных молекул,
например NHg, СН4 и т. п., появляется новая^степень
Фиг. 28а. Молярная теплоемкость
квантового ротатора как функция температуры.
(Единицы масштаба Л, 9.)
сюбоды, связанная с вращением вокруг оси симметрии. В
связи с этим в числе возможных значений энергии C4.1)
§ 36, Дополнения к теории излучения и теории твердого тела 303
появляются члены, содержащие момент инерции
относительно этой оси и зависящие еще от одного индекса
суммирования. Соответственно сумма состояний
представляется уже двойным рядом. Новая степень свободы приводит
при достаточно высоких температурах к повышению
теплоемкости одного моля на R/2. При понижении
температуры эта степень свободы вымерзает, начиная с некоторой
температуры, зависящей от значения соответствующей
характеристической температуры U'. При этом молярная теп-
:юемкость многоатомного газа с^ — ЪВ. переходит в
теплоемкость двухатомного газа с„ = 5/?/2. При больших Н'
(что соответствует малому моменту инерции относительно
оси симметрии) многоатомная молекула и при обычных
температурах ведет, себя как двухатомная.
§ 35. ДОПОЛНЕНИЯ К ТЕОРИИ ИЗЛУЧЕНИЯ
И ТЕОРИИ ТВЕРДОГО ТЕЛА
В § 20 была получена формула для средней энергии
линейного осциллятора
?^ = ,..Д'_1 * C5.1)
Такой энергией обладает осциллятор, находящийся в
полости в равновесии с падающим на него черным
излучением при температуре Т.
Выше уже было указано, что эту формулу можно
вывести гораздо проще при помощи статистики. Для
доказательства придется только вспомнить рассмотрение
линейного осциллятора в §33, п. 1.Там, правда, не было
речи о полости с излучением, однако предполагалось,
что, во всяком случае, осциллятор находится в
равновесии с окружающей средой. При этом было безразлично,
каким именно путем установилось равновесие — в
результате ли излучения, или при столкновениях с молекулами
газа. Таким образом, в согласии с квантовой гипотезой
Планка C3.3), равновесная энергия дается формулой
C3.6а), в которой следует лишь опустеть множитель L
(число Авогадро), поскольку в данном случае речь идет
не о моле, а об одном отдельном осцилляторе. Получа
ющийся при этом результат в точности совпадает с C5.1).
304 Гл. IV. Общие принципы статистики. Метод ячеек
1. Метод собственных колебаний. Для статистики
безразлично, оперирует ли она с материальными
объектами или с состояниями, с экономическими величинами
или с погрешностями измерений и т. д. Особенно
плодотворным оказалось ее применение к изучению
собственных электромагнитных колебаний полости
(параллелепипеда или куба).
В т. II (Механика деформируемых сред, § 44) было
изучено число и расположение собственных колебаний
упругого параллелепипеда и было показано, что в случае
полости, свободной от вещества или заполненной только
излучением, появляются некоторые упрощения. В
последнем случае элементарное соотношение для синусов строго
удовлетворяет краевым условиям (Ftang = 0). В первом
случае это возможно только при так называемых
смешанных краевых условиях.
К этому необходимо еще добавить, что уравнение
D4.16а) из т. II относилось к числу ? упругих
собственных K0w4e6aHMU с частотой меньше v. Чтобы применить
ого к электромагнитному полю, положим
с^ = с = скорость света, с^ = оо.
В результате получаем уравнение, выведенное Релеем
в 1900 г.,
S = ^^^ C5.2)
Здесь F —объем параллелепипеда или куба. Отсюда
находим число собственных колебаний в интервале cfv
rf5 = 5^v2^. C5.2а)
Если в соответствии с законом равномерного
распределения энергии по степеням свободы приписать каждому
собственному колебанию среднюю энергию кТ (а не А7'/2,
так как необходимо учитывать и потенциальную энергию),
то для плотности энергии на интервал частот c?v получим
«v = ^2 = ?v^*7^. C5.3)
^^ 3d Дополнения к теории излучения и теории твердого тела ЗОЯ
:.)т(> закон Релея —Джинса B0.16). Как известно, он
противоречит опыту, ибо приводит к выводу, что Mv—»оо
при V—>оо.
Если, следуя Дебаю ^), рассматривать каждое
собственное колебание как квантованный осциллятор (см. также
§•^33, п. 3) с энергией C5.1), то вместо C5.3) из C5.1)
и C5.2а) получим
_и dz _ 8тсу^ Лу /ос: /\
Т. е. закон излучения Планка B0.39). Планк привел этот
метод в четвертом издании своей книги A921 г.),
охарактеризовав его как «чрезвычайно простой вывод» закона
излучения.
2. Теория теплоемкости твердого тела по Дебаю.
Главное различие между упругим телом и полостью с
излучением состоит в том, что для упругого тела благодаря
ого дискретной структуре число степеней свободы
ограничено, в то время как для полости с излучением это
число бесконечно велико (по крайней мере, насколько это
известно в настоящее время). Так как число собственных
колебаний тела равно числу его степеней свободы, а
каждый из атомов, составляющих решетку, имеет 3 степени
свободы, то число собственных колебаний ? ограничено
сверху значением (для одного моля)
1^ = 3L (L — число Авогадро). C5.5)
Величине ^^ соответствует наибольшая возможная частота
собственных колебаний v^^. Дебай обрезает спектр
собственных колебаний (как это подробно описано в т. II)
при частоте v = v^. Значение энергии твердого тела
вычисляется тогда по формуле
M= ^ Udl, C5.6)
причем квантованное значение энергии U дается формулой
C5.1). Предельная частота v^ определяет характеристи-
^) Debye, Ann. d. Phys., 33.
300 Гл. J\. Обилие принципы статистики. Метод ячеек
ческую температуру твердого тела 9 (так называемую
дебаевскую температуру)
в=^. C5.7)
Вычисление интеграла C5.6) дает (как показано в т. II,
§ 44)
" = ?^» с, = ^/?(^|-УприГ<в, C5.8а)
й = 3/гГ, с„=3/?^6 кал/град'моль при Г > в. C5.86)
Предельный случай C5.86) приводит к закону Дюлонга
и Пти. В предельном случае C5.8а) средняя энергия
зависит от температуры так же, как и в законе Стефана —
Больцмана (в теории излучения); для с^ получается закон
Дебая: молярная теплоемкость твердого тела
пропорциональна кубу температуры. Эта зависимость блестяще
подтвердилась на опыте как качественное правило (не
учитывающее особенностей кристаллической структуры).
Тем самым были внесены необходимые поправки в метод
Эйнштейна, упомянутый в § 33, п. 2. Так же как и
формула Эйнштейна, закон Дебая удовлетворяет теореме
Нернста, но теперь при стремлении Т к абсолютному
нулю теплоемкость стремится к нулю не экспоненциально,
а по параболе третьего порядка. Причина этого ясна.
Эйнштейн рассматривает атомы твердого тела как
независимые друг от друга осцилляторы. Дебай группирует
молекулы, которые при собственных колебаниях движутся
совместно^). Длины волн этих групп тем больше, чем
ниже температура; в той же мере растет корреляция
между молекулами, что делает несостоятельным предпо-
.гожение Эйнштейна о независимости колебаний атомов.
Качественную иллюстрацию поведения й и с^ в случае,
промежуточном между предельными случаями C5.8а) и
C5.86), дает фиг. 73 в т. П.
Таким образом, отпадает и последняя из
перечисленных Кельвином трудностей статистической физики.
^) В том, что эти группы допустимо рассматривать (в
противоположность атомам решетки) как независимые друг от друга
образования, можно убедиться из факта постоянства скорости звука
для всех частот.
§ 36, Сумма состояний в V-пространстве 307
§ 36. СУММА СОСТОЯНИЙ в Г-ПРОСТРАНСТВБ
В § 29 было показано, что метод Больцмана нельзя
непосредственно применить к реальным телам. Однако,
объединяя большое число элементарных ячеек в один
элемент, можно получить уравнения, которые правильны
с практически вполне достаточным приближением.
Поскольку термодинамические зависимости являются
совершенно общими, весьма желательно иметь и строгое
обоснование. Эту проблему мы будем рассматривать
одновременно с задачей о представлении основного уравнения
статистики в Г-пространстве, что понадобится в § 37.
1. Условие Гиббса. Приводимое здесь рассмотрение,
проливающее новый свет на гипотезу Больцмана о
равновероятности одинаковых фазовых ячеек, принадлежит
Гиббсу^). Если взять два фазовых элемента в различных
участках фазового пространства, то вероятности найти
в одном из них фазовую точку какой-либо конкретной
системы могут оказаться различными и зависящими
от времени. С полной общностью для них справедлива
формула
Аг(у = /(/?!, ...,/?F, 91, ...,gF, 0^- C6.1)
Здесь AS, как и раньше, означает фазовый элемент, h^
(где F = Nf) — величину элементарной ячейки, а функция
/(/?, д, t) показывает, как меняется вероятность от точки
к точке и с течением времени. Зта общая формула для ^w
играет, например, роль при выяснении вопроса о том,
как растут с течением времени погрешности, связанные
с неточным заданием начальных условий механической
системы.
В термодинамической статисаике рассматриваются
исключительно состояния равновесия, когда /(/?, q) не
зависит явно от времени. Отсюда вытекают некоторые
следствия.
^) J. W. G i b b S, Elementare Grimdlagen der statistischen
Mechanik, Leipzig, 1905. (См. перевод: Дж. В Гиббс, Основные
принципы статистической механики, М.—Л., 1946.)
308 Г л, IV, Общие принципы статистики. Метод ячеек
Рассмотрим два фазовых элемента AQ и Д2', один
из которых представляет собой результат временной
эволюции другого. Разность соответствующих им времен
пусть будет At = t' — t. В силу C6.1) соответствующие
вероятности равны:
^, ^w^ = f{p\q\t + At)^
Aw = f{p,q,t)-^, ^w' = f{p\q\t + At)
Эти величины равны друг другу, так как во все моменты
времени данный фазовый элемент заключает одни и те же
фазовые точки. Таким образом, Аш = Аш'. Кроме того,
AQ = AQ' (согласно теореме Лиувилля). Принимая во
внимание, что в состогниях равновесия функция / не
зависит от времени, видим, что если совокупность точек (/?, д)
при движении системы переходит в (/?', д'), то
f{p.q)=f{p',q')- C6.2)
Итак, имеет место следующая теорема: в состоянии
термодинамического равновесия вероятность {отнесенная к
одной элементарной ячейке) является интегралом дви^урсения.
Канонические системы всегда имеют по меньшей мере
один интеграл движения — таковым является энергия
Е = Н {р, q). Каждая функция энергии
/ = /(Я) C6.3)
также представляет собой интеграл движения.
Наряду с энергией в аргументе могут содержаться
и другие интегралы движения, например энергия части
системы (если последняя распадается на независимые
части), момент количества дви:нсения (если система может
свободно вращаться) и т. п. Однако все интегралы движения,
кроме Ну постоянны только условно. Ничтожные
изменения системы, которые зачастую совсем не нарушают
общего баланса энергии, но играют важную роль в
установлении равновесия, устраняют случайные интегралы
движения. Вспомним, например, случай идеального газа,
когда молекулы практически изолированы друг от друга,
но при столкновениях обмениваются энергиями. Можно
привести также пример космической материи, вращение
которой не ограничивается «стенками сосуда», так что
при статистическом рассмотрении наряду с энергией
§ 36. Сумма состояний в Т-пространстве 309
должен приниматься во внимание и момент количества
движения.
Тем не менее в общем случае термодинамическая
статистика характеризуется только некоторой функцией
энергии. Надо лишь установить вид этой функции. Здесь
место гипотезы Больцмана об априорной вероятности
занимает гипотеза Гиббса: две связанные механические
системы, находящиеся в состоянии статистического
равновесия, остаются в состоянии равновесия и в предельном
случае исчезающего взаимодействия {т. е, при разделении
систем).
Если, например, два тела с разными температурами
были приведены в соприкосновение и температуры их
в результате этого вы равнялись, то равновесная
температура останется неизменной и при возможном (в
дальнейшем) разделении тел. Точнее говоря, температура заметно
не изменяется, пока в общем балансе энергии можно
пренебречь работой против сил сцепления (что, например,
имеет место для тел достаточно больших размеров).
Пусть Н^и ^2 —функции Гамильтона двух подсистем,
di Н = Н у^-{- Н ^-гЬН — интеграл движения для всей системы
с энергией связи ЬН {ЬН--^0). Тогда в силу C6.3)
функции распределения будут
fiiHi), /ЛН,) и f{H)^f{H, + H,).
Гипотеза Гиббса приводит к уравнению
f,{H,)-U{H,) = f{H, + H,). C6.4)
Действительно, вероятность найти каждую систему в
некотором определенном состоянии должна быть одной и
той же как до, так и после разделения.
2. Связь с методом Больцмана. Заметим прежде
всего, что в пределе при 8Я—>0 левая и правая части
уравнения C6.4) представляют собой интегралы
движения, так что гипотеза Гиббса (так же как и гипотеза
Больцмана) относится лишь к начальным условиям.
Более того, взяв производную от C6.4) по Hi и Н^,
получим
/' (Я,+в,) = /; (Я,). /, (Я,) = л (Яj) • /; (я,).
310 Гл. IV. Общие принципы статистики. Метод ячеек
Следовательно, отношения
/, (Н,) /2 {Но) Р
должны быть постоянны, так как в противном случае
функции 'различных аргументов не могут быть равны
друг другу. Интегрируя, сразу находим функцию
распределения Больцмана
/(Я) = е-«-Р^. C6.5)
Постоянный множитель е~* введен здесь по аналогии
с B9.10). Он определяется из условия равенства единице
суммы всех вероятностей 2^^ = 1| таким образом,
гр«= ^e-^H^^z. C6.6)
Величина Z есть сумма состояний в Г-пространстве (пока
еще она записана в виде интеграла). Множитель 1/Л^
уже предвещает квантовомеханические поправки,
рассматриваемые в п. 3.
Из суммы состояний можно вывести все величины,
характеризующие равновесие. В частности, вариация Z
по Н дает
Для средней энергии получаем, аналогично B9.16),
^^¦-Чг' C6.8)
Температура и энтропия определяются при помощи
второго начала
TbS = bU-\bH.f{p,q)'^^.
Второй член здесь характеризует изменение энергии,
как это и требуется вторым началом. Действительно,
варьируя Н при неизменном распределении молекул
в Г-пространстве, видим, что фигурирующий здесь
интеграл представляет собой не что иное, как работу, совер-
§ 36. Сумма состояний в V-пространстве 311
шаемую над системой. Пользуясь C6.7), получаем отсюда
]де вариация 81nZ учитывает изменение как Н, так и р
(вклад от последнего характеризуется третьим слагаемым).
11а основании C6.8) это равенство приводится к виду
TbS^\b^U + lnZ),
откуда, сравнивая величины, стоящие до и после знака
варьирования, и интегрируя, находим
Г = 1, S = kQnZ + ^'^. C6.9)
Неопределенный пока множитель пропорциональности к
ость не что иное, как постоянная Больцмана, как видно
из сопоставления C6.9) с уравнением Больцмана B9.1).
Из уравнений C6.6) и C6.9) следует далее
5 = А(а + р?/) = А(а + рЯ)= -klnf{p,q).
Черта здесь означает усреднение. В явном виде мы имеем
S=-k^f.\nf.^. C6.10)
Сравнивая это с уравнениями B9.5) и B9.5а), согласно
которым
i
получаем
S = klnW, C6.11)
что совпадает с B9.1).
Действительно, уравнение C6.5) из всех возможных
функций распределения /, соответствующих заданной
энергии ?/", дает как раз те, которые обращают в
максимум In Ж в C6.11). Расчет протекает здесь совершенно
laK же, как и в § 29. Таким образом, гипотезы Гиббса
11 Больцмана эквивалентны.
Пусть теперь система распадается на N одинаковых
независимых частей с / степенями свободы каждая (соот-
312 Гл. IV. Общие принципы статистики. Метод ячеек
ветствующие им координаты и импульсы обозначим
)о1» • • •» 9/» /?Г» . .., 9/, .. ., /?Т, . . ., 7^/^)- Тогда полная
энергия системы равна
Я = Я„(р', q')-\-H,{p", 0+ ... ^H,{pi^), ф^)).
Отсюда непосредственно следует, что сумма состоянии
вырождается в произведение IS сомножителей
Z=.\e-^ Но (Р', Q') ^ . .С е-9 Но (р W, а^) ^
отличающихся друг от друга только обозначением
переменной интегрирования. Поэтому все они одинаковы, и,
следовательно,
Z = Z^, C6.12)
где
Z^=\e-^Ho(p,Q)^ C6.13)
есть сумма состояний в ^-пространстве. Тем самым
устанавливается связь с прежним методом рассмотрения.
3. Переход к квантовой статистике. В общем случае
следует исходить все же из суммы состояний в Г-про-
странстве. Распадение Z на множители Z^ связано с очень
специфическими предпосылками, которые не выполняются
уже для квантового идеального газа, как это будет
показано в § 37. Переход к квантовой статистике будет
проведен в несколько этапов, первые два из которых уже
пройдены при рассмотрении ji-пространства. Третьему
этапу, на котором оказывается необходимым переход к
Г-пространству, посвящается § 37. Четвертый этап —кван-
товомеханический вывод суммы состояний — требует
такого углубления в квантовую механику, что мы
удовлетворимся здесь лишь переносом классической суммы
состояний в квантовую область и отошлем читателя к
специальной литературе ^).
^) М. D el b ги ск, G. МоИёге, Sitzungsbcr. PreuB. Ак.
Wiss., Phys.-Math. Klasse, \r. 1 A936).
§ 36. Сумма состояний в I-пространстве 31.5
Первый этап перехода к квантовой статистике состоит
в замене интеграла состояний C6.6) суммой состояни!!
Z=2]e-P^(">. C6.14)
(п)
Суммирование здесь производится по всем фазовым
ячейкам величины h^ в Г-пространстве. Результаты
получаются практически те же самые, что и в C6.6), так как под-
интегральное выражение очень мало меняется внутри
одной фазовой ячейки. Существенна лишь величина
фазовой ячейки, ибо от нее зависит значение постоянной
энтропии [ср. C1. 5в)]. Но величина фазовой ячейки уже
учтена в формуле C6.6) выбором выражения. dQ/h^ для
фазбвого элемента. Действительно, формула C6.6) есть
не что иное, как апроксимация квантовомеханической
суммы C6.14) в смысле эйлеровой формулы суммирования.
Надо еще показать, что не только сумма состояний
C6.6), но и выражение C6.14) распадается на множите-
/ш в соответствии с C6.12). Если разделить фазовое
пространство отдельных молекул на ячейки величины Л^ то
:шергия Е (п) в C6.14) представится в виде
Е{п) = ^щв^. C6.15)
Здесь п^ — число молекул в определенной фазовой ячейке
[х-пространства, е-— энергия молекулы в этой ячейке.
Суммирование проводится по всем ячейкам в ji-простран-
стве; символ {п) в C6.14) означает, что суммирование
надо проводить по всем возможным разложениям общего
числа частиц
iV = 2^i. C6.16)
i
Весовой множитель g, который встречался в C3.11),
в выражении C6.14) отсутствует. Он неявно учитывается
условием, что каждое значение энергии пишется в сумме
столько раз, сколько оно встречается. Равенство
значений Е (w) в различных фазовых ячейках Г-пространства
обусловлено совпадением энергии е^ в различных фазовых
ячейках [х-пространства (вырожденные состояния молекул).
Это вырождение и дает весовой множитель в уравнении
C3.11).
УЛА Гл. IV. Общие принципы статистики. Метод ячеек
Пока мы 1)ассматриваем (как это делается в
классической механике) молекулы одного сорта как различимые
(индивидуализированные), определенное значение энергии
Е {п) может осуществляться многократно также и потому,
что молекулы могут различным образом распределяться
110 фазовым ячейкам, так что всегда л^ молекул
находится в первой ячейке, п^ — во второй и т. д. Число
возможных распределений задается, как мы знаем, формулой
B9.3). Если в сумме состояний C6.14) каждое распреде-
,11ение N чисел заполнения учитывать только один раз
(это указывается штрихом над знаком суммы), то в нее
надо ввести в качестве весового множителя величину
B9.3). Получим
е ' . C6.17)
Z,. C6.18)
z'l^z'l'^ . . . C6.17а)
Факториальные множители как раз совпадают с
полиномиальными коэффициентами. Каждое произведение
степеней
^1 ^2 • • •
встречается в последней сумме столько же раз, сколько
в сумме всех величин z^, возведенной в Л^-ю степень.
Следовательно,
плв
Z^Z",, Zo = 2zi=Se-% C6.1<))
i i
что совпадает с выражениями C6.12), C6.13). Величина
Zf^ есть сумма состояний в (^-пространстве, но здесь она
представлена как сумма по фазовым ячейкам ЛЛ
Второй этап перехода к квантовой статистике состоит
и том, что под ?^ понимаются энергии, соответствующие
Положим
Тогда
Z =
Z--~
Zj п^\п^\...
е-''---^.
¦ 2j пу\ п,\...
§ Зв. Сумма состоянии в V-пространстве 31 Г)
ис различным фазовым яче1'1кам, а различным квантовым
состояниям. Вместо суммы по различным фазовым яче1'|-
нам ji-пространства появляется сумма по квантовым
состояниям молекулы. Гипотеза Больцмана о
равновероятности фазовых ячеек заменяется гипотезой о
равновероятности квантовых состояни11. Возникаюищс в результате
этого изменения рассмотрены в § 33 — 35.
В то время как первый этап перехода к квантовой
статистике привел к определению постоянной энтропии,
на втором этапе появляются поправки к закону
равнораспределения. Необходимо устранить еще серьезную
погрешность, которая встретилась нам при выводе
формулы Саккура C1.5в). Это будет сделано на третьем этапе
(§ 37). Кроме того, там же мы придем к представлению
о вырождении газа. Результаты § 37 окажутся в согласии
с тепловой теоремой Нерп ста.
4. Анализ гипотезы Гиббса. Вернемся еще раз к
1^ыражению C6.5). Что означает в Г-пространстве
зависимость от ZT? Если изучается конкретная полностью
изолированная система, то энергия ее имеет вполне
определенное значение и, следовательно, нет никакого
распределения /(Я). Отсюда следует, что, пользуясь
выражением C6.5), мы имеСхМ в виду не вполне замкнутую
систему.
Вывод выражения C6.5) из C6.4) показывает, что,
действительно, здесь идет речь о системе, выделенной из
некоторой большей системы. Таким образом, каноническог
распределение C6.5) справедливо для термодинамической
системы в термостате.
Для замкнутой системы следует обратиться
непосредственно к уравнению C6.3). Тогда мы получим
{const в малом интервале U — bU<C Н <U-\-bL\
О вне этого интервала.
C6.20)
'>1о так называемое микроканоническое распределение
Оно имеет место, если гипотезу равновесия Гиббса
заменить условием постоянства энергии H = U = const.
Вычисления в Г-пространстве в этом случае менее просты,
316 Гл. IV» Общие принципы статистики. Метод ячеек
однако получающиеся таким путем результаты
практически совпадают с выводами из канонического
распределения, так как отклонения от среднего значения энергии
для не слишком малых систем по большей части
совершенно ничтожны. Действительно, возмущение, вносимое
термостатом, обычно имеет порядок величины энергии
связи, которой можно пренебречь (см., однако,
задачу 1V.9).
Для ^.-пространства все обстоит совершенно так же.
Так как переход к ^.-пространству предполагает, что
каждая молекула слабо взаимодействует со своим
окружением, то каждая молекула находится как бы в
термостате, роль которого играют остальные молекулы.
Поведение каждой отдельной молекулы в среднем может тогда
описываться при помощи канонического распределения
в ji-пространстве, но это и есть распределение Больцма-
на B9.10). Безразлично при этом, начнем ли мы
рассуждения с канонического или с микроканонического
распределения.
§ 37. ОСНОВЫ КВАНТОВОЙ СТАТИСТИКИ i)
1. Квантовая статистика тождественных частиц.
Согласно квантовой механике, тождественные частицы не
отличимы друг от друга. Это относится не только к
электронам в атомных оболочках или в металле (они уже
давно признаны неиндивидуализируемыми), не только
к фотонам или любым элементарным частицам, но в
равной мере и к атомам или молекулам газа, даже если
они отличаются друг от друга каким-нибудь особым
признаком (ионизация, возбуждение). Поэтому нельзя, как
это делалось в § 29, выделять отдельные частицы из
общего числа Л^* и распределять их в соответствии с
координатами /?|, q^ по ячейкам фазового пространства Д2^.
Можно отличать друг от друга только состояния системы,
т. е. не состояния s^ отдельной частицы, а состояния
^) Мы следуем здесь краткому, но очень содержательному
изложению Шрёдипгера (Schrodinger, Statistical
Thermodynamics, Cambridge 1948. (См. перевод: Шрёдингер,
Статистическая термодинамика, М., 1948.)
§ 37. Основы квантовой статистики 317
всего газа, энергии которых даются формулой
Е{п) = 2щч. C7.1)
Поэтому с квантовой точки зрения имеет смысл не
прежняя сумма состояний B9.15), относящаяся к отдельной
молекуле
2^x = Zo = 2e-P*S C7.2)
а только «сумма состояний газа» C6.14)
Zr = Z = 2'e-P^("). C7.2а)
Ее и следует положить в основу дальнейших рассуждений.
В силу неразличимости частиц обмен двумя
частицами между двумя различными ячейками не приводит и
новому состоянию. Таким образом, каждое распределение
частиц, удовлетворяющее условию C6.16), должно
учитываться только один раз. Соответственно факториальный
множитель (статистический вес) в C6.17) заменяется
единицей. Поэтому, как и в C6.17), нужно употреблять
знак суммы 2* Теперь —без факториального множите-
ЛЯ — произвести суммирование уже не так легко. Прежде
всего теперь нельзя исходить непосредственно из суммы
состояний в J1-пространстве. Переход к ji-пространству
возможен только в случае классического идеального газа.
Прежде чем перейти к систематическому исследованию
суммы C7.2а), рассмотрим один предельный случай. Он
позволит нам понять, почему очень часто оказывается
почти достаточной сумма состояний C6.17).
В § 29 отмечалось, что в идеальном газе при
нормальных условиях примерно из 30000 ячеек максимум в одной
содержится одна молекула^) (прочие не заполнены).
В этом случае практически встречаются только числа
заполнения О и 1, так что числа п^\ в знаменателе в C6.17)
^) Среднее число частиц на одну фазовую ячейку есть е *,
максимальное его значение равно е~*. Среднее число ячеек на одну
частицу в соответствии с B9.14) и B9.15) составляет e^=Zo/N.
В частности, для одноатомного газа это дает: V/N |/ 2nmkTyh^f^30 000
при т= 1,67-10-2^ г.
318 Гл. IV. Общие принципы статистики. Метод ячеек
всегда равны 1. Это означает, что в данном предельном
случае суммы C6.17) и C7.2а) отличаются только на
множитель iV!, который не зависит от п^ и поэтому может
быть вынесен за знак суммы 2-
(п)
Отсюда вытекает, что для идеального газа при
нормальных условиях имеем
2^класс. = 2^0 ^N! 2квант. = Л'! Z. C7.3)
Следовательно, новая сумма состояний
и
hi Z ^ iV In Zo - iV (In Л^ ^ 1) C7.3a)
(если воспользоваться формулой Стирлинга для 1пЛ^!).
Таким образом, все предыдущие рассуждения
остаются в силе с точностью до аддитивной постоянной.
Последней, однако, уже достаточно, чтобы внести необходимую
поправку в формулу Саккура, так как если к значению
энтропии, даваемому формулой C1.5в), добавить
— A^(lniV —1), то получится как раз формула Саккура —
Тетроде
5 = A-iV In ^ {iTzmkTfl^ ^ . C7.4)
Выражение в правой части пропорционально iV, как это
и должно быть. Однако оно еще не удовлетворяет
теореме Нернста. Согласие с последней достигается только
при более точном вычислении суммы, которое можно
провести при помощи так называемого метода Дарвина —
Фаулера.
2. Метод Дарвина —Фаулера^). Этот метод
используется здесь для того, чтобы учесть условие C6.16).
Связывая этот раздел с именами Дарвина и Фаулера,
необходимо сделать известную оговорку. Названные
авторы применяли свой метод к классической статистике
1) С. G. Darwin, R. Н. Fowler, Phil. Mag., 44, 450,
823 A922). См. также R. Н. Fowler, Statistical Mechanics,
Cambridge, 1929.
§ 37. Основы квантовой статистики 319
(см. цитированные выше работы от 1922 г., которые
предшествуют появлению собственно квантовой
статистики). Если бы Дарвин и Фаулер проводили вычисления,
используя сумму состояний Z, то в их распоряжении было
бы только выражение C6.17). Но оно, как показывают
вычисления, йе приводит ни к чему другому, кроме
классического соотношения C6.12) между Z и Z^, т. е. не дает
никаких новых результатов. В самом деле, в
классическом случае, как видно из выражений C6.17)— C6.19)^
условие C6.16) можно учесть вполне элементарным путем.
Дарвин и Фаулер ставили себе целью показать, как
можно в статистике Больцмана учесть условие
постоянства энергии
f/==2ni8i C7.5)
в том случае, когда л^ — малые числа. Так как в их
методе существенно, чтобы слагаемые в C7.5) были
целыми числами, то значения е^ нужно «измерять столь
малыми единицами», чтобы все они приближенно могли
рассматриваться как целые числа. Обозначим эту (еще
явно не введенную) малую единицу посредством е^. Ввести
ее необходимо для того, чтобы функцию комплексного
переменного С, соответствующую нашей функции F(C)
[см. формулу C7.8)], можно было разложить по целым
степеням С и применить к ней теорему Коши. Однако
неизбежный при этом предельный переход ?о "~^ О
приводит к разбиению шкалы энергий на бесконечно малые
отрезки, что противоречит конечности фазовых
элементов. Иордан упоминает об этом в своей книге ^), но, по
его выражению, «не проводит более подробной дискуссии
по поводу этих обстоятельств, мешающих нашему
рассмотрению». Условие постоянства энергии C7.5) уже
учтено в самом выражении для суммы состояний.
Последнее было получено в § 36 другим путем, так что для
уравнения C7.5) нам больше не требуется метод
Дарвина—Фаулера. Теперь, однако, появляется еще условие
постоянства числа частиц C6.16), которое в квантовой
статистике уже не является тривиальным. Его также
1) Р. Jordan, Sammlung Wissenschaft, 2 Aufl., Bd. 87, Anm.
2, S. 33, Vieweg, 1944.
320 Гл. IV. Общие принципы статистики. Метод ячеек
МОЖНО учесть методом Дарвина — Фаулера, следуя Шрёдин-
геру^), причем в данном случае законность этого метода
даже менее проблематична, ибо все слагаемые в C6.16)
по самой сути дела являются целыми числами.
Для вычисления суммы состояний C7.2а) обратимся
к выражению C6.18). Подставив значение энергии из
C7.1), получим
Z = 2'2?^z^2... C7.6)
Если просуммировать это выражение по всем w^, то
возникнет множество членов, не входящих в сумму
состояний C7.6). Их можно уничтожить при помощи
искусственного приема: следует заменить величины z^ из C6.18)
на Сг^ и произведение
nz^^i на C'^iHz^. C7.7)
г i
Бырая^ение, возникающее при этом после суммирования
в C7.6) по всем п^, обозначим через Г(С). Разложим его
по степеням С и обратим внимание на ту группу членов,
которые умножены на С^. В силу C7.7) это и будет как
раз наша сумма состояний C7.3а). Итак, можно написать
y(C)=...4-C^Z+... C7.8)
Вторым искусственным приемом члены с С отделяются
от всех остальных. Следуя Дарвину и Фаулеру, мы
используем теорему Кош и о вычетах; тогда
Z = 2^^y(L)r^"-*rf:. C7.9)
Здесь С рассматривается как комплексная переменная:
интеграл берется по замкнутому контуру, окружающему
точку С = 0 и не содержащему внутри никаких других
особенностей подинтегральной функции. При этом
исчезают все члены степенного ряда, обозначенные в C6.8)
многоточием, и остается только вычет (член, содержащий
С"^), который, согласно формуле C7.9), непосредственно
дает Z.
1) S с h г о d i п g е г, Statistical Thermodynamics, Cambridge,
1948, Ch. VII. (См. перевод: Шрёдингер, Статистическая
термодинамика, М., 1948.)
§ 37. Основы квантовой статистики 3ZI
3. Статистика Бозе Эйнштейна и Ферми — Дирака.
Исследуем теперь подробнее вспомогательную функцию
Г (С). Заменяя в выражении C7.6) 2 на Cz и не принимая
во внимание условия C6.16), получаем
Y{Q=- 2 (^гГ^- 2 (Сг,^... C7.10)
«1=0 «2=0
Суммирование легко выполняется, если числа п^, как это
уже указывалось в C7.9), независимо принимают все
значения
ni = 0, 1, 2,...
Тогда мы имеем просто
i
С точки зрения квантовой механики собственные
функции системы являются при этом симметричными
функциями координат ее составных частей. Этот случай был
рассмотрен Бозе в 1924 г. в применении к фотонному
газу и вскоре после этого распространен Эйнштейном на
газы материальных частице).
Возможен, однако, и другой случай, также
реализующийся в природе и соответствующий антисимметричным
собственным функциям, когда
Аг^ = 1 или 0.
Этот случай был введен в квантовую механику Ферми
в 1926 г. (причем он ссылался на принцип Паули) и
независимо от него Дираком. Важнейшей областью
применения этой статистики являются электроны в металле.
Из уравнения C7.10) получаем тогда
Y{i) = l[{i + <:^,). C7.11а)
^) Мы здесь не будем подробнее останавливаться на этой связи
с симметричными или антисимметричными волновыми функциями
квантовой механики.
322 Гя. IV. Общие принципы статистики. Метод*ячеек
Оба случая можно объединить формулой
1^(^ = 11A TCzO=F^ C7.12)
i
(Верхний и нижний знаки приводят к статистике Бозе —
Эйнштейна или Ферми — Дирака соответственно.)
В обоих случаях F @) = 1 и вблизи точки С = О
функцию Y (С) можно разложить по степеням С, как это уже
предполагалось в формуле C7.8). В случае статистики
Ферми —Дирака У (С) представляет собой целую
(голоморфную) функцию, которая монотонно возрастает вдоль
положительной вещественной оси. В статистике Бозе —
Эйнштейна Y (С) — разрывная (мероморфная) функция,
обладаюш.ая полюсами во всех точках
Согласно определению z^ [см. C6.18)], эти точки
расположены на положительной части вещественной С-оси.
Если нормировать энергию так, что при любом i г^ > О,
то все С{ располагаются за точкой С=1, причем число их
постепенно возрастает до бесконечности с ростом С. При
C<(Ci)MHH. Y{(J) ведет себя монотонно и в случае
статистики Бозе — Эйнштейна.
Рассмотрим теперь логарифм подинтегрального
выражения в C7.9), обозначив его через F {(,):
/^(C) = lny(g-(iV + l)lnC. C7.13)
Функция F (С) обращается в + оо при С = О (так как
1пС= —с») и очень быстро убывает, пока в правой части
C7.13) преобладает второй член. Однако еще прежде,
чем С достигнет единицы, начинает преобладать первый
член, который, так же как и Г(ч), монотонно возрастает.
Таким образом, на положительном отрезке вещественной
оси имеется точка С©» в которой
F'(Co) = 0. C7.13а)
Значение второй производной F'^ {^о) в этой точке
положительно и очень велико, поскольку переход от монотонно
убывающей к монотонно возрастающей ветви F {(,) про-
§ 37, Основы квантовой статистики 323
исходит очень быстро, и притом тем быстрее, чем больше N,
Последнее обстоятельство будет использовано ниже.
В связи с появлением обеих «новых» статистик заметим
еще, что фактически новыми являются не статистики,
а объекты, к которым они применяются, —
тождественные частицы и их квантовые состояния симметричного
или антисимметричного типа.
4. Метод седловых точег:. Займемся вычислением
интеграла в формуле C7.9). Вводя определенный в C7.13)
логарифм подинтегрального выражения, вместо C7.9)
можно написать
Z = ^^e^(^NfC. C7.14)
Разложим F (С) в степенной ряд около точки С = Cq. Согласно
C7.13а), линейный член ряда исчезает, так что
/^(C) = i^(C„)f4F-(g(C-C„J+... C7.15)
Двумерные потенциальные функции не могут иметь
ни абсолютного максимума, ни абсолютного минимума,
так как в силу равенства АФ = 0 вторые производные
З^ФуЭж^ и д^Ф/ду^ всегда имеют разные знаки. Поэтому
поверхность и = const всегда является выпуклой в одном
направлении и вогнутой в другом, перпендикулярном
к первому. Таким образом, при дФ/дх = дФ/ду = О имеется
седловая точка.
То же самое относится и к поведению вещественной и
и мнимой V частей каждой комплексной функции F {i)
в тех точках С = Со, где F'(Co)==0. Мы видели, что если
двигаться вдоль положительной вещественной полуоси,
то в точке С = Со функция F {(,) имеет резкий минимум.
Следовательно, если двигаться вдоль линии,
параллельной мнимой оси, то в точке Со должен быть острый
максимум.
Интеграл C7.14) берется по контуру, охватывающему
нулевую точку, например по окружности. Если
последнюю провести через точку С^, то путь интегрирования
пройдет через крутой «перевал» («наиболее крутой подъем»
по одну сторону перевала и «наиболее крутой спуск»
324 Гл. IV. Общие принципы статистики. Метод ячеек
по другую его сторону). Иногда метод седловых точек
называют «методом перевала».
Существенный вклад в интеграл вносится только
малым отрезком вблизи вершины. Эту часть окружности
Фиг. 29. Плоскость С (С=а:+/г/)
вблизи точки перевала Со- Дана
качественная картина линий равной
высоты.
можно заменить отрезком касательной, а остальной частью
пренебречь (см. фиг. 29). На данном отрезке мы имеем
^ = Co + i?/, -Уо<У<+Уо'
C7.16)
Пренебрегая членами высшего порядка, получаем из
C7.14) и C7.15)
+У0
Z=Aei^(:„)J exp{-i-/"'(C„)j/='}d2/. C7.17)
-УО
Вводя новую переменную
видим, что если вторая производная /^''(Cq) достаточно
велика, то
+ 00
^=iVwh''''"' \ '-''''^=
§ 37, Основы квантовой статистики 325
Тогда, подставляя F(Co) из C7.13), находим
Очень поучительно проверить правильность этого
результата в классическом случае, хотя тогда, как мы
видели выше, сумму можно легко и совершенно строго
вычислить и не пользуясь методом Дарвина— Фаулера.
В этом случае для суммы состояний следует пользоваться
выражением C6.17а), а не C7.6). Если для суммы C6.16а)
[как и для суммы C7.6)] отбросить условие C6.16) и
внести знаменатели п-^\ п^\ ,,, под знаки частных сумм по
Wj, Wg..., то вместо F(C) получим соответствующую ей
«классическую» функцию
I'"K.nacc. = iV! П (S -^) =A^!e«^'+^2+-) = iV!e^z«. C7.19)
Как и раньше, оказывается, что применение суммы
состояний Zq в классической статистике вполне законно.
Из формул C7.13) и C7.19) вытекают соотношения
F(C) = CZo + lniV! -(Л^ + 1IпС, C7.20)
F'(:) = Zo--f-4 C7.20а)
F'{1) = ^. C7.206)
Отсюда в силу C7.13а) находим
^0 = ^^ /2^^^Р^ = (-Л^У'2о- C7.20b)
Формула C7.18) теперь принимает вид
Z = ^';^^ ^' ^ Zl C7.21)
Численные множители, согласно формуле Стирлинга
326 Гл, IV. Общие принципы статистики. Метод ячеек
B9.4а), равны единице. Действительно, при TV > 1
N\e^+^ е 1
(iV + l)^/27r(iV + l) (l+-i-Y l/l + JL
;^1. C7.21a)
Следовательно, формула C7.21) не дает ничего нового
и, как это уже отмечалось в п. 2, приводит к известной
формуле C6.19). Поэтому обращение к классическому
случаю может рассматриваться только как проверка
изложенного выше (отнюдь не простого) приближенного метода.
Кроме того, этот пример может служить заменой до сих
пор не приведенного доказательства того, что при большом
N F" (Со) — очень большая величина. Действительно,
последнее предположение существенно использовалось при
переходе от C7.17) к C7.18), и оно необходимо для
применения метода седловых точек. Из соотношения C7.206)
следует, что в классическом случае это условие выполняется:
поскольку Cq — конечное число, меньшее единицы,
величина F" (!(,q) растет пропорционально N до бесконечности.
Мы будем предполагать, что это верно также и в случае
квантовой статистики.
Пергеходя к квантовой статистике, составим логарифм
суммы состояний C7.18), воспользовавшись при этом
выражением C7.12) для У (С):
In Z = Т 2 In A Т Со^Л - {N +1) In Со -1 In [2uF" (Со)].
i
Здесь можно пренебречь единицей во втором члене,
а также последним членом. Действительно, поскольку
F"(Co) пропорционально iV, последний член по порядку
величины равен In iV и в пределе при TV —» оо становится
сколь угодно малым по сравнению со вторым членом.
Таким образом, получаем
lnZ= :р 2 ЬA Т Co2i)-A^lnCo. C7.22)
i
Величина Со определяется из уравнений C7.13) и C7.13а),
которые дают (если пренебречь единицей по сравнению с N)
^[1пГ(Со)] = -^. C7.22')
§ 38. BbipooicdeuHbie газы 327
Т. е. в силу C7.12)
^i?br^- C^-23)
Подставив в C7.22) и C7.23) выражение C6.18), получим
(полагая затем Со = ^"'")
In Z = Т S Ь A Т е-«-Р«0 + iVa C7.22а)
-ттёт =Л^- C7.23а)
I
Для больших значений а из C7.23а) получаем
e-«Zo = iV, а = In Zq - In Л^,
и выражение C7.22а) принимает вид
lnZ = A^lnZo-iV(lniV-l),
что находится в соответствии с C7.3а). Следовательно-
очень большие значения а (а > 1) соответствуют преде ль,
ному случаю обычных газов. По мере того как а становится
малой или отрицательной величиной, наступает
вырождение газа. Важнейшим примером вырожденного газа
являются электроны проводимости в металлах, к
изучению которых мы перейдем в последуюш;их параграфах.
§ 38. ВЫРОЖДЕННЫЕ ГАЗЫ
1. Распределения Бозе—Эйнштейна ц
Ферми—Дирака. Вследствие неразличимости тождественных молекул
в § 37 мы исходили из суммы состояний в Г-пространстве.
Однако результаты, заключающиеся в формулах C7.22а)
и C7.23а), содержат суммы только по ji-пространству.
Сумма в формуле C7.22а)
In F = Т S In A Т e-*-f •) =-- Ф (а, Р) C8.1)
328 Гл. IV. Общие принципы статистики. Метод ячеек
играет ту же роль, что и сумма состояний. Она
представляет собой термодинамический потенциал. Из
уравнения C7.22') следует
N=-^. C8.2)
Внутренняя энергия дается выражением C6.8)
и=-^, C8.3)
а числа заполнения - формулой C6.7)
1 дФ 1
C8.4)
Пользуясь C8.4), из C8.2) и C8.3) получаем
iV = 2n„ /7=S^iSi. C8.5)
Для энтропии в силу C6.9) имеем
8=^к{Ф^о.М + Щ. C8.6)
Дифференциал Ф равен
dф^ -Nda-Ud^-^^nidB^. C8.7)
По термодинамической терминологии Ф есть потенциал,
соответствующий независимым переменным а, р, е^.
Согласно C8.7), функция Ф, определенная
равенством C8.1), представляет собой один из видов
термодинамического потенциала. Она отличается от прежних
выражений последнего тем, что в данном случае наряду
с энергией зависимыми термодинамическими переменными
являются также и числа частиц (задача П.1). Для
совершенно изолированной системы энергия U = const и все
фазовые точки лежат на одной поверхности в фазовом
пространстве. При этом все равновеликие фазовые
элементы, расположенные между данной поверхностью
и поверхностью U+MJ, являются равновероятными.
В этом случае говорят о микроканоническом распределении
н микроканоническом ансамбле.
До сих пор мы рассматривали только канонические
ансамбли. О таковых можно говорить, если система
находится в тепловом контакте с окружающей средой. Для
§ 38. Выро(Н€денные газы 329
каждой температуры имеется каноническая функция
распределения, которая допускает флуктуации средней
энергии, обусловленные взаимодействием системы с
термостатом. Различие между результатами вычислений,
произведенных при помощи канонического и
микроканонического распределений, для систем с большим числом степеней
свободы очень незначительно, так как колебания энергии
чрезвычайно малы.
Функция состояния Ф относится к так называемым
большим каноническим ансамблям. Если а - заданная
величина, которая (в отличие от нашего дальнейшего
рассмотрения) не может быть исключена при помош;и
условия C8.2), то общее число частиц N также испытывает
флуктуации. Последние обусловлены тем, что в данном
случае система может обмениваться с окружающей средой
не только энергией, но и частицами. При этом, как
и раньше, в достаточно больших системах флуктуации
в общем очень малы, так что не возникает никаких
заметных различий. Мы удовлетворимся только этим
указанием и вернемся к каноническим распределениям,
предполагая величину а исключенной при помощи
равенства C8.2) (см. также § 40).
Из формулы C8.1) в предельном случае больших а
следует
1пУ = Ф = е-«2е-И = е-«го. C8.1')
Отсюда, согласно выражениям C8.2) —C8.4), получаем
или, исключая а,
(/=-iV-^p-, ni---^-^^, C8.8)
что находится в соответствии с B9.16). Здесь п^
—обычная больцмановская функция распределения.
Те же вычисления можно провести и при не слишком
больших значениях а, однако операция исключения а при
этом уже не элементарна. В любом случае вычисления
можно производить в J1-пространстве. Однако, в то время
как в Г-пространстве функция распределения всегда имеет
330 Гл. IV. Общие принципы статистики. Метод ячеек
ВИД б-«~Р^(«) [изменяется лишь кратность уровней
энергии Е{п)]^ в р.-пространстве мы получим вместо больцма-
новского выражения б"*"'^*! новые функции
распределения. В соответствии с C8.4) в случае Возе — Эйнштейна
и В случае Ферми —Дирака
1
е * + 1
C8.46)
Эти формулы дают возможность пользоваться ji-простран-
ством и при рассмотрении квантовых газов; для этого
следует лишь, как говорят обычно, перейти к новым
статистикам. Обычно формулы C8.4а) и C8.46)
обосновываются при помощи комбинаторных методов, в которых
уже в р.-пространствв принимается во внимание
неразличимость частиц и принцип Паули. Однако мы не будем
останавливаться на этом.
Очень показательно выражение для энтропии C8.6).
Из C8.4) следует, что
П1
Подставив это выражение в C8.6), получаем
или после элементарных преобразований
1^= - 2 \пМЪ, Т A ± «i)ln(l ± п,)\. C8.9)
i
Первое слагаемое здесь совпадает со значением InW
по Больцману [см. C6.10)]. Все же в целом представляет
собой квантовомеханическое выражение для In Ж,
поскольку именно величина
\nW^ = - S {/i In/i Т A ± /01пA ± А)} C8.10)
§ 38, Выроо*сденные вазы 331
обращается в максимум функциями распределения C8.4а)
и C8.46) при дополнительных условиях Л == 2/i = coiist,
f7= 2/i4 = const.
В случае статистики Ферми —Дирака это изменение
термодинамической вероятности легко охарактеризовать.
Заметим, что выражения
/?=1-п^, /1-л^ C8.11)
представляют собой соответственно вероятности не найти
в i-M квантовом состоянии ни одной молекулы или
обнаружить там одну молекулу. Из C8.10) получаем
1пРУ-= -S(/?b/? + /iIn/i). C8.12)
Это выражение отличается от больцмановского одним
членом под знаком суммы, определяющим больцмановскую
термодинамическую вероятность свободных мест. Отсюда
получаем распределение Ферми, если искать максимум
W' при условиях
/? + /' = !, ^fi = N, IiBji^U. C8.13)
То же самое верно и для случая Возе —Эйнштейна.
В этом случае также нужно принимать во внимание
больцмановскую термодинамическую вероятность
свободных мест. Пусть /i — вероятность того, что в i-м
квантовом состоянии находятся п молекул. Тогда
термодинамическая вероятность равна сумме выражений
больцмановского типа
lnW+=-S (Л In/? + /i In/| + /Пп/?+...). C8.14)
i
Если искать максимум выражения C8.14) при
дополнительных условиях
|/i») = l, S«/i"' = iV, Sn/l = t^. C8.15)
n=0 1уП i,n
TO получим распределение Возе — Эйнштейна C8.4а), а из
C8.9) найдем значение InW*. (Это показано в задаче
1V.7.)
332 Гл. IV, Общие принципы статистики. Метод ячеек
2. Степень вырождения газов. Рассмотрим квантовый
идеальный газ, состоящий из одноатомных молекул.
В этом случае сумму в C8.1) можно приближенно
заменить интегралом
оо
Ф= Т 5" 5 '"^^^ "^ e-«-PP2/2m)^,2^^,^ C8.16)
о
т. е.
Ф=-^(^)''5((сс), C8.16а)
где ^p^=^2mt и
)^ (а) = Т 4- ^ In A ^ е-«-0 Ytdt. C8.166)
V^ J
Поправка к формуле C8.16) приводится ниже в C8.26).
Используя формулы C8.16а) и C8.2) —C8.4), находим
число частиц
N=-^{2zmkT)'''x'{<^)< C8.17)
энергию
U = ^^{2^mkTf4Tx{^)=^^NkT [ -^J C8.17а)
и давление (учитывая, что Ф + Л''а= —ftF и dF =
= -SdT-pdV)
P-JW = -W i'^^rnkTf'^ТX(«)• C8.176)
Из двух последних равенств вытекает не зависящее от
5^ (а) соотношение
pV = ^U, C8.18)
которое, следовательно, сохраняется и в квантовой
статистике.
В силу C8.17) а является функцией аргумента
р = ^ ^!_—, C8.19)
§ 38. Выромсденные газы
333
измеряющего степень отклонения волпчины C8.17),
C8.17а) и C8.176) от соответствующих значений для
идеального газа. Назовем р степенью вырождения газа.
На основании C8.166), C8.17) и C8.19) имеем
•=-гы-М
Vtdt
в«+' ip 1
C8.20)
так что при больших а р мало. Малость р означает,
следовательно, что газ является классическим. Уже в § 37
Классический
предельный
случай
-2 ~Ч^
1
4
3
2
1
-/
^(-х/х')
-
-
о^^^^/
2
Ферми-у/дирак
1^
1пр = 1п(-Х')
3 4 5, 6 7
Бозе-Эйнштейн
Фиг. 30. Логарифм редукционного фактора энергии
и/и z=z—i/-^' в уравнении C8.17а) как функция
?>ОЛЬЦМдН
логарифма степени вырождения р =--—х'* определяемой
выражением C8.20). (Расчет Бауера.)
было показано, что для газа при нормальных условиях
р % 1/30000.
На фиг. 30 показано, как зависит от р энергия газов
Бозе — Эйнштейна и Ферми —Дирака. Эта зависимость
используется для исключения константы а из выражений
C8.17) и C8.17а).
Пример, когда значение р велико и газ сильно
вырожден, дают нам электроны проводимости в металле. В § 39
мы увидим, что электроны проводимости в первом
приближении ведут себя как свободные частицы.
Следовательно, они движутся в металле так же, как молекулы
334 Гл, IV, Общие принципы статистики. Метод ячеек
газа в сосуде. Однако электронный газ является в высшей
степени вырожденным. Для меди р ^ 5000 (при условии,
что каждый атом дает один электрон проводимости). Так
как электроны подчиняются принципу Паули,
электронный газ описывается статистикой Ферми — Диракаг. В то
время как для малых р газы Бозе — Эйнштейна и Ферми —
Дирака ведут себя одинаково, в предельном случае
больших р они заметно отличаются друг от друга, почему
и следует рассматривать их по отдельности.
3. Сильно вырожденный газ Бозе — Эйнштейна. До
сих пор из газов, подчиняющихся статистике Бозе —
Эйнштейна и заметно вырожденных, был известен только
газ световых квантов. Оказывается, однако, что
наступление сверхтекучести гелия при очень низких
температурах связано с вырождением. Во всяком случае,
сверхтекучесть возникает только у изотопа Не*, подчиняющегося
статистике Бозе — Эйнштейна. У Не^, который описывается
статистикой Ферми —Дирака, она отсутствует.
Для газа Бозе — Эйнштейна число частиц в силу C8.17)
и C8.20) пропорционально величине
Этот интеграл достигает максимума при а = 0. Значения
а < О следует исключить, поскольку тогда интеграл
расходится. Тот факт, что интеграл C8.21) при а = О еще
сходится и принимает значение С (^/2) = 2,612 (С ~ дзета-
функция), приводит к весьма неправдоподобным
следствиям. Именно, получается, что в конечном объеме можно
разместить лишь конечное число Бозе-частиц, причем это
число убывает с температурой пропорционально Т^^^ и при
абсолютном нуле обращается в нуль. Это противоречит
предположению о том, что в случае статистики Бозе —
Эйнштейна в каждом квантовом состоянии может
находиться сколь угодно много частиц. Этот вывод и в самом
деле неправилен. Он получился в результате замены
суммы C8.1) интегралом. При низких температурах, для
которых ре^ > 1, такую замену производить нельзя.
§ 38. Выроукденные газы 33j
Выберем начало отсчета энергии так, чтобы
наименьшее возможное значение ее равнялось нулю, и
пронумеруем уровни энергии согласно их величине:
О = Sq < е^ < eg < ...
Тогда средние числа частиц в различных энергетических
состояниях даются формулой C8.4)
= ^1' '^i = JтikГi ('¦ = !'2, ...). ^38.22)
Множители g^, как и в формуле C3.11), означают
кратности соответствующих уровней е^. Осдовной уровень
считается простым. Так как в пределе при а—>0 Лд
неограниченно растет, общее число частиц может быть каким
угодно. Конечность интеграла C8.21) при а = 0 означает
оо
только, что число частиц л = 2 ^i в возбужденных со-
стояниях не может превысить некоторого предела, так что
увеличение числа частиц сверх этого предела происходит
исключительно за счет основного состояния. Частицы до
известной степени «конденсируются» в основном состоянии
(конденсация Эйнштейна). Число п определяется
интегралом C8.21). Согласно формуле C8.17), определяя
степень вырождения, как и прежде, равенством C8.19),
получаем ^)
«=-fx'-(«)<yC(|) = iVo. C8.23)
Таким образом, пренебрежение низшим уровнем
энергии при определении п практически ничего не меняет
в интеграле. Можно показать, что погрешность при этом
имеет величину порядка Ьп/п = {p/Nf^^. Этим можно
пренебречь даже при значительных степенях вырождения.
Обратимся теперь к высоким степеням вырождения.
В этом случае, согласно уравнению C8.23), n<^N.
Практически все частицы находятся в основном состоянии.
^) Конечно, при этом формула C8.20) уже не имеет места, так
как она связана с равенством n = N.
330 Гл. IV. Общие принципы статистики. Метод ячеек
Следовательно, в силу C8.20) а—очень малая величина.
При этом NQ<t N м
В первом приближении [если в C8.22) подставить а «^ 0]
числа частиц в eo36yiHcdeHHbix состояниях оказываются
равными
n.^n\^-j^ . C8.25)
е * — 1
Исследуем теперь, когда можно заменить п^ на
величины w?, определенные равенствами C8.25), т. е. когда
n\--'^i_(e^^i)e^"i_ e^Vo ,, ,
Преясде всего видим, что это выражение мало при
больших значениях Ps^. Однако возможен и случай Psi <С 1,
если только для всех возбужденных состояний е-
соблюдается неравенство
Энергия первого возбужденного уровня по порядку
величины составляет
ЧТО соответствует дебройлевской длине волны порядка
линейных размеров сосуда, занимаемого газом.
Следовательно, указанное выше неравенство соблюдается, если
2ткТ ^ ^^ •
Согласно C8.19), отсюда следует условие
р|/^з»4г. C8.26)
которое выполняется в предельном случае больших р, так
как тогда Hq ^ N,
§ 38. Выро:нсде}1Ные газы ЗЬ7
Числа частиц в возбужденных состояниях сильно
вырожденного газа Бозе — Эйнштейна даются формулой
C8.25) и зависят только от температуры. В первом
приближении эти частицы описываются той я^е функцией
распределения, что и световые кванты (см. § 20). Более
точные значения в C8.22) можно получить при помощи
модифицированного потенциала
оо
Ф = -1пA--е-«) = -|^-B7:тЛ:Г)'/2-^^1пA--^-«-01/7с/^
г ^ Q
C8.16')
Первый член здесь представляет собой поправку,
вносимую в правую часть C8.16) для низшего квантового
состояния. Давление не зависит от нее. Согласно C8.176),
оно равно
Р = X (а) (J^y {кТ)" - X @) О^У" (кТ)"' . C8.27)
Получается нечто вроде формулы для давления пара.
Покажем еще, что вырождение газа Бозе — Эйнштейна
совместимо с теоремой Нернста о теплоемкости. Из
формулы C8.6) с учетом формул C8.1) —C8.3) следует
причем а определяется из соотношения C8.2)
Вблизи абсолютного нуля для всех возбужденных
состояний ^Sj » 1, так что i Ф О все члены суммы стремятся
к нулю по меньшей мере как в~^^ч Остаются только
не зависящие от температуры слагаемые
l5„=-ln(l-e-«) + ^. Л^ = - ^
е«_1 е«_1
338 Гл. IV. Общие принципы статистики. Метод ячеек
Соответственно энтропия S экспоненциально стремится
к постоянному значению
Для энтропии на одну частицу при абсолютном нуле
получаем
*о = -Ж = *Т^*0 C8.28)
(с точностью до членов, имеющих порядок величины,
которым мы уже пренебрегали ранее).
До сих пор предполагалось, что низшему уровню
соответствует энергия Sq = 0. Если е^ Ф О, то, поскольку в силу
C8.2) а зависит от температуры, можно положить а =
= а' —Psq, после чего формула C8.1) вновь примет
прежний вид. Равным образом и соотношения C8.2) и C8.4)
останутся без изменений. Вместо C8.3) мы получим
:const
Отсюда следует, что
Таким образом, если при дифференцировании по Р
считать постоянной не а, а а', то энергия уменьшится на
величину нулевой энергии Nbq,
§ 39. ЭЛЕКТРОННЫЙ ГАЗ В МЕТАЛЛАХ
1. Замечания к теории Друде. После открытия
электронов не могло быть никакого сомнения в том, что
электрический ток переносится электронами. Друде
выдвинул представление, что электроны в металле ведут
себя, как молекулы газа, и принимают участие в
термодинамическом равновесии. Наиболее крупный успех
теории Друде состоял в выводе закона Видемана — Франца
для отношения теплопроводности х к электропроводности
а в форме
^=3^^. C9.1)
§ 39. Электронный газ в металлах 339
Здесь е обозначает электрический заряд электрона. Друде
получил также формулу для электропроводности
а = ^ C9.2)
mv
(/ — длина свободного пробега электрона, л —число
свободных электронов в 1 м^у v — средняя скорость, е и m —
соответственно заряд и масса электрона), которая в
известном смысле имеет значение и в настоящее время.
Теория Друде, по крайней мере качественно, объясняла
сущность и внутреннюю связь многочисленных других
термоэлектрических и термомагнитных явлений, как,
например, термо-э. д. с, контактная разность потенциалов,
термоэмиссия электронов из металлов и т. п.
Однако лежащее в основе теории представление
противоречит данным об удельных теплоемкостях. В самом
деле, в состоянии термодинамического равновесия каждая
степень свободы электрона должна иметь среднюю энергию
уАГ, так что молярная теплоемкость электронов должна
составлять 3/?/2 = 3 кал/моль-град. Последнее, очевидно,
несовместимо с правилом Дюлонга и Пти. Кроме того,
при более точном учете максвелловского распределения
по скоростям в формуле C9.1) получается множитель 2
вместо 3, что нарушает согласие с опытами Егера и Дис-
сельхорста. Только если предположить, что число
свободных электронов значительно меньше числа атомов, можно
избежать трудностей в приведенном и в других случаях.
В связи с названными обстоятельствами вера в
представления Друде об «электронном газе» была совершенно
поколеблена. В сущности, неудача казалась понятной.
Фактически электроны в металле движутся не в
отсутствие силового полЯу а в периодическом потенциальном
поле, которое создается ионами металла. К этому
присоединяется еще взаимодействие электронов между собой.
Все же квантовомеханически представление о
свободных электронах в металлах можно до известной степени
оправдать. Во всяком случае, Зоммерфельд в 1928 г.
с успехом возобновил представления Друде о свободных
электронах и показал, что прежние трудности отпадают,
если учесть, что электронный газ является сильно вы-
340 Гл, IV. Общие принципы статистики. Метод ячеек
рожденным газом Ферми —Дирака [см. § 38, формула
C8.20)]. Рассмотрим несколько выводов из этих
представлений, следуя статье Зоммерфельда и Бете ^).
2. Полностью вырожденный газ Ферми—Дирака.
Будем исходить из уравнения C8.1). В случае Ферми —
Дирака справедлив нижний знак. Полагаем а = — РС, так
как оказывается, что в предельном случае Г—>0
конечным остается не а, а С ^). Тогда получаем
Ф = 21пA + е"'^^*^~'^^). C9.3)
i
Здесь величина С имеет простой термодинамический
смысл. Для свободной энтальпии в уравнении G.4)
получаем из C8.6) и C8.176)
В этом уравнении взаимно уничтожаются члены,
содержащие и и Ф (последние в силу того, что Ф
пропорционально V и, следовательно, УдФ/дУ = Ф). Поэтому
имеем
C=-j = 5. C9.4)
Таким образом, С представляет собой свободную
энтальпию на один электрон. Аналогичные соотношения можно
получить и для статистики Бозе —Эйнштейна.
В противоположность статистике Бозе — Эйнштейна
в данном случае величина а может принимать и
отрицательные значения, и, следовательно, величина С может
оказаться положительной. Согласно выражению C8.4),
среднее число заполнения 1-го уровня равно
1) А. Sommerfeld, Н. Be the, Elektronentheorie der Me-
lalle, Artikel in Handbuch der Physik von H. Geiger, K. Scheel,
Bd. XIV, 2 Кар. 3, I, S. 333.
*) Смешение с С из § 37 уже не является опасным.
§ 39, Электронный газ в металлах
341
В частности, в предельном случае 7* = О (Р—>оо) эта
величина будет равна О или 1 в зависимости от того,
какая из величин е^ и С больше. Следовательно, в случае
полного вырождения (фиг. 31, кривая а) получаем,
снабжая величины при абсолютном нуле индексом О,
"^ = { о
при
при
Si < Со,
h > Со.
C9.6)
Здесь Со играет роль предельной энергии. Все уровни
энергии ниже Cq заполнены, выше Со свободны. Следова-
Ф^иг. 31. Среднее число частиц на одну фазовую
ячейку.
а—при абсолютном нуле температуры, Ь—для р=10/С> с—для
р=1/; (!:=const).
тельно, при абсолютном нуле будут заняты самые
низкие уровни. Согласно принципу Паули, в каждом
состоянии могут находиться два электрона с противоположно
направленными спинами (внутренними моментами
количества движения).
Полное число электронов определяет предельную
энергию. Если определить предельный импульс Р^ при
помощи соотношений
Р1
^o-bi^ ^o=/2mCo,
то число частиц равно
N--
9 L l!! рз
C9.7)
C9.7а)
342 Гл. IV. Общие принципы статистики. Метод ячеек
а энергия составляет
ТТ —П
'Лз 5 2w
С^ = 2гз?Й- C9.76)
Множитель 2 обусловлен тем, что, согласно квантовой
механике, вследствие двух ориентации спина каждая
фазовая ячейка включает два квантовых состояния и
поэтому содержит два электрона. Вычисление предельного
импульса Pq дает
Таким образом, согласно формулам C9.76) и C8.7), днер-
гия и предельная энергия при абсолютном нуле равны
Для давления, пользуясь соотношением C8.18), находим
Po = lb^{s-Kv) • C9.10)
Для меди давление составляет р^ ^^ 3,8-10^ атм. Это
огромное значение характеризует электрическое
притяжение между электронами и ионами. Предельдая энергия
для меди составляет Сси ^ И»3 • 10""^^ эрг f^l эв. Она
сравнима с энергией ионизации атома водорода A3,54 эв).
Полная энергия на один моль составляет Uq= -^Z/Cq, что
приблизшельно совпадает с теплотворной способностью
угля. Далее, сила, приходящаяся на поперечное сечение
иона, равна электростатическому притяжению. Отсюда
находим pj''^ ^ е^1^т:г^г^, где г*^G/Л^)^Ь, что
представляет величину порядка нескольких ангстрем. Если
оставить величину г неопределенной, то, вообще говоря,
никакого равновесия сил не будет. Должно выполняться
условие
Если подставить сюда р^ из формулы C9.10), то получим
__ 471?оГ* J_ / 3 Y^8 ^*
§ 39, Электронный газ в металлах 343
ИЛИ, полагая h/mc = A (комптоновская длина волны) и
а = е2/47:е^Йс (постоянная тонкой структуры),
'^ "*" 5 \SnJ аг"^ г •
Если 7^1 (следовательно, г > lA), то давление
слишком мало, чтобы уравновесить притяжение,— электроны
и ионы сдвигаются теснее. При f > 1 {г <, ik) давление
оказывается слишком большим и металл будет
разрушаться. Отсюда видно, что силы сцепления в металлах
обусловлены в первую очередь электронами проводимости.
Указанные значения относятся к абсолютному нулю.
Фактически давление, энергия и предельная энергия
зависят от температуры. Однако • степень вырождения
настолько велика, что они лишь очень слабо изменяются
с температурой. Действительно, степень вырождения
в C8.19) станет величиной порядка единицы лишь при
температуре eupoafcdeuuH
3. Почти полное вырождение. С ростом температуры
скачок функции jii в C9.6) сглаживается (см. фиг. 31,
кривые b и с). Однако вблизи абсолютного нуля (пока
Т <С в;^ 100 000° К, т. е. кТ « Со или рСо > 1) переход от
от п^ = 1 к Аг^ = О происходит очень быстро. Это
обстоятельство позволяет с хорошим приближением вычислить как
интеграл в C8.16), так и другие связанные с ним интегралы.
Полагая в C8.17) t + a = t — ^i = x и добавляя перед
интегралом спиновый множитель 2, имеем для числа частиц
-PC
Интегрируя в C8.17а) по частям, находим также
выражение для энергии
гт __ ^^V /2т\б/2 7 {х + ?ф^с1х ^Q
-1С
344 Гл, IV. Общие принципы статистики. Метод ячеек
Далее оба интеграла преобразуются интегрированием по
частям. Из C9.11) следует
Первый множитель подинтегрального выражения
представляет собой симметричную функцию х. Второй
множитель при больших значениях РС незначительно
изменяется в интервале, в котором функция е*/(е^ + 1J
заметно отличается от нуля. Поэтому второй множитель
можно разложить по степеням х. Так как, кроме того,
подинтегральное выражение экспоненциально убывает
с увеличением абсолютной величины Ху то для больших
РС нижнюю границу интеграла можно заменить на — оо.
Таким образом, окончательно получаем
^'=w B^"^)''^ (i3^)<i +щ+т-^ • ••>• (^^-^^"^
—оо
Интеграл, содержащий второе слагаемое, исчезает в силу
нечетности подинтегральной функции. Первый интеграл
легко вычисляется и равен 1. Последний пропорционален
интегралу
+00 оо
\^^^^^dx = 2[x^e-«-2e-^- + 3e-^-- ...)dx =
—00 о
= 2.2l(l-J,+A-...)=f
Подставляя найденные значения в выражение C9.11а),
получаем
N^'4,Vi^f{i^.^i'l^^...). C9.116)
Отклонения от предельного случая полного вырождения
имеют, следовательно, порядок величины (Т/Ьу,
Соответствующий расчет для C9.12) дает
§ 39, Электронный газ в металлах 345
Первые члены в выражениях C9.116) и C9.12а),
естественно, совпадают с выражениями C9.7а) и C9.76).
Если обозначить значение С при абсолютном нуле
так же, как и выше, через Со (граничная энергия Ферми),
то из выражения C9.116), приравнивая значение N при
Г = О и Т =^0, получаем
или
'^ = '^o(l-f2iP+...)- C9-13)
Согласно выражению C9.12а),
следовательно, в силу C8.18)
Р = Ро(}+^^+ •••)• ^^^-^^^
4. Специальные проблемы. Зависимость от
температуры обусловлена тем обстоятельством, что часть
электронов, при абсолютном нуле занимающих уровни ниже
границы Ферми, теперь ее переходит. Число их, согласно
C9.11) и C9.7а), составляет
00 / 1 + Х\Ч2
N - 2 У Ки % J е^ + 1
Р Со-С)
Пренебрегая поправочным членом порядка Г/в,
величину С здесь можно заменить на CqI тогда
-Ж=ТГо\^^Ч^=Т^^^2. C9.16)
о
Заметим, что чувствительными к изменению температуры
оказываются лишь те электроны, которые переходят через
границу Ферми и, согласно уравнению C8.12), образуют
свободные места ниже границы Ферми. Таким образом,
понятно, что для представлений Друдо имеет значение
не полное число ррободных электронов N, а лишь их
346 Гл, IV, Общие принципы статистики. Метод ячеек
малая часть 8Л^. Следовательно, в данном случае мы
приходим совершенно естественно к уменьшению
эффективного числа свободных электронов.
Поэтому удельная теплоемкость электронов столь мала,
что правило Дюлонга и Пти не нарушается. Из
уравнения C9.14) для теплоемкости получаем выражение
[При расчете были использованы формулы C9.76) и
C9.9).] Учитывая C9.7а), имеем
Теплоемкость одного моля составляет (/V = L —число
Авогадро)
^Электр. ^^ ~2 Т~ "К Т~ ^Дюлонг—Пти • (оУ. J 7)
Дробь Сэлектр. / сдюлонг-пти является величиной порядка
bN/N в выражении C9.16). Это составляет, в частности,
для меди приблизительно 7'/54000°К и, следовательно,
при 7**^300° К равно ^/igo- Таким образом, электронная
теплоемкость составляет меньше 1%.
Применим выражение C9.5) или C8.8) к
рассмотрению эффекта Ричардсона —эмиссии электронов из
накаленного катода. Наше предположение о свободных
электронах означает, что внутри металла потенциал имеет
постоянное значение (фиг. 32). Однако на границе
металла имеются значительные силы, направленные
противоположно давлению электронов и уравновешивающие
его. Следовательно, там потенциал должен быстро
возрастать от отрицательного значения до нуля во внешнем
пространстве. Величина И^ —Cq представляет выигрыш
энергии, который получается, если внешний электрон,
попадая внутрь металла, приобретает там граничную
энергию Ферми. Следовательно, W есть полная разность
между потенциальной энергией электрона снаружи и
внутри металла. Естественно, реальный ход потенциала
отличается от изображенного на фиг. 32. Внутри имеют
место уже упоминавшиеся периодические изменения по-
§ 39, Электронный газ в металлах
347
тенциала, а на границе —не скачкообразный, а
непрерывный (хотя, может быть, и очень быстрый) подъем.
Однако упрощение, сделанное на фиг. 32, не нарушает
характерных черт эффекта Ричардсона.
При достаточно большой энергии электроны металла
MoiyT преодолеть потенциальный барьер на границе и
выйти наружу. Условие того, что это произойдет, состоит
в том, чтобы доля кинетической энергии электрона.
снарути-
Фиг. 32. К эффекту Ричардсона.
соответствующая движению перпендикулярно к границе,
превышала И^, т. е. (если направить ось z по нормали
к границе) должно быть
Поэтому плотность тока электронов, выходящих из металла,
определяется интегралом
Д = 2
J_ С —El
h^ i 1 + ез
dpx dpy dpz
QX^(^p^l2m--%)
Верхний предел интегрирования равен -{- оо. Нижний
предел интегрирования для р^ и Ру равен — оо и для р^
равен yimW. Отсюда, если положить (Р/2т) {р% + ру) = /,
348 Гл. IV. Общие принципы статистики Метод ячеек
^рУ2т — ^W == 5 и С «t; Cq, получим
оо
О
Так как W^ > Со и, вообще говоря, Р(И^- Cq) > 1, то
единицей в знаменателе можно пренебречь по сравнению
с экспоненциальной функцией. Тогда получаем формулу
Ричардсона
/, = i^A^r^exp(-21--io). C9.18)
Если произвести расчет с больцмановским
распределением, а не фермиевским, то вместо C9.18) найдем
I,= —.2!L Ykfe-^i^'^, C9.18а)
у 2т1т
Легко понять, почему в квантовомеханической формуле
стоит не W/kT, а {W — (,^)/кТ. Действительно, электрон
должен преодолеть лишь разность между высотой барьера
и граничной энергией Ферми. Величина Nq в классической
формуле означает число свободных электронов. Если
приравнять друг другу множители в C9.18) и C9.18а), то
для эффективного числа электронов получим
N,=^v^^^m'=iv^NY(^j^y. C9.19)
Это опять значительно меньше, чем число свободных
электронов. Однако редукционный множитель отличается
от соответствующего множителя в уравнениях C9.16) и
C9.17). Вместо кТ/С^ получается {кТ/(,^у1^. Это ясно
показывает, что переход от теории Друде к современной
электронной теории металлов достигается не просто путем
введения редуцированного числа свободных электронов.
К рассмотрению проводимости и закона Видемана —
Франца мы сможем обратиться лишь в гл. V. В
заключение покажем, что электронный газ также удовлетворяет
теореме Нернста. Согласно выражению C8.16), потенциал
§ 40. Средние квадратичные флуктуации 34^J
для электрона имеет вид
6
Интегрирование по частям приводит к выражению
Ф __ StiF -¦/¦/2m\3 Т Yt^dt
О '
ИЛИ, принимая во внимание C9.12),
Ф = |-РС/. C9.20)
Следовательно, согласно выражению C8.6), энтропия
имеет вид
S=^k^(^^U^^^, C9.21)
Кроме того, в силу C9.7а), C9.76) и C9.14) имеем
U, = \^,N, С^ = |(;„Л^(Ц-^ _^). C9.21а)
Если это выражение и выражение C9.13) для С подставить
в C9.21), то получим
или
кТ
S = ^Nk^{ = Сэлектр. при кТ « Со). C9.22)
Энтропия возрастает пропорционально Г; в пределе при
Г—>0 она обращается в нуль. Согласно C9.16),
энтропия имеет величину порядка kbN.
§ 40. СРЕДНИЕ КВАДРАТИЧНЫЕ ФЛУКТУАЦИИ
До сих пор мы вычисляли средние значения или даже
только значения, соответствующие максимальной
вероятности, предполагая, что они совпадают с макрофизическими
наблюдаемыми значениями Это предположение
оправдывается тем, что законы для средних значений совпадают
с термодинамическими законами, и, задавшись опреде-
350 Гл. IV. Общие принципы статистики. Метод ячеек
ленными модельными представлениями, можно вычислить
термодинамические функции состояния.
Это отнюдь не очевидно. Понятие среднего значения
включает возможность более или менее сильных
отклонений от яето —флуктуации. Отдельные измерения могут
дать значения, более или менее отклоняющиеся от
среднего. Однако хорошее согласие между статистическими
средними значениями и данными макрофизических
наблюдений показывает, что эти флуктуации в
термодинамической статистике обычно очень малы (следствие закона
больших чисел).
Для доказательства необходимо ввести величину,
измеряющую эти флуктуации. Средняя флуктуация всегда
равна нулю, ибо среднее значение самой величины
определено так, что отклонения в обе стороны одинаково
вероятны. Искомая мера дается лишь средней
квадратичной флуктуацией.
Средние значения мы будем снабя^ать чертой сверху,
как это уже делалось выше. Тогда, согласно C6.14),
среднее значение энергии системы в Г-пространстве равно
^E(n)i
-РЕ(П)
^=Ч.-рв(п) =-^- (^0-1)
Флуктуации представляют собой разности между
измеряемыми значениями Е{п) и средним значением Е, т. е.
АЕ{п)==Е{п)-Е.
Отсюда находим среднюю квадратичную флуктуацию
энергии
{^Ey = [^E{n)Y = [Е{п)^Ёу. D0.2)
Величина Д-Б есть корень квадратный из средней
квадратичной флуктуации. Именно ее обычно имеют в виду,
когда горорят о средней флуктуации. Формула D0.2)
в явной форме гласит
'^[Е{п)-Е]^е''^^^''^
§ 40. Средние квадратичные флуктуации 351
Так как усреднение представляет собой линейную
операцию (и каждое среднее значение по отношению к
последующему усреднению является константой) то из
формул D0.2) и D0.3) следует, что
{Щ^=^^-2ЕЖ+Е^ = Ё^^ЕК D0.4)
Средняя квадратичная флуктуация некоторой величины
равна разности меукду средним значением квадрата этой
величины и квадратом ее среднего значения. Отсюда,
в частности, следует, что эта разность всегда должна
быть положительной.
Воспользуемся формулой D0.4) для вычисления средней
квадратичной флуктуации энергии. В выражении
п
числитель получается из знаменателя двойным
дифференцированием по р. Имеем, следовательно.
Подставляя в D0.4) средние значения D0.1) и D0.5),
получаем
^ ^ 2 дЗ* Z^ \д^ J
Это выражение представляет в точности производную
дроби Z'/Zy так что окончательно находим
(ДЕJ = (Е-Ё)^ = -^^ . D0.6)
Принимая во внимание значение Р и формулу D0.1), по
следнее выражение можно переписать в виде
Таким образом, средняя квадратичная флуктуация энергии
определяется чисто термодинамическим путем. Она
пропорциональна теплоемкости С.
352 Гр, IV. Общие принципы статистики. Метод ячеек
Отсюда для одноатомного идеального газа получаем
[так как U = -^NkT, см. уравнение B2.6) 1
(Д?J = |-№Г^ ^ = У^. D0.8)
Следовательно, для одного моля вещества (Л' = L) средняя
квадратичная флуктуация составляет примерно lO"^^
средней энергии, что совершенно ненаблюдаемо. ПриЛ^ = 150
мы имели бы AE/U = 6у7%.
Тот факт, что в телах большого размера флуктуации
не играют никакой роли, а заметны только на малых
участках, объясняется действием закона больших чисел.
Колебания энергии на 6,7% при комнатной температуре
соответствуют колебаниям температуры на ± 20°. Со
значением флуктуации в малых областях мы уже
познакомились при изучении броуновского движения (см. § 24).
Формула D0.7) справедлива в общем случае. В
частности, для системы квантовых осцилляторов из
соотношения D0.6) и D0.8) получаем
N (/сеJ
(е»/Т_1J
(^^y = TW^^ D0.9)
т. е. аналогично равенству D0.8)
АЕ 1
Для твердого тела из C5.8а) и C5.86) следует, что
1 2 т/У/ в\'/2 1 гг ^^ г.
D0.9а)
АЕ
и
Yn
D0.10)
-^4=г-, если Г>в.
Отсюда видно, что в данном случае при абсолютном нуле
обращаются в нуль сами флуктуации, но отнюдь не
отношения А^ к энергии теплового движения U, Тот факт,
что при Г—»О ^Е обращается в нуль, вытекает также
из тепловой теоремы Нернста (см. § 12), так как вели-
§ 40. Средние квадратичные флуктуации 353
чишк С=^ди/дТ в формуле D0.7) пропорциональна удель«
ной теплоемкости. Для электронного газа, согласйоC9.21я),
имеем
ЧТО аналогично первому соотношению D0.10).
Совершенно таким же способом можно вычислить
средние квадратичные флуктуации чисел заполнения п^.
Аналогично уравнению C8.4) получаем
D0.12)
т. е. в случае газов Бозе — Эйнштейна или Ферми
—Дирака в силу уравнения C8.4) имеем
О«0^ = ,ж^.- (^0.13)
или
^n,=Vn,{l±n,). D0.14)
В случае газа Ферми — Дирака это выражение заметно
отличается от нуля только на границе Ферми.
Непосредственно в точке 8| = С>Со [см. уравнение C9.5)] ^п^
принимает максимальное значение А/г^ = 1/2. Для
возбуждённых состояний сильно вырожденного газа Бозе —
Эйнштейна w^ < 1, следовательно, An, = Vn,. Последнее
равенство справедливо и в предельном случае
классической статистики. Особый интерес представляет основное
состояние газа Бозе — Эйнштейна. Так как 1 < Wq(^ Л^), то
В этом случае флуктуации имеют величину порядка самого
среднего значения и, следовательно, они очень велики.
В связи с этим следует заметить, что выражение D0.14)
справедливо для больших канонических ансамблей. При
дифференцировании в выражении D0.12) считается rio-
стоянным а, а не число частиц N. Поэтому имеет место
постояйный обмен частицами между отдельными членами
354 Гл, IV. Общие принципы статистики. Метод ячеек
ансамбля, что объясняет, как могут возникнуть большие
флуктуации.
В силу малости отдельных фазовых ячеек флуктуации
в их группах имеют практически большое значение.
Если через 2' обозначить сумму по некоторой опреде-
i
ленной группе ячеек, то среднее число частиц в них,
согласно C8.4), равно
^• = 2j ^i= -jZj -JiT^
i i
И выражение для средней квадратичной флуктуации
имеет вид
D)^ = K-^,r = f 2?^. D0.15)
Так как в случае уравнения C8.4) п^ зависит только
от 8^, смешанные члены в двойной сумме выпадают, и
мы имеем
(Ч)''=-|-2'-|г=2'"Д1±«1)- (^0.16)
i ^ i
В частности, если флуктуации происходят в элементе
объема Д7, то из D0.16) получаем (без спинового
множителя 2)
О
В предельном случае статистики Больцмана это дает
Подставляя сюда вместо е* выражение C1.4), получаем
{АпГ=.^, D0.18)
или, вводя среднее число частиц в элементе объема
AV{n = N^V/V), имеем
§ 40. Средние квадратичные флуктуации 355
Такие флуктуации плотности можно наблюдать, так как
они вызывают рассеяние света. Голубой цвет неба
является следствием рассеяния солнечного света на флуктуацпях
плотности воздуха.
Конечно, можно было бы непосредственно вывести
формулу для флуктуации D0.18а), принимая во внимание
лишь постоянство средней плотности. Этот вывод
значительно проще, чем тот, который был приведен. Однако
в нашем выводе выявляется связь с основными пOw^oжeни-
ями статистической механики, утрачиваемая при других
выводах. Прямой вывод будет рассмотрен в задаче IV.8.
Для электронного газа из D0.17) следует (со спиновым
множителем 2)
/А \о О AF/2m\3/2 г е^ ^/——^dx
Интеграл вычисляется точно так же, как и в § 39. В
предельном случае полного вырождения интегрирование дает
т. е., согласно C9.7) и C9.7а),
Отсюда, учитывая, что n = iVAF/F, получаем
Флуктуации плотности Ферми-газа также исчезают при
абсолютном нуле.
В заключение отметим, что более высокие степени
флуктуации также можно вывести из суммы состояний.
Например,
{E{n)-Ef = - ^- D0.20)
Зоб Гл. tV. Общие принципы Статистики. Метод ячеек
Действительно,
¦~3?з~- \Z ) " \Z ZV ~
3
Для идеального газа lnZ= —уЛ^1пр+ ... Отсюда
следует, что
?5 9 лг«' ^- ®- ч :^ V - К 9 ^^ •
г л а в а V
ОСНОВЫ ТОЧНОЙ КИНЕТИЧЕСКОЙ ТЕОРИИ ГАЗОВ
Развитая в гл. IV статистическая механика дает
атомистическое обоснование термодинамики как теории
теплового равновесия. Более того, она дополняет
термодинамику, позволяя в принципе выразить термодинамические
функции [например, свободную энергию F B9.12а) или
сумму состояний B9.15)] через микроскопические
параметры.
Атомистическое рассмотрение неравновесных процессов
оказывается значительно менее простым.
Феноменологические законы были сформулированы в § 21. Следуя
историческому развитию, мы ограничимся рассмотрением
поведения молекул идеального газа. Таким образом, задача
состоит в строгом изложении развитой в гл. III
кинетической теории газов. В последнее время получила
заметное развитие кинетическая теория конденсированного ве-
п^ecтвa. Однако ее рассмотрение выходит за рамки этого
учебника. В этой главе придется чаще, чем раньше,
ссылаться на специальную литературу. Это относится
в особенности к методам решения кинетического
уравнения, которое мы здесь выведем^).
Неполнота этой главы обусловлена не только
ограничениями в постановке задачи. Она вытекает также из того,
что в явном виде вычисления удается провести лишь для
очень простых и грубых молекулярных моделей. Особенно
это относится к тем случаям, когда необходимо учесть
квантовые свойства молекул. В этой области успех
достигнут в электронной теории металлов, где, в частности,
1) М. Born, Cause and Chance, Anm. S. 203.
358 Гл. V. Основы точной кинетической теории газов
Зоммерфельду удалось вывести закон Видемана— Франца.
Этот вывод будет приведен в конце настоящей главы.
§ 41. КИНЕТИЧЕСКОЕ УРАВНЕНИЕ
М AKCBEvl.l Л — БОЛ ЬЦМ АН А
1. Описание состояния в кинетической теории газов.
Идеальный газ характеризуется тем, что состояния его
молекул являются независимыми (исключая акт
столкновения). Состояние отдельной молекулы можно полностью
описать, задав ее координаты и скорость в каждый
момент времени. Ограничимся только одноатомными
молекулами, рассматривая их как твердые и гладкие шарики.
Пусть координаты молекулы задаются вектором г = {х^, х^,
Жд), а скорость — вектором v = (^j, Jg, fg)^)*
Элементы объема в пространствах координат и
скоростей равны соответственно
dx = dx^dx2dx^, d% — d\^di^dK^, D1.1)
В противоположность гл. IV здесь мы везде (за
исключением § 45) будем отвлекаться от квантовой природы
этих элементов объема. Предположим их достаточно
большими, чтобы внутри их помещалось много молекул, и в
то же время настолько малыми, чтобы плотность внутри
каждого элемента можно было считать постоянной.
Математически это означает, что dx и dS, несмотря на их
величину, можно рассматривать как дифференциалы. Этим
видоизмененным определением можно оправдать
обозначения в выражениях D1.1).
Вычислим число частиц rfv в окрестности точки (г, v)
в элементе фазового ji-пространстБа. При этом заменим
импульс частицы на скорость, так что указанный элемент
равен dx d\. Тогда получим
6fv = /(r, V, t)dxd'i, D1.2)
1) Возможность рассматривать молекулу как твердый шарик
и вместе с тем говорить лишь о ее поступательном движении
обус^човлена квантовыми явлениями. См. в связи с этим
примечание в начале § 34, где упоминается о вращательной энергии
электронов.
§ 41, Кинетическое уравнение Максвелла—Вольцмана 359
Полное число частиц дается интегралом
iV=^/(r, V, t)dxdi. D1.3)
Здесь интегрирование распространяется по всему
пространству (например, заключенному внутри сосуда) и по
всем скоростям. Следовательно, полное среднее значение
некоторой функции g (г, v) имеет вид
^=^5^(г> v)/(r, y,t)dxd\. D1.4)
Однако для кинетической теории имеют значение
только локальные средние значения. Они представляют
средние значения в пространстве скоростей, которые, вообще
говоря, меняются от точки к точке. Если ср (v) - некоторая
произвольная функция скорости, то ее локальное среднее
значение дается интегралом
?(r) = -^5?(v)/(r,v)rf^ D1.5)
Здесь п обозначает локальную плотность частиц
/1=5/(г, \)d\. D1.5а)
Следовательно, величина ndx представляет число частиц
в элементе объема dx, независимо от их скоростей.
Например, среднее значение скорости равно
n = \^xf{T,y)dl. D1.6)
Будучи записано в компонентах, это выражение
представляет собой тройной интеграл
Щ @^1, ^2, д:з) = 7 S S S ^iH^v ^2' ^з; ^1» ^2» У cJ^idSgCftg.
D1.6а)
Кроме сил межмолекулярного взаимодействия, о
которых речь будет идти ниже, следует задать еще внешнюю
силу. Последнюю можно считать функцией точки
F = F(r) = (A-,(r), ^^(г), Хз(г)). D1.7)
360 Г.1,- V. Основы точной кинетической теории газов
Мы не рассматриваем здесь силы, зависящие от скорости,
которые действуют, например, на заряженные частицы
в магнитном поле.
При этих предположениях оказывается возможным
полностью обосновать термодинамику движущегося
одноатомного газа. Прекрасный нетривиальный пример этого дает
открытие Клузиусом и Вальдманом так называемого
диффузионно-термического эффекта, содержаи1,егося в более
высоких приближениях к решению кинетического
уравнения для разнородных молекул. Из термодинамики
известно, что идеальные газы смешиваются без изменения
температуры. Однако сам процесс смешивания
сопровождается термическими эффектами. Эти эффекты неявно
содержатся в расчетах Чэпмена^) и Энскога^), но их
значение для эксперимента впервые было осознано
Клузиусом и Вальдманом. Последние авторы впервые
наблюдали эти эффекты. Сгязь их с термодиффузией указана
в § 21, п. 3 (соотношения взаимности).
2. Изменение функции / со временем. Кинетическое
уравнение Максвелла — Больцмана мы получим, поставив
вопрос об изменении функции / со временем. Для этого
предположим, что функция / непрерывна и достаточное
число раз дифференцируема (это возможно в силу нашего
определения элементов объема dx и d^). Плотность в
фазовом ji-пространствс / (г, v, t) изменяется вследствие
движения частиц и при столкновениях. Рассмотрим
промежуток времени А^ относительно которого предположим,
что он велик по сравнению с продолжительностью
столкновения т^, так. что большинство столкновений,
начавшихся в этом промежутке времени, в нем же и
заканчивается, но мал по сравнению со средним временем между
двумя столкновениями т. Следовательно, путь,
пройденный за время А/, мал по сравнению со средней длиной
свободного пробега. Поэтому, вообще говоря, в промежутке
времени ^t молекула испытает столкновение не более
1) Chapman, Phil. Trans., 211,433 A911); 216, 279A916),
217, 115 A916).
*) D. E n s к о g, Kinetische Encrgie der Vorgange in ma^ig ver-
diinnten Сазеп, Inaug. Diss. (Uppsala, 1917); Arkiv fur Matem., 16,
Nr. 16 A921); Kungl. Svenska Akad., 63, 4 A922).
§ 41. Кинетическое уравнение Максвелла—Больцмдна 361
одного раза» Это предполагает малость радиуса действия
межмолекулярных сил по сравнению с расстоянием между
атомами и тем более по сравнению с длиной свободного
пробега (§ 27).
Если внутри интервала ^t вообще не произошло
столкновения, то
г—^r'=r+vA/ и V—>v' = vH—FA^
И, следовательно,
/(г, V, t)dxdi'^f{r + \^t,y + -^?M, t + U)dx'd\' =
= {/(r.v.O + A.[vg+iFK:-b§(] + ...}^x'.^r.
D1.8)
Последнее преобразование справедливо, если можно
пренебречь более высокими членами разложения, т.че.
если функция / лишь незначительно изменяется за время
^t. Заметим, что в силу неравенства А/ < т это
предположение вполне совместимо со значительными изменениями /
на длине свободного пробега. Кроме того, согласно
теореме Лиувилля (§ 28), для описанного выше движения, не
возмущенного столкновениями, имеем
dx' d^' = dxd\, D1.8а)
так что произведения дифференциалов в обеих частях
равенства D1.8) взаимно уничтожаются.
В результате столкновений часть молекул выходит из
элемента dx dl, другие же входят в него из других
элементов dx^ dii. Соответственно одни столкновения приводят
к убыли, а другие — к увеличению числа частиц. Таким
образом, изменение D1.8) числа частиц вследствие их
движения дол^^но быть равно разности между увеличением
•^gew и уменьшением /yeri числа частиц за счет
столкновений. Следовательно, для единицы времени и единицы
фазового объема имеем
К + ^1 + ^^'57 = -^«--Лсн. D1.9)
Это и есть кинетическое уравнение Максвелла^Болгцмана.
Величины /gew и Леп ВЫЧИСЛЯЮТСЯ по законам упругого
362 Гл, V, Основы точной кинетической теории газов
удара. Для краткости градиенты функции / в
пространствах координат и скоростей обозначены соответственно
через df/dr и djjds,
3. Законы упругого удара. Законы упругого удара
зависят от вида сил, действующих между молекулами.
До сих пор предполагалось лишь, что радиус действия
межмолекулярных сил мал. Это справедливо, например,
для сил, зависящих от расстояния по закону F'^l/г'^при
достаточно больших значениях п. Именно этот случай
всегда и обсуждается. При w = 5 он приводит к особенно
простым результатам. В далнейшем будет
рассматриваться только предельный случай л-'>оо, соответствующий
предположению о твердых шариках. Пусть диаметр
шарика равен S. Он представляет вместе с тем наименьшее
возможное расстояние между центрами двух шарообразных
атомов. Конечно, реальная молекула отнюдь не
представляет собой твердый шарик. В лучшем случае модель
одноатомной молекулы годится при не слишком высоких
температурах^).
Полная энергия и полный импульс должны
сохраняться при столкновении. Следовательно, если обозначить
скорости обеих частиц до и после удара соответственно
через V, Vj, и v', v'l, то должны быть справедливы
уравнения
v + Vi = v'4-Vb
v2 + vf = v'2-J-vi'. D1.10)
Обозначив через V относительную скорость частиц перед
ударом
V = Vi-v D1.11)
и разрешив уравнение D1.10) относительно скоростей после
удара, получим
V' = V -;- (Ve) е, vi = v^ ~ (Ve) е. D1.12)
1) При обычных температурах более высокие энергетические
состояния атома термическим путем практически не возбуждаются,
так что закон притяжения определяется поляризацией атомов.
§ 41, Кинетическое уравнение Максвелла—Больцмана 363
Здесь е обозначает произвольный единичный вектор.
Следовательно, относительная скорость после удара равна
У = у; - V' = V - 2 (Ve) е. D1.12а)
Таким образом, имеем
(V'e)= -(Ve). D1.126)
Разрешив теперь уравнение D1.10) относительно v и v^,
получим
V = V' + (Ve) е, Vi = vi - (Ve) е. D1.13)
Эти уравнения по форме совпадают с D1.12).
Следовательно, уравнения преобразования скоростей являются
взаимно обратными.
Соотношение D1.11) для одинаковых частиц допускает
удобное графическое представление, которое мы еще
Ф п г. 33. Векторная
диаграмма скоростей при
упругом столкновении двух
одинаковых шаров.
будем с успехом применять в дальнейшем. Точки v и v^
на фиг. 33 обозначают концы ректоров v и v^. Вектор,
их соединяющий, изображает относительную скорость V.
Если из точки V провести луч в направлении +е, а из
точки Vj— в направлении — е, то, проектируя вектор V
на оба противоположно направленных параллельных луча,
получим векторы db(Ve)e. Эти проекции проведены
304 Га, V. Основы точной кшитической теории газов
ОТ точек V и \\ к точкам v' и \[. Четыре точки v, v^,
v', v[ соответствуют углам прямоугольника (независимо
от направления вектора е). Все они лежат на окружности
с диаметром |V| и центром в середине отрезка,
изображающего вектор V; при этом точки v и Vj, а также
точки v' и \[ лежат на противоположных концах
диаметров.
Следует еще указать смысл вектора е при соударении
твердых шаров. Согласно уравнению D1.12а), имеем
V-V' = 2(Ve)e. D1.14)
При ударе имеет место передача импульса. С одной
стороны, она должна происходить перпендикулярно
к плоскости соприкосновения шаров при ударе, т. е.
вдоль линии центров. С другой стороны, передаваемый
импульс равен изменению импульса каждого из шаров,
и, следовательно, пропорционален величине
V' - V = Vi - v; = (Ve) е. D1.14а)
Отсюда следует, что вектор е направлен по линии центров.
Это совпадает с направлением биссектрисы угла между
V и V [так как, согласно D1.126), векторы V и V имеют
одинаковые составляюпше]. Наблюдателю, движущемуся
со скоростью V, paBHoii скорости одного из шаров перед
ударом, представится картина, изображенная на фиг. 34.
4. Интеграл столкновений Больцмана. При
вычислении правой части уравнения D1.9) удобно воспользоваться
фиг. 34. Она будет несколько нагляднее, если
рассматривать движение центра налетающего шара относительно
сферы де11ствия радиуса s (фиг. 35). Число молекул,
попадаюп1Их за время М в телесный угол rfo) (т. е. на
элемент плоп^ади s-diii), составляет
Первый множшель s'^ rfo) | Ve | А^ представляет объем косого
цилиндра, показанного на фиг. 35, который содержит
частицы определенного направления и определенной
скорости, приходящие за время А/; второй множитель
дает плотность этих выделенных частиц.
Фиг. 34. К кинематике упругого стол1ч-
новепия.
Фиг. 35. Сфера действия п число
столкновений.
366 г л, V, Основы точной кинетической теории газов
Величина /(г, v, t)dxdl представляет число молекул,
обладающих скоростью v. Следовательно, число
столкновений между молекулами со скоростями v и v^ и линией
центров, HanpaBwieHHofi вдоль вектора е^), равно
52cfa)|Ve|Ai/(r, Vj, t)f{T, V, t)dxdkdli. D1.15)
Интегрирование по всем скоростям Vj и всем
направлениям вектора е дает полное число столкновении, которые
изменяют траектории частиц со скоростью v. Это число,
отнесенное к единице времени и единице фазового объема
dxdi, следует подставить в уравнение D1.9); тогда
мы получим
ЛеП =у 5 l^^l^^'"' ^1» ^)^('» ^» t)di^di,. D1.16)
Множитель ^2 обязан своим происхождением тому, что
интегрирование здесь распространено по всем шарам,
в то время как физически подразумевается только
половина их, как это ясно видно из рассмотрения изменения
вектора е при параллельном сдвиге v.
Вычисление jgew протекает совершенно аналогично.
В этом случае векторы v и v^ соответствуют скоростям
после столкновения. Действительно, /gew обозначает как
раз увеличение числа частиц со скоростью v вследствие
столкновений. Пусть соответствующие скорости перед
столкновением суть v' и v(. Число столкновений на
единицу площади атомной сферы s^din равно
52da)|V'elA//(r, V', 0/(г, v;, t)dxdl'dl[. D1.15а)
Согласно D1.126), | V'e | = | Ve | и, кроме того, в силу D1.13)
(а также и вследствие теоремы Лиувилля, §28) ^$'cf$( =
= dldl-^. Действительно, составляя якобиан
преобразования D1.13), находим [полагая координаты вектора е
равными (О, О, 1)]2)
^) При таком кратком способе выражаться всегда имеется
в виду, что векторы v, v^ имеют указанные значения внутри
интервалов с?а) и dx di,
2) Изменение знака функционального детерминанта
компенсируется знаком множителя V'e. Заметим при этом, что величина
-s-1 V'e I dz' dVi получается из (V'e) c?5 rfSJ при V'e) > 0.
§ 41. Кинетическое уравнение Максвелла—Больцмана
36"
д{У, У,)
a(v', vi)"
1 . . 1 0 . .
1
.1.1.0.
. . 0 J . . 1
0 . . j 1 . .
. 0 . ] . 1 .
. . 1 1 . . 0
1. D1.16a)
Таким образом, искомое увеличение числа частиц за счет
столкновений, рассчитанное на единицу времени и
фазового объема, составляет
^ew = y 5|Ve|/(r, v + (Ve)e, t)f{r, х,-{Уе)е, t)di^d^,.
D1.166)
Величины D1.16) и D1.166) суть интегралы
столкновений Больцмана. Подставим их в уравнение D1.9), вводя
обычные обозначения:
/ =/(г, V, 0. /i = /(r, Уг, t),
/'=/(г, Y',0 = /(r, v + (Ve)e,0, D1.17)
/; = /(r, v;, 0 = /(r. v,-(Ve)e,0-
Тогда кинетическое уравнение Максвелла — Больцмана
примет вид
%-^^%'r^l^%-^\^^\{f'f[-fh)d<^dl,. D1.18)
Таким образом, математическая задача строгой
кинетической теории газов состоит в решении этого
нелинейного интегро-дифференциального уравнения.
5. Гипотеза Больцмана о молекулярном беспорядке.
Прежде чем исследовать свойства кинетического
уравнения, обсудим одно существенное предположение,
справедливость которого выше неявно подразумевалась. При
выводе интегралов столкновений необходимо было знать
вероятность Ж (г, v; r^, Vj) того, что молекула в фазовой
точке (г, v) столкнется с другой молекулой в точке (г^, у^).
368 Гл. V. Основы точной кинетической теории газов
Однако фактически нам были известны лишь вероятности
W (г, v) и VF (г^, Vi) того, что молекула находится в фазовой
точке (г, v) или (г^, Vj). Их легко вычислить из /(г, v).
Именно в силу уравнения D1.3) имеем (рассчитывая
Ж (г, v) на единицу объема в фазовом пространстве)
Ж(г, v) = ^/(r. V).
Наличие в D1.15) и D1.15а) произведения двух функций
означает, что величина И^(г, v; r^, \j) при г ^^ r^ равна
нулю, а при г = Tj имеет мультипликативный вид
PF(r, v; г, yi) = W{r, v).T^(r, vj.
Таким образом, вероятность той или иной скорости
частицы предполагается не зависящей от скоростей других
частиц.
В том, что это является особым предположением, можно
убедиться, если исходить из функции W (v, Vj). Поскольку
все молекулы равноправны, функция W (v, Vj) должна
быть симметрична относительно перестановки v и v^.
Интегрируя, находим
^ W(V, V,)dl,= ^W(v„ v)dk, = PF(v).
В силу симметрии в обоих случаях получается, очевидно;
одна и та же функция W{v). Однако отсюда отнюдь
не следует, что И^(у, \i)— W {y)W (у^). Это равенство,
вообш;е говоря, не имеет места.
Мультипликативный вид функции W {Tj^, v; r^, v^)
связан с гипотезой Больцмана о полном молекулярном
беспорядке. Последняя соответствует предположению
Максвелла о мультипликативности вероятностей (§23), гипотезе
об априорной равновероятности равновеликих фазовых
объемов (§ 28) или постулату Гиббса (§ 36). Гипотеза
молекулярного беспорядка весьма существенна для
справедливости принципа энтропии, который выводится в § 42
из уравнения D1.18). Борн^) с сотрудниками недавно
точнее проанализировал эту гипотезу.
1) М. Born, Cause and Chance, Anm. I, S. 203.
§ 42. Н'пгеорема и распределение Максвелла 369
§ 42. Л-ТЕОРЕМА И РАСПРЕДЕЛЕНИЕ МАКСВЕЛЛА
I. If-теорема. Обратимся теперь к принципу энтропии.
На основании C6.10) выражение для энтропии можно
записать в виде ^)
Я= -А:^ Inf'fdxdl D2.1)
Следует лишь иметь в виду, что здесь рассматривается
движение в ji-пространстве, а не в Г-пространстве [как
это было в C6.10)]; кроме того, теперь не будут
учитываться квантовые свойства фазовых ячеек. Выражения B9.5)
(для газа Больцмана) и C8.12) и C8.14) (для газов
Ферми — Дирака и Бозе — Эйнштейна) относятся
непосредственно к J1-пространству. В предыдущей главе,
посвященной термодинамическому равновесию, был рассмотрен
лишь вопрос о том, при каких условиях интеграл D2.1)
имеет максимум. Теперь нас интересует изменение его
со временем. Следует ожидать, что при известных из
термодинамики условиях величина Н окажется неубывающей.
В отсутствие равновесия полная энтропия менее
существенна, чем локальная. Поэтому вместо D2.1) исследуем
изменение во времени функции
Я= -к\ Inf-fdi. D2.2)
Для соответствующей производной получаем
т. е., согласно уравнению D1.18),
'»^k\ii + lnf)(^y% + lF'J^-J,)dl D2.3)
если под Jf понимать интеграл столкновений в правой
части D1.18):
Jf-i[\'^^\irri-ffi)d^di,. D2.4)
^) Мы пишем теперь Я вместо *9, следуя Больцману.
370 Гл. V. Основы точной кинетической теории газов
Первое слагаемое в правой части уравнения D2.3)
можно преобразовать следующим образом:
^ (l + ln/)v|^6fe = div^ vln/./cf?.
Интеграл
S= -А^ \In f'fdl D2.5)
представляет вектор тока, соответствующий значению Н,
определяемому выражением D2.2). Таким образом,
уравнение D2.3) принимает вид
^^ + div S = - ft ^ A + In /) J^ dl D2.6)
Здесь уже использован тот факт, что второй член
в правой части уравнения D2.3) исчезает. Действительно,
поскольку F зависит только от координат точки, мы имеем
т
F\il + lnf)%d^ = ^F[lifln/)dk.
Следовательно, этот интеграл можно преобразовать
в интеграл по поверхности бесконечно большой сферы
в пространстве скоростей. Так как на границе этой сферы
вследствие конечного значения энергии функция /
исчезает, получаем
±F^{l + \nf)%di = 0. D2.7)
Таким образом, остается только интеграл в правой
части уравнения D2.6), которая, согласно D2.4),
принимает вид
- ^^ 5 I Ve I A + In /) (/'/; - /Л) (^ш di rffe,. D2.8)
При перестановке двух троек переменных интегрирования
V и Vj значение интеграла не изменяется. Следовательно,
он равен также
- '^ 5 I Ve I A + In Л) (/'/; - /Л) di. dk d-,.
§ 42, Н-теорема и распределение Максвелла 371
При ЭТОМ следует иметь в виду, что при такой замене
V-> -V и
V' = V + (Ve) е —> Vj — (Ve) е = v^,
Vj = Vj — (Ve) e —> V + (Ve) e = v'.
Таким образом, правую часть уравнения D2.6) можно
записать в следующей, более симметричной форме:
- ^' 5 I Ve I B + In / + In Л) (/'/; - /Л) аш d% dl,. D2.9)
Вместо V и Vj можно также интегрировать по v' и v(.
Согласно уравнению D1.8а), выражение D2.9) равно
¦ 4
^ I Ve I B + In / + Ш Л) (/7; - /Л) d^ d^' d^[.
При этом надо считать исключенными не v' и v,*
[посредством формул D1.12)], а v и v^ [посредством
формул D1.13)]. Далее можно переменные интегрирования
V' и \[ переименовать в v и v^, что не приведет к каким-
либо недоразумениям, так как вначале v и Vj не
встречались. Однако после замены переменных удобно
определить новые переменные, согласно формулам D1.12),
так что для выражения D2.8) (обозначаемого через G)
получаем
D2.10)
Легко видеть, что при этом множитель | Ve | в подинте-
гральном выражении не изменяется. Ниже будет показано,
что G = 0 [см. B1.8)].
Интеграл приобретает большую симметрию, если
заменить его полусуммой двух равных выражений D2.8)
и f42.10). При этом происходит изменение знака в
последней скобке подинтегрального выражения. Следовательно,
получаем
G(r, 0= -k\^{\-^\nf)J^dl^
= -^5|Vel(ln/-blnA-ln/'-ln/;)(/7;-/A)tfa)cfU5i,
D2.11)
372 Гл, V, Основы точной кинетической теории газов
ИЛИ после простого преобразования
G(r, 0 = -?- 5 I Ve|InnL(/'/;-/A)rf«)d^Sx. D2.12)
Заметим, что это преобразование будет использовано
в дальнейшем в связи с интегралом типа D2.8) с
произвольной функцией ф(у) вместо 1-{-1п/. В этом случае
получаем вполне аналогично
S
Ve l-iCv) (/'/:-/Л) flfo) eft rf^i =
= I $ i Ve I (Ф + •>! - ф' - Ф;) (/'/; - ffi) d<» d% d\,. D2.13)
Здесь имеются в виду различные ф-функции, как в D1.17).
Прежде всего заметим, что интеграл D2.12) не может
быть отрицательным, так как функции In/'/(//Л и /7( ~//i
всегда имеют одинаковые знаки. Отсюда следует, что
-^H-divS = G>0. D2.14)
Тесная связь этого уравнения с уравнением B1.10) еще
явится предметом рассмотрения ниже. Интегрирование
по некоторому конечному объему дает
-^\ Н dx-{^\^ S^do==\Gdx>Q, D2.15)
если второй член объемного интеграла преобразовать
на основании теоремы Гаусса в интеграл по поверхности ^).
Интеграл \Hdx изменяется, во-первых, вследствие
наличия потока энтропии через поверхность и, во-вторых, так
как в объеме существует некоторое распределение
источников, дающих положительную (или нулевую) энтропию.
Если рассматривается замкнутая система, то поток через
поверхность исчезает и, следовательно, должно
выполняться неравенство
•^\^Н dx=^ \^Gdx>Q, D2.16)
^) Здесь с?а —элбхмент площади поверхности, а vSn — компонента
вектора S в направлении внешней нормали к поверхности.
§ 42. Н'тпеорема и распределение Максвелла 373
Энтропия замкнутой системы не убывает. Однако
уравнение D2.14) имеет более общее значение, чем принцип
энтропии в термодинамике. Оно дает величину
необратимого изменения энтропии //. Кроме того, согласно
выражению D2.5), оно определяет поток энтропии.
2. Распределение Максвелла. Если G = 0, то
изменение энтропии определяется только потоком ее. Так как
подинтегральное выражение в D2.12) не может быть
отрицательным, то величина G обращается в нуль только
в том случае, когда
/7; = //i. D2.17)
или, если положить
1п/=ф, D2.17а)
при условии, что
f+ Ф; = Ф + Ф1- D2.176)
Следовательно, сумма ф + фх не изменяеися при ударе.
Она является аддитивным интегралом дви:нсения.
Можно сразу указать пять функций, удовлетворяющих
условию D2.176); одна из них есть константа, а
остальные представляют собой выражения для импульса
и энергии:
Фо=1. Ф1 = ?1. 1'2 = ^2. ^z = h> 1'4 = |v2. D2.18)
Действительно, это единственные аддитивные интегралы
движения.
Для доказательства обратимся к изображению точек —
антиподов (расположенных на противоположных концах
диаметра) в § 41. Назовем функцию ф(у) антиподной,
если для всех точек-антиподов v и v^ на любой
произвольной сфере в пространстве скоростей справедливо
равенство
^(v) + ^(Vi) = const.
(Константа, конечно, может меняться от сферы к сфере.)
Так как при столкновении точки v и Vj переходят в своп
антиподы, лежащие на той же сфере, то уравнение D2.176)
удовлетворяется. Поэтому антиподная функция
эквивалентна аддитивному интегралу движения.
374 Гл, V. Основы точной кинетической теории газов
Легко показать, что антиподная функция тождественно
равна нулю, если она обращается в нуль в следующих
пяти точках^):
@,0,0), A,0,0), @,1,0), @,0,1), (-1,0,0). D2.19)
Далее, всегда можно, составляя ту или иную линейную
комбинацию пяти функций D2.18), образовать функцию,
имеющую в характеристических точках D2.19)
произвольные наперед заданные значения, т. е. совпадающую в этих
точках с любой антиподной функцией. Так как разность
между заданной и образованной таким путем антиподными
функциями сама является антиподной функцией, которая
обращается в нуль в точках D2.19), то она вообще равна
нулю. Следовательно, существуют только антиподные
функции и аддитивные интегралы движения вида
^ = a^ + av + a4V^ D2.20)
В силу D2.17а) отсюда следует (если ввести другие
обозначения для констант)
ln/ = a-7(v-uJ, D2.20а)
т. е. при е* = а получается закон распределения по
скоростям Максвелла
/ = /Jv) = ae-v(v-uJ D2.21)
с той лишь разницей, что здесь величины а, ^ и и могут
быть функциями от г и ^ В этом случае говорят о
локальном распределении Максвелла.
Надо еще доказать лемму Града. Рассмотрим сначала
только плоскость ?^, Еу, изображенную на фиг. 36, а.
Из пяти точек D2/19) в этой плоскости лежат четыре
точки: Л, В, С, D. По предположению, в них ф = 0. Это же
справедливо и для всех узловых точек квадратной решетки
на фиг. 36,6. Действительно, всегда можно найти пары
таких точек-антиподов, для трех из которых уже известно,
что ф = 0 и, следовательно, то же самое справедливо для
^) Доказательство дано Градом [Н. Grad. Commun. pure appl.
Math., 2, 311 A949I.
§ 42, Н-теорема и распределение Максвелла
375
четвертой. Например, (^, Z); 5,1), (С,/); 5, 2), (Л, С;/), 7)
и т. д.
Таким же путем можно найти и другие точки, в
которых ф обращается в нуль. Пусть на фиг. 36, б точки,
обозначенные знаком « + », представляют собой узловые
точки фиг. 36, а. Тогда ф исчезает также и в средних
точках а, Ь, с, d, . . ,у как это вытекает из рассмотрения
12
13
14
11
10 .
1
9
^ ^
1 1
8_
1
1
1
1
¦< 1
1
1
1
т
7 \б_ ,
3
4
5
16
Фиг. 36. К выводу локального распределения Максвелла.
следующих пар антиподов: (а, Ь\ 2, 5), F, d\ 5, 6), (с, d\ 5, 8),
(а, с; 4, 5), (а, d\ b, с). Из соотношений
ф(а) + фF) = фB) + фE) = 0, фF) + фН = фE) + фF) = 0,
ф(с)-Ьф(^) = фE) + ф(8) = 0, ф(а) + ф(с) = фD) + фE) = 0,
ф(а) + фИ = 'М^) + Ф@
вытекает, что
Этот процесс разбиения, очевидно, вновь приводит к
квадратной решетке (но уже с меньшими ячейками).
Следовательно, его можно продолжить. Таким образом, мы
получаем сетку сколь угодно плотно расположенных точек,
в которых функция ф исчезает. В предположении
непрерывности ф отсюда вытекает, что ф(у) = 0, что и требо*
валось доказать.
.'З7б Гл. V» Основы точной кинетической теории газов
Используя все точки D2.19), можно без особого труда
обобщить данное построение и доказательство на три
измерения.
3. Равновесные распределения. Уравнение D2.21)
содержит множество распределений, совместимых с
условием изменения энтропии только вследствие наличия
потока. Однако не все функции г и / в уравнении D2.21)
допускаются кинетическим уравнением Максвелла—Больц-
мана. Напротив, из уравнения D1.18) следует
4^+vl + i-F^ = 0. D2.22)
Из уравнения D2.22) после подстановки выражения
D2.20а) и простого преобразования получаем
i = - (uV) (а --ju^) - 7u2, D2.23)
где индексы i и к означают ж, у или z.
Первые два уравнения D2.23) можно преобразовать
к виду
Т = т@, u = f^r i-[a(Or] + b@. D2.24)
Так как и есть средняя локальная скорость, то
уравнение D2.24) представляет особую комбинацию переноса,
вращения и радиального расширения. Полное движение
всегда изотропно. Так как за время 8^ г переходит
в r-fuo/, то, следовательно, dr переходит в dr-\-dubt и
dv^-^dr^ + 2{drdu)bt
(если опустить член с 8/^). Согласно D2.24), имеем
du =~dr-{- [ac?r],
следовательно,
dv^'^f^l+l-U^dr'-. D2.25)
Все расстояния изменяются в равных отношениях.
§ 43, Основные уравнения гидродинамики 377
Последнее уравнение D2.23) определяет вид силового
поля, в котором возможно локальное распределение
Максвелла. В частности, при и = 0 из D2.23) и D2.24) получаем
а = а(г), 7 = const, b = 0, а = 0 D2.26)
и, кроме того,
F=+-Jgrada. D2.27)
Таким образом, в отсутствие локальной скорости потока
тепловое равновесие устанавливается только в не
зависящем от времени потенциальном поле (ср.
барометрическую формулу).
§ 43. ОСНОВНЫЕ УРАВНЕНИЯ ГИДРОДИНАМИКИ
1. Разложение функции распределения в ряд. Для
вычисления интеграла столкновений в уравнении D1.18)
необходимо знать функцию распределения /, являющуюся
решением кинетического уравнения. Поскольку мы
рассматриваем неравновесный случай, она будет отличаться
от локального распределения Максвелла D2.21)» Однако
эти отклонения от равновесного распределения, вообще
говоря, малы. Поэтому при определении функции /
целесообразно исходить из локального распределения Максвелла.
Без значительного ограничения общности можно положить
Действительно, отношение //Д представляет собой
по существу разложение по полиномам Эрмита от трех
переменных [эти полиномы образуют полную систему
функций (см. т. VI, Дифференциальные уравнения в
частных производных физики)], и можно ожидать, что при
незначительных отклонениях от раврювесия коэффициенты
разложения окажутся быстро убывающими.
Индексы к J I, т, . . . в уравнении D3.1) соответствуют
координатам ж, ?/, 2. Подразумевается, что по одинаковым
индексам производится суммирование, так что, например,
378 Гл. К. Основы точной кинетической теории газов
второй член суммы в подробной записи имеет вид
п^Х.а.^^а ^^'
о
Таким же образом следует понимать и следующие члены.
Коэффициенты а^, а^^, а^^;^, ..., вообще говоря, зависят
от г и ^ но, по определению, не зависят от v. Они
образуют тензоры 1, 2 п более высокого ранга, которые можно
считать симметричными по всем индексам.
Разложение функции / D3.1) будет однозначным лишь
в том случае, если определить не зависящие от скорости
коэффициенты а, а, ^ и и в локальном распределении
Максвелла
Д = е«-т (v-uJ ^ д^-т (v-uJ^ D3.2)
Эти коэффициенты можно определить, потребовав, чтобы
отдельные интегралы допускали строгое вычисление уже
с функцией Д; это следующие интегралы: плотность
частиц
n=^fd\^^f,dl = ai^^y\ D3.3а)
средняя скорость
v = lj'v/da = |^v/od$ = u D3.36)
И среднее изотропное давление [см. формулу B2.3а)]
P = ^\i^-^Ld\='^\{y-yL,d^"-^ D3.3b)
(давление на стенки малого объема, движущегося со
средней скоростью и).
Уравнение D3.30) позволяет определить коэффициент а
в выражении D3.2). Если определить температуру Т
соотношением
T = W D3.4)
И воспользоваться выражением р = пт для плотности
массы, то получим
»-i(wr- («¦*»
§ 43, Основные уравнения еидродинамики 379
Два других уравнения дают условия для определения
коэффициентов разложения D3.1). Из выражения для i-ii
компоненты уравнения D3.36) следует
Так как функция Д вместе со всеми своими
производными исчезает на поверхности бесконечно большой сферы
в пространстве скоростей, то можно произвольное число раз
интегрировать по частям, опуская при этом интегралы
по поверхности. Получаем
'^kifod'i-a^^Udi^^y.dl
В левой части остаются теперь только два слагаемых.
Так как первое из них совпадает с правой частью, то
а^ = 0. D3.5)
Аналогично из уравнения D3.Зв), подставив в него /,
определяемое выражением D3.1), и проинтегрировав
по частям, получим
= \{l-h)'Ud^-
Первое слагаемое в левой части сокращается с членом
в правой части, второе в силу D3.36) и D3.5)
тождественно равно нулю. Из рассмотрения третьего члена
следует, что а^^Ь^^^ = Оу где 8^^^ представляет собой символ
Кронекера:
I 1 при i = k,
^^^ "" 1 О при i Ф к.
Следовательно, для суммы диагональных членов а^^ имеем
fl^^=0. D3.6)
380 Гл, V. Основы точной кинетической теории газов
Поэтому уравнение D3.1) принимает следующий вид:
^ "" V "^ 2р ''fti ^5fe д'м 6р ^fe^^ d^fe д'^1 д%т ^
Для коэффициентов разложения здесь введено другое
обозначение, более удобное для дальнейшего. Из
уравнения D3.6) вытекает еще дополнительное условие
= 0 D3.7а)
(шпур тензора о^^^ равен нулю).
2. Уравнения переноса Максвелла. Здесь под
моментом мы будем понимать локальное [вычисленное по
формуле D1.5)] среднее значение некоторой степени скорости,
например
5л=-^5^л/«г5.
Более важными в дальнейшем окажутся моменты
локальной относительной скорости
c = v —v = v —U, г^ = Ji —^i = Ei —Mi. D3.8)
В этом случае моменты второй степени (при dc = d^)
имеют вид
^Л = 4 5 ^i^kfd(^'
Для моментов первой степени, т. е. средних значений,
согласно уравнению D3.8), имеем
c, = \{^,-\)fdc = 0. D3.9)
Выгода разложения в ряд D3.1) или D3.7) состоит в том,
что при вычислении моментов всегда приходится иметь
дело с конечным числом коэффициентов разложения (число
их равно степени момента).
Все без исключения моменты можно выразить через
моменты, вычисленные относительно распределения
§ 43. Основные уравнения гидродинамики 381
Максвелла. Введем д^ля произвольной функции
скорости 9(c) помимо локального среднего D1.5)
еще среднее по локальному распределению Максвелла
i» = 15/ocp«fS. D3.10)
Тогда, интегрируя по частям, получим ^)
—7-4 -о , 1 Щ^ , 1 п ^° . //о Q Ч
?(с) = ? +-2i'kie^lv^-r^~Q,un-^^^ D3.9а)
Так как распределение Максвелла симметрично
относительно отраженрш (с —» —с), то при усреднении по нему
все нечетные моменты исчезают. Далее, из
инвариантности распределения Максвелла относительно отражения
и вращения вытекают (с точностью до численных
множителей) соотношения
CiCl==fK, qc^o = -^(Sj,8^, + 8^,5„4-8,,8,.). D3.10а)
Численные множители получаются из рассмотрения
конкретных интегралов:
Таким образом, в силу D3.9а) средние моменты имеют
вид (недописанные члены в скобках получаются круго-
^) Это преобразование может привести к трудностям в более
высоких членах, если встречаются множители с = У ^Ь так как
получающиеся интегралы могут расходиться, несмотря на то, что
интеграл D3.10) сходится. В таких случаях можно опз^тить
интегрирование по частям или взять лишь конечные части интеграла.
См. L. Schwartz, Theorie des distribution, Paris, 1950).
382 Гл. V. Основы точной кинетической теории газов
вой перестановкой)
cA = ySife+7^ift. (^i4^i=jQiki^ (^3.96)
P
jki-
Моменты, вообще говоря, зависят от пространственных
координат и от времени. Они удовлетворяют
характеристическим дифференциальным уравнениям, вытекающим
из кинетического уравнения D1.18). Если умножить
последнее на ср (v) и проинтегрировать по пространству
скоростей, то получим (непосредственно или после
интегрирования по частям) уравнение переноса для величины
cp(v): _
|-(P?) + div(p^)-f(-|f) = /(cp). D3.11)
Здесь через р и f обозначены соответственно плотности
массы и силы:
р = т/г, f = -^F = nF. D3.12)
Выражения рср и pcpv представляют соответственно
плотность и плотность тока величины cp(v). /(ср) обозначает
интеграл
•^(?) = ^ $ <P(v)(/7;-//i) |Ve|rf«,d$d5i, D3.12а)
который, согласно D2.13), можно переписать в виде
«/(?)=-^5^?+?i-?'"T;)(/7;-//i)ive|cfa)d$rf?,.
D3.13)
Назовем это выражение моментом величины ср (v)
относительно интеграла столкновений.
Если в уравнение D3.11) вместо ср подставить
аддитивные интегралы движения D2.18), то правая часть
обращается в нуль. В D2.18) приведены все без
исключения аддитивные интегралы движения. Поэтому мы полу-
dt
§ 43, Основные уравнения гидродинамики 383
чаем пять уравнений, соответствующих пяти законам
сохранения: массы, энергии и трех компонент импульса.
Эти уравнения имеют вид
(pt) + div(pt^) = nF(-|i-). D3.14)
Равенство /(ф) = 0 означает, что масса, энергия и импульс
при ударе не изменяются. В присутствии внешнего поля
импульс может не оставаться постоянным.
3. Сохранение массы. Закон сохранения массы
получается при ф = фд=1. Тогда уравнение D3.14) принимает
вид
|f + div(pu)=0. D3.15)
[Здесь использовано уравнение D3.36).] Это известное из
гидродинамики уравнение непрерывности [см. т. II,
Механика деформируемых сред, уравнение E.4')]. Оно
справедливо в более общем случае, чем это соответствует
предположениям, необходимым для вывода уравнения
Больцмана.
Физический смысл уравнения D3.15) обнаруживается,
если проинтегрировать его по произсольному конечному
объему V. Так как полная масса, содержащаяся в объеме F,
равна
{pdx = Л/,
то от первого члена уравнения D3.15) получаем diM/di.
Интеграл от второго члена преобразуется, согласно
теореме Стокса:
\ div(ри) б/о; = \ pii^do.
Здесь da — элемент поверхности О, ограничивающий
объем F, и^ — компонента средней скорости в
направлении внешней нормали к поверхности. Величина pu^da
есть поток массы через элемент поверхности do] он
положителен, если поток направлен наружу, и отрицателен,
если поток направлен внутрь. Интеграл распространяется
по всей поверхности О, ограничивающей объем V, Поэтому
384 Гл. V. Основы точной кинетической теории газов
ИЗ уравнения D3.15) получаем
\ рс?ж= \ pu^da. D3.15а)
dM _ _ _rf_
dt ~~ dt
Уменьшение массы в объеме V равно потоку массы нару^нсу.
Плотность потока массы равна
s = pu, D3.156)
т. е. она определяется переносом вещества со средней
скоростью U.
4. Сохранение импульса. Для вывода закона
сохранения импульса запишем уравнение D3.14) отдельно для
каждой компоненты
1. ' ~
dt
(P'W+I.CP'^W^/.dt) D3.14а)
И положим в соответствии с D2.18) ф = ф. = ?.. Получим
4(р«Л + |л(рШ = Л- D3.16)
Первый член можно преобразовать при помощи
уравнения непрерывности. Имеем
д f ч Ьи\ , до ' ^ / \
"аг (P«i) = Р ж + "i 17 = P«i - "i ал (Р"") =
Вследствие этого уравнение D3.16) принимает вид
pf-pD-«'=S)«'=-|tp(^rS'^-«i«'')l + A-D3.17)
Здесь символ d/dt = (d/dt-\'Ui^d/dk) обозначает полную
производную, отнесенную к некоторому объему,
движущемуся со средней скоростью данного вещества [см. т. II,
уравнение A1.3)].
Выражение в квадратных скобках в силу D3.96) равно
Р EЛ - «i«ft) =*о (^i - «i) (^ft - ^h) = P^i^k = P^ik + ^ik'
§ 43. Основные уравнения гидродинамики 385
Таким образом, если вернуться к векторной записи, из
D3.17) строго получаем
p-^ = p(^-^ + uv)u= -grad;?-Divj-J-f. D3.18)
Здесь дивергенция тензора Dive представляет вектор
с компонентами
(Div<T)i = -^. D3.18а)
Уравнение D3.18) совпадает с основными
уравнениями гидродинамики. Поскольку <з^. = О, а^^
представляет тензор напряжений, описывающип только силы
сдвига (см. т. II, § 10). Условие
<'ift = 0 D3.19а)
приводит к уравнениям Эйлера. При
^ife =
i(S+??-4t^0 ^'^•'''
получаются уравнения Навье-Стокса. Эта форма тензора
^ift в § 44 выводится (в известном приближении) из
кинетического уравнения,
В случае ламинарного (куэттовского) течения (см. § 27)
u = (tt(y), О, 0) получаем уравнение B7.4), а именно:
— _ ^_
Обратно, равенство D3.196) следует из уравнения B7.4),
если учесть трансформационные свойства входяпц1х сюда
величин при вращениях системы координат.
При интегрировании по малым (!) объемам,
движущимся со средней скоростью, получаем уравнение,
аналогичное уравнению D3.15а),
^^pu,dx=-\{pn,-\-o,Jdc+^f,dx. D3.20)
Здесь III обозначает i-ю компоненту единичного вектора
п в направлении внешней нормали, а <^in=^^i}Jh-
Приращение импульса в единицу времени определяется
потоком импульса через поверхность (снаружи внутрь вслед-
386 Гл. V. Основы точной кинетической теории газов
ствие отрицательного знака) и полной силой,
приложенной к данному объему (последняя вычисляется из
плотности силы f).
5. Сохранение энергии. Закон сохранения энергии
получается, если положить
Уравнение D3.14) в этом случае принимает вид
^(^A7^^ + div(^^^T^) = fv. D3.21)
Здесь, согласно D3.8) и D3.10),
v2=(C + uJ = c2 4-u2 = -^ + u2.
Таким образом, первый член в D3.21) имеет вид
Преобразуя это выражение при помощи уравнения
непрерывности D3.15) и уравнения для импульса D3.18),
получаем
3 • 1
у /? — Y u2 div (pu) — u [р (uV) u + grad p + Div a — f ],
или
-div ('-|u2.u + [ffu])+(«)+(uf). D3.22)
Здесь произведение [<svi\ тензора 9 на вектор u
представляет собой вектор с компонентами
l««]i = <'ift«ft. D3.22а)
деформации (см. т. II, § 1), описы-
6 есть часть тензора деформации (см. т. II, § 1),
описывающая сдвиг,
§ 43. Основные уравнения гидродинамики 387
а (ае) — скалярное произведение тензоров а и е,
определяемое выражением
(„) = а,,е„ = с„^. D3.22b)
Эквивалентность двух последних выражений следует из
симметрии тензора ^{<^ik = ^ki) ^ обращения его шпура
в нуль (ау. = 0).
Таким же образом можно преобразовать среднее
значение во втором члене уравнения D3.21). При этом
получаем
у2.у= (C + uJ.(c-f-u)= c2.C-f-c2-U-b
+ 2[("cl7)u] + u2.u. D3.23)
Согласно D3.96), первый член, записанный в
компонентах, имеет вид
Если ввести вектор Q с компонентами
Qi-Y^и^' (^3.24)
то получим
c2.c = -Q. D3.24а)
Для второго члена на основании D3.96) находим
^2.u^iP_u, D3.246)
В третьем члене величина (с | с) обозначает тензор с^с^^у
так что в силу уравнения D3.96) имеем
2[^)u]=^u + }m. D3.24b)
Наконец, четвертый член остается без изменения.
Подставляя выражения D3.22) и D3.23) в уравнение
D3.21) и принимая во внимание соотношения D3.24) —
D3.24в), находим
4^-(uV)p + div[Q-(-4/>u]+(ae)-ffu = fu.
388 Гл, V. Основы точной кинетической теории газов
Элементарное преобразование дает
D3.25)
Будем рассматривать вектор Q, определенный
равенством D3.24а), как плотность потока тепла
(характеризующую энергию, переносимую движущимися капельками)
и введем в качестве плотности внутренней энергии
(кинетической энергии в движущейся системе координат)
выражение
<? = 1-^^=4/?. D3.24г)
Тогда уравнение D3.25) можно переписать следующим
образом:
f + divQ=(| + uv)(? + divQ =
= - <? div U - J3 div U - («). D3.26)
Это уравнение можно преобразовать двумя способами.
Перенося первый член справа в левую часть, получим
§4-div(Q + (?u)=-pdivu-(«). D3.27)
В правой части здесь стоит работа сжатия и работа
против сил внутреннего трения (см. § 44). Под знаком
дивергенции в левой части уравнения D3.27) фигурирует доля
локального изменения Qy обусловленная
теплопроводностью (Q) и конвекцией (^и).
Введем вместо плотности энергии энергию на единицу
массы G» т. е. положим Q = pq\ тогда получим
Й = РI + ^1 = Р Й - ? <*'^ (Р") = Р л - div(pgu).
Таким образом, из уравнения D3.27) получаем
p| + divQ = p(| + uv)<? + divQ =
=-/?divu-(«). D3.28)
^ 43. Основные уравнения гидродинамики 389
Правая часть этого уравнения имеет тот же смысл, что
п прежде. Слева дополнительно появляется только
тепловой поток, обусловленный теплопроводностью, так как
теперь, говоря макрофизически, мы рассматриваем
определенный элемент массы и следим за ним в процессе его
движения.
6. Принцип энтропии. Будем исходить из
определений плотности энтропии и потока энтропии D2.1) и D2.5)
и из уравнения D2.14) с существенно положительным
распределением источников энтропии G, определяемым
равенством D2.12).
Если 7] — энтропия единицы массы, то
Н =
Р^»
и из уравнения D2.14) следует, если принять во
внимание уравнение непрерывности D3.15),
/ д \
Я = рт] + рт] = рт] — 7] div (pu) = р ( ^^ -Ь WV J 7] — div (рщ).
Вследствие этого уравнения D2.14) принимает вид
9^, = 9(i+uv)ri=-diy(S-pr,n) + G. D3.29)
Из потока S в этом случае вычитается энтропия,
переносимая конвекцией Яи. Согласно D2.1) и D2.5), имеем
8-Ни= -А \ clnf'fdl D3.29а)
Вычислим приближенно S- Яи и Я. Будем считать,
что функции / и Д мало отличаются друг от друга, так что
можно положить
In/^ In Д.
В этом случае, согласно D2.21),
In/ ^ In а — ifc^.
Отсюда в силу D2.1) получаем
390 Гл. V. Основы точной кинетической теории газов
Т. е., согласно D3.Зв) и D3.4а),
Я= р 1пр + -17 —р In Г + const р.
Отсюда для энтропии единицы массы [ср. уравнение
E.10)] получаем выражение
7]= ^A('lnp--|lnr^+const, D3.30)
или, если взять полную производную,
rfY)_ л / 1 rfp г i dT\ Г43 30а^
rfi- ~m V7 'dt^'^ ~T Tt) • (<t^.^iJa)
Умножение на Т дает
rpd'{\ dq пкТ dp
'di~"di p2" It '
так как q =-7y nkT/p=-j{k/m)T, Если последнее
уравнение записать в дифференциалах и использовать формулу
р = пкТ, то оно примет вид
Tdr, = dq \~pdf~^ . D3.31)
Это второе начало термодинамики с dq в качестве
дифференциала внутренней энергии. Полученное
соотношение связано с апроксимацией In/^ InД, означающей, что
распределение не слишком отличается от равновесного.
Последнее ограничение аналогично условию,
накладываемому при определении обратимых процессов. Однако
фактически оно является менее жестким и допускает,
чтобы величина G в уравнении D3.29) была отлична от
нуля. Причина этого состоит в том, что здесь
приравниваются лишь логарифмы In/ и In/^, которые при
больших значениях аргументов изменяются медленно.
Величина отклонений от равновесного распределения,
допускаемых этим ограничением, была уже указана д §21, п. 6.
Обосновать это можно путем более точного решения
кинетического уравнения. Однако мы этого делать не будем
и отошлем читателя к соответствующей литературе ^).
1) D. Enskog, Phys. Zs., 12, 533 A911): J. Meixner, Zs.
Phys. Chemie, 53, 235 A943).
§ 43. Основные уравнения гидродинамики idl
Величину G можно вычислить при помощи
уравнения D3.29). Прежде всего из D3.29а) следует (если для
In/ подставить максвелловское выражение)
На основании D3.24а) выражение справа равно
^^77^ = ?-
Следовательно, получаем термодинамически естественный
результат
8-Яи = ^ . D3.32)
Подставляя это выражение и выражение D3.31) в
уравнение D3.29), найдем
Второй член в правой части можно преобразовать при
помощи закона сохранения энергии D3.28). Принимая во
внимание равенство р/Т =^пк=^{к/т)р, получаем
G=-^(Q.gradr)-l(ncr)-A(| + pdivu).
Последнее слагаемое здесь исчезает в силу уравнения
непрерывности. Следовательно, имеем
G= -^(Q» gradr)--l(ae). D3.33)
Это основное уравнение, на котором базируется
термодинамика необратимых процессов. В связи с этим сошлемся
на §21. На основании уравнения D3.196) и закона
теплопроводности Фурье B1.3)
Q= -xgradr D3.33а)
можно заключить, что величина G в уравнении D3.33)
существенно положительна:
G= +^^(gradrJ + ^e2 > 0. D3.336)
392 Гл, V. Основы /почиой кинетической теории газов
В связи с этим заметим, что закон Фурье, так же как
уравнение D3.196), можно обосновать при помощи
кинетической теории газов (см. § 44).
§ 44. К ИНТЕГРИРОВАНИЮ КИНЕТИЧЕСКОГО УРАВНЕНИЯ ]
1. Интегрирование методом моментов. Было развито
много приближенных методов интегрирования
кинетического уравнения D1.18). Относительно деталей теории
интегрирования отсылаем читателя к обзорной работе
Херцфельда ^) и к уже цитированной работе Града ^).
Из всех приближенных методов здесь будет использован
лишь один —тот, в котором применяется разложение D3.1)
по производным функции распределения Максвелла. При
этом и он будет развит лишь в той мере, какая
необходима, чтобы понять существо метода и обосновать
уравнения D3.196) и D3.33а), ведущие к уравнениям Навье —
Стокса и уравнению теплопроводности. Практически при
этом мы приходим к методу моментов.
Весьма обширный класс функций g(v) можно
разложить в ряд по производным функции распределения
Максвелла. Если положить
Siy)-(A, + A,J- + A,,^^ + ...)U D4.1)
то коэффициенты А^, А'^, A,^i, ... можно определить,
вычисляя моменты
nGo=^g{\)d^nAf„
nG,= \^i,g{y)dl = niAjl-A,), D4.2)
nG,a = 5 ^ft а, g (v) di = n {A, T^ti - a;i\ - Afil + ^„) и т. д.
Средние значения \, f^^^i, ... определяются здесь
согласно выражениям D3.10). Соотношения D4.2) представляют
^) К. F. Не г zf eld, Freie Weglange und Transport erscheinun-
gen in Gasen, Hand-u. Jahrbuch der Chemischen Physik, Bd. Ill, 2,
Abschn. IV, Leipzig, 1939.
2) H. Grad, Commun. pure appl. Math., 2, 311 A949).
§ 44. К интегрированию кинетического уравнения 393
рекурентные формулы для коэффициентов разложения,
позволяющие однозначно определить последние. Получаем
A^^G,ll-G,„ D4.3)
A, = G„m;-GJ?-G,12 + G„ и т. д.
Отсюда следует, что если моменты равны, то
коэффициенты разложения двух функций совпадают. Эта теорема
и будет использована при решении кинетического
уравнения. Ее можно применять, если удается удовлетворить
всем уравнениям моментов D3.11). Вместо того чтобы
решать кинетическое уравнение, можно проинтегрировать
совокупность уравнений моментов. Последние составляют
удобный исходный пункт для приближенного расчета.
В § 43 уже были рассмотрены первые уравнения
моментов, а именно все те, в которых вклад от интеграла
столкновений обраш;ается в нуль. Рассмотрим теперь еще
уравнения моментов для ср = ^^^^^ и ср = ^^^у?^^. Согласно
уравнению D3.11), получаем
i (Pir^h) +1 (рШ,) - {lifk + IJi) = Jiu^ D4.4)
-| {?WK) + i(РЩГ^,) - (Wi + ^"^i/,-+МЛ) = •^i;'.- D4-5)
В правых частях здесь стоят моменты относительно
интеграла столкновений
Jik = ^ \ ^i^K (/'/; - /Л) I Ve i cfu) dS d^,„ D4.4a)
Л;. = '-^5 ^iMh(/'/;-//i)|VeKu,cfUSi. D4.5a)
2. Прсобразовгние уравнений моментов. При
вычислении средних значений и моментов относительно
интеграла столкновений в уравнениях D4.4) и D4.5) следует
подставлять функцию распределения / в определенном
приближении. Простейшее нетривиальное приближение
получится, если в обеих частях уравнений сохранить
394 Гл. V» Основы точной кинетической теории газов
лишь высшие неисчезающие выражения. Это означает,
что в данном приближении мы используем в левой части
распределение Максвелла.
Таким образом, отчасти возвращаясь к прежним
вычислениям, заменим средние значения степеней Е
следующими выражениями:
pli = pKi, pE^ft ^ рЩ = /?8ift + pWiKft, D4.6)
9WJi ^ РЩГ? = f (Mii 4 ...) + /> [(8;7.«Л +•..) +
+ (8uK>Ki 4-...)] +pKiWyWftKr
При этом уравнение D4.4) принимает вид
После простого преобразования получаем
Если заметить, что в используемом здесь приближении
о.,^ = 0 и ^(==0, то, согласно уравнениям D3.15), D3.18)
и D3.25), все члены справа, кроме второго, исчезают.
Пользуясь выражением для тензора деформаций е^^ [см.
D3.226)], получаем
Л, = 2/>е,,. D4.7)
Точно так же можно преобразовать и уравнение D4.5).
Если подставить средние значения D4.6), то прежде всего
§ 44, к интегрированию кинетическоео уравнения 395
получаем (в сокращенной записи)
•^iy» = Jt ^Р ("i^-fe Н • • •) + PKi«y"fcl +
+it [7(МуИ- • • •)+/'(Vi»i+ • • •)+
+ P (SiiMyWft +.••) + рщи^и„щ ] -
-f(/iV+---)-(/i«y«ft+...)-
Вновь объединяя соответствующие члены, находим
следующее, более обозримое выражение:
+f[v(<'5-/. + fe) + ...] +
+'' [vl(f)+• ¦¦ ] +l'^ <U'(p")l».v»+
+ [«<«.(pS-/.+4f)+-] +
+''[a4+t+-]-
Отсюда на основании уравнений D3.15), D3.18) и D3..25),
в которых следует положить а^^ = 0, ^^ = 0, и принимая
во внимание, что р/р = (к/т) Г, получаем
или в силу D4.7)
J4>^=i(i^hH-r...)^-{u,J,,+ -..). D4.8)
Ниже мы увидим, что величины Jif^ и а.,^
пропорциональны друг другу. Так как в левых частях уравнений D4.4)
и D4.5) мы положили а,-^ = 0, то следует также и в
уравнении D4.8) опустить /у,,. Окончательно получаем
396 Гл. V. Основы точной кинетической теории газов
Отсюда вытекает выражение для шпура
3. Вычисление моментов относительно интеграла
столкновений. Моменты относительно интеграла
столкновений даются выражениями D3.12а) или D3.13) и
обращаются в нуль, если заменить / функцией
распределения Максвелла. Следовательно, в данном случае
необходимо учесть поправочные члены. Самые низшие члены
в разложении произведения /Д, согласно D3.7), имеют
вид
Так как первое слагаемое никакого вклада в момент
не дает и так как квадратичные члены а^^ и Q^^^
встречаются лишь в четвертом порядке, то искомый момент
представляет собой в данном приближении линейную
однородную функцию относительно о,,^ и Q^^^.
Моменты в уравнениях D4.4а) и D4.5а) точно так же,
как и коэффициенты а^^^ и Qij)^, являются
симметричными тензорами. Так как в данном случае играют роль
только тензоры о^^, Q^.^^ и единичный тензор а^^^, то
можно утверждать, что моменты относительно интеграла
столкновений имеют следующий общий вид:
D4.12)
В первом уравнении D4.12) отсутствует член,
пропорциональный 8y^J, так как в силу однородности е.. = 0.
Используя уравнения D4.7) и D4.9), можно при
помощи уравнений D4.12) вычислить коэффициенты
разложения 0|,j и Qijk. Они пропорциональны соответственно
е,, и [{dTJdi)b^^-{ Щ1д])\,^ {дТ/дк)Ь,^].
§ 44, К интегрированию кинетического уравнения 397
Обозначения для множителей пропорциональности
выберем в соответствии с D3.196) и D3.33а), тогда получим
а,,= -27]е,„ (?^=-х^. D4.13)
При этом последнее равенство в силу D3.24) вытекает
из выражения для тензора Qij^C-
Коэффициенты внутреннего трения и теплопроводно
сти 7] и X получаются из уравнений D4.12). Если в них
подставить выражения D4.13) и D4.13а), то получатся
два уравнения, которые надо отождествить с
уравнениями D4.7) и D4.9);
Отсюда находим
'^="^'7' ""= ~ 2/гF + 5с) = +Т Гь5^ ~^'^' D4.14)
Следовательно, чтобы определить т] и х, необходимо лишь
вычислить а и Ь + Ъс.
Подставляя.выражения D4.13) и D4.13а) в D3.7),
получаем для функции распределения
f^f ^±г J^4^:^f^±^^'- D415^
/ - /о р hk е-,, dik ^ 5р V. di dii) Q^) • ^^^-^^^
Выражения D4.13) и D4.13а') совпадают с D3.196) и
D3.33а) и дают обоснование уравнений Навье — Стокса и
уравнения теплопроводности, как уже было показано в
D3.336). Плотность источников энтропии D3.33)
оказывается существенно положительной (как это и должно
быть), если положительны величины х и tj (см. п. 4).
4. Коэффициенты внутреннего трения и
теплопроводности. Мы должны еще вычислить интегралы D4.4а)
и D4.5а) и связать их с выражениями D3.13). В интег-
398 Гл. V. Основы точной кинетической теории газов
ралах D4.4а) и D4.5а) можно преобразовать слагаемое
с множителем ff[. Поменяем местами переменные v', v(
и V, Vj. В силу D1.126) и D1.16а) значения d^di^-^m |Ve|
при этом не изменяются; первый множитель меняет знак
на обратный. Таким образом, можно представить D4.4а)
и D4.5а) в виде
и выполнить интегрирование в два этапа.
Так как функции распределения /и Д не зависят
от направления вектора е, то прежде всего можно
выделить интегрирование по углам:
/y = /nv, Vi)=4-^5(<p' + ?;-'f-cp,)|Velrfu,. D4.17)
Рассматривая специально /^ при ср = ^-f^ и ср = Е|5у5^,
имеем
hk= +^ \ {Ш¦\^Ы[k-liK-\гi'^,u)\'f^\d^^> D4.18)
Аа= +-? \ Ei5;$,;+a{i^/,r,ft-5iS^-?ii^^,)|Ve 1(^@.
D4.18а)
Здесь V — относительная скорость. Если ввести наряду
с этим среднюю скорость U = -2-(v-f v,), то получим
v-U-lv, v, = U + iv, D4.19а)
и на основании D1.12)
v' = U-yV', v; = U + ^V', D4.196)
где
V'-V-2(Ve)e. D4.19в)
§ 44. К интегрированию кинетического уравнения 399
После элементарного преобразования получаем
= 2 (Ve)^ е,е^ - (Ve) (F^e, 4 VJ = V,„ D4.20a)
= (U,V^, + U,V^r\-U,V\^). D4.206)
Интеграл D4.18) зависит, согласно D4.20a), только
от вектора V. Так как результат должен представлять
собой симметричный тензор со шпуром, равным нулю,
имеем
lik = ^-f-V ( V,V, -1 F^8.,) . D4.20)
Из D4.18a) и D4.206) получаем
hiH = {UiIj, + U,I,, + U,I^,). D4.21)
Постоянная f получается из рассмотрения частного
случая
/ift^i^ = '-^V^ = ^^ [(Ve)*-F«(Ve)=']|Ve|dm =
1
Таким образом, ^ = —ir и
/,, = _ ^ 7 ( F,F, -1 F^^Si,) . D4.22)
4fe-
Уравнение, определяющее момент относительно
интеграла столкновений, согласно D4.16) и D4.17), имеет вид
Здесь произведение /Д дается формулой D4.11). Первый
член ее не дает никакого вклада. Две следующие пары
слагаемых можно объединить, так как уравнения D4.21)
и D4.22) симметричны относительно v и v^. Получаем
400 Гл. V. Основы точной кинетической теории газов
При помощи интегрирования по частям производные
можно перенес1и на 7-^. Тогда вместо /Д появляется
функция
/о/ог = п'1,е '^ > 2 . D4.24)
Отсюда следует, что интеграл по нечетным полиномам
от V/|V| исчезает, так что D4.23) дает [если принять
во внимание D4.22) или D4.21)]
Остальные члены суммы получаются круговой
перестановкой индексов iy /, к.
Интеграл в первом из выражений D4.25)
симметричен относительно ik и rs, и шпур его исчезает. Отсюда
следует, что
D4.25а)
Численный множитель найдем, придавая индексам
какие-нибудь конкретные значения; пусть, например,
г == г = ж, k = s = z. Тогда
^ Т^ \ ЙУ^ЯУ. '0/01 "* ""^i
dV^dV,
Tims
Чт//^/| i^Vx-hvi vivi\,. ,, ,,
Подставляя сюда вместо /o/oi выражение D4.24), можно
выполнить интегрирование по U и по направлениям
вектора V. Следовательно, находим
О
или
/ = - ^ mnh^ О^У' = - - nV {7:ткТу1к D4.26)
§ 44, К интегрированию кинетического уравнения 401
Подставляя D4.25а) в выражения D4.25), получаем
в соответствии с формулами D4.12)
Ji,k = jIQ4k-l,HQirrhk-^ .••). D4.27)
Сравнение с D4.12) дает
а = 2-^, fc = 3-, с=-4- —• D4.27а)
Таким образом, из соотношений D4.14) и D4.26)
находим
X 5 а к 15 к
7) 26 + 5ст 4 т
{44.28а)
Для одноатомных газов измерения дают следующие
значения:
Не Ne Аг Кг Хе
4тх
ibkri
= 0,98 1,00 0,98 1,02 1,03
Согласие очень хорошее, особенно если учесть, что
представление об атомах как о твердых шарах, казалось бы,
должно быть пригодно лишь с качественной стороны.
Интересный результат получается при сопоставлении
выражений D4.13), если применить первое из них к
ламинарному течению, т. е. считать, что и = (О, м^ (а;), 0).
В этом случае мы имеем
dUy 7]_ ^Pv
^""У '^ дх ~ Ип'дх
<^^.. =
^Ч^--У-В- D4.29)
Здесь /?у —средний импульс одной молекулы в
направлении оси у и а^^ —поток импульса через единицу
площади элемента поверхности, перпендикулярной к осп х.
Во второе выражение D4.13) подставим соотношение
D4.28а) и введем вместо температуры локальную энер-
402 Гл. V» Основы точной кинетической tnuopuu ёаабб
3
гию теплового движения Q^-K-hT, Тогда получим
Следовательно, перенос тепла происходит лучше, чем
перенос импульса. Качественно это понятно. Переносу
благоприятствуют большие скорости молекул в
направлении соответствующего потока. Изменение этих
скоростей не влияет на переносимый импульс, так как при
внутреннем трении речь идет о переносе компоненты
импульса, перпендикулярной к его потоку. Однако при
переносе энергии дело обстоит иначе, так как каждая
компонента скорости дает вклад в энергию. Отсюда следует,
что большие энергии благоприятствуют переносу и что
тепловой контакт в среде является более тесным, чем
импульсный.
Для вращательной энергии только что сказанное не
имеет места. Поэтому для многоатомных молекул,
предел
ставляемых в виде твердых шаров, Эйкен заменил -к- Q
в D4.29а) на
Это приводит [при учете выражения для средней
энергии молекулы Q = {f/2)kT] к соотношению
Здесь / означает число степеней свободы. При различных
значениях / имеем
/=3 5 6
1+1 = 2,5 1,9 1,75
Измерения дают
Нг Oj СО Воздух
1+^ = 2,00 1,92 1,81 1,96 (вместо 1,9)
§ 4S. Проводимость и закон Видемана—Франца 403
и далее
СН^ COg CgHg
1+1=1,74 1,64 1,66 (вместо 1,75).
В выражении D4.28) значение j/T* пропорционально
средней скорости. Легко вычислить, что
пт ^
Отсюда следует
-- :, . D4.30)
32/2
Подставляя сюда выражение /=1/^7:5^/2 из B7.11)
(/ имеет порядок длины свободного пробега), получаем
§ 45. ПРОВОДИМОСТЬ И ЗАКОН ВИДЕМАНА- ФРАНЦА
1. Кинетическое уравнение и уравнение переноса для
электронов в металле. Кинетическое уравнение для
электронов в металле отличается от уравнения Больцмана
D1.18) следующими особенностями: во-первых, следует
учитывать только столкновения между электронами
проводимости и ионами решетки, во-вторых, поскольку
последние гораздо тяжелее первых, можно считать, что при
столкновениях ионам передается только импульс, но
практически не передается энергия. Очевидно, последнее строго
не выполняется, так как электроны, согласно § 39,
участвуют в тепловом равновесии. Однако в первом
приближении закон сохранения энергии по сравнению с законом
сохранения импульса можно не принимать во внимание.
Если V — скорость электрона перед столкновением с ионом
решетки, рассматриваемым сначала как твердый шар
(фиг. 37), то скорость v' после столкновения равна
v'=v-2(ve)e. D5.1)
404
Гл, V, Основы точной кинетической теории газов
Здесь, как и прежде, е —единичный вектор в направлении
линии центров. Изменение импульса
а изменение энергии
v) = — 2m (ve) е,
A^ = ^(v'2
у2) = 0.
D5.2)
D5.3)
Если через / обозначить функцию распределения
электронов, то левая часть кинетического уравнения D1.18)
останется неизменной. Однако интеграл столкновений
изменится. В подинтегральном выражении останется
только один множитель /.
Другой множитель
заменится вероятностью встречи
электрона с ионом решетки.
Последняя определяется
плотностью ионов tIq.
Обозначив через S радиус иона,
получим совершенно так
же, как и в § 41, п. 4,
•^verl =
Фиг. 37. К столкновениям
между электронами проводимости и
ионами решетки.
= y%s2 ^ |ve|/(r, v,/)cfa),
J ew=2"Wo^^^ |ve|/(r,v-
- 2(ve)e, 0^^. D5.4)
Если ввести длину свободного пробега I = l/riQT.s^y то
кинетическое уравнение примет вид
iT + -W + i^W = -^[\'-'^'-f)do>. D5.5)
Здесь функция /' зависит от аргумента v', определяемого
выражением D5.1), так же как / зависит от v. Согласно
нашему исходному представлению, длина свободного
пробега I должна быть постоянной. Однако это представление
бесспорно является очень грубым. Чтобы оно лучше
отражало действительное положение вещей, предположим, что /
зависит от скорости электронов и от некоторых свойств
§ 46, Проводимость и аакон Видемана—Франца 405
решетки — прежде всего от ее температуры. Более точное
определение требует привлечения методов волновой
механики, чего мы здесь делать не будем.
Уравнение переноса для функции ср (v) получается
аналогично уравнению D3.11). Умножая уравнение D5.5)
на cp(v) и интегрируя по v, находим
-|-(p^) + div(pcpv)-«F(|i) = 7(cp). D5.6)
Здесь /(ср) обозначает момент функции ср относительно
интеграла столкновений:
= ^\t^^V'^'-'^'>'^''- (^5.5а)
Очевидно, законы сохранения массы и энергии
справедливы, как и прежде, так как при ср = 1 и ср = у^/З имеем
ф' —ср = 0. Однако вместо закона сохранения импульса
появляется уравнение
-^(pv) + Div[pGhO]- nF=^54d5 5(v'-v)|ve|d«..
D5.7)
Интеграл по о), согласно D5.1), равен
\ (v' — v) I ve I rfo) = — 2 \ I ve I (ve) e rfo) = — 2'kv\. D5.8)
Отсюда следует, что правая часть представляет собой
однородную вектор-функцию степени 2 от v. Чтобы найти
численный множитель, умножим D5.8) скалярно на v
и разделим на г;^. Получим
Z_C (v'-v)! уе|йо)= -2 \ |C|3 6fCrfcp= -27:.
Поэтому уравнение D5.7) принимает вид
-(pv)-bDiv[pGhO-'^F=-p(if). D5.9)
д ,-к
dt
2. Приближенное решение кинетического уравнения.
Рассмотрим решения уравнения D5.5), близкие к равно-
406 Гл. V, Основы точной кинетической теории газов
весному решению Д. Их можно найти, если, как и в § 42,
исходить из принципа энтропии. Из выражения для
плотности энтропии C8.9)
Я= ^k\[n\nn + {l-n)\n{i-n)]^di
после дифференцирования по времени получаем
В силу уравнения D5.5) и так как f={2?n^/h^)n, правая
часть этого уравнения принимает вид
*div [ v[nlnn + (l-n)ln(l-w)]^'rf; +
, ^ f Iv| 1 /i7(l —л')/ / ч л 2тЗ ,.
' 4л: J / л/A — /г) ^ ' h^
Следовательно, получаем
-^|^+divS = G. D5.10)
Здесь плотность потока энтропии
S- -Л^ \ \п\пп^ {i - п)\п{\ - п)]'^ d^^ D5.11)
и плотность источников энтропии
G = 7- \-Ц-^ш-^;71 ^{п' — n^disi -j^dl, D5.11а)
4:: 3 / л/A —л) ^ ^ ЛЗ ^ ^ /
При равновесии плотность источников G должна обра-
П1,аться в нуль. Так как функция w/(l —w) монотонно
растет с увеличением п, то подинтегральное выражение
не может быть отрицательным. Поэтому равенство G = 0
возможно лишь при п' = п. Так как из всех интегралов
столкновени!! этому условию удовлетворяют лишь 1 и \^,
то / должно быть функцио!! ТОЛЬКО энергии:
п = п,{Е), E = ^w\ D5.12)
Этот результат является очень неопределенным. Здесь
ск'азывается пренебрежение взаимодействием электронов.
§ 45. Проводимость и закон Видемана—Франца 407
Мы должны представить себе, что в уравнении D5.11а)
к G присоединен еще один член [аналогичный
соответствующему члену в D2.12)], обусловленный столкновенпя-
ми между электронами. Обычно этот член мал. Однако
если выполняется условие D5.12), т. с. если ?г зависит
только от энергии, то остается только он один. Если бы
значение G определялось уравнением D2.12), то для Д
получилось бы распределение Максвелла. Однако известно,
что электроны подчиняются распределению Ферми. Не
входя в обсуждения вопроса о возможном изменении
выражения для плотности источников энтропии G, следует поло-
лшть, согласно C9.5),
Здесь р и С — параметры, которые в данном случае могут
еще зависеть от координат и времени; 8 = 1/АГ, С означает
свободную энтальпию, приходящуюся на один электрон
[см. уравнение C9.4)]. Только в формуле D5.12а) и
заключается различие между теориями электропроводности Зом-
мерфельда и Друде — Лорентца.
Еще в одном пункте формулы D5.12) и D5.12а)
отличаются от прежних результатов. Равновесное
распределение более уже не относится к локальной средней скорости.
Математически отсюда следует, что импульс электрона
при столкновении не сохраняется. Физически также
понятно, что распределение электронов изменяется, если система
электронов движется относительно решетки, остающейся
неподвижной. Это означает, что теперь в разложении
функции распределения / по производным от равновесного
распределения /q появляются также и производные первого
порядка:
/ — /о "ft Яй, ' 9о ^fii Я^и ЯЁ, fin ^h
D5.13)
Следуя Зоммерфельду (и Лорентцу), используем здесь
несколько иное приближение. Так как Д зависит от
энергии Е, член первого порядка можно записать в виде
-«ft-^= -^Ык)/о
408 Гл. V, Основы точной кинетической теории гааов
(дифференцирование по Е обозначается штрихом). Более
высокие члены разложения в D5.13) содержат выражения
такого же типа. Если
(или если выделить из Q^i^ часть такого вида), то в
качестве вклада от члена третьего порядка получим
-4fQ^ikr -у('?А)(/о'+|^/;").
Для этих выражений характерно, что они имеют форму
и что коэффициенты Uf^ кроме координат и времени зависят
еще от энергии Е, но не зависят от направления скорости.
Поступая таким же образом со всеми членами уравнения
D5.13), получаем ряд следующего типа:
/ = f,iE) + U^(E)i,+±U,,{E)h^h:+... D5.14)
Можно считать, что здесь более высокие коэффициенты
разложения представляют симметричные тензоры, шпуры
которых исчезают. Таким образом, имеем С/';^^ = 0, С/*,^,^^ = 0
и т. д.
Если, например, U^^^i Ф О, то этот тензор можно
представить в виде
Ниш = ^Шт + (F,8,„ + VKk + ^т8ы).
подобрав входящие сюда величины так, чтобы выполнялось
условие и^ш^^- Для это следует лишь положить
[/¦^^^^ = 57^. Поэтому первый член принял бы нужную
форму, а второй дал бы вклад в ряд D5.14) в виде
Это, очевидно, можно включить во второй член ряда D5.14).
Подставим теперь выражение D5.14) в кинетическое
уравнение D5,5), сохраняя в обеих частях уравнения
§ 46, Проводимость и закон Видемана—Франца 409
только первые неисчезающие члены. Ограничиваясь
стационарным движением {df/dt = 0)j получаем
Правая часть, согласно D5.8), равна
-lt;(Uv).
Поскольку уравнение D5.15) должно выполняться при
любой ориентации вектора v, получаем
U=-f(/;F + f^-). D5.16)
Вследствие этого функция распределения в первом
приближении имеет вид
/ = /o-<f./;F + ^)- D5.17)
3. Плотность электрического тока и потока энергии.
Плотность электрического тока J и плотность потока
энергии W даются интегралами типа
W^= J^V^S. (^5.18)
Именно,
J= -eWo, D5.18а)
W = Wi. D5.186)
Из соображений симметрии первый член в D5.17) не
дает никакого вклада в интеграл D5.18). От второго
члена после интегрирования по углам получаем
О ^
Введем обозначение
^(') = -ТТ7' ' = НЕ-<:) D5.19)
в +1
410 Гл» V, Основы точной кинетической теории газов
И выразим V из соотношения Е = B/т) v^; тогда получим
Полагая
о
D5.20)
ОО
Кп= -^W \ S'(^)lE"dE, D5.21а)
получаем выражение D5.20) в виде
yf„=^K„,,F, + K,,,F„ D5.22)
ИЛИ для интересуюш;их нас частных случаев
Исключая отсюда величину
Е'= --iPi-flgradp = E + |grad(;, D5.216)
находим
Это согласуется с формулами B1.18а) и B1.186). Отсюда
находим проводимость
о^е^К^, D5.24)
коэффициент Пельтье
П = ^, D5.24а)
абсолютную термо-э.дх,
^ = ^7EW^ D5.246)
§ 45. Проводимость и закон Видемана—Франца 411
И теплопроводность
X = ^ у . D5.24в)
4. Закон Ома. Закон Ома можно получить, если
принять, что величины С и р не зависят от пространственных
координат. ДеР1Ствительно, в этом случае из D5.216)
следует, что Е' = Е, и соотношение D5.23) дает
J = aE. D5.25)
Это —закон Ома. Если вычислить интеграл/iTj по формуле
D5.21а) для полного вырождения (§ 39, п. 2), то для
проводимости из выражения D5.24) получим
_ 167tmCo/o ^2
Здесь Со и /q — значения С и / на границе Ферми. Согласно
формулам C9.7) и C9.7а), плотность частиц равна
87гт^ з' г ^2.
Таким образом, выражение для а можно преобразовать
следующим образом:
а = — -^. D5.25а)
Это —формула Друде C9.2).
Здесь ^0 — скорость на границе Ферми, а /^ - значение
длины свободного пробега для электронов с этой
скоростью. Естественно, величина /д, определяясь свойствами
решетки, может зависеть от температуры последне!!.
В противоположность этому Vq от температуры не зависит.
Лишь в случае почти полного вырожденрш в соответствии
с § 39, п. 2 V обнаруживает слабую температурную
зависимость. Последняя дается членом порядка (kT/mvl)
и практически является ненаблюдаемой.
Впрочем, значение Iq можно вычислить из измерений
проводимости. Например, для серебра при комнатной
температуре получается значение (если считать, что
каждый атом дает по одному электрону проводимости)
/^^5-10"^ см. Таким образом, /^ значительно превышает
412 Гл . V. Основы точной кинетической теории газов
расстояние между ионами решетки, что бесспорно
указывает на необходимость квантовомеханического вычисления
длины свободного пробега.
Для определения коэффициента Пелыпъе также
достаточно (по крайней мере здесь) вычислить интеграл К^
в предельном случае полного вырождения. Из формулы
D5.21а) получаем
^n = ^W D5.21b)
И, следовательно,
П = ^. D5.26)
Удивительно, что в данном случае длина свободного
пробега выпадает. Коэффициент Пельтье "П имеет простой
смысл. Когда gradp = 0, плотность потока энергии равна
W = n-2^ V.
Очевидно, что здесь мы имеем дело с макроскопическим
переносом кинетической энергии, так как электроны,
дающие вклад в среднее значение v, лежат вблизи
границы Ферми. Относительно эффекта Пельтье см. § 21.
Его рассмотрение требует более точных вычислений.
Если плотность электрического тока J = О, но
распределение температуры не однородно, то течет тепловой
ток, возбуждающий электрическое поле. Напряженность
его определяется теплопроводностью х и абсолютной
термо-э. д. с. е. Обе величины исчезают, согласно
формуле D5.21в), в предельном случае полного вырождения.
5. Теплопроводность и абсолютная термо-э. д. с.
Чтобы определить значения х и е, следует более точно
вычислить разности в числителях. Согласно § 39, п. 4
для интеграла в выражении D5.21а)
где F^ = lE"^, получаем
^п= + W [^п(С) + б^С^/^:(С)] . D5.27)
§ 45. Проводимость и закон Видемана—Франца 413
Плотность частиц дается формулой C9.116)
Таким образом, находим
В поправочном члене можно вместо С поставить
значение на границе Ферми Cq. Для множителя перед
квадратной скобкой следовало бы воспользоваться формулой
C9.13); однако в данном случае оказывается достаточным
и здесь подставить значение С на границе Ферми.
Действительно, согласно D5.27а), имеем
rj2
^Дз-л:'.=2^'-общA-о+з-2-2.2.1),
так .что множители перед квадратной скобкой в
выражении D5.27а) умножаются только на члены более
высокого порядка. Следовательно, получаем
Полагая ^Q = {m/2)vl, находим из D5.24в) выражение
для теплопроводности
;с = 1^ 1 ^^пкТ. D5.28)
Замечательно, что фигурирующие в выражении D5.27а)
производные от /(ч) при вычислении х сокращаются.
Абсолютная термо-э. д. с. вычисляется из выражения
Таким образом, выражение D5.246) принимает вид
414 Гл. V. Основы точной кинетической теории газов
На этот раз в окончательное выражение входит первая
производная от /(С).
6. Закон Видемана —Франца. В формуле Друде
D5.25а) и в выражении D5.28) для теплопроводности
фигурирует лишь значение Iq/Vq на границе Ферми. Оно
вообще выпадает, если составить отношение х/а. Тогда
получается закон Видемана — Франца
у = у,^7'- D5.30)
В работе Лорентца с экспериментом сравнивалось
значение отношения
Л = —=— -J- (число Лорентца). D5.31)
Мы будем рассматривать не зависяи1.ее от единиц
измерения отношение
^o = a^^ = -J^ = T = 3-29. D5.32)
Таким образом, надо умножить число Лорентца на
е^/к^ = 1,344 • 10® град^ • в"'-^. Опыт показывает, что
величина Aq не постоянна, а убывает с уменьшением
температуры. Однако при высоких температурах кривая
приближается к асимптотическому значению. При температуре
100° С имеем
Си Ag Pb Pt
ЗД5 3,19 3,46 3,51
Для Си и Ag при этой температуре кривая А^{Т) ужо
близка к асимптотическому значению тг^/З. Для РЬ и Pt
она лежит значительно выше.
Друде на основании приближенного расчета принимал
величину Aq равной 3. Однако при более точном расчете
на базе классической физики Лорентц получил значение 2.
В противоположность этому квантовомеханическое
значение Aq оказывается заметно более точным. Фактически
для большинства веществ значение Л^ несколько меньше
§ 45. Проводимость и закон Видемана—Франца 415
теоретического, что, повидимому, свидетельствует о
недостаточно высокой температуре.
Однако некоторым веществам соответствуют заметно
большие значения Л^. Например, для вольфрама
(поликристаллического) при Т — 273° К имеем Л^ = 4,11. Для
висмута (мелкокристаллического) при Г = 90° К значение
Лд достигает 5,56, а при Г = 273°К Ао = 3,62. Для
висмута появляется даже температурная аномалия. Прежде
всего Aq в этом случае растет с уменьшением
температуры. Здесь уместно вспомнить, что число Лорентца
относится только к электропроводности и теплопроводности,
осуществляемым электронами. Когда вклад решетки в
теплопроводность становится заметным, отношение у^/оТ
увеличивается.
Таким образом, значение А^ = 5,56 должно означать, что
примерно ^/g потока тепла обусловлено решеткой, так что
теплопроводность се (вычисленная из полного значения
х = 0,06 кал-см'^-сек'^ -град'^) должна составлять
приблизительно 0,0241). Это не совсем невероятно. Например,
поваренная соль при 20° С представляет очень хороший
изолятор (удельное сопротивление р = 1.10^^ ом-см).
Однако теплопроводность ее при 25° С составляет
х = 0,02 каЛ'См'^ ' сек ^-град^.
В то время как рассхмотренные до сих пор отклонения
могли быть обусловлены условиям опыта (недостаточно
высокая температура, значительное участие в
теплопроводности ионов решетки), сам факт температурной
зависимости Aq бесспорно не объясняется побочными
условиями. Здесь сказывается тот факт, что интеграл
столкновений в уравнении D5.5) был вычислен согласно
классической механике.
При таком вычислении возникает ряд трудносте!!. Так
в первоначальной концепции ионы металла
рассматриваются как твердые упругие шарики определенного
радиуса s. Тогда длина свободного пробега равна
l/m:s^. Фактически это предположение не имеет большого
смысла, поскольку вычисление длины свободного пробега
1) Данные взяты из справочника J. D'ans, Е. Lax, Taschen-
buch fur Ghemiker und Physiker, 2 Aufl., Berlin, 1949, S. 112G,
1129.
416 Гл, V, Основы точной кинетической теории ecLdoe
ДОЛЖНО выполняться на основе квантовой механики^). Мы
рассматривали здесь I как неизвестную функцию
температуры решетки Т^ и энергии электрона Е, Все же следует
подчеркнуть, что само понятие длины свободного пробега
/ с точки зрения квантовой механики имеет смысл лишь
при очень высоких или (с известными ограничениями) при
очень низких температурах.
1) Н. Веthе, А. Sоmmеrfеld, Elektronentheorie der Ме-
talle, Handbuch der Physik von H. Geiger uod K. Scheel, Bd. XIV,
2, Кар. 3,1, S. 333. (Cm. перевод: Г. Бете, A. Зоммерфельд,
Электронная теория металлов, М.—Л., 1938.)
ЗАДАЧИ
К ГЛАВЕ I
1.1. Между тремя переменными ж, г/, z существует
функциональная зависимость / (ж, у, z) = О, или, если
разрешить относительно z, z = f{x,y). Доказать
справедливость соотношения
/'dz\ /дх\ /ду\ _ _.
и вывести, отождествив Ху у, z соответственно с /?, F, Г,
связь между коэффициентом теплового расширения,
термическим коэффициентом давления и сжимаемостью.
[Использовать определения A.5) и A.6).]
1.2. К вопросу об отоплении.
Какое количество тепла необходимо, чтобы повысить
температуру комнаты от О до 20°" С?
1.3. Абсолютная температура и идеальный газовый
термометр.
Показать, что абсолютная температура Г,
определенная выражением F.7а), совпадает с температурой,
фигурирующей в уравнении состояния идеального газа.
1.4. Применение второго начала к выводу одного
алгебраического неравенства.
а) Два тела с теплоемкостями С^, Cg и начальными
температурами Г^, Tg вступают в тепловой контакт при
условии постоянства объема. Какова их общая конечная
температура?
б) Для частного случая идеального газа сравнить
значения энтропии до и после выравнивания температуры
(-iS* > 0) и вывести соотношение между арифметическим
и геометрическим средними.
1.5. Один моль идеального газа обратимо расширяется
до удвоенного объема: а) при постоянном давлении.
418 Задачи
б) изотермически, в) адиабатически. Каковы работа
расширения, количество подведенного тепла и изменение
энтропии газа?
1.6. Представим себе цикл Карно с водой в качестве
рабочего вещества. Температуры холодильника и
нагревателя равны 2 и 6° С, так что при 6"^ С имеет место
изотермическое расширение, а при 2° С — изотермическое
сжатие. Следовательно, если давление является
достаточно низким, то при обеих температурах подводится
тепло [см. G.10)] и полностью превращается в работу,
что находится в противоречии со вторым началом.
Требуется разрешить это противоречие. Для этого удобно
вычертить адиабаты и изотермы в плоскости Г, v при
температуре окружающей среды 4° С.
1.7. Показать, что в общем случае отношение
изотермической сжимаемости к адиабатической сжимаемости,
как и в случае идеального газа (см. т. И, Механика
деформируемых сред), равно отношению удельных тепло-
емкостей при постоянном давлении и постоянном объеме,
т. е.
— = — , если xs= E- >^т= о- •
Для этого выразить дифференциал dq через dv и dp
и показать, что
1.8. Один килограмм воды изотермически сжимают
при 20° С от давления 1 am до давления 20 am.
Вычислить затраченную работу, отведенное количество тепла
д увеличение внутренней энергии. Средняя сжимаемость
х = 0,5-10"* на 1 am, средний коэффициент теплового
расшения а = 2-10'^ на 1°. Использовать уравнение G.7а)
и приведенное в указании к решению задачи 1.1
соотношение B).
1.9. Адиабатическое равновесие атмосферы.
При так называемом конвекционном (адиабатическом)
равновесии воздуха, которое особенно хорошо проявляется
при ветре, величина pV^ не зависит от высоты, если у —
молярный или удельный объем. Если использовать связь
к главе I 419
между давлением и плотностью, справедливую при
равновесии газа в поле тяжести, то из этого соотношения
вытекает линейное падение температуры с высотой.
Измерения дают величину 1° на 100 л/. Найти
соответствующее теоретическое значение и вычислить высоту
атмосферы, описываемой в общем случае уравнением
политропы /7у" = const, где л —степень политропы.
Вычислить, в частности, высоту адиабатической и изотермической
атмосфер (для последней л = 1) при температуре Земли 0° С.
1.10. Истечение газов.
Вычислить конечную температуру и верхнюю границу
скорости истечения перегретого водяного пара при
начальных температуре 300° С и давлении 5 am. Предполагается,
что через соответствующее сопло газ адиабатически
расширяется до давления 1 am.
При вычислении исходить из того, что кинетическая
энергия не превышает (в крайнем случае равна) разности
энтальпий сжатого и расширившегося газа (см. § 4, п. 2).
Доказать это можно на основании уравнения Бернулли
для сжимаемой среды (см. т. П, Механика деформируемых
сред, § И) при условии стационарности потока и
отсутствия завихрений.
1.11. Изотермическое равновесие атмосферы.
Газ находится в закрытом ящике в поле тяжести Земли.
В этом случае к внутренней энергии добавляется
потенциальная энергия. Последняя зависит от высоты над
поверхностью Земли.
а) Определить условия термодинамического равновесия,
разделяя газ на ячейки (с номерами i) объема V^
и высоты 2,. над поверхностью Земли. Считать, что
каждой из ячеек соответствует определенный удельный объем
Vi и температура Т^, Вычислить максимум энтропии при
заданных энергии, количестве и объеме всего газа.
б) Показать, что температура не зависит от места.
в) Показать, что потенциал Гиббса не зависит от
координат точки, если в его определение включить
потенциальную энергию (ср. электрохимический потенциал
в § 18).
г) Вычислить плотность как функцию высоты,
предполагая справедливым уравнение состояния идеального
газа.
420 Задачи
д) Вычислить разность высот, для которых разность
потенциальных энергий одного моля составляет RT,
1.12^). Дано уравнение состояния pv = oiu{TyV)y где
1^G*, у)— внутренняя энергия, а —постоянная.
1. Показать, что внутреннюю энергию и и удельную
энтропию S можно представить в виде
где ср — произвольная функция своего аргумента и ср' (х) =
= жф' (ж).
2. Показать, что гг/у= а7'(«+*)/'=^, если плотность
энергии u/v зависит только от температуры Т. Это
справедливо, например, для черного излучения при а = -з-
(см. § 20, п. 2) и для Бозе-газа из N частиц массы т
2
ниже температуры Tq f^ (h^/mk) {N/vyf^ при a = -о-
-(конденсация Эйнштейна, см. § 38).
3. Доказать, что вблизи абсолютного нуля функция
ср (Tif) имеет степенной вид
ср(Гу«) = СГ^г;«гп (т>0).
а) Определить связь между а и т, налагаемую
условием динамической стабильности {dp/dv)T<0.
б) Пусть м-^О при Г—»0. Тогда, согласно
соотношению неопределенности, v — v{T, р) > О также при Г —» 0.
Какое требование налагается неравенством v{Ty р)> О
на связь между а и т?
в) Найти выражение для внутренней энергии и вид
уравнения состояния, учитывая результаты задач 1.12.3а
и 1.12.36.
4. Какой вид имеет степенной закон (задача 1.12.3),
если при Г —»О и{Ту v) стремится к некоторому
конечному пределу?
К ГЛАВЕ И
П.1. Вывести из уравнения A4.116), что выражение pV
представляет термодинамический потенциал для
независимых переменных Г, /?, ji.. (Это обстоятельство суще-
1) См. Н. Einbinder, Phys. Rov., 74. 8С5A948).
к главе II 421
ственно в теории больших канонических ансамблей в
статической механике; относительно специального
применения см. § 40.)
II.2. Температурный коэффициент теплоты
испарения вдоль кривой давления пара.
В формуле A6.14а) было использовано
экспериментальное значение для dr/dT. Вычислить теоретическое
выражение для этой величины, образовав ее дифференциал
(исходя из определения г = А/г). Использовать уравнение
Клаузиуса — Клапейрона и некоторые соотношения,
приведенные в таблице § 7.
П.З. В теплонепроницаемом закрытом котле объемом
20 л находится 1 кг воды при 10° С, образуя
жидкую и парообразную фазы. Какую энергию следует
подвести, чтобы нагреть содержимое котла до 200° С?
Указание: удобно представить себе, что конечное
состояние достигается в результате следуюпцтх переходов:
а) изотермическое сжатие вплоть до полного сжижения,
б) изотермическое сжатие от давления пара р = 0,0125 am
при 10° С до давления /?= 15,86 am (давление пара
при 200°С), в) нагревание при постоянном давлении
без образования пара (Ср ^ 1 кал-г''^-град'^)у г)
изотермическое расширение (испарение) до первоначального объема.
Количеством тепла, подведенным в процессе «б», и
работой расширения в процессе «в» пренебречь (см. также
задачу 1.8).
При 10° С: теплота испарения г = 591,6 кал/г,
удельный объем жидкости v^ = 1,00 дм^/кг, пара v^ = 106,4ж^/«г.
При 200°С: г = 463,5 «ал/г, 1^^ = 1,16 дм^/кг,
^2 = 0,127 м^кг.
11.4. Реализация термодинамической температурной
шкалы.
Показать, что из уравнения Клаузиуса — Клапейрона
Tdp/dT = r/{vD — Vj^^i) можно вычислить абсолютную
температуру Г, если величины г, vq и Vpi известны как
функции давления пара р.
11.5. Давление паров ртути составляет:
а) при 50° С 0,0127 торр и при 60° С 0,0253 торр,
б) при 300° С 247 торр и при 310° С 305 торр.
Вычислить в этих интервалах теплоту испарения г,
предполагая ее постоянной внутри каждого отдельного
422 Задачи
интервала. Удельным объемом жидкости пренебречь;
считать, что пар ведет себя как идеальный газ. В
промежутке между двумя интервалами линейно
интерполировать г и вычислить отсюда кривую давления ртутного
пара. Экстраполировать формулу для кривой давления
пара к температурам выше 300° С и вычислить
температуру кипения при давлении 1 атм = 760 торр
(правильное значение 356,7° С).
11.6. Рассмотреть газ, молекулы которого могут
находиться в трех энергетических состояниях Sq, е^, е^ (^i —^о
и ^2 ~ ^0"~ энергии возбуждения первого и второго
возбужденных состоянирг), как смесь трех газов, молекулы
которых обладают соответственно внутренними энергиями
Sq, Ej, Sg. Вывести условия равновесия между ними,
предполагая, что постоянная энтропии одинакова для трех
газов и что возможны переходы 0^1 и 0^2. Показать,
что результат не изменится, если возможны также и
переходы 1^2.
11.7. Е принципу детального равновесия.
а) Вычислить разности химических потенциалов jij — \Iq
и F^2 "¦ F^o ^ предыдущей задаче для незначительных
отклонений чисел молей п^ от их равновесных значений л^.
б) Сделав следующие предположения об изменениях
чисел Hq, Wj, п^: изменение в секунду, например, величины
^1 происходит вследствие того, что часть частиц,
пропорциональная наличному их числу /с^ п^^, переходит в
состояния О и 2, в то время как части, пропорциональные
Uq и ^2 (Агю^о и AjoWg), переходят в состояние 1,
написать уравнения для dn^/dt. Здесь содержится
предположение, что число переходов из одного состояния в другое
зависит только от числа молекул, находящихся в первом
состоянии, и пропорционально их концентрации.
в) Показать, что из этого предположения следует
закон действующих масс [см. уравнение C) в указании
к решению предыдущей задачи], если отношения rij^/riQ
и ^2/^0 не зависят от п^ и «2. Сравнение с упомянутым
уравнением C) дает два соотношения, которым должны
удовлетворять коэффициенты /г^,^.
г) Выразить величины п^ в найденных в задаче 11.76
уравнениях для dn^/dt через разности [а^ — ^^о ^ Н-з"" 1^о
к главе III
423
при помощи соотношений, выведенных в задаче 11.7а.
Какие выводы относительно механизма реакции следуют
из соотношений взаимности Онзагера (см. § 21, п. 4)?
У//////Ш/Л
М
т
п
К ГЛАВЕ III
III. 1. Вертикально расположенный цилиндр закрыт
поршнем с массой Л/, который может беспрепятственно
двигаться под влиянием силы тяжести. В цилиндре
вертикально (вверх и вниз) со скоростью с движется шар
с массой т <С М (фиг. 38). Шар
упруго отражается от дна и от
поршня. Влиянием силы тяжести на
шар пренебречь.
а) Отвлекаясь от размеров шара,
установить условие равновесия для
поршня и сравнить его с
уравнением идеального газа.
б) Принять во внимание радиус
шара г и сравнить полученное при
этом условие равновесия с
уравнением Ван-дер-Ваальса.
в) Поршень медленно
поднимается со скоростью F < с. Сравнить
получающуюся при этом потерю
энергии шара с работой газа dW = pdV,
III.2. Пользуясь распределением Максвелла B3.9),
вычислить: а) наиболее вероятную скорость, б) среднюю
скорость н в) корень из среднего значения квадрата
скорости.
Ш.З. Вычислить число молекул водорода Kg,
ежесекундно падающих на площадь стенки о = 1 см^ с
перпендикулярной к стенке скоростью, превышающей
12 000 MJcen, Температура газа равна 0°С, полное число
молекул в 1 см^ равно 2-10^^.
111.4. Вычислить математическое ожидание выпадения
числа 6 при бросании игральной кости (число бросаний А).
Найти средний квадрат флуктуации, определяемый как
111.5. Вычислить давление идеального газа на стенку,
расположенную в плоскости а; = О прямоугольной системы
Фиг. 38.
Одномерный газ, состоящий
из одной молекулы.
424
Задачи
координат, если стенка на больших расстояниях
притягивает молекулы, а на малых отталкивает их с силой,
описываемой потенциалом U= — Ле~** +j5e~2^:
а) если радиус действия сил мал по сравнению с
длиной свободного пробега,
б) если эти две величины сравнимы между собой.
Каков должен быть радиус действия сил стенки для
гелия при нормальных условиях, чтобы они оказывали
влияние на давление на стенку?
III.6. Идеальный газ находится в двух сосудах при
одинаковой температуре Т и различных давлениях /?j
Фиг. 39. Перенос массы и энергии сквозь
узкое отверстие.
и /?2. Сосуды расположены рядом, и в перегородке между
ними имеется очень узкое отверстие с площадью о (фиг. 39).
а) Вычислить количество газа, протекающее в единицу
времени в сторону меньшего давления в стационарном
случае (/?i = const , Pg^^^o^^t;).
б) Вычислить энергию, переносимую в единицу времени.
в) Какова средняя энергия, переносимая одной частицей?
о
г) Почему она больше, чем ^ кТ?
д) Что следует сделать, чтобы условия опыта
сохранились стационарными?
III.7. В газе при температуре Т находится
подвижная пластина В между двумя закрепленными пластинами
А^ и А^. Расстояния между всеми пластинами малы
по сравнению с длиной свободного пробега, так что
столкновения между молекулами можно не учитывать.
Пластины В п Aj^ имеют температуру газа, а пластина А^
нагрета до несколько более высокой температуры
к главе IV 425
а) Какую силу испытывает подвижная пластина В,
если каждая молекула при отражении садится на стенку,
с которой она находится в тепловом равновесии? (Степка
бесконечно протяженная. Все пластины имеют одинаковые
плондади F.)
б) Как, зная эту силу, определить давление газа?
(Результаты этой задачи применяются при расчете
высоковакуумного манометра.)
К ГЛАВЕ IV
IV. 1. Пусть при каком-либо измерении возможно
большое число п одинаковых по величине и независимых
друг от друга элементарных ошибок, влияние которых
на результат измерения ( fe или — е) подчинено закону
случая. Показать, что вероятность отклонения результата
измерения на величину х от истинного значения для
больших п дается выражением W — а&х^{ — х^12пг^).
Для наглядности положить в основу вывода доску
Гальтона. Шарик, ударившийся о гвоздь, с одинаковой
вероятностью покатится налево или направо.
IV.2. Пусть ошибка отдельного измерения не остается
постоянной, как и в предыдущей задаче, а принимает
набор значений в целом интервале. Пусть f^{x)dx
вероятность того, что ошибка измерения лежит в интервале
(ж, x-\-dx).
а) Какой вид имеет в этом случае вероятность ошибки
fn{x) при наложении п независимых однородных ошибок?
б) Показать, что все функции /^(о;) суть функции
Гаусса, если таковой является функция Д. Как
изменяется ширина гауссовой кривой?
в) Как выглядит функция /^ для больших п, если
Д (х) ^ 1 при \х\ <^ г и f^{x) = Q при ] о; | > s? Изобразить
fv /2' /3 ^^^ этого случая графически и выяснить их
геометрический смысл в многомерном кубе.
IV.3. Вычислить статистический вес W для N
молекул:
а) если все молекулы имеют равные по направлению
и величине скорости +$,
б) если половина молекул имеет скорость -J-^> а ДРУ'
гая половина — скорость —- 5,
426 Задачи
в) если равные части молекул имеют скорости
соответственно ± ^, ± -Цу ± С
Показать, что каждое следующее такое подразделение
при TV—>оо бесконечно вероятнее, чем предыдущее.
IV.4. Зеркальце висит на кварцевой нити с модулем
кручения D и освещается таким образом, что его
повороты, вызванные ударами окружающих молекул газа
(броуновское движение), можно регистрировать на шкале.
Пусть положение покоя соответствует ср = 0 (ср —угол
поворота). Вероятность того, что значение угла поворота
зеркала заключено между ср и ср-[-с?ср, согласно закону
равномерного распределения энергии по степеням свободы,
дается выражением
Wd(f = Ae-^m/kTd^, E^ot = у D(f^,
Из наблюденного среднего по времени ср^ можно
вычислить постоянную Больцмана к. Какое значение для числа
Авогадро получается из измерений при Г = 287° К,
/) = 9,43-10"^ дин-см, ср2 —4,18-10'® и газовой постоянной
Л = 8,32-10^ эрг/град-моль (Капплер)?
IV.5. Пусть дан кубический кристалл, содержащий
N = 10^^ атомов. Энергия связи на один поверхностный
атом составляет 9 эв. Вычислить отношение энергии
связи к энергии теплового движения. При какой величине
кристалла они сравнимы по величине?
IV.6. 1. Показать, что из уравнения для б-функции
02(^21^^ = 22^ ^^%osBn + l)^z
71=0
можно вывестр! сумму состояний ротатора
1 .„F''-^\.''\-njd.
1 с-'-^('Ю
dz Z
О
2. При помощи трансформационной формулы
(аналогичной формуле A5.8) в т. VI для 6 = 63)
''('Ю<7У"'-"""''{^\т)-
к главе IV 427
где
+ 00
бо(г/^)= 2 {-i)"exp[ir.{n4 + 2nz)],
П——00
вывести формулу
Z (д) = ^ е'^^. (irg)V2 С * ' е-'2/, ^^
—со
Символ \ означает здесь главное значение интеграла
в смысле Коши. При выводе использовать известное
из теории мероморфных функций разложение t/smt.
3. Вычислить главное значение интеграла в
предыдущей задаче: а) разлагая 1/sin^ по степеням е^', б)
разлагая t/sint по степеням /. Вывести отсюда используемую
в предыдущей задаче трансформационную формулу 0^—^ Oq.
4. Разложение, полученное в задаче IV.6.36,
представляет собой полусходящийся ряд, который для малых
значений q дает хорошие приближенные значения. Вычрг-
слить молярную энергию и молярные теплоемкости
вращения из первого члена этого разложения и сравнить их
с результатами § 33. При каких значениях q ошибка при
вычислении с^ не превосходит 1%?
5. Оценить влияние более высоких членов разложения
на молярную теплоемкость и сравнить оба ряда при
д = 0,458, е29 = 2,5.
IV.7. Распределение Бозе — Эйнштейна.
Пусть /i, /ь /f, ..., /("^ — вероятности того, что в i-ii
фазовой ячейке находится О, 1, 2, .. ., w частиц, так что
дополнительные условия имеют вид: 2! /Г^ = 1» 2 ^fi^^ = ^^ »
п n,i
ynfi^^Bi — U. Вычислить функцию распределения
щ = 2 '^Л^^ В равновесном случае, если логарифм термо-
п
динамической вероятности дается формулой
In Т4^ = - 2 (Л In л+/Пп л f/Пп я f •..).
i
т. е. представляет сумму больцмановских членов.
428 Задачи
IV.8. Флуктуации плотности.
Вычислить пространственные флуктуации плотности
в элементе объема AF газа, заполняющего сосуд объема V.
Предполагается, что средняя плотность газа во всех
точках одинакова.
IV.9. Флуктуации плотности в критической точке.
Рассмотрим реальный газ, iV молекул которого
находятся в объеме V,
а) Каков вид уравнения Ван-дер-Ваальса в этом
случае и каковы критические значения?
б) Вычислить термодинамическим путем логарифм
суммы состояний из термического и калорического Г ^^ = -^-^3^)
уравнений состояния, учитывая, что при высоких
температурах Т и малых плотностях N/V реальный газ ведет
себя как идеальный.
в) Определить относительную среднюю флуктуацию
числа частиц в объеме AF на основании формулы
ш*=
V/NAV
Nd4nz/dm
Рассмотреть случай малых отклонений от состояния
идеального газа и критическое состояние.
г) Обосновать эту формулу для флуктуации, исходя
из уравнения D0.15).
К ГЛАВЕ V
V.I. При помощи законов сохранения энергии и
импульса вывести формулы, связывающие скорости до удара
Vj, Vg и скорости после удара у{, Vg, если массы равны
т^ и 7^2. Показать, что для этого преобразования
справедлива теорема Лиувилля о равенстве фазовых объемов.
Определить среднее отношение разностей энергий до
и после удара.
При усреднении принять во внимание, что все
направления линии центров равновероятны и что
направления скоростей Vj, v^ перед ударом статистически
независимы.
Указания к решению задач 429
УКАЗАНИЯ R РЕШЕНИЮ ЗАДАЧ
1.1. Из равенства z = z{x,y) для произвольных
приращений dXy dy получаем
^^=(-?)/-+( t). ^2/. A)
Пусть теперь приращения dx и dy связаны таким образом,
что dz исчезает. Тогда их отношение есть {ду/дх)^, и из
выражения A) находим
f ду\ ^ __ (dz/dx)y
\дх Jz^ {dz/dy)y:'
откуда легко получить доказываемое соотношение.
Подставляя в предыдущее соотношение вместо z {х, у)
уравнение состояния р = р(Т, F), получаем
или, принимая во внимание A.5) и A.6),
JI 1_
Va Vx
-/'P47w=-i. C)
Т. е. Р/? =axF^.
1.2. Для нагревания комнаты от О до 20° С необходимо
каждой единице массы сообщить количество тепла Ср-20°.
Далее, {с^^- Су)моль = Л и, кроме того, для всех
двухатомных газов (а следовательно, и для смеси их, в частности
для воздуха) отношение Ср/с^= 1,4. Отсюда следует
^р моль = «^>^-«^> V-^)
Ср = 3,5 — = 3,5 29 кал/г. град. B)
Здесь использовано значение R из D.8); для [х
подставлено среднее число молей Ng и Og. Чтобы пересчитать
формулу B) к единице объема, надо помножить это
равенство на среднюю плотность воздуха р==1,25 кг/м^. Умножая
еще на разность температур B0° С), получаем из
формулы B)
р. Ср. 20° = ^^^^'^f^'^^ ^6,0 ккал/мК C)
430 Указания к решению аадач
Заметим, что вследствие постоянства давления и
расширения воздуха при нагревании значительная доля его
выходит из комнаты. Каково должно быть отопление,
необходимое для поддержания температуры 20° С,
зависит от проницаемости и теплопроводности стенок
и окон и, следовательно, лежит за рамками нашего
расчета.
Энергия, подводимая при нагревании комнаты, согласно
Эмдену (см. § 6, п. 6), вновь уходит в виде внутренней
энергии воздуха, выходящего из комнаты. Мы не можем,
однако, не подвергнуть критике уравнение Эмдена F.19).
В нем упущен из виду тот факт, что энергия всегда
определена лишь с точностью до некоторой постоянной.
Поэтому уравнение F.19) нужно заменить следующим
уравнением:
и^и,=^с,{Т-Т,). D)
Здесь Tq может означать любую температуру выше
температуры конденсации, при которой еще справедливо
уравнение состояния идеального газа. Таким образом,
поскольку и^ = ргг, имеем
^i-=c^T + p{u,-cJ,). E)
Здесь первый член в правой части не зависит от Г в силу
уравнения состояния рТ = \ip/B, Однако этого нельзя
сказать о втором члене, который благодаря большой
теплоте испарения положителен и значительно превосходит
первый член. Следовательно, Эмден не прав,
ограничиваясь в своих уравнениях F.19) и F.20) только первым
членом. Однако можно показать, что второй член (опять
благодаря условию р = \^р/ИТ) уменьшается при
нагревании. Следовательно, плотность энергии не остается
постоянной, как полагает Эмден, а даже уменьшается при
отоплении. Тем более оказывается справедливым
достойный внимания вывод о первенствующеii роли энтропии
по сравнению с энергией.
1.3. Если рабочим веществом в цикле Карно является
идеальный газ, то для одного моля его при
изотермических переходах 1 —» 2 и 3 —> 4 справедливы следующие
Указания к решению задач 431
выражения {Q — подведенное количество тепла):
2
dii=:0, dq = pdv, Q^= {pdv = RT^\ii^ ,
1
3
cfM = 0, dq = pdv, Q^=- \pdv=^RT^\n^
4
И, следовательно,
Q, ^ T, In F2/F1 .,.
С другой стороны, для адиабатических (изоэнтропических)
переходов 2-->3 и 4—>1, согласно уравнению E.3а),
имеем
T^-'^T^vV или Г,уГ* = Г1уГ'. B)
Исключая Т^ или Tg, отсюда окончательно получаем
равенство отношений v^lv^ и vjv^^. Вследствие этого
уравнение A) переходит в F.8). Таким образом, в силу F.7)
действительно ср(9) = Г, как было постулировано в выра-
ражении F.7а).
1.4. Под теплоемкостью С понимают количество тепла,
требуемое для нагревания тела на 1°. Так как удельная
теплоемкость относится к единице массы, то при
постоянном объеме получаем C = Mcvi где Л/ —масса
рассматриваемого тела.
Во всех задачах теплопроводности объем является
заданным, следовательно, dW=^0 и dU = dQ. Поэтому
здесь можно пользоваться теорией теплорода; последняя
дает «правило смешения»
T = cL,T^\-aJ'^, а, = -^--^, а^ = -^-^. A)
Согласно уравнению E.10), для идеальных газов 1 и 2
при постоянном объеме
432 Указания к решению задач
и благодаря аддитивности энтропии имеем
^S =. ^S, ^ А^2 = (^1 + ^г) In Г - Ci In Т^ - Сз In Т^.
Согласно второму началу для замкнутой системы ^S > 0.
Отсюда, поделив на С^-^С^ и использовав равенство A),
получим
aj^ + а,Т, > rji.r»/,- где а,4 a, = i. B)
(При a;i = ag = ^/2 получаем известное правило о том, что
среднее арифметическое больше среднего геометрического.)
Неравенство B) гласит: если при образовании среднего
арифметического величины Г^, Tg входят с весами а^, ag,
то при образовании среднего геометрического они входят
в степенях, равных соответствующим весам. Только
в тривиальном случае Г^ = Т^ неравенство переходит
в равенство.
При наличии п газов, находящихся во взаимном
тепловом контакте, вместо B) получаем
a,Г,+ ...a,Г^>Tr...n^ где а,+ ...а, = 1. C)
Алгебраический вывод этой теоремы имеется в
литературе i).
Читателю предлагается попытаться применить этот
метод к исследованию других необратимых процессов.
1.5. Пусть исходное состояние задано параметрами
Tq, Vqj Pq, Вычисляя интеграл от давления по объему
в пределах от Vq до 27^, получаем при
^) Р = Ро^ б)/? = /?о-^, в)/? = /?о^
соответствующий ряд выражений для работы расширения
а) /?Го. б) RT,ln2, в)НТ,^^=^.
Количество подведенного тепла дается интегралом от
dg ^ CvdT -\ pdv= c^dT — vdp. Для него соответственно
1) Pol у а, Szego, Aufgaben und Lehrsatze, Berlin, 1925;
H a г d V, Li t 11 e wor d. P ol va. Ircniialities. Cainbridere. 1934.
Указания к решению задач 433
получаем ряд выражений:
а) ЛГо-^, б) ЛГо1п2, в) 0.
Изменение энтропии в каждом из трех случаев
определяется выражением s = с^ In Т -\-R\nv + const и равно
а) Ср1п2, б) /?1п2, в) 0.
1.6. Согласно G.8) и G.8а), имеем
ds=±{du\-pdv) = ^dT-^(^-^P^Jv,
Выражая {dp/dT)^ из соотношения B) в решении задачи
1.1 и вводя коэффициент теплового расширения а и
сжимаемость X, получаем
ds = -^dT-]-^dv.
Таким образом, поскольку для адиабаты ds = Oy наклон
ее в плоскости Г, v дается формулой
Поэтому наклон адиабаты слева от минимума на
кривой Vy Т положителен (так как там а < 0), а справа
от него (там, где а > 0) отрицателен. В минимуме
касательная к адиабате параллельна оси v. Вычертив
качественно изобары в плоскости Т, V, легко сообразить, что
не существует адиабат, соединяющих две указанные
в задаче изотермы, если только процесс проводится в
области давлений, при которых минимум объема на изобаре
лежит между 2 и 6°С.
1.7. При постоянном объеме Ихмеем dq — CxjdT =^
— С1){дТ/dp)i)dp. При постоянном давлении dq = c^dT ==
= Ср {дТ/ди)р dv. Поэтому в общем случае получаем
r^. = ^, = c,(f-) rf. + c.(f )/р. A)
434 Указания к решению задач
Адиабатическую сжимаемость получим, если в
соотношении A) положим ds = 0. Тогда имеем
V др Л ср К, др Jv\ дТ Ур ' Ср \ др Jt'
В последнем преобразовании было использовано
соотношение B) из решения задачи 1.1.
1.8. Согласно G.8) и G.8а), имеем
dq^du + pdv^c^dT + xQ^^^dv.
При изотермическом сжатии dT = 0. Далее dv={dv/dp)Tdp.
Тогда, согласно соотношению B) из решения задачи 1.1,
получаем
Таким образом, считая коэффициет теплового расширения
постоянным в данном интервале давлений, находим
подведенное количество тепла
\dq=: - ГшА/?= -293 град Л дм^^2Л0г^ град'^Л9ат =
= —1,113 Л'ат= —26,1 кал
(так как 1 л-ат = 1 дм^Л кп-см'^ =^23,АЗ кал).
Затраченная работа равна
= 1 ал«з.0,5.10-*ат-1.уD00-1)ат2 =
= 10'2 ^•ат = 0,23 кал.
1.9. См. т. II, Механика деформируемых сред, § 7.
1.10. Уравнение Эйлера [т. II, уравнение A1.5I
в отсутствие внешних сил для стационарного [(9v/9^) = 0]
и безвихревого (rotv = 0) течения гласит [если учесть
уравнение A1.6) в т. II]
grad~-= ---grad/?. (Д)
Укйданил к решению аадач 435
Фигурирующее в т. II условие несжимаемости
[уравнение A1.4)] здесь следует заменить соотношением между/?
и р для адиабатического процесса
Прсле исключения из уравнения A) р при помощи
соотношения B) уравнение A) интегрируется и мы получаем
i:! З.-Р0. Т Г1 (Р \(У-')/П ..
2 2 - ро T-U ЧРоУ J~
-»^»['-(ir""]- р)
Согласно соотношению E.3а), {р/РоУ'^^^^^'^ = Т/Т^. Таким
образом,
2 2 Р о Р •
Следовательно, разность удельных кинетических
энергий, как утверждалось, равна разности удельных
энтальпий СрГо и СрГ.
В нашем примере Ср = 0,49 кал-г~^*градг^, f = l>33.
Следовательно, из формулы C), полагая Vq ^ О, получаем
у = 880 м/сек. Читателю предлагается сравнить это
значение со средней скоростью молекул водяного пара при
300°С, равной 770 м/сек (см. § 22). Температура после
расширения равна 110° С.
1.11. Прежде всего будем исх;одить из произвольного
уравнения состояния. Состояние газа в i-й ячейке объемом
F| описывается заданием температуры Т^ и удельного
объема Vi. Тогда количество вещества, содержащегося
в 1-й ячейке, равно Vi/Vi. Пусть удельная внутренняя
энергия в i-й ячейке равна щ (Г^, Vi). Тогда полная
внутренняя энергия газа, содержащегося в этой ячейке, равна
{Vi/Vi) Ui G*1, yj, а его потенциальная энергия равна
(Vi/Vi) gZi, где g' —ускорение силы тяжести. Если (/ —
полная энергия, а iV —полное количество вещества в
системе, то
436 Указания к решению задач _^
Аналогично для полной энтропии S получаем
i
где Si{T^, 2;^) — удельная энтропия газа в i-й ячейке.
При равновесии энтропия S имеет максимум
относительно всех вариаций Г^, 1\, совместных с условиями
постоянства N и U. Таким образом, если ввести два
множителя Лагранжа X и [х, то должно иметь место равенство
i i
i
причем величины T^i, ^i, X, [i здесь считаются
независимыми. Это дает
ц(-.,+щ^;)^11^;Ц(и,+,.,-.,^^о. B)
Из уравнения A) в силу соотношения dsJdT^ =
= A/Т^)(9м^/5Г^^ [см. соотношение G.1)] следует, что
(х = 1/Г., т. е. всеГ^ одинаковы. Поэтому положим Г^ = Г.
Из уравнения B) в силу соотношений G.7) и G.8) получаем
/а^\ _fdpi-^ f^^l\ ^Т f^-lL^ -n
и после небольшого преобразования
g (Г, V,) Е=и,^ gz, - Ts, -f v,p, = - IT.
Слева здесь стоит удельный потенциал Гиббса для
состояния в i-й ячейке, причем к внутренней энергии
добавлена потенциальная энергия. Правая часть равенства не
зависит от г, следовательно, то же относится и к левой
части. Для идеального таза имеем (для простоты прини-
Указания к решению задач 437
маем, что удельная теплоемкость не зависит от
температуры)
7? Ti Т
Ui = cJ-\Uo, Si = c„lnr-f-Inc + Se, Pi = ---,
где ji — молекулярный вес.
Таким образом, поскольку g (Г, у-) не зависит от i
и, следовательно, от 2, получаем
V (г) = v^^^'IR'^y р (z) = p^e-^s'i^T^,
>ния высоты
Для искомого значения высоты Az, определяемого
равенством
окончательно находим
Дг = — = —-— = —
v-g g Pog *
Это вместе с тем средняя высота
со
I zp (г.) dz
J Р B) dz
о
При /?о=10^ дин'см-^, Ро = 0,0012939 г-см'^, g =
= 981 см-сек'^ для Az получаем
Az = 8-10^ см = 8 км.
Следовательно, если вертикальная протяженность газа
значительно меньше 8 км, то падение давления с
высотой при равновесии почти незаметно. Оно составляет для
разности высот 80 м только 1%. Однако на
метеорологических станциях при корректировке барометров
относительно высоты над уровнем моря эта величина учитывается.
1.12.1. Подстановка р из уравнения состояния в
термодинамическое соотношение {du/dv)T = - р Л-Т {др/дТ)^, дает
для и дифференциальное уравнение в частных производных
ди ^ "V^ '^ТдТ'
438 Указания к решению задач
Общее решение этого уравнения имеет вид
где CS (Гу*)—произвольная функция. В этом можно
убедиться непосредственно подстановкой в дифференциальное
уравнение. Выражение для энтропии легко получить из
соотношения
Т ds=^du-]r pdv.,
2. Если отношение u/v зависит только от Г, то
величина У""*"*ср (Гг;*) от V не зависит, т. е. ср(Гу°) =
= оG'у*)(*+1)/'=^, где а —некоторая постоянная. Отсюда
следует высказанное в задаче утверждение. Для черного
излучения р = ^/^{u/v)j следовательно, а = 1/з. Тем самым
мы приходим к закону излучения Стефана - Больцмана
и/и~оТ^. Для а = 2/з получим u/v^aT'^l^. Это известный
результат из теории конденсации Эйнштейна [см. C8.27)].
3. а) Из предыдущего следует, что /? = aC7'^t;*"^~'=^-*;
лед)вательно, условие {др/до)т <0 сводится к неравенству
а(т-1)-1<0.
б) Имеем
Чтобы объем был положителен при Г —^ О и
произвольных значениях /?, должно выполняться условие
а(т-1)-1> 0.
в) Оба приведенных выше неравенства одновременно
выполняются тогда, и только тогда, когда а (т—1) — 1=0,
т. е. когда m — 1-f-(l/a) и, следовательно,
и = СГ(«+1)/« у, /? = аСГ(«+1)/«.
4. Из соотношения и =СТ^?^*(^~^) и условия О < к < оо
при Г = О следует, что m = 0. Таким образом, и =» у~*-const
и /??;«+* = const. При ol = ^/q получаем соотношение C9.10)
между давлением и объемом электронного газа.
Указания к решению задач 439
11.1. Из уравнения A4.116) получаем
d(pV) = pdV + Vdp = SdT + pdV +^ n^d^., A)
Отсюда следует
11.2. Из определения r = ^h для дифференциала вдоль
кривой давления пара ср(/?, Т) = 0 получаем
Так как dT и dp связаны между собой уравнением ср —- О,
то, учитывая D.11) и уравнение Клапейрона, получаем
Согласно соотношениям в таблице § 7, находим dh =
= Tds-j vdp, следовательно.
@).=^(i).+".
и, согласно той же таблице, получаем соотношение вза"
имности
^др)т Urjp'
откуда находим
Здесь а обозначает коэффициент теплового расширения.
Итак, получаем
^(^'^)^ = ^o~TA{vc^). B)
Подстановка выражения B) в уравнение A) дает
440 Указания к решению задач
В ЭТОМ выводе нет необходимости делать какие-либо
предположения относительно поведения пара или жидкости.
П.З. Обозначим количество пара через ж, а
количество воды через 1 кг — х. Объем пара тогда равен xVc,,
объем воды —A «г —гг) у^. Таким образом, для х имеем
уравнение
XV^-{-{1 кг — х) 1\ = 20 дм^,
откуда
20 анз —t/i-l кг
х = .
V2 — V^
В частности, при 10° С ж = 0,169 г, при 200° С ж = 149,7 г.
Следовательно, в трех частных процессах подводятся
следующие количества тепла.
а) Конденсация О Л 69 г пара при 10° С:
-0,169 г.591,6 кал/г=гг -> 100 кал.
б) Нагревание от 10 до 200° С:
1 л:г-190 град-] кал-г^^-град~^ = 190000 кал.
в) Испарение 149,7 г жидкости при 200° С:
149,7 г-463,5 кал/г ^QQAOO кал.
Всего подводится количество тепла 259 300 кал.
Работа по изменению объема составляет:
а) +19 5^3.0,0125 am = 0,238 дм^-ат,
в) —18,84 3^3.15,86 ат= -298,8 дм^-am.
Всего-298,6 дм^-ат= -7000 кал.
Таким образом, подведенная энергия составляет
252,3 ккал.
ил. Имеем
Проинтегрируем сначала это равенство в пределах от
О до 100° С. Пусть 7*0 — абсолютная температура точки
таяния льда. Абсолютную температуру точки кипения воды
при давлении 760 торр определяют как Tq+IOO^. Пусть,
Указания к решению задач 441
далее, р^ и pj^^ —давление пара рассматриваемой
жидкости соответственно при О и 100° С. Тогда получаем
in^-^Jy--:^ A)
Интеграл в правой части при данных предположениях
можно проинтегрировать численно, получив таким образом
уравнение для определения Т^. Тогда для абсолютной
температуры Г, соответствующей какому-либо другому
давлению.пара, получаем выражение
Ро
Согласно общим теоремам термодинамики, значение
Tq не зависит от выбранной жидкости. Равным образом
выражение B) дает одну и ту же температуру Т для
различных жидкостей, если только они находятся в
тепловом равновесии.
II.5. В обоих десятиградусных интервалах давление
пара изменяется слишком сильно, чтобы можно было
заменить производную dpIdT отношением конечных
приращений ^p/^Ty поэтому необходимо строго
проинтегрировать уравнение Клапейрона
dp г
dT T(v^^vj,)
A)
в предположениях, сделанных в задаче. Будем считать г
в интервале 10° С постоянной величиной, а пар—идеальным
газом. Величины г, vd и Vpi следует отнести к одному
молю. Тогда Vd -= ^Т/р^ а объемом Vpi можно пренебречь
по сравнению с vd» Уравнение A) дает при этом rfln/? =
= -{r/R)d{l/T)n
или
Я 2',-7'Л %J ¦
442 Указания к решению задач
Подставляя указанные численные значения, получаем для
теплоты испарения в первом и втором температурных
интервалах соответственно
г = 7413/?иг = 7045Л.
Следовательно, наше предположение о постоянстве
теплоты испарения в большой температурной области уже
неправильно. Найденные значения можно рассматривать
как средние для обоих интервалов. Поэтому они
относятся к значениям температуры 55 и 305° С или 328
и 578° К. Таким образом, линейная интерполяция дает
для теплоты испарения выражение
г = G896°-1,472 Г) Л. B)
Чтобы найти температуру кипения при давлении
760 торру проинтегрируем уравнение A) более точно,
Пользуясь выражением B). Тогда получим
dlnp^ +G896°-1,472Г)^
и
ln^ = 7896°Q-l)-1.4721n|;. C)
Под /?2 здесь понимается давление пара 305 торр при
температуре 310° С = 583° К. Если подставить для р
давление 760 торр J то из соотношения C) можно определить
температуру кипения Т при этом давлении. Получаем
1^^ = 1,600-0,1864 In g^.
Это трансцендентное уравнение очень просто решается
методом последова.тельных приближений. Для этого в
правую часть подставляют некоторое приближенное
значение для искомой температуры. Вычисляемое в левой
части значение Т опять подставляют в качестве нового
приближенного значения и т. д.
Подставляя в качестве первого приближения значение
630° К, получаем Г = 630,7° К. Подстановка этого значения
вновь в качестэе црцближенной величины приводит к по-
Указания к решению задач 443
лученному уже значению температуры. Следовательно,
температура кипения равна 630,7°К, или 357,7°С.
II.6. Будем исходить из уравнения A3.3) и образуем,
лак и там, выражение
oG = '^bn,[g,iT,p)-RT\n(^^]. A)
i
Так как возможны переходы 0^1 и О ;^ 2, то величины
л^ и ^2 можно варьировать независимо. Следовательно,
оло= —8^1 —§«2. В § 13 рассматривалась только одна
химическая реакция и потому произвольно варьировалось
только одно из чисел л^, тем самым все прочие вариации
ojij оказывались определенными.
Таким образом, из выражения A) находим
g,-g, = RT\n'^^, g,-go = RT In^^, B)
В силу соотношений g^ (Г, р) = и^ (Г, р) — Tsi (Г, р) + pv^
(здесь еще следует положить v^^v^^v^ и равенства
энтропийных констант Sq^s^ = s^, получаем gi—gQ =
= Ki - Ко = ^ (^1 - ^о); §2-8о = ^2- «о = ^(^2-^0)» где L -
число молекул в одном моле. Таким образом, находим
Lin—Ч) _ Ь(б2—ер)
^ = е RT ^ ^2^g RT ^ C)
/г.
о
Это закон Максвелла —Больцмана, который здесь мы
вывели термодинамическим щ'^тем и который был получен
в § 29 методами статистической механики. Учет переходов
17^2, очевидно, ничего не меняет в рассмотрении.
Обобщение на случай произвольного числа возбужденных
состояний производится непосредственно.
II.7. а) Величины л^ относятся теперь к
произвольному составу. Равновесные значения, найденные в
предыдущей задаче, обозначим через п^. При этом имеем
^1 Til ^
444 Указания к решению задач
ИЛИ при 1^1- /1^1 <С Wi (т. е. вблизи от равновесия)
РТ / ^1~^1 ^0 — ^0 \
Fb, ^^ = лт(^^^^—^^^=^). A)
б) Изменение величин п^ со временем определяется
дифференциальными уравнениями
^ = *1о^о ~ ^ii'^i + К2 ^2» B)
Сумма всех п^ постоянна, независимо от их значений
в отдельности. Отсюда следует, что
'^ОО" "'10 + ^20» '^И ~ ^01 + ^21» "^22 ""^02 + ^12* (^)
Первое из этих соотношений показывает, что все
молекулы, покидающие состояние О в единицу времени,
количество которых равно /гоо^о» попадают в состояния 1 и 2
(в количествах соответственно К^^п^ и к^^щ). Так же
интерпретируются и остальные два соотношения.
в) При равновесии, по определению, dnjdt = 0. Таким
образом, в этом сл^'чае имеют место равенства
^00 ^0 ~ ^01 ^1 "Ь ^02 ^^2»
*11^1 = ^0^-' *12'Ч» D)
«^22 ^2 ~ '^20 '^О ""' "1 '^l*
Следовательно, равновесие характеризуется не
отсутствием всех переходов вообще, а тем, что переходы,
обусловливающие увеличение некоторой компоненты,
происходят так же часто, как и противоположные переходы.
На основании соотношения C) можно утверждать,
что из трех уравнений D) независимыми являются не
Указания к решению задач 445
более двух. Они определяют отношения п^п^ и hJuq
как функции коэффициентов к^^^, которые со своей
стороны, согласно предположеншо, не зависят от состава. Таким
образом, из результатов предыдущей задачи [формула C)
в задаче III.6] следует, что коэффициенты А|,^, помимо
условий C), связаны еще двумя соотношениями. Тот факт,
что отношения величин л^ не зависят от состава системы,
если и не доказывает уравнения B), то, по крайней мере,
показывает возможность существования последних. Если
правые части уравнений B) представляют произвольные
функции линейной комбинации величин л^, то это
следствие остается в силе (если только указанные функции
исчезают, когда аргумент равен нулю).
г) На основании уравнений A) и из условия {п^— п^) +
4 (Wj — /ii) 4- (/ig — ng) =^ О (сохранение полного количества
вещества) имеем
"о
fl \> 1. • U' п
где
Л = Ло + Л^ + ^2 = ^0 + ^1 + ^2'
Подставим эти выражения в правые части уравнений B),
заменив все п^ разностями п^ - л^, что возможно в силу
соотношений D). Тогда получим, если опять принять во
внимание соотношения D),
J — —
RT -^/ = - А-11 Til (н - ^^o) + ^12 «2 A^2 - \^о)-
446 Указания к решению задач
Сравним эти выражения с B1.26). Следовательно,
соотношение взаимности Онзагера а^^ = «gi означает, что
*12^=*21^- E)
При помоищ C) и D) получаем и остальные соотношения
Теперь А^12^2 представляет число частиц, которое
получает в единицу времени компонента 1 от компоненты
2, ^21^^ —число частиц, получаемых в единицу времени
/\
Фиг. 40. К детальному, циклическому и смешанному
равновесию.
компонентой 2 от компоненты 1. Таким образом,
соотношение взаимности Онзагера требует, чтобы при
равновесии эти выражения были равны между собой, т. е.
течение реакции 1^2 при равновесии не зависит от того,
что одновременно также текут реакции 0^1 и 0^2.
Равным образом, согласно F), каждая из двух других
реакций при равновесии течет независимо от остальных
реакций. Эта теорема имеет весьма общий характер и в
ее обш,ей редакции называется химиками принципом
детального равновесия.
Наряду с детальным равновесием можно представить
себе циклическое равновесие или смешанное равновесие.
Эти три случая изображены схематически на фиг. 40.
Длина стрелок соответствует частоте переходов. Из этих
трех случаев только детальное равновесие обладает
исключительным свойством: оно остается неизменным, если
какой-нибудь переход (например, 1^2) прервать ив
прямом и обратном направлениях, например при помопщ
Указания к решению задач
декатализатора. При циклическом равновесии после
прекращения перехода 1—>2 переходы 2—»О и 0-^1
сначала продолжались бы дальше и происходило бы
увеличение компонент О и 1 по сравнению с предыдущими
равновесными значениями. То же самое относится и к
смешанному равновесию. Однажды установившееся
незаторможенное равновесие сохраняется сколь угодно долго,
причем, согласно основным законам термодинамики, оно
не может быть сдвинуто никаким катализатором или де-
катализатором. Поэтому принцип детального равновесия
допускает также следующую формулировку: не
существует такого декатализатора, который бы различным
образом влиял на прямую и обратную реакции, т. е.
прекращал бы одну реагцию, не прекращая вместе с тем
другую.
Предположение, высказанное здесь в качестве принципа
детального равновесия для трех состояний молекул,
доказывается также в кинетической теории газов или
электронной теории металлов и в конце концов является
следствием равенства значений обоих квантовомеханиче-
ских матричных элементов для перехода между двумя
стационарными состояниями.
IIL1. а) Скорость поршня перед столкновением с
шаром равна V, Она меняется на обратную, если
Mv = mc. A)
Время подъема поршня t^ — v/gy время его обратного
возвращения tj = 2v/g, Последнее должно быть равно
времени обратного возвращения шара tr = 2l/c. Отсюда
следует, что
VC = Ig. B)
Средняя сила, действующая на поршень, равна
K = ^f = mg^=.Mg C)
[последнее — В силу уравнения A)]. Далее, уравнения
A) и B) дают
mc^ = Mvc = Mgl = ^Fl,
448 Укааанил к решению задач
где F — площадь поперечного сечения, т. е. если ввести
давление p = Mg/F и объем V = Fl, -
pV = mc^=2U D)
вместо 2/g С/*, что имеет место в случае трехмерного
идеального газа.
б) Уравнение A) и выражения для высоты подъема
и времени обратного возвращения поршня остаются
неизменными, так же как и формула C). В уравнении B)
следует заменить / на / — 2г. Из измененного уравнения
B) следует
mc^=^Mg{l- 2г),
или
p{V--2rF) = 2U. E)
Так же как и в уравнении Ван-дер-Ваальса, здесь из
общего объема газа V вычитается постоянный объем слоя
с толщиной, равной диаметру шарика, и площадью,
равной площади поперечного сечения цилиндра.
в) Если поршень считается очень тяжелым, то
справедлив закон отражения (увеличением скорости поршня
можно пренебречь). Пусть с - скорость перед ударом, с' —
скорость после удара. Тогда
c-V = c' + V.
Изменение кинетической энергии составляет
y(c'2-c2)^-27WcF.
За время ^t такая энергия будет отдана в точности
At/tj. = сМ/21 раз. Следовательно, подставив в это
выражение пройденный поршнем путь Ах = V^t, получим
- Д^Г = - 2mcV '^^ = - ^ Ад: = - Mglx = - pAV. F)
III.2. а) Вычисляя логарифмическую производную по с
от ср (с) = 4тсс2 (т/21гАГ)'/2 е-^с2/2лт ^ получаем
<D^ с 2кТ'
Указания к решению задач 449
следовательно, в соответствии с B3.10) равенство ср'==0
дает
со
б) Из определения с = \ сер с?с следует (если положить
тс^12кТ = 5*)
о
Этот интеграл сводится к интегралу Пуассона и равен
Va*!' Следовательно,
в) Из соотношения с^ = \ с^ср (с) dc находим
^ О
Вычисляя интеграл, получаем
Таким образом,
у-5/2
С2 = --. C)
Отношение квадратов скоростей составляет
с2:с2:4 = 3:-:2, D)
откуда следует B3.11).
450 Указания к решению задач
Ш.З. Из цилиндра, показанного на фиг. 23, в
единицу времени выходит
dZ = сс,-п (^У' е-^-'^"^) <'=^+'=5+^^ dc^dc,dc,
молекул. Полное число молекул получается
интегрированием по Cjg, Су от — со до -+- со и по с^ от Со = 12 000 м/сек
до 00. Вводя цилиндрические координаты, получаем
(^Ш^У^'(^ху ^у» c,) = (pcoscp, psincp. С).
Отсюда находим
Это можно выразить также, используя формулу для
средней скорости c = A{kT/2i:myi^. Получаем
4
При указанных в задаче численных значениях находим
с = 1690 л«/се«, -^ = 8,45.1023, ?"^ = 64.
Отсюда Z = 5»10"^ сек."^ Таким образом, приблизительно
через каждые 3 минуты на стенку падает молекула
с такой большой скоростью.
III.4. См. § 27, п. 4, уравнение B7.14). Вероятность
того, что число 6 встретится первый раз в 1, 2, 3, ..., к-м
бросании, соответственно равна
w. = l ' '
'»" 6 » 6 6' ve / 6' •••' ^6У 6 •
Вычисление средних значений от Л, А*, ...
выполняется простейшим образом при помощи производящей
Указания к решению задан 451
функции
fit) = iw,t'^=^^.
Здесь р = ^/j. Это дает сразу
/A)=|ж, = 1.
а искомые средние значения находягся
дифференцированием:
/'(l) = 2Wft = A.
Для вычислений удобнее представить их в виде
следующих логарифмических производных:
/(!)¦'
р_А-Г/'@]' ,/'A)
Таким образом, получаем
W = (*r^= [ -^+р^]__,+^=,^=. 30.
Следовательно, результат имеет вид
А ± ДА = 6 ± /30 = 6 ± 5,5.
III.5. Закон сохранения энергии гласит:
J (^2 + у2 ^ 22) - ^ + Ае-^ - Бе~2«х. A)
Компоненты скорости по осям у и z постоянны. Если
положить (т/2) {у^ + ^^) = -E'tang (тангенциальная энергия)
и ?^ — i^tang = -^п' '''^ -^tang в данном случае есть константа.
452 Указания к решению задач ^
Следовательно, для Е^ имеем уравнение
у ж2 = J5:„ i- Ле~«^ - Ве-2ах B)
Это уравнение можно проинтегрировать. Получаем
г _ /^ = (^^У' J {Е^ + Ае-^^ - Ве--^^^)-У2 dx,
т. е., если положить e*^ = S,
Введем новую переменную $ = С — ^ ''2 i?„ и обозначим
q = E/?:„) + (^V4^n); тогда имеем
Полагая C = CoChX, получаем
ИЛИ
т. е.
ИЛИ
. = lln[;„cha(^An)-(.-,)-4]
И
Отсюда находим изменение импульса в случае «а»
Дро = m[i(+ 00) -X (- ос)] = 2 Bmi?JV2. D)
В точности такое же выражение пол>'чается и при
ударе о твердую стенку, так что для давления не
получается никакого различия. Лишь в случае «б», когда
Указания к решению задач 453
средняя длина свободного пробега сравнима с радиусом
действия силового поля стенки, следует ожидать
отклонений. Пусть т —среднее время между двумя
столкновениями; тогда на основании соотношения C) получаем
^р=-т[х(/о + '^)-x{t^- т)] =
^^ eh а (lEnfmf^ z - (Л/2?'„ Со) '
т. е. В первом приближении (для больших х)
Д/, = Др„A+^е-«(^^"""^"^'). Eа)
Из уравнения A) следует, что 1/а имеет величину
порядка радиуса действия силового поля стенки.
Изменение импульса А/? существенно зависит от а, если
вторым слагаемым в скобках нельзя пренебречь по
сравнению с единицей. Это имеет место при условии
а ^ \ т J \тс^ у
Т. е. только тогда, когда радиус действия силового поля
стенки по крайней мере равен длине свободного пробега /.
III.6. а) Обратимся к фиг. 23. Число частиц,
падающих из изображенного там цилиндра, составляет
Интегрирование по полупространству дает
оо
Интеграл, включая множитель 2, равен \ е"ЧЛ = 1!.Та-
0
КИМ образом (вводя вместо п давление /?), имеем
v = a;?BiumA:r)-V2.
454 Указания к решению задач
Когда /?2 > /?1, то справа налево протекает больше частиц,
чем в обратном направлении. Разность составляет
Av=aB7:mAr)-V2Aj[?. A)
б) Величину AW можно вычислить, зная ^/^тсЧ^^
Расчет ведется так же, как в случае «а». Получаем
j.r т 2кТ о, /2kTyi2
W = ov-^ 2! = ат? ( ) ,
2 т ^ \7im у
откуда находим
в) Энергия, переносимая одной частицей, составляет
^ = 2кТ> ^кТ. C)
г) Отношение APF/^v превышает y^^» i^k как
частицы, обладающие большей энергией, приходят из большего
объема.
д) Вследствие течения давление будет изменяться.
Согласно неравенству C), правый сосуд охлаждается,
а левый нагревается. Следовательно, необходимо
искусственно заботиться о выравнивании давления и подводе
тепла (т. е. поместить систему в аермостат).
III. 7 Пусть плотности молекул, движущихся от
пластин ^1, Ао и В J соответственно равны щ, п^ и п^. Тогда,
согласно условию задачи, равновесие частиц
осуществляется, если п^с^ = UqC^ = n^Cz или, что в силу
изотропии сводится к тому же, если
П^С = HqC = п^с'. A)
Здесь сие' обозначают средние скорости соответственно
при температурах Т и Т'.
Давление р вычисляется при равновесии из формулы
1 -
р = ^тпс^, В данном случае п = Зп^ = 2wq, следовательно.
2 -
г^-=:^т7цс^. Отдача молекул, движущихся от В, одина-
Указания к решению задач
455
кова в обе стороны и компенсируется. Силовое воздс!!-
ствие на пластину В, которое не компенсируется,
обусловлено только приходящими молекулами. Результпру-
юш;ая сила составляет
Отсюда, согласно условию A), находим
Поэтому, принимая во внимание, что c^^YT,
получаем
B)
ИЛИ
_-2JtT
Р — рьт '
-Г,
C)
D)
IV. i. Получаются следующие частоты ошибок:
Ошибки
п=?0
1
2
3
4
-4е
1
16
— Зе
1
8
^2г
1
4
4
16
— е
•1
2
3
8
0
1
2
Т
6
16 1
+ 5
1
2
3
8
+ 2г
1
т
4
16
+ 3?
1
8
+ 4*
1
16
В общем случае вероятности ошибок даются
биномиальными коэффициентами. Пусть л —общее число
отдельных ошибок, а Л —число положительных ошибок; веро-
456 Указания к решению задач
ятность этого события равна
^п,к-2п к\{п — к)\ »
и величина ошибки составляет
f^^f^ = кг + {п — к) { — г) =^ {2к ^ п) г = X.
Если ввес1И вместо к величину ошибки а;, то
и (при dk= 1)
J _ i п\ dk 1 п\ dx
V2"^2s/ V.2 2s/
Правую часть можно преобразовать по формуле Стирлинга
Отсюда находим
п л:
Логарифмы сомножителей в знаменателе равны
lO±S)i»0±?)-l('±r.)(±s-jS>) =
""i 2i+4;ir«'
Их сумма, сяедовательно, составляет
аг«
2пв* •
Поэтому произведение в знаменателе равно е* Z^'^*^, и,
следовательно, мы получаем
Указания к решению задач 457
что И требовалось доказать. Если положить rr= S Bпе2)^2,
то интеграл от dw равен
[clw = ±\e--d^L
V
IV.2. а) При наложении двух однородных статистически
независимых ошибок измерения вероятность того, что
ошибки лежат между (х\ х* -^-dx') и (х\ x" + dx")y равна
fi{x')fi{x')dx4x\
Полная ошибка составляет х' + х" = х. Чтобы найти
интересующую нас вероятность определенной полной
ошибки, полагаем, следовательно, х' = Х'-х' и
интегрируем по х'. Получаем
fA^)=]h{x')hix-x')dx'. A)
в общем случае, если w-я ошибка налагается на (л — 1)-ю,
то получаем
fn{^)-]iiix')f,^.ii^--^')dx'. B)
б) Если /^ = (T.a^^)-^l2e-^*i'^^ f при \ f^^dx = l\ то из
уравнения B) вытекает равенство
1 = -^ + !=^. C)
Следовательно, функция Гаусса удовлетворяет
уравнению B) и значение полуширины кривой пропорционально
в) Для ответа на поставленный вопрос заметим, что
в этом случае уравнение C) принимает следующий вид:
+•
/nW=2n /n-l(^-^')^^'.
Будем искать полную систему решений в виде
458 Указания к решению задач
Отсюда при произвольном X получаем
следовательно,
^„(X) = C(X)Ei2A^)"-\ D)
Мы найдем теперь искомое решение, если представим
f-i{x) в виде интеграла Фурье
f^{x)= [ С(к)е~^'Ы1.
Отсюда, согласно интегральной теореме Фурье [см. т. VI,
§ 4, уравнение D.13)], получаем
— »
Таким образом, коэффициент Фурье для /^ имеет вид
а сама функция есть
Для больших значений п выражение (sinXe/Xs)'*
заметно отличается от нуля лишь в непосредственной
окрестности точки X = 0. Поэтому можно заменить этот
множитель колоколообразной кривой
Указания к решению задач 459
При этом интеграл F) легко вычисляется. Имеем
или
Таким образом, как и в задаче IV. 1, получается
распределение Гаусса.
Вычислим интеграл F) при п«=1, 2, 3. В общем
случае, полагая \г = t, S = х/г, имеем
+ 00
Интегрируя по частям, получаем
+сх>
, 1 i с dt rf«-i / . n, ы\
Функция под знаком производной имеет вид
при л = 1: sin ^ cos $^,
при п = 2: sin^ t cos $/ = -2A — cos 2t) cos E^
при /г = 3: sin^ / cos;/ = -4 C sin i — sin 3^) cos U,
Соответственно нулевая, первая и вторая производные
от этих выражений равны
п = 1: sin^cosE/,
л = 2: sin 2t cos ?^ — у ? A — cos 2t) sin U,
3 3
л = 3: —-^(sin^--3sin3/)cosS^—-yS (cos^ —
- cos 3/) sin U-^V^ C sin t - sin 3/) cos ^t.
Bee получающиеся интегралы принадлежат к одному
типу (разрывной множитель Дирихле). Мы выпишем
460 Указания к решению задач
результаты интегрирования для отдельных слагаемых
ряда и для интервалов, в которых они не обращаются
в нуль.
Случай л=1: +1 при |5|<1,
это наш исходный пункт.
Случай л = 2: +1 при | Е1 < 2,
+ y|?| при 1^1 > 2.
Отсюда следует
2ef^ = i—^\k\ при |а|<2.
3
Случай и = 3: — -^ при | S | < 1,
Т при |Я<3.
-4ia| при |?|>1,
+ ||М при И|>з,
-||а|« при ui<i,
4-||5|» при |S|<3.
Это аает в интервалах
0<11|<1 48/з-|-1|5р,
3<U|<cx?. 0.
Указания к решению задач
461
-/ О н
-3 -2 Ч О *1
I и:
браженные для чисел измерений
*2
•^3
Фиг. 41. Функции главных сечений для многомерных кубов, изо-
ш п=1, 2, 3.
На фиг. 41 изображены функции Д, /g, /3. Функция
fi состоит из одной горизонтальной прямой, /з — из двух
наклонных прямых, /з~из трех
параболических кривых и т. д. Можно
тотчас же указать геометрический
смысл этих функций. Так как Д
возникает при наложении двух
постоянных функций /i, то квадрат со
стороной 25 оказывается равномерно
заполненным. Найдем области, в
которых суммы ошибок постоянны. .
Они определяются отрезками пря- D^
мых, перпендикулярных к диагонали Фиг, 41а. Главной
DD квадрата, изображенного на сечение квадрата,
фиг. 41а. Длины этих отрезков даются
функцией /g. Соответственно /з приводит к плоскостям
сечения куба, перпендикулярным к его главной
диагонали, и т. д.
462 Указания к решению задач
IV.3. Распределим N частиц по л местам (iV^, N^,..., iVJ.
Тогда
W^
N.,\..,Nn\
Таким образом, применяя формулу Стирлинга B9.4а),
получаем
а) И', = ^=1,
Отсюда следуют пропорции
ЙГ = E^) -2 ->со при /V->cx>,
W'c 27 oN AT
IV.4. Имеем
+ 0O
— CO
Отсюда следует
Число Авогадро равно
L = ^ = |^^^. 10-^2 моль-1 = 6,05.1023 моль-1.
/)сра 9,43-4,18 '
IV.5. Положим N = Z^^ Z = 10'. Тогда число частиц
на поверхности равно yVo = 6Z2. Тепловая и поверхностная
энергии соответственно составляют
Указания к решению ^адач 463
Отсюда следует при Г = 290"^ К
и _Z^r _ 10^.400.10-^Д_10в .|/ппп
ио~'2эв -1,6-10-1».9" 72 "^ 1^^^.
U^Uq, если 2=10714000 = 700. Таким образом, число
частиц Л^ = 3,4-10^, а линейные размеры образца
составляют приблизительно / —2-10'^Z си* = 1,4-10"^ см.
IV.6. 1. Имеем
оо со со
О п=0 О
Отсюда и вытекает высказанное в задаче утверждение,
так как все интегралы под знаком суммы равны ir/2.
2. Если подставить ряд для 6^ в трансформационную
формулу, то получим
<^Ю = (.ТУ" i(-l)"exp[-"^(.-n)^|.
Tli=~00
Отсюда
^й)--^«"'G)"'(^")й2<-""х
П=—00
ОО
«
При одновременном изменении знаков z и п изменяются
лишь пределы интегрирования, поэтому
со —• +00
lim ^ =]im \ =-^ с *.
Таким образом, имеем
+ »
^(я)=\^"(тУ" S(-i)"x
П=—00
X f i=^exp [-^^iz-nr]dz
Указания к решению задач
Замена переменной г = 1/т: + п дает
Z{q) = jef4H^4)-''*\ *( f (-1Г^)е-'''«ЙЛ
—СО П=:—00
Отсюда в силу равенства
+00
^ ^ ' t + rnz sin t
n=—ОО
И вытекает доказываемое утверждение.
3. Формально можно написать
sint 1 —e-»*^ -^
ft=0
fe=:0
00 «o
= -^2 (e+Bft+i)u-e-Bfe+i)i0 = 2 2 sinBA+l)^
ft=0 л=о
Интеграл в Z{q) (см. выше) преобразуется следующим
образом:
+ 00 ОО
2 J * ^ sin BЛ +1) fe-'"/' rf< = 2 J * sin BЛ: +1) fe-"/' dfi,
—CO 0
T. е., полагая т = ^2, получаем интеграл типа
преобразования Лапласа, а именно:
00
2 ^ sin BЛ + 1) Ух е-^/« с/х = 5 {жд)У^ охр [ - (^ А + у Y ? ] .
о
Этот интеграл можно легко вычислить или взять из
таблиц по преобразованиям Лапласа^). Получаем
^(9)= 2 BA+l)e-Mft^i)a. A)
fe=0
*)W. Magnus, б;. Oberhettinger, Formein und Satze
fiir die speziellen Funktionen der mathematisclien Physik, Berlin, 1943.
Указания к решению задач 465
Получается в точности уравнение C3.3), выведенное
из трансформированного представления 9-функции, в связи
с чем доказательство трансформационной формулы
становится излишним. Можно, впрочем, вывести ее из
трансформационной формулы, приведенной в т. VI
[формула A5.8)].
Если q очень мало, то заметный вклад в интеграл
дает лишь малый интервал около ^ = О (величины Д^ = 7).
В этом случае можно исходить из степенного ряда
t
sin
"t = ^-(*'~ll + l!T+ '")^
¦^ ^ 6 ^ ^360^ ^ 15120 ^ ^ ' • •
Подставляя это выражение в Z(g), получаем иятег{.алы
—оо
или в специальных случаях
Отсюда следует
Разлагая также и е«/* и перемножая оба ряда, получаем
и
466
Указания к решению задач
4. Из выражения для InZ дифференцированием
получаем
dT
ди
q^ ди
= Rq
dq '
2 d^ In Z
C)
Ь dq ^ dq^ '
Отсюда находим
В первом приближении для внутренней энергии
получается выражение, с точностью до константы совпадающее
с найденным в § 33,
к = /гг-~ле, с, = 7?.
Если V45 9^<Vioo (т- е- g < 0,67), то ошибка здесь
меньше 1%.
5. Следующая таблица показывает точность,
достижимую при небольшом числе членов:
Значения членов в
выражении для Су
q=0,3
0,5
0,9
1,2
1
1
1
1
1
2
0,002
0,008
0,018
0,032
3
0,000407
0,00372
0,01263
0,0299
Уже при q = 0,6 третьим членом нельзя пренебрегать
по сравнению со вторым. Таким образом, асимптотическое
разложение пригодно вплоть до gr = 0,5 (т. е. при Т > 20).
После этого надо пользоваться выражением A).
Выражение B) прежде всего показывает, что при больших
температурах теплоемкость с^ быстро приближается к
значению /?, причем это приближение происходит сверху.
Указания к решению задач 4б7
В заключение вычислим значение с^ при похмощи обоих
разложений дляе2« = 2,5, т. е. при д = 0,458, ^2 = 0,20977,
дЗ = 0,09607. Асимптотический ряд (с учетом первых трех
членов) дает
с^ = 1,006/г.
Из формулы C3.3), используя выражение C),
получаем для Су следующее значение:
Здесь
Z = 1 + Зе-2'? + 5e-«'^ + 7e-i2^ + 9e-2o^ + 1 le-soa^
- Z' = бе-з'? + 30e-«^ + 84e-i2'^ -f ISOe'^o^ + ЗЗОе^зо^,
Z" = 12e-2^ + 180e-«^+1008e-i2^+3600e-2o« + 9900е-зо9.
При заданном значении q
Z - 2,549, Z' = 4,683, Z" = 20,836
и
с, = 1,0064/г.
Отсюда следует [так как в ряду A) учтены все члены,
дающие заметный вклад], что нужно принять во
внимание и четвертый член асимптотического ряда B). При
учете его разность достигает 0,5^/оо, так что с
небольшим числом членов обоих рядов можно количественно
вычислять молярные теплоемкости.
IV.7. При заданных дополнительных условиях InW
должен иметь максимальную величину. Учитывая
дополнительные условия методом множителей Лагранжа, нужно
искать максимум выражения
- S {/^"' In /Г> + )-i/i"^ + «n/i"^ + ^пг/Г'}.
п, i
Производные по всем /[^^ должны исчезать.
Следовательно, получаем
In /i**^ i- 1 -i- Xi + ал -I- р/ге^ = О,
Указания к решению задач
т. е.
Условие 2 ft^^ = 1 Д^^т
п
—f = 1
1-.—P-i •
так что уравнение A) принимает вид
/(^) ^ A _ е—P'i) . ^-(«-^ P«i)«. B)
Функция распределения есть
«i = S "/i"' = - A - «"'""•') Та S «"'"' "''"•
п п
Отсюда
Это распределение Бозе — Эйнштейна
что и требовалось получить.
Постоянные а и р определяются из остальных
дополнительных условий, которые теперь имеют обычный вид
IV.8. Объем сосуда V состоит из частей V^^^V и
F2 = F —Д7. Вероятность встретить молекулу в части F^
или Fg равна VJV или F2/F, так как равные элементы
объема равновероятны; Вероятность найти Л^^ частиц
в объеме F^ и N^ частиц в объеме V^ определяется
статистическим весом
•^(^..^.)-w(^)""G')"'-
Указания к решению задач 460
Отсюда обычным путем находим
N, = N^\ N,{N,~i) = N(N-l)p,.
Следовательно, получаем
или
AiV, 1 / Fo 1 / F — AF
7, V ^v, V
iVi f NVi V iVAF •
IV.9. a) Уравнение Ван-дер-Ваальса для N частиц
в объеме V имеет вид
[^+ЧгЛ(ж-«)-*^- С)
Здесь постоянные А и В вычисляются из постоянных
Ван-дер-Ваальса а и &. А именно,
^ = Т^' ^ = 1 A«)
(L —число Авогадро). Критические значения получаются,
как и в § 9, из условий
/др 2AN*\/V оЛ , 1 / , AN^\ ^
/ д^р 6AN^\ (V т^.г (dp 1AN^\_^
в которых надо положить dpjdV ^&^pldV^ = 0,
Это дает следующие критические значения (удобно
начать с последнего уравнения):
^кр. = 3iVfi, /?кр. = 27^ » *3^кр. = 275 • (^)
б) Логарифм суммы состояний есть
термодинамическая функция состояния с дифференциальной формой
rflnZ= -Ud^^^^pdV. C)
470 Указания к решению задач
Если давление р выразить при помощи уравнения
состояния, то отсюда получим
Интегрируя, находим
lnZ = A^[ln(F-A^iB) + p^^]+iV/(P). D)
Здесь / (Р) — произвольная функция р.
Из последнего уравнения, вычисляя внутреннюю
энергию согласно уравнению C), находим
^--(т).-^т-^/'(й-
Следовательно, теплоемкость при постоянном объеме
составляет
Последнее равенство выполняется асимптотически при
J5—>0. Таким образом, имеем
/ Ф) "^232 ¦
и интегрирование дает асимптотически справедливые
выражения
/'(p)-*-|+Ci.
/(P)-*-|lnp+CiP4-C,.
Отсюда получаем
1п2-*Л?[1п(Г-Л^В) + ?Л^—|]np]-f
+ NC,^-^NC,. E)
Указания к решению задач 471
Это выражение еще более упрощается в предельном случав
\nZ = N\nN + N (\n^-\\n^^+NC^^^NC^. Eа)
Это выражение должно совпадать с соответствующим
выражением для идеального газа
Aпг)идеальн. газ =N [ 1 + 1п-^-yin р +1п (^-^^ ' ] ,
откуда следует, что G^ = 0 и
N\nN-\-NC^ = N^\ + \nQ^y*^ .
Окончательно получаем
lnZ = A^[l+^P^ + ln(I-5)(^)'4g(P)].F)
Здесь g(P) представляет часть /(Р), которая исчезает при
р-^О.
в) Уравнение F) немедленно дает
(91nZ InZ
dN
_lnZ /^piV VIN Л
Повторное дифференцирование по N, если одновременно
умножить на — 7V, дает
д.ачпг inz /inz , .Qiv f \ .q iv ,
, ^iV/7 1 25iV A
f, BN ч2 /. BN\^ V
BkT
G)
В предельном случае идеального газа (Л—>0, 5—»0)
имеем
-.V^^=l. Gа)
Относительная средняя флуктуация в этом случав
вычисляется в соответствии с уравнением D0.18).
472 Указания к решению задач
Если величины А и В принять во внимание только в
первом приближении, то получим
^* ал?» ^ ' F V. вкт J' ^"^^
Подставляя в уравнение G) критические значения B),
а именно BN/V = Vs. Л/В кТ = ^V,, находим
-yV^ = 0. Gв)
В этом случае относительная средняя флуктуация
становится бесконечно большой
/ Ад\2 _ V|N^V
К:^) -^~ n?WzJW ^ ^
Большие флуктуации приводят к сильному рассеянию света
(см. т. IV, § 33), что иллюстрируется ярко выраженной
опалесценцией реального газа в критической точке,
г) В силу уравнения D0.15) имеем
М 42- ^ \\Vd4nZ\ _ 1 / у> ^ д^Ф \
i, k ' i, Л "'
так как величины а и р следует считать постоянными
[см. § 40, особенно уравнение D0.12)]. Символ Е'
обозначает суммирование по всем фазовым ячейкам в объеме AF.
В силу C8.4) последнее равенство можно переписать
в виде
i "'
Здесь п — среднее число частиц в объеме AF [п = {\V/V)N].
Теперь из уравнения C8.7) следует
i
Указания к решению 9адач 473
[величина 9 эквивалентна Ф = In У из § 38, формула
C8.1), и относится к объему AF],
1).,,.,-К§),...- <•»>
('>«)'=-S'S'--?- <9«)
Таким образом, из соотношения (9) получаем
дп{ дп
да да
i
Используя преобразование Лежандра, возвращаемся
к сумме состояний
cf (ср + ал) = ас?л—Mcfp —р 2 л^ cfe^ =-рг-с? In Z.
i
Получаем
^ AF / d\aZ\ _ / dJnZ \
"" V\dn )^, .i V dN Ур,,. •
Поэтому выражение (9а) можно переписать следующим
образом:
^ ^^ "" да/дп ^V d4nZ/dm '
Если это выражение разделить еще на квадрат п = NAV/Vy
то получится указанная формула для флуктуации.
V.I. Можно непосредственно убедиться, что из
законов сохранения импульса и энергии вытекают
соотношения
/ , 2/П2 /\т \
V, = v, -^ -^— (Ve) е,
v; = V2 ?^(Ve)e.
Здесь V = Vg — Vi — относительная скорость. Если V | е,
то удар является центральным. Чтобы убедиться в
справедливости теоремы Лиувилля о равенстве фазовых объ*
емов, надо доказать, что детерминант матрицы
преобразования шестимерного пространства скоростей v^ и Vg равен—1
474 Указания к решению задач
(для этого удобно направить вектор е по одной из
осей координат).
Разность энергий после столкновения вычисляется по
формулам A) и составляет
2 ^1 2 2 — 21 23
- G^Tp^(Vi-V2, e)(miV, + m2V2, e). B)
Далее, в силу равновероятности всех направлений
среднее по е значение произведения вида (Ue) (Ve) равно
(Ue) (Ve)^^^ = iuV.
Подставляя это в соотношение B), получаем
^;-^-^ = ^x-^.-T-(;^;S^(Vx-v„m,v, + m,v,). C)
Так как направление Vg не зависит от направления v^,
то, усредняя по направлению Vg, получим
^^^^^"^^ = 0.
Таким образом, в последней скобке в выражении C)
остается только член m{v\ — т^\\ = 2 {Е^ — Е^).
Следовательно, получаем
Ei — Ei_. __ 2 4mim2
Ei—E2 3 (mi + mo)* *
При m^ = m^ это отношение составляет 7з» при т^ < т.у
оно равно 1—8 mJSm^. Результат показывает, что
разность кинетических энергий сталкивающихся тел в
среднем всегда убывает. Таким образом, устанавливается
закон равномерного распределения по степеням свободы
средней кинетической энергии поступательного движения.
Этот закон справедлив не только для молекул одного
сорта, но также и для различных молекул.
ОГЛАВЛЕНИЕ
От редакции 3
Предисловие автора 5
Из предисловия издателей 9
Глава I. Термодинамика. Общие принципы 11
§ 1. Температура как функция состояния 11
§ 2. Работа и количество тепла 15
§ 3. Идеальный газ 19
1. Закон Бойля—Мариотта ( 19 ). 2. Закон Гей-
Люссака ( 20 ). 3. Закон Авогадро и
универсальная газовая постоянная ( 22 ).
§ 4. Первое начало термодинамики. Энергия и
энтальпия как функции состояния 24
1. Эквивалентность тепла и работы ( 25 ). 2.
Энтальпия как функция состояния ( 28 ). 3.
Соотношение между удельными теплоемкостями ср
и cv ( 30 ).
§ 5. Обратимый и необратимый адиабатические
процессы 32
1. Адиабатический обратимый процесс ( 32 ).
2. Адиабатический необратимый процесс ( 36 ).
3. Процесс Джоуля—Кельвина ( 38 ). 4. Одно
очень важное следствие ( 40 ).
§ 6. Второе начало термодинамики 41
1. Цикл Карно и его коэффициент полезного
действия ( 42 ). 2. Первая часть второго начала
термодинамики ( 45 ). 3. Вторая часть второго
начала термодинамики ( 52 ). 4. Простейший
численный пример ( 54 ). 5. Некоторые
исторические замечания ( 57 ). 6. К вопросу о
взаимоотношении энергии и энтропии ( 59 ).
§ 7. Термодинамические потенциалы и соотношения
взаимности 60
§ 8. Термодинамические равновесия 67
1. Незаторможенное термодинамическое
равновесие и максимум энтропии ( 67 ). 2. Система в
незаторможенном термодинамическом равновесии
является изотермической и изобарической ( 69 ).
3. Дополнительные степени свободы в
заторможенном равновесии ( 69 ). 4. Экстремальные свой-
476
Оглавление
ства термодинамических потенциалов ( 70 ).
5. Теорема о максимальной работе ( 73 ).
§ 9. Уравнение Ван-дер-Ваальса 77
1. Ход изотерм ( 78 ). 2. Энтропия и
калорические свойства газа Ван-дер-Ваальса ( 80 ).
§ 10. Сжижение газа по Ван-дер-Ваальсу 82
1. Интегральный и дифференциальный процессы
Джоуля—Кельвина ( 82 ). 2. Кривая инверсии
и ее техническое применение ( 84 ). 3. Границы
области сосуществования жидкой и
газообразной фаз в плоскости р, v ( 86 ).
§ И. Шкала Кельвина 92
§ 12. Тепловая теорема Нернста 96
Глава II. Применение термодинамики к конкретным
системам 103
§ 13. Смесь газов; парадокс Гиббса. Закон Гульдбер-
га и Вааге 103
1. Обратимое разделение газов ( 105 ). 2.
Увеличение энтропии при диффузии и парадокс Гиббса
( 106 ). 3. Закон действующих масс ( 108 ).
§ 14. Химические потенциалы и химические постоянные 114
1. Химические потенциалы \ч ( 115 ). 2. Связь
величин fit и gi для смесей идеальных газов
( 118 ). 3. Химические постоянные идеальных
газов ( 119 ).
§ 15. Разбавленные растворы 120
1. Общие принципы и история ( 120 ). 2.
Уравнение состояния разбавленных растворов Вант-
Гоффа ( 122 ).
§ 16. Различные агрегатные состояния воды. К теории
паровой машины 126
1. Кривая давления пара и уравнение
Клапейрона ( 126 ). 2. Фазовое равновесие между водой
и льдом ( 131 ). 3. Удельная теплоемкость
насыщенного водяного пара ( 132 ).
§ 17. Общие принципы теории равновесия фаз ... . 134
1. Тройная точка воды ( 135 ). 2. Правило фаз
Гиббса ( 139 ). 3. Закон Рауля для разбавленных
растворов ( 141 ). 4. Закон абсорбции Генри
( 1803 г. ) ( 144 ).
§ 18. Напряжение гальванического элемента 145
1. Электрохимические потенциалы ( 146 ). 2.
Элемент Даниэля ( 1836 г. ) ( 147 ). 3. Сведение
отдельных реакций к упрощенной брутто-реакции
( 149 ). 4. Уравнение Гиббса—Гельмгольца ( 151 ).
5. Численный расчет ( 152 ). 6. Интегрирование
уравнения Гиббса—Гельмгольца ( 154 ).
§ 19. Ферромагнетизм и парамагнетизм 155
L Работа намагничивания и магнитное уравне-
Оглавление
477
ние состояния ( 155 ). 2. Функция Ланжевена для
парамагнетиков ( 158 ). 3. Теория ферромагнетизма
Вейсса ( 160 ). 4. Удельные теплоемкости сн и см
( 165 ). 5. Магнитокалорический эффект ( 170 ).
§ 20. Излучение в полости 172
1. Закон Кирхгофа ( 173 ). 2. Закон Стефана—
Больцмана ( 176 ). 3. Закон Вина ( 178 ). 4. Закон
излучения Планка ( 184 ).
§ 21. Необратимые процессы. Термодинамика
неравновесных состояний 193
1. Теплопроводность и появление локальной
энтропии ( 193 ). 2. Теплопроводность в анизотропных
телах и соотношения взаимности Онзагера ( 196 ).
3. Термоэлектрические явления ( 198 ). 4.
Внутренние превращения( 206 ). 5. Общие закономерности
( 208 ). 6. Область применимости
термодинамической теории необратимых процессов ( 211 ).
Глава III. Элементарная кинетическая теория газов . . . 213
§ 22. Уравнение состояния идеального газа 213
§ 23. Распределение Максвелла 219
1. Распределение Максвелла в случае
одноатомного газа. Вывод 1860 г. ( 219 ). 2. Вычисления
и эксперимент ( 222 ). 3. Общие соображения
о распределении по энергиям. Множитель
Больцмана ( 225 ).
§ 24. Броуновское движение 227
§ 25. Статистика парамагнитных веществ ....... 235
1. Классическая функция Ланжевена ( 235 ).
2. Функция Ланжевена, видоизмененная
согласно квантовой теории ( 238 ).
§ 26. Статистический смысл постоянных в уравнении
Ван-дер-Ваальса 240
1. Собственный объем молекул и постоянная Ь
( 241 ). 2. Вандерваальсовы силы сцепления
и постоянная а ( 243 ).
§ 27. Проблема длины свободного пробега 246
1. Вычисление длины свободного пробега в
одном частном случае ( 247 ). 2. Вязкость ( 249 ).
3. Теплопроводность ( 253 ). 4. Общие замечания
к проблеме длины свободного пробега ( 255 ).
Глава IV. Общие принципы статистики. Метод ячеек . . 258
§ 28. Теорема Лиувилля. Г-пространство и
^-пространство 258
1. Многомерное Г-пространство ( 259 ). 2. Теорема
Лиувилля ( 260 ). 3. Равновероятность состояний
для идеального газа ( 262 ).
478
Оглавление
§ 29. Принцип Больцмана * . . . . 265
1. Статистический вес как мера вероятности
состояния ( 266 ). 2. Максимум вероятности как
мера энтропии ( 270 ). 3. Выводы относительно
метода элементарных ячеек ( 272 ).
§ 30. Сравнение с термодинамикой 274
1. Изохорический процесс ( 274 ). 2. Общий
случай процесса в газе в отсутствие внешнего
поля ( 275 ). 3. Газ в силовом поле. Формула
Больцмана ( 277 ). 4. Распределение скоростей по
Максвеллу— Больцману ( 278 ). 5. Смесь газов ( 280 ).
§ 31. Удельная теплоемкость и энергия газа из
абсолютно твердых молекул 281
1. Одноатомный газ ( 281 ). 2. Газ двухатомных
молекул ( 284 ). 3. Газ многоатомных молекул
и трудности его рассмотрения, отмеченные
Кельвином ( 288 ).
§ 32. Теплоемкости упругих молекул и твердых тел . 289
1. Двухатомная молекула ( 289 ). 2.
Многоатомный газ ( 291 ). 3. Твердые тела и закон Дюлонга
и Пти ( 291 ).
§ 33. Квантование энергии колебаний 292
1. Линейный осциллятор ( 293 ). 2. Твердое тело
( 297 ). 3. Обобщение на любые квантовые
состояния ( 297 ).
§ 34. Квантование вращательной энергии 298
§ 35. Дополнения к теории излучения и теории
твердого тела 303
1. Метод собственных колебаний ( 304 ). 2. Теория
теплоемкости твердого тела по Дебаю ( 305 ).
§ 36. Сумма состояний в Г-пространстве 307
1. Условие Гиббса ( 307 ). 2. Связь с методом
Больцмана ( 309 ). 3. Переход к квантовой
статистике ( 312 ). 4. Анализ гипотезы Гиббса ( 315 ).
§ 37. Основы квантовой статистики 316
1. Квантовая статистика тождественных частиц
( 316 ). 2. Метод Дарвина—Фаулера ( 318 ). 3.
Статистика Бозе—Эйнштейна и Ферми—Дирака
( 321 ). 4. Метод седловых точек ( 323 ).
§ 38. Вырожденные газы 327
1. Распределения Бозе—Эйнштейна и Ферми-
Дирака ( 327 ). 2. Степень вырождения газов ( 332 ).
3. Сильно вырожденный газ Бозе—Эйнштейна
( 334 ).
§ 39. Электронный газ в металлах 338
1. Замечания в теории Друде ( 338 ). 2.
Полностью вырожденный газ Ферми—Дирака ( 340 ).
3. Почти полное вырождение ( 343 ). 4.
Специальные проблемы ( 345 ).
§ 40. Средние квадратичные флуктуации 349
Оглавление
479
Глава V. Основы точной кинетической теории газов . . . 357
§ 41. Кинетическое уравнение Максвелла—Больцмана 358
1. Описание состояния в кинетической теории
газов ( 358 ). 2. Изменение функции / со
временем ( 360 ). 3. Законы упругого удара ( 362 ).
4. Интеграл столкновений Больцмана ( 364 ).
5. Гипотеза Больцмана о молекулярном
беспорядке ( 367 ).
§ 42. H-теорема и распределение Максвелла 369
1. H-теорема ( 369 ). 2. Распределение Максвелла
( 373 ). 3. Равновесные распределения ( 376 ).
§ 43. Основные уравнения гидродинамики 377
1. Разложение функции распределения в ряд ( 377 ).
2. Уравнения переноса Максвелла ( 380 ). 3.
Сохранение массы ( 383 ). 4. Сохранение импульса
( 384 ). 5. Сохранение энергии ( 386 ). 6. Принцип
энтропии ( 389 ).
§ 44. К интегрированию кинетического уравнения . . 392
1. Интегрирование методом моментов ( 392 ).
2. Преобразование уравнений моментов ( 393 ).
3. Вычисление моментов относительно
интеграла столкновений ( 396 ). 4. Коэффициенты
внутреннего трения и теплопроводности ( 397 ).
§ 45. Проводимость и закон Видемана—Франца . . . 403
1. Кинетическое уравнение и уравнение переноса
для электронов в металле ( 403 ). 2.
Приближенное решение кинетического уравнения ( 405 ).
3. Плотность электрического тока и потока
энергии ( 409 ). 4. Закон Ома ( 411 ). 5.
Теплопроводность и абсолютная термо-э.д.с. ( 412 ). 6.
Закон Видемана—Франца ( 414 ).
Задачи 417
К главе I 417
К главе II 420
К главе III 423
К главе IV 425
К главе V 428
Указания к решению задач 429
О ПЕЧАТКИ
Стр,
42
87
151
285
313
455
458
Строка
11 СН.
1 СВ.
3 св.
2 сн.
12 сн.
Формула ( 4 )
8 св.
Напечатано
только подъем груза
группировки
( 14.17 )
центров осей:
разложениям
к
{C( \ )e~ixxdk
Следует читать
подъем груза только
соединения
( 14.7 )
центров ) осей:
распределениям
К
{ C( K )eiXxd\
А. Зоммерфельд
ТЕРМОДИНАМИКА
И СТАТИСТИЧЕСКАЯ ФИЗИКА
Редактор Н. Л. ТЕЛЕСНИН
Технический редактор В. И. Шаповалов
Сдано в производство 25/IV.1955 г.
Подписано к печати 18/VII 1955 г.
Т-06313. Бумага 84х 1081/32=7,5
бум. л. 24,6 печ. л. Уч.-ивд. л. 22,9
Изд. М 2/2588. Цена 18 р.
Зак. М 932.
Издательство иностранной литературы.
Москва, Ново-Алексеевская, 52.
16-я типография Главполиграфпрома
Министерства культуры СССР.
Москва, Трехпрудный пер., 9.
А. ЗОММЕРФЕЛЬД
СТАТИСТИЧЕСКАЯ ФИЗИКА