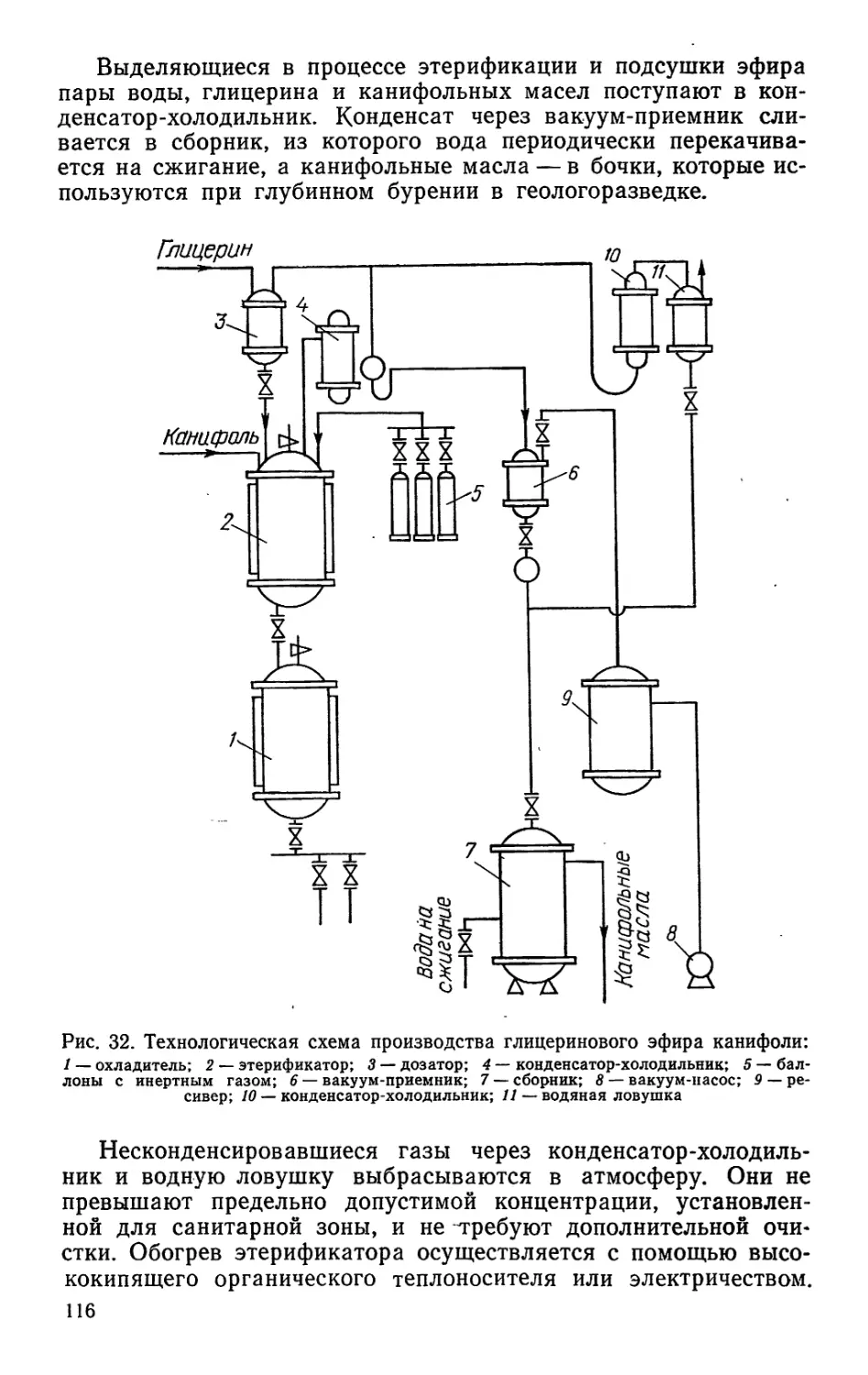
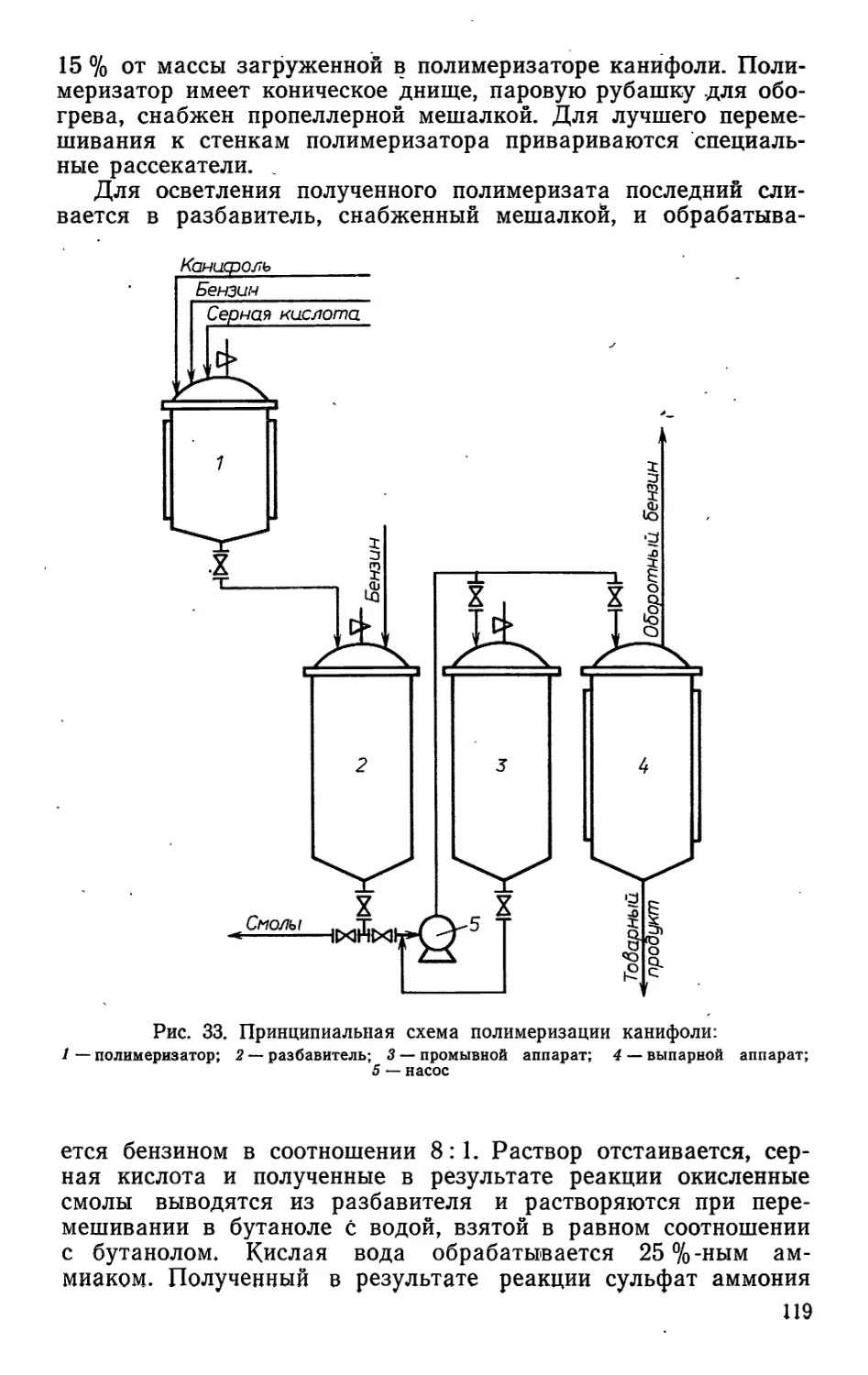
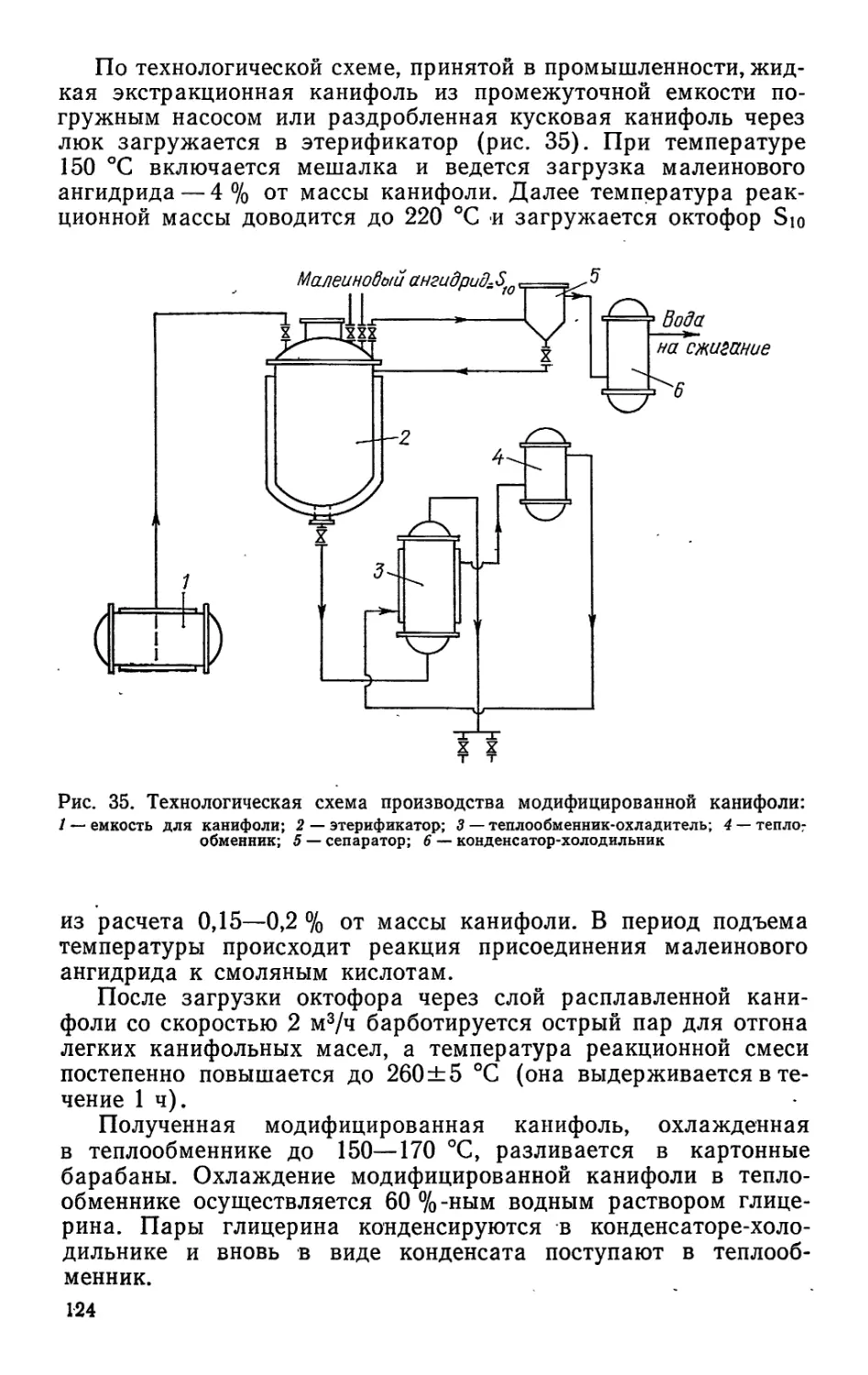
/



































































































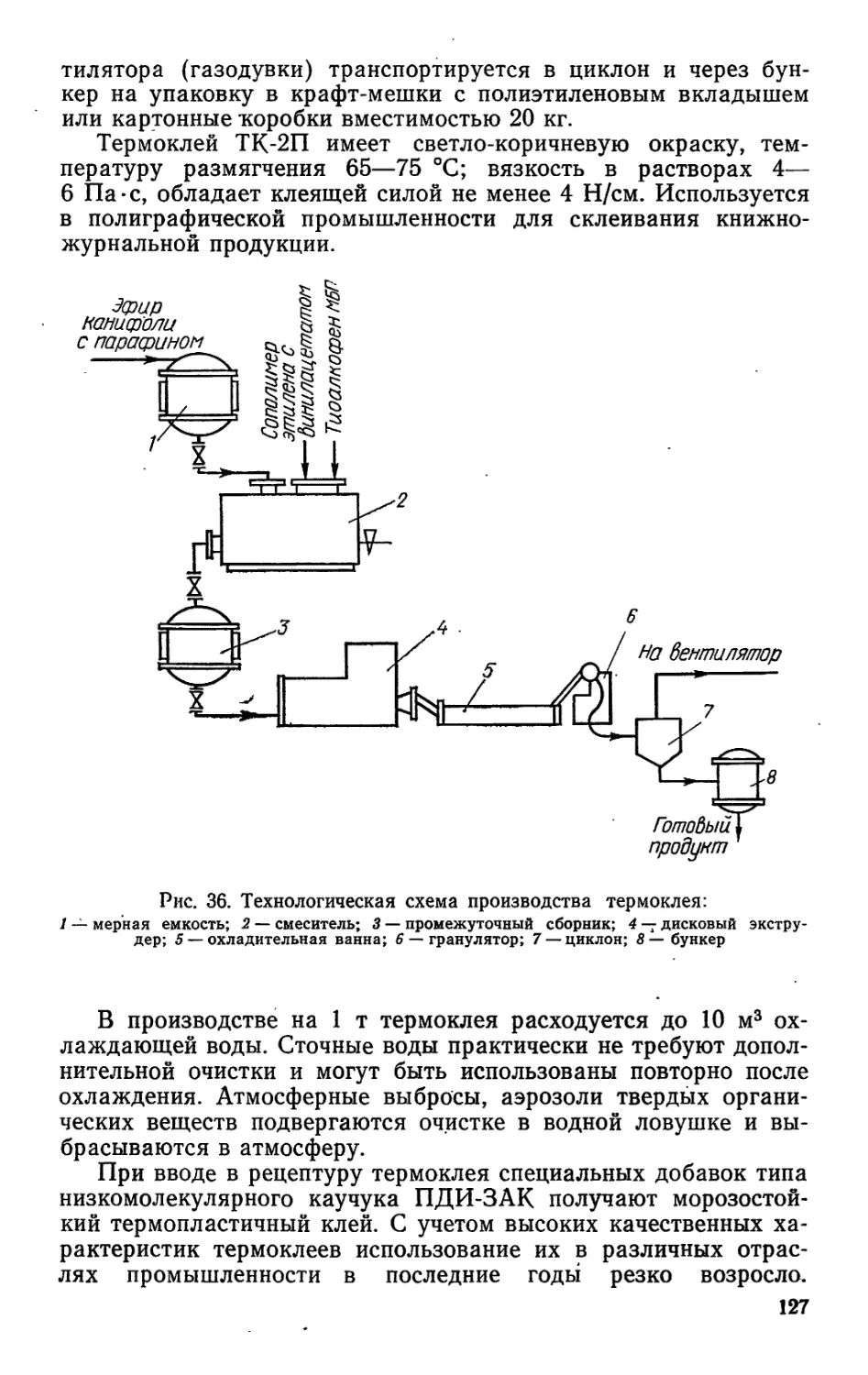


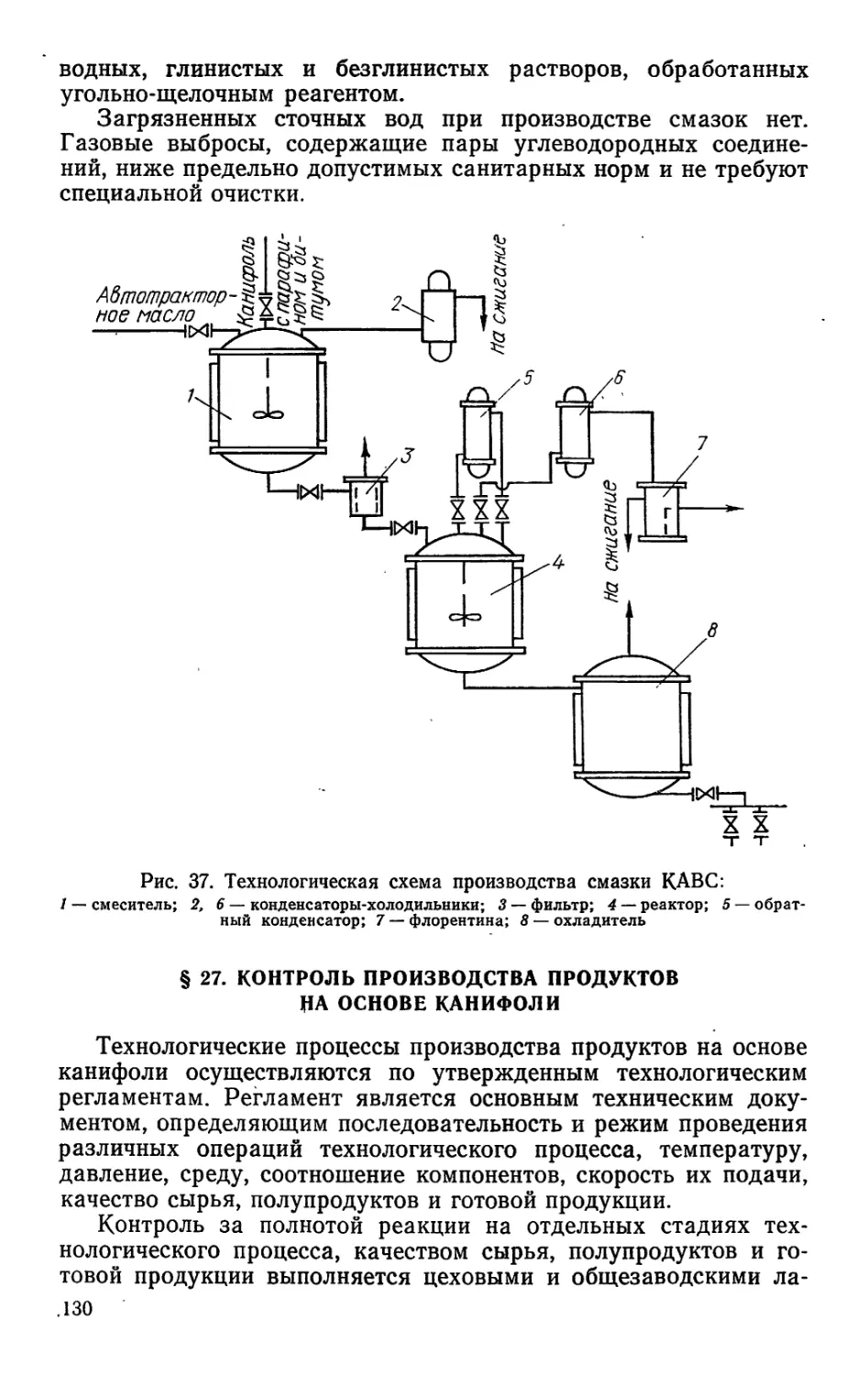



















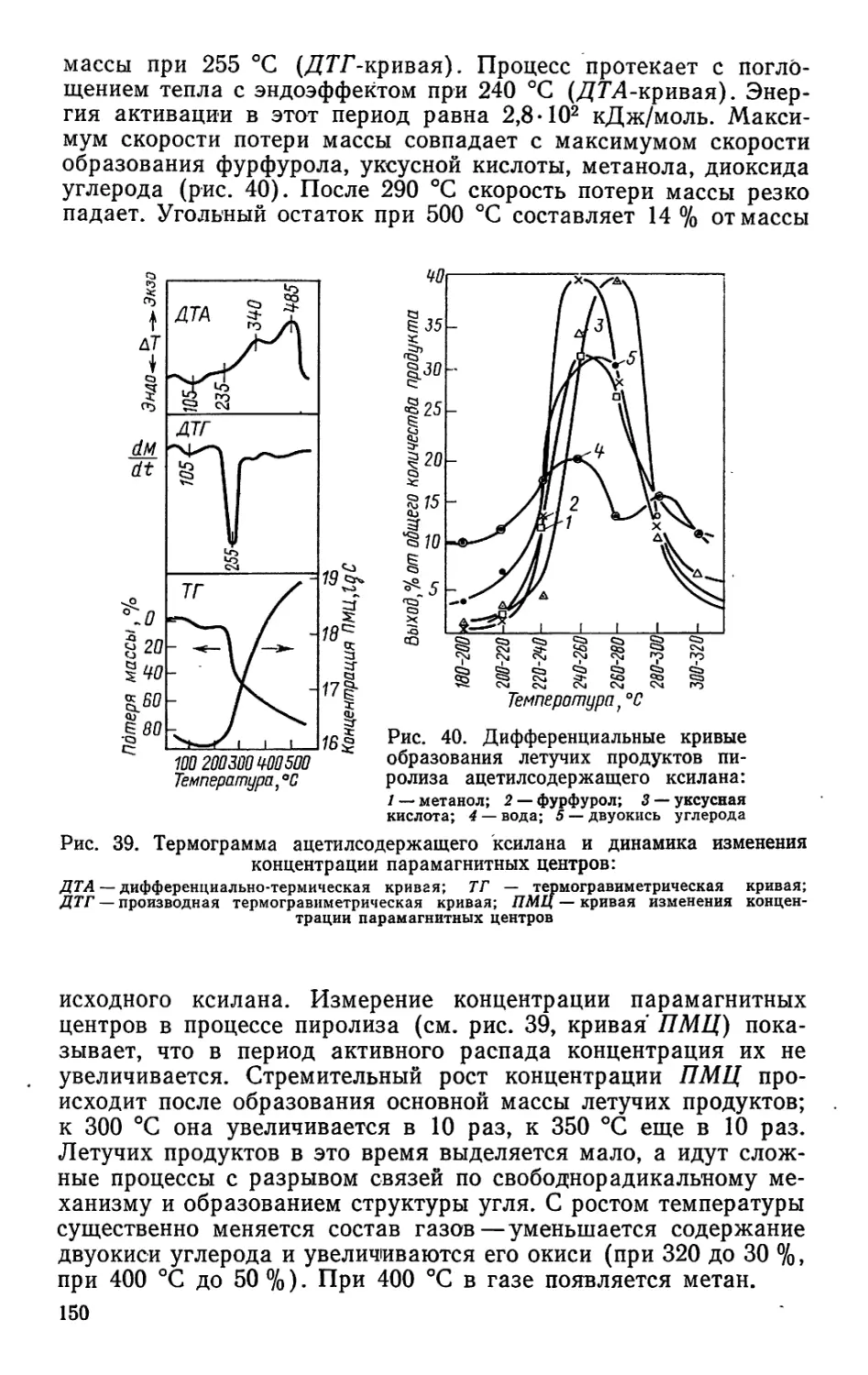



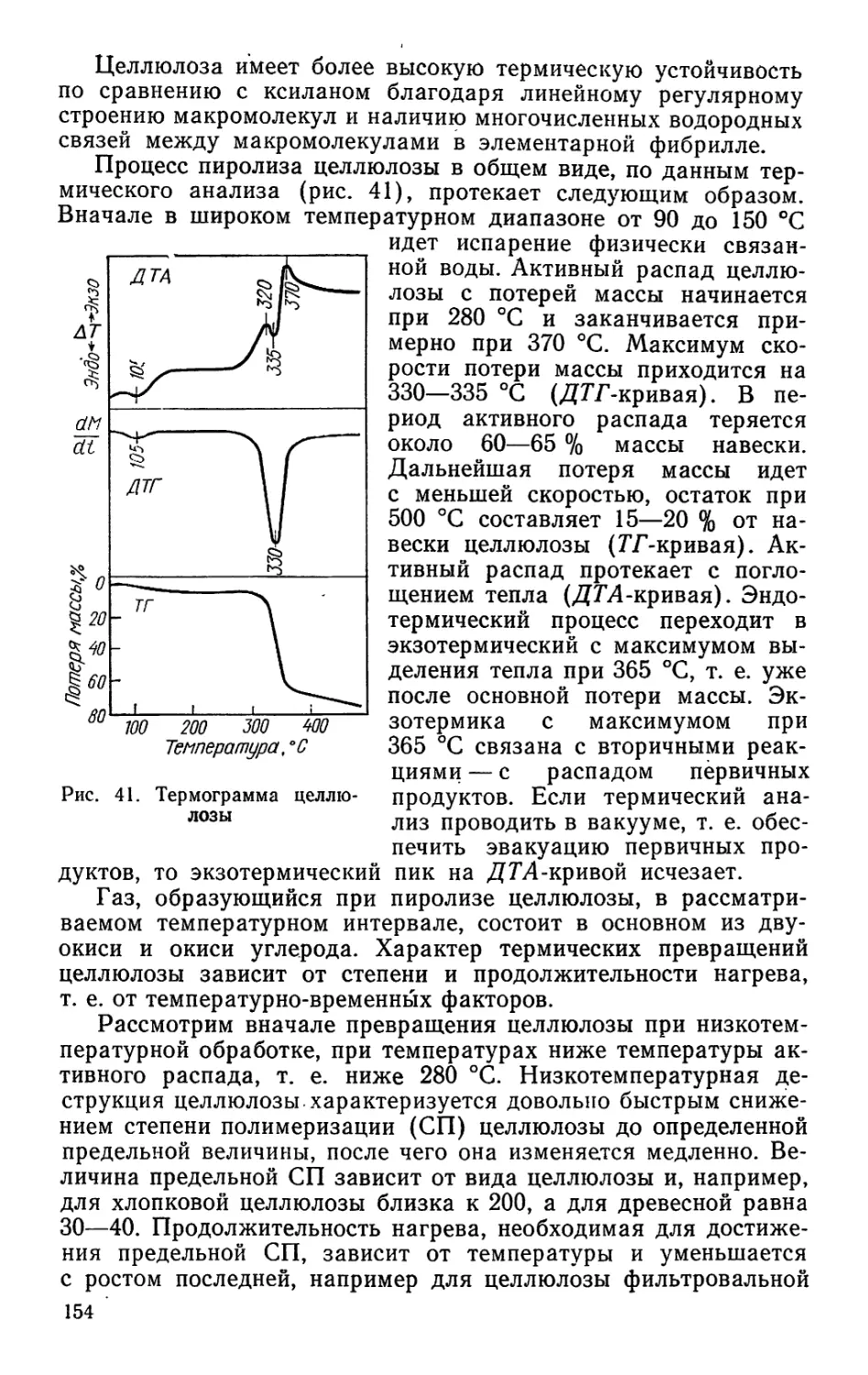























































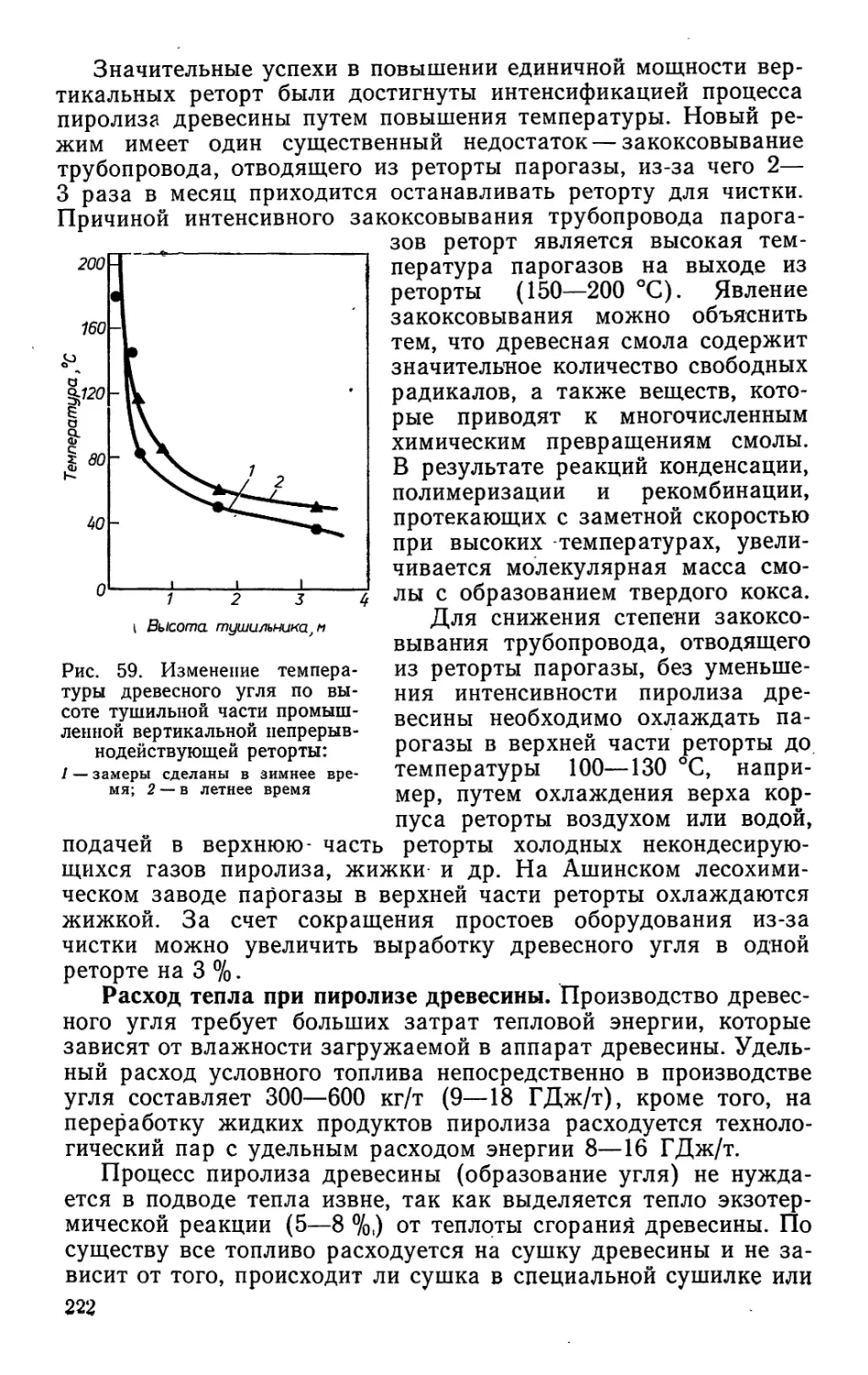


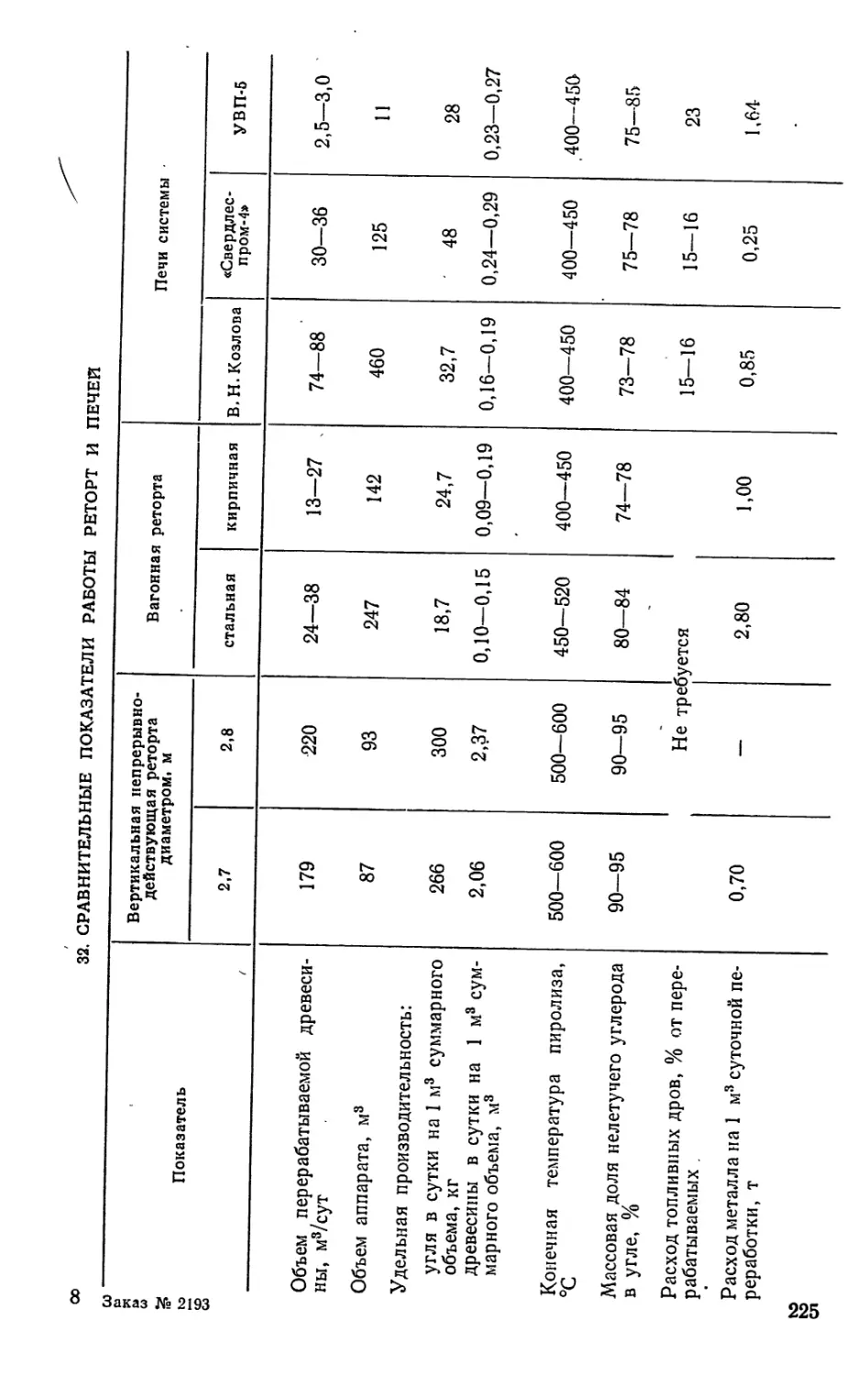





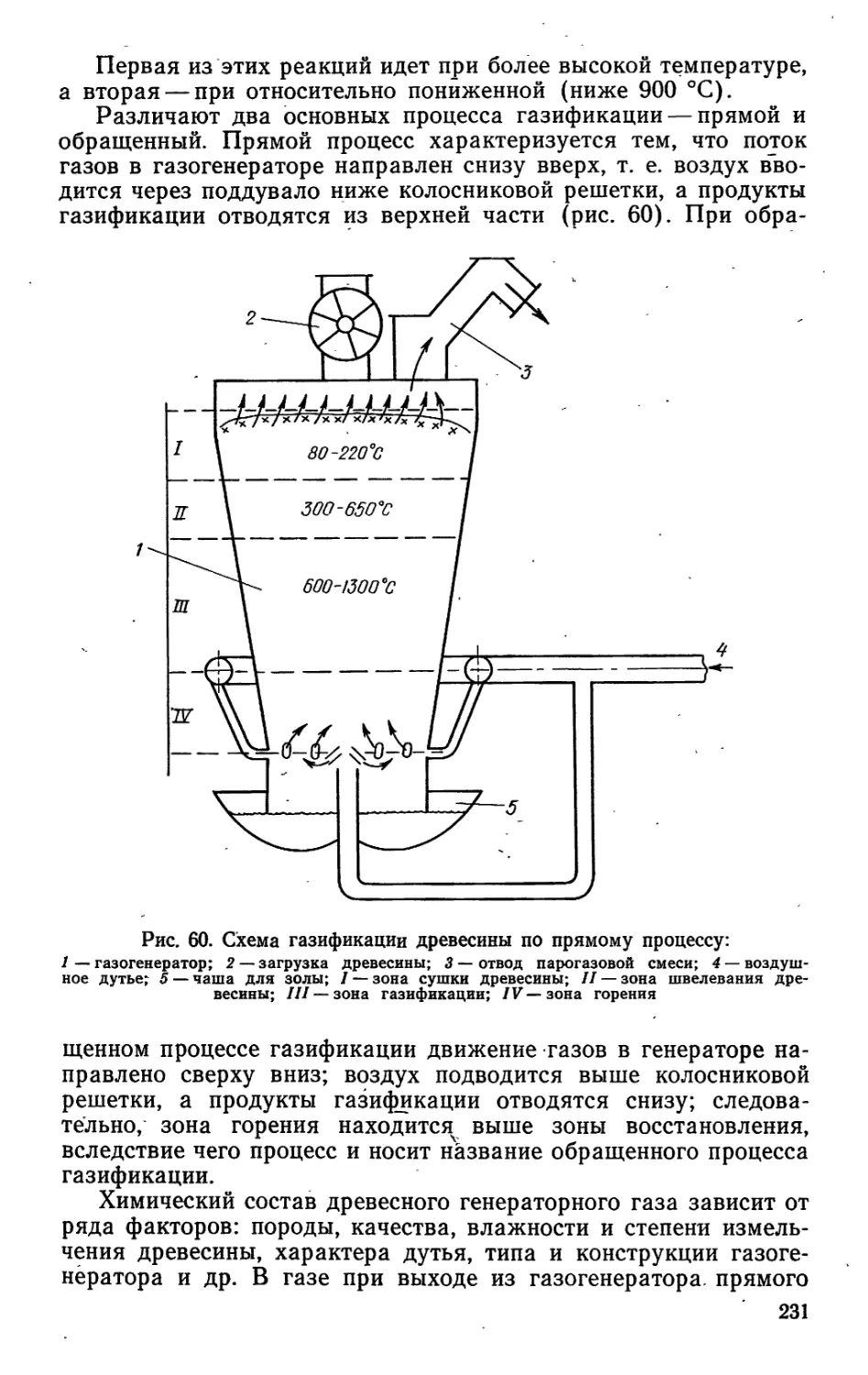


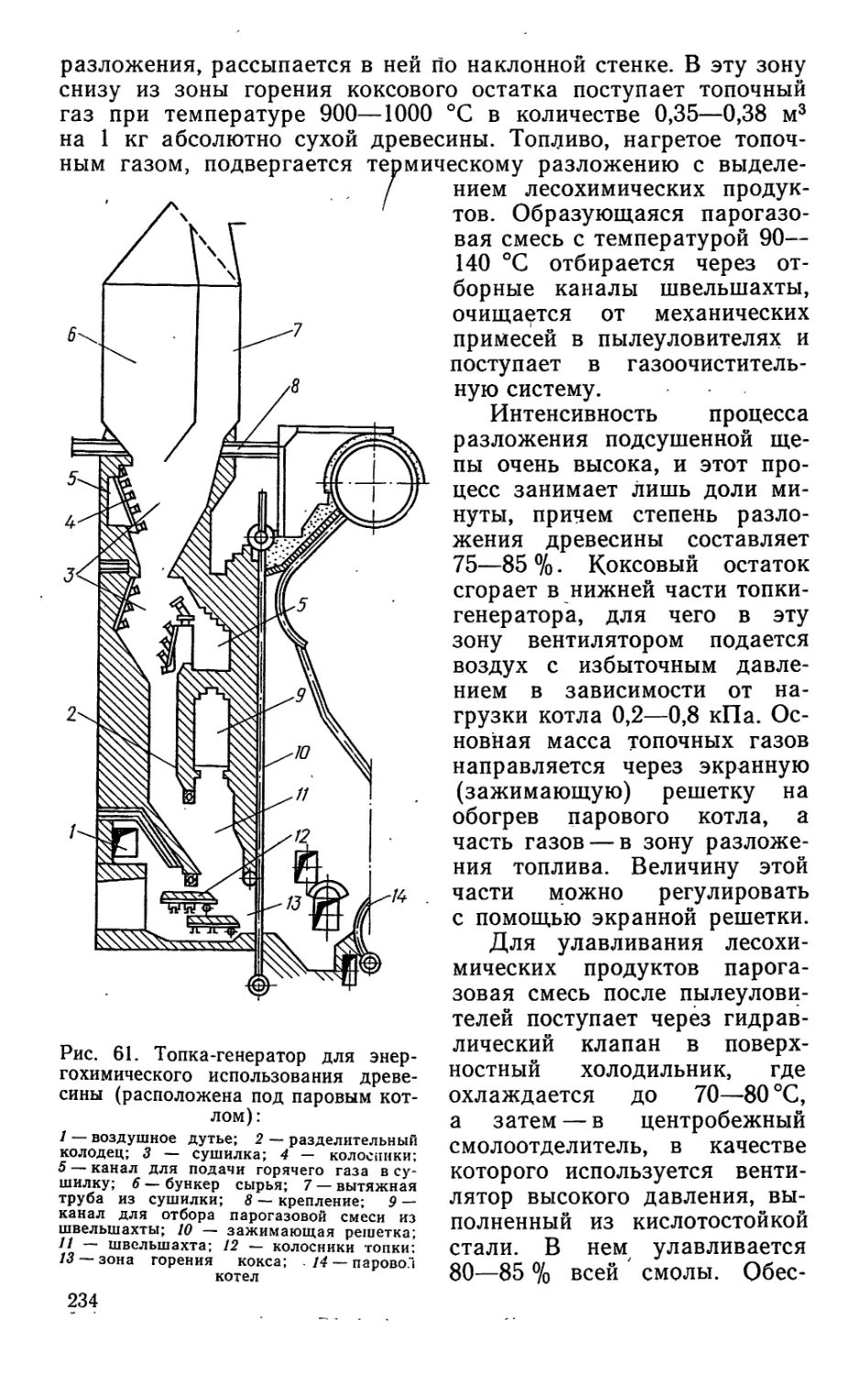





























































































Author: Bыpoдoв В.А. Кислицын А.Н. Глухарева М.И.
Tags: лесное хозяйство лесоводство лесная промышленность учебник для вузов
ISBN: 630* 86.002.2 (075.8)
Year: 1987




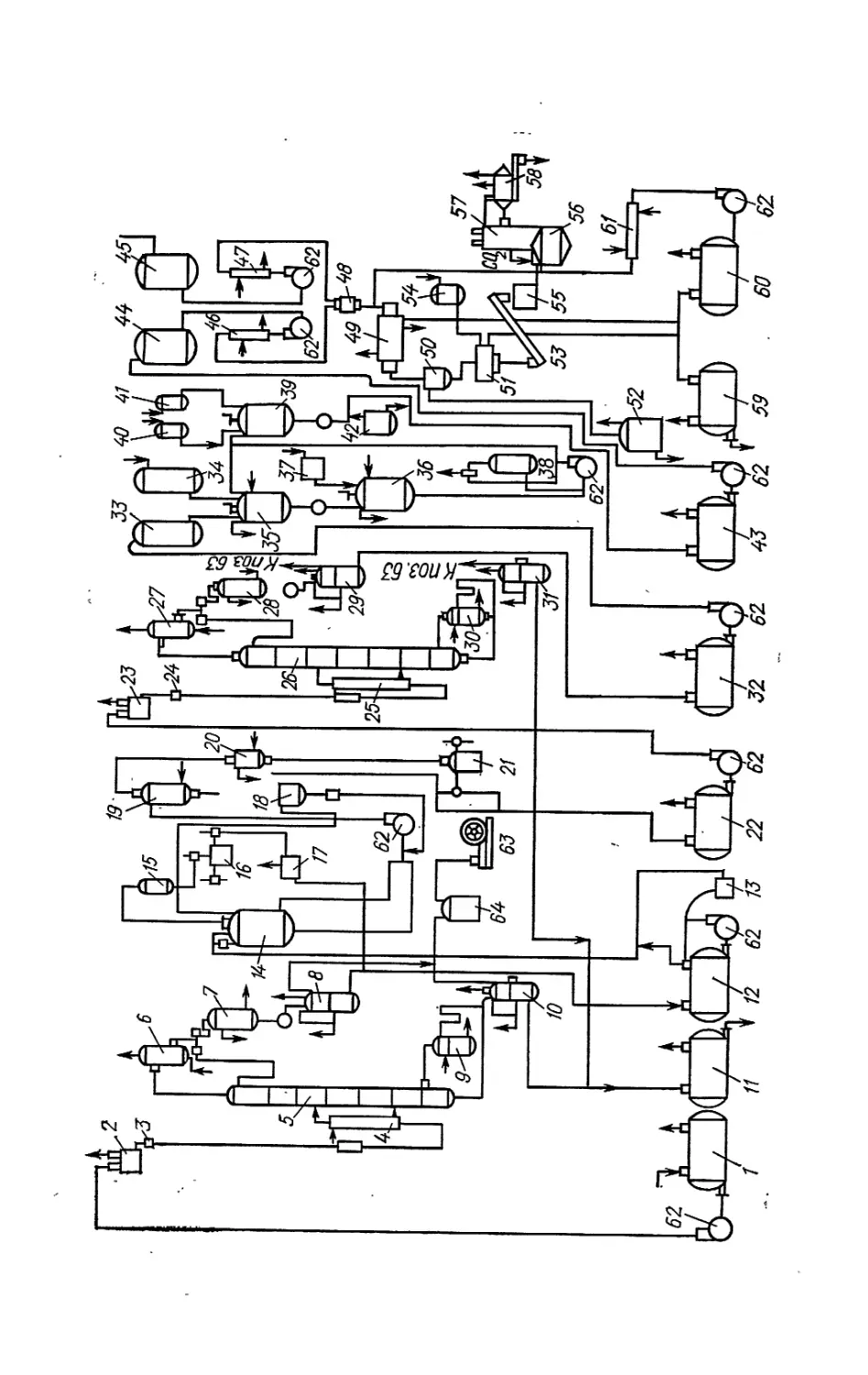
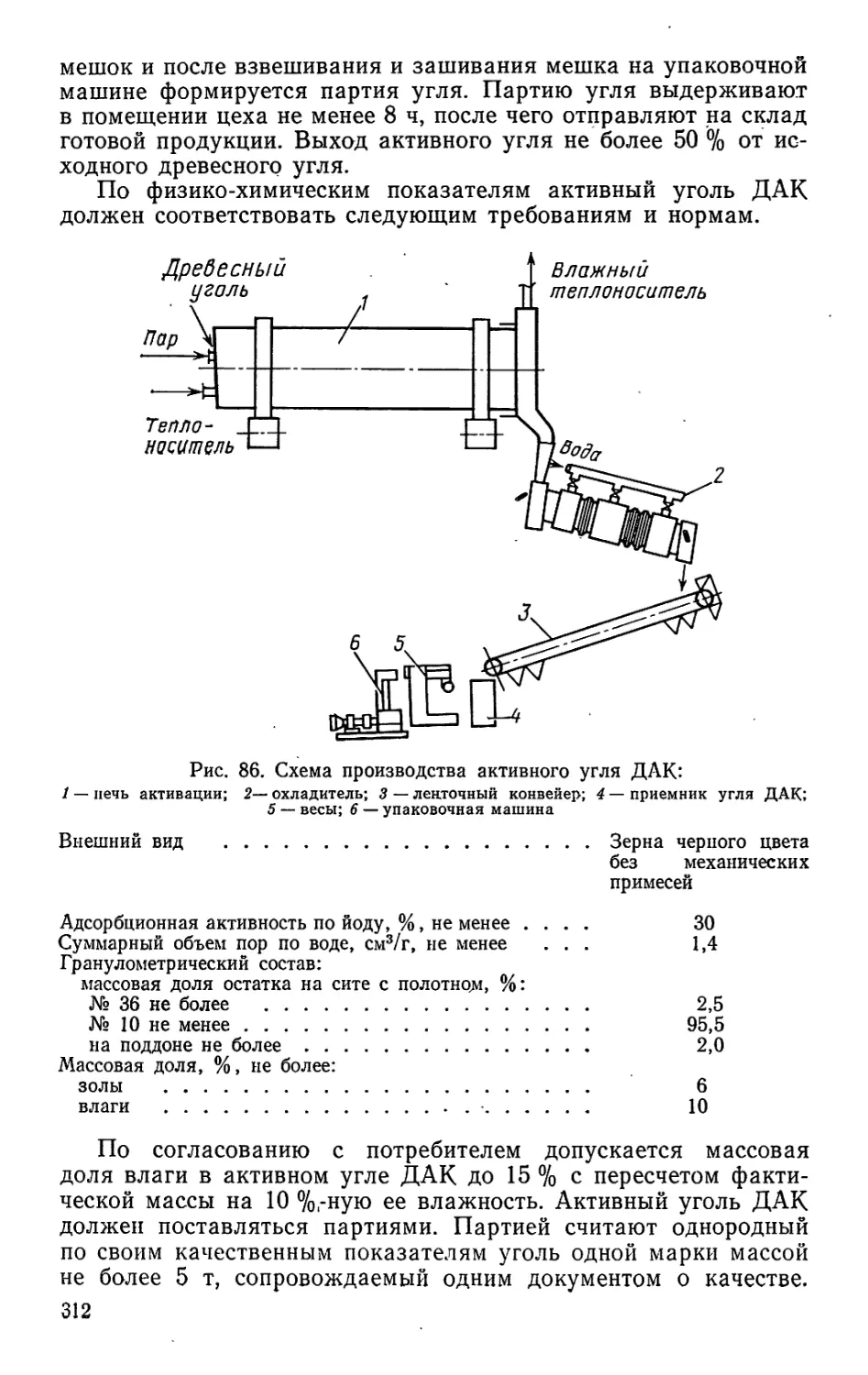
Text
ДЛЯ ВУЗОВ
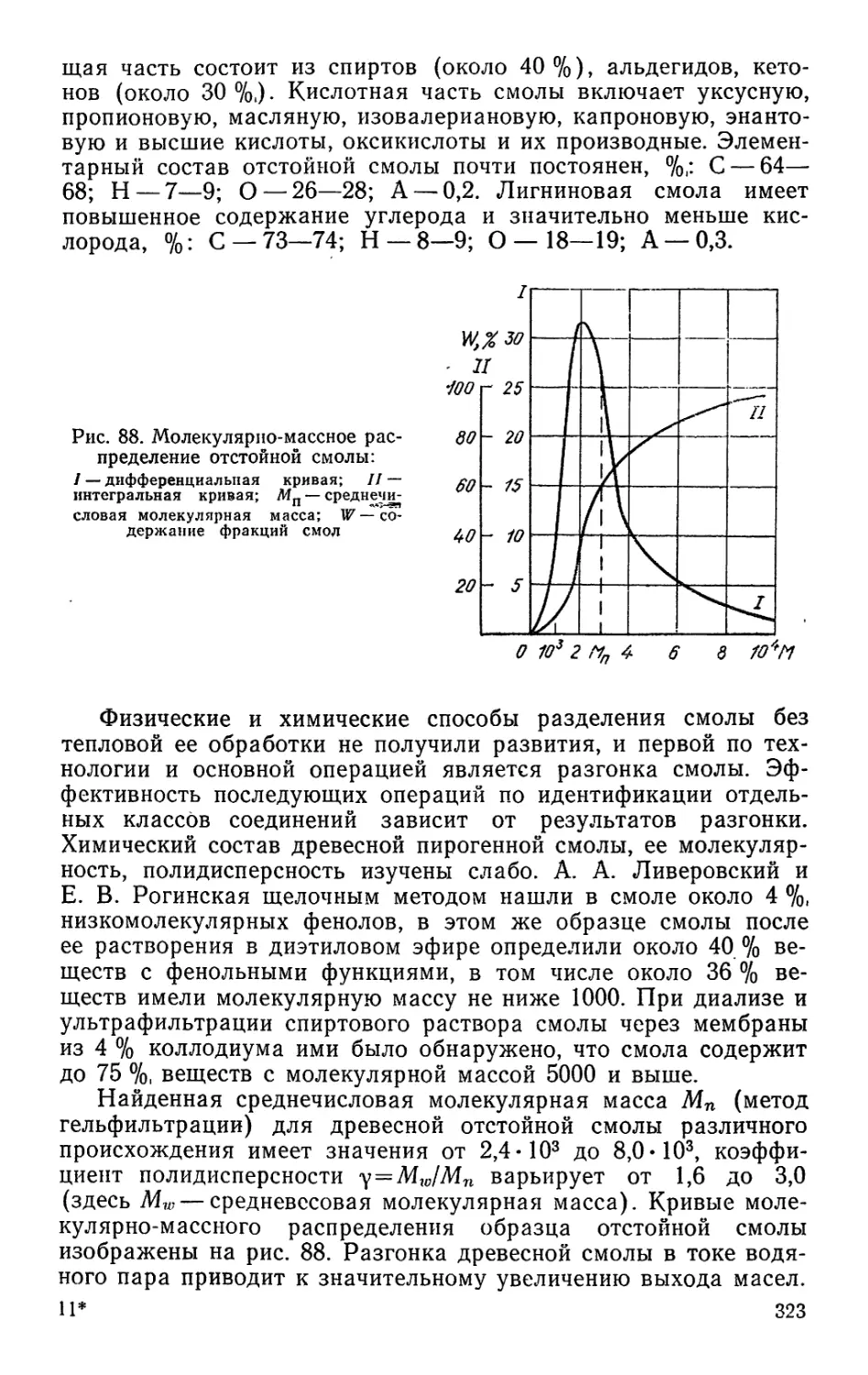
ТЕХНОЛОГИЯ
ЛЕСОХИМИЧЕСКИХ
ПРОИЗВОДСТВ
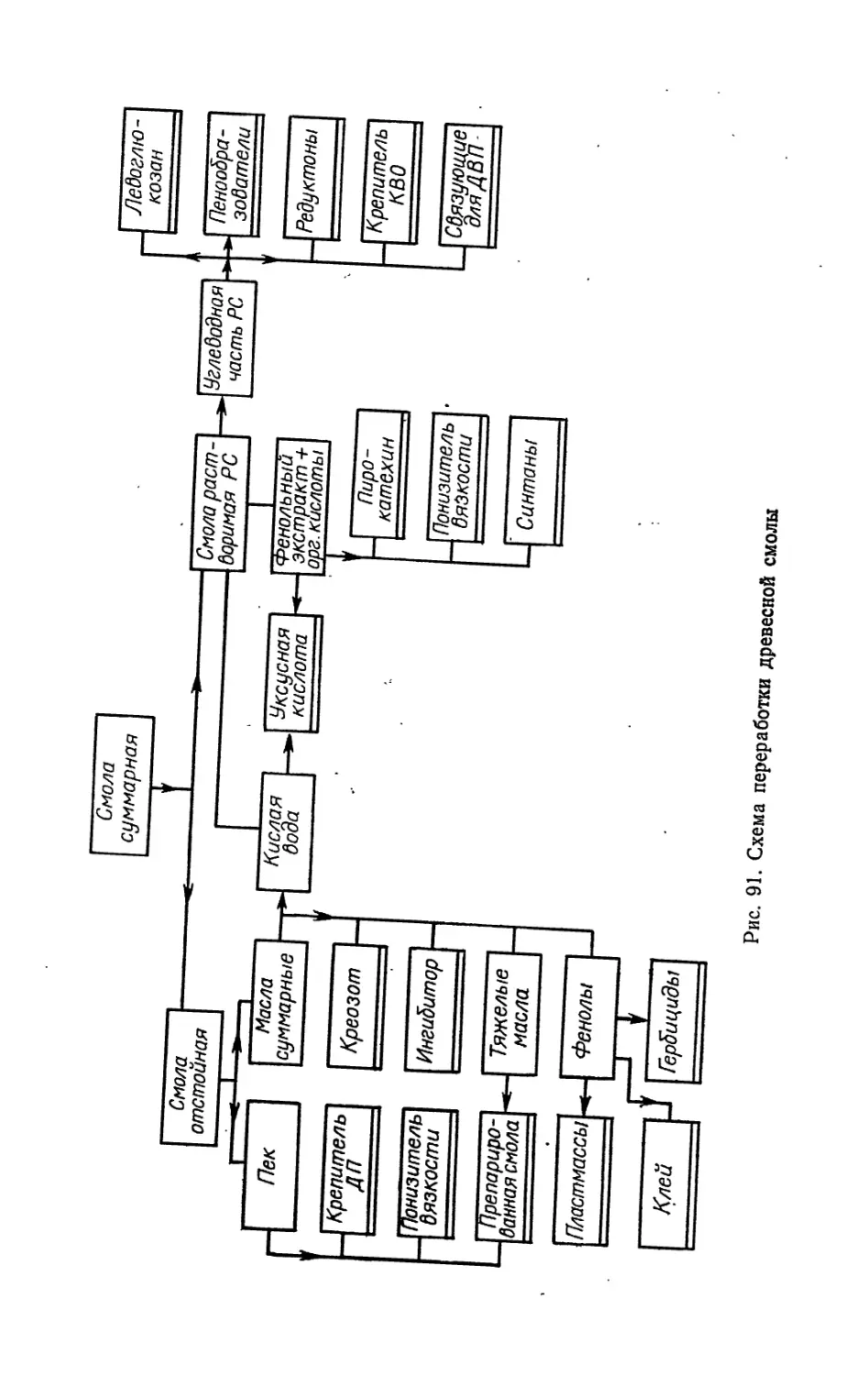
ТЕХНОЛОГИЯ
ЛЕСОХИМИЧЕСКИХ
ПРОИЗВОДСТВ
Допущено Министерством высшего
и среднего специального образования "
СССР в качестве учебника для
студентов вузов, обучающихся
по специальности "Химическая
технология древесины"
МОСКВА
"ЛЕСНАЯ ПРОМЫШЛЕННОСТЬ"
1987
УДК 630* 86.002.2(075.8)
Технология лесохимических производств: Учебник для вузов/Выродов
В. А., Кислицын А. Н., Глухарева М. И. и др.— М.: Лесная промышленность,
1987. — 352 с.
Рассмотрены вопросы теории и технологии основных лесохимических про-
изводств: канифольно-терпентинного, канифольно-экстракционного, пиролиз-
ного, а также переделочных производств по переработке первичных продук-
тов лесохимии — канифоли, скипидара, древесного угля, древесной смолы, ук-
сусной кислоты.
Для студентов лесотехнических вузов.
Табл. 48, ил. 96, библиогр.— 25 назв.
Авторы: В. А, Выродов, А. Н. Кислицын, М. И. Глухарева, А. И. Кип-
рианов, Л. М. Ефимов, П. И. Журавлев.
Рецензенты: д-р техн, наук А. М. Чащин (ЦНИЛХИ) и кафедра
химической технологии древесины Уральского лесотехнического института.
Виктор Антонович Выродов,
Алексей Николаевич Кислицын,
Маргарита Ивановна Глухарева,
Алексей Иванович Киприанов,
Леонид Михайлович Ефимов,
Петр Иванович Журавлев
ТЕХНОЛОГИЯ ЛЕСОХИМИЧЕСКИХ ПРОИЗВОДСТВ
Редактор издательства К. Г. Бурмистрова
Переплет художника О. А. Кознова
Художественный редактор К. П. Остроухов
Технический редактор О. А. Колотвина
Корректор И. Б. Ш е м а н с к а я
Вычитка Е. Н. Соколовой
ИБ № 2016
Сдано в набор 18.08.86. Подписано в печать 02.12.86. Т-23820. Формат 60Х90Л6. Бу-
мага книжно-журнальная. Гарнитура литературная. Печать высокая. Усл. печ. л. 22,0.
Усл. кр.-отт. 22,0. Уч.-изд. л. 24,28. Тираж 2900 экз. Заказ № 2193. Цена 1 р. 10 к.
Ордена «Знак Почета» издательство «Лесная промышленность».
101000, Москва, ул. Кирова, 40а.
Ленинградская типография № 4 ордена Трудового Красного Знамени Ленинградского
объединения «Техническая книга» им. Евгении Соколовой Союзполиграфпрома при Го-
сударственном комитете СССР по делам издательств, полиграфии и книжной торговли.
191126, Ленинград, Социалистическая ул., 14.
3003000000—022
Т---------------41—87
037(01)—87
© Издательство «Лесная промышленность», 1987 г.
ВВЕДЕНИЕ
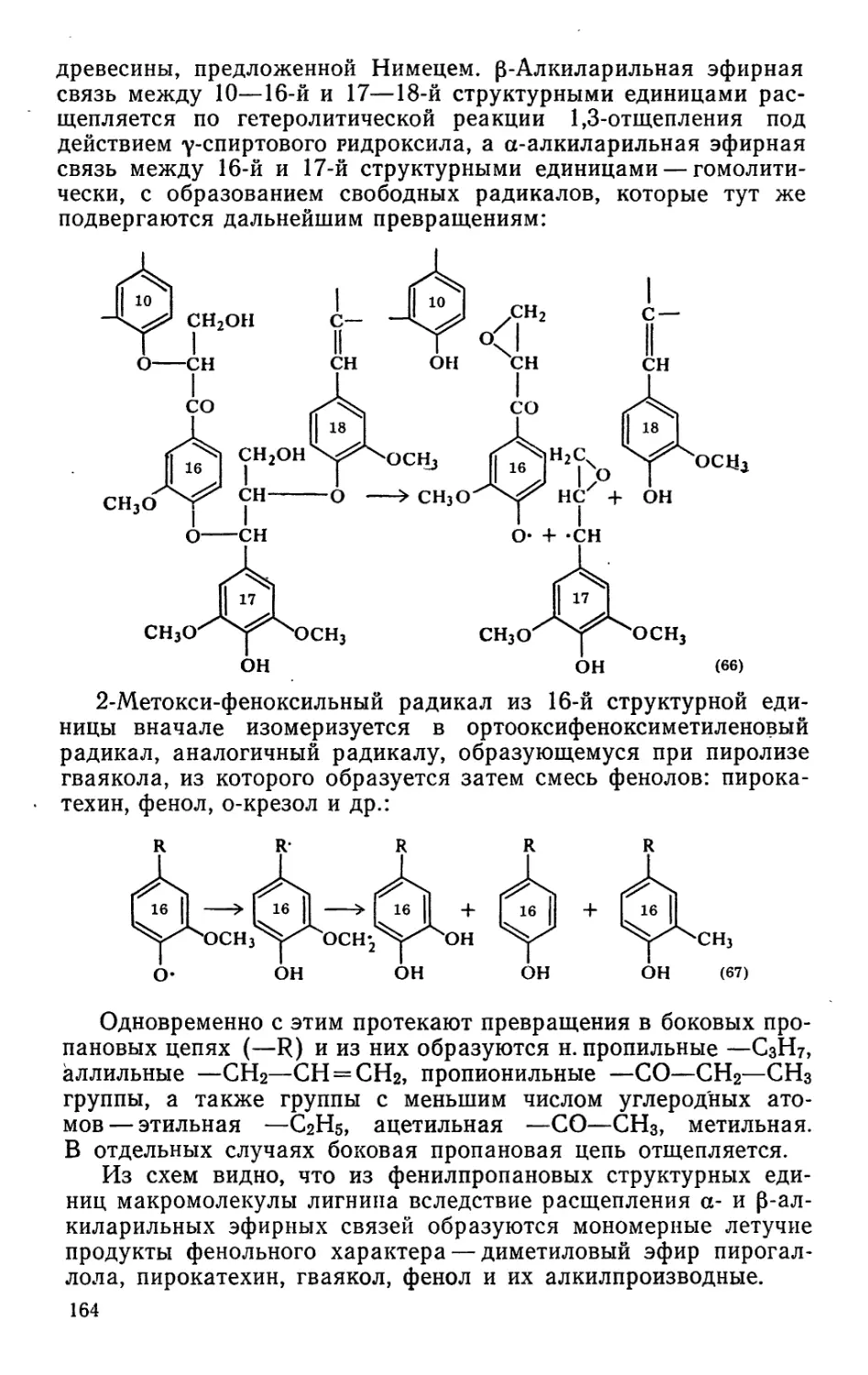
Коммунистическая партия Советского Союза настойчиво и
последовательно ставит задачи эффективного использования
сырьевых и энергетических ресурсов, направляет усилия работ-
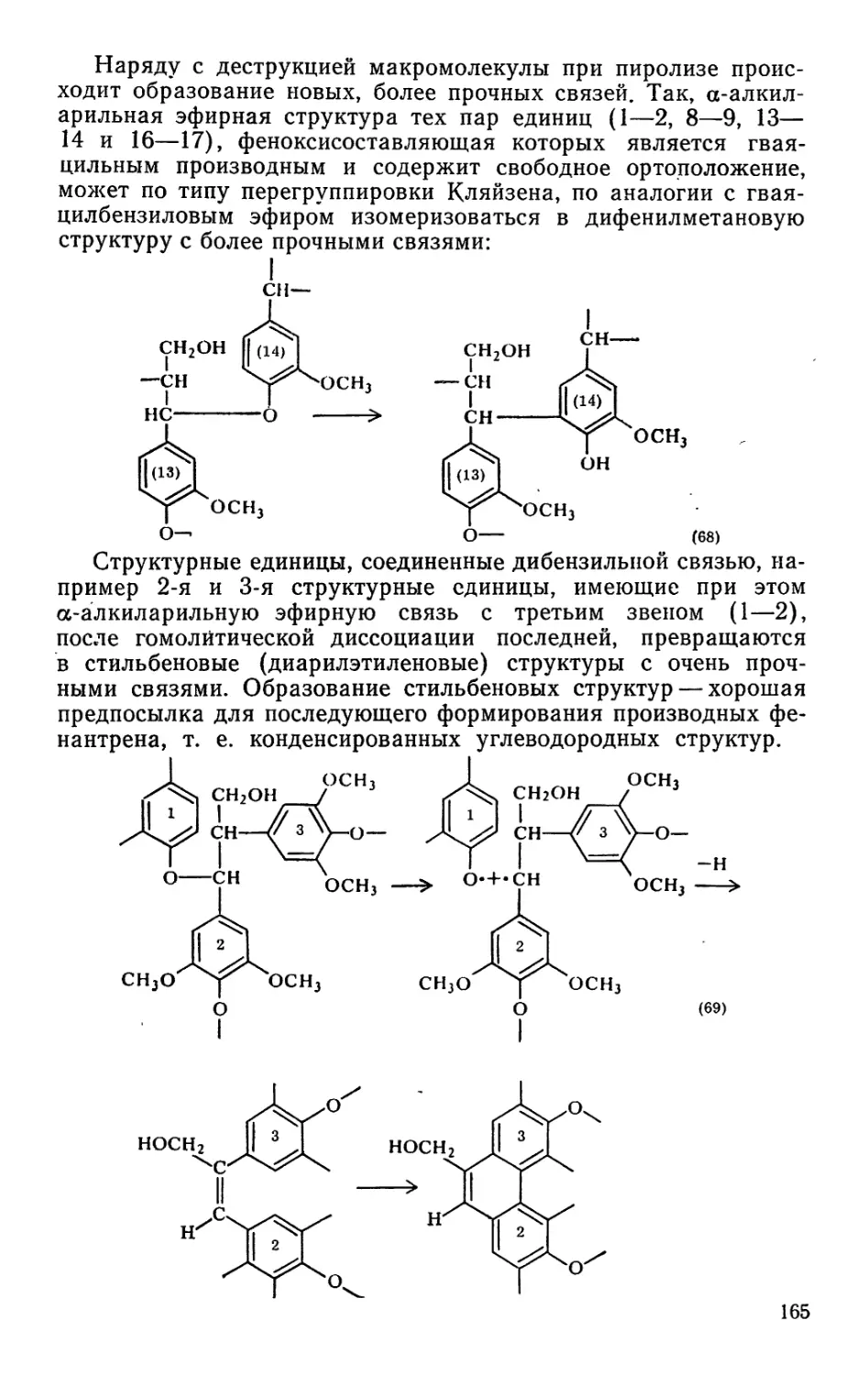
ников всех сфер деятельности на создание технологических
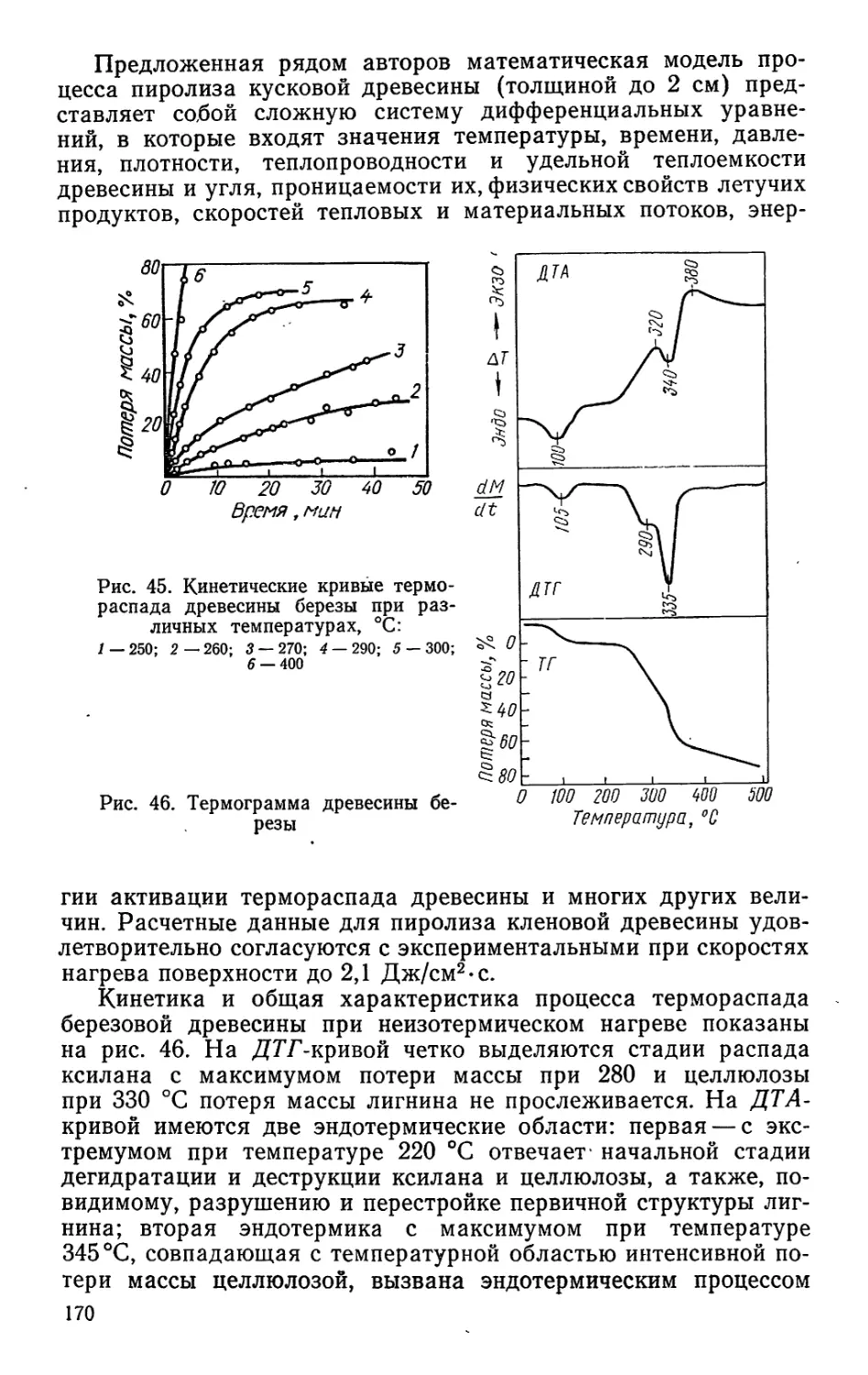
процессов безотходной технологии, которые обеспечивали бы
комплексное использование сырья и максимальную охрану ок-
ружающей среды. Для выполнения поставленных задач необ-
ходимо не только умело и бережно использовать природные
запасы древесины и других видов сырья, но и постоянно уве-
личивать научно-технический потенциал, в том числе профес-
сиональный уровень кадров, и прежде всего молодых специа-
листов, которых готовят вузы.
Специалисты лесохимической промышленности должны хо-
рошо знать отрасль, продукцию, которую она выпускает. Но-
менклатура лесохимической продукции весьма обширна, зна-
чимость ее для народного хозяйства огромна. Экстрактивные
вещества дерева — терпены, смоляные и жирные кислоты, ду-
бильные и красящие вещества, липиды, стерины и их произ-
водные являются незаменимым сырьем для получения ряда
медицинских препаратов в фармакологии, парфюмерии, сель-
ском хозяйстве (хвойная мука, биологические добавки в корм
животным и т. д.). Древесный уголь — одна из чистейших при-
родных форм углерода — используется при получении сверх-
чистых элементов, в катализе, при очистке препаратов. Пере-
работкой древесной смолы получают фенолопродукты, препа-
раты для улучшения качества бетонов, литейные крепители,
понизители вязкости бурильных растворов, антиокислители,
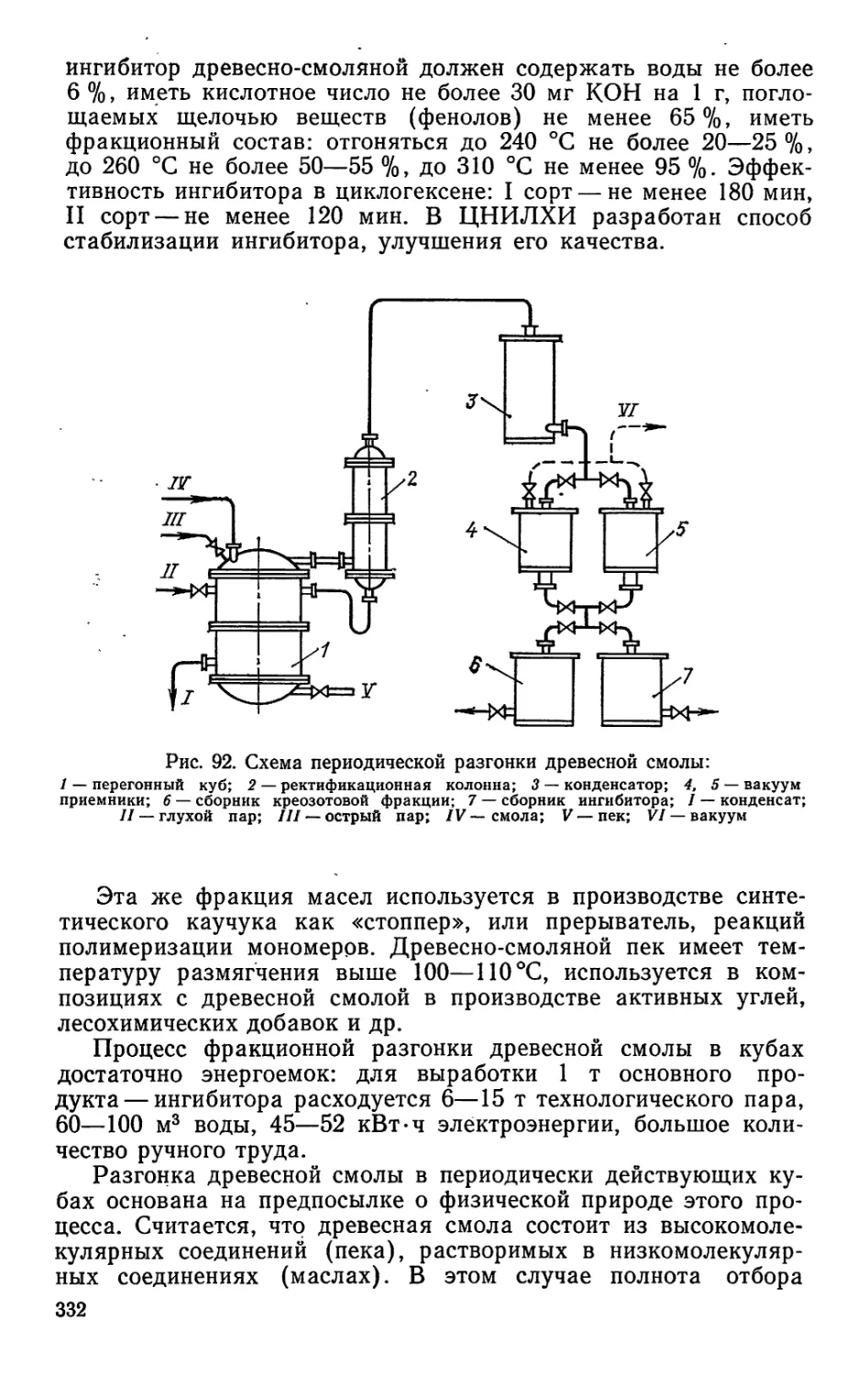
коптильные жидкости. Пищевая уксусная кислота в основном
является продуктом также лесохимической промышленности.
Для подготовки специалистов-лесохимиков и предназначен
данный учебник «Технология лесохимических производств»,
который состоит из двух самостоятельных разделов «Техноло-
гия экстрактивных веществ дерева» и «Термическая перера-
ботка древесины». В учебнике изложены теоретические основы,
техника и технология ведущих производств лесохимической
промышленности, учтены все последние достижения науки и
техники.
В учебнике не описаны устройство оборудования и проек-
тирование предприятий, а также получение талловых продук-
тов сульфатно-целлюлозного производства, биологически ак-
тивных веществ из зеленой части и коры дерева. Этот мате-
риал изложен в самостоятельных курсах учебного плана.
Однако авторы описали химизм образования промежуточных и
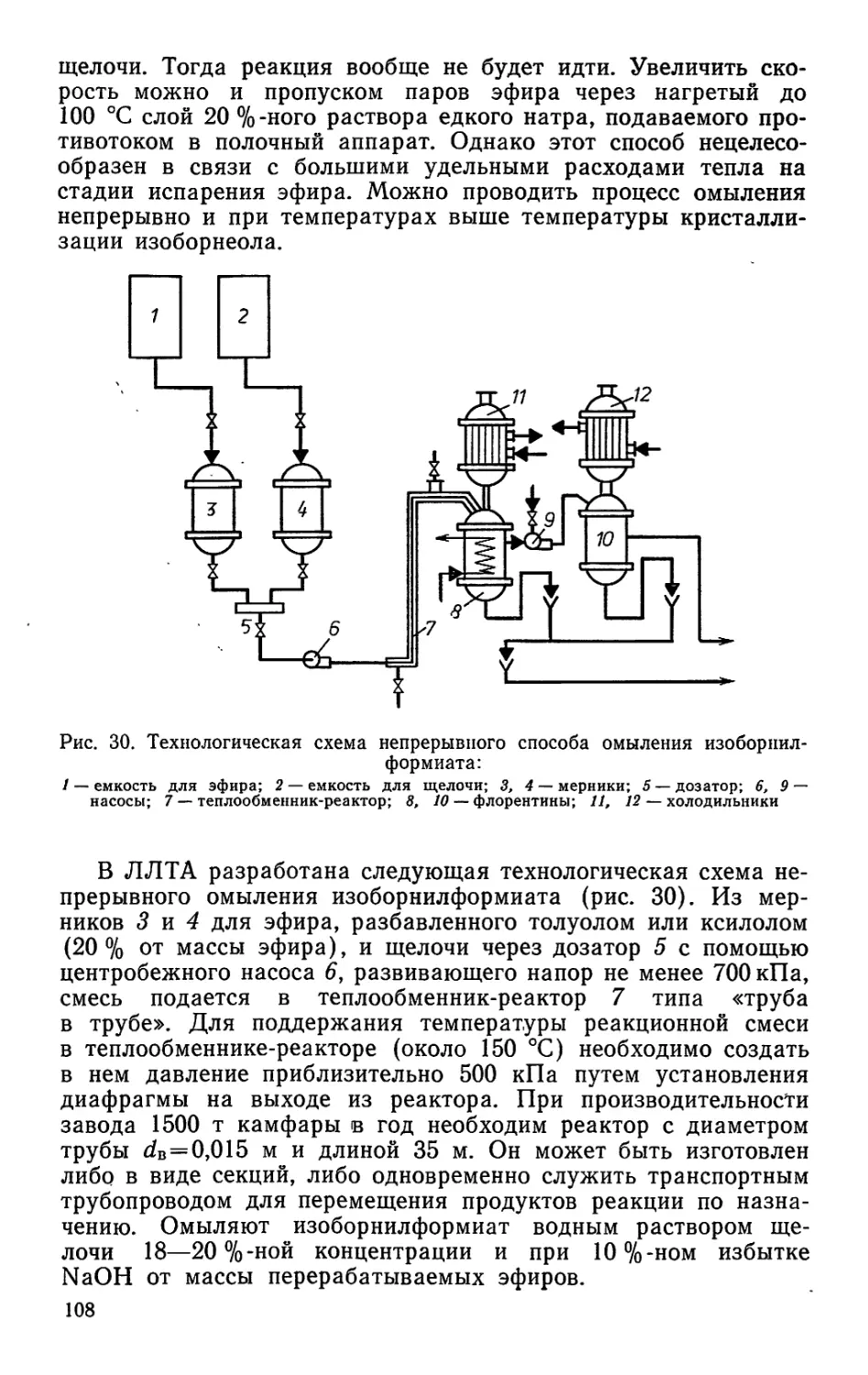
1* з
конечных продуктов пиролиза древесины и ее компонентов;
значительно расширили представления о парамагнитных и
поверхностно-активных свойствах древесного угля. С новых
позиций изложены принципы переработки древесной смолы,
рассматриваются пути использования всех компонентов дре-
весной смолы в различных отраслях народного хозяйства.
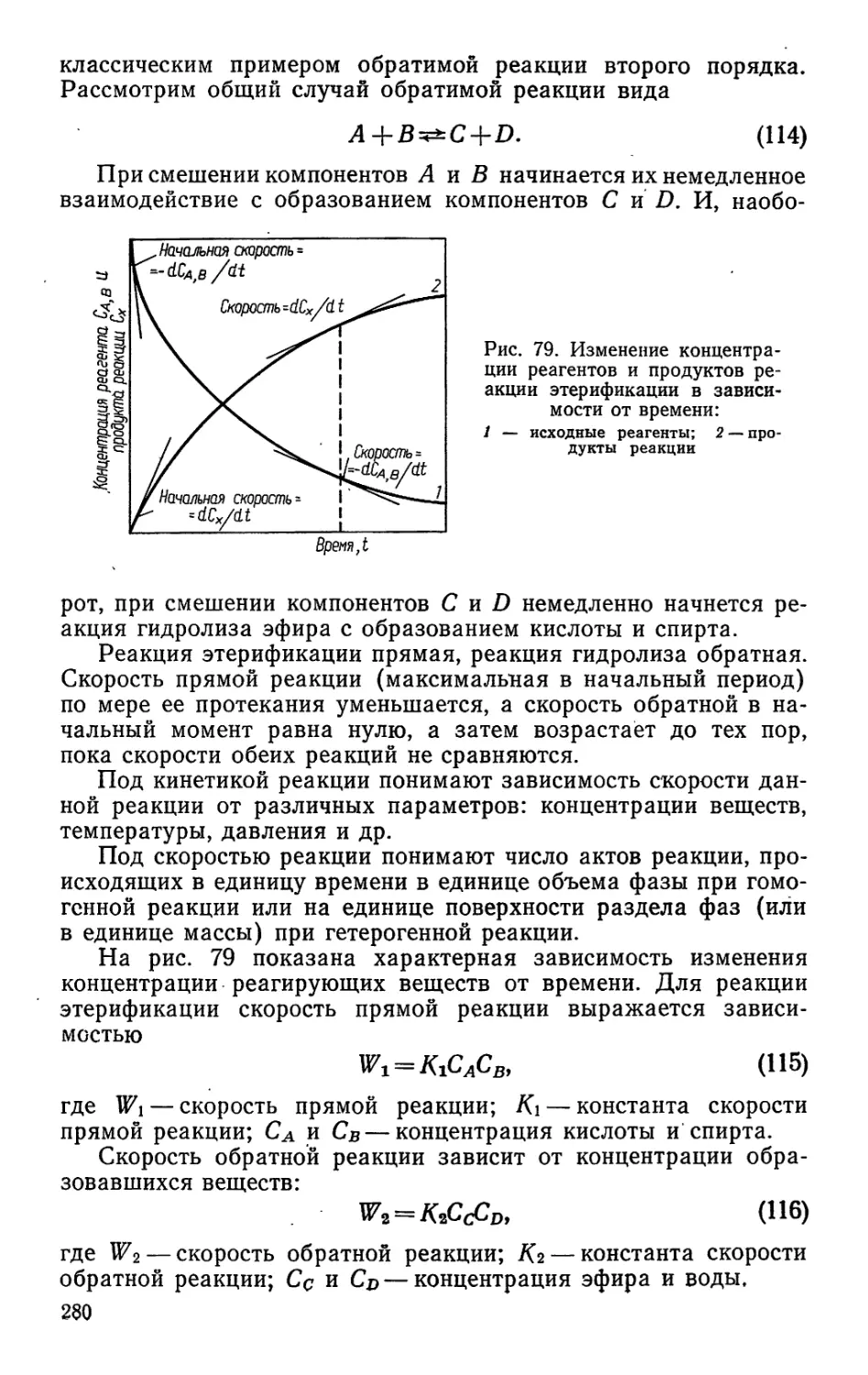
Задача учебника — не только подготовить специалиста по
данной отрасли, но научить его бережно относиться к народ-
ному богатству нашей Родины — лесу, к древесине — основ-
ному ресурсу леса.
Главы 1, 2, 3, 4 написаны д-ром техн, наук проф. В. А. Вы-
родовым, глава 5 инженером П. И. Журавлевым, главы 6 и
8 д-ром хим. наук А. Н. Кислицыным, главы 7, 9 и 14 — канд.
техн, наук Л. М. Ефимовым, главы 11, 12, 13 — канд. техн,
наук М. И. Глухаревой, главы 10, 15 — д-ром техн, наук проф.
А. И. Киприановым.
Авторы выражают глубокую признательность официаль-
ным рецензентам: д-ру техн, наук А. М. Чащину, сотрудникам
кафедры химической технологии древесины Уральского лесо-
технического института.
Раздел I
ТЕХНОЛОГИЯ ЭКСТРАКТИВНЫХ ВЕЩЕСТВ
ДЕРЕВА
Глава 1
ЭКСТРАКТИВНЫЕ ВЕЩЕСТВА ДЕРЕВА,
ИХ СВОЙСТВА И ПРИМЕНЕНИЕ
Под экстрактивными веществами дерева понимают веще-
ства, извлекаемые из различных частей деревьев хвойных и
лиственных пород с помощью воды, органических растворите-
лей, острого водяного пара, а также механическим отжимом
или подсочкой. Иногда их называют сопутствующими компо-
нентами древесины.
Производство экстрактивных веществ дерева делится на две
основные группы. Первая группа производств: канифольно-
терпентинное (сырье — живица и баррас; основные продукты —
живичный скипидар и живичная канифоль) и канифольно-
экстракционное (сырье — различные виды смолистой древе-
сины, главным образом сосновый пневый осмол; продукты —
экстракционная канифоль, флотационное масло и экстракци-
онный скипидар).
Производства второй группы основаны на переработке ка-
нифоли и скипидара. Продуктами этого производства явля-
ются модифицированные виды канифоли (полимеризованная,
диспропорционированная, гидрированная, осветленная, окис-
ленная, хлорированная), смоляные кислоты (абиетиновая, лево-
пимаровая, декстропимаровая), синтетические смолы (альбер-
толи, эфиры гликолевый, глицериновый, пентаэритритовый, ма-
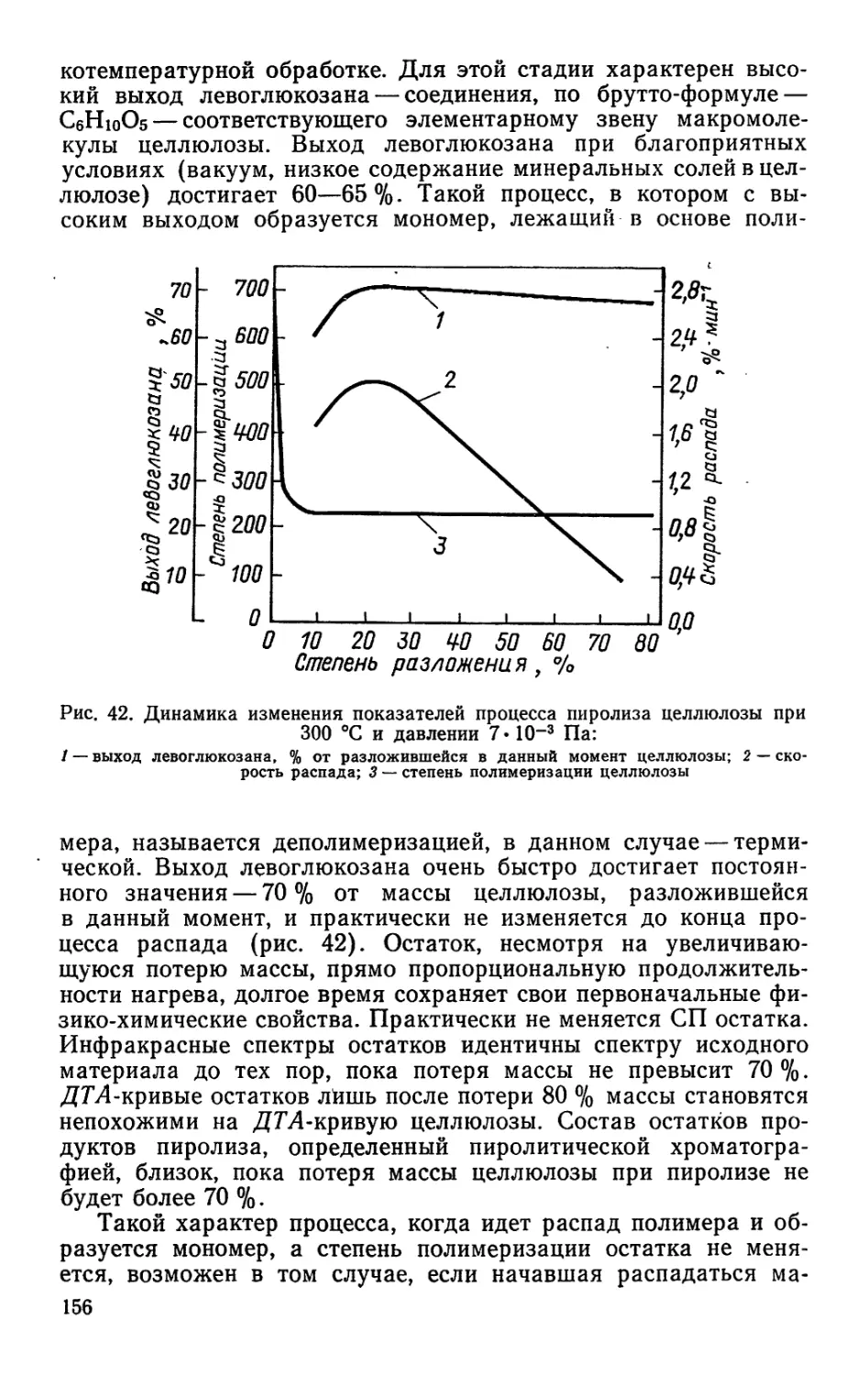
леиновая смола), резинаты (кальция, цинка, марганца, свинца,
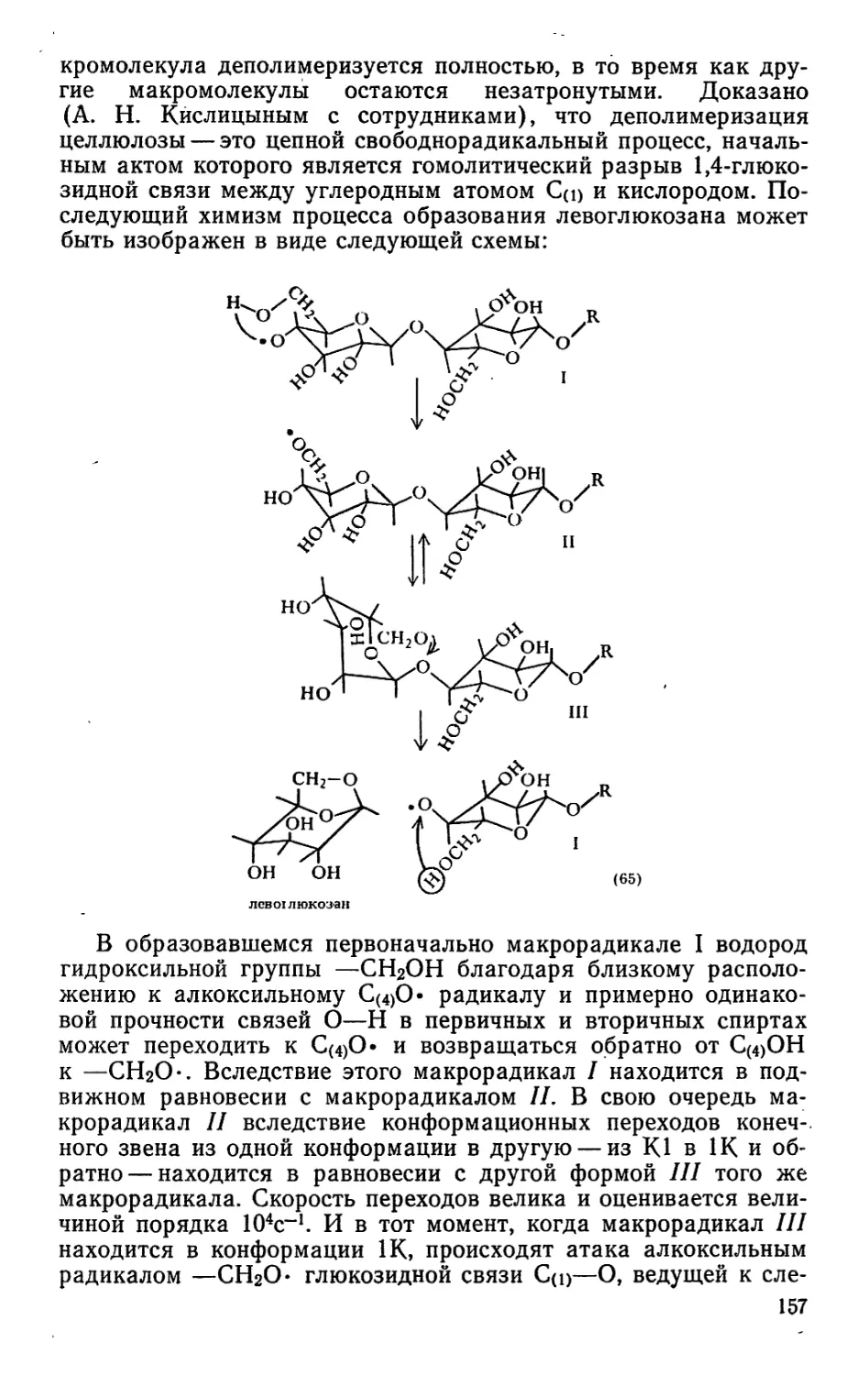
меди), эмульгаторы, олифы и др. Продуктами производства на
основе переработки скипидара являются а- и р-пинены, камфен,
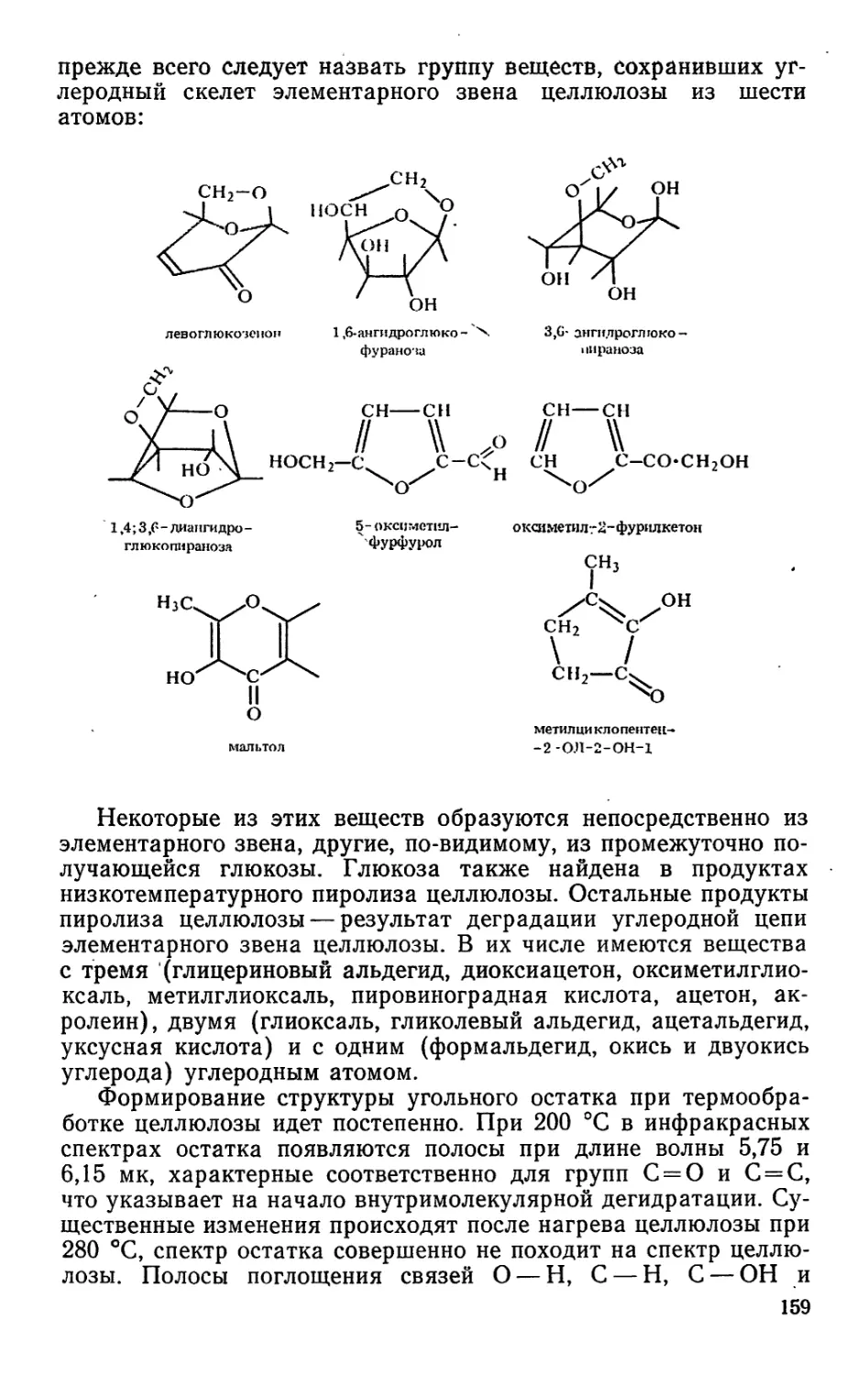
камфара, димеры и полимеры, окситерпеновые растворители и
окситерпеновые смолы, флотореагенты, синтетические души-
стые вещества и др.
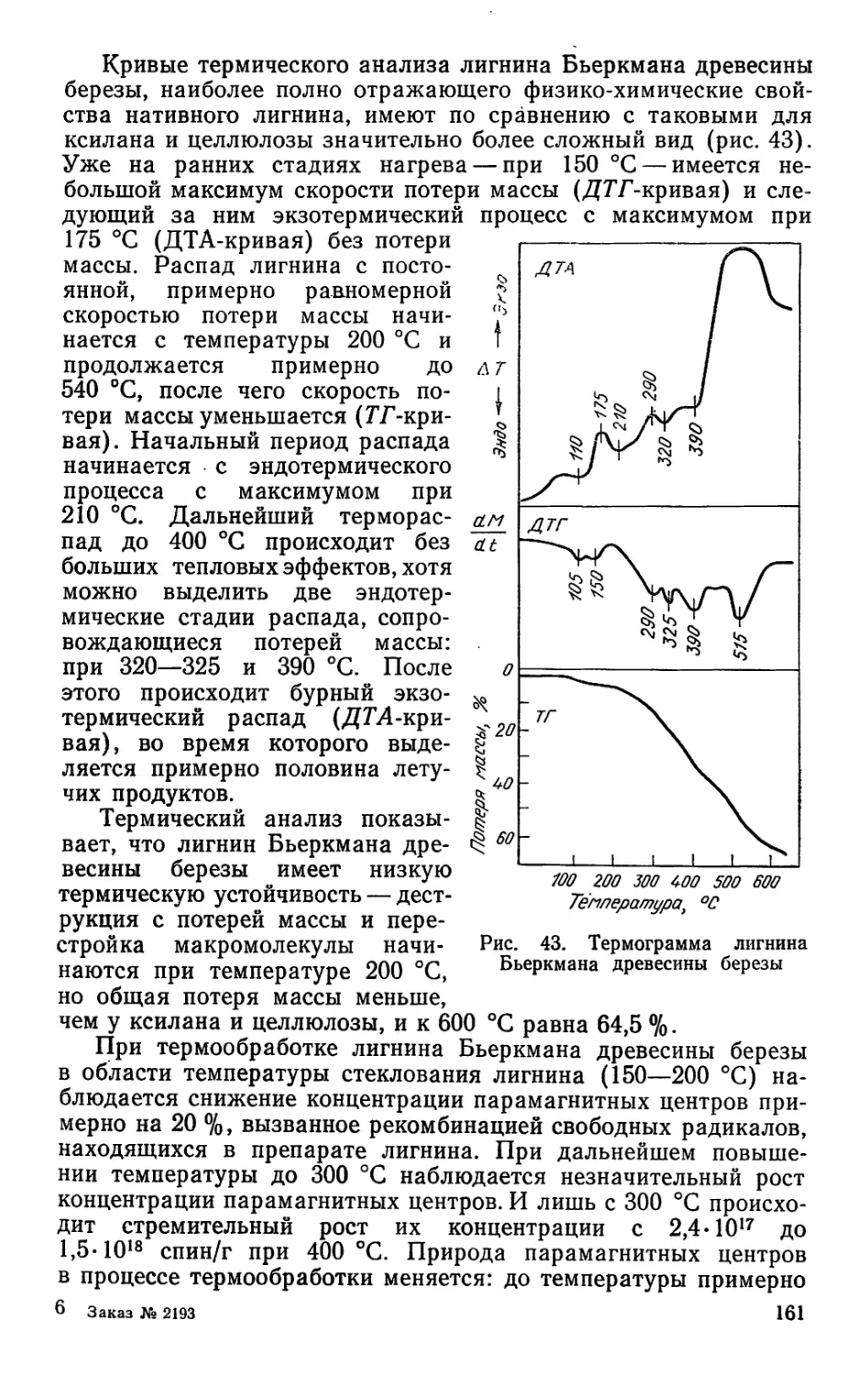
§ 1. СКИПИДАР
Летучая часть живицы представляет собой смесь терпено-
вых углеводородов (общая формула СюН16). Товарный про-
дукт, получаемый путем отгонки из живицы летучей части,
называется скипидаром. Практически все терпеновые углево-
5
1. СОСТАВ ЖИВИЧНОГО СКИПИДАРА НЕКОТОРЫХ ОТЕЧЕСТВЕННЫХ ЗАВОДОВ
Завод-изготовитель Состав скипидара» %
а-пинен камфен р-пинен мирцен X <и св X <3 дипентен Р-фел- ландрен а-терпи- нен терпино- лен
Горьковский «Оргсинтез» Борисовский Тихвинский Барнаульский 70,5 67,0 63,5 63,5 0,85 1,00 1,00 1,00 2,0 3,5 4,0 4,5 1,0 0,5 1,0 1,5 17,5 19,0 20,0 20,5 5,0 6,0 5,0 2,5 1,0 0,5 2,0 1,5 0,3 1,0 1,0 0,5 1,85 1,50 2,50 1,50
дороды, входящие в состав скипидара, обладают склонностью
к изомеризации под действием кислот, повышенной темпера-
туры, света, катализаторов и других факторов.
Для выделения индивидуальных компонентов скипидара
обычно пользуются вакуум-ректификацией, т. е. разделением
смеси при пониженных температурах, чтобы предупредить их
термическую изомеризацию. В настоящее время состав скипи-
дара определяется методом газожидкостной хроматографии
(табл. 1).
Несмотря на то, что терпеновые углеводороды имеют оди-
наковую молекулярную массу 136 и общую формулу СюН16,
тем не менее их физико-химические свойства заметно отлича-
ются, так как строение их различно (табл. 2).
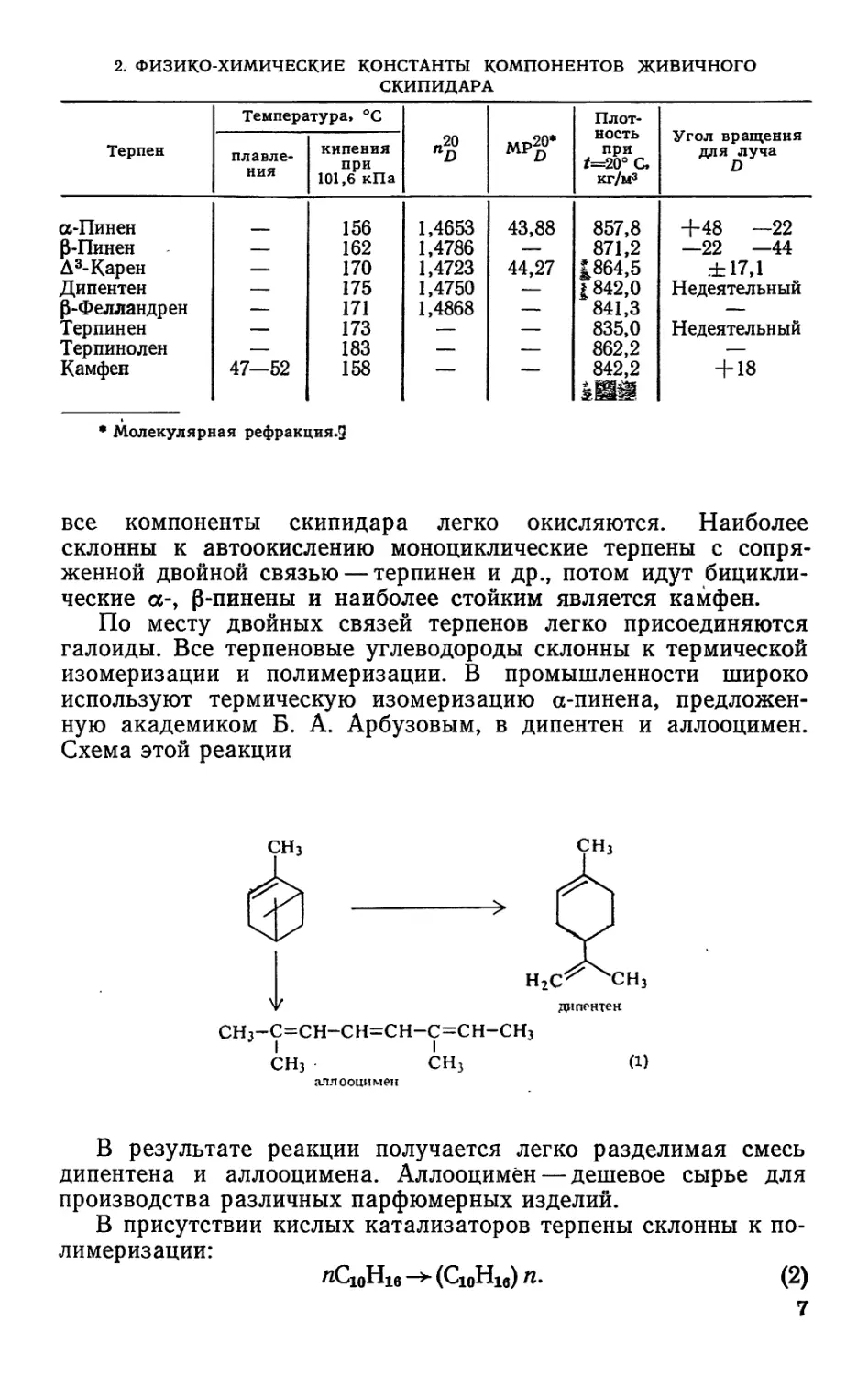
Структурные формулы терпеновых углеводородов, входя-
щих в состав живичного скипидара, имеют следующий вид:
фелландрен
дипентек
камфен
ОГ-терпинен терпинолен
Живичный скипидар представляет собой бесцветную про-
зрачную жидкость с характерным запахом, он хорошо смеши-
вается с большинством органических растворителей. На воздухе
6
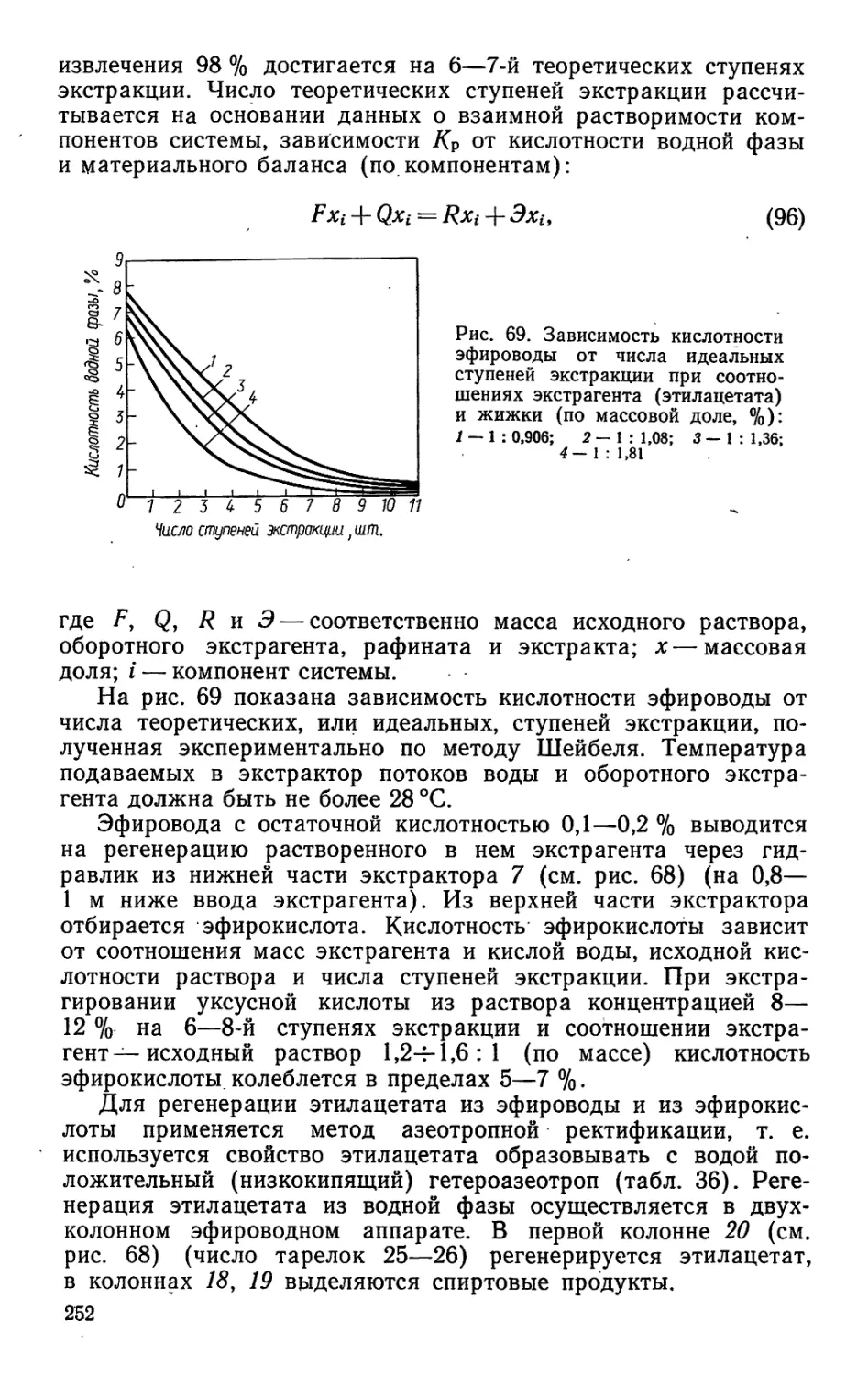
2. ФИЗИКО-ХИМИЧЕСКИЕ КОНСТАНТЫ КОМПОНЕНТОВ ЖИВИЧНОГО
СКИПИДАРА
Терпен Температура, °C „20 nD Плот- ность при /=20° С, кг/м3 Угол вращения для луча D
плавле- ния кипения при 101,6 кПа
а-Пинен 156 1,4653 43,88 857,8 +48 —22
0-Пинен — 162 1,4786 — 871,2 —22 —44
Д3-Карен — 170 1,4723 44,27 £864,5 ±17,1
Дипентен — 175 1,4750 — 1842,0 Недеятельный
Р-Фелландрен — 171 1,4868 — 841,3 —
Терпинен — 173 — — 835,0 Недеятельный
Терпинолен — 183 — — 862,2 —
Камфен 47—52 158 — — 842,2 ЬИЙ + 18
• Молекулярная рефракция.5
все компоненты скипидара легко окисляются. Наиболее
склонны к автоокислению моноциклические терпены с сопря-
женной двойной связью — терпинен и др., потом идут бицикли-
ческие а-, р-пинены и наиболее стойким является камфен.
По месту двойных связей терпенов легко присоединяются
галоиды. Все терпеновые углеводороды склонны к термической
изомеризации и полимеризации. В промышленности широко
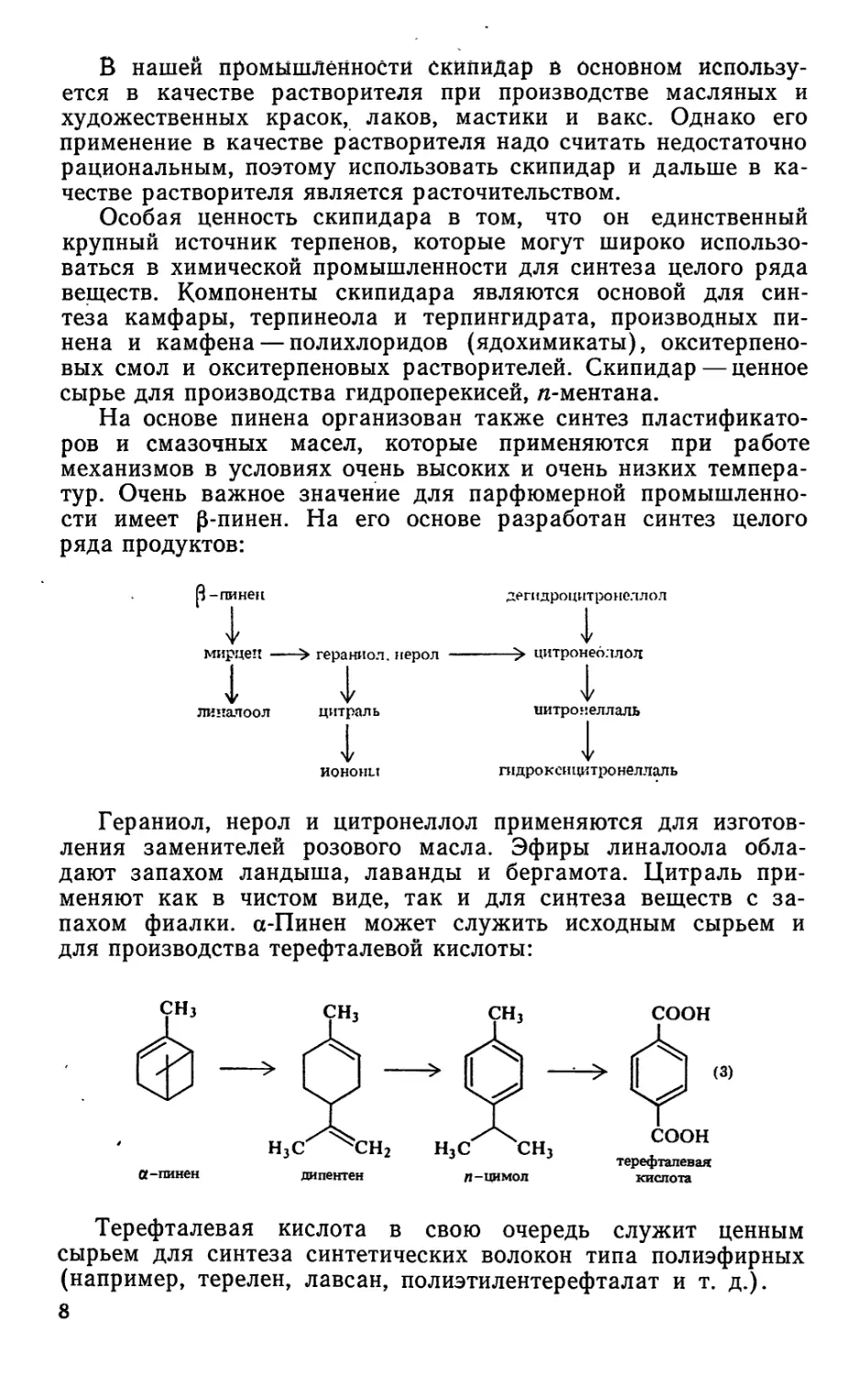
используют термическую изомеризацию а-пинена, предложен-
ную академиком Б. А. Арбузовым, в дипентен и аллооцимен.
Схема этой реакции
дипентен
сн3-с=сн-сн=сн-с=сн-сн}
СНз сн3 (1)
аллооцимен
В результате реакции получается легко разделимая смесь
дипентена и аллооцимена. Аллооцимен — дешевое сырье для
производства различных парфюмерных изделий.
В присутствии кислых катализаторов терпены склонны к по-
лимеризации:
/гС10Н1в —> (С10Н1в) п. (2)
7
В нашей промышленности скипидар в основном использу-
ется в качестве растворителя при производстве масляных и
художественных красок, лаков, мастики и вакс. Однако его
применение в качестве растворителя надо считать недостаточно
рациональным, поэтому использовать скипидар и дальше в ка-
честве растворителя является расточительством.
Особая ценность скипидара в том, что он единственный
крупный источник терпенов, которые могут широко использо-
ваться в химической промышленности для синтеза целого ряда
веществ. Компоненты скипидара являются основой для син-
теза камфары, терпинеола и терпингидрата, производных пи-
нена и камфена — полихлоридов (ядохимикаты), окситерпено-
вых смол и окситерпеновых растворителей. Скипидар — ценное
сырье для производства гидроперекисей, и-ментана.
На основе пинена организован также синтез пластификато-
ров и смазочных масел, которые применяются при работе
механизмов в условиях очень высоких и очень низких темпера-
тур. Очень важное значение для парфюмерной промышленно-
сти имеет 0-пинен. На его основе разработан синтез целого
ряда продуктов:
дегидроцитронеллол
мирцен --->
линалоол
гераниол.нерол
цитраль
I
ИОНОНЫ
цитронебллол
цитро.челлаль
гидро кси цитронеллаль
Гераниол, нерол и цитронеллол применяются для изготов-
ления заменителей розового масла. Эфиры линалоола обла-
дают запахом ландыша, лаванды и бергамота. Цитраль при-
меняют как в чистом виде, так и для синтеза веществ с за-
пахом фиалки. а-Пинен может служить исходным сырьем и
для производства терефталевой кислоты:
а-пинен
Терефталевая кислота в свою очередь служит ценным
сырьем для синтеза синтетических волокон типа полиэфирных
(например, терелен, лавсан, полиэтилентерефталат и т. д.).
8
§ 2. КАНИФОЛЬ
Живичная канифоль — хрупкое стекловидное вещество от
светло-желтого до коричневого цвета; растворима в большин-
стве органических растворителей. Представляет собой аморф-
ную смесь до 98 % смоляных кислот и нейтральных неомыляе-
мых веществ.
Живичная сосновая канифоль состоит из 92 % смеси смо-
ляных кислот, 0,4—1,6 % жирных и 6,4 %' нейтральных продук-
тов. Смоляные кислоты, входящие в состав живицы, представ-
ляют собой ненасыщенные реакционноспособные соединения
с двумя двойными связями, имеющие общую формулу С20Н30О2.
Исследование химического состава компонентов канифоли, которыми
являются изомерные смоляные кислоты, было начато в 20-х годах
XIX в. Для идентификации смоляных кислот, содержащихся в жи-
вице, использовали в различных растворителях способ кристаллизации.
Метод этот был очень трудоемким и не всегда давал желаемые резуль-
таты. В конце XIX в. Вестберг предложил применять для выделения
смоляных кислот кристаллизацию натриевых солей. С конца XIX в. и по
40-е годы XX в. русские ученые Ф. М. Флавицкий, В. В. Шкателов,
В. Е. Тищенко, Б. А. и А. Е. Арбузовы, В. Н. Крестинский
и др. изучали химический состав смоляных кислот, входящих в состав
живицы различных отечественных хвойных пород. Позднее исследованиями
И. И. Бардышева, X. А. Черчеса, Н. Ф. Комшилова и др. было показано,
что смоляные кислоты способны образовывать с аминами хорошо кри-
сталлизующиеся соли. Это позволило установить достаточно точный состав
смоляных кислот, входящих в сосновую живицу.
В последнее время для количественного и качественного
анализа состава смеси смоляных кислот широко применяются
хроматографические методы анализа в сочетании со спектро-
фотометрией. И. И. Бардышевым, X. А. Черчесом и др. были
установлены состав смоляных кислот канифоли из сосновой
живицы и основные их физические константы.
Было показано, что абиетиновая кислота является наиболее
распространенной смоляной кислотой, которая входит в состав
всех видов канифоли (табл. 3).
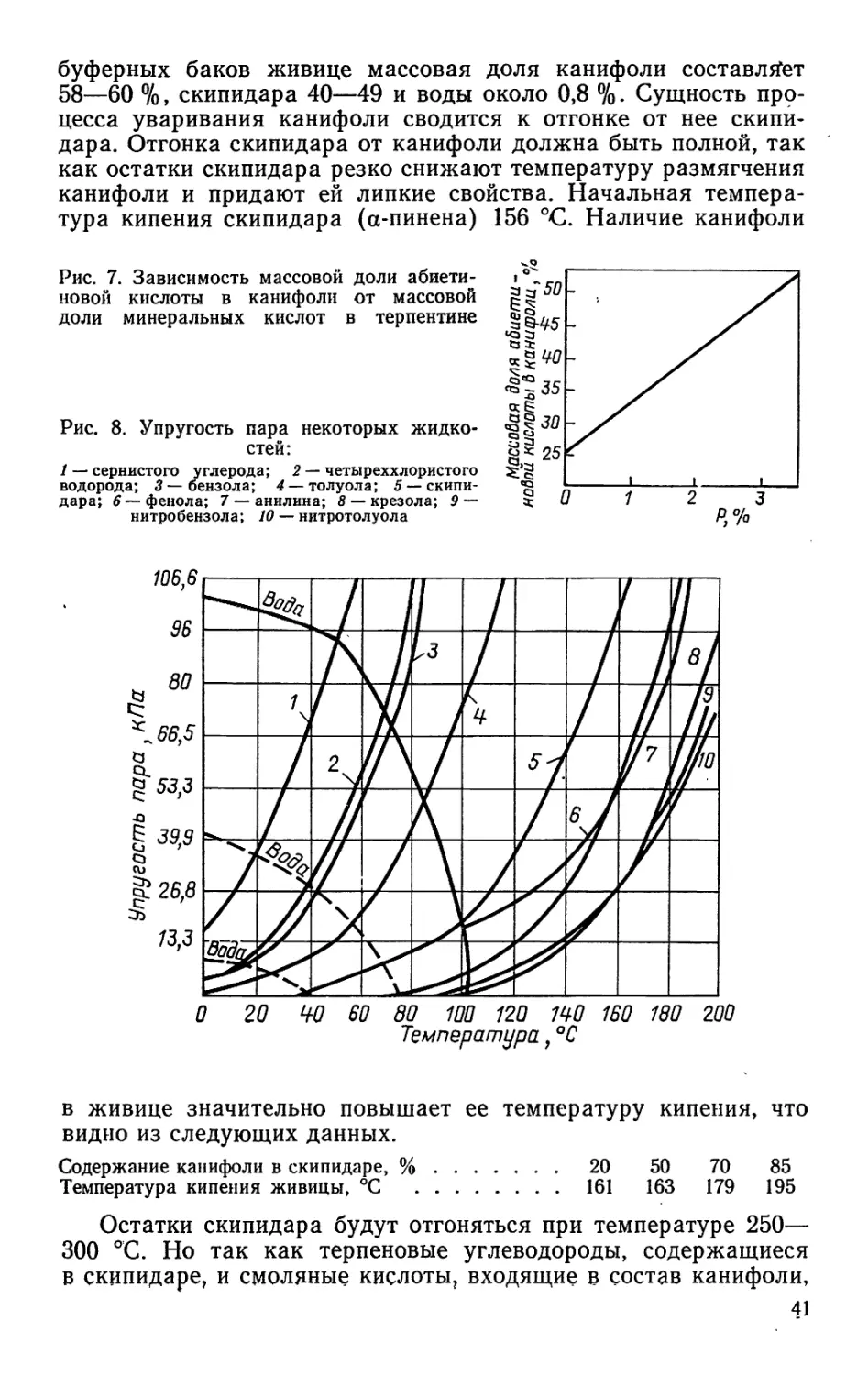
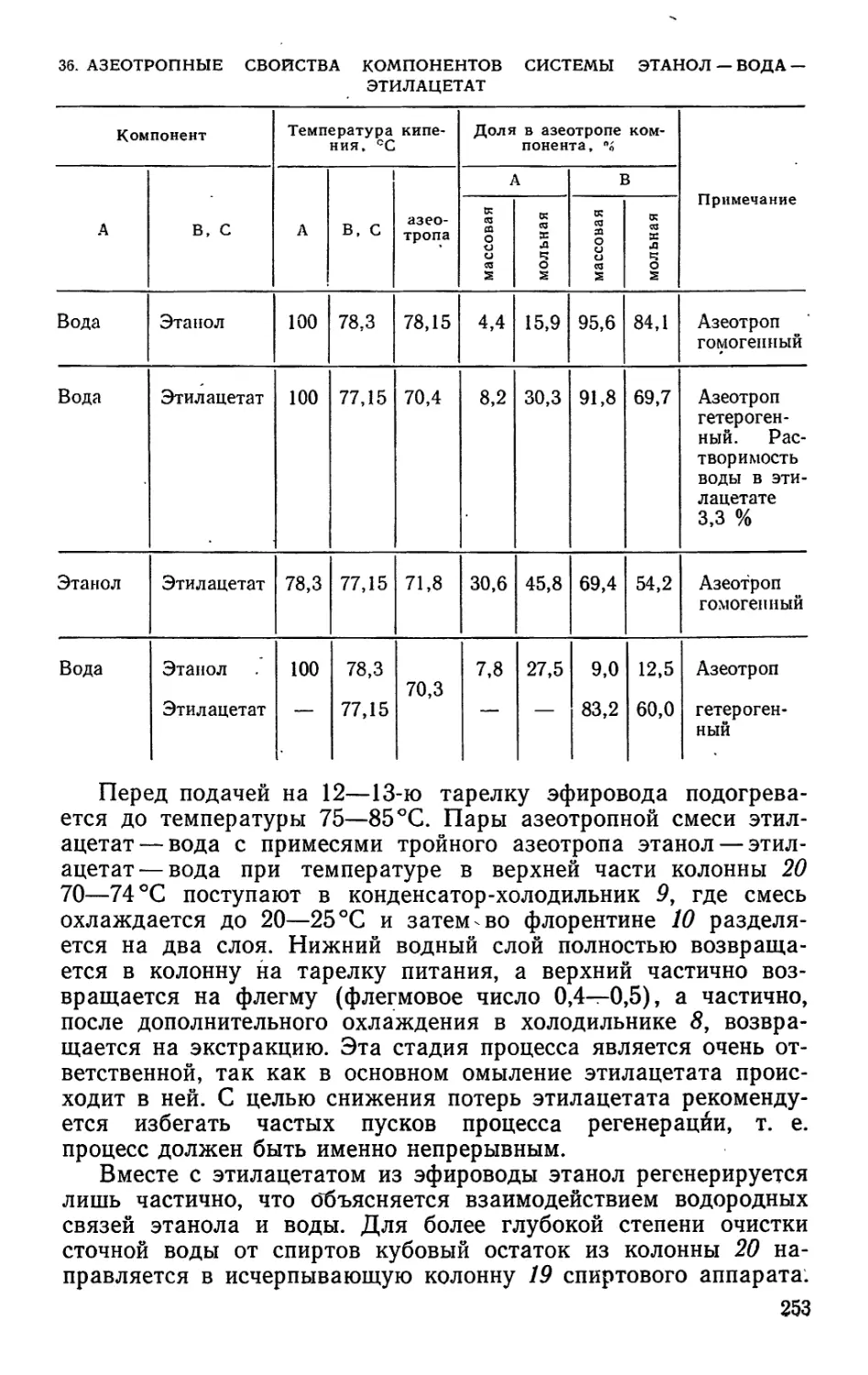
3. КОЛИЧЕСТВЕННЫЙ СОСТАВ СМЕСЕЙ СМОЛЯНЫХ КИСЛОТ, ВЫДЕЛЕННЫХ ИЗ
ЖИВИЦЫ РАЗЛИЧНЫХ ХВОЙНЫХ ПОРОД
Порода древесины Содержание кислот, %
п алюст- ровой пимаро- вой и др.* абиети- новой левопи- маровой неоабие- тиновой дигидро- абиети- новой
Сосна: обыкновенная 12 20 15 36 10 5
крымская 6 28 13 18 13 12
пицундская 19 21 37 3 2 18
Ель обыкновенная 17 17 8 37 7 14
Кедр сибирский 3 44 32 1 1 4
Лиственница сибирская 16 36 32 1 8 4
♦ Тетрагидроабиетиновая и дигидроабиетиновая кислоты.
9
Что касается состава смеси смоляных кислот, входящих
в живичную канифоль, полученного на различных канифольно-
терпентинных заводах, то он неоднороден. По данным ЦНИЛХИ,
массовая доля пимаровой кислоты в живичой канифоли колеб-
лется от 6,5 до 8,4 %, изопимаровой — от 7,2 до 9,7, левопима-
ровой+палюстровой — от 11,6 до 27,6, абиетиновой — от 33,0
до 56,0, неоабиетиновой — от 8,9 до 18,9, дигидроабиетиновой —
от 2,3 до 3,7 %. Различие количественного содержания отдель-
ных смоляных кислот объясняется спецификой технологиче-
ского режима, особенно на стадии уваривания канифоли на
различных предприятиях. Накопление абиетиновой кислоты
происходит в результате изомеризации в нее других кислот.
Левопимаровая кислота, содержание которой преобладает
в сосновой и еловой живице, легко изомеризуется под дейст-
вием повышенной температуры. Исследованиями ЦНИЛХИ по-
казано, что в результате термической изомеризации при 155 °C
левопимаровая кислота полностью превращается в палюстро-
вую (7 %), абиетиновую (85 %) и неоабиетиновую (8 %) • Со-
держание других смоляных кислот в канифоли по сравнению
с содержанием их в живице изменяется незначительно.
Кроме смоляных кислот, неомыляемых веществ и жирных
кислот, в живичной канифоли в небольших долях могут быть
танниды — окрашенные вещества, содержащиеся в коре хвой-
ных деревьев, углеводороды с высокой температурой кипения
(состав С20Н32) и резинаты, главным образом, железа и дру-
гих металлов.
Сосновая живичная канифоль вырабатывается марки А,
которая делится на три сорта: высшая категория качества, 1-й
и 2-й сорта. Качество канифоли определяется по ГОСТ 19113—
84, согласно которому устанавливаются следующие физико-
химические показатели:
1. Внешний вид — прозрачная стекловидная масса.
2. Цветность — в зависимости от цветности установлено 13
марок канифоли. Они обозначаются буквами, при этом кани-
фоль марок X, W, Wi — высшего качества и 1-го сорта; марок
WG, N — 1-го сорта; М, К, У, Н — 2-го сорта.
3. Массовая доля воды для всех сортов канифоли не дол-
жна быть выше 0,2 % -
4. Массовая доля золы в зависимости от сортности не дол-
жна превышать 0,03—0,04 % -
5. Массовая доля механических примесей. Механические
примеси в пробе канифоли обычно просматриваются на свет,
имеют вид посторонних включений, в зависимости от сорта их
должно быть не более 0,03—0,04 %.
6. Температура размягчения для канифоли высшего каче-
ства должна быть не ниже 69 °C, для 1-го сорта 68 °C, для
2-го 66 °C.
7. Кислотное число характеризует количественное содержа-
ние кислот в канифоли. Для канифоли высшего качества кис-
10
лотное число должно быть не менее 169, для 1-го сорта—168
и для 2-го — не менее 166 мг КОН на 1 г канифоли.
8. Массовая доля неомыляемых веществ. Неомыляемыми
называются вещества, не вступающие в реакцию со щелочью.
Их должно быть для канифоли высшего качества не более
6,0 %, для 1-го сорта — 6,5 и для 2-го — 7,5 %.
9. Склонность к кристаллизации для канифоли должна от-
сутствовать.
Выпускаемые нашей промышленностью образцы живичной
сосновой канифоли имеют температуру размягчения 66—71 °C,
кислотное число 166—170, массовую долю неомыляемых ве-
ществ 4,4—6,9 %; влага, зольность и механические примеси от-
вечают нормам, предусмотренным ГОСТом.
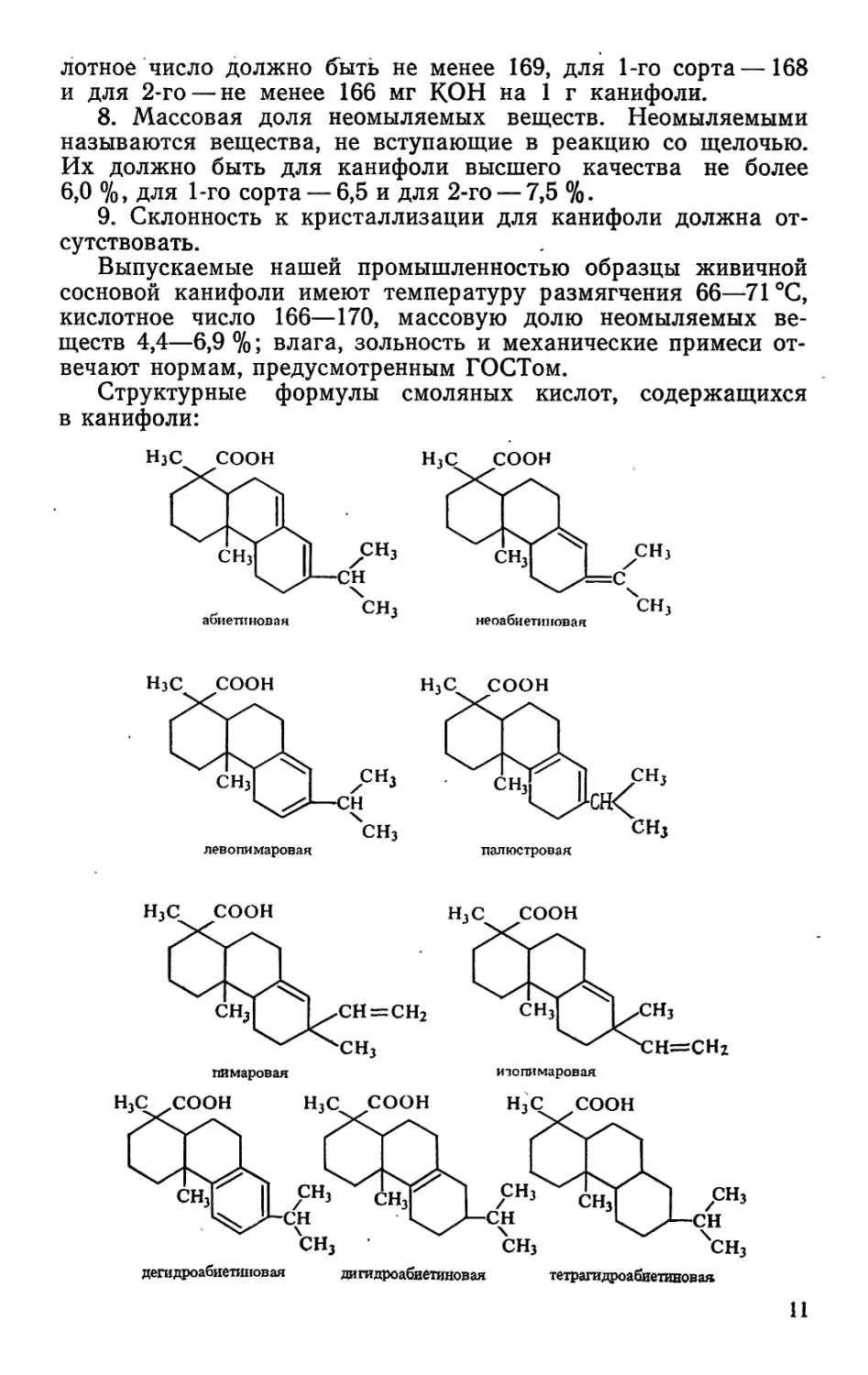
Структурные формулы смоляных кислот, содержащихся
в канифоли:
дегидроабиетшювая дигидроабиетиновая тетрагидроабиетивовая
11
Канифоль хорошо растворяется в диэтиловом эфире, аце-
тоне, метиловом и этиловом спиртах, трихлорэтилене, уксусной
и жирных кислотах, бензоле, ксилоле, толуоле и бензине.
В воде канифоль нерастворима, при нагревании частично
эмульгируется. Легко окисляется кислородом воздуха, особенно
в мелкораздробленном состоянии.
Наибольшей склонностью к окислению в чистом виде обла-
дает абиетиновая кислота, тогда как неоабиетиновая, дегидро-
абиетиновая, палюстровая, левопимаровая, изопимаровая, пи-
маровая кислоты при хранении их на воздухе более 5 лет ос-
таются неизменными.
В ЦНИЛХИ ведутся исследования по предотвращению окис-
ления канифоли путем введения в нее антиокислителей. Уста-
новлено, что наибольший интерес представляют антиокисли-
тели фенольного и дифениламинного видов.
Высокая склонность канифоли к окислению делает ее пыль
взрывоопасной. Так как канифоль представляет собой смесь
изомерных смоляных кислот, то физико-химические константы
ее имеют групповой характер. Плотность канифоли колеблется
в пределах 1070—1085 кг/м3. Теплотворная способность ее из-
меняется в пределах 38020—38426 кДж/кг. Горит канифоль
ровным коптящим пламенем, ее теплопроводность 0,4609 Вт/
/(м«К), теплота плавления 66,202 кДж/кг. Теплоемкость ка-
нифоли, кДж/(кг-К), зависит от температуры и может быть
определена по формуле
Ct = 16 927 + 0,00097 (Д/). (4)
Температура самовозгорания канифоли 850 °C. Канифоль
в обычных условиях нелетуча. Перегонку ее можно вести под
глубоким вакуумом или в токе перегретого до 200 °C водяного
пара. Зависимость температуры кипения канифоли под ваку-
умом от давления приведена ниже.
Остаточное давление
в системе, Па ... 266,6 533,2 666,5 799,8 1066 1333 1999 2666 3998
Температура, °C . . 226 244 250 256 266 174 288 300 310
При нагревании канифоли до 300°C происходит ее разло-
жение, сопровождающееся декарбоксилированием и дигидрата-
цией смоляных кислот с образованием углеводородов, например
абиетина и абиетана:
2С20Н30О2 —*• С19Н30 + С19Н28 + СО -|- СО2+Н2О. (5)
абиетан абиетин
Получающиеся при разложении канифоли абиетан и абие-
тин называют .канифольными маслами, присутствие которых
в канифоли резко снижает ее сортность, а выделившиеся СО и
СОг ухудшают условия труда, поэтому разложения канифоли
допускать нельзя.
С целью придания канифоли устойчивости к окислению кис-
лородом воздуха ее подвергают различным способам модифи-
цирования. Например, при длительном нагревании канифоли
12
при температурах ниже 250 °C в присутствии палладия, нане-
сенного на активный уголь, происходит диспропорционирование,
или межмолекулярное перераспределение, атомов водорода
в молекулах смоляных кислот с образованием из абиетиновой
кислоты дегидро- и дигидроабиетиновой кислот:
2С20Н30О2 -> CsoH^Oa + Сг()Н32О2. (6)
абиетиновая дегидроаби- дигидро-
кислота етиновая абиетиновая
кислота кислота
Дальнейшее взаимодействие абиетиновой кислоты с вновь
образовавшейся дигидроабиетиновой кислотой приводит к по-
лучению тетрагидроабиетиновой кислоты:
СгоНзо02 ~|- СэдНзгОг —> C2oH2s02 -J- С23Н34О2. (7)
абиетиновая дигидроаби- дегидроаби- тетрагидро-
кислота етиновая етиновая абиетиновая
кислота кислота кислота
Полученные в процессе диспропорционирования дегидро-,
дигидро- и тетрагидроабиетиновые кислоты практически не
окисляются кислородом воздуха.
Устойчивую к окислению канифоль получают путем присое-
динения по двойным связям молекул смоляных кислот водо-
рода. Образование дигидроабиетиновой кислоты С20Н32О2
в процессе гидрирования проходит сравнительно легко, а тет-
рагидроабиетиновой— с большим трудом. Вопросом гидриро-
вания канифоли занимались И. И. Бардышев и Е. Б. Смирнова.
Гидрирование канифоли ведут в 35 %-ном растворе ее в бен-
зине в присутствии ~0,1 % палладиевого катализатора от
массы канифоли при температуре 150 °C и избыточном давле-
нии порядка 5000—6000 кПа. Реакция идет по схеме
С1ЭН29СООН + Н2 -> С19Н31СООН + Н2 СыНззСООН. (8)
абиетиновая дигидроабие- тетрагидро-
кислота типовая абиетиновая
кислота кислота
С целью стабилизации физико-химических свойств кани-
фоли ее полимеризуют в растворах различных органических
растворителей: толуоле, бензине, бензоле, и-гексане и др. Ре-
акцию ведут в присутствии кислых катализаторов, в качестве
которых могут быть использованы серная и фосфорная кис-
лоты, двухлористый цинк и четыреххлористое олово, фтори-
стый водород и активные глины: бентонит, боксит и др. Основ-
ные продукты полимеризации — димеры абиетиновой кислоты:
2С20Н30О2 —> С40Н60О4. (9)
Строение димера абиетиновой кислоты пока не доказано.
Полимеризованная канифоль более стойка к окислению и
имеет более высокую температуру плавления, она значительно
светлее обычной канифоли и не кристаллизуется.
Так как канифоль является смесью смоляных кислот, то
для нее свойственны все реакции, характерные для кислот.
Канифоль способна вступать в реакцию с металлами, с солями
и щелочами. Соли канифоли или смоляных кислот называют
резинатами.
13
При взаимодействии канифоли и едкого натра образуется
резинат — Na-канифольное мыло:
С19Н29СООН + NaOH C19H29COON а+Н2О. (10)
Эта реакция может протекать и при взаимодействии кани-
фоли с Na2CO3:
г^НвдСООН+Na2CO3 -> 2C19H29COONa + СО2 + Н2О. (11)
Канифольное мыло представляет собой гидрофильный про-
дукт, способный образовывать в органических растворителях
коллоидные растворы, практически полностью растворимые
в воде; обладает высокими поверхностно-активными свойст-
вами, что обусловливает использование его в мыловаренной
промышленности. В хозяйственном мыле массовая доля его
достигает 40 %, в туалетном — значительно меньше.
Канифоль легко вступает в реакции этерификации с раз-
личными спиртами. Наиболее распространенным является гли-
цериновый эфир канифоли, который нашел широкое примене-
ние в лакокрасочной и полиграфической промышленности. Об-
разование глицеринового эфира канифоли идет по схеме
сн2он
I
СН-ОН + ЗС,9Н29СООН
СН2ОН
о
II
СН2-О-С-С|9Н29
» сн-о-с-с19н29+зн2о
I О
сн2-о-с-с|9н29
о
(12)
При взаимодействии с
пентаэритритовый эфир
пентаэритритом канифоль
образует
сн2он
Хсн2он
с + 4С19Н29СООН =
^СН2ОН
СН2ОН
о
II
СН2О-С-С19Н29
/ о
/ II
/,СН-О-С-С|9Н29
+4Н2О
\'СН-О-С-С|9Н29
\ II
\ °
СН2-О—С—С49Н29
О '
(13)
В масляных лаках применяется глифталевая смола, пред-
ставляющая собой сложный эфир глицерина, канифоли и фта-
левой кислоты (техническое название «пентолановая смола»);
снюн
I
сн-он
I
СН2ОН
+ 2С19Н29СООН
+ С6Н4(СООН)2
СН2ОСО-С19Н29
I
> СНОСО-СбН4СООН+ЗН2О (14)
СН2ОСО~С|9Н29 ,
14
или еще более сложный эфир глицерина, канифоли, фталевой
и линолевой кислот.
Канифоль хорошо вступает в реакцию этерификации и
с ксилитом (пентит) и сорбитом (гексит). Большие возможно-
сти имеются для использования в этих целях дульцита — од-
ного из самых дешевых многоатомных спиртов.
Качество эфиров, полученных из многоатомных спиртов,
часто намного выше качества глицеринового эфира.
С тех пор как было показано, что тутовое масло и эфиры
канифоли дают хорошие водостойкие лаки, их стали употреб-
лять для покрытия корпусов кораблей, окраски палуб, самоле-
тов, приготовления изоляционных лаков, получения огнестой-
ких и водонепроницаемых тканей, брезента и т. д.
Если эфиры канифоли многоатомных спиртов используются
в лакокрасочной промышленности в качестве пленкообразова-
телей в различных лаках, эмалях и красках, то эфиры одно-
атомных спиртов (метилового, этилового, бутилового и др.) яв-
ляются ценными пластификаторами пластмасс и служат заме-
нителями пищевых и других растительных масел.
В некоторых зарубежных странах на основе обычной или
модифицированной канифоли налажено производство аминов
канифоли. Установлена способность канифоли к конденсации
с альдегидами и ангидридами. Особую ценность представляют
продукты конденсации их с эфирами канифоли, например гли-
цериновым эфиром канифоли. При конденсации глицеринового
эфира канифоли с малеиновым ангидридом получается про-
дукт, который дает светоустойчивые пасты и совместные рас-
творы с нитроцеллюлозой. Вместо малеинового ангидрида мо-
жно применять малеиновую кислоту. При обработке живичной
канифоли малеиновым ангидридом получают аддукт левопима-
ровой кислоты — малеопимаровую кислоту и малеинизирован-
ную канифоль.
Малеопимаровая кислота представляет собой продукт кон-
денсации левопимаровой кислоты и малеинового ангидрида:
левопимаровая
кислота
малеиновый
ангидрид
мал ео пи Маров ая кислота
Получение продуктов конденсации на основе канифоли мо-
жно осуществлять на основе многих органических соединений.
Наиболее важными из них являются фенолальдегидные смолы,
модифицированные эфирами канифоли,— альбертоли. Продукт
15
конденсации канифоли с формальдегидом представляет собой
соединение вида
При конденсации /г-фенолов и канифоли в присутствии
формальдегида получается соединение
. Краски, имеющие в своей основе альбертоли, растворенные
в высыхающих маслах, задерживают обрастание ракушками
днищ морских кораблей.
Для живичной канифоли характерным свойством является
способность кристаллизоваться. Закристаллизованная канифоль
теряет практически все ценные физико-химические свойства и
фактически является браком.
В результате проведенных совместных исследований
ЦНИЛХИ и ЛТА им. С. М. Кирова было установлено, что од-
ной из основных причин, вызывающих склонность живичной
канифоли к кристаллизации, являются режимные условия пе-
реработки живицы. Было показано, что при уваривании кани-
фоли при повышенных температурах (свыше 175 °C) и в слу-
чае присутствия в терпентине даже незначительной массы
минеральных кислот и наличия влаги идет активная изоме-
ризация смоляных кислот. Основным продуктом изомеризации
является абиетиновая кислота. Исследованиями установлено,
что при накоплении в канифоли абиетиновой кислоты свыше
40 % от массы смоляных кислот канифоль практически всегда
склонна к кристаллизации.
Для предупреждения кристаллизации канифоли канифоль-
но-терпентинным заводам рекомендованы следующие правила
варки и розлива канифоли:
1. Тщательно отмыть терпентин после отстаивания от ми-
неральных кислот (H2SO4, Н3РО4), которые могут попадать
в живицу в результате подсочки и осветления ее.
2. При уваривании канифоли избегать высоких температур
(выше 175°C), при которых в результате термической изоме-
ризации идет активное образование абиетиновой кислоты.
16
3. Канифоль уваривать до полного удаления воды, так как
даже небольшая часть ее способствует процессу кристалли-
зации.
4. Не допускать попадания воды и минеральной пыли в тару,
в которую разливают канифоль.
5. В отдельных случаях рекомендуется проводить частич-.
ную нейтрализацию канифоли углекислым натрием, взятым
в количестве 2—2,5 % от массы канифоли.
6. Розлив канифоли вести таким образом, чтобы можно
было быстро охладить ее в упаковочной таре и тем самым
пройти через критическую температуру кристаллизации
(100°C). Летом розлив канифоли идет медленнее, чем зимой.
7. Для предохранения канифоли от кристаллизации реко-
мендуют добавлять в нее около 3 % малеиновой кислоты или
глицерина. —
Применение канифоли. Канифоль в настоящее время нахо-
дит весьма широкое применение; потребляют ее около 70 от-
раслей промышленности. Однако для ряда потребителей со-
вершенно неприемлемыми являются такие свойства канифоли,
как сравнительно низкая температура размягчения, высокое
кислотное число, недостаточная влагоустойчивость, мягкость,
хрупкость, склонность к кристаллизации, способность легко
окисляться кислородом воздуха и др. Поэтому канифоль
у большинства потребителей используется в виде ее производ-
ных. Чаще всего потребляют канифоль при производстве бу-
маги и картона. Для проклейки бумаги в нашей стране ис-
пользуется около 35 %, в США — 43, в Японии до 47 % всего
потребляемого объема канифоли. Как правило, бумажная про-
мышленность постоянно испытывает недостаток в канифоли,
что привело к применению в этой отрасли многих заменителей.
Писчую бумагу и бумагу для печати обязательно проклеивают
для создания на ее поверхности гидрофобной пленки, препят-
ствующей растеканию красок и чернил; для проклейки бумаги
до сих пор используют канифольный клей, изготовленный ис-
ключительно на основе живичной канифоли. Канифольный клей
представляет собой пасту или сухой порошок, состоящие из
коллоидного раствора резината натрия и содержащие до 30 %
свободных смоляных кислот. Для проклейки бумаги использу-
ется 2—5%-ный водный раствор клея, массовая доля которого
в пересчете на абсолютно сухую массу должна составлять
0,5—3 % бумажной массы. Канифоль в виде резината алюми-
ния из водного раствора клея высаживается водным раство-
ром серно-кислого глинозема концентрацией 0,7—1,2%. Обмен-
ная реакция этих веществ идет по схеме:
6C19H29COONa+Al2 (SO4)3 -> 2А1 (С19Н29СО)3 + 3Na2SO4. (18)
При сушке бумажного полотна резинат канифоли плавится
и заклеивает поры бумаги, что придает ей гидростойкость. Для
17
улучшения проклеивающих свойств канифоли ее подвергают
различным модификациям, что способствует росту сортности
бумаги. Например, использование для приготовления бумаж-
ного клея. малеопимаровой или гидрированной канифоли, по-
лимеризованной и диспропорционированной канифолей, устой-
чивых к самоокислению, обеспечивает сохранность цветности
бумаги, исключая ее пожелтение на свету и при хранении.
Высокие изоляционные свойства канифоли и ее производ-
ных обеспечили применение ее в электро- и радиотехнической
промышленности.
Одним из крупнейших потребителей канифоли в виде рези-
натов натрия и калия является производство синтетического
каучука, особенно дивинилстирольного и бутадиенстирольного.
Резинаты калия и натрия являются хорошими эмульгаторами
при эмульсионной полимеризации и обеспечивают повышенные
свойства каучука.
При использовании натриевых и калиевых солей диспро-
порционированной канифоли в производстве синтетического ка-
учука (СК) обеспечивается надежная и высокая степень био-
химической очистки промышленных стоков. Для получения
эмульгаторов при производстве СК используют только гидри-
рованную или диспропорционированную канифоль, так как
смоляные кислоты, имеющие сопряженные двойные связи, вы-
зывают длительный индукционный период и нарушают нор-
мальный полимеризационный процесс.
В резинотехнической промышленности канифоль использу-
ется как добавка для придания резине эластичности и моро-
зостойкости, для приготовления резинового клея, изготовления
линолеума и др.
В связи с бурным ростом автомобильной, тракторной и
авиационной промышленности рост потребления канифоли и ее
производных в производстве каучука и резинотехнических из-
делий будет расти.
Амины канифоли могут быть использованы для регенера-
ции каучука, пропитки бумаги, производства ядохимикатов и
гербицидов, флотореагентов и др. Из продуктов канифоли
можно вырабатывать эффективные поверхностно-активные ве-
щества — гидрофобизаторы для обработки древесноволокнистых
плит, эмульгаторы для битумных покрытий в дорожном строи-
тельстве.
Канифоль широко применяют для производства кожезаме-
нителей, креолина, эмульсионных смазок и др.
При издании многокрасочных иллюстрированных журналов,
репродукций картин, почтовых марок, географических карт и
других изданий полиграфическая промышленность широко ис-
пользует сиккативы канифоли, резинаты цветных металлов,
главным образом марганца, меди, свинца, кобальта, цинка
и др. Сиккативы улучшают пластичность красок и ускоряют
высыхание олиф, изготовленных на основе растительных масел.
18
В производстве лаков и эмалей обязательной составной
частью являлись экзотические нейтральные смолы типа ко-
пала, шеллака или даммара. В связи с резким сокращением их
производства все шире стали применять производные кани-
фоли: глицериновый, этиленгликолевый, глифталевый, пента-
эритритовый и другие эфиры канифоли.
Вместо пищевых жиров канифоль применяется в мыловаре-
нии для повышения моющих свойств мыла.
В США обычная и модифицированная канифоли широко
используются для производства термостойких связующих.
Кроме крупных потребителей, канифоль применяют в про-
изводстве сургуча и мухоловкой бумаги. Она входит в состав
хирургического пластыря и большинства лыжных мазей. Су-
ществуют и многие другие способы использования канифоли.
Из литературы известно, что смоляные кислоты, входящие
в состав канифоли, ценятся как сырье для синтеза целого ряда
биологически активных веществ и других ценных продуктов.
В настоящее время на основе сосновой живицы в ЦНИЛХИ
разработана технология получения малеопимаровой кислоты и
эфиров канифоли — компонентов лака. Эта технология может
быть осуществлена на любом канифольно-терпентинном заводе
с большим экономическим эффектом.
Глава 2
КАНИФОЛЬНО-ТЕРПЕНТИННОЕ ПРОИЗВОДСТВО
Одним из важнейших экстрактивных веществ деревьев
хвойных пород являются смолистые вещества, собираемые при
подсочке деревьев сосны, кедра, ели, лиственницы в виде свет-
лого смолистого бальзама, который заживляет рану, закрывает
ткани древесины от проникновения микроорганизмов и насе-
комых. Отсюда и происходит техническое название бальзама —
живица. Живица стерилизует и раны живого организма. При
переработке живицы получают до 70—75 % канифоли и 16—
20 % скипидара от общего объема вырабатываемых в нашей
стране таких продуктов.
Возникновение терпентинного промысла в России относится к началу
XVIII в., когда для строительства флота, организованного Петром I, тре-
бовалось много смолы. Возросшая потребность в смоле вызвала возникно-
вение промысла подсочки сосны — осмолоподсочки (или вельского спо-
соба подсочки), имевшей целью главным образом получение просмолив-
шейся древесины.
Позднее, в конце XIX и начале XX вв. многие русские ученые-химики
и лесоводы неоднократно пытались ставить опыты подсочки нашей сосны
разными методами. Большой интерес к подсочке хвойных пород проявил
великий русский ученый Д. И. Менделеев. Еще в 1892 г. он на основе
работ С. Ф. Флавицкого пытался создать в России канифольно-терпентин-
ное производство. В тот же период Д. И. Менделеев посоветовал
В. Е. Тищенко уделить особое внимание терпентинному промыслу. В 1895 г.
В. Е. Тищенко напечатал книгу «Канифоль и скипидар», которая не по-
теряла своей ценности и до настоящего времени.
19
Однако до революции подсочное производство не нашло в нашей
стране промышленного размаха и едва покрывало 4 % всей потребности
в канифоли и скипидаре, остальное ввозилось из-за границы. Причиной
такого положения являлась, с одной стороны, невысокая техника под-
сочки, дававшая низкий выход живицы, а с другой — консерватизм,
существовавший в лесном ведомстве дореволюционной России. Среди
работников лесного ведомства того времени существовало мнение, что под-
сочка вредит лесу и понижает механические свойства древесины; кроме
того, считалось, что произрастающая у нас сосна малопродуктивна и
непригодна для подсочки.
В 1919 г. опыты по подсочке возобновились. В 1925 г. постановле-
нием Совета Труда и Обороны (СТО) подсочные промыслы были взяты
государством под особое покровительство. В 1926 г. был организован
трест «Русская смола» (позднее — «Лесохим»). В сентябре того же года,
। по инициативе Ф. Э. Дзержинского, работники треста организовали сове-
• щание ведущих ученых и специалистов по подсочке леса. На этом совеща-
нии с докладами выступали академик А. Е. Арбузов, профессора
Е. Ф. Вотчал, В. И. Лебедев, И. А. Яхонтов, И. И. Орлов и др. Меры,
принятые правительством СССР, дали результаты: уже к 30-м годам СССР
перестал ввозить канифоль и скипидар и сам стал экспортировать скипи-
дар. К 1936 г. Советский Союз занял третье место в мире по добыче
живицы, а к 1953 г., увеличив добычу живицы более чем вдвое по срав-
нению с предвоенным периодом, даже вышел на второе место в мире и
первое в Европе по объему заготовки живицы.
Добывают живицу путем подсочки. В СССР наиболее при-
годна для подсочки сосна обыкновенная (Pinus silvestris L.).
В подсочку вовлечено около 2,5 млн. га сосновых насаждений.
Из сосны добывают около 99 % всей живицы, в том числе
свыше 80 % на территории РСФСР и остальное — на Украине,
в Белоруссии, Прибалтийских республиках и Казахской ССР.
Длительное время больше половины живицы добывалось на
территории европейской части страны. В настоящее время жи-
вицу добывают в районах Севера, Сибири и Дальнего Вос-
тока.
§ 3. СЫРЬЕ ДЛЯ КАНИФОЛЬНО-ТЕРПЕНТИННОГО
ПРОИЗВОДСТВА
Добыча живицы. Процесс добычи живицы складывается из
основных производственных операций: разметки карр, нанесе-
ния на них специальных ранений — подновок, сбора и затари-
вания живицы.
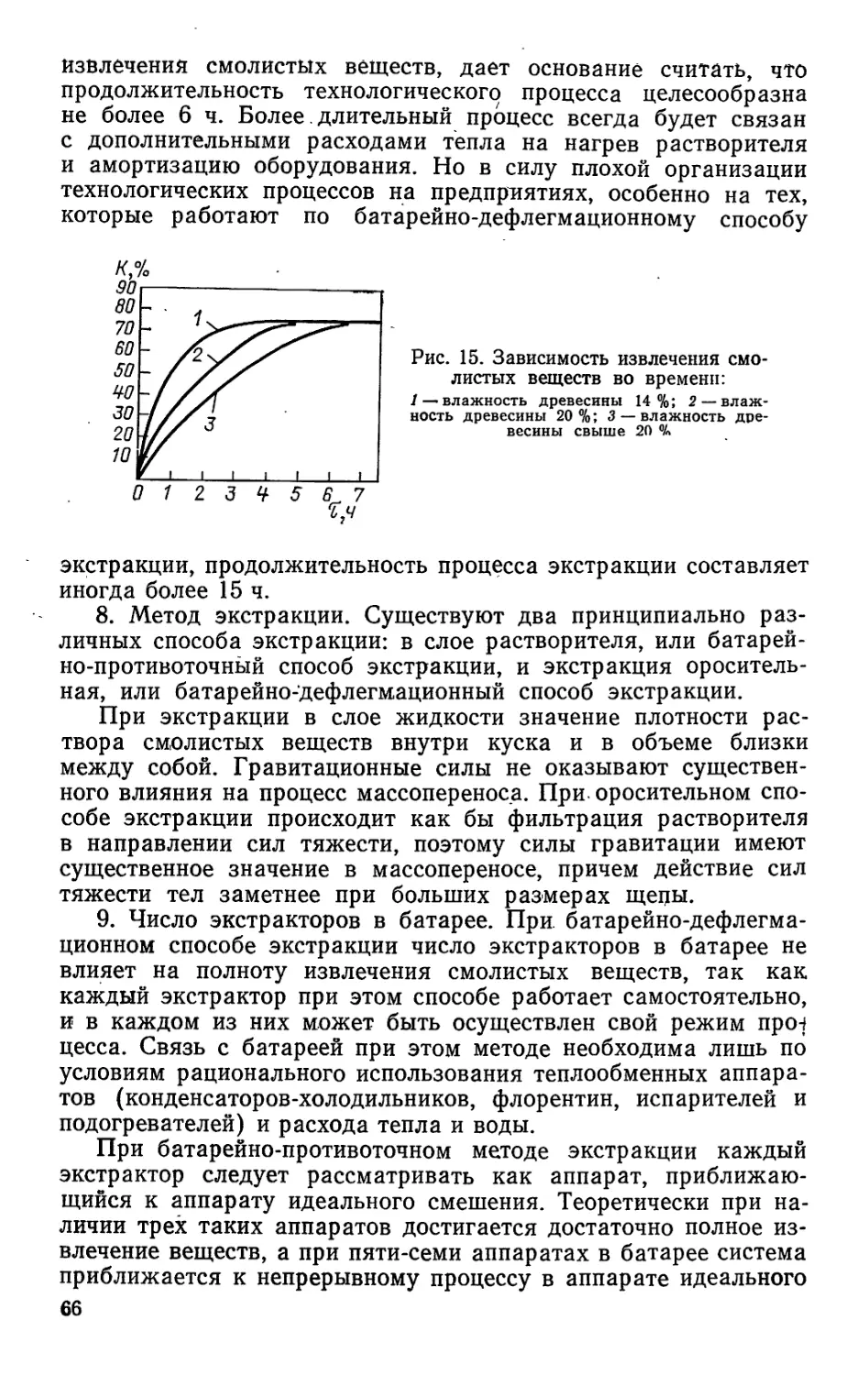
При подготовке деревьев к подсочке на той части ствола,
где будет наноситься подновка, осенью или зимой срезают
грубую кору, создавая подрумяненную поверхность. На ней
ранней весной подвешивают приемники и проводят направля-
ющие желобки для стока живицы в приемники. Участок по-
верхности ствола дерева, подготовленный для нанесения под-
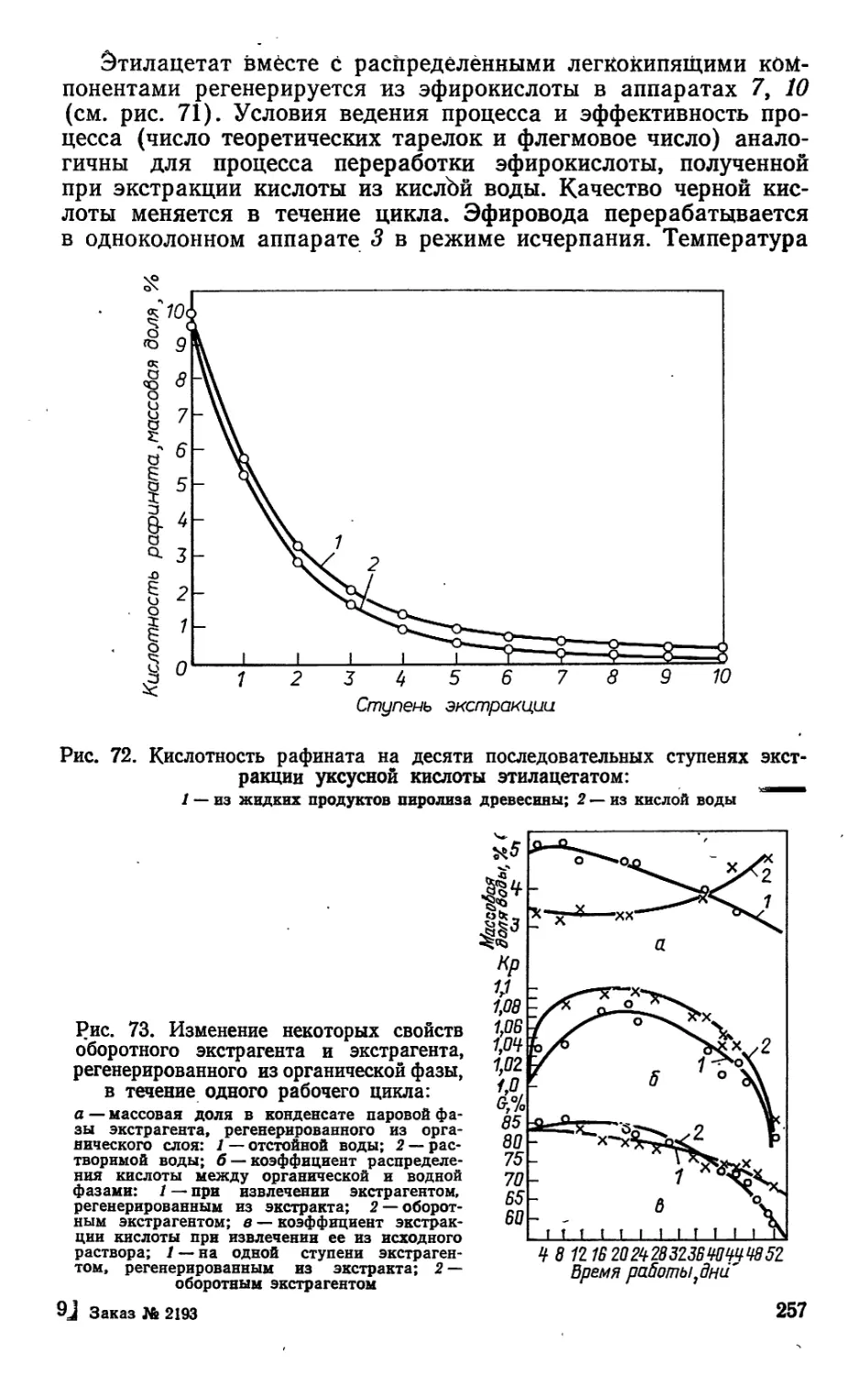
новок, называется каррой (рис. 1).
Нанесение подновок начинается весной при повышении
среднесуточной температуры до 7—9 °C й заканчивается при
наступлении устойчивых заморозков. Подновки наносят с по-
мощью специальных инструментов, которые называются ха-
20
ками, в два приема — в обе стороны от середины карры или
от желобка под углом 45° к вертикали (90° между подков-
ками). Такую двойную подновку называют карроподновкой,
а поверхность ствола, на которой нанесены подновки — зерка-
лом карры. Процесс нанесения подновок называется вздым-
кой, а рабочий, наносящий подновку, вздымщиком.
Период времени между двумя очередными подковками на-
Рис. 1. Общий вид карры при
подсочке сосны:
1 — приемник живицы; 2 — длина под-
новки; 3 — ребристая карра текущего
года подсочки; 4, 7 — шаги подновки;
5 — карроподновки; 6 — межкарровая
перемычка; 8 — рифленая карра; 9 —
длина зеркала карры; 10 — ширина
подновки; 11 — направляющий жело-
бок; 12 — подрумяненная поверхность
ствола; 13 — межкарровый ремень;
14 — глубина подновки; 15 — ширина
карры
зывают паузами вздымки и измеряют в днях. Различают два
способа нанесения подновок: восходящий, когда очередная
подновка наносится выше предыдущей, и нисходящий, при ко-
тором подновки идут сверху вниз. Расположение карр также
бывает восходящим, когда карра очередного года подсочки
закладывается выше карры предыдущего года, и нисходящим,
когда процесс ведется наоборот. Между каррами очередных
лет подсочки оставляют промежутки, на которых подновки не
наносятся, их называют межкарровыми перемычками.
Выход живицы зависит от смолопродуктивности деревьев и
размеров подновок. Длина подновок измеряется по протя-
женности линии среза, глубина — по толщине (в радиальном
направлении) срезаемого слоя без коры, а ширина — перпен-
дикулярно линии среза. Расстояние по вертикали между
верхними или нижними гранями двух смежных подновок назы-
вается шагом подновок. Ширина карры измеряется по окруж-
ности ствола между концами карроподновок, а длина — по
вертикали. В большинстве случаев на дереве закладывают две
карры. Между ними оставляют полосы нетронутой коры,
21
которые называются межкарровыми ремнями и служат для под-
вода питательных веществ из кроны в нижнюю часть ствола.
Отношение суммарной ширины всех карр, заложенных на
дереве, выраженное в процентах, к длине его окружности назы-
вается нагрузкой дерева каррами и является одним из основ-
ных показателей интенсивности подсочки.
В СССР применяют несколько систем подсочки: длитель-
ную, долгосрочную, краткосрочную и осмолоподсочку.
Длительная система подсочки наилучшим обра-
зом сохраняет жизнедеятельность деревьев и обеспечивает до-
статочный выход живицы, но применима лишь для тех дре-
весных пород, у которых поверхность ранения зарастает, что
создает возможность повторного использования для подсочки
одних и тех же участков ртвола. По этой системе подсочка
может вестись в течение 30—40 лет на таких высокопродуктив-
ных и быстрорастущих видах сосен, как крымская — Pinus
pallasiana и пицундская — Р. pityusa, а также лиственницы.
Долгосрочная система подсочки также сохраняет
жизнедеятельность деревьев и обеспечивает достаточный вы-
ход живицы в течение 10—15 лет. Эта система применяется
в насаждениях сосны обыкновенной Pinus silvestris, в тех райо-
нах, где вегетационный период довольно длительный (Укра-
ина, Белоруссия, Прибалтика и др.). Подсочка ведется по тех-
нологии, не вызывающей резкой деформации ствола.
Краткосрочная система подсочки обеспечивает
максимальное увеличение ежегодного валового сбора живицы
с дерева и с единицы площади насаждений. Эту систему в рай-
онах с коротким вегетационным периодом применяют в насаж-
дениях, предназначенных в ближайшее время для рубки. Ведут
такую подсочку до 5 лет самыми различными способами, ме-
тодами и приемами.
Осмолоподсочку как систему применяют для использования
сосновых насаждений V и Va бонитетов. Она обеспечивает
получение живицы и барраса, а также стволового осмола, яв-
ляющихся сырьем для канифольно-экстракционных и сульфат-
но-целлюлозных предприятий. Осмолоподсочка допустима
в условиях достаточной увлажненности в зоне средней и се-
верной тайги и т. п.
Смолообразование и смоловыделение в деревьях хвойных
пород. Теоретические основы смолообразования и смоловыде-
ления, которые и до настоящего времени лежат в основе ор-
ганизации технологии подсочки, были разработаны Л. И. Ива-
новым.
Живица в дереве содержится в системе смоляных ходов,
которые делятся на продольные и поперечные. Продольные
смоляные ходы образуются в поздней части годичного слоя;
они вытянуты вдоль дерева, имеют длину от 0,1 до 0,8 м и
средний диаметр 0,1 мм. Поперечные смоляные ходы постро-
ены так же, как и продольные. Диаметр поперечных ходов
22
Рис. 2. Смоляные ходы в древесине
сосны:
А — продольный; Б — поперечный
выше места ранения, что имеет
в среднем равен 0,04 мм, длина поперечных ходов зависит от
диаметра дерева; поперечные ходы встречаются только в серд-
цевинных лучах и удлиняются вместе с нарастанием слоев
древесины и луба.
Продольные ходы очень редко имеют непосредственную
связь между собой. Основное соединение продольных смоляных
ходов, расположенных в различных годичных слоях, происхо-
дит с помощью поперечных
ходов, которых имеется не-
сколько сотен на 1 см? по-
перечного сечения дерева.
При нанесении подновок
смоляные ходы перерезаются,
и из них вытекает живица.
При этом под влиянием под-
сочных ранений в наружных
слоях' заболони образуются
дополнительные патологиче-
ские продольные смоляные
ходы.
Наличие соединений про-
дольных и поперечных смоля-
ных ходов древесины позво-
ляет при вскрытии поверхност-
ных ходов получать живицу
из довольно отдаленных по
радиусу слоев заболони, рас-
положенных как ниже, так и
большое практическое значение.
Поперечный и продольный смоляные ходы имеют одинако-
вое анатомическое строение. Поперечный разрез продольного
смоляного хода показан на рис. 2. Смоляной ход представляет
собой канал /, образованный живыми выделительными клет-
ками 2, оболочки которых срастаются в сплошное кольцо, об-
разуя внешнюю стенку канала. Вокруг выделительных клеток
расположены живые клетки сопровождающей паренхимы 3,
которые играют роль хранилища запасных питательных ве-
ществ. Между выстилающими клетками и клетками сопровож-
дающей паренхимы расположен слой мертвых, сильно сплю-
щенных клеток 4. В местах разрыва мертвого слоя выдели-
тельные клетки соприкасаются с клетками сопровождающей
паренхимы, что и создает возможность притока питательных
веществ к выделительным клеткам.
В европейской части СССР масса живицы в дереве диамет-
ром 0,32 м вместе с корнями, стволом и ветвями составляет
примерно 1,7 кг. Между тем дерево указанных размеров за
один сезон в отдельных случаях дает 2—2,5 кг живицы, а при
подсочке в продолжение нескольких лет может дать во много
раз больше, чем содержится в системе смоляных ходов.
23
Это обстоятельство доказывает, что в процессе подсочки
происходит новое образование живицы в довольно больших ко-
личествах. В итоге не только целиком возмещается убыль жи-
вицы при подсочке, но при нормальной нагрузке древесина де-
рева, подсачиваемого ряд лет, как правило, заметно обогаща-
ется смолистыми веществами.
В последнее время установлено, что живица, находящаяся
в системе смоляных ходов, образуется в выстилающих клетках
самих смоляных каналов. Существовавшее мнение, что живица
образуется в хвое кроны, оказалось несостоятельным, так как
было доказано, что смоляные ходы хвои не сообщаются со
смоляными ходами древесины.
Клетки сопровождающей паренхимы также не могут
явиться местом образования смолистых веществ. Эти клетки
дальше отстоят от смоляного канала и отделены от него слоем
мертвых клеток, что не позволяет перемещаться живице в смо-
ляной канал.
Вопрос о том, как образуются смолистые вещества и эфир-
ные масла, до сих пор до конца не разрешен, хотя этому было
посвящено много исследований как отечественных, так и ино-
странных ученых. Научные работы, проведенные с примене-
нием метода меченых атомов, показали, что живица, очевидно,
участвует в обмене веществ в дереве. Известно, что исходным
материалом для синтеза живицы являются углеводы, но о ме-
ханизме образования терпенов и смоляных кислот имеются
различные, нередко противоречивые, гипотезы. Принято счи-
тать, что живица образуется из углеводов в выделительных
клетках смоляных ходов, однако многие исследователи предпо-
лагают, что биосинтез живицы является многостадийным и про-
текает в различных частях растущего дерева.
Недостаточно разработанной остается пока и теория смоло-
выделения. По представлениям многих ученых, живица в жи-
вом дереве заполняет смоляные ходы, создавая избыточное
давление до 120—200 кПа, поэтому при перерезании смоляных
ходов она сразу начинает вытекать из них. Постепенно смоло-
выделение прекращается, так как опорожнение канала смоля-
ного хода идет быстрее, чем новообразование живицы в высти-
лающих клетках и приток ее в канал. Поэтому выстилающие
клетки под действием осмотического давления начинают заса-
сывать воду из окружающей ткани, увеличиваясь в объеме,
выпячиваясь внутрь канала и закрывая его. Чаще всего про-
цесс истечения живицы прекращается из-за закупорки вскры-
той части смоляного хода закристаллизовавшейся или загус-
тевшей живицей.
Практика подсочки показывает, что эффективность этих
производств в значительной степени зависит от смолопродук-
тивности насаждений. Различают биологическую и технологи-
ческую смолопродуктивность.
Биологическая смолопродуктивность зависит от целого ряда
24
факторов. Наиболее важные из них: диаметр ствола дерева,
возраст, климатические условия, характер почвы, свет и дру-
гие факторы.
Диаметр ствола дерева — наиболее важный фактор, влияю-
щий на смолопродуктивность как отдельных деревьев, так и
насаждений в целом. Чем лучше были условия произрастания
(класс бонитета), тем больше диаметр и высота дерева и тем
лучше в таком дереве протекает процесс смолообразования и
выделения живицы при ранениях. С увеличением диаметра
ствола возрастает число смоляных ходов, а следовательно, и
суммарная емкость дерева по запасу живицы, что обеспечи-
вает более обильное выделение живицы при подсочке. Биоло-
гическая смолопродуктивность зависит и от индивидуальных
особенностей отдельных деревьев. Два внешне совершенно оди-
наковых дерева, произрастающих в одних и тех же условиях,
могут иметь разную продуктивность, причем низкая смолопро-
дуктивность сохраняется из года в год. Поэтому если дерево
в первый год дало малый выход живицы, то это повторится и
в последующие годы; в связи с этим они из подсочки исклю-
чаются вообще.
Возраст дерева оказывает большое влияние на смолопро-
дуктивность деревьев. С возрастом дерева увеличиваются его
диаметр и высота, следовательно, возрастают условия смоло-
образования и смоловыделения. Однако в разных районах
СССР минимальный возраст, при котором можно начинать
подсочку произрастающей у нас сосны, далеко не одинаков.
В южных и юго-западных районах страны с теплым и влажным
климатом рост сосны более интенсивен и она сравнительно
раньше становится пригодной для подсочки. В северных и вос-
точных районах сосна становится пригодной к подсочке го-
раздо позднее.
Влажность также является важным фактором, влияющим
на биологическую смолопродуктивность деревьев. Практикам
хорошо известно, что после дождей в теплую погоду смолопро-
дуктивность резко возрастает. С другой стороны, повышенная
влажность в почве (заболоченные места) вызывает снижение
выходов живицы. На очень сухих почвах также наблюдается
снижение выходов, но в меньшей степени, чем при излишнем
увлажнении почвы.
Температура воздуха существенно влияет на выход живицы.
Повышение температуры способствует биологической активно-
сти дерева, обмену веществ в клетках, что способствует более
эффективному смолообразованию.
Выход живицы меняется в течение сезона. В начале сезона
выход относительно низкий; постепенно он увеличивается и
достигает максимума в июле—августе, а потом падает. Объяс-
няется это влиянием средней температуры. В начале и конце
сезона подсочки температура воздуха низкая, соответственно
и жизнедеятельность деревьев невелика, вегетационные про-
25
цессы слабые и выход живицы низкий. Самый эффективный
период для наших лесов — июль и август.
В зависимости от климатических условий выход живицы
с одного дерева колеблется от 0,9 до 1 кг на севере и в лесах
Сибири, от 2 до 2,5 кг на юге и в среднем по СССР составляет
до 1,2 кг с дерева сосны.
Выход живицы при промышленной подсочке — технологиче-
ская смолопродуктивность — бесспорно зависит от биологиче-
ской смолопродуктивности, но также и от технологии и режима
подсочки.
Чем больше ширина карры и нагрузка деревьев каррами,
тем больше вскрывается продольных смоляных ходов и тем
выше, хотя и не пропорционально, выход живицы. Сильное
влияние на выход живицы оказывает величина паузы вздымки.
При коротких паузах (1—3 дня) выход живицы с каждой под-
новки снижается по сравнению с длительными паузами (7—10
дней), соответственно и ниже производительность труда вздым-
щиков, однако общий выход живицы с карры и дерева за се-
зон при коротких паузах выше. При обычной подсочке опти-
мальной паузой считается 3—5 дней, а с применением химиче-
ских и биологических стимуляторов 7—14 дней и более.
Способы подсочки. В настоящее время в СССР применя-
ются три способа подсочки.
Обычная подсочка, при которой наносится только
чисто механическое ранение — карроподновки. При обычной
подсочке выделение живицы в теплый период сезона прекра-
щается в течение суток и истечение в холодный сезон подсочки
длится до 2 суток, после чего смоляные ходы закупориваются
разбухшими выделительными клетками.
Подсочка с химическим воздействием, при ко-
торой используются химические реагенты (главным образом
серная кислота), свежие подновки смазывают серной кислотой
с помощью специальных хаков. Серная кислота медленно про-
никает в промежуток между древесиной и лубом, впитывается
в древесину и разрушает живые клетки, в том числе клетки
смоляных ходов. При этом закупорки смоляных ходов уже не
происходит и длительность смоловыделения увеличивается в не-
сколько раз: тем больше, чем больше доза кислоты. Таким
образом, такие химические реагенты, как серная кислота и
хлорная известь, являются стимуляторами смоловыделения, но
не способствуют дополнительному смолообразованию. Примене-
ние серной кислоты позволяет увеличить паузу вздымки до
3—4 нед. При этом выход живицы с каждой подновки увели-
чивается по сравнению с обычной подсочкой в 4—5 раз, а про-
изводительность труда вздымщиков в 2—3 раза, но выход жи-
вицы с карры за сезон несколько снижается.
При паузе вздымки 7—10 дней производительность труда
вздымщиков возрастает на 150—200 %, но при этом выход жи-
вицы с карры увеличивается на 20—40 % против обычной под-
26
сочки., В настоящее время в СССР обычной подсочкой и под-
сочкой с применением серной кислоты добывается до 15 % жи-
вицы от общего объема заготовок.
Подсочка сосны с применением биостимуля-
торов, из которых в настоящее время наиболее распростра-
нены вытяжка из кормовых дрожжей — сульфитно-спиртовая
барда (ССБ) и сульфитно-дрожжевая бражка (СДБ). В по-
следние годы с помощью этих стимуляторов добыто свыше
70 %' всей живицы в нашей стране.
Считается, что эти стимуляторы усиливают синтез органи-
ческих веществ в дереве и интенсифицируют процесс смолооб-
разования, а также снижают вязкость живицы. Таким образом,
они одновременно стимулируют и смолообразование и смоло-
выделение. В результате этого в среднем за 8—10 лет выход
живицы выше, чем при обычной подсочке на 36 % на Урале, на
52 % в Горьковской и на 59 % в Иркутской областях. Произ-
водительность труда вздымщиков также увеличивается на
30—40%.
При подсочке с применением ССБ и СДБ необходимо избе-
гать попадания ее в живицу. Уже при массовой доле стимуля-
торов в живице свыше 0,05 %; создаются серьезные трудности
при ее переработке и ухудшается качество канифоли.
В ряде районов страны ведутся опытные подсочные работы
по применению различных добавок к вытяжке из кормовых
дрожжей (гидреля, парафена, мочевины и др.), которые дали
в отдельных случаях положительные результаты. Наиболее
перспективным считается применение вытяжки из кормовых
дрожжей.
В настоящее время считают, что целесообразность примене-
ния различных стимуляторов при добыче живицы зависит от кли-
матических, таксономических особенностей сосны обыкновенной.
Собранную живицу доставляют на приемные пункты и зата-
ривают в бочки. В основном применяют оборотные железные
бочки вместимостью 180—190 кг с одним съемным дном.
Подсочка других хвойных. Кроме сосны, в Советском Союзе
в небольших объемах ведут подсочку кедра, ели, пихты, лист-
венницы.
На территории СССР произрастают два вида кедра: сибир-
ский и маньчжурский. Первый распространен на территории Во-
логодской области, на Урале и в Сибири до Забайкалья; вто-
рой — на Дальнем Востоке.
Основную массу кедровой живицы добывают на Алтае за
счет подсочки кедра сибирского. Смолоносный аппарат под-
сочки кедра похож на сосновый, поэтому техника подсочки
кедра сибирского не отличается от техники подсочки сосны
обыкновенной. Состав живицы кедра близок к составу живицы
сосны.
Однако живица кедра труднее кристаллизуется, чем живица
сосны, поэтому ее выделение из подновок продолжается значи-
27
тельно дольше, от 17 до 77 сут, пауза вздымки 8—10 дней. Дли-
тельность подсочки может быть до 10 лет. Выход кедровой жи-
вицы с карры за сезон значительно меньше, чем сосновой, и
составляет около 0,3 кг. Добыча кедровой живицы составляет
несколько сот тонн в год. Используется она для производства
иммерсионного масла и идет как добавка к сосновой живице.
Ведутся работы по получению из кедровой живицы более цен-
ных продуктов.
Самой распространенной породой у нас является листвен-
ница. В древесине лиственницы живица находится не только
в смоляных ходах, но и в смоляных карманах. Особенность
свойств строения древесины лиственницы требует и другой тех-
нологии подсочки, что приводит к повышению себестоимости ли-
ственничной живицы по сравнению с сосновой более чем
в 1,5 раза. При переработке лиственничной живицы встреча-
ются затруднения. В настоящее время в ЦНИЛХИ проведены
работы, обеспечивающие другие направления ее использования,
в частности выделение нейтральной части и использование ее
в парфюмерной промышленности, а остатка для приготовления
бумажного клея.
В отличие от других хвойных пород в древесине пихты нет
смоляных ходов, а находятся они только в коре, и живица
пихты накапливается в виде вздутий между корой и древеси-
ной. Для извлечения живицы из этих вздутий, их прокалывают
заостренным концом трубки, соединенной с приемником, и на-
давливанием на вздутие пережимают живицу в сборник.
Добыча пихтовой живицы возможна только при температуре
не ниже 16 вС, так как при более низких температурах она
практически теряет текучесть. Сбор пихтовой живицы с одного
дерева можно производить 1 раз в 2—3 года. С одного дерева
можно добыть 0,1—0,2 кг живицы за сезон.
Хорошо очищенную нелетучую часть пихтовой живицы (пих-
товый бальзам) применяют для склеивания оптических стекол,
в линзоскопической технике, медицине и др.
Подсочку ели пока ведут только в опытном порядке. Еловая
живица имеет очень высокую себестоимость, а раненые деревья
легко поддаются воздействию гнилостных грибков, что снижает
качество древесины.
С еловых деревьев собирают еловую серку, которая истекает
из случайных ранений деревьев. Еловая серка идет на выра-
ботку абиетиновой смолы и смолы нейтральной воздухововле-
кающей (СНВ), которая используется в качестве добавок к бе-
тонам.
Кроме того, в нашей стране собирают так называемый бар-
рас (живица III сорта), который остается на каррах после
окончания сезона подсочки или при заготовках стволового ос-
мола. В составе барраса массовая доля сора достигает 10—
15%, влаги — до 25, а скипидара падает до 7—10 %. Как пра-
вило, баррас на заводах перерабатывают совместно с живицей,
28
добавляя в нее 5—10 % барраса, причем качество канифоли не
ухудшается.
Состав живицы. Основным сырьем канифольно-терпентинных
предприятий является живица сосновая. В химическом отноше-
нии живица представляет собой раствор твердых изомерных
смоляных кислот общей формулой С20Нз0О2 и неомыляемых ве-
ществ в смеси терпеновых углеводородов общей формулой
С1оН1б. Доля в живице терпеновых углеводородов, находящихся
в смоляных каналах в момент вытекания, доходит до 36%, и
смоляные кислоты полностью в них растворены. Только что вы-
текшая из смоляных ходов живица представляет собой легко-
подвижную светлую, прозрачную жидкость. На воздухе, осо-
бенно в жаркую влажную погоду, легколетучие компоненты жи-
вицы быстро улетучиваются из образовавшегося пересыщенного
раствора, смоляные кислоты выкристаллизовываются, живица
становится густой. При длительном пребывании на воздухе жи-
вица затвердевает и превращается в баррас.
В последние годы в связи с использованием при подсочке хи-
мических реактивов и биостимуляторов паузы вздымок сбора
живицы возросли, что привело к обеднению живичного скипи-
дара легколетучими компонентами, такими, как а- и р-пинены.
Доля их в живичном скипидаре снизилась с 68—70 до 55—60 %.
В процессе добычи живицы в нее попадают сор и вода. Ка-
чество живицы и ее приемка заводами регламентируются. Как
показала практика, в живице, поступающей на канифольно-тер-
пентинные заводы, содержится 71—79 % смоляных кислот, 14—
20 терпеновых углеводородов, 3—6 воды и 0,7—1,0 % сора.
§ 4. ТЕХНОЛОГИЯ ПЕРЕРАБОТКИ ЖИВИЦЫ
Производство канифоли и скипидара из живицы организовано в нашей
стране в послереволюционный период. Основным видом живицы, поступаю-
щей на канифольно-терпентинные заводы, является живица сосны. В 20-е
годы в Советском Союзе было построено 15 предприятий по переработке
живицы мощностью 2000—6000 т живицы в год. Установки часто носили
кустарный характер, при котором использовались огневой или пароогневой
способы отделения скипидара от.канифоли. При переработке живицы этими
способами получались низкие выходы и качество канифоли и скипидара,
поэтому они не нашли широкого применения.
На более крупных предприятиях применяли паровой способ переработки
живицы. При этом были использованы технологические схемы иностранных
фирм (французской фирмы «Дюпон»). Накопленный опыт эксплуатации
первых заводов по переработке живицы позволил вскоре советским инжене-
рам и конструкторам разработать наиболее надежные и совершенные тех-
нологические схемы переработки живицы, оснащенные отечественным обору-
дованием. Большая заслуга в создании современной технологии переработки
живицы принадлежит инженерам К. П. Михееву, М. К. Жлобо, П. П. Поля-
кову, В. И. Филатову, Н. П. Позднякову и другим работникам отрасли.
Технологический процесс переработки живицы в канифоль
и скипидар состоит из следующих операций: складирования,
первичной обработки сырья, плавления, осветления, отстаивания
живицы от сора и воды, промывки терпентина водой, отгонки
скипидара и уваривания и розлива канифоли.
,29
Складирование и первичная обработка живицы. Основную
массу живицы в нашей стране добывают в течение июля — ав-
густа, в меньших объемах — в мае, июне, сентябре. Таким об-
разом, живица является сезонным сырьем. Для обеспечения
равномерной работы предприятия необходимо создавать запасы
живицы. Поэтому большую часть живицы летнего поступления
оставляют на хранение на специально оборудованных заводских
площадках. До 1963 г. живицу с места добычи на заводы тран-
спортировали в деревянных бочках вместимостью 200—250 л.
В настоящее время основная часть живицы на предприятия по-
ступает в металлических бочках той же вместимостью с одной
отъемной крышкой.
На предприятиях мощностью 6000—12 000 т/год живица хра-
нится в транспортных бочках в закрытых складских помещениях
или на открытых эстакадах под навесом. При хранении жи-
вицы в деревянных бочках или в другой недостаточно герме-
тичной таре происходит утечка наиболее богатой скипидаром
части живицы, поэтому в бетонном полу склада или эстакады
делаются углубления и специальные сточные канавки для
сбора вытекшей живицы.
На заводах большей мощности живица хранится на откры-
тых площадках с цементированным настилом. Бочки склади-
руют с помощью башенных или козловых кранов.
В последние годы на канифольно-терпентинных заводах для
хранения живицы используют металлические емкости вмести-
мостью 125—2000 м3. Такой способ хранения живицы получил
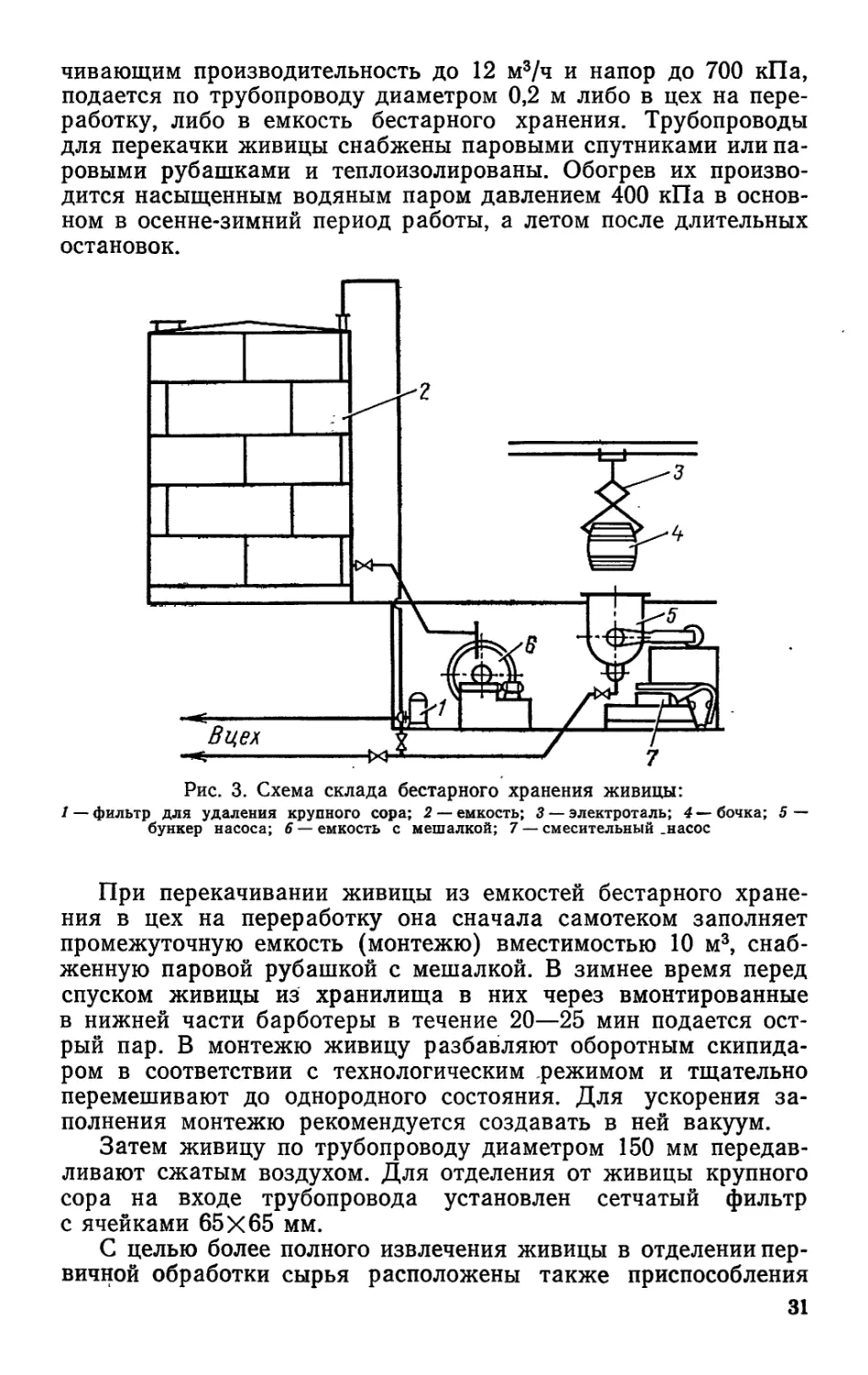
название бестарного (рис. 3). Он почти полностью исключает
потери живицы и при благоприятных условиях заготовки жи-
вицы создает предпосылки для ритмичной работы предприятия
в течение всего года.
Для лучшей организации работ при переработке живицы на
заводе организовано специальное отделение первичной обра-
ботки сырья. Пол этого отделения со стороны подъездных путей
устанавливается на уровне пола железнодорожного вагона.
Деревянные и металлические бочки с живицей выгружаются
из железнодорожных вагонов или автомашин на открытую
рампу отделения с помощью электротали с кран-балкой. Каж-
дую бочку взвешивают на специальных весах, а из каждой
десятой отбирают пробу для анализа состава живицы. После
взвешивания живица из бочек выгружается в загрузочный бун-
кер. При подаче барраса в загрузочный бункер его предвари-
тельно измельчают вручную на куски размером не более
120 мм.
Живица в загрузочном бункере с помощью ленточной ме-
шалки размешивается до однородного состояния, а в осенне-зим-
ний период разбавляется оборотным скипидаром, подаваемым
из цеха переработки живицы. Скипидара в живице содержится
25—30 %, дозируют его с помощью специального устройства.
Подготовленная живица бетононасосом марки С-252, обеспе-
30
чивающим производительность до 12 м3/ч и напор до 700 кПа,
подается по трубопроводу диаметром 0,2 м либо в цех на пере-
работку, либо в емкость бестарного хранения. Трубопроводы
для перекачки живицы снабжены паровыми спутниками или па-
ровыми рубашками и теплоизолированы. Обогрев их произво-
дится насыщенным водяным паром давлением 400 кПа в основ-
ном в осенне-зимний период работы, а летом после длительных
остановок.
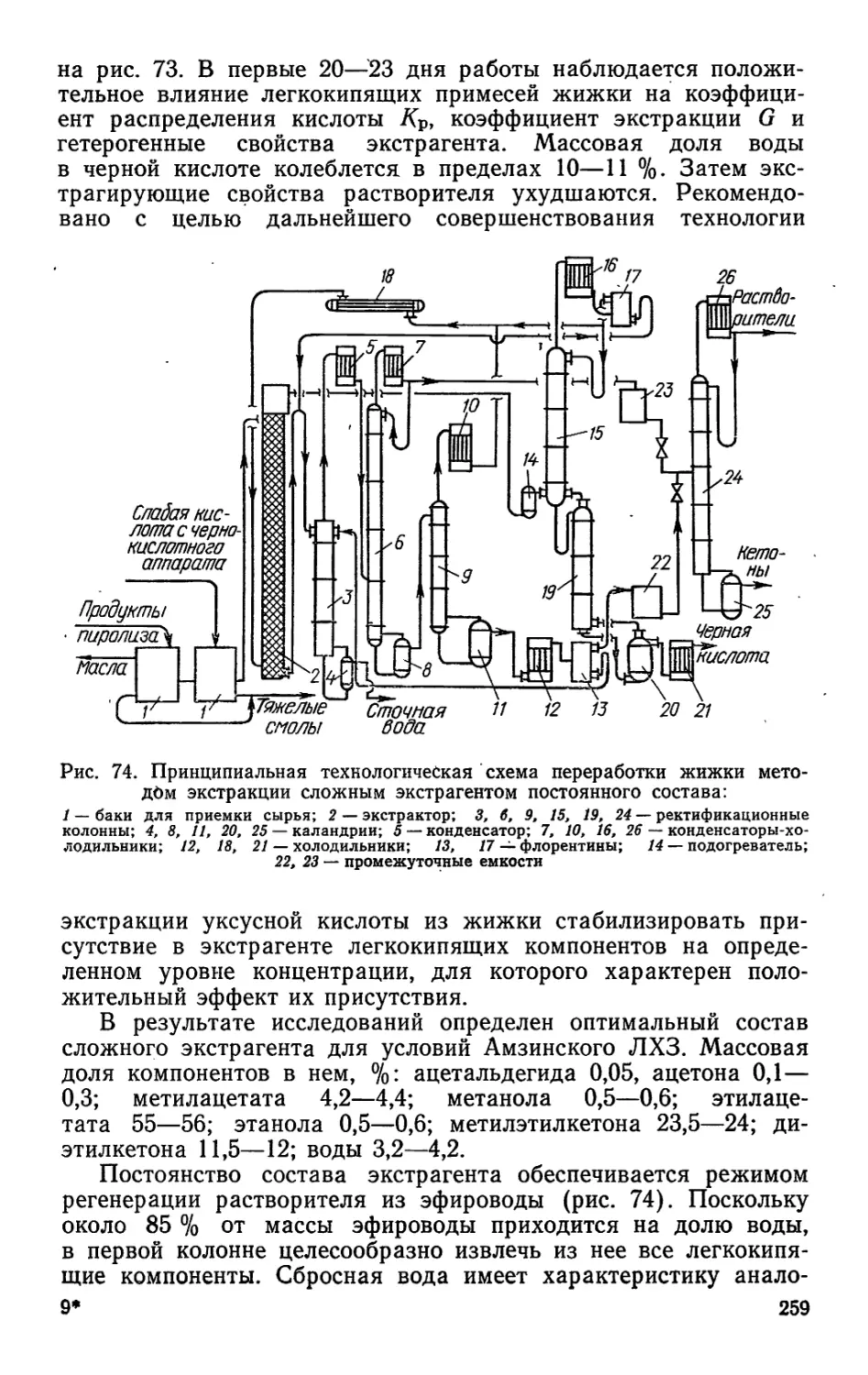
Рис. 3. Схема склада бестарного хранения живицы:
1 — фильтр для удаления крупного сора; 2 — 3 — электроталь; 4 — бочка; 5 —
бункер насоса; 6 — емкость с мешалкой; 7 — смесительный .насос
При перекачивании живицы из емкостей бестарного хране-
ния в цех на переработку она сначала самотеком заполняет
промежуточную емкость (монтежю) вместимостью 10 м3, снаб-
женную паровой рубашкой с мешалкой. В зимнее время перед
спуском живицы из хранилища в них через вмонтированные
в нижней части барботеры в течение 20—25 мин подается ост-
рый пар. В монтежю живицу разбавляют оборотным скипида-
ром в соответствии с технологическим режимом и тщательно
перемешивают до однородного состояния. Для ускорения за-
полнения монтежю рекомендуется создавать в ней вакуум.
Затем живицу по трубопроводу диаметром 150 мм передав-
ливают сжатым воздухом. Для отделения от живицы крупного
сора на входе трубопровода установлен сетчатый фильтр
с ячейками 65x65 мм.
С целью более полного извлечения живицы в отделении пер-
вичной обработки сырья расположены также приспособления
31
для очистки бочек, которые, как правило, представляют собой
установки по гидроочистке от механических примесей.
Способ этот заключается в следующем: опорожненную бочку
помещают над открытым бетонированным водоемом вмести-
мостью 6—8 м3. С помощью приводных роликов бочка в гори-
зонтальном или слегка наклонном положении вращается со ско-
ростью 5—б мин-1. Из гибкого шланга в бочку под давлением
400—500 кПа подают струю холодной воды. Налипшая живица
при этом легко отделяется от стенок бочки и в виде пены сте-
кает в водоем, где она отстаивается от воды и с помощью не-
большого винта направляется в цех на переработку. Вода ис-
пользуется многократно, поэтому расход ее незначителен. На
очистку одной бочки затрачивается не более 1,5 мин.
После очистки деревянные бочки просушивают и ремонти-
руют. Затем их либо отправляют в химлесхозы для повторного
использования под живицу, либо используют в качестве тары
для канифоли.
Плавление живицы. Живицу, поступающую в цех, перераба-
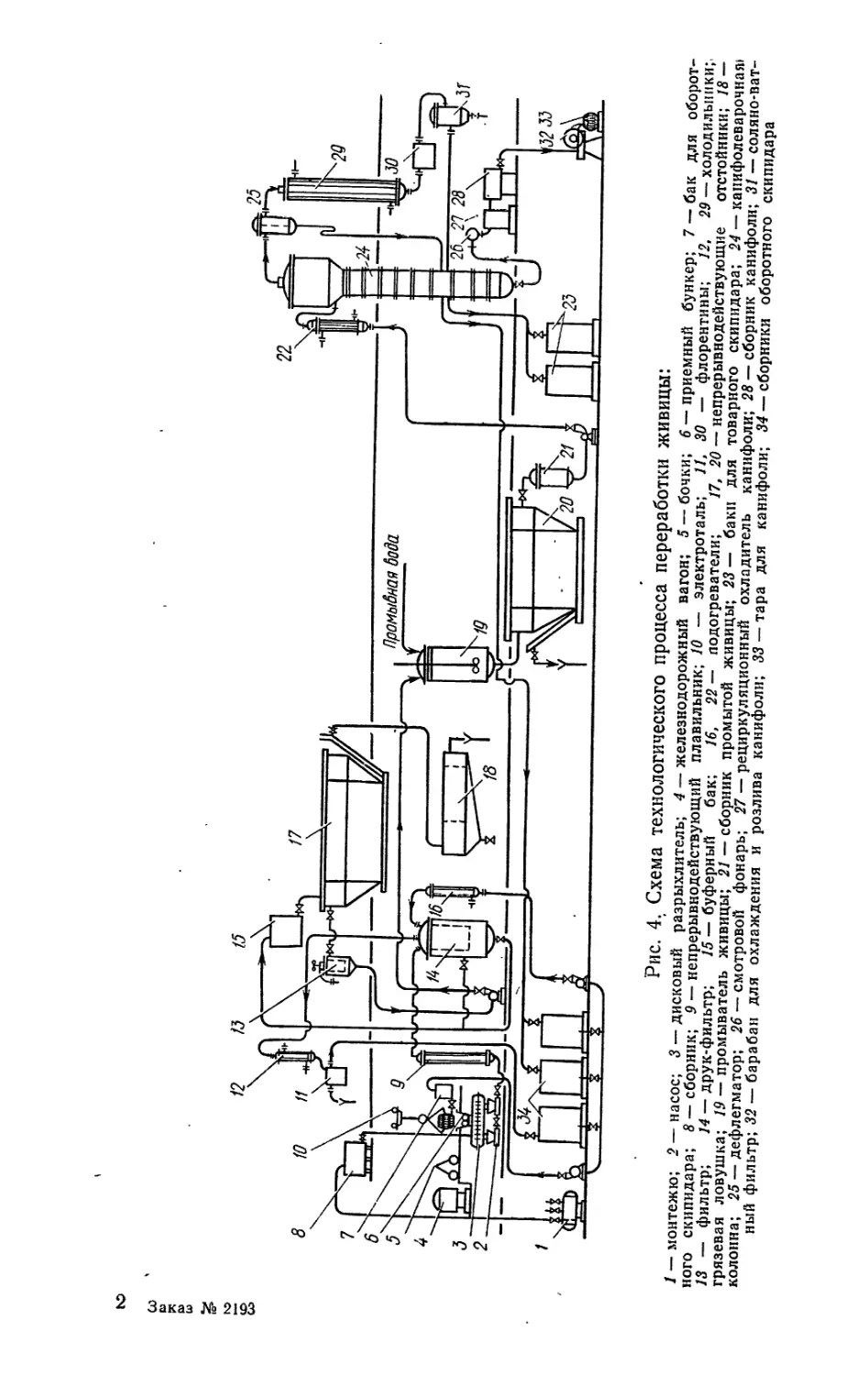
тывают по технологической схеме, изображенной на рис. 4. Эта
схема носит название декантационного способа переработки жи-
вицы. При плавлении живицу нагревают до 105—ПО °C. При
этой температуре живица становится хорошо текучей, легко
фильтруется и транспортируется по трубопроводам с помощью
насосов.
На большинстве заводов СССР живицу плавят в аппаратах-
плавильниках периодического действия вместимостью 5—6 м3.
Процесс плавления, как и все процессы, строго регламентиру-
ется. Из сборника живица подается в плавильник. Контроль за
объемом загружаемой живицы производится с помощью весо-
мера или радиоизотопного уровнемера УР-8.
Одновременно с загрузкой живицы в плавильник из бака
оборотного скипидара подается расчетный объем скипидара, об-
легчающего процесс плавки и дальнейшую переработку живицы.
Плавят живицу острым паром под давлением 400 кПа, подавае-
мым через барботер в нижней части аппарата. Пар нагревает
живицу и перемешивает ее, что ускоряет процесс плавления.
Вследствие конденсации пара содержание воды в живице повы-
шается и способствует эффективному экстрагированию водора-
створимых веществ, содержащихся в соре. Это является недо-
статком плавления острым паром, так как приводит к потемне-
нию живицы. Процесс плавки считается законченным, когда
температура расплавленной живицы достигает 100 °C, конденса-
ция пара прекращается и начинает расти давление. После того
как давление в плавильнике достигает 150 кПа, подача пара
в барботер прекращается, и расплавленная живица под дей-
ствием избыточного давления пережимается через решетку
(ложное дно), установленную в нижней части аппарата, в бу-
ферный бак. Продолжительность плавления с учетом загрузки,
выгрузки и очистки плавильника составляет 30—35 мин. После
32
Заказ № 2193
Рис. 4. Схема технологического процесса переработки живицы:
/ — монтежю; 2 — насос; 3 — дисковый разрыхлитель; 4 — железнодорожный вагон; 5 —бочки; 6 — приемный бункер; 7 — бак для оборот-
ного скипидара; 8 — сборник; 9 — непрерывнодействующий плавильник; 10 — электроталь; 11, 30 — флорентины; 12, 29 — холодильники;.
13 — фильтр; 14 — друк-фильтр; 15— буферный бак; 16, 22 — подогреватели; 17, 20 — непрерывнодействующие отстойники; 18 —
грязевая ловушка; 19 — промыватель живицы; 21 — сборник промытой живицы; 23 — баки для товарного скипидара; 24 — капифолеварочиая»
колонна; 25 — дефлегматор; 26 — смотровой фонарь; 27 — рециркуляционный охладитель канифоли; 28 — сборник канифоли; 31 — соляно-ват-
ный фильтр; 32 — барабан для охлаждения и розлива канифоли; 33 — тара для канифоли; 34 — сборники оборотного скипидара
каждых 4—5 плавок производится экстракция смолистых. Для
этого в плавильник закачивают оборотный скипидар, предвари-
тельно нагретый до 100—120 °C, и через барботер подают ост-
рый пар, пока давление в аппарате снова не достигнет 150 кПа,
после чего экстракт аналогично расплавленной живицы пережи-
мается в буферный бак. После пережима от проэкстрагирован-
ного сора острым паром отгоняют скипидар. Пары скипидара
Рис. 5. Схема удаления водой сора из плавильников:
1 — плавильники; 2 — трубопровод для воды; 3 — трубопровод для пара и сжатого воз-
духа; 4 — сетка; 5 — трубопровод для удаления сора; 6 — приемник пульпы (сора
с водой); 7 — автоприцеп; 8 — емкость
и воды конденсируются в холодильнике-конденсаторе, и конден-
сат поступает во флорентину. Во флорентине скипидар отделя-
ется от воды и поступает в бак оборотного скипидара, а под-
скипидарная вода поступает в бассейн для мойки бочек.
В настоящее время на заводах внедряется гидравлический
способ удаления сора (рис. 5), полностью исключающий физи-
ческий труд и загазованность производственных помещений.
При этом способе плавильник наполовину заполняется нагретой
водой, через барботер подается острый пар, при помощи кото-
рого перемешивается сор. Когда давление в плавильнике до-
стигнет 100 кПа, не прекращая подачи пара, открывают зад-
вижку на трубопроводе для удаления сора, и вода вместе с со-
ром выбрасывается в приемник с фильтрующей сеткой. После
отделения вода повторно используется для гидроочистки, а сор
выгружается в прицеп автомашины.
Иногда живицу плавят непрерывным способом. Плавильник
непрерывного действия представляет собой трубу диаметром
0,3 м и-высотой 6 м, снабженную паровой рубашкой, барботаж-
ным кольцом в нижней части для подачи острого пара и винто-
вой мешалкой.
Живица в плавильник подается непрерывно с помощью бе-
тононасоса по трубопроводу, снабженному паровой рубашкой.
34
В плавильник также непрерывно поступает 3 %-ный водный ра-
створ фосфорной кислоты. В результате подогрева в трубопро-
воде и энергичного разогрева и перемешивания в плавильнике
живица расплавляется и непрерывно поступает в один из двух
фильтров, в котором она отделяется от крупного сора и направ-
ляется в буферный бак. После 8—16 ч, в зависимости от сорно-
сти живицы и производительности плавильника, подача живицы
переключается на другой аппарат. Отключенный друк-фильтр
заполняют оборотным скипидаром и проводят экстракцию сора
при нагревании глухим паром. Друк-фильтр представляет собой
цилиндрический автоклав с вертикальной фильтрующей корзи-
ной, покрытой двумя сетками — частой и редкой. Такой друк-
фильтр позволяет осуществлять многократную экстракцию сора
небольшими порциями скипидара без применения острого пара,
в результате чего массовая доля смолистых в отработанном
соре снижается с 15—25 до 5—7%. После экстракции скипи-
дара и отдувки его от сора он выгружается из друк-фильтра
так же как из плавильника периодического действия. Расход
водяного пара на плавку 1 т живицы составляет примерно 100—
120 кг.
В настоящее время в ЦНИЛХИ заканчиваются работы, обес-
печивающие полностью непрерывный способ переработки жи-
вицы.
Осветление живицы. Так как при добыче живицы часто поль-
зуются железными приемниками, а транспортировку и хранение
осуществляют в металлической таре, то это приводит к образо-
ванию резинатов железа Ре(С19Н2эСОО)2. Благодаря окислению
смоляных кислот кислородом воздуха могут образовываться и
более сложные соединения. Присутствуют в живице танниды и
красители, содержащиеся в соре, и продукты их взаимодействия
с железом и другими минеральными примесями. И, наконец,
в живице имеются продукты окисления жирных и смоляных кис-
лот, которых содержится особенно много в баррасе.
Для повышения качества канифоли, главным образом цвет-
ности, живицу подвергают осветлению. На наших заводах освет-
ление живицы производится химическим способом, путем воз-
действия на нее водным раствором ортофосфорной кислоты
Н3РО4. Операцию осветления живицы совмещают с процессом
ее плавления. Техническую ортофосфорную кислоту разбавляют
водой до 8—12 %-ной концентрации и добавляют к живице при
загрузке ее в плавильник. Масса добавляемой кислоты опреде-
ляется по формуле
Р = КС!п, (19)
где Р — масса абсолютно сухой Н3РО4, кг; К — коэффициент,
значение которого для светлой живицы 0,056, а для темной
0,102; С — масса живицы, загруженной в плавильник емкостью
1000 кг; п — число миллилитров 0,5 н. раствора едкого натра,
2* 35
пошедшего на титрование 3 мл разбавленной ортофосфорной
кислоты.
Как правило, расход кислоты в пересчете на абсолютно су-
хую ортофосфорную кислоту составляет 6,5—10 кг на 1000 кг
живицы. Взаимодействие фосфорной кислоты с резинатами же-
леза проходит по схеме
3Fe (С19Н29СО2)2 + 2Н3РО4 -> Fe3 (РО4)2 + 6С19Н29СООН. (20)
Соли железа фосфорной кислоты бесцветны и хорошо раст-
воримы в воде. Они успешно отделяются от живицы при отде-
лении ее от воды и сора в отстойниках. Химическое осветление
живицы фосфорной кислотой обеспечивает выпуск канифоли по
шкале цветности не ниже марки WG.
Высокий эффект осветления и предупреждение кристаллиза-
ции достигаются путем дополнительной промывки отстоявшейся
живицы водой. Дополнительная промывка обеспечивает практи-
чески полное удаление солей железа фосфорной кислотой, тан-
нидов и красящих веществ, а также остатков фосфорной кис-
лоты. Удаление солей железа, таннидов и красящих веществ
улучшает цветность канифоли, а удаление фосфорной кислоты
предупреждает изомеризацию смоляных кислот на стадии ува-
ривания канифоли.
Отстаивание живицы. В живице любого времени сбора содер-
жатся вода, крупный сор в виде хвои, кусков коры, древесной
стружки, остатков насекомых и мелкий сор в виде песка, земли-
стой пыли и солей железа.
В процессе плавления живицы и ее перегонки через ложное
дно в плавильнике или в друк-фильтрах в буферную емкость
удается отделить крупный сор. Мелкий. сор и вода остаются
в живице. Основным способом отделения живицы от остатков
сора и воды является отстаивание. При обычных условиях от-
делить примеси от живицы невозможно, так как живица обла-
дает высокой вязкостью, и вода в ней эмульгируется.
Обычно отстаивают расплавленную и прогретую до 90 °C
живицу. При таких температурах тяжелый сор (минеральные
примеси, песок, земля) осаждается вниз, легкий сор всплывает,
вода же в зависимости от плотности живицы может находиться
сверху или снизу живицы. Известно, что движущей силой про-
цесса отстаивания является разность плотностей среды и от-
стаивающихся частиц (табл. 4).
При температуре 20 °C разность плотностей живицы и воды
значительна, но отделить их друг от друга невозможно из-за вы-
сокой вязкости среды. При повышении температуры плотность
живицы приближается к плотности воды. Таким образом, для
отделения воды нужно не только нагреть живицу, но и увели-
чить разность плотностей живицы и воды.
Производительность отстойников определяется по уравнению
Vo = Fowo, (21)
36
4. ПЛОТНОСТЬ ВОДЫ В ЗАВИСИМОСТИ ОТ ТЕМПЕРАТУРЫ И ПЛОТНОСТЬ
ЖИВИЦЫ В ЗАВИСИМОСТИ ОТ ТЕМПЕРАТУРЫ И СОДЕРЖАНИЯ В НЕЙ
СКИПИДАРА
Температура» °C Плотность воды» кг/м3 Плотность живицы» кг/м3, при содержании скипидара, %
15 го 40 50
20 998 1028 1000 978 957
40 992 1016 986 964 943
60 983 1004 974 948 927
80 972 990 956 934 911
90 966 984 948 926 903
100 959 977 940 919 895
5. ВЯЗКОСТЬ ЖИВИЦЫ В ЗАВИСИМОСТИ ОТ СОДЕРЖАНИЯ СКИПИДАРА
Температура, °C Вязкость живицы, Па«С’1О3 при содержании скипидара, %
20,4 25 30 35 40
20 390
30 — — — 500 180
40 — — 660 230 89
50 — 900 280 100 • 43
60 920 300 115 51 28
70 300 ' 130 58 30 17
80 145 65 34 19 14,54
90 — — 26 — 8,12
где Vo — объем отстоявшейся живицы, м3/с; Fo — площадь от-
стаивания, м3; wo — скорость отстаивания частиц, в данном слу-
чае капелек воды, м/с.
Таким образом, производительность отстойника определяется
площадью отстаивания, а не объемом отстойника, и скоростью
отстаивания, которая в данном случае определяется по уравне-
нию Стокса:
Wo= t (22)
18р.
где d— диаметр капелек воды, м; pi, рг — плотность воды и
живицы, кг/м3; g — ускорение свободного падения, м/с2; ц—ди-
намическая вязкость живицы, Па’С (табл. 5).
Все параметры берутся при температуре отстаивания, кото-
рая составляет 90 °C. Диаметр капелек воды в живице опытным
путем -не установлен. Предполагается, что он колеблется в пре-
делах от 0,001 до 1,0 мм.
Ранее на большинстве заводов СССР для более быстрого
отстаивания живицы в процессе плавления к ней добавляли ски-
пидар и поваренную соль. Так, например, при массовой доле
37
в живице 30 % скипидара плотность ее при температуре 90 °C
составляет 948 кг/м3, а плотность воды при добавлении к ней
10 % поваренной соли 1074 кг/м3. При такой разности плотно-
стей разделение воды и живицы идет достаточно быстро и
полно. Однако применение поваренной соли нежелательно по
двум причинам.
1. Водные растворы поваренной соли являются сильными
Рис. 6. Непрерывнодействующий отстойник для живицы:
1, 2 — штуцера; 3 — сплошная перегородка; 4 — ситчатая перегородка; 5 — труба;
6 — выступы; 7 — затвор
электролитами, которые способны вызывать сильную коррозию
основного оборудования и коммуникационных трубопроводов,
а также образовывать дополнительные промышленные стоки,
очистка которых очень затруднительна.
2. Способность образовывать накипи в теплообменных аппа-
ратах и в канифолеуваривательной колонне приводит к увели-
чению расхода греющего пара и частым остановкам основного
оборудования па чистку. Бессолевой способ отстаивания жи-
вицы приводит также к снижению зольности канифоли.
Для снижения вязкости и увеличения разности плотностей
живицы и воды массовую долю скипидара в живице доводят
до 40%. При этом наряду с улучшением условий отстаивания
опыт эксплуатации промышленных отстойников показал, что
скорость отделения воды от живицы возрастает более чем
в 3 раза, на 15—20 % снижается продолжительность плавки
живицы. Кроме того, при 40 % скипидара уменьшается содер-
жание воды в живице, поступающей в канифолеузаривательную
колонну, и увеличивается коэффициент использования смоли-
стых веществ за счет уменьшения их в отработанном соре на
25—30 %. Отстаивание живицы ведут в отстойниках как перио-
дического, так и непрерывного действия. Наиболее надежным и
хорошо зарекомендовавшим себя в эксплуатации является от-
стойник непрерывного действия конструкции М. К. Жлобо и
П. П. Полякова (рис. 6). Он представляет собой удлиненный
стальной сборник прямоугольной формы вместимостью около
60 м.3 с герметичной плоской крышкой и конусообразным дни-
щем. Отстойник внутри футеруют кислотоупорным цементом,
38
а снаружи, для поддержания постоянной температуры живицы,
тщательно теплоизолируют.
Исходная живица из буферного бака поступает в отстойник
сразу через три штуцера 1, и одновременно очищенная живица
отводится с противоположной стороны также через три шту-
цера 2. На пути движения живицы в отстойник установлены две
перегородки. На входе живицы в отстойник смонтирована
сплошная перегородка 3 на глубину 700 мм от крышки, направ-
ляющая поток живицы вниз. Для выхода из отстойника части
живицы на глубину 500 мм от крышки устанавливается перед
штуцерами вторая, ситчатая, перегородка 4. Она служит для
задержки скапливающегося в верхнем слое легкого сора — пре-
имущественно коры. В днище отстойника расположен ряд кону-
сообразных выступов 6. Их назначение — увеличить жесткость
конструкции, а также замедлить движение живицы в горизон-
тальной плоскости и улучшить условия отстоя. Они создают как
бы ряд отсеков, которые соединены с трубой 5, расположенной
непосредственно под днищем отстойника по всей его длине.
Труба заканчивается гидравлическим затвором 7 для непрерыв-
ного отвода отстоя из отстойника.
Режим работы отстойника следующий: в отстойник из бу-
ферного бака непрерывно поступает горячая (92—95 qC) рас-
плавленная живица с 40 % скипидара и 12—20 % воды (сла-
бого раствора фосфорной кислоты и ее солей). В момент по-
ступления живицы в отстойник скорость ее движения резко
падает. Вследствие этого происходит быстрое отделение от жи-
вицы основной массы водно-грязевого отстоя. При последую-
щем движении живицы в горизонтальной плоскости происходит
дальнейшее отделение ее от воды и сора. Очищенная (отстой-
ная) живица поднимается вверх, фильтруется от всплывшего
легкого сора через ситчатую перегородку и непрерывно отво-
дится из отстойника в буферный бак. Из нижней части отстой-
ника через трубу и гидравлический затвор отстой непрерывно
отводится в ловушку. Обслуживание отстойника очень простое
и заключается в периодическом наблюдении через смотровые
фонари за равномерностью поступления исходной живицы и вы-
водом очищенной живицы и отстоя. Производительность такого
отстойника составляет 200—250 т живицы в сутки.
При правильной работе отстойника живица, поступающая из
него на переработку, практически не содержит сора даже в виде
механических пылинок, а объем воды не превышает 0,6%. Ис-
пользование отстойников непрерывного действия не только спо-
собствует повышению качества продукции, но и приводит к со-
кращению объема производственных помещений и росту произ-
водительности труда почти вдвое.
В последние годы в результате низкой культуры труда при
подсочке применяемые биостимуляторы (в частности, суль-
фитно-спиртовая барда) попадают в живицу. Сульфитно-спир-
товая барда обладает высокими поверхностно-активными свой-
39
ствами, поэтому наличие ее в живице способствует образованию
устойчивой эмульсии либо живицы в воде, либо, наоборот, воды
в живице. Особенно устойчивые эмульсии образуются в период
плавления живицы в токе острого пара. Образовавшаяся эмуль-
сия вода — живица практически не расслаивается и приводит
к большим потерям живицы при отстаивании. Для обеспечения
расслоения живицы и воды на стадии плавления добавляют та-
кие деэмульгаторы, как катамин АБ (0,2—0,3 % от массы жи-
вицы) .
Катамин АБ способен связывать лигносульфонаты, попадаю-
щие в живицу вместе с сульфитно-спиртовой бардой и суль-
фитно-дрожжевой бражкой, предотвращая образование эмуль-
сий. Для улучшения свойств канифоли и снижения ее кристал-
лизации необходимо дополнительно промывать отстоявшуюся
живицу водой от катамина АБ и его комплексов с лигносульфо-
натами.
Наличие катамина в канифоли снижает ее качество. Осо-
бенно это сказывается при использовании канифоли в производ-
стве синтетического каучука, где она применяется в качестве
эмульгатора. В настоящее время установлен жесткий контроль
за содержанием сульфитно-спиртовой и сульфитно-дрожжевой
бражки в живице, что благоприятно сказалось на работе кани-
фольно-терпентинных заводов.
Промывка живицы. Одним из недостатков канифоли явля-
ется ее кристаллизация. Закристаллизованная канифоль явля-
ется браком. Работами ЦНИЛХИ и ЛТА было показано, что
основной фактор, вызывающий кристаллизацию канифоли,— со-
держание в ней абиетиновой кислоты. При массовой доле абие-
тиновой кислоты свыше 40 % канифоль практически всегда спо-
собна к кристаллизации. Исследованиями было показано, что
основное накопление абиетиновой кислоты происходит на ста-
дии канифолеварения. Под действием тепла и кислотных ката-
лизаторов происходит изомеризация левопимаровой, палюст-
ровой и неоабиетиновой кислот в абиетиновую.
Доля кислотного катализатора, поступающего на канифоле-
варение с живицей, зависит от массы в ней влаги и от кислот-
ности отстойной воды. Массовая доля абиетиновой кислоты в ка-
нифоли зависит от массовой доли кислотного катализатора
(в пересчете на Н3РО4), поступающего на канифолеварение
(рис. 7).
Следовательно, для предотвращения кристаллизации живич-
ной канифоли путем снижения в ней массовой доли абиетиновой
кислоты необходимо ликвидировать избыточное поступление
минеральных кислот в терпентине. Это достигается дополнитель-
ной промывкой водой живицы, отстоявшейся в специальном ре-
акторе, в количестве 5—10 % от массы терпентина и последую-
щим отстаиванием, что позволяет снизить содержание абиети-
новой кислоты в готовом продукте на 20—30 %.
Уваривание канифоли. В поступающей после отстойников из
40
буферных баков живице массовая доля канифоли составляет
58—60 %, скипидара 40—49 и воды около 0,8 %. Сущность про-
цесса уваривания канифоли сводится к отгонке от нее скипи-
дара. Отгонка скипидара от канифоли должна быть полной, так
как остатки скипидара резко снижают температуру размягчения
канифоли и придают ей липкие свойства. Начальная темпера-
тура кипения скипидара (а-пинена) 156 °C. Наличие канифоли
Рис. 7. Зависимость массовой доли абиети-
новой кислоты в канифоли от массовой
доли минеральных кислот в терпентине
Рис. 8. Упругость пара некоторых жидко-
стей:
1 — сернистого углерода; 2 — четыреххлористого
водорода; 3 — бензола; 4 — толуола; 5 — скипи-
дара; 6 — фенола; 7 — анилина; 8 — крезола; 9 —
нитробензола; 10 — нитротолуола
в живице значительно повышает ее температуру кипения, что
видно из следующих данных.
Содержание канифоли в скипидаре, %.......... 20 50 70 85
Температура кипения живицы, °C ............ 161 163 179 195
Остатки скипидара будут отгоняться при температуре 250—
300 °C. Но так как терпеновые углеводороды, содержащиеся
в скипидаре, и смоляные кислоты, входящие в состав канифоли,
41
при повышенных температурах склонны к изомеризации, что
приводит к снижению их качества (при температурах свыше
230—250 °C канифоль разлагается), то ведение отгонки скипи-
дара при высоких температурах недопустимо. Для снижения
температуры отгонки скипидара от канифоли этот процесс
можно вести при пониженном давлении в системе, в токе инерт-
ного газа (N2, СО2 и др.) и с водяным паром.
На наших заводах отгонку скипидара ведут только в токе
острого водяного пара. Несмотря на простоту этого способа и
его экономичность, он имеет существенный недостаток, так как
приводит к образованию дополнительного объема сточных вод,
составляющих до 30 % от общего объема промышленных сто-
ков канифольно-терпентинных заводов.
Многие взаимонерастворимые вещества при кипении обра-
зуют пары азеотропной, или нераздельно кипящей, смеси. При-
чем температура кипения азеотропной смеси всегда имеет тем-
пературу кипения ниже ее низкокипящего компонента.
В результате исследований было показано, что это положе-
ние хорошо согласуется с законом Дальтона, по которому общее
давление паров в системе F складывается из суммы парциаль;
ных давлений ее компонентов. При перегонке паров воды и ски-
пидара
F = Р^вХв -|- Р скХск, (23)
где Р — общее давление в системе, кПа; Рв, Рек — упругости
паров воды и скипидара в чистом виде при температуре кипе-
ния смеси, кПа; хв, хск — мольные доли воды и скипидара
в парах.
Температура, при которой будет идти процесс перегонки,
может быть определена на основании парциальных упругостей
перегоняемых жидкостей. Для скипидара из сосны, произрас-
тающей в СССР, температура перегонки с водяным паром при
атмосферном давлении принимается 96 °C. Это легко опреде-
ляется графическим способом (рис. 8).
Величины упругости паров жидкостей на графике нанесены
в виде кривых. По оси абсцисс отложена температура кипения,
по оси ординат — соответствующие величины упругостей. Кри-
вая упругости паров воды нанесена в обратном положении, на-
чало кривой по оси ординат лежит в точке, соответствующей
101,32 кПа. При таком положении она будет пересекать кривые
упругости всех других жидкостей в точках, соответствующих
температуре кипения азеотропа этих жидкостей.
Нетрудно убедиться в том, что перпендикуляр, опущенный
из точки пересечения кривой для воды с кривой для другой
жидкости на ось абсцисс, дает температуру кипения смеси,
а сумма отрезков на оси ординат для точки пересечения кривых
равна 10 132 кПа.
Если кривые упругости паров воды провести, начиная с ор-
динаты не 101,32 кПа, а, например, при остаточном давлении
42
в системе 39,9 или 13,3 кПа, то пересечение этих кривых с кри-
выми упругости других жидкостей дадут точки, соответствую-
щие температурам кипения смеси при этих давлениях в си-
стеме.
В случае двухкомпонентной системы в соответствии с урав-
нением Ван-Клина отношение масс перегоняемых жидкостей
равно отношению произведений молекулярных масс на парци-
альное давление этих жидкостей при температуре кипения
смеси:
Gb • GcK- в • Л4СкРСК,
(24)
где GB и Gck — массы воды и скипидара в смеси паров, кг; Мп
и ТИск — молекулярные массы воды и скипидара соответственно
18 и 136.
На рис. 8 видно, что при общем давлении в системе
101,33 кПа Рв = 86, а РСк=65,33 кПа. Подставляя в уравнение
все значения величин, получим:
Gg ; МвРв =.,18'86.’0 =о,2 кг/кг,
Gck МскРск 136-65,33
(25)
т. е. при перегонке скипидара с водой на 1 кг скипидара расхо-
дуется 0,2 кг воды.
Эффективность испарения летучего вещества зависит отего
физико-химических свойств и условий перегонки. Чем меньше
молекулярная масса вещества, тем выше эффективность пере-
гонки. Эффективность перегонки зависит и от высоты слоя пе-
регоняемой жидкости, размеров пузырьков водяного пара и от
скорости его прохождения через слой испаряемой жидкости.
Увеличение скорости прохождения паров воды способствует тур-
булизации системы и, таким образом, повышению поверхности
контакта фаз и росту коэффициента насыщения пузырьков во-
дяного пара летучим веществом. В зависимости от этих условий
эффективность испарения для чистой жидкости может изме-
няться в пределах 0,5—0,9.
В случае отгонки скипидара от живицы, когда летучий ком-
понент отгоняется от нелетучего остатка, процесс осложняется
еще и тем, что в ходе отгонки происходит сплошное обеднение
смеси летучим компонентом и вследствие этого непрерывно из-
меняется упругость паров летучего компонента и растет темпе-
ратура кипения смеси.
Упругость паров летучего вещества над его раствором с не-
летучим остатком всегда ниже, чем над слоем его в чистом
виде. В этом случае к закону Рауля вводится поправочный ко-
эффициент К.
К = А1(А+В), (26)
где А — мольное количество летучего вещества в растворе, %;
В — мольное количество нелетучего компонента в растворе, %.
43
Рис. 9. Канифолеварочйая колонна:
у — штуцер для отвода паров скипидара и воды;
2 — сепарационное устройство; 3 — штуцер для
входа терпентина; 4 — вход пара в змеевик; 5 —
выход конденсата; 6 — гидравлический затвор;
7 — переливная труба; 8 — сетчатая тарелка;
9 — колпачковая тарелка; 10 — штуцер для вхо-
да пара в барботер; 11 — штуцер выхода кани-
фоли
Из этого следует, что. с умень-
шением в живице скипидара (лету-
чего компонента) величина коэффи-
циента К будет падать, а значит, и
упругость паров скипидара будет
уменьшаться, так как закон Рауля
будет иметь вид:
Рек ~ КРСК-^СК, (27)
где рек — парциальная упругость
скипидара над раствором при тем-
пературе перегонки; Рск — упру-
гость паров скипидара над жидко-
стью в чистом виде при той же
температуре; хСк — мольная доля
скипидара в растворе.
Вместе с уменьшением К, т. е.
концентрации летучего компонента
в смеси, упругость паров скипи-
дара будет понижаться, что приве-
дет к увеличению расхода острого
водяного пара.
Методом вычисления можно по-
казать, что если вести отгонку ски-
пидара при 96 °C, то при остаточ-
ном содержании его в канифоли
(около 1 %) расход водяного пара
на 1 кг отгоняемого скипидара со-
ставит примерно 100 кг. Это обсто-
ятельство приводит не только к не-
оправданному росту расхода тепла,
но и к образованию большого объ-
ема промышленных стоков. Для из-
бежания этого уваривание ведут
при повышенных температурах,
165—170 °C. Отгонку скипидара от
живицы и уваривание канифоли
производят в канифолеварочных
колоннах преимущественно непре-
рывным способом, а на заводах ма-
лой мощности — в канифолевароч-
ных кубах.
44
Непрерывнодействующая канифолеварочная колонна изобра-
жена на рис. 9. По технологической схеме хорошо отстоявшаяся
живица с помощью насоса через фильтры поступает в бачок по-
стоянного уровня, откуда непрерывно через подогреватель на-
правляется в распределительный лоток, находящийся в верхней
части колонны. Нагрев живицы, поступающей в верхнюю часть
колонны, в подогревателе производится до 160—165 °C. Острый
водяной пар подается через барботер, расположенный в нижней
части колонны. Для поддержания необходимой температуры
(165—170 °C) в колонне вводится дополнительное количество
тепла с глухим паром, подаваемым в подогреватели и змеевики,
расположенные на тарелках по всей высоте колонны. Во избе-
жание конденсации водяного острого пара он подается в кани-
фолеварочную колонну в перегретом состоянии при 800—
1200 кПа.
При установившемся режиме на каждой тарелке колонны об-
разуется слой живицы. Перетекая через переточные^трубки, на-
гретая живица движется вниз, острый пар поднимается вверх,
нагревает слой жидкости на тарелках до температуры кипения
азеотропа скипидар — вода и увлекает его с собой в паровой
фазе. Из верхней части колонны пары скипидара и воды посту-
пают в дефлегматор, где наиболее тяжелокипящие компоненты
скипидара-конденсата и часть водяных паров конденсиру-
ются.
Конденсат из дефлегматора направляется во флорентину,
где скипидар отделяется от воды и поступает в бак оборотного
скипидара, который в свою очередь используется для разбавле-
ния живицы в плавильном отделении цеха.
Основная масса паров воды и скипидара после дефлегма-
тора поступает в конденсатор-холодильник, из которого при тем-
пературе 20—25 QC поступает во флорентину. В скипидаре после
разделения его с водой во флорентине содержится небольшая
масса эмульгированной в нем влаги, поэтому он имеет мутно-
ватый цвет.
Для освобождения от влаги скипидар поступает на сушку
в соляно-ватные фильтры. В результате связывания влаги солью
скипидар после фильтров становится прозрачной бесцветной
жидкостью и поступает в баки товарного скипидара, откуда пе-
рекачивается на склад готовой продукции.
Вода из флорентины после отделения от нее скипидара со-
бирается в промежуточные баки и используется в качестве обо-
ротной воды при приготовлении осветляющего раствора фосфор-
ной кислоты, гидравлического удаления сора из плавильников,
мойки бочек и других целей. Уваренная канифоль из нижней
части колонны через гидрозатвор с паровым обогревом при тем-
пературе 165—170 °C поступает в сборник. Перед поступлением
в сборник канифоль охлаждается в теплообменнике до 145—
155 °C. Производительность колонны по живице составляет до
200 т/сут.
45
Рис. 10. Непрерывнодействующий канифолеварочный куб:
/ — тарелка; 2 — змеевик; 3 — каплеулавливающая тарелка; 4 — каплеотбойник; 5 —
барботер
На предприятиях небольшой мощности применяют непрерыв-
нодействующие канифолеварочные кубы (рис. 10). Куб пред-
ставляет собой вертикальный цилиндрический аппарат высотой
2,5 м и диаметром 1 м. Внутри аппарата смонтировано пять-
шесть секций змеевиков глухого пара, между которыми уста-
новлены ситчатые тарелки.
Живица, пройдя предварительный нагрев в змеевиковом по-
догревателе, непрерывно подается в среднюю часть куба. В ниж-
ней его части вмонтирован барботер острого пара. Отбор паров
скипидара и воды производится через верхнюю часть куба, ка-
нифоль также непрерывно выводится из нижней части. Произ-
водительность куба при площади нагрева змеевиков 25 м1 2 со-
ставляет 40—45 т живицы в сутки.
Розлив канифоли. Канифоль представляет собой смесь изо-
мерных смоляных кислот. Ее аморфное стеклообразное состоя-
ние с характерным раковистым изломом объясняется тем, что
образующийся в процессе ее получения сплав смоляных кислот
при быстром охлаждении застывает, не успевая закристаллизо-
ваться. Так как скрытая теплота образования кристаллов при
получении аморфной канифоли не выделяется, то канифоль
всегда имеет избыток энергии и является системой термодина-
мически неустойчивой.
А-А
Рис. 11. Барабанный охладитель канифоли:
6-Б
1 — охладительный барабан; 2 — подшипник; 3 — сальник; 4 — полый вал; 5 — раз-
брызгиватель воды; 5 —шкив; 7 — ванна для канифоли; 8 — регулировочный болт; 9 —
станина; 10 — нож; 11 — охлажденная канифоль
47
Это обстоятельство может привести канифоль при опреде-
ленных условиях в более устойчивую кристаллическую форму,
которая является крайне нежелательной. Многолетний опыт
производства канифоли показал, что наибольшую склонность
к кристаллизации она имеет при температуре 80—120 °C. При-
чем при температуре 80 °C кристаллы смоляных кислот выпа-
дают в виде линз, при 90—100 °C — в виде треугольников, при
110—115 °C— прямоугольников и квадратов. При температуре
ниже 80 °C канифоль становится очень вязкой и кристаллизация
практически не идет; при температуре выше 125 °C кристаллы
находятся в расплавленном состоянии и кристаллизация невоз-
можна.
Склонность к кристаллизации чаще всего проявляется при
розливе в тару товарной канифоли. Если сразу заполнить бочку
горячей канифолью, то в ее середине канифоль будет сохранять
температуру благоприятную для ее кристаллизации. Поэтому
при розливе канифоли нужно обеспечить практически мгновен-
ное охлаждение ее до температуры ниже 80 °C. Это можно осу-
ществить, используя для розлива канифоли охладительные ба-
рабаны (рис. 11). Охладительный аппарат представляет собой
полый барабан, изготовляемый, как правило, из листовой меди.
Для лучшего охлаждения вода подается непосредственно на
внутреннюю стенку барабана с помощью специального распы-
лителя. Частота вращения барабана регулируется с помощью
редуктора вариатора; при оптимальных условиях работы она
составляет 5—6 мин-1. Применение барабанов механизирует
трудоемкие операции розлива и охлаждения канифоли. Произ-
водительность таких барабанов составляет 600—800 кг охлаж-
денной до 60—85 °C канифоли в час.
Эксплуатация охладительных барабанов очень проста. Ка-
нифоль из сборника самотеком поступает в бачок постоянного
уровня, а из него — в коробку канифолеразливочного барабана.
Поплавок-регулятор поддерживает уровень расплавленной ка-
нифоли в коробке так, чтобы нижняя часть охладительного ба-
рабана была погружена в канифоль на 15—20 мм. При враще-
нии барабан захватывает тонкий (около 2 мм) слой канифоли,
которая быстро охлаждается на его поверхности до 60—65 °C
и в виде эластичной пленки срезается неподвижно закреплен-
ным ножом в бочку. Бочки с канифолью взвешивают и вывозят
на склад готовой продукции. Площадь, необходимая для скла-
дирования канифоли, определяется из расчета времени, необ-
ходимого для ее охлаждения, формирования партии по цветно-
сти, маркировки и погрузки в железнодорожные вагоны. Прак-
тически установлено, что полезная площадь склада должна
быть такой, чтобы обеспечить размещение одновременно пяти-
дневного запаса вырабатываемой канифоли.
Однако применение их при розливе канифоли имеет целый
ряд недостатков: низка производительность, необходима частая
чистка наружной поверхности, коэффициент заполнения тары
48
a
лэвпХюоп ‘goMniBjjAd hosocIbii Аионнэждвнэ ‘AVoaoduogXdi
on иояэюивэ bVXmio ‘явд HHHdouBH a KoiaBaahBaadon woooobh
WHHJRXdjon BHimdopo ей чвофинвя Banirsod эиэхэ иоявх udy
•ww OZ saifog эн KHiodaaio ojoh
-hosXdjBE wodawEBd и ии g*o иониИнгох веэтгэж оаоннваоипиПо
йен BHOidBH ей it 001 отэошихээиа HHBgBdsg чхвао£Ч1гопэи
HdBi эахээьвл а в ‘HHtaZtfodu иоаохоа эГвеяэ вн oiiHaaxatfadoon
-ЭН 41HffOa£HOdlI K019Xfi'H9WOM9d И1ГОфиНВЯ aHITEOd О1Ч1ГЭП ИОХЕ Э
•аоиЕинвхэк и BHHBaotfXdogo wo'n'Bd xnlnoiBxogBd HHliEdgna xo
‘винэПтэиоц ojOHHgaiotfoasHodu ндэфэоихв ей ээн a эи'ппив^ви
1и1лой1яэ1Г€ — zi iHAeadodiMaire — ц !wodoii?£oV э икофинвя чтэоякэ — oi 1и1гофинвм
в1Гояя danXini — в IdtfHKifHh jjoxAim — 9 tqif9iBh0iifMi4fl0diMair€ — д ‘.ияявь’поп — g 4
tHdoiEEoV — g !и1гофинвм Kirtf udBi — z JwotfOHHdnodiMaife э dagOHHOM JjiqHqiraoXdeM — /
пигофинвм uaHifcod олояээьихеиохяв BiflT oaxoHodxo^ ‘SI ’3Hd
-ou ‘EJEirs и qiriqu ээ qx^aciqa xXjow oriniBCuififBiDUcbi иггофинвя
oauE£odudn*(gi *oud) иЕофиния aauircod Лдоэопэ Лиомээьихемох
-ав on вяаонвхэЛ кэхэЛЕатгопэи odum ээа uwada ээХпеохэен q
‘xXtfEOa ЕЭХЭЭИ1И ИЕОфиниМ ИЛНЭ1ГЦ ИИКО1ГЭ ХЯжэи мая яих
‘ияьор а шгофинвя ияаоявпЛ этгэоп и Eoxaaaciroirodn еинэеэияо
ooQTiodu •вхХ’п'Еоа woVodoirouH еэхэееэияо онаиэнахни шгофиния
HOhEdOJ ВЯНЭ1Ш ЕЕЯНОХ ‘OJOX QIMOd){ • % £6 ОХГ 06 10 КЭХЭ1Г9Э1ГОЯ
в коллектор для розлива или в автоматический дозатор. В авто-
матическом дозаторе использован принцип сообщающихся сосу-
дов. Четыре дозирующие емкости объемом по 100 л помещены
в коробку с канифолью. При включении дистанционного управ-
ления краны дозирующих емкостей становятся в положение
«Закрыто». Канифоль перетекает из коробки в дозирующие
емкости. Уровень канифоли в коробке снижается, поплавок, кон-
тролирующий наполнение коробки, опускается, открывается от-
верстие канифолепровода и коробка снова наполняется кани-
фолью. Общий уровень канифоли в дозирующих емкостях вы-
равнивается, достигая такого положения, при котором поплавок,
помещенный в одной из дозирующих емкостей, всплывает, вклю-
чает микропереключатель, от которого сигнал поступает в ко-
лонку дистанционного управления для автоматического пере-
ключения кранов в положение «Открыто». Отмеренные дозы
сливаются в барабаны, после чего приводится в действие кон-
вейер. Заполненные барабаны перемещаются, а под дозатор по-
дается очередной поддон с порожними барабанами, и цикл пов-
торяется.
§ 5. ОЧИСТКА СТОЧНЫХ ВОД И ГАЗОВЫХ ВЫБРОСОВ
Объем сточных вод при переработке живицы на канифольно-
терпентинных заводах невелик и составляет 1 м3 в пересчете на
единицу товарной продукции. В основном сточные воды обра-
зуются на стадии отстаивания живицы от сора и воды, а также
от мойки бочек. Однако загрязненность сточных вод весьма ве-
лика (загрязненность по ХПК 18000—20 000 мг/л). Сточные
воды имеют высокую кислотность: pH 1,3—2,3. При обработке
этих вод гашеной известью их загрязненность уменьшается в 2—
3 раза, в основном за счет выпадания в осадок резината каль-
ция и сорбции других смолистых веществ на поверхности осадка
и гидроокиси кальция. Сточная вода из отделения уваривания
канифоли составляет вторую часть всех стоков канифольно-тер-
пентинных заводов и представляет собой подскипидаренную
воду из флорентины. Эта вода имеет ХПК 2000—4000 мг/л и со-
держит в основном эмульгированный скипидар.
Учитывая небольшие объемы сточных вод, окончательную
очистку их на канифольно-терпентинных заводах часто ведут
методом сжигания в циклонных топках.
Газовые выбросы от аппаратов загрязнены парами скипи-
дара. Однако низкая упругость паров скйпидара и небольшой
объем отходящего воздуха обусловливают невысокое загрязне-
ние воздуха и практически укладываются в предельно допусти-
мую концентрацию (ПДК) для скипидара, равную 300 мг/м3.
Для обеспечения более полной очистки газовых выбросов и
более глубокого извлечения скипидара на воздушных линиях
устанавливают либо мощные концевые холодильники, либо
скрубберы, орошаемые минеральными маслами.
50
6. МЕТОДЫ контроля
Объект контроля Место и порядок отбора пробы Контрол и ру емый показатель
Загрузка и плавление живицы
Живица сосновая Из бочек с живицей — Полный анализ по ГОСТу,
Фосфорная кислота средняя проба от каждой партии при поступлении на склад Из пробоотборника при поступлении в живичную коробку — 1 раз в смену От каждой партии при по- лигносульфонаты То же Концентрация, плотность
Катамин АБ ступлении в цех Из емкости для слабой кислоты — после пригото- вления раствора От каждой партии при по- То же Полный анализ по ТУ
Сор ступлении на склад Из плавильников при вы- Смолистые вещества, терпе-
Терпентин грузке — среднесменная проба Отстаивание жив Из пробоотборника перед новые углеводороды, влага, сор 1 и ц ьг Влага, терпеновые углево-
Слой: водный подачей на промывку, каждые 2 ч Из отстойников при сли- дороды Кислотность, смолистые ве-
грязевой ве — 2 раза в смену Из отстойников при сливе щества, терпеновые углево- дороды Смолистые вещества, терпе-
Скипидар грязи или грязевых лову- шек — перед выводом Канифолеварен На выходе из соляноват- новые углеводороды, влага, сор и е Влага
Канифоль ного фильтра — каждые 2 ч Из сборника товарного скипидара — после запол- нения Из коробки охлаждающе- Кислотность, плотность, пре- делы перегонки, объем отгона Цвет, температура размяг-
Воздух го барабана при розли- ве — каждые 30 мин Из бочек и барабанов при формировании партии Воздушная сре Из отделений: плавильно- чения Полный анализ по ГОСТу Д а Скипидар
го, мойки бочек, канифо- леварения, загрузки живи- цы — на рабочих местах аппаратчиков 2 раза в ме- сяц Из отделения розлива ка- нифоли — 2 раза в месяц Канифоль
51
Продолжение
Объект контроля Место и порядок отбора пробы Контролируемый показатель
Сточная вода Сточные водь Из канализационных ко- лодцев—2 раза в месяц I ХПК, кислотность
§ 6. КОНТРОЛЬ ПРОИЗВОДСТВА
Контроль за соблюдением технологического режима на всех
стадиях процесса осуществляется цеховой лабораторией по рег-
ламенту. Место отбора пробы, частота, допустимые нормы и ме-
тоды контроля определяются условиями по табл. 6.
Контроль за качеством готовой продукции осуществляет ОТК
завода. Контроль за температурой, давлением, расходом пара
и воды, уровнем веществ в мерниках и сборных емкостях при
ведении технологического процесса осуществляется с помощью
контрольно-измерительных приборов.
Технико-экономические показатели. При существующем спо-
собе переработки общий коэффициент извлечения содержащихся
в живице смолистых веществ составляет 98 %, т. е. потери при
производстве составляют не более 2 %. При этом потери кани-
фоли не превышают 1%, а скипидара 6 % от их исходной
массы в живице.
Электроэнергии на 1 т перерабатываемой живицы расхо-
дуется около 14 кВт-ч, в том числе на технологические нужды
около 8,5 кВт-ч, расход тепла 1,7 ГДж и воды 7 м3.
Данные действующих лесохимических предприятий о струк-
туре себестоимости продукции канифольно-терпентинного произ-
водства свидетельствуют о высокой материалоемкости. Основ-
ные статьи расхода на 1 т канифоли, %, приведены ниже.
Сырье и основные материалы .......................................93,2
Вспомогательные материалы и тара :............................ 2,7
Энергетические затраты ........................................... 0,8
Заработная плата основная и дополнительная ....................... 0,6
Расходы на содержание оборудования ............................... 1,0
Общезаводские расходы ............................................ 1,0
Внепроизводственные расходы ..................................... 0,7
Глава 3
КАНИФОЛЬНО-ЭКСТРАКЦИОННОЕ ПРОИЗВОДСТВО
Впервые вопрос об организации производства канифоли и скипидара из
смолистой древесины поставил Д. И. Менделеев. А в 1909 г. проф. П. И. Кур-
санов разработал и предложил промышленный -способ переработки смолистой
древесины путем экстракции смолистых веществ бензином. Впоследствии
этот метод нашел применение в канифольно-экстракционных производствах
во всех странах.
52
В дореволюционный период первые заводы по извлечению канифоли
бензином из щепы соснового пневого осмола были построены в Финляндии
и во Владимирской губернии. В период первой мировой войны все работы
по получению канифоли из смолистой древесины были прекращены. Однако
накопленный в тот период опыт работы канифольно-экстракционных заводов
позволил уже в 1923 г. начать строительство крупного канифольно-экстрак-
ционного завода в поселке Вахтан Горьковской обл. В 1927 г. завод был
введен в эксплуатацию и работает до настоящего времени. В первые же
годы его работы оказалось, что себестоимость экстракционной канифоли на
25—30 % ниже, чем живичной.
В дальнейшем развитие производства смолистых веществ из осмольной
древесины пошло по направлению щелочной экстракции. Продуктами .этого
производства являлись канифольное мыло и паровой скипидар. Процесс
их получения сводился к обработке осмольной щепы слабыми водными рас-
творами едкого натра при температуре кипения. Производство это было
более простым и более безопасным в пожарном отношении, чем экстракция
бензином. Было построено пять канифольно-мыльных заводов, однако кани-
фольное мыло было продуктом низкого качества, оно трудно транспортиро-
валось, поэтому применение его в народном хозяйстве было ограниченно.
Вскоре производство его закрыли.
В то же время велись научно-исследовательские и проектные работы по
усовершенствованию технологии экстракции смолистых веществ из осмола
бензином. Результаты работ легли в основу проекта и строительства заво-
дов по батарейно-противоточному способу экстракции осмольной щепы на
Ново-Белецком (Гомельском) бумажно-лесохимическом комбинате’ в 1952 г.,
на Нейво-Рудянском лесохимическом заводе в 1954 г., по комбинирован-
ному способу экстракции на Верхотурском лесохимическом заводе в 1958 г.
В 1961 г. Вахтанский канифольно-экстракционный завод был переведен на
батарейно-дефлегмационный способ экстракции. В IX пятилетке в эксплуа-
тацию был введен еще ряд канифольно-экстракционных заводов: Братский,
Лесосибирский, Зиминский, Медвежьегорский и др. На всех этих предприя-
тиях в основе технологии извлечения канифоли из осмольной щепы исполь-
зуется метод батарейно-дефлегмационной, или оросительной, экстракции.
В настоящее время практически все операции по получению
канифоли и скипидара из смолистой древесины являются непре-
рывными, поэтому ведутся интенсивные научно-исследователь-
ские и проектные работы по созданию непрерывного способа
экстракции смолистых веществ бензином из осмольной щепы.
§ 7. СЫРЬЕ ДЛЯ КАНИФОЛЬНО-ЭКСТРАКЦИОННОГО
ПРОИЗВОДСТВА
Сырьем для канифольно-экстракционных производств слу-
жит смолистая древесина, в основном сосна. Различают сле-
дующие виды смолистой древесины:
1. Спелый сосновый пневый осмол — ядровая часть зрелых
пней и корней сосны.
2. Стволовый осмол — стволовая часть сосны, просмолив-
шаяся в результате осмолоподсочки.
3. Карровый осмол — горбыли, образующиеся на лесопиль-
ных предприятиях при распиливании сосновых стволов, подверг-
нутых прижизненной подсочке; под ним понимают также
стружку с карр, снятую примерно на глубину 1,5 мм.
4. Валежный и колодниковый осмол — естественно просмо-
лившаяся комлевая часть ветровальных, в том числе поражен-
ных легкими низовыми пожарами деревьев.
53
5. Свежий пневый сосновый осмол — пни сосны, которые
могут быть заготовлены в период проведения лесосечных работ
или через год — два после рубки насаждений.
6. Смолистая . древесина, получаемая в результате искус-
ственного прижизненного просмоления заболонной части дерева
под воздействием различных смолостимуляторов.
Из всех указанных видов смолистой древесины практическое
значение для канифольно-экстракционного производства имеет
спелый сосновый пневый осмол, который на месте заготовки
очищается от грунта и гнили. Горелый осмол, кроме того, очи-
щается от обугленной части древесины. Длина отдельных кус-
ков не должна превышать 60, а диаметр 40 см. По степени раз-
делки различают полуразделанный осмол (или осмол взрывной
заготовки без дополнительной разделки) и неразделанный ос-
мол (или осмол механизированной заготовки, тоже без допол-
нительной разделки и очистки). Массовая доля смолистых в пе-
ресчете на 20%-ную влажность осмола должна быть не ниже
13 %. Массовая доля скипидара, как правило, не регламенти-
руется, но обычно она составляет 3—4 % массы абсолютно су-
хого осмола.
Пневый осмол является своеобразным сырьем. Заготовку
его, как правило, начинают в среднем через 8—15 и более лет
после рубки деревьев. Такой разрыв во времени между рубкой
леса и заготовкой осмола обусловливается процессом естествен-
ного созревания-осмола, который состоит в относительном обо-
гащении ядровой части пня за счет разрушения менее смоли-
стой заболонной части пня в результате гниения.
Срок созревания пня будет зависеть от первоначальной смо-
листости заболони и ядра, климатических условий, характера
почвы. В более теплых и влажных районах лесонасаждений и
при суглинистых почвах период созревания гораздо короче (Ук-
раина, Белоруссия и др.). А в районах Северной Карелии, Вос-
точной Сибири, на Урале, в условиях песчаных почв и холод-
ного климата, пневый осмол может созревать более 25 лет. Как
правило, более мелкие пни сгнивают нацело, и через 8—15 лет
на вырубках остается менее половины пней.
В созревшем осмоле наиболее богата смолистыми шейка
корня. Смолистость надземной части пня по мере удаления от
шейки постепенно уменьшается. Смолистость подземной части
пня, особенно боковых корней, пониженная, однако они тоже
используются в производстве.
Начальная смолистость надземной части пней (рис. 13) в мо-
мент рубки дерева составляет в заболони 1,5—4%, а в ядре
10—20 % и более. При современных механизированных заготов-
ках спиливание стволов осуществляется заподлицо с землей, что
приводит к резкому сокращению пневого осмола.
Осмольное сырье рассредоточено на больших площадях ле-
совозрбновляющихся вырубок, с низкой концентрацией его на
1 га. На ряде площадей Белорусской ССР и центральных обла-
54
стей европейской части СССР запасы осмола составляют всего
0,5—1 м3 плотного объема на 1 га. Средний запас пневого ос-
мола на площадях осмолозаготовительных предприятий колеб-
лется от 2,5 до 7,5 м3 плотного объема на 1 га.
При такой низкой концентрации сырья осмолозаготовки рас-
пространяются на большую площадь и, естественно, на всей
площади наряду с высокосмолистым осмолом всегда попадается
Рис. 13. Разрез пня сосны:
1 — заболонь; 2 — ядро
и малосмолистый. Так, при средней смолистости всего пневого
осмола не ниже 13 % могут попадаться куски с массовой долей
смолистых от 1,5 до 22 %. А доля смолистых веществ в очищен-
ном осмоле в пересчете на 20%-ную влажность в зависимости
от давности рубки составляет, %: 1—5 лет 10—17; 6—10 лет
П—19; 16—20 лет 18—21.
Следовательно, созревание пня заканчивается в основном
через 5—10 лет. Но, как правило, корчевку пней начинают лишь
через 10—15 лет, а иногда и позже, когда осмол достигает тех-
нической спелости. К этому времени почти обгнивают заболонь
и большая часть боковых корней, благодаря чему корчевание и
очистка пня облегчаются.
Запасы и смолистость пневого осмола зависят от возраста
и качества вырубленных сосновых насаждений и возраста пня.
Наиболее богатый смолистыми пневый осмол получается при
вырубке перестойных деревьев на сухих суглинистых, супесча-
ных и песчаных почвах, а наименее смолистый — на болотистых
почвах.
Заготовка пневого осмола. Производственный процесс осмо-
лоз аготовок имеет специфические особенности, что отражает
характер заготовляемого сырья и степень концентрации его на
единице площади. К особенностям осмолозаготовительного про-
изводства относятся: большая территориальная разбросанность
сырья; чрезвычайно малый объем продукци, получаемой с еди-
ницы площади; различие природных, экономических, производ-
ственных и других условий в разных регионах заготовки ос-
мола.
Эти особенности осмолозаготовок затрудняют механизацию
труда, доставку рабочих и технологического оборудования, вы-
воз заготовляемого осмола. Для лучшей организации, и коорди-
55
нации работ по заготовке осмола созданы Всесоюзное произ-
водственное объединение «Союзлесхимпром» и «Главлес-
пром».
Для успешной работы канифольно-экстракционных заводов
необходимо ежегодно заготовлять до 2 млн. м3 плотного объема
пневого осмола. Однако из-за сложности всех видов работ по
осмолозаготовкам пока не обеспечивается нужное количество
заготовок пневого осмола.
В настоящее время заготовку осмола ведут бригадами, чис-
ленность которых и состав выполняемых работ могут быть раз-
личны. Заготовка осмола включает четыре основные фазы: кор-
чевку, трелевку, разделку и вывозку.
Корчевка пневого осмола. Основной способ корчевки пне-
вого осмола — взрывной, обеспечивающий достаточную сохран-
ность молодого подроста на вырубках. Однако этот способ очень
трудоемок. Почти 50 % всех вспомогательных работ на заго-
товке осмола взрывным способом относится к подготовке взрыва
(патронирование, охрана и перевозка взрывчатки). Куски пня
после взрыва разлетаются на значительное расстояние и их при-
ходится собирать в кучи («скучивать»); потери осмола при
этом достигают. 20—25%. К тому же взрыв является источни-
ком повышенной опасности. Средняя выработка при взрывном
способе корчевки пней на 1 чел.-день колеблется от 3 до 7 м3
плотного объема.
В последнее время при корчевке пней шире применяется ме-
ханизированный способ. Хорошие результаты механизирован-
ного способа достигнуты с применением агрегата АКП-1, разра-
ботанного Карельским- научно-исследовательским институтом
лесной промышленности на базе трактора ТДТ-55А и серийно
выпускаемого Майкопским машиностроительным заводом. Тех-
нологическое оборудование агрегата состоит из основания с ма-
нипулятором, захвата для корчевания пней и двух аутригеров.
Захват снабжен двумя челюстями с гидравлическим приводом,
гидравлическими домкратами и вибратором.
Корчевка пней агрегатом АКП-1 осуществляется на заранее
подготовленных площадях, на которых убраны опасные деревья
и намечены технологические коридоры. Корчевку пней обычно
начинают от лесовозной дороги (уса). Перемещается агрегат
в середине ленты строго по намеченному технологическому ко-
ридору и корчует все пни в зоне действия манипулятора. Опыт-
ные механизаторы выкорчевывают все пни на полосе шириной
до 16 м при вылете манипулятора от 1,5 до 8 м. При соблюде-
нии всех технологических требований на разрабатываемых уча-
стках сохраняется не менее 80 % подроста. Молодняк уничтожа-
ется в технологическом коридоре в основном гусеницами агре-
гата. Манипулятором и захватом агрегата истребляется не более
2—2,5 % подроста. Средняя производительность при корчевке
пней с помощью АКП-1 составляет около 20 м3 складочного
объема. При такой производительности трудозатраты механизи-
56
рбванного способа корчевки пней снижаются более чем в 2 раза
по сравнению со взрывным способом.
Высокая производительность механизированного способа
корчевки осмола достигнута при использовании трактора Т-100
с клином. Средняя сменная производительность при примене-
нии этого агрегата составляет более 22 скл. м3.
Однако применение высокопроизводительных тракторов-
бульдозеров на корчевке ограничивается необлесившимися пло-
.щадями. Таких площадей очень мало, а в европейской части
они почти полностью отсутствуют. Поэтому наиболее практич-
ным при корчевке пней надо считать агрегат АКП-1.
Выкорчеванный пень очищают от основной массы грунта
с помощью вибратора строго над подпневой ямой, чем дости-
гается выравнивание почвы для последующих лесовосстанови-
тельных работ.
Трелевка пневого, осмол а. На трелевке или подвозке пневого
осмола с участка заготовки на верхний склад к месту раз-
делки используют в основном машины ТПО-МЛТИ, ПЛО-1А
и ЛП-23.
Машина ТПО-МЛТИ разработана Московским лесотехниче-
ским институтом на базе колесного трактора Т-40А. Технологи-
ческое оборудование ее состоит из подъемного механизма, двух-
тонного погрузчика и лебедки трелевочного трактора ТДТ-40М.
Эта машина очень проста по конструкции и эксплуатации и ис-
пользуется в основном на подвозке пневого осмола взрывного
способа корчевки к разделочной площадке. Недостатком ма-
шины является низкий уровень механизации работ, так как она
не исключает ручного труда при выгрузке осмола в ковш и
увязки пневого осмола тросами.
Агрегат ПЛО-1А разработан Кавказским филиалом
ЦНИИМЭ и ВПКИлесмашем. Он создан на базе трактора ТДТ-
55А, оснащенного манипулятором от серийного погрузчика ПЭ-
0,8, самосвальным кузовом и захватом.
Подборщик-погрузчик ЛП-23 разработан КарНИИЛПом на
базе трактора ТБ-1 и выпускается Петрозаводским ремонтно-
механическим заводом. Технологическое оборудование подбор-
щика-погрузчика включает самосвальный кузов вместимостью
12 м3 и захват.
При сборе, погрузке и подвозке пней.машины ПЛО-1А или
ЛП-23 движутся строго по следу АКП-1, оставленному при кор-
чевке пней, чтобы исключить повреждение молодняка.
При очистке выкорчеванных пней от грунта и гнили исполь-
зуют гидравлический способ (т. е. удаление грунта и гнили под
действием струи воды) или электровзрывной. Чаще эти спо-
собы совмещаются.
При взрывном способе происходит не только удаление бал-
ласта, но и частичное раскалывание пней, что способствует сни-
жению трудозатрат при разделке осмола. Большая эффектив-
ность и высокая производительность достигаются при использо-
57
вании для очистки пней ударно-фрикционного способа в уста-
новках, созданных на базе корообдирочного барабана КБ-3.
Разделывают пни с помощью электро- и бензопил, крупные
куски раскалывают колунами. Разделанный осмол грузят в ку-
зова автомобилей и вывозят на нижний склад у железнодорож-
ной линии или водной магистрали, а иногда — на склад сырья
экстракционного завода. Более 95 % всего заготовленного ос-
мола к указанным пунктам доставляется автомобильным тран-
спортом. На вывозке используются, как правило, бортовые ма-
шины различных марок.
Корчевание из общих трудозатрат на заготовку осмола за-
нимает в среднем 25%, сбор и трелевка — 21, очистка и раз-
делка — 30, погрузка и вывозка — 23 %. С увеличением расстоя-
ния вывозки с 30 км до 100 и более доля ее в себестоимости
осмола возрастает до 32 %, что сказывается и в целом на себе-
стоимости осмола. Средняя производительность труда по всему
комплексу работ на заготовках пневого осмола составляет 0,5—
1,1 м3 на рабочего в день.
Характеристика пневого осмола. По содержанию смолистых
пневый осмол подразделяют на жирный, средний и тощий. В на-
стоящее время согласно ОСТ 13-131—82 «Осмол пневый сосно-
вый» длина кусков пней должна быть не более 0,6 м, диаметр
не более 0,4 м; они не должны иметь ответвлений. Массовая
доля канифоли не менее 130 кг на 1000 кг пневого осмола в пе-
ресчете на осмол-20 %-ной влажности1. Учет осмола ведется
по массе, при приемке осмола в лесу в поленницах — в едини-
цах объема. Полнодревесность поленниц осмола принимают рав-
ной 0,5, хотя этот коэффициент сильно колеблется и при плохой
укладке может быть значительно меньше.
Другие виды смолистой древесины. Трудности заготовки пне-
вого осмола и резкое сокращение вырубок с запасами пневого
осмола в европейской части СССР ставят вопросы об использо-
вании других видов смолистой древесины для канифольно-экс-
тракционных заводов.
Одним из источников смолистой древесины может служить
стволовый осмол, образующийся в результате осмолоподсочки
низкобонитетных сосновых насаждений и в основом использую-
щийся в осмолоскипидарных производствах. Назначение осмоло-
подсочки — обеспечить прижизненное просмоленйе заболонной
части ствола дерева с попутным сбором живицы, затвердевшей
на стволах в виде барраса.
Осмолоподсочку в основном проводят в Архангельской обл.
Ей подвергаются сосновые древостои V, Va бонитетов. В древо-
стоях V бонитета рекомендуется 8-летняя осмолоподсочка с хи-
мическим воздействием хлорной извести, в первые 3 года (в дре-
1 См. изменение к ОСТ 13-132—82 «Осмол пневый сосновый» от
28 марта 1985 г.
58
востоях Va бонитета в основном ведут 4-летнюю осмолопод-
сочку) без химического воздействия. На каждом стволе закла-
дывают лишь одну широкую карру, оставляя питательный ре-
мень шириной 15—20 см. Подновки глубиной 2—3 мм наносят
косарем или скобелем. Первые 3 года наносят по 10 подновок
высотой 5—6 см, на 4-й год— 10 подновок по 10 см, на 5-й —
8 подновок по 15 см, на 6-й год делают перерыв для лучшего
просмоления древесины. В последние 2 года наносят по одной
подновке высотой 75 см, при этом на 8-й год снимают карровый
питательный ремень.
При нанесении таких подновок происходит местное омерт-
вление и подсыхание древесины. Мертвая часть древесины иг-
рает роль естественного внутреннего приемника, куда перете-
кает живица из соседних живых частей ствола, благодаря чему
смолистость заболони значительно возрастает. Общая высота
просмоленной части ствола, которая после рубки и представляет
собою стволовой осмол, 4,5—6 м. Средняя смолистость стволо-
вого осмола в пересчете на осмол 20 %-ной влажности дости-
гает 8—12 %. По своему качеству смолистые стволового осмола
близки к качеству смолистых, содержащихся в спелом пневом
осмоле.
С гектара эксплуатируемых осмолоподсочкой древостоев по-
лучают 20—25 пл. м3 стволового осмола. Выход барраса состав-
ляет 0,3—0,6 кг с каждого дерева в год в течение первых 4—
6 лет осмолоподсочки.
В настоящее время ведутся исследовательские работы
.КирНИИЛПом и ЦНИЛХИ по искусственному прижизненному
просмолению сосновых насаждений путем обработки подновок
ранений водными растворами гербицидов. В некоторых странах
для этих целей используют гербициды паракват и дикват. В на-
шей стране наряду с ними применяют водные растворы других
гербицидов. Предварительные результаты показали, что через
3—4 года обработки ранений деревьев сосновых насаждений
массовая доля смолистых в стволовой части древесины на вы-
соте до 1,5—2 м (с учетом и подземной части дерева) состав-
ляет в среднем 13—16%. Качество смолистых соответствует
составу смолистых спелого пневого осмола. Так как качество
древесины после осмолоподсочки искусственного просмоления
стволовой части древесины сосны по своим физико-химическим
и механическим свойствам соответствует показателям свеже-
срубленного дерева, технология переработки ее должна преду-
сматривать комплексные способы.
Предварительные исследования и опытно-промышленные вы-
работки показали наиболее рациональные способы переработки
искусственно просмоленной древесины.
1. Комплексная переработка смолистой стволовой древесины
путем совмещения экстракции смолистых веществ из древесины
с последующей переработкой щепы сульфатным способом в цел-
люлозу. Причем в этом случае благодаря предварительному
59
извлечению в процессе экстракции смолистых веществ снима-
ются смоляные затруднения сульфатного производства целлю-
лозы.
2. Комплексное использование смолистой стволовой древе-
сины в сочетании канифольно-экстракционных производств
с производством древесноволокнистых плит.
3. Комплексное использование смолистой стволовой древе-
сины путем соединения канифольно-экстракционных . произ-
водств с гидролизно-дрожжевыми и гидролизно-спиртовыми за-
водами.
4. Комплексное использование смолистой стволовой древе-
сины в сочетании канифольно-экстракционных производств
с производством высококачественного древесного угля и его
производных, а также целого ряда ценных для народного хо-
зяйства жидких продуктов пиролиза древесины (уксусной кис-
лоты, понизителей вязкости, ингибиторов, консерваторов древе-
сины, коптильных препаратов и др.).
§ 8. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА ЭКСТРАКЦИИ
СМОЛИСТЫХ ВЕЩЕСТВ
Общие представления о процессе экстракции. Процесс экс-
тракции в системе твердое тело — жидкость используется в раз-
личных отраслях промышленности: сахарной, при производстве
растительных и эфирных масел, а также дубильных веществ
и др. Процесс экстракции смолистых веществ из просмоленной
древесины широко применяется в лесохимической промышлен-
ности. И хотя уже продолжительное время ведется большой
объем исследований по изучению закономерностей процесса экс-
тракции, однако вполне обоснованной теории этого процесса
пока не получено. Экстракция смолистых веществ из осмоль-
ной древесины представляет собой многостадийный процесс.
Основные стадии, определяющие скорость экстракции смоли-
стых веществ из осмольной щепы, следующие:
1. На первой стадии на поверхности раздела фаз, в частно-
сти твердое тело — жидкость, всегда существует ламинарная
пленка, или диффузионный слой, перемещение веществ через ко-
торый к поверхности твердого тела в основном осуществляется
только за счет молекулярной диффузии. Толщина этого слоя и
возможность ускоренного перемещения веществ зависят от гид-
родинамического режима движения жидкой фазы в слое твер-
дого тела (щепы). Таким образом, на этой стадии определяю-
щей будет конвективная диффузия, являющаяся функцией гид-
родинамического состояния системы (рис. 14).
2. Стадия проникновения растворителя по капиллярам, смо-
ляным ходам и порам во внутренние части щепы. Эта стадия
определяется молекулярной диффузией и будет зависеть от при-
роды растворителя (гидрофобный он или гидрофильный),
а также структуры древесины (степени спелости осмола) и ка-
чества подготовки сырья.
60
3. Стадия растворения смолистых веществ в растворителе.
Скорость этой стадии будет зависеть от состояния смолистых
веществ и содержания их в древесине. Смолистые вещества
могут быть в значительной степени окислены или заполимеризо-
ваны. Скорость растворения будет зависеть и от природы раст-
ворителя (при использовании бензина, главным образом, от тем-
пературы его кипения) и от его объема, находящегося в порах.
Рис. 14. Схема массообмена в
цессе экстракции:
/ — щепа; 2 — ламинарная пленка,
диффузионный подслой; 3 — объем
творителя
4. Стадия вывода раствора смолистых веществ в раствори-
теле из капилляров, пор и смоляных ходов к внешней поверх-
ности щепы. Эта стадия определяется молекулярной диффузией,
и скорость ее будет зависеть от концентрации раствора смоли-
стых внутри щепы и концентрации их на границе раздела фаз,
температуры процесса, а также от размеров щепы, т. е. будет
определяться градиентом концентраций.
5. Стадия отвода растворенных смолистых веществ от по-
верхности щепы через ламинарную пленку, или диффузионный
слой, в объем. На этой стадии скорость процесса также будет
зависеть от гидродинамической обстановки в системе и опреде-
ляться конвективной диффузией.
В общем виде процесс экстракции смолистых веществ из
щепы может быть представлен основным уравнением массопе-
редачи
М = ШСсрт, (28)
где М — масса извлекаемых смолистых веществ, кг; т — продол-
жительность процесса экстракции, с; К — коэффициент массо-
передачи;
Роб В р Кр. п Рраст Кем Нем Рем
здесь Роб — коэффициент, характеризующий перенос раствори-
теля из объема к ламинарной пленке, или диффузионному слою,
м/с; б— толщина диффузионного слоя, м; Dp — коэффициент
диффузии растворителя через диффузионный слой, м2/с; Кр. п—>
коэффициент массопроводности растворителя в порах; объем
растворителя, поступающего в поры через единицу сечения пор
61
в единицу времени, м3/(м2-с); рраст — константа скорости раст-
ворения смолистых веществ в растворителе, представляющая
объем растворенных смолистых веществ с единицы поверхности
за единицу времени, м3/(м2-с); Ксм — коэффициент массопро-
водности раствора смолистых в порах; jDcm — коэффициент диф-
фузии смолистых через диффузионный слой, м2/с; рсм— коэф-
фициент, характеризующий перенос смолистых веществ в объем,
м/с; F — общая площадь поверхности смолистых веществ в дре-
весине, м2; Сср — средняя разность концентраций смолистых ве-
ществ в щепе и растворе, кг/м3.
Q _____ £*П
Ср- 2,31g (Сп/Соб) ’
(30)
здесь Сп — концентрация смолистых веществ в порах щепы,
кг/м3; Соб — концентрация смолистых в объеме раствора, кг/м3.
Приближенно процесс экстракции смолистых из осмольной
щепы может быть описан и экспериментальным законом Фика:
— —-- т, (31)
д/
где D — коэффициент молекулярной диффузии смолистых ве-
ществ в растворе бензина из древесины, м2/с;
ДС-СВ-СГ, (32)
где Св — концентрация смолистых веществ в растворе на по-
верхности растворения, кг/м3; Сг — концентрация смолистых ве-
ществ в растворе на границе выхода из поры, кг/м3; А/ — длина
поры (принимается половина длины щепы, так как экстракция
идет в основном в двух направлениях), м.
Анализ уравнений (28) и (31) дает основание считать, что
процесс экстракции смолистых веществ из древесины очень сло-
жен и определяющей стадией является молекулярная диффузия
в древесине растворителя и раствора смолистых веществ.
Было установлено, что коэффициент молекулярной диффузии
при 0 °C Z>o=O,5* 10-5 м2/с, а характер его изменения в зависи-
мости от температуры определяется уравнением
£>/=£)о_М£+2!11, (33)
где Dt — коэффициент диффузии в бензине раствора смолистых
веществ, полученных из древесины, при любой температуре,
м2/с; |л0, ц/ — вязкость раствора смолистых веществ в бензине
при 0 °C и любой температуре, Па-с; t — температура проведе-
ния процесса, °C; То=273 °C.
Из этого уравнения видно, что если экстракцию вести при
/=60 °C, .D6o=l,15-10"5 м2/с, а при /=90 °C П90=1,75- 1Q-5 м2/с.
Исследованиями ЦНИЛХИ и ЛТА было показано, что коэф-
фициент массопереноса смолистых веществ, растворенных в бен-
зине, практически не зависит от начальной и конечной влажно-
62
7. ЗАВИСИМОСТЬ КОЭФФИЦИЕНТА МАССОПРОВОДНОСТИ ОТ ЭКВИВАЛЕНТНОГО
ДИАМЕТРА ЩЕПЫ
Диаметр щепы, м-103 Коэффициент массопроводности
для бензина БР-1 для бензина БЛХ
9,1 4,74-1О~8-/-0’74 17,0-10—8-/—0,83
4,4 0,95- 10-8-f0’66 1,7-10“ &-t~ °’73
2,3 6,49-10—8-/—1,05 3,4-10“9t~ °’60
сти щепы и скорости ее сушки, но в значительной мере зависит
от размеров щепы (табл. 7) и продолжительности процесса
экстракции и может быть определен по формуле
Кт=аА (34)
где а — опытный коэффициент; t — температура процесса, °C;
п — показатель степени зависимости от температуры.
Хотя процесс экстракции смолистых веществ из древесины
изучен недостаточно, для практического руководства управле-
нием процесса могут быть использованы основные факторы,
влияющие на скорость и эффективность этого процесса.
Основные факторы, определяющие скорость процесса экс-
тракции. 1. Вид, способ и степень измельчения осмола оказы-
вают существенное влияние на скорость процесса экстракции
и коэффициент извлечения смолистых веществ из древесины.
Характерной особенностью древесины как капиллярно-пори-
стого тела является ее неоднородность и анизотропность в про-
дольном, тангенциальном и радиальном направлениях. Так, че-
рез площади поперечного сечения перенос массы смолистых
веществ в 8—9 раз больше, чем через единицу площади боко-
вой поверхности. Скорость извлечения канифоли из торцовой
поверхности в 5 раз больше, чем через тангенциальную поверх-
ность, и в 16 раз больше, чем через радиальную.
Используемые в настоящее время на наших канифольно-
экстракционных заводах способы подготовки технологической
щепы влияют на ее фракционный состав (табл. 8).
8. ФРАКЦИОННЫЙ СОСТАВ ТЕХНОЛОГИЧЕСКОЙ ЩЕПЫ
Показатель Фракция щепы, мм Показатель Фракция щепы, мм
2-5 5-10 10-15 2—5 5-10 10-15
Доля В ИСХОДНОЙ 24 57 19 Толщина, мм 0,9 2,0 4,4
массе щепы, % Длина, мм Ширина, мм 8,7 2,1 18,3 3,9 25,4 7,2 Эквивалентный размер щепы, мм 2,3 4,4 9,1
63
Естественно, чём мельче 1цёпа, тем больше её поверхность.
Из закона Фика [см. формулу (31)} следует, что чем больше
поверхность, тем быстрее идет процесс экстракции.
Однако приготовление мелкой щепы удорожает операцию
подготовки осмола к экстракции. Кроме того, большое содер-
жание мелкой фракции ухудшает гидродинамику растворителя
в слое за счет его уплотнения и снижения пористости, что»
в конечном результате приводит к снижению коэффициента
извлечения смолистых из древесины.
Большая масса мелкой фракции щепы и пыли приводит к за-
сорению коммуникаций движения мицеллы и образованию на-
кипи в теплообменных аппаратах. Размеры щепы определяют
дальнейшее ее использование после экстракции (производство
древесностружечных плит, картона и др.).
Для канифольно-экстракционных заводов характерен состав
щепы, приведенный в табл. 8. С целью улучшения условий
проникновения растворителя в щепу и раствора смолистых ве-
ществ обратно ее подвергают деформированию на гладковал-
ковых дробилках. Эта операция позволяет в значительной сте-
пени изменить внутреннюю структуру щепы, вследствие чего
скорость процесса экстракции значительно возрастает. Так, при
экстракции обычной технологической щепы (влажность .14 %)
продолжительность процесса составляет 7 ч при среднем ко-
эффициенте извлечения 72,7—77,6 %, а после деформирования
при продолжительности процесса 2—3 ч коэффициент извлече-
ния смолистых из древесины составляет 84—92 % •
2. Влажность древесины является важным фактором, опре-
деляющим скорость экстракции. Так как извлечение смолистых
веществ из осмольной древесины ведут бензином, который явля-
ется гидрофобным растворителем, то наличие влаги в древе-
сине препятствует его проникновению внутрь ее куска. Поэтому
на всех канифольно-экстракционных заводах перед началом
процесса экстракции осуществляется предварительная подсушка
древесины.
Исследованиями, выполненными в ЦНИЛХИ и ЛТА,
было установлено, что оптимальная влажность осмольной щепы
14—15%. Более высокая влажность препятствует проникнове-
нию бензина внутрь куска щепы, а более низкая влажность
приводит к усыханию кусков щепы, что ведет к уменьшению
диаметра пор и капилляров и ухудшает проникновение раство-
рителя внутрь куска. Сушка щепы осуществляется непосред-
ственно в экстракторе либо с помощью паров бензина из ис-
парителя, либо парами из испарительных колонн.
3. Температура процесса экстракции. С повышением темпе-
ратуры уменьшается вязкость растворителя, увеличивается ко-
эффициент молекулярной диффузии, растет константа скорости
растворения смолистых веществ в бензине, что благоприятно
сказывается на увеличении скорости экстракции в целом и по-
вышении коэффициента извлечения смолистых веществ.
64
4. Давление в системе. С повышением давления возрастает
скорость пропитки щепы растворителем, но затрудняется об-
ратный выход раствора смолистых веществ из щепы. Поэтому
давление в системе имеет косвенное положительное значение,
так как позволяет повысить температуру растворителя и про-
цесса в целом. Но повышение давления в системе требует вы-
сокой герметичности оборудования и коммуникаций и всегда
увеличивает пожарную опасность производства и загрязнен-
ность рабочих помещений.
5. Содержание смолистых веществ в осмоле. Движущей си-
лой всех диффузионных процессов является разность между ра-
бочей и равновесной концентрацией. Так как при любом спо-
собе экстракции растворитель на выходе из экстрактора не
может иметь нулевую концентрацию по смолистым, то в щепе
всегда будет остаточная равновесная смолистость. Практика
эксплуатации канифольно-экстракционных заводов показывает,
что конечная концентрация смолистых веществ в проэкстраги-
рованной щепе составляет 2,5—5,5%. Поскольку коэффициент
извлечения
Кизв = Сн^Ск 100%, (35)
то чем больше Сн (начальная концентрация смолистых в щепе)
при одинаковой Ск (конечная концентрация смолистых в отра-
ботанной щепе), тем больше будет Кизв-
6. Гидродинамические условия. При извлечении смолистых
веществ на стадии подвода растворителя из объема к поверх-
ности щепы и на стадии отвода раствора смолистых от поверх-
ности щепы в объем важным фактором является гидродинами-
ческая обстановка в слое щепы. Поскольку основным сопро-
тивлением скорости процесса является массоперенос внутри
куска щепы, режим движения растворителя оказывает влияние
на скорость процесса только в определенных пределах. Опти-
мальной скоростью движения растворителя в слое щепы в пе-
ресчете на полное сечение экстрактора является величина
0,1 м/с. Дальнейшее увеличение скорости движения раствори-
теля в слое щепы не ведет к повышению скорости процесса.
Наоборот, в зависимости от способа экстракции это приводит
либо к увеличению расхода энергии (батарейно-противоточный
способ экстракции), либо к увеличению расхода растворителя
и тепла (батарейно-дефлегмационный способ экстракции).
Очень важной является организация слоя щепы в экстракторе,
так как от этого зависит равномерность омывания щепы рас-
творителем (особенно это важно при батарейно-дефлегмацион-
ном способе экстракции).
7. Время процесса экстракции. Между продолжительностью
процесса и полнотой экстракции веществ существует прямая за-
висимость. Однако, как видно на рис. 15, явно выраженный
асимптотический характер кривых, характеризующий скорость
3 Заказ № 2193 65
извлечения смолистых веществ, дает основание считать, что
продолжительность технологического процесса целесообразна
не более 6 ч. Более. длительный процесс всегда будет связан
с дополнительными расходами тёпла на нагрев растворителя
и амортизацию оборудования. Но в силу плохой организации
технологических процессов на предприятиях, особенно на тех,
которые работают по батарейно-дефлегмационному способу
Рис. 15. Зависимость извлечения смо-
листых веществ во времени:
2 —влажность древесины 14 %; 2 — влаж-
ность древесины 20 %; 3 — влажность дре-
весины свыше 20 %
экстракции, продолжительность процесса экстракции составляет
иногда более 15 ч.
8. Метод экстракции. Существуют два принципиально раз-
личных способа экстракции: в слое растворителя, или батарей-
но-противоточный способ экстракции, и экстракция ороситель-
ная, или батарейно-дефлегмационный способ экстракции.
При экстракции в слое жидкости значение плотности рас-
твора смолистых веществ внутри куска и в объеме близки
между собой. Гравитационные силы не оказывают существен-
ного влияния на процесс массопереноса. При оросительном спо-
собе экстракции происходит как бы фильтрация растворителя
в направлении сил тяжести, поэтому силы гравитации имеют
существенное значение в массопереносе, причем действие сил
тяжести тел заметнее при больших размерах щецы.
9. Число экстракторов в батарее. При батарейно-дефлегма-
ционном способе экстракции число экстракторов в батарее не
влияет на полноту извлечения смолистых веществ, так как
каждый экстрактор при этом способе работает самостоятельно,
и в каждом из них может быть осуществлен свой режим про-;
цесса. Связь с батареей при этом методе необходима лишь по
условиям рационального использования теплообменных аппара-
тов (конденсаторов-холодильников, флорентин, испарителей и
подогревателей) и расхода тепла и воды.
При батарейно-противоточном методе экстракции каждый
экстрактор следует рассматривать как аппарат, приближаю-
щийся к аппарату идеального смешения. Теоретически при на-
личии трех таких аппаратов достигается достаточно полное из-
влечение веществ, а при пяти-семи аппаратах в батарее система
приближается к непрерывному процессу в аппарате идеального
66
вытеснения. Поэтому можно считать, что в экстракции должно
участвовать не более пяти экстракторов. Опыт канифольно-экс-
тракционных производств при создании батареи из 12—18
экстракторов полностью подтверждает это.
10. Природа растворителя. Природа применяемого раство-
рителя существенным образом влияет на скорость и полноту
процесса экстракции. Важными физико-химическими свой-
ствами растворителя являются его растворяющая способность,
поверхностно-активные свойства, вязкость, температура кипе-
ния и молекулярная масса.
Из наиболее распространенных растворителей самой высо-
кой экстрагирующей способностью обладает диэтиловый эфир.
Если его экстрагирующую эффективность принять за единицу,
то сравнительная активность других наиболее доступных рас-
творителей такова: дихлорэтана 0,62—0,64; бензола 0,58—0,64;
ксилола 0,52—0,55; скипидара 0,46; бензина БР-1 0,30—0,32
в долях от диэтилового эфира.
В производственных условиях растворитель должен отвечать
целому ряду условий; быть недефицитным, малотоксичным, об-
ладать пониженной взрыво- и пожароопасностью, стабильностью
к химическим и температурным воздействиям, быть недорогим,
не склонным вступать в реакции с продуктами экстракции и
легко регенерируемым; поэтому в нашей стране и за рубежом
в качестве растворителя используются различные фракции бен-,
зина.
Длительное время на наших канифольно-экстракционных
заводах для резиновой промышленности использовались бензи-
новые растворители марки БР-1 или БР-2; плотность их дол-
жна быть не более 730 кг/м3, температура кипения 80—НО °C.
Так как температура процесса экстракции является одним из
важных факторов, определяющих скорость процесса экстракции
и полноту извлечения смолистых веществ, то в настоящее время
все заводы переведены на использование в качестве раствори-
теля бензина марки БЛХ — бензина лесохимического с темпе-
ратурными пределами кипения 105—130 °C.
При применении растворителя БЛХ содержание смолистых
веществ в отработанной щепе снизилось примерно на 1 %, ко-
эффициент извлечения их увеличился соответственно на 4,5 % -
Выход канифоли из 1 т осмола 20 %-ной влажности повысился
на 15 кг, а расход бензина на 1 т канифоли уменьшился на
102 кг. Кроме того, увеличилась скорость сушки, соотношение
вода — бензин в дистилляте паров сушки уменьшилось до 1:7
против 1: 15 в случае применения БР-1 или БР-2, так как тем-
пература кипения азеотропной смеси вода — бензин повыша-
ется с 70 до 95 °C, за счет чего получается дополнительная эко-
номия тепла на этом этапе процесса экстракции.
Математическое описание процесса. Четко сформулированной
теории процесса экстракции в системе твердое тело — жидкость
пока нет. Некоторые авторы предложили математическое опи-
3* 67
сание процесса экстракции смолистых веществ из осмольной
щепы, но эти описания, как правило, носят приближенный ха-
рактер и не всегда достаточно полно отражают состояние про-
цесса экстракции. В последнее время работниками ЦНИЛХИ
и ЛТА сделана попытка математического описания процесса
экстракции с учетом основных факторов, влияющих на него.
Дифференциальное уравнение одномерной молекулярной
диффузии для неустановившегося процесса массообмена, когда
скоростью взаимного перемещения фаз можно пренебречь,
имеет вид
дС D д*С
ди дХ2 ’
(36)
где D — коэффициент молекулярной диффузии смолистых ве-
ществ внутри куска щепы, м2/с.
Левая часть уравнения отражает нестационарность процесса,
а правая — распределение концентрации экстрагируемого веще-
ства в твердом теле в результате молекулярной диффузии.
Для решения уравнения принимаются начальные и гранич-
ные условия. Начальные условия допускают, что смолистые ве-
щества в момент начала экстракции распределены равномерно
внутри куска щепы, т. е.
Схо = Со,
где Со — начальная концентрация смолистых в порах.
Граничными принимаются условия третьего рода
-^-^«^(Сп-СД (37)
где К—коэффициент массопередачи; Сп, С\ — концентрации
растворенных смолистых веществ соответственно на поверхности
щепы и в объеме растворителя.
Осмольная щепа — полидисперсный материал. Отдельные ее
частицы по форме приближаются к параллелепипеду. В соот-
ветствии с основными геометрическими размерами и с учетом
разных скоростей в торцовом и боковых направлениях форма
частиц щепы приведена по общепринятому методу к шарообраз-
ной с эквивалентным радиусом по уравнению
1/Ri = lfRl+1/(8/?2)2 + 1/(8Я3)2, (38)
где Ri — эквивалентный приведенный размер i-й фракции щепы,
м; Ri — полудлина щепы вдоль волокон, м; Rz, R3—.полураз-
меры щепы по толщине и ширине, м.
Процесс экстракции смолистых веществ из древесины пока
недостаточно изучен. Наиболее приемлемым решением является
преобразование методом теории подобия уравнения (36) крае-
68
вых условий (37). Получаем обобщенное уравнение, описываю-
щее кинетику извлечения смолистых веществ:
X/Xo=f(F'o, Bh ₽), (39)
где X — средняя массовая доля смолистых веществ внутри щепы
в момент времени т; XQ— массовая доля смолистых веществ
внутри щепы при т=0; Fq^Dt/Rz — диффузионный критерий
Фурье, учитывающий, в течение какого времени установится
процесс; 1\ = KRID— диффузионный критерий Био, характе-
ризующий перенос вещества на границе раздела фаз; £ — пара-
метрическая переменная, определяющая отношение массы
жидкой фазы к массе твердого тела (гидромодуль).
При экстракции смолистых веществ гидрофобным раствори-
телем отрицательное действие на скорость процесса оказывает
влажность древесины. Поэтому для учета этого фактора в урав-
нение необходимо ввести параметрическую величину — безраз-
мерную влажность Wc.
Wc=WjWon, (40)
где IFo — влажность щепы при т=0; ТГ0П— оптимальная влаж-
ность щепы 14—15%.
Так как процесс экстракции целесообразно вести при опти-
мальной влажности, то его можно разделить на два этапа:
экстракцию, совмещенную с сушкой щепы, и экстракцию без
сушки. Тогда для первого периода обобщенное уравнение при-
мет вид:
«>
а для второго периода, поскольку процесс будет идти при оп-
тимальной влажности, т. е. Wo=jj7On=l и е = 0, уравнение'
примет вид:
=о2(-^У'(-~Ур*- (42>
Ao \ А2 Z \ 1^2 Z
Средние значения коэффициентов ai, а2 и соответствующих
показателей степеней первого и второго периодов для бензина
БЛХ следующие: ai=2,5; Z>i=0,3; Ci=—0,5; di=0,35; e=0,2;
02=6,5; 62=—0,2; c2 =—0,6; </2=0,8;
§ 9. ТЕХНОЛОГИЯ ПЕРЕРАБОТКИ СМОЛИСТОЙ
ДРЕВЕСИНЫ
Склад сырья канифольно-экстракционных заводов. Осмол и
другая смолистая древесина поставляются на канифольно-экс-
тракционные заводы железнодорожным, водным и автомобиль-
ным транспортом. Для бесперебойной работы завода необхо-
дим 3—6-месячный запас сырья.
69
Осмол, поступающий на склад сырья, принимают по массе,
взвешивание производится непосредственно в вагонах и авто-
мобилях. После выгрузки, определяют влажность, содержание
канифоли и пересчитывают массу осмола на осмол 20 %-ной
влажности. Хранится осмол на складе сырья в кучах. Макси-
мально допустимые размеры куч, м: длина 120; ширина 40,
высота 12, что соответствует объему одной кучи 35 тыс скл. м3.
Между кучами должны быть противопожарные проходы не ме-
нее 25 м. Проходы между кучами и производствами категорий
пожарной опасности А и Б должны составлять не менее 120 м,
а между производствами категорий В и Г — не менее 70 м.
Кучевое хранение осмола и использование козловых кранов
типа ККУ-7,5 с грейферным захватом, для его внутризаводской
транспортировки позволяет значительно сократить площади
склада сырья и механизировать -трудоемкие работы.
Склад сырья обычно оборудуется железнодорожными пу-
тями, по которым осмол для переработки транспортируют на
специальных вагонетках-платформах или с помощью скребко-
вых и ленточных конвейеров. При значительном удалении
склада осмол можно перевозить в отделение измельчения с по-
мощью автосамосвалов.
Подготовка осмола к экстракции. Сильно загрязненный
грунтом осмол перед измельчением целесообразно промывать
водой под высоким давлением, а крупные куски распиливать
с помощью бензо- или электропил или раскалывать механиче-
скими или гидравлическими колунами.
Измельчают осмол с помощью дисковых рубительных машин
ДР-28, производительность которых до 15 скл. м3/ч, МРН-100
производительностью до 75 м3/ч и барабанных рубительных
машин польской фирмы «Цекоп» производительностью до
12 м3/ч. Дисковые и барабанные рубительные машины оснаща-
ются электродвигателями мощностью соответственно 130, 500 и
40, 160 кВт.
Диффузия растворителя и мицеллы значительно лучше про-
ходит через торцевую поверхность, поэтому при измельчении
стараются обеспечить максимальную величину этой поверхно-
сти. Получающаяся при измельчении и доизмельчении осмола
древесная и минеральная пыль отделяется на сортировках от
щепы и удаляется при помощи аспирационных систем в специ-
альные бункера, из которых вывозится в отвалы. Осмольная
пыль является взрывоопасной, поэтому все технологическое
оборудование аспирационных систем должно быть взрывобез-
опасным,.
Отходы при измельчении составляют 5—8 % от массы ос-
мола. Технологическая щепа после измельчения, сортировки и
доизмельчения обычно имеет состав по длине волокна от 25 до
3 М1^ и по толщине от 1,5 до 3 мм. При этом щепа размером
до 15 мм по длине волокна должна составлять не менее 80 %
от общей ее массы. Чем меньше щепа, тем больше ее соприкос-
70
йовение с растворителем. Однако большое содержание пыли и
мелкой щепы уплотняет слой, затрудняет равномерное проник-
новение растворителя, забивает коммуникации батарей.
На канифольно-экстракционных заводах СССР существуют
три технологические схемы измельчения осмола. По одной из
них работает Вахтанский КЭЗ (рис. 16). Из вагонетки 1 осмол
вручную загружается в рубильную машину ДР-28 2. Полу-
Рис. 16. Технологическая схема измельчительного отделения Вахтанского КЭЗ:
1 — вагонетка; 2 — рубительная машина; 3, 12 — циклоны; 4 — молотковая дробилка;
5, 7, 15 — ленточные конвейеры; 6 — вентилятор; 8 — загрузочная воронка; 9 — сорти-
ровка; 10 — пневмопровод; 11 — дробилка ДГВ-100; 13 — ковшовый элеватор; 14 — бун-
кер; 15 — ленточный конвейер
чающаяся щепа под действием центробежной силы диска вы-
брасывается в циклон 3, где она частично отделяется от пыли и
поступает на ленточный конвейер 7. По конвейеру щепа направ-
ляется в сортировку 9, где разделяется на крупную технологиче-
скую мелочь и пыль. Мелкая фракция щепы и пыль являются
отходом производства, поэтому из циклона 3 они выводятся
в отвал. Крупная щепа с верхнего сита сортировки поступает
на дополнительное измельчение в молотковую дробилку
МРН-100 4, смешивается с технологической щепой с нижнего
сита сортировки 9 на ленточном конвейере 5 и сбрасывается
в загрузочную воронку 8. Потоком воздуха, который создается
вентилятором 6, она по пневмопроводу 10 подается в циклон 12,
а из него в дробилку ДГВ-1000 11,в которой деформируется,
снова поступает на транспортер 5 и сбрасывается в ковшовый
элеватор 13 для загрузки в экстракторы через бункер 14 и лен-
точный конвейер 15. Такая схема проста в аппаратурном ис-
полнении, но не обеспечивает удовлетворительного фракцион-
ного состава технологической щепы.
Вторая схема измельчения осмола, применяемая на Нейво-
Рудянском ЛХЗ, показана на рис. 17. Из вагонетки 1 осмол
вручную загружается в патрон рубительной машины ДР-28 2,
из нее образующаяся щепа выбрасывается в циклон 3 и ков-
шовым элеватором 5 направляется на сортировку 6. Крупная
71
щепа с верхнего сита сортировки поступает на доизмельчение
в молотковую дробилку МРН-100 4 и затем вновь на сорта-,
ровку. Технологическая щепа с нижнего сита сортировки 6 че-
рез бункер 7 и питатель 8 подается в ковшовый элеватор 9,
а затем на ленточный конвейер 10 для загрузки в экстракторы.
Мелкая щепа и пыль, проходящие через нижнее сито, направ-
ляются в отвал.
Рис. 17. Технологическая схема измельчительного отделения Нейво-Рудян-
ского ЛХЗ:
1— вагонетка; 2 — рубительная машина; 3 — циклон; 4 — молотковая дробилка; 5, 9 —
ковшовые элеваторы; 6 — сортировка; 7 — бункер; 8 —- питатель; 10 — ленточный кон-
вейер
Рис. 18. Технологическая схема измельчительного отделения Решетинского
КЗ, КЭЗ Братского ЛПК, Медвежьегорского, Лесосибирского и Зиминского
КЭЗ:
1 — куча; 2 — пластинчатый питатель; 3 — рубительная машина; 4, 7 — молотковые дро-
билки; 5, 9 — ковшовые элеваторы; 6 — сито сортировки; 8, 10, 11, 13 — конвейеры;
12 — бункер
72
По этой схеме получается более однородный фракционный
состав технологической щепы, но производительность сорти-
ровки невелика.
Третья схема измельчения осмола (рис. 18) применяется на
современных канифольно-экстракционных заводах: Медвежье-
горском, Зиминском и др. В ней используется оборудование
фирмы «Цекоп». По этой схеме осмол со склада сырья подво-
зится на автосамосвалах и сбрасывается на площадку измель-
чительного отделения в кучи 1 перед пластинчатыми питате-
лями 2, с помощью которых осмол направляется в рубительные
машины 3. Полученная щепа из рубительной машины посту-
пает на доизмельчение в молотковую дробилку 4. Затем ков-
шовым элеватором 5 щепа подается на верхнее сито сорти-
ровки 6, где разделяется на три фракции: крупную, технологи-
ческую и мелкую. Мелкая щепа вместе с пылью выводится,
в отвал. Крупная щепа с верхнего сита сортировки подается
на доизмельчение в молотковую дробилку 7, после чего по об-
щему конвейеру 8, на который поступает щепа со всех техноло-
гических потоков, направляется в ковшовый элеватор 9,
а с него сбрасывается на ленточный конвейер 10. С него по
мере необходимости она может подаваться на загрузку в экс-
тракторы с помощью ленточного конвейера 11 или в бункер
запаса сырья 12, из которого с помощью ленточного конвейера
13 она может направляться на экстракционную батарею.
Методы экстракции. В настоящее время на лесохимических
предприятиях СССР для экстракции смолистых веществ из
щепы смолистой древесины используются методы: батарейно-
противоточный, батарейно-дефлегмационный с местным упари-
ванием растворов и батарейно-дефлегмационный с централизо-
ванным упариванием растворов. Предполагается введение
в эксплуатацию на одном из лесохимических заводов непрерыв-
ного способа экстракции.
Батарейно-противоточный метод экстрак-
ции. Экстракционные батареи, работающие по противоточ-
ному методу, состоят из шести экстракторов, при этом на про-
тивоточной экстракции находятся следующие экстракторы: на
отдувке растворителя, на разгрузке отработанной щепы и за-
грузке свежей щепы. Экстрактор, в котором находится щепа
с самым низким содержанием смолистых, предназначен для
отключения от батареи. Он получил название хвостового.
А экстрактор, который после сушки щепы подключается к ба-
тарее со свежей щепой, называется головным. Промежуток
времени, через который происходит подключение очередного
головного экстрактора и отключение от батареи хвостового, на-
зывается рабочим периодом. При рабочем периоде 1 ч 20 мин.
оборот экстрактора в среднем продолжается 8 ч; экстракция
идет 5 ч 20 мин.
Подача свежего растворителя в каждый следующий хво-
стовой экстрактор и отбор раствора смолистых веществ (мис-
73
целл) из каждого последующего головного экстрактора в ба-
тарее происходит непрерывно, а отключение и подключение
экстракторов к батарее периодически. В целом такой процесс
называется полунепрерывным.
Технологическая схема этого способа экстракции показана
на рис. 19. Щепа из измельчительного отделения ленточным/
конвейером 1 подается в экстрактор 2. Через подогреватель,
В канализацию
Рис. 19. Технологическая схема экстракционной батареи КЭЗ Нейво-Рудян-
ского ЛХЗ:
/ — конвейер; 2 — экстракторы; 3, 4 — подогреватели; 5, 7 — конденсатор-холодильник;
6 — сепаратор; 8 — приемник; 9, /3 —насосы; 10, /2 — сборники; // — флорентина; 14 —
бункер; 15 — ленточный конвейер
Рис. 20. Технологическая схема экстракционной батареи Вахтанского КЭЗ:
1, 2, 3, 4 — конденсаторы-холодильники; 5, 6 — подогреватели; 7 — экстракторы; 8 —
ленточный конвейер; 9 — испарители; 10 — фильтр; 11, 16, 19 — сборники; 12, 15, 18 —
насосы; 13, 17, 20 — сборники-флорентины; 14 — скребковый конвейер
74
3 в нижнюю часть загруженного щепой экстрактора 2 острым
паром пережимается мисцелла из экстрактора, отключаемого
от батареи для отдувки растворителя из отработанной щепы,
разгрузки его и загрузки свежей щепой. В течение одного ра-
бочего периода слабая мисцелла кипятится в экстракторе, по-
догревая и подсушивая щепу. Пары растворителя и воды (пары
сушки) отбираются из верхней части экстрактора и через се-
паратор 6 направляются в конденсатор-холодильник 7. Пары
конденсируются, растворитель отделяется от воды и направля-
ется в сборник-флорентину 11. Здесь происходит дополнитель-
ное отделение воды от растворителя. Растворитель перелива-
ется в сборник 12 оборотного растворителя, а вода сбрасыва-
ется в канализацию. Растворитель из сборника 12 с помощью
насоса 13 через подогреватели 4 и 3 подается в нижнюю часть
хвостового экстрактора. Проходя сквозь слой щепы, раствори-
тель извлекает смолистые вещества и из верхней части хво-
стового экстрактора через подогреватель 3 перетекает в ниж-
нюю часть соседнего экстрактора и так далее до головного.
Такой способ поддерживает значительную разность концентра-
ций между смолистыми веществами в щепе и в растворе. Из
верхней части головного экстрактора отбирается мисцелла мас-
совой концентрацией 4—6 % и через сепаратор 6, где отде-
ляется от паров сушки, поступает в приемник 8, работающий
как флорентина.
Во флорентине мисцелла отделяется от воды и мелкого сора
и сливается в сборник 10, из которого насосом 9 направляется
на переработку. Пары из сепаратора 6 идут в конденсатор-хо-
лодильник 7. Температура в экстракторах поддерживается за
счет подогревателей 3. Гидравлическое сопротивление, возни-
кающее при перекачивании растворителя, достигающее в хвос-
товом экстракторе 350—400 кПа, приводит соответственно к по-
вышению температуры растворителя.
Таким образом, по истечении рабочего периода экстракци-
онной батареи осуществляют следующие операции: а) прекра-
щают отдувку растворителя из отработанной щепы из отклю-
ченного на разгрузку и загрузку очередного экстрактора, от-
дувку растворителя ведут острым паром давлением 490—
590 кПа; полноту отдувки растворителя контролируют индика-
тором ИС-4; б) по окончании отдувки отработанная щепа по-
ступает в бункер 14, откуда ленточным конвейером 15 в ко-
тельную на топливо; в) подключают очередной головной
экстрактор после подсушки в нем свежезагруженной щепы и
начинают отбор из него мисцеллы; г) отключают очередной
хвостовой экстрактор и пережимают из него слабую горячую
мисцеллу в экстрактор, со свежей загруженной щепой для под-
сушки; д) подают в экстрактор острый пар для отдувки рас-
творителя из щепы по окончании пережима слабой мисцеллы;
в) начинают подачу растворителя в очередной хвостовой
экстрактор.
75
Достоинства батарегшо-противоточного метода экстракции:
равномерное соприкосновение растворителя в слое щепы; по-
вышенная температура процесса, определяемая гидравлическим
сопротивлением в системе; равномерный состав мисцеллы; пол-
ная взаимозаменяемость экстракторов, что обеспечивает устой-
чивость работы батареи в целом.
Недостатки метода: повышенные требования к герметично-
сти оборудования и запорной аппаратуре; повышенная огне- и
взрывоопасность; сложная коммуникация трубопроводов и воз-
можность забивания их пылью и мелкой щепой; не использу-
ются пары растворителя с испарительных колонн для предва-
рительной сушки свежей щепы.
Батарейно-дефлегмационный метод экстрак-
ции с местным упариванием мисцеллы. Этот метод
применяется на Вахтанском канифольно-экстракционном за-
воде (рис. 20)., Полная схема экстракционной батареи состоит
из. восьми экстракторов 7, каждый из которых имеет испари-
тель 9. Экстракцию смолистых веществ ведут сразу в шести-
семи экстракторах путем постоянного орошения свежим или
оборотным растворителем, подогретым до температуры кипе-
ния в подогревателях 5 и 6 и подаваемым в верхнюю часть
экстрактора через распределительное устройство. Проходя че-
рез слой щепы сверху вниз, растворитель извлекает смолистые
вещества. Образующиеся растворы собираются в нижней ча-
сти экстракторов и поступают в испарители 9, где идет частич-
ное упаривание растворов. При этом пары растворителя под-
нимаются через слой щепы вверх противотоком стекающему
раствору. В этом случае процессы экстракции и сушки идут
параллельно во всех экстракторах. В головной экстрактор по-
дают свежего растворителя меньше, а кипячение растворов
ведут интенсивнее, чем в хвостовом экстракторе. Образуется
избыток растворов в хвостовом экстракторе, и мисцелла пе-
ретекает по коммуникациям к головному экстрактору, посте-
пенно упаривается и через фильтр 10 стекает в сборник-фло-\
рентину 13, где отделяется от воды. Вода сбрасывается в кана-
лизацию, а мисцелла переливается в сборник, откуда насосом
подается в отделение переработки мисцеллы. Массовая, концен-
трация канифоли в мисцелле достигает 10—25 кг/м3. Темпера-
тура процесса автоматически поддерживается близкой к темпе-
ратуре кипения растворителя. Пары сушки из верхней части
экстрактора последовательно поступают в конденсаторы-холо-
дильники 3, 4. Тяжелая фракция растворителя конденсируется
в конденсаторе-холодильнике 4 и стекает в сборник-флорен-
тину 17, где растворитель отделяется от воды и перетекает
в сборник 16, откуда поступает в головной экстрактор, чтобы
обеспечить более высокую температуру экстракции. Низкоки-
пящая фракция растворителя конденсируется в конденсаторе-
холодильнике 3, где происходит частичное отделение раствори-
теля от воды, и поступает в сборник-флорентину 20, дополни-
76
тельно отделяется от воды и поступает в сборник 19, из кото-
рого насосом 18 через подогреватель 5 растворитель подается
на орошение хвостового экстрактора. Вода из конденсатора-хо-
лодильника и сборников-флорентин сбрасывается в канали-
зацию.
По окончании экстракции экстрактор отключают от бата-
реи и ставят на отдувку растворителя от щепы,, разгрузку от-
работанной щепы и загрузку свежей щепы. Отдувку раствори-
теля от щепы ведут острым паром давлением 490—590 кПа,
подавая его в нижнюю часть экстрактора. Пар, проходя через
слой щепы, насыщается парами растворителя и через верхнюю
часть экстрактора направляется в конденсаторы-холодильники
1, .2, в которых пары конденсируются и растворитель разделя-
ется на фракции аналогично парам сушки в конденсаторах-
холодильниках 3, 4. Контроль за полнотой отдувки ведут также
с помощью индикатора ИС-4. Отработанная щепа выгружа-
ется и направляется на энергохимическую переработку.
При таком методе экстракции мисцелла в нижней части
экстрактора не соприкасается со щепой, это дает возможность
поддерживать концентрацию мисцеллы любой величины неза-
висимо от концентрации смолистых веществ в щепе. Однако
при повышении концентрации канифоли в мисцелле растет тем-
пература раствора, и существующая схема не позволяет повы-
сить концентрацию канифоли в мисцелле выше 250 кг/м3.
Достоинства этого метода экстракции: малая зависимость
коэффициента извлечения смолистых веществ от влажности
перерабатываемого сырья, что достигается удалением воды из
системы на всех этапах работы батареи; батарея работает при
атмосферном давлении, снижая взрыво- и пожароопасность,
улучшая гигиену производственных помещений; мисцелла по-
лучается высокой концентрации.
Недостатки метода: каждый экстрактор работает практи-
чески самостоятельно, что затрудняет поддержание устойчивого
режима в каждом из них; трудно обеспечить равномер-
ное орошение слоя щепы растворителем, что требует специ-
альной организации слоя щепы по всему сечению экстракч
тора; отсутствие избыточного давления в системе приводит
к понижению температуры, что снижает скорость экстракции
и коэффициент извлечения смолистых из щепы.
Батарейно-дефлегм ационный метод экстрак-
ции с централизованным упариванием рас-
твора. Этот метод используется на ряде канифольно-экстрак-
ционных заводов. Свежая щепа в головном экстракторе нагре-
вается и сушится за счет паров растворителя, поступающих из
испарительной колонны, получающихся при упаривании мис-
целлы в ней. В течение первого рабочего периода орошение
щепы свежим растворителем не производится, во второй рабо-
чий период сушка осуществляется совместно с частичным оро-
шением, если система хорошо прогрелась. Во всех остальных
77
экстракторах пары растворителя подаются из испарительной
установки только для компенсации тепловых потерь и поддер-
жания температуры процесса экстракции, близкой к темпе-
ратуре кипения растворителя. Одновременно щепа орошается
в пяти-шести экстракторах (три головных и два-три хвосто-
вых), причем в хвостовых — чистым растворителем, в голов-
ных— слабой мисцеллой, получающейся в хвостовых экстрак-
Рис. 21. Технологическая схема экстракционной батареи КЭЗ Братского
ЛПК, Медвежьегорского, Лесосибирского, Зиминского КЭЗ:
1 — буэкер; 2 — пластинчатый питатель; 3, 10, 15 — ленточные конвейеры; 4 — ковшо-
вый элеватор; 5, 7 — конденсаторы-холодильники; 6, 8 — сепараторы; 9 — экстракторы;
11, 12 — подогреватели; 13, 14 — фильтры; 16, 17, 18 — насосы; 19, 21, 23 — сборники;
20, 22, 24 — сборники-флорентины; 25 —флорентина-бензоловушка
торах. В процессе экстракции основные тепловые нагрузки при-
ходятся на головной экстрактор, где необходимо нагреть щепу
до температуры экстракции и удалить до 50 % исходной влаги.
Технологическая схема этого метода экстракции, осуществ-
ленная на Медвежьегорском, Лесосибирском и Зиминском
КЭЗах, рассчитана на 10 экстракторов вместимостью по
37,5 м3 каждый (рис. 21).'Щепа из измельчительного отделе-
ния по ленточному конвейеру 10 с помощью сбрасывающего
устройства загружается в экстрактор 9, который закрывается
и подключается в головное положение к экстракционной бата-
рее. Для нагрева и высушивания щепы в нижнюю часть экс-
трактора 9 подаются пары растворителя, поступающие с ис-
парительно-уваривающей установки, которые, поднимаясь вверх,
частично конденсируются; часть конденсата идет на пропитку
щепы, а другая в виде мисцеллы из нижней части экстрактора
78 . .
стекает в общий коллектор для мисцеллы и вместе с мисцел-
лой из головных экстракторов проходит через фильтр 13, сбор-
ник-флорентину 20 и поступает в сборник 19. Пары раствори-
теля, пройдя через слой щепы, увлекают с собой пары воды,
содержащейся в щепе, и выходят из верхней части экстрактора
в сепаратор 8, тд$ отделяются от капелек воды, конденсата
и пыли.
Конденсат из сепаратора 8 через фильтр 14 направляется
в сборник-флорентину 22, где слабая мисцелла отделяется от
воды и перетекает в сборник 21. Пары из сепаратора 8 конден-
сируются в конденсаторе-холодильнике 7; конденсат идет
в сборник-флорентину 24, там растворитель отделяется от воды
и переходит в сборник 23, а подсушенную щепу в головном
экстракторе начинают орошать оборотной мисцеллой. Оборот-
ная мисцелла из сборника 21 насосом 18 через подогреватель
И, где она подогревается до температуры кипения, направля-
ется в верхнюю часть экстрактора 9. Стекая вниз через слой
щепы, оборотная мисцелла экстрагирует из нее смолистые ве-
щества и из нижней части экстрактора выводится через фильтр
13, где отделяется от мелкой щепы и пыли, и поступает в сбор-
ник-флорентину 20. Там мисцелла отделяется от воды и перете-
кает в сборник 19, из которого насосом 16 перекачивается для
дальнейшей переработки на испарительно-уварительную уста-
новку. Вода из сборников-флорентин 20, 22 и 24 перетекает во
флорентину-бензоловушку 25, дополнительно отстаивается от
растворителя и сбрасывается в канализацию. Растворитель по
мере накопления в бензоловушке 25 откачивается в сборник-
флорентину 24.
Щепа в головных экстракторах орошается оборотной мис-
целлой в течение трех последующих периодов. После отвода
оборотной мисцеллы по вышеописанной схеме в течение трех
следующих рабочих периодов орошение ведут оборотным рас-
творителем. Оборотный растворитель из сборника 23 насосом
17 через подогреватель 12, где нагревается до температуры ки-
пения, подается, как оборотная мисцелла, в верхнюю часть экс-
трактора. Из нижней части экстракторов, которые в это время*
работают как хвостовые, отбирается оборотная мисцелла и
аналогично конденсату, образующемуся при сушке щепы, идет
в сборник 21. Для компенсации тепловых потерь в эти периоды
в экстракторы подаются пары растворителя из испарительной
установки. По окончании экстрагирования экстрактор отклю-
чается от батареи и ставится на отдувку растворителя от от-
работанной щепы, разгрузку ее и загрузку свежей. Процесс
отдувки и его контроль аналогичны вышеизложенным методам
экстракции. Пары отдувки из верхней части экстрактора идут
в сепаратор 6, где отделяются от капелек конденсата. Конден-
сат собирается в. сборник 21, а пары поступают в конденсатор-
холодильник 5. Образующийся конденсат направляется в сбор-
ник 23. Отработанная щепа через нижний люк экстрактора
79
выгружается на ленточный конвейер 15, потом ковшовым элева-
тором 4 сгружается в бункер 1 и поступает на топливо в ко-
тельную.
Рассматриваемый метод имеет такие же достоинства и недо-
статки, что и дефлегмационный метод с местным упариванием
растворов. Однако централизованное упаривание мисцеллы
позволяет более рационально использовать теплообменную ап-
паратуру и экономить тепло.
Рис. 22. Технологическая схема непрерывнодействующей экстракционной уста-
новки НДЭ-100:
1 — ленточный конвейер; 2 — подбункеры; 3 — основной бункер; 4 — элеватор; 5 —
скребковый конвейер; 6 — привод; 7 — винтовой питатель; 8 — экстрактор; 9 — уровне-
мер; 10 — промежуточный бункер; 11 — форсунка; 12 — отдувочный аппарат; 13 — кон-
денсатор; 14 — насос; 15, 25 — сборники; 16 — флорентина; 17 — сборник бензина; 18,
26 — насосы; 19 — подогреватель; 20, 22, 23 — винты; 21 — бензоотделитель; 24 — отстой-
ник мисцеллы; 27 — выпарной аппарат; 28 — фильтр; 29 — решетка
Непрерывные методы экстракции смолистой древесины.
В настоящее время стоит вопрос об организации непрерывных
методов экстракции смолистых веществ из древесины. Непре-
рывные процессы имеют ряд преимуществ перед периодическим
и даже перед полунепрерывными: возможность высокого уровня
механизации трудоемких работ, автоматизация технологических
операций, стабильность получаемой продукции, повышение
культуры труда и др.
Непрерывнодействующий экстрактор, разработанный
ЦНИЛХИ, мощностью 100 тыс. скл. м3 в год предполагается!
установить на одном из лесохимических заводов. Технологиче-
ская схема этого метода показана на рис. 22. Технологическая
щепа с помощью ленточного конвейера 1 поступает в элеватор
80
4, затем по скребковому конвейеру 5 в промежуточный бункер
экстрактора. Избыток щепы со скребкового конвейера попадает
в основной бункер 3, откуда при необходимости щепа вновь по-
дается в элеватор. Промежуточный бункер 10 в целях преду-
преждения слеживания щепы имеет подбункер 2, на котором
установлен электровибратор. Щепа загружается в экстрактор
с помощью винтового питателя 7, который одновременно обес-
печивает герметичность верхней части экстрактора. В верхней
части экстрактора установлен шиберный уровнемер 9, отклю-
чающий винтовой питатель при заполнении экстрактора щепой.
Для распыления мисцеллы, подаваемой на орошение, имеются
форсунки 11.
Щепа, загруженная в экстрактор 8, заполняет аппарат на
всю высоту и движется сверху вниз под действием сил тяжести.
В средней части экстрактора, в зоне отбора мисцеллы, имеется
самоочищающаяся с помощью движущегося слоя щепы фильт-
рующая решетка 29. Проэкстрагированная щепа из верхней
части экстрактора выводится с помощью пяти выгрузочных вин-
тов 22, затем поступает в коллекторный винт 23, который по-
дает ее в бензоотделитель 21. Бензоотделитель представляет
собой герметизированный ковшовый элеватор с- перфорирован-
ными ковшами, в котором в процессе транспортировки в отду-
вочный аппарат 12 отделяется основная масса бензина. Отду-
вочный аппарат — пустотелая емкость, расширяющаяся книзу,
что предупреждает зависание щепы. Выгрузку отработанной
щепы ведут тремя винтами 20.
Процесс экстракции в НДЭ-100 осуществляется в две ста-
дии. В зоне А осмольная щепа сушится парами бензина и оро-
шается слабой мисцеллой, в зоне Б щепа подвергается проти-
воточной экстракции горячим бензином. Бензин, предвари-
тельно нагретый в подогревателе 19, подается на экстракцию
нйсосом 18. Горячий растворитель в зоне Б движется снизу
вверх противотоком к щепе и заполняет экстрактор на 2/з вы"
соты.
Мисцелла с массовой долей канифоли 2—4 % отводится из
средней части экстрактора через фильтрующую решетку и на-
правляется через выносные фильтры 28 в отстойник мисцеллы
24. Отстоявшаяся мисцелла из сборника 25 насосом 26 подается
в выпарной аппарат 27, в котором она упаривается до 20—
35 %-ной концентрации. Пары бензина для подогрева и сушки
щепы направляются в зону А. Упаренная мисцелла поступает
на дальнейшую переработку, часть слабой мисцеллы подается
для орошения свежей щепы в зону А.
Пары сушки из верхней части экстрактора поступают в кон-
денсатор 13 воздушного охлаждения, из которого конденсат
поступает, во флорентину 16. Из флорентины бензин идет
в сборник бензина 17, вода — на дополнительную очистку
в сборник 15, а затем на биоочистку. В отдувочном аппарате
с помощью острого водяного пара растворитель удаляют из
81
щепы. Ввод острого пара в целях снижения его расхода про-;
изводится в двух точках. Основной объем растворителя отго-
няется за счет пара температурой 170—180 °C, подаваемого
в среднюю часть аппарата. В нижнюю часть аппарата подается
пар температурой 130—140 °C, что позволяет снизить темпе-;
ратуру выгружаемой щепы и предупредить ее самовозгорание.
Для снижения потерь паров растворителя система работает
под разрежением 10—100 Па, которое создается сборником
25. Приводы винтовых питателей и выгружателей снабжены
вариаторами, которые позволяют регулировать производитель-
ность экстрактора. НДЭ-100 имеет геометрический объем
136 м3, рабочий 125 м3, продолжительность экстракции 8 ч.
Недостатком непрерывного способа экстракции являются
большие удельные нормы объема оборудования на единицу
перерабатываемого сырья. Так, суммарный объем только экс-
трактора и отдувочного аппарата при использовании НДЭ-100
в 1,5 раза выше, чем при применении батарейно-дефлегмаци-
рнных способов экстракции. Выход из строя хотя бы одного
узла в системе НДЭ-100 приведет к полной остановке всего
производства, чего не может быть при остановке даже несколь-
ких экстракторов в батарейной экстракции.
Сравнительные показатели, достигаемые при различных ме-
тодах экстракции с применением одинаковой массы осмоль-
ной щепы, дают следующие результаты: при батарейно-проти-
воточном методе экстракции выход канифоли из 1 т щепы со-
ставляет в среднем 110—115 кг, а коэффициент извлечения
82—84 %. Батарейно-дефлегмационный метод экстракции с ме-
стным упариванием мисцеллы обеспечивает выход канифоли
100—105 кг/т, а коэффициент ее извлечения составляет 80—'
82 %. В настоящее время -при батарейно-дефлегмационном ме-
тоде экстракции с централизованным упариванием раствора
смолистых веществ выход канифоли составляет 80—100 кг/т,
а коэффициент ее извлечения 77—80 % • Заниженные показа-
тели в этом способе экстракции объясняются не совсем удач-
ной конструкцией экстрактора, в котором затруднено равно-
мерное смачивание щепы растворителем по всему сечению ап-
парата. В последнее время на Зиминском КЭЗе и на других!
предприятиях внесены предложения по более равномерному
формированию слоя щепы в экстракторах, улучшающие эти
показатели; они почти достигли уровня батарейно-противоточ-
ного метода экстракции. Опытными работами, выполненными
в ЦНИЛХИ, показано, что в случае использования НДЭ-100
коэффициент извлечения канифоли составит 87 %.
Переработка мисцеллы. Полученная в процессе экстракции
мисцелла поступает па дальнейшую переработку, в результате
которой осуществляется регенерация растворителя (с массовой
концентрацией в нем скипидара не более 2%), приводящая
к получению темной экстракционной канифоли и скипидара-
сырца. Большое содержание бензина в исходной мисцелле поз-
82
воляет применять ее в качестве флегмы при отгонке раствори-
теля. В настоящее время на канифольно-экстракционных заво-
дах используются непрерывнодействующие технологические
схемы переработки мисцеллы трех видов, различающиеся лишь
незначительными модификациями аппаратурного оформления
процесса.
Принципиальная технологическая схема испарительно-ува-
рительной установки на современных заводах показана на
рис. 23. Мисцелла из экстракционной батареи через фильтр 1
(где она очищается от механических примесей) и подогрева-
тель 2, в котором подогревается до температуры кипения, по-
ступает на среднюю тарелку ректификационной колонны 3,
Мисцелла с парами растворителя, стекая с тарелки на тарелку
в противотоке, теряя легколетучие и обогащаясь тяжелолету-
чими фракциями, по перекидной трубе идет из колонны 3
в нижнюю часть испарителей 5 пленочного типа, в которых ис-
паряется основная масса летучих веществ, и парожидкостная
смесь выбрасывается в сепаратор 4, где происходит разделе-
ние пара и жидкости. Пары растворителя из сепаратора по-
ступают в нижнюю часть ректификационной колонны 6, имекн
щей выносной испаритель 7, благодаря которому обеспечива-
ется дополнительное упаривание мисцеллы. Пары растворителя
из колонны 6 поступают в нижнюю часть колонны 3 и, пройдя
снизу вверх, подаются в экстракционную батарею. Упаренная
мисцелла с массовой долей канифоли 600—700 кг/м3 непре-
рывно отводится из верхней циркуляционной трубы, соединяю-
щей колонну 6 с испарителем 7, и через барометрический за-
твор 8 и подогреватель 9 под действием вакуума поступает
в сепарационную часть канифолеварочной колонны 10, Прин-
цип действия канифолеварочной колонны аналогичен работе
такой колонны при получении канифоли из живицы. Для улуч-
шения процесса отгонки летучих веществ от канифоли в ниж-
нюю часть колонны подается острый водяной пар давлением
980 кПа с температурой 200—220 °C. Кроме того, в канифоле-
варочной колонне создается вакуум порядка 67 кПа, смягчаю-
щий режим уваривания канифоли. Смесь паров воды и лету-
чих веществ (скипидара-сырца) из верхней части колонны
10 идет в конденсатор-холодильник 11, и после разделения
в сборник 14 скипидар-сырец поступает'на переработку. Ува-
ренная канифоль из нижней части колонны 10 при темпера-4
-> туре 165—175 °C поступает в вакуум-приемник 13, а из него
в сборник товарной продукции 14, Оборудование такой испари-
тельно-уварительной установки позволяет перерабатывать 60—
80 м3 мисцеллы в час, а при других технологических схемах —
юлько 7—25 м3. .
Ректификация скипидара-сырца. Производство товарного
экстракционного скипидара и флотомасла осуществляется на
непрерывно-действующей двухколонной ректификационной уста-
новке (рис. 24).
83
Скипидар-сырец с массовой долей бензина 30—40 % (плот-
ностью 790—810 кг/м3) из бака 1 скипидара-сырца через по-
догреватель 2 и регулятор давления 3, а также расходомер </4
поступает в первую ректификационную колонну 7. Колонна ра-
ботает при атмосферном давлении с флегмовым числом 1—2.
Контроль температуры в верхней и нижней части колонны
производится с помощью приборов 5. В процессе работы доля
Рис. 23. Технологическая схема испарительно-уварительной установки КЭЗ
Братского Л ПК, Медвежьегорского, Лесосибирского и Зиминского КЭЗ:
/ — фильтр; 2, 9 — подогреватели; 3, 6, 10 — колонны; 4 — сепаратор; 5, 7 — испарители;
8 — барометрический затвор; 11 — конденсатор-холодильник; 12 — флорентина; 13 — ва-
куум-приемник; 14 — сборник товарной продукции
Рис. 24. Схема процесса ректификации экстракционного скипидара-сырца:
/ — бак; 2 — подогреватель; 3 — регуляторы; 4 — расходомеры; 5 — приборы контроля
температуры; 6 — испарители; 7, 13 — колонны; 8 — уровнемеры; 9 — расходомеры; 10 —
дефлегматоры; // — сборник; /2 —регулятор плотности; 14 — холодильник флотомасла-
сырца; 15 — вакуум-приемник флотомасла-сырца; 16 — индикатор состава паров; 17 —
холодильник товарного скипидара; 18 — вакуум-приемник товарного скипидара; 19 —
сборник товарного скипидара
бензина в скипидаре, отбираемого из нижней части колонны,
не должна превышать 0,5—0,7 %. Отбираемый из верхней ча-
сти колонны бензин с 10 %-ным содержанием терпеновых угле-
водородов направляется в емкость для оборотного раствори-
теля.
Скипидар-сырец, содержащий терпингидрат, растворимость
которого в терпеновых углеводородах незначительна, может
привести к зарастанию коммуникаций кристаллами терпингид-
рата. Во избежание этого необходимо трубопроводы на участке
между сборником промежуточных фракций 11 и второй ректи-
фикационной колонной 13 снабжать паровыми рубашками или
паровыми спутниками.
Во второй ректификационной колонне 13, работающей под
вакуумом (остаточное давление 13 кПа), получают товарный
скипидар из верхней части, а снизу отбирают флотомасло-сы-
рец. Колонна работает с флегмовым числом 2—4. Получаемый
скипидар I сорта имеет плотность 855—864 кг/м3, коэффициент
рефракции 1,467—1,472, начальную температуру кипения не
ниже 162 °C; объем отгона до 175 °C должен составлять не
менее 90%. Массовая доля терпеновых углеводородов во фло-
томасле-сырце не должна превышать 15%.
На некоторых предприятиях организовано разделение то-
варного скипидара на индивидуальные терпены методом пе-
риодической ректификации. Например, выделение пиненовой
фракции а- и р-пинена с чистотой 90—95 % достигается при
однократной ректификации. Для получения дипентена с соот-
ветствующей чистотой необходима 3—4-кратная ректификация.
Это связано как с малым содержанием дипентена в экстрак-
ционном скипидаре (10%), так и с близкой температурой ки-
пения компонентов смеси: у-терпинен 174 °C, дипентен 180 °C.
В процессе вакуум-ректификации флотомасла-сырца в пе-
риодически действующей колонне могут быть отобраны сле-
дующие фракции: скипидар обеспененный — промежуточная
фракция, терпеновые спирты — терпингидратная фракция и се-
сквитерпеновые углеводороды. Первые две фракции отбирают
при остаточном давлении 13 кПа, остальные при 1,3 кПа. Ку-
бовый остаток представляет собой полимеры. Обеспененный
скипидар соединяют с товарным скипидаром, промежуточную
фракцию направляют в вакуум-приемник флотомасла-сырца
15 на повторную переработку. Терпингидратную фракцию по
мере накопления загружают в куб, добавляют 2—3 % фосфор-
ной кислоты и производят вакуум-перегонку. При этом терпин-
гидрат частично дегидратируется до спиртов, частично до мо-
ноциклических терпенов. Как правило, дистиллят содержит до
50 % терпеновых спиртов и его добавляют к флотомаслу.
Химический состав флотомасла весьма сложен. В его состав
входят практически все терпеновые углеводороды и ряд ве-
ществ, химический состав которых еще не установлен. По внеш-
нему виду сосновое флотомасло представляет собой светло-
85
?. ОБЪЁМ И КАЧЕСТВЕННАЯ ХАРАКТЕРИСТИКА СТОЧНЫХ ВОД, ПОЛУЧАЕМЫХ
ПРИ ПЕРЕРАБОТКЕ 1000 Т ОСМОЛА 20 %-НОИ ВЛАЖНОСТИ
Место образования сточных вод Объем, м3 Загрязненность Загрязняющий компонент
по ХПК» мг О/л по БПК, МГ Ог/л
Конденсат паров сушки и отдувки Сточная вода: 120 2 300 1 300 Бензин, органические кислоты
от уваривания кани- фоли 7,5 17 000 12 000 Скипидар, терпинги- драт
от перегонки флото- масла 6,0 29 000 20 000 Терпингидрат, терпено- вые спирты
от вакуум-насосов 30 1 200 600 Бензин, канифольные масла
желтую маслянистую жидкость. Флотирующим началом флото-
масел являются терпеновые спирты. По ОСТ 13253—84 выпу-
скают три сорта флотомасел: СМЭФ-ВС (сосновое масло экс-
тракционное флотационное высшего сорта) с массовой долей
спиртов не менее 75%, кислотным числом 1,0 и плотностью
915—935 кг/м3; СМЭФ-1 (то же первого сорта) с массовой до-
лей не менее 65%, кислотным числом 1,5, плотностью 910—
9,40 кг/м3; СМЭФ-П (то же второго сорта) с массовой долей
спиртов не менее 50 %, кислотным числом 1,5, плотностью
ЭГО—940 кг/м3.
Плотность полимеров 940—1020 кг/м3, кислотное число их
не менее 60, а вязкость при 50°C не более 0,36 Па*с. При об-
работке малеиновым ангидридом из них могут быть получены
продукты, способные заменить канифоль.
§ 10. ОЧИСТКА ПРОМЫШЛЕННЫХ стоков
И ГАЗОВЫХ ВЫБРОСОВ
Основными загрязнениями сточных вод канифольно-экстрак-
ционных производств (табл. 9) являются растворенные в не-
больших массах органические кислоты, эмульгированные и рас-
творенные бензин, скипидар, канифольные масла и канифоль.
Сточные воды, получаемые при сушке, отдувке и от вакуум-
насосов, направляются в отстойники, в которых отделяются
в основном бензин и канифольные масла, а вода поступает
на биоочистные сооружения. Сточная вода, получаемая при
уваривании канифоли и перегонке флотомасла, подвергается
очистке, из нее удаляется терпингидрат, который составляет
70—80 % всех загрязнений. Путем обработки сточных вод ми-
неральными кислотами, серной (H2SO4) 0,1 %-ной концентра-
ции в течение 1—1,5 ч или фосфорной (Н3РО4) 0,2—0,3 %-ной
концентрации в течение 6 ч терпингидрат превращается в тер-
пинеол, который практически нерастворим в воде и является
86
основой флотомасел. Следовательно, этот способ позволяет не
только очистить сточные воды, но и получить дополнительную
массу флотомасел. Несмотря на то, что очистка с помощью,
фосфорной кислоты более длительна, применение ее снижает
коррозионное воздействие на оборудование. При последующей
биохимической очистке воды нейтрализованная фосфорная кис-
лота является биогенным веществом.
В настоящее время ведутся работы по снижению объема
получаемых сточных вод при производстве экстракционной ка-
нифоли. Это достигается путем использования для отдувки бен-
зина от проэкстрагированной щепы вторичного пара, образую-
щегося за счет испарения сточной воды, выделяющейся при
высушивании осмола, проведения уваривания канифоли и рек-
тификации флотомасла под глубоким вакуумом. Для снижения
объема условно чистых сточных вод используют системы обо-
ротного водоснабжения, позволяющие утилизировать тепло.
Для уменьшения потерь растворителя и предупреждения
загрязнений производственных помещений и атмосферы все
технологические аппараты, сборники, флорентины имеют воз-
душные линии, объединенные в общую систему, из которой
воздух поступает на рекуперацию. В настоящее время на всех
канифольно-экстракционных заводах планируется очистку Та-
зовых выбросов производить с применением холодильной ма-
шины. По этой схеме оборотная вода, пройдя холодильную
машину, при температуре 1—2 °C подается в скруббер, куда
противотоком поступают газовые выбросы. Содержащиеся в воз-
духе органические вещества и вода конденсируются и увле-
каются водой, а промытый воздух при температуре 3—5 °C идет
в атмосферу. Вода, температура которой 8—10 °C, из скруб-
бера поступает во флорентину, где отделяется от растворителя,
а потом идет на повторный цикл.
Воздух от паров бензина и скипидара очищается оконча-
тельно, если его дополнительно направить в скруббер, орошае-
мый соляровым маслом, но при наличии скипидара, легко окис-
ляющегося и полимеризующегося, эта операция затрудняется.
§ 11. КОНТРОЛЬ ПРОИЗВОДСТВА
Схема химического контроля канифольно-экстракционного
производства приведена в табл. 10.
При экстракции смолистых веществ из осмольной щепы
контролируют степень ее измельчения и влажность, а также
массовую долю смолистых веществ как в исходной, так и в от-
работанной щепе.
Высокая концентрация смолистых веществ в отработанной
щепе сигнализирует об отклонениях в режиме экстракции
При упаривании мисцеллы в ней контролируют концентра-
цию канифоли до и после упаривания, а при уваривании ка-
нифоли определяют температуру ее размягчения. Процесс рек-
87
10. СХЕМА ХИМИЧЕСКОГО КОНТРОЛЯ
Объект контроля
Место и периодичность
отбора пробы
Контролируемый
показатель
Экстракция смолистых веществ из осмола
Осмол Бензин От каждой партии — при поступлении на склад То же Полный анализ по ОСТу Полный анализ по ГОСТ или ТУ
Осмольная щепа (исходная) С конвейера или с сорти- ровки — каждый час для составления среднесменной пробы Из пробоотборного крана на линии подачи в экс- трактор — каждые 2 ч Из пробоотборного крана на линии отбора мицел- лы — каждые 2 ч С конвейера — при вы- грузке экстрактора Степень измельчения, влага, смолистые вещества в пере- счете на канифоль, летучие
Рабочий раствори- тель (оборотный) Мисцелла Отработанная ще- па Температурные пределы пе- регонки, смолистые веще- ства, н-бутиловый спирт Смолистые вещества Влага, смолистые вещества, летучие
Упаривани е мисцеллы и уваривание канифоли
Мисцелла Упаренная мисцел- ла Канифоль Скипидар-сырец Из пробоотборного крана на линии подачи в испа- рительную установку — каждые 2 ч Из пробоотборного крана на линии отбора — каж- дые 2 ч Из вакуум-приемника — от каждого слива Из пробоотборного крана на сливной линии — каж- дые 2 ч' Из барабанов или бочек — при формировании партии Из пробоотборного крана на трубопроводе — каж- дые 2 ч Смолистые вещества То же Температура размягчения, внешний вид То же Полный анализ по ГОСТу Плотность, кислотное число
Ректификация скипидара-сырца
Скипидар-сырец Из сборника — каждую смену Плотность, кислотное число
Фракции: бензиновая Из пробоотборного крана Плотность
на линии отбора — каж- дые 2 ч
скипидарная То же То же
Из сборника перед филь- трацией — после заполне- ния Из сборника товарного скипидара — после запол- нения Плотность, начальная тем- пература кипения, объем от- гона до 175 °C, кислотное чи- сло Полный анализ по ГОСТу, влага, бензин
88
Продолжение
Объект контроля Место и периодичность отбора пробы Контролируемый показатель
Флотомасло Из пробоотборного крана вакуум-приемника — каж- дый час или перед филь- трацией Из сборника товарного флотомасла — после за- полнения Плотность, кислотное число Полный анализ по ГОСТу
Кубовый остаток Из колонны — перед сли- вом Плотность, кислотное число, спирты
Воздушная среда и сто чные воды
Воздух Из производственных по- мещений на рабочих ме- стах — 1 раз в 10 дней Древесная пыль, канифоль, скипидар, бензин
Сточная вода Из канализационных ко- лодцев — 1 раз в 10 дней ХПК, кислотность
тификации скипидара-сырца контролируют по плотности и со-
ставу фракций. Контроль за разгонкой скипидара на фракции
осуществляют путем определения их плотности, начальной тем-
пературы кипения, показателя преломления, а при необходимо-
сти также путем нахождения состава фракций методом газо-
жидкостной хроматографии (ГЖХ).
При переработке смолистой древесины на канифоль и ски-
пидар основные расходы на 1 т товарной продукции склады-
ваются из следующих технико-экономических показателей:
сырья (осмола) 10495 кг, бензина 318 кг, пара 25,6 т, воды
534 м3, электроэнергии 382 кВт • ч.
Глава 4
ПРОИЗВОДСТВО ВТОРИЧНЫХ ПРОДУКТОВ
НА ОСНОВЕ СКИПИДАРА
Скипидар в зависимости от способа производства делится
на живичный, экстракционный, сульфатный и сухоперегонный
(табл. 11). Основную массу производимого скипидара состав-
ляет живичный. В состав всех видов скипидара входят терпе-
новые углеводороды общей формулой Ci0Hi6. Структурные фор-
мулы терпеновых углеводородов, входящих в состав скипида-
ров, были изображены в главе 1.
Скипидары являются единственным крупным источником
терпенов, которые широко применяются в качестве исходного
сырья для синтеза разнообразных веществ. Общеизвестны та-
кие продукты химической переработки скипидаров, как синтети-
ческая камфара, технический камфен и их полихлорпроизвод-
89
11. ОСНОВНЫЕ КОМПОНЕНТЫ СКИПИДАРА И ИХ ФИЗИКО-ХИМИЧЕСКИЕ
СВОЙСТВА
Терпены Скипидар и его состав» %
живичный экстрак- ционный сульфат- ный сухопере- гонный
а-Пинен Р-Пинен Камфен Д3-Карен Дипентен (смесь d- и 1-лимоне- нов) Терпеновые спирты (а-Терпине- ол) Прочие терпены* 65—72 5—6,5 0,7—1,9 10—15 3—7 45—65 1,5—2,5 1,0-1,5 19—38 3—7 63—68 2—3 0,8—1,8 12—15 3—6 40—53 1—1,5 0,9—1,3 24—45 3—5
— 3 —9 — —
3—7 2 —7 2—5 3—5
Температура, °C Плотность, кг/мя
Терпены плавления кипения
а-Пинен р-Пинен Камфен Д3-Карен Дипентен (смесь d- и 1-лимоне- нов) Терпеновые спирты (а-Терпине- ол) Прочие терпены* 45 38—40 155—156 161—162 158 170—171 175—176 218—220 857,8 871,2 842,2 964,5 842,0
* Терпеновые углеводороды» содержащиеся в долях процентов и пока не имеющие
практического значения, например терпинены, фелландрены, мирцен и др.
ные (инсектициды), окситерпеновые смолы и растворители,
политерпены, синтетическое сосновое масло, терпинеол и тер-
пингидрат, а также продукты его ректификации: пинен, дипен-
тен и карен. При окислении скипидаров кислородом воздуха
образуются продукты, которые применяются в рецептурах ла-
ков в качестве пленкообразователей и пластификаторов, сикка-
тивов для масляных красок и заменителей пищевых жиров
в различных смазках. Окисленные скипидары обладают силь-
ными фунгицирующими свойствами, полностью подавляют рост
грибков и плесеней. При гидрировании моноциклических тер-
пенов получается параментан, а из пинена — пинан. При окис-
лении их кислородом воздуха образуются гидроперекиси, кото-
рые являются эффективными инициаторами процесса полиме-
ризации при производстве синтетического каучука, а также
применяются как добавки, ускоряющие воспламенение дизель-
90
кого топлива и др. Однако такое ценное сырье до настоящего
времени на 55—60 % применяется в натуральном виде в каче-
стве растворителя и только меньшая часть перерабатывается.
§ 12. ПОЛУЧЕНИЕ ОКСИТЕРПЕНОВОГО РАСТВОРИТЕЛЯ
И ЛАКОВ
Окситерпеновую смолу вырабатывают в соответствии с ТУ.
Ее характеризуют по внешнему виду, цвету, показателю пре-
ломления, массовой доле влаги и летучих веществ, а также
5
Изомеризо-
ванный
скипидар}
11—
Вода д канализацию
Сжатый.
Й Окситерпенобая смола,-----
с растворителем Д, /4
АЧ1!'.
5
Рис. 25. Схема получения окситерпеновых смол и растворителя:
1 — изомеризатор; 2, 7 — конденсаторы-холодильники; 3, 6, 13 — флорентины; 4 —
фильтр; 5, /4 —сборники; 8 — реактор; 9 — насос; 10 — устройство для загрузки ката-
лизатора; 11 — куб-окислитель; 12 — конденсатор-холодильник
по кислотному числу и растворимости в этил- и бутилацетате,
этиловом и бутиловом спиртах, а также в ксилоле.
Технологический процесс получения окситерпеновой смолы
и растворителя (рис. 25) состоит из трех последовательных
операций: изомеризации скипидара без пинена, окисления изо-
меризата кислородом воздуха и перегонки летучих продуктов
с отбором легких фракций окисленного скипидара и окситер-
пенового растворителя.
Изомеризацию скипидара без пинена проводят в реакторе-
изомеризаторе в присутствии титанового катализатора (0,2 %
от массы сырья) при температуре 160—180 °C и постоянном пе-
ремешивании до получения изомеризата с показателем прелом-
ления 1,489—1,492. Готовый изомеризат охлаждают до 60 °C и
фильтруют через сменные фильтры от катализатора. При по-
тере катализатором активности его вместе с остатком изомери-
зата откачивают в реактор, где от него отгоняют скипидарные
продукты, а катализатор вывозят в отвал.
В ЛЛТА разработан способ регенерации катализатора, за-
ключающийся в обжиге осмолившегося катализатора. Окись ти-
тана идет на производство катализатора по существующей тех-
91
нологии. В результате изомеризации Д3-карен и дипентен пре-
вращаются в терпинолен и а-, p-терпинены, которые легче окис-
ляются. Окисление изомеризата производится в кубе-окислителе,
в верхнюю часть которого подается изомеризат. В нижнюю
часть куба в барботер через редукционный .клапан, отрегу-
лированный на давление 0,07 МПа, подают воздух таким обра-
зом, чтобы не происходило капельного уноса продукта.
Из куба паровоздушная смесь поступает в конденсатор, и
сконденсировавшийся «легкий» окисленный скипидар через
флорентину сливается в приемник. При окислении температура
реакционной смеси поддерживается 75—95 °C. Окончание реак-
ции окисления контролируется по плотности и показателю пре-
ломления готового продукта, которые при температуре 20 °C
должны иметь плотность 1010—1100 и 1,5010—1,5090 кг/м3.
Готовый продукт содержит 80—85 % окситерпеновой смолы
и 15—18 % окситерпенового растворителя. Цвет продукта не
темнее цвета 40 мг йода по йодометрической шкале, кислот-
ность не более 6 мг КОН/г. Выход продукта 80—85 % от ис-
ходного сырья.
В промышленности окситерпеновая смола с растворителем
используется в основном в качестве компонента нитроцеллюлоз-
ного (терпеноколлоксилинового) лака НЦ-224, который приме-
няют в основном в мебельной промышленности. Технология
производства лака проста: при температуре окружающей среды
в периодически действующем реакторе вместимостью 3—5 м3,
снабженном якорной мешалкой, смешивают лаковую основу
(раствор литерного коллоксилина марки ПСВ в смеси органи-
ческих растворителей) с пластификатором (окситерпеновая
смола с окситерпеновым растворителем) и пленкообразующим
глицериновым эфиром канифоли. Готовый продукт отфильтро-
вывают от механических примесей на друк-фильтр ах.
В состав нитроцеллюлозного лака НЦ-224 входят лаковая
основа (81 %), окситерпеновая смола с растворителем (13%) и
глицериновый эфир канифоли (6 %). В смесь органических рас-
творителей, служащую для растворения литерного коллокси-
лина марки ПСВ, входят 10—12 % этилацетата, 11—13 % бу-
тилацетата, 11—13 % бутилового спирта и 35 % этилового
спирта.
Основные физико-химические свойства лака: массовая доля
летучих веществ 25—30%, вязкость не более 40 Па-с, время
высыхания не более 1,5 ч, цвет по йодометрической шкале не
темнее цвета 40 мг йода.
Окситерпеновая смола и окситерпеновый растворитель мо-
гут быть использованы при изготовлении лаков различных ком-
позиций. В ЦНИЛХИ на основе окситерпеновой смолы разра-
ботана рецептура лака горячего нанесения и тропикостойкого
лака, хорошо выдерживающего повышенную температуру,
влажный воздух и подавляющего рост грибов. Кроме того, учи-
тывая частичную совместимость окситерпеновой смолы с мас-
92
ляными красками, ее можно применять в качестве разбавителя
(пластификатора) в производстве м.асляных эмалей.
Окситерпеновый растворитель является хорошим фунгици-
дом. Покрытые растворителем деревянные изделия предохраня-
ются от разрушительного действия микроорганизмов.
Рассмотренная технология производства окситерпеновых
смол и растворителя практически не дает загрязнения окру-
жающей среды.
§ 13. ПРОИЗВОДСТВО ПОЛИТЕРПЕНОВ
Получаемые полимеры представляют собой маслообразную
жидкость светло-желтого цвета и отвечают требованиям ТУ:
плотность при 20 °C не менее 940 кг/м3 и кинематическая вяз-
кость не менее 8 * 10-4 м2/с. Сырьем для производства политер-
пенов могут служить все типы скипидаров в натуральном виде,
но рациональнее использовать скипидар без пинена.
В настоящее время производство политерпенов организовано
на Борисовском бумажно-лесохимическом заводе; исходное
сырье — обеспинененный и экстракционный скипидары.
Промышленная технологическая схема производства поли-
терпенов показана на рис. 26. Она включает три основные ста-
дии: изомеризацию скипидара, полимеризацию и отгонку неза-
полимеризованных мономеров от полимеров. На первой стадии
скипидар через подогреватель подают в аппарат-изомеризатор,
наполненный шариковым алюмосиликатным катализатором.
Изомеризацию бициклических терпенов (в основном Д3-карена
в случае скипидара без пинена, а в случае экстракционного ски-
пидара а-, р-пинена) в моноциклические проводят при темпе-
ратуре 130—150 °C. Температуру процесса регулируют за счет
отвода экзотермического тепла реакции путем подвода охлаж-
дающей воды в змеевик аппарата. Пары скипидара и воды че-
рез сепаратор поступают в конденсатор-холодильник. После
разделения конденсата во флорентине скипидар направляется
в сборник скипидара для повторной изомеризации, а вода —
на очистные сооружения.
Из нижней части изомеризатора изомеризованный скипидар
поступает в реактор-полимеризатор, также наполненный шари-
ковым алюмосиликатным катализатором. Реакцию проводят
при температуре 140—180 °C, которая устанавливается автома-
тически по мере накопления полимеров в реакционной массе.
Пары воды и терпеновых мономеров из верхней части полиме-
ризатора поступают в обратный холодильник, а после разделе-
ния конденсата во флорентине мономеры направляются в поли-
меризатор, а вода — на очистку. По окончании реакции, кото-
рая считается законченной, когда температура в реакторе
достигает 180 °C, полимеризат через холодильник поступает
в сборник. Из сборника его направляют в куб-испаритель для
отгонки мономеров. Отгонку мономеров от полимеров произво-
дят под вакуумом 86,6—93,3 кПа при температуре 80—160 °C
93
до достижения вязкости 8*105 м2/с. Собранные мономеры на-
правляют на повторную полимеризацию совместно со евежими
мономерами, поступающими на реакцию из реактора-изомери-
затора. Полученные полимеры из куба-испарителя поступают
в сборник товарной продукции. •
Полимеры терпенов применяются в качестве замасливателя
стекловолокна и стеклоткани, для приготовления твердых и
Рис. 26. Схема производства политерпенов: •
1— подогреватель; 2 — изомеризатор; 3, 11— конденсаторы-холодильники; 4, 7 — фло-
рентины; 5 — полимеризатор; 6 — обратный холодильник; 8 — холодильник; 9, 13, 14 —>
сборники; 10 — куб-испаритель; 12 — вакуум-приемник; 15 — насос
жидких мазей, пластификатора, повышающего прочность, свето-
и водостойкость пленкообразующих веществ, сиккатива, уско-
ряющего высыхание масляных красок и лаков, а также компо-
нента в производстве иммерсионного масла.
В ЛТА были проведены работы по выбору более активных
катализаторов. В результате было отмечено, что при использо-
вании катализатора КУ-23 все компоненты любых видов скипи-
дара (живичного, экстракционного и сульфатного) могут быть
заполимеризованы на 95—98 % против 45—55 % в присутствии
алюмосиликатного катализатора. Полученные полимеры отве-
чают требованиям ТУ.
§ 14. ПОЛУЧЕНИЕ ТЕРПИНГИДРАТА И ТЕРПИНЕОЛА
Терпингидрат Ci0H20O2-H2O представляет собой кристалли-
ческое вещество белого цвета с температурой плавления 116—
117 °C. Он является продуктом присоединения к терпину одной
молекулы кристаллизационной воды.
94
Технологический процесс промышленного синтеза терпин-
гидрата основан на гидратации а-пинена. Для получения тер-
пингидрата одна часть скипидара или пиненовой фракции пере-
мешивается в течение 6 ч с 1,5—2 массовыми частями 60 %-ной
серной кислоты при температуре 5 °C. Серная кислота в про-
цессе гидратации разбавляется льдом до 40 %-ной концентра-
ции с целью уменьшения тепловых явлений. При этом четырех-
членное кольцо в молекуле пинена разрывается, а по месту раз-
рыва и по двойной связи присоединяются две молекулы воды
с образованием терпина. Присоединяя одну молекулу воды, тер-
пин кристаллизуется, образуя терпингидрат. По окончании ре-
акции реакционная смесь отстаивается, верхний слой непро-
реагировавших терпенов удаляют, а нижний сливают в воду,
доводя концентрацию раствора по серной кислоте до 25%. Че-
рез 24—48 ч терпингидрат выкристаллизовывается, его отде-
ляют от кислоты, нейтрализуют 10%-ным водным раствором
каустической соды и направляют для промывки водой на цент-
рифугу и далее собирают в тару для товарной продукции.
Терпингидрат применяется в медицине при лечении дыха-
тельных путей и синтезе технического терпинеола.
В одном из методов синтеза терпинеола терпингидрат явля-
ется промежуточным продуктом, который получается гидрата-
цией а-пинена смесью серной кислоты с толуолсульфокислотой.
При этом методе одну часть а-пинена обрабатывают в аппарате
с мешалкой двумя частями серной и толуолсульфокислот в от-
ношении 1:1 при температуре окружающей среды. Реакция
гидратации протекает по уравнению
Выход терпингидрата при этом на 10—15 % выше, чем при
гидратации а-пинена в присутствии серной кислоты на холоде.
На стадии получения терпинеола в реактор с мешалкой за-
гружают щелочной терпингидрат и воду в отношении 1:0,7.
Реакционную смесь нагревают и окисленный скипидар отго-
няют с парами воды. Отгонку скипидара ведут до тех пор, пока
плотность скипидара в пробе не достигнет 900 кг/м3, после чего
водный раствор щелочи отделяют от органической части. Остав-
шуюся смесь подкисляют до массовой доли серной кислоты
0,2—0,35%, затем при интенсивном нагревании образовавшийся
терпинеол-сырец отгоняют с водяным паром.
Водную фракцию через флорентину возвращают в реактор,
а терпинеол-сырец направляют в сборник. Уровень реакцион-
95
ной смеси в реакторе поддерживается постоянным за счет по-
дачи свежей подкисленной воды. Затем терпинеол-сырец ней-
трализуют 15—20%-ным раствором едкого натра и ректифици-
руют при пониженном давлении. Отбираемый в процессе
ректификации окисленный скипидар осветляют перегонкой и ис-
пользуют в качестве растворителя. Ректификацией щелочного
терпинеола-сырца получают флотомасло. Кубовые остатки от
перегонки окисленного скипидара, ректификации терпинеола и
щелочного сырца используют для приготовления грубых отду-
шек и антисептиков.
В составе технического терпинеола, кроме основного ком-
понента а-терпинеола, присутствуют 0- и у-терпинеолы: Химизм
образования терпинеолов из терпингидрата следующий:
H2so4
-н2о
терпинеолы
Терпинеол имеет приятный запах, напоминающий запах си-
рени. Применяется в парфюмерии (при изготовлении одеколо-
нов, духов) и в мыловарении.
К терпинеолам всегда примешиваются и другие продукты
дегидратации терпингидрата: окись терпина, называемая ци-
неолом (температура кипения 176—177 °C), и терпйненол (тем-
пература кипения 208—210 °C). Их структурные формулы:
Н3С СН3 Н3С СНз
цинеол терпйненол
96
Как и терпинеолы, цинеол обладает приятным запахом.
В последние годы в промышленности применяется метод
получения терпинеола, разработанный в ЦНИЛХИ; гидратация
а-пинена в среде разбавленного водного раствора муравьиной
кислоты. В основу этого метода заложена способность а-пинена
присоединять воду с образованием одноатомных спиртов
в среде разбавленной кислоты:
(45)
C10Hlfi+ нсНо“н-^С10Н17ОН.
Рис. 27. Схема производства терпинеола:
1— этерификатор; 2, 4, 5, 9, 10, 11 — сборники; 3 — нейтрализатор; 6 — сборник тер-
пинеола-сырца; 7 — ректификационная установка; 8, 12 — флорентины; 13 — конденса-
тор-холодильник
Наряду с образованием спиртов образуются эфиры
С10Н16 + НСООН->СюН17ОСОН. (46)
Под действием щелочи эфиры омыляются до спиртов
C10Hi7(X:OH + NaOH-^C10H17OH + HCOONa. (47)
Таким образом, технология получения терпинеола сводится
к гидратации а-пинена, нейтрализации свободной кислоты, омы-
лению эфиров и ректификации терпинеола (рис. 27).
Гидратацию а-пинена проводят в реакторе, снабженном ме-
шалкой и паровой рубашкой. Реакционная смесь готовится в со-
отношении а-пинена с 70 %-ной муравьиной кислотой 1,5:2.
В ходе реакции температура реакционной массы поддержива-
ется на уровне 65 °C подачей горячей воды в паровую ру-
башку. При достижении плотности верхнего «масляного» слоя
890—900 кг/м3 реакция считается законченной. По окончании
реакции реакционную смесь отстаивают. Нижний кислый слой
4 Заказ J6 2193 97
сливают в сборник и используют для последующей гидратации
при приготовлении раствора муравьиной кислоты. Верхний слой
нейтрализуют в нейтрализаторе раствором едкого натра. Ней-
трализация считается законченной при достижении pH среды 7.
После этого содержимое нейтрализатора отстаивается, водный
раствор формиата натрия сливают и направляют на регенера-
цию муравьиной кислоты. В нейтрализаторе остается органиче-
ская масса; содержащиеся в ней эфиры подвергают омылению
40 %-ным водным раствором едкого натра при температуре ки-
пения смеси. После этого смесь снова отстаивают, щелочной
раствор формиата натрия сливают в сборник и в последующем
используют для нейтрализации свободной муравьиной кислоты
в «масляном» верхнем слое.
Оставшийся верхний слой в нейтрализаторе промывают,
промывные воды соединяют со щелочным раствором формиата
натрия, а терпинеол-сырец, составляющий основное содержание
верхнего слоя, направляют на ректификацию в куб периодиче-
ски действующей колонны. Ректификацию, как и для всех тер-
пеновых соединений, ведут под вакуумом. Углеводородную и
промежуточную фракции отбирают при температуре ПО—
145 °C, терпинеол при 145—160 °C. Углеводородную фракцию
используют, как и окисленный скипидар, в качестве раствори-
теля. Промежуточную фракцию повторно ректифицируют. Ку-
бовый остаток от ректификации используют в качестве пласти-
фикатора при изготовлении различных видов понизителей вяз-
кости при глубинном бурении скважин. Терпинеол, полученный
по этому способу, содержит в основном а-терпинеол, который
легко кристаллизуется на холоде.
При применении этого метода синтеза терпинеола практи-
чески отсутствуют промышленные стоки и газовые выбросы.
Товарный продукт «масло сосновое синтетическое», содер-
жащий смесь терпеновых спиртов, главным образом «-терпи-
неола, вырабатывается в соответствии с требованиями
ТУ 13-05-66—81. Его характеризуют по внешнему виду, плотно-
сти, массовой доле воды и спиртов, кислотному числу и темпе-
ратурным пределам перегонки.
§ 15. ПРОИЗВОДСТВО синтетической камфары
Камфара СюН^О — кетон терпенового ряда; внешне кам-
фара — белое кристаллическое вещество с характерным запа-
хом. Ее молекулярная масса 152,23; температура плавления
178,5—179,5 °C, температура кипения 207,4—209,1 °C; плот-
ность 995 кг/м3. Камфара малорастворима в воде (~1 г/л) и
хорошо растворима в большинстве органических растворителей.
сн3
к/^=°
98
Камфара входит в состав многих эфирных масел (шалфей,
розмарин) и, следовательно, довольно широко распространена
в природе. Но особенно богаты камфарой все части дерева кам-
фарного лавра, произрастающего в Японии и Китае. Техноло-
гия производства камфары из камфарного лавра заключается
в отгонке ее острым паром из измельченной древесины, кор-
ней и листьев.
Первоначально камфара использовалась в медицине для
инъекций при упадке сердечной деятельности, в качестве мазей
и спиртовых растворов для растираний при простудных заболе-
ваниях. В быту камфара используется как антисептик и ядохи-
микат в борьбе с молью.
В 70-е годы прошлого столетия было установлено, что кам-
фара хорошо растворяет нитроклетчатку, причем образующийся
твердый раствор обладает термопластическими свойствами, хо-
рошо окрашивается и механически обрабатывается. Новый ма-
териал, получивший название целлулоида, быстро нашел при-
менение в технике и быту. Из целлулоида различной расцветки
изготовляют отделочные материалы, предметы домашнего оби-
хода, детские игрушки. Эти изделия прочны, изящны и гигие-
ничны.
На основе целлулоида получают кинопленку, небьющееся
стекло «триплекс», нитроцеллюлозные лаки. Применяется кам-
фара в качестве флегматизатора и в производстве бездымных
порохов.
Мировое производство камфары в настоящее время состав-
ляет 7000—9000 т в год. На долю Советского Союза падает
примерно одна треть ее мирового производства. Камфару про-
изводят трех видов: естественную из камфарного лавра (Япо-
ния, Тайвань), из пихтового масла (СССР, Корейская Народно-
Демократическая Республика) и камфару на основе скипидара
(СССР, ГДР, ФРГ, Индия, Великобритания, США). 1
В зависимости от способа производства камфары она бы-
вает оптически деятельной или рацемической. Так, камфара,
полученная из камфарного лавра, имеет d-форму с углом вра-
щения плоскости поляризации +46,5°; камфара, полученная из
пихтового масла, имеет Z-форму с углом вращения —44°; а кам-
фара, полученная синтезом на основе скипидара, является ра-
цемической смесью d- и /-форм с углом вращения ±1,5°.
Над синтезом камфары на основе скипидара работали многие ученые
мира. Было предложено несколько технологических схем, но все.они имели
общий недостаток — большие удельные нормы расхода сырья, что делало
их неконкурентоспособными с производством естественной камфары из кам-
фарного дерева.
Промышленный способ производства синтетической камфары, распро-
страненный во всех странах мира, был разработан в 30-е годы в СССР
В. Е. Тищенко, Г. А. Рудаковым, С. Я. Коротковым. В основе этого спо-
соба лежит реакция каталитического превращения — изомеризация непре-
дельных углеводородов в присутствии алюмосиликатных катализаторов, от-
крытых Л. Г. Гурвичем. В. Е. Тищенко и Г. А. Рудаков связали изомери-
зационные превращения а-пинена, содержащегося в скипидаре, с кислыми
4* 99
свойствами алюмосиликатных катализаторов. Последние позднее уступили
место другим, более эффективным катализаторам, позволяющим значительно
повысить выход камфена. В настоящее время в СССР и других странах
в качестве катализатора применяется двуокись титана.
Химический процесс и технология производства синтетиче-
ской камфары. Технология получения камфары из скипидара
по изомеризационному методу в настоящее время состоит из
следующих операций:
1. Ректификация скипидара с получением технического пи-
нена и обеспинененного скипидара.
2. Каталитическая изомеризация пинена в камфен и три-
циклен.
3. Ректификация изомеризата с целью выделения камфена
и трициклена.
4. Формилирование камфена и трициклена муравьиной кис-
лотой с получением изоборнилформиата.
5. Вакуум-ректификация и дистилляция эфиров.
6. Омыление изоборнилформиата водными растворами ще-
лочи с выделением изоборнеола; растворы формиата натрия на-
правляются на регенерацию муравьиной кислоты.
7. Каталитическое дегидрирование изоборнеола с получе-
нием камфары.
8. Очистка камфары в этиловом спирте перекристаллиза-
цией.
Вспомогательными операциями являются приготовление ка-
тализаторов для изомеризации пиненов и дегидрирование изо-
борнеола, регенерация муравьиной кислоты и растворителей,
толуола или ксилола, этилового спирта.
Химический процесс синтеза камфары при изомеризацион-
ном способе проходит следующим образом:
тю2-н2о
-------->
140- 150°С
а-пинен камфен изоборнилформиат
сн3
NaOH
-------->
155—160°С
СН3
Н
OH+NaCOOH
изоборнеол
CuCO3,Cu(OH)2
------------->
185°С
камфара (48)
Технология производства синтетической камфары (рис. 28).
Для получения камфары, как правило, используется живичный
скипидар, но при хорошей очистке могут быть использованы
экстракционный и сульфатный скипидары.
Ректификация скипидара. Скипидар представляет многоком-
понентную смесь терпеновых углеводородов, а при производстве
100
камфары используются только а-, р-пинены и камфен, а Дика-
рей,'дипентен и другие компоненты не только не используются,
но и осложняют процесс побочными реакциями. Поэтому ски-
пидар предварительно подвергают ректификации. При ректифи-
кации многокомпонентную смесь скипидара принимают за
псевдобинарную, за легкокипящую — пиненовую фракцию (а-,
Р-пинен, камфен с температурой кипения 155—162 °C), за тя-
желокипящую — кареновую фракцию (Д3-карен, дипентен и
другие терпены с температурой кипения 171—178 °C).
Терпеновые углеводороды являются нетермостойкими соеди-
нениями и при повышенных температурах способны изомеризо-
ваться. Так, наиболее ценные компоненты, а- и р-пинены, могут
превращаться в дипентен и давать полимеры, что весьма неже-
лательно. Поэтому ректификацию ведут в вакуум-ректификаци-
онных колоннах, как правило, с колпачковыми тарелками не-
прерывного действия производительностью по пиненовой фрак-
ции 400—800 кг/ч.
Полученная пиненовая фракция — бесцветная, прозрачная,
летучая жидкость с характерным скипидарным запахом. Плот-
ность пиненовой фракции при 20 °C 858—861 кг/м3, коэффи-
циент преломления 1,465—1,467, угол вращения [а]о от +20
до —25°.
Пиненовая фракция и кубовый остаток при ректификации
скипидара имеют следующий состав, %:
Пиненовая фракция Кубовый остаток
а-Пинен .................................. 87—90 ’ 2—3
Р-Пинен ........................... 6—7 2—3
Камфен..................................... 2—3 3—4
Дз-Карен .................................. 4—5 1—2
Дипентен ................................... — 15—20
Прочие терпены.............................. — 25—40
При ректификации скипидара часть пиненовой фракции вы-
водится из процесса синтеза камфары и используется в каче-
стве товарного продукта (технического пинена), который при-
меняется как растворитель для художественных красок, сырье
для синтезов, ядохимикаты, терпингидраты, терпинеолы и др.
Кубовый остаток перегоняют и получают обеспинененный
скипидар. Это прозрачная жидкость плотностью при 20 °C
860—870 кг/м3, коэффициент преломления «д 1,475—1,482. Ски-
пидар без пинена — товарный продукт, применяемый в качестве
растворителя или сырья для получения окситерпеновых смол,
полимеров и другой продукции. Остаток от перегонки (поли-
меры) — темная малоподвижная жидкость, используется для
приготовления грубых отдушек, антисептика, понизителя вязко-
сти, флотомасла.
Основная масса пиненовой фракции на камфарных заводах
направляется на следующую стадию синтеза камфары — изо-
меризацию пиненов в камфен и трициклен. Изомеризация
101
Рис. 28. Схема синтеза камфары по изомеризационному способу:
1 — цистерна терпентинного масла; 2 — напорный бак; 3, 24 — баки постоянного уровня;
4, 25 — подогреватель для скипидара; 5, 26 — ректификационные колонны; 6, 27 — де-
флегматоры; 7, 15, 20, 23 — холодильники; 8 — вакуум-приемник пиненовой фракции;
9, 30 — каландрии; 10, 31 — вакуум-приемники кубовых остатков; 11 — сборник кубовых
остатков; 12 — сборник пиненовой фракции; 13 — соляно-ватный фильтр; 14 — изомериза-
тор; 16, 21 — флорентины; 17 — сборник к флорентине; 18 — рамный фильтр; 19 — аппа-
рат для отгонки; 22 — сборник изомеризата; 23 — напорный бак для изомеризата; 29 —
вакуум-приемник камфена; 32 — сборник камфена; 33 — мерник камфена; 34 — мерник
муравьиной кислоты; 35 — форэрификатор; 36 — окончательный этерификатор; 37 — мер-
ник для серной кислоты; 38 — сборник кислой смеси; 39 — нейтрализатор; 40— мерник
для щелочи; 41 — мерник для воды; 42 — сборник кислой воды; 43 — сборник нейтраль-
ного эфира; 44 — мерник нейтрального эфира; 45 — мерник щелочи; 46 — подогреватель
для эфира;. 47—подогреватель щелочи; 48 — диафрагменный омылятор; 49 — кристалли-
затор; 50 — промежуточная емкость; 51 — центрифуга; 52 — сборник изоборнеольных ма-
сел; 53 — наклонный винт; 54 — мерник воды; 55 — бункер для изоборнеола; 56 — испа-
ритель; 57 — контактор; 58 — конденсатор для камфары; 59 — сборник промывных вод;
60 — сборник щелоков; 61 — холодильник оборотного щелока; 62 — насосы; 63— вакуум-
насос; 64 — ресивер
пинена в камфен происходит в присутствии титанового катали-
затора, вносимого в реакционный аппарат-изомеризатор в ко-
личестве около 0,1 % от массы пиненовой фракции. Титановый
катализатор готовят из технической метатитановой кислоты
Н2ТЮз, которая используется в производстве титановых белил.
Процесс приготовления титанового катализатора сводится
к очистке кислоты и приданию ей каталитической активности
путем перевода в натриевую соль:
H2TiO3+2NaOH = Na2TiO3 + 2Н2О. (49)
Полученный метатитанат натрия промывают водой и обра-
батывают муравьиной кислотой.
Na2TiO3+2НСООН = Н2ТЮ3 + 2HCOONa. (50)
После отделения формиата натрия катализатор промывают
водой до кислотности промывных вод 0,3—0,4 мл 0,1 н. рас-
твора NaOH на 1 г катализатора. После катализатор отфиль-
тровывают и нагревают в вакуум-сушильном шкафу при посте-
пенном повышении температуры до 100 °C.
H2TiO3 нагРевание . > TiO2 H2O. (51)
Гидрат двуокиси титана измельчают на мельнице в порошок
и хранят в герметичной посуде в сухом темном месте. Актив-
ность катализатора проверяют в лабораторных условиях. Ката-
лизатор считается пригодным, если в процессе изомеризации
пинена с 1 % катализатора при температуре 135—140 °C из
каждых 100 г образуется не менее 80 % камфена.
Изомеризацию пинена обычно проводят в вертикальных
стальных аппаратах-изомеризаторах периодического действия,
снабженных эффективными мешалками пропеллерного или тур-
бинного типа и змеевиками для глухого пара и воды. Процесс
протекает при 140—150 °C. Реакция экзотермическая. При пре-
вращении 1 кг пинена в камфен выделяется 335 кДж, а в дипен-
тен — 230 кДж. Если это тепло недостаточно быстро отводится,
то превращение может ускориться из-за местного перегрева,
а диффузия продуктов реакции камфена и дипентена с поверх-
103
ностй катализатора и диффузия пииеиа к поверхности катали-
затора будет отставать. В результате дипентен и камфен под-
вергаются вторичным превращениям, в том числе полимериза-
ции. Реакция полимеризации протекает с выделением 1000—
1200 кДж на 1 кг прореагировавшего вещества. Поэтому с на-
чала реакции подогрев отключается и начинается подача
охлаждающей воды в змеевик-холодильник. Продолжитель-
ность процесса изомеризации в заводских условиях может
быть выбрана самая разнообразная (от 1 ч до нескольких су-
ток). В среднем считается, что в 1 м3 реактора реакция может
завершаться за 2—3 ч. В аппаратах-изомеризаторах вместимо-
стью 50 м3 продолжительность реакции составляет 5—6 сут.
Контроль за реакцией ведут по величине показателя пре-
ломления реакционной смеси. Когда он достигает величины
1,473—1,474, реакцию прекращают. Полученный изомеризат
охлаждают до температуры 70—80 °C и фильтруют с помощью
закрытого друк-фильтра через плотный текстильный материал.
Катализатор смывают и направляют на повторную изомериза-
цию. Кроме основной реакции образования камфена, в процессе
изомеризации протекают побочные реакции.
Схема превращения а-пинена идет по следующей формуле:
где (СюН1б)п — полимеры (п может быть 2, 3 и более).
Содержащийся в пиненовой фракции А3-карен также ча-
стично изомеризуется в дипентен и терпинолен, которые подвер-
гаются дальнейшим превращениям. Каталитическая изомериза-
ция пиненов в присутствии твердых катализаторов относится
к гетерогенно-каталитическим процессам.
104
Исследованиями в ЛТА им. С. М. Кирова было показано,
что основной причиной, вызывающей побочные реакции, явля-
ется плохой массообмен. При проведении реакции изомериза-
ции в жидкой фазе с двуокисью титана получают выход целе-
вых продуктов примерно 65—70 % от исходного сырья. Столько
камфена и трициклена содержится в профильтрованном изоме-
ризате.
Выделение камфена и трициклена из изомеризата осущест-
вляется ректификацией в вакуум-колоннах, аналогичных колон-
нам для ректификации скипидара. Полученный технический
камфен — кристаллическое вещество с полупрозрачными кри-
сталлами; температура его плавления 42—46 °C, массовое со-
держание этерифицирующихся веществ (камфен, трициклен,
фенхены) 95—98%, частично отбирается в виде товарной про-
дукции. Камфен используется для синтеза ядохимикатов и про-
изводства душистых веществ в парфюмерной промышленности.
Основная масса камфена поступает на стадию формилирования
при производстве камфары. Во избежание закупорки комму-
никаций кристаллическим камфеном трубопроводы и прием-
ники должны обогреваться до температуры 50—60 °C паровыми
рубашками или паровыми спутниками. Кубовый остаток из ко-
лонны ректификации изомеризата направляется на производ-
ство окситерпеновых смол и окситерпенового растворителя.
В ЛТА разработан непрерывный метод изомеризации пи-
нена в камфен в трехфазной системе, при которой выход кам-
фена и трициклена достигает 92—95 % против 65—70 % при
периодическом способе и исключается как самостоятельная
стадия ректификации изомеризата (рис. 29).
Пиненовая фракция (96—98 % а- и 0-пиненов) из напор-
ного бака 1 поступает в бачок 2 постоянного уровня и через рас-
ходомер в подогреватель 4. Подогретый до 135—140 °C пинен
(процесс ведется при соответствующем вакууме) из подогре-
вателя поступает в куб-испаритель 5. Пары пинена из куба-ис-
парителя поступают под нижнюю тарелку колонны-изомериза-
тора 6. Для обеспечения устойчивой турбулентности на колпач-
ковых тарелках скорость паров в полном сечении колонны
должна поддерживаться 0,6—1 м/с. На верхнюю тарелку ко-
лонны-изомеризатора подается суспензия титанового катализа-
тора в камфене. Образование суспензии происходит в фонаре
8, расположенном на флегмовой линии, в который вводится не-
обходимая масса катализатора в сухом виде из емкости 7 с по-
мощью дозирующего устройства. Туда же из бачка 9 подается
нагретый до 135—140 °C камфен. Подача камфена из емкости
9 должна производиться только в период пуска установки и
продолжаться до момента, когда процесс в колонне-изомериза-
торе установится и пары дистиллята, поступающего в дефлег-
матор 10, будут состоять в основном из паров технического
камфена. После этого бачок 9 отключается и смыв катализа-
тора осуществляется флегмой. С этого момента колонна-изо-
105
меризатор 6 работает как изомеризатор и ректификационная
колонна. Технический камфен из дефлегматора 10 и холодиль-
ника И направляется в вакуум-приемник 12, а оттуда в сбор-
ник товарного камфена 14.
Катализатор, суспензированный в кубовом остатке, собира-
ется в кубе 15, откуда направляется на изомеризацию скипи-
дара без пинена при производстве окситерпеновых смол. Ва-
куум в системе создается вакуум-насосами через ресивер 13.
Рис. 29. Технологическая схема изомеризации пинена в камфен по непре-
рывному способу:
1 — бак; 2 — бачок; 3 — расходомер; 4 — подогреватель; 5 — кубы-испарители; 6 — колон-
на; 7, 9 — емкости; 8 — фонарь; 10 — дефлегматор; 11 — холодильник; 12 — вакуум-
приемник; 13 — ресивер; 14 — сборник; 15 — сборник суспензии; 16 — приемники суспен-
зии; 17 — сборник полимеров
Формилирование камфена с целью получения изоборнилфор-
миата является следующей стадией при синтезе камфары. Фор-
милирование камфена осуществляют в две ступени: 96—
98 %-ной муравьиной кислотой; формилирование, которое про-
водится в реакторах-этерификаторах, снабженных мешалкой и
паровой рубашкой, при температуре 60—70 °C до получения
106
55—65 % изоборнилформиата без катализатора. Затем добав-
ляют 4—5 % серной кислоты от массы камфена. Так как реак-
ция
CioHie+НСООН -> СюН17ОСОН (53)
обратимая, а сдвиг ее вправо лучше проходит при пониженных
температурах, то вторую ступень проводят при охлаждении ре-
акционной массы до 15—20 °C путем подачи охлаждающей
воды в паровую рубашку или змеевик. Реакция заканчивается
при достижении в реакционной смеси 92—94 % изоборнилфор-
миата (остальное — камфен и другие терпены). Продолжи-
тельность реакции в целом составляет 15—20 ч. Эфир после
нейтрализации содержащихся в нем серной и избытка муравьи-
ной кислот и промывки направляют на вакуум-ректификацию
и вакуум-дистилляцию с целью отделения камфена и поли-
меров.
В настоящее время осваивается непрерывный способ фор-
милирования камфена, разработанный сотрудниками ЦНИЛХИ
совместно с работниками Горьковского ПО «Оргсинтез». Этот
способ будет осуществлен в колонном аппарате тарельчатого
типа, изготовленном из титановых сплавов. Процесс формили-
рования будет идти в результате контакта паров муравьиной
кислоты с жидким камфеном, подаваемых в аппарат противо-
током. В этом же аппарате будет происходить и ректификация
эфиров, которые сразу будут поступать на омыление. Омыле-
ние изоборнилформиата проводится 20%-ным водным раство-
ром едкого натра:
CwHi70COH-|-NaOH->C1oH17OH + HCOONa. (54)
Щелочь добавляют в избытке против расчета на 10 %. При
омылении эфиров после вакуум-ректификации выход изобор-
неола составляет практически 100 % от теоретического, а при
омылении изоборнилформиата, не прошедшего вакуум-ректи-
фикацию, снижается на 5—10%. Реакция омыления в течение
4—6 ч проводится в реакторах-автоклавах с мешалкой и паро-
вой рубашкой при температуре 150—160 °C и давлении 500—
600 кПа. В принципе реакция омыления изоборнилформиата
при хорошем контакте его со щелочью идет мгновенно. В про-
цессе реакции образуется изоборнеол, кристаллы которого пла-
вятся при температуре 203—206 °C, поэтому при остаточном
содержании в органической части эфиров (10—15 %) при тем-
пературе реакции 150 °C образуется твердая фаза (кристаллы
изоборнеола), которая затрудняет контакт щелочи и эфира,
отчего скорость реакции резко затормаживается и длится ча-
сами. Для ускорения реакции можно добавлять растворители,
в частности ксилол или толуол, которые используются на. ста-
дии жидкофазного дегидрирования изоборнеола в камфару. Но
при добавке растворителя необходимо значительно интенсифи-
цировать перемешивание реакционной массы, иначе произойдет
расслоение растворов эфира в растворителе и водного раствора
107
щелочи. Тогда реакция вообще не будет идти. Увеличить ско-
рость можно и пропуском паров эфира через нагретый до
100 °C слой 20%-ного раствора едкого натра, подаваемого про-
тивотоком в полочный аппарат. Однако этот способ нецелесо-
образен в связи с большими удельными расходами тепла на
стадии испарения эфира. Можно проводить процесс омыления
непрерывно и при температурах выше температуры кристалли-
зации изоборнеола.
Рис. 30. Технологическая схема непрерывного способа омыления изоборнил-
формиата:
/ — емкость для эфира; 2 — емкость для щелочи; 3, 4 — мерники; 5 — дозатор; 6, 9 —
насосы; 7 — теплообменник-реактор; 8, 10 — флорентины; 11, 12 — холодильники
В ЛЛТА разработана следующая технологическая схема не-
прерывного омыления изоборнилформиата (рис. 30). Из мер-
ников 3 и 4 для эфира, разбавленного толуолом или ксилолом
(20 % от массы эфира), и щелочи через дозатор 5 с помощью
центробежного насоса 6, развивающего напор не менее 700 кПа,
смесь подается в теплообменник-реактор 7 типа «труба
в трубе». Для поддержания температуры реакционной смеси
в теплообменнике-реакторе (около 150 °C) необходимо создать
в нем давление приблизительно 500 кПа путем установления
диафрагмы на выходе из реактора. При производительности
завода 1500 т камфары в год необходим реактор с диаметром
трубы dB=0,015 м и длиной 35 м. Он может быть изготовлен
либо в виде секций, либо одновременно служить транспортным
трубопроводом для перемещения продуктов реакции по назна-
чению. Омыляют изоборнилформиат водным раствором ще-
лочи 18—20%-ной концентрации и при 10%-ном избытке
NaOH от массы перерабатываемых эфиров.
108
Из реактора под действием перепада давления продукты
реакции (толуольный раствор изоборнеола, водный раствор
формиата натрия и избытка щелочи) поступают во флорентину
8, где происходит отделение толуольного раствора изоборнеола
от водных растворов. При перекачке толуольного раствора из
флорентины 8 во флорентину 10 к нему добавляется вода, при-
близительно 0,25 части от объема толуольного раствора. По-
дают промывную воду путем подключения всасывающего па-
трубка насоса 9 к водопроводу с технической водой. Ввод воды
необходим для отмывки от толуольного раствора изоборнеола,
эмульгированного в нем водного раствора формиата натрия и
щелочи. Промывные воды из флорентины 10 направляются
в общий поток со щелоками из флорентины 8 и после нейтра-
лизации муравьиной кислотой поступают на ее регенерацию.
Промытый толуольный раствор изоборнеола направляется на
дегидрирование.
Так как продукты омыления из реактора поступают во фло-
рентину 8 при температуре 120—150 °C, происходит сильное
самоиспарение воды, толуола и изоборнеола. Для конденсации
и охлаждения их над флорентиной устанавливается обратный
холодильник 11, а внутри ее — змеевик для охлаждающей воды.
Над второй флорентиной 10 устанавливается также обратный
холодильник 12 для предупреждения загазованности производ-
ственных помещений и окружающей среды.
Превращение изоборнеола в камфару. Получить камфару из
изоборнеола можно или его окислением, или каталитическим
дегидрированием. Однако метод окисления не может конкури-
ровать с методами каталитического дегидрирования. Преиму-
щества каталитического дегидрирования изоборнеола по срав-
нению с окислительными методами заключаются в следующем:
1) в незначительном расходовании катализатора по сравнению
с расходом окислителей, в легкости регенерации катализато-
ров и практически в отсутствии вредных сточных вод на этой
стадии; 2) в почти полном отсутствии побочных реакций и
практически количественном превращении изоборнеола в кам-
фару; 3) в более легком выделении камфары из продуктов ре-
акции.
Каталитическое превращение изоборнеола в камфару на
практике проводят как в виде периодического жидкофазного,
так и непрерывного парофазного процесса. В обоих случаях
выход камфары составляет 92—95 % от абсолютно сухого изо-
борнеола, что близко к теоретическому.
Дегидрирование изоборнеола в жидкой фазе осуществляется
в стальных аппаратах, снабженных мешалками рамного типа и
паровыми рубашками. Реакция дегидрирования изоборнеола
в камфару следующая:
С10Н17ОН —>• С10Н16О + Нг • (55)
109
Эндотермическая реакция проходит так: при превращении
1 кг изоборнеола поглощается около 400 кДж/кг тепла. По-
этому для проведения процесса необходим не только подогрев
до заданной температуры (процесс протекает при 180—200 °C),
но и постоянное восполнение поглощаемого тепла. Обычно обо-
грев аппарата осуществляется через рубашку водяным паром
давлением 2000 кПа. Можно использовать высокотемператур-
ный органический теплоноситель (ВОТ)—смесь дифенила и
дифенилоксида. При дегидрировании 1 т изоборнеола выделя-
ется 145 м3 водорода (при нормальных условиях).
В качестве катализатора используют основную углекисло-
медную соль СиСО3Си(ОН)2. Часто применяют смесь углекис-
лых солей меди и никеля, добавляя от массы 1—3 % изобор-
неола. Порядок проведения реакции состоит в следующем: вме-
сте с изоборнеолом в реактор загружают 12—15 % толуола или
ксилола от массы изоборнеола. В составе технического изобор-
неола содержится 2—4 % воды и около 2 % камфена и других
примесей. Сначала при температуре 100—120 °C отгоняется
вода, содержащаяся в изоборнеоле. Смесь паров воды, толуола,
изоборнеола и примесей конденсируется в холодильнике и на-
правляется во флорентину на разделение. Из флорентины орга-
ническая часть возвращается в реактор, а вода отводится на
очистку. После отгонки воды в дегидратор засыпают порцию
катализатора и поднимают температуру реакционной массы до
180—200 °C.
Скорость реакции зависит от многих факторов: от чистоты
изоборнеола, качества и количества катализатора, скорости его
введения в реакцию, качества (чистоты) растворителя, эффек-
тивности нагрева и перемешивания системы. За ходом реакции
следят по интенсивности выделения водорода. Очень бур-
ное течение реакции может привести к выносу из реактора во-
дородом растворителя, сырья продуктов реакции, а также пы-
левидного катализатора. По окончании реакции в аппарате
остаются камфара, растворитель и катализатор. Сначала ведут
отгонку растворителя. После этого отгоняют камфару перегре-
тым до 200 °C водяным паром, расход которого не превышает
2—4 % от массы камфары. В этих условиях отгоняемая кам-
фара не образует липких пленок на поверхности конденсато-
ров-холодильников и легко счищается с нее скребками. На
камфарных заводах используются конденсаторы-холодильники
камфары непрерывного действия, состоящие из неподвижного
корпуса с рубашкой для охлаждающей воды, в котором вра-
щается барабан из литой стали с внутренним цилиндром.
Между наружными и внутренними цилиндрами барабана цир-
кулирует холодная вода, входящая в барабан и выходящая из
него по специальным приспособлениям.
Пары камфары вводятся в пространство между корпусом и
барабаном, конденсируются и охлаждаются на их поверхно-
стях. Ножи счищают камфару, которая двухвинтовым выгру-
110
жателем-затвором непрерывно выводится в тару для товарной
продукции или для дальнейшей очистки ее.
Периодический жидкофазный процесс каталитического де-
гидрирования изоборнеола имеет целый ряд недостатков: мно-
гостадийность процесса (загрузка, растворение, отгонка воды,
загрузка катализатора, дегидрирование, отгонка растворителя,
перегонка камфары, чистка аппаратов) препятствует его меха-
Рис. 31. Технологическая схема непрерывного парофазного способа дегидри-
рования изоборнеола:
1, 8 — приемники; 2 — элеватор; 3 — винтовой питатель; 4 — испаритель с контактным
аппаратом; 5 — барабанный конденсатор; 6 — капельная ловушка; 7 — винтовой выгру-
жатель; 9 — ресивер
низации и автоматизации. Поэтому в настоящее время отдается
предпочтение непрерывному каталитическому дегидрированию
изоборнеола, который практически полностью исключает отме-
ченные недостатки жидкофазного способа (рис. 31). Изобор-
неол из приемника скребковым элеватором поднимается к вин-
товому питателю и непрерывно подается в испаритель, кон-
структивно совмещенный с контактным реактором. В испарителе
постоянно поддерживается температура 230—240°C. Это обес-
печивает эффективное испарение изоборнеола. Пары его по-
ступают в трубное пространство контактного реактора. Трубки
контактного аппарата заполняются таблетированным катали-
затором (с размером гранул 1,5Х 1,5x2 см). Проходя через
слой катализатора высотой 1,5 м при температуре 250—260 °C,
пары изоборнеола дегидрируются в камфару. Время контакта
20—30 с. Обогрев испарителя и контактного аппарата, как пра-
вило, производится теплоносителем ВОТ. Продукт реакции —
пары камфары и водород — поступает в конденсатор-холодиль-
ник непрерывного действия, аналогичный аппарату того же на-
значения, используемому при жидкофазном дегидрировании.
111
Для обеспечения взрыво- и пожаробезопасности при пуске и
останове установки аппараты заполняют углекислотой для вы-
теснения водорода из системы.
Нормальный срок службы катализатора 40—45 сут. После
этого аппарат подлежит чистке от накопившегося в испарителе
нелетучего остатка, а в контактном аппарате катализатор за-
меняется свежим. Расход катализатора около 0,3—0,35 % от
массы получаемой камфары, что в 10 раз меньше, чем при
жидкофазном способе дегидрирования. При этом способе де-
гидрирования, как правило, камфара не подлежит дополни-
тельной очистке.
Камфара, полученная при жидкофазном способе дегидриро-
вания, после перегонки не всегда является достаточно чистым
продуктом, и ее частично или полностью нужно очищать, осо-
бенно в производстве рацемической камфары для медицинских
целей. Для этого можно использовать сублимацию, перегонку
с паром или перекристаллизацию.
На заводах для очистки камфары чаще всего используют
перекристаллизацию из этилового спирта. Другие растворители
применять нецелесообразно, так как камфара загрязняется их
остатками. Спиртовые остатки в камфаре неопасны, потому что
в большинстве случаев ее используют в виде спиртовых рас-
творов.
Растворимость камфары в спирте зависит от температуры:
Температура насыщения, °C .... 50,8 40,6 ^31,4 10,4 0,8 10,6
Растворимость камфары, кг/м3 .... 314 240"W194 120 93,5 76,5
При перекристаллизации камфару растворяют при темпера-
туре кипения спирта, которого берут 58 % от массы камфары.
Отфильтрованный раствор упаривают до соотношения кам-
фара — спирт 4: 1 и охлаждают в кристаллизаторах до 20 °C.
При этом выкристаллизовывается около 63 % камфары, кото-
рая в виде кашицеобразной массы центрифугируется для отде-
ления кристаллов от маточника, направляемого на упаривание.
Отделенный маточник снова упаривается до соотношения кам-
фара — спирт 4:1 и опять выделяется кристаллическая кам-
фара. Второй маточник упаривают до соотношения 6:1, так
как кристаллизация при более низкой концентрации идет труд-
нее. Из третьего маточника камфара отгоняется перегретым
паром от нелетучих полимеров. Перекристаллизованная кам-
фара отвечает требованиям ГОСТа и составляет около 90 % от
сырой камфары. Потери спирта составляют около 10%.
Контроль за производством синтетической камфары. iVhioro-
ступенчатость процесса, сложность химических превращений,
протекающих при производстве синтетической камфары, и не-
обходимость максимального сокращения потерь ценного сырья
требуют очень тщательного химического контроля за всеми ста-
диями производства.
112
При ректификации скипидара осуществляется контроль за
качеством скипидара-сырца и определяется массовое содержа-
ние а-пинена в пиненовой фракции и в кубовом остатке.
Процесс изомеризации а-пинена контролируют измерением
угла вращения плоскости поляризации и показателя преломле-
ния изомеризата, а при ректификации изомеризата определяют
массовую долю камфена й продукте и в кубовом остатке.
Кроме того, осуществляют контроль за качеством полученного
камфена.
При формилировании камфена определяют кислотность и
массовую долю эфиров в реакционной смеси. Особенно важ-
ное значение в этом процессе имеет контроль за кислотно-
стью нейтрализованных эфиров, так как оставшиеся в изо-
борнилформиате кислоты вызывают его разложение при
ректификации.
При омылении изоборнилформиата контролируются содер-
жание эфиров в реакционной массе и свободная щелочность,
а в выделенном изоборнеоле — содержание эфиров, влажность,
нелетучий остаток и температура плавления.
В процессе каталитического дегидрирования изоборнеола
контролируют влажность, температуру кристаллизации кам-
фары, а также массовую долю формилируемых терпенов. Низ-
кая температура кристаллизации камфары, а также повышен-
ная доля содержания в ней терпеновых углеводородов и изо-
борнеола сигнализируют о неполадках в ходе процесса.
При производстве синтетической камфары контролируются
качество приготовляемых катализаторов и других вспомога-
тельных материалов (муравьиная и серная кислоты, толуол,
спирт й др.), а также состояние окружающей среды.
При непрерывных процессах измеряются температура, дав-
ление или вакуум в аппаратах, скорость подачи исходных ве-
ществ и отбора продуктов, а при периодических — масса или
объем компонентов. Постоянно контролируется температура и
давление греющего пара, ВОТ.
Камфара синтетическая выпускается марок А и Б. По
физико-химическим показателям она должна соответство-
вать требованиям ГОСТ. Синтетическую камфару характери-
зуют (по цвету, температуре кристаллизации, содержанию
этерифицируемых веществ: воды, нелетучего остатка, нерас-
творимого в воде остатка, золы, железа, а также по кислот-
ности.
Камфара медицинская рацемическая должна удовлетворять
требованиям Госфармакопеи. В медицинской камфаре опреде-
ляют температуру плавления, вращение плоскости поляризован-
ного света, прозрачность и цветность раствора, нелетучий оста-
ток и органические примеси.
Технико-экономические показатели производства камфары
(удельные расходы на I т технической камфары марки А) при-
ведены ниже.
ИЗ
Скипидар, т ....................................................... 2,14
Кислота:
муравьиная 100 %-ная, т.......................................... 0,91
серная 96 %-ная, т .............................................. 0,08
метатитановая, кг ............................................... 0,50
Сода:
каустическая 92 %-ная, т ........................................ 0,42
кальцинированная, кг ........................................... 25,00
Толуол, кг........................................................ 50,00
Медный купорос, кг .............................................., 30,00
Спирт этиловый 100 %-ный, дал ..................................... 8,00
Вода, м3 ........................................................ 307,00
Электроэнергия, кВт-ч .......................................... 1096,00
Глава 5
ПРОИЗВОДСТВО ВТОРИЧНЫХ ПРОДУКТОВ
НА ОСНОВЕ КАНИФОЛИ
Технические требования к различным видам канифоли, пре-
дусмотренные действующими стандартами, удовлетворяют ин-
тересам ее потребителей, но некоторые физико-химические по-
казатели канифоли не устраивают отдельные отрасли промыш-
ленности, а в ряде случаев даже нежелательны. Часто при
сохранении наиболее важных свойств канифоли — светлой
окраски и кислотности — потребителям необходима канифоль
с более высокой температурой размягчения, повышенной твер-
достью и влагостойкостью.
Учитывая высокую реакционную способность канифоли,
можно получить различные виды модифицированной канифоли
с заранее заданными свойствами, необходимыми потребителю.
К модифицированной измененной канифоли относятся полиме-
ризованная, гидрированная и диспропорционированная. Все они
обладают большой стабильностью к действию кислорода воз-
духа.
Как и все органические кислоты, смоляные кислоты, входя-
щие в состав канифоли, способны вступать в реакцию этери-
фикации с различными спиртами. Полученные в результате
реакции эфиры канифоли имеют высокую температуру плавле-
ния и с успехом используются в смоляных основах типограф-
ских и водоупорных красок, в производстве лаков и пластифи-
каторов. В отдельных случаях для этерификации применяется
модифицированная канифоль. Эфиры канифоли, сплавленные
с сополимером этилена и винилацетата, употребляют в произ-
водстве клеев-расплавов различного назначения и бумажной
клеевой ленты. Канифоль легко вступает в реакцию с различ-
ными металлами, образуя резинаты. Резинаты марганца и
свинца используются в качестве сиккатива, а резинаты цинка и
кальция — в рецептурах водостойких лаков и красок.
Смоляные кислоты с сопряженными двойными связями
вступают в реакцию диенового синтеза с малеиновым ангидри-
дом или фумаровой кислотой. Канифольно-малеиновые и кани-
114
фольно-фумаровые аддукты применяются в производстве ме-
бельных лаков, полиграфических красок улучшенного качества
и при проклейке бумаги писчих сортов.
Большой интерес представляют амино- и нитрилопроизвод-
ные канифоли. Известно, что за рубежом они используются
в качестве эмульгаторов, антикоррозионных составов, инсекти-
цидов, фунгицидов, гербицидов, пластификаторов и мягчителей
в производстве каучука и пластических смол. Низкие сорта ка-
нифоли в составе антивибрационных смазок применяются при
проведении буровых работ в геологоразведке.
§ 16. ПОЛУЧЕНИЕ ЭФИРОВ КАНИФОЛИ
Канифоль, потребляемая в лакокрасочной промышленно-
сти, используется после этерификации ее одно- и многоатом-
ными спиртами. Эфиры одноатомных спиртов и канифоли —
вязкие жидкости с температурой кипения 210—300 °C; эфиры
канифоли с этиленгликолем, диэтиленгликолем, глицерином,
пентаэритритом — твердые смолы. Эфиры канифоли получают
прямой этерификацией канифоли в присутствии катализаторов
или без них, причем этерификация канифоли с многоатомными
спиртами проходит легче, чем с одноатомными.
Глицериновый эфир канифоли, часто называемый эфиром
Гарпиуса, занимает одно из первых мест среди других эфиров,
вырабатываемых промышленностью. Получение глицеринового
эфира канифоли проходит по формуле (12).
В промышленной технологии раздробленная кусковая кани-
фоль через загрузочный люк или расплавленная канифоль из
мерной емкости насосом загружается в этерификатор, снаб-
женный лопастной мешалкой (рис. 32). После разогрева кани-
фоли до 140—160 °C включается мешалка, из дозировочной
емкости в этерификатор сливается глицерин—12 % от массы
загруженной канифоли. Температуру реакционной массы после
загрузки глицерина доводят до 200±5 °C и выдерживают в те-
чение 1 ч. Далее реакционную массу нагревают до 270±5 °C
и периодически контролируют на кислотное число. Реакция эте-
рификации считается законченной, когда кислотное число до-
стигнет 12, после этого подогрев массы прекращается. Под-
сушка сырого эфира производится с подключением вакуум-на-
соса при остаточном давлении 0,06—0,07 МПа, температура
плавления его при этом возрастает.
Готовый глицериновый эфир канифоли с температурой раз-
мягчения не ниже 75 °C сливается в охладитель, где он охлаж-
дается до 160—180 °C и через сливной коллектор расфасовы-
вается в картонные барабаны вместимостью 100 л.
С целью предотвращения образования взрывных концентра-
ций смеси парогазов с воздухом при сливе эфира из этерифи-
катора и охладителя в аппаратах создается «подушка» инерт-
ного газа, дозируемого из баллонов.
115
Выделяющиеся в процессе этерификации и подсушки эфира
пары воды, глицерина и канифольных масел поступают в кон-
денсатор-холодильник. Конденсат через вакуум-приемник сли-
вается в сборник, из которого вода периодически перекачива-
ется на сжигание, а канифольные масла — в бочки, которые ис-
пользуются при глубинном бурении в геологоразведке.
Рис. 32. Технологическая схема производства глицеринового эфира канифоли:
/ — охладитель; 2 — этерификатор; 3 — дозатор; 4 — конденсатор-холодильник; 5 — бал-
лоны с инертным газом; 6 — вакуум-приемник; 7 — сборник; 8 — вакуум-насос; 9 — ре-
сивер; 10 — конденсатор-холодильник; // — водяная ловушка
Несконденсировавшиеся газы через конденсатор-холодиль-
ник и водную ловушку выбрасываются в атмосферу. Они не
превышают предельно допустимой концентрации, установлен-
ной для санитарной зоны, и не требуют дополнительной очи-
стки. Обогрев этерификатора осуществляется с помощью высо-
кокипящего органического теплоносителя или электричеством.
116
Глицериновый эфир канифоли состоит в основном из три-
глицеридов смоляных кислот с примесью ди- и моноглицеридов.
Цвет его по йодометрической шкале не должен быть темнее
60 единиц для высшего и 80 для I сорта, кислотное число 11—
12, температура размягчения не ниже 77—75 °C, массовая доля
золы не более 0,1—0,15 %.
Глицериновый эфир канифоли хорошо растворяется в непо-
лярных органических растворителях, трудно омыляется, стоек
на воздухе, не имеет склонности к кристаллизации, будучи рас-
творен в спиртотолуольной смеси, совмещается с основными
пигментами; Еще более высокое качество глицериновый эфир
канифоли имеет при использовании для его приготовления мо-
дифицированных видов канифоли (полимеризованной, гидриро-
ванной).
Глицериновый эфир канифоли — основной компонент при из-
готовлении термопластичных клеев различного назначения и по-
лиграфических красок. После обработки малеиновым' ангидри-
дом или фумаровой кислотой дает канифольно-малеиновые и
канифольно-фумаровые смолы — хорошие пластификаторы-от-
вердители лаковых пленок.
§ 17. ПРОИЗВОДСТВО ДРУГИХ ВИДОВ ЭФИРОВ
КАНИФОЛИ
Пентаэритритовый эфир' получается в результате взаимодей-
ствия канифоли с четырехатомным спиртом пентаэритритом по
реакции уравнения (13). Технологический процесс производ-
ства пентаэритритового эфира канифоли аналогичен процессу
получения глицеринового эфира канифоли. Время реакции ка-
нифоли при 270±5 °C и последующей вакуумной подсушке не-
сколько продолжительнее, чем при производстве глицеринового
эфира канифоли. Пентаэритритовый эфир канифоли по сравне-
нию с глицериновым имеет более высокую температуру размяг-
чения (90—120 °C), кислотное число 10—12.
Лаковые пленки на основе пентаэритритового эфира кани-
фоли обладают повышенным блеском, прочностью, водостойко-
стью, щелочестойкостью, устойчивы к действию ультрафиоле-
товых лучей, имеют хорошую адгезию к металлу, защищая его
от коррозии. Канифольно-малеиновая смола на основе пента-
эритритового эфира применяется в качестве компонента мас-
ляных, нитроцеллюлозных эмалей и отверждающей добавки
к некоторым полиэфирным смолам.
Реакция канифоли с этиленгликолем
2С19Н29СООН + С2Н4 (ОН)2 С2Н4 (ОСОС!9Н29)2 + 2Н2О (56)
и с одноатомными спиртами
С19Н29СООН + ROH -> R (ОСОС19Н29) + Н2О. (57)
117
Этиленгликолевый эфир канифоли не имеет широкого при-
менения и используется в качестве пленкообразующего и пла-
стифицирующего агента в нитроцеллюлозных лаках и эмалях
для покрытия поверхностей мебели третьего класса.
Эфиры канифоли и одноатомных спиртов применяются в ка-
честве заменителя высыхающих растительных масел, для про-
питки текстильных изделий, в составе масс для пропитки элек-
трических кабелей, в рецептуре связующих в каучуке, латексе,
изготовлении искусственной кожи.
§ 18. ПОЛУЧЕНИЕ КАБЕЛЬНОЙ НЕКРИСТАЛЛИЗУЮЩЕЙСЯ
КАНИФОЛИ
Кабельная некристаллизующаяся канифоль является про-
дуктом частичной этерификации глицерином сосновой живич-
ной канифоли. Технология кабельной некристаллизующейся
канифоли разработана Институтом физико-органической хи-
мии Академии наук БССР и не имеет принципиального разли-
чия с технологией полной этерификации канифоли глицерином.
Загрузка глицерина составляет 5 % от загруженной массы
канифоли. Реакция этерификации осуществляется при темпера-
туре 270 ±5 °C в течение 7—8 ч, выдержка в течение 2 ч, ва-
куумная подсушка 1,5 ч. Этерификация канифоли проходит на
40—50 % • Этого вполне достаточно, чтобы получать продукт
с высокими и стабильными электроизоляционными свойствами.
Полученная в результате этерификации кабельная кристалли-
зующаяся канифоль имеет удельное объемное электрическое
сопротивление не менее 2-1012 Ом*см при напряжении 100±
±5 В и 110 °C, температуру размягчения не ниже 70 °C, мас-
совую долю золы не более 0,1 % и полное отсутствие склонно-
сти к кристаллизации.
Используется кабельная некристаллизующаяся канифоль
для изготовления пропиточных кабельных составов в электро-
технической промышленности.
§ 19. ПРОИЗВОДСТВО ПОЛИМЕРИЗОВАННОЙ КАНИФОЛИ
Одним из путей повышения качества канифоли является со-
кращение числа двойных связей в ее молекулах за счет реак-
ции каталитической полимеризации (димеризации) смоляных
кислот, в первую очередь абиетиновой. Процесс димеризации
абиетиновой кислоты в присутствии катализатора — серной
кислоты может идти по уравнению (9).
Центральным научно-исследовательским и проектным инсти-
тутом лесохимической промышленности разработан способ по-
лучения полимеризованной канифоли путем обработки серной
кислотой бензинового раствора канифоли (рис. 33). 50%-ный
раствор канифоли в бензине в течение 6—7 ч при 40±2 °C об-
рабатывается 80—85 %-ной серной кислотой, составляющей
118
15 % от массы загруженной в полимеризаторе канифоли. Поли-
меризатор имеет коническое днище, паровую рубашку для обо-
грева, снабжен пропеллерной мешалкой. Для лучшего переме-
шивания к стенкам полимеризатора привариваются специаль-
ные рассекатели. ,
Для осветления полученного полимеризата последний сли-
вается в разбавитель, снабженный мешалкой, и обрабатыва-
Рис. 33. Принципиальная схема полимеризации канифоли:
1 — полимеризатор; 2 — разбавитель; 3 — промывной аппарат; 4 — выпарной аппарат;
5 — насос
ется бензином в соотношении 8:1. Раствор отстаивается, сер-
ная кислота и полученные в результате реакции окисленные
смолы выводятся из разбавителя и растворяются при пере-
мешивании в бутаноле с водой, взятой в равном соотношении
с бутанолом. Кислая вода обрабатывается 25%-ным ам-
миаком. Полученный в результате реакции сульфат аммония
119
используется в качестве удобрения,- Раствор окисленной смолы
в бутаноле отмывается водой от остатков серной кислоты и по-
дается на отгонку растворителя и уваривание.
Окисленная смола аналогична по физико-химическим свой-
ствам абиетиновой смоле и применяется в качестве воздухо-
вовлекающих добавок в строительной промышленности.
Полимеризат из разбавителя перекачивается в промывной
аппарат с мешалкой, где подвергается трехкратной промывке
водой. Первые две промывки производятся оборотной водой
с массовой долей 5—6 % сульфата аммония, третья про-
мывка— чистой водой. Упаривание и уварка бензинового рас-
твора полимеризованной канифоли производится непрерывно
на выпарном и колонном аппаратах. Отогнанный бензин ис-
пользуется в качестве оборотного для полимеризации.
Полимеризованная канифоль по внешнему виду не отлича-
ется от живичной канифоли, кислотное число ее несколько
ниже, чем у исходной канифоли, и колеблется в пределах 150—
160, температура размягчения 90—100 °C, массовая доля не-
омыляемых 8—10%, вязкость толуольного раствора в 2—
2,5 раза выше.
Полимеризованная канифоль используется для этерифика-
ции, так как для нее требуется меньший расход глицерина,
а полученные эфиры имеют высокую температуру размягчения
и вязкость. Эфиры полимеризованной канифоли широко приме-
няются при производстве полиграфических красок для глубо-
кой печати.
В процессе получения полимеризованной канифоли образу-
ется большой объем сточных вод. На промывку полимеризата,
содержащего 1 т канифоли, необходимо до 20 т воды. Сточная
вода загрязнена серной кислотой, бензином, бутанолом и дру-
гими органическими соединениями.
Для очистки сточных вод предусматриваются очистные со-
оружения, включающие в себя отстойники общего и специаль-
ного назначения типа смолоотстойников, нейтрализация порош-
кообразной известью и доочистка на локальных установках КУ.
Для сокращения потребления воды предусматривается уст-
ройство системы оборотного водоснабжения, включающее ка-
меры холодной и горячей воды, насосную станцию, градирню и
установку для хлорирования, воды жидким хлором. С учетом
выполнения мероприятий по очистке сточных вод и оборотному
водоснабжению сброс сточной воды в процессе полимеризации
канифоли довольно значителен и составляет около 15 м3 на 1 т
вырабатываемого продукта. ХПК такой воды после очистки
800—1000 мг/л.
Выбросы в атмосферу содержат пары бензина и бутанола.
Максимальная концентрация их при соблюдении требований
к герметичности аппаратуры и перекачивающих устройств на
границе санитарной зоны составляет не более 0,3 мг/м3 и не
превышает предельно допустимой.
120
§ 20. ПОЛУЧЕНИЕ ГИДРИРОВАННОЙ КАНИФОЛИ
Гидрированная канифоль является одной из модификаций
канифоли, устойчивой к окислению кислородом воздуха. В про-
цессе гидрирования канифоли водород присоединяется по месту
двойных связей смоляных кислот, в первую очередь абиетино-
вой, с образованием насыщенных дигидроабиетиновых и тетра-
гидроабиетиновой кислот по реакции уравнения (8).
Тетрагидроабиетиновая кислота — продукт изомеризации
дигидроабиетиновой кислоты с абиетиновой. Известно не-
сколько способов получения гидрированной канифоли: перио-
дический с использованием в качестве катализатора никеля,
осажденного на кизельгуре; непрерывный, по которому рас-
плавленная канифоль, насыщенная водородом, прокачивается
через батарею реакторов, заполненных катализатором — нике-
лем Ренея. В настоящее время наиболее предпочтителен спо-
соб, по которому через расплавленную канифоль при темпера-
туре 125—300 °C и давлении 49-105 Па, в присутствии катали-
затора— палладия на 5 %-ном угле, взятом от массы
канифоли, пропускается водород. Реакция продолжается около
8,5 ч при постоянном перемешивании реакционной массы. По
окончании реакции гидрированная канифоль растворяется в то-
луоле, отфильтровывается от механических примесей и осво-
бождается от растворителя путем уваривания в колонном ап-
парате. Отогнанный толуол используется в качестве оборот-
ного. Полученный продукт имеет температуру размягчения
84 °C, кислотное число 163, неомыляемых веществ до 9 % и не-
сколько светлее исходной канифоли.
Гидрированная канифоль используется при изготовлении
светоустойчивых мыл и клея для склеивания линз, кинопленки,
липких пластырей, в качестве добавок к пропитывающим сме-
сям в электронике, пластмассам, жевательной резинке и т. д.;
в соединении с поливиниловыми продуктами — для изготовле-
ния пленок, лент и лаков. Особенно большой интерес гидриро-
ванная канифоль представляет для резиновой промышленно-
сти, использующей ее при изготовлении шлангов, ремней, кон-
вейерных лент, белых резин и других изделий, не имеющих
сажевых наполнителей. В каучуке гидрированная канифоль не
изменяет структуры полимера, служит активным наполнителем
и может быть введена в каучук до 33 % без ухудшения его
свойств.
§ 21. ПРОИЗВОДСТВО ДИСПРОПОРЦИОНИРОВАННОЙ
КАНИФОЛИ
Модифицирование канифоли возможно не только в резуль-
тате прямого гидрирования ее с помощью водорода. В опреде-
ленных условиях реакция протекает с одновременным дегидри-
рованием и гидрированием молекул смоляных кислот отщеп-
ляющимися атомами водорода. При этом дигидроабиетиновая
кислота может вступать в реакцию изомеризации с новой
121
молекулой абиетиновой кислоты с образованием дегидроабиети-
новой и тетрагидроабиетиновых кислот. Такой процесс изомери-
зации смоляных кислот получил название диспропорциониро-
вания канифоли. В промышленности диспропорционирование
канифоли осуществляется при температуре 190—270 °C на
палладии, нанесенном на активированный уголь. Реакция идет
по уравнениям (6), (7).
Рис. 34. Принципиальная схема диспропорционирования канифоли:
1 — плавильник; 2 — фильтр; 3 — форконтактор; 4 — насос
В технологическую схему производства диспропорциониро-
ванной канифоли (рис. 34) входят плавильник, фильтр и бата-
рея непрерывнодействующих форконтакторов.
Раздробленная кусковая канифоль через загрузочный люк
помещается в плавильник, снабженный лопастной мешалкой,
и плавится при температуре 150—180 °C. Расплавленная ка-
нифоль отфильтровывается на фильтре с выемной корзиной и
с помощью шестеренчатого насоса подается на батарею пере-
точных форконтакторов. Форконтактор оборудован ложным
ситчатым дном, которое засыпано на 2/з высоты аппарата ката-
лизатором — палладием, нанесенным на активированный уголь.
Температура в форконтакторе поддерживается 190—270 °C.
По мере старения катализатора один из форконтакторов бата-
реи выводится из процесса на перезарядку свежим катализа-
тором, причем после перезарядки подача канифоли произво-
дится так, чтобы она поступала в форконтактор с наименее ак-
тивным катализатором и только в конце — со свежим. Расход
катализатора составляет около 2 % от массы канифоли, массо-
вая доля активного металла в нем 0,016%. Отработанный ка-
тализатор с целью повторного использования можно подверг-
122
нуть регенерации. Расход катализатора может быть значи-
тельно сокращен, если реакцию диспропорционирования прово-
дить в атмосфере водорода. Обогрев плавильника, фильтра и
форконтакторов осуществляется парами высококипящего орга-
нического теплоносителя.
Полученная диспропорционированная канифоль содержит не
более 5 % смоляных кислот с сопряженными связями, кислот-
ное число не менее 158, температура размягчения не ниже
65 *С.
Диспропорционированная канифоль используется в произ-
водстве дивинилстирольных каучуков для приготовления кани-
фольного эмульгатора, для изготовления резинатов, эфиров и
других производных лучшего качества, чем аналогичные произ-
водные неизмененной исходной канифоли.
Производство диспропорционированной канифоли не загряз-
няет сточные воды. Выбросы в атмосферу не превышают пре-
дельно допустимых санитарных норм.
§ 22. ПОЛУЧЕНИЕ КАНИФОЛЬНО-МАЛЕИНОВЫХ
И КАНИФОЛЫЮ-ФУМАРОВЫХ АДДУКТОВ
Модифицирование сосновой живичной канифоли осущест-
вляется в этерификаторах, снабженных мешалкой. Модифици-
рующий реагент — малеиновый ангидрид загружается при тем-
пературе 140—150 °C, фумаровая кислота — при 180 °C (4—
5 % от массы канифоли). Реакция модифицирования проходит
при 180—195° С. Готовый продукт охлаждается в охладителе и
разливается через коллектор в картонные барабаны вместимо-
стью 100 л. (
Модифицированная канифоль имеет высокие качественные
показатели: кислотное число 170—180, температуру размягче-
ния 60—70 °C, не склонна к кристаллизации.
В качестве отходов производства при модифицировании об-
разуются канифольные масла, используемые в геологоразведке
при глубинном бурении. Парогазовые выбросы через конденса-
тор-холодильник и водную ловушку сбрасываются в атмосферу.
Особое значение имеет модифицирование экстракционной ка-
нифоли. Процесс при этом совмещается с осветлением канифоли
химическим реагентом. В качестве осветлителя в отечественной
лесохимической промышленности используется октофор Sio,
представляющий собой алкилфенолдисульформальдегидную
смолу такого примерного состава:
нон2с
СН2ОН
123
По технологической схеме, принятой в промышленности, жид-
кая экстракционная канифоль из промежуточной емкости по-
гружным насосом или раздробленная кусковая канифоль через
люк загружается в этерификатор (рис. 35). При температуре
150 °C включается мешалка и ведется загрузка малеинового
ангидрида — 4% от массы канифоли. Далее температура реак-
ционной массы доводится до 220 °C и загружается октофор Sio
Рис. 35. Технологическая схема производства модифицированной канифоли:
1 — емкость для канифоли; 2 — этерификатор; 3 — теплообменник-охладитель; 4 — тепло;
обменник; 5 — сепаратор; 6 — конденсатор-холодильник
из расчета 0,15—0,2 % от массы канифоли. В период подъема
температуры происходит реакция присоединения малеинового
ангидрида к смоляным кислотам.
После загрузки октофора через слой расплавленной кани-
фоли со скоростью 2 м3/ч барботируется острый пар для отгона
легких канифольных масел, а температура реакционной смеси
постепенно повышается до 260±5 °C (она выдерживается в те-
чение 1ч).
Полученная модифицированная канифоль, охлажденная
в теплообменнике до 150—170 °C, разливается в картонные
барабаны. Охлаждение модифицированной канифоли в тепло-
обменнике осуществляется 60 %-ным водным раствором глице-
рина. Пары глицерина конденсируются в конденсаторе-холо-
дильнике и вновь в виде конденсата поступают в теплооб-
менник.
124
Модифицированная сосновая экстракционная канифоль по
сравнению с исходной имеет более светлый цвет (на 4—5 марок
по йодной шкале эталонов цветности), кислотное число не ме-
нее 155 и температуру размягчения не ниже 56 °C.
Модифицированная канифоль используется для приготовле-
ния клеевых композиций, применяемых при проклейке высших
сортов писчей бумаги, ее производные — в лакокрасочной, поли-
графической и текстильной промышленности.
Модифицирование экстракционной канифоли малеиновым
ангидридом совместно с формалином позволяет получить ад-
дукт с температурой размягчения до 80 °C. Такой продукт ши-
роко используется в резиновой промышленности. При дозировке
формалина наблюдается вспенивание реакционной массы, и
слив его поэтому ведется медленно, а на линии парогазовой
смеси перед конденсатором-холодильником устанавливается
сепаратор. Сточные воды, загрязненные канифольными мас-
лами и формалином, откачиваются ,на установку сжигания.
§ 23. ПРОИЗВОДСТВО РЕЗИНАТОВ КАНИФОЛИ
При взаимодействии канифоли и едкого натра получается
резинат натрия С^НгэСООЫа, который в органических раство-
рителях образует коллоидные растворы. Он хорошо растворя-
ется в воде, растворяет жиры, размягчает мыла, изготовленные
на основе твердых жиров, и сам обладает большой моющей спо-
собностью. Эти свойства резината натрия используются при из-
готовлении хозяйственного и туалетного мыла. Эмульгирующие
свойства резинатов натрия и калия используются в производ-
стве синтетического каучука при проведении реакции полиме-
ризации мономеров в эмульсионной среде. С целью улучшения
качественной характеристики эмульгатора резинат натрия или
калия приготавливается из диспропорционированной канифоли.
На основе канифоли и ее производных промышленностью
вырабатываются резинаты марганца, свинца, кобальта, каль-
ция, цинка. Соли тяжелых металлов канифоли применяются
в различных отраслях народного хозяйства. Резинат кальция,
известный под названием отвержденной канифоли, с массовой
долей 2,5—2,7 % металла является одним из первых синтетиче-
ских продуктов, полученных из канифоли.
В настоящее время наибольший интерес представляют рези-
наты модифицированной канифоли и в первую очередь поли-
меризованной. Процесс солеобразования из полимеризованной
канифоли протекает без кристаллизации реакционной массы,
а получаемые резинаты имеют повышенное качество. Полиме-
ризованная канифоль поглощает большую массу металла, чем
обычная. Так, при взаимодействии плавленой полимеризован-
ной канифоли с гидратом окиси кальция Са(ОН)г массовая
доля кальция в последней достигает 4 %, а температура
размягчения полученного резината на 20—40 °C выше, чем
у обычного.
125
Резинат кальция применяется в производстве полировочных
масел, типографских красок, в качестве связующего для стерж-
ней в литейном деле.
Для получения резината цинка в расплавленную полимери-
зованную канифоль при 150 °C и интенсивном перемешивании
вводится уксусно-кислый цинк, температура реакционной смеси
повышается до 265±5 °C, и под разрежением отгоняются ук-
сусная кислота и канифольные масла. Сливают резинат при
подаче в реактор инертного газа. Массовая доля цинка в ре-
зинате достигает 9%, а температура размягчения 150—155 °C,
вязкость в растворе толуола 35—50 сСт.
Резинат цинка используется при приготовлении полиграфи-
ческих красок глубокой печати. Печатно-технические свойства
красок на резинате цинка значительно выше, чем у красок, из-
готовленных на «Копале-44». Кроме того, «Копал-44» раство-
ряется только в ароматических углеводородах и применяется
в толуольных красках, вредных для здоровья. Резинат цинка
растворяется в уайт-спирите и бензине, поэтому использование
его значительно улучшает условия труда на заводах, изготав-
ливающих краски, и в типографиях. Резинат цинка превосхо-
дит импортную смолу «Эркацит-165» по растворимости, вязко-
сти, блеску и скорости высыхания.
Резинаты марганца, свинца и кобальта используются в ос-
новном в качестве сиккативов, ускоряющих высыхание расти-
тельных масел.
§ 24. ПОЛУЧЕНИЕ ТЕРМОПЛАСТИЧНЫХ КЛЕЕВ
Эфир канифоли — один из компонентов, используемых в ком-
позициях термопластичных клеев различного назначения. Оте-
чественной промышленностью по разработкам Ленинградского
объединения «Печатный двор» и ЦНИЛХИ внедрена технологий
производства термопластичного клея ТК-2П, представляющего
собой композицию сополимера этилена с винилацетатом, эфи-
ром канифоли, парафином и целевыми добавками (стабилиза-
торами) .
По технологической схеме (рис. 36) расплавленная смесь
глицеринового эфира канифоли (30,5%) с парафином (20,3%)
дозируется из мерной емкости в смеситель, снабженный мощной
винтовой мешалкой, в который затем загружается сополимер
этилена с винилацетатом (49 %), тиоалкофен МБП или фенозан
(0,2 %). При температуре 70—90 °C смесь тщательно переме-
шивается и самотеком через промежуточный сборник поступает
на дисковый экструдер. Жгуты термоклея из фильер экструдера
выдавливаются в охладительную ванну и охлаждаются в по-
токе воды.
Охлажденные жгуты поступают на приемное устройство гра-
нулятора и с помощью тянущего валка подаются к режущей
фрезе. Измельченный в виде гранул термоклей с помощью вен-
126
тилятора (газодувки) транспортируется в циклон и через бун-
кер на упаковку в крафт-мешки с полиэтиленовым вкладышем
или картонные -коробки вместимостью 20 кг.
Термоклей ТК-2П имеет светло-коричневую окраску, тем-
пературу размягчения 65—75 °C; вязкость в растворах 4—
6 Па-с, обладает клеящей силой не менее 4 Н/см. Используется
в полиграфической промышленности для склеивания книжно-
журнальной продукции.
Рис. 36. Технологическая схема производства термоклея:
1 — мерная емкость; 2 — смеситель; 3 — промежуточный сборник; 4 — дисковый экстру-
дер; 5 — охладительная ванна; 6 — гранулятор; 7 —циклон; 8— бункер
В производстве на 1 т термоклея расходуется до 10 м3 ох-
лаждающей воды. Сточные воды практически не требуют допол-
нительной очистки и могут быть использованы повторно после
охлаждения. Атмосферные выбросы, аэрозоли твердых органи-
ческих веществ подвергаются очистке в водной ловушке и вы-
брасываются в атмосферу.
При вводе в рецептуру термоклея специальных добавок типа
низкомолекулярного каучука ПДИ-ЗАК получают морозостой-
кий термопластичный клей. С учетом высоких качественных ха-
рактеристик термоклеев использование их в различных отрас-
лях промышленности в последние годы резко возросло.
127
Центральным научно-исследовательским институтом бумаги
(ЦНИИБ) разработаны рецептуры термопластичных клеев для
бумажной ленты с клеящим слоем и липкой ленты; Ленинград-
ским проектно-конструкторским и технологическим бюро легкой
промышленности — рецептуры клея для обувной промышлен-
ности.
Во всех композициях термопластичных клеев используется
от 11 до 60 % глицеринового эфира канифоли. Технология про-
изводства термоклеев в принципе одинакова и отличается лишь
по условиям охлаждения и гранулирования готового продукта.
§ 25. ПРОИЗВОДСТВО УКРЕПЛЕННОГО КЛЕЯ
И КЛЕЯ-ПАСТЫ
Технология производства укрепленного клея разработана Ин-
ститутом химии древесины АН Латвийской ССР и внедрена на
предприятиях лесохимической отрасли.
Модифицирование смоляных кислот осуществляется непосред-
ственно в терпентине при температуре 80—90 °C. Условия про-
ведения реакции исключают возможность изомеризации смоля-
ных кислот и позволяют почти полностью сохранить содержа-
щуюся в живице левопимаровую кислоту. В результате реакции
левопимаровой кислоты с малеиновым ангидридом образуется
малеопимаровая кислота. Реакция идет по уравнению (15).
Расход малеинового ангидрида составляет 12—21 % от
суммы смоляных кислот в терпентине. Процесс модификации ле-
вопимаровой кислоты экзотермичен, во время реакции темпера-
тура реакционной массы повышается до 100—110 °C. По окон-
чании реакции реакционная масса нейтрализуется 37 %-ным рас-
твором едкого натра. При дозировке щелочи масса сначала
густеет, а затем разжижается с интенсивным выделением тепла
и пенообразованием.
Реакция нейтрализации считается законченной, когда про-
реагирует не менее 90 % карбоксильных групп в аддукте. После
нейтрализации острым водяным паром через барботер от
реакционной массы отгоняется скипидар, а полученную клей-
пасту разбавляют водой до 70 %-ной . концентрации и сливают
в 200-литровые железные бочки.
Укрепленный клей — пастообразная масса с массовой долей
сухих веществ 68—72 %; массовая доля свободных смоляных
кислот в пересчете на сухое вещество 7—16%, малеопимаровой
кислоты не ниже 10%, летучих веществ 1,5—2%; полностью
растворяется в воде.
Технология производства клея-пасты на основе экстракцион-
ной модифицированной осветленной канифоли разработана
ЦНИЛХИ. Технологический процесс заключается в нейтрализа-
ции смоляных кислот канифоли ЭМО 20 %-ным раствором ед-
кого натра при температуре 70—80 °C и интенсивном перемеши-
вании. Расход едкого натра составляет 12 % от массовой доли
128
загруженной канифоли. Процесс нейтрализации считается за-
конченным, когда в пробе при ее разбавлении холодной водой
не наблюдается выпадения смоляных кислот.
Массовая доля сухих веществ в клее-пасте 70 %, малеопима-
ровой кислоты 10 %, свободных смоляных кислот до 18 %, рас-
творимость в воде полная.
Укрепленный клей и клей-паста на основе экстракционной
модифицированной осветленной канифоли используется в целлю-
лозно-бумажной промышленности для проклейки бумаги, в том
числе типа «тетрапак» для упаковки пищевых продуктов.
Сточные воды при производстве укрепленного клея в объеме
42 м3 на 1 т готового продукта не содержат загрязняющих при-
месей и могут быть использованы повторно; флорентинные воды
(около 1 м3 на 1 т продукта) через отстойники сбрасываются
на очистные сооружения. Атмосферные выбросы при производ-
стве как укрепленного клея, так и клея-пасты не превышают
предельно допустимых санитарных норм.
§ 26. ПОЛУЧЕНИЕ АНТИВИБРАЦИОННОЙ СМАЗКИ
Рецептуры канифольных антивибрационных смазок разра-
ботаны ЦНИЛХИ. В зависимости от особенностей применения
их в каждом конкретном случае в рецептуре может быть ис-
пользовано от 50 до 60 % автотракторного трансмиссионного
масла, 10 % нефтяного строительного битума, 5 % нефтяного
парафина, до 35 % канифоли сосновой экстракционной, или ка-
нифольно-экстракционных полимеров, или резината цинка.
Технология производства смазок (рис. 37) включает приго-
товление смеси из твердых компонентов, ее термообработку и
розлив полученного продукта в тару. В промышленности в сме-
ситель, снабженный мешалкой и паровой рубашкой для обо-
грева, загружается порция автотракторного масла и нагрева-
ется до температуры 100—105 .°C с целью выпарки влаги. За-
тем в нагретое масло сливаются предварительно расплавленная
канифоль, битум и парафин. Смесь нагревают при перемешива-
нии до однородной массы (при 140 °C), затем сливают ее через
фильтр в реактор, обогреваемый парами высококипящего орга-
нического теплоносителя. Температуру массы в реакторе дово-
дят до 270 °C, после чего нагрев отключают, массу охлаждают
до 250 °C и сливают в охладитель.
При термообработке реакционной массы пары воды и легких
масел отбираются через конденсатор-холодильник во флорен-
тину, при охлаждении — через обратный конденсатор, при этом
канифольные масла вновь возвращаются в реактор. В охлади-
теле реакционная масса охлаждается до 80—90 °C, после чего
заливается в 200-литровые железные бочки.
Канифольная антивибрационная смазка применяется для.
смазывания поверхности бурильных труб с целью снижения тре-
ния и гашения вибрации, в качестве промывочной жидкости
5 Заказ № 2193 129
водных, глинистых и безглинистых растворов, обработанных
угольно-щелочным реагентом.
Загрязненных сточных вод при производстве смазок нет.
Газовые выбросы, содержащие пары углеводородных соедине-
ний, ниже предельно допустимых санитарных норм и не требуют
специальной очистки.
Рис. 37. Технологическая схема производства смазки КАВС:
1 — смеситель; 2, 6 — конденсаторы-холодильники; 3 — фильтр; 4 — реактор; 5 — обрат-
ный конденсатор; 7 — флорентина; 8 — охладитель
§ 27. КОНТРОЛЬ ПРОИЗВОДСТВА ПРОДУКТОВ
НА ОСНОВЕ КАНИФОЛИ
Технологические процессы производства продуктов на основе
канифоли осуществляются по утвержденным технологическим
регламентам. Регламент является основным техническим доку-
ментом, определяющим последовательность и режим проведения
различных операций технологического процесса, температуру,
давление, среду, соотношение компонентов, скорость их подачи,
качество сырья, полупродуктов и готовой продукции.
Контроль за полнотой реакции на отдельных стадиях тех-
нологического процесса, качеством сырья, полупродуктов и го-
товой продукции выполняется цеховыми и общезаводскими ла-
.130
бораториями ОТК по специально разработанным или общесоюз-
ным методикам проведения анализа.
В производстве продукции на основе канифоли определяю-
щими показателями физико-химических свойств сырья, полупро-
дуктов и продукции являются: цвет по стандартной шкале эта-
лонов цветности; температура плавления (размягчения) веще-
ства, °C; кислотное число, мг КОН на 1 г продукта; плотность,
г/см3; склонность к кристаллизации; массовая доля золы, воды,
механических примесей, неомыляемых веществ и отдельных кис-
лот, %; удельное объемное электрическое сопротивление при
110 °C, Ом-см.
Учитывая, что технология производства продуктов на основе
канифоли базируется в основном на периодически действующем
оборудовании, а процесс цикличен по времени, автоматические
средства контроля, определяющие свойства вещества, не исполь-
зуются.
Контрольно-измерительные приборы для регулирования тех-
нологических процессов выбираются с учетом особенностей кон-
кретного процесса. Например, при замере расхода жидкостей
определяют агрессивность и загрязненность измеряемого потока,
изменения плотности вещества во времени; при установке тер-
мометров — необходимость поддержания узкого диапазона изме-
ряемых температур и т. д. В зависимости от назначения и ха-
рактеристики продукта, получаемого на основе канифоли,
используются не все вышеперечисленные физико-химические по-
казатели, а только характерные для данного продукта (табл. 12).
Производство продуктов на основе канифоли относится к ка-
тегории пожароопасных, а отдельные из них и к взрывоопас-
ным. В этом случае для обеспечения противопожарной безопас-
ности в помещениях цеха устанавливаются в качестве сигнали-
заторов взрывоопасных концентраций паров бензина, бутанола,
12. СХЕМА КОНТРОЛЯ ПРОИЗВОДСТВ
Измеряемый или регулируемый параметр Датчик Вторичный прибор и регулятор
Температура реакционной массы или отдельных ве- ществ в аппаратах и пере- точных линиях Давление паров жидкостей, греющего пара, паров ВОТ Уровень плава, органиче- ских веществ, жидкостей, воды Расход жидкостей, плава органических веществ, ост- рого пара Термобаллон или термопара типа ТХ К-0083 во взрывобезопасном исполнении Уровнемер УБ Ротаметр типа РПС; диафрагма типа ДКН Манометрический термометр типа ТСТ или потенциометр типа КС Манометры типа ОБМ, МСС Вторичный прибор типаПВ; радиоизотопный прибор типа РГЭ-213 Вторичный прибор типаПВ; дифманометр типа ДМ
5*
131
толуола, скипидара или их смесей с воздухом сигнализаторы
типа СВК-ЗМ или СГГ-2М.
Лесохимическая промышленность является единственным по-
ставщиком канифоли. Объем производства канифоли в 1985 г.
увеличился по сравнению с 1970 г. более чем в 1,5 раза. В по-
следние годы основное внимание уделяется увеличению выпуска
более дешевых и менее трудоемких видов канифоли — экстрак-
ционной и талловой. Предполагается, что объем их выпуска
в ближайшем будущем составит более 50 % против 18 %
в 1970 г. Однако, несмотря на постоянный рост выработки ка-
нифоли, последняя до сих пор остается дефицитным продуктом.
Расширение ассортимента продуктов на основе канифоли и
сокращение потребления обычной канифоли позволяют снизить
ее расход и повысить качество продукции в отраслях-потреби-
телях. В структуре себестоимости продуктов на основе канифоли
определяющим показателем является материалоемкость произ-
водства. В отдельных случаях, особенно при использовании жи-
вичной канифоли, стоимость сырья в себестоимости продукта
составляет более 90%. Затраты в себестоимости производства
диспропорционированной канифоли, %, приведены ниже.
Сырье и основные материалы ...........................93,8
Вспомогательные материалы и тара ..................... 2,0
Энергетические затраты ............................... 1,4
Заработная плата ..................................... 0,5
Расходы на содержание оборудования ................... 0,6
Общезаводские расходы ................................ 1,0
Внепроизводственные расходы .......................... 0,7
Основным фактором, влияющим на экономическую эффектив-
ность производства, в данном случае остается экономное ис-
пользование сырья и энергетических затрат. Значительно весо-
мее народнохозяйственный эффект использования изделий,
содержащих в своем составе продукты на основе модифициро-
ванной канифоли. За счет улучшения физико-химических свойств
такой канифоли повышается качество изделий на ее основе.
В первую очередь это относится к резинотехническим изделиям,
каучукам, бумаге. При использовании термоклеев повышается
срок носки клееной обуви, упрощается процесс изготовления пе-
реплетов книжно-журнальной продукции. При использовании
эфиров модифицированной канифоли в красках на 1,5—2 года
увеличивается срок эксплуатации покрытий и т. д.
Раздел II
ТЕРМИЧЕСКАЯ ПЕРЕРАБОТКА
ДРЕВЕСИНЫ
Глава 6
ХАРАКТЕРИСТИКА ТЕРМИЧЕСКИХ МЕТОДОВ
ПЕРЕРАБОТКИ ДРЕВЕСИНЫ
В этой главе приведены краткие исторические сведения
о термических методах переработки древесного сырья, современ-
ном их состоянии и народнохозяйственном значении. К термиче-
ским методам переработки древесного сырья — методам пиро-
лиза в широком понимании этого термина, происходящего от
греческих слов руг — огонь и lysis — разложение, распад,— от-
носятся смолокурение, дегтекурение, углежжение, собственно
пиролиз, газификация и энергохимическое использование дре-
весины.
В учебной и научной литературе применяется несколько тер-
минов для обозначения понятия «пиролиз древесины», что
крайне затрудняет использование автоматизированных систем
научно-технической информации. Термины «термолиз» и «кар-
бонизация» хотя и раскрывают сущность процесса пиролиза,
в литературе встречаются редко и поэтому в целях унификации
терминологии от применения их следует отказаться. Термин
«швелевание» (от нем. schwelen Harz — гнать смолу), обозна-
чающий пиролиз древесины на одной из стадий процесса ее га-
зификации, также пе рекомендуется к использованию.
В настоящем учебнике принят термин «пиролиз древесины»,
полно отражающий сущность процесса и завоевавший в послед-
нее время широкое признание лесохимиков. Процесс пиролиза
древесины при температурах ниже начала температуры интен-
сивного распада с выделением тепла, например в среде жидкого
теплоносителя, принято называть предпиролизом, при темпера-
турах выше 700 °C, выше температур, поддерживаемых в со-
временных промышленных аппаратах,— высокотемпературным
пиролизом.
Краткие сведения о термических методах переработки древесного
сырья. Смолокурение — процесс получения сосновой смолы при термическом
разложении соснового пневого осмола. Возникновение смолокурения на Руси
относится к XII в. Долгое время сосновая смола была одним из основных
предметов русского экспорта. В 1912 г. за границу вывезено около 22 000 т
сосновой смолы, при общем производстве около 90 000 т. После первой
133
мировой войны спрос на сосновую смолу постоянно падал, и к настоящему
времени смолокурение почти исчезло.
Дегтекурение'—процесс получения дегтя при термическом разложении
бересты. Дегтекурение — также весьма старинный русский промысел. Бере-
стовый деготь, как и сосновая смола, в больших объемах вывозился за
границу. Производство дегтя сохранилось до сих пор, однако потребности
сельского хозяйства в нем удовлетворяются не полностью.
Углежжение — одно из типичных лесохимических производств прошлого.
Уже в глубокой древности древесный уголь применяли для выплавки метал-
лов и в качестве топлива. Еще Теофраст (372—287 гг. до н. э.) писал о вы-
жигании древесного угля. Об углежжении на Руси впервые упоминается
в Новгородской таможенной грамоте в 1571 г. Единственным продуктом
углежжения является уголь. Объем выработки угля в России был весьма
значителен, максимум производства — около 1 млн. т в год — был достиг-
нут перед первой мировой войной. Подавляющая часть угля использовалась
в металлургии для выплавки чугуна. В период гражданской войны и раз-
рухи выработка древесного угля резко упала, но в период восстановления
и развития народного хозяйства увеличилась и в 1934 г. приблизилась
к довоенному уровню. Однако затем древесный уголь в черной металлургии
был заменен более дешевым коксом и выработка его резко сократилась.
В настоящее время древесный уголь в этой промышленности практически \
не применяется. По мере общего развития техники менялся и сам процесс
углежжения: ямное и кучное углежжение постепенно заменялось углежже-
нием в разного рода печах, вначале периодического, а затем и полунепрерыв-
ного действия.
Начало использования жидких продуктов при углежжении в России
относится к концу XVIII — началу XIX в. В 1793 г. президент Вольного
экономического общества А. А. Нартов доложил об использовании «кислой
влажности из угольных куч» и о возможности применять «пригорелую дре-
весную кислоту» в ситценабивном деле, для протрав, получения ярь-медянки
и т. д. Уксусную кислоту из «сырого древесного уксуса» выделяли так назы-
ваемым порошковым способом в виде кальциевой соли. Для выделения уксус-
ной кислоты из кальциевой соли вначале использовали следующий способ.
Уксусно-кислый кальций растворяли в воде и обрабатывали сульфатом
натрия, гипс отделяли, а из раствора получали уксусно-кислый натрий, очи-
щали его кристаллизацией, разлагали серной кислотой, и уксусную кислоту
отгоняли. Позднее процесс получения уксусной кислоты значительно упро-
стили и серной кислотой стали обрабатывать непосредственно уксусно-каль-
циевый порошок.
Процесс термической переработки древесины, при котором наряду с дре- \
весным углем утилизируют жидкие продукты, в русской литературе назы- s
вался сухой перегонкой древесины. Сейчас это название считается устарев- ;
шим и заменено пиролизом древесины, а название сухоперегонного произ-
водства — пиролизным производством. 4
Вслед за уксусной кислотой в середине прошлого века началось произ-
водство метилового спирта из жидких продуктов сухой перегонки. Метило-
вый спирт шел на приготовление лаков и политур, а смесь трех частей
спирта и одной части скипидара под названием «камфин» применялась
в лампах для освещения. Позднее метанол служил в производстве форма-
лина. В конце XIX в. в связи с появлением производства нитроцеллюлозы
возникло производство ацетона из уксусно-кальциевого порошка. Долгое
время лесохимия была единственным производителем уксусной кислоты и
этилацетата, метанола, ацетона и формалина. В настоящее время лесо-
химические ацетон и формалин не вырабатываются, метанол выпускается
в небольшом объеме. Ацетатные растворители производятся в основном
из синтетической и лишь в незначительном объеме из лесохимической уксус-
ной кислоты, формалин — полностью из синтетического метанола.
Прежде заводы сухой перегонки представляли собой мелкие, полукус-
тарные предприятия. Создание современного индустриального пиролизного
производства началось в годы первых пятилеток с пуска Ашинского (1932 г.)
и Сявского (1937 г.) лесохимических заводов.
134
В настоящее время пиролизные заводы — это индустриальные предприя-
тия с высокой концентрацией производства, важная составная часть лесо-
химической подотрасли промышленности. Техника и технология пиролиза
древесины, однако, неоднородны. Наряду с современными аппаратами —
вертикальными непрерывнодействующими ретортами — в отрасли сохрани-
лись морально устаревшие реторты и печи с худшими условиями труда и
более низкими технико-экономическими показателями работы. Все построен-
ные в послевоенное время заводы, реконструируемые и вновь строящиеся,
оснащаются вертикальными ретортами. Единичная мощность таких реторт
достигает 70 тыс. м3 плотного объема перерабатываемой древесины в год.
Все лесохимические предприятия работают по схеме полной конденсации
жидких продуктов. Применявшийся в спиртопорошковом производстве по-
рошковый способ выделения уксусной кислоты, как более сложный и
дорогостоящий, изживает себя, и сейчас уксусная кислота извлекается
непосредственно из водного конденсата пиролиза древесины (жижки) пре-
имущественно методом экстракции этилацетатом. Технология переработки
жижки постоянно совершенствуется, сейчас это почти непрерывный процесс
с локальными системами автоматического регулирования на отдельных ста-
диях. Техническая лесохимическая уксусная кислота в основном перераба-
тывается в пищевую. В СССР создано не имеющее аналогов за рубежом
смолоперерабатывающее производство, продукция которого широко исполь-
зуется в народном хозяйстве.
Газификация древесины — это процесс получения из древесины газооб-
разного топлива. В России газификация древесины в газогенераторах пря-
мого процесса появилась в середине XIX в. для обеспечения горючим гене-
раторным газом печей в черной металлургии и в стекольном производстве.
Крупные газогенераторные станции, например Ижевская, оборудованные
газогенераторами производительностью 30 тыс. пл. м3 перерабатываемой
древесины в год, работали до конца 50-х годов текущего столетия. Долгое
время жидкие продукты, образовавшиеся при газификации, не использо-
вались. Лишь в 1932—1935 гг. была разработана и внедрена технология ути-
лизации жидких продуктов с получением уксусно-кальциевого порошка и дре-
весной газогенераторной смолы. За это группа работников лесохимической
промышленности во главе с А. А. Деревягиным — А. А. Ливеровский,
Н. В. Чалов, В. А. Лямин и В. И. Корякин — была удостоена Государственной
премии СССР.
Большое распространение в 1930—1946 гг. получили газогенераторы
обращенного процесса. Они устанавливались для обеспечения газообразным
топливом двигателей внутреннего сгорания на автомашинах, тракторах,
катерах. В 1950—1960 гг. интенсивно разрабатывались техника и техноло-
гия газификации различных древесных отходов, прежде всего от лесоза-
готовок, с получением энергетического газа (для котельных или газодизе-
лей) и смолы. Однако энергохимия, как ее тогда называли, вследствие
обеспечения леспромхозов дешевой электроэнергией от государственных
энергосистем не получила развития. В настоящее время во многих странах
мира снова возрос интерес к - газификации древесины с целью получения или
энергетического газа, или синтез-газа для последующего превращения его
в метанол — полупродукт для органического синтеза.
Последним из известных методов термической переработки древесины
назовем энергохимическое использование топливной древесины. Сущность
его заключается в предварительном термическом разложении поступающей
в топку древесины, с отбором парогазов и утилизацией жидких продуктов
и в последующем сжигании (но не газификации) твердого остатка — древес-
ного угля. Энергохимическое использование древесной щепы — обессмолен-
ной щепы канифольно-экстракционного производства — на практике осуще-
ствляется в скоростной топке — генераторе В. В. Померанцева, работающей
на Вахтанском канифольно-экстракционном заводе с 1950 г.
Из всех описанных методов термической переработки древе-
сины наиболее важное народнохозяйственное значение имеет
пиролиз древесины. Поэтому при изложении последующего
135
материала основное внимание будет уделено химии и техноло-
гии пиролизного производства.
Продукция пиролизного производства и ее значение для на-
родного хозяйства. Перечень товарной продукции пиролизного
и переделочного производств насчитывает несколько десятков
наименований, но главными являются древесный уголь, пище-
вая уксусная кислота, смола и различные смолопродукты.
Основными потребителями древесного угля являются цвет-
ная металлургия, производство сероуглерода и активных уг-
лей. В цветной металлургии древесный уголь используется
в качестве покровного флюса для создания защитного поверх-
ностного слоя, предохраняющего расплавы металлов от окисле-
ния и улучшающего процесс раскисления металлов, например
меди. Под слоем флюса производится плавка многих сплавов
цветных металлов — оловянных, алюминиевых, кремниевых, бе-
риллиевых и других бронз, некоторых латуней, мельхиора и
т. д. Но большая масса угля используется в производстве кри-
сталлического кремния в качестве восстановителя. Для обес-
печения необходимой чистоты получаемого продукта сырье
для производства кристаллического кремния, в том числе вос-
становитель, должно быть особенно чистым.* Древесный уголь
по своей чистоте (доле примесей минеральных веществ) и фи-
зико-химическим свойствам (высокой реакционной способности
и высокому удельному электрическому сопротивлению) более
других твердых углеродных восстановителей отвечает предъяв-
ляемым требованиям по чистоте и поэтому применяется в про-
изводстве кремния в значительных объемах. Кристаллический
кремний в свою очередь используется в производстве различ-
ных кремниевых сплавов, главным образом силумина, а также
для получения кремния высокой чистоты для полупроводнико-
вой техники.
В производстве сероуглерода (CS2) древесный уголь при-
меняется в качестве источника углерода. Получение сероугле-
рода в слоевых аппаратах основано на реакции парообразной
серы с углеродсодержащим материалом при 600—800 °C. Серо-
' углерод применяется при производстве вискозы для ксантогени-
рования целлюлозы, в производстве четыреххлористого угле-
рода и т. д.
В производстве активированного угля древесный уголь слу-
жит физической основой товарного продукта. Активированный
уголь — это уголь с высокоразвитой пористой структурой и боль-
шой внутренней поверхностью. Сущность процесса активации
заключается в газификации, выжигании части углерода акти-
вирующими агентами при 850—900 °C. В качестве последних
используются чаще всего перегретый водяной пар или парога-
зовая смесь от сжигания дизельного топлива с добавкой во-
дяного пара. Активированный уголь применяется в пищевой,
фармацевтической, химической и других отраслях народного хо-
зяйства для очистки жидких продуктов и растворов от приме-
136
сей. Перспективно использование его для очистки питьевой воды
и доочистки сточных вод.
Помимо этих отраслей промышленности, древесный уголь
применяется в производстве ферросплавов (сплавов железа
с различными элементами — кремнием, марганцем и др.), в про-
изводстве карбюризатора для цементации поверхности сталь-
ных изделий, в производстве дымных порохов, ионообменников
для очистки реактивов от примесей поливалентных металлов и
в ряде других областей.
Мелкий древесный уголь, отсеиваемый от угля-сырца, ис-
пользуется в качестве добавки в рацион животных, а также вза-
мен крупнокускового угля, когда тот служит лишь защитным
материалом в производстве цветных металлов, при изготовле-
нии оцинкованной проволоки, стекла, при термообработке
стальных изделий и т! д.
В будущем древесный уголь благодаря своим специфическим
свойствам — макропористой структуре, высокой термостойкости,
стойкости к растворам солей, щелочей, кислот и к органическим
растворителям — может еще шире использоваться как полупро-
дукт в качестве полимерной матрицы для химической модифи-
кации.
Уксусная лесохимическая кислота по объемам выработки и
по стоимости не может конкурировать с синтетической уксусной
кислотой в качестве полупродукта для синтеза в химической
промышленности. Но как пищевой продукт для консервирова-
ния и применения в быту уксусная лесохимическая кислота по
своим вкусовым х качествам превосходит синтетическую. Кроме
того, синтетическая уксусная кислота не применяется как пи-
щевой продукт из-за использования при ее производстве вред-
ных и токсичных веществ. Поэтому объемы производства и по-
требления лесохимической пищевой уксусной кислоты непре-
рывно растут.
Древесная смола используется прежде всего в виде так на-
зываемой кондиционной смолы в химической промышленности
и в качестве мягчителя при регенерации резинотехнических из-
делий.
Из продуктов смолопереработки наиболее ценными явля-
ются древесносмоляной ингибитор, применяемый в производ-
стве синтетического каучука и мономеров для пластмасс, для
стабилизации бензина, и понизитель вязкости лесохимический,
используемый в литейном производстве и при бурении нефтя-
ных скважин.
Ацетатные растворители — этил- и бутилацетат, вырабаты-
ваемые на лесохимических заводах из синтетической уксусной
кислоты, находят весьма широкое применение в химической, ла-
кокрасочной промышленности и многих других отраслях народ-
ного хозяйства.
137
Глава 7
ХАРАКТЕРИСТИКА ТЕХНОЛОГИЧЕСКОГО СЫРЬЯ
Физические свойства древесины; Физические свойства дре-
весины характеризуются плотностью, влажностью, гигроскопич-
ностью, теплоемкостью и другими параметрами.
Плотность древесины, или древесинного вещества, у всех
пород одинакова и в среднем равна 1,55 г/см3. Плотность дре-
весины как физического тела из-за наличия полостей меньше
и колеблется в значительных пределах в зависимости от по-
роды, условий произрастания, индивидуальных особенностей
каждого дерева и в большей степени от влажности древесины
(табл.13).
Из наиболее распространенных в СССР древесных пород
среднюю плотность (в воздушно-сухом состоянии) выше 0,55
имеют многие лиственные породы (дуб, ясень, клен, граб, бук,
береза), а из хвойных только одна лиственница; плотность ниже
0,55 г/см3 — все остальные хвойные породы (сосна, пихта, ель,
кедр и др.) и лиственные (ольха, осина, тополь, липа). Плот-
ность имеет существенное значение в процессе пиролиза дре-
весины при расчете массы загружаемой в аппарат древесины
и получаемых продуктов и др.
Для некоторых технологических расчетов необходимо знать
насыпную массу измельченной древесины, т. е. массу щепы или
опилок, входящую в единицу объема аппарата при свободной
загрузке. Масса 1 насыпного м3 хвойной щепы в пересчете на
абсолютно сухую древесину составляет обычно 130—160 кг, бе-
резовой щепы в среднем 191, щепы осмольной отработанной
в среднем 186, щепы из лесосечных отходов смешанных пород
120—160, опилок 100—135 кг. При искусственном уплотнении
насыпная масса опилок и щепы может быть на 30—50 % выше.
13. ПЛОТНОСТЬ НАИБОЛЕЕ РАСПРОСТРАНЕННЫХ ПОРОД ДРЕВЕСИНЫ
в АБСОЛЮТНО СУХОМ состоянии
Порода Плотность, г/см3 Средний объем клеточных стенок, % Объем полостей» %
Пределы колебаний Средняя
Пихта * к 0,31—0,50 0,38 25 75
Ель 0,30—0,56 0,42 27 73
Сосна 0,31—0,65 0,47 30 70
Лиственница .0,43—0,82, 0,63 _ 41
Осина 0,32—0,61 "0747 “30 70
Липа 0,33—0,62 0,47 30 70
Ольха 0,33—0,64 0,49 32 68
Береза 0,42—0,79 0,60 39 61
Бук 0,45—0,79 0,64 41 59
Клен 0,48—0,74 0,65 42 58
Дуб 0,46—0,88 0,68 44 56
138
Древесина содержит свободную (в полостях клеток) и свя-
занную (в оболочках клеток) влагу. Различают абсолютную и
относительную влажность древесины. Абсолютная влажность
выражается в процентах от абсолютно сухого вещества древе-
сины, а относительная — в процентах от влажной древесины.
Относительную влажность Wo можно пересчитать в абсо-
лютную W по формуле
W = 100Го/(Ю0—Го), (58)
а абсолютную в относительную
Го=Ю0Г/(100+Г). (59)
При расчетах процесса сушки древесины используют пока-
затели абсолютной влажности, в расчетах по технологии лесо-
химических производств — относительной влажности (табл. 14).
По данным табл. 14 можно определить массу абсолютно су-
хой древесины в 1 м3 древесины той или иной влажности. На-
пример, если 1 м3 березовой древесины 20 %-ной относительной
влажности весит 670 кг, то массовая доля в нем абсолютно су-
хой древесины 670 (100—20) : 100=536 кг, но не 600, как могло
бы показаться на основании цифр первой строки табл. 14. Это
расхождение — следствие того, что при высушивании древесины
ниже определенной влажности происходят ее усадка и уплот-
нение. На большинство свойств древесины оказывает влияние
изменение содержания связанной влаги. При достаточно дли-
тельной выдержке древесина приобретает равновесную влаж-
ность, которая зависит от влажности и температуры окружаю-
щей среды. Уменьшение содержания связанной влаги вызывает
сокращение линейных размеров и объема древесины — усушку.
При увеличении содержания связанной влаги, а также поглоще-
нии древесиной жидкостей происходит явление, обратное усуш-
ке — р азбухание.
Влажность различных частей растущего или свежесрублен-
ного дерева неодинакова. Средняя относительная влажность яд-
ровой древесины хвойных пород составляет 25—30%, спелой
древесины 35—40, заболони 45—65 %. У древесных пород
14. СРЕДНЯЯ МАССА 1 М3 ПЛОТНОГО ОБЪЕМА ДРЕВЕСИНЫ РАЗНЫХ ПОРОД
В ЗАВИСИМОСТИ ОТ ВЛАЖНОСТИ
Влажность, % Порода
абсолютная относитель- ная ель осина сосна береза бук
0 0 420 470 470 600 640
10 9 440 490 500 630 670
25 20 470 530 540 670 710
50 33 560 620 640 790 830
75 43 655 730 740 915 975
100 50 750 830 850 1050 1110
139
влажность древесины от периферии к центру уменьшается по-
степенно. Общая влажность свежесрубленной древесины хвой-
ных пород равна 45—50%, мягколиственных 40—50, твердо-
лиственных 30—45 %.
Способность высушенной древесины поглощать водяные пары
из воздуха до состояния равновесия называется ее гигроскопич-
ностью. Пределом гигроскопичности (точкой насыщения во-
локна) называется состояние, при котором в древесине содер-
жится максимальная масса связанной влаги, а свободная влага
отсутствует. Гигроскопичность древесины различных пород
почти одинакова и при относительной влажности воздуха 100 %
и температуре.20 °C влагоемкость древесины сосны и дуба со-
ставляет 28—30 %, при 90 % 21—22 %, при 50 % 9—10 % ит.д.
Способность древесины поглощать воду при погружении
в нее называется водопоглощением. Масса воды, которую мо-
жет поглотить древесина, зависит от объема полостей в ней.
Например, древесина березы, имеющая плотность в абсолютно
сухом состоянии 0,6 г/см3, теоретически может поглотить около
130 % воды от массы сухого вещества, при этом плотность ее
составит 1,2 г/см3. При увеличении плотности древесины выше
1 г/см3 она тонет.
Теплоемкость древесины складывается из теплоемкостей
собственно древесины и содержащейся в ней влаги, а также
смолистых веществ. При этом теплоемкость смолистых веществ
в 1,5 раза, а воды в 2,5 раза больше, чем удельная теплоемкость
абсолютно сухой древесины, которая равна 1,55 кДж/(кг-К)
при температуре 0 °C. С повышением температуры удельная
теплоемкость древесины возрастает по линейному закону и при
100 °C увеличивается примерно на 25%. Замораживание сы-
рой древесины приводит к уменьшению теплоемкости, так как
лед имеет вдвое меньшую теплоемкость, чем 'вода. Увеличение
влажности древесины от 0 до 130 % приводит к повышению теп-
лоемкости в 2 раза.
Древесина — плохой проводник тепла. Ее теплопроводность
зависит от условной плотности древесины (породы), направле-
ния потока тепла относительно оси древесного волокна, темпе-
ратуры и влажности. Коэффициент теплопроводности сухой дре-
весины колеблется в пределах 0,1—0,4 Вт/(м-К). Увеличение
плотности сухой древесины, т. е. повышение доли, занимаемой
в единице объема древесинным веществом, приводит к возрас-
танию теплопроводности. Это происходит вследствие того, что
древесинное вещество имеет в 20 раз больший коэффициент
теплопроводности, чем воздух. Теплопроводность древесины
вдоль волокон в 3 раза больше, чем поперек волокон. По ра-
диальному и тангенциальному направлениям коэффициенты
теплопроводности древесины могут несколько отличаться один
от другого, так как в тангенциальном направлении вытянуты
зоны поздней древесины годичных слоев. Поздняя древесина,
особенно хвойных пород, более плотная, а следовательно, бо-
140
15. ВЕЛИЧИНЫ УДЕЛЬНЫХ ОБЪЕМНОГО И ПОВЕРХНОСТНОГО СОПРОТИВЛЕНИИ
ДРЕВЕСИНЫ
Порода и структурное направление древесины Влажность, % Удельное сопротивление
объемное, Ом-см поверхностное, Ом
Береза: вдоль волокон 8,2 4,2- 10м 4,0-10й
поперек волокон 8,0 8,6-10“ 2,8-10“
Бук: вдоль волокон 9,2 1,7-10® 9,4-10“
поперек волокон 8,3 1,4-10“ 7,9-10“
Дуб: вдоль волокон 7,9 2,0-10“
поперек волокон 7,9 1,3-10“ 5,5-10“
лее теплопроводная. Увлажнение древесины, т. е. замещение со-
держащегося в ней воздуха водой, имеющей в 23 раза большую
теплопроводность, приводит к возрастанию теплопроводности
древесины. Повышение температуры влажной древесины при-
водит к еще большему увеличению теплопроводности.
Теплота сгорания древесины сильно зависит от ее влажности
и незначительно от породы. Теплота сгорания абсолютно сухой
древесины различных пород колеблется в пределах 20—
21-Ю3 кДж/кг. Средняя теплота сгорания свежесрубленных
дров составляет около 8,5-103 кДж/кг, а воздушно-сухих
15 -103 кДж/кг.
Способность древесины проводить электрический ток харак-
теризует ее электрическое сопротивление. Различают удель-
ное объемное и удельное поверхностное сопротивление
(табл. 15). Удельное объемное электрическое сопротивление
численно равно сопротивлению при прохождении тока через две
противоположные грани кубика из древесины размером 1X1X
X1 см и определяется по формуле
p = RS/l, (60)
где R— сопротивление образца, Ом; S — площадь основания
образца, см2; I — длина ребра образца, см.
Поверхностное электрическое сопротивление измеряется
в омах и численно равно сопротивлению квадрата любого раз-
мера на поверхности образца древесины при подведении тока
к электродам, ограничивающим две противоположные стороны
этого квадрата.
Как видно из данных табл. 15, электропроводность древе-
сины зависит от породы и структурного направления древесины.
Удельное объемное сопротивление вдоль волокон большинства
пород в несколько раз меньше сопротивления поперек волокон.
Сухая древесина относится к лучшим электроизоляционным ма-
териалам, но с повышением массовой доли влаги в древесине
ее электрическое сопротивление уменьшается.
141
16. ХИМИЧЕСКИЙ СОСТАВ ДРЕВЕСИНЫ НЕКОТОРЫХ ПОРОД, % ОТ МАССЫ
А. С. Д.
Состав Береза Betula verrucosa Осина Populus tremula Сосна Pinus Silvestris (Ленин- градская обл.) Листвен- ница Larix dauhrica
Целлюлоза 35,4 41,8 39,6 34,5
Лигнин 19,7 21,8 23,9 27,2
Легкогидролизуемые полисахариды В том числе: 25,5 20,3 17,8 24,2
пентозаны (без уроновых кислот) 24,6 16,3 7,6 5,3
уроновые кислоты 5,7 8,0 3,8 3,2
общие метоксигруппы 5,6 5,7 4,7 4,4
ацетильные группы 5,8 5,6 1,4 1,4
Древесина, находящаяся в переменном электрическом поле,
проявляет диэлектрические свойства, которые характеризуются
двумя показателями: диэлектрической проницаемостью 8 и тан-
генсом угла диэлектрических потерь tgo. Диэлектрическая про-
ницаемость целлюлозы и лигнина вдвое больше диэлектриче-
ской проницаемости воздуха (s воздуха равно 1), поэтому ди-
электрическая проницаемость абсолютно сухой древесины мало
зависит от ее плотности. Вдоль волокон диэлектрическая про-
ницаемость в 1,5—2 раза больше, чем поперек, а с повышением
влажности диэлектрическая проницаемость увеличивается (е
воды равняется 81). Тангенс угла диэлектрических потерьдре-
весины возрастает с увеличением ее плотности.
Химический состав древесины (табл. 16). Сухая древесина
независимо от породы имеет близкий элементный состав, % от
массы а. с. д.: углерод 48—52, водород б—7, кислород 43—45 и
азот 0,1—0,6. Главными составными частями древесины явля-
ются целлюлоза, лигнин и гемицеллюлозы.
Содержание основных компонентов даже в древесине одной
породы колеблется довольно значительно, но в целом в древе-
сине лиственных пород относительно больше пентозанов, одного
из представителей гемицеллюлоз, и меньше лигнина, чем в дре-
весине хвойных пород. Содержание целлюлозы колеблется при-
мерно в одних пределах.
В небольших количествах в древесине содержатся минераль-
ные и разнообразные экстрактивные, извлекаемые растворите-
лями вещества. Минеральные вещества включают в основном
углекислые и кремнекислые соли калия, натрия, кальция,
а также соединения многих других элементов, в сумме они со-
ставляют, как правило, 0,2—1,3 % от массы древесины. К числу
экстрактивных веществ относятся: танниды, терпены,, смоляные
и жирныекмслоты, стерины и т. д.
Гнилая древесина, которая попадает в технологическое
сырье для пиролиза, по химическому составу сильно отличается
142
17. ГРУППЫ ДРЕВЕСНЫХ ПОРОД
Назначение древесного сырья Группа пород
1-я 2-я З-я
Для пиролиза Для углежже- ния Береза, бук, ясень, граб, ильм, вяз, дуб, клен Береза, бук, ясень, граб, ильм, вяз, дуб, клен Осина, ольха, липа, тополь, ива Сосна, ель, кедр, пихта, листвен- ница Осина, ольха, липа, тополь, ива
18. РАЗМЕРЫ ДРЕВЕСНОГО СЫРЬЯ
Назначение древесного сырья Длина, м Отклонение по длине* см Толщина круглых поленьев, см
Для пиролиза Для углежжения 1,0 и кратные 0,75; 1,00; 1,25 и крат- ные 1,0 ±3,0 ±3,0 3—14 3—14
от здоровой. При поражении бурыми гнилями (деструктивный
тип разрушения) резко уменьшается содержание целлюлозы и
более чем в 2 раза увеличивается относительное содержание
лигнина. При поражении белыми гнилями (коррозионный тип
разрушения), напротив, уменьшается содержание лигнина и
увеличивается целлюлозы (примерно до 50 %). Пентозаны раз-
рушаются в обоих случаях, но особенно при поражении бурой
гнилью.
Сырье для пиролиза и требования к нему. Сырьем для пи-
ролиза древесины служит технологическая древесина, преиму-
щественно лиственных пород (береза, бук, дуб и др.). Древесное
сырье хвойных и лиственных пород для пиролиза и углежжения
должно соответствовать ГОСТ 24260—80. Древесное сырье для
пиролиза предназначено для переработки в аппаратах промыш-
ленного типа, для углежжения — кучным методом. Древесное
сырье должно заготавливаться из древесных пород, группы ко-
торых указаны в табл.17.
Древесина дуба может быть использована для пиролиза и
углежжения только в том случае, если она непригодна для вы-
работки дубильных экстрактов. Древесное сырье для пиролиза
2-й группы и углежжения 2-й и 3-й групп заготовляется только
по согласованию с потребителем.
Размеры древесного сырья должны соответствовать требо-
ваниям, указанным в табл. 18.
143
19. КАЧЕСТВО ДРЕВЕСНОГО СЫРЬЯ
Порок древесины Норма порока древесного сырья
для пиролиза для углежжения
Гниль ядровая и за- болонная Гниль наружная трухлявая Допускается в круглых и колотых поленьях размером не более 3 % от площади торца с вы- ходом на один или .оба торца Не допускается Допускается размером не более 15 % в круглых и не более 3 % в колотых полень- ях от площади торца с выхо- дом на один или оба торца Не допускается
Объем круглых поленьев толщиной от 3 до 6 см не должен
превышать 10 % от объема партии. По согласованию с потреби-
телем допускается заготавливать для пиролиза древесное сырье
длиной менее 1 м, для углежжения — менее 0,75 м.
Древесное сырье для пиролиза и углежжения длиной 1 м и
менее допускается заготовлять в расколотом виде, при этом
наибольшая линия раскола по торцу не должна превышать
20 см. Поленья подлежат расколке: толщиной 20—25 см —
на две части; 26—40 см — на четыре части; более 40 см —
на столько частей, чтобы наибольшая линия раскола по торцу
не превышала 20 см. Древесное сырье длиной более 1 м заго-
тавливают только в круглом виде. По качеству древесное сырье
должно соответствовать требованиям, указанным в табл. 19.
Другие пороки древесного сырья, не указанные в таблице,
допускаются. В древесном сырье сучья должны быть срублены
так, чтобы длина оставшейся части после среза не превышала
20 мм. По влажности древесное сырье делится на воздушно-
сухое (влажность 25 % и менее), полусухое (влажность 26—
50%) и сырое (влажность более 50%). Влажность древесного
сырья определяется лабораторией предприятия-потребителя. ч
Глава 8
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПИРОЛИЗА
ДРЕВЕСИНЫ
§ 28. ОБЩАЯ ХАРАКТЕРИСТИКА ПРОЦЕССА ПИРОЛИЗА
Пиролиз древесины — процесс деструкции высокомолекуляр-
ных компонентов древесины с образованием низкомолекулярных
продуктов, сопровождающийся вторичными реакциями конден-
сации, рекомбинации и т. п. реакциями усложнения молекул
с образованием нелетучего остатка, под воздействием тепла
в среде, практически не содержащей кислорода.
144
Процесс пиролиза древесины можно разделить на четыре
стадии:
1. Стадия сушки древесины, заканчивающаяся примерно при
150 °C. На этой стадии из древесины удаляется содержащаяся
в ней влага, химический состав древесины почти не изменяется
и летучих продуктов практически не образуется.
2. Начальная стадия распада древесины, протекающая
Рис. 38. Динамика изменения показа-
телей процесса пиролиза древесины:
1 — температура снаружи реторты; 2 —
температура внутри реторты; 3 — диффе-
ренциальная кривая выделения газов пи-
ролиза; 4 — дифференциальная кривая
выделения жидких продуктов пиролиза
при температуре от 150 до 270—275 °C. В этот период начина-
ется разложение менее термостойких компонентов древесины
с выделением реакционной воды, углекислоты, окиси углерода,
уксусной кислоты и некоторых других продуктов, изменяется
химический и элементный состав.
Обе стадии эндотерм'ичны и идут лишь при подводе тепла.
3. Стадия собственно пиролиза — бурного распада древе-
сины с выделением тепла (экзотермический процесс) и образо-
ванием основной массы продуктов разложения. Начинается она
при 270—275 °C и заканчивается примерно при 400 °C.
4. Стадия прокалки угля (не считая охлаждения угля), за-
канчивающаяся в зависимости от аппарата и способа пиролиза
при температуре 450—600 °C. При этом выделяется небольшой
объем жидких продуктов и значительный газов. Осуществля-
ется снова за счет подвода тепла извне.
Общая характеристика процесса пиролиза древесины в ап-
парате (реторте) периодического действия с внешним нагревом
наглядно показана на рис. 38. Кривые изменения температур
показывают, что, несмотря на начальное значительное повыше-
ние температуры снаружи реторты, температура внутри ее дол-
гое время остается в пределах 150 °C (пока не закончится
процесс сушки). Затем температура внутри реторты быстро
поднимается и, когда начинается бурное экзотермическое раз-
ложение, становится выше температуры снаружи реторты. После
завершения выделения основного объема жидких и газообраз-
ных продуктов температуру снаружи повышают, начинают про-
калку угля, выделение жидких продуктов в этот период умень-
шается, а под конец почти полностью прекращается.
В результате пиролиза древесины образуются следующие
первичные продукты: нелетучий остаток — древесный уголь и
145
парогазовая смесь, которая после охлаждения в конденсацион-
ной системе делится на водный конденсат, называемый жижкой,
и неконденсируемые в этих условиях газы.
Неконденсируемые газы. Газы пиролиза древесины содер-
жат двуокись и окись углерода, метан, непредельные углево-
дороды и водород. Состав газов, выделяющихся в ходе пиро-
лиза, сильно меняется. В начальный период выделяются газы
с преимущественным содержанием двуокиси (до 75 %) и окиси
углерода. По мере повышения температуры содержание дву-
окиси углерода падает и увеличивается содержание горючих
компонентов — вначале окиси углерода, а затем метана, непре-
дельных углеводородов и водорода; теплотворная способность
газа повышается.
Жидкие продукты. Жижка представляет собой эмульсию от-
стойной нерастворимой смолы в водном растворе органических
веществ, расслаивающуюся на отстойную смолу (нижний слой)
и прозрачный, цвета крепкого чая, резко пахнущий водный
слой — отстоявшуюся жижку (верхний слой). Иногда на по-
верхности жижки отстаиваются так называемые всплывные
масла.
Отстоявшаяся жижка — водный раствор нескольких сот ин-
дивидуальных химических веществ, представителей практически
всех классов органических соединений, содержащих углерод,
водород, кислород и даже азот. Среди них можно назвать одно-
атомные спирты (метиловый и аллиловый) и гликоли (этилен-
гликоль); альдегиды (формальдегид, ацетальдегид, фурфурол);
диальдегиды (глиоксаль) и оксиальдегиды (гликолевый альде-
гид), ацетали (диметилацеталь); кетонь! (ацетон), дикетоны
(диацетил), оксикетоны (оксиацетон) и кетоальдегиды (метил-
глиоксаль); кислоты (муравьиная, уксусная, пропионовая, ак-
риловая) и их метиловые эфиры, оксикислоты (гликолевая) и
их лактоны (у-валеролактон), ангидросахара (левоглюкозан),
фенолы (фенол, пирокатехин, гваякол, диметиловые эфиры пи-
рогаллола), метиламины, пиридин и другие соединения. Отсто-
явшаяся жижка используется для выделения из нее уксусной
кислоты. Одновременно в отдельных случаях получают так на-
зываемую растворимую смолу — часть веществ, которые не из-
влекаются из жижки органическими растворителями и при ее
упаривании остаются в кубе.
Древесный уголь. Это твердый продукт пиролиза древе-
сины— хрупкое, пористое вещество блестящего черного цвета
с синеватым отливом в изломе, сохранившее в известной мере
микро- (клеточные пустоты, поры) и макроструктуру (годичные
кольца, сердцевинные лучи) исходной древесины.
Органическая масса угля состоит из углерода, кислорода и
водорода, массовая доля которых в угле зависит от конечной
температуры пиролиза. При повышении температуры пиролиза
доля углерода возрастает, а кислорода и водорода падает.
В угле содержится до 3 % минеральных примесей, главным об-
146
разом окислов и карбонатов калия, натрия, кальция и, в мень-
шей степени, других элементов; для него характерным является
низкое содержание фосфора и серы.
Древесный уголь — высокопористое тело, общий объем пор
достигает 75 % объема угля. Подавляющим видом пор явля-
ются макропоры (поры с эффективным радиусом>100 нм),
объем переходных пор и микропор (пор с эффективным радиу-
сом <1,5 нм) невелик.
В связи с развитой пористостью плотность угля истинная
(без пор) и кажущаяся (с порами) сильно различаются. Так,
истиная плотность промышленных углей равна 1350—
1450 кг/м3, а кажущаяся в несколько раз ниже и зависит от по-
роды древесины (например, для угля из березы равна примерно
350 кг/м3).
Химическая структура угля изучена слабо. Можно лишь ска-
зать, что упомянутые выше изменения в элементном составе при
повышении температуры пиролиза ведут к накоплению в угле
углерода с sp2 и sp гибридизацией, т. е. с двойными и трой-
ными связями, и конденсированных ароматических структур.
Несмотря на это, древесный уголь относится к труднографити-
руемым материалам. Угли, полученные при 400—500 °C, рент-
геноаморфны.
Древесный уголь — диэлектрик, удельное сопротивление
угля, полученного при температуре пиролиза 400 °C, составляет
около 109 Ом/м. При повышении температуры пиролиза, по мере
обогащения угля углеродом, электрическая проводимость уве-
личивается, сопротивление уменьшается.
Древесный уголь обладает парамагнитными свойствами, т. е.
способностью, будучи помещенным в магнитное поле, погло-
щать электромагнитное излучение. Парамагнетизм обусловлен
наличием в угле свободных радикалов, стабилизированных
в твердой полимерной матрице угля, и парамагнитными свойст-
вами полисопряженных систем, природа которых недостаточно
изучена. Парамагнетизм — важное физическое свойство угля,
так как наличием свободных радикалов и их реакциями опре-
деляется высокая реакционная способность древесного угля
к низкотемпературному взаимодействию с кислородом и склон-
ность к самовозгоранию. Концентрация парамагнитных центров
в угле, полученном при 450 °C, составляет около 4-Ю19 спин/г,
или, другими словами, один парамагнитный центр приходится
примерно на 1200 углеродных атомов.
§ 29. ПИРОЛИЗ КОМПОНЕНТОВ ДРЕВЕСИНЫ И ХИМИЗМ
ОБРАЗОВАНИЯ ОСНОВНЫХ ПРОДУКТОВ
Пиролиз древесины — это сложнейший физико-химический
процесс. В изучение теории пиролиза, начатое в начале века,
большой вклад внесли многие зарубежные (Класон, Бэнбери,
Хаулей, Пальмер) и отечественные (К. И. Ногин, В. Н. Козлов,
А. К. Славянский, В. И. Корякин и др.) ученые. Однако многое
147
остается недостаточно выясненным до настоящего времени. Сле-
дует подчеркнуть, что разработка законченной теории пиро-
лиза наталкивается на серьезнейшие трудности, связанные
с многокомпонентным составом древесины, сложностью физиче-
ского и химического строения компонентов, вторичными превра-
щениями первичных продуктов распада и т. п. Тем не менее за
последние два десятилетия усилиями многих исследователей
(О. П. Головой, В. Н. Сергеевой, Г. Э. Домбург, А. Н. Кисли-
цына— СССР; М. Котика — ЧССР; К. Като — Япония и др.)
в изучении теоретических вопросов пиролиза древесины достиг-
нуты значительные успехи. Они стали возможными благодаря
общему прогрессу науки и появлению новых, преимущественно
инструментальных, методов исследования. К их числу можно
отнести: термические методы анализа, в частности дифференци-
альный термический анализ и дифференциальную сканирую-
щую калориметрию, радиоспектроскопию, особенно метод элек-
тронного парамагнитного резонанса (ЭПР), газожидкостную и
гельпроникающую хроматографию, метод меченых атомов и т.д.
Ввиду сложности строения древесины и ее состава при изу-
чении пиролиза принято вначале изучать распад изолированных
компонентов, чтобы установить связь между ними и образую-
щимися продуктами, изучить ход процесса пиролиза в чистом
виде, выяснить химизм образования отдельных продуктов, а за-
тем использовать эти данные для объяснения процесса пиролиза
древесного комплекса. В соответствии с этим подходом сначала
рассмотрим пиролиз основных компонентов древесины — геми-
целлюлоз, целлюлозы и лигнина и химизм образования из них
отдельных продуктов.
Пиролиз гемицеллюлоз. Гемицеллюлозы — это смешанные
полисахариды, построенные из пентоз и гексоз. В древесине ли-
ственных пород, основном виде сырья пиролизных предприя-
тий, преобладают пентозаны, главным представителем которых
.является 4-О-метилглюкуроноксилан
о
ОН ОАс
Ас = СОСН3
4-О-метилглюкуроноксилан — полисахарид, цепная макро-
молекула которого построена из остатков D-ксилозы (степень
148
полимеризации около 200), соединенных 1,4-0-глюкозидной
связью. Примерно к каждому седьмому элементарному звену
макромолекулы 1,2-а-глюкозидной связью присоединены ос-
татки 4-О-метилглюкуроновой кислоты. Некоторые карбоксиль-
ные группы находятся в виде метиловых эфиров. Гидроксиль-
ные группы 3 или 2 углеродных атомов каждого 1,5 элементар-
ного звена этерифицированы уксусной кислотой (массовая доля
ацетильных групп по расчету равна примерно 16%).
При пиролизе древесины из ксилана образуются газы, жид-
кий конденсат и твердый угольный остаток. Ксилан ответствен
за образование основной массы метанола, уксусной кислоты и
фурфурола, образующихся при пиролизе древесины. Все лету-
чие продукты пиролиза ксилана растворимы в водном конден-
сате; нерастворимых летучих продуктов практически не обра:
зуется. Относительный выход угольного остатка из ксилана не-
велик и в 2—3 раза ниже, чем из лигнина.
Рассмотрим процесс термораспада ксилана, содержащего
11,5 % ацетильных групп, т. е. по составу весьма близкого к на-
тивному, и химизм образования из него наиболее характерных
продуктов (по данным А. Н. Кислицына и Е. В. Ищерикова).
Ацетилсодержащий ксилан имеет низкую термическую ус-
тойчивость. Деструкция макромолекулы ксилана, как показы-
вает изучение характера изменения вязкости медно-аммиачных
растворов ксилана после термообработки, начинается при 120—
140 °C случайным разрывом цепи по ослабленным связям.
При этом образуются фрагменты постепенно уменьшающейся
по мере повышения температуры молекулярной массы. Характер
распада ксилана на начальной стадии тесно связан с особенно-
стями его химического и физического строения. Ксилан как
в изолированном виде, так и в клеточной стенке древесины
имеет аморфную структуру. Неупорядоченное расположение
макромолекул со случайными водородными связями, которые
при большом числе способствуют упрочнению структуры, и на-
личие в ксилозных звеньях электроотрицательных заместите-
лей, ослабляющих глюкозидную связь, обусловливают как низ-
кую термическую стойкость ксилана, так и случайный характер
разрыва цепи макромолекулы. Деструкция макромолекулы вна-
чале не сопровождается химическими изменениями структурных
звеньев и потерей массы.
На рис. 39 приведены кривые термического анализа ацетил-
содержащего ксилана: термогравиметрическая кривая (ГГ) по-
казывает интегральное (суммарное) изменение массы образца;
дифференциальная термогравиметрическая кривая (ДТГ) —
дифференциальное (в единицу времени) изменение массы,
а кривая дифференциального термического анализа (ДТА) сви-
детельствует о выделении или поглощении тепла в процессе
термораспада. Из графика видно, что после удаления влаги до
200 °C потерь массы не наблюдается (ТГ—кривая). Активный
распад ксилана идет при 220—290 °C с максимумом потери
149
массы при 255 °C (ДТТ-кривая). Процесс протекает с погло-
щением тепла с эндоэффектом при 240 °C (ДТЛ-кривая). Энер-
гия активации в этот период равна 2,8-102 кДж/моль. Макси-
мум скорости потери массы совпадает с максимумом скорости
образования фурфурола, уксусной кислоты, метанола, диоксида
углерода (рис. 40). После 290 °C скорость потери массы резко
падает. Угольный остаток при 500 °C составляет 14 % от массы
Рис. 40. Дифференциальные кривые
образования летучих продуктов пи-
ролиза ацетилсодержащего ксилана:
1 — метанол; 2 — фурфурол; 3 — уксусная
кислота; 4 — вода; 5 — двуокись углерода
Рис. 39. Термограмма ацетилсодержащего ксилана и динамика изменения
концентрации парамагнитных центров:
ДТА — дифференциально-термическая кривая; ТГ — термогравиметрическая кривая;
ДТГ — производная термогравиметрическая кривая; ПМЦ — кривая изменения концен-
трации парамагнитных центров
исходного ксилана. Измерение концентрации парамагнитных
центров в процессе пиролиза (см. рис. 39, кривая' ПМЦ) пока-
зывает, что в период активного распада концентрация их не
увеличивается. Стремительный рост концентрации ПМЦ про-
исходит после образования основной массы летучих продуктов;
к 300 °C она увеличивается в 10 раз, к 350 °C еще в 10 раз.
Летучих продуктов в это время выделяется мало, а идут слож-
ные процессы с разрывом связей по свободнорадикальному ме-
ханизму и образованием структуры угля. С ростом температуры
существенно меняется состав газов—уменьшается содержание
двуокиси углерода и увеличиваются его окиси (при 320 до 30 %,
при 400 °C до 50 %) • При 400 °C в газе появляется метан.
150
Рассмотрим, как при термораспаде ксилана образуются наи-
более характерные продукты — фурфурол, уксусная кислота и
метанол. Фурфурол и фурановые соединения при пиролизе,
а также при гидролизе древесины образуются в основном из
пентозанов. Однако если при гидролизе фурфурол является
главным продуктом превращения пентозанов и выход его при-
ближается к теоретическому, то при пиролизе его выход неве-
лик. Так, при пиролизе дезацетилированного ксилана получа-
ется самое большее (9%) содержание фурфурола, или около
12 % от теоретически возможного. Из нативного ксилана, на-
ходящегося в древесине, при предпиролизе в среде жидкого
теплоносителя фурфурола образуется также около 12 % от тео-
ретического выхода. Фурфурол образуется в результате трех
последовательных реакций отщепления воды от ксилозы. На
первой стадии образуется 3-дезоксиксилозон НОСН2—
СН (ОН) —СН2—СО—С ( = 0) Н, при последующей дегидрата-
ции фуранозной формы которого и получается фурфурол. Об-
разование фурфурола — это сложный, многостадийный процесс.
Низкий выход фурфурола объясняется тем, что при простом пи-
ролизе образуется мало ксилозы, а образовавшаяся ксилоза не
только дёгидратируется в ксилозон, но и по конкурирующей ре-
акции полимеризуется в олигосахара.
Незначительные количества фурановых соединений, в част-
ности 5-оксиметилфурфурол, оксиметил-2-фурилкетон и про-
дукты их распада, образуются при пиролизе целлюлозы.
Уксусная кислота при пиролизе древесины, как показано
К. Падовани и С. С. Ермолаевой, образуется из ацетильных
групп, которые связаны с ксиланом. Значительно меньшая часть
уксусной кислоты получается при пиролизе целлюлозы и лиг-
нина. Предполагалось, что образование кислоты из ксилана про-
исходит в результате гидролиза сложноэфирной связи:
с—о—СОСНз+Н2О-> —С—ОН + СНзСООН.
(61)
В настоящее время установлено, что уксусная кислота обра-
зуется не по реакции гидролиза, а путем спонтанного отщепле-
ния ацетильных групп под воздействием тепла по двум основ-
ным реакциям: 1) по молекулярной реакции р-отщепления
с участием водорода а-углеродного атома:
о=с
\
сн3
/ \
> НО н
СНу—соон
О \[ (62)
151
2) по гетеролитической реакции так называемого 1,3-отщепле-
ния с участием водорода гидроксильной группы у того же со-
седнего атома углерода:
сн^соон
(63)
Водяной пар не оказывает влияния на скорость образования
уксусной кислоты из ацетилсодержащего ксилана, но при пиро-
лизе кусковой влажной древесины, когда продолжительность
нагрева может оказаться очень большой, можно допустить и
гидролитический путь отщепления ацетильных групп.
Древесина любой породы содержит определенное число ме-
токсигрупп СН3О—, поэтому вполне естественно, что образова-
ние метилового спирта при пиролизе исследователи связали
с их наличием в древесине. Так как лигнин содержит значитель-
ное число метоксилов и дает при пиролизе метанол, вначале
считали, что последний образуется из метоксигрупп лигнина.
Однако выход метанола из древесины, например, твердолист-
венных пород, равный примерно 1,5—1,8 %, намного ниже теоре-
тического, рассчитанного на содержание метоксигрупп, равного
5—6 %. В то же время выход спирта примерно соответствует со-
держанию в древесине легкоотщепляемых метоксигрупп, свя-
занных с гемицеллюлозами. Поэтому и пришли к выводу, что
метанол образуется в основном не из лигнина, а из гемицеллю-
лоз. Исследования механизма термораспада лигнина подтвер-
дили этот вывод, показав, что в силу специфики химизма рас-
пада метоксигруппы лигнина не могут служить источником об-
разования существенной массы метанола.
Метоксигруппы в ксилане березовой древесины находятся
у четвертого углеродного атома С<4) звена метилглюкуроновой
кислоты. Группа углеродных атомов С(4), С(5> и С<6) метилглю-
куроновой кислоты по структуре аналогична р-замещенной про-
пионовой кислоте ХСНг—СНг—СООН. Характерной особен-
ностью таких кислот является отщепление заместителей при
простом нагревании при невысоких температурах (150—
250 °C) по механизму внутримолекулярного р-отщепления
|< >1 ' \оон (.«.
CII1O' СН.ОН
В метилглюкуроновой кислоте имеются еще более благопри-
ятные предпосылки для отщепления метоксигрупп, чем в р-за-
152
мещенной метоксипропионовой кислоте, благодаря наличию
у С(5> не одной, а двух сильно электроотрицательных групп —
карбоксильной и ацетильной. Две группы способствуют боль-
шему сдвигу электронной пары связи С<5>—Н в сторону угле-
рода, т. е. большей протонизации водорода, как раз и являю-
щейся причиной легкого отщепления р-заместителей.
Двуокись углерода при пиролизе ксилана образуется за счет
отщепления карбоксильных групп метилглюкуроновой кислоты.
К 240 °C в ацетилсодержащем ксилане остается всего двадца-
тая часть карбоксигрупп, а газ на 85 % состоит из углекислоты.
С небольшим выходом при пиролизе ксилана получаются од-
ноатомные фенолы — фенол, о-(основной компонент, 80 % от фе-
нолов), м- и п-крезолы. Образуются они в результате вторич-
ных реакций конденсации первичных карбонилсодержащих про-
дуктов распада ксилана. В то же время в угольном остатке
даже при 510 °C не обнаружено ароматических структур.
Обобщая данные о пиролизе ксилана и образовании отдель-
ных соединений, следует отметить, что вследствие особенностей
химизма отщепления ацетильных групп условия обычного пиро-
лиза недостаточно благоприятны для образования уксусной кис-
лоты. Еще в большей степени относится это к образованию фур-
фурола. Варьирование режимных параметров процесса, напри-
мер темпа нагрева, вида теплоносителя и т. п., существенно не
изменит выходов этих продуктов, так как не затрагивает хи-
мизма первичных процессов их образования.
Пиролиз целлюлозы. Целлюлоза — основной компонент дре-
весины и самый распространенный природный полимер. Макро-
молекула целлюлозы — это линейный неразветвленный полимер,
построенный из элементарных звеньев D-глюкопиранозы, соеди-
ненных 1,4-р-глюкозидными связями, общей формулой
[С6Н7О2(ОН)з]п -
Цепные макромолекулы, располагаясь параллельно друг
другу, образуют пучки, называемые элементарными фибрил-
лами, которые содержат примерно 80 макромолекул. Строение
элементарных фибрилл неоднородно. Они состоят из областей
с упорядоченной структурой — кристаллитов, разделенных
аморфными участками с перекрещивающимся и искривленным
расположением цепей целлюлозы. Эти физические и химические
особенности строения предопределяют характер термораспада
целлюлозы.
153
Целлюлоза имеет более высокую термическую устойчивость
по сравнению с ксиланом благодаря линейному регулярному
строению макромолекул и наличию многочисленных водородных
связей между макромолекулами в элементарной фибрилле.
Процесс пиролиза целлюлозы в общем виде, по данным тер-
мического анализа (рис. 41), протекает следующим образом.
Вначале в широком температурном диапазоне от 90 до 150 °C
Рис. 41. Термограмма целлю-
лозы
дуктов, то экзотермический
Газ, образующийся при
идет испарение физически связан-
ной воды. Активный распад целлю-
лозы с потерей массы начинается
при 280 °C и заканчивается при-
мерно при 370 °C. Максимум ско-
рости потери массы приходится на
330—335 °C (ДГЛкривая). В пе-
риод активного распада теряется
около 60—65 % массы навески.
Дальнейшая потеря массы идет
с меньшей скоростью, остаток при
500 °C составляет 15—20 % от на-
вески целлюлозы (ТГ-кривая). Ак-
тивный распад протекает с погло-
щением тепла (ДГА-кривая). Эндо-
термический процесс переходит в
экзотермический с максимумом вы-
деления тепла при 365 °C, т. е. уже
после основной потери массы. Эк-
зотермика с максимумом при
365 °C связана с вторичными реак-
циями — с распадом первичных
продуктов. Если термический ана-
лиз проводить в вакууме, т. е. обес-
печить эвакуацию первичных про-
пик на ДТД-кривой исчезает.
пиролизе целлюлозы, в рассматри-
ваемом температурном интервале, состоит в основном из дву-
окиси и окиси углерода. Характер термических превращений
целлюлозы зависит от степени и продолжительности нагрева,
т. е. от температурно-временных факторов.
Рассмотрим вначале превращения целлюлозы при низкотем-
пературной обработке, при температурах ниже температуры ак-
тивного распада, т. е. ниже 280 °C. Низкотемпературная де-
струкция целлюлозы характеризуется довольно быстрым сниже-
нием степени полимеризации (СП) целлюлозы до определенной
предельной величины, после чего она изменяется медленно. Ве-
личина предельной СП зависит от вида целлюлозы и, например,
для хлопковой целлюлозы близка к 200, а для древесной равна
30—40. Продолжительность нагрева, необходимая для достиже-
ния предельной СП, зависит от температуры и уменьшается
с ростом последней, например для целлюлозы фильтровальной
154
бумаги № 1 от 6 ч при 225 °C до 25 мин при 280 °C. Первичный
термораспад целлюлозы происходит по аморфным участкам эле-
ментарных фибрилл. Кристаллическая структура целлюлозы
при этом сохраняется. Основным продуктом распада является
вода. Но образование двуокиси углерода и низкомолекулярных
органических веществ (гликолевого альдегида, глиоксаля и др.)
свидетельствует о глубоком разложении некоторой части эле-
ментарных звеньев целлюлозы. В целом целлюлоза сохраняет
свои свойства.
Совершенно другой продукт получается, если целлюлозу
после достижения ею предельной СП нагревают при тех же тем-
пературах в течение продолжительного времени. Происходят
глубокие физико-химические изменения всей массы целлюлозы.
В остатке повышается содержание углерода, падает содержание
а-целлюлозы, разрушается кристаллическая структура. Глубина
превращений зависит от температуры и продолжительности на-
грева. Главный продукт распада, как и при кратковременном
нагреве — вода. Поэтому образующийся остаток целлюлозы на-
зывают ангидроцеллюлозой. Основной реакцией на этой стадии
является внутримолекулярная внутризвенная дегидратация, ко-
торая приводит к образованию карбонилсодержащих структур
и двойных связей. Чем выше температура обработки, тем боль-
шая доза целлюлозы подвергается глубокой деструкции с об-
разованием низкомолекулярных соединений.
По свойствам ангидроцеллюлоза сильно отличается от цел-
люлозы. На термограммах ангидроцеллюлозы отсутствуют ха-
рактерные для целлюлозы эндотермический пик, совпадающий
с максимальной скоростью потери массы, и следующий за ним
экзотермический. Изменяется состав летучих продуктов пиро-
лиза ангидроцеллюлозы: чем больше потеря массы при ее полу-
чении, тем больше выход ацетальдегида, фурана и ацетона при
пиролизе.
Таким образом, при низкотемпературном нагреве целлюлозы
вначале разрушаются аморфные области, остаток сохраняет ос-
новные физико-химические свойства исходного материала. За-
тем, при длительном нагреве, термораспад охватывает и кри-
сталлиты, в результате чего образуется продукт, по свойствам
непохожий на целлюлозу.
Бурный распад целлюлозы с интенсивной потерей массы на-
чинается примерно при температуре 280 °C. Характер протекаю-
щих при этом процессов зависит, при прочих равных условиях,
от темпа нагрева — быстрого или медленного.
При быстром подъеме температуры, например при внесении
целлюлозы в предварительно нагретую зону пиролиза, процесс
протекает следующим образом. Вначале, как показано О. П. Го-
ловой с сотрудниками, пиролизуются аморфные области эле-
ментарных фибрилл целлюлозы и СП быстро падает до предель-
ной величины. Потеря массы невелика и составляет 4—8 % •
Последующий распад целлюлозы протекает иначе, чем при низ-
155
котемпературной обработке. Для этой стадии характерен высо-
кий выход левоглюкозана — соединения, по брутто-формуле —
C6HioOs — соответствующего элементарному звену макромоле-
кулы целлюлозы. Выход левоглюкозана при благоприятных
условиях (вакуум, низкое содержание минеральных солей в цел-
люлозе) достигает 60—65%. Такой процесс, в котором с вы-
соким выходом образуется мономер, лежащий в основе поли-
fl --1-1--1----1-1----1-1____UQ0
0 10 20 30 40 50 60 70 80 '
Степень разложения, °/о
Рис. 42. Динамика изменения показателей процесса пиролиза целлюлозы при
300 °C и давлении 7 • 10-3 Па:
1 — выход левоглюкозана, % от разложившейся в данный момент целлюлозы; 2 — ско-
рость распада; 3 — степень полимеризации целлюлозы
мера, называется деполимеризацией, в данном случае — терми-
ческой. Выход левоглюкозана очень быстро достигает постоян-
ного значения — 70 % от массы целлюлозы, разложившейся
в данный момент, и практически не изменяется до конца про-
цесса распада (рис. 42). Остаток, несмотря на увеличиваю-
щуюся потерю массы, прямо пропорциональную продолжитель-
ности нагрева, долгое время сохраняет свои первоначальные фи-
зико-химические свойства. Практически не меняется СП остатка.
Инфракрасные спектры остатков идентичны спектру исходного
материала до тех пор, пока потеря массы не превысит 70%.
ДТД-кривые остатков лйшь после потери 80 % массы становятся
непохожими на ДТД-кривую целлюлозы. Состав остатков про-
дуктов пиролиза, определенный пиролитической хроматогра-
фией, близок, пока потеря массы целлюлозы при пиролизе не
будет более 70 %.
Такой характер процесса, когда идет распад полимера и об-
разуется мономер, а степень полимеризации остатка не меня-
ется, возможен в том случае, если начавшая распадаться ма-
156
кромолекула деполимеризуется полностью, в то время как дру-
гие макромолекулы остаются незатронутыми. Доказано
(А. Н. Кислицыным с сотрудниками), что деполимеризация
целлюлозы — это цепной свободнорадикальный процесс, началь-
ным актом которого является гомолитический разрыв 1,4-глюко-
зидной связи между углеродным атомом C(i> и кислородом. По-
следующий химизм процесса образования левоглюкозана может
быть изображен в виде следующей схемы:
левы люкоэан
В образовавшемся первоначально макрорадикале I водород
гидроксильной группы —СН2ОН благодаря близкому располо-
жению к алкоксильному С(4)О« радикалу и примерно одинако-
вой прочности связей О—Н в первичных и вторичных спиртах
может переходить к С(4>0» и возвращаться обратно от С(4)ОН
к —СН2О-. Вследствие этого макрорадикал I находится в под-
вижном равновесии с макрорадикалом II. В свою очередь ма-
крорадикал И вследствие конформационных переходов конеч-
ного звена из одной конформации в другую — из К1 в 1К и об-
ратно— находится в равновесии с другой формой III того же
макрорадикала. Скорость переходов велика и оценивается вели-
чиной порядка 104с~*. И в тот момент, когда макрорадикал III
находится в конформации 1К, происходят атака алкоксильным
радикалом —СН2О- глюкозидной связи C(i>—О, ведущей к сле-
157
дующему элементарному звену, и разрыв ее с замещением гли-
козидного кислорода у C(i) алкоксильным. В результате образу-
ется левоглюкозан, а свободнорадикальный центр передается на
следующее звено макромолекулы. Затем снова все многократно
повторяется.
Таким образом, термическая деполимеризация целлюлозы
квалифицируется как цепной свободнорадикальный процесс
с внутримолекулярной передачей цепи и образованием левоглю-
козана в едином реакционном комплексе. Физическая структура
кристаллитов создает благоприятные предпосылки для протека-
ния реакции деполимеризации. Во-первых, наличие многочис-
ленных водородных связей повышает термическую устойчивость
кристаллитов, что позволяет им сохраниться до той критиче-
ской температуры, когда подводимой энергии становится доста-
точно для гомолитической диссоциации глюкозидной связи и
зарождения цепного свободнорадикального процесса деполиме-
ризации. Во-вторых, благодаря протеканию деполимеризации
в твердой фазе макрорадикала и отсутствию условий для его
диффузии свободнорадикальный процесс локализуется
в «клетке» и не переходит в объем. Тем самым обеспечивается
направленность процесса в сторону образования левоглюкозана.
Деполимеризация является хотя и главной, но не единствен-
ной реакцией термораспада при высокотемпературном скорост-
ном пиролизе целлюлозы. Параллельно протекают реакции де-
гидратации и глубокого разложения ангидроглюкозных звеньев,
образования побочных продуктов деполимеризации, вторичного
распада первичных продуктов.
Соотношение между двумя основными направлениями термо-
распада целлюлозы — дегидратацией целлюлозы с последую-
щим хаотичным распадом ангидроцеллюлозы и деполимериза-
цией с образованием левоглюкозана, а следовательно, и выход
соответствующих им продуктов зависят от температуры пиро-
лиза и скорости нагрева. При медленном нагреве до темпера-
туры интенсивного распада или при предварительном изотерми-
ческом низкотемпературном нагреве реакцией дегидратации
затрагиваются значительные части целлюлозы, нарушая ее
структуру и строение. Вследствие этого нарушения деполимери-
зация и образование левоглюкозана идут в меньшей мере, при
этом увеличивается выход угольного остатка. При быстром, ско-
ростном нагреве (при наличии других условий — низкой золь-
ности целлюлозы и быстрого выноса продуктов из зоны реак-
ции) создаются благоприятные условия для деполимеризации:
процессы дегидратации не успевают развиться, выход угля
уменьшается. Например, при пиролизе при температуре 370 °C
обеззоленной фильтровальной бумаги, предварительно нагре-
той до 275 °C, выход угля составляет 27,6%, а без предвари-
тельной температурной обработки — всего 13,1 %.
Состав летучих продуктов пиролиза целлюлозы сложен,
в них найдено не менее 100 индивидуальных веществ. Среди них
158
прежде всего следует назвать группу веществ, сохранивших уг-
леродный скелет элементарного звена целлюлозы из шести
атомов:
1 ,4; 3,С- диангидро- 5- оксиметил-
глюкопнраноза 'Фурфурол
3,6- анги.лроглюко-'
пираноза
СН—СИ
С-СО-СН2ОН
мальтол
оксиметил фурилкетон
СНз
метилци кл о пентек-»
-2 -ОЛ-2-ОН-1
Некоторые из этих веществ образуются непосредственно из
элементарного звена, другие, по-видимому, из промежуточно по-
лучающейся глюкозы. Глюкоза также найдена в продуктах
низкотемпературного пиролиза целлюлозы. Остальные продукты
пиролиза целлюлозы — результат деградации углеродной цепи
элементарного звена целлюлозы. В их числе имеются вещества
с тремя (глицериновый альдегид, диоксиацетон, оксиметилглио-
ксаль, метилглиоксаль, пировиноградная кислота, ацетон, ак-
ролеин), двумя (глиоксаль, гликолевый альдегид, ацетальдегид,
уксусная кислота) и с одним (формальдегид, окись и двуокись
углерода) углеродным атомом.
Формирование структуры угольного остатка при термообра-
ботке целлюлозы идет постепенно. При 200 °C в инфракрасных
спектрах остатка появляются полосы при длине волны 5,75 и
6,15 мк, характерные соответственно для групп С = О и С = С,
что указывает на начало внутримолекулярной дегидратации. Су-
щественные изменения происходят после нагрева целлюлозы при
280 °C, спектр остатка совершенно не походит на спектр целлю-
лозы. Полосы поглощения связей О — Н, С — Н, С — ОН и
159
С — О, сохранявшиеся еще при температуре 240 °C, быстро ос-
лабевают. Основными становятся полосы при 5,87 и 6,2 мк, со-
ответствующие сопряженной с двойной связью С = О группе и
сопряженным С=С связям. Затем полоса при 5,87 мк уменьша-
ется и при 500 °C исчезает полностью. Полоса при 6,2 мк при
температуре 600 °C становится наиболее интенсивной, что сви-
детельствует о развитии систем с сопряженными связями. Ин-
тенсивное ослабление полос, характерных для С = О группы, со-
впадает с максимумом скорости выделения окиси и двуокиси
углерода. При медленном подъеме температуры уже при 200—
250 °C в остатке появляются ароматические структуры. К тем-
пературе 300 °C не менее 30 % углерода находится в ароматиче-
ских соединениях; к 700—900 °C образуются высококонденсиро-
ванные, но слабо ориентированные структуры. Графитации цел-
люлозного угольного остатка не наблюдается даже при очень
высоких температурах. Фибриллярное строение целлюлозы
в угольном остатке сохраняется.
При пиролизе древесины из целлюлозы, так же как из ге-
мицеллюлоз, образуется много воды (около 35 %) и многочис-
ленные кислородсодержащие соединения, большая часть которых
растворима в водном конденсате. Относительный выход угля
из целлюлозы, как и из гемицеллюлоз, значительно ниже, чем
из лигнина.
Пиролиз лигнина. Лигнин — последний основной компонент
древесины, представляющий собой нерегулярный сильно развет-
вленный полимер весьма сложного, неустановленного точно
строения. Макромолекулы лигнина построены из фенилпропано-
вых Се — Сз структурных единиц. Непосредственными веще-
ствами, из которых при биосинтезе строятся макромолекулы
лигнина, являются синаповый, конифериловый и п-кумаровый
(п-оксикоричный) спирты.
R
но—\-сн=сн-сн2он
«6 синаповый спирт: R = R* == ОСН3
К конифериловый спирт: R =Н; R'= ОСН3
кумаровый спирт: R= R'= Н
Этим спиртам в макромолекуле соответствуют сирингил-,
гваяцил- и п-оксифенилпропановые структурные единицы. Строе-
ние лигнинов различных пород неодинаково. Лигнин древесины
березы — основного сырья пиролизного производства — построен
из сирингил- и гваяцилпропановых единиц, а лигнин древесины
осины содержит дополнительно п-оксифенилпропановые еди-
ницы. Структурные единицы в макромолекуле лигнина соеди-
нены разнообразными простыми эфирными и углерод-углерод-
ными связями. Основными типами связи являются р- и а-ал-
киларильные эфирные связи, имеющие невысокую прочность.
160
ан
'С происходит без at
процесс с максимумом при
А
о
Рис. 43. Термограмма лигнина
Бьеркмана древесины березы
200 300 ЛОО 500 600
Температура( °C
Кривые термического анализа лигнина Бьеркмана древесины
березы, наиболее полно отражающего физико-химические свой-
ства нативного лигнина, имеют по сравнению с таковыми для
ксилана и целлюлозы значительно более сложный вид (рис. 43).
Уже на ранних стадиях нагрева — при 150 °C — имеется не-
большой максимум скорости потери массы (ДТГ-кривая) и сле-
дующий за ним экзотермический
175 °C (ДТА-кривая) без потери
массы. Распад лигнина с посто-
янной, примерно равномерной
скоростью потери массы начи-
нается с температуры 200 °C и
продолжается примерно до
540 °C, после чего скорость по-
тери массы уменьшается (ТГ-кри-
вая). Начальный период распада
начинается с эндотермического
процесса с максимумом при
210 °C. Дальнейший терморас-
пад до 400 °~
больших тепловых эффектов, хотя
можно выделить две эндотер-
мические стадии распада, сопро-
вождающиеся потерей массы:
при 320—325 и 390 °C. После
этого происходит бурный экзо-
термический распад (ДГА-кри-
вая), во время которого выде-
ляется примерно половина лету-
чих продуктов.
Термический анализ показы-
вает, что лигнин Бьеркмана дре-
весины березы имеет низкую
термическую устойчивость — дест-
рукция с потерей массы и пере-
стройка макромолекулы начи-
наются при температуре 200 °C,
но общая потеря массы меньше,
чем у ксилана и целлюлозы, и к 600 °C равна 64,5 %
При термообработке лигнина Бьеркмана древесины березы
в области температуры стеклования лигнина (150—200 °C) на-
блюдается снижение концентрации парамагнитных центров при-
мерно на 20 %, вызванное рекомбинацией свободных радикалов,
находящихся в препарате лигнина. При дальнейшем повыше-
нии температуры до 300 °C наблюдается незначительный рост
концентрации парамагнитных центров. И лишь с 300 °C происхо-
дит стремительный рост их концентрации с 2,4-1017 до
1,5-1018 спин/г при 400 °C. Природа парамагнитных центров
в процессе термообработки меняется: до температуры примерно
6 Заказ Ns 2193 161
Т.20
Г"
гй 60
370 °C парамагнитные свойства вызваны наличием реакцион-
носпособных свободных радикалов, выше — парамагнитных по-
лисопряженных систем.
Продуктами пиролиза лигнина, как и других компонентов
древесины, являются твердый нелетучий углеродистый остаток,
жидкий конденсат и газы. Жидкий конденсат расслаивается на
водный слой и нерастворимую в нем смолу. Состав смолы пред-
определяется ароматической природой лигнина. Подавляющая
доля идентифицированных в смоле веществ относится к классу
ароматических соединений. Основной группой веществ смолы
являются фенолы. Идентифицировано более 50 соединений. Они
могут быть подразделены на три следующие группы: одноатом-
ные фенолы — фенол, о-, м-, п-крезолы, ксиленолы (диметил-
фенолы), триметилфенолы, этилфенол, пропилфенол и др.; двух-
атомные фенолы орторяда: пирокатехин (ортодиоксибензол),
монометиловый эфир пирокатехина (гваякол) и, главным обра-
зом, паразамещенные производные его — метил (R=CHs)-, этил-
(R = CH2CH3)-, пропил (Е=СН2СН2СНз)- и аллил (К=СНгСН=
Z
R = C
СН2)-гваяколы, ванилин
, ацето (R=СОСНз) -
\ н/
и пропио (К = СОСН2СНз)-ванилоны и ряд других; трехатомные
фенолы, точнее 1,3-диметиловый эфир пирогаллола и его про-
изводные, аналогичные производным гваякола.
R
осн3
он
гваякол
(R=H)
сн3о
з
1.3- диме т иловый
эфир пирогаллола
(R-H)
Помимо этого, в фенолах найдены в незначительных массах
метадиоксибензол-резорцин и его алкилпроизводные, п-диокси-
бензол-гидрохинон. Содержание фенольных соединений в смоле
около 50 %.
Второй основной группой веществ смолы являются нейтраль-
ные вещества. В них идентифицированы многочисленные арома-
тические углеводороды бензольного ряда: бензол, толуол, кси-
лолы, этил-, пропил-, пропенил-, изопропилбензолы, стирол,
п-цимол, конденсированные углеводороды ряда нафталина, антра-
цена и даже 3,4-бензпирен, а также другие углеводороды, на-
пример инден, индан, дифенил, флуорен и др. Состав кислород-
содержащих нейтральных соединений изучен слабо. Массовая
доля нейтральных веществ в смоле составляет 20—30 %.
. 162
Помимо фенолов и нейтральных веществ, в смоле присут-
ствуют кислоты (10—20 % от массы смолы). Среди них обнару-
жены как ароматические, так и алифатические Ci—Сб кислоты.
В водном дистилляте, выход которого из лигнина значи-
тельно меньше, чем из целлюлозы, содержатся кислоты: му-
равьиная, уксусная, пропионовая и др., а также различные низ-
комолекулярные спирты, альдегиды, кетоны, эфиры, например
формальдегид, метанол, метилацетат, пропионовый альдегид,
ацетон, аллиловый спирт, метилпропионат, диацетил, метилпро-
пил- и метилизопропилкетон. Кроме того, в водном конденсате
присутствуют многие компоненты смолы, более или менее рас-
творимые в нем.
Основными компонентами пиролиза газов являются окись уг-
лерода и метан, углекислоты содержится меньше, чем в газах
пиролиза углеводных компонентов древесины. В еще меньших
массах найдены водород, этилен и другие углеводороды С2 — С5.
Таким образом, состав продуктов пиролиза лигнина сложен.
И связано это, как увидим ниже, с особенностями термораспада
его сложной нерегулярно построенной макромолекулы.
Для выяснения исключительно сложного химизма терморас-
пада лигнина учеными А. Н. Кислицыным, Г. Э. Домбург
(СССР), Р. Брежны (ЧССР) и др. изучен термораспад много-
численных индивидуальных соединений, моделирующих как
строение структурных единиц лигнина, так и различные типы
связей между ними. Полученные ими данные позволяют создать
картину химических превращений лигнина при пиролизе.
Особенности строения лигнина, как нерегулярного, чрезвы-
чайно разветвленного полимера, структурные единицы которого
соединены несколькими типами простых эфирных и углерод-угле-
родных связей, предопределяют характер деструкции его макро-
молекулы при пиролизе. Термораспад лигнина протекает, в от-
личие от регулярно построенной макромолекулы целлюлозы, не
по деполимеризационному типу, а путем распада по месту наи-
более слабых связей. Этими же особенностями строения лиг-
нина определяется исключительная сложность химических реак-
ций при термораспаде. Механизм первичных реакций расщеп-
ления различных типов связей между структурными единицами
неодинаков и осложняется к тому же одновременно идущими
вторичными превращениями. Поэтому невозможно описать даже
основные реакции, протекающие при термораспаде лигнина.
Можно лишь наметить пути, по которым идет деструкция макро-
молекулы, образование низкомолекулярных соединений и фор-
мирование структуры твердого остатка.
Рассмотрим вначале химизм процесса термораспада лигнина
на примере распада фрагмента, структурные единицы которого
соединены двумя основными типами связей: а- и 0-алкилариль-
пыми эфирными связями, составляющими около 80 °/о от общего
числа связей. Строение этого и других фрагментов и нумерация
структурных единиц даны по схеме строения лигнина березовой
6* 163
древесины, предложенной Нимецем. p-Алкиларильная эфирная
связь между 10—16-й и 17—18-й структурными единицами рас-
щепляется по гетеролитической реакции 1,3-отщепления под
действием у-спиртового гидроксила, а а-алкиларильная эфирная
связь между 16-й и 17-й структурными единицами — гомолити-
чески, с образованием свободных радикалов, которые тут же
подвергаются дальнейшим превращениям:
2-Метокси-феноксильный радикал из 16-й структурной еди-
ницы вначале изомеризуется в ортооксифеноксиметиленовый
радикал, аналогичный радикалу, образующемуся при пиролизе
гваякола, из которого образуется затем смесь фенолов: пирока-
техин, фенол, о-крезол и др.:
Одновременно с этим протекают превращения в боковых про-
пановых цепях (—R) и из них образуются н. пропильные —СзН7,
аллильные —СН2—СН = СН2, пропионильные —СО—СН2—СН3
группы, а также группы с меньшим числом углеродных ато-
мов— этильная —С2Нб, ацетильная —СО—СН3, метильная.
В отдельных случаях боковая пропановая цепь отщепляется.
Из схем видно, что из фенилпропановых структурных еди-
ниц макромолекулы лигнина вследствие расщепления а- и р-ал-
киларильных эфирных связей образуются мономерные летучие
продукты фенольного характера — диметиловый эфир пирогал-
лола, пирокатехин, гваякол, фенол и их алкилпроизводные.
164
Наряду с деструкцией макромолекулы при пиролизе проис-
ходит образование новых, более прочных связей. Так, а-алкил-
арильная эфирная структура тех пар единиц (1—2, 8—9, 13—
14 и 16—17), феноксисоставляющая которых является гвая-
цильным производным и содержит свободное ортоположение,
может по типу перегруппировки Кляйзена, по аналогии с гвая-
цилбензиловым эфиром изомеризоваться в дифенилметановую
структуру с более прочными связями:
сн—
Структурные единицы, соединенные дибензилыюй связью, на-
пример 2-я и 3-я структурные единицы, имеющие при этом
а-алкиларильную эфирную связь с третьим звеном (1—2),
после гомолитической диссоциации последней, превращаются
в стильбеновые (диарилэтиленовые) структуры с очень проч-
ными связями. Образование стильбеновых структур — хорошая
предпосылка для последующего формирования производных фе-
нантрена, т. е. конденсированных углеводородных структур.
165
Стильбеновая структура может образоваться также между
11-й и 12-й структурными единицами вследствие реакции деги-
дратации. Образующиеся вначале первичные свободные ради-
калы играют в дальнейших превращениях важнейшую роль.
Появление в системе активных радикалов, таких, как атомарный
кислород при распаде р-алкиларильной эфирной связи или фено-
ксильный при распаде а-алкиларильной эфирной связи, иници-
ирует распад лигнина. В их присутствии метоксильные группы
лигнина начинают превращаться уже при температурах ниже
300 °C, хотя в гваяколе отщепление их идет при температуре
выше 400 °C. В процессе свободнорадикального распада мето-
ксигруппы, как было показано на примере анизола и гваякола,
появляются новые свободные радикалы, продолжающие цеп-
ную реакцию распада. Некоторые промежуточные фенильные и
феноксильные радикалы рекомбинируют друг с другом с обра-
зованием дифенильных и дифенилэфирных структур. В резуль-
тате всей этой сложной системы последовательных и парал-
лельных реакций образуются низкомолекулярные летучие про-
дукты и, главное, происходит полная перестройка первичной
структуры макромолекулы лигнина. При этом алкиларильные
эфирные и дибензильные связи, прочность которых не превы-
шает 200 кДж/моль, заменяются более прочными связями типа
связей в дифениловом эфире (энергия гомолитической диссо-
циации £аКт = 314 кДж/моль), дифенилметане (Еакт =
= 356 кДж/моль), бензофеноне (EaKT=364 кДж/моль), дифе-
ниле (Еакт=396 кДж/моль) и стильбене (Еакт = 417 кДж/моль).
Это и предопределяет более высокий выход нелетучего уголь-
ного остатка из лигнина.
Температурно-временные параметры пиролиза оказывают
сильное влияние на направление термораспада макромолекулы
лигнина и выход продуктов. При медленном нагреве создаются
благоприятные условия для изомеризационных превращений а-
алкиларильной эфирной связи в дифенилметановые производ-
ные, для дегидратации в дибензильных структурах (11—12)
с образованием стильбена и т. д., в результате которых струк-
турные единицы сшиваются более прочными связями в нелету-
чий остаток. Выход низкомолекулярных продуктов деструкции
уменьшается. При быстром скоростном нагреве, напротив, сразу
происходит множественное расщепление а- и р-алкиларильных,
дибензильных и других связей на мономерные и димерные струк-
турные единицы. Выход смолы и фенолов в этом случае увели-
чивается, а угля уменьшается.
§ 30. ФИЗИКО-ХИМИЧЕСКАЯ ХАРАКТЕРИСТИКА
ПРОЦЕССА ПИРОЛИЗА
Общая физико-химическая характеристика процесса пиро-
лиза древесины включает данные о процессе передачи тепла
и нагрева древесины, о кинетике термораспада, тепловом эф-
166
Направление теплового потока
Рис. 44. Схема передачи тепла
к древесине при внешнем газовом
нагреве:
1 — газовый теплоноситель; 2 — стенка
реторты; 3 — парогазовая среда в ре-
торте; 4 — поверхность куска древе-
сины; 5 центр куска древесины
фекте процесса, последовательности распада компонентов дре-
весины, процессе углеобразования и т. д.
Процесс передачи тепла и нагрева древесины. Для нагрева
древесины при пиролизе применяются два способа передачи
тепла: внешний нагрев с передачей тепла через стенку и внут-
ренний нагрев с передачей тепла от греющего агента непосред-
ственно древесине. В качестве внутреннего теплоносителя могут
служить газы (парогазы), жидкости и твердые тела.
Рассмотрим процесс передачи
тепла древесине при используе-
мом сейчас в промышленных ап-
паратах внешнем и внутреннем
газовом нагреве. При внешнем
нагреве (рис. 44) процесс пере-
дачи тепла от топочных газов
древесине осуществляется в сле-
дующей последовательности: от
газа к стенке реторты, от стенки
к парогазовой среде внутри
реторты и от парогазов к древе-
сине. Тепло от топочного газа,
имеющего температуру 800—
900 °C, к стенке реторты пере-
дается в основном путем конвек-
ции, и в меньшей • степени,
лучеиспусканием. Вследствие тер-
мического сопротивления, созда-
ваемого газовой пленкой около
стенки, температура последней значительно ниже температуры
газового теплоносителя и равна приблизительно 600—650 °C.
Стенка реторты благодаря хорошей теплопроводности стали
имеет почти одинаковую температуру по всей толщине. При пе-
редаче тепла от стенки к парогазовой смеси внутри реторты
тепловой поток снова преодолевает сопротивление газовой
пленки. Так как скорость движёния газов внутри реторты ниже,
чем топочных газов около наружной стенки, а также ниже
температура теплопередающего тела, коэффициент теплопере-
дачи от стенки парогазам и конвекцией и лучеиспусканием бу-
дет меньше, чем от топочных газов стенке. Следовательно, тем-
пературный перепад будет еще больше. Практически парогазы
внутри реторты нагреваются до 450—500 °C. Последним сопро-
тивлением на пути теплового потока к древесине является газо-
вая пленка около поверхности куска древесины. От внешней
поверхности к внутренним слоям древесины тепло передается
путем теплопроводности клеточной стенки, конвекцией парога-
зов в полостях и лучеиспусканием от более нагретой клеточной
стенки к менее нагретой. Температура от наружной поверхности
к центру куска падает. Поэтому пиролиз куска древесины про-
исходит постепенно: поверхность может обуглиться, в то время
167
как внутри может происходить еще сушка. Чем мельче кусок,
тем равномернее и быстрее прогревается древесина.
При внутреннем газовом нагреве процесс передачи тепла
древесине значительно упрощается. Теплоноситель вводится
прямо в реторту и контактирует непосредственно с древесиной.
Единственным термическим сопротивлением в этом случае яв-
ляется газовая пленка около поверхности куска древесины. По-
этому эффективность подвода тепла к древесине при внутрен-
нем теплоносителе значительно выше.
Для интенсификации процесса пиролиза древесины необхо-
димо увеличить количество подводимого к древесине тепла, по-
вышая температуру теплоносителя (газовой среды) около дре-
весины или увеличивая его поток. При внешнем нагреве объем
парогазов внутри реторты не регулируется, следовательно, ин-
тенсификация процесса возможна только за счет повышения
их температуры. Но возможности повышения температуры паро-
газов внутри реторты выше 450—500 °C ограничены тепловой
стойкостью конструкционного материала — углеродистой стали.
При повышении температуры стенки выше 700 °C происходят ее
размягчение и деформация и интенсивное окисление, что приво-
дит к преждевременному износу оборудования. Измельчение
древесины с целью увеличения тепловоспринимающей поверхно-
сти не ведет к интенсификации процесса, так как поверхность
дров или тюлек и так значительно больше теплопередающей
поверхности стенок реторты. Таким образом, при внешнем на-
греве возможности для интенсификации процесса отсутствуют.
При внутреннем нагреве температура газового теплоносителя
достигает 650—700 °C, удельный расход его на единицу массы
древесины выше, чем в аппаратах с внешним нагревом, следова-
тельно, тепла подводится в аппарат значительно больше. Да и
интенсивность передачи этого тепла к древесине вследствие бо-
лее высокой скорости омывания куска также выше. Лимити-
рующим интенсификацию фактором становится величина тепло-
воспринимающей поверхности древесины. Поэтому при внутрен-
нем нагреве древесину целесообразно измельчать.
Благодаря отмеченным преимуществам внутреннего газового
нагрева удельная производительность вертикальных непрерыв-
нодействующих реторт, работающих по этому принципу, в 5—6
раз выше производительности вагонных реторт внешнего нагрева
при одинаковой степени разделки древесины.
При использовании в качестве внутреннего теплоносителя пе-
регретого водяного пара процесс теплопередачи не имеет прин-
ципиальных отличий от процесса при внутреннем газовом на-
греве. В качестве внутреннего теплоносителя можно применять
также жидкости и твердые тела.
Процесс термораспада древесины в нагретом жидком тепло-
носителе, например в керосине, при температуре не выше 275 °C,
нельзя назвать пиролизом, так как это предпиролиз. Процесс
передачи тепла от жидкого теплоносителя к древесине имеет
168
некоторые особенности. В отличие от газового теплоносителя, ко-
торый передает тепло только внешней поверхности куска, жид-
кий теплоноситель впитывается по узким сосудам в поры дре-
весины, нагревает внутреннюю часть куска и по более широким
сосудам выходит обратно вместе с вытесняемым воздухом или
продуктами распада. Устанавливается циркуляция теплоноси-
теля. Благодаря резкому увеличению тепловоспринимающей по-
верхности интенсивность процесса предпиролиза высока. При
предпиролизе в жидком керосине разлагаются наименее термо-
стойкие компоненты древесины, главным образом гемицеллю-
лозы, и в остатке образуется так называемая бурая древесина.
Хотя в промышленности процесс пиролиза в жидком теплоноси-
теле не реализован, он представляет интерес в качестве первой
ступени при разработке последовательного направленного термо-
распада древесины, так как осуществляется в очень мягких,
легко контролируемых условиях.
Процесс теплопередачи от твердого теплоносителя (напри-
мер, металлических шаров или древесного угля) к древесине
осуществляется при непосредственном контакте путем лучеиспу-
скания и теплопроводности. Древесный уголь при этом получа-
ется в том же аппарате и в раскаленном виде частично возвра-
щается в процесс. Благодаря развитым поверхностям интенсив-
ность теплопередачи также выше, чем при внешнем и даже
внутреннем газовом нагреве. Такие процессы используются
в непрерывнодействующих аппаратах для пиролиза опилок.
Кинетика термораспада древесины. Кинетические кривые тер-
мораспада сухих березовых опилок с размером частиц 0,25—
2 мм в атмосфере азота при различных температурах показаны
на рис. 45. Видно, что при 250 °C потеря массы происходит
с очень небольшой скоростью, при 270° — за 1 ч нагрева теря-
ется около 50 % массы, при 290° разложение идет со значитель-
ной скоростью и примерно через 20 мин потеря массы, достигнув
60%, прекращается, наконец, при температуре 400 °C термо-
распад протекает с большой скоростью и за 5 мин теряется бо-
лее 70 % массы древесины.
Скорость распада березовых опилок (размер частиц менее
0,6 мм) в инертной атмосфере при температурах до 700 °C под-
чиняется уравнению
dV/dT = 46e~63№(Rr)(l—V)2, (70)
где V — массовая доля выделившихся летучих; т — время, мин;
7? — газовая постоянная; Т — температура, К.
Разработка математической теплофизической модели про-
цесса пиролиза кусковой древесины наталкивается на большие
трудности из-за неоднородного строения древесины — анизотро-
пии физических свойств, наличия пустот и т. п.— и дефектов
в структуре, образующихся при сушке и пиролизе в древесине
и угле. Поэтому больших успехов в этом направлении не до-
стигнуто. .
169
Предложенная рядом авторов математическая модель про-
цесса пиролиза кусковой древесины (толщиной до 2 см) пред-
ставляет со,бой сложную систему дифференциальных уравне-
ний, в которые входят значения температуры, времени, давле-
ния, плотности, теплопроводности и удельной теплоемкости
древесины и угля, проницаемости их, физических свойств летучих
продуктов, скоростей тепловых и материальных потоков, энер-
Рис. 45. Кинетические кривые термо-
распада древесины березы при раз-
личных температурах, °C:
1 — 250; 2—260; 5 — 270; 4 — 290; 5 — 300;
5 — 400
Рис. 46. Термограмма древесины бе-
резы
гии активации термораспада древесины и многих других вели-
чин. Расчетные данные для пиролиза кленовой древесины удов-
летворительно согласуются с экспериментальными при скоростях
нагрева поверхности до 2,1 Дж/см2-с.
Кинетика и общая характеристика процесса термораспада
березовой древесины при неизотермическом нагреве показаны
на рис. 46. На ДТГ-кривой четко выделяются стадии распада
ксилана с максимумом потери массы при 280 и целлюлозы
при 330 °C потеря массы лигнина не прослеживается. На ДТА-
кривой имеются две эндотермические области: первая — с экс-
тремумом при температуре 220 °C отвечает1 начальной стадии
дегидратации и деструкции ксилана и целлюлозы, а также, по-
видимому, разрушению и перестройке первичной структуры лиг-
нина; вторая эндотермика с максимумом при температуре
345 °C, совпадающая с температурной областью интенсивной по-
тери массы целлюлозой, вызвана эндотермическим процессом
170
деполимеризации. ТТ-кривая показывает, что основная потеря
массы происходит в момент распада ксилана и целлюлозы;
после температуры 350 °C скорость потери массы резко умень-
шается. Остаток при 500 °C составляет около 24 % от древе-
сины.
Пиролиз древесины протекает в несколько стадий, характери-
зующихся различными скоростями распада (ДТГ- и ТТ-кривые)
и энергиями активации. Величина энергии активации колеблется
в зависимости от температурного интервала, скорости нагрева и
ряда других факторов от нескольких десятков до нескольких
сот (до 350) кДж/моль. Например, энергия активации термо-
распада древесины бука в интервале температур 300—340 °C
равна примерно 90—100 кДж/моль.
Тепловой эффект процесса пиролиза древесины. Тепловой
эффект пиролиза является важной статьей прихода тепла при
пиролизе древесины в промышленных аппаратах. Имеющиеся
в литературе оценки величины теплоты реакций пиролиза дре-
весины колеблются весьма значительно и отличаются не только
по величине, но и по знаку. Объясняется это различиями в ме-
тодике экспериментов, так как величина теплового эффекта пи-
ролиза древесины зависит от размера кусков, давления в си-
стеме, температуры пиролиза и т. д. При пиролизе древесины
в вакууме, когда обеспечивается хорошая эвакуация первичных
продуктов распада из зоны реакции и вторичные процессы све-
дены к минимуму, процесс пиролиза является эндотермическим.
Следовательно, первичные процессы термораспада компонентов
древесины идут с поглощением тепла. Пиролиз кусковой древе-
сины при нормальном давлении в аппарате является экзотер-
мическим процессом. Величина экзотермического эффекта
с очень большими допусками в ту или другую сторону может
быть оценена в 1000 кДж/кг. Тепловой эффект зависит от ко-
нечной температуры пиролиза и увеличивается по мере ее по-
вышения, достигая максимума при 600 °C.
Природа теплового эффекта твердо не установлена, недоста-
точно ясно, за счет каких реакций обеспечивается выделение
тепла. В тепловой эффект пиролиза древесины основную долю
вносит тепло экзотермического распада лигнина. Судя по росту
величины теплового эффекта.при повышении конечной темпера-
туры пиролиза даже после выделения основной части летучих
продуктов в момент формирования структуры угля, можно по-
лагать, что реакции перестройки структуры угольного остатка
с образованием полиядерных ароматических систем являются
важными источниками тепловой энергии. Известно также, что
если в пиролизуемом сырье содержится более 15 % кислорода,
то тепловой эффект термораспада положителен. Выделение
тепла при этом связано с перестройкой структуры компонентов
древесины, сопровождающейся образованием новых кислородсо-
держащих соединений с более выгодным энергетическим со-
стоянием — окиси и двуокиси углерода.
17)
Распад компонентов в древесном комплексе. Процесс рас-
пада сложного древесного комплекса при пиролизе, естественно,
является отражением превращений, происходящих с его основ-
ными компонентами. Рассмотрим, как и в какой последователь-
ности происходит термораспад их в самой древесине. Из термо-
граммы древесины (см. рис. 46) и данных о термораспаде кси-
лана и целлюлозы следует, что ксилан менее термостоек, чем
целлюлоза. Лигнин по термической устойчивости, если оцени-
вать ее не только по интенсивности потери массы, но и по хи-
мическим изменениям структуры, занимает промежуточное поло-
жение между ксиланом и целлюлозой. Действительно, уже
в момент плавления (при температурах около 170—180 °C)
в лигнине происходят конденсационные процессы, а при даль-
нейшем повышении температуры, начиная с 210 °C, задолго до
температуры активного распада целлюлозы, образуются летучие
продукты деструкции и, главное, за счет расщепления основных
типов связи между элементарными звеньями макромолекулы
осуществляется перестройка структуры: непрочные р- и а-алкил-
арильные связи заменяются более прочными связями. Но, если
вопрос о термостойкости рассматривать с точки зрения органи-
зации последовательного ступенчатого разложения компонентов
с получением летучих продуктов, т. е. с технологической сто-
роны, то самым термостойким компонентом окажется лигнин.
Целлюлоза в древесине в виде микрофибрилл располагается
во всех слоях клеточной стенки, образуя своеобразный каркас.
Ксилан и лигнин содержатся также во всех слоях, но в раз-
личных долях. Ксилан размещается в виде аморфной массы
между микрофибриллами целлюлозы. Лигнин в процессе лиг-
нификации, происходящей на стадии одревеснения клеточной
стенки, располагается в микропустотах между углеводными
компонентами, контактирует и образует химические связи
с ними. Учитывая такой характер размещения компонентов
в древесине, нельзя не допустить влияния ксилана и лигнина,
заполняющих пустоты целлюлозного каркаса, на термораспад
целлюлозы. Ксилан и лигнин весьма существенно, хотя и кос-
венным образом, влияют на процесс термораспада целлюлозы,
оцениваемый по выходу левоглюкозана. Вследствие того, что
целлюлоза «замурована» в аморфной массе ксилана и лигнина,
продукты разложения последних создают препятствие свобод-
ному выходу образующегося при деполимеризации левоглюко-
зана, левоглюкозан разрушается и выход его очень низок. До-
статочно удалить из древесины один из компонентов (или кси-
лан, или лигнин) и пиролизовать целлолигнин или холоцеллю-
лозу, как выход левоглюкозана увеличивается более чем в 10
раз и сравнивается с выходом его из целлюлозы в аналогичных
условиях. Лигнин в небольшой степени оказывает и непосред-
ственное, ингибирующее, отрицательное влияние на процесс тер-
мораспада целлюлозы в целлолигнине, но снижает выход лево-
глюкозана.
172
20. ЗАВИСИМОСТЬ ЭЛЕМЕНТНОГО СОСТАВА УГЛЯ ОТ ТЕМПЕРАТУРЫ
ПИРОЛИЗА
Темпера- тура пиролиза, °C Элементный состав, % на абс. сухой беззольный уголь Темпера- тура пиролиза, °C Элементный состав, % на абс. сухой беззольный уголь
углерод водород кисло- род + азот углерод водород КИСЛО- РОД+ азот
400 78,82 4,04 17,14 600 90,47 3,05 6,48
500 87,22 3,74 9,04 700 92,23 1,63 6,14
термического
распада
Процесс углеобразования. Процесс
древесины заключается, с одной стороны, в деструкции макро-
молекул компонентов древесины с образованием низкомолеку-
лярных летучих продуктов и, с другой — в образовании нелету-
чего твердого «полимерного» остатка — древесного угля.
Древесный уголь формируется .из всех основных компонен-
тов древесины, но с относительно более высоким выходом из
лигнина. Физико-химические свойства угля изменяются в про-
цессе пиролиза и в конечном итоге зависят от температуры про-
калки угля (табл. 20).
Из таблицы видно, что по мере повышения температуры пи-
ролиза содержание углерода в угле растет, а водорода и кис-
лорода падает, атомные соотношения С/Н и С/О увеличиваются.
Анализ кривых изменения атомных соотношений С/Н и С/О
(рис. 47, кривые 3 и 4) показывает, что особенно интенсивное
снижение доли водорода в угле происходит при температуре
выше 600 °C, а кислорода — до 600 °C. Удаление кислорода при
более высокой температуре замедляется. Полного удаления кис-
лорода не удается достичь даже при высокой температуре, так
как кислород незначительно отличается от углерода по атом-
ному радиусу и углу между связями и может частично заме-
щать его в углеродных монослоях, не нарушая их структуры.
По мере повышения температуры пиролиза вследствие измене-
ния элементного состава изменяется и такая важная структурно-
статистическая характеристика угля, как степень ароматизации
(кривая 2). Степень ароматизации, т. е. доля ароматического
углерода в общей массе углерода, при температуре 400 °C рав-
ная 0,77, постепенно растет и при 700 °C достигает 0,93.
Древесный уголь относится к числу твердых углеродных ма-
териалов с неупорядоченной тонкой структурой и является
аморфным углеродом, труднографитируемым при дальнейшем
повышении температуры термообработки. Препятствием для го-
могенной кристаллизации и образования графитовой структуры
служат наличие связок в виде углеродных цепей или атомов
между образующимися углеродными сетками, связок, затруд-
няющих перегруппировки и формирование компактной графито-
вой структуры. Образование неграфитирующейся структуры
173
происходит в значительной мере из-за того, что древесина раз-
лагается, не переходя в пластичное состояние, в котором созда-
ются более благоприятные условия для упорядочения структуры.
Высокое содержание кислорода в исходном материале также
способствует зарождению малоупорядочепной и труднографи-
тируемой структуры.
Температура, °C
Рис. 47. Зависимость некоторых ха-
рактеристик березового угля от тем-
пературы пиролиза древесины:
1 — относительная концентрация парамаг-
нитных центров; 2 — степень ароматиза-
ции; 3 — атомное отношение С /О; 4 —
атомное отношение С/Н
Тонкая структура углеродных материалов исследуется мето-
дом рентгеноструктурного анализа. При этом определяются
средний линейный размер плоскостей углеродных слоев La, сред-
няя толщина блоков, образованных этими слоями Lc, и рас-
стояние между плоскостями слоев dooz- Рентгеноструктурный
анализ древесного угля, полученного при температуре пиролиза
400 и 500 °C показывает, что он полностью аморфен. При
600 °C в аморфной структуре угля появляются кристаллиты —
блоки, состоящие из плоских углеродных слоев. При 600 и
700 °C блоки эти невелики (£в=2,5 и Lc=l нм) и слабо упако-
ваны, межплоскостп’ое расстояние равно 0,42 нм (в графите
^оо2=О,335 нм). В угле, прокаленном при температуре около
1800 °C, появляются так называемые турбостратные структуры,
представляющие собой блоки, слои в которых аналогичны гра-
фитовым плоскостям, но располагаются они друг над другом
хотя и параллельно, но' не строго ориентированно, как в гра-
фите, а произвольно. Трехмерная упорядоченность в них отсут-
ствует. Вследствие такого неориентированного расположения
плоскостей расстояние между ними больше, чем в графите, но
значительно меньше, чем в кристаллитах угля, полученного
при температуре 600—700 °C, и равно 0,342 нм. При прокалке
угля при температурах выше 2000 °C появляются графитовые
структуры. В этот момент древесный уголь представляет собой
слабоструктурированную фазу с вкрапленными в нее кристал-
литами с турбостратной и графитовой структурами. Содержа-
ние графитовых структур в угле, прокаленном при температурах
выше 200 °C, возрастает, но даже термообработка угля при
3400 °C не ведет к полной его графитации.
174
Такое явление, когда в аморфном углеродном материале при
термообработке появляются отдельные графитовые структуры,
называется многофазной графитацией. Причины многофазной
графитации древесного угля связаны со сложным неоднородным
строением древесины и с различиями в процессах формирования
углеродсодержащего остатка из отдельных компонентов, в осо-
бенности из твердой неплавкой целлюлозы и аморфного плав-
кого лигнина.
Процессы термического распада компонентов и образования
многих продуктов протекают по свободнорадикальному меха-
низму. Несмотря на высокую реакционную способность, часть
образующихся в процессе деструкции свободных радикалов со-
храняется в угле, благодаря стабилизации их в твердой мат-
рице угольного вещества. Концентрация парамагнитных центров
в угле в процессе термообработки непостоянна (см. рис. 47, 1).
После 400 °C, в момент, когда интенсивная потеря массы за счет
разложения гемицеллюлоз и целлюлозы закончилась и со значи-
тельно меньшей скоростью образуются тяжелокипящие фракции
древесной смолы, концентрация парамагнитных центров продол-
жает расти и при температуре 500° достигает максимума. После
500 °C концентрация парамагнитных центров начинает умень-
шаться и при 600°, т. е. к моменту появления в аморфной струк-
туре угля блоков с более или менее упорядоченным располо-
жением слоев, падает примерно в 10 раз. Концентрация ПМЦ
в древесном угле, полученном при температуре 700 °C, незначи-
тельна. С появлением в рентгеноаморфном угольном веществе
областей с определенной упорядоченной структурой совпадает
резкое повышение электронной проводимости: удельное элек-
трическое сопротивление угля при изменении температуры пиро-
лиза на 100 °C скачкообразно в 105—106 раз уменьшается.
Химизм образования плоских углеродных структур, из ко-
торых построены блоки, не ясен. Неизвестно, образуются ли они
путем последовательного присоединения небольших фрагмен-
тов, ведущего к росту размеров гексагональной сетки монослоя
ароматического углерода, или по типу реакций поликонденсации
предварительно образовавшихся ароматических соединений.
Формирование тонкой структуры угля, ее перестройка, когда
разрушаются поперечные связи между углеродными слоями,
обусловливавшие рыхлое их расположение, сопровождается уп-
лотнением угольного вещества. Об этом свидетельствует увели-
чение пикнометрической плотности угля. Например, пикномет-
рическая плотность березового угля, полученного при темпера-
туре 400, 500, 600 и 700 °C, увеличивается соответственно
с 1,374 до 1,500; 1,548 и, наконец, до 1,594 г/см3. Конечно, плот-
ность тела древесного угля намного ниже плотности графита,
равной 2,25 г/см3. Отчасти это связано с тем, что в структуре
древесного угля имеются замкнутые, недоступные для раство-
рителя поры, объем которых, например, в березовом угле, по-
лученном при 600—700 °C, около 12 % от объема тела.
175
Наряду с изменениями тонкой структуры по мере повышения
температуры пиролиза меняется и макроструктура: кажущаяся
плотность и доступная пористость в березовых углях при по-
вышении температуры пиролиза с 400 до 900 °C увеличивается
с 0,365 до 0,400 г/см3 и соответственно с 73,9 до 77,1 %.
Изменяется в процессе пиролиза и такая важная характери-
стика угля, как реакционная способность. Наибольшей реакци-
онной способностью обладают угли, полученные при темпера-
туре 400 и 500 °C. Высокая реакционная способность угля, по-
лученного при этих температурах, связана с наличием в угле
свободных радикалов, максимум содержания которых прихо-
дится на этот же температурный интервал (см. рис. 47, 1). Упо-
рядочение структуры, появление кристаллитов приводят к рез-
кому снижению реакционной способности угля, полученного при
600 °C. Однако благодаря низкому содержанию трехмерно упо-
рядоченного углерода и развитой пористости угля реакционная
способность остается значительной по сравнению с реакционной
способностью каменноугольного кокса и подобных материалов,
что и определяет высокие потребительские качества древесного
угля как восстановителя в различных процессах.
§ 31. ВЛИЯНИЕ СЫРЬЕВЫХ ФАКТОРОВ НА ВЫХОД
ПРОДУКТОВ ПИРОЛИЗА
Еще ранее исследованиями, ставшими классическими, и ана-
лизом накопленного производственного опыта установлено, что
выход продуктов пиролиза зависит от многих факторов, которые
можно объединить в две группы: факторы, связанные с харак-
теристикой сырья (сырьевые факторы), и факторы, характери-
зующие режим процесса пиролиза (режимные факторы).
К сырьевым факторам, оказывающим прямое влияние на ход
процесса пиролиза и выход продуктов, относятся: химический
состав древесины, зависящий от породы дерева, наличие коры
и гнили, размер кусков пиролизуемой древесины и ее влажность.
Выход продуктов пиролиза из различных пород древесины.
Выход продуктов пиролиза зависит от породы пиролизуемой
древесины, так как существуют отличия в физико-химическом
составе и структуре различных пород дерева. В табл. 21 приве-
дены выходы продуктов пиролиза из древесины березы — основ-
ной породы, используемой в качестве сырья в пиролизном произ-
водстве, и для сравнения — из сосны (условия пиролиза: реторта
с внешним нагревом, конечная температура пиролиза 450 °C).
Из табл. 21 видно, что выход уксусной кислоты и метанола
из сосновой древесины примерно в 2 раза ниже, чем из березо-
вой. Это закономерно, так как в древесине сосны и вообще хвой-
ных пород содержится в 3—4 раза меньше пентозанов, главным
образом ответственных за образование этих продуктов, а галак-
тоглюкомамнан — основной представитель гемицеллюлоз — со-
держит примерно в 3 раза меньше ацетильных групп, чем кси-
176
21. ВЫХОДЫ ПРОДУКТОВ ПИРОЛИЗА ИЗ ДРЕВЕСИНЫ
БЕРЕЗЫ И СОСНЫ
Продукт Выход» % от массы а. с. д. Продукт Выход. % от массы а. с. д.
березы сосны березы сосны
Уголь Смола: 31,8 37,8 Газы: углекислота 10,0 10,1
отстойная 7,9 11,7 окись углерода 3,3 3,7
растворимая 8,2 8,0 метан 0,5 0,6
Уксусная кислота Метанол 7,1 1,6 3,5 0,9 непредельные углеводороды 0,2 0,2
Вода 27,8 22,3 потери 1,6 1,2
лан древесины лиственных пород, и совсем не содержит мето-
ксильных групп. Выход угля и отстойной смолы из сосны не-
сколько выше за счет большего содержания в ней лигнина и
частично за счет образования их из смолистых экстрактивных
веществ. Выход газов одинаков из обеих пород древесины.
В дополнение к табличным данным следует добавить, что
выход жидких продуктов из твердолиственных пород, также ис-
пользуемых при пиролизе (бука, граба), несколько выше, чем
из березы, а из осины — чуть ниже. Казалось бы, использование
осины в качестве сырья для пиролиза приемлемо. Тем не менее
при существующем объемном методе учета перерабатываемой
древесины пиролиз осины крайне невыгоден, потому что масса
кубометра древесины осины, а следовательно, и выход продук-
тов пиролиза из него в 1,34 раза ниже, чем из древесины бе-
резы. Нежелательно использование осиновой древесины и по
другой причине: механическая прочность угля, получаемого из
нее, в 2 с лишним раза ниже прочности угля из березы. А это
ведет к измельчению угля при транспортировке и перевалках и
снижению товарной фракции.
Выход продуктов пиролиза из коры. Кора березы резко от-
личается по составу от стволовой древесины. В целом в наруж-
ном и внутреннем слоях коры содержится значительно больше
(примерно в 10 раз) экстрактивных веществ — жиров, восков,
тритерпенов, таннидов и т. д. и минеральных веществ и меньше
в 2 раза гексозанов (включая целлюлозу), чем в стволовой
древесине. Пентозаны содержатся также в меньшей доле, но,
главное, они отличаются по составу: содержание метоксигрупп
в 2 раза, а ацетильных в 5—6 раз меньше, чем в пентозанах
древесины. В соответствии со специфическим составом коры из-
меняется и выход основных продуктов пиролиза: выход мета-
нола в 2 раза, а уксусной кислоты в 3 раза меньше, чем при пи-
ролизе древесины березы. Выход угля и особенно смолы —
выше. Но уголь из коры имеет высокую зольность и низкую
177
механическую прочность и впоследствии истирается в мелочь,
а смола имеет специфический состав и при отстаивании жижки
переходит преимущественно в малоценные всплывные масла.
Вследствие всего этого кора оказывает отрицательное влияние
на выход и качество продуктов пиролиза березовой древесины.
Однако окорка древесины — операция трудоемкая и в практике
не применяется.
Выход продуктов пиролиза из древесины, пораженной
гнилью. Гнили, повреждающие древесину, влияют на выход и
качество продуктов пиролиза. Выход жидких продуктов зависит
от характера изменений в химическом составе древесины, выз-
ванных различными типами гнили и опцсанными выше. В соот-
ветствии с этими изменениями в составе при пиролизе древе-
сины, пораженной бурой гнилью, выход метанола и уксусной
кислоты уменьшается в 2 раза, а отстойной смолы увеличива-
ется.
При поражении белой гнилью выход уксусной кислоты па-
дает в 1,5 раза.
Особенно неблагоприятно отражается гниль на качестве дре-
весного угля: механическая прочность его снижается, повыша-
ется склонность к самовозгоранию. Поэтому и ограничивается
в технологических дровах, поставляемых на лесохимические за-
воды, доля древесины, пораженной гнилью. Вследствие этого
необходимо принимать меры по предупреждению загнивания
древесины и на заводе, при хранении, особенно в кучах.
Влияние величины кусков древесины на выход продуктов.
Величина куска .пиролизуемой древесины оказывает большое
влияние на выход жидких продуктов (табл. 22) и особенно на
продолжительность процесса.
Из табл. 22 видно, как по мере увеличения размера кусков
пиролизуемой древесины падает выход почти всех жидких про-
дуктов— уксусной кислоты, метанола, растворимой смолы.
Опыты по пиролизу древесины березы в вагонных ретортах
Ашинского лесохимического завода согласуются с данными
табл. 22: выход уксусной кислоты из чурки длиной 0,3 м на
22. ВЛИЯНИЕ ВЕЛИЧИНЫ КУСКОВ ДРЕВЕСИНЫ НА ВЫХОД ЖИДКИХ
ПРОДУКТОВ
Продукт Выход, % от массы а. с. д. березы, при длине кусков, см
3,2 6,3 12,5 25,0 50,0
Уксусная кислота 5,80 5,97 5,42 4,73 4,27
Метанол 1,65 1,73 1,58 1,38 1,30
Смола:
отстойная 7,08 6,50 7,00 7,00 5,76
растворимая 4,50 4,93 4,25 3,13 • 2,62
178
15 % выше, чем из древесины длиной 1 м. Теоретически, чем
мельче древесина, тем мягче процесс ее термораспада и тем
выше выход жидких продуктов. При пиролизе крупных кусков
древесины продукты пиролиза, выходящие из центральной части
куска, подвергающегося пиролизу в последнюю очередь, контак-
тируют с перегретыми наружными слоями угля и частично раз-
лагаются. При пиролизе крупных кусков увеличивается продол-
жительность процессов сушки и пиролиза, возрастают затраты
тепла. Механическая прочность угля из нерасколотых длинных
поленьев ниже, чем из чурок. На практике древесину разделы-
вают на чурки длиной 0,2—0,33 м. Дальнейшее уменьшение
размеров куска древесины, предназначенной для пиролиза
в действующих промышленных аппаратах, нерационально из-за
потерь древесины при распиловке и дополнительных энергетиче-
ских затрат.
Для получения максимальных выходов жидких продуктов и
резкого увеличения удельной производительности оборудования
следовало бы пиролизовать древесину в виде щепы. Однако
мелкий уголь из щепы не отвечает требованиям ГОСТа. Пере-
работка щепы станет целесообразной, если на месте будет орга-
низована переработка мелкого угля во вторичные продукты, на-
пример в активный уголь.
Влияние влажности древесины на выход продуктов. На осно-
вании длительного практического опыта в производстве опре-
делился оптимальный уровень относительной влажности, рав-
ный 15—25%, с которой древесина должна направляться на
пиролиз. Изменение влажности древесины в указанном интер-
вале практически не влияет на выход продуктов пиролиза.
Некоторые колебания в выходах, приводимые в литературе, не-
значительны и находятся в пределах ошибки эксперимента. По-
вышение влажности выше оптимального значения ведет к по-
следствиям отрицательно сказывающимся на технико-экономи-
ческих показателях процесса.
1. Увеличивается расход топлива и снижается производи-
тельность реторт.
2. Получается жижка слабой концентрации по уксусной кис-
лоте, что усложняет и удорожает процесс ее переработки.
3. Ухудшается механическая прочность угля, особенно если
процесс проводится с повышенной скоростью или древесина
находится в крупных кусках.
При пиролизе очень сухой древесины в аппаратах периоди-
ческого действия с внешним нагревом нормальный спокойный
ход процесса в период экзотермического разложения древесины
нарушается, в результате снижается выход уксусной кислоты и
метанола и получается непрочный уголь. В аппаратах с внут-
ренним нагревом благодаря мягкому нагреву и лучшим усло-
виям для эвакуации продуктов из зоны пиролиза выход продук-
тов из такой древесины остается высоким.
179
§ 32. ВЛИЯНИЕ РЕЖИМНЫХ ФАКТОРОВ НА ВЫХОД
ПРОДУКТОВ ПИРОЛИЗА
Такие параметры процесса, как способ подвода тепла, конеч-
ная температура пиролиза, скорость подъема температуры, дав-
ление в аппарате, оказывают большое влияние на выход про-
дуктов пиролиза.
Влияние способа нагрева древесины на выход продуктов.
В аппаратах с внешним нагревом разложение древесины начи-
нается около стенок аппарата и вследствие слабой циркуляции
парогазов и малой теплопроводности древесины идет неравно-
мерно. Для обеспечения полного пиролиза древесины, располо-
женной в центре аппарата, приходится перегревать древесину,
находящуюся около стенок. Выходящие из центра аппарата па-
рогазы, соприкасаясь с перегретыми стенками и древесным уг-
лем, частично разлагаются. В аппарате с внутренним обогревом
тепло от парогазового теплоносителя передается непосредст-
венно древесине, древесина разлагается равномерно во всем
объеме, одновременно обеспечиваются хорошие условия для
выноса продуктов пиролиза из аппарата. Поэтому при пиролизе
древесины в вертикальных непрерывнодействующих ретортах
с внутренним теплоносителем выход смол, особенно раствори-
мых, тяжелолетучих и нетермостойких, выше. Выход кислот,
сгХиртов и альдегидов (% от массы а. с. д.) в обоих случаях,
сильно колеблется, но можно заметить тенденцию к увеличению
выходов при внутреннем нагреве (табл. 23).
23. СОСТАВ ЖИДКИХ ПРОДУКТОВ ПИРОЛИЗА ДРЕВЕСИНЫ
Аппарат Жижка Кислоты Спирты
общие летучие
Реторта: вертикальная непре- рывнодействующая горизонтальная перио- дически действующая 42,0—48,8 68,5—70,7 5,7—8,6 6,5-7,0 5,4—7,4 5,6-6,1 0,8—3,0 1,1—2,2
Продолжение
Аппарат Альдегиды Смола
отстойная растворимая
Реторта: вертикальная непре- рывнодействующая горизонтальная перио- дически действующая 0,6—1,6 до 0,7 6,0—8,6 4,9—5,9 5,0—10.2 2,9—3,2
180
Выход угля сравнивать трудно, так как конечная темпера-
тура пиролиза сильно различается: в аппаратах с внешним на-
гревом она не выше 500 °C, а в аппаратах с внутренним тепло-
носителем не ниже 550 °C. Тем не менее замечено, что выход
угля в вертикальных непрерывнодействующих ретортах даже
при одинаковой температуре пиролиза ниже, чем в аппаратах
с внешним нагревом. Это объясняется, с одной стороны, увели-
чением выхода угля из-за частичного разложения смолистых
веществ при пиролизе в аппаратах с внешним нагревом, с дру-
гой— уменьшением выхода угля в аппаратах с внутренним теп-
лоносителем вследствие частичной его газификации газами
теплоносителя, особенно в тех случаях, когда температура теп-
лоносителя на входе в реторту превышает 600 °C.
Нелегко также сравнивать выход газов, так как газы, обра-
зующиеся при пиролизе древесины с внутренним газовым нагре-
вом, сразу смешиваются с теплоносителем и в общем потоке
выводятся из аппарата (табл. 24). Вследствие разбавления га-
зов пиролиза теплоносителем неконденсирующиеся газы из
вертикальных непрерывнодействующих реторт содержат зна-
чительно больше азота и меньше всех остальных компонентов.
Термическое разложение измельченной древесины в токе
перегретого водяного пара хотя и имеет некоторые преимуще-
ства перед обычным пиролизом — повышенный выход уксусной
кислоты, альдегидов, удобство регулирования процесса, но не
настолько, чтобы компенсировать высокие энергетические
24. ХАРАКТЕРИСТИКА ГАЗОВ, ПОЛУЧАЕМЫХ В АППАРАТАХ С РАЗЛИЧНЫМИ
СПОСОБАМИ НАГРЕВА
Аппарат Состав газа, % по объему
СО2 СО | сн4 С„Нт п tn
Реторта: вертикальная непрерывно- действующая горизонтальная периодически действующая вагонная 28,1—28,8 47,7 14,0—17,0 16,9 2,0-6,0 9,7 0,4—0,9 1,5
Продолжение
Аппарат Состав газа, % по объему Теплотворная способность, Дж/м3, при . нормальных условиях
на о2 N3
Реторта: вертикальная непрерывно- действующая горизонтальная периодически действующая вагонная 6,8—8,3 17,0 0,3—0,4 0,4 44,2 6,8 (3,9—4,9)-10е 8,3-10е
181
затраты на перегрев пара и переработку разбавленного конден-
сата.
Влияние степени нагрева на выход продуктов. Продукты пи-
ролиза древесины образуются в широком интервале темпера-
тур, но выделение основной их массы заканчивается при тем-
пературе 400 °C. Дальнейшее повышение конечной температуры
пиролиза приводит к небольшому увеличению выхода высоко-
кипящих смол и существенному — неконденсируемых газов.
В газах возрастает доля водорода, метана и непредельных уг-
леводородов. Изменяются также выход и характеристика угля:
чем выше конечная температура пиролиза, тем меньше выход
угля и тем выше содержание нелетучего углерода в нем.
Влияние скорости нагрева на выход продуктов (табл. 25).
Данные табл. 25 показывают, что увеличение продолжитель-
ности процесса пиролиза с 3 до 14 сут мало влияет на выход
метанола и уксусной кислоты, но сильно влияет на выход угля
и смол: выход смол уменьшается в 10 раз, а древесного угля
вследствие разложения смол увеличивается примерно в 1,5 раза.
При уменьшении продолжительности пиролиза до 15 мин
выход угля, по данным В. А. Коробкина, уменьшается до
15,5%.
На практике придерживаются продолжительности пиролиза,
зависящей от типа аппарата. Сокращение продолжительности
процесса пиролиза в аппаратах периодического действия уве-
личивает валовую выработку угля, но выход ценных продуктов
снижается и уменьшается срок службы реторт.
Влияние давления в аппарате на выход продуктов (табл. 26).
По мере повышения давления выход летучих кислот и смолы
дадает, а выход угля и метанола возрастает. При пиролизе
древесины под вакуумом отстойной смолы не образуется.
Указанные в табл. 26 возможности изменения выхода про-
дуктов в зависимости от давления на практике не использу-
ются из-за сложностей оформления высокотемпературного про-
цесса под вакуумом или давлением. Более того, отклонение
давления от оптимального в современных промышленных ап-
25. ЗАВИСИМОСТЬ ВЫХОДА ПРОДУК- 26. ВЛИЯНИЕ ДАВЛЕНИЯ НА
TOB ПИРОЛИЗА ОТ СКОРОСТИ НАГ- выход ПРОДУКТОВ ПИРОЛИЗА
РЕВА
Продолжи- тельность процесса, ч Выход, % от массы а.с.д. березы Давление, Па Выход, % от массы а.с.д. березы
угля лету- чих кислот мета- нола смол угля лету- чих кислот мета- нола смол
3 25,5 7,8 1,5 18,0 6,7* 102 19,5 9,4 1,2 37,2
8 30,8 7,9 1,5 17,0 9,8-104 36,6 6,3 1,4 17,0
16 33,2 7,1 1,5 10,1 8,2-105 40,5 5,4 1,5 9,1
336 39,1 6,8 1,4 1,8 8,8-10е 44,0 4,2 2,6 —
182
паратах нежелательно (особенно создание вакуума), так как
в этом случае возможен подсос воздуха и образование взрыв-
чатой смеси с неконденсируемыми газами пиролиза. Однако
в настоящее время разработаны экономически эффективные
процессы термической переработки с получением новых про-
дуктов, прежде всего левоглюкозана, в которых одна из стадий
проводится при пониженном давлении.
При переработке древесины в аппаратах с внутренним на-
гревом нагретый поток газов и паров выносит продукты пиро-
лиза из зоны реакции и создает условия, приближающиеся
к пиролизу под вакуумом.
§ 33. ВЛИЯНИЕ ХИМИЧЕСКИХ РЕАГЕНТОВ
НА ТЕРМИЧЕСКИЙ РАСПАД ДРЕВЕСИНЫ
Обобщая изложенные выше данные о химизме терморас-
пада изолированных компонентов и всего древесного комплекса,
можно сделать вывод, что изменением физических параметров
процесса пиролиза древесины трудно добиться значительного
увеличения выхода ценных продуктов, невозможно осуществить
последовательный распад ее компонентов. Компоненты древе-
сины — гемицеллюлозы, целлюлоза и лигнин — разлагаются
в близких температурных интервалах. Изменяя параметры про-
цесса, не удается вмешаться в первичные механизмы процесса
образования ценных продуктов и не удается осуществить на-
правленного термораспада древесины. Кардинально решить
проблему можно лишь применяя химические реагенты.
Многочисленные исследования и поиски химических реаген-
тов по целевому назначению можно подразделить на исследо-
вания, направленные на увеличение выхода летучих продуктов
(метанола, уксусной кислоты, фурфурола, левоглюкозана
и т. д.) и на исследования по повышению выхода древесного
угля, по формированию его структуры и свойств.
Влияние химических реагентов на выход жидких продуктов.
Первые попытки применения химических реагентов при пиро-
лизе древесины были сделаны около 60 лет назад с целью уве-
личения выхода продуктов, которые представляли тогда наи-
большую ценность,— метанола и уксусной кислоты. При пи-
ролизе древесины клена и бука, обработанных фосфорной
кислотой (7,5 % от массы древесины),выход метанола увеличи-
вается в 1,5 и 2,5 раза соответственно, но в 4—5 раз уменьша-
ется выход смолы; выход уксусной кислоты остается без изме-
нения. Выход метанола повышается и при использовании до-
бавок соды. Выход уксусной кислоты повысить не удалось.
Исследования в этом направлении развития не получили. Не-
сравнимо большие усилия были направлены на разработку
способов получения фурфурола при термическом разложении
древесины в присутствии катализаторов.
Шри обычном пиролизе древесины березы выход фурфурола
не превышает 1 % от массы а. с. д.> или не более 10 % от тео-
183
ретического выхода. Выделение его из жижки при таких низ-
ких выходах экономически не оправдано. Между тем из древе-
сины, содержащей примерно 25 % пентозанов, попутно с основ-
ными продуктами пиролиза — углем и уксусной кислотой —
можно получить фурфурол с возможным выходом около 10 %
от массы а. с. д. Поэтому так широко и проводились исследова-
ния по получению фурфурола при различных методах термиче-
ской переработки древесины — при пиролизе, при предпиролизе
в среде жидкого теплоносителя, при газификации и т. д.
Широкие исследования по получению фурфурола при пиро-
лизе древесины провел В. И. Корякин (ЦНИЛХИ). Выход
фурфурола при пиролизе кубиков 10X10X10 мм березовой
древесины, пропитанных растворами серной и соляной кислот
и бисульфата натрия, в лабораторных опытах достигает 7 %
от массы а. с. д. Способ проверен в полузаводских и промыш-
ленных условиях. При пиролизе березовой древесины в виде
чурок длиной 200 мм, предварительно высушенных до влажно-
сти не более 10 % и затем пропитанных 0,9 %-ной серной кис-
лотой, в вагонных ретортах Сявского лесохимического завода
выход фурфурола составил максимум 3,7 % от массы а. с. д.
При этом снизился выход летучих кислот (на 22%) и смолы.
Невысокий выход фурфурола не оправдывает трудовых и энер-
гетических затрат на автоклавную пропитку чурок, вторичную
сушку древесины и сложную технологию выделения и очистки
фурфурола. Повысить выход фурфурола и упростить техноло-
гию пиролиза можно при переходе на переработку измельчен-
ной древесины — щепы.
Исследования по получению фурфурола при предпиролизе
древесины проведены в Ленинградской лесотехнической акаде-
мии им. С. М. Кирова А. К. Славянским. При простом пред-
пиролизе древесины березы в среде керосина разрушаются
преимущественно пентозаны, но выход фурфурола не превы-
шает 2 % от массы а. с. д. Предпиролиз березовой щепы, пропи-
танной 6 % -ной соляной кислотой, дает выход фурфурола 6—
10%. Но при этом наряду с полным разложением пентозанов
происходит сильное разрушение целлюлозы: за первые полчаса
процесса (к 120 °C) разрушается половина ее, к 180 °C оста-
ется треть от первоначального содержания, а через 90 мин от1
начала предпиролиза (к 270°) остаток бурой древесины ее со-
всем не содержит. Таким образом, основная цель — осущест-
вить последовательный направленный процесс термического
распада компонентов древесины — не достигается. Многочис-
ленные опыты с использованием в качестве катализаторов раз-
личных солей взамен соляной кислоты не дали положительных
результатов; выход фурфурола не превысил 6 % даже при
большом расходе реагентов.
В последнее время в Институте химии древесины АН Латв-
ССР (Н. А. Ведерников, П. Н. Одинцов) разработан эффектив-
ный способ комплексной переработки древесины, включающий
184
стадию получения фурфурола при термической обработке из-
мельченной древесины водяным паром в присутствии катализа-
тора. Отличительной особенностью способа является примене-
ние небольшой массовой доли (3—5 % от древесины) катали-
затора — концентрированной серной кислоты. Катализатор
в тонкораспыленном, виде наносится на древесину в двухвалко-
вом червячно-лопастном смесителе, обеспечивающем хорошую
равномерность распределения. Выход фурфурола значительно
выше, чем при прочих методах его получения, и достигает
80 % от теоретически возможного. Весьма важно, что при этом
не происходит разрушения целлюлозы. Благодаря этому целло-
лигнин является полноценным материалом для дальнейшей хи-
мической переработки в гидролизном производстве, в произ-
водстве плитных материалов и для получения левоглюкозана.
Этот способ со значительным экономическим эффектом реали-
зован в промышленности.
Катализаторы использованы и при разработке способов по-
лучения левоглюкозана при пиролизе древесины. Выход лево-
глюкозана при обычном пиролизе березовой древесины в токе
инертного газа-теплоносителя не превышает 1 % от массы а.с.д.
или около 2—2,5 % от теоретически возможного.
Роль реагентов при получении левоглюкозана из древесины
весьма специфична. Реагентов, ускоряющих процесс деполи-
меризации целлюлозы и способствующих увеличению выхода
левоглюкозана, не найдено. Поэтому реагенты, применяемые
при пиролизе древесины с целью увеличения выхода левоглю-
козана, не влияя на сам процесс деполимеризации и вообще
на распад целлюлозы, обеспечивают условия, необходимые для
эвакуации левоглюкозана из зоны реакции.
Сильные кислоты, используемые при получении фурфурола,
для этой цели непригодны. Так, при пиролизе буковой древе-
сины в присутствии серной и соляной кислот под вакуумом вы-
ход левоглюкозана хотя и увеличивается, но остается низким —
не более 3 % от массы а. с. д.
Объясняется это тем, что вместе с ксиланом сильные кис-
лоты разлагают и целлюлозу.
По способу, разработанному в Институте химии древесины
АНЛатвССР А. Я. Калниньшем и В. Н. Сергеевой, термиче-
скую переработку древесины проводят в присутствии сернокис-
лых железа (двухвалентного) или цинка, алюмокалиевых или
железоаммонийных квасцов. На первой стадии процесса, при
обработке древесины лиственных пород, пропитанной раствором
катализатора, водяным паром, разлагается ксилан и получа-
ются фурфурол, летучие кислоты и твердый остаток — целло-
лигнин. На второй стадии целлолигнин пиролизуют в токе
перегретого водяного пара при 320 °C и остаточном давлении
4-Ю3 Па. Выход левоглюкозаиа составляет 10—11 % от массы
а. с. д. (примерно 25 % от теоретически возможного и 40 % от
максимально достигнутого при пиролизе чистой целлюлозы).
185
Таким образом, значение первой стадии сводится по существу
к получению целлолигнина.
В последнее время предложен способ получения левоглюко-
зана из древесины в одну стадию при обычном пиролизе под
атмосферным давлением (ЦНИЛХИ, А. Н. Кислицын с сотруд-
никами). Пиролиз проводится в присутствии реагентов, напри-
мер монохлоруксусной кислоты, хлористого марганца и др.;
которые сильно снижают температуру распада ксилана (с 290
до 200—250 °C) и практически не оказывают влияния на тер-
мораспад целлюлозы. Это приводит к тому, что в древесине
к началу деполимеризации целлюлозы ксилан разлагается,
а остаток фактически становится целлолигнином. Благодаря
этому при одностадийном пиролизе при атмосферном давлении
из древесины березы, пропитанной указанными реагентами, вы-
ход левоглюкозана достигает 13 % от массы а. с. д. Во всех
описанных способах получения фурфурола и левоглюкозана
при термическом разложении древесины используются ре-
агенты, обладающие или явными, или потенциальными кислот-
ными свойствами. Щелочи, напротив, подавляют процессы их
образования, но способствуют увеличению выхода отстойной
смолы и обогащению ее простыми фенолами.
Влияние химических реагентов на выход и свойства древес-
ного угля. Использование химических реагентов представляет
большой интерес для решения важнейшей задачи пиролизного
производства — увеличения выхода древесного угля. О значе-
нии этой задачи говорит тот факт, что в древесный уголь
переходит менее половины имеющегося в древесине углерода.
При исследовании влияния добавок химических реагентов
(3 % г-моля реагента на 100 г древесины) на выход древесного
угля методом термического анализа найдено, что большая
группа веществ обеспечивает увеличение его выхода более чем
на 25 % по сравнению с выходом в контрольном опыте (24,5 %
от массы а. с. д.). К их числу относятся: диаммонийфосфат —
выход угля без золы 30,8%, сульфаниловая кислота — 31, бор-
ная кислота — 32,4, моноаммонийфосфат — 32,5, фосфорная
кислота — 33 и кислотно-перекисный катализатор ЦНИЛХИ —
33,3 %. Увеличение выхода угля объясняется уменьшением по-
терь массы при термораспаде ксилана и целлюлозы. Химиче-
ские реагенты способствуют образованию на начальной стадии
процесса вследствие реакций дегидратации промежуточной
структуры с двойными связями и карбонильными группами,
при дальнейшем распаде которой развиваются конденсацион-
ные процессы образования угольного нелетучего остатка.
При пиролизе березовой щепы, содержащей около 2,8 % от
массы а. с. д. диаммонийфосфата или диаммонийфосфата и
хлористого аммония в соотношении 1:1, в вертикальной не-
прерывнодействующей реторте опытного завода ЦНИЛХИ вы-
ход угля увеличивается в среднем на 34 %. Хорошие резуль-
таты по увеличению выхода древесного угля достигнуты при
186
использовании кислотно-перекисного катализатора (ЦНИЛХИ,
А. Н. Завьялов). Он представляет собой раствор смеси серной
кислоты и перекиси водорода в соотношении 20:1. При терми-
ческом анализе древесины в присутствии кислотно-перекисного
катализатора выход угольного остатка увеличивается в 1,5 раза.
Отмечено повышение примерно в 1,3 раза теплового эффекта
экзотермического распада древесины, что весьма интересно
с позиций расхода тепловой энергии. При пиролизе древесины
в присутствии кислотно-перекисного катализатора (2,5 % от
массы а. с. д.) в вертикальной непрерывнодействующей реторте
опытного завода ЦНИЛХИ выход древесного угля (в пере-
счете на нелетучий углерод) увеличивается по сравнению
с контрольными опытами не менее чем на 30%. Весьма важно,
что удельная производительность 1 м3 реакционного простран-
ства реторты составила 500 кг/ч, что примерно в 3 раза выше
достигнутой производительности этой же реторты при пиролизе
березовой щепы. Выход растворимой и отстойной смол при
пиролизе с катализатором падает соответственно в 5 и 2 раза,
а летучих кислот в пересчете на уксусную — в 1,8 раза. Однако
снижение выхода жидких продуктов не имеет большого значе-
ния, так как по предлагаемому способу парогазы без охлажде-
ния подлежат сжиганию, а газы от сжигания предназначены
для использования в качестве теплоносителя в 'основном про-
цессе. Уголь-сырец пригоден для производства активного ос-
ветляющего угля.
Химические реагенты при пиролизе древесины влияют не
только на количественный выход жидких продуктов и древес-
ного угля, в их присутствии изменяется и физико-химическая
структура последнего. Влияние реагентов на физико-химиче-
скую структуру и свойства угля может быть, использовано для
получения угля-сырца с определенными свойствами, как полу-
продукта для дальнейшей химической переработки. Исследова-
ний в этом направлении пока немного.
Более подробно изучено влияние трех реагентов, повышающих выход
древесного угля — хлорида металла переменной валентности (К-1), диам-
монийфосфата (К-2) и кислотно-перекисного катализатора (К-3) — на
формирование его физико-химической структуры и свойств. Специфика их
действия проявляется уже в неодинаковом изменении элементного состава
при повышении температуры пиролиза. Так, в присутствии К-1 в интервале
400—600 ° С происходит интенсивная потеря кислорода, а выше 600° —
дегидрогенизация. В присутствии К-2 интенсивное удаление кислорода на-
блюдается лишь после 500 °C. Особенности их действия отражаются на из-
менении степени ароматичности: в присутствии К-1 степень ароматичности
выше, а с К-2 ниже степени ароматичности угля, полученного без реагента.
По-разному идет формирование не только химической, но и тонкой физи-
ческой структуры. К-1 способствует образованию более упорядоченной
структуры: при 700 °C образуются не очень большие кристаллиты, но с до-
статочно совепшенной внутренней структурой типа турбостратной (d002=
=0,342 мм). В присутствии К-2 при 700 °C размеры кристаллитов совсем
малы и слабо упакованы (doo2>O,46 нм). Особенности тонкой структуры
своеобразно отражаются на плотности тела угля: с К-1 получаются угли
с меньшей пикнометрической плотностью (при 700 °C—-1,470 г/сма),
187
а с К-2 — более плотные (1,677 г/см3), чем угли, полученные без реагентов
(плотность 1,594 г/см3), хотя, казалось бы, угли с большей упорядоченно-
стью структуры должны иметь большую плотность. Объясняется это тем,
что уголь, полученный с К-1 при 700 °C, содержит 33,7 % от объема тела
замкнутых пор, недоступных для растворителя, расположенных, по-види-
мому, между кострообразно лежащими кристаллитами, а уголь, полученный
с К-2, совсем не содержит замкнутых пор. Древесный уголь, полученный без
реагентов, содержит 12,8 % замкнутых пор. Эти особенности макроструктуры
имеют важное практическое значение, так как известно, что замкнутые поры
играют большую роль в развитии пористой структуры активных углей.
По-разному влияют реагенты и на электрические свойства углей: удельное
сопротивление угля, полученного с К-1, меньше, чем угля, полученного
с К-2; К-3 по своему влиянию на структуру и свойства угля по большин-
ству показателей занимает промежуточное положение.
Структурные особенности древесных углей, полученных с химическими
реагентами, предопределяют и свойства их как полупродуктов, например,
для получения сорбентов различной пористой структуры. Показана прин-
ципиальная возможность получения из них при активации водяным паром
активных углей с мезопористой (адсорбционная активность по мелассе 80 %),
микромезопористой (адсорбционная активность по йоду 80 %, по мелассе
80 %) и микропористой (адсорбционная активность по йоду 90—100 %) струк-
турой с выходом около 20 % от массы а. с. д.
Из изложенного следует, что химические реагенты могут
служить эффективным средством направленного пиролиза дре-
весины с целью увеличения выхода продуктов пиролиза и по-
лучения древесного угля с определенной физико-химической
структурой и свойствами.
§ 34. ТЕРМИЧЕСКОЕ ОЖИЖЕНИЕ ДРЕВЕСИНЫ
Термическое ожижение древесины рассматривается в на-
стоящее время как реальный перспективный способ получения
жидкого топлива — заменителя нефтепродуктов. Примерно
с 1930 г. известно, что при обработке древесины органическими
растворителями при повышенных температурах и давлении
можно добиться полного или значительного превращения ее
в жидкие продукты/ Эффективность ожижения повышается
в атмосфере водорода в присутствии катализаторов. Например,
при обработке еловой древесины двумя частями смеси этанола
и бензола (1:1) при 280 °C и давлении около 16 МПа получа-
ется 54 % жидких продуктов. Остаток составляет 17%. А при
обработке той же древесины при тех же примерно температуре
и давлении, но в атмосфере водорода и в присутствии нике-
левого катализатора древесина полностью превращается в пе-
нообразную массу; 63,5 % продукта — вязкая смола, 53 % ко-
торой растворимо в эфире.
При обработке древесины в креозотовой фракции древесной
смолы (температура выкипания 200—265 °C) вся древесина
переходит в жидкие продукты даже без применения катали-
заторов. Получается 14 % неконденсируемых газов, содержа-
щих 85 % углекислоты, 64—65 % жидкой смолы и 18,5% воды.
При постепенном нагреве древесины до 300 °C (конечное
давление около 14 МПа) в сложной смеси, содержащей 35 %
бензола, 35 % этанола, 10 % смоляных масел и 20 % жидких
188
продуктов растворения древесины, древесина растворяется пол-
ностью. Смолистые вещества (выход 68 % от массы а. с. д.) со-
держат 40—43 % фенолов. Судя по выходу последних в рас-
чете на древесину (24—27%), практически весь лигнин пере-
ходит в фенольные соединения. В качестве растворителей ис-
пытывались антраценовое масло, сланцевый дистиллят, тетра-
лин и другие продукты и полупродукты.
За рубежом, где острее ощущается энергетический кризис,
некоторые способы ожижения древесины доведены до опытно-
промышленной проверки. По одному из способов водную сус-
пензию измельченной древесины быстрорастущего тополя гид-
рируют в присутствии никелевого катализатора при темпера-
туре 325—375 °C и давлении 10 МПа. Древесина при этом-
полностью превращается в жидкие продукты и газ. Выход,
жидких продуктов достигает 31 %; По другому, похожему спо-
собу получается 40 % жидкого топлива с теплотворной спо-
собностью 35,5—36,5 МДж/кг.
Ожижение древесины производится и в две стадии: на пер-
вой стадии подкисленную водную суспензию, содержащую 25 %
древесины, нагревают при 80 °C и давлении около 1 МПа, на
второй — полученную эмульсию обрабатывают синтез-газом
(смесь окиси углерода и водорода) при 370 °C и давлении’
около 20 МПа. Выход жидкого топлива составляет 30—35 %
от древесины с теплотворной способностью 36,5 МДж/кг, мас-
совая доля фенолов в нем — около половины.
Существует еще один способ ожижения древесины. Масля-
ную суспензию измельченной древесины в присутствии катали-
затора (карбонат щелочного металла) обрабатывают смесью
окиси углерода и водорода при 350 °C и давлении около
28 МПа. Выход топливного масла с теплотворной способностью
29—33 МДж/кг составляет 70 % от древесины. Число разрабо-
ток в этом направлении растет.
Глава 9
ТЕХНОЛОГИЯ ПИРОЛИЗА ДРЕВЕСИНЫ
В главе рассмотрены организация склада для приемки и
хранения древесины, оборудование и аппараты для разделки,
сушки и пиролиза древесины. Освещены вопросы совершен--
ствования технологии получения древесного угля в. современ-
ных аппаратах лесохимической промышленности — вертикаль-
ных ретортах.
§ 35. ПРИЕМКА И ХРАНЕНИЕ ТЕХНОЛОГИЧЕСКОЙ
ДРЕВЕСИНЫ
Склад сырья. Для склада сырья выбирается ровная, сухая
площадка в непосредственной близости от предприятия, но
с соблюдением расстояний, требующихся по условиям проти-
189
вопожарной безопасности. Древесину поставляют на склады
пиролизных предприятий железнодорожным или автомобиль-
ным транспортом, равномерно снабжая предприятия сырьем
в течение года.
Железнодорожный транспорт хорошо приспособлен для пе-
ревозки длинномерной древесины, что существенно отражается
на организации перегрузочных работ, подборе средств механи-
зации и технологии подготовки сырья на складе. Выбор необ-
ходимого оборудования для обслуживания железнодорожных
поставок производят, исходя из объема поступающей древе-
сины. Древесину перевозят в открытом подвижном составе
с точным соблюдением технических условий погрузки и креп-
ления груза. Лесоматериалы укладывают вдоль оси вагона.
Для повышения использования грузоподъемности подвижного
состава погрузка древесины длиной более 3,5 м на четырехос-
ные платформы и полувагоны производится с использованием
верхней, суженной части габарита.
Короткомерную древесину перевозят россыпью в полуваго-
нах, обрешеченных по периметру вертикально установленными
стойками, или в пакетах. Для образования пакетов применяют
многооборотные полужесткие стропы или контейнеры-обре-
шетки.
При естественной сушке древесины технически обоснован-
ным является 12-месячный ее запас, так как при более дли-
тельном хранении влажность древесины практически не изме-
няется, а разрушение от грибковых поражений резко увеличи-
вается. Запас древесины при искусственной сушке должен быть
не менее 3 мес.
Древесное сырье на складах лесохимических предприятий
ранее хранилось в виде поленниц, что требовало больших за-
трат ручного труда при погрузочно-разгрузочных работах. В на-
стоящее время кучевое хранение технологической древесины
(табл. 27) позволило уменьшить площадь, занимаемую скла-
дами, и механизировать погрузочно-разгрузочные работы.
Для предприятия с мощностью по перерабатываемому сырью
около 140 тыс. пл. м3 в год общая площадь склада древесины
для хранения трехмесячного запаса должна быть около 2,5 га.
27. РАСЧЕТЫ ПЛОЩАДЕЙ СКЛАДОВ ПРИ КУЧЕВОМ МЕТОДЕ СКЛАДИРОВАНИЯ
ДРЕВЕСИНЫ
Мощность предприятия по перераба- тываемому сырью, тыс. м3 Макси- мальный объем кучи, тыс. м3 Необходимый объем древесины, тыс. м3, при запасе, мес Необходимая площадь склада, м2, при запасе, мес
3 12 3 12
70 50 17,5 70 16 300 38 300
140 50 35,0 140 24 400 65 600
350 50 87,5 350 44 200 148 400
190
При хранении древесины в кучах максимальный объем их в со-
ответствии с противопожарными нормами должен быть
50 тыс. м3 древесины при высоте до 14 м (см. табл. 27). Ширина
прямоугольных куч или диаметр круглых куч должны быть Не
более 50 м, расстояние между продольными сторонами прямо-
угольных куч — 25 м, между торцовыми сторонами, а также
между круглыми кучами— 15.
Рис. 48. Схематический план склада сырья:
/ — железнодорожный путь; 2 — кран консольно-козловой грейферный; 3 — кабельный
лоток; 4 — штабеля длинномерной древесины; 5 — разобщитель бревен; 6 — бревно-
таска; 7 — слешер для долготья; 8 — узел для сортировки и расколки древесины; 9 —
конвейер выносной; 10 — цех подготовки сырья; // — дорога для автомашин; 12 — пита-
тель тарельчатый; 13 — кучевой штабель короткомерной древесины; 14 — конвейер лен-
точный
Склад сырья для удобства эксплуатации должен быть хо-
рошо освещен и благоустроен. На рис. 48 показан схематиче-
ский план склада технологической древесины.
Приемка и учет древесного сырья. Древесное сырье прини-
мают партиями. Партией считается любой объем древесного
сырья одного назначения и оформленный одним документом.
При приемке технологической древесины следят за тем, чтобы
она удовлетворяла требованиям стандарта по породному со-
ставу, размеру, массовой доле гнили и была очищена от сучьев.
Учет древесного сырья производится по объему в складоч-
ных кубических метрах. Складочную меру в плотную перево-
дят по фактическому коэффициенту полнодревесности, который
определяют следующим образом. Выкладывают пробную по-
ленницу и с лицевой стороны вне клеток по выбору намечают
прямоугольник высотой равной высоте поленницы и длиной ос-
нования вдоль поленницы не менее 8 м. Стороны прямоуголь-
ника очерчивают углем, краской или мелом. В прямоугольнике
проводят диагональ, которая должна пересечь торцы не менее
60 поленьев. Длину диагонали измеряют с точностью до 1 см,
причем доли менее 0,5 см в расчет не принимают, а доли рав-
ные 0,5 см и более считают за целый сантиметр.
191
Сумму протяжения торцов измеряют по диагонали. Каждый
торец измеряют с точностью до 0,5 см, при этом доли менее
0,3 см в расчет не принимают, а доли равные 0,3 и более счи-
тают за 0,5 см.
Коэффициент полнодревесности, выраженный в сотых долях,
устанавливают делением суммы протяжений торцов поленьев
по диагонали на длину всей диагонали. При длине пробной
поленницы менее 8 м проводят не одну, а две диагонали. Если
длина основания намеченного прямоугольника охватывает все
протяжение между двумя соседними клетками и по диагонали
этого прямоугольника размещается менее 60 торцов поленьев,
то намечается указанным выше способом еще один прямоуголь-
ник вне клеток в той или другой поленнице. В последнем слу-
чае коэффициент плотности укладки древесного сырья уста-
навливают делением суммы протяжения торцов поленьев по
двум диагоналям на сумму длин этих диагоналей.
В поленницах сырья из круглых поленьев проверку полно-
древесности производят измерением толщины торцов поленьев
в коре на лицевой стороне кладки в прямоугольнике площадью
4 м2. Коэффициент полнодревесности определяют делением
суммарной площади торцов поленьев на площадь прямоуголь-
ника.
Значение полученного коэффициента полнодревесности срав-
нивают со значением обязательного коэффициента полнодре-
весности, приведенным в табл. 28. Если значение коэффици-
ента полнодревесности, вычисленное по диагонали, отличается
28. ЗНАЧЕНИЯ ОБЯЗАТЕЛЬНОГО КОЭФФИЦИЕНТА ПОЛНОДРЕВЕСНОСТИ
Коэффициенты полнодревесности для поленьев длиной, м
Породы 0,25 0,33 0,50 0,75 1,00 1,25 1,50 2,00 2,50 3,00
1. Круглые
Хвойные
Лиственные
Хвойные
Лиственные
Тонкие (толщиной 3—10 см)
I 0,79 I 0,77 I 0,74 I 0,71 I 0,69 I 0,67 I 0,66 I 0,64 I 0,62 I 0,61
I 0,75 I 0,72 I 0,69 | 0,65 I 0,63 | 0,61 | 0,60 | 0,58 | 0,56 | 0,55
Средние (толщиной 11—14 см)
I 0,81 I 0,79 I 0,76 I 0,74 I 0,72 I 0,71 I 0,70 I 0,68 I 0,67 I 0,66
I 0,80 I 0,78 I 0,75 | 0,72 | 0,70 | 0,68 | 0,67 | 0,65 | 0,63 | 0,62
2. Колотые (из поленьев толщиной 15 см и более)
Хвойные I 0,77 I 0,75 I 0,73 I 0,71 I 0,70 I 0,69 I 0,68 I 0,66 I 0,64 I 0,63
Лиственные | 0,76 | 0,74 | 0,71 | 0,69 | 0,68 | 0,67 | 0,65 | 0,63 | 0,62 | 0,60
3. Смесь из круглых (40 %) и колотых (60 %) поленьев
Хвойные I 0,77 I 0,75 I 0,73 I 0,72 I 0,70 I 0,69 I 0,68 I 0,67 I 0,66 I 0,65
Лиственные | 0,76 | 0,74 | 0,71 | 0,69 | 0,68 | 0,67 | 0,66 | 0,65 | 0,64 | 0,63
192
от значения, приведенного в табл. 28, на 0,02 и более, объем
партии определяют по фактическому коэффициенту полнодре-
весности, определенному замером.
Пересчет объема партии древесного сырья, выраженного
в массе, т, в плотную меру, м3, производят умножением массы
сырья каждой породы в отдельности на соответствующий пе-
реводной коэффициент с учетом влажности древесины.
Приемку долготья производят так же, как и приемку мет-
ровой древесины. В последнее время на некоторых лесохимиче-
ских предприятиях приемка технологической древесины произ-
водится весовым методом путем взвешивания ее на весах с по-
следующим пересчетом массы в плотную меру.
Требования к хранению сырья. Процесс хранения сырья ока-
зывает существенное влияние на качество полученных из него
продуктов пиролиза и технологию работ на складе. Способ
хранения древесины в кучах должен создавать условия для
механизации перегрузочных и транспортных работ. Помимо
этого, во время хранения древесины на складе должны быть
сохранены все ее технологические свойства, а также приняты
меры к защите древесины от поражения грибами и насеко-i
мыми. Среди многочисленных вредителей существует группа
особых грибов, развивающихся преимущественно в условиях
лесных складов и носящих название складских.
По виду разрушенной древесины различают два основных
типа грибных гнилей: коррозионный и деструктивный. Приме-
ром коррозионной гнили может служить гниль лиственных по-
род древесины от некоторых грибов из рода полипорус (Polypo-
rus adustus и Polyporus zonatus); примером деструктивной
гнили — гниль от трутовика (Polyporus sulphureus), березовой
губки (Fomes betulinus), окаймленного трутовика (iFomes un-
gulatus) и др. Кроме отмеченных двух основных типов гнилей,
существуют смешанные гнили. Примером коррозионно-деструк-
тивной гнили может служить белая гниль (Fomes igniarius).
По месту расположения в стволе можно различать сердцевин-
ную, периферийную и смешанную гнили.
Органические вещества древесины трудно усваиваются гри-
бами. Для превращения их в легкоусвояемые вещества, вызы-
вающие гниение древесины, вырабатываются ферменты, воз-
действующие на компоненты клеточных оболочек (целлюлоза,
гемицеллюлоза и лигнин), и на вещества, содержащиеся в клет-
ках древесины (крахмал, глюкозиды, жиры и пр.).. Фермен-
тами, превращающими целлюлозу и гемицеллюлозу в глюкозу
и другие растворимые сахара, являются целлюлаза и гемицел-
люлаза (или цитаза), а ферментом, разрушающим лигнин,—
лигниназа. Крахмал, глюкозид и жиры имеют второстепенное
значение для питания дереворазрушающих грибов.
Древесина в кучах поражается также личинками различи
ных жуков: короедов, усачей, рогохвостов и др. Условия их
развития в древесине (температура, влажность, породный
7 Заказ № 2193 193
состав) часто совпадают с условиями поражения ее грибами.
Выход продуктов пиролиза из древесины, пораженной гри-
бами, резко падает, а древесный уголь теряет при этом меха:
ническую прочность и резко повышается его способность к са-
мовозгоранию при хранении. Наименее стойка к грибам древе-
сина березы, бука, осины, липы и ольхи.
Интенсивность развития грибов и скорость гниения древе-
сины зависят от влажности древесины и микроклимата в зоне
ее хранения. При некотором минимуме и максимуме этих ус-
ловий мицелий гриба гибнет. Для большинства видов дерево-}
разрушающих грибов оптимальная температура роста 22—
29 °C при влажности древесины 40—50 %. Если влажность дре-
весины ниже 30 или выше 50 %, то рост грибов резко замедля-
ется. При температуре выше 36 °C грибы отмирают. Весной
и осенью низкая температура задерживает рост грибов, зимой
он совсем прекращается. Практически опасными для древе-
сины являются месяцы с мая по август включительно.
Таким образом, для предохранения древесины от повреж-
дения грибами необходимо содержать ее в условиях пони-/
женных температур и в сухом или очень влажном состоянии.*
В условиях лесохимической промышленности для преду-
преждения обоих видов повреждений и улучшения сушки дре-
весины внутри куч рекомендуется делать проходы или приме-
нять искусственную вентиляцию. Для этого необходимо уст-
раивать кучи высотой не более 10 м и шириной 25 м.
Подготовка сырья к переработке. Ранее лесозаготовитель-
ные предприятия поставляли для пиролиза древесину, разде-
ланную на поленья длиной 1 м. Поэтому на складе сырья
разделка древесины не предусматривалась или в соответствии
с принятой технологией разделывалась на чурки длиной 140—
330 мм. На таких предприятиях технологическая древесина со
склада поступала* в разделочное отделение.
В настоящее время технологическая древесина поступает на
предприятия в виде 2, 4 и 6-метровых кряжей или хлыстов.
В этом случае разделка древесины переносится на склад сырья
лесохимического предприятия.
Для увеличения производительности труда на складах дол-
жны широко внедряться прогрессивные технологические про-
цессы, оборудование, транспортные средства и другие научно-
технические разработки. Мощность предприятий пиролиза дре-
весины составляет 70—300 тыс. м3 технологической древесины
в год.
Распиловка и расколка древесины являются основными опе-
рациями в цикле подготовки сырья для пиролиза и произво-
дятся с целью получения нужных по длине и толщине размеров
чурок. Для распиловки больших объемов сырья применяют
многопильные станки (слешеры) с круглыми пилами, позво-
ляющие распиливать кряжи одновременно на большое число
отрезков поперечным надвиганием на комплект пил. Станки
194
этого типа обладают высокой производительностью и в усло-
виях лесохимического производства являются основным видом
оборудования для поперечной распиловки древесины (рис. 49).
Слешер представляет собой наклонный стол, ширина кото-
рого несколько больше длины распиливаемых кряжей. На столе
смонтирован поперечный цепной конвейер для подачи кряжей
на распиловку. Между цепями конвейера с захватами
опилок
в шахматном порядке вмонтированы пильные валы с пиламц
и приводными электродвигателями. Число пил слешера и рас-
стояния между пилами выбираются в зависимости от длины
распиливаемых кряжей и заданной длины отрезков. В про-
мышленности применяют слешеры с 2—7 пилами, позволяю-
щими распиливать поленья и производить оторцовку одного из
концов ствола. Для сбора образующихся при пилении опилок
в нижней части цепного стола устраивается воронка с наклон-
ными стенками. Удаление опилок от слешера производится
с помощью конвейеров или пневматических установок всасы-
вающего действия.
Слешеры монтируют в закрытых неотапливаемых помеще-
ниях. Распиловочные установки являются местом повышенной
опасности для работающих, поэтому к их расположению и уст-
ройству предъявляют требования, регламентируемые специаль-
ными правилами. Места установки пил слешера изолируются
от мест нахождения обслуживающего персонала сетчатым ог-
раждением из толстой проволоки, предохраняющим обслужи-
вающий персонал от случайных ударов. Слешерный стол также
должен иметь ограждение, чтобы обслуживающий персонал
во время работы пил не подходил к нему. Для входа ремонт-
ного персонала на стол слешера устраиваются специальные
двери, сблокированные с пусковым механизмом пил. Пилы
слешерных установок доджны быть снабжены автоматически
7* 195
действующими тормозами, сблокированными с пусковым уст-
ройством пил, позволяющими после выключения электродвига-
телей останавливать пильный диск за 4—6 с. Для смены пил
необходима установка подъемных устройств грузоподъем-
ностью, соответствующей массе пилы.
Древесина диаметром более 14 см и длиной 1 м подлежит
расколке. На лесохимических предприятиях расколку древе-
сины производят на гидравлическом дровокольном станке марки
КГ-8А, который состоит из следующих узлов: станины, рас-
калывающего устройства, толкателя, гидроаппаратуры, при-
вода, пульта управления и электрошкафа.
Станина является базовым узлом, на котором монтируются
все основные части; представляет собой жесткую сварную кон-
струкцию. В передней части станины, перед направляющей рас-
калывающего устройства, устанавливается лоток для приема
и направления древесины во время раскалывания. Раскалываю-
щее устройство состоит из основания коробчатой формы, к ко-
торому крепятся раскалывающие клинья, обеспечивающие рас-
колку древесины различного диаметра на две, четыре и шесть
частей. Оно может перемещаться вверх и вниз по направляю-
щей станины при помощи гидроцилиндра, закрепленного в спе-
циальном пазе направляющей.
Толкатель шарнирно соединен со штоком гидроцилиндра,
который перемещает его назад и вперед вдоль оси станины.
Гидроаппаратура и привод станка состоят из двух гидроци-5
линдров, один из которых шарнирно соединен с толкателем!
станка, а второй служит для подъема и опускания раскалын
вающего устройства, золотника для управления работой раска-
лывающего устройства,. двух золотниковых распределителей
для переключения на различные режимы работы, насосов низ-
кого и высокого давления, а также контрольной и измеритель-
ной аппаратуры.
Пульт управления предназначен для дистанционного управ-
ления станком и располагается в удобном для оператора ме-
сте. При годовом объеме переработки технологической древе-
сины 100' тыс. м3 в год численность работающих на Складе
сырья составляет 65 чел., в слешерной 19 чел., а! при перера-
ботке 300 тыс. м3 в год работающих на складе сырья увеличи-
вается на 3 и в слешерной на1 чел. Расход электроэнергии при
этом уменьшается на 55%, а ориентировочные затраты на пе-
реработку 1 м3 древесины — на 25 %.
§ 36. СУШКА ТЕХНОЛОГИЧЕСКОЙ ДРЕВЕСИНЫ
Главная цель сушки состоит в превращении древесины из
природного сырья в промышленный материал с улучшенными
технологическими свойствами. Сушка — процесс удаления влаги
из материала путем ее испарения. Удаляется, как правило,
влага, связанная с древесиной физико-химически (адсорбци-
196
окно и осмотически) и механически (влага макро- и микрока-
пилляров).
При сушке древесины влага внутренних слоев, прежде чем
испариться, должна переместиться к поверхности. Скорость ее
перемещения внутри древесины во много раз меньше, чем воз-
можная скорость ее испарения с поверхности. Поэтому поверх-
ностные слои древесины высыхают быстрее, чем внутренние.
Чем выше температура теплоносителя, ниже его влажность и
больше скорость движения у поверхности, тем быстрее проис-
ходит испарение, а следовательно, высушивание поверхностных
слоев древесины.
Из-за отставания процесса движения влаги из внутренних
областей древесины по сравнению с процессом испарения ее
с поверхности образуется значительная разность (перепад)
влажности между внутренними и поверхностными слоями дре-
весины. Чем больше перепад влажности в древесине, тем ин-
тенсивнее перемещение в ней влаги. Свойство древесины пере-
мещать в себе влагу под действием градиента влажности на*
зывается влагопроводностью.
Вторым побудителем перемещения влаги внутри материала
является разность температур между внутренними и наружными
слоями высушиваемого материала. Свойство древесины пере-
мещать в себе влагу под влиянием разности температуры на-
зывается термовлагопроводностью. Непосредственным побуди-
телем движения влаги в древесине при наличии разности тем-
пературы по ее сечению является перепад давления водяного
пара. Давление паров влаги на поверхности древесины с повы-
шением температуры возрастает, и пары диффундируют в по-
ток сушильного агента.
Термовлагопроводность весьма эффективно используется
при высокотемпературной сушке древесины (до 380 °C), когда
интенсивность перемещения влаги в древесине резко возрастает.
Опыт работы сушилок при высокотемпературной сушке древе-
сины показал, что сушку древесины до конечной влажности!
(10—15%) можно осуществить в течение 7—8 ч.
Способы сушки древесины. В настоящее время для сушки
древесины на пиролизных предприятиях применяется атмосфер-
ная (естественная) и камерная (искусственная) сушка.
Атмосферная сушка — способ сушки древесины в естествен-
ных условиях (без подогрева) при ее выдержке на складе. При
атмосферной сушке исключительно важно обеспечить непре-
рывное движение воздуха у поверхности отдельных поленьев
древесины. Регулировать ее можно лишь в незначительной
степени путем изменения плотности укладки куч. Интенсив-
ность атмосферной сушки зависит от климатических условий
местности и от времени года (зимой сушка практически пре-
кращается). Несмотря на ее простоту и сравнительно низкую
себестоимость, этот способ сушки применяется лишь на не-
которых лесохимических предприятиях, так как на складах
197
сырья предприятий не всегда имеется необходимый запас дре-
весины.
При естественной сушке древесины в отсутствие принуди-
тельного движения сушильного агента (свободное испарение)
процесс идет очень медленно (несколько недель); он ускоря-
ется при обтекании древесины потоком теплоносителя, т. е.
при искусственной сушке.
По методам передачи тепла различают следующие основ-
ные виды сушки: конвективная (камерная), кондуктивная (кон-
тактная), радиационная, сублимационная и высокочастотная.
Каждый из этих видов может иметь несколько разновидностей
в зависимости от типа теплоносителя и особенностей применяе-
мого оборудования. Существуют также комбинированные спо-
собы сушки, в которых одновременно применяются различные
виды передачи тепла или совмещаются другие признаки раз-
личных видов сушки.
Конвективная газовая сушка — процесс обезвоживания
древесины нагреванием для превращения влаги из жидкого
в парообразное состояние; при этом объем влаги увеличива-
ется в сотни раз и она выделяется в окружающую среду в виде
пара. Масса удаленной из древесины влаги соответствует ко-
личеству подведенного к ней тепла. Это основной промышлен-
ный способ сушки технологической древесины на пиролизных
предприятиях, осуществляемый в сушильных камерах различ-
ных конструкций. Для нагревания и циркуляции сушильного
агента камеры снабжаются специальными устройствами. При
камерной сушке сроки высыхания древесины сравнительно не-
большие (несколько часов), и при квалифицированном ее про-
ведении можно получать технологическое сырье любой конеч-
ной влажности.
Кондуктивная сушка осуществляется путем непосредствен-
ного контакта высушиваемого материала с нагреваемой поверх-
ностью. 4
Радиационная сушка основана на передаче материалу тепла
излучением от нагретых тел. Этот способ для сушки древесины
на пиролизных предприятиях не применяется, но рекомендован
для сушки материалов при деревообработке.
Сублимационная сушка — удаление влаги из материала в за-
мороженном состоянии под вакуумом.
Высокочастотная сушка основана на удалении влаги из ма-
териала под действием. электрического поля высокой частоты.
Применяется для сушки изделий из древесины. Благодаря рав-
номерному нагреву древесины по всему объему, возникновению
положительного градиента температуры и избыточному давле-
нию внутри куска продолжительность высокочастотной сушки
в десятки раз меньше, чем конвективной. Из-за сложности обо-
рудования, большого расхода электроэнергии и высокой стои-
мости сушки этот метод не находит широкого применения
в. промышленности.
198
Снижение тепловых затрат при сушке древесины. Один из
основных факторов, влияющих на производительность пред-
приятий пиролиза древесины и себестоимость получаемой про-
дукции,— влажность перерабатываемого сырья. Чем суше дре-
весина, тем быстрее проходит процесс пиролиза, повышается
кислотность жижки и, следовательно, снижаются тепловые за-
траты на ее переработку и утилизацию сточных вод. Поэтому
интенсификация процесса сушки является первостепенной зада-
чей повышения эффективности работы предприятий пиролиза
древесины.
Однако при глубокой сушке древесины растут материальные
затраты, требуются сушилки больших объемов, поскольку их
удельная производительность при низкой влажности древесины
уменьшается в несколько раз. Принято считать, что наиболее
экономичен пиролиз древесины влажностью 15—25%; но прак-
тически влажность высушенной древесины колеблется в более
широких пределах.
Сушка технологической древесины осуществляется за счет
тепла продуктов сгорания жидкого, твердого или газообразного
топлива. Свежесрубленная древесина имеет 80—100% абсо-
лютной влажности, но при длительной естественной сушке
влажность может быть снижена до 25 %. В последнее десяти-
летие все лесохимические предприятия перешли на кучевое хра-
нение древесного сырья, но вопросы естественной сушки дре-
весины в кучах изучены недостаточно. Известно, что за 11 мес
при высоте кучи 10 м и основании 18X18 м влажность древе-
сцны уменьшилась с 69 до 33%. В настоящее время склады
сырья лесохимических предприятий не приспособлены для про-
ведения естественной сушки древесины и, что самое главное,
отсутствуют необходимые запасы древесины, а в сушилки по-
ступает древесина с влажностью 70—90 %.
Искусственная сушка древесины в горизонтальных сушил-
ках производится при начальной температуре сушильного
агента 180—300 °C и массовой доле кислорода в нем 12—14%;
в вертикальных непрерывнодействующих сушилках темпера-
тура сушильного агента 160—360 °C и массовая доля кислорода
в газе 3—17%. Отработанный сушильный агент с испаренной
влагой при температуре 80—160 °C сбрасывается в атмосферу.
Конечная влажность древесины после сушки в сушилках,
помимо исключительно важного влияния на технологические
процессы пиролиза (производительность аппаратов, качество
угля и жидких продуктов пиролиза, объем сточных вод и др.),
заметно влияет на расход тепловой энергии при пиролизе дре-
весины. Анализ показывает, что за счет естественной сушки дре-
весины расход тепла на искусственную сушку может быть
уменьшен в 3—4 раза.
На многих предприятиях температура отработанного тепло-
носителя, сбрасываемого в атмосферу, 160 °C. Сбрасывать
агент сушки в атмосферу температурой ниже 100 °C при ис-
Ч'./ J99
пользовании специальной топни нецелесообразно из-за конден-
сации влаги в дымовой трубе и ухудшения качества сушки.
Охлаждением отработанного агента в специальном аппарате
до температуры 50—60 °C только за счет конденсации влаги
можно получить низкотемпературный теплоноситель с тепловой
энергией 1,2—1,5 ГДж/т абсолютно сухой древесины, или около
60 % от затраченного на сушку тепла. Однако прежде чем ре-
шить вопрос утилизации низкотемпературного теплоносителя,
необходимо найти пути его использования.
Другие резервы экономии тепла: повышение температуры
в топке сушилки, снижение подсосов воздуха в сушильный
агент, хорошая теплоизоляция сушилок и трубопроводов.
Математическая модель процесса сушки древесины. Для
описания процесса сушки технологической древесины в прямо-
точной непрерывнодействующей сушилке со сравнительно рав-
номерным распределением теплоносителя по всему объему дре-
весины можно использовать математическую модель в виде
линейных уравнений регрессии для следующих показателей:
для конечной влажности
- W* = 33,2—0,1027/н —0,145fK—3,34т—3,73ш + 0,301 Гн +
+ 207d$+56,7ft (71)
при SocTu) = 5,46% и Rw = 0,95;
для степени термического разложения древесины в процессе
сушки
Y=—1,34+0,0246/н-Ь 0,374т-0,0341FH—2,7ЫФ (72)
при Soctt = 0,72 и /?х = 0,88;
для продолжительности сушки
т=6,8—0,0181^—0,0258^—0,160^—0,374а»+
+0,0487ХО2 + 0,0705Ц7н+33,Мф + 9,36ft (73)
при Зостт=0.72 и 7?х = 0,88;
в области условий, ограниченных значениями;
200 < t№ + 380;
0,47 s£ w С 1,22;
0,08 < <1ф < 0,185;
80 С tK 175;
26 < Wh < 94;
0,14 < ft < 0,25;
3<т< 10;
0 < WK < 60;
2,5 Xq2 20,
где WB и — начальная и конечная абсолютная влажность
древесины, %; У — степень термического разложения древесины,
определяемая по убыли сухой массы древесины в процессе
сушки, %; т — продолжительность сушки, ч; tB и tK — началь-
ная и конечная температура сушильного агента, °C; w — ско-
рость теплоносителя в свободном сечении сушилки в пересчете
200
на нормальные, условия, м/с; h — высота образца древесины, м;
ХО9 — массовая доля кислорода в сушильном агенте, %;
Зостт —остаточная дисперсия выходных параметров; Л?— мно-
жественный коэффициент корреляции; — фиктивный диаметр
образца древесины, м;
с/ф = д/4?7л, (74)
здесь F — площадь поперечного сечения образца древесины
произвольной формы, м2.
Конструкции сушильных устройств. В производственных ус-
ловиях сушку древесины производят в различных установках
непрерывного и периодического действия. Конструкция и тип
сушилок определяются конструкцией реторт и печей, перераба-
тывающих подсушенную древесину. В большинстве случаев дре-
весина вводится в камеру пиролиза на вагонетках, в кото-
рых осуществлялась и. сушка. В вертикальных непрерывнодей-
ствующих сушилках и ретортах древесина движется сверху вниз
под действием силы тяжести.
Туннельные сушилки. Сушка технологической древесины
для вагонных стальных реторт периодического действия осуще-
ствляется в сушилке, схема которой показана на рис. 50. Кир-
пичные сушильные каналы длиной 33,6 м и сечением 2,3X 2,6 м2
попарно объединены в блоки и разделены общей продольной
стеной. В каждом канале помещается 8 или 12 (при удлинен-
ных сушилках) технологических вагонеток вместимостью каж-
дая. 5,6—6,1 м3 древесины, разделанной на чурки длиной 150—
330 мм. Загрузка и выгрузка сушилок происходит в разных
концах камеры. Одновременно в сушилку подают состав из
четырех технологических вагонеток с древесиной.
Технологическая вагонетка собрана на горизонтальной пря-
моугольной раме, выполненной из швеллерного железа. К раме
и стойкам, расположенным по ее углам, приварены решетки из
полос сортового железа для того, чтобы газы лучше проникали
внутрь слоя загруженной древесины. Нижняя половина боко-
вых стенок вагонетки может быть съемной (для возможной
загрузки метровых поленьев). Древесину в виде чурок длиной
150—330 мм грузят навалом. В первой половине сушилки за-
груженная древесина сохнет в течение одного оборота реторты
(в среднем 17—18 ч), после чего передвигается во вторую по-
ловину сушила, а в первую половину загружают четыре ва-
гонетки нового состава с сырой древесиной. Продолжитель-
ность сушки древесины во второй половине сушилки — также
один оборот реторты. В двухоборотных сушилках на этом
цикл сушки заканчивается, а в трехоборотных производят еще
одно передвижение вагонеток. Продолжительность сушки дре-
весины, загруженной в двухоборотные сушилки, составляет 34—
36 ч, в трехоборотных — 51—54 ч.
201
Сушка технологической древесины производится дымовыми
газами, которые вентилятором нагнетаются в распределитель-
ный боров от топок ретортных печей. Дымовые газы от котель-
ной дымососом нагнетаются в общий боров, откуда подаются
в продольные распределительные борова сушил, из которых
агент сушки поступает в сушильный канал через отверстия,
сделанные по всей длине наружной стены.
Рис. 50. Технологическая схема сушилки для стальных вагонных реторт:
/ — вытяжная труба; 2 — технологическая вагонетка; 3 —сушильная камера; 4 — вен-
тилятор; 5 — распределительный боров
Дымовые газы, поступающие в сушило от топок ретортных
печей, разбавляются воздухом, засасываемым через специаль-
ный патрубок в газопроводе перед вентилятором, чтобы снизить
их температуру до 2’20—270 °C. Из продольных распредели-
тельных боровов дымовые газы через верхние отверстия посту-
пают в сушильную камеру по всей ее длине и поднимаются
вверх между стеной камеры и вагонетками. Распределившись
над верхним сводом камеры, дымовые газы проникают в ваго-
нетки с древесиной, отдают свое тепло древесине, охлаждаясь
и увлажняясь при этом, и опускаются в нижнюю часть камеры
в канал для отвода отработанного теплоносителя, откуда че-
рез вытяжную трубу сбрасываются в атмосферу. Практически
часть газов удаляется также через неплотности загрузочной и
выгрузочной дверей сушилки.
В СССР также работают туннельные сушилки для кирпич-
ных реторт. Камеры в сушилке такого типа объединены по-
парно и связаны между собой общей системой боровов, подво-
дящих агент сушки, и размещенных на перекрытиях сушильных
каналов.
В каждом сушильном канале длиной 15 м помещается со-
став из четырех стальных вагонеток, вмещающих по 4,2 м3
202
древесины. Поперечное сечение сушильных каналов и самих ре-
торт— почти правильный круг. Диаметр поперечного сечения
сушильного канала 2220 мм, внешний диаметр- вагонеток
1900 мм, длина вагонеток 3200 мм. Дно и стенки технологиче-
ской вагонетки на 3/4 высоты обшиты листовым железом^
а в верхней части боковых и торцевых стенок высверлены круг-
лые отверстия для прохождения газа. Площадь отверстий в тор-
цевых стенках составляет 15 %, а в боковых 10 % от всей
площади листа, затрудняя доступ агента сушки к древесине.
Буковая древесина метровой длины в сушилках этого типа
выдерживается в течение одного оборота реторты, т. е. при-
мерно 24 ч, после чего все вагонетки передвигаются в реторту,
а сушилка загружается новой партией древесины.
Древесину сушат отходящими дымовыми газами топок ре-
торт и топочными газами из специальной топки, температура
которых не превышает 170—180 °C. Теплоноситель поступает
в систему боровов, уложенных на перекрытиях сушильных кана-
лов либо со стороны загрузки древесины, либо с выгрузочной
стороны. Отработанный агент сушки выбрасывается через от-
верстия в стояках в атмосферу.
Шахтные сушилки СВНД-4. В отечественной лесохимиче-
ской промышленности около 33 % технологической древесины
перерабатывается в вертикальных непрерывнодействующих ре-
тортах. Древесина для них высушивается в шахтных сушилках
(рис. 51), технологическая схема сушки древесины показана
на рис. 52.
Сушилка представляет собой шахту цилиндрической формы
диаметром 4,2 м и высотой цилиндра 16,6 м. Движение техно-
логической древесины и теплоносителя внутри сушилки осу-
ществляется прямотоком, загрузка и выгрузка древесины —
порционная.
Сырая лиственная древесина, разделанная на чурки длиной
150—330 мм, с ленты конвейера загружается в течку объемом
2,5 м3 древесины, а из нее при открытии тележечного гидроза-
твора проваливается в верхнюю часть сушилки. В сушилке
древесина прогревается и из нее испаряется влага. Высушен-
ная древесина в нижней части сушилки разгрузочными плоско-
стями направляется на цепной питатель и выгружается порци-
ями около 1,2 м.3 в ковш скипового подъемника для загрузки
в реторту.
Сушат древесину дымовыми газами температурой 180—
350 °C (в зависимости от влажности поступающей на сушку
древесины), которые образуются в топке при сжигании печного
топлива и неконденсирующихся газов реторты.
Сушильный агент, проходя сверху вниз через весь слой дре-
весины в сушилке, насыщается влагой, охлаждается и направ-
ляется к дымососу. Часть отработанного теплоносителя дымо-
сосом выбрасывается в атмосферу через дымовую трубу, остав-
шаяся часть (рециркулят) нагнетается в камеру смешения
203
топки, куда поступают и дымовые газы из камеры сгорания
для формирования теплоносителя. Объемный состав теплоноси-
теля, %, следующий: СО2 11,7—15,7; О2 1,6—5,1; СО 0,4—2,5;
Н2 0,1—1,1; СН4 0,2—1,3 и остальное N2.
Для наблюдения за режимом сушилка оснащена приборами
для контроля температуры теплоносителя на входе и выходе,
расхода топлива и воздуха в топку сушилки. При необходимо-
сти сушилка может быть переведена на автоматический режим
поддержания температуры.
Рис. .51. Схема сушилки увели-
ченного полезного объема (шахт-
ная сушилка):
1 — штуцер для ввода теплоносителя;
2 — течка для загрузки древесины;
3 — сушилка; 4 — конус; 5 — штуцер
для вывода теплоносителя; 6 — течка
для выгрузки древесины; 7 — цепной
питатель
Рис. 52. Технологическая схема
сушки древесины в шахтной су-
шилке СВНД-4:
1 — воздуходувка; 2 — топка; 3 — ды-
мосос; 4 — сушилка
204
29. ПОКАЗАТЕЛИ РАБОТЫ ШАХТНЫХ И ТУННЕЛЬНЫХ СУШИЛОК
Показатель Шахтная Амзинско- го ЛК СВ Н Д-4 (диаметр 3,6 м) СВНД-4 (диаметр 4,2 м) Стальная реторта Кирпичная реторта Печь В. Н. Козлова
Объем, м3 92 162 220 220 65 290
Производительность, кг/ч а. с. д. 1530 3070 4800 480 430 1880
Продолжительность сушки, ч Температура, °C: 12,9 5,1 6,7 38,0 20,8 26,0
на входе в сушилку 244 335 320 226 185 100
на выходе из сушилки 106 148 115 85 60 55
Скорость сушильного агента, м/с 1,72 1,16 1,20 0,38 0,30 . 0,43
Перепад давления по сушилке, Па 1350 700—1000 500—900 60-80 20 400—600
Массовая доля кислорода в сушильном агенте, % Влажность древесины относительная, %: 11,7 5,9 5,0 17,7 17,0—18,0 13-18
начальная 46,5 41,2 40,2 32,0 28,8 43,0
конечная 24,3 13,6 17,1 17,5 17,9 28,1
Отношение массы испаренной влаги к начальной 0,63 0,77 0,69 0,55 0,44 0,48
Нагрузка на полный объем по испаренной влаге, кг/(м3-ч) 9,10 10,30 10,10 0,55 1,23 2,26
Отношение теплопотерь к поступающему в систему теплу 0,05 0,27 0,05 0,23 0,15 0,16
Удельный расход тепла на испарение влаги, МДж/кг 7,2 4,3 4,1 12,5 6,6 12,9
Теоретический удельный расход на испарение при теплопотерях 10 % в том же режиме, МДж/кг 6,3 3,8 3,6 5,5 5,2 3,7
Показатели работы шахтных и туннельных сушилок приве-
дены в табл. 29.
Для сравнения с действительным в табл. 29 приведен и тео-
ретический удельный расход тепла, кДж/кг, вычисленный для
сушилок, использующих отработанные дымовые газы:
Яо =
2610 4-3,35/к
1 Ка (^к/^н)
(75)
и для сушилок со специальной топкой для генерации сушиль-
ного агента
_ 2610 + 3,35fK Z7fi
Чс---------------ГЙ------’ (76)
“ (21—ХО2)100
где К. а— коэффициент теплопотерь сушилки.
Из табл. 29 следует, что при одинаковом объеме (220 м3)
производительность шахтной сушилки на порядок выше, чем
сушилки для стальной реторты. Нагрузка по испаренной влаге
в шахтных сушилках составляет 9,1—10,3 кг/м3-ч, а в тун-
нельных 0,55—2,26; продолжительность же сушки в шахтных
сушилках сокращается в 4—8 раз по сравнению с туннельными.
Для получения и формирования сушильного агента удельный
расход тепла на испарение влаги в сушилках со специальной
топкой (СВНД-4) в 1,7—1,8 раза меньше, чем в сушилках, ра-
ботающих на отработанных дымовых газах (шахтная сушилка
Амзинского лесокомбината). Однако Амзинский лесокомбинат
использует для сушки технологической древесины отработанный
теплоноситель после котельной, поэтому утилизация сбросного
тепла представляет интерес и для других лесохимических пред-
приятий.
§ 37. ПИРОЛИЗ ДРЕВЕСИНЫ
Пиролизное производство на крупных лесохимических пред-
приятиях включает в себя пиролиз древесины с полной перера-
боткой жидких продуктов пиролиза и получением товарного
продукта — древесного угля. При этом неконденсирующиеся
газы пиролиза используются как топливо.
Аппараты для термического разложения древесины подраз-
деляются по принципу действия на непрерывнодействующие,
периодически действующие и полунепрерывного действия.
Непрерывнодействующие аппараты наиболее совершенны,
так как в них полнее используется их объем, достигается уско-
рение процесса пиролиза и экономия топлива, они могут быть
механизированы и автоматизированы; в них налажен контроль
за соблюдением режима пиролиза, уголь получается по каче-
ству более однородный с массовой долей нелетучего углерода
до 95 %. Условия труда обслуживающего персонала удовлетво-
ряют современным требованиям производства.
206
Периодически действующие аппараты обладают многими не-
достатками: у них низкий уровень механизации технологиче-
ского процесса, низкая удельная производительность; они
в ряде случаев не отвечают современным требованиям по ох-
ране окружающей среды.
Аппараты полунепрерывного действия обладают низкой
удельной производительностью. В промышленной практике ча-
сто их называют непрерывнодействующими.
По принципу обогрева аппараты пиролиза древесины разде-
ляются на аппараты с наружным и внутренним обогревом.
В аппаратах с наружным обогревом тепло от теплоносителя
к древесине передается через стенки реторт, обогреваемые го-
рячими дымовыми газами. Внутри аппаратов тепло от стенок
через прослойку газов передается древесине лучистым тепло-
вым потоком и конвекцией. Разложение древесины в аппаратах
начинается в первую очередь около стенок и вследствие малой
теплопроводности древесины проходит неравномерно. Поэтому
стенки аппаратов приходится перегревать и топочные газы вы-
ходят из аппарата с высокой температурой.
Аппараты с внутренним обогревом обладают многими пре-
имуществами: тепло от теплоносителя передается непосредст-
венно древесине путем искусственной циркуляции и конвектив-
ного теплопереноса.
В лесохимической промышленности используются аппараты
внутреннего обогрева с газообразным теплоносителем, который
получают путем сжигания жидкого или газообразного топлива
в специальных топках. Разложение древесины в этом случае
проходит в более мягких условиях и при более низкой темпеч
ратуре.
Жидкие продукты пиролиза быстрее выводятся током те-
плоносителя из аппарата и не разлагаются от соприкоснове-
ния с его перегретыми стенками, поэтому выход некоторых*
продуктов выше, чем в аппаратах с наружным обогревом.
Однако для аппаратов с внутренним вводом теплоносителя
необходимо увеличивать конденсационную систему, так как кон-
центрация продуктов пиролиза древесины в парогазовой смеси
вследствие ее разбавления циркулирующим теплоносителем
уменьшается.
Горизонтальные реторты и печи. К этим аппаратам отно-
сятся: туннельная стальная вагонная реторта, туннельная кир-
пичная вагонная реторта, углевыживательная печь системы
В. Н. Козлова, кирпичная камерная печь Шварца, углевыжи-
гательные печи «Свердлеспром-4» и УВП-5.
Туннельная стальная вагонная реторта
(рис. 53) работает по принципу внешнего обогрева дымовыми
газами. Технологический процесс пиролиза древесины в тун-
нельных стальных вагонных ретортах состоит из следующих
операций: загрузки реторт, пиролиза древесины и разгрузки
реторт.
207
Каждая реторта представляет собой камеру, склепанную
или сваренную из 10—12-миллиметровой стали. Длина реторты
16,5 м, ширина 2 м, высота 2,5 м. Реторта вмещает четыре ва-
гонетки полезной вместимостью 5,6—6,1 м3 каждая. Одновре-
менно в реторту загружается 20—21 м3 древесины, разделан-
ной на чурки длиной 150—330 мм. Реторты попарно обмуро-
ваны в кирпичной камере и образуют одну ретортную печь или
Рис. 53. Туннельная стальная реторта:
/ — вентилятор; 2 — экран; 3 — заслонка; 4 — реторта; 5 — вытяжные зонты;
6 — топка; 7 — конденсатор
блок. Каждая реторта свободно висит на подвесках, прикреп-
ленных к металлическому каркасу печи. Расстояние от свода
топки до дна реторты равно примерно 1 м. Реторты имеют мас-
сивные чугунные двери, снабженные бороздками для набивки
прографиченного асбестового шнура. По ее дну проложен рель-
совый путь. Парогазовые продукты пиролиза отводятся из ре-
торты через два патрубка, расположенных на высоте 1675 мм
от низа реторты с боковых ее сторон; эти патрубки соединены
с конденсаторами-холодильниками.
Подсушенная в сушилке древесина подается в реторту на
четырех вагонетках; после закрытия дверей начинается про-
цесс пиролиза. Реторта обогревается дымовыми газами, обра-
зованными при сжигании жидкого топлива и неконденсирую-
щихся газов пиролиза в двух топках, расположенных под
обоими концами реторты. Топки перекрыты сводом из огнеупор-
ного кирпича. Продукты сгорания топлива проходят из топки
по горизонтальному каналу (в боковых стенках свода его уст-
роены 34 небольших прогара — отверстия прямоугольной
формы), нагревают боковые стенки реторты и направляются
208
в конец печи, где поднимаются и, омывая верх реторты, венти-
лятором нагнетаются в сушилки. Между стальной стенкой ре-
торты и обмуровкой печи проложен толстый асбестовый шнур.
Разрежение в топке поддерживается 15—30 Па.
В начале процесса переугливания обогрев усиливают. Затем
по достижении под ретортой температуры 450—460 °C во из-
бежание износа стенок реторты и перегрева конденсаторов наг
грев несколько ослабляют, вновь усиливая его на 0,5—1 ч (до
температуры 500—600 °C) в конце процесса для прокалки угля.
Во избежание подсоса воздуха давление в реторте в период
разложения древесины должно быть 50—150 Па. Подсос воз-
духа в зону пиролиза древесины недопустим. Наблюдение за
равномерностью накала свода печи и низа реторты ведется по
показаниям термометрических милливольтметров. Подача
в топку реторты неконденсирующихся газов пиролиза регули-
руется задвижками, расположенными на подводящих патруб-
ках; газ должен подаваться только в достаточно раскаленную
топку.
Пиролиз древесины в реторте в зависимости от ее влажно-
сти, и степени разделки продолжается в среднем 17—18 ч. Сле-
довательно, суточная пропускная способность одной реторты
составляет около 28 м3 древесины. Далее уголь переводят в ту-
шильники и выдерживают в течение 34—36 ч. Половину этого
времени уголь находится в тушильниках первого ряда, а вто-
рую половину — второго. Охлажденный уголь до выгрузки на
склад дополнительно выдерживается на открытой площадке
в течение 17—18 ч. Выстаивание угля необходимо для того,,
чтобы полностью предотвратить возможность его самовозгора-
ния на складе. Выгружают уголь на сортировку при помощи
опрокидывателя вагонеток.
Разгрузка каждой реторты начинается с выбивания клиньев
у ретортной двери со стороны углетушильников и открытия
заслонки в вытяжном колпаке над площадкой между ретор-
той и тушильником угля. Перед тем как выбить последний
клин, обязательно закрывают задвижку у конденсаторов на га-
зомагистрали.
После открытия двери укладывают съемные перекидные
рельсы от реторты к тушильнику угля, за буфер первой ваго-
нетки прицепляют трос от электролебедки и перевозят ваго-
нетки с углем в заранее освобожденный тушильник. Уголь при
выгрузке из реторты всегда загорается, и вагонетки с углем во
время передвижения из реторты в первый ряд тушильников
орошают водой. Вагонетки передвигают в такой последователь-
ности: сначала четыре вагонетки с площадки выстаивания угля
подают на склад для разгрузки; затем из второго ряда тушиль-
ников передвигают четыре вагонетки на площадку выстаива-
ния, а из первого ряда тушильников во второй ряд — на охлаж-
дение. Далее из реторты четыре вагонетки передвигают
в тушильник, а в реторту помещают четыре вагонетки с древеси-
209
ной из сушилки. Все эти операции производят при помощи
электролебедки, расположенной между площадкой выстаива-
ния и складом угля; от электролебедки к вагонеткам идет
стальной трос. В сушилку при помощи мотовоза ввозят вновь
четыре загруженные древесиной вагонетки, одновременно пе-
редвигая находящиеся в сушилке четыре вагонетки во вторую
ее половину, ближе к ретортам. При передвижении вагонеток
из сушилки в реторту между ними также укладывают съемные
перекидные рельсы после открытия дверей реторты. Как только
передвижение вагонеток закончено и реторта загружена ваго-
нетками с высушенной древесиной, перекидные рельсы снимают,
чугунные двери реторты закрывают и плотно прижимают к чу-
гунной раме стальными клиньями. Так же закрепляют и двери
тушильников.
Выход древесного угля составляет 151—159 кг из 1 м3 бе-
резовой древесины и 142—154 кг из древесины смешанных!
пород.
Контроль процесса переугливания ведется по температуре
под ретортой. Кроме того, систематически наблюдают за ско-
ростью выделения дистиллята, его кислотностью и темпера-
турой. Температура дистиллята при выходе из ретортных кон-
денсаторов не должна подниматься выше 30 °C, а температура
отходящей из конфенсатороЪ воды — выше 80 °C. При кислот-
ности дистиллята ниже 1,5 % производят разгрузку реторты.
Туннельные кирпичные вагонетки реторты
используются на заводах пиролиза древесины с обогревом при
помощи калориферов (жаровых труб). Кирпичные реторты груп-
пируются обычно в блоки попарно, в общей кирпичной кладке.
На рис. 54 изображена кирпичная реторта с цилиндрическим
сводом и цилиндрической вагонеткой в ней. В промышленности
применяются и прямоугольные кирпичные реторты с прямо-
угольными вагонетками. Каждая реторта представляет собой
камеру длиной 15,8 м и поперечником 2,1 м, имеющую толщину
стенок в 2,5 и 3,5 кирпича. С обоих концов реторта имеет двери;
стенки реторты глухие. Кладка реторты выполнена на глине
с примесью жидкого стекла. Двери реторты бывают обычно
в виде стенки, подвешенной на специальный блок над дверным
проемом. Они имеют вид рамы каркаса, заполненного кирпи-
чом. Щели между полотном двери и конструкцией собственно
печи в период пиролиза замазывают глиной или обкладывают
асбестовым шнуром.
Вдоль всей длины реторты на уровне пола уложены на спе-
циальных опорах рельсы (колея 750 мм), по которым в реторту
вводятся сцепом три-четыре вагонетки вместимостью обычно
около 4,2 м3 древесины каждая.
Реторта обогревается продуктами сгорания смеси некон-
денсирующихся ретортных газов и генераторного газа. Указан-
ная смесь сжигается в калориферах, изготовленных из стальных
(частично жароупорных) труб> уложенных вдоль нижней части
210
рётортЫ, между рельсами и уровнем пода реторты. Диаметр
труб около 200 мм, численность их шесть-восемь в каждой ре-
торте. Трубы от приемника газа, расположенного впереди ре-
торты, проходят через всю реторту по длине и заканчиваются
в борове, который соединяется с дымовой трубой. Каждая
труба является одновременно топкой и калорифером. Воздух,
необходимый для горения газа в трубах, подсасывается
Рис. 54. Кирпичная реторта с калорифером:
1 — распределительный коллектор для газа; 2 — канал реторты; 3 — дверь реторты;
4 — люк для выпуска паров влаги в период сушки древесины; 5 — штуцер для отвода
парогазов; 6 — дымоход; 7 — калорифер
(вследствие разрежения, создаваемого тягой дымовой трубы)
через кольцевое пространство между трубой-калорифером и
трубой, подводящей газ из сборного газопровода. При таком
способе обогрева регулировать температуру внутри реторты
довольно легко.
Генераторный газ, добавляемый к неконденсирующимся га-
зам, получают в отдельных газогенераторах, работающих по
принципу обращенного процесса газификации.
Парогазовая смесь отводится из реторты через штуцер, рас-
положенный сбоку в своде передней части реторты. Для отвода
паров воды в период сушки загруженной древесины (до повы-
шения кислотности дистиллята до 1—1,5%) имеется особый
люк, соединенный с атмосферой. Лишь после сброса этих паров
кислая парогазовая смесь направляется в поверхностный кон-
денсатор, откуда неконденсирующиеся газы идут на сжигание,
а дистиллят (жижка) на переработку.
Продолжительность оборота реторты составляет в среднем
18—20 ч. При переугливании воздушно-сухой древесины рас-
ход топлива на обогрев реторт очень низок, что объясняется
хорошей системой обогрева реторт жаровыми трубами и по-
ниженными потерями тепла через кирпичные стенки по срав-
нению с его потерями через стальные.
211
Калориферный обогрев можно рассматривать как переход-
ный к внутреннему обогреву. При калориферном обогреве ус-
ловия разложения древесины мягче, чем в стальных ретортах
с наружным обогревом.
Тушильники угля к кирпичным ретортам часто устраивают
в виде обычных сушильных камер, поливаемых сверху струй-
ками отбросной воды из конденсаторов реторт, при этом про-
цесс тушения угля ускоряется вдвое.
Углевыжигательная печь системы В. Н. Коз-
лова — циркуляционная, двухканальная, вагонеточная, непре-
рывнодействующая; работает по принципу внутреннего обо-
грева и предназначена для получения древесного угля и жидких
продуктов пиролиза. Каждый канал печи имеет камеры сушки,
пиролиза древесины и охлаждения угля, конденсационную и
рекуператорную установки. Камера сушки отделена от основ-
ного канала.
Два кирпичных сушильных канала объединены в общий блок
и разделены продольной стенкой. Сушильный канал длиной
50 и шириной 2,3 м сверху перекрыт кирпичным сводом. Рас-
стояние от пода сушила до замка свода 2,43 м.
У входа и выхода кирпичной камеры сушки имеются сталь-
ные шиберы. Она вмещает 10 вагонеток, объем каждой около
7 м3 древесины, разделанной на чурки длиной 220 мм, кото-
рые загружаются насыпью. Сушка в камере противоточная,
производится смесью отработанных дымовых газов из рекупе-
раторного отделения и камеры сушки с температурой 200—
250 °C.
Продолжительность пребывания древесины в камере сушки
20—25 ч. Весь состав вагонеток передвигается одновременно
в камерах печи через 2—2,5 ч, в зависимости от рлажностш
древесины. Теплоноситель через боров, уложенный по оси пода
сушилки, вводится в сушильный канал под вагонетки с древе-
одной через 35 щелей длиной 0,8 и шириной 0,07 м. Пройдя
2/з длины сушильного канала, часть агента сушки отсасывается
для смешения с горячими газами от калорифера, а остальная
часть проходит через переднюю зону сушильного канала и вы-
водится в дымовую трубу через отверстия в поду канала. Сте-
пень заполнения сушильного канала очень низка, поэтому те-
плоноситель может проходить вдоль сушильного канала по сво-
бодной части его сечения, не контактируя достаточно хорошо
с древесиной.
Частые загорания древесины в этих сушилках, как и в тун-
нельных горизонтальных^ объясняются использованием в каче-
стве теплоносителя дымовых газов с массовой долей кислорода
около 13—18 %.
Печь состоит из приемного тамбура, камеры переугливания,
среднего тамбура, камеры охлаждения угля и выводного там-
бура. Печь вмещает 16 вагонеток: 3 в приемном, среднем и вы-
ходном .тамбурах, 7 в камере углежжения и 6 в камере охлаж-
212
дения. Пиролиз древесины осуществляется циркулирующими
парогазами, нагретыми в рекуператоре до 420—480- °C. Паро-
газы поступают в камеру пиролиза по. газоходу,., расположен-
ному в выходном конце камеры, посередине пода, длина кото-
рого соответствует длине четырех вагонеток. В своде газохода
устроены отверстия, через которые нагретые газы поступают
под вагонетки.
Парогазы проходят по камере пиролиза навстречу вагонет-
кам с древесиной и углем, отдают тепло, обогащаются продук-
тами сушки и пиролиза древесины и вместе с ними выходят из
печи в начале камеры пиролиза в систему конденсации. В кру-
говом движении, при определенной производительности печи,
объем циркулирующих газов остается все время приблизи-
тельно одинаковым.
В камере сушки и в печи вагонетки передвигаются толка-
телем, который состоит из двух лебедок с электродвигателями,
стального каната и кареток на роликах, прикрепленных к ка-
нату. (
Температурный режим камеры углежжения: в начале камеры
180—220 °C, середине 275—350 и конце 380—410 °C. Продол-
жительность охлаждения древесного угля 12 ч.
Рекуперационная установка включает две топки, в которых
сжигаются дрова, древесные отходы и неконденсирующиеся
газы (возможно сжигание жидкого топлива). В рекуператоре
циркуляционные газы при температуре 90 °C нагнетаются в ка-
лорифер, где нагреваются до 420—480. °C, затем поступают
в камеру углежжения.
Производительность одного канала печи 74—88 м3/сут, или
1500—2000 т угля в год; выход древесного угля при перера-
ботке березы до 37 %, по массе к абсолютно сухой древесине,
массовая доля нелетучего углерода в угле 73—78 %.
Кирпичные камерные печи Шварца эксплуатиру-
ются в небольшом количестве, так как условия обслуживания
печи тяжелые: загрузка древесины и выгрузка древесного угля
производятся вручную, не решен вопрос охраны окружающей
среды (газы пиролиза выбрасываются в атмосферу), печи ма-
лопроизводительны. Их используют в основном на нижних
складах леспромхозов для получения древесного угля. Камер-
ная печь (рис. 55) сложена из красного кирпича, фундамент
из бутового камня; горизонтальное сечение печи прямоуголь-
ное, свод — продольный и может* быть сферической или плоской
формы.
Вместимость печей позволяет загружать от 50 до 100 м3
древесины складочного объема. Древесина в печь загружается
вручную через боковые двёри. В нижнем слое дрова уклады-
вают горизонтально, в среднем вертикально, в верхнем — на-<
валом мелкие дрова. Теплоносителем служат дымовые газы,
получаемые от сжигания дровяного топлива в топке, располо-
женной под основанием печи. Дымовые газы из топки проходят
213
под свод пени, откуда через дрова опускаются вниз и выходят
через дымовые трубы, поставленные по углам печи. Часть
конденсирующихся продуктов (в основном смола) стекает
в сборник. После окончания переугливания загруженных
дров все отверстия в печи замазывают глиной и без доступа
воздуха оставляют печь для охлаждения. Весь период работы
Рис. 55. Камерная печь (ураль-
ская) :
1 — печь; 2 — столб шатра; 3 —
дымовая труба; 4 — горизонталь-
ные балки; 5 — шатер; 6 — углуб-
ление перед топкой; 7 — топка; 8 —
сборник смолы и жижки
Рис. 56. Установка для пиролиза древесины «Свердлеспром-4»:
1 — вытяжная труба камеры пиролиза; 2 — контейнер с загруженной древесиной; 3 —
съемная плита свода; 4 — камера сушки древесины; 5 — камера пиролиза; 6 — колодец
для подсмольной воды; 7 — канал для вывода парогазов и подсмольной воды; 8 — канал
для подвода газов от топки к сушильной камере; .9 — канал для вывода газов из ка-
меры сушки; 10 — топка камеры пиролиза
печи (от загрузки древесины до выгрузки охлажденного угля)
занимает 6—8 дней. В топке сжигается 12—18 % Дров от объ-
ема переугливаемой древесины. Качество получаемого древес-
ного угля неоднородное, массовая доля нелетучего углерода
колеблется от 65 до 75 % •
У г л евыжигате л ь н а я печь «Свердлеспром-4».
Коллектив объединения «Свердлеспром» разработал несколько
типов усовершенствованных углевыжигательных установок на
базе печей Шварца, наилучшей из которых в настоящее время
является печь «Свердлеспром-4» (рис. 56). Она отличается бо-
ги
лее высокой степенью механизации погрузочно-разгрузочных
работ и несколько улучшенными санитарными условиями об-
служивания. Сырьем для пиролиза служит древесина листвен-
ных и хвойных пород при длине кусков 150—1000 мм.
Установка состоит из трех камер, из которых две боковые
предназначены для углежжения, а третья — для сушки дров.
Вместимость каждой камеры 87,5 м3 древесины, которая загру-
жается в специальные металлические контейнеры вместимостью
8,8 м3 каждый. Всего р камеру загружается 10 контейнеров.
Размер каждого контейнера, мм: ширина 1840, высота 2100 и
дл/ина 4700.
/ Стены камер железобетонные, покатый пол выполнен из бе-
тона толщиной 50 мм; вверху установка перекрывается съем-
ными железобетонными плитами, которые так же, как и кон-
тейнеры с древесиной и древесным углем, поднимаются и транс-
портируются консольно-козловым краном. В среднюю часть
камер углежжения из топки снизу подводится теплоноситель
(горячие дымовые газы с температурой не выше 700 °C), полу-
чаемый в результате сжигания топливной древесины или дре-
весных отходов, расход которых составляет 15 % от объема пе-
рерабатываемого технологического сырья.
Для удаления парогазовой смеси из камер углежжения уста-
новлены две деревянные трубы. Средний оборот каждой камеры
углежжения 5 сут. Утилизация парогазовых продуктов отсут-
ствует. Мощность установки по переработке березовой древе-
сины около 12 тыс. м3 в год, выход угля 1980 т в год.
Углевыжигательная печь УВП-5 передвижная, пе-
риодического действия с цилиндрической двухстенной камерой
углежжения, с внутренним обогревом древесины при углежже-
нии топочными газами, с выносной топкой. Топка футерована
огнеупорным кирпичом. Внутри камеры по всей длине и над
топкой расположен газовый канал, служащий распределителем
горячих топочных газов. Выход топочных газов осуществляется
через вытяжную трубу. Диаметр печи 2,33 м, длина 6 м. Ка-
мера углежжения оборудована боковыми и верхними люками,
предназначенными для загрузки сырья и выгрузки угля. Для
удобства выгрузки древесного угля из камеры печь оборудована
механизмом поворота.
Полный технологический цикл получения угля в печи вклю-
чает следующие операции: подготовительные работы, загрузку
дров в камеру углежжения, углежжение, охлаждение угля в ка-
мере и выгрузку его. При работе камеры 60 ч (один полный
цикл) производительность печи составляет 100—120 т угля
в год. Утилизация парогазовых продуктов не ведется.
Шахтные непрерывнодействующие аппараты. Вертикаль-
ная непрерывнодействующая реторта. На новых
и реконструируемых предприятиях сооружаются только верти-
кальные непрерывнодействующие реторты как наиболее совер-
шенные. В этих аппаратах получается древесный уголь с мас-
215
совой долей нелетучего углерода не менее 88 % и зольностью не
более 2,5 % • Единичная мощность отечественных вертикальных
реторт достигает 9 тыс. т угля в год. Предусмотрены верти-
кальные реторты удвоенной единичной мощности (до 14 тыс,, т
угля в год).
На рис. 57 изображена общая схема вертикальной непре-
рывнодействующей реторты с аппаратурой для подачи тепло-
Дребесина
Рис. 57. Общая схема вертикальной непрерывно действующей реторты:
/ — вертикальная реторта; 2 — топка; 3 — турбогазодувка второго контура; 4 — контей-
нер для угля; 5 — конвейер-стабилизатор; 6 — скребковый конвейер; 7 — сборник жид-
ких продуктов пиролиза; 8 — сборник оборотной жижки; 9 — насос; 10 — турбогазодувка
основного контура; // — холодильник; 12 — скруббер; 13 — гидрозатвор
носителя в реторту, выгрузки, охлаждения и стабилизации дре-
весного угля. Реторта представляет собой стальной цилиндр,
составленный ид царг на фланцевом соединении с внутренним
диаметром 0,5—2,8 м. Наиболее распространена вертикальная
реторта с внутренним диаметром 2,7—2,8 м, толщиной стенок
14 мм, высотой с затворами 26 м, полезной высотой 15,1—18 м.
Реторта имеет вверху загрузочное устройство для загрузки
древесины, внизу — конусную часть и выгрузочное устройство
для угля. Верхний затвор реторты автоматически открывается
и закрывается (в зависимости от места, где находится ковш
216
скипового подъемника). Реторта имеет четыре штуцера: для
вывода парогазовой смеси, для ввода в реторту теплоносителя,
для вывода газов, охлаждающих уголь, а также для ввода в ре-
торту холодных газов на охлаждение угля. Для улучшения рас-
пределения теплоносителя в реторте и обеспечения более равно-
мерной прокалки угля в середине реторты имеется изготовлен-
ный из листовой стали усеченный конус. Под ним установлен
второй конус, из-под которого газы, охлаждающие уголь и на-
гревающиеся при этом, отводятся газодувкой в топку реторты.
\ Для обеспечения равномерного движения древесины в ре-
торте установлен стальной конус, обращенный основанием вниз.
Он задерживает спуск центральной части столба древесины,
уменьшая давление шихты на выгрузочный скребковый кон-
. вейер. Верхняя часть реторты изолирована пенодиатомовым кир-
пичом. Во избежание подсосов воздуха в ней поддерживается
небольшое давление.
Технологический процесс получения угля в цехе вертикаль-
ных непрерывнодействующих реторт включает пиролиз древе-
сины, охлаждение и конденсацию жидких продуктов пиролиза,
охлаждение неконденсирующихся газов и угля, стабилизацию
угля и получение теплоносителя.
Рассмотрим технологическую схему пиролиза древесины
в вертикальных шахтных ретортах Ашинского лесохимического
завода. Подсушенная в сушилке древесина в виде чурки длиной
200 мм скиповым подъемником подается в загрузочную течку
реторты, откуда при открытии верхнего тележечного гидравли-
ческого затвора загружается в реторту. Загружается реторта
только при наличии в ней свободного места, которое фиксиру-
ется механическим уровнемером. В реторте осуществляется пи-
ролиз древесины, продуктами которого являются древесный
уголь и парогазы. Древесина в реторте медленно движется
сверху вниз и проходит следующие зоны:
окончательной сушки древесины при температуре 150—
270 °C, которая сопровождается потерей главным образом воды;
разложения древесины при температуре 270—450 °C.
Полученный уголь движется вниз и проходит также зоны:
прокаливания при температуре 450—600 °C; охлаждения при
температуре 25—70 °C, где он охлаждается неконденсирующи-
мися газами пиролиза и непрерывно выгружается герметичным
скребковым конвейером на дополнительно установленный лен-
точный конвейер. На конвейере уголь охлаждается и стабили-
зируется в слое до 100 мм в атмосфере воздуха в течение 7 мин
и далее ссыпается в вагонетку объемом 8,9 м3, в которой и на-
правляется на угольный склад для сортировки и отгрузки по-
требителям.
-Степень стабилизации угля контролируется по повышению
температуры его в полностью заполненном контейнере. Ранее
уголь' при загрузке в контейнере самовозгорался, его приходи-
лось заливать водой. По новому способу стабилизации угля при
217
температуре выгрузки его из реторты от 70 до 80 °C и окру-
жающей среды выше 0 °C температура угля в контейнере не
поднимается выше 40 °C, т. е. уголь стабилизируется, и полно-
стью исключается самовозгорание его в технологической ва-
гонетке.
Из верхней части реторты парогазы отводятся с температу-
рой 100—130 °C, направляются в форконденсатор и через гид-
равлический затвор в скруббер-конденсатор, где из них улав-
ливаются жидкие продукты пиролиза, которые далее поступают
на переработку (см. главу 11). Орошение скруббера-конденса-
тора осуществляется жижкой при температуре 25—35 °C. Па-
рогазовая смесь состоит из паров и капель лесохимических про-
дуктов, паров воды, неконденсирующихся газов пиролиза и га-
зов теплоносителя. Неконденсирующиеся газы с температурой
30—50 °C турбогазодувкой основного газового контура подаются
на охлаждение угля в нижнюю часть реторты, а избыток газов
поступает в топку сушилки. Пройдя через слой угля и охладив
его, неконденсирующиеся газы нагреваются до температуры
160—250 °C, выводятся из-под нижнего распределительного ко-
нуса реторты турбогазодувкой второго газового контура и на-
правляются частично в топку реторты на сжигание (в нижнюю
часть), частично на разбавление горячих дымовых газов (в верх-
нюю часть) при формировании теплоносителя с температурой
600—700 °C и объемной долей кислорода не более 1 %.
В топку для сжигания неконденсирующихся газов воздухо-
дувкой подается воздух. Теплоноситель из топки поступает под
верхний газораспределительный конус реторты и, проходя
в верх реторты, обеспечивает прокалку угля и испарение влаги,
оставшейся после сушки в древесине. Смешиваясь с летучими
продуктами пиролиза, теплоноситель выводится из реторты
в виде парогазов, начиная новый цикл рециркуляции части га-
зов. Режимы работы реторты приведены в табл. 30.
Состав газов, используемых в качестве теплоносителя, топ-
лива и охлаждающего агента, а также их плотность и низшая
теплота сгорания приведены в табл. 31.
Установка для пиролиза древесины фирмы
«Л а м б и о т». В последние годы в СССР пущена в эксплуата-
цию установка на базе непрерывнодействующей реторты в ре-
жиме безотходного производства, разработанная бельгийской
фирмой «Ламбиот», с единичной мощностью по углю 2000 т
в год. Реторта (рис. 58) представляет собой цилиндр с перехо-
дом к нижней части в конус. Вверху реторта имеет загрузоч-
ный люк для древесины, внизу нижний шлюзовой затвор для
выгрузки древесного угля. Парогазовые продукты пиролиза сжи-
гаются как в самом процессе, так и в виде факела.
Исходным сырьем для реторты служит воздушно-сухая дре-
весина в виде чурок длиной 300 мм и влажностью 25—35 %.
Разделка древесины на чурку производится на специальной гид-
равлической установке гильотинного типа.
218
30. РЕЖИМЫ РАБОТЫ РЕТОРТЫ
Место замера Температура, °C Давление. КПа
Газоход теплоносителя в реторту 600—700 2,1—2,5
Низ реторты 25—70 1,8—3,0
ДТбд вторым конусом В зоне: 150—250 2,0—2,9
прокаливания угля 450—600 2,0—3,0
пиролиза древесины 300—500 —
подсушки древесины 150—270 —
На выходе парогазов из реторты 100—130 0,4—0,6
Газ на охлаждение угля 30—50 1,8—3,0
31. СОСТАВ ГАЗОВ, ИХ ПЛОТНОСТЬ И ТЕПЛОТА СГОРАНИЯ
Газ Объемная концентрация, % Плот- ность, кг/м3 Низшая теплота сгорания, кДж/кг
со2 о2 со Н2 сн4 n2
Газ-теплоноситель гв ре- торту 21,0 0,4 6,7 5,7 4,0 62,2 1,32 2897
Основного контура 18,3 0,6 13,0 9,7 4,8 53,6 1,24 4413
Второго контура 18,4 0,7 12,6 9,5 4,8 54,0 1,25 4341
Из узла выгрузки угля 14,8 8,7 2,3 8,2 5,4 60,5 1,25 2548
Условно реторту можно разделить на три части. Верхняя,
куда загружается древесина, является дополнительной сушил-
кой; средняя (расширенная) является зоной пиролиза и про-
калки угля и нижняя — зоной охлаждения угля. В промежутке
между верхней и средней зонами располагается топочная зона,
куда при помощи вентилятора вдуваются через специальные
штуцера горячие газы пиролиза, забираемые с низа средней
зоны из-под специального конуса. В топочную часть реторты че-
рез патрубки поступает воздух, который позволяет сжигать
часть поступающих парогазов пиролиза, повышая температуру
в этой зоне до 500—600 °C. Смесь парогазов и продуктов их
сжигания частично поступает в нижележащую зону пиролиза,
а избыток уходит в верхнюю часть реторты — сушилку. Поступ-
ление воздуха в топочную зону, а также массы горячей смеси
в зону пиролиза регулируется автоматически при помощи ми-
никомпьютера.
Горячие газы, образующиеся в топочной зоне, поступают
в сушильную часть реторты для подсушки древесины от 25—
35 %-ной влажности до абсолютно сухого состояния и выбра-
сываются через трубу в атмосферу. Эта смесь парогазов и па-
ров воды сушки еще достаточно горюча и горит в виде факела,
благодаря чему исключается попадание в атмосферу вредных
по составу парогазов: выбрасываемые продукты их горения не
содержат, кроме углекислоты, вредных примесей.
219
Образующийся в процессе пиролиза древесный уголь посту-
пает из зоны пиролиза в зону охлаждения, что осуществляется
за счет ввода в нижнюю часть реторты охлажденных инертных
газов, циркулирующих в этой зоне. Циркуляция инертных газов
производится следующим образом. При достаточно высокой
температуре инертные газы, выходящие из зоны охлаждения
угля, концентрируются под конусным сужением, расположенным
между зонами пиролиза и охлаждения угля, и направляются
в небольшой циклон, откуда попадают в конденсатор-холодиль-
ник (скруббер), орошаемый охлажденной водой. Далее холод-
ные газы вентилятором вдуваются в нижнюю часть реторты.
Некоторый избыток воды, образующийся в процессе конденса-
ции парогазов, попадающих в замкнутую систему охлаждения
инертного газа, сливается в условно чистые воды.
Весь процесс пиролиза и последующая транспортировка угля,
начиная’ от загрузки древесины в реторту и кончая загрузкой
угля в бункера, автоматизированы. Первоначальный запуск ре-
торты производят за счет сжигания в топоч-
ной зоне топлива при заполненной углем
нижней части реторты и древесиной сушиль-
ной ее части. После нагрева реторта начи-
нает работать за счет сжигания собственно
парогазов, имеющих достаточно высокую теп-
лотворную способность.
Время процесса, включая сушку, пиролиз
и охлаждение угля, составляет 14 ч. при на-
чальной температуре процесса 500 °C. Уста-
новка рассчитана на переработку 8000 т в год
воздушно-сухой древесины, что соответствует
плотному объему 10 350 м3 березовой или
12230 м3 сосновой древесины с получением
2000 т древесного угля влажностью не более
4 % и массовой долей нелетучего углерода
85—90 % (в зависимости от температуры про-
калки угля). Выход угля в пересчете на бе-
резовую древесину достигает 193 кг/м3 и на
сосновую— 163 кг/м3.
Фирмой «Ламбиот» разработаны уста-
новки производительностью 6000 т угля в год.
Установки прежнего типа, с использованием
жидких продуктов пиролиза, в настоящее
время фирмой не выпускаются из-за отсутст-
вия потребности в них.
Рис. 58. Реторта для пиролиза древесины:
/ — люк для загрузки древесины; 2 — выход парогазов в атмо-
сферу; 3— зона сушки; 4— зона сжигания парогазов; 5 —зона
пиролиза; 6 — вентилятор; 7, 14—конусы для разгрузки слоя
угля; 8— зона охлаждения; 9 — вход охлаждающего газа;
10 — люк для выгрузки угля; 11 — шибер; 12 — выход горячего
газа; 13 — конус, разделяющий зоны; 15 — впуск воздуха;
16 — коллектор для воздуха
220
Пути повышения единичной мощности непрерывнодействую-
щих реторт. На новых и реконструируемых заводах сооружа-
ются только вертикальные непрерывнодействующие реторты, по-
этому в последние годы значительная часть работ выполнена по
их усовершенствованию с целью повышения единичной мощно-
сти реторт. В этих аппаратах получается древесный уголь с мас-
совой долей нелетучего углерода не менее 88 % и зональностью
не более 2,5 %,. Единичная мощность по углю отечественных
вертикальных реторт достигает 1100 кг/ч.
Проблема повышения их мощности имеет важное народно-
хозяйственное значение. Совместными работами ЦНИЛХИ и
ЛТА методом моделирования процесса пиролиза древесины по-
лучены необходимые данные для проектирования таких реторт
удвоенной мощности, которые заложены в проекте реконструк-
ции Сявского лесохимического завода. Они обеспечивают также
повышение производительности труда.
Однако проблема повышения единичной мощности верти-
кальных реторт этим не исчерпывается. Необходимо повысить
единичную мощность и существующих вертикальных реторт. На
некоторых заводах объем реторт увеличен путем установки
в верхней' части реторт дополнительной царги вместимостью
17—18 м3, что позволило повысить единичную мощность ре-
торты на 10—12 %,. Открылась возможность повысить единич-
ную мощность вертикальной непрерывнодействующей реторты
и за счет перехода на новый, более интенсивный способ ох-
лаждения и стабилизации древесного угля, разработанный
ЦНИЛХИ.
Суть способа заключается в том, что горячий уголь в виде
отдельных кусков или в тонком слое охлаждают непосредст-
венно воздухом, причем начальная температура угля должна
быть ниже критической для данных условий. Охлаждение угля
до этого уровня должно осуществляться известными приемами
(инертными газами или через стенку).
В результате охлаждения указанным способом уголь стано-
вится малоактивным по отношению к кислороду и отпадает не-
обходимость в специальной его стабилизации с целью исключе-
ния самовозгорания. Установлено (рис. 59), что средняя темпе-
ратура угля в точке, лежащей на 1 м ниже уровня выхода ох-
лаждающего газа, составляет 70 °C. Таким образом, при работе
реторты по новому режиму для охлаждения угля достаточен
тушильник высотой 1 м (дальнейшее снижение температуры
угля происходит на конвейере). Высвобождается около 15 м3
объема реторты, который может быть использован для увели-
чения высоты зоны пиролиза на 2,5 м после переноса штуцеров
выхода из реторты газа, охлаждающего уголь, ввода теплоно-
сителя в реторту и соответствующего повышения мощности
вспомогательного оборудования. Реторта с уменьшенной зоной
охлаждения древесного угля запроектирована для Моломского
лесохимического завода.
221
Рис. 59. Изменение темпера-
туры древесного угля по вы-
соте тушильной части промыш-
ленной вертикальной пепрерыв-
нодействующей реторты:
1 — замеры сделаны в зимнее вре-
мя; 2 — в летнее время
Значительные успехи в повышении единичной мощности вер-
тикальных реторт были достигнуты интенсификацией процесса
пиролиза древесины путем повышения температуры. Новый ре-
жим имеет один существенный недостаток — закоксовывание
трубопровода, отводящего из реторты парогазы, из-за чего 2—
3 раза в месяц приходится останавливать реторту для чистки.
Причиной интенсивного закоксовывания трубопровода парога-
зов реторт является высокая тем-
пература парогазов на выходе из
реторты (150—200 °C). Явление
закоксовывания можно объяснить
тем, что древесная смола содержит
значительное количество свободных
радикалов, а также веществ, кото-
рые приводят к многочисленным
химическим превращениям смолы.
В результате реакций конденсации,
полимеризации и рекомбинации,
протекающих с заметной скоростью
при высоких температурах, увели-
чивается молекулярная масса смо-
лы с образованием твердого кокса.
Для снижения степени закоксо-
вывания трубопровода, отводящего
из реторты парогазы, без уменьше-
ния интенсивности пиролиза дре-
весины необходимо охлаждать па-
рогазы в верхней части реторты до
температуры 100—130 °C, напри-
мер, путем охлаждения верха кор-
пуса реторты воздухом или водой,
подачей в верхнюю- часть реторты холодных некондесирую-
щихся газов пиролиза, жижки и др. На Ашинском лесохими-
ческом заводе парогазы в верхней части реторты охлаждаются
жижкой. За счет сокращения простоев оборудования из-за
чистки можно увеличить выработку древесного угля в одной
реторте на 3 %.
Расход тепла при пиролизе древесины. Производство древес-
ного угля требует больших затрат тепловой энергии, которые
зависят от влажности загружаемой в аппарат древесины. Удель-
ный расход условного топлива непосредственно в производстве
угля составляет 300—600 кг/т (9—18 ГДж/т), кроме того, на
переработку жидких продуктов пиролиза расходуется техноло-
гический пар с удельным расходом энергии 8—16 ГДж/т.
Процесс пиролиза древесины (образование угля) не нужда-
ется в подводе тепла извне, так как выделяется тепло экзотер-
мической реакции (5—8 %,) от теплоты сгорания древесины. По
существу все топливо расходуется на сушку древесины и не за-
висит от того, происходит ли сушка в специальной сушилке или
222
в самом аппарате для пиролиза древесины. Приближенно удель-
ный расход топлива можно принять пропорциональным влаж-
ности исходной древесины, при этом расход топлива в периоди-
чески действующем производстве примерно в 2 раза выше, чем
в непрерывнодействующем. Для расчета примем наиболее со-
вершенную схему цеха вертикальных непрерывнодействующих
реторт Ашинского лесохимического завода.
Расход тепла на пиролиз можно определить по формуле
Цп = Get, (77)
где G — поток теплоносителя в расчете на 1 т древесного угля,
т/т; с— теплоемкость теплоносителя, кДж/(кг-К); t — темпера-
тура теплоносителя, °C.
Удельный расход тепла для пиролиза древесины в верти-
кальной реторте выражен приближенно уравнением
(96№fe + 5,3fn + 35K — 820)
<7р = ----------> V6)
4Т 4П
где Wk — влажность загружаемой древесины, %,; tn, tn — темпе-
ратура парогазов и теплоносителя, °C; К — коэффициент тепло-
передачи через стенку реторты, Дж(м2-К).
, Приняв параметры пиролиза для вертикальной непрерывно-
действующей реторты £г = 600 °C, /п=150 °C, К=1 Дж(м2-К);
№й.=25 %„ получим удельный расход тепла при 25 %-ной влаж-
ности древесины равный 3210 МДж/т.
Анализ уравнения (78) показывает, что основной расход
тепловой энергии в процессе пиролиза наблюдается при испаре-
нии оставшейся в древесине влаги. Суммарный расход тепла на
сушку древесины в сушилке и реторте прямо пропорционален
начальной влажности древесины. Коренное отличие сушки дре-
весины в реторте от сушки в сушилке состоит в том, что влага,
испаренная в реторте, не выбрасывается в атмосферу, а посту-
пает на дальнейшую переработку в составе жидких продуктов
пиролиза. Эта влага является вредным балластом, от которого
освобождаются в процессе разделения жидких продуктов за
счет затраты энергии греющего пара в химических цехах пред-
приятий. Увеличение удельного расхода энергии греющего пара
на удаление влаги из жидких продуктов пиролиза в расчете на
1 т древесного угля можно вычислить приближенно по формуле
Д<7ж = 223Гк. (79)
Для пиролиза древесины в вертикальных ретортах этот рас-
ход равен приблизительно 5580 МДж/т.
Тепло, выделяющееся при пиролизе древесины в результате
экзотермической реакции, является для вышеуказанных условий
постоянным и достаточно стабильным источником энергии. Оно
полностью расходуется на разложение древесины и нагрев про-
дуктов пиролиза и для вертикальной реторты составляет 4—
5 %, от теплотворной способности древесины, т. е. около 3,6 ГДж
на 1 т древесного угля. Для процесса пиролиза этой энергии
223
недостаточно, если влажность древесины, поступающей в ре-
торту, более 3—5 %,.
Кроме тепла экзотермической реакции, при пиролизе древе-
сины выделяются газы пиролиза — источник экономии тепло-
вой энергии. При производстве 1 т угля таких газов выделяется
около 540 м3 с теплотой сгорания 7800 МДж/т. Этой химической
энергии обычно с избытком хватает для проведения процесса
пиролиза, однако избыток неконденсирующихся газов на мно-
гих предприятиях выбрасывается в атмосферу. На Ашинском
заводе значительная часть газов пиролиза направляется
в топки сушилок, что позволяет экономить до 30 %, жидкого
топлива, расходуемого на сушку древесины, т. е. около
2610 МДж/т.
Древесный уголь в реторте охлаждается неконденсирующи- '
мися газами с температурой 40—60 °C, которые направляются
в нижнюю часть реторты противотоком к углю. Газы, нагретые
углем до температуры 150—250 °C, выводятся через штуцер
в средней части реторты и направляются в топку для формиро-
вания теплоносителя, образуя второй контур. Древесный уголь,
прокаленный при 550 °C, содержит 660 МДж/т физического
тепла, которое в основном расходуется на нагрев охлаждающих
его газов, поэтому тепло это утилизируется на 70—90%. Ско-
ростная стабилизация древесного угля, которая требует темпе-
ратуры угля при выгрузке на реторты 60—80 °C, практически
не влияет на общую массу утилизируемой энергии. Конденсация
жидких продуктов происходит при прохождении парогазов че-
рез конденсационную систему реторт. Тепло конденсации и. ох-
лаждения парогазов в системе отводится с охлаждающей водой
и в настоящее время не утилизируется.
На всех лесохимических предприятиях, оснащенных верти-
кальными непрерывнодействующими ретортами, имеются значи-
тельные резервы экономии тепла и топлива. Помимо более пол-
ного использования газов пиролиза, это — глубокая сушка дре-
весины до 10—15 %,-ной влажности, изоляция сушилок, реторт и
газоходов, снижение температуры отработанного сушильного
агента до 100 °C и температуры парогазов на выходе из ре-
торты со 150—200 до 100—130 °C.
Показатели работы реторт и печей. Для сравнения в табл. 32
приведены показатели работы реторт и печей различных типов
при пиролизе березовой древесины. Из сравнения показателей
следует, что удельная производительность вертикальной непре-
рывнодействующей реторты по перерабатываемой древесине
в 8—26 раз больше, чем удельная производительность вагонных
реторт и печей различных систем, а по углю на 1 м3 суммар-
ного объема аппарата в 6—17 раз.
Массовая доля нелетучего углерода в угле, полученном в ва-
гонных ретортах и печах, составляет 73—84 %„ в то время как
в угле, полученном в вертикальных непрерывнодействующих ре-
тортах, 90—95 %. В вагонных ретортах и печах для пиролиза
224
00
Заказ № 2193
32. СРАВНИТЕЛЬНЫЕ ПОКАЗАТЕЛИ РАБОТЫ РЕТОРТ И ПЕЧЕЙ
Показатель ( Вертикальная непрерывно- действующая реторта диаметром, м Вагонная реторта Печи системы
2,7 2,8 стальная кирпичная В. Н. Козлова «Свердлес- пром-4» УВП-5
Объем перерабатываемой древеси- ны, м3/сут 179 220 24—38 13—27 . 74—88 30-36 2,5—3,0
Объем аппарата, м3 87 93 247 142 460 125 11
Удельная производительность: угля в сутки на 1 м3 суммарного объема, кг древесины в сутки на 1 м3 сум- марного объема, м3 266 2,06 300 2,37 18,7 0,10—0,15 24,7 0,09—0,19 32,7 0,16-0,19 ' 48 0,24—0,29 28 0,23—0,27
Конечная температура пиролиза, °C 500—600 500—600 450—520 400—450 400—450 400—450 400—450
Массовая доля нелетучего углерода в угле, % 90—95 90—95 80—84 74—78 73-78 75-78 75—85
Расход топливных дров, % от пере- рабатываемых , Не тре( эуется 15—16 15—16 23
Расход металла на 1 м3 суточной пе- реработки, т > > । 0,70 — 2,80 1,00 0,85 0,25 1,64
древесины используют тепло, полученное при сжигании 10—
40 %, топливных дров от переработанного в аппарате техноло-
гического сырья. В настоящее время существует дефицит дре-
весного угля, и для удовлетворения потребности народного хо-
зяйства в нем необходимо производить уголь не только в лесо-
химической, но и в лесозаготовительной промышленности, в ко-
торой могут быть использованы печи различных систем. Глав-
ное же развитие должно получить промышленное производство
древесного угля в лесохимической отрасли.
§ 38. САМОВОЗГОРАНИЕ ДРЕВЕСНОГО УГЛЯ
А. Н. Завьялов с сотрудниками (ЦНИЛХИ) изучил процесс
самовозгорания древесного угля и предложил эффективный спо-
соб его стабилизации, который внедрен в производство на вер-
тикальных непрерывнодействующих ретортах.
Парамагнитные свойства древесного угля. Эти свойства обус-
ловлены присутствием в нем макрорадикалов, срединных R*Cp
и концевых R-K; их максимальное количество возникает при
пиролизе древесины соответственно при температурах 325 и
550 °C. Они характеризуются различными параметрами ЭПР-
спектра, разной реакционной способностью и термостойкостью.
Большой реакционной способностью обладают R*K, которые на
воздухе при обычных условиях легко переходят в R-Cp по ре-
акциям:
R • к + О2 -> RKOO •, (80)
RKOO. +RcpH->RKOOH + R.cp. ' (81)
Эти реакции с большой скоростью протекают даже при низ-
ких температурах. Возникающие перекисные макрорадикалы
(ROO •) фиксируются методом ЭПР. Перекисные группы легко
определяются йодометрически. Концевой макрорадикал R-K об-
ладает высокой термической устойчивостью вплоть до темпера-
туры 1000 °C.
Срединные макрорадикалы R • Ср не термостойки и полностью .
распадаются уже при температурах выше 300 °C на концевые
макрорадикалы и низкомолекулярные продукты — оксиды угле-
рода, водород, углеводороды:
R • ср -> R • к + СО2+СО+ Н2 + СтНя. (82)
Механизм автоокисления и самовозгорания древесного угля.
В зависимости от реакционной способности и температуры 1 кг
древесного угля в течение 1 ч поглощает от 0,001 до 0,1 кг
кислорода с одновременным отщеплением низкомолекулярных
продуктов, главным образом воды. Масса угля при этом прак-
тически не изменяется. Поглощение кислорода происходит в ре-
226
зультате непрерывного цепного повторения реакций концевых
[реакции (80) и (81)] и срединных макрорадикалов:
R • ср + Оа —> RcpOO •,
(83)
RcpOO- +RCpH->RcpOOH + R.Cp. (84)
Вода выделяется по реакции разветвления цепи:
RcpOOH RepH —> RcpO • R • Ср + Н^О.
(85) '
Как видно из приведенных реакций, процесс окисления дре-
весного угля развивается по цепному [реакции (80), (81), (83)
и (84)] разветвленному [реакция (85)] механизму. Для этого
процесса характерны критические параметры количества пара-
магнитных центров (ПМЦ), температуры, геометрических раз-
меров массы угля, концентрации кислорода.
Самовозгорание древесного угля представляет собой лавино-
образное низкотемпературное автоокисление угля, когда один
из параметров процесса (чаще это бывает масса угля или тем-
пература) превысил критическое значение для данных условий.
Теоретические основы стабилизации древесного угля. Стаби-
лизировать уголь — это значит лишить его способности само-
возгораться в условиях перевозки и хранения. Первопричина
самовозгорания угля — макрорадикалы, поэтому решение за-
дачи сводится к снижению количества ПМЦ в угле до уровня,
при котором не происходит развития процесса его окисления
кислородом воздуха до самовозгорания.
Макрорадикалы в твердом веществе гибнут не за счет.диф-
фузии, а по эстафетному механизму путем многократного чере-
дования реакций передачи цепи
R • ср + RepH —> RepH + R * ср» (86)
R-K+RcpH->RKH + Rcp (87)
до тех пор, пока два активных центра не окажутся рядом и не
произойдет их рекомбинация:
R+R'.-^R — R'. (88)
По реакциям (87) и (88) можно за 1—2 ч довести количе-
ство ПМЦ в угле до уровня, при котором он потеряет способ-
ность самовозгораться. Но для этого необходимо нагреть уголь
до температуры 700—800 °C. Следовательно, для действующих
производств древесного угля этот путь его стабилизации непри-
годен.
Активные центры древесного угля могут гибнуть, без доступа
воздуха при температурах 100—300 °C по реакциям (86) и (88).
Продолжительность стабилизации угля при этих условиях со-
ставляет несколько часов. Однако при температуре 20 °C этот
процесс длится не менее 10 сут, поэтому такой путь стабилиза-
ции древесного угля хотя и применяется на некоторых заво-
8* 227
дах, является устаревшим, малоэффективным и требует интен-
сификации.
Наиболее эффективный и в то же время интенсивный спо-
соб — стабилизация древесного угля воздухом в слое менее кри-
тическом (тонком слое). В основу его положено использование
элементарных реакций макрорадикалов (80), (81), (83), (84)\и
(88). Казалось бы, раз по тем же реакциям развивается процесс
автоокисления угля вплоть до самовозгорания, то стабилизация
его воздухом невозможна. Однако это не так. Дело в том, что
автоокисление древесного угля может проходить в двух режим-
ных областях: в условиях, когда один или несколько парамет-
ров процесса превышают критическое значение и когда все па-
раметры ниже критической величины. В первом случае ско-
рость возникновения новых ПМЦ по реакции разветвления цепи
(85) превышает скорость гибели их по реакции (88) и процесс
развивается лавинообразно, вплоть до самовозгорания угля. Во
втором — скорость гибели ПМЦ превышает скорость их обра-
зования, и процесс затухает, причем концентрация ПМЦ в угле
непрерывно снижается, т. е. уголь стабилизируется.
Таким образом, стабилизация древесного угля воздухом —
это окисление его кислородом воздуха в условиях, когда ни
один параметр процесса (концентрация ПМЦ, геометрические
размеры массы угля, температура, концентрация кислорода) не
превышает критического значения.
Общие требования к процессу стабилизации древесного угля
воздухом. Скорость и степень гибели ПМЦ в угле при прочих
равных условиях в значительной мере зависят от температур-
ных условий процесса стабилизации. При температуре —20 °C
нужная степень стабилизации достигается за несколько часов,
при 20 °C — за десятки минут, а при 90 °C за несколько ми-
нут. Однако, чем выше температура, тем ниже критические зна-
чения других параметров: геометрических размеров массы угля
и концентрации кислорода (скорости подачи воздуха). Так, для
массы угля, полученного при конечной температуре пиролиза
500 °C, с геометрической формой'куба критическими объемами
при температурах 25, 50 и 90 °C являются соответственно 120,
45 и 12 л. Наиболее благоприятна для проведения стабилиза-
ции геометрическая форма массы угля в виде пластины, т. е.
слоя, причем чем он тоньше, тем лучше.
Таким образом, при технологическом оформлении процесса
стабилизации древесного угля воздухом необходимо выполнить
следующие требования: обязательным условием стабилизации
угля воздухом в слое является непрерывная выгрузка его из ре-
торты на конвейер; высота слоя угля на конвейере должна быть
минимальной, т. е. приближаться к максимальным размерам
куска угля (50—100 мм); стабилизация угля проводится при
температуре 50—80 °C; расход воздуха для стабилизации дол-
жен быть минимальным, но не менее того, что требуется по хи-
мическим реакциям (достаточно естественной тяги); продолжи-
228
тельность процесса зависит от требуемой степени стабилиза-
ции, для исключения самовозгорания угля в объеме технологи-
ческой вагонетки (10 м3) время пребывания угля на конвейере
7 мин, в объеме железнодорожного вагона (50 м3) 30 мин.
Глава 10
ИСПОЛЬЗОВАНИЕ МАЛОЦЕННОЙ ДРЕВЕСИНЫ
На лесозаготовительных участках лесных предприятий,
а также на деревообрабатывающих предприятиях образуется
значительное (до 50 % от заготовленной древесины) количе-
ство дров и малоценной древесины (низкосортная древесина
от рубок ухода и как остаток на лесосеке, отходы от крон де-
ревьев и от раскряжевки хлыстов, пни, кора, горбыль, рейки,
опилки, фрезерная стружка и т. д.). Для получения технологи-
ческой щепы для нужд целлюлозно-бумажной и микробиологи-
ческой промышленности, а также для производства древесных
плит и пластиков используется лишь половина малоценной дре-
весины. На удовлетворение энергетических потребностей лес-
ных и деревообрабатывающих предприятий, на централизован-
ное обеспечение теплом лесных поселков расходуется только
часть малоценной древесины. Однако на эти нужды из обще-
государственных энергосистем расходуется в большом количе-
стве мазут, природный газ и электроэнергия.
Рациональным и пока единственно возможным путем приме-
нения оставшейся малоценной древесины является энергохими-
ческая переработка. Состав, порода древесного сырья и соот-
ношения компонентов древесины не оказывают влияния на
процесс переработки. Необходимым условием является качест-
венная подготовка щепы-дробленки по гранулометрическому со-
ставу и предварительная сушка до заданной влажности. Основ-
ные первичные продукты энергохимической переработки мало-
ценной древесины: кислая вода или смола и топливный газ;
в процессе сухой перегонки образуется также древесный уголь.
Энергохимическими процессами использования щепы-дроб-
ленки являются: газификация в щепяных газогенераторах, шве-
левание в топках-генераторах ЦКТИ системы В. В. Померан-
цева, пиролиз в вертикальных непрерывнодействующих ретор-
тах и двухстадийный пиролиз (в среде жидкого теплоносителя
и высокотемпературный). В энергетических целях используется
52 % теплотворной способности древесины при сухой перегонке,
42.— при газификации, 64 — при швелевании в топке-генера-
торе, 26 %,— при двухстадийном пиролизе. Генераторный газ
используется или в двигателях внутреннего сгорания, или в га-
зовых турбинах, швель-газ и продукты горения из топок-гене-
раторов— в паровых котлах.
Стационарные и транспортные газогенераторы обращенного
процесса наиболее перспективны, так как вырабатывают газ
229
для двигателей внутреннего сгорания. В этом случае 100 %, теп-
лотворной способности древесины используется в энергетических
целях..При двухстадийном пиролизе древесина (щепа) вначале
подвергается обработке при 275 °C в среде жидкого теплоноси-
теля (дизельное топливо), при этом образуется основная масса
уксусной кислоты и ее гомологов, легколетучих спиртовых про-
дуктов и низкомолекулярных смол, содержащих в основном
продукты углеводного характера. Образующийся газ состоит
в основном из СО2. После такой обработки древесина имеет
нулевую влажность и в значительной степени освобождена от
кислородсодержащих соединений. Во второй стадии процесса
эта древесина подвергается высокотемпературному пиролизу
при 650—700 °C. Здесь образуется высокопрокаленный уголь,
газ теплотворной способностью до 16,8 тыс. кДж/кг и древесная
смола, содержащая до 50 % фенолов. Газ после тонкой очистки
в среде раскаленного до 850 °C древесного угля компримиру-
ется до (18—20) • 102 кПа и используется в виде баллонного
топлива в транспортных двигателях внутреннего сгорания.
§ 39. ГАЗИФИКАЦИЯ ДРЕВЕСИНЫ
Газификация древесины, как и других видов твердого топ-
лива,— это процесс превращения ее в газообразное топливо.
Реакция образования основной горючей части генераторного
газа проходит по уравнению
2С+О2 = 2СО + 246,44 кДж. (89)
Это уравнение выражает происходящий в газогенераторе
процесс неполного окисления раскаленного угля кислородом
воздуха с образованием оксида углерода. При избытке воздуха
происходит полное сгорание топлива:
С+О2=СО2 +408,84 кДж. (90)
В отличие от сжигания газификация производится при огра-
ниченном доступе воздуха в газогенератор, достаточном только
для образования оксида углерода. При газификации, кроме ре-
акции неполного окисления углерода, идет и ряд других реак-
ций. Например, углерод взаимодействует с углекислотой:
С+СО2 = 2СО—162,41 кДж. (91)
Эта реакция обратима, при высоких температурах в слое
раскаленного угля в газогенераторе она идет в сторону образо-
вания оксида углерода. При введении водяного пара в газоге-
нератор реакция его с раскаленным углеродом идет с поглоще-
нием тепла (эндотермически) по уравнениям:
с + Н2О = Н2 + СО— 118,82 кДж; (92)
С+2Н2О = СО2 + 2Н3—75,24 кДж, (93)
. 230
Первая из этих реакций идет при более высокой температуре,
а вторая — при относительно пониженной (ниже 900 °C).
Различают два основных процесса газификации — прямой и
обращенный. Прямой процесс характеризуется тем, что поток
газов в газогенераторе направлен снизу вверх, т. е. воздух вво-
дится через поддувало ниже колосниковой решетки, а продукты
газификации отводятся из верхней части (рис. 60). При обра-
Рис. 60. Схема газификации древесины по прямому процессу:
1 — газогенератор; 2 — загрузка древесины; 3 — отвод парогазовой смеси; 4 — воздуш-
ное дутье; 5 — чаша для золы; / — зона сушки древесины; II — зона швелевания дре-
весины; /// — зона газификации; IV— зона горения
щенном процессе газификации движение газов в генераторе на-
правлено сверху вниз; воздух подводится выше колосниковой
решетки, а продукты газификации отводятся снизу; следова-
тельно, зона горения находится^ выше зоны восстановления,
вследствие чего процесс и носит название обращенного процесса
газификации.
Химический состав древесного генераторного газа зависит от
ряда факторов: породы, качества, влажности и степени измель-
чения древесины, характера дутья, типа и конструкции газоге-
нератора и др. В газе при выходе из газогенератора, прямого
231
процесса содержится следующее количество конденсируемых ве-
ществ, г: воды 450—580 на 1 м3 газа, смолы 45—130, кислот 5—
25 и спиртов 3—8. Выход газа в пересчете на сухой газ (при
нормальных условиях — 0 °C и давлении 101,3 кПа) колеблется
от 1,6 до 1,9 м3 на 1 кг абсолютно сухой древесины и он со-
держит (в объемных дол-ях процента) 21—33 %, СО, 5—11 %
СО2, 1,5—3 % СН4, 0,1—0,8 %, CnHm, 9—15 % Н2, 0,2—0,5 % О2
и 46—54 % N2. Теплота сгорания газа Q=5400—6900 кДж/м3.
Лесохимические продукты могут быть получены только при
прямом процессе газификации древесины; их улавливание из ге-
нераторного газа совмещается с процессом его очистки. Наи-
больший выход продуктов наблюдается при переработке древе-
сины, измельченной в щепу.
Газ, полученный при обращенном процессе газификации, со-
держит незначительное количество продуктов швелевания и не
требует сложной очистки. В основном обработка газа сводится
к его охлаждению и освобождению от механических примесей.
Газификация древесины проводится с целью получения го-
рючего газа, используемого в энергетических целях, например
для сжигания в двигателях внутреннего сгорания или в топках
паровых котлов для выработки электроэнергии с одновремен-
ным получением при очистке газа лесохимических продуктов.
Очистка газа от пыли обычно достигается осаждением ее
в пылевых камерах и циклонах или промывкой газа водой. Очи-
стка газа от смолы осуществляется преимущественно в центро-
бежных смолоотделителях или электрофильтрах. При электри-
ческой очистке из газа извлекается 97—98 % смолы.
Уксусная кислота улавливается из обессмоленного газа раз-
личными способами, но наиболее применим способ, основанный
на поглощении уксусной кислоты раствором уксусно-кислого
кальция. ।
Получен опыт работы газогенераторных установок на ниж-
них складах лесозаготовительных предприятий. Сырьем для га-
зификаций служит идущая в отходы и малоценная, не исполь-
зуемая в основном производстве древесина. Подготовка сырья
заключается в измельчении ее в щепу-дробленку и сушке до
20—25 %,-ной влажности. Парогазовую смесь из генератора от-
бирают при температуре 80—90 °C с последующим охлажде-
нием до 60 °C. Образующийся конденсат представляет собой
водную смесь отстойной и растворимой смолы и части летучих
кислот. В зависимости от температуры охлаждения парогазов
содержание воды в конденсате колеблется от 35 %, до 65 %.
Газ, освобожденный от смолы, направляется для сжигания
в топки паровых котлов без дополнительной очистки и осушки.
Если же газ подается в двигатели внутреннего сгорания, то он
подвергается дополнительной очистке и осушке в фильтрах и
адсорберах.
Полученный конденсат называют кислой смолой (при влаж-
ности не более 40 %) или кислой водой. Переработка конден-
232
сата осуществляется на лесохимических заводах. Мощность ти-
повых газогенераторных установок 25 тыс. и 50 тыс. м3 древе-
сины в год. Выработка из 1 м3 плотной древесины: жижки
194 кг, смолы (безводной)—86 кг, газа-,330 нм3, электроэнер-
гии — 66 кВт • ч.
§ 40. ЭНЕРГОХИМИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ ДРЕВЕСИНЫ
Твердое органическое топливо: каменный уголь, торф, древе-
сина, сланец является не только потенциальным источником
тепловой энергии, но и сырьем для получения ценных химиче-
ских продуктов. При сжигании в топках паровых котлов ука-
занные виды топлива могут использоваться комплексно, энерго-
химически: топливо вначале подвергается швелеванию в токе
инертного газового теплоносителя с отбором летучих продуктов
швелевания, затем углеродистый остаток, а также швельгаз
сжигаются в топке парового котла. Процесс осуществляется
в едином потоке. Отбираемые из швелыпахты летучие продукты
термического разложения конденсируются в очистной системе
и подвергаются дальнейшей переработке с получением химиче-
ских товаров, а очищенный швельгаз идет в топку парового
котла как газообразное топливо. Древесина как топливо обла-
дает большим содержанием летучих, поэтому нуждается в пред-
варительной подготовке перед сжиганием в топке, парового
котла.
Разработанная в Центральном котлотурбинном институте
В. В. Померанцевым топка для комплексного энергохимического
использования древесины имеет ряд особенностей. Как изве-
стно, предел форсирования дутья определяется не скоростью хи-
мической реакции горения, а механической устойчивостью слоя.
С целью форсирования процесса сжигания древесины, в топке
парового котла В. В. Померанцев предложил проводить пред-
варительную подготовку (швелевание) древесины, а для устой-
чивости слоя разместить его между двумя вертикально распо-
ложенными решетками: колосниковой и зажимающей. Таким
образом, сама конструкция топки и условия форсирования ее
вызывают необходимость предварительного освобождения от
летучих соединений топлива, направляемого в активную зону,
что создает внутреннюю органическую связь между процессами
швелевания и горения топлива.
Топка-генератор энергохимического котлоагрегата состоит из
трех основных конструктивных элементов: сушилки (над кото-
рой расположен топливный бункер), зон разложения (швель-
шахты) и активного горения (рис. 61). В двухкаскадной су-
шилке древесина высыхает до влажности 8—10%, столб топ-
лива образует затвор, препятствующий перетеканию продуктов
швелевания в сушилку и, наоборот, сушильного агента в швель-
шахту. В качестве топлива используется древесина, измельчен-
ная в щепу. Высушенное топливо, поступая в зону термического
233
разложения, рассыпается в ней по наклонной стенке. В эту зону
снизу из зоны горения коксового остатка поступает топочный
газ при температуре 900—1000 °C в количестве 0,35—0,38 м3
на 1 кг абсолютно сухой древесины. Топливо, нагретое топоч-
ным газом, подвергается те
Рис. 61. Топка-генератор для энер-
гохимического использования древе-
сины (расположена под паровым кот-
лом):
1 — воздушное дутье; 2 — разделительный
колодец; 3 — сушилка; 4 — колосники;
5 — канал для подачи горячего газа в су-
шилку; 6 — бункер сырья; 7 — вытяжная
труба из сушилки; 8 — крепление; 9 —
канал для отбора парогазовой смеси из
швельшахты; 10 — зажимающая решетка;
Н — швельшахта; 12 — колосники топки:
13 зона горения кокса; . 14 — паровой
котел
мическому разложению с выделе-
нием лесохимических продук-
тов. Образующаяся парогазо-
вая смесь с температурой 90—
140 °C отбирается через от-
борные каналы швельшахты,
очищается от механических
примесей в пылеуловителях и
поступает в газоочиститель-
ную систему.
Интенсивность процесса
разложения подсушенной ще-
пы очень высока, и этот про-
цесс занимает лишь доли ми-
нуты, причем степень разло-
жения древесины составляет
75—85 % • Коксовый остаток
сгорает в нижней части топки-
генератора, для чего в эту
зону вентилятором подается
воздух с избыточным давле-
нием в зависимости от на-
грузки котла 0,2—0,8 кПа. Ос-
новная масса топочных газов
направляется через экранную
(зажимающую) решетку на
обогрев парового котла, а
часть газов — в зону разложе-
ния топлива. Величину этой
части можно регулировать
с помощью экранной решетки.
Для улавливания лесохи-
мических продуктов парога-
зовая смесь после пылеулови-
телей поступает через гидрав-
лический клапан в поверх-
ностный холодильник, где
охлаждается до 70—80 °C,
а затем — в центробежный
смолоотделитель, в качестве
которого используется венти-
лятор высокого давления, вы-
полненный из кислотостойкой
стали. В нем улавливается
80—85 % всей смолы. Обес-
234
смоленная парогазовая смесь поступает через гидравлический
затвор в топку котла и сжигается в щелевых газовых горелках.
Смолистые конденсаты направляются на^переработку вместе
с кислой водой из гидрозатворов газового тракта. Выход с 1 т
абсолютно сухого топлива: пара 5,6 т, суммарного конденсата
180 кг, в том числе смолы 89 кг, кислот (в пересчете на уксус-
ную) 13,5 кг.
Глава 11
КОНДЕНСАЦИЯ ПАРОГАЗОВ
Парогазы, выходящие из реторты или газогенератора, со-
держат целый ряд компонентов, значительно отличающихся по
своим физическим и химическим свойствам. Принято классифи-
цировать их на жидкие и газообразные продукты пиролиза дре-
весины.’Жидкие продукты пиролиза — это гамма тех компонен-
тов, которые могут быть сконденсированы при атмосферном
давлении в случае использования охлаждающего агента с тем-
пературой не менее 5 °C.
Остальные компоненты, которые не могут быть сконденсиро-
ваны при данных условиях, именуются газообразными продук-
тами пиролиза. После отделения от жидких продуктов пиролиза
газообразные продукты используются частично для охлаждения
угля, частично для разбавления теплоносителя, подаваемого
в топки реторты и сушилок.
Жидкие продукты пиролиза древесины после конденсации и
отстаивания разделяются на три слоя (фазы): легкие всплыв-
ные масла, жижку и отстойную смолу. В состав жижки может
входить приблизительно до 14 %( растворимой смолы, но в за-
висимости от способа конденсации парогазов растворимая
смола может быть выделена и как самостоятельная фракция.
Выбор технологии конденсации парогазов должен всегда оп-
ределяться конъюнктурой смоляных продуктов и экономикой
производства. На лесохимических предприятиях страны эксплу-
атируются две технологические схемы конденсации жидких про-
дуктов пиролиза древесины: скрубберная и система конденса-
ции с использованием поверхностных теплообменников. Ни одна
из них не может быть названа оптимальной, хотя обе имеют ряд
достоинств.
Принципиальная технологическая схема конденсации паро-
газов в скрубберной системе (рис. 62)—одна из первых, внед-
ренных на отечественных заводах. Парогазы, выходящие из
реторты, как правило, содержат угольную пыль, часто непро-
газифицированные остатки древесины (опилки, кору). Поэтому
перед конденсацией парогазы пропускают через так называемый
форконденсатор 2, где наряду с частичной конденсацией смолы
отделяются все твердые включения. Смола, отводимая из гидро-
затворов форконденсаторов в сборник / — самый малоценный
235
продукт: она или выводится в отвал, или сжигается в топках
котельных. После форконденсаторов парогазы с температурой
100—130 °C поступают в первый, а затем во второй скрубберы,
орошаемые жижкой. Ранее первый скруббер орошался отстой-
ной смолой, второй — жижкой. Исследования ЦНИЛХИ пока-
зали, что при орошении первого скруббера смолой ухудшается
качество отстойной смолы, снижается выход ингибиторной фрак-
Рис. 62. Принципиальная технологическая схема конденсации парогазов
в скрубберной системе:
1, // — сборники; 2 — форконденсатор; 3, 4 — скрубберы; 5 —газодувка; 6 — каплеуло-
витель; 7 — циклон; 8 — холодильник; 9, 10, 12 — насосы
ции. Попытки снизить температуру орошаемой смолы приводили
к увеличению вязкости смолы и-забиванию запорной арматуры.
В результате выполненных исследований было предложено оро-
шать оба скруббера жижкой при температуре 35—40 °C, чтобы
улучшить конденсацию смолы и уменьшить время пребывания
смолы в горячей зоне.
Орошение скрубберов жижкой ведут по следующему циклу.
Жижка (с примесями смолы) из нижней части скрубберов са-
мотеком поступает в семикорпусный (т. е. многосекционный)
холодильник 8, где охлаждается до температуры 35—40 °C,
а затем насосом 9 подается на орошение скрубберов 5, 4. Из-
быток сконденсировавшихся жидких продуктов из нижней ча-
сти обоих скрубберов (на 40—60 см выше уровня отбора цир-
236
куляционной части конденсата) выводится в сборник 11, где
отстаивается и разделяется на три фракции (слоя). Легкие
всплывные (или дегтярные) масла (плотность 894—960 кг/м3)
собираются над жижкой и выводятся из верхней части сбор-
ника 11. Тяжелые (отстойные) смолы (плотность 1020—
1260 кг/м3) выводят из нижней части сборника 11, а жижка от-
бирается на 1—1,5 м выше места отбора отстойных смол.
Парогазы, точнее несконденсированные газы, после второго
скруббера 4 с помощью газодувки 5 отводятся в каплеулови-
тель 6, а затем для окончательной осушки — в циклон 7 и да-
лее используются, как отмечено выше. Жидкая фаза из капле-
уловителя 6 и циклона 7 поступает в сборник конденсата И.
Основное технологическое оборудование данной стадии —
скрубберы. Обычно используются насадочные скрубберы, они
удобны в эксплуатации, обеспечивают хороший контакт фаз.,
Основным критерием для выбора их конструкции и расчета яв-
ляются вопросы массообмена.
Режим работы скрубберов: температура парогазов на входе
в первый скруббер 100—130 °C, на выходе из него 60—66 °C,
на выходе из второго скруббера 35—45 °C; температура жижки,
подаваемой на орошение, 35—40 °C, на выходе из первого скруб-
бера 64—70 °C, на выходе из второго скруббера 45—55 °C.
Плотность орошения жижки на 1 м3 отбираемой жижки: в пер-
вом скруббере 3—5 м3, во втором 3—4 м3.
Холодильник 8 должен обеспечивать эффективное охлажде-
ние продуктов конденсации. Процесс этот очень ответствен,
эксплуатационные условия трудны, требуются частые чистки
поверхностей теплообмена. Поэтому требуется, чтобы сама
конструкция аппарата обеспечивала повышенную надежность
оборудования и удобства эксплуатации. Именно эти требования
должны являться критериальными при выборе оборудования
данного узла. Каждая из фракций, отбираемых из сборника 11,
имеет свое целевое назначение и перерабатывается по опреде-
ленной технологической схеме.
Легкие всплывные масла могут быть использованы для про-
изводства дегтя или масел марки ПЯ (противоящурные). От-
стойные смолы в натуральном виде находят ограниченное при-
менение. Смолу, как правило, подвергают переработке с целью
получения на ее основе таких ценных продуктов, как ингибитор,
креозотовое масло, древесно-смоляной пек и др. Сырая смола
используется лишь для пропитки железнодорожных шпал и ка-
натов.
Жижка, полученная по данной схеме конденсации парогазов,
содержит, кроме растворимой смолы, много других органиче-
ских компонентов, относящихся к самым разным классам хими-
ческих соединений: кислоты, спирты, кетоны, альдегиды и др.
Концентрация этих компонентов в жижке, кроме карбоновых
кислот и метанола, так незначительна, что при современных до-
стижениях большой химии выделение их из жижки нецелесо-
237
образно. На лесохимических пиролизных заводах жижка пере-
рабатывается исключительно для получения из нее уксусной
кислоты, и лишь на двух заводах организовано производство
и метанола и древесно-спиртовых растворителей марок А и Э.
Долгое время (особенно в 30-е годы) лесохимическая промыш-
ленность являлась ведущей отраслью по производству мета-
нола. Сегодня метанол получают в основном методом синтеза,
а доля метанола, вырабатываемого лесохимической промышлен-
ностью, составляет менее 1 %.
Скрубберная система конденсации парогазов имеет целый
ряд недостатков. Прежде всего это большая энергоемкость про-
цесса дальнейшей переработки жижки с целью получения тех-
нической уксусной кислоты. Причем большая энергоемкость
сохраняется при любой схеме переработки жижки, так как оп-
ределена ее качеством — содержанием в жижке растворимых
смолистых компонентов. Другой недостаток данной технологии
конденсации парогазов — неоднократная термическая обработка
.смоляных продуктов, ухудшающая их ингибирующую способ-
ность. К тому же при ее разгонке увеличивается выход менее
ценного продукта — пека. Однако при орошении скрубберов
жижкой выход масел из отстойной смолы несколько больше по
сравнению с технологией конденсации парогазов в поверхност-
ных теплообменниках. К недостаткам скрубберной системы кон-
денсации относится и большая металлоемкость процесса отстаи-
вания жижки (требуется эксплуатация емкостей вместимостью до
400 м3 для полного отделения от жижки масел и тяжелых смол).
Схема конденсации парогазов с использованием поверхност-
ных теплообменников лишена некоторых из этих недостатков
(рис. 63). Парогазовая смесь из реторты непрерывно выводится
в форконденсаторы 1, 2, где происходит очистка парогазов от
механических примесей (мелкой щепы, коры и т. д.). Механи-
ческие примеси вместе с высококипящей частью смолы осажда-
ются в смоляных затворах 25, 26 и периодически через 10—
12 ч удаляются как отходы производства. На участке от ре-
торты до первого конденсатора 3 происходит забивание трубо-
проводов твердыми (коксообразными) продуктами, являющи-
мися результатом химического превращения древесной смолы
с увеличением молекулярной массы. Этот процесс идет очень
интенсивно при температуре выше 130 °C. Для снижения сте-
пени закоксовывания трубопроводов рекомендуется смесь про-
дуктов термического разложения древесины и теплоносителя
охлаждать при выходе из реторты до температуры 100—130 °C.
Эта рекомендация является общей для любой схемы конденса-
ции парогазов. Пары из форконденсаторов поступают в поверх-
ностные конденсаторы 3—7. Целесообразно вначале весь объем
парогазов пропустить через один конденсатор 3, а затем раз-
делить на два потока. Рекомендуется использовать двухходовые
конденсаторы. 3—5. Сконденсировавшиеся жидкие продукты пи-
ролиза принимают в сборники 16, 24, Парогазы из конденса-
238
торов 4, 5 далее поступают в поверхностные одноходовые кон-
денсаторы 6, 7, где конденсируются более низкокипящие про-
дукты, чем в конденсаторах 3—5.
В поверхностных теплообменниках конденсируется до 98 %,
всех жидких продуктов пиролиза древесины. Более глубокая
очистка парогазов, проводить которую целесообразно при ис-
пользовании несконденсировавшихся газов в контуре охлажде-
жижка 6 химический цех
Рис. 63. Принципиальная технологическая схема конденсации парогазов с ис-
пользованием поверхностных теплообменников:
1,2 — форкондеисаторы; 3—7 — конденсаторы-холодильники; 8, 9, 12, 15 — каплеулови-
тели; 10 — пенный аппарат; 11, 14, 16, 19, 21, 23, 24 —сборники; 13 — газодувка; 17,
18, 20, 22 — насосы; 25, 26 — смоляные затворы
ния угля (что и показано на рис. 63), осуществляется в капле-
уловителях 12, 15 (работающих по типу скруббера), в пенном
аппарате 10 (с использованием для промывки технической воды
2—3 кг на 100 м3 газа) и в каплеуловителях 8, 9. Конденсат
от этих аппаратов собирается в сборнике 14 и используется для
орошения всех пяти поверхностных теплообменников, увеличи-
вая тем самым степень конденсации жидких продуктов в каж-
дом из них. В какой-то степени это мероприятие обеспечивает
более мягкие условия процесса конденсации на поверхности
теплообмена и тем самым увеличивает цикл работы от чистки
до чистки.
При необходимости на орошение может подаваться и жижка
из сборников 16, 24. Орошение жижкой бывает особенно эф-
фективно в летнее время, когда температура охлаждающей
воды более 30 °C. В сборниках конденсата 16, 24 происходит
239
разделение жидких продуктов на легкие масла, жижку и от-
стойную смолу. Целевое назначение каждой из этих фракций
такое же, как и при скрубберной системе конденсации.
Основным технологическим оборудованием этой схемы кон-
денсации являются поверхностные теплообменники. При выборе
их конструкции и расчетах теплообмена необходимо учитывать
и эксплуатационные требования.
Основной недостаток данной технологии—большая энерго-
емкость процесса переработки жижки. Процесс отстаивания
конденсата идет значительно быстрее, а смоляные продукты
претерпевают только одноразовую термическую обработку (при
их конденсации).
Смолистые вещества, получаемые при конденсации парога-
зов в скрубберной системе или с применением поверхностных
конденсаторов, претерпевая термические воздействия, контак-
тируют с кислородом воздуха, что ухудшает , их качество (при
разгонке уменьшается выход масел и снижается ингибирующая
способность).
ЦНИЛХИ совместно с МИХМом разработали принципиально
новую технологию выделения смолистых веществ из парогазов,
основанную на применении аппаратов циклонного типа и ад-
сорберов тарельчатого или насадочного типа. Схема выделе-
ния смолистых веществ в аппаратах циклонного типа позво-
ляет не только повысить качество смоляных продуктов, но и
значительно снизить энергоемкость переработки жижки, так
как по данной технологии массовая доля растворимых смол
в жижке столь незначительна, что не требуется обессмолива-
ние ее перед экстракцией. Схема конденсации парогазов в дан-
ном случае обеспечивает получение жижки с массовой долей
смолистых веществ до 1 %.
Принципиальная технологическая схема разделения компо-
нентов парогазов в циклонах и скрубберах показана на рис. 64.
Парогазы непрерывно выводятся из верхней части реторты при
температуре 100—130 °C, поступают в форконденсаторы для
отделения угольной пыли, коры, высококипящей части смолы
(на рис. этот узел не показан), а затем в первый циклон /, где
происходит отделение наиболее крупных капель отстойной
смолы (диаметром 20 мкм и более). Эта часть смолы — наиме-
нее ценная — принимается в отдельный сборник 15. Из первого
циклона парогазы поступают во второй циклон 2, в котором
почти полностью выделяется отстойная смола. Эта наиболее
ценная часть смолы собирается в сборнике 14. В двух первых
циклонах улавливается отстойная смола, находящаяся в паро-
газах в виде капель. Растворимая часть смолы присутствует
в виде тумана и паров, уловить ее в аппаратах циклонного типа
невозможно. Для ее выделения необходимо использовать ад-
сорбционный метод с орошением скрубберов непосредственно
получаемой фракцией смолы. Для снижения степени конденса-
ции воды и уксусной кислоты в адсорбер 3 на орошение пода-
240
ется растворимая смола из сборника 13, подогретая до тем-
пературы . 80—85 °C. В адсорбере одновременно осуществля-
ются два процесса: выделение основной массы растворимой
смолы и укрупнение ее частиц. Полное выделение растворимой
смолы достигается в циклоне 4. Из циклона 4 обессмоленные
парогазы отсасываются в поверхностные конденсаторы-холо-
дильники 5, 6 и 7, в которых происходит конденсация обессмо-
Рис. 64. Принципиальная технологическая схема разделения компонентов па-
рогазов с использованием аппаратов циклонного типа и скрубберов:
1, 2, 4 — циклоны; 3 — адсорбер; 5, 6, 7 — конденсаторы-холодильники; 8, 11, 13, 14,
15 — сборники; 9, 10, 12 — насосы
ленной жижки, выкачиваемой насосом 9 в химический цех для
дальнейшей переработки.
Эта схема разделения жидких продуктов пиролиза более
сложна, так как в ней используются процессы не только кон-
денсации, но и сепарации. Основными критериями расчета и
выбора оборудования являются дисперсный состав смолистых
веществ, скорость парогазов и температура конденсации инди-
видуальных компонентов. Скорость парогазов в первом циклоне
рекомендуется поддерживать 5—7 м/с, во втором 22—25, в чет-
вертом— 35—40 м/с. Схема обеспечивает улавливание (отде-
ление) смолы на 93—95 °/0|.
В табл. 33 приведены основные характеристики жижки, по-
лученной по разным схемам конденсации парогазов, кислой
воды после обессмоливания жижки в выпарных аппаратах, и
смеси кислой воды и жижки, подаваемой на экстракцию. Кис-
лотность жижки не зависит от способа конденсации. Этот по-
казатель определяется породой переугливаемой древесины и,
главным образом, ее влажностью после сушилок. Существенно
241
33. ОСНОВНЫЕ ХАРАКТЕРИСТИКИ ЖИЖКИ, КИСЛОЙ ВОДЫ И СМЕСИ КИСЛОЙ
\ ВОДЫ И ЖИЖКИ
Показатель* Жижка, полученная по схеме - Кислая вода после выпарного аппарата Смесь жижки и кислой воды, пода- ваемая на экстракцию
конденсации в скруббер- ной системе или с исполь- зованием поверхност- ных тепло- обменников разделения с использо- ванием циклонов» скрубберов и поверхно- стных тепло- обменников
Плотность при 20 °C, кг/м3 1010—1022 1010—1012 998—1002 1006—1010
Кислотность, % (в пере- счете на уксусную кис- 7,4—8,5 8,8—8,5 7,5—8,4 7,4—8,8
лоту) Массовая доля раствори- мой смолы, % 10,2—12,0 1,2—1,8 0,2—0,4 0,7—1,0
* При пиролизе древесины
влажностью 20—25 %.
характеристика жижки, полученной по различным технологиче-
ским схемам конденсации парогазов, отличается только по мас-
совой доле в ней растворимых смол.
, Если качество жижки практически не зависит от схемы кон-
денсации парогазов, то характеристика смолы прежде всего
зависит именно от способа конденсации и разделения жидких
продуктов пиролиза древесины (табл. 34). При скрубберной
схеме конденсации или конденсации парогазов в поверхностных
теплообменниках на стадии конденсации выделяется только от-
стойная смола. Растворимая смола присутствует в жижке и вы-
деляется на дальнейших стадиях ее переработки. С точки зре-
ния качества смолы предпочтительнее скрубберная система, но
при условии орошения скрубберов жижкой.
При использовании циклонов в схеме разделения компонен-
тов парогазовой смеси выделяются все смолы: отстойные и ра-
створимые, причем в . первом — преимущественно тяжелокипя-
щая часть отстойной смолы (пек), во втором — смесь отстойной
и растворимой смол, а в третьем циклоне выделяется только
растворимая смола. Это положение является особенностью
данной технологии конденсации парогазов. Разработанная тех-
нологическая схема конденсации парогазов с использованием
циклонов для отделения смолистых веществ еще не прошла
промышленных испытаний. Однако уже сейчас можно отметить
ее достоинства (компактность, малую металлоемкость, низкую
энергоемкость дальнейшей переработки жижки) и недостатки
(чувствительность к колебаниям нагрузки).
К материалам, рекомендуемым для изготовления оборудо-
вания для конденсации парогазов, предъявляются особые
242
243
34. КАЧЕСТВО СМОЛЫ В ЗАВИСИМОСТИ ОТ СПОСОБА КОНДЕНСАЦИИ И
РАЗДЕЛЕНИЯ ЖИДКИХ ПРОДУКТОВ ПИРОЛИЗА ДРЕВЁСИНЫ
Показатель Схема конденсации Отделение смол
в скрубберной системе с оро- шением жижкой в поверхност- ных теплооб- менниках в I циклоне во II циклоне в ш циклоне
Плотность, кг/м3 1068—1136 1069—1115 1150—1270 1080-1150 1060—1080
Влажность, % 14—26 10-13 5—10 5-9 20-25
Кислотность, % Фракционный состав, %: 7—10 5—6,5 9—10 8—12 8—10
масел до 240 °C 15—23 14—20 20—30 12—20 30—40
ингибиторной фракции (240—310 °C) 35—40 40—47 — 35—48 20—45
Всего масел, % 50—56 56—59 20—30 55—60 60—80
Пека, % Температура, °C: 44—50 41—44 70—80 40-45 20—40
размягчения пека 77—81 60—76 66—67 — —
конца разгонки 310—340 290—320 330—360 245—312 230—280
Ингибирующий эффект 0,9—1,1 0,8-1,0 — — —
Доля выделенных смолистых веществ, % 40—60 (отстойная) 40—60 (отстойная) 20-25 (отстойная) 50—55 4 (отстойная + растворимая) 15—20 (растворимая)
требования: стойкость к газовой коррозии, химическая стой-
кость ' к различным органическим компонентам, особенно
к одноосновным карбоновым кислотам, смолам, спиртам, аль-
дегидам. В качестве конструкционных материалов рекоменду-
ются нержавеющие стали марок 08Х21Н6Т, 12Х18Н10Т,
08Х17Т, 15Х25Т и биметаллы (например, Ст. 3+12Х18Н10Т
или Ст. 3+10Х17Н13М2Т).
Глава 12
ПРОИЗВОДСТВО УКСУСНОЙ КИСЛОТЫ
Во многих странах целевым продуктом пиролиза древесины
является исключительно уголь. В СССР в связи с ежегодным
увеличением выпуска угля и концентрированием его производ-
ства на крупных предприятиях остается весьма экономичным
выделение из жидких продуктов пиролиза древесных смол и
особенно уксусной кислоты. Эти производства способствуют
снижению себестоимости угля и увеличивают ресурсы ценных
химических продуктов. Лесохимическая уксусная кислота се-
годня — основной источник производства пищевой уксусной
кислоты. Получают ее в двух производствах: при переработке
уксусно-кальциевого порошка и при переработке жидких про-
дуктов пиролиза древесины, а точнее — жижки.
Жидкие продукты пиролиза или газификации древесины со-
держат 72—84 % воды, 8—16 % низкомолекулярных карбоно-
вых кислот Ci—С4 (из них 85 % приходится на долю уксусной
кислоты), 6—12 % водорастворимых смолистых веществ и до
6 % прочих легкокипящих компонентов (спирты, эфиры, ке-
тоны, альдегиды и др.).
Предложены самые разнообразные способы переработки
жижки с целью получения из нее уксусной кислоты. Промыш-
ленное применение нашли порошковый, экстракционный и рек-
тификационный (азеотропная ректификация) ч способы.
Длительное время основным способом извлечения из жи-
жки уксусной кислоты являлся порошковый. Конечный про-
дукт этого производства — уксусно-кальциевый порошок — да-
лее перерабатывается в уксусную кислоту путем разложения
его серной кислотой. Переработка уксусно-кальциевого поро-
шка осуществляется централизованно. Однако из-за сложности
процесса, низких выходов продукта, большой трудоемкости,
высокой себестоимости и неблагоприятных условий труда этот
метод постепенно теряет значение. Производство уксусно-каль-
циевого порошка не может являться крупным сырьевым источ-
ником уксусной кислоты, но, очевидно, останется как способ
утилизации кислот из сточных и газовых выбросов. В отдель-
ных случаях можно его рекомендовать и как способ перера-
ботки жижки на мелких установках газификации отходов дре-
весины и гидролизно-фурфурольных предприятиях.
244
Сегодня, основной метод извлечения уксусной кислоты из
жижки — экстракционный, реже используется метод ректифика-
ции. Применение того или другого способа зависит от концен-
трации уксусной кислоты в исходном сырье. Технико-экономи-
ческие исследования показывают, что способ азеотропной
ректификации целесообразно применять для извлечения кис-
лоты из водных растворов, когда массовая доля кислот в них
не менее 16%. Метод простой ректификации экономически
оправдывает себя только при переработке растворов с массовой
долей кислоты более 60%. Массовая доля уксусной кислоты
в жижке, получаемой почти на всех лесохимических предприя-
тиях пиролиза древесины, не превышает 12 %. Для ее извлече-
ния в этом случае наиболее экономичен экстракционный спо-
соб.
При переработке водных растворов кислоты крепостью
12—16 % нельзя однозначно рекомендовать один из последних
двух способов концентрирования кислоты. Большое значение
в этом случае имеют отдельные технологические решения: вы-
бор способа подготовки жижки к экстракции, выбор экстра-
гента, аппаратурное оформление процесса, мощности произ-
водства и в очень большой мере — способ очистки сточных вод,
получающихся после извлечения уксусной кислоты из исход-
ного раствора.
§ 41. ЭКСТРАКЦИОННЫЕ СПОСОБЫ ИЗВЛЕЧЕНИЯ
уксусной кислоты ИЗ ЖИЖКИ
Метод экстракции уксусной кислоты из водных растворов
основан на различной растворимости уксусной кислоты и воды
в экстрагенте. Для извлечения уксусной кислоты из водных
растворов предложены разнообразные растворители, относя-
щиеся к самым различным классам соединений. В лесохими-
ческой промышленности для экстракции кислоты долгое время
использовали диэтиловый эфир, обладающий хорошими экс-
трагирующими свойствами, но малой селективностью. Из про-
стых эфиров исследовали возможность применения для экс-
тракции диизопропилового и пропилового эфиров. Из сложных
эфиров в разное время применяли метилацетат, бутилацетат,
изобутил ацетат, изопропилацетат, этилбутират и др.
Многочисленные исследования посвящены изучению экс-
трагирующих свойств этилацетата. Р. Иглисфилд (США), изу-
чив свойства более сорока растворителей, отметил этилацетат,
как один из лучших экстрагентов для извлечения уксусной
кислоты из растворов с массовой долей ее менее 16 %.
А. Н. Брик и В. П. Сумароков (ЦНИЛХИ, г. Москва) изучили
в сравнении свойства диэтилового эфира и этилацетата и уста-
новили преимущества последнего. В результате этих исследо-
ваний на лесохимических предприятиях вместо диэтилового
эфира стали использовать этилацетат, что позволило значи-
245
тёльно улучшить тёхнико-экономичёские покйзатёли производ-
ства технической уксусной кислоты и снизить его пожароопас-
ность.
Весьма перспективно использование в качестве экстраген-
тов метилэтилкетона, диэтилкетона, метилпропилкетона, метил-
изопропилкетона. Эти растворители бесспорно имеют преиму-
щества перед этилацетатом. Однако небольшие объемы их про-
изводства сдерживают использование их в промышленном
масштабе. Хорошие результаты получены при извлечении
уксусной кислоты некоторыми аминами. Но их токсичность не
позволяет применять их в промышленности.
Выбор экстрагента для внедрения его в производство обу-
словлен совокупностью таких его свойств, как емкость, селек-
тивность по отношению к извлекаемому компоненту, коэффици-
ент распределения, плотность, межфазное натяжение, эконо-
мичность процессов его регенерации, химическая стабильность,
стоимость, промышленная доступность, токсичность, воспламе-
няемость, коррозионная активность и др. Оптимальное выпол-
нение всех требований затруднительно. Поэтому, как правило,
при выборе однокомпонентного экстрагента принимается ком-
промиссный вариант. В последнее время ведется поиск бинар-
ных или многокомпонентных растворителей, что позволяет варь-
ировать конкурирующие свойства их составляющих.
Принято считать, что лучшим , из однокомпонентных экстра-
гентов для извлечения уксусной кислоты из водных растворов
является этилацетат, хотя он и обладает рядом существенных
недостатков (легко гидролизуется, переэтерифицируется и др.).
С целью улучшения экстрагирующих свойств этилацетата пред-
лагались различные добавки: метанол, метилэтилкетон, диэтил-
кетон, ацетон, метилацетат й другие компоненты, в присутствии
которых значительно увеличивается коэффициент распределе-
ния уксусной кислоты между органической и водной фазами.
Следует ожидать, что дальнейшие поиски экстрагентов, в том
числе и для извлечения уксусной кислоты из водных раство-
ров, будут направлены именно на исследование бинарных и
более сложных растворителей.
Экстрагирующие свойства растворителя оценивают в ос-
новном по трем показателям: коэффициенту распределения из-
влекаемого компонента между фазами в условиях равнове-
сия Лр, коэффициенту экстракции G и масштабности гетероген-
ной области.
Для системы вода — уксусная кислота растворитель Лр
принято выражать как отношение концентрации уксусной ки-
слоты в фазе экстракта (т. е. органическом — верхнем слое)
к концентрации кислоты в фазе рафината (т. е. водном слое):
КР = С1/С2, (94)
где Сь С2 — массовые доли кислоты в экстракте и рафи-
нате, %.
246
Коэффициент экстракции G учитывает не только концентра-
ционное распределение кислоты между фазами, но и объемы
(или массы) фаз:
G = C^rn-J (С&п2) = Кр/И1/т2, (95)
где /пь т2 — массы экстракта и рафината.
Коэффициент распределения кислоты Кр безусловно явля-
ется важной характеристикой растворителя, но далеко не пол-
ной. Этот показатель определяет в основном число ступеней
экстракции, необходимых для достижения заданной степени
извлечения при определенном соотношении фаз. Следовательно,
этот показатель определяет габариты экстрактора, его высоту.
Чем выше Кр, тем меньше требуется ступеней экстракции, тем
меньше высота экстрактора и наоборот.
Коэффициент экстракции G дает нам представление о емко-
сти экстрагента. Сравнивая величины G для различных ра-
створителей, полученных в одинаковых условиях, можно сде-
лать вывод, что чем выше значение G, тем меньше потребуется
экстрагента для достижения заданной степени извлечения кис-
лоты, т. е. тем меньше будет соотношение потоков экстрагента
й исходного раствора, подаваемых в экстрактор, а именно эти
параметры определяют и габарит оборудования (диаметр экс-
трактора у регенерационных колонн) и энергетические затраты.
Важным показателем экстракции является взаимная раство-
римость компонентов. На рис. 65 показаны кривые раствори-
мости трехкомпонентных систем вода — уксусная кислота —
эфир (петролейный, бутилацетат, этилацетат и метилацетат).
Самая большая область существования гетерогенных фаз в си-
стеме вода — уксусная кислота — петролейный эфир.
Преимущество таких растворителей в том, что их можно
использовать для экстракции уксусной кислоты из водных
растворов практически любой крепости и после отгонки раство-
рителя из фазы экстракта получать концентрат с высокой мас-
совой долей кислоты и незначительными примесями воды. Пет-
ролейный эфир не нашел широкого применения в данной тех-
нологии из-за очень низкого коэффициента распределения /Ср.
В системе вода — уксусная кислота — метилацетат, наобо-
рот, гетерогенная область весьма незначительна, а значе-
ние Кр высокое. Но этот растворитель также не может быть
рекомендован для промышленного внедрения из-за малой ем-
кости, легкой гидролизуемости и малой разности между его
плотностью и плотностью водного раствора, что снижает ли-
нейную скорость движения экстрагента в аппарате. Его можно
применять в лабораторных целях только для экстракции кис-
лоты из водных растворов с массовой долей ее не более 18%.
Гетерогенная зона в системе вода — уксусная кислота — этил-
ацетат (табл. 35) несколько меньше, чем в системах с пе-
тролейным эфиром и бутилацетатом. Но высокое значение /Ср
247
и другие достоинства определили внедрение в производство
именно этого растворителя.
Из табл. 35 видно, что в системе вода — уксусная кис-
лота— этилацетат значение Др зависит от кислотности фаз.
Для различных экстрагентов удобно представлять зависи-
мость Др как функцию кислотности водной фазы (т. е. рафи-
ната). Характер этой зависимости может быть самый разно-
Рис. 65. Кривые растворимости и
характерное положение воды в трех-
компонентных системах:
А — вода; В — уксусная кислота; С —
эфир; Ni, Сь Na, С2 — равновесные со-
ставы; 1 — метил ацетат; 2 — эти л ацетат;
3 — бутилацетат; 4 — петролейный эфир
Массовая доля кислоты в водной сраъе°/о
Рис. 66. Зависимость коэффициента
распределения уксусной кислоты Др
от кислотности водной фазы при эк-
стракции ее из жижки:
1 — диэтилкетоном; 2 — метилэтилкетоном;
3 — этил ацетатом; 4 — бинарным раство-
рителем этилацетат (92 %) — ацетон (8%)
Массовая доля в экстрагенте второго
компонента {х),%
Рис. 67. Сравнение зависимо-
стей от состава бинарных раст-
ворителей:
а — коэффициента экстракции G;
б — коэффициента распределения
образный (рис. 66), знать о ней необходимо для расчета числа
ступеней экстракции для конкретной системы.
При выборе бинарных или псевдобинарных экстрагентов
рекомендуется выполнять сравнительный анализ зависимо-
стей G=f(x) и Лр=/(х) во всем диапазоне концентраций
(рис. 67). Если кривой / на графике зависимости Kp=f(x) со-
248
35. ДАННЫЕ ПО РАСПРЕДЕЛЕНИЮ УКСУСНОЙ КИСЛОТЫ В СИСТЕМЕ ВОДА—
УКСУСНАЯ КИСЛОТА—ЭТИЛАЦЕТАТ
Органический слой Коэффициент распределе- ния кр Водный слой
уксусной кислоты этилаце- тата воды уксусной кислоты этилаце- тата воды
12,25 77,5 10,25 1,038 11,8 11,7 76,5
10,0 81,0 9,0 1,020 9,8 11,2 79,0
7,5 85,3 7,2 0,97 7,7 10,0 82,3
5,0 89,3 5,7 0,95 5,25 8,75 86,0
2,5 93,0 4,5 0,91 2,75 7,95 89,3
0,107 — — 0,68 0,16 — —
ответствует кривая 1 на графике зависимости G=f(x), то во
всем диапазоне концентраций этот растворитель эффективнее
однокомпонентного экстрагента, взятого за основу. Но опти-
мальная область составов бинарных растворителей лежит в пре-
делах присутствия второго компонента 65—75 %.
Если для исследуемого бинарного экстрагента характерны
зависимости 2, то этот бинарный экстрагент вряд ли можно
рекомендовать взамен основного однокомпонентного, несмотря
на то, что в области его концентрации до 85 % наблюдается
некоторое повышение коэффициента распределения. И, наобо-
рот, экстрагент, для которого характерны зависимости 3, более
предпочтителен по сравнению с предыдущим, хотя в области
его концентраций и отмечается некоторое снижение значе-
ний /Ср. Выбор этого экстрагента не однозначен, но при опре-
деленных технологических условиях он может конкурировать
с исходным однокомпонентным растворителем. Окончательный
выбор экстрагента делают на основе технико-экономических
расчетов, учитывающих все факторы ведения технологического
процесса.
На лесохимических предприятиях страны внедрены различ-
ные технологические схемы извлечения уксусной кислоты мето-
дом экстракции, отличающиеся способом подготовки жижки
к экстракции. Среди них выделим два способа: экстракция из
обессмоленной и обесспиртованной жижки с применением
этилацетата и переработка необессмоленной и необесспирто-
ванной жижки экстрагентом сложного многокомпонентного со-
става на основе этилацетата и легкокипящих компонентов
жижки.
Экстракция уксусной кислоты этилацетатом из обессмолен-
ной и обесспиртованной жижки. Принципиальная технологиче-
ская схема этого производства показана на рис. 68. Жидкие
продукты пиролиза или газификации древесины после частич-
ного отстаивания в ретортных цехах принимаются в сборник 1
химического цеха и направляются в обесспиртовывающий ап-
парат на верх колонны 25 для отгонки всех легколетучих ком-
понентов. На этой стадии выделяется фракция так называе-
249
мого спирта-сырца, которая долгое время являлась сырьем для
получения технического метанола и спиртовых растворителей.
Спирт-сырец содержит до 60 % метанола, 18—30 % воды и
10—42 % прочих компонентов (в основном ацетон и метил-
ацетат). В настоящее время только два лесохимических пред-
приятия перерабатывают эту фракцию с целью получения ме-
танола. На всех других заводах она утилизируется методом
Рис. 68. Принципиальная технологическая схема экстракции уксусной кис-
лоты из обесспиртованной и обессмоленной жижки:
/, 22, 23 —сборники; 2, 14, 15, 18, 19, 20, 25 — ректификационные колонны; 3, 5, 9, 11,
12 — конденсаторы-холодильники; 4, 8 — холодильники; 16, 24 — каландрии; 6 — выпар-
ной аппарат; 7 — экстрактор; 17, 21 — подогреватели; 10, 13 — флорентины
сжигания или используется для приготовления спиртовых ра-
створителей марки А или Э.
Для снижения осмоления контактных элементов исчерпы-
вающей части колонны 25 вместе с жижкой в аппарат подают
отстойную смолу (2—6 % от жижки). В обесспиртовывающем
аппарате достигается степень выделения метанола 92—96 %.
Ректификацию ведут в колонне с числом практических тарелок
(колпачковых) 40 (в исчерпывающей части 15, в укрепляю-
щей— 25) при флегмовом числе 4—5. Температура в верхней
части колонны 2 70—75 °C, внизу колонны 25 102—106 °C.
Обесспиртованная жижка после отстаивания от тяжелых
отстойных смол в специальном буферном сборнике 23 направ-
ляется в выпарной аппарат (двух- или трехкорпусный) 6, где
происходит выделение растворимых смол. Принцип работы вы-
парных аппаратов может быть самым различным. Эксплуати-
250
руются три вида выпарных аппаратов: с принудительной цир-
куляцией (в трубчатых или пленочных испарителях с циркуля-
цией увариваемой смолы с помощью насосов), двухкорпусные
и трехкорпусные с использованием соковых паров.
В трубчатку первого корпуса выпарного аппарата 6 непре-
рывно подается обесспиртованная жижка, в межтрубное про-
странство этого корпуса — насыщенный пар. Температура
в первом корпусе 102—ПО °C, давление атмосферное. Пары
жижки из первого корпуса направляются в межтрубное про-
странство второго корпуса выпарного аппарата, где они служат
уже теплоносителем для второй ступени аппарата. Частично
упаренная в первом корпусе жижка направляется в трубное
пространство второго корпуса, где создается вакуум
(40—53) • 103 Па. Этому давлению соответствует температура
кипения упаренной жижки 85—90 °C. Третий корпус работает
по принципу второго. Вакуум в этом аппарате поддерживается
(80—82,5) • 103 Па, температура 60—70 °C. Конденсат из меж-
трубного пространства второго и третьего корпусов аппарата и
пары из третьего корпуса направляются в конденсатор-холо-
дильник 5, а затем обессмоленная и обесспиртованная жижка
(или кислая вода) тщательно отстаивается в сборнике 22 от
легких всплывных масел.
В разное время и разными исследователями доказано отри-
цательное влияние легких всплывных масел на процесс экс-
тракции. В присутствии легких всплывных масел резко увели-
чивается растворимость этилацетата в системе, что приводит
к увеличению соотношения экстрагент — исходный раствор, и
возрастанию потерь этилацетата за счет гидролиза (в основ-
ном на стадии регенерации его из рафината).
Кислая вода (или смесь кислой воды и жижки на одном из
лесохимических заводов) кислотностью 7—12 % направляется
на верх экстрактора 7 для извлечения уксусной кислоты этил-
ацетатом, который подается в нижнюю его часть. Экстрагент
диспергируется в барботажных прорезях и в виде капель под-
нимается в верхнюю часть аппарата благодаря разности плот-
ностей, извлекая из исходного раствора уксусную кислоту.
В практике широко используются ситчатые колонные экстрак-
торы (провального типа или с организованным переливом тя-
желой фазы). Эти экстракторы характеризуются достаточно
высокой единичной мощностью: например, экстрактор диамет-
ром 1,2 м имеет нагрузку по двум фазам до 27—30 м3/ч.
Эффективность таких аппаратов существенно зависит от
степени горизонтального отклонения ситчатых тарелок. Такие
массообменные элементы требуют очень строгого горизонталь-
ного их расположения при монтаже оборудования. В против-
ном случае в массообмене будет участвовать не вся тарелка,
а только ее часть.
Для системы уксусная кислота —вода — этилацетат при на-
чальной кислотности исходного раствора 10—12 % степень
251
извлечения 98 % достигается на 6—7-й теоретических ступенях
экстракции. Число теоретических ступеней экстракции рассчи-
тывается на основании данных о взаимной растворимости ком-
понентов системы, зависимости Лр от кислотности водной фазы
и материального баланса (по.компонентам):
Fxi + Qxi = Rxi + 3xit
(96)
Число ступеней зкстракици. t mm.
Рис. 69. Зависимость кислотности
эфироводы от числа идеальных
ступеней экстракции при соотно-
шениях экстрагента (этилацетата)
и жижки (по массовой доле, %):
1 — 1 : 0,906; 2 — 1 : 1,08; 3 — 1 : 1,36;
4— 1 : 1,81
где F, Q, R и Э — соответственно масса исходного раствора,
оборотного экстрагента, рафината и экстракта; х—массовая
доля; i — компонент системы.
На рис. 69 показана зависимость кислотности эфироводы от
числа теоретических, или идеальных, ступеней экстракции, по-
лученная экспериментально по методу Шейбеля. Температура
подаваемых в экстрактор потоков воды и оборотного экстра-
гента должна быть не более 28 °C.
Эфировода с остаточной кислотностью 0,1—0,2 % выводится
на регенерацию растворенного в нем экстрагента через гид-
равлик из нижней части экстрактора 7 (см. рис. 68) (на 0,8—
1 м ниже ввода экстрагента). Из верхней части экстрактора
отбирается эфирокислота. Кислотность эфирокислоты зависит
от соотношения масс экстрагента и кислой воды, исходной кис-
лотности раствора и числа ступеней экстракции. При экстра-
гировании уксусной кислоты из раствора концентрацией 8—
12 % на 6—8-й ступенях экстракции и соотношении экстра-
гент — исходный раствор 1,24-1,6:1 (по массе) кислотность
эфирокислоты. колеблется в пределах 5—7 % -
Для регенерации этилацетата из эфироводы и из эфирокис-
лоты применяется метод азеотропной ректификации, т. е.
используется свойство этилацетата образовывать с водой по-
ложительный (низкокипящий) гетероазеотроп (табл. 36). Реге-
нерация этилацетата из водной фазы осуществляется в двух-
колонном эфироводном аппарате. В первой колонне 20 (см.
рис. 68) (число тарелок 25—26) регенерируется этилацетат,
в колоннах 18, 19 выделяются спиртовые продукты.
252
36. АЗЕОТРОПНЫЕ СВОЙСТВА КОМПОНЕНТОВ СИСТЕМЫ ЭТАНОЛ —ВОДА —
ЭТИЛАЦЕТАТ
Компонент Температура кипе- ния, СС Доля в азеотропе ком- понента, % Примечание
А в, с А в, с азео- тропа А в
массовая мольная массовая мольная
Вода Этанол 100 78,3 78,15 4,4 15,9 95,6 84,1 Азеотроп гомогенный
Вода Этилацетат 100 77,15 70,4 8,2 30,3 91,8 69,7 Азеотроп гетероген- ный. Рас- творимость воды в эти- лацетате 3,3 %
Этанол Этил ацетат 78,3 77,15 71,8 30,6 45,8 69,4 54,2 Азеотроп гомогенный
Вода Этанол Этилацетат 100 78,3 77,15 70,3 7,8 27,5 9,0 83,2 12,5 60,0 Азеотроп гетероген- ный
Перед подачей на 12—13-ю тарелку эфировода подогрева-
ется до температуры 75—85°C. Пары азеотропной смеси этил-
ацетат— вода с примесями тройного азеотропа этанол — этил-
ацетат— вода при температуре в верхней части колонны 20
70—74 °C поступают в конденсатор-холодильник 9, где смесь
охлаждается до 20—25°C и затем во флорентине 10 разделя-
ется на два слоя. Нижний водный слой полностью возвраща-
ется в колонну на тарелку питания, а верхний частично воз-
вращается на флегму (флегмовое число 0,4—0,5), а частично,
после дополнительного охлаждения в холодильнике 8, возвра-
щается на экстракцию. Эта стадия процесса является очень от-
ветственной, так как в основном омыление этилацетата проис-
ходит в ней. С целью снижения потерь этилацетата рекоменду-
ется избегать частых пусков процесса регенерацйи, т. е.
процесс должен быть именно непрерывным.
Вместе с этилацетатом из эфироводы этанол регенерируется
лишь частично, что объясняется взаимодействием водородных
связей этанола и воды. Для более глубокой степени очистки
сточной воды от спиртов кубовый остаток из колонны 20 на-
правляется в исчерпывающую колонну 19 спиртового аппарата.
253
Число тарелок- в исчерпывающей колонне должно быть не ме-
нее 15, в укрепляющей 14—16. Процесс ведут при флегмовом
числе 5—6. Температура в верхней части колонны 18 65—
68 °C, в нижней части колонны 19 102—104 °C. Спиртовая
фракция после конденсации и охлаждения используется для
приготовления низкоквалифицированных растворителей, т. е.
выводится из экстракционного процесса. Сточная вода, выходя-
щая из эфироводного аппарата, при переработке кислой воды
имеет загрязненность по ХПК 4000—6000 мг/л.
При переработке смеси жижки и кислой воды ХПК значи-
тельно выше за счет присутствия органических компонентов
растворимой смолы (до 30 000 мг/л). Массовая доля этанола
в сточной воде не должна быть выше 0,15%, этилацетата не
выше 0,08 %, уксусной кислоты 0,15—0,20 %.
Регенерация этилацетата из эфирокислоты осуществляется
в одноколонном ректификационном аппарате. Эфирокислота
перед подачей в нижнюю часть колонны 14 подогревается до
температуры 70—80 °C. Пары этилацетата и воды в виде азео-
тропной смеси выводятся из колонны 14 в конденсатор-холо-
дильник 12 и с температурой 20—25 °C поступают во флорен-
тину на разделение. Водный слой из флорентины направляется
вместе с эфироводой из экстрактора на переработку в эфиро-
водный аппарат (в колонну 20), а эфирный слой разделяется
на два потока: флегму (флегмовое число процесса зависит от
кислотности эфирокислоты, рис. 70) и оборотный экстрагент,
который дополнительно охлаждается в холодильнике 8 (см.
рис. 68) и вместе с эфиром, регенерированным из эфироводы,
возвращается на экстракцию. Основное требование к этому
процессу — получение регенерированного эфира кислотностью
не выше 0,006%. При большей кислотности оборотного эфира
не может быть достигнута высокая степень извлечения уксус-,
ной кислоты из кислой воды. Черная кислота, с массовой долей
этилацетата до 4 % и воды до 5 % выводится из каландрии 16.
Температура в верхней части колонны 14 70—72 °C, в калан-
дрии 16 122—130 °C. Число тарелок в исчерпывающей части
колонны 16, в укрепляющей — 20.
Экстракция уксусной кислоты из необессмоленной и необес-
спиртованной жижки эстрагентом сложного состава. Впер-
вые в 1953 г. работами В. П. Сумарокова и 3. М. Володуцкой
была доказана возможность экстрагирования уксусной кис-
лоты из необессмоленной и необесспиртованной жижки. При
этом качество технической уксусной кислоты не отличалось от
качества кислоты, полученной при переработке кислой воды,
а затраты пара на производство 1 т 100%-ной технической ки-
слоты снизились почти на 20 %. Недостатком этого способа
переработки жижки является большая загрязненность сточных
вод, получаемых после регенерации экстрагента из эфироводы.
За счет присутствия в ней водорастворимых неэкстрагируемых
смолистых веществ ХПК сточных вод достигает 70—90 г/л.
254 ,
Сточная вода в этом случае может быть обезврежена лишь ме-
тодом сжигания или каталитического окисления, так как при-
менение биохимических методов очистки неэкономично.
Ранее обесспиртовывание жижки считалось обязательным
прежде всего из-за необходимости получения метанола, про-
дукта крайне дефицитного. К тому же считалось, что легко-
кипящие примеси, выделяемые со спиртом-сырцом, мешают
•Рис. 70. Зависимость флегмового
числа процесса переработки эфиро-
кислоты от ее кислотности (при ох-
лаждении конденсата до 20 °C):
1—теоретическая (или расчетная) зави-
симость для чистой системы вода — ук-
сусная кислота — этилацетат; 2 — практи-
ческие данные
16
Кислотность Эфирокислоты, %
Рис. 71. Принципиальная схема извлечения уксусной кислоты из необессмо-
ленной й необесспиртованной жижки методом экстракции:
/, 1а — сборники для отстаивания жижки; 2 — экстрактор; 3— эфироводная колонна;
4, 8 — конденсаторы-холодильники; 5, 9 — флорентины; 6 — подогреватель; 7 — укрепля-
ющая часть эфирокислотного аппарата; 10 — исчерпывающая часть эфирокислотного
аппарата; 11 — каландрия; 12, 13 — холодильники
процессу экстракции кислоты, т. е. снижают ее коэффициент
распределения.
Работами ЦНИЛХИ в 70-х годах было показано, что при
переработке необессмоленной и необесспиртованной жижки на
255
снижение Кр большее влияние оказывают не легкокипящие
примеси, а смолистые вещества. Опыт одного лесохимического
предприятия, освоившего схему переработки необесспиртован-
ной и необессмоленной жижки, доказал ее экономическую
целесообразность.
В последнее время пиролиз древесины в вертикальных
непрерывнодействующих ретортах осуществляется при высоко-
температурном режиме. Массовая доля метанола в жижке сни-
зилась до 0,8—0,9 %. При экстракции уксусной кислоты из не-
обессмоленной и необесспиртованной жижки в процессе мас-
сообмена участвуют и смолистые вещества и те легкокипящие
компоненты, которые отгоняются в спирт-сырец при обесспир-
товывании продуктов пиролиза (в основном ацетальдегид,
метилацетат, ацетон, метанол, метилэтилкетон, этанол, ди-
этил кетон).
Некоторые компоненты спирта-сырца ранее были рекомен-
дованы как экстрагенты для извлечения уксусной кислоты из
водных растворов (метилэтилкетон, диэтилкетон, метилацетат),
некоторые как добавки к различным экстрагентам для улучше-
ния экстрагирующих свойств однокомпонентных растворите-
лей. Это предопределяет и теоретическую и практическую целе-
сообразность экстракции кислоты из жижки непосредственно
после тщательного отстаивания от нее легких всплывных масел
и тяжелых смол.
Принципиальная технологическая схема, которая эксплуа-
тируется на Амзинском ЛХЗ, показана на рис. 71. В сборниках
1 и 1а жидкие продукты пиролиза тщательно отстаиваются от
легких всплывных масел и тяжелых смол. В сборник 1а направ-
ляется слабая уксусная кислота от переработки черной кис-
лоты. Жижка из сборника 1а (на 1,5—2 м выше уровня отбора
тяжелых смол) подается на экстракцию. В начале процесса
экстрактор заполняется этилацетатом марки А. В процессе
экстракции спиртовые примеси жижки вместе с уксусной кис-
лотой распределяются между водной и эфирной фазами сог-
ласно закону фазового равновесия. При кислотности жижки
10—12 % соотношение масс экстрагента и жижки поддержи-
вается равным 1,4—1,7:1. Число идеальных ступеней экстрак-
ции в данном случае определить трудно из-за многокомпонент-
ное™ системы. В экстракторе степень извлечения уксусной
кислоты 96—98 %. Минимальная кислотность эфироводы, кото-
рая может быть достигнута по данной технологии, 0,3—0,4%.
Это объясняется тем, что при проведении анализа вместе
с карбоновыми кислотами (массовая доля их не более 0,15 %)
титруются и трудно экстрагируемые оксикислоты и их лак-
тоны.
На 10 ступенях экстракции (рис. 72) кислотность эфироводы
при экстракции жижки достигается не ниже 0,3 %, в то время
как кислотность эфироводы при переработке кислой воды
в аналогичных условиях может быть менее 0,1 % (0,08 %).
256
Этилацетат вмёсте с распределёнными легкокипящими ком-
понентами регенерируется из эфирокислоты в аппаратах 7, 10
(см. рис. 71). Условия ведения процесса и эффективность про-
цесса (число теоретических тарелок и флегмовое число) анало-
гичны для процесса переработки эфирокислоты, полученной
при экстракции кислоты из кислЪй воды. Качество черной кис-
лоты меняется в течение цикла. Эфировода перерабатывается
в одноколонном аппарате 3 в режиме исчерпания. Температура
Рис. 72. Кислотность рафината на десяти последовательных ступенях экст-
ракции уксусной кислоты этилацетатом:
/ — из жидких продуктов пиролиза древесины; 2 — из кислой воды
Рис. 73. Изменение некоторых свойств
оборотного экстрагента и экстрагента,
регенерированного из органической фазы,
в течение одного рабочего цикла:
а — массовая доля в конденсате паровой фа-
зы экстрагента, регенерированного из орга-
нического слоя: 1 — отстойной воды; 2 — рас-
творимой воды; б — коэффициент распределе-
ния кислоты между органической и водной
фазами: 1 — при извлечении экстрагентом,
регенерированным из экстракта; 2 — оборот-
ным экстрагентом; в — коэффициент экстрак-
ции кислоты при извлечении ее из исходного
раствора; 1 — на одной ступени экстраген-
том, регенерированным из экстракта; 2 —
оборотным экстрагентом
9J Заказ № 2193
4 8 121В 202428323BWM4B52
Время работы ^дна
257
& верхней части колонны 64—69 °C, внизу 102—104 °C. Флег-
мовое число по эфиру 0, по водному слою 0,2—0,3. Число таре-
лок колпачкового типа 16—17. ХПК сточной воды 5,9—7,5 г/л.
Регенерированный из водной и эфирной фаз экстрагент объеди-
няется, дополнительно охлаждается и возвращается на экс-
тракцию. При этом в процессе экстракции происходит постоян-
ное накопление легкокипящих примесей в системе.
Цикл процесса экстракции прерывается через 1,5—2 мес
после накопления в оборотном экстрагенте массы легкокипя-
щих компонентов, достаточной для наступления момента гомо-
генизации фаз. В этом случае регенерированный экстрагент не
возвращается на экстракцию, а выводится из системы. В си-
стему вновь закачивается этилацетат марки А. Цикл экстрак-
ции возобновляется.
Основными недостатками процесса являются его периодич-
ность, непостоянство состава всех материальных потоков и,
следовательно, экстрагирующих свойств оборотного экстра-
гента, температурных режимов, повышенная массовая доля
воды в черной кислоте. К преимуществам данной технологии
следует отнести простоту аппаратурного оформления процесса,
незначительные потери этилацетата, малую энергоемкость и
низкую себестоимость технической уксусной кислоты.
В табл. 37 приведены сравнительные технико-экономические
показатели производства уксусной кислоты экстракционным
способом с применением этилацетата (экстракция кислоты из
кислой воды) и экстрагента сложного состава (экстракция кис-
лоты из необессмоленной и необесспиртованной жижки).
Характер влияния легкокипящих компонентов на процесс
экстракции кислоты из жижки в течение одного цикла показан
37. ТЕХНИКО-ЭКОНОМИЧЕСКИЕ ПОКАЗАТЕЛИ ПРОИЗВОДСТВА УКСУСНОЙ
КИСЛОТЫ МЕТОДОМ ЭКСТРАКЦИИ ЭТИЛАЦЕТАТОМ И ЭКСТРАГЕНТОМ
СЛОЖНОГО СОСТАВА
Показатель Экстракция
этил ацетатом экстрагентом сложного состава
Массовая доля уксусной кислоты в исход- ном растворе, % 7,4 10,7
Объемное соотношение экстрагент — исход- ный раствор 1,4 1,9
Степень извлечения кислоты (при одинако- вом числе идеальных ступеней экстракции), % 98,1 98,8
Массовая доля воды в черной кислоте, % Флегмовое число процесса переработки эфи- рокислоты 3—5 8—10
1,6 0,9
Расход этилацетата на 1 т 100 %-ной техни- ческой кислоты, кг 72-120 12—17
258
на рис. 73. В первые 20—23 дня работы наблюдается положи-
тельное влияние легкокипящих примесей жижки на коэффици-
ент распределения кислоты Кр, коэффициент экстракции G и
гетерогенные свойства экстрагента. Массовая доля воды
в черной кислоте колеблется в пределах 10—11 %. Затем экс-
трагирующие свойства растворителя ухудшаются. Рекомендо-
вано с целью дальнейшего совершенствования технологии
Продукты
• пиролиза\
Масла
Спадая кис-
лота счерно-
кислатного
аппарата
Рис. 74. Принципиальная технологическая схема переработки жижки мето-
дом экстракции сложным экстрагентом постоянного состава:
/ — баки для приемки сырья; 2 — экстрактор; 3, б, 9, 15, 19, 24 — ректификационные
колонны; 4, 8, 11, 20, 25 — каландрии; 5 — конденсатор; 7, 10, 16, 26 — конденсаторы-хо-
лодильники; 12, 18, 21 — холодильники; 13, 17 — флорентины; 14 — подогреватель;
22, 23 — промежуточные емкости
экстракции уксусной кислоты из жижки стабилизировать при-
сутствие в экстрагенте легкокипящих компонентов на опреде-
ленном уровне концентрации, для которого характерен поло-
жительный эффект их присутствия.
В результате исследований определен оптимальный состав
сложного экстрагента для условий Амзинского ЛХЗ. Массовая
доля компонентов в нем, %: ацетальдегида 0,05, ацетона 0,1—
0,3; метилацетата 4,2—4,4; метанола 0,5—0,6; этилаце-
тата 55—56; этанола 0,5—0,6; метилэтилкетона 23,5—24; ди-
этилкетона 11,5—12; воды 3,2—4,2.
Постоянство состава экстрагента обеспечивается режимом
регенерации растворителя из эфироводы (рис. 74). Поскольку
около 85 % от массы эфироводы приходится на долю воды,
в первой колонне целесообразно извлечь из нее все легкокипя-
щие компоненты. Сбросная вода имеет характеристику анало-
9* 259
гичную сточной воде, полученной по технологии экстракции
экстрагентом переменного состава. Во второй колонне выде-
ляют все компоненты, температура кипения которых ниже, чем
у этилацетата (ацетальдегид, ацетон, метанол, метилацетат).
Кубовый продукт второй колонны поступает на верх третьей
колонны, в которой выделяются в виде кубовой жидкости все
компоненты с температурой кипения выше 77,15°С (этанол,
метилэтилкетон, диэтилкетон и др. — кетоно-спиртовая фрак-
ция). На экстракцию возвращается только дистиллят из тре-
тьей КОЛОННЫ;
Легкокипящие компоненты могут использоваться для произ-
водства древесно-спиртовых растворителей марки А или Э,
а кетоноспиртовая фракция — для получения индивидуальных
кетонов, продуктов в настоящее время дефицитных и весьма
нужных для народного хозяйства.
Усовершенствованная схема извлечения кислоты из жижки
экстрагентом постоянного состава позволяет: сократить на ста-
дии экстракции соотношение экстрагент — жижка на 10—12%,
что повлечет за собой снижение объемов эфирокислоты, т. е.
снижение энергозатрат и повышение производительности обо-
рудования; снизить расход этилацетата на 1 т 100%-ной тех-
нической кислоты до 4—6 кг; получать черную кислоту с массо-
вой долей воды не более 2 %, что повлечет снижение энерго-
затрат на переработку черной кислоты.
В табл. 38 приведены условия ведения процесса переработки
эфироводы по предложенной технологии и корректировка ре-
жима переработки эфирокислоты.
38. УСЛОВИЯ ВЕДЕНИЯ ПРОЦЕССА ПЕРЕРАБОТКИ ЭФИРОВОДЫ И КОРРЕКТИ-
РОВКА РЕЖИМА ПЕРЕРАБОТКИ ЭФИРОКИСЛОТЫ
Регенерация экстрагента Показатели процесса ректификации
Число теоретических тарелок, шт. Флегмо- вое число Температура, °C
в исчер- пываю- щей части в укреп- ляющей части верха колонны низа колонны
Из водной фазы по стадиям: выделение всех легкокипя- 7 0 0 65—71 100-102
щих компонентов выделение компонентов с 8 14 18—22 50—50,5 74—75
температурой кипения ниже 77,15 °C выделение компонентов 6 0 0,2—0,3 70—71,5 92—94
с температурой кипения выше 77,15 °C Из эфирокислоты 12 6 0,3—0,4 72—74 128—130
260
§ 42. АЗЕОТРОПНЫЙ СПОСОБ ИЗВЛЕЧЕНИЯ УКСУСНОЙ
кислоты из жижки
Процессы ректификации основаны на фазовых равновесиях
между жидкой и паровой фазами. Распределение компонентов
между фазами выражают величиной относительной летучести
о О
Pt и Pk—давление паров
(97)
Рис. 75. Фазовые равновесия жид-
кость—пар в системе уксусная
кислота — вода при 101,3 кПа ».
Массовая доля уксусной, кислоты
б жидкой (разе, % .
от состава смеси, значения
где
чистых компонентов; yz и у&—
коэффициенты активности, харак-
теризующие степень отклонения
поведения компонентов от иде-
ального.
Коэффициент относительной
летучести является очень важной
функцией, так как характеризует
степень легкости разделения дан-
ной пары компонентов; при ма-
лых ее значениях ректификация
затруднена (к таким и относится
система вода — уксусная кислота;
рис. 75), и, наоборот, чем выше
значение тем легче могут
быть разделены компоненты ме-
тодом простой ректификации.
Величины Р? и Р^ не зависят
их определяются только температурой В табл. 39 приведены
данные по упругостям паров некоторых органических соедине-
ний, присутствующих в жижке.
Значения у/ и yk зависят от массовых долей всех компонен-
тов системы, т. е. от состава жидкой фазы. Изменяя состав жид-
кой фазы, мы можем менять и величину относительной летуче-
сти компонентов. Это свойство и лежит в основе азеотропной и
экстрактивной ректификации. Азеотропная ректификация осно-
вана на подборе такого увлекателя (антренера),'в присутствии
которого увеличивается a,ik за счет повышения коэффициента
активности легколетучего компонента у/, а экстрактивная—за
счет уменьшения коэффициента активности труднолетучего ком-
понента у*.
К антренерам предъявляются следующие основные требо-
вания.
1. Они должны образовывать с извлекаемым компонентом
азеотроп, температура кипения которого должна быть как
можно меньше температуры кипения отделяемого компонента;
а массовая доля увлекаемого компонента в азеотропе — как
можно больше.
261
39. УПРУГОСТЬ ПАРОВ НЕКОТОРЫХ ОРГАНИЧЕСКИХ
Компонент Упругость паров (Па-IO-’)
0 10 20 30 40 50
Вода Кислота: 0,61 1,22 2,32 4,21 7,32 12,26
муравьиная 1,43 2,52 4,41 6,96 11,01 16,78
уксусная 0,47 0,84 1,57 2,65 4,53 7,49
пропионовая Спирт: 0,10 0,20 0,40 0,76 1,37 2,40
метиловый 2,53 6,80 11,86 19,99 32,26 51,05
этиловый 1,68 3,-15 5,85 10,50 18,03 29,62
н. пропиловый 0,46 0,96 1,93 16,97 6,68 11,60
изобутиловый — — — — — —
н. бутиловый — 0,26 0,59 1,27 2,48 4,49
Ацетальдегид 40,03 66,50 100,41 152,95 220,78 —
Диэтиловый эфир 23,99 38,26 63,98 84,38 121,04 163,96
Ацетон 6,66 14,66 23,99 37,55 55,99 81,18
Метилэтилкетон 2,00 5,60 9,73 15,73 24,79 37,59
Метилформиат 25,99 41,24 63,50 94,36 137,16 92,98
Этилформиат 9,65 16,03 25,66 39,66 59,54 86,56
Бутил формиат 0,93 ' 1,66 3,06 4,65 8,05 13,03
Метилацетат 8,28 13,97 22,64 35,43 53,37 78,47
Этилацетат 3,20 5,73 9,73 15,73 24,79 37,59
Пропилацетат 0,93 — 3,32 — 9,43 —
Изобутилацетат — — — -— — —
Бутилацетат 0,29 0,57 1,06 1,92 3,32 5,44
Метил пропион ат 2,91 5,16 8,80 14,34 22,52 34,12
Этилпропионат 1,10 2,06 3,68 — 10,36 —
Метил бутир ат 0,97 1,83 3,26 5,57 9,20 14,58
2. Они не должны вступать в реакцию ни с одним из компо-
нентов, не должны разлагаться, т. е. они должны быть химиче-
ски неактивными и термически стойкими.
3. Теплота испарения и теплоемкость антренера должны
быть как можно ниже.
4. Антренеры должны быть доступными и дешевыми. Жела-
тельно, чтобы антренер с извлекаемым компонентом, имел как
можно меньшую взаимную растворимость и образовывал с ним
гетероазеотроп. Это относится к системам, где вода является
извлекаемым компонентом или выбрана в качестве антренера.
Выбор антренера определяет технологическую схему про-
цесса, его параметры, эффективность способа разделения. При
разделении смеси вода — уксусная кислота подбирается антре-
нер для вывода из системы воды.
Метод азеотропной переработки жижки внедрен только на
одном лесохимическом заводе (Великобычковском). Характери-
стика перерабатываемой на этом предприятии жижки: плот-
ность 1020—1033 кг/м2 3 4, кислотное число 98—135 мг КОН/г,
эфирное число 32—37 мг КОН/г, массовая доля метанола 3,2—
2,6%,
262
СОЕДИНЕНИЙ, ПРИСУТСТВУЮЩИХ В ЖИЖКЕ
при температуре, сС
60 70 80 90 100 по 120 130 140
19,83 31,07 47,27 70,4 101,3 — — — —
25,29 37,27 53,07 73,59 100,42 — — — —
11,7 18,27 26,94 39,03 55,52 77,66 105,60 171,98 188,06
4,05 6,62 10,52 16,26 24,47 36,0 52,3 73,3 98,6
77,31 114,10 164,76 — —
47,01 72,35 108,32 158,23 — — — —
19,55 31,79 50,01 76,34 112,05 160,40 — — —
13,18 21,08 33,10 51,15 77,58 117,91 159,20 221,98 •—
7,87 14,93 33,10. 51,15 77,60 112,42 159,20 221,84
114,64 158,89 — — — — — —
54,39 79,05 110,77 151,69 — — — — —
122,36 168,76 —
16,73 29,26 42,56 61,18 85,12 146,30 — — —
111,64 153,56 — — — — — — —
55,32 79,47 117,70 151,03 — — — — —
22,86 — 49,61 68,82 96,26 129,82 171,70 224,20 288,86
— — — — — — 113,48 149,95 195,54
8,89 13,87 21,20 31,46 45,73 65,19 90,93 122,0 150,0
50,58 72,88 102,54 139,38 187,26 — — — —
25,00 — 53,68 — 104,40 — 183,94 — —
22,28 33,25 48,07 67,43 93,19 125,15 165,85 217,63 279,23
В качестве разделяющего агента используются так называе-
мые древесноспиртовые масла, получаемые при переработке
спирта-сырца, имеющие следующие показатели: плотность 890—
896 кг/см3, растворимость в воде при 20—25 °C 6—10 % (по
объему), растворимость воды в маслах 5—7 % (по объему),
теплота испарения 335—343 кДж, массовая доля эфиров 20—
40 %, кетонов и альдегидов, образующих гетероазеотропы с во-
дой, 30—40%. При нормальном давлении масла перегоняются
с водой при температуре 85—95 °C, массовая доля воды,
отогнанной с гетероазеотропами, 25—33 %.
Технология переработки жижки состоит из следующих ста-
дий: отстаивания тяжелых смол и легких всплывных масел от
жижки; обесспиртовывания и обессмоливания жижки; азеотроп-
ного укрепления кислоты с целью получения черной кислоты;
регенерации антренера из водных фаз; переработки черной
кислоты с целью получения технической уксусной кислоты (и
далее пищевой) и из побочной стадии — переработки спирта-
сырца для получения фракции древесно-спиртовых масел.
На рис. 76 показана принципиальная технологическая схема
азеотропного извлечения кислот из жижки. Стадия отстаивания
263
тяжёлых смол и Легких ВСплЫйнЫх мйсёЛ имеет Те же Цёлй, ЧТО
и при переработке жижки экстракционным способом. То же
можно сказать и о стадии обесспиртовывания (эти стадии на
рис. 76 не показаны).
Обесспиртованная жижка из бака 1 непрерывно поступает
в три последовательно соединенных трубчатых испарителя 12,
предназначенных для выделения из жижки растворимых смол,
Рис. 76. Принципиальная технологическая схема азеотропного спо-
соба извлечения уксусной кислоты из жижки
которые отбираются только из последнего испарителя. Темпе-
ратура в первом корпусе поддерживается 108—110 °C, во вто-
ром 128—135 °C, в третьем 150—160 °C. Растворимая смола
поступает в сборник 11, откуда выкачивается на дальнейшую
переработку. Пары кислой воды из всех трех испарителей по-
ступают в нижнюю часть ректификационной колонны 8 (число
колпачковых тарелок 21—23). На верхнюю тарелку колонны 8
подается антренер из флорентины 7 (или из бака 3 для воспол-
нения потерь и в начале процесса). Температура паров вверху
колонны 82—86 °C. Флегмовое число изменяется в пределах
1,5—3. Пары воды и антренера конденсируются в конденсато-
рах 2, 4, охлаждаются в холодильнике 6 до температуры 40—
45 °C и поступают на разделение во флорентину 7. Верхний
264
слой полностью возвращается в колонну, а нижний — водный —
выводится в колонну 5 с целью регенерации из него растворен-
ного антренера. Кислотность водного слоя должна быть не
более 0,3%. Температура вверху колонны 5 колеблется в пре-
делах 82—94 °C, флегмовое число 0, число тарелок колпачко-
вого типа 25—26. Регенерированный антренер после охлажде-
ния в холодильнике 6 поступает на разделение также во фло-
рентину 7. Отгонка антренера ведется с помощью острого пара.
ХПК сточной воды до 5000 мг/л. Исчерпание воды и антренера
из кислоты ведется в колонне 10 (число тарелок 16—18, темпе-
ратура в нижней части колонны ПО—115 °C). Из каландрии 9
выводится концентрированный раствор кислоты в виде так на-
зываемой черной кислоты; ее плотность 1058—1067 кг/м3, кис-
лотное число 416—428 мг КОН/г; эфирное число 60—
70 мг КОН/г, массовая доля воды в ней 33—40%, смолистых
веществ 22—30 %.
Черная кислота, полученная азеотропным способом перера-
ботки жижки, содержит по сравнению с черной кислотой, полу-
ченной в процессе экстракции кислоты этилацетатом или слож-
ным экстрагентом, значительно больше воды и смолистых ве-
ществ. Это относится к недостаткам способа азеотропного ук-
репления жижки с помощью древесно-спиртовых масел. К тому
же состав масел меняется в процессе ректификации, меняется
состав гетероазеотропа, неустойчивы все технологические пара-
метры.
Более стабильным становится процесс при добавлении к дре-
весно-спиртовым маслам бутилацетата. Бутилацетат можно ис-
пользовать как антренер, но в связи с тем, что он относительно
дорог, склонен к гидролизу и переэтерификации, он не нашел
широкого применения в качестве антренера в лесохимической
промышленности (в жижке содержится до 2—2,5 % муравьиной
кислоты).
§ 43. ПОРОШКОВЫЙ СПОСОБ ПЕРЕРАБОТКИ ЖИЖКИ
На мелких установках газификации древесины и заводах су-
хой перегонки малой мощности организация азеотропного или
экстракционного способа извлечения уксусной кислоты из
жижки нерентабельна. В таких случаях применяют порошковый
способ переработки жижки, основанный на реакции нейтрали-
зации уксусной кислоты известью,
2СН3СООН+Са (ОН)2 = (СН3СОО)2 Са + 2Н2О. (98)
Образовавшийся раствор уксусно-кальциевой соли упари-
вают До пастообразного состояния, а затем' высушивают. Для
нейтрализации уксусной кислоты используется раствор гашеной
извести (известковое молоко) концентрацией окиси кальция
около 115 г/л.
265
Технологическая схема процесса проста, а аппаратурное
оформление разнообразно. Технологический процесс состоит из
отделения отстойной смолы; приготовления водной суспензии
извести; нейтрализации жижки способом холодного или горя-
чего насыщения с отгонкой спирта-сырца; отстаивания раствора
кальциевых солей органических кислот от шлама и удаление
шлама; упаривания раствора уксусно-кальциевой соли до пасто-
образного состояния; сушки пасты до порошкообразного состоя-
ния уксусно-кальциевой соли.
Уксусно-кальциевые порошки перерабатываются централизо-
ванно в основном с целью получения пищевой уксусной кислоты.
Массовая доля уксусно-кислого кальция изменяется в зависи-
мости от сорта порошка от 57 до 80 %. Массовая доля воды не
более 12 % (10—12%). Кроме того, в порошке содержится до
8 % избыточной извести и до 30 % органических примесей.
Под действием серной кислоты кальциевая соль уксусной
кислоты разлагается по реакции
(СН3СОО)2 Са + H2SO4 = 2СН3СООН + CaSO4. (99)
Принципиальная технологическая схема переработки по-
рошка показана на рис. 77.
Разложение уксусно-кальциевого порошка ведут в периоди-
ческом режиме, в специальном аппарате (аппарат Линде); У нас
в стране используются аппараты Линде диаметром 2370 мм,
высотой 850 мм (геометрический объем 3,75 м3). Аппарат с ме-
шалкой имеет паровую рубашку (тепло подводится не только
через боковые-стенки, но и через днище), боковой люк для вы-
грузки гипса (окшары), люк в крышке аппарата для загрузки
порошка. Обогревается аппарат паром давлением 70-10—80Х
Х104 Па. После загрузки порошка в аппарат 1 закрывают
люк, включают мешалку (частота вращения 4—5 мин-1) и на-
чинают порциями подачу серной кислоты из сборника 2 в тече-
ние 40—60 мин. После загрузки серной кислоты в паровую ру-
башку начинают подачу пара, и аппарат осторожно разогре-
вают.
Кроме основной реакции идут побочные:
СаО+H2SO4 = CaSO4+Н2О; (100)
H2SO4— 1 /2О2 -> H2SO3 -> SO2 4- Н2О; (101)
НСООН СО+Н2О; (102)
Са (СН3СОО)2 СаСО3 + (СН3)2 СО. (103)
Очень важно, чтобы перерабатываемый порошок был мел-
ким. Крупные зерна или куски порошка остаются неразложив-
шимися под действием серной кислоты, а под действием темпе-
ратуры идут процессы его разложения с образованием ацетона
и углекислого кальция. По мере отгонки уксусной кислоты ре-
акционная масса густеет. В этот период возможно затвердение
266
всей массы (как следствие гидратации гипса). Гидратации пре-
пятствует серная кислота, поэтому она всегда берется в из-
бытке по отношению к теоретически необходимой массе. Избы-
ток серной кислоты должен быть больше, чем больше в по-
рошке смолистых веществ и гомологов уксусной кислоты.
Отгонка уксусной кислоты протекает неравномерно. Вна-
чале кислота отгоняется быстро, массовая доля кислоты в дис-
Рис. 77. Принципиальная технологическая схема разложения уксусно-каль-
циевого порошка:
/ — аппарат Линде (РПД); 2, 7, 14, 15, 21, 22 —сборники; 3, /7 — ректификационные ко-
лонны; 4, 16 — конденсаторы-холодильники; 5, 9, 11 — циклоны; 6, 19 — холодильники;
8, 10 — абсорберы; 12, 13 — нейтрализаторы; 18 — куб-испаритель; 20 — насосы
тилляте достигает 85 %, под конец отгонка протекает очень
медленно, а концентрация кислоты падает до 30—35%. Пары
уксусной кислоты конденсируются и частично охлаждаются
в конденсаторе-холодильнике 4 (см. рис. 77), кислота-сырец по-
ступает в сборник 22. Разложение порошка ведут под вакуумом.
В аппарате поддерживают давление около 92,5-10—95,7-103Па,
при этих условиях легче и полнее извлекается уксусная кислота
из реакционной массы, повышается производительность аппа-
рата.
Вместе с газами из сборника 22 в вакуумную систему по
воздушным линиям увлекаются и пары уксусной кислоты, кото-
рая улавливается в системе циклонов и абсорберов. Сернистый
газ, плохо растворимый в воде, из абсорбера 10 после цик-
лона И поступают в нейтрализатор 13, тдр улавливается ра-
267
створом каустической соды. При достижении концентрации
22,5—25,5 % раствор бисульфита натрия выводится из системы
и реализуется как товарный продукт. Связывание сернистого
газа осуществляется по реакциям
SO2+2NaOH = Na2SO3 + Н2О; (104)
Н2О+Na2O3+SO2 = 2NaHSO3. (105)
Уксусная кислота из сборника 22 поступает в ректификаци-
онную колонну 17 для отгонки SO2, который возвращается
в производство бисульфита натрия, а очищенная кислота-сырец
отбирается из куба 18 через холодильник 19 и направляется па
дальнейшую переработку. Колонна 17 имеет 10—12 тарелок
колпачкового типа. Температура в верху колонны 50—60 °C.
Массовая доля уксусной кислоты в кислоте-сырце зависит
от концентрации серной кислоты, от влажности порошка и ко-
леблется в пределах 60—75 %. В неочищенной кислоте-сырце
присутствует до 1,5% легких смоляных масел, альдегидов, ке-
тонов и прочих органических веществ, до 0,6 % сернистой кис-
лоты, до 0,1 % серной кислоты. Массовая доля карбоновых
кислот — гомологов уксусной кислоты в кислоте-сырце может
быть до 8 % (в том числе муравьиной кислоты до 3 %).
Полученный при разложении порошка гипс в зависимости от
вида перерабатываемого порошка загрязнен многими вещест-
вами, в промышленности его называют окшарой. В составе ок-
шары 60—65 % сульфата кальция (безводного), 12—15 % воды
(гидратной и свободной), 10—12 % серной кислоты, 15 % ук-
сусной кислоты и ее гомологов, около 0,5—1,5% неразложив-
шегося порошка, 4—7 % смолистых веществ, 4—6 % посторон-
них механических примесей (землистое вещество, уголь и пр.).
Выход окшары составляет до 2500 кг на 1 т уксусной кислоты-
сырца (в пересчете на 100%-ную кислоту).
Окшара является многотоннажным отходом производства,
пока не находит сбыта и вывозится в отвал. Предлагались
различные способы ее переработки. Были предложения о полу-
чении на ее основе алебастра, экстрихгипса, серной кислоты,
цемента, сульфата аммония и др.
В ГДР и ФРГ окшару в гранулированном виде используют
главным образом в качестве удобрения. Ее смешивают с из-
мельченным шлаком, золой, известью или фосфитной мукой,
смачивают 5—7%-ным раствором аммиака, гранулируют при
влажности до 10 % и сушат до массовой доли воды в гранулах
2-3 %.
У нас в стране еще в 1923—1924 гг. были проведены испы-
тания по использованию нейтрализованной окшары в качестве
удобрения для зерновых культур. Результаты были положи-
тельными. В последнее время в ЦНИЛХИ совместно с Сиб-
НИИСхозом ведутся расширенные поиски использования ок-
шары в качестве мелиоранта для солонцовых почв.
268
§ 44. ТЕХНОЛОГИЯ ПЕРЕРАБОТКИ ЧЕРНОЙ КИСЛОТЫ
С ЦЕЛЬЮ ПОЛУЧЕНИЯ ТЕХНИЧЕСКОЙ УКСУСНОЙ КИСЛОТЫ
Черная кислота, полученная по различным технологическим
схемам, значительно различается по содержанию карбоновых
кислот, воды и смолистых веществ (табл. 40). Наибольшую
долю воды (до 35 %) содержит черная кислота, полученная
методом азеотропной ректификации с использованием в каче-
стве антренера древесных масел, а наибольшую долю смоли-
стых веществ — кислота, полученная методом экстракции кис-
лоты из необессмоленной и необесспиртованной жижки. Черная
кислота является сырьем для производства технической уксус-
ной кислоты, которая затем почти в полном объеме (до 80%)
перерабатывается в пищевую уксусную кислоту — дефицитный
продукт широкого потребления.
Извлекают уксусную кислоту из черной методом ректифи-
кации. Черная кислота в зависимости от состава перерабаты-
вается на непрерывнодействующих чернокислотных аппаратах
по различным технологическим схемам. Она представляет со-
бой весьма сложную многокомпонентную систему. Организация
процесса ректификации таких смесей требует глубоких и пол-
ный знаний свойств системы (табл. 41) и ее составляющих,
прежде всего фазовых равновесий жидкость—пар.
Технические схемы ректификации черной кислоты на различ-
ных предприятиях отличаются друг от друга. Универсальной^
позволяющей получать максимальный выход технической кис-
лоты первого сорта, следует считать схему, изображенную на
рис. 78.
Черная кислота при температуре 105—110 °C из сборника 1
непрерывно подается в ректификационную колонну 5. Колонна 5
имеет 25 колпачковых тарелок (в том числе в укрепляющей
части 15). Температура в верхней части 104—110 °C (в зависи-
мости от массовой доли воды в черной кислоте), в нижней
160±2 °C. В этой колонне происходит отделение смолистых
веществ от уксусной кислоты. Процесс ведут при флегмовом
числе 1. Головная фракция этой колонны содержит карбоновые
кислоты Ci—С4, воду, органические примеси с температурой
кипения до 120 °C и высококипящие компоненты, образующие
положительные азеотропы с водой. Массовая доля всех органи-
ческих веществ, кроме карбоновых кислот, колеблется в преде-
лах 3—7 %. Кубовой продукт колонны 5 содержит смолы и
до 30 % кислот. Дистиллят колонны 5 направляют на ректифи-
кацию в колонну 7 с целью отгонки воды, масел и муравьиной
кислоты. Колонна 7 имеет 45 тарелок колпачкового типа (в том
числе в укрепляющей части 25). Процесс ведут при флегмовом
числе (по водному слою) 9—12; температуру вверху колонны
поддерживают 101—104 °C, внизу каландрии 119—122 °C. Кон-
центрация кислот в дистилляте колонны 7 не должна превышать
20%. В этих условиях дистиллят ограниченно растворим
269
to 40. СОСТАВ ЧЕРНОЙ КИСЛОТЫ, ПОЛУЧЕННОЙ ПО РАЗЛИЧНЫМ ТЕХНОЛОГИ-
О ЧЕСКИМ СХЕМАМ
Способ переработки жижки ПЛОТНОСТЬ при 20 °C» кг/м3 Общая кислот- ность (в пере- счете на уксус- ную), % Эфирное число, мг КОН/г Массовая доля, % Массовая доля индивидуальн ых кислот, %
воды смолис- тых ве- ществ муравьи- ной уксус- ной пропио- новой масляной и изомас- ляной
Азеотропный (с использованием древесных масел) 1050—1060 45—55 61—68 32—35 18—22 0,8—1,7 40—42 2,5—3,0 0,4—0,7
Экстракционный: из необессмоленной и необес- спиртованпой жижки экстраген- том переменного состава 1125—1130 45—60 150—165* 8—20 16—30 1-1,5 42—50 2—4 2-3
из обесспиртованной и обессмо- ленной жижки с применением этил ацетата 1065—1070 60—70 32—40 6—10 6—9 0,5—1,5 56—64 5-6 1,5-2,5
из обесспиртованной, не пол- ностью обессмоленной (50 %) жижки с использованием этил- ацетата 1075—1082 58—63 35—38 5—8 17—23 0,5-1,0 52-57 4—5 1,5-3,0
♦Массовая доля этилацетата при этом 0,5 — 0,8 %.
41. АЗЕОТРОПНЫЕ СВОЙСТВА СИСТЕМ, КОМПОНЕНТЫ КОТОРЫХ ПРИСУТ-
СТВУЮТ В ЧЕРНОЙ КИСЛОТЕ
Компонент Температура ки- пения» °C Массовая доля в азеотропной смеси» %
А Б компо- нента Б азеотроп- ной смеси компе А шента Б
Вода Вода Вода Вода Вода Вода Вода Вода Вода Вода Вода Вода Вода Вода Кислота: муравьиная уксусная пропионовая масляная изомасляная валериановая изовалериановая Фурфурол Фенол Ацетон Метилэтилкетон Диэтилкетон Диацетил Пиридин 100,7 118,1 141,1 163,5 154,4 186,4 176,5 161,5 181,75 56,2 79,6 101,7 ' 87,5 115,6 107,6 Неазео' 99,2 99,6 98,8 99,8 99,5 97,5 99,6 Неазео1 73,5 82,9 78,5 92,6 22,3 гропна 83,6 82,5 71,8 89,0 81,6 65 90,8 тропна 36,9 56,1 <10 43 77,7 16,4 17,5 28,2 11,0 18,4 . 35 9,2 63,1 43,9 >90 57
Вода Кислота: муравьиная уксусная 100,7 118,1 107,1 39,3 48,2 12,5
Вода Кислота: муравьиная пропионовая 100,7 141,1 107,20 37,9 57,4 4,7
Вода Кислота: муравьиная изомасляная 100,7 163,5 | 107,62 38,8 59,3 1,9
Вода Кислота: муравьиная изомасляная 100,7 154,4 } 107,02 34,2 57,8 9,0
Вода Этанол Этилацетат 78,3 77,15 | 70,3 7,8 9,0 83,2
Вода Пропанол Пропилацетат 97,2 101,6 } 82,2 21,0 19,5 59,5
Вода Бутанол Бутилацетат 117,75 126,2 J 89,4 37,3 27,4 35,3
Вода Бутанол . Бутилформиат 117,75 106,8 }вЗ,6 21,3 10,0 68,7
271
Продолжение
Компонент Температура ки- пения» °C Массовая доля в азеотропной смеси» %
А Б компо- нента Б азеотроп- ной смеси компонента
А Б
Вода Этанол 78,3 78,15 4,4 95,6
Вода Пропанол 97,26 87,4 18,45 81,55
Вода Бутанол 117,75 92,4 38,0 62,0
Вода Этил ацетат 77,15 70,4 8,2 91,8
Вода Пропилацетат 101,6 82,76 16,2 83,8
Вода Бутилацетат 126,2 90,2 28,7 71,3
в воде, т. е. представляет собой гетерогенную смесь. После раз-
деления во флорентине 9 водный слой частично выводится из
системы, а частично возвращается в колонну, Органический
верхний слой (масла) полностью выводится из системы.
Из каландрии 10 обезвоженная уксусная кислота поступает
на следующую стадию переработки — в колонну 11 для выделе-
ния из нее технической уксусной кислоты и фракции высококи-
пящих гомологов уксусной кислоты. Колонна 11 имеет 30—
35 колпачковых тарелок (в укрепляющей части 15—18). Про-
цесс ведут при флегмовом числе 1,0—1,2. Температура вверху
колонны поддерживается 116—118 °C, в нижней части 153—
Рис. 78. Принципиальная технологическая схема переработки черной кис-
лоты с целью получения технической уксусной кислоты:
1 — сборник; 2 — насос; 3 — подогреватель; 4, 10, 13,16 — каландрии; 5, 7, 11, 14 — рек-
тификационные колонны; 6, 8, 12, 15 — конденсаторы-холодильники; 9 — флорентина
272
159 °C. Сверху колонны 11 отбирают техническую уксусную
кислоту с массовой долей органических соединений карбониль-
ного характера (в пересчете на муравьиную кислоту) 0,2—
0,3 %, воды не более 1,5%.
Смолистый остаток из каландрии 13 возвращается в чер-
ную кислоту или вместе со смолистым кубовым остатком ко-
лонны 5 (отбор из каландрии 4) подается на пятую тарелку
колонны 14 для более полного выделения кислоты. Колонна 14
имеет 10 тарелок колпачкового типа, работает без флегмы, под
вакуумом 48—50 кПа. Температура вверху колонны 138—
142 °C, в нижней части каландрии 16 162—164 °C. Дистиллят
колонны 14 возвращается в черную кислоту. В смоле, отбирае-
мой из каландрии 16, остаточная массовая доля кислот не дол-
жна быть более 10 % (доля уксусной кислоты в ней 20—30 %).
Четырехколонная схема ректификации рекомендуется для
переработки черной кислоты, содержащей значительную долю
смолистых веществ и воды. При переработке черной кислоты
с массовой долей смолистых веществ менее 10 % можно исклю-
чить из технологического процесса стадию обессмоливания (т. е.
исключить колонну 5).
В табл. 42 приведено распределение уксусной кислоты по
технологическим потокам. Энергетические затраты на перера-
42. РАСПРЕДЕЛЕНИЕ УКСУСНОЙ КИСЛОТЫ, %, ПО ТЕХНОЛОГИЧЕСКИМ. ПО-
ТОКАМ
Статья материального баланса кислоты и технологический поток Экстракция уксусной кислоты Азеотропное укрепление древесно- спиртовыми маслами
из кислой воды этила- цетатом из смеси кислой воды и жижки (50 %) этил ацетатом из жижки сложным экстрагентом переменного состава
Поступило КИСЛОТЫ С ЖИЖ- КОЙ в химический цех на переработку 100 100 100 100
Отобрано на стадии обес- спиртовывания со спир- том-сырцом 0,2—0,5 0,4-0,6 — 0,3—0,7
Поступило кислоты на экстракцию или азео- тропное укрепление 88—90 90—92 100 89—90
Получено кислот с тех- нической уксусной кис- лотой (выход кислот) 72—74 69—72 67—70 69—72
Потери кислот со сточной водой 2—4 5-6 4—5 2-4 •
Отобрано кислот со смо- ляными продуктами (сум- марно), % 9—10 12—13 14—17 11—12
Потери кислот* 6—9 7—9 9—11 6—8
* Кроме технологических потерь учитываются ошибки расчетов, связанных с
определением группового состава на условный продукт (уксусную кислоту).
273
ботку I т жижки зависят от технологической схемы получения
технической уксусной кислоты. При переработке 1 т необессмо-
ленной и необесспиртованной жижки кислотностью 8—10 % за-
трачивается около 1,6 ГДж пара, 2,2 кВт-ч электроэнергии,
25—30 м3 технической воды. При получении технической кис-
лоты из обессмоленной и обесспиртованной жижки затраты на
переработку 1 т жижки составляют: по пару около 2 ГДж,
электроэнергии 4,7—5 кВт-ч, воды 35—40 м3. Наиболее энерго-
емким является азеотропный способ извлечения кислоты из
жижки. Расход пара на переработку 1 т жижки в этом случае
составляет около 2,8 ГДж, электроэнергии около 1 кВт-ч, воды
50—60 м3.
§ 45. ТЕХНОЛОГИЯ ПРОИЗВОДСТВА ПИЩЕВОИ
УКСУСНОЙ кислоты
Сырьем для производства пищевой уксусной кислоты явля-
ется кислота-сырец, полученная при разложении уксусно-каль-
циевых порошков, и техническая уксусная кислота. Эти два
вида сырья перерабатываются по различным технологическим
схемам.
Кислоту-сырец с массовой долей основного вещества 60—
85%, освобожденную от сернистого газа, подвергают ректифи-
кации в периодически действующей колонке колпачкового типа
с 28—30 тарелками. Кислоту-сырец разгоняют на четыре фрак-
ции. Первую фракцию отбирают при флегмовом числе 10—15 до
прекращения расслаивания масел. Эта фракция содержит в ос-
новном соединения некислотного характера, образующие с во-
дой гетероазеотропы. Выход фракции 2—6 %. Затем отбирают
слабую кислоту при флегмовом числе 3-*-5. С этой фракцией
отгоняют воду и основную долю муравьийой кислоты (до кре-
пости уксусной кислоты в кубе 88—90 %). Первая фракция
утилизируется методом сжигания или вместе со слабой кисло-
той используется в производстве эфиров (БЭФ). Выход второй
фракции определяется качеством сырья и составляет около
30%. Третью фракцию, так называемую Переходную, отбирают
при флегмовом числе 2—3 до массовой доли в кубовой жидко-
сти органических веществ в пересчете на муравьиную кислоту
не более 0,8 % (проба из куба не должна давать опалесценции
при смешении с водой). Выход этой фракции 2—3 %. По мере
накопления ее возвращают на ректификацию. Четвертая фрак-
ция— полуфабрикат пищевой кислоты. Отбор ее ведут при
флегмовом числе 1—2 в начале отгонки и без флегмы в конце
дистилляции. Выход полуфабриката из кислоты-сырца не пре-
вышает 70 %.
Для более глубокой очистки полуфабриката от муравьиной
кислоты и веществ с карбонильной группой его обрабатывают
при температуре 132—135 °C контактной ёмесью, состоящей из
30—40 % серной кислоты, 45—55 % уксусной, 10—15 % воды.
274
С этой целью полуфабрикат из ректификационной колонны
подают в испаритель, откуда пары поступают в контактный
аппарат, проходят через слой контактной смеси и направляются
в конденсатор-холодильник. Концентрация муравьиной кислоты
после контактного аппарата снижается в 2—2,5 раза. Продукты
конденсации, полимеризации и окисления постоянно накаплива-
ются в контактной жидкости. Поэтому после пропускания через
нее 60—80 объемов кислоты смесь заменяют. Отработанная кон-
тактная смесь используется для разложения уксусно-кальцие-
вых порошков.
Дистиллят паров, выходящих из контактного аппарата, да-
лее обрабатывают раствором перманганата калия в уксусной
кислоте. Процесс окисления ведут в аппарате с мешалкой в те-
чение 1—2 ч. На одну загрузку перманганата калия делают
три загрузки кислоты. Обработанную перманганатом калия кис-
лоту перекачивают в куб-дистиллятор, где происходит отгонка
пищевой уксусной кислоты от солей тяжелых металлов и окис-
лов марганца. Расход перманганата калия составляет 7—9 кг на
1 т пищевой кислоты. На последней стадии получения пищевой
уксусной кислоты необходимо использовать коррозионноустой-
чивое оборудование, так как в пищевой кислоте допускается
присутствие меди не более 0,0005 %, железа не более 0,001 %,
солей тяжелых металлов не более 0,0005 %. Конденсатор-холо-
дильник к кубу-дистиллятору изготавливают, как правило, из
серебра или титана. Емкости для готового продукта используют
эмалированные. Обычно на стадии дистилляции получают 90—
98 %-ную уксусную кислоту. Ее разбавляют до требуемой кон-
центрации дистиллированной водой. Выход пищевой кислоты от
загрузки кислоты-сырца составляет около 65 %.
При окислении перманганатом калия протекают реакции:
5НСООН + 2КМпО4+6СН3СООН = 5СО2 + 8Н2О+
+ 2СН3СООК+2 (СН3СОО)2 Мп; (106)
5RCHO+2КМпО4 + 6СН3СООН = 5RCOOH + 2СН3СООК +
+ 2 (СН3СОО) Мп + ЗН2О. (107)
Кубовые остатки из окислителя после трех-четырех операций
выгружают и перерабатывают вместе с уксусно-кальциевыми
порошками на стадии разложения.
Пищевая уксусная кислота выпускается с массовой долей
основного вещества 80, 70 и 30%. Техническая уксусная ки-
слота, полученная экстракционным или азеотропным способом,
почти полностью перерабатывается с целью получения пищевой
уксусной кислоты. Качество этого вида сырья значительно выше,
чем кислоты-сырца, поэтому технология ее переработки более
проста. Процесс ректификации ведут в непрерывном режиме
в колонне с 35—40 тарелками колпачкового типа. При флегмо-
вом числе 10 и температуре вверху колонны ПО—114 °C отби-
275
рают головную фракцию, вместе с которой выводятся почти вся
муравьиная кислота и другие вещества карбонильного харак-
тера и окисляющиеся примеси. Выход этой фракции составляет
9—12 %. Она может быть использована для выделки кож, в про-
изводстве растворителя БЭФ или после дополнительной обра-
ботки окислителями возвращена в производство. С 12-й тарелки
из паровой фазы отбирают полуфабрикат пищевой уксусной
кислоты, который не требует обработки контактной смесью.
Остаточные примеси легко могут быть разрушены обработкой
перманганатом калия. Благодаря исследованиям, выполненным
в ЦНИЛХИ, в последние годы разработана технология разло-
жения муравьиной кислоты с помощью перманганата калия при
температуре 60—80 °C в непрерывнодействующем аппарате.
Выход пищевой уксусной кислоты из технической несколько
больше, чем из кислоты-сырца и составляет 82—85 % (в пере-
счете на 100 %-ную кислоту).
§ 46. СТОЧНЫЕ ВОДЫ И СПОСОБЫ ИХ УТИЛИЗАЦИИ.
ОТХОДЫ ПРОИЗВОДСТВА. КОРРОЗИЯ
ОБОРУДОВАНИЯ И РЕКОМЕНДАЦИИ ПО ПРИМЕНЕНИЮ
КОНСТРУКЦИОННЫХ МАТЕРИАЛОВ
В полном цикле производства технической или пищевой
уксусной кислоты образуются большие объемы сточных вод и
производственных отходов. Это определено главным образом
качеством сргрья. При извлечении уксусной кислоты из жижки
на 1 т технической кислоты приходится до 10 т сточных вод, так
как доля воды в сырье составляет 80—85%. При разложении
ускусно-кальциевых порошков на 1 т пищевой кислоты образу-
ется до 2,5 т окшары. Вместе с вопросами организации произ-
водств основного назначения необходимо решать и вопросы пе-
реработки сточных вод, газовых выбросов и технологических от-
ходов.
В лесохимической промышленности используются три ме-
тода обезвреживания или переработки сточных вод: сжигание,
биохимическая (или биологическая) очистка и очистка в пру-
дах-накопителях. Выбор способа определяется загрязненностью
сточной воды и ее объемами. Загрязненность стоков оценивают
по показателям ХПК (масса кислорода, необходимая для хими-
ческого окисления органики; этим методом определяют общую
или суммарную загрязненность органическими веществами) и
БПК (масса кислорода, требуемая только для биохимического
разложения органики), в миллиграммах или граммах кисло-
рода на 1 л стоков.
При ХПК более 50 000 мг/л и при больших объемах стока
рекомендуется применять метод сжигания в циклонных печах
или в специальных топках. Этим методом обезвреживаются
сточные воды от производства уксусной кислоты методом экс-
тракции из необесспиртованной и необессмоленной жижки, ма-
276
ела, получаемые при переработке черной кислоты, кислоты-
сырца и др. Стоимость очистки 1 м3 сточных вод этим способом
15—28 р.
Биохимическим (или биологическим) способом обезврежи-
ваются сточные воды, имеющие загрязненность по ХПК не бо-
лее 2000 мг/л. При этом БПК должно быть 1000—1200 мг/л.
Сточные воды, имеющие загрязненность более 2000 мг/л, перед
подачей в аэротенки или биофильтры разбавляют очищенной
водой до установленных норм загрязненности. Биохимическая
очистка основана на биоокислении органических веществ ми-
крофлорой активного ила или биопленки. Этот метод включает
четыре стадии очистки: механическую (удаление грязи, всплы-
вных слоев), физико-химическую (нейтрализация кислот до pH
стоков перед подачей в аэротенки 6,5—8,5), биохимическую и
доочистку (с использованием активированного угля, доаэра-
цией, использованием жизнедеятельности высших водных расте-
ний и т. д.). Этот метод очистки весьма распространен. Им обез-
вреживаются сточные воды после регенерации экстрагента из
эфировод, получаемых в экстракционном и азеотропном спосо-
бах извлечения уксусной кислоты из кислой воды (и из смеси
кислой воды и жижки). Стоимость очистки 1 м3 сточных вод за-
грязненностью по ХПК 2000 мг/л 20—30 к.
Иногда очистку сточных вод ведут в прудах-накопителях,
т. е. в искусственных водоемах. Пруды-накопители выполняют
роль усреднителя с естественными процессами окисления орга-
нических веществ за счет контакта с кислородом воздуха на по-
верхности воды. Их можно рекомендовать для очистки сточных
вод малой загрязненности по БПК.
Объемы газовых выбросов в производстве уксусной кислоты
незначительны. Вопросы их очистки решаются совместно с тех-
нологическими. Создание для этих целей специальных стацио-
нарных установок не требуется.
Среды уксусно-кислотного производства на всех технологи-
ческих стадиях весьма агрессивны в отношении коррозии обо-
рудования. Коррозия металлов усиливается под действем тем-
ператур, в присутствии муравьиной и серной кислот. Особенно
неблагоприятно влияет кислород воздуха. Именно поэтому ре-
комендуется вести процессы в непрерывном режиме и эксплуа-
тация оборудования с нарушенной герметизацией недопустима.
Коррозия резко усиливается при неправильно выполненных
монтажно-сборочных работах. Нельзя, например, допускать кон-
такта разнородных металлов.
Медь в средах уксусно-кислотного производства стойка
только при полном исключении контакта с кислородом воздуха,
т. е. под вакуумом. Ее сплавы с оловом, цинком, алюминием
стойки в средах этого производства лишь до температуры 50 °C
и используются для изготовления вентилей, кранов, задвижек.
В средах, содержащих до 10 % уксусной кислоты, при тем-
пературе до 50 °C хорошую стойкость имеет сталь марки
277
12Х18Н10Т. При повышенных температурах и в средах, где
массовая доля муравьиной кислоты более 3%, оборудование
должно быть изготовлено из стали марок 10Х17Н13М2Т
ОХ23Н28МЗДЗТ (06ХН28МДТ) или титана марок ВТ1-1, ВТ 1-0.
В средах, содержащих серную кислоту, стойки кислотостойкие
эмали и стали марки 0Х23Н28МЗДЗТ.
Из прокладочных материалов рекомендуются паронит марки
ПОН (до 40 °C), кислотостойкий паронит ПК и ПМБ-1 при тем-
пературах до 160 °C. В последнее время хорошо зарекомендо-
вали себя спирально-навитые прокладки СНП. В качестве на-
бивочных материалов используют фторопластовый уплотнитель-
ный материал (ФУМ), асбестовый, пропитанный фторопластовой
эмульсией с тальком (АФТ) и асбестовый, пропитанный гра-
фитом (АГ).
Глава 13
ПРОИЗВОДСТВО АЦЕТАТНЫХ РАСТВОРИТЕЛЕЙ
Основными представителями сложных эфиров уксусной ки-
слоты и низкомолекулярных спиртов — ацетатных растворите-
лей, которые вырабатываются в лесохимической промышленно-
сти в значительных объемах, являются этил- и бутилацетат.
Эфиры уксусной кислоты — активные растворители и. широко
используются в народном хозяйстве или как таковые, или в ком-
позиционных составах. Основными потребителями этил- и бу-
тилацетата являются лакокрасочная промышленность, кожевен-
но-обувная, текстильная, радиоламповая, электротехническая,
оборонная, полиграфическая и медицинская.
Впервые крупнотоннажное производство этил- и бутилаце-
тата было освоено лесохимической промышленностью. До 50-х
годов для производства эфиров использовали исключительно ле-
сохимическую ускусную кислоту. В настоящее время на лесо-
химических заводах доля синтетической уксусной кислоты,
используемой для производства ацетатных растворителей, со-
ставляет 90—95 %.
Около 70 % бутилацетата вырабатывается в лесохимической
промышленности, 30 % — в химической. Весь бутилацетат полу-
чают методом этерификации уксусной кислоты бутанолом.
Этим же способом получают бутилацетат и за рубежом.
Выпуск этилацетата, получаемого в лесохимической про-
мышленности, составляет около 60 %, около 40 % вырабатывает
химическая промышленность. До 60-х годов весь этилаце-
тат так же, как и бутилацетат, получали способом этерифи-
кации. В начале 60-х годов организовано производство этила-
цетата методом конденсации ацетальдегида (метод В. Е. Ти-
щенко). Сегодня этим способом производят до 25 % этилацетата.
За рубежом его также получают или способом этерификации,
278
или методом конденсации ацетальдегида. Образование слож-
ных эфиров методом этерификации протекает по реакции
RCOOH + R'OH=^RCOOR' + H2O. (108)
Реакцию этерификации обычно проводят в присутствии раз-
личных катализаторов, в паровой или жидкой фазе, в избытке
спирта или кислоты. Синтез сложных эфиров из альдегидов осу-
ществляется по реакции
2RCHO^RCOOCH2R. (109)
В качестве катализатора используют алкоголят алюминия,
активированный хлоридом железа или хлоридом алюминия и
окисью цинка. Метод нашел применение для получения этила-
цетата. Процесс в данном случае протекает при температуре от
—10 °C до +10 °C. Этилат алюминия загружают в реактор
в виде раствора в этилацетате.
Для получения сложных эфиров может быть рекомендован
метод переэтерификации. Различают два вида этой реакции —
реакция обмена кислотными радикалами:
RCOOR' + R"COOH R"COOR' + RCOOH (110)
и реакция обмена спиртовыми радикалами (реакция алкого-
лиза).
RCOOR' + R"OH=₽± RCOOR" + R'OH. (Ill)
\
Обе реакции катализируютсй кислотами.
Одним из способов получения сложных эфиров может быть
способ дегидрогенизации (дегидрирования) спиртов. Принци-
пиально процесс протекает в две стадии:
RCH2OH~^RCHO (реакция Сабатье); (112)
2RCHO->RCOOCH2R (реакция Тищенко). (113)
Механизм сложноэфирной конденсации спиртов очень тру-
ден, до сих пор остается до конца не выясненным, и многие ис-
следователи для объяснения предлагают различные схемы. Хо-
рошими катализаторами этой реакции являются медно-торие-
вый и медно-урановый. Селективность катализаторов зависит от
температуры. Например, превращение этанола при 180—200 °C
идет до альдегида, при 200—250 °C до этилацетата, выше
250 °C — до кетонов.
§ 47. ТЕОРЕТИЧЕСКИЕ ОСНОВЫ И МЕХАНИЗМ
РЕАКЦИИ ЭТЕРИФИКАЦИИ
Кинетические законы реакции этерификации кислот спир-
тами являются общими для реакционных систем при наличии
специфичных особенностей. Реакция этерификации является
279
классическим примером обратимой реакции второго порядка.
Рассмотрим общий случай обратимой реакции вида
A+B^C+D. (114)
При смешении компонентов А и В начинается их немедленное
взаимодействие с образованием компонентов С и D, И, наобо-
Рис. 79. Изменение концентра-
ции реагентов и продуктов ре-
акции этерификации в зависи-
мости от времени:
1 — реагенты; 2 —про-
дукты реакции
рот, при смешении компонентов С и D немедленно начнется ре-
акция гидролиза эфира с образованием кислоты и спирта.
Реакция этерификации прямая, реакция гидролиза обратная.
Скорость прямой реакции (максимальная в начальный период)
по мере ее протекания уменьшается, а скорость обратной в на-
чальный момент равна нулю, а затем возрастает до тех пор,
пока скорости обеих реакций не сравняются.
Под кинетикой реакции понимают зависимость скорости дан-
ной реакции от различных параметров: концентрации веществ,
температуры, давления и др.
Под скоростью реакции понимают число актов реакции, про-
исходящих в единицу времени в единице объема фазы при гомо-
генной реакции или на единице поверхности раздела фаз (или
в единице массы) при гетерогенной реакции.
На рис. 79 показана характерная зависимость изменения
концентрации реагирующих веществ от времени. Для реакции
этерификации скорость прямой реакции выражается зависи-
мостью
W1 = K1CACB, (П5)
где Wi — скорость прямой реакции; Ki — константа скорости
прямой реакции; СА и Св — концентрация кислоты и спирта.
Скорость обратной реакции зависит от концентрации обра-
зовавшихся веществ:
TT2 = K2CcCD, (116)
где W2— скорость обратной реакции; Кг— константа скорости
обратной реакции; Сс и Со— концентрация эфира и воды.
280
Скорости реакций в данный момент времени определяются
для прямой реакции
dCA.B __ v (а — х)(Ь — х) _
------------------у*------• (117)
для обратной реакции
dCct р ___ (с —|— -х) (tf —|— х) у 11 qv
dt 2 V2 ' '
где х—число молей прореагировавшей кислоты (равно числу
молей прореагировавшего спирта); V — объем смеси, л; а, Ь, с
и d — соответственно число молей кислоты, спирта, эфира и
воды в начальный момент.
Общая скорость реакции этерификации равна 1Г=1Г1—1Г2
(Н9).
dx _ ЛСа, в dCc,p _ „ (а — х)(Ь — х) ______„ (с — x)(d — х)
dt dt dt 1 V2 2 ya *
(120)
При достижении химического равновесия ITi = IF2.
При повышении температуры скорость химической реакции
возрастает. Считается, что с увеличением температуры на 10 °C
скорость реакции увеличивается в 2—3 раза.
При изучении кинетики химических-реакций, как правило,
исследуют влияние различных факторов не на скорость реакции,
а на ее константу К. Физический смысл константы скорости ре-
акции заключается в численном равенстве ее скорости химиче-
ской реакции при условии равенства в начальный момент кон-
центрации (или произведения концентрации). реагирующих ве-
ществ единице.
Размерность константы скорости меняется с порядком реак-
ции. Под ним подразумевается сумма степеней при концентра-
циях тех компонентов, которые входят в кинетическое уравне-
ние. Например, W—KCA. Это реакция первого порядка. Размер-
ность константы скорости реакции с-1. Реакции второго порядка
имеют кинетические уравнения вида W=KCACB, W=K.CA2. Раз-
мерность К в этом случае л/(моль-с). И так далее. Могут быть
реакции с дробным порядком. Иногда целесообразно отмечать
порядок реакции только по какому-то одному веществу. Тогда
это условие оговаривается: реакция имеет такой-то порядок по
такому-то компоненту. Нет смысла сравнивать константы реак-
ций разных порядков.
Для очень большого числа реакций значение константы ско-
рости определяется прежде всего выбранным катализатором
(табл. 43). Роль катализаторов и состоит в увеличении' скоро-
сти химической реакции, а значит, и ее константы. Во всех опы-
тах катализатора взято 1 % от массы 100%-ной уксусной кис-
лоты, соотношение реагентов 1:1 (в молях).
281
43. ЗНАЧЕНИЯ КОНСТАНТ СКОРОСТИ РЕАКЦИИ атЕРИФЙКАЦИИ УКСУСНОЙ
КИСЛОТЫ ЭТИЛОВЫМ СПИРТОМ ПРИ 70 °C С РАЗЛИЧНЫМИ КАТАЛИЗАТОРАМИ
Катализатор Константа скорости реакции К» моль/(л»мин) Катализатор Константа скорости реакции К» моль/(л»мин)
Серная кислота Катиониты: КУ-23 (10/60) КУ-2-8 3,78-10“» 1,83-10“® 1,54-10-з КБ-41 К (10/60) КУ-23 (20/100) Фосфорная кис- лота КФ-П 1,4Ы0*3 1,26-1О"3 1,02-10-* 7,68-10-5
44. ЗАВИСИМОСТЬ КОНСТАНТЫ СКОРОСТИ РЕАКЦИИ ОТ КОНЦЕНТРАЦИИ КА-
ТАЛИЗАТОРА
Концентрация катализатора в зоне реакции» мг»экв/л Константа скорости реакции К» л/(моль*мин) Концентрация катализатора в зоне реакции» мг-экв/л Константа скорости реакции К» л/(моль-мин)
Катализатор Серная кислота Катализатор КУ-2-8 (формованный)
58 4,57-10“® 87 6,56-10“®
87 6,49-10“® 116 7,51-10“4
116 8,38-10“® 146 9,00-10“4
146 10,46-10“® 175 1,12-10“»
Катализатор КУ-2-8 (мелкозернистый)
116 9,35-10“4 233 1,31-10“»
175 1,25-10“® 291 1,65-10“»
233 1,46-10“® 349 1,88-10“»
407 1,92-10“»
291 1,65-10“» 524 2,42-10-з
Из табл. 43 видно, что максимальная скорость реакции бы-
вает при использовании в качестве катализатора серной ки-
слоты.
Константа скорости реакции зависит не только от природы
катализатора, но и от его концентрации в зоне реакции, хотя и
в меньшей степени (табл. 44).
Константа скорости реакции этерификации, как и всех реак-
ций, зависит от температуры и описывается уравнением Арре-
ниуса:
К = Ле-Е/йГ, (121)
где А — предэкспоненциальный множитель; — газовая посто-
янная [8,3144 Дж/(моль»К)]; Т — температура, К; Е— энергия
активации, кДж/ (моль • К).
Определены константы скорости реакции этерификации не-
которых кислот бутиловым спиртом (без катализатора) при
мольном соотношении реагентов 1:1. При 30 °C для уксусной
кислоты /(=0,0168» 10-4 л/(моль»мин), а при 90 °C K=l,37»
•10-4 л/(моль»мин) (т. е. на 2 порядка выше). Для муравьиной
282
кислоты: при 30 °C К=9,6-10-“ л/(моль-мин), при 70 °C
К=60.9-10-4 л/(моль • мин) • (т. е. на порядок выше).
Существенное влияние на скорость реакции оказывает избы-
ток одного из компонентов против их мольного соотношения.
Если один из реагентов берется в очень большом избытке, то его
концентрация во время реакции практически не изменяется, т. е.
скорость реакции в таком случае будет определяться только
концентрацией одного компонента. Это один из примеров, когда
условия проведения реакции могут изменить ее порядок.
Избыток одного из компонентов всегда увеличивает кон-
станту скорости реакции. Например, при этерификации уксус-
ной кислоты бутиловым спиртом при 70 °C на формованном ка-
тализаторе КУ-2-8 (50 мг-экв на моль кислоты) значение кон-
станты скорости (л/(моль-мин) • 10-3) равно: при мольном из-
бытке спирта 4 и 5 боответственно 1,31 и 1,40, а при мольном
избытке кислоты 2, 3, 4 и 5 соответственно 1,25; 1,58; 1,82 и 1,93.
Эти зависимости показывают, что для реакции этерификации
уксусной кислоты предпочтительнее избыток кислоты, чем
спирта, так как конбтанта скорости реакции, например, при
пятикратном мольном избытке кислоты выше [(1,93 •
• 10-3 л/(моль-мин)], чем при пятикратном мольном избытке
спирта [(1,40• 10~3 л/(моль-мин)].
В условиях равновесия скорость прямой реакции равна ско-
рости обратной реакции, т. е. скорость суммарной реакции
равна нулю.
Константа равновесия реакции Кс обычно выражается урав-
нениями через концентрации реагирующих и образующихся ве-
ществ. Если уравнение этерификации имеет вид аА+ЬВ^сС+
+dD, то
к = [CcF[CD]d
С КмГКв]6 ’
где [Сс]; [Сд], [Сд] и [Св] — концентрация компонентов в момент
равновесия, моль/л.
Или
к __ lCc + x]c [CD^-x]d
[Са-х]а-[Св —х]ь ’
где Сл, Св, Сс и Сд—начальные концентрации соответствую-
щих компонентов, моль/л; х — концентрация образовавшегося
по реакции компонента (воды или эфира), моль/л.
Величина константы равновесия реакции зависит от способа
ее выражения. Обычно константу равновесия выражают через
мольные концентрации, но можно, например, выразить ее через
число молей веществ, участвующих в реакции, или через пар-
циальные давления реагентов и продуктов реакции.
Еще в работах Бертло показано, что константа равновесия
реакции этерификации уксусной кислоты этанолом равна 4. Это
говорит о том, что, если в реакцию этерификации ввести по
283
1 молю кислоты и этанола (100 %-ных), то в момент равновесия
в системе будет по 0,67 моля эфира и воды и по 0,33 моля ки-
слоты и спирта.
Для реакции этерификации уксусной кислоты этанолом или
бутанолом в жидкой фазе в качестве катализатора использу-
ется в основном серная кислота. Соляная кислота также явля-
ется активным катализатором, но она летуча и вызывает силь-
ную коррозию оборудования. Фосфорная кислота обладает
гораздо меньшей коррозионной активностью по сравнению с сер-
ной и соляной, но она менее каталитически активна и дороже
других кислот.
Серная кислота в отличие от соляной и фосфорной наиболее
доступна, дешева и активна, поэтому хотя и имеет ряд сущест-
венных недостатков, получила широкое применение. К недостат-
кам серной кислоты как катализатора следует отнести способ-
ность сульфировать ненасыщенные соединения, вызывать про-
цессы конденсации и полимеризации непредельных органиче-
ских соединений, способствуя тем самым засмолению поверхно-
стей теплообмена реакторов-этерификаторов, и, безусловно, по-
вышенную коррозионную активность.
Хорошими катализаторами реакции этерификации, мало от-
личающимися по каталитической активности, но лишенными
недостатков серной кислоты, являются бензол- и п-толуолсуль-
фокислоты. Но они мало применяются из-за сравнительно боль-
шой стоимости, а главное из-за трудностей, связанных с загруз-
кой их в систему, определяемых свойствами этих кислот
(например, бензолсульфокислота при температуре до 63 °C
представляет собой аморфное твердое вещество, т. е. не обла-
дает текучестью).
С точки зрения снижения коррозии оборудования интерес
представляют или способы этерификации в паровой фазе, или
процессы этерификации в жидкой фазе, комбинирующие аппа-
раты идеального вытеснения (форэтерификация) и идеального
смешения (доэтерификация). И в том и в другом случаях при-
меняются твердые катализаторы, а реакция идет в гетероген-
ной фазе. Парофазная этерификация интересна eiije и тем, что
резко сдвигается равновесие в сторону эфирообразования (кон-
станта равновесия достигает значений до 400—450).
§ 48. ТЕХНОЛОГИЯ ПРОИЗВОДСТВА ЭТИЛАЦЕТАТА
В технологии производства ацетатных растворителей важ-
ное значение имеют процессы ректификации и дистилляции.
Уже на первой стадии (этерификации) для вывода из зоны ре-
акции образовавшихся продуктов используется метод ректифи-
кации,способствующий достижению более полного превращения
исходных компонентов в целевой продукт. Полученный на ста-
дии этерификации эфир-сырец далее перерабатывают с целью
получения товарного эфира также способом ректификации.
284
И, наконец, процессы ректификации лежат в основе технологии
регенерации легколетучих ценных компонентов из водных рас-
творов или кубовых остатков. Правильная организация процес-
сов дистилляции и ректификации во многом определяет тех-
нико-экономические показатели отдельных предприятий, выра-
батывающих этил- и бутилацетат. Оптимальные технологические
режимы могут быть разработаны на основе глубоких знаний не
только химических процессов, но и процессов массо- и теплооб-
мена.
Основными технологическими стадиями производства этил-
и бутилацетата являются: 1. Приготовление этерификационной
смеси. 2. Этерификация уксусной кислоты спиртами с целью по-
лучения эфира-сырца. 3. Обесцвечивание, промывка и нейтрали-
зация эфира-сырца. 4. Ректификация эфира-сырца с целью
получения товарной продукции. 5. Переработка побочных про-
дуктов (кубовых остатков и головных погонов) с целью созда-
ния безотходной технологии. 6. Регенерация легколетучих ком-
понентов из водных растворов. 7: Обезвреживание сточных вод
и газовых выбросов.
Организация каждой из этих стадий производства строго ин-
дивидуальна в зависимости от вида производства, качества то-
варной продукции и перерабатываемого сырья, характеристики
эксплуатируемого оборудования и др.
На всех лесохимических заводах производство этилацетата
организовано по единой принципиальной технологической схеме:
используется метод этерификации уксусной кислоты этанолом
в жидкой фазе, в качестве катализатора применяется серная
кислота, реакцию этерификации ведут в избытке кислоты непре-
рывным способом и т. д.
Однако в результате эксплуатации оборудования различной
характеристики, отличия качества перерабатываемого сырья,
разных требований к качеству готовой продукции на каждом
предприятии технологические режимы каждой стадии производ-
ства имеют свои преимущества, свои недостатки, т. е. строго
индивидуальны.
Приготовление этерификационной смеси. В производстве
этилацетата эта стадия считается обязательной. Смесь гото-
вится в периодически действующих смесителях. Соотношение
реагентов определяется массовой долей этилацетата в товарном
эфире и свойствами системы этанол — вода — этилацетат. Пере-
мешивание смеси до однородности осуществляется насосом по
методу «циркуляция на себя». Смесь считается однородной,
если кислотность в нижней и верхней частях сборника отлича-
ется менее чем на 0,5 %.
В качестве сырья используется уксусная кислота синтетиче-
ская и регенерированная с массовой долей основного вещества
97—99,5%, лесохимическая техническая уксусная кислота кис-
лотностью 93—96 % (доля этого вида кислоты не более 12 % от
общей массы этого сырья), спирт синтетический этиловый собъ-
285
емкой долей алкоголя 92—92,5 % и спирт этиловый гидролиз-
ный с объемной долей алкоголя 94—95%. Емкость баков для
приготовления смеси должна быть достаточной для обеспечения
8—10 ч непрерывной работы установки.
В промышленности эксплуатируются одноколонные аппа-
раты непрерывного действия, где синтез этилацетата осущест-
вляют в избытке уксусной кислоты (рис. 80). В начале цикла
в куб-этерификатор 2 загружают уксусную кислоту, этиловый
Рис. 80. Непрерывнодействующий аппарат этерификации уксусной кислоты
этанолом:
/ — сборник; 2 — куб-этерификатор; 3 — испаритель; 4 — ректификационная колонна; 5 —
конденсатор-холодильник; 6 — холодильник; 7 — флорентина
спирт (20 % от массы теоретически необходимой для этерифи-
кации загруженной кислоты, что обеспечивает в зоне реакции
пятимольный избыток кислоты) и верную кислоту (2—3 % от
массы 100%-ной уксусной кислоты). После разогрева аппарат
30—45 мин работает в режиме полного орошения с целью сни-
жения кислотности дистиллята до 0,05—0,1 %, после чего начи-
нают подачу исходной этерификационной смеси в испаритель 3.
Образовавшиеся в результате реакции этилацетат и вода с при-
месями этанола в виде смеси азеотропов (около 90 % бинарного
азеотропа этилацетат — вода и 10 % тройного азеотропа эта-
нол— этилацетат — вода) непрерывно выводятся из колонны 4.
Температура гетерогенного дистиллята, поступающего во фло-
286
рентину 7 после холодильника 6, должна быть 20—25 дС
с целью более полного использования азеотропных свойств раз-
деляемой системы.
С этой же целью рекомендуется подавать , на флегму в ко-
лонну 4 не расслаивающийся во флорентине 7 эфир-0ырец,
а сухой этилацетат, полученный после азеотропного обезвожи-
вания эфира-сырца. Это позволяет увеличить производитель-
ность этерификатора в 1,7 раза по сравнению с режимом по-
дачи на флегму сырого эфира-сырца.
Высокая степень превращения уксусной кислоты (до 99,5 %)
и спирта (до 95 %) в кубе-этерификаторе достигается за счет
большого избытка в зоне реакции уксусной кислоты и непре-
рывного вывода из зоны реакции продуктов синтеза. Процесс
этерификации в данном случае совмещен с процессом ректифи-
кации.
Ректификационная колонна 4 должна иметь 28—30 практи-
ческих тарелок. Флегмовое число процесса определяется мас-
сой воды, которую надо вывести из системы, и возвращае-
мой в колонну с флегмой. Предположим, что процесс идет
в идеальных условиях и степень превращения этанола достиг-
нута 100%, в таком случае дистиллят представляет собой азео-
троп этилацетат — вода, в котором массовая доля воды 8,2%.
После расслаивания дистиллята около 4 % воды растворяется
в этилацетате и возвращаетця в колонну. Поэтому во флорен-
тине отделяется только около 50 % воды и выводится из зоны
реакции. Если на флегму в колонну 4 подавать эфир-сырец из
флорентины 7, то в зависимости от массовой доли кислоты в ис-
ходной уксусной кислоте, при получении эфира-сырца кре-
постью 93 % рекомендуются следующие флегмовые числа.
Массовая доля кислоты в исходной уксусной кис-
лоте, % ............................. 80 85 90 95
Флегмовое число................... . 6,8 5,8 5,0 4,5—4,0
При меньших флегмовых числах в реакционной зоне накап-
ливается вода, что приводит к снижению степени превращения
реагентов в эфир, т. е. к снижению массовой доли эфира в
эфире-сырце, а это в свою очередь ухудшает азеотропные свой-
ства дистиллята, так как увеличивается растворимость воды
в эфирном слое. Процес становится неуправляемым.
Производительность этерификаторов определяется в основ-
ном качеством исходного сырья, площадью поверхности тепло-
обмена, типом и размерами ректификационной колонны. На
лесохимических заводах получают с 1 м2 сечения колонны
около 600—900 л/ч эфира-сырца крепостью 93—95%, кислот-
ностью 0,05—0,1 %. Температура в верхней части ректифика-
ционной колонны поддерживается 68—70 °C, в кубе-этерифика-
торе 102—104 °C.
Процесс этерификации с отгонкой этилацетата и воды осу-
ществляется непрерывно до тех пор, пока в реакторе не нако-
пятся смолистые вещества или другие продукты конденсации
287
(особенно при использовании синтетического Спйрта или Пере-
работке лесохимической уксусной кислоты), которые оседают на
поверхностях теплообмена змеевика куба-этерификатора и ка-
ландрии, ухудшают теплопередачу и снижают производитель-
ность установки. Длительность непрерывного цикла работы эте-
рификатора составляет 3 мес и более при работе на сырье хо-
рошего качества и 25—40 сут при использовании сырья низкого
качества. При накоплении смолистых примесей цикл работы за-
канчивают, содержимое куба-этерификатора перекачивают на
отработку в специальный аппарат. При отработке в аппарат
подают не этерификационную смесь, а только этиловый спирт
до прекращения процесса образования эфира. Эфир-сырец, по-
лучаемый при отработке, содержит значительно меньше основ-
ного вещества, имеет повышенную кислотность и рекомендуется
для возврата на стадию этерификации.
Стабильность режима этерификации обеспечивается систе-
мами автоматической стабилизации и регулирования некоторых
параметров. Стабилизируются подача этерификационной смеси
в систему и эфира на флегму в колонну. Регулируются давление
в аппарате воздействием на расход пара; температура воды,
отходящей из конденсатора-холодильника, воздействием на ее
расход; уровень в кубе-этерификаторе воздействием отбора
эфира-сырца.
Обесцвечивание, промывка и нейтрализация эфира-сырца.
Эфир-сырец, полученный при переработке лесохимической тех-
нической уксусной кислоты, имеет желто-зеленую окраску, ко-
торая не исчезает после ректификации эфира-сырца. В этих
случаях стадии обесцвечивания и промывки обязательны. Ино-
гда в технологическую схему включают только промывку и
нейтрализацию. Обычно это делают при получении на стадии
этерификации эфира-сырца массовой долей основного вещества
менее 88%. Эта операция увеличивает не только объем сточ-
ных вод, но и расход пара на производство товарной продук-
ции. Нейтрализация эфира-сырца обязательна при переработке
эфира-сырца кислотностью более 0,05 %.
Исследованиями последних лет показано, что цветность эфи-
ра-сырца обусловлена присутствием в нем производных а-дике-
тенов (диацетила и др.) и производных кетонов с функциональ-
ной группой — СН = С = О. Для обесцвечивания эфира-сырца
используют бисульфит натрия. Есть данные о применении для
обесцвечивания эфиров перекиси водорода. После обработки
эфира-сырца бисульфитом, перед его нейтрализацией обяза-
тельна стадия промывки водой, так как плохо промытый обес-
цвеченный эфир вновь на стадии нейтрализации приобретает
желто-зеленую окраску.
На некоторых заводах стадия нейтрализации заменена про-
мывкой водой, что оправдывает себя при переработке эфира-
сырца кислотностью менее 0,1 %. При кислотности эфира 0,1 %
и более для нейтрализации используют 8—12 %-ный раствор
288
кальцинированной соды. На рис. 81 показана (в объеме полной
принципиальной технологической схемы производства этилаце-
тата) схема стадии обесцвечивания, промывки и нейтрализации.
Эфир-сырец из сборника 10 насосом подается в напорный бак,
а из него непрерывно в нижнюю часть бисульфитной колонны
12, заполненной кольцами Рашига и раствором бисульфита на-
трия плотностью 1140—1200 кг/м3. Отработанный бисульфит
натрия через гидравлик выводится в сборник эфироводы 9,
а обесцвеченный эфир-сырец, пройдя через слой бисульфита
натрия, поступает на промывку водой в шаровые смесители 14
и затем во флорентину 16. Эфир-сырец из флорентины непре-
рывно поступает в нижнюю часть нейтрализатора колонного
типа 18. При соотношении потоков содового раствора и эфира
1:10—8 (по массе) происходит нейтрализация эфира-сырца до
остаточной кислотности 0,004—0,008 %.
Азеотропная сушка и ректификация. Сушка этилацетата-
сырца азеотропным способом рекомендуется только в том слу-
чае, если массовая доля в нем эфиров более 87%. При доле
этилацетата в эфире-сырце 83—87 % процесс возможен, но тех-
нико-экономические показатели процесса значительно ухуд-
шатся. Азеотропная сушка невозможна при массовой доле эти-
лацетата менее 83 % из-за гомогенности тройного азеотропа.
В случае использования для синтеза этилацетата лесохимиче-
ской уксусной кислоты процесс азеотропной сушки осложняется
присутствием в системе альдегидов, кетонов, метилформиата,
которые отгоняются в эфир-сырец, ухудшая азеотропные свой-
ства дистиллята. В этих случаях рекомендуется перед азеотроп-
ной сушкой выделить в отдельной ректификационной колонне
с достаточно большим числом тарелок (28—33) все легкокипя-
щие примеси (при температуре вверху колонны 50—64 °C и
флегмовом числе 20—25).
Обесцвеченный и нейтрализованный этилацетат-сырец
с всплываемостью не менее 88 % (при смешении равных объ-
емов эфира-сырца и воды должно отслоиться 88 % эфирного
слоя от начального объема) из сборника 20 (см. рис. 81) непре-
рывно через напорный бак 31 поступает в подогреватель 32 и
затем на 20-ю тарелку колонны 34. Всего в колонне 42—44 та-
релки. Конденсат в виде смеси тройного (этанол — вода — эти-
лацетат) и бинарного (вода — этилацетат) азеотропов из де-
флегматора 35 поступает на охлаждение в холодильник 36, где
охлаждается до 25—30 °C, а затем во флорентину 37. Водный
слой из флорентины направляется в сборник 9 эфироводы,
а эфирный слой разделяется на два потока: основная часть
эфирного слоя возвращается в колонну на флегму, а меньшая
(не более 8 %) выводится в сборник 40 головной фракции и ис-
пользуется для приготовления менее квалифицированных рас-
творителей типа АКР, Э-80. Часть сухого, т. е. обезвоженного
эфира, из испарителя 33 или из нижней части колонны 34 вы-
водится через холодильник 38 в сборник 39 сухого эфира и
Ю Заказ № 2193 289
Рис. 81. Технологическая схема получения товарного этилацетата:
1, 11, 13, 15, 17, 21, 31 — напорные баки; 2, 9, 10, 20, 39, 40, 46 — сборники; 3 — реактор;
4, 24, 33, 42 — испарители; 5, 23, 29, 34, 41, 48 — ректификационные колонны; 6, 25, 28,
35, 43, 49 — дефлегматоры; 7, 26, 27, 30, 36, 38, 44, 45 — холодильники; 8, 16, 19, 37,
50— флорентины; 12 — бисульфитная колонна; 14—шаровые смесители; 18 — колонна
нейтрализации; 22, 32 — подогреватели; 47 — куб-испаригель
далее подается насосом в качестве флегмы в колонну 5 этери-
фикационного аппарата, а часть подается на 10-ю тарелку рек-
тификационной колонны 41. Колонна 41 предназначается для
отделения от этилацетата тяжелокипящих компонентов, в основ-
ном от высокомолекулярных эфиров: этилпропионата, этилбу-
тирата и др. Товарный этилацетат получают в виде дистиллята
колонны 41. Температура вверху колонны 41 74—78 °C, число
тарелок 28—30, флегмовое число процесса 1,5—1,6. Из испари-
теля 42 при температуре 96—100 °C выводится фракция эфи-
ров высших кислот, около 4—5 % от массы товарного этилаце-
тата; С целью более полного извлечения из этой фракции
этилацетата, она дополнительно ректифицируется в колонне пе-
риодического действия 48,
Узел азеотропной сушки и ректификации имеет контуры ре-
гулирования: уровень в испарителях 33 и 42 регулируется в за-
висимости от расхода эфира, давление в этих аппаратах с по-
мощью расхода греющего пара, температура подогрева эфира-
сырца с помощью подачи греющего пара в подогреватель 32,
Состав паров в верхней части испарителя 33 определяется с по-
мощью индикатора состава типа ИС-4 и регулируется расходом
греющего пара в этот аппарат.
По действующей нормативно-технической документации на
предприятиях, страны вырабатывается этилацетат двух марок:
марки Б (массовая доля эфира 91± 1 %, воды не более 0,2%)
и марки А (массовая доля эфира не менее 99 %, воды не более
0,1%).
Регенерация эфира и спирта из эфироводы. Эфировода со
стадии этерификации, азеотропной сушки, отработанный рас-
твор бисульфита натрия, промывные воды и содовые растворы
после нейтрализации содержат до 3% этилацетата и 8—15%
этилового спирта. Регенерация эфира и спирта из эфироводных
растворов обязательна не только из-за экономических сообра-
жений, но и из-за необходимости очистки сточных вод перед их
подачей на биохимическую очистку.
Возможны два варианта схемы регенерации эфира и спирта
из водных растворов: в одноколонном и двухколонном аппа-
рате. Первый вариант рекомендуется для производств неболь-
ших объемов, второй для производства этил ацетата более 14—
16 тыс. т в год. При двухколонной схеме регенерации в первой
колонне отгоняют этилацетат, во второй этиловой спирт.
Все водные растворы принимаются в сборник 9 эфироводы
(см. рис. 81), откуда насосом их закачивают в напорный бак
21. В подогревателе 22 эфироводу подогревают до температуры
10* 291
60—65 °C, а затем подают на 11-ю тарелку эфирной колонны
23 с числом тарелок колпачкового типа 23—25. Вверху дефлег-
матора 28 отбирают этилацетат и частично этанол. Температура
вверху колонны 68—72 °C, флегмовое число 5—6. Вода из
куба или испарителя 24 перетекает в колонну 29 на 15-ю та-
релку. Всего в колонне 23—25 тарелок. Спиртовая фракция от-
бирается при температуре 75—80 °C и флегмовом числе 2—3.
Температура внизу колонны 29 102—103 °C. Часто для отгонки
спирта используют острый пар, целесообразнее вести отгонку
спирта глухим паром с помощью кожухотрубного испарителя
с целью снижения объема сточных вод. Сточная вода перед
сбросом в канализацию охлаждается до 30—40 °C. Спиртовая
фракция крепостью 50—70 % используется на стадии этерифи-
кации, а эфироспиртовая фракция первой колонны регенераци-
онного аппарата крепостью 89—92 % перерабатывается вместе
с эфиром-сырцом. При одноколонной схеме регенерации голов-
ная фракция используется на стадии этерификации.
Переработка хвостовых фракций. Кубовые остатки, полу-
чающиеся после отработки кубов-этерификаторов и при ректи-
фикации сухого этилацетата-сырца, перерабатывают по мере
их накопления отдельно с целью более глубокого извлечения
из них этилацетата. Этилацетатная фракция отбирается при
флегмовом числе 2—3, температуре вверху колонны 68—78 °C.
Затем до 125 °C при флегмовом числе не более 3 отбирают
фракцию, обогащенную этилпропионатом и этилбутиратом. Эту
фракцию используют для приготовления растворителя АКР.
Смолистые остатки после ректификации обезвреживают мето-
дом сжигания.
При использовании 100%-ной уксусной кислоты теоретиче-
ский расход ее на 1 т 91 %-кого эфира равен 620,5 кг, этанола
555,6 кг. Для производства этилацетата с массовой долей ос-
новного вещества 99 % теоретически требуется затратить кис-
лоты 675, этанола 527,5 кг. Коэффициент использования уксус-
ной кислоты 93—95 %, этанола 90—94 %.
Доля сырья в себестоимости этилацетата составляет 85—
88%. Себестоимость этилацетата, полученного по методу
В. Е. Тищенко, ниже полученного методом этерификации, так
как в последнем случае используется менее дешевое сырье.
Себестоимость товарного продукта увеличивается при исполь-
зовании сырья плохого качества, так как при этом возрастают
затраты на переработку рециклов, побочных продуктов и сни-
жается коэффициент использования сырья. Например, коэффи-
циент использования технической лесохимической уксусной ки-
слоты равен 91—92%, а синтетической 94—95%. На производ-
ство 1 т этилацетата марки А расходуется пара 16,7—18,4 ГДж,
воды технической 140—180 м3, электроэнергии 35—44 кВт*ч.
При использовании сырья низкого качества рекомендуется на
предприятиях вырабатывать вместе с этилацетатом другие рас-
творители (например Э-80, АКР, РМЕ, 647).
292
§ 49. ТЕХНОЛОГИЯ ПРОИЗВОДСТВА БУТИЛАЦЕТАТА
Технологии производства бутилацетата и этилацетата
в принципе аналогичны. Основными стадиями производства бу-
тилацетата также являются этерификация, подготовка эфира-
сырца к ректификации, ректификация нейтрализованного бути-
лацетата-сырца с целью получения товарного эфира с массовой
долей бутилацетата (91 ±1) %- (марки Б) или 99 % (марки А)
и регенерация легкокипящих компонентов из водных растворов.
Показатели производства и параметры отдельных техноло-
гических стадий обусловлены физико-химическими свойствами
исходных компонентов и продуктов синтеза. В отличие от си-
стемы этилацетат — этанол — вода взаимная растворимость
в системе бутилацетат—бутанол — вода в сочетании с азео-
тропными свойствами системы бутилацетат — вода и тем более
в системе бутилацетат — бутанол — вода более благоприятны
для отгонки воды из зоны реакции в совмещенном реакционно-
ректификационном процессе на стадии этерификации. В про-
цессе непрерывной этерификации уксусной кислоты этанолом
даже при использовании абсолютно безводных исходных компо-
нентов недопустимо на флегму в колонну этерификационного
аппарата возвращать водный слой, а для увеличения произво-
дительности этерификаторов рекомендуется даже подавать на
флегму сухой этилацетат. Это объясняется тем, что масса реак-
ционной воды при взаимодействии 1 моля кислоты и 1 моля
этанола составляет 17%, а массовая доля воды в азеотропе
этилацетат — вода 8,2%. Из-за взаимной растворимости эти-
лацетата и воды эффективность азеотропного выделения воды
на стадии этерификации снижается еще больше.
Совсем другие закономерности характерны для системы
производства бутилацетата. При полном превращении 1 моля
уксусной кислоты и 1 моля бутанола в продуктах реакции со-
держится около 13,5 % воды, а в азеотропе бутилацетат — вода
28.9%. Растворимость воды в бутилацетате в присутствии до
10% бутанола изменяется незначительно и не превышает 2%.'
Поэтому на стадии этерификации в производстве бутилацетата
в ректификационную колонну с целью получения бутилацетата-
сырца с низкой кислотностью (0,10 % и менее) рекомендуется
возвращать водный слой на флегму, если массовая доля воды
в кислоте менее 32%. Массовая доля воды в тройном азеот-
ропе бутанол — вода — бутилацетат больше, чем в двойном
(37,3 % по сравнению с 28,9%), поэтому его присутствие не
ухудшает, а, наоборот, способствует выводу воды из системы.
Для синтеза бутилацетата используют синтетическую, реге-
нерированную и, очень редко, лесохимическую уксусную ки-
слоту. Бутиловый спирт перерабатывают в синтетический, полу-
ченный методом брожения (очень ограниченные объемы), и
спирт СК, выделенный из отходов производства заводов синте-
тического каучука.
293
сокомолекулярных смолистых
Черный
эфир-сырец
Рис. 82. Аппарат периодического дейст-
вия для этерификации уксусной кислоты
бутанолом:
/, 6, 7 — сборники; 2 — куб-этерификатор; 3 —
ректификационная колонна; 4 — конденсатор-
холодильник; 5 — флорентина
Длительное время для производства бутилацетата в основ-
ном использовали бутанол СК. Это обстоятельство надолго за-
держало перевод процесса этерификации с периодического на
непрерывный, так как спирт СК содержит до 9 % непредельных
соединений (в пересчете на кротиловый спирт), которые под
действием серной кислоты конденсируются с образованием вы-
ств. Эти смолистые вещества
осаждаются на греющих по-
верхностях и ухудшают теп-
лопередачу. Применение
лесохимической уксусной
кислоты, содержащей так-
же смолистые компоненты,
усложняет положение. При
работе на плохом сырье
уже после двух-трех опера-
ций требуется чистка грею-
щих поверхностей кубов-
этерификаторов. В против-
ном случае резко снижается
производительность аппа-
рата. В процессе непрерыв-
ной этерификации происхо-
дит накопление смолистых
компонентов в кубе-этери-
фикаторе, что и вызывает
трудности в ведении техно-
логического процесса и экс-
плуатации оборудования,
делает бессмысленным ре-
жим непрерывности. Неод-
нократные попытки перево-
да основной стадии процес-
са — этерификации на не-
прерывный режим при
работе на сырье плохого
качества оказывались без-
успешными.
В последние годы доля использования бутанола СК снизи-
лась до 20 %. Значительно улучшилось качество регенерирован-
ной и лесохимической уксусной кислоты. Благодаря этому стало
возможным осуществлять процесс этерификации в непрерывном
режиме. Однако большинство предприятий, вырабатывающих
бутилацетат, осуществляют процесс этерификации в аппаратах
периодического действия.
Периодический процесс этерификации уксусной кислоты бу-
танолом. Периодический способ этерификации уксусной ки-
слоты бутанолом заключается в следующем (рис. 82). В куб-эте-
рификатор 2 с помощью насоса загружают уксусную кислоту и
294
бутанол. Одновременно йз напорного бака дозируют катализа-
тор — серную кислоту из расчета 0,5 % от массы уксусной ки-
слоты. Целесообразно серную кислоту загружать по одной за^
грузочной линии с уксусной кислотой. Первоначальную за-
грузку реагентов производят с таким расчетом, чтобы масса
жидкости в кубе-этерификаторе 2 составляла 80 % от его гео-
метрического объема. Загрузку уксусной кислоты определяют,
исходя из объема куба-этерификатора, массовой доли основного
вещества в сырье и требований к качеству эфира-сырца. После
загрузки в куб-этерификатор всей массы уксусной кислоты за-
гружают бутанол, обычно 50—60 % от определенной по расчету-
его массы. Аппарат разогревают с помощью глухого пара дав-
лением 0,6—0,8 МПа. После разогрева колонны 3 и заполнения
флорентины 5 в куб-этерификатор подают еще порцию бута-
нола, равную объему жидкости, задержанной в колонне, фло-
рентине, трубопроводах. В это время аппарат работает 20—
30 мин в режиме полного орошения, что необходимо для уста-
новления гидродинамического равновесия и снижения кислот-
ности дистиллята. При кислотности дистиллята не более 0,5 %
начинают отбор образовавшейся эфироводы из флорентины 5
в сборник 6. Температура паров в верхней части колонны из-
меняется от 86 до 90 °C. Органический слой из флорентины 5
полностью возвращается в колонну 3 на орошение. По мере от-
бора эфироводы в куб-этерификатор 2 добавляют бутанол из на-
порного сборника 1 до расчетного количества (1,08—1,13 моля
спирта на 1 моль загруженной кислоты). После полной за-
грузки бутанола и отгонки основной массы воды в верхней ча-
сти колонны 3 поднимается температура до 100 °C, и аппарат
переводят на режим полного орошения для концентрирования
легкокипящих примесей и избыточного бутанола и доэтерифи-
кации остаточной уксусной кислоты. Кислотность черного
эфира-сырца в кубе-этерификаторе 2 должна быть в этот мо-
мент не более 2 %, а массовая доля эфира не менее 84 %. Если
кислотность эфира-сырца более 2%, в реактор догружают бу-
танол; если в эфире-сырце бутилацетата менее 84 %, а кислот-
ность менее 2 %, в систему догружают уксусную кислоту. Аппа-
рат работает в режиме полного орошения еще 1—1,5 ч. К концу
цикла отключают флорентину, из нижней части колонны 3 вы-
водят легкокипящие компоненты в. сборник 7 для использова-
ния их или на стадии этерификации, или в производстве раство-
рителя БЭФ. При использовании лесохимической уксусной
кислоты в состав легкокипящего эфира входит до 12 % бутил-
формиата, до 25 % бутанола и до 60 % бутилацетата.
Черный эфир-сырец перед выкачкой из системы имеет кис-
лотность не более 0,5 %, эфиров не менее 89 %. Эфир-сырец та-
кого качества перерабатывается с целью получения товарного
бутилацетата марки Б.
Продолжительность операции определяется качеством сырья
и габаритом оборудования. При эксплуатации кубов-этерифи-
295
каторов объемом 13 м3 и использовании синтетической уксусной
кислоты и синтетического бутанола операция длится 6—8 ч,
при переработке слабой кислоты время цикла увеличивается.
Большое значение для достижения максимальной произво-
дительности аппарата имеет чистота греющих поверхностей. Ре-
комендуется при применении бутанола СК или лесохимической
уксусной кислоты чистку поверхности змеевика производить
через 8—10 операций.
Рис. 83. Схема синтеза бутилацетата методом этерификации
уксусной кислоты в избытке бутилового спирта
Непрерывный способ этерификации в избытке бутилового
спирта. Схемы непрерывной этерификации уксусной кислоты
бутиловым спиртом принципиально могут различаться прежде
всего условием ведения реакции, т. е. в зависимости от того,
в избытке какого реагента протекает процесс. Отсюда возни-
кают различия в аппаратурном оформлении. Для ведения про-
цесса в избытке спирта требуется двухколонная установка, про-
цесс этерификации в избытке кислоты осуществляется в одно-
колонном аппарате. Применение различных катализаторов
также определяет специфичность аппаратурной схемы. Схема
синтеза бутилацетата методом этерификации уксусной кислоты
в избытке бутилового спирта показана на рис. 83. Смесь уксус-
ной кислоты и бутилового спирта (как и для производства этил-
ацетата; кислотность смеси 38—43 %) .в мольном соотношении
1: (1,13-4-1,14) через напорный бак 1 подается в реактор 3,
в который перед началом процесса загружают бутанол (55 %
от геометрического объема), уксусную кислоту (30 %) и серную
296
кислоту. Концентрация катализатора в зоне реакции должна
быть 0,2—0,4%. Если для приготовления смеси используется
60—85 %-ная уксусная кислота, то смесь поступает не-в реак-
тор, а в азеотропную колонну 4 для отгонки основной массы
воды. В реакторе при температуре 112—114 °C уксусная ки-
слота почти нацело этерифицируется. Пары воды, бутилацетата,
бутилового спирта в виде смеси бинарного и тройного азеотро-
пов, а также частично уксусной кислоты поступают в нижнюю
часть колонны 4 с температурой 110—112 °C. Колонна 4 пред-
назначена для вывода из системы реакционной и введенной
с сырьем воды. Азеотропная смесь через конденсатор 5 посту-
пает во флорентину 6, где разделяется на два слоя: верхний —
эфирный полностью возвращается в колонну 4 в виде флегмы,
нижний — эфироводный поступает в сборник 7 для последую-
щей регенерации из него эфира и спирта. Серная кислота
в этерификатор подается периодически, порциями через каждые
полчаса из расчета 5 кг на 1 т бутилацетата.
За счет вывода из куба-этерификатора воды и избытка
в реакционной зоне бутанола создаются условия для доста-
точно полного превращения уксусной кислоты в эфир. Из эте-
рификатора смесь бутилацетата и бутилового спирта с массо-
вой долей воды 0,6—1 %, уксусной кислоты (не более 2%) и
катализатора непрерывно поступает на верхнюю (11-ю) та-
релку исчерпывающей колонны 116, где происходит доэтери-
фикация кислоты. Пары воды, бутилацетата и избыточного бу-
•тилового спирта сверху исчерпывающей колонны поступают
с температурой 118—122 °C в укрепляющую колонну Па. Из-
быточный бутиловый спирт в виде смеси, близкой к составу
двойного азеотропа бутилацетат—бутиловый спирт, вместе
с небольшой массой воды из верхней части колонны Па при
температуре паров 110 °C направляется в конденсатор 10,
а затем во флорентину 9. Часть конденсата в виде флегмы воз-
вращается в колонну Па, а остальная масса его возвращается
в реактор 3 для создания в зоне реакции избытка бутилового
спирта. Легкокипящие компоненты, накопившиеся в системе,
выводятся в бак 8. Вода', накапливающаяся во флорентине,
периодически выводится в сборник 7. Число тарелок в колонне
Па 20—22, флегмовое число процесса 2—2,5.
Из подкубка колонны 116 бутилацетат-сырец непрерывно
поступает в испаритель 12, откуда основная масса эфира-сырца
через холодильник 13 выводится в сборник 14 для последую-
щей его переработки в товарный продукт. При использовании
бутилового спирта СК эфир-сырец содержит смолистые веще-
ства, окрашивающие его в желто-коричневый или черный цвет
и осложняющие дальнейшую переработку эфира. В связи
с этим целесообразно парожидкостную смесь из испарителя 12
направлять в другой испаритель (на схеме не показан), а пары
светлого эфира-сырца после конденсации и охлаждения выво-
дить из системы на нейтрализацию. Испаритель периодически
•297
чистят от накопившихся смолистых веществ. Двухколонная
установка достаточно устойчива в работе, обеспечивает высо-
кий коэффициент использования сырья: по кислоте 97,8%, по
спирту 98,9%. Однако наличие серной кислоты в производст-
венных потоках не позволяет решить проблему коррозии обору-
дования. Испарители 2 и 12, реактор 3, колонна 116 и холо-
дильник 13 работают в жестких условиях, особенно испарители
12 и 2, колонна 116, поэтому их необходимо изготовлять из
стали марки 0Х23Н28МЗДЗТ. Сталь марки Х18Н9Т имеет низ-
кую коррозионную стойкость в этих условиях. Установка высо-
копроизводительна и позволяет перерабатывать уксусную ки-
слоту любой концентрации, но весьма металлоемка и доста-
точно сложна в аппаратурном оформлении.
Метод непрерывной этерификации бутанола в избытке кис-
лоты. Этот метод более перспективен по сравнению с этерифи-
кацией в избытке спирта. Принципиальная схема заключается
в совмещении процессов этерификации и ректификации. Исход-
ные компоненты: бутиловый спирт и уксусная кислота непре-
рывно подаются в зону реакции, в реактор, а продукты реакции
(эфир и эфировода) непрерывно отбираются из верхней части
ректификационной колонны в виде дистиллята. Как и для всех
непрерывных процессов, главным условием стабильности ре-
жима непрерывной этерификации является строгое соблюдение
материального баланса за любой промежуток времени. Если
можно предположить, что реакция этерификации в колонне не
протекает, то материальный баланс в процессе непрерывной
этерификации следует рассматривать в двух вариантах в зави-
симости от того, какая из операций конкретно рассматривается.
При контроле за процессом этерификации необходимо поддер-
живать равенство (при постоянном составе жидкости в зоне
реакции) массы веществ, введенных в реакционную зону, массе
веществ, образовавшихся в результате реакции и выведенных
из аппарата. Материальный баланс процесса ректификации
(ректификация в колонне без исчерпывающей части) следует
рассматривать как равенство массы паров, поступающих из ре-
актора в колонну, сумме масс дистиллята (эфир-сырец и эфи-
ровода) и жидкости, возвращающейся из колонны в реактор
(реакционная смесь).
Вторая особенность этого процесса — возможность получе-
ния эфира-сырца с массовой долей эфира в нем от 70 до 98 %.
При этом оптимальный состав кубовой жидкости и соотноше-
ние исходных компонентов, подаваемых в систему, определя-
ются качеством эфира-сырца, т. е. оптимальный состав жидко-
сти в реакторе и ее массу нужно определять, исходя из требо-
ваний к процессам этерификации и ректификации в их тесной
взаимосвязи.
Технологическая схема процесса непрерывной этерификации
уксусной кислоты бутанолом в избытке кислоты показана на
рис. 84. В начале работы в реактор 7 загружают уксусную кис-
298
лоту и бутиловый спирт в соотношении 6—5:1 (молей) и сер-
ную кислоту (2 % от уксусной кислоты). После разогрева и ра-
боты установки около 1,5 ч в режиме полного орошения в зоне
реакции наступает равновесие, характеризующееся следующим
составом кубовой жидкости, % • уксусной кислоты 55—65, эфира
22—30, воды 8—16; бутилового спирта 2—4; катализатора 0,4—
0,6. Колонна 8 работает в режиме полного орошения до дости-
д Сододый раствор
марки А
Рис. 84. Технологическая схема процесса непрерывной этерификации уксус-
ной кислоты бутанолом в избытке кислоты:
/, 2, 12, 13, 19, 20, 25, 27, 30, 36, 38, 41, 42, 48 — сборники; 3, 4, 5, 18, 24, 40 — напорные
баки; 6 — испаритель; 7—реактор; 8, 31, 44 — ректификационные колонны; 9, 33, 45,
49 — дефлегматоры; 10, 35, 47 — флорентины; //—холодильник; 14, 15, 16, 17, 22, 26,
29, 37, 39, 50 — насосы; 21, 23 — нейтрализаторы; 28, 43 — подогреватели; 32, 46 — кубы-
испарители; 34 — конденсатор-холодильник
жения кислотности дистиллята 0,05—0,1 %. После этого в си-
стему подают исходные реагенты и отбирают продукты реакции,
строго соблюдая часовой материальный баланс. Ранее исход-
ные реагенты подавали в реактор в виде смеси. Для этого
в специальных емкостях готовили этерификационную смесь
кислотностью 40—42 % и затем закачивали ее в сборник. Через
299
бак постоянного уровня смесь подавали в нижнюю часть испа-
рителя 6. Как показала практика работы, исходные компоненты
лучше подавать в систему раздельно. Операция приготовления
этерификационной смеси трудоемка, требует специального об-
служивания и контроля. Режим раздельной подачи исходных
реагентов имеет определенные преимущества: снижается тру-
доемкость процесса, нарушения режима работы реактора или
колонны могут быть более оперативно выправлены за счет еди-
новременного изменения соотношения подаваемых в реактор
кислоты и спирта. Уксусная кислота и бутиловый спирт в соот-
ношений 1,25—1,33 (массовые доли) из напорных баков 4 и 3
подаются в нижнюю часть испарителя 6, откуда вместе с флег-
мой из колонны 8 в виде парожидкостной смеси через барботер,
углубленный в жидкую фазу реактора 7, поступают в зону ре-
акции. Температура в реакторе 7 поддерживается 106—ПО °C.
Пары воды, эфира, спирта и уксусной кислоты поступают из
реактора 7 в нижнюю часть ректификационной колонны 8 на
разделение. Вверху колонны температура должна быть 90,0—
90,5 °C. После конденсации паров и охлаждения конденсата до
60—65 °C дистиллят разделяется на два слоя во флорентине
10. Эфирный слой частично возвращается в колонну в виде
флегмы (поддерживается флегмовое число 1—1,5 по эфиру) и
частично отбирается после дополнительного охлаждения в хо-
лодильнике /7 до 20—30 °C в сборник 13 светлого эфира-сырца.
Кислотность эфира-сырца 0,005—0,1 %, массовая доля эфира
92—97%. Водный слой из флорентины также частично возвра-
щается в колонну для азеотропной отгонки образовавшегося
бутилацетата и частично выводится из системы в сборник 12.
Через 40—60 дней непрерывной работы аппарат останавли-
вают на чистку и переводят в периодический режим работы.
С целью уменьшения загрязнения греющих поверхностей испа-
рителя и получения эфира-сырца с массовой долей эфиров 95—
97 % рекомендуется использовать синтетическую уксусную ки-
слоту и синтетический бутиловый спирт. Особый контроль не-
обходимо осуществлять за массовой долей в зоне реакции сер-
ной кислоты или другого катализатора. Катализатор вносится
в систему периодически через 20—25 дней.
Для управления процессом непрерывной этерификации ис-
пользуются контуры автоматического регулирования в зависи-
мости: от давления паров в реакторе 7 изменяется подача
греющего пара в змеевик реактора (при увеличении давле-
ния подача пара уменьшается); от уровня жидкости в реак-
торе 7 изменяется подача в испаритель 6 уксусной кислоты
(при увеличении уровня жидкости в кубе подача кислоты
уменьшается); от расхода уксусной кислоты зависит подача
бутилового спирта (при увеличении расхода уксусной кислоты
повышается подача бутилового спирта); от температуры на
контрольной тарелке изменяется отбор эфира-сырца (при уве-
личении температуры отбор уменьшается).
300
Кроме того, осуществляется контроль за расходом вводимых
в систему кислоты и спирта с помощью ротаметров, за темпе-
ратурой в верхней части колонны, в жидкой фазе испарителя,
после дефлегматора 9 с помощью манометрических термомет-
ров. Контролируется также уровень жидкости в баках кислоты,
спирта, эфира-сырца, эфироводы, серной кислоты с помощью
гидростатических уровнемеров. Замеряется температура кон-
денсата пара из змеевика реактора 7 и испарителя 6 с по-
мощью потенциометров.
Описанный выше процесс непрерывного синтеза бутилаце-
тата в избытке кислоты обладает многими преимуществами
перед другими вариантами: достигается высокая степень кон-
версии реагентов, улучшается качество бутилацетата-сырца,
снижаются расходные нормы, сырья, упрощается дальнейшая
переработка эфира-сырца. Однако, как и в других способах,
осуществляемых с применением серной кислоты в качестве ка-
тализатора, слабым местом является коррозия оборудования,
вызванная как самой серной кислотой, так и продуктами ее
разложения (двуокисью серы).
Существенно снизить коррозию оборудования в этом про-
цессе можно за счет применения других катализаторов, напри-
мер бензолсульфокислоты.
Обесцвечивание, промывка и нейтрализация бутилацетата-
сырца. Бутилацетат-сырец, полученный периодическим спосо-
бом этерификации, обычно содержит, %: 88—91 бутилацетата,
6—8 бутанола, 0,5—1,5 бутилформиата, 0,2—0,7 бутилпропио-
ната; 0,5—Ю,9 уксусной кислоты, 0,6—2,9 смолистых веществ,
0,15—0,3 серной кислоты, 0,1—0,15 воды и до 0,6 прочих ве-
ществ. Светлый эфир-сырец, т. е. эфир-сырец, полученный
в процессе непрерывной этерификации, имеет несколько иной
состав, %: основного вещества 92—97, бутанола 1—5, уксусной
кислоты 0,005—0,2, воды 0,7—1,5 и до 1 различных гомологов
эфиров уксусной кислоты. В светлом бутилацетате отсутствуют
серная кислота и смолистые примеси.
Для снижения кислотности до требований нормативно-тех-
нической документации на товарную продукцию эфир-сырец
нейтрализуют раствором кальцинированной соды. Эта стадия
обязательна. При использовании лесохимической уксусной ки-
слоты эфир-сырец дополнительно обесцвечивают раствором би-
сульфита натрия, затем промывают водой и только после этого
направляют на нейтрализацию.
Технология обесцвечивания и промывки бутилацетата-сырца
проста в аппаратурном оформлении и осуществляется подобно
обесцвечиванию и промывке этилацетата-сырца. Необходимо
также отметить, что эти технологические операции включаются
крайне редко, так как доля использования лесохимической кис-
лоты весьма незначительна.
В зависимости от технологии получения бутилацетата-сырца
используются два способа нейтрализации. Технологическая
301
В скруббер
марки, б использование остатки на
ила на сброс переработку
Рис. 85. Технологическая схема получения и переработки черного эфира-
сырца:
1,2 — емкости; 3, 4, 17, 25, 30, 49 — напорные баки; 5, 10, 11, 18, 20, 24, 25, 37, 38, 43,
44, 46, 47, 56 ~ сборники; 6 — реактор; 7, 33, 40 — ректификационные колонны; 8, 34,
41, 53 — дефлегматоры; 9, 19, 22, 23, 35, 55 — флорентины; 12—16, 26—29, 57—60 — насосы;
21 — нейтрализатор; 31, 36, 39, 52 — кубы-испарители; 32, 51 — насадочные ректифика-
ционные колонны; 42, 45, 54 — холодильники; 48, 53 — конденсаторы-холодильники; 50 —
подогреватель
схема нейтрализации черного эфира-сырца при производстве
бутилацетата марки Б показана на рис. 85.
При использовании на периодической стадии этерификации
бутанола СК эфир-сырец содержит смолистые вещества, окра-
шивающие его в темно-коричневый цвет. Но, несмотря на то,
что эфир-сырец имеет темный цвет, нет необходимости в его
обесцвечивании, так как смолистые вещества легко отделяются
на стадии ректификации.
Нейтрализацию эфира-сырца ведут раствором кальциниро-
ванной соды, пропуская эти два потока с большой скоростью
через перемешивающие устройства (сопла или диафрагмы), со-
здающие устойчивый турбулентный поток (Re» 10000), что не-
обходимо для быстрой и полной нейтрализации уксусной и сер-
ной кислот. Режим турбулентности создается с помощью насо-
сов. Расслаивание эфира и содового раствора происходит во
флорентинах.
Рекомендуется трехступенчатая схема нейтрализации. На
первой ступени осуществляют промывку и частичную нейтрали-
зацию эфира-сырца дважды отработанным содовым раствором
с остаточной щелочностью 2%, на второй — используют содо-
вый раствор с третьей ступени (щелочностью 5—6 %.) и на тре-
тьей ступени дважды нейтрализованный эфир-сырец смешивают
со свежим содовым раствором щелочностью 10—12%. Отноше-
ние эфир — содовый раствор зависит от кислотности исходного
эфира-сырца. При кислотности эфира-сырца 0,5—0,6 % расход
содового раствора на 1 т эфира равен 200—250 кг. Принцип ра-
боты узла нейтрализации ясен из рис. 85. Трижды отработан-
ный содовый раствор с остаточной щелочностью 0,5—0,8 % вы-
водится из сборника 20 на регенерацию. Эфир-сырец кислотно-
стью менее 0,005 % непрерывно поступает в сборник 24, откуда
перекачивается на стадию ректификации.
Светлый эфир-сырец может быть нейтрализован по более
простой технологии, в насадочных нейтрализаторах колонного
типа, в режиме противотока. Принципиальная технологическая
схема нейтрализации светлого эфира-сырца показана на рис. 84
в объеме схемы производства бутилацетата марки А. Светлый
эфир-сырец с кислотностью 0,008—0,2 % из напорного бака 18
подается в нижнюю часть первой ступени нейтрализации —
в нейтрализатор 21. Пройдя слой содового раствора, эфир-
сырец частично нейтрализуется и из верхней части нейтрализа-
тора 21 направляется на вторую ступень нейтрализации в нейт-
рализатор 23, Нейтрализованный до кислотности 0,003—0,005 %
303
эфир-сырец выводится на дальнейшую переработку. Содовый
раствор готовится отдельно, закачивается в сборник 24, откуда
подается в верхнюю часть нейтрализаторов 21 и 23 или непре-
рывно, или периодически—1 раз в 2—3 дня. Нейтрализаторы
перед началом работы заполняются на 80—85 %, объема содо-
вым раствором крепостью 10—12 %. Отработанный содовый ра-
створ из нижней части каждого нейтрализатора через гидрав-
лик выводится на регенерацию. Нейтрализаторы должны иметь
отстойные зоны в верхней части с целью полного расслаивания
эфира и содового раствора. Особенно важно иметь отстойную
зону на второй ступени нейтрализации. Для создания хорошего
массообмена оба нейтрализатора заполняются кольцами Ра-
шига размером 25x25 мм, а эфир-сырец в оба нейтрализатора
подается через кольцевой барботер с отверстиями, расположен-
ными только на его нижней поверхности.
Опыт эксплуатации показывает, что нейтрализаторы диамет-
ром 1200 мм и высотой 4200 мм обеспечивают эффективный ре-
жим нейтрализации эфира-сырца кислотностью до 0,4 % при
производительности по эфиру 2200—2500 кг/ч. Расход соды со-
ставляет 4—6 кг на 1 т эфира. Объем сточных вод от процесса
нейтрализации светлого эфира-сырца во много раз меньше, чем
при нейтрализации черного эфира-сырца.
Ректификация бутилацетата-сырца. Для очистки бутилаце-
тата-сырца от воды, бутилформиата, эфиров высших кислот, бу-
танола и смолистых используется метод ректификации. В зави-
симости от объемов производства и качества эфира-сырца (чер-
ный или светлый) процесс ведут в периодическом или непрерыв-
ном режимах, в одно-, двух- или трехколонных аппаратах.
Периодическая ректификация проводится в одноколонном
аппарате, состоящем из куба-испарителя, ректификационной ко-
лонны с числом тарелок 30—34, конденсатора-холодильника,
флорентины, холодильников для товарного эфира и фракции
легкокипящих компонентов и емкостного оборудования. После
разогрева аппарата и установления гидродинамического равно-
весия отбирается при температуре 98—108 °C легкокипящий
эфир с водой, при температуре 108—118 °C и флегмовом числе
10—16 — промежуточная фракция, а в интервале температур
118—125 °C при флегмовом числе 0,5—1,0 — товарная фракция
бутилацетата. В кубе колонны остаются тяжелокипящие эфиры
и смолистые вещества. Кубовой продукт перерабатывается от-
дельно с целью извлечения эфиров, которые могут быть исполь-
зованы для производства растворителя БЭФ. Смолистые веще-
ства пока не находят постоянного сбыта и чаще всего сжига-
ются. Легкокипящая фракция после отделения от водного слоя
и промежуточная фракция повторно используются на стадии' эте-
рификации. Эфироводный слой направляют на регенерацию вме-
сте со всеми водными растворами. Выход фракции товарного
эфира марки Б при ректификации эфира-сырца с массовой долей
основного вещества 88—89 % составляет не более 80 %.. Перио-
304
дическая ректификация оправдывает себя при объемах произ-
водства не более 2 тыс. т в год. При больших объемах рекомен-
дуется непрерывная технология всех стадий производства бутил-
ацетата. Чем выше массовая доля бутилацетата в эфире-сырце,
тем проще аппаратурное оформление стадии его ректифи-
кации.
При ректификации светлого бутилацетата-сырца, получен-
ного из синтетической уксусной кислоты и синтетического бу-
танола, его перерабатывают в непрерывном режиме, отбирая то-
варный продукт из середины колонны на 8—10 тарелок ниже
тарелки питания. Схема ректификации светлого бутилацетата-
сырца показана на рис. 84 в объеме технологической схемы про-
изводства бутилацетата марки А. Для производства бутилаце-
тата марки Б в эфире-сырце массовая доля основного вещества
должна быть 89—90 %;, а для получения бутилацетата марки А
массовая доля бутилацетата в эфире-сырце, подаваемом на рек-
тификацию, должна быть не менее 94 %, (рекомендуется 95—
97%). Вторым обязательным условием ведения процесса рек-
тификации является отсутствие в светлом эфире отстойной воды.
В противном случае так же, как и при переработке черного
эфира-сырца, содержащего отстойную воду, будет наблюдаться
закисание эфира и в товарном продукте доля воды будет
выше нормы требования. Светлый эфир-сырец из сборника 25
подается в подогреватель 28, а затем с температурой 102—106 °C
поступает на 15-ю тарелку ректификационной колонны 31, име-
ющей 35 практических тарелок колпачкового типа. При темпе-
ратуре вверху колонны 104—108 °C, если перерабатывается 89—
90%-ный эфир-сырец, и 110—114 °C, если перерабатывается
94—97 %-ный эфир-сырец, отбирается фракция легкокипящих
компонентов и вода в виде смеси азеотропов. Массовая доля го-
ловной фракции составляет 5—6 %. Во флорентине 35 при тем-
пературе не выше 65 °C фазы расслаиваются. Эфироводный
слой полностью выводится из системы, а верхний слой легко-
кипящего эфира только частично, а основная доля возвраща-
ется на флегму. Флегмовое число процесса ректификации 4—10.
Из середины колонны из паровой или жидкой фазы с 5—6-й
тарелки отбирается товарный продукт. Эфир охлаждается в хо-
лодильнике 34 до 25—35 °C и направляется в сборник товарной
продукции 41 или 42, Легко кипящий эфир возвращается в тех-
нологический процесс на стадию этерификации. В состав легко-
кипящей фракции входит 65—75 %, бутилацетата, 3—5% воды,
19—25 % бутанола и до 4 %, легкокипящих компонентов, в ос-
новном бутилформиата. Через 20—30 дней работы, после того
как температура в нижней части колонны поднимется выше
132 °C, из каландрии или куба-испарителя 32 выводятся тяже-
локипящие компоненты, которые перерабатываются отдельно
с целью выделения бутилпропионата; бутилбутирата и дру-
гих тяжелокипящих эфиров, используемых в производстве
БЭФ,
305
Управление процессом ректификации в одной колонне
с тремя точками отбора затруднительно, требуется высокая
культура обслуживания, строгая стабилизация всех технологи-
ческих параметров.
Черный эфир-сырец, полученный по периодической схеме
этерификации, перерабатывают с целью получения бутилацетата
марки Б, но он долгое время являлся сырьем и для производ-
ства бутилацетата марки А. Принципиальная технологическая
схема ректификации черного эфира-сырца с целью получения
бутилацетата марки Б показана на рис. 85 в объеме полной
технологической схемы производства бутилацетата марки Б.
Из напорного бака 30 нейтрализованный эфир с массовой
долей эфира 90—91 %, непрерывно подается в верхнюю часть
насадочной колонны 32, заполненной кольцами Рашига разме-
ром 25x25 мм. Высота’слоя насадки 2,5—3 м. Присутствие от-
стойной воды недопустимо. В колонне 32 бутил ацетат освобож-
дается от основной массы смолистых веществ, концентрирование
которых идет в кубе-испарителе 31 при температуре 132—
135 °C. При повышении температуры более 135 °C смолистые
выводятся из куба-испарителя 31. Перед началом работы реко-
мендуется в куб-испаритель 31 загрузить 10—15 кг кальцини-
рованной соды для предотвращения закисания эфира в первый
момент. Пары из колонны 32 поступают на 15-ю тарелку ко-
лонны 33. Избыточный бутанол, вода и легкокипящие примеси
отгоняются в колонне 33, имеющей 30—32 тарелки, на флегму
в колонну 33 возвращается только органический слой из фло-
рентины 35. Флегмовое число 5—8, температура в верху колонны
33 102—106 °C. Легкокипящий эфир из сборника 37 использу-
ется на стадии этерификации. Товарный бутилацетат марки Б
отбирается с 6—7-й тарелки из паровой фазы. Для производ-
ства бутилацетата марки А эфир из колонны 33 подают на 15-ю
тарелку колонны 40 с числом практических тарелок 28—30.
В этом случае товарный бутилацетат марки Б получают в виде
дистиллята колонны 40. Температура вверху колонны 40 120—
124 °C, процесс ведут при флегмовом числе 3—6. Бутилацетат
марки А отбирают с 5—6-й тарелок из жидкой фазы.
Регенерация эфира и спирта из эфироводы. Из эфироводы,
полученной на стадии этерификации, прбмывки, обесцвечива-
ния, нейтрализации и ректификации, регенерируют легкокипя-
щие компоненты, в основном бутанол и бутилацетат. Принци-
пиальная технологическая схема переработки эфироводных
фракций показана на рис. 85. Перед подачей в колонну эфиро-
вода подогревается до температуры 80—85 °C. Ректификацию
ведут в эмульгационном режиме. В насадочную ректификацион-
ную колонну 51 на флегму возвращают только водный слой.
Температуру вверху колонны 51 поддерживают 88—92 °C,
в кубе 102—105 °C. После регенерации в сбросной воде допу-
скается массовая доля спирта не более 0,i %, эфиров не более
0,05 %,. Загрязненность сбросной воды по ХПК зависит от ка-
30$
чества исходного сырья и колеблется в пределах 8—17 г/л. При
больших объемах производства бутилацетата рекомендуется
разделять потоки эфироводы. Сбросную воду, полученную от ре-
генерации эфироводы этерификационной и отобранной на ста-
дии ректификации, рекомендуется использовать для приготов-
ления содового раствора, а сточную воду от регенерации эфиро-
воды, полученной на стадиях нейтрализации, обесцвечивания и
промывки, направлять на дальнейшую очистку.
Технико-экономические показатели производства бутилаце-
тата. Бутилацетат вырабатывается двух марок: марки Б с мас-
совой долей основного вещества 91 ±1 % и -марки А с массо-
вой долей эфира не менее 99 %,. Теоретический расход уксус-
ной кислоты на производство 1 т бутилацетата марки Б
(91 %-ный эфир) составляет 470,7 кг, бутанола 670,5 кг. На про-
изводство бутил ацетата марки А (99 %,-ный эфир) теоретически
расходуется уксусной кислоты 512 кг, бутанола 631,5 кг. Данные
приводятся для сырья, содержащего 100 % основного вещества.
Коэффициент использования сырья в производстве бутилацетата
высок: для уксусной кислоты он составляет 96—98 °/о, для бута-
нола 97—98%. Энергетические затраты на производство 1 т
бутилацетата марки Б составляют: пара 5,0—5,25 ГДж, воды
технической 35—40 м3, электроэнергии 22—24 кВт • ч.
С целью повышения эффективности производства при объ-
еме выработки бутилацетата более 10 тыс. т в год рекоменду-
ется организовывать выработку таких менее квалифицирован-
ных растворителей, как БЭФ, АМР-Зм и др. Кроме того, целе-
сообразно выпускать композиционные растворители 646, 647,
648 и др.
§ 50. СТОЧНЫЕ ВОДЫ И ГАЗОВЫЕ ВЫБРОСЫ
В ПРОИЗВОДСТВЕ АЦЕТАТНЫХ РАСТВОРИТЕЛЕЙ.
СПОСОБЫ ИХ ПЕРЕРАБОТКИ.
КОРРОЗИЯ ОБОРУДОВАНИЯ И РЕКОМЕНДАЦИИ
ПО ПРИМЕНЕНИЮ КОРРОЗИОННОСТОЙКИХ МАТЕРИАЛОВ
Производство ацетатных растворителей можно отнести к ма-
лоотходным. На 1 т этилацетата образуется около 250—270 кг
сточных вод, 12—13 кг кубовых смолистых отходов и около
10—20 кг легкокипящей фракции, используемой для производ-
ства других растворителей (Э-80 или АКР).
В производстве бутил ацетата на 1 т товарной продукции
приходится 180—200 кг сточных вод, около 25—30 кг смолистых
кубовых остатков при использовании сырья низкого качества
и 8—15 кг легкокипящих компонентов, перерабатываемых для
производства БЭФ. Смолистые кубовые остатки не находят пока
сбыта и обезвреживаются методом сжигания.
Сточные воды после регенерационного аппарата имеют за-
грязненность по ХПК не более 12—18 мг/л и обезврежива-
ются или в прудах-накопителях, или биохимическим методом
307
(см. главу 12). Коррозия основного и вспомогательного обору-
дования, трубопроводов и т. д. вызывает большие производ-
ственные затраты как на ремонт оборудования, так и на пол-
ную или частичную их замену. Поэтому необходимо тщательно
подходить к выбору материала в зависимости не только от
основных компонентов среды, но еще и в очень большой сте-
пени от примесей.
Кубы-этерификаторы, изготовленные из малостойких мате-
риалов, рекомендуется футеровать углеграфитовыми плитками
ГМ.З, ГМ-1, пропитанными фенолформальдегидной смолой, или
ATM (пластмассы на основе фенолформальдегидной смолы
с наполнителем из мелкодисперсного графита) с использова-
нием замазки арзамит-5. В смесях синтетической уксусной кис-
лоты с серной наиболее стойка сталь 06Х28М.ДТ. Коррозия этой
марки стали не превышает 0,002 мм/год. В средах эфира-сырца
этил- и бутилацетатного производства и эфироводы при повы-
шенных температурах рекомендуется применять титан ВТ1-0,
сталь 08Х22Н6Т и 10Х17Н13М2Т. Дефлегматоры, флорентины,
холодильники, емкостное оборудование для легкокипящего
эфира, эфироводы и уксусной кислоты следует изготавливать
из стали 12Х18Н10Т, 10Х17Н13М2Т. Оборудование стадий рек-
тификации и регенерации может быть изготовлено из стали
08Х22Н6Т, 12Х18Н10Т, 08Х17Т, 20X13. Низколегированные стали
08Х22Н6Т, 08Х21Н6М2Т и безникелевые стали 08Х17Т и 20X13
используются для изготовления трубопроводов, емкостного обо-
рудования для уксусной кислоты, эфироводы щдругих неагрес-
сивных сред при температуре не более 80 °C.
Срок службы прокладочных материалов (в основном паро-
нита и асбестового полотна), применяемых для уплотнения
фланцевых соединений, составляет в производстве ацетатных
растворителей не более 1 мес. Наиболее стойки паронит ПК,
ПМБ (срок службы 4 мес и более), паронит Бр-1 (срок службы
4—5 мес). Спирально-навитые прокладки СНП пригодны для
многократного применения при температуре до 250 °C, они слу-
жат 8 мес и более.
Из набивочных материалов для сред ацетатного производ-
ства (кроме черного эфира) можно использовать набивку АСФ,
пропитанную суспензией фторопласта, асбестовую набивку
АГ-1. В средах черного эфира следует применять ФУМ (он ре-
комендуется для всех сред).
Глава 14
ПЕРЕРАБОТКА ДРЕВЕСНОГО УГЛЯ
В зависимости от групп перерабатываемой древесины, пред-
назначенной для пиролиза и углежжения по ГОСТ 24260 — 80,
древесный уголь вырабатывают трех марок: А — уголь, получае-
мый при пиролизе древесины твердолиственных пород и березы;
308
Б — уголь, получаемый при пиролизе смеси древесины твердо-
лиственных и мягколиственных пород; В — уголь, получаемый
при пиролизе древесины мягколиственных и хвойных пород.
Древесный уголь с размером частиц более 12 мм применяют
в производстве кристаллического кремния, активных углей и се-
роуглерода. В указанных производствах уголь используется
в процессах, осуществляемых при высоких температурах (выше
800 °C) в слое самого угля или шихты. В соответствии с назна-
чением к углю предъявляются определенные технические требо-
вания по гранулометрическому составу, зольности и влажности.
Так, для обеспечения нормальной гидродинамики процесса
в слое при получении сероуглерода, кристаллического кремния
и активного угля необходимо, чтобы уголь имел определенный
размер кусков. Крупный уголь потребитель измельчает и мелочь
отсеивает. Поэтому массовая доля в товарном древесном угле
частиц размером менее 12 мм ограничивается.
Во всех вышеупомянутых процессах при нагреве угля до
рабочих температур из него удаляется некоторое количество
летучих веществ (газов и смол). Эти вещества, по меньшей
мере, являются балластом, а иногда затрудняют ведение техно-
логического процесса, конденсируясь в газопроводах и загряз-
няя отходящие газы и продукты. Поэтому желательно, чтобы
древесный уголь содержал как можно больше неулетучиваю-
щегося при нагревании углеродного остатка. Эта технологиче-
ская характеристика товарного древесного угля оценивается
массовой долей так называемого нелетучего углерода — остатка
(за вычетом золы) от прокалки угля при температуре 860 °C
в течение 7 мин. Чем выше конечная температура пиролиза, тем
качественнее уголь по этому показателю.
В древесном угле содержание золы регламентируется. Зола
в угле — нежелательная примесь потому, что при переработке
угля она или переходит в товарные продукты и загрязняет их,
затрудняя очистку, или переходит в шлак и создает технологи-
ческие трудности, уменьшая период работы оборудования между
чистками. Беззольный уголь получить при пиролизе древесины
невозможно, так как неорганические соли входят в клеточный
сок и при испарении влаги при сушке остаются в древесине.
Регламентируется и массовая доля в угле воды, являющейся
балластом. В отдельных случаях, например при получении се-
роуглерода, вода — вредная примесь, которая, взаимодействуя
с серой, образует побочные продукты — сероводород и тиоокись
углерода (S = C = O), увеличивая тем самым ее расход и за-
трудняя очистку сероуглерода. На испарение влаги расходуется
также дополнительное тепло. При соблюдении технологии полу-
чения древесного угля свежий уголь содержит 1—4 % влаги.
Но вследствие наличия пор уголь способен поглощать влагу
из воздуха и при хранении в закрытом помещении его влаж-
ность повышается до 6 %,. При прямом контакте с водой уголь
поглощает большую массу воды, иногда превышающую его соб-
309
ственную. Поэтому так важно правильно организовать'хранение
угля в закрытых складах и транспортировку в крытых вагонах.
Последним требованием, предъявляемым к углю потребите-
лями, является порода древесины, из которой он получен, так
как от этого, при прочих равных условиях, зависит реакционная
способность и механическая прочность угля. Весьма нежелате-
лен для применения в некоторых областях, например в произ-
водстве активного угля, уголь, полученный из разнопородной
древесины. Для этого производства требуется уголь из твердо-
лиственных пород древесины (березы, бука). Это требование
к товарному углю контролируется кажущейся плотностью и мас-
сой 1 дм3, которые для березового угля должны быть не менее
0,37 г/см3 и 210 г/см3 соответственно.
С учетом требований различных потребителей пиролизные
предприятия вырабатывают товарный уголь нескольких марок
и сортов. Уголь высшей категории качества — уголь марки А
высшего сорта, получаемый из твердолиственных пород древе-
сины, должен иметь нелетучего углерода не менее 90 %„ золы
не более 2,5, воды не более 6 и угля с размером частиц менее
12 мм в местах погрузки не более 5%. Наименьшая допусти-
мая массовая доля нелетучего углерода в угле равна 67 %..
Кроме названных производств, древесный уголь с размером
частиц более 12 мм применяют также в производстве силико-
кальция, электроугольных изделий, ферросплавов, дымных по-
рохов, в машиностроении для получения графитовых смазок,
как наполнитель пластмасс и др. Технологию этих производств
изучают в специальных курсах.
Лесохимические предприятия вырабатывают также уголь
древесный мелкий. нефракционированный с размерами частиц
менее 12 мм (ТУ 13-4000177-86 — 85), подкормку древесно-
угольную (ТУ 81-05-139 — 77) с размером частиц 4—12 мм и
уголь древесный дробленый (ТУ 13-4000177-189 — 84) с разме-
ром частиц 4—12 мм.
Подкормку древесно-угольную применяют в качестве .мине-
ральной добавки в рацион при выращивании животных, в ос-
новном свиней. Массовая доля нелетучего углерода в подкормке
должна быть не менее 75 %„ а воды не более 10. Уголь дре-
весный дробленый используется в производстве цветных метал-
лов, а также в производстве стали, чугуна, оцинкованной про-
волоки, стекла, при термообработке стальных деталей. Массо-
вая доля нелетучего углерода в угле должна быть не менее
77 %„ золы и воды соответственно не более 5 и 6 %. Уголь мел-
кий древесный нефракционированный применяют в производстве
сталей. Массовая доля нелетучего углерода в нем должна быть
не менее 70 %, воды и золы соответственно не более 15 и 10 %,-
В лесохимической промышленности древесный уголь пере-
рабатывается на следующие основные продукты: активный
уголь марки ДАК, углеродные ионообменники, карбюризатор
древесно-угольный и древесно-угольные брикеты.
310
§ 51. ПРОИЗВОДСТВО АКТИВНЫХ УГЛЕЙ
Активный уголь — пористый адсорбент, скелет которого по-
строен из рыхлых и неправильно упакованных пачек, состоящих
из сеток 6-членных углеродных колец, ковалентно связанных
с углеродными радикалами, с водородом и реже с кислородом.
Активные угли хорошо адсорбируют органические вещества.
Они обладают неоднородной поверхностью и пористостью. Раз-
личают микропоры размерами 1—2 нм. (1 нм = 10~9 м) с сильно
развитой поверхностью, поры переходных размеров (5—100 нм)
с поверхностью до 100 м2/г и макропоры с размерами более
100 нм и малой поверхностью — около 1 м2/г.
Активный уголь получают из ископаемых или древесных уг-
лей удалением из них смолистых веществ и созданием разветв-
ленной сети пор. Применяют его в сорбционной технике для
улавливания и возвращения в производство ценных органиче-
ских растворителей, для разделения газовых смесей, в проти-
вогазовой технике, как адсорбенты и как основу для каталити-
ческих и хемосорбционно-активных добавок, для очистки воды
и других сильно полярных жидкостей от примесей органиче-
ских веществ, в медицине для поглощения газов и различных
вредных веществ при желудочно-кишечных заболеваниях.
В лесохимической промышленности вырабатывается уголь
активный древесный дробленый марки ДАК, предназначенный
для очистки парового конденсата от масла и других примесей.
Сырьем для его производства служит угольная мелочь с разме-
ром частиц 1,2—3,5 мм (отход карбюризаторного производства).
Технологический процесс производства активного угля со-
стоит из следующих стадий: транспортировки его к печи акти-
вации, активации, охлаждения и упаковки активного угля
(рис. 86). Уголь с размером частиц 1,2—3,5 мм подается в печь
активации — стальной цилиндр, футерованный жароупорным
кирпичом. Угол наклона печи активации 2°, частота вращения
0,5—1,2 мин-1. В этой печи уголь прокаливается в течение 45—
60 мин под воздействием парогазовой смеси, состоящей из про-
дуктов сгорания дизельного топлива и перегретого пара. Акти-
вирующим началом является входящий в состав парогазовой
смеси химически связанный кислород, в результате взаимодей-
ствия с которым смолистые и другие органические загрязнения
исходного угля превращаются в газообразные соединения или
образуют твердые углеродные вещества. Температура в печи ак-
тивации поддерживается 800—950 °C, так как при повышении
её более 950 °C ухудшается качество активного угля, а темпе-
ратура менее 800 °C приводит к снижению интенсивности про-
цесса активации.
Активный уголь из печи активации непрерывно поступает
в охладитель — вращающийся барабан, орошаемый сверху во-
дой. Охлажденный до температуры не более 30 °C уголь затем
идет на конвейер с резиновой лентой, с него ссыпается в крафт-
311
мешок и после взвешивания и зашивания мешка на упаковочной
машине формируется партия угля. Партию угля выдерживают
в помещении цеха не менее 8 ч, после чего отправляют на склад
готовой продукции. Выход активного угля не более 50 % от ис-
ходного древесного угля.
По физико-химическим показателям активный уголь ДАК
должен соответствовать следующим требованиям и нормам.
Древесный
Влажный
Рис. 86. Схема производства активного угля ДАК:
1 — печь активации; 2— охладитель; 3 — ленточный конвейер; 4 — приемник угля ДАК;
5 — весы; 6 — упаковочная машина
Внешний вид ........................................Зерна черного цвета
без механических
примесей
Адсорбционная активность по йоду, %, не менее .... 30
Суммарный объем пор по воде, см3/г, не менее ... 1,4
Гранулометрический состав:
массовая доля остатка на сите с полотном, %:
№ 36 не более .................................. 2,5
№ 10 не менее.................................. 95,5
на поддоне не более............................ 2,0
Массовая доля, %, не более:
золы .................................................... 6
влаги .............................. ........... 10
По согласованию с потребителем допускается массовая
доля влаги в активном угле ДАК до 15 % с пересчетом факти-
ческой массы на 10 %,-ную ее влажность. Активный уголь ДАК
должен поставляться партиями. Партией считают однородный
по своим качественным показателям уголь одной марки массой
не более 5 т, сопровождаемый одним документом о качестве.
312
Для проверки качества активного угля на соответствие его по-
казателей требованиям стандарта пробы отбирают от 10 единиц
продукции (но не менее чеАм от 10, если в партии менее 100
единиц продукции). При получении неудовлетворительных ре-
зультатов анализа хотя бы по одному из показателей проводят
повторный анализ пробы, отобранной от удвоенного количества
единиц продукции той же партии. Результаты повторного ана-
лиза являются окончательными и распространяются на всю
партию.
Активный древесный уголь ДАК пожароопасен. Нижний кон-
центрационный предел воспламенения 170 г/м3, температура
воспламенения 195 °C, температура самовоспламенения 457 °C.
§ 52. ПРОИЗВОДСТВО УГЛЕРОДНЫХ ИОНООБМЕННИКОВ
Ионообменники — твердые, нерастворимые в воде вещества
или материалы, способные к ионному обмену. Из растворов
электролитов ионообменники могут поглощать положительные
или отрицательные ионы, выделяя в раствор эквивалентное ко-
личество других ионов, имеющих заряд того же знака. По знаку
заряда обменивающихся ионов ионообменники делят на катио-
ниты (для обмена катионов) и аниониты (для обмена анионов)
в зависимости от того, какие свойства они проявляют: кислот-
ные или основные. По химической природе ионообменники бы-
вают неорганические и органические.
Важнейшее свойство ионообменников — поглотительная спо-
собность, так называемая ионообменная емкость. Ее выражают
максимальным числом миллиграмм-эквивалентов (мг • экв)
ионов, поглощаемых единицей массы или объема ионообмен-
ника в условиях равновесия с раствором электролита (статиче-
ская ионообменная емкость), или в условиях фильтрации рас-
твора через слой ионообменников до «проскока» ионов в фильт-
рат (динамическая ионообменная емкость). Кроме высокой ионо-
обменной емкости, к ионообменникам предъявляют ряд других
существенных требований: селективности к определенным ионам,
термической и химической стойкости, механической прочности
и срока службы.
Применяют ионообменники при водоподготовке, в гидроме-
таллургии в процессах обогащения сырья, в химической, коже-
венной, гидролизной, фармацевтической, пищевой промышленно-
сти, в производственной практике, науке и быту. Использование
ионообменников позволило усовершенствовать методы качест-
венного и количественного анализа многих неорганических и
органических веществ.
В ЦНИЛХИ (А. Н. Завьялов с сотрудниками) совместно
с институтом физической химии АН УССР было показано, что
в определенных условиях проходит эффективное поверхностное
окисление древесных углей, приводящее к получению древесного
окисленного угля (ДОУ)—углеродного ионообменника, на
313
поверхности которого имеются различные функциональные
группы: карбоксильные, спиртовые, фенольные, карбонильные,
хинонные, лактонные, перекисные, энольные и др. Кажущаяся
плотность ДОУ 0,45—0,52, истинная плотность 1,5—1,9 г/см3,
пористость 75—82 %, удельное электрическое сопротивление
2,1 • 1084-1,5• 10й Ом• см. Элементный состав, %: углерода 55—
75, водорода 1,5—4, кислорода 18—40, минеральных примесей
2,5—0,4.
Свойства ДОУ определяются характером поверхностных
функциональных групп, их количественным соотношением и фор-
мой (водородной или катионозамещенной). В зависимости от
этого ДОУ проявляет комплексообразующие, ионообменные,
электронообменные или каталитические свойства.
Технология получения ДОУ позволяет путем изменения ре-
жима окисления и последующих обработок целенаправленно
формировать химическую структуру поверхности ДОУ и полу-
чать углеродные ионообменники различного назначения. Разра-
ботана технология получения ионообменников: ДОУ-1, ДОУ-2,
ДОУ-Зс, ДОУ-4с, ДОУ-5, ДОУ-6 и др.
В сравнении с ионообменными смолами ДОУ имеет ряд пре-
имуществ: более термостоек (до 300 °C), обладает высокой ра-
диационной и химической устойчивостью (не растворяется, не на-
бухает и не слипается во всех средах), отличается исключитель-
ной избирательностью, нетоксичен, что позволяет использовать
его в производстве особо чистых веществ и в пищевой промыш-
ленности. ДОУ можно использовать не менее 5 лет. Его регене-
рируют путем обработки минеральными кислотами. После дли-
тельного срока эксплуатации он может быть легко ликвидирован
путем сжигания, так как в отличие от ионообменных смол он сго-
рает до двуокиси углерода и воды без образования ядовитых
веществ. Технология получения ионообменников различна. Для
наглядности опишем процесс выработки ионообменников ДОУ-2
и ДОУ-Зс. Процесс для ДОУ-2 периодический, состоит из сле-
дующих основных стадий: окисления древесного угля, охлаж-
дения окисленного угля и его термообработки, окисления тер-
мообработанного угля, обеззоливания ионообменника, сушки и
упаковки ионообменника.
Сущность процесса выработки ионообменника ДОУ-2 заклю-
чается в следующем. Древесный уголь с размером гранул 2—
5 мм окисляют кислородом воздуха, предварительно подогре-
того и равномерно продуваемого через слой угля до ионообмен-
ной емкости 1—1,5 мг-экв/г, а затем охлаждают. Охлажден-
ный окисленный уголь термообрабатывают без доступа воздуха
и возвращают на повторное окисление до ионообменной емкости
0,8—1 мг-экв/г. После второго окисления уголь снова термооб-
рабатывают и окисляют в третий раз до ионообменной емкости
не менее 1 мг-экв/г. Полученный ионообменник обеззоливают
минеральной кислотой, промывают дистиллированной водой, су-
шат и упаковывают.
314
Ионообменник ДОУ-Зс также получают по периодическому
процессу, включающему окисление древесного угля, охлаждение
окисленного угля и его перевод в солевую форму, окисление со-
левой формы окисленного угля, обеззоливание ионообменника,
сушку и упаковку ионообменника.
Древесный уголь-сырец с размером гранул 2—5 мм окис-
ляют до ионообменной емкости 1—1,5 мг*экв/г и охлаждают.
Затем окисленный уголь переводят в солевую форму раствором
едкого натра, промывают дистиллированной водой до pH рас-
твора 8—9, сушат и направляют на повторное окисление до
ионообменной емкости 2—2,5 мг*экв/г. После второго окисле-
ния уголь повторно переводят в солевую форму раствором ед-
кого натра, промывают, сушат и окисляют в третий раз до ионо-
обменной емкости 3—3,5 мг • экв/г, после чего окисленный уголь
обеззоливают раствором соляной кислоты, промывают дистилли-
рованной водой до нейтральной реакции и в третий раз перево-
дят в солевую форму. ДОУ-Зс в солевой форме промывают ди-
стиллированной водой, сушат и упаковывают.
Основные аппараты для получения углеродного ионообмен-
ника следующие: для окисления древесного угля, для термооб-
работки окисленного угля и для обеззоливания, промывки и
сушки обеззоленного угля. Аппарат для окисления древесного
угля, термообработанного окисленного угля и окисленного угля
представляет собой металлическую камеру. С целью исключения
загрязнения ДОУ ионами металла при его коррозии аппарат
для окисления и термообработки угля должен быть изготовлен
из нержавеющей стали. В камеру окисления подается подогре-
тый в калориферах воздух. Охлаждение угля после окисления
производится на воздухе. Аппарат для термообработки окислен-
ного угля представляет собой шахтную печь с электрообогре-
вом, снабженную снизу секторным питателем, из которого уголь
поступает на ленту конвейера камеры окисления. Аппарат для
обеззоливания, промывки и сушки обеззоленного угля — эмали-
рованный реактор с паровой рубашкой для подогрева, к кото-
рому подведены линия сжатого воздуха для перемешивания рас-
творов и вакуумная линия для сушки обеззоленного угля. Ап-
парат имеет верхний штуцер для загрузки угля и нижний для
выгрузки обеззоленного угля.
§ 53. ДРЕВЕСНО-УГОЛЬНЫЕ БРИКЕТЫ
На лесохимических предприятиях страны скапливается боль-
шая масса древесно-угольной мелочи, которая не находит сбыта
и может быть переработана в брикеты. Древесно-угольные бри-
кеты представляют собой высококачественное топливо, а также
могут быть использованы в производстве кристаллического
кремния для замены части крупнокускового древесного угля.
Брикеты обладают большой механической прочностью (6,9—
9,8 МПа), повышенной плотностью (0,9—1 г/см3), высокой теп-
315
лотворной способностью (30 000—32 000 кДж), малой гигроско-
пичностью.
Брикетирование древесно-угольной мелочи производится
с применением связующих веществ. В качестве связующих мо-
гут быть использованы продукты термической переработки твер-
дых топлив и нефтепереработки, продукты переработки расти-
тельных материалов — декстрин, крахмал, меласса, лигносуль-
фонаты, талловый пек и др.
В настоящее время на Сявском лесохимическом заводе ра-
ботает установка по брикетированию древесно-угольной мелочи
малой мощности. В ЦНИЛХИ выполнен проект установки для
брикетирования древесно-угольной мелочи производительностью
2000 т брикетов в год для Амзинского лесокомбината, на кото-
ром намечается строительство цеха по производству брикетов
из древесно-угольной мелочи с размером частиц менее 4 мм..
Процесс производства древесно-угольных брикетов состоит
из следующих стадий: измельчения угля, приготовления
брикетной массы, прессования брикетов, сушки их и прокали-
вания.
Древесно-угольная мелочь с угольного склада автотранспор-
том доставляется к установке, затем элеватором загружается
в дисковую дробилку. В дробилке древесно-угольная мелочь из-
мельчается до фракции менее 1 мм и винтовым конвейером пе-
риодически загружается в бегуны, куда одновременно подается
связующее — лигносульфонаты, нефтебитумы или древесная
смола, а из водопроводной сети — определенный объем воды.
Полученная брикетная смесь в чаше бегунов перемешивается,
растирается и поступает на прессование. Сформованные сырые
брикеты ленточным конвейером подают в сушилку, обогревае-
мую дымовыми газами, а затем прокаливают в печи. В настоя-
щее время Сявский лесохимический завод реализует брикеты по-
сле стадии сушки (без прокалки) для использования в кузнеч-
ном производстве и для отопления железнодорожных вагонов.
Прокаливание брикетов производится теплоносителем, по-
лученным от сжигания неконденсирующихся газов с присадкой
солярового масла в топке-смесителе. В процессе прокаливания
брикетов образуются парогазы, которые поступают в конденса-
тор, где конденсируются и охлаждаются. Полученный конденсат
возвращается в бегуны на приготовление следующей порции
брикетной массы или направляется на сжигание в топку. В слу-
чае использования в качестве связующего древесной смолы кон-
денсат поступает в сборник-отстойник, откуда верхний слой
(вода) подается в бегуны на приготовление брикетной смеси,
а нижний (масло) — на сжигание в топку вместе с соляровым
маслом.
Брикеты охлаждаются неконденсирующимися газами, полу-
чающимися в процессе прокаливания. Прокаленные брикеты ох-
лаждают до 30 °C и транспортируют на склад готовой продук-
ции (табл. 45).
316
45. ТЕХНИЧЕСКАЯ ХАРАКТЕРИСТИКА ДРЕВЕСНО-УГОЛЬНЫХ БРИКЕТОВ
Показатель Связующее
лигносульфонаты нефтебитум
Прочность на сжатие, МПа Истирание, % Массовая доля, %: золы . влаги летучих Влагоустойчивость, % Пористость, % Реакционная способность, % Электросопротивление, Ом-см 6,9—9,8 1 b 93 4,2—6,2 2,5—3,6 3,3—4,5 ;юо 54 91 5 1,2-105—6,1-10б 9,8—15,0 96 2,9—5,2 1,7—2,4 2,3—5,2 100 52 88,0 9,7-105—8,2-106
Теплотворная способность неконден- сирующихся газов, полученных при прокаливании брикетов, кДж/м3 18 800 10 000
Оптимальными условиями получения брикетов являются:
массовая доля связующего 15—20%, воды 40 % от массы аб-
солютно сухого сырья, время приготовления смеси в бегунах
60—90 мин, давление прессования 25 МПа, температура прока-
ливания 500—550 °C.
Древесно-угольные брикеты транспортируют по железной до-
роге в крытых сухих вагонах или автотранспортом под брезен-
том, хранят в закрытых сухих складских помещениях.
§ 54. ПОЛУЧЕНИЕ КАРБЮРИЗАТОРА
Карбюризатор — композиция из технического углерода и не-
органического или органического катализатора, применяемая
для поверхностной цементации стальных изделий. В качестве
технического углерода может быть использован каменный уголь,
древесный уголь, каменноугольный и нефтяной кокс или полу-
кокс.
Цементация — химико-термическая обработка поверхностных
слоев металлических изделий диффузионным насыщением уг-
леродом при температуре более 900 °C. Цель цементации — по-
вышение твердости, износоустойчивости или усталостной проч-
ности деталей. Глубина цементационного слоя металла состав-
ляет 0,5—3 мм.
При использовании одного технического углерода при тем-
пературах более 900 °C цементация идет очень медленно/ Для
ускорения процесса используются катализаторы — карбонаты
щелочных и щелочно-земельных металлов, хлориды и цианаты
этих металлов, а также некоторые органические соединения.
При цементации стальных изделий разложение, например,
углекислого бария протекает по следующей химической реакции:
ВаСО3’-> ВаО+СО2. (124)
317
Образующаяся двуокись углерода взаимодействует с угле-
родом древесного угля:
со2+ С(угОЛЬ) - 2СО. (125)
Насыщение стали углеродом происходит за счет актив-
ной диффузии атомарного углерода из окиси углерода с поверх-
ности внутрь металла:
2СО = СО2 + Сатом. (126)
Рис. 87. Схема производства карбюризатора:
1 — бункер; 2 — опрокидыватель; 3 — транспортер; 4 — дробилки (/ ступени, II ступени,
III ступени, IV — доизмельчение); 5 — элеваторы; 6 — сортировка; 7 — бункер; 8 — мер-
ник; 9 — обмазочный аппарат; 10 — барабанное сито; 11, /2—бачки; /3 —дозатор;
14 — смеситель; 15 — бункер; 16 — конвейер; 17 — топка; 18 — барабанная сушилка; 19 —
конвейер-стабилизатор; 20 — приемник; 21 — весы; 22 — упаковочная машина; 23 —
циклон; 24 — труба
Лесохимической промышленностью выпускается карбюриза-
тор древесно-угольный, представляющий собой зерна светло-се-
рого цвета, состоящие из частиц угля с нанесенной на их по-
верхность смеси углекислого бария и крахмала или тапиоко-
вого декстрина.
Технологический процесс . производства карбюризатора
(рис. 87) состоит из следующих стадий: транспортировки и
дробления древесного угля-сырца, обмазки угля суспензией уг-
лекислого бария с крахмалом, сушки сырого карбюризатора,
его охлаждения, стабилизации и упаковки в мешки. Древесный
уголь-сырец, полученный при конечной температуре пиролиза
около 500 °C, из опрокидывателя поступает в бункер и далее
ленточным конвейером подается в дробилку первой ступени, где
измельчается до размеров кусков не более 50 мм. После этого.
318
уголь элеватором подается в дробилку второй ступени, в кото-
рой измельчается до размеров кусков не более 30 мм. Из вто-
рой дробилки уголь поступает на сортировку, где разделяется
на четыре фракции: крупную (более 14 мм), полезную (3,5—
14 мм), мелкую (1,2—3,5 мм) и очень мелкую (менее 1,2 мм),
которая поступает на производство мелкозернистого карбюри-
затора или брикетов и в виде исключения на сжигание в топки
ретортного цеха. Уголь с размером частиц 1,2—3,5 мм предна-
значен для производства активного угля ДАК. Крупная фрак-
ция угля направляется в дробилку третьей ступени, оттуда эле-
ватором на вторую половину рассева, где также получаются че-
тыре фракции. Доизмельчёние крупной фракции угля может
производиться в дробилке четвертой ступени.
Полезная фракция угля элеватором подается в специальный
бункер, откуда через мерник направляется на обмазку в аппа-
рат типа бетономешалки. Для обмазки угля готовят суспензию
углекислого бария и крахмала. Крахмал размешивают в бачке
в холодной воде и выливают в другой бачок, в котором его за-
варивают. Крахмальный клейстер через мерник-дозатор на-
правляют в смеситель, куда также загружают рассчитанную
массу углекислого бария, и перемешивают до получения одно-
родной массы. Готовую суспензию подают в обмазочный аппа-
рат для нанесения ее на уголь. После перемешивания получают
сырой карбюризатор, который через бункер ленточным конвейе-
ром направляют для сушки во вращающуюся сушильную печь.
Сушильная печь представляет собой вращающийся барабан
с лопастями внутри. Угол наклона барабана печи составляет
2—3°, частота вращения 10 мин-1. В барабан сушильной печи
прямотоком подается теплоноситель при температуре 570—
700 °C, который получают в топке от сжигания солярового
масла. Отработанный теплоноситель через циклон сбрасывается
в дымовую трубу. Сухой карбюризатор с температурой 120—
150 °C выгружают на ленточный конвейер. Там он охлаждается
и стабилизируется в токе воздуха в течение 120 с при высоте
слоя 40—50 мм до температуры 30—45 °C и далее подается
в бумажные мешки. Затем после взвешивания на весах и за-
шивки мешка на упаковочной машине карбюризатор направ-
ляют на склад готовой продукции.
На древесно-угольный карбюризатор распространяются тре-
бования ГОСТ 2407 — 83. По физико-химическим показателям
древесно-угольный карбюризатор должен соответствовать нор-
мам, указанным ниже:
Массовая доля, %:
углекислого бария .................................................20±2
углекислого кальция, не более......................................2,0
общей серы, не более...............................'.............0,04
двуокиси кремния, не более.........................................0,2
летучих веществ, не более..........................................8,0
воды, не более.....................................................4,0
319
Гранулометрический состав:
массовая доля остатка, %:
на сите с полотном № 100, не более................................ 6
на сите с полотном № 35, не менее................................ 92
на поддоне, не более.............................................. 2
По согласованию с потребителем допускается массовая доля
углекислого бария в карбюризаторе менее 18 %. Нормы даны
в пересчете на безводный карбюризатор.
При работе с древесно-угольным карбюризатором необхо-
димо соблюдать меры предосторожности. В производственных
условиях контроль загрязнения воздуха рабочей зоны должны
производить по углекислому барию. Предельно допустимая кон-
центрация аэрозоля углекислого бария в воздухе рабочей зоны
производственных помещений составляет 0,5 мг/м3. Помещения,
в которых проводят работы с древесно-угольным карбюризато-
ром, должны быть оборудованы приточно-вытяжными вентиля-
ционными установками общего и местного назначения. Работу
с карбюризатором необходимо проводить в спецодежде, приме-
няя средства индивидуальной защиты. Образующаяся при про-
изводстве и применении карбюризатора древесно-угольная пыль
пожароопасна и имеет нижний концентрационный предел вос-
пламенения в воздухе 128 г/м3.
Древесно-угольный карбюризатор массой не более 20 кг упа-
ковывают в бумажные мешки. По согласованию с потре-
бителем допускается упаковка карбюризатора в полиэтилено-
вые мешки.
Предложен также древесно-угольный мелкозернистый карбю-
ризатор, который представляет собой смесь древесного березо-
вого угля с размером частиц до 2 мм и массовой долей нелету-
чего углерода не менее 78 %., углекислого калия и активаторов
процесса насыщения. По эффективности действия мелкозер-
нистый карбюризатор полностью заменяет импортный карбюри-
затор типа «Park № 20» и обеспечивает скорость цементации
0,3 мм/ч. В отличие от карбюризатора, в состав которого входит
углекислый барий, он не содержит ядовитых веществ. Производ-
ство такого карбюризатора налажено на Сявском лесохимиче-
ском заводе. Имеются технические условия на продукт (ТУ
13-4000177-199 — 85).
Имеется карбюризатор, состоящий из 90 % древесного угля
и 10 %, карбоната натрия. Недостаток этого карбюризатора —
низкая скорость цементации, составляющая при температуре
900 °C около 0,1 мм/ч. С целью повышения скорости цементации
в состав такого карбюризатора предложено вводить 5—25 %,
добавки порошкообразного наводороженного железа или 3—
6 % карбамида в виде порошка или заменить обычный древес-
ный уголь активированным углем.
Предложены также твердые карбюризаторы, где в качестве
углеродсодержащих компонентов входят древесный уголь и по-
лукокс, а для интенсификации процесса в карбюризатор добав-
320
ляют углекислые натрий или барий, тиокарбамид, хлористый
аммоний и др.
Кроме цементации твердыми углеродсодержащими карбюри-
заторами на предприятиях массового производства часто при-
меняют цементацию в смесях газов, содержащих углеводороды
(природный газ), либо оксид углерода и углеводороды (про-
дукты пиролиза керосина), а также жидкую цементацию в рас-
плаве солей углекислого и хлористого натрия и 6—10 % кар-
бида кремния. Приведенными примерами не исчерпываются спо-
собы получения новых продуктов на основе древесного угля и
области их использования.
Глава 15
ПЕРЕРАБОТКА ДРЕВЕСНЫХ СМОЛ
Древесная пирогенная смола пользуется спросом во многих
отраслях народного хозяйства. Продукты ее переработки благо-
даря специфическим свойствам часто не имеют заменителей.
В нашей стране организована промышленная выработка инги-
биторов окисления моторных топлив, понизителей вязкости бу-
рильных растворов, антисептирующих составов, литейных кре-
пителей, воздухововлекающих и пластифицирующих добавок
в бетоны, коптильных жидкостей для бескаицерогенной обра-
ботки мясных и рыбных изделий и для дубления белково-кол-
басных оболочек. Выполненные под- руководством Д. В. Ти-
щенко в ЛТА, в ЦНИЛХИ, в других научно-исследовательских
институтах, на заводах работы по изучению химического состава
древесных пирогенных смол и по изысканию путей их превра-
щения в товарные продукты позволили создать схемы получе-
ния индивидуальных лесохимических фенолов и на их основе
синтетических дубителей, пластмасс, связующих, гербицидов,
левоглюкозана, оксикислот. При пиролизе древесины можно по-
лучить до 2 % суммарных фенолов (от массы древесины), тогда
как из каменноугольной смолы получают лишь 0,1 %, фенолов
от массы исходного угля.
Древесная смола образуется при пиролизе древесины, при
энергохимической переработке ее газификацией и швелеванием.
Наибольший объем составляет продукт, вырабатываемый при
переугливании лиственных пород древесины; основные аппа-
раты— вертикальные реторты с внутренним нагревом и реторты
различных конструкций с внешним и внутренним нагревом.
В Советском Союзе накоплен опыт эксплуатации крупных
газогенераторных станций древесного питания на металлурги-
ческих, стекольных, целлюлозно-бумажных предприятиях. Раз-
витие газификации также идет по пути энергохимического ис-
пользования малоценной древесины и древесных отходов
в условиях лесозаготовительных и деревообрабатывающих
предприятий.
П Заказ № 2193 321
Древесная смола вырабатывается одновременно с сжига-
нием древесины в топке-генераторе ЦКТИ системы В. В. Поме-
ранцева. Высокие технико-экономические показатели комплекс-
ного сжигания древесины, а также технологического процесса
и конструкционное совершенство комбинированной топки-гене-
ратора позволили перенести этот процесс на другие виды топ-
лива. На целлюлозно-бумажных и деревообрабатывающих ком-
бинатах в отходах окорки древесины накапливается до 10 %,
(от перерабатываемой древесины) коры. Большинство предприя-
тий имеют под паровыми котлами энергетические топки ЦКТИ
системы В. В. Померанцева. Перевод этих топок на энергохи-
мический процесс технически освоен, благодаря этому имеется
возможность в короткий срок, при небольших капиталовложе-
ниях организовать производство древесной пирогенной смолы
в значительных количествах.
Источник получения пирогенной смолы — гидролизный лиг-
нин, который используется для выработки активных и специаль-
ных углей. Независимо от способа улавливания жидких продук-
тов пиролиза древесины древесную смолу идентифицируют
промывкой, отстаиванием или дробной конденсацией паров пи-
ролиза, получая отстойную древесную смолу. В пироконденсате
или жижке содержатся водорастворимые нелетучие или труд-
нолетучие вещества, образующие растворимую древесную
смолу. При перегонке жижки растворимая смола образует не-
перегоняемый остаток и подвергается дальнейшей переработке.
Существует экстракционная древесная смола — остаток при
ректификации черной уксусной кислоты.
§ 55. СОСТАВ И СВОЙСТВА ДРЕВЕСНОЙ
ОТСТОЙНОЙ СМОЛЫ
В древесной смоле присутствуют значительные количества
кислородсодержащих соединений (до 90%.). Групповой состав
древесной смолы весьма сложен и зависит от ее происхождения.
В отстойную смолу входят, %: фенолов (включая смоляные,
высшие жирные кислоты и фенолокислоты) 45—60; нейтраль-
ных веществ 25—30; карбоновых кислот 10—15.
Состав фенольной части смолы мало зависит от существую-
щих промышленных способов пиролиза древесины, но в большей
степени зависит от породы древесины. Фенолы отстойной смолы
содержат неполные метиловые эфиры: при пиролизе хвойной
древесины — не менее 40 %, при пиролизе лиственной древе-
сины — не менее 60 %,. Причем в фенолах смолы лиственных
пород преобладают производные пирогаллола, а в фенолах
смолы хвойных пород — производные пирокатехина. Нейтраль-
ные вещества отстойной смолы на 75 %, состоят из кислородсо-
держащих соединений и на 25 % из углеводородов. Углеводо-
родная часть содержит ароматические соединения: нафталин,
диметилнафталин, дегидроретен, парафины. Кислородсодержа-
322
щая часть состоит из спиртов (около 40%), альдегидов, кето-
нов (около 30%.). Кислотная часть смолы включает уксусную,
пропионовую, масляную, изовалериановую, капроновую, энанто-
вую и высшие кислоты, оксикислоты и их производные. Элемен-
тарный состав отстойной смолы почти постоянен, %; С — 64—
68; Н — 7—9; О — 26—28; А — 0,2. Лигниновая смола имеет
повышенное содержание углерода и значительно меньше кис-
лорода, %: С —73—74; Н— 8—9; 0 — 18—19; А —0,3.
Рис. 88. Молекулярно-массное рас-
пределение отстойной смолы:
I — дифференциальная кривая; II —
интегральная кривая; Мп — среднечи-
словая молекулярная масса; W — со-
держание фракций смол
Физические и химические способы разделения смолы без
тепловой ее обработки не получили развития, и первой по тех-
нологии и основной операцией является разгонка смолы. Эф-
фективность последующих операций по идентификации отдель-
ных классов соединений зависит от результатов разгонки.
Химический состав древесной пирогенной смолы, ее молекуляр-
ность, полидисперсность изучены слабо. А. А. Ливеровский и
Е. В. Рогинская щелочным методом нашли в смоле около 4 %,
низкомолекулярных фенолов, в этом же образце смолы после
ее растворения в диэтиловом эфире определили около 40 % ве-
ществ с фенольными функциями, в том числе около 36 % ве-
ществ имели молекулярную массу не ниже 1000. При диализе и
ультрафильтрации спиртового раствора смолы через мембраны
из 4 % коллодиума ими было обнаружено, что смола содержит
до 75 %, веществ с молекулярной массой 5000 и выше.
Найденная среднечисловая молекулярная масса Мп (метод
гельфильтрации) для древесной отстойной смолы различного
происхождения имеет значения от 2,4 • 103 до 8,0 * 103, коэффи-
циент полидисперсности y=Mw/Mn варьирует от 1,6 до 3,0
(здесь Мю — средневесовая молекулярная масса). Кривые моле-
кулярно-массного распределения образца отстойной смолы
изображены на рис. 88. Разгонка древесной смолы в токе водя-
ного пара приводит к значительному увеличению выхода масел.
11* 323
46. РЕЗУЛЬТАТЫ ПЕРЕГОНКИ СМОЛЫ СЕРНОЙ КИСЛОТОЙ ИЛИ БИКАРБОНА-
ТОМ НАТРИЯ
Смола Выход, %
фенолов нейтральных веществ всего
Натуральная 12,6 6,5 19,1
Смола + 0,25 % H2SO4 18,4 7,6 26,0
Смола -j- N аНСОз 12,6 4,6 17,2
Перегонка предварительно обработанной смолы серной кисло-
той или бикарбонатом натрия дала следующие результаты
(табл. 46).
Результаты приведенных в табл. 46 исследований говорят
о том, что древесная отстойная смола весьма полидисперсный
материал: встречаются соединения с молекулярной массой от
100—150 до (3—8) • 103. Высокомолекулярные компоненты
смолы — продукты термического распада лигнина — представ-
ляют собой сетку линейных цепей, сшитых межцепными свя-
зями, и по конфигурации приближаются к несвободно про-
пускаемому Гауссову клубку. Функциональные (фенольные,
карбоксильные) группы не целиком доступны при обработке
реагентами, что приводит к занижению показаний содержания
групп соединений в смоле. Так, при определении группового
состава смолы щелочным методом в разных образцах смолы
более 30 % веществ не определяется. Можно полагать, что
определяемая часть смолы — конечные продукты термического
распада лигнина, целлюлозы и гемицеллюлоз (они могут быть
идентифицированы), а неопределяемая часть — высокомолеку-
лярные соединения термораспада лигнина. Эти соединения яв-
ляются источником образования масел при тепловой обработке
смолы, например при ее разгонке. Селективной экстракцией
органическими растворителями дополнительное количество фе-
нолов и других соединений из высокомолекулярных компонен-
тов смолы получить не удается.
§ 56. СПОСОБЫ ПЕРЕРАБОТКИ ДРЕВЕСНЫХ СМОЛ
Отстойная смола. При тепловой обработке смолы в усло-
виях разгонки в присутствии воды и кислоты высокомолекуляр-
ные соединения ее претерпевают реакции деполимеризации и
гидролиза, приводящие к образованию масел. Параллельно идут
реакции уплотнения с образованием пека. Таким образом, про-
цесс разгонки смолы — процесс физико-химический: образование
низкомолекулярных соединений, составляющих древесно-смоля-
ные масла, посредством химических реакций деструкции и от-
гонки образовавшихся масел. Изменяя условия тепловой обра-
ботки смолы, можно регулировать полноту образования масел
324
и пека. Течение химических реакций, приводящих к образова-
нию масел, зависит от кислотности среды, температуры раз-
гонки, продолжительности реакции; полнота отгонки образовав-
шихся масел зависит от относительной летучести масел и пар-
циальной упругости их над жидкостью. Наиболее благоприятные
условия для процесса маслообразования могут быть созданы
в трубчатом реакторе. В этом аппарате вода и органические кис-
лоты проходят вместе со смолой всю нагревательную зону, обес-
печивая течение реакций кислотного гидролиза: нагрев смолы
длится непродолжительное время; температура разгонки, ско-
рость подачи сырья, продолжительность нагрева смолы хорошо
регулируются. Для избежания закоксовывания труб обязательно
соблюдение развитого турбулентного режима потока смолы на
всем реакционном участке трубы при равномерном ее нагреве.
Принципиальная схема реактора изображена на рис. 89.
Выход масел при разгонке образца газогенераторной смолы
влажностью 17,4 %, и кислотностью 4,7 % при различной тем-
пературе и остаточном давлении 0,3 • 102 кПа представлен
в табл. 47. Для сравнения в этой же таблице приведен выход
фракций масел при периодической (производственной) разгонке
смолы.
Подобная закономерность выхода масел наблюдается для
всех видов древесной отстойной смолы. В смоле содержится
значительное количество низкомолекулярных соединений, пере-
гоняемых при ее тепловой обработке и образующих масла. Од-
нако основное количество масел образуется в результате кис-
лотного гидролиза при температуре выше 200 °C. Разгонка газо-
генераторной смолы при разной кислотности дала зависимость
содержания масел от кислотности смолы, показанную на рис. 90.
Положительное влияние кислот на образование масел опреде-
лено и в других видах смолы. Увеличение кислотности более
4 % не приводит к дальнейшему возрастанию выхода масел.
Природа кислоты не оказывает заметного влияния (муравьи-
47. ВЫХОД МАСЕЛ ПРИ РАЗГОНКЕ ОБРАЗЦА ГАЗОГЕНЕРАТОРНОЙ СМОЛЫ И
ФРАКЦИЙ МАСЕЛ ПРИ ПЕРИОДИЧЕСКОЙ РАЗГОНКЕ СМОЛЫ
Разгонка Выход продуктов разгонки, % к безводной смоле Температура размягчения пека, °C '
креозотовых масел> «кип 180—240°С Ингибитора; «кип240-310°с в том' числе фенолов
Периодическая Непрерывная при £кип, °C: 7—9 18—19 12—14 80—110
180 13 9 12 —
200 16 19 17 38
220 18 22 21 44
240 18 29 26 52
260 19 40 33 68
280 18 59 37 98
325
ная, уксусная, ортофосфорная кислоты, а также смесь органи-
ческих кислот с одинаковым эффектом действуют на процесс
образования масел). В реакционной зоне трубчатки присутст-
вует двухфазная смесь (жидкость — пар) исходного сырья, про-
дуктов реакции, кислоты и воды, поэтому реакции масло- и цено-
образования идут в гетерогенной среде. Последовательно-парал-
лельные реакции указанных типов возможны в кинетической и
диффузионной области. Определяющими факторами протекания
реакций, при данной температуре, а следовательно, и количест-
венного выхода продуктов разгонки являются: время контакта
продолжительность пребывания
Рис.. 90. Содержание масел
в смоле т, %, после термо-
обработки при различной ее
кислотности Кук, %, при тем-
пературе разгонки, °C:
/ — 260; 2 — 240
Рис. 89. Принципиальная схема ре-
актора для термогидролиза и раз-
гонки древесной смолы:
1 — подача смолы; 2 — реактор; 3 — огне-
вой нагрев; 4 — сепаратор; 5 — пары масел
и воды; 6 — острый пар; 7 — пек
трубчатки) — в кинетической области и степень турбулизации
потока — в диффузионной области. При разгонке смолы в про-
мышленных трубчатых печах при температуре 260—280 °C кри-
терий Рейнольдса изменяется в нагреваемом участке от 1,4Х
Х104 до 3‘105. Оптимальное время пребывания смеси в реак-
ционной зоне составляет (14-2) • 10-2 с. Процесс идет в кинети-
ческой области. Найденные расчетным путем размеры трубча-
того реактора и сопротивление системы для разной производи-
тельности представлены в табл. 48.
В зависимости от породы переугливаемой древесины, от спо-
соба пиролиза, а также выделения древесной смолы и сроков
48. РАЗМЕРЫ ТРУБЧАТОГО РЕАКТОРА И СОПРОТИВЛЕНИЕ СИСТЕМЫ ДЛЯ РАЗ-
НОЙ ПРОИЗВОДИТЕЛЬНОСТИ
Размеры Производительность, т/ч
0,5 1,0 2,0 3,0 4,0 5,0 6,0
Длина, м 100 200 280 320 380 430 460
Диаметр, мм 25 40 55 68 76 86 94
Сопротивление, кПа-10—2 5,5 6,0 6,7 7,2 7,3 7,5 7,6
326
хранения химический состав ее неодинаков. При разработке ре-
жимов непрерывной разгонки смолы в трубчатой печи учтены
следующие обстоятельства: aj образование масел при разгонке
смолы идет в присутствии воды и кислот, поэтому целесооб-
разно перерабатывать смолу после естественного отстоя с влаж-
ностью 15—18 % и кислотностью 4—8 %,; б) наиболее легко
осуществим на отечественных смолоперерабатывающих заводах
вакуум порядка (0,74-0,8) • 102 кПа, поэтому для промышленной
разгонки рекомендуется остаточное давление в системе (0,24-
4-0,25) • 102 кПа.
Состав и свойства продуктов разгонки древесной смолы —
масел и пека. Увеличение выхода масел с температурой раз-
гонки наблюдается за счет высококипящих фракций. Выход фе-
нолов растет пропорционально выходу масел. Значительная
доля фенолов и продуктов фенольного характера содержится
в пеке. Методика разделения пека на составляющие его классы
соединений разработана Д. В. Тищенко и А. Н. Кислицыным.
В промышленных образцах пека, полученного при непрерывной
разгонке, содержатся, %: нейтральных веществ 10—12; фенолов,
фенолокислот 62—67, карбоновых кислот 10—12; водораствори-
мых веществ 4—10.
В результате разгонки смолы в трубчатой печи количество
фенолов увеличивается в 1,5—1,8 раза. Фенолокислоты, содер-
жащиеся в пеке, являются продуктами вторичных процессов;
их молекулярная масса равна (8—10) • 103, а такие соединения
нелетучи: в случае образования фенолокислот при пиролизе
древесины они должны были находиться в нелетучем остатке —
древесном угле.
При непрерывной разгонке смолы наблюдается изменение
самих фенолов, заключающееся в отщеплении метоксильных
групп, их рекомбинации. Так, содержание фенола увеличивается
в 1,5 раза, пирокатехина с 3,9 % до 5,2 %(, диметиловых эфиров
метил-, этил- и пропилпирогаллола с 1,5% до 1,9%. (в пере-
счете на безводную смолу).
Получаемые при переработке тяжелых фракций нефти свет-
лые виды топлива, особенно бензин, химически нестабильны,
так как содержат значительное количество ненасыщенных угле-
водородов, склонных к окислению при соприкосновении с кисло-
родом воздуха уже при обычной температуре. Окислительные
процессы в крекинг-топливе приводят к образованию высокомо-
лекулярных кислородсодержащих соединений, ухудшающих
эксплуатационные свойства этого топлива. Цепной радикальный
механизм реакций окисления является причиной высокой чув-
ствительности их к различным инициаторам и ингибиторам.
Присадки — антиокислители или ингибиторы окисления — спо-
собны вступать в реакцию со свободными радикалами, связывая
их и тем самым обрывая цепь окисления или ограничивая ее
развитие. Стабилизирующий эффект достигается при использо-
вании некоторых фенолов и аминов. Из одноатомных фенолов
327
наибольшее влияние оказывают орто- и параизомеры. Много-
атомные фенолы обладают наибольшим стабилизирующим эф-
фектом в том случае, когда гидроксильные группы расположены
рядом, например пирокатехин (индукционный период 1890 мин),
пирогаллол (1440 мин). Остальные двух- и трехатомные фенолы
дают более низкий стабилизирующий эффект. Фенолы древесно-
смоляных масел на 50—60 % состоят из неполных метиловых
эфиров, стабилизирующий эффект которых незначителен. От-
сюда вытекает важная с практической стороны возможность по-
вышения стабилизирующего эффекта древесно-смоляных масел
путем замены в молекулах эфиров лесохимических фенолов ме-
токсильной группы на свободный гидроксил.
Фенолы как сырье для синтеза различных продуктов харак-
теризуются способностью к реакциям присоединения или чис-
лом реактивных положений в молекуле. Для получения про-
странственных полимеров каждая молекула фенолов должна
иметь не менее трех реактивных положений. Лесохимические
фенолы имеют ограниченное применение из-за недостаточной
способности к реакциям присоединения. Среднее значение числа
реактивных положений лесохимических фенолов, происхожде-
ние которых — общие промышленные способы пиролиза, равно
2,2—2,4.
Атомы водорода в ядре молекулы фенола, находящиеся
в орто- и параположениях по отношению к свободным феноль-
ным гидроксилам, активизируются ими и способны к реакциям
присоединения. Число таких атомов соответствует числу реак-
тивных положений в молекуле того или иного фенола:
о-крезол п-крезол м-крезол
Число реактивных положений некоторых лесохимических фе-
нолов: пирокатехин — 4; метил-, этил- и пропилпирокатехин-3;
гваякол-3; метил- и пропилгваякол-2; 1,3-диметиловый эфир
пирогаллола-3; 1,3-диметиловый эфир метилпирогаллола-2;
1,3-диметиловый эфир пропил пирогаллола-2.
Низкая реакционная способность суммарных метиловых эфи-
ров многоатомных фенолов объясняется значительным содер-
жанием замещающих — метильных, этильных и пропильных
групп, а также метоксила. Повышение реакционной способно-
сти фенолов также, как и повышение стабилизирующего эф-
фекта ингибитора, достигается заменой метоксильной группы
на гидроксильную и отщеплением замещающих групп в ядре
фенола:
328
Разложение эфиров фенолов с образованием свободных фе-
нолов проводится: а) гидролизом в присутствии галоидоводо-
родных кислот, фосфорной кислоты, хлористого алюминия, со-
ляно-кислого анилина, щелочей; б) каталитическим крекингом
на алюмосиликатах или с другими катализаторами; в) терми-
ческим крекингом.
Промышленные способы гидролиза и каталитического кре-
кинга разработаны недостаточно из-за трудностей регенерации
катализатора, предотвращения его отравления, защиты аппа-
ратуры от коррозии. Деметоксилирование эфиров фенолов тер-
мическим крекингом может быть осуществлено на нескольких
стадиях пирогенетической переработки: путем высокотемпера-
турного пиролиза древесины, пиролизом древесной смолы, дре-
весно-смоляных масел или фенолов.
Наиболее перспективным является парофазный пиролиз сум-
марных древесно-смоляных масел с целью повышения качества
ингибитора и увеличения реактивных положений фенолов.
Энергия связи О—СНз составляет около 160—180 кДж/моль.
С—СНз в бензольном кольце — около 300, С—ОН в спиртах —
около 340 и бензольного кольца — около 480 кДж/моль. Из
этого следует, что при пиролизе масел возможен режим, когда
произойдет замена метоксильных групп на гидроксильные и от-
щепление алкильных групп с сохранением фенолов.
Пиролиз древесно-смоляных масел, осуществленный в тер-
моконтактном реакторе с твердым теплоносителем, а также
в трубчатом реакторе, показал одинаковые результаты: опти-
мальная температура пиролиза 530—540 °C, продолжительность
реакции или время контакта 0,4—0,7 с, выход пиролизата 53—
55 % к исходной смоле, содержание метоксильных групп сни-
зилось с 5,6 % до 2,5—2,4 % •
Стабилизирующий эффект пиролизата как антиокислителя
возрос в 1,6—1,9 раза по сравнению с маслами непрерывной
разгонки, которые показывают индукционный период 565 мин
(для сравнения: ингибитор, вырабатываемый на Сявском ле-
сохимическом заводе, показывает индукционный период
505 мин).
Состав и свойства древесной смолы определяют многообраз-
ные возможности использования ее или продуктов, полученных
из нее. Традиционно древесная отстойная (осадочная) смола
329
применяется для консервации древесины, в судостроении, при
просмолке канатов и пеньки, в качестве мягчителя при регене-
рации резины. Модифицированная древесная смола использу-
ется по новым направлениям. Так, уваренная отстойная смола
лиственных пород после омыления применяется в чугунолитей-
ном производстве, в строительном деле как воздухововлекаю-
щая добавка в бетоны. Однако основная масса вырабатываемой
лесохимическими заводами древесной смолы подвергается раз-
гонке. Схема переработки древесной смолы по основным на-
правлениям представлена на рис. 91.
В предыдущих главах описаны способы улавливания, кон-
денсации и сбора жидких продуктов пиролиза древесины. Во
всех случаях отстойную смолу требуется освободить от водо-
растворимых продуктов. С этой целью суммарную жижку от-
стаивают; отстойную смолу, в зависимости от дальнейшей пе-
реработки, или промывают и отстаивают, или обезвоживают
с получением уваренной смолы 7—12 % влажности или конди-
ционной смолы 4 %-ной влажности. Обезвоживание проводится
в колонных непрерывнодействующих аппаратах.
Уваренная смола подвергается фракционной разгонке в пе-
риодически действующем смолоразгонном кубе с паровым или
огневым нагревом (рис. 92). Смола поступает в чугунный куб 1,
снабженный змеевиком для глухого перегретого пара и барбо-
тером для острого перегретого пара. В некоторых случаях куб
имеет топку для внешнего обогрева. Водяной пар перегревают
до 320—400 °C, острый пар расходуется в 2—3-кратном коли-
честве к получаемым маслам. Система работает при остаточном
давлении (0,254-0,3) • 102 кПа. При разгонке пары поступают
в ректификационную колонну 2, а затем —в конденсатор 3 и
далее, через вакуум-приемник 4, в соответствующие сборники
5 и 6. В сборниках 5, 6 происходит отстаивание полученных
продуктов от кислой воды и промывка ингибитора. Пей выпу-
скают в специальные открытые приемники, где он застывает.
Оборот куба емкостью 8—10 м3 составляет 24 ч. В продолже-
ние цикла разгонки последовательно отбираются следующие
фракции: 1) кислая вода и легкие масла с температурой кипе-
ния до 180 °C; 2) креозотовые масла с температурой кипения
в пределах 180—240 °C; 3) ингибитор (антиокислитель) с тем-
пературой кипения в пределах 240—310 °C.
Выход кислой воды составляет 8—12%, в зависимости от
влажности исходной смолы; выход легких масел — до 4%. Эти
масла всплывают над водным слоем. Выход креозотовых масел
7—9%. Эти масла используются как вспениватели при обога-
щении металлических руд флотацией, антисептики древесины,
а также при выделке кожи и как сырье для медицинского
креолина — дезинфицирующих и антисептических средств в ме-
дицине и ветеринарии.
Выход ингибитора на смолоперерабатывающих заводах не-
одинаков и колеблется от 14 до 30 %. По требованиям ГОСТа
330
Рис. 91. Схема переработки древесной смолы
ингибитор древесно-смоляной должен содержать воды не более
6 %, иметь кислотное число не более 30 мг КОН на 1 г, погло-
щаемых щелочью веществ (фенолов) не менее 65%, иметь
фракционный состав: отгоняться до 240 °C не более 20—25%,
до 260 °C не более 50—55 %, до 310 °C не менее 95 %. Эффек-
тивность ингибитора в циклогексене: I сорт — не менее 180 мин,
II сорт — не менее 120 мин. В ЦНИЛХИ разработан способ
стабилизации ингибитора, улучшения его качества.
Рис. 92. Схема периодической разгонки древесной смолы:
1 — перегонный куб; 2 — ректификационная колонна; 3 — конденсатор; 4, 5 — вакуум
приемники; 6 — сборник креозотовой фракции; 7 — сборник ингибитора; I — конденсат;
II — глухой пар; III— острый пар; IV— смола; V—пек; VI — вакуум
Эта же фракция масел используется в производстве синте-
тического каучука как «стоппер», или прерыватель, реакций
полимеризации мономеров. Древесно-смоляной пек имеет тем-
пературу размягчения выше 100—ПО °C, используется в ком-
позициях с древесной смолой в производстве активных углей,
лесохимических добавок и др.
Процесс фракционной разгонки древесной смолы в кубах
достаточно энергоемок: для выработки 1 т основного про-
дукта— ингибитора расходуется 6—15 т технологического пара,
60—100 м3 воды, 45—52 кВт-ч электроэнергии, большое коли-
чество ручного труда.
Разгонка древесной смолы в периодически действующих ку-
бах основана на предпосылке о физической природе этого про-
цесса. Считается, что древесная смола состоит из высокомоле-
кулярных соединений (пека), растворимых в низкомолекуляр-
ных соединениях (маслах). В этом случае полнота отбора
332
масел определяется температурой разгонки и парциальной уп-
ругостью паров масел. С целью уменьшения парциальной
упругости паров масел, т. е. с целью увеличения их выхода
в куб подается острый водяной пар, разгонка ведется под по-
ниженным остаточным давлением (снижение температуры ки-
пения масел).
Каждый вид древесной отстойной смолы содержит опреде-
ленное количество низкомолекулярных соединений, которые при
нагреве отгоняются, образуя масла. Однако при нагреве смолы
происходят термические превращения ее составляющих. В ус-
ловиях разгонки смолы в периодически действующих кубах
вначале удаляются вода и летучие кислоты, в процессе нагрева
в течение 18—24 ч происходит дальнейшая конденсация угле-
водов, фенолов, фенолокислот с образованием пека. Постоянная
подача острого водяного пара создает условия для реакций
гидролиза высокомолекулярных соединений, что способствует
увеличению выхода масел. Однако для образования масел пу-
тем кислотного гидролиза при разгонке смолы в периодически
действующих кубах условий нет, поэтому выход древесно-смо-
ляных масел низкий.
Инженерные изыскания непрерывных способов разгонки дре-
весной отстойной смолы начаты со времени организации про-
изводства древесно-смоляного ингибитора (1936—1938 гг.) на
Ветлужском лесохимическом заводе под руководством проф.
М. Д. Тиличеева.
Гипотеза кислотного гидролиза высокомолекулярных компо-
нентов смолы сформулирована Д. В. Тищенко на основании
длительных наблюдений за поведением смолы при разгонке ее
в условиях различной кислотности. Д. В. Тищенко утверждает,
что термические воздействия в совокупности с действием воды
и кислот приводят к деполимеризации и гидролизу высокомо-
лекулярных соединений смолы. На основании этих соображе-
ний был предложен способ непрерывной разгонки смолы. Сна-
чала это была насадочная колонна, в которой разгонка смолы
велась под вакуумом с присадкой острого пара. Однако выяс-
нилось, что для образования масел достаточно той влаги, что
содержится в смоле после 2—4-месячного ее хранения, поэтому
был продложен способ разгонки в трубчатом испарителе (типа
испарителя Кестнера). При температуре паров на выходе из
аппарата 230 °C и остаточном давлении (0,10-^0,15) • 102 кПа
выход масел составлял 45%. Достигнуть приемлемых сроков
непрерывной работы не удалось ввиду быстрого закоксовывания
труб.
Авторы не создали развитого турбулентного режима потока
смолы в трубах, в результате чего пристенный ламинарный слой
смолы подвергался длительному нагреву и коксованию. В на-
учно-исследовательских лабораториях Ашинского и Сявского
лесохимических комбинатов пытались организовать фракцион-
ное разделение смолы в токе перегретого водяного пара в спе-
333
циальных смолоразгонных колоннах. На Ветлужском лесохими-
ческом заводе работники ЦНИЛХИ осуществляли разгонку
смолы в пенном аппарате в токе дымовых газов при темпера-
туре 400—600 °C. Во всех случаях работа закончилась неуда-
чей из-за закоксовывания рабочих поверхностей аппаратов.
Большой вклад в разработку непрерывных способов разгонки
смолы внесли инженеры опытного завода ЦНИЛХИ, в част-
ности В. И. Гусаков. Им разработан и испытан в производ-
ственных условиях способ разгонки смолы с газовым теплоно-
сителем, заключающийся в нагреве тонкодисперсного смоляного
тумана бескислородным дымовым газом (/=600 °C) с после-
дующей ректификацией смоляных масел на товарные фракции.
Однако промышленный образец смолоразгонной камеры ока-
зался непригодным к эксплуатации. Причина — коксование
смолы на стенках камеры.
Работниками ЦНИЛХИ создан и испытан способ разгонки
древесной смолы и пиролиза ее компонентов в термоконтактном
аппарате с твердым теплоносителем. Этот способ наряду с раз-
гонкой смолы позволяет повышать реакционную способность
фенолов и качество ингибитора путем деметоксилирования и
деалкилирования фенолов при температуре 500—530 °C.
Термоконтактные аппараты широко используются для пиро-
лиза легких нефтепродуктов, каменноугольных и сланцевых
смол. В частности, на сланцеперерабатывающем комбинате
им. В. И. Ленина в г. Кохтла-Ярве (Эст. ССР) был испытан
термоконтактный аппарат для переработки тяжелого и легкого
нефтяного сырья. Разгонка и пиролиз газогенераторной смолы
влажностью 14—16 % выполнены на этом аппарате (рис. 93).
Подогретое сырье вводится в верх зоны пиролиза В через фор-
сунки в распыленном состоянии и далее направляется сверху
вниз в прямотоке с раскаленным движущимся теплоносителем,
поступающим в реакционную зону из вышерасположенного
регенератора А по переточным щелям 5. При контакте с рас-
каленным теплоносителем сырье подвергается пиролизу, в ре-
зультате которого образуются летучие продукты и кокс, отла-
гающийся на поверхности теплоносителя. Парогазовая смесь
в конденсационной системе разделяется на суммарную жидкую
часть (пиролизат)' и неконденсирующиеся газы. Теплоноситель
из нижней части зоны пиролиза В поступает в питатель 2 пнев-
моподъемника 4, куда из топки пневмосистемы поступают ды-
мовые газы с температурой до 600 °C; с помощью дымовых га-
зов осуществляется подъем теплоносителя по пневмоподъемнику
4 в сепаратор 5.
В сепараторе происходит отделение твердого теплоносителя
от дыма: дым выбрасывается в атмосферу, а теплоноситель на-
правляется в регенератор Д. Нагрев теплоносителя в регене-
раторе осуществляется частично за счет сгорания кокса, отло-
жившегося на поверхности теплоносителя в процессе пиролиза,
и частично за счет сжигания газа в специальной топке 6,
334
7
в
10
о
Рис. 93. Схема термокон-
тактного аппарата для раз-
гонки и пиролиза древесной
смолы:
1 — топка пневмосистемы; 2 —
питатель; 3 — бункер теплоно-
сителя; 4 — пневмоподъемник;
5 — сепаратор; 6 — топочно-
распределительное устройство;
7 — переточная щель; 8 — рас-
пределитель сырья; 9 — реак-
ционная зона; 10 — отбор паро-
газов; I — газ в топку; // —
воздух в топку; III — пневмо-
агент; IV — дымовые газы из
зоны регенерации; V — сырье
на пиролиз; VI — парогазовая
смесь продуктов пиролиза; А —
зона регенерации; Б — нейтраль-
ная зона; В — зона пиролиза
(реакционная зона)
вмонтированной в центральной части регенератора А. Регене-
ратор и реактор представляют собой секции одного аппарата,
разобщенные нейтральной зоной — переточными щелями 7. Ско-
рость рециркуляции теплоносителя регулируется винтовым кон-
вейером, установленным в верхней части регенератора, режим-
ные показатели которого следующие:
Температура в зоне пиролиза, °C .............................. 500—520
Кратность циркуляции теплоносителя к сырью, т/т .............. 42—48
Выход продуктов пиролиза, % к безводной смоле:
пиролизата ................................................... 50—52
газа .......................................................... 11—13
воды разложения............................................. 7—8
кокса ......................................................23—24
потерь . . _................................................ 5—7
Теплотворная способность газа пиролиза, кДж/м3 (2,5-=-
ч-2,7)/104, влажность пиролизата 6—8%. Фракционный состав:
выкипает до 180°—5%, до 240° — 28, до 300° — 58, до конца
кипения — 85 % • Содержание: фенолов 40 %, групп — ОСН3
0,6—0,9 % • В качестве теплоносителя используются алюмоси-
ликат и шамот размером гранул 4X4 мм.
В термоконтактном аппарате имеют место два процесса:
отгонка имеющихся низкомолекулярных компонентов смолы и
термический крекинг, или пиролиз высокомолекулярных со-
единений. Температура процесса определяется оптимальными
условиями реакций деметоксилирования эфиров фенолов. Зна-
чительное тепловое напряжение реакционного объема, возмож-
ность создания высокотемпературного режима процесса исклю-
чая передачу тепла через стенку, возможность применения твер-
дого катализатора, регулируемость процесса по температуре и
продолжительности контакта присущи для термоконтактных
аппаратов с движущимся теплоносителем. Однако высокая
энергоемкость, сложность аппаратурного оформления, неста-
бильность процесса ограничивают возможность применения
этих аппаратов, особенно при сравнительно небольших объе-
мах производства.
Итак, основная операция переработки древесной отстойной
и суммарной смолы — разгонка на большинстве лесохимических
заводов осуществляется периодическим способом. Основные не-
достатки его: низкий выход товарных продуктов, низкая про-
изводительность труда, тяжелые условия труда рабочих, огра-
ниченная номенклатура вырабатываемой продукции.
Существующий, взгляд на разгонку смолы как на физиче-
ский процесс разделения летучей и нелетучей частей дистилля-
цией тормозит дальнейшее развитие ее технологии.
После неудачных попыток организации процесса разгонки
смолы в многотрубных аппаратах и в камерах с газовым теп-
лоносителем гипотеза кислотного гидролиза подвергалась кри-
тике и ставилась под сомнение.
336
Анализ работы смолоразгонных аппаратов показал, что при-
чиной неустойчивой работы этих систем была недостаточная
турбулизация потока в нагревательной части смолоразгонного
аппарата, что приводило к образованию кокса в ламинарном
пристенном слое смолы. Промышленная проверка разгонки дре-
весной смолы в трубчатой печи на сланцеперерабатывающем
комбинате им. В. И. Ленина (г. Кохтла-Ярве ЭССР) показала,
что этот процесс технически осуществим. На этой основе раз-
работан и внедрен в промышленность способ разгонки древес-
ной смолы и дальнейшая переработка смолопродуктов.
Цех разгонки древесной смолы в трубчатой печи с выпуском
товарной продукции — ингибитора, креозотовых масел и омы-
ленного пека (смолы) успешно работает с 1969 г. Принципи-
альная технологическая схема процесса разгонки смолы с вклю-
чением основного оборудования изображена на рис. 94. Сум-
марная смола из сборников смолохранилища 1 насосом 2 через
подогреватель 3 подается в трубчатую печь 4. Топливом в печи
является природный газ или мазут. . Парожидкостная смесь
пека, воды, масел и кислот поступает в сепаратор 5. В сепара-
тор по мере надобности подается острый пар. Слив пека осу-
ществляется непрерывно через барометрическую трубу или
в сборник 6 и далее на расфасовку в крафт-мешки или в один
из двух реакторов для получения омыленного пека или же омы-
ленной смолы СДО. Пары масел и воды поступают в ректифи-
кационную колонну 7. Колонна снабжена пятью тарелками кол-
пачкового типа. Пары масел и воды из сепаратора 5 в колонну
7 поступают в перегретом состоянии, поэтому на нижних та-
релках расположены змеевики с проходящей внутри труб водой
для охлаждения паров до температуры их конденсации. Инги-
битор отбирается снизу колонны, а также из жидкой фазы ниж-
них трех тарелок, пары креозотовых масел и воды — через верх
колонны. Регулируя температурный режим колонны 7, добива-
ются получения тяжелой фракции, отвечающей требованиям
ГОСТ на ингибитор. Тяжелая фракция (ингибитор) из колонны
7 через вакуум-приемник 9 поступает в сборник 10. Пары лег-
ких масел (креозотовая фракция) и воды конденсируются в хо-
лодильнике 11, где происходит охлаждение конденсата; через
сборник 12 креозотовая фракция поступает в сборник 13.
Вакуум в системе создается вакуум-насосом 14. Конструкция
печи, принятая для разгонки древесной смолы, изображена на
рис. 95. В печи имеется топочное пространство и шахта, отде-
ленная от топочного пространства перевальной кирпичной стен-
кой, не доходящей до свода. Продукты горения газа или жид-
кого топлива из топочного пространства через перевальную
стенку поступают в шахту, а оттуда в дымоход и в дымовую
трубу. Нагревательные трубы, по которым движется смола, по-
мещают в шахту (конвекционную часть), под сводом (потолоч-
ный экран), и на передней стенке (фронтовой экран). Смола
поступает в трубы конвекционнной части, затем — в трубы по-
337
толочного, фронтового экранов и далее. Концы труб выходят
через кладку наружу печи, где последовательно соединяются
ретурбентами. Ретурбенты помещаются в закрытых коробах
для уменьшения теплопотерь. Применяют только цельнотяну-
тые трубы из легированной стали; ретурбенты изготовлены
также из стали или кремнистого чугуна.
Диаметр трубчатого реактора зависит от производительно-
Рис. 94. Схема технологического процесса непрерывной разгонки древесной
смолы в трубчатой печи:
1 — смолохранилище; 2 — сырьевой насос; 3 — подогреватель; 4 — трубчатая печь; 5 —
сепаратор; 6 — сборник пека; 7 — ректификационная колонна; 8 — конденсатор легких
масел; 9 — холодильник; 10 — вакуум-приемники ингибитора; //-вакуум-приемники
легких масел; 12 — сборник ингибитора; 13 — сборник легких масел; 14 — вакуум-насос
сти и скорости подачи сырья. Скорость йодачи должна быть
такой, чтобы на всем нагреваемом участке трубы был развитой
турбулентный режим. При разгонке смолы влажностью 15—
17 % скорость подачи ее 0,2—0,3 м/с обеспечивает скорость
парожидкостной смеси на выходе из трубчатки при темпера-
туре 260—180 °C до 115—120 м/с; критерий Рейнольдса имеет
значение в конвекционной части (84-9) • 104, в радиационной ча-
сти он (4-т-5)-105. Длина трубчатки определяется не только
требуемой поверхностью нагрева смолы, но и необходимым вре-
менем реакции химических превращений ее компонентов.
В табл. 48 приведены размеры трубчатого реактора, а также
сопротивление системы в зависимости от производительности.
Методика расчета приведена в работе А. И. Киприанова «Про-
цесс разгонки древесной смолы в трубчатых печах» (М„
ВНИПИЭИлеспром, 1970).
338
Сборники сырья, трубчатая печь, сепаратор, барометриче-
ская труба и сборник пека расположены вне зданий на специ-
альной площадке. Материальные трубопроводы теплоизолиро-
ваны, линии смолы и пека имеют, паровые рубашки. Сырьем
является суммарная (отстойная, кубовая, растворимая) сухо-
перегонная смола Свалявского и Перечинского лесохимических
заводов влажностью 12—16%. Из смолы такого качества вы-
Рис. 95. Конструкция трубчатой печи:
1 — газовая горелка; 2 — радиационная часть; 3 — вывод парожидкостной смеси; 4 —
потолочный экран; 5 — перевальная стенка; 6 — конвекционная часть; 7 — ввод смолы;
8 — дымоход
ход ингибитора при периодической разгонке составляет 11—
14%. Оптимальный режим работы смолоразгонного производ-
ства следующий:
339
Температура, °C:
на входе смолы в трубчатку ............................... 60—70
на выходе из трубчатки ................................. 240—250
газов над перевальной стенкой ...........,.............. 500—550
паров масел в сепараторе................................ 220—230
низа колонны ........................................... 220—230
верха колонны........................................... 120—130
Остаточное давление в системе, кПа.......................(0,3—0,4)-102
Давление смолы на входе в трубчатку, кПа..................(6,0—6,5)* 102
Скорость, м/с:
подачи смолы в трубчатку................................ 0,2—0,25
парожидкостной смеси на выходе из трубчатки ............ 90—110
Критерий Re на выходе из трубчатки........................ 3-105
Теплонапряженность поверхности труб, кВт/м2 .............. 15
Тепловой КПД установки ................................... 60 %
Выход креозотовой фракции составляет 10—12 % к безвод-
ной смоле, выход ингибитора 50 % к безводной смоле. Каче-
ственные показатели ингибитора:
Плотность, кг/м3 .....................-.....................1100—1170
Влажность, % ........................................... 4—6
Содержание фенолов, % (по Каттвинкелю) ..................... 60—70
Фракционный состав, %:
до 110° С . . . . :.............................................. 4
до 180 °C ................................................... 6
до 240 °C ................................................... 26
до 260 °C ................................................... 60
до 310 °C ................................................... 94
Кислотное число ............................................... 30
Содержание фактических смол увеличивается до 1,5 мг/100г
бензина.
Стабилизирующий эффект.
Индукционный период . .
Поглощение кислорода
Продукт Свалявского
лесохимзавода
565 мм
6 мл
Продукт Сявского лесо-
химкомбината
505 мм
9,5 мл
Температура размягчения пека 70—75 °C.
Основные экономические показатели работы смолоперераба-
тывающего производства после реконструкции следующие: сни-
жение себестоимости продукции на 40 %; увеличение произво-
дительности труда работающих в данном производстве более
чем в 2 раза. Для улучшения показателей смолоперерабатываю-
щего производства целесообразно организовать четкое разде-
ление отстойной и растворимой смолы в конденсационной си-
стеме ретортных цехов, ограничивать подготовку смолы для
разгонки в трубчатых печах естественным отстоем, а также не-
обходимо поддерживать более глубокий вакуум в трубчатой си-
стеме печи.
Переработка древесной растворимой смолы. Выход раство-
римой смолы при пиролизе древесины составляет 7—8%, при
газификации и швелевании 16—25%. Растворимая смола от
340
газификации древесины (ее органическая часть) включает 15—
20 % органических летучих кислот, 30—35 % оксикислот и лак-
тонов оксикислот, до 30 % левоглюкозана, 15—20 % фенолов.
В меньших количествах содержится формальдегид, этиленгли-
коль, метилциклопентенолон, редуктоны, уроновые кислоты. Со-
держание фенолов в растворимой смоле пиролиза лиственных
пород древесины составляет 40—45%, причем основная часть
водорастворимых фенолов присутствует в свободном виде.
Растворимая смола после разделения на классы соединений
дает углеводную часть, фенольный экстракт и уксусную кис-
лоту-сырец. Каждый из этих полупродуктов может быть пере-
работан в товарные продукты. Фенольный экстракт служит
сырьем для получения технического пирокатехина, для синтеза
дубителей (синтанов) и понизителя вязкости глинистых буриль-
ных растворов. Углеводная часть растворимой смолы, упарен-
ная до влажности 25—30 %, используется в качестве литейного
крепителя обесфеноленного. Кроме того, тот же продукт может
быть использован в качестве связующего для производства дре-
весно-волокнистых плит сухим способом. При более глубокой
переработке из углеводной части растворимой смолы может
быть извлечен левоглюкозан — трехатомный спирт, устойчивый
при повышенной температуре против действия кислот и щело-
чей. Используется как основа для получения полиэфиров, об-
ладающих повышенной термостойкостью. Кальциевые соли ок-
сикислот, выделенные при идентификации левоглюкозана, об-
ладают высокими пенообразующими свойствами, что позволило
их применить в качестве добавок вместо канифоли, лакричного
корня или боенской крови в производстве пенобетона.
На лесохимических заводах растворимая смола чаще всего
перерабатывается совместно с отстойной смолой, образуя сум-
марную древесную смолу; лишь на некоторых предприятиях ор-
ганизована выработка продуктов из растворимой смолы. Так,
на Амзинском лесохимическом заводе из обескислоченной рас-
творимой смолы, содержащей 50—55 % фенолов (от органиче-
ской части), вырабатывается полифенольный лесохимический
понизитель вязкости бурильных растворов (ПФЛХ).
Упаренная растворимая смола в присутствии Нг5О4 обра-
батывается формалином в реакторе при t 804-90 °C. Образу-
ется новолак. Например,
341
Продукт реакции промывается горячей водой, далее раство-
ряется в 42 %-ном растворе едкого натра. Параллельно гото-
вится 25%-ный раствор оксиметансульфоната натрия раство-
рением сульфита натрия в горячей воде и смешением раствора
с формалином:
Na2SO3 + НгО+СН2О НО—СН2—SO3Na 4- NaOH.
Щелочной раствор промытого новолака смешивается с окси-
метансульфонатом натрия — идет реакция сульфометилирова-
ния (при /100 °C):
с образованием двухкольчатого водорастворимого новолака
(новолакметансульфонат). Раствор полученного новолака упа-
ривается до влажности не более 7%. После охлаждения твер-
дый продукт размалывается и упаковывается; этот продукт —
ПФЛХ.
Подготовленная для получения ПФЛХ растворимая смола
содержит смесь лесохимических фенолов, поэтому конечный
продукт представляет собой многокольчатый новолакметан-
сульфонат следующего строения:
где п=0—4; 7?=—СНз; —С2Н5; —С3Н7; —ОСНз.
Использование ПФЛХ при бурении скважин (добыча нефти
и газа) обеспечивает улучшение тиксотропных свойств промы.
вочных глинистых растворов, приготовленных на пресной и на
морской воде, а именно: снижает вязкость этих растворов и
предельное напряжение сдвига. На 1 т готового продукта рас-
ходуется примерно 1050 кг экстракционной смолы, 250 сульфита
натрия, 210 формалина, 40 серной кислоты и 100 кг каустиче-
ской соды.
Модификации ПФЛХ успешно прошли испытания как до-
бавки в глинисто-угольные смеси в литейном производстве,
342
а также в керамической промышленности в качестве присадок
к сухому каолину с целью повышения текучести шликеров и
улучшения качества фарфоро-фаянсовых изделий.
На Моломском лесохимическом заводе из древесной раство-
римой смолы, упаренной до плотности 1230—1280 кг/м3, выра-
батывается поверхностно-активная добавка для строительных
материалов (ЛХД). Поверхностно-активная добавка ЛХД от-
носится к полифункциональным композициям, оказывает пла-
стифицирующее и воздухововлекающее действие на бетонные
смеси, улучшает вязкость, уменьшает расслаиваемость бетон-
ной смеси, что обеспечивает возможность ее перевозки на зна-
чительные расстояния. Добавка ЛХД увеличивает водонепрони-
цаемость и морозостойкость бетона, что позволяет вести строи-
тельство гидротехнических сооружений в сложных гидрологиче-
ских и климатических условиях. Технология получения ЛХД
заключается в последовательной обработке упаренной раство-
римой смолы едким натром и гидроокисью кальция до pH
11,0—12,5 при температуре 80—90 °C. ЛХД изготавливается
в жидком виде, содержание сухих веществ составляет не ме-
нее 50 %. Расход сырья на 1 т ЛХД следующий, кг: смола дре-
весная растворимая 560, едкий натр 42 %-ный 340, известь не-
гашеная 20, вода 80. На канифольно-экстракционном заводе
«Вахтан» суммарный пироконденсат отстаивается; отстойная
смола влажностью 15 % и плотностью до 1200 кг/м3 использу-
ется в качестве мягчителя ДС в резинорегенератной промыш-
ленности. Отстоянная жижка или растворимая смола с содер-
жанием органических веществ 22—25 % подвергается упари-
ванию.
Дистиллят представляет собой коптильный препарат «Вах-
толь». Дистиллят — прозрачная жидкость от желтого до
светло-коричневого цвета с характерным запахом копчености.
Плотность—1010—1025 кг/м3. Содержит летучих кислот не бо-
лее 2,8 %, фенолов не более 0,6 %, метилового спирта в свобод-
ном состоянии и в виде эфиров не более 0,15%; не содержит
тяжелых углеводородов типа бенз (а) пирена. Применяется для
жидкостного копчения мясных и рыбных продуктов и сыра;
а также в качестве составляющего компонента для приготов-
ления дубителей белковой колбасной оболочки. В зависимости
от степени упаривания растворимая смола (остаток), содержа-
щая углеводы и лактоны оксикислот, используется по следую-
щим направлениям.
1. Коптильный препарат МИНХ. Плотность 1270—1300 кг/м3.
Используется для жидкостного копчения мясных продуктов/Пе-
ред обработкой разбавляется водой в соотношении 1:7 или
1:10.
2. Связующее ОКСИЗАН. Плотность 1270—1300 кг/м3, со-
держание сухих веществ не менее 65%. Используется как свя-
зующее при изготовлении литейных стержней и форм, отверж-
даемых в оснастке.
343
3. Связующее литейное необессмоленное КВ. Плотность не
ниже 1300 кг/м3, содержание сухих веществ не менее 70%.
После упаривания в специальном сборнике-аэраторе продува-
ется воздухом. Применяется как связующее при изготовлении
стержней 3-го и 4-го классов сложности и литейных форм,
а также как противопригарная добавка в формовочных смесях
для чугунного и цветного литья.
§ 57. ПРИМЕНЕНИЕ ДРЕВЕСНОГО ПЕКА
В древесный пек входят 60—80 % фенолокислот и продуктов
конденсации фенолов и альдегидов, содержащихся в перераба-
тываемой смоле. Молекулярная масса пека, полученного при
непрерывной разгонке древесной смолы в трубчатом реакторе,
МП=(8-И2).1О3, а молекулярная масса пека, полученного при
периодической разгонке, составляет более (1,5ч-1,8) • 104.
Древесный пек используется для получения двух- и трех-
компонентного древесно-пекового крепителя (ДП), для получе-
ния связующей смеси при выработке активных углей, а также
для получения смолы древесной омыленной (СДО). Практиче-
ский интерес представляет СДО, которая идет в качестве воз-
духововлекающей добавки при приготовлении строительных рас-
творов и легких бетонов: СДО позволяет снизить на 50—
250 кг/м3 плотность бетона, улучшает его теплофизические свой-
ства, снижает его теплодеформацию, придает пластичность. Рас-
ход СДО от 0,1 до 0,3 % от массы цемента. СДО также выра-
батывают из древесной отстойной смолы, после отгонки из нее
воды и легких масел.
На Свалявском лесохимическом комбинате выработка СДО
идет следующим порядком. Из сепаратора смолоразгонной уста-
новки по барометрической трубе пек непрерывно поступает
в один из двух реакторов. При наполнении реактора на V2 по-
дачу пека переключают на параллельный аппарат. В охлаж-
денный до 80—100 °C пек добавляются креозотовые масла и
20 %-ный раствор едкого натра до заполнения реактора (при-
мерно 100 частей раствора на 100 частей пека), включается
мешалка. Перемешивание ведется несколько часов. Горячая
омыленная древесная смола расфасовывается в бумажные
мешки.
§ 58. ПРЕДЛАГАЕМЫЕ СХЕМЫ ПЕРЕРАБОТКИ
СУММАРНОГО ДРЕВЕСНОГО ПИРОКОНДЕНСАТА
Традиционная технология переработки жижки предусматри-
вает максимальное извлечение уксусной кислоты и ее гомоло-
гов, а также спиртовых продуктов. Ради этого вся смесь мно-
гократно подвергается термической обработке при длительном
344
нагреве, чаще всего в периодически действующей аппаратуре.
Присутствующие в жижке углеводы, кислоты и спирты, фенолы
и альдегиды претерпевают реакции этерификации, поликонден-
сации, уплотнения, в результате чего выход и качество основ-
ных продуктов падает.
Пиролиз древесины в различных модификациях процесса
в определенной степени управляем при получении таких конеч-
ных продуктов, как древесный уголь и горючий газ, однако не
управляем по качеству образующихся жидких продуктов и по-
луфабрикатов. Даже в одном процессе различные факторы
в значительной мере влияют на качество пироконденсатов.
Дальнейшая переработка жижки приводит к еще более глубо-
ким изменениям термолабильных и реакционноспособных ее
компонентов.
На ряде лесохимических заводов внедрены отдельные опе-
рации по рациональной первичной переработке жижки. Основ-
ные признаки предлагаемой технологии: а) четкое разделение
водорастворимой и водонерастворимой частей жижки; б) мяг-
кая термическая обработка сырья с минимальным пребыванием
в нагреваемой зоне.
Жижка состоит из водорастворимых веществ: углеводов, лак-
тонов, оксикислот, кислот, спиртов, эфиров, водорастворимых
фенолов и т. д. Углеводная часть термонеустойчива, при нагре-
вании претерпевает значительные изменения.
В отстойной смоле содержится также значительное количе-
ство термонеустойчивых углеводных продуктов и нейтральных
веществ. Целесообразно при первичной обработке суммарной
жижки максимально отделить водорастворимые вещества от
продуктов фенольного характера и других высокомолекулярных
соединений.
Предлагаемая схема переработки суммарной жижки изо- -
бражена на рис. 96. Суммарная жижка хорошо перемешива-
ется с дополнительной промывкой отстойной смолы водой, под-
вергается непрерывному разделению на отстойную смолу и
отстоянную жижку в гравитационном отстойнике оригинальной
конструкции. Отстойная смола подвергается термогидролизу и
разгонке в трубчатом реакторе непрерывного действия, в ре-
зультате чего выход фенолов возрастает на 40—50 % против
выхода при обычной разгойке, при этом качество фенолов как
сырья для синтеза становится выше. О возможности использо-
вания полученных древесно-смоляных масел в качестве ингиби-
тора или антиполимеризатора сказано ранее.
Древесно-смоляной пек в зависимости от глубины отбора
масел имеет разную температуру плавления и используется для
получения СДО в качестве воздухововлекающей добавки в бе-
тоны, в качестве пластификатора, древесно-пекового крепителя.
Отстоянная жижка (кислая вода) подвергается разгонке
в однотрубном проточном дистилляторе с временем пребыва-
ния в зоне нагрева не более 1,5—2 с. Полученный дистиллят
345
Рис. 96. Принципиальная схема переработки суммарной жижки в непрерывнодействующей аппаратуре (вариант)
полностью освобожден от нелетучих и высококипящих примесей
и далее перерабатывается в общем потоке получения древесно-
спиртовых продуктов, уксусной кислоты и ее гомологов из
обессмоленной жижки.
Освобожденная от летучих продуктов растворимая смола со-
держит значительное количество углеводов и других первичных
продуктов пиролиза древесины и перерабатывается на лево-
глюкозан, лесохимические фенолы, литейные крепители и др.
В зависимости от условий пиролиза древесины, систем улавли-
вания жидких продуктов организуются различные варианты
безотходной технологии получения лесохимических продуктов,
имеющих постоянный сбыт и высококвалифицированное приме-
нение.
СПИСОК РЕКОМЕНДУЕМОЙ ЛИТЕРАТУРЫ
1. Афанасьева В. В., Смирнова Е. Б. Производство и использование
канифоли за рубежом за последние годы.— М.: ВНИПИЭИлеспром,
1984.— 10 с.
2. Бедрин А. К:, Падерин В. Я. Производство экстракционной канифоли
и продуктов на ее основе в СССР и за рубежом.— М.: ВНИПИЭИлеспром,
1977. _ 54 с.
3. Выродов В. А. Канифольно-терпентинное производство/ЛТА. — Л.,
1982. —68 с.
4. Выродов В. А. Канифольно-экстракционное производство/ЛТА. — Л.,
1983. —58 с.
5. Выродов В. А. Методы расчета основного оборудования камфарного
производства/ЛТА.— Л., 1978. — 38 с.
6. Демеш К. А., Исмаилева Е. И. Заготовка пневого осмола предприя-
тиями Минлеспрома СССР.— М/ ВНИПИЭИлеспром, 1978.— 24 с.
7. Древесный уголь. Получение, основные свойства и области примене-
ния древесного угля/О. В. Бронзов, Г. К. Уткин, А. Н. Кислицын и др.—
М.: Лесн. пром-сть, 1979. —137 с.
8. Жаров В. Г., Серафимов Л. А. Физико-химические основы дистилля-
ции и ректификации. — Л.: Химия, 1975. — 239 с.
9. Житков А. В;, Мазарский С. М. Хранение и подготовка древесного
сырья в целлюлозно-бумажной промышленности. — М.: Лесн. пром-сть,
1980. —224 с.
10. Журавлев П. И. Получение - производных скипидара. — М.: ВНИПИ-
ЭИлеспром, 1983.— 36 с.
11. Кречетов Н. В. Сушка и защита древесины. — М.: Лесн. пром-сть,
1975. —400 с.
12. Крылатов Ю. А., Падерин В. Я. Канифоль экстракционная модифи-
цированная— новый вид проклеивающего материала для бумаги. — М.:
ВНИПИЭИлеспром, 1979. — 32 с.
13. Левин Э. Д. Теоретические основы производства древесного угля.—
М.: Лесн. пром-сть, 1980.— 152 с.
14. Медников Ф. А. Биологические основы и техника подсочки. — М.:
Лесн. пром-сть, 1980.— 170 с.
15. Радбиль Б. А., Кушнир С. Р. Кристаллизация живичной канифоли
и ее предупреждение.— М.: ВНИПИЭИлеспром, 1983.— 45 с.
16. Рудаков Г. А. Химия и технология камфары.—М.: Лесн. пром-сть,
1976. —208 с.
17. Санников Ю. Г. Карровый осмол как сырье для канифольно-экстрак-
ционного производства.— М.: ВНИПИЭИлеспром, 1983.— 30 с.
18. Теоретические и практические вопросы производства и переработки
канифоли и скипидара//Сб. науч. тр. ЦНИЛХИ.— Горький, 1982.— 150 с.
19. Совершенствование термической переработки древесины/Сб. науч. тр.
ЦНИЛХИ. —М.: Лесн. пром-сть, 1980. —125 с.
20. Справочник лесохимика/М. И. Глухарева, Н. П. Дроздов, Л. А. Ер-
макова и др. — М.: Лесн. пром-сть, 1974. — 373 с.
21. Технология и оборудование лесохимических производств/Л. В. Гор-
дон, В. В. Фефилов, С. 6. Скворцов, В. И. Лисов. — М.: Лесн. пром-сть,
1979.-288 с.
22. Химико-технический контроль, лесохимических производств/Л. В. Гор-
дон, А. М. Чащин, Б. А. Радбиль и др.—М.: Лесн. пром-сть, 1978. — 352 с.
23. Фролов Г. М., Шабуров М. А. Производство уксусной кислоты.—
М.: Лесн. пром-сть, 1978. — 240 с.
24. Чащин А. М., Глухарева М. И. Производство ацетатных раствори-
телей в лесохимической промышленности. — М.: Лесн. пром-сть, 1984.—
240 с.
25. Шульгин Ю. Н., Билюба В. Ф. Оптимизация режимов сушки техно-
логической древесины.— М.: ВНИПИЭИлеспром, 1975.—45 с.
348
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абиетан 12
Абиетин 12
Аддукты малеиновые 15
Азеотроп 261
Альбертоль 15
Аппараты
— сушки
----непрерывнодействующие 201
— — периодически действующие 201
— пиролиза
— непрерывнодействующие 206, 215
----периодически действующие 206, 207
----полунепрерывного действия 206, 207
Бальзам (см. живица)
Баррас 22, 28
Брикеты древесно-угольные 315
Бутилацетат 278
— применение 278
— сырец 304
---- нейтрализация 301
— — обесцвечивание 309
Г азогенератор
— обращенного процесса 231
— прямого процесса 231
Газы пиролиза древесины 146
— плотность 219
— состав 181, 219
— теплота сгорания низшая 219
Гербициды 231, 331
Деггекурение 134
Древесина
— абсолютно сухая 138
— анализ термический 170
— влажность
----абсолютная 139
---- относительная 139
---- равновесная 139
— водопоглощение 140
— газификация 135, 227, 321
— гниль 144
— диэлектрическая проницаемость 142
— ожижение 188
— пиролиз (см. Пиролиз древесины)
— плотность 138
— подготовка к пиролизу 194
— полнодревесность 192
— состав химический 142
— теплоемкость 140
— теплопроводность 140
— теплота сгорания 141
— усушка 139
— хранение 193
— электропроводность 141 *
— энергохимическая переработка 229, 233
Дубитель белковых колбасных оболочек
321, 343
Живица
— вязкость 37
— добыча 20
— еловая 28
— кедровая 27
— лиственничная 28
— осветленная 35
— отстаивание 36
— плавление 32
— плотность 37
—- сосновая 29
Жидкость коптильная
— Вахтоль 343
— МИНХ 343
Жижка 146, 235
Ингибитор окисления крекинг-топлив
321, 331, 337, 340, 346
Ионообменники древесно-угольные 313
Камфара
— натуральная 99
— синтетическая 100
Канифоль
— гидрированная 12, 121
— диспропорционированная 13, 121
— кабельная 118
— модифицированная 123
— осветленная 123
— полимеризованная 13, 118
— резинаты 13, 125
— эфиры
---глицериновый 14, 115
---пентаэритритовый 14, 117
Карбюратор древесно-угольный 317
Карра
— восходящая 21
— нисходящая 22
— подготовка 20
Кислота
— абиетиновая 11
— дегидроабиетиновая 11
— левопимаровая 11
— • неоабиетиновая 11
— палюстровая 11
— пимаровая И
— смоляные 11
— тетрогидроабиетиповая 11
— уксусная
---химизм образования 151
---этерификация 279
--- техническая
--- — переработка 274
—------получение 269
--- черная
------- переработка 269
-------получение 249, 263
Контроль производства
— ацетатов 291, 300
— камфары 112
— канифольно-терпентинного 51
— канифольно-экстракционного 87
— пиролизного 210, 217
Ксилан
- г анализ термический 149
— пиролиз 149
Левоглюкозан
— способы получения 185
— химизм образования 157
Лигнин
— анализ термический 161
— пиролиз
--- продукты 162
---химизм процесса 163
Метанол
— выделение 250, 256
— химизм образования 152
Масла древесно-смоляные 324
Очистка промстоков и газовых выбросов
производства
— ацетатов 307
— канифольно-терпентинного 50
— канифольно-экстракционного 86
— уксусной кислоты 276
Парогазы
— конденсация
---в поверхностных теплообменниках
238
349
--- в скрубберах 235
---‘В циклонах 240
— состав 145, 180
Пек древесно-смоляной 344
Пиролиз древесины
— высокотемпературный 133
— кинетика 169
— нагрев
---внешний 167
--- внутренний 168
— распад компонентов 172
— продукты
---выход 182, 235
--- области применения 136
--- характеристика 146
— стадии процесса 145
— тепловой эффект 171
— углеобразование 173
Подсочка сосны
— длительная 21
— долгосрочная 21
— краткосрочная 21
— обычная 26
— подновка 20
— с биостимуляторами 27
— с химвоздействием 26
Понизитель вязкости полифенольный лесо-
химический 341
Разгонка
— смолы 325
— масел 327
— непрерывная 326, 328
— периодическая 332
Реактор термоконтактный трубчатый
331, 337
Скипидар
— живичный 6
— применение 8
— состав 6
— экстракционный 83, 85
Склад сырья
— древесины 190
— осмола 69
Смола древесная
— гидролиз при разгонке 325, 333
— лигниновая 323
— молекулярная масса 324
— отстойная 321
— растворимая 321, 324
Смолокурение 133
Сушилки
— туннельные 201
— шахтные 203
Сушка
— атмосферная 197
— вакуумная 198
— высокочастотная 198
— камерная 197, 198
— кондуктивная 198
— радиационная 198
— сублимационная 198
Терпеновые углеводороды
— аллоцимен 7
— камфен 6
— Дз-карен 6
— ментан 6
— а- и р-пинены 7
— а-терпинен 6
— терпинолен 6
— Р-фелландрен 6
— п-цимол 8
Технико-экономические показатели произ-
водства
— бутилацетата 307
— камфары 113
— канифольно-терпентинного 52
— канифольно-экстракционного 89
— этилацстата 292
Углежжение 134, 143
Уголь древесный
— автоокисление 226
— активность 311
— влажность 309
— выход 210, 213, 220
— зольность 309
— охлаждение 209, 218
— переработка 308
— применение 136
— самовозгорание 226
— стабилизация 227
— технические требования 308
— углерод нелетучий 225, 309
— характеристика 146
Факторы, влияющие на пиролиз
— влажность древесины 179
— гниль 178
— давление в аппарате 182
— кора 177
— порода древесины 176
— размер куска 178
— скорость нагрева 182
— способ нагрева 180
— степень нагрева 182
— химические реагенты 183
Фурфурол
— способы получения 183
— химизм образования 151
Целлюлоза
— анализ термический 154
— деполимеризация термическая 155
— деструкция низкотемпературная 154
— продукты пиролиза 158
— структура угля 159
Экстракция смолистых веществ
— основные факторы процесса 63
— сырье 53
— теория процесса 60
Этилацетат
— применение 278
— сырец 278
---нейтрализация 288
--- обесцвечивание 288
---промывка 288
ОГЛАВЛЕНИЕ
Введение
3
Раздел I
ТЕХНОЛОГИЯ ЭКСТРАКТИВНЫХ ВЕЩЕСТВ ДЕРЕВА
Глава 1. Экстрактивные вещества дерева, их свойства и применение . 5
§ 1. Скипидар................................................................. 5
§ 2. Канифоль....................................................................9
Глава 2. Канифольно-терпентинное производство.......................................19
§ 3. Сырье для канифольно-терпентинного производства............................20
§ 4. Технология переработки живицы..............................................29
§ 5. Очистка сточных вод и газовых выбросов.....................................50
§ 6. Контроль производства......................................................52
Глава 3. Канифольно-экстракционное производство.....................................52
§ 7. Сырье для канифольно-экстракционного производства .... 53
§ 8. Теоретические основы процесса экстракции смолистых веществ . 60
§ 9. Технология переработки смолистой древесины.................................69
§ 10. Очистка промышленных стоков и газовых выбросов ...... 86
§ 11. Контроль производства.....................................................87
Глава 4. Производство вторичных продуктов на основе скипидара . . 89
§ 12. Получение окситерпенового растворителя и лаков.91
§ 13. Производство политерпенов........................93
§ 14. Получение терпингидрата и терпинеола...........94
§ 15. Производство синтетической камфары..............98
Глава 5. Производство вторичных продуктов на основе канифоли . .114
§ 16. Получение эфиров канифоли..............................115
§ 17. Производство других видов эфиров канифоли..............117
§ 18. Получение кабельной некристаллизующейся канифоли . . . .118
§ 19. Производство полимеризованной канифоли.................118
§ 20. Получение гидрированной канифоли.......................121
§ 21. Производство диспропорционированной канифоли...........121
§ 22. Получение канифольно-малеиновых и канифольно-фумаровых
аддуктов..................................... 123
§ 23. Производство резинатов канифоли........................125
§ 24. Получение термопластичных клеев........................126
§ 25. Производство укрепленного клея и клея-пасты ...........128
§ 26. Получение антивибрационной смазки......................129
§ 27. Контроль производства продуктов на основе канифоли . . .130
Раздел II
ТЕРМИЧЕСКАЯ ПЕРЕРАБОТКА ДРЕВЕСИНЫ
Глава 6. Характеристика термических методов переработки древесины 133
Глава 7. Характеристика технологического сырья....................138
Глава 8. Теоретические основы пиролиза древесины..................144
§ 28. Общая характеристика процесса пиролиза..................144
§ 29. Пиролиз компонентов древесины и химизм образования основ-
ных продуктов............."...................................147
§ 30. Физико-химическая характеристика процесса пиролиза . . . 166
§ 31. Влияние сырьевых факторов на выход продуктов пиролиза . . 176
351
§ 32. Влияние режимных факторов на выход продуктов пиролиза . 180
§ 33. Влияние химических реагентов на термический распад древесины 183
§ 34. Термическое ожижение древесины...........................188
Глава 9. Технология пиролиза древесины.............................189
§ 35. Приемка и хранение технологической древесины.............189
§ 36. Сушка технологической древесины..........................196
§ 37. Пиролиз древесины........................................206
§ 38. Самовозгорание древесного угля...........................226
Глава 10. Использование малоценной древесины.......................229
§ 39. Газификация древесины....................................230
§ 40. Эпергохимическое использование древесины.................233
Глава 11. Конденсация парагазов....................................235
Глава 12. Производство уксусной кислоты............................244
§ 41. Экстракционные способы извлечения уксусной кислоты из жижки 245
§ 42. Азеотропный способ извлечения уксусной кислоты из жижки . 261
§ 43. Порошковый способ переработки жижки......................265
§ 44. Технология переработки черной кислоты с целью получения тех-
нической уксусной кислоты......................................269
§ 45. Технология производства пищевой уксусной кислоты .... 274
§ 46. Сточные воды и способы их утилизации. Отходы производства.
Коррозия оборудования и рекомендации по применению конструк-
ционных материалов.............................................276
Глава 13. Производство ацетатных растворителей.....................278
§ 47. Теоретические основы и механизм реакции этерификации . . 279
§ 48. Технология производства этнлацетата......................284
§ 49. Технология производства бутил ацетата....................293
§ 50. Сточные воды и газовые выбросы в производстве ацетатных рас-
творителей. Способы их переработки. Коррозия оборудования и ре-
комендации по применению коррозионностойких материалов . . . 307
Глава 14. Переработка древесного угля..............................308
§ 51. Производство активных углей..............................311
§ 52. Производство углеродных иоиообменников...................313
§ 53. Древесно-угольные брикеты................................315
§ 54. Получение карбюризатора..................................317
Глава 15. Переработка древесных смол...............................321
§ 55. Состав и свойства древесной отстойной смолы..............322
§ 56. Способы переработки древесных смол.......................324
§ 57. Применение древесного пека...............................344
§ 58. Предлагаемые схемы переработки суммарного древесного пиро-
конденсата ....................................................344
Список рекомендуемой литературы....................................348
Предметный указатель...............................................349