/









































































































Author: Мышляева Л.В. Краснощеков В.В.
Tags: неорганическая химия аналитическая химия химия
Year: 1972




Text
АКАДЕМИЯ НАУКСССР
ОРДЕНА ЛЕНИНА ИНСТИТУТ ГЕОХИМИИ И АНАЛИТИЧЕСКОЙ ХИМИИ им. В. И. ВЕРНАДСКОГО
Серия: «АНАЛИТИЧЕСКАЯ ХИМИЯ ЭЛЕМЕНТОВ»
АНАЛИТИЧЕСКАЯ ХИМИЯ КРЕМНИЯ
Л. В. Мышляева, В. В. Краснощеков
ИЗДАТЕЛЬСТВО «НАУКА»
Москва 1972
УДК 546.28:543/545
Серия: «Аналитическая химия элементов»
Главный редактор академик А. П. Виноградов
Редакционная коллегия:
И. П. Алимарин, А. И. Бусев, А. П. Виноградов, А. Н. Ермаков, Ю. А. Золотов, А. В. Карякин, П. Н. Палей, С. Б. Саввин, И. В. Тананаев, М. П. Волынец (ученый секретарь)
Редактор тома «Аналитическая химия кремния»
А. И. Бусев
Адрес редколлегии:
117334. Москва В-334, Воробьевское шоссе, 47а Ордена Ленина Институт геохимии и аналитической химии им. В. И. Вернадского
Академии наук СССР
2-5-5 522-1972
ОТ РЕДКОЛЛЕГИИ
Институт геохимии и аналитической химии им. В. И. Вернадского АН СССР осуществляет издание серии монографий по аналитической химии отдельных элементов. Серия «Аналитическая химия элементов» составит около пятидесяти томов. Надобность в подобного рода издании давно назрела. Вместе с тем у нас накопился огромный опыт многочисленных лабораторий и теперь стало возможным и необходимым его подытожить.
Аналитическая химия любого элемента и его соединений в настоящее время представляется чрезвычайно разнообразной как вследствие сложности современных объектов исследования и широты диапазона концентраций, которые бывает необходимо определить, так и вследствие разнообразия использующихся методов.
В связи с этим для монографий был разработан общий план как в смысле содержания, так и последовательности изложения материала. В них содержатся общие сведения о свойствах элементов и их соединений. Затем рассматриваются химические реакции, являющиеся основанием для аналитических методов. Методы, как физические, так и физико-химические и химические, излагаются применительно к количественному определению данного элемента, начиная с анализа сырья, далее — типичных полупродуктов производства и, наконец, конечной продукции — металлов и сплавов, окисей, солей и других соединений и материалов. Как правило, приводятся принципы определения и, где это необходимо, дается точное описание всего процесса определения. Необходимое внимание уделяется быстрым методам анализа. Самостоятельное место занимает изложение методов определения так называемых элементов-примесей в чистых материалах.
Обращается внимание на точность и чувствительность методов в связи с общей тенденцией повышения чувствительности методов определения следов элементов-примесей.
Монографии содержат обширную библиографию, включая литературу последних лет; они рассчитаны-на широкий круг химиков, в первую очередь на химиков-аналитиков научно-исследовательских институтов и заводских лабораторий различных отраслей хозяйства, а также на преподавателей и студентов химических вузов.
К составлению монографий привлечены крупнейшие советские специалисты, имеющие опыт работы в области аналитической химии того или иного химического элемента.
Отдельные тома серии «Аналитическая химия элементов» будут выходить самостоятельно, по мере их подготовки. Вышли в свет монографии, посвященные торию, таллию, урану,рутению, молибдену, калию, бору, цирконию и гафнию, кобальту, бериллию, редкоземельным элементам и иттрию, никелю, технецию, прометию, астатину и францию, ниобию и танталу, протактинию, галлию, фтору, селену и теллуру, алюминию, плутонию, нептунию, трансплутониевым элементам, готовятся к печати монографии по аналитической химии радия, платиновых металлов, германия, магния, кадмия и других элементов.
Мы обращаемся с просьбой ко всем читателям присылать свои замечания и отзывы о монографиях.
ПРЕДИСЛОВИЕ
Кремний — второй по распространенности в природе и весьма важный в практическом отношении элемент. Определение кремния— обязательная и очень ответственная операция в анализе естественных и искусственных силикатов, руд, черных и цветных металлов и сплавов, кремнийорганических соединений и других кремнийсодержащих веществ, в контроле производства и готовой продукции. Несмотря на широкую распространенность кремния в природе и почти повсеместное нахождение в самых разнообразных материалах, вопросы анализа соединений кремния и методы аналитического определения этого элемента разработаны недостаточно. До сравнительно недавнего времени это было легко объяснить: требованиям промышленности и исследовательской практики отвечал достаточно надежный, хотя и крайне длительный, гравиметрический метод определения кремния. Определение кремния гравиметрическим методом не вызывало возражений еще и потому, что отделение его было необходимо для выполнения дальнейших аналитических операций в ходе анализа веществ сложного состава. Однако с развитием силикатной промышленности, возникновением промышленного производства кремнийорганических соединений и полупроводниковых материалов перед аналитической химией кремния возникли новые задачи. Подготовка вещества к анализу, методы разложения проб, удаление мешающих элементов — все это должно быть пересмотрено и заново решено по отношению к определению кремния.
Все большее значение начали приобретать вопросы так называемого фазового, или вещественного, анализа, когда требуется не только количественно определить кремний, но и установить, в виде каких соединений он присутствует в анализируемых материалах.
Возросли требования не только к увеличению точности результатов аналитических определений, но и к сокращению времени определения и повышению чувствительности аналитических методов, возникла задача определения весьма малых количеств посторонних примесей в основном веществе. Широкое применение в аналитической химии кремния нашли физические и физико-химические методы анализа.
5
Большой литературный материал, относящийся к аналитической химии кремния, до последнего времени не был исчерпывающе критически обобщен. Настоящая монография является попыткой систематизации литературных данных по аналитической химии кремния с учетом личного опыта авторов. В книге изложены теоретические основы аналитической химии кремния, описаны химические, физико-химические и физические методы его определения в природных и технических объектах, методы отделения кремния от сопутствующих элементов, а также методы определения примесей в элементном кремнии и его соединениях высокой чистоты.
Монография написана по общей схеме, рекомендуемой для серии «Аналитическая химия элементов». Л. В. Мышляевой написаны главы 1—5, 10, 11, а также первый, второй и третий разделы 9-й главы; остальные разделы 9-й главы, а также главы 6—8 написаны В. В. Краснощековым.
Авторы считают своим приятным долгом выразить глубокую благодарность рецензентам — доктору химических наук С. В. Бруевичу, доктору химических наук А. А. Немодруку, доктору химических наук Ф. М. Шемякину и кандидату химических наук Е. Н. Егоровой. Особенно благодарны авторы профессору А. И. Бу-севу за постоянную помощь, ценные указания и поддержку при написании монографии.
Авторы заранее благодарят всех читателей, которые выскажут свои критические замечания и пожелания.
Л. В. Мышляева, В. В. Краснощеков
Глава 1
СВОЙСТВА КРЕМНИЯ И ЕГО СОЕДИНЕНИЙ
ОБЩАЯ ХАРАКТЕРИСТИКА ЭЛЕМЕНТА
Кремний — химический элемент IV группы периодической системы Д. И. Менделеева. Его порядковый номер 14, атомная масса 28,086 1710]. Ранние данные по определению атомной массы приведены в [837, 946], библиография — в работах [580, 610, 660, 786].
В 1811 г. Гей-Люссак и Тенар выделили кремний по реакции SiF4+4K->Si+4KF, но не изучили его свойств. Берцелиус в 1823 г. получил элементный кремний из кремнефторида калия аналогичным путем по реакции K2SiFe+4K->6KF+Si, установил его элементную природу, изучил свойства и превратил сожжением в SiO2, доказав сложность состава кремнезема. Берцелиус дал новому элементу название silicium, производное от латинского silex — кремень. Русское название «кремний» было принято в 1834 г.
Электронное строение атома кремния в основном состоянии ls22s22pe3s23p2. Содержание естественных изотопов кремния в элементе и его соединениях составляет (в %): 28Si 92,27; 29Si 4,68; 30Si 3,05. Период полураспада искусственных радиоизотопов: 27Si 4,5 сек; 31Si 2,65 час; 32Si около 100 лет [474, 479]. Радиус иона Si (IV) по Полингу 0,44 А [435]. Радиус атома при четверной координации и ковалентной связи 1,175 А. Для расчетов длины связей правило аддитивности неприменимо, так как во многих соединениях проявляется ионный характер связей [435]. Ионизационные потенциалы (ваз) равны: Si(I) 8,15; Si(II) 16,34; Si(III) 33,46; Si(IV) 45,13; Si(V) 166,73; сродство к электрону 1,22 эв [688]. Энергии связей кремния с другими элементами см. в [270]. В соединениях, устойчивых при обычных условиях, для кремния характерна степень окисления 4+. Свойства кремния см. в [293, 610, 623, 688].
РАСПРОСТРАНЕННОСТЬ И ПРИМЕНЕНИЕ
Кремний — второй по распространенности (после кислорода) элемент земной коры. Из многочисленных расчетов Адлер [23] приводит данные Аренса [6351 и Виноградова 1129]. По данным Ви
7
ноградова, в земной коре имеется (в вес. %): О 47, Si 27,6, Al 8,6, Fe 5, Са 3,5, Na 2,5, Mg 2, К 2,5, H 0,15, Ti 0,6. Перечисленные элементы обычно входят в состав природных соединений кремния и искусственных силикатов. Аналогичные данные приводит и Кларк [704, 7051, упоминая, что в составе метеоритов кремний также занимает второе место по весу и по числу атомов (см. также [688, 972]).
Кремний не встречается в природе в элементном состоянии, он распространен главным образом в виде двуокиси, ее гидратов и силикатов и алюмосиликатов — солей кремневых и алюмокремне-вых кислот, SiO2 имеет большое число кристаллических и аморфных разновидностей, из которых следует упомянуть кварц (наиболее чистая его природная разновидность — горный хрусталь), тридимит, кристобалит, яшмы, опал. Существуют многие другие природные и искусственно полученные разновидности двуокиси кремния, например коэсит [709] — разновидность двуокиси кремния с высокой плотностью, стишовит [495] и др. [524, 610, 651, 708, 714, 721, 751, 761, 762, 855, 856, 935, 996, 1026]. Термодинамически устойчив до 870°С кварц, в интервале 870 — 1470°С — тридимит, выше 1470°С — кристобалит. Теплоты образования и фазовых превращений в системе SiO2 см. в [828, 1025]. Кремнийсодержащие минералы — полевые шпаты, слюды, оливины, пироксены и многие другие — присутствуют во всех важнейших горных осадочных и изверженных породах. Кремний содержится во всех водах, как пресных, так и соленых; спектрально кремний обнаружен на солнце. Соединения кремния входят в состав тканей растений и животных [1060], они содержатся в костях позвоночных животных [820, 1073], накапливаются в больших количествах морскими простейшими водорослями и животными организмами (диатомеи, радиолярии, кремневые губки). Состав соединений кремния, в виде которых он входит в растительные и животные организмы, а также роль их в жизнедеятельности растений и животных выяснены недостаточно [4, 178, 879].
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ СВОЙСТВА
Элементный кремний — кристаллическое металловидное тело. Долгое время считалось, что кремний может быть получен в аморфном и кристаллическом состоянии. Однако «аморфный» кремний лишь мелкокристаллическая разновидность кубической модификации кремния. Кремний. кристаллизуется в правильной системе и имеет кубическую гранецентрированную кристаллическую решетку с периодом а = 5,4297А [621]. Существует высокотемпературная гексагональная модификация [71,1083]. Плотность кремния 2,328 г/см?, температура плавления 1423°С, температура кипения около 2600°С. Твердость по шкале Мооса равна 7, по Бриннелю — 240 кГ/см*.
8
Кремний — полупроводник и используется как один из важнейших полупроводниковых материалов. Нормальный обратимый потенциал кремния не измерен из-за исключительно малой растворимости его соединений в воде. По расчетам Латимера [3231, стационарный потенциал кремния в кислых растворах равен —0,84 в, в щелочных—1,73 в. Экспериментально найдены несколько более положительные значения [194]. Элекрические свойства кремния очень сильно зависят от наличия примесей [18]. Дороговизна очистки кремния препятствует его использованию. Высокочистый кремний используют для солнечных батарей, для нужд радиоэлектроники и для других целей [137, 247, 293, 416, 698]. Получение чистого кремния см. в [71, 137, 247, 293, 416, 623].
Кремний окисляется при повышенных температурах, что затрудняет его введение в сплавы. С галогенами кремний дает- галогениды, причем фтор реагирует уже при комнатной температуре, с серой образует соединение SiS2 [1090], с азотом —Si3N4 [1093]. Соединения кремния с водородом •— силаны — неустойчивы, самовоспламеняются на воздухе, высший их представитель SieH14 [178]. Кремний образует силициды металлов состава Mg2Si, Ca2Si, MnSi2, FeSi, CrSi, TiSi2 и др. [645]. О соединениях кремния с бором см. в [468]. С кислородом кремний дает один окисел SiO2. Существование соединений, в которых кремний двухвалентен, подвергается сомнению. Однако полимеры (—SiO—)х известны [584, 863]. О кратности связи Si и ее свойствах см. в [139, 1128].
Кремний растворяется в разбавленных растворах щелочей при нагревании с выделением водорода, а также в смеси фтористоводородной и азотной кислот и анодно — в щелочах, фтористоводородной кислоте и ее солях. В присутствии органических растворителей растворение идет быстрее [194, 195,367]. При высоких температурах кремний растворяется во многих расплавленных металлах: олове, алюминии, литии, свинце, серебре и др.
Заряд ядра атома кремния экранирован вследствие большого радиуса атома, поэтому кремний отдает свои валентные электроны. В соединениях кремний четырехковалентен, по отношению к атомам и группам, имеющим большой заряд ядра й малый объем, его координационное число равно шести (ОН-, F- и др.).
В некоторых обменных реакциях (например, гидролиза хлор-силанов) кремний, по мнению Бажанта и др. [52|, образует промежуточные соединения, в которых он пятиковалентен.
Высокая энергия связи кремний — кислород [50] делает термодинамически выгодными реакции образования двуокиси кремния и других кислородсодержащих соединений. Именно поэтому кремний из соединений с углеродом, водородом, серой и другими элементами в присутствии кислорода переходит в SiO2. Многообразны его соединения с кислородом [325, 616].
9
ОСОБЕННОСТИ СТРОЕНИЯ СО ЕДИНЕНИЙ КРЕМНИЯ
Большинство природных и многие синтетические силикаты имеют кристаллическую структуру. Имеется определенная связь между кристаллооптическими свойствами соединений кремния и их химическим составом.
Основные представления о составе и строении силикатов и алюмосиликатов были сформулированы еще Бутлеровым в его работах о полисоединениях в минеральной химии [101, 140, 299, 381]. Рассматривая соединения кремния как производные ортокремне-вой кислоты (полного гидрата), Бутлеров характеризует поликрем-невые кислоты как ангидрогидратные соединения, образующиеся в результате потери «п» молекулами полного гидрата — Si(OH) 4 — последовательно (n—1), п, (п+1) и т. д. до 4п/2 молекул воды. В результате получаются линейные полимеры нескольких рядов. Ряд А имеет общий вид 31яОя_1(ОН)2я+2,ряд В —Si„O„(OH)2n и т. д. вплоть до SinO2n. На основании этой теории ангидрогидратов Бутлеров высказал впервые правильные взгляды на строение природных алюмосиликатов как производных не кремневых, а алюмокремне-вых кислот. Вернадский [126] развил эти положения, подтвержденные дальнейшими исследованиями.
Работами Бреггов по исследованию силикатов методами интерференции рентгеновских лучей было установлено, что кремний в них находится в большинстве случаев в виде звеньев (SiO4)4-[677, 678]. Каждое такое звено — тетраэдр с атомом кремния в центре. Координационное число кремния по кислороду таким образом составляет 4. Однако Полинг и Гольдшмидт вычисляли ионный радиус кремния в предположении шестерной координации кремния по кислороду. Вследствие этого были получены сравнимые численные значения для радиусов других ионов. О координации кремния с шестью ионами кислорода см. также в [886, 982].
Плотно упакованный тетраэдр (SiO4)4- может быть заменен тетраэдром (А1О4)5- в структуре алюмосиликатов и другими структурно аналогичными группами.
Классификация силикатных минералов выполнена Бреггом исходя из принципов, близких к теории ангидрогидратов. Основные группы силикатных минералов, по Бреггу, следующие:
I. Минералы, содержащие изолированные силикатные радикалы:
а) минералы, содержащие (SiO4)4-, например форстерит Mg2SiO4 или ортосиликат натрия (ряд А Бутлерова; n = 1)
б) минералы, содержащие звено (Si2O7)6- (ряд А Бутлерова; п=2);
в) минералы, содержащие кольцо (Si3О9)в-, например бентонит; г) минералы, содержащие кольцо (Si4O12)8-;
д) минералы, содержащие кольцо (SieO18)12-.
II. Цепные силикаты, построенные из соединенных между собой через кислород звеньев (SiO4)4-:
10
а) линейные силикаты эмпирического состава (SiO|_)„. К ним относятся минералы группы пироксенов (ряд В Бутлерова Si„O„(OH))2„;
б) двойные цепи — поперечно соединенные линейные цепи (SiO3)2~ (ряд С Бутлерова Si„O„+1(OH)2n_2). Основная группа — амфиболы, например тремолит.
III. Сетчатые структуры, состоящие из гексагональной сетки тетраэдров (SiO4)4-. Структурная единица (Si4О10)4- (ряд D Бутлерова Si„O„+3(OH)2ZI_ 4). При замещении Si(IV)Ha А13+ получим (AlSi3O10)5-. К этой группе относятся пластинчатые минералы, обладающие спайностью по базопинакоиду, — слюды, тальк, глинистые минералы.
IV. Силикаты, образующие трехмерную сетку тетраэдров (SiO4)4-. Типичный представитель — кварц (SiO2)„. При замещении в тетраэдрах атома кремния получаются различные минералы этой группы, например полевые шпаты (AlSi3Og)~ [713].
Разнообразие форм силикатов очевидно вследствие большого количества возможных структур.
Прочность связи Si—О, плохая растворимость кремневой кислоты и ее склонность к полимеризации — причины того, что большинство минералов перечисленных групп практически нерастворимы в кислотах, за исключением горячей Н3РО4. Говоря о растворимых в кислотах (вернее — разлагаемых кислотами) минералах, не следует забывать, что разложение происходит медленно. Оно тормозится образованием на поверхности растворяемого вещества коллоидной гелеобразной кислоты, преграждающей доступ кислоты внутрь частицы. На структуре продуктов разложения силикатов кислотами отражается первоначальная структура силиката. Ортосиликаты, производные ряда А Бутлерова [101], куда входят изолированные радикалы, дают при разложении низкомолекулярные продукты — моно- и дикремневую кислоты. Силикаты линейной структуры, слоистой структуры (с двухмерной сеткой тетраэдров (SiO4)4- [5241), а также силикаты с пространственной сеткой тетраэдров [60] в лучшем случае при обработке кислотами могут дать полимеры кремневой кислоты или же вообще не будут разлагаться кислотами. Отнесение силиката к определенной группе часто зависит от содержания в нем воды. Например, силикат натрия с эмпирической формулой Na2SiO3-9H2O, по-видимому, содержит в своей структуре изолированные группы (SiO4)4- (имеются также предположения о шестерной координации кремния в этом соединении). Безводный силикат натрия Na2SiO3 при растворении не дает в растворе мономерных форм кремневой кислоты, а сохраняет цепочки тетраэдров линейного строения.
Представления о строении природных силикатов в какой-то мере переносятся и на строение синтетических силикатов.
Некоторые неорганические производные кремния несиликатного характера встречаются в сплавах и в виде примесей в элементном кремнии. В сплавах на основе железа, меди и алюминия кремний
11
находится либо в элементном состоянии, либо в виде силицидов, карбидов, нитридов, полимерного моноксида. В виде силикатов кремний в сплавах может присутствовать на поверхности в шлаках или внутри — в виде шлаковых включений. При обработке металлов кислотами в процессе их анализа возможно окисление кремния (или разложение силикатов) с переходом в кремневую кислоту, но не исключены и потери за счет образования летучих соединений.
Установление правильных взглядов на состав и строение кремневых кислот и их производных — природных силикатов — связано с изучением свойств эфиров ортокремневой кислоты — тетраалкоксисиланов. Начиная с самых ранних работ по синтезу и исследованию кремнийорганических соединений — исследований Эбельмена [731] и до работ Айлера и его сотрудников [4], Андрианова, Соколова [28, 486] этому вопросу было посвящено много исследований, нашедших свое отражение и обобщение в литературе. Строение кислот, выделить которые в чистом виде не представляется возможным, изучено на их эфирах, выделение которых успешно проведено многими исследователями, хотя первоначально это были, по-видимому, полимерные продукты.
Принцип конденсационной полимеризации, изложенный Менделеевым и Бутлеровым применительно к ортокремневой кислоте и ее производным общего вида Si(OR)4, мог быть и был распространен на область химии эфиров ортокремневой кислоты. Менделеев еще в 50-х годах прошлого столетия высказал мысль о полимерности кремнезема [348]. Он впервые сравнил свойства углерода и кремния и подчеркнул своеобразие кремния. По этому вопросу существуют более поздние публикации [52, 140, 178, 196, 978].
После работ Бутлерова наибольшее значение для выяснения строения кремневых кислот имеют исследования Конрада, Бехле, Зигнера и Гросса [870, 1017], Кинга [861, 862], Милиуса и Грошуф-фа [931] и других исследователей.
В работах Конрада, Бехле, Зигнера [870], Зигнера и Гросса [1017] и Кинга [861, 862] представлено все многообразие состава возможных продуктов поли конденсационных процессов, имеющих место при гидролизе эфиров ортокремневой кислоты и соответственно при дегидратации самой кремневой кислоты. Аналогичным образом протекает гидролиз алкилзамещенных эфиров [28] и ре-' акции пиролиза. По Кингу, может быть получено несколько типов комплексных полимерных алкоксисиланов, аналогичных по составу ангидрогидратам Бутлерова, где R~C2H5, например R2n+2 Si„O3n+1, R2„Si„O3„, R2n_2Si„O3„_1 и т. д. Многие природные силикаты и алюмосиликаты являются производными соответствующих кислот (где R=H].
Авторам принадлежит трактовка механизма реакций гидролиза и конденсации тетраалкоксисиланов с образованием полимеров линейной, циклической и пространственной структуры. Омылением этих полимеров получены и соответствующие кислоты. О выделении кремневых кислот как конечного продукта гидролиза эфиров ор-12
такрёмневой кислоты сообщалось во многих работах [28, 178, 381, 731, 870, 1002, 1017].
В аналитической практике следует учитывать своеобразие кремнийорганических соединений, заключающееся прежде всего в легкой растворимости многих мономерных и полимерных кремнийорганических соединений в органических растворителях, в их гидрофобности и устойчивости к действию окислителей. Это затрудняет их анализ в водных средах и облегчает использование в аналитических целях неводных растворителей. Методы анализа кремнийорганических соединений в значительной степени близки к методам анализа элементоорганических соединений, каковыми они по существу и являются. Однако после выполнения операций разложения кремнийорганических соединений методы определения самого кремния в конечном счете ничем не отличаются от определения его в неорганических соединениях кремния.
ХИМИКО-АНАЛИТИЧЕСКАЯ ХАРАКТЕРИСТИКА СОЕДИНЕНИЙ КРЕМНИЯ
Число соединений кремния, свойства которых используются для его качественного обнаружения, отделения от сопутствующих элементов и количественного определения, невелико. Сюда относятся кремневая кислота и продукты ее конденсационной полимеризации, растворимые силикаты, кремнефтористоводородная кислота, гетерополикислоты, содержащие в свем составе кремний. Для отделения кремния от других элементов, а также для определения его прямыми и косвенными путями используются свойства фторида кремния.
Свойства кремневой кислоты
Существование кремневой кислоты в водном растворе в мономерной форме в настоящее время считается бесспорным. Это доказано определением молекулярного веса криоскопическим методом [636, 830, 831, 875, 1092], изучением диализа и диффузии [464, 684, 931L Состав кремневой кислоты соответствует формуле Si(OH)4 или H4SiO4. Кремневая кислота — очень слабый электролит. Значение pH ее водного раствора составляет около 4,0—4,5, значение констант ионизации Ki = 10-9’8; Д2 = 10-12'16 [980]. Данные по константам ионизации H4SiO4 см. в работах 758, 1047].
Изображение кремневой кислоты в виде H2SiO3 в настоящее время считается неправильным, так как доказано, что координационное число кремния по кислороду равно четырем. В щелочных средах возможно существование иона [Si(OH)e]2~ по аналогии с ионом SiF2— и на основании данных рентгеновских исследований щелочных силикатов [1087]. Поэтому предложено кремневую кислоту изображать как Н23](ОН)в[или Si(OH)4-2H2O], однако достаточных доказательств правильности такого изображения пока нет.
13
Истинный раствор кремневой кислоты может быть получен растворением двуокиси кремния в воде, обработкой кислотами растворов щелочных силикатов с последующим удалением Посторонних ионов диализом (или ионным обменом) или гидролизом эфиров ортокремневой кислоты [например, Si(OCH3)4] и галогенидов кремния (например, SiCl4).
Растворимость кремневой кислоты, полученной одним из перечисленных способов, по данным многих исследователей, обобщенным Айлером [4] иШеллом [1011], составляет в расчете на SiO2 величину 0,01—0,017% при комнатной температуре. При повышении температуры растворимость кремневой кислоты возрастает, достигая величины 0,04% при 94°С [8751. В случае охлаждения насыщенных растворов кремневой кислоты, а также при получении ее действием кислот на растворы щелочных силикатов или гидролизом хлорсиланов или тетраалкоксисиланов кремневая кислота образует пересыщенные растворы. В этих растворах она может в течение некоторого времени оставаться в мономерном состоянии, что имеет большое значение для анализа.
Растворимость кремневой кислоты и скорость ее растворения зависят от нескольких факторов, из которых важнейшие — взятая модификация двуокиси кремния, степень ее дисперсности и температура. При встряхивании с водой геля кремневой кислоты равновесие растворимости устанавливается за несколько часов, в случае кварца нужны многие месяцы и даже годы.
Кварц практически не растворяется в воде до 150°С; при дальнейшем повышении температуры наблюдается прямая пропорциональность между ростом температуры и растворимостью двуокиси кремния.
Растворимость двуокиси кремния зависит от размеров частиц, что может быть выражено упрощенным уравнением Оствальда — Фрейндлиха [8271
In Sr/S= 2 EV/пТг)
vpe S — растворимость крупной частицы; Sr — растворимость частицы радиуса г; Е — поверхностная энергия, эрг/см2; V — молярный объем, см3; R— газовая постоянная 8,31-107, эрг/моль-град; Т — абсолютная температура.
Расчеты Айлера [4] показывают, что растворимость кварца резко возрастает, если размер частиц становится меньше 10 нм. Растворимость частиц кварца сг=5нл« больше растворимости крупных аморфных частиц гидратированной кремневой кислоты.
В зависимости от значения pH содержание кремния в водном растворе меняется [табл. 1].
Однако Айлер на основании литературных данных и результатов собственных исследований пришел к выводу, что растворимость самой кремневой кислоты практически не зависит от pH. Повышение содержания кремния в растворах с высоким значением pH
Пента- ® й «• в» ст о 4L о о о о •J. Ю tn _ in чэ "1 <75 <75 <75 <75 ст w • mi ст 'TS '-S щ ю ю X X X X X X ст ст ст ст ст о, у. У, У, У, У,
Тетра- СО СТ w о о о о «9 «9 <75 <75 <75 <75 w 00 «О «в ст '“^А ‘'То in tn in X X X X X сч ст ст ст ст у, у, у, у. у.
Три- о 1-f OS 00 О о О л .Д’ со «о СО GO С/) У оо СО '^О Л НМ мм Н-> tn Um Um Um мм Ст СТ Ст Um у, у, о. о
Ди- ООО вЧ СТ _СТ <0 «в ст iA in XXX СТ СТ у, у, у.
О X О о Q со 00 «в СТ 'Тп ю X X ст ст у, у
Т ип —1 СТ СО 41 + 1 1 1 1 £ £ £ « £ СО СО «о СО СО О е О О О о _е г? <75 е <75 <75 <75 ст > Ст * + <Л 1 1 1 1 е е е е е е Ст СТ СТ СТ ст ст iA ''"То in iA in X X X X X X ст еч ст ст ст ст у, у, у. у, у. у.
14
связано с переходом кремневой кислоты в силикат-ион; этйм объсяняется присутствие в растворе дополнительных количеств кремния.
Растворимость кремневой кислоты существенно повышается в присутствии некоторых оксикислот (например, лимонной, винной [649]), ацетона и других органических веществ [366, 771, 773, 781]. В процессе конденсационной полимеризации кремневой кислоты не происходит изменения pH раствора. На этом основании Александер [636] сделал вывод, что поликремневые кислоты также кислоты слабые. Из пересыщенного раствора кремневой кислоты она не выделяется в мономерном состоянии, а претерпевает процесс конденсационной полимеризации. Первая стадия его может быть выражена уравнением
2Si(OH)4-> (HO)3SiOSi(OH)3 + Н2О.
Дальнейшее взаимодействие образующихся молекул с молекулами Si(OH)4 и между собой дает полимеры с различной длиной силок-санной цепи
Si(OH)4 + (НО)3 SiOSi(OH)3 -+(HO)3SiOSi(OH)2 OSi(OH)3 + Н2О, общего вида Si„O„_1(OH)2n+2.
Образование полимерных продуктов линейного строения, а также форм дегидратированной кремневой кислоты и ее производных при гидролизе и конденсации Si(OR)4 (где R может быть водородом или другим органическим или неорганическим радикалом) впервые описано Бутлеровым в его теории образования полисоединений в минеральной химии. Производные ряда Si„O„_1(OR)2n+2, гдеР=СН3, получены Конрадом, Бехле и Зигнером [870] при гидролизе тетраметоксисилана Si(OCH3)4. Несколько позднее Кингом [861, 862] описаны типы полимерных полиэтоксисилоксанов, представленные в табл. 2. При сопоставлении данных этих ученых с классификацией природных силикатов оказывается, что имеется полная аналогия в структуре эфиров поликремневых кислот и природных силикатов.
Процесс конденсационной полимеризации ортокремневой кислоты и ее производных не ограничивается образованием линейных полимеров. Возможно образование продуктов циклического строения, образование разветвленных цепей и сетчатых пространственных структур. Такие продукты выделены Айлером [832], они получаются также при дегидратации кремневой кислоты в процессе ее аналитического определения.
Реакция конденсационной полимеризации кремневой кислоты катализируется ионами Н+ и ОН-, вследствие чего на кривой зависимости скорости полимеризации от pH среды имеется четко выраженный максимум (рис. 1), соответствующий значению pH около 2. Таким образом, в области pH от 1 до 3 пересыщенный раствор кремневой кислоты обладает максимальной устойчивостью [4, 405].
16
Мйнимум устойчивости кремневой кислоты наблюдается при pH около 6 [676 , 796].
Александер [638] исходя из термодинамических представлений обосновывает превращение монокремневой кислоты через промежуточные полимерные формы до одной из форм двуокиси кремния как переход системы в состояние, отвечающее минимальному запасу свободной энергии (минимальная поверхность). Объяснение механизма полимеризации кремневой кислоты Тредвелл и Виланд [1054] связывают с координационной ненасыщенностью кремния
Рис. 1. Зависимость устойчивости золей кремневой кислоты от pH раствора (т — время образования геля)
в Si(OH)4 по отношению к ионам ОН-. Эти взгляды развиты в работах Вейля и Айлера [4, 830, 831]. ИонОН- имеет размеры, сходные с размерами Р--иона, он способен замещать его в некоторых природных фторсодержащих силикатах и алюмосиликатах (например, в слюдах). Рентгенографическими исследованиями доказано существование 51(ОН)2“-иона в гидратированных щелочных силикатах. Согласно гипотезе Айлера, в слабощелочных и кислых средах координационное число кремния по кислороду возрастает от четырех до шести
Si(OH)4 н- Н2О + ОН- —* [(H2O)Si(OH)5] ,
Si(OH)4 + Н2О + HF —*
H,OV
)Si(OH)
FZ
Образование дисиликат-иона по схеме
НОН но„ 1 .он
НО ; он
но
-,2-
ОН .-ОНч ОН
НО. I X X I Л)Н
;SiC .Si. + 2Н2О
HO-' • X ; ХОН
ОН ''ОН- он
при низком значении pH (от 3 до 5) идет с незначительной скоростью, так как содержание силикат-ионов и гидроксильных ионов в растворе
17
мало. Стадии процесса представляются следующим образом:
НОЧ но/
ОН
+ он~
+ HjO
ОН но. ; хон“ \ I Z ж НО'' I ''ОКОН
он
но, : ,он7 ''ж'
но' 'он, дн
+
он он
но, ,он но, । дн, : ,он ''su--oh—*
''он но' • 'он/ 'он он он
-2ОН~
НО, ZOH
—НО—^Si—О—Si6—ОН + Н+ + Н2о
H0,Z 'он
Таким образом, Вейль [1087] считает, что в присутствии ОН-- и Р--ионов кремневая кислота дает активированный промежуточный комплекс с координационным числом кремния, равным пяти и даже шести, при присоединении молекулы воды.
При более высоких значениях pH присутствие ионов ОН- ускоряет как полимеризацию, так и диспропорционирование полимеров, сопровождающееся образованием полимеров более высокого молекулярного веса и выделением мономера или низкомолекулярных полимеров. Это обстоятельство имеет значение в анализе. Так, например, в случае проведения полимеризации (дегидратации) кремневой кислоты в нейтральной или слабощелочной среде в растворе вследствие диспропорционирования будет оставаться значительное количество низкомолекулярных форм кремневой кислоты. Для низких значений pH диспропорционирования практически не наблюдается; таким образом, ион ОН- ускоряет не только образование, но и разрушение связи Si—О—Si. Присоединение ОН "-иона к группе SiOSi(OH)3 приводит к образованию SiOSi(OH)4; сопутствующее этому процессу перераспределение электронной плотности влечет за собой ослабление связи Si—О—Si и дальнейший ее разрыв.
В зависимости от способа аналитического определения кремния возникает необходимость смещения равновесия полимеризации — деполимеризации:
а) при гравиметрическом определении в форме SiO2 необходимо по возможности полное выделение кремневой кислоты в виде вы-сокополимеризованных форм;
б) при колориметрических и титриметрических определениях нужно сохранить кремневую кислоту в мономерном состоянии или в виде низкомолекулярных форм, легко подвергающихся деполимеризации с повышением pH среды. Установлено, что реакционно
18
способными при необходимости перевода кремневой кислоты в гетерополианион [Si(Mo3O10)4]4- можно считать только мономерную и димерную формы кремневой кислоты и силикат-ионов.
Существование сложного многоступенчатого процесса полимеризации — деполимеризации препятствует быстрому установлению равновесия в системе, что усложняет и затягивает операцию аналитического определения кремния. (Вопросы кинетики полимеризации см. в [781, 791, 795].) Однако это обстоятельство может быть использовано в анализе: потери кремневой кислоты вследствие растворимости ее уменьшаются, так как при обработке осадка кремневой кислоты водными растворами кислот состояние истинного равновесия не достигается.
Деполимеризация кремневой кислоты при повышении pH раствора с образованием силикат-иона идет с заметной скоростью при рН>9,7. При болеее низких значениях pH по существу единственной мономерной формой кремневой кислоты в водных растворах может быть Si(OH)4.
Изучение поведения силикатов щелочных металлов показывает, что при рН<10,5, когда отношение Si:Na становится равным 2:1, образуются агрегаты кремневой кислоты более сложного строения, чем силикат-ион и кремневая кислота. Установлено наличие в растворе дисиликат-иона. Для условного обозначения силикат- и дисиликат-ионов как SiO2—и Si2О2—на основании расчетов Роллера и Эрвина [980] Айлер [4] вывел следующее соотношение:
[si2o52-] [он-]
= 10_'°>34'
Расчеты показывают, что в растворе 1 М относительно SiO2 при pH 13,8 мономерная и димерная формы силикат-иона существуют в равных количествах. При pH 13,5 при установившемся равновесии уже около 97% кремневой кислоты находится в форме дисиликат-иона. Приведенное соотношение не учитывает существования равновесий между моно-, ди- и полисиликат-ионами:
Si2O2- + SiO2-+H2O->Si3O72-+2OH-.
Принимая координационное число кремния по кислороду равным шести, можно написать
2Si(OH)g
-I^-
OH он но, ! дн^ ! дн ';Sk' гон"
йо' ; 'он' ; 'он он он
19
он он он
- , -р- Г ->2- Н(Х i -он- ! ощ ! он
Si2(OH)10J + [Si(OH)6] —';Sif 'ySi;'
но'' ; ''он'' i ''он'' ; ''он он он он
2—
+ 2OH"i
далее в общем виде
[Si(OH)6(Si(OH)4)„ Si(OH)5]2- + [Si(OH)e]2--> ->[Si(OH)6(Si(OH)4)n+1Si(OH)5]2- + 2OH-.
При дальнейшей дегидратации образуются сложные полимерные агрегаты, которые могут существовать в водных растворах продолжительное время. В щелочном растворе образуются устойчивые коллоидные частицы, несущие отрицательный заряд и окруженные ионами Na+, образующими диффузный слой. Со временем размер частиц растет, падает, агрегативная устойчивость. В нейтральной среде поверхность частиц кремневой кислоты, образующихся при дегидратации, практически не заряжена, так как Н+-ионы находятся в виде ОН “-групп, входящих в состав кремневой кислоты или ее полимеров. Однако связь О—Н все же слабее связи Si—О, поэтому поверхность кремневой кислоты имеет слабокислый характер. Полимерные частицы кремневой кислоты в нейтральной и тем более кислой среде быстро агрегируются вследствие того, что они не имеют заряда. Продуктом агрегации частиц является гидратированный гель SiO2 — трехмерная сетка тетраэдров SiO*-.
Следует отметить разницу, которая существует в структуре кремневых кислот, выделенных из кислых и основных растворов: из кислых растворов выделяются волокнистые полимеры или сетчатые, имеющие характер гранул. В слабощелочных растворах получаются коллоидные частицы, дающие при дегидратации порошкообразную кремневую кислоту.
Поведение кремневой кислоты в условиях ее аналитического определения
Поведение кремневой кислоты и образуемых ею ионов в водных растворах имеет большое значение для выполнения аналитического определения кремния. Следует отметить поэтому еще раз несколько важных для аналитика фактов.
1. Низкая растворимость кремневой кислоты и ее способность давать при дегидратации полимерные малорастворимые продукты используются для гравиметрического определения кремния в форме SiO2. Равновесие процесса растворения — осаждения, а также процесса полимеризации—деполимеризации, связанного с ним, 20
проходит через большое число промежуточных стадий. Скорость растворения и деполимеризации низка. В условиях анализа, когда высушенный осадок, представляющий собой смесь поликрем-невых кислот, смачивают кислотой и обрабатывают горячей водой, процесс растворения не приходит к состоянию равновесия. Раствор кремневой кислоты не будет насыщенным, и потери от растворения вследствие этого снижаются. Однако поскольку потери от растворения являются величиной абсолютной, Шелл [1011] рекомендует для уменьшения относительной ошибки определения вследствие растворимости использовать большие навески (не менее 0,5 г, желательно до '2,0 г в анализе силикатов).
Выпаривание фильтрата после отделения основной массы кремневой кислоты позволяет выделить дополнительное ее количество. Однако увеличение числа таких обработок не дает возможности полного выделения кремневой кислоты вследствие ее заметной растворимости, которая, кроме того, повышается при нагревании. Поэтому не имеет смысла при гравиметрическом определении выпаривать раствор больше двух раз, тем более что при длительной обработке возможно значительное выщелачивание кремневой кислоты из посуды и искажение результатов. Истинно растворенная кислота может быть определена в фильтрате колориметрическим методом [188, 836, 1011].
2. Существует разница в свойствах осадка частично дегидратированной кремневой кислоты в зависимости от способа подготовки раствора перед его выпариванием. Аналитик стремится получить плотный гранулированный осадок, который при дальнейшей обработке, прокаливании и взвешивании не будет распыляться. Такой осадок удается получить при разложении анализируемых соединений кислотами сравнительно высокой концентрации. При обработке плавов, получаемых при сплавлении соединений кремния со щелочными плавнями (NaOH, Na2CO3), частично обезвоженная кремневая кислота обычно получается в виде очень тонкодисперсного, объемистого порошка, очень гигроскопичного и легкоподвижного. Вследствие этого потери при обработке осадка более вероятны — работа с такими осадками затруднительна.
3. Полная дегидратация кремневой кислоты достигается при 358°С [726]. Однако гигроскопичность двуокиси кремния, полученной при таком прокаливании, очень высока, и получение хороших результатов невозможно. Понижение гигроскопичности двуокиси кремния достигается прокаливанием при высоких температурах, когда заметно сокращается ее поверхность. Гранулированная двуокись кремния менее гигроскопична, чем порошкообразная. Прокаливание желательно заканчивать при температуре около 1200°С.
4. Обладая высокоразвитой поверхностью, гель кремневой кислоты адсорбирует катионы, находящиеся в растворе. Дальнейшим промыванием осадка они частично удаляются. Однако прокаленный осадок двуокиси кремния все же содержит заметные их количества.
21
Искажение результатов может быть устранено при дальнейшей обработке осадка смесью фтористоводородной и серной кислот. Эта операция не дает возможности освободиться от ошибок в том случае, если осадок плохо отмыт от ионов щелочных металлов. При прокаливании сульфатов щелочных металлов они не переходят в окислы, как это происходит с сульфатами железа, алюминия и других элементов, но остаются в форме сульфатов. Вследствие этого можно получить заниженные значения содержания кремния.
5. Кремневая кислота очень слабая. Поэтому она не может быть с достаточной точностью оттитрована растворами щелочей в водных растворах. Кроме того, наличие в растворах, содержащих кремний, даже при значениях pH около 13,5 смеси моно- и диси-ликат-ионов затрудняет использование для определения кремневой кислоты методов, основанных на реакциях осаждения.
6. При колориметрическом определении кремния в виде окисленных или в осстановленных форм кремнемолибденового или кремне-ванадиймолибденового комплексов, а также при осаждении кремния в виде комплексных продуктов присоединения органическими основаниями можно получить правильные результаты только в том случае, если в анализируемом растворе весь кремний содержится в виде мономерной кремневой кислоты, силикат- и дисиликат-ионов. При наличии форм кремния, содержащих более двух атомов Si в молекуле, количественный переход кремния в гетерополикислоту, например по реакции
Na4SiO4+12 (NH4)2MoO4 + 14H2SO4-+
->Н4 [Si (MosO10)4] + 12 (NH4)2 SO4 + 2Na2SO4 + I2H2O,
невозможен, так как скорость деполимеризации низка. При подготовке растворов кремневой кислоты к переводу кремния в гетерополианион с целью дальнейшего колориметрирования или осаждения обычно прибегают к длительной обработке раствора щелочными реагентами, а также сплавлению со щелочами или карбонатами щелочных металлов. При необходимости сколько-нибудь длительного сохранения кремния в мономерном состоянии в растворе в кислой среде следует помнить, что Si(OH)4 наиболее устойчива в интервале pH 1—3.
7. Для перевода кремния в комплексный ион SiFg— или кремнефтористоводородную кислоту H2SiF6 могут быть использованы любые формы растворенной кремневой кислоты. Кремнефторид-ион гидролитически неустойчив, поэтому его получить можно только в кислой среде. Обработка аморфной кремневой кислоты HF также позволя ет количественно перевести кремний в кремнефтористоводородную кислоту. На модификации двуокиси кремния, обладающие высокой плотностью (например, кварц, коэсит и др.), фтористоводородная кислота практически не действует.
8. Переход кремневой кислоты в силикат-ионы, принимая координационное число кремния по кислороду в щелочных средах 22
равным шести, может быть представлен уравнениями
Si (ОН)4 + Н2О + ОН" -> [H2OSi (ОН)6]
[H2OSi (ОН)Б]" + ОН" -> [Si (ОН)в]2- + Н2О.
Достоверно установлено существование только двух простых силикат-ионов (е отношением SiO2:Na2O=l:l и 2:1) [806, 839]. С понижением значения pH раствора щелочного силиката могут образоваться агрегаты гидратированного кремнезема высокого молекулярного веса. Высказано предположение, что полисили-катный ион имеет сходство с гетерополикислотным анионом, например [SiO4Si12O24(OH)12]ie- [652].
Реакции монокремневой кислоты
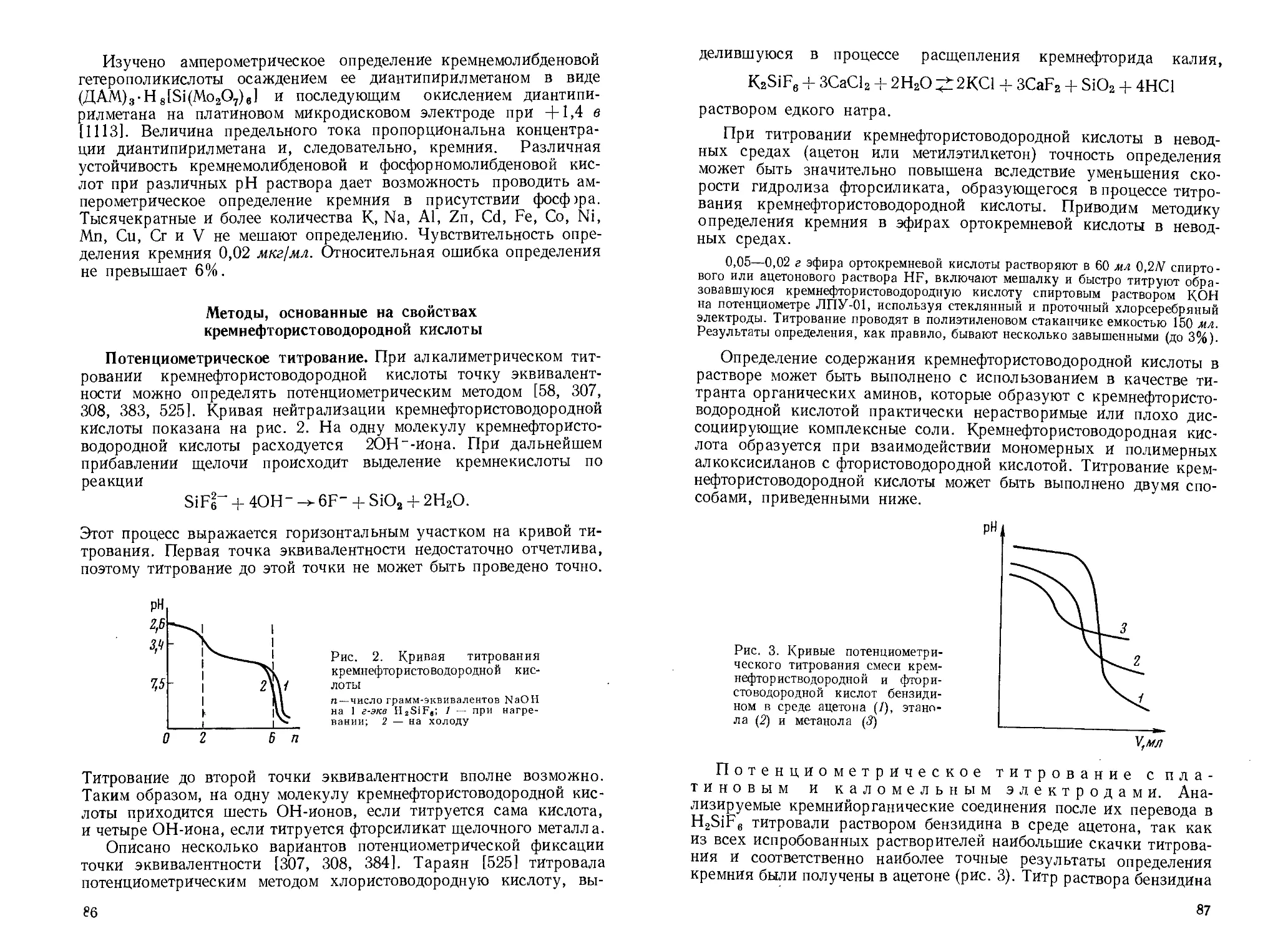
Реакция нейтрализации. Реакция нейтрализации кремневой кислоты аналитического применения не имеет: очень слабую, практически недиссоциированную кислоту в водных растворах не удается оттитровать растворами оснований, скачок титрования недостаточно резок [1097].
Реакции осаждения. Кремневая кислота и силикат-ионы могут быть осаждены действием солей бария, цинка, кобальта, кадмия, свинца [74, 312, 420, 846] (см. также главу 2). В виде силикатов бария, кадмия и цинка кремний обнаруживают и отделяют в качественном и количественном анализе. Для количественного определения кремния используют реакции образования силикатов кобальта [846], свинца [74, 420] и бария [525, 1103].
Широкого использования в анализе реакции осаждения малорастворимых силикатов не находят.
Реакции комплексообразования [165]. Реакции образования гетерополи соединений [16, 268, 406, 407, 436, 508, 720, 806, 842, 843, 859, 1034, 1035, 1072, 1098]. Образование гетерополисоединений с дальнейшим аналитическим применением их для качественного и количественного гравиметрического, оксидиметрического или спектрофотометрического определения кремния может быть проведено только исходя из низкомолекулярных форм кремневой кислоты (мономера и димера) и растворов молибдатов и ванадатов в средах с низким значением pH. Эту реакцию кремневой кислоты и силикат-ионов следует считать одной из самых важных. Возможности ее аналитического использования не исчерпаны. Для аналитического определения кремния чаще всего применяют превращение мономерной кремневой кислоты в кремне- 12-молибденовую кислоту или кремневанадиймолибденовую кислоту. Условия выполнения реакции и ее химизм подробно описаны в главе 7. См. также использование этой реакции для определения кремневой кислоты с помощью кинетических измерений [880].
Образование кремнефтористоводородной кислоты и кремнефторидов. Для образования кремнефтористоводородной кислоты H2SiFe и кремнефторидов мо-
23
гут быть использованы монокремневая и поликремневые кислоты. Поэтому эта реакция не может быть рассмотрена только как характерная реакция монокремневой кислоты. Кремнефтористоводородная кислота и кремнефториды образуются при действии фтористоводородной кислоты на кремневую кислоту и ее полимеры, силикаты, некоторые кремнийорганические соединения [292, 572, 852, 896, 989] фтористоводородной кислоты или фторидов при низких значениях pH растворов. При высоких значениях pH гекса-фторидный комплекс кремния гидролитически неустойчив и разлагается с образованием кремневой кислоты и фторидов (см. главу 6).
Образование
Образование пирокатехинкремневого гт - НО^\
комплекса. При действии пирокатехина на рас-
на рас-
творы, содержащие силикат-ион, образуется комплексная пиро-
катехинкремневая кислота Н2 Si
, которая мо-
жет быть оттитрована щелочью [655, 982].
Реакция конденсационной полимеризации. Эта реакция, уже рассмотренная нами (см. стр. 16), применяется для выделения и отделения кремневой кислоты. Реакция идет ступенчато, продукты ее обладают различным молекулярным весом. Длина цепи полимергомологов и строение сетчатых и пространственных решеток полимеров во многом зависят от pH раствора, скорости выпаривания, присутствия других элементов. Процесс полимеризации в некоторой степени обратим [149, 187, 210, 211, 643, 767, 931, 1000, 1001, 1092, 1097], поэтому полное выделение кремневой кислоты из раствора при переводе ее в полимерное состояние в условиях анализа не достигается [25, 38, 149, 161, 188, 601, 660, 1011]. Реакция конденсационной полимеризации кремневой кислоты при гравиметрическом определении кремния завершается полной дегидратацией кремневой кислоты с образованием полимера (SiO2)„. Дегидратация завершается при 358°С [727]. Однако осадок гигроскопичен. Длительное прокаливание с частичным образованием кристобалита снижает гигроскопичность. В форме SiO2 кремневую кислоту взвешивают. Потери при дальнейшем прокаливании SiO2 в отсутствие влаги ничтожны вплоть до точки плавления (1710°С) [1025].
Реакции кремнефтористоводородной кислоты и кремнефторид-ионов
Реакции нейтрализации. Комплексная кремнефтористоводородная кислота H2SiF6 проявляет свойства сильной кислоты. Взаимодействие кремнефтористоводородной кислоты с основаниями протекает в две стадии
H2SiF6 + 2NaOH -> Na2SiF6 + 2Н2О;
Na2SiFe + 4NaOH 6NaF + Si (OH)4.
(1)
(2)
24
Используя гидролитическую неустойчивость кремнефторид-иона, определяют кремний в кремнефторидах по реакции (2). Количественный перевод двуокиси кремния в кремнефтористоводородную кислоту по реакции SiO2+6HF->H2SiF6—2Н2О осуществим при достаточном избытке HF и охлаждении [572, 1011]. В результате реакции получается смесь HF hH2S1F6. При титровании ее щелочью на процесс нейтрализации H2SiF6 накладывается в некоторой степени гидролиз иона SiF|“и нейтрализация избытка HF. Кроме того, адсорбционные свойства образующегося осадка (SiO2)„(H2O)m делают нечетким переход окраски индикатора. Использование неводных сред [292, 298, 304, 382, 384, 852], а также отделение кремнефтористоводородной кислоты от фтористоводородной (обычно осаждением в виде K2SiF6 с дальнейшим титрованием полученного кремнефторида) дает возможность количественно определять кремний в форме кремнефторид-иона методом нейтрализации. Метод предложен первоначально Штолба [1031, 1032] и впоследствии уточнен и видоизменен различными авторами (см. главу 6, а также [574]).
Реакции осаждения. Для' обнаружения кремния в форме крем-нефторид-ионов последние осаждают ионами бария и натрия.
Действием солей калия в присутствии спиртов кремнефторид-ионы могут быть осаждены количественно [506, 507, 772, 954, 993, 998, 1018, 1031, 1032]. Осаждением бензидином
H2N </ NH2 (и другими основаниями [292,
300, 302, 383]) с образованием продуктов присоедине-
ния, например C12H12N2-H2SiF6, кремнефтористоводородную кислоту количественно отделяют от фтористоводородной кислоты. Полученный осадок может быть взвешен после высушивания при
гравиметрическом определении или разложен гидролитически — для определения кремнефтористоводородной кислоты методом нейтрализации. Осаждение кремнефтористоводородной кислоты аминами см. в [307, 308].
При осаждении кремнефтористоводородной кислоты титрованным раствором бензидина и других органических оснований [178, 307, 308] точку эквивалентности устанавливают потенциометрическим и кондуктометрическим методами.
Реакции термического разложения. Кремнефтористоводородная кислота при нагревании разлагается с образованием фторида кремния и фтористоводородной кислоты. В виде паров H2SiF6 не существует. Этот процесс с использованием дегидратирующих агентов (обычно H2SO4) применяют для косвенного определения кремния в веществах, содержащих двуокись кремния, кремнефториды, и в осадках кремневой кислоты после их прокаливания:
25
Реакции кремне-12-молибденовой кислоты
Реакции нейтрализации и гидролитического расщепления. Кремнемолибденовую кислоту изображают как H4[Si(Mo3O10)4] или H8[Si(Mo2O7)6], полагая ее четырех-или восьмиосновной и рассматривая ее как производное H4SiO4 или H8SiO6 (иначе H2Si(OH)e). Несмотря на то что для определения основности гетерополикислот, в том числе и кремнемолибденовой, выполнено много работ, полной ясности в этом вопросе пока нет. Иногда кремнемолибденовую кислоту изображают как H4[Si(Mo3O10)4]-2H2O, что суммарно соответствует формуле H8[Si(Mo2O7)e],
Можно считать точно установленной возможность количественного осаждения и титрования кремнемолибденовой кислоты как кислоты четырехосновной. Дальнейшая нейтрализация ее связана с большими трудностями, процесс нейтрализации сопровождается расщеплением гетерополианиона. Большинство аналитиков в последнее время принимают формулу кремнемолибденовой кислоты H4[Si(MosO10)4] [404, 601, 843, 1011]. Однако при изображении смешанных гетерополикислот, например кремневанадиймолиб-деновой Н 8[Si(Mo2O7)5(V2O6)], исходят все же из формулы H8[Si(Mo2O7)6], Наиболее основательные доказательства восьмиосновности кремнемолибденовой кислоты приведены в монографии Никитиной [404].
Кремнемолибденовая кислота реагирует с неорганическими (и некоторыми органическими) основаниями по уравнениям
Н4 [Si (Мо3О10)4] + 4NaOH -+Na4 [Si (MosO10)4] + 4H2O; (1)
H4[Si(Mo3O10)4] + 24NaOH-^I2Na2MoO4 + H4SiO4+ I2H2O. (2)
Реакция (2) используется для определения кремния титрованием по остатку [1043]. Как четырехосновная кремне- 12-молибденовая кислота может быть оттитрована органическими основаниями с кондуктометрической фиксацией точки эквивалентности [386, 387]. Титрование идет в присутствии значительных количеств других кислот.
Реакции осаждения. Кремнемолибденовая кислота может быть осаждена действием солей рубидия, ртути(1), таллия(1), цезия [426, 1011] в форме четырехзамещенной соли. Реакция используется для качественного обнаружения кремния. Многие органические основания (например, хинолин, пиридин, 8-ок-сихинолин), а также алкалоиды (например, цинхонин, кокаин и др.) осаждают кремнемолибденовую кислоту. Форма осаждения с хинолином — 4C9H4N-H4SiMo12O40, с 8-оксихино-лином — 4C9H7ON-H4SiMo12O40. Реакции используются для определения кремния, кремнемолибденовой кислоты и других гетерополикислот гравиметрическим (см. главу 5) и бромид-броматным (см. главу 6 ) методами. Эти реакции служат и для количественного определения органических оснований с помощью гетерополи кислот [404, 843].
26
Реакции окисления-восстановления. Молибден, входящий в состав кремне-12-молибденовой кислоты, может быть восстановлен. Образующиеся при этом продукты восстановления имеют весьма интенсивную окраску, что используется для спектрофотометрического определения кремния (см. главу 7). Восстановление молибдена в кремне-12-молибденовой гетерополикислоте может быть использовано и для косвенного комплексонометрического определения кремния, содержащегося в гетерополикислоте. Кроме кремне-12-молибденовой кислоты для дальнейшего определения в форме восстановленного гетерополикомплекса используют также крем-неванадиймолибдеиовую кислоту H8[Si(Mo2O7)6(V2O6)]-xH2O [47, 232].
Восстановление кремнемолибденовой кислоты действием иоди-да калия с одновременным образованием двух окрашенных продуктов реакции (восстановленной формы гетерополикислоты и иода) было предложено для дальнейшего колориметрического определения кремния [123]. Эта же реакция предложена для йодометрического определения кремния. Другие редокс-методы см. в [1042].
Бромирование 8-рксихинолина, входящего в состав комплекса 4C9H7ON-H4SiMo12O40, служит основой бромид-броматного метода определения кремния в его соединениях [69, 134].
Восстановленная форма кремнемолибденовой кислоты может быть осаждена органическими основаниями, например хинолином, и кремний определен гравиметрически [564].
Реакции термического разложения. Гетерополикислоты и продукты их взаимодействия с органическими основаниями при нагревании в определенных пределах температур имеют постоянный состав, что служит для гравиметрических определений. Так, например, соединение 4C9H7ON-(H4SiMo12O40)-xH2O при нагревании может иметь устойчивые весовые формы:
4C9H7ON-SiO2-12МоО3-2Н2О; SiMo12O38-4C9H7ON и SiO2-I2MoO3 (см. главу 3).
Для количественного определения кремния используют физические свойства его соединений и сплавов. (Подробно см. главу 8.)
Глава 2
МЕТОДЫ качественного ОБНАРУЖЕНИЯ кремния
Для качественного обнаружения кремния используют химические и физико-химические методы. Из физических методов преимущественно применяют эмиссионный спектральный метод анализа. Для обнаружения кремния эмиссионным спектральным методом используют линии кремния 2506,90; 2516,12; 2528,52; 2881,59 А. Приходится учитывать возможность наложения на линии кремния близких по длине волн линий других элементов или линий основного элемента пробы. Одновременное наложение для всей группы линий маловероятно, поэтому практически всегда можно выбрать условия, в которых кремний может быть обнаружен спектрально с достаточной надежностью хотя бы по одной из «последних» линий. Библиография приведена в [64, 99, 243, 339, 920, 968], а также в главе 7. С помощью стилоскопа кремний обнаруживают при искровом возбуждении по линии 6346,69 Айв дуге (с некоторыми затруднениями) по линии 3905,52 А при содержании —1% [253].
Химическими методами кремний обнаруживают в растворах или реже — реакциями, выполняемыми при сплавлении. В водных растворах кремний может присутствовать в виде Si(OH) 4, а также ионов кремневой, кремнефтористоводородной, кремнемолибденовой и других гетерополикислот при соответствующих значениях pH раствора. Известны также растворимые в воде комплесные кислоты с двухатомными фенолами [655, 982].
Ввиду того что из неорганических соединений кремния значительно растворимы в воде только силикаты щелочных металлов, гетерополисоединения, кремнефтористоводородная кислота и кремнефториды, значение которых в общем не так уж велико, определению кремния обычно предшествуют операции разложения его соединений: сплавление или кипячение с различными щелочными реагентами, кислотами и окислительными смесями. Эти же операции применяют и при качественном определении кремния в кремнийорганических соединениях. Для анализа сооединений крем-28
ния, разлагаемых кислотами, к сплавлению прибегают редко, так как кремний в них может быть обнаружен по образованию полимерной кремневой кислоты, которая сравнительно легко идентифицируется.
Подготовка вещества для качественного обнаружения кремния описана в соответствующих работах и руководствах, например [72, 275, 481, 513, 517, 518, 552, 624]. Специфические особенности анализа кремнийорганических соединений отражены в работах [85, 178, 295—297, 612]. Качественное определение имеет некоторые особенности также и в том случае, если обнаружить кремний необходимо в готовых изделиях. В этом случае используют приемы бесстружкового растворения действием на поверхность анализируемого объекта растворами кислот и щелочей [316, 411, 509, 512, 624].
Как общий прием разложения соединений кремния, нерастворимых в воде и не разлагаемых кислотами, используется сплавление или спекание с карбонатами, щелочами, окислами и их смесями. При сплавлении или спекании анализируемые вещества переходят в соединения, растворимые в воде или разлагаемые кислотами. Для сплавления и спекания используют Na2CO3, К2СО3, NaOH, Na2O2, PbO, NH4Cl+CaCO3 [72, 513, 552]. Сплавление производят в платиновых, серебряных, никелевых тиглях, а также на кусочке древесного угля и в петле платиновой проволоки [513].
Полученный плав по охлаждении обычно разлагают водой или водными растворами кислот или последовательно водой и кислотами. Образования характерного осадка гидратированной кремневой кислоты при разложении соединений кремния или продуктов их сплавления или спекания кислотами часто бывает совершенно достаточно для качественного определения больших количеств кремния. Для получения более точных результатов, а также для обнаружения малых количеств кремния рекомендовано несколько реакций, большинство из которых описано в руководствах [72, 624].
РЕАКЦИИ СИЛИ КАТ-ИОНА
Образование поликремневых кислот. Действие разбавленных растворов кислот. При прибавлении разбавленных растворов кислот — хлористоводородной, серной, азотной и других — к растворам силикатов щелочных металлов до создания в растворе pH 4—6 выделяется студенистый или хлопьевидный осадок кремневой кислоты. Реакцию применяют для обнаружения и отделения кремневой кислоты.
Действие аммонийных солей. При действии растворов аммонийных солей сильных кислот на растворы силикатов щелочных металлов при кипячении также выделяется осадок кремневой кислоты.
Образование силиката цинка. При действии аммиаката цинка на растворы силикатов щелочных металлов образуется белый осадок силиката цинка, менее растворимый, чем кремневая кислота.
29
Образование силиката кадмия. При действии гидроокиси кадмия кремневая кислота осаждается в форме силиката кадмия.
Образование силиката бария (кальция). При действии хлорида бария (кальция) на растворы силикатов щелочных металлов образуется малорастворимый осадок силиката бария (кальция). Реакция не специфична, поэтому ее используют для отделения сили-кат-иона вместе с другими анионами.
Образование адсорбционных соединений гидратированной кремневой кислоты с растворами некоторых основных органических красителей. При действии основных красителей на аморфную кремневую кислоту происходит их адсорбция, и они прочно удерживаются на поверхности SiO2. Для выполнения этой реакции предложен фуксин [72, 624], малахитовый зеленый [72, 624, 970], метиленовый синий [72, 624, 970], сафранин, родамин [72], метиловый фиолетовый [970]. Реакция является проверочной после получения осадка кремневой кислоты, который сам по себе может быть плохо заметным.
Осадок кремневой кислоты на фильтре или на предметном стекле обливают разбавленным (обычно 1%-ным) водным раствором красителя и после нескольких минут выдержки осторожно смывают избыток реактива водой. В присутствии гидратированной кремневой кислоты осадок приобретает яркую окраску.
Реакция образования кремневой кислоты при сплавлении. Погружая нагретую платиновую проволочку с петлей в твердый NH4NaHPO4 и нагревая ее, получают в петле «перл» метафосфата натрия. Полученным в петле еще теплым шариком прикасаются к порошку исследуемого вещества, нагревают и сплавляют его. В шарике появляется характерное волокнистое помутнение — «скелет» SiO2.
РЕАКЦИИ КРЕМНЕФТОРИД-ИОНА
Реакции могут быть использованы для обнаружения кремнефторидов, фтора и кремния. Однако для обнаружения кремния, если он не находится в форме кремнефторид-иона, его нужно сначала перевести в ион [SiFe]2-. Для этого соединения кремния обрабатывают фтористоводородной кислотой или фторидами в кислой среде. В присутствии водоотнимающих средств, обычно H2SO4, кремний переходит в летучий фторид кремния, который улавливают водой или водными растворами реактивов и идентифицируют кремний в виде кремнефторид-ионов или кремневой кислоты, образующихся по реакции 3SiF4+3H2O->2H2SiFe+H2SiO3.
Этим методом кремний может быть отделен от всех примесей, мешающих его определению, за исключением бора, который должен быть предварительно удален выпариванием с метанолом и серной кислотой [734].
Для выделения SiF4 пробу вещества или продукта сплавления обрабатывают в платиновой, свинцовой или тефлоновой посуде (тиг-30
ле, ложке, чашке), выделившийся газ улавливают каплей воды или водного раствора реактива, помещенной на крышку платинового тигля, на предметное стекло, покрытое слоем вазелина, коллодия или асфальтового лака, или на пластинку из органического стекла.
Образование кристаллического осадка кремнефторида натрия [72, 275, 623]. Каплю исследуемого раствора помещают на предметное стекло и вносят маленький кристалл NaCl. В присутствии кремнефторид-иона под микроскопом хорошо различимы бледно-розовые кристаллы кремнефторида натрия гексагональной формы в виде шестигранников и розеток [721.
Образование Na2SiF6 можно наблюдать, если к 1—2 мг вещества, в котором предполагается наличие кремния, на защищенном или органическом стекле прибавить последовательно каплю 5%-ного раствора NH4F в 10%-ной НС1 (или каплю HF и кристаллик NaCl). Под микроскопом видны кристаллы Na2SiF6. Этой реакцией можно открыть силикат-ион и кремнефторид-ионы в присутствии сульфат-ионов.
Образование кристаллического осадка кремнефторида бария. Реакцию выполняют с раствором, содержащим кремнефторид-ионы [72]. При добавлении соли бария образуются кристаллы ромбоэдрической формы — иголочки и удлиненные призмы.
Образование кристаллического осадка кремнефторида хинина (кодеина, стоваина, стрихнина) [275, 983]. К I—2 каплям раствора после подсушивания на предметном стекле прибавляют каплю 1%-ного водного раствора сульфата хинина. При этом образуются иглы кремнефторида хинина. Образование кремнефторидов органических оснований см. в [178].
Образование кристаллического осадка кремнефторида бензидина [301, 303]. К капле раствора исследуемого вещества на предметном стекле прибавляют каплю 1%-ного раствора бензидина в метаноле или ацетоне. Образуются характерные иглы кремнефторида бензидина. Другие микрореакции см. в [624, 958, 984].
Образование осадка ]Co(NH3)5C1] SiF6. При действии аммиаката кобальта образуется кристаллический осадок [Со (NH3)5Cl]SiF6 в виде мелких прямоугольников. Открытие ионов SiFjT в присутствии фторид-,тетрафторборат- и сульфат-ионов см. в [743].
Образование кремневой кислоты в капле воды [72, 749]. Твердое вещество или осадок, содержащий кремний, смешивают в ступке с тройным количеством KNaCO3 и сплавляют. Охлажденный сплав выщелачивают водой, разлагают серной кислотой, выпаривают досуха. Прибавляют фторид кальция, немного окиси магния и серной кислоты до образования кашицы. Выделяющийся SiF4 улавливают каплей воды на крышке платинового тигля, покрытой асфальтовым лаком. Капля воды мутнеет. Каплю воды можно помещать на стеклянную палочку или пластинку, покрытую лаком, или в ушко платиновой проволочки [6241. Помутнение капли объясняется образованием осадка кремневой кислоты по реакции
3SiF4 + ЗН2О -> 2H2SiFe + H2SiOs.
31
РЕАКЦИИ КРЕМНЕМОЛИБДАТ-ИОНА
Для обнаружения кремния в виде кремнемолибдат-ионов [Si(MosO10)4]4- и продуктов их восстановления кремний необходимо перевести в эти ионы. Это возможно в том случае, если кремний присутствует в истинно растворенном состоянии, т. е. в форме силикат-ионов. Для использования растворов кремнефторидов необходимо предварительное удаление или связывание фторид-ионов, что достигается прибавлением солей алюминия или соединений бора (см. главу 5).
Образование кремнемолибденовой кислоты и восстановление ее хлоридом олова (II) [949]. 1—2 капли щелочного раствора, полученного при растворении сплава исследуемого соединения с карбонатами натрия или калия, помещают в микротигель или на капельную пластинку, прибавляют 2 капли 10%-кого водного раствора (NH4)2MoO4 и 4А СН8СООН до кислой реакции. Затем сюда же прибавляют 3 капли 1%-ного раствора SnCl2 и избыток раствора NaOH. В присутствии кремния образуется синее окрашивание. Определению мешают ионы РО^- и AsO^-. Кремний может быть отделен от них в форме SiF4.
Образование кремнемолибденовой кислоты и восстановление ее бензидином [513, 748, 750, 849]. Метод основан на окислении бензидина до продукта хиноидного строения синего цвета кремнемолибденовой кислотой, одновременно восстанавливающейся до молибденовой сини. Реакция может быть выполнена в микротигле или на фильтровальной бумаге. В последнем случае она менее чувствительна.
Вариант 1.В микротигель помещают каплю слабокислого исследуемого раствора, каплю молибдата аммония [100 мл Н2О, 5 г (NH4)2MoO4, 35 мл 6N HNOS], нагревают до начала кипения и затем охлаждают. К охлажденному раствору прибавляют каплю 0,5%-ного раствора бензидина в 10%-ной СН8СООН и несколько капель насыщенного раствора ацетата натрия. Синее окрашивание раствора указывает на присутствие кремневой кислоты.
Вариант 2. На фильтровальную бумагу помещают каплю исследуемого раствора, каплю раствора (NH4)2MoO4 (см. вариант 1). Бумагу слегка нагревают, помещают на пятно каплю 0,5%-ного раствора бензидина и затем держат в парах аммиака. Появление синего окрашивания указывает на присутствие кремневой кислоты.
Обнаружение кремневой кислоты в присутствии фосфорной кислоты (250-кратный избыток последней) этим методом предусматривает предварительное удаление больших количеств фосфорной кислоты в виде фосфорномолибдата аммония. В фильтрате после отделения осадка открывают кремневую кислоту реакцией с бензидином, разрушая небольшие количества неосажденной фосфорной кислоты щавелевой кислотой. Обнаружение кремния в водах см. в [316], в растворах щелочей — в [316,552]. Реакцию осаждения крем-32
немолибденовой кислоты для обнаружения кремния обычно не используют. Открытие SiO*~ в присутствии РО^- методом кольцевой бани см. в [776].
ИДЕНТИФИКАЦИЯ НЕКОТОРЫХ ВЕЩЕСТВ, СОДЕРЖАЩИХ КРЕМНИЙ
Кроме обнаружения кремния с применением разложения сплавлением и обработки кислотами описаны способы надежной или ориентировочной идентификации веществ, содержащих кремний, — природных и искусственных силикатов, сплавов и т. п. [624, 664, 774]. Идентификация веществ на основании одних химических реакций не всегда бывает надежной, поэтому ее обычно сопровождают определением физических констант — плотности, коэффициента преломления и др. Основные сведения по этому вопросу имеются в руководствах и монографиях [98, 316, 363, 394, 513, 552, 624, 728, 786, 799, 1021].
Обнаружение свободной аморфной кремневой кислоты. Беренс и Клей [72] рекомендуют для обнаружения аморфной кремневой кислоты в шлифах минералов и в порошкообразных материалах использовать адсорбцию красителей, например метиленового синего. Та же реакция рекомендована для ориентировочного обнаружения групп минералов после травления шлифа концентрированными растворами хлористоводородной и серной кислот и смывания кислоты водой. Такие минералы, как ортоклаз, альбит, роговая обманка, эпидот, гранат, не дают окрашивания. Лабрадорит, лейцит, оливин по краям шлифа окрашиваются сильнее, чем в центре; серпентин, хлорит, слюды, тальк, стеатит окрашиваются и без травления.
Имеются данные по качественному исследованию стекол и эмалей [72, 692]. Реакция с метиленовым синим или с сафранином позволяет обнаруживать кремневую кислоту в присутствии окисей кальция и алюминия, которые не окрашиваются этими красителями. Много данных по идентификации кремнесодержащих веществ приведено в монографиях [227, 316, 624]. См. также [967].
Отличие аморфной кремневой кислоты от ее кристаллических разновидностей [316, 552, 596, 745, 747, 759, 792, 1059]. В ячейке капельной пластинки или микротигле смешивают 1—5 мг исследуемого вещества с 1—2 каплями реактива (насыщенный раствор све-жеосажденного хромата серебра в 6 N растворе аммиака). В присутствии аморфной кремнекислоты немедленно выделяется красный осадок хромата серебра [316].
Обнаружение силикатов, разлагаемых кислотами, действием насыщенного раствора диметилглиоксимата никеля [316, 552, 596, 745]. В ячейке капельной пластинки смешивают 1—5 мг мелкорас-тертой пробы минерала и 1—2 капли реактива (2—3 г NiSO4-• 7Н2О растворяют в 300 мл воды, смешивают с насыщенным раствором диметилглиоксима в этаноле и через 30 мин фильтруют).
2 Аналитическая химия кремния
33
Если силикат разлагается кислотами, масса через несколько минут окрашивается в яр ко-алый цвет.
Приведены также реакции, которыми можно отличить тальк от каолина 1316, 747], кварц от кальцита [316, 747] и др. [227].
Реакции комплексов хромата серебра с аммиаком или этилендиамином используются для обнаружения кислотной или основной природы вещества [316, 596, 745, 746].
Маркировка сплавов, содержащих кремний. Для качественного обнаружения сплавов используют обычные методы работы, основанные на растворении или разложении пробы вещества, а также бес-стружковые методы, с тем чтобы не разрушать исследуемое изделие [410, 411, 509, 521]. Для растворения или разложения изделия с поверхности наносят каплю или порцию реактива и изучают состав полученного раствора и характер изменения поверхности изделия. Если требуется поместить несколько капель растворителя, то делают это с помощью лунки из парафина [411].
Обработка поверхности сплава обычно состоит в очистке ее наждачной бумагой. Подробные сведения по маркировке сплавов и обнаружению кремния в сплавах см. в [72, 261, 316, 624].
Качественное обнаружение кремния в кремнийорганических соединениях. Большинство реакций разработано Крешковым, Борк, Шемятенковой и обобщено в монографиях и работах [85, 178, 295—297, 612].
Для выполнения реакций на кремний в кремнийорганических соединениях прежде всего проводят минерализацию кремнийорга-нического соединения, а затем выполняют обычные ранее описанные реакции обнаружения кремния.
Минерализация может быть выполнена путем осторожного озоления [1027], гидролитического расщепления кремнийорганических соединений или сплавления. Тетрафункциональные и некоторые трифункциональные кремнийорганические соединения, например p-хлорзамещенные алкилтрихлорсиланы, легко разлагаются на холоду или при небольшом нагревании их с водными растворами кислот или щелочей с образованием кремневой кислоты. Для определения кремния в тетраалкоксисиланах, например тетраметоксисилане [294], несколько капель исследуемого соединения осторожно нагревают и выпаривают досуха с 0,5 мл разбавленной (1:1) НС1. Слегка прокаленный осадок смывают водой на фильтр, омачивают 0,1%-ным раствором метиленового синего в 10%-ной СН8СООН и затем снова промывают 1—2 раза водой. В присутствии кремневой кислоты осадок окрашивается в интенсивно-синий цвет.
Для разложения соединений, которые гидролизуются с большим трудом, гидролиз проводят 50%-ным раствором NaOH в серебряном тигле на водяной бане; смесь разбавляют водой, подкисляют НС1, выпаривают, слегка прокаливают и далее повторяют операции открытия кремния с метиленовым синим.
Для разложения кремнийорганических соединений применяют и другие гидролизующие агенты, окислители, окислительные 34
смеси: хлорную кислоту, олеум, смесь хромового ангидрида и йодата калия в среде серной и фосфорной кислот, смесь концентрированных серной и азотной кислот, смесь серной кислоты с перманганатом и др. [296, 297, 962].
Для обнаружения кремния в нелетучих кремнийорганических соединениях используют методы минерализации, основанные на сплавлении. Пробу кремнийорганического соединения смешивают с 5—6-кратным количеством смеси карбоната и перекиси натрия и осторожно нагревают в платиновом или никелевом тигле, постепенно увеличивая нагрев до получения однородного плава. Плав обрабатывают разбавленной НС1 (1:1), переносят содержимое тигля в фарфоровую чашку, выпаривают досуха, смачивают осадок разбавленной НС1 и водой, фильтруют и после промывания открывают кремний реакцией с метиленовым синим или другими красителями основного характера.
35
Глава 3
МЕТОДЫ ОТДЕЛЕНИЯ КРЕМНИЯ ОТ СОПУТСТВУЮЩИХ ЭЛЕМЕНТОВ
Для отделения кремния используют реакции осаждения, фракционного растворения, отгонку соединений кремния или мешающих элементов, электролиз, ионный обмен, экстракцию.
Обзоры литературы по методам отделения имеются в работах [16, 185, 268, 372, 404, 601, 624, 737, 786, 843, 10111.
МЕТОДЫ ОСАЖДЕНИЯ
Осаждение в форме /zSiO2zraH2O. Для отделения кремния от сопутствующих элементов в аналитической практике чаще всего используется осаждение в виде гидратированной полимерной кремневой кислоты. Метод основан на низкой растворимости в воде как самой кремневой кислоты, так и ее полимеров, а также на малой скорости деполимеризации и растворения при обработке водными растворами кислот осадка дегидратированной кремневой кислоты. Состояние равновесия процесса перехода кремневой кислоты из осадка в раствор в условиях анализа не достигается, что позволяет несколькими последовательно проводимыми операциями дегидратации, растворения примесей и отделения кремневой кислоты фильтрованием выделить кремневую кислоту из раствора почти полностью. Полученный осадок в большей или меньшей степени загрязнен со-осажденными примесями, которые при прокаливании не удаляются или могут быть удалены. Методы осаждения кремневой кислоты дегидратацией ее кислотами и другими реактивами рассмотрены в главе 5.
Во время дегидратации кремневой кислоты выпариванием с кислотами происходит отделение ее от некоторых элементов. При выпаривании с хлористоводородной кислотой полностью удаляются германий и мышьяк [615]. При пропускании тока сухого хлористого водорода в трубчатой печи при 700° С над осадком кремневой кислоты, загрязненной примесями соединений сурьмы, мышьяка, ртути, олова, хрома, висмута, германия, молибдена, ванадия, скан
36
дия, эти элементы удаляются в виде хлоридов [817]. Метод используется в случае анализа гетерополикислот [392, 404], стали [764].
Выпаривание с хлорной кислотой позволяет выделить сурьму, ниобий, тантал, олово и вольфрам [1011], со смесью хлористоводородной и хлорной кислот — хром в виде СгО2С12 [817].
Для уменьшения соосаждения ионов железа(Ш) с кремневой кислотой применяют комплексон III [365]. Соосажденную вольфрамовую кислоту можно растворить в гидроокиси аммония. Соосаж-дение титана, тория, олова и циркония предотвращают, обезвоживая кремневую кислоту в сильнокислых растворах, причем для удерживания в растворе олова используют серную килоту, образующую с ним комплексный сульфат. Введением большого избытка серной кислоты можно также освободить осадок кремневой кислоты от малорастворимых фосфатов циркония и титана.
Отделению кремневой кислоты в форме nSiO2-mH2O мешают фторид-ионы, образующие летучие соединения SiF4 и H2SiF6. Их связывают добавлением соединений алюминия или бора [811, 817, 841, 1004, 1010, 1011] или отделяют кремний в форме K2SiF6 (см. главу 6). Отделение от фосфора см. в [1079].
Осаждение в форме солей кремнефтористоводородной кислоты. Из неорганических солей кремнефтористоводородной кислоты наименьшей растворимостью обладают кремнефториды бария и калия.
В анализе обычно используется осаждение кремния в форме K2SiFe, иногда Na2SiF6 или BaSiF6 [373, 624]. Растворимость кремнефторида калия в воде значительна (0,40%). Ее понижают введением избытка хлорида калия и спирта. Вследствие этого растворимость понижается настолько, что метод можно использовать для осаждения и отделения кремния в анализе с дальнейшим алкалиметрическим определением кремния в осадке различными методами (см. главу 6, а также [172, 514, 525, 813]). Отделение кремния в форме K2SiF6 от бериллия см. в [595], PbSiF6 от хрома — в [971].
Разработан метод отделения кремния в форме кремнефторидов органических оснований (бензидина, п фенилендиамина и др.) [301, 384, 441]. Кремнефторид бензидина после отделения может быть высушен и взвешен; содержание кремния в нем можно определить колориметрически по бензидину [411]. Кремнефторид бензидина осаждают из растворов кремнефтористоводородной кислоты (после перевода в нее кремния из анализируемого раствора) в полярных органических растворителях и их смесях с водой. Процесс осаждения кремнефторидов органических оснований может быть использован для аналитического, определения кремния химическими и электрохимическими методами [302, 307, 308, 384].
Осаждение в форме солей гетерополикислот, содержащих кремний. Гетерополи кислоты — кремнемолибденовая, кремневольфрамовая, кремневанадийвольфрамовая дают малорастворимые соединения с ионами щелочных металлов — калия, рубидия и цезия [404, 494, 1011], а также таллия (I) и ртути (I) [1011]. Процесс образования этих нерастворимых соединений может быть использован
37
для качественного обнаружения кремния [624]; для количественного его отделения он не нашел применения. Однако для извлечения и количественного отделения рубидия и цезия он используется.
Кремнемолибденовая кислота осаждает хелатные комплексы (с этилендиамином, уротропином, тиокарбамидом) многих катионов (хрома, кобальта, никеля, меди, цинка, серебра, кадмия, олова, ртути) [404].
Кремнийсодержащие гетерополикислоты образуют малорастворимые соединения со многими органическими основаниями. Отделение кремния в форме кремнемолибдата хинолина [76, 180, 291, 563, 564, 578, 644, 673, 702, 788, 1099], оксихинолина [69, 530, 727, 788, 907, 966, 1004, 1039], пиридина [40, 694, 1099], уротропина [726, 768], 2,4-диметилхинолина [915] и других органических оснований [197, 404, 440, 843, 1099] используется для отделения и гравиметрического определения кремния [76, 404, 440], а также для определения соответствующих органических оснований. В полученных соединениях после их отделения кремний может быть определен и другими методами. Процесс осаждения кремнесодержащих гетерополи кислот используется для количественного определения самих гетерополикислот кремния и соответствующих органических оснований электрометрическими методами (сводку литературы см. в [404]).
Для удаления железа и фосфора при определении кремния в виде желтой кремнемолибденовой кислоты предложено осаждать их последовательно действием гидрофосфата натрия и карбоната кальция [557]. Отделение в форме силикатов см. в [846, 930, 1082].
МЕТОДЫ дистилляции
Очень многие соединения кремния могут быть переведены при обработке фтористоводородной кислотой в кремнефтористоводородную кислоту и тетрафторид кремния. В форме этих соединений кремний может быть отделен от большинства элементов. Температура кипения SiF4 составляет —65° С, кремнефтористоводородная кислота отгоняется с водяным паром при температурах порядка 135° С [506, 507, 735, 805, 981]. Четыреххлористый кремний обычно отгоняют в присутствии концентрированной серной или хлорной кислоты [805, 817, 819].
Процесс дистилляции фторида кремния используется для концентрирования примесей при анализе веществ, содержащих кремний в больших количествах (см. главу 10), и для косвенного определения двуокиси кремния, в том числе и в прокаленных осадках SiO3, загрязненных примесями, адсорбированными в процессе осаждения (см. главу 5). На поглощении отогнанных SiF4 и H2SiFe основаны также методы определения кремния [691, 735, 825, 981].
Конструкции приборов для отгонки с последующим поглощением отогнанных соединений и аналитическим определением кремния 38
описаны в [543, 779 , 819 , 825]. Описаны диффузионный метод отделения SiF4 и определение кремния [640, 825], отделение от вольфрама [392, 408], отгонка в форме летучих силанов и их поглощение [847], отделение от германия [4151.
Поскольку при обработке фтористоводородной и серной кислотами вместе с фторидом кремния улетучивается и фторид бора, при анализе соединений, содержащих кремний и бор, должно быть предусмотрено предварительное удаление или связывание бора обычно в форме борнометилового эфира и комплексов с различными органическими веществами (см. в главу 5, а также [402, 950, 1004]). Отделение кремния в виде SiCl4 и SiBr4 см. в [446, 531, 885], в виде SiCl4 при одновременном осаждении бора в форме (СвН5)3СС1 • ВС13 см. в [8851. Отделение от кремния молибдена в виде МоО3-2НС1 см. в [646]. При определении кремния в стали большие количества железа также отделяют отгонкой в виде FeCl3 при 400—• 700° С [1841. Отделение органической части анализируемых веществ сжиганием с дальнейшим определением кремния в золе см. в [360, 401]. Определение SiF4 в газовых смесях см. в [611].
ЭКСТРАКЦИОННЫЕ МЕТОДЫ
Обзоры и сводки литературы общего характера см. в [7, 15, 16, 43, 179, 221, 372, 622, 843, 925, 1110].
Кремний может быть отделен от сопутствующих элементов экстракцией в форме гетерополикислот в кислой среде. Экстракция изучена главным образом для кремнемолибденовой кислоты и ее восстановленной формы [16 , 49, 179 , 497, 613 , 703, 843 , 951, 1044, 1056].
Гетерополикислоты, содержащие кремний, хорошо растворимы в кислородсодержащих органических растворителях (спиртах, эфирах, кетонах) и некоторых аминах. Для экстракции кремнемолибденовой кислоты используют главным образом нормальные и изобутанол и пентанол [7, 613, 642, 766, 843, 985]. Соответствующим подбором растворителей и их смесей кремнемолибденовая, а также кремневанадиймолибденовая [47, 699] кислоты могут быть отделены от сопутствующих им гетерополикислот фосфора, мышьяка и германия [565, 642, 703, 766, 843, 951, 952, 953, 1112]. После отделения экстракцией кремнемолибденовая кислота может быть определена колориметрически (см. главу 7, [7, 565, 1033]) и другими методами, например титрованием [385, 386]. Коэффициент экстракции имеет высокое значение [7, 16]. Экстракция кремния аминами в форме гетерополикислот и продуктов их восстановления описана в работах [315, 497, 1024, 1100]. Экстрагирование гетерополикислот в виде ионных ассоциатов с органическими основаниями также может служить для концентрирования и отделения кремния от мешающих элементов [497, 1024, 1129, 1134].
Экстракция органическими растворителями позволяет отделить кремнийорганические соединения от неорганических соединений,
39
в том числе и от неорганических соединений кремния. Это используется для качественного определения кремнийорганических соединений в органической фазе (см. главу 3).
Коэффициент распределения при экстракции кремния аминами в форме кремнефтористоводородной кислоты невелик. Метод использован для определения кремния в кремнийорганических соединениях и силикатах [292] и для отделения кремния [1118].
Многие мешающие определению кремния ионы могут быть отделены от него экстракцией их в форме дитизонатов, карбамина-тов, купферонатов и других соединений хлороформом и другими растворителями [606, 642, 876, 1011]. Методика экстракционного отделения купферонатов сурьмы, висмута, железа, молибдена, ниобия, тантала, олова, титана, вольфрама, ванадия и частично меди хлороформом описана в [918]. Другие работы по экстракции соединений кремния и экстракционному отделению примесей см. в [385, 776, 994, 1081, 1138].
ХРОМАТОГРАФИЧЕСКИЕ МЕТОДЫ
Хроматографическое отделение кремния в форме кремневой кислоты от сопутствующих катионов предусматривает как последующее определение кремневой кислоты, так и анализ смеси поглощенных катионов. Отделение производят после перевода кремния сплавлением в истинно растворимое состояние и последующей обработки кислотами на катионитах КУ-2, СДВ-3 и др. [171, 172, 306, 576]. В элюате кремний определяют в форме K2SiFe. Отделение кремния от катионов щелочных металлов на катионите КУ-2 позволяет в дальнейшем определить его по реакции образования кремнефторида бензидина [306]. Кремний отделяют хроматографически от мешающих катионов при ускоренном анализе минерального сырья [171, 576].
Отделение силикат-иона от мешающих ионов на анионитах используется для отделения кремния от фосфора [502, 503, 905, 1053], бора [498, 1040] и других элементов. Кремневая кислота адсорбируется смолами сильноосновного характера. Слабоосновными анионитами могут быть поглощены и отделены от кремневой кислоты хлористоводородная, серная и угольная кислоты [932].
Поглощенную кремневую кислоту (например, смолой Дауэкс-2 [948]) отмывают 0,5 N водным раствором едкого натра. Работы обобщающего характера по этому вопросу приведены в [171, 634].
Ионообменный метод может служить также для концентрирования малых количеств кремния, содержащихся в природных водах, в высокочистой воде [1070]. Ионный обмен здесь может быть использован с целью очистки воды или дальнейшего определения кремния.
Использование ионного обмена для отделения кремния от сопутствующих элементов см. в [172, 643, 823, 974, 1040, 1053, 1117], для концентрирования — в [928, 1094]. Отделение кремния отРОз-,
40
C0^_, S0^~ и AsC>3- методом хроматографии на бумаге описано в [400, 670, 776, 900], газовая хроматография фторидов представлена в [738]. Использование хроматографии для выделения примесей из чистого кремния отражено в главе 11.
ЭЛЕКТРОЛИЗ
Электролиз используется для выделения шлаковых включений (содержащих кремний) из черных металлов. При этом основное количество металла удаляется [182, 184, 261, 470, 660]. Электролизом на ртутном катоде можно выделить из разбавленного сернокислого раствора большие количества As, Sb, Bi, Cd, Сг, Co, Cu, Ga, Ge, In, Fe, Pb, Hg, Mo, Ni, Re, Se, Ag, Те, Sn, Au, Zn и платиновых металлов (кроме рутения).
Глава 4
ПОДГОТОВКА ВЕЩЕСТВА К АНАЛИЗУ
ОТБОР И ИЗМЕЛЬЧЕНИЕ СРЕДНЕЙ ПРОБЫ ВЕЩЕСТВА
Отбор средней пробы вещества для количественного определения в нем кремния производится по общим правилам отбора средней пробы, предусмотренным стандартами, и описанным в руководствах по анализу силикатов, сплавов, кремнийорганических соединений. Твердые анализируемые вещества перед анализом измельчают: грубое дробление обычно выполняют в ступке из нержавеющей стали или с помощью пресса, дальнейшее измельчение — последовательно в фарфоровой, стеклянной или агатовой ступках. Для уменьшения загрязнения анализируемых веществ в процессе измельчения рекомендуется использовать ступки из муллита, корунда и карбида бора. Загрязнение жидких кремнийсодержащих соединений (кремнийорганических, элементокремнийорганических и т. п.) возможно в процессе хранения веществ вследствие коррозии стенок сосудов. Поэтому желательно хранить кремнийсодержащие жидкости и их растворы в полиэтиленовой посуде. При анализе металлов и сплавов возможно небольшое загрязнение их в процессе снятия стружки.
Величина навески и техника ее взятия зависят от природы, агрегатного состояния вещества, содержания в нем кремния и выбранного метода определения. Для твердых веществ определение содержания гигроскопической влаги обычно предшествует разложению вещества.
РАСТВОРЕНИЕ ИЛИ РАЗЛОЖЕНИЕ ВЕЩЕСТВ
Без предварительного растворения или разложения анализируемого объекта кремний определяют химическими и физико-химическими методами лишь в водах и разбавленных растворах силикатов щелочных металлов. Кремнийсодержащие соединения по признаку растворимости могут быть классифицированы как:
42
1) растворимые в воде; 2) нерастворимые в воде (разлагаемые кислотами и неразлагаемые кислотами [536]).
Перед выделением и определением кремния необходимо перевести анализируемое вещство в водный раствор. Кремний и кремнийсодержащие функциональные группы могут быть определены в неводных растворах [296, 298].Об особенностях разложения кремнийорганических соединений см. главу 9.
Использование физических методов анализа (спектрального, рентгеноспектрального, радиометрии) позволяет обходиться без предварительного разложения пробы.
В самом общем случае анализируемое кремнийсодержащее соединение переводят в раствор сплавлением и разложением плава водой, водными растворами кислот или концентрированными кислотами. Для материалов и веществ, количественно разлагаемых кислотами, операция сплавления исключается из хода анализа. Однако соединения, содержащие кремний и фтор одновременно, обычно сплавляют, так как при разложении их кислотами возможны потери кремния вследствие улетучивания его в виде SiF4. Силикаты истинно или коллбиднорастворимые в воде в зависимости от способа определения в них кремния обычно обрабатывают кислотами или щелочами.
СПЛАВЛЕНИЕ
Выбор плавня в каждом отдельном случае зависит от состава анализируемого образца и производится по возможности с учетом дальнейших аналитических операций в ходе анализа после отделения кремния. При сплавлении кремний и его соединения, а также другие составные части анализируемого вещества или смеси реагируют с плавнем с образованием соединений, растворимых в воде или разлагаемых кислотами. Дальнейшее выполнение аналитических операций принципиально не отличается от анализа соединений, растворимых в воде или разлагаемых кислотами. При использовании в качестве плавней карбонатов щелочных или щелочноземельных металлов аналитик в самом простейшем случае имеет дело с реакциями в системах R2O — SiO2 — СО2 и RO — SiO2 — СО2. Основные сведения по этому вопросу имелись еще у Ле Шателье [325]. Система Na2O — SiO2 — СО2 изучена Ниггли [940], обнаружившим в ней орто-и метасиликаты натрия, между которыми существует равновесие
Na2CO3 + Na2SiO3^± Na4SiO4 + СО2.
Сдвиг этого равновесия в сторону образования более щелочного силиката при повышении температуры имеет первостепенное значение в анализе, поскольку при дальнейшем растворении в воде или разложении кислотами ортосиликаты дают мономерную форму кремневой кислоты, а метасиликаты — полимеры.
43
Реакция частиц полимерной двуокиси кремния с карбонатом натрия сопровождается распадом цепочечных молекул на группы SiO4-.
Нагревание карбоната калия с кремневой кислотой при постоянных температуре и давлении двуокиси углерода показывает [940], что реакции
К2СО3 + SiO2 -> K2SiO3 + СО2 и
2K2SiO3 + СО2 —> K2Si2O5 + К2СО3
перекрываются; вторая реакция подчиняется закону действия масс. Реакция образования дисиликата калия имеет место еще в твердом состоянии, причем наблюдается спекание.
В системе К2О — А12О3 — SiO2 — СО2 образуется калиофилит К2О-Al2O3-2SiO2, плохо растворимый в расплаве карбонатов. Аналогичная система изучена и для соединений натрия.
Сплавление с карбонатами щелочных металлов. Для сплавления чаще всего применяют безводный химически чистый карбонат натрия (или бикарбонат, который легче получить чистым). С карбонатом натрия сплавляют силикатные минералы и горные породы, искусственные силикаты, руды, агломераты, шлаки, высококремнистые материалы — пески, кварцевое стекло, сплавы, содержащие кремний.
В некоторых случаях, например, при анализе фтор- и хлорсодержащих веществ, используют смесь карбонатов калия и натрия или реактив KNaCO3, обладающий более низкой температурой плавления, чем карбонат натрия [717]. Гиллебранд [149] указывает, что в целом ряде случаев смесью карбонатов натрия и калия пользоваться не следует, потому что из-за легкоплавкости она не может обеспечить достижения температур, необходимых для разложения многих минералов. Кроме того, работа с полученными плавами может осложняться, так как соли калия в большей степени адсорбируются осадками, чем соли натрия. Исключается также возможность дальнейшей обработки плава хлорной кислотой, что нередко рекомендуется.
Применяют как плавень и химически чистый карбонат калия, руководствуясь главным образом более высокой растворимостью продуктов сплавления (по сравнению с натриевыми солями), однако он имеет некоторые недостатки кроме уже упомянутых — он гигроскопичен и перед применением приходится прибегать к обезвоживанию прокаливанием. Сплавление с карбонатами щелочных металлов и их смесями рекомендуется выполнять в платиновой посуде. Количество плавня обычно шестикратное. Иногда рекомендуется применять и большее количество плавня (до 20: 1 и даже выше) [93, 94, 1041]. Карбонат натрия используют также и для спекания.
Если в процессе сплавления необходимо окислить некоторые составные части анализируемого вещества, к карбонатным плавням прибавляют окислители, например нитрат или хлорат калия [418,
44
576]. Количество окислителя может быть различным, составляя иногда до трети количества плавня. Даже в отсутствие добавок окислителей при сплавлении идут процессы окисления кислородом воздуха, скорость которых под влиянием высокой температуры в условиях высокощелочной среды и при тонком измельчении реагирующих веществ вполне достаточна для того, чтобы можно было не вводить специальных окислителей. Введение окислителей нежелательно еще и потому, что оно может усилить коррозию платиновой посуды.
Во избежание порчи тигля при наличии в анализируемом веществе восстановителей (например, сульфидов железа или углистых веществ) пробу исследуемого вещества перед сплавлением можно прокалить в фарфоровом тигле.
Сплавление с гидроокисями щелочных металлов. Сплавление производят при значительно более низких температурах, чем с карбонатами, причем реакции силикатообразования протекают с большей скоростью. Платиновую посуду использовать нельзя. Обычно применяют никелевые или железные тигли. Применение золотых и серебряных тиглей и чашек требует большой осторожности, так как они не выдерживают нагревания до высоких температур. При сплавлении со щелочами следует сначала расплавить сам плавень до прекращения вспенивания и разбрызгивания и только после этого внести анализируемое вещество, помещая его на поверхность уже охлажденного до затвердевания плавня, и затем нагреть до сплавления.
Весовые отношения вещество: плавень обычно составляют 1 : 10 и более низкие — 1 : 30 и даже 1 : 100. При сплавлении со щелочами стекол предложено предварительно смачивать навеску спиртом или спиртовым раствором щелочи [988, 990]. Для определения кремния в силицидах сплавление со щелочами — лучший способ перевода веществ в растворимое или разлагаемое кислотами состояние [155, 269, 358, 359]. Сплавление со щелочами используется в анализе нелетучих кремнийорганических соединений (см. главу 9) для разложения фторсодержащих веществ (см. главу 5). Предложено использовать в качестве плавня смесь едкого натра и едкого кали. Эта смесь при соотношении NaOH : КОН = 5 : 7 с температурой плавления 150° С разлагает очень многие природные и синтетические силикаты [919]. Смесь до 200—250° С практически не реагирует с платиной, серебром и никелем. Для сплавления используют также смеси едких щелочей с карбонатами и щелочей с окислителями, например KNO3, Na2O2.
Сплавление с перекисью натрия. Перекись натрия в качестве плавня используется одна и в смеси со щелочами. Применяют ее для сплавления кремния, его сплавов, некоторых силикатов, кремнийорганических соединений — соединений и веществ, где требуется перевести в более высокую степень окисления или сам кремний (например, в силицидах или кремнийорагнических соединениях) или сопутствующие ему элементы. Сплавление обычно производят
45
в железных, никелевых, циркониевых тиглях, стальных или никелевых бомбах, иногда используют фарфоровые тигли, помещая на дно и стенки их подкладки из СаСО3, CaSO4, Na2CO3, Na2SO4, K2SO4.
Сплавление co щелочными металлами. Щелочные металлы в качестве плавней применяют в анализе кремнийорганических соединений (см. главу 9).
Сплавление с тетраборатом натрия. В качестве плавня тетраборат натрия чаще всего используют в смеси с карбонатом натрия, иногда вводят в небольших количествах окислители. Таким образом удается разложить трудносплавляемые минералы. Применяются также смеси Na2B4O7+Na2CO3+K2CO3 (или KNaCO3).
Использование тетрабората натрия (а также борной кислоты и борного ангидрида) вносит в анализ осложнение: после сплавления бор приходится удалять в виде борнометилового эфира, так как он мешает выполнению последующих аналитических операций, загрязняя образующиеся осадки. Борную кислоту и борный ангидрид более полутораста лет применяют в тех случаях, когда необходимо в дальнейшем ходе анализа определять щелочные металлы или при анализе веществ, содержащих фтор — в этом случае потерь SiO2 в виде SiF4 не происходит. Фтор выделяется в виде BFз. Кроме борной кислоты и борного ангидрида из плавней кислого характера используют бисульфаты, пиросульфаты и бифториды щелочных металлов и их смеси. Сплавление с кислыми плавнями в присутствии фторидов обычно не предусматривает дальнейшего определения SiO2 из данной навески.
Применение в качестве плавня бифторида калия рекомендуется при условии дальнейшего алкалиметрического определения кремния [522]. Плавень Na2CO3+Na2B4O7+Al2O3 применяется для анализа образцов, содержащих фосфаты и фториды [715]. Силикаты, содержащие бор, сплавляют со смесью K2CO3+Na2CO3+ZnO [128, 577], фторсиликаты — со смесью Na2CO3+ZnO [1013]. Другие плавни см. в [25, 161, 576, 593, 654, 778, 813, 869].
В большинстве случаев рекомендуется использовать безводный карбонат натрия; другие плавни следует применять только в случае крайней необходимости, не забывая о подборе соответствующей посуды, температурного режима и других условий анализа [815, 1085].
СПЕКАНИЕ
Метод спекания применяется для разложения большой группы кремнийсодержащих материалов — руд, шлаков, силикатов. Метод имеет определенные преимущества по сравнению с методом сплавления. На сплавление образца и обработку плава затрачивается много времени; кроме того, при сплавлении анализируемый раствор загрязняется большим количеством солей щелочных металлов. В случае спекания вещества количество плавня может быть сильно уменьшено, время обработки плава сокращается.
46
Основные данные по системам, входящим в состав плавней, можно найти в монографиях Эйтеля [621], Будникова и Гинстлинга [92] и ряде других работ [63, 546, 579].
При нагревании анализируемого вещества с небольшим количеством безводного карбоната натрия (или другого плавня) в течение нескольких минут образуется прочная пористая масса (спек), которая хорошо разлагается кислотами. Дальнейшее повышение температуры и продолжительности нагревания снова снижает реакционную способность вещества. Нагревание должно быть кратковременным, температуру не следует поднимать выше определенных пределов. Таким образом, при взаимодействии твердых веществ возникает некоторое промежуточное, разрыхленное переходное состояние.
Причину наблюдаемой повышенной реакционной способности следует искать в процессах, протекающих при разрушении старой и образовании новой структуры кристаллической решетки [92], 546]. Разрыхлению решетки способствуют диффузия ионов щелочного реагента и повышение температуры. При нагревании возрастает число дефектов кристаллической решетки. Деформация решетки не ограничивается местом проникновения чужеродного иона, а в какой-то степени распространяется в глубь решетки. «Расшатывание» решетки способствует увеличению способности вещества растворяться в кислотах. Реакционная способность реальных кристаллов тем выше, чем больше энергия их решетки отличается от энергии решетки идеального кристалла [92, 929]. Влияние дефектов в кристаллах на скорость термического разложения твердых веществ см. в [84].
Отрицательное влияние длительного нагревания объясняется тем, что при более полном протекании реакций завершается процесс образования новой кристаллической структуры, которая обладает меньшим запасом свободной энергии. Полученная монолитная масса значительно медленнее поддается действию кислот. Установлено также, что проникновение в решетку разлагаемого вещества ионов натрия идет быстрее, чем проникновение ионов калия. В связи с этим для разложения рекомендуется использовать спекание с карбонатом натрия, а не с К2СО3 или смесью карбонатов. Необходимо подобрать такие условия спекания, при которых легко разрушается старая кристаллическая решетка, а новая если и образуется, то с максимальным количеством искажений. Такое метастабильное состояние решеток замораживается быстрым охлаждением спекшейся массы; вещество становится высокореакционноспособным и легко разлагается кислотами. Спекание сопровождается реакциями нагреваемых твердых веществ между собой. По вопросам механизма и кинетики твердофазных реакций имеется литература, рассматривающая поведение и свойства кристаллического тела при нагревании [63, 70, 92, 465, 466, 546, 621], влияние степени измельчения веществ [248] в условиях нагревания. Теории твердофазных реакций оценены критически [433, 546]. Иссле
47
дование процесса сплавления и спекания минералов см. в [432, 548].
При разложении анализируемых веществ спеканием соотношение количества вещества и плавня, как правило, значительно выше, чем при сплавлении: оно составляет обычно величину 1:1. Установлено, что в смесях Na2CO3+SiO2 скорость взаимодействия тем выше, чем больше содержание SiO2 в смеси. Для выполнения спекания использование платиновой посуды не обязательно. Спекание можно проводить в фарфоровых и корундизовых тиглях, на никелевой или серебряной пластинке. Техника спекания отличается от техники сплавления. Постепенное повышение температуры здесь обычно не применяется — чаще всего смесь плавня с анализируемым веществом кратковременно нагревают в разогретой печи.
Навеска силиката переводится в раствор в течение 12—15 мин, а продолжительность определения составляет 1—1,5 час. Анализ можно ускорить, используя метод коагуляции кремневой кислоты желатиной.
Для спекания кроме Na2CO3 применяют и другие плавни: Ы2СО3, К2СО3 [546], LiBO2 [1037], а также смеси Na2CO3+K2CO3, MgO+Na2CO3 [546], но чаще всего используется все же Na2CO3 как наиболее пригодный.
ПРЕДВАРИТЕЛЬНОЕ ПРОКАЛИВАНИЕ
Применение предварительного прокаливания анализируемых материалов во многих случаях способствует их разложению кислотами. Этот метод применяют при анализе карбонатных пород [25, 161, 510, 515, 700, 882], глин [161, 212, 816].
При прокаливании карбонатных пород с большим содержанием окиси кальция идут реакции в системе СаО — SiO2 — СО2. Эта система при нагревании претерпевает изменения, в результате которых уже при сравнительно низких температурах в достаточно уплотненном материале начинается образование силикатов в твердой фазе. Кинетика этих реакций, изученных в различных условиях (температура, время, размер частиц), описана в [840]. Найдено, что соотношение между толщиной слоя продукта реакции у, средним радиусом зерен кристаллической фазы г, присутствующей в меньшем количестве в реагирующей смеси, и количеством продуктов реакции х описывается простым уравнением
(. 100 —
У—у юо )*
где х и у зависят от времени. При различной дисперсности реагирующих веществ отношения более сложные.
Твердофазные ракции в системе СаО — SiO2 — СО2, а также реакции, идущие при спекании, когда частично образуется жидкая фаза, приводят к образованию силикатов щелочноземельных ме
48
таллов [510]. Кроме того, наличие дефектов в кристаллической структуре кремнесодержащего вещества облегчает диффузию щелочного расплава и дальнейшее разложение анализируемого вещества. Именно протекание твердофазных реакций при предварительном прокаливании карбонатных пород позволяет в дальнейшем прибегать к сплавлению для перевода кремнеземистых и глинистых примесей в такое состояние, когда их удается разложить обработкой кислотами.
РАЗЛОЖЕНИЕ И РАСТВОРЕНИЕ ПРОДУКТОВ СПЛАВЛЕНИЯ И СПЕКАНИЯ
Обычно плавы разлагают последовательным действием воды и кислот. После разложения продуктов сплавления водой кремний может быть определен в полученном растворе колориметрическим методом (см. главу 9). Таким образом, разложение кислотами плавов (а также некоторых силикатов, сплавов и кремнийорганических соединений) — операция по существу общая для анализа соединений кремния. Для разложения плавов соединений кремния используют главным образом хлористоводородную кислоту; применяются также серная, азотная, хлорная кислоты, иногда уксусная. Эти же кислоты применяют при анализе материалов, растворимых в воде и разлагаемых кислотами. Смеси кислот используют обычно при разложении металлов и сплавов, содержащих кремний [26, 184, 185, 324, 482, 545, 553, 583]. Выбор кислоты и методики разложения во многом зависит от состава анализируемого вещества и способа выполнения определения кремневой кислоты. Для выполнения определения кремневую кислоту отделяют от сопутствующих ей элементов или обходятся без отделений. Здесь возможно несколько вариантов.
1. При обработке раствора кислотами с последующим выпариванием (или другими способами обезвоживания) происходит дегидратация и полимеризация кремневой кислоты, вследствие чего понижается скорость ее растворения, что способствует отделению от хорошо и быстро растворяющихся в кислотах примесей сопутствующих элементов. Определение кремневой кислоты выполняется далее гравиметрическим методом. Существует много вариантов этого метода определения кремния, он считается наиболее изученным, а получаемые результаты при тщательном выполнении операций достаточно надежны.
2. Раствор обрабатывают фтористоводородной кислотой или фторидами в присутствии солей калия для осаждения кремния в форме кремнефторида калия (или кремнефторидов органических оснований) или, наконец, для отгонки в форме SiF4 с дальнейшим улавливанием фторида кремния с использованием посуды из пластмасс [578, 779, 1057].
3. Кремневую кислоту из щелочного раствора, полученного обработкой плава водой, переводят в соответствующую гетерополи
49
кислоту (кремнемолибденовую или кремневанадиймолибденовую) для дальнейшего выполнения определения спектрофотометрическим или гравиметрическим методами (см. главы 5 и 7).
4. Кремневая кислота в щелочном растворе может быть оттитрована комплексонометрическим или амперометрическим методами.
5. Кремневую кислоту из раствора переводят в комплексную пирокатехинкремневую кислоту, которая может быть оттитрована после добавления избытка щелочи ацидиметрически с потенциометрической индикацией конца титрования (см. главу 6).
Разложение кислотами. Хлористоводородную кислоту применяют для разложения металлов и сплавов, содержащих кремний, природных и искусственных силикатов, разлагаемых кислотами, и для обработки плавов тех материалов, которые кислотами не разлагаются. Испол'ьзуют концентрированную и умеренно разбавленную кислоту.
В некоторых работах для ускорения разложения рекомендуется обработка концентрированной хлористоводородной кислотой под давлением [212]. Кроме того, применяют серную, азотную, хлорную, реже уксусную кислоты. Применяется последовательная обработка кислотами, например хлористоводородной и серной, азотной и хлорной — в анализе черных металлов [434].
Используются в различных комбинациях смеси кислот серной, хлористоводородной, фосфорной, фтористоводородной.
Смеси кислот используют для разложения металлов, сплавов, силикатов, кремнийорганических соединений и других материалов, содержащих кремний. Применяя смесь кислот, можно добиться большей полноты или большей скорости разложения анализируемого материала, замены сплавления кислотным разложением (например, бокситов [908] сплавов [874], стали [182, 338]). Создаются лучшие условия разложения, дегидратации или окисления [912], а также выделения кремния в форме SiF4, кремнефтористоводородной кислоты или ее солей в различных материалах.
Другие методы разложения анализируемых веществ. Для разложения сплавов на основе алюминия и некоторых пород, содержащих его в больших количествах, используют щелочные растворители, например водные растворы щелочей или карбонатов щелочных металлов [80, 347]. Этот же метод применим и к некоторым силицидам, например ферросилицию [80, 182]. Другие способы разложения анализируемых веществ см. в [182, 306, 774, 878, 991, 1019]. О потерях при разложении силицидов кислотами см. [119]. Теория бес-стружкового разложения кислотами изложена в [509, 521], щелочами — в [334].
Глава 5
ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ
ОПРЕДЕЛЕНИЕ В ФОРМЕ SiO2
Определение кремния в его соединениях взвешиванием в форме SiO2 — самый старый, но до настоящего времени не потерявший своего значения по точности и воспроизводимости результатов анализа метод.
Метод основан на выделении кремния в процессе конденсационной полимеризации в виде полимеризованной кремневой кислоты, ее дальнейшей дегидратации высушиванием и прокаливанием и взвешивании полученной безводной полимерной двуокиси кремния SiO2.
Метод применяют для определения значительных количеств кремния. При выделении кремневой кислоты из истинных или коллоидных растворов одновременно происходит и отделение ее от сопутствующих элементов, которые можно определить в полученном фильтрате. Поэтому при анализе веществ сложного состава аналитики до сих пор отдают предпочтение гравиметрическому методу определения кремния.
Гравиметрическому определению кремния в форме SiO2 обычно предшествует разложение соединений кремния с переводом определяемого элемента в виде кремневой кислоты в коллоиднорастворимое или истиннорастворимое состояние. Цель дальнейшей обработки состоит в том, чтобы коллоиднорастворенную кремневую кислоту дегидратировать и коагулировать, истиннорастворенную полимеризовать и дегидратировать. Поскольку истиннорастворен-ная кремневая кислота способна давать пересыщенные растворы, процесс ее дегидратации и коагуляции требует специальных приемов.
Так как потери вследствие растворимости — величина абсолютная, Шелл [1009, 1011, 1012] настоятельно рекомендует для уменьшения этого источника ошибок брать большие навески, чтобы снизить относительные потери. По этой же причине для определения малых количеств кремния гравиметрический метод используют редко.
51
Основной метод, применяемый для гравиметрического определения кремния,— это метод дегидратации в кислой среде. Выделение кремневой кислоты дегидратацией из нейтральных или слабощелочных растворов применяют лишь в специальных случаях — при анализе материалов, содержащих значительные количества фтора. В этом случае возможны потери кремния из-за улетучивания его в присутствии кислот в виде тетрафторида SiF4.
Обстоятельное обсуждение вопросов, связанных с дегидратацией кремневой кислоты, проводится последовательно в ряде работ и монографий Гиллебрандом [149], Григорьевым [161], Ленделем, Гофманом и Брайтом [324], Егоровой [186—188], Шеллом [1011] и другими авторами [166, 184, 185, 335, 520, 815, 883, 1015, 1080].
Е. Н. Егорова так характеризует основные операции гравиметрического метода в применении к анализу силикатов [188]: 1) сплавление навески тонкоизмельченного силиката и разложение плава кислотой (или разложение кислотой самого анализируемого силиката); 2) выпаривание кислого раствора и полученного сухого остатка для более полной дегидратации кремневой кислоты и перевода ее в «труднорастворимое» состояние; 3) обработка сухого остатка кислотой и растворение солей при разбавлении водой; 4) фильтрование и промывание выделенной кремневой кислоты; 5) вторичное выпаривание полученного фильтрата для выделения оставшейся в растворе кремневой кислоты (далее повторение операций 2,3 и 4); 6) прокаливание обоих осадков кремневой кислоты до постоянного веса; 7) контрольная обработка двуокиси кремния смесью фтористоводородной и серной кислот; 8) выделение кремневой кислоты, соосажденной с полуторными окислами в ходе анализа.
Из перечисленных операций процессы сплавления, спекания и разложения плавов уже рассмотрены нами в главе 4. Для дегидратации кремневой кислоты в кислой среде чаще всего используют хлористоводородную кислоту, находят себе применение также серная, азотная, хлорная и уксусная кислоты. Используются и другие дегидратирующие вещества — глицерин, ацетилхлорид.
После разложения плава или анализируемого образца кислотой необходимо выделить кремневую кислоту из раствора и отделить ее от сопутствующих элементов. Для этой цели кислый раствор выпаривают досуха. В процессе выпаривания происходят конденсационная полимеризация кремневой кислоты за счет реакции
—SiOH-J-HOSi-----> —SiOSi—Н2О (см. главу 1), разрушение
гидратной оболочки геля и дальнейшая дегидратация геля упариванием и высушиванием кислого раствора.
В полученном осадке наряду с глубокополимеризованными формами имеются формы низкополимеризованные; растворимость их в воде различна. Процесс дегидратации — гидратации обратим, поэтому при обработке полученных осадков разбавленными кисло
52
тами и водой кроме растворения низкополимеризованных форм кремневой кислоты возможна частичная деполимеризация и пептизация геля кремневой кислоты. Некоторое количество кремневой кислоты останется вследствие этого неосажденным и попадет в фильтрат.
После отделения осадка фильтрат повторно обрабатывают кислотой при нагревании, выпаривают раствор ( а иногда и нагревают остаток до температур порядка 120—130° С [149, 185, 269, 370, 685, 884], вследствие чего значительная часть оставшейся в растворе кремневой кислоты снова осаждается. Однако нельзя забывать, что полного осаждения не удается достигнуть не только при втором, но и при дальнейших упариваниях [161, 643, 1011]. Операцию дегидратации повторять больше двух раз не имеет смысла, тем более что при этом возможно введение в раствор кремневой кислоты из реактивов и посуды.
Осадок кремневой кислоты адсорбирует примеси, содержащиеся в анализируемом растворе. Соосаждаются значительные количества борной кислоты, вольфрама, .ниобия, тантала и другие примеси. Осадок промывают обычно разбавленным раствором кислоты для освобождения от соосажденных катионов и предотвращения гидролиза солей, которые они образуют при выпаривании раствора анализируемого вещества или плава после обработки растворами кислот. Некоторое количество адсорбированных примесей в осадке все-таки остается.
Результаты определения кремния могут быть искажены и при дальнейшей обработке осадка. При излишне быстром озолении фильтра без достаточного доступа кислорода воздуха образуется карбид кремния S1C вместо SiO2. В осадке содержится некоторое количество соосажденных катионов щелочных металлов, а также бор, если он присутствует в значительных количествах в анализируемом образце или вводится в виде плавня. При дальнейшей обработке осадка прокаленной и взвешенной двуокиси кремния небольшим количеством фтористоводородной и серной кислот кремний удаляется вследствие образования летучего тетрафторида. Кремний, находящийся в осадке в виде SiC, улетучивается лишь частично, что обусловливает занижение полученных результатов. Вместе с SiF4 удаляется бор в виде BF3, вследствие этого происходит завышение результатов определения кремния. При прокаливании остатка после обработки кислотами соосажденные примеси щелочных металлов образуют сульфаты. Это также искажает результаты определения кремния.
Кремневая кислота из фильтратов в ходе анализа силикатов может быть выделена из раствора при дальнейшем осаждении аммиаком полуторных окислов [149, 188, 763, 1011]. При этом соосаждение кремневой кислоты происходит главным образом в присутствии значительных количеств алюминия [149, 188, 829, 1011]. Осадок полуторных окислов после прокаливания сплавляют с пиросульфатом калия или натрия и выделяют из него кремневую кислоту
53
дегидратацией. Тем не менее ни эта обработка, ни многократная дегидратация не могут все же выделить кремневую кислоту количественно.
Истинно растворимая кислота может быть определена колориметрически — из осадка с R2O3 или фильтрата после отделения SiO2—такое решение вопроса наиболее удовлетворительно [188, 845, 894, 1011, 1105].
Добиваясь полноты выделения кремневой кислоты из раствора и высокой точности определения кремния, никогда не следует забывать, что в процессе анализа аналитик имеет дело с фарфоровой и стеклянной посудой, которая может вследствие разъедания ее агрессивными кислотными и щелочными растворами посылать в раствор значительные количества кремния. Это особенно заметно при пользовании новой посудой. Влияние этих загрязнений тем больше, чем меньше навеска определяемого вещества. Использование платиновой посуды позволяет освободиться от этих ошибок, однако в больших количествах она недоступна. Рекомендуется [894] вести выпаривание в танталовых чашках, использовать посуду из полиэтилена и тефлона [292, 384, 707].
Методы выделения кремневой кислоты дегидратацией при выпаривании кислотных растворов
Методы с хлористоводородной кислотой. Основные операции: 1) разложение силиката или плава водой (применяется не всегда); 2) обработка образца или плава хлористоводородной кислотой (концентрированной или разбавленной); 3) выпаривание досуха на водяной или паровой бане; 4) обработка сухого остатка (дегидратированная кремневая кислота и соли) разбавленным водным раствором хлористоводородной кислоты и удаление растворимых солей фильтрованием.
При обработке образцов и плавов хлористоводородной кислотой происходит улетучивание As(III) и Ge(IV) в виде хлоридов; хром переходит в Сг(Ш) и марганец в Mn(II); Se, Те, V — в четырехвалентное состояние. Кроме того, могут образоваться нерастворимые соединения анионов, содержащих серу, хлор, бром, иод и фосфор с катионами свинца, бария, титана, циркония, гафния, серебра и др. Поэтому количественному анализу должно предшествовать качественное исследование образца.
Предпочтение, отдаваемое аналитиками хлористоводородной кислоте при обезвоживании осадка кремневой кислоты, объясняется тем, что она безопасна в обращении, сам процесс дегидратации не требует особого внимания со стороны аналитика.
Растворенный в воде или разлагаемый кислотами анализируемый образец помещают в платиновую или фарфоровую чашку (обычно емкостью 100—200 мл). Если образец подвергали сплавлению, то в чашку переносят плав из тигля. Приемы извлечения плава из тиг
54
ля описаны в учебных руководствах [149, 161, 269, 324, 1011]. Обычно поступают следующим образом. Тигель вынимают щип-щами из печи (или снимают с горелки), осторожно поворачивая, дают плаву тонким слоем распределиться по стенкам и затем погружают тигель в холодную воду (вода не должна попадать внутрь тигля). Плав растрескивается и легко извлекается из тигля.
При перенесении плава некоторые аналитики пытаются соскребать его частицы со стенок тигля палочкой, мнут тигель для облегчения отделения плава — эти приемы недопустимы, так как применение их резко сокращает срок службы платины.
При нагревании плава с 10% -ной НО обильно выделяется двуокись углерода. После окончания ее выделения снимают часовое стекло и обмывают его водой; раствор упаривают. Образующаяся студнеобразная масса при помешивании и растирании палочкой переходит в тонкий порошок. Если предполагается выполнение многократной дегидратации (обычно двукратной) [39], то ограничиваются выпариванием досуха. При однократной дегидратации (с выделением примерно 98—99% имеющейся SiO2) выпаривают до исчезновения запаха НО [161, 166, 1011]. Этот способ применяют чаще всего.
Некоторые авторы рекомендуют вести выпаривание до состояния «влажных солей» [439], другие требуют нагревания в сушильном шкафу [149, 269, 685, 783] или на банях с высококипящими жидкостями [853]. Недостатки и достоинства таких способов выполнения процесса дегидратации кремневой кислоты обсуждены в литературе [149, 161, 187, 188, 1011]. Сушка в шкафу загрязняет осадок кремневой кислоты и может способствовать его пептизации при дальнейшем фильтровании [161] вследствие образования растворимых силикатов [188, 335, 418].
При двукратном выделении кремневой кислоты выпаренный досуха остаток в чашке охлаждают, смачивают 10—12 мл концентрированной НС1 и накрывают часовым стеклом. После 15—20-минутного стояния (время некоторыми авторами незначительно изменяется [161, 188, 1011 ]) обрабатывают массу 50—70 мл горячей воды из промывалки. Жидкость декантируют через фильтр с белой лентой, 2—3 раза промывают остаток в чашке декантацией горячей водой, затем более или менее нацело переносят осадок на фильтр и там промывают до отрицательной реакции на хлорид-ион. Обычно рекомендуют вести промывание разбавленным раствором НС1 (при первой дегидратации 5 : 95, при второй 1 : 99 [1011 ]). Фильтрат с промывными водами количественно переносят в фарфоровую чашку с остатками кремневой кислоты после первой дегидратации, упаривают раствор на водяной бане до исчезновения запаха НС1, охлаждают, смачивают осадок конц. НС1 и повторяют описанные операции с той разницей, что осадок кремневой кислоты переносят из чашки на фильтр обязательно количественно, применяя для этой цели протирание кусочками фильтра с помощью палочки и многократное обмывание водой или внесение бумажной пульпы.
Оба фильтра с осадками (после первой и второй дегидратации) помещают в фарфоровый или платиновый тигель, осторожно подсушивают, озоляют, избегая воспламенения, и прокаливают до постоянного веса (30—40 мин и повторно 15—20 мин). Температура прокаливания в большинстве случаев рекомендуется 1000—1100° С [913] или до 1200° С [1011 ]. Сжигание в муфеле при постепенном его нагревании дает хорошие результаты. Некоторые авторы рекомендуют прокаливание при более низких температурах, но это вряд ли обоснованно.
55
В работе [72] прокаливание ведут до 358° С, но осадок взвешивают при этой же температуре, что позволяет избежать ошибок вследствие его гигроскопичности.
Для ускорения охлаждения тигля с осадком перед переносом в эксикатор рекомендуется ставить его на поверхность металлической или каменной плитки на 30—40 сек, взвешивание выполнять по возможности быстрее во избежание адсорбции осадком влаги.
Для исключения ошибок, связанных с соосаждением посторонних ионов [1010], осадок (в платиновом тигле) смачивают водой, прибавляют 5—10 капель конц. H2SO4, несколько миллилитров HF, осторожно выпаривают и прокаливают в течение нескольких минут. По разности весов до и после обработки смесью кислот находят содержание SiO2 в исследуемом веществе. Соосаж-денние посторонних ионов предотвращают введением комплексона III [365].
В результате слишком быстрого сожжения кремневая кислота может получиться с сероватым оттенком вследствие образования карбида кремния при прокаливании без достаточного доступа кислорода воздуха. Это приводит к получению ощутимых ошибок при дальнейшей обработке осадка смесью кислот и его прокаливании: содержание кремневой кислоты получается заниженным. Ошибки при обработке смесью кислот возможны и при недостаточно полном промывании осадка вследствие того, что адсорбированные им соли щелочных металлов дают в результате сульфаты; содержание SiO2 также получается заниженным.
Количество кремневой кислоты, остающееся в растворе после второй дегидратации, сравнительно невелико, и при последующих повторных операциях дегидратации оно существенным образом не изменяется. Обычно этой величиной пренебрегают. Однако, как справедливо отмечают некоторые авторы, введение поправки после обработки смесью кислот без введения поправки на потери от растворения кремневой кислоты методически не оправдано. Выделение или определение истиннорастворимой кремневой кислоты из фильтрата возможно двумя путями [188, 1011]: соосаждением с полуторными окислами при обработке фильтрата аммиаком или колориметрически.
Метод с серной кислотой. Методика в общем аналогична методике с использованием хлористоводородной кислоты. Основные операции:
1) обработка образца или плава раствором серной кислоты (концентрированной или разбавленной); 2) выпаривание до образования густых белых паров серного ангидрида; 3) охлаждение раствора и обработка водой для растворения солей при нагревании; 4) фильтрование и промывание осадка кремневой кислоты.
К возможным осложнениям при использовании серной кислоты для дегидратации относятся: осаждение свинца и бария в виде нерастворимых сульфатов, медленное растворение сульфатов никеля, алюминия, кобальта, железа (II), требующее нагревания раствора (что способствует также переходу в раствор и самой кремневой кислоты). Кроме того, возможно соосаждение с кремневой кисло
56
той соединений сурьмы, германия и олова. Определение выполняют в платиновой, фарфоровой или стеклянной посуде.
Особенности методики: после разложения образца или сплава нагревание ведут до образования белых паров и продолжают при парообразовании еще 2—4 мин. Более длительное выпаривание не рекомендуется, так как оно может привести к образованию осадков безводных труднорастворимых сульфатов железа, никеля, кобальта, хрома, алюминия. После охлаждения пробы воду следует прибавлять очень осторожно. Операции фильтрования, промывания и дальнейшей обработки осадка аналогичны таковым в методе с использованием хлористоводородной кислоты.
Метод с хлорной кислотой. Метод имеет преимущества вследствие высокой растворимости большинства солей, большей степени выделения SiO2 [700, 1006, 1096]. Обычно применяют хлорную кислоту в смеси с другими кислотами, например с азотной кислотой. Кислоты применяют последовательно: сначала анализируемое вещество или продукт, полученный при его сплавлении с карбонатом натрия или другими плавнями-, разлагают, как обычно, водой и кислотами — хлористоводородной или азотной — и уже потом добавляют хлорную кислоту.
Основные операции: 1) обработка образца или плава раствором хлорной кислоты или добавление хлорной кислоты к раствору, полученному разложением образца действием других кислот (обычно хлористоводородной); 2) выпаривание до появления густых белых паров хлорной кислоты и кратковременная выдержка при условии их конденсации; 3) охлаждение раствора и обработка водой с последующим нагреванием для растворения солей; 4) фильтрование и промывание осадка разбавленным раствором хлористоводородной кислоты.
Выпаривание, как и в случае метода с использованием серной кислоты, ведут до выделения густых паров хлорной кислоты. Затем стакан или чашку накрывают стеклом и греют еще 15—20 мин при конденсации паров НС1О4, не доводя до кипения, так как при кипении количество осажденной SiO2 уменьшается. Количество НС1О4 должно быть таким, чтобы в горячей концентрированной НС1О4 не образовывалось осадка. Обычно при двукратной дегидратации первую дегидратацию выполняют с НС1 или с HNO3, а вторую—с НС1О4 [654, 908, 1009, 1012].
При остывании раствор перхлоратов затвердевает. К нему прибавляют 4—5-кратное по объему количество воды, нагревают до кипения, и дальнейшую обработку ведут аналогично обработке солянокислого раствора, применяя для промывания НС1 (1 : 99) и затем воду, с тем чтобы хлорную кислоту из осадка удалить по возможности нацело, иначе при прокаливании будут небольшие взрывы (потрескивание), что может вызвать потери. Комбинирование обработки азотной и хлорной кислотами благоприятно в отношении безопасности выполнения определения, так как восстановители будут окислены азотной кислотой к тому времени, когда в раствор вводится НС1О4.
Преимущества использования хлорной кислоты состоят в быстроте дегидратации, уменьшении толчков при выпаривании, хорошей растворимости перхлоратов в воде при большой скорости растворения. Недостатки — малая растворимость перхлоратов щелочных металлов, что очень важно при разложении плавов, высокая окис.
57
лительная способность хлорной кислоты, делающая ее крайне опасной.
Необходимо соблюдать осторожность при работе с хлорной кислотой, потому что взрывы при малейшем недосмотре весьма вероятны. Для предотвращения возможности взрывов хлорную кислоту обычно применяют после обработки образца азотной кислотой или при вторичной дегидратации. Для промывания осадков используется соляная кислота, промывание должно быть очень тщательным, иначе возможны потери осадка от небольших взрывов (потрескивание) в процессе озоления и прокаливания осадка кремневой кислоты. Общие правила при работе с хлорной кислотой — осторожное ее хранение, наличие легкопромываемой тяги и всех вытяжных коммуникаций, тщательное и продуманное выполнение аналитических операций при работе с веществами, содержащими восстановители. Работу с хлорной кислотой можно доверять только высококвалифицированным аналитикам. Эти условия, конечно, вызывают возражения в отношении использования НС1О4 в анализе соединений кремния, особенно с точки зрения техники безопасности.
Особенности поведения некоторых элементов при использовании хлорной кислоты для дегидратации кремневой кислоты: хром переходит в Cr(VI); возможно частичное соосаждение соединений молибдена, германия, ванадия, марганца и висмута [1011].
Дегидратация в присутствии других кислот. Значительно реже для дегидратации используют азотную и уксусную кислоты. Это объясняется, по-видимому, тем, что они не имеют особых преимуществ перед другими кислотами при наличии определенных недостатков: некоторые ацетаты легко гидролизуются и в процессе выпаривания дают труднорастворимые оксиацетаты [железо (III), алюминий и др.]; в присутствии азотной кислоты также возможен гидролиз и соосаждение соединений титана, ванадия, ниобия, молибдена, тантала, вольфрама, германия, олова и сурьмы. Пономарев считает, однако, что HNO3 полнее выделяет SiO2 [438, 439]. Часто используют смеси кислот (см. главу 4). Предложено использовать смесь NH4C1 + НС1 [94, 208, 871].
Сравнение действия хлористоводородной и азотной кислот на карбонатные плавы [515] показало, что в соответствии с оптимальными условиями получения крупнозернистых осадков желательно применение концентрированных кислот для разложения плавов [438, 510]. Кремневая кислота выделяется более полно при дегидратации азотной кислотой с плотностью 1,4—1,5 г/см3, однако при этом наблюдается [515] значительное загрязнение ее примесями.
Определение малых количеств кремневой кислоты, оставшихся в фильтрате после отделения в форме п SiO2 • т Н2О
Попытки выделить нацело кремневую кислоту путем многократной ее дегидратации, несмотря на тщательность выполнения, обречены на неудачу. Это объясняется тем, что скорость растворения 58
и скорость деполимеризации кремневой кислоты — величины в достаточной степени большие с точки зрения аналитика, даже в условиях кратковременного воздействия воды и кислот. Если учесть при этом возможность поступления кремневой кислоты из реактивов и выщелачивания ее из посуды, то многократная дегидратация может только исказить результаты.
Возможно, однако, почти количественное выделение неосажден-ной при двукратной дегидратации кремневой кислоты соосажде-нием в общем ходе анализа с полуторными окислами. Полнота выделения зависит от количества полуторных окислов: если оно мало (<0,1 а) — кремневая кислота безвозвратно теряется. Этот вопрос специально исследован, и данные обобщены [188, 1011].
Егорова [188] изучила соосаждение малых количеств кремневой кислоты с гидроокисями алюминия, железа и титана на синтетических растворах. Количество А12О3, которое необходимо для практически полного выделения SiO2, зависит от абсолютного количества последней. Для выделения 1 мг SiO2 требуется количество А12О3 в 3,5 раза большее по весу, для 25 мг — лишь в 1,7 раза. С гидроокисями железа и титана кремневая кислота соосаждается неполностью даже при 5—7-кратном избытке осадителя.
Соосажденная кремневая кислота из осадка полуторных окислов может быть выделена обычной дегидратацией. Кроме того, прокаленный осадок полуторных окислов может быть обработан HF и H2SO4 и содержание SiO2 найдено по разности весов до и после прокаливания. Разработаны методы фотометрического определения малых количеств кремневой кислоты, оставшейся в растворе [188, 845].
Сочетание однократной дегидратации с колориметрическим определением кремневой кислоты в фильтрате дает хорошие результаты при значительной экономии времени на выполнение определения. Методики описаны в нескольких работах [188, 784, 845, 1010, 1015, 1105].
Определение кремневой кислоты в силикатных породах комбинированным — гравиметрическим и колориметрическим — способом [846]. 1 г минерала или породы сплавляют с Na2CO3 и после разложения водой и НС1 выпаривают досуха, к сухому остатку прибавляют 10 мл конц. НС1 и горячую воду для растворения солей, осадок фильтруют, промывают и прокаливают. Фильтрат и промывные воды собирают в мерную колбу емкостью 200 мл, охлаждают, доводят водой до метки, и аликвотную пробу — 5 мл — колориметрируют. Определение выполняют по синему кремнемолибденовому комплексу, как описано в главе 7.
Ускоренные методы определения кремния в форме SiO2
Сокращение затрат времени на гравиметрическое определение кремния может быть достигнуто несколькими путями: 1) при косвенном гравиметрическом определении, когда анализируемый образец обрабатывают смесью серной и фтористоводородной кислот. Метод
69
используется при анализе веществ с высоким содержанием кремния для экспресс-определений и в некоторых других случаях [691, 10801; 2) при сокращении расхода времени на подготовительные операции — замена сплавления спеканием, замена сплавления разложением кислотами под давлением (см. главу 4); 3) при ускорении разложения кислотами за счет введения в них различных добавок, например NH4C1 [94, 161, 208, 325, 438]; 4) путем исключения повторных дегидратаций с использованием наиболее эффективных дегидратирующих средств — серной или хлорной кислот, глицерина, ацетилхлорида, уксусного ангидрида [210, 211, 403, 891, 892, 939, 950, 963]; 5) ускорением процесса выпаривания с кислотами повышением температуры бани, для чего используются высоко-кипящие жидкости [853], термоизлучатели [592]; 6) при выделении кремневой кислоты с использованием коагулянтов, главным образом желатины [38, 162, 164, 188, 228, 379, 438, 515, 519, 529, 788, 1030]; 7) ускорением фильтрования, промывания, сушки и прокаливания осадка, например при фильтровании под давлением или в вакууме, заменой сушки промыванием органическими растворителями [169, 326, 626].
Другие ускоренные методы см. в главах 6, 7 и 9, а также в [1106].
Использование для ускорения выделения кремневой кислоты коагулирующего действия растворов желатины применяется чаще всего.
Определение кремневой кислоты дегидратацией с желатиной. Выделение кремневой кислоты коагуляцией ее органическими соединениями с большим молекулярным весом давно используется в технологии и в аналитической практике [4, 188, 439, 529]. В качественном анализе кремневую кислоту осаждают с целью идентификации растворами малахитового зеленого, метиленового синего, сафранина и других красителей (см. главу 3). Альбумин выделяет из растворов кремневые кислоты с большим молекулярным весом (гексакремневую и т. д.).
В практику гравиметрического определения кремния коагулянты вошли после работы Тимофеюка [529], который использовал желатину сначала для облегчения фильтрования в технологии переработки апатитовых руд, а потом для отделения кремневой кислоты в ходе анализа соединений кремния. В качестве преимуществ метода автор указывает на замену трехкратного выпаривания однократным, а также на то, что осадок кремневой кислоты получается чище и не приходится прибегать к дальнейшей его кислотной обработке. Кроме того, значительно ускоряются фильтрование и промывание осадка.
Другие коагулянты — столярный клей, альбумин, казеин, агар-агар и другие вещества [162, 164, 519, 780, 1091] — также дали удовлетворительные результаты. Однако в общем желатиновый метод Тимофеюка сохранил свою основу. И хотя следует критически воспринимать высказывания некоторых аналитиков о том, что желатиновый метод «по точности не уступает классическому», его 60
преимущества в отношении быстроты все же неоспоримы. По заключению Егоровой, сделанному на основании большого экспериментального материала, содержание SiO2, полученное ускоренным методом, всегда ниже установленного классическим методом [188]. Разность колеблется в пределах от 0,25 до 1%.
Изучая желатиновый метод, авторы работ ставили перед собой различные цели, поэтому метод имеет многочисленные варианты. В большинстве случаев цель состояла в ускорении определения, были попытки разработки способов более полного выделения кремневой кислоты' из растворов, а также получения чистых осадков.
Ускорение определения кремневой кислоты желатиновым методом достигается благодаря: 1) однократной предварительной дегидратации выпариванием с кислотами или исключению выпаривания с кислотами вообще; 2) быстроте фильтрования и промывания коагулированного осадка; 3) чистоте осадка, вследствие чего нет необходимости обрабатывать его смесью HF и H2SO4.
При действии желатины обеспечивается коагуляция коллоид-норастворенной кремневой кислоты, которая могла бы забивать поры фильтра и замедлять фильтрование. Вследствие того что фильтрование и промывание проходят быстро, время соприкосновения промывных вод с осадком сокращается. Это уменьшает потери осадка от растворимости. Однако истиннорастворенная молекулярнодисперсная кремневая кислота желатиной не осаждается. Поэтому потери неизбежны.
Уменьшение потерь от растворимости в различных вариантах желатинового метода достигается за счет: 1) применения для разложения плавов концентрированных кислот [147, 438, 515]; 2) предварительного выпаривания раствора или досуха, или до состояния влажных солей [25, 188, 438], или просто до небольшого объема; 3) использования выпаривания с серной или хлорной кислотами [36]; 4) промывания растворами электролитов (например, 2%-ным раствором NH4C1) для предотвращения пептизации геля.
Осаждение кремневой кислоты желатиновым методом чаще всего выполняют из солянокислых растворов, получаемых разложением плавов или анализируемых образцов, причем в большинстве случаев в этот раствор вводят желатину без предварительного выпаривания [125, 162, 529]. Экспериментальное изучение условий выделения кремневой кислоты желатиной из солянокислых растворов или из смеси HC1+H2SO4 см. в [36, 515, 1091], литературный обзор — в [188, 485, 1091].
Оптимальная концентрация хлористоводородной кислоты перед добавлением желатины составляет 20 г на 100 мл раствора, оптимальная температура выделения кремневой кислоты 70—75° С, минимальное количество желатины около 40 мг на 0,2818 г SiO, [1091].
Выполнение сравнительных определений в различных кислотах показывает, что уксусная и фосфорная кислоты не могут быть ис
61
пользованы в качестве сред для выделения кремневой кислоты желатиной. Из уксуснокислых растворов кремневая кислота выделяется недостаточно полно [485, 1091], фосфорная кислота, как правило, сильно загрязняется кремневой кислотой в процессе хранения, и применение ее искажает результаты анализа. Наиболее полно из обследованных кислот выделяют кремневую кислоту концентрированные серная и азотная кислоты [585].
Механизм действия желатины как коагулянта кремневой кислоты не нашел еще достаточного объяснения. Чаще всего считают [188], что отрицательно заряженные частицы коллоидной кремневой кислоты коагулируют вследствие прибавления положительно заряженного коллоида — желатины, однако избыток желатины не оказывает пептизирующего действия. По мнению Швайгера [607], действующим началом желатины следует считать органические аминокислоты. В качестве коагулянта он предлагает антраниловую кислоту.
Изучение различных вариантов выделения кремневой кислоты желатиной из кислотных растворов, полученных разложением карбонатных плавов, показало, что кремневая кислота остается в растворе главным образом благодаря переходу геля в раствор при растворении солей и промывании осадка. В качестве наиболее точного и одновременно простого варианта желатинового метода определения кремния на основании большого сравнительного экспериментального материала рекомендован следующий [188].
Навеску тонкоизмельченного анализируемого материала (глина, полевой шпат, кварцевый песок, слюда) около 0,5 г тщательно перемешивают с равным по весу количеством безводного карбоната натрия и спекают при 800° С в течение 1,5 час в платиновом тигле. Спек разлагают при нагревании 10 мл конц. НС1, раствор выпаривают до состояния влажных солей и вводят 3 мл 2%-ного раствора желатины. Далее растворяют соли 50 мл воды (без нагревания), фильтруют осадок и промывают холодным раствором НС1 (5 : 95). Операции высушивания, озоления фильтра и прокаливания осадка ведут, как обычно.
Определение кремния в присутствии фторидов
При определении кремневой кислоты гравиметрическим методом после дегидратации кислотами в присутствии фторид-ионов наблюдаются значительно потери кремния. Это объясняется улетучиванием кремния в виде SiF4, как отметил еще Берцелиус, который предусмотрел и предотвращение потерь кремния [662, 663]. В дальнейшем вопрос был дополнительно изучен [188, 817, 1013, 1049], и длительный метод Берцелиуса упрощен [188, 376, 817, 1010, 1013, 1014], а также предложены другие методы определения кремния в присутствии фтора [413, 674, 797, 997].
Методы определения кремния в присутствии фтора можно подразделить на несколько групп: 1) методы, основанные на выделении кремневой кислоты из растворов с высоким значением pH [715, 817, 1013]; 2) методы, основанные на связывании фторид-ионов в комплексные ионы с алюминием или бором [674, 841, 997, 1004,
62
1010, 10281; 3) алкалиметрические, колориметрические и другие методы, которые рассмотрены в главах 6 и 7.
Для осаждения кремния в щелочных средах растворенный в воде образец или разложенный водой щелочной плав отделяют от осадка фильтрованием и затем несколько часов обрабатывают раствором (NH4)2CO3 при небольшом нагревании (около 40° С), оставляют стоять на ночь и на следующий день отфильтровывают осадок кремневой кислоты, промывая его холодной водой с небольшим количеством карбоната аммония. Фильтрат упаривают почти досуха и, разбавив небольшим количеством воды, нейтрализуют 2N НС1 (в отсутствие фосфатов и хрома) или HNO3 [149] по фенолфталеину. Затем фильтрат обрабатывают аммиачным раствором окиси цинка (приготовление см. в [161]) или аммиачно-карбонатным раствором окиси ртути [149], выпаривают досуха на водяной бане, извлекают водой, фильтруют и промывают водой.
Осадки кремневой кислоты и силиката цинка после смывания с фильтра [149, 161] или сжигания фильтров помещают в фарфоровую чашку и проводят дальнейший анализ методом двойной дегидратации с НС1. Осадок кремневой кислоты обрабатывают смесью HF и H2SO4 и по разности весов до и после обработки находят содержание SiO2.
Метод описан в руководствах [149, 188]. Он был упрощен последовательно несколькими исследователями [188, 817, 1013, 10141. Было предложено исключить осаждение карбонатом аммония при сохранении обработки аммиачным раствором окиси цинка или со-, кратить длительность обработки, заменяя стояние выпариванием; предложено сплавление со смесью окиси цинка и карбоната натрия [10131, и в конечном счете сформулированы методики, значительно сокращающие длительность определения.
Сравнительная оценка методов и детальный разбор отдельных стадий методов даны в работе [188]. Установлено, что в растворе после выделения кремневой кислоты аммиачным раствором окиси цинка по Берцелиусу [662, 6631 оставалось от 1 до 3,9 мг SiO2, причем остаток практически не зависел от количества первоначально взятой кремневой кислоты, но был тем меньше, чем точнее выполнялась нейтрализация перед введением окиси цинка. Автор считает [188], что при замене выдерживания с карбонатом аммония выпариванием почти досуха при точной нейтрализации раствора по метиловому красному [149] замена аммиачного раствора окиси цинка нитратом цинка не имеет преимуществ.
Ускоренное определение содержания двуокиси кремния во фторсодержащих силикатах (в редакции автора [188]). Метод разработан с использованием синтетических смесей (SiO2-|-+Al2O3+CaF2) и образцов полевого шпата и слюды с добавкой фторида кальция.
Навеску образца 0,5—1 г сплавляют с 4 г карбоната натрия, плав выщелачивают в платиновой чашке водой, нерастворимый остаток отфильтровывают и промывают 2—3 раза 0,5%-ным раствором Na2CO3. Затем нерастворимый остаток обрабатывают при кипячении 50 мл 2%-ного раствора Na2CO3, фильтруют и промывают 0,5%-ным раствором Na2CO3. Оба фильтрата помещают в платиновую чашку и прибавляют в нее 10 г твердого (NH4)2CO3; после растворения его раствор постепенно нагревают и выпаривают почти досуха.
Влажную массу разбавляют 30—40 мл воды, отфильтровывают выделившуюся кремневую кислоту и промывают ее 0,5%-ным раствором (NH4)2CO3. Фильтрат и промывные воды собирают в мерную колбу емкостью 250 мл и доводят водой до метки. В аликвотной части раствора устанавливают количество НС1, необходимое для нейтрализации по метиловому оранжевому, а в другой ча
63
сти раствора после нейтрализации определяют содержание кремневой кислоты, недовыделенной карбонатом аммония, колориметрически по желтому кремнемолибденовому комплексу.
Ранее полученные осадки кремневой кислоты смывают с фильтров последовательно небольшим количеством воды и разбавленной НС1 в фарфоровую чашку, сами фильтры озоляют и золу присоединяют к содержимому чашки. Раствор выпаривают на водяной бане до состояния влажных солей и вводят при перемешивании 3 мл 2%-ного раствора желатины. После прибавления 30 мл веды и растворения солей выделившийся осадок кремневой кислоты отфильтровывают и промывают разбавленным раствором НС1 (5 : 95). Ошибка вследствие потерь кремнезема составляет около 0,5%.
Преимущество метода кроме быстроты — возможность одновременного определения фтора из той же нагески.
Метод сплавления с окисью цинка [1013]. Для сплавления со смесью ZnO и Na2CO3 анализируемый образец должен быть очень тщательно измельчен. Навеску образца 0,5—2,0 г смешивают с 6 г смеси ZnO и Na2CO3 (1:5) (даются и другие соотношения [1011]) в платиновом (можно никелевом) тигле и помещают в холодный муфель, доводя за 30 мин температуру до 950— 1050° С (здесь же описана методика сплавления на горелке). Плав (или тигель с плавом, если плав не отделился при охлаждении) помещают в стакан из нержавеющей стали емкостью 500 мл и прибавляют 200 мл холодной воды, спек размельчают и оставляют стоять для растворения солей (обычно на ночь).
Вынув и обмыв водой тигель и крышку, медленно прибавляют в стакан при перемешивании металлическим шпателем (никель или нержавеющая сталь) 25 мл раствора аммиачной окиси цинка [1,0г ZhOh 13г (NH4)2CO3 и 2 мл конц. NH4OH растворяют в 10 мл воды и доводят до 25 мл]. Раствор нагревают до кипения, накрывают и ставят на горячую плитку в более широкую посуду (например, в алюминиевую чашку) для предотвращения разбрызгивания.
После десятиминутного кипячения стакан снимают с плитки, дают ему стоять 5 мин и фильтруют содержимое, собирая фильтрат в мерную колбу емкостью 500 мл. Осадок после тщательного промывания декантацией переносят количественно на фильтр и промывают горячей водой до отсутствия реакции на фторид-ионы. Объем фильтрата в мерной колбе доводят водой до метки, и после перемешивания в аликвотной пробе (25 мл) устанавливают pH 1,3—1,4 с помощью НС1 (контроль на pH-метре) и определяют кремний в форме восстановленного кремнемолибденового комплекса.
Отфильтрованный осадок, содержащий кремний, вместе с фильтром переносят в платиновую чашку, фильтр озоляют, к осадку прибавляют 20 мл НС1 и определяют кремневую кислоту после разложения и двукратной дегидратации как обычно. Для второй дегидратации рекомендуется употребление 20 мл НС1О4 и 3_j«.i1HNO3.
Общее содержание кремневой кислоты в образце получают, суммируя результаты гравиметрического и колориметрического определений. Поправка на потери кремневой кислоты вследствие неполноты выделения при двукратной дегидратации составляет около 0,2%.
Разработаны методы определения кремния в присутствии фтора, основанные на введении ионов-комплексообразователей для связывания фтора. В качестве таких ионов изучены Al(III), Ti(LV), Zr(IV) и В(Ш). Результаты, представляющие интерес, получены с бором и алюминием.
Нашли практическое применение методы с использованием А1С13, причем обе описанные методики [188, 1010] предполагают гравиметрическое определение основного количества кремневой кислоты и фотоколориметрическое — кремневой кислоты, оставшейся в растворе после дегидратации по желтому [1881 и по синему, гетерополи комплексам [1010].
64
Еще в ранних работах было замечено, что присутствие сравнительно небольших количеств фторидов не вызывает потерь кремневой кислоты в ходе анализа. Эти количества составляли около 2% фтора. Дальнейшее изучение показало, что в анализируемых образцах присутствовали значительные количества алюминия. Влияние добавления А1(Ш) на извлечение кремневой кислоты из фторсиликатов по обычной гравиметрической методике изучено в [188, 1011].
Определение кремния во фторсиликатах в присутствии хлорида алюминия [1011]. 0,5—1,0 г образца сплавляют с 5 г Na2CO3 в платиновом или корундизовом тигле, плав растворяют в 200 мл НС1 (1 : 3), содержащей не менее 500 мг А1(Ш) (при введении большего количества могут быть задержки в фильтровании из-за забивания пор фильтра), упаривают под нагревательной лампой, подкисляют, фильтруют и промывают как обычно [149]. После второй дегидратации для облегчения собирания осадка прибавляют бумажную массу. Результаты определения равноценны результатам, полученным методом с окисью цинка [1015]; потери составляют около 0,2 мг. На эту величину рекомендуется вводить поправку.
Определение кремния и фтора при совместном присутствии см. в [413, 797, 1010, 1013], кремния в присутствии фтора и фосфора — в [715, 805], фтора и бора — в [1004, 1014].
Определение кремния в присутствии бора
При гравиметрическом определении кремния в присутствии соединений бора осадок кремневой кислоты в некоторой степени загрязняется ими, и результаты получаются завышенными. Ошибка неизбежна и при обработке осадка кремневой кислоты смесью фтористоводородной и серной кислот, потому что фторид бора улетучивается вместе с фторидом кремния. Соединения бора могут присутствовать в качестве составной части анализируемых образцов (например, в стеклах или глазурях), а также вносятся при использовании плавней — тетрабората натрия, борного ангидрида, борной кислоты.
В некоторых работах [811,950] определение кремния и последующие аналитические операции ведут без отделения бора. Пренебрежение возможностью загрязнения осадка кремневой кислоты бором может внести серьезные искажения в результаты определений [999, 1011].
Удаление борной кислоты из анализируемого раствора основано на реакции образования летучего борнометилового эфира [1004]
Н3ВО3 + ЗСН3ОН -> В (ОСН3)3 + ЗН2О.
Сухой остаток, полученный при дегидратации выпариванием солянокислого раствора, обрабатывают абсолютным метанолом, насыщенным хлористым водородом. Раствор выпаривают досуха на водяной бане, повторяя операцию обработки от двух до четырех раз. Пределы применимости этой методики с использованием про-
3 Аналитическая химия кремния
65
дажного метанола и этанола определены в работе [1881. Найдено, что эффективное удаление борной кислоты продажным этанолом и метанолом по существу не достигается и применять можно только обезвоженный метанол с четырехкратной обработкой осадков.
Предложен метод определения кремния в присутствии бора, основанный на связывании борной кислоты в борноглицериновую кислоту [963]. Автором дана методика обработки плавов, получаемых сплавлением анализируемых веществ с окисью бора. Другие работы по определению кремния в присутствии бора приведены в [188, 439, 950, 1011], а также бора и фосфора — в [439].
ДРУГИЕ ГРАВИМЕТРИЧЕСКИЕ МЕТОДЫ
Разработано несколько методик гравиметрического определения кремния, основанных на осаждении кремнесодержащих гетерополикислот (главным образом кремне-12-молибденовой кислоты) органическими основаниями. Возможность такого осаждения упоминается Бергом [69] и впервые осуществлена Волынцом [134—136] при определении кремния в силикатных породах осаждением кремне-12-молибденовой кислоты 8-оксихинолоном. Метод требует обязательного перевода кремневой кислоты в мономерную форму, что обычно осуществляется сплавлением образца со щелочами или карбонатами щелочных металлов [151, 406, 530, 674, 788, 843].
8-Оксихинолин образует с желтой кремне-12-мо-
НО N
либденовой кислотой соединение состава H4SiMo12O40-4C9H7ON при pH 1,3—1,5. Фтор может быть связан прибавлением борной кислоты.
Определение двуокиси кремния в горных породах, содержащих фтор [674]. Навеску образца, содержащую не более 30 мг SiO2 или SiO24-P2O6 (если не предполагается работать с аликвотными частями раствора), сплавляют с NaOH. Плав растворяют в воде и прибавляют Н3ВО3 (от 0,5 до Юг в зависимости от величины навески и предполагаемого содержания фтора), индикатор тимоловый синий и по каплям конц. НС1 до перехода цвета индикатора от желтого к красному. Затем прибавляют последовательно при перемешивании 8 мл НС1 (1 : 9), 5 мл СН3СООН (1 : 2) и 20 мл 10%-ного раствора молибдата аммония и дают стоять 15 мин. Приливают к раствору 40 мл НС1 (1 : 1) и сразу же осаждают кремнемолибденовую кислоту добавлением 60 мл раствора 8-оксихинолина (14 г реактива растворяют в 20 мл QN НС1 и доводят водой до 1 л) из бюретки при перемешивании. Стакан с осадком нагревают на водяной бане при 60° С в течение Ю мин при периодическом помешивании.
После охлаждения раствор сливают с осадка во взвешенный фильтрующий тигель со стеклянным фильтром № 4, осадок дважды промывают декантацией насыщенным водным раствором кремнемолибдата 8-оксихинолина и количественно переносят в тигель, где промывают осадок этим же промывным раствором еще 10 раз и в заключение — еще один раз 5 мл воды.
Осадок сушат 1 час при 140° С, охлаждают в эксикаторе и взвешивают. Весовая форма 4CeH7ON• SiO2- 12МоО3-2Н2О. Фактор пересчета на SiO2 равен 0,025. При нагревании вплоть до 200° С этот состав сохраняется [1011]. Затем осадок теряет 2 молекулы воды и в пределах 225—250° С может быть взвешен как
66
4CeH7ON-S102-12Mo03. В пределах 593—813° С состав осадка соответствует Si02-12Mo03 и выше 813° С начинается возгонка МоО3. В присутствии фосфора следует производить пересчет после определения его другими методами.
Для пород, содержащих 40—70% SiO2, очень точные результаты были получены полу микрометодом [915] при использовании для осаждения 2,4-диметилхинолина. Применяют навески вещества с содержанием SiOa до 2 мг. Определению мешают мышьяк и германий. Фосфор осаждается вместе с кремнием. На основании сообщения Волынца [134, 135] разработаны методики микро- и полумикроопределений кремневой кислоты с 8-оксихинолином [907, 1004]. Установлены оптимальные условия для выполнения этого определения: кислотность по НО 2—3N, температура около 25° С. Разработана методика для анализа летучих соединений кремния [1004] с навесками 2—20 мг (иногда до 100 мг). Образцы сплавляют с металлическим калием в никелевой бомбе с дальнейшим разложением водным раствором метанола. Стеклянную посуду (чашки, стаканы, воронки) настоятельно рекомендуется заменять никелевой, платиновой или полиэтиленовой.
При анализе кремнийорганических соединений применяют также и полумикровариант методики Волынца [135, 907], причем подготовку образца к определению выполняют сплавлением в бомбе с перекисью натрия.
Навеску 50—200 мг анализируемого кремнийорганического соединения сплавляют с Na2O2 [907 ] (2—3 г) и сахаром (0,05—0,1 г) в никелевой бомбе Парра [907]. Плав растворяют в воде, слегка подкисляют НС1 (проба на лакмус), и полученный раствор разбавляют водой до 1 л в мерной колбе. Аликвотную часть (50 мл} раствора переносят в колбу емкостью 250 мл, добавляют 15 мл НС1 (1 : 1), 50 мл воды и 15 мл 20%-ного раствора молибдата аммония. Затем колбу закрывают, содержимое нагревают при 75° С в течение 10 мин и после охлаждения до комнатной температуры прибавляют 20 мл НС1 (1 : 1).
Далее в колбу из бюретки прибавляют 25 мл 0,4/1’ раствора 8-оксихинолина, одновременно встряхивая содержимое колбы. При этом выпадает осадок оксин-кремнемолибденового комплекса. Затем колбу с содержимым нагревают при 65° С в течение 10 мин. Охлажденный до комнатной температуры раствор с осадком фильтруют.
Осадок на фильтре промывают раствором 8-оксихинолина в НС1. Отмытый осадок сушат при НО—120° С в течение часа, затем сжигают и прокаливают при 500° С до постоянного веса. Весовая форма SiO2-12MoO3.
Возможно, что осадок кремнемолибдата 8-оксихинолина имеет состав, несколько отличающийся от теоретического вследствие со-осаждения молибдата 8-оксихинолина, но фактор для расчета SiO2 настолько низок, что на результатах определения кремния это практически не отражается.
Кремне-12-молибденовая кислота может быть осаждена пиридином [40, 41, 694, 911], хинолином [180, 291, 644, 657, 659, 673, 702, 788, 1099], амидопирином [788, 860], гексиламином [725], диантипирилметаном [440].
Определение кремния в форме кремне-12-молибдата хинолина рекомендовано для силикатных материалов при самых разнообразных содержаниях SiO2 [189, 659]. Продолжительность определения
3*
67
—3 час; фосфор, ванадий и мышьяк мешают определению и должны быть удалены.
0,5 г образца, измельченного до прохождения через сито 200 меш (при содержании SiO2 больше 65% навеска 0,25 г), помещают в никелевый тигель на слой предварительно нагретого до сплавления и охлажденного плавня (7 г NaOH) [659]. Образец смачивают этанолом во избежание распыления и после непродолжительного нагревания для испарения спирта плавят на пламени горелки, вращая тигель и постепенно повышая температуру. Сплавление длится около 5 мин, тигель с прозрачным плавом осторожно охлаждают до затвердевания погружением в холодную воду.
Еще горячий тигель помещают в никелевый стакан емкостью 400 мл и, накрыв стеклом, наливают в тигель из промывалки (приподняв стекло) горячую воду. Когда плав растворится, тигель вынимают щипцами, ополаскивают горячей водой тигель и стекло.
Полученный раствор доводят до объема около 175 мл, количественно переносят в коническую колбу емкостью 500 мл, куда предварительно помещено 20 мл конц. НС1, промывая стакан несколькими маленькими порциями воды. Раствор быстро охлаждают до комнатной температуры, количественно переводят в мерную колбу емкостью 250 мл и доводят водой до метки.
Отбирают аликвотную пробу раствора, содержащую 5 мг SiO2, разбавляют в полиэтиленовом стакане до объема около 250 мл и вносят около 3 г гранулированного NaOH, растворяя его перемешиванием. К раствору прибавляют 10 капель раствора индикатора* и по каплям НС1 до перехода окраски индикатора от синего через желтый до красного цвета.
Полученный раствор переносят в стеклянный стакан емкостью 800 мл, прибавляют 10 мл НС1 (1:9) и доводят объем раствора до 400 мл. К полученному раствору прибавляют 50 мл 10%-ного раствора молибдата аммония при перемешивании; после 10-минутной выдержки прибавляют 50 мл конц. НС1 и осаждают кремне-12-молибденовую кислоту, прибавляя 50 мл 2%-ного раствора хинолина из бюретки при перемешивании. Стакан нагревают около 10 мин до 80° С с выдержкой при этой температуре 5 мин.
Охладив раствор с осадком до 20° С (или ниже), фильтруют осадок через взвешенный тигель со стеклянным фильтром № 4. После количественного переноса осадок промывают на фильтре шесть раз 0,05%-ным раствором хинолина (25 мл осадителя доводят до 1 л водой), затем сушат при 150° С, охлаждают и взвешивают. Весовая форма SiO2-12MoO3-4CeH7N-2H2O. Фактор пересчета на SiO2 равен 0,0256.
Гравиметрическое микроопределение кремния в органических материалах в виде хинолинкремнемолибдата см. [702], в растворимых силикатах [624]. Обзор методов определения кремния осаждением кремне-12-молибденовой кислоты органическими основаниями с гравиметрическим окончанием имеется в работах Жана [842, 843], основные данные цитированы Никитиной [404, 406, 407]. Даются указания на применение амидопирина [788, 860], пиридина [40, 41, 911], 8-оксихинолина [151, 788, 901, 907, 966, 1004], триметиламина [843, 911], хинолина [76, 291, 644, 659, 788, 1099], 2,4-диметилхинолина [915]. О применении производных пиридина и хинолина см. в [530, 788, 899, 907], диантипирилметана [197, 440], уротропина [725, 726, 788].
* Индикатор: а) 0,1 г крезолового красного растворяют в 5,3 мл 0,1 N раствора NaOH и разбавляют водой до 100 мл\ б) 0,1 а тимолового синего растворяют в 20 мл этанола, прибавляют 2,1 мл раствора NaOH и доводят водой до 100 лы; растворы а) и б) смешивают.
68
Определение кремневой кислоты в силикатных породах с диантипирилметаном (полумикрометод) [440]. 0,05 г пробы с содержанием 10—50% SiO2 сплавляют с 1 г Na2CO3, плав выщелачивают водой, по охлаждении прибавляют 30 мл \N НС1, раствор разбавляют до 250 мл. К 25 мл полученного раствора приливают 7 мл \N НС1, 5 мл 10%-ного раствора молибдата аммония, нагревают на водяной бане 15 мин, разбавляют вдвое водой (при большом содержании разбавляют до 1000 мл), вводят 7—14 мл конц. НС1 и по каплям 3 мл 2%-ного раствора диантипирилметана в 0,5W НС1.
Выделившийся осадок соли кремнемолибденовой кислоты с диантипирилметаном отфильтровывают на стеклянном фильтре № 3 или 4, промывают холодной водой, слабо подкисленной НС1, высушивают при 125—130° С и взвешивают. Фактор пересчета 0,01986. Ошибка определения около 0,9%, продолжительность определения 3—3,5 час.
Харцдорф [810] изучил реакции большой группы органических оснований различного состава с кремнемолибденовой кислотой и рекомендовал в качестве осадителя для гравиметрического определения кремния пиридин. Получаемый при этом осадок, по данным автора, обладает низкой растворимостью, хорошо фильтруется и оголяется. В виде кремнемолибдата пиридина еще можно определять с достаточной точностью 0,1 мг кремния в виде SiO2 в 400 мл раствора. На этом основании метод рекомендован для определения кремния в различных соединениях и материалах, в том числе HF, криолите, фториде кальция, кремнийорганических соединениях.
Автор подчеркивает необходимость тщательного соблюдения условий осаждения для сохранения кремневой кислоты в мономерном состоянии и количественного перевода ее в кремнемолибденовую кислоту. Подготовку раствора проводят после сплавления образца с карбонатами щелочных металлов или со щелочами, а также после кипячения с растворами щелочей — при анализе водных растворов. Подкисление раствора для перевода кремния в кремнемолибденовую кислоту возможно только после прибавления молибдата аммония.
Пробу щелочного раствора, содержащего 0,1—100 мг SiO2, помещают в полиэтиленовый стакан, прибавляют 45 мл раствора молибдата аммония (200 г продажного препарата растворяют в воде, доводят объем раствора до 1 л и нейтрализуют раствором NH4OH по тимоловому синему), доводят общий объем раствора до 400 мл и прибавляют по каплям раствор конц. НС1 до pH 2,5 (контроль по pH-метру со стеклянным электродом).
После часового стояния прибавляют при перемешивании 75 мл раствора конц. НС1 и 65 мл раствора хлорида пиридиния (к 158 г пиридина прибавляют 400 мл 8N НС1 и доводят водой до 1 л). Образовавшийся светло-желтый хлопьевидный осадок выдерживают в растворе 1 час и затем отфильтровывают через стеклянный фильтр № 2. Для промывания достаточно 150 мл промывного раствора (к 60 мл осаждающего раствора хлорида пиридиния прибавляют 120 мл 8N НС1 и доводят водой до 1 л).
После кратковременного просушивания осадка при 120° С его озоляют и прокаливают при температуре не выше 500° С. Весовая форма SiO2-12МоО3.
При анализе фторсодержащих материалов к пробе раствора перед добавлением молибдата аммония прибавляют 7 г Na2B4O7- ЮН2О. Это позволяет маскировать до 1 г фтора.
Разработан метод определения кремния осаждением синего кремнемолибденового комплекса хинолином [563, 564]. Метод
69
позволяет определять кремний в присутствии фосфора, мышьяка и ванадия.
Определение кремния в сталях [564]. Образец стали (~0,5 г) растворяют при нагревании в 20 мл 5%-ной H2SO4, фильтруют, промывая осадок раствором H2SO4; полученный фильтрат доводят водой до 50—60 мл и прибавляют 20 мл 5%-ного раствора молибдата аммония (при этом появляется синяя окраска раствора). Через 10 мин вводят 10 мл конц. НС1 и 30 мл 2%-ного раствора хинолина [20 мл хинолина растворяют в 50 мл НС1 (1 : 1) и доливают водой до 1 л], нагревают до 70—80° С, хинолинкремнемолибдат осаждается.
Раствор с осадком охлаждают до 20° С, фильтруют через стеклянный фильтр № 4, промывают 5—6 раз промывным раствором (25 мл 2%-ного раствора хинолина в 1 л раствора), сушат 1 час при 150° С и взвешивают. Фактор пересчета 0,02566.
Определение кремневольфрамовой кислоты в ее препаратах предложено выполнять с помощью 8-оксихинолина [121, 1086]. Осаждение гетерополикислот органическими основаниями используют в анализе силикатов [41, 291, 440, 673, 899, 915], чугуна и сталей [563, 564], фтористых соединений [674, 810], шлаков [93, 151], кремнийорганических соединений [292, 296, 384], клинкера и портландцемента [93], стекла [76, 564].
Для гравиметрического определения кремния в кремнийорганических соединениях предложено осаждать кремнефтористоводородную кислоту бензидином [292 , 384 , 441].
20—50 мг тетраэтоксисилана помещают в полиэтиленовый стакан, содержащий около 50 мл 0,1—0,ЗА^ спиртового или ацетонового раствора фтористоводородной кислоты. После двухминутного перемешивания содержимого стакана (покачиванием) добавляют постепенно при перемешивании 20 мл 1%-ного спиртсвого или ацетонового раствора бензидина. Выпавший осадок кремнефтористоводородного бензидина количественно переносят в фильтрующий тигель № 3 и фильтруют с применением вакуума (водоструйный насос). Осадок промывают на фильтре охлажденным спиртом или ацетоном и сушат тигель при 100° С до постоянного веса. Весовая форма C12H12N2.H2SiF6. Время определения 2—3 час [292, 384].
Глава 6
ТИТРИМЕТРИЧЕСКИЕ МЕТОДЫ
Прямое титрование кремневой кислоты растворами гидроокиси натрия и калия неприменимо, так как кремневая кислота слабая и очень трудно подобрать индикатор, который дает резкий переход окраски вблизи точки эквивалентности. Кроме того, выделение в процессе титрования студенистого осадка кремневой кислоты при концентрации ее в растворе выше 0,01 N существенно искажает результаты определения.
Существует ряд методов титриметрического определения кремния, однако наиболее часто для определения кремния используют методы, основанные на свойствах кремнемолибденовой и кремнефтористоводородной кислот.
МЕТОДЫ, ОСНОВАННЫЕ НА СВОЙСТВАХ КРЕМНЕМОЛИБДЕНОВОЙ КИСЛОТЫ
Методы окисления-восстановления
Кремний может быть определен титрованием желтой кремнемолибденовой кислоты восстановителями. Количество восстанавливающего агента, требуемого для восстановления кремнемолибденовой кислоты, пропорционально количеству кремния, идущего на образование гетерополикислоты. По количеству восстановителя, пошедшему на образование молибденовой сини, судят о содержании кремния. Вейцман [1231 предложила восстанавливать образующуюся в ходе анализа кремнемолибденовую кислоту иодидом калия с последующим титрованием выделившегося иода (в количестве, эквивалентном содержанию кремния) раствором тиосульфата.
В последнее время появились указания о возможности определения кремния титрованием восстановленного молибдена, входящего в состав кремнемолибденовой кислоты [1123, 1132]. Кутейников предложил использовать этот метод для определения кремния в тугоплавких соединениях. Для восстановления кремнемолибденовой
71
кислоты использовали растворы сульфита натрия, а для титрования восстановленных соединений — перманганата калия [1111]. Приводим методику определения кремния предлагаемым методом.
Навеску анализируемого вещества 0,1—0,3 г сплавляют в платиновом тигле с содой при 1000° С. Плав выщелачивают горячей водой и переносят в мерную колбу емкостью 250 мл. Отбирают аликвотную часть раствора, содержащую 1,1— 15 мг кремния, добавляют 25 мл 10%-ного раствора молибдата аммония, устанавливают pH 2—3, добавляют 20 мл 10%-ного Na2SO3, кипятят 7 мин для восстановления кремнемолибдата до молибденовой сини. Увеличивают концентрацию H2SO4 до 2N, кипятят еще 15 мин для разложения избытка сульфита и титруют 0,05У раствором КМпО4 до исчезновения синей окраски. Железо и ниобий мешают определению кремния предлагаемым методом.
Из числа органических реактивов, используемых для определения кремния, широко применяется оксихинолин. Оксихинолин ведет себя как амфотерное соединение, т. е. как слабое основание и очень слабая кислота 2(КИСл=2-10-10, Досв= 1 • 10~10 [135].
Как основание о-оксихинолин образует с кислотами продукты присоединения, в которых один эквивалент кислоты связывает одну молекулу основания. Свойства гетерополикислот образовывать с оксихинолином труднорастворимые продукты присоединения использованы для определения кремния. Этот метод обладает высокой точностью, так как молекулярный вес молибдосиликатов органических оснований велик по сравнению с атомным весом кремния, связанного в комплекс.
Волынец первый предложил использовать это свойство оксихинолина для определения кремния [134, 135]. Метод основан на получении кремнемолибденовой кислоты при взаимодействии молибденовокислого аммония с подкисленным раствором силиката, последующем осаждении ее избытком оксихинолина C9H7ON и титровании избытка оксихинолина бромит-броматным методом. Волынец использовал метод для определения кремния в динасах, кварцитах, глинах и т. д.
Навеску 0,25 г высушенного при 105—110° С кварцита, динаса, глины или шамота сплавляют с 0,5 г NaOH в никелевом тигле (кварциты и динасы можно сплавлять с 3 г соды в платиновом тигле). Плав выщелачивают водой, разбавляют до 200 мл, хорошо перемешивают, приливают 30 мл конц. НС1, нагревают почти до кипения и охлаждают. Полученный раствор переносят в мерную колбу емкостью 1000 мл, разбавляют водой и тщательно перемешивают.
Отбирают пипеткой 100 мл раствора в мерную колбу емкостью 500 мл, приливают 20 мл раствора молибденовокислого аммония (20 г на 1000 мл воды), затем 5 мл НС1 (1 : 1), закрывают колбу каучуковой пробкой (во избежание изменения объема жидкости вследствие испарения) и нагревают на водяной бане 10 мин при температуре не выше 80° С. Затем колбу с раствором охлаждают, вынимают пробку, приливают из бюретки еще 20 мл НС1 (1 : 1). Осаждают кремнемолибденовую кислоту, приливая из бюретки раствор оксихинолина (14 г оксихинолина растворяют в 22 мл НС1 (1 : 1), разбавляют до 1000 мл, перемешивают и устанавливают титр полученного раствора броматометрическим титрованием).
При содержании SiO2 в анализируемом образце от 40 до 70% приливают 27,5 мл осаждающего реактива, при содержании свыше 70% — 32,5 мл. После осаждения кремнемолибденовой кислоты колбу закрывают пробкой и нагревают на водяной бане в течение 10 мин при 60—70° С. Охлаждают раствор, осадок отфильтровывают через сухой беззольный фильтр (12 см).
72
К 25 мл фильтрата приливают 30 мл раствора НС1 (1 : 1), 30 мл 8%-ного раствора щавелевой кислоты и 130 мл воды. Затем к фильтрату приливают 12,5 мл 0,1Л1 раствора бромат-бромида, 5 мл 10%-ного раствора йодистого калия и 5 мл 10%-ного раствора крахмала, после чего титруют выделившийся иод 0,05jV раствором тиосульфата. Это титрование предварительное.
Второе, более точное титрование производят со 100 мл фильтрата, который переносят пипеткой в эрленмейеровскую колбу, приливают 120 мл 8%-кого раствора щавелевой кислоты, 100 мл НС1 (1 ; 1), 225 мл воды и А мл 0,11V раствора бромата. Величину А находят по формуле: А=(12,3—0,5 В) -4-]-1,5 мл, где В —• расход тиосульфата при первом титровании. Закрывают колбу пробкой, оставляют стоять на 2 мин, приливают 5 мл 10%-ного раствора KJ, 5 мл раствора крахмала и титруют тиосульфатом. Содержание кремния находят по формуле
SiO2, % =
m — (Л — 0,5-Bj)-Т-1,175 0,025
где т — количество SiO2, соответствующее введенному до осаждения количеству оксихинолина (0,45 г оксихинолина эквивалентны 0,046578 г SiO2); А — количество бромид-бромата, израсходованное па титрование 100 мл раствора; Bt — количество тиосульфата, израсходованное на обратное титрование; Т — титр бромата, пересчитанный на SiO2.
Оксихинолиновые методы применяются для определения кремния в шлаках, силикатах, кремнийорганических. соединениях [135, 297]. Методы длительны, трудоемки, но отличаются высокой точностью. Ошибка определения не превышает ±0,2% [966].
Методы кислотно-основного титрования
Хасэгава разработал косвенный титриметрический метод определения кремния, основанный на его осаждении в виде (C9H7ON)4-• H4SiO4-12МоО3, разложении осадка раствором едкого натра и титровании избытка щелочи. Кремний, содержащийся в алюминиевом сплаве, действием едкого натра сначала переводили в Na2SiO3 по реакции
Si + 2NaOH + Н2О = Na2SiO3 + 2Н2.
Действием хлористоводородной кислоты Na2SiO3 переводили в H4SiO4, реагирующую с молибдатом натрия при pH 1,1 с образованием желтой кремнемолибденовой кислоты Hg[Si(Mo2O7)e]-H2O.
При дальнейшем подкислении раствора и введении оксихинолина происходит образование (C9H7ON) 4-Н 4SiO4-12МоО3, который при взаимодействии со стандартным раствором едкого натра разлагается по уравнению
(C9H7ON)4 • H4SiO4 • 12MoO3+24NaOH
-^4C9H,ON -I- H4SiO4 + 12H2O + 12Na2MoO4.
Избыток едкого натра оттитровывали и определяли содержание кремния, принимая во внимание, что 1 г-экв едкого натра соответствует V24 г-экв кремния. Продолжительность анализа 70 мин. При содержании кремния в анализируемом образце от 0,4 до 13% стандартное отклонение составляет 0,006—0,02% [578]. Определению мешают фосфор, мышьяк и германий.
73
Методы комплексонометрии
Предложен комплексонометрический метод определения кремния, основанный на переводе его из силикат-иона в кремнемолибденовую кислоту H8[Si(Mo2O7)6], ее экстракции, разрушении экстрагированного соединения действием щелочей с образованием ионов МоО2- и на последующем определении ионов молибдена комплексонометрическим методом.
Метод использован для определения кремния в кремнийорганических и элементокремнийорганических соединениях [385], а также в силицидах железа, алюминия и марганца [1107]. Для экстракции кремнемолибденовой кислоты используют изоамиловый или бутиловый спирты. Приводим методику определения кремния в силицидах комплексонометрическим методом.
Навеску силицида 0,1 г помещают в платиновую чашку, прибавляют .3 мл конц. HNO;1 и 10 капель HF. После прибавления каждой капли раствор с осадком перемешивают вращением чашки и затем оставляют стоять 5—10 мин (можно раствор оставить на ночь). Переносят раствор в мерную колбу емкостью 100 мл, рръоцт объем раствора до метки водой и затем хранят его в сосуде из тефлона.
К 10 мл раствора прибавляют на шпателе 0,02—0,03 г комплексона III, 5 мл воды и нейтрализуют раствор 30%-ным раствором NaOH по индикаторной бумаге до pH 4—5. Нагревают раствор и приливают к нему кипящую смесь, состоящую из 5 мл 5%-ного раствора молибдата аммония, 4 мл 4.V H2SO4 и 6 мл воды. Оставляют раствор на 5—7 мин. Еще теплый раствор переносят в делительную воронку, смывая стенки чашки 15 ma4N H2SO4. Прибавляют 5 мл изоамилового спирта и экстрагируют кремнемолибденовую кислоту. Экстракцию повторяют трижды.
Объединенные экстракты промывают в делительной воронке 20 мл4\' H2SO4, отделяют спиртовой слой, приливают к нему 20 мл воды и взбалтывают. Нижний желтоокрашенный слой сливают в колбу Эрленмейера емкостью 250 мл. Реэкстракцию проводят 3—4 раза до обесцвечивания органической фазы. Приливают к раствору в колбе 10 мл конц. НС1 и кипятят 5 мин. Добавляют осторожно к кипящему раствору 5 мл 10%-ного раствора солянокислого гидразина и упаривают раствор до объема 5—10 мл (раствор при этом приобретает ярко-зеленую окраску).
Раствор переносят в колбу Эрлейнмейера емкостью 500 мл, приливают 20—25 мл 0,05 М раствора комплексона III, доводят раствор водой до объема 150—200 мл, прибавляют сухой эриохром черный Т, после чего нейтрализуют раствор аммиаком до перехода окраски раствора в зеленый или синий цвет. Приливают еще 2—3 мл раствора аммиака и кипятят раствор 5 мин. Охлаждают раствор до 50—70° С и титруют 0,05 М раствором ZnCl2 до перехода окраски из сине-зеленой в красную. (Если после кипячения раствора окраска его изменилась, то перед титрованием следует добавить немного раствора аммиака и эрио-хрома черного Т.)
Определению мешают германий, фосфор и мышьяк, образующие в аналогичных условиях гетерополикислоты. Относительная погрешность при содержании кремния в анализируемом образце 40—60% равна ±0,3—0,6%. Продолжительность определения 3 час.
Предложены комплексонометрические методы определения кремния, основанные на свойствах кремневой кислоты. О косвенном микроопределении от 0,5 до 5 мг кремневой кислоты сообщает Пеник [847].
74
Кремневую кислоту в щелочном ацетоновом растворе осаждают в виде CoSi4O9 раствором Co(NO3)2. Осадок центрифугируют, многократно промывают водным раствором метанола и растворяют в аммиачном растворе ЭДТА. Добавляют буферный раствор с pH 10 и эриохром черный Т (иногда тропеолин 00 для улучшения перехода окраски). Обратное титрование избытка ЭДТА проводят раствором соли магния. Таким способом определяют содержание кобальта в осадке. Определение продолжается 20 мин и дает стандартное отклонение лишь 2,7%.
Быстрый косвенный комплексонометрический метод определения кремния в доменных шлаках описан в работе Даценко [1108]. Для определения кремния в доменных шлаках необходимо знать содержание окиси кальция в шлаке и его основность. Содержание кремнезема находят по формуле
SiC>2, % =СаО, %/основность шлака.
Для определения окиси кальция используют комплексонометрический метод с индикатором мурексидом. Определение ведется без отделения железа, алюминия и марганца. Влияние их устраняется добавлением солянокислого триэтаноламина. Основность шлака устанавливают по калибровочной кривой, определив предварительно свободную окись кальция. Длительность определения 18—20 мин. Относительная ошибка около 1,4%.
МЕТОДЫ, ОСНОВАННЫЕ НА СВОЙСТВАХ КРЕМНЕФТОРИСТОВОДОРОДНОЙ КИСЛОТЫ
Кремний проявляет способность образовывать с ионами, являющимися сильными донорами электронов, комплексные соединения. Вследствие этого четырехфтористый кремний способен присоединять еще два фторид-иона, обладающих электроотрицательными свойствами, с образованием кремнефтористоводородной кислоты или ее солей [461, 572, 846, 852].
Склонность кремнефтористоводородной кислоты и ее солей к гидролизу послужила предпосылкой для разработки алкалиметрических методов определения кремния. Почти все алкалиметрические методы сравнительно просты и удобны, однако отличаются недостаточной точностью.
При определении кремния алкалиметрическим методом приходится иметь дело со смесью кремнефтористоводородной и фтористоводородной кислот и раздельным их определением. Несмотря на наличие обширной литературы по этому вопросу, подобная задача тем не менее остается сложной. Сложность определения кремнефтористоводородной и фтористоводородной кислот при их совместном присутствии состоит в том, что при титровании H2SiF6 щелочью на процесс найтрализации накладывается процесс гидролитического расщепления S1F2--hohob. Процесс гидролиза сопровождается
75
выделением коллоидного осадка SiO2-nH2O, адсорбционные свойства которого делают нечетким переход окраски индикатора. Точность определения снижается также вследствие повышения в процессе титрования концентрации фторид-ионов, увеличивающих прочность комплекса SiF^— [601].
При выполнении определения алкалиметрическим методом обычно прибегают к разделению H2SiF6 и HF осаждением кремнефтористоводородной кислоты в виде ее калиевой или натриевой соли.
Штолба [1031, 1032] первым предложил метод определения кремния, основанный на его осаждении в виде кремнефторида калия или натрия. Растворимость кремнефторида понижали добавлением спирта. После отделения образующегося осадка фильтрованием его титровали щелочью в соответствии с реакцией
K2SiF6 + 4КОН 6KF + SiO2 + 2Н2О.
Впоследствии различные авторы [152, 246] предложили большое количество методов определения кремния, которые в какой-то степени являются видоизменениями и уточнениями метода Штолба. Хотя каждый метод в отдельности учитывал недостатки предыдущего, большинство из них в конечном счете сводилось к количественному осаждению кремния в виде кремнефторидов натрия или калия с использованием в качестве реактивов фтористоводородной кислоты и ее солей.
Некоторые алкалиметрические методы определения кремния были основаны на осаждении кремния в виде кремнефторида натрия, отделении его от мешающих примесей и титровании фтористоводородной кислоты, выделившейся при гидролизе Na2SiFe, стандартными растворами оснований [375]. Однако кремнефторид натрия обладает значительной растворимостью в воде (0,65%), поэтому для осаждения кремния чаще используют кремнефторид калия, обладающий значительно более низкой растворимостью (0,12%).
Для получения K2SiF6 обычно используют фтористоводородную кислоту с солями калия (KNO3, КО) или другие неорганические кислоты (НО, HNO3, Н3РО4) с фторидами калия.
Фтористоводородную кислоту применяют обычно в тех случаях, если анализируемый объект хорошо растворяется в ней, что имеет место при анализе соединений, образующих с фторид-ионами прочные комплексные соединения (например, титана, циркония, алюминия и т. д.). Следует учитывать, что фтористоводородная кислота, как правило, загрязнена кремнефтористоводородной кислотой, вследствие чего необходимо или проводить холостой опыт, или очищать HF от H2SiF6. Очистка фтористоводородной кислоты производится путем ее насыщения хлоридом калия с последующим отделением осадка кремнефторида калия.
Для промывания осадков кремнефторида натрия или калия обычно используют насыщенные водные растворы хлористого натрия или калия [665]. Однако кремнефториды в некоторой степени рас
76
творимы в воде, поэтому для уменьшения растворимости Na2SiF6 и K2SiF6 следует использовать 50% -ный этанол, насыщенный хлоридом калия [896].
Промывную жидкость готовят растворением 70 г КС1 в 1000 мл 50% -ного этанола. Приготовленный таким образом раствор оставляют на два дня, затем добавляют к нему 5 мл 0,5% -ного раствора метилового красного и точно нейтрализуют 0,1jV растворами NaOH и НС1 до промежуточного оранжевого цвета.
Раствор можно регенерировать 10 раз. Для этого к 1000 мл использованного раствора добавляют 5,0 г СаО, тщательно перемешивают раствор и оставляют стоять на два дня. Затем сливают раствор с осадка и нейтрализуют его вначале 4% -ным раствором КОН, после чего более точно 0,1W раствором НС1 до оранжевого цвета.
Осадок кремнефторида калия или натрия после отделения от избытка фто-рид-ионов подвергают гидролизу, который наиболее полно протекает при 90— 100° С по реакции
K2SiF6 + 4Н2О -> 2KF + Si (ОН)4 -|- 4HF.
Выделившуюся в процессе гидролиза HF титруют обычно по фенолфталеину. Одиако фенолфталеин в присутствии пульпы бумаги в мутных средах дает недостаточно четкий переход окраски.
Луврие и Войновичем [896] были испробованы в качестве индикаторов пурпурно-красный бромкрезол, феноловый красный и смесь 0,5%-ного раствора бромтимолового голубого и 0,5%-ного фенолового красного в этаноле. Переход окраски при использовании этой смеси от ярко-желтой к фиолетовой более заметен, чем при титровании по фенолфталеину.
Фтористоводородная кислота, выделившаяся при гидролизе кремнефторида калия, может быть оттитрована со смешанным индикатором (1 г 1 %-ного водного раствора бромтимолового синего и 1 г 1 %-ного водного раствора фенолового красного). Точность определения при содержании кремния менее 1% не превышает 7 отн.%, а при содержании кремния больше 1% —менее 1 отн.%.
Для ускорения гидролитического расщепления K2SiF6 применяют СаС12. Гидролиз усиливается вследствие образования труднорастворимого осадка CaF2 (произведение растворимости CaF2 равно 3,4-10-11). Освобождающуюся при этом хлористоводородную кислоту в количестве, эквивалентном содержанию фтористоводородной кислоты, оттитровывают стандартным раствором щелочи по фенолфталеину или с индикатором Ташира [665].
Определение кремния в шлаке. Тонкоизмельченную навеску анализируемого шлака около 0,2 а растворяютв платиновой чашке в 20 мл HNO3 (1 : 1), прибавляют еще 20 мл HNO3 той же концентрации и охлаждают раствор. Затем при постоянном перемешивании вводят 5 г NH4F, через 1—2 мин 15 г KNO3 и немного бумажной массы.
Осадок кремнефторида калия переносят на фильтр и промывают насыщенным раствором KNO3. Фильтр с осадком переносят в коническую колбу, содержащую 200—300 мл кипящей воды и 10 мл 40%-ного раствора СаС12, нейтрализованного по метиловому красному, и титруют выделившуюся хлористоводородную кислоту 0,2N раствором NaOH по индикатору Ташира (раствор 0,125 г метилового красного и 0,083 г метиленового голубого в 100 мл этанола) до светло-зеленой окраски.
77
Для получения K2SiF6 Танино Каити при анализе кислых шлаков использовал сплавление с KHF2. Для извлечения кремнефторида калия из плавов их обрабатывали растворами кислот и солей: азотная кислота с нитратом калия, хлористоводородная кислота с хлоридом калия, бисульфатом калия, муравьиной, азотной и лимонной кислотами. При анализе шлаков для извлечения кремнефторида калия из плава использовали 4N HNO3.
После растворения плава осадок кремнефторида калия переносят на фильтр и промывают несколько раз 2%-ным раствором КС1. Далее помещают осадок с фильтром в коническую колбу, добавляют 100 мл холодной воды (10±2° С) и нейтрализуют 0,2М раствором КОН по фенолфталеину. Нейтрализованный раствор нагревают почти до кипения и титруют 0,2М раствором NaOH. Компоненты шлаков мало влияют на точность определения [522 ].
Основное преимущество описанных выше методов определения кремния заключается в том, что эти методы меньше зависят от влияния сопутствующих примесей. Точность определения кремния в данном случае зависит от двух факторов: выбора промывной жидкости и условий отмывки осадка кремнефторида калия от кислоты. Однако последняя операция несколько удлиняет определение.
В некоторых вариантах этого метода осадок кремнефторида калия не отделяют от раствора, а после его осаждения избыток кислот в растворе точно нейтрализуют, затем обрабатывают осадок горячей водой и титруют фтористводородную кислоту, выделившуюся при гидролизе K2SiF6, раствором едкого натра в присутствии всех компонентов раствора.
Так, при определении кремния в силикатах кислый раствор силиката нейтрализуют по метиловому оранжевому и осаждают алюминий и железо в виде криолита. Затем раствор подкисляют и осаждают кремнефторид калия, после чего избыток кислот точно нейтрализуют по фенолфталеину, разбавляют раствор горячей водой и титруют фтористоводородную кислоту раствором едкого натра [200].
При проверке этого метода на цементах Жуковская и другие [200] получили заниженные и плохо воспроизводимые результаты вследствие частичного гидролиза осадка кремнефторида калия при нейтрализации раствора по фенолфталеину. Поэтому авторы проводили процесс нейтрализации и последующего титрования фтористоводородной кислоты по индикатору с более низким интервалом перехода pH, чем фенолфталеин. Из проверенных индикаторов наиболее приемлемыми оказались бромкрезоловый пурпурный (pH 5,2—6,8) и бромтимоловый синий (pH 6,0—7,6). Вредное влияние железа и марганца было устранено с помощью комплексона III, удерживающего эти катионы в растворе.
Шир и Комере [1018] впервые применили алкалиметрический метод для анализа кремнийорганических соединений. После минерализации кремнийорганических соединений образующуюся кремневую кислоту сплавляли с карбонатом натрия и калия, выщела-78
чивали плав и определяли кремний алкалиметрическим методом. Авторы нашли, что при алкалиметрическом определении кремния в кремнийорганических соединениях в качестве индикатора лучше использовать смесь метилового красного и бромкрезолового зеленого, так как другие индикаторы часто обесцвечиваются в среде, богатой F- или SiF^-ионами. Но вследствие того что интервал окраски перехода этой смеси лежит как раз в области pH, когда гидролиз SiF2~.HOHOB уже начался (pH 3,5), необходимо при титровании предотвратить гидролиз. Для этого требуется соблюдение следующих условий: 1) по возможности сохранить минимальный объем титруемого раствора; 2) понизить растворимость K2SiF6 насыщением раствора твердым хлоридом или нитратом калия; 3) титрование проводить по возможности быстро.
Авторы определяли кремний в самых различных мономерных и полимерных кремнийорганических соединениях.
При анализе кремнийорганических соединений наибольшее распространение нашли методы, основанные на образовании крем-нийфторсиликата аммония в .присутствии хлористоводородной кислоты. По количеству хлористоводородной кислоты, вступившей в реакцию, судят о содержании кремния, т. е. определение сводится к титрованию избытка кислоты щелочью.
Терентьев, Сявцилло и Лускина при анализе технических этилполисилоксанов после окисления их смесью серной и хромовой кислот заканчивали определение алкалиметрическим методом. Впоследствии этим же методом авторы определяли кремний в некоторых элементоорганических соединениях после их минерализации смесью серной кислоты с персульфатом аммония или перекисью натрия [528].
Навеску анализируемого соединения 20—30 мг (взятую по разности) помещают в реакционную колбу, в которую предварительно наливают 10 мл конц. H2SO4. Из мерного цилиндра в ту же колбу при непрерывном перемешивании добавляют по каплям 2—3 мл конц. хромовой кислоты. Колбу помещают в нагретую до 150'—160° С баню и при этой температуре выдерживают в течение 30 мин. Затем колбу охлаждают на воздухе. К охлажденному раствору добавляют 30— 40 мл воды, кипятят 5 мин при 110—120° С, добавляют 1,0—1,5 мл свежеприготовленного раствора желатины и оставляют стоять на 5—10 мин для коагуляции кремневой кислоты.
Выделившуюся кремнекислоту отфильтровывают через беззольный фильтр и промывают дистиллированной водой. Осадок вместе с фильтром переносят в полиэтиленовый стакан, содержащий 5—7 мл 30%-ного раствора NaOH, 5—6 капель индикатора (индикатор готовят смешением 6 ч. 0,1%-ного спиртового раствора метилового красного с 5 ч. 0,1%-ного водного раствора бромкрезолового зеленого, к 100 мл такой смеси добавляют 0,5 мл 0,\N раствора NaOH), нейтрализуют НС1 (1 : 1), а затем подкисляют до слабокислой реакции разбавленной НС1 (1 : 10).
Полученный раствор переводят в коническую колбу, точно нейтрализуют 0, W раствором щелочи так, чтобы общий объем раствора в колбе не превышал 50 мл. Далее раствор насыщают твердым КС1, опять точно нейтрализуют, вносят 10 мл 1% -ного раствора NH 4F, 20 мл 0, 1W НС1 и оттитровывают избыток кислоты O,1N раствором щелочи. В точке эквивалентности окраска изменяется от красной до зеленой. Ошибка определения не превышает 2%.
79
Вайль использовал этот метод для определения кремния в растворимых силикатах. Кальций, бораты и фосфаты мешают определению кремния [1088]. При помощи ионного обмена кремний может быть отделен от мешающих примесей. Хализова [576] на катионите КУ-2 отделяла таким образом кремний от примесей А1, Fe, Ti, Са, Mg, Мп, Сг, Ni, Си, Zn и Be.
Из других методов отделения кремния от мешающих элементов следует остановиться на его отгонке в виде кремнефтористоводородной кислоты из кислых растворов (серная или фосфорная кислоты). Розенбуш использовал этот метод для определения кремния в гольевых солях и кожах [981]. В соответствии с этой методикой отогнанную кремнефтористоводородную кислоту поглощали насыщенным раствором хлорида калия. При этом выделялось эквивалентное количество хлористоводородной кислоты, которую после отделения от осадка кремнефторида калия титровали по феноловому красному. Кремнефтористоводородную кислоту отгоняли при 135+5° С, поднимая перед окончанием отгонки температуру до 160° С. Для разложения образцов применяли 38%-ную серную или 50%-ную фосфорную кислоты. Лучше для разложения использовать нелетучую фосфорную кислоту, так как в случае использования серной кислоты при 160° С частично отгоняются пары SO3, что приводит к завышенным результатам определения.
При определении кремния в тетраэтоксисиланах и этоксиполисилоксанах Кальман и Ваго предложили ацидиметрический метод определения кремния, принципиально отличающийся от ранее описанных. Метод основан на способности фтористоводородной кислоты при взаимодействии с анализируемыми соединениями в среде органических растворителей образовывать кремнефтористоводородную кислоту, которую далее можно оттитровать едким натром [852]. Указанным методом кремний может быть определен во всех мономерных и полимерных алкоксисиланах [304].
Определение кремния в алкоксисиланах. Для определения кремния в полиэтиленовый стакан емкостью 150 мл, содержащий около 50 мл 50%-ного этанола и 1—2 мл 40%-ной HF, помещают навеску анализируемого вещества около 0,05 г. Содержимое стакана перемешивают покачиванием стакана. Через 3—5 мин в раствор прибавляют из бюретки 10% -ный раствор КОН до появления розовой окраски по фенолфталеину и затем нейтрализуют раствором 0,21V НС1. При этом концентрация спирта должна быть не ниже 50%-ной.
Раствор переносят в коническую колбу емкостью 1000 мл, разбавляют до 300 мл кипящей дистиллированной водой и титруют 0,21V раствором NaOH до неисчезающего розового окрашивания по фенолфталеину.
Титр едкого натра устанавливают в тех же условиях по тетраэтоксисилану.
Определение можно закончить йодометрическим методом. Точность определения повышается, однако время определения увеличивается в 2—3 раза.
Для определения кремния в моно- и полимерных алкоксисиланах предложен метод, основанный на осаждении кремнефтористоводородной кислоты, образующейся в процессе обработки кремнийорганических соединений фтористоводородной кислотой,
80
органическими аминами и на последующем титровании образующегося комплексного соединения [301, 303]. Наименьшей растворимостью в спиртовых и ацетоновых растворах обладает кремнефто ристоводородный бензидин. После осаждения кремнефтористоводородной кислоты по реакции
ROH
H2SiF6 + C12H12N2 C12H12N2 • H2SiF6
нон
определение кремния проводят двумя путями: прямым алкалиметрическим титрованием либо алкалиметрическим титрованием по остатку.
Прямое алкалиметрическое титрование. В полиэтиленовый стакан емкостью 200 мл, содержащий 10 мл 0,31V спиртового раствора HF, помещают 0,03—0,05 г исследуемого кремнийорганического соединения, перемешивают в течение 2 мин и добавляют 10 мл 1%-ного спиртового раствора бензидина. Образующийся осадок отфильтровывают через плотный беззольный фильтр на полиэтиленовой воронке в полиэтиленовый стакан емкостью 400 мл. Осадок промывают спиртом. Фильтр помещают в стеклянную коническую колбу емкостью 500 мл, добавляют туда 200 мл кипящей воды и титруют выделившуюся кремнефтористоводородную кислоту-0,1 Д' раствором КОН в присутствии фенолфталеина. Параллельно проводят холостой опыт.
Алкалиметрическое титрование по остатку. При определении HF по остатку анализ проводят по описанной выше методике до получения осадка кремнефторида бензидина. После промывания осадка фильтрат и промывную жидкость собирают вместе в полиэтиленовый стакан емкостью 400 мл и затем титруют избыток кислоты 0,11V раствором КОН по фенолфталеину. Ошибка определения кремния не превышает ±0,2%. Время определения 30 мин.
Титриметрические методы широко применяются для определения кремния в чугунах и сталях [991], растворимых и нерастворимых силикатах [523, 814], минеральном сырье [665], карбиде кремния [813], кремнийорганических соединениях [528, 1018], рудах [988], хромовых ваннах [854] и других материалах [301, 303, 960, 966, 1043, 1095].
При титриметрических вариантах определения кремния точку эквивалентности можно определять также электрохимическими методами: кондуктометрическим, потенциометрическим и полярографическим.
Большинство электрохимических методов определения кремния также основано на титровании кремнемолибденовой или кремнефтористоводородной кислот.
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ
Методы, основанные на свойствах кремнемолибденовой кислоты
Потенциометрическое титрование. Предложены потенциометрические методы определения кремния, основанные на титровании кремнемолибденовой кислоты различными восстановителями. Ко
81
личество восстановителя, необходимое для восстановления кремнемолибденовой кислоты, пропорционально количеству кремния, содержащегося в анализируемом объекте. В качестве восстановителей предложен целый ряд соединений с различной окислительно-восстановительной способностью: хлорид и оксалат олова, 1-амино-2-нафтол-4-сульфокислота, аскорбиновая кислота и др. [1109]. При определении кремния в объектах с малым содержанием его расхождение между потенциометрическими и фотометрическим методами не превышает 1—2%.
Использование процесса нейтрализации кремнемолибденовой кислоты в водных растворах для определения кремния ограничено вследствие одновременного гидролитического расщепления гетерополианиона в процессе его нейтрализации. Перевод кремнемолибденовой гетерополикислоты в неводный раствор с помощью экстракции позволяет не только предотвратить гидролиз Н 8[Si(Mo2O7)e], но и отделить кремнемолибденовую кислоту от мешающих примесей: избытка молибдата аммония, посторонних солей и т. д.
Условия отделения кремнемолибденовой кислоты экстракцией рассмотрены в литературе [7, 16, 1116, 1120]. Установлено [1120], что при экстракции гетерополикислот изо- и w-бутанолом в органическую фазу одновременно переходят и неорганические кислоты (серная, хлористоводородная, азотная, хлорная), используемые для разложения анализируемых образцов. Таким образом, в органической фазе присутствует смесь кислот, и определение кремния сводится к их дифференцированному титрованию.
Титрование H8[Si(Mo2O7)e] в смеси с НС1, HNO3 и H2SO4 раствором гидроокиси тетрабутиламмония и калия в ацетон-изобу-танольной среде позволяет сделать вывод, что хлористоводородная и азотная кислоты титруются суммарно с кремнемолибденовой кислотой, а при титровании смеси кремнемолибденовой и серной кислот получены два хорошо выраженных скачка. Количественные расчеты показывают, что реакция может быть выражена следующими уравнениями:
Н8 [Si (Мо2О,)6] + 4 (С4Н9)4 NOH
-Ч(С4Н9)4 N]4-H4 [Si (Мо2О7)6] + 4Н2О;
H2SO4 + (С4Н9)4 NOH [(С4Н9)4 N] HSO4 + Н2О
(первая точка эквивалентности);
[(C4H9)4N] HSO4 + (С4Н9)4 NOH [(С4Н9)4 N]2SO4 + Н2О (вторая точка эквивалентности).
При использовании в качестве титранта метанолового раствора едкого кали серная, азотная, хлористоводородная и кремнемолибденовая кислоты титруются суммарно. Таким образом, экстракционно-потенциометрический метод может быть использован для определения кремния, если для разложения анализируемых образцов используется H2SO4, а в качестве титранта—(C4H9)4NOH.
82
Кондуктометрическое титрование. После перевода в неводный раствор кремнемолибденовая кислота может быть оттитрована не только потенциометрическим, но и кондуктометрическим методом, причем в этом случае она может быть оттитрована дифференцированно не только в смесях с серной, но и с азотной и хлористоводородной кислотами [386, 387]. Органические и неорганические основания образуют с кремнемолибденовой кислотой в ацетоне и его смесях с бутанолом и пентанолом малорастворимые или мало-диссоциирующие комплексные соли. Поэтому в процессе титрования кремнемолибденовой кислоты неводными растворами органических аминов электропроводность уменьшается до точки эквивалентности, а затем становится постоянной при титровании слабыми основаниями или резко возрастает при титровании сильными основаниями. Приводим методику кондуктометрического определения кремния в некоторых силикатах и кремнийорганических соединениях.
Определение кремния в кремневой кислоте и растворимых силикатах. Навеску вещества, содержащую 4—5 мг Si, помещают в полиэтиленовый стакан емкостью 100 мл, обрабатывают 10 мл 2% -ного раствора КОН и нагревают на кипящей водяной бане до полного растворения. Охладив стакан, переносят содержимое количественно 40 мл воды в колбу емкостью 250—300 мл и нейтрализуют 4Л' H2SO4 по фенолфталеину (раствор 1).
В другой колбе готовят смесь 10 мл 20%-ного раствора молибдата аммония, 25 мл воды и 15 мл 4N H2SO4 (раствор 2). Колбы с растворами 1 и 2 нагревают на кипящей водяной бане 5 мин. Приливают раствор 2 к раствору 1, при этом образуется кремнемолибденовая кислота. Нагревают ее на кипящей водяной бане 5 мин, охлаждают до комнатной температуры, прибавляют еще 35 мл 4N H2SO4 и экстрагируют 50 мл изобутилового спирта.
Аликвотную часть органической фазы (10 мл) помещают в ячейку кондуктометра, прибавляют 50 мл ацетона и титруют стандартным раствором гидроокиси тетраэтиламмония. Титрант стандартизуют по кристаллическому кремнию по аналогичной методике.
Определение кремния в кремнийорганических и элементокремнийорганических соединениях. Навеску вещества, содержащую 4—5 мг Si, помещают в термостойкий стакан емкостью 20 мл, обрабатывают 1 мл конц. H2SO4 и нагревают раствор до начала выделения белых паров. К охлажденному раствору прибавляют при нагревании порциями по 0,2 г персульфат аммония до полного обесцвечивания. После охлаждения содержимое стакана количественно переносят с помощью 50 мл воды в колбу емкостью'250—300 мл, осторожно нейтрализуют по фенолфталеину твердым КОН и добавляют его избыток (1 г). Смесь нагревают на кипящей бане 5 мин, нейтрализуют по фенолфталеину 4N H2SO4 (раствор 1). Далее анализ ведут, как указано выше.
Определение кремния в сплавах. Навеску сплава, содержащую 40—50 мг Si, помещают в полиэтиленовый стакан, обрабатывают 10 мл 30%-ного раствора КОН, нагревают на кипящей водяной бане до растворения, стакан охлаждают и содержимое количественно переносят в мерную колбу емкостью 250 мл. Для определения кремния берут аликвотные части (по 25 мл). Едкое кали нейтрализуют по фенолфталеину 4N H2SO4 (раствор 1). Далее анализ ведут, как указано в первой методике.
Кондуктометрическое титрование пробы раствора проводят на установке, состоящей из звукового генератора ЗГ-1, реохордного моста Р-38, осциллографа НО-ЗМ, магнитной мешалки, ячейки емкостью 100 мл и микробюретки. Точку эквивалентности находят графически, откладывая на оси абсцисс объем титранта, а на оси ординат — сопротивление раствора.
83
Методы полярографии и амперометрии. Ионы кремния не восстанавливаются на ртутном капельном электроде, поэтому для установления количества этого элемента предлагаются косвенные методы, которые связаны с полярографическим определением кремнемолибденовой гетерополикислоты [793, 804, 819, 1038].
При полярографировании кремнемолибденовой и молибденовой кислот в зависимости от характера посторонних электролитов наблюдается несколько ступеней восстановления. В литературе отсутствует единое мнение о числе ступеней восстановления, различных значениях pH получения и восстановления кремне молибденовой гетерополикислоты. Расхождение в опубликованных данных объясняется различием в условиях получения гетерополикислот [1114]. Наиболее подробно полярографическое поведение кремнемолибденовой гетерополикислоты рассмотрено в работах Жана [842—844].
Молибденовая кислота, сопутствующая обычно в ходе анализа кремнемолибденовой кислоте, в присутствии молочной, щавелевой и азотной кислот дает две волны при — 0,35 и —0,50 в. В сернокислой среде наблюдается одна волна при —0,82 в [1007]. Парамолибдат аммония в среде однозамещенного фосфата и лимонной кислоты (pH 2,8) в присутствии КС1 образует две волны — при —0,23 и —0,58 в.
Кремнемолибденовая кислота в присутствии ацетата аммония, уксусной кислоты и хлорида калия при pH 3,5 дает две волны при —0,35 и —0,85 в, высота которых пропорциональна концентрации кремнемолибденовой кислоты в растворе. При pH 4,55 образуется одна неярко выраженная волна.
Кристаллическая кремнемолибденовая кислота при концентрации 10-4 Л4 в среде 0,1 Л4 сульфата калия при pH 3,5 дает одну волну (—0,25 в), в то время как для |3-кремнемолибденовой кислоты характерная волна отсутствует. Для кремнемолибденовой кислоты, образованной из метасиликата натрия и молибдата аммония, в среде нитрата аммония и 0,18 N азотной кислоты возникает только одна волна [719].
В среде сульфата натрия и серной или уксусной кислоты парамолибдат аммония дает при pH 1,8 также только одну волну. Для кремнемолибденовой кислоты в тех же условиях наблюдается полярографическая волна с хорошо выраженным максимумом.
В азотнокислой среде при pH 1 в присутствии нитрата аммония щелочные растворы силиката образуют с парамолибдатом аммония кремнемолибденовую кислоту, полярограммы которой содержат одну волну при потенциале примерно —0,1 в. На этом методе основано полярографическое определение растворимой двуокиси кремния в нитрате уранила [1037].
Полярограммы кремнемолибденовой кислоты изучены в различных средах [1007]. Кремнемолибденовая кислота в присутствии 0,1 N раствора хлорида калия дает две волны, для которых потенциалы полуволны соответственно равны —0,48 и —1,2 в.
84
Величина этих волн пропорциональна концентрации гетерополикислоты при содержании последней от 0,2 до 1,0 мМ. В присутствии винной или щавелевой кислот получаются две волны (с потенциалами полуволны —0,3 и —0,55 в соответственно). В присутствии серной кислоты (от 0,05 до 6,0 Л4) на полярограмме образуются три максимума, два из которых при концентрации серной кислоты 0,05 Л4 характеризуются = —0,26 и —0,37 в. Максимум
тем менее резкий, чем более высокая концентрация кислоты. В присутствии фосфорной, азотной, хлористоводородной кислот получаются кривые, близкие к полярографическим кривым в серной кислоте.
Восстановление кремнемолибденовой гетерополикислоты происходит по двум ступеням: по первой ступени образуется пятивалентный молибден, что соответствует первой полуволне, затем образуется трехвалентный молибден, что соответствует второй полуволне.
Грассхоф и Хан [793] исследовали полярографическое поведение кремнемолибденовой кислоты и нашли, что в растворе лимонной кислоты возникают три волны, из которых первая и третья соответствуют восстановлению молибденовой гетерополикислоты, а вторая — кремнемолибденовой.
Измерение количества свободной молибденовой и кремнемолибденовой кислоты в растворе дает возможность сделать вывод о том, что кремнемолибденовая гетерополикислота разлагается на 50% согласно уравнению
Н4 [Si (Мо12О40)] + 6H2O^Si (ОН)4 + 2Н3 (Мо6О21Н3)*.
Таким образом, три волны на полярограмме относятся к кремнемолибденовой кислоте и ионам молибденовой кислоты, которые получаются при ее разложении.
Восстановление на капельном ртутном электроде протекает по следующему уравнению:
[Si (Мо12О40)]4- + 4Н+ + 4е = [SiMo12O36 (0Н)4Г.
а-Кремнемолибденовая кислота имеет потенциал полуволны, равный —0,47 в, т. е. на 90 мв более отрицательный, чем потенциал Р-кремнемолибденовой кислоты.
Грассхоф и Хан использовали полярографическое восстановление p-кремнемолибденовой кислоты для определения кремния в алюминиевых сплавах [793]. Авторы указывают на преимущества полярографического метода по сравнению с фотометрическим, так как определение возможно проводить в окрашенных растворах и влияние посторонних ионов незначительно.
Применение переменнотоковой полярографии для определения кремния в силикатах меди рассмотрено в работе [1114].
* Это относится к (3-кремнсмолибденовой кислоте.
85
Изучено амперометрическое определение кремнемолибденовой гетерополикислоты осаждением ее диантипирилметаном в виде (ДАМ)з-Н8[Si(Mo2O7)e] и последующим окислением диантипи-рилметана на платиновом микродисковом электроде при +1,4 в [1113]. Величина предельного тока пропорциональна концентрации диантипирилметана и, следовательно, кремния. Различная устойчивость кремнемолибденовой и фосфор но молибденовой кислот при различных pH раствора дает возможность проводить амперометрическое определение кремния в присутствии фосф)ра. Тысячекратные и более количества К, Na, Al, Zn, Cd, Fe, Co, Ni, Мп, Си, Сг и V не мешают определению. Чувствительность определения кремния 0,02 мкг/мл. Относительная ошибка определения не превышает 6%.
Методы, основанные на свойствах кремнефтористоводородной кислоты
Потенциометрическое титрование. При алкалиметрическом титровании кремнефтористоводородной кислоты точку эквивалентности можно определять потенциометрическим методом [58, 307, 308, 383, 525]. Кривая нейтрализации кремнефтористоводородной кислоты показана на рис. 2. На одну молекулу кремнефтористоводородной кислоты расходуется 2ОН~-иона. При дальнейшем прибавлении щелочи происходит выделение кремнекислоты по реакции
SiFi- + 4ОН- -> 6F" + SiOa + 2Н2О.
Этот процесс выражается горизонтальным участком на кривой титрования. Первая точка эквивалентности недостаточно отчетлива, поэтому титрование до этой точки не может быть проведено точно.
Рис. 2. Кривая титрования кремнефтористоводородной кислоты
п—число грамм-эквивалентов NaOH на 1 г-экв H2SiFe; 1 — при нагревании; 2 — на холоду
Титрование до второй точки эквивалентности вполне возможно. Таким образом, на одну молекулу кремнефтористоводородной кислоты приходится шесть ОН-ионов, если титруется сама кислота, и четыре ОН-иона, если титруется фторсиликат щелочного металла.
Описано несколько вариантов потенциометрической фиксации точки эквивалентности [307, 308, 384]. Тараян [525] титровала потенциометрическим методом хлористоводородную кислоту, вы
86
делившуюся в процессе расщепления кремнефторида калия, K2SiF6 + ЗСаС12 + 2Н2О 2КС1 + 3CaF2 + SiO2 + 4НС1
раствором едкого натра.
При титровании кремнефтористоводородной кислоты в неводных средах (ацетон или метилэтилкетон) точность определения может быть значительно повышена вследствие уменьшения скорости гидролиза фторсиликата, образующегося в процессе титрования кремнефтористоводородной кислоты. Приводим методику определения кремния в эфирах ортокремневой кислоты в неводных средах.
0,05—0,02 е эфира ортокремневой кислоты растворяют в 60 мл 0,27V спиртового или ацетонового раствора HF, включают мешалку и быстро титруют образовавшуюся кремнефтористоводородную кислоту спиртовым раствором КОН на потенциометре ЛПУ-01, используя стеклянный и проточный хлорсеребряный электроды. Титрование проводят в полиэтиленовом стаканчике емкостью 150 мл. Результаты определения, как правило, бывают несколько завышенными (до 3%).
Определение содержания кремнефтористоводородной кислоты в растворе может быть выполнено с использованием в качестве титранта органических аминов, которые образуют с кремнефтористоводородной кислотой практически нерастворимые или плохо диссоциирующие комплексные соли. Кремнефтористоводородная кислота образуется при взаимодействии мономерных и полимерных алкоксисиланов с фтористоводородной кислотой. Титрование кремнефтористоводородной кислоты может быть выполнено двумя способами, приведенными ниже.
Рис. 3. Кривые потенциометрического титрования смеси крем-нефтористводородной и фтористоводородной кислот бензидином в среде ацетона (7), этанола (2) и метанола (5)
Потенциометрическое титрование с платиновым и каломельным электродами. Анализируемые кремнийорганические соединения после их перевода в H2SiFe титровали раствором бензидина в среде ацетона, так как из всех испробованных растворителей наибольшие скачки титрования и соответственно наиболее точные результаты определения кремния были получены в ацетоне (рис. 3). Титр раствора бензидина
87
устанавливали по тетраэтоксисилану (Ткип = 166,5°С, d=0,933, «2° = 1,3852) потенциометрическим методом. Титрование производили на установке, состоящей из потенциометра ППТВ с зеркальным гальванометром (с чувствительностью до 10“ 9 а), магнитной мешалки, микробюретки, полиэтиленовой ячейки для титрования (емкостью 150 мл) с платиновым и каломельным электродами (рис. 4). Применяли вынесенный каломельный электрод, соединенный с ячейкой для титрования полиэтиленовым мостиком, заполненным насыщенным спиртовым раствором КС1.
Определение кремния в некоторых кремнийорганических соединениях. В полиэтиленовый стакан емкостью 150 мл, содержащий 15—20 мл 0,2—0,4 N ацетонового раствора HF, вносят навеску (0,03—0,05 г) исследуемого кремнийорганического соединения. К раствору добавляют 25 мл ацетона, и после предварительного перемешивания погружают в ячейку платиновый электрод и солевой мостик каломельного электрода. После 5-минутной выдержки начинают титрование кремнефтористоводородной кислоты 0,5% -ным раствором бензидина в ацетоне. Ошибка определения не превышает 5% .
1
Рис. 4. Схема установки для потенциометрического определения кремния с применением каломельного и платинового электродов
/ — потенциометр ППТВ; 2 — ила’ тиновый электрод; 3 — полиэтиленовая ячейка; 4 — мостнк для хлорида калия; 5 — полиэтиленовая пробка; 6 — каломельный электрод сравнения
Рис. 5. Схема установки для потенциометрического некомпенсационного титрования с биметаллическими электродами 1 — полиэтиленовая ячейка; 2 — платиновый электрод; 3 — мешалка; 4 — вольфрамовый электрод; 5 — гальванометр; 6 — источник тока; 7 — выключатель; 8 — рео-хордный мост
Способ некомпенсационного титрования с биметаллическими электродами. Установка для некомпенсационного титрования (рис. 5) состоит из микробюретки, аккумулятора, полиэтилетовой ячейки для титрования (емкостью 150 мл) с электродами, магнитной мешалки и микробюретки. В качестве индикаторного электрода применяют платиновый электрод, в качестве стандартного электрода сравнения — вольфрамовый.
88
В полиэтиленовый стакан емкостью 150 мл, содержащий 15—20 мл 0,2—0,4 N метанольного раствора HF, вносят навеску 0,05—0,20 г исследуемого соединения. К раствору добавляют 25 мл ацетона и погружают биметаллические электроды. Титрование производят 0,5%-ным раствором бензидина в метаноле при тщательном перемешивании. Точку эквивалентности фиксируют по изменению направления движения стрелки гальванометра.
Метод характеризуется невысокой точностью (ошибка определения достигает 7%), однако он прост и быстр. Титрованию мешают титан, железо, германий и другие элементы, образующие с фтористоводородной кислотой в среде неводных растворителей растворимые комплексные соединения.
Безрогова [58] разработала потенциометрический метод определения суммы кремнефтористоводородной и серной кислот в смеси с фтористоводородной кислотой. Титрование проводят в среде ацетона этанольным раствором едкого натра с использованием графитового и каломельного электродов. Ошибка определения суммы H2SO4 и H2SiFe составляет +3,0%, суммы всех кислот ±0,2%. Описано потенциометрическое определение кремневой кислоты титрованием ее растворами фторидов с использованием в качестве индикатора титанового электрода, чувствительного к изменению концентрации фторид-ионов в растворе [1124].
Кондуктометрическое титрование. Кондуктометрическим методом можно непосредственно титровать кремнефтористоводородную кислоту в среде органических растворителей органическими и неорганическими основаниями [305, 306, 382, 383].
Титрование растворами органических аминов. При титровании кремнефтористоводородной кислоты в неводных растворах органическими аминами электропроводность раствора уменьшается в результате связывания высокоподвижного иона водорода в малодиссоциирующее соединение амин-Н2Б1Ре (рис. 6) вплоть до точки эквивалентности, после чего она остается почти постоянной. Органические амины — слабые основания, поэтому электропроводность их растворов крайне низка и прибавление аминов в некотором избытке к раствору практически не изменяет его электропроводности (см. рис. 6).
В процессе определения кремния в виде кремнефтористоводородной кислоты последней, как правило, сопутствует фтористоводородная кислота. При кондуктометрическом титровании раствора фтористоводородной кислоты органическими аминами электропроводность в процессе титрования увеличивается. Фтористоводородная кислота слабая (р/С = 3,4), и электропроводность ее мала. При добавлении амина к фтористоводородной кислоте в среде неводных растворителей образуется хорошо диссоциирующая комплексная соль (типа солей аммония), что может быть отражено реакцией
RNH2 + HF^>RNHt ± F~.
Электропроводность смеси кремнефтористоводородной и фтористоводородной кислот при добавлении амина уменьшается по мере
89
Электропроводность s« Электропроводность-. Электропроводность
V.mo .
Рис. 6. Кривые кондуктометрического титрования фтористоводородной (3), кремнефтористоводородной (7) кислот и метанола (2) бензидином
Рис. 7. Кривые кондуктометрического титрования смеси кремнефтористоводородной и фтористоводородной кислот бензидином в среде метанола (7), этанола (2) и ацетона (3)
Рис. 8. Кривые кондуктометрического титрования смеси кремнефтористоводородной и фтористоводородной кислот бензидином в метаноле с различным содержанием СН3ОН: 99% (7), 87% (2), 50% (3)
Рис. 9. Схема установки для кондуктометрического определения кремния
1 — реохордный мост;
2 — полиэтиленовая ячейка;
3 — платиновые электроды;
4 — звуковой генератор;
— стандартные сопротивления;
С — емкость;
V — нуль-инструмент
связывания кремнефтористоводородной кислоты. После достижения момента эквивалентности, т. е. полного оттитровывания кремнефтористоводородной кислоты, электропроводность раствора, как правило, начинает возрастать (рис. 7).
Из исследованных титрантов следует отдать предпочтение бензидину. Его применение дает наиболее резко выраженное изменение электропроводности вблизи точки эквивалентности и соответственно наиболее точные результаты.
Авторы исследовали влияние растворителей на характер кривых кондуктометрического титрования. Наиболее резкое изменение электропроводности имело место в растворителях с наименьшей диэлектрической проницаемостью, так как увеличение диэлектрической проницаемости растворителя связано с увеличением ионизирующих свойств среды и соответственно с увеличением диссоциации получаемых в них комплексных соединений. Этим же объясняется закономерное изменение характера кривых кондуктометрического титрования смеси кремнефтористоводородной и фтористоводородной кислот аминами в органических растворителях с различным содержанием воды (рис. 8).
Кондуктометрический метод применяется также для определения кремния в некоторых кремнийорганических соединениях [302]. Измерение электропроводности производили на установке (рис. 9), состоящей из высокочастотного генератора ЗГ-1, реохордного моста Р-38, нуль-инструмента, магнитной мешалки, микробюретки, полиэтиленовой ячейки с электродами. Все стеклянные части электродов покрывали полиэтиленом. В качестве ячейки использовали полиэтиленовый стакан емкостью 200 мл. Так как реохордный мост рассчитан на измерение максимального сопротивления среды 30 ком, а при измерении электропроводности в ряде случаев имеют дело со средами с сопротивлением до 200 ком, то в схеме Р-38 последовательно к сопротивлению в 1 ом присоединяли магазин сопротивлений на 100 ком. Приводим методику определения кремния в полимерных и мономерных кремнийорганических соединениях.
В полиэтиленовую ячейку с кремнефтористоводородной кислотой, образующейся при обработке кремнийорганических соединений раствором HF, погружают платиновые электроды, соединенные с общей мостовой схемой. Титруют полученный раствор 0,5%-ным метанольным раствором бензидина из микробюретки до момента эквивалентности. В качестве среды для титрования используют метанол, этанол или ацетон. Титр раствора бензидина устанавливают по тетраэтоксисилану этим же методом. Измерение электропроводности проводят на частоте 3000 гц. По точности кондуктометрический метод несколько превышает потенциометрический. Ошибка определения =tl,5%.
Титрование растворами щелочей. Кондуктометрическое титрование кремнефтористоводородной кислоты можно проводить метанольным раствором едкого кали. Метод использован для определения кремния в алкоксисиланах и некоторых силикатах.
01
При обработке силикатов фтористоводородной кислотой образуется смесь кремнефтористоводородной кислоты, кремнефторидов металлов и избытка фтористоводородной кислоты. Кондуктометрическое титрование по описанному выше способу применять нельзя из-за наличия сопутствующих катионов.
Пропуская раствор силиката через катионит СДВ-3 в Н-форме, получают раствор кремневой кислоты, который после обработки избытком фтористоводородной кислоты количественно переходит в кремнефтористоводородную кислоту. Последнюю определяют титрованием метанольным раствором КОН. Метод может быть использован для определения кремния в некоторых щелочных и щелочноземельных силикатах.
Высокочастотное кондуктометрическое титрование. Обычный метод кондуктометрического титрования имеет недостатки, которые могут влиять на точность определения кремния. В процессе титрования образующийся осадок комплексных солей кремнефтористоводородной кислоты с органическими аминами, налипая на электроды, меняет их поверхность, что приводит к некоторым ошибкам при нахождении точки эквивалентности. Поэтому определение кремния целесообразнее проводить на установке для высокочастотного кондуктометрического титрования. Определение проводят на приборе, состоящем из высокочастотного анализатора, выносного микроамперметра со шкалой на 50 мка, полиэтиленового стаканчика (емкостью 150 мл с внешним диаметром, равным внутреннему диаметру ячейки), вынесенных электродов, микробюретки и механической мешалки. Кривые высокочастотного кондуктометрического титрования аналогичны кривым титрования при низких частотах.
Безрогова [58] определяла кремнефтористоводородную кислоту в сумме с серной кислотой в присутствии фтористоводородной кислоты высокочастотным методом. Титрование проводят в водно-ацетоновой среде (30—40% Н2О) этанольным раствором едкого натра. Ошибка определения «суммы H2SiFe и H2SO4 составляла 3,8% , суммы всех кислот — 0,4% .
Амперометрическое титрование. Описано амперометрическое определение свободной кремнефтористоводородной кислоты и кремне-фторид-ионов в гальванических ваннах для хромирования с использованием в качестве титранта азотнокислого свинца [971]. Определение проводят по току восстановления РЬ2+. Метод применим для образцов, в которых содержание фторид-ионов не превышает 6 мг.
Метод определения кремния титрованием рестворов кремнекислого натрия нитратом свинца [597] Беркович [74] использовал для определения кремния в силикатах. В качестве фона автор применял раствор нитрата калия. Титр раствора азотнокислого свинца устанавливали по кремнию.
Амперометрическое титрование кремнекислых растворов азотнокислым свинцом возможно только в том случае, если содержание 92
кремния в пробе не менее сотых долей миллиграмма SiO2 и не более 0,5 мг. В пробах, содержащих около 1 мг SiO2, наблюдается расхождение с данными весового анализа, так как образуется объемистый осадок силиката свинца, затрудняющий титрование. Точку эквивалентности находят графически. Характер кривых титрования меняется в зависимости от концентрации растворов. Для растворов, содержащих десятые доли миллиграмма SiO2, получаются две пересекающиеся прямые; для растворов, содержащих сотые доли миллиграмма SiO2, левая часть ниспадающая, а затем идет прямая линия.
Определению мешают сульфат-ионы. Хлорид-ионы не мешают, если их содержание не превышает в 10 раз содержание кремнекислых солей. Титрование кремнекислых солей в присутствии кар-бонат-ионов может быть проведено лишь в нейтральной или слабокислой среде. Титрование в щелочной среде непосредственно проводить нельзя. Щелочной раствор силиката нагревают до 80° С, прибавляют 2 капли фенолфталеина и горячий раствор нейтрализуют хлористоводородной кислотой до обесцвечивания индикатора, затем прибавляют 1—2 капли щелочи-той же нормальности, что и кислота, до появления слабо-розовой окраски. После такой подготовки титрование протекает нормально. Ошибка определения не превышает 7%.
Чумаченко, Коршун и другие [597] применили этот метод для определения кремния в кремнийорганических соединениях. Они использовали восстановительный способ разложения кремнийорганических соединений путем их нагревания с металлическим калием в герметически закрытых стальных микробомбах при 850— 900° С. В процессе разложения соединений кремния в таких условиях происходит восстановление кремния до элементного состояния.
В этом случае кремний растворим даже в слабых щелочах, и не происходит потерь его в виде летучего тетрафторида в присутствии фторид-ионов в анализируемом растворе.
Глава 7
ФОТОМЕТРИЧЕСКИЕ МЕТОДЫ
Фотометрические методы определения кремния широко используются при анализе различных соединений. Особенно большое значение эти методы имеют при определении небольших количеств кремния.
Фотометрические методы основаны на способности кремневой кислоты в кислом растворе образовывать с ионами молибдата, ванадата, вольфрамата и другими соответствующие комплексные гетерополи кислоты, окрашенные в желтый цвет. Гетерополикислоты образуются в результате химической реакции между координационно ненасыщенной кислотой и водой (например, H4SiO4 + + 2Н2О = H8SiO6) и последующего взаимодействия образовавшейся координационно ненасыщенной кислоты с анионами молибденовой, вольфрамовой или ванадиевой кислот в присутствии водородных ионов
H8SiO6 + 12 (NH4)2 МоО4 + 24НС1 ->
->Н8 [Si (Мо2О7)6] + 24NH4C1 + 12Н2О.
Наиболее часто фотометрически определяют кремний в виде желтого кремнемолибденового комплекса, максимум поглощения которого расположен при 352 нм. Измерения при длинах волн менее 345 нм проводить нельзя. Обычно работают в области 345— 400 нм [268]. Максимум оптической плотности всегда наблюдается при соотношении csi : сМо = 1: 12, что было установлено еще в 1882 г. [949] и далее подтверждено другими авторами [508, ИЗО].
Скорость взаимодействия молибдата с кремневой кислотой зависит от степени полимеризации последней. Реакция между молиб-дат-ионом и мономерной кремневой кислотой при 20° С полностью заканчивается за 75 сек, а при нагревании до 80° С за 1 сек [701, 720, 806, 1091, 1092]. Дикремневая кислота полностью реагирует примерно за 10 мин. Для взаимодействия поликремневых кислот необходимо время, пропорциональное степени их полимеризации. При некоторой степени полимеризации кремневая кислота уже
04
практически не взаимодействует с молибдатом с образованием соответствующей гетерополикислоты, поэтому в процессе определения кремния процесс полимеризации необходимо предотвратить.
При действии на желтые гетерополикислоты восстановителей образуются продукты восстановления молибдена, окрашенные в интенсивно-синий цвет. Это явление также лежит в основе фотометрических методов определения кремния, еще более чувствительных, чем определение в виде желтых гетерополикислот. Чувствительность определения по синей гетерополикислоте примерно в пять раз выше, чем по желтой [268].
СТРОЕНИЕ И НЕКОТОРЫЕ СВОЙСТВА КРЕМНЕМОЛИБДЕНОВОЙ КИСЛОТЫ
Строение, основность, степень ионизации и некоторые другие характеристики кремнемолибденовой кислоты, хотя и изучались в многочисленных работах, до сих пор еще не вполне уточнены. Наиболее подробно связанные с этим вопросы разобраны в работах [7, 124, 165, 404].
Со времени открытия гетерополикислот было выдвинуто несколько точек зрения на их строение. Ранние представления о строении кремнемолибденовой кислоты сводились к изображению их состава в виде цепных формул типа
0 0 О ОН
II II II I
НО —Мо—О —Мо—... —Мо—О— Si -ОН
II II II |
0 0 о он
или
- О —МоО2—. . . —О —МоО2—ОН
/О —МоО2 —
Si(
\0 —МоО2 —
- О —МоО2 —
.. -О-МоО2-ОН
. ,-О-МоО2-ОН . . -О-МоО2-ОН
Согласно теории цепного строения, основность гетерополикислоты должна соответствовать основности исходной кислоты, т. е. кремнемолибденовая кислота как производная ортокремневой кислоты должна быть четырехосновной.
Исходя из координационной теории, гетерополикислоты являются продуктами замещения кислорода в кислотах с общей формулой НпЭОе. Если в гипотетической кислоте с формулой Нп[ЭОв] кислород замещен димолибдат-ионами, а Э — представляет собой атом кремния, то получается кремнемолибденовая кислота. У кремнемолибденовой кислоты, как это следует из формулы, на один атом комплексообразователя приходится двенадцать координированных окислов. Основность кремнемолибденовой кислоты, пред
95
ставляющая собой разность между суммой валентности лигандов и валентностью комплексообразователя, равна восьми. Более того, удалось выделить соли серебра и ртути кремнемолибденовой кислоты, основность которых соответствовала восьми.
Строение внутренней сферы восьмиосновной кремнемолибденовой гетерополикислоты представляется следующим образом. Центральный атом кремния окружен атомами кислорода. Эти атомы кислорода неравноценны, часть их, связанная с двумя атомами водорода, обозначена О2' (атомы кислорода из аквогруппы); остальные атомы кислорода из гидроксильных групп связаны с одним атомом водорода и обозначены О-. Атомы кислорода, являясь координационными центрами второго порядка, объединяют вокруг себя 12 молекул окислов металлов (МоО3). Строению кремнемолибденовой гетерополикислоты соответствует формула
(О-)4
Н8
Si
(О2-)2
(МоО3)12
хН2О.
Пространственная модель кремнемолибденовой кислоты с координационным числом шесть для центрального атома представляется в виде двух кубов, помещенных один в другом и взаимно параллельно расположенных. В центре первого куба находится атом-комплексообразователь — кремний, в центрах граней расположены атомы кислорода (октаэдрическая группировка), середина ребер второго куба занята двенадцатью группами МоО3.
Пфейффер вполне закономерно предполагал существование двух геометрически изомерных форм, обусловленных неодинаковым расположением групп ОН- и Н2О. Подобная изомерия формально возможна. Однако вследствие легкого перемещения ионов водорода подобные изомеры должны легко переходить друг в друга.
Большой вклад в дело изучения строения гетерополи кислот внесли кристаллохимические и рентгеновские исследования этих соединений. Согласно этим исследованиям основность гетерополикислот не должна превышать основность кислоты, отвечающей центральному атому комплекса, т. е. основность кремнемолибденовой кислоты не должна превышать четырех.
Некоторые исследователи пытались согласовать существование солей высокоосновных гетерополикислот с данными рентгеноструктурных и кристаллохимических исследований, выдвинув теорию о неравноценности ионов водорода. Согласно этой теории, например, кремневольфрамовая кислота (а аналогично ей, видимо, и кремнемолибденовая) диссоциирует не ступенчато, а одновременно отделяя четыре иона водорода. Кроме «сильной» кислотной функции, отвечающей отщеплению четырех ионов водорода, гетерополикислоты кремния характеризуются еще наличием «слабой» кислотной функции, отвечающей отщеплению остальных ионов водорода. Отщепляющиеся в первую очередь ионы водорода со-
96
ответствуют основности исходной ортокремневой кислоты, а ионы водорода, отвечающие слабой кислотности, возникают из присоединившихся молекул воды. Неравноценность ионов водорода может быть отражена с помощью формулы типа
Н4
77- [Si (Мо2О7)61 Н4
Таким образом, строение, основность, механизм образования гетерополисоединений до настоящего времени остаются невыясненными окончательно.
Наличие во внутренней координационной сфере кремнемолибденовой гетерополикислоты лигандов двоякого рода дает возможность предполагать существование изомеров кремнемолибденовой кислоты [743, 1035].
цис-Изомер (а-форма) — обычная кремнемолибденовая кислота, получаемая в промышленных условиях, достаточно устойчива. транс-Изомер (P-форма) еще не выделен в кристаллическом состоянии. Феррари первый указал на существование различных форм кремнемолибденовой кислоты [753—756]. Он установил, что а- и p-формы имеют различные окраски. а-Форма имеет более слабую окраску в сравнении с p-формой. Светопоглощение растворов P-формы более чем в два раза превышает светопоглощение ос-формы. Страйклэнд установил, что а- и P-формы имеют одну и ту же эмпирическую формулу [1034]. а-Форма устойчива в растворах, содержащих 1,45—1,50 эквивалента хлористоводородной кислоты на 1 г-ион МоС)|~. P-Форма получается при наличии в растворе более чем двух эквивалентов хлористоводородной кислоты на то же количество МоО|". Однако P-форма неустойчива и за несколько часов переходит в а-форму [843]. В присутствии избытка молибдена превращение p-формы в a-форму замедляется, и полупериод реакции составляет около 25 час, повышение температуры и присутствие посторонних электролитов не столь значительно ускоряет этот переход. Присутствие этанола и ацетона также повышает устойчивость р-кремнемолибденовой гетерополи кислоты [696, 697].
Кемуля и Росоловский предполагают существование еще у-формы кремнемолибденовой кислоты. При pH от 0 до 4 при 30-минутном кипячении а- и p-формы целиком превращаются в у-форму, отвечающую формуле H4[Si(Mo3O10)4]-H2O, которая является более стабильной, чем а- и P-формы кремнемолибденовой кислоты [404, 1133]. P-Комплекс, который чаще используется для фотометрического определения, получается, таким образом, в более кислой среде при строгом соблюдении требуемых условий. Приводим оба метода определения кремния с получением а- и р-комп-лексов [601].
Образование P-комплекса. Большое значение имеет соблюдение требуемого pH раствора и проведение фотометрического определения через определенное время. Чем ниже величина pH, тем мед-
4 Аналитическая химия кремния 97
леннее происходит образование кремнемолибденового комплекса. Предлагается поддерживать значение pH раствора в границах 1,5—1,7 (хотя имеются и другие данные, например 1,5—2,0 и т. д.). После образования комплекса следует подождать не менее 20 мин (но после 45 мин становится уже заметным превращение р-комп-лекса в слабее окрашенный a-комплекс). В сильноразбавленных растворах желтая окраска развивается более медленно.
К Ю мл анализируемого раствора, содержащего 0,4—1,0 мг кремнекислоты, прибавляют 1 мл 9N H2SO4, 5 мл раствора персульфата аммония и 5 мл раствора молибдата аммония. Приводят значение pH раствора к 1,6±0,1, контролируя с помощью потенциометра, и разбавляют до 50 мл раствором H2SO4, величина pH которого равна 1,6. Через 20 мин измеряют оптическую плотность. Получаемые результаты воспроизводимы с отклонениями порядка 0,2—0,5%. Если указанные границы pH соблюдаются не так строго (pH колеблется в пределах от 1,25 до 2,2), то ошибка может достигать 5%.
Образование «-комплексов. Определение проводят при pH от 2,5 до 3,9, нагревая раствор до кипения. Этот фотометрический метод несколько менее чувствителен, чем предыдущий, но получаемая окраска более устойчива, и определение может быть выполнено с точностью ±0,1%. Температура, при которой проводят измерение, имеет большое значение, отклонение не должно превышать 0,4% на Г С.
Определение кремния в горной породе, содержащей 20—70% двуокиси кремния, проводят следующим образом [601 ]. Навеску 100— 200 мг горной породы сплавляют с 10 г NaOH в никелевом тигле в течение 5—10 мин и обрабатывают плав 40 мл раствора 0,05 М комплексона III и небольшим количеством воды. Затем раствор разбавляют до 500 мл в полиэтиленовой посуде. Этот раствор должен весить 509,4 г при 20° С, поэтому вместо разбавления до объема 500 мл можно разбавить до достижения этого веса. Отобрав 25 мл полученного раствора, прибавляют 10 мл буферного раствора (получают путем смешивания равных объемов 2 М раствора монохлоруксусной кислоты и 2 М раствора хлорацетата аммония) и 10 мл 3,53%-ного раствора молибдата аммония. После проделанных операций значение pH раствора должно составлять 3,5—3,7. Раствор помещают в кипящую водяную баню на 5—10 мин, охлаждают, разбавляют до 50 мл водой и измеряют оптическую плотность.
Кремнемолибденовая кислота хорошо растворима в воде, разбавленных кислотах, спиртах, эфирах и нерастворима в бензоле, хлороформе и сероуглероде. В присутствии хелатообразующих агентов (например этилендиамина) кремнемолибденовая кислота осаждает Cr, Ni, Со, Си, Zn, Ag, Cd, Sn и Hg.
Кремнемолибденовая гетерополикислота легко восстанавливается в кислом растворе с образованием синих продуктов восстановления. При восстановлении а-кремнемолибденовой кислоты получается сначала зеленое окрашивание, обусловленное присоединением четырех электронов к аниону, затем синее окрашивание, связанное с присоединением еще нескольких электронов.
Из всех описанных восстановителей только хлорид олова образует восстановленную форму а-кислоты сине-зеленого цвета. Другие восстановители всегда образуют только синий продукт восстановления — а-кремнемолибденовую кислоту. При восста-98
давлении Р-кремнемолибденовой кислоты получается только синяя форма, которая на воздухе очень неустойчива, вследствие чего необходимо поддержание постоянной кислотности. Оптическая плотность восстановленной Р-кремнемолибденовой кислоты на 66% слабее, чем восстановленной а-кремнемолибденовой кислоты той же концентрации. Таким образом, а- и p-изомерные кремнемолибденовые кислоты дают различные продукты восстановления. Однако оба эти продукта при окислении азотной кислотой, бромной водой, перекисью водорода и другими окислителями снова переходят в исходные гетерополикислоты.
Механизм восстановления и строение восстановленных продуктов изучены еще недостаточно хорошо. При восстановлении а-крем-немолибденовой кислоты до молибденовой сини электролитическим методом образуются два соединения различного состава Mo(V): :Mo(VI) = l:5 и Mo(V):Mo(VI) = 1:2. Первый из них имеет максимум при 750 нм, а второй — при 800 нм. При восстановлении Р-крем-немолибденовой гетерополикислоты получается соединение состава Mo(V) : Mo(VI) = 1:1 с максимумом светопоглощения при 830 нм [842—844]. Примерно тот же состав имеют продукты восстановления кремнемолибденовой гетерополикислоты, полученные с использованием различных восстановителей, но наряду с этими продуктами в ряде случаев образуются продукты восстановления с отношением Mo(V) : Mo(VI) = 1:3 и максимумом светопоглощения при 680 нм.
В аналитической практике более глубокое восстановление ведет к образованию восстановленных окислов молибдена с более высоким коэффициентом светопоглощения, что значительно повышает чувствительность метода. Однако степень восстановления очень сильно зависит от условий проведения эксперимента. Результаты, полученные при использовании сильных восстановителей, часто менее воспроизводимы, чем в тех случаях, когда применяются более мягкие восстановители и получаются менее сильно восстановленные соединения.
При использовании методов определения кремния, связанных с образованием синих продуктов реакции, необходимо соблюдать ряд требований к восстановителям, применяемым в этой реакции. Так как восстановленную кремнемолибденовую гетерополикислоту не отделяют от избытка реактива, то необходимо подобрать такой восстановитель и такие условия реакции, чтобы восстанавливался только связанный в комплекс молибдат и в то же время избыток свободного молибдата оставался без изменения. Это необходимо учитывать при определении кремния, так как сильные восстановители взаимодействуют не только со связанным, но и со свободным молибдатом, что приводит к завышенным результатам при определении кремния. Слабые восстановители не действуют на избыток молибдата, однако кремнемолибденовую кислоту они восстанавливают чрезвычайно медленно и часто неколичественно. В связи с этим к выбору восстановителя необходимо относиться чрезвычайно
4*
99
внимательно и строго соблюдать рекомендуемые условия проведения реакции.
В различных методиках применяются следующие восстановители: хлоргидраты-2,4-фенола, метол [930), гидразин [671], фенил-гидразин, бензидин [774, 1079], восстанавливающие сахара типа глюкозы, пирамидон, n-фенилендиамин, нитрозо-Р-нафтол, резорцин, аскорбиновая кислота, соль Мора [8, 409, 547, 605, 672],хлорид олова [30, 42, 428, 570, 571, 590, 695], гидроксиламин, гидразин, тиосульфат, хинон, сульфит, гидрохинон, иодистый калий, оксалат олова и др. [8, 14 , 30, 322, 409, 428, 443 , 444 , 526 , 533, 547, 556, 570, 571, 590, 614, 639, 671, 672, 695, 842—844]. Условия восстановления зависят от химической природы восстановителя и применяемых реактивов.
Хлорид олова является одним из наиболее «старых» восстановителей. В ряде случаев в качестве добавки, стабилизирующей хлорид олова, применяется сульфаминовая кислота [647]. Показано, что при использовании SnCl2 в сернокислой среде кислотность не должна быть ниже 0,8 N [42J. При более низкой кислотности происходит восстановление избытка реактива, т. е. свободного молибдата. Хлорид олова как восстановитель имеет ряд недостатков [428, 533, 543, 590, 695]. Воспроизводимые результаты получаются лишь при использовании химически чистого хлорида олова. Кроме того, матовый налет оловянной кислоты, появляющейся на стенках кювет, значительно искажает результаты определения [202].
Бабко и Шановская при определении фосфора в виде фосфорно-молибденовой кислоты нашли, что в системе хлорид олова — серная кислота наблюдается высшая чувствительность. Они предположили, что и кремнемолибденовый комплекс в этой системе будет давать наиболее интенсивную окраску [47]. Однако хлорид олова настолько сильный восстановитель, что даже при очень тщательном контроле может показать завышенные результаты. Более воспроизводимые результаты получаются при использовании оксалата олова [732].
Растворы оксалата олова устойчивее, чем растворы хлорида, вследствие стабилизирующего действия на окислительно-восстановительную реакцию Sn(II) — 2e~~Sn(IV) оксалат-ионов, об-, разующих комплексные соединения с ионами олова. Продукты восстановления кремнемолибденовой гетерополикислоты оксалатом олова обладают большей устойчивостью, чем продукты восстановления ее хлоридом олова. Это обстоятельство некоторыми авторами объясняется наличием в растворе оксалат-ионов, обладающих слабыми восстановительными свойствами. Следовательно, в этом случае на кремнемолибденовую кислоту оказывают влияние сразу два восстановителя [Sn(II) и С2О|~], что приводит к получению продуктов, обладающих несколько большей стабильностью.
Многие исследователи предпочитают использовать в качестве восстановителя соль Мора [8, 42, 202, 297, 338, 520, 533, 547, 548, 100
553, 570, 571]. Закисное железо в отличие от хлорида олова в слабокислой среде не восстанавливает молибдат-иона как в свободном виде, так и в составе мышьяковисто- и фосфорномолибденового комплексов. Таким образом, при использовании в качестве восстановителя соли Мора влияния фосфора и мышьяка можно не опасаться [547, 548]. Однако при этом часто наблюдаются значительные отклонения от среднего результата определения кремния, поэтому удобнее пользоваться в ходе анализа более мягкими восстановителями, такими, как аскорбиновая кислота [266, 530, 672, 782] или 1-амино-2-нафтол-4-сульфокислота [590, 670, 690, 706]. При использовании в качестве восстановителя аскорбиновой кислоты максимальная окраска раствора развивается в течение 15 мин. Продукты восстановления неустойчивы на воздухе. Более устойчивые продукты восстановления образует 1-амино-2-нафтол-4-сульфокислота, но интенсивность их окраски зависит от кислотности раствора.
Матвеев и Глудина установили, что при использовании в качестве восстановителя сульфита натрия в ряде случаев возникают ошибки вследствие изменения интенсивности и характера образующейся при восстановлении окраски кремнемолибденовой гетерополикислоты, что обусловлено непостоянством окислительно-восстановительного потенциала сульфита при хранении. В случае применения в качестве восстановителя насыщенного раствора сульфита и сульфата натрия, обладающего постоянным окислительно-восстановительным потенциалом, результаты получаются более стабильные [344].
При определении кремния в карбиде кальция в качестве восстановителей были использованы смесь гидрохинона с карбонатом натрия и сульфит натрия в уксуснокислой среде. В первом случае иногда наблюдалось выпадение осадка карбоната кальция, во втором — окраска растворов получалась различных тонов от синего до зеленого. Наилучшие результаты в данном случае были получены при использовании в качестве восстановителя смеси гидрохинона и сульфита натрия в ацетатном буферном растворе [232].
Если применять в качестве восстановителя мочевину или тиомочевину [614], то процесс восстановления нужно вести в присутствии ионов меди, играющих роль катализатора [338], так как восстановление кремнемолибденовой гетерополи кислоты тиомочевиной протекает крайне медленно, а в присутствии меди ускоряется в семь раз. Тиомочевина и мочевина используются в качестве восстановителя при определении кремния в меди и медных сплавах [614].
Вейцман воспользовалась иодидом калия в качестве восстановителя при фотометрическом определении кремния в стали [123]. Окислительно-восстановительные потенциалы систем Мое+/Мо5+ и J2/2J~ очень близки между собой (0,50 и 0,54 в), поэтому шестивалентный молибдат-ион не должен восстанавливаться иодид-ио-ном, если кислотность раствора не слишком высока. Комплексный
101
ион молибдата восстанавливается легче, по-видимому, вследствие того, что лиганд в образующейся восстановленной форме прочнее связан с центральным атомом, чем в окисленной. В принятых автором условиях восстановление желтого кремнемолибденового комплекса происходило количественно.
Другие восстановители представляют гораздо меньший интерес и используются редко. По-видимому, невозможно остановиться на каком-либо одном универсальном восстановителе. Каждый из применяемых восстановителей имеет свои специфические преимущества и недостатки и применяется в зависимости от природы анализируемого объекта и условий проведения реакции. Однако 1-амино-2-нафтол-4-сульфокислота наиболее перспективна при использовании ее в качестве восстановителя. Растворы ее устойчивы, а реакция характеризуется высокой чувствительностью. При определении кремния в железе, чугунах и сталях в качестве восстановителя следует использовать соль Мора.
Одним из основных факторов, влияющих на образование кремнемолибденовой гетерополикислоты, является кислотность раствора. Кремнемолибденовая кислота устойчива в определенном интервале pH. Оптимальное значение pH зависит от природы кислоты и состава среды. В присутствии хлористоводородной кислоты и в отсутствие других солей оптимальный интервал pH находится в пределах от 2,6 до 2,77. В присутствии солей алюминия для существования кремнемолибденовой кислоты оптимальное значение pH равно 1,6 [803]. В зависимости от среды оптимальные значения pH меняются от 0,8 до 1,7 и даже 7,5 [732, 965].
Небезразлично, применяется ли в качестве реактива молибдат натрия или молибдат аммония. Первый из этих реактивов образует кремнемолибденовую кислоту в присутствии хлористоводородной кислоты в интервале pH от 1,8 до 2,5, а в сернокислой среде при pH 2. Молибдат натрия требует для образования кремнемолибденовой гетерополикислоты более высокой кислотности. Бабко и Шановская указывают [46, 47], что кислотность раствора в различных методиках колеблется от 0,01 до 0,30 N [13, 46, 124, 145, 407, 410, 429, 470, 526, 533, 556, 565, 570, 571, 590, 732, 835, 1102, 1125].
При фотоколориметрическом определении кремния в виде жел-' той или синей кремнемолибденовой гетерополи кислоты верхний предел кислотности обусловлен разложением гетерополикислоты. Понижение кислотности ниже известного предела также недопустимо, так как при этом будет восстанавливаться избыток реактива, т. е. свободный молибдат [47]. Верхний предел кислотности (в отличие от нижнего) не зависит от химической природы восстановителя. Максимальная степень кислотности определяется устойчивостью полученного желтого кремнемолибденового комплекса и его способностью взаимодействовать с восстановителями. Если кремне молибдат уже получен, то он не разрушается даже в S&5M H2SO4.
102
Бабко считает, что Главным условием определения крёмнИй является достаточно низкая кислотность (|Н+]<; 0,16 N) перед прибавлением молибдата аммония и достаточно высокая кислотность ([Н+ ]>0,16 Д1) при восстановлении [47]. Последнее условие особенно существенно для устранения влияния фосфора. В большинстве описанных методик рекомендуется кислотность 1,6—1,7 N, но восстановление можно вести и при более высокой кислотности.
Другим важным фактором является концентрация молибдата аммония. Кремнемолибденовый комплекс сильно диссоциирован, поэтому требуется избыток молибдата. На 10 мг кремнекислоты при конечном объеме раствора 100 мл требуется 5 мл 10%-ного раствора молибдата. В ряде случаев для определения кремния применяют недостаточный избыток молибдата аммония для полного связывания кремния в окрашенный комплекс. Тогда следует обращать внимание на то, чтобы в стандартных и испытуемых растворах была одинаковая концентрация реактива молибдата. При большой концентрации молибдата, сильно подкисленный раствор приобретает зеленоватый оттенок вследствие частичного восстановления желтой гетерополикислоты. Это мешает измерению интенсивности окраски. При связывании кремния в синий гетерополикомплекс отношение Mo:Si не должно превышать 100:1 [407, 590].
В процессе образования кремнемолибденовой гетерополикислоты на оптическую плотность раствора сильно влияет порядок сливания реагирующих компонентов и длительность их взаимодействия. Как указывают Бабко и Шановская [46, 47], наиболее слабая окраска и плохо воспроизводимые результаты получались в тех случаях, когда силикат смешивали с молибдатом, а затем через некоторое время прибавляли кислоту. Наиболее воспроизводимые результаты наблюдались в случае предварительной обработки силиката кислотой с последующим прибавлением молибдата. Оптическая плотность при этом в течение часа усиливается. Время достижения наибольшей оптической плотности несколько сокращается при увеличении избытка молибдата, но через 24 часа оптическая плотность во всех случаях уменьшается.
ИОНЫ, МЕШАЮЩИЕ ОПРЕДЕЛЕНИЮ КРЕМНИЯ
Систематического изучения действия мешающих ионов при фотометрическом определении кремния не проводилось. Однако в процессе анализа обычных объектов могут образовываться различные гетерополикомплексы, в которых место молибдена может быть занято вольфрамом, ураном, ванадием, ниобием, галлием, а место кремния—фосфором, мышьяком, германием, никелем, кобальтом, марганцем и другими элементами. Это, естественно, приводит к изменению оптической плотности раствора и сказывается на точности определения. Ряд ионов при определенных условиях обра-
103
Зуют с кремнемолибденовой кислотой осадки, что также приводит к ошибкам определения.
Существуют три принципиально различных метода устранения влияния мешающих ионов: 1) подбором соответствующих условий исключается влияние мешающих ионов, и определение кремния проводят без отделения от них; 2) мешающие ионы отделяют от раствора, содержащего кремний, экстракцией, отгонкой или сорбционным .способом; 3) кремний отделяют от мешающих ионов дистилляцией или экстракцией.
Определение кремния в присутствии мешающих ионов без отделения от последних
Определение кремния в присутствии мышьяка и фосфора. Фосфор, мышьяк и другие элементы реагируют с молибдатом аммония аналогично кремнию [404]. Это необходимо учитывать при определении кремния. На оптическую плотность кремнемолибденового комплекса оказывают влияние и другие ионы, но в значительно меньшей степени. Фосфорномолибденовая гетерополикислота образуется значительно медленнее, щелочные соли ее менее растворимы в воде и с течением времени выпадают из раствора в виде осадка. Образование мышьяковистомолибденовой гетерополикислоты происходит лишь при температуре кипения, причем образующаяся гетерополикислота сразу выпадает в осадок в виде аммонийной или натриевой соли.
Задача определения кремния в присутствии мышьяка и фосфора вследствие сходства их свойств чрезвычайно трудна и, несмотря на большое число работ, посвященных этому вопросу, до сих пор не получила полного разрешения. В литературе описан ряд методов определения кремния в присутствии фосфора и мышьяка [45, 361, 526, 556, 565, 697, 917].
Группа методов определения кремния основана на различной прочности образующихся гетерополикислот при различных величинах pH. Хотя некоторые авторы и указывают в своих работах на большую устойчивость фосфорномолибденового комплекса по сравнению с кремнемолибденовым, в большинстве работ отмечается обратное соотношение прочности этих комплексов [47, 470, 571]. Так, в работах [47, 265] было показано, что при подкислении раствора до [Н+] = 2 N и выше фосфорномолибденовый комплекс разрушается, а кремнемолибденовый остается устойчивым. Это подтверждается и другими данными. На этой основе разработана группа методов определения кремния в присутствии фосфора.
В другой группе методов также используется большая устойчивость кремнемолибденового комплекса, однако мешающие гетерополикислоты в этом случае разрушают действием различных комплексообразующих агентов [14]. Так, например, при введении избытка РС)3--ионов фосфорномолибденовый комплекс разрушается с образованием бесцветных комплексов с меньшим числом коорди-104
нированных ионов молибдата [14]. Кремнемолибденовый комплекс при этом не разрушается. Наиболее благоприятный избыток cp:csi = = 1:2. Мышьяковистая кислота при небольшом избытке, так же как и РС)з_-ионы, обесцвечивает фосфорномолибденовый комплекс. Однако большой избыток мышьяковистой кислоты приводит к ослаблению окраски кремнемолибденового комплекса. В качестве комплексообразующих веществ также используют лимонную, щавелевую, виннокаменную кислоты и их соли. Если прибавить щавелевую (3 мл 10%-ного раствора), лимонную или винную (4 мл 10%-ного раствора) кислоту, то окраска фосфорномолибденового комплекса становится настолько слабой, что ею можно пренебречь, если содержание фосфора не больше, чем в пять раз, превышает содержание кремния.
В присутствии ионов трехвалентного железа лимонная и щавелевая кислоты дают окрашенные соединения, что следует учитывать при анализе кремния. Щавелевая кислота со временем разрушает также-и кремнемолибденовую кислоту, поэтому в ходе анализа после прибавления щавелевой кислоты или ее солей нужно по возможности быстрее прибавлять восстановитель [570].
Определение кремния в низкоуглеродистых и высокоуглеродистых сталях. Для растворения углеродистых и низколегированных сталей применяют 3,5%-ный раствор H2SO4; для растворения высоколегированных сталей — смесь 8%-ной НС1 и 12%-ной HNO3 (2 : 1), для растворения чугунов — смесь 10%-ной HNO3 и 12%-ной H2SO4 (1 : 1,2). При содержании кремния в анализируемом образце до 2% навеску 0,25 г растворяют при умеренном нагревании в 50 мл соответствующей кислоты в колбе емкостью 250 мл. Разложение анализируемых образцов заканчивается после прекращения выделения пузырьков газа.
При анализе углеродистых, низколегированных сталей и чугунов к полученному раствору добавляют 0,225 г КМпО4 и кипятят в течение 2 мин. Добавляют Н2О2 для разрушения образовавшейся МпО2 и обесцвечивания избытка КМпО4. Раствор охлаждают, разбавляют до метки дистиллированной водой и тщательно перемешивают. При анализе высоколегированных сталей раствор кипятят до удаления паров NO2, затем охлаждают до комнатной температуры, разбавляют дистиллированной водой до метки и тщательно перемешивают.
При содержании кремния в анализируемом образце от 2 до 4% навеску 0,25 г помещают в мерную колбу емкостью 500 мл. Далее работают, как описано выше, увеличив лишь количество прибавляемых реактивов вдвое. В том случае, если раствор содержит нерастворимые примеси карбидов, их предварительно отделяют фильтрованием.
Далее в мерную колбу емкостью 50 мл помещают 20 мл испытуемого раствора и 10 мл 2,5%-ного раствора молибдата аммония. Перемешивают, оставляют на 10 мин, добавляют 10 мл 5%-ной щавелевой кислоты и 5 мл восстановителя (6 г сульфата железа и 1 мл 3,5%-ной серной кислоты разводят в небольшом количестве воды и разбавляют до 100 мл), доводят водой до метки, перемешивают и фото-метрируют. В качестве раствора сравнения используют раствор, полученный в аналогичных условиях, но вместо 20 мл испытуемого раствора используют дистиллированную воду.
Содержание кремния находят по калибровочной кривой. Калибровочную кривую строят по той же методике, что и при определении кремния в сталях по стандартным образцам.
Определение кремния в присутствии фторид-ионов. Фторид-Ионы мешают определению кремния, вызывая ослабление окрас
105
ки вследствие связывания кремния в комплексные ионы гексафторида. Влияние фторид-ионов устраняется обычно прибавлением комплексообразующих веществ. Наиболее часто для этой цели используют борную кислоту [173]. Если прибавить борную кислоту (20 в. ч. Н3ВО3 на 1 в. ч. F"), то окраска от кремния восстанавливается почти количественно.
Карлсон и Бенке использовали фтористоводородную кислоту для растворения циркония и бериллия, а затем связывали избыток фтора борной кислотой перед получением кремнемолибденового комплекса. Кремний из водной фазы не терялся ни во время растворения образца, ни во время образования окраски. Авторы пришли также к заключению, что фторид-ионы способствуют сохранению кремния в мономерной форме [690].
0,5 г титанового сплава помещают в полиэтиленовый стакан, добавляют 40 мл воды и 5 мл 15%-ной HF. Стакан закрывают полиэтиленовой крышкой и оставляют стоять на ночь для растворения образца. Добавляют 100 мл воды, 4 г борной кислоты и перемешивают раствор до полного растворения борной кислоты. При постоянном перемешивании добавляют по каплям 3%-ный раствор КМпО4 до слабо-розовой окраски. Раствор выдерживают на кипящей водяной бане в течение 1,5 часа, охлаждают до комнатной температуры, фильтруют и промывают осадок дистиллированной водой.
Фильтрат и промывные воды собирают в мерной колбе емкостью 250 мл, разбавляют водой до 200 мл и выдерживают в течение 1 часа. После этого добавляют 10 мл 5%-ного раствора молибдата аммония и перемешивают раствор. Оставляют на 10 мин для появления желтой окраски кремнемолибденовой кислоты. Затем прибавляют 5 мл 20%-ного раствора щавелевой кислоты и 3 мл восстановителя. Раствор доводят дистиллированной водой до метки, перемешивают и через 1 час измеряют интенсивность окраски при 700 нм. Содержание кремния находят по калибровочной кривой. Восстановитель готовят растворением в 175 мл воды 30 г бисульфита натрия, 1 г сульфита натрия, 0,5 г 1-амино-2-нафтол-4-сульфокислоты. Разбавляют раствор до 200 мл и фильтруют через бумажный фильтр.
Калибровочную кривую строят, используя стандартный раствор,содержащий 0,05 мг/мл кремния (для анализа сплава с содержанием кремния менее 0,18%) или 0,5 мг/мл кремния (для анализа сплава с содержанием кремния от 0,18 до 1,5%).
Титан, цирконий и другие элементы также образуют с Е~-ионами комплексные соединения. Однако устойчивость этих комплексов значительно выше, чем устойчивость кремнефторида, поэтому при прибавлении борной кислоты они не разрушаются. Вместо борной кислоты можно использовать соли алюминия, который образует с фторид-ионами гексафторид алюминия [118, 639]. При анализе алюминиевых сплавов количество алюминия во много раз превышает содержание фтора и устраняет его влияние.
Определение кремния в присутствии алюминия, железа, кальция и других элементов. При спектрофотометрическом определении кремния в силуминах с использованием в качестве восстановителя 1-амино-2-нафтол-4-сульфокислоты Толмачев и Затучная исследовали влияние различных ионов на определение кремния [533].
Влияние посторонних элементов (сме, мг/мл) при определении кремния (csi=7-10~4 мг/мл) охарактеризовано следующим
106
образом:
Элемент min з GMe * iU ’ мг/мл Элемент с^п-ю-мг/мл
А1 35,0 Мп 160,0
Fe(III) 15,4 Си 14,0
Са 77,0 Ti 0,30
Zn 0,02 Bi 0,02
РЪ 7,00
Как видно, цинк, титан и висмут, если они присутствуют в несколько больших количествах, чем это обычно бывает в силуминах, мешают определению кремния. Влияние же других элементов, как и следовало ожидать, незначительно.
Присутствие в растворе солей олова, свинца, железа и алюминия не влияет существенно на точность определения кремния в медных сплавах, если концентрация кремневой кислоты не выше 1 мг в 100 мл азотнокислого раствора. Присутствие солей марганца (2 мг на 100 мл азотнокислого раствора) и никеля (Змг на 100 мл раствора) приводит к полимеризации кремневой кислоты уже при комнатной температуре. Однако со временем (4—5 суток) в азотнокислых растворах при комнатной температуре происходит обратный процесс деполимеризации кремневой кислоты. Таким образом, при анализе бронз типа БАЖМ и БАЖН определение кремния фотометрическим методом становится возможным лишь на четвертый или пятый день после растворения навески.
Авторами было установлено, что персульфат аммония способствует деполимеризации кремневой кислоты как при комнатной температуре, так и при нагревании до 100° С. Ранее персульфат аммония был использован при определении кремния в магниевых сплавах [412]. При определении кремния по желтому кремнемолибденовому комплексу нет необходимости отделять его, так как области поглощения голубого раствора азотнокислой меди и желтого раствора кремнемолибденовой кислоты не. перекрываются.
Определение кремния в бронзах типа БАЖМ и БАЖН. Навеску сплава 0,1 г помещают в полиэтиленовый стакан, содержащий 15 мл 25%-ного раствора персульфата аммония и 3 мл конц. HNO3. Растворяют навеску вначале на холоду, затем при -нагревании в течение 5—10 мин при 100° С. ' Раствор охлаждают и через 30 мин переносят в мерную колбу емкостью 100 мл, разбавляют примерно до 40 мл бидистиллятом и добавляют 25 мл 10%-ного молибдата аммония. Через 10 мин добавляют 20 мл H2SO4 (1 : 5), доводят бидистиллятом до метки и измеряют интенсивность окраски раствора с синим светофильтром. Содержание кремния находят по калибровочной кривой.
Калибровочную кривую строят аналогичным, путем. Навеску меди высокой чистоты, эквивалентную количеству меди в навеске сплава 0,1 г, растворяют в смеси персульфата аммония и азотной кислоты. Растворы охлаждают, переносят в мерную колбу емкостью 100 мл и через 30 мин добавляют 0; 0,1; .... 1,0 мг кремния в виде стандартного раствора. Доводят объем раствора примерно до 40 мл и далее работают, как описано выше.
Соли железа в количестве 0,5 мг на 20 мл раствора не мешают фотометрическому определению кремния, если содержание последнего составляет от 0,25 до 0,50 мг. При большом содержании желе
107
за растворы приобретают зеленоватый оттенок, и результаты определения кремния получаются заниженными [276]. Железо может быть удалено в виде фосфата в уксуснокислой среде, а избыток фосфата —• в виде фосфата кальция.
Другой источник ошибок при определении кремния в присутствии большого количества железа заключается в том, что железо (III) с молибдатом аммония может при определенных условиях образовывать труднорастворимую соль, осаждение которой связано с адсорбцией значительного количества кремнемолибденовой кислоты из раствора [642[.
Такие элементы, как Ba, Bi, РЬ и Sb, могут давать в присутствии гетерополикислот осадки или мути и поэтому должны быть предварительно удалены из раствора.
Определение кремния в присутствии анионов. Присутствие анионов существенно влияет на окраску гетерополисоединений. Бабко и Шановская показали, что хлорид-, сульфат- и нитрат-ионы при рН>2 снижают интенсивность окраски кремнемолибденовой гете-рополикислоты.
Галахов при определении кремния в корунде обнаружил, что увеличение концентрации сернокислых солей калия и натрия приводит, наоборот, к усилению окраски кремнемолибденовой гетерополикислоты [145].
Висинтин и Монтериоло при определении кремния в природных водах установили, что определению кремния не мешают хлорид-ионы при содержании их в растворе <2,5 г/л, нитрат-ионы при содержании <2,5 г/л иЗО4-ионы при концентрации <1,7 г/л [1072].
Как правило, в ходе анализа сульфат-, хлорид-, нитрат-ионы присутствуют в растворе в количествах, не оказывающих существенного влияния на интенсивность окраски, вследствие чего их влияние не учитывается.
Методы определения кремния, основанные на отделении его от мешающих примесей экстракцией или дистилляцией
Методы, основанные на отделении кремния от мешающих приме, сей в виде тетрафторида кремния или кремнефтористоводородной кислоты, известны давно. Холт описал метод определения микро-граммовых количеств кремния в плутонии, урановых сплавах, сталях и в фосфорной кислоте [819]. Кремний количественно отгоняли в виде тетрафторида в равномерно нагреваемом дистилляционном платиновом приборе (рис. 10). Дистиллят поглощался раствором борной кислоты и молибдата аммония, и в нем определяли кремний фотометрическим методом.
В платиновый тигель, содержащий навеску анализируемого вещества, помещают 5 мл НС1О4. Нагревают тигель до полного разложения вещества. При разложении образцов урана или плутония навеску обрабатывают последовательно 1 мл 10%-ной НС1О4, 2 мл НС1О4 (1 : 1) и, наконец, 4 мл конц. НС1О4.
108
Рис. 10. Дистилляционный при. бор для отгонки кремния 1 — нагреватель;
2 — платиновый тигель;
3 — трубка для ввода кислот;
4,6— тефлоновые трубки;
5 — воздуходувка;
7 — пробка
После разложения образца тигель охлаждают, помещают в платиновый дистилляционный сосуд в нагреватель и закрывают кусочком алюминиевой фольги. Соединяют платиновую трубку дистилляционного сосуда с тефлоновой трубкой абсорбционного сосуда, содержащего 25 мл абсорбционного раствора, и пропускают воздух со скоростью 125 мл!мин.
Через верхнюю трубку быстро вводят в тигель 0,3 мл смеси HF и HNO3 (5 мл 50%-ной HF4-25 мл конц. HNO3). Нагревают дистилляционный сосуд первые 10 мин энергично, следующие 10 мин — медленно поднимая температуру до 200° С. Через 20 мин нагрев прекращают, отсоединяют сосуд с абсорбционным раствором (значение pH раствора должно быть 1,2—1,3).
Раствор оставляют стоять на 10 мин для образования (J-кремнемолибденовой гетерополикислоты. Добавляют 5 мл смеси 55%-ной H2SO4 и 2,6%-ной винной кислот (1 : 1), 0,8 мл восстановителя и разбавляют водой в колбе емкостью 50 мл др метки. Через 20 мин измеряют оптическую плотность раствора при 815 нм, используя в качестве раствора сравнения дистиллированную воду. Содержание кремния находят по калибровочному графику.
Для приготовления восстановителя 0,1 г 1-амино-2-нафтол-4-сульфокисло-ты и 0,4 г гидроокиси натрия растворяют в 25 мл воды, добавляют 5,4 г бисульфита натрия, доводят объем раствора до 50 мл и перемешивают. Метод позволяет определять до 50 мкг кремния.
Этим же методом Ульянов [543] определял кремний во фторидах и железных сплавах. Кремний отгонялся при 140° С из раствора серной кислоты в виде тетрафторида кремния, и последний поглощался раствором смеси молибдата аммония с борной кислотой и далее определялся фотометрически. Метод позволяет определять от 10 до 140 мкг кремния.
Дистилляционные методы требуют для их выполнения сложной и дорогой аппаратуры и поэтому не нашли широкого применения.
Кремний может быть отделен от мешающих примесей экстракцией его в виде кремнемолибденовой кислоты. При определении примеси в некоторых полупроводниковых металлах Назаренко и Флянтикова для отделения кремния от примесей удаляли основные элементы в виде летучих соединений: сурьму — в виде трибромида, галлий — в виде ортооксихинолината, индий — в виде трихлорида, таллий — в виде окиси при сплавлении его с едким натром [395]. В ходе анализа галлия, таллия и индия определению кремния мешает платина, попадающая из посуды, вследствие нало
109
жения желтой окраски платинохлористоводородной кислоты на окраску кремнемолибденовой сини, поэтому определение кремния проводят после предварительной экстракции кремнемолибденовой кислоты.
Экстракционно-фотометрические методы определения кремния в последнее время находят все большее применение, что, несомненно, связано с их преимуществами перед фотометрическими методами в водных растворах. Недостаток всех аналитических методов, основанных на фотометрировании кремнемолибденовой кислоты в водных растворах (особенно при фотометрировании восстановленных форм), заключается в мешающем влиянии избытка молибдата. Применение экстракции позволяет легко устранять это влияние и получать надежные воспроизводимые результаты. Перевод кремнемолибденовой кислоты в органическую фазу позволяет существенно повысить ее устойчивость во времени.
Основные затруднения при определении кремния в виде р-крем-немолибденовой кислоты связаны с неустойчивостью этого соединения вследствие перехода в a-форму. Экстрация-Р-кремнемолиб-деновой кислоты органическими растворителями устраняет это затруднение, так как в среде органических растворителей устойчивость р-формы значительно выше.
Общий вид спектров поглощения экстрагированной кремнемолибденовой кислоты мало отличается от спектров водных растворов, но максимумы поглощения экстрагированных кислот несколько смещены в коротковолновую область спектра.
Коэффициент распределения кремнемолибденовой кислоты зависит от кислотности раствора, концентрации экстрагируемого соединения, присутствия нейтральных солей и температуры. Этот вопрос детально рассмотрен в работах Шкаравского [613], в обзоре [16] даны обобщающие данные.
Чувствительность определения кремния может быть значительно повышена в случае применения для экстракции органических оснований и фотометрирования их солей с кремнемолибденовой кислотой. При использовании в качестве экстрагента полиокси-этиленлауриламина возможно определение 1 мкг кремния (в пересчете на SiO2) в 1 л раствора. Об использовании для экстракции кремнемолибденовой кислоты тридециламина, лауриламина, триоктил-и тринониламина см. в работах [179, 642, 1024].
Преимущество экстракционных методов определения кремния заключается также в возможности отделения кремнемолибденовой кислоты от других мешающих элементов (Р, As, Ge), гетерополикислоты которых имеют отличные от кремнемолибденовой кислоты коэффициенты распределения.
Для отделения кремния от фосфора и мышьяка проводят последовательную экстракцию мешающих элементов в виде гетерополикислот [565, 1102]. Фосфорномолибденовая, мышьяковистомолиб-деновая и кремнемолибденовая кислоты растворимы в бутиловом, изобутиловом, изоамиловом спиртах, диэтиловом эфире, в эфирах
110
уксусной кислоты значительно лучше, чем в воде. Исходные компоненты растворимы в этих растворителях значительно хуже. В органических растворителях, не содержащих кислорода (бензол, хлороформ и т. д.), гетерополикислоты нерастворимы. Растворяющая способность кислородсодержащих экстрагентов может быть снижена прибавлением неполярных растворителей (хлороформ, бензол), причем снижение зависит от вида гетерополикислот. При этом следует отметить, что извлечение соответствующей гетерополикислоты из сферы реакции сдвигает равновесие реакции в сторону образования соответствующей гетерополикислоты, и скорость ее образования увеличивается.
Вначале извлекают фосфорномолибденовый комплекс с помощью бутанола и хлороформа (1:3), затем мышьяковистомолибденовый — бутанолом и этилацетатом (1:1) и, наконец, кремнемолибденовый комплекс — бутанолом. В процессе работы следует строго поддерживать нужную концентрацию ионов водорода, ибо с ее изменением меняется скорость расслаивания жидкостей.
С использованием экстракции удается определить тысячные и десятитысячные доли процента кремния в воде. На полноту экстракции гетерополикислоты оказывает влияние ряд факторов: солевой состав водной фазы, температура экстракции и т. д. Однако неполное извлечение определяемого комплекса не оказывает существенного влияния на точность определения кремния, если построение калибровочной кривой и само определение проводить в одних и тех же условиях. Экстракцию проводят не только с целью отделения кремния от мешающих ионов, но и с целью повышения его концентрации.
Методы определения кремния, основанные на удалении мешающих примесей экстракцией, дистилляцией
или с использованием ионного обмена
Мешающий определению мышьяк (а также германий) может быть удален в виде хлорида: при кипячении солянокислых растворов образующиеся хлориды германия и мышьяка полностью улетучиваются [268]. Мышьяк(Ш) и мышьяк(У) также полностью улетучивается в процессе перегонки с НВг и НСЮ4 при 200° С.
Германий улетучивается в каждой из этих смесей при условии, что смесь добавляется перед нагреванием, что предотвращает образование двуокиси германия. При этих же условиях улетучиваются сурьма(Ш) и (V) и олово(П) и (IV). При нагревании с НС1 и НС1О4 полностью улетучивается также хром(Ш). Галлий может быть удален в виде ортооксихинолината.
Большим неудобством указанных методов отделения мешающих ионов является происходящая в процессе отделения дегидратация кремния. Поэтому перед фотометрированием он должен быть сплавлен со щелочами.
Ill
Для удаления примесей также применяют ионный обмен (1122]. После перевода кремния в растворимую форму мешающие катионы отделяются на ионообменной колонке, й в полученном фильтрате определяют кремний фотометрическим методом.
Большинство описанных фотометрических методов определения кремния применяют для определения небольших количеств кремния (до 15—20%), причем с увеличением содержания кремния в анализируемом образце точность определения снижается. Применение дифференциального метода позволяет значительно повысить точность определения [531]. Сущность дифференциального метода заключается в том, что в «нулевой» раствор кроме реактивов вводят известное количество определяемого элемента и по отношению к нему измеряют оптическую плотность анализируемого раствора. Концентрацию кремния находят по калибровочному графику или вычисляют по формуле
сх ~ с0 + DXF,
где сх — концентрация кремния в исследуемом растворе; с0 — концентрация кремния в «нулевом» растворе; Dx — оптическая плотность исследуемого раствора; F~kclD, Лс-щ—с2.
Тихонов и Чернышева определяли дифференциальным методом кремний в материалах титанового производства [531]. Этим же методом Матросова и Зубкова определяли кремний в алюминиевых сплавах при содержании его около 16% [347]. В качестве стандартного раствора авторы использовали раствор с содержанием кремния около 15%.
При дифференциальном методе точность определения, как правило, повышается в 2—4 раза. Разность оптических плотностей между «нулевым» и испытуемым растворами при дифференциальном фотометрическом методе измеряют с помощью различных спектрофотометров или фотоколориметров. По данным ряда авторов, при определении различных элементов использование фотоколориметров Люмтрона, Спеккера и ФЭК-М для дифференциальных измерений не дает выигрыша в точности по сравнению с абсолютным методом. Лучшие результаты получаются при использовании прибора ФЭК-56 с ртутной лампой.
Фотометрический метод широко применяют для определения кремния в сталях [428, 547, 666], железе и чугунах [8], шлаках и силикатах [470, 520, 767], сплавах [410, 693], водах [88, 322], в алюминии [118, 347, 719], кремнийорганических [295—297] и элементокремнийорганических соединениях [296], биологических объектах [1079], никеле и меди [565], ферросилиции, материалах титанового производства [835], полупроводниковых металлах [395], фторидах и фтористоводородной кислоте [173], морских водах [1035] и других материалах.
Определение кремния по скорости образования кремнемолибденовой кислоты описано в [ИЗО, 1131]. Так, при низких кислотностях (0,03—0,07 М НС1О4) и при постоянной концентрации молиб-112
дата начальная скорость изменения оптической плотности растворов, содержащих Si(OH)4, НС1О4 и Na2MoO4, прямо пропорциональна концентрации кремния. При анализе поддерживают постоянную ионную силу раствора. Начальную скорость определяют графически по наклонам прямых в координатах «оптическая плотность — время». Содержание кремния находят по калибровочному графику. Ошибка определения кремния при его концентрациях >3 и 0,Зжг/ли составляет соответственно 1 и 4%. Длительность определения 10— 30 сек. Определению мешают ионы железа, фтора, фосфора и мышьяка.
ФЛУОРЕСЦЕНТНЫЙ МЕТОД
Флуоресцентный метод пока не нашел широкого применения для определения кремния [81, 736]. Эллиот и Радлей разработали флуоресцентный метод определения кремния, основанный на реакции силиката натрия в среде формамида с бензоином с образованием соединения, дающего интенсивно-зеленую флуоресценцию. Бор также образует с бензоином комплексное соединение состава 1:1 [736], которое дает интенсивную флуоресценцию в области 450—520 нм с максимумом поглощения при 370 нм. Для устранения влияния бора в анализируемый раствор добавляют маннит.
Интенсивность флуоресценции изменяется во времени, максимального значения свечение достигает через 60 мин после смешения растворов. Пропорциональная зависимость между интенсивностью флуоресценции и содержанием в растворе кремния сохраняется в интервале концентраций от 2,0 до 10,0 мкг/мл. Присутствие в растворе фосфора и мышьяка в микрограммовых количествах определению кремния не мешает.
Кремний выделяют в виде силиката натрия, и водный раствор осторожно выпаривают в платиновой посуде досуха. Твердый остаток осторожно растворяют в 1 мл дистиллированной воды и 0,5 мл 15%-иого раствора NaOH (свободного от кремния), вводят 0,03 г маннита, и раствор переносят в мерную колбу емкостью 25 мл. В эту же колбу добавляют 15 мл формамида, 0,5 мл насыщенного раствора трехкратно очищенного бензоина в этаноле и 0,5 мл 2%-ного водного раствора солянокислого гидроксиламина. Полученный раствор разбавляют формамидом до метки.
Аналогично готовят холостую пробу и эталонные растворы н спустя 40 мин через каждые 5 мин до получения максимального отсчета измеряют интенсивность флуоресценции каждого из полученных растворов. По полученным максимальным отсчетам строят калибровочную прямую и находят содержание кремния в анализируемом растворе.
Высокое значение интенсивности флуоресценции холостой пробы зависит в основном от флуоресценции продуктов окисления бензоина, который в щелочной среде легко окисляется кислородом воздуха. Флуоресцентная реакция на кремний является более специфичной, чем определение по кремнемолибденовой кислоте. Определяемый минимум 2 мкг. Авторы подчеркивают, что при использовании других восстановителей чувствительность определения можно увеличить до 1 мкг, что особенно важно для определения кремния в чистых металлах и полупроводниках,
Глава 8
ПРОЧИЕ МЕТОДЫ
СПЕКТРАЛЬНЫЕ МЕТОДЫ
В настоящее время спектральный анализ широко применяется для определения кремния. Основные методы анализа и их теоретическое обоснование изложены в ряде руководств и монографий [30, 146, 206, 241, 242, 251, 319, 320, 332, 459, 460, 491, 633, 675, 744, 807—8091.
Кремний широко распространен в природе, поэтому его линии почти всегда присутствуют на спектрограммах любых исследуемых образцов, образуя характерные группы, которые облегчают ориентировку в спектре. Спектрографический метод в большинстве случаев применяется для определения малых количеств кремния, например при анализе карбонатных пород, солей, некоторых природных вод и т. д. Вместе с тем имеются методы, позволяющие определять большие концентрации кремния (до 40%) [3, 127, 332].
Спектральное определение кремния выполняют по следующим наиболее чувствительным линиям (в А):
2435,159
2438,782
2443,378
2506,899
2514,331
2516,123
2519,207
2524,118
2528,516
2631,310
2881,578
2970,347
2987,648
3905,528
6347,010
6371,090
При определении кремния в рудах его легко обнаружить по появлению наиболее интенсивной линии 2881,578. При небольших концентрациях кремния в рудах появляется также весьма характерная группа из шести линий: 2506,899; 2514,331; 2516,123;
2519,207; 2524,118; 2528,516 А, из которых самой интенсивной линией является линия 2516,123.
Линии других элементов, присутствующих наряду с кремнием, могут мешать его определению. Например, линия 2528,516 А перекрывается интенсивной линией сурьмы 2528,52 А, а линия кремния 2514,331 А почти сливается с очень слабой линией железа
2514,38 А.
114
Таблица 3
Внутренние стандарты для спектрального определения кремния
Анализируемое соединение X, А для кремния Элемент сравнения 0 k, A для элемента сравнения
Ферросилиций 2435,159 А1 2652,5
Чугун 2506,899 Fe 2507,9
Магнезит 2514,331 Си 2492,1
Сталь 2516,123 Fe 2518,1
Фтористые соли 2516,123 Си 2492,1
Никель 2516,123 Ni 2551,0
Пудра для корунда 2516,123 А1 2552,5
Теллур 2516,123 Cd 2677,6
Минеральные масла 2516,123 Cd 2571,1
Чугун - 2516,123 Fe 3053,09
Свинцовая пудра 2516,123 Al 2552,5
Трехокись вольфрама 2519,207 Сг 2492,1
Оловянистые шлаки 2528,516 Ni 2545,9
2528,516 Си 2492,1
Магнезит 2881,578 Mg 2938,5
Магниевые сплавы 2881,578 Mg 5528,5
Феррованадий 2881,578 Fe 2880,7
Ферросилиций 2881,578 Fe . 2858,3
Алюминий 2881,578 Al 3057,2
Мартеновские шлаки 2881,578 Sn 2850,0
Мартеновские руды 2881,578 Ni 2943,9
Титановый концентрат 2881,578 Ni 2881,3
2881,578 Fe 2929,0
Цементный шлам 2881,578 Си 2882,9
Сплавы 2881,578 Ni 3414,0
Йодистый натрий 2881,578 Na 2660,4
Силикаты 2987,648 Ba 3071,6
3905,528 Co 3995,3
Сталь, силумины 6371,360 Fe 6303,0
Чугун 6346,7 Fe 6400,0
Шлак 6346,7 Zn 6362,3
Аналитические линии кремния (в А), совпадающие с линиями других элементов, приведены ниже:
2516,123 2881,578 2881,58
2515,58 2880,72 2881,94
2515,69 2880,77 2882,04
2515,81 2880,80 2882,09
2516,12 2880,73 2882,22
2516,25 2881,23 2882 54
115
В зависимости от характера анализируемого объекта в качестве внутреннего стандарта применяют линии различных элементов: алюминия, железа, хрома, меди и т. д. (табл. 3).
Ошибка определения кремния спектральным методом колеблется от 2 до 20% 1105, 106, 158, 2631.
Спектральный метод рекомендуется для определения кремния в самых различных материалах: сталях [2, 36,55,56,64, 65, 77, 86, 91, 104, 280, 333, 480, 492], чугунах [1, 2], шлаках [102, 103, 106], ферросплавах [36, 105, 127, 274], хромистых сталях [104], железных рудах [277—279, 544], мартеновских шлаках [127], сварных швах [314], силуминах [2, 59, 154, 473], металлическом хроме [245], никеле [109, 562], алюминии [107], магнии [24], фольге из никеля [59], редких землях [689], трехокиси вольфрама [729, 916], свинцовой пудре [3361, пудре для корунда [337], алюминиевых сплавах [53, 122], хромокобальтовых сплавах [201], керамических материалах на никелевой основе [288], магниевом сплаве [24, 977], глиноземе [181], оловянистых шлаках [431], магнезите [423], латуни [419, 473], медных и свинцовых шлаках [895], глинах [273], микрообразцах на основе нитрата стронция [424], концентратах плавикового шпата [87], углях [734], иодистом натрии [2631, водах [424], строительных материалах [722], графите [158], полевом шпате [209], фтористых солях [289], цементе и цементном шламе [3 , 252], трех- и пятихлористом фосфоре [311] и других материалах [41, 42, 158, 159, 241, 242, 244, 265, 1071, 1074, 1076].
ОПРЕДЕЛЕНИЕ ПО ПЛОТНОСТИ
Для сплавов, в которых плотности соответствующих компонентов Значительно отличаются друг от друга, определение кремния можно проводить по измерению этого параметра. В литературе имеются данные по изучению зависимости плотности сплава от содержания в нем кремния.
Бабаев [36] разработал метод определения кремния в силико-хроме (сплав кремния, хрома, железа и углерода) и ферросилиции. Плотность кремния значительно отличается от плотности хрома и железа. Плотность сплава зависит от присутствия в нем углерода, но обычно содержание этого элемента в сплавах достаточно постоянно, поэтому ошибка определения кремния по плотности не превышает 1 %.
При определении кремния в ферросилиции (сплав алюминия, железа, кальция, марганца, фосфора и серы) примеси алюминия и кальция, плотность которых близка к плотности кремния, влияют на точность анализа. Например, 44%-ный ферросилиций, содержащий 2,0% А1 и 0,5% Са, имеет плотность ^—5,04; тот же ферросилиций с содержанием 0,5% А1 и 0,1% Са имеет плотность 5,18. Поэтому ошибка определения в ряде случаев может достигать 3%. Вследствие этого при построении калибровочных кривых необходимо учитывать влияние алюминия и кальция.
116
ОПРЕДЕЛЕНИЕ МЕТОДОМ МИКРОТВЕРДОСТИ
Для исследования процесса насыщения поверхности различных сплавов кремнием могут быть использованы металлографический, рентгенографический и другие методы. Эти методы, однако, чрезвычайно сложны, длительны и трудоемки. Метод определения содержания кремния на определенной глубине путем испытания на микротвердость быстр и несложен. Сведения о зависимости твердости поверхностных слоев от изменения их химического состава (вследствие образования твердых растворов) весьма ограниченны, что связано с трудностями определения твердости по глубине слоя обычными методами. В табл. 4 представлены результаты изменения
Таблица 4
Зависимость чисел твердости (ркГ[ммг) железокремневых сплавов от содержания кремния
Содержание кремния, % Нагрузка, кГ Содержание кремния, % Нагрузка, кГ
20 50 20 50
0 192 185 6,5 507 417
1,07 210 201 12,5 743 597
3,5 232 272 18,5 1037 797
4,78 396 324
микротвердости железокремнистых сплавов с содержанием кремния от 1,07 до 18,5%. Испытания проводят на приборе Хрущова и Берковича [589].
Как видно, с увеличением содержания кремния в растворе a-железа твердость последнего возрастает.
ТЕРМОЭЛЕКТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
Термоэлектрический метод определения кремния в железе, сталях [5, 54, 667, 1029], чугунах [733, 873], сплавах [277] основан на измерении величины термоэлектрического потенциала, возникающего при нагревании анализируемого образца и одного из двух стандартных электродов, между которыми он помещен. Термоэлектродвижущая сила (ТЭДС) зависит от разности температур горячего и холодного конца, процентного содержания составляющих компонентов или их соединений, обработки (термической или механической) и т. д. Если ТЭДС измеряется в определенном интервале температур, термическая и механическая обработка образцов одинакова, а изменение концентраций некоторых компонентов не влияет заметно на ТЭДС. Если установлена зависимость ТЭДС от процентного содержания этих компонентов, то в этом случае можно определить концентрации тех компонентов, которые значительно влияют на величину ТЭДС.
117
На величину ТЭДС углеродистой стали заметно влияет содержание кремния и слабее — содержание углерода, если количество каждого компонента не превышает 2% . Влияние кремния ТЭДС объясняется изменением энергии электронов атомов железа при внедрении кремния в кристаллическую решетку.
Если количество углерода и кремния в бинарных сплавах железа не превышает 2%, то ТЭДС таких сплавов изменяется по линейному закону. Эмпирическая зависимость может быть выражена формулой
Si, % — В// — Л/С,
где I — термоэлектродвижущая сила анализируемой стали по отношению к определяемому металлу; А и В — постоянные, устанавливаемые опытным путем; Си Si — содержание углерода и кремния, %.
Имеется и другой метод учета влияния углерода на ТЭДС при определении кремния в углеродистой стали. Подбирается несколько групп эталонов углеродистой стали так, чтобы для каждой группы содержание углерода было постоянным, а содержание кремния изменялось бы в анализируемом интервале концентраций. Для каждой группы эталонов строят график в координатах «7 —Si, %». Количество кремния в анализируемом образце находят по графику, для которого содержание углерода ближе всего подходит к таковому в анализируемом образце.
При анализе простых сталей влияние одинаковых количеств углерода и марганца на изменение ТЭДС примерно равнозначно. При этом изменение содержания углерода или марганца на 0,1% соответствует по влиянию на ТЭДС изменению содержания кремния в среднем на 0,01%. Этим объясняется кажущееся иногда на практике отсутствие влияния на ТЭДС углерода и марганца в простых сталях, в которых с повышением содержания углерода увеличивается содержание марганца. Хром при содержании от 0,1 до 1,2% не влияет на ТЭДС.
В передельном чугуне на ТЭДС влияют кремний и марганец. Влияние марганца невелико. При содержании в образце 0,41% кремния влияние марганца в 12 раз меньше влияния кремния; при содержании кремния 0,69%— в 7—8 раз. Установление эмпирической зависимости ТЭДС от содержания кремния и марганца в передельном чугуне невозможно, так как при различных содержаниях марганца в чугуне его влияние на ТЭДС неодинаково. Остается один метод — метод подбора.
В качестве термопар могут быть использованы самые различные материалы. При определении кремния в чугуне в качестве холодного элемента использовали медный цилиндр, охлаждаемый термостатированной (20° С) водой; в качестве нагреваемого электрода служил серебряный стержень, снабженный надежной теплоизоляцией, исключающий потери в окружающую среду [873]. В качестве горячего элемента может быть использована латунь [278]. Разность
118
температур между холодными и горячими спаями в различных методиках колеблется от 100 до 300° С.
Обычно точность термоэлектрического определения кремния составляет + 1%.
ОПРЕДЕЛЕНИЕ ПО УДЕЛЬНОМУ СОПРОТИВЛЕНИЮ
Зависимость удельного сопротивления железа от содержания присадочных элементов описана в ряде работ [657, 716, 941]. Для чистых низколегированных железнокремниевых соединений соотношение между содержанием кремния и удельным сопротивлением описывается уравнением р=0,099+0,12% Si.
Исследования зависимости удельного сопротивления железа от содержания кремния в кремнистых сталях с различным содержанием углерода, марганца, фосфора и серы показали, что на удельное сопротивление основное влияние оказывает растворенный углерод. Его влияние значительно превосходит влияние всех других присадочных элементов. Однако содержание растворенного углерода во многих сплавах незначительно, поэтому на удельное сопротивление основное влияние оказывает кремний.
Другие присадочные элементы находятся в незначительных количествах, поэтому их влиянием можно пренебречь. Исходя из этого возможно быстрое определение кремния в трансформаторных сталях [752, 821].
Глава 9
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В ПРИРОДНЫХ И ТЕХНИЧЕСКИХ ОБЪЕКТАХ
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В НЕКОТОРЫХ ПРИРОДНЫХ И СИНТЕТИЧЕСКИХ СИЛИКАТАХ И ГОРНЫХ ПОРОДАХ
Выбор метода анализа зависит от состава образца и содержания кремния. Содержание кремния в материалах неизвестного состава качественно и полуколичественно может быть определено спектральным методом. Это позволяет установить размер навески, выбрать способы разложения, отделения от мешающих элементов и определения (см. главы 4, 5 и 7).
При значительном содержании кремния (порядка 5% и выше) обычно используют гравиметрический метод. Все более широко применяется титриметрический метод, основанный на гидролитическом разложении иона SiF^~ (см. главу 6). При малом содержании кремния его определяют чаще всего фотоколориметрическим методом (см. главу 7).
Анализ горных пород и силикатного минерального сырья описан в монографиях и руководствах (17, 25, 75, 161, 166, 243, 353, 355, 356, 490, 569, 581, 628, 632, 728, 789, 1078]. Условия определений и методы анализа приведены в ГОСТах. Обзоры методов анализа силикатов и новые методы см. в [79, 100, 157, 160, 198, 199, 252, 313, 342, 354, 376, 377, 484, 686, 765, 1016, 1030, 1076, 1077].
Определение кремния в растворимом стекле. Растворимые стекла — смесь силикатов и полисиликатов щелочных металлов — характеризуются высоким содержанием кремневой кислоты (порядка 60—75%). При действии воды на измельченное растворимое стекло образуется раствор, в котором кремневая кислота присутствует в виде силикат- и полисиликат-ионов, а также частично в виде монокремневой кислоты, т. е. в истинно- и коллоиднорастворимом состоянии [4]. Поэтому в зависимости от выбранного способа определения кремневая кислота в полученном растворе под-120
вергается дальнейшей полимеризации (например, выпариванием с кислотами) или, наоборот, деполимеризации (например, обработкой растворами гидроокисей или карбонатов щелочных металлов).
Разработаны методы анализа для твердых стекловидных силикатов и растворов. Основные методы см. в [163, 1060].
Определение кремния в растворимом стекле гравиметрическим методом [163]. Среднюю пробу 2—3 г гидратированного стекловидного силиката измельчают в корундизовой или агатовой ступке до получения порошка, проходящего через сито 900 отв! см2. Навеску 1—1,5 г полученного порошка помещают в платиновую чашку, заливают пятикратным количеством воды и выдерживают на кипящей водяной бане 30—40 мин, помешивая стеклянной палочкой и прибавляя воду по мере ее испарения. После окончательного растворения в чашку прибавляют 50 мл горячей воды, выдерживают 10—15 мин на кипящей водяной бане, приливают 10—15 мл НС1 (1 : 1) и выпаривают, растирая твердый остаток палочкой до исчезновения запаха НС1.
Чашку охлаждают, сухой остаток смачивают конц. НС1, накрывают чашку часовым стеклом и дают постоять 5—10 мин без нагревания. Затем приливают 50 мл горячей воды, перемешивают палочкой осадок и после отстаивания декантируют раствор и переносят осадок на фильтр; промывают горячей НС1 (1 : 20) и затем 2—3 раза горячей водой. Фильтр с осадком переносят в тигель, осторожно озоляют и прокаливают до постоянного веса в электропечи при 1000° С.
Рекомендуется применять для ускоренных определений фотоколориметри-ческий метод, а также метод, основанный на расчете содержания SiO2 по разности после определения гидратной влаги и щелочности.
Предложены ускоренные алкалиметрические методы определения кремневой кислоты в растворимых стеклах [346, 488, 489, 650].
Определение кремневой кислоты в растворимых силикатах полумикрометодом [506, 507]. В колбу емкостью 50—100 мл, покрытую внутри тонким слоем парафина (или в полиэтиленовый стакан), вводят анализируемый раствор в количестве, соответствующем 1,5—7 мг SiO2. К раствору добавляют 1-—2 капли раствора метилового красного,нейтрализуют 0,017V раствором НС1 и вводят 2—8 мл раствора, содержащего 2,5 г NaF и 25 г КС1 в 100 мл. Из бюретки добавляют 1—55 мл 0,01 JV НС1 (в зависимости от предполагаемого содержания SiO2), через 20 мин вводят 5—10 мл спирта и еще через 10 мин титруют избыток не вошедшей в реакцию хлористоводородной кислоты 0,01 N раствором NaOH. Для внесения поправки на индикатор проводят холостой опыт.
Удовлетворительные результаты получаются при анализе проб, содержащих Mn2+, Fe2+, Сг3+, МоО2~, WO2- и VO~. Присутствие Fe3+ или А13+ мешает определению.
Ускоренное алкалиметрическое определение кремния в растворимом стекле [488]. 5 мл силиката натрия (d=l,19) или 10 мл (d= 1,093) помещают в мерную колбу емкостью 100 мл, доводят водой до метки и тщательно перемешивают. 15—20 мл полученного раствора титруют в присутствии метилового красного 0,17V НС1 до исчезновения желтого окрашивания индикатора и переносят в парафинированную фарфоровую (или полиэтиленовую) чашку.
Затем в раствор добавляют 3—4 г кристаллического NaF, несколько капель индикатора метилового красного, перемешивают пластмассовой палочкой и титруют 17V НС1 до исчезновения желтого окрашивания индикатора. Титрование заканчивают, когда последние капли кислоты придают раствору постоянное бледно-розовое окрашивание. Абсолютная ошибка по сравнению с весовым методом составляет ±0,8%. Продолжительность определения 4—5 мин.
121
Описано определение малых количеств силикат-ионов кинетическим методом [880], кондуктометрическим в растворах солей кремневой кислоты [420], комплексонометрическим микрометодом в среде ацетона [846]. Растворимые силикаты определяют в морской воде [680, 930] колориметрическим микрометодом [1011], гравиметрически в форме кремнемолибдата хинолина [564],Определение кремния в алюминатных растворах описано в [118], См. также [344, 624, 670, 719, 760, 788, 1048, 1060, 1072, 1091, 1098].
Определение кремния в карбонатных породах. Содержание кремневой кислоты в карбонатных породах изменяется в широких пределах; это влияет на выбор метода разложения пробы и выполнения определения. В монографии [25] приведено пять схем анализа карбонатных пород в зависимости от содержания в них кремния. Приводим краткое изложение методик определения кремния из трех схем, наиболее интересных, с точки зрения авторов. Выбор метода анализа связан с предварительным определением нерастворимого остатка, величина которого позволяет судить о том, насколько велико содержание кремния в веществе. Содержание нерастворимого остатка оценивается как малое, значительное и очень высокое при обработке пробы раствором НС1 (1:1).
Анализ образцов с малым содержанием SiO2. Навеску измельченной породы 0,5—0,6 е помещают в фарфоровую чашку емкостью 30—40 мл, смачивают водой, покрывают чашку стеклом и прибавляют по каплям НС1 (1 : 1) до прекращения выделения СО2. Стекло снимают и обмывают, приливают еще 2—3 мл НС1 и выпаривают раствор досуха. Сухой остаток смачивают НС1 и после 5 мин стояния обрабатывают горячей водой. К полученному раствору с осадком прибавляют при помешивании NH4OH до запаха, осадок отфильтровывают, промывают 2%-ным раствором NH4NO3, нейтрализованным аммиаком по метиловому красному.
Осадок растворяют на фильтре в небольшом количестве горячей разбавленной HNOa (1 : 10). Из фильтрата после прибавления 1 мл конц. HNO3 снова осаждают полуторные окислы аммиаком и фильтруют через тот же фильтр. После промывания и высушивания фильтр с осадком помещают в платиновый тигель, озо-ляют, прокаливают и взвешивают. Далее обрабатывают осадок HF и H2SO4, выпаривают досуха, прокаливают и взвешивают. По разности в весе находят содержание SiO2.
Анализ образцов со значительным содержанием SiO2. Навеску карбонатной породы предварительно прокаливают при высокой температуре и, охладив, количественно переносят в фарфоровую чашку. Далее разлагают НС1, как описано в предыдущей методике. Раствор выпаривают досуха, остаток смачивают хлористоводородной кислотой, прибавляют горячую воду, осадок отфильтровывают, промывают водой, подкисленной НС1, высушивают и прокаливают. Далее обрабатывают HF и H2SO4 по предыдущей методике.
Анализ образцов с высоким (а иногда и знач и -тельным) содержанием SiO2. В этом случае сплавляют навеску образца с карбонатом натрия, причем в отличие от обычных соотношений плавень:вещество это отношение берется несколько пониженным (оттрех до шести). Дальнейшее выполнение процедуры см. в главе 5.
В карбонатных породах с низким содержанием двуокиси кремния рекомендуется определение выполнять колориметрическим методом (см. главу 6, а также [38, 700, 959]). Иногда в карбонатных породах с малым содержанием кремния его не определяют; в этих случаях ограничиваются определением «нерастворимого 122
остатка». Образцы разлагают также кислотами с последующей обработкой отфильтрованного «нерастворимого остатка» смесью HF и H2SO4. Вместо сплавления рекомендуется разложение спеканием, обработка НО под давлением, смесью HC1+HNO3, смесью HNO3+HC1O4 [ 700].
Ускоренные методы см. в [120, 375], ускорение разложения при введении комплексонов — в [975], спектрофотометрический микрометод — в [445].
Определение кремния в цементах. Для определения кремния в портландцементе и в клинкере используют гравиметрический метод, его ускоренные варианты, а также титриметрический и колориметрический методы. Методы анализа имеются в руководствах [38, 149, 161, 377, 549] и ГОСТах. Для арбитражных анализов рекомендуется гравиметрическое определение с двойной дегидратацией и обработка осадка смесью HF с H2SO4. Выделения неосаж-денной кремневой кислоты из фильтрата не производят.
Гравиметрическое определение по ГОСТ 5382-6 7. Навеску 0,5 г цемента помещают в фарфоровую чашку, смачивают 10 мл воды, прибавляют при помешивании 10 мл НС1 (1 : 1) и выпаривают досуха на водяной бане. По охлаждении смачивают содержимое чашки 5—7 мл конц. НС1, накрывают чашку часовым стеклом и оставляют стоять 5 мин. Затем добавляют 15—20 мл горячей воды, накрывают чашку часовым стеклом, дают раствору отстояться в течение 10 мин на кипящей бане и фильтруют отстоявшийся раствор над осадком через неплотный фильтр. Осадок в чашке промывают 2—3 раза горячим раствором НС1 (5 : 95) вначале декантацией, а затем переносят на фильтр и промывают горячей водой до исчезновения реакции на хлорид-ионы (проба с раствором AgNO3, подкисленным HNO3). Осадок сохраняют.
Фильтрат с промывными водами переносят в ту же чашку, в которой производилось выпаривание, и вторично выпаривают на водяной бане до исчезновения запаха НС1. По охлаждении сухой остаток в чашке смачивают 5—7 мл НС1 и вновь выделяют кремневую кислоту, как указано выше, отфильтровывают осадок через неплотный беззольный фильтр и промывают его 2—3 раза горячим раствором НС1 (5 : 95), а затем горячей водой.
Фильтрат и промывные воды сохраняют для дальнейших определений, а осадок на фильтре присоединяют к ранее выделенному осадку кремневой кислоты, помещают в предварительно прокаленный и взвешенный платиновый тигель и озоляют вначале при низкой температуре, накрыв тигель во избежание потерь при обугливании фильтров, затем приоткрывают крышку и прокаливают при 1050—1100° С в течение 30 мин. Прокаливание повторяют до постоянного веса.
Осадок в тигле смачивают несколькими каплями воды, прибавляют 3—4 капли H2SO4 и 7—10 мл HF и выпаривают содержимое тигля на песчаной бане до прекращения выделения паров. После этого тигель прокаливают при 1050— 1106° С в течение 3—5 мин, охлаждают в эксикаторе и взвешивают, вычисляя содержание SiO2 по разности в весе тигля до и после обработки смесью кислот.
Гравиметрическое определение кремния в цементе в комбинации с ускоренными методами определения других компонентов см. в [78, 377, 379, 380, 539, 550, 882]. Для ускорения гравиметрического определения кремневой кислоты в цементе предложено несколько способов. Осадки лучшего качества — крупнозернистые, легкофильтруемые — получаются при выделении кремневой кислоты действием концентрированных кислот [511]; коагуляции кремневой кислоты способствует добавление хлорида или нитрата
123
аммония [93, 94, 208, 511], желатины [37, 162, 378, 835, 882] и других коагулянтов [162].
Определение с желатиной [379]. 0,5 г тонкоизмельченного клинкера или цемента помещают в стакан емкостью 250 мл, смачивают несколькими каплями воды, приливают 20—30 мл конц. НС1, нагревают до кипения и кипятят 5—6 мин. После охлаждения до 70±5° С к раствору при перемешивании прибавляют 3—4 мл нагретого до 70° С 2% -ного раствора желатины. Раствор выдерживают на бане при 70° С в течение 10 мин, затем фильтруют через неплотный фильтр. Осадок промывают, высушивают, прокаливают до 1000° С и взвешивают. Если необходима большая точность, то осадок обрабатывают HF и H2SO4.
Другие варианты ускоренного гравиметрического определения кремния см. [37, 38, 379, 380, 511, 546, 757, 835, 949].
Алкалиметрический метод основан на переводе кремния в KaSiFe с титрованием его после отделения фильтрованием или без отделения.
Определение S1O2 в клинкере [200 ]. Навеску клинкера 0,25 г, пропущенную через сито № 85, помещают в чашку из платины или фторопласта-4 и растворяют при нагревании в 10 мл НС1 (1 : 2). К полученному раствору прибавляют 5 мл 4%-ного раствора комплексона III и нейтрализуют по бумажке конго 40%-ным раствором КОН (NaOH) до перехода окраски ее из синей в красную.
Для осаждения алюминия в форме криолита чашку нагревают, к горячему раствору приливают 15—-20 мл 4%-ного раствора фторида натрия (40 г NaF растворяют в 1 л горячей воды, прибавляют 80 г КС1 и после отстаивания [фильтруют в парафинированную или полиэтиленовую склянку) и греют еще'З—5 мин при помешивании. Объем раствора не должен превышать 50—60 мл, иначе изменение окраски индикатора будет нечетким. Затем приливают в небольшом избытке НС1, быстро охлаждают раствор и, непрерывно помешивая, насыщают хлоридом калия до небольшого остатка его на дне чашки.
Содержимое чашки количественно переносят в стакан емкостью 500 мл, разбавляют небольшими порциями холодной воды, насыщенной КС1, так чтобы объем раствора был не больше 75 мл, прибавляют к раствору 10—15 капель 0,1%-ного раствора индикатора бромтимолового синего и осторожно нейтрализуют, приливая растворы по каплям из бюретки, сначала \N, а затем 0,252V раствором КОН (NaOH) до ясного голубого окрашивания, сохраняющегося в течение 1 мин.
После этого разбавляют раствор до 250 мл кипящей водой, нагревают до начала кипения, прибавляют еще около 1,5 мл того же индикатора и титруют 0,252V раствором NaOH до голубого окрашивания раствора. Титр раствора щелочи устанавливают по стандартному образцу с известным содержанием кремния.
Другие методы титриметрического определения кремния в цементах см. в [38, 153, 200, 990, 992, 993, 1103].
Для определения содержания кремния в цементе разработан дифференциальный колориметрический метод [79]. Применение его позволяет сократить время, затраченное на анализ, при сохранении вполне удовлетворительной точности. Метод основан на образовании комплексной кремнемолибденовой кислоты, ее последующем восстановлении или аскорбиновой кислотой, или метолово-сульфитным восстановителем, или эйкоген-сульфитным восстановителем и измерении оптической плотности раствора по сравнению с раствором, полученным аналогичной обработкой стандартного образца.
124
Стандартный раствор для фотометрического определения кремния готовят иа основе стандартного шлама, состав которого проверен в нескольких лабораториях. Сухую навеску шлама 0,3 г сплавляют в платиновом тигле с 1,5 г смеси (2 : 1) безводного карбоната натрия с безводным тетраборатом натрия при 1000° С в течение 3—5 мин.
Охлажденный тигель помещают в стакан, содержащий 100 мл теплого раствора НС1 (1 : 3), и выщелачивают плав при постоянном перемешивании до полного растворения. Затем разбавляют водой до 150—200 мл, переносят прозрачный раствор в мерную колбу емкостью 500 мл, охлаждают до комнатной температуры, доводят до метки водой и хорошо перемешивают. Раствор пригоден в течение месяца для фотометрирования, построения и проверки градуировочных графиков по определению двуокиси кремния и других компонентов цемента.
Приготовление растворов восстановителей см. в [377], а также в главе 9. Для приготовления метолово-сульфитного восстановителя 20 г метола (параметиламинсульфата), 12 г лимонной кислоты и 12 г безводного сульфита натрия растворяют в 800 мл воды и доводят объем раствора водой до 1000 мл.
Для построения градуировочного графика отбирают пипеткой 3, 5 и 7 мл стандартного раствора, переносят в мерные колбы емкостью 100 мл, добавляют по 25—30 мл воды, по 5 мл 5%-ного водного раствора молибдата аммония, перемешивают, дают стоять в течение 10 мин для полноты образования кремнемолибденовой кисло-ты. Затем в каждую колбу добавляют по 20 мл метолово-сульфит-ного восстановителя, доводят объемы растворов водой до меток, перемешивают и снова дают постоять 15 мин для получения восстановленного комплекса. Растворы колориметрируют, пользуясь красным светофильтром, в кюветах с толщиной слоя 10 мм.
Аликвотную часть 5 мл считают основной и сначала фотометрируют кювету с этим раствором. Затем относительно основного раствора промеряют оптические плотности растворов из двух других аликвотных частей (3 и 7 мл). По полученным величинам относительных плотностей и соответствующих им концентраций растворов строят градуировочный график.
Для выполнения определения содержания кремния в цементе (а также в шламе, клинкере, глине) навеску образца (0,3 г шлама, 0,2 г клинкера или цемента, 0,05 г глины) сплавляют и выщелачивают, как описано выше. В мерные колбы емкостью 100 мл отбирают аликвотные пробы по 5 мл от анализируемого и стандартного растворов, добавляют все необходимые реактивы и выдерживают растворы, как при построении калибровочного графика, и затем фотометрируют. В соответствии с полученной величиной оптической плотности по графику находят содержание кремния.
Определение кремния в глинах. Анализируемый материал должен быть тонко измельчен. Разложение обычно выполняют сплавлением с карбонатом натрия в платиновых тиглях. В литературе рекомендуются и другие плавни — борный ангидрид,Дифторид калия, тетраборат натрия, окись свинца, окись висмута, перекись натрия, борат лития (см. главу 4).
Рекомендуется спекание с Na2CO3 [510, 546]. Разложение хлористоводородной кислотой может быть выполнено под давлением [212, 816] или после предварительного прокаливания [212, 510, 546, 816]. Для арбитражных и маркировочных анализов ГОСТы предусматривают двукратную дегидратацию и определение кремния в осадке полуторных окислов.
Определение SiO2 в глинах при сплавлении [212]. Навеску глины помещают в платиновый тигель емкостью 25—30 мл, прибавляют шестикратное количество безводного Na2CO8, тщательно перемешивают смесь палочкой, утрамбовывают ее и обтирают палочку над тиглем небольшим количеством плавня. Плавление ведут в муфельной печи или на горелке таким образом, чтобы не происходило сильного вспучивания и вспенивания массы. После
125
того как расплавленная масса опустится на дно тигля, нагревают ее до прекращения выделения пузырьков газа и еще 8—10 мин. Масса в конце сплавления имеет вид прозрачной жидкости, если содержание двуокиси кремния значительно, или в ней имеются хлопьевидные включения — при большом содержании окиси алюминия. Весь процесс сплавления на горелке длится 30—40 мин.
После извлечения из тигля плав переносят в фарфоровую чашку, прикрытую часовым стеклом, приливают 10%-ную НС1. Когда выделение пузырьков СО2 прекратится, снимают и обмывают часовое стекло и выпаривают раствор на водяной бане досуха, растирая комочки стеклянной палочкой. Чашку снимают с бани, охлаждают, смачивают сухой остаток полностью конц. НС1 и, накрыв часовым стеклом, дают стоять на холоду 20 мин. Затем приливают в чашку 60—70 мл кипящей воды, перемешивают и после отстаивания сливают раствор иа фильтр средней плотности. Осадок промывают 2—3 раза декантацией, затем переносят на фильтр и промывают горячей водой до отрицательной реакции на хлорид-ионы в промывной воде.
Фильтрат и промывные воды помещают в ту же чашку, где производилось первое выпаривание, выпаривают до исчезновения запаха НС1 (сушка до более высоких температур не рекомендуется) и повторяют операции растворения, фильтрования и промывания аналогично первой дегидратации. Оба фильтра с осадками кремневой кислоты помещают в платиновый тигель, осторожно подсушивают, обугливают, озоляют и прокаливают до постоянного веса при 1000— 1100° С.
Осадок смачивают несколькими каплями воды, несколькими каплями конц. H2SO4, приливают 10 мл конц. HF, выпаривают и прокаливают до постоянного веса. По разности находят содержание SiO2.
Гравиметрическое определение SiO2 в каолиновых глинах без сплавления [161]. 0,5—1,0 г тонкоизмельченного вещества прокаливают в муфельной печи при 700—750° С 1,5—2 час, охлаждают и количественно переносят в фарфоровую чашку, смывая остатки 2/V НС1. Добавляют еще 100—120 мл НС1, накрывают чашку часовым стеклом и ставят на водяную баню на 5—6 час. Нерастворимый остаток отфильтровывают, тщательно промывают водой до отрицательной реакции на хлорид-ионы.
Для выделения кремневой кислоты, оставшейся в растворе, фильтрат вместе с промывными водами выпаривают в той же чашке досуха и далее обрабатывают, как обычно. Оба осадка прокаливают вместе и обрабатывают после взвешивания смесью H2SO4 и HF, находя после повторного прокаливания содержание SiO2 по разности весов. Замена при повторном выпаривании НС1 на H2SO4 или НС1О4 упрощает и сокращает время выполнения определения [594, 1006].
Литяну [893] предложен ускоренный метод определения SiO2 в глинах, бокситах, каолинах (см. также [890—892]).
Анализируемое вещество сплавляют с KHSO4 или K2S2O7 до прекращения выделения SO3. Первоначально расплавленная масса затвердевает в процессе нагревания. Ее выдерживают 3—4 мин при 800° С, охлаждают, обрабатывают разбавленной НС1 (2 : 1) или H2SO4 (3 : 1), кипятят 2—3 мин, осадок отфильтровывают и промывают с отсасыванием. Фильтр с осадком сушат, озоляют и прокаливают до постоянного веса и обрабатывают HF-|-H2SO4. Продолжительность определения не более 2 час. Метод по быстроте и точности соответствует методу с желатиной и хлорной кислотой [893].
Спектральное определение SiO2 в глинах описано в литературе, однако химические методы используются значительно чаще, именно для определения SiO2, при выполнении определений других компонентов спектральным методом. Ускоренные методы анализа глинистых материалов см. в [626, 835].
При анализе бокситов выбор метода разложения зависит от минералогического состава: для бокситов гематито-гидраргилитово-
126
го состава возможно разложение смесью кислот (H2SO4 и НО); бокситы гематито-диаспорового и белитового состава рекомендуется сплавлять с пиросульфатами калия или натрия. Разложение бокситов смесью H2SO4, НС1 и HNO3 с последующим гравиметрическим определением см. в [908].
Описано несколько методов микроопределения кремневой кислоты в силикатах, не разлагаемых кислотами, например в [41, 8741.
Гравиметрическое микроопределение кремнекисл о т ы [274]. В платиновый тигель отвешивают около 10 мг тонкоизмель-ченного силиката и сверху помещают около 200 мг безводного Na2CO3. Тигель закрывают платиновой крышкой и сплавляют, постепенно нагревая тигель в муфельной печи до 1000° С. По охлаждении смывают крышку горячей водой в тигель и еще вводят воду, заполняя ею тигель на одну треть. Тигель закрывают часовым стеклом с вплавленным капилляром, через который пропускают газообразный НС1 в течение часа. Конец капилляра не должен касаться жидкости. По окончании пропускания НС1 нижнюю поверхность стекла и капилляр обмывают над тиглем горячей водой, накрывают тигель обычным часовым стеклом и нагревают 30 мин на водяной бане. По окончании выделения СО2 часовое стекло обмывают над тиглем горячей водой, выпаривают раствор в тигле досуха и еще дважды досуха после прибавления по 1 мл конц. НС1.
К остатку добавляют 4—5 мл НС1 (1 : 3), нагревают на водяной бане до растворения солей. Раствор отсасывают через платиновую трубку для фильтрования во второй платиновый тигель и кремневую кислоту промывают три раза по 0,5 мл горячей НС1 и затем водой до тех пор, пока промывная жидкость не перестанет давать реакцию на хлорид-ионы.
После этого тигель и трубку с осадком сушат сначала на водяной бане, затем 10 мин в сушильном шкафу или на алюминиевой колодке и выпаривают с тремя каплями H2SO4, затем прокаливают 3—4 час при 950—1000° С. Охлаждают, взвешивают, затем прокаливают еще раз и взвешивают повторно. Вводят в тигель 0,5 мл конц. HNO3 и 3—4 мл HF и выпаривают полностью на водяной бане; прибавляют 1 мл HF и 0,1 мл конц. H2SO4. После удаления HF нагревают тигель и трубку в алюминиевой колодке для испарения H2SO4. По разнице в весе остатка после прокаливания и ранее полученного загрязненного осадка находят содержание SiO2.
Перманганатометрическое микроопределение кремневой кислоты [41]. Навеску в 2—3 мг силиката сплавляют 5—15 мин в платиновом тигле с 0,5 г смеси равных частей карбонатов натрия и калия. После охлаждения выщелачивают горячей водой и раствор собирают в конической колбе емкостью 100 мл. Раствор нагревают до 50—60° С, добавляют 10 мл 10%-ного раствора молибдата аммония и затем по каплям подкисляют конц. НС1 до просветления и пожелтения раствора и через 1—2 мин добавляют еще небольшой объем НС1.
Раствор нагревают до 40—60° С и осаждают кремнемолибденовую кислоту добавлением по каплям при взбалтывании 10 мл 10%-ного раствора пиридина в НС1 (1 ; 5). Через 1—2 мин, когда осадок станет крупнозернистым, фильтруют и 4—5 раз промывают 1%-ным раствором пиридина в 2%-ной НС1. Осадок смывают горячей водой с фильтра обратно в колбу, растворяют в 50 мл H2SO4 (1 : 4), нагревают и пропускают через редуктор с кадмием. Раствор собирают в колбу с углекислотой, где его титруют 0,1/V КМпО4. 1 мл 0,1/V КМпО4 соответствует 0,1667 мг SiO2.
Для определения кремния в различных породах кроме обычного гравиметрического рекомендуется ускоренный метод с желатиной [36, 262, 438, 848].
Определение кремния в стеклах. Содержание кремния в оконном, посудном, тарном стекле обычно определяют гравиметричес
127
ким методом [657, 779, 882]. Подготовке проб посвящена работа [853].
В платиновый тигель, предварительно прокаленный и взвешенный, берут навеску подготовленного порошка стекла и тщательно перемешивают с плавнем-смесью 5 г Na2CO3 и 0,1—0,2 г KNO3, добавляя плавень частями в 5—6 приемов [882]. Порошок, оставшийся на шпателе, обтирают остатком плавня и ссыпают в тигель.
Нагревают тигель на газовой горелке, постепенно увеличивая пламя, чтобы процесс выделения газов и плавление шли спокойно. Температуру доводят до 1000° С и после прекращения выделения газов вращением тигля (с помощью тигельных щипцов) распределяют сплав тонким слоем по стенкам тигля. Затем тигель охлаждают погружением в воду, следя за тем, чтобы вода не попала внутрь тигля.
Для выщелачивания плава в тигель наливают горячую воду и несколько капель конц. НС1. Плав выщелачивают в фарфоровую чашку, тигель и крышку обмывают над чашкой 2N НС1, в чашку добавляют, прикрыв ее часовым стеклом, конц. НС1. Для ускорения разложения плав время от времени разминают стеклянной палочкой, после окончания выделения СО2 добавляют еще 3—5 мл конц. НС1 и полученный раствор выпаривают на водяной бане до исчезновения запаха НС1 и еще два часа.
Охлажденный сухой остаток смачивают 2—3 мл конц. НС1, накрывают чашку часовым стеклом и через 15—20 мин прибавляют около 50 мл горячей воды, помешивая осадок стеклянной палочкой. Раствор сливают на фильтр, осадок промывают несколько раз декантацией и затем на фильтре до отрицательной реакции на хлорид-ионы. Фильтрат переносят в чашку, где проводилось первое выпаривание, и повторяют операцию выпаривания, сокращая количество НО для смачивания остатка и выдерживая осадки с кислотой 5—10 мин. Осадок отфильтровывают, заботясь на этот раз о количественном переносе кремневой кислоты на фильтр.
Оба осадка соединяют, фильтры обугливают и озоляют, осадки прокаливают до постоянного веса при 1000—1200° С. Чистоту прокаленного осадка проверяют обработкой смесью HF и H2SO4 с последующим прокаливанием и взвешиванием.
Для ускоренного определения кремния в стекле применяется фотоколориметрический метод.
Навеску стекла 0,1—0,2 г помещают в никелевый тигель емкостью 50 мл, добавляют 1 г NaF и 10 мл 5%-ной СН3СООН [343 ]. Тигель накрывают крышкой, ставят на асбестовую сетку и нагревают на слабом пламени горелки до кипения; раствор кипятят 20—25 мин, затем тигель охлаждают, добавляют горячей воды и переносят его содержимое в мерную колбу емкостью 500 мл, куда предварительно помещено 20 мл 30%-ного раствора A12(SO4)3. Колбу нагревают 5 мин на кипящей водяной бане, затем охлаждают и доливают до метки бидистиллятом.
Из колбы отбирают пипеткой 5 мл раствора и помещают в мерную колбу емкостью 100 мл; сюда же приливают 15 мл бидистиллята и 5 мл 5%-иого раствора молибдата аммония в 5%-ной СН3СООН. Раствор выдерживают 5 мин, затем нагревают на кипящей водяной бане 5 мин и, добавив 5 мл сульфит-сульфатного восстановителя, дальнейшее определение ведут, как описано в главе 7.
В натрийборалюмосиликатных стеклах раствор после выщелачивания плава хлористоводородной кислотой выпаривают несколько раз в присутствии метанола [850]. При выделении кремнекисло-ты действием НО результаты получаются заниженными [894]. Рекомендуется выпаривать в танталовой чашке и дегидратацию выполнять дважды, извлекая кремнекислоту также из осадка полуторных окислов.
128
В анализе свинецсодержащих стекол ускорение процесса дегидратации кислотами достигается при использовании для бань высо-кокипящих растворов или жидкостей [853]. Ускоренные методы определения SiO2 в стеклах см. в [271, 290, 291, 784], капельно-колориметрическое определение кремния — в [266], комбинирование гравиметрического и колориметрического методов —в [1105].
Объемный метод определения кремневой кислоты в стекле после осаждения в форме K2SiFe и отделения (вариант метода [1032]) разработан Тананаевым и Бабко [514]. Авторы предлагают использовать два индикатора для фиксации точки эквивалентности —• метиловый оранжевый и нейтральный красный (см. главу 6).
Бесстружковое определение SiO2 в известковонатриевом стекле см. в [266, 893]; методы расчета —в [516]. Сравнительные определения кремния в стеклах приведены в [739, 740]. Применение физико-химических методов определения кремния см. в [784, 1039].
Определение кремния в керамических и огнеупорных материалах. Методы анализа описаны в руководствах [191, 399] и предусмотрены ГОСТ ами.
Гравиметрическое определение кремния в форме SiO2 с двойной дегидратацией рекомендуется для арбитражных маркировочных анализов. Операции подготовки образца к анализу обычные (см. главу 4); выбор плавня определяется составом образца. Чаще всего используют 5—6-кратное количество безводного карбоната натрия. Для высокоглиноземистых огнеупоров требуется очень тонкое измельчение, причем даже оно не всегда обеспечивает полноту сплавления с такими плавнями, как Na2CO3, KHSO4 (или K2S2O7) [654]; рекомендуется сплавление со смесью Na2CO3+CaO (6:1) при соотношении вещество: плавень, равном 2:7. Температура сплавления 1200° С. Смесь для сплавления NaCO3+CaO не ком-куется, при сплавлении нет разбрызгивания, плав легко разлагается НС1 (1:3). Применение этой смеси позволяет разложить полностью такие огнеупоры, как силлиманит или муллит.
Огнеупоры и огнеупорное сырье с высоким содержанием алюминия сплавляют с NaOH [848], со смесью Na2B4O7+KNaCO3 [1011], спекают. Плавни, содержащие СаО, применяют также для высококремнеземистых образцов.
При двукратной дегидратации и больших навесках (до 2 а) потерями SiO2 в фильтрате пренебрегают. Первую дегидратацию выполняют с хлористоводородной, вторую с хлорной кислотами. Это обеспечивает достаточную полноту выделения SiO2, безопасность работы и отсутствие потерь вследствие толчков и взрывов. Осадок SiO2 обрабатывают смесью HF и H2SO4, операцию выделения SiO2 из полуторных окислов обычно опускают.
Гравиметрическое определение кремния в огнеупорных материалах [1011 ]. Навеску воздушносухого тонкоизмель-ченного образца около 2 г помещают в платиновый ригель или чашку емкостью от 30 см3, прибавляют 1 г свежепрокаленной х. ч. СаО (для высококремнеземистых образцов 2 г) и 5 (или 6) г безводного Na2CO3. Содержимое тигля тщательно
5 Аналитическая химия кремния
129
перемешивают стеклянной палочкой с расплющенным концом, размельчая комочки, и уплотняют смесь. Палочку протирают над тиглем небольшим количеством плавня.
Сплавляют в муфельной печи, вначале нагретой не выше 500° С, а еще лучше совсем охлажденной, или на окислительном пламени горелки, постепенно увеличивая пламя или обогрев. Плавление после прекращения выделения пузырьков газов продолжают при температуре около 1200° С еще некоторое время (15 мин для глин, 30 мин для кианита).
Охлажденный плав переносят в фарфоровую или платиновую чашку, тщательно удаляя с крышки и тигля остатки плава обработкой несколькими каплями НС1 и горячей водой. В чашку приливают 75 мл НС1 (1 : 3) и, закрыв ее часовым стеклом, ставят на водяную баню. При необходимости кусочки плава размельчают стеклянной палочкой; после прекращения выделения газов смывают часовое стекло и прикрывают им чашку теперь уже неплотно.
Раствор выпаривают досуха, дают чашке остыть и смачивают сухой остаток конц. НО (25—30 мл). Через 2 мин прибавляют 15 мл холодной воды и, накрыв часовым стеклом, снова ставят на 5—8 мин на водяную баню. После этого чашку снимают с бани, прибавляют 60—75 мл кипящей воды, перемешивают палочкой до растворения солей и фильтруют через фильтр средней плотности (белая лента), перенося содержимое чашки на фильтр количественно. Промывание ведут 6—8 раз НС1 (5 : 95), объем промывной жидкости обычно составляет около 75 мл. Фильтрат и промывные воды собирают в стакан емкостью 400 мл, прибавляют 10 мл HNO3 и 35 мл НС1О4, перемешивают и, прикрыв неплотно часовым стеклом, нагревают на плитке до появления паров НС1О4. Нагревают еще 2—3 мин и затем закрывают стакан стеклом плотно и нагревают еще несколько минут. Сняв стакан с плитки, дают ему остыть, прибавляют 125—150 мл горячей воды и немного бумажной массы, перемешивают и фильтруют в мерную колбу емкостью 500 см3.
После того как содержимое стакана перенесено на фильтр количественно с помощью горячей воды, осадок на фильтре промывают 8—10 раз горячей водой. Оба фильтра с осадками переносят в тигель для прокаливания (желательно использовать тигель, в котором производилось сплавление), обугливают фильтры, озо-ляют и прокаливают 30 мин при 1200° С. После доведения осадка до постоянного веса его обрабатывают 5 каплями 18N H2SO4 и 15 мл конц. HF; кислоты осторожно выпаривают, нагревают для разложения сульфатов, прокаливают при 1000 С° 1—2 мин и после охлаждения взвешивают. По разности находят вес SiO2.
Для ускоренных определений дегидратацию проводят один раз, осадок SiO2 не обрабатывают смесью HF+H2SO4. Используется также метод с желатиной [848, 1030], титриметрическое определение [343, 896, 988, 989].
Определение кремния в хромсодержащих огнеупорах см. в [374], физико-химические методы — в [343, 1039].
Определение кремния в шлаках. Способы разложения анализируемых образцов зависят от их состава. Разложение доменных шлаков обычно выполняют сплавлением с Na2CO3, действием кислот с последующим сплавлением [324]; для разложения мартеновских шлаков применяют кислоты и их смеси [1095]. Дальнейшее определение выполняют гравиметрическим (в форме SiO2), а также тит-риметрическими, спектрофотометрическим и другими методами [169, 470, 520, 643, 665, 922, 950, 990, 993, 1095].
Определение кремния в шлаках никелевой и свинцовой плавки см. в [553], в шлаках медеплавильного производства —в [107], метод ускоренного определения термометрическим титрованием — в [1121].
130
Сравнение и критическая оценка методов определения кремния в силикатах
Сравнение методов и отдельных прописей аналитического определения кремния в силикатах выполнялось с целью установления их точности, воспроизводимости, быстроты и трудоемкости.
Для гравиметрического определения обобщающие сведения имеются в работах [148, 149, 188, 995, 1015, 1078, 1085]. Многие авторы указывают на необходимость совершенствования методов химического анализа силикатов, кремнийорганических соединений и других веществ и материалов, содержащих кремний [207, 292, 296, 848, 872, 914, 1080]. При сравнении четырех методов дегидратации (солянокислотного с двукратным выпариванием, солянокислотного с желатиной, солянокислотного с добавлением глицерина и сернокислотного) предпочтение из-за скорости выполнения отдано последнему. Он рекомендован для массовых определений кремния в растворимых стеклах и органических силикатах [403].
На основе сравнения различных методов (гравиметрического, титриметрического и рентгенографического) определения кремния в природном кварце, стекле, лаве отмечено [1126], что модифицированный гравиметрический метод Армана и Берту [644] является наиболее быстрым (2,5 час) и точным (ошибка 0,2%). Однако для массовых анализов в производстве стекла, цемента и керамики более удовлетворительным является рентгенографический метод.
При сравнении нескольких методов определения кремния в карбонатных и силикатных материалах была установлена наибольшая надежность метода с хлорной кислотой [700]. Для кварцитов и кварцевых изделий рекомендована (по результатам сравнения пяти методов) обработка смесью азотной и фтористоводородной кислот [1080]. Два метода дегидратации (хлористоводородной кислотой и глицерином, а также хлорнокислотного с желатиной) показали сходные результаты [892].
Обзоры гравиметрических и титриметрических методов определения кремневой кислоты в силикатах опубликованы в [658, 1075, 1077]. Оценка ускоренных химических и физико-химических методов приведена в [909, 1074]. Абсолютная разница в результатах, полученных методами дегидратации хлористоводородной кислотой и желатинового [884], составляет 0,23—0,72%, причем результаты, полученные по второму методу, всегда несколько ниже.
Предложено [188, 643, 784, 845, 1011, 1015, 1105] для повышения точности определения кремния комбинировать гравиметрическое и спектрофотометрическое определение кремния.
Многочисленные межлабораторные исследования по определению кремния в силикатах и по анализу силикатов [41, 643, 668, 904, 914, 1015, 1075] показывают, как отмечает Бабко [41], что большинство ошибок имеет систематический характер и поэтому статистическая обработка результатов нецелесообразна [787].
5*
131
При определении содержания двуокиси кремния гравиметрическим методом наиболее вероятны следующие ошибки: 1) неполнота выделения кремневой кислоты из раствора при коагуляции геля, дающая отрицательную ошибку (относительная ошибка порядка 1 %); 2) загрязнение осадка кремневой кислоты вследствие гидролиза соединений алюминия, железа, титана при промывании, вызывающее положительную ошибку; 3) загрязнение остатка примесями в HF и неполнота образования фтористого кремния, обычно приводящие к отрицательной ошибке.
К этим основным ошибкам могут прибавляться и другие систематические и случайные ошибки. Автор рекомендует усилить внимание к отдельным этапам методики, указывает на необходимость выпуска более разнообразного ассортимента стандартных образцов, улучшения качества реактивов, широкой постановки научных исследований по сравнению аналитических методов (см. также [914, 1127]).
Отмечается [904] значительный разброс результатов определений в межлабораторных анализах. Факторы, влияющие на точность анализа силикатов, обсуждены в работах [182, 185 , 631, 848, 904, 914].
Результаты определения кремния гравиметрическим методом в различных материалах — стеклах, огнеупорах, шлаках и т. п., изучены с использованием методов математической статистики [739—741, 858]. Найдена зависимость ошибки определения от содержания кремния в анализируемых материалах и установлено, что результаты получаются систематически заниженными по SiO2 и завышенными по А12О3 [1015]. Сравнительная характеристика методов с применением желатины приведена в [37, 438, 909]. Обзор и критическая оценка методов определения кремния в металлургических шлаках см. в [1119].
Проверена точность и воспроизводимость быстрых методов анализа силикатов [909]. Изучение колориметрического метода определения кремния в силикатах в форме восстановленной кремнемолибденовой кислоты проведено в работе [909]; рекомендован метод [1008]. Исследованы условия выделения и гравиметрического определения кремния в силикатах [186, 188].
Вопросы унификации методов анализа силикатов см. в [41, 653, 914]. Критика методов определения кремния в материалах стекольного производства дана в [872]; обсуждению и критической оценке методов анализа силикатных материалов посвящены работы [522, 807], экспериментальная проверка нескольких методов анализа силикатов и рекомендации приведены в [1075—1077].
Проведена сравнительная оценка методов определения кремния в других объектах: кремнийорганических соединениях [292, 296, 297], почвах [29, 700]. Разработаны международные стандарты [183]. В других работах также рассматриваются ошибки.определения кремния [883].
132
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В СИЛИЦИДАХ МЕТАЛЛОВ И ДРУГИХ ТУГОПЛАВКИХ СОЕДИНЕНИЯХ
В монографии [469] собран литературный материал по анализу тугоплавких соединений кремния. Сведения обобщающего характера по этому вопросу см. также в работах [357—359, 467, 645].
Силициды переходных металлов, отличающиеся тугоплавкостью (Ti, Zr, Hf, V, Nb, Ta, Cr, Mo, W), карбиды и нитриды кремния обычно сплавляют со щелочами, карбонатами щелочных металлов, перекисью натрия и другими плавнями [155, 269, 287, 469, 813]. Некоторые силициды удается разложить фтористоводородной кислотой или ее смесями с другими минеральными кислотами [430 , 720]. Карборундовые огнеупоры разлагают сплавлением, так как, кроме концентрированной Н3РО4 (при высоких температурах), другие кислоты и их смеси на карбид кремния не действуют [358, 359, 425, 467, 469].
Способы аналитического определения кремния для силицидов титана, циркония, ванадия, тантала, ниобия, хрома, молибдена и вольфрама в общем аналогичны и описаны в упомянутой выше монографии [469]. Используют главным образом гравиметрическое определение в форме SiO2. Приводим методику определения кремния в силициде титана [155].
0,1 г силицида сплавляют в железном тигле с 10-кратным количеством Na2O2. Плав выщелачивают водой, приливают 15—20 мл НС1 до растворения гидроокиси железа. Затем дают раствору охладиться, приливают 30—40 мл НС1О4 и выпаривают до появления густых белых паров НС1О4, которым дают выделяться в течение 10—15 мин. Если раствор покрывается коркой до появления паров хлорной кислоты, необходимо прибавить еще некоторое количество НС1О4, предварительно охладив раствор, и продолжать выпаривание.
К холодному раствору прибавляют 15 мл конц. НО, 100 мл горячей воды, нагревают до растворения солей и отфильтровывают осадок кремневой кислоты. Осадок промывают несколько раз горячей НС1 (5 : 95) и водой. Промытый осадок помещают в платиновый тигель, высушивают и прокаливают при 1000— 1050° С до постоянного веса. Взвешенный осадок обрабатывают 3—5 мл HF и 2—3 каплями конц. H2SO4, выпаривают, прокаливают и взвешивают, находя содержание SiO2 по разности.
При сплавлении силицида титана с Na2O2 в никелевом тигле можно выделять кремневую кислоту сернокислотным методом. В этом случае ТЮ2 не выпадает .в осадок совместно с SiO2. Метод характеризуется также большей безопасностью.
При определении общего содержания кремния в силицидах других металлов рекомендуются следующие условия: способ разложения, навеска, количество плавня и материал тигля одинаковы (силицид титана может быть сплавлен со щелочью в серебряном тигле [155], силицид циркония — с безводным карбонатом натрия в платиновом тигле [786]). Для разложения силицидов во всех случаях используют едкий натр; для силицидов титана и хрома, кроме того, перекись натрия; для силицидов хрома — карбонат натриш Сплавы разлагают последовательным действием воды и серной кис-; лоты; для силицида титана предложено, кроме того, разложение
133
последовательным действием воды, хлорной и хлористоводородной кислот, для силицида молибдена — воды и НС1.
Разложение силицидов кислотами может быть использовано для различных силицидов: титана — смесью фтористоводородной и азотной, а также смесью серной и фосфорной кислот; циркония — фтористоводородной кислотой, смесью серной кислоты и бифторида калия; ванадия — фтористоводородной и ее смесью с азотной кислотой; тантала и ниобия — смесью фтористоводородной и азотной кислот; хрома — фтористоводородной и концентрированной хлористоводородной кислотами; молибдена — смесью фтористоводородной и хлористоводородной кислот.
Во избежание соосаждения двуокиси циркония охлажденный раствор, полученный при выпаривании разложенного плава силицида циркония с серной кислотой, разбавляют 100 мл 50%-ного раствора лимонной кислоты [287]. Аналогичным образом поступают при анализе силицидов ниобия, тантала и вольфрама, приливая к охлажденному сернокислому раствору 150 мл насыщенного раствора щавелевой кислоты [425]. Свободный кремний в силицидах титана, тантала и хрома определяют по одинаковой методике [469].
Навеску силицида около 0,5 г обрабатывают в платиновой чашке при нагревании 60 мл 1%-ного раствора NaOH в течение 45—60 мин. Отфильтровывают раствор в мерную колбу емкостью 250 мл, промывая осадок силицида дважды раствором NaOH и затем водой. Из полученного раствора после подкисления серной кислотой отбирают 5 мл в мерную колбу емкостью 50 мл и затем определяют кремний фотоколориметрическим методом по желтому кремнемолибденовому комплексу. Эту методику нельзя использовать для анализа силицидов ниобия и вольфрама ввиду их растворимости в разбавленных растворах щелочей.
Общий и фазовый анализы карбида кремния и изделий из него освещены с достаточной полнотой в работах [359, 448, 469]. В карборундовых огнеупорах находят общее содержание кремния, а также проводят фазовый анализ, определяя свободный углерод и кремний, двуокись кремния, карбид и нитрид кремния. Методика фазового анализа разработана Всесоюзным институтом огнеупоров [448].
Определение свободного кремния. Навеску измельчен-иого карборунда около 1 г помещают в платиновый тигель и прокаливают при 450—500° С до постоянного веса (определение свободного углерода). Прокаленный остаток количественно переносят в платиновую чашку емкостью около 150 мл, смачивают водой и прибавляют 50 мл 0,5W NaOH. Чашку накрывают ча- • совым стеклом и ставят на кипящую водяную баню на 10—15 мин. Полученный раствор осторожно сливают с осадка на двойной плотный фильтр. При содержании кремния до 10% фильтрат собирают в мерную колбу для дальнейшего фото-колориметрического определения, при большем содержании — в фарфоровую чашку для гравиметрического определения методом дегидратации НС1.
Обработку остатка в чашке щелочью и сливание раствора на фильтр повторяют дважды. При последней обработке остаток из чашки количественно переносят на фильтр, промывают 0,0W NaOH и водой до исчезновения щелочной реакции в промывных водах. Колориметрическое определение выполняют по желтой окраске кремнемолибденового комплекса, см. [286, 287].
Для определения суммы SiO2-f- SiC-|- Si3N4 фильтр с осадком помещают во взвешенный платиновый тигель, озоляют и прокаливают при 700° С до постоянного веса.
134
Для определения SiO2 прокаленный осадок переносят в платиновую чашку емкостью около 70 мл, смачивают водой н прибавляют 50 мл 12,5—15 N HF. После двухчасового стояния без нагревания в раствор переходит практически вся S1O2 и небольшое количество Si3N4. Осадок переносят на плотный фильтр, помещенный на парафинированную (или полиэтиленовую) воронку, и промывают несколько раз холодной дистиллированной водой. Затем фильтр с осадком помещают в стакан и нагревают 25—30 мин на плитке со 100 мл 1,45'lVHCl для извлечения соединений алюминия, железа и кальция.
Нерастворившийся остаток отделяют фильтрованием через плотный фильтр, озоляют и прокаливают фильтр с осадком при 700° С. По разности результатов взвешиваний определяют сумму SiC и Si3N4. При расчете содержания SiO2, SIC и Si3N4 используют результаты определения азота, полуторных окислов и кальция, а также учитывают поправку на растворимость Si3N4 в HF.
Быстрый титриметрический метод определения общего содержания кремния в карбиде кремния см. в [813].
Навеску анализируемого вещества около 1 г сплавляют в платиновом тигле с 5—6 г смеси, содержащей 85—90% К2СО3 и 10—15% Н3ВО3. Полученный плав разлагают разбавленной НС1 и переводят в мерную колбу. Аликвотную часть раствора кипятят с 8—10 мл конц. НС1, охлаждают до 30—40° С, помещают в пластмассовый стакан и прибавляют 4 г твердого NaF или KF (до насыщения). Осадок KaSiF« отфильтровывают под вакуумом, промывают раствором, содержащим 7 г КС1, 50 мл Н2О и 50 мл С2Н5ОН, переносят в коническую колбу, содержащую 400—500 мл горячей воды, перемешивают и оттитровывают выделившуюся HF 0,15 A NaOH в присутствии фенолфталеина. Продолжительность определения 1,5 час.
Другие методы определения кремния в тугоплавких соединениях см. в [286, 287, 722, 775].
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В МЕТАЛЛАХ И СПЛАВАХ
Содержание кремния в металлах и сплавах обычно незначительно и не превышает 1—2%. В металлах высокой чистоты содержание кремния не превышает обычно сотых и даже тысячных долей процента.
Кремний в металлах и сплавах может находиться в различных формах. Например, в железных сплавах кремний содержится в трех формах [185]: 1) в виде твердого раствора кремния в железе; 2) в виде силицидов железа, марганца и других элементов (FeSi2, FeSi, FeMnSi, где нормальные валентные связи не проявляются); 3) в виде силикатов (окисленная форма кремния): 2FeO-SiO2, 2МпО-• SiO2, А12О3-51О2идр. (так называемые неметаллические включения). В ходе анализа обычно определяют общее содержание кремния. Подробно условия определения кремния в металлах и сплавах указаны в работах [184, 185, 553, 1122].
Сталь, чугун, железо. Для определения кремния в этих объектах предложен ряд методов. Некоторые не требуют предварительного разложения анализируемого вещества. В основном эту группу методов составляют спектральные [2, 36, 55, 56, 64, 65, 77, 86, 91, 158, 253, 255, 280, 333, 480]. Химические методы определения кремния требуют предварительного разложения анализируемого материала.
135
Методы разложения металлов и сплавов зависят от состава последних и от применяемого метода определения кремния. Для разложения сталей, чугунов и железа с целью последующего определения кремния фотометрическим методом навеску растворяют в серной кислоте при нагревании. Для растворения применяют также серную кислоту с добавлением азотной кислоты, перекиси водорода или персульфата аммония. В ряде случаев образцы растворяют в смеси хлористоводородной, азотной кислот с добавками перекиси водорода и персульфата аммония.
Широко применяется для растворения сталей, чугунов и железа хлорная кислота, которая при температуре >200° С является энергичным окислителем. Разложение хлорной кислоты при нагревании происходит по реакции
4НС1О4 = 2С12 + 7О2 + 2Н2О.
Применение хлорной кислоты при определении кремния в высоко-хромистых материалах особенно удобно, так как при этом не образуются труднорастворимые соединения хрома. Растворение анализируемого образца происходит быстро. Кремневая кислота количественно выделяется в осадок уже при однократном выпаривании. Осадок получается чистый и хорошо фильтруемый. Вольфрамовая, танталовая и ниобиевая кислоты также содержатся в осадке.
При определении кремния с использованием хлористоводородной кислоты раствор выпаривают до полного удаления воды и хлористоводородной кислоты, в случае применения хлорной или серной кислоты нагревают до выделения паров хлорной (200° С) или серной (300° С) кислот. Нагревание необходимо для перевода кремневой кислоты из коллоидного состояния в осадок. Так как осадок вместе с кремневой кислотой может быть загрязнен нерастворимыми и плохо растворимыми веществами (графитом, окисью алюминия, окисью титана, вольфрамовой кислотой, карбидами и т. д.), его проверяют на чистоту обработкой смесью фтористоводородной и серной кислот. Последняя добавляется для предупреждения гидролиза SiF4 и связывания воды, выделяющейся при реакции
SiO2 + 4HF = SiF4 f +2Н2О.
После разложения образца определение кремния заканчивается весовым, титриметрическим или фотометрическим методами. Об определении кремния в сталях, чугунах и железе весовым методом см. в [470, 553, 564, 570], титриметрическим [988], фотометрическим — в [8, 428, 547, 666].
Алюминий и алюминиевые сплавы. Кремний является составной частью алюминиевых сплавов многих марок. В алюминиевых сплавах он находится в виде химического соединения, твердого раствора или в свободном состоянии. Разложение сплава обычно проводят кислотами или щелочным способом.
При растворении сплава в хлористоводородной кислоте свободный кремний не растворяется и может быть отделен фильтрованием.
136
Однако при этом осадок кремневой кислоты адсорбирует на своей поверхности значительное количество других элементов. В случае применения для разложения серной, азотной и фосфорной кислот сплав разлагается полностью и получается абсолютно прозрачный раствор.
Алюминий и его сплавы легко разлагаются едким натром. Приводим методику определения кремния в металлическом алюминии.
Навеску 0,5 г металлического алюминия помещают в никелевый тигель и обрабатывают 5 мл раствора NaOH. Охлаждают, прибавляют 5 мл 3%-ной Н2О2, кипятят 10 мин, разбавляют до 50 мл и фильтруют, собирая фильтрат в склянку, содержащую 15 мл 9N H2SO4. Остаток промывают.
Фильтрат переносят в мерную колбу и разбавляют водой до 500 мл. Отобрав аликвотную порцию (50 мл), помещают ее в мерную колбу емкостью 100 мл, прибавляют 5 мл раствора молибдата, перемешивают и оставляют на 5 мин. Затем приливают 15 мл 9N H2SO4, 10 мл 4%-ного раствора щавелевой кислоты, 10 мл 2,5%-ного раствора соли Мора в 0,4М H2SO4 и разбавляют водой до 100 мл. Определяют оптическую плотность полученного раствора при 820 нм. Окраска устойчива в течение 1 часа. Проводят холостой опыт.
Магний и его сплавы. При разложении магниевых сплавов следует учитывать, что в случае растворения сплава в кислотах магний может образовывать с кремнием легколетучие соединения; это приводит к потерям последнего. Во избежание таких потерь проводят разложение в присутствии окислителей — персульфата аммония, азотной кислоты и т. д.
В зависимости от содержания кремния применяют фотометрический или весовой методы. Для сплавов, содержащих сотые и тысячные доли процента кремния, рекомендуется применять фотометрический метод. Анализ сплавов, содержащих большие количества кремния (более 1,5%), следует проводить весовым путем.
Приводим методику фотометрического определения кремния.
Навеску сплава в 1 г растворяют в 5 мл персульфата аммония и 20—35 мл 5N HNO3 в стакане емкостью 300 мл, накрытом часовым стеклом, на холоду. Раствор переводят в мерную колбу емкостью 100 мл, приливают 10 мл раствора молибденовокислого аммония, перемешивают и оставляют стоять на 30 мин. Через 30 мин вводят 10 мл 8N H2SO4, перемешивают, доводят водой до метки и переносят раствор в делительную воронку емкостью 200 мл, прибавляют 5 мл бутанола, взбалтывают, затем вновь вводят 15 мл бутанола и вновь перемешивают. Дают жидкости расслоиться, затем водный раствор смывают в другую воронку, а окрашенный раствор кремнемолибденового комплекса — в градуировочный цилиндр с притертой пробкой.
Экстракцию бутанолом повторяют до получения прозрачного экстракта. Все окрашенные экстракты собирают в один цилиндр. В другой градуировочный цилиндр вводят бутанол в объеме, равном объему испытываемого раствора, и из микробюретки стандартный раствор А или Б (в зависимости от содержания кремния) до одинакового оттенка окраски в обоих цилиндрах.
Содержание кремния в сплаве определяют по формуле
Si, %=a7’-100/g,
где a — количество раствора Б, затраченного на титрование, мл\ Т — титр стандартного раствора кремния Б, выраженного в граммах кремния; g — навеска, г.
Об определении кремния в металлах и сплавах см. также в 12, 36, 55, 56, 64, 410, 419, 473, 565, 693, 977, 9911.
137
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В ПРИРОДНЫХ ВОДАХ И КОНДЕНСАТЕ
Кремний в природных водах находится обычно в виде коллоидальной кремневой кислоты, в виде различных форм гидратированного кремнезема, и только в щелочных водах одновременно с коллоидальным кремнеземом содержатся также ионы HSiO^ и SiO^~. Наряду с гидратами кремнезема в водах иногда содержатся также и силикаты в коллоидальном состоянии.
Вопрос о соотношении различных форм кремнезема изучен очень слабо. Это соотношение зависит главным образом от pH среды. Например, в минеральных водах с рН<0,5 кремневая кислота содержится в недиссоциированном состоянии в виде H2SiO3; с повышением pH увеличивается содержание в воде ионов HSiO^- и SiO|~. Обычно содержание кремневой кислоты в природных водах незначительно и не превышает 25—50 мг!л [57, 90, 322, 463]. Лишь в водах термальных источников содержание кремневой кислоты достигает 150—200 мг!л. В таких случаях кремний определяют весовым методом в виде двуокиси кремния [6, 451].
В фарфоровой чашке на водяной бане почти досуха выпаривают 250—300 мл исследуемой воды. Затем чашку накрывают часовым стеклом и через ее носик осторожно вводят с помощью пипетки раствор НС1 (1 : 1) до прекращения выделения пузырьков СО2, обмывают внутреннюю сторону стекла водой, снова выпаривают раствор на водяной бане досуха и оставляют стоять остаток на кипящей водяной бане в течение 2 час.
Высушенный остаток обрабатывают 10 мл НС1 (1 : 1), накрывают часовым стеклом и оставляют на 3—5 мин, после чего, прибавив 100 мл дистиллированной воды, фильтруют осадок через небольшой беззольный фильтр. Осадок сначала промывают 1%-ной НС1, затем горячей водой до отрицательной реакции на хлорид-ионы.
Фильтр с осадком переносят во взвешенный платиновый тигель, озоляют, прокаливают при 800° С и взвешивают. Затем проверяют кремневую кислоту на чистоту, для чего к осадку добавляют 0,1—0,2 мл H2SO4 (1 : 1) и 1 мл 40%-ной HF, содержимое тигля выпаривают досуха, прокаливают и взвешивают тигель. Разность в весе — содержание двуокиси кремния в пробе.
Однако для массовых определений использование весового метода нецелесообразно. При содержании кремневой кислоты порядка 1—50 мг/л определение кремния проводят по желтому кремнемолибденовому комплексу. Некоторые авторы определяют кремний визуальным методом [6, 451], способом уравнивания или способом эталонных растворов. Эти методы обычно используют для массовых анализов в гидрохимических исследованиях либо по желтому кремнемолибденовому комплексу (менее чувствительный метод), либо по предложенному позднее синему восстановленному комплексу (более чувствительный метод).
Для ускорения процесса анализа визуальное колориметрирование по первому способу производилось не с натуральными стандартами, а с имитирующими цветными растворами пикриновой кислоты и хромовокислого калия. Однако в настоящее время ре-
138
комендуется сравнивать окраски, пользуясь только натуральными стандартами Na2SiO3 или Na2SiF6, основные растворы которых стабильны в течение по крайней мере шести месяцев.
Приводим методику определения кремния в морских водах с использованием в качестве стандартного раствора силиката натрия. При содержании кремния в воде >0,2 мг!л определение проводят по желтому комплексу.
В цилиндры Несслера емкостью по 50 мл вливают пробы испытуемой воды. Затем последовательно во все пробы прибавляют по 1 мл подкисленного 10% -ного раствора, молибдата аммония, который готовят смешением 9 мл 11%-ного раствора молибдата аммония с 1 мл 50%-ной (по объему) H2SO4. Пробы закрывают стеклянными пробками и быстро перемешивают.
Через 20 мин сравнивают окраски в цилиндрах Гейера или на фотоколориметре с эталонными растворами силиката натрия. Последние готовят разведением в цилиндрах Несслера 0,25; 0,50; 1,00; 2,00; 4,00 мл рабочего стандарта.
Для приготовления стандартного раствора в платиновом тигле осторожно сплавляют 1,07 г тонконзмельченного спектрально чистого горного хрусталя с 6 г химически чистой соды, плав выщелачивают в мерную колбу емкостью 1 л. Приготовленный таким образом раствор разбавляют еще в 10 раз дистиллированной водой; 1 мл полученного раствора содержит 50 мкг кремния. Цилиндры доливают до метки (50 мл) «бескремневой» морской или дистиллированной водой при работе с пресными водами. «Бескремневая» морская вода готовится из поверхностной морской воды, медленно профильтрованной через слой прокаленной окиси алюминия. Содержание кремния в «бескремневой» морской воде определяют методом добавок. Если стандарты приготовлены на морской воде, то вводить поправки на содержание солен не нужно. Для устранения мешающего влияния фосфат-ионов обычно используют неустойчивость фосфорномолибденового комплекса при pH 1,6—2,0.
Определению кремния в водах кроме Р2О6 мешают ионы NH+, F- (дают мутный раствор), С2С)2_, а также Н2О2, As2O3, C4H4O6-As2O3 предлагают экстрагировать смесью бутанола и этилацетата. Отрицательное влияние F"-ионов устраняют прибавлением 20-кратного избытка борной кислоты. При определении кремния в окрашенных водах последние необходимо предварительно обесцветить коллоидальным осадком фосфата кальция следующим образом.
К 100 мл испытуемой воды, налитой в мерную колбу емкостью 200 мл, прибавляют 1 мл 2,5%-ного раствора Na2HPO4, 1 мл 10%-ного раствора СаС12 и 1 мл 2,5%-ного раствора NH4OH. Раствор доводят до 200 мл дистиллированной водой и после перемешивания оставляют на 20 мин.
В 100 мл фильтрата, отвечающего 50 мл испытуемой воды, определяют кремневую кислоту. Если этим методом полного обесцвечивания достичь не удается, то дополнительно обрабатывают окислителем, для чего 100 мл фильтрата нагревают до кипения, прибавляют несколько миллилитров НС1 (1 : 1) и немного персульфата аммония. Раствор кипятят до обесцвечивания и, если нужно, прибавляют еще персульфат аммония. После охлаждения в нем определяют содержание кремния по желтому или синему кремнемолибденовому комплексу. При содержании кремния в анализируемых водах <1мг/л определение проводят по синему комплексу.
Носкова и Пика [9421 опреде ляли кремний в питьевой воде по синему кремнемолибденовому комплексу с использованием в качестве восстановителя соли Мора. При определении 0,1—0,8 мг/л
139
SiO2 получаются хорошо воспроизводимые и достаточно точные результаты; при определении 0,01 мг/л SiO2 ошибка ±30%. Авторы указывают, что фосфаты в количестве 2,7—6,8 мг/л Р2О5 определению кремния не мешают. Моррисон и Уилсон [927] при определении кремния в воде определяли оптическую плотность а- и 0-кремнемо-либденовой кислоты при 742 и 810 нм. Полученные авторами результаты свидетельствуют о возможности исключения различных источников ошибок путем подбора условий анализа. Для гх-кремнемолибденовой кислоты наиболее подходящим восстановителем является SnCl2, для 0-кремнемолибденовой кислоты — 1-амино-2-нафтол-4-сульфокислота.
а-Кремнемолибденовая кислота нечувствительна к высоким концентрациям солей, но она неустойчива во времени и обладает высокой чувствительностью к фосфат- и хлорид-ионам.
0-Кремнемолибденовая кислота более чувствительна к высоким содержаниям солей, однако она малочувствительна (в присутствии винной или щавелевой кислот) к фосфат-ионам. Приводим методику определения кремния в воде по синему комплексу.
К 50 мл анализируемой воды в мерной колбе емкостью 100 мл добавляют 1 мл НС1 (1 : 1) и 2 мл 10%-ного раствора молибдата аммония. Раствор в колбе перемешивают после добавления каждого реагента. Через 5 мин добавляют еще 1,5 мл 10%-ного раствора щавелевой кислоты, 2 мл восстановителя, перемешивают и разбавляют дистиллированной водой до метки.
Восстановитель готовят растворением 15 г бисульфита натрия, 0,5 г сульфита натрия и 0,25 г 1-амино-2-нафтол-4-сульфокислоты в 100 мл воды. Через 1 мин определяют оптическую плотность раствора примерно при 815 нм. Содержание кремния находят по калибровочному графику. Вводят поправку на холостой опыт.
При определении микроколичеств кремния в высокочистой воде его предварительно концентрируют на анионите или определяют после экстракции кремнемолибденового восстановленного комплекса. Приводим методику определения кремния в конденсате пара с использованием экстракции кремнемолибденовой кислоты [179].
100 мл исследуемого раствора помещают в полиэтиленовую колбу и добавляют 2,5 мл 10%-ного молибдата аммония в 5%-ной H2SO4. Через 10 мин добавляют 2,5 мл 28%-ного раствора винной кислоты и еще через 5 мин 2 мл восстановителя. Восстановитель готовят растворением в 100 мл воды 0,2 г 1-амино-2-наф-тол-4-сульфокислоты, 2,4 г сульфита натрия и 14 г метабисульфита калия. Переносят раствор в делительную воронку, добавляют 15 мл конц. H2SO4 и 25 мл изоамилового спирта. Органическую фазу отделяют и переносят в колбу емкостью 25 мл, разбавляют до метки органическим растворителем и измеряют оптическую плотность при 800 нм. Содержание кремния находят по калибровочному графику.
При определении кремния в морской воде было установлено, что соли изменяют интенсивность окраски кремнемолибденовой кислоты [1008]. Зависимость между оптической плотностью и содержанием солей в растворе установлена Бином [666].
Для исключения ошибки при определении кремния в морской воде ряд авторов рекомендует готовить растворы для построения калибровочных графиков на морской воде, содержащей минималь
14П
ное количество кремния и обладающей той же соленостью, что и анализируемая вода [89].
Разработана методика спектрального определения очень малых количеств кремния в природных и промышленных водах в дуге переменного тока [218]. Количественный анализ проводится по методу «трех эталонов». Градуировочные графики, по которым велось определение кремния, строились для каждой пластинки по трем эталонам в координатах s — 1g с (с — концентрация кремнезема в растворе). Каждая аналитическая линия использовалась только в области нормальных почернений, при этом графики получались прямолинейными.
Почернения измеряли на микрофотометре МФ-2. Линия Si 2516,123 А применялась при анализе в интервале концентраций кремнезема 7-10“ 7 4-1 • 10“ 5 г/мл. Линия 2519,207 А—при концентрациях 5-10“ 5 4-3-10“4 г/мл SiO2. Кроме того, с успехом использовались линии: 2881,576° А (интервал концентраций 1,5-10“ 6 4-1 • 10“ 5 г/мл SiO2) и 2506,899 А (3-10"64-2-10“5 г/мл SiO2). Определение концентраций меньших, чем 7-Ю-7 г/мл, велось по линиям кремния 2516,123 и 2881,578 А. Абсолютная чувствительность количественных определений составляет 4-10~8 г кремния, нанесенного на электроды. Стандартная ошибка±Н%.
ОПРЕДЕЛЕНИЕ КРЕМНИЯ
В КРЕМНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Большинство существующих в настоящее время методов определения кремния в кремнийорганических соединениях предусматривает предварительную полную минерализацию анализируемого соединения. Минерализация вещества с целью перевода атома кремния из его элементоорганической связи в ионное состояние достигается различными методами, становясь тем более простой, чем более полярной является связь. Для кремнийорганических соединений минерализация вещества требует химической обработки в жестких условиях. Только в случае соединений, в которых кремний связан с углеродом посредством других атомов, кремний переводится в соединение с полностью ионогенными связями при действии мягких агентов — воды, водных растворов щелочей и кислот. Однако следует отметить, что эти соединения относятся к числу кремнийорганических соединений условно.
Методы разложения кремнийорганических соединений могут быть основаны на различных принципах: гидролитическом расщеплении, мокром окислении, разложении сухим способом и сожжении.
Методы определения кремния, основанные на гидролитическом расщеплении кремнийорганических соединений. Многие кремнийор-ганические соединения подвергаются гидролитическому расщепле
141
нию с количественным образованием кремневой кислоты [461, 7691, например, по следующим реакциям:
S iC 14 + ЗН2О -> SiO2 • Н2О + 4НС1,
CISi (ОСН3)3 + 4Н2О SiO2 • 2Н2О + ЗСН3ОН + НС1,
FSi (NCO)3 + 4Н2О SiO2 • 2Н2О + HF + 3HCNO,
Cl 2SiS + 3H2O SiO2 • H2O + 2HC1 + H2S,
Si (OC2H5)4 + 2H2O-> SiO2 + 4C2H5OH,
Si (SCN)4 +2H2O->SiO2 + 4HSCN и т. д.
Скорость гидролитического растепления зависит от многих факторов: строения исходных продуктов, температуры, pH среды и т. д. В щелочных и кислых средах скорость гидролиза увеличивается, поэтому в аналитической практике редко пользуются для гидролиза водой, а применяют небольшие количества кислоты или щелочи [296, 297]. Для разложения соединений, которые реагируют с водой, образуя кислоту, например четыреххлористого кремния или трихлорсилана, добавлять дополнительно кислоту или щелочь не требуется. Скорость гидролиза в данном случае возрастает по мере накопления хлористого водорода, действующего катализирующим образом.
Соединения, содержащие в своем составе связи Si—Si, Si—N, Si — H, разлагаются в процессе гидролиза с выделением водорода
^Si-Si^ + H2O^ = Si-O-Si = + H2.
Количество водорода, выделяющегося в результате этой реакции, эквивалентно содержанию кремния. Для определения кремния в этом случае может быть измерено количество водорода, выделившегося при реакции [1003].
Кремний определяют также весовым методом в виде двуокиси после соответствующей обработки щелочных растворов хлористоводородной, азотной или другими кислотами.
Для анализа соединений, которые очень бурно реагируют с водой, во избежание потерь вещества от разбрызгивания разработаны специальные методы проведения гидролиза [27, 641]. Например, навеску вещества вводят в абсолютный спирт, в котором оно подвергается этерификации, а затем уже получают продукты, гидролизующиеся спокойно. Установлено, что при анализе изоцианат-силанов этот метод дает лучшие результаты, чем при гидролизе водой [641].
Некоторые авторы [296, 297] при анализе эфиров ортокремневой кислоты, трихлорсилана и других соединений, имеющих связи Si — Н, Si — N, Si — Cl, Si — OR и т. д., использовали для гидролиза водные или спиртовые растворы аммиака. Разложение эфи
142
ров растворами аммиака происходит быстрее, чем кислотами, так как не получается геля кремневой кислоты, который затрудняет диффузию гидролизующей среды в глубокие слои еще не разложенного эфира. В случае гидролиза в аммиачных растворах образуются растворимые соли аммония [769]. Однако недостатком этого метода является сильное разбрызгивание частиц геля в конце выпаривания.
Позднее было установлено, что гидролиз в аммиачных растворах протекает быстро лишь при анализе тетраметоксисилана и метоксиполисилоксанов [292]. С увеличением же длины углеводородного радикала гидролиз в аммиачной среде сильно замедляется. В этом случае анализ целесообразнее проводить в сильнокислых средах. Фридель и Ладенбург [769] при определении кремния в кремнийорганических соединениях проводили гидролиз разбавленным раствором аммиака в толстостенных стеклянных ампулах. По окончании гидролитического расщепления содержимое трубки количественно переносили в платиновую чашку и выпаривали на водяной бане досуха. Промытый, отфильтрованный и взвешенный осадок прокаливали до постоянного веса. В случае частичного образования карбида кремния последний сплавляли с карбонатом натрия, плав разлагали водой и после обработки плава хлористоводородной кислотой и хлоридом аммония получали чистую кремневую кислоту.
При определении кремния в этоксифторсилане, четыреххлористом кремнии, феноксихлорсилане и других соединениях [296, 297] для гидролиза применяли водный раствор едкого кали.
Анализ алкоксифторсиланов трудоемок и длителен. К щелочным продуктам гидролиза прибавляют карбонат аммония. Выпавший через 24 часа осадок фильтруют, промывают и сушат. Фильтрат упаривают на водяной бане для разложения избытка карбоната аммония. Далее к фильтрату добавляют раствор аммиака и окись цинка и после упаривания до исчезновения запаха аммиака. Осадок обрабатывают хлористоводородной кислотой, фильтрат снова выпаривают досуха и выдерживают несколько часов при 150° С. Выпаривание и сушку повторяют еще два раза. Полученный осадок SiO2 прибавляют к осадку двуокиси кремния, полученному ранее.
Андреев при определении кремния в некоторых сравнительно легколетучих кремнийорганических соединениях со связью Si—С использовал свойство последних давать при гидролизе нелетучие полисилоксаны сетчатой структуры [27]. Гидролиз проводят под слоем 95—96%-ного спирта. В некоторых случаях к спирту для ускорения гидролиза добавляют несколько миллилитров 25%-ного аммиака. Затем содержимое тигля упаривают и прокаливают до постоянного веса. Метод пригоден для анализа веществ с небольшим содержанием углерода типа Cl 3SiCH2SiCl 3 или полимеров, полученных на основе метилтрихлорсилана. В случае анализа веществ с большим содержанием углерода возможно образование карбида кремния. Для предотвращения этого в случае образования темного осадка последний обрабатывают смесью азотной и серной кислот.
143
Глудина при анализе алкоксисиланов после их гидролитического расщепления в уксусной кислоте определяла в них кремний фото-колориметрическим методом [153].
Кальман и Ваго предложили определять кремний объемным методом титрованием кремнефтористоводородной кислоты, образующейся при гидролитическом расщеплении тетраэтоксисилана и этоксиполисилоксанов фтористоводородной кислотой, раствором едкого натра [852].
В полиэтиленовый стакан емкостью 150 мл с крышкой помещают около 50 мл 50%-ного этанола и добавляют 1—2 мл 40%-ной HF, свободной от кремнефтористоводородной кислоты. Затем из капельницы вводят 0,1—0,2 г анализируемого вещества, стакан закрывают крышкой и содержимое стакана перемешивают, слегка покачивая его в течение 3—5 мин. К полученной таким образом смеси кремнефтористоводородной и фтористоводородной кислот прибавляют из бюретки 10%-ный раствор КОН до появления малинового окрашивания по фенолфталеину и затем нейтрализуют 0,2М H2SO4 или НС1. В процессе этой операции раствор должен содержать не менее 50% спирта, поэтому в случае необходимости добавляют нужное количество спирта.
Раствор переносят в коническую колбу емкостью 1000 мл, разбавляют до 500—700 мл горячей дистиллированной водой и титруют 0,2М NaOH до неисчезающего розового окрашивания по фенолфталеину. Длительность определения 15—20 мин. Ошибка не превышает 0,2%.
Методы определения кремния, основанные на гидролитическом расщеплении, просты и удобны. Однако применимы они для определения кремния лишь в сравнительно небольшой группе кремнийорганических соединений и почти неприменимы для соединений, содержащих связь Si — С. В этом случае проведение анализа предусматривает использование других методов, описанных ниже.
Методы определения кремния, основанные на мокром окислении.
Одним из самых простых способов количественного определения кремния в кремнийорганических соединениях является окисление последних концентрированной серной кислотой [176, 296, 297, 865]. Навеску анализируемого вещества в тигле или кварцевой колбе обрабатывают серной кислотой, которую затем медленно испаряют, а тигель или кварцевую колбу прокаливают до постоянного веса. Однако эти методы не могут быть применены для анализа легколетучих веществ, которые улетучиваются с парами SO3 [865]. Кроме того, при анализе многих веществ, содержащих ароматические радикалы, результаты определения кремния получаются заниженные вследствие неполного окисления и неизбежного образования карбида кремния.
Описан метод определения кремния в нелетучих кремнийорганических соединениях при разложении веществ 98%-ной серной кислотой [296]. Для анализа легколетучих веществ авторы навеску брали в тигель, уже содержащий несколько миллилитров серной кислоты, в результате чего в первый момент происходил гидролиз с образованием нелетучих кремнийорганических продуктов, а потом уже — окисление.
Некоторые соединения, например тетрафенилсилан и тетра-этилсилан [865], не окисляются серной кислотой до SiO2, а ди-
144
фенилдиэтилсилан не окисляется до двуокиси кремния даже смесью серной и азотной кислот. В некоторых случаях к серной кислоте прибавляют металлическую ртуть, катализирующую процесс окисления [687]. Бажант, Хваловски и Ратоуски [52] в качестве катализатора при окислении кремнийорганических соединений серной кислотой использовали СпО.
Полис получил хорошие результаты при разложении кремнийорганических соединений смесью серной кислоты и перманганата калия [962]. В ходе окисления наряду с кремневой кислотой образуется двуокись марганца, которую растворяют в хлористоводородной кислоте и затем отделяют от двуокиси кремния фильтрованием.
Киппинг [864, 865] предложил вместо перманганата калия использовать концентрированную азотную кислоту. Это сокращает время анализа и повышает точность, так как исключается операция фильтрования и промывания. Азотную кислоту использовали также для окисления тетраметилсилана [768] и эфиров ортокремневой кислоты [742]. При анализе большой группы кремнийорганических соединений применяют в качестве окислительной смеси серную и азотную кислоту [141, 296, 297].
В том случае, если после испарения кислот и прокаливания осадок имеет серый цвет, содержание кремния находят после обработки осадка смесью серной и фтористоводородной кислот по разности в весе, обусловленной образованием летучего тетрафторида кремния.
Андрианов при определении кремния в алкилтриэтоксисиланах использовал концентрированную азотную кислоту [28]. Фалькен-бург, Тристер и Кован [742] применяли для окисления эфиров ортокремневой кислоты и высших жирных кислот, имеющих в цепи 18 атомов углерода, разбавленную азотную кислоту. Крешков, Сявцилло и Шемятенкова [309] разлагали как нелетучие кремнийор-ганические соединения, так и легколетучие галогенсиланы смесью олеума и дымящей азотной кислоты. Авторы перед окислением га-логенсиланов переводили последние в ацетоксипроизводные (ал-килтрихлорсиланы, алкилдихлорсиланы и четыреххлористый кремний) или сульфопроизводные (диалкилдихлорсиланы).
Навеску 0,5 г исследуемого вещества помещают в предварительно прокаленную до постоянного веса при 800° С кварцевую колбу емкостью 75—100 мл. Затем в колбу вводят 1,5 мл 25%-ного олеума и 2—3 капли конц. HNO3, содержащей 20% окислов азота. Содержимое колбы осторожно нагревают на электроплитке в вытяжном шкафу. Добавление HNO3 по каплям повторяют несколько раз до тех пор, пока органическая часть навески не окислится.
После этого прибавляют еще 2—3 капли HNO3 и содержимое колбы упаривают, усиливая нагревание, для удаления избытка кислот. При этом следят, чтобы не было сильного вспенивания. После того как выделение паров закончено, колбу помещают в муфель и прокаливают при 800° С до постоянного веса. Метод характеризуется высокой точностью, но очень длителен.
Смесь алкилхлорсиланов анализируют по методу, который используется для определения компонента, содержащегося в исследуемой смеси в преобладающем количестве. При этом впоследствии было установлено [304], что при анализе этилдихлорсилана, метил
145
и этилтрихлорсилана, примесь диэтил- и диметилхлорсилана до 25% существенно не влияет на точность результатов. Этим же методом кремний был определен в гексаалкилдисиланах [143], алкилметоксисиланах [144], алкилхлорсиланах и ацетоксисиланах [177].
При определении кремния в ариламиносиланах перед окислением серной кислотой их растворяют в ледяной уксусной кислоте.
Воронков и Романов [142] при определении кремния в алкил-трихлорсиланах навеску вещества предварительно гидролизовали под слоем 90%-ного спирта и добавляли разбавленную хлористоводородную кислоту. После испарения содержимого тигля почти досуха окисление дальше проводили, как обычно, смесью олеума и азотной кислоты. Этот метод был использован для определения кремния в фенилцианатсиланах [641].
Гурецкий [168] определял кремний в продуктах конденсации эфиров ортокремневой кислоты с содержанием кремния (в пересчете на5Ю2)до 52%. Навеску вещества вначале гидролизовали 5%-ной серной кислотой, а затем окисляли вещество концентрированной азотной кислотой. Определение кремния заканчивали весовым методом. Точность определения очень высока, однако возможны потери вследствие разбрызгивания в конце выпаривания.
Интересный метод определения кремния в летучих кремнийорганических соединениях предложил Смит [1020].
Согласно Смиту, навеску вещества берут в ампуле, конец которой запаивают. Затем конец ампулы быстро обламывают и погружают в высокий тигель емкостью 10 мл, содержащий смесь 60%-ного олеума и дымящей HNO3. Этот тигель погружают в другой тигель емкостью 50 мл с ледяной водой. Широкий конец ампулы осторожно нагревают, окисляя содержимое тигля, медленно вытекающее под слой олеума и дымящей азотной кислоты.
Однако метод неприменим для анализа легколетучих галоген-силанов и большой группы кремнийорганических соединений, которые осмоляются в процессе окисления (например, содержащих фур-фурокси-группы) [304].
Быстро и точно определяется кремний в различных типах нелетучих кремнийорганических соединений путем их окисления 60%-ной хлорной кислотой. В этом случае окисление протекает вначале не очень бурно, но с повышением температуры повышается окислительный потенциал кислоты, в результате чего окислительный процесс ускоряется. В некоторых случаях в качестве окислителя используют смеси хлорной с азотной и серной кислотами [669]. Недостатком использования НС1О4 является опасность воспламенения и взрыва.
Гурецкий [168] описал полумикрометод определения кремния в различных типах кремнийорганических соединений путем их окисления бромом в азотнокислой среде.
После разложения кремнийорганических соединений мокрым окислением кремний определяют, как правило, весовым методом. Однако в работах последних лет замечается тенденция к использованию объемных методов.
146
Шир и Комере [1018] после окисления кремнийорганических соединений смесью олеума и азотной кислоты заканчивали определения кремния объемным алкалиметрическим методом. Объемные методы обладают достаточной точностью, а по скорости определения значительно превышают весовые методы.
Методы определения кремния, основанные на разложении кремнийорганических соединений сухим способом. Методы определения кремния в кремнийорганических соединениях с использованием окисления в процессе разложения веществ перекисью натрия, нитратами, смесью перекиси натрия с карбонатами и другими смесями [296, 297] применяют для анализа самых различных кремнийорганических соединений как твердых, так и жидких низко-кипящих. Впервые метод разложения сухим способом с перекисью натрия предложил Парр (см. библиографию в 297). Хард, Сервайс и Кларк [907] окисляли кремнийорганические соединения в никелевых бомбах смесью перекиси натрия с сахаром.
Навеску 50—200 мг анализируемого кремнийорганического соединения сплавляют с 2—3 г Na2O2 и 0,05—0,1 г сахара в никелевой бомбе Парра. Плав растворяют в воде, слегка подкисляют НО (проба на лакмус) и полученный раствор разбавляют водой до 1 л в мерной колбе.
Аликвотную часть (50 мл) раствора переносят в колбу емкостью 250 мл, добавляют 15 мл НС1 (1 : 1), 50 мл дистиллированной воды и 15 мл 20%-кого раствора молибдата аммония. Затем колбу закрывают, содержимое нагревают при 75° С в течение 10 мин и после охлаждения до комнатной температуры прибавляют 20 мл НС1 (1 : 1).
Далее в колбу из бюретки прибавляют 25 мл 0,4А раствора оксихинолина, одновременно встряхивая содержимое колбы. При этом выпадает осадок оксин-кремнемолибденового комплекса.
Затем колбу с содержимым нагревают при 65° С в течение 10 мин. Охлажденный до комнатной температуры раствор с осадком фильтруют. Осадок на фильтре промывают раствором оксихинолина в НС1. Отмытый осадок сушат при НО— 120° С в течение 1 часа, после этого сжигают и прокаливают при 500° С до постоянного веса.
Весовая форма SiO2-12MoO3 [135].
Метод определения кремния, основанный на сплавлении в металлических бомбах, дает хорошие результаты при анализе кремнийорганических соединений различных типов. Окисление веществ в бомбе происходит быстро. Однако этот метод имеет некоторые недостатки, а именно — неизбежное разрушение материала бомбы и загрязнение вследствие этого анализируемых продуктов. Это значительно снижает его ценность.
Гурецкий предложил более простую технику окисления кремнийорганических соединений сплавлением их со смесью перекиси натрия, нитрата калия и карбоната калия, взятых в отношении 3 : 3 : 10 [168].
Навеску анализируемого соединения около 0,2 г берут в никелевый тигель, уже содержащий слой окислительной смеси высотой около 5 мл, и засыпают той же окислительной смесью до краев тигля. Тигель с шихтой покрывают другим тиглем большего размера и оба тигля переворачивают. Зазор между тиглями засыпают окислительной смесью слоем толщиной 15—20 мм. Нагревают дно тигля до расплавления смеси. После застывания тиглей плав выщелачивают и в фильтрате определяют кремний любым из ранее описанных методов.
147
Предложен метод определения кремния в кремнийорганических соединениях, содержащих фтор [822]. После разложения навески перекисью натрия с этиленгликолем и растворения плава SiO^-осаждают аммиачным раствором окиси цинка в виде ZnSiO3, который отфильтровывают, озоляют и определяют кремний весовым методом после обработки смесью азотной и хлористоводородной кислот. Метод кропотлив и длителен.
Метод сплавления с перекисью натрия и ее смесями с другими веществами обладает одним существенным недостатком: вводится огромное количество окислителя по сравнению с навеской вещества, что приводит к значительным ошибкам вследствие загрязнения анализируемого вещества посторонними ионами. В связи с этим в последнее время получили распространение методы определения кремния в кремнийорганических соединениях, основанные на разложении последних металлическим натрием или калием при 800—1000° С. Марклоу разлагал кремнийоргацические соединения, содержащие фтор, сплавлением их с металлическим натрием в никелевых бомбах и затем определял кремний фотоколоримет-рическим методом [903].
Коршун и сотр. разработали метод определения кремния в кремнийорганических соединениях, основанный на их разложении металлическим калием при 800—850° С в стальных бомбах [258, 284]. Метод позволяет наряду с кремнием определять и другие элементы. Бондаревская, Сявцилло и Кузнецова [296] определяли кремний в большой группе кремнийорганических соединений разложением их металлическим калием в стальных бомбах, предварительно продутых кислородом при температурах порядка 900—1000° С. Время разложения колеблется от 40 до 60 мин, возрастая с увеличением количества фенильных радикалов у кремния. После выщелачивания плава кремний определяли алкалиметрическим методом.
Методы определения кремния, основанные на сожжении. При разработке методов определения кремния в кремнийорганических соединениях, основанных на сожжении последних, вначале были предприняты попытки перенести без изменений на кремнийоргани-ческие соединения обычные методы органического анализа [962, 964]. Однако они были неудачными: в процессе сожжения кремнийорганических соединений обычно образуется карбид кремния, что приводит к заниженным данным при определении кремния.
В 1941 г. Рохов описал микрометод каталитического сожжения кремнийорганических соединений в токе кислорода для одновременного определения в них углерода, водорода и кремния [13, 41, 979]. Позднее он дал полумикромодификацию своего метода. В предлагаемом методе анализируемое вещество вначале медленно испаряется при 300—500° С, затем после смешения с кислородом поступает на платиновую сетку, нагретую до 850° С, где и окисляется. Двуокись кремния улавливают в трубке при помощи стеклянной ваты и о содержании кремния судят по привесу трубки.
Метод Рохова может быть применен для определения кремния в кремнийорганических соединениях полимеров различных типов, однако каждый тип полимера должен быть сожжен при определенной температуре. Превышение оптимальной температуры, как правило, приводит к воспламенению вещества и образованию карбида кремния, вследствие чего результаты определения кремния получаются заниженными. Поддержание определенной температуры по всей длине трубки затруднительно. Образующаяся при сожжении двуокись кремния, оседая на поверхности платины, снижает ее активность. Естественно, что к ошибкам приводит и сам процесс взвешивания громоздкой трубки длиной 18 см с очень небольшим количеством образовавшейся двуокиси кремния. Однако несмотря на все указанные недостатки, при осторожном сожжении и медленном поднятии температуры авторы получили вполне удовлетворительные результаты как при одновременном определении водорода, углерода и кремния, так и при одновременном определении углерода, водорода, кремния и серы [256, 257].
При анализе кремнийорганических соединений, содержащих серу [257], результаты определения кремния этим методом получались заниженными вследствие образования карбида кремния. Во избежание этого авторы проводили анализ в присутствии катализатора — окиси меди, нанесенной на асбест.
Метод Рохова применен для одновременного определения кремния, водорода, углерода, галогенов и кислорода (по разности) в летучих и газообразных кремнийорганических соединениях [296, 297]. Принцип метода Рохова был использован также при определении кремния в летучих кремнийорганических соединениях 1681].
Был разработан новый ускоренный метод элементного микроанализа [258, 259, 285], основанный на быстром пиролитическом разложении исследуемых кремнийорганических соединений почти без доступа кислорода при 850—900° С. Продукты разложения смешиваются с большим избытком кислорода и окисляются им при 900—950° С (скорость тока кислорода 30—50 мл/мин). При анализе кремнийорганических соединений некоторых классов авторам не удалось исключить образование карбида кремния и обеспечить полноту пиролиза, поэтому сожжение следует производить в присутствии катализатора — окиси хрома на асбесте, — помещаемого непосредственно в стаканчик для разложения. В дальнейшем разработанный метод авторы применили для анализа кремнийорганических соединений различных классов, использовав его также для определения наряду с кремнием фосфора, серы и др. [258, 259, 284].
По мнению многих исследователей, методы сухого сожжения трудоемки и редко дают хорошие результаты [978].
149
ОПРЕДЕЛЕНИЕ КРЕМНИЯ В ЭЛЕМЕНТОКРЕМНИЙОРГАНИЧЕСКИХ СОЕДИНЕНИЯХ
Для определения кремния в элементокремнийорганических соединениях применяют те же методы, что и для анализа кремнийорганических соединений. Однако одновременное присутствие ионов металлов (наряду с кремнием) определенным образом отражается на минерализации элементоорганических соединений. Методы минерализации кремнийорганических соединений, основанные на гидролитическом расщеплении, для определения кремния в элементокремнийорганических соединениях, содержащих Si—С-свя-зи, не применяются. Методы, основанные на сожжении, также почти не применяются, поскольку в процессе сожжения наряду с двуокисью кремния образуется окисел соответствующего металла. Таким образом, возможно лишь определение суммы окислов.
Для разложения элементокремнийорганических соединений чаще всего используют методы, основанные на мокром окислении, реже — на сплавлении. Для минерализации элементокремнийорганических соединений обычно используют те же окислители и окислительные смеси, что и для минерализации кремнийорганических соединений, например смесь концентрированной серной кислоты с персульфатом калия или аммония, йодатом калия, дымящей азотной кислотой, хромовой кислотой, олеум. Персульфат аммония с концентрированной серной кислотой является наиболее удобным окислителем, так как методе его использованием отличается быстротой выполнения и никаких осложнений при последующем определении кремния не возникает. После минерализации кремний определяют гравиметрическим, титриметрическим или фотометрическим методами. Минерализацию оловокремнийорганических и титан-кремнийорганических соединений проводят также смесью концентрированной серной кислоты с йодатом калия. Определение кремния заканчивали гравиметрическим методом в форме SiO2 [296].
В ряде работ после минерализации элементокремнийорганических соединений методами мокрого окисления определение кремния заканчивали титриметрическим методом. Лускина, Терентьева и Градскова [5281, разложив алюминийфосфоркремнийорганические соединения смесью персульфата калия и концентрированной серной кислоты и выделив кремний в виде кремнефторида калия, заканчивали определение алкалиметрическим методом. Аналогичным образом определяли кремний в полиалюмоорганосилоксанах [5281 и титанкремнийорганических соединениях после их разложения смесью серной и хромовой кислот.
Методы минерализации, основанные на сплавлении, применяются реже, чем методы мокрого окисления. В качестве плавней используют соду с добавками нитрата калия, тетраборат калия, гидроокись и перекись калия или натрия и т. д. После сплавления в металлических бомбах определение кремния обычно заканчивают спектрофотометрическим методом. Загрязнение продуктов материалом бомб является одним из основных недостатков этого метода.
Глава 10
МЕТОДЫ ВЕЩЕСТВЕННОГО (ФАЗОВОГО) АНАЛИЗА СОЕДИНЕНИЙ КРЕМНИЯ
Методы вещественного (фазового, рационального, минералогического) анализа, несмотря на большое практическое и теоретическое их значение, разработаны все же недостаточно. Общие принципы фазового анализа изложены в работах [216, 222, 226, 352, 587, 1084]. Значение выполнения некоторых определений с точки зрения технологии производства и требований техники безопасности также освещено в литературе [92,167, 174, 225, 417, 434, 455, 536, 601, 682].
Методы количественного определения вещественного состава соединений кремния подразделяются на три группы: 1) химические методы, основанные на избирательном действии растворителей по отношению к химическим соединениям, входящим в состав анализируемого образца; 2) физические и физико-химические методы, основанные на зависимости свойств анализируемых веществ от их фазового состава; 3) расчетные методы, основанные на зависимости фазового состава от содержания отдельных элементов, найденного валовым анализом.
Все три группы методов часто используются совместно. Имеется литература общего характера по этому вопросу, касающаяся выбора растворителей [352, 372, 587, 588], расчетов [133, 317, 371], физических методов определения фазового состава [226, 254, 321, 568, 727, 1084, 1101].
ОПРЕДЕЛЕНИЕ ДВУОКИСИ КРЕМНИЯ
Определению двуокиси кремния как фазы посвящено много работ. Вопрос рассматривается с нескольких точек зрения: определение модификаций SiO2 при совместном присутствии и наряду с другими веществами; определение свободного кварца; определение аморфной кремневой кислоты. Общие сведения о формах SiOa см. в главе 1.
151
Для определения различных форм SiO2 (кварц и кристобалит, кварц и тридимит) используют главным образом физические методы. Определение в горных породах по дифракционной интенсивности с использованием в качестве регистрирующего прибора счетчика Гейгера — Мюллера выполнено с погрешностью 5% [887]. В другой работе ошибка оценивается в 5—14% [349]. В качестве внутреннего стандарта использован флюорит. Для оценки правильности метода дифракции рентгеновских лучей применительно к определению кварца метод был проверен на смесях кварца с графитом, бериллом, слюдой и другими веществами [224]. Установлены пределы применимости метода. Продолжительность определения 2 час.
Проверка, проведенная на двух-, трех- и четырехфазных системах (кварц, кристобалит, тридимит, стекло), показала, что для главных интерференционных линий каждой модификации наблюдается линейная зависимость интенсивности интерференционных линий от количества фазы в смеси. Подсчет количества фазы в смеси производился как среднее по трем интерференционным линиям. Ошибка определения меньше, чем +2%.
В работах [798, 1005] сделана попытка применения рентгеновского ионизационного анализа для количественного определения твердых фаз двуокиси кремния. Сравнены интенсивности интерференционных линий чистых модификаций SiO2 с линиями в смеси.
Оценка изменения линейных размеров образцов динасового кирпича при различных температурах позволяет количественно определить содержание в нем кварца, тридимита и кристобалита [95, 921]. Для кварца наиболее резкое изменение удлинения лежит в интервале 500—600° С, для тридимита — при 100 и 200° С, для кристобалита — 200—300° С.
Для измерения из середины кирпича выпиливают стержень размером 20 X 20 X 100 мм, отмечают на нем положение точек для измерения, помещают образец в электрическую печь и через каждые 100° С измеряют удлинение с помощью катетометра. На основании измерений рассчитывают фазовый состав.
Измерение плотности также позволяет судить о фазовом составе динаса, а следовательно, и о его качестве. Определение свободного кварца в руде и породах петрографическим методом можно произвести путем подсчета числа его зерен из общего количества зерен минералов при содержании SiO2>10%. Определение разновидностей SiO2 в динасе методом количественного рентгеновского анализа см. в работе [818].
Количественное определение кристаллических модификаций двуокиси кремния дифференциальным термическим методом основано на изменении количества поглощаемого тепла при фазовых переходах [471, 898]. В качестве эталонных веществ при снятии термограмм используют смесь, состоящую из SiO2 в количестве, соответствующем предполагаемому содержанию в породе, и материала, не испытывающего превращений, например окиси алюминия [826].
152
Метод характеризуется как ориентировочный. Содержание SiO2 находят сравнением кривых дифференциального термического анализа (ДТА) исследуемой пробы и известных смесей чистого SiO2 с инертным материалом. Оценка содержания кварца, тридимита и кристобалита в работе [471] выполнена с использованием в качестве стандартных веществ хрусталя и кристобалита, полученного при 1530° С из геля кремневой кислоты.
При определении содержания кварца в разнообразном керамическом сырье методом ДТА используют обратимый переход а-~Р-форм кварца около 573° С [534, 535, 969]. Однако этот переход при нагревании маскируется эндотермическим эффектом от разложения каолинита. Поэтому используют экзотермический эффект при охлаждении и сравнивают с градуировочной кривой для количественной оценки содержания кварца по величине площади пика.
Описание прибора для определения свободной SiO2 см. в работе [535]. В качестве эталонов используют 6—7 смесей, содержащих различные количества SiO2 и А12О3. Операции автоматизированы. Пробы, содержащие уголь, должны быть предварительно прокалены.
Для определения содержания кварца в тефлоне также используется термогравиметрический метод [888]. Определение кварца и опала в горных породах см. в работе [31]. Изучение структуры кварца, кристобалита и стекловидного кремнезема по отражению в ИК-области проведено в работе [889]; о формах существования SiO2 в расплавленных шлаках см. [193]; определение SiOH-rpynn в SiO2 см. в [857].
Количественное определение свободной SiO2 становится возможным, по данным разных авторов, при содержании >5% [224, 349], но наиболее надежные результаты получаются при содержании 30—70%.
Определение кварца. В группе химических методов различаются два направления: 1) подбор растворителя или группы растворителей для всех веществ, кроме кварца; 2) изыскание избирательного растворителя для кварца. Большинство работ выполнено в первом направлении. Определение кварца в силикатных породах и материалах предусматривает растворение силикатов в присутствии кварца при действии Na2S [227, 617], H2SiFe [167], НС1 + NH4F [501, 558, 559], HBF4 [824, 1011], H4P2O7 или H3PO4 [174, 175, 345, 679, 973, 1011, 1045, 1046], NaHSO4 [31] и др. Перечень растворителей см. в [203, 227, 1055].
В сборнике [4171 имеется сравнительная оценка основных методов определения свободной кремневой кислоты. Сведения по этому вопросу см. также в работах [167, 205, 227, 318, 618, 933, 1036, 10451.
Чаще всего для растворения силикатной составляющей используют фосфорную и пирофосфорную кислоты [174, 175, 453, 669, 679, 973, 1045, 1046].
153
Определение свободного кварца в силикатах пирофосфатным методом [ 1045]. Фосфорную кислоту предварительно обезвоживают, медленно нагревая 8—10 мл в кварцевой чашке До 250° С и выдерживая при этой температуре около 30 мин. Анализируемую пробу измельчают и пропускают через шелковое сито с диаметром отверстий 100 мк. Пробу вещества около 100 мг помещают в тигель и вливают при помешивании приготовленную, нагретую до 250° С кислоту; помещают тигель в цилиндрический медный блок, нагретый до 275° С. Тигель накрывают часовым стеклом с отверстием для термометра, доводят температуру до 250° С и выдерживают 10—20 мин. Большая часть силикатов разлагается нацело.
Охладив тигель снаружи помещением в чашку с холодной водой, прибавляют к его содержимому 50—60 мл горячей воды и перемешивают. Для предотвращения выделения кремневой кислоты добавляют 20 мл тартрата аммония и кристаллик винной кислоты. Горячий раствор фильтруют. Если фильтрат получается мутным, то обработку повторяют. При разбавлении водой добавляют 10 мл 40%-ной фтороборной кислоты и выдерживают 1 час.
Осадок отфильтровывают, промывают горячей водой, подкисленной уксусной кислотой, сушат и озоляют в платиновом тигле и прокаливают при 800° С в муфеле. Прокаленный осадок обрабатывают смесью HF и H2SO4 и по разности вычисляют содержание кварца. В присутствии больших количеств щелочноземельных металлов и полуторных окислов их предварительно растворяют в НС1; вещества с высоким содержанием углерода озоляют.
В работе [679] изучено отношение к фосфорной кислоте аморфных и кристаллических разновидностей двуокиси кремния и установлено, что растворимость молотого кристаллического кварца прямо пропорциональна поверхности порошка. При равном размере частиц аморфная кремневая кислота растворялась в фосфорной кислоте гораздо быстрее, чем кристаллический кварц, тридимит и кристобалит. О взаимодействии модификаций кремнезема с фосфорной кислотой см. также [1045]. Химическая устойчивость стекол по отношению к фосфорной кислоте описана в [345]. Автор работы не рекомендует применять способ с фосфорной кислотой для определения свободного кварца в обожженных глинах, потому что там содержатся различные кристаллические и аморфные разновидности SiO2, скорость растворения которых в фосфорной кислоте различна.
Микрохимический вариант метода дан в работе [669] для определения содержания кварца в рудничной пыли (навеска 10 мг). Продолжительность определения около 8 час. Для повышения точности при серийных определениях пользуются калибровочным графиком, построенным на эталонных смесях кварца и полевого шпата. В Н3РО4 нацело растворяются лабрадорит, периклин, оливин, мусковит, флогопит, тальк, роговая обманка, каолинит, калиевый полевой шпат. Точность пирофосфатного метода характеризуется ошибкой ±1,0% [973] при определении свободного кварца в горных породах.
Уточняя метод определения свободного кварца в пылях рудников цветной металлургии, авторы работы [554] рекомендуют пыли с высоким содержанием свинца и других металлов, образующих сплавы с платиной, предварительно обрабатывать азотной кислотой. Абсолютная ошибка определения свободной SiO2 колеблется в пределах от 0,5 до 2% при содержании ее от 9 до 60%.
154
В настоящее время для определения свободного кварца пирофосфатным методом в пыли используется вариант методики, разработанный Добровольской [174, 175]. Двуокись кремния, нера-створившаяся в пирофосфорной кислоте, определяется весовым методом, если количество ее >10 мг, и колориметрическим — в форме восстановленной кремнемолибденовой кислоты — при низких содержаниях.
Пыль, собранную на ватном фильтре, после взвешивания помещают в фарфоровый тигель и озоляют при 600—650° С. К остатку прибавляют 5 мл конц. НС1 и 2 мл конц. HNO3, закрывают крышкой и кипятят 8—10 мин. Содержимое тигля количественно переносят в стакан емкостью 150 мл, прибавляют 50 мл горячей воды, фильтруют и промывают остаток на фильтре 4—5 раз горячей водой.
Высушив и озолив фильтр в платиновом или кварцевом тигле, прибавляют к остатку свежеприготовленную, нагретую До 250° С пирофосфорную кислоту и нагревают на масляной бане 15 мин в пределах 248—253° С, помешивая платиновой проволочкой. После охлаждения содержимое тигля переносят количественно в стакан с помощью 50 мл горячей воды (50—60°). Если после 1,5—2 час стояния имеется неоседающая коллоидная кремневая кислота, к раствору прибавляют 20 мл 0,5N NaOH и помещают на 20 мин в водяную баню.
После этого раствор отделяют от осадка фильтрованием, промывают горячей водой, НС1 (1 : 9) и снова водой до отсутствия реакции на РО34_-ионы с молибдатом. Фильтр с осадком сушат, озоляют, прокаливают в платиновом тигле и взвешивают.
Определение заканчивают проверкой на чистоту с HF и H2SO4, если вес SiO2>0,01 г. При меньшем количестве осадок сплавляют с пятикратным количеством смеси K2CO3+Na2CO3, растворяют плав в теплой воде и далее выполняют определение кремневой кислоты колориметрическим методом. Продолжительность определения 6—7 час.
Автором изучена также растворимость в пирофосфорной кислоте кварца и 20 минералов. Плохо растворяются в пирофосфорной кислоте берилл, топаз, циркон; хорошо — полевошпатовые породы, слюды [175].
Для повышения точности определения кварца в пыли внесены изменения в технику отбора пробы пыли с помощью ватных фильтров [4961 — предлагается обеззоливать вату смесью НС1 и HF и обрабатывать ватный фильтр перед сожжением концентрированной хлористоводородной кислотой.
Предложено заменять платиновую посуду кварцевой [97, 203] и использовать кристаллическую пирофосфорную кислоту с температурой плавления 61° С [97]. Установлено, что для пород с высоким содержанием силикатов [454] по методике [174, 175] получаются плохо воспроизводимые результаты. Предлагается после кислотной обработки вводить едкий натр до 10%-ной избыточной концентрации или же прибавлять 15—20 капель 40%-ной HF в воду для разбавления полученного раствора [96]. Абсолютная ошибка составляет + 0,5%. Рекомендуется проводить холостой опыт. Определение S1O2 по методике [175] в витающей угольной пыли очистных забоев шахт проведено на большом количестве образцов [450].
В другом варианте методики предлагается использовать Н3РО4 или Н3РО4, частично обезвоженную (нагреванием до 200° С, а не до 250° С [203]). Для связывания поливалентных металлов вводят комплексон III; кремневую кислоту, полученную при разло-
155
женим силикатов, растворяют в 20%-ном NaOH или КОН, которые кварц практически не растворяют. Автор характеризует метод как ускоренный, хотя определение длится 6—7 час. Определение свободного кварца при растворении силикатов в кремнефтористоводородной кислоте описано в работах [167, 170, 617]. Для определения кварца в горных породах и рудничной пыли после сравнительной проверки и небольших изменений [617] принята методика [170]. Щекатуриной и Петрашень [617—620] разработаны методы определения кварца в горных породах в присутствии силикатов. Изучена растворимость силикатов кальция и алюминия в различных условиях (температура, концентрация реактива, время обработки) в сульфиде натрия [620]. См. также [51, 225, 496].
Для отделения свободного кварца от других минералов проводят [60] последовательную обработку смесью хлористоводородной и азотной кислот при нагревании, остаток сплавляют с K2S2O7. Двуокись кремния из силикатов растворяют обработкой раствором едкого натра. Двукратное сплавление с NaHSO4 с дальнейшей обработкой щелочью при нагревании применено для определения кварца и опала в горных породах [31]. Разложение смесью НО и HF с последующей обработкой остатка щелочью см. в [501, 537, 558, 559]. Мешают определению минералы берилл, сердолик, хризопраз и др. Время определения 16 час, точность ±5% [537]. Метод применен для определения кварца в сульфидных рудах [559]. В работе [532] свободный кварц определяют в остатке после обработки руд и сопутствующих пород концентрированной НС1 и H2SO4 (1 : 4) и 5%-ным раствором Na2CO3 (см. также [536]). Метод, основанный на переводе свободного кварца в растворимое состояние в присутствии силикатов, предложен Полежаевым [434] и изложен с небольшими изменениями в работах [538, 560]. Для выполнения определения производят сплавление со смесью КНСО3 и КС1 (1 : 1) или NaHCO3 и NaCl (1 : 1) в 50-кратном количестве.
0,05 г измельченного материала помещают в стакан емкостью 20—25 мл, смачивают 1—2 каплями воды и обрабатывают 10 мл смеси равных объемов НС1 (1 : 1) и HNO3 (1 : 1) [434]. После двухминутного кипячения охлажденный раствор фильтруют через плотный беззольный фильтр и промывают последовательно нагретыми до кипения растворами — 2%-ным раствором NH4C1 (дважды) и 20мл 10%-него раствора Na2CO3. Фильтр с осадком осторожно подсушивают и прокаливают в стальном тигле на пламени горелки. Эта обработка применяется для материалов, содержащих несиликатные соединения металлов.
Охладив тигель, прибавляют 2,5 г тщательно растертого плавня (КС1-|--|-КНСО3) и перемешивают металлической палочкой. Смесь высыпают в агатовую ступку, растирают и снова количественно переносят в тигель, удаляя остатки из ступки небольшим количеством плавня. Тигель начинают осторожно нагревать, затем (через 3—5 мин) постепенно доводят до красного каления и расплавления массы. Вращая тигель, выдерживают его в пламени около 2 мин, охлаждают и к застывшей массе в тигле прибавляют постепенно 40 мл 5% -ного раствора Na2CO3, нагревают до кипения, сливают по палочке раствор на фильтр, собирая фильтрат в мерный цилиндр для дальнейшего колориметрирования.
Метод рекомендуется применять, если содержание SiO2 составляет >10% [538]. При более низких содержаниях предпочтительнее использовать метод с пирофоефорной кислотой.
156
Определение свободной аморфной кремневой кислоты. Это определение необходимо для оценки качества керамического сырья [61, 147, 205, 434, 682, 910]. Метод [147] основан на обработке исследуемого материала при нагревании на водяной бане 5%-ным раствором Na2CO3 с последующим фильтрованием и колориметрированием фильтрата [910].
Для определения аморфной кремневой кислоты, по Полежаеву [434], помещают 0,05 г вещества в металлический (например, железный) тигель емкостью 20—25 мл, прибавляют 15 мл 4%-ного раствора КОН, кипятят 3—5 мин, затем фильтруют в мерный цилиндр. Остаток промывают на фильтре 12,5 мл 20%-ного раствора смеси КНСО3 и КС1, ополаскивая им тигель. Затем тигель ополаскивают 10 мл 2%-ного раствора КОН и промывают осадок на фильтре. Фильтрат доводят до определенного объема и далее колориметрируют кремневую кислоту в форме желтого кремнемолибденового комплекса.
Разработана и рекомендуется методика [205, 310] для ускоренного определения аморфной двуокиси кремния в глинах. Метод основан на обработке определяемого образца глицерином и последующем титровании комплексной кремнеглицериновой кислоты водно-глицериновым раствором гидроокиси бария.
Определение аморфной кремневой кислоты в природных силикатах [205]. Для выполнения определения готовят из насыщенного водного раствора Ва (ОН)2 разбавлением смесью воды и глицерина (1 : 1) 0,01М раствор гидроокиси бария с дальнейшей стандартизацией его по раствору НС1. Индикаторы (внешние) —0,05%-ные спиртовые растворы ализаринового желтого или фенолфталеина. Сначала определяют расход титранта на титрование глицерина: 5 мл глицерина помещают в стакан емкостью 50 мл и титруют водно-глицериновым раствором Ва (ОН)2 при помешивании. Для определения момента эквивалентности на белую фарфоровую пластинку помещают каплю индикатора и к ней прибавляют каплю титруемой смеси. Капли перемешивают стеклянной палочкой и по появлению фиолетовой окраски (в случае ализарина) и розовой (в случае фенолфталеина) определяют конец титрования.
Затем определяют расход титранта на титрование навески двуокиси кремния. Для этого 10 мг гидратированной двуокиси кремния тщательно растирают с 5 мл глицерина в течение 15—20 мин в стаканчике емкостью 50 мл и титруют стандартным раствором Ва (ОН)2: приливают такое количество миллилитров титранта, которое соответствует 5 мл глицерина, перемешивают 3—4 мин и далее титруют медленно, добавляя титрант по 3—5 капель до изменения цвета капли индикатора на фарфоровой пластинке. Расход титранта на 10 мг двуокиси кремния определяют по разности объема титранта, пошедшего на титрование смеси 10 мг двуокиси кремния с 5 мл глицерина, и объема титранта, пошедшего на титрование 5 мл глицерина.
Навеску образца 20—30 мг обрабатывают глицерином при тщательном перемешивании, затем титруют водно-глицериновым раствором гидроокиси бария аналогично титрованию гидратированной кремневой кислоты. Содержание свободной аморфной двуокиси кремния в образце рассчитывают по формуле
SiO2, % = 10-У3-ЮО/О^ — V,)-a,
где а — навеска вещества, г; — объем Ва(ОН)2, пошедший на титрование 10 мг SiO2 с 5 мл глицерина, мл; У2 — объем Ва(ОН)2, пошедший на титрование 5 мл глицерина, мл; V3 — объем Ва(ОН)2, пошедший на титрование навески образца, мл.
Параллельно авторы определяли аморфную кремневую кислоту по методу Гедройца [147] с хорошей сходимостью результатов.
157
Предложенный метод вполне пригоден для определения «SiO2 • • тН2О в технических продуктах. Преимущество его — быстрота выполнения (30—40 мин по сравнению с 4—5 час по методу Гед-ройца). Определение свободной SiO2 в горных породах и порошкообразных материалах см. в [792, 965, 973].
Кроме химических методов для количественного определения кварца и других модификаций свободной кремневой кислоты используют рентгенографический метод [223, 224, 349, 887, 1005], метод дифференциального термического анализа [471, 534, 535, 826, 888, 969] и др. [227, 449, 901]. Полуколичественное определение коллоиднодисперсной кремневой кислоты при пропитке бумаги может быть выполнено по интенсивности окраски с некоторыми алкиламинозамещенными трифенилметанлактона [1058].
Количественный рентгеновский фазовый анализ соединений кремния основан на зависимости интенсивности дифракционного отражения от содержания определяемой фазы в исследуемом образце. Однако эта зависимость не выражается прямой пропорцией, поэтому необходимо пользоваться серией эталонных образцов с известным содержанием фаз [216, 222, 452]. Ценность рентгеновского метода для определения кварца особенно велика в тех случаях, когда материал характеризуется высокой степенью дисперсности и применение фосфатных методов дает большие ошибки [92].
Ивойлов и Денисова [223] определяли свободный кварц в рудничных пылях измерением массового коэффициента ослабления излучения пробы без внутреннего стандарта. Абсолютная ошибка определения 0,5—2,5%. Время определения 2 час. В качестве внутренних стандартов для определения кварца в пылях используют флюорит [887], окись бериллия [501], сульфид цинка [222, 501].
Полуколичественное определение свободной двуокиси кремния проводят различными методами: методом бумажной хроматографии [901], ПК-спектрометрии [801], адсорбционно-фотометрическим [933]; расчетный метод определения содержания свободной SiOa на основании усреднения данных химических анализов приведен в [51].
Выполнены работы по сравнительной оценке методов определения свободной кремневой кислоты (кварца) в пыли дробильных цехов [501], в горных породах [453]. Проведены межлабораторные испытания метода определения кварца в пыли [933]. Установлено [97, 501], что данные петрографического метода близки к данным рентгеноспектрального; химические и термические дают гораздо более высокие результаты. В работе [203] предпочтение отдается химическим методам. Другие работы по определению свободной SiO2 см. в [73, 504, 600, 734, 792].
Использование химических методов для фазового анализа сравнительно ограниченно, причем очень часто они применяются в сочетании с расчетными методами. Выбор селективных растворителей затрудняется малой растворимостью силикатов. Метод использования селективной растворимости пригоден для решения частных 158
задач, когда минералогический состав анализируемого образца сравнительно прост. Для продуктов, образующихся в системе MgO— Сг2О3 — SiO2 [260], применяют 15%-ный раствор NH4C1 с добавкой NH4OH, избирательно растворяющий MgO; форстерит растворяется в 0,2 N НС1, энстатит — в смеси HNO3 и HF, хромит магния — при сплавлении со смесью тетрабората и карбоната натрия. Содержание свободной кремневой кислоты находят расчетным путем.
В присутствии ZnO и SiO2 силикат цинка определяют [427] после растворения окиси цинка в смеси Лоу (200 мл 10%-ного раствора NH4OH и 32 г NH4C1), которая растворяет природный гидратированный силикат цинка и не растворяет безводный силикат. Далее ортосиликат цинка разлагается 6%-ной СН3СООН, 21 %-ной НС1 или 10%-ным раствором едкого натра. Свободная кремневая кислота практически не реагирует с этими растворителями и определяется в остатке по разности. Избирательное растворение силиката цинка в НС1 описано в работе [812].
Фазовый анализ кремне-алюминиевых сплавов, основанный на фракционном растворении, см. в [204].
Минералогический анализ пород дистена и андалузита и продуктов их обогащения проведен путем селективного растворения в смеси НС1 и NaF с экстракцией нерастворимого остатка тяжелыми жидкостями [87].
Для исследования фазового состава цементного клинкера использованы в качестве растворителей уксусная и борная кислоты [35, 281, 388, 389, 455—458, 555]. В 5%-ном водном растворе борной кислоты растворяются C3S и C2S в кристаллической фазе и C2S в стекловидной фазе; 1М СН3СООН полностью разлагает трехкальциевый алюминат и силикаты кальция и лишь на 10—11% C4AF. Вследствие того что действие растворителей не вполне селективно, авторы вводят поправочные коэффициенты [455]. В работе [35] дан метод определения С3А прокрашиванием шлифа образца органическими красителями. Метод используется и в фазовом анализе глин [493].
Предложены схемы вещественного анализа сочетанием экстракционных и расчетных методов. Для глинистых пород без применения сплавления определены главным образом несиликатные составляющие [956]; для агломератов никелевых руд силикатная часть извлекается растворением NaOH и НС1 с последующим делением на фракции в бромоформе (по плотности). Содержание различных минеральных форм (кварц, клиноэнстатит и другие формы) находили расчетом.
Нефелин в нефелиновых рудах [249] в отличие от полевого шпата и эгерина разлагается при кипячении с 5%-ным раствором НС1 за 1 мин (или с 5%-ным раствором H2SO4 — за 45 мин, 2,5%-ным раствором лимонной кислоты — за 45 мин, 2,5%-ным раствором щавелевой кислоты — за 1—2 мин); его определяют расчетным путем по количеству А12О3, перешедшей в раствор.
Содержание ортоклаза и альбита в полевых шпатах, глинах, керамических массах вычисляют по содержанию окислов щелочных металлов (К2О и Na2O). Приближенно содержание нефелина в хибинской руде рассчитывается по среднему содержанию кремневой кислоты, если принять состав нефелина 6Na(K)AlSiO4 • nSiO2, где п ~ 1. Способ рекомендован для быстрого технического анализа. Содержание А13О3 в глинистом веществе определяют вычитанием количества А13О3, связанного в полевых шпатах, из общего содержания. Содержание двуокиси кремния также рассчитывают вычитанием из 100% суммы процентных содержаний глинистого вещества и полевых шпатов.
В работе [976] дан критический обзор методов определения рационального состава керамических масс и сырья. Автор работы [712] указывает, что расчетные методы по данным химического анализа дают хорошие результаты для полевых шпатов и лишь удовлетворительные — для других видов керамического сырья. Метод определения муллита в керамических изделиях на основании данных анализа описан в работе [800].
Фазовый состав портландцементного клинкера, глиноземистого шлака и доменного шлака на основании данных химического анализа рассчитывают по числу миллимолей соответствующих окислов, содержащихся в ста весовых единицах анализируемого вещества [33, 472]. Расчет для портландцементного клинкера ведут по окиси кальция исходя из представлений об установившемся в системе равновесии и полной кристаллизации [785]. Содержание свободной окиси кальция определяют отдельно и вычитают из общего количества СаО. Затем по содержанию SO3 рассчитывают количество CaSO4, по содержанию Fe2O3 — количество C4AF, затем количество С3А — по А12О3, по SiO2 — количество C2S и остаток СаО пересчитывают на C3S, вычитая его из рассчитанного содержания C2S. Для глиноземистого шлака расчет также начинается с малых составляющих — серы и окиси магния, полагая, что они целиком войдут в состав соответственно сульфида кальция и шпинели. Остальные расчеты ведут в зависимости от положения состава шлака на тройной диаграмме. Количественное определение главных фаз портландцемента на основании данных химического анализа и рентгенографии см. в [711]. В доменном шлаке сера связывается в сульфид кальция (и марганца), окись магния — в окер-манит 2СаО • MgO • 2SiO2. Оставшиеся окислы рассчитывают так же, как и в случае глиноземистого шлака, исходя из положения состава шлака на тройной диаграмме.
Расчетным путем в сочетании с химическими методами [388, 455, 458] определяют состав плавленого трехкомпонентного клинкера (СаО, А12О3, SiO2), основываясь на представлениях о неравновесном ходе кристаллизации клинкерных расплавов [551]. Предложены пересчетные факторы для вычисления молекулярных соотношений, ионных весовых процентов и других факторов на основании данных химического анализа [794]. Для расчета содержания
160
каолинита по А12О3 применен комплексонометрический метод [718]. Даны условия определения каолинита и слюды в глинах и каолинах на основании только определения А12О3.
Анализ водной и солянокислой вытяжки позволяет рассчитать состав минеральной части бурого угля [586]. Сопоставляя данные химического состава нерастворимого остатка и стеклофазы в огнеупорах с результатами рентгенографического определения количества корунда на дифрактометре, авторы работы [156] находят расчетным путем количество и состав муллитной фазы в высокоглиноземистых огнеупорах. Абсолютная ошибка определения корунда и муллита составляет ±3%.
Метод скоростного количественного фазового анализа осадочных пород с использованием реакций газообразования (главным образом при действии карбида кальция на водные силикаты) разработан и применен к многочисленным объектам Бергом [67]. Метод комбинируется с термографическими исследованиями. Другие работы по использованию дифференциального термического анализа для определения фазового состава горных пород см. в [897, 1068].
Выделение и определение нитрида кремния см. в [607, 608]. В работе [442] определение свободного кремния выполняют после растворения его в 1%-ном водном растворе NaOH. При изучении действия различных реагентов (кислот и щелочей) на силициды IV, V и VI групп периодической системы [287] были разработаны методы определения в технических продуктах общего содержания кремния, свободного кремния и металла. Общее содержание кремния определяют после сплавления навески вещества с NaOH в никелевом или железном тигле с последующей обработкой 10%-ной НС1 или H3SO4 или смесью H2SO4 и Н2С2О4. Свободный кремний переводят в раствор действием 1 %-ного NaOH и затем определяют колориметрическим методом.
Фазовый химический анализ карборунда и кремнистых сплавов на его основе также предусматривает [358, 448, 926] определение свободного кремния, извлекаемого действием 1 %-ного раствора щелочи.
Определение (удаление) свободной кремневой кислоты можно выполнить действием HF и H2SO4 [267,358] или последовательным действием HF, HNO3h НС1О4. Другие работы по анализу технического SiC для определения его фазового состава см. в [286, 566, 567].
Проведение операции сплавления и разложения плава непосредственно в тигле ускоряет анализ [358]. Результаты рассчитывают, вводя эмпирическую поправку в связи с однократным выпариванием. Отделение и определение фаз в шлаках (силикатная фаза), SiO2, Si и SiC производят на основании фракционного растворения в 85%-ной Н3РО4, концентрированной HF, смеси HF и HNO3. Оставшийся нерастворенным SiC сплавляют с KNaCO3 и определяют как обычно.
*/46 Аналитическая химия кремния
161
В обзоре [955] наиболее эффективными из методов количественного и полуколичественного определения отдельных минералов с помощью дифракции рентгеновских лучей признаны: метод внутреннего стандарта; метод, основанный на определении массового коэффициента поглощения различных фаз, и метод, включающий оценку интенсивности дифракционного излучения, которые равноценны по точности. Большое внимание уделяется подготовке пробы к анализу. Метод используют для количественного определения каолинита [683] с относительной ошибкой <14%.
В монографии [452] рассмотрено изучение структуры глинистых минералов рентгеновскими методами. Описаны основные методы идентификации и количественный анализ смесей глинистых минералов. В методе дифракции рентгеновских лучей измеряется интенсивность дифракционных отражений. Последняя связана с содержанием компонента в смеси, но эта связь не может быть охарактеризована как прямая пропорция. Метод не позволяет оценить содержание аморфных компонентов.
Для количественного рентгеноспектрального анализа смесей используют сравнение со стандартными бинарными или многокомпонентными смесями, а также при определении отдельных компонентов — метод добавок с измерением интенсивности отражения в зависимости от количественного содержания определяемого компонента в образце до и после введения определенных добавок его в смесь. Стандартные минералы для определений — слюды, каолинит, кварц, хлориты и др. Приближенно содержание кварца и полевого шпата в каолине может быть выполнено методом Дебая. После термической обработки изучены линии муллита — кварца-и муллита — кристобалита.
Определение кристаллических фаз в системе стекол Ы2О — А12О3 — SiO2 основано на сравнении интенсивности линий определяемой фазы и NaCl. Продолжительность анализа 1,5 час, относительная ошибка определения около ±10%. Определены фазы: кварц, а-тридимит, а-кристобалит, сподумен, а также силикаты и алюминаты лития. Определение индивидуальных силикатов см. в [937].
Дифференциально-термическим методом исследуются также сложные смеси. Возможности практического использования термографии для количественного фазового анализа природных и синтетических силикатов, технических продуктов отражены в работах [63, 66, 68, 898, 957]. О комплексном термическом анализе силикатов см. работу [251]. Продолжительность анализа 15—20 проб — трое суток. Приближенно состав пород устанавливают на основании термогравиметрических определений. Точность определений дифференциально-термическим методом составляет для каолинита при определении его в глинах 5% [1069].
Для определения минеральных компонентов горных пород используют изучение ИК-спектров порошков, наносимых в виде пасты в изопропиловом спирте на стандартное окошечко из NaCl.
162
Сцинтилляционный спектральный метод минералогического анализа [449] основан на счете частиц заданного атомного состава по эмиссионному спектру элементов. Подсчет производится с помощью электронной счетной машины.
Петрографический метод имеет большое значение для фазового анализа материалов, содержащих кремний (керамика, абразивы, стекло, цемент, шлаки). Методике исследования посвящено много работ. Вместе с данными справочного характера результаты этих исследований приведены в монографиях и руководствах [32, 62, 83, 138, 227, 272, 350, 452, 582, 866, 1101], где с петрографической точки зрения охарактеризованы огнеупоры, шлаки, вяжущие вещества, керамика, камни в технических стеклах, неметаллические включения в сталях.
Для качественной и полуколичественной оценки содержания отдельных фаз используются различные методы: изучение тонких шлифов в проходящем свете, исследование порошкообразных минералов иммерсионным методом, изучение полированных шлифов в отраженном свете.
Сложность химического и фазового состава, обилие полиморфных превращений затрудняют количественное определение (а иногда даже и идентификацию) состава технических продуктов, содержащих соединения кремния.
Количественный микроскопический анализ основан на подсчете числа частиц определяемой фазы в тонких шлифах. На основании подсчета вычисляют площадь, занимаемую зернами определяемого вещества, и его содержание. Погрешности метода связаны с возможным несовершенством конструкции прибора, искажениями при изготовлении шлифа (так как данный участок может быть непредставительным), ошибками при определении компонента по его оптическим характеристикам и статистическими погрешностями.
Методика подсчета зерен в тонких шлифах минералов впервые предложена Солласом [1023]. В настоящее время для этой цели используют интеграционные микрометренные столики или интеграционные микрометры. Количественному определению в каждом случае обязательно предшествует точная идентификация каждого зерна определяемой фазы. Это обычно делается путем сравнения с литературными данными на основании определения показателей преломления, силы двулучепреломления, цвета, характерных особенностей зерен (трещины, спайности, рельеф, наличие двойников). Используются приемы окрашивания зерен различными реактивами: например, зерна полевого шпата окрашиваются раствором кобальтинитрита натрия [32]. Библиография по вопросам идентификации фаз имеется в работе [227].
Подсчет числа зерен позволяет найти содержание определяемой фазы в объемных процентах; для расчета весовых процентов необходимо знать плотность определяемого компонента. Подсчет числа зерен определяемой фазы можно выполнять в шлифе и в дисперсионной среде — для порошкообразных материалов (например, для
7 Аналитическая химия кремния
163
определения кварца в тальке). Количественный петрографический анализ используют для определения качества продукции промышленности силикатов, а также в исследовательской практике [34].
Микроскоп для изучения портландцементного клинкера впервые применен Ле Шателье; представления о фазовом составе даны Торнебомом [1051, 1052], использование интеграционного столика см. в [833]. При микроскопическом изучении стекол удается по показателю преломления определить тип стекла, а следовательно, его качественный и ориентировочно-количественный состав [833], а также природу свилей и камней [867]. Данные по минералогии глин и библиографию см. в [516].
Точность и воспроизводимость результатов микроскопических исследований зависит во многом от правильной подготовки образцов. Кроме поляризационного микроскопа используют также стереоскопический микроскоп, металлографический, электронный, инфракрасный — с целью получения дополнительных данных о количественном и качественном составе образцов. Одновременно образцы изучают методом дифференциального термического анализа, абсорбционной спектроскопии, рентгенографическим.
Имеются работы по количественному петрографическому анализу гидратированных цементов [625, 1022], цементных растворов [282, 283]; относительная ошибка определения ±30% [283]. О сочетании электронномикроскопических исследований с результатами химического анализа для целей вещественного анализа см. [868].
Определение различных форм растворимой кремневой кислоты описано в работе [927], а их разделение—в [1139, 1140].
О фазовом анализе технических объектов, содержащих кремнефтористоводородную кислоту и кремнефториды, см. работы [213, 595, 854, 902, 1031].
Глава 11
ОПРЕДЕЛЕНИЕ ПРИМЕСЕЙ В КРЕМНИИ И НЕКОТОРЫХ ЕГО СОЕДИНЕНИЯХ
Выбор метода определения примесей в кремнии и некоторых его соединениях зависит от степени чистоты анализируемых веществ. Кремний технической чистоты (95—98% основного вещества), применяемый в металлургии для получения легированных сталей, чугунов, сплавов с медью и алюминием и для других целей, анализируют на содержание примесей (Al, Fe, Са, Ti) без определения основного вещества.
Для определения по ГОСТ 2178-54 среднюю пробу 10—15 г, отобранную после дробления вещества на стальной плите молотком из марганцовистой стали, растирают в агатовой или яшмовой ступке до прохождения через сито № 75. Материал не отмагничивают; 2 г вещества помещают в платиновую чашку емкостью 200 мл, смачивают водой и прибавляют по каплям~20—25 мл HF и по каплям 20—25 мл конц. HNO3.
По окончании бурной реакции тщательно обмывают стенки чашки и крышку водой, прибавляют 5 мл конц. H2SO4, выпаривают досуха, прибавляют еще 3 мл H2SO4, нагревают до густых паров, охлаждают, разбавляют осторожно водой и в полученном растворе определяют сумму полуторных окислов осаждением аммиаком, железо — хроматометрически, кальций — перманганатометрически или комплексонометрически, титан — фото колориметрически.
Описан вариант анализа, основанный на растворении растертой и отмагниченной пробы в 50%-ном растворе щелочи с отделением кремния в виде двуокиси после дегидратации обычным методом [190].
Неметаллические включения в кремнии определяют методом хлорирования: при этом основа удаляется в виде S1C14, а включения, имеющие характер силикатов, анализируют как обычно, используя методы сплавления с карбонатными плавнями с последующим разложением растворами кислот.
Для определения примесей в кремнии, карбиде кремния и природных разновидностях двуокиси кремния используют эмиссионный спектральный метод анализа [112, 113, 432, 475].
Методы определения примесей в кремнии высокой чистоты очень специфичны, и поэтому в пределах данной монографии можно привести лишь самые общие сведения по этому вопросу и примеры
7*
165
отдельных методов определений. Эти методы во многом сходны при анализе группы веществ, необходимых для производства полупроводниковых материалов: кремния, карбида кремния, двуокиси кремния, трихлоргидросилана, хлорсилана, иодсилана. Требования к материалам высокой чистоты изложены в работах [18, 130, 229, 250]. Проблемы анализа веществ высокой чистоты и методы определения малых концентраций примесей освещены в работах [9—11, 18—22, 41, 130, 131, 220, 351, 353, 364 , 390, 391, 437, 464, 483, 561, 627, 923, 1104, 1115].
Химические методы неприменимы к анализу кремния и его соединений высокой чистоты, в которых содержание примесей в ряде случаев не должно превышать 10~9—10~и%. Использование химических реактивов (даже высокочистых) вообще крайне ограниченно. Специально изучены условия работы, которые необходимо соблюдать при этом: чистота рабочих помещений, специфичность подготовки пробы для анализа, выбор посуды, очистка и хранение реактивов [ПО, 364, 421], проверка чистоты реактивов и воды [111, 364, 498, 603]. Очищенные кислоты рекомендуется хранить в полиэтиленовой, фторопластовой и кварцевой посуде не более 30 дней.
Поскольку некоторые примеси попадают в кремний из воздуха, специально исследованы источники загрязнений в процессе анализа [116]. Сравнение результатов работы в боксе и на открытом воздухе показывает, что в последнем случае реактивы и анализируемые вещества загрязняются примесями А1, В, Fe, Са, Си, Mg, Мп, Ni, Sn, Pb, Ti, Cr.
Строгого соответствия между электрофизическими свойствами полупроводников и их химическим составом пока не установлено. Однако для кремния и карбида кремния наибольшее влияние оказывают примеси элементов III и V групп периодической системы. «Универсально» вредны Си, Fe и А1. Прежде всего необходимо количественно определять бор и фосфор, поскольку их дозировка как легирующих элементов определяет тип проводимости.
В зависимости от назначения полупроводниковых материалов в них определяют до 15—20 примесей, иногда до 30—40 [364]. Физическими и физико-химическими методами анализа можно определить примеси более 70 элементов [598]. Обычно используют несколько методов (спектральный, рентгеноспектральный, полярографический, масс-спектрометричес кий, радиоактивационный)[ 181.
ФИЗИЧЕСКИЕ МЕТОДЫ
Ни один из применяемых методов не может быть охарактеризован как универсальный, однако чаще всего применяют спектрографический, радиоактивационный и масс-спектрометрический методы. Точность и чувствительность этих методов при контроле чистоты веществ обсуждены в работах [10, 11, 19, 20, 218, 220, 364, 1062, 1065].
166
Спектральный метод
Чтобы исключить возможность загрязнения образцов при подготовке их к спектральному определению, разработаны приемы измельчения кремния и подготовки электродов [218]. Было изучено измельчение в ступках из агата, пьезокварца, лейкосапфира и между пластинами монокристаллического кремния. Рекомендовано истирание между пластинами или в ступке из монокристалла кремния после протравливания HNO3 и HC1,HNO3 и HF [115]. Грубое дробление производят между пластинами из фторопласта. При истирании первые порции материала отбрасывают.
Эталонные растворы для определения примесей готовят из нитратов или окислов Си, Са, Al, Mg, Fe, Ni, Ag, Pb, In, Mn, Ga и хранят в полиэтиленовой посуде до 7 месяцев [413]. Определение выполняют обычно после предварительного химического концентрирования примесей [114, 116], хотя имеются работы по определению примесей без обогащения [117, 609, 834, 1063].
При анализе особочистой SiO2 обычно концентрируют примеси, удаляя основу обработкой фтористоводородной кислотой [340, 422, 476, 477, 527] или ее парами [114, 216]. Описаны и другие методы выделения примесей из двуокиси кремния: высоковольтный электродиализ [214], экстракция [802].
Загрязнение образцов при обогащении химическими способами изучено в работе [368]; авторами разработаны способы разложения кремния парами кислот. Например, для выполнения спектрального определения образец помещают на тефлоновую пленку, разлагают парами HF и HNO3, а затем пленку с концентратом переносят в канал угольного анода и сжигают в дуге постоянного тока. Для получения чистой фтористоводородной кислоты, необходимой для обогащения, предложен метод ультраочистки, основанный на изотермической дистилляции [877].
В полиэтиленовую чашку помещают два полиэтиленовых стакана, в один из которых наливают 150 мл технической концентрированной HF, в другой — около 150 мл деминерализованной воды, и чашку плотно накрывают другой полиэтиленовой чашкой. Через два дня получают чистую HF с концентрацией около 12 М.
Перегонкой еще одной порции технической HF в течение двух дней получают 24 М HF. Спектрохимически показано, что содержание примесей в очищенной HF составляет 0,001—0,01 мкг/мл.
Очистка HF вымораживанием описана в работе [1137].
При подготовке к спектральному анализу летучих полупродуктов для производства высокочистого кремния — хлорсилана, трихлоргидросилана — применяются специфические приемы концентрирования примесей — сорбция угольным порошком [340, 422], осаждение примесей с испарением основы [114, 446, 629, 1066], экстракция [802].
Чаще всего спектральным методом определяют бор [114, 340, 446, 802, 1066]. Чувствительность метода определения бора на ИСП-22 составляет 10-2—10-7%. Определение может быть выпол
167
нено с предварительным обогащением или без него. Этим методом определяют бор в двуокиси кремния, связывая его маннитом во избежание улетучивания при обработке образца фтористоводородной кислотой [114, 4761.
1 г SiO2 разлагают 6 мл 45%-ной HF, добавляя две капли 1%-ного раствора маннита [476]. Концентрат упаривают почти досуха и наносят на торцы электродов, предварительно обожженных в дуге постоянного тока и пропитанных 3% -ным раствором полистирола в бензоле. Подсушенный осадок на электродах фиксируют каплей 0,6%-ного раствора полистирола.
Спектры снимают на ИСП-28 при экспозиции 40 сек в дуге переменного тока при 8 а.
Градуировочный график строят по абсолютным почернениям линии В I 2497,7 А, применяя в качестве эталона водный раствор борной кислоты.
Средняя квадратичная ошибка среднего результата трех параллельных определений 10%. Достигнутая чувствительность 2 • 10~6% ^'лимитируется загрязнением реагентов, применяемых для концентрирования бора.
Определяют бор в кремнии на ИСП-22 с одним конденсором освещения щели, с использованием угольных электродов и дуги постоянного тока в качестве источника возбуждения и без предварительного обогащения пробы [609]. Электроды обжигают в дуге постоянного тока на воздухе в течение 1 мин. Кратер анода заполняют порошком кремния и зажигают дугу в токе азота. Спектры регистрируют на фотопластинке после 5 сек обжига электродов с пробой. Определение ведут по аналитической паре В 2497,73 А/ Si2563,67 А. Наименьшая определяемая концентрация бора (1,5— —2) • 10~4%, вероятная ошибка определения ±15 отн.%. Авторами описаны также условия возбуждения и регистрации спектров для определения Mg, Си, Al, Fe, Ti. Использование тока азота позволяет устранить влияние молекулярных спектров SiO и CN.
Усиление линии бора и ослабление фона может быть достигнуто при выполнении спектрального определения в токе аргона [834]. Чувствительность метода повышается до 10" 5% с использованием дуги постоянного тока.
Используя обрывную дугу переменного тока на ИСП-22, определяют примеси As, Al, В, Fe, Sb и W с чувствительностью порядка 10"5—10~4% [1063]. Готовят две серии эталонов из тонкоизмель-ченного кремния высокой чистоты, используя одну из них для определения А1, В, Fe и Ti, а другую — для определения As, Sb и W. Относительная ошибка определений также не превышает ±15%.
Зильберштейн с сотрудниками и другие авторы проводили спектральное определение примесей в кремнии и его соединениях с использованием разряда в горячем полом катоде [217, 219, 421], причем была достигнута абсолютная чувствительность метода 10~9—10"10 г при определении Ag, Мп, Си, Ga, In, Al, Ni, Mg, Fe.
Понижение летучести примесей и ускорение процесса обогащения кремния обработкой образцов в камере из фторопласта-4 парами кислот достигнуто в работе [219].
Определение примесей в кремнии, двуокиси кремния и кремнемедном сплаве см. в [216, 235, 369, 629, 770, 1063, 1064].
168
Особенности спектрального определения примесей в жидких летучих полупродуктах для производства высокочистого кремния сМ. в [523, 591, 1062, 1067].
Радиоактивационные методы
Радиоактивационные методы используют чаще всего для определения содержания микропримесей элементов, оказывающих наибольшее влияние на электрофизические свойства полупроводниковых материалов — бора, золота, меди, марганца и других элементов. Эти методы характеризуются высокой чувствительностью и специфичностью [233], которая, однако, снижается вследствие флуктуаций радиоактивного распада и наличия фона. Они применяются в комбинации с другими методами, например масс-спектрометрическим. Повысить чувствительность можно, увеличивая время облучения и измерения, а также размер пробы и детектора излучения, разумеется, в определенных пределах. Используются также методы обогащения проб, но количество химических процедур во избежание введения загрязнений должно быть минимальным.
Без использования химических методов разложения образца можно определить микропримеси в кремнии по у-спектрам их радиоактивных изотопов [192]. При этом имеются затруднения при идентификации с помощью сцинтилляционного у-спектрометра из-за собственного у-излучения кремния при превращении 31Si —>-31Р и переходе 31Р из возбужденного в основное состояние. Чувствительность метода для различных примесей составляет от 10~6 до 10-11 г.
Активация тепловыми нейтронами возможна для элементов с атомными номерами ^83. Чувствительность нейтроноактивационного анализа для различных элементов 10~5—10_10% [20]. Для изучения локализации примесей используют метод авторадиографии.
При нейтроноактивационном анализе, включающем нейтронное облучение и лучевую спектроскопию [934], часто оказывается невозможным спектрофотометрическое разрешение фотопиков целого ряда элементов, так как они лежат в очень узком интервале (например, для Си, Sb, Cd и As этот интервал составляет 0,51—0,57 А4эв). Изменением продолжительности облучения образца нейтронами можно уменьшить совпадение фотопиков соответствующих изотопов с различными периодами полураспада. Например, для определения нанограммовых количеств примесей 27 элементов в полупроводниковом кремнии разработано три схемы активационного анализа с использованием ионного обмена в зависимости от периода полураспада, который равен соответственно 0,3—15 час, 12—90 и > 90 час [777]. Использование ионообменной хроматографии хлор-комплексов для нейтроноактивационного анализа описано в работе (934].
169
Методика одновременного определения 14 элементов без химического разложения пробы дана в работе [851] и 29 элементов — в работе [1050].
Определение следовых количеств элементов в поверхностных слоях полупроводникового кремния может быть осуществлено после травления поверхности смесью HNO3 и HF с последующим облучением в потоке нейтронов вместе со стандартами. Метод позволяет определить до 2 • 10~9 а Си и 3-10~9 г Au.
При радиоактивационном анализе двуокиси кремния также применяют ионообменную хроматографию [215, 233—2401.
Пробу двуокиси кремния прокаливают 3 час при 1000° С, запаивают в кварцевую илн полиэтиленовую ампулу и облучают 6—15 час в потоке (0,5—!)• • 1044 нейтр/см*-сек [233].
Облученный образец протравливают царской водкой и разлагают смесью HNOS и HF. Полученный раствор выпаривают до полного удаления SiF4 и HNOS, остаток растворяют в воде и последовательно пропускают через полиэтиленовую колонку с анионитом АВ-17 н катионитом КУ-2. Последовательно элюируют и определяют соответствующие элементы, измеряя активность элюата без разделений, если имеется достаточная разница в периодах полураспада радиоизотопов, или после разделения химическими методами. Разработаны способы определения 27 элементов.
Чувствительность нейтроноактивационного определения микропримеси бора в полупроводниковом кремнии и полупродуктах для его получения по реакции 10В (п, а)-> 7 Li облучением проб в потоке тепловых нейтронов 108 нейтр/см^-сек при регистрации у-квантов с энергией 470 кэв с помощью сцинтилляционного спектрометра составляет 10_Б%. Такая же чувствительность получена при регистрации а-частиц с помощью тонкослойной эмульсии [328].
В работе [1061] на масс-спектрометре измеряется количество гелия, образующегося при облучении нейтронами кристаллов кремния, содержащих бор. Масс-спектрографическое определение следов бора описано в работах [936, 1061]. В работе [936] использован метод изотопного разбавления. Другие работы, посвященные определению бора, см. в [327, 328, 936, 9451.
Радиоактивационное определение микроколичеств кислорода в кремнии [945, 987] облучением а-частицами со средней энергией 40 Мэв производят путем измерения у-активности 18F, образующегося по реакциям:
1вО (a, a)18F; 1вО(а, pn)18F и 16О(а, 2n)18Ne-^18F.
Образец (30X17X1 мм) полируют алундом, промывают водой, этанолом и петролейным эфиром, закрепляют в алюминиевом держателе, охлаждаемом водой, н облучают 20 мин в циклотроне пучком а-частнц интенсивностью около 10 мка. Облученный образец взвешивают, стирают верхний слой образца (15 мг) с помощью смеси порошка алунда (--—400 меш; 0,1 г) н воды (0,2 мл) и собирают образовавшуюся пасту кремния и алунда в полиэтиленовую пробирку для измерения у-актнвностн. Затем образец промывают этанолом, сушат, взвешивают и снова снимают часть поверхностного слоя.
После удаления ~100 мг Si с поверхности образца его измельчают, сплавляют со смесью 25 г КОН н 100 мг NaF в никелевом тигле, плав растворяют в смеси 30 мл воды н 150 мл конц. H2SO4 н отгоняют HF при 150—160° С, поглощая
170
ее разбавленным раствором NH4OH. Из отгона осаждают Р~-ноны (вместе с CaCOs) действием СаС12, измеряют в осадке активность 18F (Г 1/2= 1,8 час) по аннигиляционному у-пику 0,51 Мэв на сцинтилляционном у-спектрометре и снимают кривую распада при этой энергии.
Для определения химического выхода фтора ('—•20%) осадок после измерения активности обрабатывают уксусной кислотой, остаток отфильтровывают, прокаливают в платиновом тигле, обрабатывают смесью HF и H2SO4 и взвешивают в виде CaSO4. Продолжительность определения около 2 час.
Для определения в кремнии примеси марганца, оказывающего значительное влияние на его свойства как полупроводника, сопоставлены четыре метода активационного анализа: по (3-активности, по (3-активности с фильтром 0,63 г)см\ по у-спектру и по у-спектру без разрушения образца [329]. Предпочтение отдано последнему методу как наиболее простому и быстрому.
Сравнительное изучение методов определения фосфора и бора (10~й—10~7%) в полупроводниковом кремнии несколькими методами, в том числе радиоактивационными, дано в работе [661]; определение мышьяка и серебра см. в [790, 838]; галогенов — в [944], натрия в кремнии и двуокиси кремния — в [938, 986], алюминия в кварце — в [331]. .
Радиоактивационное определение следов элементов с последующим у-спектрометрированием выполнено для трихлоргидросилана после гидролиза основы и испарения раствора [117, 150]. Определяемые количества элементов (в мкг): Na — 0,009; Си — 0,006; Ga — 0,004; As — 0,002; Sb — 0,002 и Мп — 0,03. Применение радиоактивационных методов для анализа кремния и его соединений описано также в работах [192, 230, 231, 235, 330, 447, 573, 924, 926, 943, 947, 1135, 1136].
Другие физические методы
Для определения примесей донорного или акцепторного характера в летучих соединениях кремния высокой чистоты предложен метод, основанный на измерении п- и р-сопротивления слоя кремния, свежеосажденного на монокристаллической кремневой пластинке [906]. Метод проверен при определении РС13 в SiHCl3; установлено, что понижение сопротивления прямо пропорционально концентрации РС13 [575]. Измерение электрических свойств полупроводников для определения примесей описано в работах [362, 478, 1089]. Метод электронного парамагнитного резонанса применен для определения содержания азота в монокристаллах a-SiC [330]. Определение кислорода в кремнии физическими методами и сравнительная характеристика этих методов приведены в работах [723, 730].
ФИЗИКО-ХИМИЧЕСКИЕ И ХИМИЧЕСКИЕ МЕТОДЫ
Обзор физико-химических методов оценки степени чистоты газообразных и жидких продуктов дан в работе [505].
Для определения микроколичеств бора в кремнии предложен спектрофотометрический метод, основанный на предварительном
171
отделении бора от кремния в виде борнометилового эфира и дальнейшей обработке дистиллята раствором куркумина. Описан также капельный вариант этого метода, позволяющий определять 0,05 мкг бора в 0,05 мл раствора реакцией отогнанного борнометилового эфира с куркумовой бумагой или кур кумином [487].
Методом отгонки в форме AsH3 определяют мышьяк с дальнейшим поглощением бромидом ртути (II) полученного дистиллята [274, 542].
Экстракционно-спектрофотометрическое определение фосфора в хлориде кремния выполняют после его окисления до Н3РО4, переведения в фосфорнованадиймолибденовую кислоту и экстракции последней [881].
Для определения РЬ, Си и Bi в полупроводниковых материалах применяют полярографический метод [108]. Метод амальгамной полярографии с накоплением применен для определения микропримесей Bi, Sb и Т1 в трихлоргидросилане [132].
К 2 г двуокиси кремния добавляют 100 мг особочистого КС1, 3 мл конц. HNO3 и эту смесь во фторопластовой чашке вносят во фторопластовую камеру, содержащую 20 мл особочистой конц. HF и нагревают 3 час при 200° С (разложение 10 г SiHCl3 проводят в присутствии 100 мг КС1 после выпаривания пробы досуха при 80° С). Через 2 час в чашку прибавляют 0,5 мл конц. HNO3 и 2 мл конц. HF и выпаривают досуха. К остатку прибавляют полярографический фон, переносят в ячейку и проводят электролиз на ртутной капле, подвешенной на серебряном контакте. Определение висмута проводят на фоне 11V НС1, свинца и таллия — на фоне 0,1 М КС1 с добавлением 10~в М комплексона III. Ошибка определения каждой из примесей = 10%.
Метод применяется также для определения микропримесей в SiCl 4 [ 248].
Фотометрические методы описаны для определения азота [500], фосфора [661, 961], иода [396, 540], тантала [12, 429].
В работе [630] рассмотрены перспективы применения кинетических методов для анализа веществ высокой чистоты, метод применен для определения иода в кремнии [540, 541]. Микроколичества меди (5-Ю-6—5-10-8%) можно определять каталиме-трическим методом [82, 341].
Электрометрическими и другими методами изучено поведение ультрамикропримесей в соединениях кремния в неводных средах [132, 248, 604].
Флуориметрическое определение примесей в кремнии и его соединениях описано в работах [13, 393], экстракционные методы в сочетании с флуориметрией см. в [13, 724].
Определение большой группы примесей в элементном кремнии, иодиде и хлориде кремния химическими методами описано в работе [397].
Другие методы определения микропримесей в твердых, жидких и газообразных продуктах, содержащих кремний, см. в работах [12, 44, 341, 398, 414 , 478, 499 , 591, 599 , 602].
ЛИТЕРАТУРА
1. Абрамов В. В., Богданова Е. Т., Таганов К. И. Зав. лаб., 16, 1218 (1950).
2. Авербух М. М., ЕринаИ. И., Стрельцов И. Г. Зав. лаб., 14, ПО (1948).
3. Айдаров Т. К. Цемент, № 3, 22 (1950).
4. Айлер Р. К. Коллоидная химия кремнезема и силикатов. М., Госстройиз-дат, 1959.
5. Акимов Г. В., Певзнер Л. Э. Зав. лаб., 8, 1273 (1939).
6. Алекин О. А. Химический анализ вод суши. Л., Гидрометеоиздат, 1954.
7. Алексеев Р. И. Зав. лаб., 11, 122 (1945).
8. Алексеенко И. С. Зав. лаб., 18, 178 (1952).
9. АлимаринИ. П. Вести. МГУ, серия II, Химия, № 5, 3 (1963).
10. Алимарин И. П. Ж. аналит. химии, 18, 1412 (1963).
11. Алимарин И. П. Сб. «Качество материалов для полупроводниковой техники (Труды коллоквиума 1957—1958 г.)». М., Металлургиздат, 1959.
12. Алимарин И. П., ГибалоИ.М., Головина А. П., Митцель Ю. А. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 213.
13. Алимарин И. П., Головина А. П., Гибало И. М., Митцель Ю. А. Ж- аналит. химии, 20, 339 (1965).
14. Алимарин И. П., Зверева В. Е. Труды Ин-та прикладной минералогии, 63, 3 (1934).
15. Алимарин И. П., Золотов Ю. А. Зав. лаб., 28, 1285 (1962).
16. АлимаринИ. П., Судаков Ф. П., Клитина В. И. Успехи химии, 34, 1368 (1965).
17. Алимарин И. П., Фрид Б. И. Количественный микрохимический анализ минералов и руд. М., Госхимиздат, 1961.
18. Алимарин И. П., Яковлев Ю. В. Зав. лаб., 26, 915 (1960).
19. Алимарцн И. П., Яковлев Ю. В. Изотопы и излучение в химии. М., Изд-во АН СССР, 1958, стр. 141.
20. Алимарин И. П., Яковлев Ю. В. Сб. «Современные методы анализа». М., «Наука», 1965, стр. 7.
21. Алимарин И. П., Яковлев Ю. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 7.
22. Алимарин И. П., Яковлев Ю. В., Жабин А. И. Сб. «Применение меченых атомов в аналитической химии». М., Изд-во АН СССР, 1955, стр. 58.
23. Аллер Л. Распространенность химических элементов. М., ИЛ, 1963.
24. Алпатов М. С. Сб. «Цветные сплавы», т. 2, вып. 4. М., Оборонгиз, 1949, стр. 52.
25. Анализ минерального сырья, изд. 2. Под ред. Ю. Н. Книпович и Ю. В. Мо-. рачевского. Л., Госхимиздат, 1959.
26. Анализ черных металлов. Под ред. С. В. Липина. М. — Свердловск, Металлургиздат, 1951.
27. Андреев Д. И. Ж- прикл. химии, 28, 641 (1955).
28. Андрианов К. А. Кремнийорганические соединения. М., Госхимиздат, 1955.
29. Аринушкина Е. В., Болтенко Т. П. Вести. МГУ, серия II, Химия, № 3, 105 (1955).
173
30. Арнаутов И. В., Андреева Л. И., Изюмова Л. Г., Симонова В. И, Справочные таблицы основных спектральных линий для полуколичественного анализа минерального сырья. Новосибирск, Изд-во СО АН СССР, 1965, стр. 171.
31. Астафьев В. П. Сб. «Определение свободного кремнезема в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 51.
32. Астреева О. М. Петрография вяжущих материалов. М., Госстройиздат, 1959.
33. Астреева О. М. Справочные таблицы для расчета минералогического состава клинкера и шлака по данным химического анализа. М., Промстройиздат, 1954.
34. Астреева О. М. Цемент, № 3, 30 (1953), № 6, 11 (1953).
35. Астреева О. М., Лопатникова Л. Я. Новое в петрографии цементного клинкера. М., Промстройиздат, 1951.
36. Бабаев М. В. Зав. лаб., 6, 645 (1937); 7, 308 (1938); 7, 601 (1938); 14, 661 (1948) 15, 1193 (1949); 17, 738 (1951); 22, 1294 (1956).
37. Бабачев Г. И. Ж- аналит. химии, 13, 716 (1958).
38. Бабачев Г. Н., Лукашев Л. К. Ръководство за химични и физико-механични наследования на силикатни и карбонатни материали. София, «Техника», 1963.
39. Бабачев Г., Николова А. Строит, материали и силикатна пром-ст. (Бълг.), 7, 18 (1966).
40. Бабко А. К. Ж. прикл. химии, 10, 374 (1937); 11, 41 (1938).
41. Бабко А. К- Зав. лаб., 7, 1121 (1938); 13, 9 (1947); 20, 518 (1954); 21, 269 (1955); 29, 518 (1963).
42. Бабко А. К-, Евтушенко Л. М. Зав. лаб., 23, 423 (1957).
43. Бабко А. К-, Жаровский Ф. Г. Зав. лаб., 25, 42 (1959).
44. Бабко А. К., Маркова Л. В., Цыбина Т. С. Зав. лаб., 30, 648 (1964).
45. Бабко А. К., Пилипенко А. Т. Фотометрический анализ. М., «Химия», 1968.
46. Бабко А. К.., Шановская С. С. Ж. общ. химии, 23, 380 (1953).
47. Бабко А. К., Шановская С. С. Зав. лаб., 18, 1417 (1952).
48. Бабко А. К., Шкаравский Ю. Ф. Ж. неорган. химии, 7, 1565 (1962); 8, 934, 2668 (1963).
49. Бабко А. К.., Шкаравский Ю. Ф. Изв. вузов, Химия и хим. технология, 4, 370 (1961).
50. Бабушкин В. И., Матвеев Г. М., Мчедлов-Петросян О. П. Термодинамика силикатов. М., Госстройиздат, 1962.
51. Баев X. А. Сб. «Определение свободной двуокиси кремния в'горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 96.
52. Бажант В., Хваловски В., Ратоуски Ц. Силиконы. М., Госхимиздат, 1960.
53. БажулинП.А., Баскакова А. А., Строганов А. Р. Ж. техи. физики, 4, 578 (1934).
54. Базилевич А. С. Зав. лаб., 4, 685 (1935).
55. Барьаианская Ф. С. Зав. лаб., 11, 681 (1945).
56. Барышанская Ф. С. Изв. АН СССР, серия физ., 9, 657 (1945).
57. Бахман В. И., Крапивина С. С. Анализ минеральных вод. М., Медгиз, 1963.
58. Безрогова Е. В. Ж- аналит. химии, 19, 1498 (1964).
59. Белецкий М. С. Изв. АН СССР, серия физ., 5, 277 (1941); 9, 627 (1945).
60. Белов И. В. Хим. наука и промышленность, 3, 46 (1958).
61. Белостоцкая И. С. Стекло и керамика, 20, 22 (1963).
62. Белянкин Д. С., Иванов Б. В., Лапин В. В. Петрография технического камня. М., Изд-во АН СССР, 1952.
63. Белянкин, Лапин В. В., Торопов И. А. Физико-химические системы силикатной технологии. М., Промстройиздат, 1954.
64. Белькевич Я. П. Руководство по спектральному анализу металлов. Л., Судпромгиз, 1950.
65. БелькевичЯ- П., Брук Л. Е., СвентицкийН. С. Зав. лаб., 10, 617 (1941)
66. Берг Л. Г. Введение в термографию. М., Изд-во АН СССР, 1961.
67. Берг Л. Г. Скоростной количественный фазовый анализ. М., Изд-во АН СССР, 1952.
174
68. Берг Л. Г., Николаев А. В., РодэЕ.Д. Термография. М.—Л., Изд-во АН СССР, 1944.
69. Берг Р. Применение о-оксихинолина в аналитической химии. М.—Л., ОНТИ, 1937, стр. 18.
70. Бережной А. С. Кремний и его бинарные системы. Киев, Изд-во АН УССР, 1958.
71. Бережной А. С. Огнеупоры, 13, 256 (1948).
72. Беренс Г., Клей П. Д. Микрохимический анализ, ч. I. Л-, Химтехиздат, 1928, стр. 96.
73. Беркович. М. Т. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 69.
74. БерковичМ. Т., Васильева Л. П. Зав. лаб., 18, 179 (1952).
75. Библиографический указатель работ научных сотрудников Ин-та химии силикатов за 1948—1961 гг. Л., Промстройиздат, 1963.
76. Бильтюкова Э. П., Колбасникова А. И., Савицкий М. Р. Стекло и керамика, 22, 47 (1965).
77- Богданова В. В. Цветные сплавы, т. 2. М., Оборонгиз, стр. 57.
78. Богданова И. В., Нещадимова Н. М. Фотометрические и другие экспрессные методы анализа цементных материалов для контроля производства. М., Промстройиздат, 1954.
79. Богданова И. В., Савченко Г. Н. Труды Гипроцемент, 25, 57 (1962).
80. Богданченко А. Г. Зав. лаб., 7, 732 (1938).
81. Божевольнов Е. А. Люминесцентный анализ неорганических веществ. М., «Химия», 1966, стр. 415.
82. Божевольнов Е. А., К Рейнгольд С. У. Сб. «Методы анализа химических реактивов и препаратов», вып. 2. М., Изд. ИРЕА, 1965, стр. 65.
83. Бокий Г. Б. Иммерсионный метод. М., Изд-во АН СССР, 1950.
84. Болдырев В. В. Влияние дефектов в кристаллах на скорость термического разложения твердых веществ. Изд. Томского ун-та, 1963.
85. Борк В. А. Качественный анализ кремнийорганических соединений. Авто-реф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1949.
86. Боровик С. А., Бабаева А. В., Ушакова Н. И., Рудый Р. И. Ж. аналит. химии, 13, 580 (1958).
87. Брадинская Е. М. Сб. научных трудов Иркутского НИИ редких металлов, № 9, 46 (1961).
88. Бруевич С. В. Труды Ин-та океанологии АН СССР, 26, 241 (1958).
89. Бруевич С. В. Химия вод, осадков морей и океанов. М., «Наука», 1964.
90. Бруевич С. В. Таблицы пересчета содержания элементов и ионов в природных водах. М., Изд-во АН СССР, 1962, стр. 16.
91. Брук Л. Е. Изв. АН СССР, серия физ., 5, 331 (1941).
92. Будников П. П., Гинстлинг А. М. Реакции в смесях твердых веществ. М., Стройиздат, 1965, стр. 71.
93. Будников П. П., Жуковская С. С. Ж- прикл. химии, 10, 1560 (1937); 13, 311 (1940).
94. Будников П. П., Жуковская С. С. Зав. лаб., 7, 1124 (1938).
95. Будников П. П., Кассиян Н. С. Зав. лаб., 1, № 4, 39 (1932).
96. Булычева А. И., Мельникова П. А. Сб. «Методы определения запыленности воздуха и свободного кварца в пыли промышленных предприятий». М., Профиздат, 1956, стр. 49.
97. Булычева А. И., Мельникова П. А. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 23.
98. Буракова Т. Н. Кристаллооптические константы и их использование в микрохимическом анализе. Изд-во ЛГУ, 1964.
99. Бусев А. И. Аналитическая химия. Литература на русском языке (1941 — 1952 гг.). М., Изд-во АН СССР, 1956.
100. Бут Т. С., Виноградов Б. Н., Гаврилова Т. И., Горшков В. С., Долгополов Н. Н., Мягкова М. А., Сироткина Н. Л., Фадеева В. С. Современные методы исследования строительных материалов. М., Госстройиздат, 1962.
175
101. Бутлеров А. М. ЖРФХО, 12, 37 (1880).
102. Буянов Н. В. Зав. лаб., 14, 565 (1948); 19, 554 (1953).
103. Буянов Н. В. Изв. АН СССР, серия физ., 12, 439 (1948).
104. Буянов И. В. Труды ЦНИИ черных металлов, вып. 31, 67 (1963).
105. Буянов Н. В., Иванова Л. А., Сухова Н.П, Там же, стр. 29.
106. Буянов Н. В., ИвановаЛ.А., Сухова Н.П., Т имошенко Н. Н. Там же, стр. 19.
107. Бычков М. К-, Иванова А. И. Зав. лаб., 5, 500 (1936).
108. Вайнштейн Ю. И., Гинзбург К. Я- Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 64.
109. Варшавская Л. И., Иванцов Л. И., Полякова В. В. Зав. лаб., 16, 48 (1950).
НО. Василевская Л. С. Сб. «Методы анализа веществ высокой чистоты». М.,
«Наука», 1965, стр. 16.
111. Василевская Л. С.,
112. Василевская Л. С., Там же, стр. 71.
113. Василевская Л. С., Сб. «Получение и стр. 128.
114. Василевская Л. С.,
Жукова Л. К- Там же, стр. 493.
Кондрашина А. И., Макарова Г. А., Панарина Н. А.
Кондрашина А. И., Макарова Г. А., ПанаринаН.А.
анализ веществ особой чистоты». М., «Наука», 1966,
Кондрашина А. И., Шифрина Г. Г. Сб. «Методы ана-
лиза веществ высокой чистоты». М., «Наука», 1965, стр. 84, 86.
115. Василевская Л. С., Муравенко В. П., Кондрашина А. И. Ж- аналит. химии, 20, 540 (1965).
116. Василевская Л. С., Муравенко В. П., Кондрашина А. И. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 76.
117. Василевская Л. С., Садофьева С. А. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 121.
118. Васильев К. А., Баринова О. Д. Зав. лаб., 8, 916 (1939).
119. Васильев К- А., ВегринМ.Л. Зав. лаб., 7, 263 (1938).
120. Васильев П. И. Методы ускоренного анализа карбонатных пород. М., Гос-геоллитиздат, 1955.
121. Ватанабэ X. Бунсэки кагаку, 12, 1196 (1963).
122. Введенский Л. Е., Мендельштам С. Л., Райский С. М., Смирнов В. Ф., Строганов А. Р., Сухенко Н. А. Методы спектрального анализа металлов. М., Гостехиздат, 1940.
123. Вейцман Р. М. Зав. лаб., 23, 153 (1957).
124. ВеприцкаяВ. Зав. лаб., 4, 1514 (1935).
125. Веприцкая В. Огнеупоры, 45, 284 (1936).
126. Вернадский В. И. Очерки геохимии. М., Горногеолнефтьиздат, 1934.
127. Веселовская И. М. Зав. лаб., 13, 219 (1947); 15, 940 (1949).
128. Винковецкая С. Я. Бюлл. н.-техн. информации Гос. геологич. комитета СССР. Отд. техн, информации ВИЭМС, № 3 (53), 92 (1964); РЖХим., 1965, 20 Г 114.
129. Виноградов А. П. Геохимия, № 1, 6 (1956).
130. Виноградов А. П. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 5.
131. Виноградов А. П. Сб. «Методы определения и анализа редких элементов». М., Изд-во АН СССР, 1961, стр. 10.
132. Виноградова Е. И., Каменев А. И. Труды Комиссии по аналит. химии АН СССР, 15, 175 (1965).
133. Висьневский Я- С. Универсальные таблицы для пересчета минералогического состава горных пород на химический состав и химического состава на минералогический. Ташкент, «Наука», 1965.
134. Волынец М. И. Зав. лаб., 5, 162 (1936).
135. ВолынецМ.И. Укр. хим. ж., 11, 18 (1936).
136. Волынец М. И., Бернштейн С. С. Зав. лаб., 5, 1071 (1936).
137. Вопросы металлургии и физики полупроводников. Полупроводниковые соединения и твердые сплавы (сб. статей). М., Изд-во АН СССР, 1957.
138. «Вопросы минералогии глин» (сб. статей). М., ИЛ, 1962.
139. Воронков М. Г. Труды конференции по химии и практическому применению кремнеорганических соединений, вып. 6. Л., изд. ЦБТИ, 1958, стр. 136.
176
140. Воронков М. Г. Химия кремнеорганических соединений в работах русских и советских ученых. Изд-во ЛГУ, 1952.
141. Воронков М. Г., Карпенко Г. В. Ж- общ. химии, 27, 325 (1957).
142. Воронков М. Г., Романова Н. Г. Ж- общ. химии, 8, 2122 (1958).
143. Воронков М. Г., ХудобинИ.Н. Ж- общ. химии, 26, 584 (1956).
144. Воронков М. Г., Якубовский А. И. Ж. общ. химии, 25, 1124 (1955).
145. ГалаховФ.Д. Зав. лаб., 6, 1011 (1937).
146. Гаррисон Д., ЛордР., Луфбуров Д. Практическая спектроскопия. М., ИЛ, 1950.
147. Гедройц К. К- Химический анализ почв. М.—Л., Сельколхозгиз, 1932,
148. Гиллебранд В. Ф. Химия силикатов. Методы анализа силикатных и карбонатных пород. Л., Химтехиздат, 1929.
149. Гиллебранд В. Ф., Лендель Г. Э., Брайт Г. А., Гофман Д. И. Практическое руководство по неорганическому анализу. М., «Химия», 1966.
150. Гильберт Э. Н., Пронин В. А., Артюхин П. И., Николаев А. В. Докл. АН СССР, 172, 853 (1967).
151. Гинзбург Л. Б. Зав. лаб., 7, 1041 (1938).
152. Гинзбург В. Л., Глуховецкая Н. П. Зав. лаб., 26, 559 (1960).
153. Глубина Н. И. Фотоколориметрический метод определения кремния в силикатах и кремнийорганических соединениях. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1955.
154. Глухов А. Д. Изв. АН СССР, серия физ., 9, 619 (1945).
155. Голутвин Ю. М. Ж- физ. химии, 30, 2251 (1956).
156. Гончаров В. В., Шмитт-Фогелевич С. П., Бородачева М. Е. Зав. лаб.,
30, 729 (1964).
157. Горбатый Ю. Е., Эпельбаум М. Б. Сб. «Металлургические шлаки и их применение в строительстве». М., Госстройиздат, 1962, стр. 438.
158. Горбунова Л. Б., Конькова Е. С., Кутейников А. Ф. Зав. лаб., 30, 38 (1964).
159. Горбунова Л. Б., Конькова Е. С., Кутейников А. Ф. Сб. «Конструкционные углеграфитовые материалы». М., «Металлургия», 1964, стр. 302.
160. Горшков В. С., ТимашевВ.В. Методы физико-химического анализа вяжущих веществ. М., «Высшая школа», 1963.
161. Григорьев П. Н. Химический анализ строительных материалов. М.—Л., ОНТИ, 1932.
162. Григорьев П. Н., Кожина Е. И. Зав. лаб., 6, 751 (1937).
163. Григорьев П. В. Матвеев М. А. Растворимое стекло. М., Госстройиздат, 1956.
164. Григорьев П. Н., Пожарская П. И. Зав. лаб., 5, 1443 (1936).
165. Гринберг А. А. Введение в химию комплексных соединений. М.—Л., «Химия», 1971.
166. Гровс А. Анализ силикатов. М., ИЛ, 1953.
167. Гурвиц С. С., ПодгайцВ.В. Зав. лаб., 14, 935 (1948).
168. Гурецкий И. Д. Определение кремния в кремнийорганических соединениях. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1951.
169. Давыдов А. Л., КочуговаЕ.И. Зав. лаб., 8, 1308 (1939).
170. Данильченко Е. П., Репа А. Г. Стекло и керамика, № 8, 10 (1950).
171. Дегтяренко Д. А. Применение метода ионного обмена для определения бора, фосфора и кремния. Автореф. канд. дисс. Киев, ИОНХ АН УССР, 1954.
172. Дегтяренко Д. А., Ощаповский В. В. Научн. докл. Львовского политехи, ин-та, 22, 107 (1956).
173. Добкина Б. М. Зав. лаб., 14, 755 (1948).
174. Добровольская В. В. Сб. «Материалы совещания по промсанхимии». М., Изд-во АМН СССР, 1954.
175. Добровольская В. В. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 15.
176. Долгов Б. Н. Ж. общ. химии, 10, 1771 (1940).
177. Долгов Б. И. Успехи химии, 1, 626 (1940).
178. Долгов Б. Н. Химия кремнеорганических соединений. М.—Л., ОНТИ, 1933.
177
179. Дорохова Е. Н. Химико-аналитическое изучение условий образования и восстановления кремнемолибденовой кислоты. Автореф. канд. дисс. МГУ, 1965.
180. Дорохова Е. Н., Шахова З.Ф., Чуян Н. К.. Вести. МГУ, серия «Химия»? № 4, 85 (1966).
181. ДруцкаяЛ. В. Изв. АН СССР, серия физ., 19, 103 (1955).
182. Дымов А. М. Зав. лаб., 1,№10, 45 (1932); 6,359 (1937); 10,661 (1941); 21, 504 (1955).
183. Дымов А. М. Зав. лаб., 23, 1523 (1957).
184. Дымов А. М. Технический анализ. М., «Металлургия», 1964, стр. 116.
185. Дымов А. М. Технический анализ руд и металлов. М, Металлургиздат, 1949.
186. Егорова Е. Н. Изв. АН СССР, ОХН, 3, 419 (1953).
187. Егорова Е. И. Изв. АН СССР, ОХН, 1, 16 (1954).
188. Егорова Е. Н. Методы выделения кремневой кислоты и аналитического определения кремнезема. М.—Л., Изд-во АН СССР, 1959.
189. Егорова Е. И., Голубева Л. Г. Огнеупоры, № 9, 51 (1967).
190. Енгалычев И. М., Ефанова М. Д. Зав. лаб., 18, 1089 (1952).
191. Ермолаева Е. В. Методы химического анализа огнеупорного сырья и изделий. Харьков, Металлургиздат, 1954.
192. Ерохина К. И., Лемберг И. X., Макашева И. Е., Маслов И. А., Обухов А. П. Зав. лаб., 26, 821 (1960).
193. ЕсинО.А., Гаврилов Л. К.., Ленинских Б. М. Докл. АН СССР, 88, 713 (1953).
194. Ефимов Е. А., Ерусалимчик И. Г. Электрохимия германия и кремния. М., Госхимиздат, 1963.
195. ЕфимовЕ.А., Ерусалимчик И. Г., Соколова Г. П. Ж. физ. химии, 36, 1005(1962).
196. Жданов А. А. Исследования в области полиэлементоорганосилоксанов. Автореф. докт. дисс. М., ИНЭОС АН СССР, 1968.
197. Живописцев В. П. Зав. лаб., 31, 1043 (1965).
198. Жуковская С. С. Труды Южгипроцемент, 4, 153 (1963).
199. Жуковская С. С., Водолаженко Н. И., Левина К. Д. Бюлл. техн, информации Южгипроцемент, 25 , 43 (1958).
200. Жуковская С. С., Левина К. Д., Водолаженко Н. И. Цемент, № 3, 21 (1961).
201. Журавлев Г. И., Терещенко П. Н. Зав. лаб., 14, 1101 (1948).
202. Журавская В. И. Зав. лаб., 16, 873 (1950).
203. Заварзина Е. И. Стекло и керамика, 21, 20 (1964).
204. Завьялова Л. Л., ИвойловА. С., Шаронова А. В. Зав. лаб., 33, 303 (1967).
205. Загоровская А. А. Исследование продуктов взаимодействия некоторых кремнийсодержащих соединений с солями неорганических кислот в глицериновых растворах. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1963.
206. Зайдель А. Н., Прокофьев В. К., Райский С. М. Таблицы спектральных линий. М., ГИТЛ, 1952.
207. ЗайковскийФ. В., Садова Г. Ф., Фуртова Е. В. Зав. лаб., 27, 1474 (1961).
208. Зайцев М. И. Зав. лаб., 3, 492 (1934).
209. Закирова Ф. 3., Овсянников В. Н., Фишман Н. С. Труды 4-го Уральского совещания по спектроскопии. Свердловск, «Металлургия», 1963, стр. 157.
210. Занько А. А., Сердюкова О. К- Докл. Львовского политехи, ин-та 1, 96 (1955).
211. Занько А. А., Сердюкова О. К. Докл. Львовского политехи, ин-та, 5, 165 (1963).
212. Занько А. М., БутенкоГ.А. Зав. лаб., 4, 1188 (1935).
213. Заринский В. А., Гурьев И. А. Ж. аналит. химии, 18, 1306 (1963).
214. Заринский В. А., Фарафонов М. М., ЗатееваВ. В. Ж. аналит. химии, 12, 677 (1957).
215. Засухин Э. Н., Калинин А. И., Кузнецов Р. А., Моисеев В. В. Сб. «Радиохимические методы определения микроэлементов». М.—Л., «Наука», 1965, стр. 168.
216. Зевин Л. С., Хайкер Д. М. Рентгеновские методы исследования строительных материалов. М., Стройиздат, 1965.
178
217. Зельдин Н. О. Ускоренные методы химического анализа огнеупорных изделий, материалов и сырья. М.—Харьков, Металлургиздат, 1946.
218. Зильберштейн X. И. Зав. лаб., 19, 443 (1953).
219. Зильберштейн X. И., Калитеевский И. И., Разумовский А. Н., Федоров Ю. Ф. Зав. лаб., 28, 43 (1962).
220. Зильберштейн X. И., ПирюткоМ.М., Никитина О. И., Федоров Ю.Ф. Зав. лаб., 28, 680 (1962); 29, 1266 (1963); 25, 1474 (1959).
221. ЗолотовЮ. А. Труды Комиссии по аналит. химии АН СССР, 15, 3 (1965).
222. ИвойловА. С. Рентгенографическое определение содержания кварца в рудной пыли. Изд. Иркутского НИИ редких металлов, 1954.
223. Ивойлсв А. С., Денисова. Л. Л. Зав. лаб., 28, 700 (1962).
224. Ивойлов А. С., Денисова Л. Л. Научн. труды Иркутского НИИ редких металлов, 10, 61 (1961).
225. Ильин А. И. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 72.
226. Ильин Н. И. Сб. «Современные методы, анализа». М., «Наука», 1965, стр. 59.
227. Инсли Г., Фрешетт В. Д. Микроскопия керамики, цементов, стекол, шлаков и формовочных песков. М., Госстройиздат, 1960.
228. ИольсонЛ.М., Дубовицкая Э. И., Граф Э. К. Зав. лаб., 5, 1053 (1936).
229. Иоффе А. Ф. Физика полупроводников. М., Изд-во АН СССР, 1957, стр. 200.
230. Исаева Е. А., Макашева И. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 103.
231. Исаева Е. А., Макашева И. Е., Маслов И. А. Там же, стр. 100.
232. Йоу Д. Колориметрия. М., ИЛ, 1935, стр. 240.
233. Калинин А. И. Нейтроноактивационное определение микропримесей в кремнии и его соединениях с применением ионообменной хроматографии. Автореф. канд. дисс. Л., Ин-т химии силикатов им. И. В. Гребенщикова, 1965. Р
234. Калинин А. И., Кузнецов Р. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 93.
235. Калинин А. И., Кузнецов Р. А., Моисеев В. В. Радиохимия, 4 (5), 575 (1962).
236. Калинин А. И., Кузнецов Р. А., Моисеев В. В. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 108.
237. Калинин А. И., Кузнецов Р. А., Моисеев В. В. Сб. «Радиохимические методы определения микроэлементов». М.—Л., «Наука», 1965, стр. 171, 176.
238. Калинин А. И., Кузнецов Р. А., Моисеев В. В., Мирин А. И. Докл. АН СССР, 141, 98 (1961).
239. Калинин А. И., Кузнецов Р. А., Моисеев В. В., Соколова М. И. Сб. «Радиохимические методы определения микроэлементов». М.—Л., «Наука», 1965, стр. 180.
240. Калинин А. И., Кузнецов Р. А., Моисеев В. В., Цепурнек В. Э. Сб. «Радиохимические методы определения микроэлементов». М.—Л., «Наука», 1965, стр. 161.
241. Калинин С. К., Марзуванова В. Л., ФайнЭ.Е. Спектральные линии для анализа минерального сырья. Алма-Ата, Изд-во АН КазССР, 1957.
242. Калинин С. К-, НаймаркЛ.Э., Марзуванова В. Л., И смогу лова К. И. Атлас спектральных линий для стеклянного спектрографа. М., Госгеолтех-издат, 1956.
243. Калинин С. К., Файн Э. Е. Спектральный анализ минерального сырья. Алма-Ата, Изд-во АН КазССР, 1962.
244. Калинин С. К., Декель А. А., Алексеева А. И., НаймаркЛ.Э. Атлас спектральных линий для кварцевого спектрографа (2050—6900 А). М., Гостехиздат, 1952.
245. Калинский Д. М. Зав. лаб., 24 , 755 (1955).
246. Калужская В. М. Зав. лаб., 3, 908 (1934).
247. Карасюк Б. А., Грибов А. И. Полупроводники — германий и кремний. М., Атомиздат, 1961.
248. Карбаинов Ю. А., Стромберг А. Г. Изв. Томского политехи, ин-та 128, 75 (1964).
179
249. Кацнельсон Э. М. Обогащение руд, № 1 (49), 38 (1964).
250. Качество материалов для полупроводниковой техники (сб. статей). М., Металлургиздат, 1959.
251. Келер Э. К., Кузнецов А. К. Докл. АН СССР, 88, 1031 (1953).
252. КельцеваЗ. А. Изв. АН СССР, серия физ., 19, 100 (1955).
253. Кирин И. С., Свентицкий Н. С. Зав. лаб., 9, 1270 (1940).
254. Китайгородский А. И. Рентгеноструктурный анализ мелкокристаллических и аморфных тел. М.—Л., Гостехиздат, 1952.
255. Клер М. М. Изв. АН СССР, серия физ., 18, 291 (1954).
256. Климова В. А.,
257. Климова В. А.,
258. Климова В. А., 11, 223 (1956);
259. Климова В. А., микроанализа.
Березницкая И. Г. Ж. аналит. химии, 11, 292 (1956).
Березницкая И. Г. Ж. аналит. химии, 12, 424 (1957).
Коршун М. О., Березницкая И. Г. Ж. аналит. химии, Докл. АН СССР, 96, 81 (1954).
Коршун М. О., Гельман Н. Е. Новый метод элементарного М., Госхимиздат, 1949.
260. Ключаров Я- В., Хлебникова И. Ю. Ж. прикл. химии, 38, 1139 (1965).
261. Клячко Ю. А., Атласов А. Г., Шапиро М. М. Анализ газов, неметаллических включений и карбидов в стали. М., Металлургиздат, 1953.
262. КобякГ. Г., ПоплевинаЛ. В. Уч. зап. Пермского ун-та, № 111, 126 (1964).
263. Ковалев Н. А., Жиглов Ю. С. Зав. лаб., 19, 179 (1963).
264. Козлова А. В., КоржП.Д. Сб. научи, трудов Магнитогорского горно-металлургич. ин-та, вып. 16, 132 (1958).
265. Кокорин А. И., Васильев И. Д. Зав. лаб., 12, 123 (1946).
266. Колобова К- К., Герасимова В. А. Зав. лаб., 22, 286 (1956).
267. Колобова К- К-, Яковлева В. С. Огнеупоры, № 2, 56 (1966).
268. Колориметрические (фотометрические) методы определения неметаллов. М., ИЛ, 1963.
269. КольтгофИ. М., Сендэл Е. Б. Количественный анализ. М.—Л., Госхимиздат, 1948.
270. Кондратьев В. Н. Успехи химии, 26, 861 (1957).
271. Кониси Ю. Ёгё кёкайси, 66, 155 (1958).
272. Коновалов П. Ф., Волконский Б. В., Хашковская А. П. Атлас микроструктур цементных клинкеров, огнеупоров и шлаков. М.— Л., Госстройиздат, 1962.
273. Конопелька И. А., Ткачев Л. И., Раецкая Д. Я- Инж.-физ. ж., 1, 109 (1958).
274. Коренман И. М. Количественный микрохимический анализ. М., Госхимиздат, 1948, стр. 283.
275. Коренман И. М. Микрокристаллоскопия. М., Госхимиздат, 1947.
276. Коренман И. М., Кожухин А. А. Зав. лаб., 9, 43 (1940).
277. Корж П. Д. Зав. лаб., 10, 1215 (1950); 11, 319, 1140 (1945); 13, 65 (1947);
15, 170 (1949); Изв. АН СССР, серия физ., 14, 685 (1950).
278. КоржП. Д., Ершова А. П. Зав. лаб., 18, 441 (1952).
279. КоржП.Д., Козлова А. В. Зав. лаб., 15, 937 (1949).
280. Корицкий В. Г. Изв. АН СССР, серия физ., 9, 647 (1945).
281. Корниенко Г. Г. Исследование фазового состава портландцементного клинкера и высокоглиноземистого шлака методами химического анализа. Автореф. канд. дисс. М., ВНИИ стекла, 1955.
282. Корниенко Г. Г. Сб. трудов ВНИИ новых строительных материалов, 4, 88 (1961).
283. Корнилович Ю. Е., Дымченко В. Г. Зав. лаб., 29, 201 (1963).
284 Коршун М. О., Гельман Н.Э., Шевелева Н. С. Ж. аналит. химии, 13, 6952 (1958).
285. Коршун М. О., Климова В. А., Березницкая И. Г. Докл. АН СССР, 84, 1175 (1952).
286. Косолапова Т. Я-, Котляр Е. Е. Зав. лаб., 24, 1442 (1958).
287. Косолапова Т. Я-, КугайЛ.Н., Модылевская К. Д-, Радзиковская С. В., Серая О. Г. Бюлл. Ин-та металлокерамики и спецсплавов АН УССР, № 6, 69 (1961).
288. Костецкий Б. И., Белицкий М. Е., Натансон М. А. Порошковая металлургия, № 2, 40 (1962).
180
289. Кошелева М. М., Чернецова В. И. Зав. лаб., 21, 460 (1955).
290. Красновский О. В. Инструкция по ускоренному анализу кремнеалюмо-
291.
292.
293.
294.
295.
296.
297.
298.
299.
300.
301.
302.
303.
304.
305.
306.
307.
308.
309.
310.
311.
312.
313.
314.
кальций-магний-натриевого стекла и силикатных сырьевых материалов. М., Промстройиздат, 1952.
Красновский О. В., Гастева Е. В., Герасимова Г. И. Стекло. Бюлл. ВНИИ стекла, № 4 (117), 38 (1962).
Краснощеков В. В. Определение кремния в кремнийорганических соединениях. Канд. дисс. М., МХТИ им. Д. И. Менделеева, 1964.
Кремний (сб. статей). М., ИЛ, 1960.
Крешков А. П., Борк В. А. Ж- аналит. химии, 6, 78 (1951).
КрешковА. ГЕ, Борк В. А. Успехи химии, 28, 576 (1959).
Крешков А. ГЕ, Борк В. А., Бондаревская Е. А., Мышляева Л. В., Сявцил-ло С. В., Шемятенкова В. Т. Практическое руководство по анализу мономерных и полимерных кремнийорганических соединений. М., Госхимиздат, 1962.
КрешковА. ГЕ, Борк В. А., Мышляева Л. В., Нессонова Г. Д. Анализ кремнийорганических соединений. М., Госхимиздат, 1954.
Крешков А. П., Быкова Л. ЕЕ, Казарян И. А. Кислотно-основное титро-
вание в неводных растворах. М., «Химия», 1967. Крешков А. ГЕ, Мышляева Л. В. Труды МХТИ им. 25, 33 (1957).
Крешков А. П., МышляеваЛ. В., ГеншафтЮ.С., Ж. неогран, химии, 9, 183 (1964).
Крешков А. П., Мышляева Л. В., Краснощеков В. В.
№ 151858; Бюлл. изобр., Ns 22, 50 (1962).
Крешков А. ГЕ, МышляеваЛ. В., Краснощеков В. В.
Ns 161956. Бюлл. изобр., № 8, 2 (1964).
Крешков А. ГЕ, Мышляева Л. В., Краснощеков В. В.
(1963).
Крешков А. ГЕ, Мышляева Л. В., 51 (1962).
Крешков А. ГЕ, МышляеваЛ. В., 65 (1964).
Крешков А. ГЕ, МышляеваЛ. В.,
Столбов В. Ф.,
Д. И. Менделеева, Краснощеков В. В. Авт. свид. СССР, Авт. свид. СССР, Зав. лаб., 29, 924
Краснощеков В. В. Пластмассы, № 12, Краснощеков В. В. Пластмассы, № 1, Саюшкина Е. ЕЕ, Краснощеков В. В.,
Седова И. В. Вести, технической экономической информа-
ции, № 9, 35 (1964).
Крешков А. ГЕ, Мышляева Л. В., Хачатурян О. Б., Краснощ/еков В. В.
Ж. аналит. химии, 18, 1375 (1963).
Крешков А. ГЕ, Мышляева Л. В., ХачатурянО. Б., Краснощеков В. В.
Изв. вузов, Химия и хим. технология, 7, 198 (1964).
Крешков А. ГЕ, Сявцилло С. В., Шемятенкова В. Т. Зав. лаб., 22, 1425 (1956).
Крешков А. ГЕ, Чивикова А. ЕЕ, Загоровская А. А. Ж. аналит. химии, 20, 1253 (1965).
Кротова И. К-, Кошелева М. М. Сб. «Новые методы анализа и исследования в основной химии». М., изд. НИИУИФ, 1962, стр. 3.
КрюковаТ.А., Синякова С. И., АрефьеваТ. В. Полярографический анализ. М., Госэнергоиздат, 1959.
КугайЛ. ЕЕ, Косолапова Т. Д. Металлокерамические материалы и методы их исследования. Информ, материалы АН УССР, 1959, стр. 7.
Куделя Е. С. Автогенная сварка, 10, 99 (1950).
315. Кузнецов В. И. Успехи химии, 23, 654 (1954).
316. Кульберг Л. М., Альтерзон Г. С., Велыпман Р. П. Капельный анализ-
М.—Л., Госхимиздат, 1951.
317. Купфер С. М. Стекло и керамика, № 4, 35 (1959).
318. Кутателадзе К- С., Меликадзе В. Д. Научн. сообщения ВНИИ цементной промышленности, 3 (34), 18 (1958).
319. Кучкарев Е. А. Исследование в области элементарного и молекулярного эмиссионного спектрального анализа неводных растворов органических
8 Аналитическая химия кремния
181
<5-2,
и элементоорганических соединений. Канд. дисс. М., МХТИ им. Д. И. Менделеева, 1966.
320. Ландсберг Г. С., Мандельштам С. А., Райский С. Н. Ж. техн, физики, 3, 771 (1933).
321. Лапин В. В. Труды Ин-та геол, наук АН СССР, вып. 2, 5 (1956).
322. Лаптев Ф. Ф. Анализ воды. М., Госгеолтехиздат, 1955.
323. Латимер В. М. Окислительные состояния элементов и их потенциалы в водных растворах. М., ИЛ, 1954, стр. 258.
324. Лендель Г., Гофман Д., Брайт Г. Анализ черных металлов. М.—Л., ОНТИ, 1934.
325. Ле Шателье А. Кремнезем и силикаты. Л., ОНТИ, 1929, стр. 24.
326. Лисицкий Н. Н. Зав. лаб., 5, 366 (1936).
327. Лобанов Е. М., Звягин В. И., Зверев Б. П. Сб. «Радиационные эффекты в конденсированных средах». Ташкент, «Наука», 1964, стр. 74.
328. Лобанов Е. М., Звягин В. И., Зверев Б. П., Блинков Д. И. Там же, стр. 64.
329. Лобанов Е. М., Звягин В. И., Диет А. А., Зверев Б. П., Свиридова А. И., Московцева Г. А. Ж- аналит. химии, 18, 1349 (1963).
330. Лобанов Е. М., Леушкина Г. В. Сб. «Ядерная физика и ее применение», ч. 1. Ташкент, «Фан», 1966, стр. 89.
331. Лобанов Е. М., Чанышев А. И., Дутов А. Г., Худайберганов А., Аширов М. Г. Труды Всес. координац. совещания по активационному анализу, 1962. Ташкент, «Наука», 1964, стр. 91.
332. Лопатникова Л. Д. Труды ВНИИ цементной промышленности, 18, 75 (1963).
333. Лохов П. В. Зав. лаб., 14, 628 (1948).
334. Лошкарев А. Г. Ж. прикл. химии, 31, 1175 (1958).
335. Ляссьёр А. Анализ силикатов. М., ИЛ, 1954.
336. Малкова О. П., Рудневский Н. Д. Ж. аналит. химии, 11, 135 (1956).
337. Малкова О. П., Рудневский Н. Д. Изв. АН СССР, серия физ., 19, 224 (1955).
338. Мальцев В. Ф. Зав. лаб., 22, 1044 (1956); 24, 537 (1958).
339. Мандельштам С. Л. Введение в спектральный анализ. М., Гостехиздат, 1946.
340. Мартынов Ю. М., Дорнблит И. И., Смирнова Н. П., Джагацпанян Р. В. Зав. лаб., 27 , 839 (1961).
341. Мартынов Ю. М., Дрейнгольд Е. А., Маевская Б. М. Зав. лаб., 31, 1447 (1965).
342. Массильон Т. Д. Спектральный анализ в цементной промышленности. М., Промстройиздат, 1956.
343. Матвеев М. А., Г луд ина Н. И. Сб. «Физико-химические основы керамики». М., Промстройиздат, 1956, стр. 556.
344. Матвеев М. А., Глудина Н. И. Труды МХТИ им. Д. И. Менделеева, 21, 49 (1956).
345. Матвеев М. А., Мазо Э. Э., Дачур Ф. Т. Стекло и керамика, 20, № 10 (1963); № 11, 9 (1963).
346. Матвеев М. А., Рабухин А. И. Ж. ВХО им. Д. И. Менделеева, 6, 592 (1961).
347. Матросова Т. В., Зубкова 3. А. Зав. лаб., 31, 945 (1965).
348. Менделеев Д. И. Основы химии, ч. I. Сочинения, т. 13. М., 1949, стр. 88.
349. Менковский М. А., Чурбаков В. Ф. Зав. лаб., 28, 1102 (1962).
350. Методическое руководство по петрографическому изучению глин. М., Госгеолтехиздат, 1957.
351. Методы анализа веществ высокой чистоты (сб. статей). М., «Наука», 1965.
352. Методы изучения вещественного состава и их применение, вып. I. Новосибирск, «Наука», 1965.
353. Методы определения и анализа редких элементов (сб. статей). М., Изд-во АН СССР, 1961.
182
354. Методы химического анализа и химический состав минералов (сб. статей). М., «Наука», 1967.
355. Методы химического анализа минерального сырья, вып. 6. М., Гостеолтех-издат, 1960.
356. Методы химического анализа огнеупорного сырья и изделий (сб. статей). М., Металлургиздат, 1954.
357. Миклашевский А. И. Зав. лаб., 6, 1209 (1937).
358. Миклашевский А. И. Зав. лаб., 7, 168, 1433 (1938).
359. Миклашевский А. И. Карборунд. М.—Л., ГОНТИ СССР, 1938.
360. Миронов Д. П. Зав. лаб., 29, 438 (1963).
361. Мисуми С., Тарутани Т. Бунсэки кагаку, 10, 1113 (1961).
362. Митиель Ю. А., Алимарин И. П., Гибало И. М., Головина А. П. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 57.
363. Михеев В. И., Сальдау Э. П. Рентгенометрический определитель минералов, т. 2. Л., «Недра», 1965.
364. Михеева Л. М., Михеев Н. Б. Радиоактивные изотопы в аналитической химии. М., Госатомиздат, 1961.
365. Момоки К., Сэкино Д. Бунсэки кагаку, 12, 560 (1963).
366. Морачевский Ю. В., Егорова Е. Н. Докл. АН СССР, 122, 612 (1958).
367. Морачевский Ю. В., Егорова Е. Н. Ж- прикл. химии, 32, 1925 (1959).
368. Морачевский Ю. В., Зильберштейн X. И.’, Пирютко М. М., Никитина О. Н. Вести. ЛГУ, № 22, 140 (1962).
369. Морачевский Ю. В., Зильберштейн X. И., Пирютко М. М., Никитина О. Н. Ж. аналит. химии, 17, 614 (1962).
370. Морачевский Ю. В., Пирютко М. М. Изв. АН СССР, ОХН, 8, 834 (1956).
371. Морачевский Ю. В., Христофоров Б. С., Винке Л. К., Шенфельд М. С., Шабанов В. Н. Сб. «Фазовый анализ руд и минералов». Изд-во ЛГУ, 1962, стр. 17.
372. Моррисон Д., Фрейзер Г. Экстракция в аналитической химии. Л., Гос-химиздат, 1960.
373. Мустафин И. С., Молот Л. А. Сб. «Методы контроля химического состава неорганических и органических соединений». Куйбышев, изд. Средневолжского ЦБТИ, 1966, стр. 182.
374. Мышкин С. Н. Огнеупоры, № 6, 286 (1960).
375. Мышкин С. Н. Огнеупоры, № 5, 239 (1961).
376. Мышляева В. В. Химия и индустрия (Бълг.), 34, 101 (1962).
377. Мышляева В. В., Мышляева Л. В., Лукина М. Н. Современные ускоренные методы химического анализа материалов цементного производства (обзор). М., изд. ЦНИИТЭСТРОМ, 1969.
378. Мышляева В. В., Нагерова Э. И. Труды ВНИИ цементной промышленности, 16 , 66 (1962).
379. Мышляева В. В., Нагерова Э. И. Ускоренный химический анализ материалов цементного производства. М., изд. ВНИИ цементной промышленности, 1959.
380. Мышляева В. В. Нагерова Э. И., Лукина М. Н. Научн. сообщ. ВНИИ цементной промышленности, 12 (43), 39 (1961).
381. Мышляева Л. В. Исследование связующих свойств эфиров ортокремневой кислоты. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1948.
382. Мышляева Л. В., Краснощеков В. В. Труды МХТИ им. Д. И. Менделеева, 42, 178 (1963).
383. Мышляева Л. В., Краснощеков В. В., Седова И. В. Техническая и экономическая информация, серия «Методы анализа и контроля производства в химической промышленности», вып. 5. М., изд. НИИТЭХИМ, 1964, стр. 30.
384. Мышляева Л. В., Краснощеков В. В., Седова И. В. Труды МХТИ им. Д. И. Менделеева, 44, 132 (1963).
385. Мышляева Л. В., Максимова Т. Г. Техническая и экономическая информация, серия «Методы анализа и контроля производства в хим. промышленности», вып. 4. М., изд. НИИТЭХИМ, 1968, стр. 12.
386, Мышляева Л. В., Седова И. В. Зав. лаб., 34, 263 (1968).
в*
183
387. Мышляева Л. В., Седова И. В., Кузина Л. В., Краснощеков В. В., От-рощенко И. Г., Пекшева В. В. Техническая и экономическая информация, серия «Методы анализа и контроля производства в химической промышленности, вып. 4. М., изд. НИИТЭХИМ, 1966, стр. 19.
388. Нагерова Э. И. Труды 3-го Всес. совещания зав. лабораториями цементной промышленности. Л., Промстройиздат, 1945, стр. 81.
389. Нагерова Э. И., Лебедева А. Д. Цемент, № 2—3, 23 (1941).
390. Назаренко В. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 34.
391. Назаренко В. А. Хим. наука и промышленность, 4, 235 (1959).
392. Назаренко В. А., Винковецкая С. Д. Зав. лаб., 19, 386 (1953).
393. Назаренко В. А., Винковецкая С. Д., Равицкая Р. В. Укр. хим. ж., 28, 726 (1962).
394. Назаренко В. А., Полузктов Н. С. Полумикрохимический анализ минералов и руд. М., Госхимиздат, 1950.
395. Назаренко В. А., Флянтикова Г. В. Зав. лаб., 24, 663 (1958).
396. Назаренко В. А., Шустова М. Б. Зав. лаб., 27, 15 (1961).
397. Назаренко В. А., Шустова М. Б., Лебедева Н. В., Шитарева Г. Г., Бирюк Е. А., Равицкая Р. В. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 40.
398. Назаренко В. А., Шустова М. Б., Флянтикова Г. В., Никонова М. П., Равицкая Р. В. Там же, стр. 234.
399. Назаренко 3. Л. Труды H.-техн. общ-ва черной металлургии. Укр. республ. правление, 4 , 93 (1956).
400. Накано. Нихон кагаку дзасси, 75, 328 (1954).
401. Негина В. Р., Шуванова Н. В., Крашенинникова Е. П., Блинова Р. В. Зав. лаб., 28, 671 (1962).
402. Немодрук А. А., Каралова 3. К- Аналитическая химия бора. М., «Наука», 1964, стр. 132.
403. Нессонова Г. Д., Турковская Д. В., Болтунова Н. И. Зав. лаб., 25, 1069 (1959).
404. Никитина Е. А. Гетерополисоединения. М., Госхимиздат, 1962.
405. Никитина Е. А. Зав. лаб., 24, 398 (1958).
406. Никитина Е. А. Ж- общ. химии, 10, 779 (1940).
407. Никитина Е. А. Успехи химии, 8, 889 (1939).
408. Никитина Е. А., Кокурина А. С. Ж- общ. химии, 23, 1263 (1953).
409. Никитина Е. А., Парташникова М. 3. Труды ИРЕА, 22, 119 (1958).
410. Никитина Е. И. Зав. лаб., И, 231 (1945); 24, 398 (1958).
411. Никитина Е. И. Сортировка цветных и черных сплавов капельным методом без взятия стружки. М., Оборонгиз, 1949.
412. Никитина О. Н. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 67.
413. Николаев Н. С., Суворова С. Н., Гурович Е. И., Пека И., Корчемная Е. К-Аналитическая химия фотра. М., «Наука», 1970.
414. Николаева К- И., Божевольнов Е. А. Сб. «Методы анализа химических реактивов и препаратов», вып. 11. М., изд. ИРЕА, 1965, стр. 43.
415. Нисида. Бунсэки кагаку, 5, 47 (1956).
416. Новое в получении монокристаллов и полупроводников (сб. статей). М., ИЛ, 1962.
417. Определение свободной двуокиси кремния в.горных породах и рудничной пыли (сб. статей). М., изд-во АН СССР, 1958.
418. ПанасюкВ.И. Химический контроль производства стекла. М., Гизлег-пром, 1952, стр. 84.
419. Панченко Г. Е. Изв. АН СССР, серия физ., 5, 313 (1941).
420. Пасовская Г. Б. Труды Туркменского гос. мед. ин-та, № 5-6, 384 (1955).
421. Певирв Г. А., Красильщик В. 3., Скузоватова Т. П. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 88.
422. Певцов Г. А., Манова Т. Г. Там же, стр. 88.
423. Педан Г. А. Изв. АН СССР, серия физ., 19, 102 (1955).
184
424. Пейзулаев Ш. И., Карабаш А. Г., Крауз Л. С., Костарева Ф. А., Бабина Ф. Л., Смирнова-Аверина Н. И., Кондратьева Л. И., Воронова Е. Ф., Мешкова В. М. Зав. лаб., 24, 733 (1958).
425. Пенькова Е. Ф., Яковлев П. Я- Зав. лаб., 16, 1495 (1950).
426. Перельман Ф. М., Зворыкин А. Я-, Якубовская Т. Н. Ж. неорган. химии, 3, 1374 (1958).
. 427. Петренко А. Г., Христофоров Б. С., Леонардова Л. А. Сб. «Методы изучения вещественного состава и их применение», вып. 1. Новосибирск, «Наука», 1965, стр. 66.
428. Петрова В. И. Зав. лаб., 16, 98 (1950).
429. Петухова Н. И. Зав. лаб., 25, 86 (1959).
430. Пирютко М. М. Зав. лаб., 29, 1179 (1963).
431. Писарев В. Д., Корнилов А. В., Кострова 3. П. Зав. лаб., 22, 198 (1956).
432. Пихтин А. Н. Зав. лаб., 31, 559 (1965).
433. Позин М. Е., Гинстлинг А. М. Ж- прикл. химии, 24, 37 (1951).
434. Полежаев И. Г. Сб.: «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 33.
435. Полинг Л. Природа химической связи. М., Госхимиздат, 1947, стр. 81.
436. Полотебнова Н. А. Гетерополикислоты с центральными атомами — кремнием и германием. Автореф. канд. дисс. Кишиневский ун-т, 1955.
437. Получение и анализ веществ особой чистоты (сб. статей). М., «Наука», 1966.
438. Пономарев А. И. Зав. лаб., 6, 691 (1937).
439. Пономарев А. И. Методы химического анализа силикатных и карбонатных горных пород. М., Изд-во АН СССР, 1961.
440. Поплевина Л. В., Кобяк Г. Г. Уч. зап. Пермского ун-та, 111, 128 (1964).
441. Попов В., Дерябин А., Мышляева Л. В., Краснощеков В. В. Труды МХТИ им. Д. И. Менделеева, 46, 9 (1964).
442. Попова О. И., Кабанник Г. Т. Бюлл. Ин-та металлокерамики и спецспла-вов АН УССР, № 6, 64 (1961).
443. Проскурякова Г. Ф. Труды Свердловского с.-х. ин-та, 7, 375 (1960).
444. Проскурякова Г. Ф. Там же, стр. 389.
445. Пчелинцев Д. А. Сб. «Методы химического анализа и химический состав минералов». «Наука», 1967, стр. 12.
446. Пчелинцева А. Ф., Раков Н. А., СлюсареваЛ. П. Зав. лаб., 28, 677 (1962).
447. Радиохимические методы определения микроэлементов (сб. статей). М.— Л., «Наука», 1965.
448. Разживина А. Л. Труды ВНИИ огнеупоров, 29, 191 (1961).
449. Райхбаум Я- Д--, Стахеев Ю. И. Ж- аналит. химии, 20, 299 (1965).
450. Райхлин Н. Т., Альбертон Н. И., Колышевская Э. М. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 99.
451. Резников А. А., Муликовская Е. П., Соколов И. Ю. Методы анализа природных вод, 2-е изд. М., Госгеолтехиздат, 1963.
452. Рентгеновские методы изучения и структура глинистых минералов (сб. статей). М., «Мир», 1965.
453. Родионова А. Е., Ковалев А. Г., Овчаренко П. П. Изв. вузов. Химия и хим. технология, 6, 518 (1963).
454. Романенко Е. Г., Зубрицкая А. А. Зав. лаб., 30, 164 (1964).
455. Рояк С. М., Нагерова Э. И., Корниенко Г. Г. Научн. сообщ. ВНИИ цементной промышленности, 2, (33), 34 (1958).
456. Рояк С. М., Нагерова Э. И., Корниенко Г. Г. Труды ВНИИ цементной промышленности, 5, 59 (1952).
457. Рояк С. М., Нагерова Э. И., Корниенко Г. Г. Труды Совещания по химии цемента. М., Промстройиздат, 1956, стр. 42.
458. Рояк С. М., Нагерова Э. И., Корниенко Г. Г. Цемент, № 2, 22 (1959).
459. Русанов А. К- Спектральный анализ руд и минералов. М., Госгеолиздат, 1948.
460. Русанов А. К-, Ильясова Н. В. Атлас пламенных, дуговых и искровых спектров элементов. М., Госгеолтехиздат, 1958.
461. Рысс И. Г. Фтор и его неорганические соединения. М., Госхимиздат, 1956.
185
462. Сабанеев С. ЖРФХО, II отд., 24, 69 (1892).
463. Савченко Г. С., Дятловицкая Ф. Г., Ярошенко В. А., Альбова Е. А. Методы химического и микробиологического анализа воды. Киев, Госмедиз-дат, 1961, стр. 11.
464. Сажин Н. П. Сб. «Методы определения и анализа редких элементов» М., Изд-во АН СССР, 1961, стр. 11.
465. Сакаино, Мория. Ёгё кёкайси, 61, 145, 199, 475 (1953).
466. Сакаино, Мория. Ёгё кёкайси, 62 , 243, 319 (1954).
467. Самсонов Г. В. Свойства тугоплавких соединений (справочник). М., Металлургиздат, 1962.
468. Самсонов Г. В., Латышева В. П. Докл. АН СССР, 105, 499 (1955).
469. Самсонов Г. В., Пилипенко А. Т., Лазарчук М. А. Анализ тугоплавких соединений. М., Металлургиздат, 1962.
470. Сапир А. Д. Зав. лаб., 18, 23 (1952); 19, 1035 (1953).
471. Сата, Киёура. Ёгё кёкайси, 62, 449 (1954).
472. Сато С., Тагаи X. Ёгё кёкайси, 69, 102 (1961).
473. Свентицкий И. С. Ж- техн, физики, 14, 605 (1944).
474. Селинов И. П. Атомные ядра и ядерные превращения, т. 1. Таблицы по физике атомного ядра. М.—Л., Гостехиздат, 1951.
475. Семенов Н. И. Спектральный анализ кварцевого сырья. М., Промстройиздат, 1957.
476. Семов М. П. Зав. лаб., 29, 1450 (1963).
477. Семов М. П. Сб. «Методы анализа химических реактивов и препаратов», вып. 12. М., изд. ИРЕА, 1966, стр. 52.
478. Серебрякова Г. В., Божевольнов Е. А. Сб. «Методы анализа химических реактивов и препаратов», вып. 11. М., изд. ИРЕА, 1965, стр. 58, 89.
479. Сиборг Г., Перлман И., Холлендер Д. Таблицы изотопов. М., ИЛ, 1956, стр. 26.
480. Смирнов В. Ф. Изв. АН СССР, серия физ., 5, 325 (1941).
481. Смышляев С. И. Труды Уральского политехи, ин-та, 94, 168 (1960).
482. Современные методы анализа (сб. статей). М., «Наука», 1965.
483. Современные методы анализа в металлургии (сб. статей). М., Металлургиздат, 1955.
484. Современные методы исследования силикатов и стройматериалов (сб. статей). М., Госстройиздат, 1960.
485. Соколов Н. М. Бюлл. ВХО им. Д. И. Менделеева, 4, 37 (1940).
486. Соколов Н. И. Методы синтеза полиорганосилоксанов. М., Госэнергоиздат, 1959.
487. Соколова А. Л., Махарашвили Н. А. Ж- аналит. химии, 14 , 745 (1959).
488. Соколович В. Е. Зав. лаб., 29, 1178 (1963).
489. Соколович В. Е. Сб. трудов НИИ оснований и подземных сооружений Госстроя СССР, № 54, 135 (1964).
490. Сочеванов В. Г., Волкова Г. А., Волкова Л. П., Мартынова Л. Т., Пахомова К- С., Попова Т. П., Розбианская А. А., Розовская Г. В., Шмакова И. В. Методы химического анализа минерального сырья, вып. 2. Полярография. М., Госгеолтехиздат, 1956, стр. 55.
491. Спектральный анализ. Аннотированный указатель советских работ 1931 — 1950 гг. М., «Металлургиздат», 1960.
492. Спектральный и химический методы анализа материалов. Сб. методик. М., Металлургия, 1964.
493. Спиро Н. С., Гоголева М. В. Труды НИИ геологии Арктики, 119, 65 (1961).
494. Спицын Викт. И., Бабаев Н. Б. Ж- неорган. химии, 5, 580 (1960).
495. Стишов С. М., Белов Н. В. Докл. АН СССР, 143, 951 (1962).
496. Стогний Н.П. Зав. лаб., 25, 420 (1959).
497. Судаков Ф. П., Клишина В. И., Маслова Н. Т. ВесТн. .МГУ, серия «Химия», № 1, 98 (1966).
498. Судузки Т. Нихон кагаку дзасси, 82, 696 (1961).
499. Супин Г. С. Сб. «Методы анализа химических реактивов и препаратов» вып. 13. М., изд. ИРЕА, 1966, стр. 132,
186
500. Сухов Г. В., Гризик А. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 55.
501. Сысоева Р. С. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 103.
502. Такаги Т., Хасимото Ц., Сасаки М. Бунсэки кагаку, 12, 618, (1963).
503. Такаги Т., Хасимото Ц., Сасаки М. Тоё сода кэнкю хококу, 7, 84 (1963).
504. Такаяма. Бунсэки кагаку, 5, 102 (1956).
505. Такзути Ц. Дэнки кагаку, 33, 900 (1965).
506. Тананаев И. В. Ж- прикл. химии, 6 , 834 (1933).
507. Тананаев И. В. Ж- прикл. химии, 7, 729 (1934).
508. Тананаев И. В. Зав. лаб., 11, 246 (1945).
509. Тананаев И. А. Бесстружковый метод. Свердловск, Металлургиздат, 1948, стр. 219.
510. Тананаев Я. А. Весовой анализ. М.—Л., ГОНТИ, 1938.
511. Тананаев Н.А.З-лъ. лаб., 1, №8—9, 24 (1932).
512. Тананаев Н. А. Зав. лаб., 13, 389 (1947).
513. Тананаев Я. А. Капельный метод. М.—Л., Госхимиздат, 1954.
514. Тананаев Н. А., Бабко А. Д. Укр хим. ж., 5, 71 (1930).
515. Тананаев Н. А., Бычков М. Д. Зав. лаб., 4, 648 (1935).
516. Тананев Н. А., Ганаго Л. И. Зав. лаб., 21, 9 (1955).
517. Тананаев Н. А., Смышляев С. И. Труды Уральского политехи, ин-та, 69, 101 (1957).
518. Тананаев И. А., Смышляев С. И. Ж- неорган. химии, 1, 1943 (1956).
519. Тананаев Н. В. Зав. Лаб., 12, 248 (1946).
520. Тананаев Н. В., Дубова М. И. Зав. лаб., 18, 22 (1952).
521. Тананаева А. И. Труды Уральского политехи, ин-та, 69, 143 (1957).
522. Танино Д. Когё кагаку дзасси, 65, 926, 931, 1032, 1036, (1962); 68, 1034 (1965).
523. Тарасевич И. И., Железнова А. А. Труды Комиссии по аналит. химии АН СССР, 15, 121 (1965).
524. Тарасов В. В. Докл. АН СССР, 46, 122 (1945); Новые вопросы физики стекла. М., Госстройиздат, 1959.
525. Тараян В. М. Зав. лаб., 7, 176 (1938).
526. Тартаковский В. Д. Легкие металлы, 5, 7 (1936); 5, 24 (1936).
527. Татаркер Г. Б. Силикаты в советской литературе. Библиография за 1941— 1952. М., Промстройиздат, 1948, 1949, 1952, 1953, 1957.
528. Терентьев А. П., Лускина Б. М., Сявцилло С. В. Ж- аналит. химии, 16, 635 (1961); 19, 1251 (1964); 20,990 (1965).
529. Тимофеюк Д. М. Зав. лаб., 2, 19 (1933).
530. Тиновская Е. С. Сб. «Химия, технология и применение производных пиридина и хинолина». Рига, Изд-во АН ЛатвССР, 1960, стр. 253.
531. Тихонов В. И., Чернышева А. Н. Зав. лаб., 31, 164 (1965).
532. Ткаченко Н. С., Хрипач С. М. Зав лаб., 14, 357 (1948).
533. Толмачев В. Н., Затучная Л. А. Зав. лаб., 23, 152 (1957).
534. Торопов С. А. Сб. «Определение двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 61.
535. Торопов С. А., Гольдина Ц. А. Зав. лаб., 22, 1118 (1956).
536. Тредвелл Ф., Голл В. Курс аналитической химии, т. 2, ч. 1. М., ОНТИ, 1935, стр. 447.
537. Троицкий А. А. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 58.
538. Трощенко Д. Д. Сб. «Лекарственные сырьевые ресурсы Иркутской области и их врачебное применение». Иркутское обл. изд-во, 1961, стр. 200.
539. Труды Всес. совещания зав. лабораториями цементного производства. М., Госстройиздат, 1959.
540. Туманов А. А., Сидоренко А. И., Доренман Я- И. Зав. лаб., 30, 1058 (1964).
541. Туманов А. А., Сидоренко А. И., Доренман Д. И. Сб. «Получение и анализ веществ особой чистоты». М., «Наука», 1966, стр. 247.
542. Туманов А. А., Сидоренко А. И., ТараденковаФ. С. Зав. лаб., 30, 652 (1964).
543. Ульянов А. И. Зав. лаб., 19, 1154 (1953).
187
544. Унифицированные методы химического анализа железных и марганцевых руд. Свердловск, изд. Минчермет, 1943.
545. Унифицированные методы химического анализа ферросплавов. Свердловск, изд. Минчермет, 1944.
546. Усатенко Ю. И. Ускоренные химические и физико-химические методы
анализа агломератов железных руд, силикатов и материалов, получаемых на основе высокоогнеупорных окислов металлов. Докт. дисс. М. — Днепропетровск, МХТИ им. Д. И. Менделеева, 1954.
547. Усатенко Ю. И., Орлова Ю. >7. Зав. лаб., 15, 1365 (1949).
548. Усатенко Ю. И., Федаш И. П. Зав. лаб., 24, 1180 (1958).
549. Ускоренные методы химического анализа в цементной промышленности (сб. статей). М., Промстройиздат, 1957.
550. Ускоренный химический анализ на основные окислы сырьевой смеси цементного производства (сб. статей). М., Промстройиздат, 1954.
551. Фазовый состав трехкомпонентных клинкеров (сб. статей). Челябинск, изд. Уральского филиала Академии строительства и архитектуры СССР, 1962.
552. Файгль Ф. Капельный анализ. М., ОНТИ, 1937, стр. 393.
553. Файнберг С. Ю. Анализ цветных металлов. М., Металлургиздат, 1956.
554. Файнберг С. Ю., Бляхман А. А. Сб. научи, трудов Гос. НИИ цветных металлов, № 12, 111 (1956).
555. Фатеева Н. И., Козлова В. К- Цемент, № 4, 13 (1966).
556. Федоров А. А., Линкова Ф. В., Озерская Ф. А., Соколова Г. П., Кричевская А.М. Зав. лаб., 26, 1357 (1960).
557. Федоров П. Ф. Зав. лаб., 4, 747 (1935).
558. Федорова. М. Н., Клименко Ю. В. Зав. лаб., 19, 11 (1953).
559. Федорова М. Н., Клименко Ю. В. Труды Н.-и. проектного ин-та «Урал-механобр», 3, 150 (1958).
560. Фидельман Ф. М. Сб. «Новое в области санитарно-химического анализа». М., Медгиз, 1962, стр. 254.
561. Физические методы анализа следов элементов (сб. статей). М., «Мир», 1967.
562. Филимонов Л. Н., Хандрос В. О. Зав. лаб., 24, 712 (1958).
563. Филипов Д. Докл. Болг. АН, 13, 543 (1960), 16, 377 (1963).
564. Филипов Д. Машиностроене (болг.), 10, 17(1961); 11, 40 (1961).
565. Филиппова И. А., Кузнецова Л. И. Зав. лаб., 16, 536 (1950).
566. Филоненко Н. Е., Алферов В. А., Боровкова Л. А. Огнеупоры, № 7, 320 (1954).
567. Филоненко Н. Е., Боровкова Л. А. Абразивы, № 9, 3 (1953).
568. Филоненко И. Е., Лавров И. В. Петрография искусственных абразивов. М.—Л., Машгиз, 1958.
569. Финкельштейн Д. И., Борецкая В. А. Методы анализа минерального сырья. М., Геолтехиздат, 1958.
570. Фогельсон Е. И. Зав. лаб., 22, 163 (1956).
571. Фогельсон Е. И., Казачкова Ф. С. Зав. лаб., 13, 565 (1947); 23, 24 (1957).
572. Фтор и его соединения (сб. статей), т. 1, 2. М., ИЛ, 1953.
573. Фудзии И. Бунсэки кагаку, 13, 1054 (1964).
574. Фукамаути X., Идэно Р., Мацубара Т. Бунсэки кагаку, 13, 313, 316 (1964).
575. Фукуси Н. Бунсэки кагаку, 15, 868 (1966).
576. Хализова В. А., Алексеева А. Д-, Смирнова Е. П. Зав. лаб., 30, 530 (1964);
32, 928 (1966).
577. Хара С. Осака когё гидзюцу сикэнсё кихо, 12, 182 (1961).
578. Хасэгава М. Нихон киндзоку гаккайси, 24, 493, 654 (1960).
579. Хауффе К- Реакции в твердых телах и на их поверхности, ч. 2. М., ИЛ, 1963.
580. Химическая литература. Библиографический справочник (1920—1952 гг.).
М. — Л., Госхимиздат, 1953.
581. Химические и физико-химические методы анализа минерального сырья (сб. статей). М., Госгеолтехиздат, 1955.
582. Химический и петрографический методы контроля цементного производства. Л., Стройиздат, 1939.
188
583. Химический контроль производства в металлургической и металлообрабатывающей промышленности (сб. статей). Изд. СНХ Днепропетровского экономии. р-на, 1960.
584. Химия высокотемпературных материалов (сб. статей). Л., «Наука», 1967.
585. Химия стекла (сб. статей). М., ИЛ, 1950.
586. Хризман И. А. /К. аналит. химии, 10, 373 (1955).
587. Христофоров Б. С. Избирательные растворители в вещественном анализе. Новосибирск, Изд-во СО АН СССР, 1964.
588. Христофоров Б. С. Сб. «Методы изучения вещественного состава и их применение», вып. 1. Новосибирск, «Наука», 1965, стр. 5.
589. Хрущов М. М., Беркович Е. С. Микротвердость, определяемая методом вдавливания. М., Изд-во АН СССР, 1943.
590. Цап М. Л. Зав. лаб., 21, 10, 281 (1955).
591. Цеховольская Д. И., Заварицкая Т. А. Труды Комиссии по аналит. химии АН СССР, 13, 399 (1963).
592. Цой Р. И. Стекло и керамика, 18, 40 (1961).
593. Цубаки И. Бунсэки кагаку, 12, 849 (1963).
594. Цыбулевский X. И. Зав. лаб., 7, 489 (1938).
595. Чернихов Ю. А., Гульдина Е. И. Зав. лаб., 4, 487 (1935).
596. Чулановский В. М. Введение в молекулярный спектральный анализ. М., Гостехиздат, 1951.
597. Чумаченко М. Н., Коршун М. О., Бурлака В. П., Симонова В. Н. Докл. АН СССР, 133, 138 (I960).
598. Чупахин М. С. Сб. «Современные методы анализа». М., «Наука», 1965, стр. 33.
599. Чупахин М. С., Главин Г. Г. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 104.
600. Шановская С. С. Сб. «Методы определения свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 44.
601. Шарло Г. Методы аналитической химии. Количественный анализ неорганических соединений. М., «Химия», 1965.
602. Шафран И. Г., Взорова И. Ф., Доросинская М. И., Фидлон Л. К-, Юрьева В. А. Сб. «Методы анализа веществ высокой чистоты». М., «Наука», 1965, стр. 61.
603. Шафран И. Г., Взорова И. Ф., Доросинская М. И., Фидлон Д. К., Юрьева В. А. Там же, стр. 81.
604. Шафран И. Г., Взорова И. Ф., Доросинская М. И., Фидлон Л. К-, Юрьева В. А. Труды ИРЕА, 30, 223 (1967).
605. Шахова 3. Ф., Дорохова Е. И. Вести. МГУ, серия Химия, № 2, 77 (1965).
606. Шахова 3. Ф., Моторкина Р. К-, Мальиева И. И. Ж. аналит. химии, 12, 95 (1957).
607. Швайгер М. И. Зав. лаб., 26, 1223 (1960).
608. Швайгер М. И. Зав. лаб., 29, 890 (1963).
609. Швангирадзе Р. Р., Мозговая Т. А. Ж- аналит. химии, 12, 708 (1957).
610. Шварц Р. Успехи химии, 26, 923 (1957).
611. Шеметихина А. А. Сб. «Физико-химическое и технологическое исследование минерального сырья». Ташкент, «Наука», 1965, стр. 116.
612. Шемятенкова В. Т. Качественный анализ кремнийорганических соединений. Автореф. канд. дисс. М., МХТИ им. Д. И. Менделеева, 1953.
613. Шкаравский Ю. Ф. Укр. хим. ж., 30, 670 (1964); Изв. вузов, Химия и хим. технология, 3, 370 (1961).
614. Шмулевич Е. Д. Зав. лаб., 28, 811 (1962).
615. Шпирт М. Д., Сендульская Т. И., Тананаев И. В. Ж- неорган. химии, 8, 2611 (1963).
616. Шубников А. В. Кварц и его применение. М., Изд-во АН СССР, 1940.
617. Щекатурина Л. Г., Кондратова В. П., Петрашень В. И. Труды Новочеркасского политехи, ии-та, 31, 79 (1955).
618. Щекатурина Л. Г. Детрашень В. И. Там же, 97, 137 (1959).
619. Щекатурина Л. Г., Петрашень В. И. Сб. «Определение свободной двуокиси кремния в горных породах и рудничной пыли». М., Изд-во АН СССР, 1958, стр. 54.
189
620. Щекатурина Л. Г., Петрашень В. И. Сб. «Передовые методы химической технологии и контроля производства». Изд-во Ростовского ун-та, 1964, стр. 348.
621. Эйтель В. Физическая химия силикатов. М., ИЛ, 1962.
622. Экстракция в аналитической химии и радиохимии (сб. статей). М., ИЛ, 1961.
623. Электрофизические свойства германия и кремния (сб. статей). М., ИЛ, 1957.
624. Эмих Ф. Микрохимический анализ. Л., Госхимтехиздат, 1932, стр’. 135.
625. Юнг В. Н., Бутт Ю. М., Николаев Ю. Н. Зав. лаб., 10, 518 (1941).
626. Юрист И. М., Короткова О. И. Зав. лаб., 27, 274 (1961).
627. Яковлев Ю. В. Зав. лаб., 28 , 643 (1962).
628. Якубович А. А. Ускоренный анализ минерального сырья с применением сцинтилляционной аппаратуры. М., Госатомиздат, 1963.
629. Ямагути Н., Сакаи И., Хата А., ХасэгаваМ. Бунсэки кагаку, 11, 997 (1962).
630. Яцимирский К- Б. Труды по химии и хим. технологии (Горький), 1, 49, (1963).
631. Ahrens L. Н. Mineral Mag., 30, 467 (1954).
632. Ahrens L. H. Quantitative Spectrochemical Analysis of Silicates. London, 1954.
633. Ahrens L. H. Spectrochemical Analysis. Cambridge, Addison-Wesley Press, 1950.
634. Ahrens L. H., Edge R. A., Brooks R. R. Analyt. chim. acta, 28, 551 (1963).
635. Ahrens L.H., Taylor S.K. Spectrochemical Analysis. Mass. Addison — Wesley, 1961, Table 8.
636. Alexander G. B. J. Am. Chem. Soc., 75, 2887, 5655 (1953); 76, 2094 (1954).
637. Alexander G. B., Heston W. M.., Iler R. K. J. Phys. Chem., 58, 453 (1954).
638. Alexander I. J. Am. Chem. Soc., 43, 434 (1921).
639. Alimarin I., Zverev V. Mikrochemie, 16, 89 (1937).
640. Alon A., Bernas B., Frenkel M. Analyt. chim. acta, 31, 279 (1964).
641. Anderson H. H. J. Am. Chem. Soc., 66, 934 (1944); 69, 30 (1947); 70, 1220 (1948).
642. Anderson L. H. Acta chem. scand., 13, 1743 (1959); 14, 1571 (1960).
643. Anderson L. H. Arkiv kemi, 19, 223, 249, 257 (1962).
644. Armand M., Bertoux J. Analyt. chim. acta, 5, 380 (1951); 8, 510 (1953).
645. Aronsson B., Torsten L., Rundqust S. Borides, Silicides and Phosphides. London, Methuen, 1965.
646. Asch W. Z. anorg. allgem. Chem., 28, 273 (1901).
647. Backsfrom N. Rev. Metall., 52, 82 (1955).
648. Backsfrom N. lernkontor. Ann., 137, 849 (1953).
649. Bacon F. R., Raggon F. C. J. Am. Chem. Soc., 42, 199 (1959).
650. Bacon L. R., Wills J. H. Soap and Chem. Spec., 30, 85, 89 105 (1954).
651. Badalucco A. Ceramica, 11, № 3, 41; № 6, 46; № 11, 47 (1956).
652. Baker C. L., due L. R., Wills J. H. J. Am. Chem. Soc., 72, 5369 (1950).
653. Baron J., Debras-Guedon J., Kiehl J.-P. Bull. Soc. fran?. ceram., № 58, 57 (1963).
654. Barret E. P., Schroeder F. W. J. Am. Chem. Soc., 8 (1), 69 (1925).
655. Bartels H., Erlenmeyer H. Helv. chim. acta, 47, 13 (1964).
656. Behr A., Blanchet M. L., Malaprade L. Chim. analyt., 42, 501 (1960).
657. Bennet H. J. Soc. Glass Technol., 43, 59 (1959).
658. Bennet H. Trans. Brit. Ceram. Soc., 54, 319 (1955).
659. Bennet H., Hawley W. G., Eardley R. P. Trans. Brit. Ceram. Soc., 57, 1 (1958).
660. Berl-Lunge. Chemisch-technische Untersuchungsmethoden, 2 Aufl., Berlin, 1931.
661. Berthel K--HDoge H.-G., Ehrlich G., Ko the A., Schmidt A. Mikrochim. acta, 1963, 702,
662. Berzelius J. J. J. Schweigg., 16, 426 (1816).
663. Berzelius J. J. Pogg. Ann., 1, 169 (1824).
664. Beutelspacher H., Marel H. W. Tonind. Ztg., 85, 517 (1961).
665. Bieber B., Vecera Z. Hutn. listy, 15, 397 (1960).
190
666. Bien G. Analyt. Chem., 30, 1525 (1958).
667. Bierwirth G. Giesserei, 45, 546 (1958).
668. Blanke M. Silikattechnik, 6, 247 (1955).
669. Blanzat A., Barbe M. Arch, malad. profess., 14, 349 (1953).
670. Blasius E., Czekay A. Z. anal. Chem., 147, 1 (1955); 156, 81 (1957).
671. BoltzD. Colorimetric determination of non-metals. N. Y.— London, Inter-science 1958
672. Boulin R. Ind. chim. Belg., 1, 142 (1959).
673. Brabson J. A., Duncan R. D., Murphy I. J. Analyt. Chem., 35, 1102 (1963).
674. Brabson J. A., Mattraw H. C., Maxwell G. E., Darraw A., Needham M. F. Analyt. Chem., 20, 504 (1948).
675. Brade W. Chemical spectroscopy. N. Y., J. Willey, 1945.
676. Brady A. P., Brown A. G., Huff H. J. Coll. Sci., 8, 252 (1953).
677. Bragg tt7. H. Proc. Roy. Soc., London, 114, 450 (1927).
678. Bragg W. L. Z. Kristallogr., 74 , 237 (1930).
679. Brehler B. Ber. Dtsch. keram. Ges., 33, 329 (1956).
680. Brewer P. G., Riley J. P. Analyt. chim. acta, 35, 514 (1966).
681. Brill. Mikrochim. acta, 1966, 1047.
682. Brindley G. W. J. Am. Ceram. Soc., 1961, 42.
683. Brindley G. W., Rurtossy S. S. Am. Mineralogist, 46, 1205 (1961).
684. Brintzinger H., Brintzinger W. A. anorg, allgem. Chem., 196, 44 (1931).
685. Bucherer H., Meier F. Z. anal. Chem., 82, 35 (1930).
686. Burglen L., Longuet P. Rev. mater, constr. et trav. publ., № 474, 59 (1955).
687. BygdenA. C. A., 14, 1974 (1920).
688. Colas R. Nouveau traite de chimie minerale, v. 8. 2-th Publ. Paris, 1965, p. 271—682.
689. Capdevila Perez C., Alvarez Gonsalez F. Ann. Real. soc. esp. fis. у quim., В 58, 13 (1962).
690. Carlson A. B., Banks С. V. Analyt. Chem., 24, 472 (1952).
691. CarronM.R., Cuttitta F. Geol. Surv. Prof. Paper, № 45-0-B, 78 (1962).
692. Carron M. R., Stevens R. E. Analyt. Chem., 25, 1782 (1953):
693. CaseO., Catani R. A. Ind. Eng. Chem. Anal., Ed., 16, 309 (1944).
694. Cavallero L., Tani A. Att. Accad. Sci. Terrere, 24, 216 (1946).
695. Celechovsky J. Chem. listy, 48, 391 (1954).
696. Chalmers R. A., Sinclair A. G. Analyt. chim. acta, 33, 384 (1965).
697. Chalmers R. A., Sinclair A. G. Analyt. chim. acta, 34, 412 (1966).
698. ChapinD. M., Fuller C. S., Pearson G. L. Сб. «Кремний». M., ИЛ, 1960, стр. 19.
699. Charreton В., Chauveau F., Bertho G., Courtin P. Chim. analyt., 47, 17 (1965).
700. Chichilo P. J. Assoc, off. Agric. Chemists, 45, 6 (1962); 46, 603 (1963); 47,
1019 (1964).
701. ChowD.T. H7., Robinson R. J. Analyt. Chem., 25, 646 (1953).
702. Christopher A. J., Fennell T. R. Taianta, 12, 1003 (1965).
703. Clabaugh W. S., Jackson A. J. Res. Nat. Bur. Stand., 62, 201 (1959).
704. Clarke F. W. U. S. Geol. Surv. Bull., № 770 (1924).
705. Clarke F. W., Washington H. S. U. S. Surv. Prof. Papers, 127, 1 (1924).
706. Codell M., Clemency C., Norwitz G. Analyt. Chem., 25, 1432 (1953).
707. Codell M., Norwitz G. Analyt. chim. acta, 16, 327 (1957).
708. CoenenM. Silic. ind., 28, 147 (1963).
709. Coes L. Science, 118, 131 (1953).
710. Comptes rendus XXIII Conference of International Union of pure and Applied Chemistry. London, 1965, p. 175—180.
711. Copeland L. E., Brunauer S., RantroD.L., Schulz E.G., Weise С. H. Analyt. Chem., 31, 1521 (1959).
712. Coraducci P. Ceramica, 9, № 3,43 (1954).
713. Csajaghy G., Emszt M., Szepesi R. Acta geol. Acad. sci. hung., 5, 157 (1958).
714. Cypris R. Sprechsaal, 97, 5 (1964).
715. Czech F. W., HrycyshynT. P., Fuchs R. J. Analyt. Chem., 36, 2026 (1964).
716. DaevesR. Werkstoffhandbuch Stahl und Eisen, 3. Aufl. 1953, Bd. 11, S. 4.
191
717. Debras-Guedon J. Ind. Ceram., № 555, 345 (1963).
718. Dempir J. Stavivo, 41, 183 (1963).
719. De-Sesa M. A., Rogers L. B. Analyt. Chem., 26, 1278 (1954).
720. Dienert F., Wandenbulcke F. C. r. Acad. Sci., 176, 1478 (1929).
721. Dinger K., Jebsen-Marwedel H. Sprechsaal, 91, № 7,134 (1958).
722. DohrH., WiebkeG. Tonind. Ztg., 87, 87 (1963).
723. Donovan P. D., Evans J. L., BushG.H. Analyst, 88, 771 (1963).
724. Ducret L., Cornet C. Analyt. chim. acta, 25, 542 (1961).
725. Duval C. Analyt. chim. acta, 1, 33 (1947).
726. Duval С. C. r. Acad, sci., 218, 119 (1944).
727. Duval C. Inorganic Thermogravimetric Analysis. N. Y., Elvier, 1953, p. 123.
728. Duval C. Traite de microanalyse minerale qualitative et quantitative, v. 3. Paris 1956
729. Dvorak J., Wanek IE. Chem. analit., 7, 201 (1962).
730. Eardley R. P., Clarke H. S. Appl. Spectroscopy, 19, 186 (1965).
731. Ebelmen J. Ann., 57, 334 (1846).
732. Eckert G. Z. anal. Chem., 161, 421 (1958); 162, 408 (1958).
733. Egen W. Giesserei, 49, 849 (1962).
734. Ehrlich G., GerbatschR., JaetschK., Scholze H. Reinstoffe Wissenschaft und Technik. Berlin, 1963, S. 421.
735. Ehrlich P., KeilT. Z. anal. Chem., 166, 154 (1959).
736. Elliot G., Radley J. A. Analyt. Chem., 33, 1623 (1961).
737. ElvingP.J., Horton C. A., Willard H.H. Fluorine Chemistry. N. Y., 1954, p. 45.
738. Engelbrecht A., Nachbaur E., Mayer E. J. Chromatogr., 15, 228 (1964).
739. Fairbairn H. W. Geochim. et cosmochim. acta, 4, 143 (1953).
740. Fairbairn H. W., Schairer J. F. Am. Mineralogist, 37, 744 (1952).
741. Fairbairn H. W., Schlecht W. G., Stevens R. E., Demen W. H., Ahrens L. H., Chayes E. U. S. Geol. Surv. Bull., № 980 (1951).
742. Falkenburg L., Treeter H., Cowan J. J. Am. Chem. Soc., 69 , 486 (1947).
743. Falkner P. R., BurnsD. T. Mikrochim. acta, 1965, 318, 322.
744. Fawcett B., Jones B., Wilson R. Proc. Phys. Soc., 78, 1223 (1961).
745. FeiglF. Chem. Anal., 32, 52 (1943); 33, 76 (1944).
746. FeiglF. Ind. Eng. Chem., Anal. Ed., 14, 316, 519 (1942).
747. FeiglF. Laboratory Manual of Spot Test. N. Y., Nordeman, 1943.
748. FeiglF. Spot Tests, N. Y., Nordeman, 1939, p. 219.
749. FeiglF., Krumholz P. Ber., 62, 1138 (1929).
750. FeiglF., Krumholz P. Mikrochemie, Pregl-Festschrift, 1929, S. 82.
751. Fenner C. N. J. Wash. Acad. Sci., 2, 471 (1912); Am. J. Sci., 36, 331 (1913).
752. Fenzke H., Hesse E. Neue Hiitte, 7, 752 (1962).
753. Ferrari С. C. A., 46, 1413 (1952).
754. Ferrari C. Gazz. chim. ital., 81, 692 (1951).
755. Ferrari C. Mikrochim. acta, 1956 , 550.
756. Ferrari C., LugoP. Ann. sperim. agrar., 7, 369 (1953).
757. Ferrari F. Cemento, № 7, 2 (1955).
758. Flint E. P., Well L. S. J. Res. Nat. Bur. Stand., 12, 751 (1934).
759. Florentin D., Heros M. Bull. Soc. chim., 14, 213 (1947).
760. Florinskaja W. Silikattechnik, 11, 364 (I960).
761. FlorkeO. W. Ber. Dtsch. keram. Ges., 33, 319 (1956); 38, 89 (1961).
762. FlorkeO. W. Naturwiss., 43, 419 (1956).
763. Fowler R. M. Analyt. Chem., 24, 196 (1952).
764. Fowler R. M. Ind. Eng. Chem., Anal. Ed., 4 , 382 (1932).
765. Fratini N. Ann. chimica, 44, 709 (1954).
766. Fresenius W., Schneider W. Z. anal. Chem., 207, 16 (1965); 214, 341 (1965).
767. Fricke R., Huttig G. F. Handbuch der allgemeine Chemie. Leipzig, 1937, S. 146.
768. Friedel C„ Crafts J. Ann., 136, 203 (1865).
769. Friedel C., Ladenburg A. Ann., 147, 355 (1868); 143, 118 (1867).
770. Friedman H. A. U. S. Atom. Energy Commiss., Y-962-Y-1059 (1957).
771. Frydrich R. Chem. Ber., 1, 151 (1964).
192
TI2. Fuchs W., VeiserO. Arch. Eisenhiittenwesen, 27, 429 (1956).
773. FunkH. Kolloid Z., 184, 154 (1962).
774. Funk H. Z. Naturforschung, 176, 199 (1962).
775. FunkH., Schauer H. Chem. Technik, 6, 432 (1954).
776. Gawargious Y. A., Ottendorfer L. J., Bishara S. W. Analyt. chim. acta, 34, 111 (1966).
777. Gebauhr IE., Martin J. Z. anal. Chem., 200, 266 (1964).
778. Geilmann IE., Ganssle A. Glastechn. Ber., 28, 16 (1955).
779. Geilmann IE., Tblg G. Glastechn. Ber., 30, 335 (1957); 33, 245 (1960).
780. German A., Geanta G. Rev. Chim. (Bucharest), 14 (7), 424 (1963).
781. Ghosh B. N„ Pal. P. K. J. Ind. Chem. Soc., 39, 795 (1962).
782. Gibson M. AERE, CR-521; M. Jean. Chim. analyt., 44, 195 (1962).
783. Gilbert J. Analyst, 15, 176 (1890).
784. Glass Technol., 6, №4, 103 (1965).
785. Glauser A. Zement — Kalk—Gips, 18, 536 (1965).
786. Gmelin Kraut’s Handbuch der anorganische Chemie, Bd. 3, Abt. 1. Heidelberg, Verlag Chemie Weinheim, 1912, S. 101, 120; 1932, S. 208; 1959. S. 121.
787. Coldich S. S., Oslund E. II. Bull. Geol. Soc. Am., 67, 811 (1956).
788. GonzalesC. J. An. Real. soc. esp. fis. у quim., B54, 677 (1958).
789. Garlich E. Analiza krzemianow. Warszawa, Wad. Geol., 1958.
790. GotoH., Amano H., Inoue J. Nippon Kindzoku Gakkaishi, 24, 85 (1960).
791. Goto R. J. Phys. Chem., 60, 1007 (1956).
792. Graham E.K. Chem. Anal., 44, 37 (1955).
793. Grasshoff K-, HahnH. Z. anal. Chem., 168, 247 (1959); 173, 29, 198 (1960).
794. Green J. Ann. N. Y. Acad. Sci., 62, № 13, 295 (1955).
795. Greenberg S. A. J. Polimer. Sci., 27, 523 (1958).
796. Greenberg S. A., SinclairD. J. Phys. Chem., 59, 435 (1955).
797. Grimaldi F. S., Ingram B., CuttittaF. Analyt. Chem., 27, 918 (1955).
798. GrylickiM. Silikattechnik, 7, 325 (1956).
799. Guellemin G. Microanalyse qualitative appliquee a la determination des es-peces minerales. Paris, 1953; РЖХим., 1954, №22, 48567K.
800. Gupta H. N. D., Joshi V. Indian J. Appl. Chem., 26, 53 (1963).
801. Guyotieannin C., HerosM., Le Lay J. Bull. Soc. pharmac. Bordeaux, 102, 197 (1963).
802. HaasC. S., Pellin R. A., Everingham M. R. Analyt. Chem., 36, 245 (1964).
803. HahnH., Grasshoff К. Z. anal. Chem., 173, 29 (1960).
804. HahnH., Wagenknecht E. Z. anal. Chem., 182 , 343 (1961).
805. Harel S., Herman E. R., Talmi A. Analyt. Chem., 27, 1144 (1955).
806. Harman R. IE. J. Phys. Chem., 31, 616 (1927); 32, 44 (1928).
807. Hartley F. Proc. Internal. Comm. Glass, 2, 45 (1955).
808. Harvey C. A method of semiquantitative spectrographic analysis. Glendale, California, Appl. Res. Labor. 1947.
809. Harvey C. Spectrochemical Procedures. Detroit, Michigan, Appl. Res. Labor., 1950.
810. HarzdorfC. Z. anal. Chem., 227, 96 (1967).
811. Hasel IE. M. Analyt. Chem., 24, 196 (1952).
812. Hedvall J. A., Schiller G. Z. anorg. Chem., 221, 97 (1934).
813. HeltaiF., Beke E. Rev. chim., 9, 311 (1958).
814. Herman A., Sedlackova 0. Chem. zvesti. 10, 375 (1956).
815. Hoffman J. I. J. Res. Nat. Bur. Stand., 25, 379 (1940).
816. Hoffman J. I., Leslie R. T., Caul H. J., Clark L. J., Hoffman J. D. J. Res. Nat. Bur. Stand., 37, 409 (1946)
817. Hoffman J. I., Lundell G. E. F. J. Res. Nat. Bur. Stand., 3, 581 (1929);
22, 465 (1939).
818. Holmquist S. B., Berry T. F., Zwell L. Am. Ceram. Soc. Bull., 37, 317 (1958).
819. HoltB.D. Analyt. Chem., 32, 124 (1960).
820. HoltP.F., Yates D. M. Biochem. J., 54, 300 (1953).
193
821. Holthaiis С. Ber. Chemikeraussch. VdEh., № 39, 1 [цит. no: Neue Hiitte, 7, 752 (1S62)].
<'75. Holzapfel L., Gottschalk G. Z. anal. Chem., 142, 115 (1954).
823. Honda M. J. Chem. Soc. Japan, 70, 103 (1949).
824. Honda M., Jost F. Coll. Czech. Chem. Comm., 31, 776 (1966).
825. HozdicC. Analyt. chim. acta, 33, 567 (1965); Analyt. Chem., 38, 1626 (1966).
826. HrskaK- Oesterr. Bibliogr., № 18, 14 (1654).
827. Hulett G. A. Colloid Chemistry. N. Y., 1926, p. 637.
828. Humphrey G. L-, KingE. G. J. Am. Chem. Soc., 74, 2041 (1952).
829. IlerR.K- J. Am. Ceram. Soc., 47, 194 (1964).
830. IlerR.K. J. Phys. Chem., 56, 678, 680 (1952).
831. IlerR.K- J. Phys. Chem., 57, 604, 654 (1953).
832. IlerR.K.., Pinkney P. Ind. Eng. Chem., 11, 1379 (1947).
833. Insley H., Flint E. P., Newman E. S., Swenson J. A. J. Res. Nat. Bur. Stand., 21, 355 (1938).
834. Ishino T. Technol. Repts Osaka Univ., 14, 963 (1964).
835. Iversen E. Kemisk, 36, 75, 78 (1955); 37, 1 (1956).
836. Iwasaki J. Japan Analyst, 9, 184 (1960).
837. Jaeger F.M. Z. Elektrochem., 32, 328 (1926).
838. James J. A., RicardsD.H. Nature, 175, 769 (1955).
839. Jander G., Heukeshoven W. Z. anorg. allgem. Chem., 201, 301 (1931).
840. Jander W. Z. anorg. allgem. Chem., 163, 1 (1927).
841. Jannasch P., Weber H. Ber., 32, 1670 (1899).
842. JeanM. Ann. chim. Paris, 3, 470 (1948).
843. Jean M. Chim. analyt., 37, 125 (1955); 38, 37 (1956); 44, 195 (1962).
844. Jean M. Courrier normalis., 33, 810 (1966).
845. Jeffery P. G., Wilson A. D. Analyst, 85, 478 (1960).
846. Jenik J. Chem. prumysl., 11, 188 (1961).
847. Jenik J., Juracek M. Coll. Czech. Chem. Comm., 26, 967 (1961).
848. Jeszalik A. Przegl. geol., № 8, 336 (1954); № 2, 82 (1955); № 3, 130 (1955); № 8, 382 (1955).
849. Jolies A., NeurathF. Z. angew. Chem., 11, 315 (1898).
850. J. Soc. Glass Technol., 40, 53 (1956).
851. Juvanczne U. V., OrdoghM. Kozp. fis. kutato int. kozl., 12, 365 (1964).
852. Kalman L., Vdgd A. Magyar. Kern., folyoirat, 64, 123 (1958).
853. Karell Z. Szklo i ceram., 8, 263, 297 (1957); 9, 122 (1958).
854. Karsten F. Metalloberflache, 8, B55 (1954).
855. Kautsky H., Haase L. Ber., 86, 1226 (1953).
856. Kautsky H., Haase L. Z. Naturforsch., 86, 45 (1953).
857. Kellum G. E., Smith R. C. Analyt. Chem., 39, 341 (1967).
858. Keram. Z., 5, 29 (1953).
859. King E. J., Stacy B.D., Holt P. F., Yates D. M., Pickles D. Analyst, 80, 441 (1955).
860. KingE. J., Watson J. L. Mikrochemie, 20, 49 (1936).
861. KingG. J. Oil Colour Chem. Assoc., 13, 28 (1930).
862. KingG. Paint Manuf., 1, 52 (1931).
863. Kipping F. S. J. Chem. Soc., 123, 2590 (1923).
864. Kipping F., Doeuvre J. Trarti de Chimie Organique, 14, 553 (1939).
865. Kipping F., Loyd L. Proc. Chem. Soc., 13, 174 (1899).
866. Klimboff M. Indian Ceram., 2, 331 (1956).
867. Knapp 0. Silikattechnik, 14, 75 (1963).
868. Koch W. Z. anal. Chem., 209, 105 (1965).
869. Koenig E. W. Ind. Eng. Chem., Anal. Ed., 11, 532 (1939).
870. Konrad E., BachleO., Signer R. Ann., 474, 276 (1929).
871. Korac V. Cement (Jugosl.), 8, 114 (1964).
872. Koudela S. Sklar a keramik, 6, №9—10 228 (1956).
873. KrajinaA., Dolezal I. Taianta, 11, 1127 (1964).
874. Kral S. Hutn. listy, 10, 94 (1955).
194
875. Krauskopf К- В. Geochim. et cosmochim. acta, 10, 1 (1956).
876. KukeC.L. Analyt. Chem., 25, 148 (1953).
877. Kwestroo W., Visser J., Analyst, 90, 297 (1965).
878. Kwiatkowski E., Szychlinski J. Chem. analit. (Polska), 5, 65 (1960).
879. LadenburgA. Ber., 41, 966 (1908).
880. Lafurna J. Rev. mater, constr. et trav. publ., № 457, 275 (1953).
881. Lancaster W. A., Everingham M. R. Analyt. Chem., 36 , 246 (1964).
882. Langmyhr F. J., Saether B. Zement — Kalk— Gips, 9, 429 (1956).
883. Larsen E. S. Am. J. Set., 35, 94 (1938).
884. LaskeH. Wissen. Zeitung. Martin-Luther-Univ. Halle, 11, 873 (1962).
885. LeibigerH. Telefunken-Rohre, № 36, 111 (1959).
886. LeviG.R., Peyronel G. Z. Kristallogr., 92, 190 (1935).
887. LibertlA., Collottl G. Ann. chimica, 44, 454 (1954).
888. Light T. S., Fitzpatrik L. F., Phaneuf J. P. Analyt. Chem., 37, 79 (1965).
889. Lipinski D., Schwiete H. E. Gias— Email — Keramo — Techn., 14, 325 (1963).
890. Liteanu. C., Bran S. Rev. roumane chim., 11, 869 (1966).
891. Liteanu C., Bran S. Studia Univ. Bades-Bolyai. Chem., № 2, 127 (1959); № 2, 89 (1961).
i 892. Liteanu C., Rusu G., Strusievici C. Studii .?i cercetari chim., 3, 55 (1955).
893. LiteanuC., Strusievici C., Rusu G. Ibid., p. 61.
894. Loffler J. Glastechn. Ber., 27, 321 (1954).
895. Lounamaa N. Spectrochim. acta, 7 , 358 (1956).
896. Louvrier J., Voinovitch I. A. Ind. ceram., № 510, 243 (1959).
897. MacKenzie R. C. The differential thermal investigation of clays. London, Mineral. Soc., 1957.
898. MacKenzie R. C., Mitchell B.D. Analyst, 87, 420 (1962).
899. Majumdar A. K-, Banergjee S. Analyt. chim. acta, 13, 424 (1955).
900. MalyE. Chem. zvesti, 15, 918 (1961).
901. MalyE. J. Chromatogr., 19, 206 (1965).
902. ManM. Rev. chim., 9, 466 (1958).
903. Marklow R., Haszeldine R. J. Chem. Soc., 1956, 962.
904. Maurel P. Bull. Groupe frang. argiles, 15, 65 (1964).
905. Maynes A. D. Analyt. chim. acta, 32, 211 (1965).
906. McAleer W. J., Kozlowski M. A., Pollak P. I., Denkewalter R. G. J. Elect-rochem. Soc., 109, 1, 138 (1962).
907. McHard J. A., Servais P. C., Clark H. A. Analyt. Chem., 20, 325 (1948).
908. Medicus K- Z. anal. Chem., 145, 337 (1955).
909. Mercy E. L. P. Geochim. et cosmochim. acta, 9, 161 (1956).
910. Merten T. F. ZiegelIndustrie, 9, 442 (1956); 9, 613 (1956).
911. Merz W., Mika J. Chim. analyt., 37, 125 (1955).
912. MeyerS., KochO.G. Mikrochim. acta, 1963, 929.
913. Miehr W., Koch P., Kretzert J. Z. angew. Chem., 43 , 250 (1930)
914. MieldsM., Schering G. Silikattechnik, 6, № 4, 142; № 6, 241 (1955).
915. Miller C. A., Chalmers R. A. Analyst, 78, 24 (1953).
916. Millner T., Horkay K- Acta techn. Acad. sci. hung., 33, 201 (1961).
917. Milton R. F. Analyst, 76, 431 (1951).
918. Minster J. T. Analyst, 71, 428 (1946).
919. Mituzas J. Liet. TSR auStpjp mokyklu mokslo darbai. Chemija ir chem. technol., 3, 21 (1963); РЖХим, 1963, 20 M 3.
920. MoenkeH. Spektralanalyse von Mineralien und Gesteinen. Eine Anleitung zur Emissions- und Absorptionsspektroskopie. Leipzig, 1962.
921. Morand M., KielM. Silicates ind., 23, 248 (1958).
922. Morris A. G. C. Analyst, 90, 325 (1965).
923. Morrison G. H. Trace analysis: physical methods. N. Y. — London— Sydney, Interscience, 1965.
924. Morrison G. H., Cosgrove J. F. Analyt. Chem., 27, 810 (1955).
925. Morrison G. H., Freiser H. Analyt. Chem., 34 , 64 (1962).
195
926. Morrison G. H., Rupp R. L. Analyt. Chem., 29, 892 (1957); U. S. Dept Com. Off. Techn. Serv. PB Rept., 153, 682 (1960).
927. Morrison I. R., Wilson A. L. Analyst, 88, 100 (1963).
928. Morrison I. R., Wilson A. L. Analyst, 88, 446 (1963).
929. Mtschedlow-Petrossian О. P., Babuschkin IF. /. Silikattechnik, 9, 209 (1958).
930. Mullin J. F., Riley J. P. Analyt. chim. acta, 12, 162 (1955).
931. Myllius F., GroschufE. Ber., 39, 116 (1906).
932. NachodF. C. Ion Exchange, N. Y., Acad. Press, 1949, p. 134.
933. Nagelschmidt G. Analyst, 81, 210 (1956).
934. Nakai T., Yajima S., Fujiil., Okada M. Bunseki kagaku, 8, 367 (1959).
935. NemeczE. Epitoanyag, 10, 250 (1958).
936. Newton D. C., Sanders J., Tyrrell A. C. Analyst, 85, 870 (1960).
937. Nicolas J., Legrand C. Bull. Soc. fran;. ceram., № 92, 113 (1959).
938. Niese S. Kernenergie, 7, 105 (1964).
939. NigamRamesh Chandra. Lab. Pract., 15, 863 (1966).
940. Niggly P. Z. anorg. allgem. Chem., 84, 229 (1914).
941. Norburry A. L. J. Iron Steel Inst., 101, 621 (1920).
942. Noskova M., Pika L. Energetika, 11, 503 (1961).
943. NozakiT., BabaH., ArakiH. Bull. Chem. Soc. Japan, 33, 320 (1960).
944. NozakiT., RawashinaT., Baba FL, ArakiH. Bull. Chem. Soc. Japan, 33, 1428 (1961).
945. NozakiT., ShimadaJ. Rev. Elec. Commun. Lab. (Tokyo), 9, 403 (1961)-
946. OdlingM. IF. Rep. de chim. pure, 1, 49 (1858).
947. Ordogh M., JuvanczneU. V. Tavkoslesi kutato int. kosl., 7, 113 (1962).
948. Osmun R., Wirth L. Ind. Eng. Chem., 43, 1085 (1951).
949. Par'mentierF. C. r., 94, 214 (1882).
950. Pa-sztor L. C. Analyt. Chem., 33, 1270 (1961).
951. Paul J. Analyt. chim. acta, 23, 178 (1960); 35, 200 (1966).
952. Paul J. Mikrochim. acta, 1965, 836.
953. Paul J., Pover IF. F. R. Analyt. chim. acta, 22, 185 (1960).
954. Penfield S. L. J. Chem. Soc., 1879, 27.
955. Petruk IF. Canad. Mineral., 8, 68 (1964).
956. PetzoldA. Silikattechnik,8, 511 (1957).
957. PetzoldA., Gohlert I. Tonind. Ztg., 86, 228 (1962).
958. Pfeil E. Chem. Labor, und Betrieb, 5, 580 (1954).
959. Picasso G. Metallurgia ital., 54, 394 (1962).
960. Piskos R. Wiad. chem., 14, 1 (1960).
961. Pohl F. A., Bonsels IF. Mikrochim. acta, 1962, 97.
962. Polis A. Ber., 19, 1012, 1024 (1886).
963. Poster L. Analyt. Chem., 33, 1270 (1961).
964. Potter G. Analyt. Chem., 22 , 927 (1950).
965. Prentice A., Ritchie P.D. J. Appl. Chem., 6, 21 (1956).
966. PykasT., GrabinskaR. £esz. nauk. Politechn. slqskiej, № 12,93 (1957).
967. Radczewski О. E. Ber. Dtsch. Keram. Ges., 38, 389 (1961).
968. RadellJ., Hunt P. D., Murray E.C., Burrows IF. D. Analyt. Chem., 30, 1280 (1958).
969. Ram A., Banerjee J. C., Nandi D. N. Trans. Indian Ceram. Soc., 14, 169 (1955).
970. Ramachandran V. S., Racker R. P., Patwardhan N. R. Trans. 8th Internet. Ceram. Congr. Copenhagen, 1962, p. 293.
971. Ramachandran S., Natarajan S. R. J. elektroanalyt. Chem,, 11, 230 (1966).
972. Rankama R., SahamaT. G. Geochemistry. Chicago, Univ. Chicago Press» 1950.
973. Rhedey P., RobozJ. Banyaszati lapok, 11, 402 (1956).
974. Riedel V. Chem. Techn., 18, 567 (1966),
975. RingbomA., Ahlers P. E., Siitonen S. Analyt. chim. acta, 20, 78 (1959).
976. Ristic M. Hem. ind., 14, 149 (1960).
977. Roca Adell M. Ann. Real. soc. esp. fis. у quim., B59, 345 (1963).
196
978. RochowE. G. An Introduction to the Chemistry of the Silicones. N. Y., Wiley, 1951, p. 162.
979. Rochow E., Gilman W. J. Am. Chem. Soc., 63, 798 (1941).
980. Roller P. S., Ervin G. J. Am. Chem. Soc., 62, 468 (1940).
981. Rosenbusch K. Leder, 4, 108 (1953).
982. Rosenheim A., Raibmann B., Schendel G. Z. anorg. allgem. Chem., 196, 160 (1931).
983. Rosenihaler L. Mikrochemie, 14, 363 (1934).
984. Rost R. Mikrochemicke urcovani nerostu, Praha, SPN, 1956.
985. RufE. Z. anal. Chem., 151, 169 (1956); 161, 1 (1958).
986. Rybach L. Radiochim. acta, 2, 138 (1964).
987. Saito K-, NozakiT., TanakaS., FurukamaM., Cheng H. Intern. J. Appl. Radiat. and Isotopes, 14, 357 (1963).
988. Sajo I. Acta chim. Acad. sci. hung., 6, 233 (1955); 28, 259 (1961).
989. Sajo I. Gyors modszer szilikatok kozetek, ercek, ?alakok, tiizalloanyagok stb. elemzesere. Budapest, 1954.
990. Sajo I. Kohasz. lapok, 9 , 353 (1954); 9, 400 (1954).
991. Sajo I., Barna L. Acta chim. Acad. sci. hung., 10, 19 (1956).
992. Sajo/., SiposB. Z. anal. Chem., 222, 23 (1966).
993. Sajo!., Vjvaril. Z. anal. Chem., 202, 177 (1964).
994. SchinkD.R. Analyt. Chem., 37, 764 (1965).
995. Schlecht W. G. Analyt. Chem., 23, 1568 (1951); 161, 1 (1958)
996. Schramke E. Keram. Z., 11,'№ 1, 15; № 2, 58 (1959).
997. Schrenk W. T., Ode W. H. Ind. Eng. Chem., Anal. Ed., 1, 200(1929).
998. Schucht L., Moller W. Ber., 39, 3693 (1906).
999. Schwartz M. C. Ind. Eng. Chem., Anal. Ed., 14, 893 (1942).
1000. Schwarz R. Angew. Chem., 67, 117 (1955).
1001. Schwarz R. Z. anorg. allgem. Chem., 276, 33 (1954).
1002. Schwarz R., Kessler A. Z. anorg. allgem. Chem., 263, 15 (1950).
1003. Schwarz R., Sexauer W. Ber., 59, 333 (1926).
1004. Schwarzkopf 0., Heinlein R. Wright Air Devel. Center, Techn. Rept., 56, 19 (1957).
1005. Schwiete H. E., Stollenwerk H. Arch. Eisenhiittenwesen, 28, 17 (1957).
1006. Seiser H. Sprechsaal, 86, 337 (1953).
1007. Shamy H., IssaR. Egyp. J. Chem., 1, 247 (1958); 2, 91 (1959).
1008. Shapiro L., Brannock W. M. U. S. Geol. Surv. Circ., № 165 (1952).
1009. Shell H. R. Am. Ceram. Soc. Bull., 28, 349 (1949).
1010. Shell H. R. Analyt. Chem., 26, 591 (1954); 27, 2006 (1955).
1011. Shell H.R. Treatise cn analytical chemistry, pt. 2. Analytical chemistry of the elements, v. 2. N. Y.— London, Interscience Publishers, 1962, p. 107—206.
1012. Shell H. R. U. S. Bur. Mines Rept. Invest., Ks 4420 (1949).
1013. Shell H.R., Craig R. L. Analyt. Chem., 26, 996 (1954).
1014. Shell H. R., Craig R. L. U. S. Bur. Mines Rept. Invest., № 5158 (1956).
1015. Shell H. R., Martin G. W. U. S. Bur. Mines Rept. Invest., № 5557 (1959).
1016. Shugart P. L. Rock. Prod., 57, 92, 94 (1954).
1017. Signer R., Gross H. Ann. 488, 56 (1931); 499, 158 (1932).
1018. Sir Z.}_ Romers R. Chem. listy, 50, 88 (1956).
1019. Smith G. F. Mixed Perchloric, Sulfuric and Phosphoric Acids and their Applications in Analysis. Columbus, Ohio, 1942.
1020. Smith B. Acta Chem. Scand. 11, 579 (1957).
1021. SmithO. C. Identification and qualitative chemical analysis of minerals. N. Y., 1953.
1022. Smolczyk H. G. Tonind. Ztg., 86, 261 (1962).
1023. Sollas W. J. Trans. Roy. Irish Acad. (Dublin), 29, 471 (1887).
1024. Sonnenschein W. Z. anal. Chem., 168, 18 (1959).
1025. Sosman R. B. The Properties of Silica. N. Y., 1927.
1026. Sosman R. B. Trans. Brit. Ceram. Soc., 54, 655 (1955).
1027. Spialter L., BallesterM. Analyt. Chem., 34, 1183 (1962).
197
1028. Spielhaczek H. Z. analyt. Chem., 119, 1 (1940).
1029. StadelerD. Giesserei., technk.-wiss. Reih., 16, 853 (1956).
1030. Staufenberger 0. Sprechsaal, 94, 9 (1961); 96, 155 (1963).
1031. StolbaF. J. prakt. Chem., 89, 129 (1863); 96, 175 (1865).
1032. StolbaF. Z. anal. Chem., 12, 390 (1863).
1033. Stoll K- Z. anal. Chem., 112, 81 (1938).
1034. Strickland J. H. D. J. Am. Chem. Soc., 74, 862 (1952).
1035. Strickland J. H. D., Parsons T. R. A Practical Handbook of Sea Water Analysis. Ottawa, 1968.
1036. Stubican V. Arh. hig. rada i toksikol., 8, 166 (1957).
1037. Suhr N.H., I ngamells С. 0. Analyt. Chem., 38, 730 (1966).
1038. Sundaram А. К., Sundaresan M. Analyt. chim. acta, 19 , 601 (1958).
1039. Su Yao-Sin, Campbell D. E., Williams J.P. Analyt. chim. acta, 32, 559 (1965).
1040. Suzuki T. Nippon Kagaku Zasshi, 82, 696 (1961).
1041. Takahashi J. Bunseki kagaku, 13, 193 (1964).
1042. Takahashi T., Miyake S. Bunseki kagaku, 9, 579 (1960).
1043. Takahashi T., Miyake S. Taianta, 4, 1 (1960).
1044. Talasek V., Ocenasek M. Energetika (CSSR), 13, 608 (1963).
1045. Talvitie N. A. Analyt. Chem., 23, 623, 664 (1951).
1046. Talvitie N. A. J. Am. Hyg. Assoc., 25 (2), 169 (1964).
1047. Thilo E., FunkH., Wichman E. U. Ab. Dtsch. Akad. Wiss. Berl., KI. Mat. allg. Naturwiss., 4, 1 (1950).
1048. Thomann H., Gerstmann 0., Beyer B. Giessereitechnik, 6, 244 (I960); РЖХим., 1961, 8 К 309.
1049. Thomas F. Silikattechnik, 10, 129 (1959).
1050. Thompson В. A., Strause В. M., Leboeuf M.B. Analyt. Chem., 30, 1023, (1958).
1051. Tornebohm A. E. Tonind. Ztg., 21, 1148 (1897).
1052. Tornebohm A. E. Zement, 4, 287 (1903).
1053. ToyC.H., Van SantenR. T. Analyt. Chem., 36, 151 (1964).
1054. Tredwell W. D., Wieland W. Helv. chim. acta, 13, 842 (1930).
1055. Trostel L. J., Wynne D. J. J. Am. Ceram. Soc., 23, 1 (1940).
1056. Trudell L., BoltzD.F. Analyt. Chem., 35, 2122 (1963).
1057. Tuma J. Mikrochim. acta, 1962, 513.
1058. Turner V. L. US Pat., № 3032401 (1.05.1962).
1059. VahlF. Tonind. Ztg., 80, 23, 397 (1956).
1060. Vail J. G. Soluble Silicates. N. Y., Reinhold, 1952.
1061. Vanderslice T. A., Whetten N. R. J. Phys, and Chem. Solids, 25, 513 (1964).
1062. Ve^sernyes L. Acta chim. Acad. sci. Hung., 28, 111 (1961).
1063. Veqsernyes L. Acta geol. et geogr. Univ. Comenianae. Geol., № 6, 428 (1961).
1064. Veqsernyes L. Magyar Kern, folyoirat, 72, 377 (1966).
1065. Ve^sernyds L. Tavkozlesi kutato int. kozl., 11, 121 (1966).
1066. Veessernyds L., Hangosl. Z. anal. Chem., 208, 407 (1965).
1067. Vezsernyds L., Zombori V. Tavkozlesi kutato int. kozl., 7, 428 (1962).
1068. VenialeF. Ceramica, 16, № 7, 55 (1961).
1069. VenialeF. Ceramica, 16, № 8, 49 (1961).
1070. Verbestel M. Chim. ind., 57, 41 (1947).
1071. Vilnat J., Voinovitch I. Publ. Group, avanc. method, spectrogr., 389 (1961); 307 (1962).
1072. Visintin B., Monteriolo S. Ann. chimica, 51, 266 (1961).
1073. VoinarA.O. Biokhimija, 2, 19 (1946).
1074. Voinovitch 1. A. Bull. Group, fran?. argiles, 12, 3 (1960).
1075. Voinovitch I. A. Bull. Soc. franf. ceram., № 33, 69 (1956).
1076. Voinovitch I. A. Bull. Soc. fran;. ceram., № 39, 65 (1958).
1077. Voinovitch I. A. Chim. analyt., 39 , 454 (1957); 40 , 332 (1958).
1078. Voinovitch I. A., Debras-Guedon J., Louvrier J. L’analyse des silicates. Paris, Hermann, 1962.
1079. Volk R. J., Weintraub R. L. Analyt. Chem., 30, 1011 (1958).
198
1080. Wacykiewicz R. Hutnik, 21, 5 (1954).
1081. WadelinC., Mellon M. G. Analyt. Chem., 25, 1668 (1953).
1082. Wakamatsu S. Japan Analyst, 9, 22 (1960).
1083. WartenbergH. Z. anorg. allgem. Chem. 283, 372 (1956).
1084. Washington H. S. Am. J. Sci., 4, 61 (1900).
1085. Washington H. S. The chemical analysis of rocks. N. Y., Wiley, 1930.
1086. Watanabe H. Bunseki kagaku, 12, 1196 (1963).
1087. ll'W/ IT. A. A new Approach to Surface Chemistry and to Heterogeneous Catalysis. N. Y., 1951, p. 46—48.
1088. Weil W. A. Penn. State Univ. Bull., № 57, 47 (1951).
1089. Weisberg L. R. Analyt. Chem., 38, 31 (1966).
1090. Weiss A., Weiss A. Z. anorg. allgem. Chem., 276, 95 (1954).
1091. Weiss G., Sieger H. Z. anal. Chem., 119, 245 (1940).
1092. Weitz E., Frank H., Schuhard M. Chem. Ztg., 74, 256 (1950).
1093. Wiberg E., Michaud H. Z. Naturforsch., 96, 500 (1954).
1094. Wickbold R. Z. anal. Chem., 171, 81 (1959).
1095. WilkP. Hutnik, 23, 1 (1956).
1096. Willard H. H., Cake W. E. J. Am. Chem. Soc., 42, 2208 (1920).
1097. Willstatter R., Kraut H., Lobinger K. Ber., 58, 2462 (1925); 61, 2280, 2291 (1928); 62, 2027 (1929).
1098. Wilson A. L. Analyst, 90, 270 (1965).
1099. Wilson H. N. Analyst, 74, 243 (1949).
1100. Wilson H. N., Skinner J. M. Recucil trav. chim., 79, 574 (1960).
1101. Winchell A. N., Winchell H. Elements of optical mineralogy. N. Y., Wiley, 1951.
1102. Woodman J. Am. Chem. Soc., 23, 96 (1901).
1103. Zymny E. Cement. Wapno, Gips, 12, 152 (1956).
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА *
1104. Аналитическая химия редких металлов и полупроводниковых материалов (сб. статей). М., Изд. Моск, дома научно-техн, пропаганды, 1970.
1105. БилыпюковаЭ. П., Шапиро К. Э. Стекло. Труды НИИ стекла, № 1 (137).
М., Промстройиздат, 1969, стр. 53.
1106. Богданова В. И. Ж. аналит. химии, 25, 332 (1970).
1107. Васильева М. Г., Бешикдашван М. Т. Ж- аналит. химии, 25, 1592 (1970).
1108. Доценко О. В. Зав. лаб., 28, 279 (1962).
1109. Дорохова Е. И., Опарина Л. И. Ж- аналит. химии, 25, 544 (1970).
1110. Золотов Ю. А., Кузьмин Н. М. Экстракционное концентрирование. М., «Химия», 1971.
1111. Кутейников А. Р., Киревина Г. П., Степанова А. Н. Зав. лаб., 35,
7 (1969).
1112. ЛебедеваЛ. И., Птушкина М. И. Сб. «Применение органических реагентов в аналитической химии». Изд-во ЛГУ, 1969, стр. 138.
1113. Луговой С. В., Рязанов И. П. Зав. лаб., 33, 688 (1967).
1114. Ляликов Ю. С., Аронина И. В. Ж- аналит. химии, 25 , 978 (1970).
1115. Методы получения и анализа веществ особой чистоты (сб. статей). М., «Наука», 1970.
1116. МышляеваЛ. В., Седова И. В., Отрощенко И. Г., Пекшева В. В. Труды МХТИ им. Д. И. Менделеева, 58 , 262 (1968).
1117. Немодрук А. А., Палей П. И., Безрогова Е. В. Ж- аналит. химии, 25,
319 (1970).
1118. Палыиин Е. С., Палей П. И., Давыдов А. В., Иванова Л. А. Ж- аиалит. химии, 24, 797 (1969).
* В этот список включены в основном издания, выщедшие в свет после сдачи рукописи в Издательство.
199
1119. Силаева Е. В., Петрова Н.П,, Курбатова В. И., Степин В. В., Сту-денская Л. С., Барбаш Т. Л. Труды ВНИИ стандартных образцов и спектральных эталонов, 5, 136 (1969).
1120. Хачатурян О. Б., Мышляева Л. В., Седова И. В., Микуленок Г. Е. Ж. прикл. химии, 43, 1617 (1970).
1121. ШашураМ.В., Яковлев П.Я-, Коротеева Н. Г. Зав. лаб., 35 1442 (1969).
1122. Шемякин Ф. М., Степин В. В. Ионообменный хроматографический анализ металлов. М., «Металлургия», 1970.
1123. Щербаков В. Г., Вейцман Р. М., Антонова Р. А. Труды Всес. и.-и. и проекта. ин-та тугоплавких металлов и твердых сплавов, № 10, 192 (1970).
1124. Ackermann L., Lange Y. Taianta, 17, 693 (1970).
1125. Boltz О. F., De-Vries T., Mellon M. G. Analyt. Chem., 21, 563 (1949).
1126. Campos Oritiz M. An. Fac. guim. у farm. Univ. Concepcion, 19, 21 (1969).
1127. Debras-Guedon J. Chim. analyt., 52, 145 (1970).
1128. Eckert H. J., KonigH., Match. D., ThanE. Z. anal. Chem. 9, 464 (1969).
1129. Golkowska A. Chem. analit. (Polska), 14, 803 (1969); 15, 59 (1970).
1130. Hargis L. G. Analyt. Chem., 42, 1497 (1970); Anal. chim. acta, 52, 1 (1970).
1131. Ingle I. D., Cronch S. R. Analyt. Chem., 43, 47 (1971).
1132. Kazuyoshi O. Japan Analyst, 15, 347 (1966).
1133. KemulaW., Rosolovsky S. Roszn. chem., 36, 1417 (1962).
1134. Kollar R., Plichon V., Saulnier J. Analyt. chim. acta, 50, 457 (1970).
1135. Nozaki T., Yatsurugi X., Akiyama N. J. Radioanalyt., 4 , 87 (1970).
1136. Schweikert E. A., RookH.L. Analyt. chem., 42, 1525 (1970).
1137. Tatsumoto M. Analyt. Chem., 41, 2088 (1969).
1138. VldcilF., Bock R. Z. anal. Chem., 249, 19 (1970).
1139. Wicker W., HoebbelD. Z. anorg. allgem. Chem., 366, 139 (1969).
1140. WuF.F.H., GbtzJ., Jamieson W. D., Masson C. R., J. Chromatogr., 48, 515 (1970).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адсорбционные соединения кремневой кислоты 30, 60
Адсорбция примесей 21, 53
Азотная кислота 50, 57, 58, 76—78, 84 , 99, 105, 107, 109, 146
Алкалиметрическое определение кремния в карборунде 135 кремнийорганических соединениях 79, 144 растворимых стеклах 121 силикатах 83 стеклах 129 цементе 124
Алкалоиды 26
Алюминия хлорид 64, 65
Алюмосиликаты природные 8, 10 11 структура 11
Альбумин 60
Амидопирин 68
Амины 25, 39, 81, 89
Аморфная кремневая кислота избирательные растворители 157 обнаружение в присутствии силикатов 33
других окислов 33 определение 157
Аниониты 40, 170
Анодное растворение кремния 9, 41
Арбитражные методы 125, 129
Бензидин 25, 31, 32, 37, 70, 81, 87, 91, 100
Бесстружковый анализ 28, 34, 129 Бор
определение в полупроводниковых материалах 166—171
отделение от кремния 39, 65, 172
Борная кислота 46, 106, 109, 135
Борнометиловый эфир 65, 172
Бромид-броматный метод 27, 72
Бутлерова ряды 10,16
теория полисоединений 10, 16
Весовые формы определения кремния 51, 66
двуокись кремния 20, 55
кремнемолибдаты органических оснований 66—69
условия получения 67,68 кремнефториды органических ос-
нований 70, 89
Восстановители для кремнемолибденовой кислоты
1 - амино - 2 - нафтол - 4 - сульфокислота 100, 102, 106, 124, 140
аскорбиновая кислота 100, 124 бензидин 32, 100
гидразин 100 гидразинсульфат 100 гидроксиламин 100 гидрохинон 100 глюкоза 100 калия иодид 100, 105, 135 метол 100, 124 метолово-сульфитный 124 олова оксалат 100 олова хлорид 32, 98, 100 натрия тиосульфат 100 а-нитрозо-Р-нафтол 100 сульфит-сульфатный 101, 128 фенилгидразин 100 эйкоген-сульфитный 100, 106, 124, 140
Высокочистые соединения кремния 166
иодсилан 166
двуокись 166 карбид 165 особенности их анализа 165, 166 применение в полупроводниковой технике 8, 9, 165
содержание примесей 166 трихлоргидросилан 166, 172 хлорсилан 166, 172
201
Гексаметилентетрамин 38, 68
Гексиламин 68
Гетерополисоединения кремния 23 аналитическое применение 23, 37, 66, 71, 81, 94
качественный анализ 23
реакция образования 23 спектрофотометрия 23, 94 титриметрия 23, 71, 81
Гидролиз кремнийорганических со-
единений 16, 141
Глицерин 66, 157
Глицеринкремпевая кислота 157
Гравиметрическое определение кремния 52
в присутствии
бора 65
фтора, предотвращение потерь 62, 64, 66, 69
в форме двуокиси кремния 51
с желатиной 60
в форме
кремнемолибдатов органических оснований 66, 69
кремнефторидов органических оснований 70
Дегидратация выпариванием
в кислой среде 51, 54, 129
в присутствии желатины 60, 129
осложнения 53
основные операции 52
в щелочной среде 63
Дегидратирующие агенты 52, 54, 56, 60
Диализ 13
Диантипир илметан 69, 86
2,4-Диметилхинолин 38, 67, 68
Диметилглиоксимат никеля 33
Дитизон 40
Дифференциальная колориметрия 124
Желатина, механизм действия 62
Желатиновый метод 60, 124, 130
Идентификация веществ, содержащих кремний 33
ИК-спектроскопия в фазовом ана-
лизе 153, 158, 162
Ионный обмен 40, 111
Калия
бисульфат 126
бифторид 46
гидроокись 76—78, 83, 92, 144
карбонат 29, 44, 47, 135
кремнефторид 7, 25, 76
растворимость 76
титрование 25, 76
нитрат в плавнях 44, 128 пиросульфат 126, 137, 150 хлорат в плавнях 44
Капельные реакции на кремний 32, 33
Каталиметрические методы определения примесей 172
Катиониты концентрирование 40, 170 удаление примесей 40, 111 Качественные реакции в растворах 28 кремнемолибдат-иона 28, 31, 32 кремнефторид-иона 25, 28, 30 силикат-иона 29 в сухом состоянии 30
Кварц 11, 14, 153 определение
в горных породах 153, 154
в керамическом сырье 153
в рудничной пыли 154, 155, 162
в сульфидных рудах 156 методами
дифференциального термине ского анализа 152, 162
ИК-спектроскопии 153 петрографическим 152, 157 термогравиметрическим 153 рентгеновским 152, 157 химическими 153
Кинетический метод 23, 112, 122, 172
Коагулянты 60, 123
Коагуляция золя кремневой кислоты 60
Кобальта аммиакат 31, 75
Кодеин 31
Колориметрическое определение
кремния 22, 56, 122, 124, 128, 155
Комплексные этилсиликаты 15
Комплексон III, применение для определения кремния 74, 75, 122 разложения образцов 37, 124, 155, 161, 172
Кондуктометрическое титрование • кремнемолибденовой кислоты 83, 122
кремнефтористоводородной кислоты 89, 122
Концентрирование примесей 166 ионным обменом 170 испарением основы 165, 167, 170
Коррозия
стеклянной посуды 42 тиглей 45
Коэсит 8, 22
Кремневанадиймолибденовая кислота 23, 27, 38
202
Кремневольфрамовая кислота 38 осаждение 8-оксихинолином 70
Кремнемолибденовая кислота
гидролиз 2G, 82
изомерия 97
определение в присутствии других
кислот 26, 82
основность 26, 95
получение 22
реакции
восстановления 27
кислотно-основного взаимодействия 26, 73
осаждения 26, 73
свойства, применение для определения кремния 26, 38, 64, 66
гравиметрическим методом 26, 66
формы осаждения 26, 66
весовые формы 27
кислотно-основным титрованием
• в воде 26
в неводных растворах 73 редокс-методами 26, 27, 71, 127 спектрофотометрическими методами 27, 64, 124
термическое разложение соединений 27
Кремнефториды
обнаружение 25, 31
образование 22, 23
реакции
гидролиза 25, 75
кислотно-основного взаимодействия 25
комплексообразования 37
осаждения 37, 76
свойства, применение для опреде-
ления кремния 25, 37, 75 Кремнефтористоводородная кислота образование 23, 75 определение
в смеси с фтористоводородной кислотой 25, 31, 75, 80, 81, 86, 91, 92
кондуктометрическим титрованием 25, 83
потенциометрическим титрованием 25, 81
реакции
осаждения
неорганические осадители 25, 76
органические осадители 25, 81 кислотно-основного взаимодей-
ствия 24, 25, 81, 91
термического разложения 25, 38
Кремний 7
изотопы природные и искусственные, период полураспада 7 исторические сведения 7 координационное число 7, 9—11, 17, 19
потенциалы ионизации 7
природные соединения 8, 9
в биологических объектах 8
в земной коре 8
в метеоритах 8
радиус атома 7
распространенность 7
растворимость 9
свойства 8
степень окисления 7, 9
элементный, получение высокочистого кремния 7, 9 модификации 8 применение 8, 9, 165
Кремнийорганические соединения аналитическая характеристика 13 методы разложения 34, 141 минерализация 34, 39, 140 определение кремния в летучих
КОС 143, 145, 146
полумикрометодом 67
реакции
кислотно-основного взаимодействия 81, 83, 141
поликонденсации 12, 17
термического разложения 12
Кремния аналитическое определение качественное 28
в объектах
воде 122
глинах 125
каолине 126
карбонатных породах 122
карборунде 134
кварцитах 72
керамических материалах 129 кремнийорганических соеди-
нениях 67, 74, 79, 81, 83, 141
криолите 69
металлах 70
плавиковом шпате 69
растворимом стекле 120
силикатах и горных породах 66, 83, 98, 127
силицидах металлов 133
сплавах 136
стеклах 70, 128
цементах 70, 123
шлаках 70, 75, 77, 130
спектральным методом 28
химическими методами в рас-
творах в виде
кремнемолибдат-ионов 28
203
кремнефторид-ионов 28 силикат-ионов 28 при сплавлении 30 количественное
гравиметрическими методами в форме двуокиси кремния 51 кремнемолибдатов органических оснований 66, 92 кремнефторидов органиче-
ских оснований 70, 81
комплексонометрическим методом 74, 122
методами кислотно-основного титрования 73, 81
редокс-методами 71 Кремния двуокись гидратация 14 модификации 22, 151 — 154 определение в вещественном анализе
методом ДТА 152
ИК-спектроскопии 153 петрографическим 163 по плотности 152 рентгеновским 152 термогравиметрическим 153 химическими 155, 157 полимерность 12 растворимость
зависимость от условий 14, 16 модификации 14
- pH 15, 17 температуры 14 твердофазные реакции 47 термодинамические данные 9, 17 Кремния карбид 13, 53, 56 Кремния соединения
с азотом 9 водородом 9 кислородом 9 металлами 9 углеродом 13
Кремния элементного определение в карборунде 134, 161 в нитриде 161 в силицидах 34, 161 сплавах 34, 161
Кристобалит 8, 152 Купферон 40 Кур кумин 172
Малахитовый зеленый 30, 60 Масс-спектрометрия 167
Метанол 65, 67, 75, 87, 90, 128, 172 Метиленовый синий 30, 33, 34, 60 Методы определения кремния 81, 94, 131, 132 физико-химические амперометрический 84, 92 кондуктометрический 83, 98, 122
потенциометрический 81 фотометрические 94, 124 физические 114
по микротвердости 117
по плотности 116 спектральные ИЗ по ТЭДС 117 химические
гравиметрические 52, 66 титриметрические 71
Микропримеси, методы определения 165 физико-химические 171 физические 166, 172 химические 171
Микрореакции на кремний 31
Минералогические данные о соединениях кремния 10
Минералы разложение кислотами 11, 43, 50 сплавлением 43 растворами щелочей 50 растворимость 11
Молибденовая синь, получение 32 применение в анализе 32, 98 свойства 98
Натрий металлический, плавень 46 Натр ия
гидроокись 73, 76, 80, 87, 105, 133, 144, 146
карбаминат 40 карбонат 44, 48, 125, 127, 133 кремнефторид 31, 76
перекись 67, 79, 122, 133, 147
Неводные растворители, применение в качестве среды 13, 25, 43, 81, 122, 172
для экстракции 39, 74, 82, НО, 137, 140
Нейтроноактивационный метод применение 169, 170 чувствительность 169
Нерастворимый остаток 122
Обнаружение кремния в водах 32 кремнийорганических соединениях 34 растворах щелочей 32 силицидах 34 сплавах 34
Обнаружение свободной кремневой кислоты
в присутствии других окислов 33 силикатов 33, 157
в различных материалах 151, 158 8-Оксихинолин 26, 38, 66, 68, 72
204
Олова хлорид 32, 98, 100
Определение кремния количественное в
водах 32, 138
кремнийорганических соедине-
ниях 74, 79, 83, 140
металлах 135
растворах щелочей 32
силикатах 120
сплавах
цветных металлов 137
черных металлов 136
элементокремнийорганических со-
единениях 74, 83, 150
Ортокремневая кислота 10
выделение из растворов 53
дегидратация 21, 24, 52
константы ионизации 13, 22
мономер 10, 13, 19, 23
поведение в условиях анализа 20,
24, 41
полимеры 10, 20
растворимость 14
зависимость от pH 15, 16
потери 21, 51, 54, 55
растворы
истинные 14, 51, 54, 120
коллоидные 14, 51, 120
термодинамические данные 17
устойчивость 17
реакции 23
кислотно-основного взаимодей-
ствия 23, 71
комплексообразования 23, 49, 64
конденсационной полимеризации 16, 24
осаждения 23, 29
титрование
амперометрическое 84, 92
кислотно-основное 22, 23
комплексонометрическое 50, 75
Основания органические 22, 26, 29,
38, 80, 89
Оствальда—Фрейндлиха уравнение 14
Отбор проб 42
Отделение кремния
осаждением в виде
двуокиси кремния 36, 49
кремнефторида
калия 36, 49, 76
органических оснований 37, 49, 81
кремнемолибдатов органических оснований 37, 81
примесей 32, 38
отгонкой в виде
силанов 39
фторида кремния 25, 30, 36, 38,
48, 56, 108, 127, 109, 129, 167
соединений сопутствующих элементов 36, 39, 111, 172 хроматографическое 40 экстракцией
гетерополисоединений 39, 74, 82, НО, 140
кремнефтористоводородной кислоты 39
электролизом
анодным растворением основы 41 выделением примесей на ртутном катоде 41
Ошибки определения кремния, потери от растворимости 18, 21 систематические 131, 132 статистическая обработка результатов 131, 132
Парра бомба 67, 147
Пиридин 26, 38, 69, 127
Пирокатехин 24
Пирокатехинкремневая кислота 24, 28, 50
Плавни, выбор 29, 35, 44, 48, 161 карбонатные 31, 44, 63, 122, 125, 129 133
кислые ’ 46, 126, 127, 129, 156 смешанные 44—46, 64, 129, 156 щелочные 21, 44, 129, 133
Полимерная кремневая кислота обнаружение 29 поведение в растворах 52, 53
Полупроводниковые материалы 9, 166
Полярография 92, 172
Потенциометрическое титрование 81
Примеси в кремнии высокой чистоты, методы определения 165 физико-химические 171 физические 166—168 химические 171
Прокаливание анализируемых материалов 48, 122, 126
осадков 21, 24 осложнения 24, 53
Посуда для
спекания 46, 47 сплавления 29, 44—46 хранения анализируемых веществ и реактивов 42, 166
материалы железо 46, 133, 157 золото 46 кварц 155, 166 кремний 167 никель 46, 67 платина 44, 54, 67, 124, 155 политетрафторэтилен 54, 124,166 полиэтилен 42, 54, 67, 121, 166
205
серебро 46 сталь 156, 165 стекло 42 тантал 54, 128
Радиоактивационные методы применение 169 сравнение 171 чувствительность 169—171
Разложение соединений кремния кремнийорганических 34, 43, 141 неорганических 28, 57 плавов 49, 57 потери при разложении 57 реактивы для разложения
кислоты 28, 48, 50, 127, 156, 172
щелочи 50, 156 спеков 49
Растворение соединений кремния 42 плавов 49 спеков 49, 78
Растворимость соединений кремния 43, 54
Реактивы
очистка 42, 167 хранение 42, 167 чистота 166
Родамин 30
Сафранин 30, 33, 60
Серебра хромат 33
Серная кислота 37, 50, 56, 82, 83, 92, 107, 137, 144
Силаны 39, 144
Силикат-ион, качественные реакции 29
Силикаты классификация 10 разложение 156 растворимость 43, 54, 156 растворимые 23
Силициды, разложение 74, 133
Силоксаны 16
Соосаждение примесей осложнения в анализе 53, 54, 57 предотвращение осложнений 37, 53 растворение 37
Спекание 46, 125
Спектральные линии кремния 28,115
Спектральные методы
ИК-спектроскопия 153, 158, 162 у-спектрометрия 169 подготовка проб 171 эмиссионный 120, 126 подготовка проб 168 стандарты внутренние чувствительность 167, 168 у-Спектрометрия 169 Спектрофотометрия 94, 171 Сплавление соединений кремния
аппаратура 29
биологические материалы 45
в бомбах 45, 141, 147
посуда 29, 45
применение ЗДМ 44
реакции 44
с металлами 46
силикатов 44
силицидов 45
фторсиликатов 46, 62
Сплавы
обнаружение кремния 34
определение кремния 136
Стилоскоп 28
Стишовит 8
Стоваин 31
Стрихнин 31
Структура соединений кремния 10
Твердофазные реакции 47
влияние дисперсности 47
Термический анализ 152
дифференциальный 152, 162
термогравиметрия 153
Тетраалкоксисиланы, гидролиз 15,
80, 141
Тетрабутиламмония гидроокись, тит-
рант 82
Тетраэтил аммония гидроокись 83
Тиокарбамид 38
Титриметрические методы определе-
ния кремния 71
кислотно-основное титрование 73, 75, 81, 123, 129
комплексе но метр ия 74, 122
Тридимит 152
Триметиламин 68
ТЭДС, измерение 117
Ускоренные методы определения
кремния 59
алкалиметрический 121, 124, 129
гравиметрический
в виде двуокиси кремния 126
с желатиной 59, 63, 123
комплексонометрический 74
отгонкой, косвенные 80, 109
спектрофотометрические 59, 94, 121
Фазовый анализ
методы 151
расчетные 157, 159, 160
физико-химические 157
физические 162, 163
химические 157
объектов
глин 157, 162
горных пород 159, 162, 163
динаса 152
карборунда 134, 161
206
клинкера 159
кремневых сплавов 135
огнеупоров 161, 162
силикатов 157, 162
силицидов 161
стекол 162, 164
цементов 163
шлаков 161
селективные растворители 153, 157, 159, 161
я-Фенилендиамин 37, 100
Физические методы определения
кремния 27, 43, 114
Флуоресцентный метод 113, 172
Фосфор, отделение от кремния 40,
172
Фосфорная кислота 153, 154
Фото колориметрия, осложнения 103
Фториды
осложнения в анализе 37, 62, 105
предотвращение осложнений 37, 64, 105
Фтористоводородная кислота применение 22, 62, ‘74, 75, 80, 81, 86, 91, 92, 123
ультраочистка 166
Фуксин 30
Хелатные комплексы 38
Хинина сульфат 31
Хинолин 26, 38, 68, 69
Хлористоводородная кислота 50, 54, 72, 74, 76, 79, 80, 87, 93, 123, 126, 144, 146
Хлорная кислота 50, 57, 108,
130, 133, 136, 146
Хлороформ 40, 111
Хроматография
газовая 41
ионообменная 111, 170
Цинк 37 Цинка аммиакат 63 окись 63
Цинхонин 26
Экстракционно -титриметрические методы 74, 81
Экстракционно-фотометрические методы НО, 172
Экстракционные методы разделения 39, 40
Экстракция гетерополисоединений 39, 74 81, НО, 137, 172
кремнефтористоводородной кислоты 39
кремнийорганических соединений 39
сопутствующих элементов 40
Электролиз 41
Электронный парамагнитный резонанс 171
Этанол, удаление бора 66
Этилендиамин 34, 38, 98
ОГЛАВЛЕНИЕ
От редколлегии ......................'.............................. 3
Предисловие ........................................................ 5
Глава 1
Свойства кремния и его соединений .................................. 7
Общая характеристика элемента ........................... 7
Распространенность и применение ................ 7
Физические и химические свойства ............................... 8
Особенности строения соединений кремния ...................... 10
Химико-аналитическая характеристика соединений кремния .... 13
Глава 2
Методы качественного обнаружения кремния 28
Реакции силикат-иона .......................................... 29
Реакции кремнефторид-иона ..................................... 30
Реакции кремнемолибдат-иона ................................... 32
Идентификация некоторых веществ, содержащих кремний .... 33
Глава 3
Методы отделения кремния от сопутствующих элементов................ 36
Методы осаждения .............................................. 36
Методы дистилляции ............................................ 38
Экстракционные методы ......................................... 39
Хроматографические методы ..................................... 40
Электролиз .................................................... 41
Глава 4
Подготовка вещества к анализу ..................................... 42
Отбор и измельчение средней пробы вещества..................... 42
Растворение или разложение веществ ............................ 42
Сплавление .................................................... 43
Спекание ...................................................... 46
Предварительное прокаливание ................................
Разложение и растворение продуктов сплавления и спекания . .
Глава 5
Гравиметрические методы .........................................
Определение в форме SiO2 ....................................
Другие гравиметрические методы ..............................
209
Глава 6
Титриметрические методы .......................................... 71
Методы, основанные на свойствах кремнемолибденовой кислоты . 71
Методы, основанные на свойствах кремнефтористоводородной кислоты 75
Электрохимические методы ..................................... 81
Глава 7
Фотометрические методы ........................................... 94
Строение и некоторые свойства кремнемолибденовой кислоты ... 95
Ионы, мешающие определению кремния ............103
Флуоресцентный метод ...........................................ИЗ
Глава 8
Прочие методы .....................................................114
Спектральные методы ...........................................114
Определение по плотности ................116
Определение методом микротвердости ...........................117
Термоэлектрическое определение ...............................117
Определение по удельному сопротивлению .......................119
Глава 9
Определение кремния в природных и технических объектах............120
Определение кремния в некоторых природных и синтетических силикатах и горных породах .....................................120
Определение кремния в силицидах металлов и других тугоплавких соединениях ..................................................133
Определение кремния в металлах и сплавах......................135
Определение кремния в природных водах и конденсате............138
Определение кремния в кремнийорганических соединениях .... 141
Определение кремния в элементокремнийорганических соединениях 150
Глава 10
Методы вещественного (фазового) анализа соединений кремния........151
Определение двуокиси кремния .................................151
Глава 11
Определение примесей в кремнии и некоторых его соединениях .... 165
Физические методы ............................................166
Физико-химические и химические методы ........................171
Литература .......................................................173
Предметный указатель .............................................201
Аналитическая химия кремния (серия «Аналитическая химия элементов»). М ы ш-л я е в а Л. В., Краснощеков В. В. М., «Наука», 1972, стр. 212.
В книге изложены теоретические основы аналитической химии кремния, отражено ее современное состояние. Обобщены исследования последних лет, направленные на разработку новых химических, физико-химических и физических методов определения, отмечены преимущества и недостатки отдельных методов. Критически рассмотрен большой литературный материал по вопросу аналитической химии кремния.
Приведенная в книге библиография дает возможность использовать книгу как справочник.
Киига предназначается для химиков-аналитиков научно-исследовательских и заводских лабораторий, для специалистов в области геохимии, химии и технологии элементо-оргаиических соединений, силикатов, высокочистых веществ, металлов и сплавов.
Таблиц 4. Иллюстраций 10. Библ. 1140 казн.
Лидия Васильевна Мышляева, Валентин Васильевич Краснощеков
Аналитическая химия кремния
Утверждено к печати
Ордена Ленина Институтом геохимии и аналитической химии им. В. И. Вернадского Академии наук СССР
Редактор М. П. Волынец
Редактор издательства Р. А. Баранова. Художественный редактор Н. Н. Власик Технический редактор Р. Г. Грузинова
Сдано в набор 30/VI 1972 г. Подписано к печати 30/Х 1972 г.
Формат 60X90716. Уел. печ. л. 13,25. Уч.-изд. л. 15,2. Бумага № 2. Тираж 2600. Тип. зак. 1446. Т-17728. Цена 1 р. 15 к.
Издательство «Наука». 103717 ГСП, Москва, К-62, Подсосенский пер., 21
Набрано в Чеховском полиграфкомбинате Главполиграфпрома Государствеииого комитета Совета Министров СССР по делам издательств, полиграфии и книжной торговли г. Чехов, Московской области
Отпечатано во 2-й типографии издательства «Наука».
121099, Москва, Г-99, Шубинский пер. 10