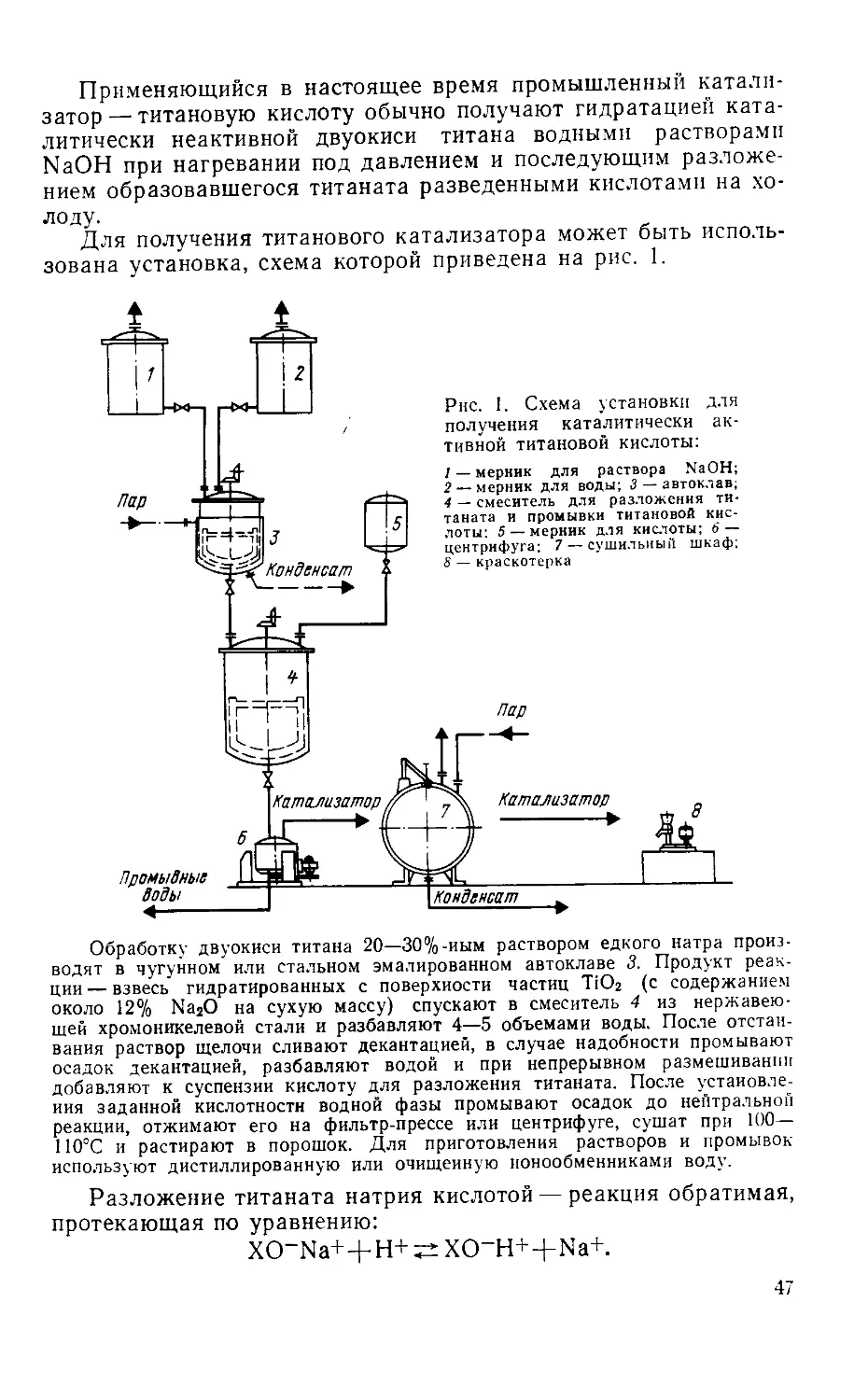
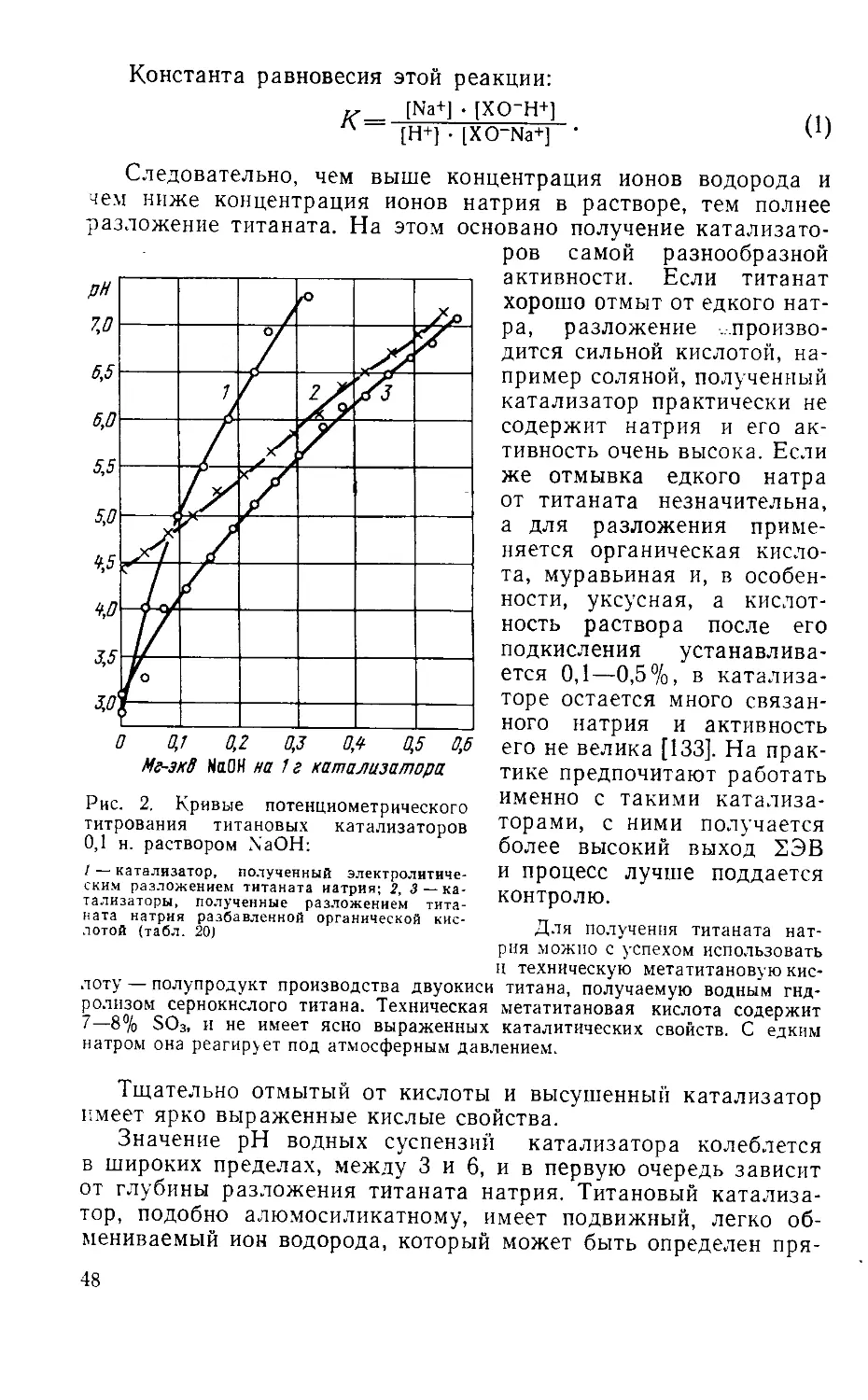
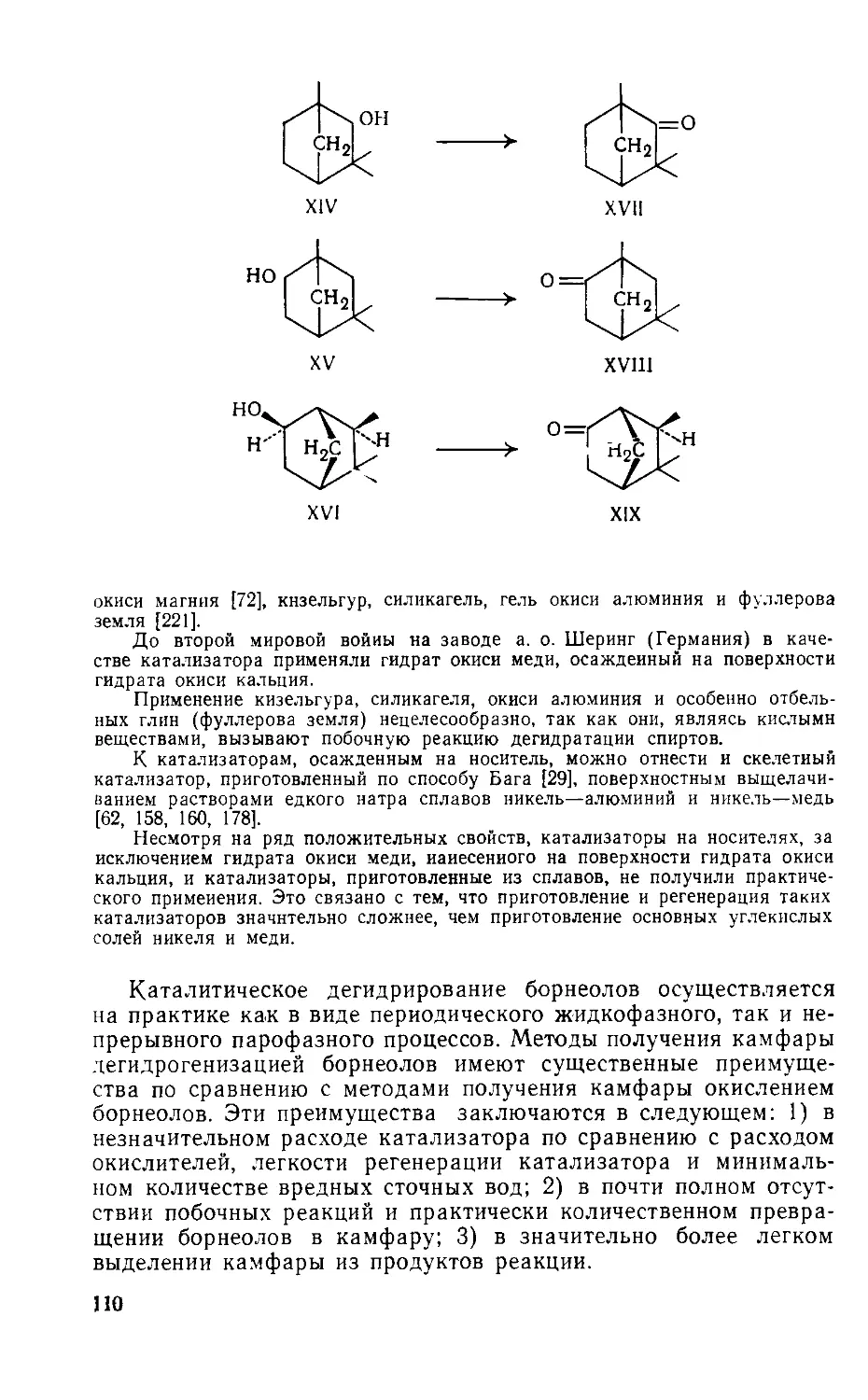
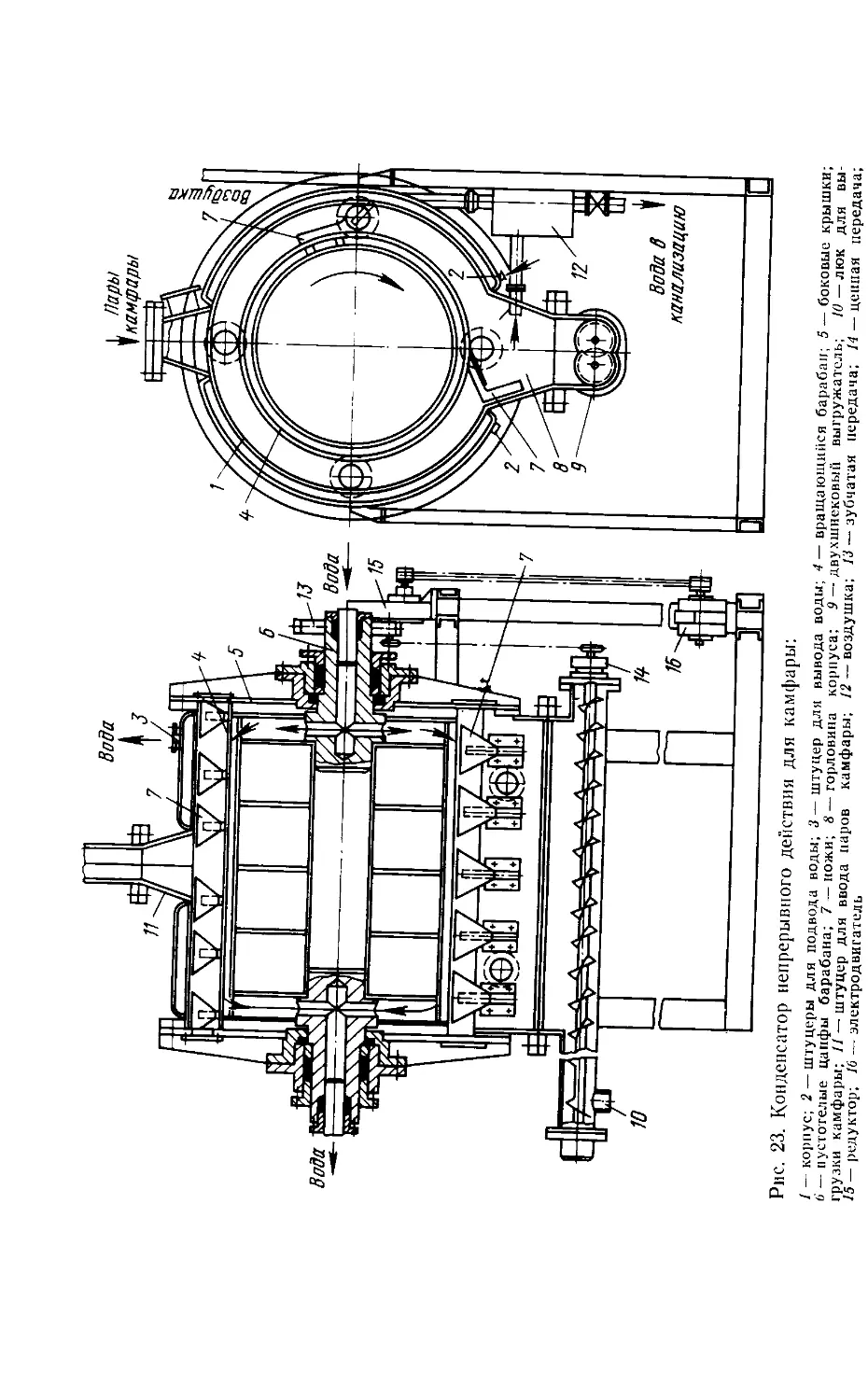
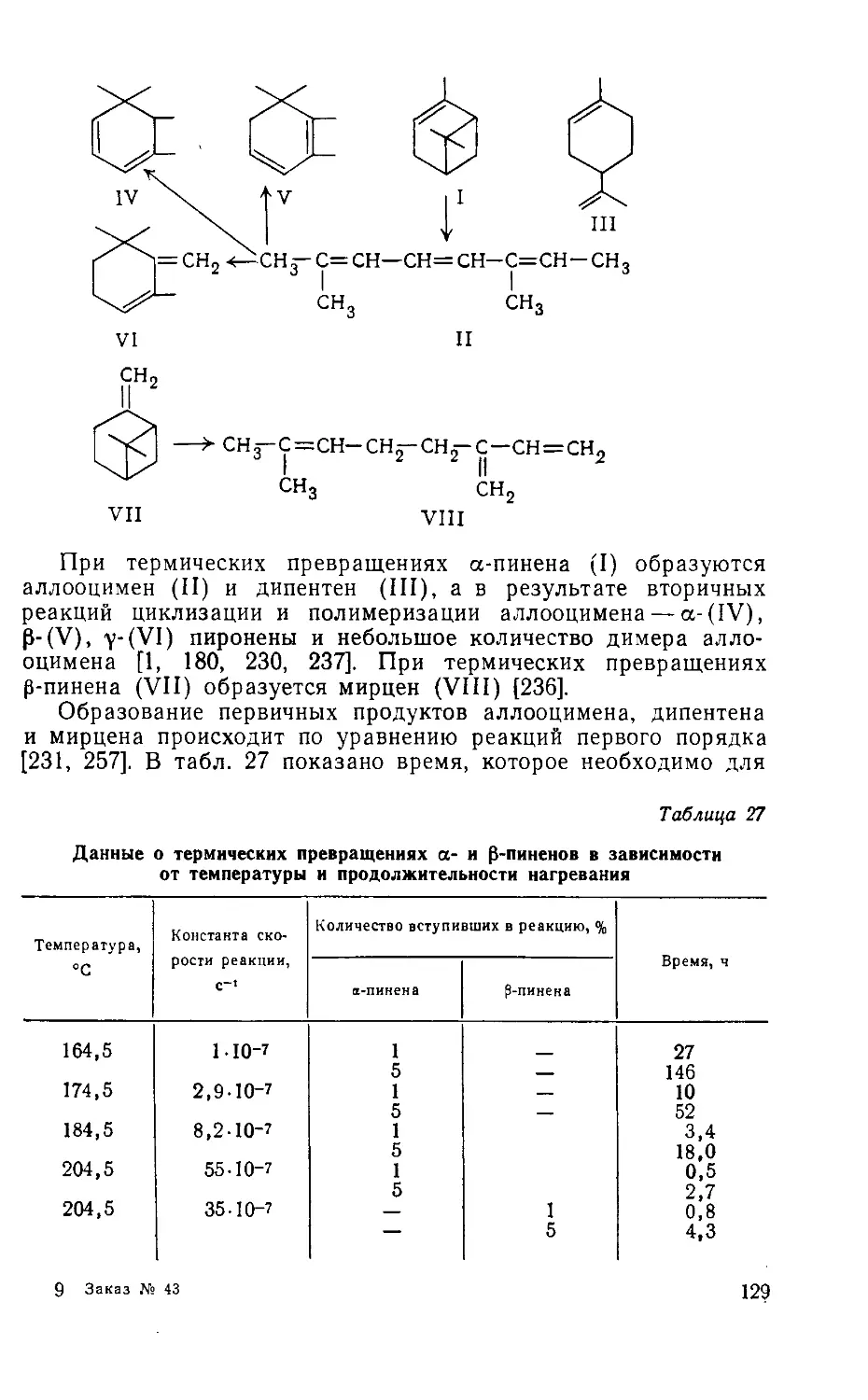
/











































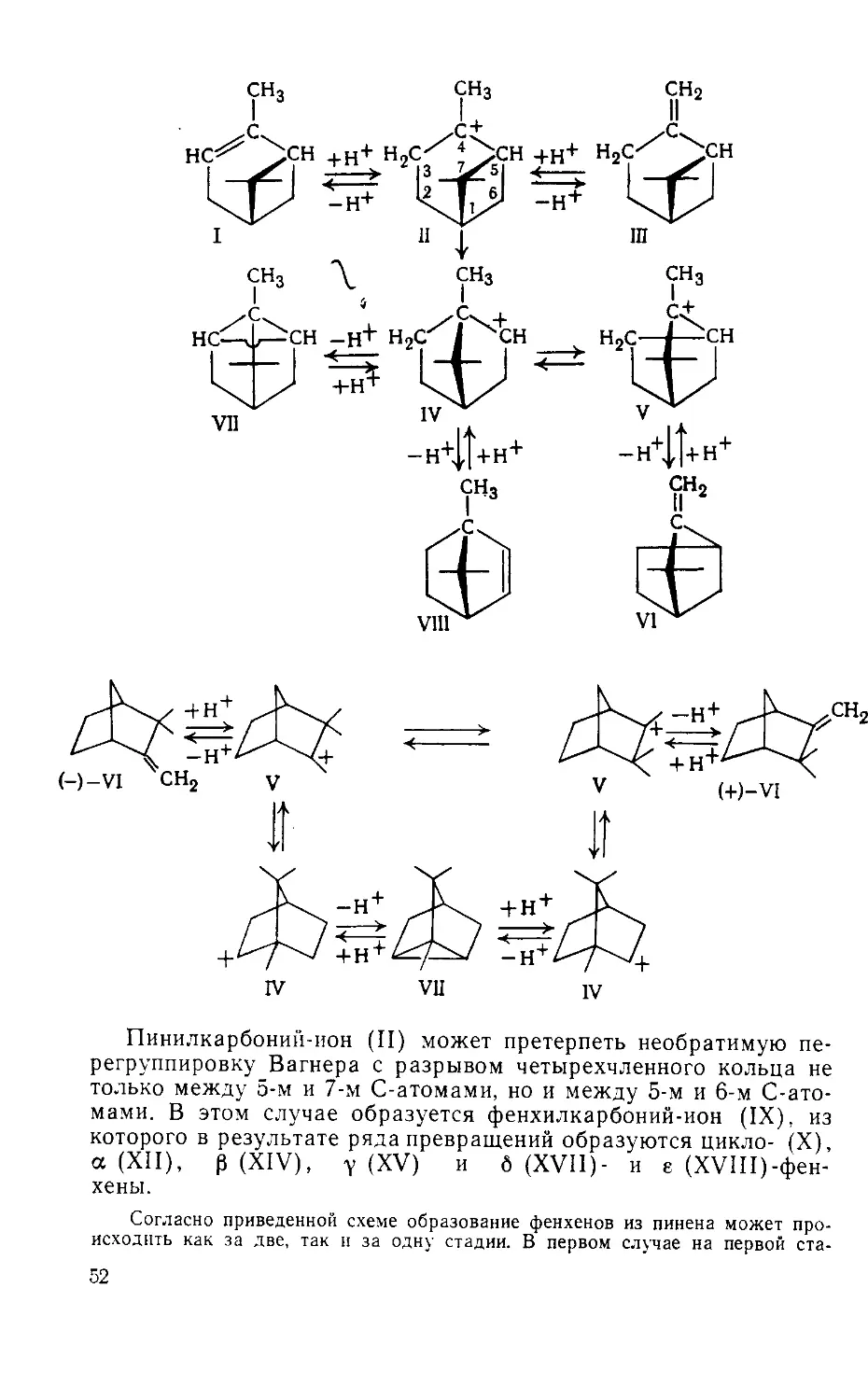

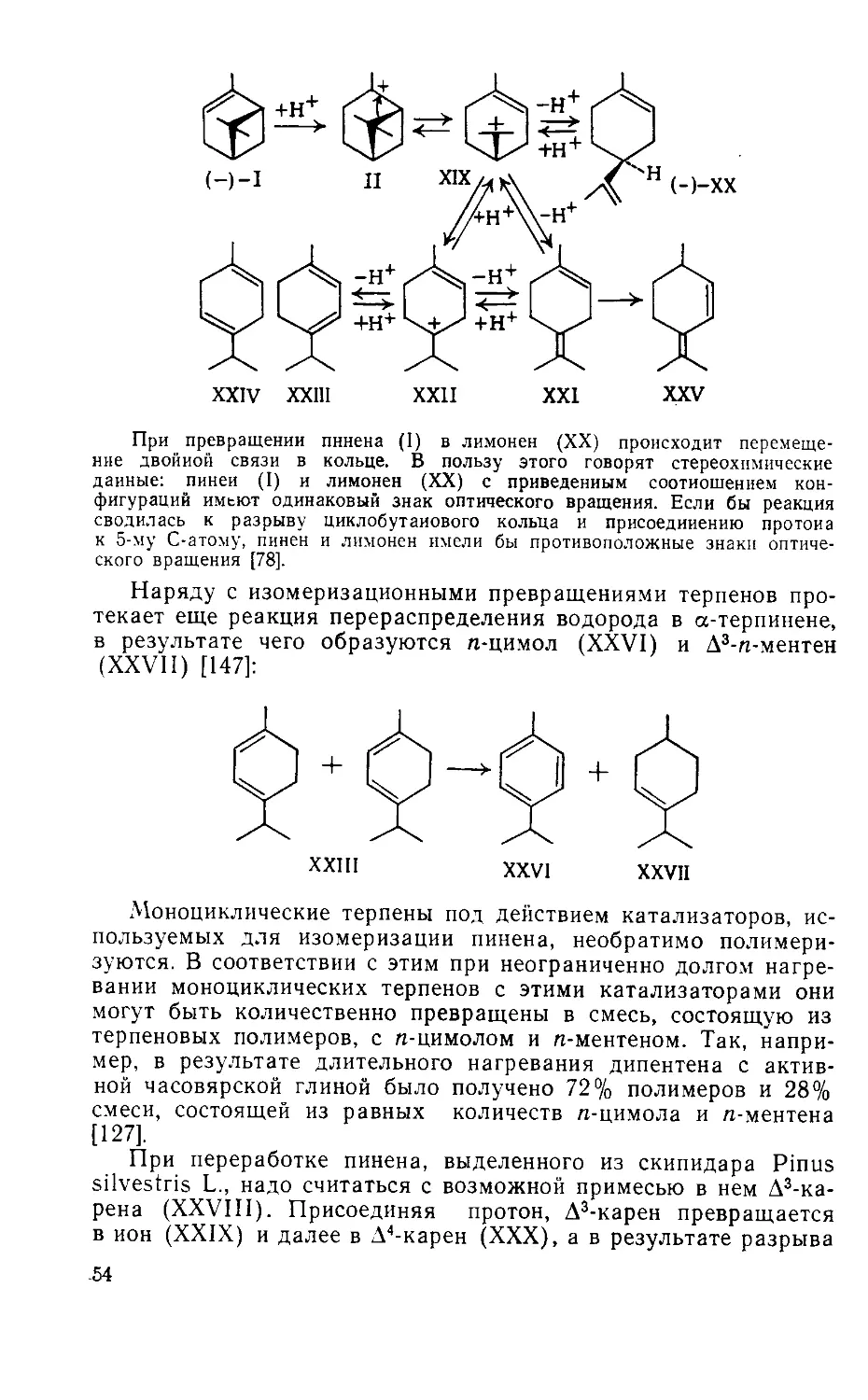




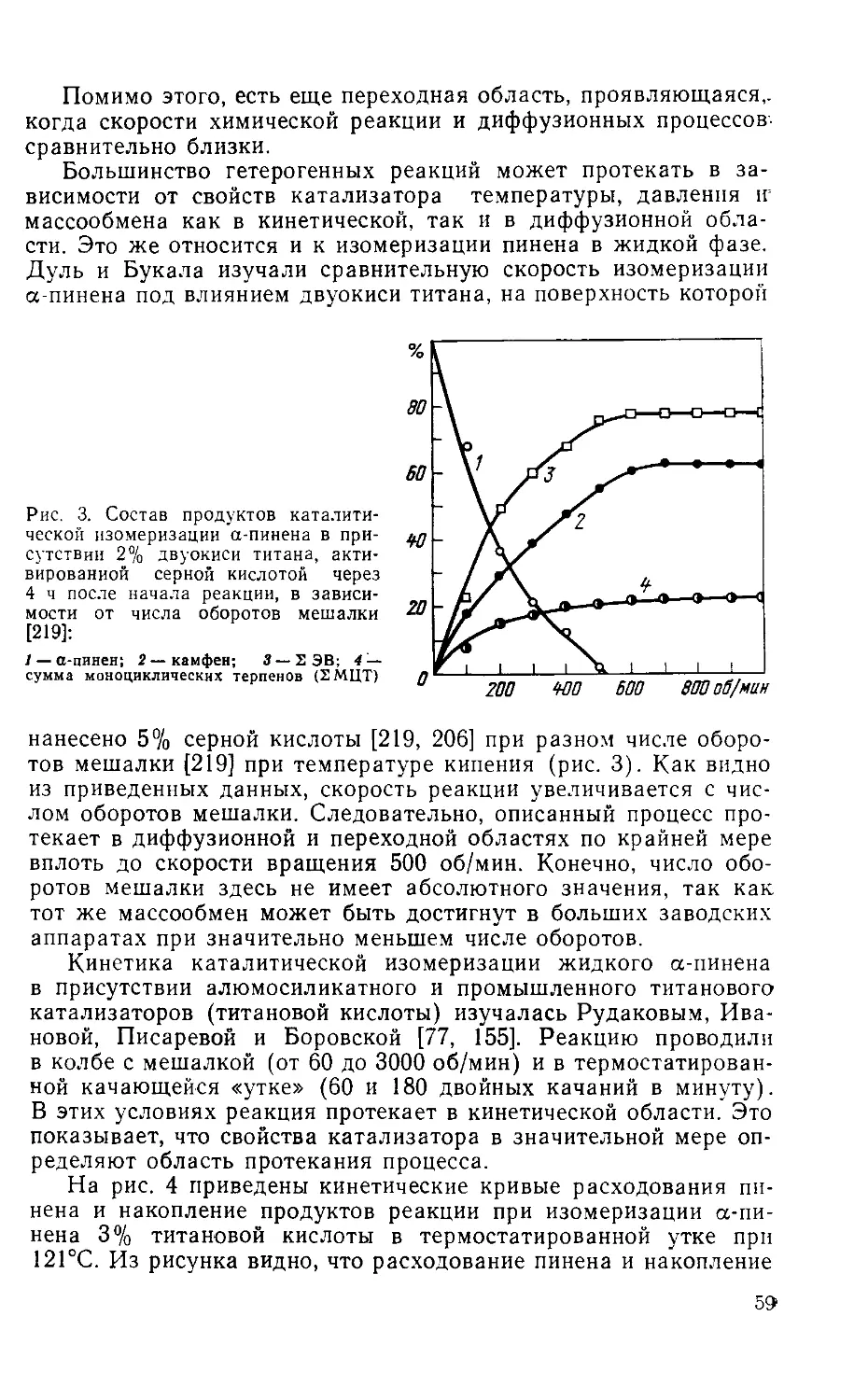
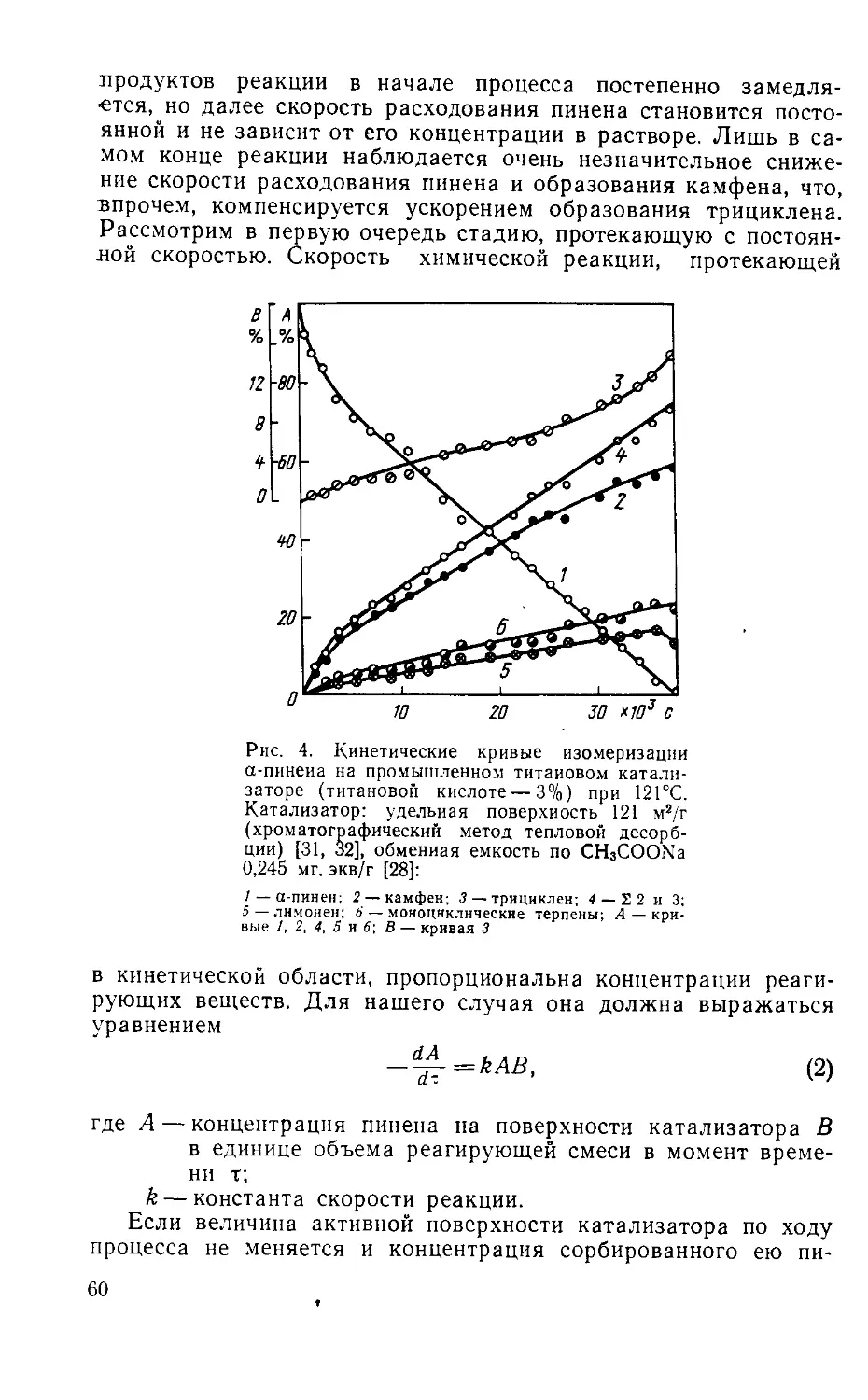

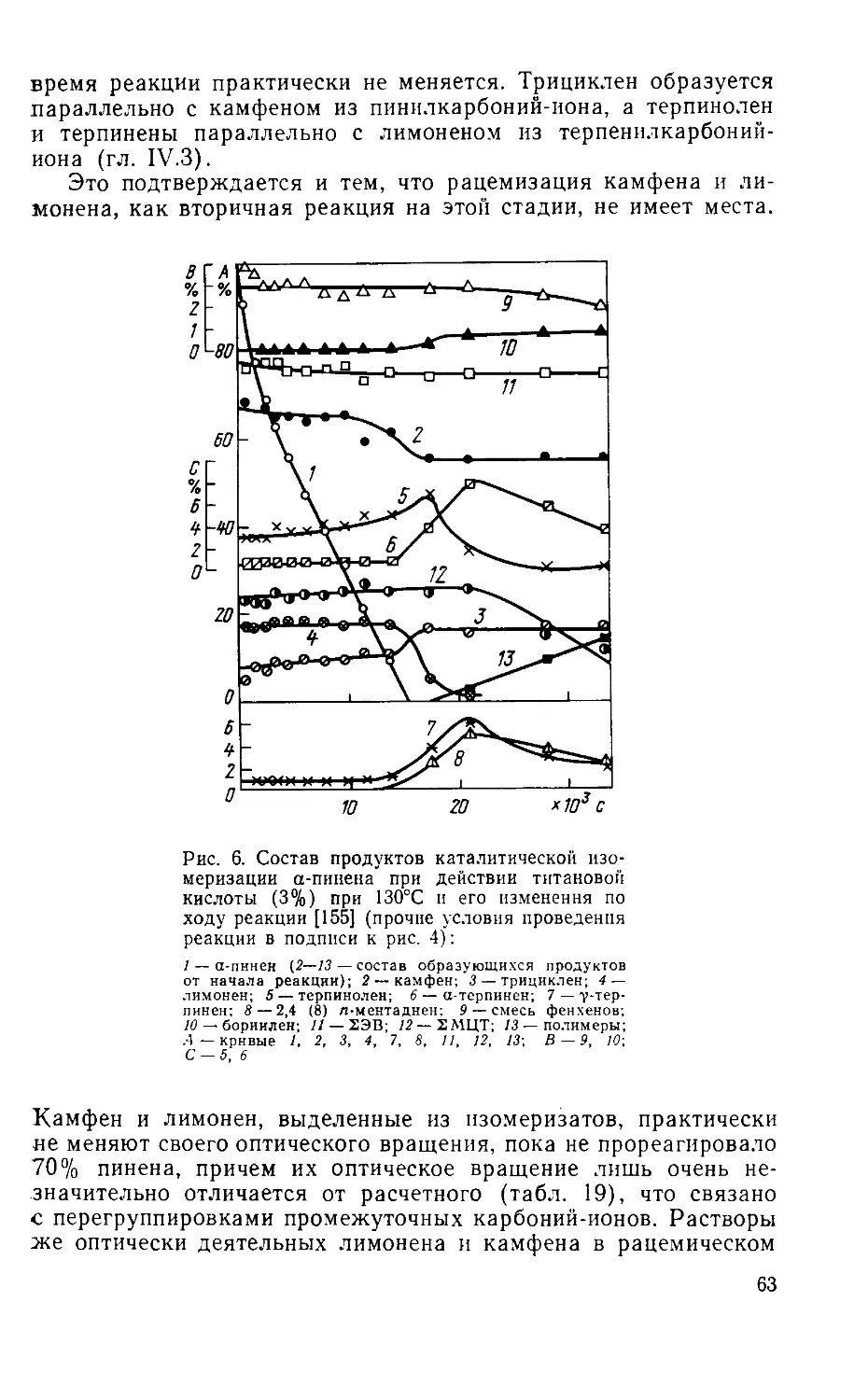




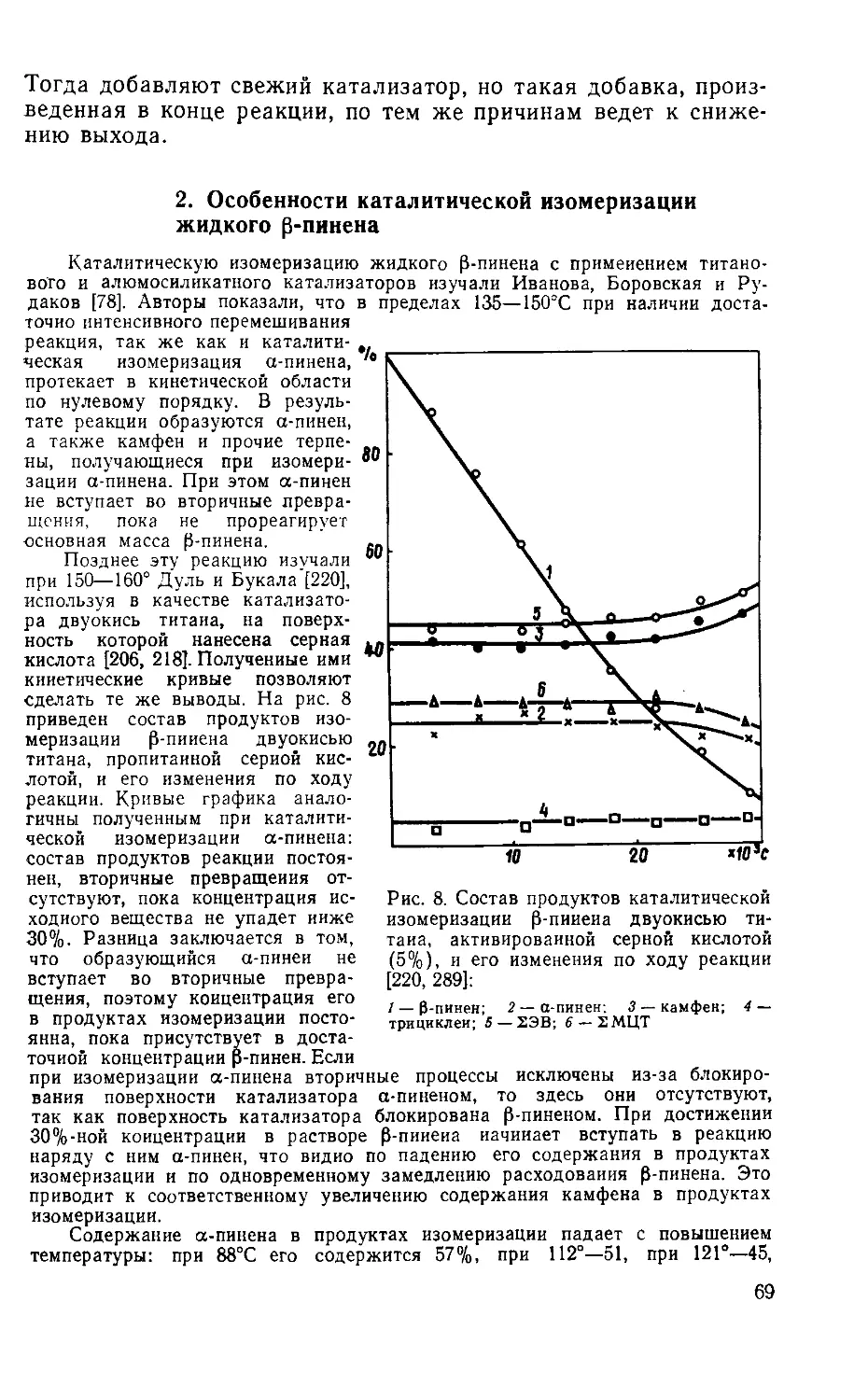























































































































Author: Рудаков Г.А.
Tags: органическая химия химическая промышленность лесная промышленность лесохимическая промышленность
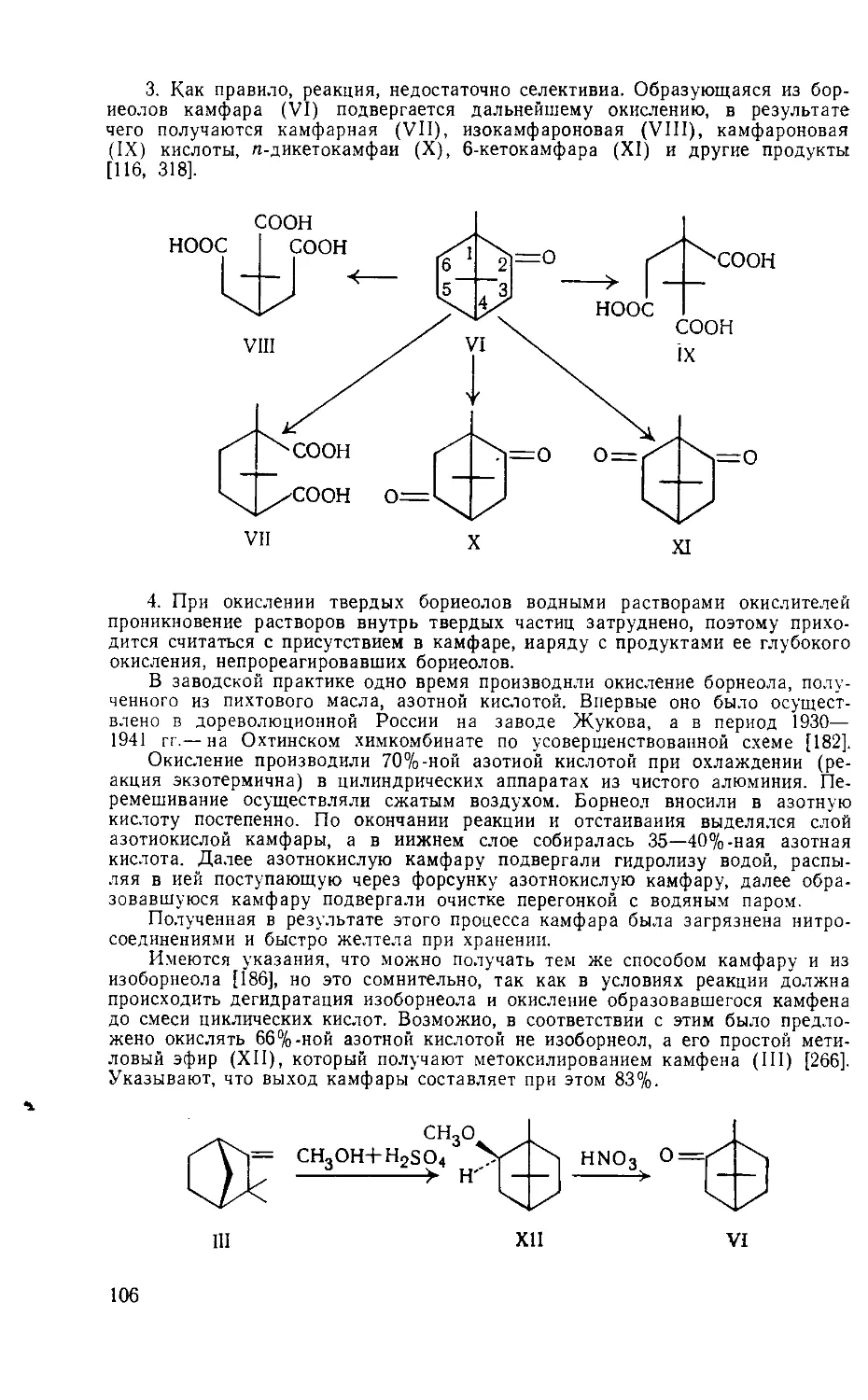
Year: 1976




Text
______Г. A. Ру д а к о в
ХИМИЯ И ТЕХНОЛОГИЯ
КАМФАРЫ
ИЗДАНИЕ 2-е,
ИСПРАВЛЕННОЕ И ДОПОЛНЕННОЕ
ИЗДАТЕЛЬСТВО
«ЛЕСНАЯ ПРОМЫШЛЕННОСТЬ»
Москва 1976
УДК 547.599.6
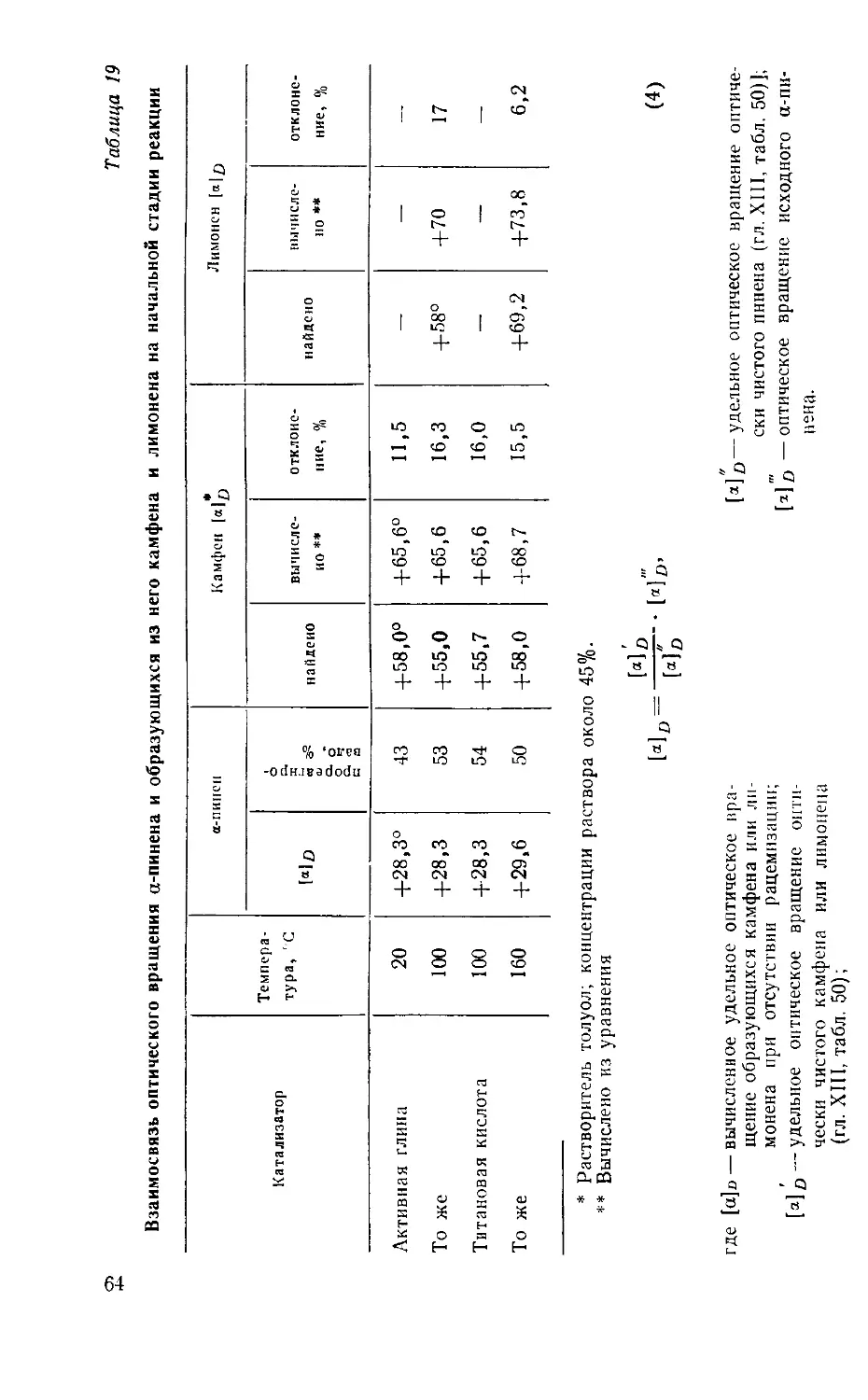
Химия и технология камфары, изд. 2-е, исправленное и дополненное.
Г. А. Рудаков. М„ «Лесная промышленность», 1976, с. 208.
Рассмотрены химические процессы, на которых основано производство
камфары, и описана технология этого производства. Приведены данные спра-
вочного характера о производстве и составе скипидаров, пихтового масла,
о составе естественной и синтетической камфары различного происхожде-
ния, о наиболее достоверных свойствах и физических константах терпенов.
Большое внимание уделено методам исследования, в том числе газожидкост-
ной хроматографии.
Монография предназначена для научных и инженерно-технических работ-
ников лесохимической промышленности.
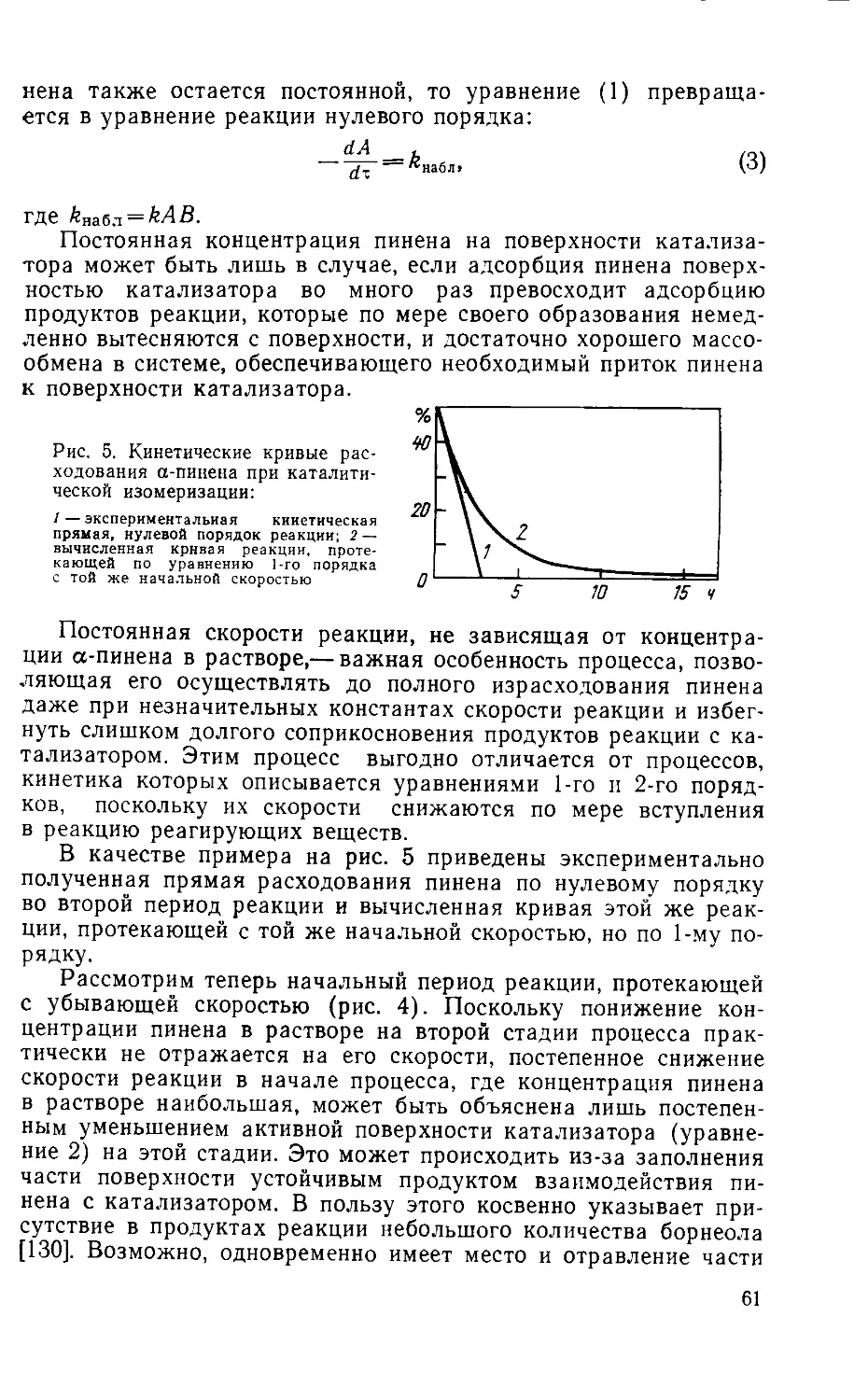
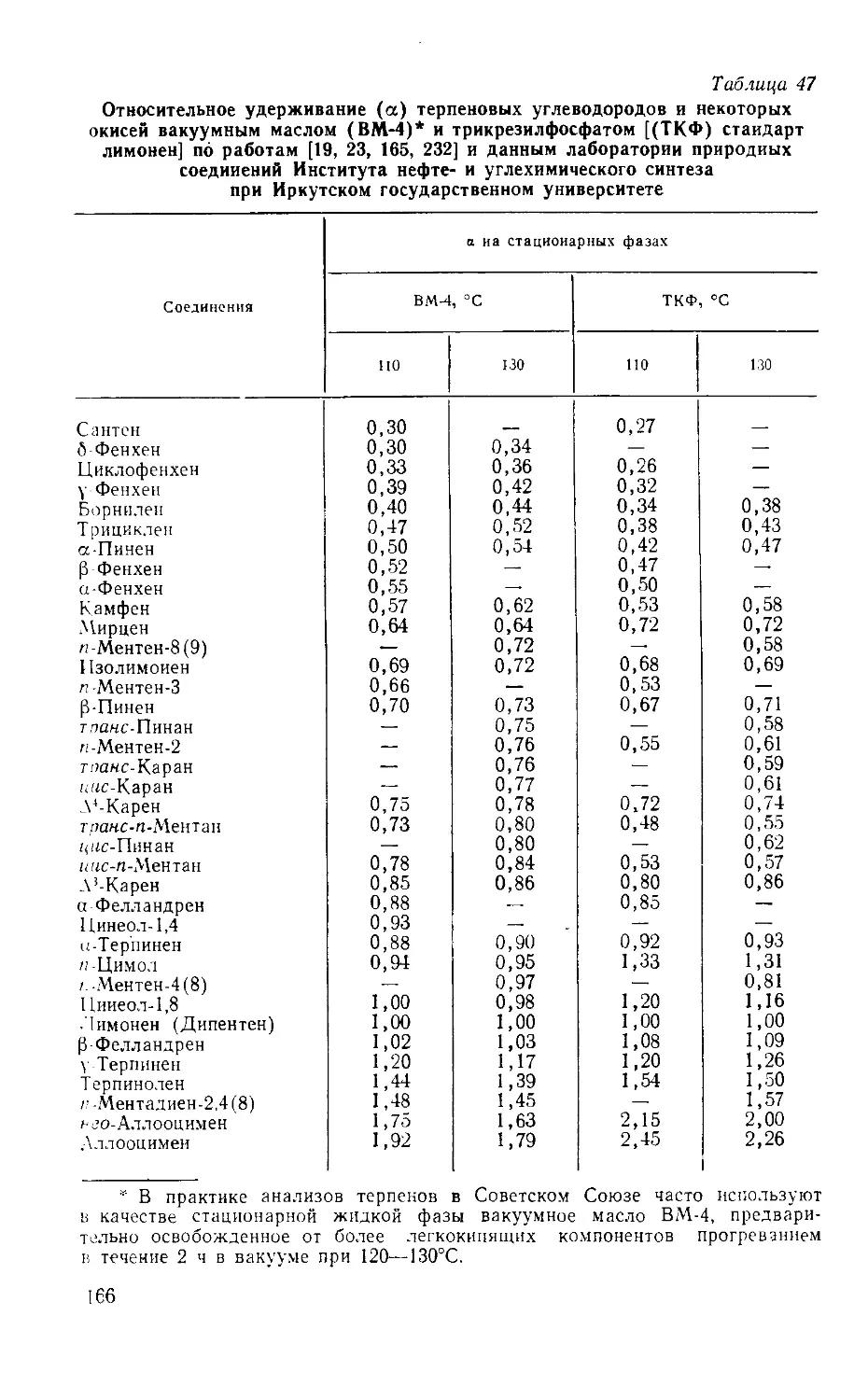
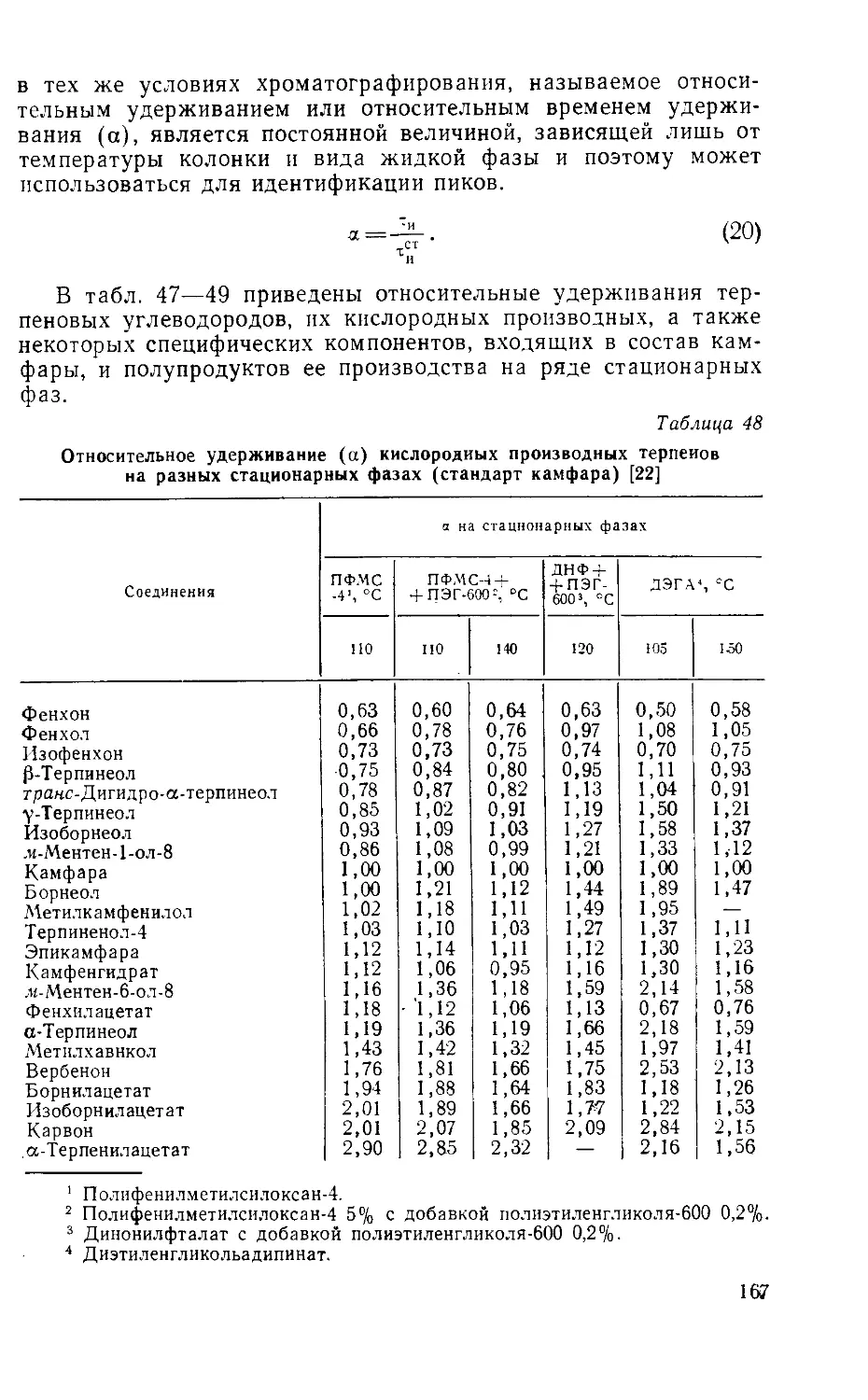
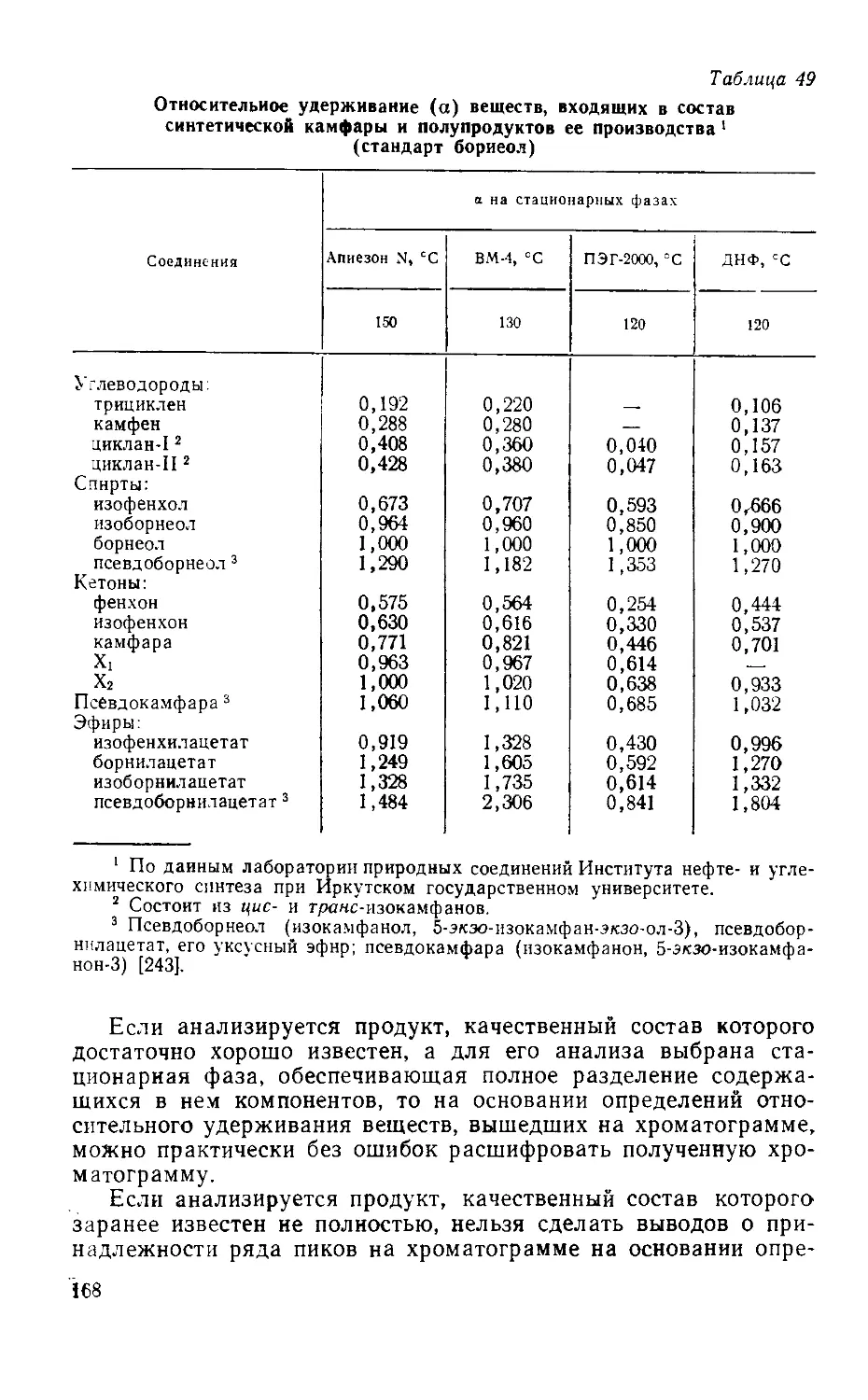
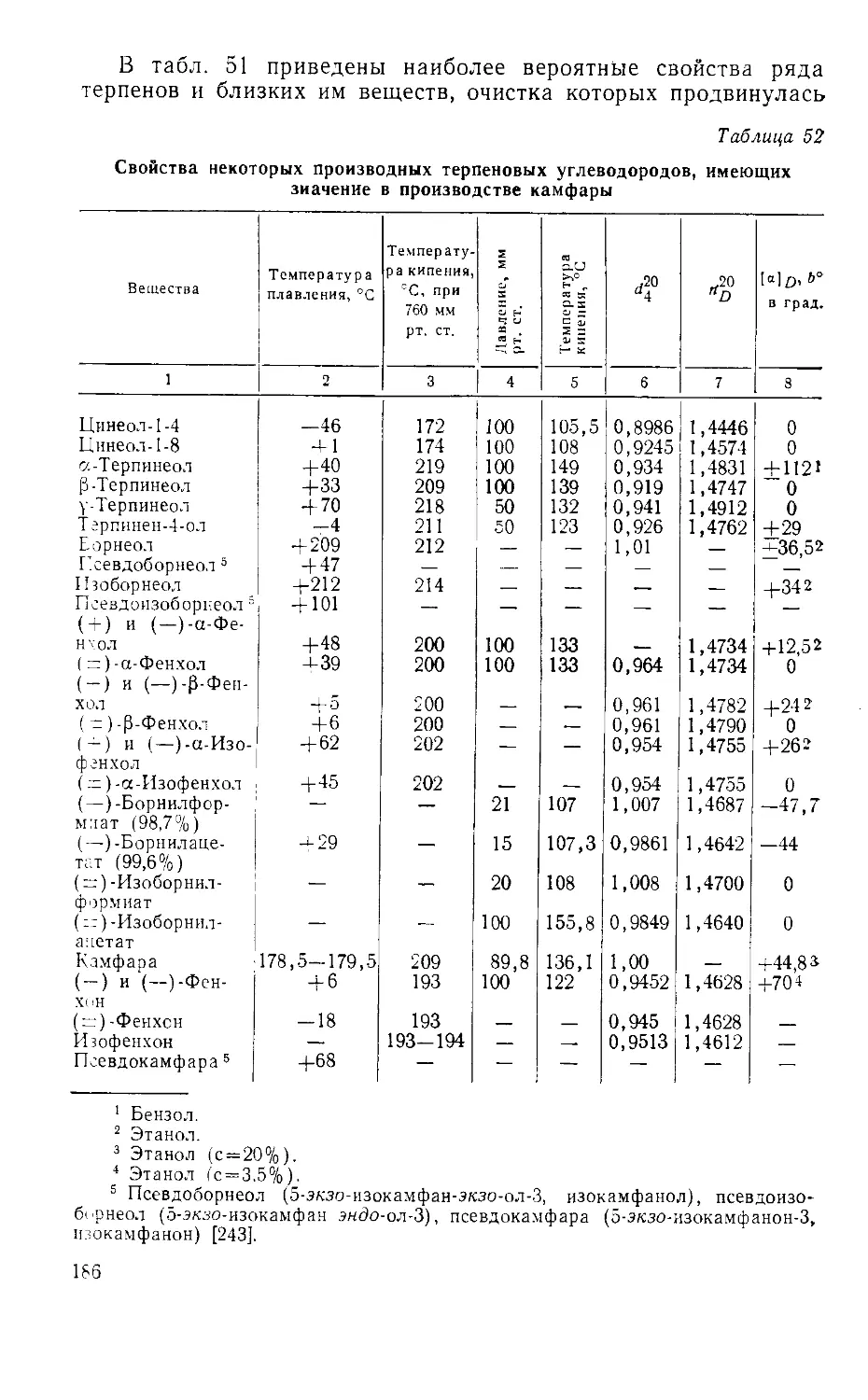
Табл. 53, ил. 31, библиогр.— 340 назв.
р 31414—084
037(01)-76
104—76
© Издательство «Лесная промышленность», 1976
ВВЕДЕНИЕ
Последнее десятилетие на мировых рынках ощу-
щается недостаток скипидаров и восполнение де-
фицита в ближайшие годы вряд ли возможно. Это
неизбежно должно привести к перемещению производ-
ства ряда весьма необходимых продуктов, основанного
на химической переработке скипидара, в страны,
имеющие собственную сырьевую базу. Поэтому эти
производства должны привлекать к себе внимание на-
шей страны, вырабатывающей значительное количе-
ство канифольно-скипидарных продуктов. К указан-
ным производствам относится и производство синтети-
ческой камфары. В то же время современная литера-
тура по синтезу камфары и химической переработке
терпенов на русском языке практически отсутствует.
Это вызвало необходимость переиздать настоящую
книгу, выпущенную небольшим тиражом в 1961 году.
За этот период накопился большой материал, по-но-
вому освещающий химизм отдельных стадий производ-
ства синтетической камфары, разработаны новые тех-
нологические процессы производства.
Настоящая книга окажется полезной не только
ученым и специалистам, непосредственно связанным
с производством камфары, студентам, изучающим это
производство, но и широкому кругу лесохимиков
и химиков-органиков, работающих в области кани-
фольно-скипидарной промышленности, а также химии
и технологии терпенов.
Глава XI настоящей книги написана автором сов-
местно с научным сотрудником руководимой им лабо-
ратории природных соединений Института нефте-и уг-
лехимического синтеза при Иркутском университете
им. Жданова В. И. Гармашовым, а глава XII — сов-
местно с заведующим лабораторией хроматографии
того же института канд. хим. наук Р. И. Сидоровым
и В. И. Гармашовым.
1*
3
Большую помощь оказали, прислав различные ма-
териалы и оттиски статей, работники заводов:
Е. Б. Богданов, Л. П. Метешкина, 3. Л. Чистякова,
ст. научный сотрудник Центрального научно-исследо-
вательского и проектного института лесохимической
промышленности Г. В. Нестеров, ст. научный сотруд-
ник Всесоюзного института пластмасс В. И. Любоми-
лов, доцент Ленинградской лесотехнической академии
им. Кирова В. А. Выродов, чл.-корр. АН БССР
И. И. Бардышев, профессор Томского медицинского
института А. С. Саратиков, профессор Вроцлавского
политехнического института М. Букала, зав. произ-
водством камфары фирмы Хехст (ФРГ) доктор
Г. Кейхер, глава Невель-Сторес лаборатории департа-
мента земледелия США Рэй В. Лауренс. Всем им и
канд. техн, наук Л. Г. Сливкину, оказавшему большую
помощь в подготовке рукописи, автор приносит иск-
реннюю благодарность.
ГЛАВА I. ОБЩИЕ СВЕДЕНИЯ О ПРОИЗВОДСТВЕ
КАМФАРЫ
1. Камфара, ее свойства, производство
и потребление
Камфара (I) (камфанон-2, борнанон-2 или 1,7,7-триметил-
бицикло 1,2,2-гептанон-2)—терпеновый бициклический кетон,
который находит широкое применение в медицине, промышлен-
ности и в быту:
8
I
Внешне камфара белое кристаллическое вещество с харак-
терным, приятным запахом. Ее эмпирическая формула СюЬЬбО,
молекулярная масса 152,23, т. пл. 178,5—179,5°С, т. кип. 207,4—
209,1°С. Камфара мало растворима в воде (~1 г/л) и хорошо
растворима в большинстве органических растворителей: спирте,
диэтиловом эфире, бензоле, петролейном эфире, сложных эфи-
рах, кетонах, галоидопроизводиых углеводородов, уксусной кис-
лоте и других, а также в крепкой серной кислоте и жидком
сернистом ангидриде.
Кристаллы камфары обладают значительной летучестью,
легко слеживаются и под небольшим давлением превращаются
в прозрачную глыбу.
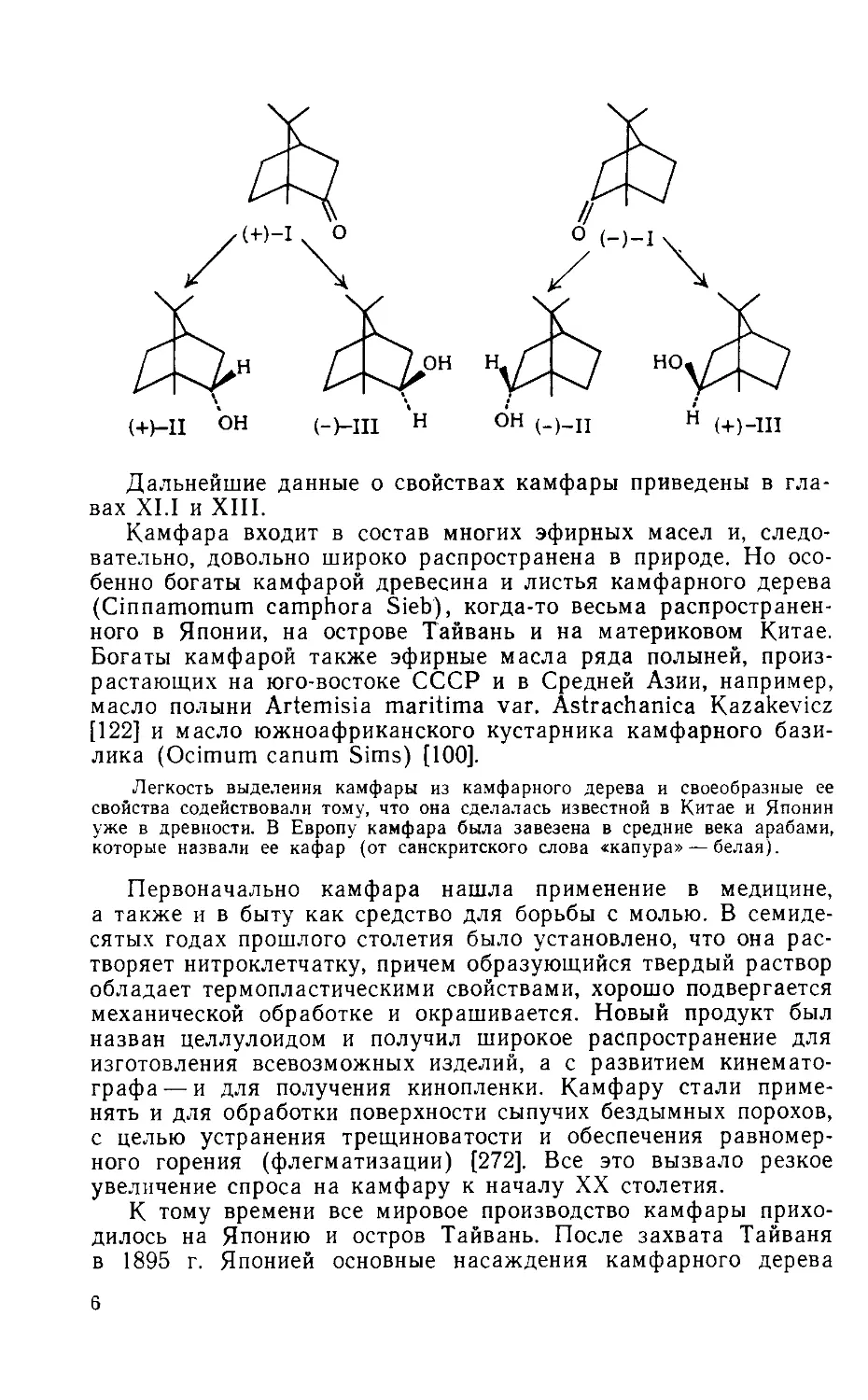
Камфара (I) известна в виде правого ( + ) и левого ( —)
оптических изомеров. На схеме показаны их абсолютные кон-
фигурации [95, 196, 267].
При восстановлении водородом камфара превращается в изо-
мерные спирты: борнеол (II) и изоборнеол (III). Эти спирты
различаются пространственным расположением атомов водорода
и гидроксильных групп при 2-м С-атоме: у борнеола группа ОН
направлена внутрь кольца (эндо), а у изоборнеола — наружу
(экзо). Из ( + )-камфары получается ( + )-борнеол и (—)-изо-
борнеол, а из (—)-камфары (—)-борнеол и ( + )-изобор-
неол.
5
Дальнейшие данные о свойствах камфары приведены в гла-
вах XI.I и XIII.
Камфара входит в состав многих эфирных масел и, следо-
вательно, довольно широко распространена в природе. Но осо-
бенно богаты камфарой древесина и листья камфарного дерева
(Cinnamomum camphora Sieb), когда-то весьма распространен-
ного в Японии, на острове Тайвань и на материковом Китае.
Богаты камфарой также эфирные масла ряда полыней, произ-
растающих на юго-востоке СССР и в Средней Азии, например,
масло полыни Artemisia maritima var. Astrachanica Kazakevicz
[122] и масло южноафриканского кустарника камфарного бази-
лика (Ocimum canum Sims) [100].
Легкость выделения камфары из камфарного дерева и своеобразные ее
свойства содействовали тому, что она сделалась известной в Китае и Японии
уже в древности. В Европу камфара была завезена в средние века арабами,
которые назвали ее кафар (от санскритского слова «капура» — белая).
Первоначально камфара нашла применение в медицине,
а также и в быту как средство для борьбы с молью. В семиде-
сятых годах прошлого столетия было установлено, что она рас-
творяет нитроклетчатку, причем образующийся твердый раствор
обладает термопластическими свойствами, хорошо подвергается
механической обработке и окрашивается. Новый продукт был
назван целлулоидом и получил широкое распространение для
изготовления всевозможных изделий, а с развитием кинемато-
графа— и для получения кинопленки. Камфару стали приме-
нять и для обработки поверхности сыпучих бездымных порохов,
с целью устранения трещиноватости и обеспечения равномер-
ного горения (флегматизации) [272]. Все это вызвало резкое
увеличение спроса на камфару к началу XX столетия.
К тому времени все мировое производство камфары прихо-
дилось на Японию и остров Тайвань. После захвата Тайваня
в 1895 г. Японией основные насаждения камфарного дерева
6
оказались в руках этой страны. Японское правительство ввело
государственную монополию на право приобретения сырой кам-
фары и камфарного масла у кустарей, дальнейшую переработку
этих продуктов и их сбыт. Монополия была введена на Тайване
с 1899 г. и в Японии с 1903 г. Государство устанавливало пла-
новые цифры производства камфары, сдаточные и отпускные
цены.
Введение монополии позволило Японии резко поднять цены
на камфару, которые почти удвоились за первые 7 лет ее суще-
ствования. Однако высокие цены явились одновременно стиму-
лом для других стран в разрешении проблемы изыскания соб-
ственных источников получения камфары.
При решении этой проблемы были использованы два пути:
организация плантаций камфароносов и химический синтез.
Первый путь не привел к ощутимым результатам, второй же
после долгих усилий и ряда неудач привел к созданию про-
мышленности синтетической камфары в ряде стран.
Первые предприятия, получающие синтетическую камфару,
возникли в период с 1899 по 1907 г. в США, Германии, Фран-
ции, Англии и России. На заграничных предприятиях в качестве
исходного продукта для синтеза камфары был использован тер-
пеновый углеводород пинен, содержащийся в качестве основного
компонента в большинстве скипидаров. В России на заводе
Жукова в качестве исходного продукта был использован бор-
нилацетат, содержащийся в эфирном масле пихты сибирской
(Abies sibirica L.).
Технологические процессы производства камфары на первых
предприятиях были весьма несовершенны, а качество оставляло
желать лучшего. Все же возникновение этих предприятий вы-
звало опасение в Японии, так как успешное их развитие грозило
потерей ее монопольного положения, а поэтому в 1907 г. Япония
значительно снизила цену на естественную камфару [182]. Боль-
шинство предприятий, выпускающих синтетическую камфару,
не выдержало конкуренции и прекратило существование. Про-
изводство синтетической камфары сохранилось только в Герма-
нии на заводе акционерного общества Шеринг в Шарлоттен-
бурге (район Большого Берлина) и к началу первой мировой
войны достигло годовой выработки 600 т камфары в год, а к на-
чалу второй мировой войны достигло 2000 т в год.
В результате исследований, проведенных в разных странах,
производственный синтез камфары был значительно улучшен
и к двадцатым годам текущего столетия он начал успешно кон-
курировать с производством естественной камфары.
Перспективы развития производства синтетической камфары
особенно укрепились в связи с тем, что к этому времени про-
изводство естественной камфары уже не могло покрыть всей
мировой потребности в камфаре без риска почти полного уни-
чтожения за короткий срок насаждений камфарного дерева.
7
В настоящее время производство синтетической камфары
улучшилось в такой степени, что с ним уже не может конку-
рировать производство естественной камфары.
Все же естественная и синтетическая камфара не вполне
идентичные продукты: естественная камфара оптически дея-
тельна, тогда как синтетическая большей частью лишена опти-
ческой деятельности (смесь ( + ) и ( —) изомеров). Кроме того,
естественная и синтетическая камфара различаются по количе-
ству и составу примесей. Естественная камфара выпускается
на рынок в виде очень чистого продукта, содержащего не более
0,5—1% примесей, и имеет температуру кристаллизации 176—
178°С. Техническая синтетическая камфара, полученная из ски-
пидара, выпускается в виде значительно менее чистого продукта.
Ее температура кристаллизации колеблется в пределах 159—
170°С, а содержание примесей (в основном терпеновые кетоны)
составляет 5—10%. Это не мешает ей заменять естественную
камфару в большинстве областей применения.
До сих пор остается спорным вопрос о применении синтети-
ческой камфары в медицине. Существовало мнение, что физио-
логическое действие на организм оказывает только (+)-кам-
фара. Впоследствии это мнение было опровергнуто, поскольку
фармакопеями большинства стран было разрешено применять
надлежащим образом очищенную синтетическую камфару на-
ружно и подкожно [96, 200, 210, 217, 291, 292]. Однако советская
фармакопея [56] разрешает применять синтетическую камфару
только наружно. В то же время она допускает применение для
подкожного введения наряду с ( + )-камфарой ( — )-камфару,
получаемую синтетическим путем из эфирного масла пихты си-
бирской (пихтового масла). Поскольку рацемическая синтети-
ческая камфара, получаемая из скипидара, представляет собой
смесь равных частей ( + ) и (—)-оптических изомеров (см.
гл. XI), каждый из которых разрешен к применению советской
Государственной фармакопией в отдельности, и поскольку син-
тетическая камфара может быть очищена до любой желаемой
степени, запрещение применять синтетическую камфару под-
кожно представляется не обоснованным.
В настоящее время камфара сохранила перечисленные об-
ласти своего применения, но так как производство целлулоида
имеет тенденцию к снижению [192, 325] (табл. 1), мировое про-
изводство камфары несколько снизилось (табл. 2), в основном
за счет производства естественной камфары.
Вряд ли приходится рассчитывать на увеличение производ-
ства целлулоида в мировом масштабе, хотя это вполне воз-
можно в отдельных странах. Поэтому увеличение мирового про-
изводства камфары может произойти лишь в случае, если ей
будут найдены новые области применения, например, в качестве
сырья для химического синтеза, в качестве пластификатора и пр.
Такие поиски ведутся, иногда в самых неожиданных областях,
8
так, например, недавно был взят патент на применение камфары
в качестве компонента твердого ракетного топлива совместно
с декабораном [170].
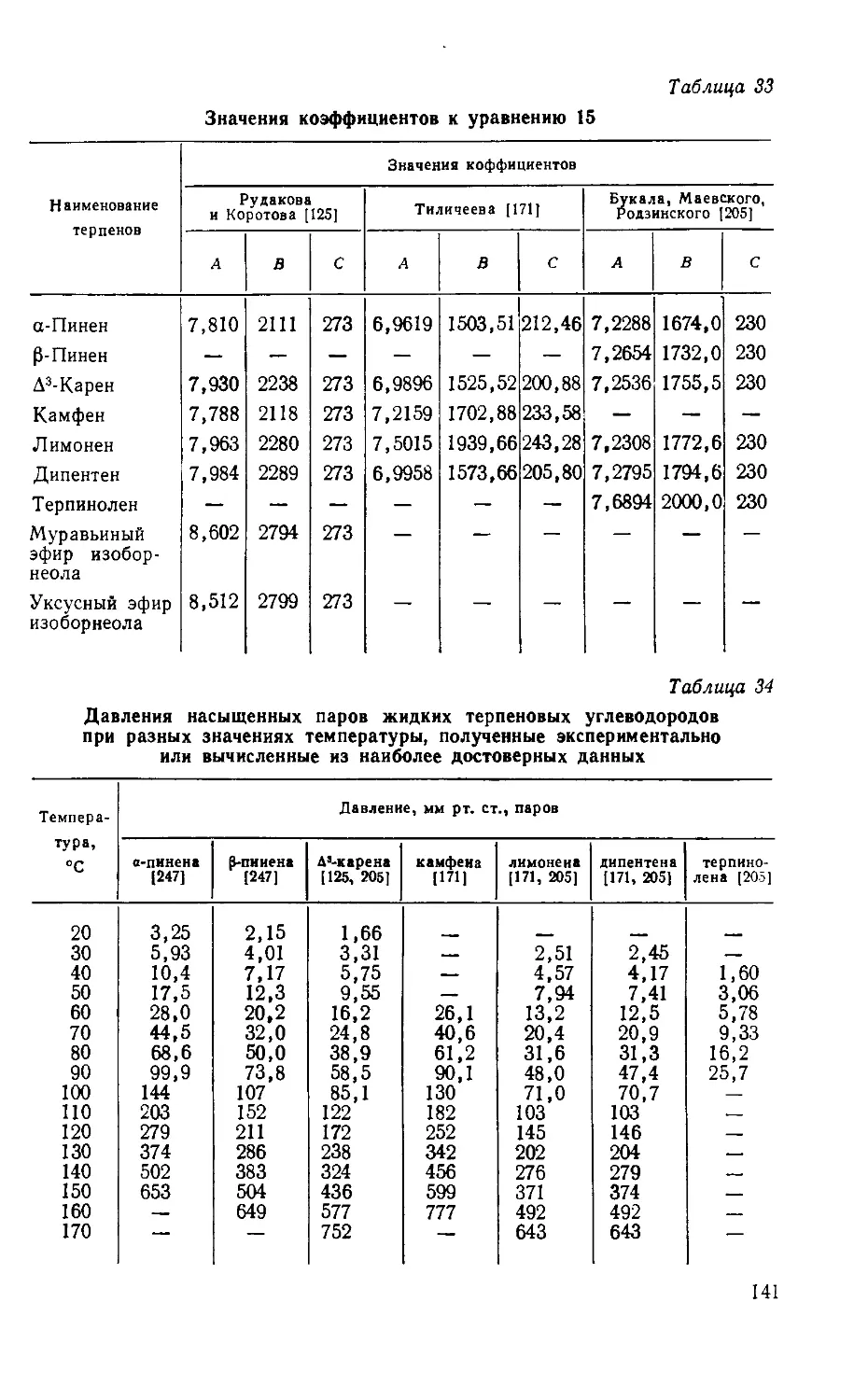
Таблица 1
Мировое производство целлулоида *
и примерный расход на него камфары
Год Производство целлулоида [192], т Примерный расход кам- фары, т
1900 15000 4 250
1914 25 000 6 250
1928 40000 10000
1939 30 000 7 500
1950 25 000 6 250
* Без СССР.
Таблица 2
Мировое производство камфары
в тыс. т [182, 191, 212, 260, 261, 263,
273, 331]
Гои Всего В том числе
естествен- ной синтети- ческой
1905 3,3 3,0 0,3
1914 6,6 6,0 0,6
1928 10,5-11,5 4,5 6-7
1936 11,3 6,3 5
1957 8-9 1,8 7-8
1972 8-9 0,8-1.3 7-8
В то же время надо считаться с тем, что в медицине и
в ряде других областей камфара остается необходимым продук-
том: производство целлулоида в сокращенном объеме продол-
жает существовать и запасы натуральной камфары почти ис-
черпаны, а ее выработка стала нерентабельной (гл. 1.2). В силу
этого производство синтетической камфары, даже если ей не
будет найдено новых областей применения, сохранится на суще-
ствующем уровне.
Многие полупродукты производства синтетической камфары:
пинен, камфен, изоборнилацетат, моноциклические терпены
в настоящее время получили самостоятельное значение, и пред-
приятие, прежде выпускавшее только синтетическую камфару,
ныне выпускает значительно больший ассортимент, продукции,
что повышает его рентабельность даже при отсутствии расши-
рения производства камфары.
Несомненно, на производство синтетической камфары оказы-
вает сильнейшее влияние наметившийся за последние годы на
международных рынках дефицит в скипидарах (гл. II.1). Это
неизбежно приведет к перераспределению производства между
отдельными странами.
В 1957 г. производство синтетической камфары из скипидара было пре-
кращено в Италии (200—500 т/год), а в 1958 г. в США фирма Дюпон за-
крыла завод производительностью до 4000 т камфары в год [191]. Италия
не имеет своего скипидара, а США, хотя и являются главным его производи-
телем, испытывают в нем острый недостаток для других химических произ-
водств. Свои потребности в камфаре США стали покрывать в основном за
счет импорта около 1000 т камфары в год [197]. В то же время в 1964 г.
было организовано производство синтетической камфары в Индии [212, 260].
За рубежом синтетическая камфара производится на следующих пред-
приятиях: ГДР, Chemische Fabrik Finowtal (Finow), ФРГ, Farbwerke Hochst
(Werk Gersthofen), производительность до 3000 т камфары в год [261],
9
Англии. ВХ Plastics Ltd. (Brantham, Manningtree Essex), производительностью
около 1500 т камфары в год [273], Индии, Camphor and Allied Products Ltd.
(Clutterbockganj, Uttar Pradesh), производительностью около 1000 т камфары
в год [260].
Рассматривая в свете вышеизложенного перспективы произ-
водства камфары в перечисленных странах, следует сказать, что
они наименее благоприятны в ФРГ, ГДР и Англии, которым
приходится импортировать необходимый для производства ски-
пидар, а также в Индии, где скипидар очень беден пиненом.
Наоборот, в СССР и КНР перспективы развития производства
камфары благоприятны, поскольку там скипидар производится
в большом количестве и он достаточно богат пиненом. Спрос
на камфару в Советском Союзе за последние годы настолько
возрос, что стоит вопрос об увеличении производственных мощ-
ностей.
Цена на синтетическую камфару иа международных рынках находится
в зависимости от цены на скипидар. В 1960 г. техническая камфара стоила
3500—4000 западногерманских марок за 1 т (800 руб.) [191]. В 1968 г.
1100 американских долларов (1100 руб.) [197], а к 1973 г. достигла 4500—
5000 западногерманских марок за 1 т (1000 руб.). Медицинская камфара це-
нится иа 25—50% дороже.
2. Получение естественной камфары
Почти все мировое производство естественной камфары ба-
зируется на эфирном масле, которое содержится в древесине,
корнях, сучьях, ветвях и листьях камфарного дерева.
Камфарное дерево (Cinnamomum camphora Sieb) встречается в трех фи-
зиологических формах, которые, сохраняя морфологию своего вида, продуци-
руют эфирные масла разного состава. Эти формы по японской терминологии
называются: Хои-шо (истинное камфарное дерево), Хо-шо, или Шиу-шо (ду-
шистое камфарное дерево) и Ю-шо (масляное дерево).
Эфирное масло Хон-шо наиболее богато камфарой и содержит ценный
для парфюмерной промышленности сафрол (метиленовый эфир л-аллилпиро-
катехина). При отгонке эфирного масла из щепы этого дерева часть кам-
фары выделяется в кристаллическом виде. Эфирное масло Хо-шо значительно
беднее камфарой, при отгонке эфирного масла из щепы Хо-шо твердая кам-
фара не выделяется. Несмотря на это, масло Хо-шо ценится, так как содер-
жит важный для парфюмерной промышленности терпеновый спирт линалоол.
Эфирное масло Ю-шо еще беднее камфарой. Оно содержит много цинеола
и а-терпииеола, ио содержание сафрола и линалоола невелико. Поэтому это
масло ценится меньше, чем масла Хон-шо и Хо-шо.
В табл. 3 приведены данные о составе камфарного масла разных физио-
логических форм камфарного дерева.
В Японии почти исключительно росла форма Хои-шо, иа Тайване росли
формы Хон-шо и Хо-шо, а на материковом Китае в основном Ю-шо. К мо-
менту окончания второй мировой войны запасы камфарного дерева и в Японии
и на Тайване сильно истощились. Имеющиеся в Японии насаждения по Хи-
раицуми [254] к 1947 г. содержали около 40 000 т камфары и камфарного
масла, столько же содержалось в насаждениях Тайваня. По данным того же
автора, иа материковом Китае камфарное дерево встречается на площади
около 30 млн. га, запас древесины составляет 9,6 млн. т н запасы камфарного
масла 150 000 т. Однако 90% произрастающих в материковом Китае деревьев
относится к бедной камфарной форме Ю-шо.
10
Таблица 3
Состав эфирных масел из разных физиологических форм камфарного дерева
Компоненты Содержание компонентов в маслах деревьев, %
Хон-шо 1 (Япония) Хо-шо 2 (Тайвань) Ю-шо 2 (материковый Китай)
Монотерпены 14 7,2 18,5
Цинеол 4,6 4,1 21,6
Камфара 45,6 39,6 33,3
Терпеновые спирты 9,9 36,8 3 19,3 4
Сафрол 18,1 7,8 0,8
Сесквитерпены 6,2 2,0 3,0
Прочие 1,2 2,0 1,8
Смолистые вещества 0,4 0,5 1,7
' Данные относятся к жидкой части масла.
2 Данные относятся ко всему маслу.
3 Главным образом линалоол.
4 Главным образом а-терпинеол.
Процесс выделения камфары из камфарного дерева состоит
из двух стадий. Первая стадия начинается с заготовки сырья
и заканчивается отгонкой острым паром сырой камфары и кам-
фарного масла от щепы камфарного дерева. Ее осуществляют
кустари, используя самую примитивную технику. Вторая стадия
заключается в дальнейшей переработке и очистке этих продук-
тов. Это производится на заводских установках.
Выход камфары и камфарного масла зависит от вида и физиологической
формы камфарного дерева, его возраста и от того, какие части дерева пере-
рабатывались. Твердая камфара получается только при переработке камфар-
ного дерева Хон-шо. В табл. 4 приведены данные о выходах камфары и кам-
фарного масла при переработке древесины Хон-шо в зависимости от возраста
дерева.
Сырая камфара (сорт В), полученная кустарями, содержит
около 2,5% воды, около 2% масел и около 0,05% нерастворимых
в органических растворителях примесей. Сырую камфару и кам-
форное масло подвергают дальнейшей переработке на крупных
заводских установках.
Очистка сырой камфары достигается однократной или дву-
кратной сублимацией. Однократно сублимированная камфара
(сорт ВВ) содержит 99% камфары и 1% масел, а дважды суб-
лимированная (сорт RC)—99,5% камфары и 0,5% масел.
Камфарное масло подвергают ректификации на многоколон-
ных установках непрерывного действия. При этом получают
камфару, по своему качеству примерно соответствующую сорту
ВВ, которую направляют на дальнейшую очистку, а также ряд
11
Таблица 4
Выход камфары и камфарного масла при переработке древесины Хон-шо
в зависимости от возраста дерева
Возраст, лет Размеры дерева Выход от массы древесины, %
высота, м диаметр, см объем древесины, пл. м3 камфары камфар- ного масла всего
5 1,75 1,19 0,0006 1,00 1,00
10 4,68 5,92 0,0095 0,13 1,08 1,21
20 8,78 15,37 0,1170 0,50 1,50 2,00
30 11,52 24,82 0,3473 0,82 1,40 2,22
40 13,48 34,22 0,7255 1,00 1,30 2,30
50 14,94 43,72 1,2760 1,10 1,30 2,40
100 — 78,10 — 1,60 1,10 2,70
жидких продуктов, выпускаемых под названиями: белое, корич-
невое (красное), синее масла и терпинеол. Некоторые из этих
масел представляют значительную ценность, превосходящую
ценность камфары. Особо ценными являются коричневое масло,
содержащее до 50—60% сафрола, используемого для синтеза
гелиотропина, и масло «Хо», получаемое при перегонке камфар-
ного масла дерева Хо-шо и содержащее до 80—85%линалоола.
Само собой разумеется, что крайне примитивный технический
уровень первой стадии производства, связанный с большой за-
тратой рабочей силы и, вероятно, с большими потерями про-
дуктов, не может обеспечить рентабельного производства. По-
этому падение производства естественной камфары объясняется
не только истощением запасов камфарного дерева, но и этой
причиной.
Уже к моменту окончания второй мировой войны производство естествен-
ной камфары в Японии было бы нерентабельным, если бы наряду с камфа-
рой не получали сафрол, используемый в парфюмерии для получения гелио-
тропина. Поэтому делались новые посадки камфарного дерева с расчетом,
что в этих условиях производство просуществует еще многие годы. Эти рас-
четы не оправдались в связи с быстрым развитием производства сассафрасо-
вого масла в Бразилии.
В результате этого производство камфары в Японии пришло к полному
упадку. Есть предположение, что оно в ближайшие 5—10 лет полностью пре-
кратится [242]. На Тайване производится до 500—1000 т камфары в год [241].
Здесь производство имеет более прочную основу, чем в Японии, так как
в качестве отхода переработки древесины Хо-шо получают дорогой линалоол.
Однако и здесь можно ждать спада производства в близком будущем.
На Тайване же была открыта новая физиологическая форма камфарного
дерева Хо-шо, названная линалооловым деревом (Cinnamomum camphora
Sieb., subsp. formosana, var. ofientalis, subvar. Linaloola Hirota), в листьях
которого находится масло (масло «Хо») с 85% линалоола.
Линалооловое дерево в кустящейся форме стали разводить в Японии на
труднодоступных землях и на Тайване. Несомненно, с этим источником лина-
12
лоола будет трудно конкурировать переработке древесины Хо-шо, что приве-
дет к дальнейшему спаду производства камфары.
До войны в Советском Союзе была сделана попытка культивации афри-
канского кустарника — камфарного базилика (Ocimum canum Sims.) с целью
получения оптически деятельной камфары для медицинских целей. Посевы
этой культуры проводили на Северном Кавказе и на Украине. С 1 га план-
таций собирали 8—10 т зеленой массы, а из нее получали 40 кг масла, со-
держащего 30—35 кг камфары [100]. Однако в результате освоения производ-
ства значительно более дешевой синтетической камфары культивация и пере-
работка камфарного базилика была прекращена.
3. Получение синтетической камфары [182, 189, 226,
263, 268, 331]
Под синтетическими подразумеваются продукты, которые
получены с помощью химических реакций из элементов: угле-
рода, водорода, кислорода и т. д. Комппа, а также Перкин мл.
и Торп осуществили такой синтез камфары [99, 116, 318]. Он
имел большое научное принципиальное значение, но был не
применим на практике из-за сложности и низких выходов.
Предпочтительнее использовать для получения камфары уже
созданные природой и доступные терпены, в первую очередь
а- и р-пинены, основные компоненты скипидаров, камфен и бор-
нилацетат, содержащиеся в хвойном эфирном масле пихты си-
бирской (пихтовом масле) и, возможно, борнеол, содержащийся
в высших фракциях экстракционного скипидара (Пайн-оле).
Таким образом, термин «синтетическая» в отношении камфары
не совсем точен. В патентной литературе указывалось на воз-
можность применения в качестве исходного продукта для син-
теза камфары ароматического углеводорода п-цимола [316].
Если бы получали камфару из этого сырья, она имела бы
больше основания называться синтетической.
Способы, предложенные для синтеза камфары из а-пинена
по первой стадии этого процесса (она самая важная), можно
разделить на четыре группы.
К первой группе можно отнести способы синтеза камфары
(I) из а-пинена (IV) через борнилхлорид (V). Борнилхлорид об-
разуется при взаимодействии а-пинена с сухим хлористым водо-
родом. При нагревании со щелочами борнилхлорид (V) претер-
певает изомеризацию и отщепляет хлористый водород, превра-
щаясь в камфен (VI). Камфен действием органической кислоты
в присутствии серной кислоты превращают в эфир изоборнеола
(VII), который омыляют едким натром и образовавшийся изо-
борнеол (III) превращают в камфару (I) каталитической дегид-
рогенизацией. До середины тридцатых годов синтез камфары из
пинена осуществляли по описанной схеме, которую часто назы-
вают классической.
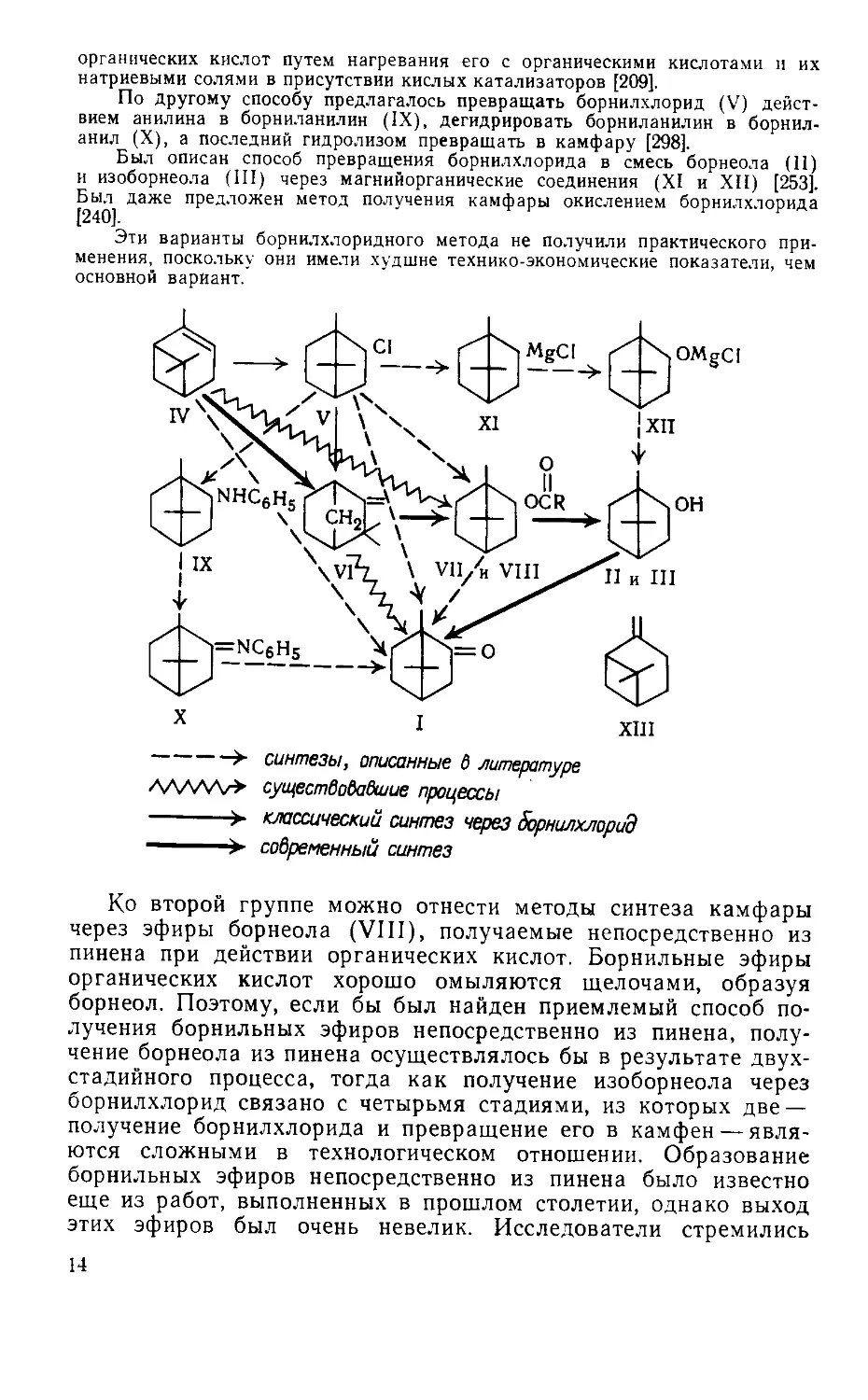
Кроме синтеза камфары через борнилхлорид — камфен, в литературе
были описаны и другие методы, при которых камфен в качестве промежуточ-
ного продукта не получается. Так, было предложено непосредственно пре-
вращать борнилхлорид в борнильные (VIII) и изоборннльные (VII) эфиры
13
органических кислот путем нагревания его с органическими кислотами и их
натриевыми солями в присутствии кислых катализаторов [209].
По другому способу предлагалось превращать борнилхлорид (V) дейст-
вием анилина в борниланилин (IX), дегидрировать борниланилин в борнил-
анил (X), а последний гидролизом превращать в камфару [298].
Был описан способ превращения борнилхлорида в смесь борнеола (11)
и изоборнеола (III) через магнийорганические соединения (XI и XII) [253].
Был даже предложен метод получения камфары окислением борнилхлорида
[240].
Эти варианты борнилхлоридного метода не получили практического при-
менения, поскольку они имели худшие технико-экономические показатели, чем
основной вариант.
синтезы, описанные б литературе
существовавшие процессы
--------> классический синтез через 5орнилхлорид
- > современный синтез
Ко второй группе можно отнести методы синтеза камфары
через эфиры борнеола (VIII), получаемые непосредственно из
пинена при действии органических кислот. Борнильные эфиры
органических кислот хорошо омыляются щелочами, образуя
борнеол. Поэтому, если бы был найден приемлемый способ по-
лучения борнильных эфиров непосредственно из пинена, полу-
чение борнеола из пинена осуществлялось бы в результате двух-
стадийного процесса, тогда как получение изоборнеола через
борнилхлорид связано с четырьмя стадиями, из которых две —
получение борнилхлорида и превращение его в камфен — явля-
ются сложными в технологическом отношении. Образование
борнильных эфиров непосредственно из пинена было известно
еще из работ, выполненных в прошлом столетии, однако выход
этих эфиров был очень невелик. Исследователи стремились
14
к тому, чтобы подобрать такие органические кислоты и условия
реакции, которые обеспечили бы высокий выход эфиров.
По получению борнильных эфиров непосредственно из пинена
были взяты многочисленные патенты. Для взаимодействия с пи-
неном предлагались: борная [252], уксусная [340], салициловая
[208], себациновая [213], щавелевая [185], тетрахлорфталевая
[227] и многие другие кислоты. Отличительной особенностью
этих способов является получение оптически деятельного бор-
неола, а следовательно и камфары. Предлагалось осуществлять
взаимодействие пинена с кислотами также в присутствии ката-
лизаторов, например, взаимодействие с уксусной кислотой
в присутствии борного ангидрида [314] и взаимодействие со ща-
велевой кислотой в присутствии хлористого алюминия [304].
В этом случае получались эфиры изоборнеола. Предлагалось
также использовать направляющее действие растворителей [214].
Эти исследования не привели к положительным результатам,
так как выход эфиров борнеола получался низкий из-за побоч-
ных реакций, приводящих к образованию больших количеств
моноциклических терпенов. Кроме того, в этом случае одновре-
менно с эфирами борнеола образуется значительное количество
эфиров фенхилового спирта [215], что осложняет получение кам-
фары, соответствующей по своему качеству международным
стандартам.
Предприятие, впервые в мире наладившее производство синтетической
камфары на Ниагарском водопаде (США, 1905 г.), получало щавелевый эфир
борнеола действием щавелевой кислоты на пинен. Предприятие просущество-
вало недолго, что и понятно, так как выход щавелевого эфира борнеола не
превышал 30%.
После первой мировой войны во Франции существовало производство
синтетической камфары через тетрахлорфталевый эфир борнеола, получае-
мый действием тетрахлорфталевой кислоты на пинен. Тетрахлорфталевый
эфир борнеола труднолетуч и легко отделяется от терпеновых углеводородов.
Камфара, полученная этим методом, сохраняла оптическую деятельность, но
имела низкую температуру кристаллизации (157—161°С), по-видимому из-за
значительной примеси фенхона. Производство просуществовало недолго, так
как не могло конкурировать с предприятиями, получающими синтетическую
камфару другими методами.
Начиная с тридцатых годов исследования по получению эфи-
ров борнеола действием органических кислот на пинен практи-
чески прекратились. Из работ этого периода заслуживает вни-
мания попытка установить зависимость между константами
диссоциации кислот и выходом эфиров борнеола при взаимодей-
ствии этих кислот с пиненом [265].
К третьей группе относятся методы синтеза камфары, первой
стадией которых является каталитическая изомеризация пинена
в камфен. Эти методы получили название изомеризационных.
Изомеризационные методы позволили значительно упростить
классическую схему производства камфары, так как с их внед-
рением две наиболее сложные стадии классической схемы:
получение борнилхлорида (V) из пинена (IV) и получение
15
камфена (VI) из борнилхлорида (V) были заменены на одну,
технологически не сложную стадию изомеризации пинена (IV)
в камфен (VI). Дальнейшая переработка камфена осуществля-
ется через изоборнильные эфиры (VII) и изоборнеол (III).
Изомеризационный метод был разработан в результате иссле-
дований, проведенных в СССР [63, 172, 173] и Германии [313],
и внедрен в промышленную практику этих двух стран перед
второй мировой войной. В настоящее время производство кам-
фары во всем мире осуществляется изомеризационным методом,
который связан не только с более простой технологией, но и
обеспечивает получение продукта с тем же или даже лучшим
выходом, но более высокого качества, чем классический метод
производства через промежуточную стадию борнилхлорида. Кам-
фара, полученная изомеризационным методом, не содержит
хлора и содержит меньше изофенхона, чем камфара, полученная
через борнилхлорид.
В патентной литературе упоминается еще четвертый путь получения кам-
фары из пинена, в результате его прямого окисления на солнечном свету и
некоторыми окислителями [268]. Этот путь не получил развития и, по-види-
мому, основан на ошибочном эксперименте, так как образование камфары при
окислении пинена не подтверждено последующими публикациями.
Наряду с а-пииеном для всех описанных методов синтеза
камфары может быть использован также 0-пинен (XIII). По-
добно а-пинену, р-пинен при действии хлористого водорода об-
разует борнилхлорид, при действии органических кислот превра-
щается в эфир борнеола и путем каталитической изомеризации
может быть превращен в камфен.
В дальнейшем будут подробно рассмотрены борнилхлорид-
ный и изомеризационный методы получения камфена, первый
как основной метод производства камфары в прошлом, второй
как общепризнанный в технике метод. Кроме того, будут рас-
смотрены методы переработки камфена в камфару.
ГЛАВА II. СЫРЬЕ ДЛЯ ПОЛУЧЕНИЯ
СИНТЕТИЧЕСКОЙ КАМФАРЫ
1. Производство скипидаров
Скипидарами называются летучие вещества (эфирные масла),
содержащиеся в древесине хвойных. В основном они состоят из
монотерпеновых углеводородов общей формулы СюНщ и, как
правило, содержат лишь небольшие примеси сесквитерпенов и
кислородных производных терпенов. В древесине скипидар рас-
творяет генетически связанные с терпенами смоляные кислоты.
Этот раствор сосредоточен в особых вместилищах — смоляных
ходах и при надрезе коры и верхних слоев древесины (под-
16
новке) вытекает из места надреза. Вытекающий из дерева рас-
твор называется живицей.
По способу производства скипидары подразделяют на жи-
вичные, экстракционные, сульфатные, сульфитные и сухопере-
гонные.
Живичные скипидары получают из живицы разных видов
сосен. Живицы других видов хвойных, например ели, исполь-
зуются в незначительном количестве.
Живица представляет собой вязкую, приятно пахнущую бесцветную жид-
кость, из которой вскоре после выделения ее из дерева начинают выкристал-
лизовываться смоляные кислоты. После этого живица превращается в про-
питанную жидкостью кристаллическую массу, напоминающую по консистенции
мед. В живице, в зависимости от вида сосны, содержится 20—30% скипи-
дара. Остальная часть почти целиком приходится на смоляные кислоты.
Выделение живицы из надреза продолжается несколько дней. Если сделать
новый надрез, оно возобновляется, так как это ведет к быстрому биосинтезу
живицы в месте надреза. Этот способ добычи живицы называется подсочкой.
При подсочке на стволах сосен через регулярные промежутки времени де-
лают подновки в виде тонких срезов, расположенных непосредственно над
или под ранее сделанными. Живица стекает в приемники, из которых ее
время от времени выгружают и направляют на канифольно-скипидарные за-
воды для переработки на канифоль и скипидар.
В разных странах разработаны различные способы подсочки, которые
применяются в соответствии с местными условиями. В результате подсочки
некоторый участок ствола оголяется, этот участок называется каррой.
Средине выходы живицы (иа карру или на дерево) зависят от целого
ряда факторов: вида сосны, климата, длительности подсочного сезона, спо-
соба подсочки, возраста и состояния самого дерева. В табл. 5 приведены
данные о средних выходах живицы, получаемые в разных странах при про-
мышленной подсочке сосен разных видов [302].
Таблица 5
Средние выходы живицы, получаемые при промышленной подсочке
разных видов сосен
Страны Вид сосны Выход за сезон, кг
на карру на дерево
СССР, Польша, ГДР, ФРГ Р. silvestris L. 0,7 1,5
Австрия То же 1-1,5 —
» Р. nigra var austriaca Hoss 2-3 —
Франция Испания и Португалия США P. pinaster Sol. P. pinaster Sol. с при- месью P. halepensis Mill u P. nigra Arn. P. eliotti Eng., P. palus- tris Mill. 1,5-2,7 2—4 2,5—4 7-11
Большая затрата рабочей силы заставляет искать пути раци-
онализации подсочки. Было предложено создавать длительные
2 Заказ № 43
17
подсочные хозяйства на искусственно выращенных плантациях
из высокопроизводительных видов сосен, выводить особо высоко-
продуктивные деревья путем селекции [302] и воздействовать на
подновку стимуляторами смоловыделения. Этот метод использу-
ется в СССР и США. Сущность метода заключается в том, что
если свеженанесенную подновку смазать соответствующим хими-
ческим реагентом, например серной кислотой или биологическим
стимулятором, то живица выделяется значительно дольше. При
этом выход живицы на единицу площади ствола несколько по-
вышается и, что самое важное, из-за более редких подновок
на 30—50% и более сокращаются трудозатраты [52, 76, 245].
Эти и некоторые другие способы позволили рационализовать
подсочку и снизить трудозатраты. Механизировать подсочку
с тем, чтобы довести ее до уровня индустриального производ-
ства, пока не удалось, она по-прежнему осталась трудоемким
производством. В результате начался отлив рабочей силы с под-
сочного промысла в высокоразвитых индустриальных странах
(США, Франции). Они не могли конкурировать со странами,
использующими дешевый труд (Португалия, Индия, КНР), что
привело к перемещению подсочного промысла в эти страны и
почти полному прекращению его в США (табл. 6), где разви-
ваются более прогрессивные методы получения канифольно-ски-
пидарных продуктов (см. ниже). За период с 1965 по 1971 г.
количество живичного скипидара, выработанного в СССР, тоже
упало. Тем не менее в СССР принимаются меры, чтобы поднять
производство живицы до уровня 1965—1966 гг. [87] и даже не-
сколько более высокого [119], в результате чего с 1973 г. начался
рост добычи живицы в нашей стране. Мировое производство
Таблица 6
Мировое производство живичного скипидара [2, 87, 119, 283, 284]
Выработано по годам, тыс. т
Страны 1950 1965 1966 1967 1968 1959 1970 1971 1972
США 44,5 18,2 13,7 11,0 8,1 5,7 4,2 4,6 4,3
Франция 15,2 7,3 7,1 6,5 5,3 4,0 3,7 3,6 2,2
Португалия 10,0 16,6 16,6 17,5 17,3 17,5 19,6 20,4 22,7
Испания 9,0 8,4 9,0 8,6 9,8 8,7 9,0 6,9 7,9
Мексика 5,4 6,2 7,0 7,5 8,0 8,0 8,0 7,3 7,9
Греция 17,0 5,0 3,6 3,0 2,1 2,0 2,1 3,8 3,4
Индия —. 6,0 7,0 8,0 7,3 6,8 6,5 5,4 5,4
Польша — 5,7 4,9 5,0 5,0 5,2 5,3 5,6 5,7
КНР — 22,2 23,6 21,8 24,0 28,0 30,0 31,7 33,8
СССР 16,2 29,2 29,0 29,4 28,3 25,9 22,8 20,8 18,7
Прочие 3,3 7,2 7,8 7,0 5,5 6,0 6,2 6,7 6,6
Всего 120,6 132,0 129,3 125,3 120,7 117,1 117,4 116,8 118,6
18
живичного скипидара с 1950 г. держится на более или менее
постоянном уровне— 120—130 тыс. т и составляет в настоящее
время около 50% от общего производства скипидаров.
Процесс переработки живицы сводится к отделению сора,
Отгонке скипидара с острым паром и охлаждению остатка от
перегонки (канифоли) в условиях, при которых он застывает
в стекловидную массу не кристаллизуясь.
Экстракционные скипидары получают из сильнопросмоленной
сосновой древесины, в основном сосновых пней (пневый осмол).
Оставшиеся на месте сплошных рубок сосновых насаждений пни посте-
пенно накапливают смолистые вещества. Происходит ли это в результате
синтеза смолистых веществ или же это накопление только кажущееся, про-
исходящее из-за разрушения древесины вследствие гниения при одновремен-
ном сохранении находящихся в пне смолистых веществ, точно еще не выяс-
нено. По прошествии 10—15 лет после рубки содержание смолистых веществ
в пнях поднимается иногда до 25—30% и более. Накопление смолистых ве-
ществ хорошо происходит лишь в пнях крупных деревьев на сухих почвах.
На болотистых почвах, в пнях мелких деревьев накопление смолистых ве-
ществ происходит слабо и пни сгнивают без остатка.
Пневый осмол заготовляют путем корчевания специальными тракторами
или взрывным способом, разделывают на куски, после чего направляют для
переработки на канифольно-экстракционные заводы. Процесс переработки
осмола сводится к его измельчению на рубильной машине до размеров щепы
3—30 мм (вдоль волокна), последующей экстракции смолистых веществ и
выделению скипидара и канифоли из экстракта. Экстракция, как правило,
производится бензином прямой гонки.
В отличие от живичного, сырой экстракционный скипидар содержит неко-
торое количество кислородных соединений, главным образом терпеновых
спиртов, а также небольшое количество бензина. Поэтому скипидар подвер-
гают дополнительной ректификации, выделяя спиртовую фракцию (так назы-
ваемое сосновое флотационное масло, или пайн-оль) и скипидар, по своим
свойствам мало отличающийся от живичного.
Производство экстракционных скипидара и канифоли полу-
чило широкое (особенно после второй мировой войны) развитие
в США, где имелись большие запасы пневого смола, оставше-
гося от вырубки девственных лесов в Южных штатах (табл. 7).
Таблица 7
Выработка экстракционного скипидара в СССР и США [2, 87, 119, 283, 284]
Страна Выработано по годам, тыс. т
1950 1965 1966 1967 1968 1969 1970 1971 1972
США 38,5 27,4 25,2 22,8 21,0 17,7 15,4 14,0 12,2
СССР 0,7 4,3 4,3 4,3 4,3 4,5 4,7 5,1 5,3
Рост канифольно-экстракционного производства был обус-
ловлен меньшими затратами труда на единицу продукции, чем
2*
19
при подсочке. Но с 1950 г. объем канифольно-экстракционного
производства начал сокращаться, поскольку внедрялось более
выгодное направление получения канифольно-скипидарных про-
дуктов из отходов сульфатно-целлюлозного производства.
В СССР канифольно-экстракционное производство сущест-
вует с 1927 г., но размер его очень невелик. В силу острого-
недостатка в стране канифоли было запланировано увеличение-
производства экстракционной канифоли с 16 тыс. т в 1971 г.
до 70 тыс. т в 1975 г. [36, 87], что должно привести к соответст-
венному увеличению производства экстракционного скипидара
с 4 тыс. т до 18—19 тыс. т. В европейских странах, за исклю-
чением Польши, производство экстракционного скипидара не
получило развития из-за отсутствия сырьевой базы.
Сульфатный скипидар является отходом производства цел-
люлозы по сульфатному способу, обычно применяемому для
получения целлюлозы из смолистой древесины хвойных пород.
Скипидар отгоняется из варочных котлов при сдувках в количестве 8—
15 кг на 1 т целлюлозы. Отличительной особенностью этого скипидара яв-
ляется отвратительный запах, который ему придают сопутствующие серни-
стые соединения. Поэтому этот скипидар подвергают обязательной очистке-
от дурнопахнущих веществ.
Очищенный сульфатный скипидар по своему составу мало
отличается от живичного и поэтому применяется в технике на-
ряду с живичным и экстракционным скипидарами. Так как по-
лучение такого скипидара связано с минимальными затратами,
а производство сульфатной целлюлозы быстро развивается,
сульфатный метод получения следует рассматривать как наибо-
лее перспективный из всех существующих.
Производство сульфатного скипидара чрезвычайно быстро
развивалось в США, Швеции и Финляндии, т. е. в странах, где
имеется крупное производство сульфатной целлюлозы. В настоя-
щее время 80% скипидара, выпускаемого в США, и 40% ми-
рового производства скипидара составляет сульфатный скипи-
дар (табл. 8), так как трудозатраты на его получение самые
низкие. По Полякову и Лисову [119], трудозатраты на получение
1 т канифоли, включая заготовку сырья, составляют (чел.-дни):
живичной 71; экстракционной 36; талловой 3.
В Советском Союзе вырабатывают около 2 млн. т сульфат-
ной целлюлозы, следовательно, имеется возможность выпускать
от 16 до 30 тыс. т сульфатного скипидара-сырца, или соответ-
ственно от 13 до 24 тыс. т очищенного сульфатного скипидара.
К сожалению, эти возможности используются лишь на 17—30%.
Это связано с недостаточным вниманием, уделяемым целлюлоз-
но-бумажной промышленностью использованию отходов произ-
водства, и значительными потерями скипидара при внедряемой
в СССР непрерывной варке целлюлозы по сравнению с перио-
дической. По этой же причине последние годы наблюдается
некоторое снижение производства сульфатного скипидара
20
Таблица 8
Выработка очищенного сульфатного скипидара 1 в различных странах
[2, 119, 283, 284]
Страна Выработано по годам, тыс. т
1940 1950 1960 1965 1966 1967 1968 1969 1970 1971 1972
США 8,8 31,6 52,5 68,5 69,5 68,2 77,0 78,5 74,4 74,0
Швеция — 1,5 1,5 15,0 10,7 11,2 11,8 12,0 8,6 8,3 —
Финляндия — — —. 7,6 8,5 9,3 8,9 9,6 8,7 7,2 7,4
СССР — — — 3,9 3,9 4,2 3,9 4,2 4.1 4,1 4,0
Всего 8,8 31,1 54,0 95,0 92,6 93,6 101,6 104,3 95,8 93,6
1 Опубликованные данные по сырому скипидар}' пересчитывались иа очи-
щенный путем умножения на фактор 0,8.
в США и Скандинавии, несмотря на увеличение производства
сульфатной целлюлозы. Высказывается мнение, что в ближай-
шее время непрерывный процесс варки целлюлозы будет упо-
рядочен, после чего начнется новый рост производства сульфат-
ного скипидара [119, 320]. Снижение выходов скипидара из-за
использования наряду с древесиной сосны древесины других
пород должно перекрываться ростом производства.
Сухоперегонные скипидары получают при сухой перегонке пневого
осмола. Процесс осуществляется иа кустарных установках. Эти скипидары
производятся в сравнительно небольшом количестве, а состав их непостоянен.
Наряду с терпенами они содержат продукты термического разложения дре-
весины. В СССР производят 3—4 тыс. т сухоперегонного скипидара.
Сульфитные скипидары в небольшом количестве получают в качестве
отхода при производстве сульфитной целлюлозы.
Последнее десятилетие использование скипидара для химической перера-
ботки чрезвычайно расширилось, он перерабатывается на синтетический пайн-
оль, инсектициды, терпеновые смолы, душистые вещества и другие продукты.
По этой причине в прошлом крупнейший экспортер скипидара — США почти
полностью прекратил его экспорт в Европу. В результате на международных
рынках обнаружился дефицит в скипидаре и цены на него поднялись со 136
американских долларов за 1 т в 1965 г. до 300—350 долларов в 1972 г.
Создавшийся дефицит в скипидарах на международном рынке тормозит
развитие химического синтеза на их основе в странах, не производящих
скипидар. Это же создает благопрятные условия для синтеза камфары в Со-
ветском Союзе.
Дефицит в скипидарах, как это уже отмечалось, обусловлен не отсутст-
вием сырьевой базы, а отсутствием способов достаточно эффективно ее ис-
пользовать. Многие страны ведут работы по устранению дефицита, и не
исключено, что производство канифольно-скипидарных продуктов вновь нач-
нет увеличиваться [299, 320].
21
2. Состав скипидаров и требования к скипидару
как сырью для производства синтетической
камфары
Состав живичных скипидаров разных видов хвойных варьи-
рует в широких пределах, состав скипидаров индивидуальных
деревьев одного вида колеблется [11], но средний состав круп-
ных партий скипидара, полученных с большого числа деревьев
одного вида, более постоянен. В табл. 9 приведены данные
о составе живичных скипидаров разных видов сосен, подсачи-
ваемых в достаточно широких масштабах в разных странах.
Таблица 9
Состав живичных скипидаров, получаемых из разных видов сосен
Страна Виды сосны
СССР [15, 21, 24] Р. silvestris L.
Польша [202, 275] США [315] Р. palustris Mill,
Франция [315] Р. elliotti Eng. Р. pinaster Sol.
Португалия [315] P. pinaster Sol.
Греция [315] P. halepensis Mill, u P. nigra Arnold P. halepensis Mill.
КНР [315] P. masoniana Lamb.
Мексика [315] Ряд видов
Индия [280] P. longifolia Roxb.
Состав скипидаров, %
55-70 2—7 1 15-30 3-6
45—57 6-9 25-37 3—5
60-65 25—30 1 — 3-5
70-75 20-25 1 —_ 1-5
75-80 15-20 1 — 3
95 1 1 1-2
85—90 3-5 1-2 — 1-2
85-90 3-5 1-2 — —
25 10 — 38 2
Помимо перечисленных, в живичных скипидарах содержится в небольших
количествах целый ряд других монотерпеновых углеводородов, в том числе
трициклен, мирцен, а-терминен, терпинолен, Р-фелландрен и другие их произ-
водные: борнилацетат, терпенилацетат, борнеол, терпинеол и т. д., а также
сесквитерпены. В индийском скипидаре содержание сесквитерпенов (лонги-
фолена) достигает 25%.
Как видно из табл. 9, состав скипидаров сосны Pinus Sil-
vestris колеблется особенно сильно. Это может быть объяснено
наличием нескольких подвидов указанной сосны [120], имеющей
исключительно большой ареал произрастания, и различной эво-
люционной продвинутостью отдельных популяций [118]. Уста-
новлено, что колебания в составе скипидаров Pinus silvestris
подчиняются следующей закономерности. Содержание пиненов
в скипидарах из Восточной Сибири и из некоторых районов
центра европейской части СССР находится на верхнем пределе.
Содержание пиненов в скипидарах из Белоруссии — на нижнем.
В Польше и ГДР содержание пиненов еще ниже.
22
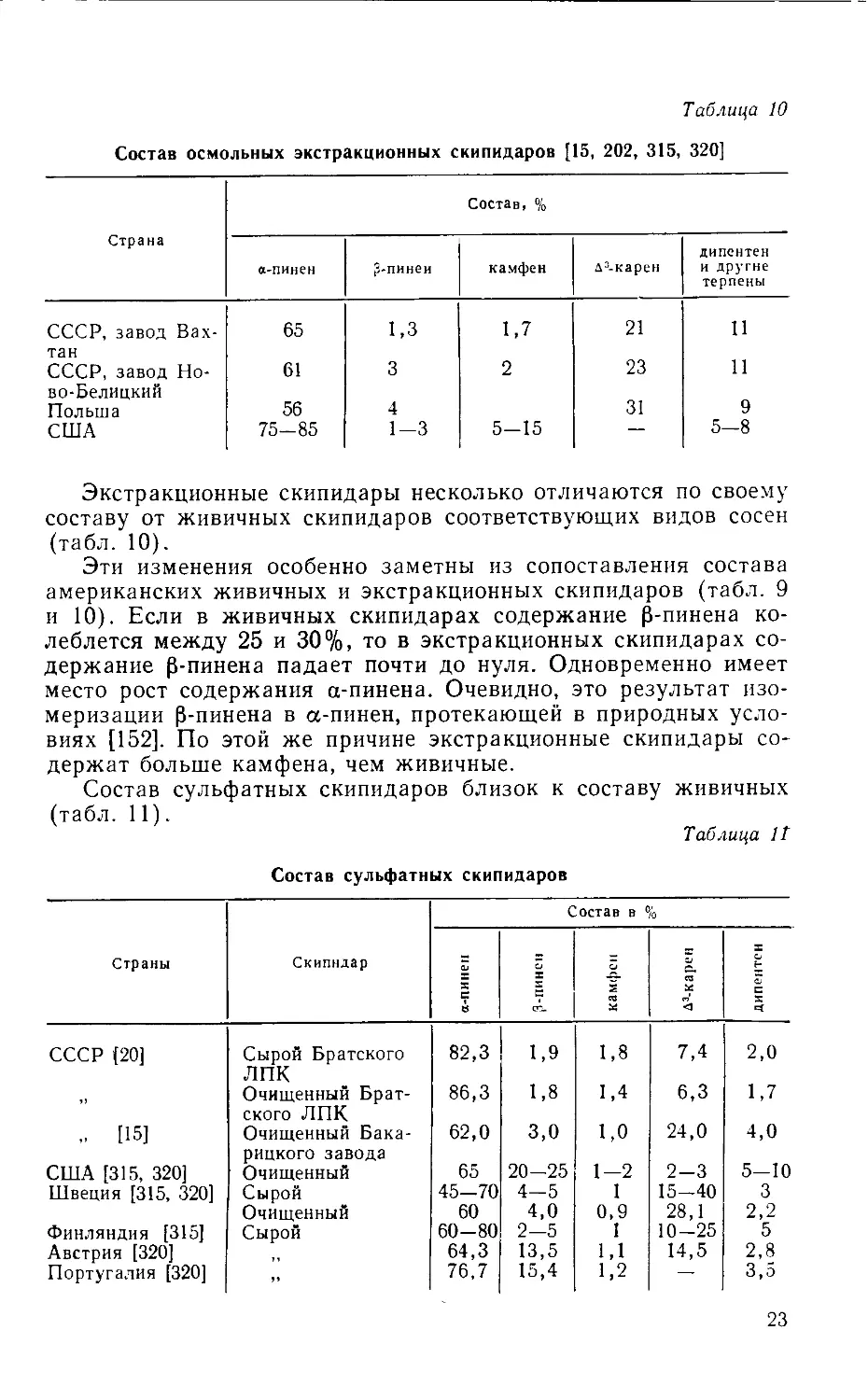
Таблица 10
Состав осмольных экстракционных скипидаров [15, 202, 315, 320]
Страна Состав, %
а-пинен 3-пинеи камфен д3-карен дипентен и другие терпены
СССР, завод Вах- 65 1,3 1,7 21 11
тан СССР, завод Но- 61 3 2 23 11
во-Белицкий Польша США 56 75-85 4 1-3 5-15 31 9 5-8
Экстракционные скипидары несколько отличаются по своему
составу от живичных скипидаров соответствующих видов сосен
(табл. 10).
Эти изменения особенно заметны из сопоставления состава
американских живичных и экстракционных скипидаров (табл. 9
и 10). Если в живичных скипидарах содержание 0-пинена ко-
леблется между 25 и 30%, то в экстракционных скипидарах со-
держание р-пинена падает почти до нуля. Одновременно имеет
место рост содержания а-пинена. Очевидно, это результат изо-
меризации р-пинена в а-пинен, протекающей в природных усло-
виях [152]. По этой же причине экстракционные скипидары со-
держат больше камфена, чем живичные.
Состав сульфатных скипидаров близок к составу живичных
(табл. 11).
Таблица It
Состав сульфатных скипидаров
Страны Скипидар Состав в %
а-пинен 3-ПИНС11 камфен Д3-карен | дипентсн
СССР [20] Сырой Братского лпк 82,3 1,9 1,8 7,4 2,0
Очищенный Брат- ского ЛПК 86,3 1,8 1,4 6,3 1,7
[15] Очищенный Бака- рицкого завода 62,0 3,0 1,0 24,0 4,0
США [315, 320] Очищенный 65 20-25 1-2 2-3 5-10
Швеция [315, 320] Сырой 45-70 4-5 1 15-40 3
Очищенный 60 4,0 0,9 28,1 2,2
Финляндия [315] Сырой 60-80 2-5 1 10-25 5
Австрия [320] 64,3 13,5 1,1 14,5 2,8
Португалия [320] 76,7 15,4 1,2 — 3,5
23
Очищенный сухоперегонный скипидар содержит те же компоненты, ио пи-
ненов в нем значительно меньше [15].
Сульфитный скипидар не содержит пинена и в своей основной части
состоит из п-цимола.
Для производства камфары можно использовать с одина-
ковым успехом как а-, так и р-пинен, но так как р-пинен нахо-
дит применение в других областях производства и он дороже,
предпочитают использовать а-пинен или а-пинен с небольшой
примесью р-пинена. Учитывая, что современные методы ректи-
фикации позволяют выделять достаточно чистые пинены из
любых скипидаров, можно считать, что все живичные, экстрак-
ционные и сульфатные скипидары, содержащие пинен, пригодны
для этой цели. Все должно решаться экономическими соображе-
ниями. Например, при переработке таких бедных пиненом ски-
пидаров, как индийский, получаются отходы в виде смеси терпе-
нов, уже не содержащие пинена. Такой продукт под наимено-
ванием скипидар живичный без пинена ТУ 81-05-74-69 ценится
в СССР в два раза дешевле живичного скипидара. Поэтому
перерабатывать бедные пиненом скипидары имеет смысл лишь
при невысокой их цене. Очищенный сухоперегонный скипидар со-
держит на 50% меньше пинена, чем живичный, а цена на него
высокая, поэтому использовать его для синтеза камфары нецеле-
сообразно. Благодаря низкой цене и высокому содержанию пи-
нена для переработки на камфару особенно целесообразно
использовать сульфатные скипидары. Скипидары, используемые
для синтеза камфары, должны иметь возможно более однород-
ный состав, чтобы было возможно осуществлять их ректифика-
цию при постоянном режиме.
Многие зарубежные фирмы выпускают выделенные нз ски-
пидаров а- и jj-пинены различной степени чистоты. В 1960 г.
в США выпускали 35 000 т а- и 7500 т р-пинена [216]. а-Пинен
выпускают также в ФРГ, Финляндии и Франции, а р-пинен—
во Франции. Эти продукты могут быть с успехом использованы
для синтеза камфары. Однако их приобретение вряд ли оправ-
дано. Предприятия, выпускающие камфару, применяют ректи-
фикацию на разных стадиях производства, имеют соответствую-
щий опыт и поэтому с успехом могут осуществлять ректифи-
кацию скипидара на месте и наряду с камфарой выпускать
а- и р-пинены. Так поступают и в СССР, где большая часть
вырабатываемого пинена производится предприятиями, выпу-
скающими синтетическую камфару. На выпускаемый нашими
предприятиями живичный пинен имеется ГОСТ 11956—66, со-
гласно которому технический пинен должен иметь ид 1,465—
1,467 и не менее 97% продукта должно перегоняться в пределах
151,5—162°. В среднем он имеет следующий состав: а-пинен 85%,
Р-пинен 4%, камфен 5%, Д3-карен и другие терпены 6%. Наряду
с а-пиненом в небольшом количестве выпускается р-пинен
ТУ 81-05-08-71 с содержанием основного вещества не менее
24
85% • За рубежом а-пинен выпускается с содержанием 90—95%
основного вещества, а fj-пинен — двух сортов, с содержанием
75—80% основного вещества, который используют для получе-
ния терпеновых смол, и рафинированный с содержанием 95%
основного вещества, который используется для синтеза душистых
веществ.
3. Скипидар и пинен как технические продукты
[262, 263, 285, 302]
Живичный, экстракционный и сульфатный скипидары пред-
ставляют собой жидкости с ^4° ~0,86. Рефракция и фракцион-
ный состав зависят от происхождения скипидара. Температура
вспышки (открытый тигель) около +35°С; плотность паров от-
носительно воздуха 4,7, нижняя граница взрываемости смеси
паров скипидара с воздухом (760 мм рт. ст., 20°С) 0;8% объ-
емных (45 г/м3), температура воспламенения 220°С. Технические
условия на живичный скипидар в Советском Союзе установлены
ГОСТ 1571—66, на сульфатный очищенный ОСТ 81—6—70, экст-
ракционный— ГОСТ 16943—71.
Скипидар, так же как и многие индивидуальные терпены,
производит раздражающее действие на кожу и слизистые обо-
лочки. Характерно его сенсибилизирующее действие, поэтому
лица, работающие со скипидаром, должны остерегаться попа-
дания его на кожные покровы. Для лиц, не соблюдающих пред-
осторожности, характерны профессиональные экземы и аллер-
гические заболевания. Раздражающее действие на кожу Д3-ка-
рена значительно сильнее, чем пиненов. Действие на кожу
продуктов окисления скипидара более пагубно, чем действие
самих терпенов. Так как Д3-карен окисляется легче пинена,
можно предположить, что это и есть причина его токсичности.
а-Пинен, молекулярная масса 136,23, бесцветная легко по-
движная ^жидкость чс характерным (скипидарным) запахом,
легко окисляющаяся на воздухе. Температура кипения 156°С,
d? 0,8582, пц 1,4654. Вязкость (20°С) ~ 1,7 • 10~3 (Н-с)/м2
(1,7 сп), диэлектрическая постоянная 2,1787, легко адсорбирует
СО2, SO2, NH3, теплоемкость (20°С) 1,8 кДж/(кг• град), теплота
испарения 289 кДж/кг, теплота сгорания — ДН (25°С при посто-
янном давлении) 6204,9±1 кДж/моль (1483,0 ккал/моль), кри-
тическая температура /Кр 346°С, критическое давление Ркр
20,5 атм, dl46 0,251, температура вспышки (открытый тигель)
33°С, температура воспламенения 263°С (ASTM-D 286—30),
верхняя граница взрываемости смеси паров пинена с воздухом
6% по объему (340 г/м3), нижняя граница 0,8% по объему
(45 г/м3). Токсичность пинена аналогична или несколько меньше
токсичности скипидара.
Хранить скипидар и пинен можно в стальных цистернах,
заполненных не более чем на 93%, предохраняя от света и
25
воздуха. Во избежание окисления прибавляют ингибиторы 2,6-ди-
трет-изобутилкрезол [197], 1,4-дифенилдиамин, пирокатехин [103].
Чтобы обеспечить постоянный состав сырья, хранение скипидара
желательно осуществлять в больших цистернах.
4. Пихтовое масло
Пихта Сибирская (Abies sibirica L.) имеет широкий ареал,
охватывающий северо-восток европейской части СССР, Запад-
ную, Центральную и отчасти Восточную Сибирь, встречается
в горах Казахстана и Тувинской области.
В качестве сырья для получения пихтового масла используются кончики
пихтовых веток, длиной 15—25 см, так называемая пихтовая лапка. Заго-
товка пихтовой лапки производится на лесосеках или с живых растущих
деревьев за 1—2 года до рубки [88]. Заготовленную лапку подвозят к близко-
расположенным кустарным установка,м, где отгоняют эфирные масла острым
паром. Выход масла в среднем составляет около 1—1,5% от массы лапки
[182], производительность пихтоваренной установки 3—6 т масла в год [88].
Состав пихтового масла колеблется в довольно широких
пределах (гл. Х.2), поскольку оно выпускается мелкими парти-
ями и не усредняется в больших емкостях. По ГОСТ 11699—66
содержание суммы борнилацетата и борнеола в пихтовом масле
должно быть не ниже 32%.
Таблица 12
Состав отдельных образцов пайн-оля [22, 197]
Компоненты Состав пайн-оля, %
экстракционного сульфатного
СССР США СССР
Терпеновые углеводороды 20,0 4,0 41,7
фенхон 0,5 0,6 0,4
Камфара 3,4 3,6 1,5
Транс-дигидро-а-терпинеол 16,2 5,8 1,5
а-Фенхол 2,5 — 2,0
а-Фенхол + терпинеи-1 -ол 10,6
Терпинен-4-ол 11,8 3,7 7,8
Р-Терпинеол 0,1 1,5 0,1
Эстрагол (метилхавикол) 1,6 0,6 0,5
у-Терпинеол 3,1 — 1,7
Изоборнеол 0,1 1,6 0,1
а-Терпинеол 9,5 59,0 9,8
Борнеол 1,9 7,4 3,5
Анетол цис- и транс- 0,9
.и-Ментен-6-ол 13,3 — 5,7
Прочие вещества 16,0 0,7 23,7
В том числе высококипящие веще- 12,0 — 21,4
ства
26
5. Пайн-оль
Пайн-оль, или сосновое масло, получают при экстракции
пневого осмола при очистке сульфатного скипидара и синтети-
чески из пинена. Общее производство пайн-оля достигло в США
к 1971 г. 51 000 т {283, 284]. В Советском Союзе сосновое масло
производится в незначительном количестве. В табл. 12 приве-
дены данные о составе и некоторых свойствах пайн-оля, выра-
батываемого в СССР и США.
В США используют пайн-оль по разным назначениям, в том числе и для
получения терпинеола, эстрагола и анетола, которые применяют в парфюмерии.
В качестве побочных продуктов получают борнеол, фенхиловый спирт и кам-
фару [288]. Борнеол и фенхиловый спирт дегидрируют и разделяют феихон
и камфару. Количество получаемой таким образом камфары, учитывая раз-
меры производства терпинеола в США, не может превышать 100—200 т/год.
Если в производстве используется природный пайн-оль. камфара получается
оптически деятельная.
ГЛАВА III. ПОЛУЧЕНИЕ КАМФЕНА ПО СХЕМЕ
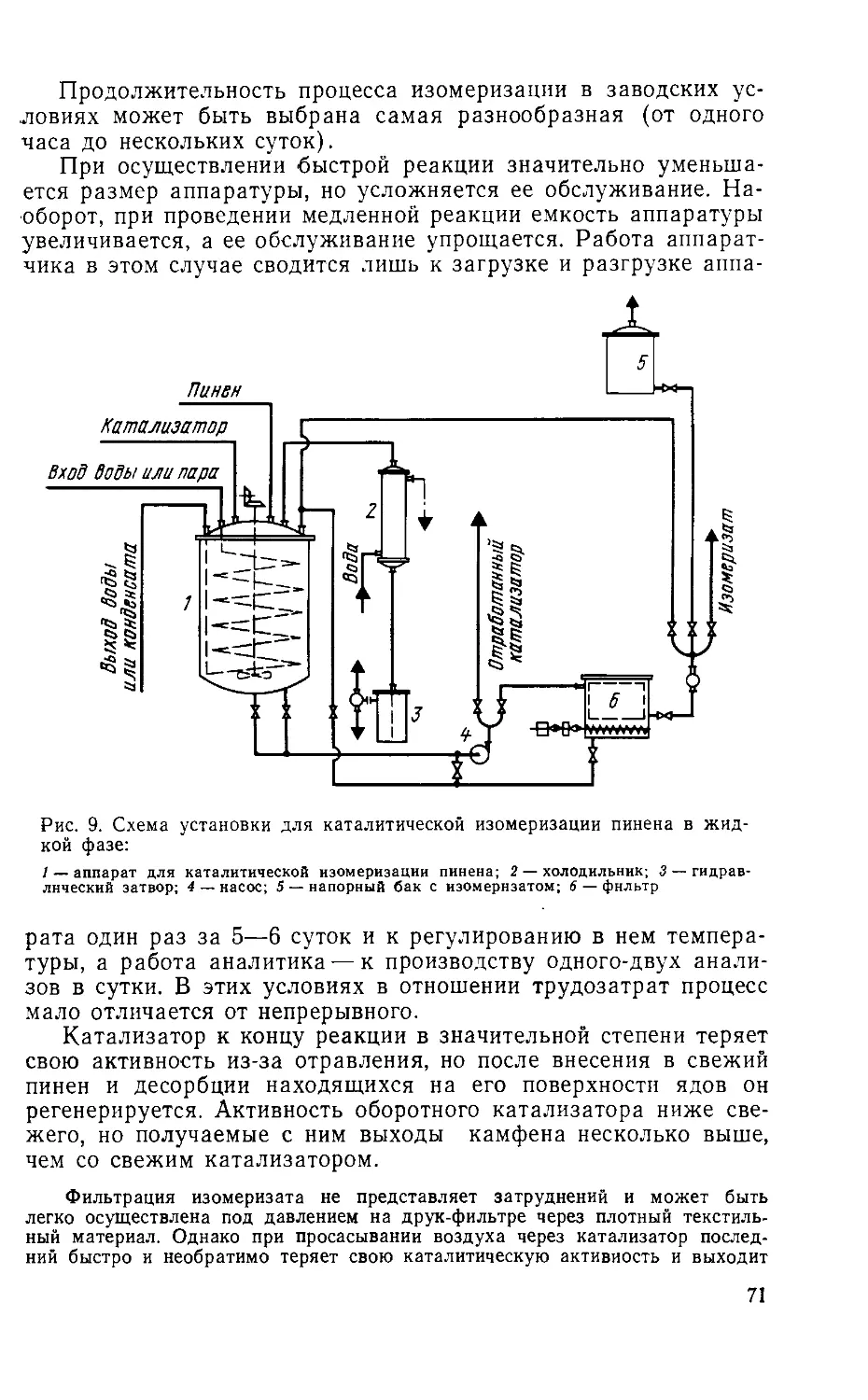
ПИНЕН-БОРНИЛХЛОРИД-КАМФЕН
В настоящее время промышленный синтез камфена через
борнилхлорид нигде не ведется, но в течение первой трети те-
кущего столетия синтез камфена через борнилхлорид был един-
ственным промышленным методом его получения.
Основные принципы, заложенные в химизм борнилхлоридного
метода, распространяются и на современный изомеризационный
метод синтеза камфена, поэтому химизм изомеризационного ме-
тода становится значительно яснее после ознакомления с хи-
мизмом получения камфена через борнилхлорид.
1. Получение борнилхлорида
Борнилхлорид (I) образуется из а-(II)- и 0- (III)-пиненов
при насыщении их сухим хлористым водородом. Хлористый во-
дород присоединяется по двойной связи, образуя цис-гидрохло-
рид пинена (IV), который изомеризуется в борнилхлорид [189,
276, 277].
Борнилхлорид — белое кристаллическое вещество с прият-
ным, слегка напоминающим камфару запахом. Температура
плавления чистого борнилхлорида 132°С.
Гидрохлорид пинена (IV)—чрезвычайно неустойчивое соединение, которое
быстро изомеризуется в борнилхлорид. В условиях получения борнилхлорида,
при температурах выше 0°, скорость изомеризации гидрохлорида пинена
в борнилхлорид превышает скорость его образования, поэтому его не удается
обнаружить в продуктах реакции. Но если насыщение пинена проводить при
температуре —70°С [276, 277], его удается выделить. Гидрохлорид пинена пред-
ставляет собой кристаллическое вещество. При действии на гидрохлорид воды
или спирта он немедленно отщепляет хлористый водород, а поэтому титруется
в присутствии этих растворителей как свободная кислота.
27
Образование из пинена борнилхлорида сопровождается не-
сколькими побочными реакциями.
(-)-Ш
Первая побочная реакция сводится к присоединению хлори-
стого водорода к молекуле пинена, сопровождающемуся раскры-
тием четырехчленного кольца и образованием moho-(V) и ди-
(VI)-гидрохлоридов лимонена.
II V
VI
Моногидрохлорид лимонена (V)—жидкость с т. кип. 97—98°С при И —
12 мм рт. ст.
Дигидрохлорид лимонена (VI)—кристаллическое вещество с т. пл. 50“С.
Если насыщение пинена хлористым водородом осуществляют
при полном отсутствии влаги, то в качестве побочного продукта
реакции образуется лишь около 10—15% моногидрохлорида
лимонена, но если реакция ведется в присутствии влаги или
применяется недостаточно хорошо высушенный хлористый во-
дород, выход борнилхлорида резко снижается за счет образова-
ния дигидрохлорида лимонена. При неблагоприятных условиях
выход борнилхлорида может дойти до нуля, поэтому при по-
лучении борнилхлорида уделяют особое внимание сушке хлори-
стого водорода и пинена и созданию условий, при которых даже
небольшое количество влаги не может попасть в реакционную
смесь.
Вторая побочная реакция вызывается образованием наряду
с quc-гидрохлоридом пинена (IV) тра«с-гидрохлорида (VII),
28
который, претерпевая аналогичную изомеризацию, превращается
в жидкий фенхилхлорид (VIII).
VII (+)-УШ
Наряду с борнилхлоридом (I) всегда образуются кристал-
лические изоборнилхлорид (IX) и гидрохлорид камфена (X).
Все эти три вещества образуют равновесную смесь [276, 277].
Взаимосвязь этих соединений наиболее понятна из их простран-
ственных формул:
Превращение борнилхлорида в изоборнилхлорид даже при
нагревании протекает очень медленно. При комнатной темпера-
туре это превращение не происходит. В некоторых растворите-
лях и в присутствии катализаторов взаимные изомеризационные
превращения ускоряются. Взаимные превращения изоборнилхло-
рид—камфенгидрохлорид протекают с заметной скоростью уже
при комнатной температуре и ускоряются кислотами. Переход
от камфенгидрохлорида к изоборнилхлориду сопровождается
выделением тепла, поэтому при высоких температурах равно-
весие смещается в сторону гидрохлорида камфена, а при низ-
ких температурах — в сторону изоборнилхлорида [277].
Все три гидрохлорида: борнилхлорид, изоборнилхлорид и
камфенгидрохлорид, образующие равновесную смесь, использу-
ются для получения камфена.
Продукты, полученные при взаимодействии пинена с хлори-
стым водородом, представляют собой кашицеобразную массу,
29
состоящую из твердого борнилхлорида и из раствора борнил-
хлорида в фенхилхлориде и гидрохлоридах лимонена. Выде-
ленная твердая фаза тоже не является чистым борнилхлори-
дом и содержит гидрохлорид лимонена и фенхилхлорид.
Выход на производстве кашицеобразной смеси гидрохлори-
дов составлял 120% на исходный пинен, или 95% от теорети-
ческого. Так как полное выделение чистого борнилхлорида из
продукта реакции связано со значительными затруднениями, то
хлористый водород отщепляли от всех хлоридов, образовавшихся
во время реакции, и из полученной при этом смеси терпенов
выделяли камфен.
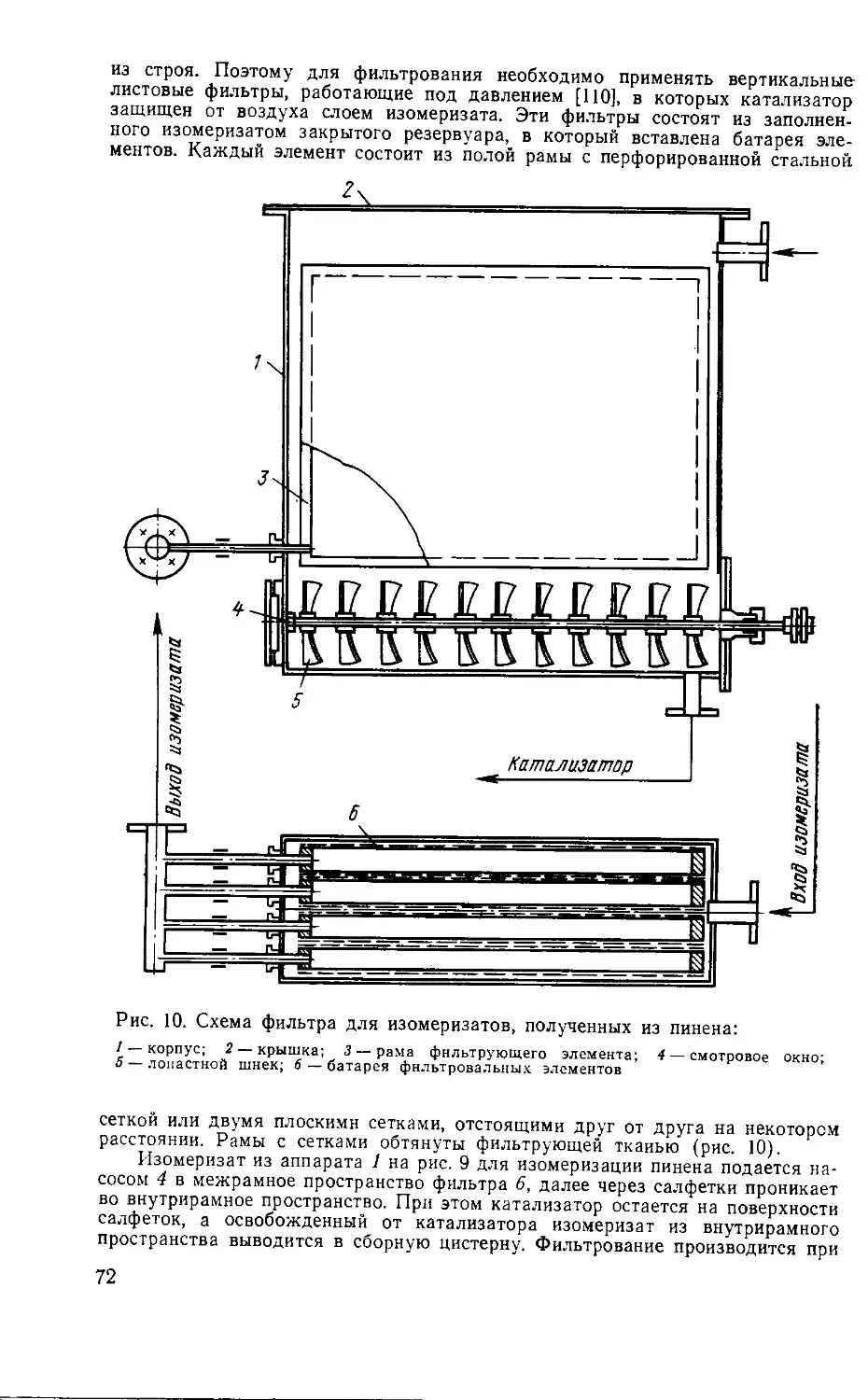
2. Превращение борнилхлорида в камфен
Превращение борнилхлорида в камфен осуществляется путем
нагревания его со щелочными агентами. Сам по себе борнил-
хлорид (I) отщепляет хлористый водород с ничтожной скоро-
стью, образуя борнилен (XI) и трициклен (XII), но находя-
щийся с ним в равновесии гидрохлорид камфена (X) отщепляет
хлористый водород очень быстро, превращаясь в камфен
(XIII). Скорость реакции определяется наиболее медленной
ее стадией — изомеризацией борнилхлорида в изоборнилхло-
рид (с. 29).
Другие гидрохлориды, присутствующие в сыром борнилхло-
риде, тоже реагируют со щелочами. Так, фенхилхлорид (VIII)
превращается в сложную смесь фенхенов. В этой смеси присут-
ствуют а-(XIV) и цикло (XV)-фенхены, образующиеся в резуль-
тате непосредственного отщепления хлористого водорода от
фенхилхлорида или от продукта его изомеризации — третичного
а-фенхенгидрохлорида (XVI). Имеются данные о наличии в сме-
си р-, у- и d-фенхенов, образование которых связано с вторич-
ными реакциями (см. гл. IV.3).
Гидрохлорид лимонена (V) при отщеплении хлористого во-
дорода превращается в лимонен (XVII) и терпинолен (XVIII).
30
Одновременно с основной реакцией образования терпеновых
углеводородов образуются терпеновые спирты. Например, из
гидрохлорида лимонена (V) образуется терпинеол (XIX), а из
гидрохлорида камфена (Х)-камфенгидрат (XX).
На получение камфена из борнилхлорида были взяты многочисленные
патенты [182, 268, 331]. Основная идея, заложенная во многих патентах, за-
ключается в применении для отщепления хлористого водорода от борнилхло-
рида гидроокисей металлов первой и второй групп в присутствии эмульгато-
ров, например, натриевых солей стеариновой кислоты при температуре 180—
200° под давлением. В других патентах предлагается использовать органиче-
ские основания, растворяющие борнилхлорид, например, анилин или фенолят
натрия в растворе фенола.
В Германии ставили опыты по непрерывному отщеплению хлористого во-
дорода от борнилхлорида перегретым водяным паром [310]. В связи с внедре-
нием изомеризационного метода исследования были прекращены.
Для получения камфена на предприятии Шеринг в Эберсвальде (Герма-
ния) брали 1500 кг сырого борнилхлорида, 520 кг стеариновой кислоты,
1350 кг 37%-ного раствора NaOH (17% избытка). Смесь нагревали при 180°
в автоклаве при интенсивном размешивании в течение 12—15 ч. После этого
реакционную массу передавливали в перегонный куб, откуда терпены, после
подкисления массы, отгоняли с острым водяным паром. Остаток стеариновой
кислоты возвращали в производство. Выход терпенов составлял около 75—
76% от массы сырого борнилхлорида (95—96% от теоретического).
На Рейнской камфарной фабрике в Дюссельдорфе отщепление хлористого
водорода производили гидроокисями кальция и магния [309] с добавлением
сульфитного щелока в качестве эмульгатора при 160° в течение 10—15 ч.
В этих условиях значительная часть дипентенгидрохлорида (V) превращалась
в а-терпииеол (XIX).
В СССР, иа сравнительно небольшой установке по получению камфена,
отщепление хлористого водорода от борнилхлорида осуществляли едким
натром в присутствии анизола, нафтеновых кислот или канифоли.
Продукты, полученные после отщепления хлористого водорода от смеси
гидрохлоридов, подвергали ректификации для выделения камфена и трицик-
лена *. Отделение этих последних от дипентена и других моноциклических
терпенов не встречало особых затруднений, но отделение их от фенхенов
было сопряжено с практически непреодолимыми препятствиями, так как т. кип.
гх-фенхена совпадает с т. кип. камфена, а т. кип (3-фенхена лишь на 3° выше
т. кип. трициклена. В то же время феихены желательно было возможно пол-
нее отделить от камфена, так как при дальнейшей переработке они образуют
изомерный камфаре жидкий кетон—изофенхон, который остается в камфаре
в виде примеси.
Любомилов, Рутовский и Шереметева [90] установили, что скорость обра-
зования камфена из борнилхлорида превышает скорость образования фенхе-
нов из фенхилхлорида. Поэтому, прерывая реакцию иа определенной стадии,
можно получить смесь терпенов, содержащую значительно меньше фенхенов,
чем при полном превращении. Это значительно упрощает процесс выделения
камфеиа и получения камфары с высокой температурой плавления, ио вместе
с тем осуществление процесса связано с неизбежными потерями. Кроме того,
имеется постоянная опасность загрязнения камфена незначительными количе-
ствами борнил- и фенхилхлоридов даже при тщательной ректификации. Это
может привести к получению камфары с содержанием хлора, что делает ее
непригодной для производства целлулоида [263].
* Трициклеи, получающийся в количестве нескольких процентов, так же
как и камфен, может быть превращен в камфару.
31
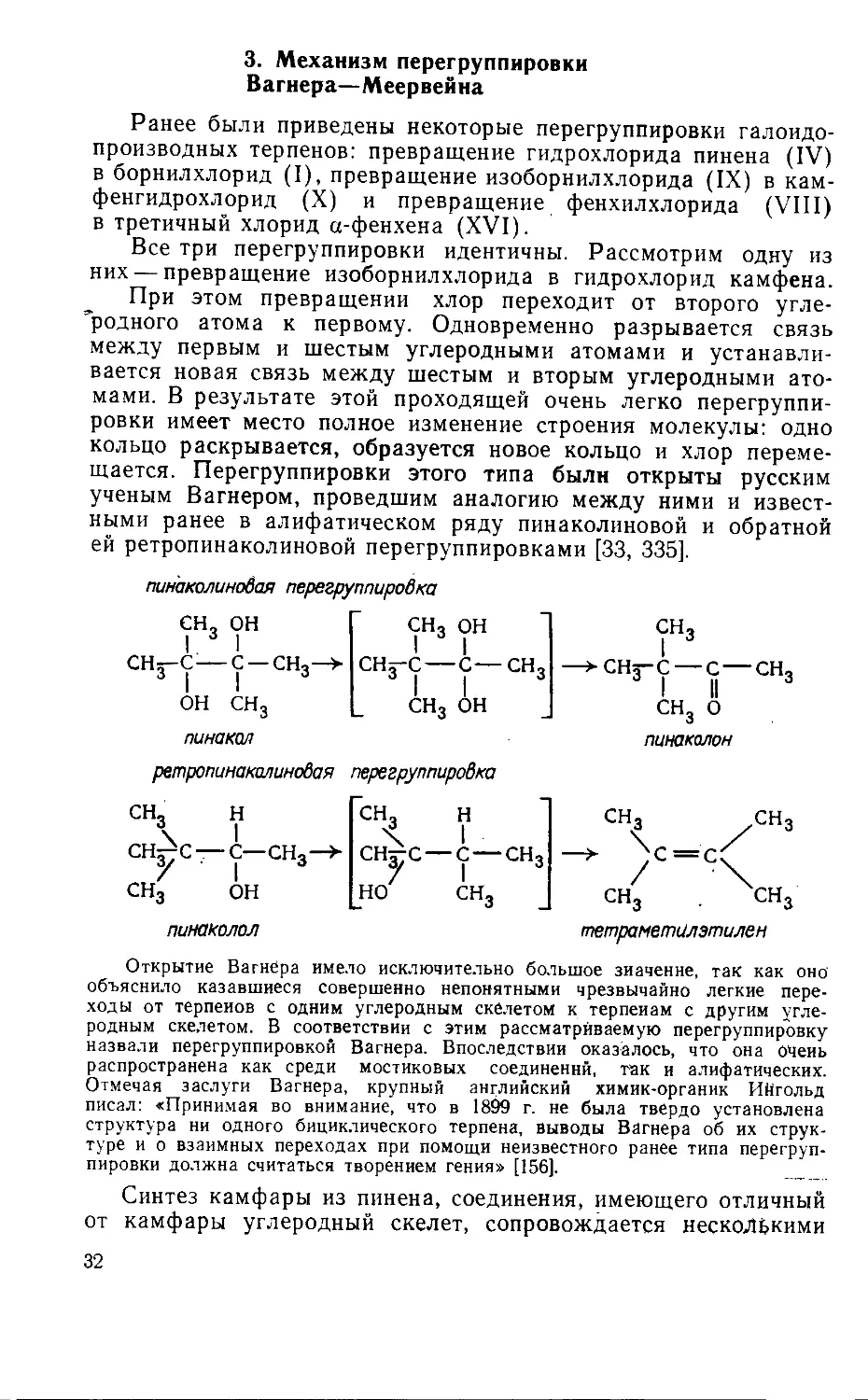
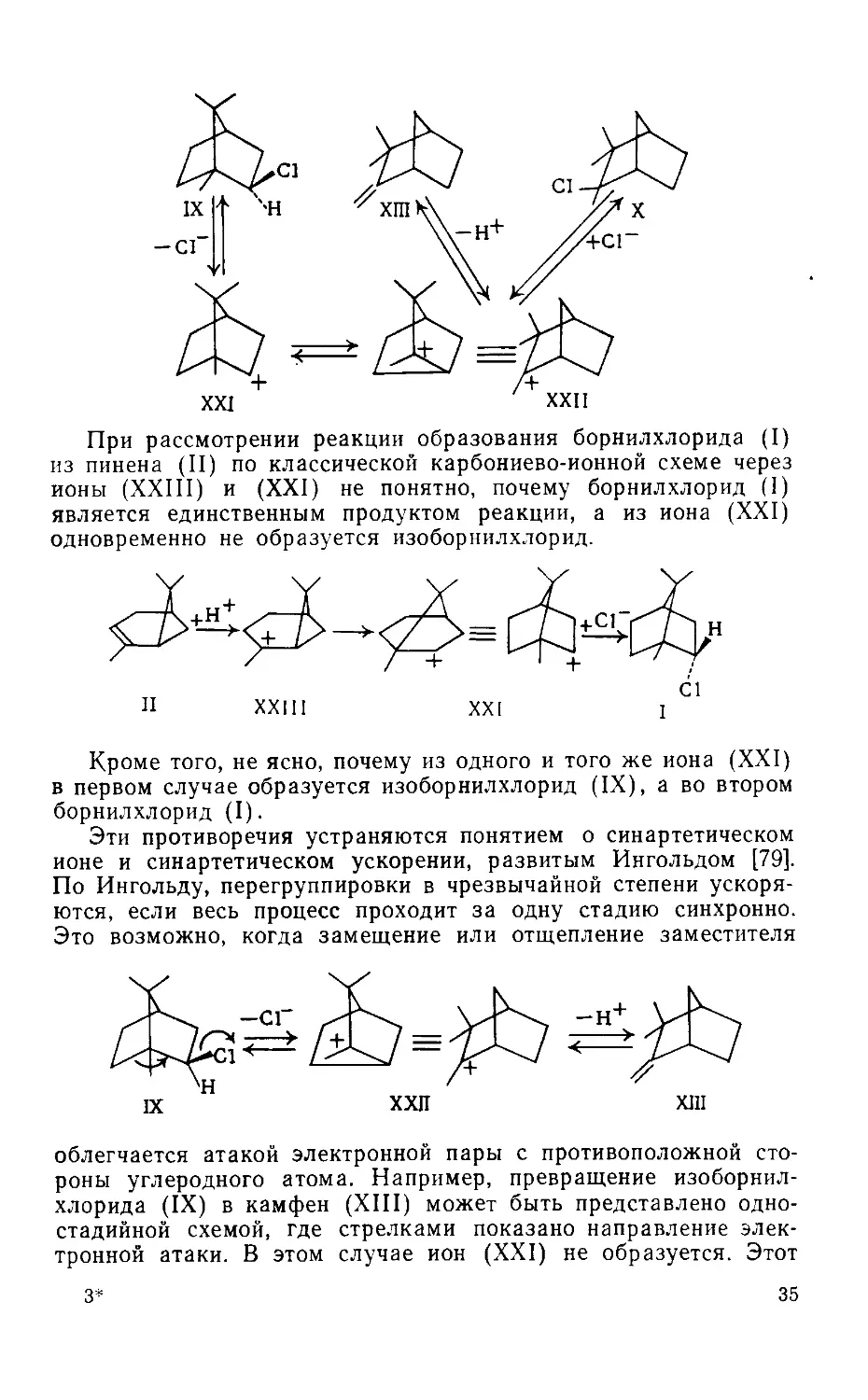
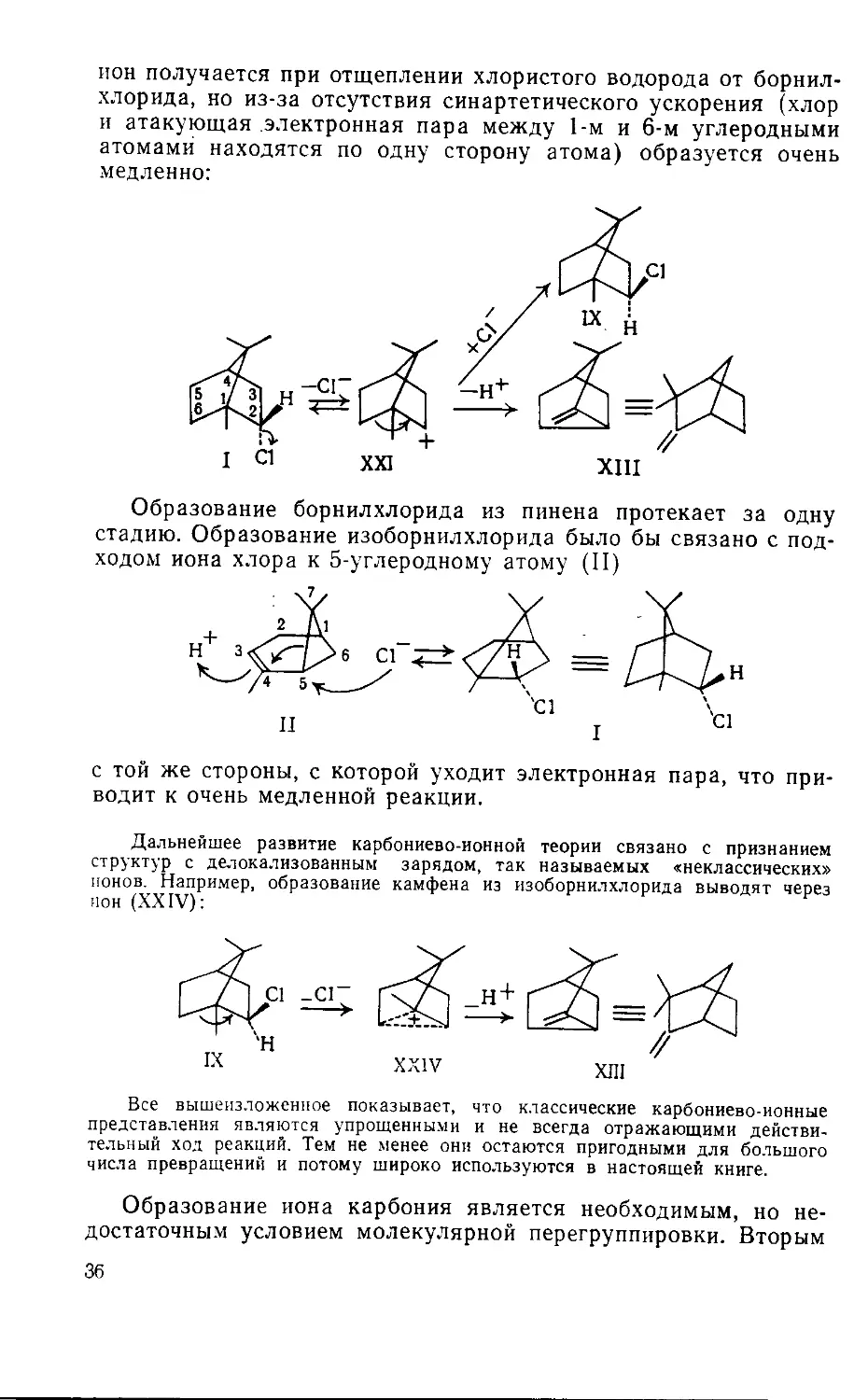
3. Механизм перегруппировки
Вагнера—Меервейна
Ранее были приведены некоторые перегруппировки галоидо-
производных терпенов: превращение гидрохлорида пинена (IV)
в борнилхлорид (I), превращение изоборнилхлорида (IX) в кам-
фенгидрохлорид (X) и превращение фенхилхлорида (VIII)
в третичный хлорид а-фенхена (XVI).
Все три перегруппировки идентичны. Рассмотрим одну из
них — превращение изоборнилхлорида в гидрохлорид камфена.
При этом превращении хлор переходит от второго угле-
~родного атома к первому. Одновременно разрывается связь
между первым и шестым углеродными атомами и устанавли-
вается новая связь между шестым и вторым углеродными ато-
мами. В результате этой проходящей очень легко перегруппи-
ровки имеет место полное изменение строения молекулы: одно
кольцо раскрывается, образуется новое кольцо и хлор переме-
щается. Перегруппировки этого типа были открыты русским
ученым Вагнером, проведшим аналогию между ними и извест-
ными ранее в алифатическом ряду пинаколиновой и обратной
ей ретропинаколиновой перегруппировками [33, 335].
пинаколиновая перегруппировка
СН
I. '
ОН
I
с — сн3—>
он сн3
сн3 он
I I
сн^-с—С—СН
3 I I
сн3 он
сн3
—>СНз-С — с — СН
3 I II
снз 0
пинакал пинаколон
ретропинакалинобая перегруппировка
сн,
о
сн^с —
сн3
н
I
с—сн3—>
он
СН н
СНчуС— с — сн3
у I 3
но сн3
тетраметилэтилен
пинаколол
Открытие Вагнера имело исключительно большое зиаченне, так как оно
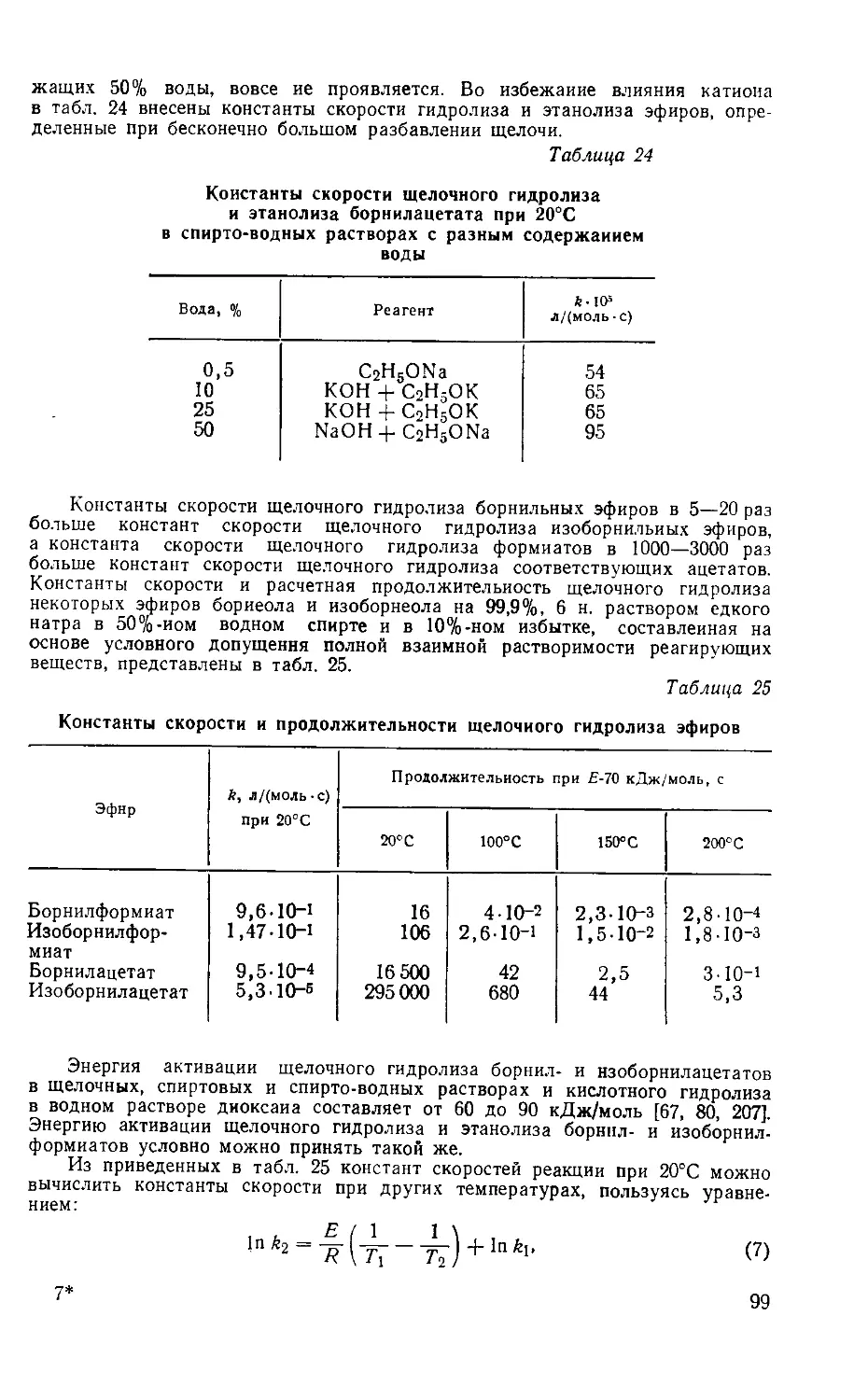
объяснило казавшиеся совершенно непонятными чрезвычайно легкие пере-
ходы от терпенов с одним углеродным скелетом к терпенам с другим угле-
родным скелетом. В соответствии с этим рассматриваемую перегруппировку
назвали перегруппировкой Вагнера. Впоследствии оказалось, что она очень
распространена как среди мостиковых соединений, так и алифатических.
Отмечая заслуги Вагнера, крупный английский химик-органик Ийгольд
писал: «Принимая во внимание, что в 1899 г. не была твердо установлена
структура ни одного бициклического терпена, выводы Вагнера об их струк-
туре и о взаимных переходах при помощи неизвестного ранее типа перегруп-
пировки должна считаться творением гения» [156]. ____
Синтез камфары из пинена, соединения, имеющего отличный
от камфары углеродный скелет, сопровождается несколькими
32
подобными перегруппировками. Поэтому следует более подробно
остановиться на их механизме. Этим вопросом занимаются уже
около 80 лет. За это время опубликованы сотни научных работ,
обобщенных в ряде обзоров [79, 99, 193, 256], очень многое разъ-
яснено, но все же последнее слово еще не сказано.
Особенно большой вклад в разъяснение механизма перегруп-
пировки Вагнера сделал 5\еервейн [276—279], поэтому эту пе-
регруппировку в настоящее время называют перегруппировкой
Вагнера—Меервейна. Меервейн, изучив кинетику взаимных пре-
вращений изоборнилхлорид—камфенгидрохлорид, привел убе-
дительные доводы, показывающие, что эта перегруппировка яв-
ляется результатом ионизации гидрохлорида и что изменения
углеродного скелета происходят не в молекуле гидрохлорида,
а в образовавшемся из нее ионе.
Для подтверждения своих взглядов Меервейн привел сле-
дующие доказательства.
1. Скорость перегруппировки (реакция первого порядка) за-
висит от ионизирующей способности растворителя. В раствори-
телях, вызывающих ионизацию, она увеличивается, в раствори-
телях, уменьшающих ионизацию, уменьшается.
2. Перегруппировка ускоряется некоторыми хлоридами, на-
ример SnCU, SbCU, которые могут соединяться с хлором в ком-
плексные ионы, что благоприятствует ионизации растворенного
вещества.
3. Скорость превращения эфиров камфенгидрата в эфиры
изоборнеола возрастает вместе со склонностью их кислотных
остатков образовывать самостоятельные ионы, т. е. эта скорость
велика для эфиров сильных кислот (H2SO4, НВг) и мала для
эфиров слабых кислот (С6Н5СООН, СН3СООН).
Хотя в своих работах Меервейн убедительно показал, что
внутримолекулярная перегруппировка хлоридов происходит
в образовавшихся из них ионах, он подробно не коснулся тех
движущих сил, которые вызывают перегруппировку. Эту сторону
проблемы развил Уитмор [181, 336].
Карбониево-ионная теория, развитая Уитмором, внесла по-
нятие о ионе карбония и его превращениях. При диссоциации на
ионы какого-либо хлорида R1R2R3CCI согласно этой теории дол-
жен получиться отрицательно заряженный ион хлора и положи-
тельно заряженный углеводородный ион, так называемый ион
карбония:
Ri Г R;
r2:c:ci r2:c
R3 R3
Ион карбония может быть уподоблен сложным ониевым ионам,
например
С1
3 Заказ № 43
33
Н 1+
иону аммония
H:N:H
Н
иону сульфония R : S : R
R
иону гидрония ГН:О:Н
', иону оксония R : б:Н
Н
Н
но отличается от них тем, что один из атомов углерода, входя-
щий в состав ионов, окружен лишь шестью электронами вместо
нормальных восьми. При такой неустойчивости возникают силы,
стремящиеся окружить атом углерода нормальным октетом
электронов. В результате этого атом с недостаточным числом
электронов может оторвать от соседних углеродных атомов сое-
диненные с ними атомы и целые группы атомов вместе с ва-
лентными электронами, образующими связь, что приводит к пе-
регруппировке внутри карбониевого иона. Это не стабилизирует
систему. Стабилизация может произойти путем соединения кар-
бониевого иона с анионом. Тогда образуется молекула исходного
вещества (если карбониевый ион за время своего существова-
ния не успел перегруппироваться) или молекула нового вещества
(если перегруппировка успела произойти). Стабилизация может
произойти и за счет потери карбониевым ионом протона (во-
дородного атома лишенного электронов). В этом случае обра-
зуется двойная связь, в результате чего получается непредель-
ный углеводород.
Карбониевые ионы могут возникать не только в результате
диссоциации хлоридов, но и в целом ряде других случаев, в том
числе при действии протонов на непредельные углеводороды.
При этом протон присоединяется к более богатому водородом
углеродному атому (правило Марковникова). Наиболее легко
образуются и наиболее устойчивы карбониевые ионы при трех-
замещенном углероде. Продолжительность периода полупревра-
щения обычного иона карбония составляет 10-5 с [181]. При
обозначении иона карбония ставится знак плюс у заряженного
атома углерода, окруженного шестью электронами.
Карбониево-ионную теорию, в том виде как она здесь изло-
жена, называют классической и ионы с локализованным заря-
дом— классическими ионами.
В свете изложенного образование камфена (XIII) и его гид-
рохлорида (X) из изоборнилхлорида (IX) может быть пред-
ставлено схемой, согласно которой превращение осуществля-
ется через ионы (XXI) и (XXII).
Такого рода схемы для написания перегруппировок Вагне-
ра—Меервейна позволяют понять причины, вызывающие пере-
группировки, но в то же время объясняют далеко не все.
В частности, не понятно, почему из иона (XXI) при обратной
реакции образуется почти исключительно изоборнилхлорид (IX),
а не более стабильный борнилхлорид (I).
34
При рассмотрении реакции образования борнилхлорида (I)
из пинена (II) по классической карбониево-ионной схеме через
ионы (XXIII) и (XXI) не понятно, почему борнилхлорид (I)
является единственным продуктом реакции, а из иона (XXI)
одновременно не образуется изоборнилхлорид.
Кроме того, не ясно, почему из одного и того же иона (XXI)
в первом случае образуется изоборнилхлорид (IX), а во втором
борнилхлорид (I).
Эти противоречия устраняются понятием о синартетическом
ионе и синартетическом ускорении, развитым Ингольдом [79].
По Ингольду, перегруппировки в чрезвычайной степени ускоря-
ются, если весь процесс проходит за одну стадию синхронно.
Это возможно, когда замещение или отщепление заместителя
облегчается атакой электронной пары с противоположной сто-
роны углеродного атома. Например, превращение изоборнил-
хлорида (IX) в камфен (XIII) может быть представлено одно-
стадийной схемой, где стрелками показано направление элек-
тронной атаки. В этом случае ион (XXI) не образуется. Этот
3* 35
ион получается при отщеплении хлористого водорода от борнил-
хлорида, но из-за отсутствия синартетического ускорения (хлор
и атакующая электронная пара между 1-м и 6-м углеродными
атомами находятся по одну сторону атома) образуется очень
медленно:
Образование борнилхлорида из пинена протекает за одну
стадию. Образование изоборнилхлорида было бы связано с под-
ходом иона хлора к 5-углеродному атому (II)
с той же стороны, с которой уходит электронная пара, что при-
водит к очень медленной реакции.
Дальнейшее развитие карбониево-ионной теории связано с признанием
структур с делокализованным зарядом, так называемых «неклассических»
ионов. Например, образование камфена из изоборнилхлорида выводят через
ион (XXIV):
Все вышеизложенное показывает, что классические карбониево-ионные
представления являются упрощенными и не всегда отражающими действи-
тельный ход реакций. Тем не менее они остаются пригодными для большого
числа превращений и потому широко используются в настоящей книге.
Образование иона карбония является необходимым, но не-
достаточным условием молекулярной перегруппировки. Вторым
36
необходимым условием является соответствующее изменение
свободной энергии системы (термодинамическое условие). Так,
например, образование иона карбония из борнилхлорида имеет
место, а перегруппировка его в гидрохлорид пинена не проис-
ходит.
ГЛАВА IV. ИЗОМЕРИЗАЦИОННЫЙ СПОСОБ
ПОЛУЧЕНИЯ КАМФЕНА
1. Сущность метода, механизм и развитие
взглядов на роль катализатора
В начале нашего столетия русский химик Гурвич [63, 64]
обнаружил, что при введении сухого флоридина (американская
отбельная глина) в жидкий пинен начинается самопроизвольная
реакция с сильным выделением тепла. В продуктах реакции на-
ряду с моноциклическими терпенами и политерпенами он нашел
небольшое количество камфена.
Работа Гурвича открывала перспективу создания нового ме-
тода синтеза камфары, в котором двухстадийный и сложный
синтез камфена через борнилхлорид мог быть заменен односта-
дийным каталитическим процессом. Однако реакция, открытая
Гурвичем, почти 20 лет не находила практического применения
потому, что и Гурвич и последующие исследователи [89, 172, 332]
считали, что они получают камфен с низким выходом. До из-
вестной степени это могло быть и так, поскольку камфен может
полимеризоваться под действием того же катализатора. Но,
по-видимому, дело заключалось не только в этом, айв непра-
вильной оценке состава изомеризатов, так как в распоряжении
исследователей отсутствовали надежные методы анализа. Обычно
они принимали за камфен лишь тот, который им удавалось вы-
делить в результате дробной разгонки изомеризатов из колбы
Фаворского. Разумеется, они выделяли при этом лишь незна-
чительную часть камфена, так как для полного его выделения
нужна колонка с 30—50 теоретическими тарелками. Колба Фа-
ворского эквивалентна лишь 1—2 теоретическим тарелкам. Точ-
ная лабораторная ректификация тогда еще не применялась
в лабораторной практике.
С 1931 г. изомеризацию пинена под действием глин начали
изучать Тищенко и Рудаков. Они ставили целью использовать
эту реакцию для получения камфена. Все свои выводы авторы
строили на количественном определении его в продуктах реак-
ции. Разработанный ими метод анализа был основан на пре-
вращении камфена в изоборнилацетат и последующем омыле-
нии этого эфира [175]. В результате проделанной работы
Тищенко и Рудаков показали, что при проведении реакции в наи-
более благоприятных условиях выход ацетилирующихся терпе-
нов, которые они принимали за камфен, достигает 55—62% [174].
37
Позднее было показано, что при каталитической изомериза-
ции пинена, помимо камфена, образуются другие ацетилирую-
щиеся терпены: трициклен и фенхены (131]. Трициклен при
взаимодействии с уксусной кислотой в присутствии серной об-
разует эфир изоборнеола, следовательно, он является таким же
полноценным полупродуктом для получения камфары, как и
камфен. Фенхены в тех же условиях образуют эфир изофенхи-
лового спирта. Этот эфир трудно отделим от изоборнильноп>
эфира, вследствие чего образующийся при их переработке изо-
фенхон переходит в камфару. Выход технической камфары уве-
личивается, но содержание в ней основного вещества снижается
(гл. IV и XI), однако образование фенхенов не препятствовало
внедрению изомеризационного метода в промышленную прак-
тику, так как оно было того же порядка, что и при получении
камфена через борнилхлорид.
Тищенко и Рудаков показали, что параллельно с изомериза-
цией пинена в камфен и моноциклические терпены происходит
полимеризация продуктов реакции, которая протекает медленнее
их образования, поэтому для получения хорошего выхода кам-
фена необходимо своевременно прервать реакцию. Если этого не
сделать, выход камфена может снизиться.
На основе работы Тищенко и Рудакова в СССР была пу-
щена первая заводская установка по получению камфена ката-
литическим способом. Способ получил название изомеризацион-
ного. Изомеризации подвергали жидкий пинен при непрерывном
размешивании в аппаратах периодического действия. Катализа-
тор подвергали предварительной активации (гл. IV.2,6).
Механизм превращений терпенов при получении камфена
изомеризационным способом и роль катализатора привлекли
внимание многих исследователей.
Гурвич [63, 64], открывший каталитические превращения непредельных
углеводородов в присутствии алюмосиликатных катализаторов, предполагал,
что при изомеризации пинена катализатор играет чисто физическую роль:
на его поверхности происходит сорбция терпенов, сопровождающаяся сжатием,
вызывающим полимеризацию этих углеводородов, а под влиянием выделяю-
щегося тепла происходит деполимеризация с образованием новых изомеров.
Последующие исследователи [172, 332] показали, что полимеры, получаю-
щиеся при нагревании пинена с алюмосиликатами, не образуют камфена при
деполимеризации и, следовательно, схема образования камфена и моноциклн-
ческих терпенов, данная Гурвичем, неверна, а образование камфена и моно-
циклических терпенов при каталитической изомеризации пинена следует счи-
тать первичным процессом.
Тищенко и Рудаков [174] связали изомеризационные превра-
щения пинена с кислыми свойствами алюмосиликатных ката-
лизаторов.
Кислые свойства алюмосиликатов были открыты в конце
прошлого столетия Вернадским [35] и Земятченским [73, 74]. Эти
ученые показали, что глины и образующие их алюмосиликаты
38
не являются алюминиевыми солями кремниевой кислоты, как
это думали раньше, а представляют собой сильные алюмокрем-
невые кислоты.
Основываясь на этих работах, Тищенко и Рудаков пришли
к выводу, что реакции, возникающие при взаимодействии пинена
с алюмосиликатами, протекают по тому же механизму, что и
реакции взаимодействия пинена с кислотами. К такому типу
реакций относится, например, взаимодействие пинена с хлори-
стым водородом, рассмотренное в главе III. В этом случае ос-
новными продуктами реакции являются борнилхлорид, фенхил-
хлорид и гидрохлорид лимонена. При отщеплении от этих сое-
динений хлористого водорода при действии щелочей образуется
камфен, сложная смесь фенхенов и смесь моноциклических тер-
ненов. Если бы в условиях реакции происходило самопроизволь-
ное отщепление хлористого водорода от образовавшихся гидро-
хлоридов, она превратилась бы в каталитический процесс
образования камфена, фенхенов и моноциклических терпенов
непосредственно из пинена. Такого рода превращения испыты-
вают при нагревании эфиры борнеола, получающиеся при дей-
ствии на пинен пикриновой [271] и 2-хлорцимол-5-сульфокислот
{278], образуя камфен.
Такого же рода реакции, по Тищенко и Рудакову, претерпе-
вает пинен под действием алюмосиликатов. Это было сформу-
лировано следующим образом: «На поверхности коллоидных
частиц сложных минеральных кислот, образующих глину, про-
исходит их взаимодействие с пиненом. Образующиеся при этом
эфиры борнеола и терпинеола вследствие непрочности разруша-
ются с образованием камфена и дипентена» [174].
Представления о механизме изомеризации пинена в присут-
ствии алюмосиликатов, развитые Тищенко и Рудаковым, легли
в основу общей теории алюмосиликатного катализа, которая
объясняет не только каталитические превращения пинена,
но в равной степени относится к каталитическим превращениям
других непредельных углеводородов. Эта теория и в настоящее
время продолжает оставаться общепризнанной.
Механизм изомеризации эфиров пиненгидрата в эфиры бор-
неола, а этих последних в эфиры камфенгидрата изложен
в гл. III.3. Первой стадией реакции является диссоциация эфира
с образованием иона карбония, второй — изомеризация этого
иона, третьей—ассоциация иона карбония с кислотным анио-
ном. Все эти стадии протекают синхронно. При действии на не-
предельные углеводороды кислых катализаторов, подобных алю-
мосиликатному, ионы карбония образуются не в результате дис-
социации эфира, получающегося на поверхности катализатора,
а в результате захвата молекулой непредельного углеводорода
протона, отщепляемого катализатором. Так, например, образо-
вание из а-пинена (I) пинил-карбоний-иона (II), по этим пред-
ставлениям, должно протекать по схеме:
39
По-видимому, в действительности имеет место образование
ионов карбония по более сложной схеме, так как имеющиеся
данные о реакциях ионов карбония в условиях гетерогенного
катализа не позволяют рассматривать эти ионы как кинетически
свободные частицы. Вероятно, это сильно поляризованные по-
верхностные соединения, которые могут реагировать и как ионы
карбония, и как молекулы сложных эфиров. Поэтому приведен-
ная выше схема образования иона карбония путем захвата про-
тона молекулой непредельного углеводорода должна рассмат-
риваться как упрощенная. Однако в силу ее наглядности она
будет использована в дальнейших рассуждениях.
Независимо и практически одновременно те же выводы
о кислой природе катализаторов, вызывающих изомеризацию
пинена, были сделаны в Германии Меервейном, Ульферсом и
Эрбе [313].
2. Катализаторы
Для получения камфена из пинена, кроме алюмосиликатных
катализаторов, были предложены многие другие. Почти все они
являются кислотами. Большая часть катализаторов — твердые
нерастворимые в пинене вещества, в том числе кремневая,
борная, титановая, ванадиевая, мышьяковая, молибденовая,
сурьмяная, вольфрамовая кислоты, борновольфрамовая, кремне-
вольфрамовая, кремневанадиевая, борнофосфорная, фосфорно-
оловянная, фосфорновольфрамовая, фосфорномолибденовая,
мышьяк-молибденовая, борноуксусная, щавелевоалюминиевая и
щавелевохромовая комплексные кислоты и гетерополикислоты,
кислые соли, в том числе кислые соли фосфорновольфрамовой
кислоты, кислые фосфаты магния, марганца, урана [311, 313],
катионообменные смолы [54, 112], нейтральные сернокислые соли
двухвалентных металлов, кристаллизующиеся с одной молекулой
воды [312, 323], а также восстановленные водородом никелевые,
цинковые и медные соли молибденовой кислоты [225, 339].
Изомеризация пинена в камфен была также описана в при-
сутствии следующих растворимых в пинене катализаторов: пик-
риновой кислоты [271], смоляных кислот [13], трибромфенола
[290], сульфоуксусной кислоты [183, 274]. К последним работам
до известной степени примыкает старое наблюдение Армстронга
и Тильдена об образовании небольшого количества камфена
при действии серной кислоты на скипидар [188].
40
Среди перечисленных катализаторов названы сернокислые соли двух-
валентных металлов, содержащие одн\ модек\л\ кристаллизационной воды.
Они не подходят к обычному понятию о кислотах как о веществах, диссо-
циирующих в водном растворе с образованием водородных ионов и всту-
пающих в реакцию со щелочами с выделением воды и образованием солен.
Но такое представление о кислотах распространяется только на водные рас-
творы. В неводных же средах, по Бернстеду. кислотами называются веще-
ства, содержащие водород, и способные при реакциях отщеплять протон.
Возможно, что отщепление протона от нейтральных сернокислых солеи двух-
валентных металлов происходит за счет содержащейся в них молекулы кри-
сталлизационной воды п объяснение действия этих катализаторов остается
в рамках кислотной теории.
Значительно труднее сделать выводы о природе и механизме каталити-
ческого действия восстановленных водородами молибдатов никеля, пинка и
меди, так как публикаций по этому вопросу очень мало и они допускают
не однозначное их толкование.
К катализаторам, применяемым для изомеризации пинена,
предъявляются следующие требования.
1. Должен быть обеспечен высокий выход камфена.
2. Образование фенхенов должно быть минимальным.
3. Катализаторы должны быть достаточно дешевы пли, по
крайней мере, расходоваться в небольших количествах, или
легко регенерироваться.
Далеко не все перечисленные катализаторы удовлетворяют
указанным требованиям.
В довоенное время германской фирмой Шеринг была про-
ведена большая работа по подбору катализаторов для изоме-
ризации пинена. Результаты этих исследований, выполненных
Ульферсом и Эрбе, стали известны по окончании второй миро-
вой войны. В табл. 13 приведены максимальные выходы техни-
ческого камфена, т. е. суммы камфена, трициклена и фенхенов,
получающиеся при проведении реакции с разными катализато-
рами. При действии некоторых катализаторов, например щаве-
левой кислоты, наряду с камфеном получается эфир борнеола.
Образующийся в этих случаях борнеол приплюсован к камфену.
Точность анализов, вероятно, лежит в пределах ±5%.
Эти данные показывают на значительное преимущество ти-
танового катализатора по сравнению с рядом других изученных
катализаторов. С титановым катализатором получают не только
наиболее высокий выход технического камфена, но и имеют
наименьшее образование фенхенов. Это позволяет получать кам-
фару лучшего качества, чем с другими катализаторами и по
борнилхлоридному методу. На ранних стадиях развития изоме-
ризационного способа для промышленного получения камфена
применяли алюмосиликатные катализаторы, одноводный серно-
кислый магний и (в опытном порядке) вольфрамовую кислоту.
Со временем алюмосиликаты и сернокислый магний были остав-
лены и лишь титановая кислота в качестве катализатора для
изомеризации пинена стала применяться во всем мире. Тем не
менее мы рассмотрим наряду с действием титановой кислоты
на пинен действие на него алюмосиликатных катализаторов,
41
поскольку они применялись в заводской практике Советского
Союза.
Таблица 13
Выходы технического камфена, полученные из пинена на разных
катализаторах, по данным лаборатории завода а. о. Шеринг
в г. Эберсвальде
Катализатор Выход, % Катализатор Выход, %
Титановая кислота 85 Фосфорновольфрамовая, крем- 54
Вольфрамовая кислота 83 невая кислоты, фосфорномо-
Сурьмяная кислота 83 либденово-кислый аммоний
Магний сернокислый моногид- 77 Кремневольфрамовая кислота, 53
рат Диаспор и глина 70 силикагель Молибденовая кислота 52
Борнофосфориая кислота 69 Борноуксусная и борноволь- 49
Ванадиевая кислота 68 фрамовая кислоты
Щавелевокислый алюмииий+ 65 Каолин не гидратированный 48
-(-щавелевая кислота Пористая не обожженная ке- 47
Вермикулит 63 рамика 46
Щавелевая кислота 62 Пермутит
Каолин гидратированный 61 Мышьяковомолибденовая кис- 44
Марганец фосфорнокислый 60 лота
КИСЛЫЙ Фосфорномолибдеиовая кис- 41
Фосфорноурановая кислота 59 лота 35
Фосфорнооловянная кислота 55 Борная и мышьяковая кислоты
Для использования в качестве катализаторов брали природ-
ные глины, обладающие высокой дисперсностью, предпочти-
тельно с крупных разработок, чтобы обеспечить постоянные
свойства материала.
Глину измельчали, просушивали при температуре 125—150°С, после чего
использовали в качестве катализатора. Активность такого катализатора
колеблется в довольно широких пределах. Более постоянны каталитические
свойства глин, активированных нагреванием с 10%-ной соляной кислотой 1174].
Целью активации является удаление с поверхности катализатора сорбирован-
ных катионов и замена их ионами водорода. Активирование ведут обычно
при нагревании в течение 6—8 ч, после чего катализатор промывают декан-
тацией, фильтруют на фильтр-прессах, сушат на воздухе, измельчают и затем
окончательно высушивают в сушильном шкафу.
Алюмосиликатные катализаторы имеют резко выраженные кислые свой-
ства: их суспензии в воде имеют кислую реакцию, кроме того, они вытесняют
уксусную кислоту из водных растворов уксуснокислого натрия. Активность
катализаторов повышается по мере увеличения их кислотности. Последнюю
определяют по количеству мг-эквивалентов уксусной кислоты, вытесненной
1 г катализатора [28], или по количеству мг-эквивалентов едкого натра, кото-
рое нужно затратить для доведения pH суспензии катализатора до 7 [133].
Активность катализаторов сравнивают по скорости изомеризации пинена.
Свойства некоторых алюмосиликатных катализаторов, применяемых для изо-
меризации пинена, приведены в табл. 14.
42
Таблица 14
Свойства некоторых алюмосиликатных катализаторов,
используемых для получения камфена из пинена
Расход NaOH в мг-эквива-
лентах
Наименование катализаторов
на доведение
pH суспензии
I г катали-
затора в воде
до 7
на нейтрали-
зацию
СНзСООН,
выделившейся
при взаимо-
действии 1 г
катализатора
с CHsCOONa
Часовярская глина, высушен- 62 6,48 — 0,038
ная при 12532 То же, активированная 80 2,67 0,183 0,223
То же 88 3,49 0,101 0,138
Определена с помощью
метиленовой синей (см. гл. XII. Б.
1)
В противоположность алюмосиликатам, кремневая кислота и водная окись
алюминия совершенно лишены каталитической активности [146]. Это явление
объясняют строением решеток перечисленных веществ [107].
Заместители вокруг атома кремния располагаются подобно заместителям
вокруг атома углерода и занимают положение по углам тетраэдра. В крем-
невой кислоте, которую можно рассматривать как полимер, имеет место со-
единение неопределенного числа мономеров тетраэдрических ячеек кремния
через кислородные атомы. На следующей схеме приведено двухмерное изо-
бражение трехмерной решетки соединения кремния с кислородом:
1
— Si Si 1 1 Si —
1 1 О о 1 h О 1
11 I — о —Si—О—Si—О— 1 Si— О —
О О О
— Si— — Si— — Si —
Каждый атом кремния имеет четыре валентности, каждый атом кислорода
две валентности. Все валентности полностью насыщены. Можно представить
в такой структуре и наличие атомов водорода, соединенных с атомами кис-
лорода. Здесь электростатическое правило валентности соблюдено, все ва-
лентности полностью насыщены и связь носит гомополярный характер, проч-
ность связи равна 1. Условия отщепления протона здесь крайне затруднены,
поэтому кремниевая кислота является слабой кислотой.
43
Алюминий способен изоморфно замещать атом кремния в решетке сили-
ката, занимая место кремния. Алюминий трехвалеитен, однако, заняв место
кремния, он становится окруженным четырьмя кислородными атомами:
— Si — — Si — — Si —
Il I 3/4 II
— О —Si ^4-0 — Al — 0-4 Si — О —
l| 1^ Il
О I О Ю
— Si— —Si Si '
В этих условиях прочность связи кислород—алюминий составит 3/4,
а валентность каждого атома кислорода окажется ненасыщенной на ‘/4. Та-
ких атомов кислорода 4, следовательно, каждый тетраэдр, составленный ато-
мом алюминия и четырьмя атомами кислорода, ненасыщен на одну полную
валентную связь, а так как кислород в решетке заряжен отрицательно,
то в ячейке, окружающей алюминий, создается отрицательный заряд, кото-
рый компенсируется протоном. Ионный характер связи между протоном
и алюминиевым комплексом ячейки придает всему образованию свойства
сильной кислоты.
Помимо изложенного, имеются и другие объяснения кислотности алюмо-
силикатов [10, 98, 104].
Наблюдавшаяся некоторыми авторами каталитическая изомеризация
пинена под влиянием силикагелей объясняется наличием в этих гелях при-
меси алюминия [146].
Для проведения каталитической изомеризации вносят ката-
лизатор при интенсивном размешивании в жидкий пинен и на-
гревают реакционную смесь при заданной температуре. Реакция
протекает уже при 20°С и даже при более низких температурах
[174], но для уменьшения количества катализатора, который сор-
бирует значительное количество терпенов, процесс ведут при
температуре между 100 и 160—170°С (температура кипения ре-
акционной смеси). Из изомеризата отгоняют технический камфен,
выход около 60%. Камфен сильно загрязнен фенхенами. При
дальнейшей переработке из этого камфена получают техниче-
скую камфару с низкой температурой кристаллизации (156—
163°С) из-за примеси изофенхона.
Ряд исследователей проводили работы по улучшению каталитического
способа получения камфена с использованием тех же алюмосиликатных ката-
лизаторов. Значительная часть исследований была посвящена поискам лучших
катализаторов среди еще не испробованных для этой цели алюмосиликатов.
Исследования Гурвича [63] — изомеризация пинена флоридином, Тищенко и
Марта [172] — применение каолина и боксита и Тищенко и Рудакова [174] —
применение глин чрезвычайно сузили возможности патентования, тем более
что Тищенко и Рудаков писали не о глинах как таковых, а об образующих
их алюмокремневых кислотах, что, по-существу, распространяло их публи-
кацию на все алюмосиликаты. Тем не менее в СССР и в США были полу-
чены авторские свидетельства и патенты на изомеризацию пииена рядом алю-'
44
мосиликатных материалов. Эти авторские свидетельства и патенты, по-су-
ществу, не внесли ничего нового (табл. 15). Заявленные выходы камфена
практически не отличались от выходов, полученных Тищенко и Рудаковым.
Таблица 15
Выход технического камфена при каталитической изомеризации пинена
на разных алюмосиликатных катализаторах
Катализатор Год пуб- 1 ликации Темпера- тура, СС Выход, % Авторы
Активированные 1932- 20-160 55-62 Тищенко В. Е., Рудаков Г. А.
ГЛИНЫ 1933 [173, 174]
«Сиштоф» 1 1936 30 до 65 Дашкевич Б. Н., Вольнов Ю. Н. где]
«Сиштоф»1 1947 100-155 50-55 [ObJ Рутовский Б. Н., Любомилов
В. И. [161]
Пирофилит 1948 158 61 Керзон [251]
Галлуазит 1945 150 59-63 Кирпатрик [250]
Хлорит 1945 156 60 Керзон [249]
Вермикулит 1938 156 56-71 Хенке, Этцель [222]
1 Авторы называют «сиштофом» нерастворимый остаток от извлечения
алюминия из глин с помощью серной кислоты. Он состоит из кремнезем;.
неразложениого алюмосиликата и может содержать сернокислый алюминий
и серную кислоту.
Значительно большего внимания заслуживают работы, предпринятые для
изменения свойств алюмосиликатных катализаторов с целью повышения вы-
ходов камфена и улучшения процесса изомеризации пинена в целом.
Главными недостатками активных глин, приготовленных по
описанному выше способу, были:
1. Очень высокая каталитическая активность, в результате
чего продолжительность реакции до полного исчерпывания пи-
нена составляла лишь 1—2 ч при температуре кипения. Это
делало невозможным осуществлять надлежащий контроль за
процессом. Колебание активности катализатора приводило или
к слишком глубоко зашедшей реакции и потере части образо-
вавшегося камфена из-за лолимеризации или к неполному пре-
вращению пинена, что крайне отрицательно влияло на после-
дующие стадии производства.
2. Образование наряду с камфеном значительного количе-
ства неотделимых от них фенхенов, которые при последующей
переработке оставались в камфаре в виде изофенхона и сни-
жали ее качество.
45
Оставлял желать лучшего и выход этерифицирующихся ве-
ществ (2ЭВ) *.
Чтобы снизить активность катализатора, Любомилов [92] предложил
обрабатывать активированную глину, полученную по Тищенко и Рудакову,
равной массой пинена при 120—150°С. После отжима от образовавшихся
полимеров глину промывают спиртом и сушат. Реакция с таким «травлен-
ным» катализатором протекает значительно меиее энергично, а выход 2ЭВ
повышается до 64%.
Камфен при нагревании с алюмосиликатным катализатором полимери-
зуется значительно медленнее, чем одновременно с ним образующиеся моно-
циклические терпены и фенхены [130, 131]. На этой основе был описан способ
очистки камфена от фенхенов [93, 128]. Способ позволяет получать камфен
с температурой кристаллизации 42—43°С, который при дальнейшей перера-
ботке превращается в камфару с температурой кристаллизации 165—169°С.
Очистка сопровождается потерей части камфена.
Остаток, получающийся после извлечения из глины алюминия серной кис-
лотой при производстве квасцов, состоящий из кремнезема и иеразложениого
алюмосиликата («сиштоф»), изомеризует пииен подобно глине. Выходы Z3B
получаются примерно одинаковые [65, 161]. Однако, если на поверхности
катализатора остаются сернокислый алюминий и серная кислота, выходы
увеличиваются и при соотношении между ними 1-: 1 мол выходы S3B дости-
гают 75—78%. Катализатор этого типа действует на пииен меиее энергично,
чем активная глина или остаток, отмытый от солей алюминия. Образующийся
камфеи сильно загрязнен фенхенами. Выход 63—68% 2ЭВ был достигнут
и на синтетических силикатах алюминия, хрома и железа, полученных из рас-
творимого стекла и соответствующих солей [162].
Описанные улучшения алюмосиликатных катализаторов в основном не
дошли до промышленности, так как алюмосиликатные катализаторы уступили
место титановой кислоте.
Титановый катализатор впервые упоминается в патенте
фирмы Шеринг [313]. Он был внедрен в германскую промышлен-
ность до второй мировой войны, а после войны его получение и
применение были описаны в работах [167, 199, 224].
Успех титанового катализатора обусловлен следующими при-
чинами:
1. Высоким выходом 2ЭВ.
2. Низким содержанием фенхенов в продуктах реакции.
3. Малой полимеризацией терпенов, которая не превышает
2% при правильном ведении процесса.
4. Возможностью получения катализаторов высокой и пони-
женной активности, что позволяет вести процесс достаточно
медленно. Это обеспечивает надлежащий контроль за ходом ре-
акции, а в случае надобности дает возможность форсировать
процесс.
5. Малым расходом катализатора и простотой его регене-
рации.
* Этерифицирующимися, формилирующимися или ацетилирующимися на-
зываются вещества, образующие сложные эфиры при взаимодействии с му-
равьиной или уксусной кислотой в присутствии катализатора, например сер-
ной кислоты. В изомеризатах, полученных из пинена,— это сумма камфена,
трициклена, борнилена и фенхенов.
46
Применяющийся в настоящее время промышленный катали-
затор— титановую кислоту обычно получают гидратацией ката-
литически неактивной двуокиси титана водными растворами
NaOH при нагревании под давлением и последующим разложе-
нием образовавшегося титаната разведенными кислотами на хо-
лоду.
Для получения титанового катализатора может быть исполь-
зована установка, схема которой приведена на рис. 1.
Рис. 1. Схема установки для
получения каталитически ак-
тивной титановой кислоты:
Пар
Конденсат
I — мерник для раствора NaOH;
2 — мерник для воды; 3 — автоклав;
4 — смеситель для разложения ти-
таната и промывки титановой кис-
лоты: 5 — мерник для кислоты; 6 —
центрифуга; 7 — сушильный шкаф;
8 — краскотерка
Пар
к
Катализатор
В
ПромыВныв П
Воды
Катализатор
Конденсат
Обработку двуокиси титана 20—30%-иым раствором едкого натра произ-
водят в чугунном или стальном эмалированном автоклаве 3. Продукт реак-
ции— взвесь гидратированных с поверхности частиц ТЮ2 (с содержанием
около 12% Na2O на сухую массу) спускают в смеситель 4 из нержавею-
щей хромоникелевой стали и разбавляют 4—5 объемами воды. После отстаи-
вания раствор щелочи сливают декантацией, в случае надобности промывают
осадок декантацией, разбавляют водой и при непрерывном размешивании
добавляют к суспензии кислоту для разложения титаната. После установле-
ния заданной кислотности водной фазы промывают осадок до нейтральной
реакции, отжимают его на фильтр-прессе или центрифуге, сушат при 100—
110°С и растирают в порошок. Для приготовления растворов и промывок
используют дистиллированную или очищенную ионообменниками воду.
Разложение титаната натрия кислотой — реакция обратимая,
протекающая по уравнению:
XO-Na++H+^XO~H++Na+.
47
Константа равновесия этой реакции:
„ [Na+] • 1ХО-Н+]
л [Н+] • [XO-Na+] •
(О
Следовательно, чем выше концентрация ионов водорода и
чем ниже концентрация ионов натрия в растворе, тем полнее
разложение титаната. На этом основано получение катализато-
Мг-экв НаОИ на 1 г катализатора
Рис. 2. Кривые потенциометрического
титрования титановых катализаторов
0,1 н. раствором ХаОН:
ров самой разнообразной
активности. Если титанат
хорошо отмыт от едкого нат-
ра, разложение .произво-
дится сильной кислотой, на-
пример соляной, полученный
катализатор практически не
содержит натрия и его ак-
тивность очень высока. Если
же отмывка едкого натра
от титаната незначительна,
а для разложения приме-
няется органическая кисло-
та, муравьиная и, в особен-
ности, уксусная, а кислот-
ность раствора после его
подкисления устанавлива-
ется 0,1—0,5%, в катализа-
торе остается много связан-
ного натрия и активность
его не велика [133]. На прак-
тике предпочитают работать
именно с такими катализа-
торами, с ними получается
более высокий выход 2ЭВ
/ — катализатор, полученный электролитиче-
ским разложением титаната натрия; 2, 5 —ка-
тализаторы, полученные разложением тита-
ната натрия разбавленной органической кис-
лотой (табл. 20)
и процесс лучше поддается
контролю.
Для получения титаната нат-
рия можно с успехом использовать
ц техническую метатитановую кис-
лоту — полупродукт производства двуокиси титана, получаемую водным гид-
ролизом сернокислого титана. Техническая метатитановая кислота содержит
7—8% SO3, и не имеет ясно выраженных каталитических свойств. С едким
натром она реагирует под атмосферным давлением.
Тщательно отмытый от кислоты и высушенный катализатор
имеет ярко выраженные кислые свойства.
Значение pH водных суспензий катализатора колеблется
в широких пределах, между 3 и 6, и в первую очередь зависит
от глубины разложения титаната натрия. Титановый катализа-
тор, подобно алюмосиликатному, имеет подвижный, легко об-
мениваемый ион водорода, который может быть определен пря-
48
мым потенциометрическим титрованием водной суспензии ката-
лизатора раствором NaOH или титрованием уксусной кислоты,
выделившейся в результате обменной реакции катализатора
с 1 н. раствором уксуснокислого натрия [28, 133].
Свойства некоторых образцов катализатора приведены
в табл. 16, а кривые их потенциометрического титрования рас-
твором NaOH — на рис, 2.
Таблица 16
Свойства титановых катализаторов
0,655
0,565
Разложение тита-
ната натрия кис-
лотой
То же
Электролитиче-
ское разложение
титаната натрия
9,5
7,9
8,7
0,23
2,62
4,38
0,49
3,06
4,44
6,1
2,9
0,56
0,53
0,105
0,30
132 160 0,2 0,75
180 / 135 1,0 1,0
[ 160 0,2 0,75
200 135 1,0 8,0
114 135 2,0 2,0
Из приведенных данных видно, что pH и обменная способ-
ность катализаторов по NaOH и СНзСООИа колеблются в ши-
роких пределах. Активность катализаторов, измеренная по коли-
честву времени, которое нужно затратить на полную изомери-
зацию пинена, зависит в первую очередь не от его pH, а от
количества способных к обмену водородных ионов, отнесенных
к единице массы катализатора. При одинаковой обменной спо-
собности у катализаторов с большой удельной поверхностью pH
всегда сдвинут в кислую область меньше, чем у катализаторов
с малой удельной поверхностью.
Так же, как и кремневая, твердая титановая кислота пред-
ставляет собой частично гидратированный полимер двуокиси
титана, и ей ни в какой степени не соответствуют мономолеку-
лярные формулы Ti(OH)4 или H2TiO3.
Кислые свойства титановой кислоты обусловливаются не со-
держанием в ней сорбированной кислоты, использованной для
разложения титаната, а так же, как и свойства алюмосиликат-
ных катализаторов,— строением ее решетки. Это подтверждается
4 Заказ № 43
49
тем, что активный титановый катализатор может быть по-
лучен электролитическим расщеплением титаната натрия, т. е.
без применения кислот в процессе его получения [133].
В составе титанового катализатора обязательно присутствие конституци-
онной воды; с удалением воды он так же, как и алюмосиликат, теряет свои
каталитические свойства.
Рудаков, Шестаева н Иванова исследовали возможность придания ката-
литических свойств каталитически неактивной двуокиси титана путем нане-
сения на ее поверхность серной кислоты. Такой катализатор по своим свой-
ствам напоминает титановый, полученный разложением титаната натрия кис-
лотами [149] (гл. IV.4). Это исследование развил Букала с сотрудниками,
в результате чего был опубликован новый способ приготовления титанового
катализатора [206, 218]. Для получения катализатора по этому способу
100 массовых частей двуокиси титана обрабатывают 20—200 массовыми
частями 5—10%-ного водного раствора серной кислоты. Полученную пасту
после хорошего размешивания подсушивают в тонком слое при 60—100°С,
после чего подвергают активации 2—6 ч при 130—160°С.
Реакция с катализатором, полученным по описанному способу, протекает
более медленно, чем с титановым катализатором, полученным через титанат
натрия, поэтому авторы рекомендуют вести процесс при 160°С в присутствии
2% катализатора, содержащего 5% H2SO4. В этих условиях продолжитель-
ность реакции составляет 4 ч [219], тогда как с 0,2% катализатора, получен-
ного пз титаната натрия, при 160°С реакция проходит за 1 ч. Состав про-
дуктов реакции, полученных с обоими катализаторами, практически одина-
ковый.
Получить катализатор нанесением серной кислоты на каталитически неак-
тивную двуокись титана проще, чем разложением титаната кислотами, но его
применение, вероятно, встретило бы осложнения из-за необходимости подвер-
гать регенерации сравнительно большое количество катализатора после одно-
разового использования [219]. Процесс регенерации сводится к экстракции
сорбированных катализатором терпенов и повторением процесса приготовле-
ния катализатора вновь, начиная с нанесения серной кислоты. Катализа-
тор же, полученный из титаната натрия, применяется многократно без реге-
нерации, отчего его расход очень мал, порядка 0,6 кг/т переработанного
пинена.
Двуокись титана поступает в продажу в виде двух модификаций, имею-
щих разные решетки: рутила и анатаза. По Дулю и Букала, из общих моди-
фикаций может быть получен как активный, так и иеактивиый катализатор.
В своей работе авторы ограничиваются оценкой активности катализаторов,
полученных из различных торговых марок двуокиси титана [219].
Применяемый в нашей промышленности катализатор, судя по рентгено-
граммам, имеет структуру анатаза или аморфен. Степень дисперсности по-
рядка 10-5—10-6 см.
Большинство катализаторов крекинга теряют каталитическую активность
при взаимодействии с перегретым водяным паром [121].
Потери каталитической активности катализатора сопровождаются сокра-
щением его удельной поверхности и исчезновением мелких пор. Титановая
кислота теряет свою каталитическую активность при длительном кипячении
ее водных суспензий. При этом поверхность катализатора сокращается при-
мерно в 2—2,5 раза. Если в катализаторе содержалось много натрия (сус-
пензия превращается из кислой в щелочную), активность катализатора пол-
ностью теряется. Если же катализатор до кипячения содержал мало натрия,
то суспензия катализатора имеет кислую ракцию и у катализатора сохра-
няется некоторая, однако сильно пониженная, активность. Очевидно, потеря
активности связана с сокращением удельной поверхности катализатора и
уменьшением обменной способности. При большом содержании натрия в ка-
тализаторе в результате сокращения обменной способности все сохранив-
шиеся, способные к обмену протоны замещаются натрием, в результате чего
50
имеет место полная потеря каталитической активности. После удаления
«атрия из такого катализатора он приобретает незначительную каталити-
ческую активность.
3. Реакции, протекающие при получении камфена
методом каталитической изомеризации
а- и р-пиненов
При получении камфена из пинена через борнилхлорид на
обеих стадиях процесса получают продукты, достаточно устой-
чивые к присутствующим реагентам: борнилхлорид не изменя-
ется при действии на него хлористого водорода, а камфен прак-
тически не изменяется под влиянием щелочного агента, приме-
няемого для отщепления хлористого водорода от борнилхлорида.
При изомеризационном методе получения камфена первич-
ные продукты изомеризации под влиянием присутствующего ка-
тализатора вступают во вторичные реакции, которые приводят
к новым превращениям. Рассмотрим эти превращения.
Присоединяя протон по двойной связи, оба пинена (I) и
(ПГ) превращаются в ион карбония (II). Реакция обратима,
и она приводит к равновесию между а- и 0-пиненами [137] (с. 52
сверху). Так как в равновесной смеси при температуре реакции
находится около 98% а- и 2% 0-пиненов, то применение в каче-
стве исходного продукта 0-пинена или смеси а- и р-пиненов с
большим содержанием 0-пинена на начальном этапе реакции дает
значительное накопление а-пинена в изомеризатах, в продуктах
же изомеризации а-пинена 0-пинен обнаруживается лишь в не-
большом количестве.
Пинилкарбоний-ион (II), кроме того, претерпевает несколько
необратимых перегруппировок. Его электронно недостаточный
4-й С-атом может оттянуть электронную пару, образующую
связь между 5-м и 7-м С-атомами и образовать борнилкарбо-
ний-ион (IV) (перегруппировка Вагнера, гл. III.3). Борнилкар-
боний-ион, претерпевая вторичную перегруппировку Вагнера,
превращается в изокамфилкарбоний-ион (V), а этот последний
отщепляя протон, превращается в камфен (VI). Кроме того,
борнилкарбоний-ион с отщеплением протона превращается
в трициклен (VII) и борнилен (VIII). Изомеризация борнилкар-
боний-иона (IV) в изокамфилкарбоний-ион (V) и образование
камфена (VI) и трициклена (VII) из этих ионов являются обра-
тимыми реакциями. Вероятно, обратимым является и образова-
ние борнилена (VIII), поэтому камфен, трициклен и борнилен
имеют равновесные концентрации (с. 52 сверху).
Камфен, трициклен и борнилен под влиянием катализаторов
подвергаются полимеризации [130].
Во много раз быстрее, чем полимеризация, протекает рацемизация кам-
фена (с. 52 снизу). Оиа может происходить как из-за перегруппировки Намет-
кина, так и из-за обратимых превращений камфен-трициклен [141]. Этот про-
цесс не имеет существенного значения для последующей переработки кам-
фена иа камфару и не снижает выхода.
4*
51
IV vu IV
Пинилкарбоний-нон (II) может претерпеть необратимую пе-
регруппировку Вагнера с разрывом четырехчленного кольца не
только между 5-м и 7-м С-атомами, но и между 5-м и 6-м С-ато-
мами. В этом случае образуется фенхилкарбоний-ион (IX), из
которого в результате ряда превращений образуются цикло- (X),
а (XII), ₽ (XIV), у (XV) и 6 (XVII)- и е (ХУШ)-фен-
хены.
Согласно приведенной схеме образование фенхенов из пинена может про-
исходить как за две, так и за одну стадии. В первом случае на первой ста-
52
дии образуются цикло-(Х)- и а (ХП)-фенхены, иа второй 0 (XIV), у (XV>
и б (XVII) и е (XVIII)-фенхены. Промежуточным продуктом является цик-
лофенхен. По более новым представлениям допускают возможность взаимных
превращений ионов (IX), (XI), (XIII) и (XVI) или образование соответст-
вующих резонирующих мостиковых ионов. В этом случае все фенхены обра-
зуются в качестве первичных продуктов [234]. Из (—)-а-пинена и соответ-
ствующего ему нона (II) образуются (—)-цикло, (—)-0- и ( + )-а-фенхены.
При нагревании с катализатором фенхены не образуют но-
вых продуктов изомеризации и реакция ограничивается взаим-
ными превращениями фенхенов по приведенной схеме, однако
происходит полимеризация фенхенов, протекающая необратимо
и быстрее, чем полимеризация камфена, что позволяет очищать
камфен от части сопутствующих ему фенхенов [93, 128, 131].
Третья необратимая перегруппировка пинилкарбоний-иона
(II) вызывается захватом электронной пары, образующей связь
между 5-м и 7-м С-атомами для образования двойной связи
между 4-м и 5-м углеродными атомами. При этом образуется
терпенилкарбоний-ион (XIX). Отщепляя протон, этот ион пре-
вращается в смесь лимонена (XX) и терпинолена (XXI). При
изомеризации иона (XIX) в карбоний-ион (XXII) и отщеплении
от последнего протона получается терпинолен (XXI), а (XXIII)-
и у (ХХ1У)-терпинены [135, 147, 220].
Первичный продукт реакции состоит в своей основной массе
из лимонена (XX) и терпинолена (XXI), образование же а- и
у-терпиненов незначительно. Но терпинолен в результате вто-
ричных реакций превращается в а (XXIII)- и у (ХХ1У)-терпи-
нены и п-метадиен-2,4 (8) (XXV).
5Я
XXIV XXIII XXII XXI xxv
При превращении пннена (I) в лимонен (XX) происходит перемеще-
ние двойной связи в кольце. В пользу этого говорят стереохимические
данные: пинеи (I) и лимонен (XX) с приведенным соотношением кон-
фигураций имеют одинаковый знак оптического вращения. Если бы реакция
сводилась к разрыву циклобутаиового кольца и присоединению протона
к 5-му С-атому, пинен и лимонен имели бы противоположные знаки оптиче-
ского вращения [78].
Наряду с изомеризационными превращениями терпенов про-
текает еще реакция перераспределения водорода в а-терпинене,
в результате чего образуются n-цимол (XXVI) и Д3-п-ментен
(XXVII) [147]:
XXIII XXVI XXVII
Моноциклические терпены под действием катализаторов, ис-
пользуемых для изомеризации пинена, необратимо полимери-
зуются. В соответствии с этим при неограниченно долгом нагре-
вании моноциклических терпенов с этими катализаторами они
могут быть количественно превращены в смесь, состоящую из
терпеновых полимеров, с n-цимолом и n-ментеном. Так, напри-
мер, в результате длительного нагревания дипентена с актив-
ной часовярской глиной было получено 72% полимеров и 28%
смеси, состоящей из равных количеств n-цимола и п-ментена
[127].
При переработке пинена, выделенного из скипидара Pinus
silvestris L., надо считаться с возможной примесью в нем Д3-ка-
рена (XXVIII). Присоединяя протон, Д3-карен превращается
в ион (XXIX) и далее в Д4-карен (XXX), а в результате разрыва
54
циклопропанового кольца образует сложную смесь моноцикли-
ческих терпенов пара- и мета-рядов [16, 132]:
(+)-XXVIII XXIX (+)- XXX
Образующиеся при каталитической изомеризации пинена и
скипидара терпеновые полимеры представляют собой вязкие
жидкости, состоящие на 75% из димеров и на 25% из полиме-
ров большей молекулярной массы, главным образом тримеров.
Димеры кипят при атмосферном давлении при температуре выше 300“С,
а при 17 мм рт. ст. 187—190°С. Димер Р-фенхена (XXXI)—кристаллическое
вещество с температурой плавления оптически деятельной формы + 83°С,
а рацемической +57°С [329].
Терпеновые полимеры практически не перегоняются с водя-
ным паром. Это облегчает их отделение от камфена и других
монотерпенов:
XXXI
Вопрос о химическом строении терпеновых полимеров, получающихся
в качестве побочных продуктов при каталитической изомеризации терпенов,
остается до сих пор открытым, за исключением димера 0-феихена (XXXI),
строение которого было установлено [329]. Димеры терпенов, получаемые при
каталитической изомеризации пинена, неоднородны и состоят из димеров
с одной двойной связью, вероятно, димеров пинена и камфена и димеров
моноциклических терпенов с двумя или тремя двойными связями. Димер
Р-феихеиа образует несколько кристаллических производных: гидрохлорид,
гидробромид, дихлорид. Кристаллические производные других димеров не
описаны.
Терпеновые полимеры получили широкое (около 10 тыс.
в год) применение в США как адгезионное средство. Ценятся
в основном продукты более высокой степени полимеризации, чем
полимеры, получающиеся при изомеризации пинена [239].
4. Некоторые более тонкие стороны механизма
изомеризации пинена
Катализаторы, вызывающие изомеризацию пинена, действуют специ-
фично. Одни из них направляют реакцию преимущественно в сторону образо-
вания камфена, другие в сторону образования моноциклических терпенов.
Это специфическое действие катализаторов не находит объяснения в рамках
55-
карбониево-ионных представлений, так как непонятно, почему пиннлкарбоний-
ион (II) в одних случаях дает один, а в других случаях другие продукты.
Поэтому нельзя сводить реакцию только к действию протона на молекулу
пинена, а нужно считаться и с анионом при действии растворимых кислот
и со строением решетки твердого катализатора.
Для растворимых в пинене кислот была сделана попытка найти зависи-
мость между константами их диссоциации в водных растворах и выходами
эфиров борнеола и камфена при действии этих кислот на пинен [265].
Рудаков, Шестаева и Иванова [149] изучали влияние структуры поверх-
ности твердого катализатора на направление реакции. Было установлено, что
серная и фосфорная кислоты, изомеризующие пинен почти исключительно
в моноциклические терпены, после нанесения иа поверхность некоторых
каталитически неактивных носителей начинают изомеризовать его и в камфен.
При этом было показано, что соотношение между образующимися при реак-
ции моноциклическими терпенами и суммой камфена и фенхенов зависит
не от природы кислот, нанесенных на поверхность носителя, а от специфиче-
ских свойств самого носителя. Например, выход камфена при каталитической
изомеризации пинеиа под влиянием серной, фосфорной кислот и алюмосили-
ката, нанесенных на поверхность каталитически неактивной двуокиси кремния,
совершенно одинаков. Однако выход камфена возрастает после нанесения
серной и фосфорной кислот на двуокись титана и падает после нанесения
фосфорной кислоты иа древесный уголь (табл. 17). Это показывает, что
образование тех или иных продуктов реакции определяется ие только спо-
собностью твердого катализатора отщеплять протон, но и структурой его
поверхности.
Таблица 17
Выход камфена при изомеризации пинена кислотами, нанесенными
на поверхность каталитически неактивных веществ при 155°
Носитель Нанесено на носитель^ % % катали- затора Продолжи- тельность реакции в часах 1 Прореагиро- вало пинена : в % Превратилось пинена в кам- фен и фснхсн В % 1
Кремневая кислота безводная — 5 2,5 0 0
То же А120з (17) 0,25 3 100 43,9
H2SO4(4,2) 2 7 74 44,3
.. Н3РО4 (10) 3 6 70 42,8
А12О3 (прокален- ная) — 2 4 0 0
То же H,SO4 (4,2) 0,75 13 100 65
Т1О2 — 2 4 0 0
То же H,SO4 (4,2) 1 7 100 68
Уголь древесный Н3РО4(Ю) 2,5 5,5 100 63
Н3РО4(Ю) 5 1 100 9
Исследования показали, что специфическое влияние иа реакцию поверх-
ности твердого катализатора не связано с наличием и размерами его пор.
Катализаторы, полученные нанесением окиси алюминия (0,1%) иа поверх-
ность силикагелей с различным радиусом пор (от 11 до 70А), а также на
аэросил-силикагель, вовсе лишенный пор, хотя и отличались по каталитиче-
ской активности, обладали одинаковой специфичностью. В катализатах, полу-
ченных в присутствии этих катализаторов, соотношение между моноцикличе-
скими и бициклическими терпенами было одинаковым [77].
Таким образом, специфическое действие твердых катализаторов надо ско-
рее искать в структуре их решеток и, возможно, в образовании комплексов
.56
углеводород—катализатор с разной степенью поляризации, зависящей от
степени кислотности и структуры решетки [77, 149]. Эти выводы до известной
степени подтверждены в работе [113], где показано, что увеличение пористо-
сти катиоиообменных смол и придание этим порам определенной структуры
не приводит к увеличению выхода камфена при изомеризации пинена в при-
сутствии указанного катализатора.
ГЛАВА V. ОСНОВНЫЕ ПРИНЦИПЫ
ПРОМЫШЛЕННОЙ ТЕХНОЛОГИИ
ИЗОМЕРИЗАЦИОННОГО СПОСОБА
ПОЛУЧЕНИЯ КАМФЕНА
1. Научные основы существующего процесса
В промышленной практике подвергают изомеризации жидкий
пинен. Реакцию ведут в аппаратах периодического действия
(изомеризаторах) при непрерывном размешивании. По оконча-
нии реакции изомеризат отделяют от катализатора и направ-
ляют на дальнейшую переработку, а изомеризатор загружают
свежим пиненом. Катализатор используют многократно. Пара-
метры технологического процесса: количество и свойства ката-
лизатора, температура реакции и массообмен, а следовательно и
продолжительность процесса, могут быть самые разнообразные.
Однако эти параметры должны обеспечивать получение изоме-
ризатов, богатых камфеном, содержащих возможно меньшее
количество фенхенов и несодержащих непрореагировавшего пи-
нена. Последнее требование вызвано тем, что дальнейшая пере-
работка изомеризатов, имеющих даже небольшую примесь
пинена, осложняется, а качество полученных из них камфена и
камфары значительно ухудшается.
На первый взгляд это трудно разрешимая задача, так как
все катализаторы, образующие камфен, вызывают и вторичные
превращения терпенов, в том числе и полимеризацию камфена.
Поэтому представляется, что, добиваясь полного использования
пинена, неизбежно придется потерять часть образовавшегося
камфена и других монотерпенов, превратив их в малоценные
димеры. Этого можно избежать, если при осуществлении про-
изводственного процесса учитывать особенности реакции.
Все терпены полимеризуются под действием катализаторов,
вызывающих изомеризацию пинена, но скорость полимеризации
различных терпенов разная. Моноциклические терпены, особенно
терпинены, полимеризуются намного быстрее камфена (табл. 18).
Это позволяет не опасаться полимеризации камфена, если ко-
личество катализатора не слишком велико и продолжительность
процесса соответствует скорости превращения.
Изомеризация жидкого пинена в присутствии твердых ката-
лизаторов протекает в гетерогенной среде. Подобные процессы
состоят из ряда стадий: диффузии молекул реагирующего
57
Таблица 18
Сравнение скоростей каталитических превращений терпенов
в присутствии катализаторов, применяемых для получения камфена
из пинена
Наименование терпенов Катали- затор, % Темпера- тура, °C Продолжи- тельность, ч Полиме- ризация, % Изомеризация, перераспре- деление водо- рода и другие превращения, %
Катализатор — активная глина
Пинен 0,5 160 1 8,2 91,8 а
Камфен 0,5 160 1 1 Нет
5 160 I 33,5 2б
20 100 2 44,0 Нет
Дипентен 0,5 170 1 23,6 38 в
Д3-Карен 0,5 170 1 10,9 45 г
Катализатор — титановая кислота
Пииен 2 135 1,5 2 98а
Камфен 10 135 5 16,9 12—16 д
Дипентен 10 135 6 67 33 е
0,5 135 0,5 9,6 64 ж
Терпинолен 0,5 135 1,5 22 633
Д3-Карен 1 135 2 5 42 г
а Камфен, трициклен, моноциклические терпены; 6 моиоциклические тер-
пены, п-цимол, n-ментеи и другие углеводороды (130); в терпинолен, а- и у-
терпинены, небольшое количество я-цимола и п-ментена [127]; г Д4-карен, мо-
ноциклические терпены, главным образом п-ряда [129, 132]; я трициклен [141,
324]; е п-цимол и п-ментен; терпинолен, а- и у-терпинены, немного п-цимола
и п-меитена [134, 147]; 3 дипеитен, а- н у-терпинены, 3,5% смеси п-цимола
с п-ментеном [147].
вещества к поверхности катализатора, сорбции их на поверхно-
сти, химической реакции, десорбции продуктов реакции, диффу-
зии продуктов реакции в объем. Скорость процесса в целом опре-
деляется скоростью самой медленной его (лимитирующей) ста-
дии. Если скорость химической реакции намного меньше скоро-
стей диффузионных процессов,— реакция протекает в кинетиче-
ской области. Если скорость диффузии молекул к поверхности
катализатора протекает много медленнее химической реакции,—
процесс протекает в диффузионной области.
Процесс, протекающий в диффузионной области, не ускоря-
ется с повышением температуры, но ускоряется от увеличения
массообмена в системе. Его энергия активации равна нулю.
И наоборот, процесс, протекающий в кинетической области, не
ускоряется с увеличением массообмена, но его скорость возра-
стает с увеличением температуры. Обычно скорость реакций,
протекающих в кинетической области, возрастает в 2—3 раза
при повышении температуры на 10°С.
-58
Помимо этого, есть еще переходная область, проявляющаяся,,
когда скорости химической реакции и диффузионных процессов-
сравнительно близки.
Большинство гетерогенных реакций может протекать в за-
висимости от свойств катализатора температуры, давления ir
массообмена как в кинетической, так и в диффузионной обла-
сти. Это же относится и к изомеризации пинена в жидкой фазе.
Дуль и Букала изучали сравнительную скорость изомеризации
а-пинена под влиянием двуокиси титана, на поверхность которой
Рис. 3. Состав продуктов каталити-
ческой изомеризации а-пинена в при-
сутствии 2% двуокиси титана, акти-
вированной серной кислотой через
4 ч после начала реакции, в зависи-
мости от числа оборотов мешалки
[219]:
/ — а-пинен; 2—камфен; 3 — S ЭВ; 4 —
сумма моноциклических терпенов (2МЦТ)
нанесено 5% серной кислоты [219, 206] при разном числе оборо-
тов мешалки (219] при температуре кипения (рис. 3). Как видно
из приведенных данных, скорость реакции увеличивается с чис-
лом оборотов мешалки. Следовательно, описанный процесс про-
текает в диффузионной и переходной областях по крайней мере
вплоть до скорости вращения 500 об/мин. Конечно, число обо-
ротов мешалки здесь не имеет абсолютного значения, так как
тот же массообмен может быть достигнут в больших заводских
аппаратах при значительно меньшем числе оборотов.
Кинетика каталитической изомеризации жидкого а-пинена
в присутствии алюмосиликатного и промышленного титанового
катализаторов (титановой кислоты) изучалась Рудаковым, Ива-
новой, Писаревой и Боровской [77, 155]. Реакцию проводили
в колбе с мешалкой (от 60 до 3000 об/мин) и в термостатирован-
ной качающейся «утке» (60 и 180 двойных качаний в минуту).
В этих условиях реакция протекает в кинетической области. Это
показывает, что свойства катализатора в значительной мере оп-
ределяют область протекания процесса.
На рис. 4 приведены кинетические кривые расходования пи-
нена и накопление продуктов реакции при изомеризации а-пи-
нена 3% титановой кислоты в термостатированной утке при
121°С. Из рисунка видно, что расходование пинена и накопление
59
продуктов реакции в начале процесса постепенно замедля-
ется, но далее скорость расходования пинена становится посто-
янной и не зависит от его концентрации в растворе. Лишь в са-
мом конце реакции наблюдается очень незначительное сниже-
ние скорости расходования пинена и образования камфена, что,
впрочем, компенсируется ускорением образования трициклена.
Рассмотрим в первую очередь стадию, протекающую с постоян-
ной скоростью. Скорость химической реакции, протекающей
Рис. 4. Кинетические кривые изомеризации
а-пинеиа на промышленном титановом катали-
заторе (титановой кислоте — 3%) при 121°С.
Катализатор: удельная поверхность 121 м2/г
(хроматографический метод тепловой десорб-
ции) [31, 32], обменная емкость по CH3COONa
0,245 мг. экв/г [28]:
/ — а-пинен; 2 — камфен; 3— трициклен; 4 —S2 и 3;
5 —лимонен; 6 — моноцнклнческие терпены; А — кри-
вые 1, 2. 4, 5 и 6; В — кривая 3
в кинетической области, пропорциональна концентрации реаги-
рующих веществ. Для нашего случая она должна выражаться
уравнением
(2)
где А — концентрация пинена на поверхности катализатора В
в единице объема реагирующей смеси в момент време-
ни т;
k — константа скорости реакции.
Если величина активной поверхности катализатора по ходу
процесса не меняется и концентрация сорбированного ею пи-
60
йена также остается постоянной, то уравнение (1) превраща-
ется в уравнение реакции нулевого порядка:
-^ = *на6л, (3)
ГДе ^набл— kAB.
Постоянная концентрация пинена на поверхности катализа-
тора может быть лишь в случае, если адсорбция пинена поверх-
ностью катализатора во много раз превосходит адсорбцию
продуктов реакции, которые по мере своего образования немед-
ленно вытесняются с поверхности, и достаточно хорошего массо-
обмена в системе, обеспечивающего необходимый приток пинена
к поверхности катализатора.
Рис. 5. Кинетические кривые рас-
ходования а-пинена при каталити-
ческой изомеризации:
/ — экспериментальная кинетическая
прямая, нулевой порядок реакции; 2 —
вычисленная кривая реакции, проте-
кающей по уравнению 1-го порядка
с той же начальной скоростью
Постоянная скорости реакции, не зависящая от концентра-
ции а-пинена в растворе,— важная особенность процесса, позво-
ляющая его осуществлять до полного израсходования пинена
даже при незначительных константах скорости реакции и избег-
нуть слишком долгого соприкосновения продуктов реакции с ка-
тализатором. Этим процесс выгодно отличается от процессов,
кинетика которых описывается уравнениями 1-го и 2-го поряд-
ков, поскольку их скорости снижаются по мере вступления
в реакцию реагирующих веществ.
В качестве примера на рис. 5 приведены экспериментально
полученная прямая расходования пинена по нулевому порядку
во второй период реакции и вычисленная кривая этой же реак-
ции, протекающей с той же начальной скоростью, но по 1-му по-
рядку.
Рассмотрим теперь начальный период реакции, протекающей
с убывающей скоростью (рис. 4). Поскольку понижение кон-
центрации пинена в растворе на второй стадии процесса прак-
тически не отражается на его скорости, постепенное снижение
скорости реакции в начале процесса, где концентрация пинена
в растворе наибольшая, может быть объяснена лишь постепен-
ным уменьшением активной поверхности катализатора (уравне-
ние 2) на этой стадии. Это может происходить из-за заполнения
части поверхности устойчивым продуктом взаимодействия пи-
нена с катализатором. В пользу этого косвенно указывает при-
сутствие в продуктах реакции небольшого количества борнеола
[130]. Возможно, одновременно имеет место и отравление части
61
поверхности примесями к пинену. Переход к постоянной скоро-
сти на второй стадии реакции соответствует окончанию этих:
процессов.
В соответствии с изложенным реакция изомеризации пинена
имеет две константы скорости: начального момента k', которая
определяется как тангенс угла касательной к кинетической кри-
вой в момент времени т = 0, и второй стадии, протекающей с по-
стоянной скоростью k". Соотношение k"/k' показывает долю ак-
тивной поверхности, участвующей в реакции на второй стадии.
С понижением температуры доля активной поверхности, участ-
вующей в реакции на второй стадии, падает и при 90°С практи-
чески достигает нуля. Поэтому реакция при этой температуре,
начавшаяся после внесения катализатора, вскоре приостанавли-
вается. Это соответствует заполнению поверхности катализатора
устойчивым продуктом присоединения.
Энергия активации начального периода реакции составляет
£'=120 кДж/моль (29 ккал/моль), второй стадии процесса £" =
= 168 кДж/моль (40 ккал/моль). Е', по-видимому, близка к £ИСт,
а £" — значительно отличается от нее, так как зависимость k'r
от температуры определяется не только изменением скорости
реакции, но и изменением величины активной поверхности ката-
лизатора, не занятой устойчивым продуктом присоединения.
Выход 2ЭВ * и суммы камфена и трициклена (SKT) в пределах
100—130°С практически не зависят от температуры реакции.
Следовательно, энергии активации образования камфена, три-
циклена, лимонена и фенхенов приблизительно одинаковы,
и разница в константах скоростей должна быть приписана лишь
разнице в энтропиях активации образования перечисленных про-
дуктов.
Следует остановиться на вторичных превращениях камфена,
лимонена и других терпенов, образовавшихся в качестве пер-
вичных продуктов при каталитической изомеризации пинена.
На рис. 6 показан состав продуктов каталитической изоме-
ризации а-пинена под действием титановой кислоты при 130°С
и его изменение по ходу реакции в условиях интенсивности пере-
мешивания. Под продуктами изомеризации подразумеваются
все вещества, содержащиеся в анализируемой пробе изомери-
зата, за вычетом а- и р-пиненов (небольшое количество р-пи-
нена образуется во время реакции) и примесей камфена и три-
циклена, содержащихся в исходном продукте. Как видно из
рис. 6, состав образующихся из пинена продуктов остается почти
постоянным, пока не прореагирует около 70% содержащегося
в растворе пинена. На этой стадии продукты реакции состоят из
65% камфена, 8—9 трициклена, 17 лимонена, 3 фенхенов, 3—
4 терпинолена, 1 —1,5% а- и у-терпиненов. Все эти продукты
можно считать первичными, поскольку их концентрация во
* См. примечание на с. 46.
62
время реакции практически не меняется. Трициклен образуется
параллельно с камфеном из пинилкарбоний-иона, а терпинолен
и терпинены параллельно с лимоненом из терпенилкарбоний-
иона (гл. IV.3).
Это подтверждается и тем, что рацемизация камфена и ли-
монена, как вторичная реакция на этой стадии, не имеет места.
Рис. 6. Состав продуктов каталитической изо-
меризации а-пинена при действии титановой
кислоты (3%) при 130°С и его изменения по
ходу реакции [155] (прочие условия проведения
реакции в подписи к рис. 4):
1 — а-пннен {2—13— состав образующихся продуктов
от начала реакции); 2 — камфен; 3 — трициклен; 4 —
лимонен; 5 — терпинолен; 6 — а-терпинен; 7 — v-тер-
пинен: 8 — 2,4 (8) л-ментаднен; 9 — смесь фенхенов;
10— бориилен; 11— 2ЭВ; 12 — 2МЦТ; 13 — полимеры;
.4 — кривые 1. 2, 3, 4, 7, 8, 11, 12, 13\ В — 9, 10\
С —5, 6
Камфен и лимонен, выделенные из изомеризатов, практически
не меняют своего оптического вращения, пока не прореагировало
70% пинена, причем их оптическое вращение лишь очень не-
значительно отличается от расчетного (табл. 19), что связано
с перегруппировками промежуточных карбоний-ионов. Растворы
же оптически деятельных лимонена и камфена в рацемическом
63
о
Таблица 19
Взаимосвязь оптического вращения а-пинена и образующихся из него камфена и лимонена на начальной стадии реакции
Катализатор Темпера- тура, 'С а-пинен Камфен [а]^ Лимонен [а|£>
прореагиро- вало, % найдено вычисле- но *♦ отклоне- ние, % найдено вычисле- но ** отклоне- ние, %
Активная глина 20 +28,3° 43 +58,0° +65,6° 11,5 — — —
То же 100 +28,3 53 +55,0 +65,6 16,3 +58° +70 17
Титановая кислота 100 +28,3 54 +55,7 +65,6 16,0 — — —
То же 160 +29,6 50 +58,0 +68,7 15,5 +69,2 +73,8 6,2
* Растворитель толуол; концентрации раствора около 45%.
* Вычислено из уравнения
lal D
[а1о = , ‘ ' 1а] D’ (4)
la J D
где [а]/> — вычисленное удельное оптическое вра-
щение образующихся камфена или ли-
монена при отсутствии рацемизации;
[а]о — удельное оптическое вращение опти-
чески чистого камфена или лимонена
(гл. ХШ, табл. 50);
[а] о—удельное оптическое вращение оптиче-
ски чистого пннена (гл. ХШ, табл. 50)],
[т] о —оптическое вращение исходного а-пи-
цена-
пинене при нагревании с катализатором практически полностью
сохраняют свое вращение, пока не прореагирует около 70% ра-
цемического пинена [134, 135, 141].
Такое течение процесса находится в полном соответствии
с его кинетикой. Вся поверхность катализатора покрыта реаги-
рующим пиненом, продукты реакции по мере своего образо-
вания немедленно десорбируются и, следовательно, не подверга-
ются вторичным превращениям.
После того как прореагировало 70% пинена, начинается
медленная, но все ускоряющаяся обратимая реакция превраще-
ния камфена в трициклен. Как только содержание пинена в ра-
створе упадет до 10%, эта реакция протекает уже быстро и
к моменту израсходования пинена останавливается из-за дости-
жения равновесия.
Состав моноциклических терпенов практически постоянен,
пока не прореагирует 90% пинена. Они состоят в основном из
лимонена (75%) и терпинолена (17%), но с этого момента на-
чинается быстрое превращение лимонена во вторичные про-
дукты в такой последовательности: лимонен->терпинолен-►
-►а- и у-терпинены и 2,4 (8)-п-ментадиенполимеры. Послед-
ние образуются в основном за счет полимеризации а-терпинена,
а так как содержание а-терпинена в продуктах реакции в при-
сутствии пинена очень мало, то предпосылки для образования
полимеров до полного израсходования пинена отсутствуют. Ско-
рость превращения лимонена в другие монотерпены по крайней
мере в четыре раза больше скорости его образования, и если бы
лимонен подвергался вторичным превращениям в присутствии
пинена, его концентрация в продуктах реакции была бы низка на
протяжении всего процесса, а концентрация продуктов изоме-
ризации лимонена была бы высока и создались бы условия
для значительной полимеризации.
Из изложенного следует, что трициклен, терпинолен, а- и у-
терпинены образуются и как первичные, и как вторичные про-
дукты реакции.
Содержание фенхенов в изомеризате составляет около 3%
(4% на 2ЭВ). Пока в растворе присутствует пинен, в состав
фенхенов входит около 80% а-фенхена и около 10% циклофен-
хена. После того как содержание пинена в изомеризате снизится
до 10%, начинается быстрое превращение а-фенхена в 0- и у-
фенхены. В момент окончания реакции фенхены состоят на
70—75% из а- и 0-фенхенов, остальное приходится на цикло- и
у-фенхены; б- и е-фенхены в условиях реакции практически не
образуются, для их образования нужны более жесткие условия
процесса.
Таким образом, после того как прореагировало 70% пинена,
десорбция камфена начинает замедляться и пинен перестает
покрывать всю поверхность катализатора, хотя продолжает по-
крывать ее большую часть. После того как прореагировало 90%
5 Заказ № 43
65
пинена, начинает задерживаться десорбция всех продуктов ре-
акции и они подвергаются вторичным превращениям.
Это должно было бы вызвать некоторое замедление расхо-
дования пинена если не после 70% прореагировавшего пинена,
то, во всяком случае, после того, как он прореагировал на 90%.
Но этого не наблюдается, возможно, из-за недостаточной чув-
ствительности методов анализа или увеличения активной по-
верхности катализатора в конце реакции, в связи с уменьшением
площади, занятой устойчивым соединением пинена с титановой
кислотой, из-за снижения
Рис. 7. Состав продуктов каталитиче-
ской изомеризации в момент их образо-
вания из жидкого а-пинена в присут-
ствии титановой кислоты (3%) при 130°С
(прочие условия проведения реакции
в подписи к рис. 6):
1 — камфен; 2 — трициклен; 3— 2 1, 2; 4 —
2МЦТ
содержания пинена в рас-
творе.
Сама по себе реакция
превращения части камфена
в трициклен не вредна, так
как оба эти терпена с почти
равным успехом превраща-
ются в камфару. Но неже-
лательна она потому, что
протекает на тех же участ-
ках поверхности катализа-
тора, где образуется камфен
из пинена, что ведет к заме-
длению образования камфе-
на, в то время как скорость
образования моноцикличе-
ских терпенов начинает тор-
мозиться значительно позже.
В результате этого содер-
жание 2ЭВ в продуктах
изомеризации пинена не-
значительно снижается от начала реакции к ее окончанию.
В рассмотренном на рис. 6 примере снижение составляет
2,5% от (77,5 до 75%). Это находится практически в пределах
точности анализа, поэтому целесообразно рассмотреть измене-
ние состава продуктов изомеризации в момент их образования
по мере расходования пинена (рис. 7).
Как видно из рисунка, обратимые превращения камфена
в трициклен начались уже после того, как прореагировало 50%
пинена, однако эти превращения занимали столь малую часть
активной поверхности катализатора, что не повлияли на суммар-
ную скорость образования SKT относительно образования 2МЦТ
вплоть до того момента, пока не прореагировано 80% пинена.
После этого скорость образования SKT начинает падать относи-
тельно скорости образования моноциклических терпенов, в ре-
зультате чего содержание SKT составляет лишь 65% в порции
изомеризата, образовавшейся после того, как прореагировало
90% пинена.
66
Общий выход 2КТ равен площади очерченной кривой 3, де-
ленной на 100, что составляет 72% против выхода на начальной
стадии процесса 74%, т. е. потери выхода SKT и соответственно
2ЭВ из-за неблагоприятного заключительного периода неболь-
шие. Однако это снижение выхода при неправильно поставлен-
ном процессе может быть весьма значительным.
Адсорбция пинена и продуктов его превращения титановой
кислотой и другими катализаторами, вызывающими его изоме-
ризацию, остается не изученной. Но исходя из общих законов
адсорбции известно, что в области низких концентраций раст-
воренного вещества количество его, поглощенное адсорбентом,
увеличивается с ростом концентрации раствора. В области боль-
ших концентраций оно не зависит от концентрации раствора,
так как в этом случае достигнуто полное насыщение поверхно-
сти адсорбента.
Используемые катализаторы значительно лучше адсорбируют
пинен, чем продукты его превращения. К тому же в начале ре-
акции концентрация пинена очень велика по сравнению с кон-
центрацией продуктов реакции. Поэтому адсорбционное равно-
весие на поверхности полностью сдвинуто в сторону пинена. Од-
нако к концу реакции содержание пинена падает настолько, что
его концентрация уже перестает обеспечивать полное насыще-
ние поверхности катализатора, а концентрация продуктов ре-
акции возрастает настолько, что они уже могут сорбироваться
в заметном количестве на поверхности катализатора. Когда
в растворе останется 30% пинена, в нем окажется около 45%
камфена, что дает основание для адсорбции заметного количе-
ства камфена катализатором. Содержание лимонена в данный
момент будет лишь около 12—13%, его адсорбция остается не-
значительной, поэтому вторичные превращения лимонена не про-
исходят. Когда концентрация пинена понизится до 10%, концент-
рация лимонена возрастает до 15—16%, в результате чего соз-
даются условия для его адсорбции наряду с адсорбцией пинена
и начинаются вторичные превращения. Помимо этого, надо счи-
таться и с тем, что адсорбция пинена на участках поверхности
катализатора, где образуются камфен и моноциклические тер-
пены, различная, т. е. эти участки имеют разные изотермы ад-
сорбции. Полное насыщение участков поверхности, где обра-
зуются моноциклические терпены, происходит при более низких
концентрациях пинена в растворе, чем насыщение тех уча-
стков поверхности, где образуется камфен. Это общая особен-
ность протекания многих параллельных реакций [30, 82, 123].
Все вышеизложенное не учитывало тот факт, что в слое жид-
кости, окружающей частицу катализатора, может быть более
низкая концентрация пинена, чем среднее его содержание в изо-
меризате, и более высокое содержание продуктов реакции, а это
может происходить в целом ряде случаев, и чем больше будет
разница между средней концентрацией пинена в растворе и
5*
67
в слое, окружающем частицы катализатора, тем более отрица-
тельное значение это окажет на выход 2ЭВ.
Разница в концентрации пинена у поверхности частиц ката-
лизатора по сравнению со средней концентрацией пинена в реа-
гирующей массе будет увеличиваться под влиянием следующих
факторов:
1. Плохого массообмена.
2. Большой активности катализатора.
3. Меньшей удельной поверхности катализатора по сравне-
нию с удельной поверхностью катализатора, имеющего ту же об-
менную емкость.
4. Большого количества катализатора, введенного в реакцию,
увеличения температуры в общем форсировании процесса.
Несомненно, большое значение имеют и адсорбционные свой-
ства катализатора относительно отдельных терпенов, но этот
вопрос остается не изученным.
Все эти факторы взаимосвязаны, и выбор технологических
параметров по одному из них вызывает необходимость учесть
и другие. Так, например, увеличение количества катализатора
или его активности заставляет решить — не потребуется ли во
избежание снижения выхода улучшить массообмен и т. д.
При проведении изомеризации пинена с малыми количест-
вами катализатора к концу процесса сказывается отравление
катализатора, в результате чего процесс замедляется. Замедле-
ние это, по сравнению с замедлением реакций 1-го и 2-го поряд-
ков, обычно невелико. Отравление происходит из-за сорбции
продуктов окисления и окислительной полимеризации, которые
образуются из терпенов в ходе основной реакции. Количество
этих продуктов зависит от величины раздела фаз терпены—воз-
дух и продолжительности реакции. При проведении реакции
в маленьких сосудах, например в лаборатории, где величина
поверхности раздела фаз очень велика по отношению к массе
реагирующего вещества, накопление продуктов окисления сказы-
вается особенно сильно. Чтобы снизить отравление, вводят повы-
шенное количество катализатора, прибавляют антиокислители
гидрохинон [134] и другие двух- и трехатомные фенолы [223]
или ведут реакцию под защитой инертного газа. В крупных за-
водских аппаратах, куда загружают 20—30 и даже 40 т пинена,
поверхность раздела фаз терпены—воздух по отношению к массе
реагирующего вещества невелика. Поэтому можно проводить
реакцию, ограничиваясь установкой предохранительного кла-
пана.
Отравление катализатора к концу реакции оказывает поло-
жительное влияние на процесс в целом, так как замедление ре-
акции на этом этапе обеспечивает более высокую концентрацию
пинена у поверхности катализатора при данном массообмене и,
следовательно, ведет к улучшению выхода. Однако возможна и
полная потеря активности катализатора до окончания реакции.
68
Тогда добавляют свежий катализатор, но такая добавка, произ-
веденная в конце реакции, по тем же причинам ведет к сниже-
нию выхода.
2. Особенности каталитической изомеризации
жидкого р-пинена
Каталитическую изомеризацию жидкого Р-пинена с применением титано-
вого и алюмосиликатного катализаторов изучали Иванова, Боровская и Ру-
даков [78]. Авторы показали, что в пределах 135— 150°С при наличии доста-
даков [78]. Авторы показали, что :
точно интенсивного перемешивания
реакция, так же как и каталити-
ческая изомеризация а-пинена,
протекает в кинетической области
по нулевому порядку. В резуль-
тате реакции образуются а-пинен,
а также камфен и прочие терпе-
ны, получающиеся при изомери-
зации а-пинена. При этом а-пинен
не вступает во вторичные превра-
щения, пока не прореагирует
основная масса р-пинена.
Позднее эту реакцию изучали
при 150—160° Дуль и Букала [220],
используя в качестве катализато-
ра двуокись титана, на поверх-
ность которой нанесена серная
кислота [206, 218]. Полученные ими
кинетические кривые позволяют
сделать те же выводы. На рис. 8
приведен состав продуктов изо-
меризации р-пииена двуокисью
титана, пропитанной серной кис-
лотой, и его изменения по ходу
реакции. Кривые графика анало-
гичны полученным при каталити-
ческой изомеризации а-пинена:
состав продуктов реакции постоя-
нен, вторичные превращения от-
сутствуют, пока концентрация ис-
ходного вещества не упадет ниже
30%. Разница заключается в том,
что образующийся а-пинеи не
вступает во вторичные превра-
щения, поэтому концентрация его
в продуктах изомеризации посто-
янна, пока присутствует в доста-
точной концентрации р-пинен. Если
при изомеризации а-пинена вторичные процессы исключены из-за блокиро-
вания поверхности катализатора а-пиненом, то здесь они отсутствуют,
так как поверхность катализатора блокирована р-пиненом. При достижении
30%-ной концентрации в растворе р-пииеиа начинает вступать в реакцию
наряду с ним а-пинен, что видно по падению его содержания в продуктах
изомеризации и по одновременному замедлению расходования р-пинена. Это
приводит к соответственному увеличению содержания камфена в продуктах
изомеризации.
Содержание а-пинена в продуктах изомеризации падает с повышением
температуры: при 88°С его содержится 57%, при 112°—51, при 121°—45,
7о
80
60
W
20
10
20
xfo’e
Рис. 8. Состав продуктов каталитической
изомеризации р-пииеиа двуокисью ти-
тана, активированной серной кислотой
(5%), и его изменения по ходу реакции
[220, 289]:
1— 3-пинен; 2 — а-пинен; 3 — камфен; 4 —
трициклен; 5 — ЕЭВ; 6 — S МЦТ
69
при 160°С — 26%. Очевидно, для образования а-пинеиа требуется более низ-
кая энергия активации, чем для образования камфена и лимонена.
Выход камфена и трициклеиа при каталитической изомеризации р-пинена
с титановым катализатором примерно на 10% ниже, чем при каталитической
изомеризации а-пинеиа 78, 220].
Дорогой и более дефицитный, чем а-пинен, Р-пинеи для синтеза камфары
использовать не следует.
По данным работы [220], константа скорости расходования Р-пииена на
реакцию примерно в два раза меньше соответствующей константы скорости
расходования а-пниена. По данным автора, наоборот, константа скорости
расходования Р-пинена в равных условиях на один порядок выше константы
скорости расходования а-пииена. Это подтверждают ие только данные экс-
периментального изучения скорости изомеризации а- и Р-пиненов, но и равно-
весие между а- и Р-пииенами. В равновесной смеси Р-пинен Z^a-пинеи содер-
жится лишь 3—6% Р-пииеиа [197]. Это значит, что константа скорости пре-
вращения Р-пииеиа в a-пинен по крайней мере в 20 раз больше константы
скорости обратной реакции. Поэтому свободная энергия у Р-пинена выше
свободной энергии a-пинеиа на величину
5
ДО = -RT In(5)
УО
что составляет для температуры Т=408°К и при газовой постоянной /? =
= 8,315 кДж/(град-моль) 10 кДж/моль (2,4 ккал/моль) и совпадает с экспе-
риментальными данными [246] (гл. XIII, табл. 53). Это неизбежно ведет
к большей реакционной способности р-пинена.
3. Изомеризация пииена в аппаратуре
периодического действия на заводских установках
В заводских условиях изомеризацию пинена осуществляют
обычно в вертикальных стальных аппаратах — изомеризаторах,
снабженных эффективными пропеллерными или турбинными ме-
шалками.
Подогрев аппарата осуществляется змеевиком, в который
подается глухой пар. В начальной стадии подогрев доводят до
температуры начала реакции. С этого момента выделяется
столько тепла, что надобность в подогреве отпадает и возникает
необходимость отвода излишка тепла из аппарата. Для отвода
избыточного тепла аппарат снабжен вторым змеевиком, через
который пропускают воду.
При расчете количества отводимого тепла могут быть исполь-
зованы данные, приведенные в гл. XIII, табл. 53. Если реакцию
проводят при температуре кипения, аппарат для изомеризации
соединяют с обратным холодильником. Если реакцию проводят
при более низкой температуре, целесообразно применять предо-
хранительный клапан, исключающий засасывание воздуха из
атмосферы. Изомеризацию пинена обычно проводят при атмо-
сферном давлении, но к аппарату следует предъявлять те же
требования, что и к автоклаву на 8—10 атм давления. На рис. &
приведена схема аппарата для проведения процесса изомериза-
ции пинена.
70
Продолжительность процесса изомеризации в заводских ус-
ловиях может быть выбрана самая разнообразная (от одного
часа до нескольких суток).
При осуществлении быстрой реакции значительно уменьша-
ется размер аппаратуры, но усложняется ее обслуживание. На-
оборот, при проведении медленной реакции емкость аппаратуры
увеличивается, а ее обслуживание упрощается. Работа аппарат-
чика в этом случае сводится лишь к загрузке и разгрузке аппа-
Рис. 9. Схема установки для каталитической изомеризации пинена в жид-
кой фазе:
/ — аппарат для каталитической изомеризации пинена; 2 — холодильник; 3 — гидрав-
лический затвор; 4 — насос; 5 — напорный бак с изомеризатом; 6 — фильтр
рата один раз за 5—6 суток и к регулированию в нем темпера-
туры, а работа аналитика — к производству одного-двух анали-
зов в сутки. В этих условиях в отношении трудозатрат процесс
мало отличается от непрерывного.
Катализатор к концу реакции в значительной степени теряет
свою активность из-за отравления, но после внесения в свежий
пинен и десорбции находящихся на его поверхности ядов он
регенерируется. Активность оборотного катализатора ниже све-
жего, но получаемые с ним выходы камфена несколько выше,
чем со свежим катализатором.
Фильтрация изомеризата не представляет затруднений и может быть
легко осуществлена под давлением на друк-фильтре через плотный текстиль-
ный материал. Однако при просасывании воздуха через катализатор послед-
ний быстро и необратимо теряет свою каталитическую активность и выходит
71
из строя. Поэтому для фильтрования необходимо применять вертикальные
листовые фильтры, работающие под давлением [НО], в которых катализатор
защищен от воздуха слоем изомеризата. Эти фильтры состоят из заполнен-
ного изомеризатом закрытого резервуара, в который вставлена батарея эле-
ментов. Каждый элемент состоит из полой рамы с перфорированной стальной
Рис. 10. Схема фильтра для изомеризатов, полученных из пинена:
Г — корпус; 2 — крышка; 3 — рама фильтрующего элемента; 4 — смотровое окно;
5 — лопастной шнек; 6 — батарея фильтровальных элементов
сеткой или двумя плоскими сетками, отстоящими друг от друга на некотором
расстоянии. Рамы с сетками обтянуты фильтрующей тканью (рис. 10).
Изомеризат из аппарата 1 на рис. 9 для изомеризации пинена подается на-
сосом 4 в межрамное пространство фильтра 6, далее через салфетки проникает
во внутрирамное пространство. При этом катализатор остается на поверхности
салфеток, а освобожденный от катализатора изомеризат из внутрирамного
пространства выводится в сборную цистерну. Фильтрование производится при
72
температуре около 90°, так как в этих условиях производительность фильтра
значительно выше, чем при обычной температуре, и достигает 0,4 т изомери-
зата в час с 1 м2 фильтрующей поверхности.
Коммуникация предусматривает возможность производить многократную
циркуляцию изомеризата от аппарата для изомеризации пииена через фильтр
обратно в аппарат для изомеризации или из межрамиого во внутрирамное
пространство фильтра и обратно в течение начального периода фильтрова-
ния, пока крупные поры в салфетках не заполнятся частицами катализатора
и не будет получаться прозрачный фильтрат. При разгрузке фильтра отклю-
чают насос и соединяют напорный бачок 5 с внутрирамным пространством
фильтра. В результате давления, возникающего во внутрирамном простран-
стве, салфетки несколько раздуваются, катализатор отваливается с поверх-
ности рам и размельчается шнеком, после чего суспензия катализатора через
нижний штуцер фильтра под давлением изомеризата, находящегося в напор-
ном бачке, и с помощью вакуума, создаваемого в аппарате для изомеризации
пинеиа, возвращается в аппарат. При таком способе фильтрования катализа-
тор долго сохраняет свою активность и может многократно использоваться
часто с добавкой свежего. Расход катализатора составляет 0,5—1 кг/т пере-
работанного пинена. В случае, если катализатор потерял свою активность,
суспензия подается на друк-фильтр, где катализатор отделяют от изомери-
зата и выводят из системы.
Фильтр б всегда доверху залит изомеризатом. Необходимость его переза-
рядки возникает редко.
При эксплуатации установок для каталитической изомериза-
ции пинена надо иметь в виду, что изомеризация 1 кг пинена
в камфен связана с выделением около 335 кДж (80 ккал) тепла,
а в лимонен около 290 кДж (70 ккал) (гл. ХШ, табл. 62). Если
это тепло недостаточно быстро отводится с поверхности частиц
катализатора, то превращение может ускориться из-за местного
перегрева и скорость диффузии пинена к поверхности катализа-
тора окажется меньше скорости реакции. В результате задер-
жавшиеся на поверхности частицы камфена и лимонена подвер-
гаются вторичным превращениям, в том числе полимеризации.
А эта последняя реакция протекает с выделением 1000—
1200 кДж (230—280 ккал) на 1 кг прореагировавшего вещества,
что приводит к дальнейшему ускорению реакции, которая может
принять исключительно бурный характер.
Эти явления могут проявляться при недостаточном массооб-
мене, плохом теплоотводе, применении катализаторов высокой
активности и в больших количествах.
Примером такого процесса может служить явление, наблюдаемое при
внесении в 10 см3 свежеперегнаниого пинена 5 г алюмосиликатного катали-
затора. При наличии у катализатора высокой активности в течение несколь-
ких минут смесь нагревается от комнатной температуры до температуры
кипения. Значительная часть терпенов при этом превращается в полимеры
1174].
4. Оценка существующего процесса
и перспективы его развития
Преимущества изомеризационного метода получения кам-
фена перед прежним «классическим» через борнилхлорид на-
столько очевидны, что писать о них нет надобности.
73
Современная химическая технология стремится использовать,
высокопроизводительные непрерывные процессы, поддающиеся
автоматизации и, следовательно, требующие минимальной за-
траты рабочей силы. Для изомеризации пинена используется
периодический процесс. Однако этот процесс построен так, что-
он медленно протекает во времени (оборот изомеризаторов со-
ставляет несколько суток), требует минимального наблюдения
и потому не связан с большими трудозатратами, характерными
для периодических процессов, особенно процессов с короткими
оборотными циклами, т. е. быстро протекающими во времени.
Медленное течение реакции не влечет за собой и дополнитель-
ных затрат пара, так как процесс протекает с равномерным
выделением тепла. Главное отличие от современных процессов за-
ключается в низкой производительности аппаратуры при перио-
дическом процессе изомеризации пинена. Но этот фактор не мо-
жет считаться решающим, так как 80% себестоимости камфена
определяется стоимостью сырья. Само по себе производство кам-
фары невелико и общая емкость изомеризаторов даже в 100—
120 м3 у крупного камфарного завода очень мала по сравнению
с емкостями аппаратуры, используемой в крупных химических и
нефтехимических производствах.
Значительно более серьезное замечание в адрес изомериза-
ционного метода может быть сделано в отношении выхода кам-
фена, не превышающего 80%, тогда как последующие стадии
его переработки протекают почти количественно [315].
Поэтому наибольшую выгоду для производства могло бы
дать повышение выхода камфена против существующего, и вслед
за этим его перевод на непрерывный процесс, который рассма-
тривался бы как прогрессивный лишь при условии (как мини-
мум) получения того же выхода камфена, что и при периодиче-
ском процессе, полного отсутствия пинена в изомеризатах и ра-
зумного расхода катализатора.
5. Исследования по разработке непрерывных
методов каталитической изомеризации пинена
По непрерывной изомеризации пинена опубликовано лишь
небольшое число работ. И хотя работы эти далеко не закон-
чены и носят характер предварительных сообщений, на них сле-
дует остановиться ввиду важности вопроса. На первый взгляд
осуществить непрерывную изомеризацию пинена очень просто,
достаточно пропустить жидкий пинен или его пары через слой
катализатора. Однако это не так. Протекание реакции в зоне
с высокой концентрацией катализатора, при ограниченном мас-
сообмене, приводит к значительному снижению выхода целевого
продукта (гл. V.1). Это обстоятельство в первую очередь должно
приниматься во внимание при разработке любого непрерывного
метода каталитической изомеризации пинена. При этом воз-
74
можны два решения: осуществление процессов при малой тур-
булентности с применением катализаторов малой активности и
осуществление процессов с катализаторами нормальной актив-
ности и при очень высокой турбулентности.
По первому пути велись исследования в Германии до второй мировой
войны. Опыты сводились к пропусканию жидкого пинена через две последо-
вательно соединенные U-образные трубки, заполненные гранулированным ка-
тализатором. Последний приготовляли путем затирки влажного катализатора
с гипсом и продавливания тестообразной массы через пластинку с отверстиями.
После затвердевания и сушки получали цилиндрические кусочки катализатора
длиной 10—15 мм и около 2 мм толщины.
В первой трубке находился катализатор, содержащий 20% титановой кис-
лоты и 80% гипса, во второй — 40 и 60% соответственно. Объем, занятый
катализатором, составлял 160 см3 (~70 г). Через описанный проточный аппа-
рат при 135—140°С пропускали пинен со скоростью 13,3 см3/ч в течение полу-
года. В результате было получено 45 л изомеризата с содержанием около
80% камфена. Опыт был прекращен из-за начала разрушения зерен катали-
затора.
Благодаря применению малоактивного катализатора и его разбавлению
гипсом снижения выхода камфеиа не последовало, несмотря на неблагоприят-
ные условия массообмена и высокую концентрацию катализатора в реакцион-
ной зоне. Съем продукта реакции с единицы полезной емкости реактора со-
ставил ~70 см3/(л. ч) против 10 см3/(л. ч) на аппаратах периодического
действия на том же предприятии. Если учесть, что в заводском аппарате
катализатор, во избежание разрушения от собственной тяжести, пришлось бы
разместить на ряде фильтров или иа тарелках колоииы, производительность
реактора сократилась бы по крайней мере в два раза. Исследования, связан-
ные с разработкой описанного процесса, не были доведены до конца.
По второму пути разработки непрерывного процесса пошла
группа советских исследователей [3—5, 40, 41, 334].
Изомеризацию в этом случае осуществляют, пропуская
с большой скоростью в условиях так называемого пенного ре-
жима пары пинена через суспензию катализатора в камфене
находящуюся в противотоке. Для снижения температуры кипе-
ния пинена и увеличения турбулентности разбавляют его пары
азотом. Создание высокой концентрации катализатора в зоне
реакции позволяет осуществлять процесс в реакторах малой ем-
кости, а использование пинена в паровой фазе позволяет значи-
тельно ускорить его диффузию к поверхности катализатора и
тем самым устранить побочные реакции из-за задержки прореа-
гировавших камфена и лимонена на поверхности, в условиях
большой концентрации катализатора. Наличие жидкой фазы
позволяет непрерывно удалять с поверхности катализатора от-
равляющие его вещества.
Схема установки для проведения реакции приведена на
рис. 11. В качестве реактора использована колонна с 20 сетча-
тыми тарелками. Верхние тарелки колонны орошаются камфе-
ном из дефлегматора и суспензией катализатора в камфене.
Парогазовая смесь пинена и азота подается на одну из средних
тарелок. Нижняя часть колонны работает на исчерпывание. Ско-
рость паров в свободном сечении колонны составляет 0,45—
>0,7 м/с, а при выходе из сеток 4—6 м/с, что обеспечивает пенный
75
режим. В описанном аппарате наряду с изомеризацией происхо-
дит и ректификация изомеризата. Геометрический объем уста-
новки составляет 20% периодически действующей установки
равной производительности.
Рис. 11. Технологическая схема опытно-промышлеиной уста-
новки по изомеризации пииена в камфеи в трехфазовой си-
стеме [4]:
1 — нзомеризатор-колонна; 2 — испарители пииена; 3 — напорный бачок
для суспензии; 4 — напорный бачок для пинена; 5 — дефлегматор-хо-
лодильник; 6 — газгольдер «рессивер»; 7 — «компенсатор»; 8 — прием-
ник отработанной суспензии; 9 — «компрессор»; 10— подогреватели
азота; 11— ротаметр для пинеиа; 12— ротаметр для азота; 13 — доза-
тор суспензии; 14 — подогреватель пинена
Скорость образования продуктов изомеризации с единицы
массы катализатора в трехфазовой системе несколько выше, чем
в жидкой фазе (табл. 20). Однако значительное сокращение
76
емкости аппаратуры по сравнению с емкостью аппаратуры пе-
риодического процесса вызвано тем, что при непрерывном спо-
собе в реакционной зоне отсутствуют избыток сырья и не скап-
ливаются продукты реакции.
Таблица 20
Сравнение скоростей образования продуктов изомеризации пинена
периодическим методом в жидкой фазе и непрерывным
в трехфазовой системе
Объемная емкость ката- лизатора, мг•=кв/г Температура, °C Условия реакции Изомеризуется пинена, г/кг катализатора, с
0,2451 0,46 2 130 145 В жидкой фазе начало реакции В трехфазовой системе начало реак- ции 6,7 16,5
1 По CH3COONa (28].
2 По NaOH [4].
Авторы утверждают, что в результате описанного процесса
выход камфена и трициклена достигает 90—95%, т. е. в среднем
на 15% выше выхода по существующему процессу, несмотря на
то, что процесс в трехфазовой системе протекает, казалось бы,
в неблагоприятной диффузионной области. Авторы указывают,
что осуществление разработанного ими процесса в промышлен-
ности встретилось с трудностями, которые пока не преодолены
[4]. Другой аналогичный процесс, но с применением твердого
таблетированного титанового катализатора, разрабатывается
теми же авторами [5]. Работа не вышла за пределы лаборатор-
ных испытаний. По предварительным данным, удельная произ-
водительность установки при этом не меняется, но выход кам-
фена мало отличается от выхода при периодическом процессе.
Расход катализатора значительно более высокий, чем при ис-
пользовании его в виде суспензии.
ГЛАВА VI. ПОЛУЧЕНИЕ БОРНЕОЛА
И ИЗОБОРНЕОЛА
Важный полупродукт производства синтетической камфары —
изоборнеол получают из камфена, трициклена или из их смесей.
Этот синтез в технике осуществляют в две стадии: сначала кам-
фен действием органической кислоты в присутствии катализа-
тора (например, серной кислоты) превращают в эфир изобор-
неола [194, 195], а затем полученный эфир омыляют едким нат-
ром. Попытки превратить камфен в изоборнеол, минуя стадию
эфира, промышленного развития не получили.
77
1. Получение изоборнильных эфиров из камфена
и трициклена
В качестве исходного продукта для промышленного получе-
ния изоборнильных эфиров используют технический камфен. Его
выделяют ректификацией из продуктов каталитической изомери-
зации пинена. В ограниченном количестве его можно выделять
из продуктов каталитической изомеризации пинен-камфеновой
фракции пихтового масла (гл. X).
Состав и свойства технического камфена могут колебаться
в зависимости от способа получения. В техническом камфене,
полученном изомеризацией пинена титановой кислотой, содер-
жится: камфена -~75°/о, трициклена ~20, фенхенов 4—4,5%.
Фенхены являются нежелательной примесью, так как при после-
дующей переработке они превращаются в жидкий изофенхон,
примешивающийся к камфаре. Большая часть фенхенов (~70%)
состоит из а- и 0-фенхенов, имеющих температуры кипения
столь близкие к температуре кипения камфена (гл. XIII), что их
отделение от камфена ректификацией технически нецелесооб-
разно. Поэтому предпочитают получать камфен, используя ка-
тализаторы, с которыми образование фенхенов при хорошем вы-
ходе камфена минимально. Присутствие пинена в техническом
камфене недопустимо, так как оно ведет к побочным реакциям.
Не желательна и примесь моноциклических терпенов, так как
она усложняет последующую переработку эфира. Присутствие
трициклена не противопоказано, он, как и камфен, при действии
органических кислот в присутствии катализаторов превраща-
ется в эфир изоборнеола.
Если камфен не содержит примеси моноциклических терпе-
нов и содержание фенхенов находится на уровне 4—4,5%, его
температура кристаллизации должна быть около +45°С [263],
что может служить мерилом качества камфена. Иногда допу-
скается небольшая примесь моноциклических терпенов. В этом
случае температура кристаллизации камфена более низкая. Со-
гласно ГОСТ СССР 15039—69 на камфен технический, содержа-
ние в нем ЕЭВ должно быть не ниже 95%, а температура на-
чала кристаллизации не менее 37°С.
Изоборнильные эфиры образуются из камфена при действии
многих органических кислот, но на практике применяются лишь
муравьиная и уксусная кислоты, как наиболее дешевые и до-
ступные. К тому же они имеют минимальную молекулярную
массу, вследствие чего их расход на реакцию более низок, чем
расход любых других органических кислот. Взаимодействие осу-
ществляется при температуре от 0 до +70°С.
В качестве катализаторов применяются доноры протонов.
До последнего времени применялась только серная кислота, хотя
и указывалось на возможность применения ряда других реаген-
тов: хлористого цинка [270], фтористого бора [228], фосфор-
78
них, щавелевой, борной и других слабых кислот, работа с ко-
торыми будто бы не сопровождается побочными реакциями
[307].
Преимущества этих катализаторов по сравнению с серной
кислотой весьма сомнительны, так как серная кислота дешева
и при ее применении в качестве катализатора в определенных
условиях побочные продукты вовсе не образуются.
Муравьиная кислота реагирует с камфеном и в отсутствие ка-
тализаторов [201, 306], но реакция протекает медленно.
Позднее было описано применение в качестве катализаторов
нерастворимых в реагирующих веществах ионообменников, в том
числе катионообменных смол, содержащих сульфогруппы [136,
142, 145, 269]. Такого рода катализаторы перспективны для при-
менения в непрерывном процессе, а их применение в периодиче-
ском процессе позволяет избежать образования отходов — от-
работанных смесей кислот.
Взаимодействие камфена с кислотами часто называют этерификацией,
но это неправильно, так как этерификацией называется реакция, имеющая
место при взаимодействии кислоты и спирта, ведущая к образованию эфира
и сопровождающаяся выделением воды. При взаимодействии же камфена
с органической кислотой происходит присоединение кислоты по двойной связи
углеводорода и одновременная Вагнеровская перегруппировка. Поэтому реак-
цию правильнее назвать превращением камфена в изоборнильные эфиры или
по использованной для реакции органической кислоте — формилированием,
ацетилированием и т. д.
Механизм реакции сводится к следующему: камфен (I), при-
соединяя протон, образует изокамфилкарбоний-ион (II), кото-
рый, претерпевая перегруппировку Вагнера и взаимодействуя
с органической кислотой, образует эфир изоборнеола (III)
(свыбросом протона).
Одновременно с этой основной реакцией протекают побоч-
ные:
1. Изокамфилкарбоний-ион (II), взаимодействуя с органиче-
ской кислотой, может образовать эфир камфенгидрата (IV).
Соответствующий этому эфиру спирт камфенгидрат (V) не мо-
жет быть превращен в камфару методом каталитического дегид-
рирования, и поэтому образование эфира камфенгидрата неже-
лательно. При действии на камфен уксусной и муравьиной ки-
слот эфир камфенгидрата практически не образуется.
2. Изокамфилкарбоний-ион (II) из-за гидридного переноса
может превратиться в ион (VI), соответствующий экзо-изо-
камфену (VII). Реагируя с органической кислотой, ион (VI)
превращается в уксусный эфир 5-зкзо-изокамфан-зкзо-ола-З
(псевдоборнеол) (VIII), который в процессе дальнейшей пере-
работки технического изоборнеола превращается в кетон Ь-экзо-
изокамфанон-3 (псевдокамфара) (IX), загрязняющий камфару
(X) [243]:
79
3. Под действием серной кислоты и других протононосителей,
применяемых в качестве катализаторов, происходит полимериза-
ция терпенов. Полимеризация хорошо очищенного технического
камфена при умеренных температурах, в присутствии неболь-
шого количества серной кислоты, незначительна и не превы-
шает 1%.
4. Превращение камфена в уксусный и муравьиный эфиры
изоборнеола сопровождается полной рацемизацией (из-за про-
текающей в кислом растворе перегруппировки Наметкина).
Примеси, содержащиеся в техническом камфене, при взаимо-
действии с кислотами тоже подвергаются различным превра-
щениям. Трициклен (XI) превращается в эфир изоборнеола,
а (XII)-, р (ХШ)-, у (XIV)-, б (XV)- и цикло-(XVI) фенхены об-
разуют смесь эфиров фенхилового (XVII) и изофенхилового
(XVIII) спиртов, с преобладанием последнего. Промежуточ-
ными продуктами являются ионы (XIX), (XX), (XXI) и (XXII)
(с. 81).
Для разделения эфиров изоборнеола и изофенхилового
спирта технических методов не имеется. Поэтому фенхены, со-
держащиеся в техническом камфене, переходят в виде изофен-
хилового спирта в изоборнеол и, далее, в виде изофенхона в кам-
фару.
Если в техническом камфене в виде примеси содержались
моноциклические терпены, то последние под действием серной
кислоты изомеризуются и полимеризуются. Поэтому при прев-
вращении в изоборнильные эфиры камфена, содержащего при-
месь моноциклических терпенов, количество образующихся по-
лимеров значительно увеличивается.
80
XX XVII XIV XV
Если в техническом камфене присутствовал пинен, то пос-
ледний в условиях реакции на 10—20% превращается в смесь
эфиров терпеновых спиртов [175], в моноциклические терпены н
полимеры. Образовавшиеся из пинена эфиры терпеновых спир-
тов трудно отделить от изоборнильных эфиров, поэтому при-
сутствие пинена недопустимо.
Если взаимодействие камфена с уксусной и муравьиной кислотами (в при-
сутствии серной кислоты) осуществляется при температуре около 0е или ниже
0сС, то в результате реакции получаются изоборнильные эфиры, которые при
дальнейшей переработке превращаются в камфару с высокой температурой
плавления. Если же взаимодействие осуществляется при более высокой тем-
пературе, то получают изоборнильные эфиры, которые при дальнейшей пере-
работке превращаются в камфару, загрязненную примесями, снижающими
температуру ее плавления [308].
В процессе взаимодействия камфена с муравьиной кислотой при темпе-
ратуре выше 30сС наряду с эфиром изоборнеола образуются неидентифици-
рованные эфиры другого вторичного терпенового спирта (по-видимому псевдо-
борнеола), которые в процессе дальнейшей переработки превращаются в кетон,
остающийся в виде примеси в камфаре и значительно снижающий температуру
ее кристаллизации. При температуре ниже 30°С указанные побочные продукты
не образуются. Данные, иллюстрирующие влияние температуры формплирова-
ния камфена на температуру кристаллизации полученной из него камфары,
показаны графически на рис. 12.
Температурные границы образования побочных продуктов определяются
применяемым катализатором. Например, при превращении камфена в уксусный
эфир нзоборнеола в присутствии катионообменных смол побочные продукты
не образуются даже при 70°.
Взаимодействие камфена с уксусной и муравьиной кисло-
тами в присутствии безводной серной кислоты может происхо-
дить как в гомогенной, так и в гетерогенной средах. Камфен и
6 Заказ № 43
81
97—98%-ная уксусная кислота взаиморастворимы, поэтому аце-
тилирование камфена высококонцентрированной уксусной кисло-
той протекает в гомогенной среде, если только уксусная кислота
взята в большом избытке. Водная уксусная кислота (напри-
мер, 80- или 90%-ная) не растворяет камфен, поэтому ацетили-
рование камфена водной уксусной кислотой протекает в гетеро-
генной среде. В этом процессе участвуют две фазы: раствор
серной кислоты в водной уксусной кислоте и раствор уксусной
кислоты в камфене. Если высококонцентрированная уксусная
кислота взята в небольшом избытке, то к концу реакции, по мере
Рис. 12. Влияние температуры формилирова-
ния камфена на температуру кристаллизации
полученной из него камфары:
1 — камфен, полученный из очищенного перекристал-
лизацией нзоборнеола; 2 — технический камфен за-
водского производства
использования уксусной кислоты, ее концентрация падает на-
столько, что происходит выделение водной уксусной кислоты из
раствора в виде отдельного слоя и процесс из гомогенного пе-
реходит в гетерогенный.
Формилирование камфена отличается от ацетилирования тем,
что камфен почти нерастворим в муравьиной кислоте. Поэтому
формилирование камфена даже значительным избытком высоко-
концентрированной муравьиной кислоты начинается в гетероген-
ной среде. Однако если взять достаточный избыток муравьиной
кислоты, то по мере образования изоборнилформиата взаимная
растворимость компонентов увеличивается и через некоторое
время реакция превращается в гомогенную. Если муравьиная
кислота взята в небольшом избытке, то к концу реакции, по
мере падения концентрации муравьиной кислоты, процесс мо-
жет вновь перейти в гетерогенный. Растворимость моноцикли-
ческих терпенов в смеси кислот хуже растворимости камфена.
Поэтому гомогенность реакционной смеси легче нарушается,
когда используется камфен, содержащий примесь моноцикличе-
82
ских терпенов, а если эта примесь значительна, то гомогенность
среды вообще отсутствует даже при большом избытке муравьи-
ной кислоты.
Скорость превращения камфена в изоборнильный эфир при
взаимодействии его с высококонцентрированной муравьиной
кислотой при 15—20°С в 10—20 раз превышает скорость превра-
щения камфена в уксусный эфир в присутствии одного и того
же количества катализатора [176].
С переходом из гомогенной среды в гетерогенную скорость
реакции резко замедляется. Это вполне понятно, так как в ге-
терогенной среде реакция идет только на поверхности раздела
фаз, тогда как в гомогенной среде — во всей толще раствора.
1 2 3 4 5 6 7 8 9 1011 1213141510 17181920
Время, мин
Рис. 13. Сравнение скоростей ацетилирования
камфена и трициклена смесью уксусной и сер-
ной кислот:
1 — камфен; 2 — трициклен
Трициклен, при прочих равных условиях, вступает в реакцию значительно
медленнее камфена. Наблюдения такого рода были сделаны Меервейном
и ван Емстером [276] при изучении взаимодействия камфена и трициклена
с монохлоруксусной кислотой в отсутствие катализатора. То же самое наблю-
дал и автор этой книги при изучении взаимодействия камфена и трициклена
с уксусной кислотой в присутствии серной кислоты (рис. 13) [150]. Поэтому
состав смеси терпенов, не вступившей в реакцию, по ходу процесса непре-
рывно обогащается трицикленом и его превращение в эфир изоборнеола
в значительной степени происходит в самый неблагоприятный период реакции,
когда концентрация кислоты понизилась, а процесс перешел в гетерогенный.
Это обстоятельство может служить основанием для раздельной переработки
камфена и трициклена.
Превращение камфена в изоборнильные эфиры — обратимая
реакция [201]. Тищенко и Рудаков [176] определили константу
равновесие формилирования и ацетилирования камфена при раз-
ной температуре. При расчете константы равновесия исходили
из предположения, что в равновесии участвуют камфен, органи-
ческая кислота и эфир изоборнеола. Наличие в равновесии три-
циклена во внимание не принималось. Результаты работы
сведены в табл. 21. Из констант равновесия можно вычислить
количество камфена, вступившего в реакцию с органической
кислотой при том или ином режиме процесса.
6* 83
Таблица 21
Константы равновесия ацетилирования и формилирования камфеиа 97 %-ной
уксусной и 95 %-ной муравьиной кислотами
Температура, СС Загружено Продолжи- тельность реакции, ч Количество камфена, вступившего в реакцию, % Констаита равновесия К*
органической кислоты моль, на 1 моль камфена H,SO4. 1,84 на 1 г камфеиа, г
50-60 1,47 СНзСООН 0,02 84 8,3
50-60 1,98 То же 0,02 4 90 8,3
50-60 5,65 ,, 0,02 2 97 16,9
15-20 0,99 „ 0,10 4 90,5 112
15-20 1.40 „ 0,10 4 98 116
50-60 1,66 НСООН 0,02 — 92,5 19,2
50-60 2,10 То же 0,02 — 95 16,7
15-20 1,28 „ 0,10 0,25 91,5 29,4
15-20 1,48 „ 0,10 0,25 94 29,4
* Вычислена из уравнения
________12_____
(1— т) (п—т)
где m — доля камфена (моль), вступившая в реакцию с органической кисло-
той;
п — количество органической кислоты (моль), взятой в реакцию на 1 моль
камфена.
Из данных таблицы видно, что с понижением температуры
реакции содержание эфира в равновесной смеси растет.
С увеличением содержания воды в реакционной смеси пони-
жается выход изоборнильных эфиров. Например, при формили-
ровании 1 моль камфена стандартной 85%-ной муравьиной кис-
лотой, содержащей 1,25 моль НСООН, в присутствии серной
кислоты удается достигнуть лишь 86—87%-ного превращения
камфена в эфир.
Превращение камфена в уксусный эфир изоборнеола сопро-
вождается выделением 30 кДж/моль (7,2 ккал/моль) тепла,
а превращение трициклена 37,2 кДж/моль (8,9 ккал/моль) [150],
2. Основы технологии формилирования
и ацетилирования камфена
в аппаратуре периодического действия
Периодический процесс формилирования и ацетилирования
камфена осуществляется обычно в освинцованных или футеро-
ванных керамиковыми плитками цилиндрических стальных ап-
паратах при интенсивном перемешивании. Принимая во внима-
ние выделение свыше 30 кДж (7 ккал) тепла на каждую г-мо-
лекулу прореагировавшего камфена и необходимость поддержа-
84
ния температуры реагирующих веществ около 20°С (но не выше
30°), в аппараты ставят приспособления для поддержания за-
данной температуры.
Особое внимание необходимо уделять конструкции мешалок,
так как реакция протекает обычно в гетерогенной среде при
наличии двух жидких фаз, из которых в одной находится 4—
5%-ный раствор органической кислоты в смеси камфена и изо-
борнильного эфира, а в другой — раствор катализатора в орга-
нической кислоте. Второй раствор имеет значительно большую
плотность, чем первый, и в случае, если реакция ведется с не-
большим избытком высококонцентрированной органической кис-
лоты, его объем к концу реакции резко сокращается и состав-
ляет лишь 5—6% общей загрузки аппарата. Это еще ухудшает
условия для хорошего диспергирования катализатора.
При загрузке аппарата можно добавлять катализатор к заранее приго-
товленному раствору камфена в органической кислоте или, наоборот, доба-
влять камфен к заранее приготовленной смеси кислот. Технический камфен
хранят и транспортируют в расплавленном состоянии при температуре около
50°С. Прибавление расплавленного камфена к заранее приготовленной смеси
кислот влечет за собой разогрев реакционной смеси в начале процесса до
температуры, превышающей 30°, что приводит к ухудшению качества про-
дукта. Поэтому прибавление серной кислоты к заранее приготовленному рас-
твору камфена в органической кислоте предпочтительнее, особенно при при-
менении высококонцентрированных органических кислот. Как уже отмечалось,
камфен растворяется в ледяной уксусной кислоте. В муравьиной кислоте кам-
фен не растворяется, но при 40—70°С реагирует в отсутствии катализатора
даже с 85%-иой муравьиной кислотой. Через несколько часов образуется
около 50—60% эфира. Дальше продолжать реакцию в отсутствие катализа-
тора нецелесообразно, так как она протекает слишком медленно. Поэтому
смесь охлаждают и вводят катализатор. Твердый камфен при охлаждении
смеси не выделяется из-за присутствия в смеси муравьиного эфира изобор-
неола.
Взаимодействие камфена с муравьиной кислотой в отсутствие катализа-
тора не сопровождается образованием вредных побочных продуктов даже при
70°С. Растворение камфена в уксусной кислоте и взаимодействие с муравьи-
ной кислотой в отсутствие катализатора до получения некристаллизующейся
при охлаждении смеси целесообразно осуществлять в отдельном аппарате.
Если в реакцию берут камфен, содержащий около 10% моноциклических
терпенов, то такой раствор может транспортироваться в неподогретом виде
и его можно загружать в заранее приготовленную смесь органической и сер-
ной кислот.
Взаимодействие камфена с органическими кислотами — об-
ратимая реакция. Из табл. 21 и уравнения (5) видно, что при
взаимодействии камфена с высококонцентрированной муравьи-
ной или уксусной кислотой в эквимолекулярных количествах
камфен вступает в реакцию лишь на 80—90% и соответственно
в продукте остается 10—20% непрореагировавшего камфена и
10—20% непрореагировавшей органической кислоты. При взаи-
модействии камфена с полутора-, двукратным избытком му-
равьиной или уксусной кислоты камфен практически полностью
превращается в эфир, но при этом только 50—65% органической
кислоты используется в реакции.
85
Если процесс осуществлять по первому варианту, то следует
выделять из продуктов реакции непрореагировавшие камфен и
органическую кислоту и возвращать их снова в производство.
Второй вариант вызывает необходимость регенерировать только
органическую кислоту, но в значительно большем количестве,
чем при работе по первому варианту.
Продукты, получающиеся при взаимодействии камфена с ук-
сусной или муравьиной кислотой, содержат растворенный или
взмученный катализатор (серную кислоту), в присутствии кото-
рого нельзя отогнать непрореагировавшие органическую кислоту
и камфен, так как по условиям равновесия, определяемым урав-
нением (6), по мере удаления органической кислоты изоборниль-
ный эфир распадается на камфен и органическую кислоту. Поэ-
тому непрореагировавшую кислоту вымывают из продуктов фор-
милирования или ацетилирования камфена водой, растворами
едкого натра или соды в специально предназначенных для этой
цели конических отстойниках с мешалками. В результате полу-
чают разбавленные органические кислоты или водные растворы
солей этих кислот, которые перерабатывают обычными спосо-
бами в высококонцентрированные кислоты. Отмытый, нейтрали-
зованный и высушенный эфир подвергают ректификации для
выделения непрореагировавшего камфена. Как ректификация
эфира, так и регенерация органических кислот из их водных
растворов представляют обременительные дополнительные опе-
рации. На рис. 14 приведена технологическая схема установки
для получения эфиров изоборнеола из камфена.
Существуют другие варианты процесса, ставящие цель обойти описанные
затруднения.
Бертрам и Вальбаум осуществляли ацетилирование камфена большим
избытком уксусной кислоты в присутствии небольшого количества 50%-ной
серной кислоты и по окончании реакции осаждали серную кислоту в виде
нерастворимой бариевой соли, после чего отгоняли не вошедшую в реакцию
уксусную кислоту [194, 195].
Ацетилирование камфена на заводе И. Г. Фарбениндустри в Герстгофеие
(Бавария) осуществляли примерно по тому же принципу. Реакцию проводили
в присутствии большого количества уксусной кислоты (~5 мол. на 1 мол.
камфена) и небольшого количества (около 1,5—2% по отношению к кам-
фену) крепкой серной кислоты при 80°С. По окончании реакции (в гомоген-
ной среде) к реакционной смеси добавляли уксуснокислый натрий, который
вступал в обменное разложение с серной кислотой, после чего отгоняли
уксусную кислоту и не вступивший в реакцию камфен [263].
По-видимому, от описанного способа можно ожидать хороших результа-
тов, лишь используя безводную уксусную кислоту, так как при применении
даже высококонцентрированных (96—98%-ных) органических кислот повы-
шается содержание воды в непрореагировавшей кислоте. В этом случае необ-
ходимо подкреплять оборотную кислоту до исходной концентрации, что все же
проще, чем регенерировать ее из разбавленных водных растворов или раство-
ров ее солей.
Иногда вместо высококонцентрированных 97—98% -ных уксусной и мура-
вьиной кислот применяют более дешевые водные 80—85%-ные кислоты. Усло-
вия равновесия в этом случае менее благоприятны, поэтому обычно прихо-
дится концентрировать эфиры после реакции. Кислотная смесь, содержащая
значительное количество воды, легко отделяется от эфира и может быть
86
возвращена в производство, но полезное использование органических кислот
все же остается на уровне 75—80%. Это происходит потому, что часть орга-
нической кислоты экстрагируется эфиром, откуда она может быть извлечена
только водной или щелочной промывкой в виде слабого водного раствора
или водного раствора солей.
Рис. 14. Схема установки
для получения эфиров,
изоборнеола из камфена:
1 — мерник для серной кис-
лоты; 2 — мерник для орга-
нической кислоты; 3 — аппа-
рат для получения эфиров
изоборнеола; 4 — аппарат
для промывки эфира изо-
борнеола и отделения его от
отработанной смеси кислот;
5 — сборник для отработан-
ной смеси кислот; 6 — на-
сосы; 7 — сборник для эфи-
ров изоборнеола
Способ ацетилирования камфена большим избытком водного раствора
уксусной и серной кислот с подкреплением раствора после реакции до исход-
ной концентрации путем добавки высококонцентрированной органической кис-
лоты описан в работе [333]. В качестве примера приведено взаимодействие
100 массовых частей камфена со смесью 100 массовых частей ледяной уксус-
ной кислоты (2,3 моль уксусной кислоты на 1 моль камфена) и 450 массовых
частей 60—66%-ной серной кислоты при 20—30°С. По окончании реакции
к кислотной смеси добавляется свежая ледяная уксусная кислота. И здесь
вследствие экстракции уксусной кислоты изоборнильным эфиром она исполь-
зуется не полностью. Кроме того, оборотная кислотная смесь постепенно
накапливает смолистые вещества, поэтому после ряда циклов неизбежен ее
вывод из производства. Вместо уксусной кислоты может быть использована
муравьиная кислота.
Взаимодействие камфеиа с водными органическими кислотами (особенно
уксусной кислотой) протекает значительно медленнее, чем с безводными или
87
высококонцентрированными (97—98%-ными) муравьиной и уксусной кисло-
тами, что заставляет увеличивать количество катализатора (серной кислоты).
В связи с этим возникают трудности, связанные с необходимостью использо-
вать отходные водные растворы, содержащие наряду с органической кисло-
той значительное количество серной кислоты.
3. Исследования по разработке непрерывных
методов формилирования
и ацетилирования камфена
Способы формилирования и ацетилирования камфена в ап-
паратуре периодического действия имеют существенные недо-
статки.
К этим недостаткам следует отнести неполное использование
камфена и органических кислот, которое составляет обычно 75—
80%, трудоемкость процесса и, в случае применения водных ор-
ганических кислот, получение в виде отхода значительного ко-
личества отработанных кислотных смесей, состоящих из серной
и органической кислот и воды.
Непрерывные методы ацетилирования и формилирования
камфена могут быть в значительной степени лишены этих недо-
статков. Сделанные в этом направлении разработки основаны
на том, что раствор камфена в органической кислоте непрерывно
пропускают через слой твердого или жидкого катализатора не-
растворимого в реагирующих веществах. Продукты реакции
свободны от катализатора, следовательно можно (без опасения
вызвать разложение эфира) отогнать от них непрореагировав-
шие камфен и органическую кислоту и вернуть в производство.
Рудаковым с соавторами было предложено использовать
в качестве катализаторов твердые ионообменники [142]. Осо-
бенно пригодными оказались сульфированные фенол-формальде-
гидные смолы, выпускавшиеся в СССР под марками МСФ и
Эспатит I [136, 145]. Позднее у сополимера стирола и дивинил-
бензола с сульфогруппами и макроретикулярной (сетчатой)
структурой были обнаружены еще лучшие свойства [269]. Реак-
цию вели в термостатированной колонне. Катализатор предва-
рительно обезвоживали сушкой при 150°С или вымачиванием
в ледяной уксусной или крепкой муравьиной кислоте. Последнее
можно осуществлять в том же реакторе, при непрерывном взму-
чивании катализатора потоком жидкости снизу. Взмучивание не-
обходимо, так как объем катализатора от набухания увеличива-
ется на 30—60%, в результате чего может создаться давление
на стенки реактора и произойти спекание катализатора.
Для предупреждения слеживания и спекания катализатора
во время реакции подачу сырья в колонну целесообразно произ-
водить снизу или периодически взрыхлять катализатор. Без пе-
ремешивания реакция может протекать лишь в случае, если
в колонну подается гомогенный раствор камфена в органической
кислоте. Это, в свою очередь, возможно только при применении
88
96—97%-ных и более крепких уксусной и муравьиной кислот,
взятых в значительном избытке, и при применении технического
камфена, не содержащего примеси моноциклических терпенов.
Раствор камфена в органической кислоте приготовляют в от-
дельном сосуде. Камфен хорошо растворим в уксусной кислоте,
но почти нерастворим в муравьиной кислоте. Поэтому при фор-
милировании камфена его растворяли не в муравьиной кислоте,
а в смеси муравьиной кислоты и изоборнилформиата. Для этого
часть готового продукта, содержащего изоборнилформиат и из-
быточную муравьиную кислоту, возвращали в цикл, используя
в качестве растворителя для реагирующих веществ. Например,
на 100 массовых частей камфена и 67,5 массовой части муравьи-
ной кислоты необходимо взять для получения устойчивого го-
могенного раствора 380—450 массовых частей растворителя. При
использовании 98%-ной муравьиной кислоты направляемый
в реакцию раствор имеет примерно следующий состав: камфена
и трициклена 20%, изоборнилформиата 52—55, муравьиной кис-
лоты 25,5—26,5, воды 0,8% [145].
Катионообменные смолы катализируют реакцию менее интен-
сивно, чем серная кислота, поэтому ацетилирование камфена
ведут при температуре 50—70°С. Так как в этих условиях равно-
весие сдвигается в сторону камфена, применяют большой избы-
ток уксусной кислоты (4—5 молей на I моль камфена).
Данные о сравнительной активности разных катионитов при
ацетилировании камфена приведены в табл. 22 [136]. Исходный
технический камфен подавали в реактор в виде 28,5%-ного рас-
твора в ледяной уксусной кислоте. Для сравнения также приве-
дены данные об активности сульфокатионита с макроретику-
лярной структурой [269].
Таблица 22
Сравнительная активность различных катионитов при ацетилировании
технического камфена
Катиониты Количество кам- фена, превратив- шегося в эфир, % Объемная ско- рость, обеспечи- вающая данную степень превра- щения, ч~! Количество кам- фена (г), превра- щенного в эфир в л реактора, в ч
МСФ * 89 2,3 600
Эспатит 1 * 86 1,9 490
СБС * 86 0,13 33
Сульфоуголь ** 77 0,50 110
С макроретикулярной — — 3000
структурой
* Размер зерен 0,25—2 мм.
** Размер зерен 0,25—1 мм.
89
Из приведенных данных видно, что полезная емкость реакто-
ров, необходимых для превращения в изоборнилацетат 10 т
камфена в сутки, составляет лишь 0,15 м3 в случае применения
в качестве катализатора катионита с макроретикулярной
структурой.
Важным фактором является срок службы катализатора. При
проведении опытных работ по ацетилированию камфена на 35 г
катализатора МСФ было проацетилировано 5,2 кг камфена.
Опыт длился 345 ч, заметного снижения скорости реакции не на-
блюдалось, но к концу опыта началось разрушение зерен ката-
лизатора.
Не вступившие в реакцию уксусная кислота и камфен могут
быть отогнаны от эфира. При давлении 100 мм рт. ст. камфен
образует азеотропную смесь с уксусной кислотой, содержащую
20 % камфена [143]. Это позволяет использовать практически
весь камфен и получать в результате реакции изоборнилацетат
очень высокой степени чистоты.
Формилирование камфена в присутствии катионообменных
смол можно вести уже при 20°С; однако реакция в этих усло-
виях идет слишком медленно. При 50°С реакция идет быстро, но
условия равновесия менее благоприятны (рис. 15). Поэтому це-
лесообразно вести реакцию в двух последовательно соединенных
реакторах, из которых первый имеет температуру 50°, а второй
20°С. Выходящие из первого реактора продукты частично по-
ступают во второй реактор, частично используются для раство-
рения камфена и муравьиной кислоты (рис. 16). При использо-
вании схемы, состоящей из двух реакционных колонн или за-
крытых катионообменных фильтров, работающих под давлением
с подачей реагирующих веществ снизу и заполненных катиони-
том эспатит I, в 1 л емкости реакторов превращают в изоборнил-
формиат около 250—300 г камфена в час. Как видно из табл. 22,
указанная производительность может быть значительно увели-
чена путем применения катионитов с макроретикулярной струк-
турой.
Не вошедшая в реакцию муравьиная кислота может быть
отогнана от эфира.
При отгонке муравьиной кислоты от изоборнилформиата воз-
можна обратная реакция: распадение эфира на камфен и му-
равьиную кислоту. Однако эта реакция протекает медленно и,
если разгонка ведется в аппаратуре непрерывного действия (где
продукты реакции находятся в подогретом состоянии ограничен-
ное время) при умеренной температуре (например, перегонку
проводят при 100—150 мм рт. ст.), заметного разложения эфира
не происходит [145].
Отогнанные от продуктов реакции уксусная и муравьиная
кислоты имеют более низкую концетрацию, чем исходные, по-
тому, что в реакцию вступают только уксусная и муравьиная
кислоты, а содержащаяся в исходной кислоте вода остается
90
Время контакта, мин
Рис. 15. Состав продуктов формилирования
камфена в зависимости от времени контакта
с катализатором — катионообменной смолой
«Эспатит I» с размером зерен от 0,25 до 1 мм:
1 — при 20°С; 2 — при 45—50’6
Зрир изоборнеола
Рис. 16. Схема установки для получения муравьиного эфира изобор-
неола непрерывным методом с использованием катионообменных смол
в качестве катализаторов:
1 — смеситель для растворения реагентов; 2 — насосы; 3 — первая катионитная
колонна; 4 — промежуточный сборник; 5 — ротаметры; 6 — теплообменник; / — вто-
рая катионитная колонна
91
в непрореагировавшей кислоте. Отогнанные от эфира непрореа-
гнровавшие кислоты перед возвращением в цикл необходимо
укреплять до исходной концентрации. Эта задача технологически
проще решается в отношении уксусной кислоты (гл. XI.7), что
дает преимущество непрерывному ацетилированию камфена
перед формилированием.
В качестве катализатора для непрерывного ацетилирования
камфена предложена жидкая полифосфорная кислота [229]. Ре-
актор (колонна емкостью 1,5 л с мешалкой на нижнем конце)
заполняется смесью, состоящего из 2 массовых частей полифос-
форной кислоты 2,06) и 7 массовых частей уксусной кислоты
общей массой 750 г. В колонну снизу подают раствор, состоя-
щий из 136 г камфена в 75 г уксусной кислоты и 50 г гексана,
со скоростью 300 г/ч. Продукты реакции собирают из верхней
части аппарата, они содержат 60,5% изоборнилацетата, 10,1%
непрореагировавшего камфена, 10,2% уксусной кислоты и 0,1%
фосфорной кислоты, что соответствует превращению в эфир 80%
камфена. После перегонки получают 98%-ный изоборнилацетат
и раствор камфена в уксусной кислоте, который возвращают
в цикл.
На камфарном заводе в Манингтри (Англия) осуществлен
другой вариант непрерывного ацетилирования камфена, при ко-
тором в качестве катализатора применяется серная кислота.
Первая стадия процесса осуществляется в непрерывно действую-
щем реакторе с мешалкой, а его окончание — в насадочной ко-
лонне, по которой медленно стекает реагирующая смесь [273].
4. Сравнительная оценка процессов
формилирования и ацетилирования камфена
Превращение камфена в изоборнеол осуществляют через изоборнилфор-
миат и изоборнилацетат. Каждая из этих возможностей имеет свои достоин-
ства и свои недостатки, которые к тому же по разному дают о себе знать
в данных конкретных экономических условиях.
Превращение камфена в нзоборнеол через его муравьиный эфир долгое
время представлялось более целесообразным, чем превращение через уксус-
ный эфир. Это вытекало из более дешевой цены на муравьиную кислоту,
более низкой! молекулярной массы, что позволяло ее вводить в реакцию
в меньшем количестве, чем уксусную кислоту, а также более быстрого омыле-
ния изоборнилформиата, чем омыление изоборннлацетата.
В настоящее время стали предпочитать получение изоборнеола через
его ацетат. Это объясняется рядом причин. В связи с развитием синтетиче-
ского способа получения уксусной кислоты цена на нее снизилась. Стойкость
изоборнилацетата позволила вести отгонку не вошедшей в реакцию уксусной
кислоты от продуктов ацетилирования после связывания серной кислоты.
Уксусная кислота может быть более успешно использована в непрерывном
процессе, укрепление оборотной кислоты может быть произведено азеатроп-
иыми методами (гл. XI.7), но что самое главное, изоборнилацетат получил
широкое применение в парфюмерии вместо дорогого борнилацетата. Так, на-
пример, в США выпускают для этой цели около 600 т изоборнилацетата,
он выпускается в значительных количествах в ГДР и ФРГ. Это производство
сочетается с производством камфары, поэтому вряд ли целесообразно для
предприятий получать оба эфира параллельно.
92
5. Свойства муравьиного
и уксусного эфиров изоборнеола
Муравьиный и уксусный эфиры изоборнеола — подвижные
жидкости с приятным запахом. Запах изоборнилацетата напоми-
нает запах пихтовой хвои. В зависимости от способа получения
и методов очистки свойства изоборнильных эфиров колеблются
довольно значительно. В их состав входят: изоборнильные и бор-
нильные эфиры (до 15%), эфиры фенхилового и изофенхилового
спиртов, эфиры псевдоборнеола, терпеновые углеводороды, кам-
фен и трициклен (при использовании в качестве исходного про-
дукта чистого камфена), моноциклические терпены (при исполь-
зовании загрязненного ими камфена) и терпеновые полимеры, ко-
торые содержатся в количестве 1 % при использовании чистого
камфена. Количество их резко возрастает при применении кам-
фена, загрязненного моноциклическими терпенами. В изоборнил-
формиате содержится свободный изоборнеол, образующийся
в результате омыления эфира при щелочной промывке.
При содержании суммы эфиров и свободного изоборнеола
в технических изоборнильных эфирах свыше 96% их направляют
на омыление без дальнейшего укрепления, при меньшем содер-
жании их подвергают ректификации (гл. IX.4). Изоборнилаце-
тат для парфюмерной промышленности обязательно перегоняют,
отделяя хуже пахнущие фракции [263]. По американским стан-
дартам, содержание основного вещества в изоборнилацетате,
применяемом для парфюмерии, должно быть не ниже 97%,
0,980—0,984, 1,460—1,465 [197].
Поверхностное натяжение изоборнилацетата (20°С) 28,97X
ХЮ'3 Н/м (28,97 дин/см), вязкость (20°С) 8,19-10“3 (Н-с)/м2,
упругость паров изоборнильных эфиров см. в гл. IX.5, прочие
физические свойства — в гл. XIII.
6. Гидролиз (омыление) борнильных
и изоборнильных эфиров
В технике борнеол (XXIII) получают гидролизом борнилаце-
тата (XXIV), выделенного из пихтового масла (гл. X), а изо-
борнеол (XXV) — гидролизом его уксусного и муравьиного эфи-
ров (III), полученных из камфена (схема 26).
Гидролиз муравьиных и уксусных эфиров борнеола и изо-
борнеола всегда осуществляют растворами едкого натра. Его
можно произвести и водой в присутствии кислого катализатора,
но реакция сопровождается дегидратацией изоборнеола и по-
тому в промышленности не применяется. Щелочной гидролиз
осуществляют спиртовым раствором щелочи в гомогенной среде
и водным раствором щелочи в гетерогенной среде, но в технике
его производят лишь водными растворами щелочи.
93
NaOH
1H -j-RCOONa
XXIV О
I
О=с— R
XXIII OH
Гидролиз муравьиных и уксусных эфиров борнеола и изобор-
неола обычно производят в вертикальных или горизонтальных
автоклавах при 130—160° и при непрерывном размешивании. Во
избежание отложения твердого борнеола (или изоборнеола) на
стенки аппарата его обогревают через «рубашку». Для гидро-
лиза применяют 20—30%-ные растворы едкого натра. Продол-
жительность реакции составляет отЗдо 8 часов. Реакция сопрово-
ждается значительным тепловым эффектом. Например, гидролиз
изоборнилформиата сопровождается выделением 67 кДж/моль
тепла, или соответственно 342 кДж/кг (88 ккал/кг). Поэ-
тому автоклавы должны быть рассчитаны на повышенное дав-
ление.
Борнеол и изоборнеол выделяются при этом в виде масла,
которое постепенно кристаллизуется.
Во избежание забивки штуцеров кристаллами при загрузке
автоклава прибавляют иногда небольшое количество (около 1%)
растворителя, например толуола или ксилола.
При медленном (в течение нескольких часов) охлаждении
и непрерывном размешивании содержимого аппарата продукт
выделяется в крупнокристаллическом виде. Иногда эту операцию
осуществляют в специальном кристаллизаторе.
Если применяют для гидролиза концентрированные растворы
щелочи, загрузку разбавляют водой из расчета получения рас-
творов муравьинонатриевых или уксуснонатриевых солей, со-
держащих не более 25—30% сухой соли, для предупреждения
выпадения в осадок уксуснонатриевых или муравьинонатриевых
солей при охлаждении.
Затем нужно отделить и отмыть борнеол или изоборнеол от
раствора солей, хорошо отжать его от водной фазы и одновре-
менно получить возможно более полный выход раствора натрие-
вой соли органической кислоты. Для получения более концент-
рированных растворов органических солей (до 20—25%) ведут
94
ступенчатую противоточную промывку борнеола декантацией,
а окончательный отжим осуществляют на центрифуге.
На рис. 17 приведена схема установки для гидролиза эфиров борнеола
и изоборнеола. Омыление производят в горизонтальном автоклаве 3 с ме-
шалкой. По окончании реакции омыления и кристаллизации борнеола при
медленном охлаждении раствор солей отделяют декантацией, пропускают
через флорентийский сосуд 5 для отделения незначительного количества бор-
неолов и собирают в приемнике 6, откуда раствор солей направляют на вы-
парку.
Рис. 17. Схема установки для щелочного гидролиза эфиров борнеола и
изоборнеола:
1— мерник для раствора NaOH; 2— мерник для воды; 3 — автоклав для щелочного
гидролиза эфиров; 4 — центрифуга; 5, 7, 9 — флорентийские сосуды; 6, 8, 10 — при-
емники для растворов солей органических кислот и промывных вод; 11 — приемник
для изоборнеольных масел
Промывные воды от ступенчатой противоточной промывки эфиров бор-
неола и изоборнеола отделяют от жидких побочных продуктов во флорен-
тийских сосудах 5, 7, 9 и собирают в приемниках 6, 8, 10. Для первой про-
мывки используют промывание воды от второй промывки предыдущей опе-
рации, после чего раствор направляют на выпарку. Промывные воды от
последней промывки (промывают чистой водой при отжиме борнеола или изо-
борнеола на центрифуге) направляют для предпоследней промывки при сле-
дующей операции. Жидкие побочные продукты (бориеольиые и изобориеольные
масла) из флорентийских сосудов, отделяемые в основном при отжиме бор-
неолов на центрифуге 4, собирают в приемнике 11.
В Англии гидролиз изоборнильных эфиров ведут в присут-
ствии большого количества легкого растворителя нефтяного про-
исхождения и получают по окончании реакции раствор изобор-
неола, от которого количественно может быть отделен не раст-
воримый в нем крепкий водный раствор натриевой соли
95
Рис. 18. Кривая растворимости изобор-
неола в ксилоле
органической кислоты. Далее из раствора изоборнеола отгоняют
растворитель, кристаллизуют и отжимают выделившийся изобор-
неол [211, 273]. По другим сведениям, полученный после гидроли-
за раствор изоборнеола в нефтяном растворителе направляют на
следующую операцию (дегидрирование), не выделяя из него
изоборнеол [191]. Второй вариант, исключающий относительно
грязную и трудоемкую операцию фуговки изоборнеола, более
привлекателен, он исключает также унос солей органических
кислот вместе с промывными водами. Однако исключение от-
жима изоборнеола на центрифуге оставляет в нем значительную
часть примесей. В резуль-
тате снижается качество
сырой камфары. По ана-
логии с растворимостью
изоборнеола в ксилоле
(рис. 18) добавка раство-
рителя должна произво-
диться из расчета полу-
чения 50%-ного раствора.
Наоборот, при работе по
первому варианту пере-
кристаллизация изобор-
неола повышает качество
камфары, растворы солей органических кислот хорошо реге-
нерируются, но процесс в целом усложняется. Растворитель,
используемый в процессе, по обоим вариантам загрязняется при-
месями к изоборнеолу и его необходимо перед возвращением
в процесс очищать ректификацией.
Гидролиз эфиров борнеола и изоборнеола не сопровождается
побочными процессами и практически происходит количественно.
Однако примеси, содержащиеся в эфирах борнеола и изоборнео-
ла, влияют на процесс гидролиза и на последующее выделение
спиртов.
В эфирах изоборнеола содержится примесь эфиров 5-экзо-изо-
камфан-экзо-ола-3 (псевдоборнеола) (VIII), фенхилового (XVII)
и изофенхилового (XVIII) спиртов. При нагревании с едким
натром эти эфиры образуют соответствующие низкоплавкие
спирты (гл. XIII, табл. 52).
Терпеновые углеводороды во время процесса гидролиза осо-
бых изменений не испытывают, но частично растворяют изобор-
неол и изофенхиловый спирт. Этот раствор отжимается от ос-
новной массы изоборнеола на центрифуге и на производстве
именуется изоборнеольным маслом.
В изоборнеольном масле содержится от 30 до 50% терпено-
вых спиртов, в основном изоборнеола. В соответствии с этим
при гидролизе эфиров, содержащих значительную примесь тер-
пеновых углеводородов, потери изоборнеола в маслах могут
оказаться значительными. В табл. 23 приведены расчетные дан-
96
ные о количестве изоборнеольных масел и о потерях изоборне-
ола в маслах при гидролизе муравьиного эфира изоборнеола
разной концентрации.
Таблица 23
Количество изоборнеольных масел и потери изоборнеола в маслах
при гидролизе технического изоборнилформиата разной концентрации
Содержание эфира в исход- ном продукте, % Выход в массовых частях из 100 массовых частей исходного продукта Потери изоборнеола в маслах Образуется масел в мас- совых частях на 100 массо- вых частей изоборнеола
изоборнеола изоборнеоль- ных масел массовых частей %
70 44,2 45 15 25 100
80 57,6 30 10 15 52
85 64,5 22,5 7,5 10,5 35
92 74,0 12 4 5 16
95 79,5 6 2 2,5 7,5
Изоборнеольные масла полностью от изоборнеола не отжи-
маются и на практике изоборнеольные масла получают в не-
сколько меньшем количестве, чем указано в табл. 23. Изофен-
хиловый спирт лучше изоборнеола переходит в изоборнеольные
масла, поэтому эти масла богаче изофенхиловым спиртом, чем
отжатый от них изоборнеол. Таким образом, происходит свое-
образная перекристаллизация изоборнеола в процессе его
получения и повышается его качество и качество полученной
из него камфары. Поскольку извлечение изоборнеола из изо-
борнеольных масел связано с затруднениями, то для гидролиза
берут эфиры высокой концентрации, что уменьшает количество
масел.
Отжатый на центрифуге изоборнеол содержит 3—5% влаги.
Изоборнеол обладает небольшой оптической активностью (вы-
ражается несколькими десятыми долями градуса), имеющей об-
ратный знак по отношению к оптической активности камфена,
и обусловливается примесью изофенхилового спирта. Основные
примеси, содержащиеся в изоборнеоле,—терпеновые углеводо-
роды, главным образом камфен и трициклен, изофенхиловый
спирт и псевдоборнеол. В нем содержится также небольшое ко-
личество не отмытого формиата натрия и смолистых веществ.
Эти вещества затрудняют проведение дегидрирования изобор-
неола, поэтому промывка его должна вестись возможно более
тщательно.
7 Заказ № 43
97
7. Некоторые кинетические закономерности
гидролиза борнильных и изоборнильных эфиров
и пути улучшения процесса
Гидролиз муравьиных и уксусных борнильных и изоборниль-
ных эфиров 20—30%-ными растворами едкого натра при 120—
160°С (под давлением) продолжается в заводских аппаратах
от 3 до 8 ч. Длительность реакции — результат недостаточного
массообмена в системе и препятствий, возникающих при диф-
фузии раствора едкого натра через корку кристаллов изобор-
неола, образующуюся на поверхности раздела фаз эфир—
раствор едкого натра [37]. На некоторых предприятиях для
ускорения реакции прибавляли при загрузке эфира и щелочи
эмульгаторы с целью получить более тонкую эмульсию и этим
форсировать процесс.
Выродов и Коротов показали, что значительного ускорения
гидролиза изоборнилформиата можно достичь без эмульгаторов,
осуществляя процесс при температуре выше температуры плав-
ления изоборнеола в прямом турбулентном потоке [37, 39, 84]
(гл. VI.8). В этом случае продолжительность полного гидролиза
изоборнилформиата достигает примерно 100 с [39].
Предел возможного сокращения продолжительности гидролиза борнил и
изоборнилформиатов и ацетатов в гетерогенной среде устанавливается кине-
тикой гидролиза названных эфиров в гомогенных растворах. Эти эфиры
практически нерастворимы в воде, однако растворимы в спирте и в спирто-
водных растворах, содержащих до 50% воды, что дает возможность просле-
дить за изменением скоростей реакции под влиянием увеличения содержания
воды в растворах и на основании этого получить приблизительные данные
о скорости гидролиза борнильных и изоборнильных эфиров в водных рас-
творах.
Щелочным гидролиз сложных эфиров обычно протекает по уравнению
реакций второго порядка:
d А
= *набл АВ. (2)
где dAfdx — скорость реакции в момент времени т;
А, В—концентрации эфира и щелочи в тот же момент;
k — постоянная, называемая константой скорости реакции. Она не за-
висит от концентрации реагирующих веществ, но зависит от тем-
пературы и растворителя.
Щелочной гидролиз и этанолиз ацетатов и отчасти формиатов борнеола
и изоборнеола в безводном спирте и спирто-водных растворах изучали Руда-
ков с сотрудниками [34, 67, 80, 81, 151]. Было показано, что реакция проте-
кает по уравнению второго порядка, а константа скорости реакции мало
изменяется с изменением содержания воды в растворе (табл. 24). Это дает
основание считать, что константа скорости реакции в водном растворе
была бы того же порядка, что и в растворе, содержащем 50% воды.
Гидролиз и этанолиз бориил- и изобориилацетатов в безводном спирте
и в спирто-водных растворах, содержащих меиее 50% воды, каталитически
ускоряется катионами щелочных металлов [34, 67, 81, 151]. В результате
этого с повышением концентрации раствора щелочи константы скорости реак-
ции растут. В растворах, содержащих 25—30% воды, каталитическое действие
катиона значительно слабее, чем в безводном спирте, а в растворах, содер-
98
жащих 50% воды, вовсе ие проявляется. Во избежание влияния катиона
в табл. 24 внесены константы скорости гидролиза и этанолиза эфиров, опре-
деленные при бесконечно большом разбавлении щелочи.
Таблица 24
Константы скорости щелочного гидролиза
и этанолиза борнилацетата при 20°С
в спирто-водных растворах с разным содержанием
воды
Вода, % Реагент й-103 л/(моль • с)
0,5 C2HsONa 54
10 кон + с2н5ок 65
25 КОН + С2Н5ОК 65
50 NaOH 4- C2H5ONa 95
Константы скорости щелочного гидролиза борнильных эфиров в 5—20 раз
больше констант скорости щелочного гидролиза изоборнильиых эфиров,
а константа скорости щелочного гидролиза формиатов в 1000—3000 раз
больше констант скорости щелочного гидролиза соответствующих ацетатов.
Константы скорости и расчетная продолжительность щелочного гидролиза
некоторых эфиров борнеола и изоборнеола на 99,9%, 6 н. раствором едкого
натра в 50%-иом водном спирте и в 10%-ном избытке, составленная на
основе условного допущения полной взаимной растворимости реагирующих
веществ, представлены в табл. 25.
Таблица 25
Константы скорости и продолжительности щелочного гидролиза эфиров
Эфир к, л/(моль-с) при 20°С Продолжительность при £-70 кДж/моль, с
20° С 100°С 150°С 200°С
Борнилформиат 9,6.10-1 16 4-10-2 2,3-10-3 2,8-10-4
Изоборнилфор- миат 1,47-10-1 106 2,6-10-1 1,5-10-2 1,8-Ю-з
Борнилацетат 9,5-10-4 16 500 42 2,5 3-10-1
Изоборнилацетат 5,3-10-в 295000 680 44 5,3
Энергия активации щелочного гидролиза борнил- и нзоборнилацетатов
в щелочных, спиртовых и спирто-водных растворах и кислотного гидролиза
в водном растворе диоксаиа составляет от 60 до 90 кДж/моль [67, 80, 207].
Энергию активации щелочного гидролиза и этанолиза борнил- и изоборнил-
формиатов условно можно принять такой же.
Из приведенных в табл. 25 констант скоростей реакции при 20°С можно
вычислить константы скорости при других температурах, пользуясь уравне-
нием:
£ / 1 1 \
In - yj + In k[,
(7)
7*
99
где ki и k2— коистаиты скорости реакции, л/(моль-с), при температурах 7"i
и Т2, °К;
Е — энергия активации (Дж/моль);
R— газовая постоянная (8,31 Дж/моль • град).
Зная константу скорости реакции при данной температуре, можно вычи-
слить время, необходимое для той или иной степени глубины гидролиза
эфира при этой же температуре, пользуясь уравнением:
т = 1 In /яч
Й(С1-С2) С1(С2-Х) ’
где т — время, с;
С[ и С2 — исходные концентрации эфира и щелочи, моль/л;
X— заданная концентрация эфира в конце реакции (моль/л);
k — константа скорости реакции, л/(моль-с).
Из приведенных в табл. 25 данных следует, что гидролиз борнильных и
изоборнильных эфиров в условиях высокой турбулентности с целью сокра-
щения продолжительности реакции вполне обоснован. Достигнутая Корото-
вым и Выродовым продолжительность реакции около 100 с [39] для гидро-
лиза изоборнилформиата на 99,9% примерно в 200 раз меньше продолжи-
тельности процесса в периодически действующих автоклавах, но в 105 раз
больше продолжительности реакции в гомогенной среде при тех же темпера-
турных условиях. Это понятно, если принять во внимание, что даже самый
интенсивный массообмен и диспергирование веществ в гетерогенной среде не
превращают ее в гомогенную. В соответствии с этим реакция в условиях вы-
сокой турбулентности продолжает протекать на поверхности раздела фаз,
правда, значительно увеличенной по сравнению с той, которая получается по
существующему процессу. Тем ие менее достигнутая скорость реакции в пря-
мом турбулентном потоке позволила разработать непрерывный, высокопроиз-
водительный процесс гидролиза изоборнилформиата.
8. Непрерывный способ гидролиза
изоборнилформиата
Разработанный Коротовым и Выродовым способ гидролиза
изоборнилформиата осуществляется в прямоточном (диафраг-
менном) реакторе с 40 диафрагмами при 200°С. Процесс был
опробован в производственных условиях. Реактор представляет
собой трубу длиной 1,5 м, разделенную на равные участки диа-
фрагмами, снабженными переточными отверстиями. Диаметры
переточных отверстий в диафрагмах по ходу движения рабочей
смеси составляют 5, 4 и 3 мм. Скорость движения рабочей смеси
в переточном отверстии диафрагмы достигает 10 м/с, что вызы-
вает образование высокодисперсной эмульсии реагирующих ве-
ществ. Между каждой парой диафрагм имеется емкость, в кото-
рой за счет потери скорости потока происходит частичное раз-
рушение эмульсин, а при прохождении рабочей смеси через
следующую диафрагму эмульгирование возобновляется. По мне-
нию авторов, такое состояние системы позволило вести реакцию
в «подвижной эмульсии», что обеспечивает интенсификацию
диффузионного процесса. Схема опытного диафрагменного реак-
тора, использованного авторами, приведена на рис. 19.
В последующих работах [69] указывается, что в качестве ре-
актора можно использовать теплообменник типа «труба в тру-
100
бе». Диаметр внутренней трубы выбирается таким образом,
чтобы значения критерия Рейнольдса для реакционной смеси,
протекающей по ней, были не меньше 10 000. Для обеспечения
производительности установки, соответствующей производитель-
ности завода 1000 т камфары/год, диаметр трубчатого реактора
Рис. 19. Опытный диафрагменный реактор для щелочного гид-
ролиза изоборнилформиата [39]:
/—диафрагменный реактор; 2 — напорные бачки для эфира и щелочи;
3— подогреватели эфира и щелочи; 4 — игольчатые вентили; 5 — приемник
продуктов омылеиня; 6 — штуцер для отбора проб; 7 — манометр; 8 — бал-
лон сжатого азота; 9-— термометры
составляет 12 мм при длине 20—25 м. Для поддержания необ-
ходимого давления и температуры в реакторе на выходе продук-
тов омыления установлена диафрагма диаметром 3—4 мм.
При движении рабочей смеси в подогревателе и реакторе
возникает сопротивление от давления паров воды над раствором
едкого натра и сопротивление теплообменника и реактора. Сум-
марное значение этих сопротивлений при 200°С достигает 20—
23 атм. Это сопротивление преодолевается дозировочными
101
плунжерными насосами. При быстром охлаждении образующе-
гося изоборнеола он получается в виде труднофугуемой мелко-
кристаллической пасты, медленная же кристаллизация в про-
межуточных аппаратах в значительной степени уменьшает
преимущества быстрого непрерывного процесса гидролиза. Для
устранения этих трудностей авторы рекомендуют осуществлять
гидролиз изоборнилформиата разбавленного толуолом или
ксилолом. О переработке такого раствора см. гл. VI.6.
Рис. 20. Технологическая схема щелочного гидролиза изоборнилфор-
миата [69]:
/. 2 — мерники для раствора изоборнилформиата в толуоле и растворе едкого
натра; 3 — дозирующее устройство; 4, 8 — насосы; 5 — трубчатый реактор; 6, 9—
флорентины; 7 —обратные холодильники; 10 — линия выходящего раствора нзо-
борнеола
Технологическая схема непрерывного процесса омыления
изоборнилформиата по Коротову и Выродову приведена на
рис. 20. Авторы пишут об успешном опробовании в производ-
ственных условиях разработанного ими способа гидролиза изо-
борннлформната [85].
Осуществить этим способом гидролиз изоборнилацетата не
удалось [37], несмотря на то что пребывание веществ в реакторе
в 20 раз превышало время, необходимое для гидролиза изобор-
нилацетата в гомогенной среде. Для гидролиза изоборнилаце-
тата в прямом турбулентном потоке, по мнению авторов, по-
требовалось бы значительно увеличить длину реактора и повы-
сить температуру процесса [38].
102
9. Регенерация органических кислот
Используемые для получения эфиров органические кислоты возвращаются
из процесса в виде слабощелочных растворов натриевых солей. Часть орга-
нических кислот вместе с серной кислотой в виде разбавленных водных рас-
творов возвращается после промывки эфиров. Этими слабыми кислотами
нейтрализуют избыточную щелочь, содержащуюся в растворе натриевых со-
лей. Из натриевых солей органические кислоты получают обычными мето-
дами.
10. Пути непосредственного превращения камфена
в борнеол и изоборнеол
В технике многие спирты получают гидратацией соответствующих непре-
дельных углеводоров (алкенов) серной кислотой.
Например, из пропилена (XXVI) в этих условиях получают вторичный
пропиловый спирт (XXVII). Промежуточными продуктами реакции являются
карбоний-ион (XXVIII) и пропилсерная кислота (XXIX):
H2SO4 <=> н++ [hso4]
Э-Н^ + ч-ГнзоЛ +н2о
CHj-CH=CH2 СН3-СН — сн3 I >J CHj-CH—сн3 <----->
-Н+ I —Н2О
XXVI XXVIII о—SO2OH
XXIX
сн3—СН—СН3+ H2SO
4
он
XXVII
Равновесие обратимой реакции сдвинуто в сторону пропилсерной кислоты
лишь при низкой температуре. Часто реакция сопровождается изомеризацией,
тогда получают спирты с иным углеродным скелетом, чем у исходного алкена.
В соответствии с изложенным из-за легких переходов в кислой среде от
скелета камфена к скелету борнеола при гидратации камфена (I) серной кис-
лотой можно получить не только соответствующий камфену спирт камфен-
гидрат (V), но и изоборнеол (XXV). Промежуточными продуктами реакции
должны быть изокамфил (II) и борнил-(XXX) карбоний-ионы и соответст-
вующие им алкилсерные кислоты (XXXI) и (XXXII). Если бы были найдены
I II XXXI V
XXX XXXII XXV
103
условия реакции, при которых гидратация была полной, а изоборнеол един-
ственным продуктом, эта реакция, несомненно, получила бы промышленное
применение.
Проведенные до сего времени исследования, однако, ие дали обнадежи-
вающих результатов.
При нагревании с серной кислотой разной концентрации равновесие сдви-
нуто в сторону камфена и выход изоборнеола составляет лишь несколько
процентов [189].
При интенсивном размешивании камфена с 65%-иой серной кислотой
при температуре ниже 0° в качестве основного продукта гидратации автором
книги был получен камфенгидрат.
Такие же результаты получили ранее при гидратации камфена сульфатом
диэтилоксония [189]. Значительно лучшие результаты были достигнуты при
гидратации камфена водными растворами сульфонафтеновых кислот (контакт
Петрова) [111]. В этом случае значительная часть камфеиа была превращена
в изоборнеол, однако н здесь наряду с изоборнеолом получился камфенгидрат.
ГЛАВА VII. ПРЕВРАЩЕНИЕ БОРНЕОЛОВ*
В КАМФАРУ
Для получения синтетической камфары используют техниче-
ский изоборнеол, не подвергнутый специальной очистке, но тща-
тельно отмытый от солей органических кислот и хорошо отжа-
тый от воды и масел. В ограниченном количестве используют и
борнеол, получающийся при переработке пихтового масла. О со-
ставе и свойствах технического изоборнеола см. гл. VI.6, а о со-
ставе технического борнеола — гл. Х.5.
Превращение борнеола и изоборнеола в камфару может быть,
осуществлено или окислением, или каталитическим дегидрирова-
нием. Однако методы окисления не могут конкурировать с мето-
дами каталитического дегидрирования. По-видимому, методы
окисления имели малое применение и в прошлом, так как появ-
ление промышленных методов каталитического дегидрирования
борнеола и изоборнеола относится к началу текущего столетия
и совпадает с периодом возникновения промышленности синтети-
ческой камфары.
1. О некоторых различиях в химических свойствах
борнеола и изоборнеола
Борнеолы — стереизомерные терпеновые спирты, различаю-
щиеся лишь пространственным расположением атома водорода
и гидроксильной группы при 2-м углеродном атоме. Гидроксиль-
ная группа у борнеола (I) направлена внутрь шестичленного
кольца, а у изоборнеола (II) наружу. Свойства этих спиртов на-
столько различны, что в прошлом некоторые авторы их считали
не стереизомерами, а структурными изомерами.
* Здесь и в дальнейшем изложении под термином «борнеолы» следует
понимать оба стереизомера борнеол и изоборнеол.
104
Изоборнеол под действием кислых реагентов легко претерпе-
вает перегруппировку Вагнера и, отщепляя воду, превращается
в камфен (Ш); борнеол устойчив к тем же дегидратирующим
реагентам или лишь медленно изменяется под их влиянием. Это
происходит потому, что отрыв гидроксильной группы у изобор-
неола (III) облегчен давлением электронной пары, образующей
связь между 6-м и 1-м углеродными атомами в направлении
отщепляющейся гидроксильной группы с противоположной сто-
роны 2-го углеродного атома. При отщеплении гидроксильной
группы у борнеола (I) это давление отсутствует, поскольку
электронная пара между 6-м и 1-м углеродными атомами нахо-
дится со стороны отщепляющейся группы (гл. III.3). В резуль-
тате превращение (I) в ион (IV) происходит за две стадии,
из которых первая протекает медленно:
Способность изоборнеола легко дегидратироваться с образо-
ванием камфена необходимо учитывать при его переработке
в камфару.
2. Получение камфары окислением борнеолов
По получении камфары окислением борнеола и изоборнеола в начале сто-
летия были выполнены довольно многочисленные исследования и взято значи-
тельное количество патентов. Сводка литературы дана в ряде обзоров [182,
268, 331].
Предлагались окисление воздухом в присутствии металлической меди и
ее окиси, активированного угля, окислов ванадия, молибдена и хрома, элек-
трохимическое окисление и окисление химическими реагентами: хромовой и
азотной кислотами, хлорной водой, солями хлорноватистой кислоты, перман-
ганатом, озоном. Большинство из перечисленных способов окисления не дает
приемлемых результатов.
1. Кислые реагенты непригодны для окисления изоборнеола, так как реак-
ция сопровождается его дегидратацией. Исключением является хромовая кис-
лота, которая окисляет изоборнеол, растворимый в уксусной кислоте, до кам-
фары с хорошим выходом. Но хромовая кислота окисляет до камфары и кам-
фен, поэтому окислять хромовой кислотой изоборнеол не имеет смысла.
2. Применение реагентов, содержащих хлор, недопустимо, так как оно
может привести к получению камфары, содержащей хлор.
105
3. Как правило, реакция, недостаточно селективна. Образующаяся из бор-
неолов камфара (VI) подвергается дальнейшему окислению, в результате
чего получаются камфарная (VII), изокамфароновая (VIII), камфароновая
(IX) кислоты, п-дикетокамфаи (X), 6-кетокамфара (XI) и другие продукты
[116, 318].
4. При окислении твердых борнеолов водными растворами окислителей
проникновение растворов внутрь твердых частиц затруднено, поэтому прихо-
дится считаться с присутствием в камфаре, наряду с продуктами ее глубокого
окисления, непрореагировавших борнеолов.
В заводской практике одно время производили окисление борнеола, полу-
ченного из пихтового масла, азотной кислотой. Впервые оно было осущест-
влено в дореволюционной России на заводе Жукова, а в период 1930—
1941 гг.— на Охтинском химкомбинате по усовершенствованной схеме [182].
Окисление производили 70%-ной азотиой кислотой при охлаждении (ре-
акция экзотермична) в цилиндрических аппаратах из чистого алюминия. Пе-
ремешивание осуществляли сжатым воздухом. Борнеол вносили в азотную
кислоту постепенно. По окончании реакции и отстаивания выделялся слой
азотнокислой камфары, а в иижнем слое собиралась 35—40%-ная азотная
кислота. Далее азотнокислую камфару подвергали гидролизу водой, распы-
ляя в ией поступающую через форсунку азотнокислую камфару, далее обра-
зовавшуюся камфару подвергали очистке перегонкой с водяным паром.
Полученная в результате этого процесса камфара была загрязнена нитро-
соединениями и быстро желтела при хранении.
Имеются указания, что можно получать тем же способом камфару и из
изоборнеола [186], но это сомнительно, так как в условиях реакции должна
происходить дегидратация изоборнеола и окисление образовавшегося камфена
до смеси циклических кислот. Возможно, в соответствии с этим было предло-
жено окислять 66%-ной азотной кислотой не изоборнеол, а его простой мети-
ловый эфир (XII), который получают метоксилированием камфена (III) [266].
Указывают, что выход камфары составляет при этом 83%.
СН.
। V1= ch3oh+h2so4 111 X HNO3^ 11 VI
106
При окислении борнеолов водными растворами окислителен по окончании
реакции получают гетерогенную систему, состоящую из камфары, побочных
продуктов реакции и водного раствора восстановленного окислителя. Чтобы
превратить камфару в товарный продукт, ее необходимо в первую очередь
перегнать. Перегонку осуществляют обычно с водяным паром.
Перегонка камфары с водяным паром связана со значительными трудно-
стями. Сложность процесса состоит в том, что камфара вследствие высокой
температуры плавления осаждается иа поверхности конденсатора в виде лип-
кой мазеобразной пленки, которую не удается удалить скребками и другими
аналогичными приспособлениями. В результате конденсаторы перестают отво-
дить тепло, что ведет к потерям продукта, ухудшает условия труда и вызы-
вает необходимость часто отключать конденсаторы для чистки. Производи-
тельность поверхностных конденсаторов в этом случае очень низка, особенно
если принять во внимание, что прн перегонке камфары с насыщенным водя-
ным паром с 1 кг камфары по расчету (см. табл. 26) должно перегоняться
4 кг водяных паров, которые также надо конденсировать и охлаждать. Факти-
чески же перегоняется воды значительно больше, поскольку водяной пар, про-
ходя мимо кусков камфары, не успевает насытиться ее парами.
Вместо поверхностных конденсаторов используют конденсаторы смешения,
в которых пары камфары и воды конденсируются холодной водой. Для умень-
шения потерь камфары (из-за растворения в воде) используют оборотную
воду. Перегнанную камфару обезвоживают на центрифугах до содержания
влаги около 4—5% и на гидравлических прессах — до содержания влаги
1-2% [182].
3. Каталитическое дегидрирование борнеолов
При нагревании расплавленных или растворенных в индиф-
ферентных растворителях борнеола (I) и изоборнеола (II) с не-
которыми катализаторами они отщепляют водород и превра-
щаются в камфару (VI). То же происходит при пропускании па-
ров борнеола и изоборнеола через слой нагретого катализатора.
Реакция дегидрирования борнеола и изоборнеола обратима:
при нагревании камфары с водородом в присутствии тех же ка-
тализаторов происходит гидрирование камфары, она присоеди-
няет водород и превращается в смесь борнеола и изоборнеола.
Реакции гидрирования и дегидрирования применимы к боль-
шому кругу веществ и имеют большое значение в органической
химии и технологии. Дегидрирование борнеола и гидрирование
камфары — частные случаи этих реакций.
Дегидрирование и окисление борнеолов приводят к одному
и тому же конечному продукту — камфаре, но это совершенно
различные процессы. При дегидрировании от каждой молекулы
борнеола или изоборнеола отщепляются два атома свободного
водорода, при окислении же водород окисляется до воды
(с. 108).
Если борнеолы подвергать дегидрированию в жидкой фазе,
то выделяющийся водород непрерывно удаляется из сферы
реакции и, следовательно, процесс может быть доведен до конца.
Если борнеолы подвергаются дегидрированию в паровой фазе,
выделяющийся водород остается в соприкосновении с парами
камфары, в результате чего наступает подвижное равновесие
между камфарой и водородом, с одной стороны, и смесью борне-
ола с изоборнеолом — с другой. С повышением температуры это
107
равновесие сдвигается в сторону камфары, в соответствии с чем
и здесь может быть достигнуто практически полное превращение
борнеола в камфару.
Первый патент на получение камфары парофаэным дегидрированием бор-
неолов был взят «Британской ксилонитовой компанией» в 1906 г. [238]. Од-
нако процесс дегидрирования борнеола связывают с именем финского ученого
Аскана, разработавшего жидкофазный процесс дегидрирования бернеолов, на
основе которого в Германии вскоре после первой мировой войны было поста-
влено крупное заводское производство камфары. Аскан опубликовал подроб-
ное описание процессов дегидрирования борнеола и нриготовления катализа-
торов [189, 305]. В Советском Союзе процесс был первоначально разработан
Жарковым с сотрудниками применительно к борнеолу из пихтового масла
[68, 164] и осуществлен в 1934 г. на промышленной установке в Новосибирске
[108]. Позднее он был усовершенствован на основе работ В. Е. Тищенко,
М. А. Грехнева, Ю. С. Залкинда и др. и с 1937 г. применяется в промышлен-
ности для дегидрирования изоборнеола, полученного из скипидара.
В качестве катализаторов для дегидрирования борнеолов
были предложены: никель, кобальт и медь, восстановленные во-
дородом из свежеосажденных окисей [189, 305], смешанные ка-
тализаторы одного или нескольких металлов с их окисями,
а также с окисью железа [61, 189, 305], скелетные никелевые и
медные катализаторы [62, 158, 160, 178], а также углекислые
соли меди и никеля [26, 71, 72, 179]. Указывалось на применение
в качестве катализаторов цинка, серебра и кадмия [221], однако
обоснованность применения этих катализаторов остается не-
ясной.
Первоначально в процессе дегидрирования в качестве ката-
лизаторов применяли никель, кобальт и медь, восстановленные
из их окисей водородом при 300—400°С. Сложность приготовле-
ния и пирофорные свойства (самоокисление на воздухе с само-
разогреванием, ведущим к изменению дисперсности и потере
активности) этих катализаторов привели к вытеснению их ос-
новными углекислыми солями никеля и меди, которые получают
осаждением содой из растворов сернокислых или азотнокислых
солей никеля и меди. При нагревании с расплавленными борне-
олами основные углекислые соли превращаются в мелкодисперс-
108
ные окиси с высокой каталитической активностью и по ходу
реакции восстанавливаются до закисей и металлов. Такой ката-
лизатор до сих пор применяется при промышленном дегидри-
ровании борнеолов в жидкой фазе.
Некоторые авторы указывают на повышение активности ка-
тализаторов путем применения в качестве промоторов катионов
натрия, калия, бария и железа [177, 179].
Дегидрирование борнеолов может сопровождаться побочной
реакцией образования изокамфана [97]. Она имеет место при
наличии у катализатора кислых центров, способных вызвать де-
гидратацию борнеолов. Появление таких кислых центров может
произойти из-за наличия в составе основных солей меди
и никеля сульфогрупп, не вступивших в обменное разложение с со-
дой при их осаждении из раствора медного купороса или серно-
кислого никеля. Возможно, имеются и другие не вполне разъяс-
ненные причины. В результате дегидратации борнеолов образу-
ется камфен (Ш), который гидрируется до изокамфана (XIII)
водородом, выделяющимся при основной реакции. Так как
III
XIII
борнеол более стоек, чем изоборнеол, с образованием изокам-
фана приходится считаться в большей степени при дегидрирова-
нии изоборнеола, чем при дегидрировании борнеола.
Чтобы предотвратить образование изокамфана из-за дегид-
ратации изоборнеола во время его дегидрирования, осаждение
и промывку углекислых солей никеля и меди ведут с расчетом
сохранения у них незначительной щелочности. Для улучшения
качества катализатора к нему также добавляют окиси кальция
или магния или негашеную известь. Эти мероприятия позво-
ляют практически предотвратить дегидратацию борнеолов или
снизить ее до 1—2%, но не избавляют от образования изокам-
фана из-за гидрирования камфена, содержащегося в качестве
примеси в изоборнеоле.
В техническом изоборнеоле в качестве примеси присутствуют
фенхиловый (XIV), изофенхиловый (XV) спирты и 5-экзо-изо-
камфан-экзо-ол-3 (псевдоборнеол) (XVI). Все они дегидриру-
ются одновременно с изоборнеолом, образуя фенхон (XVII).
изофенхон (XVIII) и 5-экзо-изокамфанон-З (псевдокамфара)
(XIX) (с. НО).
Иногда дегидрирование рекомендуется проводить катализаторами, нане-
сенными на носители. Применение таких катализаторов позволяет экономить
расход металла и особенно полезно в непрерывном процессе. В качестве носи-
телей в литературе упоминаются древесный уголь [61, 164, 177, 179], гидрат
109
окиси магния [72], кизельгур, силикагель, гель окиси алюминия и фуллерова
земля [221].
До второй мировой войны на заводе а. о. Шеринг (Германия) в каче-
стве катализатора применяли гидрат окиси меди, осажденный на поверхности
гидрата окиси кальция.
Применение кизельгура, силикагеля, окиси алюминия и особенно отбель-
ных глин (фуллерова земля) нецелесообразно, так как они, являясь кислыми
веществами, вызывают побочную реакцию дегидратации спиртов.
К катализаторам, осажденным на носитель, можно отнести и скелетный
катализатор, приготовленный по способу Бага [29], поверхностным выщелачи-
ванием растворами едкого натра сплавов никель—алюминий и никель—медь
[62, 158, 160, 178].
Несмотря на ряд положительных свойств, катализаторы на носителях, за
исключением гидрата окиси меди, нанесенного на поверхности гидрата окиси
кальция, и катализаторы, приготовленные из сплавов, не получили практиче-
ского применения. Это связано с тем, что приготовление и регенерация таких
катализаторов значительно сложнее, чем приготовление основных углекислых
солей никеля и меди.
Каталитическое дегидрирование борнеолов осуществляется
на практике как в виде периодического жидкофазного, так и не-
прерывного парофазного процессов. Методы получения камфары
дегидрогенизацией борнеолов имеют существенные преимуще-
ства по сравнению с методами получения камфары окислением
борнеолов. Эти преимущества заключаются в следующем: 1) в
незначительном расходе катализатора по сравнению с расходом
окислителей, легкости регенерации катализатора и минималь-
ном количестве вредных сточных вод; 2) в почти полном отсут-
ствии побочных реакций и практически количественном превра-
щении борнеолов в камфару; 3) в значительно более легком
выделении камфары из продуктов реакции.
НО
Роль катализатора и механизм реакции при каталитическом гидрировании
и дегидрировании иные, чем в ранее рассмотренных карбониево-ионных реак-
циях изомеризации терпенов. Они хорошо объясняются мультиплетной тео-
рией катализа [9, 66].
По представлениям мультиплетной теории сорбированная на поверхности
катализатора молекула реагирующего вещества может оказаться под воздей-
ствием сил притяжения отдельных атомов решетки катализатора, которые
как бы растягивают молекулу в разные стороны. В результате деформации
отдельные связи молекулы рвутся и устанавливаются новые. Например, если
молекула спирта окажется под воздействием двух активных центров (дублета),
при котором водород гидроксильной группы и один из водородов, соединенных
с тем же углеродным атомом, что и гидроксильная группа, окажутся под
воздействием одного активного центра, а кислород гидроксильной группы и
соединенный с ним углерод под воздействием другого активного центра,
то превращение осуществляется по схеме:
R-
^2 ^2
С—О —►R,—С'—О—►
II ’ll
н^.н
I2.
R,— С = О
н—н
н н
Пунктиром обозначены связи в переходном комплексе, точками — актив-
ные центры. Реакция может иметь место только при определенном соответ-
ствии расстояний между активными центрами решетки катализатора и реаги-
рующими атомами, которые должны достаточно точно накладываться на
поверхность решетки. На основании этих данных мультиплетная теория позво-
ляет подойти к сознательному подбору катализаторов.
Кинетику каталитического дегидрирования борнеолов в жидкой фазе
с применением медного, кобальтового и медно-никелевого катализаторов изу-
чали Грехиев и Брошевский [59, 60]. Авторы установили, что при наличии
в растворе от 0,8 до 6% катализатора скорость реакции пропорциональна
его количеству и не зависит от концентрации борнеола в растворе, а по-
скольку концентрация катализатора постоянна, то она описывается уравне-
нием реакций нулевого порядка:
-4? = *о. (3)
dA . , .
где —-----скорость реакции (моль/л. с) в момент времени т;
k0 — наблюдаемая константа скорости реакции (моль/л. с), равная про-
изведению k'B, где k' — истинная константа скорости (моль/с г
катализатора), а В — концентрация катализатора в единице объема
(г катализатора/л).
После того как прореагирует около 70% борнеола, скорость реакции на-
чинает меняться в зависимости от его концентрации в растворе, подчиняясь
уравнению первого порядка:
dA .
— = ktA,
at
(9)
где А — концентрация борнеола (моль/л);
ki — наблюдаемая константа скорости реакции (с-1);
При содержании в растворе 5—6% катализатора реакция описывается
уравнением первого порядка на всем своем протяжении, но при дальнейшем
увеличении количества катализатора ее скорость перестает возрастать. Позд-
нее Головин, Выродов и Коротов [45, 46] подтвердили полученные Грехневым
и Брошевским результаты.
111
Течение реакции по нулевому порядку при большой концентрации бор-
неолов в объеме и последующий ее переход в первый порядок можно объяс-
нить так же, как это было сделано в отношении реакции изомеризации пи-
нена (гл. V.1). Скорость реакции по уравнению (9) пропорциональна не кон-
центрации борнеола в объеме, а концентрации борнеола на поверхности
катализатора. Количество же вещества, сорбируемого поверхностью при его
малых концентрациях, в объеме растет приблизительно пропорционально кон-
центрации, а при больших — почти не зависит от изменений концентрации.
Отсюда получается нулевой порядок в начале реакции при больших концент-
рациях борнеола в объеме и первый порядок при его малых концентрациях
в конце реакции.
Что же касается прекращения роста скорости реакции с увеличением
количества катализатора в объеме свыше 6%, то наиболее вероятно, что
в этих условиях реакция выходит из кинетической области.
Протекание дегидрирования борнеолов по уравнению реакций первого
порядка в заключительном периоде приводит к тому, что в камфаре, как
правило, остается немного непрореагировавшего борнеола (гл. V.1, рис. 5).
В работах, посвященных изучению кинетики дегидрирования борнеолов
в жидкой фазе, при 150—180°С на медном и медно-никелевом катализаторах
(1 моль Ni и 3,5 моля Си), полученных восстановлением их углекислых со-
тен, Головин, Выродов, Коротов [46] приводят данные о том, что на медном
катализаторе константа скорости и энергия активации дегидрирования изо-
борнеола, больше, чем константа скорости и энергия активации борнеола.
На медно-никелевом же катализаторе энергии активации дегидрирования
обоих спиртов равны, но константа скорости дегидрирования борнеола
в 4 раза больше, чем у изоборнеола. На этом основании авторы делают вы-
вод, что медный катализатор целесообразно применять для дегидрирования
изоборнеола, а медио-никелевый для дегидрирования борнеола.
Кинетику дегидрирования борнеолов в паровой фазе при 180—280° С на
медно-никелевых катализаторах того же состава изучали Нестеров, Радбиль
и Рудаков [101] и получили иное соотношение между константами скоростей
дегидрирования борнеолов. По данным этих авторов, константа скорости
дегидрирования изоборнеола при 180°С больше, чем константа скорости де-
гидрирования борнеола. В то же время энергия активации дегидрирования
борнеола составляет 98,8 кДж/моль (22,2 ккал/моль), а соответствующая
энергия активации изоборнеола 76,6 кДж/моль (18,3 ккал/моль). В резуль-
тате этого с увеличением температуры обе константы сближаются и при
280°С наступает равенство констант. При более высокой температуре кон-
станта скорости дегидрирования борнеола должна превысить константу ско-
рости дегидрирования изоборнеола.
Из данных [101] следует, что для 99%_-ного превращения изоборнеола
в камфару' при" 271ГС время' контакта паров изоборнеола' с медно-никелевым
катализатором должно составить около 15 с.
Одновременно с реакцией дегидрирования были отмечены обратимые
взаимные превращения борнеол Z7 изоборнеол [101].
4. Технология периодического жидкофазного
метода дегидрирования борнеолов
Каталитическое дегидрирование борнеолов осуществляется
в стальных аппаратах, снабженных мешалками. Мешалки дол-
жны перемешивать плав, взмучивать катализатор, в то же время
преодолевать значительное сопротивление, вызванное не только
вязкостью плава, но и наличием твердой фазы в начальный пе-
риод разогрева. Обычно применяют рамные мешалки или ме-
шалки с лопастями и скребками.
112
При дегидрировании 1 т борнеолов выделяется 145 м3 водо-
рода (при нормальных условиях). Чтобы уменьшить скорость
газа над поверхностью испарения, связанную с уносом реаги-
рующих веществ и пылевидного катализатора, часто применяют
горизонтальные аппараты.
Дегидрирование борнеолов проводят при температуре 180—
200°С. Реакция сопровождается поглощением тепла в количе-
стве около 55—60 кДж (13—14,5 ккал/моль) прореагировавшего
борнеола или соответственно около 350—400 кДж/кг (85—•
95 ккал/кг). Поэтому для обеспечения достаточно быстрого про-
цесса важен не только подогрев аппарата до заданной темпера-
туры, но и восполнение поглощенного тепла. Обычно обогрев
аппарата осуществляют через рубашку водяным, паром с давле-
нием около 20 ат или другим теплоносителем. В качестве тепло-
носителя одно время применяли тяжелые цилиндровые масла
с высокой температурой вспышки, ныне применяют высокотем-
пературный органический теплоноситель (ВОТ)—смесь дифе-
нила и дифенилоксида. Во избежание кристаллизации камфары
в трубах, кранах и других соединениях, их также обогревают до
температуры около 200°С.
На рис. 21 показана заводская схема установки для дегидрирования бор-
неола. В стальной аппарат 1 с горизонтальной мешалкой емкостью около
10 м3, обогреваемый посредством паровой рубашки, загружают 5—6 т бор-
неола или изоборнеола через загрузочное приспособление 13 и около 1 т рас-
творителя из мерника 8. В качестве растворителя обычно применяют арома-
тические углеводороды (толуол, ксилол). После того как содержимое аппа-
рата под влиянием разогрева примет полужидкую консистенцию, пускают
в ход мешалку. Содержащаяся в борнеолах вода отгоняется в виде азеотроп-
ной смеси с растворителем через полую колонну 2 высотой около 7 м и диа-
метром 800 мм и холодильник 3 в сборник 4 и далее во флорентийский
сосуд 5, откуда вода выводится из системы, а растворитель возвращается
в колонну. В колонне пары борнеолов, до поступления в холодильник, не-
сколько обогащаются растворителем. Кроме того, колонна служит буфером,
предохраняющим от перебросов плава и катализатора при последующей пере-
гонке. После того как содержащаяся в борнеолах вода удалена, в аппарат
постепенно вводят катализатор н одновременно отгоняют часть растворителя
в сборник 10 с таким расчетом, чтобы температура кипения плава поднялась
до 180—190°С. Выделяющийся при реакции водород выводят из системы
через буферный сосуд (промывалку) 7 и газовый счетчик 14. Контроль за хо-
дом реакции осуществляют путем учета выделяющегося водорода и анализом
плава на содержание борнеолов.
В течение всего процесса в аппарате должна оставаться часть раствори-
теля, так как конденсирующийся в холодильнике растворитель предохраняет
его от закупорки твердыми борнеолами и камфарой. Таким образом, раство-
ритель выполняет несколько назначений: он растворяет борнеолы, с его по-
мощью удаляется вода, он предотвращает осаждение борнеола и камфары
на холодных частях аппарата и трубопроводах, по которым отводится водо-
род. Без растворителя немыслимо проведение жидкофазного процесса дегид-
рирования борнеолов.
Скорость реакции зависит от многих факторов: от чистоты
борнеолов, качества и количества катализатора, скорости его
введения в реакцию, качества растворителя, компенсации по-
терь тепла, расходуемого на реакцию, массообмена.
8 Заказ № 43 ИЗ
В аппаратах сравнительно небольшой вместимости компен-
сацию теплопотерь удается осуществлять быстрее, а контакт
борнеолов с катализатором получается более тесный, поэтому
реакция в таких аппаратах протекает быстрее, чем в аппаратах;
большой вместимости, подобных описанному. Однако слишком
быстрый процесс дегидрирования нежелателен, так как он свя-
зан с большой скоростью выделения водорода, что влечет за
собой унос продукта. В частности, в описанном выше аппарате
скорость выделения водорода стараются поддержать не более
Рис. 21. Схема установки для дегидрирования борнеола:
1 — аппарат для дегидрирования; 2 — полая колонна; 3 — холодильник; 4 — сборник;
5, 6 — флорентийские сосуды; 7 — буферный сосуд для водорода; 8 — мерник для раство-
рителя; 9 — сборник для растворителя; 10— сборник для оборотного растворителя; 11 —
сборник для хвостовых погоиов растворителя, подлежащих регенерации; 12— насос; 13—
загрузочное приспособление; 14 — газовый счетчик; 15 — шибер
25—30 м3 в час. При нормальном течении процесса через 24 ч
после начала реакции содержание борнеола в плаве составляет
50%, а через 48 ч — 10%. Продолжительность дегидрирования
в аппаратах разной конструкции в зависимости от условий ре-
акции 12—60 ч. В лабораторных условиях, при хорошем мас-
сообмене и быстром восполнении теплопотерь, реакция с тем
же количеством катализатора заканчивается быстрее чем
за час.
114
В качестве катализатора вводят 1—3% углекислых солей
меди и никеля. Применяя гидрат окиси меди, осажденный на
гидрате окиси кальция, удается значительно снизить расход
меди на реакцию без существенного снижения скорости про-
цесса.
По окончании реакции в аппарате находятся камфара, ка-
тализатор и растворитель.
Выделение камфары из плава может быть осуществлено не-
сколькими путями.
На английском камфарном заводе в Л1анингтри (Эссекс)
плав разбавляют дополнительным количеством растворителя и
полученный раствор перегоняют из куба непрерывного действия.
Камфара и растворитель при этом отделяются от катализатора
и смолистых веществ. Раствор камфары в углеводородном рас-
творителе подвергают кристаллизации в аппарате непрерыв-
ного действия, после чего камфару отжимают на центрифуге
[211, 273].
Обычно камфару отделяют от катализатора и смолистых ве-
ществ перегонкой с насыщенным или перегретым водяным па-
ром. До начала перегонки камфары необходимо из плава ото-
гнать растворитель. Даже в случае применения в качестве
растворителя толуола, кипящего при сравнительно низкой тем-
пературе, без колонны эту операцию осуществить невозможно, не
получая некоторого количества промежуточной фракции, состоя-
щей из растворителя, камфары и терпеновых углеводородов,
присутствующих в виде примесей в борнеолах. Несмотря на это,
отгонку растворителя обычно ведут не применяя ректификаци-
онных колонн, так как опасаются закупоривания их твердой
камфарой. В этих условиях небольшое количество растворителя
остается в камфаре и отгоняется с головной фракцией при пе-
регонке.
Перегонку камфары ведут с перегретым водяным паром, так
как в противоположность перегонке с насыщенным водяным па-
ром при 100°С (гл. VII.2) перегонка камфары с перегретым
водяным паром технологически решается вполне успешно. Пе-
регонку проводят в условиях, при которых содержание воды
в парах не превышает 3—4% по массе. В этом случае отгоняе-
мая камфара не образует липких пленок на поверхности кон-
денсаторов и легко счищается с нее скребками. Это позво-
ляет использовать для конденсации камфары поверхностные
холодильники, механизировать очистку их и получать сразу
же товарную камфару, не требующую отжима на центри-
фугах.
В табл. 26 приведены вычисленные данные о соотношении
между камфарой и водяным паром при разных температурах
перегонки и при условии полного насыщения водяного пара па-
рами камфары.
Из данных табл. 26 видно, что необходимое соотношение
8* 115
между камфарой и водяным паром достигается при температуре
около 200°С.
Таблица 26
Соотношение между камфарой и водяным паром при перегонке камфары
при условии полного насыщения водяного пара парами камфары
Температура, °C Упругость паров камфары, мм рт. ст., Р 1328, 338] С 1 кг камфары перегоняется воды, кг (Ь) * Содержание воды в парах
в массовых, % в объемных, %
100 21,9 3,95 80 97,1
130,9 74,5 1,09 52 90,1
140,8 108,4 0,71 42 86,3
150,8 152,3 0,47 32 80,0
160,9 216,8 0,30 23 71,6
171,1 295,1 0,19 16 61,5
180,1 390 0,11 10 48,2
191,5 514,5 0,056 5,3 32,0
201,7 664,8 0,017 1,7 12,7
* Вычислено из уравнения по закону Дальтона:
_ (760 -Р)-18
— Р-152
где 18 и 152 — молекулярные массы воды и камфары.
Массовое содержание камфары в единице объема паров
в этих условиях примерно в 20 раз больше, чем при 100°С. Из
этого следует, что при той же скорости паров производитель-
ность перегонной установки при 200°С в 20 раз выше, чем при
100сС, а это очень важно, так как максимально допустимая ско-
рость паров над поверхностью испарения, при которой пылевид-
ный катализатор не уносится с камфарой, примерно одинакова
как при 100, так и при 200°С.
Перегонку камфары с перегретым паром обычно ведут из
аппаратов для дегидрирования. Для этого колонну 2 отделяют
от холодильника 3 шибером 15 и открывают задвижку на линии
в конденсаторы для камфары (см. рис. 21). Подачу пара в рас-
плавленную камфару, нагретую до 200°С, осуществляют через
игольчатый вентиль. С 1 м2 поверхности испарения аппарата
снимают до 40—50 кг камфары в час.
Состав отгоняющейся камфары меняется по ходу перегонки.
В начальных погонах содержится небольшое количество остав-
шегося после дегидрирования растворителя. Терпеновые уг-
леводороды и изокамфан отгоняются в основном с первыми
50% погона (рис. 22), изофенхон распределяется равно-
мерно, а содержание псевдокамфары имеет тенденцию увеличи-
ваться к концу перегонки.
116
Камфару можно перегонять в токе инертного газа, но это нецелесооб-
разно, так как, во-первых, при перегонке камфары в токе инертного газа теп-
лопотери от испарения восполняются только за счет внешнего обогрева, тогда
как при перегонке с перегретым водяным паром теплопотери восполняются
и за счет перегрева пара. Во-вторых, при перегонке камфары в токе инерт-
ного газа имеют место потери продукта с отходящими неконденсированными
газами.
Для очистки камфара может быть также подвергнута возгонке под атмо-
сферным давлением или в вакууме.
Для конденсации паров камфары применяются разнообраз-
ные поверхностные холодильники.
На одном из предприятий, вырабатывавших камфару, в качестве конден-
сатора использовалась вертикальная алюминиевая труба высотой 8 м, диа-
метром 1,25 м, снабженная рубашкой для поверхностного водяного охлажде-
ния. Внутри трубы вращался бронзовый вал, подпятник которого находился
Порядковый номер пробы
Рис. 22. Содержание примесей в камфаре по ходу ее перегонки:
/ — цикланы; 2 — камфен и трициклен; 3 — изофенхон; 4 — экзоизокамфа-
нон-3
на трехконечной крестовине, а верхний конец проходил через сальник
в крышке. На валу было закреплено 40 лопастей из силумина, из которых
каждая вторая была снабжена скребковым ножом из бронзы или хромонике-
левой стали. Вал делал около 10 оборотов в минуту. Камфара с влажностью
около 3% непрерывно ссыпалась на ленту транспортера сушилки, где содер-
жащаяся в ней влага удалялась.
Конденсатор непрерывного действия (рис. 23) состоит из
неподвижного корпуса с рубашкой для охлаждения, в которой
вращается барабан из литой стали с внутренним цилиндром.
Между наружным и внутренним цилиндрами барабана цирку-
лирует холодная вода, входящая в барабан и выходящая из него
через пустотелые цапфы с боковыми отверстиями. На вращаю-
щемся барабане, а также в нижней части корпуса к горловине
крепятся срезывающие ножи. Они установлены таким образом,
что при вращении барабана могут проходить между ножами,
И?
Рис. 23. Конденсатор непрерывного действия для камфары:
/ — корпус; 2— штуцеры для подвода воды; 3 — штуцер для вывода воды; 4—вращающийся барабан; 5 — боковые крышки;
6— пустотелые цапфы барабана; 7 — ножи; 8 — горловина корпуса; 9— двухшнековый выгружатсль; 10 — люк для вы-
грузки камфары; 11— штуцер для ввода паров камфары; 12 — воздушка; 13— зубчатая передача; 14— ценная передача;
15 — редуктор; 16 — электродвигатель
укрепленными на корпусе аппарата. Нижняя часть горловины
соединена с двухшнековым выгружателем.
Пары камфары вводятся в пространство между корпусом и
барабаном и конденсируются на их холодных поверхностях.
Ножи счищают камфару, которая выгружателем-затвором не-
прерывно выводится из аппарата. Барабан, а также выгружа-
тель приводятся в движение электромотором через редуктор и
зубчатую цепную передачу.
Этот конденсатор используется в СССР для конденсации
камфары с 3—4% влаги при перегонке с перегретым водяным
паром и для конденсации ее паров при парофазном процессе.
Эти конденсаторы впервые были применены в Германии и ис-
пользовались для улавливания паров камфары при вакуум-суб-
лимации. В этом случае срезаемая с вращающегося барабана
камфара падает в периодически выгружающийся вакуум-прием-
ник. В СССР конструкция конденсаторов была слегка измене-
на— введен шнек для непрерывной выгрузки камфары.
Отогнанный во время сушки изоборнеола и дегидрирования растворитель,
за исключением хвостовых погонов, перед началом перегонки камфары может
быть возвращен в процесс без очистки. Хвостовые погоны растворителя, кроме
камфары, содержат камфен, моноциклические терпены, продукты их гидрирова-
ния и другие примеси, содержащиеся в борнеоле. По мере накопления эти
погоны перегоняют, выделяя при этом терпены и очищенный растворитель.
Растворитель используют вновь.
Оставшийся в аппарате после перегонки камфары катализатор, вследствие
восстановления его до металлической меди, обладает пирофорными свойствами.
В целях противопожарной безопасности (в период охлаждения аппарата) ка-
тализатор изолируется от атмосферного воздуха подушкой из углекислого газа,
затем его выгружают из аппарата через специальный люк после каждой или
после четырех-пяти операций. Выгрузка катализатора осложняется, если исход-
ные борнеолы были плохо отмыты от солей органических кислот, так как
в этом случае он шлакуется на стенках аппарата. Регенерация катализатора
не сложна. Сначала из него возгонкой удаляют небольшое количество кам-
фары, затем выжигают в токе воздуха органические вещества и растворяют
окиси в серной или азотной кислотах. Далее следует осаждение углекислых
солей меди или никеля содой. Регенерацию катализатора проще производить
в отсутствие носителя.
Дегидрирование борнеола протекает без побочных реакций,
а механические потери при перегонке камфары сравнительно не-
велики, следовательно, выход камфары получается высоким: из
100 массовых частей технических борнеолов получают 92—95
массовых частей камфары в пересчете на безводные продукты.
Если перегонка камфары ведется нерационально, выход иногда
падает до 82—85% от массы борнеолов [26]. Так как превраще-
ние камфена в эфир изоборнеола и гидролиз эфира протекает
почти количественно, то, как правило, выход камфары состав-
ляет 100—103% от массы переработанного камфена, или 89—
92% от теоретического.
Периодический жидкофазный процесс каталитического дегидрирования
борнеолов имеет крупные преимущества по сравнению с процессами окисле-
ния, но вместе с тем не свободен от ряда недостатков. Многостадийность
119
процесса (загрузка, растворение, отгонка воды, дегидрирование, отгонка рас-
творителя, перегонка камфары) препятствует его механизации и автоматиза-
ции и влечет за собой повышенные трудозатраты. Недостатком периодиче-
ского жидкофазного процесса каталитического дегидрирования является
присутствие растворителя. Растворитель не удается полностью отделить от
камфары, и он переходит в первые ее погоны. Эту камфару приходится подвер-
гать дополнительной очистке (отжиму, вторичной перегонке). Немалые непри-
ятности возникают из-за пылевидной структуры катализатора, который при
форсированной перегонке камфары легко уносится вместе с ее парами, что
приводит к браку н загрязнению конденсационной и приемной систем.
5. Технология непрерывного парофазного метода
дегидрирования борнеолов
При каталитическом дегидрировании в паровой фазе пары
борнеолов пропускают через реактор, заполненный катализато-
ром. Процесс осуществляется непрерывно: борнеолы испаря-
ются в испарителе, их пары проходят через слой катализатора,
образующиеся камфару и водород направляют в конденсатор,
откуда осаждающуюся на холодных поверхностях камфару уда-
ляют.
Непрерывный процесс дегидрирования борнеолов в паровой
фазе имеет большие преимущества перед периодическим про-
цессом. Эти преимущества сводятся к следующему:
1) непрерывный процесс лишен многостадийности, харак-
терной для периодического процесса, и поэтому может быть ме-
ханизирован и автоматизирован;
2) улучшаются условия и снижаются затраты труда;
3) непрерывный процесс может быть осуществлен без рас-
творителя, в результате чего отпадают дополнительные опера-
ции, связанные с отгонкой и регенерацией растворителя;
4) катализатор применяют в виде зерен или кусков, что
исключает увлечение его парами камфары.
При дегидрировании борнеолов в паровой фазе реакция за-
канчивается не полным превращением борнеолов в камфару,
а подвижным равновесием между борнеолами, с одной стороны,
камфарой и водородом — с другой. С повышением температуры
равновесие сдвигается в сторону камфары. Судя по анализам
камфары, полученной парофазным методом, можно утверждать,
что при 250—260°С реакция практически количественно сдви-
нута в сторону камфары.
Парофазный процесс дегидрирования борнеолов был изве-
стен уже в начале столетия, изучался многими исследователями
и имеет явные преимущества перед жидкофазным, но промыш-
ленное внедрение его произошло почти на 50 лет позднее внедре-
ния жидкофазного процесса. Это объясняется тем, что долгое
время основное внимание исследователей уделялось изучению
действия на борнеолы разных катализаторов, а такие кардиналь-
ные вопросы, как разработка способов испарения борнеолов, не-
прерывной подачи их паров на катализатор и непрерывного
120
улавливания камфары, оставались нерешенными. Борнеолы —
легко слеживаются, разница между температурами плавления
и кипения борнеолов составляет всего несколько градусов,
в результате чего они испаряются, практически не переходя
в жидкое состояние. При подаче твердых борнеолов в испаритель
возможно увлечение вместе с ними воздуха, который может
создать с водородом взрывоопасную смесь.
В 1958 г. была пущена первая установка по парофазному
дегидрированию борнеола производительностью 1,5 т камфары
в сутки, разработанная коллективом авторов [25, 26]. Эта уста-
новка показала принципиальную возможность использования
метода и эксплуатировалась в течение нескольких лет. Эксплуа-
тация установки позволила вскрыть ряд ее недостатков, проис-
ходящих в основном от неустойчивой температуры реакционного
пространства, что приводило к побочным реакциям, особенно
при дегидрировании изоборнеола. В 1967 г. была введена в дей-
ствие значительно более совершенная установка, разработанная
В. И. Филатовым (рис. 24), где для устранения колебания тем-
пературы при обогреве реактора использована смесь дифенила
и дифенилоксида (ВОТ), а катализатор помещен в 56 трубках
сравнительно небольшого диаметра (75 мм). Эти трубки вваль-
цованы в две плиты, которые делят аппарат на три части.
Непрерывный ввод в аппарат борнеолов осуществлен посред-
ством ленточного элеватора 1 и двух питателей: лопастного 2.
где происходит взрыхление борнеола, и шнекового 3. Последний
является одновременно затвором. Борнеолы попадают в кольце-
вой испаритель 4, отделенный от верхней камеры и нижнего ко-
нуса плитами 5, в которые ввальцованы трубки с катализатором.
Пространство между трубками заполнено парами ВОТ. Борне-
олы испаряются за счет тепла, поступающего через внутреннюю
стенку кольцевого испарителя. Пары борнеолов из кольцевого
испарителя по трубе 6 и змеевику 7 поступают в обогретую ниж-
нюю коническую часть аппарата и оттуда проходят через трубки,
заполненные катализатором в обогретую верхнюю камеру 8.
Отсюда по магистральной трубе 9 пары камфары выводятся на
вращающийся барабан конденсатора непрерывного действия 10.
Снятую ножами с поверхности барабана непрерывного конден-
сатора камфару с помощью шнека-затвора выгружают в ваго-
нетку 12.
Пары ВОТ подают в рубашку верхней камеры, откуда через
отверстия в верхней плите поступают в среднюю часть аппарата
для обогрева трубок катализатора и нагрева кальцевого испари-
теля, а далее выводятся в виде конденсата. Для обогрева ниж-
него конуса и змеевика сделан второй ввод паров ВОТ.
Катализатор (смесь углекислых солей меди и никеля) загру-
жают в виде таблеток. В процессе работы углекислые соли мели
и никеля восстанавливаются до металлов, таблетки уменьша-
ются в объеме и теряют прочность. Были предложены различные
121
Рис. 24. Схема установки для парофазпого дегидрирования борнеолов:
/ — ленточный элеватор; 2 — питатель лопастной; 3 — питатель шнековый; 4 — кольцевой испаритель;
5 — плиты с вальцованными трубками; 6 — труба для вывода паров борнеолов из испарителя; 7— змее-
вик; 8 — верхняя камера; 9 — магистральная труба; 10 — конденсатор непрерывного действия; 11 —
шнековый выгружатель; 12 — вагонетка; /J—предохранительная мембрана
способы сохранения прочности, например путем добавки к угле-
кислым солям гидроокиси кальция [153].
Нормальный срок службы катализатора 40—45 суток. После
этого аппарат подлежит чистке от накопившегося в испарителе
нелетучего остатка, а катализатор заменяется свежим. Расход
катализатора около 0,3—0,35% от полученной камфары.
В техническом изобориеоле содержатся примеси терпеновых углеводоро-
дов, которые в основном гидрируются во время процесса дегидрирования бор-
неолов. Кроме того, некоторое количество изокамфаиа образуется в резуль-
тате дегидратации изобориеола и гидрирования образовавшегося камфена.
При дегидрировании изобориеола в жидкой фазе часть этих примесей уда-
ляется с отгонкой растворителя и в головных погонах камфары. При паро-
фазном дегидрировании все примеси, содержащиеся в изоборнеоле, остаются
в камфаре. Поэтому камфара, полученная из одного н того же сырья паро-
фазным методом, всегда имеет несколько худшее качество, чем камфара, по-
лученная жидкофазным методом.
При парофазиом дегидрировании, несмотря на непрочность зерен катали-
затора, уноса его не наблюдается, вероятно в связи с малой скоростью паров
(8 см/с по сечению не заполненной части трубок). Время контакта — около
20 с.
ГЛАВА VIII. ПОЛУЧЕНИЕ КАМФАРЫ
ОКИСЛЕНИЕМ КАМФЕНА
В начале текущего столетия камфару получали окислением
камфена хромовой кислотой, но со временем этот способ был
заменен синтезом камфары из камфена через изоборнеол.
Поскольку окисление камфена достаточно долго и широко
осуществлялось в промышленности, его следует рассмотреть.
1. Получение камфары окислением камфена
хромовой кислотой
Окисление камфена производится водными растворами дву-
хромовокислого натрия, подкисленными серной кислотой, при
90°С, т. е. при температуре более высокой, чем температура
плавления камфена. В начале процесса реакция идет на поверх-
ности раздела двух жидких фаз — расплавленного камфена и
водного раствора окислителя. По мере образования камфары
температура плавления фазы, состоящей из камфена и образо-
вавшейся из него камфары, повышается и во второй половине
реакции эта фаза превращается в твердую. Поэтому в дальней-
шем реакция протекает на поверхности раздела жидкой и твер-
дой фаз.
Образование камфары при окислении камфена хромовой кис-
лотой представляется на первый взгляд неожиданным, так как,
исходя из аналогии с окислением других непредельных углево-
дородов и учитывая строение камфена (I), при его окислении
123
следовало бы ожидать образование не камфары, а, в зависимо-
сти от глубины окисления, камфенгликоля (II), камфениловой
кислоты (III) и камфенилона (IV):
I
III
IV
Образование камфары объясняют тем, что камфен, взаимо-
действуя с хромовой кислотой, образует изоборнильный эфир
хромовой кислоты (V). Реакция катализируется протонами или
ионами гидроксония (НзО+), присутствующими в растворе,
и протекает по той же схеме, что и реакция превращения кам-
фена в уксусный и муравьиный эфиры изоборнеола (гл. VI.1).
Изоборнильный эфир хромовой кислоты (V) в кислой водной
среде претерпевает внутримолекулярное окисление — восстанов-
ление, разлагаясь на камфару (VI) и соединения четырехва-
лентного хрома [189]. Последние неустойчивы и превращаются
частично в хромовую кислоту, частично в соединения трехвалент-
ного хрома. Так как скорость образования изоборнильного
эфира хромовой кислоты значительно превышает скорость окис-
ления камфена по двойной связи, камфара образуется в каче-
стве основного продукта реакции:
ЗНСгОзН- 3H3O+-|-3H2SO4 —Н2 СгО4+- Сг2 (SO4 )3 + 8Н2О
Образование качества промежуточного продукта изоборниль-
ного эфира хромовой кислоты было доказано. [189].
Окисление камфеиа в камфару сопровождается рядом побочных реакций.
Эти реакции можно разделить на три группы.
К первой группе относятся реакции окисления камфена по двойной связи.
К продуктам этой группы реакций, помимо названных камфенгликоля (II),
камфениловой кислоты (Ill) и камфенилона (IV), можно отнести окись кам-
феиа (VII) и камфенилаиовый альдегид (VIII). Последний образуется в ре-
зультате изомеризации окиси камфеиа (с. 125).
Ко второй группе относятся вторичные реакции окисления камфары,
образовавшейся на первой стадии процесса. Эти реакции, ведущие к образо-
ванию камфарной, камфароновон и изокамфароновой кислот, п-дикетокамфаиа
и 6-кетокамфары, уже были рассмотрены (гл. VI 1.2).
124
(VII)
(V III)
К третьей группе относятся реакции сульфатирования терпенов за счет
присутствующей в водном растворе серной кислоты. Эти реакции (хотя в них
вступает и сравнительно небольшое количество вещества) очень вредны, так
как образующиеся водорастворимые сульфопродукты вызывают серьезные
затруднения при последующей регенерации хромовой кислоты из восстанов-
ленных растворов хромовых солей.
Как видно из уравнений реакций, для окисления 1 моля
камфена в камфару необходимо затратить 7з моля двухромово-
кислого натрия, или на 100 массовых частей камфена соответст-
венно 64 массовые части двухромовокислого натрия. Фактиче-
ский же расход двухромовокислого натрия значительно больше
и составляет от 150 до 250% от теоретически необходимого ко-
личества. Такой расход двухромовокислого натрия является ре-
зультатом нескольких причин. Во-первых, для полного окисле-
ния камфена необходимо вводить в реакцию некоторый избыток
окислителя, который остается невосстановленным до самого
конца реакции, во-вторых, часть окислителя расходуется на по-
бочные реакции. Расход двухромовокислого натрия на них до-
вольно значителен, так как эти реакции связаны с более глубо-
ким окислением, чем окисление камфена в камфару. Так, если
на окисление 1 моля камфена до камфары затрачивается
1 г-атом кислорода, то на окисление 1 моля камфары до камфар-
ной кислоты затрачивается 3 г-атома кислорода, а на окисление
1 моля камфары до камфароновой кислоты 8 г-атомов кислорода.
Особенно сильно возрастает расход окислителя, если в камфене
содержится примесь других терпенов. Последние могут окис-
ляться вплоть до простейших органических кислот и углекислого
газа.
При наличии двух фаз, из которых одна твердая, трудно до-
биться полного окисления камфена до камфары, так как кам-
фен, находящийся внутри твердых комков, теряет контакт
с окислителем. Поэтому обычно камфара, полученная окисле-
нием камфена, содержит 2—3% неокислившегося камфена, что
снижает ее качество. Чтобы устранить эти недостатки, к кам-
фену иногда добавляют парафиновое масло или бензол. Но и
при работе в присутствии растворителя диффузионное торможе-
ние сохраняется, особенно в конце- реакции, поскольку окисле-
ние проходит значительно быстрее, чем диффузия камфена на
поверхность раздела фаз. Так как в конце реакции почти вся по-
верхность раздела фаз занята камфарой, то в это время созда-
ются благоприятные условия для ее окисления. Многие из
125
побочных продуктов растворимы в воде. Переход этих продуктов
в раствор создает благоприятные условия для их дальнейшего
окисления, что влечет за собой большой перерасход двухромово-
кислого натрия.
Чтобы уменьшить диффузионное торможение реакции, уско-
рить окисление камфена и замедлить образование вторичных по-
бочных продуктов, предлагалось вести процесс в присутствии
2,5% нафтеновых сульфокислот, которые играют роль эмульга-
тора [27]. Продолжительность реакции при этом снижается в два
раза при одновременном увеличении выхода камфары с 80 до
88%. К сожалению, в работе не указано о последующей регене-
рации хромовых растворов. В то же время не исключено, что
наличие в этих растворах растворенных сульфокислот может
вызвать значительные затруднения при электрохимическом окис-
лении хрома.
На практике окисление производили при 90°С в вертикальных или гори-
зонтальных аппаратах, выложенных диабазовой плиткой и снабженных освин-
цованными мешалками. Чтобы уменьшить побочные реакции, применяли раз-
бавленные растворы окислителя и вводили в процесс минимальное количество
серной кислоты. По Аскану [189], 100 кг камфеиа с температурой кристалли-
зации около 40°С окисляли раствором 123 кг двухромовокислого натрия в пе-
ресчете на безводный (что составляет 190% от теоретически потребного коли-
чества) в 2194 л воды, подкисленной 200 кг серной кислоты. Количество
серной кислоты употребляли, исходя из расчета получения 2% свободной сер-
ной кислоты в растворе сернокислого натрия и трехвалеитиого хрома в слу-
чае полного восстановления хромовой кислоты. Так как восстановление хро-
мовой кислоты было неполное, кислотность раствора получалась несколько
большей. Камфару отсасывали на нутче, промывали водой или раствором
щелочи и перегоняли. Выход сырой неперегнанной камфары в пересчете на
безводную составлял 88—90% от теоретического количества. Последующая
перегонка сопровождалась дополнительными потерями. Перегнанная кам-
фара имела температуру кристаллизации 152—153°С и содержала 2—3%
камфена. Низкая температура кристаллизации в основном зависела от низ-
кого качества исходного камфеиа, который получали борнилхлоридным спо-
собом. ‘
Окисление камфена хромовой кислотой в довоенный период осуществля-
лось на одном из заводов Советского Союза. Режим окисления позволял
получать камфару с температурой кристаллизации 167—169°С, с незначитель-
ным содержанием камфена за счет более жесткого режима окисления, при
котором присутствующие в камфене феихеиы окисляются до углекислого газа
и водорастворимых кислот. При этом частично окислялась и камфара, в ре-
зультате чего ее выход был значительно ниже, чем в условиях, описанных
Асканом.
Для окисления 100 кг камфена с температурой кристаллизации 35°С
брали 152 кг двухромовокислого натрия (в пересчете на безводный, что со-
ставляет 230% от теоретически необходимого количества) и 308 кг крепкой
серной кислоты, растворенных в 2190 кг воды. Реакцию вели при 90°С, при-
чем окислитель вводили не сразу, а за три-четыре приема. После того как
химическим анализом устанавливали, что введенная в реакцию хромовая кис-
лота использована, раствор солей хрома выводили из аппарата и вводили
свежий окислитель. Продолжительность окисления достигала 3 суток. По
окончании реакции камфару отсасывали на нутче, дважды обрабатывали
в смесителе 30%-ным раствором едкого натра для отделения образовав-
шихся при окислении органических кислот, перегоняли с водным паром и
отжимали иа центрифуге.
126
2. Использование восстановленных
хромовых растворов
При получении камфары окислением камфена хромовой кис-
лотой на каждую тонну камфары получают в качестве отхода
25—35 т разбавленного раствора сернокислых солей трехвалент-
ного хрома, содержащего некоторое количество невосстановлен-
ной хромовой кислоты. Рациональное использование этого от-
хода было необходимо не только из экономических, но и из
санитарных соображений.
Раствор имеет примерно следующий состав: соединения
хрома в пересчете на Сг2 (804)3 7—9%; Na2SO4 2,5—3,3; H2SO4
2-5%.
Возможно несколько направлений использования восстанов-
ленных хромовых растворов [182]:
1) переработка в хромовые квасцы, которые применяются
в большом количестве для дубления кожи и электролитического
хромирования;
2) переработка в хромовокислые и двухромовокислые соли
путем окислительного обжига, осажденного из растворов
гидрата окиси хрома;
3) электрохимическое окисление растворов с целью превра-
щения солей трехвалентного хрома в хромовую кислоту.
Получение хромовых квасцов из восстановленных хромовых растворов
встречает известные затруднения. Во-первых, растворы мало концентриро-
ваны; среднее содержание Cr2(SO4)3 (8 г в 100 г раствора) соответствует
содержанию 20 г хромовокалиевых квасцов KjSOcC^SO^s^HjO в этом же
количестве раствора, что лежит в пределах их растворимости. Во-вторых,
сернокислый хром в восстановленных хромовых растворах находится в виде
зеленой формы, из которой хромовые квасцы некристаллизуются *. Поэтому
для получения хромовых квасцов необходимо сначала восстановить содержа-
щуюся в небольшом количестве в растворе хромовую кислоту до сернокислой
соли трехвалентного хрома (для чего может быть использован сернистый газ),
затем осадить трехвалентиый хром содой в виде гидрата окиси, осадок
отфильтровать и растворить в серной кислоте. Следует отметить, что гидрат
окиси хрома выпадает в виде осадка, который трудно отфильтровывается.
По-видимому, в связи с изложенными трудностями получение хромовых квас-
цов из восстановленных хромовых растворов не нашло практического приме-
нения в технике.
На первый взгляд кажется возможным использовать в качестве дубителя
восстановленный раствор хромовых солей после нейтрализации содержащейся
* Сернокислый хром в кристаллическом состоянии и в растворах изве-
стей в двух формах (фиолетовая и зеленая), которые принципиально раз-
личны по своему строению. Фиолетовая форма сернокислого хрома раство-
ряется легче зеленой в воде (30 г Сг2 (ЗО4)з в 100 г раствора). Она полу-
чается путем растворения гидрата окиси хрома в серной кислоте или при
восстановлении хромовой кислоты при комнатной температуре. При нагревании
до 40°С фиолетовая форма переходит в зеленую. В случае прибавления к рас-
твору фиолетовой формы сернокислого хрома раствора сернокислого калия
выпадают хромовые квасцы. Учитывая высокую растворимость фиолетовой
формы сернокислого хрома в воде и сравнительно низкую растворимость
хромовых квасцов, получают их высокий выход при осаждении [297].
127
в нем серной кислоты и некоторого упаривания раствора. Однако в связи
с перевозкой растворов это неудобно.
Переработка осажденного из растворов гидрата окиси хрома в соли хро-
мовой кислоты путем окислительного обжига в присутствии соды н извести
(для рыхления массы) также не получила практического применения, хотя
этим способом н осуществляется почти все мировое производство солей хро-
мовом кислоты нз тонкоизмельченного хромистого железняка. По-видимому,
проведение этих операций оказалось экономически невыгодным.
В промышленности осуществляется электрохимическое окис-
ление трехвалентного хрома, содержащегося в отходах от окис-
ления органических веществ. Однако и этот процесс имеет свои
отрицательные стороны.
Главное затруднение связано с образованием водораствори-
мых побочных продуктов окисления камфена. Летучие продукты,
например уксусная кислота, удаляются при выпаривании
раствора. Нелетучие, как, например, сульфокислоты терпенов,
непрерывно накапливаются в растворах. Вследствие накопления
этих продуктов при электрохимическом окислении хромовых
растворов наблюдается сначала образование пенообразных пле-
нок на поверхности свинцовых электродов, а затем их разру-
шение.
По данным Аскана, себестоимость камфары, полученной
окислением камфена хромовой кислотой, была выше себестои-
мости камфары, полученной дегидрированием изоборнеола [189].
Это послужило причиной отказа от окисления камфена хром-
пиком, тем более, что в санитарном отношении этот процесс
оставляет желать много лучшего.
3. Окисление камфена другими окислителями
Процесс окисления камфена — не обычный окислительный
процесс, так как хромовая кислота действует на камфен
не только как окислитель, но и как гидратирующий агент. Поэ-
тому образование камфары при окислении камфена характерно
именно для случая применения хромовой кислоты и не проис-
ходит при окислении камфена другими окислителями.
ГЛАВА IX. РЕКТИФИКАЦИЯ СЫРЬЯ
И ПОЛУПРОДУКТОВ
1. Общие вопросы ректификации терпенов
Ректификацию терпенов вследствие их термической неустой-
чивости осуществляют в вакууме. Чтобы разобраться, в какой
мере это обосновано, рассмотрим в качестве примера термиче-
скую устойчивость а- и р-пиненов и возможные потери этих
терпенов вследствие термических превращений при ректифика-
ции скипидара под атмосферным давлением;
128
II
—> СНз-С=СН-СН^-СН„-С—сн=сн„
I 2 2 II 2
СН3 СН2
VIII
При термических превращениях а-пинена (I) образуются
аллооцимен (II) и дипентен (III), а в результате вторичных
реакций циклизации и полимеризации аллооцимена — а-(IV),
0-(V), у (VI) пиронены и небольшое количество димера алло-
оцимена [1, 180, 230, 237]. При термических превращениях
р-пинена (VII) образуется мирцен (VIII) [236].
Образование первичных продуктов аллооцимена, дипентена
и мирцена происходит по уравнению реакций первого порядка
[231, 257]. В табл. 27 показано время, которое необходимо для
Таблица 27
Данные о термических превращениях а- и р-пиненов в зависимости
от температуры и продолжительности нагревания
Температура, °C Константа ско- рости реакции, С'1 Количество вступивших в реакцию, % Время, ч
а-пинена р-пинена
164,5 1 10-7 1 27
5 146
174,5 2,9-10-7 1 — 10
5 —- 52
184,5 8,2-10-7 1 3,4
5 18,0
204,5 55-10-7 1 0,5
5 2,7
204,5 35-10-7 — 1 0,8
— 5 4,3
9 Заказ № 43
129
термического превращения одинаковых долевых количеств а-
и р-нинена (%) при 164,5, 174,5, 184,5 и 204,5°С. Продолжи-
тельность превращений а-пинена и р-пинена при 184,5 и 204,5°С
вычислена из опубликованных в литературе констант скоростей
реакции по уравнению реакций первого порядка
т = * • 2 31т ЮР (11)
k • 3600 ’ s 100 - X ' 1 '
где т — время, ч;
k— константа скорости реакции, с-1;
2,3 — множитель для перехода от натуральных логарифмов к десятичным;
х—количество пинена, вступившего в реакцию за время т, %.
Константы скорости термической изомеризации а-пинена при 164,5 и
174,5°С, приведенные в табл. 27, получены экстраполяцией.
В состав скипидаров входят а-пинен (т. кип. 156°С), р-пинен
(т. кип. 166°С), Д3-карен (т. кип. 170°С) и моноциклические
терпены (т. кип. 176°С). В соответствии с этим температура
в кубах периодически действующих аппаратов должна
колебаться между 160°С в начале гонки и 170—175°С в конце
гонки, когда содержание пинена в кубе уже невелико. Как
видно из табл. 27, при условии многосуточной перегонки это
может привести к потере заметного количества пиненов. Наобо-
рот, в условиях работы непрерывно действующей колонны, учи-
тывая пребывание скипидара в колонне не более 12 ч, потери
пиненов от термических процессов не могут превысить 1%.
Основная масса пинена находится в укрепляющей части ко-
лонны, где температура не превышает 160°С отчего потери из-за
термических процессов уменьшаются.
Термическая устойчивость другого основного компонента
скипидаров Д3-карена превосходит термическую устойчивость
а- и р-пиненов [13].
Термическая устойчивость камфена и трициклена также зна-
чительно превосходит термическую устойчивость пиненов,
поэтому и ректификацию продуктов изомеризации пиненов на ко-
лоннах непрерывного действия можно было бы проводить при
атмосферном давлении без опасения вызвать значительные по-
тери из-за термических превращений камфена и трициклена.
Препятствием в этом случае может служить только наблюдаю-
щаяся при нагревании полимеризация терпинолена [317], кото-
рый содержится в изомеризатах (~6°/о). Термическая неустой-
чивость терпинолена при температуре ректификации количест-
венно не изучена.
Из вышеизложенного следует, что многие терпены довольно
устойчивы по отношению к нагреванию, а поэтому во многих
случаях термическая неустойчивость терпенов не может служить
препятствием для ректификации их смесей под атмосферным
давлением (если последняя производится на колоннах непре-
рывного действия).
130
Рассматривать вопрос только с точки зрения термической
устойчивости терпенов нельзя, нужно считаться и с возмож-
ностью каталитических влияний стенок аппаратуры и насадки
на разгоняемые материалы. Сами по себе металлы, из которых
построена аппаратура для ректификации (сталь, медь и т. д.),
не являются катализаторами изомеризации и полимеризации
терпенов. Однако терпены являются прекрасными переносчи-
ками кислорода, поэтому образование окислов на металличе-
ских поверхностях, особенно при высокой температуре ректифи-
кации, вполне возможно, а за этим могут последовать и ката-
литические превращения терпенов.
В качестве примера может служить каталитическое разложение изобор-
нилформиата на камфен и муравьиную кислоту под влиянием окиси олова,
образовавшейся на поверхности медных луженых колец, служивших насад-
кой в ректификационной колонне на одном из заводов. Это разложение,
очень незначительное в первый период эксплуатации колонны, по мере обра-
зования окиси на поверхности насадки достигло таких размеров, что ректи-
фикационный агрегат был остановлен для замены насадки.
Используемые в качестве насадки керамиковые кольца
могут также служить катализаторами таких реакций, как изо-
меризация пиненов и разложение изоборнильных эфиров.
С точки зрения исключения возможных каталитических влияний
ректификация в вакууме (поскольку она проводится при более
низких температурах) несомненно имеет значительные преиму-
щества перед ректификацией под атмосферным давлением.
В то же время вакуум-ректификация имеет ряд существен-
ных недостатков. К недостаткам следует отнести в первую
очередь прососы воздуха. Прососы воздуха влияют на скорость
паров в колонне, что ухудшает погоноразделение, и приводит
к существенным потерям терпенов, а также вызывает окисли-
тельные процессы, которые сопровождаются отложением в рек-
тификационных колоннах твердых продуктов окисления.
В качестве примера можно указать на образование в недостаточно гер-
метизированных вакуум-ректификационных колонках, в которых разгоняется
скипидар Pinus Silvestris L., твердого полимерного вещества, по-видимому,
лактонного характера, нерастворимого в органических растворителях н отве-
чающего формуле (CioHu02)n.
В состав полимерного вещества входит около 2—3% минеральных ве-
ществ: железа и меди. Это говорит об образовании на первой стадии про-
цесса органических кислот, вызывающих коррозию аппаратуры. Возможно,
при каталитическом действии солей железа и меди в дальнейшем происходит
полимеризация продуктов окисления. Накопление полимерного вещества в ко-
лонне приводит к закупорке тарелок, а следовательно, и к необходимости их
механической чистки, что связано с длительной остановкой аппарата. По-
этому при ректификации терпенов под вакуумом нужно уделять исключитель-
ное внимание герметизации установок.
В связи с изложенным большое значение имеет правильный выбор мате-
риала для прокладок, применяемых в ректификационной аппаратуре.
В ректификационных колоннах непрерывного действия про-
дукт нагревается менее продолжительно, чем при переодическом
9* 131
процессе. Кроме того, ректификация ведется при постоянном
точно установленном режиме, который может быть автомати-
зирован.
Соображения о недостаточно постоянном составе исходных
продуктов, выдвигаемые иногда против применения непрерывно
действующих колонн, работающих на постоянном режиме,
в большинстве случаев неосновательны, так как и сырье и боль-
шинство полупродуктов синтеза камфары (продукты катали-
тической изомеризации пинена, продукты формилирования и
ацетилирования камфена) имеют достаточно постоянный
состав.
Колебание состава в довольно широких пределах можно
наблюдать только у исходного сырья — скипидара, но, приме-
няя скипидары одного происхождения, можно использовать
колонны непрерывного действия [117].
Как известно, ректификационные колонны бывают Двух типов: тарельча-
тые и насадочные. С равным успехом могут применяться те и другие, но та-
рельчатые колонны применяются в практике ректификации терпенов чаще.
К положительным сторонам тарельчатых колонн следует отнести то, что
в них в меньшей степени может проявляться каталитическое влияние твер-
дых поверхностей, поскольку эти поверхности малы.
Для создания вакуума в ректификационных агрегатах могут применяться
любые насосы. Однако применяя поршневые насосы, удобнее улавливать тер-
пены из выхлопных газов.
Скипидар и подвергаемые ректификации полупродукты кам-
фарного производства представляют собой многокомпонентные
смеси терпенов. В задачу ректификации обычно входит не пол-
ное разделение смеси на компоненты, а выделение из нее одного
или двух технически чистых веществ, например а- и р-пиненов
из скипидаров или группы веществ; смеси а- и р-пиненов из
скипидаров или смеси камфена; трициклена и фенхенов (ХЭВ)
из изомеризатов. При расчетах ректификационных колонн раз-
гоняемые сложные смеси условно рассматривают как бинарные
смеси выделяемого терпена с ближайшим к нему по темпера-
туре кипения компонентом при условии, если этот компонент
содержится в достаточно большом количестве в разгоняемой
смеси. Так, например, при расчете колонн для ректификации
скипидаров советского производства из сосны Pious silvestris
их рассматривают как бинарные смеси а-пинена и Д3-карена.
В этих скипидарах содержатся также в небольшом количестве
камфен, р-пинен и мирцен, температуры кипения которых лежат
между температурами кипения а-пинена и Д3-карена, однако
они содержатся в скипидарах в незначительном количестве,
поэтому расчет ведется по основным компонентам. Компо-
ненты, температура кипения которых лежит между темпера-
турами кипения основных компонентов, частично переходят
в отгон в качестве примеси к выделяемому веществу, а частично
переходят в остаток от перегонки.
132
2. Выделение а- и р-пиненов из скипидаров
Ректификация может ставить своей задачей раздельное вы-
деление а- и р-пиненов из скипидаров или выделение смеси
обоих изомеров.
Так как цена на р-пинен на международных рынках выше,
чем на а-пинен, и он находит более квалифицированное приме-
нение (см. гл. II.2), раздельное выделение пиненов всегда целе-
сообразно, когда содержание р-пинена в скипидаре достаточно
высоко, как это имеет место в американских живичных и суль-
фатных скипидарах. Для этой цели могут быть использованы
двухколонные аппараты непрерывного действия. Первый рас-
считывается на бинарную смесь а-пинен—р-пинен, второй на
смесь р-пинена с более высококипящим компонентом, обычно
лимоненом. Количество тарелок в первой колонне дости-
гает 70 [263].
В используемых в Советском Союзе скипидарах сосны Pinus
silvestris L. содержание р-пинена не превышает 7—10% от со-
держания суммы пиненов. Поэтому их ректификацию осущест-
вляют на одноколонном аппарате, который рассчитывается на
ректификацию бинарной смеси а-пинен—А3-карен. При этом
отбирают а-пинен с примесью нескольких процентов р-пинена.
Часть содержащегося в скипидаре р-пинена остается в кубовом
остатке с А3-кареном и другими высококипящими компонентами
скипидара и в случае надобности его выделяют оттуда.
Ректификация скипидаров была подробно изучена за последнее время
[6—8]. Изучались закономерности процесса с целью установления зависимости
коэффициентов полезного действия (КПД) ректификационных установок
от режимных условий процесса: скорости пара и высоты слоя жидкости на
тарелке.
Было показано, что при небольших скоростях парового потока до 0,6—
0,8 м/с в свободном сечении колонны КПД тарелки падает с увеличением
скорости паров, а увеличение высоты жидкости на тарелке сдерживает это
падение; при значительных скоростях парового потока от 1,8 и более м/с
в свободном сечении колонны и турбулентном режиме КПД тарелки растет
с увеличением скорости паров, а высота слоя жидкости на тарелке сказы-
вается отрицательно. При переходном режиме при скоростях парового потока
0,8—1,8 м/с в свободном сечении колонны, КПД практически не зависит от
изменения скорости паров и высоты жидкости на тарелке и составляет
0,55-0,65.
На опытно-промышленной колонне с 37 колпачковыми тарелками диамет-
ром 300 мм изучено распределение концентрации компонентов живичных ски-
пидаров при ректификации при различных флегмовых числах и нагрузках
колонны по пару. Показано, что для получения технического пинена, свобод-
ного от Д3-карена, укрепляющая часть колонны должна иметь около 30 таре-
лок. Сравнение распределения компонентов скипидара по высоте ректификаци-
онной колонны с распределением а-пинена и Д3-карена при ректификации их
бинарной смеси показало, что приравнение скипидара к бинарной смеси а-пи-
нен—Д3-карен при расчете колонны оправдано.
Первая непрерывно действующая установка для ректификации скипидара
в Советском Союзе была разработана н введена в действие в 1952 г. [117].
Колонна имела 22 тарелки, в том числе 10 в укрепляющей части. Аппарат
позволял выделять из скипидара технический пинен, содержащий 95% суммы
133
а- и Р-пиненов. В остатке от ректификации оставалось 5% пиненов, что
составляло 2°/о от их общего количества [144].
Скипидары обычно имеют незначительную кислотность от
присутствия в них органических кислот. Поэтому целесообразно,
во избежание коррозии аппаратуры, добавлять в кубы периоди-
чески действующих колонн небольшое количество соды или ед-
кого натра, а скипидар, подлежащий ректификации на непре-
рывно действующих колонных аппаратах, подвергать предвари-
тельной нейтрализации.
В скипидарах, находящихся продолжительное время на воз-
духе (особенно на солнечном свету), накапливаются продукты
окисления, в том числе перекиси. При перегонке таких скипида-
ров могут происходить сильные взрывы. Эти взрывы неодно-
кратно наблюдались в лабораторной практике. Перекисные со-
единения, вызывающие взрывы, могут быть удалены из засмолив-
шегося скипидара промывкой едким натром. В заводской
практике взрывы при перегонке скипидара никогда не наблю-
дались. Это связано с тем, что возможность накопления пере-
кисных соединений сильно понижается при хранении скипидара
в больших емкостях, где поверхность соприкосновения с воз-
духом по сравнению с общей массой вещества мала, а влияние
солнечной радиации исключено.
3. Выделение камфена и трициклена из продуктов
каталитической изомеризации пинена
Состав продуктов каталитической изомеризации пинена в ко-
личественном отношении обычно колеблется в довольно широ-
ких пределах в зависимости от применяемых катализаторов и
условий реакции. Однако в качественном отношении он до-
вольно постоянен. В соответствии с этим принципиальные во-
просы ректификации продуктов изомеризации пинена могут
рассматриваться вне зависимости от того, какие катализаторы
были применены для его изомеризации.
Из данных табл. 28 видно, что камфен может быть доста-
точно хорошо отделен от моноциклических терпенов, так как
разность температур кипения между этими компонентами со-
ставляет 16°, но отделение камфена от а- и [S-фенхенов практи-
чески невозможно, так как разность в температурах кипения
составляет всего 1—2°С. Поэтому а- и р-фенхены являются не-
избежными примесями в техническом камфене.
В литературе обычно указывают на отделение легкокипящих
фенхенов: б-, у- и циклофенхена от камфена [273]. Однако
в присутствии значительного количества трициклена, как это
бывает в изомеризатах, полученных с титановым катализато-
ром, эта задача не только технически сложна (разница в т. кип.
трициклена и у-фенхена 5°С), но и не дает существенного эф-
фекта, поскольку содержание легких фенхенов (6-, у- и цикло-
134
Таблица 28
Состав изомеризатов, получаемых из а-пииеиа с титановым катализатором
и температура кипения содержащихся в нем компонентов
Компонент Содержа- ние, % Т. кип. при 760 мм рт. ст., СС Компонент Содержа- ние, % Т. кип. при 760 мм рт. ст., СС
б-Фенхен 0,02-0,05 139 а-Терпинен 3-6 175
Циклофенхен 0,3-0,4 143 Дипентен 10 176
у-Фенхен 0,4—0,5 148 у-Терпинен 3-5 183
Борнилен ~1 150* Терпинолен 5-6 186
Трициклен 10-17 153 2,4(8) -п-Мента- 2-4 188-189
Р-Фенхен 1-1,5 157 диен
а-Фенхен 1-1,5 158 Полимеры 1-2 >300
Камфен 55-65 159
* Для борннлена прежде принимали т. кип. 146°С.
фенхена) составляет лишь около 20—25% от общей суммы
фенхенов, содержащихся в изомеризате. Поэтому рациональнее
освобождаться от фенхенов или их производных на других ста-
диях синтеза.
О раздельном получении камфена н трициклена в литературе ничего не
упоминается, хотя есть основания считать, что такое разделение могло бы
быть полезным при последующем превращении камфена в эфиры нзоборнеола
(гл. VI.1), тем более, что при изомеризации пинена в присутствии некоторых
катализаторов трициклен образуется в значительном количестве.
Из изложенного следует, что ректификация продуктов ката-
литической изомеризации пинена на титановом катализаторе
может производиться на одно-, двух- и трехколонном аппаратах
непрерывного действия.
В первом случае отгоняется смесь камфена, трициклена
и фенхенов с температурой кристаллизации около 45°С (техни-
ческий камфен), а в остатке остаются моноциклические тер-
пены и полимеры. На двухколонном аппарате можно раздельно
получить камфен и трициклен, а на трехколонном — отогнать
также и моноциклические терпены от полимеров. В последнем
случае третья колонна должна работать лишь на исчерпывание.
Если применяются катализаторы, ведущие к образованию значительного
количества б- и циклофенхенов, например MgSO4H2O [323], может оказаться
целесообразным использовать четырехколонный ректификационный агрегат.
При выделении камфена и трициклена следует иметь
в виду, что при обычной температуре они — твердые вещества
(температура кристаллизации чистого камфена составляет 48,5°,
а чистого трициклена 68°С). В соответствии с этим необходимо
поддерживать в дефлегматорах и холодильниках температуру,
135
при которой не происходит кристаллизация этих терпенов,
а также подогревать все приемники и трубопроводы, по кото-
рым проходят камфен и трициклен. Высокая температура в хо-
лодильниках и приемниках заставляет поддерживать в системе,
во избежание потерь, давление не менее 100—150 мм рт. ст.
При расчете колонн для совместной отгонки камфена и три-
циклена продукты изомеризации можно условно рассматривать
как бинарную смесь камфена и лимонена, упругости паров ко-
торых известны. Данные о зависимости упругости паров три-
циклена от давления в литературе отсутствуют, что затрудняет
расчет колонны для раздельного выделения трициклена.
Если каталитической изомеризации подвергать не смесь а- и Р-пииенов,
а неразогнанный скипидар, то в продуктах изомеризации содержатся Л3- и Д4-
карены [132]. Температура кипения Д4-карена составляет 167,5—168°С при
760 мм рт. ст. В результате разрыв в температурах кипения камфена и выше-
кипящего компонента сокращается с 18 до 9°С и, следовательно, создаются
значительно менее благоприятные условия разгонки, чем при переработке
продуктов каталитической изомеризации чистых а- и Р-пииенов.
4. Выделение изоборнильных эфиров из продуктов
формилирования и ацетилирования камфена
Ректификация продуктов формилирования и ацетилирования
камфена не является неизбежным процессом при синтезе кам-
фары. Эту операцию производят лишь тогда, когда сырой эфир,
полученный после ацетилирования камфена, содержит значи-
тельное количество непрореагировавшего камфена и других
примесей. В результате ректификации получают изоборнильный
эфир, содержащий 98% суммы изоборнильного эфира и изобор-
неола и фракцию непрореагировавшего камфена в смеси
с моноциклическими терпенами, которую или присоединяют
к изомеризату, или непосредственно направляют на формилиро-
вание или ацетилирование.
При ректификации уксусных и, особенно, муравьиных эфи-
ров изоборнеола приходится считаться с их сравнительно лег-
ким разложением на камфен и органическую кислоту при
нагревании. Однако разложение связано не с термической
нестойкостью эфиров, а обусловливается каталитическими влия-
ниями.
Разложение эфиров в первую очередь катализируется минеральными кис-
лотами. Поэтому эфиры, полученные формнлированием или ацетилированием
камфена, перед ректификацией тщательно нейтрализуют раствором соды или
едкого натра в отстойниках с мешалками. За нейтрализацией следует отстаи-
вание. Во время нейтрализации едким натром муравьиный эфир нзоборнеола
может частично омылиться, что не имеет особого значения.
Для предотвращения каталитического разложения изоборнильных эфи-
ров во время ректификации важно правильно подбирать материалы для изго-
товления аппаратуры и насадки колонн. Применение насадки из керамико-
вых колец совершенно недопустимо, так же как и применение обычной стали
и луженой аппаратуры. Наиболее подходящий материал для изготовления
аппаратуры — нержавеющая сталь и медь.
136
Чтобы уменьшить возможность разложения эфиров, ректификацию ведут
при глубоком вакууме в аппаратуре непрерывного действия.
5. Исходные данные для расчета
ректификационных колонн, предназначенных
для разделения терпенов
Для расчета ректификационных колонн необходимо знать
количественную зависимость между составом находящихся
в равновесии жидкой и паровой фаз для ряда бинарных смесей
терпенов и их производных.
В литературе имеются данные по равновесиям в системах
а-пинен—р-пинен [203, 330], а-пинен—А3-карен, а-пинен—ли-
монен (дипентен) [204, 126] а-пинен—терпинолен [204], кам-
фен—А3-карен, камфен—дипентен, камфен—муравьиный эфир
изоборнеола, камфен—уксусный эфир изоборнеола, дипентен—
муравьиный эфир изоборнеола и дипентен—уксусный эфир
изоборнеола [126]. К сожалению, все эти данные, за исключе-
нием данных по равновесию а-пинен—р-пинен, как показывает
их проверка с помощью методов, основанных на термодинами-
ческих закономерностях [83], не верны или содержат настолько
большие систематические погрешности, что использование их
может привести к серьезным ошибкам при расчете ректифика-
ционных колонн. Эти погрешности вызваны в первую очередь
наличием дефлегмации в использованных приборах, что привело
к получению значительно более выпуклых кривых, чем действи-
тельные.
Равновесие жидкость—пар в бинарном растворе а-пинен—
р-пинен изучено значительно точнее с применением исходных
продуктов высокой чистоты [203, 330]. Эти исследования позво-
ляют сделать некоторые выводы, которые могут быть распро-
странены и на другие бинарные растворы терпеновых угле-
водородов (табл. 29).
Приводимые в таблицах (см. также табл. 37, 38) коэффициенты активно-
сти у, и у2 находят из уравнений:
...У1р ... У2р
71 XiPi ’ 72 Х2Р2 ’
(12)
где Xi, xs — концентрации первого и второго компонентов в жидкости;
Уь Уг — концентрации первого и второго компонентов в парах;
Pi, Р2 — давления паров чистых компонентов прн температуре системы;
Р — общее давление паров в системе.
У идеальных систем коэффициенты активности равны еди-
нице, при положительных отклонениях от закона Рауля коэффи-
циенты активности больше единицы. При отрицательных
меньше единицы. В соответствии с этим можно считать, что
бинарная смесь а-пинен—р-пинен практически ведет себя как
идеальная.
137
Таблица 29
Температура кипения, состав пара и коэффициенты активности для бинарных
растворов а-пинен—р-пинеи по Туккеру и Гокинс [330] *
Давление 100 мм рт. ст. *♦ Давление 750 мм рт. ст. **
Содержание а-пинена, % Температура, °C _ i Коэффициенты активности Содержание а-пинена, % Температура, °C Коэффициенты активности
жид- кость пар 11 12 жид- кость пар 71 7;
0,0 0,0 98,34 1,000 0,0 0,0 165,32 1,000
6,7 6,2 98,02 0,697 1,018 4,8 11,7 164,63 2,066 0,945
14,4 18,0 97,28 0,967 0,997 24,1 30,3 163,16 1,094 0,973
25,0 30,5 96,44 0,972 0,995 34,8 41,0 161,60 1,055 0,998
35,6 41,0 95,49 0,945 1,023 43,7 51,2 160,60 1,039 0,981
45,3 50,7 94,81 0,947 1,029 51,7 58,5 160,08 1,043 0,986
66,4 72,8 92,93 0,996 0,994 65,7 70,6 158,67 1,028 1,003
73,8 80,4 92,15 1,025 0,929 74,0 80,1 157,73 1,045 0,934
87,0 88,6 91,40 0,980 1,142 86,3 88,1 156,84 1,004 1,084
95,3 95,8 90,60 0,986 1,201 100 100 156,08 1,000 ——
100 100 90,37 1,000 — — — —• — —
* Измерения выполнены в приборе Кольбурна [143, 258].
** Там же имеются данные равновесия при 15, 20, 35, 55, 300, 500 и 700 мм
рт. ст.
Поэтому по аналогии со свойствами бинарной смеси а-пн-
нен—р-пинен правомерно допустить, что и другие бинарные
смеси терпенов близки к идеальным, и для расчета колонн можно
пользоваться идеальными кривыми равновесия, построенными
на основе зависимости упругости паров индивидуальных терпе-
нов от температуры [124].
Наиболее полные данные по давлению насыщенных паров
жидких а- и р-пиненов опубликовали Гокинс и Армстронг [247]
(табл. 30), которые использовали в своей работе 99,6 %-ный а-
и 98,7%-ный р-пинены.
Давление паров жидких а- и Р-пиненов изучали также Букала [205] с со-
авторами, а давление паров а-пинена — Рудаков и Коротов [125] (табл. 31),
но все перечисленные работы были выполнены в более узкой области темпе-
ратур, чем это было сделано Гокинс и Армстронгом. Данные всех исследо-
вателей совпадают.
Рудаков и Коротов [125] изучили динамическим методом
в эбулиоскопе Свентославского давления паров жидких А3-ка-
138
Таблица 30
Давление насыщенных паров жидких а- и 0-пиненов по Гокинс и Армстронгу
а-Пинен З-Пинен
давление, мм рт. ст. температу- ра, °C давление, мм рт. ст. температу- ра, °C давление, мм рт. ст. температу- ра, °C давление, мм рт. ст. температу- ра, СС
3,06 19,40 66,79 79,70 1,89 18,67 30,11 68,70
3,43 21,25 81,45 84,66 2,13 20,00 35,03 72,18
4,91 27,20 86,53 86,18 2,59 23,04 40,38 75,38
5,93 29,67 93,85 88,37 3,35 26,77 46,06 78,54
9,01 37,02 107,25 91,96 3,88 29,40 51,08 80,83
15,11 46,87 121,24 95,22 4,44 31,63 52,12 81,18
21,24 53,64 155,39 102,20 5,95 36,73 62,00 85,81
21,58 54,13 175,74 105,95 6,03 37,04 74,88 90,75
24,26 56,52 181,64 106,71 6,97 39,36 85,10 94,07
24,82 57,04 205,80 110,48 7,78 41,29 96,22 97,28
25,45 57,48 226,03 115,02 9,67 45,47 119,62 103,36
31,66 62,23 296,11 122,06 11,85 49,36 144,67 108,76
36,44 65,48 322,25 124,97 12,33 50,06 180,89 115,45
38,41 66,49 371,31 129,82 13,59 52,27 209,81 120,01
41,66 68,22 440,12 135,67 17,01 56,52 248,11 125,28
45,55 70,40 605,53 147,43 17,56 56,89 298,24 131,34
55,73 75,05 754,57 155,72 19,13 58,81 338,32 135,97
57,74 75,99 756,01 155,71 19,65 59,43 488,06 149,50
60,25 77,07 22,92 23,13 26,51 62,57 62,93 65,87 672,31 755,97 759,02 161,17 165,75 165,87
Таблица 31
Давление насыщенных паров жидких терпеновых углеводородов
по Рудакову и Коротову (опытные данные)
а-Пинен Д3-Карен Камфен Лимонен Дипентен
ф S S Я J iu а,0 ф - Ф о S Я J яО а,0 ф » ф" у S Я bi ф <- Ф Q X Я J i и а.0 Ф г- ф” о я Я а.2 ф
Si Е Я S О« ф >» ч °- Я S Я S Е ТО S О. Ф >i ч °- Si темп тура ч °- то S Ч S Е ТО S О. Ф Ч О' ® S si темп тура
49,0 72,0 50 86,8 28,5 61,6 50 91,0 31 79,5
73,5 82,6 75 97,4 50,0 75,0 75 101,1 50 91,3
100,0 90,5 100 104,5 75,0 85,0 100 109,4 75 101,5
150,0 101,7 150 116,0 100,0 92,4 150 120,8 100 109,8
200,0 110,3 200 124,5 150,0 103,8 200 129,7 150 121,0
761,0 156,0 254 132,0 200,0 112,8 760 176,0 200 129,4
752 170,0 242,0 118,8 739 175,5
748,0 158,5
рена, камфена, лимонена, дипентена *, муравьиного и уксусного
эфиров изоборнеола в пределах от 21 до 760 мм рт. ст. (табл. 31
* Лимонен и дипентен должны иметь одинаковое давление паров при
одной и той же температуре, так как дипентен является рацемической фор-
мой лимонена. Незначительная разница в результатах иллюстрирует погреш-
ности из-за различной степени очистки образцов и точности измерения.
139
и 32). Позднее Букала, Маевский и Родзинский [205] изучили
статическим методом давления насыщенных паров Д3-карена,
лимонена, дипентена и терпинолена при температурах между 20
и 90°С, т. е. в основном в области более низких температур, чем
предыдущие исследователи.
Таблица 32
Давление насыщенных паров жидких муравьиного и уксусного
эфиров изоборнеола по Рудакову и Коротову (опытные данные)
Муравьиный эфир изоборнеола Уксусный эфир изоборнеола
давление, мм рт. ст температура, °C давление, мм рт. ст. температура, сС
21,0 110,8 38,0 130,8
44,0 128,5 47,5 136,5
66,4 139,1 71,5 147,4
89,0 147,0 96,5 155,8
135,5 158,9 145,0 167,7
181,5 167,5 193,0 176,6
Зависимость между давлением насыщенного пара (Р) мм рт.
ст. н температурой (/) °C для а- и 0-пнненов Гокинс и Арм-
стронг выразили уравнениями:
для а-пинена 1g Р=26,40174 — 273^2 + / ~ 6.16045 1g [273,2 + /]; (13)
3318,845
для Р-пинена 1g Р=28,77768— 273'2 + г—6,94243 1g [273,2 + /]. (14)
Рудаков и Коротов, а также Букала, Маевский и Родзинский предло-
жили зависимость между упругостью насыщенного пара н температурой вы-
разить уравнением
lg Р = Л - ' (15>
Значения коэффициента А, В и С приведены в табл. 33.
Тнличеев [171], использовав для обработки экспериментальных данных
Рудакова и Коротова уравнение Антуана, получил иные коэффициенты
(табл. 33).
С помощью приведенных уравнений можно вычислить упругости насы-
щенных паров всех рассмотренных терпеновых углеводородов и эфиров изо-
борнеола по заданной температуре и, наоборот, по заданному давлению —
температуру пара. Следует лишь помнить, что коэффициенты, предложенные
тем или иным автором, не могут быть использованы для температур и дав-
лений, выходящих из области экспериментальных измерений.
Для удобства пользования данными определений упругостей паров тер-
пеновых углеводородов в табл. 34 сведены наиболее достоверные величины
давления паров терпеновых углеводородов.
Упругости насыщенных паров камфары в интервале 0—
24,7°С определены Зильберман-Грановской [75], а в интервале
65—232°С де Вильде [338] (табл. 35).
140
Таблица 33
Значения коэффициентов к уравнению 15
Наименование терпенов Значения коффициентов
Рудакова и Коротова [125] Тиличеева [171] Букала, Маевского, Родзинского [205]
А в С А в с А в с
а-Пинен 7,810 2111 273 6,9619 1503,51 212,46 7,2288 1674,0 230
(J-Пинен — — — — — — 7,2654 1732,0 230
Д3-Карен 7,930 2238 273 6,9896 1525,52 200,88 7,2536 1755,5 230
Камфен 7,788 2118 273 7,2159 1702,88 233,58 — — —
Лимонен 7,963 2280 273 7,5015 1939,66 243,28 7,2308 1772,6 230
Дипентен 7,984 2289 273 6,9958 1573,66 205,80 7,2795 1794,6 230
Терпинолен — — — — — — 7,6894 2000,0 230
Муравьиный эфир изобор- неола 8,602 2794 273 — — — — — —
Уксусный эфир изоборнеола 8,512 2799 273 — — — — — —
Таблица 34
Давления насыщенных паров жидких терпеновых углеводородов
при разных значениях температуры, полученные экспериментально
или вычисленные из наиболее достоверных данных
Темпера- тура, °C Давление, мм рт. ст., паров
а-пинена [247] 8-пииена [247] А’-карена [125, 205] камфеиа [171) лимонена [171, 205] дипентена [171, 205] терпино- лена [205]
20 3,25 2,15 1,66
30 5,93 4,01 3,31 — 2,51 2,45 —
40 10,4 7,17 5,75 4,57 4,17 1,60
50 17,5 12,3 9,55 — 7,94 7,41 3,06
60 28,0 20,2 16,2 26,1 13,2 12,5 5,78
70 44,5 32,0 24,8 40,6 20,4 20,9 9,33
80 68,6 50,0 38,9 61,2 31,6 31,3 16,2
90 99,9 73,8 58,5 90,1 48,0 47,4 25,7
100 144 107 85,1 130 71,0 70,7 —
ПО 203 152 122 182 103 103 —
120 279 211 172 252 145 146 —
130 374 286 238 342 202 204
140 502 383 324 456 276 279 —
150 653 504 436 599 371 374
160 — 649 577 777 492 492 —
170 — — 752 — 643 643 —
141
Таблица 35
Давление насыщенных паров камфары [75, 338]
Камфара твердая
Камфара расплавленная
температура, °C давление, м.м рт, ст. температура, °C давление, мм рт. ст. температура, °C давление, мм рт. ст.
0* 16,5* 20,3* 65,14 0,05867 0,30477 0,36240 3,9 24,7* 30** 40** 140,84 0,54624 1,186 2,311 108,4 178,04 368,1
69,84 6,1 146,54 127,8 179,14 379,3
75,54 8,0 150,84 152,3 180,14 390,0
79,94 8,9 152,94 165,6 181,14 398,4
85,14 10,5 155,84 182,1 182,24 408,3
88', 84 14,0 157,14 190,0 183,24 419,5
92,94 15,9 158,04 196,2 186,34 456,1
95,14 17,0 160,94 216,8 188,54 481,6
100,04 21,9 163,14 231,6 191,54 514,1
102,54 23,9 165,94 252,6 191,64 517,0
103,24 25,7 168,24 272,7 193,54 544,1
105,14 27,3 168,94 274,2 195,74 587,6
109,24 31,7 171,14 295,1 198,74 619,5
110,34 32,7 173,44 318,4 201,74 664,8
115,54 41,6 173,74 321,1 203,94 699,2
117,04 42,8 176,24 346,2 206,74 749,1
120,54 50,5 177,64 359,7 211,04 817,8
125,74 61,6 178,54 368,1 215,64 920,3
129,74 71,9 179,44 379,3 220,84 1027,9
130,94 74,5 180,34 390,0 225,44 1143,4
136,14 89,8 225,84 232,34 1150,4 1290,8
* По работе [75].
* * Вычислено по уравнению (16).
Зависимости упругости паров в мм рт. ст. (Р) от абсолютной
туры (Т) описываются уравнениями:
для интервала от 0 до 40°С
1g Р= 11.35196 __343^21
гемпера-
(16)
для твердой камфары в интервале 65—180°С lgP = 8,4382—; (17)
для расплавленной камфары в интервале 178—232°С
lg Р = 7,7046 - ДЯ .
(18)
Упругости насыщенных паров борнеола [328] при разных тем-
пературах сведены в табл. 36. Упругости насыщенных паров
изобориеола были также изучены [58], однако данные не вну-
шают доверия, так как отсутствует линейная зависимость
log Р от Т.
142
Таблица 36
Давление насыщенных паров борнеола при определении статическим
и динамическим методами [328]
Статический метод Динамический метод
температура, °C давление, мм рт. ст. температура, °C давление, мм рт. ст.
78,0 2,30 77,9 2,16
95,1 6,67 96,8 6,55
110,5 15,70 110,0 15,00
130,2 40,40 131,0 40,92
150,2 96,60 156,6 115,16
158,4 127,20
6. Отгонка уксусной и муравьиной кислот
от продуктов формилирования
и ацетилирования
Отгонка органических кислот от изоборнильных эфиров,
образовавшихся при формилировании и ацетилировании кам-
фена, возможна только в случае отсутствия в перегоняемом
продукте катализатора, применяемого при получении изобор-
нильных эфиров, так как он катализирует не только прямую
реакцию образования эфира, но и обратную реакцию его рас-
щепления.
В продуктах формилирования и ацетилирования камфена,
кроме эфира изоборнеола и органической кислоты, всегда со-
держится небольшое количество камфена и трициклена.
Эти терпены желательно собирать при перегонке и возвра-
щать в процесс.
Отгонку органических кислот целесообразно проводить под
вакуумом в аппарате непрерывного действия. Это сокращает
продолжительность пребывания эфиров в горячем состоянии и
тем уменьшает возможность их расщепления.
Данные по равновесиям жидкость—пар для бинарных рас-
творов: уксусная кислота—камфен и уксусная кислота—уксус-
ный эфир изоборнеола, необходимые для расчета аппаратуры,
приведены в табл. 37 и 38 [143].
Из табл. 37 видно, что камфен при давлении 100 мм рт. ст.
образует с уксусной кислотой азеотропную смесь с минимумом
температуры кипения, содержащую 80% уксусной кислоты
(массовых). Наличие азеотропии создает благоприятные усло-
вия для выделения непрореагировавшего камфена и возвраще-
ния его в реакцию. Чрезвычайно большая разница между со-
ставами жидкой и паровой фаз в системе уксусная кислота —
уксусный эфир изоборнеола облегчает отделение уксусной
кислоты от эфира изоборнеола.
143
Таблица 37
Равновесие жидкость—пар для бинарного раствора уксусная кислота—
камфеи при 100 мм рт. ст.*
Равновесное содержание уксусной кислоты, % Температура, °C Коэффициенты активности
массовых молярных T1 72
жидкость пар жидкость пар
0 0 0 0 91,9
3,2 31,6 7,1 52,2 77,5 4,05 0,92
5,2 42,1 И,1 62,2 73,0 3,64 0,94
10,2 53,0 20,5 71,9 66,0 3,05 1,01
17,6 63,5 32,6 79,8 64,2 2,31 0,93
45,7 71,7 65,3 85,2 61,4 1,34 1,48
80,3 79,8 90,3 90,0 60,6 1,10 3,75
92,6 88,2 96,6 94,4 60,8 1,07 5,95
100 100 100 100 61,9 — —
* Исходная уксусная кислота содержала 99,2% СНзСООН. Измерения
выполнены в приборе Кольбурна [258].
Таблица 38
Равновесие системы жидкость—пар для бинарного раствора
уксусная кислота—уксусный эфир изоборнеола при 100 мм рт. ст.*
Равновесное содержание уксусной кислоты, % Температура, °C Коэффициенты активности
массовых молярных Tj 72
жидкость пар жидкость пар
0 0 0 0 154,5
2,7 60,7 8,3 85,3 119,7 1,28 0,74
7,6 75,4 21,1 90,9 100,3 1,12 1,08
12,0 85,4 30,8 95,0 89,0 1,09 1,18
23,0 91,8 49,4 97,3 78,9 1,01 1,48
48,4 95,9 74,9 98,7 71,6 0,90 2,11
82,5 98,4 93,3 99,5 64,1 1,01 4,6
100 100 100 100 62,5 — —
* Исходная уксусная кислота содержала 99,6% СНзСООН. Измерения
выполнены в приборе Кольбурна.
Данные по равновесию жидкость — пар для бинарных рас-
творов муравьиная кислота — муравьиный эфир изоборнеола и
муравьиная кислота — камфен не были опубликованы. Однако
известно, что разделение этих смесей может быть осуществлено
при давлении 100—150 мм рт. ст. без разложения эфира при
условии, если продукт находится в нагретом состоянии не более
5—6 ч [145]. Для расчета колонны в этом случае могут быть
использованы данные по равновесию в системе уксусная кис-
лота— уксусный эфир изоборнеола, которые не должны
144
существенно отличаться от данных по равновесию между жид-
костью и паром в системе муравьиная кислота — муравьиный
эфир изоборнеола.
7. Укрепление уксусной и муравьиной кислот
В случаях, когда при формилировании и ацетилировании
камфена уксусная и муравьиная кислоты вводятся в избытке,
а затем отгоняются из продуктов реакции, их концентрация при
этом падает. Падение концентрации происходит за счет части
кислоты, расходуемой на реакцию, вода остается в отработан-
ной кислоте. Например, при формировании камфена 100%-ным
избытком 98%-ной муравьиной кислоты в присутствии катио-
нообменных смол непрореагировавшая кислота возвраща-
ется в виде 96%-ной. При ацетилировании камфена наблюда-
ется аналогичное явление. Отогнанные кислоты по возвращении
в новый цикл необходимо укреплять до исходной концентрации.
Процесс может быть построен таким образом, чтобы одновре-
менно с крепкой высококонцентрированной кислотой получалась
50—80%-ная органическая кислота. Полученные после разгонки
разбавленные органические кислоты можно использовать для
нейтрализации щелочных растворов формиата и ацетата нат-
рия, получаемых после гидролиза эфиров, перед выпаркой этих
растворов с целью получения сухих солей.
Муравьиная кислота образует с водой азеотропную смесь
с максимумом температуры кипения (табл. 39).
Таблица 39
Состав азеотропных смесей муравьиная кислота—вода
при разных давлениях [105]
Давление, мм рт. ст. Температура, °C Содержание муравьиной кислоты в азео- тропной смеси, %
массовых молярных
50 42,3 66,0 43
100 55,9 69,0 46
116 60,0 69,0 46
200 71,5 72,0 50
278 80,0 73,0 51
750 — 76,5 56
760 107,3 77,5 57
1800 130 84,5 68
Наличие азеотропии не позволяет обезводить ректификацией
при атмосферном давлении разбавленную муравьиную кислоту,
но не мешает разделить ректификацией кислоту, концентрация
которой превышает концентрацию азеотропной смеси, и полу-
чить при этом высококонцентрированную муравьиную кислоту
и азеотропную смесь. Данную операцию можно рассматривать
10 Заказ № 43
145
как ректификацию бинарной системы муравьиная кислота —
азеотропная смесь, т. е. азеотропную смесь муравьиной кислоты
с водой можно принимать за один компонент. Легколетучим
компонентом в данном случае является избыточная (сверх не-
обходимой для образования азеотропной смеси) муравьиная
о 62
ё во
£ 44
42
40
Зв
56
54
52
50
28
Y08
106
104
102
100
72
70
68
\бб
Пар
о жидкость
кислота.
Так как азеотропная смесь
муравьиная кислота — вода
при 760 мм рт. ст. содержит
77,5% по массе муравьиной
кислоты, укрепление муравьи-
ной кислоты ректификацией на
одноколейном аппарате может
быть целесообразным лишь
в случае укрепления высоко-
концентрированной муравьи-
ной кислоты. Иначе выход
укрепленной кислоты будет
слишком низким.
Состав азеотропной смеси
муравьиная кислота — вода за-
висит от давления, под кото-
рым находится система. С по-
вышением давления содержа-
ние муравьиной кислоты в азе-
отропной смеси увеличивается
(см. табл. 39). Поэтому при
осуществлении ректификации
муравьиной кислоты в вакууме
можно увеличить выход кон-
центрированной муравьиной
О 10 20 30 40 50 60 70 80 90 100 йентРиРовзннои муравьиной
Содержание муравьиной кислоты кислоты. Кроме того, равнове-
8 молярных*/» сие жидкость — пар в рассма-
Рис. 25. Диаграмма «состав-темпе-тРиваемой системе более бла-
ратура кипения» бинарного растворавОПриятнО ДЛЯ ректификации
муравьиная кислота—вода при дав-в вакууме, чем при атмосфер-
лении, мм рт. ст.: ном давлении (рис. 25). Под-
1 — 760-, 2- 200; з-5о робные данные о равновесии
приведены в работе [83].
Смещение азеотропной точки системы муравьиная кислота —
вода при изменении давления позволило Гельперину и Новико-
вой [42, 43] разработать метод укрепления муравьиной кислоты
любой концентрации до желаемой крепости, пользуясь двумя
колонными аппаратами, из которых один работает при атмо-
сферном давлении, а второй — при давлении 50 мм рт. ст.
На колонном аппарате, работающем при атмосферном давле-
нии, отгоняется вода из слабой кислоты, а азеотропная смесь,
содержащая 77,5% муравьиной кислоты (как остаток),
146
направляется на вторую колонну, работающую в вакууме. Здесь
отгоняется высококонцентрированная муравьиная кислота,
а остаток, состоящий из 66%-ной муравьиной кислоты, возвра-
щается на первую колонну. Расчет показывает, что первая колон-
на должна иметь 8—10 теоретических тарелок в укрепляющей
части и 8,5—12 — в исчерпывающей части, т. е. всего около
20 теоретических тарелок. Вторая колонна, работающая в ваку-
уме, должна иметь 5 теоретических тарелок в укрепляющей
и 5 в исчерпывающей частях.
Уксусная кислота с водой не образует азеотропных смесей,
и ее укрепление можно производить обычным методом. Данные
о равновесии жидкость—пар для бинарного раствора вода —
уксусная кислота приведены в работе [83]. Обезвоживание
уксусной кислоты может быть произведено также посредством
азеотропной перегонки, используя в качестве антренера н-про-
пилацетат, образующий азеотроп, кипящий при 81°С с содержа-
нием 14% воды, и н-бутилацетат, образующий азеотроп, кипя-
щий при 90,7°С с содержанием 27% воды [105, 287].
ГЛАВА X. ПРОИЗВОДСТВО КАМФАРЫ
ИЗ ПИХТОВОГО МАСЛА
1. Некоторые исторические сведения
Первые исследования пихтового масла были выполнены в Питербурге
Голубевым в конце XIX начале XX столетия. Было установлено, что пихто-
вое масло содержит до 40—45% (—)-борнилацетата, (—)-борнеол и (—)-
камфен. Эти вещества были выделены практически чистыми даже с точки
зрения современных требований [47, 50, 51]. Голубев же разработал способ
превращения борнилацетата в камфару, который сводился в выделению бор-
нилацетата, его гидролизу, окислению борнеола азотной кислотой и возгонке
камфары [48, 49].
В 1907 г. на заводе Жукова был осуществлен разработанный Голубевым
синтез камфары. Производство просуществовало недолго, в связи с резким
снижением цены на японскую камфару.
Тот же синтез, но на основе несравненно более совершенной технологии,
был осуществлен в Ленинграде на Охтинском химическом комбинате в 1929 г.
[182]. Производство просуществовало вплоть до начала второй мировой войны.
В 1934 г. производство синтетической камфары из пихтового масла было
организовано в Новосибирске *. Здесь превращение борнеола в камфару осу-
ществляли уже не окислением азотной кислотой, а каталитическим дегидри-
рованием [164]. В результате стали получать камфару без примеси нитро-
соединений. Камфара, полученная этим способом из пихтового масла, имела
левое оптическое вращение и была практически оптически чистой.
В результате работ Вершинина с сотрудниками [163] в Со-
ветском Союзе было разрешено применять (—)-камфару, по-
лученную из пихтового масла, для перентерального введения
* Со временем производство было перенесено на Урал.
10*
147
в организм наряду с ( + )-японской камфарой и, в настоящее
время, в нашей стране для этой цели используют почти исклю-
чительно камфару, полученную из пихтового масла.
2. Особенности пихтового масла как сырья
для производства камфары
В состав пихтового масла входит около 100 компонентов,,
большинство из них содежится в очень небольшом количестве,
порядка сотых долей процента, главные же сантен 3—4%, три-
циклен 2—4, а-пинен 10—30, (—)-камфен 10—25, (—)-р-пинен
1—2, мирцен ~1, ( + )-Д3-карен 5—10, (—)-р-фелландрен и
(—)-лимонен в сумме 5—7, терпинолен ~1, (—)-борнеол
1—6, (—)-борнилацетат 30—40, сесквитерпены и сесквитерпе-
новые спирты 2—4% [50, 108, 152, 182]. Состав сесквитерпенов
сложен и до конца не изучен, в их состав входят углеводороды
и спирты. Главными компонентами среди сесквитерпеновых
углеводородов являются: кариофилен (I) ~40%, гумулен (II)
и безаболен (III) в сумме около 45%. Кислородные производ-
ные составляют около 30% от общего содержания сесквитер-
пенов, т. е. около 1% от массы пихтового масла [108, 109]:
Поскольку в пихтовом масле содержатся камфен и борнил-
ацетат, а при получении синтетической камфары из пинена по-
лучают камфен и изоборнилацетат в результате ряда химиче-
ских превращений, на первый взгляд может показаться, что
переработка в камфару пихтового масла представляет собой
как бы упрощенную переработку в камфару скипидара. Однако
это не совсем так. Из-за наличия в пихтовом масле сесквитер-
пенов, имеющих высокие температуры кипения, сравнительно
близкие к температуре кипения борнилацетата, их не отделяют
от борнилацетата при ректификации масла или отделяют лишь
частично. В результате сесквитерпены попадают во все полу-
продукты производства и в камфару, которая должна подвер-
гаться специальной очистке для их отделения. Это приводит
к существенным отличиям в переработке борнильного эфира,
выделенного их пихтового масла, от переработки изоборниль-
ного эфира, полученного из скипидара.
148
Выделить ректификацией из пихтового масла камфен не уда-
ется из-за присутствия в нем а-пинена, кипящего при темпера-
туре на 2°С ниже, чем камфен. Поэтому для получения рацеми-
ческой камфары из камфена целесообразно выделять ректифи-
кацией пинено-камфеновую фракцию и подвергать ее
изомеризации [159]. Следует, однако, иметь в виду, что в пихто-
вом масле содержится сантен (IV) (т. кип. 140°С), который
необходимо отделять от пинено-камфеновой фракции или от
полученного из нее изомеризата, так как в процессе дальней-
шего синтеза он превратится последовательно в эфир сантенола
(VI) через ион V, сантенол (VII) и сантенон (VIII) т. пл. 58—
61°С, который будет загрязнять камфару. Содержание
3—4% сантена в пихтовом масле, если его не отделять при
ректификации, поднимается в пинено-камфеновой фракции до
8—10, а в техническом камфене до 10—15%.
3. Ректификация пихтового масла
Пихтовое масло ректифицируют с целью выделить (—)-бор-
нилацетат и пинено-камфеновую фракцию. В качестве побочных
продуктов получают сантеновую головку, фракцию моноцикли-
ческих терпенов и небольшой остаток от перегонки, содержа-
щий борнилацетат и значительное количество сесквитерпенов.
Отбор пинено-камфеновой фракции нецелесообразно произво-
дить на той же колонне, где выделяют борнилацетат, требуются
более эффективные колонны. Поэтому при выделении борнила-
цетата сначала отгоняют всю смесь монотерпеновых угле-
водородов от борнилацетата, а затем эту смесь подвергают
самостоятельной ректификации, разделяя ее на сантеновую
головку, пинен-камфеновую фракцию и фракцию, содержащую
149
А3-карен, моноциклические терпены и пр. Сырой (—)-борнила-
цетат содержит примесь борнеола, сесквитерпенов и смолистых
веществ, от которых его освобождают дистилляцией в вакууме.
В кубовом остатке от вакуум-дистилляции содержится еще бор-
нилацетат, который можно извлечь отдувкой водяным паром,
однако получаемый при этом продукт в такой степени загрязнен
сесквитерпенами (до 25—30%), что его использование в произ-
водстве камфары вряд ли целесообразно, тем более что содер-
жание борнилацетата в этой фракции не превышает 3—3,5% от
его общего количества. Вследствие сравнительно небольшого
объема производства и значительных колебаний в составе пих-
тового масла ректификацию пихтового масла обычно произ-
водят в аппаратуре периодического действия.
При расчете колонны для выделения борнилацетата пихтовое масло мо-
жно рассматривать условно как бинарную смесь борнилацетата с лимоненом
(гл. IX.4 и 5); колонны же для выделения пинено-камфеновой фракции
из монотерпенов пихтового масла и камфена из изомеризата, полученного
из пинен-камфеновой фракции, подобны колоннам для выделения пинена из
•скипидара и камфена из продуктов изомеризации пинена (гл. IX 3 н 5).
4. Использование пинен-камфеновой фракции
В целях использования пинена, содержащегося в пинен-кам-
феновой фракции, и облегчения выделения из нее камфена было
предложено превращать в камфен и моноциклические терпены
содержащийся во фракции пинен путем ее нагревания с соот-
ветствующими катализаторами [159]. Использование в качестве
катализатора активированной глины поднимало содержание
2ЭВ во фракции на 5—6% (до 80%) и при последующей рек-
тификации практически количественно выделялся камфен
(т. крист. +45—( + 48)°С). Вероятно, при применении титано-
вого катализатора удалось бы получить еще лучшие результаты.
Пинен-камфеновая фракция пихтового масла содержит камфен с очень
высоким оптическим вращением: ([а]о—92° [51], [а]о — 106° [152], что при-
ближает его к оптически чистому (гл. XIII). Поэтому пинен-камфеновая
фракция может быть использована для получения практически уникального
реактива — камфена с очень высокой оптической активностью. В этом случае
камфен следует выделять, не прибегая к изомеризации присутствующего пи-
нена, так как она сопровождается рацемизацией.
Пинен-камфеновая фракция может быть использована и как раствори-
тель, заменяющий скипидар в некоторых областях его применения.
5. Переработка борнилацетата в камфару
Дистиллированный борнилацетат, выделенный из пихтового
масла, имеет примерно следующий состав: монотерпены 1—3%,
борнилацетат 85—90, борнеол 2—8, сесквитерпены 5—7%.
Внешне процесс переработки борнилацетата в камфару ничем
не отличается от уже описанной переработки изоборнилацетата и
изоборнилформиата (гл. VI и VII). Борнилацетат гидролизуют
150
водным раствором едкого натра, отжимают на центрифуге
и подвергают дегидрированию в жидкой или паровой фазе. Усло-
вия этих реакций более благоприятны, чем при переработке
изоборнилацетата. Константа скорости щелочного гидролиза
борнилацетата на один порядок выше, чем соответствующая
константа скорости гидролиза изоборнилацетата (гл. VI), и от-
носительно большая устойчивость борнеола к реакциям дегидра-
тации позволяет не опасаться этой реакции при дегидрировании
в паровой фазе. Неблагоприятным фактором является более
медленная реакция дегидрирования борнеола, чем изоборнеола,
но для такого дорогого продукта, как камфара из пихтового
масла, это не имеет существенного значения.
Основная разница в переработке борнилацетата из пихто-
вого масла и изоборнилацетата, полученного из скипидара, про-
исходит из-за присутствия в борнилацетате сесквитерпенов.
После гидролиза часть сесквитерпенов отделяется при отжиме
борнеола на центрифуге в виде масла, содержащего около 25%
борнеола и около 50% сесквитерпенов, однако полученный бор-
неол содержит все же 4—5% сесквитерпенов.
После дегидрирования в паровой фазе получают сырую кам-
фару, содержащую около 90% основного вещества, около 20—
30 компонентов различных примесей, в том числе около 3%
различных сесквитерпенов.
Отжим на центрифуге повышает содержание основного ве-
щества в камфаре до 91—92%, а содержание сесквитерпенов
снижается примерно до 1 %.
Такая камфара должна пройти специальную очистку (рафи-
нацию), чтобы оказаться пригодной для использования в меди-
цине. Очистка может быть осуществлена двумя путями: пере-
кристаллизацией из спирта (см. гл. XI. 4) или перегонкой с на-
сыщенным водяным паром. Последний способ очистки основан
на том, что сесквитерпены плохо растворяются в камфаре и
образуют отдельную фазу на поверхности ее кристаллов *.
Упругость пара систем, состоящих из нескольких фаз, склады-
вается из упругостей пара каждой фазы в отдельности, незави-
симо от других и независимо от количественного соотношения
между отдельными фазами. Если в перегонном кубе находится
твердая камфара и вода, то фактически присутствуют три фазы:
вода, раствор терпенов и их кислородных производных в кам-
фаре и раствор монотерпенов и их кислородных производных,
в том числе камфары, в сесквитерпенах. Перегоняются все три
фазы в соотношении,' пропорциональном упругости их паров,
т. е. камфара, жидкая смесь моно- и сесквитерпенов (камфар-
* При долгом хранении камфары с примесью сесквитерпенов последние
скапливаются в нижнем слое у днища тары. Так, после продолжительного
хранения образца с большим содержанием сесквитерпенов содержание кам-
фары в верхнем слое оказалось 95%, а в нижнем 69%. В верхнем слое сескви-
терпенов не оказалось, а в нижнем их было 19%.
151
ное масло) и вода в постоянном соотношении, до тех пор, пока
не отгонится все камфарное масло, после чего перегоняется
чистая камфара. Если исходная камфара была хорошо отжата,
выход головных фракций не превышает 20—25%. От них отжи-
мают жидкое масло и отжатую твердую камфару возвращают
в процесс. Этот способ очистки камфары связан со всеми недо-
статками, разобранными при описании перегонки камфары
с насыщенным водяным паром (гл. VII. 2).
ГЛАВА XI. СВОЙСТВА КАМФАРЫ И ЕЕ ОЧИСТКА*
1. Свойства камфары как индивидуального
химического соединения
При температуре около 0°К рацемическая камфара пред-
ставляет собой молекулярное соединение ( + ) и (—) камфары
в соотношении 1:1, которое не образует смешанных кристаллов
с оптически деятельной камфарой. В этих условиях кристаллы
рацемической и оптически активной формы образуют механи-
ческую смесь. С повышением температуры это молекулярное
соединение начинает диссоциировать и при комнатной темпера-
туре рацемическое соединение перестает существовать; крис-
таллы камфары в этих условиях представляют собой твердый
раствор антиподов, не связанных химически [303]. По этой при-
чине температура плавления оптически активной, рацемической
камфары и их смесей одинакова и лежит в пределах 178,5
и 179,5°С [166]. Разница между данными разных авторов обус-
ловлена разной степенью чистоты исследованных образцов,
а возможно, зависит и от использованной методики определения
температур плавления.
При комнатной температуре камфара имеет гексагональную
структуру кристаллов, при нагревании до 75—100°С они превра-
щаются в кубическую.
Оптическое вращение расплавленной ( + ) -камфары [а]о179°с +
+ 70,85° [а]р193°с+70,88°. Оптическое вращение камфары в рас-
творе зависит от растворителя и концентрации раствора, в аб-
солютном спирте имеет место следующая зависимость: [а]о20°с +
+ 41,4° (с. 1), +43,6° (с. 5), +44,8° (с. 20), +48,4° (с. 50) [295].
Оптическое вращение камфары в гексане и диэтиловом эфире
не зависит от ее концентрации. Добавка воды к растворителю
снижает оптическое вращение. Так, для раствора камфары
в 75%-ном спирте были получены следующие данные: [а]оагс+
+ 34° (с. 1); +36,9° (с. 5); +39,8° (с. 20) [295].
Плотность камфары: d°Q 1,000, d® 0,994—0,996, d^ 0,992,
с^05'3 0,81 1 [263], насыпная масса 0,5 кг/л [263]. О зависимости
* Написана совместно с В. И. Гармашовым.
152
давления насыщенных паров камфары от температуры см.
гл. IX. 5. Скрытая теплота испарения камфары 50,75 кДж/моль
[338], скрытая теплота испарения при температуре кипения
59,54 кДж/моль [338], скрытая теплота плавления 34,45 кДж/кг
[263], 44,93 кДж/кг [328], теплоемкость 20—34°С 1,733 кДж/(кгХ
Xград), 20—164°С 1,846 кДж/(кг-град) [263], теплота сгора-
ния 5906,6 кДж/моль [264], тройная точка 180,1°С при 385,8 мм
рт. ст. [338], криоскопическая постоянная 40°С (1 моль на 1000 г
камфары), температура вспышки 66°С [263], температура
воспламенения 466°С [263]. Нижний предел температуры само-
произвольного взрыва смеси паров камфары (26% массовых)
с воздухом при 1 атм 485°С. Взрываемость текущей смеси паров
камфары с воздухом от электрической искры при 200°С наблю-
дается, начиная от концентрации паров камфары 9% массовых.
Взрываемость той же текучей смеси от нагретой до 870—980°С
нихромовой проволоки в пределах концентраций камфары 6—
16% массовых [259]. Взвешенная в воздухе пыль камфары
фракции 850 мкм имеет нижний предел взрываемости 10,1 г/м3,
температуру искрения 460°С, температуру самовоспламенения
850°С. Осевшая пыль пожароопасна [55]. Насыщенный раствор
в воде содержит при 14—17°С 1,67, при 39°С — 1,60 г камфары/л
[235, 263]. Растворимость камфары в спирте (с разным содержа-
нием воды), бензине и ксилоле см. в гл. XI. 5; влияние отдель-
ных примесей на температуру плавления — гл. XI. 2 и 3. По от-
ношению к кислороду воздуха камфара устойчива. Главнейшие
производные, служащие для идентификации и для количествен-
ного определения: оксим — т. пл. 120°С, семикарбазон ( + )-
камфары — т. пл. 248°С, (—)-камфары — т. пл. 238°С *, (±)-
камфары— т. пл. 236—238°С; 2,4-динитрофенилгидразон ( + ) и
(—)-камфары — т. пл. 177°С, (±)-камфары — т. пл. 164°С [263].
Подробные данные о химических свойствах камфары приве-
дены в работах [235, 318].
2. Состав и свойства товарной камфары
Товарная камфара не является индивидуальным веществом,
а содержит ряд примесей, состав и количество которых зависит
от ее происхождения. Содержание примесей в японской естест-
венной камфаре очень невелико: порядка 1 % в однократно суб-
лимированной камфаре (сорт ВВ) и лишь около 0,5% в дважды
сублимированной (сорт RC). Наоборот, содержание примесей
в технической синтетической камфаре составляет 5—10%. Это
не мешает применять синтетическую камфару в производстве
целлулоида наравне с естественной. Для назначений, требую-
щих более чистого продукта, синтетическая камфара подверга-
ется очистке (гл. XI. 4).
* Температура плавления семикарбазонов ( + ) и (—)-камфары должны
быть одинаковые.
153
Основные примеси в синтетической камфаре — изофенохон,
5-экзо-изокамфанон-З (изокамфанон, псевдокамфара) и угле-
водороды, в том числе трициклен, камфен и два циклана (гид-
рированных терпена), из которых один изокамфан [97], а второй
неидентифицирован. Предполагается, что оба циклана являются
цис- и транс-изокамфанами [154]. Кроме того, в синетической
камфаре содержатся небольшие количества борнеола, изооор-
неола, фенхилового и изофенхилового спиртов, а также псевдо-
борнеол (5-экзо-изокамфан-экзо-ол-З) и целый ряд очень незна-
чительных неидентифицированных примесей.
Камфара, полученная из борнилацетата пихтового масла
(в отличие от синтетической камфары, полученной из скипи-
дара), является практически оптически чистой (—)-камфарой,
в ней отсутствуют примеси изомерных кетонов: изофенхона и
изокамфанона. Примесь изокамфана также более низкая, чем
в синтетической камфаре из скипидара, полученной в равных
условиях. Наоборот, примесь борнеола в ней, как правило, бо-
лее высокая.
В табл. 40 приведены данные анализов товарной камфары,
выработанной в разных странах. Там же для сравнения приве-
ден анализ японской естественной камфары. Эти анализы
не отображают среднего состава продукции названных стран,
а относятся к отдельным образцам. Тем не менее заслуживает
внимания близкое сходство проанализированных образцов, за
исключением образца, выработанного в Англии, где содержание
камфары ниже, чем в продукции других стран, а содержание
терпеновых углеводородов и изофенхона значительно выше.
Возможно, это является результатом того, что в Англии гидро-
лиз изоборнильного эфира ведут в углеводородном раствори-
теле и полученный раствор, не выделяя из него изоборнеола,
непосредственно направляют на дегидрирование. В этих усло-
виях исключается отжим изоборнеола на центрифуге, во время
которого удаляется часть содержащихся в нем примесей (см.
гл. VI. 6).
Температура кристаллизации у синтетической камфары бо-
лее низкая по сравнению с чистым продуктом, что является
результатом влияния примесей. Лишь примесь борнеолов повы-
шает температуру кристаллизации камфары, примеси же изо-
фенохона и других терпеновых кетонов, а также примесь угле-
водородов снижают ее. Зависимости температур плавления би-
нарных систем камфара — изоборнеол, камфара — изокамфанон
и камфара — изокамфан от их состава в первом приближении
описываются прямыми линиями, соединяющими температуры
плавления чистых компонентов. В соответствии с этйм примесь
к камфаре 1% изокамфана или изокамфанона снижает ее тем-
пературу плавления на 1,1°С. Примерно также снижает темпе-
ратуру плавления примесь камфена. Зависимость температуры
кристаллизации камфары от примеси изофенхона и изоборнеола
154
Таблица 40
Аналитическая характеристика камфары, выпускаемой за рубежом *
Показатели Синтетическая камфара Естественная камфара
ГДР ФРГ, ; техническая Англия КНР, техническая Индия, техническая порошко- образная, Япония
о X =5 к н « форма- конейная
Температура кри- 168,5 172 170,5 166,5 171,5 173,0 178,5
сталлизации, °C **
Состав (%):
камфара 92,7 94,7 94,9 90,3 94,9 94,3 99,1
трициклен 0,05 0,1 — 0,2 — — —
камфен 0,15 0,2 0,15 3,0 0,05 0,1 0,1
цикланы 1,0 0,9 — 0,3 — Следы —
фенхон 0,2 — 0,3 0,3 0,2 — —
изофенхон 1,5 1,4 1,8 2,6 1,6 0,9 —
фенхол — — — 0,1 0,05 — ——
изофенхол 0,1 —- 0,6 0,1 0,05 0,2 —-
изоборнеол Следы Следы 0,25 0,7 0,6 0,3 —
борнеол Следы Следы 0,6 0,7 — 0,4 0,5
5-экзо-изокам- 1,7 1,4 0,5 1,4 2,3 2,5
фанон-3
Прочие примеси 2,6 1,3 0,9 0,3 0,25 1,3 0,3
1-терпеновых ке- 97,2 97,5 97,5 94,6 99,0 97,7 99,1
ТОНОВ
* Образцы камфары производства ФРГ, Англии, КНР, Индии н Японии
были любезно предоставлены доктором Г. Кейхером (завод фирмы Хехст
Герстгафен). Образцы камфары из ГДР получены с завода Финовталь непо-
средственно.
** По паспортным данным.
показана на рис. 26, из которого видно, что примесь 1 % изо-
фенхона снижает температуру плавления камфары на 1,9—2°С,
а примесь изоборнеола повы-
шает ее на 0,3—0,35°С.
В табл. 41 приведены
данные анализов типичных
образцов камфары, выпус-
каемой в Советском Союзе.
Рис. 26. Температура кристалли-
зации бинарных систем камфара—
изоборнеол , камфара—изофенхои:
1 — камфара—изоборнеол; 2 — камфа-
ра— изофенхои
155
Таблица 41
Аналитическая характеристика синтетической камфары, вырабатываемой
в Советском Союзе
Показатели Камфара
из скипидара ИЗ пихтового масла 3
марка А 1 марка В 2 рацемиче- ская 2
Температура плавления, °C 4 175,8-176,7
Температура кристаллизации, 169 164 172 —
°C4
[а]а (95% спирт, с. 10) Состав (%): — — — —40,4°
камфара 92,0 89,9 94,6 96,9
монотерпеиы 0,5 0,2 0,2 0,3
цикланы 3,3 1,6 1,0 0,8
фенхон 0,05
изофенхон 0,9 2,4 0,9 —
изоборнеол Следы Следы Следы 0,3
борнеол То же То же То же 1,0
изокамфанон 2,0 2,2 2,1 —
сесквитерпены — — Следы
прочие примеси 5 1,3 3,7 1,2 0,7
S-терпеновых кетонов 94,9 94,5 97,6 96,9
1 Единичный образец.
2 Среднее из анализов 5 образцов.
3 Среднее из анализов 10 образцов.
4 По паспортам производителей.
5 В том числе вода и этанол, оставшиеся после перекристаллизации
камфары.
3. Технические требования к камфаре
Промышленность выпускает камфару различных сортов и марок, отли-
чающихся по степени чистоты и по внешнему виду. Согласно ГОСТ 1123—72
синтетическая камфара, выпускаемая в Советском Союзе для технических на-
значений, должна соответствовать нормам, указанным в табл. 42, а камфара,
используемая в медицине,— нормам, предусмотренным Государственной фар-
макопеей СССР [56], сведенным в табл. 43.
К сожалению, и те и другие нормы плохо обеспечивают надлежащее
качество продукта. Показатели качества и согласно ГОСТ 1123—72 и со-
гласно Госфармакопеи СССР ясно оговаривают допустимые границы механи-
ческих загрязнений, ио не оговаривают границы допустимых химических за-
грязнений. Согласно ГОСТ 1123—72 о наличии химических примесей в кам-
фаре следует судить по содержанию этерифицирующихся веществ (борнеолы
и камфен) и по температуре кристаллизации камфары. Но борнеолу и кам-
фен вовсе не главные примеси (см. табл. 40 и 41). Основными примесями
в технической камфаре являются цикланы, изофенхон и 5-экзо-изокамфаион-З,
а они по-разному влияют на температуру ее кристаллизации (см. гл. Х1.2).
Соотношение между примесями колеблется в довольно широких пределах,
в результате чего деление камфары на марки А и Б часто получается искус-
ственным.
156
Таблица 42
Технические требования к синтетической камфаре по ГОСТ 1123—72
Показатели Марка А Марка Б
Цвет по хромпиковой шкале № 1 № 1
Температура кристаллизации, °C, ие меиее 169 164
Содержание этерифицирующихся веществ в пе-
ресчете на борнеол, %, не более 2 3
Содержание воды, % не более 1,5 2
Содержание нелетучего остатка, %, не более 0,05 0,1
Содержание нерастворимого в спирте остатка,
%, ие более 0,01 0,05
Содержание золы, %, не более 0,01 0,02
Кислотность Раствор 10 г камфары
в 100 см3 нейтрального
спирта показывает ще-
лочную реакцию по фе-
нолфталеину после вне-
сения 0,1 см3 0,1 н. рас-
твора едкого натра
Содержание железа, %, не более* 0,001 0,001
* Для камфары, применяемой в производстве целлулоида и фото-киио-
промышленности.
Еще хуже обстоит дело с оценкой качества медицинской камфары. О на-
личии в ней примесей согласно требованиям Госфармакопеи СССР можно
судить только по оптическому вращению и температуре плавления. Главной
примесью в медицинской камфаре, полученной из бориилацетата пихтового
масла, является борнеол. Борнеол имеет оптическое вращение, близкое к оп-
тическому вращению камфары (гл. XIII), а его присутствие в количестве
до 5—7% при одновременном присутствии 1—1,5% углеводородов не повы-
шает температуру плавления продукта выше предусмотренной нормами.
Таблица 43
Технические требования к камфаре Государственной фармакопеи СССР
Показатели (+)-Камфара (—)-Камфара (±)-Камфара
Температура плавления, °C [а] 10%-раствор в 95%-ном спирте, град. 174-180 174-180 171-178
+41 +44 _39 ч- —44 -1ч-+1
Нелетучий остаток, %, ие более 0,05 0,05 0,05
Проба на отсутствие влаги Раствор 1 г камфары в 10 см3 петро- лейного эфира прозрачен
Проба на цветность Раствор 1 г камфары в бесцветен и прозрачен 4 см3 спирта
Проба на отсутствие масел Отсутствие пятен при отжимании ме- жду листами белой писчей бумаги
Проба иа присутствие органических Сравнение окраски раствора 0,5 Г
примесей камфары в 5 см3 крепкой серной кис- лоты с окраской 5 см3 эталонного раствора 0,033 г К2СГ2О7 в 100 см5 воды
157
В то же время считается, что примесь борнеола в камфаре, используемой
для переитерального введения в организм, вредна [163].
Значительно правильнее было бы оценивать качество кам-
фары по содержанию в ней основного вещества при применении
современных методов газо-хроматографического анализа. Это
определение может быть выполнено достаточно быстро и на-
дежно (гл. XII. 4) [154]. Если данные количественного определе-
ния камфары дополнить определением температуры кристалли-
зации или плавления, можно составить представление и о том,
какие примеси в ней содержатся.
Требования к камфаре, предъявляемые за рубежом, приведены в табл. 44
и 45. Синтетическая камфара используется в зарубежной медицине наравне
с естественной не только наружно, но и для введения под кожу. Технические
условия зарубежных фармакопей предусматривают содержание в камфаре
основного вещества, определенного в виде 2,4-динитрофенилгидразона не
ниже определенного предела. Но и это для правильной оценки качества про-
дукта недостаточно, так как, во-первых, стандартное отклонение метода
велико <т=1,5% (гл. XII.4) и, следовательно, воспроизводимость результата,
единичного анализа лежит в пределах ±3% при доверительной вероятности
0,95; во-вторых, 2,4-динитрофенилгидразоном определяется ие камфара,
а сумма всех терпеновых кетонов, содержащихся в камфаре, что приводит
к завышенным результатам определения камфары в синтетической кам-
фаре [154].
4. Очистка камфары
Существуют различные способы очистки камфары: перегонка
с паром, сублимация, ректификация и перекристаллизация [263].
Так как температуры кипения изофенхона и камфары доста-
точно близки (193—194°С и 209°С соответственно), методы раз-
деления изофенхона и камфары перегонкой с водяным паром
или сублимацией малоэффективны. Эти процессы могут очистить-
камфару от смолистых веществ. Наверное ректификацией можно
довести чистоту камфары до любой заданной степени, но этот
процесс трудно осуществить технически.
В противоположность ректификации, перекристаллизация
камфары проста и дает вполне удовлетворительные результаты.
Очистку синтетической камфары перекристаллизацией широко-
осуществляли и осуществляют в промышленности в Советском
Союзе, в Германии [167] и до известной степени в Англии, хотя,
по некоторым данным [273] там предпочитают очищать перекри-
сталлизацией не камфару, а изоборнеол, направляемый на де-
гидрирование.
Для очистки перекристаллизацией камфару растворяют в ка-
ком-либо растворителе. Присутствующие в камфаре нераствори-
мые вещества отфильтровывают или отделяют декантацией;
после этого раствор медленно превращают в пересыщенный,
в результате чего часть камфары постепенно выделяется из
раствора в виде кристаллов. Пересыщенный раствор получают
постепенным изменением его температуры или методом отгонки
158
Таблица 44
Главнейшие требования к естественной камфаре за рубежом
Показатели Техническая камфара [263] Медицинская камфара
ГДР [217] Франция [291] Англия [200] ООН [96] США [292]
Т. пл. °C [а]о20, град.* Камфара в %, не меиее** 176—178 +39 ч-+44,5 от 93 до 99,5 175-178 +43 ч- +44,5 98 174-179 +42 ч- +44 96 174—179 4-40 ч-+43 96 174-179 +40 ч- +43 96 174-179 +41 ч- +43
* Этанол, ГДР — 20%-ный раствор, прочие страны—10%-иый раствор.
* * Определенная в виде 2,4-динитрофенилгидразона.
Таблица 45
Главнейшие требования к синтетической камфаре за рубежом
Показатели Техническая камфара [263] Медицинская камфара
ГДР |217] Франция (291 [ Аиглия [200] ООН [96] США [292]
Т. пл. °C, не менее 160 170-179 170 174-179 174-179 174-179
[а] о20, град.* 0 —0,4 +1,0 ~0 -1,5ч- +1,5 —1,5 ч- +1,5 ~0
Камфара, %, не менее** Нелетучий остаток, %, не бо- лее 92 96 0,05 96 0,1 96 0,05 96 0,05 —
* Этанол, ГДР — 20%-ный раствор, прочие—10%-ный раствор.
** Определенная в виде 2,4-дппптрофспилгидразопа.
части растворителя. Медленное охлаждение раствора обеспечи-
вает образование крупных кристаллов. Содержание примесей
в камфаре сравнительно невелико, но они не переходят целиком
в маточный раствор, а частично остаются в виде твердого рас-
твора в выпавшей камфаре (смешанные кристаллы). Концент-
рация каждой примеси в жидкой и твердой фазах определяется
коэффициентом распределения К из уравнения
-^=^4’ <19>
dX
где -----соотношение между чистым веществом и загрязне-
нием в выпавших кристаллах;
X
—---соотношение между чистым веществом и загрязне-
нием в равновесной жидкой фазе.
Чем выше коэффициент распределения, тем эффективнее рас-
творитель.
Выпавшие из раствора кристаллы камфары отжимают от ма-
точника на центрифуге. Полученные после отжима маточные
растворы упаривают и выделяют из них камфару с понижен-
ной температурой кристаллизации, которую направляют на вто-
ричную перекристаллизацию. Первый маточный раствор может
быть также использован в качестве растворителя для перекри-
сталлизации «сырой» камфары. Процесс переработки маточни-
ков обычно продолжается до тех пор, пока в остатке не оста-
нется некристаллизующееся масло, носящее название камфар-
ного масла, которое является отходом производства.
В состав камфарного масла входят камфара, изофенхон,
изокамфанон, изоборнеол, терпеновые углеводороды и некоторое
количество растворителя. Правильный выбор растворителя —
одно из существенных условий для успешного проведения про-
цесса перекристаллизации. Помимо высокого коэффициента рас-
пределения примесей между твердой и жидкой фазами (урав-
нение 19), растворимость камфары должна быть высокой при
повышенной температуре и низкой при охлаждении. Только
в этом случае можно ждать высокого выхода при каждой от-
дельной операции.
В промышленности СССР с 1938 г. в качестве растворителя
для перекристаллизации камфары успешно применяют этиловый
спирт. Этот же растворитель применяли и в Германии [167].
Спирт в качестве растворителя удобен благодаря значительной
разнице растворимости в нем камфары в зависимости от темпе-
ратуры и умеренной его летучести, что позволяет работать без
больших потерь растворителя.
На рис. 27 показана зависимость растворимости камфары
в этиловом спирте от температуры [169].
160
Пользуясь графиком, нетрудно выбрать режим перекристал-
лизации, пригодный для того или иного случая, обеспечивающий
осаждение из раствора заданного количества камфары, или,
наоборот, вычислить выход камфары, который получится при
охлаждении ее насыщенного раствора до заданной температуры.
Рис. 27. Растворимость камфары в спирте и водно-
спиртовых растворах. Концентрация спирта, %;
1 — 40; 2 — 50; 3 — 60; 4 — 70; 5 — 80; 6—90; 7 — 95;
8 — 100
Для перекристаллизации камфары можно почти с равным успе-
хом применять спирт концентрацией 75—100%. Это позволяет
вести перекристаллизацию технической камфары, содержащую
Таблица 46
Эффективность отделения различных примесей при перекристаллизации
камфары из 95%-ного этанола
Показатели Камфара *
исходная после двух перекристал* лизаций исходная 1 после одной перекристал* лизацин исходная 1 после одной перекристал- । лизацин после двух перекристал- лизаций 1
Температура кри- сталлизации, °C Состав (%): камфара трициклен и камфен цнкланы изофенхон изокамфанои Прочие примеси, в том числе влага и спирт 171 94 0,2 0,8 1,9 2,0 1,1 176 98,6 0,1 0,5 0,2 0,3 0,3 162,0 90,1 Следы 1,5 2,3 4,7 1,2 171,5 93,9 Следы 1,0 0,6 3,1 1,4 162,9 86,0 0,7 5,3 2,1 2,4 3,5 168,8 91,3 0,5 4,7 0,8 1,9 0,9 171,4 93 0,4 4,0 0,3 1,1 1,2
* Выход перекристаллизованной камфары при каждой перекристаллиза-
ции около 65%.
11 Заказ № 43
161
влагу. Например, в случае растворения 4 массовых частей кам-
фары с влажностью 4% в одной части 95%-ного спирта его кон-
центрация снизится до 82%, т. е. останется в допустимых пре-
делах. Из графика можно также видеть, что глубокое (ниже
+ 10---I- 15°С) охлаждение при перекристаллизации камфары
из спирта не нужно, так как дает незначительный эффект.
Для перекристаллизации камфары с целью очистки ее от
изофенхона весьма эффективным растворителем оказывается
этанол, в меньшей степени он эффективен для очистки от изо-
камфанона и не эффективен для очистки от углеводородов. Это
иллюстрируется данными табл. 46. Возможная примесь спирта
к камфаре при применении ее в медицине безвредна, чего
нельзя сказать об углеводородных растворителях. Тем не менее
перекристаллизация камфа-
ры от углеводородного раст-
ворителя может оказаться
целесообразной при необхо-
димости очистить ее от твер-
дых цикланов (камфан, изо-
камфан). Судя по некоторым
литературным источникам,
это делают в Англии [211,
273]. На графике (рис. 28)
приведена зависимость рас-
творимости камфары от тем-
пературы в двух образцах
бензина и в ксилоле.
Конечный выход очищенной
камфары прн переклисталлизации
Растворяется каншары в 100 е
растворителя, г
Рис. 28. Растворимость камфары в бен-
зине и ксилоле:
1 — бензин, фракция 90—100°С. d=0,737; 2 —
бензин, фракция 100—135°С, d=0,743; 3 — кси-
лол
зависит от качества исходного про-
дукта н желаемой степени очистки.
Если исходная камфара имеет
температуру кристаллизации 166—
167°С, а после перекристаллиза-
ции получают камфару с тем-
пературой крнсталлизацин 169—
170°С, выход достигает 92—95%.
Одновременно в качестве отхода получается 2—3% камфарного масла. Если
исходная камфара имеет температуру кристаллизации 160°С, выход камфары
(т. крнст. 169—170°С) снижается до 85—88%, а выход камфарного масла
соответственно увеличивается. Выход первой фракции кристаллов (т. крист.
169—170°С) в обоих случаях составляет около 60—65%.
Аппаратура для перекристаллизации камфары состоит из смесителя для
ее растворения, нутч-фильтра для отделения кристаллов камфары от маточ-
ника, кристаллизаторов непрерывного или периодического действия, центри-
фуги для отжима камфары, сборников для маточников, куба для -отгонки
растворителя из маточников и ректификационной колонны для рекуперации
растворителя.
Отжатая на центрифугах камфара содержит небольшое количество спирта.
В некоторых странах такую камфару перед выпуском в продажу освобо-
ждают от спирта в вакуум-сушилках.
162
ГЛАВА XII. МЕТОДЫ ИССЛЕДОВАНИЯ,
СВЯЗАННЫЕ С ПРОИЗВОДСТВОМ КАМФАРЫ
В настоящей главе описываются методы исследований, кото-
рыми можно пользоваться при проведении повседневного
фабрично-заводского контроля, а также методы, необходимые
для решения более сложных задач. Наряду с описанием методик
отдельных исследований разбираются их воспроизводимость,
точность и границы применения.
А. Исследование с применением
газо-жидкостной хроматографии *
В производстве камфары наиболее важное место занимают
анализы многокомпонентных смесей терпенов. Сюда относятся
анализы скипидара, технического пинена, пихтового масла, изо-
меризатов, технического камфена и др. Некоторые из этих ана-
лизов должны производиться постоянно, другие спорадически.
В первой четверти текущего столетия такая задача пред-
ставлялась практически не выполнимой. Даже качественный
анализ любого из перечисленных продуктов следовало оценивать
как трудное и продолжительное исследование, в результате ко-
торого можно было получить весьма ограниченную информацию.
Данные же о количественном составе можно было получить
в основном лишь для тех или иных групп соединений, например,
данные о содержании 2ЭВ, сложных эфиров, кетойов и т. д.
В 1930—1940 гг. была разработана аналитическая ректифика-
ция. С ее помощью исследуемый продукт Делили на большое
число фракций приблизительно равного объема, рассчитывая
таким путем получить чистые компоненты и ряд бинарных сме-
сей с тем, чтобы потом установить их состав физическими ме-
тодами. Результаты анализов отдельных фракций суммировали.
В дальнейшем аналитическая ректификация непрерывно совер-
шенствовалась. Физические методы анализа стали распростра-
нять и на тройные смеси терпенов [148, 322]. Внедрение анали-
тической ректификации в практику химических лабораторий
позволило выполнить большинство перечисленных задач. Однако
трудоемкость метода и продолжительность каждого из пере-
численных исследований ограничивали его применение.
С 1952 г. в практике анализа смесей органических веществ
получил широкое распространение новый метод исследования —
газо-жидкостная хроматография (ГЖХ). Этот метод основан
на продвижении молекул вещества в потоке газа-носителя через
твердый сорбент с нанесенной на него жидкой фазой. Разделе-
ние веществ осуществляется за счет многократного перераспре-
деления вещества между жидкой и газовой фазами в хромато-
графической колонке. Вещества, имеющие различные коэффици-
* Написана совместно с Р. И. Сидоровым и В. И. Гармашовым.
И*
163
енты распределения, передвигаются вдоль колонки с различной
скоростью и определяются иа выходе из нее детекторами.
По сравнению с ранее использовавшимися методами иссле-
дования ГЖХ имеет ряд крупных преимуществ:
1. Трудоемкость и продолжительность исследования сокра-
щается в десятки раз.
2. Высокая эффективность хроматографических колонн и
чувствительность детекторов увеличивает количество полученной
информации о составе анализируемых веществ за счет их луч-
шего разделения и определения незначительных примесей,
ускользающих от определения при аналитической ректификации.
3. Возможность исследования с минимальным количеством
анализируемого вещества (10—0,001 мг).
ГЖХ стала широко использоваться при анализе органиче-
ских веществ, в том числе при исследовании эфирных масел,
скипидаров и других смесей терпенов. Количество исследований
в области ГЖХ очень велико, и они продолжаются в связи с ее
непрерывным совершенствованием. Лишь по анализу терпенов
методом ГЖХ за истекшие 20 лет опубликовано более 800 ра-
бот. Теоретические основы и практика ГЖХ изложены во мно-
гих специальных монографиях, например [94, 157], к которым мы
отсылаем читателя, поскольку материал слишком велик, чтобы
войти в настоящую книгу. Здесь уместно остановиться лишь на
некоторых специальных вопросах применения ГЖХ при иссле-
дованиях, связанных с производством камфары.
1. Аппаратура
Анализ методом ГЖХ проводится на хроматографах различных марок,
отличающихся друг от друга главным образом способом детектирования
и типом разделяющих колонок.
Детекторов различных типов, используемых в ГЖХ, насчитывается более
десятка, но наиболее широкое распространение нашли катарометры н пла-
менно-ионизационные детекторы. Принцип работы катарометра основан на
различии в теплопроводности таких газов, как гелий, водород и азот (газы-
носители), с одной стороны, и паров органических соединений — с другой.
Пламеино-ионизациоиные детекторы основаны на способности органических
веществ давать ионы в водородном пламени, количество которых пропорцио-
нально содержанию вещества в газе-носителе. Чувствительность пламенно-
ионизационных детекторов на 2—3 порядка выше чувствительности катаро-
метров.
Колонки для ГЖХ можно разделить на два типа — насыпные и капилляр-
ные. Первые обычно небольшой длины — 0,1—10 м при внутреннем диаметре
2—10 мм. Материалом для их изготовления служат: сталь, медь, алюминий,
стекло. Насыпными они называются в соответствии с тем, что заполняются
твердым носителем, несущим на себе стационарную жидкость, называемую
неподвижной фазой. В отличие от них капиллярные колонки содержат ста-
ционарную жидкость, нанесенную на внутреннюю поверхность капилляра
диаметром 0,1—0,5 мм. Длина капиллярной колонки может достигать 300 м.
Материалом для изготовления капиллярных колонок служит нержавеющая
сталь, стекло, медь и ее сплавы, полимеры.
Разделяющая способность капиллярных колонок по сравнению с насып-
ными выше в 10 раз н более. В то же время количество вещества, вводимое
в капиллярные колонки, примерно в 100—1000 раз меньше количества, кото-
164
рое можно ввести в насыпную колонку. Поэтому для определения микропрп-
месей предпочтительнее насыпная колонка при условии достаточно хорошего
разделения анализируемых веществ и использования пламенно-нони ра-
ционного детектора. Практика анализов сложных смесей терпеновых углево-
дородов и их производных (эфирные масла, скипидары, продукты и сырье
камфарного производства) показывает, что использование капиллярных коло-
нок с различными стационарными фазами дает большую информацию о со-
ставе вышеназванных продуктов, по сравнению с применением насыпных.
В свою очередь, простота работы с насыпными колонками делают их приме-
нение удобным в условиях производства.
Применяются также микронасыпные колонки. Например, колонка диа-
метром в 1 мм и длиной 2 м, заполненная халькомидом-18, нанесенным на
хромосорб, была использована для анализа продуктов каталитической изоме-
ризации пинена на титановом катализаторе [289]. Объем информации уступал
объему информации, получаемому на капиллярных колонках. Основным преи-
муществом микронасыпных колонок является быстрота анализа. Однако им
присущи и отрицательные свойства других колонок: невысокая эффективность
насыпных колонок н малая емкость капиллярных.
2. Качественный анализ
Качественный состав продуктов, с которыми имеет дело про-
изводство синтетической камфары, в основном известен. Обычно
производственника интересует не качественный, а количествен-
ный состав. Однако для того, чтобы сделать правильные коли-
чественные выводы из хроматограмм, необходимо знать, каким
соединениям соответствуют полученные пики, т. е. необходимо
произвести расшифровку записи самописца или осуществить ка-
чественный анализ.
Выводы о качественной принадлежности того или иного пика
на хроматограмме делают исходя из относительного удержива-
ния образующего его вещества а.
Объем газа-носителя, необходимый для элюирования компо-
нента исследуемой смеси из хроматографической колонки, на-
зывается удерживаемым объемом, а необходимое на это время —
временем удерживания т.
Времени удерживания элюируемого компонента соответст-
вует отрезок на хроматограмме от момента ввода пробы до мо-
мента появления максимума соответствующего пика. Эта вели-
чина зависит как от условий анализа, так и от особенностей
прибора, поэтому она несопоставима с аналогичными величи-
нами, полученными на других приборах.
Чтобы сделать поправку на мертвый объем прибора, отсчет
ведут не от момента ввода пробы, а от момента выхода несор-
бируемого вещества, воздуха или метана, вводимых вместе
с пробой. Полученное таким образом исправленное время удер-
живания (ти), хотя и не зависит от мертвого объема прибора,
продолжает зависеть от прочих условий хроматографирования.
Однако отношение исправленного времени удерживания (ти)
к исправленному времени удержания вещества — стандарта (^7)
165
Таблица 47
Относительное удерживание (а) терпеновых углеводородов и некоторых
окисей вакуумным маслом (ВМ-4)* и трикрезилфосфатом [(ТКФ) стандарт
лимонен] по работам [19, 23, 165, 232] и данным лаборатории природных
соединений Института нефте- и углехимического синтеза
при Иркутском государственном университете
Соединения а иа стационарных фазах
ВМ-4, °C ТКФ, °C
ПО 130 по 130
Сантен 0,30 0,27
б-Фенхен 0,30 0,34 — —
Циклофенхен 0,33 0,36 0,26 —
у Фенхен 0,39 0,42 0,32 —
Борнилеи 0,40 0,44 0,34 0,38
Трициклен 0,47 0,52 0,38 0,43
а-Пинен 0,50 0,54 0,42 0,47
Р Фенхен 0,52 —. 0,47 —
«-Фенхен 0,55 —. 0,50 —
Камфен 0,57 0,62 0,53 0,58
Мирцен 0,64 0,64 0,72 0,72
«-Ментен-8 (9) «— 0,72 0,58
Изолимоиен 0,69 0,72 0,68 0,69
п -Ментен-3 0,66 — 0,53 —
Р-Пинен 0,70 0,73 0,67 0,71
тланс-Пинан — 0,75 — 0,58
«-Ментен-2 — 0,76 0,55 0,61
тмнс-Каран — 0,76 — 0,59
(</гс-Каран — 0,77 — 0,61
Л4-Карен 0,75 0,78 0,72 0,74
транс-п-Ментан 0,73 0,80 0,48 0,55
/<;гс-Пинан — 0,80 — 0,62
/лгс-н-Ментан 0,78 0,84 0,53 0,57
Л’-Карен 0,85 0,86 0,80 0,86
а Фелландрен 0,88 0,85 —
Цинеол-1,4 0,93 —. — —
о-Терпинен 0,88 0,90 0,92 0,93
п-Цимол 0,94 0,95 1,33 1,31
t -Ментен-4(8) — 0,97 — 0,81
Цинеол-1,8 1,00 0,98 1,20 1,16
Лимонен (Дипентен) 1,00 1,00 1,00 1,00
Р Фелландрен 1,02 1,03 1,08 1,09
у-Терпинен 1,20 1,17 1,20 1,26
Терпинолен 1,44 1,39 1,54 1,50
г -Ментадиен-2,4 (8) 1,48 1,45 — 1,57
есо-Аллооцимен 1,75 1,63 2,15 2,00
Дллооцимен 1,92 1,79 2,45 2,26
* В практике анализов терпенов в Советском Союзе часто используют
в качестве стационарной жидкой фазы вакуумное масло ВМ-4, предвари-
тельно освобожденное от более легкокипящих компонентов прогреванием
и течение 2 ч в вакууме при 120—130°С.
166
в тех же условиях хроматографирования, называемое относи-
тельным удерживанием или относительным временем удержи-
вания (а), является постоянной величиной, зависящей лишь от
температуры колонки и вида жидкой фазы и поэтому может
использоваться для идентификации пиков.
(20)
В табл. 47—49 приведены относительные удерживания тер-
пеновых углеводородов, их кислородных производных, а также
некоторых специфических компонентов, входящих в состав кам-
фары, и полупродуктов ее производства на ряде стационарных
фаз.
Таблица 48
Относительное удерживание (а) кислородных производных терпенов
на разных стационарных фазах (стандарт камфара) [22]
Соединения а на стационарных фазах
ПФМС -41 2, °C ПФМС-4-f- + ПЭГ-600°C ДНФ + + ПЭГ- 6003, °C ДЭГА4, СС
по по 140 120 105 150
Фенхон 0,63 0,60 0,64 0,63 0,50 0,58
Фенхол 0,66 0,78 0,76 0,97 1,08 1,05
Изофенхон 0,73 0,73 0,75 0,74 0,70 0,75
Р-Терпинеол 0,75 0,84 0,80 0,95 1,11 0,93
транс- Дигидро-а-терпинеол 0,78 0,87 0,82 1,13 1,04 0,91
у-Терпинеол 0,85 1,02 0,91 1,19 1,50 1,21
Изоборнеол 0,93 1,09 1,03 1,27 1,58 1,37
-и-Ментен-1-ол-8 0,86 1,08 0,99 1,21 1,33 1,12
Камфара 1,00 1,00 1,00 1,00 1,00 1,00
Борнеол 1,00 1,21 1,12 1,44 1,89 1,47
Метилкамфенилол 1,02 1,18 1,И 1,49 1,95 —
Терпиненол-4 1,03 1,10 1,03 1,27 1,37 1,11
Эпикамфара 1,12 1,14 1,11 1,12 1,30 1,23
Камфенгидрат 1,12 1,06 0,95 1,16 1,30 1,16
л-Ментен-6-ол-8 1,16 1,36 1,18 1,59 2,14 1,58
Фенхилацетат 1,18 1,12 1,06 1,13 0,67 0,76
а-Терпинеол 1,19 1,36 1,19 1,66 2,18 1,59
Метплхавнкол 1,43 1,42 1,32 1,45 1,97 1,41
Вербеной 1,76 1,81 1,66 1,75 2,53 2,13
Борнилацетат 1,94 1,88 1,64 1,83 1,18 1,26
Изоборнилацетат 2,01 1,89 1,66 1,77 1,22 1,53
Карвон 2,01 2,07 1,85 2,09 2,84 2,15
а-Терпенилацетат 2,90 2,85 2,32 — 2,16 1,56
1 Полифенилметилсилоксан-4.
2 Полифенилметилснлоксан-4 5% с добавкой полиэтиленгликоля-600 0,2%.
3 Динонилфталат с добавкой полиэтиленгликоля-600 0,2%-
4 Диэтиленгликольадипинат.
167
Таблица 49
Относительное удерживание (а) веществ, входящих в состав
синтетической камфары и полупродуктов ее производства 1
(стандарт бориеол)
Соединения а на стационарных фазах
Апиезон N, СС ВМ-4, °C ПЭГ-2000, °C ДНФ, сс
150 130 120 120
Углеводороды:
трициклен 0,192 0,220 — 0,106
камфен 0,288 0,280 — 0,137
циклан-1 2 0,408 0,360 0,040 0,157
циклан-П 2 0,428 0,380 0,047 0,163
Спирты:
изофенхол 0,673 0,707 0,593 0,666
изоборнеол 0,964 0,960 0,850 0,900
борнеол 1,000 1,000 1,000 1,000
псевдоборнеол 3 1,290 1,182 1,353 1,270
фенхон 0,575 0,564 0,254 0,444
изофенхон 0,630 0,616 0,330 0,537
камфара 0,771 0,821 0,446 0,701
Xi 0,963 0,967 0,614
х2 1,000 1,020 0,638 0,933
Псевдокамфара 3 1,060 1,110 0,685 1,032
Эфиры:
изофенхилацетат 0,919 1,328 0,430 0,996
борнилацетат 1,249 1,605 0,592 1,270
изоборнилацетат 1,328 1,735 0,614 1,332
псевдоборнилацетат 3 1,484 2,306 0,841 1,804
1 По данным лаборатории природных соединений Института нефте- и угле-
химического синтеза при Иркутском государственном университете.
2 Состоит из цис- и транс-изокамфанов.
3 Псевдоборнеол (изокамфанол, 5-экэо-изокамфан-экзо-ол-З), псевдобор-
нилацетат, его уксусный эфир; псевдокамфара (изокамфанон, 5-экзо-изокамфа-
Если анализируется продукт, качественный состав которого
достаточно хорошо известен, а для его анализа выбрана ста-
ционарная фаза, обеспечивающая полное разделение содержа-
щихся в нем компонентов, то на основании определений отно-
сительного удерживания веществ, вышедших на хроматограмме,
можно практически без ошибок расшифровать полученную хро-
матограмму.
Если анализируется продукт, качественный состав которого
заранее известен не полностью, нельзя сделать выводов о при-
надлежности ряда пиков на хроматограмме на основании опре-
168
деления относительных удерживаний на одной стационарной
фазе, так как многие вещества имеют близкие, а часто совпа-
дающие относительные удерживания. Во многих случаях пра-
вильные выводы могут быть сделаны после определений отно-
сительных удерживаний на 2—3 колонках со стационарными
фазами разной полярности. Удерживание зависит от строения
хроматографируемых веществ и характера входящих в них
функциональных групп и при переходе от одной стационарной
фазы к другой для разных соединений меняется по-разному.
Поэтому совпадение относительных удерживаний на нескольких
фазах разной полярности с табличными данными позволяет сде-
лать идентификацию пиков достаточно надежной. Это же по-
зволит убедиться в том, что те или иные пики образованы од-
ним веществом. При образовании пика на данной фазе двумя
веществами с одинаковым удерживанием есть все основания
ожидать, что на фазе другой полярности эти пики разделятся.
Во многих случаях для идентификации веществ, содержащихся в неиз-
вестной смеси, приходится их выделять с помощью препаративной ГЖХ или
ректификации и устанавливать их природу спектральным анализом, изучением
физических свойств и получением химических производных. Это не обычные
случаи, ио они могут встретиться при проведения исследовательских работ,
связанных с производством синтетической камфары.
3. Количественный анализ
Количественный анализ методом ГЖХ основан на зависи-
мости величины сигнала детектора или площади пика, записан-
ного самописцем, от массового количества вещества, поступив-
шего в детектор с газом-иосителем.
При проведении количественного анализа должны быть
исключены химические превращения в испарителе и хромато-
графической колонке (например, термическое разложение или
изомеризация), должны соблюдаться точное термостатирование
колонки и постоянная скорость газа-носителя.
К хроматограммам, используемым для количественных рас-
четов, предъявляется ряд требований: нулевая линия должна
быть стабильной, наложение пиков различных компонентов
должно быть исключено, что достигается соответствующим под-
бором жидкой стационарной фазы, пики должны быть симмет-
ричны, приближаясь своей формой к гаусовской кривой [94, 157].
Метод внутренней нормировки. Расчет процентного состава
смеси при применении этого метода производится делением из-
меренной площади каждого пика, умноженной иа 100, на сумму
площадей всех пиков.
Непременное условие возможности применения этого ме-
тода — отсутствие в смеси веществ, не образующих пиков на
хроматограмме, например, примесей высококипящих терпеновых
полимеров, смол или воды. Иначе метод приводит к неверным
результатам, если только указанные вещества не определяются
169
отдельно и на их присутствие не вносится поправка в расчет.
В практике анализов, связанных с производством камфары, на-
личие таких примесей в анализируемых смесях явление доста-
точно частое.
Некоторые компоненты содержатся в анализируемых смесях
в столь малых количествах, что не регистрируются самописцем
при работе с катарометром. Если этих компонентов много и их
содержание составляет в.общей сложности несколько процентов,
в результаты определения компонентов, поддающихся учету,
опять-таки вносится заметная ошибка.
При применении метода внутренней нормировки по отноше-
нию к смесям, содержащим вещества, различающиеся по своей
природе, необходимо применять коэффициенты относительной
чувствительности, так как величина сигнала детектора зависит
не только от массового содержания детектируемого вещества
в газе-носителе, но и от его природы. В связи с тем, что разные
детекторы работают по различному принципу, коэффициенты
относительной чувствительности должны определяться для каж-
дого детектора. Так, например, для катарометра и пламенно-
ионизационного детектора применяются разные коэффициенты
относительной чувствительности, причем для последнего они
значительно выше. При анализе смесей веществ близкой при-
роды с применением катарометра, например смесей терпеновых
углеводородов, коэффициенты относительной чувствительности
можно не применять. При применении пламенно-ионизационного
детектора в этом случае в результаты анализа будут вноситься
уже заметные систематические погрешности (несколько процен-
тов), а при анализе смеси веществ разной природы, таких, как
техническая камфара, пихтовое масло и т. п., погрешности мо-
гут оказаться очень значительными.
При применении метода внутренней нормировки следует
иметь также в виду, что линейная зависимость площадей пиков
от концентрации соответствующих веществ в газе-носителе со-
блюдается достаточно точно в ограниченных пределах [157],
поэтому при расчете хроматограмм смесей, в которых содержа-
ние компонентов очень сильно различается (скипидар, техни-
ческий камфен, камфара), вносятся дополнительные погреш-
ности.
При измерении площадей малых пиков возникает погреш-
ность, для устранения которой прибегают к переключению чув-
ствительности прибора, что связано с изменениями сопротивле-
ний в электрической схеме. Хотя это и увеличивает точность
измерений, но и вносит дополнительные погрешности.
Метод внутренней нормировки при анализе многокомпонент-
ных смесей осложняется трудоемким измерением площадей всех
пиков. В то же время метод внутренней нормировки очень полезен
в случаях, когда надо получить полные данные о составе иссле-
дуемой смеси.
170
г
Метод внутреннего стандарта. При производстве количест-
венного определения по этому методу к навеске анализируемого
материала добавляют навеску несодержащегося в нем вещества-
стандарта. Смесь хроматографируют и вычисляют отношение
площадей пиков стандарта и определяемого вещества. После
этого из предварительно составленного калибровочного графика
находят, какому массовому соотношению между стандартом и
определяемым веществом соответствует данное соотношение
площадей, а из него находят
процентное содержание опре-
деляемого вещества в анализи-
руемом материале.
К используемому в ка-
честве стандарта веществу
предъявляются следующие тре-
бования: оно должно быть
очень чисто, его пик должен
выходить близко к пику опре-
деляемого вещества и в то же
время не перекрывать других
пиков, оно не должно образо-
вывать асимметричного пика
и иметь летучесть, близкую
к летучести определяемого ве-
щества.
В качестве примера рассмо-
трим определение основного
вещества в товарной камфаре,
используя в качестве стан-
дарта нафталин [154].
К навеске товарной камфары
1,052 г добавили навеску нафталина
0,848 г. Величина навески нафталина
подбирается с расчетом получения со-
отношения площадей пиков стандарта
Рис. 29. График относительно!! ка-
либровки для количественного опре-
деления камфары с использованием
нафталина в качестве внутреннего
стандарта:
sH/sK — отношение площадей пиков наф-
талина и камфары; >тк — они пение
масс нафталина н камфары
и определяемого вещества в сонзме-
римых пределах, чтобы повесить точность анализа. После хроматографирова-
ния раствора смеси в серном эфире было получено соотношение межд> пло-
щадями пиков, нафталина и камфары SH/SK= 1,022. Из графика рис. 29
следует, что этому соотношению площадей соответствует соотношение между
массами нафталина й камфары (тк) в анализируемом материале-^-—=0,860,
откуда процентное содержание камфары в исследуемом образце сост; вляет
0,848-100 _qo70,
0,860-1,052 '°'
Результаты анализов, выполненные методом внутреннего
стандарта, надежнее, чем результаты анализов, выполненных ме-
тодом внутренней нормировки, так как при применении метода
внутреннего стандарта исключаются систематические погреш-
ности из-за присутствия в анализируемой смеси компонентов,
171
не регистрируемых детектором. Ошибка в определении одного'
из компонентов смеси не влияет на точность определения других
компонентов, как это имеет место при применении метода внут-
ренней нормировки. Применение метода внутреннего стандарта
значительно упрощает обсчет хроматограмм в случае, когда
нужно узнать не полный анализ смеси, а определить в ней
1—2 компонента. Тогда дело ограничивается измерениями пло-
щадей пиков стандарта и определяемого вещества. При приме-
нении этого метода отпадает и необходимость в определении
коэффициентов относительной чувствительности всех компонентов,,
содержащихся в смеси. Подбором условий хроматографирования
можно уменьшить и погрешности в измерении площадей пиков.
Отрицательными сторонами метода является трудность подбора
стандартного вещества, удовлетворяющего изложенным требо-
ваниям.
Воспроизводимость и точность анализа. ГЖХ получила ши-
рокое применение в количественном анализе, но, к сожалению,
метод часто применяют без учета погрешностей определений.
Поэтому необходимо кратко остановиться на погрешностях
метода.
Возможные источники ошибок хроматографического метода
располагают в такой последовательности [94}: а — ошибки в ме-
тодике ввода пробы; б — адсорбция или разложение пробы
в хроматографе; в — неправильная оценка характеристики де-
тектора; г — неправильная оценка характеристики самописца;.
д — неточность метода интегрирования (определение площадей).
О ряде возможных крупных систематических ошибок указана
выше. Остановимся на наиболее «критическом этапе» с точки
зрения внесения случайных (не систематических) погрешно-
стей— на определении площади хроматографического пика
в виде числа, связанного с составом пробы [94].
Для измерения площадей хроматографических пиков исполь-
зуются различные методы: планиметрирование, вырезание и
взвешивание, определение площади треугольника, расчет с по-
мощью электронного цифрового интегратора. Иногда вместо
площадей измеряют пропорциональные им величины: произве-
дение высоты пика на время удерживания (отрезок на хрома-
тограмме от запуска до максимума пика), произведение высоты
пика на его ширину на половине высоты или просто высоту
пика (при обязательной калибровке).
Сравнение различных методов интегрирования показывает,
что стандартное отклонение или средняя квадратичная погреш-
ность [70, 168] (о) расчета с помощью электронного интегратора
лежит в пределах меньше 0,5% [94]. Одиако лишь незначитель-
ная часть хроматографов снабжена интеграторами, в результате
чего обсчеты обычно производятся на основе ручных измерений
по одному из перечисленных способов. Воспроизводимость полу-
ченных результатов в среднем лежит в пределах стандартного
172
отклонения (а) 2—4% относительных [94, 157]. При измерении
площадей малых пиков погрешность значительно возрастает и
может достигнуть нескольких десятков процентов. Наоборот,
когда измеряются площади достаточно больших пиков, зани-
мающих по высоте 50—70% ширины ленты самописца, погреш-
ность обычно лежит в пределах 1—1,5%. При хроматографиро-
вании методом внутреннего стандарта стандартное отклонение
может быть доведено до 0,5% за счет получения наиболее бла-
гоприятных для обсчета пиков определяемого вещества и стан-
дарта, что требует соответствующей опытности эксперимента-
тора.
Из теории распределения погрешностей известно, что 68%
результатов анализа лежат в пределах стандартного отклонения
о, 95% в пределах 2а и 99,7% в пределах За [70, 168]. Поэтому,
рассматривая единичный анализ, можно считать, что в лучшем
случае его результаты лежат в пределах ±1% отн. при довери-
тельной вероятности 0,95, а обычно в пределах ±2—3% отн.
При определении примесей, содержащихся в анализируемом
материале в количестве меньшем 1%, результаты могут лежать
в пределе ±20% отн. при названной доверительной вероятности.
Погрешности могут быть уменьшены путем проведения ряда
параллельных анализов и вычисления среднего арифметического
результата. Стандартное отклонение среднего арифметического
(S) составляет:
5 = -^, (21)
Ул
где п — число параллельных определений.
Так, например, произведя 10 параллельных анализов, можно
уменьшать относительную погрешность единичного результата
±2% отн. до 0,7%. Однако это приведет к неприемлемому для
практики увеличению продолжительности исследования.
Примеры применения ГЖХ в камфарном производстве. ГЖХ
практически можно применять для анализа всех смесей терпе-
нов, встречающимися в производстве камфары, с ее помощью
можно получить ответы о составе сырья, полупродуктов и гото-
вой продукции производства. ГЖХ в принципе позволяет вести
наблюдение и за ходом процессов на основании анализа проб,
взятых в разное время из реакторов. Впрочем, не во всех слу-
чаях такое широкое применение ГЖХ целесообразно, так как
иногда можно получить ответы на выдвигаемые производством
вопросы быстрее, а иногда и точнее иными методами. Кроме
того, газохроматографические методы еще не разработаны для
решения всех возможных конкретных задач производства даже
там, где их применение в принципе вполне возможно.
Рассмотрим отдельные примеры применения ГЖХ в произ-
водстве камфары.
173
Анализ камфары [154]. Товарная синтетическая камфара содержит SO-
95 % основного вещества н ряд примесей, из них главные: терпеновые угле-
водороды (камфен и трициклен), гидрированные терпены (изокамфан), терпе-
новые спирты (борнеол, изоборнеол), терпеновые кетоны (фенхон, изофен-
хои, изокамфанон), см. гл. XI, табл. 47 и 48. Кроме -того, камфара может
содержать влагу и небольшое количество этанола, оставшееся в ней после
перекристаллизации. Такой состав продукта требует производства анализа
методом внутреннего стандарта. Определение основного вещества в камфаре
может быть произведено на насыпной колонке, что удобно в условиях произ-
водственной лаборатории; используют колонку длиной 2 м с внутренним диа-
метром 4 мм. Твердый носитель полихром I (0,25—0,5 мм), стационарная
жидкая фаза апиезон L или N (10%), газ-носитель водород, предпочтительнее
елий, при скорости 80 см3/мин. Детектор катарометр. Температура испари-
теля 240—260°С, температура колонки 170±0,5°С. В качестве стандарта при-
меняют нафталин [184, 244], который можно иметь в виде очень чистого про-
дукта.
Для построения калибровочного графика необходимо использовать чистую
камфару, получаемую из не содержащих примесей других кетонов японской
естественной камфары или камфары, полученной из пихтового масла, очищая
их через семикарбазоиы: 20 г камфары растворяют в 40 мл ледяной уксус-
ной кислоты, добавляют 20 г солянокислого семнкарбазида и 30 г свежеплав-
енного уксуснокислого натрия. При перемешиваиин греют 3 ч при темпера-
туре 70°С. По охлаждении добавляют 300 .см3 дистиллированной воды и пере-
мешивают до полного растворения солей. Осадок семикарбазона промывают
на стеклянном фильтре водой, петролейиым эфиром и перекристаллизовы-
вают из этилового спирта.
Просушенный семикарбазон разлагают 10%-ной серной кислотой при на-
гревании иа водяной бане с обратным холодильником в течение 4 ч. Выделив-
шуюся камфару перегоняют с водяным паром, отделяют от воды, отжимают
п сушат в эксикаторе до постоянной массы. Чистоту продукта проверяют хро-
матографически.
Достоверность анализов в значительной степени определяется правильно-
стью калибровочного графика. Поэтому каждая точка для построения гра-
фика должна быть средним арифметическим нескольких определений. Прямая
проводится иа основании расчета методом наименьших квадратов.
Стандартное отклонение при определении камфары о=±0,5% относитель-
ных, следовательно, анализ может быть выполнен с воспроизводимостью
± 1 % относительных при доверительной вероятности 0,95. Увеличение коли-
чества проб соответственно увеличивает точность анализа.
Для анализа с успехом может быть использована аналогичная колонка
с пламенно-ионизационным детектором [184] и капиллярная колонка длиной
50—100 м с той же стационарной жидкой фазой или с полипропилеиглико-
лем (завод фирмы Хехст в Герстгофене, ФРГ).
Метод внутреннего стандарта следует признать единственно надежным
методом и для определения примесей в камфаре.
Для анализа можно использовать как насыпную колонку (150—170°С),
так и капиллярную (120—150°С), последнюю предпочтительнее. Прочие усло-
вия хроматографирования те же, что и при определении камфары. В качестве
стандарта для определения изомерных кетонов также используют нафталин
и, исходя из близкой природы камфары, нзофенхона и нзокамфанона, поль-
зуются тем же калибровочным графиком, что и для определения камфары.
В качестве стандарта для определения терпеновых углеводородов и цикланов
используют ундекан, который можно иметь в виде очень чистого вещества.
Стандартное вещество в данном случае вводится в значительно меньшем
количестве, с таким расчетом, чтобы образованные им пики были соизмеримы
с пиками примесей, а в колонку вводят большее количество вещества, чтобы
обеспечить достаточно большие пикн примесей. Оба стандартных вещества
могут быть прибавлены к навеске камфары одновременно.
Описанный анализ позволяет получить более полную и более правильную
информацию о составе камфары, чем ранее использовавшиеся химические
174
методы анализа. Применявшееся в ряде стран массовое определение кам-
фары в виде 2,4-динитрофенилгидразона [106] при анализе синтетической
камфары приводило к завышенным результатам, так как одновременно
с 2,4-динитрогидразоном камфары осаждались 2,4-динитрофенилгидразоны
изофенхона и 5-экзо-изокамфаиона-З [154]. При определении этим методом
основного вещества в камфаре из пихтового масла получают те же результаты,
что и при определении методом ГЖХ, но воспроизводимость результатов
худшая (а=—1,5) [106] при более трудоемком анализе. Кроме того, метод
ГЖХ дает исчерпывающую информацию о примесях к камфаре, что не дают
прежние методы анализа.
Анализы борнеолов, борнильных и изоборнильных эфиров и пихтового
масла с целью определения борнилацетата производятся тем же методом
с применением тех же стандартных веществ. Однако, если анализ ставит
своей задачей лишь суммарное определение эфиров, особенно формиатов
в техническом изоборнилформиате или борнилацетата в пихтовом масле, он
может быть выполнен быстрее и точнее общеизвестным методом гидролиза
титрованным раствором щелочи. Если же анализ ставит своей задачей полу-
чение более подробной информации, например, получить данные не о сум-
марном содержании эфиров, а об их раздельном содержании, то только метод
ГЖХ позволяет дать надлежащий ответ.
Определение а-пинена в скипидарах, техническом пинене и кубовых остат-
ках от ректификации производится с применением неполярных или с.табо-
поляриых фаз: вакуумного масла ВМ-4, апиезонов L и N, трикрезилфосфата.
Обычно используют насыпные колонки длиной 4—6 м, а расчеты ведут мето-
дом внутренней нормировки. Анализируемые продукты содержат высококипя-
щие примеси (скипидар, кубовые остатки от ректификации). Некоторые
компоненты содержатся в них в очень небольшом количестве (примеси к тех-
ническому пинену, камфен, мирцен, p-феландрен, терпинолен, п-цимол в ски-
пидарах). Поэтому н здесь предпочтительнее было бы пользоваться методом
внутреннего стандарта.
Анализы продуктов изомеризации пинена, технического камфена и кубо-
вых остатков от его ректификации осложняются наличием веществ с очень
близкими временами удерживания: трициклен, а-пииен, а- и Р-фенхены, кам-
фен (гл. XIII, табл. 50, 51), поэтому при применении насыпных колонок воз-
можно взаимное наложение пиков. Разделение пииена и трициклена удается
осуществить, пользуясь колонками длиной 4 м. Однако избежать неточностей
анализа за счет совпадения пиков а-пинена и Р-феихена, а также а-фенхена
и камфена обычно не удается. Поэтому при определении пииеиа и камфена
на насыпных колонках надо считаться с систематическими погрешностями.
На капиллярных колонках длиной 100 м с применением в качестве стацио-
нарной жидкой фазы вакуумного масла ВМ-4 все компоненты смеси, за ис-
ключением политерпенов, хорошо разделяются. Количество политерпеиов при
нормальном процессе изомеризации пинена не превышает 2—3%, что позво-
ляет применить для анализа метод внутренней нормировки. Однако в слу-
чаях, когда процесс отклонялся от нормы и полимеры образовались в значи-
тельном количестве, погрешность в результатах анализа увеличивается, по-
скольку политерпены не выходят из колонки и не регистрируются самописцем.
Производя анализ изомеризатов, необходимо параллельно определять поли-
меры и вносить поправку в результаты анализа; если нужно произвести не
полный анализ изомеризатов, а определить в них основные компоненты, целе-
сообразнее применять метод внутреннего стандарта, используя в качестве
стандарта декан. Относительное удерживание декана по лимонену на вакуум-
ном масле ВМ-4 0,742 и его пик не накладывается на пики продуктов изоме-
ризации пинена. Для определения камфена и трициклена в техническом кам-
фене также предпочтительнее применять метод внутреннего стандарта,
поскольку прнмеси других терпенов в нем незначительные. Что касается
оценки качества технического камфена в условиях производства, то оно
может быть сделано достаточно надежно по его температуре плавления.
Стандартное отклонение при определении пинена и камфена в изомеризатах
находится в пределах около 0 = 2% относительных.
175
Контроль за ходом процесса изомеризации а-пинена можно осуществлять,
наблюдая за соотношением сумм площадей пиков камфена и трициклена
к площади пика пинена и по исчезновению а-пинена в конце реакции. Важно
при этом ие спутать пик а-пинена с пиком 0-фенхена и трициклена.
Контроль за ходом процесса дегидрирования изоборнеола может также
производиться на основе сравнения площадей пиков камфары и непрореаги-
ровавших борнеола и изоборнеола в пробах, отбираемых по ходу процесса.
Б. Химические и физические методы исследования
1. Исследования исходных продуктов
Испытание катализаторов для изомеризации пинена. Испы-
тание катализаторов, применяемых для изомеризации пинена,
обычно производится путем проведения пробных опытов изо-
меризации пинена в лаборатории, если стоит вопрос только
о проверке активности катализаторов. Однако когда необходи-
мо выяснить, почему данный катализатор не активен, или если
необходимо произвести испытание нового, прежде не применяв-
шегося катализатора, возникают новые подлежащие выяснению
вопросы. В частности, при отсутствии каталитической активности
у катализаторов следует поинтересоваться, не происходит ли это
из-за недостаточно развитой поверхности или из-за отсутствия
кислотности. При лабораторных испытаниях новых катализато-
ров необходимо установить, не образуется ли наряду с камфе-
ном значительное количество фенхенов.
Испытание катализаторов на активность. Для испытания катализаторов
на активность применяют круглодонную трехгорлую колбу. Через централь-
ное горло помещают в колбу мешалку с затвором, через боковые горла —
термометр и обратный холодильник, обогрев — масляная баня. В колбу загру-
жают 100—200 г свежеперегианиого пинена; 0,01% гидрохинона, который при-
меняется в качестве антиокислителя, что позволяет вести процесс, не защи-
щая поверхность пинена от окисления. После нагревания пинена до темпе-
рату ры примерно на 20° ниже, чем заданная, вводят катализатор. Контроль
за ходом процесса ведут по изменению физических констант продукта и с по-
мощью газо-хроматографического анализа. Отбор проб удобно производить
тонкой пипеткой, которую вводят в колбу через трубку обратного холодиль-
ника. Чтобы получать сравнимые результаты, необходимо проводить процесс
со строго определенным количеством катализатора для данных условий
опыта и всегда использовать свежеперегнанный пинен.
Неверные представления об активности катализатора могут получиться,
если пинеи содержит продукты окисления, так как изомеризующее действие
катализаторов в присутствии продуктов окисления тормозится, а в некото-
рых случаях и полностью подавляется.
При испытании новых катализаторов необходимо определять также и их
полимеризующее действие.
Определение удельной поверхности катализаторов. Удельная поверхность
катализаторов может быть определена различными способами, но даже про-
стейшие из них достаточно сложны [31, 32]. Прост и удобен метод определе-
ния по адсорбции метиленовой голубой. Этот способ основан иа том, что
метиленовая голубая сорбируется из водного раствора твердыми сорбентами
по всей их поверхности, причем 1 мг красителя сорбируется 1 м2 катализа-
тора [133]. Правда, при определении абсолютных величин удельных поверхно-
стей по адсорбции метиленовой голубой возможны значительные ошибки, так
как на разных поверхностях ориентации молекул красителя может меняться
176
и, следовательно, меняется площадь, занятая его молекулой [57]. Когда срав-
нивают катализаторы одного и того же происхождения, метод вполне при-
емлем.
Для производства определения к 60 см3 0,15% раствора красителя при-
бавляют около 0,1 ±0,0002 г катализатора, после чего смесь встряхивают на
машине в течение 2 ч. Через 12 ч раствор декантируют, разбавляют водой
в 100 раз и замеряют его оптическую плотность на фотоколориметре. Кон-
центрация раствора определяется по градуировочной кривой. Из убыли кон-
центрации находят количество красителя, поглощенного навеской, и вычи-
сляют поверхность катализатора.
Обменная способность катализаторов [28]. К 5 г (±0,001 г) катализатора
прибавляют пипеткой 100 см3 1 н. раствора уксусно-натриевой соли и при
периодическом перемешивании выдерживают смесь 1—2 ч. Затем фильтруют
через сухой фильтр, отбирают пипеткой 40 см3 раствора и титруют 0,1 н. рас-
твором едкого натра. Во избежание изменения концентрации раствора (из-за
адсорбции уксусной кислоты фильтром) первые 5 см3 фильтрата отбрасываю:.
Обменные свойства катализатора выражают в мг-экв уксуснокислого натри г,
вступившего в реакцию обмена с 1 г катализатора, или в см3 1 н. раствора
едкого натра, затраченного на титрование уксусной кислоты, выделенной 1 г
катализатора.
Испытание катализаторов для дегидрирования борнеолов.
Испытание катализаторов, применяемых для дегидрирования
борнеолов, в жидкой фазе на активность. За основу активности
катализаторов обычно принимают скорость дегидрирования изо-
борнеола в присутствии постоянного количества катализатора.
Скорость реакции определяют по измерению количества выде-
ляющегося водорода.
Следует указать на два варианта этого испытания. По пер-
вому варианту, используемому на заводах СССР, учитывают
время, необходимое на выделение 2200 см3 водорода (при атмо-
сферном давлении и комнатной температуре) при дегидрирова-
нии 15 г перекристаллизованного из спирта изоборнеола, что со-
ответствует превращению 95% изоборнеола в камфару. Основ-
ная углекислая медь пригодна в качестве катализатора, если
продолжительность реакции в присутствии 3% катализатора не
превышает 45 мин; однако предпочитают те катализаторы, с ко-
торыми реакция проходит за 35 мин и менее. По другому
варианту, применявшемуся в Германии, учитывали лишь про-
должительность начального периода реакции и считали, что
в присутствии 1% углекислой меди 7% изоборнеола должны
прореагировать за 2—8 мин, а 40% —за 5—25 мин. Испытания
велись на техническом, предварительно проверенном изобор-
неоле.
На заводах СССР дегидрирование ведут в широкой пробирке или кругло-
донной колбочке, соединенной с ловушкой Дина и Старка (для улавливанил
выделяющейся воды) и обратным холодильником. Улавливание и учет водо-
рода производятся в перевернутых заполненных водой стеклянных цилинд-
рах, погруженных в ванну с водой.
В Германии испытания катализаторов производили в приборе, изобра-
женном на рис. 30. Технический изоборнеол в количестве 55 г и 40 г рас-
творителя загружали в колбу, после чего содержащуюся в изоборнеоле воду
отгоняли вместе с избытком растворителя. По достижении температуры плава
182—183° вносили катализатор и наблюдали за скоростью выделения
12 Заказ № 43
177
водорода при указанной температуре, которую поддерживали постоянной
в течение всего испытания.
Оценка активности и селективности катализаторов дегидри-
рования борнеола в паровой фазе [102]. Условия дегидрирования
борнеола в паровой фазе резко отличаются от условий дегидри-
рования в жидкой фазе, поэтому катализаторы, используемые
для парофазного процесса, испытываются по иной методике.
Установка схематически изображена на рис. 31.
Рис. 30. Установка для проведения лабораторных работ по дегидрирова-
нию борнеолов:
/ — колба емкостью 250 см3; 2 —приемник для растворителя и воды; 3 — мерный ци-
линдр для учета водорода; 4 — насос; 5 — масляная баня; 6—термометры; / — элек-
трическая плитка; 8 — ванна для воды (размеры, мм)
Дозирующим насосом 1 подают 120 см3 10%-ного раствора изоборнеола
в толуоле со скоростью 186 см3/ч в куб-испаритель 2 из нержавеющей стали,
снабженный электрообогревом и заполненный нихромовой насадкой для уве-
личения поверхности испарения. Из куба-испарителя пары поступают в элек-
трообогреваемый медный контактный аппарат 3, представляющий собой реак-
тор типа труба в трубе, длиной 400 мм с межтрубным пространством 7 мм,
в котором находится 16 см3 предварительного прокаленного в течение 2 ч
при 300°С катализатора с зернением 1—2 мм. Свободное пространство реак-
тора под катализатором тоже заполняется нихромовой насадкой. По выходе
из реактора пары конденсируются в холодильнике 5. Раствор собирается
в приемнике 7. Образовавшийся во время реакции водород выводят в атмо-
сферу через счетчик 6. Температура испарителя 260—300°С, температура
178
контакта 250±1°С. Конденсат по мере отбора делят на три части. Первую —
20 см3 отбрасывают. Вторую и третью, по 50 см3 каждая, подвергают ана-
лизу методом ГЖХ. Катализатор можно считать активным, если не меиее
70% изоборнеола превратилось в камфару.
Рис. 31. Схема установки для испытания катализаторов дегидри-
рования борнеолов в паровой фазе:
1 — дозирующий иасос; 2 — куб-испаритель; 3 — контактный аппарат; 4 —
термопара; 5 — холодильник; 6 — газовый счетчик; 7 — приемник
Селективность катализатора рассчитывают из уравнения
С =_________
32-Bi ’
(22)
где С — селективность катализатора;
А — изоборнеол, превращенный в камфару, %;
12*
179
Bi—Bi — разность между процентным содержанием терпеновых углеводоро-
дов в продукте реакции и исходном борнеоле. Если С^30, катали-
затор считается селективным.
Для испытания катализатора используют перекристаллизованный изобор-
неол с температурой плавления не менее 208°С и толуол «особой чистоты»
(ГОСТ 11144-65).
Прокаливание катализатора ведут в реакторе, после чего пропускают
через него пары толуола до получения прозрачного конденсата, и лишь после
этого при установившейся температуре начинают пропускать через реактор
раствор изоборнеола в толуоле.
2. Исследования полупродуктов
Определение суммы камфена, трициклена и фенхенов в изоме-
ризатах (22ЭВ). В настоящее время количественный состав
продуктов каталитической изомеризации пинена наиболее ра-
ционально устанавливать с помощью ГЖХ. Прежде в них опре-
деляли количество терпенов, образующих эфиры при взаимодей-
ствии с муравьиной или уксусной кислотами в присутствии
катализаторов (2ЭВ). С названными кислотами вступают в ре-
акцию камфен, трициклен и фенхены, т. е. терпены, содержа-
щиеся в техническом камфене, поэтому это определение часто
называли также определением камфена.
Количественное определение 2.ЭВ по методу В. И. Любомилова [9/].
Навески (около 1 г продуктов каталитической изомеризации пинена и около
1.2 г (1 см3) формилнрующей кислотной смеси), взятые с точностью до
— 0,0002 г, помещают в колбу с пришлифованной пробкой, перемешивают
в течение 6 мин, после чего к содержимому добавляют 50 мл насыщенного
раствора поваренной соли и оттитровывают избыток кислоты 0,5 н. раство-
ром едкого натра в насыщенном растворе хлористого натрия * в присутствии
фенолфталеина до приобретения раствором розового цвета. Концом титрова-
ния считается продержавшееся в течение минуты розовое окрашивание.
Содержание камфена (X) вычисляют по уравнению
_ (ау - й) • 0,068 • 100
ЛК ’ (
где а — количество 0,5 н. раствора едкого натра, см3, которое затрачивается
на нейтрализацию 1 г кислотной смеси;
b — количество 0,5 н. раствора NaOH, затраченное на обратное титрова-
ние при производстве анализа, см3;
q — навеска кислотной смеси, г;
Р—навеска анализируемого продукта, г;
0,068 — титр 0,5 н. раствора едкого натра в граммах, камфена.
К — поправочный коэффициент на неполноту формилирования (0,94—0,97).
Кислотная смесь для формилирования состоит на 90% по массе из 96—
98%-ной муравьиной кислоты и на 10% из серной кислоты (d^ 1,84)—моно-
гидрата. Следует учитывать, что кислотная смесь при хранении медленно
изменяет свой титр вследствие разложения муравьиной кислоты. Поэтому ее
не следует хранить дольше 10—15 дней. Соотношение между кислой смесью
и раствором едкого натра а проверяют перед анализом. Присутствующий
в анализируемом материале пинен под действием кислотной смеси претерпе-
вает ряд реакций, в том числе частично превращается в эфир изоборнеола.
* В этих условиях не происходит гидролиза эфира.
180
Поэтому, анализируя пииен по описанной методике, находят в нем около
10% несуществующего камфена. Это надо иметь в виду при анализе продук-
тов неполной изомеризации пинена, взятых по ходу процесса, и вносить по-
правки в результаты анализа, если содержание пинена в анализируемом про-
дукте может быть установлено.
Этим методом также определяют содержание 2ЭВ в камфаре по
ГОСТ 1123—72, но пересчитывают их на изоборнеол.
Определение полимеров в изомеризатах и в изоборнильных эфирах. Для
количественного определения полимеров 25 г изомеризата илн изоборнильного
эфира перегоняют при давлении 6—15 мм, нагревая содержимое колбы до
120—130°С, и остаток, состоящий из полимеров, взвешивают.
Терпеновые полимеры, образующиеся прн каталитической изомеризации
пинена, в основном состоят из димеров, имеющих высокий показатель пре-
ломления (пд -1,52). Поэтому можно вычислить приблизительно процентное
содержание полимеров в изомеризате, пользуясь уравнением
1,520Х+1,472(100-Х) = 100л^, (24)
где X — процентное содержание полимеров;
1,472 — показатель преломления (20°С) изомеризата, не содержащего поли
меров;
20
nD—показатель преломления исследуемого изомеризата.
Исследование технического камфена. Выводы о качестве технического
камфена обычно могут быть сделаны иа основании его физических свойств,
в особенности его температуры кристаллизации.
Температура кристаллизации определяется по температурной кривой охла-
ждения в пробирке, в которую помещено 4—5 г технического камфена. Сна
чала камфен расплавляют, а затем следят за изменением температуры прн
медленном его охлаждении на воздухе. Отсчеты производят каждые 15 с и
наносят результаты на график. Температуре кристаллизации соответствует
горизонтальный участок кривой.
Анализ муравьиных и уксусных эфиров борнеола и изоборнеола. В про
дуктах, полученных после формилирования или ацетилирования камфена, со-
держание эфиров обычно определяют путем их гидролиза спиртовым раство-
ром едкого натра. Чтобы обеспечить более быстрый гидролиз, спиртовой рас-
твор щелочи должен содержать 5—10% воды. Муравьиные эфиры борнеол.,
и изоборнеола гидролизуются достаточно быстро даже при ком; атной темпе-
ратуре, поэтому при определении этих эфиров навеску анализируемого про-
дукта заливают избыточным количеством титрованного раствора щело н ,
выдерживают около 20—30 мнн в закрытом сосуде, после чего избыток ще-
лочи оттитровывают кислотой.
Уксусные эфиры омыляются медленно, а поэтому реакцию омыления
проводят при температуре кипения спиртовой щелочи в колбе с обратным
холодильником в течение двух часов.
При производстве анализа к навеске 1 —1,5 г исходного вещества добав-
ляют 20 см3 титрованного 0,5 н. раствора NaOH в 90%-иом спирте. По окон-
чании реакции избыток щелочи оттитровывают 0,2 н. водным раствором кис-
лоты.
При расчете процентного содержания эфира X в анализируемом продукте
пользуются уравнением
где а — количество спиртовой щелочи, затраченной на гидролиз, см3;
Т — тнтр раствора щелочи в граммах соответствующего эфира;
Р — навеска анализируемого продукта.
Если эфир содержит свободную органическую кислоту, то сначала отмы-
вают кислоту водой, а потом раствором соды, сушат эфир сернокислым
натрием и после этого анализируют, или к навеске эфира, содержащего
181
свободную кислоту, прибавляют 10 см3 спирта, две капли фенолфталеина, отти-
тровывают свободную кислоту, после чего производят гидролиз эфира обычным
способом. При расчете вычитают из навески содержащееся в ней количество
свободной кислоты.
Исследование борнеола и изобориеола. В борнеоле и изоборнеоле опре-
деляют воду, что важно знать при учете выходов и для проверки качества
отжима борнеолов на центрифуге. Определяют нелетучий остаток для про-
верки качества промывки, содержание неомыленных эфиров, температуру
кристаллизации. Кроме того, проверяют способность борнеолов достаточно
легко дегидрироваться.
Влажность, нелетучий остаток и температуры кристаллизации борнеолов
определяют по методикам ГОСТ 1123—72 на камфару. Для определения эфи-
ров используют описанную выше методику, с той лишь разницей, что навеску
соответственно увеличивают. Для определения способности к дегидрирова-
нию используют ту же методику, что и для испытания катализатора и рас-
творителя, но предварительно проверив растворитель и катализатор.
3. Контроль отдельных процессов
Качество а-пинена, получаемого при ректификации, можно контролиро-
вать по его показателю преломления.
Наблюдение за ходом изомеризации пинена можно вести по изменению
угла вращения плоскости поляризации реагирующего раствора или по изме-
нениям его показателя преломления. Угол вращения реагирующего раствора
по мере расходования а-пинена растет, достигает полуторного значения
исходного, после чего начинает падать. Реакцию заканчивают в момент паде-
ния угла вращения до исходной величины. Этот метод вполне надежен для
контроля за ходом изомеризации чистого а-пинена, смесей (—)-а- н (—)-£-
пиненов и (4-)-а-пинена с небольшой примесью (—)-fl-пинена, но не пригоден
дтя контроля за ходом изомеризации ( + )-а-пннена со значительной приме-
сью (—)-Р-пинена, так как углы вращения продуктов реакции, образую-
щихся из а-пинена, в этом случае растут в положительную сторону, а из
Р-ппнена — в отрицательную и изменения угла вращения реагирующей смеси
могут оказаться самыми разнообразными. Показатель преломления по ходу
реакции увеличивается с Пд 1.4654 (чистый а-пинен) до 1,472—1,474 в конце
реакции. Этими простейшими методами целесообразно контролировать про-
цесс на его начальных и средних стадиях, а в конце применять ГЖХ.
4. Анализ готовой продукции
Предприятия, выпускающие камфару, параллельно могут вы-
пускать целый ряд других продуктов. В Советском Союзе вы-
пускаются: пинен технический ГОСТ 11956—66, камфен техни-
ческий ГОСТ 15039—69, р-пинен технический ТУ 81-05-109—70,
Д3-карен технический ТУ 81-05-110—70, дипентен технический
ТУ 81-05-06, скипидар живичный, не содержащий пинена (очи-
щенный от полимеров кубовой остаток от ректификации скипи-
дара и изомеризатов) ТУ 81-04-74—69.
Свойства технических пинена, камфена и камфары приве-
дены в гл. II, VI, XI.
Методы испытаний камфары, пинена, камфена и других пере-
численных продуктов, изложенные в ГОСТах, ТУ и Государст-
венной фармакопее СССР, мы считаем возможным не приво-
дить.
182
Следует остановиться лишь на определении температуры
плавления и температуры кристаллизации или, что то же самое,
температуры затвердевания камфары. Возникает вопрос, как
взаимосвязаны эти константы и какая из них может быть
определена надежнее. Определение температуры кристаллиза-
ции предусмотрено в СССР для технической камфары ГОСТ
1123—72, а определение температуры плавления для медицинской
камфары — Государственной фармакопеей СССР. Температуру
плавления наблюдают визуально по полному расплавлению
пробы в капилляре. Температуру кристаллизации — по пощадке
на температурной кривой охлаждения. Несоменно, последний
метод более объективен, особенно в отношении камфары, кото-
рая из-за содержания значительного количества примесей пла-
вится в интервале нескольких градусов. Только по кривой
охлаждения температуру кристаллизации такой камфары можно
установить достаточно четко в пределах нескольких десятых
градуса. При определении температур плавления часто настолько
быстро нагревают прибор, что показание термометра отстают
от действительной температуры бани, в которой находится ка-
пилляр, в результате чего определяется заниженная температура
плавления.
Для чистых веществ температура плавления практически
равна температуре затвердевания, но у веществ, содержащих
значительное количество примесей, в том числе у камфары,
температура затвердевания всегда несколько ниже.
Из изложенного следует, что надо отдать предпочтение опре-
делению температуры кристаллизации камфары перед опреде-
лением температуры плавления.
5. О применении ректификации
для исследования продуктов
камфарного производства
С развитием газо-хроматографического анализа аналитиче-
ская ректификация потеряла свое значение. Но ректификация
как препаративный метод в практике исследований, связанных
с производством камфары, его полностью сохраняет. Вопросы
лабораторной ректификации подробно освещены в [86, 124].
ГЛАВА XIII. ФИЗИЧЕСКИЕ КОНСТАНТЫ
ИНДИВИДУАЛЬНЫХ ТЕРПЕНОВ
В производстве синтетической камфары и исследованиях,
связанных с этим производством, часто приходится пользоваться
такими физическими константами индивидуальных терпенов, как
температуры плавления и кипения, плотности, показатели
преломления и углы оптического вращения. Накопленный за
183
длительный период работы в этой области огромный фактический
материал может быть использован лишь после его серьезного
критического рассмотрения и выбора наиболее достоверных ве-
личин. Такого рода данные сведены в табл. 50, 51, 52. Они со-
ставлены на основании просмотра и критической оценки боль-
шого количества журнальных статей, обзоров и собственного
опыта автора. В них вошли данные обзоров [171, 197, 233—235,
263, 294, 300, 301, 317, 318, 321, 322] и журнальных статей [12,
14, 17, 18, 44, 80, 114, 125, 131, 132, 138—140, 147, 190, 198, 205,
246—248, 255, 293, 296, 319, 326, 327, 337].
В табл. 50 приведены физические свойства терпенов, инди-
видуальность которых не вызывает сомнений.
Таблица 50
Физические свойства индивидуальных терпенов
Н аииено- 1 ание Тс рпенов Достигну- тая чистота в .моль, % Темпера- тура кри- сталлиза- ции, ЭС Температу- ра кипения при 760 м.м рт. ст., °C 20 nD 1 '4 D оптически чистого терпена в 1 рад 1“1о терпенов в эфирных м а сл а х хвойных в град
Лимонен 98 -74 176,5 0,8421 1,4726 + 126 Разный
Дипентен 99 -89 176,5 0,8421 1,4726 0,0 0,0
А3-Карен — — 170,1 0,8645 1,4722 + 18,0 + 18,0
а-Пинен 99,6 -75 155,9 0,8582 1,4654 - 50,0 Разный
Р-Пинен 98,7 -61 166,0 0,8710 1,4790 +22,6 -22,6
К. мфен — + 48,5 158,9 0,8421* 1,4564** + 117,5** * Разный
54
4 '
54
О-
П
*** Растворитель толуол (с= 19).
Определен ie плотности и показателен преломления веществ в некоторых
зарубежных странах, главным образом в США, производят при 25°, а в СССР
пр I 20°С. В соответствии с этим в таблице значения плотности и показате-
ле I преломления, определенные при температурах, незначительно отличаю-
щ-'хся от 2(ГС, приведены к температуре 20°С, принимая поправки на каж-
дт й Г для плотности 0,0008, а для показателей преломления 0,00045. Исключе-
ние сделано лишь для камфена, так как определение плотности и показателя
преломления этого терпена приходится вести при температуре, превышаю-
щей его температуру плавления, а в этих условиях приведение указан-
ных констант к 20°С связано с погрешностями. Такого рода пересчет можно
пр шзводить при физическом анализе смесей терпенов, содержащих в каче-
стве одного из компонентов камфен, в случае если не требуется слишком
высокая точность. Тогда для камфена можно принимать civ 0,8693 и
n-l 1,4717.
Достоверность приведенных в таблице температур кипения
находится в пределах ±0,1 — ±0,2°С.
Температура кристаллизации лимонена, дипентена, а- и 0-пи-
ненов требует дальнейшего подтверждения.
184
Таблица ~1
Наиболее вероятные физические свойства ряда терпенов и близких им веществ
Наименование терпенов Температура кристаллиза- ции, °C Температура кипения при 760 мм рт. ст., °C Давление, мм рт. ст. Температуря кипения, °C Гуг с । |»1*( » град.
1 2 3 4 5 6 8
Аллооцимен -21 — 20 89 0,8060 1,7466 0
(нео) Аллооцимен -35 — 20 91 0 81 И 1,544G 0
Мирцен — 167 20 65,5 0,7887 1+701 0
Терпинолен — 186,5 100 120 0,8596 1, 1896 0
а-Терпинен — 175 100 108 0,8341 1,4733 0
у-Терпинен — 183 100 116 0,8498 1, 1735 0
п^Ментадиен-2,4 (8) — 188-189 100 120 0,8561 1,5020 -49
п-Ментаднен-3,8(9) — 183 100 117,5 0,8538 1,1897 -140
а-Фелландрен — 175-176 10 53 0,8358 1, 1710 -217
р-Фелландрен __ 176 9 53 0,8425 1,1882 -16
Изолимонен — 173 — — 0,832 1, 166 -149
Д4-Карен — 167,5-168 — — 0,8620 1,1752 -91
Трициклен + 68 153,3 100 85 0,8417'° 1,1378'° 0
Борнилен + 113 150 746 146 — -
сс-Фенхен 158,5-158,8 109 91,5 0,8697 1,1740 -44
Р-Фенхен — 156,8-157,1 — 0,8611 1,4695 -84. )
у-Фенхен — 148,4—148,9 — 0,8521 1,4579 —45, '55
б-Фенхен — 138,6-138,7 — 0,8375 1,4463 + 65.9
е-Фенхен — 152,7-153,3 — — 0,8567 1,4636 0
£-Фенхен — 148,5-148,8 — — 0,8562 1,4602 -г 36.04
Циклофенхен — 142,5-14'2,9 — — 0,8588 1,4581 чО.ОЗ
Сабинен — 163-165 — 0,8415 1,4676 + 101,1
Ту йен — 152 — 0,8301 1.4515 -37.2
Сантен — 140 — — 0,8672 1,4675 0
тр.м-Ментан — 170 100 103 0,7928 1,4367 0
цпс-м-Мептан -90 172 100 105 0,8002 1,4431 0
1(пс-Пинан -53 168 100 101 0,8575 1.4629 -23
Камфан + 154 158 — — —
Изокамфан +65 166 100 100 — —
л-Цимол -73 177 109 но 0,8570 1,4905 0
* Если приведен знак «±», это означает вращение оптически чист гл
терпена. Знак « + » или «—» — вращение выделенного терпена.
** Растворитель бензол.
В табл. 51 приведены наиболее вероятные свойства ряда
терпенов и близких им веществ, очистка которых продвинулась
Таблица 52
Свойства некоторых производных терпеновых углеводородов, имеющих
значение в производстве камфары
Вещества Температура плавления, °C Температу- ра кипения, СС, при 760 мм рт. ст. Давление, мм рт. ст. Температура кипения, °C +° -20 rtD в град.
1 2 3 4 5 6 7 8
Цинеол-1-4 —46 172 100 105,5 0,8986 1,4446 0
Цинеол-1-8 4-1 174 100 108 0,9245 1,4574 0
а-Терпинеол +40 219 100 149 0,934 1,4831 + 112»
Р-Терпинеол +33 209 100 139 0,919 1,4747 0
у-Терпинеол + 70 218 50 132 0,941 1,4912 0
Т грпинен-4-ол —4 211 50 123 0,926 1,4762 +29
Борнеол + 209 + 47 212 — — 1,01 — -36,52
Псевдоборнеол 5 — — — — — —
Изоборнеол +212 214 — — — — +342
Псевдоизоборкеол = ( + ) и (—)-а-Фе- + 101 — — — — — —
Н ХОЛ +48 200 100 133 — 1,4734 +12,52
( = )-а-Фенхол ( —) и (—)-[}-Фен- 4-39 200 100 133 0,964 1,4734 0
+ 5 200 — — 0,961 1,4782 +242
( -) -Р-Фенхол +6 200 — — 0,961 1,4790 0
(—) и (—)-а-Изо- фенхол + 62 202 — — 0,954 1,4755 + 262
( = )-а-Изофенхол + 45 202 — — 0,954 1,4755 0
(—)-Борнилфор- млат (98,7%) — — 21 107 1,007 1,4687 -47,7
(—)-Борнилаце- тат (99,6%) -+29 — 15 107,3 0,9861 1,4642 —44
(~) - ИзоборНИЛ- формиат — — 20 108 1,008 1,4700 0
(1=)-Изоборнил- ацетат — — 100 155,8 0,9849 1,4640 0
Камфара 178,5-179,5 209 89,8 136,1 1,00 — +44,8 а
( —) и (—)-Фен- Х(>Н + 6 193 100 122 0,9452 1,4628 +704
(тс) -Фенхен -18 193 — 0,945 1,4628 —
Изофенхон — 193-194 — — 0,9513 1,4612 —
Псевдокамфара 5 +68 — — — — — —
1 Бензол.
2 Этанол.
3 Этанол (с=20%).
4 Этанол (с = 3.5%).
5 Псевдоборнеол (5-экзо-изокамфан-экзо-ол-З, изокамфанол), псевдоизо-
борнеол (5-зкзо-изокамфан эндо-ол-3), псевдокамфара (5-экзо-изокамфанон-З,
изокамфанон) [243].
186
достаточно далеко. По-видимому, среди них есть и индивидуаль-
ные вещества. В первую очередь это относится к сантену, трп-
циклену, аллооцимену, 0-фелландрену, а-, ₽- и циклофенхенам,
терпинолену, 2,4(8)-п-ментадиену. Свойства терпенов, сведенные
в табл. 51, названы наиболее вероятными потому, что можно
считаться с возможностью их дальнейшего уточнения.
Для трициклена показатель преломления и плотность даны при 70"С,
так как приведение этих констант к 20°С связано с погрешностями. Одна :о
при исследовании смесей терпенов, содержащих трициклен, не требующем
слишком большой точности, можно пользоваться приведенными к £0’С вели-
чинами: d2® 0,8817 н «д 1,4603.
В табл. 52 приведены свойства ряда производных терпенов,
имеющих значение в производстве камфары.
Большое количество ИК-спектров моно- и сесквитерпенов
приведено в работах [53, 281, 282, 286, 294, 296], а по КР-спект-
рам в работах [12, 115, 187], но надо сказать, что многие работы
по КР-спектрам были выполнены тогда, когда не умели должным
образом очищать терпены, и поэтому их данные противоре-
чивы [53].
Теплоты сгорания некоторых терпенов и их производных
представлены в табл. 53.
Таблица 53
Теплоты сгорания некоторых терпенов и их производных
Вещества Чистота в моль, % Физическое состояние Теплота сгора- ния в кДж, моль Литератур а
Аллооцимен 93,7 Жидкое 6197,5
Мирцен — То же 6235,9
Лимонен 98 6166,9 г 94.А 1
Дипентен 99,3 6170,2 [240]
а-Пинен 99,6 6204,9
(З-Пинен 98,7 6213,7
Камфен — Твердое 6148,5 ।
Борнеол — То же 6151,7 [264]
Камфара — 5906,6 1
Приведенные теплоты сгорания камфена, борнеола и камфары были
опубликованы до того, как были установлены термодинамические стандарты
и учтены многие ошибки эксперимента, поэтому их нельзя считать вполне
достоверными, несмотря на то, что они были признаны Международным
Союзом чистой и прикладной химии в 1923 г.
187
СПИСОК ЛИТЕРАТУРЫ
1. Арбузов Б. А. Об изомеризации а-пинена в алифатический терпен.
<Журн. общей химии», 1933, т. 3, с. 21—34.
2. Производство живичной канифоли в СССР и за рубежом. М., 1972,
; 6 с. Авт.: Г. Д. Атаманчуков, А. И. Головин, В. Я. Падерин, В. М. Селихов.
3. Афанасьева Е. В., Выродов В. А., Коротов С. Я. Изомеризация пи-
: ена в камфен в трехфазовой системе.— В кн.: Процессы химической техно-
логии древесины и продуктов ее переработки.— Научные труды Ленингр.
. есотехн. академии нм. Кирова. № 135, вып. 1. Л., 1970, с. 11—18.
4. Афанасьева Е. В., Выродов В. А., Коротов С. Я. Изомеризация пи-
। ена в камфен в трехфазовой системе на опытно-промышленной установке.—
~ам же. с. 20—25.
5. Афанасьева Е. В., Выродов В. А. Некоторые закономерности изоме-
ризации пинена в присутствии таблетированного титанового катализатора.—
ам же. с. 26—31.
6. Бабурин С. В., Выродов В. А., Коротов С. Я. Изучение кинетики
I роцссса .массоиередачи при ректификации терпеновых углеводородов. Ле-
1 ингр. лесотс.хн. академия им. Кирова. Материалы к научно-технической
1 онференции 1968 г., вып. 1, Л., 1968, с. 84—89.
7. Кинетические параметры процесса ректификации терпеновых углево-
дородов в тарельчатых аппаратах в области низких нагрузок по паровой
фазе.— Там лее, с 89—93. Авт.: С. В. Бабурин, Г. Ф. Трушков, В. А. Выродов,
Я. Коротоз.
8. Бабурин С. В., Выродов В. А., Чирков П. К- Распределение концент-
раций компонентов при ректификации терпеновых углеводородов по высоте
слытно-промышленной колонны.—В ки.: Технология экстрактивных веществ
дерева.— Труды Ленингр. лесотехн, академии им. Кирова, № 114. Л., 1969,
с 81—84.
9. Баландин А. А. Мультиплетная теория катализа, ч. 1, М., 1963, 102 с.;
ж 2, М.. 1964. 243 с.
10. Ба.члод А. П., Топчиева К. В. Природа каталитического действия
алюмосиликатов.— «Успехи химии», 1951, т. 20, с. 161 —175.
11. Бардышев И., Пирятинский А., Бардышева К. Свойства и состав
скипидаров, полученных из живицы индивидуальных деревьев Pinus silvestris.
Докл. АН СССР, 1950, т. 75, с. 75—77.
12. Бардышев И. И., Шорыгин П. П., Шигорин Д. Н. Спектры комбина-
ционного рассеяния света некоторых терпеновых углеводородов.— «Жури,
общей химии», 1952, т. 22, с. 568—571.
13. Бардышев И. И., Ефименко В. И. Изомеризационные превращения
т ’рпенов в присутствии смоляных кислот. Докл. АН БССР, 1958, т. 2,
с 323—326; 1959, т. 3, с. 150—153.
14. Бардышев И. И., Шашкина М. Я.. Куликов В. И. Изомеризация
(3-фелландрена в а-фелландрен.— «Жури, органической химии», 1966, т. 2,
с 1039—1043.
188
15. Бардышев И. И., Куликов В. И., Перцовский А. Л. Хроматографиче-
ский анализ промышленных образцов отечественных скипидаров.— «Гидролиз-
ная и лесохимическая промышленность», 1966, № 8, с. 16—17.
16. Изомеризация А3-карена с кислыми катализаторами. Докл. АН БССР,
1966, т. 10, с. 873—876. Авт.: И. И. Бардышев, Ж. Ф. Лайко, Л. В. Сионская,
Р. Г. Шляшинский.
17. Бардышев И. И., Шашкина М. Я- О свойствах ^-фелландрена.—
Докл. АН БССР, 1966, т. 10, с. 391—393.
18. Бардышев И. И., Лайко Ж. Ф., Куликов В. И. Кислотная изомери-
зация Д4-карена.— «Жури, общей химии», 1967, т. 37, с. 1248—1252.
19. Бардышев И. И., Куликов В. И. Относительные времена удерживания
терпенов на различных жидких фазах.— «Журн. аналитической химии», 1968,
т. 23, с. 1081—1084.
20. Бардышев И. И., Веденеев К. П., Струнникова Э. Ф, Изучение со-
става сульфатных скипидаров методом газожидкостной хроматографии,—
В кн.: Сб. трудов Центр, н.-и. и проекта, ин-та лесохим. пром. (ЦНИЛХИ).
М., 1968, с. 28—32.
21. Бардышев И. И., Зенько Р. И., Горбачева И. В. Свойства и химиче-
ский состав скипидаров, выделенных из живицы сосны обыкновенной, произ-
растающей в различных районах Советского Союза.— «Гидролизная и лесо-
химическая промышленность», 1969, № 7, с. 17—18.
22. Бардышев И. И., Перцовский А. Л. О природе кислородсодержа-
щих соединений, входящих в состав скипидаров.— В кн.: Синтетические про-
дукты из канифоли и скипидара.— Труды II Всесоюзного научно-техниче-
ского совещания. Горький, 1970, с. 70—82.
23. Бардышев И. И., Куликов В. И. Газожидкостная хроматография тер-
пеновых углеводородов.— «Гидролизная и лесохимическая промышленность»,
1972, № 5, с. 17—19.
24. О составе скипидаров из живицы, добываемой в различных районах
страны.— «Гидролизная и лесохимическая промышленность», 1972, №8.
с. 21—22. Авт.: И. И. Бардышев, А. Н. Булгаков, Л. В. Ударов, Я. В. Эпш-
тейн, Л. В. Гордон.
25. Белослудцева Е, И., Гиневич Г. И., Накрохин Б. Г., Шустерман 3. Г.,
Соколов Б. Г., Нехаев И. П. Непрерывно действующий аппарат для паро-
фазного дегидрирования.— Авторское свидетельство № 117582 (1958). Бюлле-
тень изобретений, 1959, № 2.
26. Белослудцева Е. И., Гиневич Г. И. Непрерывный процесс парофаз-
ного дегидрирования борнеолов и его аппаратурное оформление.— «Гидро-
лизная и лесохимическая промышленность», 1959, № 3, с. 15—17.
27. Берлин А. А., Даванков А. Б., Калиопин Л. Е. Окисление камфена
в эмульсиях.— «Журн- прикладной химии», 1945, т. 18, с. 217—220.
28. Биттепаж Ю. А. О связи между каталитической активностью и об-
менной способностью алюмосиликатов.— «Журн. общей химии», 1947, т. 17,
с. 199—207.
29. Богословский Б. М., Казакова 3. С. Скелетные катализаторы, нх свой-
ства п применение в органической химии. М., 1967. 144 с.
30. Де Бур. Динамический характер адсорбции. М., 1962, с. 228—232.
31. Буянова Н. Е., Гудкова Г. Б., Карнаухов А. П. Исследование физико-
химических характеристик катализаторов методом газовой хроматографии.—
В кн.: Методы исследований катализаторов и каталитических реакций, т. 2.
Новосибирск, 1965, с. 56—66.
32. Буянова Н. Е., Гудкова Г. Б., Карнаухов А. П. Определение удель-
ной поверхности методом тепловой десорбции.— «Кинетика и катализ», 1965.
т. 6. с. 1085—1091.
33. Вагнер Е. Е. О строении камфена.— ЖРФХО, 1899, т. 31, с. 680—684.
34. Влияние катиона на скорость гидролиза и этанолиза борнилацетата.—
«Жури, органической химии», 1970, т. 6, 1379—1385. Авт.: А. А. Верещагина.
О. Н. Долгополов, А. Ф. Маркова, Г. А. Рудаков.
35. Вернадский В. И. О группе силиманита н роли глинозема в силика-
тах. М., 1891. 100 с.
189
36. Внедрять непрерывный метод экстракции канифоли.— «Гидролизная
и лесохимическая промышленность», 1973, № 5, с. 5—6.
37. Выродов В. А., Коротов С. Я. К вопросу об омылении эфиров изобор-
нсола водной щелочью.— «Изв. вузов. Лесной журнал», 1959, № 2, с. 150—154.
38. Выродов В. А. Омыление изоборнилацетата в жидкой фазе.— Там же,
1959, № 6, с. 132—138.
39. Выродов В. А., Коротов С. Я. Непрерывный способ омыления изо-
б< рнилформиата.— Там же, 1962, № 5, с. 148—154.
40. Выродов В. А., Коротов С. Я., Афанасьева Е. В. и др. Способ полу-
чения камфена.— Авторское свидетельство № 238541. Открытия. Изобретения.
Промышленные образцы. Товарные знаки. 1969, № 10.
41. Выродов В. А., Афанасьева Е. В., Коротов С. Я. К вопросу непре-
рывной изомеризации пинена в камфен. Теоретические основы процесса.—
В кн.: Процессы химической технологии древесины и продуктов ее перера-
ботки.— Научные труды Ленингр. лесотехн, академии им. Кирова, № 135,
вып. 1, Л., 1970, с. 11 —18.
42. Гельперин Н. И., Новикова К. Е. Концентрирование муравьиной кис-
лоты.— «Химическая промышленность», 1951, № 9, с. 267—277.
43. Гельперин Н. И., Новикова К- Е. Разделение азеотропных бинарных
смесей методом ступенчатой ректификации при двух различных давлениях.—
«7Курн. прикладной химии», 1953, т. 26, с. 912—920.
44. Годлевский И., Рожаиович К. Лимонен из бромистого лимонена.—
ЖРФХО, 1899. т. 31, с. 209—211.
45. Математическое моделирование процесса дегидрирования изоборнеола
в жидкой фазе.— В кн.: Синтетические продукты из канифоли и скипи-
дара.— Труды 11 Всесоюзного научно-технического совещания. Горький, 1970,
с. 446—454. Авт.: А. И. Головин, В. А. Выродов, Н. С. Гурфейн, С. Я- Ко-
рстов.
46. Головин А. И., Выродов В. А., Коротов С. Я. Кинетические законо-
мерности при дегидрировании борнеола п изоборнеола—В кн.: Процессы
химической технологии древесины и продуктов ее переработки.— Научные
труды Ленингр. лесотехн, академии им. Кирова, № 135, вып. 1. Л., 1970,
с. 51—56.
47. Голубев П. Г. Эфирное масло из сибирской пихты.— ЖРФХО, 1888,
т. 20, с. 585.
48. Голубев П. Г. Об исследовании эфирного масла из сибирской
пихты.— Там же, 1903, т. 35, с. 1005.
49. Голубев П. Г. О камфаре из борнеола, содержащегося в эфирном
масле сибирской пихты.— Там же, 1904, т. 36, с. 776—777.
50. Голубев П. Г. О кристаллических продуктах эфирного масла сибир-
ской пихты.— ЖРФХО, 1904, т. 36, с. 1096—1108.
51. Голубев П. Г. К характеристике 1-камфена.— Там же, 1909, т. 41,
с. 1004—1013.
52. Гордеев А. В. Системы и методы эксплуатации сосновых лесов для
потучения канифольно-скипидарных продуктов.— В кн.: Синтетические про-
дукты из канифоли н скипидара.— Труды 11 научно-технического совещания,
Горький, 1970, с. 14—24.
53. Горяев М. И., Плива И. Методы исследования эфирных масел. Алма-
Ата, 1962, с. 116—196.
54. Горяев М. И., Шостак Ф. Т., Джандосова К. Д., Петелина Л. П.
Изомеризация пинена с образованием камфена.— Авторское свидетельство
№ 232257. Открытия. Изобретения. Промышленные образцы. Товарные знаки,
1969, № 1.
55. ГОСТ 1123—72. Камфара синтетическая.
56. Государственная фармакопея СССР, изд. 10, М., 1968.
57. Грет С., Синг К. Адсорбция, удельная поверхность. М., 1970, с. 330.
58. Грехнев М. А. Определение упругости паров изобориеола.— «Лесохи-
мическая промышленность», 1936, As 9, с. 11 —12.
190
59. Грехнев М. А., Ерошевский И. Г. О порядке реакции гидрогенизации
и дегидрогенизации.— «Жури. обшей химии», 1940, т. 10, с. 2005—2013.
60. Грехнев М. А., Ерошевский И. Г. К вопросу о зависимости скорости
каталитических реакций от количества взятого катализатора.— «Журн. об-
щей химии», 1945, т. 15, с. 146—150.
61. Грехнев М. А. Применение смешанных катализаторов к процессу
дегидрогенизации изоборнеола.— «Журн. прикладной химии», 1949, т. 22,
с. 207—213.
62. Грехнев М. А. Дегидрирование изоборнеола в паровой фазе.— «Жури,
прикладной химии», 1953, т. 26, с. 231—233.
63. Гурвич Л. Г. О действии флоридиновой земли на непредельные со-
единения.— ЖРФ-ХО, 1915, т. 47, с. 827—830.
64. Гурвич Л. Г. К теории гетерогенного катализа.— Там же, 1916, т. 48,
с. 837—856.
65. Дашкевич Б. Н., Вольнов Ю. Н. Способ каталитической полимериза-
ции и изомеризации органических соединений.— Авторское свидетельство
№ 48306. Вестник Комитета по изобретательству, 1936, № 8.
66. Долгов Б. Н. Катализ в органической химии. Изд. 2-е, Л., 1959. 807 с.
67. Долгополов О. Н., Казачкова Л. Н., Рудаков Г. А. Термодинамиче-
ские характеристики этанолиза борнилацетата.— «Журн. органической хи-
мии», 1971, т. 7, с. 1897—1903.
68. Жарков П. М. О каталитическом методе получения камфары из бор-
неолов.— «Журн. химической промышленности», 1929, т. 6, с. 784—787.
69. Журавлев П. И., Чирков П. К-, Выродов В. А. Непрерывный способ
омыления изоборнилформиата. «Реферат, информ. Лесохимия и подсочка-,
1970, № 4. 7 с.
70. Зайдель А. Н. Элементарные оценки ошибок измерений. Л., 1968. 96 с.
71. Залькинд Ю. С., Михелис Ю. Л., Стоянов М. А. Способ получения
камфары.— Авторское свидетельство № 42076. Вестник Комитета по изобре-
тательству, 1935, № 3.
72. Каталитическое дегидрирование борнеола (изоборнеола).— В кн.:
Пластические массы, сб. 2, Ин-т пластических масс. Л., 1937, с. 263—280.
Авт.: Ю. С. Залькинд, Ю. Л. Михелис, М. А. Стоянов, Р. М. Левин-Коган.
73. Земятченский П. А. Поглотительные свойства русских глин. Часть
экспериментальная. Петроград, 1919. 67 с.
74. Земятченский П. А. Каолинитовые образования Южной России. СПБ.,
1896. 324 с.
75. Зильберман-Грановская А. А. Измерение малых давлении пара. Дав-
ление пара нафталина, камфары и глицерина.— «Журн. физической химии»,
1940. т. 14, с. 759—767.
76. Иванов Л. А. Биологические основы добывания терпентина в СССР.
М,—Л., 1961. 292 с.
77. Иванова Л. С., Боровская А. Г., Рудаков Г. А. Влияние пористой
структуры катализаторов на процесс кислотной гетерогенной изомеризации
а-пинена. Докл. АН СССР, 1965, т. 163, с. 113—115.
78. Иванова Л. С., Боровская А. Г., Рудаков Г. А. Кислотная изомери-
зация жидкого р-пинена в присутствии твердых катализаторов.— «Журн.
органической химии», 1967, т. 3, с. 2162—2167.
79. Ингольд К. Теоретические основы органической химии. Изд. 2-е. М.,
1973, с. 584—587, 600—648.
80. Калиновская Е. А., Рудаков Г. А. Кинетика щелочного омыления
эфиров борнеола и изоборнеола в спирто-водных растворах.— «Гидролизная
и лесохимическая промышленность», 1960, № 4, с. 1—3.
81. Калиновская Е. А., Рудаков Г. А. Некоторые особенности кинетики
щелочного гидролиза борнил- и изоборнилацетатов в водно-спиртовых рас-
творах.— «Журн. органической химии», 1967, т. 3, с. 307—311.
82. Кипперман С. Л. Введение в кинетику гетерогенных каталитических
реакций. М„ 1964. 600 с.
191
83. Коган В. Б., Фридман В. М. Справочник по равновесию между жид-
костью и паром в бинарных и многокомпонентных системах. Кн. 1 н 2. Л..
1966. 1426 с.
84. Коротов С. Я., Выродов В. А. Омыление изоборнилформиата в жид-
к >й фазе. «Изв. вузов. Лесной журнал», 1959, № 3, с. 151—158.
85. Коротов С. Я., Выродов В. А. Освоение непрерывного способа омы-
ления изоборнилформиата.— «Гидролизная и лесохимическая промышлен-
ность», 1963, № 4, с. 16—18.
86. Крель Э. Руководство по лабораторной ректификации. М., 1960.
627 с.
87. Кропотов В. И. Обеспечить народное хозяйство канифолью.— «Гидро-
лизная и лесохимическая промышленность», 1972, № 6, с. 27—30.
88. Куломзин Ю. В. Перспективы пихтоварения.— «Гидролизная и лесо-
хгмнческая промышленность», 1966, № 4, с. 30—31.
89. Лебедев С. В. Выступление по докладу Тищенко В. Е. и Марта В. Я-—
«Протокол № 6 ЖРФХО». 1928, т. 60, с. 10С9.
90. Любомилов В. И., Рутовский Б. Н., Шереметева Т. В. Исследование
борнилхлорида и его изомеров.— «Жури, общей химии», 1939, т. 9, с. 2067—
2074.
91. Любомилов В. И. Быстрый метод определения камфена для контроля
производственных операций.— «Промышленность органической химии», 1939,
т. 6. с. 167—169.
92. Любомилов В. И., Лесин Л. М., Васильева А. В., Плотникова А. Ф.,
Рутовский Б. Н. Способ получения камфена.— Авторское свидетельство
№ 59004. Бюллетень бюро изобретений Госплана при СНК СССР, 1941, № 2.
93. Любомилов В. И. Получение активированной глины—катализатора
для превращения пинена в камфен.— Авторское свидетельство № 64839. Свод
изобретений СССР, 1945. № 5.
94. Мак-Нейр Г., Бонелли Э. Введение в газовую хроматографию. М.,
1970. 277 с.
95. Де Майо П. Терпеноиды. М.. 1963. 494 с.
96. Международная фармакопея. Всемирная организация здравоохране-
ния. Женева. 1967—1969. с. 99—100.
97. Метешкина Л. П., Медведева А. В., Чиркова Р. В. Совершенствова-
ние газо-жидкостной хроматографии терпенов.— «Гидролизная и лесохимиче-
ская промышленность». 1969, № 6. с. 25—26.
98. Миесеров К- Г. Природа активных центров алюмосиликатных ката-
ли гаторов.— «Успехи химии», 1953. т. 22. с. 279—290.
99. Неницеску К. Д. Органическая химия. Т. 1. М., 1962. 863 с.; Т. 2. М.,
19i 3. 1047 с.
100. Нестеренко П. А. Камфарный базилик. «Бюллетень Государственного
Никитского ботанического сада». Ялта. 1934, № 11. 54 с.
101. Нестеров Г. В., Радбиль Б. А., Рудаков Г. А. Парофазное катали-
тп'-еское дегидрирование борнеолов.— > Гидролизная и лесохимическая про-
мышленность», 1973, № 8. с. 6—8.
102. Нестеров Г. В., Радбиль Б. А., Рудаков Г. А. Оценка активности
и селективности катализаторов дегидрирования борнеолов в паровой фазе.—
В хи.: Сб. трудов Центр, и н.-и. и просктн. ин-та лесохим. пром. ЦНИЛХИ.
М.. 1975, вып. 24.
103. Новикова Е. Н. Влияние заместителей в молекулах ингибиторов на
их защитные свойства при автоокислегни а-пинена.— «Журн. общей химии»,
1958, т. 28. с. 1993—1997.
104. Облад А., Милликен Т. мл., Мильс Г. Химическая характеристика
и структура катализаторов крекинга.— В кн.: Катализ в органической химии.
М„ 1953, с. 185—241.
105. Огородников С. К., Лестева Т. М., Коган В. Б. Азеотропные смеси. Л.,
1971. 1406 с.
106. Осанов Б. П., Зараковская А. И. Методы исследования камфары.—
«Лесохимическая промышленность», 1939. № 12, с. 27—35.
107. Паулинг Л. Природа химической связи. М., 1947. 381 с.
192
108. Изучение состава пихтового масла, получаемого из веток (лапок)
сибирской пихты.— В кн.: Труды хим. металлургия. ин-та Западносиб. фи-
лиала АН СССР, 1953, т. 7, с. 33—51. Авт.: А. П. Пентегов, М. А. Чиркова,
Л. И. Сухорукова, О. П. Кузнецова.
109. Сесквитерпеновые углеводороды живиц некоторых видов хвойных
Сибири,—Изв. СО АН СССР, 1968, № 4, вып. 2, с. 114—118. Авт.: В. А. Пеи-
тегова, Ж. В. Дубовенко, Л. Н. Вольский, С. М. Василюк, М. А. Чиркова,
Э. Н. Шмидт.
НО. Перри Дж. Г. Справочник инженера-химика, т. 2. Л., 1969, с. 189—
192.
111. Песин Л. П., Белянина Е. Т., Павловская В. А. О гидратации камфена
в изоборнеол.— «Журн. прикладной химии», 1943, т. 16, с. 129—133.
112. Петелина Л. П., Горяев М. И., Джандосова К. Д. Каталитическая
изомеризация а-пинена в присутствии анкелнта КТ-3.— Изв. АН Каз. ССР, сер.
хим.. 1967, т. 17, № 2, с. 93—98.
113. Петелина Л. П., Джандосова К. Д., Горяев М. И. О влиянии обмен-
ной емкости н набухаемости сульфакатионита анкелит КТ-3 на изомериза-
цию пинена к камфен.— Изв. АН Каз. ССР, сер. хим., 1969, т. 19, № 2,
с. 53—57.
114. Пигулевский Г. В., Кожина И. С. Исследование углеводорода изоли-
монена.— «Журн. прикладной химии», 1955, т. 25, с. 416—422.
115. Пигулевский Г. В., Рыскальчук А. Т. Спектры комбинационного рас-
сеяния света соединений, встречающихся в эфирных маслах.— В кн.: Труды
Ботанич. ин-та им. Комарова АН СССР (V). 1955. т. 3. с. 149—257.
116. Пигулевский Г. В. Химия терпенов. Л.. 1949. 286 с.
117. Поздняков Н. П., Жлобо М. К. Непрерывно действующая установка
для ректификации скипидара.— «Деревоперерабатывающая и лесохимическая
промышленность», 1952, № 7, с. 13—15.
118. Полтавченко Ю. А., Рудаков Г. А. Эволюция биосинтеза терпенов
в семействе сосновых.— «Растительные ресурсы», 1973, т. 9, с. 481—493.
119. Поляков П. П., Лисов В. И. Сырьевые ресурсы и перспективы раз-
вития производства канифоли, скипидара и вторичных продуктов на их >с-
нове.— В кн.: Синтетические продукты из канифоли и скипидара. Труды II
научно-технического совещания, Горький, 1970, с. 4—13.
120. Правдин Л. Ф. Сосна обыкновенная. М.. 1964. 190 с.
121. Рис Г. Структура и спекание крекинг-каталпзаторов ч подобных нм
материалов.— В кн.: Катализ, катализаторы органических реакций. М., 1955,
с. 37—107.
122. Новая полынь Нижнего Поволжья, дающая камфару, как главную
составную часть масла.— «Жури, опытной агрономии Юго-Востока», 1927,
т. 4, с. 410—421. Авт.: А. А. Рихтер, Л. И. Казакевич, О. Ю. Соболевская,
К. Т. Сухорукова.
123. Рогинский С. 3., Яновский М. И., Берман А. Д. Основы применения
хроматографии в катализе. М.. 1972, с. 70—75.
124. Розенгарт М. И. Техника лабораторной ректификации. М.—Л., 1951.
194 с.
125. Рудаков Г. А., Коротов С. Я. Об упругости пара некоторых терпе-
нов.— «Журн. прикладной химии», 1937, т. 10. с. 314—318.
126. Рудаков Г. А., Коротов С. Я. О равновесии паз- жидкость некото-
рых бинарных смесей веществ терпенового ряда.— Там же. с. 319—326.
127. Рудаков Г. А. О каталитических превращениях дипентека над акти-
вированной глиной.— «Журн. общей химии». 19‘.0, т. 10. с. 1673—1681.
128. Рудаков Г. А., Бланк М. Л. Способ очистки камфена.— Авторское
свидетельство № 60247. Бюллетень бюро изобретений Госплана при СНК
СССР, 1941, № 6.
129. Рудаков Г. А., Артамонов Г. А. Каталитические превращения Д3-ка-
рена над активированной глиной.— «Журн. общей химии», 1945, т. 15,
с. 75—85.
130. Рудаков Г. А., Гуляева Л. И. Каталитические превращения камфена
над активированной глиной.— «Журн. общей химии», 1946, т. 16, с. 251—260.
13 Заказ .Vs 43
193
131. Рудаков Г. А. Каталитические превращения а-пинена над активиро-
ванной глиной. Там же, с. 261—276.
132. Рудаков Г. А., Марчевский А. Т. Каталитические превращения Д3-
карена над метатитановой кислотой.— «Журн. общей химии», 1953, сб. 2,
с. 1432—1445.
133. Рудаков Г. А., Хоменко 3. С. К вопросу о каталитической активно-
сти титановой кислоты.— «Журн. общей химии», 1954, т. 24, с. 337—343.
134. Рудаков Г. А., Хоменко 3. С., Шестаева М. М. Механизм взаимо-
действия пинена, камфена и лимонена с катализаторами, вызывающими их
изомеризацию титановой кислоты и активированной глиной. Там же, с. 549—
557.
135. Механизм образования терпинолена и терпиненов при каталитиче-
ской изомеризации а-пинена на титановой кислоте. Там же, с. 1452—1457.
Авт.: Г. А. Рудаков, М. М. Шестаева, А. Т. Марчевский, 3. С. Хоменко.
136. Рудаков Г. А., Хоменко 3. С., Арбина Т. Ф. Непрерывный способ
получения уксусного эфира изоборнеола из камфена.— «Гидролизная и лесо-
химическая промышленность», 1955, № 2, с. 3—4.
137. Рудаков Г. А., Шестаева М. М. Каталитическая изомеризация а-пи-
нена в р-пинен.— «Журн. общей химии», 1955, т. 25, с. 627—631.
138. Рудаков Г. А., Шестаева М. М. К вопросу применения реакции дие-
нов эго синтеза с малеиновым ангидридом.— «Журн. прикладной химии», 1955,
т. 28, с. 1199—1204.
139. Рудаков Г. А., Шестаева М. М. Получение оптически чистого а-пи-
нена из Р-пинена.— «Журн. общей химии», 1956, т. 26, с. 2357—2361.
140. Рудаков Г. А., Шестаева М. М. О физических свойствах камфена.—
Там же, с. 2362—2364.
141. Рудаков Г. А., Шестаева М. М. Механизм рацемизации камфена на
титановой кислоте.— Там же, с. 2426—2433.
142. Рудаков Г. А., Хоменко 3. С., Арбина Т. Ф. Способ получения
уксусного и муравьиного эфиров изоборнеола из камфена.— Авторское сви-
детельство № 102445. Бюллетень изобретений, 1956, № 1.
143. Рудаков Г. А., Калиновская Е. А. О равновесии жидкость—пар
в бинарных растворах камфен—уксусная кислота и изоборнилацетат—уксус-
ная кислота.— «Гидролизная и лесохимическая промышленность», 1957, №2,
с. 8—10.
144. Рудаков Г. А., Шестаева М. М. О работе непрерывно действующей
установки для ректификации скипидара на Нейво-Рудянском лесохимическом
заводе.— «Гидролизная и лесохимическая промышленность», 1958, № 1,
с. 6—8.
145. Рудаков Г. А. Непрерывный способ получения муравьиного эфира
изоборнеола из камфена.— «Гидролизная и лесохимическая промышленность»,
1958, № 7, с. 10—11.
146. Рудаков Г. А., Шестаева М. М. О каталитических свойствах крем-
ниевой кислоты.— «Журн. общей химии», 1959, т. 29, с. 2062—2068.
147. Рудаков Г. А., Шестаева М. М. Изомеризационлые превращения тер-
пинолена в присутствии титановой кислоты. Там же, с. 2096—2100.
148. Рудаков Г. А. Химия и технология камфары. Изд. 1-е. М.—Л., 1961.
224 с.
149. Рудаков Г. А., Шестаева М. М., Иванова Л. С. О влиянии носите-
лей на направление кислотной каталитической изомеризации пинена.— Докл.
АН СССР, 1965, т. 162, с. 1320—1322.
150. Рудаков Г. А., Калиновская Е. А. Теплоты изомеризации камфена
в трициклен и перегруппировки Вагнера при превращении камфенгидрата
в пзоборнеол.— «Журн. органической химии». 1965, т. 1, с. 1199—1205.
151. Рудаков Г. А., Верещагина А. А. Третий порядок щелочного гидро-
лит изоборнилацетата в водно-спиртовых растворах.— «Журн. органической
химии», 1967, т. 3, с. 311—316.
152. Рудаков Г. А., Полтавченко Ю. А., Иванова Л. С. О биогенезисе
монотерпенов а эфирных маслах хвойных.— В кн.: Синтетические продукты
194
из канифоли и скипидара.— Труды II Всесоюзного научно-технического со-
вещания, Горький, 1970, с. 156—172.
153. Рудаков Г. А., Чудинов С. В., Нестеров Г. В., Богданов Е. Б., Се-
меновых Я- Н., Машкии Н. В. Способ получения катализатора для парофаз-
ной дегидрогенизации борнеола.— Авторское свидетельство № 386600. Откры-
тия, изобретения, промышленные образцы, товарные знаки, 1973, № 27.
154. Хроматографические методы исследования камфары, полученной из
скипидара и пихтового масла.— В кн.: Хроматографический анализ в химии
древесины. Рига, 1975, с. 125—131. Авт.: Г. А. Рудаков, В. И. Гармашов,
Т. Н. Писарева, А. Г. Боровская, Н. П. Зубричева.
155. Рудаков Г. А., Иванова Л. С., Писарева Т. Н., Боровская А. Г.
Кинетика каталитической изомеризации жидкого а-пннена на титановом ка-
тализаторе.— «Гидролизная и лесохимическая промышленность», 1975, № 5,
с. 7—9.
156. Ружичка Л. Значение теоретической органической химии для химии
терпеновых соединений.— В кн.: Перспективы развития органической химии.
М„ 1959, с. 187—222.
157. Руководство по газовой хроматографии. М., 1969. 503 с.
158. Рутовский Б. Н., Лосев Н, П., Берлин А. А. Дегидрирование бор-
неола.— «Промышленность органической химии», 1938, т. 4, с. 410—416.
159. Рутовский Б. Н., Любомилов В. И., Плотникова А. Ф. О выделении
камфена из эфирных масел.— «Журн. прикладной химии», 1940, т. 13,
с. 576—578.
160. Рутовский Б. Н., Муляр П. А. Дегидрирование борнеола в газовой
фазе с промотированным никелевым катализатором.— «Журн. прикладной
химии», 1941, т. 14, с. 173—180.
161. Рутовский Б. Н„ Любомилов В. И. О природе некоторых катализа-
торов для изомеризации пинена. Сообщение 1.— «Журн. прикладной химии»,
1947, т. 20, с. 516—520.
162. Рутовский Б. Н., Любомилов В. И. Сообщение 2. Там же, с. 625—
629.
163. Саратиков А. С. Камфара. Томск. 1966. 273 с.
164. Сивов Ф. К., Коротаева М. М,, Кочнева М. П. Получение камфары
путем дегидрогенизации борнеола и изоборнеола с металлическими катали-
заторами.— «Журн. химической промышленности», 1933, т. 10, № 3, с. 52—56.
165. Сидоров Р. И., Хвостикова А. А., Рудаков Г. А. Качественный ана-
лиз смесей терпеновых углеводородов эфирных масел.— «Гидролизная и ле-
сохимическая промышленность», 1969, № 6, с. 13—15.
166. Скоу Э. Л., Вейкгельм Г. Определение температур плавления и за-
мерзания.— В кн.: Физические методы органической химии. Под ред. Вейс-
бергер А. т. 1. М., 1950, с. 9—57.
167. Скворцов С. О. Лесохимическая промышленность Германии. М., 1949.
120 с.
168. Снесарев К. А., Зараковская А. И., Воробьева М. Т. Метрологиче-
ские основы аналитического контроля химических производств. М., 1960,
с. 12—55.
169. Справочник по растворимости, т. II, кн. 2, М.—Л.. 1963. 1181 с.
170. Танака Тоёсукэ, Мацуда Цунзо, Кимидзима Кануори и др. Твердое
ракетное топливо.— Японский патент 48—27881 (1973); РЖХим, 1974, 12П,
44П.
171. Тиличеев М. Д. Физико-химические свойства индивидуальных угле-
водородов, вып. 4. М.—Л., 1953, с. 159—170, 311—315.
172. Тищенко В, Е., Марта В. Я. О действии флоридина, каолина и бок-
сита на пинен.— ЖРФХО, протокол № 6, 1928, т. 60, с. 1009—1011.
173. Тищенко В. Е., Рудаков Г. А. К вопросу о производстве синтетиче-
ской’камфары.— «Лесохимическая промышленность», 1932, № 5—6, с. 24—25.
174. Тищенко В. Е., Рудаков Г. А. Каталитический способ получения
камфена из скипидара.— «Журн. прикладной химии», 1933, т. 6, с. 691—701.
13* 195
175. Тищенко В. Е., Рудаков Г. А. О применении реакции Бертрама и
Вальбаума для количественного определения камфена, находящегося в смеси
с пиненом и дипентеном. Там же, с. 701—703.
176. Тищенко В. Е., Рудаков Г. А. О превращении камфена в уксусный
и муравьиный эфир изоборнеола.— «Журн. прикладной химии», 1934, т. 7,.
с. 369—372.
177. Тищенко В. Е., Грехнев М. А. Дегидрирование борнеола и изобор-
неола с активированным никелевым катализатором.— «Промышленность орга-
нической химии», 1940, т. 7, с. 238—240.
178. Тищенко В. Е., Грехнев М. А., Елисеева А. А. Дегидрирование бор-
неолов на катализаторе из сплава металлов.— «Журн. прикладной химии»,
1941, т. 14, с. 395—399.
179. Тищенко В. Е., Грехнев М. А. Дегидрирование изоборнеола с мед-
ным катализатором. Там же, с. 889—893.
180. Тищенко Д., Сумм Н. О строении пнроненов.— «Журн. общей хи-
мии», 1957, т. 27, с. 379—384.
181. Уитмор Ф. Теоретические основы современных процессов перера-
ботки нефти (алкилирование, изомеризация, димеризация).— В кн.: Синтез
моторного топлива, т. 1. М., 1949, с. 7—37.
182. Ушаков С. Н., Песин Л. М. Камфара. Л., 1931. 188 с.
183. Фунахаси Т. Изомеризация пинена в камфен. Ч. 1, 2, 3, 4.— РЖХим,
1956, № 61622.
184. Чмиль В. Д. Количественное определение камфары методом газо-
жидкостной хроматографии.— «Фармация», 1971, т. 20, № 2, с. 33—35.
185. The Агг.реге Electro Chemical Со Jersey.— Герман, пат. 134553 (1900);
Ch. Zbl„ 1902, В. 2, S. 975.
185. Ando S. Oxidation of Isoborneol with Nitric Acid.— Ch. A., 1959, v. 53,
p. 18976.
187. Angus W. R. Raman Spectra of Terpenes.— Proceedings of the Indian
Academy of Science, 1938, v. 8A, p. 529—565.
188. Armstrong H. E., Tilden W. A. Uber die Einwirkung der Schwe-
fels.iure auf Kohlenwasserstoffe der Formel СюН!в.— Ber., 1879, B. 12, S. 1752—
175".
189. Aschan O. Naphtenverbindungen, Terpene und Campherarten. Berlin,
1929. 378 S.
190. Bain J. P., Best A. H., Hampton B. L., Hawkins G. A., Kitchen L. J.
The Preparation of Optically Pure L-Camphene.— J. Am. Ch. Soc., 1950, v. 72,
p. 3124—3127.
191. Bean N. E. Camphora-Curriculum Vitae of a Perverse Terpene.—
Chemistry in Britain, 1972, v. 8, p. 386—388.
192. Beckmann W. Celluloid.— In Ullmanns Encyklopadie der technischen
Chemie, 3 Aufl., B. 5, Munchen, 1954, S. 141—156.
193. Berson J. Cabonium Ion Rearrangements in Bridged Bicyclic Systems.—
In Molecular Rearrangements, ed. de Mayo P., v. 1, New-York, 1963, p. Ill—232.
194. Bertram J., Walbaum H.— Герман, пат. 67255 (1892); Ch. Zbl. 1893,
B. 1, S. 1000.
195. Bertram J., Walbaum H. Uber Isoborneol.— J. pr. Ch., 1894, B. 49 (2),
S. 1-15.
196. Birch A. J. Aliphatic Compounds.—In Annual Reports on Progress
of Chemistry for 1950, London, Chem. Soc., 1951, v. 47, p. 190—199.
197. Booth A. B. Terpenes and Terpenoids.— In Kirk-Othmer Encyclopeadia
of Chemical Technology, 2d ed.. New-York, 1969, v. 19, p. 803—834.
198. Borowiecki L., Chretien-Bessiere Y. Etude du bornylene.—Bull. Soc.
Ch.. 1967, p. 2364—2369.
199. British Intelligence Objectives Subcommitee (BIOS). German Camphor
Industry. London, 1946, N 1240, p. 21.
200. Britch Pharmacopacia. London, 1964, p. 127—128.
201. Brus G., Vebra J. Sur la transformation du camphene en estere d'iso-
borneole.— Bulletin de 1’Institut du Pin, 1935, N 6, 109—120; N 9, 187—192.
196
202. Bukala M. Badania nad skladem Polskich terpentyn z Pinus silvestris.
Wroclaw, 1952, 86 s.
203. Bukala M, Majewski J., Rodzinski W. Stan rawnowagi miedzufazowej
ukladu ciecz-para dla rzeczywistych mieszanin dwuskladnikowych. Weglewodory
terpenowe I.— Przemysl Chemiczny, 1953, t. 9, s. 513—520.
204. Bukala M., Majewski J., Rodzinski W. Stan rawnowagi miedzufazowej
ukladu ciecz-para dla rzeczywistych mieszanin dwuskladnikowych. Weglowodory
terpenowe II.— Przemysl Chemiczny, 1954, t. 10, s. 197—201.
205. Bukala M., Majewski J.. Rodzinski W. Preznosc par nasyconych weglo-
wodorow terpenowych, stalych skladnikow krajowych terpentyn z sosny pospo-
lity.— Przemysl Chemiczny, 1954, t. 10, s. 6—11.
206. Bukala M., Dul M, Przondo J.— Польский пат. 65172 (1972); РЖХим,
1973, 17 Л, 171 П.
207. Bunton С. A., Khaleeluddin К., Wittaker D. The Hydrolysis of Bornyl
and Isobornyl Acetates.— J. Ch. Soc., 1965, p. 3290—3299.
208. Chem. Fabrik von Heyden.— Герман, патенты 175097 (1904), 178934
(1904); Ch. Zbl, 1906, B. 2, S. 1589, 1907, B. 1, S. 198.
209. Chem. Fabrik vor Heyden.— Герман, патенты 184635 (1905), 185933
(1905), 187684 (1906); Ch. Zbl., 1907, B. 2, S. 434, 2000.
210. Christensen В. V., Lynch H. J. A comparative study of the Pharma-
cological action of Natural and Synthetic Camphor.— Journal of the American
Pharmaceutical Association, 1937, 786—794.
211. Coates W. M. Camphor Natural and Synthetic — Perfumery and Es-
sential Oil Record, 1952, v. 43, p. 331—335, 360—363.
212. Dastur S. P. Synthetic Camphor from Turpentine.— Chemical Age of
India, 1961, v. 12, p. 317—324.
213. Darasse L., Darasse E., Dupont L.— Франц, пат. 509680 (1919).
214. Darasse L., Dupont L.— Герман, пат. 425789 (1926); Ch. Zbl. 1923,
B. 1, S. 2841.
215. Delepine M. Sur 1’origine du fenchol dans la reaction de Bouchardat
et Lafont.— Comt. rend., 1924, v. 178, p. 2087—2089.
216. Demand will Determine Naval Stores Output.— Chemical and Engin.cr-
ing News, 1962, v. 40, N 47, p. 32—37.
217. Deutsches Arzneibuch, 7-te Aufl, Berlin, 1964, S. 99—107.
218. Dul M, Bukala M. Ustalenie optymalnych warunkow preparatyki im-
pregnowanego katalizatora kamfenizacyjnego.— Chemica Stosowana, 1971, v. 15,
s. 75—98.
219. Dul M, Bukala M. Wplyw parametrow procesu na przebieg i wyoaj-
nose reakcji isomeryzacji a-pinenu.— Chemia Stosowana, 1971, v. 15, s. 317—140.
220. Dul M., Bukala M. Badania reaktywnosci weglowodorow monote pe-
nowych skladnikow krajowych terpentyn sosnowych w obecnosci katalizato.ow
impregnowanych.— Chemia Stosowana, 1973, v. 17, s. 19—48.
221. Du Pont de Nemours.— Англ. пат. 392134 (1932); Ch. Zbl., 1933, В 2,
S. 1583.
222. Du Pont de Nemours (Henke С. O., Etzel G.).— Пат. CHIA 2129323
(1938); Ch. Zbl, 1939, B. 1, S. 3072.
223. Du Pont de Nemours (Henke С. O, Etzel G.).— Пат CHIA 2318391
(1943); Ch. A, 1943, v. 37, p. 5988.
224. Du Pont de Nemours (Etzel G.).—Пат США 2551795 (1951); Ch. A,
1951, v. 45, p. 9082.
225. Du Pont de Nemours (Arnold H. A, Carnahan J. E.).— Пат. CHIA
2571977 (1951); Ch. A, 1952, v. 46, p. 5088.
226. Etzel G. Camphor.— In Kirk E, Othmer D. F. Encyclopeadia of Che-
mical Technology It ed, v. 2, New-York, 1948, p. 808—818.
227. Fabrique de produits Chimiques de Thann et Mulhouse.— Франц, пат.
510002 (1919).
228. I. G. Farbenindustrie (Bilcken V.).—Пат. США. 1902364 (1933): Ch.
Zbl, 1933, B. 1, S. 3366.
229. Fernholz H, Schmidt H. J. (Fabr Werke Hochst).— Пат ФРГ 119(190
(1965); Ch. A, 1965, v. 63, p. 11627.
197
230. Fuguitt R. E., Hawkins J. E. The Liquid Phase Termal Isomerisation
of a-Pinene.— J. Am. Ch. Soc., 1945, v. 67, p. 242—245.
231. Fuguitt R. E., Hawkins J. E. Rate of Termal Isomerisation of a-Pinene
in the Liquid Phase.— J. Am. Ch. Soc., 1947, v. 69, p. 319—322.
232. Geger S., Zieger W., Holm S., Mayer R. Zur Gaschromatographie der
Monoterpene.— Zeitschrift ftir Chemie, 1965, B. 5, S. 309—310.
233. Gildemeister E., Hoffmann Fr. Die alherischen Ole, 4 Auff., B. 3-a,
Berlin. 1960. 626 S.
234. Gildemeister E., Hoffmann Fr. Die atherischen Ole, 4 Aufl., B. 3-b,
Berlin, 1962. 436 S.
235. Gildemeister E., Hoffmann Fr. Die atherischen Ole, 4 Aufl., B. 3-c,
Berlin, 1963. 510 S.
236. Goldblatt L. A., Palkin S. Vapor Phase Termal Isomerisation of a-
and р-Pinene,— J. Am. Ch. Soc., 1947. v. 63, p. 3517—3522.
237. Goldblatt L. A., Palkin S. The Production of a- and p-Pyronene from
allo-Ocimene.— J. Am. Ch. Soc., 1944, v. 66, p. 655—666.
238. Goldshmith J. K.—Англ. пат. 17573 (1906).
239. Gonzenbach С. T., Jordan M. A., Yunick R. P. Terpene Resins.— In
Encyclopedia of Polymer Science and Technology, v. 13, New-York, 1970,
p. 575—596.
240. Gowalowski A., Kutter L.— Англ. пат. 178797 (1921).
241. Guenther E. The Camphor and Ho-oil Industry of Taiwan.— Amer.
Perfumer and Cosmetics, 1969, v. 84, № 3, p. 51—56.
242. Guenther E. The Camphor and Ho-oil Industry of Japan.— Amer. Per-
fumer and Cosmetics, 1969, v. 84, № 4, p. 31—36.
243. Hacker K., Keicher G., Miltenberger К. H. Die Konstitution von Pseu-
doborneol.— Tetrahedron Letters, 1965, № 34, p. 2987—2989.
244. Hanada Y., Kitajima M. Eine gaschromstographische Bestimmung von
Campher.— J. Ch. Soc. Japan, pure Chem. Sect., 1959, v. 80, p. 1272—1274; Ch.
Zbl., 1961, S. 8416.
245. Harrington T. A. Production of Oleoresin from Southern Pine Trees.—
Forest Products Journal, 1969, v. 19, № 6, p. 31—36.
246. Hawkins J. E., Eriksen W. T. The Heats of Combustion of some Ter-
pene Hydrocarbons.— J. Am. Ch. Soc., 1954, v. 76, p. 2669—2671.
247. Hawkins J. E., Armstrong С. T. The Vapor Pressures of a-Pinene and
P-Pinene.— J. Am. Ch. Soc., 1954, v. 76, p. 3756—3758.
248. Herout V. О Terpenech XLY.— Chemicke listv, 1952, v. 46. p. 438—444.
249. Herkules Powder (Carson W. E.).— Пат. США 2382397 (1945); Ch. A.,
1946, v. 40, p. 363.
250. Herkules Powder (Kirpatrick W. J.).— Пат. США 2385711 (1945);
Ch. A.. 1946, v. 40, p. 607.
251. Herkules Powder (Carson W. E.).—Пат. США 2450119 (1948); Ch. A.,
1949, v. 43, p. 2234.
252. Hertkorn J.— Англ. пат. 11248 (1908).
253. Hesse А,—Герман, пат. 182943 (1904); Ch. Zbl., 1907, B. 1, S. 1470.
254. Hiraizumi T. Camphor Oil.— In the book Guenther E., The Essential
O;ls, v. 4, New-York, 1950, p. 256—328.
255. Hiickel W., Doll W., Eskola S., Weidner H. Camphenilon, Camphen-
h\drat und Methylcamphenvlol.— Lieb. Ann., 1941, B. 549, S. 186—208.
256. Hiickel W. Theoretische Grundlagen der organischen Chemie. 9 Aufl.,
В. 1, Leipzig, 1961, S. 376—427.
257. Hunt H., Hawkins J. E. The Rates of the Termal Isomerisation of a-
ard P-Pinene in the Liquid Phase.— J. Am. Ch. Soc., 1950, v. 72, p. 5618—5620.
258. Jones C. A., Schoenborn E. M., Colburn A. P. Equilibrium Still for
miscible Liquids.— Ind. Eng. Ch., v. 35, p. 666—672.
259. Jone W., Watanabe T., Okubo N. Explosion von Campher.— Ch. Zbl.,
1960, S. 1791.
260. Joschi D. P. Resin Industry in India.— Indian Forester, 1969, v. 95,
p. 513—525.
193
261. Jubilaum bei den Farbwerken in Gersthofen.— Augsburger Allgemeine
1972, 28 Avr.
262. Keicher G. Terpentinol.— In Ullmanns Encyklopadie der technischen
Chemie, 3 Aufl., B. 16, Munchen, 1965, S. 768—775.
263. Keicher G., Terpentinolprodukte.— In Ullmanns Encyklopadie der tech-
nischen Chemie, 3 Aufl., B. 17, Miinchen, 1966, S. 1—63.
264. Kharasch M. S. Heats of Combustion of Organic Compouds.— Bureau
of Standards Journal of Researches, 1929, v. 2, p. 359—430.
265. Kharasch M. S., Reinolds W. B. Isomerisation and Esterification of
a-Pinene.— Journal of Organic Chemistry, 1944, v. 9, p. 148—154.
266. Kitajima M., Noguchi M. A New Synthetic Method for Camphor.—
Ch. A., 1959, v. 53, p. 18976.
267. Klein E. Die Zusammenhange in der Monoterpenreihe. Tafel, Holzmin-
den, ,,s. a“.
268. Klimont J. M. Der technisch-synthetische Campher. Leipzig, 1921,
132 S
269. Klose W., Bochow К.—Пат. ГДР 69586 (1969); РЖХим., 1970, 15 H.
192 П.
270. Kondakov I. Synthesen unter Einwirkung von Zinkchlorid in der
hydroaromatischen Reihe.— J. pr. Ch., 1902, B. 65 (2), S. 201—238.
271. Kondakov I. L., Isomerisation des pinenes en camphene par les pl.e-
nols.— Parfumerie moderne, 1926, v. 19, p. 181—185, 212—223.
272. Kiihne S. Sprengstoffe, rauchloser Pulver.— In Ullmanns Encyclopa lie
der technischen Chemie, 3 Aufl., B. 16, Munchen, 1964, S. 99—107.
273. The Manufacture of Camphor.— Industrial Chemist. 1950, v. 26,
p. 113—119.
274. Masumoto B., Funabashi R. Camphene by Isomerisation of Pine ie.
Ch. A., 1956, v. 50, p. 5032.
275. Matowowski A., Tomaszewska L., Zacharewicz W. Badania rad
zmiennoscia skladu polskiego balsamicznego olejku terpentynowego.— Przem . si
Chemiczny, 1954, t. 10, s. 11 —18.
276. Meerwein H., van Emster K. Uber den Reaktionsmcchanismus der
Isoborneol 5^ Camphen Umlagerung.— Ber., 1920, B. 53, S. 1815—1829.
277. Meerwein H., van Emster K. Uber die Gleichgewichts—Isomeric
zwischen Bornylchlorid, Isobornylchlorid und Camphen-chlorhvdrat.— Ber., 1922,
B. 55, S. 2500—2528.
278. Meerwein H., Hammel O.. Serni A., Vorster J. Untersuchungen fiber
intermolekulare Atomverschiebungen in der Campherreihe.— Lieb. Ann., 1927,
B. 453, S. 16—47.
279. Meerwein H., Vorster J. Uber das Pinenchlorhydrat.— J. pr. Ch., 19 56,
B. 147 (2), S. 83—92.
280. Mirov N. T. Composition of Gum Terpentines of Pines — Forest Ser-
vice USA, 1961, Techn, Bull. N 1239, 158 p.
281. Mitzner В. M., Theimer E. T., Freeman S. K. The Infrared Spectra of
Monoterpenes and Related Compounds.— Applied Spectroscope; 1965, v. 19.
p. 169—186.
282. Mitzner В. M„ Mancini V. J., Lemberg S., Theimer E. T. Infrared
Spectra of Monoterpenes and Related Compounds. II. Terpene Alcohols.— Ap-
plied Spectroscopy, 1968, v. 22, p. 34—53.
283. Naval Stores Statistical Data.— Naval Stores Annual Reports, Wa-
shington, 1968—1972.
284. Naval Stores Statistical Data.— Naval Stores Review, International
Yearbook, 1972—1973.
285. Oettel H. Terpentinol Gewerbetoxikologie.— In Ullmanns Encyklopadie
der technischen Chemie, 3 Aufl., B. 16, Munchen, 1965, S. 775.
286. O’Conner R. T., Goldblatt L. A. Correlation of Ultraviolet and Infrar-
ed Spectra of Terpene Hydrocarbons.— Analytical Chemistry, 1954, v. 26,
p. 1726—1737.
199
287. Othmer D. Azeotropie and Azeotropic Destination.— In Kirk—Othmer
Encyclopeadia of Chemical Technology, 2-d ed., v. 2, New-York, 1963,
p. 839—859.
288. Palmer R. C. Solvents from Pine.— Ind. Eng. Ch., 1943, v. 35,
p. 1023—1025.
289. Pasternak A., Bukala M., Dul M. Analityczna kontrola procesow iso-
mery zacyjnych, zachodzacych w czasie otrzymywania kamfenu, przy zastoso-
wani-j chromotografii gazowey.— Chemia Stosowana, 1971, t. 15, s. 181—194.
290. Peufaillit L.— Франц, пат. 545368 (1922).
291. Pharmacopee Francaise, 8 ed., Paris, 1965, p. 247—250.
292. The Pharmacopeia of USA, 18th ed., New York, 1970, p. 97—98.
293. Pines H., Eschinazi H. E. Sodium-Catalyzed Double Bands Migration
and Dehydrogenation of d—Limonene, 1—a-Phellandrene and 2, 4 (8) and 3, 8
(9)-p-Menthadiene.— J. Am. Ch. Soc., 1955, v. 77, p. 6314—6321.
294. Pliva J., Horak M., Herout V., Sorm F. Die Terpene. Sammlung von
Spektren und phisikalischen Eigenschaften. Teil 1, Sesquiterpene. Berlin, I960,
228 Taf.
295. Poe C. F., Plein E. M. Optical Activity of Camphor in Alcoholic So-
lution.— Journal of the Physical Chemistry, 1934, v. 38, p. 883—887.
296. Pulkkinen E. Uber die Dehydratation des a- und (3-Fenchols.— Annales
Academie Scientiarum Fennicae, Helsinki, 1956, В. A74, S. 1—87.
297. Remy H. Lehrbuch der Anorganischen Chemie. 9 Aufl., B. 2, Berlin,
1959. S. 172—174.
298. Ritter J. A New Camphor Synthesis.— J. Am. Ch. Soc., 1933, v. 55,
p. 3322—3326.
299. Roberts D. R., Joye N. M., Proveaux A. T., Peters W. J., Lawrence R. V.
A Niw and more Efficient Method of Naval Stores Production.— Naval Stores
Review, International Yearbook. 1972—1973, p. 6—7.
300. Rodds Chemistry of Carbon Compounds. 2d ed., edited by Coffey S.,
Amsterdam, v. 2 B, 1968, p. 176—208, v. 2 C, 1969, p. 136—255.
301. Ropp W. S., Hawkins J. E., Reitz E. C., de Mayo P., Harris G. C.
Monuterpenoids-cyclic.— In Kirk E., Othmer D. F. Encyclopeadia of Chemical
Technology, 1st ed., v. 13, New York, 1954, p. 705—771.
302. Sandermann W. Naturharze, Terpentinol-Tallol. Chemie und Technolo-
gic. Berlin, 1960. 483 S.
303. Schafer K-> Wagner U. Uber die Umwandlung in System d-l-Cam-
pher.— Zeitschrift fur Elektrochemic, 1958, B. 62, S. 328—335.
304. Schering.—Герман, пат. 208487 (1907); Ch. Zbl, 1909, B. 1, S. 1282.
305. Schering.— Герман, патенты 219043 (1908), 219044 (1908), 271147
(1908), 271157 (1908); Ch. Zbl, 1910, B. 1, S. 972, 1914, B. 1, S. 1130.
206. Schering-Kahlbaum.— Герман, пат. 573797 (1933); Ch. Zbl, 1933, В. 1,
S. 3366.
, 07. Schering-Kahlbaum.— Франц, пат. 613806 (1926); Ch. Zbl, 1927, B. 2,
S. 1086.
308. Schering.— Герман, патенты 508093 (1930), 510429 (1930); Ch. Zbl,
1930. B. 2. 2959; 1931, B. 1, S. 160.
209. Schering.— Франц, пат. 716515 (1931); Ch. Zbl, 1932, B. 2, S. 617.
210. Schering.— Франц, пат. 703355 (1930); Ch. Zbl, 1931, B. 2, S. 1195.
.'-11. Schering.— Герман, патенты 570957 (1933), 578569 (1933), 582043
(193.5). 584965 (1933); франц, пат. 739770 (1932); Ch. Zbl, 1933, В. 1, S. 3112,
4032. В. 2, S. 1431, 2056, 3760.
312. Schering.—Англ. пат. 391073 (1933); Ch. Zbl, 1933, В. 2. S. 1431.
313. Schering (Meerwein H., infers F., Erbe R.).— Герман пат. 610402
(1935); Ch. A., 1935, v. 29, p. 3692.
314. Schmidt L.— Герман, патенты 401870 (1924), 406768 (1924), 532395
(1928), 543429 (1928); Ch. Zbl, 1925, B. 1, S. 299, 1266, 1931, B. 2, S. 3654,
1932. B. 1, S. 1855.
2,15 . Schubert H. Terpentinol und Kolophonium.— In Winnacker H„ Kiich-
ler L. Chemische Technologie, 3 Aufl., B. 3, Munchen, 1971, S. 493—510.
200
316. Schwarz F. —Пат. США 2389389 (1945); Ch. A., 1946, v. 40, p. 1837.
317. Simonsen J. L.. Owen О N.— The Terpenes, 2d ed., v. 1, Cambridge,
1947, 479 p.
318. Simonsen J. L., Owen O. N.— The Terpenes, 2d. ed., v. 2, Cambridge,
1949, 618 p.
319. Snatzke G., Kovats E., Ohloff G. Circulardichroismus von a-Pl.cl-
landren.— Tetrahedron Letters, 1966, № 38, p. 4551—4553.
320. Stonecipher W. D. Turpentine.— In Kirk-Othmer Encyclopeadia of
Chemical Technology, 2d ed., v. 20, New York, 1970, p. 748—755
321. Sutherland M. D. A Review of the Densites and Refractive Indices
of the Terpenes.— University Queensland Papers, 1948, v. 34, № 1, p. 1—21.
322. Sutherland M. D. The Refractive Index-Density Chart in the Separation
of Terpenoids.— Perfumery and Essential Oil Record, 1952, v. 43, p. 453—459.
323. Swann G. The Catalytic Isomerisation of a-Pinene to Bicyclic Terpe-
nes.— Industrial Chemist, 1948, v. 24, p. 141—145..
324. Swann G., Cripwell F, J, The Catalytic Isomerisation of Camphene.—
Industrial Chemist, 1948, v. 24, p. 573—579.
325. Synthetic Camphor growth slowed by Celluloid demise.— European
Chemical News, 1972, Febr. 25, p. 19.
326. Thomas G, A., Hawkins J. E. The Dielectric Constant, Refractive Index
and Dencity of some Terpenes. J. Am. Ch. Soc., 1954, v. 76, p. 4856—4858.
327. Thurber F. H., Thielke R. C. Optically Active Alpha-Pinenes.— J. Am.
Ch. Soc., 1931, v. 53, p. 1030—1032.
328. Timmermans J. Physico-Chemical Constants of pure Organic compo-
unds. New-York, 1950, 693 p.
329. Toivonen N. V., Alfthan V., Book L. N., Erich M. T., Heino E. K. Zur
Chemie der synthetischen Diterpene I.— J. pr. Ch., 1941, B. 159 (2), S. 70—111.
330. Tucker W, S., Hawkins J, E. Vapor-Liquid Equilibria of Alpha-Bcta-
Pinene System.— Ind. Eng. Ch., 1954, v. 46, p. 2387—2390.
331. Ullmann F, Campher.— in Encyklopadie der technischen Chemie. 2
Aufl., B. 3, Berlin, 1929, S. 60—83.
332. Venable C. The Effect of Fuller's Earth on Pinene and other Terne-
nes.— J. Am. Ch. Soc., 1923, v. 45, p. 728—734.
333. Verley A., Urbain F., Feige A.— Герман, пат. 207156 (1907); Ch. Zbl,
1909, B. 1, S. 961.
334. Vyrodov V, A., Korotov S. Ya., Afanas’eva E. V. and others.— Англ,
пат. 1118499 (1968); Ch. A., 1969, v. 70, p. 29118.
335. Wagner G., Brickner W. Uber die Beziehung der Pincnhaloidhydr ite
zu den Haloidenhydriden des Borneols.— Ber., 1899, B. 32, 2302—2325.
336. Whitmore F. The Common Basis of Intermolecular Rearrangements.—
J. Am. Ch. Soc., 1932, v. 54, p. 3274—3283.
337. Widmark G. A-3Carene and a-Pinene from Swedish Sulfate Terpen-
tine.— Acta Chemica Scandinavica. 1955, v. 9, p. 925—937.
338. De Wilde J, H. Damfdruck von Campher.— Zeitschrift fur anorganische
und allgemeine Chemie, 1937, B. 233, S. 411—414.
339. Wystrach V. P„ Barnum L, N., Garber N. J. Liquid Phase Catalytic
Isomerisation of a-Pinene.— J. Am. Ch. Soc., 1952, v. 79, p. 5786—5790.
340. Zeitschel О,—Герман. пат. 204163 (1906); Ch. Zbl, 1908, B. 2, S. 1751.
201
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Анализ готовой продукции 184, 185
— смесей терпенов, историческое раз-
витие 165, 166
—физический: контроль за ходом
изомеризации пинена 184
----определение полимеров в изоме-
ризатах 183
----технического камфена 183
— химический: борнеолов 184
----изомеризатов 182
----камфары 158, 159, 177
----эфиров борнеолов 183, 184
Антиокислители 26, 68, 178
Борнеол и изоборнеол из камфена
103, 104
-------строение и свойства 5, 6, 97,
104, 105, 152, 189
Борнильные и изоборнильные эфиры
гидролиз: в растворителе 95, 96, 102,
155
----------- кинетика 98—100
-----------технология периодиче-
ского процесса 93—97
---------из пинена 14, 15
Борнилхлорид 13, 14, 27—36
— побочные реакции при получении
28. 29
— получение 27—30
— состав 29, 30
Гс.зо-жидкостная хроматография, ана-
лиз камфары 173, 176, 177
----скипидаров 177
----смеси терпенов 165—177
----— анализы борнеолов, их эфи-
ров, пихтового масла 177
---------изомеризатов, камфена, ку-
бовых остатков 177
------- воспроизводимость и точность
анализов 174—177
------- контроль дегидрирования бор-
неолов 178
---------за ходом изомеризации пи-
ненов 178
-------- метод внутреннего стандарта
173—177
---- ----внутренней нормировки
171, 172, 177
-------относительное удерживание
кислородных производных терпенов
169, 170
-----------компонентов синтетиче-
ской камфары 170
-------——определение понятия 167,
169
-----------терпеновых углеводоро-
дов 168
-------расшифровка хроматограмм
170, 171
-------требования к хроматограм-
мам 171
Давление насыщенных паров борне-
ола 143, 144
-------изоборнеола 143
-------камфары 143
-------терпеновых углеводородов
138—142
-------эфиров изоборнеола 140
Живица 17—18
— переработка 19
Изоборнеольные масла 96, 97
Изоборнилацетат и формиат из кам-
фена 78—93
-----------в присутствии воды 82,
84, 86—88
----------катализаторы 78, 79, 88,
89, 92
-----------константы равновесия
83, 84
-----------механизм реакции 79, 80
-----------непрерывные способы
88—92
202
---------------------образование камфенгид-
рата 79
--------------эфира изофенхола 80
---------------------5-экзо-изокамфан-
экзо-ола-3 (псевдоборнеола) 79
—----------побочные реакции 79—81
---------------------полимеризация 80
---------------------регенерация избытка кис-
лот 86, 88—92
---------------------скорость реакции 83,
89, 90
-------— — сравнение процессов 92
-------температура реакции
81, 82, 84, 89, 90
-------технология 84—88
--------------трициклена 83
-------свойства 93, 189
— из камфена и трициклена, сравне-
ние скоростей реакции 83
---------------------тепловой эффект реак-
ции 84
Изоборнилформиат, непрерывный гид-
ролиз 100—102
Изоборнилхлорид 29—36
Изофенхиловый спирт (эфиры) 38, 80,
93, 96, 97, 109, 110, 155, 156, 170, 189
Изофенхон 31, 38, 45, 80, 109, 110,
155—157, 170, 176, 189
Ионы карбоння 33—36
Камфара, анализ 173, 176
— в медицине 5, 6, 8, 10, 158—160
----порохах 6
---- ракетном топливе 9
----целлулоиде 6, 8, 9, 31
----эфирных маслах 6
— дегидрированием борнеолов 107—
123
-------в жидкой фазе 112—120
-------— паровой фазе 108, 120—123
•------история 108
—------кинетика 111, 112
—------образование изокамфана 109
-------теплота реакции 113
----изоборнеола , образование изо-
фенхона НО
---------5-экзо- изокамфанона-3
(псевдокамфары) ПО
— естественная из камфарного бази-
лика 13
---------дерева 6, 10—12
---------масла 11
-------камфарной полыни 6
— — перспективы производства 8—9
----состав и свойства 8, И, 154—156
— зарубежная аналитическая харак-
теристика 155, 156
----технические требования 160
— из пайн-оля 13, 27
----пихтового масла аналитическая
характеристика 157, 158
---------в медицине 8, 148, 158
---------влияние сантеиа на произ-
водство 150
-----------сесквитерпенов на произ-
водство 149, 151—153
—--------использование камфена
149-151
---------история 148
-----— — особенности сырья 149, 150-
---------очистка 152, 153
---------переработка борнилацета-
та 151, 152
— история производства 6—10, 12—
16, 37, 38, 148
— критика методов исследования 159,
177, 185
— мировое производство 9
— окислением борнеолов 105—107
— окислением камфена 123—126, 128
----- механизм реакции 124
-----побочные реакции 124, 125
-----метилового эфира изоборнеола
106
-----пинена 16
— очистка 11, 152, 159—164
— перегонка с водяным паром И, 107,
115-119, 126, 152, 153
— перспективы производства 8—10
— растворимость 5, 154, 162, 163
— свойства 5, 153, 154. 189
— синтетическое влияние примесей на
температуру кристаллизации 155—157
-----ГОСТ 158
-----из пинена, способы получения
13—16
-----п-цимола 13
-----элементов 13
-----производители 9
— советского производства аналити-
ческая характеристика 157
— фармакопейная в СССР 8, 158, 159
— цены 10
Камфен, выделение ректификацией
31, 37, 134—136
— гидрохлорид 29, 30, 34
— изомеризационный способ получе-
ния 15, 16, 37—77, 150, 151
---------история 37, 38
---------механизм 38—40, 51—57
---------сравнение с борнихлорид-
ным 15, 38, 41, 73
— каталитическая изомеризация 58
— очистка полимеризацией примесей
46, 53
— полимеризация 37, 38, 46, 53, 58
— получение из борнилхлорида 13, 30,
----------- селективным расщеплени-
ем 31
203
— равновесие с трицикленом 51, 52,
>65
— технический 78, 183
Д3-карен в сырье 22—24, 130, 133, 136,
149
— каталитическая изомеризация 54
— и Д4-карен в изомеризатах 54, 136
Катализ, мультиплетная теория 111
Катализаторы для дегидрирования
борнеолов 108—НО, 121, 122
-----испытания 179
-----в паровой фазе, испытания
180. 181
-----изомеризации пиненов, актив-
ность 42, 43, 45—51, 68, 71, 75
-----определение 178
----- —алюмосиликаты 37—40,
41—46, 56
-----------------------------------------------------------модифицированные 46
-----------------------------выход камфеиа и 2ЭВ 37,
38, 42—46, 56, 62, 66, 67, 74, 77
-----------------------------кислотность 38—40, 42—44,
48-50
---------обменная способность 42,
43, 49, 77
--------------определение 179
---------требования к ним 41
------- — удельная поверхность 43,
49, 50, 68
-------определение 178
-------роль носителей 56
-------силикагели 44
---------серная кислота на ТЮ2
50, 56
—--------титановая кислота, дезак-
тивация при кипячении с водой 50
—-------------отравление 68
--------------1 олучение 47
--------------свойства 46—50, 62—
68
--------------таблетированная 77
Ка -алитическая изомеризация а-пи-
иен 1. изменение состава продуктов
реакции по ходу процесса 62—67
—------кинетика 57—62
--------------непрерывные методы 74—77
-------образование борнилена 46,
51, 53
-------- — лимонена 53, 60, 62—65,67
-------2,4(8)-п-ментадиена 53, 63,
65
---------Д3-п-ментена 54, 58
--------- моноциклических терпенов
37, 54, 58—60, 63, 65—67
---------полимеров 37, 38, 45, 46,
51—55, 57, 58, 63, 65, 73
---------а-терпинена 53, 54, 57, 62—
65
--------- у-терпинена 53, 54, 57,
62-65
204
---------терпинолена 53, 54, 57, 62—
65
---------трициклена 38, 46, 51, 52,
58, 60, 62—67
---------фенхенов 38, 46, 52, 53,
62—65
---------п-цимола 54, 58
-------отсутствие рацемизации 63—
65
-------оценка существующего про-
цесса 73, 74
-------первичные и вторичные реак-
ции 51—55, 62—65
-------тепловой эффект 73
-------технология 70—73
-------энергия активации 62
----р-пинена, изменение состава про-
дуктов реакции по ходу процесса
69—70
-------кинетика 69
-------образование а-пинена 23, 51,
69, 70
----пиненов равновесие между а- и
р-ппненами 51, 70
Лимонен (дипеитен), гидрохлориды 28
— из гидрохлоридов 30
— каталитическая изомеризация 53,
54, 58, 63 -67
Моноциклические терпены, каталити-
ческая изомеризация 65
•---перераспределение водорода 54
----полимеризация 54, 65
Муравьиная кислота — вода состав
азеотропных смесей 146
----укрепление 147, 148
Органические кислоты, отгонка от
изоборнильных эфиров 86, 88, 90, 92,
144—146
----регенерация 103
Пайп-оль 26, 27
Перегруппировка Вагнера—Меервейна
32—37, 51, 52, 79, 105
------- синартетическое ускорение 35
— Наметкина 51, 52, 80
Пиненгидрохлорид 27
а-Пинен как технический продукт
24—26
— термическая изомеризация 128—130
P-Пинен для синтеза камфары 13, 16,
24, 70
—использование 133
— как технический продукт 24
— термическая изомеризация 129
Пихтовое масло для получения кам-
фары 13, 26, 148—153
----получение 26
----состав 26, 149
Подсочка 17
— в разных странах 18
— выход на карру 17
— применение стимуляторов 18
Пневый осмол и его переработка 19
Равновесие жидкость—пар в бинар-
ных растворах терпенов 137, 138
-----------системе муравьиная кисло-
та—вода 146—148
-----------а-пинен — В-пинен 137,
138
----------- уксусная кислота—вода
148
----------------изоборнилацетат
144, 145
----------------камфен 144, 145
Ректификация изоборнильных эфи-
ров 131, 136
— изомеризатов 44, 134—136
— как метод исследования 165, 185
— пихтового масла 150, 151
— скипидара 128—134
— терпенов, возможность каталитиче-
ских влияний 131
-------окислительных процессов 131
—------термических превращений
128—130
----общие вопросы 128—132
----расчет колонн 132, 133, 137, 138
Скипидар как технический продукт 25
Скипидары, анализ 171
— дефицит на рынках 9, 21
— живичные, производство 17—19
----состав 22
— как сырье для производства кам-
фары 24
— сульфатные, производство 20, 21
---- состав 23
— сульфитные 21, 24
— сухоперегонные 21, 24
— цены 21
— экстракционные, производство 19,
20
----состав 23
Терпеновые полимеры 55
Терпены и их производные, спектры 190
---------физические свойства 185—
190
— теплоты сгорания 70, 190
— сравнение скоростей каталитиче-
ской изомеризации 58, 65
Трициклен, образование вместе с кам-
феном 29, 38, 51, 52, 60, 62—66, 69
— выделение ректификацией 135
Уксусная кислота, укрепление 148
Фенхены из фенхнлхлорида 30
— каталитическая изомеризация 52,
53, 65
— полимеризация 46, 53, 55
— примесь к камфену 31, 38, 46, 52,
53, 78, 134, 135
Фенхиловый спирт (эфиры) 15, 26, 27,
80, 93, 96, 109, 110, 155, 156, 169, 189
Фенхилхлорид 29, 30
а-Фепхилхлорнд третичный 30
Фенхон 26, 27, 109, НО, 157, 169, 170,
176, 189
Хром (3) соли, использование 127, 128
Целлулоид 6, 8, 9, 31
5-Экзо-изокамфанон-З (псевдокамфа-
ра) 79, 80, 109, НО, 155—157, 170,
176, 189
5-Экзо-изокамфан-экзо-ол-З (псевдо-
борнеол) в изоборнеоле 79—81, 93
Эфирное масло «хо» 12, 13, 96, 97,
109, 110, 170, 189
ОГЛАВЛЕНИЕ
Введение ........................................................... 3
Глава /. Общие сведения о производстве камфары..................... 5
1. Камфара, ее свойства, производство и потребление............. 5
2. Получение естественной камфары.............................. 10
3. Получение синтетической камфары............................. 13
Глава //. Сырье для получения синтетической камфары............... 16
1. Производство скипидаров..................................... 16
2. Состав скипидаров и требования к скипидару как сырью для
производства синтетической камфары.............................. 22
3. Скипидар и пинен как технические продукты................... 25
4. Пихтовое масло ............................................. 26
5. Пайн-оль ................................................... 26
Глава III. Получение камфена по схеме пинен-борнилхлорид-камфен 27
1. Получение борнилхлорида .................................... 27
2. Превращение борнилхлорида в камфен.......................... 30
3. Механизм перегруппировки Вагнера—Меервейна.................. 32
Глава IV. Изомеризационный способ получения камфена.............. 37
1. Сущность метода, механизм и развитие взглядов на роль ката-
лизатора ....................................................... 37
2, Катализаторы ............................................... 40
3. Реакции, протекающие при получении камфена методом катали-
тической изомеризации а- и р-пиненов........................ 51
4. Некоторые более тонкие стороны механизма изомеризации пи-
нена ........................................................... 55
Г лава V. Основные принципы промышленной технологии изомеризаци-
оиного способа получения камфена................................ 57
1. Научные основы существующего процесса .................... 57
2. Особенности каталитической изомеризации жидкого Р-пинена 69
3. Изомеризация пинена в аппаратуре периодического действия на
заводских установках ........................................... 70
4. Оценка существующего процесса и перспективы его развития 73
5. Исследования по разработке непрерывных методов каталитиче-
ской изомеризации пинена ....................................... 74
Глава VI. Получение борнеола и изоборнеола......................... 77
1. Получение изоборнильных эфиров из камфеиа и трициклена . . 78
2. Основы технологии формилирования и ацетилирования камфена
в аппаратуре периодического действия ....................... 84
206
.3. Исследования по разработке непрерывных методов формилиро-
вания и ацетилирования камфена................................ 88
4. Сравнительная оценка процессов формилирования и ацетилиро-
вания камфена................................................... 92
5. Свойства муравьиного и уксусного эфиров изоборнеола .... 93
6. Гидролиз (омыление) борнильных и изоборнильных эфиров . . 93
7. Некоторые кинетические закономерности гидролиза борнильных
и изоборнильных эфиров и пути улучшения процесса.............. 98
8. Непрерывный способ гидролиза изоборнилформиата.............. 100
9. Регенерация органических кислот............................. 103
10. Пути‘непосредственного превращения камфена в борнеол и изо-
борнеол ........................................................ 103
Г лава VII. Превращение борнеолов в камфару....................... 104
1. О некоторых различиях в химических свойствах борнеола и изо-
борнеола ...................................................... 104
2. Получение камфары окислением борнеолов...................... 105
3. Каталитическое дегидрирование борнеолов..................... 107
4. Технология периодического жидкофазного метода дегидрирова-
вания борнеолов................................................ 112
5. Технология непрерывного парофазного метода дегидрирования
борнеолов ..................................................... 120
Глава VIII. Получение камфары окислением камфена................... 123
1. Получение камфары окислением камфена хромовой кислотой . . 123
2. Использование восстановленных хромовых растворов........... 127
3. Окисление камфена другими окислителями...................... 128
Глава IX. Ректификация сырья и полупродуктов....................... 128
1. Общие вопросы ректификации терпенов......................... 128
2. Выделение и- и (3-пиненов из скипидаров..................... 133
3. Выделение камфена и трициклена из продуктов каталитической
изомеризации пинена ........................................... 134
4. Выделение изоборнильных эфиров из продуктов формилирова-
ния и ацетилирования камфеиа................................... 136
5. Исходные данные для расчета ректификацноииых колонн, пред-
назначенных для разделения терпенов ........................... 137
6. Отгонка уксусной и муравьиной кислот от продуктов формили-
рования и ацетилирования камфена............................... 143
7. Укрепление уксусной и муравьиной кислот..................... 145
Глава X. Производство камфары из пихтового масла.................. 147
1. Некоторые исторические сведения.............................. 147
2. Особенности пихтового масла как сырья для производства кам-
фары .......................................................... 148
3. Ректификация пихтового масла................................. 149
4. Использование пинеи-камфеновой фракции ...................... 150
5. Переработка борнилацетата в камфару.......................... 150
Г лава XI. Свойства камфары и ее очистка........................... 152
1. Свойства камфары как индивидуального химического соедине-
ния ............................................................ 152
2. Состав и свойства товарной камфары........................... 153
3. Технические требования к камфаре............................ 156
4. Очистка камфары.............................................. 158
207
Глаза XII. Методы исследования, связанные с производством камфары 163
А. Исследования с применением газо-жидкостной хроматографии 163
1. Аппаратура ............................................. 164
2. Качественный анализ ..................... 165
3. Количественный анализ ..................... 169
Б. Химические и физические методы исследования...... 176
1. Исследования исходных продуктов ........................ 176
2. Исследования полупродуктов ............................. 180
3. Контроль отдельных процессов............................ 182
4. Анализ готовой продукции................................ 182
5. О применении ректификации для исследования продуктов
камфарного производства.................................... 183
Г ла за XIII. Физические константы индивидуальных терпенов....... 183
Список литературы................................................. 188
Предметный указатель.............................................. 202
Георгий Александрович Рудаков
ХИМИЯ и ТЕХНОЛОГИЯ КАМФАРЫ
Редактор издательства Н. Р. Казарина
Художественный редактор В. Н. Журавский
Технический редактор А. П. Малахова
Корректор Е. Е. Ярина
Переплет художника И. Д. Богачева
Сдано в набор 24/XII 1975 г. Подписано в печать 20/V 1976 г. Т-07980.
Формат 60ХЭО'/.-з. Бумага типографская № 1. Усл. печ. л. 13,0. Уч.-изд. л. 14,32.
Тираж 930 экз. Издат. № 101/75. Заказ № 43. Цена 1 р. 65 к.
Издательство «Лесная промышленность», 101000, Москва, ул. Кирова, 40а.
Ленинградская типография № 8 Союзполиграфпрома при Государственном
комитете Совета Министров СССР по делам издательств, полиграфии
и книжной торговли.
190000, Ленинград, Прачечный пер., 6.