






































































































































































































































































































































































































































Author: Франке З.
Tags: оружие вооружение артиллерийско-техническое имущество бронированные машины и специальные средства транспорта стрелковое оружие личное оружие боеприпасы и боевые отравляющие вещества управляемые и неуправляемые ракеты и реактивные снаряды органическая химия химическая технология химические производства химия
Year: 1973




355.728
УДК 623.459.4'547
Ф 83
Ф 83 3» Франке
Химия отравляющих веществ. Т. 1. Перевод с нем М., «Химия», 1973.
440 стр., 50 табл., 30 рис., 207 ссылок.
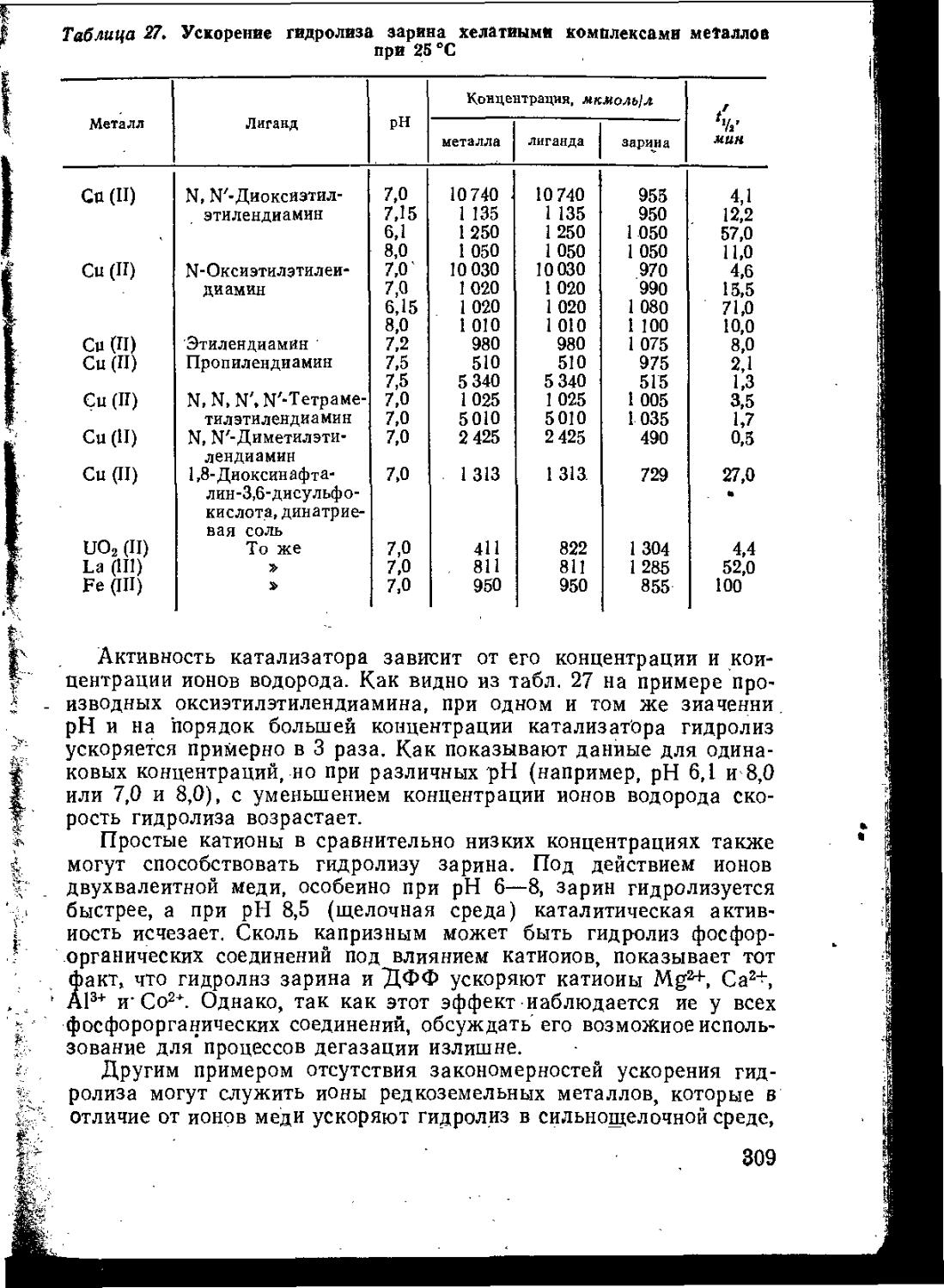
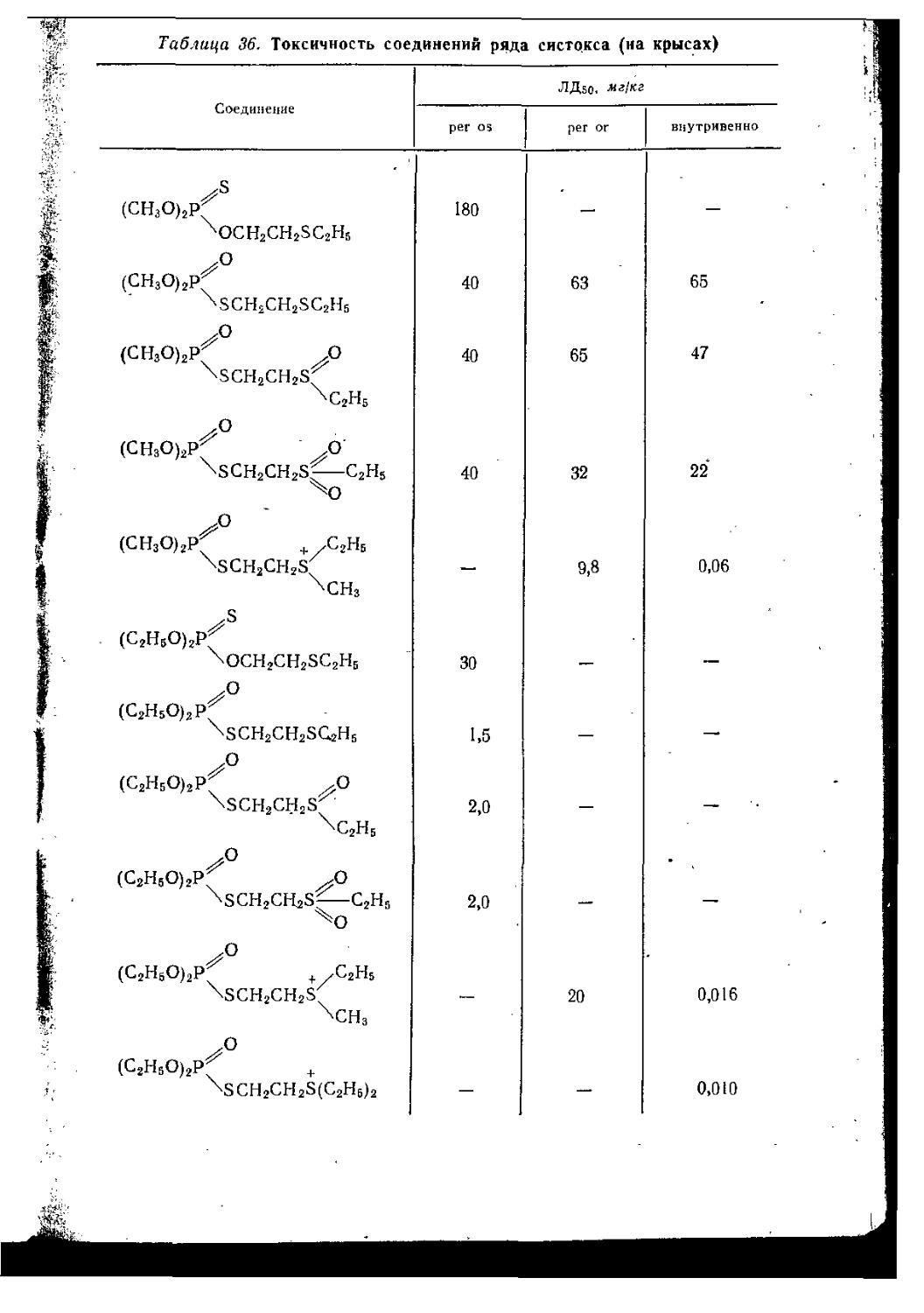
Монография, вышедшая в ГДР в 1967—1969 гг., представляет собой одно из наиболее полных современных руководств по химии отравляющих веществ. Состоит она из двух томов.
Первый том посвящен рассмотрению физических, химических и токсических свойств основных отравляющих и ядовитых соединений, а также характеристике их возможного боевого значения. Описаны способы получения наиболее важных веществ. Приводится достаточно полная библиография.
Книга представляет интерес для преподавателей и студентов высших учебных заведений, личного состава подразделений гражданской обороны, а также может быть полезна широкому кругу химиков, деятельность которых связана с химией физиологически активных веществ.
Л 0251-121 147О ф--------------14-73
050 (01)-73
355.728 + 547
©Перевод на русский язык, Издательство «Химия», 1973.
СОДЕРЖАНИЕ
От издательства.............................................................12
Предисловие к немецкому изданию.................* ................13
ХИМИЯ БОЕВЫХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
Введение ............................................................. W
1. Общие свойства боевых отравляющих веществ . .
- f.l. Физические свойства ОВ....................
> 1.1.1. Давление пара .......................
1,1.2. Температуры кнпення н плавления ОВ . .
1.1.3, Максимальная концентрация ОВ ....
1.1.4. Стойкость ОВ ........................
1.1.5. Относительная плотность пара.........
1.1.6. Способность ОВ давать аэрозоли ....
1.1.7. Растворимость и растворы ОВ..........
1.1.7Л. Понижение температуры застывания ОЕ 1Л.7.2. Изменение давления пара ОВ . . .
• 1.1.7.3. Растворимость ОВ...............
1.1.8. Единицы измерения концентрации . . .
1.2. Химические свойства ОВ.....................
1.2Л. Термическая устойчивость.............
1.2.2. Устойчивость к гидролизу.............
1.2.3. Отношение к химическим реагентам . . .
1.2.4. Устойчивость во время хранения ....
1.2.5. Отношение к различным материалам . .
1.3. Токсические свойства ОВ.......... . .
1.3.1. Пороговая концентрация...............
к 1.3.2. Предел переносимости................. .
1.3.3. Произведение Ct......................
' 1.3.4. Летальная доза.......................
1.3.5. Летальная концентрация...............
Контрольные вопросы ............
Литература .....................................
Я*. Классификация ОВ ........................ . .
'2.1. Классификация по физическим признакам . . . .
2.2. Классификация по химическим признакам . . <
Классификация по токсикологическим признакам
2.4. Военно-тактическая классификация.............
2.5. Учебно-методическая классификация . . . . .
Контрольные вопросы
. 19
. 20 . 20 . 23 . 24 . 27 . 28 . 29 . 30 . 32 . 33 . 36 . 37 . 39 . 40 . 41 . 44 . 45 . 46 . 47 . 50 . 51 . 54 . 56 . 57 . 58 . 59
. 60
. 60
. 61
. 62
. 63
. 64
. 64
5
3. Раздражающие OB ... ..............................................65
3.1. Слезоточивые OB................................................ 66
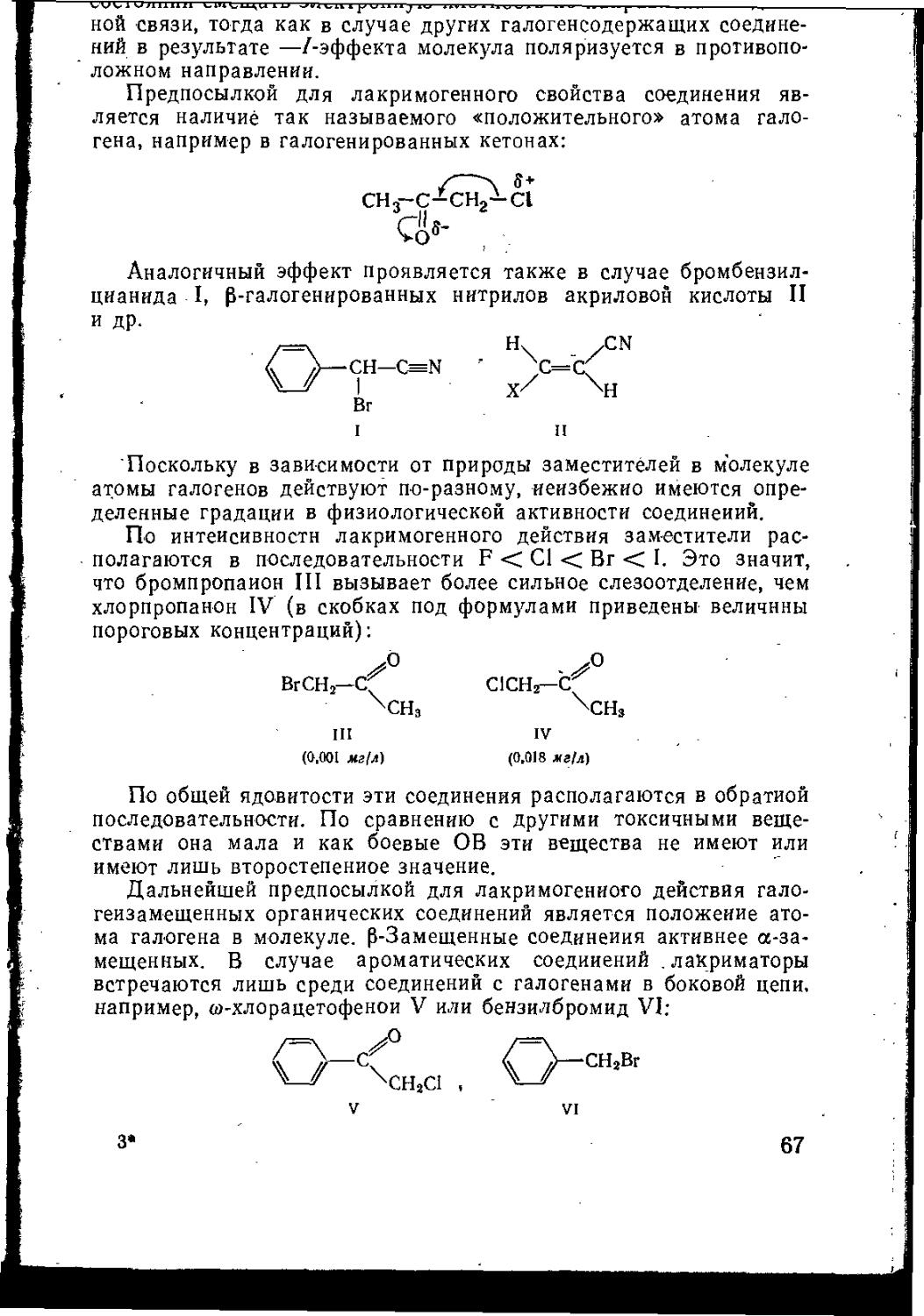
3.1.1. Химическое строение и биологическая активность.............66
3.1.2. Галогенированные кетоны ...................................69
3.1.2.1. Общие свойства .... ................69
З.1.2.2. Галогенированные алифатические кетоны 69
З.1.2.З. Галогенированные ароматические кетоны. Хлорацетофенон . 71
3.1.2.4. Бром- и фторацетофенон................................76
3.1.3. Азотсодержащие слезоточивые ОВ.......................... .77
3.1.3.1. Бромбензилцианид .....................................77
3.1.4. Физиологические свойства слезоточивых ОВ................. 81
3.2. Раздражающие носоглотку ОВ.......................................81
3.2.1. Химическое строение и биологическая активность . .........81
3.2.2. Ароматические мышьякорганические соединения................86
3.2.2.1. Общие свойства .......................................86
3.2.2.2. Дифенилхлор- и дифенилцианарсин.......................87
3.2.2.3. 10-Хлор-9,10-дигидрофенарсазин .......................92
3.2.2.4. Физиологические свойства........................ . . 97
3.2.3. Свинецорганические соединения .............................98
3.2.3.1. Методы получения......................................98
3.2.3.2. Свойства . 99
3.2.4. Морфолид пеларгоновой кислоты........................... 102
3.2.5. 2-Хлорбензилиденмалононитрил .............................103
3.3. Боевое применение раздражающих ОВ ............. 105
3.4. Защита от раздражающих ОВ и их дегазация...................... 106
Контрольные вопросы................................................. 107
Литература ......................................................... 108
4. Удушающие ОВ.....................................................109
4.1. Галогенпроизводные угольной кислоты.............................110
4.1.1. Химическое строение и биологическая активность.............НО
4.1.1.1. Связь химической структуры с биологической активностью НО
4.1.1.2. Характер и механизм действия , 111
4.1.2. Общие свойства ...... ....................................113
4.1.3. Дихлорангидрид угольной кислоты . ,......................114
4.1.3.1. Методы получения ....................................114
4.1.3.2. Физические свойства..................................115
4.1.3.3. Химические свойства..................................116
4.1.3.4. Токсические свойства .......... *. . . .119
4.1.4. Трихлорметиловый эфир хлоругольной кислоты ........ 120
4.1.4.1. Методы получения . ..................................120
4.1.4.2. Физические свойства..................................120
4.1.4.З. Химические свойства .................................121
4.1.4.4. Токсические свойства............................... 122
4.1.5. Бис-трихлорметиловый эфир угольной кислоты................123
4.2. Галогенированные иитроалканы ... .........................123
4.2.1. Химическое строение и биологическая активность........., . 124
4.2.2. Общие свойства............................................125
4.2.3. Трихлорнитрометан .................. • ...................126
4.2.3.1. Методы получения.....................................126
4.2.3.2. Физические свойства .................................127
4.2.3.3. Химические свойства ................................128
4.2.3.4. Токсические свойства..............................' . 130
4.2.4. Тетрахлордииитроэтан и другие соединения..................130
4.3. Соединения фтора с галогенами и серой..........................131
4.3.1. Трехфтористый хлор........................................131
4.3.2. Пятифтористая сера................................... . 133
6
4.4. Боевое применение удушающих ОВ . . . . ,..................... • 1^3
4.5. Защита от удушающих ОВ и их дегазация....................... 134
Контрольные вопросы................................................135
Литература ....................................................... 135
5. Кожио-иарывные О В ...................................................136
5.1. Галогенированные тиоэфиры.......................................... 138
5.1.1. Химическое строение и биологическая активность.............138
5.1.1.1. Связь химического строения с физиологической активностью 138
5.1.1.2. Принцип и механизм действия...........................НО
5.1.2. Общие свойства ............................................... 143
5.1.3. Бис-2-хлорэтиловый тиоэфир.................................144
5.1.3. 1. Методы получения .......................................145
5.13.2. Физические свойства . . ............................... • 151
5.1.3.3 , Химические свойства ....................................155
5.1.3f4 . Токсические свойства.................................. 165
5.1.4. Аналоги бис-2-хлорэтилового тиоэфира.......................166
z 5.1.4.1. Бис-2-фторэтиловый тиоэфир ..............................166
5.1.4.2. Бис-2-бромэтиловый тиоэфир...........................167
5,1.4.3. 2,2'-Бис-(2-хлорэтилтио)-диэтиловый «эфир («кислородный
иприт»)................................................. . 167
< 5.1.4.4. Бис-(2-хлорэтилтио)-алканы............................. 168
5.2. Галогенированные третичные алифатические алканы................. • 170
5.2.1. Химическое строение и биологическая активность ....... 171
5.2.1. Г. Связь, между химическим строением и биологической активностью ..................... . ..............................171
5.2.1. 2. Характер и механизм действия............................172
5.2.2. Общие свойства ... 174
5.2.3. Трис-(2-хлорэтил)-амин.........................................175
5.2.З.1. Методы получения.............-...........................175
5.23.2. Физические свойства , •..................................177
5.2.3.3. Химические свойства......................................178
5.2.3.4. Токсические свойства.....................................187
5.2.4. Другие М-алкил-М,Ы-бис- (2-хлорэтил)-амины.....................188
5.3. Галогенированные алифатические арсины...............................191
г 5.3.1. Химическое строение и биологическая активность...................191
5.3.1.1. Связь между химическим строением и биологической активностью ...........................................- . '.........191
5.З.1.2. Характер и механизм действия ............................192
53.2. Общие свойства ................................................195
5.3.3. 2-Хлорвинилдихлорарсин . . ....................................196
5.3.3.1. Методы получения........................................ 196
5.3.3.2. Физические свойства......................................198
? $.3.3.3. Химические свойства..................................... 199
5.33.4. Токсические свойства ....................................201
5.3.4. Метил-, этил- и фенилдихлорарсины -............................201
ДА Галогенированные оксимы...............................................203
5.4.1. Физиологическая активность.....................................204
5.4.2. Общие свойства ....................•«..........................205
5.4.3. Дихлорформоксим................................................206
5.4.З. 1. Методы получения........................................206
5.43.2. Свойства .............................................. 207
5.4.4. Моиохлорформоксим и трихлорметилформоксим (перхлорапетальд-оксим) ............................................................ 208
55. Боевое применение кожно-нарывных ОВ ............................... 208
* -
5.6. Защита от кожно-нарывных ОВ и их Дегазация..................... 211
5.6.1. Защита.....................................................211
5.6.2. Дегазация . •.......................................... 213
Контрольные вопросы.................................................. 214
Литература ...................................................... 215
6. Общеядовитые ОВ .................................................217
6.1. Синильная кислота и галогенцианы................................217
6.1.1. Химическое строение и биологическая активность............213
6.1.2. Цианистый водород (синильная кислота) .................221
6.1.2.1. Методы получения............................... .... 222
6.1.2.2. Физические свойства.............................. 224
6.1.2,3. Химические свойства.................................226
6.1.3. Галогенцианы ........................................... 228
6.1.4. Токсические свойства .....................................231
6.2. Мышьяковистый и фосфористый водород ............ 232
6.3. Окись углерода и карбонилы металлов........................... 235
6.3.1. Окись углерода ........................................ 236
6.3.2. Карбонилы металлов...................................... 238
6.4. Тетраэтилсвинец .............................................. 240
6.5. Боевое применение общеядовитых ОВ.............................. 243
6.6. Защита от общеядовитых ОВ и их дегазация...................... 244
Контрольные вопросы................................................ 246
Литература ........................................................ 246
7. Фосфорорганические соедииеиия.....................................247
7.1. Классификация и номенклатура фосфорорганических соединений . . . 248
7.1.1. Фосфорорганические соединения — производные кислот пятивалентного фосфора.................................................... 249
7.1.2. Фосфорорганические соединения — производные пнрофосфорной (дифосфорной) кислоты ............................................253
7.2. Биологическая активность фосфорорганических соединений.........254
7.2.1. Химическое строение и биологическая активность ....... 254.
7.2.2. Механизм действия ...... .................... , 256
7.2.3. Общий характер действия..................................261
7.3. Диалкилацилфосфаты . . . . . 263
7.3.1. Общие сведения...........................................263
7.3.2. ДиалКилфторфосфаты .................................... 264
7.З.2.1. Связь между химическим строением и токсичностью * . . 264
7.3.2.2. Методы получения ..................................266
7.3.2.3. Физические и химические свойства...................267
7.3.3. Диизопропилфторфосфат (ДФФ) 271
7.З.З.Т. Методы получения...................................271
7.3.3.2. Физические свойства .............................. 273
7.3.3.3. Химические свойства.............................. 274
7.3.3.4, Токсические свойства ..............................283
7.4. N.N-Диалкиламидо-О-алкилацилфосфаты ... -......................284
7.4.1. Общие сведения...........................................284
7.4.2. NjN-ДиалкйлаМйдо-О-алкилцианфосфаты . ...................286
7,4.3. N,N-Диметиламидо-О-этилцианфосфат 288
7.4,3,1. Методы получения ..................................288
8
74.3.2. Физические свойства , . > ...........................290
7.4.3.3. Химические свойства ...................................291
? 7.4.34. Токсические свойства . ................................292
О-Алкилалкилфторфосфонаты..........................................293
г/ 7.5.1. Общие сведения , 293
fe. 7.5.2. Связь между химической структурой и токсичностью . • . • • 294
\ 7.5.3. Методы получения ......................................... 295
и ' 7.5.4. Свойства ...................................................296
7.5,5. О-Изопропилметилфторфосфонат ............................. 298
7.5.5.1. Методы получения - . ..................................298
7.5.5.2. Физические свойства ................................. . 303
’ 7.5.5.3. Химические свойства ..................................305
7.5.5.4. Токсические свойства................................. 312
7.5.6. 3,3-0,О-Диметилизобутилметилфторфосфонат....................314
7.5.6.1. Физические и химические свойства...................314
7.6. Фосфор ил хол ины и фосфорилтиохолины . . . .................л . 315
7.6.1. Общие сведения 315
7.6.2. Связь между химической структурой и токсичностью.............316 .
7.6.3. Алкилфторфосфорилхолйны (Ь1,Ь1,Ь1-триметиламмонийалкоксиалкил-фторфосфонаты) ....................................................318
7.6.3.1. Методы получения .....................................318
7.6.3.2. Физические и химические свойства..................... . 319
7.6.4. Фосфорилтиохолины......................................... 320
7.6.4. 1. Методы получения . ........................ . . . 7 321
7.64.2. Физические и химические свойства ......................322
7.64.3. Токсические свойства................................. 323
77. Боевое применение фосфорорганических ОВ............................325
7,8. Защита от фосфорорганических ОВ и их дегазация....................327
7.8.1. Защита................................7.....................327
7.8.2. Дегазация ................................................ 328
Фосфорорганические инсектициды как ОВ ............................329
7.9.1. Производные типа систокса...................................330
7.9.1.1. Методы получения.......................................330
, 7.9.1.2. Физические и химические свойства......................331
7.9.1.3. Системное действие и токсические свойства..............335
7.9.14. Аналогичные соединения................................ 338
"7.9.2. Дйсистон й тимет.......................................... 339
7.9.2.1. Методы получения.......................................339
t 7.Э.2.2. Физические и химические свойства .........................339
7.9.2.3. Инсектицидные и токсические свойства...................340
7.9.3. Тетрам ................................................... 342
.7.9.3.1. Методы получения......................................342
7.9.3.2. Физические и химические свойства.......................342
If 7.9.3.3. Инсектицидные и токсические свойства..................343
7.9.4. Паратион и параоксон........................................344
f 7.94.1. Методы получения.................................... 344
7.94.2. Физические и химические свойства........................344
: ? 7.94.3. Инсектицидные и токсические свойства................. 346
Димёфокс ................................................... 348
f 7.9.5.1. Методы получения..................................... 348
7.9.5.2. Физические и химические свойства.......................348
£9.6. Фосдрин .............................. . . *.................349
0.7. ТЭПФ ....................................................... 349
£9.8. Военное значение инсектицидов.............................. 351
рольные вопросы...................................................352
Р&гура .........................,.................................353
8. Психохимические О В (психояды)...................................355
8.1. Производные индола............................................. 358
8.1.1. Производные лизергиновой кислоты..........................358
8.1.1.1. Диэтиламид (+)-лизергиновой кислоты (LSD, LSD-25) . . 359
8.1.1.2. Другие производные лизергиновой кислоты..............360
8.1.2. Производные триптамина................................... 361
8.1.2.1. Г^,1Ч-Диметилтриптамин...............................361
8.L2.2. Буфотенин 361
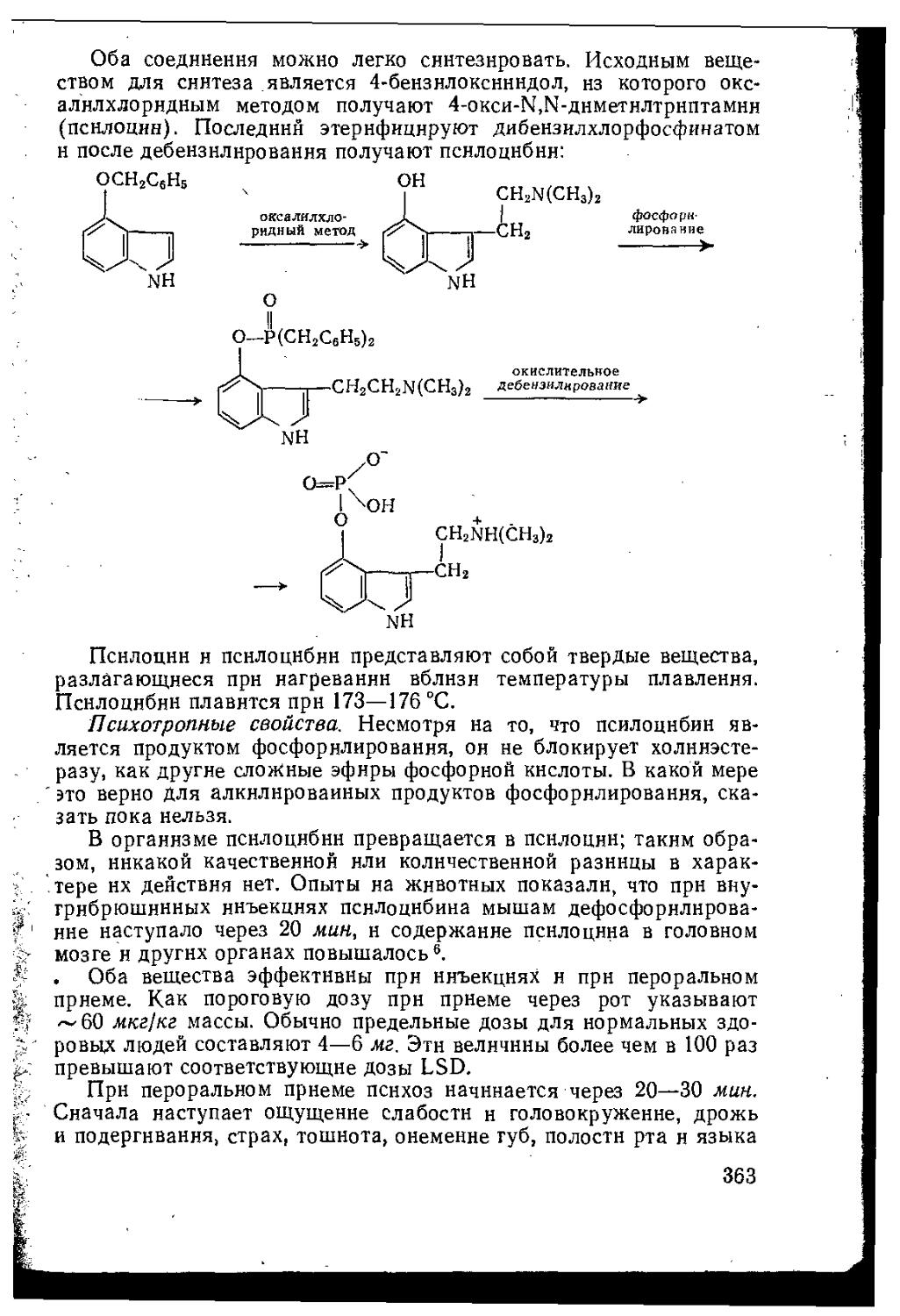
8.1.2.3. Псилоцин и псилоцибин................................362
8.2. ФеНилалкиламины.................................................364
8.2.1. Мескалин .................................................365
8.2.2. 3,4,5-Триметоксифениламинопропан (ТМА) ...................366
8.3. Производные пиперидина......................................... 366
8.3.1. Сернил ................................................. 367
8.3.2. Пиперидилбензилаты и пиперидилгликоляты...................367
8.3.2.1. N-Алкил-З-пиперидилбензилаты ........... 368
8.3.2.2. Дитран.................. ;......................... 368
8.4. Боевое применение и защита.................................... 369
Контрольные вопросы................................................. . 370
Литература ......................................................... 370
9. Фитотоксические О В (яды растений) ..............................371
9.1. Производные фенокснкарбоновых кислот............................373
9.1.1. 2,4-Дихлорфеноксиуксусная кислота.........................374
9.1.2. 2,4,5-Трихлорфеноксиуксусная кислота и ее производные .... 374
9.2. Другие карбоциклические фитояды ................................376
9.2.1. Фенилалкиловые эфиры .....................................376
9.2.2. Производные фен ил моче вины..............................377
9.2.3. Производные фенола........................................377
9.2.4. Гибберелины...............................................379
9.3. Гетероциклические соединения с фитотоксическнми свойствами .... 379
9.3.1. Индолил-З-карбоновые кислоты............\. . ............379
9.3.2. Производные 1,3,5-триазина..............*............... 380
9.4. Прочие фитотоксические вещества................................ 380
9.5. Военное применение фитотоксических ОВ...........................381
Контрольные вопросы..................................................382
Литература ......................................................... 382
10. Диверсионные яды (яды и токсины, которые могут быть использованы для заражения продовольствия, воды и др.) . .........................383
10.1. Фторорганические соединения . .............................384
. 10.1.1. Химическое строение и биологическая активность..........385
10.1.2. (о-Фторкарбоновые кислоты................................386
10.1.3. Алкиловые эфиры фторуксусной кислоты................... 388
10.1.4. Алкиловые эфиры со-фторкарбоновых кислот.................391
10.1.5. со-Фторированные спирты................................. 393
10.2. Природные яды ..................... 394
10.2.1. Яды растительного происхождения. Алкалоиды...............395
10.2.1.1. Никотин (3-метилпироллидин-2'-пиридин).............395
10.2.1.2. Алкалоиды чилибухи..................................396
10.2.1.3. Курарин.............................................397
10.2.1.4. Атропин и (—)-гирсциамин.......................... 398
10.2.1.5. Аконитин ..........................................400
10
10.2.2. Токсины грибов ...............................................401
10.2.3. Яды животного происхождения...................................402
10.2.3.1. Кантаридин..............................................402
10.2.3.2. Яды змей................................................403
10.2.3.3. Токсин скорпиона........................................403
10.2.3.4. Яды жаб ...................................... 403
10.2.3.5. Яды морских животных ...................................404
10.2.4. Бактериальные токсины ...................................... 407
10.2.4.1. Ботулинический токсин ................................ 407
10.2.4.2. Токсин столбняка........................................409
10.3. Неорганические яды..................................................410
Литература ............................................................. 414
„Дополнительная литература . . .*..........................................415
ПРИЛОЖЕНИЯ
- Приложение 1. Методы работы с химическими ОВ ............................416
-Приложение 2. Важнейшие дегазирующие растворы, применяемые в лабора- > торных условиях ........................................................ 417
Приложение 3. Меры первой доврачебной помощи при отравлениях химическими ОВ............................................................. 417
Приложение 4. Летальные концентрации и дозы некоторых ОВ и ядов . . .449 Приложение 5, Летальные дозы (ЛД50) некоторых ОВ и ядов...................420
Приложение 6. Физические свойства некоторых химических ОВ . . . . . . 423
Предметный указатель.................................................... 425
Иностранные обозначения некоторых инсектицидов и ОВ.......................435
К '
4
ОТ ИЗДАТЕЛЬСТВА
Начиная с первой мировой войны, во время которой химические вещества впервые были использованы как оружие массового поражения, они прочно заняли одно из первых мест в военном арсенале империалистических государств. Все прошедшие с того времени годы это оружие постоянно совершенствовалось и иыие эффективность его столь возросла, что Всемирная Организация Здравоохранения в 1970 г. вынуждена была отметить, что массовое, а в некоторых случаях даже ограниченное его применение могло бы нанести такой ущерб здоровью человечества, с которым невозможно было бы справиться существующим средствам здравоохранения.
Многие примеры применения отравляющих и ядовитых веществ в разных точках земного шара в последние годы свидетельствуют о реальной опасности этого оружия.
Социалистические государства в течение ряда лет неизменно и последовательно прилагают усилия к полному запрещению химического и бактериологического оружия. Весной 1972 г. по инициативе социалистических стран была принята конвенция о полном запрещении бактериологического и токсинного оружия, которая была подписана многими государствами. В то же время ряд стран — участниц НАТО препятствует решению проблемы запрещения химического оружия.
Естественно, что в создавшейся ситуации, в пла'не защиты мирного труда социалистических стран, важной государственной задачей является организация защиты от химического оружия, для чего требуется большая и серьезная подготовка подразделений гражданской обороны и всего населения страны.
В предлагаемой читателям книге рассмотрены исторические аспекты применения отравляющих веществ, начиная с глубокой древности н до последнего времени. Для широкого круга отравляющих веществ приведены физические свойства, способы индикации и дегазации. Подробно рассмотрены меры первой помощи и лечения при поражениях отравляющими веществами, основанные на серьезном рассмотрении биохимических механизмов токсического действия ряда фосфор-, фтор-, азот- и других элементооргаиическнх соединении и некоторых природных ядов.
Все это делает книгу, с одной стороны, полезным пособием для широкого круга лиц, изучающих в курсах гражданской обороны вопросы защиты от оружия массового поражения, и, с другой стороны, она может помочь в пропаганде запрещения химического оружия— одного из срёдств массового упичтожепия людей.
«ПРЕДИСЛОВИЕ К НЕМЕЦКОМУ ИЗДАНИЮ
@Й®ДСТва массового уничтожения занимают главное место в воору-Si^aии современных армий. Ядерное оружие, бактериологическое ле, боевые отравляющие вещества .являются важными факто-которые определяют формы и методы ведения современной
г.
[Мическое оружие впервые применили германские нмпериа-[ в 1915 г. С тех пор оно получило дальнейшее всестороннее тие —были открыты н приготовлены многие новые препараты :ой токсичности, Современные^ средства применения химиче-оружня позволяют использовать такие яды в любой метеоро-еской обстановке. Химическое оружие более, чем когда-либо стало веянной силой, которую нельзя недооценивать.
Угрозу применения его армиями империалистических государств,— .Что для них все средства хороши показывает варварское нападете США на Вьетнам, — требует высокой обороноспособности ро-5ины — Германской Демократической Республики.
^Учебник химии ОВ должен быть надежным подспорьем всем Й&’кто во время обучения пли по роду занятий имеет дело с хи-Вей боевых веществ. Настоящий учебник составлен ввиду появ* |рния общей потребности в едином изложении сведений о всех бое-дЫ: отравляющих веществах. При этом осветить всю проблему |ийической войны было совершенно невозможно. Например, в за-Оу этой книги не могло нходить подробное оп нс а пне средств и “Особое применения боевых химических веществ, Группы таких даЦеств, как тумаио- н дымообразователн, зажигательные средства О- Д- также остались вие рассмотрения, поскольку эти и многие ййгиё химические проблемы военного дела в настоящее время -|рцио рассматривать лишь как особые разделы химии.
^Настоящий учебник химии ОВ ограничивается описанием физиках, химических н токсических свдйств боевых отравляющих ('и ядов, эффективных для военного применения. Рассма-ся способы их обезвреживания (дегазации), качественного ествеиного определения и получения. При этом предпола-что читатель знаком с общими курсами неорганической и еской химии.
ник состоит из двух томов. В первом томе описываются i боевых отравляющих веществ, их действие и способы по-. Для некоторых веществ приводятся методики получения аториых масштабах. Содержание второго тома составляют £Дения о способах дегазации боевых отравляющих веществ и
13
дегазирующих средствах, требуемых для этой цели, и аналитическая часть, посвященная методам индикации ОВ и их количественному определению.
Обращение с такими веществами отнюдь не безопасно. Оно должно быть осторожным, хорошо продуманным, внимательным и аккуратным; при этом следует использовать' все средства защиты. Новичок должен проводить эксперименты только под руководством опытных экспериментаторов. Одновременно следует заметить, что такие эксперименты можно проводить только в хорошо оборудованных лабораториях и что экспериментирование в «домашних лабораториях» категорически запрещается хотя бы из-за большой опасности.
Автор благодарен Немецкому военному издательству, а также учащимся и слушателям, которые стимулировали создание этой книги и дали ценные советы.
Особенно признателен автор Эриху Хаазе за критические замечания при рецензировании рукописи.
ЗИГФРИД ФРАНКЕ ГЁРЛИЦ, 30.4.66 г.
химия БОЕВЫХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
ВВЕДЕНИЕ
^Знание химии боевых отравляющих веществ (ОВ) необходимо в этслоинях возможной химической войны. Одно из главных задач «той науки — изучение соединений; могущих найти военное прнме-теенве.
g.’ Цель применения боевых ОВ н ядов заключается и выселении Ч&ротивцака из строя о результате причинения ущерба его здоровые, оз действия па психику или его уничтожения. Эта цель достигается помощью различных средств применения ОВ — прн артакаеркй-Шок И воздушном нападении, при использовании химических фуга-’еов, а также ср и применении ОВ для заражения предметов снабжения" и сельскохозяйственных культур. Одновременно целью .Применении ОВ является усиление (в течение более ня к менее дли-^тельПого времени) физических и психологических нагрузок на про-^ЕЙвиикз—-необходимость принятия мер зашиты, проведения сани-Норной обработки и дегазапин.
|1яменение боевых ОВ приводит также к затруднению манев-ротивника и предъявляет большие требования к тактическим }ностям командира.
иду того что боеные ОВ обладают: самыми разнообразными ескими, химическими и токсическими свойствами, для пра-бго представления всей проблемы в целом требуется изучить ополннт&пьных проблем: возможности применения ОВ; пове-ОВ в момент применения н после него; нпдикапня и нденти-(,Ия ОВ; действие ОВ; поиск противоядий и средств защиты; ине и дегазапня ОВ.
с эти вопросы не являются чисто химическими, и дли их реще-эебуются знания из многочисленных смежных областей.
евые ОВ н яды представляют собой вещества, которые при анис тем нли иным путем в оргвнизм могут повлечь за собой ление н, в результате, заболев а и ня, которые по их тяжести, <энню н общим симптомам вряд ли можно четко отличить от еезннй, вызываемых другими причинами.
военной точки зрения невозможно провести строгое различие Г боевыми ОВ, бактериологическими возбудителями болезней «в. '
* практическим соображениям живых возбудителей болезней, >ных к размножению, рассматривают и изучают отдельно от шых> ядов. Причинами заболеваний, вызванных микробами
П
или вирусами, являются выделяемые нмн токсины. Изолированные кристаллические токсины можно производить в виде концентратов по особой технологии. Эти токсины могут иметь военное применение. Так как в широком смысле они также являются «неживыми» ядами, с военно-химических позиций их отнесли к категории биологических ядов.
В химии боевых отравляющих веществ имеют дело и с такими соединениями, как яды растений (фнтояды), которые могут служить и гербицидами и стимуляторами роста растений, а также веществами, влияющими на нормальную психику человека н вызы-' вающнмн у него неестественные, не свойственные данной личности представления н поступки.
1. ОБЩИЕ СВОЙСТВА БОЕВЫХ ОТРАВЛЯЮЩИХ ВЕЩЕСТВ
По сравнению с миллионами известных химических соединений число боевых ОВ н ядов, которые могут иметь военное применение, незначительно.
Хотя в тридцатых годах думали, что вряд лн следует ожидать появления новых ОВ, развитие этого'иаправления, начиная с 1940г., показало, что не только появились, ио и приняты иа вооружение империалистических армий весьма эффективные боевые ОВ. Если раньше «королем ядов» называли иприт, то этот титул ныне подходит ко всем современным отравляющим веществам типа зарина или V -газов, одиако при этом значение иприта для современного химического боя также сохраняется.
Требования, которые предъявляются к. боевым ОВ в отношении их свойств и применимости, отнюдь не нормированы. Разумеется, определенные свойства нужно считать главными. Общую характеристику получают иа основании совокупности всех свойств, факторов и обстоятельств. Оии образуют единое целое, в котором физикохимические свойства имеют ие меньшее значение, чем свойства' токсические.
Эффективность ОВ иа поле боя, при использовании их в целях диверсии, для уничтожения сельскохозяйственных культур, растений и т- д-, находится в непосредственной связи с этими свойствами, а также с техникой применения ОВ.
Кроме того, для принятия химического соединения иа вооружение в качестве боевого ОВ необходимы известные экономические предпосылки, например дешевизна промышленного производства н доступность сырья.
В империалистических армиях к боевым ОВ предъявляют следующие тактико-технические требования:
Тактические требования. Высокая токсичность, вызывающая смертельное или тяжелое поражение противника;
разностороннее токсическое действие, т. е. ОВ должны действовать иа различные органы, отравление должно быть комбинированным;
быстрота и «коварство» действия (скрытый, бессимптомный начальный период действия);
отсутствие органолептических характеристик (ОВ должны быть бесцветными и лишенными запаха);
19
возможно бблыпая продолжительность заражающего действий, обусловленная удачным сочетанием фнзнко-химических и токсических свойств;
поддающееся контролю и предвидению распространение в воздухе при заданных атмосферных условиях и местности;
летучесть — быстрое или медленное испарение в соответствии с тактической задачей;
удерживание на местности (стойкость)—кратковременное или длительное заражение в зависимости от тактической задачи;
повышенная проникающая способность через обмундирование, защитное снаряжение, кожу и др.;
трудность распознания с помощью химических реакций' или фн-Зико-хнмическнх методов исследования.
Технические требования. Дешевое промышленное производство из отечественного сырья;
высокая чистота продукта;
химическая устойчивость по отношению к атмосферным, гидролитическим н другим химическим воздействиям, особенно по отношению к дегазирующим средствам;
устойчивость к детонации;
соответствующее тактической задаче давление пара;
низкая температура застывания;
взаимная смешиваемость и растворимость;
способность образовывать устойчивые аэрозоли.
1.1. ФИЗИЧЕСКИЕ СВОЙСТВА ОВ
Физические свойства боевых ОВ, их растворов и смесей являются главными факторами, определяющими их применение, поведение в атмосфере и на местности, их эффективность, возможность дегазации и способы идентификации.
Для идентификации можно использовать физические константы соединений — температуры плавления и кипения, показатели преломления, плотность и др.
Летучесть и удерживаемость ОВ иа местности зависят от давления нх паров, скорости испарения, атмосферных воздействий, величины капель и др.
Растворимость и смешиваемость отравляющих веществ являются свойствами, важными для их применения и обезвреживания.
1.1.1. Давление пара
Это одна из важнейших физических характеристик боевых ОВ. Наряду с внешними факторами давление пара вещества определяет его летучесть и, соответственно, стойкость на местности. Более того, этим показателем определяется способ применения любого ОВ. -
20
Рис. 1. Зависимость давления пара от температуры.
Твердые нлн жидкие ОВ, обладающие низким давлением пара, можно успешно применять только в виде аэрозоля. «Идеальными» и универсально применимыми являются ОВ или нх смеси, давление пара которых н при низкой температуре достаточно высоко, чтобы Испарялось количество вещества, необходимое для создания боевой Концентр а дни.
Молекулыг попавшие в газовую фазу, ведут себя как молекулы газа н подчиняются соответствующим законам. Образовавшийся при этом слой пара оказывает давление р, является мерой числа молекул, испаряющихся и конденсирующихся за единицу времени в замкнутой системе. При этом жидкая и газообразная фазы находятся в динамическом' равновесии:
Пар над жидкой фазой является насыщен-
ным.
Давление пара не зависит от количества жидкости н пара, а зависит от природы вещества и от температуры. При постоянной температуре давление пара вещества постоянно: р = const при Т = const.
Повышение нлн понижение температуры изменяет среднюю скорость молекул (закон Больцмана о распределении молекул по энергии),'что приводит к изменению числа молекул над жидкой фазой и, следовательно, к изменению давления пара:
Р=Ч(Т)
(I)
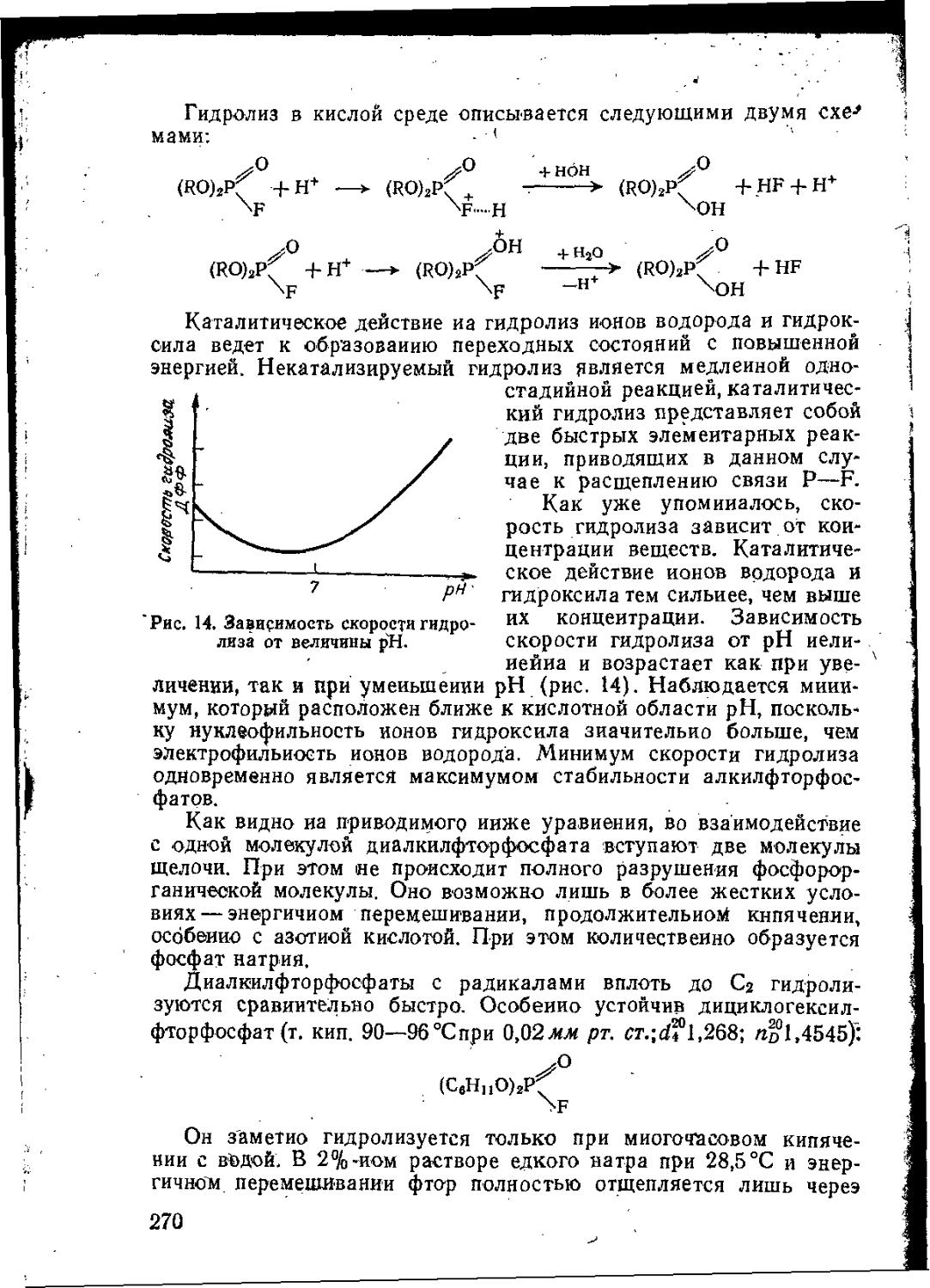
В результате повышения температуры конденсированную фазу покидает больше молекул с избыточной энергией, что влечет за собой увеличение давления пара (рис. 1). Кривая давления пара подчиняется экспоненциальному закону.
Приближенно зависимость давления пара от температуры можно рассчитать по формуле Ренье:
lgp-А—| (2)
где А и В— индивидуальные константы, значении которых можно вычислить по двум различным значениям температуры кипения Л й Тг, прн давлениях pi н если температуры кипения различаются по крайней мере на 70 °C:
lg Pl = Л — у-
1 л В
lg Р2 = А — Пр-
J г
21
В табл. 1 приведены основные числовые значения констант ряда ОВ, необходимые для расчета их давления пара.
Таблица Константы А и В ряда ОВ дли расчета давления пара по формуле Ренье
Название ОВ А В
Бис-2-хлорэтиловый тиоэфир (иприт) 9,4819 1 3117,2-
Бромчиан 10,3282 2457,5
Дифенилхлорарсин 7,8930 3288
Диизопропилфторфосфат (ДФФ) 8,872 2671
Метилдихлора рсин 8,6944 2281,7
Фосген 7,5595 1326
Изопропоксиметилфторфосфат 9,8990 2850,9
Трихлорнитрометан (хлорпикрин) 8,2424 2045,1
Термодинамическую зависимость давления пара от температуры можно описать уравнением Клаузиуса — Клапейрона1:
dp______/ о.
ат ~ г(7г-/ж)
где ЛЯисп — энтальпия (мольная теплота) испарения; Vr и V;K — мольные объемы газообразной и жидкой фаз.
При малых значениях давления пара это выражение можно уп* ростнть, если пренебречь мольным объемом жидкой фазы и принять, что для Vr справедливо общее уравнение состояния идеальных газов:
TZ DT Т7
pVr — RT-, 7Г =
Р
В таком случае получают следующее уравнение:
1 dp ___ исп
р * ат ~ R1'2 ИЛИ
d In р
ат rt2 ''
так как dp/p == яЛпр. После подстановки значения R (1,987 кал-молъ~'[ • градус^} и перехода к десятичным логарифмам получаем:
d 1S Р _____А^ИСП____
dT 2,303. 1,987-Г2
Поскольку в правую часть уравнения входят только положительные величины, производная положительна ( > 0), т. е. с возрастанием температуры давление пара увеличивается.
22
1.1.2. Температуры кипения и плавления ОВ
Температуры кипения и плавления ОВ являются важными свойствами, на основании которых можно делать заключение относительно их применения, продолжительности заражающего действия, .способов хранения и идентификации.
Как известно, температура кипения (т. кип.) —это температура, при которой давление насыщенного пара жидкости равно внешнему давлению. Она непосредственно связана с давлением пара, из чего можно сделать вывод о летучести данного вещества.
Чем ниже температура кипения ОВ, тем выше его давление пара при обычной температуре и тем выше его летучесть.
Вследствие этого, при хранении низкокипящих ОВ, например синильной кислоты (т. кип. 26,5°C), встречаются некоторые особенности. Из-за высокого давления пара внутреннее давление в герметически закрытых сосудах (бомбах, минах) с ростом внешней температуры значительно увеличивается. Это принимается вовнимайие при конструировании и выборе материалов для изготовления средств применения подобных ОВ. В случае низкокипящих ОВ уже при обычной температуре затруднено снаряжение и переснаряжение средств применения.
Все больше вырисовывается тенденция выбирать в качестве ОВ только такие соединения, температуры кипения которых лежат выше 100°C. Из сопоставления температур кипения ОВ (см. Приложение 6) видно, что некоторые соединения, которые пригодны в качестве ОВ, кипят ниже 20 °C.
Поскольку такие ОВ нельзя превратить в жидкость, для их тактического применения необходимы специальные методы. Однако в какой мере подобные методы еще сохраняют свое значение в современном бою, пока не совсем ясно. Такие ОВ, обычно обладающие заметной токсичностью, можно применять в связанном виде. Например, СО илн РНз могут применяться в виде соединений, способных постепенно генерировать эти газы; тем самым становится возможным длительное заражение воздуха.
Температура кипения может также характеризовать стойкость ОВ; по ее величине можно оценить продолжительность заражающего действия. Разумеется, в этом случае действует и множество других факторов. Кратковременным заражающим действием будут обладать лишь такие ОВ, температуры кипения которцх .очсиьтшзкш ОВ с ем дер ату.рами... кипения..испаряются. медлен:,
иее. БолылаяГчасть жидких ОВ, кипящих выше 130 °C, представляет собои~соедннеиия, поражающее действие которых проявляется не только при вдыхании паров, но и при попадании на кожу. Многие из этих ОВ применяются для заражения местности. В этой связи определенное значение имеет также величина температуры плавления или застывания.
Жидкие ОВ должны иметь возможно более низкие температуры застывания. Относительно высокие температуры кипения и низкие
23
температуры застывания позволяют применять ОВ в любое время года. Зарин является жидкостью в интервале температуры от —57 до 4-147 °C, и может применяться и летом и зимой. Напротив, иприт застывает при 14,5°С, и в зимних условиях его применение Возможно лишь в смесях с веществами, понижающими его температуру застывания.
В случае твердых ОВ, применяемых в виде аэрозолей, температура плавления должна быть не слишком низкой, ио и не слишком высокой. Так, например, температура плавления днфенилцнаиар-сина 31 °C. В жаркое летнее время это ОВ плавится внутри боеприпасов. Особенно отрицательно это сказывается иа качестве шашек ядовитого дыма. Такое свойство снижает значение этого вещества в качестве ОВ. Напротив, адамсит имеет более высокую температуру плавления (т.пл. 195°C). Его можно легко расплавить, а также испарить (т. кип. 410°С). Это удобное для применения-ОВ может использоваться в виде аэрозоля как с помощью дымовых шашек, так и гранат.
Температуры плавления и кипения веществ являются индивидуальными константами и их можно использовать для идентификации ОВ.
1.1.3. Максимальная концентрация ОВ
Максимальная концентрация См —это количество вещества, содержащееся в единице объема насыщенных паров при данной температуре (раньше эту величину называли ^летучесть»). Поскольку она имеет размерность концентрации (мг}л или лга/лг3), возможно установить определенные соотношения между этой величиной и боевой концентрацией, летальной концентрацией и т. д.
Максимальная концентрация зависит от природы ОВ (молекулярного веса Af), наружного давления, температуры, а значит и от давления пара. Она является функцией давления пара и температуры:
CM==f(p, 0 (6)
Приближенно максимальную концентрацию можно рассчитать по формуле (в леа/л):
М-273'.p.lO» , Мр
м 22,4- Т -760 . Т
Разумеется, понятие максимальной концентрации относится к замкнутой системе, когда жидкая и газообразная фазы находятся в равновесии. На практике такой системы нет, внешние воздействия, такие как ветер, воздушные потоки, атмосферная диффузия и гидролиз непрерывно отводят пары ОВ. В результате жидкую фазу покидают все новые молекулы, причем давление пара снова восстанавливается, а ОВ улетучивается или испаряется. Количество теп
24
лоты, необходимое для испарения 1 Л1 вещества, представляет собой мольную теплоту испарения, которая заимствуется из среды. Величина мольной теплоты испарения может оказаться столь большой, что быстроиспаряющиеся ОВ при испарении частично застывают. Это происходит, например, в случае синильной кислоты.
Максимальную концентрацию ин в коем случае нельзя отождествлять с боевой концентрацией. Для создания эффективной концентрации в случае некоторых ОВ требуются более высокие боевые концентрации, чем вообще можно достигнуть за счет естественной летучести.
Концентрации, которые достигаются за счет испарения прн применении ОВ в боевой обстановке, в зависимости от внешних условий примерно иа 1—2 порядка ниже, чем максимальные концентрации. Однако во многих случаях эти концентрации достаточны для продолжительного заражения воздуха иа срок от нескольких дней до нескольких месяцев. Хотя ни одно ОВ в пределе не отвечает требованиям по скорости испарения, на этот параметр можно влиять
искусственно.
Подобно давлению пара и температурам кипения значения максимальных концентраций разных ОВ весьма различны. ОВ, имею-
; щие столь низкую максимальную концентрацию, что даже при очень f высокой температуре воздуха их летучесть слишком мала для за-рражения атмосферы, могут применяться только в виде аэрозолей | (например, адамсит). Напротив, летучесть уже упоминавшейся си-£ нильной кислоты столь высока, что при применении даже очень [.больших ее количеств сколь-нибудь значительную концентрацию
£ можно поддерживать лишь в течение нескольких минут.
L Быстро испаряющиеся ОВ применяются преимущественно вие-рапно, обычно, как подготовка немедленно следующей атаки. Более ^медленно испаряющиеся ОВ предназначены для продолжительного ^заражения воздуха или местности, что затрудняет и мешает дей-: стйиям войск в этих районах, а при соответствующих обстоятельствах Г делает их даже невозможным^ В то время как в случае быстро ( испаряющихся ОВ может быть не нужна дегазация, для менее ле-туч их ОВ дегазация личного состава и материалов необходима.
Располагая данными о летучести ОВ при разной температуре, можно рационально производить разведку зараженных участков '< местности. Выше 20 °C ОВ типа ипритз столь летучи, что их можно
>; распознать уже в воздухе и производить индикацию заражения на марше. При более низкой температуре индикация этих ОВ возмож-г иа лишь непосредственно над поверхностью грунта или путем от-: бора пробы. Для этих и подобных случаев полезно помнить простое правило, сформулированное Хербстбм2, — в интервале между 10 и
-,30°С повышение, температуры на 1 °C вызывает повышение летуче-сти примерно иа 10%.
Для ОВ с температурой кипения ниже 230 °C понижению температуры кипения на 10 соответствует повышение летучести при 20°С в 1,5—1,6 раз; для ОВ с температурой кипения выше 230°С,
25
ио ниже 300 °C при таком же понижении температуры кипения следует ожидать повышения летучести в 2 раза.
Соотношение между температурами кипения и максимальными концентрациями представлено на рис. 2. Подразделение ОВ на летучие и стойкие можно производить на основании этой зависимости. Обычно ОВ с температурами кипения ниже 130 °C рассматривают как летучие, а ОВ с температурами кипения между 150 и 300°C как стойкие; ОВ, температуры кипения которых лежат в граничных областях, в зависимости от конкретных условий могут рассматриваться или как стойкие или как летучие.
Максимальная концентрация при 20 °C, ms/л
Рис. 2. Взаимосвязь между т. кип. (760 л л рт. ст.) и максимальной концентрацией ОВ (по данным Taschenbuch Luft-schutz, Teil 2, Leipzig, 1962).
Дифосген летом на Открытой местности эффективен около 30 лшн. Зимой его летучесть .меньше,'и поэтому он обладает большей стойкостью; заражающее действие длится около 12 ч. Аналогичные соотношения справедливы и для зарина, однако при этом нужно учитывать высокую токсичность этого вещества. Летом пары зарина эффективны еще через 20 ч, а зимой воздух и местность остаются зараженными в течение многих дней.
Современные ОВ типа зарина имеют сравнительно высокую скорость испарения. Из-за их высокой токсичности боевая концентрация невысока н создается она быстро путем испарения ОВ.
Воздушные потоки распространяют достаточные количества ОВ, которые образуют зараженное облако. Оно возникает сразу после применения ОВ и содержит в зависимости от способа применения наряду с газообразными молекулами ОВ также аэрозоль, который образуется при взрыве. Устойчивость такого облака ОВ зависит от метеорологических условий и местности. Оно может быть отнесено
26
ветром на много километров от места возникновения, причем отравляющее действие сохраняется вплоть до полной диффузии.
Разведка и определение глубины распространения зараженного облака ОВ составляют важную задачу службы химической разведки.
1.1.4. Стойкость О В
Стойкость ОВ находится в непосредственной связи сего летучестью. Летучесть ОВ явно ие зависит от времени, но давление и объем газа влияют на скорость испарения. Скорости испарения веществ можно сравнивать при совершенно одинаковых условиях. Точный расчет этих величин возможен только с помощью термодинамики для идеальных условий.
Стойкость ОВ на местности зависит не только от метеорологических условий и природы ОВ, но и от характера местности. Многие из этих факторов, которые выявляются уже из метеорологических данных (скорость ветра, температура, устойчивость воздушных слоев, влажность воздуха н др.) и из характера местности (растительность, структура почвы, рельеф местности и др.), можно лишь приблизительно учесть с помощью номограмм.
Стойкость ОВ можно также определить по формуле, выведенной Лейтнером3:
е_ °di _ Pi
Р2
где oDl и — скорости испарения воды при 1\ (15°С) и ОВ при температуре воздуха Г; р\ — давление пара - воды- при 15 °C (12,7л/л/рг. ст.), р2 — давление пара ОВ при температуре воздуха Г; Afj и М2 — мол. в. воды и ОВ.
Таким образом, стойкость ОВ является величиной, обратной отношению скоростей испарения вещества &d2 при температуре воздуха и воды oD1 при 15°С, принятой за единицу.
Приводим значения стойкости численные по этой формуле: Хлорпикрин . . . 0,23
Дифосген .... 0,4
Зарин.......... 3,13
некоторых ОВ (при 20 С), вы-
Люизит......... 9,6
Табун........' . 58,9
Иприт.......... 67
Эти величины позволяют сравнивать разные ОВ. Из этих данных видно, что при 20 °C иприт испаряется медленнее воды, и что при этой температуре его стойкость примерно в 21 раз превышает стойкость зарина.
Стойкость указывает на время, за которое испаряется ОВ по сравнению с водой, но она ничего не говорит о продолжительности действия ОВ, поскольку оно определяется не только летучестью и стойкостью ОВ, но и его токсичностью.
Иприт поражает органы дыхания и кожиые покровы. Для него расчет ведется преимущественно на контактное воздействие, однако в определенной ситуации учитывают также его действие на органы
27
дыхания, например при высокой температуре воздуха или лри применении иприта в виде аэрозоля. При пониженной температуре количество иприта, попадающего в воздух в результате испарения, недостаточно для значительного заражения воздуха. Этим объясняется относительно малая токсичность иприта при воздействии через органы дыхания. В случае применения зарина или зомана уже при очень низких температурах в воздухе создаются эффективные токсичные концентрации, которые делают незащищенный личный состав небоеспособным. Из-за высокой токсичности некоторые ОВ могут при известных условиях обладать большей продолжительностью заражающего действия, чем ОВ меньшей летучести и сравнительна большей стойкости.
Ниже приведены данные о стойкости ипрнта прн разных температурах и скорости ветра4:
Температура воздуха, °C Скорость ветра, м/сек, . , Стойкость ОВ, я , . . ,
О 0 -10 -10
2 3 2 3
20 17 72 60
Из приведенных величин видно, что стойкость ОВ прежде всего определяется температурой воздуха и почвы, хотя и другие метеорологические факторы при этом играют немаловажную роль.
1.1.5. Относительная плотность пара
Эффективность паров ОВ в воздухе определяется их относительной плотностью, которая представляет собой отношение плотности пара ОВ к плотности воздуха при постоянных температуре и давления:
Относительную плотность можно вычислить по плотности соответствующего вещества в газовой фазе; средняя газовая плотность воздуха б равна 1,2929 каДи3. Соединения, относительная плотность пара которых меньше 1, быстро диффундируют в верхние слои воздуха и через короткое время становятся неэффективными, тогда как вещества, относительная плотность пара которых больше 1, дольше удерживаются у поверхности почвы.
Плотность пара синильной кислоты составляет 0,93,. она легче воздуха. Синильная кислота может применяться только при помощи специальных методов, например путем использования специальных устройств — контейнеров или мин Особой конструкции. Чтобы в этом случае достигнуть эффективных боевых концентраций, нужно было бы создавать концентрации во много грамм на кубометр воздуха *. Несмотря на современные средства применения такие конй
* Рассуждения автора по поводу плотности паров ОВ справедливы для замкнутых объемов, Нужно учесть, что при концентрации HCN' в воздухе 2 же/л (смертельная концентрация) плотность зараженной атмосферы изменится меиее, чем на 0,1 %, что мало скажется на поведении зараженного облака. — Прим. ред.
28
V цеитрации нельзя поддерживать долго — синильная кислота быстро I: диффундирует в верхние воздушные слои. Без специальных средств £ применения создать достаточную боевую концентрацию синильной I* кислоты в воздухе невозможно.
? За исключением синильной кислоты и окиси углерода пары всех 8 других ОВ тяжелее воздуха.
5 Принимая во внимание закон Авогадро, можно вычислить относительную плотность пара по молекулярному весу вещества. Для воздуха средний молекулярный вес принят равным 28,9; отсюда плотность рассчитывают по следующей формуле:
' 6 с» ^QB Л?ов
; Мвозд 28,9
(10)
М.6. Способность О В давать аэрозоли
Большая часть соединений, относящихся к ОВ, являются твердыми | веществами или жидкостями. Их физические свойства, особенно ле-тучесть, не позволяют за сравнительно короткое время создавать за f счет нормальной летучести ОВ боевые концентрации, требующиеся б; для уничтожения противника.
Г Современные средства применения химических ОВ, будь то гра-иаты, бомбы и др., сконструированы таким образом, что можно р" осуществлять очень тонкое распыление ОВ в воздухе, т. е. Приме-O';,' нять ОВ в виде аэрозоля. Этот способ применения ОВ наиболее Ж эффективен.
Цг Твердые ОВ, такие как фосфорилтиохолины, хлорацетофенон, В; высокомолекулярные психохимические яды, адамсит, образуют В аэрозоли труднее, чем жидкости. Сравнительно термостабильиые г ОВ применяют, наряду с другими способами, в дымовых шашках, в В которых ОВ плавится, а затем испаряется. За счет конденсации Ц паров в воздухе из дисперсных частиц образуются аэрозоли.
Твердые ОВ можно применять в растворах. Растворитель испа-Ц}' ряется, и в аэрозоле остаются мелкие частицы твердых веществ. К Аэрозоли представляют собой квазистабильную систему из ча-стиц твердого вещества или жидкости в газообразной диспергирую-Ц щей среде, в данном случае в воздухе. Радиус частиц составляет от ~10-4 до 10~8 см. В воздушных аэрозолях, полученных различными способами, жидкие или твердые ОВ распылены до высокой степени fc' дисперсии.
Аэрозоли отличаются большой устойчивостью в воздухе, высокой ж-' проникающей способностью и большим радиусом действия. Из-за Ж малости размеров частиц аэрозоли ОВ попадают даже в тончайшие |г альвеолы легких. Чем мельче частицы, тем быстрее и глубже про-Ц иикают оии в организм, н тем легче оии всасываются. Скорость иа-Ц7 ступления отравления при вдыхании аэрозолей сравнима со ско-Ц ростью отравления при внутривенной инъекции.
Причины квазистабидьности аэрозолей определяются ме*гео])о-логическими факторами и большой подвижностью частиц. На
29
аэризиль одновременно действуют две силы — ороуновское движе-ние молекул н сила тяжести. Чем мельче частица, тем больше роль ее собственного (молекулярного) движения. С ростом скорости движения частиц стабильность аэрозоля возрастает.
Скорость седиментации частиц в основном определяется нх размерами. Вследствие малой вязкости воздуха и собственной подвижности частиц имеются хорошие условия для соударений и агрегации частиц. Недостаточный или противоположный по знаку электрический заряд способствует коагуляции.
Под действием силы тяжести все частицы аэрозоля седиментируют. Скорость седиментации возрастает с увеличением размера частиц. Например, если частицы с радиусом КН см осаждаются со скоростью около 43 см]ч, то для частиц с радиусом 10'2 см скорость седиментации составляет 4,3 л/ч. Обычно при увеличении радиуса частиц в 100 раз скорость седиментации возрастает в 10 раз.
За счет агрегации и коагуляции получается более грубодисперс-иая система, стабильность которой падает с увеличением размера частиц.
Метеорологические факторы, такие как ветер, конвекционные потоки и температура, оказывают большое влияние на устойчивость облаков аэроозлей ОВ. По отношению к этим воздействиям аэрозоли ведут себя подобно газообразным веществам.
При создании аэрозолей ОВ стремятся достигнуть возможно большей степени дисперсности. Чем тоньше получаются аэрозоли, тем они стабильнее. При благоприятных условиях аэрозоли могут существовать в воздухе дни и недели. Поэтому в случае некоторых ОВ, применяемых в виде аэрозолей, например ряда V-газов, нужно учитывать, что в зависимости от внешних условий нх действие продолжается много дней н даже недель.
Аэрозоли можно разрушить, например при помощи ультразвука или переменных электрических полей.
1.1,7. Растворимость н растворы ОВ
Растворимость или смешиваемость ОВ имеет большое значение для их хранения, применения, идентификации и определения, а.также для возможности повышения эффективности их дегазации.
Между растворами и смесями имеется различие. В растворах один компонент, т. е. растворитель, содержится в избытке по отношению к другим компонентам. Компоненты, содержащиеся в растворах в малых количествах, в чистом виде часто имеют другое агрегатное состояние.
Многие ОВ, наряду с другими способами, применяются в растворах, например, твердый хлорацетофенон (в виде так называемых слезоточивых растворов), производные фосфорилтиохолина, психохимические яды. Большая часть фитотоксических ОВ может эффективно применяться только в растворах.
30
-----о смесях компоненты содержатся в произвольных сиотнише-ниях. Различают следующие типы смесей жидкостей: системы, смешивающиеся в любых соотношениях (например зарин и вода); ограниченно смешивающиеся системы (например иприт и нефть); практически несмешивающиеся системы (например азотистый иприт и вода).
Растворы твердых и газообразных ОВ могут в свою очередь смешиваться с другими жидкостями (жидкие смеси). Из практических соображений в химии ОВ концентрацию растворов принято выражать в основном только отношением массы растворенного вещества к массе или к объему растворителя, т. е. а/100 см3, г/л, г/100 г растворителя. Концентрацию можно также выражать в объемных или мольных процентах, в единицах молярности или нормальности.
Растворимость (смешиваемость) ОВ с ростом температуры улучшается, т. е. увеличивается и скорость растворения, и количество растворенного вещества. При пониженной температуре растворимость иприта в нефти и подобных растворителях меньше, чем при повышенной. Примерно около 30°C иприт и нефть смешиваются во всех отношениях и образуют гомогенную смесь. Ниже этой температуры они смешиваются лишь ограниченно, причем образуются две жидкие фазы.
Способность ОВ проникать в организм сильно зависит от их растворимости в липидах и от температуры. Растворимость кожно-иарывных ОВ, например типа иприта, с увеличением температуры на (0° увеличивается вдвое. Результатом этого является более быстрое проникновение ОВ через кожу.
В случае жидкостей, не смешивающихся ни при каких соотношениях, растворяемое ОВ распределяется между двумя фазами. Отношение концентраций вещества в каждой из двух фаз всегда постоянно и не зависит от абсолютного количества вещества (Нернст):
Сф!
—«=* const (11)
Ьф2 ч
ч
Большая часть ОВ нерастворима либо лишь ограниченно растворима в воде.
Возможность воздействия на состояние равновесия- в растворах имеет большое значение, например при применении дегази-. рующих растворов.
Физические свойства ОВ, препятствующие их боевому применению, можно отчасти компенсировать путем приготовления растворов и смесей ОВ, пригодных для применения в любое время года V (так называемые тактические смеси). Это — смеси или растворы ОВ друг с другом или с органическими растворителями. Обычно пользуются двухкомпонентными смесями, однако известны также смеси, состоящие из большего числа компонентов.
31
При приготовлении тактических смесей ОВ в основном преследуют следующие цели:
понижение температуры плавления тех ОВ, Которые обладают относительно высокой температурой плавления;
воздействие иа давление пара, вследствие чего уменьшается летучесть и изменяется стойкость ОВ;
изменение агрегатного состояния ОВ в области температур, при которых его требуется применять;
повышение вязкости ОВ для улучшения его способности диспергироваться н прилипать, его стойкости, а также затруднения последующей дегазации;
понижение вязкости для улучшения способности ОВ образовывать аэрозоли; <
повышение боевой эффективности, особенно для достижения комбинированного отравляющего действия.
1.1.7.1. Понижение температуры застывания ОВ. Слишком вы* сокие температуры плавления некоторых ОВ, например иприта и азотистого иПрита, затрудняют их применение зимой. Чтобы сделать возможным боевое применение таких ОВ при низкой температуре, добавляют вещества, снижающие температуру застывания, например другое ОВ или органический растворитель.
Температуры застывания смесей ОВ определяют чаще всего экспериментально, после чего подбирают наиболее подходящую смесь. Температуры застывания некоторых смесей с ипритом приведены в табл. 13, стр. 154.
Многие смеси ОВ ведут себя как идеальные растворы, так что к ним можно условно применять закон Рауля. Имеющееся ограничение заключается в том, что смешиваемые компоненты должны обладать сходными свойствами.
Из относительного понижения давления пара над растворами, вычисляемого по закону Рауля, определяют повышение температуры кипения или понижение температуры застывания раствора. Эти величины можно использовать для вычисления примерной температуры застывания смесей однотипных ОВ.
Понижение температуры застывания вычисляют следующим образом:
ьт «2’Ю00
заст = & м ~ m М2'ГН]
(12)
Где £— мольная константа понижения температуры застывания—• криоскопическая постоянная, являющаяся константой вещества;
— количество растворенного вещества с мол. в Л12; Ш1— количество растворителя (в тех же единицах). •
Криоскопическую постоянную можно вычислить из удельной теплоты плавления I чистого растворителя,-
RT3
1000 • I
(13)
32
?ГДе 1 —темпераiypa uaawiuinn— j _ ____*
^Кельвина; R= 1,987 кал-моль-1 - град'1; I — удельная теплота >' плавления, кал/г.
; Для примера приведем величины криоскопических постоянных , некоторых растворителей:
е, град-кг*моль
Вода...................... 1,86
Бензол.......................... 5,07
Четыреххлористый углерод . 29,8
Иприт........................... 6,57
Нитробензол .................... 6,89
Чем меньше молекулярный вес растворенного вещества, тем больше понижение температуры плавления раствора, т. е. наибольшее понижение температуры застывания смесей ОВ достигается, если добавляемое вещество имеет возможно меньший молекулярный вес.
Как видно из данных, приведенных на стр. 154, требуются большие количества ДФФ (диизопропилфторфосфата), чтобы в результате добавления его снизить температуру застывания иприта- от 4-14,5 до —15 °C. . . .
Наиболее низкую температуру застывания получают, если компоненты смеси имеют возможно более близкие температуры плавления и их смешивают примерно в равных количествах. Например, смесь иприта с бензолом (т. пл. бензола 5,5 °C) в отношении 1: 1 застывает при —18,1 °C.
Не всегда выгодно готовить смеси с очень низкими температурами застывания. Особенно это невыгодно для смесей ОВ с нейтральными растворителями, так как при этом уменьшается содержание активного компонента (ОВ). Для достижения необходимых боевых концентраций требуются большие количества таких смесей. Поэтому обычно склоняются к тому, чтобы применять в качестве компонентов смесей также отравляющие вещества, например ОВ одинакового физиологического действия (смесь иприта и люизита) или ОВ с разными токсическими свойствами (смесь иприта н зарина).
Вследствие комбинированного характера поражающего действия последней смеси достигается исключительная боевая эффективность, которая сильно затрудняет разведку, защитные мероприятия, дегазацию и лечение пораженных.
1.1.7.2. Изменение давления пара ОВ. В растворах или смесях ОВ давление пара компонентов отличается от давления пара индивидуального вещества. Давление пара смесей ОВ является очень важным параметром, которым руководствуются при изготовлении и употреблении таких смесей. Основная задача — понижение давления пара ОВ для уменьшения его летучести и повышения стойкости.
Повышение давления пара возможно, когда силы притяжения между молекулами компонентов смеси меньше, чем между
33
одинаковыми молекулами. Это обусловлено природой таких компонентов, которые уже сами по себе обладают сравнительно высоким давлением пара, например сероуглерод, эфир, ацетон, гексан, гептан. Смесн с такими веществами при определенных соотношениях компонентов обладают максимумом давления пара. Для смесей ОВ такие соотношения недостижимы, несмотря на то, что это, по-видй-мому, способствовало бы повышению летучести.
В смесях ОВ между молекулами компонентов проявляются силы притяжения, Которые нужно преодолеть, вследствие чего понижается давление пара. Как уже упоминалось, некоторые бинарные смеси ОВ ведут себя подобно идеальным растворам, так что к ним применим закон Рауля.
Ряс. .3. Зависимость давления пара от температуры для разного состава смесей иприта с хлорпикрином (по данным PV-66/6).
В табл. 2 и на рис. 3 приведены вычисленные и полученные экспериментально значения давления пара смесей иприт — хлорпикрин, из которых видно, что поведение смесей приближается к поведению идеальных смесей.
Относительное понижение давления пара представляет собой отношение абсолютного понижения давления пара к давлению пара растворителя рр. Согласно закону Рауля относительное понижение давления пара равно мольной доле растворенного вещества х и не зависит от температуры и природы вещества:
= —Др = ppXi (14)
где «1 — число молен растворенного вещества; «2— число молей растворителя,
34
Таблица 2. давление пара смади ипрш----таирииир
Содержание хлорпикрина, МОЛ, И Парциальное давление, вычисленное по закону Рауля. мм рт. ст. Давление пара смеси. . мм рт. ст.
хлорпикрина р. иприта р2 вычисл. эксперим.
1 49,9 0 49,9
0,9 44,91 0,05 44,96 46,2
0,8 39,9 0,09 40,0 42,0
0,7 34,9 0,14 35,1 37,7
0,6 29,9 0,18 30,1 33,0
0,5 24,99 0,23 25,2 28,2
0,4 19,96 0,276 20,24 23,0
0,3 14,97 0,32 15,29 18,0
0,2 9,98 0,368 10,35 12,7
0,1 4,99 0,41 5,40 6,8
0 0 0,46 0,46 —
В случае бинарных растворов давление пара отдельных компонентов определяется их мольной долей в растворе и давлением пара компонента в чистом состоянии р0, таким образом, давление компонентов 1 и 2 будет равно:
Pi =* „ г»'*Ан= Xlp°i О5)
«I «2
/12 , - .
р2 = “ 1-- * Роз ~ Х2р02 U 5а)
. П] + П-2
Так как сумма парциальных давлений равна давлению пара смеси, получаем:
рем = Pi + Рг = -П!_ - ‘ Poi + „"2 «" * Рог О6)
Щ -f- Л-2 М] -f- П2
Рем = х1р01 + %2Р02
Сумма мольных долей компонентов равна 1, а при Xi = 1—х2 уравнение упрощается:
Рем = Poi + (рог — Pol) хг (17)
Для неидеальных смесей понижение давления пара больше. При этом силы притяжения между молекулами компонентов более значительны, чем в рассматриваемом случае. Из-за большего притяжения переход молекул в газовую фазу затруднен, вследствие чего парциальное давление пара компонентов меньше, чем в идеальных смесях:
pi<Xjpoi; рг<ХаРо2
Как видно из рис. 4, при некотором определенном составе подобной смеси общее давление пара может иметь минимальное значение.
Путем приготовления смесей ОВ стремятся достигнуть максимального понижения давления пара, которое, как показывает
2*
35
рисунок, зависит от определенного состава смеси и от природы выбранных компонентов.
Смеси ОВ должны испаряться так, чтобы состав пара, образующегося иад смесью, по возможности совпадал с составом самой смеси. Испарение смеси происходит иначе, чем испарение индивидуального вещества. Составы пара и жидкости обычно различны, и в случае неидеальных растворов и многокомпонентных систем их нельзя предсказать на основе только теоретических расчетов, а при-
ходится устанавливать экспериментально.
Легколетучий компонент покидает жидкость быстрее, чем труднолетучий, и поэтому накапливается в паровой фазе. В случае смеси фосген — иприт сначала испаряется фосген, а иприт остается в жидком или твердом состоянии, вследствие этого концентрация иприта в смеси увеличивается.
При смешении различных ОВ с некоторыми фосфорорганическими соединениями подобные изменения концентрации проявляются в меньшей степени. Смеси, в которых концентрации компонентов находятся в определенном отношении к ско-
Рис. 4. Кривая давления пара идеальной смеси (пунктир) и реальной смеси (сплошная линия) при постоянной тем-
пературе.
ростям их испарения, сохраняют в процессе испарения почти такое же соотношение концентраций компонентов в паровой фазе.
Повышение вязкости ОВ и их смесей путем введения высокомолекулярных веществ увеличивает их стойкость и понижает летучесть. Через поверхность загустителя ОВ испаряются меньше; их пары лишь медленно диффундируют через вязкую массу.
1.1.7.3. Растворимость О В. Сведения о растворимости ОВ имеют значение также для их дегазации. Дегазация с помощью дегазирующих средств наиболее эффективна тогда, когда она происходит в гомогенной фазе. Кроме того, некоторые хорошие дегазирующие средства лишь ограниченно растворимы в воде (дихлорамии, гексахлорамин и др.), и их можно применять только в соответствующих органических растворителях, чаще всего в дихлорэтане. Органические растворители применяют для дегазации в основном при низких температурах, когда взаимодействие между молекулами ОВ и дегазатора во многих случаях недостаточно энергично, и дегазация заканчивается лишь через долгое время. Между тем, если создать гомогенную среду, в которой растворяются ОВ и дегазирующее средство, то скорости реакций увеличиваются и дегазация заканчивается быстрее.
36
11 ид вер /лепныекирризии—ivia ivptiu^iui,—ij uvmn —m....
тельные приборы н подобные устройства нельзя обрабатывать дегазирующими средствами. Их дегазируют растворителями, которыми смывают ОВ с зараженного объекта. ’
Для идентификации н определения содержания ОВ в различных пробах прибегают к экстракции. Для этого выбирают растворители, обладающие возможно большей растворяющей способностью, чаще всего дихлорэтан, этанол и петролейиый эфир.
Поглощающая способность растворителя используется для взятия проб воздуха. Воздух, зараженный ОВ, пропускают через растворитель, который поглощает ОВ. На экстракции и поглощении растворителем основаны некоторые методы разделения, с помощью которых возможно распознавание н определение многих ОВ.
1.1.8. Единицы измерения концентрации
Концентрация ОВ (Сов) — это количество ОВ в объеме заражеи-ного воздуха. Ее выражают в единицах массы (массовая концентрация)
с Масса ОВ________ . lg.
ОВ *=* Объем зараженного воздуха * '
или объема (объемная концентрация):
с =__________Объем ОВ________ '
ов Объем зараженн^-о воздуха^ * }
Массовая концентрация обычно измеряется в мг]л или в г(м3. В большей части полевых аналитических методов концентрация ОВ определяется в мг/л.
Объемная концентрация измеряется в см31м3, объемных процентах, объемных промиллях. Для расчета объемной концентрации нужно привести все данные к стандартным условиям (760 ммрт. ст., 0°С). Международными единицами концентрации газов являются миллионные доли, что соответствует выражению в см3/м3. Общепринятые в промышленности индикаторные трубки дают концентрации в миллионных долях (м.д. или р. р. ш.).
В табл. 3 приведены формулы для пересчета наиболее употребительных единиц для выражения концентрации газов в воздухе.
Концентрация, необходимая для достижения определенного физиологического или токсического действия, которое приводит к смерти или потере боеспособности противника, называется боевой концентрацией (Сов).
Плотность заражения (ПЗов)— количество ОВ, находящееся иа единице площади, выражают в г[м? или в кг[га:
Зараженная площадь
37
Таблица 3. Формулы для пересчета одних единиц концентрации газов в воздухе в другие (по Дрюсу) (а — измеренная величина; М — мол. вес; Р — атмосферное давление, мм рт. ст.', Т — температура в °К)
Единицы измерения Приведение к стандартным условиям (760 мм рт. ст,, 0°С) г!м\ мг/л мг[мг', мкг! л Объемн. % Объемн. % о; см3! л см*!м*\ м. д. мм'/м* р. р. Ь. Число разбавления, Z
—т
г/jw3; мг]л 2,784 а Т а-103 6,236 аТ 62,36 а Т 62 360 аТ 62 360 000 аТ 16,035 МР
Р РМ РМ МР МР аТ
мг}м3\ мкг!л 2,784 а Т в - 1(Г3 аТ аТ 62,36 а Т 62 360 аТ 16 035 МР
Р 160,35 РМ 16,035 МР МР МР аТ
Объемн. % а 0,16035 аМР Т 160,35 а М? Т — а • 10 а • 104 а-107 10а а
Объемн. %Q; см3!л а 0,016035 аМР 16,035 аМР а • 10"J а • Ю3 а* 10е 103
Т Т а
ем3/м\ м. д. аМР аМР 1П~3 а - 103 10®
d 62 360 Т 62.360 Т а • Ю а • 10 а
мм3/м3\ р. р. b. аМР аМР а- 10-т а- 10-в а - 10-3 10э
62 360 000 Т 62 360 Т а
Число разбавления, Z а 16,035 МР ZT 16 035 МР ZT 10s Z 103 Z 10» Z 10э Z —
1.2. ХИМИЧЕСКИЕ СВОЙСТВА ОВ
Большая часть ОВ является органическими соединениями. Онн ие образуют отдельную группу соединений, а принадлежат к самым различным классам веществ и не имеют общих свойств. Их реакционная способность обусловлена специфическими свойствами, присущими соединениям этих классов, и имеющимися в молекуле заместителями.
Знание химических свойств ОВ требуется для правильного их хранения и применения, для индикации н дегазации, для понимания поведения ОВ в воздухе, механизма поражающего действия и для применения противоядий.
При выборе соединений в качестве ОВ существенны не только высокая токсичность и соответствующие физические свойства, но также и максимально возможная химическая устойчивость. Имеются соединения высокой реакционной способности, которые могут быть использованы как ОВ специфического действия. Например, фториды галогенов, которые вследствие своей огромной агрессивности разрушающе действуют на материалы и в то же время обладают высокой токсичностью. Решающим для выбора того или иного соединения в качестве ОВ является цель применения.
Принятые на вооружение империалистических армий ОВ сравнительно устойчивы к гидролизу и не реагируют или только частично реагируют со многими дегазирующими средствами. Вновь разрабатываемые ОВ должны предъявлять новые высокие требования к средствам защиты противника и к эффективности его средств дегазации. Принятый на вооружение немецкой фашистской армией иприт лишь слабо реагировал с хлорной известью и другими обычными для того времени дегазирующими средствами. Хотя это свойство иприта является положительным для ОВ, другие факторы, такие как неудовлетворительные физические свойства, плохая устойчивость при хранении, термическая нестабильность, при известных условиях препятствуют его боевому применению.
Токсичность ОВ и ядов предполагает определенную реакциои ную способность. Молекулы яда принимают участие или влияют на протекание специфических биохимических процессов и в этом*за-ключается их действие. Достаточно малого количества высокотоксичного вещества, чтобы вызвать эффективное действие. Для очень небольшого числа ядов механизмы поражения выяснены- полностью. Многие химико-биофизические процессы, идущие как параллельно, так и во взаимосвязи, представляют собой в основном каталитические реакции. Вряд ли их можно сравнивать с какими-либо процессами, происходящими вне этой среды. Яды, которые очень реакционноспособны в организме, не обязательно активны вне его.
Индикация ОВ и ядов, эффективных в чрезвычайно малых количествах, требует быстрых, специфичных и чувствительных качественных реакций. Эти три условия не всегда выполнимы.
39
Специфичность реакции часто идет в ущерб ее чувствительности и наоборот. При распознавании ОВ с целью предостережения часто жертвуют специфичностью реакции в пользу ее чувствительности.
1.2.1. Термическая устойчивость
Как почти все органические соединения, ОВ разлагаются при повышении температуры. Поведение таких ОВ при нагревании определяет способ их применения. Поэтому термически нестабильные ОВ могут применяться в виде аэрозолей только особыми способами. Это главным образом относится к пснхохимическим ядам и некоторым фосфорорганическим соединениям.
Хотя все ОВ в какой-то мере термически неустойчивы, в большинстве своем они устойчивы к детонации и могут применяться в артиллерийских снарядах. Количество тепла, кратковременно выделяющееся при взрыве, разлагает лишь небольшую часть ОВ, которой можно пренебречь.
При применении иприта, зарина и других ОВ в артиллерийских боеприпасах, бомбах и т. п. эффективный коэффициент полезного действия составляет в среднем 95—99% (при нагревании разлагается 1—5%). Многие ОВ, применявшиеся в первую мировую войну, имели значительно меньший к. п. д., часто ниже 75%, например бромбеизилциаиид. Изменяя конструкцию средств применения, можно улучшить к. п. д. безотносительно к термической -устойчивости ОВ. Поэтому ОВ применяются в минах, бомбах и других снарядах особой конструкции.
.Если какое-либо нестабильное вещество разлагается с образованием ядовитых продуктов, то вполне возможно его применение в качестве ОВ. К таковым ОВ относятся, например, хлорпикрин (трихлорнитрометан) или дифосген (трихлорметиловый эфир хлоругольной кислоты).
Наибольшей термической стабильностью должны обладать ОВ, применяющиеся в дымовых шашках. При горении шашки ОВ подвергаются длительному воздействию тепла. Ни одно из ОВ, применявшихся до сих пор в виде ядовитого дыма, не удовлетворяет требованию абсолютной термической устойчивости. Рецешуралпа-шек такова, что максимальный к. п.д. достигается при минимальном выделении тепла за ограниченное время горения. Адамсит (фе-нарсазинхлорид) разлагается выше 320 °C; хлорацетофенон при 750 °C за 15 мин разлагается на 32%, а дифеиилхлорарсии—на 48%. Однако оии могут применяться в дымовых шашках со специальной рецептурой шихты.
Обычно термическое разложение ОВ при нормальном давлении происходит уже до достижения температуры кипения, поэтому очистку ОВ перегонкой можно производить только при пониженном давлении. Идентификация ОВ по температуре кипения возможна в очень редких случаях.
Типичные во многих случаях запахи ОВ отчасти принадлежат продуктам разложения. Запах иприта, напоминающий чеснок, гор-40
ЧИЦуИЛИ хрен, воЗникае i 1 данным—uupu jum—при—и о„ разложении; аналогично, гераниевый запах люизита объясняется образованием так называемого третичного люизита. Высокая степень очистки, добавка так называемых термостабилизаторов и использование современных средств применения ОВ уменьшают во многих случаях разложение, и тем самым количество продуктов разложения, являющихся специфическими носителями запаха.
Упоминавшиеся ранее раздражающие ОВ — хлорацетофенон и адамсит наиболее устойчивы к детонации. Они могут быть использованы в смеси со взрывчаткой в боеприпасах бризантного действия.
1.2.2. Устойчивость к гидролизу
Гидролиз ОВ под действием воды в подавляющем большинстве случаев представляет собой обратную реакцию его прямого (или косвенного) способа получения. Степень гидролиза ОВ зависит от их природы. При небольшом избытке или эквивалентном количестве воды гидролиз может быть обратим. При этом устанавливается равновесие, как, например, при гидролизе иприта [бис-(2-хлорэтил^сульфида]:
уСН3СН2С1 +НгО //СН2СН2ОН
\ЗН2СН2С1 < + HCI ^СН2СН2С1
Прн большом избытке воды гидролиз не останавливается на первой стадии, а приводит к образованию неядовитого бис-(2-окси-этил)-сульфида:
уСН2СН2ОН + Нз0 /СН2СН3ОН
\ЗН2СН2С1 -HCI ^СН2СН2ОН
Хотя в данном случае образуется неядовитый продукт, эта реакция не пригодна для дегазации иприта, поскольку в обычных условиях, при которых приходится проводить дегазацию на практике, потребовался бы слишком большой избыток воды, и, кроме того, скорость гидролиза слишком мала.
Большая часть современных табельных ОВ (см. стр. 60) в значительной мере устойчива к гидролизу. Это свойство отвечает военному требованию. Устойчивость ОВ к гидролизу является одним из важнейших факторов, определяющих условия его хранения, поведение в воздухе, стойкость и вообще эффективность. Оиа является мерой отношения ОВ к дегазирующим средствам гидролитического действия и играет не последнюю роль в анализе.
Технические ОВ загрязнены в основном кислотными примесями галоидирующих агентов, которые гидролизируются или. разлагают-си в присутствии влаги. В результате стенки контейнеров корродируют, и это приводит к образованию соответствующих соединений, которые при известных условиях вызывают разложение ОВ.
41
Например хлористый водород, содержащийся в“виде примеси в тех-иическом иприте, в присутствии воды реагирует с железом с образованием хлорида железа, который способствует разложению'иприта. Выделяющийся при этом водород может быть причиной повышения давления в контейнере.
Влажная синильная кислота полимеризуется почти сразу после получения. Этот не поддающийся контролю процесс сопровождается взрывом.
Подобные отрицательные свойства сглаживаются, если получать ОВ максимально возможной чистоты нлн добавлять стабилизаторы.
При применении ОВ находятся в воздухе в виде пара или тончайшего аэрозоля. Влагой воздуха ОВ гидролизуются, и в ходе гидролиза теряют эффективность. Влияние происходящего в атмосфере гидролиза ОВ на его концентрацию зависит от природы вещества. Фосген очень быстро гидролизуется по следующей схеме:
СОС1г + НгО —> СО2 + 2НС1
Таким образом, причинами быстрой потери эффективности это-rq ОВ в воздухе следует считать не только физические свойства, но и быструю гидролизуемость *.
В общем такие ОВ, как иприт, зарин, V-газы и психохимические яды, достаточно устойчивы к гидролизу. Чем меньше вещество гидролизуется, тем продолжительнее его заражающее действие в воздухе и на местности. Следует учитывать, что при относительной влажности воздуха 60—70% начальная концентрация фосфорорганических соединений в течение 24 ч понижается в среднем на два порядка, а иприта — на один порядок.
Гидролизу ОВ способствует дождь, влажная почва, роса и др. Очень устойчивые к гидролизу ОВ, которые лишь в малой степени гидролизуются в жестких условиях (нагревание, присутствие щелочей), сохраняют свою эффективность на местности долгое время. Так, например, хлорацетофенон может выдержать весеннее таяиие снега и при благоприятных почвенных условиях, препятствующих смыванию, вновь стать эффективным.
Некоторые ОВ предназначены для заражения систем водоснабжения, продуктов питания, посевов и др. Предпосылкой для такого применения ОВ является их высокая устойчивость к гидролизу или образование при гидролизе продуктов такой же ядовитости. Зарин неограниченно растворим в воде и гидролизуется лишь медленно, равно как фторорганические соединения, предложенные в качестве диверсионных ядов. Другие яды или ОВ растворяются в воде лишь ограниченно, однако их растворимость и устойчивость к гидролизу достаточна, чтобы обеспечить эффективное заражение.
* Это утверждение ошибочно; фосген влагой воздуха практически не гидролизуется. — Прим. ред.
42
При гидролизе различных азотистых—иириыв—[п-алкил-хч,-Ы-бис-(2-хлорэтил)-амины] образуются столь же ядовитые, как и исходные ОВ, соединения, которые даже в водных растворах еще обладают кожно-нарывным действием, а прн попадании в желудочно-кишечный тракт вызывают тяжелейшие отравления, чаще всего со смертельным исходом. Поэтому во время второй мировой войны немецким командованием были специально изготовлены для заражения воды солеобразные производные таких галогенирбван-ных третичных аминов.
При применении фосфорорганических соединений системного действия типа систокса, тимета, тетрама (см. раздел 7.9) для поражения растений они должны обладать определенной устойчивостью к гидролизу. Яд, попадающий с током питательных веществ во все части растения, не должен при этом гидролизоваться. Устойчивость большей части ОВ к гидролизу делает непригодной для дегазации воду без добавки других веществ. Будучи слабым нуклеофилом вода неспособна взаимодействовать с реакционнособными центрами молекул ОВ. Только повышение нуклеофильности, достигаемое введением ОН-ионов или других нуклеофильных реагентов, приводит к ускорению гидролиза и позволяет быстро дегазировать данное ОВ.
Так, большую часть фосфорорганических ОВ можно количественно дегазировать водными растворами щелочей. Из приводимого уравнения следует, что гидролиз фосфорорганического соединения просто водой и водным раствором щелочи приводит к одному и тому же продукту:
+ HgO
-HF
+ NaOH
—NaF
Различие между этими двумя реакциями заключается лишь в скорости, гидролиз щелочью идет значительно быстрее. Проведение реакции в более жестких условиях, например, концентрированными растворами щелочи (иногда при нагревании), может привести к полному гидролизу и разложению молекулы:
4NaOH
2ROH + NaF + Na3PO4 + Н2О
Гидролизу способствует нагревание. Если гидролиз иприта в насыщенном водном растворе при 20 °C протекает несколько часов, то при 100 °C он заканчивается за несколько минут. Воду, зараженную ипритом, можно дегазировать кипячением, если количество ОВ было невелико. Такую воду можно употреблять в пищу только после соответствующего анализа.
43
Знание поведения ОВ в водных растворах необходимо' при проведении аналитических реакций. Если в данных условиях ОВ гидролизуются, то их уже нельзя распознать, если только не был выбран метод анализа, при котором определяются и продукты гидролиза.
1.2.3. Отношение к химическим реагентам
С военно-химической точки зрения прежде всего представляет интерес отношение ОВ к химическим реагентам, применяющимся для дегазации и в анализе.
Как уже отмечалось, при принятии нового ОВ на вооружение одним из требований к нему является наличие таких свойств, которые делают, непригодными имеющиеся табельные средства дегазации и индикации ОВ; все это должно затруднить его идентификацию и дегазацию.
Ввиду того, что химическое строение и, следовательно, химические свойства ОВ различны, существование универсальных средств дегазации и обнаружения невозможно. Для ОВ сходного строения можно использовать аналогичные реакции с одним и тем же реагентом. Для обнаружения многих ОВ известен ряд реакций, из которого следует выбрать наиболее подходящую. Для дегазации при известных условиях также пригодны разные вещества, однако при выборе большую роль играет экономическая сторона вопроса. Например, фосфорорганические соединения и другие ОВ можно быстро дегазировать различными хелатными комплексами металлов и производными гидроксамовой кислоты. Однако применение этих веществ может быть оправдано только для дегазации в малых масштабах. При дегазации больших площадей важнейшим дегазирующим средством является раствор щелочи и разновидности этого способа.
Реакционная способность ОВ по отношению к окислителям и восстановителям очень разнообразна. При окислении иприт сравнительно быстро превращается в неядовитый продукт. Этим пользуются для дегазации иприта, причем применяют такие окислители, как хлорную известь и моиохлорамин. Органические фосфаты в общем очень устойчивы к действию окислителей. То же наблюдается и для азотистых ипритов, которые превращаются в окиси аминов лишь в жестких условиях, практически недостижимых прн дегазации в полевой обстановке.
В зависимости от реакционной способности функциональных групп в молекулах ОВ механизм действия вещества, применяемого для дегазации, различен. Например, сульфид натрия действует на одни ОВ как восстановитель, в случае других ОВ его Дегазирующий эффект основан на облегчении гидролиза. Химически весьма устойчивый трихлорнитрометаи при восстановлении сульфидом натрия полностью разлагается с образованием СО2, N2, окислов азота, НС1 и др., тогда как в случае азотистых ипритов сульфид натрия
44
^-способствует гидролизу, в резулыdie- UUpU-JJ V 11.Л-п.ии ------
Жг /СН2СН2С1 /СН2СН3 у
Ж R—b/ 4- Na2S —> R—+ 2NaCl
Ж \CH2CH2C1 \СН2СН2 '
BL; Определенные реагенты оказывают на ОВ стабилизующее дей-ствие, что относится даже и к дегазирующим веществам. Так, моно-ЖЬ хлорамин стабилизует некоторые органические фосфаты вследствие дК образования амидов. Подобные явления существенны для дегаза-К ции, особенно если для заражения применяли несколько ОВ или их К1 смесь. Эти явления определяют последовательность применения Ж' средств дегазации.
ж/ Большая часть известных ОВ обладает достаточной реакцион-К пой способностью, чтобы их можно было идентифицировать и дега-зировать соответствующими средствами. Очень часто уже неболь-Ж того изменения молекулярного строения ОВ достаточно для его Ж дегазации. Одиако при этом предполагается, что соотношения меж-ду токсичностью или физиологическим действием ОВ и его струк-Ж турой известны, так что по характеру изменений молекулы, происхо-№ дящих при химической реакции, можно сделать вывод о физиологи-Ж ческой активности или неактивности продуктов реакции.
Ж 1.2.4. Устойчивость во время хранения
Ж Химические вещества должны оставаться устойчивыми при хране-Ж иии. Оии должны быть устойчивыми к колебаниям температуры, не Ж должны разлагаться и разрушать материал контейнера.
Ж Очень немногие ОВ достаточно устойчивы, чтобы их можно было ЦТ * хранить неограниченно долго. Совершенно устойчивы в процессе Ж- хранения ОВ типа адамсита, хлорацетофенона, психохимические Ж яды и некоторые другие. Для нх хранения обычно достаточно при-Ж митнвиого контейнера из импрегиированиого картона или древе-Ж сины. Такие ОВ, как иприт, большинство органических фосфатов Ж и галогеиоксимы, лишь ограниченно устойчивы при хранении. Их Ж устойчивость прежде всего зависит от степени их чистоты. Чем Ж чище вещество, тем оно более устойчиво. Производство их в абсо-Ж лютно чистом состоянии иногда экономически невыгодно, поэтому Ж часто ОВ представляют собой сильно загрязненные технические В продукты.
К Для улучшения устойчивости при хранении к ОВ и их смесям Ж добавляют соответствующие стабилизаторы, которые препятствуют аутоокислению, гидролизу, полимеризации и коррозии. В случае Ж некоторых ОВ действие стабилизаторов ограничено. Например, не-возможно с помощью стабилизаторов сделать жидкую синильную кислоту устойчивой при хранении иа неограниченно долгое время, Ж одиако для иприта это осуществимо.
45
---гггприртгпттвги—апллиписсдпн—лин I роль—спз—гга—слладал; иуд» они в боеприпасах или в контейнерах, обязателен и проводится с помощью физико-химических методов. В общем, запасы нестабильных ОВ на складах империалистических армий обновляются через каждые 15—20 лет. Поскольку ОВ не находят пока применения, их уничтожают или перерабатывают н заменяют новыми.
1.2.5, Отношение к различным материалам
По отношению к различным материалам, таким как металлы, древесина, резина и пластмассы, ОВ ведут себя очень по-разному. Эта проблема имеет большое значение для хранения ОВ, организации защиты и дегазации.
Обычные металлы разрушаются прежде всего такими ОВ, от которых легко отщепляются галогены или которые содержат в виде примесей реакционноспособные побочные продукты. Образующиеся производные металлов, чаще всего соединения железа, катализируют разложение ОВ. Газообразные продукты, образующиеся в процессе коррозии, повышают давление в контейнерах, что неблагоприятно сказывается на прочности последних, особенно в случае снарядов и бомб.
Во время первой мировой войны наряду с другими мерами предосторжности корродирующие ОВ помещали в боеприпасы в оболочках из свинца, олова или фарфора. Добавкой антикоррозионных средств можно парализовать, корродирующее действие современных табельных ОВ. Средства применения ОВ, которые должны продолжительное время храниться заполненными соответствующим содержимым, изготовляют нз специальной стали. В случае же средств применения ОВ, предназначенных к использованию сразу после заполнения, довольствуются более дешевыми металлическими материалами.
То, что при использовании соответствующих материалов ОВ могут десятилетиями находиться в средствах применения совершенно без разложения, показали найденные ипритные боеприпасы времен первой мировой войны. На внутренних стенках снарядов после более 30-летнего воздействия иприта не обнаружено никаких явлений коррозии, мины были полностью пригодны к боевому использованию.
Материалы, применяемые для пошива защитного обмундирования, должны быть по возможности универсальными — устойчивыми против всех ОВ в любом агрегатном состоянии. При этом требуется ие только устойчивость по отношению к чистым или техническим ОВ, эти материалы должны противостоять широкому ассортименту возможных растворов и смесей ОВ. Благодаря развитию химии пластмасс за последние десятилетия появилось большое число полимеров, которые в определенных сочетаниях представляют собой средства защиты, обладающие максимальной устойчивостью против известных ОВ и их смесей.
46
меняются сополимеры акрилонитрила с бутадиеном, полналкил-акрилаты, фторопласты и полиэтилен.
Материалы, вырабатываемые для изготовления защитного обмундирования и снаряжения, не должны растворяться в органических и неорганических растворителях. Они должны быть газонепроницаемыми, не должны ни набухать, ни размягчаться. От современных средств защиты требуется сверх того негорючесть, хорошая способность отражать световое излучение и поглощать инфракрасные лучи.
Для успеха дегазации важно знать, как ведут себя различные материалы по отношению к ОВ. Дегазировать приходится .вещества самого разного типа — резину, пластмассы, металлы, древесину, стройматериалы, кожу, дорожные покрытия, ткаии. В эти материалы ОВ проникают в большей или меньшей степени в зависимости от способности растворяться, проникать, поглощаться и диффундировать. При этом имеет значение и состояние поверхности материала. Эти факторы определяют вид и способ дегазации. Они затрудняют индикацию заражения материалов. Приходится отбирать соответствующие пробы и подвергать их специальной обработке.
1.3. ТОКСИЧЕСКИЕ СВОЙСТВА ОВ
Из предыдущих разделов очевидно, что эффективность ОВ зависит от их физических и химических свойств, имеющих большое значение для достижения максимального эффекта, который должен привести либо к уничтожению противника, либо к выводу его из строя. Не каждое высокотоксичное вещество пригодно в качестве ОВ. Оно должно обладать определенными физическими и химическими свойствами, которые делают возможным его применение. Руководящим критерием для оценки ОВ или яда является фармакологическая и токсикологическая эффективность.
Среди ОВ имеется ряд малотоксичных соединений, которые при их боевом применении отнюдь не могут уничтожить противника. Однако их воздействие на человеческий организм вызывает определенные фармакодинамические процессы, которые в свою очередь обусловливают физиологические эффекты, неизбежно ведущие к потере боеспособности. К категории ОВ, ие уничтожающих противника, а приводящих только к потере его боеспособности, следует отнести раздражающие ОВ и психохимические яды. В случае всех остальных ОВ возможно создание смертельных концентраций или доз, к которым стремятся при применении ОВ в соответствии с тактической задачей.
Фармакологические и токсические свойства ядов определяются их природой5. Существуют тесные связи между химическим строением и биологической активностью ОВ. Их действие специфично и является характеристикой их свойств. Механизм поражающего
47
действия — влияние на определенные биохимические процессы в организме —зависит от реакционной способности яда в дайной среде. В основе таких реакций лежат как чисто химические процессы, так н физико-химические изменения.
Условием эффективности яда является наличие в его молекуле определенных активных полярных функциональных групп. Механизмы поражающего действия ядов часто представляют собой очень сложные, комбинированные процессы, которые выяснены отнюдь не во всех случаях. Их изучение имеет исключительное значение для отыскания противоядий, для оказания первой помощи и лечения отравлений, и в настоящее время представляет чрезвычайно важную проблему ввиду возможности применения современных высокотоксичных ОВ.
Очень часто собственно токсичные соединения образуются лишь после попадания яда в организм. Различные ОВ и яды действуют по-разному. Из-за сходства молекулярной структуры и наличия аналогично действующих функциональных групп или атомов некоторые токсичные соединения вызывают одинаковую или по крайней мере сходную картину отравления. Нет ОВ или яда, обладающего только одним единственным действием. Все отравления сопровождаются побочными явлениями.
Классификация ядов по определенным типам, таким как раздра- -жающие, кожно-нарывные, нервно-паралитические, производится иа основании их первичного действия. Такое подразделение не является абсолютно строгим, например, вторичное действие кожно-иарывных ОВ ведет к отравлениям общего характера, которые при известных обстоятельствах оканчиваются смертью. Очень часто бывает трудно отнести тот или иной яд к определенному типу, так как он в одинаковой степени обладает разными действиями. Чтобы ' вызвать отравление, достаточное количество яда должно попасть в соответствующие органы, по отношению к которым он специфичен и где он может оказать свое первичное действие.
В то время как в распоряжении медиков имеется множество способов введения лекарства для скорейшего достижения определенного фармакологического эффекта, ОВ и яды военного назначения могут попадать в организм, главным образом, посредством вдыхания, приема через рот или кожной резорбции.
Для быстрого развития поражающего действия требуется, чтобы ОВ попадало в организм всеми возможными путями.
Современные высокотоксичиые фосфорорганические ОВ типа зарина или V-газов сравнительно быстро проникают через неповрежденную и особенно через поврежденную кожу. При известных условиях количества ОВ, проникающего через кожу, достаточно, чтобы вызвать тяжелое, даже смертельное отравление. Их относят к ОВ, которые могут попадать в организм всеми путями. Другие ОВ, например фосген, могут действовать только через органы дыхания, оии лишены какой-либо способности к резорбции через кожу,
48
Для оценки яда недостаточно сведений о характере и мела-иизме поражающего действия, значительно важнее охарактеризовать интенсивность их действия. По такой характеристике можно сравнивать разные ОВ независимо от многообразия их поражающего действия. Для этого надо располагать данными о физиологической или токсической активности ОВ, которые, разумеется, имеют ценность лишь в том случае, если они определены в одинаковых или сопоставимых условиях. Такие данные получают в результате эксперимента на живых объектах, а именно на животных. Тем не менее известно, что в исследовательских учреждениях СС в гитлеровской Германии летальные характеристики токсичности опре-целялись на людях, подобно тому, как теперь в империалистических странах это производится на приговоренных к смертной казни. Большая часть данных о токсичности по отношению к людям была получена в первую мировую войну или в результате случайных отравлений.
Техника эксперимента на живых объектах до такой степени изобилует ошибками, что характеристики токсичности, определенные таким путем, могут быть лишь ориентировочными. На эти величины большое влияние оказывает собственно подопытное животное, его конституция, состояние здоровья и упитанность. Наконец, решающим является вид животного, причем часто недостаточно брать определенные породы. Для точных определений используют животных одной и той же линии. Кроме того, в этих определениях играют большую роль поколение и возраст животного. Объективные трудности для получения точных данных возникают и из-за процессов обмена веществ в организме. Активность некоторых ядов в организме увеличивается, другие яды, напротив, обезвреживаются.
Из всего этого следует, что токсичность или эффективность яда никоим образом не зависит только от его количества. Конечно, между эффективностью и количеством яда существует некоторая пропорциональность, но это ие простое соотношение, так как перечисленные факторы оказывают влияние, которое нельзя выразить простыми математическими формулами. Чтобы в значительной мере исключить это влияние, определяют средние значения из ряда опытов. При этом учитываются средние ошибки соответствующих экспериментальных результатов.
Для характеристики физиологических и токсических свойств ОВ и ядов военного значения представляют интерес данные о максимальных их количествах, безопасных или вызывающих поражение или смертельный исход. В военной химии приняты следующие понятия: безопасная, опасная, очень'опасная и смертельная концентрации. Для всех этих градаций наряду с концентрацией приводится время экспозиции. Таких сведений обычно достаточно, чтобы оцеинтЬ обстановку и быть в состоянии принять необходимые для практических действий решения. Более точными данными,
49
по з в оляющими сравнивать Ub и~йды Друг ~с другим, являютсл, пороговая концентрация, предел переносимости, произведение Ct, летальная доза и летальная концентрация.
1.3.1. Пороговая концентрация
Пороговая концентрация — это минимальная эффективная концентрация или доза, т. е. наименьшее количество ОВ, которое может вызвать ощутимый физиологический эффект. Дозы илн концентрации ниже пороговых значений являются неопасными и не приводят к заметным физиологическим изменениям, если только не подвергаться их постоянному воздействию.
Пороговые концентрации можно приводить как для раздражающих ОВ, так и для упомянутых выше ядов, независимо от способа Применения, но для каждого способа свою. Эти значения определяют опытным путем на людях и выражают в следующих единицах:
ОВ, используемые в виде паров и аэрозолей, мг/л воздуха
ОВ, попадающие через рот, мг/кг массы
ОВ, попадающие путём кожной резорбции или при помощи инъекции, мг/см2 поверхности кожи
Последний способ особенно применим для кожно-нарывных ОВ при обозначении наименьшего раздражающего количества ОВ на единицу поверхности кожи.
Для раздражающих ОВ пороговая концентрация обозначает так называемую нижнюю границу или порог раздражения, т. е. дозу, промежуточную между эффективной и не эффективной, при которой для слезоточивых ОВ начинается лакримогенное действие,. а для раздражающих носоглотку ОВ — их характерные симптомы поражения. Нижние пределы раздражающего действия слезоточивых ОВ приведены в табл. 6, стр. 80. Нижние пределы наиболее сильных раздражителей имеют порядок величины 10-3—10~4 мг/л.
Пороговые концентрации ОВ могут предупреждать об отравлении лишь в тех случаях, когда сама картина отравления начинается с симптомов, которые в известной степени служат предостережением. Однако это возможно только для очень небольшого числа ОВ и ядов, поскольку многие из них безболезненно попадают в организм и первые признаки отравления наступают лишь тогда, когда процесс уже в полном разгаре. Тем не менее, знание величин пороговых концентраций не лишено интереса, так каи прн необходимости с их помощью можно оценить степень опасности ОВ по соответствующим признакам на поле боя или по результатам анализа проб. z
Небольшие концентрации некоторых ОВ вызывают определенные рефлекторные симптомы, например кашель или чихание. Так, трихлорнитрометан в концентрации 2* Ю3 мг]л сильно раздражает глаза, но уже при концентрациях порядка 0,1 мг/л наступают небольшие поражения легких. Напротив, связанный с действием фосгена кашель начинается лишь при концентрациях, которые уже
50
этот кашель не может рассматриваться как предостерегающий симптом. Разумеется для людей восприимчивых, с больными легкими порог раздражения может быть ниже, чем установленная для данного ОВ токсическая концентрация. Пороговые концентрации некоторых психохимических ядов имеют величину порядка 10-2 .лга/ка массы и ниже. Они вызывают у людей многочасовые психозы. Это минимальные дозы, вызывающие психоз; меньшие количества не приводят к физиологическим изменениям, при больших количествах вызываемый психоз более продолжителен и интенсивен. Различные фосфорорганические соединения вызывают сужение зрачков (миоз). Это явление иногда наступает прежде других характеристичных симптомов, т. е. в известной степени является предостерегающим сигналом, наблюдаемым при определенной пороговой концентрации. Диизопропилфторфосфат уже в концентрациях Ю’3 ма/л вызывает многодневное сужение зрачков; количества, превышающие 5* 10"3 ма/д, приводят к явлениим отравления, следующим за миозом.
Из пороговых концентраций кожно-нарывных ОВ особый интерес представляют данные для газообразных веществ или аэрозолей. Под этим понимают концентрации, при которых становятся заметными первые признаки поражения кожи, хотя дальше поражение и не развивается. Обычно это зуд или покраснение поверхности кожи. С фармакологической точки зрения больший интерес представляют данные о колгГчестве ОВ, выраженные в количестве вещества на единицу поверхности кожи.
Нежные участки кожи поражаются ипритом в концентрации 10~3 мг!л, хотя тяжелые поражения при этом не наступают. Концентрации выше 10"1 ма/л приводят уже к тяжелым последствиям. Образуются волдыри, и это ведет к некротическим распадам.
Определение пороговых концентраций затрудняют внешние факторы, такие как температура и влажность воздуха, природа индивидуума. Поэтому имеющиеся данные изобилуют ошибками и дают лишь относительные величины, которые можно рассматривать как ориентировочные.
1.3.2. Предел переносимости
Предел переносимости есть концентрация ОВ или яда в мг{л, которую человек может выдерживать определенное время без устойчивого поражения. Это — максимально возможное или допустимое количество ОВ. Для применимых в военных целях веществ пределы переносимости часто выше концентраций, ведущих к потере боеспособности. Наступающие при таких концентрациях физиологические явления могут оказаться уже столь сильными, что в результате соответствующих симптомов пораженные уже не в состоянии выполнять возложенные на них задачи и становятся небоеспособными,
51
В литературе опубликованы предельно допустимые концентрат ции ряда веществ, встречающихся в промышленной практике. Эти величины представлены в табл. 4. В ней приведены нормы предельно допустимых концентраций, принятые в СССР (ПДК)), ГДР и США (МКРМ) для некоторых веществ. Концентрации можно выражать не только в л<г//и3, как это сделано в табл. 4, но и в м. д.
Таблица 4. Предельно допустимые (ПДК) и максимальные (МКРМ) концентрации некоторых реагентов (в жг/ж3)
Вещество СССР7 (ПДК) США11 (МКРМ) ГДР’ (МКрм)
Акрилонитрил 0,5 45 20
Аммиак 20 70 50
Бензин 300 1000
Бензол 5 80 50
Бром — 7 1
Гексахлоран 0,1 0,5 0,2
Г ексоген 1
Двуокись серы 10 13 10
1,2-Дихлорэтан 10 400 50
Метанол 5 260 100
Мышьяк (порошок) — 0,5 0,3
Мышьяковистый водород 0,3 0,2 0,2
Нефть * 2000 ——
Нитробензол » 5 5
Нитроглицерин 0,5
Окись углерода 20 110 55
Пиридин 5 30 10
Ртуть 0,01 0,1 0,05
Сероуглерод 10 60 50
Серная кислота 1 1 1
Сероводород 10 30 15
Синильная кислота 0,3 11 5
Тетракарбоннл никеля — 1 —
Тетрахлорэтан 5 5 10
Тетраэтилсвинец 0,005 —' 0,05
Т олуол 50 750 200
Тринитротолуол 1 1,5 1,5
Т рихорннтрометан 7 —
Трихлорэтан — 1050 250
Уксусная кислота 5 25 20
Фенол 5 19 20
Фосген 0,5 4 0,5
Фосфор белый 0,03 0,1 0,1
(желтый)
Фосфористый водород 0,1 0,07 0,1
Фосфорорганические инсектициды 0,5—0,02 0,5—0,02 0,5—0,02
Фтористый водород 0,5 2 1
Хлор 1 3 1
Хлорбензол 50 350 50
Хлористый водород 5 7 10
Этанол 1000 1900 1000
Этиловый эфир (диэтиловый) 300 1200 500
52
(р. р. mJ. данные таилицы нимлвюоюry- 11L1 V1LLJ«« LJllk/— R—_ -_ му показателю различны в разных странах.
Для токсичных соединений можно приводить аналогичные дан-* иые. Из практических соображений их составляют так, чтобы по ним можно было судить о времени пребывания людей без противогаза или средств защиты кожи.
Максимально допустимая концентрация составляет, например, для азотистого иприта (без противогаза) около 10-3 мг/л при экспозиции 15 мин; без средств защиты кожи, но с надетым противогазом возможная экспозиция достигает 2 ч. Разумеется, эти величины не абсолютны, и могут встретиться отдельные случаи, когда наблюдаются некоторые симптомы отравления н ранее. Однако в общем боеспособность личного состава, подвергнутого воздействию таких концентраций за указанные промежутки времени, не снижается.
Максимально допустимая концентрация зарина, по американским данным, составляет от 0,001 до 0,005 мг-мин}л, а для иприта от 0,001 до 0,025 мг-мин/л, т. е. при времени экспозиции 1 мин максимально допустимая концентрация иприта в 5 раз больше, чем зарина.
В последние годы на промышленных предприятиях почти всех стран для безопасности рабочих введены и узаконены предельно допустимые максимальные концентрации на рабочем месте МКРМ (международное обозначение МАС, т. е. maximum allowable concentration). На.международном симпозиуме в Праге (1959 г.) Летавет6 определил эти концентрации как максимально допустимые концентрации, которые при постоянном воздействии на человека в течение рабочего дия не могут вызвать через длительный промежуток времени патологических изменений или заболеваний, обнаруживаемых при помощи современных Методов диагностики. Сюда относятся возможности интоксикации ие только за счет поражения через органы дыхания, ио и за счет кожной резорбции и через пищеварительный тракт, а также случайное кратковременное воздействие сравнительно высоких концентраций, комбинированное воздействие нескольких веществ, соматические явления утомления, пониженная сопротивляемость, повышенная восприимчивость и др.
Как правило, МКРМ относятся к восьмичасовому рабочему дню. Эти нормативы постоянно подвергаются критической проверке на основании новых экспериментальных и клинических данных, и, при необходимости, пересматриваются. За последние годы определены МКРМ для сотеи встречающихся в промышленности вредных веществ, которые могут действовать в виде газов, паров, аэрозолей или пыли.
Для военно-тактических целей МКРМ использовать нельзя, так как оии не дают сведений о концентрациях, эффективных в бою за короткое время.
53
1.3.3. Произведение Ct
До введения характеристик токсичности, рассматриваемых в следующих разделах, в токсикологии, особенно в военной, для сравнения отдельных ОВ между собой большую роль играло произведение Ct. Этот параметр был введен Габером (габеров фактор смертельности) и выражал соотношение между смертельной концентрацией газов, паров нлн аэрозолей ОВ и временем их воздействия. Степень токсичности Т отравляющего вещества зависит от концентрации С, экспозиции t, объема легких V и массы тела G. Прн таком подходе не учитываются все индивидуальные особенности подопытного объекта.
Таким образом, с помощью перечисленных факторов степень токсичности Т можно выразить следующим соотношением:
Поскольку степень токсичности, как правило, определяется на одинаковых подопытных объектах, в частности, на высших животных (собаках и обезьянах), отношение V/G можно считать постоянным, и поэтому выражение (21) упрощается:
T = Ct
Если, концентрация возрастает, а продолжительность воздействия постоянна, или наоборот, продолжительность экспозиции увеличивается при постоянной концентрации ОВ, то величина Т должна стремиться к значению, при котором следует абсолютный смертельный исход для подопытного объекта. Наименьшее возможное значение степени токсичности носит название фактор смертельности (U7) нравно (к мг • мин/м3):
= = (22)
. Таким образом, W является постоянной величиной, специфичной [для вида ОВ, вида подопытного объекта, условий опыта и др. Однако эта константа относится только к сравнительно кратковременным экспозициям; в случае синильной кислоты —это лишь несколько минут, в случае фосгена — самое большое до 1 ч. При более продолжительных воздействиях или при очень низких концентрациях измеренное значение произведения Ct больше, особенно в случае ядов, которые в известной мере выводятся из организма (с выдыхаемым воздухом, с мочой), обезвреживаются или удаляются другими способами и не накапливаются (например, в случае синильной кислоты). Для этого ОВ при высоких концентрациях величина Ct равна 1000, а при низких —4000.
Чтобы учесть естественные потери ОВ за счет обезвреживания в организме или выведения из него, вводят поправочный множитель е, тогда для однотипных ОВ, таких как синильная кислота и окись углерода, выражение для W принимает вид:
W = (С - е) t (23)
54
Если с <; е, то отравление может воиище не нриизиига, i(n\ пал яд обезвреживается или выводится из организма до величины е.
Ниже приводим величины Ct из работы Оттара9:
w
для ряда ОВ, заимствованные
w
Зоман.................. 80
Зарин ................ 150
Табун ................. 450
Хлорциан............. 4 000
Фосген.............. 5'000
Дифенилхлорарсин . . 15 000
Хлорпикрин......... 20 000
Значения Ct сильно колеблются. Чем меньше С/, тем токсичнее соединение. Так, по Ct зоман в 62 раза более ядовит, чем фосген, и почти в 2 раза, чем зарин. Уже 1/10 величины Ct достаточно, чтобы повлечь за собой потерю боеспособности незащищенного личного состава 9, Это ие относится к раздражающим ОВ, которые уже в очень малых концентрациях вызывают раздражение, приводящее к потере боеспособности, и в случае которых в боевой обстановке никогда нельзя достигнуть смертельных концентраций, поскольку их токсичность во много раз меньше токсичности упомянутых токсичных соединений,
В американской армии степень токсичности (мг-мин/м3) используют для оценки степени опасности, причем определенные области концентраций обозначаются следующим образом.
Незначительно опасная —боеспособность подразделения не снижается, хотя у части личного состава могут появиться незначительные признаки отравления;
средне опасная — боеспособность подразделения несколько понижена в результате случаев отравления, но оно еще в состоянии выполнять поставленные задачи;
очень опасная — боеспособность подразделения вследствие больших потерь сильно понижена.
Области концентрации иприта и зарина для указанных степеней опасности таковы (в мг-мин!м3):
- Иприт Зарин
Незначительно опасная . . . 1—25 1—5
Средне опасная........' . 20—50 6—10
Очень опасная........... 51 — 100 11 — 15
Приведенные значения отвечают степени токсичности Т для экспозиции 1 мин, так что по ним можно оценить возможную продолжительность пребывания при более высоких и более низких концентрациях, Например, при концентрации зарина 30 мг[м3 уже через 22—30 сек достигается «очень опасная» степень токсичности и соответствующее состояние небоеспособностн, что можно рассчитать, подставив соответствующие значения в формулу Т = Ct-.
Il =30Zi; /1=0,37 мин
15 = 30/z; С = 0,5 мин
55
Чтобы достигнуть степени «средне опасная» за 5 мин и степени «очень опасная» за 10 мин воздействия, требуется концентрация зарина от 1,1 до 1,5 мг/м\
1.3.4. Летальная доза
Летальная доза (ЛД)—есть приводящее к смерти абсолютное количество ОВ, попавшее в организм. Эта величина характеризует отношение количества ОВ, вызывающего смертельное поражение, к массе тела подопытного объекта и выражается в мг[кг массы.
Летальная доза позволяет лучше сравнивать яды между собой и служит критерием оценки не только ОВ и ядов, но и любых фармацевтических препаратов, применяемых в медицине. Она определяется в результате опытов иа животных.
Значения ЛД позволяют сравнивать яды между собой лишь в ' тех случаях, когда они определены на животных одинаковой породы, по возможности одной и той. же линии н при одинаковом способе введения. Чтобы исключить влияние индивидуальных факторов, искажающее эти величины, пользуются средней летальной дозой (ЛДзо), которую определяют из нескольких серий опытов и оценивают математическими и графическими методами10. Эта величина показывает, что при данной дозе погибает 50% подопытных животных. Если задается другой процент смертности, то соответственно заменяют цифры в индексе. Так, при 100%-ной гибели подопытных объектов приводят величину ЛДют.
Значения ЛД, определенные в опытах над животными, нельзя пересчитывать по отношению к людям (пересчет к '70 кг}. Очень часто смертельные дозы для людей значительно ниже, чем для животных. В 1938 г. такого рода пересчет повлек за собой гибель около 100 человек при применении диэтилеиглнколя п.
Разумеется, при использовании подходящих животных, например, обезьян, -можно сделать вывод о приближенной величине токсичности для людей. Некоторые такие данные будут приведены в соответствующих разделах этой книги.
Как видно из Приложения 5, имеются большие различия в величинах доз, установленных при разных способах введения.
При внутривенной инъекции большая часть ядов достигает места поражения быстрее, чем при внутримышечной инъекции или при приеме через рот. В качестве примера приведем средние летальные дозы, полученные на кроликах при разных способах введения диизопропнлфторфосфата ДФФ (в мг/кг):
Перорально (гг. о.)......... 4,0—9,8
Внутримышечно (в. м.).............. 0,75
Подкожно (п, к.) .............. I
Внутриглазно (в. г.)........... 1,15
Внутрибрюшинно (в. б.)............... 1
Внутривенно (в. в.) 0,34+0,01
56
на основании величин легальной ДЦДЫ визмилша гишигирши----------------
ция ОВ по токсичности. Ниже приведены такие данные, получен- t
ные в опытах с крысами при однократном приеме через рот:
ЛДм, мг/кг
Чрезвычайно токсичные .... I Высокотоксичные.............. 1—5* 10*
Умеренно токсичные...........5’Ю1—5*102
Малотоксичные................5* Ю2—5* Ю3
Практически нетоксичные . . . 5* 103—1,5< 104
Сравнительно безвредные . . . . >1,5* Ю4
Вещества, для которых ЛДм > 100 мг/кг, считают неядовитыми.
Фосфорорганические ОВ, а также фтороргаиическне соединения, предназначенные в качестве диверсионных ядов, и другие ОВ, применимые в военных целях, имеют летальные дозы менее 1 мг!кг. Их называют также сверхъядами.
1.3.5. Летальная концентрация
После второй мировой войны для характеристики ядов, действующих через органы дыхания, все больше н больше привлекают летальную концентрацию (ЛК). Этот параметр выражает количество яда в определенном объеме воздуха (обычно в мг/л или в мг/м3), которое вызывает смерть. По тем же причинам, что и в случае летальной дозы, вводится средняя летальная концентрация (ЛКбо), г. е. концентрация, при которой погибает 50% подопытных животных. В этом случае, как и в произведении С/, концентрация зависит от конкретного времени воздействия, которое обязательно надо указывать при значении летальной концентрации. Если спе-i цйально не оговорено, то, как правило, летальная концентрация относится к экспозиции 10 мин.
Наряду с необходимыми сведениями о породе животного особое значение имеет такая характеристика, как промежуток времени до наступления смерти (табл. 5).
Таблица 5. ЛК5о диизопроиилфторфосфата (ДФФ) при разной продолжительности его воздействия на мышей
Концентрация ДФФ Экспозиция, Период до наступления смерти, ч Концентрация ДФФ Экспозиция, мин Период до наступления смерти, «
мг/л М. д. (р. Р. т) мг/л м. д. (р. р. т)
5 664 1 2 0,6 80 10 1
2,65 330 2 2 0,44 58,4 10 2
0,75 99,6 5 2 0,185 24,6 30 2
Точно так же, как и для ЛД, нельзя переносить значения летальных концентраций на людей, но некоторые выводы из таких цифр делать можно. Как показывают данные табл. 5, которые были
57
L
получены йа мышах, йи в коем случае Нельзя использовать отдельные значения для пересчета к другим экспозициям и концентрациям, как это было возможно в случае грубой оценки по величине произведения Ct, Это было бы фальсификацией точных величин, определенных экспериментально.
Из приведенных данных следует, что при той же экспозиции (в данном случае 10 мин) при более высоких концентрациях смерть наступает часом раньше, чем при меньших количествах. При создании боевых концентраций токсичных ОВ, которые имеют целью уничтожение личного состава, стремятся к наивысшей технически возможной концентрации, чтобы добиться наибольших потерь при меньшей экспозиции, Поскольку для современных ОВ боевая концентрация может значительно превышать летальную как из-за высокой токсичности этих ОВ, так и из-за наличия необходимых средств применения, то экспозиция, требуемая для уничтожения или выведения из строя, достигает от нескольких минут до секуид. Постоянная готовность средств защиты к немедленному использованию и непрерывные тренировки являются важнейшими факторами сохранения живой силы при применении ОВ.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие тактике-технические требования предъявляются к химическим ОВ?
2, Нарисуйте диаграмму давления пара бис-2-хлорэтилового тиоэфира (иприта), трихлорнитрометана (хлорпикрина) и диизопропилфторфосфата (ДФФ) в интервале температуры 0—30 °C.
3. От каких факторов зависит предельная концентрация ОВ?
4. Рассчитайте по значениям давления пара, полученным из ответа на вопрос 2, предельную концентрацию сходных ОВ в одном и том же интервале температуры и начертите диаграмму зависимости предельной концентрации от температуры,
5. Какие факторы определяют стойкость ОВ?
6. Какие существуют соотношения между температурой кипения и предельными концентрациями ОВ?
7. Что такое аэрозоль?
8. Для чего приготовляют тактические смеси ОВ?
9. Вычислите температуру застывания смеси из 100 кг иприта и 20 кг бензола или 20 кг нитробензола, Какие выводы можно сделать на основании полученных результатов?
10. Почему при приготовлении определенных смесей ОВ стремятся к наибольшему понижению давления пара?
II. Как влияет устойчивость ОВ к гидролизу на его поведение на местности и в воздухе?
12. Почему только немногие химические вещества пригодны в качестве универсальных дегазирующих средств?
13. Как развивается поражающее действие яда?
14. От каких факторов зависит токсичность вещества?
15. Что такое пороговая концентрация и какое значение она имеет?
16. Какие факторы влияют на определение токсикологических и физиологи-, ческих данных?
17. Что такое средняя летальная концентрация?
68
I, Naser, Physikalische Chemie fur Techniker u, Ingenieure, Leipzig, 1966,
2. Herbst, Kolloidchem. BeiheEte, 23, 340 (1926).
3. Leitner, Militarwiss, u, technische Mitteilungen, 1926, 662.
4. Conrad R., Militartechnik, 2, 16 (1962).
5. H a u s c h i 1 d F., Pharmakologie u, Grundlagen der Toxikologie, Leipzig, 1958.
6. Zbl. E, Arbeitsmed. u. Arbeitsschutz, 9, 171 (1959).
7, Санитарные нормы проектирования промышленных предприятий. СН 245-71. М., Стройиздат, 1972. — Прим. ред.
8. Atemschutz-Information, 4, 60 (1961).
9. 011 а г В., Т. Kjem Bergwesen о[ Metall, 5, 73 (1954).
0. Roth Z., Ceskoslov. Fysiol., 10, 408 (1961).
11. Oe 11 e 1 H., Arch. Toxikol., 15, 50 (1954).
12. Zbl. f. Arbeitsmed. u. Arbeitsschutz, 11, 63 (1961).
2. КЛАССИФИКАЦИЯ ОВ
ОВ классифицируют по разным принципам. Решающими для классификации являются физические, химические, токсикологические свойства и тактические и методические соображения.
В странах, производящих ОВ, стало общепринятым в зависимости от уровня производства подразделять запасы ОВ н нх эффективность иа следующие группы:
Табельные ОВ — сюда относят производимые в больших количествах и состоящие на вооружении империалистических армий эффективные ОВ, боевое применение которых определяется соответствующими уставами. В США таковыми являются V-газы, зарин, иприт, адамсит, хлорацетофенон, психохимические яды н др., включая их всевозможные тактические смеси.
Резервные ОВ — включают такие вещества, которые не производятся непосредственно в качестве ОВ, ио при необходимости могут быть изготовлены развитой химической промышленностью в достаточном количестве. В США к этой группе наряду с другими относят синильную кислоту, галогеицианы, мышьякоргаиическне соединения и фосген,
ОВ ограниченного значения — токсичные вещества, свойства которых лишь удовлетворительны для их применения в качестве ОВ, но которые производятся промышленностью в большом масштабе для других целей, например фосфорорганические инсектициды, мышьяковистый и фосфористый водород. Это могут быть и соединения, которые имеются в распоряжении лишь в сравнительно малых количествах, так как по разным причинам нет достаточных про изводственных возможностей.
2.1, КЛАССИФИКАЦИЯ ПО ФИЗИЧЕСКИМ ПРИЗНАКАМ
Пожалуй, простейшей классификацией ОВ является их деление по агрегатному состоянию (при нормальных условиях) иа твердые, жидкие и газообразные. Бблыпая часть современных ОВ находится в жидком или твердом состоянии и применяется в таком виде, причем в соответствии с их физическими свойствами н техникой применения оии эффективны в виде газов или аэрозолей. При 20 °C ряд ОВ имеет следующие агрегатные состояния:
газообразное — фосген, окись углерода, мышьяковистый н фосфористый водород; жидкое — синильная кислота, иприт, азотистый
60
иприт, хлорпикрин, зарин, зоман; твердое — адамсит, хлорацетофенон, диэтиламид лизергиновой кислоты (LSD), 2,4-дихлор-феноксиуксусная кислота, фторацетат натрия*.
По способу заражения воздуха целесообразно различать газообразные ОВ и ОВ в форме видимых или невидимых аэрозолей, а по их давлению пара и по стойкости — летучие и стойкие ОВ (см. разд. 1,1,3).
2.2. КЛАССИФИКАЦИЯ ПО ХИМИЧЕСКИМ ПРИЗНАКАМ
Непременной предпосылкой для чисто химических исследований ОВ является их классификация по классам органических соединений, т. е. в соответствии с их строением. Как и всякая классификация, такое деление на практике имеет для исследователя и преимущества и недостатки.
Для оценки физических и химических сво.йств ОВ, относящихся к соответствующему классу, чисто химическая классификация удобна. Ряд характерных для веществ этого класса превращений используют в качестве аналитических реакций и для дегазации. Используя сведения из органической химии, можно делать определенные заключения о химической стойкости ОВ, вследствие чего работа с такими соединениями облегчается.
Номенклатура, применяемая для обозначения ОВ, должна быть аналогична номенклатуре, которая принята для органических соединений данного класса, что даст возможность исключить ошибки при идентификации ОВ. По тому же принципу можно производить отнесение еще неизвестных ОВ, вследствие чего существенно облегчается оценка их свойств.
Поскольку в пределах узких химических групп соединений существуют определенные соотношения между строением и токсичностью, можно, зная эти соотношения, сделатывывод о токсичности относящихся к этой группе соединений и, сопоставляя их с физическими и химическими свойствами, заключить — следует или не следует искать среди них отравляющие вещества. Разумеется, отнесение ОВ или яда к определенной группе органических соединений не позволяет уверенно говорить о фармакологических закономерностях. Для этого необходима классификация, учитывающая биохимические процессы. Хотя по этому вопросу уже высказывалось много предложений, проблему следует считать еще нерешенной, поскольку в настоящее время еще недостаточно изучены биохимические процессы, протекающие при многих отравлениях. Кроме того, классификация такого рода выходила бы за рамки химии ОВ.
Многие реакции идентификации и дегазации ОВ основаны на свойствах заместителей, имеющихся в молекуле ОВ. Поэтому они не характерны или лишь частично характерны для Тйких групп ОВ.
* Последние два вещества, как правило, к ОВ не относят.—Прим. ред.
61
Однако это не принижает значения классификации ОВ по химическим признакам для аналитиков, разведчиков, дегазаторов и др.
ОВ -встречаются среди разных классов органических соединений. Ими могут быть алифатические углеводороды, спирты, кетоны, амины, арсины, сложные эфиры, производные карбоновых кислот, тиоэфиры, оксимы, алкалоиды, карбо- и гетероциклические соединения, фосфорорганические, металлоорганические и другие соединения.
2.3. КЛАССИФИКАЦИЯ ПО ТОКСИКОЛОГИЧЕСКИМ ПРИЗНАКАМ
Очень целесообразной классификацией ОВ является их подразделение по наиболее выраженным явлениям отравления. По основному действию на организм человека различают:
раздражающие, к которым относятся слезоточивые ОВ — лакриматоры (например, хлорацетофенон) и раздражающие носоглотку ОВ — стерниты (иапример, адамсит); удушающие (фосген, трехфтористый хлор и др.); кожно-нарывные (иприт, люизит и др.); общеядовитые (синильная кислота, зарин, V-газы*); психохимические ОВ (диэтиламид лизергиновой кислоты LSD, дитран).
Имеется общая тенденция специализировать защитные мероприятия, санитарную обработку, первую помощь, а также дегазацию для каждой определенной группы ОВ. Простая, понятная и легко запоминающаяся классификация по главному действию ОВ является незаменимым подспорьем для гражданского населения, военнослужащих и врачей.
При такой классификации не учитываются многосторонние побочные действия ОВ. Многочисленные органические и неорганические соединения, применимые в качестве ОВ или ядов, например соли тяжелых металлов, алкалоиды, и биологические яды, нельзя охватить такой классификацией.
Возможна более жесткая систематизация иа основе патологических реакций, вызываемых действием ОВ. В соответствии с такой систематизацией ОВ подразделяются следующим образом:
вещества, вызывающие острую кислородную недостаточность (удушающие ОВ). Они действуют посредством блокирования доступа кислорода (дыхательная аноксемия, иапример хлорпикрин, фосген); блокирования переноса кислорода (сосудистая аноксемия, например окись углерода, мышьяковистый и фосфористый водород), блокирования тканевого дыхания (тканевая аноксемия, например синильная кислота и цианистые соединения);
вещества, вызывающие воспалительные процессы (возбудители воспаления), а именно: серозные и гнойные воспаления (например иприту некротические распады с сильным общеядовитым действием (например азотистый иприт и люизит);
* Зарин и V-газы составляют особую группу нервно-паралитических веществ.— Прим. ред.
62
йсщества, вызывающие патологические рефлексы (рефлекторные яды) органов зрения (например слезоточивые ОВ), органов дыхания и пищеварения (иапрнмер адамсит, днфенилхлорарсин, соли триалкнлсвинца), мышц (к ним относятся все ингибиторы холинэстеразы, например фосфорорганические ОВ);
вещества, которые вмешиваются в высшие функции центральной нервной системы (например мескалин, диэтиламид лизергиновой кислоты LSD, днтран, тетраэтилсвинец).
Такая классификация тоже не учитывает все факторы. Она представляет собой классификацию по основному действию и ограничивается важнейшими ядами, применимыми в качестве химических ОВ.
Другие классификации ядов читатель найдет в соответствующей литературе по токсикологии. Токсикологи пытаются классифицировать яды отчасти по органам поражения. Например, различают яды крови, желудка, кишечника, печени, почек, сердца, нервов, глаз, кожи и клеток, или, поскольку большинство ядов действует как ингибиторы энзимов, — по специфическому действию энзимов.
2.4. ВОЕННО-ТАКТИЧЕСКАЯ КЛАССИФИКАЦИЯ
Способы классификации ОВ, рассмотренные в предыдущих разделах, не удовлетворяют воеино-тактическнм требованиям для оценки обстановки после применения ОВ.
Для того чтобы можно было возможно более точно оценить обстановку и принять решение, следует в принципе учитывать тримомента: характер действия ОВ, поражающее действие, продолжительность заражающего действия.
По характеру действия различают ОВ, быстро ведущие к уничтожению противника (например фосфорорганические соединения, синильная кислота, фосген*) и ОВ, временно выводящие из строя (например иприт**, раздражающие ОВ, психохимические яды).
Но быстроте наступления поражающего действия следует различать быстродействующие ОВ (без скрытого периода) н медленно действующие ОВ. Быстродействующие ОВ за несколько минут приводят к иебоеспособности либо по причине смерти, либо в результате временного поражения противника. Таковыми являются фосфорорганические соединения, синильная кислота, раздражающие ОВ, некоторые психохимические яды и др. Медленно действующие ОВ (со скрытым периодом) приводят к полной иебоеспособности только после скрытого периода от одного до нескольких часов. Оии либо уничтожают, либо временно выводят из строя противника. К таким веществам относятся, например, фосген, иприт, некоторые психохимические яды.
* Фосген может быть отнесен к этой группе веществ только в том случае, если он будет применен в концентрации >10 мг/л.— Прим. ред.
** При достаточно больших концентрациях или плотностях заражения иприт способен вызывать смертельные поражения. —- Прим. ред.
63
По продолжительности заражающего действия можно различать короткодействующие (летучие) и долгодействующие (стойкие) ОВ. Поражающее действие короткодействующих ОВ длится лишь Минуты (например фосген, синильвая кислота). Действие долгодействующих ОВ может в зависимости от метеорологических условий продолжаться от нескольких часов до иесколькнх недель (иапример зоман, иприт, V-газы).
Имеется классификация, по которой вещества систематизируют в соответствии с задачами и способами применения.
Различают ОВ н иды для наступательных действий, обороны, диверсий, засад, заражения местности, удаления листвы и уничтожении посевов (фитояды), разрушения материалов, применяемые в виде аэрозолей.
2.5. УЧЕБНО-МЕТОДИЧЕСКАЯ КЛАССИФ ИКАЦИЯ
Подготовка солдат, обучение населения и воспитание квалифицированных кадров вызывает необходимость классифицировать изучаемые вещества, пользуясь дидактическими принципами. Так, при Подготовке солдат следует отдавать предпочтение токсикологической классификации ОВ, поскольку она проста и понятна и при пользовании ею можно в принципе охватить все типичные ОВ. За этим сразу должна следовать тактическая классификация, которая, одиако, при обучении более широких кругов населения излишня.
При обучении руководящих кадров химической службы нужно рассматривать все проблемы, связанные с применением ОВ. Поэтому в данной книге будет использована классификация, которая в наибольшей степени учитывает как токсикологические, так и химические признаки. В этой связи все разделы книги, за исключением материала, посвященного фосфорорганическим соедииеииим, классифицированы по основному токсическому действию ОВ, а затем следует дальнейшее подразделение по химическим признакам иа основе классификации органических соединений.
Изложение конкретного материала начинается с рассмотрения связи между токсическим действием и строением вещества с учетом его основного токсического действия, далее следует изложение возможных методов получения, физических, химических и Токсических свойств конкретных ОВ, возможности их применения, защита от них и их дегазация.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. По каким трем признакам производится классификация ОВ, Исходя из тактических требований?
2. Какое различие между быстродействующим и летучим ОВ?
3. Почему классификация ОВ по токсикологическим признакам является неполной?
j
О. Э/ДГ rt/lvniviLUTT
Сразу после начала первой мировой войны слезоточивые ОВ были применены как немецкой, так н французской стороной. Это были уже известные соединения, синтезированные в прошлом веке, ио не нашедшие практического применения. До войны некоторые из них использовались полицейскими. Хотя от употребления таких веществ ожидали многого, их эффективность оказалась ниже предполагаемой. Соединения, примененные сначала на открытой местности, не затрудняли или мало затрудняли действия противника; в распоряжении не имелось нужного количества ОВ; не были достигнуты достаточные боевые концентрации; среди применявшихся ОВ были очень нестабильные соединения.
Несмотря на совершенствование средств применения (вначале пользовались даже ружейными гранатами) удалось достигнуть лишь временного успеха, так как с течением времени армии вооружились средствами защиты. Позже тактическую роль ОВ стали усматривать в том, чтобы в результате систематического н длительного применения ОВ вынудить вражеские части находиться в средствах защиты, стеснять их маневры и ограничивать их боеспособность. Затем в распоряжение военных поступили более подходящие соединения, такие как бромпропанон (бромацетон), трихлорнитрометан (хлорпикрин) и галогенцианы. Эти вещества были не только слезоточивыми, ио и ядовитыми. Их применение предполагало не только выведение из строя, но и уничтожение противника. Однако материальные затраты на создание и поддержание смертельных боевых-концентраций были слишком велнки, поэтому применение этих веществ ограничилось лишь отдельными попытками и вряд ли сулило успех. Принятие на вооружение мышьякор-ганических соединений, сильно раздражающих носоглотку, очень резко изменило ситуацию. Вступили в действие соединения, которые применялись в виде аэрозолей и проникали через противогаз. Фильтры противогазов содержали только импрегнированный активированный уголь, который не задерживал взвешенные частицы этих новых ОВ. Пораженные срывали с лица защитные маски.
Примененные немецкой стороной мышьякорганические соединения, дифенилхлор- и дифенилциаиарсин, так называемые ОВ с синим крестом, из-за сильного «пробивного» действия были названы «вредителями противогазов». Их применяли в сочетании с такими токсичными ОВ, как фосген и дифосген, против которых
3 Зак. 448 55
иого раздражающего действия они были уже небоеспособны и деморализованы.
Тактическая установка применять комбинированно ОВ с различным физиологическим действием приобрела значение только в результате принятия на вооружение раздражающих носоглотку ОВ.
За немногими исключениями раздражающие ОВ, применявшиеся в первую мировую войну, теперь уже не имеют большого военного значения. После войны в качестве раздражающих ОВ стали известны два.эффективных и подходящих по своим физико-химическим свойствам соединения — адамсит и хлорацетофенон.
Во время второй мировой войны в Англии исследовали ряд сви-иецорганических соединений, которые по их раздражающему действию на поЛости носа и глотки сравнимы с мышьякоргаиическими. Они отличаются от других раздражающих ОВ высокой биологической активностью и стабильностью.
Применение раздражающих ОВ в современном бою спорно. Их можно эффективно применять только в тех случаях, когда оии приводят к иебоеспособности в невероятно малых концентрациях, аналитически не распознаваемых или распознаваемых лишь с трудом, когда атакованные не имеют противогазов или надевают их слишком поздно.
Особое значение имеют раздражающие ОВ для запугивания и деморализации беззащитного населения. Слезоточивые ОВ в настоящее время относятся к важнейшим полицейским средствам подавления в капиталистических странах.
Какие катастрофические последствия может иметь применение полицией слезоточивых газов против скопления людей показали действия перуанской полиции в мае 1964 г. во время бурно проходившего спортивного мероприятия. В результате возникшей паники погибло 350 человек,
3.1. СЛЕЗОТОЧИВЫЕ ОВ
3.1.1. Химическое строение и биологическая активность
При рассмотрении всей совокупности слезоточивых ОВ обращает на себя внимание то обстоятельство, что все оии являются главным образом галогенпроизводными разных классов органических соединений. Так как ие все органические галогенсодержащие соединения обладают таким физиологическим действием, то, по-видимому, лакримогенное действие этого типа веществ зависит ие только от числа атомов галогена, но и от их природы и положения по отношению к центральному атому и к функциональной группе.
Причину лакримогенного действия функциональных групп однозначно можно выяснить ие во всех случаях. Одиако в большей части таких соединений атомы галогенов обладают электронодонорным характером, т. е. действуют как заместители, которые в
66
ной связи, тогда как в случае других галогенсодержащих соединений в результате —/-эффекта молекула поляризуется в противоположном направлении.
Предпосылкой для лакримогенного свойства соединения является наличие так называемого «положительного» атома галогена, например в галогенированных кетонах:
/—> 5* СНЧ-С-СН2— С1
W
Аналогичный эффект проявляется также в случае бромбензил-цианида I, p-галогеннрованных нитрилов акриловой кислоты II и др.
fL zCN
4 2)—си—c=n ‘
1 у/ \н
Вг
I II
Поскольку в зависимости от природы заместителей в молекуле атомы галогенов действуют по-разному, неизбежно имеются определенные градации в физиологической активности соединений.
По интенсивности лакримогенного действия зам-естители располагаются в последовательности F < С1 < Вг < I. Это значит, что бромпропаион III вызывает более сильное слезоотделение, чем хлорпропанон IV (в скобках под формулами приведены величины пороговых концентраций):
BrCH2- Cf
\снэ
-
ClCHr-CT
\СН3
III IV
(0.001 мгМ)
(0,018 жг/л)
По общей ядовитости эти соединения располагаются в обратной последовательности. По сравнению с другими токсичными веществами она мала и как боевые ОВ эти вещества не имеют или имеют лишь второстепенное значение.
Дальнейшей предпосылкой для лакримогенного действия гало-геизамещенных органических соединений является положение атома галогена в молекуле. р-Замещенные соединения активнее а-за-мещенпых. В случае ароматических соединений . лакриматоры встречаются лишь среди соединений с галогенами в боковой цепи, например, w-хлорацетофенои V или бензилбромид VI;
v
VI
3*
67
---соединения,—замещенные—в—ядре ^хлириензил—и—п—д./, неактивны. С увеличением числа атомов галогенов в молекуле слезоточивое действие уменьшается, а другие физиологические эффекты усиливаются. Так, 1,3-дибромпропанон VII склонен к сильному кожио-раздражающему (крапивному) действию. Сильное слезоточивое ОВ — монохлорметиловый эфир хлоругольной кислоты VIII в результате дальнейшего замещения водородов атома хлора становится удушающим ОВ (см. раздел 4.1.1,1).
Z t Z
ВгСН2—С1СН2О—с/ ^СН2Вг \с!
VII VIII
Каково бы ни было их химическое строение, все слезоточивые ОВ действуют на чувствительные нервные окончания слизистой оболочки глаз. Их физиологическое действие сводится к ингибированию определенных ферментов, содержащих сульфгидрильные (меркапто-) группы, например таких, как уреаза, гексокииаза и папаин.
Первыми симптомами отравления являются жжение и резь в глазах, слезотечение, ощущение инородного тела, смыкание век; следствиями более высоких концентраций — временная слепота и конъюнктивиты. Эти симптомы быстро проходят. Обычно оии длятся ие более 20 ч. Поражения глаз являются доброкачественными и сравнительно быстро заживают. Необычайно высокие концентрации и попадание в глаза жидких слезоточивых ОВ могут привести к постоянной слепоте.
Высокие концентрации при вдыхании раздражают верхние дыхательные органы или весь дыхательный тракт. Картина отравления подобна отравлению фосгеном. В зависимости от концентрации-и времени ингаляции отек легких выражен более или меиее сильно..
Нейв1 описал случай отравления хлорацетофеноном со смертельным исходом. В подвале (около 34 л3) юноша 24-х лет получил тяжелое, отравление от разрыва двух гранат с хлорацетофено-, ном (по 2,7 г ОВ). Через 20 мин он был найден без сознания, а через 7,5 ч отравленный, несмотря на усилия врачей, скончался от отека легких и остановки кровообращения.
При соприкосновении с поверхностью кожи жидких или парообразных слезоточивых ОВ в высоких концентрациях частично развивается эритема. Жжение и зуд кожи являются первыми признаками, которые наступают сразу после контакта. Особенно чувствительна к парам потная или разгоряченная кожа.
Раздражения кожи являются доброкачественными и обычно не требуют серьезного лечения.
Хлорацетофенон, кроме того, проявляет хотя и слабое, ио неприятное крапивное действие.
68
3.1.2.1. Общие свойства. Слезоточивыми ОВ могут быть как алифатические, так и ароматические галогензамещенные кетоны. С военной точки зрения особый интерес представляют ароматические кетоны. По сравнению с алифатическими кетоиами они имеют лучшие физико-химические свойства. Они химически устойчивы, обладают более интенсивным физиологическим действием и легче получаются,
•Галогенированные алифатические кетоны являются жидкостями. По своему химическому поведению они аналогичны негалоге-нированиы’м кетонам, но более склонны к полимеризации. Без добавки стабилизаторов эти кетоны при хранении разлагаются. Так как оии реагируют со сталью, их можно хранить в свинцовых, стеклянных или керамических контейнерах.
Ароматические соединения представляют собой твердые, кри-* сталлические вещества, которые можно испарить без разложения. Их можно неограниченно долго хранить без добавки стабилизаторов даже в примитивных резервуарах, например, в деревянных или жестяных бочках.
Среди алифатических соединений наиболее известны бромпро-панон и 1-бромбутанон-2 (III, IX). Важнейшими представителями ароматического ряда являются хлорацетофенон и бромацетофенон (V, X)
ВгСНа—СО—СН2—СНз \^/—СО—СН2Вг
IX X
Их можно получить прямым галогенированием соответствующих кетонов. Бром- и хлорацетофенон проще и быстрее получать по реакции ФрЦделя— Крафтса.
З.1.2.2. Галогенированные алифатические кетоиы [бромпропа-нои (бромацетон) BrCIJ2COCH3 и 1-бромбутанон-2 (бромметил-этилкетон) ВгСН2СОСН2СН3]. В первую мировую войну они применялись в снарядах и ручных гранатах, отчасти в смеси с другими галогенированными кетонами, получающимися в качестве побочных продуктов («Ве»-газ, «В»-газ, мартониты, гомомартоииты, то-ииты и др.).
Методы получения. Бромкетоны в лабораторном и промышленном масштабе получают галогенированием соответствующих алканонов.
При действии жидкого брома на ацетон получается бромпропа-ион (реакция I), а из бутаноиа-2 — смесь 1-бром- и 3-бромбутано-нов (реакция 2):
СН3СОСНз + Вг3 —► ВгСН2СОСН3 + НВг (1)
+ Вг2
,СН3СОСН2СН3 ---> ВгСНаСОСНгСН3 + СНзСОСНВгСНз (2),
69
сразу окисляют в реакционной смеси добавкой хлоратов в бром и вновь используют. Вместо брома иногда применяют бромистый натрий и концентрированную серную кислоту. Образующийся при этом бромистый водород затем превращают в бром действием эквивалентного количества хлората натрия;
ЗСНзСОСНз + 3NaBr + 3H2SO4 + NaC103 —>
—► ЗВгСН2СОСН3+3NaHSO4 +ЗН2О+NaCl
Физические и химические свойства. Чистые бром-пропаион и 1-бромбутаион-2 представляют собой бесцветные жидкости, а технические продукты — желто-коричиевые жидкости с резким запахом. При нагревании до температуры кипения оии разлагаются. К детонации малоустойчивы.
Оба соединения склонны к легкой полимеризации с отщеплением бромистого водорода. Физиологическая активность при этом теряется. В качестве стабилизаторов пригодны вещества, связывающие кислоту, ио ие образующие щелочную среду; чаще всего применялась для этой цели окись магния. Стабилизованное вещество может храниться в соответствующих условиях хранения (при невысокой температуре, в темноте) более 20 лет.
Оба эти вещества обладают следующими физическими свойствами (в круглых скобках приведено давление в мм рт. ст.):
Т. кип., °с......................
Т. пл„ °C .......................
Давление пара при 20° С, мм рт. ст. Максимальная концентрация (20 °C), мг/л ............................
Относит, плотность пара............................
жидкости ....................
Вромпропанон ЬВ ром б у та нон -2
31,4 (8) —
136 (760) 145-145 (760)
-54 —
9 6,5
75 52
4,75 5,2
1,634 1,43
В воде они практически нерастворимы, в органических растворителях (спирты, ацетон) —легко растворимы.
Для дегазации этих ОВ пригодны водные или спиртовые растворы сульфида натрия:
+ NaaS
2R—С—СН2Вг ———> R— С— СН2—S—СН2—С— R
II —2NaBr || * || '
О 0 0
Образующийся тиоэфир неядовит.
Химические свойства их аналогичны свойствам незамещенных алифатических кетонов. Они образуют кетоксимы (реакция 1), с бисульфитом натрия дают кристаллические продукты при^оедиие-
70
ния (реакция 2), с цианистым водородом — жидкие циангидрины (реакция 3):
ВгСН2/ /ОН
RZ ^SOgNa
(2)jNaHSO3
nh2oh hcn BrCH2x /ОН
H2O + ВгСН2—C—R <— ------ ВгСН2—C—R ------>-
II (I) 1 (3) - r/ \cn
NOH О
Атомы брома могут быть легко замещены на другие атомы илн группы.
При действии спиртового раствора щелочи образуются соответствующие оксикетоны
+ но-
ВгСН2—С—R ------> HOCHr-С—R
А -вг А
а при обработке иодидами щелочных металлов образуются физио* логически активные иодпронзводные:
4-1
ВгСН2—С—R -----> ICH2—С—R
А -вг' А
3.1.2.3. Галогенированные ароматические кетоны. Хлорацето* фенон.
0
C6HS—С—СН2С1
Синонимы: «-хлорацетофенон, а-хлорацетофенон, фенилхлорметилкетои, фенацилхлорид.
Военные обозначения: нем. — О-Saiz; англ. — САР; амер. — CN.
Хлорацетофенон был исследован во время первой мировой войны в США, где были также созданы технологические предпосылки для его промышленного производства. До его боевого применения в первую мировую войну дело не дошло, так как промышленные предприятия в конце войны еще только строились.
•Во время второй мировой войны хлор- и бромацетофеиои были заготовлены в больших количествах многими воюющими государствами. Наряду с возможным военным применением оба соединения применялись и применяются в капиталистических странах в качестве полицейского химического оружия.
Методы получения. В промышленности и в лаборатории (й-хлорацетофеноя можно получать двумя способами: хлорированием
71
ацетофенона и по реакции Фриделя — Крафтса из бензола и хлорацетилхлорида.
По первому способу ацетофенон хлорируется при непосредственном взаимодействии с хлором в уксуснокислом растворе:
с6н5—с—сн3
+ Cig
—> СеН5—С—СН2С1
-НС1 II
О
По новому способу2 хлорирование можно проводить хлорокисью селена SeOCU в бензольном растворе.
Исходный ацетофенон получают взаимодействием бензола с ацетнлхлорндом в присутствии катализатора Фриделя — Крдфтса.
По второму способу бензол вводят в реакцию с хлорацетил-хлоридом в присутствии хлористого алюминия:
О О
II (А1С1з) II
ед + ci—с—ch2ci ——> ед—с—сн2с1
—*-НС 1
Этот способ осуществлен в промышленном масштабе.
С точки зрения электронной теории механизм реакции заключается в следующем:
Хлорацетнлхлорид получают методами, описанными в органической химии.
Лабораторный синтез. В двугордой колбе емкостью-1 л с мешалкой и обратным холодильником смешивают 100 мл сероуглерода с 0,3 моль хлорацетилхлорида и 0,4 моль бензола и постепенно при перемешивании или встряхивании прибавляют 0,25 моль тонкоиз мельченного хлористого алюминия. После присоединения обратного холодильника колбу помещают в водяную баню при 50—55 °C, перемешивают около 1 ч реакционную смесь и оставляют при той же температуре еще на 1—2 ч до прекращения выделения хлористого водорода. Обратный холодильник заменяют на нисходящий и удаляют сероуглерод откачиванием при пониженном давлении (взрывоопасно!).
Для разложения комплекса кетона с хлористым алюминием еще теплый раствор осторожно выливают на «250 см3 льда или в воду со льдом. Операция проводится в колбе емкостью 2 л. Осаждающуюся иногда окись алюминия растворяют в небольшом количестве концентрированной НС1, органический слой отделяют при помощи делительной воронки и экстрагируют 2—3 раза эфиром.
Объединенные вытяжки повторно промывают водой, затем 2%-ным раствором NaOH и снова водой. Эфирный экстракт сушат 8—10 ч над СаС12 или К2СО3. Затем осторожно отгоняют эфир на водяной бане, при этом целесообразно использовать маленькие перегонные колбы (200 мл), время от времени доливая перегоняемый раствор из капельной воронки. Наконец остаток, содержащий хлорацетофенон, перегоняют, нагревая небольшим голым пламенем; предгон до 240 гС отбрасывают, хлорацетофенон отгоняется при 245—250 °C. При длительном стоянии из дистиллята выпадает кристаллический продукт, который перекристаллизовывают из разбавленного спирта.
Физические свойства. Хлорацетофенон представляет собой бесцветное кристаллическое вещество, технический продукт окращен в серый цвет. Физические его свойства следующие:
Т. кип., °C................... 139—141 (при 14 мм рт. ст.)
245—247 (при 760 мм рт. ст.)
Т. пл., °C............................ 58-59
Давление пара при 20 °C, мм рт. ст. 0,013
Максимальная концентрация (20 °C), мг/л ................................ 0,105
Относит, плотность, пара .........................‘ 5,3
твердого вещества.................. 1,321
Теплота испарения, кал/моль ... 89
Хотя давление пара хлорацетофенона сравнительно иизкое, его летучесть при 20 °C составляет 0,11 мг/л, и если для фактора ослабления летучести на местности принять максимальное значение 10“2, то создаваемая концентрация паров достаточна, чтобы сделать зараженную хлорацетофеноном местность непреодолимой без противогаза. Нижний предел раздражения для хлорацетофенона составляет 5 • 10-4 мг/л.
Эффективность паров хлорацетофенона зависит от внешней температуры. Ниже приведены данные о зависимости давления пара хлорацетофенона от температуры и соответствующие величины летучести ОВ:
Температура, оС Давление пара, мм рт. ст. Летучесть, мг!л Температура, °C Давление пара, мм рт. ст. Летучесть, мг[л
—20 1,7. 10“3 20 1,3- 10~2 i,i •ю-;
-10 5,6-10- 5-Ю’ 30 3,1 • 10~2 2,5-10"
0 1,7-Ю"3 1,5-Ю“2 40 7,2- 10-а 5,7-10 1
10 4,8- 10~3 4,2’ 10 50 1,6’10 1 1,2
Как видно из приведенных величин, летучесть хлорацетофенона ниже 10 °C слишком мала, чтобы можно было создать эффективные концентрации. Однако в холодное время года хлорацетофенон сможет быть применим в виде аэрозоля. Хлорацетофенон достаточно термостабилен н поэтому может применяться не только в мииах н ручных гранатах, но и в дымовых шашках. Преимущественное боевое применение хлорацетофенона может быть в виде аэрозолей.
Устойчивость хлорацетофенона к разложению при температуре кипения позволяет заполнять гранаты непосредственно жидким продуктом, смешивать его с дымообразователями и даже со
73
взрывчаткой (например, 2,4,6-трннитротолуолом), особенно если их плотности примерно одинаковы.
Хлорацетофенон практически нерастворим в воде (0,1%), но хорошо растворяется в обычных органических растворителях — хлоралканах, CS2, алифатических спиртах, эфирах, кетоиах и в бензоле; в некоторых ОВ, например, иприте, фосгене, хлорпикрине и хлорциаие, ои растворяется в определенных соотношениях; слабо растворяется в SnCh и T1CI4.
Растворы хлорацетофенона в зависимости от плотности заражения, местных и метеорологических условий могут быть стойкими в течение часов и дней. Раствор хлорацетофенона в хлорпикрине в смеси с хлороформом в летнее время в лесу стоек в течение 2 ч, а зимой даже до недели; на открытой местности летом примерно 1 ч, а зимой 6 ч.
Химические свойства. По своему строению хлорацетофенон является смешанным жирноароматическим кетоном, н как таковой сравнительно стабилен и обладает малой реакционной способностью. Он вступает в реакции по карбонильной группе, свойственные алифатическим кетонам, например, с гидроксилами-иом и гидразином, однако ие образует бисульфнтного производного.
Термическая стабильность хлорацетофенона высокая, ои устойчив к детонации, разлагается лишь при длительном нагревании (15 мин); при 300°C — на 1,5%, при 600 °C — на 9% и при 900° С —-примерно иа 30%.
Гидролиз. Даже при кипячении с водой хлорацетофенон не гид-' ролизуется или гидролизуется лишь незначительно.
Находящийся на поверхности почвы под снежным покровом, а также впитавшийся в почву хлорацетофенон не теряет своих свойств и после таяния снега при соответствующей благоприятной температуре вновь физиологически эффективен.
Скорость гидролиза увеличивается при действии щелочи, но при обычной температуре столь мала, что эта реакция не имеет значения для дегазации. Количественное превращение достигается лишь при кипячении с раствором щелочи, особенно в тех случаях, когда применяются спиртовые растворы. В качестве продукта гидролиза образуется оксиметилфеиилкетон (ок си ацетофенон), кристаллическое вещество, плавящееся при 86°C:
+ NaOH
СвН5—С—СН2С1 ---—С6Н6—С—СН2ОН
II “Nacl II
о о
Другие реакции атома хлора. Атом хлора в молекуле хлорацетофенона можно заместить и в результате других реакций. При действии иодидов щелочных металлов в спиртоводиых растворах образуется иодацетофенон — игольчатые кристаллы:
+1
СеН5—С—СН2С1 -—> С6Н5—С—СН21
А А
74
Подобно алифатическим галогенированным кетонам, реакция с сульфидом натрия в спиртовых или спиртоводных растворах и в случае хлорацетофенона оказывается наиболее пригодной для возможной дегазации:
4-NaaS
2C6HS—С—СН2С1 /CeH6-C-CH2\S
|| I н 1
о к о А
Образующийся бис-фенациловый тноэфнр (т. пл. 74 °C) не проявляет физиологической активности.
При кипячении хлорацетофенона с тиосульфатом натрия получается натриевая соль фенацилтиосерной кислоты
C§Hs—С—СН2—S2O3Na.
О
Реакции конденсации. Как кетон хлорацетофенон реагирует также с гидроксиламииом. При взаимодействии с гидрохлоридом гидроксиламнна в водном метиловом спирте при комнатной температуре образуется оксим ш-хлорацетофенона (хлорметнлфенил-кетокснм):
+ nh2oh
СвН5—С—СН2С1 •------> С6Н5—С—СНгС1
II - н2о |[
О NOH
Это бесцветное кристаллическое соединение (т. пл. 88,5—89 °C), которое обладает сильным лакримогенным действием и, подобно галогенированным оксимам (см. раздел 5.4), вызывает продолжительное раздражение кожи (крапивное действие). В качестве возможного ОВ это соединение в литературе до сих пор почти не упоминалось.
С семикарбазидом хлорацетофенон образует соответствующий семикарбазон.
Окисление. С кислородом воздуха хлорацетофенон не реагирует. Сильные окислители, например, хромовая кислота, перманганат калия, двуокись селена и гипохлориты в соответствующих растворителях (бензол и диоксан) окисляют хлорацетофенон до бензойной кислоты:
+ зо /ОН
Свн5—С—СН2С1 -------> С6Н5 с
|| -С02; -HCI Чо
О
Наряду с другими в качестве побочных продуктов при этом образуются фенилуксусиая кислота и фенилглиоксаль.
Азотная кислота действует иа хлорацетофенон отчасти как окислитель, отчасти как нитрующий агент.
75
Хлорирование. При соответствующих условиях возможно дальнейшее хлорирование ацетофенона как в боковую цепь, так и в ядро. Такие хлорпроизводные обладают ослабленным слезоточивым действием, но отличаются сильным кожно-нарывным действием и потому неоднократно принимались в расчет в качестве ОВ.
При использовании хлорного железа как катализатора хлорирование приводит к образованию дихлорацетофенона XI (т. пл. 20—21,5 °C) и (0,3,4-трихлорацетофеноиа XII; трихлорацетофенон XIII получают при повышенной температуре и при УФ-облучении (т. кип. 145°C при 25 мм рт. ст.)
\:h2ci
с6н5—с—СНС12
С1
XII
с6н5—с—ecu
XIII
3.1.2.4. Бром- и фтор ацетофенон. Бромацетофенон образует бесцветные ромбические кристаллы; т. пл. 50—51 °C, т. кип. 260 °C при 760 мм рт. ст. (разл.), 131—135°C при 12 мм рт: ст. Технический продукт сильно окрашен в желтый цвет.
Соединение нерастворимо в воде, ие гидролизуется даже при нагревании до кипения. По своим химическим свойствам и методам получения бромацетофенон совершенно аналогичен хлорацетофенону.
По аналогии с хлорацетофеноном в лаборатории его можно получить из ацетофеиоиа и брома 3.
Раствор 0.5 моль ацетофенона в 100 мл ледяной уксусной кислоты в трех-горлой колбе емкостью 0,5 л, снабженной мешалкой, капельной воронкой и хлор-кальциевой трубкой, обрабатывают несколькими каплями раствора бромистого водорода в уксусной кислоте. Добавляют 0,5 моль брома, следя за тем, чтобы температура реакции не поднималась выше 20 °C. По окончании реакции выливают реакционную смесь в воду со льдом. Бромацетофенон отделяется в виде желтого масла и в виде кристаллов. После полной кристаллизации жидкость удаляют и вещество перекристаллизовывают из сцирта.
и-Фторацетофенон (т. кип. 98 °C при 8 мм рт. ст.) и его ди- и трихлорпроизводные можно получить реакцией Фриделя — Крафтса из бензола и соответствующего фторацетилхлорнда 4:
ю-Фторацетофенон обладает лакрнмогенным действием.
76
3.1.3. Азотсодержащие слезоточивые ОВ
Средн ОВ встречаются н некоторые азотсодержащие соединения — ннтропроизводные, амины, оксимы, нитрилы. К последней группе относится не только снльно токсичная синильная кислота, но н ряд соединений с лакрнмогенным действием.
Токсичность соединений, содержащих ннтрнльные группы, в большинстве случаев снижается в присутствии других атомов н групп, например, уже при введении второй ннтрнльной группы (дн-циан). За счет введения атомов галогенов уменьшается общая токсичность ннтрйлов, но приобретается лакрнмогенное действие, например, каг?у галогенцнанов. То же относится н к более высокомолекулярным соединениям. Акрилонитрил I ядовит, но не является слезоточивым веществом, тогда как галогенакрилонитрилы II — слезоточивые ОВ. То же относится к бензнлцнаннду III н бром-бензнлцианиду IV:
С И,---С И—C-N
HalCH=CH—C=N •
II
С6Н5—C1L--C-N ш
ZBr
Сс1-15—С1-/ ^C=N
IV
I
С военйо-хнмнческой точки зрения, пожалуй, наиболее важным является бромбензнлцнаннд IV, который обладает самым сильным раздражающим действием из всех производных бензола, содержащих в боковой цепи атомы галогенов (ср. табл. 6, стр. 80). Это соединение неоднократно применялось в качестве ОВ во время первой мировой войны.
Другими азотсодержащими слезоточивыми ОВ являются: феннлкарбнламннхлорнд V, трнхлорннтрометан (хлорпикрин) VI (раздел 4.2.3), галогенированные оксимы (раздел 5.4), транс-$-бромакрнлоннтрнл VII.
C6HsN=CC12
СС1з—NO2
VI
Их ZCN
хс-с^
Вг^ \н
VII
З.1.З.1. Бромбеизилцианид
С6Н5—СНВг (In
Военные обозначения: амер. — СА (в первую мировую войну), ВВС (современное); франц.«—Canute (в первую мировую войну),
77
Перед концом первой мировой войны бромбензнлцнаннд был применен французской н американской стороной. Это эффективное слезоточивое ОВ, обладающее большой стойкостью н высокой стабильностью по отношению к атмосферным воздействиям. Еще во время второй мировой войны считалось, что его можно будет использовать для заражения местности н маскировки нападения кожно-нарывными ОВ, Для современного боя его значение проблематично.
Получение. Бромбензнлцнаннд получается по схеме:
+ Вгг
С6Н5—CHSCN ----> С6Н5—СНВг
-НВг ]
CN
Физические .свойства. В инетом виде бромбензилцианнд существует в виде беловато-желтых (частично бледно-розовых) кристаллов, Технический продукт представляет собой коричневую маслянистую жидкость, Из-за своей малой летучестН бромбензнл-цианнд обладает длительной стойкостью: летучесть при 20 °C равна 0,13 мг!л^ а соответствующее давление пара составляет 1,2 • 10-2 мм рт. ст. Вследствие низкой термнческой стабильности его применение ограничено; он может быть использован, например, в снарядах с малым зарядом взрывчатки^
При нормальном давлении .бромбензилцианнд перегоняется в интервале 242—247°C (разл.); т. пл. 25,4°C; технический бромбензнлцнаннд застывает при 15—22 °C.
Малая термическая стабильность не позволяет употреблять его в виде аэрозоля, так как для создания аэрозоля потребовались бы большие заряды взрывчатки. Использование в дымовых шашках полностью исключается. Бромбензилцианнд применим в виде технического продукта (жидкость) нлн в виде раствора в других ОВ. Наряду с другими была предложена смесь бромбензнлцнанида с хлорпикрином. Добавка 20% хлорпнкрнна снижает температуру вастывання на 1 °C.
Кроме того, бромбензилцианнд растворяется в иприте, фосгене, дифосгене и других ОВ, а также хорошо растворим в органических растворителях — горячих спиртах, бензоле, эфире, галогеналканах, алифатических кетонах, уксусной кислоте; в воде практически не-
растворим.
Ниже приведены некоторые физические константы бромбензил-
цианнда при разных температурах или давлениях:
Т, кип., °C
при 12 мм рт, ст..............
при 15 мм рт. ст..............
при 760 мм рт. ст.............
Т. пл., °C........................
Давление пара, мм рт. ст.
при 0 °C ...................?
при 10 °C.....................
при 20 °C.....................
при 30 °C.....................
132-134
137-139
242—247 (разл.)
25,4
1.9-10’
З-ПГ?
1,2-10 2
2,8-10 2
78
Максимальная концентрация, мгЬг
при 20 °C...................... 0,13
при 30 °C...................... 0,23
Относит. ПЛОТНОСТЬ пара................................... 6,6
твердого вещества d^ .... 1,516
Теплота испарения» кал!моль . . . 58,7
Химические свойства. Бромбензилцианид химически очень устойчивое соединение. Термически он очень не стабнлеи — выше 150°С он уже заметно разлагается; при 200°С образуется бромистый водород, 1,2-дифенил-1,2-дицианэтилеи (дицианстиль-бен) и смолообразный продукт:
/Вг C6Hg4 zc6h5
2CeHs—Clf —> /С=С\ +2HBf
^CN NCZ \CN
Бромбензилцианид неустойчив к детонация. -К его недостаткам относится и то, что ои разрушает все металлы, кроме свинца. Поэтому его хранят в стеклянных, эмалированных, фарфоровых или пластмассовых сосудах или в металлических емкостях с соответствующими защитными покрытиями, например, лаками из хлоркаучуков.
Гидролиз. Водой при обычной температуре бромбензилцианид гидролизуется лишь незначительно. Медленный гидролиз начинается только прн нагревании. При кипячении в щелочных растворах, например, в 20%-ном растворе NaOH, возможен количественный гидролиз. Спиртовые растворы щелочей или карбонатов щелочных металлов реагируют с бромбензилцианидом уже на холоду с образованием солей дифеиилмалеиновой (ццс-дифенилэтиленди-карбоновой) кислоты и выделением аммиака и бромистого водо-
рода:
у-Вг 2С0Н6—С1\
+ <но- с«н'\с=с/с-н«
-2Вг-: -2NH3 -00С/ \СОО“
При подкислении реакционной смеси выпадает кристаллический ангидрид дифеиилмалеиновой кислоты (т. пл. 150°С):
с6н5—с--соо с6н5—с-cf
1 |j /О
C6HS—С—соо“ с6н5—с—с/
Другие реакции. При действии спиртового раствора сульфида натрия от бромбензилцианида отщепляется бромид и образуется кристаллическое вещество, представляющее собой бис-фенилацетонитриловый тиоэфир (т. пл. 150—152°C), который
79
Таблица 6. Физиологические характеристики некоторых слезоточивых О В и других раздражающих слизистые оболочки соединений
Название Порог раздра" жения. Предел переносимости, мг!л Примечания
Акролеин (пропеналь-2) 7- 10'3 7. 10“2 —
Бедзилбромид 3,5 • 10“3 5* 10“2 —
Бензилхлорид 1,6. нГ2 8,5- 10“2 При 1 мг/л опасен для органов дыхания
Бензилиодид 2* Ю'3 2,6- 10“г —
Бромацетофенон 3,1 . ю“4 Уже при низких концентрациях вызывает появление болезненных волдырей
Бромбензилцианнд 1,5 • 10’’ 5-Ю"3 >Ю-1 л/г/д действуют удушающе
1-Бромбутанон-2 1,1 • ю~3 1,1 • ю"а —
Бромпропанон 1,5 - 10-3 ю"2 При 5,6 * 10“1 л/а/л вызывает тяжелое поражение органов дыхания
Бромуксусная кислота, этиловый эфир 5- Ю"3 4- 10'2 Токсичен
Бромциан 5 • I0"3 4- 10'2 Сильно токсичен
Дибромдиметиловый эфир 2- 10"2 5-Ю"2 —
Дихлордиметиловый эфир 1,4 - 10~2 4- 10'2 —
Иодуксусная кислота, этиловый эфир 1,4. 1СГ3 3,3- ю“2 При 1,7-10"2 мг!л через 1—2 мин тяжелое поражение органов дыхания
Ксилилбромид 3,8. 10~3 5- 10'2 Удушающий
о-Нитробензилхлорид 1,8. 10~3 * —
Трибромнитрометан 3 ю'2 — Удушающий
Трихлорнитрометан 1,4 - КГ2 5 • Ю'2 То же
Фенилкарбиламинхлорид з. 10-3 3. 10'? При 8 • 10~’ мг!л через 1 —2 мин поражает органы дыхания
Хлорацетофенон з. I0-4 2- Ю"3 При 2- 1(Г3 мг!л раздражает кожу лица
Хлорпропанон 1,8 - Ю 2 1,1. 10"1
Хлорциан <2,5- Ю-3 <5- Ю~2 Сильно токсичен
80
при нагревании разлагастоя на цис-1,2-дифепйЛДИЦианэтилен—и сероводород:
уВг 2Сон5—сн(
\CN
+ NaaS
—2NaBr*
С6н5—CH—CN
S --->
| -H2S
C6H5—CH—CN
c6h5-c-cn
СЙН5—C—CN
Эту реакцию можно использовать для дегазации. Водные растворы сульфида натрия реагируют слишком медленно и менее пригодны в качестве дегазирующих растворов.
По отношению к окислителям бромбензилцианид устойчив. Лишь такие сильные окислители, как перманганат калия, перекись водорода и другие перекиси заметно реагируют с ним при продолжительном воздействии.
Галогениды тяжелых металлов, например хлорид железа (II) нли хлористый цинк, способствуют каталитическому разложению бромбензилцианида до неидентифицируемых смолообразиых продуктов. Такое каталитическое разложение является одной из причин неустойчивости бромбензилцианида при хранении в металлической таре.
3.1 *4. Физиологические свойства слезоточивых ОВ
Слезоточивые ОВ практически не обладают токсическим действием, имеющим тактическое значение. Исключением являются соединения, упоминающиеся среди ОВ других групп, например, галогенцианы, хлорпикрин, галогенированные оксимы. При достижении порога раздражения они вызывают более или менее сильное слезоотделение. Некоторые вещества сверх того обладают, даже уже в малых концентрациях, определенным кожно-раздражающим действием, которое, хотя и неприятно воспринимается, все же следует считать вторичным. С военной точки зрения представляют .интерес соединения, которые, в минимальных, концентрациях затрудняют противнику выполнение боевой задачи и принуждают его надеть противогаз.
В табл. 6 приведены физиологические данные для слезоточивых ОВ, а также для веществ, больше не имеющих значения как ОВ, но с которыми часто приходится иметь дело на химических заводах и в лабораториях.
3.2 . РАЗДРАЖАЮЩИЕ носоглотку ов
3.2.1. Химическое строение и биологическая активность
Из ОВ, раздражающих носоглотку, военный интерес представляют мышьяк- и свинецорганические соединения. Хотя подобный физиологический эффект можно ожидать (и он известен) также и для
81
других металлоорганических соединений, как возможные ОВ оии почти не упоминаются в специальной литературе.
В английских руководствах раздражающие носоглотку ОВ объединяются в группу так называемых стернитов.
Из мышьякорганических соединений, в которых атом мышьяка может быть трех- или пятивалентным, большой физиологической активностью обладают соединения трехвалентного мышьяка. Их возможному использованию в качестве ОВ также уделяется большое внимание.
Привлекает внимание, что во многих известных мышьякоргаии-ческнх ОВ атом мышьяка связан с одним или двумя атомами галогена или нитрильными группами. Для того чтобы соединения обладали физиологической активностью в военном смысле, нужно, чтобы две валентности атома мышьяка были насыщены двумя одинаковыми атомами или группами, а третья валентность — другим атомом или иной группой:
х\ ;as—z у/
где X может быть равно Y, Z =# X, Y..
Некоторые органические соединения мышьяка обладают кожно-нарывными свойствами. Так, кожио-иарывиое действие проявляют соединения типа I и IT.
R—As( ;As—X
Ху R/
i п
где X и Y—атомы галогена (а для соединения II —также CN); R— алкил или арил. Например:
CH3AsC12 (C6H5)3As (C8H5)2AsC1 C8H5AsC12 (C8H5)2AsCN Ш IV V VI VII
Метилдихлорарснн III — раздражающее носоглотку OB; трифе-инларсин IV не представляет военного интереса, ои физиологически мало активен; дифенилхлорарсин V—-сильно раздражающее носоглотку ОВ; фенилдихлорарсин VI — обладает кожно-нарывным действием, а также раздражает носоглотку.
Впрочем здесь нет строгого правила, как это следует из свойств соединения VI; вещества того и другого типа могут проявлять разное физиологическое действие.
Ароматические арсины оказывают более сильное раздражающее действие на носоглотку, чем алифатические, независимо от того, к какому типу они принадлежат. Например, приведенные 82
выше отравляющие вещества имеют следующие пределы переноси-мости (в мг!л):
CH3AsC12 . . * . 2. 10~2
C8HbAsC12 . . . 1,6-ю';
(CeH6)2AsCl . . . 10~3
Зависимости биологической активности мышьякоргаиических соединений типа RAsXY от их химического строения см. раздел 5.3.1.1.
Влияние различных атомов галогенов на физиологическую активность мышьякоргаиических соединений противоположно тому, какое оии оказывают на слезоточивые ОВ, т, е. активность падает в последовательности I < Вг < С1. Присутствие CN-групп усиливает раздражающее действие и токсичность, например порог раздражения для дифенилхлорарсина V равен 10'4 жа/л, а для дифе-нилцианарсина VII — 10-5 мг]л.
Если вместо фенильного остатка в соединение вступает высший арил, то физиологическое действие ослабляется. Таким об-разрм, число физиологически активных мышьякоргаиических соединений ограничено.
К группе ОВ, раздражающих носоглотку, относятся только ароматические арсины типа КгАзХ, важнейшими представителями которых являются дифенилхлор- и дифеиилциаиарсин, а также дн-гидрофенарсазиихлорид (10-хлор-9,10-дигидрофеиарсазни) VIII:
VIII
Относящиеся к группе стернитов свинецоргаиические соединения отвечают соединениям типа IX:
R\
R —Pb—X
R"Z
/х
Они представляют собой соединения триалкилсвинца, у кото-рых'четвертая валентность атома свинца связана с остатком неорганической или органической кислоты. В настоящее время сообщения о таких соединениях ограничиваются лишь данными об нх раздражающем действии, поэтому пока ничего нельзя сказать о токсичности этих соединений по сравнению с сильно ядовитыми соединениями тетраалкилсвннца, как например
РЬ(С2н6)* РЬ(СН2СН2С1)4
X XI
83
t
которые действуют, как нервно-паралитические яды.. Пользуясь аналогиями, можно считать, что производные триалкилсвинца должны быть меиее токсичны. Это предположение было подтверждено опытами с соединениями диалкилсвинца, которые оказались практически неактивными — их раздражающее действие в параллельных опытах равнялось нулю.
Физиологическая активность солей триалкилсвинца зависит от природы алкильных или арильных радикалов в молекуле. Как видно из табл. 8, соединения три-н-пропилсвиица XIII являются самыми активными, в то время как этильные и бутильные производные XII и XIV действуют слабее.
(С2НБ)3РЬ—X (C3H7)3Pb—X (С4Н9)зРЬ—X
XII XIII XIV
Аналогичные метильные производные обладают лишь небольшим раздражающим действием.
В вышеупомянутых соединениях группа X представляет собой (в порядке убывающей активности): метансульфамид, бензолсульфамид, н-толуолсульфамнд, о-сульфобензимид, сульфанилид, ацетат, хлорацетат, н-бутират, о-толуолсульфоиат, п-толуолсульфоиат, изовалерат, акрилат, бромид, хлорид и др.
Активность соответствующих алкилсвиицовых солей уменьшается примерно в той же последовательности. Соли органических кислот действуют сильнее, чем соли неорганических кислот. Наиболее активным соединением является метансульфамид три-н-пропилсвинца XV, который уже в разбавлении 1:2,5-107 абсолютно непереносим. В разбавлении 1: 107 бензосульфамидные XVI и п-толуолсульфамидные XVII производные становятся непереносимыми при экспозиции 40 сек.
(C3H7)3Pb—NH—SO2CH3 R3Pb—NH—SO2C6HB R3Pb— NH—SO2CeH4CH3
XV XVI XVII
Наиболее активные соли триалкилсвинца по своему действию примерно сопоставимы с действием дифенилхлорарсииа V, а иногда даже превосходят его.
В небольших количествах ОВ этой группы раздражают слизистую оболочку носа, глотки и глаз. Биохимическое их действие выяснено пока недостаточно.
Предполагают, что мышьякорганические соединения, с одной стороны, влияют на определенные ферментативные процессы, с другой — вызывают поражение клеток капилляров и нервных клеток. Частички аэрозоля, на центрах слизистой оболочки создают импульсы раздражения чувствительной периферической нервной системы. Раздражающее действие ие ограничивается локальными участками пораженной слизистой оболочки, а распространяется в область так называемых придаточных пазух носа. После удаления
84
ов на слизистой оболочке сохраняется продолжительное последействие, что объясняют поражением клеток чувствительных нервов.
Миллер6 на основе экспериментов с дифенилхлорарсином установил, что мышьякорганические соединения этого типа после многочасового воздействия уже при пороговом разбавлении 1 :109 вызывают увеличение числа видимых митозов в эпителиальной ткани, и что прн делении клеток наблюдается аномальное расположение хромосом. Эти явления дают право считать такие соединения сильными митотическими ядами.
При действии мышьякоргаиических соединений симптомы поражения наступают позже, чем в случае слезоточивых ОВ. Скрытый период составляет максимум 20—30 мин. Он зависит от концентрации и продолжительности воздействия ОВ. Обычно при концентрации 10 3 мг/л первые симптомы наблюдаются уже через 0,5—5 .мин, максимум действия наступает через несколько минут и спадает через 4—6 ч.
Действие заключается: в раздражении верхних дыхательных путей, поражении верхних дыхательных путей, раздражении глаз, воздействии на центральную нервную систему и в поражении поверхности кожи.
В результате раздражения полости носоглотки появляются позывы к кашлю, неудержимый приступ чихания, усиление выделений из ибса и слюнных желез. Одновременно наступает очень сильное лакримогенное действие, обильное слезотечение, а при высоких коицертрациях также продолжительные конъюнктивиты.
Такне сопутствующие явления, как тошнота, позыв к рвоте, головные боли, затем боли в челюстях и зубах, ощущения давления в ушах, боли в грудине, удушье и состояние страха указывают на вовлечение в процесс придаточных пазух носа.
При высоких концентрациях яастуйают поражения дыхательного тракта. В тяжелых случаях развивается острый токсический отек легких, особенностью которого является образование на слизистой пленок экссудата (псевдомембран) из фибринозных выделений. Если боли в груди через 2 ч не утихают, то можно предположить поражение легких. Испытываемая боль столь мучительна, что пораженные едва в состоянии переводить дыхание и заболевают психозами страха, которые производят впечатление помрачения рассудка.
Следствиями воздействия на нервную систему являются неуверенность походки, слабость в ногах, боли в суставах и мышцах, головокружение, в тяжелых случаях подергивание мышц и судороги, временная потеря сознания, иногда паралич различных групп мышц.
Кожное действие наступает в результате .соприкосновения с ОВ яли пребывания в атмосфере с высоким содержанием ОВ. Возникают эритемы, возможно появление опухолей на порвжен-иых участках кожи (зуд и жжение), а при определенных обстоя
85
тельствах— образование волдырей. Порой волдыри сильно распространяются. Некротические распады более глубоких слоев клетчатки не наступают, поэтому, несмотря на мучительные ощущения, поражения кожи хорошо поддаются лечению. Через двое суток образуется Струп, который через семь суток отпадает.
В Отличие от кожно-нарывных ОВ, раздражающие носоглотку ОВ не вызывают заболевания общего характера.
3.2.2. Ароматические мышьякорганические соединения
3,2.2.1. Общие свойства. Галогенированные ароматические арсины типа VII являются кристаллическими веществами. Они ие растворимы или почти ие растворимы в воде. Хотя их термическая стабильность инже, чем хлорацетофенона, их можно применять в дымовых шашках и снарядах со сравнительно большими зарядами взрывчатки. Исключительной термической стабильностью, даже превосходящей хлорацетофенон, отличается дигидрофенар-сазиихлорид X, который по своей структуре является производным фенарсазина XV1II:
Эти соединения обладают свойствами вторичных арсинов *. Как галогенарсины они окисляются и гидролизуются. Под влиянием арильных групп в довольно жестких условиях возможен почти количественный гидролиз, однако с дигидрофенарсазинхлоридом этого не происходит.
Они имеют чрезвычайно низкое давление пара и малую летучесть, недостаточную, чтобы путем испарения создать минимальную раздражающую концентрацию. Поэтому их можно применять только в виде аэрозоля, будь то в дымовых смесях, в виде растворов или в гранатах. Длительность заражения воздуха зависит от свойств аэрозоля. ' .
Ввиду их малой летучести о стойкости этих ОВ на местности говорить не приходится. При оседании на поверхность почвы аэрозоли теряют свое физиологическое действие, если только в результате каких-либо воздействий, например, образования пыли и т. п., частички снова не поднимутся в воздух.
Их хорошая растворимость в некоторых ОВ допускает применение этих соединений в тактических^ смесях* Значение таких смесей в современном бою сомнительно.
* К дигидрофенарсазинхлориду это не относится, подробнее см. стр. 94. — Прим, ред.
86
3.2.2.2. Дифеиилхлор- и дифеиилциаиарсии
CgHs—As—CgHg
С6НБ—As—CeHg
д иф ей и л х лора р с и н
Военные обозначения: нем. — Blaukreutz, Clark I; англ.— DA; амер. — DA; франц. — S ternite.
дяфенилцианарснн
нем. — Blaukreutz, Clark II; англ.— DA; амер.—DA; франц. — Sternite.
Дифенилхлорарсин был применен в 1917 г. немецкими войсками в сочетании с фосгеном и дифосгеном. В 1918 г. он был вытеснен более физиологически активным дифенилциаиарсином и применялся в виде смеси с этим ОВ.
Методы получения. Внервые эти-соединения были получены Ла Костом и Михаэлисом (1880 г.).
В дальнейшем было разработано много методов, часть которых может иметь промышленное значение. Оии различались способами арилироваиня мышьяка. Это влекло за собой известные технологические трудности, так как процессы очень длительные и в некоторых требовалось многократно переводить мышьяк из одной степени окисления в другую. Из-за множества побочных реакций это приводило к снижению выхода.
Методы, в которых в качестве промежуточного продукта выступает трифениларсин, больше не имеют промышленного значения. Потеряли свое значение и первые немецкие способы, в которых для арилироваиня мышьяка использовали реакцию хлористого феиилдиазония с арсенитом натрия (схема 1).
c6h5n2ci
Na3AsO3 ------:----
О
AsCl3 As2O3 у
. - CeH5As(ONa)a «----------
C6H5MgBr
N6aHsCI: * |hCI;SO2
* (CeH5)3As CeH5NaCl
। CeHsAs=O -----------
AsCl3
+ n
HC1; SOa
(C6H5)2AsC1 4------- (C6H6)2As^ -----
Схема 1. Устаревшие способы синтеза дифенилхлорарсина
Не исключено, что кое-где из-за яаличия соответствующего промышленного оборудования пользуются одним из старых методов.
В методе, применявшемся в крупном промышленном масштабе использована реакция треххлористого мышьяка с бензолом в присутствии хлористого алюминия (реакция Фриделя — Крафтса):
+ AlCIa
2C6He + AsCl3 _2HCf> (CeHs)2AsCl
8?
f
Для введения двух арильных групп реакцию проводят при нагревании.
Сравнительно простым и экономичным является метод Изацеску’ и Круеску. Для его проведения ие требуется сложного оборудования. По этому методу дифенилхлорарснн получают с хорошим выходом взаимодействием треххлористого мышьяка с феиилмер-курхлоридом:
2CsHsHgCl +9^SG‘> (C6H5)2AsC1
Фенилмеркурхлорид синтезируют следующим способом: из хлорной ртути получают ацетат ртути(II), который затем используют для получения ацетата фенилртути. Обработкой последнего хлористым натрием-получается феийлмеркурхлорид:
C6Ha + Hg(OCOCH3)2 —> CaH5HgOCOCH3 + СНзСООН
CaH5HgOCOCH3 + NaCl —> C6H5HgCl + CH3COONa
Лабораторный синтез. Для лабораторного получения дифенилхлорарсина пригоден предложенный Сартори видоизмененный метод Михаэлиса, который в сущности соответствует первоначальным промышленным методам, принятым западными державами.
Сущность метода заключается в следующем: на первой стадии процесса треххлористый мышьяк арилируется хлорбензолом в присутствии металлического натрия до трифениларсина, который на второй стадии при нагревании с дополнительным количеством треххлористого мышьяка превращается в дифенилхлор-арсин:
+ 3C6H5CI; 6Na
AsCl3 -------—---------> (C6Hs)3As
—oNaCl
+ AsClg 2(C6H5)3As --------> 3(C6Hb)2AsC1
В трехгорлую колбу емкостью 1 л, снабженную мешалкой, обратным холодильником и капельной воронкой, вносят 75 г нарезанного кусочками натрия и 300 мл бепзола, к которому добавлено 1—2% этилацетата. Содержимое выдерживают 30 мин для активации натрия и затем медленно приливают из капельной воронки 136 г хлорбензола и 85 г AsCl3. Через несколько минут, когда реакция становится более энергичной, колбу помещают на 12 ч в охлаждающую смесь и первые 2 ч ведут реакцию при перемешивании. После этого реакционную смесь фильтруют, промывают осадок теплые бензолом и промывную жидкость соединяют с фильтратом. Перегонкой отделяют бензол от трифениларсина, а последний перегоняют затем при 200 °C в охлаждаемый приемник (желтое масло). Помещают 30 г полученного трифениларсина в широкогорлую колбу, нагревают до 350—360 °C и прибавляют по каплям из делительной воронки с оттянутым капилляром 25,5 мл AsCl3. Темно-коричневый жидкий дифенилхлорарснн перегоняют при пониженном давлении и перекристаллизовывают из бензольного раствора.
Дифенилцианарсин можно получить из дифенилхлорарсина нагреванием с цианидами щелочных илн тяжелых металлов
(CeH₽)2AsCl
+ CN
----=-> (C6H5)2AsCN
-Cl
88
или (что более экономично), обработкой окиси дифениларсина синильной кислотой:
+ 2HCN
[(CeHgJaAsJjjO __н2о * ^CeHe^AsCN
Физические свойства. Дифенилхлор- и дифенилцнанарсин представляют собой бесцветные кристаллические вещества; технические продукты имеют окраску от серой до темно-коричневой. В тактических смесях чаще всего применяют жидкие темно-коричневые неочищенные продукты. Через длительное время неочищенные продукты превращаются в вязкую кристаллическую массу. Оба соединения при перегонке при нормальном давлении разлагаются.
Приводим физические характеристики обоих соединений:
Дифенил-хлорарсин Дифенил-цианарсин
Т. кип., °C
при 10 мм рт. ст 180 191
при 13,5 мм рт. ст «— 200
при 21 мм рт. ст — 213
при 30 мм рт. ст. ...... . 205 —
при 102 мм рт. ст 245 —
при 760 мм рт. ст 333 (разл.) 3346 (разл.)
Т. пл., °C Давление пара, мм рт. ст. 38-40* 2-35 (31,5)
при 0 °C . 10 4
при 20 °C 5-10_* 2*10
при 40 °C 3,3.10 J *—
при 60 °C 10 —
Максимальная концентрация ю~4
при 20 °C, мг/л ........ 6,8*10
Относ, плотность d2? ....... 4 1,422 1,45
Показатель преломления Пр
при 20 °C — 1,60922
при 52 °C — 1,6153
при 56 °C 1,6332 (1, 6236) —
* T. лл. лабильной модификации 13,4 °C.
Поскольку температура плавления технических продуктов ниже 30°С, они летом плавятся в контейнерах.
Из-за термической нестабильности и низкой температуры плавления дифенилцнанарсин мало пригоден для дымовых шашек. В случае дифенилхлорарсина этот недостаток устраняется соответствующим пористым наполнителем, однако такой прием не удовлетворяет требованиям, предъявляемым к подобным ядовитодымным шашкам.
Как видно из приведенных данных, давление пара (и летучесть) днфенилцианарсина ниже, чем у дифенилхлорарсина. Летучесть и того И другого соединения составляет величину порядка 10~4 мг!л, пороги раздражения — величины того же порядка. Летучесть этих ОВ недостаточна для заражения воздуха,
89
Давление пара дифенилхлорарсина можно вычислить по формуле (в мм рт. ст.):
lg р = 7,8930--^
Дифенилхлор- и днфенилцианарсии' практически нерастворимы в воде (0,2%), но хорошо растворяются в ряде растворителей — бензине, галогеналканах, бензоле, ацетоне, этаноле, эфире и др. О растворимости дифенилхлорарсина имеются следующие данные (в г/100 мл растворителя):
С3Н6ОН (абсол.) . . 20 СеНв........... 100
СС14........... ♦ 20 Нефть........... 50
Оба соединения растворяются в некоторых ОВ, например, в три-хлорннтрометане, иприте и дифосгене. Для приготовления смесей с другими ОВ его вторую мировую войну применялся только технический дифенилхлорарснн.
Химические свойства. Химическое поведение дифенил-хлор- и дифеинлцианарсина аналогично, из них более устойчиво хлорпроизводное; Оба соединения термически нестабильны, выше 230 °C дифенилхлорарснн желтеет, через 15 мин нагревания прн 600 °C разлагается 22%, а прн 750 °C — 48% вещества. Тем не менее, стабильность обоих соединений достаточна, чтобы их можно было применять в снарядах без заметного разложения. Металлы этими соединениями не разрушаются. Оба ОВ устойчивы при хранении.
Гидролиз. При обычной температуре дифенилхлор- и дифенил-цнанарснн медленно гидролизуются водой:
+ н2о
, 2(CeH6)2AsX —> {(СвНв),АзЬО
Нагревание и добавка щелочей способствуют реакции. Особенно это относится к спиртовым растворам.
Так как образующаяся при гидролизе окись дифеииларсина (т. пл. 93 °C) является сравнительно сильным раздражающим ОВ, то гидролиз аэрозолей этих ОВ влагой воздуха несущественен и эффективность их не снижается. По этой же причине реакция гидролиза не пригодна для проведения дегазации.
Другие реакции атома галогена или CN-группы. Безводный аммиак с дифеинлхлорарсином в бензольном растворе образует дифениларсииамид (т. пл. 53°C):
(CeHB)2AsCl -> (CaHsbAs-NH2
—IN Л<и
Это вещество также действует раздражающе и на носоглотку и на кожу. Дифениларсииамид — неустойчивое соединение, на воздухе превращающееся в окись дифениларсина1
90
при црииуилсшии ссриоидирида в ширившиpact виры дифенил'-------------------
хлор- и дифеиилцианарсииа образуются игольчатые кристаллы дифеииларсинсульфида (т. пл. 67°C):
+ H2S 2(C0Hs)2AsX ---->• [(C5Hs)2As]2S )
-2НХ
В этой реакции можно использовать и спиртовые или бензольные растворы сульфида натрия.
Ацетоновые растворы иодидов щелочных металлов при взаимодействии с дифеиилхлорарсином дают дифенилиодарсии, желтые кристаллы, т. пл. 41 °C:
+ Г
(C6H6)2AsC1 ---> (C6He)2AsI
-с Г
Окисление. Для возможной дегазации производных дифенил-арсина пригодно окисление до дифенилмышья'ковой кислоты:
’ + о- + н2о
(C0Hs)2AsX -------> (CeH6)2As^
-НХ ,\он
Она представляет собой физиологически- неактивные игольча- 1
тые кристаллы, т. пл,. 175 °C. '
В качестве окислителей можно использовать азотную кислоту, перекись водорода, бромную воду, перманганат калия, хлорную воду, гипохлориты, хлорамин и др.
В безводных растворах, например, в четыреххлористом углероде, дифенилхлорарсин окисляется хлором в дифеииларсеитри-хлорид (т. пл. 189°C):
+ С12 1
(C6Hs)2AsC1 --->- (C6H6)2AsC13
Аналогично действует очевидно и сухая хлориая известь.
Дифсниларсентрихлорид неустойчив и легко гидролизуется с образованием дифеиилмышьяковой кислоты:
(C,Hf),AsCl, *2Н,0> (С,Нв)гА< .
-ЗНС1 \он
При действии хлора на дифенилцианарсин в беизольиом растворе образуется дымящая иа воздухе окись дифенилдихлор- I
арсония I
+ 2С12 +2Н2О I
2(C0Hs)2AsCN ------> 2(C0H6)2AsC12CN _2HCN>
—* 2(C0H5)2AsC12OH ----> [(CeH5)2AsCl2]2O I
—H2O I
91
лоты:
+ зн2о х
[(CeH5)2AsClz]2O -2(C6H6)2Asf
“4HC1 \он
Из других представителей ОВ этого типа можно еще назвать следующие:
дифенилбромарсин— белые кристаллы, т, пл. 54—56 °C, раздражающее действие более слабое, чем у дифенилхлорарсина;
дифенилфторарсин — бесцветная жидкость, т. кип. 157,5 °C (8 мм рт. ст.), такое же раздражающее действие, как у дифенил-хлорарснна;
дифенилхлорстибин— белые кристаллы, т. пл, 68 °C, аналогичное раздражающее действие;
дифенилцианстибин — кристаллическое вещество, т. пл. 115°С, аналогичное раздражающее действие.
3.2.2.3. 10-Хлор-9,10-дигидрофеиарсазин
Синонимы: фенарсазинхлорид, адамсит.
Военные обозначения: нем. — Azin; амер. — Adamsit, DM.
Это вещество было разработано и получено во время первой мировой войны в США и по некоторым имеющимся данным применено итальянской армией. Адамсит (назван по имени американского военного химика Адамса) является одним из самых эффективных и дешевых химических ОВ.
Первые производные фенарсазина были описаны в одном немецком патенте еще в.1913 г. В период между первой и второй мировыми войнами производные фенарсазина были предметом обширных исследований многих ученых разных стран. Во время второй мировой войны адамсит был заготовлен в больших количествах некоторыми воюющими государствами. Еще и теперь он входит в арсеналы химического вооружения империалистических армий.
Методы получения. Из многих возможностей препаративного получения адамсита особое значение для промышленного и лабораторного производства приобрели следующие два метода: конденсация треххлористого мышьяка с дифениламином (метод Виланда) и конденсация трехокиси мышьяка с гидрохлоридом дифениламина (метод Контарди).
92
NH
+ AsC!3 (C6H5)2NH ——>
—'2HG1
As
Cl
Выход адамсита хороший.
Аналогично происходит синтез’из трехокиси мышьяка:
NH
+ As2Og 2(C6H5)2NH • НС1 - . > 2
—origU
I
Cl
Гидрохлорид дифениламина получают нагреванием дифениламина с соляной кислотой.
Лабораторный синтез (по Сартори). В фарфоровом тигле емкостью около 300 мл при перемешивании нагревают 42 г дифениламина и 21 мл НС1 (d 1,19) до полного испарения воды. Образовавшийся гидрохлорид дифениламина сушат 2 ч при 55 °C, смешивают с 25 а трехокиси мышьяка и при помешивании доводят до плавления, (необходима хорошая тягаУ). Расплав нагревают далее при 140—200 °C для завершения реакции (до прекращения выделения паров воды) и растворяют в ксилоле или уксусной кислоте. Выкристаллизовывается неочищенный продукт, окрашенный в зеленый цвет. Повторной кристаллизацией получают желтое кристаллическое вещество.
Физические свойства. Чистый 10-хлор-9,10-дигидрофен-арсазин представляет собой светло-желтые игольчатые кристаллы, лишенные запаха. Зеленая окраска технического продукта принадлежит красителям, образующимся при побочных реакциях.
Адамсит кристаллизуется в трех формах: в стабильной ромбической (т, пл. 195°С), в метастабнльной моноклинной (т. пл. ~ 186°C), и в нестабильной триклинной (т. пл. ~183 °C) модификациях.
Ниже приведены физические свойства адамсита:
Т. кип., °C
при 10 мм рт. ст............
при 760 мм рт. ст...........
Т. пл., °C.....................
Давление пара, мм рт. ст.
при 0 °C....................
при 20 °C...................
при 40 °C...................
при 100 °C..................
Максимальная концентрация при 20 °C, мг/л ................
Относит, плотность d™..........
120
410
195
5-10’и 2.1°2 2-10 11 2* Ю-6
2 - Ю"5
1,648
Технический продукт начинает частично плавиться уже при 160 °C,, основная масса — при 193—195°C.
93
При пониженном давлении соединение bujiuhmcich ucj рдзли’ жения. Вычисленная по кривой давления пара температура кипения равна 410 °C. При длительном нагревании при этой температуре и нормальном давлении адамсит лишь немного разлагается.
В воде адамсит практически нерастворим; на холоду он мало растворим в бензоле, ксилоле, четыреххлористом углероде, уксусной кислоте, спиртах; при нагревании растворим в ксилоле и толуоле, ледяной уксусной и муравьиной кислотах (фиолетовое окрашивание); практически нерастворим в теплом иприте. При растворении в коицеитрироваииой серной кислоте появляется вишнево-красиое окрашивание, при растворении в AsCU—темио-зеленое.
Как видно из приведенных данных, давление пара и летучесть адамсита при обычной температуре столь малы, что заражение воздуха посредством испарения невозможно. Адамсит может быть применен только в виде аэрозоля. В результате оседания иа поверхность почвы ои становится неэффективным.
Ограниченная растворимость в некоторых органических растворителях и ОВ вряд ли позволит применять адамсит в тактических смесях. Во время второй мировой войны были разработаны дымовые шашки, в которых находилась смесь адамсита и хлорацетофенона, Известны были также гранаты, снаряженные смесью адамсита и хлорацетофенона, которые, естественно, плавились порознь.
Химические свойства. По своему химическому поведению. адамсит проявляет большое сходство с дифеиилХлорарсииом, от которого он отличается наличием NH-мостика, связывающего два фенильных ядра в орто-положеииях. Из-за своей феиарсазино-вой структуры ои является чрезвычайно стабильным и малореак-ционноспособиым соединением *.
* Свойства 10-хлор-9,10-дигидрофенарсазина (адамсита) значительно отличаются от свойств вторичных галогеиарсинов, в том числе и дифенилхлорарсина. Так, адамсит представляет собой твердое желтое вещество, тогда как вторичные арсины — либо бесцветные жидкости, либо бесцветные низкоплавкие кристаллы. Адамсит медленно гидролизуется, а вторичные галогенарсины — хорошо. -
При окислении адамсит с трудом превращается в фенарсазииовую кислоту, а образование диалкил (диарил) мышьяковой, кислоты протекает легко. Все эти особенности адамситу могут быть объяснены своеобразием структуры его молекулы, которая может быть изображена следующим образом:
Действительно, в соответствии со структурой 11 адамсит должен представлять собой высокоплавкое солеобразное соединение хиноидной структуры, которая и ответственна за его окраску. Эта медленно гидролизующаяся «соль» II уже содержит окисленный атом мышьяка. — Прим. ред.
94
становясь сначала зеленым, а затем темно-коричневым. При крат- i
ковременном нагревании он почти не разлагается; устойчив к де-
тонации, !
Технический адамсит незначительно разрушает сталь, а также - латунь и бронзу. Другие материалы, такие как пластмассы, дерево и защитные покрытия — устойчивы по отношению к адамситу. Взаимодействие адамсита с металлами не является аутокаталитическим, поэтому им можно снаряжать боеприпасы.
Гидролиз. Прн нормальной температуре и нагревании адамсит почти не реагирует с водой. Гидролиз столь ничтожен, что не имеет практического значения. Адамсит, долгие годы пролежавший в воде или подвергавшийся метеорологическим воздействиям, ие изменяется и сохраняет все свойства.
Гидролизу способствуют растворы едких щелочей. В качестве продукта гидролиза образуется окись 9,10-дигидрофенарсазина, раздражающее ОВ почти той же силы, что н исходное соединение:
+ ИдО
' 2HN(C8H4)2AsC1 {NH(CeH4)2As]2O + 2ИС1
При действии хлористого водорода окись снова превращается в адамсит.
Другие реакции атома хлора. При действии различных реагентов в соответствующих растворителях атом хлора можно заместить. Однако путем добавления брома в уксуснокислый раствор -адамсита аналогичное бромпроизводиое получить нельзя, происходит разложение с образованием тетрабромдифениламииа: i
+ 4Вг2 • ;
HN(C,H,),ASC1 „ » HN(C,H,Br,),-
Прн пропускании аммиака в кипящий ксилольный раствор |
адамсита образуется бесцветное соединение — трифенарсазинамин j
[HNfCeH^AsbN, т. пл. 295—300-°C. С аминами реакция в обычных 1
условиях ие идет. Лишь при кипячении адамсита с такими ами- j
нами, как пиридин, диметиланилии или хннолин, образуются coot- |
ветствующие трифенарсазиихлориды, например, в случае пириди- |1
на — тридигидрофенарсазиихлорнд следующего состава |
HN(C6H4)2As—N(CsH4)2As—N(CeH4)2AsCl I
Это оранжево-желтые кристаллы (т. пл. 260—263°C), которые - I
при действии воды разлагаются с образованием окнси дигидрофеи- 1
арсазина. I
С сероводородом образуются желтые кристаллы дигидрофенар- |
сазинсульфида [HNfCeH^AsbS, соединение, которое при обра- I
ботке хлористым водородом превращается опять в адамсит. I
95
Окисление. Адамсит в соответствующих "растворителях мшпнтт окислить в дигидрофенарсазиновую кислоту даже мягкими окислителями:
NH
HOZ
Эта кислота представляет собой игольчатые кристаллы, плавящиеся выше 300 °C, В качестве окислителей пригодны перекись водорода, щелочные растворы перманганата калия, спиртовые растворы хлорамина и др. В зависимости от условий реакции азотная кислота оказывает окисляющее или нитрующее действие.
При осторожной обработке адамсита дымящей азотной кислотой в ледяной уксусной иислоте мышьяк остается трехвалентным и происходит нитрование в Орто- и пара-положение. Оба изомерных нитрофенарсазинхлорида — кристаллические соединения, В качестве побочного продукта при нитровании образуется 3,6-ди-нитроднгидрофенарсазин. Это нитропроизводное оказывает сильное раздражающее действие. За счет одновременного окисления образуются орто- и пара-нитропроизводные, а также динитропроизводные дигидрофенарсазиновой кислоты. Эта реакция используется в аналитических целях.
Прочие реакции. Обработка адамсита хлористым или иодистым водородом при 100—160°C приводит к образованию дифениламина:
+ 2НХ HN(C6H4)2AsC1 ——> HN(C6HS)2
Идентификация дифениламина в этой реакции может служить косвенным методом индикации адамсита.
Если адамсит растворять в концентрированной H2SO4 или в теплой муравьиной кислоте,✓ То растворы приобретают интенсивную красную нлн, соответственно, красно-фиолетовую окраску. Появление окрашивания можно объяснить комплексообразованием за счет кватернизации атомов мышьяка и азоТа *:
* Как уже отмечалось (стр. 94), окраска адамсита, очевидно, обусловлена хиноидной структурой. Изменение окраски в серной или муравьиной кислотах можно объяснить вероятным изменением системы сопряженных связей хиноидной структуры под влиянием разных заместителей, — Прим, ред.
96
ио-кислого раствора исчезает, а при подогревании снова появляется.
3.2.2.4. Физиологические свойства. Ароматические мышьякор-гаиические соединения — адамсит, дифенилхлор- и дифенилциаи-арсин — являются самыми сильными в группе ОВ, раздражающих носоглотку. В Сравнительно высоких концентрациях, которые не встречаются в боевой обстановке, они могут привести к поражениям со смертельным исходом.
Явления отравления, которые помимо раздражающего действия могут привести к жалобам на затруднение дыхания и поражениям верхних дыхательных путей и легких, длятся лишь ограниченное время и, как правило, не оставляют серьезных последствий. Явления раздражения наступают не сразу после вдыхания, а через 5—10 мнн, в зависимости от концентрации и продолжительности воздействия ОВ,
В табл. 7 приведены данные о пороге раздражения и пределах переносимости некоторых мышьякорганическнх ОВ.
Таблица 7. Физиологические данные некоторых раздражающих носоглотку мышьикорганнческих ОВ
Название Порог раздражения, мг/л Предел переносимости, мг/л Данные для экспозиции 1—2 «Мн
Адамсит 10“4 10-3—2- Ю“4 —
Дифеннлхлорарсин ю-4 . ю“3 При I0-2 ПОЗЫВ к рвоте, при 2 мг/л поражение дыхательных путей
Дифен илциа на рс ин 10 5 10 3—5-10 4 Согласно Флюри, при концентрации >2,5 мг/л даже один вдох едва переносим; немедленное раздражающее действие, боли в груди, поражение дыхательных путей
Метилдихлора рсин 2- 10 3 2,5- 10 3 См. также разд. 5.3.4
Этилдихлорарсин 1,5- 10 3 5-10~3 То же
Фенилднхлорарсин 5- 10 3 1,6. io-z
2-Хлорэтилдихлорарсин 8. 10 4 —- Ср. также разд. 5.3.3.4
Порог раздражения для адамсита равен 10-4 мг/л, а для ди-феиилциаиарсииа — 10~5 мг/л; предел переносимости для времени экспозиции 1—2 мин, в общем, на порядок выше, чем порог раздражения.
4 Зак. 448
97
----- .........-Г"-* ------«--------->-Г ---—-----,— г-феиилхлор- и дифеиилцнанарсина. Однако адамсит в любом состоянии ие действует на кожу, в то время как дифенилхлорарсин и дифеиилцианарсин в виде аэрозолей вызывают на поверхности кожи покраснения и припухлости. При соприкосновении с твердыми, а особенно с жидкими техническими продуктами или с растворами образуются волдыри. Согласно данным Флюри и Церни-ка, первые признаки поражения кожи появляются в случае дифе-иилхлорарсина при количестве 0,05 мг на I см2 поверхности кожи, болезненной является доза 0,5 мг/см2.
В литературе описаны некоторые случаи отравлений при попадании мышьяксодержащих раздражающх ОВ через желудочно-кишечный тракт. Такие отравления возникают при употреблении зараженной воды для приготовления пищи, для мытья или чистки зубов. Последствиями были боли в области желудка и рвота. Несмотря на сравнительно высокое содержание мышьяка такие отравления лишь в немногих случаях приводили к смертельному исходу.
3.2.3. Свииецоргаиические соединения
К началу второй мировой войны в Англии группой ученых Кэмб-риджского университета (Саундерс, Стеси, Хип и др.) проводились исследования свииецорганических соединений, раздражающих слизистую оболочку7. Целью этих исследований была замена мышьякоргаиических ОВ другими равноценными соединениями вследствие недостатка мышьяка в стране.
Поскольку соединения диалкилсвинца из-за их слабого раздражающего действия вряд ли могли приниматься в расчет как ОВ, работы концентрировались вокруг соединений триалкилсвии-ца типа ЕзРЬХ, в которых X мог быть остатком как неорганической, так и органической кислоты. Было получено и проверено иа физиологическую активность свыше нескольких сот свинец-оргаиических соединений этого типа.
С точки зрения промышленного производства соединения с высшими алкильными группами представляют незначительный интерес, поэтому перечень возможных ОВ ограничивается лишь низшими членами ряда, т. е. соединениями с этильными, пропильными, и бутильными группами. Из-за неудовлетворительного раздражаю-щего действия были исключены также соли триметилсвиица.
3.2.3.1. Методы получения. Получить соли триалкилсвиица можно разными способами. В зависимости от кислотного остатка получение проводится в определенных условиях, на которых здесь мы ие будем останавливаться.
Во .всех методах исходят из соответствующих тетраалкильных производных свинца. В принципе эти методы можно свести к следующим двум: к непосредственному взаимодействию тетраалкил-свиица с кислотами н к взаимодействию хлористого триалкилсвиица с солями кислот.
98
непосредственное взанмидеиывис—Kipuu<uuwvUuuUu—____
тами проводят в присутствии силикагеля:
R4Pb 4- НХ —> R2PbX + RH
Метод удобен н из-за своей простоты мог бы быть использован для промышленного получения этих соединений. Недостатком, однако, является то, что с высшими тетраалкнлпроизводиыми эта реакция идет сравнительно медленно и с малыми выходами, особенно если другой участник реакции слабая кислота.
К недостаткам метода относится также образование солей диалкнлсвница в жестких условиях реакции. Это особенно касается получения амнлпроизводиых свинца.
При взаимодействии хлористого триалкилсвннца с солями кислот происходит замещение атома хлора на аииои натриевой или калиевой соли данной кислоты:
R3PbCl + NaX RspbX-f-NaCl
В этом методе, особенно при осуществлении его в промышленном масштабе, представляет трудности удаление избыточного хлорида триалкилсвннца. Более подходящим является взаимодействие последнего с серебряной солью:
RaPbCl + AgX —► R3PbX-f-AgCl
Этот метод вряд ли пригоден для крупного производства.
Очень удобным представляется метод, впервые упомянутый Пфейфером, а также Краузе и Поландом8, заключающийся во взаимодействии очень реакционноспособной гидроокиси триалкил-свинца с соответствующими кислотами:
R3PbOH + НХ —► R3PbX + H,0
В определенных случаях отщепление гидроксильной группы возможно уже при реакции с солями щелочных металлов:
. R3PbOH 4-ЬГаХ —> R3PbX -f- NaOH
Гидроокись трнэтилсвиица получают действием 30%-иого раствора едкого иатра иа эфирный раствор хлористого триэтил* свинца, а гидроокись три-н-пропилсвинца— из водной суспензии хлористого три-н-пропилсвинца и влажной окиси серебра-
Хлористые триалкильные производные свинца готовят действием хлористого водорода на соответствующие тетраалкильные производные в подходящих растворителях (эфир, петролейиый эфир).
3.2.3.2. Свойства. Соли триалкилсвннца являются кристаллическими веществами с характерными температурами плавления. Онй представляют собой, в основном, очень стабильные соединения. Они не растворяются иди только ограниченно растворяются в воде и, в зависимости от природы кислотного остатка, лишь мало или вовсе не гидролизуются. Физические н физиологические свойства важнейших соедиений приведены в табл. 8,
4:
99
Таблица 8. Физические и физиологические свойства некоторых солей триалкилсвинца
* Экспозиция 40 сек; ** при разбавлении 1 : 10е; *** Непереносимо через 90 сек:
1 — неприятное, но еще переносимое раздражающее действие; 2 —очень неприятное, но все еще переносимое действие; 3 — непереносимое действие; + при цифре —выше указанной величины
Кислотный остаток P==C2HS RsM-CsH, й==н-С4Н0
т. пл„ °C раздражающее действие при разбавлении 1 : 107, баллы т, пл., °C раздражающее действие при разном разбавлении, баллы т. пл, °C раздражающее действие при разбавлении 1 : Iff, баллы
I : 107 1 :2,5-107
Акрилат 120 2 123 2+ 1
Антранилат 96 .2 57-58 3 2+ —1 —
Ацетат 160 2 126-128 3 2+ 86 3
Бензолсульфамид 132 3 * 96 3 — —
Бромацетат 120 1 93-94 2 — 54—55 1 +
Бромид —1 — 76-78 3 1 + 1— —
н-Бутират — — 105—106 3 2+ — —
Изовалерат — — 110-1И 3 1 + — —
Иодацетат — — 88-89 2 83 1 +
Кротонат 135-136 2 135 2+ —— 119 2+
К-сантат — — 57,1 2 — — 2+
Метансульфамид 97 — 67 з+ — —
Нафталин-2-суль- 152 — 126-127 2+ — —
фон ат
Оксалат 55 2 ** — — — — —
Пропионат 141 1 121-122 2 — 79-80 2+
о-Сульфанилид 115,5 3 — -— — — —
о-Сульфобенз- 135 з *** 130 — —
имид
Тиоцианат 35 2 — — — — —
о-Толуолсульфо- 189 — 86-87 3 2 — —
нат
л-Толуолсульфо- 170 — 82-83 3 1 + 81—82 2
нат
л-Толуолсульф- 127 3 * 100-101 з+ — — , —
амид
о-Толуолсульф- 133 — — — — —
амид
Трихлорацетат 141 I 139-140 2 — 119 2
Фталимид 131 — — -— — —
Хлорацетат 147 — 109-110 3 2+ 60 1 +
Хлорид —* 1 133-134 2+ ——ч 106,5-108,5 3
2-Хлорпропионат 106 2 99-100 3 — — 2+
Цианид 189 2 — 2+ — — —
Физиологическая активность этих соединений была установлена на людях Саундерсом и др. Она оценивалась по сравнению с хлористым диэтилсвинцом, активность которого в тех же условиях равна нулю. Обычно концентрация- составляла 1 : 107, т. е. на Ю7 моль воздуха приходился 1 моль вещества. Для этого в камере объемом 10 м3 распределяли около 20 мг соответствующего соединения, более активные соединения испытывали в большем
100
разбавлении (1 :2,5-107). Требуемое количество вещества растворяли в примерно 15 лл растворителя (обычно спирта) и раствор впрыскивали в опытную камеру. Время экспозиции составляло 10 мин.
Приведенные в табл. 8 баллы от 1 до 3 выражают степень раздражающего действия.
Как показывает сопоставление, наиболее активные соединения нужно искать в группе сульфамидов триалкилсвиица I
R3Pb-N(R/)SO2R"
I
где R = С2Н5, н-С3Н7; R' = Н, СеН5, м-С0Н4С1, п-С8Н4Вг;
Rz/ =: СН3, С0Н31 о-СН3С6Н4, я-СН3С0Н4, л-МН2С0Н4
Эти соединения в разбавлении 1 : 107 уже через 40 сек вызывают мучительное, непереносимое раздражение носоглотки, связанное с ощущениями стеснения в груди. Они исключительно стабильны, хорошо кристаллизуются.
Дальнейшие исследования привели к получению и изучению серусодержащих производных алкилсвиица, в том числе тиоэтилата II и тиофенолята триэтплсвница III
(C2H5)3PbSC2H5 (C2H5)3PbSCeH5
II III
(т, кип, 76—78 °C (т, кип. 125 °C
при 0,075 мм рт. ст.1 при 0,3 мм рт, ст.)
Соединение II в чистом виде — бесцветная жидкость, медленно разлагающаяся на свету. Ее запах напоминает запах хрена. Соединение III — желтая маслянистая жидкость, ие растворимая в органических растворителях. При действии соляной кислоты она разлагается с образованием хлористого триэтилсвинца и тиофенолята свинца (СбН55)гРЬ.
Делались попытки получать соединения, в которых бы токсическое действие фторацетатов комбинировалось с раздражающим носоглотку действием солей триэтилсвинца. Так, из фторуксусной кислоты и тетраэтилсвинца при кипячении в эфире в присутствии силикагеля был получен фторацетат триэтилсвинца:
(СгН5)4РЬ + FCH2COOH —> (C2HB)3PbOCOCH2F + С2Не
Это соединение плавится с разложением при 180,5 °C и по своему раздражающему действию сравнимо с большей частью солей триалкилсвиица. При концентрации 1,7-10‘3 мг/л (1 : 10“7) уже в течение первых минут наступает сильное раздражение. Из 10 испытуемых пятеро жаловались на боли в груди. При подкожной инъекции мышам (ЛД5о= 15 мг/кг} возникали симптомы отравления; характерные для отравления фторацетатами.
Стремление объединить раздражающее действие производных триалкилсвиица с сильным нервно-паралитическим действием
101
фосфорорганических соединений привело к получению7 бис-(три-этилсвииец)-фторфосфата IV
(С2нБ)3РЬСХ
(СгН^зРЬО/ iv
кристаллического вещества, плавящегося ьыше 260° С, растворимого в ацетоне. Уже при разбавлении 1 : 108 оно оказывает .заметное раздражающее действие, разбавление 1 : 106 абсолютно непереносимо. При разбавлении 1 : 108 сужение зрачков ие наблюдалось.
Соли триалкилсвннца являются типичными ОВ, раздражающими носоглотку, они сильно действуют на слизистые оболочки полостей иоса и глотки, нарушают дыхание и вызывают ощущение стеснения в груди. Оии могут применяться только в виде аэрозолей, так как для заражения воздуха их летучесть недостаточна и давление пара сравнительно невелико. Поскольку некоторые из этих соединений термически стабильны, возможно получение аэрозолей с помощью дымовых шашек. Одиако до сих пор все опыты проводились с распылением растворов.
3.2.4. Морфолид пеларгоновой кислоты
Морфолид пеларгоновой кислоты9 известен с 1962 г. как возможное раздражающее ОВ. Он имеет следующее строение:
О
Hsc/ ^СНз
н2(}^ ^сн2
N
o=i—(сн2)7—сн3
В виде аэрозоля ои оказывает сильное раздражающее действие иа глаза н органы дыхания. По своей активности он примерно в 5 раз сильнее хлорацетофенона. При вдыхании симптомы выражаются в ощущении жжения в полости иоса и глотки, выделениях из носа, сильном позыве иа кашель, иногда в тошноте, в сильном слезо- и слюнотечении, потливости. На свежем воздухе эти симптомы проходят быстрее, чем при отравлениях хлорацетофеноном или адамситом.
Морфолид пеларгоновой кислоты — бесцветное соединение, нерастворимое в воде, растворимое в полярных органических раство-' рителях. Ои имеет такие физические константы:
Т. кип., вС при 0,1 мм рт. ст. . ........105—П4
при 0,5 мм рт. ст......... 120—130
Относит, плотность d*5........ 0,965
Показатель преломления ....... 1,4684
102
VjULHUninril—ИП77ТПТО—гточггу-гггтп при—I uuu—rrv-да пеларгоновой кислоты с морфолином:
X) /СНгСНак
СН3—(СН2)Т—+ HN^ ‘ ;о ----------->
\С1 ^СН2СН/ ~нс‘
ц /СН2СН2\
—> СНз—(СН2)7—С—\)
ХСН2СН/
Раздражающая концентраций, вызывающая физиологические явления у 50% испытуемых, для времени экспозиции 1 мин составляет 39 мг- MunjM3; эффективность хлорацетофенона в одинаковых условиях — 213 Мё'Мин[м3.
3.2.5 2-Хлорбензилиденмалонодиннтрил (Си-Эс)
Военное обозначение: амер. — CS.
В 1960 г, в армии США было принято на вооружение новое ОВ — 2-хлор (или о-хлор-) бензилиденмалонодинитрил. Это сильное раздражающее ОВ, которое в последние годы предпочитают применять в США и других странах в качестве полицейского химического оружия для разгона и подавления демонстраций, митингов и т. п. Военные силы США применяли CS в больших количествах и во Вьетнаме.
В виде аэрозоля CS оказывает сильное раздражающее действие на глаза и верхние органы дыхания. За несколько секунд возникает тяжелый конъюнктивит, которому сопутствуют ощущение жжения, сильные боли и слезотечение. За исключением конъюнктивита это действие проходит через 5—15 мин.
Как правило, пораженные через 5—10 мин снова работоспособны, хотя порою и появляются некоторые последействия (светобоязнь, ощущение «усталости» глаз). Интенсивность конъюнктивита через 25—30 мин уменьшается.
Чаще всего воздействие на дыхательные пути приводит к потере боеспособностии работоспособности. Сначала появляется ощущение жжения в полости глотки, затем мучительное жжение в груди и ощущение «стягивания». При сильном отравлении возникает страх, который усугубляет общую симптоматику и пострадавший ие может сделать ни вдоха, ни выдоха. На свежем воздухе эти симптомы быстро проходят.
Другими проявлениями отравления CS являются сильные слюнотечение и выделения из носа, а также сильные раздражения в
103
полости носа. Ирн работах с нногДа начинаются носовые кровотечения. Головные болн, тошнота н летаргия не характерны н наблюдались не во всех случаях.
На пораженных участках кожи возникает сильное жжение, которое усугубляется влагой (пот, слезы н т. д). Даже после промывания раздражение кожи сохраняется несколько часов.
Прн длительном воздействии нлн прн высоких концентрациях образуются эритемы и волдыри.
Концентрации 2-хлорбензилнденмалонитрнла в 0,005 мг!л воздуха прн экспозиции 1 мин непереносимы, концентрации в 0,01 мг/л —- переносимы в течение около 12 сек, а 0,034 мг}л— в течение 6—9 сек. Концентрация, приводящая к потере боеспособности, составляет 0,001—0,005 jna/л, смертельных концентраций в условиях боя вряд ли можно ожидать.
2-Хлорбеизнлнденмалоиоднннтрнл получают из 2-хлорбензаль-дегида и малонитрнла:
+ CH2(CN)a
+ н2о
Он представляет собой белое кристаллическое вещество, хорошо растворяющееся в бензоле, хлороформе, ацетоне, хуже — в спирте, четыреххлористом углероде и в воде; т. пл. 95° С, т. кип. 310— 315° С (с почернением).
2-Хлорбензилиденмалонодинитрил Довольно устойчив к гидролизу. Период полупревращения прн гидролизе в смеси из 95% спирта и 5% воды составляет: прн 30° С — 95 мин, прн 40° С — 40 мин\ гидролиз на 90% проходит при 30° С за 635' мин.
Скорость гидролиза уменьшается в присутствии ионов водорода, а ноны ОН~ гидролиз ускоряют. Реакция протекает по следующей схеме:
CH=^C(CN)2
CI
+ н2о
Окислители затрагивают двойную связь, причем образуется ряд продуктов окисления. Прн действии гипохлорита образуется эпоксид такого строения:
CH=C(CN)2
Cl
4- CIO’
104
3,3. БОЕВОЕ ПРИМЕНЕНИЕ РАЗДРАЖАЮЩИХ ОВ
Современные раздражающие ОВ, как правило, представляют собой твердые вещества, наиболее эффективные в виде аэрозолей, В течение нескольких минут они приводят к потере боеспособно-ности и поэтому с точки зрения тактики их нужно рассматривать как быстродействующие ОВ.
Они не обладают большой стойкостью. Исключение составляют бромбензилцианид CeHgBrCN и фенилкарбиламинхлорид C(;H5N = CCI2. Эти соединения, а также хлорацетофенон достаточно летучи, чтобы заразить воздух при благоприятных метеорологических условиях; летучесть мышьяк- и свннецорганическнх соединений слишком мала, чтобы они могли остаться эффективными после оседания аэрозолей.
В результате введения в противогаз противодымного фильтра мышьякорганические соединения потеряли значение как аэрозольные ОВ; это значение они имели в первую мировую войну, поскольку проникали через угольный фильтр, предназначенный для поглощения газообразных ОВ. В современном бою раздражающие ОВ могут быть использованы лишь ограниченно, например, для нанесения внезапного удара, для подавления опорных пунктов, гарнизонов долговременных огневых точек, танковых экипажей, в уличном бою н для выведения из строя наблюдательных пунктов.
Быстрое действие раздражающих ОВ в малых концентрациях принуждает атакованных немедленно надевать средства защиты, затрудняет выполнение боевых задач и дает значительные преимущества нападающему. .
Раздражающие ОВ применимы при помощи дымовых шашек, артиллерийских мин, ручных гранат и средств воздушного нападения, Наряду с другими средствами специально для уличного боя разработаны ружейные гранаты, снаряженные адамситом и хлорацетофеноном.
Распыление ОВ с помощью бризантных мин требует больших зарядов взрывчатки, чем обычно принято для химических боеприпасов. Однако поскольку при этом испаряется и оседает в воздухе во взвешенном состоянии лишь часть вещества, а большая часть его диспергируется лишь грубо и поэтому менее эффективна, то специальной конструкцией мии стремятся нагреть ОВ, например подогреть мины перед выстрелом или при помощи обогревательной системы внутри мин. Наиболее эффективно применение ОВ с помощью дымовых смесей, чем достигается наибольшее распыление ОВ в воздухе. Такие ядовито-дымовые шашки, особенно с хлорацетофеноном, применяются во многих армиях капиталистических стран в учебных целях и входят в общее табельное снаряжение. Американская армия располагает многими так называемыми горючими смесями (burning mixtures). По конструкции и способу обращения с ними они соответствуют обычным дымовым шашкам. Их можно применять только при благоприятном направлении ветра и
105
скорости ие более 4 м[сек. Восходящий поток ветра быстрее, рассеивает вещество и тем еамым снижает его эффективность. Тем не меиее, при благоприятных условиях можно при больших расходах ОВ достигнуть глубин заражения от 10 км и более.
По миеиию западных военных специалистов, раздражающие ОВ играют большую роль для запугивания гражданского населения. Применение их в глубоком тылу противника с помощью средств воздушного нападения неизбежно вызывает у неподготовленного населения панику, потерю работоспособности и деморализацию.
Все средства применения раздражающих ОВ в американской армии, независимо от вида ОВ, помечены красным кольцом. Кроме того, применяется следующая маркировка:
9-Хлор-9,10-дигидрофеиарсазин (адамсит)..............DM-GAS
Дифенилхлорарсин......................................DA-GAS
Бромбензилцнанид......................................CA-GAS
Раньше средства применения бромбеизнлциаинда маркировали GAS-CA и двумя зелеными кольцами, для адамсита пользовались обозначением DM TOXIC, для хлорацетофеиоиа иногда применяли два красных кольца, а для дифенилхлорарсииа — одно белое кольцо.
Смеси ОВ обозначаются двойным символом. Например, дымовые смеси с хлорацетофеноном и адамситом имеют обозначение CM-DM-GAS.
Для растворов раздражающих ОВ к символу ОВ прибавляют буквы S или В, иапример растворы хлорацетофенона обозначают CNS и CNB.
3.4. ЗАЩИТА ОТ РАЗДРАЖАЮЩИХ ОВ И ИХ ДЕГАЗАЦИЯ
Надежной защитой от раздражающих ОВ является противогаз. Одевание защитной одежды становится необходимым только при чрезвычайно высоких концентрациях илн при работе с этими ОВ.
Противодымные фильтры современных противогазов задерживают взвешенные частички размером 1 — 5 ммк. Решающими для задержания аэрозоля в противодымном фильтре являются процессы передачи в самом фильтре. Они зависят от размера частиц, скорости воздуха, времени пребывания в противогазе и других факторов.
Для изготовления противодымных фильтров применяют обычно смесь прочных и тончайших волокон целлюлозы, хлопка н асбеста. Их укладывают в фильтр чередующимися толстыми, но рыхлыми слоями или тонкими, плотными, волокнистыми листами с большой поверхностью. В результате задержки материалом фильтра частички аэрозоля отделяются от вдыхаемого воздуха. Мощность противодымных фильтров возрастает с количеством уже задержанной пыли, при этом, разумеется, увеличивается и сопротивление дыханию.
106
Раздражающие ин на местности оиычни не дегазирую!. пели же они были применены в капельно-жидком состоянии, то при известных обстоятельствах может потребоваться дегазация, особенно для бромбензилцианида и хлорацетофенона. Ее производят с помощью водных растворов сульфида натрия. Мышьяк- и свинецорга-иические раздражающие ОБ этим способом дегазировать нельзя. В дегазации такизГ ОВ нет необходимости, так как они нелетучи; при оседании на поверхность почвы они становятся неэффективными.
Хлорацетофенон можно дегазировать. также окислителями, которые" пригодны и для дегазации мышьякоргаиических ОВ.
Предметы обмундирования дегазируют выбиванием, проветриванием или вывешиванием их на солнце. Если степень заражения высока, то обмундирование дегазируют обдувкой горячим воздухом. Оружие, материальную часть и другие предметы обтирают тряпкой, смоченной растворителем.
Воду, зараженную упомянутыми раздражающими ОВ, можно спокойно использовать для технических целен, но вследствие кожно-нарывного действия некоторых раздражающих ОВ ношение защитных перчаток при этом обязательно. Воду, зараженную мышьяк- и свинецорганическими соединениями, нельзя употреблять в пищу. Ее нельзя очистить обычными средствами. Воду, зараженную хлорацетофеноном, можно после тщательной проверки ограниченно использовать для пищевых целей.
Так как большая часть раздражающих ОВ применяется в виде аэрозолей, они лишь незначительно проникают в содержащие и иесодержащие жиры пищевые продукты и, после снятия верхних слоев (20—30 лип), эти продукты можно считать съедобными.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Объясните, почему такие галогеналканы, как дихлорэтан, четыреххлори-стыЙ углерод и другие, ие вызывают слезоточивого действия.
2. Почему галогензамещенные алифатические кетоны потеряли свае значение как слезоточивые ОВ?
3. Опишите способы получения хлорацетофенона,
4. Почему воздух заражается парами хлорацетофенона, но не заражается парами адамсита?
5, Как целесообразнее всего можно дегазировать галогензамещенные кетоны?
6. Как образуется хлорметилфенилкетоксим и какими свойствами он обладает?
7. Напишите схему основного гидролиза бромбензилцианида.
8. Какая зависимость между структурой и физиологической активностью раздражающих носоглотку мышьякорганических’ОВ?
9. От каких факторов зависит физиологическая активность раздражающих свинецоргаиических соединений?
10. К какой группе Вы отнесете раздражающие ОВ по тактической классификации?
11. Какие симптомы отравления возникают при вдыхании, паров мышьяк, органических соединений?
12. Как ведут себя мышья кор га ннческие ОВ в воде?
107
13. Какую реакцию следует ожидать при действии сухой хлорной известью на дифенилхлорарснн?
14. Чем объясняется красно^фиолетовое окрашивание теплого раствора адамсита в муравьиной кислоте?
15. Почему адамсит обладает меньшей реакционной способностью, чем ди-фенилхлорарсин?
16. Перечислите возможные способы получения солей триалкилсвннца.
17. Для какой цели и как был получен фторацетат триэтилсвинца?
18. Напишите уравнение реакции гидролитического разложения морфолида пеларгоновой кислоты.
19. Как устроен противодымный фильтр?
Л И ТЕРАТУРА
1. N a eve W., Arch. Toxikol., 18» 165 (i960).
2. S c h a e 1 e r J. P., Sonneberg F., J. Org. Chem., 28, 1128 (1963).
3. Organikum— Organisch-chemisches Grundpraktikum, 5 AulL, VEB Verlag, Berlin, 1965.
4. Bergmann J. F., Kalmus A., J. Am. Chem. Soc., 76, 4137 (1954).
5. Bull. Soc. chim. France, 15, 593 (1948’).
6. Mil 11 er H. H., Naturwiss., 33, 253 (1946).
7. J. Chem. Soc., 1949, 919, 2983; 1950, 684.
8. Ber„ 49, 2445 (1916); 55, 1282 (1922).
9. Am. Ind. Hyg. Assoc. J., 23, 194, 199 (1962).
4. УДУШАЮЩИЕ ОВ
Учитывая условия современного боя и наличие высокотоксичных фосфорорганических соединений, в настоящее время пока еще принимаются в расчет некоторые из соединений, применявшихся в первую мировую войну в качестве удушающих ОВ. Газообразный хлор, первым из ОВ этой группы примененный в апреле 1915 г., в результате принятия на вооружение средств защиты уже во второй половине войны больше не годился как ОВ, тем более, что его применение можно было обеспечить только методом газобаллонной атаки.
К концу 1915 г. газообразный хлор все больше и больше вытеснялся фосгеном, который до окончания войны сохранял наибольшее значение. К началу 1916 г. фосген применяли в артиллерийских боеприпасах и затем при помощи минометов, что было гораздо эффективнее, чем при газобаллонной атаке.
Общее количество фосгена, произведенное в период с 1915 по 1918 гг., оценивается в 150000т. Какое значение он имел в то время, видно из того, что около 80% личного состава погибших от ОВ приходится на долю отравления фосгеном. Позднее приобрели значение также дифосген и хлорпикрин. Хлорпикрин применяли преимущественно в смесях с другими ОВ.
Хотя в империалистических странах, прежде всего в США, эти ОВ (фосген, дифосген и хлорпикрин) как табельные ОВ не изготовляются и не хранятся, есть предпосылки для их применения и в дальнейшем. Необходимые средства применения держатся наготове, а предусмотренные планом мобилизации производственные мощности химической промышленности в данном случае можно будет подчинить военным нуждам.
Фосген и его производные являются важными исходными продуктами для производства пластмасс, синтетических волокон, красителей и т. д. Из-за большой потребности в фосгене производство этого соединения во всех странах с ведущей химической промышленностью за последние годы возросло.
Хотя хлор больше не имеет значения как ОВ, его ключевое положение в крупной химической промышленности достаточно известно. Один только факт, что большинство ОВ являются галогенированными соединениями, показывает, что этот продукт немыслимо исключить из производства ОВ. Свойства хлора известны из курса неорганической химии, поэтому в этой книге можно на ннх не останавливаться.
109
Предпринятые во время второй мировой войны в Германии и затем в'США исследования фторгалогенированных и фторированных соединений элементов главной подгруппы VI группы, в частности, фтористых соединений серы, повлекли за собой проверку пригодности их в качестве ОВ. Высокая токсичность, большая реакционная способность (некоторые хлорфториды воспламеняются при соприкосновении с органическими веществами) и поражающее действие не исключают применения этих соединений в качестве ОВ с многосторонним действием. Их. использование в химической промышленности в качестве фторирующих средств, которое предполагает крупное производство, несмотря на их агрессивность технологически осуществимо. Тем самым практически имеются определенные предпосылки, которые обычно необходимы для принятия ОВ на вооружение.
Удушающими ОВ, которые принимаются в расчет, являются следующие вещества: галогенпроизвсздные угольной кислоты, галогенированные нитроалканы, фторхлориды и фтористые соединения серы.
4.1. ГАЛОГЕНПРОИЗВОДНЫЕ УГОЛЬНОЙ КИСЛОТЫ
Некоторые токсичные соединения можно рассматривать как производные угольной кислоты О=С(ОН)2. Военный интерес представляют прежде всего хлорангидриды угольной кислоты, особенно дихлорангидрид — фосген и галогенированные сложные эфиры хлоругольиой кислоты, к которым относится дифосген.
4.1.1. Химическое строение и биологическая активность
4.1.1.1. Связь химической структуры с биологической активностью. Галогеипроизводиые угольной кислоты обладают разными физиологическими свойствами. Они определяются числом атомов хлора в молекуле.
В то время как дихлорангидрид угольной кислоты I является, выраженным удушающим ОВ и в начальный период отравление не вызывает в организме никаких защитных рефлексов, достаточно заместить один атом хлора на алкоксигрулпу, например, как при получении метилового эфира хлоругольиой кислоты (II), чтобы получилось сильное слезоточивое ОВ, которое, кроме того, обладает небольшим удушающим действием:
О--=С .. о=с^
\ci Х)СН?
I II
Предпосылкой физиологической активности таких соединений является наличие связи галоген — карбонильная группа. Замещение второго атома галогена на водород, алкильную нли алкокси^ 110
группу ведет к утрате физиологической активности. Для этих СО-единений, так же как и для монобром- и дибромпропанона, установлено, что увеличение числа атомов галогена имеет следствием изменение физиологических свойств. У бромпропаиоиов появляется побочное действие, а у галогенпроизводных угольной кислоты — ослабляется лакримогенное действие* и усиливается удушающее. С увеличением числа атомов хлора раздражающее действие снижается. В качестве примера следует назвать моно- и трихлорметн-ловые эфиры хлоругольной кислоты (III и IV), для которых пороги раздражения соответственно равны 0,002 и 0,005 мг/л.
X! XI
о=с о=с
\зСН2С1 ^ОСС13
III IV
В некоторых случаях военный интерес представляют также фтор- и бромсодержащие аналоги. Замена атома хлора в дихлор-ангидриде угольной кислоты атомом брома или фтора изменяет токсичность лишь незначительно. Вследствие большой неустойчивости дифтораигидрида угольной кислоты ои как ОВ вряд ли может иметь даже второстепенное значение.
4.1.1.2. Характер и механизм действия. По характеру своего действия на организм человека фосген и дифосген не различаются, тем более что принято считать, что токсическое действие дифосгена объясняется расщеплением его молекулы на две молекулы фосгена:
XI XI
О=су —> 2О=с/ ,
\оСС13 XI
Вообще фосген дает характерную картину удушающего действия. Симптомы отравления другими аналогичными соединениями те же.
Несмотря на широкие исследования, нет единой точки зрения на механизм отравления фосгеном. Установлено, что за токсичность этого соединения ответственны многие факторы и что фосген действует на огр а н из м в целом. Наиболее выраженным явлением при отравлении фосгеном является отек легких — просачивание плазмы крови в альвеолы. Это явление с физико-химической точки зрения можно объяснить либо повышением гидростатического давления крови в капиллярах легких, либо изменением проницаемости стенок капилляров1. Расширение капилляров легких и сжатие кле,-ток слизистой альвеол увеличивают проницаемость и способствуют скоплению жидкости. В результате таких изменений нарушается газообмен, жидкость плазмы мешает дальнейшей диффузии кислорода. Недостаток кислорода в легочной ткани и повышенная растворимость двуокиси углерода в экссудате способствуют повышению проницаемости стенок капилляров. Содержание двуокиси
углербда в крови увеличивается, а содержание кислорода падает.
Существует много теорий, отчасти противоположных друг Другу, объясняющих механизм действия удушающих ОВ.
Сначала отстаивали точку зрения, что происхождение отека легких можно объяснить эндоплазматическим гидролизом молекул фосгена. Образующиеся при гидролизе молекулы хлористого водорода якобы нарушают кислотно-осиовиое равновесие и таким образом изменяют проницаемость стенок капилляров. Однако экспериментальные исследования действия хлористого водорода показали, что для возникновения отека легких требуется почти в 800 раз большее количество хлористого водорода, чем образуется при гидролизе фосгена.
Более убедительной кажется теория, утверждающая, что при действии фосгена идут реакции с различными продуктами обмена веществ и ферментами, функциональные группы которых ацилируются и связываются с группой >С=О, в результате чего жизненно важные процессы обмена веществ прекращаются. С другой стороны, с тем же успехом можно допустить ацилирование важных компонентов тканей легких.
Токсический отек легких, возникающий после вдыхания паров фосгена или дифосгена, проявляется лишь после скрытого периода в несколько часов. В этот период почти отсутствуют симптомы, по которым можно диагностировать отравление; отравленный чувствует себя хорошо и, как правило, вполне дееспособен. У восприимчивых людей в это время появляется в качестве первого симптома сладкий, часто противный привкус во рту, иногда тошнота н рвота. В большинстве случаев возникают незначительные позывы к кашлю (который спустя некоторое время становится защитным рефлексом), першение и жжение в носоглотке, небольшие иаруше- . ния ритма дыхания и пульса. Эти явления дают себя зиать преимущественно у лиц с больными легкими.
После латентного периода наступает сильный кашель, одышка, синюшность лица и губ. Из-за мышечных напряжений отек легких может развиваться быстро и привести к летальному исходу.
Прогрессирующий отек легких ведет к сильному удушью, мучительному давлению в грудной клетке, ритм дыхания увеличивается от 18—20 в мин (норма) до 30—50 в мин, в кризисе — до 60—70 в мин. Дыхание судорожное. Содержащая белок отёчная пенистая и вязкая жидкость, выбрызгиваемая из альвеол и бронхиол в более широкие дыхательные пути, ведет к затруднению дыхания. Отравленный отхаркивает большие количества этой жидкости, часто смешанной с кровью. При токсическом отёке легких примерно от до У2 общего количества крови переходит в легкие, которые в результате этого опухают и значительно увели-» чиваются в массе. В то время как нормальное легкое весит около 500—600 г, можно было наблюдать «фосгеновые» легкие весом до 2500 е.
112
Гипоксия крови, потеря плазмы, повышенное содержание гемоглобина и эритроцитов почти вдвое увеличивают вязкость крови. Эти изменения замедляют кровообращение и ведут к опасной перегрузке сердечной мышцы. Кровяное давление резко падает. Отравленный пребывает в сильном возбуждении, дышит с шумом, хватает ртом воздух.
Встречаются также случаи, когда отравленный избегает любого лишнего движения и для облегчения дыхания выбирает какое-то наиболее удобное положение. Губы у таких отравленных серые, лот холодный и липкий. Несмотря на удушье, мокрота у них не отделяется. В подобных случаях наряду с отёком легких происходит нарушение кровообращения. Через несколько дней отравленный умирает,
Иногда через 2—3 суток может наступить улучшение состояния, которое через 2—3 недели может закончиться выздоровлением, но часто осложнения в результате вторичных инфекционных заболеваний могут привести к смертельному исходу.
При очень высоких концентрациях ОВ отёк легких не развивается. Отравленный делает глубокие вдохи, падает на землю, корчится и бьется в судорогах, кожа на лице становится от фиолетово-синей до темно-снней, н очень быстро наступает смерть2.
4.1.2. Общие свойства
Галогенангндриды угольной кислоты и большая часть эфиров галоген угольных кислот, получающихся в результате алкоголиза ангидридов, являются газообразными или жидкими соединениями.
Высшие члены ряда, как например, бнс-трнхлорметнловый эфир угольной -кислоты — твердые вещества. Пожалуй, важнейшим и наиболее известным представителем галоген ангидридов является днхлорангндрнд угольной кислоты I (стр. 114).
Аналогичное фтористое соединение V получено Руффом в 1934 г. Оно было исследовано во время второй мировой войны наряду с другими фторпронзводными в Англин. Теперь днфторангид-рнд угольной кислоты получают реакцией фосгена с фтористой сурьмой. Из-за неблагоприятных физико-химических свойств он столь же мало пригоден в качестве ОВ, как и смешанный галоген-ангидрнд VI.
О=С
ZF \F
о=с
VII
zOCH3 о=с(
VIII
Бромистый аналог —нестабильное соединение. Он был отвергнут как ОВ уже во время первой мировой войны.
Из сложных эфиров галогенугольных кислот наиболее ценными для ОВ свойствами обладает трнхлорметиловый эфир хлоругольной кислоты. Хотя моно- и днхлорметиловые эфиры являются
ИЗ
сильными раздражающими ОВ, вызывающими известное удушающее действие, возможное применение их в качестве боевых ОВ приходится отрицать. Метиловый эфир хлоругольной кислоты VIII оказывает чрезвычайно сильное слезоточивое действие. Одно время его добавляли в качестве предостерегающего вещества к высокотоксичным средствам борьбы с вредителями сельского хозяйства, например, к синильной кислоте (циклон В).
Из-за наличия связи углерод — хлор н подвижности атома галогена как фосген, так и эфиры хлоругольиой кислоты в соответствии с их строением О —СХ2 следует считать галогенангидридами угольной кислоты. Водой они быстро гидролизуются с образованием двуокиси углерода и соответствующих продуктов разложения. При взаимодействии с алифатическими спиртами оии дают сложные эфиры, с аминами образуют амиды угольной кислоты и применяются (особенно фосген) для введения группы СО в другие соединения.
4.1.3. Дихлорангидрид угольной кислоты
/Ci
О-С\с|
Синонимы: карбон ил хлорид, хлорокись углерода, фосген.
Военные обозначения: нем, — Griinkreuz, D-Stoff; англ. — PG-Mixture (в смеси с хлорпикрином); амер. — CG; франц. — Collongite (в смеси с четыреххлористым оловом).
4.1.3.1. Методы получения. Как в лаборатории, так и в крупнопромышленном масштабе фосген можно синтезировать по одному и тому же принципу, заключающемуся в присоединении хлора к окиси углерода в присутствии катализатора:
С12 4- СО СОС1Й 4- 26 ккал
Впервые это соединение получил Деви (1812 г.) выдерживанием иа свету смеси окиси углерода с хлором (фотокатализ).
Промышленный способ. Вследстие большого значения фосгена для химической промышленности существует много технологических схем его промышленного получения, основанных на приведенной выше реакции.
Во избежание образования хлористого водорода исходные газообразные хлор и окись углерода приходится предварительно тщательно сушить, а для достижения максимального выхода — очищать. В смесителе приготовляют соответствующую газовую смесь, которую затем при 20 °C вводят в контактные печи.
Обычно контактные печи типа колонн имеют высоту 10 я н диаметр 1 я. В качестве катализатора применяется активированный уголь, находящийся иа колосниковых решетках этих колонн. Вместо реакционных колонн часто используют реакторы в виде последовательно соединенных коробов. Для отвода теплоты реак-
114
реакции не должна превышать 150°С, оптимальные выходы получают при 125 °C.
Фосген, загрязненный окисью углерода и хлором, пропускают через охладительную систему, состоящую из водяного и глубокого ( —15°С) охлаждения, при этом конденсируется 90% образовавшегося фосгена. Остаток фосгена удаляют из газовой смеси промыванием ее тетрахлорэтаном в скрубберах. Из раствора в тетра-хлорэтане фосген выделяют в перегонных аппаратах и пропускают в охладительную систему. Тетрахлорэтан также охлаждают и вновь используют в производстве. Остаточные газы отводят. Так как онн обычно содержат значительные количества хлора, их часто используют для получения некоторых продуктов.
Лабораторный способ. Оба исходных газа — окись углерода и хлор — предварительно сушат порознь (рис. 5), сначала пропуская через промывную склянку 2 с серной кислотой, а затем через осушительную колонку 4 с хлористым кальцием. Смесителем служит трехгорлая склянка Вульфа 5, снабженная двумя
Рис. 5. Прибор для получения фосгена;
i — предохранительные склянки-, 2, 3 — промывные склянки; 4 —осушительные колонки! 5 —склянка Вульфа; 6 — холодильник Либиха; 7, 8 —ловушки.
доходящими до дна трубками для ввода газов и одной отводной более короткой трубкой. В качестве реактора применяют длинный холодильник Либнха 6, трубка которого, заполненная крупнозернистым активированным углем, охлаждается водой для отвода тепла реакции.
Фосген в основном конденсируется в соединенных друг с другом ловушках 7, помещенных в охладительную смесь (лед с солью). Остаточные газы можно дегазировать, поглощая их раствором гидроокиси натрия в ловушке 8.
Хлор вводят в смесь со скоростью 5—6 пузырьков в секунду, а окись углерода со скоростью 8—9 пузырьков в секунду. Для точного определения выхода нужно измерять расход газов с помощью ротаметров, помещаемых перед смесителем. Измерение объема газов производят только после насыщения катализатора газовой смесью. Расход газов должен составлять 8—10 л/ч.
При использовании баллонов со сжатыми газами нужно после баллона ста-ййть предохранительную склянку 1.
4.1.3.2. Физические свойства. Фосген представляет собой бесцветный газ, который ниже-8,2 °C конденсируется в бесцветную жидкость. Его запах напоминает прелые фрукты или сено. Технический продукт имеет слегка желтую или красновато-желтую окраску. Фосген примерно в 3,5 раза тяжелее воздуха. Из-за высокого
115
давления пара он даже при низких температурах обладает большой летучестью. Ниже приведены физические свойства фосгена:
Т. кип., °C
при 137 мм рт. ст.......... —30
при 760 мм рт. ст.......... 8,2
Т. пл., °C.................... -118
Давление пара при 20 °C, мм рт. ст. .. . 1173
Максимальная концентрация при 20 °C, мг)л ................. 6370
Относит, плотность пара .........»............. 3,48
жидкости d™ ............ 1,38
Критическая температура, °C ...... 183(181,7)
Критическое давление, ат...... 56 (55,3)
Летучесть фосгена достаточна для достижения токсических концентраций в зимнее время. Стойкость при —20 РС составляет около 3 ч, в летние месяцы она чрезвычайно мала — ие более 30 мин. Летучесть при —20°С равна 1,4 г/л, при -f-20oC—-около 6,4 г/л. Вследствие метеорологических воздействий фактическая концентрация фосгена в воздухе меньше и едва ли превышает 1 г/л *.
В интервале температуры от —15°С до 4-25°С давление пара фосгена (в мм рт. ст.) можно вычислить по формуле:
1g р = 7,5595 -3^ Температура, °C ....................—10 —5 0 8.2 10 20 25
Давление, мм рт. ст. . ........... 365 452 555 760 840 1173 1379
Фосген можно легко конденсировать сжатием, его критическая температура составляет 183°С, критическое давление 56 ат.
Ниже —118°C фосген застывает в белую кристаллическую массу. По некоторым данным т. пл. фосгена— 126 °C.
В холодной воде фосген растворяется мало —0,9%. Он легко растворим в органических растворителях, например, в галогенал-канах, бензине, толуоле, ксилоле, уксусной кислоте.
С военной точки зрения представляет интерес хорошая растворимость фосгена в хлорпикрине, иприте, арил- и алкилхлорарси-нах и в кислотных дымообразователях — четыреххлористых крем; нии, олове и титане; Смеси фосгена с дымообразователями Применялись в первую мировую войну и были заготовлены в больших количествах во время второй мировой войны.
4.1.3.3. Химические свойства. При обычной температуре И в отсутствие влаги фосген — стабильное соединение. При сильном нагревании он частично разлагается на хлор и окись углерода:
СОС12 С12 + СО
* Такие концентрации практически никогда в полевых условиях не достигаются.— Прим. ред.
116
СЫШЙ OUU СГП riUJinULlDtU—диииицршр^ Cl .-1\UJ1H ЧСС 1 PU—lipUAV п-
тов разложения, образующихся при взрыве, ничтожно, поэтому возможно применение фосгена в артиллерийских снарядах и т.п.
Большая часть металлов не разрушаются сухим фосгеном, исключение составляют цинк, алюминий и медь. Эти металлы образуют с фосгеном соответствующие хлориды. В присутствии A1CU с фосгеном реагируют и другие металлы. Влажный фосген вызывает коррозию всех металлов.
При хранении в стальных емкостях, например, при длительном нахождении в минах, образуется карбонил железа Fe(CO)s.
Это — красновато-желтая жидкость, тяжелее фосгена. Карбонил железа менее токсичен, чем фосген, но фотокаталитически он разлагается с образованием весьма опасной окиси углерода:
2Fe(CO)s —> СО + Fe3(CO)e
По этой причине пентакарбонил железа был в 30-х годах предложен в качестве ОВ (см. раздел 6.3.2).
Гидролиз. Фосген, растворенный в воде, быстро гидролизуется даже при низкой температуре:
СОС12 + Н2О —> 2НС1 + СО2
При 0°С в 100 г воды за 20 сек полностью гидролизуется 1 г фосгена.
Газообразный фосген почти не гидролизуется парами воды, поэтому концентрация фосгена, созданная в воздухе, заметно изменяется лишь через долгое время (см. табл. 9, стр. Н8). При сравнительно высокой влажности воздуха облако фосгена за счет частичного гидролиза может приобрести беловатый отсвет. Добавка активированного угля и щелочей ускоряет гидролиз:
СОС12 4- 4NaOH —> Na2CO3 + 2NaCl + 2ЩО
Одно время для гидролитического разложения фосгена в коробку противогаза помещали щелочные вещества, например, натронную известь. Современные противогазы содержат активированный уголь, импрегиированный основными веществами. Фосген разлагается каталитически, а освобождающаяся при этом соляная кислота связывается основаниями.
Реакция с аммиаком и аминами. Газообразный и жидкий аммиак энергично реагируют с фосгеном с образованием диамида угольной кислоты (мочевины):
/С1 o=cf
+ 4NH3
-2NH1C1
о=с
^nh2
Способ дегазации зараженного фосгеном воздуха посредством выпуска аммиака из баллонов, особенно при обработке казарм, блиндажей и др., является спорным. По данным фирмы «Дрегер-верке»,- скорость гидролиза фосгена водяным паром и скорость
117
реакции с аммиаком меньше, Чем можно иали ожидать11, ^равни-тельно быстрее идет реакция с аммиаком в присутствии водяного пара (табл. 9). Опыты проводились примерно при 20°С, исходные смеси газов приготавливались в баллонах емкостью 50 л. Разумеется, речь идет не столько о реакции в газовой фазе, сколько о разложении иа стенках сосуда.
Таблица 9. Кинетика разложения фосгена водяным паром и аммиаком (Смесь 1 — 12.5 м.д, СОС1г. 20 000 м. д. Н2Ог; смесь II-12.5 м.д. СОС12, Ю0 м. д. NH».
7 000 м.д, HjOr; смесь III —12,5 м.д, COCla, 100 М.Д. NH3, 20 000 м. д. Н2Ог)
Время, ч Непрореагировавший фосген.% Время, ч Непрореагировавщий фосген,и
смесь I смесь II смесь III смесь 1 смесь II смесь III
0 100 100 100 6 60 46
0,5 94 94 90 10 56 50 34
I 88 88 82 16 48 40 . 24
2 80 72 22 42 38 18
4 70 68 - 56
С первичными аминами фосген реагирует с образованием соответствующих производных мочевины, напримор при. употреблении анилина образуется дифенил мочевина, реакция протекает количественно:
/С1 zNHCeH3
О=С\ + 4C6HbNH2 —г О=С + 2C6HsNH2’ НС1
\ci ^NHCfiHe
При взаимодействии фосгена с 2,4,6-трихлоранилином при 100—120°С в растворе нитробензола получается симметрическая бис-(2,4,6-трихлорфеннл)-мочевина, которая в уксуснокислом растворе под давлением и повышенной температуре хлорируется 4 гипохлоритом натрия до N, N'-дихлор-бис-(2,4,6-трихлорфеннл)-мочевины:
Долгие годы этим веществом в американской армии импрегни-ровали обмундирование для защиты от кожно-нарывных ОВ (см, раздел 5.1.3.2),
118
Вторичные амины дают соответствующие тетразамещенные производные, третичные в такую реакцию с фосгеном не вступают. Некоторые третичные амины, например гексаметилентетрамин, образуют с фосгеном продукты присоединения:
COCI2 4- N4(CH2)e —► N4(CH2)6 • СОС12
Поэтому этот тетр амин применяли в жидкостных респираторах. Аналогично, пиридин образует аддукт 2CeH5N*COCl2. Оба продукта присоединения разлагаются при действий воды.
Алкоголиз. Фосген реагирует с первичными и вторичными спиртами, а также с фенолами, с образованием соответствующих эфиров угольной кислоты. Первый атом хлора замещается быстро (реакция 1), а вторичное замещение происходит значительно медленнее (реакция 2):
-Cl
<1
+ HOR
-НС1
(О
O=C,
+ HOR -HCl
-OR
(2)
Монозамещениые продукты, метиловый или этиловый эфиры хлоругольной кислоты, получаемые при действии на фосген метиловым или этиловым спиртом, являются сильными слезоточивыми веществами, служившими в качестве ОВ в первую мировую войну, Диметиловый эфир угольной кислоты — устойчивое соединение. Из него хлорированием получают бис-трихлорметиловый эфир угольной кислоты (см. раздел 4.1.5). /
Способность фенола или фенолятов давать сложные эфиры с фосгеном дала возможность использовать эти вещества в качестве поглотителей в противогазах.
Алкилхлоругольиые эфиры, промежуточно образующиеся в ре-. акции с третичными спиртами, разлагаются с образованием соответствующих хлоралканов:
Г1
о=сх 4- КзСОН
Cl
COa 4- RgCCi
Другие реакции фосгена, в частности, введение СО-группы, используются в промышленности и в анализе.
4.1.3.4. Токсические свойства. Фосген ядовит только при вдыхании паров. Первые отчетливые признаки отравления появляются после скрытого периода от 4 до 8 ч; наблюдались даже периоды в 15 ч.
По различным данным вдыхание фосгена в концентрации 0,004 мг[л в течение 60—90 мин ие приводит к отравлению. Пребывание в атмосфере, содержащей до 0,01 \ме/л, возможно
119
максимально в течение I ч. При этом восприимчивые лица могут получить легкое отравление. Концентрации в 0,022 мг/л являются смертельными уже через 30.тин экспозиции. В 50% случаев отравление при вдыхании 0,1 мг/л в течение 30—60 мин приводит к смерти. Остальные 50% оставшихся в живых длительно небоеспособны в результате тяжелейших отравлений. Даже при малом времени воздействия таких концентраций могут произойти сильные отравления, при известных обстоятельствах заканчивающиеся смертельным исходом.
Концентрация 1 мг/л при времени экспозиции 5 мин в 50—75% случаев отравления ведет к смерти; меньшие концентрации (0,5— 0,8 мг/л) приводят к тяжелым отравлениям. Концентрация 5 мг/л смертельна уже через 2—3 сек.
Малые концентрации фосгена влияют иа вкусовые ощущения. Так, например, курить сигарету в зараженном фосгеном воздухе неприятно или вовсе невозможно.
Запах фосгена еще ощутим в концентрации 0,004 мг/л. Однако иа обонятельный нерв фосген влияет так, что в дальнейшем обоняние притупляется и перестают ощущаться даже более высокие концентрации.
4.1.4. Трихлорметиловый эфир хлоругольиой кислоты
/С1 о=с% ^ОСС13
Синонимы: трихлор метиловый эфир хлормуравьиной кислоты, трихлор-метилхлорформиат, трихлорметилхлоркарбопат, дифосген.
Военные обозначения: нем. — Perstoff, Griinkreuz; англ. — Superpa-lite, Diphosgen; амер. — DP; франц. — Surpalite.
4.1.4,1. Методы получения. Хлорированием метилового эфира хлоругольной кислоты при УФ-освещении можно получить все три хлорметиловых эфира хлоругольной кислоты:
Xе1 +С1г zcl. +ci2
О-=с ——О=с; —
\<эсн3 нс \эсн2а нс
/С* +с12 /^1
—* °=< О=С
Х)СНС12 н \ОСС13
Образовавшуюся смесь разделяют фракционной перегонкой. Из-за близости температур кипения разделение моно- и днхлор-производных затруднено (монохлорметиловый эфир кипит при 107 °C, дихлорметиловый при 110 °C, трихлорметиловый при 128 °C).
4.1.4.2. Физические свойства. Дифосген — бесцветная легкоподвижная, маслянистая жидкость. Его запах напоминает запах фосгена, он кипит при 128 °C и застывает при —57 °C. Технический 120
приду К1 UWllX 4VLVL 11U /V UJ<,1U11I nu Ainpuv^i^, ----------
ные количества моно- и дихлорпроизводных, бис-трихлорметило-вый эфир угольной кислоты, хлористый водород, хлор и др.
Ниже приведены физические свойства дифосгена:
Т. кип., °C при 18 мм рт. ст........................ 41
при 760 мм рт. ст...................127,5—128
Т. пл., °C.................................. -57
Давление пара, мм рт. ст. при 0 °C................................. 3
при 10 °C................................. 5
при 20 °C............................' 10,3
при 30 °C................................ 16,3
Максимальная концентрация при 20 °C, мг/л ........... .120
Относ, плотность пара d*5................... 1,644
Показатель преломления .................... 1,4566
Вследствие малого давления nafa дифосген дольше удерживается на местности, чем фосген. При благоприятных условиях приходится считаться с эффективностью дифосгена в течение примерно 3 ч. На открытой местности летом стойкость составляет около 0,5 ч, а зимой действие может продолжаться до 10—12 ч. Возможно найдет применение в виде аэрозоля.
Концентрация, создаваемая за счет испарения, достаточна, чтобы вызвать тяжелое отравление и поэтому пребывание на зараженной местности без противогаза невозможно.
Высокая летучесть и сравнительно высокая при благоприятных условиях стойкость позволяют отнести дифосген к полустойким ОВ.
В воде он почти нерастворим, растворимость в органических растворителях, например, в галогеналканах и хлорбензоле, высока. Хорошая смешиваемость с хлорпикрином, фосгеном, дифенилхлор-арсином и другими ОВ, а также с жидкими дымообразователямн определяет важность дифосгена для приготовления тактических смесей. Растворим в четыреххлористых олове, кремнии н титане.
Устойчивость дифосгена во время хранения мала. Хлорное железо вызывает его каталитическое разложение. В качестве стабилизаторов, наряду с другими, применяют фенол в количестве 1— 2%. Однако фенол не может воспрепятствовать распаду, а лишь на время задерживает его, поэтому его приходится периодически добавлять. Образование хлорного железа можно успешно подавить введением соответствующих ингибиторов коррозии, поэтому возможно длительное хранение дифосгена без больших затрат.
4.1.4.3. Химические свойства. При нагревании дифосген разлагается на две молекулы фосгена:
С1С—ОСС13 —> 2СОС12
121
активированный уголь, окислы железа, глнна, AICI3, FeCl3 и SnCh, разлагают дифосген уже при обычной температуре. В присутствии АЮз и ZnCl2 образуются преимущественно двуокись углерода и четыреххлористый углерод:
>ОСС13 А1С1а
О=С ----------:---> СО2 + СС14
\С1
Дифосген устойчив к детонации.
Химические свойства дифосгена определяются его строением. Он дает реакции, характерные для хлорангидрида и сложного эфира. В большей-части реакций дифосген ведет себя аналогично фосгену.
Гидролиз. Гидролитическое расщепление дифосгена происходит медленно, ему способствуют нагревание илн присутствие щелочей. Прн кипячении в водных растворах дифосген полностью гидролизуется в течение нескольких минут:
/С1
+ 2НаО 2СО2 + 4НС1 ^ОСС13
XI
О=(/ + 8NaOH —► 2Na2CO3 + 4NaCl + 4Н2О
ч)СС13
✓Cl
О=с/ + 2Na2CO3 —> 4NaCI + 4СО2
Х)СС1з
При действии водного раствора сульфида натрия наряду с другими продуктами образуется сероуглерод:
/С1
O=CZ + 2Na2S —> СО2 + CS2 + 4NaCl ^OCCla
В случае крайней необходимости нагретые растворы сульфида натрия можно использовать для дегазации дифосгена.
Реакции с аммиаком и аминами. Подобно фосгену, дифосген реагирует с аммиаком с образованием мочевины. Эта сильно экзо-термнчная реакция при применении концентрированных растворов аммиака заканчивается в течение нескольких секунд.
Аналогично фосгену протекают и реакции дифосгена с анилином, гексаметилентетрамином и пиридином. Так, с анилином образуется днфенилмочевина. Другие реакции можно здесь не обсуждать.
4Д.4Л. Токсические свойства. Дифосген ядовит при вдыхании паров. В виде жидкости он через кожу не проникает. Возникающие на поверхности кожи признаки раздражения несущественны и почти не принимают характера ожога.
Дыхательные пути лишь слабо раздражаются парами дифосгена. Картина заболевания соответствует отравлению фосгеном.
Порог раздражающего действия дифосгена на глаза составляет 0,005 мг{л. Концентрация в 0,04 мг}л еще переносима при длительности пребывания 1 мин-, концентрации около 1 мг}л при времени экспозиции Ьмин смертельны. Для экспозиции около 15лшн смертельная концентрация составляет 0,5—0,7 мг}л.
4.1,5. Бис-трихлорметиловый эфир угольной кислоты /ОСС18
Синонимы: гексахлордиметиловый эфир угольной кислоты, гексахлорди* метилкарбонат, трифосген.
Это бесцветное кристаллическое вещество, т. пл. 78—79 °C, т. кип. 205—206 °C при 760 мм рт. ст. и 124 °C при 50 мм рт. ст.\ ^4°1,6. Он растворим в органических растворителях, таких как бензол, эфир н галогеналканы.
При нагревании до температуры кипения ои частично разлагается с образованием фосгена и дифосгена:
/ОСС18 ZCI
Х)СС18 хОСС1з
Трифосген получают либо хлорированием диметнлового эфира угольной кислоты, либо в качестве побочного продукта при получении галогенированных алкиловых эфиров хлоругольной кислоты.
В химическом и физиологическом отношении трнфосгеи ведет себя, как фосген илн дифосген. Гидролитическое расщепление до соляной кислоты и двуокиси углерода идет медленнее, чем в случае дифосгена. Реакции могут также способствовать нагревание и Катализаторы.
4.2. ГАЛОГЕНИРОВАННЫЕ ННТРОАЛКАНЫ
Галогенированные ннтроалканы являются производными насыщенных углеводородов, у которых атомы водорода замещены на ннт-рогруппы и галогены.
В первую мировую войну из веществ этой группы в больших количествах применялся всеми воюющими странами трнхлорннтро-метан (хлорпикрин) и отчасти — аналогичное бромистое соединение.
Как известно из курса органической химии, получение простых ннтроалканов представляет трудности, так как прямое нитрование алканов, возможное для циклических соединений, осуществимо только для высших алифатических гомологов. Обычно при получении низших гомологов исходят нз галогеналканов нлн галогеикар-боиовых кислот, которые обрабатывают нитритами.
123
Во время первой мировой войны трихлорнитрометан получали из пикриновой кислоты и хлориой извести. Сильное физиологическое действие и удовлетворительные физико-химические свойства позволили оценить его как ОВ, и явились достаточным поводом, чтобы получать и применять его в больших количествах.
Хлорпикрин и другие соединения этой группы в настоящее время производятся в промышленном масштабе для синтеза различных органических соединений, например, для получения средств борьбы с вредителями сельского хозяйства,
4.2.1. Химическое строение и биологическая активность
Уже простое введение нитрогрупп в молекулу органического соединения обычно придает ему ядовитые свойства. Здесь следует вспомнить о таких соединениях, как нитробензол, нитротолуол н тринитротриметиламнн, которые при небрежном обращении с ними могут вызвать тяжелые последствия для здоровья.
Прн введении нитрогрупп в галогенированные соединения физиологическая активность, по-видимому, изменяется н увеличивается. Так, галогеналканы, например трнхлорэтаи, тетрахлор-этан н хлороформ, известны как сильные яды для почек и печени, вызывающие как острые, так и хронические отравления. В результате ^ведения нитрогрупп в галогеналканы их токсические свойства расширяются — они приобретают сильное слезоточивое и отчасти удушающее действие.
То, что это верно не во всех случаях, можно показать иа примере двух изомерных соединений: 1-хлор-2-ннтроэтана I и 1-хлор-1-нитроэтаиа II
zNO2 cich2ch2no2 сн3сн^
I II
Соединение II обладает некоторым, хотя и слабым, лакрнмо-генным действием, тогда как соединение I не является лакриматором. Очевидно, для того чтобы физиологическая активность в смысле отравляющего действия сохранилась, необходимо, чтобы и иитрогруппа, н атом галогена были связаны с одним и тем же атомом углерода, как это можно видеть на примере трихлорнитрометана III и его производных.
Токсичность производных этана, очевидно, определяется числом нитрогрупп. Если соединение II является лишь слабым раздражающим ОВ, то сщиле-тетрахлординитроэтан IV, напротив, сильно ядовит и по своему действию превосходит хлорпикрин III. Так же обстоит дело в случае спл/л/-тетраннтродихлорэтана V
CI3CNO2 С12С—СС12 (NO2)2C—С (NO2)3
o2n no2 ci ii
III IV V
124
Замена атомов хлора на бром ведет к постепенному ослаблению физиологических свойств. Порог раздражения для хлорпикрина III составляет от 0,001 до 0,002 мг)л, а для трибромнитрометана — 0,03 мг/л.
О поведении фторсодержащих аналогов пока известно немного. Однако, согласно имеющимся публикациям, они должны быть более токсичны, чем хлористые соединения.
Галогенированные иитроалканы— это прежде всего относится к трихлорннтрометану— хорошо растворимы в липидах и в этом отношении оии подобны галогеналканам. Они действуют как ингаляционные яды н как слезоточивые ОВ, в жидком виде вызывают на коже сильные ожоги, которые обычно приводят к образованию волдырей и к сильным некротическим распадам тканей.
Как слезоточивые ОВ они уже в самой малой концентрации вызывают смыкание век; жжение и боль в глазах, слезотечение наступают без предшествующего скрытого периода.
Как ингаляционный яд хлорпикрин (при одинаковых концентрациях) обладает более коротким латентным периодом, чем фосген и дифосген. Очевидно, что вследствие иной природы соединения иной и механизм действия, ио, как и в случае отравления фосгеном, он вызывает отек легких.
В качестве доказательства этого положения Гнллерт2_ приводит в своей книге результаты внутривенных инъекций фосгена, дифосгена и хлорпикрина; инъекция хлорпикрина дает отёк легких, а инъекции фосгена н дифосгена не вызывают никаких последствий. Кроме того, этн соединения устойчивы к гидролизу, поэтому теории, выдвинутые для объяснения отравления фосгеном, могут быть неверными в случае галогенированных ннтроалканов. Устойчивость к гидролизу нужно считать причиной сильного резорбтивного проникновения галогенированных ннтроалканов в другие органы. Посредством разрушения клеток этн соединения действуют на печень, почки и сердце. Последствиями являются нарушения определенных процессов обмена веществ, нарушения кровообращения, легкая дефекация нли стабильные запоры, боли в области желудка н др.
Первые признаки ингаляционного отравления хлорпикрином заключаются в раздражениях полости глотки, Кашле, рвоте (отсюда название «рвотный газ»), легком наркотическом действии (подобном хлороформу) и в депрессиях.
4.2.2. Общие свойства
Галогенированные ннтроалканы представляют собой как -жидкие, так и кристаллические соединения. Они неустойчивы к нагреванию.
С точки зрения их строения эти вещества можно рассматривать и как нитроалканы, и как галогеналкаиы. Как таковые оии нерастворимы в воде и не гидролизуются. Поскольку гидролиз может происходить на границе раздела фаз, то для проведения гидролиза
125
необходима гомогенизация смеси. Но даже при гомогенизации (‘например, в водно-спиртовом растворе) нуклеофильного действия воды недостаточно для такого смещения электронной плотности, которое необходимо для отщепления атома хлора. Гидролиз возможен после добавки раствора едкой щелочи, поскольку нуклеофильность ионов ОН- значительно больше, чем воды.
Из-за присутствия в этих соединениях нитрогрупп добавление щелочи оказывается еще более необходимым, чем, скажем, в случае просто галогеналканов, так как группа NO2 относится к наиболее сильным акцепторам электронов, и в результате ее —/-эффекта атомы галогена более прочно связаны с атомом углерода:
В таких случаях гидролиз может осуществиться только при повышенной температуре. Вследствие содержания спирта-в реакционной смеси, например в спиртовых растворах щелочи, в то же время происходят побочные реакции.
Так же как в нитроалканах, нитрогруппу в. данном случае можно восстановить сильными восстановителями, причем одновременно отщепляются атомы хлора. В результате активации а-атомов водорода в случае -неполностью галогенированных гомологов проявляются другие возможности замещения.
4,2.3. Трихлорнитрометан
С13с—no2
Синонимы:- хлорпикрин, нитро хлороформ.
Военные обозначения: нем, —Klop (смесь с хлором), Griinkreuz-l (смесь с дифосгеном); англ. — PG-Mixture (смесь с фосгеном); амер. — PS,. NC-Mixture (смесь со SnCl4), Vomiting Gas; франц. — Aquinite (смесь со SnCh).
4.2.3.1. Методы получения. Стенхауз (1848 г.) получил трихлор ни громе тан при взаимодействии 2,4,6-грннитрофенола (пикриновой кислоты) с хлориой известью. Позднее этот способ был использован в промышленных процессах производства, при помощи которых в первую мировую войну хлорпикрин получали для военных целей.
Кроме синтеза хлорпикрина из пикриновой кислоты и гипохлорита кальция его можно получать следующими способами: хлорированием нитрометана; нитрованием хлорированных углеводородов, например хлороформа, трихлорэтана, а также трихлорацетальдегида; нитрованием алкаиов с последующим хлорированием.
Действие гипохлоритов кальция (к этим методам получения относится прежде всего применение дешевой хлориой извести) приводит к расщеплению цикла 2,4,6-тринитрофенола и одновременно
126
ключается в следующем:
N02
4-11CIO---------------> 3CC13NO2
-2СГ; -ЗНО-;
-ЗС0з~
no2
Вместо трудно растворимой в воде пикриновой кислоты применяли легкорастворимый пнкрат кальция, который получали смешением пикриновой кислоты с окисью кальция, и к смеси добавляли кашицу хлорной извести. Теплоты реакции достаточно для испарения образующегося трихлорнитрометана, пары которого затем конденсируют. Хлорпикрин можно легко перегнать с водяным паром.
В реакции можно использовать не только хлорную известь, но и непосредственно хлор, который пропускают в щелочной раствор пикриновой кислоты или других нитрофенолов. Все эти реакции протекают в общем по приведенному выше уравнению.
При применении гипохлоритов кальция, хлора и т. п. работают в щелочном растворе, который одновременно связывает кислоту. Соляная кислота ингибирует расщепление цикла.
4.2.3.2. Физические свойства. Хлорпикрин — бесцветная, маслянистая, сильно преломляющая свет жидкость с резким специфическим запахом. На свету ои становится зеленовато-желтоватым, что ‘можно объяснить частичным его разложением с образованием хлора и окислов азота. Он обладает следующими физическими свойствами.
Температура кипения хлорпикрина 112—113°С (760 мм рт. ст.) и 49°C (40 мм рт. ст.), температура плавления — 69,2 (64) °C. Его давление пара (в мм рт. ст.) можно рассчитать по формуле:
, оолол 2045,2
Ig р = 8,2424--if—
В табл. 10 приведена зависимость от температуры давления пара, максимальной концентрации и плотности. Относительная плотность пара хлорпикрина составляет 5,7.
Таблица.10. Зависимость давления иара, максимальной концентрации и плотности хлорпикрина от давления
Температура, °C Давление пара, мм рт. ст. Максимальная концентрация, мг/л Относительная плотность 4
0 5,91 57 1,693 1
10 10,87 104 1,6755
20 16,91 184 1,6579
30 30,50 295 —*
127
штгриктги температурный ивiервал, б ширим ллириикрин существует в жидком состоянии, и его высокая летучесть даже при низких температурах позволяют применять его в любое время года. В зимние месяцы достигается концентрация, меньшая абсолютно токсичной, но достаточная для подавления противника.
Стойкость на местности, лишенной растительности, составляет при обычной температуре около 6 ч. В этот период выход на зараженную местность или ее преодоление без противогаза невозможны.
Растворимость в воде прн 25 °C составляет 0,16%. Хлорпикрин хорошо растворим в органических растворителях — галогеналка-нах, алифатических спиртах, бензоле, бензине, слабо растворим в эфире. Из неорганических соединений он растворяется в четыреххлористых кремнии и олове, сульфурил хлориде.
Следует подчеркнуть, что хорошая смешиваемость с ОВ, например с ипритом, азотистым ипритом, дифосгеном, фосгеном, фосфорорганическими ОВ, делает хлорпикрин важным компонентом для тактических смесей, особенно в случае сравнительно ннз-коплавкнх ОВ, в результате чего становится возможным нх применение в зимнее время.
4.2.3.3. Химические свойства. Прн нагревании до температуры кипения хлорпикрин частично разлагается на фосген и ннтрознл-хлорнд:
CC1sNO2 —> coci2 + noci
Нитрозилхлорид может разлагаться далее на хлор н окись азота, которая кислородом воздуха окисляется до двуокиси.
В противоположность другим ОВ хлорпикрин сравнительно неустойчив к детонации. Термическая неустойчивость не позволяет применять хлорпикрин в качестве учебного ОВ посредством возгонки. Он заметно разлагается на свету, а также под влиянием дымящей серной кислоты.
Сухой хлорпикрин не разрушает нли лишь незначительно разрушает металлы.
Гидролиз. Трнхлорннтрометан водой не гидролизуется. Даже в водно-спиртовых растворах гидролиз идет чрезвычайно слабо н только в присутствии щелочей, прежде всего при нагревании, он проходит полностью:
CCI3NO2 -|- 6NaOH - —> 3NaCl -J- NaNO2 -p Na2COg -p 3H2O
В результате побочной реакции с образующимися в таких растворах алкоголят-ионамн получаются соответствующие эфиры ор-тоугольной кислоты:
cci3no2 + 4ro_ —> C(OR)4 +no; + за-
при применении чистых алкоголятов щелочных металлов эту реакцию, как главную, можно использовать в аналитических целях.
128
DUClsl UHUtfЛЧН11Ч. I\dK-1ТИ J 'pUUJlAfUl,-1 piAJluyuii i ^v.uviun-—
восстановить подходящими восстановителями, причем одновременно затрагиваются и отщепляются атомы хлора. Без специального контроля течения реакции конечным продуктом восстановления является метиламин V. При соответствующих условиях реакцию можно задержать на промежуточных стадиях. Одним из промежуточных соединений является дихлорформоксим (фос-геноксим) III, который таким путем можно получать нз хлорпикрина. Это вещество является кожно-нарывным ОВ (см. раздел 5.4.3).
В качестве восстановителя пригоден водород in statu nascendi, который можно получать действием соляной кислоты на порошок железа или цинковую пыль. Большой интерес представляет катодное восстановление.
Восстановление трихлорнитрометаиа происходит по следующей схеме (указаны только промежуточные продукты, которые удалось пока идентифицировать) 5:
4-2Н+ 4-Н+ перегруппировка
cci3no2 -——> CC13NO ——> chci2no -----------------:-----—>
—HjO ^HCI
I II
4-6H* ' 4-2H +
—> cci2=noh ——> CH3—NHOH ——> ch3nh2 — fHU —H2U
TH IV V
Трнхлорнитрозометан I—неустойчивая жидкость, раздражающая глаза н органы дыхания. Соединения, образующиеся из дн-хлорформокснма III, физиологически безвредны.
Особенно энергично происходит восстановление сульфидом натрия в водном или, лучше, в спиртовом растворе; оно приводит к полному разрушению молекулы хлорпикрина. Кроме нитрита натрия в качестве продуктов реакции образуются — окись азота, окись и двуокись углерода, азот, сероуглерод, сера и хлористый натрий. Приводимые уравнения лишь приближенно воспроизводят механизм реакции:
3NaaS
2CC13NO2 -------> 3S + N2 + 2CO2 + 6NaCI
. 3Na2S
2CC13NO2 -------> 3S + 2CO + 2NO + 6NaCl
IlNasS 7CC13NO2 ------->
—2N2 + NO2 + NO + 7S + 2CO2 + 2CO + 2COS + CS2 + NaNO2 + 21NaCl
Взаимодействие co спиртовыми растворами сульфида натрия следует считать единственно возможной реакцией для дегазации хлорпикрина. Вместо спиртовых растворов сульфида натрия при-, годны так же .водные растворы с добавками поверхностно-активных веществ.
5 Зак. 448
129
При восстановлений двуххлористым оловом в солянокислом растворе наряду с другими продуктами образуется хлорцнаи:
CCl3NO3 + 3SnCl2 + 4HCl —> C1CN + 3SnCl4 + 2Н3О
Из других восстановителей, применимых для аналитических определений, следует назвать амальгаму натрия, гидразин и уже упоминавшиеся при гидролизе алкоголяты щелочных металлов, в частности этилат натрия.
4.2.3.4. Токсические свойства. Хлорпикрин раздражает кожу и слизистые оболочки. Ои вызывает слезотечение, смыкание век, бронхит и отёк легких. Это сильное удушающее ОВ. Жидкий хлорпикрин причиниет тяжелые поражения кожи.
У большинства людей концентрация 0,002 мг/л за 3—30 сек вызывает слезотечение и смыкание век, концентрация 0,05 мг/л— непереносима. Более высокие концентрации ведут к болям в области желудка, рвоте и потере сознания. Концентрация около 0,2 мг/л за несколько секунд или минут приводит к полной утрате боеспособности.
Поражения органов дыхания появляются при концентрациях выше 0,1 мг/л. В качестве смертельной указывают концентрацию 2 мг/л при экспозиции 10 мин. Прн этой концентрации смерть наступает в течение нескольких минут.
4.2.4. Тетрахлордииитроэтаи и другие соединения
Тетрахлординитроэтан 1 — кристаллическое вещество, которое плавится с разложением в интервале 130—140°С. В воде он нерастворим н по своему химическому поведению подобен трнхлориитро-метану. Ои примерно в 6 раз токсичнее трихлоринтрометана, а по раздражающему действию превосходит его в 8 раз. Наряду с другими соединениями он получается при действии дымящей азотной кислоты на тетрахлорэтан.
o2n no2
ci2d—tci2 cf3no2 cf2cino2
I II III
Трифторнитрометан (фторпикрин) II описан в литературе как вещество, вызывающее смертельное отравление. Вдыхание его паров приводит к чрезвычайно сильным приступам кашля, и даже оставшиеся сначала в живых затем неизбежно умирают. Это жидкость, кипящая прн —31,1 °C. Ее можно получить окислением сравий*гельно легкодоступного трифторнитрозометана двуокисью свинца, окисью хрома и двуокисью марганца6;
окисление
CF3NO • > CF3NO2
Аналогично можно получить следующие соединения:
Дифторхлорннтрометан CC1F2NO2, т. кип. 24—25 °C (при 760 мм рт, ст);
130
1,1,2-ТрифТ0р-2-ХЛ0р-1,2-ДИНИТр0ЭТаН, U2hJCl.lt1 — 1. КИН.
98—99 °C (при 750 мм рт. ст.), d?!,674;
силж-дифтордихлординитроэтан, O2NCCIF—CCIFNO2, т. кип. 103°C (при 81—82 мм рт. ст.), т. пл. 12—18°С; ^451,646;
силж-тетрафтординитроэтан, O2NCF2—CF2NO2, т. кип. 57—58 °C (при 750 мм рт. ст.), т. пл. —41,8°С; ^451,595.
В какой море эти соединения могут иметь военное значение — неизвестно. Последнее из названных соединений производится в США в промышленном масштабе.
4.3. СОЕДИНЕНИЯ ФТОРА С ГАЛОГЕНАМИ И СЕРОЙ
Во время второй мировой войны в Германии была проверена возможность военного использования различных неорганических соединений фтора. Особое внимание было уделено галогенфторидам и фторидам серы.
Галогенфториды— очень реакционноспособные вещества, кото-, рые наряду с сильной ядовитостью обладают зажигательными свойствами и разрушающе действуют на материалы. По токсичности они превосходят фосген. В высоких концентрациях при достаточно малых экспозициях они действуют абсолютно смертоносно.
Трехфтористый хлор во время второй мировой войны получали в Германии в больших масштабах в качестве зажигательного средства, обладающего одновременно токсичным действием7. В последнее время в США организовано крупное промышленное производство треххлористого фтора. Это соединение считают более пригодным в качестве ОВ, чем другие галогенфториды.
Из фторидов серы в литературе наряду с другими упоминалась пятифтористая сера, очень токсичное вещество.
Эти соединения приобретают все большее значение как фторирующие агенты для получения неорганических и органических соединений фтора. Некоторые из них как сильные окислители пригодны для создания высоких температур, например при резании металлов или в качестве ракетного топлива.
4.3.1. Трехфтористый хлор
Однохлористый фтор был впервые получен и исследован в 1929 г. Руффом и Лаосом8 Ими было доказано также существование треххлористого фтора.
Трехфторйстый хлор получают при реакции хлора со фтором в медном сосуде при 200 °C.
При нормальных условиях трехфтористый хлор представляет собой бесцветный газ. В жидком виде он бесцветен или имеет желто-зеленую окраску. Он обладает слегка сладковатым запахом и уже в малых концентрациях раздражает слизистые оболочки. Физические свойства: т. кип. 11,2—11,75°C (12,1 °C), т. пл. —80°С (-83 °C), с/4Э1,8403; й?4°1,7936; критическая температура 154 °C.
5;
131
Согласно Гризарду и сотр,э, давление пара трехфтористого хлора (в мм рт. ст.) можно вычислить по формуле:
1g р = 7,36711 — 1096,917 (i 4-232,75)
Давление пара и летучесть соединения велики.
Трехфтористый хлор растворяется в воде с разложением. В химическом отношении ои ведет себя как сильный окислитель, действует на металлы с образованием фторидов. Реакция с металлами зависит от температуры и от того, образуется ли защитная пленка фторида, как, например, в случае меди; благодаря образованию защитной пленки фторида меди трехфтористый хлор можно получать только в медных сосудах.
Неметаллы защитные пленки не образуют. Иногда они реагируют с трехфтористым хлором столь энергично, что происходит воспламенение. Даже некоторые окислы, такие как двуокись серы, окись магния, окись алюминия, а также аммиак, реагируют с трех-фтористым хлором. Энергично реагируют с F3CI окислы натрия.
Бурно реагирует трехфтористый хлор с органическими соединениями. Одна капля F3C1 воспламеняет ткань, дерево, бумагу и др- Он энергично реагирует с содержимым коробки противогаза и проникает через защитную одежду.
На Западе военные специалисты считают трехфтористый хлор выдающимся ОВ, которое разрушает материалы и пригодно для уничтожения людей несмотря на средства защиты.
Трехфтористый хлор действует иа органы дыхания как ингаляционный яд. Последствиями являются вздутие и опухание легких, сужения бронхов, ожоги верхних дыхательных путей, гнойный бронхит. Высокие концентрации вызывают рефлекторный кашёль и отделение мокроты и затем приводят к тяжелейшим поражениям легких. Отравление чаще всего смертельно.
Сильно-'поражаются глаза — опухают веки, а при известных обстоятельствах происходит помутнение роговицы.
Поражения иа поверхности кожи проявляются в сильном покраснении из-за увеличенного содержания крови в пораженных участках, образовании волдырей, абсцессов и некротических распадов более глубоко лежащих тканей.
Приводим результаты опытов над крысами7 при концентрации F3C1 480 м. д.:
Наблюдавшийся эффект
Раздражение слизистой носа........
Одышка у всех подопытных животных . . Ожоги на глазах, веки смыкаются, опухают или покрываются коркой ......
Слюнотечение; все животные почти обессилены, пораженная роговица глаз становится молочно-белой .............
Смерть животных после непродолжительных конвульсий ....................
Экспозиция, Хин
2
4
5
19
76
132
Были исследованы другие—ансиняичные— л; Ш 1 д
(т. кип. 135 °C, т. пл. 8,8 °C); IF& (т. кип. 98°C, т. пл. 8,5°C); получены также IF7> BrFs, CIF2.
4.3.2. Пятифтористая сера
Из известных фтористых соединений серы, SF6, SF4 и др., в качестве возможного ОВ рассматривается пятифтористая сера S2F10. Это бесцветная жидкость с высоким давлением пара и большой летучестью, т. кип. 29 °C; т. пл. — 92°C; плотность 2,08 г/см3 (при 0°С).
Пятифтористая сера, видимо, более ядовита, чем фосген, и в высоких концентрациях уже при кратковременном воздействии является смертоносной. Водой заметно гидролизуется. .
Фториды серы очень реакционноспособны и применяются в препаративной органической химии как фторирующие агенты.
4.4. БОЕВОЕ ПРИМЕНЕНИЕ УДУШАЮЩИХ ОВ
В результате принятия иа вооружение во много раз более ядовитых фосфорорганических соединений типа зарина или V-газов, такие удушающие ОВ, как фосген, дифосген и хлорпикрин в значительной мере утратили свое значение для современного боя. Но поскольку эти вещества в мирное время производятся в крупном промышленном масштабе для изготовления пластмасс, красителей, химикалиев и т. д., имеющиеся производственные мощности составляют известный резерв для получения ОВ такого типа.
Из-за латентного периода действия фосген и дифосген с тактической точки зрения считают медленно действующими ОВ, а хлорпикрин и соединения фтора с галогенами — быстродействующими. Последние отличаются комбинированным характером действия, они являются и удушающими, и зажигательными, и разрушающими материалы средствами.
Летучесть' фосгена и дифосгена велика, поэтому эти соединения особенно пригодны для наступательных действий. Дифосген обладает известной стойкостью, которая сильно зависит от метеорологических условий. Его можно рассматривать как полу стойкое ОВ.
После второй мировой 'войны дифосген и хлорпикрин были неоднократно обнаружены в немецких боеприпасах, притом в смеси с дымообразователями, например, с четыреххлористыми титаном или оловом. Из-за невысокой токсичности и сильного раздражающего действия хлорпикрин не рассматривают как ОВ, пригодное для внезапного нападения. Скорее ои является подходящим компонентом для усиления эффективности тактических смесей, особенно смесей раздражающих ОВ. С другими ОВ хлорпикрин никогда ие используют, поскольку его раздражающее действие практически дало бы возможность сразу обнаружить ОВ. Благодаря
133
раздражающему действию и простоте обращения хлорпикрин пригоден в качестве учебного ОВ и как таковое применяется в неко-то-рых армиях.
Удушающие ОВ применяются при помощи бомб, артиллерийских боеприпасов и иногда мин. С их возможным применением следует особенно считаться в случае создания дымовых завес.
Старые американские боеприпасы, заполненные хлорпикрином, обозначались одним красным и одним белым кольцом. Во вторую мировую войну для маркировки боеприпасов использовали два зеленых кольца и обозначение PS-GAS. Средства применения, содержащие смесь хлорпикрина с четыреххлористым оловом, обозначались двумя зелеными кольцами и маркировкой GAS-NC. До второй мировой войны боеприпасы с фосгеном помечались в армии США двумя белыми кольцами, после войны — одним зеленым кольцом и маркировкой CG-GAS.
4.5. ЗАЩИТА ОТ УДУШАЮЩИХ ОВ И ИХ ДЕГАЗАЦИЯ
От удушающих ОВ защищает противогаз. Некоторые добавки к поглотителям активируют каталитические реакции соединений типа трехфтористого хлора, в результате чего ОВ теряет способность реагировать с главными компонентами поглотителя.
Одевание защитной одежды для предохранения от ОВ типа фосгена вряд ли бывает необходимо, тогда как в случае галогеи-фторидов такие средства защиты необходимы, ио защищают оии лишь в увлажненном состоянии. Некоторые пластмассы сравнительно устойчивы к действию таких веществ, например политри-фторхлорэтилен. Их можно применять как в виде накидок, так и в качестве пористых материалов в фильтрах.
Вследствие сравнительно высокой летучести дегазация удушающих ОВ на открытой местности ие нужна. Казармы, блиндажи и т. п. достаточно проветрить; иногда требуется для дегазации протопить помещение или обработать водяным паром. Хлорпикрин можно дегазировать только водными растворами сульфида натрия с добавкой поверхностно-активных веществ или спиртовыми растворами сульфида натрия.
Продукты питания заражаются фосгеном или дифосгеном только при непосредственном контакте. После прогрева или разбавления водой их можно снова употреблять в пищу в той мере, насколько образовавшаяся соляная кислота не испортила их вкусовые качества. -Продукты, соприкасавшиеся с хлорпикрином, выбрасывают. В боевой обстановке фосген и дифосген почти не заражают воду.
Предметы обмундирования и снаряжения самодегазируются, если их достаточно долго проветривать. Процессу дегазации при этом также содействует тепло, иапример, при дегазации горячим воздухом или паром.
134
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Почему газообразный хлор потерял свое значение как ОВ?
2. Опишите картину отравления фосгеном.
. 3. Каковы химические свойства фосгена и эфиров хлоругольной кислоты?
4 цто такое фотокатализ?
5. Какие факторы могут способствовать гидролизу фосгена?
6. Почему гексаметилентетрамин применяли в первую мировую войну в противогазах?
7. Как в силу своих физических свойств ведет себя фосген после применения на местности?
8. Напишите уравнение реакции дифосгена с растворами щелочей.
9. Почему реакцию фосгена и дифосгена с анилином можно использовать в аналитических целях?
10. Какая связь между химическим строением и токсическими свойствами галогенированных нитроалканов?
11. Почему галогенированные нитроалканы лишь в малой степени или совсем не гидролизуются водой?
12. Как ведет себя трихлорнитрометан по отношению к восстановителям?'
13. Какие свойства имеет трехфтористый хлор?
14. Дайте тактическую оценку фосгену как ОВ.
15. Что такое тактическая смесь и для каких целей она служит?
ЛИТЕРАТУРА
1. Jentzsch W., Med. КИп., 52, 1589 (1957).
2. G i 11 е г t Е., Die Kampfstofferkrankungen, Berlin — Wien, 1938.
3. Dragerwerke Lubeck, Mitteilungen zum Gasspur ger atr 19/31, 38. Folge (Mai, 1960).
4. Пат. США 2936322 (1963).
5. Ztschr. .Elektrochem. angew. Phys. Chem., 53, 109 (1949).
6. H a s z e 1 d i n e R. N., J. Chem. Soc., 1953, 2075.
7. Hom H. J., Wei ss R. J., A. M. A. Arch. Ind. Health, 1955, 515.
8. R u f f O., L a s s F., Z. anorg. a 11g. Chem., 183, 214 (1929).
9. J. Am. Chem. Soc., 73, 5725 (195Г).
7Г VJ-il AKD1D ri Dl Е UB
В июле 1917 г. возле Ипра английские войска были обстреляны немецкими минами, содержавшими маслянистую жидкость. Через несколько часов после соприкосновения с этим маслом у пораженных появились до тех пор неизвестные симптомы. Новое ОВ вызывало иа коже трудноизлечимые ожоги, быстро проникало через одежду и заражало местность. Начался новый этап ведения химической войны — боевое применение долгодействующих, стойких химических ОВ. В то время они имели значение скорее для обороны, чем для наступления.
Англичане установили, что новое ОВ является 2,2'-дихлорди-этилсульфидом — веществом, полученным в 1886 г. Виктором Мейером в Гёттингене, описавшим также и его специфические токсичные свойства. Высказывания В. Мейера по этому поводу следует привести дословно Г
«Сначала я склонялся к тому, что явления, наблюдающиеся при действии хлорида, следует объяснить особой восприимчивостью экспериментатора; однако в результате опытов, выполненных по моей просьбе в здешнем физиологическом институте, я уяснил нечто более важное. Согласно этим опытам, это соединение обладает в высокой степени опасными свойствами, как можно было заключить на основании предварительного, ограничивающегося наиболее важными и бросающимися в глаза наблюдениями, сообщения.
Каждого из кроликов средней величины дважды помещали на 3—4 ч в запертую клетку, вентилируемую сильным потоком воздуха. Перед попаданием в клетку поток воздуха проходил через стеклянную трубку, в которой находились полоски фильтровальной бумаги, смоченные 2,2/-дихлордиэтилсульфидом. Животные были возбуждены, часто касались лапами иоса и морды, которые имели характерную сильную красную окраску. Конъюнктива тоже покраснела, а глаза были очень влажными. Выделение влаги кожей заметно увеличилось. На следующий день глаза сильно воспалились, веки склеились гнойными выделениями. Появился сильный насморк, уши сильно опухли и в слуховом проходе появилось гнойное воспаление. К вечеру третьего дня животные умерли от острой пневмонии, распространившейся на оба легких. Один очень сильный кролик, который несколько часов вдыхал пары вещества через отверстие воздуховодной трубки, так'что они не действовали на поверхность тела, умер уже вечером того же дня от развившейся 136
пневмонии, поэтому времени для проявления других симптомов уже не было.
У кроликов, которым с помощью тонкой кисточки на неповрежденную кожу кончиков ушей было нанесено немного дихлордиэтил-сульфида, на месте нанесения совершенно не появилось следов поражения, однако все ухо сильно опухло, а в одном случае от основания слухового прохода до наружной части уха возникло обильное гнойное воспаление. Возможность затекания препарата в слуховой проход была исключена отчасти из-за незначительности нанесенного кисточкой количества вещества, отчасти вследствие того, что препарат был нанесен на наружную поверхность уха. В случае, когда кожу предварительно обнажали сбриванием шерсти с кончиков ушей, нанесенный кисточкой препарат, разумеется, вызывал преимущественное нагноение в этом месте, но одновременно и более сильное опухание всего уха н воспаление глаз. При подкожном введении примерно двух капель препарата в царапину на коже спины кролика возникало воспаление обоих глаз, очень сильный насморк и на третий день наступала смерть вследствие пневмонии. На месте нанесения препарата никаких признаков поражения не было. Так как пары вещества оказывали на экспериментаторов вредное действие, подобное вкратце описанному выше, эти опыты пришлось прекратить».
Во время первой мировой войны это соединение было исследовано и выбрано в качестве ОВ немецкими военными химиками Ломмелем и Штейнкопфом.
Маркировка немецких мин желтым крестом вскоре привела к общему названию «ОВ с желтым крестом» или, сокращенно, «желтый крест». Это обозначение впоследствии применяли ко всем ОВ с аналогичным биологическим действием, например для обозначения полученного в США 2-(хлорвинил)-дихлорарсина (люизита) или синтезированных в тридцатых годах Ь1-алкил-бис-(2-хлорэтил)-аминов (азотистых ипритов).
, Кожно-нарывные ОВ, наряду с современными фосфорорганическими ОВ, еще и теперь являются важными средствами ведения химической войны. Они могут применяться как в чистом виде, так и в виде жидких и высоковязких тактических смесей.
Соединения, которые рассматриваются и обсуждаются в качестве кбжно-нарывных ОВ, относятся к разным химическим классам. Они имеют различное химическое строение, это — тиоэфиры, амины, арсины и оксимы. Общим для них является то, что они содержат в молекуле атомы галогенов и обладают сходными биологическими свойствами.
Из галогенированных тиоэфиров особое значение имеет бис-2-хлорэтиловый тиоэфир (иприт), из галогенированных алифатических третичных аминов — ГО-алкил-бис-М,1Ч- (2-хлорэтил)-амины (азотистые иприты)-, среди моноалкнлднхлорарсннов —- 2-(хлорвинил)-дихлорарсин (люизит), а среди галогенированных оксимов — дихлорформоксим (фосгеноксим).
137
Этот перечень никоим образом не может претендовать на полноту; эти и аналогично действующие соединения в настоящее время все еще интенсивно изучаются. Исследования имеют целью найти более эффективные и, может быть, более стабильные соединения, которые при известных условиях могут обладать комбинированным действием, как, например, сочетанием кожио-нарывного с нервно-паралитическим действием и т. п.
5.1. ГАЛОГЕНИРОВАННЫЕ ТИЦЭФИРЫ
Сравнительно неядовитые тиоэфиры I в результате замещения атомов водорода иа галогены, особенно в ю-положении *, становятся токсичными. В таком случае они действуют и как ингаляционные яды, и на поверхность кожи, вызывая на ней трудно, излечимые поражения.
СдН2^ +1—S—С/Г1В2/И +1 С1СН2СН2—S—CH2CH2CI
I II
Наиболее интересным соединением из галогенированных тноэфи-ров является бис-2-хлорэтиловый тиоэфлр II, который известен под общим названием иприт, горчичный газ н др. и является важнейшим ОВ этой группы.
Во время второй мировой войны было получено и исследовано большое число аналогов иприта, обладающих кожно-нарывным действием; наряду с другими были получены ^-галогенированные бис-(р-хлорэтилмеркапто)-диалкиловые эфиры, важнейшим представителем которых является 2,2'-бис-(2"-хлорэтилтио)-днэтиловый эфир Ш, а также бис-(2-хлорэтилтио)-алканы IV, бис-2-бромэтНло-вый тиоэфир V и аналогичные фтористые соединения.
XHsCHaSCHsCHjCl /SCHaCHjCl ,
о; (сн2)«
^СНгСНгйСНгСНгС! ^SCH2CH2C1
Ш ’ IV
уСН2СН2Вг \сн2сн2вг. v
5.1.1. Химическое строение и биологическая активность
51.1.1. Связь химического строения с физиологической активностью, Биологическая активность серы в неорганических и органических соединениях зависит от степени ее окисления и положения в молекуле. Многообразие соединений серы не позволяет сделать общих выводов о взаимосвязи между строением серосодержащих соединений н их фармакологическими и токсическими
* Точнее было бы написать «особенно в р-л сложен и и». — Прим, ред.
138
свойствами. Доказано, что атом серы тиогруппы не является ответственным за разрушение клетки некоторыми производными тиоэфиров, поскольку незамещенные тиоэфиры не проявляют физиологической активности. Даже введение .другого заместителя (кроме галогена) в молекулу тиоэфира не приводит к соединениям, представляющим интерес с военной точки зрения.
Необходимым условием кожно-нарывного действия является присутствие атомов галогенов в молекуле тиоэфира. Наиболее эффективные представители — со-замещенные соединения, к которым относится и бис-2-хлорэтиловый тиоэфнр И. Такой же физиологический эффект вызывает бнс-2,3-дихлорпропиловый тиоэфир VI.
Менее или совсем не эффективны соединения, дизамещенные в положениях- 1,1'-, например бис-1-хлорэтиловый тноэфир VII, или монозамещенные продукты, подобные, этиловому эфиру VIII хщснасща S\
\зН2СНС1СНгС1
VI
иапример, 2-хлорэтилтио-
/CHClGHg Sz
^снс1сн3
VII
/СН3СН2С1 ^сн2сн3
VIII
/СНС1СН2С1 sz
, ^СН2СН2С1 '
IX
Хотя бис-2-бромэтиловый тиоэфир V и бис-2-иодэтиловый тиоэфир обладают теми же физиологическими свойствами, что и бис-2-хлорэтиловый II, их токсичность несколько ниже. Об аналогичном фтористом соединении данных нет, однако можно предполагать, что оно действует так же *.
Дальнейшее введение атомов галогенов, которое влечет за собой асимметрию молекулы галогенированного тиоэфира, снижает токсичность. Такие соединения не обладают или обладают лишь слабым нарывным действием, как, например, 1,2,2'-трихлор-днэтиловый тиоэфир IX.
Из-за наличия в молекуле двух или большего числа связанных друг с другом атомов серы тиогрупп (соединение X) физиологическая эффективность уменьшается. Напротив, введение между двумя атомами серы одной или больше метиленовых групп (максимум п = 5) ведет к усилению кожно-нарывного действия (соединение XI), которое при п > 5 снова уменьшается.
SCH2CH2C1 I
(SM
SCH2CH2CI
X
sch2ch2ci
I
(CH2)rt
SCH2CH2C1
XI
* Это утверждение, вероятно, не совсем точно. Значительная прочность С—F-связи должна привести к малой подвижности атома фтора, а следовательно, к инертности подобного рода соединений. — Прим. ред.
139
Продукты окисления ю-галогеийрованных тиоэфиров — сульфоксиды и сульфоны — проявляют известную токсичность и некоторое, хотя и слабое, кожно-нарывное действие.
5Л.1.2. Принцип и механизм действия*. Кожио-иарывиые галогенированные тиоэфиры действуют иа организм разносторонней Причиной этого является их высокая растворимость в липидах, которая способствует всасыванию яда во все части тела. Их локальное действие в качестве клеточных и капиллярных ядов сопровождается общим отравляющим действием. Единой точки зрения иа механизм действия этих соединений не существует. Из множества разрозненных данных следует рассмотреть лишь некоторые.
Первоначально высказанное объяснение разрушения клетки медленным внутриклеточным гидролизом указанных соединений ныне отвергается. Внутриклеточный гидролиз является лишь одним из совокупности процессов, составляющих механизм действия, ио не играет решающей роли для всего -механизма в целом. Точку зрения, что первичиое'действие оказывают как неизмененная молекула тиоэфира, так н продукты ее биохимического разложения, до сих пор в достаточной мере подтвердить не удалось.
Экспериментально было подтверждено иигибируюШ^е действие галогенированных тиоэфиров на некоторые жизненно важные ферменты. Например, бис-2-хлорэтиловый тиоэфир подавляет, наряду с другими, гексокиназу, лнпазу, -сывороточную пептидазу и сукцн-нилдегидрогеназу. С достоверностью установлено непосредственное связывание молекулы тиоэфира с амнно- и сульфгидрильными группами протеинов, например с фосфопротеидом казеином, хромопротеидом оксигемоглобином, альбумином и глобулином сыворотки крови, а также с цистином, лизином, аргинином и гистидином.
Согласно Бурбаху2, все кожно-нарывные ОВ нарушают энергетический баланс клеток, следствием чего является подавление Синтеза аденозинтрнфосфата.
Другие данные о механизме действия рассмотрены Штаде3.
Галогенированные тиоэфиры, представляющие военный интерес, действуют в любом агрегатном состоянии. В частности, они эффективны в виде жидкости, аэрозоля и пара. Симптомы отравления вызываются непосредственным контактом, вдыханием и попаданием в желудочно-кишечный тракт и наступают только после скрытого периода. Наряду с общеядовитым действием прежде всего поражаются Кожа, глаза, органы дыхания и пищеварительный тракт.
* Большая часть кожно-нарывных ОВ представляет собой алкилирующие средства. Не исключено, что действие их может быть направлено и на аппарат нуклеиновых кислот, результатом этих реакций может оказаться депуринизация (депиримидинизация) ДНК (РНК) и, следовательно, нарушение генетического кода. — Прим, ред,
140
Действие на кожу. При попадании кожно-нарывных иь на кожу в виде жидкости, аэрозоля или пара сначала не проявляются никакие защитные рефлексы. В зависимости от воздействующего количества скрытый период обычно составляет 4—6 ч. При сравнительно малых количествах, как, например, при воздействии газообразных ОВ, он может достигать 24 ч н более.
Первые признаки проявляются в зуде, жженнн и покраснении пораженных участков кожи (возникновение эритемы). Наощупь кожа натянутая, сухая и горячая. Затем ткани опухают. Через 20—24 ч на периферии эритемы образуются маленькие пузырьки, так называемые бусы, которые являются предшественниками сливающихся пузырей, заполняющихся затем бесцветным, иногда желтым экссудатом. Содержимое волдырей неядовито; если его выпустить, то волдыри могут заполниться снова.
При удалении оболочки пузыря обнаруживаются тяжелые разрушения тканей (некрозы). Из-за вторичных инфекций раны гноятся. При таких поражениях бросается в глаза распространение некротических распадов вширь н вглубь. Язвы очень болезненны. В зависимости от тяжести поражения нагноение длится несколько недель. Молодая грануляционная ткань, образующаяся через неделю, неоднократно опять погибает. Раны, прн известных условиях заживающие через месяц, оставляют после себя пигментированные рубцы. Суставы вблизи ран, особенно суставы пальцев рук и ног, могут стать неподвижными. Менее чувствительна кожа лица.
Очень чувствительна потная и разгоряченная кожа. В результате ускоренной разорбцин ОВ латентный период сокращается. Действие на кожу, вызываемое газообразными ОВ, почти, не идет дальше образования эритем. При высоких концентрациях образуются маленькие пузырьки, которые обычно не сливаются в более крупные и сравнительно хорошо излечимы.
Действие на глаза. Глаза особенно чувствительны к кожно-нарывным ОВ. Прн попадании в глаз жидкого ОВ происходят тяжелые некротические поражения, которые быстро распространяются вглубь и обычно приводят к потере глаза.
При действии парообразных ОВ первые признаки (ощущение инородого тела, слезотечение, жжение, опухоль конъюнктивы века, чрезвычайная светобоязнь, спазм века) появляются через 4—8 ч. Вязкие выделения склеивают веки. Помутнения роговицы ведут к потере зрения. Прн тяжелых поражениях остаются непроходящие следы.
В зависимости от тяжести поражения заболевания глаз длятся до 2 месяцев, в более легких случаях — 2—3 недели.
Действие на органы дыхания. В результате вдыхания небольших количеств ОВ через 6—8 ч появляется легкое воспаление верхних дыхательных путей, насморк, сухой кашель, першение в горле и тяжелый бронхит. Общие симптомы отравления в результате резорбции ОВ выражаются в позыве на кашель, слабости и
141
головных болях. Эти явления катаррального происхождения проходят через 3—4 суток.
Вдыхание более высоких концентраций приводит через 3 ч к сильному и мучительному кашлю, позыву на рвоту, болям в груди, потере голоса, затруднению глотания, некротическим распадам слизистой оболочки, кровоизлияниям в дыхательные пути, гнойным очагам во рту, в носу и в бронхах и, наконец, к отёку легких.
Общеядовнтое действие .проявляется в. повышении температуры тела, в апатий, потере аппетита, болях в животе, поносе, упадке сил.
Тяжелые отравления обычно на третий-четвертый день кончаются смертью. При отравлениях средней тяжести летальный исход отодвигается к концу первой или, соответственно, третьей или четвертой недели. Очень высокие концентрации ведут к острым отравлениям, напоминающим отравления фосгеном.
Действие на желудочно-кишечный тракт. При пероральном приеме кожно-нарывных ОВ отравление протекает очень тяжело. Уже через 15 20 мин появляются резкие боли в желудке, сильное слюнотечение, частая рвота, тошнота, кровавые поносы, полная потеря аппетита, жажда, бледность кожи.
При резорбции ОВ через слизистую оболочку рта и глотки образуются эритемы и пузырьки (в том числе и на губах). Быстро наступающая общая интоксикация обычно оканчивается летально. Смерть наступает через 2 суток или несколько позже.
Более или менее сильно резорбтивное действие проявляется цо всех органах. Галогенированные тиоэфиры быстро разносятся по всему организму, особенно в результате их попадания в кровь. Лишь около 12% попавшего на кожу количества ОВ остается в коже, большая же часть быстро переходит в систему кровообращения. Капиллярные кровеносные сосуды очень нежны, повреждение' нх в печени, почках, сердце, желудке, селезенке, кишечнике, мозге и т. д. ведет к кровоизлияниям в соответствующие органы и к воздействию на'Сердце (изменения пульса).
Вследствие распада белка наступает похудение, скованность движений, безразличие, упадок сил, появляются дрожь, головокружение, шум в ушах, расстройства нервной системы.
Расстройства вестибулярного аппарата, судороги мышц и потеря сознания объясняются токсическим действием на центральную нервную систему. Это первые признаки летального исхода' отравления.
При поражениях кожи, ограничивающихся стадией образования эритем, общеядовитое действие почти отсутствует.'
Если площадь пораженной кожи .мепьше 20"см2, то общеядовитое действие за счет резорбции в другие органы ие наступает. Опыты с газообразным и жидким ипритом, проводившиеся в тропических и полевых условиях англичанами во время второй мировой войны над более чем 400 человеками, показали4, что помимо локальных поражений кожи такие симптомы общего отравления, 142
как кашель, рвота, головные болн, слабость, бессонница н понос, появились у 65% через 24 ч, у 81% —через 48 ч и еще иа шестой день у 94% испытуемых.
5.1.2. Общие свойства
Галогенированные тноэфнры практически нерастворимы в воде. Они обладают характерным резким запахом, присущим также и незамещенным соединениям.
Их химические свойства определяются наличием атома двухвалентной серы н галоидными заместителями.
Обладая неподеленной парой электронов,-сульфидная сера, подобно атому кислорода в простых эфирах, способна вступать в реакции с электрофильными реагентами. По аналогии с соединениями оксоння образуются сульфонневые производные. Атом серы можно окислить, продуктами окисления являются сульфоксиды и сульфоны:
R\ Rx
)s=0 /С RZ RZ ZO
сульфоксид
сульфон
Прн взаимодействии тноэфнров с солями тяжелых металлов образуются комплексные соединения. Этн реакции можно использовать для идентификации тиоэфиров.
В галогенированных тноэфнрах атомы галогена способны замещаться. Связь углерод—хлор, например в молекуле бнс-2-хлорэтн-лового тноэфнра (иприт), наиболее полярная, и атомы галогена в этом случае более реакционноспособны, чем в галогенированных простых эфирах. Атом хлора в тиоэфнре в 20 раз более подвижен, чем атом хлора в бнс-2-хлорэтнловом эфире 5.
Скорость гидролиза бис-2-хлорэтнлового тноэфнра в водио-дн-оксаиовой смесн в 1000 раз больше, чем аналогичного эфира6. Реакционная способность галогенированных тноэфиров зависит от положения заместителей в молекуле. С увеличением размера алкильных групп легкость отщепления со-заместителей уменьшается. Феннл-2-хлорэтнловый тноэфнр XII в приведенных выше условиях реагирует в 200 раз быстрее, чем фенил-4-хлорбутнловый тио-
эфир XIII.
zc6Hs zcfiH5
s\ \
\СН2СН2С1 ZCH2CH2CH2CH2CJ
ХИ XIII
Исключение составляют соединения, замещенные в положении 3, реакционная способность которых меньше, чем в случае соединений, замещенных в положении 4.
143
Причиной более быстрого гидролиза 4-галогентиоэфиров считают промежуточное образование солей сульфоиия, которое теоретически возможно для любых галогенированных тиоэфиров с радикалами с четным числом атомов углерода (ср. раздел 5.1)
+ /СН2
R—S—СН2СН2С1 R—| + СГ
\сн2
Бром- и иодпроизводные более реакционноспособны, чем хлор-замещенные тиоэфиры, фтористые соединении являются наиболее устойчивыми.
5.1.3. Бис-2-хлорэтиловый тиоэфир
,СНгСН2С1 - s\ \СН2СН2С1
Синонимы: 2,2'-дихлордиэтнловый тиоэфир; 2,2'-дихдордиэтилсульфид; РФ'-дихлор диэтил сульфид, 1-хлор-2-(2-'-хлорэтилтио)-этан, иприт.
Военные обозначения: нем. — Schwefelyperit, Yperlt, Lost, Gelbkreuz, Senfgas; англ. — Mustard Gas, Mustard, Yellow Cross liquid, HS; амер. — Mustard HD, HS (теперь H), G. 34 и M. О. (в первую мировую войну); франц.— Yperite, Gas Mustarde, Yc., Yt.
Из так называемых OB с желтым крестом бис-2-хлорэтиловый тиоэфир под названием «лост» был впервые применен немецкими войсками в ночь с 12 иа 13 июля 1917 г. в ожесточенном сражении за Ипр. Особое значение этого ОВ заключалось не только в том, чтобы на долгое время сделать пораженных небоеспособными. С этим новым ОВ было принято на вооружение средство поражения, от которого не предохраняли средства защиты органов дыхания от уже известных ОВ. Из-за особых свойств иприта проникать через обмундирование и обувь и вызывать поражения кожи уже в небольших количествах атакованные были вынуждены защищать от его воздействия все тело.
Химическая стабильность и исключительные физические свойства, которые обеспечивали высокую стойкость иприта, возводили это соединение в ранг мощного оборонительного средства, которое стесненная немецкая армия могла долгое время эффективно противопоставлять наступательным замыслам союзников.
В марте 1918 г. немецкие войска выпустили по частям 3-й,аиг-лийской армии около 250 000 ипритных мин. Союзникам удалось применить освоенный промышленностью ипрнт по рубежам немецкой обороны только в июне 1918 г.
Потери, понесенные от иприта, были больше, чем от других ОВ. По английским данным, их потери от иприта составили 80% от общего числа отравленных ОВ; остальные 20% включали пораженных удушающими ОВ и ОВ, раздражающими носоглотку.
144
Полученный в чистом виде Мейером в 1886 г. бнс-2-хлорэтило-вын тиоэфир * еще раньше был получен другими химиками: в 1882 г. во Франции — Дюпре, в 1860 г. в Англин —Гутрн и в Германии— Ниманом. Они получали это соединение при действии этилена на моно- нлн двухлорнстую серу. Уже Гутрн н Ниман установили физиологическую активность этого вещества, хотя онн получили его лишь в неочищенном виде. Оба хнмнка отмечали, что действие этой жидкости и ее паров на кожу вызывает образование волдырей. В 1891 г. немецкий окулнст Лебер сообщил о проведенных нм опытах по исследованию действия нпрнта на глаза и о возникающих прн этом воспалениях.
Незадолго до первой мировой войны англичанин Кларк, ученик Эмиля Фншера, в химическом институте Берлинского университета изучал некоторые реакции синтезированного им самим бнс-2-хлор-этилового тиоэфира.
С тех пор как иприт стал известен в качестве ОВ, он явился предметом научных исследований. Кроме, технологических изменений, методы его получения в принципе изменились несущественно. Описанные Мейером, Гутрн н др. методы получили дальнейшее развитие н вндонзмененне для использования в промышленном масштабе.
С тех пор итальянские фашисты применили нприт прн оккупации Эфиопии, а во время второй мировой войны многими воюющими державами были заготовлены большие запасы этого ОВ. Он ин в коей мере не потерял свое военное значение, несмотря на существование более эффективных ОВ. Возможности его применения в бою для обеспечения нлн затруднения боевых действий, а также для постановки заграждений разнообразны, особенно прн помощи современных средств применения.
Ипрнт считается одним из важных ОВ н принадлежит к табельным ОВ империалистических армий.
5.1.3.1. Методы получения. Получить бис-2-хлорэтнловый тио-эфир можно разными способами: хлорированием бнс-2-окснэтнло-вого тиоэфнра (тноднглнколя) соответствующими хлорирующими агентами; прямым синтезом , из этилена н хлоридов серы; фотохимическим синтезом из хлористого этилена н сероводорода или 2-хлорэтнл меркаптана.
Методом получения из тноднглнколя пользовался еще Мейер1, который в качестве хлорирующего агента применял треххлорнстый фосфор:
3S(CH2CH2OH)2 + 2РС13 —> 3S(CH2CH2C1)2 + 2Н3РО3
Позже Кларк7 заменил треххлорнстый фосфор хлористым водо-родом
S(CH2CH2OH)2 + 2НС1 -> S(CH2CH2C1)2 + Н2О
* Иприт в чистом виде был впервые получен Н Д. Зелинским, работавшим в то время в лаборатории В. Мейера. — Прим. ре().
145
а Штейнкопф и corp.8 предложили пользоваться сульфурилхлори-дом или тионилхлоридом:
S(CH2CH2OH)2 + SOC 12 —> S(CH2CH2C1)2 + so2 + Н2О
Тиодигликоль получают по аналогии с общим методом синтеза тиоэфиров взаимодействием алкилгалогенидов, в данном случае этиленхлоргидрина, с сульфидом щелочного металла 2С1СН2СН2ОН+ Na2S —> S(CH2CH2OH)2 + 2NaCl
или из окиси этилена и сероводорода:.
2Н2С---CH2 + H2S —> S(CH2CH2OH)2
Этиленхлоргидрии получают действием хлорноватистой кислоты на этилен или, с таким же успехом, из окиси этилена и хлористого водорода:
CTL—CTC + HOCI —> С1СН2СН2ОН
Н2С----СН2 + НС1 —> С1СН2СН2ОН
Прямой синтез иприта из этилена и хлоридов серы (развитие экспериментальных исследований Гутри и Нимана) был избран во время первой мировой войны союзниками как самый удобный и быстрый. Они применяли различные смеси как моно-, так и двухлористой серы и получали при этом большое число продуктов, смесь которых оии принимали за чистое соединение.
Поуп осуществил синтез иприта из однохлористой серы и этилена при 70 °C и равномерном энергичном перемешивании с применением активированного угля в качестве катализатора,-
S2C12 + 2СН2—СН2 —> S(CH2CH2C1)3 + S (1)
Конант объяснял механизм реакции как взаимодействие эти-. лена с дихлоридом, образующимся из однохлбристой серы
S2C12 ► SC12„-]- S (2)
Этилен реагирует с дихлоридом с образованием монозамещен-ного продукта (реакция 3), атом серы в котором затем подвергается* непосредственному алкилированию (реакция 4), но одновременно образуются и политиоэфиры (реакция 5) :
SC12+CH2=CH2 —> C1SCH2CH2CI (3)
C1SCH2CH2C1 + СНг=СН2 —> S(CH2CH2Ci)2 (4)
. 2C1SCH2CH2C1 + nS —> C1CH2CH2—(S)n—CH2CH2C1 (5)
«
На основании проведенных исследований Перно9 считал более вероятным другой механизм реакции. По его представлениям при реакции этилена с однохлористой серой промежуточно образуется
146
сесквннпрнт I, который тотчас реагирует с однохлорнстон серой, давая бнс-2-хлорэтнловый тноэфнр II н трнтноэфнр III. Последний расщепляется одиохлорнстой серой. Образующийся р-хлорэтнл-сульфенхлорид IV прн действии этилена превращается в бис-2-хлорэтиловый тиоэфир II и полнтиоэфиры V. Политиоэфиры либо остаются и представляют собой побочные продукты, либо расщепляются далее (реакция 7).
+си2=сн2 +s2ci2
S2C12 -------> C1CH2CH2SCH2CH2SCH2CH2C1 -------->
I
—> S(CH2CH2C1)2+ C1CH2CH2S—S—CH2CH2C1 (6)
II III
/СН2СН2С1 +s2Cl2
s=s( -------> s; +<s)4; (?)
XSCH2CH2C1 \CH2CH2C1 \CH2CH2C1
III IV
I I
ch2=ch2
S(CH2CH2C1)2 + (S)n(CH2CH2Cl)2 <—-
II V
Использование чистой двухлористой серы снижает образование политиоэфиров, так как в этом случае ие происходит выделение коллоидной серы, что в конечном счете приводит к повышению выхода бнс-2-хлорэтнлового тноэфира:
SC12 + 2СНд=СН2 —> S(CH2CH2C1)2 (8)
Рилли 10 для получения бис-2-хлорэтнлового тноэфнра использовал четыреххлорнстую серу."*
Фотохимический синтез нз сероводорода н хлористого этилена происходит прн УФ-облученнн (300 нм) в присутствии органической перекиси как катализатора11:
2С1СН=СН2 + H2S —> S(CH2CH2C1)2 (9)
Время пребывания жидких продуктов в реакторе прн 15—25 СС составляет около 10 мин.
Возможен фотохимический синтез с применением 2-хлорэтнл-меркаптана вместо сероводорода:
С1СН=СН2+C1CH2CH2SH —> S(CH2CH2C1)2 (10)
Выходы в этой реакции, для которой в качестве катализаторов применяют диалкилднтиоэфиры, например бис-амнлдитноэфнр (CsHh)2S2, достигают около 99%, тогда как в реакции с сероводородом — лишь 75%.
147
Фотохимические синтезы имеют преимущество перед другими методами в том, что продолжительность реакции меньше, реакция идет прн обычной температуре и с количественным выходом. Реакцию можно проводить в таких растворителях, как метанол или беизол.
На схеме 2 изображены различные методы получения иприта.
Схема 2. Методы получения иприта.
Промышленные способы получения. В крупном промышленном масштабе производство иприта осуществляется по методам Мейера и Гутри. Возможен промышленный фотохимический синтез, который и был внедрен в ведущих странах по производству ОВ в связи с реорганизацией таких предприятий.
В то время как в Германии и в первую и во вторую мировую войну применяли преимущественно так называемый тнодигликоль-ный (оксольный) процесс (модицифированиый метод Мейера), производство иприта во Франции, Англии, США базировалось иа прямом синтезе из этилена и хлоридов серы,
В оксольном процессе для замещения оксигрупп пропускают в тиоднгликоль медленный ток сухого хлористого водорода. Реакторы и вся система коммуникаций установки изготовлены из свинца или освинцованы. Тем самым исключается образование солей
148
Железа. Процесс хлорирования вёдут при 50 6С, поддерживая температуру при помощи систем нагревания и охлаждения. После полного поглощения хлористого водорода полученный нпрнт, обладающий большой плотностью, выводят нз реактора и направляют на вакуумную перегонку, где прн пониженном давлении отгоняется вода. Ипрнт очищают перегонкой в вакууме илн обработкой органическими растворителями.
По сравнению с прямым синтезом производство иприта тнодн-гликольным процессом более трудоемко, но приводит к более высоким выходам (95%) продукта прн лучшем качестве.
Прямой синтез нпрнта, применявшийся союзниками, подвергался после первой мировой войны постоянным усовершенствованиям. Процесс состоит из четырех стадий: 1) получение двухлористой серы нз элементов; 2) приготовление раствора двухлорнстой серы в четыреххлорнстом углероде; 3) присоединение двухлори-стон серы к этилену путем пропускания этилена в раствор, прн этом получается разбавленный раствор нпрнта; 4) отделение нпрнта от растворителя для повторного его использования в процессе.
Все эти четыре производственные операции можно проводить непрерывно в одни прием. Применявшиеся французами реакторы имели высоту 18 м. Изготовлялись они целиком из свинца и были снабжены сложной системой змеевиков охлаждения. У каждой такой гигантской колонны был свой массивный змеевиковый конденсатор из серебра.
Во время пропускания этилена в раствор четыреххлорнстого углерода температуру реакции поддерживали 60—70 °C. Этилен должен быть предварительно тщательно высушен и очищен от спирта. Выход составлял около 80%.
Американцы применяли более низкие цилиндрические реакторы (высота 1,3 я,чднамстр 1 м). Этилен пропускали при 35 °C в одно-хлорнстую серу, одновременно в реактор малыми порциями добавляли однохлорнстую серу. Продукт получался загрязненным полнтиоэфирами и коллоидной серой; сера выпадала в осадок и ее отделяли.
Хотя при использовании чистой двухлористой серы нельзя ожидать образования коллоидной серы, следует учитывать, что этот днхлорид обычно распадается на хлор н однохлористую серу н в химическом отношении ведет себя как раствор хлора в смеси одно- и двухлорнстой серы. Если днхлорнд действительно существует в растворе, то он будет реагировать с этиленом по реакции 8, в то время как однохлористая сера взаимодействует с этиленом по реакциям 1—7 (см. выше).
Под действием избыточного хлора полнтноэфнры, в частности трнтноэфнр, превращаются в однохлористую серу н 0-хлорэтил-сульфенхлорнд
2S3(CH3CH2C1)2 + ЗС12 —> S2C12-|-4C1SCH3CH2C1 (11)
149
который с этиленом образует бис-2-хлорэтиловый тиоэфир (см. реакцию 4)9.
Как установил Перно, сырой продукт, который получают из однохлористой серы (для отличия от чистого бис-2-хлорэтилового тиоэфира он носит название прохлерит), устойчив. Ои содержит около 70% бис-2-хлорэтилового тиоэфира. Небольшую примесь бис-2'хлорэтилового дитиоэфира и нерастворимого в спирте остатка можно выразить брутто-формулой S8(CH2CH2C1)2. Возможно, они представляют собой смесь политиоэфиров общей формулы
С1СН2СН2—S—S—S—СН2СН3С1
(S)n
По американскому патенту12, бис-2-хлорэтиловый тиоэфир получают с примерно 92%-ным выходом путем распыления двухло-. ристой серы в избытке этилена прн 50—80 °C. Этот метод был разработан еще Майерсом и Стефеном, которые впрыскивали в газообразный этилен смесь одно- и двухлористой серы.
Эти процессы позволяют осуществлять непрерывное получение иприта с высокими выходами.
Для улучшения стабильности продукта и повышения его эффективности стремятся к возможно более высоким выходам н чистоте продукта.
Технический иприт (прохлерит), который может содержать до 30% примесей, очищают следующим способом13. Сырой продукт промывают водой и сушат. Высушенное вещество перед перегонкой нагревают при нормальном давлении до температуры ниже температуры кипения и затем отгоняют при 100 мм рт.ст. (т. кип. 130— 140°С). Очищенный иприт еще содержит 2—5% примесей.
К техническому и чистому иприту, как правило, добавляют стабилизаторы для придания устойчивости к длительному, даже неограниченно долгому хранению.
Лабораторный способ. В лаборатории бис-2-хлорэтиловый тиоэфир получают прямым синтезом из однохлористой серы и этилена. Однохлористая сера не должна содержать избыточного хлора, а этилен должен быть как можно более чистым. Его либо получают непосредственно в лаборатории, либо отбирают из стальных баллонов. Этилен очищают и сушат (целесообразно очищать также и газ из баллонов) концентрированной H2SO4, 20%-ным раствором NaOH и снова концентрированной H2SO4, пропуская газ через расположенные последовательно промывные склянки.
В качестве реактора используют круглодонную трехгорлую колбу емкостью 250 мл, которая снабжена подвижной трубкой для ввода газа, капельной воронкой и трубкой для отвода газа. В реакционную колбу, помещенную в водяную баню, вносят 20—30 а однохлористой серы и нагревают баню до примерно 50 °C. Температура реакции не должна подниматься выше 60 °C. Постоянный ток этилена пропускают через однохлористую серу со скоростью около 60 л/ч. Этилен начинает поглощаться не сразу, а через 1—2 ч; в течение этого времени добавляют из капельной воронки примерно еще 10 г однохлористой серы, а в ходе реакции еще 10—20 г.
Реакция продолжается несколько часов и заканчивается, когда вся однохлористая сера израсходбвана и этилен больше не поглощается. После этого реак-150
ционную смесь оставляют на некоторое время и отфильтровывают от коллоидной серы. (Внимание! Применяемая фильтровальная бумага пропитывается ипритом!) Этот технический продукт можно использовать для дальнейших опытов.
Чистый бис-2-хлорэтиловый тиоэфир получают перегонкой в вакууме. Иприт высокой чистоты можно получить 14 перекристаллизацией из спирта или петролей-ного эфира при —75 °C.
Чистый бесцветный бис-2-хлорэтиловый тиоэфир получают также обработкой тиодигликоля концентрированной НС1'при —б °C; при О °C иприт выпадает-в осадок 15.
5.1.3.2. Физические свойства. Чистый бис-2-хлорэтиловый тиоэфир— бесцветная маслянистая жидкость, не обладающая запахом. Технические продукты окрашены примесями в цвета от желтого до темно-коричневого и обладают характерным сладковатым запахом. Ниже температуры застывания (13,5 °C) тиоэфир кристаллизуется в виде длинных бесцветных ромбических кристаллов. С помощью прецизионных измерений установлена его температура плавления 14,44—14,45 °C; обычно данные колеблются в пределах 14,1 —14,5°C. Технический иприт застывает при температуре от 5 до 10 °C.
При нагревании до температуры кипения (около 217°С) иприт разлагается с образованием зловонных продуктов, напоминающих по запаху не то горчицу, не то чеснок.
Иприт американского производства, так называмый HD, имеет т. кип. 228 °C, т. пл. 14 °C.
Приводим температуру кипения иприта при разных давлениях:
Давление, Т. кип , Давление, Т. кип..
ММ рТ* ст. °C мм рт, ст. °C
760 216-218 5 84
200 180 1 54-55
20 - НО 0,45 40 ‘
15 107 0,09 30
10 95-97 01025 0
Иприт тяжелее воды (dl° 1,2741). Вследствие некоторой поверхностной активности он уменьшает поверхностное натяжение воды и в небольшой мере растекается по ней тонким слоем, как пленка масла. Добавлением более поверхностно-активных веществ, которые растворяются в иприте, эффективное растекание можно увеличить. Например, в результате добавления 1 % высокомолекулярного амина16 C22H38O2NH2 растекание иприта по воде увеличивается на 39°/о.
Давление пара бис-2-хлорэтилового тиоэфира мало. В интервале температуры 0—40°C его можно рассчитать по следующей формуле17 (в мм рт. ст.)-.
1g р = 8,3937 --^£
Бент и сотр.18 предложили другое уравнение для расчета температурной зависимости давления* пара бис-2-хлорэтилового тиоэфира:
, ПЛ£ИП 3117,2
1g р = 9,4819----—
151
По этой формуле давление пара имеет более низкое значение (см. табл. 11).
Таблица 11, Зависимость давления пара, летучести, относительной плотности и показателя преломления иприта от температуры
Температура. Давление пара, мм рт, ст. Летучесть по Веддеру. мг!л Относительная плотность d^ Показатель преломления
по Мемфорду по Бенту
-20 0,00017 —
. -10 0,010 — — W —
0 0,024 0,012 0,28 1,362 —
5 0,036 0,019 — — —
10 0,054 0,03 — — —
15 0,079 0.045 0,401 1,28 1,53125
(1,5313, 1,5278)
20 0,115 0,069 0,625 1,274 1,529
25 0,165 0,105 0,958 1,264 1,52489
30 0,23 0,15 0,443 — —
Выше 40°C 1gр уже больше не является линейной функцией от 1/Т, До температуры 140°С давление пара можно вычислить по другой формуле 19:
1g р = 38,525 - - 9,86 lg Т
В этой же таблице приведена температурная зависимость летучести, относительной плотности и показателя преломления.
Максимальная концентрация иприта равна ~0,6 мг/л при 20 °C, что указывает на высокую его стойкость. Иприт — самое стойкое из общепринятых ОВ. Его стойкость на местности составляет самое меньшее от 1,5 до нескольких суток. Соглдсно американским наставлениям, прн обычной температуре без защитной одежды запрещается вступать на открытую местность (луга и т. п.) с плотностью заражения 10—50 г/м2 до истечения первых суток, а в лесистой местности — до истечения четырех суток можно находиться с надетым противогазом не более 2 ч.
Как и у каждого ОВ, стойкость иприта зависит от метеорологических условий. С повышением температуры воздуха она очень сильно уменьшается. При температуре 20 °C она в 2—3 раза .меньше, чем при ~5°C. При скорости ветра 6 мкек она в 4 раза меньше, чем при безветренной погоде.
Хотя с возрастанием температуры воздуха стойкость уменьшается, но за счет испарения иприта воздух обогащается парами иприта. В жаркие безветренные дни создается концентрация, почти в 50 раз превышающая обычно необходимую токсическую концентрацию. Даже при скорости ветра 2 л^сек и плотности заражения
152
порядка 5 г/jn2 на открытой местности пары иприта еще эффективны в течение 3—4 ч.
При температуре ниже 15 °C, особенно на покрытой растительностью или очень лесистой местности, зараженность держится более недели. Ниже 4 °C при скорости ветра свыше 8 м/сек пары иприта в воздухе уже не эффективны.
Чтобы повысить стойкость нпрнта н, тем самым, его эффективность, вносят добавки, которые повышают его вязкость (так называемые вязкие рецептуры). Эти добавки гарантируют ие только большую продолжительность действия, ио и затрудняют дегазацию, которая в этом случае вряд ли осуществима водными дегазирующими растворами. Повышение вязкости одновременно подавляет образование аэрозоля и способствует образованию капелек большего размера (а ие дыма) при поливке с самолетов.
- В качестве повышающих вязкость добавок пригоден ряд полимеров, в частности полиметилметакрилаты с мол. в. 40 000—50 000; их добавляют в количестве 4—8%. Вязкость таких смесей* при 10°C составляет от 30 до 600 спз. Такие смеси особенно пригодны для поливки с большой высоты, поскольку форма капель при этом сохраняется, а поливка почти не заметна с земли.
Добавки больших количеств загустителей приводят к образованию .иеупругих смесей, которые нужно применять при помощи мни и бомб со сравнительно большими зарядами взрывчатки. Соли тяжелых металлов, в частности FeCl2, могут понижать или же повышать вязкость. Добавки соединений — ингибиторов коррозии противодействуют уменьшению вязкости.
Растворимость бис-2-хлорэтилового тиоэфира в воде мала, примерно 0,8 г/л. В органических растворителях — галогеиалкаиах, бензоле,'хлорбензоле — он растворяется столь же хорошо, как и в растительных или животных жирах. В то время как растворимость ОВ в абсолютном этаноле выше 16 °C составляет почти 100%, в 92%-иом этаноле она достигает едва £5%. С такими растворителями, как нефть и дизельное топливо, ои смешивается ограниченно, имеются области несмешиваемости. Критическая температура растворимости для системы нефть — иприт лежит около 30 °C, выше этой температуры^ образуется гомогенный раствор. В табл. 12 приведена растворимость иприта в воде, бензине и нефти. Эти данные показывают, что при низкой температуре для растворения иприта можно использовать только иизкокипящие фракции,- например легкий бензин и петролейиый эфир, которые являются превосходными средствами для экстракции иприта.
Растворимость иприта в различных органических растворителях можно использовать для понижения его температуры застывания. Только благодаря этому возможно применение ОВ при температурах ниже их температур плавления. В качестве соответствующих растворителей имеют значение хлорбензол, нитробензол,
* Динамическая вязкость иприта при 20 °C равна 4,50 спз.
153
Таблица 12 Растворимость иприта в воде, нефти и бензине
Температура, °C Растворимость Температура, °C Растворимость
в воде, а/л в нефти, объемп. % в бензине, объемп. % в воде, е/л в нефти, объемы. % в бензипе, объемн.%
-10 — <3 <4 10 0,7 21 77
-5 —- 4 5 15 28 ОО
0 0,3 6 7 20 0,8 38 —
5 — 10 14 30 — оо —
бензол, четыреххдористый углерод и др. Их добавляют к ОВ в количестве до 25%. В табл. 13 приведены температуры застывания некоторых из таких смесей.
Таблица 13. Температуры застывания смесей некоторых веществ с ипритом
Содержание компонента в смеси, % Температура застывания смесей, бС
с тетрахлор-этаном с хлорбензолом с хлорпикрином
10 9,8 8,4 9,8
20 6,6 6,4 6,3
30 8,1 -1,0 2,6
Со многими общепринятыми ОВ бнс-2-хлорэтнловый тноэфнр смешивается во всех отношениях. Так, например, возможны смеси его с дифосгеном, хлорпикрином, люизитом и другими алкилди-хлорарсннами, пригодными в качестве ОВ, а также с фосфорорганическими соединениями, например с диизопропилфторфосфатом (ДФФ), зарином и зоманом. Ниже приведены определенные Саундерсом20 температуры застывания смесей иприта с ДФФ:
ДФФ, я T. застыв., °C ДФФ, я T. застыв,, °C
9,5 ' 8,9 57,2 -7,5
24 4,8 69 -15
34,3 2,1 77 —22
45,5 -3,3 87 -36
Во время второй мировой войны в Германии средн прочих была приготовлена смесь иприта с так называемым кислородным ипритом.
Вследствие своей высокой диффузионной способности нприт быстро проникает через.ряд веществ и материалов. Так, жидкий нпрнт быстро просачивается через ткани, кожу, картон, бумагу и тонкую резину. Он быстро впитывается в пористые неоднородные материалы, такие, как кирпич, бетон, необработанная древесина н старые потрескавшиеся масляные покрытия. По материалам с
154
однородной поверхностью он'растекается, например по стеклу, глазурованной черепице, кафелю, гладким Масляным покрытиям. Одиако вещества, которые растворяются в этом ОВ, иапрнмер резина и воск, .поглощают его.
Сравнительно стойкими (резистентными) материалами, которые уже прн толщине слоя в миллиметры задерживают ОВ на протяжении часов и дней, являются некоторые полимеры типа неопрена, а также поливиниловый спирт, фторкаучук, оппанол, полиэтилен, тиокол. Эти вещества применяют для изготовления защитных материалов.
Для изготовления защитной одежды от воздействия иприта н других ОВ из множества веществ, пригодных для нмпрегннрова-ння тканей, бумаги и прочих материалов, выбраны некоторые.
Хлопчатобумажные ткани в результате пропнткн натриевой1 солью альгиновой кислоты, которую растворяют в воде вместе с солями медн нлн цинка, и после высушивания дополнительно обработанные водной эмульсией сополимера метил- н этилакрнлата, становятся на некоторое время непроницаемыми для иприта н подобных ОВ21. Импрегннрованные таким образом тканн служат для пошива защитного обмундирования. ' в
В США для импрегннровання применяют, в частности, производные мочевины, например сцлии-1У,М/-днхлор-бнс-'(2,4,6-трихлор-феннл)-мочевину, которую используют с добавкой ацетата натрия, связывающего соляную кислоту22. В другом патенте пользуются водной суспензией N, Ы'-днфенил-Ы'-хлор- (2,6-диметнлфеинл) -мочевины. Диспергирующим средством служит поливиниловый спирт23.
В других способах импрегннровання исходят нз продуктов окисления и полимеризации ненасыщенных карбоновых кислот, например льняного нлн тунгового масел, н других полимеров; кроме того, используют желатиновые прокладки24.
Современные импрегннрованные материалы ие впитывают илн слабо впитывают влагу; они являются водоотталкивающими.
Из других пропиток известны казеин-глнцернновая, желатнн-глицериновая, казени-полнбутнленовая, клее-глнцериновая, продукты конденсации многоатомных спиртов (глицерин, гликоль) с миогоосиовными карбоновыми' кислотами (адипиновая кислота). Онн могут служить как для импрегннровання тканей, так н для изготовления бумажных защитных накидок. После пропнткн нх стабилизуют соответствующими отвердителями.
5.1.3.3. Химические свойства. При обычной температуре бнс-2-хлорэтнловын тноэфир представляет собой устойчивое соединение. Прн нагревании выше 170°C он разлагается с образованием неприятно пахнущих ядовитых продуктов различного состава. Выше 500°С происходит полное термическое разложение. Кратковременное нагревание даже выше 300°С почти не приводит к образованию продуктов разложения, поэтому иприт считается относительно устойчивым к детонации.
155
По отиошеиню к металлам прн обычной температуре нпрнт инертен, он почти не действует на свинец, латунь, цинк, сталь, алюминий; при повышении температуры сталь разрушается.
Загрязненный нпрнт, содержащий обычно воду и хлористый водород, вызывает коррозию стали. Образующиеся соли железа способствуют коррозии. Из-за выделяющихся газов — водорода, сероводорода, этилена и других продуктов разложения — следует считаться с повышением давления в закрытых емкостях, минах, бомбах и контейнерах для перевозки.
Ингибиторы коррозии и антиоксиданты препятствуют разложению прн хранении. Такими веществами могут быть, например, галогениды тетраалкиламмония (бромистый тетраметнламмоинй), гексаметилентетрамин, пиридин, пиколки, хинолин и другие органические амннопронзводные.
Гидролиз. В водных растворах бис-2-хлорэтиловый тноэфир'гидролизуется с замещением атомов хлора на гидроксильные группы. В присутствии избытка соляной кислоты реакция обратима. Обычно она идет с образованием неядовитого тнодиглнколя:
4-H2O' .*СН2СН2ОН j-H2O
S(CH2CH2C1)2 < t sf < ..'.'.t S(CH2CH2OH)2
+HCi \ch2ch2ci +HCf
На основании способности тноэфнров давать соединения суль-фоиня некоторые исследователи25 пришли к заключению, что во время гидролиза промежуточно образуются циклические ноны суль-фоиия, поэтому реакция должна проходить по следующей схеме:
С1СН2СН2—S—СН2СН2С1
S—СН2СН2С1
Н2С—-сн2
S—СН2СН2ОН н2<^— сн2
сг
уСН2СН2С1 (3) 7—>
^СН2СН2ОН
^(4)-
НОСН2СН2—S—СН2СН2ОН
Реакции (1) н (2), а также (3) н (4), по-внднмому, проходят независимо друг от друга. В циклических переходных состояниях +
связь S—С сильно полярна, что н определяет ее дальнейшую реакционную способность.
Другие исследователи связывают возможность образования сульфониевых производных с последующими реакциями димеризации либо прн взаимодействии негидролизованного иприта с про-
156
дуктамн его гидролиза (см. выше), либо дродуктов гидролиза между собой, а также с другими процессами роста молекулы:
'Н2С. +
I Ч—СН2СН2С1
+нго /СН2СН2С1 +На0 ,СНгСН2ОН сг -<—>• s —>- s;
\сн2сн2он \СН2СН2ОН
II ш
+Н2о
/СН2СН2—S(CH2CH2OH)2'
^СН2СН2С1
сг
+н2о
---->
СН2СН2—S(CH2CH2OH)2’ sz
_ \сн2сн2он
' /СН2СН2—S(CH2CH2OH)2"
_ ^СН2СН2—S(CH2CH2OH)2_
+и20
2СГ
Продукты этих реакции гидролизуются с образованием конечного продукта гидролиза, тноднглнколя III. Некоторые из этих соединений были получены независимо.
После завершения гидролиза иприта в водном растворе (1 :50) содержание продукта реакции I + III достигает 22 %
Данные о продолжительности гидролиза в водных растворах иприта очень разноречивы. В насыщенных растворах (0,08% прн 15°C) гидролиз заканчивается через 2 ч.
Насыщенный раствор иприта через 2 ч при 15 °C еще содержит 15% (120 мг!л) негндролнзованного тноэфнра. Растворы такой концентрации уже не должны проявлять кожно-нарывного действия. Гидролиз иприта при - разных температурах приведен в табл. 14 (данные Гопкинса).
Таблица 14. Состав водных растворов, иприта после 2 ч гидролиза насыщенного его раствора *
при разных температурах
Температура, Гидролизованный иприт, % Негидролнэованный иприт, мг!л
0,6 41 197*
10,0 78 154*
15,0 85 120*
20.5 — 10
Найдено экстраполяцией.
157
Как видно из табл. 14, скорость гидролиза увеличивается с повышением температуры. Воду, содержащую небольшие количества (~ 1 %) иприта, можно дегазировать кипячением в течение 15 мин. При температуре ниже О °C гидролиз продолжается несколько суток. Более сильно зараженную воду приходится подвергать обычным приемам дегазации и очистки.
Вследствие гидролиза концентрация иприта в воздухе через сутки уменьшается почти на порядок.
Из-за малой растворимости бнс-2-хлорэтилового тиоэфнра, в зависимости от присутствующего его количества, вода заражается на длительный срок, поскольку взамен гидролизованного нпрнта в воду все время постепенно диффундируют новые порции иприта. Опыты показывают, что находящееся под водой ОВ остается эффективным годами, если совсем нлн почти отсутствует перемешивание.
Так как ОВ практически нерастворимо в воде, гидролиз может протекать только на самой границе раздела фаз. Для быстрого гидролиза обязательно'требуется энергичное перемешивание. Если же перемешивание недостаточно, то, особенно при применении технических сортов нпрнта, из-за содержащихся в нем загрязнений и примесей в результате побочных реакций образуется третья фаза, в которой иприт мало растворим н которая защищает и «консервирует» его от дальнейшего гидролиза.
Из всего сказанного выше ясно, что для достижения полноты гидролиза требуются большой избыток воды н энергичное перемешивание либо нагревание, а еще лучше, если действуют все три фактора.
Для того чтобы вывести гидролиз за поверхность раздела фаз гетерогенной системы, нужно гомогенизировать смеси, скажем, добавкой растворителей, которые могут смешиваться н с водой, и с ипритом (метанол н др.). Такими добавками пользуются, в частности, при употреблении водных дегазирующих растворов.
Скорость гидролиза бис-2-хлорэтилового тиоэфира зависит от концентрации ионов водорода — способствует ему щелочная среда, а кислая — препятствует и замедляет гидролиз.
Ионы гидроксила смещают равновесие гидролиза вправо (см. стр. 156). При избытке щелочи реакция является количественной и протекает по уравнению:
+2ОН”
S(CH2CH2C1)2 ------> S(CH2CH2OH)2
-2Ci“
В сильно щелочной области скорость гидролиза возрастает примерно на 20% по сравнению с нейтральной.
Прн взаимодействии с эквимольными количествами раствора щелочи образуется днвиниловый тиоэфир:
+20 н~
S(CH2CH2C1)2 ----------* S(CH=CH2)3
-2СГ; -2Н2О
158
Аналогично реагируют С нНрнТом соли щелочных металлов Низших карбоновых кислот, например ацетат калия:
S(CH2CH2C1)2+СНзСООК —> S(CH2CH2OCOCH3)2 + 2KC1
Реакция бнс-2-хлорэтнлового тноэфнра с аммиаком даже прн температуре выше 150°С идет очень медленно. Однако под давлением уже прн 60°C образуется 1,4-тназан— бесцветная, дымящаяся на воздухе жидкость:
сн2сн2
S(CH2CH2C1)2+ NH3 —► ^NH + 2HC1
, сн2Сн2
с первичными аминами в спиртовых растворах прн обычных условиях происходит аналогичная циклизация тноэфнра:
СН2СН2
S(CH2CH2C1)2 + RNH2 —> ^NR+2HC1
СН2СН2
4-Алкнл-1,4-тназаны, образующиеся прн взаимодействии с алифатическими аминами, представляют собой бесцветные жидкости.
Вторичные н третичные а-мнны дают соответствующие соединения, но при этом образуются линейные продукты:
S(CH2GH2C1)2 + 2HNR2
S(CH2CH2C1)2-|-2NR3 —>
' yCH2CH2NHR2-sz
_ \CH2CH2NH7?2
+
2СГ
" zCH2CH2NR3"
sz
„ \ch2ch2nr3 _
2СГ
Получающиеся четвертичные аммониевые производные физиологически активны. Обе реакции идут сравнительно медленно н не применяются для дегазации.
В работе27 описано замещение атомов хлора в бнс-2-хлорэтн-ловом тноэфире на фосфорорганические остатки. Прн реакции тио-эфира с дн ал кил фосфитам и в толуольном растворе прн 106 °C авторы получили соответствующие соедниення типа
(RO)2PCH2CH2—S—CH2CH2P(OR)3
представляющие собой высококнпящне устойчивые к нагреванию жидкости.
Окисление. Как типичный представитель тноэфнров^ иприт можно легко окислить в сульфоксид нлн в сульфон согласно уравнению
J-O о
S(CH2CH2C1)2 -->- (C1CH2CH2)2SO ---(C1CH2CH2)2SO2
160
---D—к а чес i Bt икислшслси—ирш иди di—неуемна—видиуидд,—азснпдл кислота, перманганат калия, хромовая кислота, хлорная известь, гипохлориты и др.
Хотя продукты окисления иприта — сульфоксид и бис-(2-хлор-этил)-сульфон — сильно ядовитые соединения, а сульфоксид обладает даже кожно-нарывным действием, окисление иприта является одной из важных реакций дегазации. Продукты окисления представляют собой твердые кристаллические вещества, и поэтому они не обладают кожно-резорбтивными свойствами.
Сильное электроноакцепторное влияние атомов серы и кислорода в продуктах окисления, которое индуктивно передается атомам галогена, делает невозможным гидролиз этих соединений по схеме, аналогичной для гидролиза самого бис-2-хлорэтилового тиоэфира (см. стр. 156).
При действии сильных окислителей или при окислении в более жестких условиях реакция идет дальше образования сульфоксида и сульфона и завершается полным разложением тиоэфира.
Бис-2-хлорэтиловый тиоэфир окисляется при 150°С избытком концентрированной HNO3 в сульфоксид. Реакция быстрая и экзотермическая. Она может идти н дальше — с выделением двуокиси азота и образованием в конечном счете сульфона 28:
+2HNO3
3S(CH2CH2C1)2 ———> 3(C1CH2CH2)2SO
—NO; —Н2О
+2HNO3
(C1CH2CH2)2SO ——----—(C1CH2CH2)2SO2
——rijU
При действии дымящей азотной кислоты и нагревании образуется, иапример, 2-хлорэтаисульфокислота C1CH2CH2SO2OH.
Для получения бис-(2-хлорэтил)-сульфоксида бнс-2-хлорэтило-вый тиоэфир прибавляют по каплям при 20 °C к концентрированной HNO3. Если смесь разбавить водой, то сульфоксид выпадает в виде белого осадка, который перекристаллизовывают из 60%-ного этанола (бесцветные чешуйки, т. пл. 110°C).
При действии на иприт хромовым ангидридом в уксусной кислоте при 100 °C (20 мин нагревания) образуется чистый бис-(2-хлорэтил)-сульфон, т. пл. 55—56 °C.
Сухая хлорная известь преимущественно окисляет иприт, но одновременно происходит и хлорирование. При определенных условиях эта быстрая экзотермическая реакция сопровождается воспламенением непрореагировавшего тиоэфира. В качестве продуктов реакции были идентифицированы — хлористый водород, двуокись углерода, двуокись серы, галогеналканы (например, дихлорэтан, хлороформ), хлораль, хлориды серы и др. При использовании кашицы хлорной извести реакция идет спокойнее с образовании в основном сульфоксида и сульфона
Гипохлориты в водных растворах частично окисляют, частично хлорируют бис-2-хлорэтиловый тиоэфир. Как показали исследования Молера, расход гипохлорита в несколько раз больше, чем
6 Зак, 448
161
объясняется реакциями хлорирования и каталитическими реакциями разложения гипохлорит-иоиов, вызываемыми тиоэфиром или продуктами его превращения. Хлорирующими агентами могут являться образующиеся по реакции соответствующие хлорвиниль-иые производные сульфоксида и сульфона.
По данным Холста30, иприт полностью разлагается избытком гипохлорита по следующему уравнению:
+ 140
S(CH2CH2C1)2 ----+ SO3 4-4СОг 4-2НС1 4-ЗН2О
Хлорирование ионами гипохлорита происходит только в нейтральной или слабокнслой среде. В щелочной среде оно невозможно и поэтому в основном преобладают окислительные процессы.
Согласно Штампе и сотр., насыщенные водные растворы иприта можно дегазировать хлором из расчета 350 мг хлора на 1 л воды при 20 °C. После фильтрования через активированный уголь эту воду можно употреблять в пищу26.
Хлорирование. При действии хлора и, отчасти, других хлорирующих агентов, например днхлорамнна и сульфурилхлорида,-получают различные хлорированные тиоэфиры и сульфоксиды. Ниже приведены формулы соединений, получающихся при хлорировании иприта:
С1^ /СНгСНгС! С1/ \СН2СН2С1 ^СС12СС13 \ш2СН2С1
уСС12СНС12 sz
\chcichci2
ХНС1СН2С1 sz
\ch2ch2ci -уСС12СН2С1 \:hcich2ci
✓СС1=СН2 sz
\сН2СН2С1
.СНС1СНС12
\СН2СН2С1 уСС12СН2С1 \СНС1СНС12
^CII=CIICi
\ch2ch2ci
Все они, кроме первого соединения, являющегося твердым веществом (белые неустойчивые иглы), представляют собой жидкости. В зависимости от числа атомов хлора и от их положения в молекуле эти продукты хлорирования обладают лишь слабой, либо вовсе не обладают физиологической активностью.
При действии хлора на иприт идут главным образом реакции (1-8).
Продукт присоединения, образующийся в результате реакции (I), как уже упоминалось, при обычной температуре неустойчив, и его можно выделить в кристаллическом виде только при действии хлора на раствор иприта в четыреххлористом углероде при —5°C. При дальнейшем хлорировании (2) из него образуются различные полихлорпроизьодные либо он преимущественно реаги
162
рует по направлению (3) с образованием i ,z,z -трихлирдиэтило-вого тиоэфира:
O=S(CH2CH2C1)2
|+Н2О
,СН2СН^! + ch Ск /СН3СН2С1 + ci2 _ / __> ПОЛИХЛОр-
\СН2СН2С1 <» C/\CH2CH2C1 *> производные
(3) | + С12 ' —
/СНС1СН2С1 \ch2ch2cj
В результате дегидрогалогенирования этот трихлордиэтиловый тиоэфир превращается в изомерные хлорвинильные соединения:
уСН=СНС1 (4) /СНСЛСНгС! (5) ZCC1=CH2
sf -<— > s;
\CH2CH2C1 “HCI XCH2CH2C1 -HC1 \CH2CH2C1
A I +
m J"” и “”c*
ХСНС1СН2С1 ~H2°
—:--------o=sz —----------
\ch2ch2ci
Так же, как и в случае реакций с гипохлоритом, в результате хлорирования достигается дегазация иприта.
При дегазации сульфурилхлоридом протекает в основном следующая реакция:
ХНС1СН2С1
S(CHaCH2Cl)24- SO2C12 —> Sz 4-SO24-HCI
\ch2ch2ci
Дальнейшее действие сульфурилхлорида ведет к описанным выше реакциям (1—8).
Реакции с хлораминами. Моно- и дихлорамины легко реагируют с бис-2-хлорэтиловым тиоэфиром. Дихлорамин имеет то преимущество перед монохлорамином, что реакция идет в гомогенной среде, так как дихлорамин сравнительно легко растворим в органических растворителях. Продукты реакции неядовиты.
Монохлорамин Т реагирует с ипритом с образованием N-(n-TO-луолсульфо) -бис-(2-хлорэтил) -сульфимипа, белого кристаллического соединения, т. пл. 143—144°C. Под влиянием ионов ОН-воды, сравнительно слабо полярная связь N—С1 монохлорамина поляризуется в двух направлениях, поэтому хлорамин действует и как аминирующий, и как хлорирующий агент. В обычных условиях
е*
163
дегазации важнейшей реакцией является аминирование. Хотя среди продуктов реакции были найдены и различные хлорпроизводиые, хлорирование является лишь второстепенной реакцией. Предполагается, что реакция'протекает по следующей схеме:
|| _ ХНгСНгС!
СН3С6Н4—S—N—Cl Na+ + —>
\СН2СН2С1
О
II ,СН2СН2С1
—> СН3С6Н4— S—N=S + NaCl
II \ch2ch2ci
О
N- (n-Толуолсульфо) -бис- (2-хлорэтил) -сульфимии получают по методу Сартори прибавлением иприта (17,1 а) к водному раствору монохлорамина (28 а). Примерно через 1 ч из раствора выпадает кристаллический продукт.
Согласно Россу31, в водно-ацетоновом растворе гидроокиси натрия этот сульфимин превращается в дивинилсульфимии (т. пл. 94—96 °C):
о о
II II
СН3СеН4—S—N=S(CH2CH2C1)2 ----> СН3С6Н4—S—N=S(CH=CH2)2
II -2HC1 II
о о
Дихлорамины действуют на иприт только как хлорирующие агенты (см. стр. 162).
Органические хлораминопроизводные являются'превосходными средствами дегазации иприта. Кроме уже.упомянутых соединений пригодны, например, днхлорамиды метан-, изогексан- и беизол-сульфокислот и гексахлормеламин, которые, в соответствии с их свойствами, действуют как окисляющие, аминирующие или хлорирующие агенты *.
Реакция с -гексаметилентетрамином. Одной из реакций, важных для дегазации кожи, является предложенное Брюэром и Бушеро32 воздействие гексаметилентетрамином на гидролиз иприта. Выделяющийся при гидролизе хлористый водород реагирует с гексаметилентетрамином с образованием формальдегида, который, благодаря своему дубящему действию, содействует локализации и лучшему заживлению ипритных поражений на коже. Применение гексаметилентетрамина в качестве стабилизатора этого ОВ объясняется аналогичной реакцией.
Реакция выражается следующим уравнением:
2S(CH2CH2C1)2 + ЮН2О + (CH2)6N4 —> 2S(CH2CH2OH)2 + 4NH4C1 + 6НСНО
* Все эти соединения, в основном, действуют только как хлорирующие агенты. — Прим. ред.
164
Реакции с солями металлов. Подобно незамещенным тиоэфирам, иприт обладает способностью образовывать аддукты с солями металлов, так называемые солн сульфония. Образующиеся комплексы иногда трудно растворимы в воде и имеют характерную окраску, вследствие чего они нашли применение в качественном и количественном анализе.
Чаще всего используют галогениды тяжелых металлов — иодид и хлорид ртути, хлориды меди, цинка, титана, платины, палладия и золота.
С хлорным золотом бис-2-хлорэтиловый тиоэфир образует комплексное соединение33:
S(CH2CH2C1)2 4- AuCl3 —> [C12AuS(CH2CH3C1)2]C1-
Хлориды меди(I) и ртути(1) образуют устойчивые комплексы типа С12Ме2- S (СН2СН2С1)2, а диаммйнплатинахлорид дает комплекс типа C12(NH3)2 Pt-S(CH2CH2C1)2. Согласно Бндербаху34 иприт с аминоферрипентацианидом натрия дает сине-зеленый комплекс Na3[Fe(CN)5.S(CH2CH2Cl)2J.
5.1.3.4. Токсические свойства. Иприт действует как контактный и ингаляционный яд; пары и капли иприта поражают глаза. Как уже упоминалось, вследствие высокой растворимости иприта в липидах или его резорбции во все органы наступают интоксикации обшего характера, которые являются причиной его общеядовитого действия.
Способность иприта проникать через кожу зависит от температуры. Скорость резорбции пропорциональна повышению температуры 35: при 21—23 °C она составляет около 1,4-10-3 мг/см2-мин, при более высокой температуре — около 2,7-10-3 мг)см2’мин. Во время этого опыта, проводившегося на предплечья, относительная влажность насыщенного парами иприта воздуха составляла около 46%. Количества ОВ порядка 6-10-3 мг, проникавшие через кожу, в 50% случаев вызывали образование волдырей.
В 100% случаев образование эритем наступает при дозе иприта 0,01 мг!см2 поверхности кожи, маленькие пузыри образуются при 0,1—0,15 MejcM2, большие — при 0,5 мг!см2.
Высокая чувствительность половых органов к парам .иприта проявляется в том, что эритемы образуются уже прн концентрациях 0,012 мг]л и времени экспозиции 5 мин, тогда как тот же физиологический эффект на коже плеча заметен лишь при вдвое большей концентрации (см. табл. 15).
Глаза поражаются малыми количествами ОВ. Так, концентрация 1,2-10~3 мг}л после 45 мин экспозиции вызывает светобоязнь, спазм век, и воспаление конъюнктивы, длящееся до 4 недель. Выздоровление наступает через 3 месяца. Более высокие концентрации при меньшей экспозиции приводят к опасным поражениям глаз.
' 165
Таблица 15. Действие паров иприта при различных концентрациях и разной экспозиции
Концентрация паров, мг/л, при экспозиции Действие
1 мин 5 мин 10 мин 15 мин 30—60 60 мин 180 мин
— — — — — 0,001 — Серьезных поражений нет
— — —' —‘ — 0,001 Потеря боеспособности
— 1—г —- 0,005 0,001 — Легкие поражения глаз через 12 ч
—- —- 0,01 — — Опасное, сильные поражения глаз
— 0,012* 0,025 ** —- 0,02 0,003 0,005*** 0,006 0,002 Образование эритем
0,1-0,2 — -—h — “— Продолжающийся неделями зуд на нежных участках кожи
— — — — 0,1 Опасные поражения дыхательных путей
1,0 — 0,4 — 0,3 0,1 Образование маленьких пузырьков
•— - 2,0 — 0,8 (0,75) 0,3 Образование волдырей
— 3,0 . (1,5) 2,0 0,07 — Смертельный исход
•На половых органах, •* На
коже плеча. ••• У восприимчивых.
При времени экспозиции 2—5 мин концентрация 0,03 яг/л смертельна, концентрации уже в 0Д1 мг/л при воздействии 15 мин очень опасны, они ведут к поражениям кожи и к сильным воспалениям слизистой оболочки глаз.
- При подкожной инъекции летальной дозой является 40— 60 мг/кг массы. На американских ипритных заводах значение МКРМ. при восьмичасовом рабочем дне принято равным 5-10-5мг/л.
Другие данные о действии паров иприта см. табл. 15. Эти величины следует рассматривать, как приближенные. Оии не учитывают влияния температуры, предрасположенности к отравлению и другие факторы, которые могут привести к увеличению токсичности.
5.1.4. Аналоги бис-2-хлорэтилового тиоэфира
5.1.4.1. Бис-2-фторэтиловый тиоэфир
. /СН2СНгЕ
\ch2ch2f
В 1952 г. Малатеста и Д’Атри 36 был получен бис-2-фтпрэтило вый тиоэфир, т. кип. 95—96°C (30 мм рт. ст.), d 1,2354 г/см\ Точных данных о его токсичности нет. Получают этот тиоэфир с
16G
S(CH2CH2C1)2
образованием При нагрева-окисляется до
Жхорошцм выходом (84%) обменом атомов хлора в бнс-2-хлорэти-Яяовом тиоэфире на фтор действием избытка фтористого серебра Жри нагревании в течение 2 ч при 40—50°C:
+ AgF _2AgC> 3(СН2СВД2
Ж С азотной кислотой этот тиоэфир реагирует с дбне-(2-фторэтил)-сульфоксида (т. пл. 102—103°С).
Кии-в течение 4 ч с хромовой смесью при 200 °C он 1|бис-(2-фторэтил-)-сульфона (т. пл. 41—42°C).
Я Прн стоянии (8 ч) смеси водного раствора хлорамина Т с бис-Ж2-фторэтиловым тиоэфиром образуется М-(п-толуолсульфо)-бис-(2-Лфторэтил)-сульфимин (т. пл. 135—136°C).
р Число других описанных фторированных тиоэфиров мало.
5.1.4.2. Бис-2-бромэтиловый тиоэфир
L ' zCH2CH2Br
В"
\сн2сн2вг
^Физические свойства: т. кип. 240°C разд. (760 мм рт. ст.), 134°C i (17 мм рт. ст.); т. пл. 31—34°C; бЛ52,05; давление пара при 20°C — f’X),03 мм рт. ст.; предельная концентрация при 20°С — 0,4 мгл.
После первой мировой войны бис-2-бромэтиловый тиоэфир был /Исследован как возможное ОВ. Во время второй мировой войны ои ^упоминался кое-где в литературе под названием «бромлост». По Свойствам ои соответствует иприту. Физиологическое действие этих соединений одинаково, ио бромид менее токсичен.
< Его получают по реакции тиодигликоля с трехбромистым фосфором или с бромистым водородом.
Соединение практически нерастворимо в воде, но гидролизуется Руэдой быстрее, чем бис-2-хлорэтиловый тиоэфир. Все другие соот-.фетствующие реакции также идут быстрее.
Как ОВ бис-2-бромэтиловый тиоэфир уже почти ие приии-мается в расчет.
Смешанный бромхлорид, — 2-хлорэтил-2^бромэтиловый тио-Йир, С1СН2СНг—S—СН2СН2Вг, имеет т. кип. ,125—132 °C
3 мм рт. ст.) т. пл. 24 °C. Бис-2-иодэтиловый тиоэфир — желтое кристаллическое вещество, т. пл. 62—70°С; его свойства аналогич-свойствам уже рассмотренных соединений.
й; 5.1.4.3. 2,2'-Бис-(2-хлорэтилтио)-диэтиловый эфир (< кислород-* , *;ВЫЙ ИПрИТ»)
CH2CH2SCH2CH2C1
- ^CH2CH2SCH2CH2C1
^Физические свойства: т. кип* 174° С (2 мм рт. ст.), 120° С VXJ1 мм рт. ст.); т. пл. от —30 до —38° С; ^4°1,2311.
167
Как и соединения, рассматриваемые ниже, 2,2'-бис-(2-хларэтиЛ-тно)-диэтиловый эфир был получен и исследован в Англии во время второй мировой войны 37. Это соединение обладает значительно более сильным кожно-нарывным действием, чем иприт. В общем, его физиологическая активность примерно в 3,5 раза превышает активность иприта.
При действии концентрированной HCI на тиодигликоль (ПО°C) образуется смесь, содержащая 60% иприта и 40% кислородного иприта (так называемая смесь НТ). Отделить кислородный иприт из этой смеси перегонкой нельзя.
В чистом виде его получают хлорированием 2,2'-бис-(2-окси-этилтио) -диэтилового эфира тионилхлоридом в растворе хлороформа при температуре ниже 40°C:
zCH2CH2SCH2CH2OH +SOCIa zCH,ClbSCH2(JTCl
о; ------> о;
\CHaCH2SCH2CH2OH "s°2; “HC1 \CH2CH2SCH2CH2C1
Исходное оксисоединение получают из бис-2-хлорэтилового эфира , и спиртового раствора натриевой соли 2-меркаптоэтаиола (2 моль): '
/СН2СН2С1 +2NaSCH2CH2oH /СН2СН2ьСН2СН2ОН
О -------------> О
\ch2ch2ci ' \ch2ch2sch2ch2oh
Полученный продукт плавится при 32 °C; т. кип. 215 °C (2,5 мм рт. ст.).
Кислородный иприт — бесцветная жидкость, легко растворимая в обычных органических растворителях (бензол, ацетон). В спирте она малорастворима, в воде нерастворима.
В химическом отношении кислородный иприт ведет себя как бис-2-хлорэтиловый тиоэфир. В результате идентичных реакций обычно образуются те же продукты; так например, при взаимодействии с хлорамином получается Ы-(п-толуолсульфо)-бис-(2-хлор-этил) -сульфимин.
Кислородный иприт предполагали применять в тактических смесях.
Аналогичные иоднстое и бромистое соединения неустойчивы, особенно 2,2'-бмс-(2-иодэтилтио)-диэтиловый эфир.
5.1.4.4. Бис-(2-хлорэтилтио)-алканы
.SCIbCIUCi
(СН2)Д
\SCH2CH2C1
Кожно-нарывное действие бис-2(-хлорэтилтио)-алканов, у которых я = 1—5, сильнее, чем у иприта (см., табл. 16).
По данным Вудворда и сотр. 1,2 бис-(2-хлорэтилтио)-этан (п == 2, сесквииприт) получают взаимодействием смеси 2-меркап-168
Таблица 16. Свойства бис-(2-хлорэтилтио)-алканов (СН2;й(ьсп2ьп2ы/2 Кожно-нарывное действие приведено по сравнению с действием иприта, принятым за 1, в круглых скобках дано давление в мм рт. ст.
п Т. кип., °C Т. пл., °C Кожно-нарывное действие п Т. кип,, °C Г. пл., °C Кожно-нарывное действие
0 150 (30) от —2 до 0 0,01 5 104 (0,003) -6 2
1 80-81 (0,03) 30,7 2 6 112 (0,03) 14-15 0,5
2* 140 (2) 57 5 8 134 (0,045) 21-22 0,01
3 86 (0,04) 11,5 4-5 9 — 24 1—
4 104 (0,06) -1 4 10 —► 32
* Так называемый сесквинприт, Sesqul-H.
Таблица 17. Аналоги иприта
В круглых скобках приведено давление в мм рт. ст.
Соединения T, кип., °C T. ГГЛ., °C
S(CH2C1)2 52-55 (14) —
S(CH2Br)2 82 (11) 1—
S (CH2CH2Br)2 240 (760) 31—34
S(CH2CH21)2 — 62—70
S(CH2CH2F)2 95-96 (30) '—
S(CH2CH2CN)2 — 26
S(CH2CHC1CH3)2 100 (7)
S(CH2CH2CH2C1)2 162 (43)
уСН2СНгС1
105 (12) —
XCH2CHC1CH3
S[CH2CHC1(CH2)3CH3]2 — 52
ZCH2CH2C1
sz 125-132 (13) 24
XCH2CH2Br
ZCH2CH2C1
sz — 125-126
XCH2CH2I
f3c-s-ch2f 1 (760) —-
F3C—S—CF3 —22,2 (760) —
F3C-S-CH2C1 63,5 (740) —
f2ciccf2—s—CH2CH3C1 85 (100) —
C1(C2H4SC2H,O)«C2H4SC2H4C1
n = 1 174 (2) от —30 до —38
n = 2 — 18
n = 3 — 28,5
n = 4 — 32-33
o=5 ‘ 37
S(CH2CH2OCH2CH2CI)2 145(1)
S(CH2CH2SCH2CH2C1)2 —— 73-75
S(CH2CH2SCH2CH2Br)2 — 87,5—88,5
тоэтанола и тиодигликоля (10%) с хлористым водородом при 90 °C. В общем эти соединения можно получать, действуя на соответствующие (й,(о'-дибромалканы 2-меркаптоэтанолом или соответствующим тиОлятом натрия с последующей обработкой бис-(2-оксиэтилтио)-алкана тионилхлоридом:
+2NaSCH2CH20H /SCH2CH2OH
Br(CH2)ttBr ----—-------> (CH2)n;
~Mr \sCH2CH,OH
/SCI I2 СI ДО 11 -]-2Soci2 /ЬСН2СН2С1
(CH2)/ --------> (СН,)Я;
\SCH2CH:OH "2S°£: "hc1 xsch2ch2ci
Физические свойства этих соединений сопоставлены в табл. 16.
Растворимость сесквииприта в воде составляет 0,3 а/л; он сравнительно медленно гидролизуется.
Аналогичное сесквннприту фторсодержащее соединение/ 1,2-бис-(2-фторэтилтио)-этан (CH2)2(SCH2CH2F)2, но данным Саундерса представляет собой жидкость, не обладающую ни кожно-нарывными свойствами, ни физиологической активностью токсических фторорганическнх соединений.
Некоторые свойства аналогов и гомологов иприта приведены в табл. 17.
5.2. ГАЛОГЕНИРОВАННЫЕ ТРЕТИЧНЫЕ АЛИФАТИЧЕСКИЕ АМИНЫ
В 1934—35 гг. Уорд исследовал хлорированные этиламины и среди третичных аминов этого ряда обнаружил новый тип веществ, вызывающих образование волдырей. В результате дальнейших ис-. следований, которые носили преимущественно военную направлен-ность, оказалось возможным использовать некоторые из этих соединений в качестве ОВ. Впервые с военно-теоретической точки зрения трис-(2-хлорэтил)-амин и его гидрохлорид рассматривал ’ Влассо-Поулос. Затем Моллер предложил отнести это соединение к разряду так называемых крапивных ОВ, которое он, пользуясь принятой для ОВ маркировкой разноцветными крестами, предложил обозначить красным крестом. Однако сам Моллер снял свое предложение, поскольку такая маркировка совпадала с символом Международного Красного Креста.
, В результате дальнейших исследований было обнаружено, что некоторые соединения ряда Ы-алкил-Ы.М-бис- (2-хлорэтил)-аминов по своему действию оказались эквивалентными иприту. Они предназначались для заражения местности. Кроме того, выяснилась возможность использования их четвертичных солей для заражения воды и продовольствия. Именно поэтому предметом интенсивных исследований явился сравнительно медленный гидролиз N-алкнл-М,М-бис-(2-хлорэтил)-аминов, который приводит к образованию почти столь же ядовитых продуктов, что и исходные вещества,
17Q
Во время второй мировой войны в Германии ОВ этой группы производили в производственном масштабе и держали наготове в соответствующих средствах применения, в частности, в цистернах машин для заражения местности. Обнаруженные в 1945 г. в Германии запасы трис-(2-хлорэтил)-амииа составляли около 2000 т.
Из-за сходства молекулярной структуры этих соединений с ипритом и аналогичного физиологического действия эти соединения называют азотистыми ипритами.
Хотя с современной точки зрения свойства К-алкил-К,К-бнс-(2-хлорэтил) -аминов не удовлетворяют требованиям, предъявляемым к ОВ, в частности, имеется в виду нх малая летучесть, нестабильность и другие отрицательные факторы, использование их в качестве ОВ це исключено. Это тем более вероятно, что совершенствование технических средств применения ОВ в империалистических армиях открывает широкие возможности их использования (например, в виде аэрозоля), а полная их дегазация некоторыми средствами довольно затруднительна. -Соли этих соединений с различными кислотами исключительно устойчивы при хранении, они пригодны для заражения воды.
Фармакологическое исследование так называемых азотистых ипритов, интенсивно проводившееся после второй мировой войны, имело своей целью, в частности, применение этих веществ для лечения злокачественных опухолей, лейкозов н др. Эти исследования, проводившиеся во всем мире, достигли значительных успехов.
5.2.1. Химическое строение и биологическая активность
5.2.1.1. Связь между химическим строением и биологической активностью. Из галогенированных аминов как 'ОВ имеют значение лишь производные третичных аминов. Для кожно-нарывного действия этих* соединений и поражения клеток других органов (общеядовитое действие) требуется наличие в молекуле* вещества двух 2-галогеналкильных групп. Наиболее эффективны соединения общей формулы R—Ь1(СН2СН2На1)2.
Удлинение углеродной цепи галогеналкильных групп ведет к уменьшению токсичности, которая понижается и в том случае, когда атомы галогена занимают иное положение, чем положение 2.
Длина цепи третьего алкильного остатка, пока речь идет о небольших алкильных группах, ие оказывает большого влияния (см. табл. 18). Оказалось, что токсичность прн накожном применении сильнее всего выражена у трис-(2-хлорэтнл)-амина.
В общем введение арильных групп вместо алкильных приводит К уменьшению токсичности. Если же на место атомов галогена вступают другие заместители, например в результате гидролиза оксн-группы—то токсичность падает; замена всех атоьщв галогена ведет к полной потере токсичности.
Промежуточный продукт гидролиза гидрохлорида Ы-метил-Ы,Ы-бис-2-хлорэтнламина — Ь1-метил-М,М-2-окси-2'-хлордиэтиламин все
171
же имеет ЛД50 (на мышах подкожно) 16 мг!кг и еще обладает кожно-нарывным действием, хотя и в очень ослабленной форме по сравнению с исходным основанием.
В табл. 18 приведены величины ЛД50 некоторых азотистых ипритов.
Таблица 18. Летальные дозы ЛДй0 (в мг!кг массы) некоторых азотистых ипритов
Формула На мышах На крысах
накожно подкожно накожно ПОДКОЖНО внутривенно
CH3-N(CH2CH2C1)2 29 2,6 22 1,9 ы
CsHs-N(CH2CH2C1)2 13 1,2 17 1 0,5
N(CH2CH2C1)3 7 2 4,9 0,7
Наивысшей токсичностью и наибольшей эффективностью в группе галогенированных аминов обладает по-видимому, трис-(2-хлорэтил)-амин.
5.2.1.2. Характер и механизм действия. Высокую общую токсичность Ь1-алкил-Ь1,Ь1-бис-(2-хлорэтил)-аминов и их локальное поражающее действие на кожу, как н в случае бис-2-хлорэтилового тиоэфнра, следует объяснить высокой реакционной способностью этих соединений. Хотя симптомы поражения в случае веществ обеих групп примерно одинаковы, галогенйлкиламнны (азотистые ипрйты)- обладают более сильным общеядовитым действием. Причиной этого является, по-видимому, более интенсивное блокирование ферментативных процессов.
Специфическое действие этих соединений заключается в изменениях ядер клеток, особенно хромосом. Азотистые иприты действуют подобно рентгеновским лучам, которые оказывают на клетки столь же специфическое «радиомнметнческое» действие. Установлено, что разрушение клеток вызывают прежде всего соединения с двумя 2-хлорэтильными группами (независимо от природы третьего остатка).
Хотя механизм биологического действия еще не вполне выяснен, доказано, что эти соединения способны в результате внутримолекулярного алкилирования давать гетероциклические соединения со структурой ионов этнленимония I. То же самое происходит и в случае 2-галогенироваиных тиоэфиров (соединение II):
С1СН2СНгч ХСН2 + хн2
I С1СН2СНг—|
r/ \сн2 \СН
СГ сг
I II
Эти циклические промежуточные соединения вследствие своей высокой полярности, особенно связей N—С или S—С, склонны к разнообразным реакциям с разными реакционноспособными группами.
172
' Это, в частности, проявляется в воздействии на активность раз-; личных ферментов, например на ферменты обмена холина (холин-оксидаза, холинэстераза) и.переноса фосфатов (гексокиназа). По-fe. жалуй, наиболее важное влияние оказывают галогеналкиламины t на нуклеопротеиды, особенно дезоксирибонуклеиновую кислоту, функция которой подавляется.
Так, Секстон усматривает причину этого в способности 2-хлор-этильных групп и атома азота к полимеризации, причем полимеризуется именно ион имония. В образующейся цепи каждая вторая функциональная группа располагается регулярно:
Расстояния между этими группами примерно соответствуют расстояниям между аминокислотами в вытянутой полипептидной цепи либо расстояниям между пуриновыми й пиримидиновыми остатками в нуклеиновых кислотах.
Вторая функциональная группа, имеющая строение нона имония, обладает достаточными возможностями для образования связи с активными участками протеидов или нуклеиновых кислот. По своим размерам образующиеся соединения соответствуют таким нуклеопротеидам, функция которых блокируется. Кроме того, реакционноспособная вторая функциональная группа способствует образованию сшитых макромолекул, что является еще одной причиной токсического действия таких соединений на клеточное ядро.
Следует еще раз сослаться на превосходную монографию Штаде, в которой подробно рассматривается этот круг проблем.
М-Алкил-М,М-бис- (2-хлорэтил) -амины действуют как контактные, ингаляционные и глазные яды. Их общеядовитое действие сильнее, чем у бис-2-хлорэтнлового тиоэфира.
Кожно-нарывное действие проявляется также, как и при действии иприта. В случае жидких галогеналкиламинов оно наступает после скрытого периода в 6—12 ч. Наряду с субъективными ощущениями (жжение и болн на пораженных участках кожи), при определенных обстоятельствах после эритем образуются волдыри. Окраска эритем фиолетовая. Меньшие количества галогеналкиламинов вызывают отёчные опухоли. По сравнению с ипритными, волдыри, получающиеся при действии азотистого иприта, меньше размером и быстрее заживают. Более значительные поражения кожи обусловливают быстрое ухудшение состояния здоровья, потерю в весе и другие явления.
Вряд ли следует ожидать, что пары азотистого иприта вызовут поражения кожи, тогда как при воздействии аэрозолей могут возникать эритемы.
173
Галогеналкиламины в парообразном н особенно в аэрозольном состоянии сильно поражают верхние и нижние дыхательные пути, легкие отравления проявляются в насморке, катарральном рините, воспалениях гортани и трахеи, при тяжелых отравлениях затрагиваются ткани легких.
Легкие поражения глаз сравнительно доброкачественны и быстро излечимы. Вторичные инфекции могут усугубить поражения глаз и привести к потере глаза.
Общеядовитое дейстие проявляется в возбуждении и беспокойстве, в ускорении ритма дыхания и пульса. Клонические и тонические сц^змы (симптомы поражения центральной нервной системы) ведут к явлениям паралича и нарушениям сознания. При таком поражении смерть наступает примерно через 5 ч.
Как и в случае иприта, попадание азотистого иприта через рот вызывает тяжелые воспаления в пищеводе и в желудочно-кишечном тракте. Первыми признаками такого отравления являются боли в желудке, тошнота и рвота.
5.2.2. Общие свойства
Галогенированные третичные амины в виде оснований представляют собой трудно растворимые в воде маслянистые жидкости. Сразу после перегонки они бесцветны и почти лишены запаха. -В органических растворителях они растворяются хорошо.
Их химическое поведение определяется присутствием аминного атома азота н находящимися в р-положенин к нему атомами галогенов.
Из-за наличия у атома азота неподеленной пары электронов эти соединения проявляют координационно-ненасыщенный характер. Они обладают щелочной реакцией, образуют четвертичные аммониевые основания н соли;
уСНгСН2С1 +НС1 Rv + /СН2СН2С1
R—N >- N *
\сн2сн2с1 н/ \сн2сн2С1
СГ
Эти солн столь же токсичны, как и исходные основания. Оии хорошо раствЬримы в воде и могут применяться для заражения воды. Для идентификации галогенированных аминов служат их солн с пикриновой, фосфорномолнбденовой, вольфрамовой кислотами, так как они представляют собой хорошо кристаллизующиеся вещества, обладающие четкой температурой плавления.
В отличие от атома серы в иприте, атом азота азотистых ипритов проявляет большую электроотрицательность. В результате этого реакции с электрофильными реагентами проходят медленнее. С третичными галогеналкнламинами способны реагировать лишь сильные окислители, при этом образуются окиси аминов. С солями
174
тяжелых металлов они,, образуют комплексные соединения, которые в ряде случаев можно идентифицировать, как например комплексы с хлористым золотом и калийвисмутнодндом.
Атомы галогена можно заместить. Оии столь же подвижны, как н у бис-2-хлорэтилового тноэфнра. Во время гидролиза онн могут отщепляться с образованием четвертичных этилеиимониевых производных. При взаимодействии с амниопроизводными, фенолятами и другими реагентами происходят процессы обмена.
Деалкилирование третичных галогеналкиламинов возможно при действии сильных окислителей и хлорирующих агентов.
Реакционная способность третичных галогенированных аминов убывает с увеличением размера алкильных групп. Такие соединения также менее активны и в физиологическом отношении и не имеют значения как ОВ,
5.2.3. Трис-(2-хлорэтил)-амин
уСН2СН2С1
C1CH2CH*—N^
\ch2ch2ci
Синонимы: 2,2',2"-трихлорэтиламин, р^'^-трихлорэтиламин, азотистый иприт. ч
Военные обозначения: нем. — Stickstoffyperit, Stickstofflost, Stick-stoffsenfgas, C6-Base, C6-Salz; англ, — Nitrogen Mustard, ТВА; амер. — Nitrogen Mustard, HN-3.
Трис-(2-хлорэтил)-амии в военных целях еще. не применялся. у В 1945 г. на немецких складах и на заводах были найдены большие запасы этого ОВ.
. Физиологическое действие этого соединения подробно описал , Уорд; на метод его получения был еще раньше39 выдан патент в США. Американский патент на получение гидрохлорида этого соединения предусматривал его применение для изготовления лечебного пластыря. Последующие исследования были посвящены разработке методов получения и изучению химических свойств. Во время войны его производила в основном немецкая фирма «Оргацит» (Аммеидорф). Он предназначался для заражения местности.
5.2.3.1. Методы получения. Азотистый иприт получают хлорнро-} ваннем триэтаноламина тноннл- или сульфурнлхлоридом, трех- или пятнхлорнстым фосфором, хлорокисью фосфора или однохлористон •серой.
'Эти процессы можно осуществить как без, так н с применением .растворителя. В качестве растворителей кроме бензина применяют - хлорированные алканы — хлороформ и четыреххлористый углерод. '
Хлорирование тионнлхлорндом происходит по уравнению:
N(CH3CH3OH)3 + 3SOC13 —> N(CH2CH2C1)3 -h 3SO2 + 3HC1
175
Вследствие неустойчивости самого амина обычно получают его гидрохлорид путем хлорирования гидрохлорида триэтаноламина:
HN(CH2CH2OH)3 cr + 3SOCl2 —> HN(CH2CH2C1)3 Cr+3SO2 + 3HC1
Триэтаноламин изготовляют в промышленности взаимодействием окиси этилена с аммиаком; действуя на него избытком хлористого водорода превращают его в гидрохлорид:
NH3 + 3H2C—СН2 —> N(CH2CH2OH)3
N(CH2CH2OH)3 + HC1 —> HN(CH2CH2OH)3 Cl'
Чистый трис-(2-хлорэтил)-амин (основание) получают затем действием эквимольных количеств гидроокиси или карбоната щелочного металла иа гидрохлорид с последующей перегонкой при пониженном давлении.
В лабораторном методе Мэзона и Геша40 используется предложенный Уордом 41 способ хлорирования триэтаноламина тионилхлоридом.
В 150 ма бензола растворяют 2 моль (238 г) тионилхлорида и раствор вливают в круглодонную колбу емкостью 1 л, снабженную обратным холодильником. После того, как к раствору прибавлено 0,5 моль (92,7 а) триэтаноламина, смесь нагревают 3 ч при 55 °C. Избыток тионилхлорида и бензол отгоняют при 50 °C и пониженном давлении. Для разложения остатков тионилхлорида достаточно добавить около 50 мл этанола и нагревать раствор 10—15 мин. Этанол отгоняют, а остаток перекристаллизовывают из кипящего ацетона. Выход гидрохлорида трис-(2-хлорэтил)-амина составляет 88—92%. Действием гидроокиси или бикарбоната натрия получают свободное основание в виде маслянистой жидкости; ее отгоняют при 143—144°C (15 мм рт. ст.).
По методу Мак-Комби и Парди 42 растворяют 100 г чистого триэтаноламина в 150 мл сухого хлороформа и в круглодонной колбе емкостью’ 1 л с обратным холодильником медленно смешивают с раствором 270 г тионилхлорида в 150 мл хлороформа (охлаждение!). Реакционную смесь кипятят 4—5 ч с обратным холодильником на водяной бане. Реакция заканчивается, когда хлористый водород больше не выделяется.
Выпадающую после охлаждения соль отфильтровывают или отсасывают и промывают хлороформом. Выход кристаллического гидрохлорида трис-(2-хлорэтил)-амина составляет 140—150 а.
Свободное основание можно выделить растворением гидрохлорида в воде (концентрированный раствор) и обработкой эквимольным количеством 30%-ного раствора N a ОН с последующей перегонкой отделяющейся маслянистой жидкости при пониженном,давлении; т. кип. 1-37—138 °C (15 рт. ст.).
Наивысшей степени чистоты трис-(2-хлорэтил)-амин получают многократной перекристаллизацией гидрохлорида из спирта и перегонкой амина в более высоком вакууме, например при 103—105 °C и 3 мм рт. ст.
Технический продукт, который обычно имеет темно-коричневую окраску, как правило, содержит 95—97% трис-(2-хлорэтил)-амина.
5.2.3.2. Физические свойства. Азотистый иприт —бесцветная и лишенная запаха маслянистая жидкость. Свежеперегнанный продукт через 3—4 суток приобретает окраску от желтой до коричневой. Желто-коричневый технический продукт очень неустойчив, особенно при высоких температурах.
176
Приводим основные физические показатели этого вещества:
Т. кип., °C при 760 мм рт. ст............................. 230—235 (разл.)
при 722 мм рт. ст......................... 219 (разл.)
при 20 мм рт. ст.......................... 130
при 15 мм рт. ст.......................... 137—138 (143—144)
при 10 мм рт. ст.......................... 124—126
при 1 мм рт. ст........................... 94
Т. пл., °C............................: . . . . —4
Давление пара при 20 °C, мм рт. ст............ 0,007
Максимальная концентрация, мг)л при 20 °C..................................... 0,07
при 22 °C................................. 0,094
Относит, плотность пара.......................................... 7,0
чистого вещества d™........................... 1,2348
техн, продукта d™............................. 1,24
Прн нормальном давлении азотистый иприт нельзя перегнать без разложения; по экстраполяции его т. кип. 230—235 °C, при которой он полностью разлагается. Соединение застывает между —4 и —2 °C.
Низкое давление пара азотистого иприта указывает на чрезвычайно малую летучесть этого ОВ. В интервале 0—60°C давление пара можно вычислить по следующей формуле (в мм рт. ст.) 43:
1 ППМ1 3393’4
1g р = 9,41621 — —р—
Эта формула дает лишь приближенные величины, так как получить это ОВ в чистом виде очень сложно — оно очень быстро претерпевает превращения.
Приводим давление пара азотистого иприта в интервале 0— 30 °C, вычисленное по приведенной формуле:
Температура, °C Давление пара, ММ рт. ст. Температура, Давление пара, мм рт. ст.
0 0,0009 20 0,0069
5 0,0016 25 0,0106
10 0,0026 30 0,0164
15 0,0042
Вследствие малой летучести этого вещества (при 20°С она составляет 0,04 мг/л) опасные концентрации в воздухе не возникают. Эффективное применение этого ОВ для заражения воздуха возможно только в виде аэрозоля. Аэрозоль азотистого иприта получить можно.
Растворимость этого ОВ в воде меньше, чем растворимость иприта. Она составляет при 20°C по одним данным 0,16 г/л, а по другим — 0,5 г/л.
В органических растворителях — галогенированных углеводородах, бензоле, хлорбензоле, сероуглероде, эфире, ацетоне, метаноле— трис-(2-хлорэтил)-амин растворяется очень хорошо; в
177
этаноле он растворим ограниченно; растворим в растительных н животных маслах и жирах. Растворы ОВ в полярных растворителях неустойчивы. Под действием тепла и света они темнеют вследствие димеризации амина, тогда как неполярные растворители оказывают стабилизующее влияние. С такими ОВ, как иприт и хлорпикрин, азотистый иприт смешивается во всех отношениях.
Способность трис-(2-хлорэтил)-амина к диффузии выражена не столь сильно, как у иприта. В кожу, резину и подобные материалы азотистый иприт проникает медленнее иприта, но так же быстро впитывается в древесину, текстильные ткани, картой и др.
5.2.3.3. Химические свойства. При действии света и при нагревании трис-(2-хлорэтил)-амин неустойчив. Уже при температуре выше 50°C происходит заметная димеризация амина, выше 100°C он разлагается, вблизи температуры кипения термическое разложение полное.
По отношению к металлам азотистый иприт индифферентен. Сталь практически не затрагивается даже прн повышенных температурах.
Димеризация. Даже многократно очищенный трнс-(2-хлорэтнл)-амин со временем окрашивается при хранении в теплоизолированных стеклянных сосудах. Через месяц выпадают кристаллы. Скорость образования кристаллов возрастает с температурой. Полярные растворители, особенно содержащие оксигруппу, также ускоряют этот процесс.
Продуктом димеризации являетси двухлорнстый N,N,N',N'-тетракис- (2-хлорэтил) -пиперазоний следующей Структуры:
'С1СН2СН2ч + ,СН2СН2Ч + ,СН2СН2СГ
X X
.ClCHjjCH/ ^СНгСН/ \СН2СН2С1_
2СГ
Если трис-(2-хлорэтил)-амин кипятить несколько часов с концентрированной муравьиной кислотой, получается димер с т. пл. 331 °C. При плавлении хлорид пиперазония разлагается.
В метанольных растворах главным процессом является не димеризация, а скорее замещение с образованием хлористого Ь1,№-бнс-(2-хлорэтил)-Ь1-(2'-метокснэтил)-амииа (т. пл. 133—134 °C):
+сн3он +
N(CH2CH2C1)3 ------> 1(C1CH2CH2)2NHCH2CH2OCH3] cr
При хранении в обычных условиях из метанольных растворов был выделен также двухлористый N,N'-6hc- (2-хлорэтил).-N,N'-6hc-(2'-метоксиэтнл)-пиперазоний (т. пл. 258°С)
С1СН2СН2. + уСН2СН2ч /СН2СНгОСН3 /Л
СН3ОСНгСН/ \СНгСН/ \СНгСН?С1
2С1"
?.0i
О 50 100 150 200 250
Врепя3.ч
Рис. 6. Отщепление хлор-иопов при гидролизе трис-(2-хлорэтил)-амина.
Трис-(2-хлорэтил)-амин более склонен к реакциям замещения, чем к димеризации, что приписывают малой основности этого соединения, следствием которой является пониженная способность атома азота к комплексообразованию. Соединения, аналогичные азотистому иприту, например Ь1-метил-Ь1,Ь1-бис-(2-хлорэтил)-амин, не проявляют склонности к замещению.
Образование двухлористого Ь1,Ь1'-бис-(2-хлорэтил) -N,N'-6hc-(2'-метоксиэтил) -пиперазония объясняется повышенной основностью метоксизамещенного продукта.
Димеризация не Идет в неполярных растворителях — бензоле, диоксане и даже в четыреххлорн-стом углероде. Добавки тиомочевины препятствуют димеризации.
Гидролиз. Водой трнс- (2-хлор-этил)-амин лишь медленно гидролизуется. Как показывают многочисленные исследования, гидролиз азотистого иприта происходит медленнее, чем гидролиз иприта.
Из 1 °/о-ного водного раствора азотистого иприта примерно через
20 ч на первой стадии гидролиза выделяется 1 экв хлористого водорода. Следующие стадии гидролиза происходят медленнее, что выражается в меньшем отщеплении соляной кислоты; через 24 ч при 25 °C отщепляется около полутора эквивалентов соляной кислоты.
На рис. 6 показана кинетика гидролиза этого вещества по данным потенциометрического титрования нитратом серебра отщепляющихся хлор-ионов44.
Гидролиз идет в три стадии:
+ Н20(1) + +Н20(2)
N(CH2CH2C1)3 -------> [(C1CH2CH2)2NHCH2CH2OH] сг _НС1>
+ +Н2о (3) +
—> [C1CH2CH2NH(CH2CH2OH)2] СГ -----—[HN(CH2CH2OH)3] сг
—-^НС1
За счет выделяющейся соляной кислоты образуются соответствующие гидрохлориды. Из продуктов гидролиза через 20 ч был выделен гидрохлорид N,N-6hc-(2-хлорэтил)-Ь1-(2'-оксиэтил)-амина (стадия 1); через 72 ч — преимущественно гидрохлорид N-2-хлор-этил-Ь1,Ь1-бис-(2'-оксиэтил)-амина (стадия 2) и, в небольшом количестве гидрохлорид триэтаноламина (стадия 3). Кроме того, был обнаружен в количестве около 4°/о продукт димеризации — двухлористый Ы,Ы,Ы',Ы'-тетракИС-(2-хлорэтил)-пиперазоний. Значительная часть непрореагировавшего исходного основания находилась в водном растворе в виде гидрохлорида.
179
В процессе гидролиза образуются четвертичные производные азота45, в основном производные этилеиимония типа
Г 1Г + >СН2 1 I _R/ ХСН3_
СГ
Таким образом, механизм гидролиза в целом довольно’ сложен. Поскольку по такому механизму идет гидролиз всех N-алкил-N,N-6hc-(2-хлорэтил)-аминов с короткой цепью, полезно рассмотреть его более подробно.
Стадия 1. Быстрая перегруппировка М-алкил-М,М-бис-(2-хлорэтил)-амина в хлористый Ь1-алкил-Ь1-(2-хлорэтил)-Ь1-этиленнмоний:
ХНгСНгС!
R-N(
ХСН2СН2С1
сг
Стадия 2. В.ходе дальнейшего гидролиза из хлористого этилеиимония за счет выделяющейся соляной кислоты образуется некоторое количество гидрохлорида исходного основания:
Г./М
Ж1
C1CH2CH2Z ХСН2 _
••-НС1
С Г
’ R4+/CH2CH2C1' /N\
_ Hz ХСН2СН2С1
сг
Однако основным продуктом является гидрохлорид N-алкил-N- (2-хлорэтил) -N- (2'-окснэтил) - амина:
R\ + /СН21
С1СН2СН/ ХСН2 _
сг
+н2о
" Ич + уСН2СН2ОН‘ XNX
_ н/ ХСН2СН2С1 _
сг
Стадия 3. Дальнейшее замещение атомов хлора хлорэтильных групп на гидроксильные группы происходит с промежуточным образованием других производных этилеиимония:
’ + /Сн2сн2°н '
xn;
н/ \СН2СН2С1
+н2о
-HCI
н2о
"R4 +/СН2СН2ОН‘
XNX
ХСН2СН2ОН^
Одной из дальнейших стадий является образование димера, который может получиться как из 2 моль исходного соединения, так и из 2 моль хлористого этилеиимония:
2R—N(CH2CH2C1)2 — р-
+ уСН2 '
2 XN\ |
_С1Сн2сн2/ \сн2_
СГ
’С1СН2СН2ч + уСН2СН2х ГНгСНгСП z\ Xn<
R/ ^СНаСНг^ \R
2СГ
180
Другим путем образования димера является взаимодействие 1 моль исходного основания с 1 моль этиленимониевого комплекса:
СН2Х /СН2СН2С11
r-n(ch2ch2ci)2 + | ^n:
сг
Rx zCH2CH2x L ZCH2CH2C11 X + У х + У
X X
2СГ
Гидролиз М-алкил-М,М-бис-(2-хлорэтил)-аминов согласно Сартори 46 приведен на схеме 3.
rx+/ch2ch2x +
•СН2СН2С1
2СГ
или 4-II
ХН2СН2С1 r—n;
ХСН2СН2С1
R-
•сн2
_C1CH2CH/Z \сн2^
II
СГ
FRv л:
СН2СН2С1"
сг
ш
4*
V
Rx + /CH2CH2OH-;n;
рх+ л
iv
•СН2СН2ОН сг
v VI
Схема 3. Гидролиз Ы-алкил-Ь!, Ь!-бис-(2-хлорэтил)-амина по Сартори4®.
Гидролиз в водном растворе бикарбоната натрия (pH 8) идет с образованием триэтаноламина. За 24 ч в реакцию вступает около 90—95% исходного основании. Первый эквивалент иона хлора освобождается сравнительно быстро (за 15 мин). Как удалось установить Голамбику и сотр., гидролиз на первой стадии сопровождается промежуточным Ъбразованием производного этиленимония. По мнению Контарди и сотр.47, при этом образуется скорее всего не триэтаноламин, а (2-оксиэтил)-морфолин
хсн2сн2х
OZ ^N—СН2СН2ОН
^СН2СН2/ у
С ростом pH раствора гидролиз ускоряется, но становится количественным лишь при температуре выше 80°C.
181
Хлориды этиленимония в небольших количествах можно выделить в качестве продуктов гидролиза трис-(2-хлорэтил)-амина в водио-ацетоновых растворах. Они ядовиты. Только продукт последней стадии гидролиза не ивляется ядовитым веществом.
В кислой среде образуется трис-(2-хлорэтил)-аммониевая соль соответствующей кислоты.
Отношение к кислотам. Склонность трис-(2-хлорэтил)-амина как третичного амина образовывать с кислотами комплексные соединения положительно сказывается на некоторых реакциях, в которые свободное основание не вступает, или реагирует лишь медленно. Растворимые в воде комплексные соединения обычно взаимодействуют легче, чем основания.
При действии концентрированной НС1 на азотистый иприт образуется белый кристаллический гидрохлорид трис-(2-хлорэтил)-+
амина (HN(CH2CH2Cl)3jCr. Это — лишенные запаха ромбические пластинки, т. пл. 131 °C, легко растворимые в воде, в разбавленной НС1 и в ацетоне, менее растворимые в. метаноле, галогеналканах, петролейном эфире, бензине и бензоле, ие растворимые в эфире и гексане.
При взаимодействии гидрохлорида с эквивалентными количествами щелочей регенерируется исходное основание, которое, например из раствора соды выделяется в виде маслянистых капель.
Гидрохлорид трис-(2-хлорэтил)-амина —стабильное соединение, которое можно хранить годами. Он обладает такой же токсичностью, что и свободный амнн, и его можно эффективно применять для заражения продовольствия и систем водоснабжения.
С другими минеральными кислотами амин образует такие же солеобразные комплексы, которые благодаря четкой температуре плавления могут быть использованы для его идентификации.
Для этой цели наряду с другими кислотами используют пикриновую Кислоту, при добавлении которой из растворов трис-(2-хлорэтил)-амина в органических растворителях, в воде или лучше из солянокислых растворов осаждается желтый пикрат (C1CH2CH2)3N-C6H3N3O7, т. пл. 137 °C.
С хлорной кислотой образуется землисто-серый кристаллический продукт присоединения (т. пл. 145,5°C), который при нагревании в запаянных капиллярах разлагается со взрывом. Кроме того, продукты присоединения образуются при действии фосфорновольфрамовой, фосфорносурьмяиой, фосфорномолибденовой, пик-ролоновой кислот и др.
С кислыми солями сильных кислот, например с бисульфатом натрия, идут аналогичные реакции.
Превращение азотистого иприта в его водорастворимые соли при действии разбавленных растворов минеральных кислот или кислых солей можно использовать для дегазации крупной боевой техники, улиц, лабораторных установок и даже защитного обмундирования. При этом следует помнить о корродирующем действии
182
таких растворов, а также заботиться об удалении чрезвычайно токсичных водных растворов солей азотистого иприта. Последующее промывание дегазируемых объектов большим количеством воды требуется во всех случаях.
Для дегазации материалов достаточно 5%-ного рдствора соляной кислоты илн бисульфата натрия, тогда как для дегазации местности или улиц считается достаточным расход 500 г NaHSC>4 на 1 л/2. Обильную поливку водой следует производить примерно через 30 мин.
Окисление. Азотистый иприт чрезвычайно устойчив по отношению к окислителям. Обычные дегазирующие средства—хлорная известь и монохлорамин — в водных растворах при нормальной температуре лишь слабо или совсем не реагируют с азотистым ипритом. Наряду с неядовитыми продуктами, частично образующимися при хлорировании, в реакционной смеси остается большое количество непрореагировавшего амина.
В результате медленной реакции с солянокислым раствором перманганата калия отщепляется хлорэтильная группа и образуется неядовитый бнс-(2-хлорэтил)-амин или его гидрохлорид H2N(CH2CH2C1)2C1-, т. пл. 213 °C.
Дымящая азотная кислота при обычной температуре быстро реагирует с азотистым ипритом с образованием подробно не идентифицированных продуктов. Аналогично реагирует и хромовая • смесь. Обе кислоты пригодны для дегазации лабораторной посуды.
Окись трис-(2-хлорэтил)-амииа наряду с другими продуктами получается по реакции азотистого иприта с перекисью водорода при нагревании, или при озонировании:
о
N(CH2CH2C1)3 —> (C1CH2CHJ3N->O
На холоду перекись водорода с амином не реагирует.
Легче и быстрее окись амина образуется при взаимодействии амина со слабо щелочными растворами органических перкислот, например, надуксусной, надбензойиой, мононадфталевой48. Гидрохлорид окиси трис- (2-хлорэтил)-амина имеет т. пл. 91—92 °C. Окись амина ядовита, поэтому окисление, приводящее к ее образованию, не имеет значения для дегазации. Окисление надкислотой в кислой среде проходит медленнее.
Окиси азотистых ипритов применяют для терапии рака.
Хлорирование**. При хлорировании трис-(2-хлорэтил)-амина протекают сложные реакции, которые приводят к образованию многочисленных продуктов.
В отлнчне от иприта, чтобы ввести третичное азотистое основание полностью в реакцию, нужно применять избыточное количество хлорирующего агента. Самой медленной стадией этой многостадийной реакции, по-видимому, является первая.
Уже на первой стадии третичный амин деалкилируется. Наряду С другими продуктами разложения образуются соответствующие
183
вторич-ные амины. Выделяющийся при деалкилировании хлористый водород, если его сразу не нейтрализовать, связывается с частью исходного амина, давая гидрохлорид. Гидрохлориды устойчивы к хлорированию.
Хлорирование в щелочной среде ведет почти к полному разложению азотистого иприта. Важной для дегазации является реакция с ионами гипохлорита в щелочной среде; последние способны реагировать и с основанием, и с возможно образовавшимся гидрохлоридом.
Хлорирование трис-(2-хлорэтил)-амина в небуферных растворах ведет к образованию гидрохлорида исходного амина, который при действии избытка хлора может превращаться в хлорацетальдегид и бис-(2-хлорэтил)-амин. Кроме того, образуются трихлор-ацетальдегид, бис-(2-хлорэтил)-амин и глиоксаль.
В бикарбонатном буферном 1 растворе гипохлорита (pH 8) трис-(2-хлорэтил)-амин или, соответственно, его гидрохлорид, разлагаются на бис-(2-хлорэтил)-хлорамин и хлорацетальдегид:
сю“ хО
N(CH2CH2C1)3 ---> C1N(CH2CH2C1)2+ С1СН2СГ
\н
При обработке хлористым водородом бис- (2-хлорэтйл) -хлорамин с отщеплением хлора превращается в гидрохлорид бис-(2-хлорэтил)-амина:
-B2HCI +
СШ(СН2СН2С1)2 ----> [H2N(CH2CH2C1)2]C1“
— Cl2
В действительности процессы хлорирования гораздо более сложны н вряд ли выяснены во всех деталях. Так, например, в. реакции трис-(2-хлорэтил)-амина с М-хлор-2,4,6-трихлорбензанилидом наряду с бис- (2-хлорэтил)-амином образуется глиоксаль, 3 экв N-хлорамида полностью разлагают исходное основание. Одновременно с хлорированием происходит атака 2-хлорэтильной группы в положения 1 и 2, которая ведет к деалкилированию или к полному распаду молекулы. При действии иа трис-(2-хлорэтил)-амин раствора гипохлорита кальция образуется глиоксаль, (2-хлорэтил)-дихлорамин и хлорат кальция.
При обработке третичных галогенированных аминов сухим гипохлоритом происходят вспышки. Таким способом можно сравнительно быстро и надежно дегазировать азотистый иприт. Реакция с хлорной известью протекает более спокойно и не приводит к полному превращению ОВ в неядовитые продукты. Для более эффективной дегазации хлорно.й известью обычно рекомендуется добавлять кислые соли сильных кислот, например, бисульфат'натрия, или даже разбавленные минеральные кислоты. За счет образующегося хлора in statu nascendi можно достигнуть 95—99%-ной дегазации. Дегазирующие смеси, которые готовят непосредственно
184
перед использованием или прямо на дегазируемой поверхности, должны состоять примерно из 1 части хлорной извести и 3 частей бисульфата натрия.
Сульфурилхлорид энергично реагирует с азотистым ипритом с образованием смеси гидрохлоридов исходного основания и бис-(2-хлорэтил)-амина. В этом случае также не происходит полная дегазация.
Монохлорамины даже в щелочной среде при обычной температуре реагируют медленно и неполностью с трис-(2-хлорэтил)-амином и другими третичными галогенированными аминами. Хлорамины с высоким содержанием активного хлора в соответствующих органических растворителях реагируют полностью, причем образуются неядовитые продукты.
Реакции с алокоголятами, фенолятами и алкилмеркаптидами. Атомы хлора трис-(2-хлорэтил)-амина в соответствующих условиях можно легко заместить алктиогруппами.
При действии алкилмеркаптида и нагревании через сравнительно короткое время образуется соответствующее тиоэфирное производное:
+3RSNa '
N(CH2CH2C1)3 _> N(CH2CH2SR)3 “иГч а V- L
Как уже упоминалось, азотистый иприт в метанольном растворе при обычной температуре образует N.N-бис-(2-хлорэтил)-N-(2'-метоксиэтил)-амин. В спиртовых растворах щелочей, особенно при нагревании, происходит замещение всех трех атомов хлора:
+3ROH; +3NaOH
N(CH2CH2C1)3 —„МяС. N(CH2£H2OR)3
“□lyavi*,
. Образующийся трис-(2-метоксиэтил)-амии представляет собой неядовитое, не растворимое в воде масло.
Аналогично реагируют феноляты, карбаматы, тиокарбаматы и подобные соединения.
Реакции с аминами и аммиаком46’50. Даже в Присутствии большого избытка аминов азотистый иприт вступает в реакцию только с 2 моль первичного амина.
При кипячении с анилином образуется 1-(2-фениламиноэтил)-4-фенилпнперазин (т. пл. 60°С):
ZCH2CH2X 2c6h5nh2 + n(ch2ch2ci)3 —> c6h5nhch2ch2—n; ;n—c6hs
-HC1 \ch2chZ
При нагревании до 100°C азотистого иприта или его гидрохлорида с аммиаком и добавкой этанола образуется 1-(2-амиио-этил)-пиперазин (желтое масло, т. кип. 200°C), имеющий следующую формулу;
/СН2СН2\
h2nch3ch2—tZ ;nh
\ch2chZ
185
Аналогичные соединения наряду с Другими образуются с пиперидином, этнлендиамином, анилидами и аминокислотами.
С триметилентетрамином, гексаметилентетрамином и другими метилентетраминами в водно-спиртовых растворах при обычной температуре образуется множество продуктов коиденсации, которые, пока более точно не охарактеризованы. Из гексаметилентетраминовых производных некоторых ХГ-алкил-ХГ,М-бнс- (2-хлорэтил) -аминов.были выделены соединения следующего строения:
СН2 С1'
N ^n-ch2ch2-n-ch2ch2ci
Н2СЧ/СН2 К
N
Н2С СН2 сн2
Реакции с сульфидом натрия. Прн действии на трис-(2-хлор-этил)-амин спиртовых растворов сульфида натрия отщепляются атомы хлора и образуются тиоэфирные мостики, в результате чего получается неядовитое соединение следующего типа:
/СН2СН3. s;
СН2СН2Х
,s
Спиртовые растворы сульфида натрия наиболее пригодны для дегазации азотистых ипритов.
Образование комплексных соединений. Подобно негалогениро-ванным аминам или алкиламмониевым производным трис-(2-хлорэтил)-амин реагирует с многими солями н комплексными соединениями металлов с образованием двойных соединений, которые, отчасти из-за их характерной окраски, можно использовать для идентификации ОВ.
С хлорным золотом гидрохлорид трис-(2-хлорэтил)-амина или свободное основание в растворе четыреххлористого углерода дает желто-ораижевый продукт присоединения [т. пл. 165°С (разл.)]:
HN(CH2CH2C1)3 + AuC13 —> [НМ(СН2СН2С1)з]АиС1;
С Г
С хлориой платиной образуется аналогичное комплексное соединение.
Хлористый иод осаждает из раствора амина в четыреххлористом углероде желтый, легко разлагающийся кристаллический продукт [т. пл. 82°C (разл.)] следующего строения:
[HN(CH2CH2C1)3] [1С12]_
Из других солей, реагирующих с азотистым ипритом, можно привести, например, пятихлористую сурьму, калийвисмутиодид,
186
и-одмеркураты калия и кадмия, ураиилацетат, реактив Несслера, соль Рейиеке и платинациаиат калия.
При действии ацетатов и тиосульфатов замещаются атомы хлора азотистого иприта:
+ЗСН3СОО"
N(CH2CH2C1)3 --------> N(CH2CH2OCOCH3)3
-ЗС1
+ NaS2Oj
N(CH2CH2C1)3 -----N(CH2CH:O3S2Na)3
Трис-(2-ацетоксиэтил)-амин почти неядовит. Ои представляет собой маслянистую жидкость. Продукт реакции с тиосульфатом натрия кристаллизуется из 80%-ного этанола в виде соединения CfrHiaN - Na3O9S6 • 4,5Н2О. Пользуясь тиосульфатом натрия можно определять содержание трис- (2-хлорэтил)-амина в водных растворах.
5.2.3.4. Токсические свойства. Трис-(2-хлорэтил)-амии оказывает такое же действие, как иприт. Он является контактным и ингаляционным ядом, а также поражает глаза. Особого внимания заслуживает возможность применения аммониевых солен, в частности гидрохлорида трис-(2-хлорэтил)-амииа, для заражения воды и продуктов питания.
Поражения кожи, вызываемые соприкосновением с азотистым ипритом, похожи на те, которые причиняет иприт. Растворимость трис-(2-хлорэтил)-амина в липидах меньше, поэтому проникающие дозы меньше по сравнению с дозами иприта.
Биологические эксперименты на предплечьи человека, проведенные в воздухе; насыщенном азотистым ипритом или ипритом, показали (ср. раздел 5.1.3.4), что при одинаковых условиях скорость проникновения азотистого иприта меньше, тогда как аналогичный \;-этил-Лт,\т-бис- (2-хлорэтил)-амин диффундирует быстрее, чем иприт35. Так, при 21—23°C и относительной влажности воздуха 46% скорость проникновения достигала следующих величии (в мг)см2 • мин):
М-Этил-М,М-бис-(2-хлорэтил)-амин..................... 2,8- 10-3
Бис-2-хлорэтиловый тиоэфир .......................... 1,4* 10~3
Трис-(2-хлорэтил)-амин.............................. 10,8* 10-3
При более высоких температурах соотношения примерно такие же.
Эритемы образуются уже при концентрациях 10“3—5-10-3 мг/см2 поверхности кожи. Более высокие дозы (выше 0,1 Jwa/сл*2) ведут к сильным поражениям кожи и образованию волдырей.
Появление тяжелых поражений кожи при действии па-ров азотистого иприта в боевой обстановке невероятно, так как для этого требуются слишком высокие концентрации. Некоторые поражения, такие как покраснения кожи и «доброкачественные» эритемы, появляются при концентрациях 0,4—0,6 мг[л и экспозиции 15—20 мин.
187
Образование волдырей исключено, оно наступает только при непосредственном контакте с жидким ОВ. Чрезвычайно чувствительны половые органы.
Тяжелые поражения кожи причиняет также гидрохлорид трнс-(2-хлорэтил)-амнна. Сильным действием обладают 10%-ные водные растворы, но даже 3%-ные растворы ведут к серьезным поражениям.
Воздействие через органы дыхания следует ожидать именно при применении ОВ в виде аэрозоля. Патологический эффект соответствует действию иприта, но проявляется сильнее.
Пребывание в течение 15—20 мин в атмосфере с концентрацией примерно 2 • 10-3 жа/л ОВ ведет к потере боеспособности, а концентрации порядком выше очень опасны и прн длительном воздействии приводят к отравлениям, опасным для жизни. Концентрации 0,25—1,0 л/г/л через короткое время (5 мин) действуют смертоносно.
Поражения глаз появляются уже .при низких концентрациях. При экспозиции 15 мин уже 7 • Ю"4 мг/л приводит к легким поражениям. Для такого же эффекта в случае иприта потребонйгянсь бы в 10 раз более высокие концентрации.
Смертельная кожно-резорбтивная доза для людей составляет 10—20 мг/кг массы.
5.2.4. Другие №алкил-М,М-бис-(2-хлорэтил)-амииы
Уже во время второй мировой войны в качестве других возможных • ОВ из группы галогеналкиламинов особо обсуждались М-метил-(1) н М-этнл-М,М-бис- (2-хлорэтнл) -амин (II).
/СН2СНгС1 /СЩСНзС!
СНз—C2HS— ХСН2СН2С1 ^СН2СНаС!
I п
Военные обозначения ОВ:
англ. — MBA; англ. — ЕВА;
амер. — HN-2. амер.— HN-1.
Значение, которое эти и подобные соединения приобрели в ме- • дицнне, побудило химиков и медиков синтезировать и исследовать большое число так называемых азотистых ипритов. Известно около 500 таких соединений, которые, однако, никоим образом не пригодны в качестве ОВ. Физические свойства наиболее важных соединений этого ряда, специально изучавшихся как ОВ 4б, приведены в табл. 19.
В химическом отношении N-метнл- н М-этнл-М,М-бнс-(2-хлор-этнл)-амнны ведут себя подобно трис-(2-хлорэтил)-амину. Они подвергаются димеризации и гидролизу, аналогичным образом хлорируются. Метильное производное более реакционноспособно, чем
188
Таблица 19. Физические свойства М-алкил-51,М-2,2'-днгалогендиалкиламинов RNRR"
В круглых скобках приведено давление в мм рт. ст.
Заместители Т. кип,, °C Т. пл., ”С Относ, плотность d4 t nD
R R' R’ основания гидрохлорида
сн3 СН2СН2С1 СН2СН2С1 71(9) от —65 до —60 но 1,1300го —
с2н5 СН2СН2С1 СН2СН2С1 85,5(12) -34,4 141 1,086123 1.465325
н-С3Н7 СН2СН2С1 СН2СН2С1 ( 96(10) — — 1.059323 1.462927
изо-С3Н7 СН2СН2С1 СН2СН2С1 90 (8) — 216 — —
СН2СН2С1 СН2СН2С1 ' СН2СН2С1 94(1) от —4 до —2 131 1.209325 1,4957го
СН2СН2С1 СН2СНС1СН3 СН2СНС1СН3 117 (3) — — — —
СН3 ch2ch2f ch2ch2f 123—124 (762) — — — —
ch2ch2f СН2СН2С1 СН2СН2С1 115 (13) — — — —
ch2ch2f ch2ch2f ch2ch2f 73-74 (25) — — — —
CH2CH2Br СН2СН2Вг СН2СН2Вг 145 (0,05) — — — —
сн=сн—сн3 СН2СН2С1 СН2СН2С1 80 (3) — — — —
трис-(2-хлорэтил)-амин, это прежде всего проявляется в поведении в водных растворах.
В соответствии со схемой гидролиза (схема 3, стр. 181) 1%-ный водный раствор N-mctwi-N,N-6hc-(2-хлорэтил)-амина через 2 суток при комнатной температуре имеет следующий состав (в %) 45:
Непрореагировавший амин....................................11
Л-Метил-Л-(2-хлорэтил)-N-(2'-оксиэтил)-амии гидрохлорид (V) 58
М-Мртил-М,М-бис-(2-оксиэтил)-амин гидрохлорид (VI)........ 2
Продукт димеризации ...................................... 22
На третий день содержание продукта V падает до 35%, а содержание конечного продукта VI повышается до 20%.
Растворимость И-метил-1Ч,1Ч-бнс- (2-хлорэтил)-амина в воде очень велика, она составляет около 12 г/л, тогда как для азотистого иприта приводилось значение 0,2—0,5 г/л. Повышенная растворимость является причиной более быстрого гидролиза этого соединения. Аналогичное этильное производное по своим химическим свойствам примерно соответствует трис-(2-хлорэтил)-амииу.
По своей токсичности оба соединения существенно не отличаются от азотистого иприта,, показатели токсичности этих соединений сравнимы между собой.
5.3. ГАЛОГЕНИРОВАННЫЕ АЛИФАТИЧЕСКИЕ АРСИНЫ
Как уже говорилось в разделе 3.2.1, кроме галогенарсинов типа' I, которые отличаются сильным раздражающим действием, имеется галогенарснны типа II, причем некоторые из этих соединений известны как кожно-нарывные ОВ.
X—As
R—As^
С1СН=СН—As
in
Важнейшим представителем алкилдихлорарсинов как ОВ является 2-хлорвиннлднхлорарснн III (люизит). Уже во время первой мировой войны неоднократно применялись метил- и этилдихлора рейны: Из-за общеядовитого действия эти вещества сначала относили к разряду «ОВ зеленого креста». Их часто применяли в смесях с другими ОВ, затем причислили к «ОВ желтого креста».
В настоящее время еще проявляется некоторый интерес к мышьяксодержащим ОВ. Во время второй мировой войны в распоряжении немецкого фашистского вермахта имелись большие запасы мышьяксодержащнх ОВ, которые предназначались для приготовления тактических смесей. Кроме люизита были заготовлены метил-, этил- и фенилдяхлорарсины.
Делалось достаточно попыток повысить эффективность органических дихлорареннов путем синтеза аналогичных соединений, од-190
ним из которых является полученный в 1948 г. 4-(2-хлорэтилтио)-феннлдихлорарсин51
C1CH2CH2S
АзС1г
Это соединение обладает меньшим кожно-нарывным действием, чем иприт, ио является сильным раздражающим ОВ.
5.3.1. Химическое строение и биологическая активность
5.3.1.1. Связь между химическим строением и биологической активностью. Атом мышьяка в мышьякоргаиических соединениях оказывает большее токсическое действие, чем, скажем, атом азота или серы в физиологически активных аминах и тиозфирах. В фармакологическом отношении органические производные пятивалентного мышьяка менее ядовиты, чем производные трехвалёнтного. Мышьяк-оргаиические соединения типа II обладают, кроме обще ядов итого, еще кожно-нарывным действием.
Соединения этого типа были исследованы в качестве вероятных ОВ. Примером ОВ такого рода служит ряд 2-хлорвиниларси-нов, в который могут входить первичные, вторичные и третичные соединения (III—V):
уС1 С1СН=СН—As^
\С1
III
2-хлорвиннлдихлорарснн (люизит)
С1СН=СН—
C1CH=CIL
yAs—С1
С1СН=С1И
IV
би с -(2-х ло р в и ни л )-х л ср а р с и н
yCH^CHCl
Х^СГ1=СГ1С1
V
Т рис-(2-х ло р в и ни л)-а реин
Люизит III является типичным кожно-нарывным ОВ, которое в совершенно чистом .виде вызывает лишь небольшое раздражение, в то время как соединение IV обладает сравнительно сильным раздражающим, но слабым кожно-нарывным действием. Соединение V хотя еще и достаточно ядовито, но производит во много раз меньшее раздражающее действие, чем соединение IV. Оба эти соединения содержатся в виде примесей в люизнте III. Подобное изменение токсических свойств характерно и для таких галогенированных алкиларсинов, как метилдихлор-, диметилхлор- и триметнл-арснны.
Как правило, органические производные трехвалентного мышьяка обладают очень сильным кожно-нарывным действием в том случае, если они содержат группировку — AsHal2; и особенно, если галогеном является хлор. Самым сильным кожно-нарывным
191
действием обладают Соединения, раДйкйЛЫ которых замещены id-логеном в положении 2. По-видимому, наиболее эффективным представителем является люизнт; содержащий 2-хлорвииильную группу. Если R — арил, то раздражающий характер соединения усиливается, но при этом несколько уменьшается кожно-иарывное действие. Ослабление кожно-нарывного действия происходит н с ростом длины алкильной цепн. При замещении атомов хлора другими атомами и группами уменьшается кожно-нарывное действие. Исключение составляют соединения, которые, помимо негалогени-рованного алкила, в качестве второго н третьего заместителя содержат ^-галогенированные алкильные группы. По своему строе' нию такие производные соответствуют М-алкил-М,№бис-(2-хлор-этил)-аминам.
5.З.1.2. Характер и механизм действия. Алкилдихлорарсииы действуют на незащищенную кожу и — будучи ингаляционными ядами— поражают органы дыхания. Онн обладают сильным общеядовитым действием. Следует полагать, что образование волдырей обусловлено, в первую очередь,‘ингибированием пируватоксидазы. Алкилдихлорарсииы реагируют с функциональными сульфгидрильными группами фермента:
zSH Ch zSv
Фермент^ + ^As—R —► Фермент ^As—R + 2HC1
Особенно чувствительными по отношению к мышьякоргаииче-скнм соединениям, образующим волдыри, являются также ферментные системы дегидрогеназы янтарной кислоты и гексокиназы.
Типичный представитель алкилдихлорарсинов — люизит — действует как контактный яд подобно иприту и азотистому иприту, правда, в кожу он диффундирует быстрее, чем эти ОВ. Сразу после попадания на поверхность кожи без скрытого периода развиваются те же симптомы, что и в случае иприта-.
Из первоначально желто-красной эритемы через 10—15 ч, в зависимости от степени поражения, образуется первый волдырь, заполненный желтоватой жидкостью. Маленькие пузырьки сливаются в один большой волдырь, который через 4—5 суток опадает.
Хотя вызванные люизитом некротические распады глубже, чем в случае иприта, эпидермис сравнительно быстро восстанавливается, поэтому процесс заживления протекает быстрее и опасность инфекции меньше. Поражения от люизита иа 5-ые сутки уже заметно заживают.
Бюшер52 в одном из экспериментов на предплечье человека зафиксировал различие в действии и-прнта и люизита в одинаковых условиях. Ниже вкратце приведены сделанные им наиболее важные наблюдения (см. табл.).
Пары люнзита и других аклилдихлорарсииов в сравнительно высоких концентрациях примерно через 5 ч вызывают на коже 192
Действие люизита и иприта в одинаковых условиях
Условия Люизит Иприт
В момент нанесения Легкое жжение Никаких ощущений
капли Через 5 мин Капля полностью диффунди- Медленная резорбция
Через 14 мин ровала в кожу Образование эритемы Резорбция еще не закончилась
Через 30 мин Жжение и распространение Никаких симптомов
Через 1 ч 45 мин эритемы Образование отека; на пери- То же
Через 2 ч 15 мин ферии выделяется покраснение Кожа становится темно-крас- Появление отдельных
ной красноватых пятен
Через 3 ч 15 мин Сильное жжение; площадь эритемы 12\15 см Желтовато-красное ок-
рашивание; площадь
эритемы 3,5 X 4 см
Через 4 ч 30 мин Усиление жжения Отсутствие чувствитель-
Через 5 ч 15 мин Очаг воспаления примерно Сильная эритема; ок-
в 3 раза больше, чем от раска столь же ин-
иприта тенсивна, как и от
Через 6 ч Набухание ткани люизита Отечная опухоль; пе-
Через 13 ч В середине очага вишнево- риферия уплотненная Очаг воспаления более
красные волдыри, окру- сильно отграничивает-
женные маленькими тонки- ся от непораженной
ми пузырьками ткани; на перифе-
рии — слабо красный
ободок
Через 15 ч Слияние волдырей; волдыри Резкое отграничение от
желтые, по краям с зеле- здоровой ткани; вол-
новатым оттенком дыри отсутствуют;
очаг воспаления жел-
Через 24 ч Слияние всех волдырей в товато-белый Образование пузырь-
один большой (длина ков на периферии
12 см, ширина 7 см, высо- (ожерелье); пузыри
та 4 ел); кожная ткань сливаются друг с дру-
вокруг интенсивно красная, ГОМ
Через 32 ч пузырящаяся Содержимое волдырей изли- Кожа покрыта тесно
вается, прозрачная бело- расположенными друг
желтоватая жидкость возле друга пуЗыря-
На 2-ые сутки Поверхность раны от уда- ми, окруженное пузырями поле бледное Оболочка пузырей сгла-
ленного пузыря красная и живается; в центре
воспаленная пузырей еще нет
На 3-и сутки Поверхность раны покрыта Картина та же; рука
грязно-желтым налетом отёчная, опухает (как
тесто)
7 Зак. 448
Продолжение
Условия. Люизит Иприт
На 4-ые сутки Гнойные выделения уменьшаются Пузыри продолжают сглаживаться; вокруг очага воспаления сильное покраснение (синевато-красное)
На 6-ые сутки Начинается заживление Пигментация периферии (фиолетовая)
На 8-ые сутки Сильное отслоение поверхности раны, легкое кровотечение Пигментация темнеет (коричневая)
На 10-ые сутки Дальнейшее заживление; поверхность под невскрывши-мися волдырями подсыхает П уз ыр и легко вскры- ваются; распространение пигментации
На 12-ые сутки Дальнейшее отторжение некротической ткани; у нев-скрывшихся волдырей периферия черноватая Под легко ранимым эпидермисом—разбухшая, вязкая, липкая некротическая масса
На 14-ые сутки Заживление прогрессирует; оболочка волдырей отслаивается, под ней здоровая кожа Тяжелейший некротический распад
С 15-ых по 20-ые сутки Образование верхнего слоя кожи; в середине встречаются некротические островки; дальнейшее заживление; наклонность к образованию фурункулов Кульминация поражения; некротический распад усиливается
На 26-ые сутки Выздоровление « Некротические ткани еще не отделяются
первые признаки поражения. Обычно оии ие выходят за стадию образования эритемы и через 2 4 суток исчезают.
Глаза поражаются уже при низких концентрациях алкилди-хлорарслнов. Последствиями являются жжение, болевые ощущения, боязнь света, смыкание глаз. Более высокие концентрации вызывают спазм век. Попадание капель.ОВ в глаза приводит через 7—10 суток к потере стекловидного тела.
Как ингаляционные яды алкилдихлорарсииы действуют прежде всего на верхние дыхательные пути. Симптомы те же, что и в случае ОВ, раздражающих носоглотку. После сравнительно короткого скрытого периода появляются кашель, насморк, зуд, чихание и т. п. При слабых отравлениях эти явления проходят через несколько часов, но при тяжелых они длятся несколько дней. Тяжелые отравления проявляются, кроме того, в тошноте, головных болях, потере голоса, рвоте и общем недомогании. В. зависимости от степени Отравления оии ведут к бронхопневмонии. Одышка и астматические явления служат признаками тяжелого отравления, которое может окончиться летально.
194
Отравления при приеме алкилдихлорарсинов через рот могут оказаться смертельными через несколько часов. Через короткое время появляется сильное слюнотечение и очень мучительная рвота. Коликообразные боли и кровавые поносы указывают на тяжелые отравления. При повторных мышьяковых интоксикациях общего характера во всех случаях следует ожидать тяжелых отравлений. Они проявляются в слабости, головокружении, рвоте и т. д, В тяжелых случаях следуют отказ от приема пищи, апатия, снижение кровяного давления и разнообразные поражения других органов (почек, печени, селезенки, центральной нервной системы).
5.3.2. Общие свойства
В соответствии с их химическим строением, алкилдихлорарсины следует считать моноалкнлироваиными производными галогенида (хлорида) мышьяка; их химические свойства определяются наличием трехвалентного атома мышьяка н свойствами, присущими галогениду мышьяка.
Как арсины они легко окисляются. Как галогениды элемента пятой главной группы периодической системы они легко гидролизуются с образованием соответствующих окисей арсинов:
+н2о
R—AsC12 ----R—As---О
-2HCI
В отличие от аминов они лишены основных свойств н потому не способны образовывать соли с кислотами.
Если радикалом является алкенил, как в случае 2-хлорвииил-дихлорарсниа, то ненасыщенный характер соединения проявляется также и в легкости присоединения атомов галогена, и в сравнительно легком отщеплении алкенила от центрального атома мышьяка.
Алкилдихлорарсины, рассматриваемые как ОВ, представляют собой жидкости с резким запахом. С водой они смешиваются лишь ограниченно.
Методы их получения из треххлористого мышьяка, алкилхло-ридов и натрия, нз треххлористого мышьяка и алкильных производных ртути, из хлора и первичных арсинов53 устарели н уже не используются в промышленном производстве.
2-Хлорвииилдихлорарсин получают непосредственной реакцией ацетилена с треххлористым мышьяком в присутствии катализатора.
Как ОВ заслуживают внимания следующие соединения: 2-хлор-винилдихлорарсин, метилднхлорарсии, этилдихлораренн и фенил-дихлррарсии, который целесообразно также отнести к этой труп Пё ОВ.
7*
195
XI С1СН=СН—AsZ \ci
Синонимы: 2-хлорэтенилдихлорарсин, р-хлорвиннлдихлорарсин, люизит.
Военные обозначения: нем. — Lewisit; амер. — Lewisite, М-1 (во время второй мировой войны), L.
Перед концом первой мировой войны люнзит был заготовлен американцами в качестве ОВ, но не был применен. Он представлял собой сильно загрязненный продукт, являющийся смесью первичного, вторичного и трнс-(2-хлорвнннл)-арсина, так называемых люизитов А, В и С.
Работы с хлорвиннларсинамн были предприняты в 1917 г. как в США, так и в Германии. Название «люнзит» ему было присвоено, по имени американского химика У. Ли Люиеа, исследования которого в конце концов выдвинули это ОВ на первый план; в Германии придерживались иных взглядов.
Военные круги в США возлагали на люизит большие надежды, особенно в связи с его быстрым и сильным кожно-нарывиым действием. В 1918 г. в пригороде Кливленда за короткий срок была построена промышленная установка.
Из-за возможности применять это ОВ посредством поливки с самолетов оно получило название «роса смерти». Неустойчивость полученного тогда продукта вынудила американцев после воины уничтожить свои запасы ОВ (около 150 т), утопив их в море.
После первой мировой войны исследования этих соединений и поиски улучшенных технологических процессов не прекратились. Во время второй мировой войнй армия США, как и фашистский вермахт, имели в своем распоряжении значительные- запасы люизита. Принимая во внимание наличие более эффективных ОВ, физико-химические свойства которых значительно лучше, люнзит уже не занимает среди ОВ привилегированного положения. Правда, существуют сравнительно быстрые н, к тому же дешевые методы его производства, поэтому для стран с не столь развитой химической промышленностью он еще может иметь некоторое значение.
5.3.3.1. Методы получения. Самый распространенный метод получения 2-хлорвинилдихлсУрарсина заключается во взаимодействии AsC13 с ацетиленом в присутствии хлористого алюминия. Без ката^ лнзатора Фриделя — Крафтса реакция не идет.
Прн действии ацетилена на AsCl3 в соответствующих условиях образуется в основном 2-хлорвннилднхлорарсин, а также некоторые количества бнс-(2-хлорвнннл)-хлорарснна н трис-(2-хлор-винил)-арсина. Реакция протекает по следующей схеме:
AsCl3 + НС=СН —> Cl2As—СН=СНС1
2C12As—СН=СНС1 + ЗНС=СН —> C1As(CH=CHC1)2 + Аэ(СН=СНС1)л
As(CH=CHCl)3 + AsCl3 —> C1As(CH=CHC1)2 + C12As—СН=СНС1
196
На промежуточной стадии образуется маслянистая коричневая жидкость, состоящая из смеси соответствующих комплексных соединений алюминия и мышьяка, которые затем в ходе процесса разлагаются разбавленной соляной кислотой до хлорвиниларсинов. Без учета побочных реакций образование 2-хлорвнннлднхлорарсина протекает по следующей схеме:
AsCl3 + AlGb —► AsCi3 + AlCi;
HC=CH+AsCl3 —► HC=CH—AsCl3
HC=CH—AsCl3 + AlCi; CI3AI—CH=CH^AsCl3 гидролиз^
Cl
—► AlCSfe + C1CH=CH- AsCl3
Фракционной перегонкой отделяют 2-хлорвинилдихлорарсии от побочных продуктов. Конечно, фракционная перегонка не очень удобна, так как при продолжительном нагревании 2-хлорвниил-дихлорарсии диспропорциейирует с образованием треххлористого мышьяка и вторичного бис-(2-хлорвинил)-хлорарсииа:
2С1СН = CH—AsC12 —> AsCl3+(ClCH=CH)3AsCl
В процессе, разработанном во время второй мировой войны Хьюттом и сотр.54, вместо хлористого алюминия применяется сулема или одиохлорнстая медь в 20%-иом растворе НС1 или в гидрохлориде этаноламина. В этом процессе нз двух стереоизомеров 2-хлорвинилдихлорарсина преимущественно с хорошим выходом образуется транс-изомер (ср: раздел 5.3.3.2).
Технология производства люизита несложная. В соответствующих реакторах пропускают при энергичном перемешивании ток очищенного и высушенного ацетилена в реакционную смесь, состоящую из треххлористого мышьяка и катализатора. Температуру реакции поддерживают ниже 50°С охлаждением или регулировкой подачи газа. Образовавшуюся маслянистую смесь жидких продуктов в зависимости от примененного катализатора обрабатывают разбавленной (1:1) соляной кислотой либо гидрохлоридом этанол-амииа. Процесс очистки заканчивается отделением 2-хлорвинил-дихлорарсииа перегонкой прн пониженном давлении.
По новому способу получают почти чистый н лишенный запаха продукт с выходом 85—90%. В техническом продукте содержится окрло 10% вторичного арснна н лишь небольшие количества третичного арсина в смесн с AsCl3.
Лабораторный синтез. Исходными веществами являются ацетилен и треххлористый мышьяк, катализатором служит хлористый алюминий.
В реакционную колбу емкостью 250 мл, снабженную мешалкой и трубками для ввода и вывода газа, помещают 45 г AsCls и 15 г безводного A1CU. При перемешивании и охлаждении пропускают через реакционную смесь 6—-8 л аце-
197
стым кальцием. Температура реакции не должна подниматься выше 35 °C. Затем реакционную массу медленно вливают при О °C в ~200 лл концентрированной соляной кислоты и перемешивают 10—15 мин. Рекомендуется еще раз промыть соляной кислотой органический слой. Отделяющийся маслянистый слой пере-
гоняют при давлении 20—22 мм рт. ст. Первая фракция (до 60 °C) содержит треххлористый мышьяк; при 80—100 °C перегоняется 2-хлорвинил дихлор арсин, от 120—140 °C — вторичный арсин, выше 140 ЬС — третичный.
5.3.3.2. Физические свойства. Имеющиеся в старых работах данные о физических свойствах 2-хлорвннилднхлорарсина противоречивы, так как долгое время не удавалось получить чистое вещество. Для военных нужд довольствовались стабилизованным техническим продуктом.
Технический продукт обладает сильным, навязчивым запахом гераии, очищенный продукт не имеет запаха. Технический люизит— маслянистая темно-коричневая жидкость, застывающая в интервале от —10 до —15°С. Уже ниже 10°С технический люизит становится заметно более вязким.
Из-за наличия в этом соединении двойной связи для него возможно существование двух пространственных изомеров:,
Нч ZH CL ZH
ХС=С 4C=CZ
Cl/ \AsC12 и/ ^AsC12
iuc- люизит
[изо люизит)
т ра кс-лю йзнт (обычный)
Оба изомера 2‘хлорвннилднхларарснна выделены и для них определены физические ’константы5^.
транс-Изомер
ц«с-Изомер
Т. кип., °C.............................. 169,8 .196,6
Т. пл., °C...........................к . . -44,7 -2,4
Давление пара при 25 °C, мм рт. ст.- . . 1,562 0,40
Относ. ПЛОТНОСТЬ
d™............................... 1,8598 . 1,8793
d* .................. 1,9018—0,00168 • f 1,9210—0,00167 • г
Показатель преломления ............... . 1,5898 1,6076
Летучесть при 20 °C, мг/л .' ....... 2,3 (смесь) 4,5
Выше 196°С (т. кип. обычного люизита) наступает разложение.
Растворимость люизита в воде мала, она составляет околр. 0,5 г/л. В органических растворителях — галогеналканах, спиртах и бензине он растворим хорошо; легко растворяется в растительный и животных жирах.
Вследствие хорошей смешиваемости с другими ОВ люизит пригоден для приготовления тактических смесей. Он смешивается, например с нпрнтом, дифосгеном н различными фосфорорганическими ОБ.
Способность проникать через материалы, такие как кожа, резина, древесина и текстильные ткани, у люизита выражена сильнее, чем у иприта.
5.3,3,3. Химические свойства. При термическом разложении 2’хлорвинилдихлорарсина образуются вторичный и третичный арсины, которые при известных условиях распадаются далее. Даже при кратковременном нагревании, например, при взрывах, разложение люизита значительно.
Люизит сравнительно неустойчив, что объясняется наличием группировки —AsC12 и его ненасыщенным характером.
При длительном хранении в металлических емкостях нужно избегать появления кислых продуктов разложения. Железо катализирует диспропорционирование люизита, в результате которого образуются вторичный и третичный арсины. Высококачественные стали устойчивы к действию люизита. Алюминий и его сплавы сильно корродируют прн контакте с люизитом.
В присутствии стабилизаторов или ингибиторов коррозии люизит может храниться в артиллерийских снарядах и бомбах.
Между обоими изомерами люизита нет существенной разницы в химических свойствах. Их можно отличить*по разному поведению в холодных растворах едкой щелочи; транс-изомер более реакционноспособен, чем цис-нзомер.
Гидролиз. Гидролиз люизита . определяется хлорангидриднымн свойствами группы —AsCU. Люизит сравнительно быстро гидролизуется с отщеплением соляной кислоты и с образованием окиси 2-хлорвиниларсина:
С1СН=СН—AsC12 ‘ - * и* > С1СН=СН—As=O
Склонность к быстрому гидролизу относится к наиболее существенным недостаткам этого ОВ. Отстаивавшееся ранее мнение, что вследствие образования ядовитой окиси 2-хлорвнниларснна люизит, якобы, можно применять для заражения воды, в настоящее время отпало. Теперь известны более эффективные соединения для этой цели.
Склонность к легкому гидролизу позволяет применять люизит только при чрезвычайно благоприятных метеорологических условиях.
Окись транс-2-хлорвиниларснна— белое кристаллическое вещество, т. пл. 143 °C; окись цис-изомера плавится прн 131 °C. Она обладает общеядовитым действием, а в соответствующих растворителях проявляет еще и значительное кожно-нарывное действие.
Уже слабые основания способствуют гидролизу. Достаточно разбавленных растворов аммиака, чтобы перевести 2-хлорвинилди-хлорарсин в его окись. Воздействие щелочей уже на холоду ведет к полному разложеию люизита:
+6NaOH
ClCH=CHAsCla -------> Na3AsO3 + 3NaCl + HCsCH + ЗН2О
На холоду транс-люизит разлагается щелочью с выделением ацетилена, цис-изомер разлагается при слабом нагревании с выделением винилхлорида. Это свойство служит для различения обоих
199
Изомеров. Выше 40 °C лЮизит также полностью разлагается. Люизит реагирует с едкими щелочами количественно, эту реакцию можно использовать для дегазации и для анализа.
Водными растворами сульфида натрия люизит разлагается с образованием сульфида мышьяка и ацетилена:
+3NajS
2С1СН=СН—AsC12 — —As2S3 + 2СН=СН
Другие реакции группы — AsCU. При действии сероводорода на спиртовый раствор 2-хлорвинилднхлорарсина образуется арсин-сульфид, обладающий сильным раздражающим действием:
+ H2S
CICH=CH— AsCI2 ——С1С№=СН— As=S
Реакцией, наиболее важной для дегазации, является окисление. Перекись водорода, азотная кислота и хлорная известь окисляют арсин в 2-хлорвиниларсоновую кислоту:
+0; +2Н2О
С1СН=СН—AsC12 -------> С1СН=СН—As(OH),
-2HC1
Эта кислота представляет собой игольчатые кристаллы, т. пл. 130—131 °C. Ее можно сравнительно легко получить действием хлора на водный раствор люизита. Раствор упаривают и кристаллы выделяют из воды, iftzc-Изомер кислоты имеет более низкую температуру плавления (90—91 °C). Арсоновая кислота не обладает физиологической активностью (исключение составляет прием per os).
Хлорирование. Направление хлорирования 2-хлорвинилдихлор-арснна зависит от среды. Если соединение растворить в органических растворителях, лучше всего в галогеналканах, и обработать хлором или бромом, то атомы галогена присоединяются к двойной связи винильной группы.
Хлорирование в водных растворах приводит к образованию очень неустойчивого тетрахлорарсииа. При действии воды он превращается в арсоновую кислоту; при соответствующих условиях возможно его разложение на треххлористый мышьяк и дихлорэти-леи, причем треххлористый мышьяк гидролизуется далее до окиси мышьяка:
+ С12
С1СН=СН—AsCh -----> С1СН=СН—AsCh
+знао
С1СН=СНС1 + AsCl3 <-----1 ' ^4НС1» С1СН=СН—As(OH)g
----* AsaOa-f-3HCl
200
Хлорирование приводит к образованию сравнительно неядовитых продуктов, поэтому эту реакцию можно использовать для дегазации.
5.3.3.4. Токсические свойства. Подобно рассмотренным выше кожно-нарывным ОВ, люизит действует не только как контактный, но н как ингаляционный яд, а также поражает глаза.
Кожно-нарывное действие проявляется без скрытого периода. Эритемы на коже образуются при дозах около 0,05—0,1 мг/см2 поверхности кожи. Дозы в 0,2 мг/см2 неизбежно ведут к образованию волдырей. От действия парообразного люизита в концентрации 10 Л(г/л на поверхности кожи через 15 мин экспозиции образуются волдыри.
Согласно Веддеру, концентрации порядка 0,05 мг/л при экспозиции 30 мин, а также концентрации 0,3—0,5 мг/л при экспозиции 5 мин действуют смертельно. Концентрации 0,05 мг/л при экспозиции 15 мин ведут к тяжелым отравлениям, которые имееют следствием потерю боеспособности на несколько недель. Меиыпие концентрации, порядка 0,01 мг/л, через 15 мин вызывают покраснение глаз и опухание век. Такие поражения глаз протекают мягче, чем поражения, вызываемые ипритом^ или азотистым ипритом.
Смертельная доза цри введении через кожу составляет 20 мг/кг массы.
5.3.4. Метил-, этил- и фенилдихлорарсины
XI XI XI
СН3—As С2Н5—As CeH5—As
XI XI XI
Эти и подобные соединения бцли получены и исследованы еще во второй половине XIX века Байером, Ла Костом, Деиом и др. Уже тогда стали известны их физиологическое действие и токсичность, поэтому во время первой мировой войны оии снова явились предметом широких исследований. Этилдихлорарсии был применен немецкой стороной весной 1918 г. под кодовым наименованием DICK и классифицирован сначала как ОВ «зеленый крест-3», а затем как ОВ «желтый крест-1». В США считали, что в качестве ОВ применимо метильное производное.
Фенилдихлорарсин уже в первую мировую войну применяли как растворитель для дифенилхлорарсина. Во время второй мировой войны в итальянской и немецкой фашистских армиях это ОВ служило компонентом зимних ипритных смесей. В качестве потенциальных ОВ были заготовлены метильное и этильное производные.
Методы получения. Для промышленного получения алкилди-хлорарсинов легкодоступную натриевую соль алкиларсоновой кислоты восстанавливают сернистым ангидридом до окиси алкил-арсина, которую далее действием хлористого водорода переводят
201
в соответствующий алкйлдихлорарсин:
R—As(ONa)2 ——> R—As=O
И ' '* — NagSOa
0
+2HC1 ZC1
R—As=O tttt* R—As^
-H,o \C1
Натриевые соли алкнларсоновых кислот получают алкилирова-нием арсенита натрия диалкилсульфатом
As(ONa)s + R2SO4 —> R—As(ONa)2 + RNaSO4
О
а этильное производное, — действуя хлористым этилом:
As(ONa)3 + С2Н5С1 —> C2H5As(ONa)2 + NaCl
О
Во время второй мировой войны для получения этилдихлорар-сина был разработан процесс56, основанный иа взаимодействии тетраэтилсвинца с треххлористым * мышьяком. На первой стадии при комнатной температуре нз I моль тетраэтилсвинца и 2 моль треххлористого мышьяка получается 2 моль этилдихлорарсииа; образующийся при этом хлористый диэтилсвииец на второй стадии алкилирует еще 1 моль треххлористого мышьяка:
Pb(C2Hs)4 + 2AsCl3 —>- 2C2H5AsC12+(C2H5)2PbCl2
100°C
(C2H5)2PbCl2 + AsCl3 -> C2H6AsCl2+C2HsCl + PbCl2
Свойства. Метил-, этил- и фенилдихлора рейны в чистом виде бесцветные жидкости; технические продукты темного цвета, иногда слегка желтые.
За исключением представителя ароматического ряда, летучесть этих соединений очень высока и составляет 74 мг/л для метильного и 20 мг/л для этильного производного. Фенилдихлорарсии по летучести сравним с ипритом и является самым стойким из соединений типа RAsCh.
Все соединения отличаются низкими температурами плавления. Это свойство, а также их хорошая растворимость в других ОВ дали возможность использовать их в качестве компонентов смесей ОВ.
Эти соединения плохо растворимы в воде и малоустойчивы. Исключение составляет фенилдихлорарсии. Последний является самым устойчивым представителем этого типа ОВ. Он практически не гидролизуется даже при нагревании в воде.
Химическое поведение этих соединений аналогично поведению 2-хлорвинилдихлорарсина. Водой они гидролизуются до соответствующих окисей алкил- иди ариларсинов.
202
При действии окислителей образуются метил-, этил- и феиил-арсоиовые кислоты, представляющие собой кристаллические или жидкие вещества.
Метил-, этил- и феиилдихлорарсины оказывают сильное раздражающее действие иа слизистую оболочку иоса или глотки и в этом отиошеийи сравнимы с ОВ соответствующей группы.
Их кожио-иарывное действие выражено ие столь сильно, как у люизита. Поражения кожи наступают без скрытого периода и ведут к образованию волдырей.
При вдыхании паров оии действуют как удушающие О В, вызывая бронхопневмонию и отёк легких. В зависимости от концентрации тяжелые отравления могут иметь летальный исход.
Поражения глаз по своей интенсивности превосходят поражения, причиняемые ипритом, но сравнительно легче поддаются лечению.
Данные о физических и физиологических свойствах QB приведены в табл. 20.
Таблица 20. Физические и физиологические свойства мышьякорганических соединений R—AsCl2
В круглых скобках приведена температура
Свойство СНз-AsCla С2Н5—AsCh CgHs—AsCla
Т. кий., °C 132,5 155,3 256
Т. пл., °C -42,5 -65 -20
Относит. ПЛОТНОСТЬ ^4° 1,8359 1,6595 1,654
Давление пара, мм рт. ст. 10,83 (25 °C) 2,29 (21,5 °C) 0,014 (15 °C)
Максим, концентрация прн 20 °C мг!л 74,4 20 0,4
Растворимость в воде, г/л < 1 < 1 Не растворим
Порог раздражения, мг/л 0,002 0,005 0,005
Предел переносимости, мг/л Доза, мг/см2 0,025 0,01 0,016
вызывающая поражения кожи 0,1—0,5 0,1-0,5 —
вызывающая образование волдырей Степень токсичности при экспозиции 15 мин, мг/л 3-5 2
несмертельная 0,1 0,05
смерть в отдельных случаях 0,3 0,2 —-
смертельная 0,4—0,5 0,3 0,3
5.4. ГАЛОГЕНИРОВАННЫЕ ОКСИМЫ
Во время второй мировой войны в Германии и, вероятно, и в других капиталистических странах в результате исследований, начатых еще до войны, арсенал химического оружия был пополнен так называемыми ОВ крапивного действия.
203
Рассматриваемые ниже соединения представляют собой галогенированные оксимы (альдоксимы) строения
Х\
V=N—ОН
у/
где X или Y—галогены, галогеиалкильные или нитрильные группы.
Соединения этого общего типа отличаются комбинированным характером физиологического действия. Они представляют собой сильнейшие раздражающие ОВ, но обладают в то же время удушающим и кожно-нарывным действием.
Еще в 1894 г. Неф и Шолл при проведении исследований гремучей кислоты имели дело с одним из представителей этого ряда соединений— монохлорформоксимом 1, Тридцатью годами позже Прандтль и Зенневальд57 получили другой представитель этой-группы — дихлорформоксим II
С1\
XC=N—ОН Н^
I
С1
C=N—ОН
п
Хакманн58 указал на возможность применения дихлорформок-сима в качестве ОВ. Ошеломленный его исключительной физиологической активностью, он писал: «В органической химии найдется мало веществ, которые проявляют столь сильное действие на человеческий организм, как это соединение».
Молер намеревался отнести это вещество, как и азотистый иприт, к группе ОВ «красного креста».
О возможностях применения галогенированных оксимов в качестве ОВ известно мало. Однако очевидно, что в силу их быстрого физиологического действия и малой химической стойкости они пригодны для внезапного нападения и в наступлении. Рентабельность промышленного производства увеличивает вероятность их применения.
5.4.1. Физиологическая активность
Галогенированные оксимы действуют как раздражающие, ингаляционные и кожные яды.
Уже в чрезвычайно малых концентрациях пары н аэрозоли галогенированных оксимов раздражают верхние дыхательные пути, что проявляется в немедленных приступах кашля, боли в горле и т. п. Вследствие раздражения глаз возникает сильное слезотечение, боли в глазах, воспаление роговицы, снижение остроты зрения; при сильных поражениях — временная слепота.
По удушающему действию они сравнимы с фосгеном, что можно объяснить сходством химического строения этих соединений как
204
| галогенпроизводных угольной кислоты:
I Ck Ck
f XC=O 'C=N—OH
J CI// Cp/
| фосген дихлорформоксим
(фосгенокеим)
. Ингаляционное действие приводит к отеку легких. Тяжелые отравления вызывают расстройства кровообращения и функций центральной нервной системы.
Как в твердом, так и в жидком виде галогенокснмы более или менее сильно и без скрытого периода действуют на кожу, давая ; чрезвычайно болезненные воспаления, которым предшествует сильное жжение. Характерным является крапивное действие. Крапивница распространяется по всему телу, даже если не все оно находилось в контакте с ОВ. Ожог очень сильный, неприятный и приводит к полной потере боеспособности. Обычно крапивное действие » аэрозолей начинается на коже рук и лица и распространяется затем по всему телу. Пораженные участки кожи отекают и в дальнейшем образуются болезненные белые волдыри, осложняющиеся еще и тяжелыми некротическими распадами. Раны заживают плохо и являются очагами вторичных инфекций.
В результате резорбции галогенированных оксимов даже при кожных поражениях наступают явления общего отравления, которые выражаются в головных болях, чувстве страха и т. п.
5,4.2, Общие свойства
Важнейшими представителями галогенированных оксимов являются моно-и дихлорформоксим (1иП),а также трихлорметилхлор-формоксим III. Кроме того, в специальной литературе упоминаются и некоторые другие соединения — хлорметилхлорформоксим IV, бис-(хлорметил)-формоксим V, цианхлорформоксим VI, дибром-формоксим VII, фторхлорформокснм VIII.
С13СХ
NOH ciz
III
C1CH2S
V=NOH
С г
IV
С1СН2ч
Y=NOH
С1СН/
V
Br\
V=fNoh
Br
F\ V=.\OH
C1Z
VI
VII - VIII
Соединения (I—III) — кристаллические вещества, За исключением соединения JII они хорошо растворяются в воде, Растворимость галогеналкилпроизводиых в воде ограничена.
Их можно перегонять без разложения при атмосферном давлении; оии очень летучи. Кристаллический монохлорформоксим очень быстро испаряется.
205
В химическом отношении неалкилированные соединения менее устойчивы, чем алкилированные. При длительном хранении гало-геноксимы полимеризуются. Водой они более или менее быстро разлагаются с образованием двуокиси углерода, галогеноводородной кислоты и гндроксиламина:
+2Н2О x2c=noh ----------> nh2oh-hx
-СО2; -нх
Гидролиз облегчается щелочами и кислотами.
5.4.3. Дихлорформоксим
Ск \l=NOH
СИ
Синонимы: лихлорформальдоксим, фосгеноксим.
5.4.3.1, Методы получения. В промышленности дихлорформоксим получают восстановлением трихлорнитрозометана или трихлорнитрометана (хлорпикрина). Можно пользоваться восстановителями либо электрохимическим восстановлением.
Химическое восстановление. В качестве сырья для синтеза фос-геноксима Прандтль применял трихлорнитрозометан,. который он получал, действуя азотной кислотой на натриевую соль трихлор-метилсульфиновой кислоты. Трихлорнитрозометен можно восстановить в метанольном или сероуглеродном растворе сероводородом или амальгамой алюминия до дихлорформоксима. (выход очень низкий);
+H2S
CChNO - - > Cl^NOH
—о; —НС1
Возможно получение окснма в одну стадию восстановлением легкодоступного трихлорнитрометана порошком олова илц железа. Лучшие выходы получают по способу, модифицированному Гриш-кевич-Трохимовской 59, применявшей для восстановления олово и соляную кислоту с добавкой эфира.
Реакция сопровождается образованием нескольких промежуточных соединений, которые можно выделить:'
4-2Н 4-2Н . перегруппировка
CI3CNO2 ----> CI3CNO ---> С13СН—NO ~ Д.: CI2C=NOH
—Н2О -НС1
Электрохимическое восстановление 69 Трихлорнитрометан можно восстановить электрохимически до дихлорформоксима при соответствующем составе электролита, применяя охлажденный ниже 5 °C оловянный катод;
Подобно описанному в разделе 4.2.3.3, восстановление. идет^ в несколько стадий. Регулируя напряжение, можно выделить проме-206
жуточные соединения. При дальнейшем электролизе восстанавливается м дихлорформоксим, конечным продуктом восстановления является метиламин.
Лабораторный синтезSI. В соответствующем реакторе смешивают 2 моль (329 г) трихлорнитрсциетана, 1 л концентрированной HCI (d 1,19) и 0,5 л эфира. При сильном перемешивании прибавляют при 0°С малыми порциями 240 г тонко-измельченного порошка металлического олова и охлаждением поддерживают температуру реакции 0°С. Появляющаяся сначала фиолетовая окраска указывает на образование трихлорнитпозометана, ее исчезновение является признаком того, что реакция закончилась. Продолжительность реакции около 4—5 ч, Эфирный раствор фильтруют, обезвоживают над хлористым кальцием и после отгонки эфира кристаллизуют дихлорформоксим, выход 70—75%.
5.4.З.2. Свойства. Чистый дихлорформоксим — бесцветные призматические кристаллы. Промышленный продукт — желто-коричневая жидкость со своеобразным пронзительным запахом. Кристаллическое соединение имеет т. пл. 39—43°С, т. кип. 129°С при 760 мм рт. ст. и 53—55 °C при 29 мм рт. ст.
Дихлорформоксим очень быстро улетучивается, промышленный продукт имеет высокое давление пара. Летучесть составляет 20—25 мг)л. Соединение гигроскопично; при —15 °C без доступа влаги его можно хранить без разложения несколько месяцев, тогда как при обычных условиях (особенно под действием света) оно склонно к полимеризации.
В органических растворителях — эфире, спирте и галогенал-канах — дихлорформоксим легко' растворим и такие растворы устойчивы. Особенно устойчивы растворы в этилацетате.
В воде он растворяется медленно и не полностью. В водных растворах дихлорформоксим гидролизуется.-
Гидроокиси и карбонаты щелочных металлов и аммиак быстро реагируют с водными растворами' дихлорформоксима. При действии растворов аммиака из эфирных растворов получают неядовитый и лишенный кожно-нарывиого действия цианамидхлорформ-оксим в виде бесцветных кристаллов:
N=C—NH
4-14NH3 \
6C12C=NOH —- - - M-> 3 }C=NOH
—9NH4CJ; —ЗН2О; — Np У
С1
Таким образом, дихлорформоксим взаимодействует с аммиаком иначе, чем фосген; кроме того, он не реагирует с аминами (анили- ном и диметиланилином), Поэтому его нельзя считать оксимом фосгена. В присутствии одного из этих соединений он, в зависимости от растворителя, в большей или меньшей степени диспропорцио-иирует, давая дихлор фу роксан:
2C12C==NOH —► 2НС1 4-(C1CNO)2
С дымящей азотной кислотой образуется эфир С12С — N—ONO2, При нагревании с обратным холодильником дихлорформоксим разлагается на хлорциаи и хлорноватистую кислоту.
207
Дихлорформоксим даже в газообразом состоянии действует на л резину. - 1
Концентрации уже в 0,025 мг/л вызывают сильные раздражения ] глаз. Более высокие концентрации порядка 1—35 мг/л дают пора- ' жения кожи и при вдыхании приводят к сильным отравлениям. Дозы в 30 мг/кг массы должны быть смертельными. Опытами на животных была определена летальная дЪза в 10 мг/кг.
Наиболее сильно действует дихлорформоксим в капельном со- : стоянии.
5.4.4. Монохлор форм оке им и трихлорметилформоксиу (перхлорацетальдоксим)
Ck С13С,
V=NOH ^C=NOH
н/ С г
Монохлорформоксим — очень неустойчивое соединение, выше 40°C он разлагается со взрывом, при обычной температуре — с образованием двуокиси углерода и гидрохлорида гидроксиламина. В водных растворах быстро гидролизуется. Если эти растворы об-’' работать гидроокисью натрия или аммиаком, то образуется гремучая кислота, которая затем распадается.
Хотя монохлорформоксим — сильное раздражающее ОВ, обладающее сильным крапивным действием и вызывающее образование волдырей, в качестве ОВ он, по-вндимому, непригоден. Более стабильным и подходящим соединением является трцхлорметил-хлорформоксим — белые игольчатые кристаллы. Его можно годами хранить без разложения; т. пл. 60°C, т. кип. 100°С при \5ммрт.ст. Запах этого вещества напоминает запах окиси азота. В воде три-хлорметилхлорформоксим не растворим, но в обычных органических растворителях растворяется хорошо. Водой это соединение лишь мало гидролизуется, при действии растворов щелочи он разлагается с образованием неядовитых продуктов.
5.5. БОЕВОЕ ПРИМЕНЕНИЕ КОЖНО-НАРЫВНЫХ ОВ
Из кожно-нарывных ОВ во время первой мировой войны в больших количествах применялись в военных целях только иприт и метил-и этилдихлорарсины. Позднее иприт также использовался и в других военных операциях, особенно при нападении Италии на Эфиопию.
Иприт применяли при помощи артиллерии, в том числе и при помощи обычных и так называемых химических минометов. Уже в первую мировую войну для расширения возможностей боевого применения к иприту добавляли растворители, такие как нитро- и хлорбензол или четыреххлористый углерод. Иприт пригоден для приготовления тактических смесей, что дает возможность приме
208
нять его в широком интервале температур, и даже при низких температурах.
Во время второй мировой войны нейтральные растворители были заменены рядом ядовитых соединений, которые сами по себе являются ОВ, например такими как этил- и фенилдихлорарсин, люизит, фосфорорганические соединения.
Применение кожно-нарывных ОВ типа иприта и люизита совершенствовалось с разработкой новых средств применения, например, фугасов, выливиых авиационных приборов, машин и приспособлений для заражения местности, метательных средств.
Неблагоприятные физико-химические свойства таких ОВ в известной степени компенсировались как самими средствами применения, обеспечивающими образование аэрозолей ОВ, так и введением добавок, особенно загустителей. За исключением азотистого иприта, все кожно-нарывные ОВ применимы в любое время года.
На их стойкость оказывают влияние метеорологические факторы, прежде всего дождь, температура и ветер. Самым устойчивым ОВ по отношению к дождю и другим осадкам считается иприт.. Люйзит и галогеиоксимы гидролизуются сравнительно быстро и в результате теряют свою эффективность, в то время как иприт при благоприятных почвенных условиях, т. е. когда почва не песчаная и не пористая, может после дождя опять стать активным.
Кожно-нарывные ОВ являются стойкими ОВ. Их летальное действие ограничено. Лишь в исключительных случаях — на нетренированные или лишенные средств защиты войска — они действуют летально.
Тем не менее, их токсическое действие как ингаляционных ядов велико и при известных обстоятельствах приводит к длительной утрате боеспособности. Их стойкость (за исключением галогено-ксимов) и большая способность проникать через такие материалы, как текстильные ткани, кожу и резину, определяют значение кожно-нарывных ОВ для заражения местности, боевой техники и предметов вооружения. Их сравнительно высокая стойкость принуждает к принятию защитных мер и таким образом способствует подавлению противника. Маневренность подразделений затрудняется. В случае применения этих ОВ требуется дегазация или обход зараженных участков; при благоприятных условиях постоянное и длительное ношение защитного обмундирования изматывает противника.
Наибольшей стойкостью обладают иприт и азотистый иприт. Даже при редком растительном покрове и плотности заражения 25 г/м2 приходится считаться с продолжительностью отравляющего действия до 24 ч при нормальной температуре, и до нескольких суток при низких температурах. По американским инструкциям, без защитного обмундирования й без противогаза можно вступать на местность, зараженную от 100 до 500 кг иприта (HD) на гектар, при температуре 20°C, как правило, лишь через 4 суток, а на лесистую местность — лишь через 6 суток. При температуре
209
выше 27 °C это время сокращается до I суток. При плотности заражения 75 г Ди2 на открытой местности летом люизит эффективен только 2—4 ч, а зимой — несколько суток.
В зависимости от метеорологических условий стойкость вязких рецептур кожно-нарывных ОВ составляет несколько суток'. Чтобы существенно повысить устойчивость таких смесей к дождю и влаге, в их состав обычно вводят водоотталкивающие средства.
Для опрыскивания из выливных авиационных приборов к ОВ, особенно к иприту и люизиту, добавляют вещества, которые придают им вязкость, необходимую для эффективного разбрызгивания. Размер капель, скорость оседания, плотность облака ОВ и другие факторы подбираются в зависимости от высоты полета таким образом, чтобы обеспечить эффективное заражение.
В распоряжении американских ВВС имеются выливиые авиационные приборы различных типов, которые можно приводить в действие на пилотируемых н непилотируемых самолетах, а также на самолетах-снарядах дальнего действия. При помощи специальных вертолетов можно за короткое время заразить большие участки местности. Выливные авиационные приборы служат для применения не только иприта, но н других стойких ОВ.
Применение кожно-нарывных ОВ при помощи ракет по экономическим причинам маловероятно. Особого внимания заслуживают многоствольные установки для запуска реактивных снарядов, поскольку при их помощи в течение нескольких минут можно эффек-. тивно заразить большие площади.
Таблица 21. Боеприпасы армии" США, снаряженные ипритом (по состоянию на 1962 г.; данные заимствованы из FM-3-10)
Тип боеприпасов Средство применения Максимальная дальность стрельбы (полета), кл< Масса боеприпаса, кг Масса ов, кг Степень эффектна НОСТИ боеприпаса, % '
Мины
М2А1 4,2-дюймовый миномет * 3*93 10,8 2,72 99
мбо 105-лл гаубица 11,14 15,2 1,22 99
М110 155-лк гаубица 14,95 45,9 2,95 99
М104 155-мм пушка 16 43,0 5,31 —
Реактивные снаряды МК40, MOD Установка М105; ракета М40, MOD 4,2 —
Бомбы М70 А1 Бомбардировщик; истребитель — — 272
Химические фугасы 5,45 4,5 —
* I дюйм равен 2,54 сл<.
210
Уже^в первую мировую войну выявился такой недостаток иприта,, как его характерный запах, который противник использовал для индикации ОВ. То же относится и к люизиту, обладающему еще более сильным запахом. Этот недостаток иприта был устранен получением чистого продукта. Типичный запах прежних технических образцов ОВ вряд ли еще встретится, и эта возможность индикации теперь уже маловероятна.
Гидролитическая устойчивость азотистого нпрнта определяет его значение для заражения систем водоснабжения.
Высокая летучесть и, отчасти, низкая устойчивость ограничивают боевое применение галогеноксимов. Они нестойки, и их относят к быстродействующим ОВ. Применение галогеноксимов при помощи обычных средств, за исключением выливных авиационных приборов, возможно, и его следует ожидать.
В армии США уделяют наибольшее внимание иприту. Особое значение имеет HD (перегнанный иприт), для применения которого пригодны все средства (см. табл. 21).
Ниже приведены данные62 о площадях заражения ипритом при помощи средств применения ОВ американских вооруженных сил (длительность огня 15 мин):
Площадь Подразделение заражения,
га
Дивйзион 106,7-и.и минометов.............. 160
Дивизион 105-.и .и гаубиц.................. 13,4
Дивизион 155-.и .и гаубиц........... . , 22,4
Звено истребителей-бомбардировщиков ... 8
Звено легких бомбардировщиков ...... 16
Звено средних бомбардировщиков............. 65
Звено тяжелых бомбардировщиков 112
В американских боеприпасах кожно-нарывные ОВ типа иприта обозначаются буквой Н и двумя зелеными кольцами (раньше маркировались тремя красными кольцами).
Применяется следующая маркировка:
иприт — H(HS), азотистый иприт — HN-I, очищенный иприт — HD, ипритная смесь, преимущественно с кислородным ипритом — НТ.
Люизит обозначается буквами L или М-1 и также как иприт — двумя зелеными кольцами.
5.6. ЗАЩИТА ОТ КОЖНО-НАРЫВНЫХ ОВ И ИХ ДЕГАЗАЦИЯ
5.6.1. Защита
Полная защита от кожно-нарывных ОВ достигается лишь в том случае, когда нм закрыт доступ в организм или отсутствует возможность соприкосновения с поверхностью кожи.
211
Средствами защиты от кожно-нарывных ОВ является противогаз и средства защиты кожи.
Для защиты органов дыхания от кожно-нарывных ОВ достаточно фильтрации вдыхаемого воздуха. Частично нмпрегнирован-ные слои актнвированого угля в коробках противогазов задерживают пары н аэрозоли кожно-нарывных ОВ, В отличие, например, от ОВ типа адамсита, для защиты от кожно-нарывных ОВ, одновременно обладающих сильным раздражающим действием на носоглотку, как например технический люизит н другие алкнлднхлор-арснны, а также дихлорформоксим, не требуются противодымные фильтры.
Чрезвычайно высокие концентрации ОВ, при которых будет исчерпана защитная мощность противогаза, в случае кожно-нарывных ОВ вряд лн можно создать, онн могут возникнуть лишь в непосредственной близости от места взрыва боеприпасов с ОВ.
Собственно шлем-.маска защищает кожу лица от воздействия ОВ н поэтому одновременно является частью средств защиты кожн.
Защитная мощность' обычного обмундирования практически равна нулю. Иприт прн комнатной температуре проникает через обыкновенную ткань обмундирования почти за 3 мин. Посредством нмпрегннровання текстильных тканей (ср. раздел 5.1.3,2) достигается фильтрующее защитное действие. Различают абсорбирующие н хемосорбирующие защитные пленки.
Методы нмпрегннровання сводятся к получению тех и других пленок, дополняющих действие друг друга н повышающих эффективность защиты. Хемосорбирующим действием обладают прежде всего соединения, содержащие активный хлор, например хлорамин-и N-хлоркарбамнд, которые и вводят в абсорбирующие защитные пленки. В качестве абсорбирующих веществ пригодны днбутнл-фталат, соли полимерных карбоновых кислот и др.
Защитное действие нмпрегннрованного обмундирования, включая ннжнее белье, ограничено. Оно зависит от концентрации ОВ ц от метеорологических факторов. Полная защита от воздействия газообразных ОВ, особенно от высокотокснчных, скажем, типа зарина, не гарантируется.
Сравнительно лучше защищает изолирующая защитная одежда. Ее изготовляют из материалов, как можно дольше препятствующих проникновению ОВ.
Поскольку такие защитные материалы должны быть универсальными, т. е. должны не только защищать от ОВ, но и быть механически прочными, огнестойкими, поддающимися дегазации и т. д., их изготовляют по усложненным рецептурам, которые,' разумеется, во всех странах держатся в секрете.
Для этой цели пригодны галогенсодержащие полимеры, например, полнхлорпропнлен, фторкаучук, натуральный каучук с наполнителями, смеси поливинилхлорида с бутаднен-акрилоннтрнльным каучуком.
212
5.6.2. Дегазация
Несмотря на различное химическое строение, все кожно-нарывные ОВ можно дегазировать одинаковыми дегазирующими средствами. Некоторое исключение составляет азотистый иприт, который не всегда количественно взаимодействует с обычными дегазаторами; при дегазации этого ОВ применяют удвоенное, по сравнению с нормальным, количество дегазатора.
Для дегазации кожно-нарывных ОВ пригодны дегазирующие средства окислительного и хлорирующего действия. Щелочные дегазаторы применяются лишь ограниченно. Если щелочные дегазаторы и вступают в количественные реакции с ОВ, то скорости взаимодействия в полевых условиях слишком малы.
Для дегазации наряду с хлорной известью можно использовать другие гипохлориты, моно- и дихлорамины, а для азотистого иприта, в частности, хлорамины и гипохлориты с высоким содержанием активного хлора, а также сульфурилхлорид.
Хлорную известь можно применять для дегазации местности, улиц и других поверхностей в сухом виде, ио лучше в виде суспензии. При средних плотностях заражения продолжительность дегазации составляет около 30 мин. Спустя зто время на зараженные участки можно вступать без средств защиты. Поскольку ниже 5 °C скорость реакции с хлорной известью мала, дегазацию местности зимой следует проводить смесью сульфурилхлорида с дихлорэтаном.
Участки местностя и улиц, зараженные азотистым ипритом, дегазируют хлорной известью или суспензией хлорной извести. Улицы можно дегазировать также разбавленными минеральными кислотами или растворами кислых солей. При этом все еще ядовитые солеобразные продукты смывают водой.
Боевую технику, вооружение и приборы дегазируют растворами моио- или днхлорамииа или щелочными растворами гипохлоритов. Вязкие ОВ обезвреживают только раствором дихлорамина в дихлорэтане. Этот дегазирующий раствор, .по сравнению с водным, обеспечивает значительно большую скорость реакции в гомогенных условиях.
Монохлорамин пригоден также для дегазации кожи, зараженной кожно-нарывными ОВ.
Защитное обмундирование дегазируют растворами хлорной извести или монохлорамина. Для этой цели ни в коем случае нельзя применять раствор днхлорамииа в дихлорэтане, так как некоторые защитные материалы неустойчивы к дихлорэтану.
Обмундирование, нательное белье и предметы вооружения можно дегазировать паро-аммиачным способом, либо длительным кипячением в растворе соды, иногда с добавкой стирального порошка. При этом следует учитывать, что алкилдихлорарсины реагируют только при продолжительном кипячении, а мышьяксодержащие остатки приходится удалять многократным полосканием,
213
С поверхности материалов и приборов, чувствительных к коррозии, кожно-нарывные ОВ удаляют растворителями.
Лабораторное оборудование целесообразнее всего дегазировать растворами дихлорамииа в дихлорэтане. Пригодны также растворы монохлорамина, перекиси водорода или азотной кислоты.
Галогенированные оксимы дегазируют упомянутыми выше дегазирующими средствами, однако из-за нх высокой летучести дегазация может и не потребоваться. В лаборатории пригодны растворы щелочей и минеральных кислот; в этом случае, конечно, следует ограничиться азотной кислотой, обладающей универсальным действием.
КОНТРОЛЬНЫЕ ВОПРОСЫ
I. Опишите действие кожно-нарывных ОВ на организм человека.
2. Что является причиной более быстрого гидролиза галогенированных тиоэфиров по сравнению с галогенированными эфирами?
3. Как получают бис-2-хлорэтиловый тиоэфнр из этилена и хлоридов серы?
4. Как, в силу своих физических свойств, ведет себя иприт после применения его па открытой местности?
5. Для чего и какими средствами увеличивают естественную вязкость иприта?
6,_Какие существуют точки зрения на поведение бис-2-хлорэтилового тио-эфира”в водных растворах?
7. Какими способами можно повлиять на гидролиз бис-2-хлорэтилового - тиоэфира?
8. Напишите уравнение реакции окисления галогенированных тиоэфиров.
9. Как проходит реакция бис-2-хлорэтилового тиоэфира в растворах гипохлоритов?
10. Почему хлорирование молекулы иприта можно использовать для дегазации и какие дегазирующие средства пригодны для этой Цели?
11. Объясните механизм реакции бис-2-хлорэтилового тиоэфира с монохлорамином Т.
12. Как реагирует бис-2-хлорэтиловый тиоэфир с треххлористым золотом и для чего применяют эту реакцию?
13. Что такое «кислородный иприт»?
14. Какая имеется связь между ипритом и азотистым ипритом с точки зрения их строения и физиологической активности?
15. Какими химическими свойствами обладают М-алкил-N, N-бис-(2-хлор-этил) -амины?
16. Исходя из величин давления пара (см. стр. 177), рассчитайте предельные концентрации трис-(2-хлорэтил)-амина в интервале температуры 0—25 °C и сравните эти результаты с данными для бис-2-хлорэтилового тиоэфира.
17. Почему трис-(2-хлорэти>л)'амин неустойчив во время хранения?
18. Как ведут себя N-алкнл-М, N-бис-(2-хлорэтил)-амины в водных растворах?
19. Сравните отношение иприта и азотистого иприта к окислителям. Какие можно сделать практические выводы?
20. Как ведут себя N-алкил-М, N-бис-(2-хлорэтил)-амины по отношению к хлорирующим агентам?
21. Как получают люизит?
22. Напишите уравнение реакции гидролиза люизита сильными щелочами.
23. Какими средствами можно эффективнее все’го дегазировать люизит?
24. Какой физиологической активностью обладают галогенированные оксимы?
25. Почему ниже 5 °C для дегазации кожно-нарывных ОВ вместо хлорной извести применяют смесь сульфурилхлорида с дихлорэтаном?
214
ЛИТЕРАТУРА
1. Meyer V., Chem. Вег., 19, 326 (1886).
2. В urbach J, В. E., Dermatologica (Basel), 22, 120 (1961),
3. S t a d e K,, Pharmakologie u. Rlinik Synh Gilte, Berlin, 1964.
4. S i n clair D. C., Brit. Med. J„ 1948(11), 290.
5. C. R. hebd. Seance Acad. Sci., 230, 600 (1950).
6. Bohme H„ Chem. Ber., 81, 123 (1948).
7. Cla rke H. T., J. Chem. Soc., 101, 1583 (1910),
8. Steinkopf, Chem. Ber., 53, 1007 (1920).
9. Per not B., Ann. chim., 12, 626 (1946).
10. J. Org. Chem., 27, 2651 1962).
II. Пат. США 2398480; С, A., 40, 3766 (1946).
12. Пат. США 2669587 (1937).
13. Пат. США 2691626 (1945).
14. Vigneaud V. du, Stevens С. М., J. Am. Chem. Soc., 69. 1808 (1947).
15. Bull. Soc. Sci; Cluj. Romm,, 9, 359 (1940),
16. R u e g g e be г g W. H. C,, Science, 105, 532.
17. J. Chem. Soc., 1932, 589.
18. J. Am. Chem. Soc., 70, 634 (1948).
19. Trans. Faraday Soc., 43, 42 (1947),
20. S a u n de r s В. C., J. Chem. Soc. (London), 1948, 699. .
21. Пат. США 2984584 (1963).
22. Пат. США 2968668 1963).
23. Пат. США 2988526 (1963).
24. Итал. пат. 366503.
25. Price С. С., Wake fie Id L. B„ J, Org. Chem., 12, 232 (1947).
26. Gas- und Luftschutz, 1938, 195.
27. Петров К. А., Ж0Х, 30 (92), 1909 (I960),
28. Goheen D. W., Bennett C. F., J, Org. Chem., 26, 1331 (1961).
29. Mitt. Geb. Lebensmittelunters. Hyg., 31, 115 (1940).'
30. Holst G., Swensk Kem. Tidskr., 53, 319 (1941).
31. R о s s W. C. J., Biochem. Pharm., 2, 215 (1959).
32. В r u ё г e P., В о u c h e r e a u P., J. Pharrri. chim., (8), 28, (130); 490 (1938).
33. Ann. Fisica chim,, 37, 478.(1941).
34. Sudd. Apoth.-Ztg:, 1949, 17.
35. J. Gen. Physiol., 29, 441 (1946).
36. M a 1 a t e s t a P., D’A r t r i B., Ricerea sci,, 22, 1589 (1952).
37. Woodward F. N., J.'Chem, Soc. (London), 1948, 35.
38. J- Chem. Soc. (London), 1948, 44.
39. Пат. США 2072348 (1934).
40. Ma son J. P., Gasch'P. J., J. Am, Chem. Soc., 60, 2816 (193$).
41. W a r d K. J. Am, Chem. Soc., 57, 914 (1935).
42. McCombie H., Purdie D., J. Chem. Soc. (London), 1938, 1217,
43. J. Am. Chem. Soc., 70, 1648 (1948).
44. С г a n e C W. Ry do n H. N., J. Chem. Soc.. (London), 1947, 527.
45, J. Org. Chem., 11, 550 (1946).
46. S a r t о г i M, F„ Chem, Rev., 48, 257 (1951).
47. Cont ar di A., Du mon tel E., Chimica et Industrie, 29, 169 (1947).
48. S t a h m a n п M. A., В e r g m a n n M,, J. Org. Chem,, 11, 586 (1946).'
49. J. Chem, Soc. (London), 1946, 827; J, Org, Chem., 12, 308 (1947).
50, Mann F. G. Baker F. C,, J, Chem. Soc. (London), 1957, 1881; J. Org. Chem.
12, 612 (1947).
51. Openshaw H. T., Todd A. R,, J, Chem. Soc. (London), 1948, 374,
52. Buscher H., Grim- und Gelbkreuz, Leipzig, 1923.
53. La CosteW., Ann., 208, 33 (1881); Dehn W. M„ Wilcox В. B, Am. Chem. J„ 35, 16 (1906); D e h n W. M„ Am, Chem, J., 40, 88 (1908).
215
54. Hewett C. L., J. Chem. Soc., 1948, 1203; J. Soc. Chem, Ind., 68, 258, 263 (1949).
55. J. Chem Soc. 1948, 1206; Whiting G. H., J. Chem. Soc., 1948, 1209,
56. Пат. США 2636893; пат. США 2636894.
57. Вег., 62, 1754, 1768 (1929).
58. Hackmann J. Th,, Chem. WeekbL, 31, 366 (1934).
59. Gryszkiewicz-trochimowski E., Bull. Soc. chim. France Mem,, 15, 597 (1948),
60. Z, Elektrochem. Angew. Phys. Chem., 53, 109 (1949). .
61, S e h e r A., Chem. Ber„ 83, 400 (1950).
62. Co-nrad R,, Militarwesen, 6, 586 (1962).
6. ОБЩЕЯДОВИТЫЕ ОВ
С тактической точки зрения общеядовнтые ОВ являются быстродействующими летучими ОВ.
Их токсическое действие заключается в ингибировании определенных специфических ферментов и во влиянии на некоторые процессы обмена веществ. Симптомы отравлений очень разнообразны, поэтому единая картина отравления, как в случае удушающих или кожно-нарывных ОВ, для этой группы отсутствует.
Все зти Соединения без исключения обладают определенными, неподходящими для ОВ свойствами, которые ставят под сомнение нх военное применение. Использование их возможно в результате усовершенствования технических средств применения и вообще методов применения, а также путем воздействия на их физико-хямн-ческие свойства. Наибольшее значение ямеет синильная кислота. За исключением тетраэтилсвинца, общеядовнтые ОВ являются ядами крови, которые с физиологической точки зрения следует отнести к группе «удушающих» веществ. Их общим симптомом является смерть от остановки дыхания.
Тетраэтилсвинец является нервным ядом, однако механизм его действия на организм иной, чем у фосфорорганических соединений.
Производство этих соединений химической промышленностью для покрытия нормальных мирных потребностей для некоторых веществ, например, для синильной кислоты н тетраэтилсвинца, за последние десятилетия небывало возросло, поэтому в мирное время не стоит даже учитывать нх производство как производство потенциальных ОВ.
6.1. СИНИЛЬНАЯ КИСЛОТА И ГАЛОГЕНЦИАНЫ
Высокая токсичность синильной кислоты побудила французов применить ее в 1916 г. в качестве ОВ под названием Forestite. Французский фармаколог Мажендье обратил внимание на исключительную ядовитость этого соединения, от одной капли которого, нанесенной на язык или конъюнктиву собаки, животное умирало на месте Ьоеле 2—3 вздохов.
Еще перед началом первой мировой войны Синильную кислоту неоднократно предлагали в качестве химического оружия, но до серьезного ее применения дело не дошло.
Результаты военного применения синильной кислоты не соответствовали представлениям, которые возникли на основании ее силь
217
ного токсического действия. Серьезные поражения не имели места, поскольку нельзя было создать достаточно высокие боевые концентрации. Во время первой мировой войны французы применили 4000 т синильной кислоты без заметного военного успеха.
Неудовлетворительные физико-химические свойства, особенно низкая плотность пара, вынуждала добавлять сравнительно более тяжелые вещества. Применялась смесь, содержавшая 50% HCN, 30% AsCl3, 15% SnCl4 и 5% СНС13 — так называемый винсенит, а также различные смеси синильной кислоты с треххлористым мышьяком (манганит) и др. Позднее применяли смесь синильной кислоты с хлорцианом (мангвинит).
Раздражающее действие хлорциана должно было заставить солдат снять противогаз и тем самым подвергнуть нх токсическому денствияю обоих ОВ. Впоследствии хлорциан применяли в смеси с другими веществами, особенно с треххлористым мышьяком, без синильной кислоты. Кроме хлорциана еще во время перлон мировой войны применяли бромциан и другие цианиды, например, бром-бензилцианид (раздел 3.1.3.1).
После первой мировой войны, особенно после 1939 г., были разработаны соответствующие средства применения сильной кислоты, при помощи которых за короткое время можно было у поверхности земли создавать достаточно высокие концентрации синильной кислоты, при которых все живое погибало бы в течение нескольких секунд. На фашистском полигоне (лагерь близ Мюнстера) был испытан один из выливных авиационных приборов для синильной кислоты, который с бреющего полета создавал столь высокую концентрацию синильной кислоты, что от нее не защищал имевшийся в то время общевойсковой противогаз.
Среди цианистых соединений синильная кислота еще сохраняет определенное» значение и для современного боя. Оиа и хлорциан стали незаменимыми полупродуктами крупной химической промышленности— в производстве органических нитрилов, которые являются исходными веществами для изготовления многих пластмасс и искусственных волокон, в металлургии как комплексообра-зователи, в производстве средств борьбы с вредителями сельского хозяйства, для получения аминокислот и т. д. В настоящее время в США синильную кислоту применяют в газовых камерах для приведения в исполнение смертных приговоров.
Все страны с ведущей химической промышленностью, в силу огромного развития последней, имеют в своем распоряжении соответствующие производственные мощности для получения синильной кислоты и галогенцианов,
6.1.1. Химическое строение и биологическая активность
Первым членом ряда нитрилов и изонитрилов является цианистоводородная (синильная) кислота. Она существует в двух таутомерных формах, как цианистоводородная I и как
218
изоциаиистоводородиая 11 кислоты: -и+ +н* _ +
HC^N [feN]- С=гК-Н +Н+ -Н*
I И
Следует полагать, что равновесие смещено в сторону нитрил ь-ной формы, и синильная кислота существует в этой форме.
Соединения, содержащие иитрильиую или изонитрильную группу, токсичны; более токсичны изонитрилы.
Замещение атома водорода синильной кислоты иа алкил или арил уменьшает ядовитость; так же влияет и замещение водорода на галоген нлн вторую нитрильную группу (дициан). Так, акрилонитрил менее токсичен, чем синильная кислота, причем и механизм биологического действия оказался иным. Пока этому нет объяснения. Симптомы отравления акрилонитрилом сопоставимы с симптомами отравления цианидами В результате введения атомов галогенов получают соединения с лакримогеиным действием. Хлор- или бромциаи еще остаются токсичными веществами, их относят также к общеядовитым соединениям. Однако они обладают сильным и чрезвычайно неприятным слезоточивым действием. Такой эффект, когда в результате введения атомов галогенов в соединение, содер-
жащее нитрильную группу, получилось слезоточивое вещество, мы уже наблюдали на примере бромбензилцианида.
Если атом водорода синильной кислоты замещен определенными фосфорорганическими остатками, как иапример, в случае N-диметиламидо-О-этилцианфосфата (табуи)
; ' (CH3)2Nx хО
С2Н5СИ XCN
f ill
то такие соединения более ядовиты, чем синильная кислота, при-£ чем механизм их действия на организм совершенно иной и это яв-у ляется причиной повышенной токсичности.
i Отравляющее действие синильной кислоты и ее солей (цианидов) одинаково. Отравляющее действие галогенцианов объясняет-' ся выделением цианистого водорода в организме, поэтому им i нужно приписать одинаковый механизм действия.
Общеядовитые цианистые соединения, имеющие значение как I ОВ, действуют цренмущественно как ингаляционные яды. В жид-* ком виде они способны диффундировать через кожу. Капля си-иильцой кислоты, попавшая в организм через слизистую оболочку . £ или через рану, при известных условиях убнвает человека. Сильная I' токсичность при действии через кожу установлена опытом на кры-саХ. Если хвост крысы окунуть в^синильную кислоту, то в резуль-*. тате резорбции смерть наступает в течение 10—70 мин.
; Пероральные отравления происходят в резлуьтате употребления в пищу содержащих цианиды продуктов питания или воды,
219
обычно это цианиды щелочных металлов. 11ри этом симптомы отравления, в сущности, такие же, как при отравлении через органы дыхания, но часто, в зависимости от принятого количества, отравление может быть более опасным.
Считается точно установленным, что механизм действия синильной кислоты заключается в необратимом ингибировании железосодержащих дыхательных ферментов. Вследствие сильного сродства циаинд-иона к иону Fe*3 цитохромоксидазы активность этого фермента уменьшается. В результате этого прекращаются процессы клеточного окисления, управляемые этим ферментом, которые составляют свыше 90% всей дыхательной деятельности клетки2.
Цитохромоксидаза является ферментом, содержащим железо-порфирин, в основе строения которого лежит скелет порфина
Железопорфирин координационно соединяется с цианид-ионом Подобно гемоглобину функцией этого фермента является обратимое связывание кислорода и двуокиси углерода. Цитохромоксидаза содержится во всех клетках (!).
Следствием блокирования фермента является недостаток кислорода в клетках. Обратимое связывание кислорода и двуокиси углерода дыхательным ферментом нарушается, поэтому подающийся в клетки кровью кислород уже больше не может ими восприниматься. Этим объясняется повышенное содержание оксигемоглобина в венозной крови, а отсюда н покраснение кожных покровов пострадавших.
Опытами на кошках установлено, что отравления . цианистым натрием зависят от степени ингибирования тканевого дыхания головного мозга. Для наступления смерти должно в среднем блокироваться 70% тканевого дыхания (доза 2—2,2 мг/кг). Если же оно блокировано лишь иа 40% (доза 1,7—1,8 мг/кг), то смерть не наступает3.
Прежде чем уничтожить клетки, кислородное голодание вызывает их раздражение. Этот период раздражения определяет различные картины отравления синильной кислотой. Особенно сильно поражаются наиболее чувствительные нервные клетки. Опыты на животных показали, что при повторном вдыхании воздуха с одинаковой концентрацией HCN нельзя было обнаружить никаких поражений центральной нервной системы. Поражения головного мозга появляются при чрезвычайно сильном отравлении4,
220
Организм способен самостоятельно обезвреживать некоторые количества цианидов, будь то посредством выдыхания или в результате образования тиоцианатов и циангидринов.
Смерть при отравлении синильной кислотой наступает в результате паралича дыхательного центра.
Отравление синильной кислотой происходит быстро. Отравленный очень скоро ощущает запах и вкус горького миндаля, а также металлический привкус во рту. В начальной стадии возникает чувство жжения в горле, ощущение окоченения и потери чувствительности нёба и языка, затем наступает затруднение и расстройство речи, гул в голове и в ушах, слюнотечение, тошнота, рвота и чувство слабости.
Вторая стадия начинается усиливающимся удушьем—так называемая астматическая стадия. Чувство стеснения в груди, страх, неуверенная походка, расширение зрачков, потеря зрения и слуха и потеря сознания сменяются тоническими и клоническими' судорогами с отделением мочи и дефекацией. Кожа и слизистая оболочка краснеют. Дыхание замедляется и затем прекращается вовсе. Сердечная деятельность еще продолжается несколько минут после остановки дыхания.
В зависимости от концентрации смерть при отравлении синильной кислотой наступает через 15—30 мин, абсолютно смертельны концентрации в 0,4 мг!л. При высоких концентрациях отравленные либо мгновенно падают замертво, либо шатаются, ловят ртом воздух, кричат (иногда до хрипоты), затем умирают через 3—5 мин после короткой агонии. Цвет кожи отравленных от красного до фиолетового. Такая же картина отравления проявляется при приеме через рот больших'доз цианида.
Спустя длительное время после перенесенных отравлений синильной кислотой, измеряемое годами, могут проявиться последствия, главным образом, неврологического характера.
* Галогенцианы как ингаляционные яды действуют подобно синильной кислоте, кроме того они раздражают глаза и органы дыхания.
6.1.2. Цианистый водород (сииильиая кислота)
Н—CsN
Военные обозначения: англ — VN; амер. — АС; франц.—Forestite
Синильная кислота была выделена шведским ученым Шееле в 1872 г. (Стокгольм) Предполагают, что он стал первой жертвой своего открытия, так как в 1876 г. внезапно скончался во время работы в лаборатории.
Синильная кислота как ОВ имеет определенные недостатки, однако их компенсируют ее высокая токсичность, способность к быстрому действию, дешевизна н простота производства. Все же применение синильной кислоты в современном бою сомнительно в виду наличия более ядовитых фосфорорганических соединений.
221
6.1.2.1. Методы получения. В виду большого промышленного значения синильной кислоты разработано и внедрено в промышленность большое число методов ее получения и производства.
Свободную синильную кислоту получают в лаборатории обычно из ее солей действием серной кислоты Преимущественно применяют цианистый иатрий и ферроцианид калия:
+ H2SO4
2NaCN —-—2HCN - Na2SO4
+3H2SO4
K4[Fe(CN)e] —— > 6HCN
— геЬСЦ; — 2K.JSO4
Из процессов промышленного производства следует привести лишь наиболее известные (см. схему 4)..
Схема 4. Методы промышленного производства синильной кислоты.
К ним относятся: прямой синтез из аммиака и окиси углерода; косвенный синтез из аммиака и окиси углерода через формамид; косвенный синтез из аммиака, окиси углерода н карбоната иатрия через цианистый натрий; каталитическое сожжение аммйачно-ме1-та но-воздушной смеси; получение нз карбида кальция н азота через цианамид кальция .и циаиистый кальций или натрий.
Другим немаловажным источником получения синильной кис ' лоты являются сильно загрязненные синильной кислотой газы, образующиеся при перегонке каменного угля. Синильную кислоту’ накапливают в соответствующих поглотителях и затем выделяют из иих или обычно переводят в Fe(CN)a.
222
Можно получать синильную Кислоту путем перегрева сйеколь-ной барды, содержащей до 4% азота в виде бетаина. Кроме синильной кислоты прн этом образуется аммиак, который удаляют из смеси пропусканием ее в серную кислоту (образование сульфата аммония).
Прямой синтез HCN из аммиака и окиси углерода проводят в присутствии соответствующего катализатора (активированный уголь, окислы тория, циркония, ванадия и др.) при 500—800 °C
NH3 + СО —> HCN + H2O
Косвенный синтез HCN из аммиака и окиси углерода сводится к образованию формамида, от которого каталитически (силикагель, А120з, фосфаты алюминия и др.) при 350—450°С (в некоторых процессах уже при 200 °C) отщепляют воду:
н\
NH2 ----► HCN
О'? —н2о
При получении цианистого натрия из аммиака и окиси углерода смесь обоих газов и водорода (NH3: СО: Н2 = 2 : 6 : 2) пропускают через нагретый до 620°C расплав углекислого и цианистого натрия (10: 1):
2NH3 + 2СО + Na2CO3 —> 2NaCN + СО2 + ЗН2О
Образующийся в небольших количествах (8%) цианат натрия полностью восстанавливается до цианистого натрия окисью углерода при 700 °C, поэтому выход сравнительно хороший.
По методу Андрусова смесь метана или других низших алканов (Ci — С5), аммиака и воздуха при 1000—1200 °C можно превратить в синильную кислоту на тех же самых контактах, которые применяются для сожжения аммиака (Pt, Pt — Ir, Os):
2CH4 + 2NH3 + 3O2 —► 2HCN + 6H2O
Этот процесс особенно широко применяют в странах с развитой нефтеперерабатывающей промышленностью. После удаления избытка аммиака синильную кислоту вымывают холодной водой, затем выделяют из воды и отгоняют.
Впоследствии этот метод был усовершенствован, после чего стало возможным получать синильную кислоту непосредственно из аммиака и метана, без добавки воздуха5. В противоположность первоначальному процессу в основу этого способа положена эндотермическая реакция; которую можно проводить в электрической дуге при атмосферном давлении:
CH4 + NH3 —> HCN + 3H2
При получении HCN из цианамида кальция его вводят в реакцию с углеродом и хлористым натрием при нагревании. В резуль
223
тате реакции образуется цианистый натрий с примесью цианистого кальция:
+ С + 2NaCl
CaCN2 ----ь Ca(CN)2 --2NaCN
—LaGIa
Из этих солей действием кислоты выделяют свободную синильную кислоту. Обычно этот метод служит для получения цианистого натрия.
Синильную кислоту можно получить непосредственно из цианамида кальция, окнси углерода и водорода с добавкой пылеобразного карбида в псевдокипящем слое:
CaNCN+CO + Ha —> 2HCN + СаО
. Содержание синильной кислоты в газообразных продуктах такое же, как и при прямом синтезе ее из окнсн углерода и аммиака6.
6.1.2.2. Физические свойства. Чистая синильная кислота —бесцветная, прозрачная жидкость, застывающая при —13 °C в волокнистую кристаллическую массу; т. пл. —15°C. Она обладает своеобразным дурманящим запахом, который в сильном разбавлении напоминает запах горького миндаля и часто, вследствие паралича обонятельных нервов, не чувствуется (!). Ниже приведены физич ские свойства синильной кислоты:
Т. кип., °C ................................. 25,6—26,5
Т. пл., °C....................................от —15 до — 14,5
Т. застыв., °C................................от —13 до —13,4
Давление пйра при .20 9С, мм рт. ст.......... 612
Максимальная концентрация при 20 °C, мг}л . . 837—1100
Относит, плотность
пара *.......................................... 0,947
жидкости ...................................... 0,6969
Критическое давление, ат . ............... 53,3
Критическая температура, °C........................ 183,5
Показатель преломления ............... 1,2675
Синильная кислота находится в жидком состоянии'в узком интервале температур. Из-за низкой температуры кипения и высокого давления пара она при обычной температуре очень летуча.
Приводим значения давления пара и максимальных концентраций при разных температурах (см. также рис. 7):
Температура, р. мм рт. ст. Максимальная концентрация, мг,'л Температура. °C р, ММ рт. ст. Максимальная концентрация, ’ мг/л
-10 165 20 612 837—1100
0 264 343 30 893 ОТО
10 410 — 40 1270 —
На воздухе капля синильной кислоты' испаряется столь быстро, что за счет поглощения теплоты часть капли застывает. Это малоподходящее для ОВ свойство, нз-за которого применение синильной кислоты в первую мировую войну оказалось столь неэффек-
224
Рис. 7. Зависимость давления пара синильной кислоты от температуры.
местности около 5 мин, в лесистой местности —
зимой — до 1 ч. Она пригодна только для заражения воздуха.
Газообразная синильная кислота обычно бесцветна.
Высокое давление пара синильной кислоты при хранении ее в герметически закрытых контейнерах ведет к значительному повышению внутреннего давления.
Учитывая ее объемный коэффициент расширения (в интервале температуры от —10 до 4~15РС ои составляет 1,9-10'3), жидкую синильную кислоту можно хранить и транспортировать в соответствующих контейнерах; например жидкую чистую синильную кислоту (99,5% HCN) в США перевозят в вагонах-цистернах.
Жидкая синильная кислота горит почти бесцветным или слегка окрашенным в фиолетовый цвет пламенем.
С водой сииильиая кислота смешивается во всех отношениях, ее можно выделить нз растворов перегонкой или пропусканием тока воздуха. Она легко растворима в органических растворителях — спиртах,
эфире, галогеиалкаиах, беизние и жидких углеводородах, фосфорорганических соединениях и в таких ОВ, как хлор-1 н бромциаи, фосген, а также треххлористый мышьяк. Подобные смеси в первую мировую войну 'применяли для повышения относительной плотности пара синильной кислоты.
Текстильные волокна и пористые материалы очень легко адсорбируют пары синильной кислоты. Проветриванием можно удалить лишь около 75% адсорбированной кислоты. Ниже приведены данные о способности некоторых материалов адсорбировать синильную кислоту:
Адсорбционная способность, мг HCN иг 100 г материала
Адсорбционная способность, мг HCN на 100 г материала
Резина......... 2
Полотно........ 13,6
Кожа................. 36s4
Конский волос . . 47,6
Перо..... 49,6
Фетр..... 74,4
Солома сухая . . 93
Солома влажная 126,3
Поверхности жидкостей, пищевые продукты, а также кирпич, бетон и древесина тоже адсорбируют пары синильной, кислоты. Интересно, Фго пары синильной кислоты очень сильно диффундируют через пористые стенки и другие подобные -материалы, Цианистый водород диффундирует даже через неповрежденную яичную скорлупу.
8 Зак. 448
225
бы с вредителями сельского хозяйства. Ее применяют так называемым циклонным методом, при этом наносят кислоту на носитель, обычно кизельгур. В качестве предостерегающих веществ к ней добавляют лакриматоры. Применяемый препарат циклон Б может содержать в качестве предостерегающего вещества метиловый эфир хлоругольиой или бромуксусной кислот, трихлоринтро-метаи, хлорциан, либо смесь этих веществ.
В фашистских лагерях смерти циклон Б служил для уничтожения людей. .
6.1.2.З. Химические свойства. В разделе 6.1.1. уже было сказано, что синильная кислота существует в двух изомерных формах, находящихся в равновесии друг с другом. Вклад изоциани-стоводородиой кислоты составляет не более 0,5%, поэтому для синильной кислоты, по-видимому, справедлива формулаН—CsN.
Синильную кислоту в отсутствие воздуха можно хранить долгое время — месяцами и даже годами. Однако иногда происходит ее разложение, сопровождающееся взрывом. Взрывы, происходящие во время хранения синильной кислоты, являются результатом быстрой, неуправляемой ее полимеризации. Тепло, выделяющееся в начальной стадии полимеризации, разлагает синильную кислоту до газообразных продуктов, следствием чего является повышение давления и взрыв. Полимеризации способствуют небольшие количества воды, аммиака и щелочей. Влажная синильная кислота в железных емкостях полимеризуется тотчас. Начало полимеризации; внешне проявляется в том, что прозрачная жидкость становится желтой, затем коричневой и, наконец, выпадает коричневый осадок, который превращается в буро-черную массу и заполняет весь объем жидкости.
Полимерная синильная кислота представляет собой конгломерат буро-черных, аморфных на вид хлопьев. При действии раствором щелочи выделяется аммиак и образуется раствор глубокой коричневой окраски. После появления желтой или коричневой окраски полимеризация может проходить в определенных условиях столь быстро, что обращение с таким образцом синильной кислоты может стать * опасным.
Для хранения жидкую синильную кислоту стабилизуют соответствующими веществами. Кислотные стабилизаторы — серная, фосфорная, уксусная и щавелевая кислоты и хлораигидриды, а при известных условиях также галогенцианы — стабилизуют синильную кислоту иа неограниченный срок. Их стабилизующее действие основано иа связывании аммиака или цианистого аммония, образующихся при аутокаталитическом гидролизе, а также иа подавлении ионизации синильной кислоты.
Синильную кислоту, стабилизованную минеральными кислотами, нужно постоянно контролировать, особенно, если она подвергалась воздействиям повышенной температуры. Контроль возможен посредством измерений температуры н электропроводно
226 ‘
сти — неуклонное повышение температуры или электропроводности указывает на развитие процесса полимеризации. Электропроводность достигает максимума с появлением окраски синильной кислоты, т. е. когда израсходован весь стабилизатор7.
В качестве стабилизаторов применяют также оксалаты кобальта и никеля, добавляемые в количестве 0,5—1%. Эти стабилизаторы особенно пригодны при хранении сииильиой кислоты в металлических емкостях. Их преимущество в том, что они не вызывают коррозии. В стальных баллонах с необработанной поверхностью синильная кислота разлагается. Из иных стабилизаторов следует назвать n-толуолсульфонат и р-нафтилсульфонат кобальта, фосфат никеля. Из металлов оказались 'пригодными серебро, медь, кобальт и никель. Обычно применяют различные сочетания стабилизаторов, например оксалат никеля и оксалат кобальта, которыми также покрывают изнутри металлические емкости8.
Кроме упомянутых выше металлов, жидкая синильная кислота ие действует на алюминий, циик и хромо-никелевую сталь. Сталь, литейный чугун, хромовая сталь и свинец разрушаются и непригодны для изготовления контейнеров для хранения. Применяются еще контейнеры из керамики и хлоркаучука.
Кислота HCN может реагировать двояко — как нитрил и как кислота. Это очень слабая кислота. Степень диссоциации 0,1 М раствора HCN составляет 0,001. Оиа вытесняется из солей уже при действии углекислоты воздуха:
+ СО2; + Н2О
2NaCN -----—------> 2HCN
—Na2CO3
Соли сииильиой кислоты, которые получают пропусканием цианистого водорода в растворы соответствующих гидроокисей, обычно хорошо растворимы в воде. Они столь же ядовиты, как и свободная кислота, и могут применяться в качестве ядов для диверсий. Цианиды щелочных металлов в водных растворах имеют щелочную реакцию.
Гидролиз. Синильная кислота гидролизуется в водных растворах уже при обычной температуре. Конечным продуктом гидролиза является муравьиная кислота:
+ н2о Нх +н2о Нх Нх
HCN NH2 ^С—ONH4 ^С—OH4-NH3.
ОТ
В процессе гидролиза водные растворы сииильиой кислоты перестают быть ядовитыми. Гидролизу способствуют щелочи и кислоты, особенно при нагревании. Жидкая синильная кислота реагирует энергично с растворами щелочей, а в закрытых сосудах— со взрывом.
Вода сииильиой кислотой почти не заражается'. По американским сообщениям, питьевую воду, содержащую 25 ли. д. HCN, можно после обычной водоочистки употреблять в пищу.
8*
227
Окисление. Синильная кислота мало устойчива к окислителям. При действии перекиси водорода образуется оксамид — неядовитое, кристаллическое вещество:
+ Н2О2
2HCN ----->- H2NCO—CONH2
Окисление перекисью водорода пригодно для дегазации небольших количеств воды, содержащей цианиды. Для дегазации 1 г NaCN требуется коло 2,8 г перекиси водорода (в пересчете на 100%-иую Н2О2). Наряду с другими продуктами при этой сильно экзотермической реакции образуются аммиак, окислы азота и, вероятно, азот; около 1,6% NaCN остается неокислеиным 9.
Галогенирование. Галогенирование синильной кислоты галогенами в водных растворах приводит к соответствующим галоген-циаиам:
HCN + С12 —> CICN4-HC1
Обычно галогенцианы получают из цианидов.
Образование комплексных соединений. Синильная кислота и многие ее соли являются отличными комплексообразователями. В щелочной среде они реагируют с солями тяжелых металлов, например, с сульфатами железа и меди, с образованием соответствующих комплексных соединений. Комплексные соединения дают также соли Ag, Au, Сг, Ni, Pt, Zn, Mo, W, Mn, Ir.
Комплексообразование объясняется тем, что образовавшиеся цианиды тяжелых металлов растворяются в растворах цианидов щелочных металлов и соединяются с ними:
1. 2CN”4-Fe2+ —> Fe(CN)2
4NaCN + Fe(CN)2 —> Na4[Fe(CN)el
11. 3CN’4-Fe3+ —> Fe(CN)3
3NaCN + Fe(CN)3 —> Na3[Fe(CN)el
Образование таких комплексов можно использовать для дегазации жидкой синильной кислоты и воды, содержащей цианиды, так как образующиеся соединения неядовиты и нелетучи.
В фильтрах противогазов для освобождения вдыхаемого воздуха от синильной кислоты обычно применяют соли меди и цинка.
Синильная кислота является очень реакционноспособным соединением, широко использующимся в препаративной химии. Большая часть этих реакций не представляет военно-химического интереса. Познакомиться с ними можно в учебниках органической химии. ‘
6.1.3. Галогеициаиы
Из четырех возможных, галогеицианов в первую мировую войну иашли применение хлор- и бромциан, обычно в смеси с другими ОВ, например с синильной кислотой или бромацетоном.
Вг—CN Cl—CN
(амер. — BK) * .(амер. — СК)
228
Для современного боя онн вряд ли еще могут иметь значение. Важнейшим представителем этих ОВ, который еще перед второй мировой войной состоял на вооружении армии США, является хлорциан. Фторциан, вероятно, более ядовитый, был получен в 1931 г. Косслеттом. Как показали дальнейшие исследования10, описанный способ синтеза фторциаиа вызывает сомнения. Известные в настоящее время методы получения этого соединения не пригодны для промышленного производства.
Бром- и хлорциан можно получить действием соответствующих галогенов на сииильиую кислоту
HCN + С12 —> Cl—CN + HC1
или пропусканием галогенов в водные растворы цианидов щелочных металлов:
NaCN + Ch —> CI—CN + NaCI
В промышленности хлорциан получают пропусканием хлора в распыляемый 15%-ный раствор цианистого натрия. Раствор NaCN приготовляют аналогично пропусканием HCN в тоикораспылен-ный раствор гидроокиси натрия11.
Физические и химические свойства. Хлорциан — бесцветная жидкость. Бромциан при обычной температуре — кристаллическое вещество (игольчатые кристаллы). Приводим физические константы хлор- и бромциана:
Cl-CN Br-CN •
Т. кип., °C ............................... 12,5 61,3
Т. пл., °C.........’......................... -6 52
Давление пара прн 20 °C, мм рт. ст. . . . 1002 120 (при 25 °C)
Максимальная концентрация, мг/л прн 15 °C ...............................2,6 103 1,5 • 102
(при 16 °C)
при 20 °C............................3,3-103
Относ, плотность пара................................ . 2,1 3,4
жидкости d* .......................... 1,222 2,02
(при 0 °C) (при 20 °C)
Оба соединения очень летучи. Значения давления пара хлор и бромциана при различных температурах следующие;
Температура Cl—CN Br-CN Температура, C1-CN Br-CN
-10 271 20 1002 —
0 444 21 25 —- 120
10 682 — 30 1427 —
15 ' — 63
Их растворимость в воде ограничена, но в некоторых органических растворителях (бензин, эфир, жирные спирты) Они хорошо растворимы. Оба смешиваются с другими ОВ, например с HCN, хлорпикрином; хлорциаи смешивается и с ипритом.
229
При хранении эти галогеициаиы полимеризуются подобно синильной кислоте. В отличие от синильной кислоты, полимеризации способствуют добавки кислот, остаточный хлор, вода и др. При этом образуются тримеры (CNX)3:
Хч N /X ЧС/
II I
С
X
при Х = С1 (т. пл. 145 °C) при Х = Вг (т. пл. 300 °C)
Для хранения необходимо добавлять стабилизаторы. Складские запасы нужно держать под непрерывным контролем.
Галогеициаиы следует рассматривать как галогеиаигидриды циановой кислоты НО—C=N, либо как продукты замещения водорода сииильиой кислоты галогеном, что находит отражение в их химических свойствах.
В воде при обычной температуре оии гидролизуются'с образованием неустойчивой циановой кислоты, из которой при действии галогеиоводородиых кислот получаются двуокись углерода и галогенид аммония:
“НО—C-N "
+ ИгО + Н2О
X—CN ----* || 4-НХ -----COs4-NH,X
_O=C==NH
Выше 100°С гидролиз протекает быстро.
С растворами гидроокисей щелочных металлов образуются ие- ' ядовитые циаиаты, вследствие чего реакция пригодна для дегазации:
+ 2НО“
XCN --------> OCN-
—X’; —Н2О
При действии аммиака в эфирном и водном растворах' количественно образуется цианамид: *
+ 2NH3
XCN NHi0N
Эту реакцию можно также использовать для дегазации.
Третичные амины при действии на них галогеициаибв разлагаются. Сначала образуются продукты присоединения, которые при нагревании разлагаются иа 'Диалкилцианамид и алкнлгалоге-иид:
+ XCN
R'—N—£N
Х~ CN + R"X
*230
Диалкилцианамиды легко гидролизуются с образованием вторичных аминов:
+ 2Н2О
RaN—CN —> R2N—СООН ---------> R2NH
—NH3 —СО2
Галогеициаиы присоединяются к пиридину. Продукт присоединения — галогенид цианпиридиния — при гидролизе расщепляется с образованием таутомерного цианамида глутакоиового альдегида» который гидролизуется далее до глутакоиового диальдегида:
+ Н2О
NC—NH—СН=СН—СН=СН—(Г
,0
/ОН'
«=> NC—N=CH— СН*=СН--СН=(/
°ч /ОН
^с—сн=сн—сн=с( н/ \ н
Это особый случай расщепления циклических дминов.
Циклические амины часто применяют в фильтрах противога-аор.
В США активированный уголь для противогазных коробок пропитывают производными пиридина, иапример, 2-амииопириди-иом, N-этилморфолииом, у-пиколином. Для улучшения поглощающей способности на уголь дополнительно наносят окислы меди, хрома, ванадия или молибдена12.
Галогеициаиы взаимодействуют с большей частью металлов, однако при этом .образуются защитные пленки, которые препятствуют дальнейшему разрушению. Особенно сильно их действие на медь, алюминий и цинк.
Подобно синильной кислоте, галогеициаиы образуют комплексные соединения с TiCU, SnCh, АиСЬ, SbCh ,и FeCU-
6.1.4. Токсические свойства
Рассматриваемые циаипроиз водные действуют как яды клеточного дыхания. Галогеициаиы обладают более или менее сильным слезоточивым действием, которого полностью лишена синильная кислота.
Отравление синильной кислотой может произойти при вдыхании паров, резорбции через кожу и приеме через рот. -
Обычно концентрации синильной кислоты ниже 0,05 мг/л неопасны. Даже после пребывания в такой атмосфере в течение 1 ч ни сразу, ни впоследствии признаков отравления ие наблюдается. При многочасовом вдыхании такая концентрация небезопасна и ведет к отравлению. Абсолютно безопасными при времени экспозиции 6 ч должны быть концентрации порядка 0,02 мг/л.
Концентрации в 0,08 мг/л HCN эффективны независимо от экспозиции. Пребывание в течение 15 мин в атмосфере,
231
содержащей 0,1 мг/л HCN, ведет к тяжелейшим отравлениям, иногда со смертельным исходом; при воздействии свыше 15 мин такие концентрации безусловно смертельны. Концентрации 0,2 мг/л при времени воздействия 10 мин и 0,3 мг/л при времени воздействия 5 мин также считаются смертельными.
Галогенцианы в малых концентрациях действуют раздражающе на слизистую оболочку глаз и верхних дыхательных путей. Порог раздражения для хлорциана составляет около 2,5-10-3 мг/л. При более высоких концентрациях наступают усиливающийся кашель, расстройства пищеварения, конъюнктивиты и т. д. Концентрации, вызывающие отравление при экспозиции 30—60 мин примерно равны таковым для синильной кислоты. При более кратковременном воздействии они выше, поэтому в общем можно утверждать, что хлорциан примерно вдвое менее ядовит, чем синильная кислота.
Так, смертельная концентрация хлорциана для времени воздействия 10 мин составляет 0,4 мг/л, для 30 сек — около 7 мг/л (прн этой экспозиции смертельная концентрация синильной кислоты составляет примерно 2 мг/л).
Смертельная доза синильной кислоты для людей составляет 1,0 мг/кг массы. Для NaCN оиа равна примерно 1,8 мг/кг, а для KCN около 2,4 мг/кг.
Через кожу синильная кислота всасывается как в газообразном, так и в жидком состоянии. При длительном пребывании в атмосфере с высокой (выше 0,5 мг/л) концентрацией синильной кислоты без защитной одежды, несмотря иа наличие противогаза могут произойти отравления в результате резорбции. Особенно опасно соприкосновение с жидкой синильной кислотой, однако такой случай в боевой обстановке мало вероятен.
Из несчастных случаев иа производстве известно, что при немедленном оказании помощи в первые минуты отравления и при последующем лечении оставались в живых даже те люди, которые были облиты синильной кислотой и в течение 1 ч не подавали признаков жизни. При запоздалом оказании помощи такие отравления обычно кончаются смертью.
Определенные количества цианидов детоксицируются и выводятся самим организмом.
6.2. мышьяковистый и фосфористый водород
Мышьяковистый и фосфористый водород менее /ядовиты, чем синильная кислота. Их неудовлетворительные физико-химические свойства не позволяют применять их как ОВ. Перед второй мировой войной не раз указывали иа их возможное использование, причем имелись в виду их металлические производные. Эти предложения, по-видимому, выразились в использовании металлфосфи-яов для борьбы с вредителями сельского хозяйства.
232
Физические свойства мышьякоиисют и цюсфирниш и Ви дорода следующие:
ASH3 РН3
Т. кип., °C............................ —55 —87,8
Т. пл., °C............................-113,5 —133,8
Относит, плотность пара............ 2,69 1,17
Мышьяковистый водород (арсин, AsH3) представляет собой бесцветный газ с запахом чеснока. Он в 2,7 раза тяжелее воздуха, плохо растворим в воде и щелочах. Растворимость в галогенал-канах, углеводородах и бензоле умеренная; с воздухом он дает взрывчатые смеси (4,5—68% AsH3).
Мышьяковистый водород — яд крови и нервный яд. В зависимости от концентрации и времени воздействия симптомы отравления наступают через латентный период от 2 до 15 ч\ при высоких концентрациях — через 30 мин.
Симптомы выражаются в оцепенении, ознобе, чувстве страха, тошноте, болях под ложечкой, рвоте, головных болях, жаре, желтухе. Верным признаком отравления является окрашивание мочи в результате распада кровяных телец, сначала, за счет гемоглобина — в красный цвет, затем за счет гематина — в темно-коричневый. В результате распада гемов (гемолиза) поражаются селезенка и печень (опухают). Причиной гемолиза является ингибирование каталазы в эритроцитах. Образующаяся перекись водорода разрушает кровяные тельца. Почечные каналы закупориваются и разрываются распавшимися кровяными тельпами, и функция почек ослабляется. Паралич центральной нервной системы в результате кислородной недостаточности в крови приводит через 2—4 либо через 8 суток к смерти.
Опасны концентрации мышьяковистого водорода выше 0,1 мг/л. Такие концентрации через 5—10 мин приводят к сильному отравлению, а при воздействии в течение 1 ч — к смерти. Смертельными являются 0,6 Aia/л при экспозиции 15 мин, 1,3 мг/л прн экспозиции 5 мин и 2—5 мг/л — через несколько вдохов.
Концентрации ниже 0,01 мг/л безопасны и даже после многочасового пребывания совсем не приводят к отравлениям либо к очень слабым.
Продолжительность латентного периода говорит о степени отравления. Если она составляет менее 3 ч, то следует ожидать тяжелого отравления, если 6 ч— то средней тяжести или легкого.
Мышьяковистый водород можно получить из трехокиси мышьяка и водорода
f
АзгО3 + 6Zn + 6H2SO, —► 2AsH3 + 6ZnS(\ + 3H2O
либо гидролизом арсенидов неблагородных металлов:
2АзА14-ЗНгО —> 2AsH?4rAl8O
233
на этой реакции основано возможное применение мышьяковистого водорода в виде арсенидов, которые затем при достаточной влажности воздуха разлагаются и таким образом создается длительное заражение.
Едкие щелочи разлагают арсениды.
Арсениды получают сплавлением мышьяка с металлами. Существующие в природе арсениды железа, никеля и кобальта очень устойчивы ц по своим свойствам соответствуют интерметаллическим соединениям. Для военных целей могли бы подойти арсениды щелочных и щелочноземельных металлов. Их можно дегазировать окислителями.
Фосфористый водород (фосфин, РНз) — бесцветный газ с запахом карбида кальция. При получении фосфина одновременно образуется днфосфин Р2Н4, бесцветная самовоспламеняющаяся жидкость.
Симптомы отравления фосфином примерно соответствуют симптомам отравления мышьяковистым водородом. Наиболее, явные из ннх — головная боль, одышка, слабость, головокружение и рвота; в тяжелых случаях — потеря сознания и расширение зрачков. Смерть от отёка легких и паралича сердечной мышцы наступает через несколько суток.
В несмертельных случаях отравления констатировали поражение печени и легких.
Концентрация фосфина в 0,2 жг/л в течение 1 ч переносима без последствий. Абсолютно смертельными являются концентрации порядка 3\иг/л. Неядовиты количества ниже 0,01 мг!л.
Фосфористый водород можно получить синтезом из элементов при высоком давлении и около 300 °C, либо из фосфора и водорода in statu nascendi
Р1в + зн РН3
либо путем гидролитического разложения фосфидов:
Са3Р2-|-бН2О —> 2РН3 + ЗСа(ОН)2
Особенно пригодны для этой цели фосфиды магния, кальция, алюминия и цинка.
Фосфиды можно получить взаимодействием фосфора с металлами или их солями. Военный, интерес представляют только солеобразные продукты (II-я и 111-я группы Периодической системы элементов). Фосфиды щелочных металлов слишком неустойчивы.
Для борьбы с вредителями сельского хозяйства предлагали фосфиды металлов обволакивать при нагревании в оболочку иа твердого парафина, воска и т. п. После измельчения на мельнице добавляют «запал», например карбамат аммония NH2COONH4 или NH4CN и формуют смесь прессованием. Формованную смесь можно хранить длительное время13. Очень возможно, что в таком виде фосфиды могут найти и военное применение.
234 >
6.3. ОКИСЬ УГЛЕРОДА И КАРБОНИЛЫ МЕТАЛЛОВ
Окись углерода до сих пор в качестве ОВ не использовалась, хотя оиа и обладает всеми необходимыми свойствами, которые могли бы обеспечить ее военное применение.
Преимущества окиси углерода заключаются, во-первых, в ее высокой токсичности, во-вторых, она лишена раздражающего действия, что увеличивает ее коварность. Оиа обладает известной химической стабильностью и требует специального противогаза, поскольку коробка обычного противогаза не задерживает окись углерода. Не последиим'ее достоинством является дешевизна и масштаб промышленного производства. т
Однако некоторые физико-химические свойства ие допускают применения окиси углерода как ОВ, иапример, высокая летучесть. Кроме того? по плотности окись углерода легче воздуха и ее очень трудно хранить. Для производственных и технических целей её хранят в стальных баллонах (под давлением; при обычной температуре окись углерода невозможно сконденсировать в жидкость.
В период между двумя мировыми войнами, преимущественно во Франции, проводились широкие исследования применимости окиси углерода в качестве ОВ, в том числе соединений, отщепляющих окись углерода (карбонилы металлов) и растворов окиси углерода.
Смесь пента карбонила железа и синильной кислоты считали особенно эффективной, так как оиа способна проникать через коробки обычных противогазов. Подобные смеси способствовали ослаблению поглощения других газообразных токсичных веществ и быстро исчерпывали защитную мощность поглотителя.
В распоряжении фашистского вермахта имелись фосген-карбонильные смеси, которыми были заполнены артиллерийские боеприпасы. Такие боеприпасы, найденные после войны, составляли небольшую часть запасов боеприпасов с ОВ. Оказалось, что уже малые концентрации таких смесей «пробивали» немецкий общевойсковой противогаз, даже с защитным слоем от окиси углерода (фильтр CO-FE), а стандартные СО-фильтры становились непригодными через короткое время.
При современном состоянии технических средств применения ОВ недостаточные боевые свойства окиси углерода могут быть компенсированы, поэтому ее использование в полевых условиях все же возможно.
В бою часто случались отравления окисью углерода посредством СО-содержащих газообразных продуктов, образующихся при стрельбе и взрывах; возможны и отравления при пожарах или выхлопными газами двигателей внутреннего сгорания. Немецкие фашисты использовали выхлопные газы для массового уничтожения людей; для.этого применяли специальные газовые камеры («душегубки»), установленные на шасси автомобиля.
235
6:3.1. Окись углерода
Окнсь углерода СО — газ без цвета и запаха. Он обладает следующими физическими свойствами:
Т. кип., °C............................. —191,6
Т. пл., °C.............................. —205,1
Относит, плотность пара............ . . 0,967
Плотность газа при 0 °C и 760 мм рт. ст. . 1,25001
Теплота испарения, кал]моль............... 1443
Критическое давление, ат.................. 34,5
Критическая температура, °C...............—140,2
Он мало растворим в воде (23,2 сЛ13/л), лучше — в органических растворителях (200 см3 в 1 л спирта). Особенно хорошо ои растворяется в сжиженных газах, например, в аммиаке и аминах. Считалось 14, что такие растворы можно использовать для применения окиси углерода как ОВ. В соответствии с критическими .параметрами окись углерода нельзя превратить в жидкость при обычной температуре и не очень высоком давлении. При охлаждении ниже—191,6°С и атмосферном давлении окись углерода превращается в бесцветную жидкость.
Смеси окиси углерода с воздухом взрывают. При комнатной температуре взрывоопасными являются смеси, содержащие от 16,2 до 73Д % СО.
Окнсь углерода легко диффундирует через пористые материалы н, следовательно, через' кирпичные стены, грунт и т. п. В литературе описано много случаев отравления в результате диффузии окиси углерода на большие расстояния от мест ее образования (разрывы газопроводов, неплотности и т. д.). Сродство окиси углерода к гемоглобину в 300 раз больше, чем у кислорода. Связь гемоглобина с окисью углерода прочнее, чем с кислородом. Образуется устойчивый СО-гемоглобин (СО-Hb), вследствие чего нарушается н даже прекращается нормальная’ функция гемоглобина, состоящая в обратимом связывании кислорода.
Окись углерода дезактивирует некоторые ферменты, содержащие порфириновое ядро и железо (гемины). Эти и все прочие факторы приводят к обеднению кислородом крови и клеток, особенно клеток мозга.
Отравление окисью углерода возможно только при вдыхании. Токсичность и признаки отравления у разных людей очень различны; это нужно отнести иа счет таких факторов, как различия в газовом обмене, индивидуальных особенностях (возраст, состояние здоровья, физическое и психическое состояние) и др.
Симптомы отравления соответствуют симптомам острой кислородной недостаточности — головокружение, слабость в ногах, тошнота, рвота, мелькание в глазах, ухудшение слуха, нарушения координации движений, потеря сознания, судороги, смерть от остановки дыхания. ,
236
В виду множества факторов, влияющих на отравление окисью углерода, Токсичность и симптомы отравления сопоставляют с содержанием СО-Hb в крови: содержание СО-Hb выше 60% —смертельно, концентрации 0,5 объемн.% СО (5 мг/л) при экспозиции 5—10 мин смертельны (содержание СО-Hb при этом равно 70%); концентрации 0,2 объемн. % СО (2,3 мг/л) при экспозиции 1 ч — опасны. Пребывание в атмосфере, содержащей 0,01 объеми.% СО (0,2 мг/л) при наличии физической нагрузки допустимо, самое большее, лишь в течение 1 ч, после этого появляются признаки отравления.
r-l I ' i-г—Г-1-Г-f-T "Г ' 1 I.-!—П—П-!-nPU nrJf,'ie
723^56783 70 72 7$ 76 78 20
" • j, >—r-rj,—;—I—з—'при pC!dome
“^7 J j J -т т-
время, v
Рис. 8. График токсичности окиси углерода.
На рис. 8 представлена зависимость между концентрацией окнсн углерода во вдыхаемом воздухе, содержанием СО-Hb в крови, временем экспозиции при различных физических нагрузках и токсичностью 15. Эта номограмма опубликована фирмой «Дрегерверке» и является результатом обработки литературных данных.
В качестве основы для графического выражения зависимости периодов экспозиции при разных режимах нагрузки приняты следующие объемы вдыхаемого воздуха: «при покое»—~ 5 л/мин; «при работе»— ~ 20 л/мин и «при тяжелой работе» — ~30 л/мин. Штриховкой обозначены границы областей токсичности, по-су-ществу, переходы от одной характерной картины отравления к другой. Для каждой данной концентрации СО кривая стремится к величине предельного насыщения крови (эти величины вычислены).
Эта номограмма позволяет судить о воздействии окиси углерод апри разнообразных режимах нагрузки и дает приближенную оценку максимально допустимой продолжительности ^безопасного пребывания в заданных пределах концентраций и времени. Кроме
237
того, она позволяет оценивать по данным , анализа возможную степень отравления.
Методы получения, химические свойства и применение окиси углерода, в общем, известны и здесь нет нужды их обсуждать. Ответы на все этн вопросы можно найти в монографии Шмидта16.
Вдыхаейый воздух освобождают от окиси углерода при помощи принятого в 1922 г. в США на вооружение гопкалита — смешанного окисного катализатора, состоящего из 60% двуокиси марганца и 40% окиси меди. Действие катализатора заключается в накапливании окиси углерода за счет адсорбции на пористой поверхности катализатора,.причем катализатор восстанавливается
СО + МпО2 —> СО2 + МпО
и регенерации катализатора в результате аутоокислеиия окружающим воздухом:
2МпО + Ог —> 2МпО2
Гопкалит пригоден также для аналитических определений окиси углерода.
6.3.2. Карбонилы металлов
Металлическими производными окиси углерода являются так называемые карбонилы металлов. Из множества известных карбонилов металлов военный интерес представляли пеитакарбонил железа и тетракарбоннл никеля, так как они легко разлагаются с отщеплением окиси углерода и их можно сравнительно легко получить. Оба карбонила применяются для получения чистого железа н никеля, так что для военных нужд нашлись бы определенные производственные мощности. Одно время пентакарбонил железа служил антидетонатором. Карбонилы металлов используются в некоторых процессах химической промышленности.
Физические свойства этих карбонилов следующие:
Fe(CO)5 . NI(CO)4
Т. кип.,°C..........; . . 102,7 42,8
Т, пл., °C -.......... -20(21,2) -25
Относит, плотность......1,466 (rf*8) 1,31 (^4°)
В воде оба карбонила нерастворимы.
Пентакарбонил железа, Fe(CO)s, — желтая жидкость со сравнительно высокой т. кип. (102,7°С) и низкой температурой застывания (т. пл. — 20°C). Летучесть при 18°С составляет около 310 мг/л. Выше 200 °C, а при обычной температуре — прн облучении или при контакте с активированным углем (!) пентакарбо-ннл железа разлагается на окись углерода и железо (окись железа) или, соответственно, на карбонилы железа Fe2(CO)g и Fe(CO)4.
В смесях с летучйми ОВ, например, с такими как синильная кислота иди фосген, его летучесть увеличивается, и это отрица-
238
тельно влияет на каталитическую активность гопкалита в коробке противогаза. Вледствие этого эффективность карбонила н других компонентов смеси значительно увеличивается.
Получают пентакарбоннл железа действием окиси углерода на тонкоизмельченное железо, либо непосредственно на железную руду илн железосодержащие исходные продукты прн избыточном давлении 100—200 ат и 150—200 °C:
Fe + 5СО —> Fe(CO)5
Пентакарбонил железа—сильный восстановитель, и как таковой разлагается окислителями. Он реагирует с хлором с образованием хлористого железа и окиси углерода:
Fe(CO)5 + С12 —> 5CO + FeCl2
При действии разбавленного раствора перекиси водорода выделяется коллоидное железо и образуется двуокись углерода:
Fe(CO)5 + 5Н2О2 —► Fe + 5СО2 + 5Н2О
Спиртовыми растворами пентакарбоннла железа можно восстановить, например кетоны в спирты, нитробензол в анилин. При взаимодействии с четыреххлористым углеродом наряду с железом или хлористым железом образуются фосген, окнсь углерода и гек-сахлорэтан.
Пентакарбонил железа вытесняет металлы из солей, например, серебро из азотнокислого серебра и палладий из хлористого палладия. С другими солями металлов, например с хлорной ртутью, иногда образуются комплексные соединения.
Тетракарбонил никеля Ni(CO)4 — бесцветная, не растворимая в воде жидкость, т. кип. 43 °C. Он обладает высокой летучестью, его пары в 6 раз тяжелее воздуха. Тетракарбонил никеля — неустойчивое соединение, которое постепенно окисляется на воздухе и выше 60 °C заметно разлагается.
В химическом отношении он ведет себя аналогично пентакарбонилу железа. Военное применение тетракарбонила никеля из-за его фнзнко-химическнх свойств представляется сомнительным.
Карбонилы железа и никеля действуют как ингаляционные яды. Вследствие своей высокой растворимости в липидах они могут проникать в организм и через кожу.
В некоторых случаях картина отравления и механизм токсического действия иные, чем при отравлении окисью углерода. Невидимому, нх отравляющее действие как ОВ зависит от того, вдыхают ли собственно карбонил металла, или продукт его разложения, т. е. окись углерода.
Уже в малых концентрациях пары карбонилов металлов поражают верхние и нижние дыхательные пути. Это выражается в рефлекторных явлениях — кашле, головных болях, головокружении н временном уДушьи. Повышенные концентрации затрудняют дыхание вследствие болевых ощущений в груди. Затем наблюдалось повышение температуры Тела, сильное головокружение, бред и
239
судороги. Через 10—15 ч после вдыхания развивается отёк легких, который при определенных обстоятельствах является смертельным.
Ответственным за механизм действия нужно считать блокирование некоторых ферментативных процессов, в которых принимают участие молекула карбонила и продукты ее разложения — очень реакционноспособный металл и окись углерода.
6.4. ТЕТРАЭТИЛСВИНЕЦ
РЬ(С2Н6)4
Военные обозначения; нем. — ВТА; амер, (англ.)—TEL.
Свинецорганические соединения все больше и больше применяются в современной химической промышленности в качестве алкилирующих агентов.
Важнейшим из тетраалкилпроизводных свинца является тетраэтилсвинец, который с 1923 г. нашел применение в качестве антидетонатора. За последние два десятилетия его производство небывало возросло. Свыше 90% расходуемого во всем мире горючего этилировано тетраэтилсвинцом.
После многочисленных случаев отравления, имевших место в 20-е годы, была установлена чрезвычайная токсичноть этого соединения, н поэтому тетраэтилсвинец вскоре стали считать возможным ОВ. Хотя для людей, лишенных средств защиты, тетраэтилсвинец является опасным ядом, его физические свойства не отвечают требованиям, предъявляемым к ОВ. В общем его военное применение отрицают, так как, во-первых, для защиты от него достаточно имеющегося противогаза, а во-вторых, для воздействия через кожу потребовались бы слишком высокие концентрации 17.
Исследования, продолжавшиеся до начала второй мировой войны, оставили открытым вопрос о возможности его военного применения. По .программе англо-американских военнохимнческих исследований область свинецорганических соединений была изучена систематически, в результате чего были получены соли триалкил-свинца, раздражающие слизистую оболочку (см. раздел 3.2.3) и подробно исследован тетракис-(2-хлорэтил) :свинец.
Физиологические свойства. Тетраэтилсвинец действует и как ингаляционный яд и за счет кожной резорбции. Это —опасный яд для центральной нервной системы. Он воздействует на нервные клетки коры головного мозга и вызывает сильные нарушения сосудистой системы, поскольку он поражает клетки гипоталамусной части межуточного мозга. Изменения высшей нервной деятельности в результате интоксикации тетраэтилсвинцом считают доказанными 18. Особая чувствительность нервной системы к действию тетраэтилсвинца объясняется легкой растворимостью тетраэтилсвинца в липидах. Он блокирует ферменты, причем образуются определенные стабильные комплексы свинца с белками,
240
В зависимости от концентрации и продолжительности воздействия признаки отравления появляются через 1 —12 ч, а при хронических отравлениях нли при однократном воздействии в малых концентрациях —через несколько суток.
Даже очень малые количества и те приводят к изменениям в головном мозгу, следствием чего является обычно продолжительная болезнь, заканчивающаяся смертью.
: Легкие отравления проявляются в головных болях, бессоннице,
кошмарах, потере аппетита, нарушениях координации движений, головокружениях, болях в области желудка, рвоте, двоенни в глазах, бледности и дрожании рук. Отравления длятся обычно несколько недель.
При тяжелых отравлениях симптомы выражены сильнее. Отравленный впадает в состояние возбуждения (психозы страха); он - проявляет пониженную активность, пребывает в сумеречном состоянии шизофренического типа, не контактабнлен, обливается потом, сознание нарушено, сильно теряет в весе. После возбужденного и сумеречного состояния обычно наступает смерть.
> Перенесенные отравления обычно оставляют последствия иевро-' логического типа.
' Методы получения. Крупное промышленное производство тетраэтилсвинца основано на этилировании хлористым этилом сплава свинца с натрием под давлением и при нагревании:
100 °C
4NaPb + 4С2Н5С1 --> Pb(C2H5)4 + ЗРЬ + 4NaCI
Стабилизованный высокомолекулярными соединениями, особенно ароматическими, тетраэтилсвинец очищают перегонкой. Выходы можно увеличить введением в реакционную массу треххлористого железа 19.
Из других процессов большое значение имеет разработанный ; Циглером и corp, электрохимический метод получения тетраалкил-J производных элементов главных подгрупп III — V групп и побоч-£ ной подгруппы II группы20.
| Этот процесс основан на электролизе соответствующих комплек-I. сов алюминия и щелочных металлов общей формулы MetAlR^) с применением анода из металла, алкильное производное которого
s нужно получить. Из щелочных металлов (Me) применяют смесь натрия и калия. Для получения тетраэтилсвинца используют свинцовые аноды, в качестве электролита — смесь калиевого и натрне-вого производных тетраэтилалюминия. Процесс протекает примерно по следующему уравнению:
1 РЬ + 4Ме{А1(С2Н5)4) —> 4Ме + 4А1(СгН5)з + РЬ(С2Н5)4
После отделения тетраэтилсвинца смесь электролитов после сравнительно простой переработки опять используют для электролиза,
241
Физические и химические свойства. Тетраэтилсвинец — бесцветная, легколетучая жидкость с приятным ароматичным запахом. Физические свойства его следующие:
Т. кип., °C при 13 мм рт. ст.......................
при 19 мм рт. ст...................
при 760 мм рт. ст..................
Т. пл., °C.............................
Давление пара при 20 °C, мм рт. ст. . . Максимальная концентрация, лг/л . . Относит, платность d™ ........ Показатель преломления. Пр . . ' . .
82-83
91-92
200 (разл.)
-130,2(-136)
0,261
4,6 (5,0)
1,6529 (1,650)
1,5198
Соответствующими стабилизаторами можно воспрепятствовать термическому разложению. Применение известных в настоящее время стабилизаторов дает возможность повысить температуры нагрева ОВ только до 160—180°С. Тетраэтилсвинец не застывает при охлаждении до —130,2 °C. Он во всех соотношениях растворяется в спирте, Эфире, ацетоне, бензине й других органических растворителях, растворим в животных и растительных жирах. В интервале О—35°С растворимость в воде доставляет 0,2—0,3 мг}л, с понижением температуры растворимость возрастает.
Давление пара тетраэтилсвинца (мм рт. ст.) можно рассчитать по формуле21:
1 а ЧЛбгr 2908,43 lg р = 9,34226--т
Тетраэтилсвинец очень устойчив, он относится к группе металлоорганических соединений, устойчивых к действию, воды. Водой он лишь медленно гидролизуется. Только при большом избытке воды последовательно отщепляются алкильные группы. Хотя ще- -лочи способствуют гидролизу, для целей дегазации эта реакция не имеет практического значения.
Дегазация тетраэтилсвинца, в результате которой он превращается в неорганические соединения, возможна при действии растворов хлористого или йодистого водорода в дихлорэтане илн четыреххлористом углероде. При этом тетраэтилсвинец количественно превращается в хлористый свинец. Особенно удобно применять для этой цели сульфурилхлорид в смесях с нефтью или дихлорэтаном.
Со свободными галогенами, например, с бромной водой, тетраэтилсвинец реагирует количественно. В случае концентрированных растворов тетраэтилсвинца реакция идет со взрывом. Бромная вода является подходящим дегазирующим средством для лабораторной посуды, в которой находился тетраэтилсвинец. Растворы хлорной извести также пригодны для дегазации,
242
Из других тетраалкилпроизводных свинца следует назвать: тетраметилсвинец, РЬ(СН3)4, бесцветная жидкость, т. кип. НО °C, т. пл. —30,3 °C; d? 1,995;
тетра&инилсвинец, РЬ(СН=СН2)4, т. кип. 50°C (4 мм рт. ст.)-, диэтилдивинилсвинец, (С2Н5)2РЬ(СН=СН2)2, т. кип. 30 °C (1 мм рт. ст.)\
тетракис-(2-хлорэтил)-свинец, РЬ (СН2СН2С1) 4.
6.5. БОЕВОЕ ПРИМЕНЕНИЕ ОБЩЕЯДОВИТЫХ ОВ
Большая часть соединений, рассмотренных в предыдущих разделах, применимы в качестве ОВ только в’виде смесей или раствр-ров. Эти вещества уже нельзя причислить к ОВ первостепенной важности, поскольку в настоящее время на вооружение приняты высокотоксичиые фосфорорганические соединения.
Из рассмотренных веществ наибольшее значение имеет синильная кислота. Из-за быстроты ее действия внезапное применение синильной кислоты в современном бою и при подготовке наступления может оказаться решающим с тактической точки зрения. Средства применения HCN в имлериалистических армиях значительно усовершенствованы по сравнению с имевшимися во время первой мировой войны. При их помощи за короткое время можно создать в воздухе концентрации порядка г/м3 воздуха. Чрезвычайно высокие концентрации синильной кислоты абсолютно смертельны и сильно истощают поглотитель противогаза (!).
Хотя синильная кислота по плотности легче воздуха, концентрации её, создаваемые с помощью современных средств применения, обеспечивают стойкость заражения воздуха да же летом самое большее на Юлин, а зимой при благоприятных условиях местности (растительность)—на период до 1 ч.
Синильной кислотой можно заражать только воздух. Ее могут применять при помощи специальных средств, к которым прежде всего относятся реактивные снаряды, бомбы и выливиые авиационные приборы. Возможно применение одной синильной кислоты либо ее смеси с соответствующими добавками, которые увеличивают продолжительность действия; эти добавки в то же время долины способствовать снижению поглощающей способности противогаза.
Сильное слезоточивое действие хлорциана является большим недостатком этого сравнительно токсичного соединения, так как служит индикатором н предупреждает атакованных.
Применение быстродействующих ОВ в современном бою имеет целью быстрое уничтожение или подавление противника. Предпосылкой этому является не только внезапность нападения, но и трудность распознавания примененного средства. Оно должно быть эффективным, не вызывая предварительных защитных рефлексов. Хлорциан таким требованиям не удовлетворяет, поэтому его боевое применение представляется спорным.
243
Указанным условиям почти идеально удовлетворяет окись угле- | рода. Отсутствие запаха, высокая токсичность, коварность дей- л ствия, потребность в особых средствах защиты (специальные по- I
глотители) и прочие высокие боевые качества окиси углерода ди- i
скредитируюгся, однако, большой летучестью этого вещества и ; другими неблагоприятными физико-химическими свойствами. ;
Боевое применение окиси углерода ие исключено, однако ие как таковой, а в виде веществ, отщепляющих окись углерода, или * их растворов, например в виде раствора пентакарбонила железа « в синильной кислоте, который был предложен и испытан. j
Отравления окисью углерода в боевых условиях являлись частой причиной потери боеспособности и наступали в результате I
воздействия содержащих окись углерода пороховых газов и -про- i
дуктов взрыва, так называемая «пороховая болезнь»? Пороховые газы содержат до 40 объемн.% СО. Опасность скопления СО су- | ществует особенно в закрытых помещениях. Путем определенного | сочетания видов взрывчатки, вероятно можно увеличить содер- j жание окиси углерода в газовом балансе продуктов взрыва, ио ; отнюдь не заразить воздух преднамеренно. В этой связи интерес представляет взрывчатое вещество из смеси тетранитрометаиа и | тетраэтилсвинца22. При соответствующем соотношении компоиеи- | тов смеси ожидают полного превращения всего имеющегося угле-рода в окись углерода. ?
Газообразные мышьяковистый и фосфористый водород почти ие пригодны в качестве ОВ. Применение их в бою возможно только в виде легко разлагающихся металлических производных, которые, разлагаясь, заражали бы на длительиое'время слой воздуха, иахо- дящийси в непосредственной, близости от почвы. Считают, что мышьяковистый водород можно применять в бомбах большого калибра. * • .
Современные средства обеспечивают боевое применение тетраэтилсвинца в качестве ОВ. Это — чрезвычайно сильный яд, который может применяться в соответствующих тактических смесях, чему благоприятствует его легкая растворимость почти во всех ОВ. Его исключительно коварное, обычно необратимое отравляющее ; действие заставляет считать тетраэтилсвинец страшным оружием. .
6.6. ЗАЩИТА ОТ ОБЩЕЯДОВИТЫХ ОВ И ИХ ДЕГАЗАЦИЯ i
За исключением тетраэтилсвинца, общеядовитые ОВ — соединения с низким молекулярным весом. В газообразном состоянии оии мало или совсем ие адсорбируются активированным: углем. Особенно низка адсорбционная способность активированного угля по отношению к окиси углерода и синильной кислоте. В случае мышья- ковистого и фосфористого водорода она также недостаточна. Очистка воздуха от этих газов даже при помощи хемосорбционных слоев в обычных противогазных коробках неудовлетворительна и почти не является защитой.
244
Синильную кислоту, присутствующую во вдыхаемом воздухе, связывают химически путем образования 'комплексных соединений с солями цинка и меди; окнсь углерода, мышьяковистой и фосфористый водород удаляются в результате каталитических процессов. Для очистки вдыхаемого воздуха от окиси углерода служит гопкалит.
Эти разнообразные свойства, которыми должен обладать поглотитель в противогазовой коробке, чтобы стать универсальным, отрицательно влияют на защитную мощность противогаза и способствуют быстрому истощению поглотителя или соответствующего слоя. Поглотительная коробка МО-4, а также противогазы специального й особого назначения удовлетворяют этим высоким требованиям и защищают от таких ОВ.
Во всех случаях при длительном пребывании при высоких концентрациях синильной кислоты или окиси углерода рекомендуется применять изолирующие противогазы, например противогаз ИП-46.
Синильная кислота и тетраэтилсвинец впитываются через кожу. При работе с этими веществами безусловно требуется защитная одежда. Защитную одежду совершенно необходимо надевать при работе с тетраэтилсвинцом, так как его пары также легко проникают через кожу. В боевых условиях вряд ли следует ожидать концентраций синильной кислоты, способных действовать через кожу.
Защитную одежду, зараженную тетраэтилсвинцом, нужно дегазировать, так как тетраэтилсвинец, особенно в жидком виде, очень быстро диффундирует через резину и каучукоподбные материалы. В промышленности для защиты от тетраэтилсвинца пользуются специальной защитной одеждой, которая содержит определенные наполнители и окрашена в белый цвет, чтобы загрязнения были лучше заметны.
Дегазация общеядовитых ОВ на местности не нужна ввиду их высокой летучести, она требуется в закрытых помещениях, блиндажах и т. д. Особенно в случае «тяжелых» газов, таких как мышьяковистый и фосфористый водород, она становится крайне необходимой. Обычно достаточно уже основательного проветривания. Ввиду сильной склонности пористых материалов к адсорбции синильной кислоты следует принимать соответствующие меры, чтобы затруднить последующую десорбцию газа. Помещения, зараженные синильной кислотой, можно дегазировать опрыскиванием формалином.
Для впитывания больших количеств жидкой синильной кислоты или тетраэтилсвинца применяют песок, кизельгур, опилки и др. Впитывающий материал либо дегазируют дегазирующими средствами, либо оставляют на воздухе до окончания десорбции. Остатки синильной кислоты и растворы, содержащие цианиды, следует дегазировать растворами сульфата железа, а затем обработать растворами щелочи.
245
Тетраэтилсвинец дегазируют иа местности и предметах снаряжения раствором сульфурилхлорида в дихлорэтане (1:1), керосине или других растворителях. Растворы хлориой извести и растворы галогенов, например, хлорная и бромная вода, пригодны только для дегазации лабораторных приборов.
КОНТРОЛЬНЫЕ ВОПРОСЫ
I. Чем отличаются между собой по ядовитости синильная кислота, хлоринам и табун, и как это можно объяснить?
2. Чем объясняется сильное токсическое действие синильной кислоты?
3. Назовите промышленные способы производства синильной кислоты.
4. Напишите уравнение реакции получения синильной кислоты из аммиака и окиси углерода через формамид.
5. Чем объяснить неустойчивость синильной кислоты на местности?
6. Почему нужно постоянно проверять синильную кислоту во время хранения?
7. Назовите способы стабилизации синильной кислоты.
8. Как реагирует синильная кислота с водой?
9. Почему воду, содержащую цианиды, можно дегазировать сульфатом железа с последующей обработкой едким натром?
10. Какое вещество очищает вдыхаемый воздух от синильной кислоты в хлорциана в противогазовой коробке?
11. Каким путем можно было бы применять мышьяковистый и фосфористый водород как ОВ?
12. Как протекает отравление окисью углерода?
13. На чем основано применение гопкалита как поглотителя?
14. Перечислите признаки отравления тетраэтилсвинцом.
15. Почему окись углерода могла бы быть «идеальным» ОВ и .почему она таковым не является?
ЛИТЕРАТУРА
1. Pau let G., Desflos L., Arch, intern. Pharmakodyn. Therap., 131, 54 (1961).
2. P au 1 et G., Rev. de Corps de Santfc (Paris), 3, 971 (1962). ,
3. К в а с e и к о О. Ф., Фармакол. и токсиколог., 25, 74 (1962).
4. Levine S., Weinstein В., J. Am. Pharm. Assoc. Sci. Ed., 48, 224 (1959).
5. E n d t e r F., Chem. Ing. Techn., 30, 305 (1958).
6. Пат. ФРГ 938125 KI. 12k' (1952).
7. S p о r z у n sk 1 A., Salter H. L., Rec. trav. phim., 69, 619 (1950).
8. Англ. пат. 608885 (1948).
9. В г о u c e k J., Korose Ochrana Mater, 1962, 95.
10. J. Chem. Soc., 1953, 3709.
11. Пат. США 3011864 (1959).
12. Пат. США 2963441 (1944).
13. D. A. S. 1127633; D. A. S. 131943 K.1.45I (i960).
14. Ha nne H„ Ind. Chim., 22, 322 (1985).
15. Dragerwerke Lubeck, Mitteil. zum Gasspiirgerat, 19/31, 30, Folge. (Sept., 1959).
16. Schmidt J., Das Kohlenmonooxid, Leipzig, 1950.
17. Mielenz W_, Gas- und Luftschutz, 2, 13 (1932).
18. Весников А. Б., Толгская M. С., Арх. пат. 13, 100 (1951).
19. Авт. свид. 131350 (1959); Бюлл. Изобр. № 17, 20 (I960).
20. Ziegler К., Angew. Chem., 67, 4£4 (1955); D. A. S. 1153754 KI. 120 (1958). ’
21. Feldhake C. J., Stevens C. D., J. Chem. Eng. Data 8, 196 (1963).
22. Пат. США 2486773 (1945),
7. ФОСФОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
Фосфорорганические соединения стали известны в прошлом веке. Свыше 100 лет назад в лаборатории*Вюрца был получен тетраэтилпирофосфат — соединение, токсичность которого была обнаружена лишь в 30-ые годы нашего столетия. В конце прошлого века Михаэлис получил ряд соединений, которые по своей структуре были аналогичны полученному позднее Шрадером ОВ, носящему название табун. Впервые (1932 г.) о токсических свойствах группы соединений из класса фосфорорганических производных сообщили Ланге и фон Крюгер1. Они установили, что пары алкиловых эфиров моиофторфосфориой кислоты уже в очень малых количествах после вдыхания в течение нескольких минут вызывают многочасовое удушье, легкое затемнение сознания и «явления слепоты с болезненно повышенной чувствительностью глаза к свету».
В Германии систематические исследования под руководством Шрадера, сотрудника концерна «ИГ-Фарбениидустри», привели к открытию высо'котоксичных фосфорорганических соединений. В условиях строжайшей секретности в рамках немецкой военно-химической программы были созданы предпосылки для промышленного производства некоторых соединений, пригодных в качестве ОВ.
В начале исследований проявляли особый интерес к алкиламидоацилфосфатам, из которых Ы,Ы-диметиламидо-0-этилцианфос-фат (табун) был рекомендован в качестве ,• ОВ. Планомерное изучение производных этого типа привело к группе эфирогалоид-ангидридов алкилфосфоиовых кислот, среди которых самыми токсичными представителями оказались изопропилметилфторфосфонат (зарин) и втор-неогексйлметилфторфосфоиат (зоман).
Основы для промышленного производства табуна были заложены уже в 1939 г. Немедленно было начато строительство почти полностью автоматизированной производственной установки в Ди-хернфурте на Одере. Одновременно там же были построены вспомогательные цеха для получения полупродуктов — хлора, хлоридов фосфора, диметиламииа и т. д. В 1942 г. строительство завода было завершено. По имеющимся данным, в Дихернфурте до конца войны было изготовлено 12 000 т табуна. Позже на этом же заводе была создайа установка для производства зарина, которая в июне 1944 г. начала давать продукцию.
Независимо от работ, проводившихся в Германии, к началу второй мировой войны в Англии, а затем и в США были начаты
247
военно-хнмические исследования в области фосфорорганических соединений. Пригодным в военном отношении оказался диизопро-пилфторфосфат (ДФФ), промышленное производство которого также было налажено.
Когда после окончания второй мировой войны стали известны результаты исследований фосфорорганических соединений, работы в этой области интенсифицировались либо из чисто военных соображений, либо для того, чтобы среди множества таких соединений выбрать представляющие интерес для мирных нужд. К настоящему времени получены средн соединений этого типа эффективные инсектициды, смазкн и т. п.
Область фосфорорганических соединений развилась в особую отрасль химии, стали известны десятки тысяч соединений, как токсичных, так и нетоксичных. Их фармакологические свойства привлекли внимание не только военных токсикологов, но и биохимиков, врачей и фармацевтов. С помощью фосфорорганических соединений удалось получить новые сведения о передаче раздражений нервной системой и объяснить определенные закономерности в действии некоторых биокатализаторов. Наконец, это привело и к тому, что были Найдены еще более токсичные соединения, которые по своему строению подобны природным субстратам. За последние годы средн таких соединений были обнаружены ОВ, во много раз более токсичные, чем табун нлн зарин.
Развитие хнмни фосфорорганических соединений отнюдь не завершено. Среди этих соединений можно ожидать появления еще более токсичных веществ. Большие возможности комбинаций, заметное изменение свойств при небольших изменениях строения создают богатое поле деятельности для военных химиков, фармацевтов; врачей, биологов, физиологов н др.
В настоящее время табуи можно считать уже классическим'ОВ; на первый план выдвинулись некоторые представители ряда фос-форилтиохолинов. Не исключено, что впредь мы встретим среди фосфорорганических соединений еще более токсичные вещества,
7.1, КЛАССИФИКАЦИЯ И ЙОМЕН КЛАТУРА ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Химия фосфорорганических соединений за последние годы стала особой областью органической химии, а также биохимии; общее количество производных фосфора практически необозримо.
Развитие химии фосфорорганических соединений все больше и больше вызывает необходимость введения единой номенклатуры этих соединений. Этот уже Часто ставившийся вопрос пока не привел к единой точке зрения, поскольку мнения отдельных исследователей об основах такой номенклатуры часто очень сильно расходится друг с другом.
В различных странах существуют свои принципы номенклатуры фосфорсодержащих соединений, в которых в значительной степени отражены вкусы самих исследователей н обозначения, при
246
меняемые в их публикациях. В 1952 г. американские и английские ученые пришли к соглашению о единой системе номенклатуры, которая, одиако, не получила признания за пределами этих стран. Так, например, номенклатура, принятая в Западной Германии, значительно отличается от нее. Это в равной мере относится к системе наименований, предложенной шведскими учеными2.
В данной книге будет использована в основном американо-английская номенклатура, принятая в монографиях и учебниках по органической химии*.
Наиболее часто встречающиеся названия основных представителей соединений этого класса приведены в таблице на стр. 250.
Следует заметить, что для многих фосфорсодержащих соединений стали общеупотребительными н, как правило, вошли в разные языки тривиальные названия н особенно сокращения, возникшие от химических названий.
Различают органические соединения трех- и пятивалентного фосфора. Поскольку трехвалентные фосфорсодержащие соединения практически не представляют военного интереса, в дальнейшем оии не будут рвссматриваться.
7.1.1. Фосфорорганические соединения — производные кислот пятивалентного фосфора **
Как правило, в этой группе фосфорорганических соединений встречаются следующие три основных типа:
RO\PZ
RCT
I
К типу I относятся соединения, у которых пять валентностей атома фосфора связаны с кислородом (фосфаты). К типу II относятся соединения, у которых одна или больше валентностей фосфоре связаны с углеродом (фосфоиаты, фосфинаты, окиси фосфинов). К типу III — соединения, у которых одна или, самое большее, две валентности атома фосфора связаны с кислотным остатком (ацилом); у атома фосфора могут находиться еще и алкильные группы (ацилфосфаты, ацилфосфоиаты, ацилфосфинаты). 4 Все описанные соединения вместо алкнльиых илн алкоксигрупп могут содержать арильные или арокснгруппы.
* Для детального ознакомления с этой номенклатурой можно рекомендовать книги: Р. О’Брайн, Токсические эфиры кислот фосфора, перев. с англ., Изд. «Мир»; 1964 и Д. Пурдела, Р, Вылчану, Химия органических соединений фосфора, перев. с рум., Изд. «Химия», 1972. — Прим, ред:
** Имеются в виду только соединения, рассматриваемые или упоминаемые в следующих разделах. — Прим. ред.
249
Кислота
формула название общее название
НО^ нет -он Фосфористая Фосфиты Phosphite
НОч но/Р \он Фосфорная Фосфаты PhospEate
R\ ЧР нс/ —он Алкилфосфон-нстая Фосфониты Phosphonite
R\ хр нс/ \он Алкнлфосфон-ов'ая Фосфонаты Phosphonate
R\ хр-r/ -он Ди алкилфосф инистая Фосфнниты :Phosphinite
R\ ' ^0 юн Ди алкил фосфиновая Фосфинаты Phosphinate
• •
Производные кислот
галовд ангидриды
формула название английская транскрипция
(RO)2PC! Ди ал кнлхлор фосфит Dialkylphosphorochfo-
ridite
Дналкилхлорфосфат Dialkylphosphorochlori-
(RO)2p< ХС1 date
RPCIs Алкилдихлорфосфин Alkyl dichlor phosphine
СНз^^О Z-CsHtCZ Изопропиловый эфир метилфторфосфоно-вой кислоты, изопропил метилфтор фосфо-нат Isopropyl ester rtiethyf-phosphonfluoridic acid, isopropyl methyl-phosp honf luori date
R2PCI Диалкилхлорфосфин Di al kyl chlorphosphine
^0 R1PXc Хлорангидрид ди а лкилфосфинов ой кислоты Dialkylphosphinic chlo-ride
I. Фосфаты. В зависимости от числа алкоксигрупп встречаются моно-, ди- или гриалкилфосфаты. Приведем некоторые примеры:
(С2Н6О)2Р^
диэтил-(4-нитрофенил)-фосфат; £-600
(С1СН2СН2О)3Р==О (СН3СвН4О)3Р=О трис-<2-х лорэтил )-фосфа т трис-о-то ли лфосф а т;
трнс-(2-крезил)-фосфат
(С2н5О)2р^°
\он
диэтилфосф ат
Соединения, содержащие группу фосфатов, например:
NR2, носят название амидо-
(СНзМ\ /р с2н5сх \освн5
N, N - диметила м идо-О-этил-О-феиилфосфат
(CH3)aNx
(CH3)2N/ \ос2н8
6hc-(N, N-диметиламидо)-О-этилфосфат
. II, Окиси фосфинов, фосфинаты, таких соединений следующие:
фосфориты. Общие формулы
Р3Р=О
окиси триалкилфосфиноз
О-a лкн лд«а лки лфосф ин аты
О, О-диялкилалкилфосфоиаты
III. Ацилфосфонаты и ацилфосфаты. Общие формулы этих соединений следующие:
диалкила цилфосф ат
О-алкилалкилацилфосфонат
Аминопроизводиые ацилфосфатов носят следующие названия;
N, N-диалкиламндо-О-алкялацилфосфат
6hc-(N, N-ди алкил амйдс)-ацилфосф ат
Соединения, в которых Х-галогены или CN-группы называют, например, так:
RO
R
О-a лки л а дки лх л ор(фто р)фосф о н а т ы
ди а л ки лф то рфос ф а ты
N, N-диалкиламидо О-алкилцианфосфаты
К числу ацилфосфатов относят не совсем строго фосфорилхоли-ны и фосфорилтиохолины, хотя остаток холина не является ацилом. В качестве примеров можно привести:
СНзЧ хО
C2HSOZ \OCH2CH2N(CH3)2
этокс имети лф осфори лхо ли и
С2Н5Ск .о
С2Н3о/ \>CH2CH2N(CH3)2
диэто ксифосфори лхо л ин
Если в молекуле кроме того имеется еще одни ацил, то название этого ацила пишут перед окончанием фосфърилхолин:
СН3. .0 ХР
f/ ZOCH2CH2N(CH3)2
м етилфторфосфори лхолин
Соединения, содержащие связь Р—S. Если вместо связи Р—О имеется связь Р—S, такие соединения называют аналогично: тиофосфаты (соответствуют фосфатам); ацилтнофосфаты (соответствуют ацнлфосфатам), фосфинсульфиды (соответствуют окисям фосфинов).
В том случае, когда атом серы образует связь P=S вместо связи Р —О (это не относится к фосфинсульфидам), применяется суффикс тиои. При наличии Р—S-связен каждая нз них указывается в названии.
Некоторые примеры поясняют принципы номенклатуры таких соединений.
R0\pz°
R0Z ZSR
О, О-диалкил-Э-алкил тиофосфат
Р\ Л X '
RSZ Z0R
О-алкнл-S, S-диалкнлдитиофосфат
252
Если связь Р=0 замещена связью P=S, то применяются следующие названия:
(RO)3P=S (RO)2p;
XSR триалкилтионфосфат О, О-диалкил-З-алкилтионфосфат
*\ z roAor О, О-диалкилалкилтионфосфонат
Общие формулы ацнлтно- н ацнлтнонфосфатов таковы:
RCX rox ,S
X' \SR у/ \SR
О-алкил-З-алкилацилтиофосфаты О-алкил-З-алкилацилтионтиофосфаты
Подобно фосфорнлхолннам, исключения в этой группе составляют фосфорилтнохолнны.
7.1.2. Фосфорорганические соединения — производные пнрофосфорной (дифосфорной) кислоты
Из этого ряда соединений для данной книги представят интерес только пирофосфаты типа
RO/ /OR
R° О o^OR
в которых на- месте алкоксигрупп могут находиться н диалкнламн-ногрунпы. Например
CgHgOv /ОС2Н5
V-o-p;
с2н5о/|| 11ХОС2Н5
тетраэтилпирофосфат (ТЭПФ)
(CH3)aNx /N(CH3)2
^Р—О—р/
II \N(CH3)2
TpHC*N, И,-(диметиламидо)-О-этилпирофосфат
Если вместо атома кислорода- в молекулу вступает двухвалентная сера, то для составления названия можно пользоваться теми же правилами, что н в разделе 7.1.1, в случае связи P=S —
253
тион, в случае связи Р—S—тио, например:
С2Н5О\ /ОС2Нв
)р-о-р< сгн,о/1 1ХОСЛ,
о о
тетра этилд итион пирофосфат C2H6S4 ySC2He C2HSO^ ^Х°С2Н‘
симм-О, О-диэтил-S, S-диэтилдитиопврофосфат
Особенности номенклатуры фосфорорганических соединений будут поясняться, по мере надобности, в соответствующих разделах книги. Кроме того, для всех важнейших соединений будут приведены и синонимы, и общеупотребительные обозначения.
7.2. БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ
ФОСФОРОРГАНИЧЕСКИХ СОЕДИНЕНИИ
7.2.1. Химическое строение и биологическая активность
Токсическое и фармакологическое действие некоторых фосфорорганических соединений на организм определяется физико-химическими свойствами этих веществ.
Так же как и в случае большей части уже рассмотренных ОВ, эти вещества должны содержать некоторые активные полярные группы, чтобы могли происходить определенные фармакологические реакции.
Несмотря на различия в химической структуре, в результате чего развиваются специфические побочные явления, фосфорорганические соединения обладают общим свойством дезактивировать холинэстеразу путем ее фосфорилирования. Последствия этого сходны с действием, которое оказывает физостигмин (эзерин) I, соединение совсем иного молекулярного строения?
Блокирование холинэстеразы физостигмином обратимо, а фосфорорганическими соединениями — необратимо, что объясняется ангидридной структурой (II) эти- соединений, приводящей к образованию прочной связи с ферментом.
Согласно Шрадеру3, фосфорорганические соединения биологически активны в том случае, если их строение удовлетворяет Сле-
254
ajkmuhm уыювимм. rj центральный агом фосфора пятивалентен; 2) у центрального атома находится двоесвязный атом кислорода нлн серы н, кроме того, две группы — алкнл, арил и др., либо моно- или диалкиламнногруппы; 3) пятая аалентность фосфора насыщена ацильным остатком.
Этим требованиям удовлетворяет выведенная Шрадером общая формула III
Хх ^O(S) X X^Z \у
in
где X, X'= R, RO(S), RNH, R2N; Y = галоген, CN, SCN, OCN, остатки фенола, енола, холина, тнохолина и др.
Сколь разнообразные . возможности, не говоря уже об изменениях X и X', дает изменение заместителя Y, можно показать иа двух приводимых ниже примерах.
Диэтнл-4-ннтрофенилтионфосфат IV
IV
по своему строению соответствует V и VI:
C2HSOX ,s
/р\
С2НЕСг ^ОН
v
О, О-диэтилтиоифосфорная кислота
(сильная кислота)
смешанному ангидриду кислот
VI
4-ннтрофенол (сильная кислота)
Введение одной или большего числа метиленовых групп между атомом фосфора и остатком нитрофенола снижает биологическую активность. Это показывает, что внешне незначительное изменение структуры может оказать существенное влияние иа фармакологическую активность.
Продукт конденсации двух молекул О,О-диэтилтионфосфор-иой кислоты — 0,0,0',0'-тетраэтилдитионпнрофосфат VII (суль-фо-ТЭПФ, сульфотеп, дитион)
С2НБОч .S S4 /ОСгНа РХ
С2н&су\п/ \ос2н5
VII
также является ангидридом. Это соединение биологическиактивно.
Хотя для органиков, занимающихся синтезом фосфорорганических соединений, ключевая формула Шрадера является важным**
подспорьем для поиска биологически акз'инных гиединени-н, -----
всей совокупности фосфорорганических соединений нельзя вывести общих закономерностей изменения токсичности в зависимости от структуры. Разумеется, в пределах определенной группы можно сформулировать некоторые соотношения между изменениями токсичности и структуры или применить их к другим группам. Эти взаимосвязи рассматриваются в следующих разделах.
X -
7.2.2. Механизм действия
Механизм действия биологически активных фосфорорганических соединений был выяснен уже во время второй мировой войны в Германии и Англии. Было установлено, что эти соединения препятствуют и подавляют биокаталитическую активность холинэстеразы (ХЭ). Последующие очень интенсивные исследования показали, что ответственным за действие фосфорорганических соединений нужно считать ингибирование не только холинэстеразы» но также и ряда других ферментов, таких, как эстераза печени, липаза материнского молока, фосфатаза почек, амилаза и др.
Общепризнано, что первичное действие фосфорорганических соединений заключается в блокировании холинэстеразы. Чтобы понять значение этого процесса, нужны некоторые сведения о механизме передачи нервного импульса.
Хотя нервную систему следует рассматривать как единое целое, из соображений целесообразности проводят различие в локализации и функции между центральной (ЦНС) и периферической (ПНС) нервной системами.
Функция нервной системы заключается в следующем: воспринять раздражение в области рецепторов (чувствительные нервные окончания); энергетически сформировать волну возбуждения или импульс в рецепторах^ передать специфическую энергию возбуждения ЦНС и там преобразовать ее; в ответ иа раздражение передать импульс к периферии, чтобы вызвать определенную реакцию исполнительных органов.
В ЦНС имеется два типа нервов — сенсорные (чувствительные) и моторные (двигательные).
Сенсорные нервы воспринимают специфическое для них раздра- . жеиие и проводят импульсы в ЦНС или по периферическим трактам к другим участкам.
Моторные нервы переносят сигналы ЦНС к периферийным исполнительным органам. Кроме этой системы, поддающейся волевому контролю, имеется вегетативная нервная система, значительно менее зависимая от участия сознания, по которой периферическое раздражение поступает в ЦНС и, после его преобразования, отсылается к периферии. Эти автономные системы простираются почти во все органы, которые связаны друг с другом через ЦНС. Это та часть нервной системы, которая стимулирует гладкие мышцы, сердечные мышцы и железы.
256
разделение автономной системы на симпатическую н парасимпатическую нервную системы.
Для наших целей более целесообразно классифицировать разновидности нервов по трансмиттерам (медиаторам)—веществам, переносчикам нервного импульса.
По виду таких трансмиттеров [важнейшими являются ацетилхолин (АХ) и адреналин] различают адренэргическне и холин-эргнческне нервы.
Ад ренэ рваческими являются периферические трак*гы постганглио-нариых симпатических волокон, холинэрёическими — центральные
и периферические постганглно-парные парасимпатические н некоторые симпатические тракты (например инервнрующне потовые железы и др.) к исполнительным органам, либо преганглио^ иарные парасимпатические и симпатические тракты к автономным ганглиям и к надпочечникам. Сюда относятся также моторные волокна, идущие к мышцам, контролируемым сознанием, или к их
Рис. 9. Схема передачи моторного импульса по нервному волокну.
концевым пластинкам.
Большая часть постганглнонарных симпатических нервных волокон прн раздражении выделяет адреналин (VIII), а также норадреналин (IX):
Эти соединения воздействуют на исполнительные органы, железы и мышцы и стимулируют их деятельность.
Как уже сказано, при возбуждении постганглионарные парасимпатические нервные волокна выделяют преимущественно ацетилхолин (АХ), вследствие чего возникают определенные воздействия на исполнительные органы.
На пути к нервным окончаниям моторный импульс попадает в преобразователи (синапсы), в которых имеются тесные контак-' ты ганглиев через рецепторы с моторными концевыми пластинками. Рецептор концевой пластинки находится в покое в заря-женном, возбудимом состоянии (рис. 9,а). В противоположность внутренней, заряженной отрицательно, внешняя сторона нервного
9 Зак. 448
257
ВОЛОКНа ИЛИ МЫШЦЫ заряжена пилижигельни. напряжение в njiei* ке равно 100 мв.
При появлении импульса в одном участке моторной концевой пластинки имеющийся потенциал нарушается и поверхность возбужденного участка становится отрицательной (рис. 9,6). Деполяризация ведет к появлению тока, который возбуждает новые участки (рис. 9,в). Ток может идти в обоих направлениях, но, поскольку возбуждение возникает только на конце волокна, эта индукция распространяется лишь в одном направлении (рис. 9,г).
Моторная концевая пластинка
Y Импульс
Моторная концевая пластинка
Рис. 10. Схема действия ацетилхолина на возбужденную мышцу; а — невозбужденное состояние перед поступлением импульса; ^ — возбужденное состоя*' ине после поступления импульса (образуется АХ, мышца сокращается); в—связывание субстрата е ферментом; по мере связывания сокращение мышцы ослабевает (переход в состояние а).
Скорость распространения импульса в человеческом организме при температуре тела составляет около 120 м/сек. Импульсы раздражения быстро чередуются, примерно с частотой 30 Импульсов в секунду.
Деполяризованная концевая пластинка не поддается возбуждению. Для того чтобы стало возможным воспринять следующий импульс, нужно удалить из системы трансмиттер.„Под каталитическим -влиянием а цетил холинэстеразы (АХЭ) ацетилхолин (X) (АХ)
(CH3)3N—СН2СН2—О—С—СН3 . i-
X гидролитически разлагается на холии (X—ОН) и уксусную кислоту (НА) I -
[АХЭ]
АХ + ЙОН НА 4-Х—ОН (1)
После того как выделившийся при возбуждении свободный АХ своим воздействием иа рецепторы в ткани полосатой мышцы вызвал ее сокращение, он связывается е ХЭ посредством функциональных групп фермента (рис. 10). Связывание происходит с участием четвертичного атома азота и положительного атома угле
258
рода ацетильной группы АХ. Последний взаимодействуй и «эне-разным» центром с образованием ацетилированного фермента и холииа (рис. 11).
Ацетилированный фермент легко разлагается гидролитически на исходный фермент и уксусную кислоту. Следующий импульс снова высвобождает АХ. Если при помощи ХЭ разложение АХ идет недостаточно быстро (оно происходит за несколько миллионных долей секунды), то накопление АХ вследствие непрерывной деполяризации ведет к постоянному раздражению нервных клеток, что влечет за собой длительную стимуляцию периферийных элементов нервной системы (нервных волокон, мышечных клеток, желез и др.). Таким образом, для передачи импульса возбуждения необходим быстрый гидролиз АХ.
сн3 I СН3 .
I I.
N СН2—— СН2-----О -г— С1 О
СН3
Дни онный Эстеразный
центр центр
Рис. 11. Связывание холинэстеразы с ацетилхолином.
Несмотря иа то, что известны разные виды холинэстеразы, речь идет, главным образом, об ацетилхолииэстеразе (АХЭ). По существу нет разницы в распределении и активности моторных нервных волокон в отдельных участках Сплетений нервных окончаний в самых различных органах. После гидролиза АХ в передаче импульса к мышечным волокнам принимает участие все сплетение нервных окончаний в целом.
С функциональными группами АХф реагирует множество соединений, которые путем вытеснительной конкуренции затрудняют связывание субстрата' с ферментом и таким образом препятствуют и снижают каталитическую активность АХЭ. Кроме биологически активных фосфорорганических соединений, которые вызывают необратимое ингибирование ХЭ, известны многие другие органические соединения (антихолииэстеразы), по большей части это четвертичные основания, которые действуют сходным образом, но их действие обычно обратимо.
Холинэстеразы, независимо от их происхождения (ХЭ сыворотки, мозга, эритроцитов, спинного мозга), реагируют с так называемыми аитихолинэстеразами очень по-разному. Это позволяет
9:
259
провести различие между самими ферментами и одновременно классифицировать ингибиторы. Так, различают:
ингибиторы, которые связываются с анионным центром фермента, например четвертичные органические соли аммония, холин, метиленовый голубой;
ингибиторы, которые связываются с эстеразным центром фермента, сюда относятся фосфорорганические соединения;
ингибиторы, которые связываются с обоими центрами; предполагают, что это происходит в случае неостигмина, эзерина, а также фосфорилхолинов и фосфорилтиохолинов.
КО >OR
Р=Ю
ОН
с -----
'+НОН
Рис. 12. Схема блокирования холинэстеразы фосфорорганическим ОВ и ее регенерация:
а — в остеразиом» положении фермента имеется отрицательно заряженный центр, рядом с ним реакционноспособный атом водорода ОН- или- NH-групп, образующий водородную сьрязь с молекулой фосфорорганического вещества; b — нуклеофильная атака поляризованного атома фосфора отрицательно заряженным центром, при этом происходит разрыхление связи Р—F и становится возможным ее 'разрыв; с —возможное деблокирование фермента в результате гидролиза.
Ингибирование ХЭ фосфорорганическими соединениями происходит, прежде всего, посредством фосфорилирования фермента. Однако считается доказанным, что ответственным за блокирование ХЭ следует считать не только фосфорилирование, а множество еще подробно не выясненных реакций, которые изменяют структуру и активность фермента 4.
При действии фосфорорганического ОВ образуется соответствующий фосфорилированный фермент, например, по следующей схеме (G — кислород оксигруппы серина):
R°x .О । X)
+:G—Н —> Р^ 4-H+4-F" (2)
RO' \F ROZ Vi
окружение ✓ |
эстеразного
центра ХЭ
Для быстрого блокирующего действия определяющей является высокая скорость этой реакции. Регенерация активной ХЭ являет-
.260
ся реакцией, на скорость которой можно воздействовать катализаторами. В естественных условиях она идет очень медленно, неделями и месяцами (реакция 3):
RCk Я +н2о ------------->
RCr Vi
RO\
: G -H
RCK ХОН I
(3)
Процесс связывания ХЭ н ее регенерации изображены на рис. 12. Связывание АХЭ с молекулой фосфорорганического соединения препятствует гидролитическому расщеплению АХ (реак-- ция 1, стр. 258), поэтому в результате накопления АХ сохраняется непрерывное раздражение (ср. рис. 13).
Ряс. 13. Схема действия ацетилхолина после блокирования ацетилхолннэстеразы фосфорорганическим ОВ.
Подавление ХЭ, вызванное фосфорорганическими соединениями, отличается от подавления другими ингибиторами как количественно, так и своей продолжительностью и плохой обратимостью. Разумеется, такого рода различия имеются и между отдельными фосфорорганическими соединениями.
На основании имеющихся исследований в настоящее время еще невозможно в полной мере сформулировать непосредственные взаимосвязи между химическим строением фосфорорганических соединений, их ядовитостью и более или менее сильным подавлением ими холинэстеразы, хотя в пределах некоторых групп имеются ценные наблюдения о связи этих факторов.
7.2.3. Общий характер действия
Характер действия фосфорорганических соединений, в сущности, ^ определяется вмешательством их в сложные функции ацетилхолина посредством подавления АХЭ. Ингибирование эстеразы неизбежно ведет к накоплению АХ, следствием чего являются дли-• тельные и сложные воздействия на организм.
Помимо других причин, симптомы отравления фосфорорганическими соединениями сводятся к стимуляции холинэргнческой и
261
центральной нервной систем. Чрезмерные стимуляции ведут к параличам, причем симптомы поражения очень сложные.
Воздействия на холинэргическую нервную систему в результате накопления АХ заключаются в следующем5:
сердце — частота сокращений уменьшается, мощность падает;
глаза — сосуды сужаются, аккомодационная мышца сокращается, глаз аккомодируется к ближнему зрению;
кровеносные, соеудц сердца, легких, кожи и мыш,ц — расширяются, бронхов — сужаются;
желудочно-кишечный тракт— перистальтика и цикл усиливаются, секреция увеличивается;
* мочевой пузырь — мускулатура стенок сокращается, сфинктер ослабевает;
кожа — увеличивается секреция потовых, слюнных, слезных и носовых желез.
Воздействие на центральную нервную систему проявляется в состояниях возбуждения и параличах, расстройствах психики и действии на- дыхательный центр. .Для большинства отравлений фосфорорганическими соединениями характерно быстрое*появле-ние симптомов отравления, которые, в основном, совпадают.
Отравление может быть вызвано: вдыханием паров или аэрозоля, резорбцией жидкости или паров в высокой концентрации через кожу, резорбцией через конъюнктиву глаз, попаданием в пищеварительный тракт.
Признаки отравления могут появиться даже при очень малых концентрациях, которые не обнаружцмы или почти ие обнаружи-мы аналитически. Следует учесть, что токсичность фосфорорганических ОВ увеличивается примерно вдвое при повышении температуры на 10°С.
Обычно клиническую картину отравления подразделяют па три степени:
1. Легкие отравления: концентрация ОВ — около 1/10 от смертельной боевой концентрации; латентный период 10,—15 мин, при очень легких отравлениях — до 2, ч; продолжительность отравления 1—5 суток.
' Сймптомы: сужение зрачков, выделения из иоса, усиленное слюнообразование, головные боли, умеренная одышка с ощущением давления в груди, потеря аппетита, чувство тоски и страха, потливость, бессонница, рассеянность.
2. Отравления средней тяжести. Концентрация ОВ — около 1/5 смертельной боевой концентрации; латентный период 5 мин, самое большее — 2 ч; продолжительность отравления 1—2 недели, полное возобновление активности ХЭ' лишь через 4— 6 недель.
Симптомы: как в случае легкого отравления, ио интенсивнее; кратковременные судороги, сужение зрачков (до размеров булавочной головки), рвота, очень сильное слюнотечение, приступы астмы, периодические спазмы в гортани, учащенное иеравно-
262
; мер ное дыхание, сильный кашель, выделение мокроты в виде лин-кой пенистой слизи, судороги мышц (особенно ног) и др.
Отравления средней тяжести прн несвоевременном лечении смертельны, в-период до полной реактивации ХЭ отравленный сверхвоспрннмчив к тому же ОВ и подобным ядам.
3. Тяжелые отравления. Концентрация ОВ — от 1/3 до 1/2 смертельной боевой концентрации; латентный период отсутствует или очень короткий; смерть иатсупает через 6—12 ч.
Симптомы: как в случаях 1 и 2, но в более быстрой последовательности; зрачки не реагируют, мучительное давление в глазах, сильные головные боли, рефлексивная отрыжка и рвота, тяжелейшее удушье, сильнейшие пароксизмы судорог, нарушение сознания, по мере изнурения оргаинзйа переход судорог в параличи бессилия, смерть.
При смертельных боевых концентрациях смерть после вдыхания наступает через 5 мин. Причина смерти паралич дыхательного центра, остановка кровообращения' н сердечной деятельности, прн больших дозах — немедленная смерть от удушья.
7.3. ДИАТГКИЛАЦИЛФОСФАТЫ
7.3.1. Общие сведения
Диалкилацилфосфаты можно рассматривать как диэфнры заме щениой фосфорной кдслоты, у которых X = Hal, CN, SCN:
RO\PZ
Особое значение имеют диалкилфторфосфаты, которые являются биологически самыми активными соединениями этой группы. Их получают фторированием хлорангидридов кислот фосфора. .
Ланге н фои Крюгер1 получили диметнл- н диэт ил фтор фосфаты очень сложным способом. Они были первыми исследователями, обнаружившими чрезвычайную токсичность этих соединений.
Во время второй мировой войны в институтах Кембриджского университета по заданию Министерства снабжения английские Мученые Мак-Комби, Саундерс, Стесн н др. исследовали дналкнл-^фторфосфаты и аналогичные соединения. Они обнаружили исключительно высокую токсичность некоторых веществ и разработали ^различные методы получения; их эксперименты показали возможность военного применения некоторых ди ал кил фтор фосфатов, в частности, динзопропнлфтор фосфата (Д ФФ).
263
132. Диалкилфторфосфаты
Диалкилфторфосфаты являются диэфирами фторфосфориой кис-//Q лоты (RO)2P<< .
'F
7.3.2.1. Связь между химическим строением и токсичностью. Связь между химическим строением диалкилфторфосфатов и аналогичных соединений и их токсичностью была подробно изучена Саундерсом и сотр. во время второй мировой войны.
Ключевая формула Шрадера полностью верна для соединений этой группу. Как и для фосфорорганических соединений других групп, в этом случае также оказалось, что в зависимости от природы различных заместителей и строения алкоксигрупп наблюдаются ослабления и усиления токсичности.
Условием высокой токсичности является наличие незамещенных вторичных алкоксигрупп, причем вторичный атом углерода должен быть связан с атомом кислорода:
RR'CHO^
RRZCHC)/
(СН3)2СНСН2СН20х .О
(СНз)2СНСН2СН2О/ \f и
(СН3)2СНО. X)
(СН3)2СНо/. XF пт
сн3
«-c3h7£hc\j
к-С3Н7СНО//
(С2Н6)2СНОХ
(С2н5)2снс/ \f
Например, бис-(3-метилбутил)-фторфосфат II не проявляет мистического действия и обладает лишь незначительной токсичностью по сравнению с диизопровилфторфосфатом (ДФФ) III или с бис- (4-метилизопеитил) -фторфоефатом IV.
СН3
(СНзЬСНСНгСНО^
(СН3)2СНСН2СНО// \f
СН3
IV
2Н3 VI
С удлинением боковой цепи R (I) при вторичном углероде или при введении неразветвлениых н-алкоксигрупп токсичность уменьшается. Так, ди-3-пеитилфторфосфат V значительно меиее токсичен, чем изомерный диизопеитилфторфосфат VI.
Токсичность этих соединений снижается от ДФФ 1Ц через ди-изобутилфторфосфат II к ди-3-пеитилфторфосфату V, так же, как она падает от ДФФ через диэтил- к ди-«-пропилфторфосфату (VIII и IX):
СН3
CaHg^HO^ С2Н3О\ ^;О «-С3Н7О^
с2н5снс/ V С2Нес/ -\f н-С3Н7с/
СН3 VII VIH IX
264
Аналогичные данные получены и для соединений с циклическими радикалами.
Дифенилфторфосфат X нетоксичен и не обладает мистическим действием, тогда как дициклогекснлфторфосфат XI более ядовит, чем ДФФ III, а вызываемый им миоз столь же интенсивен. Напротив, токсичность бис-(2-метилциклогексил)-фторфосфата XII
ослаблена.
В качестве доказательства повышенной токсичности дициклр-гексилфторфосфата XI по сравнению с ДФФ III Сауидерс приводит для них значения ЛД5о (0,11—0,14 и 0,36—0,5 мг/л соответственно, на мышах, крысах и кроликах). Повышенную токсичность соединения XI по сравнению со нсеми другими диалкнлфторфос-фатами можно объяснить только наличием вторичного атома углерода, связанного с атомом кислорода.
Замещение атомов водорода алкоксигрупп галогенами, например фтором или хлором, даже в случае вторичных алкоксигрупп приводит к уменьшению токсичности. В некоторых случаях Эти соединения проявляют совершенно иные физиологические свойства.
Если между атомами фосфора и фтора вводить в молекулу метиленовые группы (такие соединения принадлежат тогда к группе О,О-диалкнлалкилфосфонатов XIII)
RCk <0 RCk .0
RO/ \r Rd/ \cH2)nF
XIII
то с увеличением числа метиленовых групп п токсичность уменьшается, равно как и в случае, когда в молекуле содержатся атомы серы вместо атомов кислорода.
Военный интерес может представлять лишь небольшое число диалкилфторфосфатов. При этом может идти речь только о самых ядовитых и самых устойчивых соединениях, к которым кроме соединений III и XI относятся диизобутилфторфосфат II и бис-(4-метилизопентнл) -фторфосфат IV.
Важнейший представитель этой группы диизопропилфторфос-фат III рассмотрен в разделе 7.3.3. Выводы о свойствах других соединений можно сделать на основании данных, приведенных для ДФФ.
265
7.3.2.2. Методы получения. Фторфосфаты можно легко получить фторированием соответствующих диалкнлхлорфосфатов фтори-
стым натрием:
(RO)2p;
ХС1
+ NaF ------>
—NaCi
(RO)2p^
Диалкилхлорфосфаты можно получать следующими а) Реакцией треххлористого фосфора со спиртом ствии пиридина получают триалкилфосфнт, который хлором, причем одновременно идет и хлорирование:
+ зс5н6ы '
PC1’ + 3ROH ^CsHsN.HC1-> (R0)1P
•
(RObP + Cl. (RO)2Pf +RC1
методами: в присут-окисляют
б) Реакцией треххЛористого фосфора с соответствующим спиртом в отсутствие третичного основания получают диалкилфосфит и затем обрабатывают егохлором:
PCU4-3ROH —> (RO)2P—OH + RC14-2HC1
(RO)2P-OH + C12 —> (RO)2P^ Н-НС1
ХС1
в) Если исходным веществом является диалкилфосфит, то можно в качестве хлорирующего агента применять сульфурил-хлорид
(RO)2P—OH-|-SO2C12 —> (RO)2P^ +HC14-SO3
\С1
или, лучше, N-хлорсукцинимид:
ГО—ГII2 ,о го—сн2
(RO)2P—ОН 4- CINZ | —> (RO)2P\ + HNZ |
\:о-сн2 \ci \со-сн3
г) Хлорокись фосфора в присутствии пиридина реагирует со спиртом с образованием триалкилфосфата, который при дальнейшем действии хлорокнсй фосфора наряду с дихлорпроизводным дает диалкилхлорфосфат:
+ 3C5H5N.
poci3 + 3roh _3C5H5N.HC1> (RO)3P=O
*°\ '
(RO)3P=O 4- 2РОС13 ^ 3 ЧР^
C’lz ХС1
R°\
2(ro)9p=o + poci3 —> з ;р'
roz чз
266
WL- bee эти методы получения диалкилхлорфосфатов включают три Я® стадии. Кроме того, обычно приходится выделять промежуточные у продукты. Саундерсу и сотр.6 удалось разработать одностадийный яВлроцесс промышленного производства диалкилфторфосфатов на Жрснове метода (б), исключив таким образом применение слишком дорого го для промышленного производства пиридина [см, методы
W: При действии треххлористого фосфора иа раствор соответствующие щего спирта в четыреххлористом углероде с последующим хлори-Ж рованием и фторированием фтористым натрием и нагреванием реакционной смеси получают после очистки и перегонки диалкил-ж&фторфосфат, выход 70—75%.
Ж Применение в качестве хлорирующего агента вместо хлора «kN-хлор сукцинимида в одностадийном процессе было предложено |||Толдуайтом и Саундерсом для получения диалкилфторфосфатов, Ж'£дувствительных к кислотам. Этот прием считают наиболее целе-Жсооб разным для получения дициклогекс ил фтор фосфата.
Очевидно, можно получать диалкилфторфосфаты прямым взаи-||р.Модействием галогеифторфосфатов с соответствующими спиртами. Ж-:Было предложено использовать для этой цели дихлор фтор окись ^^лЬосФопа и трифторокись фосфора:
® ' Z
в? 2R0H + C1’P\ -2НС? 2<R°)=P\
Г XF XF
II-
Ж 2ROH + POF3 ---> 2(RO)J<
Ж -2HF \p
-У'В
В исключительных случаях для связывания галогеноводорода этих реакциях применяют амииы.
Дихлорфторокись фосфора получают взаимодействием хлор-в’Окиси фосфора с фтористой сурьмой в присутствии питифтористой сурьмы как катализатора:
SbF3(SbF5)
Ж, РОС13 -------> POC12F
Ok Ни та, ии другая реакция не пригодны дли промышленных про-жЕдессов. Их скорость слишком мала, а получение фторокиси фосфора 1а|;требует сложного оборудования, которое осуществимо только при ЙВШооизволстве в малом масштабе.
жр 7.3.2.3. Физические и химические свойства. Диалкилфторфосфа-др-ты — устойчивые бесцветные жидкости со слабым запахом.
иЖ Диметилфторфосфат растворим в воде, в то время как соеди-ж|неиия вплоть до С5 или С6 (общее число атомов углерода в моле-Ж|жуле) растворимы умеренно. Некоторые соединения С6 и выше, якрключая соединения с циклическими радикалами и галогеналкиль-ЖЯыми группами (последние иногда и < С6), — нерастворимы в Жррде (табл. 22).
267
Таблица 22. Физические и физиологические свойства диалкилфторфосфатов (RO)2P(O)F
В круглых скобках приведено давление в мм рт. ст,’, Р —растворим, Нр — нерастворим; Ур —умеренно растворим
R S Т. кип., °C Растворимость ' Показатель преломления „25 nD Токсичность по сравнению с ДФФ
ингаляционное действие миотическое действие
СН3 149(760); 50(10) р 1,3553 Меньше Меньше
сан5 171 (760); 62(11). Ур 1,3708 Меньше Меньше
w-C3H7 100 (20); 62 (2) Нр 1,3875 Меньше Незначительное
(СН3)3СН 183(760); 71 (10) Ур 1,3795 — —
я-Сь4Н9 128 (30); 80 (5) Нр 1,3993 Меньше Незначительное
СН3СН2СНСН3 91 (12) Нр — Сравнимое Сравнимое
(С2Н5)2СН 98 (2) нр — Меньше Меньше
С*Н3(СН2)8СН2 102(5); 102(4) Нр . 1,4080 Меньше Незначительное
(СН3)2СНСН2СН2 • 138 (23) Нр — Незначительное Отсутствует
(СН3)2СНСН2СНСНЭ 106 (1) Нр — Сравнимое Более сильное
ед 118(0,4); 142(3) Нр — ‘ Не ядовит Отсутствует
Чикло-С6Нп 116(0,3); 120(3) Нр 1,4532 Больше Сравнимое
4-сн3ед0 120(0,1); 174(3) Нр Меньше Меньше
ch2cich2 143(15); 123(5) Нр -1,437 (при 18 °C) Незначительное —
ch2fch2 102 (0,8) — 1,397 (при 19 °C) Меньше —
«летучесть соединений этой группы мала; они имеют низкие дав-ление паров и высокие температуры кипения.
Водой диалкилфторфосфаты гидролизуются:
(RO)2Pf
+ н2о
77^ (*0)гР\
С увеличением числа атомов углерода в молекуле скорость гидролиза постепенно убывает. Этильное производное7 гидролизуется примерно в 18 раз быстрее, чем диизопропилфторфосфат, 1%-ный раствор которого полностью разлагается7 через 72 ч.
Гидролитический разрыв связи Р—F водой представляет собой нуклеофильное замещение фтора ионом гидроксила. Разумеется, нуклеофильность и поэтому смещение электронной плотности в фосфорорганической молекуле малы. На гидролиз можно повлиять изменением температуры, pH, соответствующими катализаторами и растворителями.
Щелочные агенты ускоряют разрыв связи Р—F, поскольку нуклеофильность гидроксильных ионов значительно больше, чем воды; атом фтора замещается ионом гидроксила:
+но-
(ro)2p; —(ro)2p;
\F -F \qh
Скорость гидролиза пропорциональна концентрации гидроксильных ионов и фосфорорганического соединения. Это справедливо, для всех галогензамещенных фосфорорганических соединений.
Более подробное рассмотрение гидролиза показывает, что электрофильный заместитель фтор является электроотрицательным, что ведет к поляризации молекулы и к увеличению электрофильности атома фосфора. Ион гидроксила подходит с электростатически более выгодной стороны молекулы и вступает с ней во взаимодействие на некотором удалении. Одновременно увеличивается длина связи Р—F. Образуется переходное состояние, в котором связь с гидроксил-ионом еще не образовалась, а с фтором еще не разорвалась. Из-за наличия электроотрицательных элементов вокруг атома фосфора переходное состояние очень неустойчиво и распадается по наиболее сильно поляризованной связи Р—F;
RO\+/O~
НО~ + X
ROZ 4F
но-p;
I 4F
L or J
RO4 + ZO~ P\
Ro/ \QH
-f- F
Гидролиз диалкилфторфосфатов в равной мере катализируется /„ионами водорода, образовавшимися на первой стадии реакции8. / Как и при катализе гидроксильными ионами, реакция протекает по механизму Sn2. Полагают, что обе реакции идентичны.
269
Гидролиз в кислой среде описывается следующими двумя схе-* мами: - ( ’
и?0 +HQH
<roj2p; 4-н —> <ro)2p^+ ------> (ro)2p^ + hf + h+
'F V...H XOH
(RO)2Pf + H+ —► (RO)2P< -----—> (RO)2P^ + HF
\f ~h \dh
Каталитическое действие и а гидролиз ионов водорода и гидроксила ведет к образованию переходных состояний с повышенной энергией. Некатализируемый гидролиз является медленной одно-
Рис. 14. Зависимость скорости гидролиза от величины pH.
стадийной реакцией, каталитический гидролиз представляет собой две быстрых элементарных реакции, приводящих в данном случае к расщеплению связи Р—F.
Как уже упоминалось, скорость гидролиза зависит от концентрации веществ. Каталитическое действие ионов водорода и гидроксила тем сильнее, чем выше
их концентрации. Зависимость скорости гидролиза от pH нели-
нейна и возрастает как при увеличении, так и при уменьшении pH (рис. 14). Наблюдается минимум, который расположен ближе к кислотной области pH, поскольку нуклеофильность ионов гидроксила значительно больше, чем электрофильность ионов водорода. Минимум скорости гидролиза одновременно является максимумом стабильности алкилфторфос-фатов.
Как видно иа приводимого ниже уравнения, во взаимодействие с одной молекулой диалкилфторфосфата вступают две молекулы щелочи. При этом не происходит полного разрушения фосфорорганической молекулы. Оно возможно лишь в более жестких усло-
виях— энергичном перемешивании, продолжительном кипячении, особенно с азотной кислотой. При этом количественно образуется фосфат натрия.
Диалкилфторфосфаты с радикалами вплоть до Сг гидролизуются сравнительно быстро. Особенно устойчив дициклогексил-фторфосфат (т. кип. 90—96°Спри 0,02мм рт. CT.\d?l,20&, п2£1,4545):
(свн11о)2р;
Он заметно гидролизуется только при многочасовом кипячении с водой. В 2%-иом растворе едкого натра при 28,5 °C и энергичном, перемешивании фтор полностью отщепляется лишь через 270
90 мин. При продолжительном встряхивании гидролиз происходит по следующему уравнению:
Л Z
(С6НИО)Ж + 2NaOH —> (СвНнО)2Р^ + NaF+H2O
^ONa
Дициклогексилфторфосфат— бесцветная, очень ядовитая жидкость. Его получают реакцией циклогексанола с дихлор фторокисью фосфора:
2С8НИОН+С12Р\ —> (С6НИО)2Р^ +2НС1
\f ^F
За некоторыми исключениями (см. табл. 22), диалкилфторфос-фаты — сильно токсичные соединения. Они ингибируют холинэстеразу и иногда проявляют сильное мистическое действие.
Несмотря на превосходные инсектицидные свойства, оии из-за своей высокой токсичности находят лишь ограниченное применение в качестве инсектицидов.
Некоторые соединения, особенно ДФФ, начали применять в качестве лекарственных препаратов.
7.3.3. Диизопропилфторфосфат (ДФФ)
(СНЭ)2СНО
(СН3)2СНО
Военные обозначения: англ. — DFP; амер. — PF-3.
Токсичность и свойства ДФФ были впервые сообщены Саундерсом 7 в декабре 1941 г. на конференции в Лондоне. Это очень эффективное соединение может быть применено в военных целях. В результате появления более токсичных фосфорорганических ОВ его военное применение сомнительно. Во время второй мировой войны в Англии и США были заготовлены большие количества этого соединения в качестве ОВ.
7.3.3.1. Методы получения. Промышленные способы. Наиболее подходящим является «английский одностадийный процесс», разработанный и испытанный как модификация процесса синтеза кислых фосфитов. Этот-процесс основан на следующих реакциях:
РС13 + ЗНОСН(СН3)2 —> 1(СН3)2СН012РОН4-(СН3)2СНС14-2НС1 (1)
+ С12
((СН3)2СНО]2Р—ОН ---> t(CH3)2CHO]2Pf (2)
—НС1 \ri
[(СН3)2СНО]2Р^
+ NaF
—Na Cl
(3)
271
В результате взаимодействия треххлористого фосфора с изопропиловым спиртом выделяется, значительное количество тепла, поэтому без интенсивного наружного охлаждения выход продукта по этой реакции мал. Если исходить из раствора изопропилового спирта в четыреххлористом углероде и вводить в него треххлорн-стый фосфор, то температуру реакции можно регулировать без специального охлаждения, выход составляет около 90%.
В трехстадийном' процессе последующим хлорированием (реакция 2) получают хлорфосфат (выход 80—90%). Фторирование хлорфосфата фтористым натрием (реакция 3) проводят при нагревании бензольного раствора (выход ДФФ составляет 90%); общий выход при таком процессе составляет 60—70% ДФФ.
В модифицированном и приспособленном для промышленного производства процессе использована возможность понижения температуры реакции путем применения четыреххлористого углерода (см. выше). В растворе четыреххлористого углерода протекают все приведенные выше реакции, благодаря чему на стадии фторирования нет надобности добавлять бензол для растворения диизопропил ф торф осф ат а.
Одностадийный процесс состоит в том, что в раствор изопропилового спирта в четыреххлористом углероде вводят треххлористый фосфор и полученную реакционную смесь хлорируют хлором при.0°C. Фторирование фтористым натрием проводят при кипячении с обратным холодильником. Процесс идет при энергичном перемешивании реакционной смеси. После фильтрования реакционного раствора и отгонки четыреххлористого углерода выделяют ДФФ перегонкой при пониженном давлении. Выход чистого продукта составляет около 75%.
Отработанный четыреххлористый углерод снова вводят в непрерывный процесс, благодаря чему этот способ очень рентабелен, тем более, что в качестве исходных продуктов применимо сравнительно дешевое сырье технической степени чистоты. Важно, чтобы исходные вещества были совершенно безводными, то же относится и к самому процессу реакции.
Вода или попадание влаги способствует диссоциации образующегося хлористого водорода. Неизбежному в таком случае гидролизу подвергается прежде всего диизопропилфторфосфат. Поэтому выходы значительно уменьшаются, причем следует заметить, что кипячение, необходимое для фторирования, привело бы в этом случае к исключительно сильному разложению.
Лабораторный синтез ДФФ. В А боратории ДФФ получают одностадийным процессом. Хлорирование целесообразно проводить N-хлорсукцинимидом, разумеется, можно использовать и хлор.
При применении N-хлор сукцинимида в качестве хлорирующего агента 9 все реагенты, а также применяемые азот и воздух нужно очищать и сушить. Четыреххлористый углерод после перегонки следует сушить над пятиокнсыо фосфора; изопропиловый спирт и треххлористый фосфор рекомендуется перегонять несколько раз (3—4 раза); воздух и азот целесообразно сушить концентрированной серной кислотой, причем нужно избегать захвата капель серной кислоты.
272
Приготовляют раствор 36 г (0,6 моль) изопропилового спирта в 60 мл четыреххлористого углерода и помещают его в трехгорлую круглодонную колбу емкостью 250 мл, снабженную мешалкой и термометром. Очень медленно и при -'Сильном перемешивании из капельной воронки приливают по каплям раствор 27,5 г (0,2 моль) треххлористого фосфора в 40 мл четырех хлор истого углерода. Температуру реакции следует поддерживать между 50 и 60 °C, при необходимости нужно подогревать.
Затем капельную воронку заменяют обратным холодильником с осушительной трубкой, заполненной хлористым кальцием, и 60—90 мин кипятят смесь для удаления хлористого водорода. После этой операции вместо термометра вставляют доходящий до дна колбы капилляр для ввода газа и при пониженном давлении через продукт в течение примерно 2 ч пропускают тщательно высушенный воздух. Следует обращать внимание на то, чтобы по возможности был удален весь хлористый водород. Испарившийся четыреххлористый углерод восполняют новой порцией (около 30 лл).
Последующее хлорирование проводят при сильном перемешивании, добавляя 27 г (0,2 моль) N-хлорсукцинимида небольшими порциями (примерно по .0,2 а). Вставленный к этому времени снова термометр позволяет контролировать температуру реакции, которая должна лишь незначительно повышаться. Время от времени требуется охлаждение (!). По завершении хлорирования смесь охлаж-' дают ниже 5 °C, выделяющийся сукцинимид быстро отфильтровывают и осадок промывают небольшим количеством холодного четыреххлористого углерода. Фторирование проводят в двугорлой круглодонной колбе путем прибавления 42 г (1 моль) фтористого натрия и кипячения реакционной смеси с обратным холо-. дильником при интенсивном перемешивании по крайней мере в течение 3 ч. После 'охлаждения реакционную массу фильтруют и фильтрат сушат над безводным сульфатом натрия или магния. Четыреххлористый углерод отгоняют при пониженном давлении. Оставшийся раствор следует перегонять при пониженном давлении в токе азота. Выход ДФФ составляет около 75%.
7.3.3.2. Физические свойства. Диизопропилфторфосфат представляет собой бесцветную жидкость со слабым фруктовым запахом. Широкий интервал температуры, в котором соединение остается .жидким (от +183 до —82°С), наводит на мысль о его возможном военном применении при низких температурах, особенно в качестве компонента тактических смесей ОВ специального военного назначения. Примечательна хорошая растворимость ДФФ в других ОВ и в органических растворителях. В воде при 20 °C растворяется 1,5% ДФФ.
Ниже приведены физические константы ДФФг
Т. кип., °C при 760 мм рт. ст.........................
при 25 мм рт. ст.......................
при 21 мм рт. ст.......................
при 16 мм рт. ст.......................
при 2 мм рт. ст................. Т. пл., °C................................
Давление пара при 20 °C, мм рт. ст........
Максимальная концентрация прн 20 °C, мг}л . ... . Относит, плотность
пара...................................
жидкости d\
при 20 °C............................
при 25 °C............................
при 27 °C................. .'.......
Вязкость при 20 °C, спз. . . .............
183 84—85 78-84
73 42—43
-82 0,57 (0,21)
5,6
6,4
1,0611 I.057I
1,055 1,65
273
Термическая нестабильность ДФФ , вблизи его температуры кипения не является очень серьезным недостатком. Хотя прЦ быстром нагревании ДФФ разлагается, ио потерн его при этом незначительны.
Величины давления пара и летучести в интервале температуры О—30 °C приведены в табл, 23, В интервале 0—60 °C их можно рассчитать по формуле:
_ __о 2671 , ,
1g р = 8,872----- (мм рт. ст.)
2671 1g С3 • 12,342 - (мг!л)
Давление пара', Составляющее при 20 РС 0,57 ммрт.ст., придает ДФФ летучесть (около 5,6 кг/л), обеспечивающую ему известную стойкость иа местности. Образование смертельной концентрации за счет летучести можно ожидать только в некоторых случаях, принимая во внимание естественные факторы ослаблений. Концентрация испарившегося ДФФ в воздухе почти не бывает выше 0,1 мг[л. Она достаточна, чтобы вызвать длительное отравленне,-
Таблица 23. Величины давления пара и летучести ДФФ в интервале 0—30 °C
В круглых скобках приведены данные Шрадера ю
Температура, °C Давление пара, ММ рт. ст. Летучесть, ме!л Температура, Давление пара, ММ рт. СТ. Летучесть, мг[л
0 0,12 (0,03) 1,3 (0,355) 20 0,57 (0,21) 5,6 (2,117)
5 0,18 2 25 0,81 8,0
10 0,27 (0,08) 2,8 (0,835) 30 Г, 14 (0,5) 11,1 (4,873)
15 0,4 4,1
Неограниченная смешиваемость ДФФ с ипритом явилась поводом, чтобы приготовить смеси этих двух ОВ и определить их температуры плавления (см. стр. 154). Смесь, состоящая из 87% ДФФ и примерно 13% иприта, имеет температуру застывания —36°С. Эту смесь могут применять при различных климатических условиях.
7.3.3.3. Химические свойства. ДФФ является сравнительно стабильным соединением, которое можно хранить долгий срок. При длительном хранении в стальных контейнерах, главным образом в присутствии влаги, наблюдается значительная корразия в результате отщепления фтористого водорода. Добавление соответствующих стабилизаторов, .связывающих фтористый водород, обеспечивает почти неограниченное хранение,
274
Гидролиз, ДФФ гидролизуется водой по следующей реакции:
(изо-С3Н7О)2Р/
+ н2о
_нр > (изо-С3Н7О)2Р/ ХОН
(I)
' При 15°С гидролиз 1%-ного водного раствора .ДФФ заканчивается лишь через 72 ч. Устойчивость к гидролизу проявляется Гакже в том, что малые количества ДФФ перегоняются с водяным jjapoM без образования сколько-нибудь значительных количеств фтористого водорода.
На гидролиз можно воздействовать. Температурная зависимость юнстаиты скорости гидролиза подчиняется уравнению Аррениуса
ь = ь _.й“л//гг к — кмакс е
ли, в дифференциальной форме: din fe А
' dT = RT* "
О каталитическом влиянии ионов гидроксила иа гидролиз уже оворилось. Строго говоря, в противоположность кислотному ката-изу в данном случае процесс ие является истинно каталитическим, «скольку участвующие в процессе ионы гидроксила расходуются.
Скорость гидролиза пропорциональна количеству гидроксил faux ионов и не зависит от ионной силы раствора п, из чего следует, то гидролиз ДФФ под влиянием оснований является обычной ре-кцией замещения атома фтора иа гидроксильную группу. Она за-Лючается в нуклеофильной атаке гидроксильных ионов иа атом фсфора, в результате которой становится возможным отщепление |£ила от атома фосфора. Механизм реакции основного гидролиза ,Йл приведен ранее иа стр. 269.
/ Константы скорости реакции k для ДФФ и некоторых других Соединений, приведенные иа стр. 297, позволяют сравнить гидролитическую устойчивость ДФФ с другими фосфорорганическими сочинениями.
Йо
По данным Саундерса, гидролиз ДФФ едким натром протекает следующему уравнению:
+2NaOH
(цзо-С3Н70)2Р/ —-—>• (изо- С3Н7О)2Р/
XF -NaF;—нар \(Жа
(2)
Проводились опыты и с избыточным количеством едкого нат-г Д — иа 1 моль ДФФ расходовалось 4 моль NaOH. Кроме того/ яло показано, что основному гидролизу значительно способствует <льиое перемешивание. Так, при 17 °C и встряхивании гидролиз ФФ проходил количественно по приведенной схеме в течение
.18*
275
"зи мин. Ъ параллельном опыте без встряхивания за это время про-гидролизовалось лишь около' 16% ДФФ и оставался неизмененный нерастворениый ДФФ, который исчезал после 5 мин встряхивания. При 2542 основной гидролиз за 15 мин приводит к полному отщеплению фтора8.
При действии концентрированных растворов щелочи возможен полный гидролиз ДФФ, приводящий к полному разрушению молекулы фосфорорганического соединения:
+ 4NaOH
(изо-СаН7О)2Р^ ---------> NaaPO4 + NaF + НаО -J- 2 азо-С3Н7ОН
(3)
• Поскольку при основном гидролизе даже по реакции (2) образуются физиологически неактивные продукты, для дегазации ДФФ в большинстве случаев достаточно применять разбавленные растворы щелочи.
Гидролиз ДФФ и других фосфорорганических соединений ускоряется рядом реагентов. Одним из наиболее эффективных катализаторов является гипохлорит-ион. Сходное действие проявляют анноны хромовой, вольфрамовой и молибденовой кислот.
Вагиер-Яурегг12 и Куртни13 обнаружили каталитическую активность некоторых внутрикомплексиых (хелатных) соединений медн, никеля, олова, титана, тория, уранила, цирконила и др. Наивысшей каталитической активностью обладают хелаты иона двухвалентной меди. Эти каталитические эффекты проявляются также и при гидролизе других фосфорорганических соединений, так что сказанное ниже можно перенести и на аналогичные соединения.
Каталитическое действие хелатных комплексов меди. Из изученных хелатных соединений металлов производные меди(П) наиболее эффективно способствуют гидролизу ДФФ. Хелаты железа и марганца каталитически неактивны. Реагенты — образующие хелаты—с понижением заряда меди меньше способствуют гидролизу. Имеются в виду соединения, которые связываются с центральным атомом меди ионной связью, а их нейтральная группа, кроме того, взаимодействует координационно, как, например, в хелатах, образованных 3-аминопропионовой I (р-алаиин) и (±)-2-амиио-3-оксипропионовой кислотой II [(±)-серин].
Значительно лучшие каталитические свойства проявляют хелаты, у которых лиганды связаны с центральным атомом меди
276
| только координационно. Например хелаты, образованные этилен-| диамином III, имидазолом IV и 2,2'-дипиридилом V:
NH2
/ ^СНг
С I
\ /СН2
nh2
N----СН
';с<' %н |
NH----СН
ш
IV
I; Особенно важны хелаты типа Cu2+X (X — лигаид). Хелаты типа Си2+Х2 — плохие катализаторы, это показано на примере этилен-
| диаминтетрауксусиой кислоты VI, которая с двухзарядными иона-ми металлов дает комплексы
типа VII:
ОН
О
нооснгс сн2соон N I сн2 сн2
yN
норсн2с сн2соон VI
сн2 сн2\
СН2
II
О
ГН 2
сн2
VII
/СН2 с
I он
Такие комплексы мало или ди а л кил фтор фосфатов.
Причиной каталитического т. меди типа Си2+Х, по-видимому,
совсем ие
катализируют гидролиз
хелатов двухвалентной
действия ----является образование с ДФФ про-
Д1 межуточного хелатного соединения VIII:
О
(СНОСНО,. °
;Си**Х
<5-
VIII
Ф, Отщепление атома фтора объясняется тем, что в результате ^.образования комплекса VIII полярность связей Р —О и Р—F уве-^личивается, что облегчает нуклеофильную атаку гидроксильного ||;иоиа на атом фосфора. Скорость гидролиза увеличивается с ростом Жжонцеитрации ионов гидроксила или хелата меди, ио эти увеличе-ж'Мия не пропорциональны друг другу.
ж?' Как установил Вагнер-Яурегг, в кислой среде два вакантных «координационных места хелата меди занимают молекулы воды образуется диаквокомплекс, в котором при увеличении
277
концентраций ЙОНОВ гидроксила молекулы воды замещаются ио нами гидроксила:
’ хн .
' ХСн2+ —► XCu2+f Н ,
\ /Н
+ но_ /ОН +но~
XCu2+(H2O)2 XCu2+; XCu3+(OH)2
4-H* \QH +H+
H
Активность катализатора приписывают дигидроксильиому комплексу двухвалеитиойдиеди/
Теория катализа фосфорорганических соединений хелатами меди до конца еще не выяснена. Очевидно, происходит несколько параллельных к встречных реакций. , . .
Ниже приводятся данные, полученные Вагиером-Яуреггом, иллюстрирующие ускорение гидролиза ДФФ хелатными комплексами меди(II). Без катализатора гидролиз ДФФ завершается в течение 2500 мин. Для проведения гидролиза к 10 мкмоль фосфата добавляли смесь C11SO4 и аминокислоты (по 50 мк моль каждого) и 2,2 мл буфера pH 7,6 (NaHCO)a и нагревали смесь при 38°С.
Лиганд Время 50и-ного гидролиза,
мин
Метионин (2-амино-4-тио метилмасляная кислота). ................................... 100
(±)-Сернн (2-амнно-З-оксип роп ионов ая
кислота) .......................... ’ 37
Р-Алании (3-аминопропионовая кислота) . 29
(±)-А ланин (2-аминопропионовая кислота) ........................................ 27 v
Глицин (2-амино уксусная кислота) . ... 27
(—)-Аспарагиновая (2-аминоянтарная)
кислота.................................... 26
Глутаминовая (2-амниоглутаровая) кислота 25.
Лизин (2,6-диамниокапронОвая кислота) . . 23
Этнлендиамин . ................................ 16
Имидазол . .................................... 14
4ИГ-Диметнл-2,2'-дипиридил...................... 9
(—)-Гистиднн [2-амяно-3-(имидазолил-4")-
пропноновая кислота] 8
2,2,-Днпнридил . . ........................... 4,5
N,N,N',N -Тетраметилэтилендиамин . . . 3—4 (экс трап.) .
Как можно видеть, хелаты меди(П) ускоряют гидролиз ДФФ во много раз. Наиболее подходящими катализаторами для гидролиза ДФФ считаются хелаты, образованные этилендиамином III, 2,2'-дипиридилом V и тетраметилэтилеидиамином IX:
(CH3)2N^CH2—СНг—N(CH3)2
IX
278
Хотя для дегазации в крупном масштабе хелаты меди вряд ли имеют значение, для обезвреживания мелких объектов они играют большую роль.
Реакции ДФФ с гидроксиламином и его производными. ДФФ и его аналоги, например зарин, реагируют с такими нуклеофильными реагентами, как гидроксиламии и его производные — гидроксамовые кислоты н оксимы, а также с перекисью водорода, перекисями и фенолами. Многие из этих реакций в более обобщенном смысле Можно рассматривать как каталитический гидролиз,
Гидроксиламии, и в первую очередь его производные—оксимы и гидроксамовые кислоты, с давних пор находятся в центре широких исследований. Эти соединения представляют большой интерес благодаря своей способности восстанавливать активность холинэстеразы, ингибированной фосфорорганическими соединениями. Наиболее хорошо известным соединен нем этого ряда является порученный в 1955 г. иоднстый метилпиридиинй-2-альдоксим.
При действии гидроксиламина ДФФ превращается в диалкилфосфат, Эта реакция приводит к тому же результату, что н гидролиз, и протекает в две стадии, причем образуется нестабильное промежуточное соединение14. Определяющей скорость процесса является первая стадия. Реакция идет в слабощелочной среде (pH 7,5) по следующей схеме:
+ nh2oh
---—> (C3H7O)2P^
~HF \onh2
/>
(C3H7O)2P^ X)H
из 4,5-10-3 моль ДФФ и 22,5-10-3 моль
щдроксиламина при pH 7,6 и температуре 38 °C ДФФ реагирует
(С3Н7О)2Р' \f.
В реакционной
смеси
+ 2NH2OH
—n2. —nh3, —2Н2О
Йа 50% за 108 мин 15,
;; Оксимы реагируют с
(Продуктов гидролиза. Особое значение имеют монооксимы а-днке-
онов16. Оии реагируют с ДФФ с образованием промежуточного Уединения, которое очень быстро разлагается. Отщепление атома ^тора и его замещение ионами гидроксила „объясняется катали-кческим действием оксим-аинонов, вследствие чего становится возможной нуклеофильная атака гидроксильных ионов. Реакция идет по Следующей схеме, которая приведена в общем виде, так Как она .справедлива и для других фосфорорганических соединений:
R. (RO)2PF + R'C—CR"
- АД >
0 0 N—O
(RO)2P—o— n о +HO“ -
(!) R"C—CR'
ДФФ и его аналогами с образованием
-> (RO)2P~ О- N о 4-F"
(!) R"(!—Cir
(RO),P -О’ + 1ГСОО’*+ R"CN + н+
279
Суммарное уравнение:
+ но"
(R0)2pf + R'C—CR" ----->-
О О N—О"
f
(RO),.P О- + R'COO” + R"CK + HF
О
Увеличение скорости гидролиза ДФФ действием оксимов пропорционально концентрации ионизированного оксима. Некоторые оксимы способны реактивировать ингибированную холинэстеразу. Это, в частности, такие О’ксимы, в молекуле которых имеется четвертичный атом азота, иапример иодистый N-метилпиридиний-2-альдокснм I, а также двучетвертичные соли алифатических диаминов, такие как дибромид пропан-1,3-бис- (М-пиридиний-4'-аль-доксима) II или дихлорид 2-оксапропан-1,3-бис-(Ь1-пиридииий-4'-альдоксим (токсогенин) III и простые соединения, как моноксим диацетила IV
CH=NOH
CH=NOH
N—CH2CH2CH3—N
2Br- •
CH=NOH CH=NOH
2СГ
ш IV
Гидроксамовые кислоты17 реагируют с ДФФ и его аналогами с образованием неустойчивых фосфорилированных гидроксамовых кислот. Эти кислоты сравнительно быстро разлагаются, давая продукты гидролиза фосфорорганического соединения и соответствующие изоцианаты. Подобно оксимам, активность гидроксамовых кислот приписывают их анионам. Определяющей скорость процесса является первая стадия, в которой анион кислоты выступает в качестве нуклеофильного агента. Замещение становится возможным лишь благодаря поляризации фосфорорганической молекулы анионом гидроксамовой кислоты в результате расшатывания связей Р=О или Р—F.
Высокую реакционную способность гидроксамовых кислот приписывают их таутомерным формам, так называемым гидрокснмо-вым кислотам, либо их аииоиам. Исследования показали, что ани- '
280
Генная форма более активна. Это проявляется в том, что скорость . реакции в слабощелочной среде (pH 7,5—7,6) значительно больше, Г чем при низких pH.
Возможный механизм реакций гидроксамовых кислот с ДФФ ^заключается в следующем. Прн действии аниона гидроксамовой ^кислоты отщепляется фторид-нон и образуется соответствующая ^фосфорилированная гидроксамовая кислота:
Z Z
(С3Н7О)2Р^ +~О— NH—(Г --------> (С3Н7О)2Р^ ,0
\F \R -F" Хо— NH—С/
г
В присутствии гидроксильных ионов это производное гидроксамовой кислоты перегруппировывается в гндроксимовую форму:
(С3Н7О)2Р +но
\о—nh—с; —>
\R -н2о
». В результате перегруппировки Лоссеня из гндрокснмового про-- изводного образуется продукт гидролиза ДФФ и соответствующий изоцианат:
(СзН7О)2Р^ /О' —> (С3Н7О)2Р^ +RN=C=O
\q—N=<Z ЧГ
Ж
При дальнейшем взаимодействии изоцианата с ионом гидроксамовой кислоты получается в качестве конечного продукта соответствующий уретан: ? /° i; £№0=0+"О— NH—cZ —> RNH—С—О—N-C—R
I ! 4
ДФФ, который в отсутствие катализатора гидролизуется на 50% течение 2500 мин и более, прн действии, например, бензгндрокса-^мовой кислоты реагирует на 50% уже за 22 мин, £ Наряду с другими, эффективными катализаторами гидролиза ^ДФФ являются никотннгндроксамовая н пиколингндроксамовая ^кислоты. Как показано ниже,’ реакционная способность азотсодержащих соединений сильно снижается в результате образования четвертичных солей (данные Хаклн, цитируемые по книге
281
О’Брайна 18). Опыты проводились в присутствии 8,5-гидр* оксамовой кислоты при pH 7,6 и температуре 30 °C:
Катализаторы Время 50%-ного
Гидролиза ,мин
Без катализатора........................ 2500 (3000)
Бенз гидроксамовая кислота ....... 22*
Никотингидроксамовая кислота........... 20
иодметилат . .... г................ 68
Изоянкотянгндроксамовая кислота .... - 28
иодметилат . ...................... 73
• В присутствии 22,5; 10 3 моль кислоты и 38 °C —только
9.5 мин.
Реакции с аминами™. В неводных растворах ДФФ реагирует' с аминами с образованием устойчивых соединений. При комнатной температуре, в реакцию вступают только первичные амины. Аналогичный хлор фосф ат в тех же условиях вступает во взаимодействие и с вторичными аминами. Образующиеся продукты нерастворимы в воде.
В' растворе четыреххлористого углерода при действии этилен-имина на ДФФЧ получается 0,0-диизопропил-№-этилеииминфосфатл т. кип. 76—78 °C (1—2 мм рт. ст.): >
/СН2 /Сн2
(«30-c3h7o)2pf + hn; I —> («до-с3н7о)ар—n; I
£ Vh, -HF Д \сн,
В результате реакции ДФФ с бензиламином можно' выделить гидрофторид бензиламина (т. пл. 149—150°C). В водных растворах реакция идет по следующей схе^е:
+н2о
(йзо-С3Н7О)2Р^ + CaHBCH2NH2—(изо-С3Н7О)2Р^ + C6HBCH2NH-lfF XF >ОН
При реакции ДФФ с циклогексиламином в водной ’суспензии окиси кальция (pH 10) за 150 мин отщепляется атом фтора~ и получается кристаллическая соль ДФФ и циклогексиламииа (т. пл. 193°C): . .
(изо-С3Н7О)2Р^° 4- HiNC.Hu + Н2О (азо-С3Н7О)2Р^°
XF- ~hf XOH-H2NCeHn
Реакции с фенолом. Механизм реакцйи с фенолом н его производными обсуждается при рассмотрении свойств зарина- (см. раздел 7.5.5.3); В данном случае речь идет о реакции ионизированного фенола с ДФФ. Реакция протекает по следующему уравнению:
(С3Н7О)2Р 4- С3Н6О — * > (С3Н7О)2Р.
XF ХОСвН5 *
282
Аналогично реагируют резорцин и гидрохинон. При pH 7,6 ДФФ Г гидролизуется фенолом на 50% примерно за 320 мин. При дей-? ствии ди- и триоксибеизолов гидролиз ДФФ протекает значительно ^быстрее. Так, период 50%-нопо гидролиза ДФФ составляет в слу-£'.чае 1,2-диоксибензола 27 мин, а в случае 1,2,3-триоксибензола— с-лишь 6 мин (температура 38°C).
Э Фенолы пригодны для дегазации ДФФ.
' 7.3.3.4. Токсические свойства. ДФФ — типичный ингибитор хо-
Глинэстеразы. Ои незаметно проникает в организм при вдыхании \ паров или в результате-резорбции через кожу. Малейшие количества ДФФ без'раздражения глаз вызывают сильное сужение зрачков. Приводим краткое описание наблюдений, полученных ^Саундерсом в проведенных им опытах.
ь- При пребывании в помещении объемом 10 м3, содержащем 20,0082 мг/л ДФФ через 5 мин наступало сильное сужение зрачков, (-.длившееся часто до 7 суток. При той же -концентрации и экспозиции менее 5 мин последействия иного рода ие наблюдались. Мио-этическое действие не ослабевает до 72 ч и приводит к полной понтере работоспособности подопытного. В отдельных случаях миоз 'можно было наблюдать до 3 месяцев. Этот эффект вызывают низ-кие концентрации; достаточно самых малых количеств паров, ад-t’;сорбированных одеждой, чтобы их действие проявлялось еще ие-сколько часов.
Действие на глаза характеризовалось следующим образом:
а) у всех подопытных сужение зрачков; часто зрачки величиной йдшь с булавочную головку, в глаз попадает очень мало света; с уменьшением интенсивности освещения зрение ухудшается;
б) индивидуальные изменения остроты зрения;
в) уменьшение степени аккомодации (ближнее зрение);
г) светобоязнь, головные боли и боли в заглазье; зрачковый рефлекс отсутствует,
г.- Опыты иа животных показали, что действие ДФФ при вдыхали паров и при инъекции существенно не различается. У крыс и [ышей, подвергнутых воздействию смертельных концентраций, начала наступало сужение зрачков и слюнотечение. Затем следо-&ло ослабление мышц, потеря мышечной координации, удушье, [оное и, наконец, остановка дыхания. При экспозиции 10 мин ,1Д50 составляла 0,36 мг/л для крыс и 0,44 мг/л для мышей. Наущение воздуха ДФФ приводило к смерти животных через 1 мин. Йыхание останавливалось до того, как переставало биться сердце. £ Легкие отравления следует ожидать при концентрациях порядка- 5* Ю 3—10“2 мг/л при экспозиции 5 мин, а концентрации выше б* 10 2 мг/л ведут к тяжелым и смертельным отравлениям.
Симптомы отравления, вызываемые у человека, совпадают с ’^мптомами у животных. У людей проявлялись потеря аппетита, шазмы внизу живота, понос, рвота,’ усиленное потоотделение и (люиотечсние, водянистые выделения из носа, сужение зрачков и бронхов, учащенное мочеиспускание, замедление пульса как- при
283
действии мускарина. Никотиноподобное действие вызывает тремор, мышечные судороги и паралич. Воздействия на ЦНС проявляются в резко гипертрофированных видениях, кошмарах, головных болях и бессоиице.
7.4, КЛ-ДИАЛКИЛАМИДО-О-АЛКИЛАЦИЛФОСФАТЫ
7.4.1. Общие сведения
Перед второй мировой войной в Германии под руководством Шрадера начались исследования фосфорорганических соединений, содержащих алкиламиногруппы. Кроме ряда ценных инсектицидов были найдены такие соединения, как Ь1,М-диметнламидо-О-алкил-цианфосфат (табун), представляющие интерес как ОВ, которые затем стали производиться в промышленном масштабе и были заготовлены в военных целях. •
Полученные сначала соединения типа I, не имевшие ацильных групп, оказались биологически неактивными. И только введение в молекулу кислотного остатка позволило Шрадеру и сотр, в период между 1937—1939 гг. получить активные соединения;
RO'7 \)R RO'7 RO'7 4n
I II HI
RHN< / R2N ,0
XP
RHn/ \f(CN) RsN^ \f(CN).
iv v
В центре внимания были вещества, которые из-за своей сильной токсичности для теплокровных в то время браковались как инсектициды; это особенно касалось Ы,М-диалкиламидо-О-алкил« фторфосфатов и N-N-диалкиламидо-О-алкцлцианфосфатов типа II и III и аналогичных бис-(алкиламидо)- и бис-(дналкиламидо)-производных типа IV и V.
Независимо от работ, проводившихся в Германии, в 1942 г. в Англии и в США Саундерсом, Баргом, Хипом и др. были изучены те же группы соединений и, в том числе, те же самые вещества20* Например Соединения VI—VIH:
(CH3)2N\ ,о р\
(CH3)2n/ \f vi
(CH3)2N
;0
(т, кип. 86 °C при (R —алкил, арил, Н;
15 мм рт. ст.) R*, R" — алкил, арил)
VIII . , 4
(т. кип. 50 °C при . 2 мм рт, ст.)
Диамидофторфосфаты типа IV и V представляют собой жидкие или кристаллические вещества. Они сильно токсичны при вдыхании
284
аров. Самым ядовитым соединением является бис-(диметил-мидо)-фторфосфат VI. Это неограниченно растворимая в воде жидкость. Для мышей (подкожно) ЛД50 составляет 1 мг!кг, для роликов (внутривенно) — 3 м.г!кг. Для мышей приводится ЛК50 ;09 мг{л. Как и все соединения этой группы, соединение VI не вызывает миоза, и, кроме высокой'токсичности, обладает превосход-ыми инсектицидными свойствами.
На живые растения бис-(диметиламидо)-фторфосфат действует ак системный яд (см. раздел 7.9.5); растения, обработанные этим >еществом, длительное время остаются токсичными как для насе-£омых, так и для теплокровных (!).
L Диамидофторфосфаты химически очень устойчивы, они не гидролизуются водой. По имеющимся данным21, ни один из полученных диамидофторфосфатов не пригоден в качестве ОВ.
Моноамидоалкилфторфосфаты VII более ядовиты, чем диамино-ТронзвоДные и, в отличие от них, вызывают миоз.
Особое внимание привлек М,М-диметиламидо-О-этилфторфос->ат VIII, свойства которого были изучены в Германии н в Аиг-фи. Это соединение примерно в 2 раза более ядовито, чем ДФФ. го ЛД50 для мышей составляет 2,5 мг!кг (подкожно). Это растворимая в воде и очень летучая жидкость; она была получена по хеме:
РОС13
+С2Н5ОН
---->
—НС1
CI о +2(CH3)2NH
С1/ \ос н —(CH3)2NH-HC1
(CH3)2Nk хО +kf (CH3)2N\ ^;О
—> X
с2н5о/ \С1 ~KC1 c2hsoz \f
I Исследовались также моиоалкиламино производные, средн них ^-фениламидо-О-этилфторфосфат IX
C6H5NH4 хО
с2н5о/ \f
IX
рдниенне, обладающее меньшей токсичностью (ЛД5о для мышей '.мг[кг, подкожно).
( Большое значение во время второй мировой войны приобрел N-диметиламидо-О-этилцианфосфат (табуи).. Несмотря на то, а табун из-за своей’меньшей по сравнению с другими ОВ токсич-Сти утратил значение как химическое ОВ ряда фосфорорганиче-ЙХ соединений, ои и теперь еще является предметом многочис-йных исследований.
В табл. 24 приведены некоторые представители ряда моно- и $лкилдиам идо фтор- и диалкилдиамидоциаифосфатов.
285
Таблица 24. Свойства некоторых дналкнламндофтор-и диалкнламндоцнанфосфатов (XX'P(O)Y]
В круглых скобках приведено давление в мм рт. ст.
Заместители т. КИП,, °C T. пл., °C Токсичность ЛДй, же/ка
X X' Y
(CH3)2N (CH3)2N F 86(15) — 1
(C2Hb)2N (C2H5)2N F . 124 (20) 160
(C4H9)2N (c4h0)2n F 59,5 16
C6HSNH csh5nh F — 145 90
C6HnNH C6H11NH - F 127 9
(CHO2N CeH5NH C2H6O C2H5O F F 76-78 (18) 100—150 (0,2) 50 2,5 10
(CHa)zN CH3O CN 60—70(1,5) 1,9
(CH3)?N c2Hso CN 108(12) 1 0,6
(CH3)2N изо-C3H7O CN 93-94 (4,3) — • 0,4
(CH3)2N C6HnO CN 140 (2) — —
(CH3)2N с2н5о CN 94-95 (2) 4,0
7.4.2. К,К-Диалкиламидо-О-алкилциаифосфаты
Соединения типа Ы,М-диалкиламидо-0-алкилцнанфосфатов следует рассматривать как алкиловые эфиры диалкиламндоцианфосфорной кислоты. _
Общим методом их получения может быть, согласно Шрадеру22, реакция диалкнламндодихлорфосфатов с эквнмольнымн количествами цианидов .щелочных металлов и соответствующих спиртов, в инертном растворителе, например в хлорбензоле. Реакция идет по следующей схеме:
X + NaCN + R'OH
R2N. X)
)P( 4-NaC14-HCi R'OZ \CN
Все известные до настоящего времени соединения такого типа, получают по этому методу. Даже взаимодействие с циклогексане- • лом не встречает никаких затруднений.
Необходимым условием ‘для успеха реакции является связывание выделяющегося хлористого водорода, которое достигается добавлением второго моля цианида щелочного металла.
Нужный для синтеза диалкнламндоднхлорфосфат получают по реакции хлорокиси фосфора с дналкиламнном:
XI
. POCi34-2RaNH —> R2N—pf + R2Nh - HC1
f\cl о
М,М-ДиалкиламидО'О-алкилцианфосфаты — умеренно растворимые в воде жидкости. Они более устойчивы, чем аналогичные дн-286
djjKH.mHdtupoLipd 1ы, чю можно ииъясчгить присутствием диалкил-. аминогрупп.
Как видно из рис. 15, скорость гидролиза изменяется с числом > : атомов углерода в молекуле. Увеличение размеров алкильных групп Н:приводит к стабилизации этих соединений 22ал
Рис. 15. Отщепление CN-ионов при гидролизе диалкиламидоалкил-цианфосфатов при pH 7,2 по данным Хол мстед та 22а;
/—О, О-диэтил циаифосфат; 2 — N, N-диметил амидо-О-метил циаифосфат; 3-N.N-диметиламидо-О-этил цианфосфат; 4— N, N-диметил амидо-О-проп ил цианфосфат;
। 5—Г4,М-диэтиламидо-0 -эти лци ан фосфат.
F,.
? Г
В нейтральных растворах гидролиз идет не только по схеме
R2N4 R2N4 .О
р; + сьг + н*
к + н2о
I;;а|Го уже на первой стадии затрагивается связь Р—N и происходит ^отщепление диал1киламниогрупп.
В случае основного гидролиза реакция протекает по уравнению;
Ь. R2N4 А) +но~ R2N4 хО
< Ro/ \cN ~cn* rq/ XqH c
, Предполагают, что образуется неустойчивое промежуточное coll единение следующего строения:
F СГ
р:
Как и следует ожидать, механизм кислотного гидролиза ^^N-дналкнламидо-О-алкилцианфосфатов иной, чем в случае ди-Ойкил ацил фосфатов. Причина этого кроется в природе этих
В 287
соединений. Остаток CN оказывает оолее слаЬое элемрифильние влияние, чем атом фтора или хлора. Ему противодействует нуклеофильное влияние диалкиламииогруппы, которая совсем ие обладает или может обладать лишь слабым электрофильным действием по сравнению с более сильным электрофильным влиянием алкоксигруппы. Электроположительная поляризация атома фосфора в таких соединениях будет слабее, чем в случае диалкилацилфосфатов. Вследствие этого атом фосфора менее склонен подвергаться нуклеофильной атаке, скажем, гидроксильных ионов.
Как установил Ларссон23, положительный мезомерный эффект атома азота аминогруппы устраняется добавкой ионов водорода в первой фазе гидролиза. В этом случае в результате возможной нуклеофильной атаки молекулы воды на атом фосфора может происходить лишь расщепление связи Р—N:
Скорость реакции в целом определяется первой стадией, на которой она пропорциональна концентрации ионов водорода.
Подобно ДФФ, М,М-дналкйламидо-0-алкилцианфосфаты при определенных условиях вступают во взаимодействие с такими нуклеофильными реагентами, как фенолы и гидроксиламииы.
7.4.3. NjN-Диметиламидо-О-этилциаифосфат
(СН3)г!\
х
СгН.СК \CN
Сии'сиимы: этиловый эфир диметила мндощаифосфорной кислоты, табун.
Военные обозначения: нем. — Tabun, Trilon 83, Т83; Gelan; амер. — GA.
7.4.З.1. Методы получения. По Шрадеру М,М-диметиламидо-О-этилцианфосфат получают из диметиламидодйхлорфосфата, который синтезируют по реакции хлорокиси фосфора с диметиламином:
2(CH3)2NH.+ РОС13 —> (CH3)2NP(O)C12 4- (CH3)2NH . НС1
При действии иа этот дихлорид цианистого натрия и этанола получается табун:
(СН2)2ЬЕ (CH3)2N<
+ 2NaCN + Сан5он —> + 2NaC14-HCN
Сг \С1 C2HacZ \CN
288
В Англии это соединение получают несколько необычной реак* дней из эфиров хлорфосфористой кислоты и амина:
2(CH3)2NH + (С2НБО)2РС1 —> (CH3)2N—Р(ОС2Нб)2 + (CH3)2NH*HC1
(CH3)2N4 .о
(ch3)2n—p(oc2hb)2 + icn —-> )р; + c2h5i
С2Н5с/ \CN
Промышленный способ. В «ИГ-Фарбениндустри» табун получали в две стадии: сначала синтезировали дйметнламндодихлор-фосфат, а затем замещали атомы хлора в днхлорнде иа цнан- и этокси группы.
На первой стадии пропускают газообразный днметиламин в хлорокись фосфора. Реакция экзотермнчна и приводит к образованию гидрохлорида диметнламнна (во избежание загустевания реакционной смеси применяют избыток хлорокиси фосфора, что позволяет осуществить эндотермический процесс):
(CH3)2Nx
(CH-jhNFbHCl + РОС13 —► Р^ +2НС1
С1Х ХЯ
В реакционной смеси содержится около 60% диметнламидодн-хлорфосфата, который фракционной перегонкой выделяют при пониженном давлении (степень чистоты ~ 99%),
На второй стадии происходит замещение атома хлора дн-хлорнда, для: чего это соединение вводят в раствор абсолютного этанола (стехиометрическое количество) в хлорбензоле и добавляют избыток цианистого натрия. Охлаждением поддерживают температуру реакции ниже 40 °C, Реакционную смесь подвергают фракционной разгонке в вакууме, причем хлорбензол возвращают в процесс. Выход табуна составляет около 80%,
Лабораторный способ. Лабораторный синтез табуна аналогичен промышленному процессу. Для получения диметиламидодихлорфосфата в двугорлую колбу емкостью 2 л, снабженную мешалкой и капельной воронкой, помещают раствор 77 г (0,5 моль) хлорокЯси фосфора в 500 мл безводного эфира. При перемешивании и охлаждении (—10 °C) медленно прибавляют раствор 45 г (1 моль) диметиламина в 300 мл холодного безводного эфира. Эфир отгоняют на водяной баие при небольшом вакууме. После удаления эфира для введения в реакцию образовавшегося гидрохлорида диметиламина прибавляют еще 665 г (4,35 моль) хлорокиси фосфора и реакционную смесь нагревают с обратным холодильником 20 ч в атмосфере двуокиси углерода. (
Избыток хлорокиси фосфора можно отогнать при атмосферном давлении (т. Кип. 105 °C). Остаток подвергают двукратной перегонке при 72 ?С (10 ммрт.'ст.). Выход диметиламидодихлорфосфата 104 г (0,64 моль). Это бесцветная жидкость с запахом фруктов; растворима в эфире, бензоле, этаноле и четыреххлористом углероде; в воде мало растворима.
Для получения N, N-диметиламидо-О-этилцианфосфата в двугорлую колбу емкостью 1 л помещают смесь из 23' г (0,5 моль) абсолютного этанола, 54 а (1,1 моль) цианистого натрия и 400 мл безводного хлорбензола. Из капельной вороики при охлаждении (0°С) и перемешивании прибавляют примерно в течение 25 мин по каплям 81 а (0,5 моль) диметиламидодихлорфосфата. Реакционную смесь нагревают 30 мин на водяной бане прн 40 °C, при этом образовавшийся при реакции цианистый водород отгоняется. После этого смесь нужно
1Q Зак. 448 289
перемешивать еще 90—120 мин. Хлорбензол удаляют при пониженном давлении (14 Мм рт. сг.) при 31 °C. Коричневатый остаток подвергают фракционной перегонке, причем собирают последнюю фракцию с т. кип. 100—108 v (9 мм рт. ст^). Выход продукта 59,5 г (0,36 моль).
7.4.3.2. Физические свойства. В чистом виде NjN-ДИметилами-до-О-этилцианфосфат — прозрачная . как вода жидкость; промышленный продукт имеет коричневатую окраску. Чистый препарат обладает запахом фруктов. Неочищенный продукт или частично разложившийся табун вследствие образования цианистого водорода пахнет горьким миндалем, д в высоких концентрациях — аминами.
Ра створи Af ость ОВ в органических растворителях — галогеиал-Каиах, бензоле и его гомологах, ацетоне и метаноле — хорошая; растворяется он также в некоторых ОВ. Табун ие растворим либо лишь плохо растворим в жидких углеводородах. Прн встряхивании с нефтью он дает эмульсию, которая при стоянии снова расслаивается. Растворимость в воде при 20°С составляет ~12%. Плотность табуиа больше воды.
Физические свойства табуиа следующие:.
Т. кип., °с
при 760 мм рт. ст.............. 237—240 (разл;) при 12 мм рт. ст.......... .... 108
при 10 мм рт. ст............... 101 — 103
при 7—8 мм рт. ст.............. 97—102
при 2 мм рт. ст»............. . 95—96
Т. пл., °C.................. . . —48 (—14)
Давление пара при 20 °C, мм рт. ст. ..... . 0,073
Максимальная концентрация при 20°С,. . . 0,6
Относит, плотность
пара........................ 5,6
*жидкасти ....................... 1,077—1,0971
Показатель преломления п^ . . ...... 1,4250
Вязкость прй 20 °C, спз . ... . .... 2,60
Табун нельзя перегнать без разложения при атмосферном давлении. Приводимые в литературе значения температуры кипения колеблются между 212 и 240 °C; найденная экстраполяцией величина лежит между 237 и 240°С.
В противоположность сравнительно высокой .летучести ДФФ, та- • бун, вследствие низкого давления пара (при 20°С 0,073 мм рт. ст.), обладает лишь небольшой летучестью, которая при 20 °C. составляет около 0,6 мг!л. По летучести он примерно соответствует иприту. Отсюда видно, что в случае табуна речь, идет об очень стойком ОВ, которое при благоприятных метеорологических факторах, хороших местных условиях н при температуре 18 °C из-за высокой токсичности может быть эффективным в воздухе в качестве ингаляционного яда до 24 ч.
Вследствие малой летучести табуиа немецкая фашистская армия предусматривала его применение преимущественно в вдде 290 . ..
аэрозоля, чтобы достигнуть возможно более массового уннчтоже-иия противника. 1
В литературе имеются различные данные о температуре плавления табуна (—14, —48°С и др.). Эти расхождения можно объяснить тем, что исследования часточпро«водили лишь с промышленными продуктами, достаточно чистыми для военного применения. То же можно сказать и о температуре кипения. Оказывается, что табуи можно применять при низких температурах. Для стабилизации его смешивают с органическими растворителями.
Фашистский вермахт вначале располагал раствором табуна, который содержал 5% хлорбензола (так называемый табуи А), затем содержание хлорбензола было увеличено до 20% (табун В), что
Рис. 16. Отщепление CN-ионов при гидролизе табуна при различных pH; значения ЛДэо водных растворов определяли иа белых мышах (по данным Холм-стедта 22а).
- повлекло за собой улучшение стабилизации ОВ. Хотя того же эффекта можно достигнуть и с другими растворителями, по экономическим причинам остановились иа хлорбензоле, который применяли в процессе получения табуна и затем регенерировали.
7.4.3.3. Химические свойства. Табуи неустойчивое соединение. При нагревании выше 200 °C ои отщепляет цианистый водород. При достаточной защите от корразии табуи может долгое время -краииться в металлических контейнерах; это также касается боеприпасов, заполненных табуиом. Сильное корродирующее действие . оказывает технический табуи, так как ои содержит значительные ;; ^количества диметиламидодихлорфосфата.
Гидролиз. Как уже отмечалось, гидролиз табуна22а происходит ие только с отщеплением иитрильиой группы, ио одновременно атакуется и связь Р—N. Только таким образом объясняется резкое уменьшение токсичности 'водных растворов табуиа (ср. рис. 16).
Ю* 291
---□—HeuyipepHUH—системе концентрация ионОв водородав ходе гидролиза увеличивается, поэтому связь Р—N полностью разрывается уже на первой стадии реакции. На отщепление CN-группы особенно влияют ионы гидроксила, тогда как образование амииа нужно объяснять влиянием ионов водорода:
(CH3)2N\^O
C2H5OZ ^CN
+Н0"
(CH3)2Nx Л)
+CN~
C2H5OZ ^ОН
(CH3)2NH\ я /р\ —>
С2Н5(Г 'CN
Н0\ +
;р; + (ch3)2nh3
c2hsoz \cn
В дистиллированной воде через 9 ч табун гидролизуется иа 50% с образованием синильной кислоты. Как показывает рис. 16, скорость расщепления связи Р—N зависит от концентрации ионов водорода. Вообще не ясно в какой мере в сильно кислой среде образуются CN-ноны. Установлено, что с увеличением концентрации ионов водорода отщепление диметиламнна ускоряется.
Скорость гидролиза табуна значительно увеличивается при нагревании. При ~ 95°С уже через 10 мин отщепляется 50% цианида. Для дегазации воды, зараженной табуном, достаточно длительного кипячения. Через 30 мин кипячения 1%-ный водный раствор табуна полностью гидролизуется. Если такая вода предназначена для пищевых целей, то, разумеется, нужно принять меры, чтобы вода не содержала цнаннд-ионов. Для технических целей такую воду, безусловно, можно применять.
По американским данным, предельная допустимая концентрация табуна в воде составляет 1,5—2 м. д./л прн максимальном расходе воды 5 л в сутки в течение 3 суток.
Большие количества воды можно дегазировать добавками гипохлорита нлн хлорированием в табельных системах додоочнсткн.
Растворами щелочей табун полностью разлагается:
(CH3)2Nk Л
+ 4NaOH —> (CH3)2NH + C2HSOH + Na3PO4 + HaO + NaCN C2H5O/ ZCN
Эта реакция пригодна для дегазации. При не слишком сильном заражении вместо растворов щелочи достаточно растворов гидроокиси аммония или теплых растворов соды.
7.4.3.4. Токсические свойства. Табун является типичным иервио-паралнтнческим ОВ, токснчноеть которого примерно в 5 раз превышает токсичность ДФФ. Отравление происходит в результате
292
вдыхания паров или резорбции через кожу. Очень легко табун проникает через слизистые оболочки, особенно через конъюнктиву.
Отравляющее действие, в зависимости от дозы, наступает сразу, без латентного периода, самое позднее, через 10 мин. Оно проявляется в беспокойстве, оживленности, тошноте, рвоте, сужении зрачков и бронхов, слюно- н потоотделении и других симптомах описанных для ДФФ. В случае отравления табуном преобладают тонические и клонические судороги, которые можно принять за паралич. К этому добавляются сильное беспокойство и страх, отравленный впадает в состояние шока. В дальнейшем наступают длительные явления подавленности, замедление ритма дыхания и сердечной деятельности. Смерть может наступить уже через 20 мин, при меньших дозах — через 24 ч.
Концентрации от 0,005 до 0,01 мг/л при воздействии в течение 2 мин приводят к легким отравлениям, которые, в сущности, заключаются в сужении бронхов и зрачков. Дыхание затруднено в течение 24 ч.
Тяжелые отравления прн том же времени воздействия наступают при концентрациях от 0,05 до 0,2 мг/л. Через несколько минут это приводит к затруднению дыхания, возбуждению, тоническим и клоническим судорогам, которые повторяются через нерегулярные отрезки времени. Они продолжаются примерно 2—4 мин, в некоторых случаях даже часами. Тяжелые отравления обычно смертельны.
Согласно Штаде, при нормальном дыхании, а также прн отсутствии физического напряжения, абсолютно смертельными являются концентрации:
Экспозиция, Летальная доза, мин мг/л
15 0,3
5 0,5-0,75
2 I
.1 1,25—1,5
0,5 1,75—2,0
Обычно Приводят среднюю летальную концентрацию (дозу)'для людей при экспозиции 1 мин в покое — 0,4 мг/л, в работе 0,1 мг/л.
Токсичность табуна при резорбции через кожу составляет 50—70 мг/к.г массы. Пероральная летальная доза равна 5 мг/кг массы.
7.5. О-АЛКИЛАЛКИЛФТОРФОСФОНАТЫ
7.5.1. Общие сведения '
Систематические военно-химические исследования, проводившиеся в фашистской Германии уже перед началом войны, привели к открытию О-алкнлалкилфторфосфонатов — соединений, токсичность которых значительно превосходила токсичность диалкнлфтор-
293
фосфатов. Некоторые представители этой группы оказались мощными химическими ОВ; одно из таких соединений, О-изопропил-метнлфторфосфоиат (зарни} как ОВ представлял больший интерес, чем табун. Завод для его производства был построен в Германии в конце войны.
На основе трофейной документации американцам удалось после 1945 г, за сравнительно короткий срок наладить промышленное производство зарина н усовершенствовать технологию процесса, ~
Вместе с так называемыми V-газамн О-алкилалкилфтор-фосфонаты в настоящее время относятся к важнейшим боевым ОВ.
В соответствии с их структурой, О-алкилалкилацнлфосфоиаты I нужно рассматривать как алкиловые эфиры ацилзамещеииой алкил фосфоновой кислоты, а фторпроизводные II — как алкиловые эфиры алкил фтор фосфоновой кислоты
7.5.2. Связь между химической структурой и токсичностью
Как н в случае дналкилфторфосфатов, существуют аналогичные взаимосвязи между молекулярным строением О-алкнлалкнлфтор-фосфоиатов н нх токсической активностью. Как и там, соединения с алкокснгруппой нормального строения проявляют меньшую токсичность по сравнению с соединениями с разветвленными цепями. Очевидно, в этом случае также необходим вторичный атом углерода при атоме кислорода алкокснгруппы, чтобы получилось высо-котокснчное соединение. Сравнение двух важнейших представителей этой группы, О-нзопропнлметнлфторфосфоиата III и 0-3,3- днметилизобутн л метил фторфосф он ат а IV показывает, что наличие, наряду со вторичным, третичного атома углерода ведет к значительному возрастанию токсичности.
(СНзЬСН-О^^О сн/ V
зарин
(ЛД60 0,1 мг!кг на мышах, подкожно)
(СН8)зС—сн-о^^о СН3/
IV
зомаи
(ЛД50 0.06 мг!кг да мышах, подкожно)
ш
с6н5о
СНд/' \р
цнклозарие
294
‘ -В результате изменения строения алкокси- или алкильной группы в молекуле следует ожидать более ядовитых соединений.
По сравнению с диалкилфторфосфатами О-алкилалкилфтор-фосфоиаты проявляют большую токсичность, что можно объяснить тем, что алкокси гр у пЦа заменена алкильной. Это явление можно наблюдать и иа примере других фосфорорганических соединений.
7.5.3. Методы получения
^Заключительные стадии синтеза О-алкилалкилфторфосфоиатов мо-j гут быть осуществлены двумя способами: а) этерификацией и фторированием соответствующего алкилдихлорфосфоиата; б) фторированием О-алкилалкилхлорфорфоната.
_ Хотя способ (а) является двуст адийиым, возможна одновременна я^этериф икания и фторирование.'
+R'OH;+P~ /OR, r-p; ------------>• r-p;
^XCl -Н+;-2СГ 1 \F
Ь В качестве (фторирующих агентов применяют фториды щелоч-&иых металлов или фтористый водород. Чтобы воспрепятствовать ^Образованию нежелательных побочных 'продуктов, фторирующий £агеит или спирт берут в избытке. В некоторых методах исполь-Оуются растворители или третичные основания.
Ё . В немецком «салин-процессе» зарин получали по реакции метил-Й|ихлорфосфоната с избытком фтористого натрия и изопропилового |а|пирта, тогда как в «перекрестном» процессе этерификацию проводили после фторирования. Возможна и обратная последовательность обеих реакций. Промежуточные продукты при этом ие выделяли.
Ь' При взаимодействии фторирующего агента с алкилдихлор-|фосфоиатом образуется адйилднфторфосфонат;
|Б Его можно вводить в дальнейшую реакцию отдельно или в |ймеси с алкилдихлорфосфонатом.
ft:_ Образующийся при реакции дифторида со спиртом фтористый Овдо род (реакция 1) действует и а продукт этерификации дихло-1фида (реакция 2) и в результате фторирования образуется другая ^молекула О-алкилалкилфторфосфоиата (реакция 3). В качестве
В _ 295
побочного продукта (реакция 4) образуется 0,0-диаЛкилалкнл-
фосфонат24,
^\р^° +^'Он R^^O
F/ \F —hf \F AV
R\ zsO +R'OH Rk zO
(2)
Clz \C1 “HC1 R'OZ \ci
RxZ +m, R\fZ° (3)
R'O/ \C1 -HCI R'O^
R< zO +r'OH R4 zO
Ap-/ >. (4)
R'CZ ZC1 ~HCI R'OZ ZOR'
По способу (б) О-алкилалкилхлорфосфонат, получаемый действием фосгена на соответствующий О,О-диалкилалкилфосфоиат
R\ +coci2 R\.
далее фторируют фторидами щелочных металлов или фтористым водородом (см. выше, реакция 3). В том и другом способе для по* лучения необходимых полупродуктов требуется ряд разнообразных превращений (см. раадел 7.5.5.1).
7.5.4. Свойства
О-Алкилалкилфторфосфонаты — бесцветные жидкости, иногда со слабым запахом. Низшие члены ряда хорошо растворимы в воде. Так, О-изопропилметилфторфосфонат'смешивается с водой во всех отношениях. Как и следует ожидать, с ростом числа атомов углерода растворимость уменьшается. То же происходит и при наличии арильных групп в молекуле. Так, 3,3-диметилизобутильное производное IV (зоман) лишь умеренно растворимо в воде, а фенильное производное V — почти нерастворимо.
Рассматриваемые соединения обладают малой летучестью. Однако вследствие их сильной токсичности, предельная концентрация при благоприятных условиях и соответствующей плотности заражения достаточна для эффективного заражения' воздуха.
Водой О-алкилалкилфторфосфонаты гидролизуются по следующей схеме:
ROZ \f —hf ro/
Как и в случае диалкилфторфосфатов, у фосфорорганических Соединений этой группы с увеличением размеров алкильных групп наблюдается постепенное повышение устойчивости к гидролизу.
296
В табл. 25 приведены определенные Ларссоном константы скорости k реакции второго порядка для гидролиза некоторых О-алкилалкилфторфосфонатов при pH 8—9. Они демонстрируют влияние строения этих соединений на их способность к гидролизу. Большая величина константы скорости k соответствует неустойчивой молекуле. Для сравнения приводим подобные данные для ДФФ и табуна. Так величины k для этих соединений равны 0,83 н 7,49 л • моль~х • сект1, а энергия активации табуна равна 10,1 ккал) моль.
Таблица 25. Константы скорости и энергии активации гидролиза О-алкнлалкилфторфосфонатов [R'O(R)P(O)F]
R R' k при 25 °C, — I _ —I л* моль *сек Энергия активации двА ккал/моль
СН8 сн8 106 10,5
СНа сгн6 60,7 11,2
СН8 я-С3Н7 54,0 10,4
сн3 (CH3)SCHCH2CH2 49,3 12,0
сн8 (СНз)аСН 25,8 9,1
с2н5 (СНз)2СН 9,35 10,7
(СН3)2СН (СН3)2СН 2,03 9,2
Механизм гидролиза и воздействующие иа него факторы, по существу, такие же, как и в случае дналкнлфторфосфатов. Спонтанный гидролиз с заметной скоростью начинается прн pH 7. С увеличением концентрации гидроксильных ионов скорость гидролиза сильно возрастает. Так, прн pH 12 гидролиз, зарина происходит почти мгновенно.
Наряду с соединениями III—V в качестве потенциального ОВ рассматривался так называемый этилзарни VI:
изо-С3Н70
С2Н8
VI
Считается, что этилзарин(О-нзопропилэтилфторфосфонат) можно легко получать в промышленности из треххлористого фосфора реакцией с этаном и кислородом через этилдихлорфосфонат25:
С2Н8\
РС13 + СНзСНз + 72о2 )р^ 4- НС1
Сг \С1
Этилдихлорфосфонат вводят в дальнейшую реакцию, как описано в разделе 7.5.3 (метод а).
297
z Этилзарни— жидкость, обладающая меньшей летучестью, чем зарин; его токсичность составляет примерно у8 от токсичности зарина. Ои обладает следующими физическими свойствами:
Т. кип., °с........................ .
Давление пара при 20 °C, мм рт. ст . .
Максимальная концентрация1 при 20 °C, мг/л ............. ... .
Относит, плотность d^° .........
Показатель преломления Яд...............
67—68 (18 мм рт. ст.)
0,0959
8,0
1,0552
1,3817
7.5.5. О-Изопропил метил фторфосфоиат
СН3к >0
/р\ «зо-С 3НТ(Х 'F .
Синонимы: изопропиловый эфир фторфосфоцовой кислоты, зарин.
Военные о б озн а че и и я; нем. — Sarin, Trilon 144, Т144, Trilon 46, Т46; амер. — GB.
Согласно точке зрения американских военных, зарин, наряду с так называемыми V-газами, имеет большое значение в химической войне для заражения воздуха, продовольствия, систем водоснабжения н др. ’ . •
Зарнн был получен в Германии в 1938—1939 гг. Перед окончанием войны на ее территории в стадии строительства находились две промышленные установки для его производства общей производительностью 600 т в месяц. По французским данным, в 1944— 1945 гг. на пилотных установках было изготовлено около 30 т зарина. В настоящее время зарнн (GB) занимает значительное место в американском производстве ОВ,
7.5.5.1. Методы получения. На завершающей стадии, процесса зарнн получают уже описанными методами (см. раздел 7.5.3). В методе (а) используют метилднхлорфосфонат, а в методе {б) -в дальнейшие превращения вводят О,О-дннзопропнлметнлфосфо-нат. Для обоих способов справедливы ,следующне^уравнеиня реакций: . •
+3NaF; +озо-С3Н7ОН
—2NaCl; — NaHFj
СНзх .О
«э0'С3НтСг
(а)
+COCI2 -----------------> —В30-С3Н7О; —СОа
с113\р^° ияо-С3Н7О^
<б)
Метилднхлорфосфонат получают разными Методами из трех-хлорнстого фосфора в несколько стадий.
298
Шрадер26 в своем синтезе исходил из диметил фосфита, который i действием натрия превращали в натриевое производное диметил-Гфосфита (реакция 1). Дальнейшее взаимодействие с хлористым 'метилом привело к 0,0-диметилметилфосфоиату (реакция 2), из ^.которого обработкой хлором в присутствии треххлористого фое-<• фора был получен метилдихлорфосфонат:
(СН3О)гР—ОН -3^* 1(СН,0),P-О] Na+ (О
[(СН3О)2Р—О]“ Na+ СН3—Р(ОСН3)2 —NaCI || 0 (2)
СН3—Р(ОСН3)2 +2CI*+2PCI1 , СНз-PCl, —2POCl3; —2СН3С1 1| (3)
I О,0-Диметилметилфосфоиат получают также, взаимодействием fметанола с хлором и треххлористым фосфором.
; Возможно27 превращение триметилфосфита в О,О-днметнл- метилфосфонат с помощью метилгалогенида в результате перегруппировки Арбузова:
(СН3О)зР (СНзО)аР^
к ХСН3
L Перегруппировка происходит при нагревании. По-видимом у, при этой перегруппировке - речь идет 6 каталитической реакции, особенно в том случае, когда алкильные группы фосфита и алкил-галогенида одинаковы. Достаточно уже небольшой добавки алкил-галогенида, чтобы происходила изомеризация значительного количества фосфита. Вместо йодистого метила можно применять Метилсульфат. Скорость перегруппировки Арбузова зависит от числа атомов углерода алкильной группы и с ростом этого числа уменьшается. Механизм реакция Арбузова следующий:
(ЦО)зР
+R'X
RO—Р
ROZ
RC\
Р +RX
RO^ ^0
; , Подобные реакции протекают и с другими аналогичными сое-лДииеииямн. Так, реакцию диметилфосфита с метилатом натрия в : Присутствии хлористого метила, при которой тоже образуется 0,0-диметилметнлфосфО'нат, следует считать также реакцией Арбузова:
(СН3О)2Р—ОН
+СН»О№; +CH3Ci .
—NaCI;—СН3ОН ,\С]
299
Эта реакция послужила основой промышленного производства 0,0'Диметилмстилфосфоната для синтеза зарина. Диметилфосфит получают по реакции треххлористого фосфора с метанолом без добавки третичного амина:
РС1э + ЗСН3ОН —> (СН3О)2Р—ОН + СН3С1 + 2НС1
Та же реакция в присутствии амина приводит к тр и метилфосф ит у:
РС13+ЗСН3ОН > (CH3O)3P+3R3N-HC1
Из-за дороговизны амина она оказалась непригодной для промышленного производства фосфита.
В так называемом «перекрестном» промышленном процессе производства зарина применялся другой метод получения окиси метил-дихлорфосфоната из диметилфосфита, полученного из треххлористого фосфора и метанола без третичного амина.
Выше 100°С диметилфосфит неустойчив. Ои изомеризуется в метиловый эфир метилфосфоиовой кислоты:
СНзОк >0 (СН3О)2Р—ОН —> )Р(
СН/ ^ОН
Две молекулы этого эфира с отщеплением диметилового эфира конденсируются с образованием диметилпирофосфоновой кислоты:
О 0 0
II л И л 1
2СН3—Р—ОСН3 —> СН3ОСН3 + СН3Р—О—РСЦз он он он
С повышением температуры, ио ие выше 300 °C, реакция идет главным образом по этой схеме с образованием пирофосфоната. Оба продукта реакции при обработке хлором и треххлористым фосфором дают метилдихлорфосфонат:
3^Р^ ----+2?_г:-+2РС1а-> СН3—РС12
СНзО^ \)Н -снзс1: -2РОС1з>—нш
0 0
II II +ЗС12; +3PCJ3
СН3Р—О—РСН3 > 2СН3-РС12
। । —ЗРОС1з; —2nd г?
он он о
В производстве зарина можно использовдть смесь, обоих соединений.
Кроме того метилдихлорфосфонат можно получать по реакции треххлористого фосфора с хлористым метилом и безводным-хлористым алюминием2®:
РС13 + А1С13 4-СН3С1 —> [СНзРОД* [А1С11Г
300
Раствор 'комплексной соли в хлористом метилене разлагается при добавлении воды, а еще лучше концеитрироваииой соляной кислоты при —30 °C в результате гидролиза с образованием метил-дихлорфосфонага:
[СН3РС13]+[А1С14Г - +7Н?.% СНз—РС12 + А1С13 • 6Н2О + 2НС1
После отделения хлористого алюминия хлористый метилен отгоняют при пониженном давлении. Полученный метилдихлорфос-. фонат превращают в зарин упомянутыми выше методами.
Этот способ получения зарина и других фосфорорганических соединений, разработанный впервые Киниером и Перреном29, был в некоторых странах использован для промышленного производства. '
Переработка остатков от гидролиза заключается в нагревании, отделении летучих компонентов, кальцинировании и хлорировании. Из газовой смеси выделяют треххлористый фосфор в виде комплекса с хлористым алюминием. Можно также получать из него чистую окись алюминия. Таким образом, рентабельность процесса обеспечивается переработкой остатков после гидролиза'30.
Вместо соляной кислоты для гидролиза промежуточного комплекса с хлористым алюминием пригодны такие соединения, как метоксиметилтетраэтиленгликоль СН3О(СН2СН2О)4СНз, более индифферентный к образующемуся метилдихлорфосфоиату.
Динзопропилметнлфосфоиат можно получить тремя способами:
1. Этерификацией метилдихлорфосфонатаизопропнловым спиртом в присутствии третичного амина:
гчд DPI Ч-2 изо-С3Н7ОН; +2R3N fTT D/Af и .
СН3РС12 --------*д.-----------> СНэР(ОС3Н7-«эо)2
—2RSN-HC1
Для промышленного производства зарина эта реакция, п©-видимому, почти ие имеет значения, так как более рентабелен непосредственный синтез его из метнлдихлорфосфоиата.
2. Превращением диизопропилфосфнта в натриевую соль ди-метилфосфита, которую далее вводят в реакцию с алкилгалоге-индом:
(изо-С3Н7О)2Р
> [(изо-С3Н7О)2РО]' Na*
—1/2Н2
+сн3х_
—Nax’>
(изо- С3Н7О)2Р
3
301
3. По реакции триизопропил фосфита с иодистым метилом (реакция Арбузова):
+сн31 (изо-С3Н7О)3Р -------> (цзо-С3Н70)2Р^
—нзо-с3н71 ^СНз
Триизопропилфосфит получают взаимодействием изопропилового спирта с треххлористым фосфором в присутствии третичного амина. Как уже упоминалось, без третичного основания при одновременной обработке хлором образуется диизопропилфосфит.
Промышленный способ получения зарина. В Германии зарин получали так называемыми салииовым и перекрестным процессами. И тот, и другой процессы идут в четыре стадии.
Салин-процесс можно схематически представить следующим рядом реакций:
+ CH3ONa; +CI2;
рсь (сн3о)2р~он .+CHLCI> (сн3о)2рсн3
(Н (2) || . (3)
о
+NaF; СН34 /О
___w +нзо-С3Н7ОН 3\_^
--> С Г1 з г С1 j ----> jF.
II м) «зо-С3Н70^ \F
Первую стадию салин-процесса (взаимодействие абсолютного спирта с треххлористым фосфором) проводят при интенсивном охлаждении. Эта реакция сильно экзотермическая, поэтому смесь охлаждают во избежание изомеризации и конденсации. На стадии (2) полученный диметилфосфит в бензольном или метанольном растворе вводят при 30 °C в реакцию с метилатом натрия в присутствии хлористого метила. На стадии (3) алкоксигруппы заменяют иа атомы хлора обработкой хлором в присутствии треххлористого фосфора; иа стадии (4) при 60 °C атомы хлора замещают иа фтор избытком фтористого натрия и на изопропоксигруппу — изопропиловым спиртом.
Схема «перекрестного» процесса следующая:
РС13 -+£Нз°> (СН3О)2Р—-ОН (и
СНз°\
СН/хОН
О о
С Н3—Р—Q—р—СН3
он он
СН3РС12 —
II
0 +изо-с3н7он
+hf| И)
+С12; +РС13
---------->
(3)
chZ
CH3PFa —
А
302
Первая стадия процесса — та же, что и при салин-процессё. Но /если в салин-процессе интенсивным охлаждением противодействуют изомеризации и конденсации, то в перекрестном методе к
;этим процессам стремятся и поэтому реакцию ведут при иагрева-нии выше 100“С. Смесь продуктов изомеризации и конденсации обрабатывают на стадии (3) хлором и треххлористым фосфором при 70-°С для получения метилдихлорфосфоиата. Действием безводного фтористого водорода получают смесь равных количеств дихлорида и дифторида, из которой иа стадии (4) получают зарин обработкой избытком изопропилового спирта.
В США синтез зарина был упрощен: метилдифторфосфонат
стали получать сразу после стадии изомеризации-конденсации, .мииун стадию получении метилдих/йэрфосфоиата. Дифторид получают 31 реакцией О,О-диметилметилфосфоиата или метилового ^афира пирофосфориой кислоты с безводным фтористым водородом £дри 100—200°С под давлением 3—25 ат. Далее дифторид превращают в зарин действием спирта в присутствии третичного амина. ?-'• Зарии. очищают от кислот и других фосфорорганических соеди-нёний растворением в эфире и пропусканием через колбнны, заполненные насыщенным водой силикагелем. Примеси задерживаются
в колоиие, их вымывают водой, а зарин — эфиром.
На некоторых американских заводах зарин получают через соответствующий комплекс алкилхлорфосфоната с хлористым алю-
минием (стр. 300). Сравнительно дорогие катализаторы Фри-
деля— Крафтса регенерируют или перерабатывают в другие цеи?
ные алюминиевые соединения. Комплексообразование происходит 'ft' растворителе, например, в хлористом метилене, 1,1,2-трихлор-
’ЭТилене, тетрахлорэтане и других галогенированных углеводородах.
Л. 7.5.5.2. Физические свойства. ЭЬрии — жидкость без цвета и без запаха. Ои гигроскопичен и смешивается с водой во всех отношениях. При нагревании до температуры кипения он разлагается. К кратковременному воздействию высоких температур, развиваю-
щихся при взрыве, он более устойчив, чем табун.
'£. Ниже приведены физические константы зарииа:
при 760 мм рт.ст. . ................ 147,3 (разл.); 151,5
при 15 мм рт. ст............................. 56,5—57
при 12. мм рт. стг ♦ .................. 50
$7 при 10 мм рт. ст................................... 48—49
. Т. пл., °C ................................ -57
> . Давление пара при 20 °C, мм рт. ст........ 1,48 (1,75; 1,57; 1,43)
»£' Максимальная концентрация при 20 °C, мг/л 11,24 (12,0)
. Относит. ПЛОТНОСТЬ
пара................................... 4,86
К’/ ЖИДКОСТИ
Л- при 20°С............................. 1,1102—1,0943
“ при25°С. ......................... . 1,0941-1,0887
при 30 °C.................................. 1,0829
303
Показатель преломления
при 20 °C...................... 1,3830
при 25 °C................... 1,3790
Вязкость при 20° С, спз ........... 1,82
Опубликованные в литературе физические константы зарина очень между собой различаются. Так, например, для температуры плавления приводят значение — 57 °C. При этой температуре зарин застывает в стеклообразную массу, иногда даются другие значения, которые, по-видимому, определены для неочищенного зарина или для его растворов.
Давление пара зарина (в мм рт. ст.) в интервале температур 0—60/С можно вычислять по формуле:
1g р = 9,8990 --^1
Хотя летучесть зарина сравнительно высока, ввиду большой токсичности это ОВ можно считать как летучим, так и стойким.
В табд. 26 приведены величины давления пара и соответствующие значения летучести, вычисленные по формуле:
lg cs Т = 13,2505 —
2850,9 Т
Таблица 26. Давление пара и летучесть зарина в интервале 0—30 °C
Температура, °C Давление пара, мм рт. ст. Летучесть, мг/л Температура, °C Давление пара, мм рт. ст. Летучесть, ме[л
0 0,29 2,4 20 1,48 11,3
5 0,44 3,6 25 2,17 16,3
10 0,67 5,4 30 3,11 23,1
15 1,01 7,9
Те же данные приведены на рис. 17.
При благоприятных метеорологических условиях чистый зарин сохраняется иа местности в жидком виде летом до 5 ч, а его пары еще могут быть эффективными через 20 ч; в зимних условиях считается, что зарин стоек до 2 суток. Если сопоставить смертельную концентрацию паров зарина, которая при экспозиции 2—5 мин составляет самое большее 0,05 мг}л, с его летучестью, то при учете естественных факторов .ослабления оказывается, что при ]0°С и более низких температурах в воздухе образуются безусловно смертельные концентрации зарина.
При благоприятных метеорологических условиях вследствие высокой токсичности зарина в направлении распространения облаков отравляющих веществ возникают зоны чрезвычайной опасности.
304
Смертельные или очень опасные концентрации зарина могут возникать даже на расстоянии 15—25 км от места применения. Растворимость зарина в органических растворителях хорошая, он во всех отношениях растворим в спиртах, сложных эфирах, кетонах, Талогеналканах, бензоле, толуоле и др. Зарин смешивается с такими ОВ, как иприт и ДФФ.
Пары зарина легко адсорбируются различными материалами, например, текстильными тканями, особенно шерстяными, древесиной, пористым кирпичом и бетоном.
Рис. 17. Давление пара и летучесть зарина в интервале 0—30 °C.
♦ В результате последующей десорбции зарина в ранее- незаряженном воздухе могут создаться столь высокие концентрации десорбированных паров зарина, что при известных условиях они -могут оказаться смертельными. Это особенно касается закрытых • помещений, в которых была сложена одежда, зараженная зарином.
7.5.5.3. Химические свойства. О-Изопропилметилфторфосфонат сравнительно устойчив. Для длительного хранения его нужно стабилизовать, особенно прн хранении в металлических сосудах. Стабилизующее действие оказывают аминни нлн такие растворители, -как метанол, галогеиалканы.
В химическом отношении зарцн ведет себя почти аналогично Динзопро'пи л фторфосфату. Он гидролизуется с заметной скоростью. На гидролиз оказывают влияние те же реагенты, что и в случае ДФФ. Рассмотренные для ДФФ механизмы каталитических реак-Ций применимы и для реакций зарина.
- Разрыв связи Р—С возможен только в жестких условиях. Легче : Всего разрывается связь Р—F.
/. • у Гидролиз. В водных растворах зарин гидролизуется по следую-> щей схеме:
U3O~ С3Н7О4
+на0
-HF
изо-С3Н7Ск хО
СН,/ '''ОН
305
Скорость гидролиза зависит от температуры и от концентрации ионов водорода. С повышением температуры иа 10°С скорость гидролиза в нейтральной среде увеличивается почти вдвое. Хотя в течение долгого времени отрицали спонтанный гидролиз зарина в воде, исследования Эпштейна и сотр.32 показали, что эта реакция с заметной скоростью начинается при pH 7. '
По мере гидролиза токсичность остающегося раствора зарина непрерывно уменьшается. В результате образования фтористого
водорода, что равносильно повышению концентрации ионов водорода, сначала наступает стабилизация скорости гидролиза (pH 4) и только в сильно кислой среде проявляется каталитическая активность ионов водорода33 (рис. 18). Как видно из графического изображения хода гидролиза, минимум скорости гидролиза лежит в области pH 6,5—4,0, при этом время полупревращения составляет 175 ч.
Чтобы поддерживать при спонтанном гидролизе концентрацию ионов водорода выше 10“*, требуется, чтобы начальная концентрация зарина составляла по крайней мере 5 мг/л. Если вследствие малых концентраций зарина или присутствия в воде буферных веществ поддерживаются пределы указанного интервала
Рис. 18. Гидролиз зарина при 25 °C в зависимости от pH по данным Лоса33.
pH, то спонтанный гидролиз зарина подавляется и вода иа длительный период остается зараженной. Предельная допустимая концентрация зарина в питьевой воде составляет 0,5 м. д./л при максимальном расходе воды 5 л в сутки в течение 3 суток.
В щелочной среде зарин ведет себя подобно ДФФ. Существует пропорциональность между скоростью гидролиза и концентрацией гидроксильных ионов. Время полупревращения (в ч) для гидролиза при 25 °C в области pH от 7 до 13 можно вычислить по формуле:
t',lt = (б,4* Ю“8) • Ю"ри •
Бич и Сасс34 приводят следующие данные о продолжительности осуществления 1%-иого и 99%-иого гидролиза 0,001 М раствора зарина в воде при 25°C (в ч): ‘
pH lK-ный гидролиз 99К-НЫЙ гидролиз
1 2 100
3 15 6000
5-6 120 100000
9,5 0,14 66
11,5 0,003* 1,3
* Экстраполировано.
306
г: Из этих данных видно; что степень гидролиза зарина зиачй-
£ тельиа даже в слабощелочной среде, поэтому зарии быстро гидро-лизуется в разбавленных растворах щелочей или в растворах Ж аммиака или соды. Продукты гидролиза неядовиты. В щелочных В;7 растворах реакция идет по уравнению:
С3Н70ч хО +2NaOH C3H7Ck хО /Рх --------------> /к
L . СН3/ XF -NaFi-H2O СНз/ XQNa
I? В сильно концентрированных растворах щелочей в результате r-Г. гидролиза происходит расщепление связи Р—О:
С3Н7О. Ь +3NaOH
-------> C3H7OH + CH3P(ONa)2,
CHZ 4 —NaF; —h20 I
Связь Р—С ие разрывается даже при длительном нагревании с концентрированными щелочами.
Каталитическое влияние иа гидролиз зарниц оказывает ряд Ч анионов. К наиболее эффективным катализаторам относятся гипо-хлорит-ионы, которые по активности превосходят гидроксильные ионы.
Предполагается, что на первой стадии атом фтора обменивается непосредственно с гипохлорит-иоиом (реакция 1). Это превращение определяет скорость реакции в целом. Затем происходит быстрый гидролиз образовавшегося (реакция 2):
С3Н7ОХ ,0
CH3Z \f
j С3Н70ч Xi
промежуточного соединения
сн/ \ОС1
+C1O-
—F”
+H2o
Р CH3Z ХОС1
С3Н7СХ £)
(1)
—Н+; -0СГ CHZ \0Н
(2)
$
Причиной высокой реакционной спосцрности гипохлорит-иоиов считают образование циклического переходного состояния I или П при атаке фосфорорганической молекулы. Это приводит к увеличе-иию положительного заряда иа атоме ^фосфора.. Промежуточные Продукты очень реакционноспособны и склонны к нуклеофильному ^\|ВаМещеиию атома фтора гидроксильными ионами воды.
«зв-С3Н7О О
Р С1
CH3. F O-
изо-С3Н70 О
р—......О
СНз^ ^F—С1
П
Скорость гидролиза, которая определяется первой стадией ^/реакции, пропорциональна концентрации гипохлорит-иоиов.
307
Каталитическое действие лииейио зависит от содержания активного хлора:
«гидр « k 1зарин] 1С12]
Каталитическая активность гипохлорита зависит от концентрации иоиов водорода, она особенно велика в щелочной области. Для дегазации зарина* при pH 7 требуется лишь Vs концентрации активного хлора, необходимой при pH 6, а при pH 8 — лишь ’А концентрации активного хлора, нужной при pH 7. В то же время это показывает, что ответственными за каталитический эффект следует считать гипохлорит-ионы.
Превосходное каталитическое действие растворов гипохлоритов, при помощи которых можно быстро и надежно проводить дегазацию зарина и других фосфорорганических ОВ, например ДФФ, табуиа и зомаиа, привело к тому, что растворы гипохлорита приняты некоторыми армиями в качестве дегазирующего средства.
Сходный каталитический эффект оказывают хромат,-, молибдат-и вольфрамат-ионы (СгО^, MoOt", WO^), ио их активность меньше, чем гипохлорит-иоиов.
Причиной их каталитического действия считают комплексообразование аниона с молекулой зарина, вследствие чего облегчается электрофильная атака молекулы воды иа атом фосфора. Интересно при этом, что речь идет об анионах, которые к тому-же склонны давать- нерастворимые комплексы с фосфат-иоиами.
Тиосульфат-ион S2O3A хотя и обладает большой нуклеофильностью, ие ускоряет гидролиза зарина и табуиа. То же относится к иону МпОТ, каталитическое действие которого недостаточно, чтобы его можно было применять в качестве дегазатора.
На гидролиз зарина оказывает каталитическое влияние также ряд катионов-. Самыми активными катализаторами являются некоторые хелатные комплексы металлов, из которых, по исследованиям Куртни и сотр.13, самыми эффективными, за некоторыми исключениями, опять-таки оказываются хелаты двухвалентной меди.
Причиной каталитического-действия в этом случае (ср. раздел 7.3.3.3) также является образование активного промежуточного комплекса, в котором полярность связей Р—О и Р—F больше, в результате чего атом фосфора становится менее устойчивым к действию молекул воды.
В табл. 27 приведено время полупревращения при гидролизе зарйиа в присутствии различных хелатов металлов13.
В порядке убывания активности хелаты располагаются в следующий ряд (приводятся лиганды): Н,1Ч,1Ч',1Ч'-тетраметилэтилеиди-амии, М,М'-диметилэтилеидиамии, пропилеидиамин, этилеидиамии, N, М'-диоксиэтилэтилеидиамии, N-оксиэтилэтилендиамии.
• Более подробно этот вопрос рассматривается в т. 2 дайной книги.
308
Таблица 27, Ускорение гидролиза зарина хелатными комплексами металлов при 25 °C
Металл Лиганд pH Концентрация, мкмоль1л Ч’ мин
металла лиганда зарина
Си (II) N, N'-Диоксиэтил- 7,0 10 740 10 740 955 4,1
этилендиамин 7,15 1 135 1 135 950 12,2
6,1 1250 1 250 1 050 57,0
8,0 1 050 1 050 1 050 11,0
Си (II) N-Оксиэтилэтилеи- 7,0' юозо 10 030 970 4,6
диамин 7Д 1020 1 020 990 15,5
6,15 1 020 1 020 1 080 71,0
8,0 1 010 1 010 1 100 10,0
Си (П) Этилендиамин ' 7,2 980 980 1 075 8,0
Си (II) Пропилендиамин 7,5 510 510 975 2,1
7,5 5 340 5 340 515 1,3
Си (II) N, N, N'. N'-Тетраме- 7,0 1025 1 025 1 005 3,5
тил эти лендиа ми н 7,0 5010 5010 1 035 1,7
Си (11) N, N'- Ди метил эти- 7,0 2 425 2 425 490 0,5
лендиамин
Си (II) 1,8-Диоксин афта- 7,0 1313 1 313 729 27,0
лин-3,6-дисульфо-
кислота, динатрие-
ван соль
UO2 (II) То же 7,0 411 822 1 304 4,4
La (111) 7,0 811 811 1 285 52,0
Fe (III) 7,0 950 950 855 100
|ч Активность катализатора зависит от его концентрации и кои-центрации ионов водорода. Как видно из табл. 27 на примере про-* - изводных оксиэтилэтилендиамина, при одном и том же значении.
pH и на порядок большей концентрации катализатора гидролиз jp ускоряется примерно в 3 раза. Как показывают данные для одина-
ковых концентраций, но при различных pH (например, pH 6,1 и 8,0 Ж или 7,0 и 8,0), с уменьшением концентрации ионов водорода ско-J рость гидролиза возрастает.
* Простые катионы в сравнительно низких концентрациях также могут способствовать гидролизу зарина. Под действием ионов
. двухвалентной меди, особенно при pH 6—8, зарин гидролизуется .; быстрее, а при pH 8,5 (щелочная среда) каталитическая актив-1 иость исчезает. Сколь капризным может быть гидролиз фосфор-* органических соединений под влиянием катионов, показывает тот
факт, что гидролиз зарина и ДФФ ускоряют катионы Mg^, Са24-, „ * А13+ и’Со2". Однако, так как этот эффект наблюдается ие у всех
фосфорорганических соединений, обсуждать его возможное исполь-зование для’процессов дегазации излишне.
Другим примером отсутствия закономерностей ускорения гидролиза могут служить ионы редкоземельных металлов, которые в отличие от ионов меди ускоряют гидролиз в сильнощелочной среде, I - 309
что при непременном условии применения жесткой воды можно использовать для дегазации.
Реакция с фенолами. Зарин реагирует с фенолом с образованием фтористого водорода. Необходимым условием этой реакции является щелочная среда; это значит, что фенол участвует в реак-' ции только в ионизированной форме — в виде фенолят-иона. В результате реакции образуется производное фенола
ызо-С3Н7Ок <^0 U3O-C3H7O4 £)
+’ОС6Н5 —> + F"
СН/ XF СН/ \OC6HS
т. е. происходит замещение атома фтора фенолят-ионом.
Хотя эта реакция в водном растворе при комнатной температуре идет сравнительно медленно, она даже в таких условиях принципиально пригодна для дегазации. Лучшие результаты достигаются, например, в водно-спиртовых растворах.
1,2-Диоксн- или 1, 2, 3-триоксипроизводные бензола (пирокатехин, адреналин, пирогаллол) более реакционноспособны, чем фенол или м- или п-диоксипроизводные. Группа Р—О способна образовывать водородные связи, что приводит к ассоциации молекулы зарина с о-оксифенолят-ионом, поэтому второй атом водорода этого о-оксипроизводного в результате связывания с кислородом полностью теряет свою активность. Ответственным за реакцию является, как и в реакции с фенолом^ однозарядный ион оксифенолята. Приводим схему механизма реакции зарина с пирокатехином:
Замещение атома фтора однозарядным ионом оксифенолята происходит аналогично замещению алкоголят-ионами.
Другие фосфорорганические ОВ также реагируют с фенолами, причем в этом случае освобождается кислота, соответствующая связанному с фосфором ацилу.
Реакция с перекисью водорода. Зарин и другие фосфорорганические соединения легко реагируют с перекисью водорода и с перекисями. Эти превращения могут служить как для дегазации, так и для определения фосфорорганических соединений.
Взаимодействие перекиси водорода с зарином протекает аналогично щелочному гидролизу. Скорость этой реакции зависит от
310
концентрации ионов водорода. Оптимальные условия реакции — щелочная среда (pH > 8,4).
Определяющей скорость и направление реакции является нуклеофильная атака иона перекиси водорода на атом фосфора, причем образуется перкислота:
изо-С3Н7О^
СНз^5\f
+ноо_ изо-С3Н7Ск ----->
-F" СНГ ^ООН
U3O~ С3Н7 Ок
у' + н
Перкислоты разлагаются
по следующим схемам:
изо-С3Н7Ок ,0 +н2о2
X ------------->
СНГ хоо------н2°;~О2
азо-С3Н7Ок X) + сн3/ \осг
изо-С3Н7(Х ^0
Хр/ СН3/
«зо-С3Н7О\
СНГ
+он“ изо-С3Н7ОХ хО
--------> 2 >
-HF; —’/2О2 СНз/ \q-
Несмотря на более высокую основность гидроксильных ионов, скорость этих реакций примерно в 50 раз превышает скорость щелочного гидролиза.
Это показывает, что гидроперокснионы обладают большей способностью к расщеплению связи Р—F, чем гидроксильные ионы. По аналогии с реакцией фенолят-нонов в этом случае также можно предполагать, что сначала образуются переходные состояния, а затем возникают водородные связи между гндропероксиионом и атомом кислорода группы Р=О:
д-
R(O)4evO--H
R(o/ I'ч0-0
Г
Образование водородной связи между гндропероксиионом й' атомом фтора считают невозможным.
Синтезированные к настоящему времени некоторые фосфорорганические перекиси являются неустойчивыми соединениями, легко отщепляющими кислород33, Сложные эфиры общего строения
(ro)2p^°
\о-0-С(СН3)3 ,
получаемые по реакции гидроперекиси т/эет-бутила с диалкилхлор-Гфосфатами, оказались нестабильными соединениями, воспламенявшимися после кратковременного стояния.
Гидролиз зарина, сравнительно медленный прн обычных условиях, очень сильно катализируется небольшими добавками пере-кнси водорода. Так, 0,1%-иая добавка Н2О2 к водному" раствору бикарбоната натрия (pH 7,4) сокращает продолжительность
i ' зп.
гидролиза зарина30 с 8 ч до 1 ч 24 мин (время полупревращения), к водному раствору фосфата (pH 8,4) —с 1 ч 24 мин до 12 мин, к водио-ацетоновому раствору с фосфатным буфером (pH 11,0) — с 6,5 ч до 1,8 сек.
Для дегазации зарина и других органоацилфосфонатов рекомендуют 3°/о-ные щелочные растворы перекиси водорода, при необходимости можно применять более концентрированные растворы перекиси водорода.
Определение фосфорорганических соединений по перкислотам, образующимся при действии ионов перекиси водорода, обусловлено высокой реакционной способностью перкислот, которые окисляют вводимые ароматические амины и диамины в характерные красители. Эти красители служат для обнаружения фосфорорганических соединений, а также для количественных определений.
Вместо перекиси водорода в аналитических реакциях обычно применяют щелочные пербораты, перекись мочевины, перекиси бария, натрия и др. В качестве реагентов можно использовать, иапример, бензидин, о-толуидин, о-дианизидии, индол, люминол и имин диизонитрозоацетоиа. Реакции перкислот с аминами протекают по следующей схеме:
R(Ok R(Ok X)
2 "Х + 2NH2r —” Z + 2Н2О + RN=NR
R(OK ХО-0" R(O)Z \О"
7.5.5Л. Токсические свойства. Зарин в 4—5 раз более ядовит, чем табун. Отравление может быть вызвано вдыханием паров и резорбцией через неповрежденную, а особенно, через поврежденную кожу. Резорбция паров через кожу возможна при высоких Концентрациях ОВ, что в полевых условиях нереально, вследствие чего отравление таким путем маловероятно. Зарин легко просачивается через слизистую оболочку глаз и дыхательных путей.
Мистическое действие при отравлениях зарином проявляется не столь интенсивно, как при отравлении ДФФ, тем не менее, и в этом случае достаточно небольших концентраций, чтобы сильно повредить зрение.
Легкие отравления, которые ведут к утрате боеспособности на 4—о суток и сопровождаются сужением зрачков, затруднением дыхания, потливостью и усиленным слюнотечением, наступают при концентрациях от 0,0002 до 0,002 мг/л зарина при экспозиции 2 мин. Эти же концентрации при воздействии более 15 мин могут оказаться смертельными, если лечение начать несвоевременно.
Тяжелые отравления, которые иногда приводят к смерти, наступают при концентрациях от 0,01 до 0,005 мг/л при экспозиции 5 мин. Миоз сохраняется от нескольких дней до нескольких недель.
Другими симптомами отравления являются обильная потливость, очень сильное слюнотечение, головокружение, замедление речи, мьцпечиые судороги ит, д.
312
Смерть От остановки работы сердца наступает через 10—20 мин при концентрациях 0,02—0,05 мг/л и при экспозиции 2—5 мин. Симптомы быстро сменяют д]эуг друга. Через несколько минут отравленные теряют сознание.
Как было показано опытами иа животных37, учащенное дыхание, Особенно после физического напряжения, ведет при отравлении фосфорорганическими соединениями к повышенной интоксикации. Опыты с зарином, поставленные иа крысах во вращающейся клетке, показали, что при мышечной работе токсичность ОВ увеличивается примерно иа 20% и более по сравнению с показателями в отсутствие физического напряжения.
Значения ЛД50 для крыс при экспозиции 10 мин в отсутствие физической нагрузки составляют 0,0267 мг/л, а после нагрузки — 0,0096 мг/л.
Было установлено, что люди вообще по-разиому и индивидуально реагируют иа яды в смысле их дозы или концентрации. По мнению американских специалистов, это относится и к действию зарина, поэтому смертоносное действие, которой является целью военного применения ОВ, может и ие быть абсолютно молниеносным, а в зависимости от индивидуальных особенностей наступить через некоторое время.
В табл. 28 приведены данные38 о количестве умерших (в %) из числа отравленных в зависимости от летальной концентрации (доза составляет примерно 0,02 мг^мин/л, экспозиция 30 сек).
Таблица 28. Влияние числа принятых доз зарина ( — 0,02 мг* мин/л) на исход отравления
Смертельный исход, % Время после отравления, мин
одной дозой двумя дозами' '
10 4 3
48 15 6
95 — 15
Токсичность жидкого зарина через кожу для людей составляет 30—50 мг/кг массы. Для паров зарина средняя летальная доза через кожу составляет 8,4. мг^мин/л (на обезьянах) и 15 мг-мин/л для людей. Резорбция зарина через кожу происходит сравнительно быстро. Норма абсорбции39 т. е. количество, попавшее в ток крови, составляет ие более 0,001—0,002 мг/см2-мин. Одной капельки за-й- рииа, попавшей на кожу человека, по-видимому, достаточно, чтобы иа несколько дней сделать пораженного небоеспособным, в то время как большие капли вызывают смертельные отравления. Ре-; зорбции зарина значительно способствует потная кожа.
Летальная доза при приеме через рот составляет 0,14 мг/кг массы, а по некоторым источникам — 0,04—0,06 мг/кг. При
313
одноразовом приеме 0,022 мг/кг происходит легкое Отравление. В этом случае симптомы отравления проходят в течение 6 ч. Чуть большие дозы вызывают опасные, а при известных обстоятельствах смертельные отравления.
Летальная доза при попадании жидкого зарина в глаза, по-видимому, составляет 0,01 см3.
7.5.6. 3,3-0-Диметилизобутилметилфторфосфоиат
СН3
(СНз)зС—
СН3/
С и н о н и м иг/ етор-О-неогексилметилфторфосфонат, пинаколиловый эфир метилфторфосфоновой кислоты, зоман.
Военные обозначения: нем. — Soman, Trilon; амер. — GD.
В 1945 г. к концу войны в Германии зоман находился в стадии лабораторных испытаний; была разработана технология его производства.
Это ОВ производится в США и принадлежит к табельному химическому вооружению американской арМии.
Зоман получают аналогично зарину. Удобным является синтез из метилдихлорфосфоната, который фторируют и этерифнпнруют^ либо непосредственная этерификация метилдифторфосфата. Пинаколиловый спирт (3,3-Диметилбутанол-2) получают из ацетона или 2,2-днметнлпропионового альдегида по реакции Гриньяра. Трудность синтеза зомана заключается в нерентабельности получения Чистого пинаколилового спирта.
7.5.6.1. Физические н химические свойства. Зоман — бесцветная жидкость, промышленный продукт окрашен в желто-корнчневый цвет. Его термическая стабильность сравнима со стабильностью зарина. Выше 150 °C зомаи заметно разлагается..
.Физические константы зомана следующие:
Т. кип., °C
прн 760 лсл рт. ст. ....... , при 20 мм рт. ст, .............
при 15 мм рт. ст...............
при 0,1—0,2 мм рт. ст..........
Т. пл., °C .......................
Давление пара при 20 йС, мм рт. ст. . Максимальная концентрация
при 20 ®С, мг/л................
Относят, плотность пара..............................
жидкости d™.............. . , .
Показатель преломления ...........
190 (167-200)
95
85
42-43
-80
0,92 (0,8; 0,5; 0,26; 0,207)
10
6,33
1,0131 (1,026)
1,408
Летучесть, вычисленная по давлению пара, при 20 °C составляет
10 мг/л (по другим источникам — 3 мг/л}. Он более стоек, Чем
ЗИ
£
зарин, и поэтому особенно пригоден для заражения местности. Продолжительность заражающего действия зимой составляет от 10 до 15 ч.
Зоман, к которому добавлены стабилизаторы, можно хранить длительное время. Растворимость зомана в воде мала™менее 1,5%. Он растворим в органических растворителях, например в спиртах, сложных эфирах, кетонах, галогеналканах, а также в некоторых ОВ, например в иприте. Пористые материалы поглощают зоман сильнее, чем зарин.
Зоман -стабильное соединение, более устойчивое к действию воды, чем зарин. На гидролиз зомана влияют те же факторы (см. выше). Возможные реакции дегазации протекают с зоманом также медленнее.
С концентрированными водными или спиртовыми растворами щелочей и растворами аммиака зоман количественно превращается в ди натриевую соль метил фосфоновой кислоты:
СНа
CH3P(ONa)2 + (СНз)зССНОН (!)
+3NaOH
—NaF;
-нао
в ди натриевую соль метил фосфоновой кислоты: СН3 (СН3)3С(!нС\ ^0 zP\ СН3/ V
Реакции с фенолятами, перекисями, оксимами и гидроксиламинами идут так же, как с зарином.
к 7.5.6.2. Токсические свойства. Зоман примерно в 3 раза более < токсичен, чем зарин.. Симптомы отравления у них одинаковые, но <. отравление зоманом хуже поддается лечению. Неопасные переио-. симые концентрации составляют менее 7-10-7 мг/л, концентрации выше 2-10-5 мг/л при экспозиции 15 мин опасны.
Симптомы слабого отравления, такие, как сужение зрачков, | слюнотечение и потливость, появляются уже при концентрациях 10~4 мг/л после 1—2 мин воздействия. При более длительной экспо-!' зиции наступают легкие отравления, которые влекут за собой по-терю боеспособности на несколько суток.
? Концентрации от 2-10~3 до 4-10 3 мг/л вызывают тяжелые от-F/ равления, порою со смертельным исходом. Абсолютно смертельны t концентрации выше 2-10 2 мг/л при экспозиции 5 мин.
| Зоман обладает большой токсичностью' при резорбции через кожу, опасные концентрации составляют 10—20 мг/кг массы. Дозы 10,02-0,04 мг/кг при приеме через рот смертельны.
7.6. ФОСФОРИЛХОЛИНЫ И ФОСФОРИЛТИОХОЛИНЫ*
7;6.1. Общие сведения
В последнее время на вооружение армии США были приняты новые фосфорорганические ОВ, так называемые V-газы. Их
* См. такгке раздел 7.9.3.
&1&
токсичность для теплокровных во много раз превышает токсичность нервно-паралитических ОВ (зарин, табун и т. д.).
Новые V-газы являются следующим поколением семейства соединений, синтезированных и изученных в 1957 г. Таммелииом 40 в шведском Исследовательском институте национальной обороиы. Из-за высокой токсичности, удачных для ОВ физических и химических свойств и легкости получения V-газам, наряду с зарином, придают большое значение в качестве химических ОВ. Достаточно уже того, что их к; п. д. очень велик, ибо химические ОВ оценивают по количеству боеприпасов, расходуемому для достижения эффективных боевых концентраций и заражения местности. По имеющимся оценкам, при применении V-газов для создания необходимых боевых концентраций требуется почти в 100 раз меньше вещества, чем при применении других ОВ.
Введение в качестве ОВ фосфорилированных производных холина со всей очевидностью демонстрирует злоупотребление энзимологическими познаниями во имя военных целей. На основании теоретических данных и сведений о биохимических процессах, вызываемых каталитической активностью ацетилхолинэстеразы при расщеплении ацетилхолина, которым принадлежит решающая роль в передаче возбуждения вегетативной нервной системой, были созданы токсичные вещества, подобные по структуре продуктам естественного обмена веществ. Они вступают иа место этих продуктов, ио ие перенимают их биологические функции.
Соединения, полученные Таммелином, представляют собой фосфорилированные производные холина, которые по своему строению родственны ацетилхолину I и способны более или' менее сильно дезактивировать холинэстеразу. Этим соединениям можно приписать общую структуру II:
О Х\^°
СНзС—О—CH2CH2N(CH3)3 Yz >О—(СН2)а—z
(S)
I и
+ где X = OR, R, NR2; Y «» OR, Hal (особенно F); Z=NR2, NR3
Выводы о характере заместителей были сделаны по аналогии с другими фосфорорганическими соединениями, представляющими военный интерес.
Поскольку в этом случае, может идти речь как о производных холина, так и о производных тиохолина, проведено принципиальное различие между фосфорилхолинами и фосфорилтиохолинами, причем фосфорилтиохолииы имеют большее военное значение.
7.6.2. Связь между химической структурой и токсичностью
Блокирующее холинэстеразу действие производных фосфорил-холина определяется их структурным сходством с природным субстратом ацетилхолином, что является предпосылкой способности
316
этих соединений присоединяться к функциональным центрам молекулы холинэстеразы.
По сравнению с алкоксиалкилфторфосфатами фосфорилированные производные холниа проявляют иногда меньшую, иногда большую токсичность. Если в О-алкилалкилфторфосфонатах заместить +
атом фтора остатком холина OCH2CH2N(CN3)3, то как видно из табл. 32 (стр. 325), токсичность сильно уменьшается. Аналогичное зарину производное холина изопропоксиметилфосфорилхолин III имеет ЛД50 210 мг}кг массы (зарин 0,4 л/а/кг);
«зо-С3Н7О^ «зо-С3Н7О^
СНз^ \OCH3CH2N(CH5)3 CHZ ^SCH2CH2N(CH3)3
Ш IV
С точки зрения норм фармакологии это соединение неядовито. Аналогично, дналкокснфосфорнлхолины также обладают меньшей токсичностью.
В противоположность алкоксналкилфосфорнлхолинам, в результате введения в соединения типа зарина тиохолнновой группы иа место атома фтора токсичность увеличивается; значение ЛД50 для изопропоксиметилфосфорилтиохолина IV составляет 0,12 мг/кг.
Уменьшение размеров алкоксигруппы, невидимому, имеет следствием увеличение токсичности, так как четвертичная соль это-ксиметилфосфорилтиохолииа V в 4 раза более ядовитая, чем соединения IV:
. с2н5ох ,0
Р;
г
в О-алкил-имеет ЛД50
К v vi
Ж Резкое увеличение токсичности установлено и в том случае, Ж когда холнновым остатком замещают алкоксигруппу ж метилфторфосфонатдх. Метилфторфосфорнлхолнн VI ж '0,10 мг(кг и, таким образом, в 4 раза более ядовит, чем зарин.
Г Увеличение размеров холинового остатка также приводит к уси-ленню токсичности, как, например, было установлено для метил- '
Г фторфосфорилгомохолныа VII (ЛД50 0,05 л/г/кг) и метилфтор-
~ фосфорил 2-метилхолина VIII (ЛДИ 0,07 мг/кг):
' СН3ч X) СНзк хО -
X - Z\
f F/ ^OCH3CH2CH2N(CH3)3 XOCHCH2N(CH3)3
I ’ VII VHI CH3
Чрезвычайно высокая токсичность фторфосфорнлхолннов объ-? ясняется, во-первых, сходством с ацетилхолином, а во-вторых,— L реакционной способностью связи Р—F, которая определяет ско-L рость фосфорилирования холинэстеразы.
317
Сопоставление алкоксиалкилфосфорнлхолннов с аналогичными ' фосфорилтиохолинами ясно свидетельствует о быстром изменении биологической .активности фосфорорганических соединений в результате малейших изменений строения. Можно полагать, что среди производных тиохолина могут встретиться еще более токсичные соединения, скажем, при наличии фтора в молекуле. Увеличение размера алкильной группы при четвертичном атоме азота, например введение группы C2Hs, мало сказывается иа токричиости этих соединении (ср. раздел 7.9.3).
Каково влияние четвертичной аммониевой группы на токсичность соединения, видно из сравнения с аналогичным метилфтор-фосфорилкарбохолином IX
СН3^
^ОСН2СНаС(СН3)3
IX '
для которого ЛД50 равна 0,8 жа/ка (внутрибрюшинно) или 0,1 жа/кг (внутривенно), и, таким образом, он в 8—10 раз меиее токсичен, чем соединение VH.
В общем, можно констатировать, что в результате введения оииевой группы в боковые цепи различных фосфорорганических соединений получаются высокотокснчные вещества (ср. раздел 7.9.1); это справедливо как в случае аммониевых, так и сульфоииевых групп. Так, например, тетрам X (ЛД50 0,5 мг]кг иа мышах, внутрибрюшинно, см. раздел 7.9.3) менее ядовит, чем его производное с четвертичным азотом XI (ЛД50 0,17 мг/кг):
Я /°
(с2н5О)2р; (с2н6о)2р^ + -
\SCH2CH3N(C2H5)2 XSCH2CH2N(C2H5)8
X XI
Представляющие военный Интерес фосфорилированные производные холина возможны прежде всего среди фторфосфорилхоли-иов и фторфосфорилтиохолинов либо среди фосфорилтиохолинов. " Подобные соединения описаны в разделе 7.9, из них некоторые могут быть применены в-военных целях.
7.6.3. Алкилфторфосфорилхолииы
(ИЛ,М-триметиламмоиийалкоксиалкилфторфосфоиаты)
Из соединений группы
F^ ^O(CH2)„N(CH3)3 в результате работ Таммелниа стали известны фосфорилированные метилпроизводные холина, гомохолина и 2-метилхолииа.
7.6.3.1. Методы получения. Метод получения метилфосфорил-холинов аналогичен синтезу зарина. На заключительной стадии ис
318
ходят из метилфторхлорфосфоиата, на который действуют соответ* ствующим диалкиламиноспиртом в присутствии третичного амина (для связывания выделяющегося хлористого водорода). Образуется неустойчивое диалкиламинопроизводное:
СН3ч ,0 СНЭЧ ,0
+ ho(ch2)„n(ch3)2 —> + на
FZ \С1 F/ Х)(СН2)Л(СН3)2
Реакция идет в инертном растворителе (безводный эфир) прн кипячении с обратным холодильником в течение 1 ч. Выпадающий в осадок гидрохлорид амина отфильтровывают и фильтрат перегоняют при пониженном давлении.
Далее амин превращают в четвертичную соль аммония обработкой метилгалогенидом в эфирном растворе при комнатной температуре:
СН3\ X) +ch3i ГСНз\
, X > —* + 1
F/ XO(CHs)rtN(CH3)2 L F/ XO(CH2)„N(CH3)3_
Метилфторхлорфосфонат получают теми же способами, что и при синтезе зарина. Аминоспирты можно синтезировать, например, взаимодействием соответствующих хлорзамещенных спиртов с аминами либо путем присоединения содержащих воду аминов к окисям алкиленов.
Применяя соответствующие алкоголяты щелочных металлов, можно проводить фосфорилирование диалкиламиноспиртов в отсутствие третичного амина,
7.6.3.2. Физические и химические свойства. Соединения, полу* ченные на первой стадии синтеза алкилфторфосфорилхолииов, не содержащие четвертичного атома азота, представляют собой высо-кокипящие маслянистые жидкости. Они сравнительно неустойчивы, растворимы в воде и в результате внутримолекулярного образования четвертичной соли в течение нескольких дней превращаются в твердые вещества следующего строения
"СН- X)
/Р\ + . о; . л(сн8),
F~
’в
Представляющие собой растворимые в воде кристаллические веще-- ства; идентифицировать эти соединения можно по их четким тем-пературам плавления.
Неустойчивость диалкиламинопроизводных заставляет получать сразу четвертичные соединения, которые столь же токсичны/ но более устойчивы. Некоторые физические свойства диалкиламиио-В" производных приведены в табл. 29.
319
Таблица 29. Физические свойства некоторых мстилфторфосфорилхолииов
СНзЧ .О FZ XOR
В круглых скобках приведено давление в мм рт. ст.
к Т. КИП., °C Относительная плотность .25 ' d4 Показатель преломления „25 Т, пл, иодметилата, °C
—CH2CH2N(CH3)2 40 (0,2) 1,14 1,4130 152
-CHCH2N(CH3)2 40(0,1) 1,06 1,4150 84
СН3 /
Как следует из величин констант скорости гидролиза (табл. 31), метилфторфосфорилхолины гидролизуются значительно быстрее, чем зарин или рассматриваемые ниже производные тиохолина. Из этих данных видно, что чем ближе четвертичный атом азота расположен к атому фосфора, тем больше скорость гидролиза. Причиной этого является сильный — /-эффект холнновой группы, который облегчает нуклеофильное замещение.
При их гидролизе отщепляется атом фтора, а не холииовый остаток41
СНзх
F^₽XO(CH2)„N(CH3)3
+ОН' ------> —н+; —F-
(CH3)3N(CH2)
При pH 8 период полупревращения метнлфторфосфорилхолина составляет 9 мин, а для зарина в тех же условиях он равен 320 мин. Хотя гидролиз происходит быстрее по сравнению с зарином, водные растворы метнлфторфосфорилхолина в течение 3 ч н дольше остаются более ядовитыми, чем растворы зарина и других фосфорорганических соединений. Это обстоятельство особенно важно учитывать в случаях заражения воды.
Вследствие неустойчивости и легкости расщепления связи Р—F фторфосфорилхолины можно легко дегазировать. Дегазация возможна как с помощью щелочей, так и растворов гипохлоритов и подобных катализаторов.
> 7.6.4. Фосфорилтиохолииы
R(Ok ,0
X
R(O/ \S(CH2)ftN(CH3)3
Два типа соединений из этой группы уже были рассмотрены выше — это диалкоксифосфорилтиохолины (тиохолиновые эфиры диалкоксифосфорных кислот) и алкоксиалкилфосфорилтиохолины.
320
Нужно полагать, что в этой группе имеется еще много других соединений, о которых пока нет данных в литературе. Физические константы описанных к настоящему времени фосфорилтиохолииов приведены в табл. 30.
7.6.4.1. Методы получения. Фосфорилтиохолины получают следующими способами: а) прямым синтезом из диметиламинотиоэтанола, подобно производным холина; б) изомеризацией соответствующих производных тиофосфорной кислоты; в) присоединением триметиламина к О,О-диэтил-5-(2-бромэтил)-тиофосфату.
Способ а). Прн взаимодействии диметиламинотиоэтанола с соответствующими галоидангидридами фосфоновых или фосфорных кислот для нейтрализации образующегося хлористого водорода применяют третичный амин, либо проводят реакцию с тиолятами: R(°)\ - R(O)\ +сн3х
+ SCH2CH2N(CH3)2 ---> . ------->
R(Or ХС1 —СГ Р(ОГ \SCH2CH2N(CH3)2
R(O)s^O
R(O)/ \SCH2CH2N(CH3)3 X"
Таким путем получаются продукты очень высокой степени чистоты, что не всегда требуется по условиям их применения,'поэтому чаще всего пользуются вторым, более рентабельным способом.
Способ б). Изомеризация представляет собой перегруппировку производного тиофосфорной кислоты в соответствующий тиоэфнр, Т. е. атом серы связи P = S превращается в тиоатом связи Р—S (тнои-тиольная перегруппировка):
Изомеризация облегчается нагреванием или освещением и приводит к соединениям с иными химическими и токсическими свойствами.
Тиохолиновые эфиры получают изомеризацией соответствующих тноифосфорилхолинов примерно при 100°С, для которых перегруппировка происходит быстрее, чем для других веществ, что объясняют образованием более устойчивых промежуточных соединений, ускоряющих перегруппировку. Промежуточное соединение, образующееся при этой перегруппировке, в известном смысле представляет собой ионное образование (иои имония)42:
С2Н6О^
C2H5OZ \)CH2CH2N(CH3)?
’С2Н5ОЧ Н2Сх +
+ I zN(CHah
_С2Н5Сг ХО" Н2(У
С2Н5О\
С2Н6С>/
Н2С\ +
+ | zN(CH3)2
Н2С/
с2н5ох
С2Н3О^ X,SCH2CH2N(CH3)2
11 3aiL 448
321
Производные ТИОфОСфОрНОИ"КИСЛОТЫ ПилуЧЭТВ'Г Я3~ IHUHtpuupupHJr* хлоридов взаимодействием с диалкиламиноспиртами.
Способ в). По Шрадеру10, четвертичные аммониевые соединения можно получать присоединением триметиламииа к соответствующим О,О-диалкил-5-(2-бромэтИл) -тнофосфатам:
(RO)2p^ -+(СНз)^> (RO)2p^
\SCHPCH2Br \SCH2CH2N(CH3)3 Вг"
7.6Л.2. Физические и химические свойства. Соединения без четвертичного атома азота — жидкие, а с четвертичным атомом азота— устойчивые твердые кристаллические вещества, которые лишь умеренно растворяются в воде. Их летучесть чрезвычайно мала. Поэтому они могут применяться только в виде аэрозолей. Прн помощи приспособлений’для образования аэрозолей эти вещества могут применяться как в растворах, так и в дымовых смесях, Фос-форилтиохолииы пригодны в качестве ядов для диверсий.
Физические константы фосфорнлтиохолинов и некоторых аналогичных соединений приведены в табл, 30.
Таблица 30. Физические свойства фосфорилтиохолинов
R\ О
и аналогичных им соединений типа
R'/ XS—X
В круглых скобках приведено давление в мм рт. ст.
Заместители T, кип., °C ’ Относительная Плотность л25 d4 Показатель преломления Пр Т. пл. иод-мети-лата, °C
R R' X
сн3 c2hso (CH3)2N(CH3)2 (CH2)2N(CH3)2 80 (0,06) 1,0725 1,478 ПО
СН3 ИЗО-С3Н70 77 (0,1) 1,0501 1,4675 164
сн3 с2н5о , (CH2)2N(C2Hg)2 80(0,01) —' — -—
С2Н8О С2Н8О (CHa)2N(CH3)2 85(0,1) 1,0772 1,4655 138
едо С2Н5О (CHa)3N(CH3)2 85 (0,005) <—►
С2Н5О С2Н8О CH(CH3)CH2N(CH3)2 94 (0,08) —И- —
с2н5о С2Н5О (ch2wch2)8 110 (0,012) —— ——
с2н5о С2Н8О (CH2)2N(CH2)4 108 (0,01) <— —
с2н5о СаН8О (CH2)2N(C2Hg)2 97 (0,2) — 1,4732 (при 21 °C) —
с2н8о сн3 (CH2)2N(C2Hg)2 80 (0,01) —— '— —
с2н8о с2н5 (CH2)2N(C2H5)2 94 (0,01) '— —
с2н6 с2н8 (CH2)2N(C2Hg)2 81 (0,02) '— —
изо-с 3Н 7 О изо- С 3Н7 О (CH2)2N(CH3)2 85 (0,1) 1,0296 1,4570 154
Их высокая токсичность и склонность к легкому образованию аэрозолей при благоприятных метеорологических условиях делают возможным заражение различных объектов на много дней и даже недель. Необходимые для этого плотности заражения составляют 10~3—10-4 от плотности заражения ипритом.
322
1\,аК ВИДНО ИЗ ДанНыА i duji. оi, сриирирнл iiiuAUdunim—цичj - --
чивы к гидролизу, чем зарии и другие фосфорорганические соединения. Даже в снльнощелочной среде при pH 10 гидролиз идет очень медленно. Период полупревращения фосфорнлтнохолинов при гидролизе составляет около 10 ч при 20 °C, а для зарина в тех же условиях лишь 1 мин.
Таблица 31. Константы скорости гидролиза некоторых
фосфорилированных производных холина и тиохолина Р , R'Z XX
а также зарина, ДФФ и табуна
Заместители A, л.лоль-1
К R' X
СНз ОСН2СН2К(СИ3)з F 935
СНз O(CH2)3N(CH3)3 F 30!
СН3 OCH(CH3)CH2N(CH3)3 F 381
СНз ОС2Н5 OCH2CH2N(CH3)3 0,033
ОС2Н, ОС2Н3 ОСН2СН2Й(СНэ)з 0,030
СНЭ ОС2Н5 SCH2CH2N(CH3)3 0,17
ОСаН> ОС2Н5 SCH2CH2N(CH3)3 0,52
СНз (зарин) 0C3H7-U30 F’ 25,8
ОС3Н7-нзо (ДФФ) ОС3Н7-ызо F" 0,83
N(CH3)2 (табун) ОС2Н5 CN’ 7,49
При гидролизе отщепляется тиохолиновая группа с одновременным образованием соответствующей кислоты.
В результате взаимодействия электронной пары атома серы с положительно заряженным атомом азота связь Р—S ослабляется и становится возможным ее разрыв. Количественная реакция возможна только с сильными щелочами в разбавленных вдвое растворах.
7.6.4.3. Токсические свойства. Алкилфторфосфорилхолины и ал-килфосфорилтиохолины являются сильными ингибиторами холинэстеразы. По своей молекулярной структуре они приближаются к ацетилхолину.
Согласно представлениям Таммелина, онн присоединяются к холинэстеразе и по «анионному», и по «эстеразному» центрам молекулы фермента. Предполагаемый механизм такого присоединения, при котором не образуется истинная химическая связь с анионным центром, представлен на рис. 19/ В то время как фтор-фосфорилхолины, по--видимому, связываются с молекулой субстрата в целом, в результате чего отщепляется нуклеофильный атом фтора (рис. 19 а), в случае фосфорилхолииов и фосфорнлтнохолинов,
11*
323
не содержащих фтора, в результате взаимодействия с анионным центром отщепляется остаток холнна или тиохолина и становится возможной связь атома фосфора с эстеразным центром (рис. 19 6).
Связь холиновых эфиров с ацетилхолинэстеразой, по-видимому, прочнее, чем связь с другими фосфорорганическими соединениями.
Рис. 19. Схема взаимодействия фосфорилхолииов с холинэстеразой.
Проведенные иа животных и in vitro эксперименты по реактивации ингибированного фермента действием диацетилмонооксима и йодистого 2-пиридинальдоксима (стр. 280) и др. показывают, что они, в отличие от ингибирования другими фосфорорганическими соединениями, *в данном случае ие столь эффективны. При лечении пострадавших от отравления производными фосфорилхолина возникают порою совсем иные проблемы, чем в случаях отравления орга-
824
иическими фосфатами, рассмотренными ранее. Это также придает этим соединениям значительный интерес как химическим ОВ.
Симптомы отравления те же, что и при отравлении другими ’ фосфорорганическими соединениями. Оии проявляются через определенные периоды, продолжительность которых, в зависимости от типа холинового эфира и от дозы или концентрации при вдыхании, составляет от нескольких минут до нескольких часов.
Имеются сведения, что принятые на вооружение американцами V-газы в 100—1000 раз более ядовиты, чем зарии. Однако тогда сответствующне летальные дозы н концентрации должны быть очень малы, что противоречит данным Таммелина и Фредериксона, приведенным в табл. 32. Из подобных сравнений можно заключить, что концентрации выше 5-Ю-4 мг/л при вдыхании в течение 2 мин смертельны.
Таблица 32. Токсичность некоторых фосфорнлхолиновых и фосфорилтиохолиновых эфиров43 по сравнению с токсичностью зарина
Соединение ЛД50, мг!чг
иа белых мышах, внутрибрюшинно на кроликах, мышах, внутривенно
Этоксиметилфосфорилхолин 375,0 —
Диэтоксифосфорилхолин 495,0 —
Изопропоксиметилфосфорилхолин 210,0 —
Метил фторф о сфорил холин 0,1 0,01
Метнлфторфосфорил-2-метилхолин 0,07 0,008
Метил ф торф о сфо ри лго мо хо л ии 0,05 0,006
Этоксиметилфосфорилтиохолин 0,03 —*
Диэтоксифо сфо рилтиохолии 0,14 —
Изопропоксиметилфосфорнлтиохолин 0,12 —
О-Изопропилметилфторфосфонат (зарии) 0,4 0,02
Поразительна большая токсичность при проникновении через кожу некоторых фосфорнлхолиновых эфиров, принимаемых во внимание в качестве ОВ; летальные дозы ЛД50 составляют 0,2— 1,0 мг/кг веса. Это объясняется быстрой резорбцией этих соединений через кожу и далее в организм.
7.7. БОЕВОЕ ПРИМЕНЕНИЕ ФОСФОРОРГАНИЧЕСКИХ ОВ
Фосфорорганические ОВ могут применяться в жидком виде, опрыскиванием н в виде аэрозолей. Длительное заражение воздуха возможно вследствие нх летучести, склонности к-легкому образованию аэрозолей и высокой токсичности. В армиях империалистических государств предусматривается их боевое применение, прежде всего посредством артиллерии, ракет тактического и оперативно-тактического назначения, залповых ракетных установок, авиационных бомб и других образо'Вателей аэрозолей, а также химических и реактивных мин. Возможно применение этих ОВ при помощи выливиых приборов.
Фосфорорганические ОВ действуют быстро и смертоносно. Особое значение придают зарину и V-газам. Они предназначены для применения как в наступательных, так н в оборонительных операциях. В американской армии предполагают применять V-газы при внезапных атаках. Считают, что в результате молниеносного огневого налета противник окажется уже не в состоянии одеть средства защиты, либо прежде чем он их оденет, он уже будет отравлен. V-Газы вполне пригодны для длительного заражения местности и воздуха.
ОВ типа зарина и V-ряда могут применяться в виде аэрозолей прн любых метеорологических условиях. Продолжительность их действия с понижением температуры воздуха увеличивается. Зимой на участках с застоем воздуха нх действие может сохраняться несколько недель.
При благоприятных условиях продолжительность действия зарина прн средних плотностях заражения и 20°C составляет около 2 суток, для V-газов считается, что продолжительность заражения может достигать 10 суток.
В случае фосфорорганических ОВ-при благоприятных метеорологических и местных условиях смертоносные облака отравляющих веществ могут распространяться иа расстояние до 30 км от места возникновения. Даже на большем удалении еще могут быть концентрации ОВ, приводящие тГпотере боеспособности.
Для людей без средств защиты места взрывов и окружающая площадь являются опасными для жизни в течение нескольких суток, а в случае V-газов — порой до нескольких недель.
Наибольшая эффективность фосфорорганических ОВ достигается в результате максимально быстрого применения нх в наивысших концентрациях и на наименьшем участке. Огневые налеты должны длиться 30—60 сек. Это возможно при массированном применении артиллерии, оперативно-тактических ракет, в боеголовках которых содержится около 200 кг ОВ, или при комбинированном использовании артиллерии и ракет, прн использовании бомбардировщиков н истребителей-бомбардировщиков, а также многоствольных пусковых установок для реактивных снарядов, как, например, американские 45-ствольные пусковые установки М91.
О тактических возможностях заражения местности фосфорорганическими ОВ типа зарина артиллерийскими и военно-воздушными подразделениями армии США можно привести следующие данные44 (при продолжительности атаки 30 сек)'.
Заражаемая
Подразделение площадь,
196,6-.и.м минометный дивизион........ 21
Дивизион 105-к>и гаубиц............ . 3,3
Дивизион 155-леи гаубиц............... 6
Звено истребителей-бомбардировщиков . , 5,2
Звено легких бомбардировщиков .... Ю,4
Звено средних бомбардировщиков .... 66
Звено тяжелых бомбардировщиков . . . 144
326
При этом более чем на 50%обстреливаемой площади создаются смертельные концентрации.
На вооружении армии США состоит преимущественно зарнн (GB) и ОВ V-ряда (обозначение VX). В табл. 33 приведены по состоянию.иа 1962 г. важнейшие виды боеприпасов американских сухопутных войск, снаряженные этими ОВ, и средства их применения.
Таблица 33. Боеприпасы армии США, снаряженные зарином (GB) и V-газом (VX)
Виды боеприпасов ов Средства применения Максимальный радиус действия. км Масса боеприпаса, кг Масса ОБ, кг Эффективный к. п. д, боеприпаса, %
Мины М 360 М 121 М 122 Т 174 Реактивные снаряды М55, 115 мм Ракетные боего- GB GB/VX GB GB/VX GB/VX 105-мм гаубица 155-мм гаубица 155-лм/ пушка 8-дюймовая гаубица Установка М91 11,1 15 23,5 16,9 11 16,1 45,9 45,9 97,0 26,4 (26,2) 0,74 2,95 2,95 7,12 4,80 (4,54) 99 99 99 99 99
ловки E19R2,762 AiJH Е20318 мм Е21 GB/VX GB GB/VX «Honest John» «Little John» «Sergeant» 33,8 18,3 139' 568 119 744 210 30 190 95
Бомбы М34, А1, 1000 фунтов МС-1,750 фунтов Химические мины М23 GB GB VX Бомбардировщик, истребитель Бомбардировщик, истребитель 513 322 10,5 89,6 99,9 5,23 90
7.8. ЗАЩИТА ОТ ФОСФОРОРГАНИЧЕСКИХ ОВ
И ИХ ДЕГАЗАЦИЯ
7.8.1. Защита
Высокая токсичность фосфорорганических ОВ, быстрота их лея ствня в низких концентрациях и трудность их индикации в сочета нии с разнообразием возможностей их применения требуют максимально коротких сроков для оповещения об опасности и ппивеле ния средств защиты в боевую готовность. г
Необходимым требованием для боеспособности и боеготовности войск явлиется: постоянная готовность всех средств защиты не медленное применение защитных средств при любом нападении даже если химическое нападение, казалось бы, не имеет места’ пользование средствами защиты до тех пор, пока окончательно не
327
установлено, что химического нападения не было, либо пока не исчезнут следы химических ОВ; постоянная тренировка со средствами защиты, пока войска полностью не овладеют их применением в любой ситуации.
Противогаз и защитная одежда защищают от фосфорорганических ОВ в виде аэрозолей и паров, при непременном условии, что они находятся в безупречном состоянии.
При длительном пребывании в зараженном воздухе, особенно в случае ОВ типа зарина и V-газов, защитную одежду следует время от времени менять или дегазировать иа себе. Если ОВ попало на защитное снаряжение или оружие, его следует тотчас дегазировать с помощью индивидуального противохимического пакета. -
Через обычное обмундирование фосфорорганические ОВ проникают быстро. Пораженных нужно как можно быстрее подвергнуть санитарной обработке. Специальная пропитка обмундирования обеспечивает только временную защиту от аэрозолей и паров, а также капель ОВ.
Соблюдение личной гигиены есть высшая обязанность каждого военнослужащего. Это особенно относится к химическому бою. Через чистую сухую кожу ОВ проникает медленнее, чем через потную и загрязненную. Потрескавшиеся места кожи и ранки нужно заклеить пластырем, чтобы предотвратить быструю резорбцию фосфорорганических ОВ.
„Воду, продовольствие и сельскохозяйственные продукты можно употреблять только после химического контроля.
7.8.2. Дегазация
Фосфорорганические ОВ дегазируют растворами следующих веществ: щелочей, аммиака, карбонатов щелочных металлов, щелочных гипохлоритов, щелочной хлорной извести, фенолятов и крезо-лятов, сульфида натрия, сульфурилхлорида, перекиси водорода.
Для дегазации местности служат растворы щелочей и гипохлоритов или суспензии хлорной извести. Транспорт, боевую технику и т. п. дегазируют растворами щелочей или гипохлоритов. Растворы аммиака, соды, фенолята, а также извести применяют для материалов, легко корродирующих при действии щелочей.
Для дегазации кожи и предметов личного снаряжения пригодны хелаты двухвалентной меди, гидроксамовые кислоты, разбавленные растворы перекиси водорода и слабощелочные растворы.
Водные растворы сульфида -натрия с добавками поверхностно-активных веществ лишь ограниченно пригодны для дегазации местности.
. Лабораторные приборы дегазируют водно-спиртовыми растворами щелочей. Для этой же цели применимы растворы перекиси водорода и насыщенные растворы гипохлоритов. Обмундирование и снаряжение дегазируют паро-аммиачным способом или кипячением в растворах соды или других дегазирующих растворах.
328
Хлорирующие дегазаторы, например хлорная известь или суль-фурилхлорид, при взаимодействии с табуном образуют ядовитый хлорциан. Фосфорорганические ОВ не подлежат дегазации хлораминами.
7.9. ФОСФОРОРГАНИЧЕСКИЕ ИНСЕКТИЦИДЫ КАК ОВ
В последние 10 лет проводилось исследование фосфорорганических инсектицидов, особенно таких, которые на длительное время защищают растения от поражения вредителями. Уже в 1936 г. были получены .инсектицидные препараты, которые растение воспринимает через корневую систему и листья вместе с питательными веществами и гормонами. Растение разносит инсектицид теми же путями, что и все другие, нужные для его развития вещества. В результате заражаются все части растения. Такие вещества имеют общее название системных инсектицидов. Кроме производных типа систокса, системной эффективностью обладают также действующие начала препаратов тимет, тетрам, дисистон, параоксон, диме-фокс и др.
Бблыпая часть этих' соединений превращается в растении в иные, иногда более токсичные вещества, которые в свою очередь действуют как инсектициды, но не безопасны и для теплокровных. Растение само через определенное время разрушает эти вещества до неядовитых продуктов.
Предпосылкой системного действия и условием применения таких средств являются: инсектицидная активность; хорошая растворимость в растительных соках И воде; быстрая диффузия через растительные ткани — корни, листья, стебли травы—в сок растения; максимальная устойчивость при pH 4,5—6,5 (область pH растения); поддающееся контролю разложение в растении до неядовитых продуктов.
Для мирного использования инсектицидов требуется, чтобы они были возможно более безвредны нли мало ядовиты для теплокровных или чтобы до окончания уборки урожая в растении завершалось их разложение до безвредных продуктов, поскольку растения, зараженные системными инсектицидами, представляют непосредственную опасность для человека и животных. >
Исследования американских военных химиков были направлены на создание веществ, ие поддающихся дегазации, которые, попадая в растительные культуры, могли бы наносить ущерб и вызывали бы смерть людей. Оказалось, что такие качества присущи фосфорорганическим соединениям, обладающим системным действием, высокой токсичностью, медленной'гидролизуемостью и способностью к диффузии. Хорошую способность вещества к диффузии через растительную ткань нужно связывать с хорошей растворимостью в липидах, так как речь обычно идет о соединениях, обладающих высокой токсичностью при действии через кожу,
329
Благодаря определенному сочетанию химических, физических и токсических свойств соединения типа систокса и некоторые аналогичные вещества имеют военное значение. Отличие их от других препаратов системного действия заключается в том, что они легко усваиваются растением и поэтому более устойчивы к влиянию погоды. В результате проникновения в растение на иих труднее воздействуют метеорологические факторы. Так, на заводах фирмы «Байер» работающим на производстве способного проникать через кожу систокса полагается носить противогаз и защитную одежду.
В следующих разделах рассматриваются также некоторые инсектициды, не обладающие системным действием, но имеющие общее значение.
7.9.1. Производные типа систокса
Производные типа систокса были разработаны в лабораториях фирмы «Байер» в Леверкузене. Название систокс образовано из слов «системный» и «токсичный». По Шрадеру45, они представляют собой продукты конденсации диалкилгалогентионфосфатов с тиоалкоксиэтанолами.
Наиболее известными соединениями этого типа являются метиловые и этиловые эфиры. Оба гомолога существуют в виде изомеров. Продажные препараты являются смесью тиониой I и тиоль-' ной П форм
R0\zs
R'O/ \ЗСН2СН28К"
тип систокса
I
RO\PZ
R'q/ \sch2ch2sr"
тип изосистокса
II
где R, R' = алкил; R" = алкил, арил.
Тионная форма обладает сравнительно слабым системным действием. Образующаяся в результате тион-тиольной изомеризации тиольная форма отличается более сильным системным действием.
Выпускаемое в ФРГ метильное производное носит название метасистокс, а этильное — систокс.
7.9.1.1. Методы получения. При промышленном производстве соединений типа систокса получают смесь форм I и П. Каждую из форм можно также приготовить в отдельности.
Тионную форму получают конденсацией диэтилхлортионфое-фата с 2-тиоэтоксиэтаиолом в присутствии веществ, связывающих кислоту, например пиридина или карбонатов калия или натрия:
(с2н5о)2рх + hoch2ch2sch2ch3 —> (с2н5О)2р;
\ci HC1 XOCH2CH2SCH2CHj
330
Диэтилхлортионфосфат получают взаимодействием тиотреххло-ристого фосфора с этилатом натрия
S=pCl3 + 2NaOC2H5 —> (С2Н5О)2Р^ + 2NaCl
\ci
а 2-тиоэтоксиэтанол можно легко получить из окиси этилена и этилмеркаптана:
Н2С---CH2 + HSCH2CH3 —> hoch2ch2sch2ch3
Более простым методом получения систокса является переэтерификация триэтилфосфита 2-тиоэтоксиэтаиолом с последующим присоединением серы:
(С2Н6О)3Р + hoch2ch2sc2h5 ------>
—С2Н5ОН
+s
(C2HSO)2P—OCH2CH2SC2Hs (C2HSO)2P(
\och2ch2sc2h5
Тиольная форма образуется при (см. раздел 7. 6. 4. 1, метод б); при полностью в течение 2,5 ч:
(C2HSO)2P^ \OCH2CH2SC2H5
тиояяая форма систокса
изомеризации тионной формы 130 °C изомеризация проходит
(С2Н5О)2Р^
xsch2ch2sc2h5
тиольиая форма систокса
Чистую тиольную форму получают взаимодействием аммониевой соли диэтилтиофосфорной кислоты с 2-хлорэтилтиоэтиловым эфиром по следующей схеме:
C2H5SCH2CH2C1 +
О
- II
S-P(OC2II5)2
NHt
------->
-NH4CI
(C2HsO)2P
^SCH2CHaSC2Hs
2-Хлорэтилтиоэтиловый эфир получают аналогично иприту из тионилхлорида и 2-тиоэтоксиэтанола:
+SOC12
C2H5SCH2CH2OH ---------> c2h5sch2ch2ci
-SO2; -НС!
7.9.1.2. Физические и химические свойства. Чистые соединения типа систокса как в тионной, так и в тиольной форме — бесцветные, легкоподвижные маслянистые жидкости. При 20 °C растворимость тионной формы систокса в воде меньше (0,06 г/л), чем для
331
тиольной (2 г/л). Обе формы растворимы в обычных органических растворителях. Они малолетучи. Продажные препараты содержат эмульгаторы, иапример беизилоксидифениловый эфир полиэтилеи-гликоля.
Устойчивость соединений ряда систокса увеличивается с величиной алкильных остатков. Как при нагревании, так и при хранении разбавленных или неразбавленных растворов систокса происходит непрерывная изомеризация тионной формы в тиольную:
(Г?О)2Р\ (RO)aP^
XOCH3CH2SR ^sch2ch2sr
Равновесие этой реакции сдвинуто вправо, поэтому содержание тиольной формы (болёе устойчивой) по мере установления равновесия в смеси возрастает.
Систокс является смесью обоих изомеров. Физические свойства • тиониых и тиольных форм систокса и метасистокса приведены в табл. 34.
Таблица 34. Физические свойства соединений типа систокса (по данным Шрадера)
Соединение Т. кип. (при 1 мм рт. ст.), °C Давление пара при 20 °C, мм рт, ст. Максимальная концентрация при 20 °C, ма/м3 Показатель преломления «20 nD Относительная плотность ,20 Растворимость в воде при 20 ЬС, е/л
Систокс
(С2НВО)2Р^ \och2ch2sc2h5 тиоиная форма 123 2,48- 10~4 3,5 1,4900 (при 18 °C) 1,119 (при 21 °C) 0,06
(С2НеО)2Р^ 4sch2ch2sc2h6 тиольная форма Метаснстокс 128 2,6- 10 ~4 3,67 1,5000 (при 18 °C) 1,132 (при 21 °C) 2,0
Z (СНаО),Р' ^OCH2CH2SC2H5 тиоиная форма 106 18,5- 10"“ 23,3 1,5063 1,1904 0,33
(СН3О)2Р^° xsch2ch2sc2hb тиольная форма 118 3,6 - ю~4 4,5 1,5065 1,207 3,3
332
Скорость изомеризации зависит от природы растворителя. В полярных растворителях, например в этаноле, она увеличивается, а в неполярных — уменьшается.
Степень изомеризации систокса в разных растворителях при 37,2 °C, по данным Фуку то и Меткалфа46, имеет следующие величины’:
Степень изомеризации
Растворитель через 39 суток, %
2,2,4-Триметилпентан 1,8
Бензол............................ 4,1
Этил ацет ат..................... 4,3
Диоксан.......................... 4,8
Без растворителя................. 6,0
Метилэтилкетон................... 8,6
Хлороформ........................ 23,5
Этанол........................... 56
Сильнее всего изомеризацию катализирует вода. Это наблюдение привело к предположению, что изомеризация сопровождается образованием промежуточных ионных соединений:
(RO)2PZ =₽=* (RO)2P^ + | )SR
xoch2ch2sr чг h2cz
jO Н2Ск + £>
*=* (ro)2p^ + | ;sr (ro)2p(
\s' H2CZ \sch2ch2sr
Данные о продолжительности изомеризации неразбавленного систокса очень различаются между собой. Шрадер и сотр. иашли, что время изомеризации на 50% при 90°C составляет 38 ч, а при 105°С—10 ч', Фукуто и сотр. приводят для 95°C время полупревращения 87 ч. При комнатной температуре изомеризация должна продолжаться около 4 лет. О’Брайи 'предполагает, что причиной этих различий являются реакции переалкилироваиия этих соединений, которые в данном случае играют важную роль.
Хит и Ваидекар47 при использовании тщательно очищенного препарата метасистокса в тиольной форме (1%-иый раствор) показали, что летальная доза ЛД50 для крыс в течение дня изменяется от 60 до 2 лг/кг; токсичность неочищенного препарата в ти-оииой форме за 1 ч изменилась от 220 до 2
За увеличение токсичности, по-вндимому, ответственны две реакции, в том числе самоалкилироваиие, которое происходит как в самом продукте, так и в водных его растворах:
sP +Н2О ПО\ хР
2(RO3)PZ ----—> X + (RO)2PZ
ZSCHaCH2SR *но ROz xSCH2CH2SR XsCHaCHaSRa
По сравнению с исходными соединениями производные сульфонил ингибируют холинэстеразу в 1000 раз сильнее.
333
Переалкилированне метоксипроизводных соединении происходит быстрее, чем этоксипроизводных.
В то время как в результате изомеризации в равновесии находятся тиольная и тиоииая формы, в результате переалкилированИЯ появляется еще одни изомер I, а по реакции тиольной формы с промежуточным соединением образуется соединение II:
(RO)2p^ + /R' (RO)2pz +zR
xocii2cii2s( ^sch2ch2s^
\r ^ch2ch2sr
I II
В нейтральных водных растворах тиольная форма устойчивее, чем тионная. Это объясняется более сильным электрофильным влиянием атома кислорода связи Р=О по сравнению с атомом серы связи Р—S. Гидролиз тиольной формы происходит по следующей схеме;
+н2о
(RO)2p; -------> (ro)2p^ + hsch2ch2sr
zsch2ch2sr \он
За 6 месяцев 0,2%-ный водный раствор тиольной формы систокса гидролизуется иа 50%. Суммируя все рассмотренные химические свойства систокса, можно схематически так изобразить превращения систокса в водных растворах:
переалкилированне R^\
> (ROhPf , + Р
/0 (RO)2p(" \SCH2CH2Sr .[[изомеризация zsch2ch2sr2 iioz zsch2ch2sr гидролиз > (RO)2P^ + hsch2ch2sr ZOII
(RO)2P + I si н2с/ 1 1 1 1 (RO)2P^ 1 zoch2ch2sr 1 реакция переходного Г) комплекса Л? \ > (RO)2P - +/R \SCH2CH2S' \ch2ch2sr переалки- о ИЛ Я лироваиие ни\ > до)Х + + xO(CH2)2SR2 ROz 4)(CH2)2SR
334
По отношению к щелочам тионная форма систокса иолее устий- чива, чем тиольная. Периоды полупревращения при гидролизе в 1 н. растворах щелочей (pH 13) при 25 °C составляют 75 и 0,85 мин соответственно. В кислой среде соотношения обратные. Период полупревращения при гидролизе тионной формы в 1 и. растворах Кислот при 84,5°C составляет 330 мин, а тиольной — 550 мин. При гидролизе в нейтральной среде (как и в щелочной) более устойчива тионная форма.
При избытке щелочи тиольная группа отщепляется:
+2NaOH
(RO)2p; ---—> (RO)2p; + NaSCH2CH2SR
^SCH2CH2SR ~ 5 XONa
Под влиянием окислителей систокс окисляется в сульфоксид и сульфон:
(rO)2p; —> (RO)2P^ —>
\sch2ch2sr ^sch2ch2s^
XR
сульфоксид
-Л
—► <ro)2p;
\SCH2CH2S—R 40
- сульфон
Продукты-окисления более устойчивы, чем тноэфир, а по своей токсичности онн мало отличаются от тиоэфиров. Они обладают системным действием и их можно получить отдельно.
В организме систокс подвергается окислительным превраще-. ниям, ведущим к образованию сульфоксидов и сульфонов. Вследствие того, что эти продукты превращения столь же сильно ядовиты, токсическое действие производных систокса объясняют сочетанием действия исходных соединений и продуктов их окисления48.
7.9.1.3. Системное действие и токсические свойства. Воспринятый корнями, листьями и другими частями растений систокс в результате диффузии попадает в поток питательных веществ растения и разносится им во все его части. Это не препятствует и не причиняет вреда развитию самого растения. Вследствие системного или, иначе, «внутритерапевтического» действия фосфорорганических соединений обработанные стандартными препаратами в установленных концентрациях (!) растения приобретают защиту от сосущих насекомых на срок до 4 недель. Продолжительность защитного действия зависит от концентрации раствора для опрыскивания нли поливки, от внешней температуры и от возраста растений. Старые растения усваивают препарат медленнее, чем молодые. Тионные формы инсектицида не обладают или обладают лишь слабым системным действием,
335
' В результате окисления систокса в соответствующие продукты (сульфоксид и сульфон) характер токсического н системного действия не меняется, одиако из-за большей устойчивости сульфоксида и сульфона их системное действие более продолжительно.
Чрезвычайно длительным действием обладает сульфоксид, являющийся истинным носителем системного действия систокса.
В табл. 35 показано системное действие тиольной формы метасистокса и продуктов его окисления в концентрации 0,05% через различные промежутки времени. Сульфоксидное производное даже через 19 суток обладает 100%-иым системным действием (1).
Таблица 35. Системное действие тиольной формы метасистокса и продуктов его окисления (испытания на Mycus persicae) по данным Шрадера45
Соединение Степень системного действия, К
через 2 СутОК через 6 суток через 10 суток через 13 суток через суток через 19 суток через 26 суток
Мета систокс 100 90 30
сульфоксид 100 100 100 '100 100 100 90
сульфон 100 100 40 — — — —
Систокс находит применение преимущественно в плодоводстве и разведении кормовых культур. В овощеводстве его применение запрещено (1). Его следует применять, самое позднее, за 42 дня до уборки урожая.
Производные ряда систокса, будучи фосфорорганическими соединениями, являются типичными ингибиторами холинэстеразы. На Организм человека и животных они действуют с характерными для Таких соединений симптомами.
Типичными признаками отравления являются головокружение, покраснение кожи лица, сужение зрачков, потливость, рвота, желудочные Спазмы, понос, подергивания и судороги мышц, боли в икроножных мышцах, затруднение дыхания и отек легких.
Как уже отмечалось, носителями токсических и системных Свойств препаратов типа систокса являются тиольные производные. Тиоиные формы представляют собой как бы «резерв» — в результате изомеризаций они восполняют расход тиольных производных и продуктов их окисления.
Отравления через кожу препаратами типа систокса встречаются чаще, чем отравления другими фосфорорганическими инсектицидами. Растворимость в липидах у тионных производных выражена сильнее.
В табл. 36 приведена токсичность соединений типа систокса, определявшаяся на крысах,
336
Таблица 36. Токсичность соединений ряда систокса (иа крысах)
Соединение ЛДйо. Л г/кг
per os per or внутривенно
(СН3О)2Р^ 180 — —
XOCH2CH2SC2Hs (СН3О)2Р^ 40 63 65
\sch2ch2sc2h5 (СН3О)2Р^ 0 40 65 47
\SCH2CH2S\ ^С2Н5 /° (СН3О)2Р^ О' \SCH2CH2S^—С2Н5 40 32 22
Чо (СН3О)2Р^ +/СгН5 \SCH2CH2S\ 9,8 0,06
Vh3 XS (С2Н5О)гР^ \och2ch2sc2h5 30
/° (C2HSO)2P^ \SCH2CH2SG2H6 1,5 -
(C2H5O)2P^ A} \SCH3CH2S/r 2,0
^СгНй (C2H6O)2P( .0 \SCH2CH2S—C2HS 2,0 •
о (C2H6O)2P^ +/С2н5 \SCH2CH2S/ 20 0,016
Vh3 /° (C2HSO)2P^ \SCH2CH2S(C2H5)2 — 0,010
7.9.1.4. Аналогичные соединения. В случае соединений, приведенных ниже, замещение алкокснгрупцы на алкильную также приводит к повышению токсичности. Соединение I в 3 раза более токсично тиольной формы систокса. При увеличении размера одной или обеих алкоксигрупп в соединениях ряда метасистокса токсичность возрастает и достигает максимума в случае производных систокса. Введение более длинной нормальной или разветвленной цепи (соединения II и III) приводит к менее токсичным веществам. Ниже приведены летальные дозы для ряда соединений (иа крысах, per os):
ДДба.
«г/кз
c2hs'ox ,о
i. ;р^ о,5
сн/ 4sch2ch2sc2h5
С2Н5ОХ ,0
П. 20
(СН3)2СНС/ \SCH2CH2SC2H5
адо .о
ш. 10
CH3CH2CH2OZ \SCH2CH2SC2H5
z
IV. (С2НеО)2р; 0,5
4sc2HB
/3
V. . (С2НбО)2р( 2
XSCH2SC2Hs
При удалении из систокса одной или обеих метиленовых групп (соединения IV и V) получают вещества, обладающие более сильным системным действием, применение которых для защиты растений проблематично из-за их высокой токсичности. Соединение IV по токсичности равно соединению I. Если вещество содержит меньше на одну СНг-группу (соединение V) и, кроме того, имеется еще атом тнониой серы, то оно обладает большей системной активностью (см. раздел 7.9.2), хотя токсичности тиольной и тионной форм примерно равны.
Вещества с разветвленной углеродной цепью тиоэфнрной группы менее устойчивы, они быстро гидролизуются в водных растворах. Напротив, и.х сульфоксидные производные исключительно устойчивы, а по активности их можно сравнить с соединениями, рассмотренными выше.
Если S-алкилыную группу в производных тиольной формы систокса заменить диметиламиногруппой, то получаются фосфорил-тиохолины (см. раздел 7.6,4).
338
7.9.2. Дисистон и тимет
Дискетой (дитиосистокс) и тимет являются превосходными препаратами системного действия, которые можно отнести к группе О, О-диалкил-S- (алкилтиоалкил) -тионфосфатов:
R0\ Z
\s(CH2)„SR
Дисистон J производится в ФРГ фирмой «Байер», тимет (П) — американской фирмой «Америкен Цианамид Ко»:
С2н5О ,S
C2Hsc/ \sCHaCH2SCaH6
с2н5с\ .s /р
с.>н6си \sch2sc2h5
Синонимы: О, О-диэтил-5-2-(этилтиоэтил)-тионфосфат, О, О-ди-этил-5-2-(этилмеркаптоэтил)-дитио-фосфаг, дитиосистокс
Синонимы: О, О-диэтил-S- (этилтиометил)-тионфосфат, О, О-диэтил-S- (этилмеркаптометил)-дитиофосфат, -* тимет
7.9.2.1. Методы получения. И то, и другое соединение можно получить по Шрадеру взаимодействием со-хлоралкилтиоэтилового эфира с натриевой солью О,0-днэтилдитиофосфорной кислоты при 100°С:
Z А
(с2н5о)2р; + ci(ch2)„sc2h6 —* (С2н5о)2р;
\SNa ^S(CH2)rtSC2Hs
7.Э.2.2. Физические и химические свойства. Дисистои и тимет — маслянистые жидкости, мало растворимые в воде. Они легко растворяются в большей части органических растворителей и в жирах. Летучесть тимета довольно высока по сравнению с другими инсектицидами и составляет 12,4 ла/л*3 прн 20 °C. Приводим некоторые физические характеристики обоих препаратов:
Т. кип., °C (при I мм рт. ст.)
Т. пл., °C................
Давление пара при 20 °C, мм рт. ст.......... Максимальная концентрация
при 20 °C, мг/м3........
Относит, плотность d\ . . . .
Показатель преломления
Растворимость в воде прн 20 °C, г/л •..............
Дисистои
Тимет
128 114
— -15
1,8 • 10~4 8,4-10”
2,7 12,4
1,144 (при 20 °C) 1,167 (при 25 °C)
1,5348 (при 20 °C) 1,5349 (при 25 °C)
0,025 0,05—0,08
Тимет менее устойчив к гидролизу, чем дисистон. Период полупревращения при гидролизе при 70 °C и pH 7 составляет для
339
дисистона 27,6 ч, а для тимета 2,8 ч. При 70 °C и pH 9 дисистон гидролизуется на 50% за 7,2 ч.
Химически оба соединения ведут себя как и производные ряда систокса. Оии окисляются по тиоэфирной группе до сульфоксида и сульфона. Окисление происходит также и в растении, причем одновременно идет превращение связи P=S в связь Р = О:
(С2Н5О)2Р^
XS(CH2)„SC2H5
(С2Н6О)2Р Л) (С2Н5О)2р( ,0
XS(CH2)nS^ XS(CH2)„S(
C2Hg ХС2Н5
(с2н50)2р; (С2Н5О)2Р(
xS(CH2)nS-C2H5 xS(CH2)ftS~C2HB
чо
I Z
(С2Н5О)2Р^ —> Н3РО4
хон
Образующиеся метаболиты являются сильными ингибиторами холинэстеразы. Растениями они разлагаются до фосфорной кислоты с промежуточным образованием неядовитой дналкилфосфор-ной кислоты.
7.9.2.3. Инсектицидные и токсические свойства. При применении в качестве средств защиты растений дисистон и тимет сначала проявляют контактно-иисектицидное действие. Резорбция внутрь растения происходит медленно. Поэтому их используют преимущественно для обработки посевного материала, а также в смеси с углем вносят под корни растений. Согласно инструкции по их применению, системное действие продолжается в течение 4—6 недель, при необходимости его можно продлить.
Оба соединения очень токсичны для теплокровных; тимет более ядовит, чем дисистон. Ниже приведены показатели токсичности этих соединений, их метаболитов и аналогов. По данным Шрадера, ЛД50 (ла/ка) на крысах per os составляют:
340
Тимет, его производные и аналоги
(С2Н5О)2Р^ ... 2
\SCH2SC2H5 (С2Н5О)аР^ ,0 ... 1
xsch2s(' хед (С2Н5О)2Р( ^0 1
XSCH2S— C2HS Ч0 z (С2Н6О)2Р^ лэ . . . 0,5—1
XSCH2S^ хс2н6 Z (c2Hso)2p; SCH2S—*C2HB 40 C2H64 .s ‘ ...... ... 1 ... 1
C2H5OZ ^SCH2SC2HB CH84 .s ;p; ... 2,5
(CH3)2CHCZ \SCH2SC2Hs Дисистои, его производные и (С3Н5О)2Р^ ан а л о г и . . . 12
XSCH3CH2SC2H6 (C2H5O)2ZS ,0 ... 5
XSCH2CH2S^ (С2НвО)2р(^ ^0 ...... S CH2CH2S—С2НВ z (Сгн6о)2р; л . . . 5 , . . 1,5
XSCH2CH2S^ С2НВ z (C2H6O)2P^ ^0 XSCH2CH2S—C7HB . . . 1,5
341
с2н5у л
ХРХ ............... 2,0
c2h5oz \sch2ch2sc2h5
СНзх ;р( ............. 2,5
(СНз)2СНСК xSCH2CH2SC2Hs
7.9.3. Тетрам
С2НбО, \р/
C2HgOZ ^SCHaCHaN (C2Hg)2
Синонимы: О,О-ДИэтил-5-(2-диэтиламино)-этилтиофосфат, 0,0-диэтил-S- (2-диэтиламиноэтил) -тиофосфат, амитон.
Это соединение родственно фосфорилтиохолинам.
7.9.3.1. Методы получения. Тетрам получают взаимодействием 2-диэтиламиноэтилата натрия с О,О-диэтилхлортионфосфатом, сопровождающимся немедленной изомеризацией тионпроизводиого:
------>
(С2НЙО)2Р\ -г NaOCH2CH2N(C2H6)2 -NaCi
И Z
—> (С2Н5О)2Р^ —> (C2HSO)2P^
\OCH2CH2N(C2H5)2 \sCH2CH2N(C2H6)2
В лаборатории его можно получать по следующему способу. Растворяют в 350 мл бензола 35,1 г 2-диэтиламиноэтанола и к раствору прибавляют 4,6 г натрия. После растворения всего, натрия при длительном кипячении с обратным холодильником получается этилат. При сильном перемешивании прибавляют 37,7 г О,О-диэтилхлортионфосфата и выдерживают смесь 4 ч при 70—80 °C для завершения изомеризации. После отделения хлористого натрия (фильтрованием) раствор перегоняют в вакууме (0,2 мм рт. ст.).
Четвертичное аммониевое производное можно получить по Шрадеру присоединением триэтиламина к 0,0-диэтил-Э-(2-бром-этил)-тиофосфату [см. раздел 7.6.4.1; метод (в)].
7.9.3.2. Физические и химические свойства. Тетрам умеренно растворим в воде, легко растворяется в органических растворителях. Физические константы тетрама следующие:
Т. кип., °C при 0,01 мм рт. ст. ....... 80
при 0,2 мм рт. ст............ 97
при 2 мм рт. ст............. 134
Показатель преломления п™ ........ 1,5075
Вследствие основного характера он с кислотами образует соответствующие аммониевые соли. Как и для фосфорилхолинов, возможно получение четвертичных аммониевых солей при действии алкилгалогенидов, что приводит к сильно токсичным соединениям. Четвертичные соли растворимы в воде (!).
342
Тионная форма при нагревании в водных растворах или в иных полярных растворителях изомеризуется в тиольную форму с промежуточным образованием циклических имониевых иоиов [см. раздел 7.6.4.1, метод (б) и раздел 7.9.1.2].
Находивший до сих пор ограниченное применение для защиты растений кислый оксалат
[(C2H5O)2P(O)SCH2CH2NH(C2H5)2] [НООССОСГ]
имеет т. пл. 100—101 °C; токсичность этого соединения через кожу (ЛД50 на крысах) составляет 2 л/а/ка.
7.9.3.3. Инсектицидные н токсические свойства. Тетрам обладает продолжительной и превосходной системной активностью. Однако ввиду его очень высокой токсичности для теплокровных возможно лишь ограниченное применение тетрама для защиты растений.
Токсичность тетрама через кожу высока (ср. раздел 7.6.4.3); средняя смертельная доза ЛДбо для крыс (per os) составляет 3—7 л<а/ка, для мышей (внутрибрюшинно) — 0,5 мг/кг.
Как и в случае систокса и тимета, замена алкоксильной группы на алкильную приводит к более ядовитым соединениям (например, соединениям I и II, для которых ЛД50 0,25 мг/кг):
СНз\ C2Hs\
хр
C2H5OZ \sCH2CH2N(C2Hs)3 C2HsOZ \sCH2CH2N(C2H5)2
1 11
При известных обстоятельствах такие соединения могут быть применены как боевые ОВ. При нормальных метеорологических условиях они очень долго эффективны на местности.
С удлинением углеродной цепи, особенно при наличии арильной группы, токсичность уменьшается (соединение III, ЛД5о 25 мг/кг). Замена обеих алкоксигрупп алкильными также увеличивает токсичность (соединение IV, ЛД50 1 мг/кг):
п-СН3С6Н4х .О ,О
X (сгн5)2р;
С2Н5СИ XSCH2CH2N(C2H5)2 \SCH2CH2N(C2H5)2
II1 IV
Соединения с диалкиламииогруппой в молекуле по токсичности сравнимы с тетрамом, особенно если сера находится в тнонной форме (соединение V, ЛДбо 2,5 мг/кг):
(CH3)2N\
C2HSZ ZSCH2CH2N(CaHs)2 v
343
7.9.4. Паратион и параоксон
(С2Н5О)2Р^ ^ОСбНЛОгП
I
Синонимы: О,О-диэтил-(4-нитро-фенил)-тнонфосфат, парафос, тнофос, Е 605
(С2Н5О)2Р^
\0C6H4N02-n и
С инонимы: О,О-диэтид-(4-ннтро-феннл)-фосфат, миитакол, Е 600
Паратион I и параоксон II получены в 1944 г. Шрадером и первоначально были названы Е 605 и Е 600 соответственно. Паратион очень распространен как контактный инсектицид. В начале 50-х годов он широко применялся в Западной Германии.
Параоксон обладает превосходным контактным и системным ядовитым действием, ио высокая токсичность для теплокровных допускает лишь ограниченное его применение.
7.9.4.1. Методы получения. На заключительной стадии синтеза паратион и параоксон получают реакцией О,О-диэтилхлортионфос-фата с 4-нитрофенолятом натрия:
----->
—NaCl
(С2Н5О)2р
Соответствующие О,О-диэтилхлорфосфаты можно получить из треххлористого фосфора через хлорокись фосфора либо через тиотреххлористый фосфор. Этерификация этих соединений избытком этанола дает соответствующие алкилдихлорфосфаты, которые в присутствии связывающих кислоту веществ этерифици-руются другим молем этанола или этилата натрия с образованием 0,0-диалкилхлорфосфатов:
+о2
-----•> 2О=РС13
2РС13 - +5Я
------> 2S=PC13-
-(S)O—РС1з + носгн5 ——> С2Н5О-РС12
—ИС! ||
O(S)
^O(S)
С2Н5О-РС12 + NaOC2H5 —> (C2H5O)2Pf
и "NaC1 \ci
O(S)
7.9.4.2. Физические и химические свойства. Паратион и параоксон— жидкости без запаха. Оии хорошо растворяются в обычных
344
растворителях, ио мало растворимы в нефти и минеральных маслах. Параоксон лучше растворим в воде, чем паратион, чем объясняется его способность к быстрой резорбции в растения; паратион не проникает в растения.
Физические свойства паратиона и параоксона следующие:
Паратион
Параоксон
Т. кип., °C (при 6 мм рт. ст.). 150
Т. пл., °C..................... 6,1
Давление пара при 20 °C, мм рт. ст. 0,57 • I0-5
Максимальная концентрация при 20 °C, мг/м3 ............... 0,09
Относит, плотность d*.........1,265 (25 °C)
Растворимость в воде при 25 °C, г/л . . 0,024
144
1,2- I0’5
0,18
1,2667 (20 °C)
2,4
В табл. 37 сопоставлены периоды полупревращения при гидролизе паратиона и параоксона в зависимости от концентрации ионов водорода. Для параоксона можно констатировать то же явление, что и для диалкоксифторфосфатов и алкоксиалкилфторфосфоиатов, у которых в слабокислой среде устойчивость к гидролизу увеличивается. В данном случае область устойчивости соответствует интервалу pH 2—5.
Таблица 37. Периоды полупревращения при гидролизе паратиона и параоксона при 70 °C в зависимости от pH (по данным Шрадера)
pH Период полупревращения, ч । РН Период полупревращения, ч
параоксон паратион параоксон паратион
I 18,5 34 1 6 18 13
2 22,7 27 7 11,5 7,8
3 23 21 8 9,2 4,1
4 24,4 17,5 9 2,1 2,7
5 ’ 24,4 19,5
Параоксон — сильный системный яд. В связи с хорошей растворимостью в воде и сравнительно высокой устойчивостью к гидролизу ои может быть пригоден как диверсионный яд для заражения воды, продовольствия и напитков.
Устойчивость гомологов параоксона к гидролизу возрастает с длиной и характером разветвления углеродных цепей.
Самыми устойчивыми являются соединения с углеродными цепями, разветвленными вблизи атома кислорода. Так, для параоксона при pH 13 период полупревращения при гидролизе (25 °C) составляет 10 мин, О,О-диизопропил-(4-нитрофснил)-фосфата
он равен 108 мин, а для вторичного бутоксипроизводиого : -264 мин.
345
При действии окислителей паратион превращается в параоксон с отщеплением тиоиной серы:
(С2Н5О)2Р^
\о-
NO2
+ 2О2; +Н2О
-hsso4
(С2Н5О)2Р
Эта реакция объясняет действие паратиона на живой организм, заключающееся в блокировании холинэстеразы. Согласно этим представлениям истинным ингибитором является параоксон.
Фосфорилирование фермента происходит под влиянием кислотного 4-нитрофенолятного остатка. Если 4-нитрофенильную группу заменить фенильной
(С2Н5О)2Р
то такое соединение уже не оказывает фосфорилирующего действия. Оно не отвечает ключевой формуле Шрадера и не является биологически активным.
Паратион и параоксон могут легко вступать в реакцию с алко-голятами и фенолятами с образованием неядовитых продуктов:
7.9.4.3. Инсектицидные и токсические свойства. Паратион как инсектицид обладает контактным и ингаляционным отравляющим действием, а также пригоден для заражения приманок. В отличие от параоксона он лишен системного действия. Как инсектицид он эффективен I—2 суток после применения, но не дольше 8 суток.
Из-за чрезвычайной токсичности параоксон, пожалуй, вряд ли можно применять как инсектицид. По сравнению с паратионом ои во много раз более токсичен, как видно из сопоставления величин ЛД50 обоих препаратов (табл. 38).
Малые дозы параоксона уже в разбавлении 1:10 000 оказывают мистическое действие, на этом основано применение его сильно разбавленных растворов для лечения глаукомы.
Согласно исследованиям Химического центра армии США49, аэрозоли параоксона со средним диаметром частиц 0,9 лл очень токсичны. При одинаковых условиях параоксон в виде аэрозоля лишь в 3,5 раза менее токсичен для крыс, чем зарин. Так, при экс-
346
Таблица 38. Сравнение токсичности паратиона и параоксона
Препарат ЛД50. мг/кг
на мышах, подкожно на кроликах, через кожу на крысах, перорально
Паратион 10-12,5 40-50 13
Параоксон 0,6-0,8 5 3
позиции 10 мин значение ЛД50 составляет для параоксона 0,089 мг/л, а для зарина 0,027 мг/л.
При физической нагрузке, при которой увеличивается объем вдыхаемого воздуха за едйницу времени и усиливается резорбция
аэрозоля в дыхательных путях, токсичность параоксона увеличивается почти в 2 раза. В этом случае (опыты на животных во вращающейся клетке) ЛД50 составляет для параоксона 0,032 мг/л, а для зарина 0,01 мг/л.
Соотношения между токсичностью и химической структурой, установленные для диалкоксиацилфосфатов и алкоксиалкилацил-фосфонатов соблюдаются и в данном случае.
Если RO-группу заменить алкильной, сохранив нитрогруппу в положении 4, то токсичность, в общем, увеличивается. О-Этил-О-(4-нитрофенил)-этилфосфонат (армии) III обладает в 10 раз большей биологической активностью, чем параоксон. Токсичность этого соединения ЛДбо (на крысах, per os) составляет 1 мг/кг. Он оказывает и более сильное мистическое действие, проявляющееся уже в разбавлении 1:20 000. Вторичная RO-группа с разветвлением вблизи атома кислорода, например, в О-изопропил-О-(4-нитрофе-нил)-метилфосфонате IV влечет за собой дальнейшее повышение токсичности (ЛД50 равно в тех же условиях 0,5 жг/кг):
Токсичность веществ с группой NO2 в положении 2 (соединение V) уменьшается (ЛД5о 3,8 мг/кг), а с группой NO2 в положении 3 (соединение VI) такое же, как у соединения III (ЛД50 1 мг/кг);
347
7.9.5. Днмефокс
(CH3)2N4 хО
(CH3)2NZ
Синои нм ы: бис-(ЬГ,ЬГ-днметиламидо)-фторфосфат, пестокс14, ханав,
CR-409.
В соответствии с программой военио-хнмических исследований димефокс был получен в 1940 г. в Германии Шрадером и в 1942 г. в Англии Хитом и Саундерсом. Он относится к группе бис-Ы.М-ди-алкиламидофторфосфатов и является самым токсичным ее представителем (ср. раздел 7.4.1).
Димефокс обладает сильным системным действием. Его применяют даже для защиты деревьев от насекомых-грызунов. Для этого днмефокс в желатиновых капсулах помещают под корни дерева. Через 24 ч капсулы растворяются и растение усваивает димефокс через корни.
Военное применение этого вещества в качестве ОВ отрицается21.
7.9.5.1. Методы получения. В английском способе50 получения . используют реакцию диметиламина с дихлорфторфосфатом в бензольном или эфирном растворе:
+4НЖСН.Ь (CHs)jNy О ---------------> - р
—2HN(CHg)2-HCI (CH3)2N^ ^F
7.Э.5.2. Физические и химические свойства. Димефокс — бесцветная жидкость со слабым запахом. Он обладает следующими физическими свойствами:
Т. кип., °C
при 1 мм рт ст.............. 38
при 10 мм рт. ст............. 80
Давление пара при S0 °C, мм рт. ст...... 0,11
Летучесть при 20 °C, мг!л .......... 0,93
Относит, плотность ........... 1,1151
Показатель преломления ......... 1,4267
Димефокс смешивается с водой во всех отношениях и при этом лишь слабо гидролизуется. Степень гидролиза за 30 мин составляет 9%, за 500 ч — 30%.
В слабощелочных растворах при комнатной температуре димефокс очень устойчив. При кипячении с обратным холодильником с гидроокисью натрия он полностью гидролизуется за 30 мин:
,О [(CH3)2N]2Pf^
+2NaOH
[<CHlhN1<ONa
348
В кислой среде в результате расщепления связи Р—N гидролиз идет быстрее:
Л +2Н+; +2Н2О + л®
[(CH3)2N]2p; -----------> 2[(CH3)2NH2] + (HO)2PZ
* ' \R \F
г; V
V Димефокс — сильный ингаляционный яд. Средняя летальная
I концентрация для мышей составляет 0,095 мг/л. Мистическим дей-
? ствием ои ие обладает, резорбция через кожу слабая. Средняя ле-тальиая доза ЛД50 для крыс (перорально) ’ составляет 7,5 мг/кг.
| Подобно другим амидофосфатам, холииэргически активное веще-£ ство образуется из димефокс# только в организме.
L ’ Столь же токсичен английский препарат NC7; действующим Ha-В.; чалом этого вещества является бис-(Ы,Ы-диметиламидо)-азидФос-
I' Фат:
В (CH3)2Ny .О ,
Рх
у (CH3)2NZ \n3
7.9.6. Фосдрнн
СН3О. /О
СН.о/ \о—с=сн— cf
. U 0CHs
jy Синоним: диметил-(1-карбметоксн-1-пропен-2-ил)-фосфат. $;;
Препарат фосдрнн является контактным инсектицидом, ио одно-ВС /временно обладает и системным действием.
Это жидкость, смешивающаяся с водой во всех отношениях. |Г| В нейтральных водных растворах при комнатной температуре фос-дрии гидролизуется медленно (период полупревращения около !С 30 суток), в щелочных растворах гидролиз происходит очень быстро. Товарные препараты обычно содержат два изомера — цис-Е . н транс-фосдрии в соотношении 3 : 2.
| Фосдрнн сильно токсичен (ЛД50 для мышей перорально соста-„ вляет 3,7 мг/кг), обладает способностью к сильной .резорбции через кожу.
7.9.7. ТЭПФ
С2Н5О ОС2Н6
ХР—О—РХ
C2H6OZJj| ^ОС2Н5
Синонимы: тетраэтилпиррфосфат, бладан, нифос Т, НЕТ, ТЕРР.
ТЭПФ наряду с паратионом относится к наиболее известным ^фосфорорганическим инсектицидам. Ои принадлежит к группе тет-^раалкилпирофосфатод..
349
Впервые ТЭПФ был получен в 1850 г. Мошниным. С тех пор его синтезировали и исследовали многие химики, не зиая о его сильной токсичности (фои Клемонт в 1854 г., Аббот в 1879 г., Давалье в 1906 г., Найлен в 1930 г. н др.).
Самым распространенным способом получения ТЭПФ является контролируемый гидролиз днэтнлхлорфосфата;
2(C2HSO)2P
^С1
+н2о
„нг1> (С2Н5О)2Р-О-Р(ОС2Н6)2 —JnCl 11 и
Образующийся хлористый водород можно удалять откачиванием в вакууме или нейтрализовать рассчитанными количествами веществ, связывающих кислоту, например пиридина, бикарбоната натрия.
ТЭПФ — бесцветная нлн слабожелтая жидкость. Ее физические константы следующие:
Т. кип., °C
при 1 jhjh рт. ст........... 124
при 2 мм рт. ст........... 135
Давление пара при 20°C, мм рт. ст. . . 1,55* 10—4
Летучесть при 20 °C, мг!м3 ....... 2,5
Относит, плотность ............ 1,185
♦
Показатель преломления ........ 1,4196
Он смешивается с водой во всех отношениях. В обычных органических растворителях легко растворим, в бензине, петролейном эфире и др.— с трудом. Прн нагревании выше 270 °C ои термически диссоциирует, прн 230 °C,— полностью разлагается с отщеплением этилена.
ТЭПФ быстро гидролизуется водой, при 25 °C период полупревращения равен ~ 7 ч (кислоты и щелочи ускоряют его гидролиз):
+н2о
* (С2Н50)2Р-0-Р(ОС2Н6)2 “--> 2(С2Н5О)гР,
II II ' \0Н
ОО-
Скорость гидролиза гомологичных соединений с увеличением числа атомов углерода уменьшается.
ТЭПФ — сильный ннгнбнтор холинэстеразы, эффективен прн ингаляции, при приеме через рот, а также в результате кожиой резорбции. Скорость резорбции велика. Одноразовая кожно-резорбтивная доза в 400—600 мг должна быть безусловно смертельной для человека. Пероральная летальная доза составляет 100 мг. Для крыс (per os) ЛД50 равна 1,1 мг/кг.
ТЭПФ является контактным ядом и не обладает системным действием. Поскольку он гигроскопичен и быстро гидролизуется, его можно применять незадолго до уборкн урожая. По-внднмому, ТЭПФ почти не представляет военного интереса.
350
7.9.8. Военное значение инсектицидов
Рассмотренные фосфорорганические инсектициды не во всех случаях пригодны для военного применения в качестве химических ОВ. Производство и ассортимент фосфорорганических инсектицидов за последние годы сильно возрос. Во многих странах появились заводы для нх производства. В настоящее время американская промышленность в состояинн производить около 14—15 тыс. т паратиона в год наряду с другими инсектицидами.
Инсектициды, кроме нх большого значения для защиты растений, имеют известный военный интерес. Это прежде всего соединения с системным действием, которые иа определенное время делают растения ядовитыми н для теплокровных'. В некоторых случаях системное действие может сохраняться от посева до уборки урожая. В этом смысле определенную опасность представляют наиболее токсичные представители, иапример соединения типа систокса, тетрама, тнмета н днмефокса.
Хотя токсичность этих соединений в общем меньше, чем у зарина, иужио принять во внимание последствия, которые наступают в результате употребления в пнщу зараженных таким образом растений и плодов. Заражение живых растений является предметом военных исследований в империалистических государствах. Они развиваются в двух направлениях:
а) уничтожение самих растений, с тем чтобы лишить противника продовольственных ресурсов (фнтояды, см. раздел 9);
б) отравление н уничтожение противника в результате потребления зараженных растительных культур.
Работы над созданием других соединений с лучшим системным действием и токсическими свойствами еще продолжаются. Как известно, в результате незначительных изменений строения фосфорорганических соединений нх биологическая активность меняется скачкообразно, поэтому и средн системных ядов это может дать большие возможность вариаций. Например, в бельгийских военно-химических лабораториях исследовались 51 аналоги тетрама; из них некоторые оказались сильными ингибиторами холинэстеразы. Подобные исследования проводятся в военно-химических лабораториях н других стран.
Некоторые нз рассмотренных в этом разделе соединений могут применяться для заражения систем водоснабжения и жизненно важных продуктов питания. Во всех случаях целью такого коварного применения является отравление противника.
Применение системных ядов для заражения растений не представляет трудностей. Имеющимися средствами можно в кратчайший срок заразить большие посевные площади.
В принципе существует возможность заражать фосфорорганическими инсектицидами растительные культуры, потребление которых повлечет за собой тяжелые последствия для здоровья.
351
КОНТРОЛЬНЫЕ ВОПРОСЫ -
1. Напишите формулы следующих соединений:
О-(3,3-диметилбутил)-пропилфторфосфоната; бис- (N.N-этилметиламидо) -цианфосфата; тетра-(NjN-диметиламидо)-пирофосфата.
2. Что выражает выведенная Шрадером общая формула?
3. Что следует понимать под ингибитором холинэстеразы?
4. Как классифицируют ингибиторы.холинэстеразы?
5. В чем проявляется блокирование холинэстеразы?
6. Какие токсические действия вызывают фосфорорганические соединения?
7. Опишите свойства дициклогексилфторфосфата.
8. Как получают диалкилфторфосфаты без применения третичного амина?
9. 'Как происходит гидролиз диалкил фтор фосфатов в кислой, нейтральной и щелочной средах?
10. Напишите уравнение реакции ДФФ с концентрированными растворами щелочей.
11. Какие хелаты меди проявляют наилучшую каталитическую активности при гидролизе фосфорорганических соединений?
12. На чем основана каталитическая активность хелатов меди при гидролизе фосфорорганических соединений?
13. Напишите уравнение реакции ДФФ с гидроксиламином.
14. Какое значение имеют некоторые оксимы, например метилпиридин-2-альд-оксим или токсогенин?
15. Напишите схему реакций гидроксамовых кислот с диалкилфторфосфа-тами.
16. Как действует ДФФ на глаза?
17. По какой реакции получают М,М-диалкиламидо-О-алкилцианфосфаты?
18. Как ведут себя М.М-дналкнламйдо-О-алкнлцианфосфаты в водных растворах?
19. Напишите схему гидролиза табуна.
20. Какие взаимосвязи имеются между строением диалкилцианфосфатов и О-алкилалкилцианфосфонатов и их токсичностью?
21. Какие реакции происходят при фторировании и этерификации алкил-дихлорфосфонатов?
22. Что такое реакция Арбузова?
23. Нарисуйте схему всех методов получения зарина.
24. Чем объясняется большая глубина распространения облака зарина?
25. Какой области pH соответствует наименьшая скорость гидролиза зарина?
26. На чем основано каталитическое действие гипохлорит-ионов при гидролизе фосфорорганических соединений?
27. На чем основана каталитическая активность лерекисей при гидролизе .фосфорорганических соединений?
28. Какие связи существуют между стррением О-алкилалкилфторфосфоватов и строением производных фосфорил- и фосфорилтиохолина?
29. Какими способами можно получать фосфорилхолины?
30. Что такое изомеризация? Напишите механизм превращения тионфосфо-рилхолинов в тиофосфорилхолииы.
31. Обоснуйте способность алкилфторфосфорилхолинов к быстрому гидролизу.
32. Как происходит щелочной гидролиз алкилфторфосфорилхолинов и ди-алкоксифосфорилтиохолинов?
33. Объясните существенное различие йежду механизмами биохимического действия О-алкилалкилфторфосфонатов, алкилфторфосфорил- и алкилфосфорил-тиохолинов.
34. Назовите принятые в США сокращенные обозначения зарина, зомана и табуна.
35. Сопоставьте возможности дегазации фосфорорганических ОВ. ‘ _
36. Что такое соединения с системным действием?
37. Какие требования предъявляют к системным ядам с гражданской и военной точек зрения?
352
. 38. Чем отличаются друг от друга тиин- и гиилснсгиксыг-----------
;; t .39. Каким превращениям подвергаются тимет и дисистон в растении?
40. Что такое параоксон?
41. Что такое димефокс? Чем ои отличается от табуна?
42. Каков состав фосдрина?
ЛИТЕРАТУРА
I. Lange W., von Kruger, Вег., 65, 1598 (1932).
2,iBo Holmstedt, Acta physiol. Scand., 25, SuppL. 90, Stockholm (1951).
3/Schrader G. Die Entwicklung neuer Insektizide auf Grundlage organischer Fluor- und Phosphorverbindungen, Weinheim, 1952.
4. Мельников H. H„ Химия, 1961, 684.
5. Rasmussen H. B„ Terp P., Acta Pharm, Org. Chem., 58 (108), 268 (1951).
6. McCombie, Saunders В. C.. Nature, 1946, 287; Англ. пат. 601210.
7. SaundersB. C., Stacey, J. Chem. Soc., 1948, 695.
& Waters, de Worms, J, Chem. Soc., 1949* 926.
>9. Goldwithe, Saunders В. C., J. Chem. Soc., 1955, 2040.
10. Шрадер Г. Новые фосфорорганические инсектициды. Перев. с нем., «Мир», Г 1965.
11. L а г s s о n L., Acta chem. Scand., И, 1131 (1957).
12. J. Ащ. Chem. Soc., 77, 922 (1955) ; Arznelmittelforschung, 4, 527 (1954),
13. J. Am. Chem. Soc., 79, 3030 (1957k
14. J a h n d о r f, J. Am. Chem. Soc., 78, 3686 (1956).
15. Wagner-Jauregg Th:, Arzneimittelforschung, 6, 194 (1956).
16. J. Chem. Soc., 1958, 1583.
17. J. Am. Chem. Soc., 77, 3651 (1955); 78, 3594 (1956); 79, 2618 (1957).
18. О’Б p а й и, Токсичные эфиры кислот фосфора, пер. с англ., М., «Мир» 1954,
19. J. Am. Chem. Soc., 73, 5202 (1951). .
20. Saunders В. C., Heap R., J. Chem. Soc., 1948, 1313.
21. S a r t о r i M., Chim. et ind., 34, 212 (1952).
22. Нем. пат. 767830 KL 122 (1939).
22a. H о 1 m s t e d t B., Synthesis and Pharmacologie of Dimethylamidoethoxyphosphoryl Cyanide (Tabun), Stockholm, 1951, p. 35.
23. Larsson L, Acta Chem. Scand., 6, 1470 (1952 ; 7, 306 (1953).
24. J. Am. Chem. Soc., 82, 3843 (I960).
25. Sto hr R., Militdrwesen, 2, a Belneft, 55 (1958).
26. Schrader G., BIOS, 1947, 714, 41.
.27. J, Chem. Soc., 1948, 702.
i'28. Ottar В., T. Kjem Bergvesen og Metall, 5, 73 (1954); C. A., 48, 2342 (1954);
Canad. J. Chem., 38, 1416 (1960).
29. К i n n e а г A. M., P e г г e n E. A., J. Chem. Soc., 1952, 3437,
30. Пат. США 3014778 (1953); 3014781 (1953).
>31. Пат. США 2994715 (1949 .
*;32. J. Am. Chem. Soc., 78, 4068 (1956).
33. Lohs K, Synthetische Gifte, Berlin, 1963, S. 308.
34, Beach R. S., Sass N. J„ Anal. Chem., 33, 901 (1961).
>35. Chem. Ber., 95, 381 (1962).
'36. Mar sh D. J., N e a 1 e E„ J. Appl. Chem., 1958, 324.
t.37. Arch. Industr. Health, 17, 34 (1958).
38. Armed Forces Chem. J., 24, 4 (I960).
>39. Frederikson T., Acta derm.-venerol,T 38, 1 (1958).
40. Tanimelin L. E., Svensk. Ketri. Tidskr., 70, 157 (1958).
41. J a r s s о n L„ Acta Chem. Scand., 11, 1131 (1957).
.42. Tammelin L. E., Acta Chem. Scand., 11, 1738 (1957); Fukuto T. R„ S t a f f о r d E. M., J. Am. Chem. Soc., 79, 6083 (1957),
12 Зак. 448
353
43. Tammelin L. E., Arkiv f. Kemi, 12, 287 (1958); Svensk. Kem. Tidskr., 70, 157 (1958); Frederikson T„ Arch, intern. Pharmacodyn. Therap., 113, 101 (1957); 115, 474 (1958).
44. Conrad R., Militarwesen, 6, 586 (1962).
45. Schrader G., Angew. Chem., 69, 86 (1957).
46. Fukuto T. R-, Metkalf R. L., J. Am. Chem. Soc., 78, 5182 (1956).
47. H eat h D. F., V a ndekar M,, Biochem. J., 67, 187 (1957).
48. Wirth W., Naunyn-Schmiedbergs Arch, exper. Path., 234, 352 (1958),
49. Arch. Industr. Health, 17, 34 (1958).
50. Saunders В. C., Heap R,, J. Chem. Soc., 1948, 1313.
51. P оchetA„ de Vleeschhou wer G. R., Arch, intern. Pharmacodyn. Therap., 140, 669 (1962),
8. ПСИХОХИМИЧЕСКИЕ ОВ (ПСИХОЯДЫ)
В 1958 г. стало известно, что в некоторых военно-химических исследовательских центрах США проводятся исследования так называемых психохимических веществ — соединений, которые могут оказывать воздействие иа психику человека. Некоторые из этих соединений в последние годы были испытаны в полевых условиях; 'разумеется, результаты таких испытаний не опубликовываются. Тем не менее, эти опыты демонстрируют стремление руководства американской армии иметь на вооружении наряду с высокотоксичными фосфорорганическими соединениями и такое химическое оружие, которое в самых малых концентрациях ведет лишь к утрате противником активности и боеспособности, .
Представляющие военный интерес психояды наряду с известными недостатками имеют и преимущества перед другими ОВ. Так, к потере боеспособности приводят чрезвычайно малые дозы и концентрации этих веществ, не обнаруживаемые обычными методами индикации. Психозы, вызываемые этими веществами, длятся определенное время, ие оставляя никаких последствий.
Психоактивные соединения, вызывающие необратимые психозы, вряд ли представляют военный интерес.
Психохимические ОВ лишены цвета, запаха и вкуса. Их можно применять как в бою, так и в качестве диверсионных ядов для заражения воды, продовольствия и напитков,. Некоторые соединения чрезвычайно устойчивы иа воздухе и к действию воды.
Среди полученного за последние годы значительного числа психоактивных веществ имеются такие, которые обладают определенными качествами ОВ и доступны для получения в больших количествах. Крупнотоннажное промышленное производство некоторых важных соединений пока еще сопряжено с известными трудностями. Для того чтобы ликвидировать разрыв между возможностями производства и военного применения, в США ассигнуются крупные суммы на разработку и испытание технологических процессов. Считается, что психохимические ОВ пригодны «для боевого применения при локальных столкновениях, когда военные действия ограничены по своим масштабам, оперативной глубине и задачам».
Однако, по этим взглядам, психояды могут иметь и стратегическое значение, как яды для диверсий. При помощи сравнительно малых количеств ОВ можно добиться значительных последствий в глубоком тылу противника. Считают, что заражение систем
12*
355
водоснабжения н другие мероприятия могут на определенный промежуток времени сделать недееспособными широкие круги населения. Последствиями этого были бы ощутимые нарушения производства, страх, неуверенность и непредвидимые поступки.
Психояды отличаются от Других ОВ своей ’ эффективностью,. В то время как в случае фосфорорганических ОВ к потере боеспособности приводит доза, в 2 раза меньшая смертельной, в случае, психохимических ядов соответствующая доза составляет порою лишь 1/1000 смертельной.
По сравнению с другими ОВ, например с синильной кислотой н зарином, величины боевых концентраций синильной кислоты, зарина и психояда относятся друг к другу как 1:0,1:0,001, т. е. чтобы сделать противника небоеспособным, требуется количество, психряда в 1000 раз меиьшее, чем соответствующее количество синильной кислоты.
Действие разных психоядов на людей различно?Однако эти различия ие Столь характерны, чтобы были возможны какие-либо заключения о взаимосвязях между химическим строением ядов и их токсическим действием. Психояды обладают широким спектром действия. С точки зрения структурного типа, среди иих встречаются, например, производные индола, феиалкиламинов и некоторые производные пиперидина. Это либо природные, либо синтезированные соединения. '
Вещества, рассмотренные в следующих разделах, дают возможность ознакомиться с проблематикой психохимических ОВ. Область исследования психоядов ни в коем случае.не завершена; более того, это начало развития1 исследований, направленных к подчинению человеческого сознания всестороннему воздействию и контролю.
Психояды вызывают у нормальных людей психические аномалии, так называемые модельные психозы. Психика человека изменяется, ио какие-либо устойчивые поражения организма прн этом не проявляются в отличие, иапример, от действия тетраэтилсвинца и других подобных соединений. Хотя психические изменения могут быть очень разнообразными, на передний план выдвигается галлюциногенное действие.
Галлюциногены приводят к чрезмерному возбуждению некоторых клеток головного мозга, которое проявляется, в частности, в зрительных и слуховых галлюцинациях. В' воображении отравленного предстают вещи, предметы, которые он ранее совершенно не ощущал органами чувств. В результате психических нарушений сознания естественные ощущения притупляются и отображаются в совершенно ином виде. Сознание затемняется либо выключается совсем.
Иллюзии, возникающие вследствие психоза, нарушают привычную систему восприятий и могут привести к утрате чувства реальности. Настроение и эмоции отравленного полностью определяются психозами, вызванными галлюциногенами. Такие изменения имеют отчасти шизофренический характер.
356
С точки зрения механизма действия между естественной шизи-i френией и искусственными психозами, по-видимому, имеется опре-деленное сходство. У шизофреников были обнаружены те же или ^аналогичные продукты распада, которые получаются и прн приеме пснхоядов. Еще несколько лет назад полагали, что ответственным ; за естественные психические расстройства нужно считать некий / специфический «возбудитель», который, якобы, выделили нз крови V больного шизофренией («тараксеин»). Как естественные, так и вы-^аваниые искусственно психозы определяются множеством факторов, t. которые во всей совокупности можно выявить только в результате е длительной, трудоемкой работы специалистов в разных отраслях | ананий.
£ Фильм Химической службы армии США (о лабораторных опы-р-тах с кошкой и мышью), показанный 3 декабря 1958 г. на конференции в Нью-Йорке, продемонстрировал действие веществ такого S рода: если нормальная кошка в соответствии с природными ии-гйстинктамн убивала мышь, то та же самая кошка под действием Гпснхояда вела себя совершенно иначе. Она пугалась при виде мышн и принимала оборонительную позу.
Обычно галлюциногены оказывают также эйфорическое дей-^ ’< ствие, которое выражается в приятной и радостной безмятежности, > анакомой по состоянию опьянения. Некоторые психояды воздей-ствуют на волю, что ведет к апатии, снижению психической приспо-^собляемости, к неустойчивости, скованности и к большей нерешительности, т. е. оказывают на пораженных депрессивное действие.
Считают, что среди применяемых в последние годы для лечения ^Психических расстройств «психиатрических» препаратов имеются ^некоторые соединения из числа так называемых транквилизаторов, пропускающие военное использование-. Транквилизаторы обладают ^успокаивающим (седативным) действием. Они усыпляют, вызывают ацатню и безразличие н отрицательно действуют и а мыслительные способности и способность к сосредоточению.
£ Первостепенный военный интерес представляют две группы пси-ухохнмических соединений — пснходислептики и транквилизаторы.
Соединениям первой группы уделяют наибольшее внимание, £ среди них имеются самые эффективные вещества. Они приводят «раздвоению» личности, нарушают чувственные восприятия, угнетают сознание, дезориентируют, вызывают галлюцинации, видения И» страх.
Транквилизаторы предназначены подавлять волю, рассудок и ^психическую деятельность. Наступающее равнодушие,у вялость т. п. должны помешать выполнению боевой задачи.
. По своей химической природе средн пснхоядов имеются производные иидола, феналкиламнны, производные пиперидина.
Психояды — соединения сложного строения. Однако даже Heit которые простые соединения проявляют определенные психические воздействия, например диметилацетамид, который в дозах свыше 400 мг/кг массы.-(по сравнению с другими психоядами это большая
357
доза) вызывает депрессии, потерю ориентации, летаргию, зрительные галлюцинации, бредовые представления и т. п. По имеющимся данным ’, психоз длился около 7 суток и в пяти из описанных 15 случаев привел к смерти.
В упомянутых группах как возможные психоактивные ОВ представляют военный интерес диэтиламид лизергиновой кислоты из группы производных индола и гликоляты из группы производных пиперидина.
8.1, ПРОИЗВОДНЫЕ ИНДОЛА
Психояды, относящиеся к этой- группе, имеют конденсированную гетероциклическую структуру индола I.
|НО.
CH2CH2NH2
NH
Индол является структурным фрагментом некоторых жизненио-важных биогенных аминов, таких, как триптофан, триптамин н серотонин.
Серотонин, или 5-окситриптамин II оказывает существенное влияние на нервную деятельность человека. Функция мозга находится в тесной взаимосвязи с метаболизмом этого соединения.
Общепризнано, что родственные по структуре психояды вытесняют серотонин, но не перенимают его функцию, состоящую в передаче нервного импульса центральной нервной системе.
Ингибирование фермента аминооксидазы нарушает метаболизм серотонина, что ведет к известным аномалиям и психическим расстройствам. Психохимические ОВ, родственные серотонину, действуют на пока еще неизвестные рецепторы нервной системы.
ч
8.1.1. Производные лизергиновой кислоты
Лизергиновая кислота III является главным структурным фрагментом алкалоидов спорыньи (эргоалкалоидов) группы эрготамина и эрготоксина:
соон
Некоторые амиды лизергиновой кислоты в самое последнее время были выделены (швейцарская фирма Сандоц А. Г., Базель)
358
из семян различных вьющихся растений (Rivea corymbosa, Ipomea tricolor) и мексиканского ритуального снадобья «ололиукви». Это вещество использовали во время религиозных торжеств, чтобы вызвать состояние опьянения. Выделенные из него производные индола является галлюциногенами2, содержание которых в семенах составляет от 0,01 до 0,05%.
Из алкалоидов спорыньи лизергиновую кислоту выделяют действием метанольных растворов щелочей.
Возможными психоядами из числа производных лизергиновой кислоты называют следующие: диэтиламид, этиламид и морфолид лизергиновой кислоты. Наибольшее внимание уделяется диэтил-амиду лизергиновой кислоты, который можно получить синтетически.
8.1.1.1. Диэтиламид( + )-лизергииовой кислоты (LSD, LSD-25)
O = C-N(C2H5)2 AAj-GHjj
NH
В молекуле диэтиламида лизергиновой кислоты ядро индола сконденсировано с частично гидрированным ядром хинолина Хотя возможен полный синтез этого соединения, его получают превращением в диэтиламид лизергиновой кислоты, выделенной .из алкалоидов спорыньи. Спорынью выращивают на ржи.
В настоящее время мировое производство алкалоидов спорыньи ие превышает нескольких килограмм. Получение этих веществ пытаются увеличить за счет культивирования спорыньи на питательных средах. Одновременно ведутся разработки соответствующей, экономически рентабельной технологии синтеза производных лизергиновой кислоты.
Общие свойства. Диэтиламид лизергиновой кислоты — кристаллическое, легко разлагающееся при нагревании вещество. Разложение начинается уже при температуре плавления (т. пл. 83°C). Из-за нерастворимости амида в воде на практике применяют легко растворимый тартрат лизергиновой кислоты (т. пл. 198—200°C). Диэтиламид химически очень устойчив.
Психотропные свойства. Диэтиламид лизергиновой кислоты даже в чрезвычайно малых количествах вызывает у людей психозы. Пороговая доза для здорового человека составляет3 от 0,260 до 0,295 мкг!к.г массы; минимальная доза для людей — 10—30 мкг!кг., меньшие дозы не вызывают психозов.
При дозах между 20 и 125 мкг! кг первые признаки отравления человека появляются через 15—60 мин.
359
Перед началом психоза отравленный чувствует легкую тошноту, зрачки расширяются. Психоз, вызываемый LSD, выражается в беспокойстве, расстройстве зрения, ослаблении внимания, беспричинном смехе, затруднениях речи, зрительные восприятия кажутся искаженными — предметы и вещи деформируются, искажаются, увеличиваются или уменьшаются в размерах и принимают неестественную окраску. Отравленный теряет ощущение времени и скорости; в то время как ои сидит в быстро мчащемся автомобиле, ему кажется, что ои едва перемещается. Реакции сильно замедляются.
Зрительные галлюцинации проявляются в виде фантастических ярко окрашенных и пестрых образов, они дополняются слуховыми галлюцинациями, которые, в свою очередь, вызывают определенные зрительные иллюзии. Галлюцинации часто воспринимаются мучительно.
Отравленные впадают в состояние страха и страдают манией преследования. Они настроены враждебно и недоверчиво, повы-шейио чувствительны к прикосновению и иногда реагируют иа него импульсивно и злобно. Они некоитактабельны и их поведение сравнимо с поведением шизофреников.
Психоз достигает максимума интенсивности через 2—4 ч после отравления и продолжается 5—12 ч, в зависимости от принятой дозы. В отдельных случаях наблюдаются запоздалые реакции через 24 ч и даже через неделю и больше.
Перенесенный психоз никаких последствий ие оставляет. Человек нормален и может вспомнить большую часть галлюцинаций. В больших дозах LSD может привести к смерти. При смертельных дозах наблюдаются те же симптомы отравления, что и при меньших количествах, но более сильные. ’
Расширение зрачков выражено сильнее, расстройства координации приводят к неуверенности походки, а также хватательных движений и других действий. Конечности и голова кажутся невероятно тяжелыми, начинаются рвота, слюнотечение, параличепо-добиая слабость, увеличение чувствительности кожи, озноб, невнятная и нечленораздельная речь и выкрики. Смерть наступает от остановки дыхания.
Средине летальные дозы (ЛДзо) при внутривенных инъекциях составляют: для мышей 46 мг/кг; для крыс 16,5 мг/кг и для кроликов 0,3 мг/кг.
8.1.1.2. Другие производные лизергиновой кислоты. Этиламид лизергиновой кислоты (LAE) по своему психотропному действию примерно в 10 раз слабее, чем LSD. Галлюцинации выражены не столь сильно.
Гидразид лизергиновой кислоты (LSH). Производство этого соединения было подготовлено в Германии концерном «ИГ Фарбен Иидустри» в 1944 г. Это вещество считается пригодным для применения в качестве химического оружия.
360
: Морфолид лизергиновой кислоты (LSM). Эффективность этого Ш^г-вещества составляет около 1/3 от эффективности LSD, Вызываемое Ш нм действие аналогично действию LSD.
8.1.2. Производные триптамина
\ Различные производные триптамина упоминаются в специальной литературе в качестве возможных психоядов. Некоторые из этих 1 соединений относительно просто получить синтезом. Эти вещества । были открыты и исследованы с военной точки зрения в конце пяти-; . де ся ты х год о в.
Ж Триптамин V — амин белкового происхождения, который обра-Зуется при аминокислотном обмене белка в результате декарбокси-Ж лнрования триптофана IV:
СН2СНСООН ----—
„ nh2 NH
IV
ch2ch2nh2
NH v
.4"
8.1.2.1. ^№-Диметилтриптамин
CH,CH2N(CH3)e
Ж NH
Синонимы: 3- (Р-диметиламиноэтил) -индол, 3-ин долил-0-диметила мино-этан, DMT.
Ж N.N-Диметилтриптамии можно получить из индола. Это соеди-иение было выделено из южноамериканского наркотического расте-Ж ния Piptadenia peregrina. Индейцы Ориноко приготовляли из ли-стьев и семян-этого растения нюхательный табак, который употреб-ляли перед битвой, чтобы сделать себя нечувствительными
.J к боли.
Он представляет собой кристаллическое вещество, которое в до-зах порядка 70 мг вызывает у людей галлюцинации, беспечное ^ настроение, сопровождающееся ощущением изменения личности. .Ж Картина психоза сходна с психозом от LSD. Эффективная одно-разовая доза составляет 1 мг{кг массы. v
8.1.2.2. Буфотенин
Il но
СН2СН2М(СНз)2
Ж! ’NH
Синонимы: 5-окси-З- (0-диметил аминоэтил) -индол, 5-оксиипдолил-р-диме-тиламнноэтан, 5-окси-М,М-диметилтриптамин,
Буфотенин является оксипроизводным М,М-диметйлтриптамина.
IL Наряду с другими соединениями его выделяют из секретов жаб и из различных растений,
Его можно синтезировать из 5-метоксииндола путем получения цианпронзводного, восстановления, метилирования и затем омыления (в бензоле):
CH2CH2N(CH3)£
NH
Буфотенин — кристаллическое вещество с т. пл. 146 °C. Из-за наличия фенольной гидроксильной группы он проявляет слабо кислые свойства. Атом азота аминогруппы можно превратить в четвертичный. Пикрат и оксалат буфотенина имеют т. пл. 178 н 94 °C соответственно. Подобно физостигмину, буфотенин подавляет холинэстеразу и поэтому оказывает парализующее действие на двигательные центры нервной системы (миоз, слюнотечение). Его психотропное действие примерно соответствует действию LSD.
8,1.2.3. Псилоции и псилоцибин
он
l^jl—JpCH2CHaN(CH3)2
псилоцин
псилоцибин
Псилоцин (4-окси-М,К-диметнлтриптамин) изомерен буфотенину. Наряду с псилоцибином (фосфат псилоцина), он был выделен Гофманом н Геймом4 из мексиканского ритуального грнба тео-нанкате (Psylocybe mexicana Heim). Этот грнб ацтекн употребляли в качестве наркотика прн различных культовых обрядах. В ходе выполнения американской программы военно-химических исследований эти соединения были подробно изучены и их действие испытано иа добровольцах. О масштабах эксперимента свидетельствует то, что в нем приняло участие 400 человек5.
362
Оба соединения можно легко синтезировать. Исходным веществом для синтеза является 4-бензнлоксннндол, нз которого окс-алнлхлорндным методом получают 4-окси-№,М-днметнлтрнптамнн (псилоцин). Последний этернфицнруют дибензнлхлорфосфинатом и после дебензилирования получают пснлоцнбнн:
о кс а лил хлорид ный метод
фосфо рн-ли рова иве
О
О—Р(СН2С6Н5)2
CH2CH2N(CH3)2
NH
CH2NH(CH3)2
сн2
NH
окислительное дебензилирование
у;
$
Пснлоцнн н пснлоцнбнн представляют собой твердые вещества, разлагающиеся прн нагревании вблизи температуры плавления. Пснлоцнбнн плавится прн 173—176 °C.
Психотропные свойства. Несмотря на то, что псилоцибин является продуктом фосфорилирования, он не блокирует холинэстеразу, как другие сложные эфиры фосфорной кислоты. В какой мере это верно Для алкилированных продуктов фосфорилирования, ска-
зать пока нельзя.
В организме пснлоцнбнн превращается в пснлоцнн; таким образом, никакой качественной нли количественной разницы в характере их действия нет. Опыты на животных показали, что прн внутрибрюшинных инъекциях псилоцибина мышам дефосфорнлнрова-нне наступало через 20 мин, н содержание псилоцина в головном мозге н других органах повышалось6.
. Оба вещества эффективны прн инъекциях н прн пероральном приеме. Как пороговую дозу прн приеме через рот указывают ~60 мкг}к.г массы. Обычно предельные дозы для нормальных здоровых людей составляют 4—6 мг. Эти величины более чем в 100 раз превышают соответствующие дозы LSD.
Прн пероральном приеме психоз начинается через 20—30 мин. Сначала наступает ощущение слабости и головокружение, дрожь и подергивания, страх, тошнота, онемение губ, полости рта н языка
363
и расстройства речи. Позже, примерно через 1 ч, наблюдаются зрительные эффекты: яркие краски, сны — сначала спокойные, переходящие затем в кошмары, галлюцинации, теряется способность к сосредоточению н мышлению, окружающий мнр зрительно и на слух воспринимается лишь расплывчато, теряется ощущение времейи, расстояния и пространства. Через 2 ч эти явления становятся сильнее и продолжительнее. Психоз достигает кульминации. Дальнейшие симптомы отравления псилоцибином заключаются в нарушениях координации, усилении сухожильных рефлексов и расширении зрачков.
Начало н продолжительность психоза зависят от дозы и способа введения яда. Первые симптомы прн подкожных инъекциях от 40 до 200 мкг/кг массы проявляются через ‘5 мин, прн пероральном приеме от 60 до 210 мкг]кг — через период от 30 мин до 2 ч. Прн таких дозах психоз длится примерно 3—5 ч.
После перенесенного психоза пострадавшие обычно жалуются на головные боли и утомление, реакции еще замедлены. Через некоторое время проходят н эти явления.
8.2. ФЕН АЛ КИЛАМИ НЫ
Соединения типа феналкнламинов играют очень важную роль в определенных процессах обмена, веществ в организме. Два важнейших пснхохнмическнх вещества этого ряда, мескалин н 3,4,5-три-метокснфенэтиламин, родственны феиэтнламину VI, который можно считать родоначальником этой группы.
Фенэтнламни — основной структурный фрагмент таких соединении, как адреналин VII н норадреналин VIII, эфедрин IX, первитин X и др., которые оказывают влияние на обмен азота в организме, а также воздействуют иа нервную системы.
периферическую и центральную
НО-
СН2СН2№
VI
ОН
chch2nh2
VIII
364
Средн этих очень ценных в фармакологическом отношении веществ, обладающих несколько отличными от адреналина свойствами, мескалин и 3,4,5-трнметокснфеннзопропнламин занимают особое положение как по своему, строению, так и по активности в качестве пснхояда. Подобно LSD,, они родственны эндогенным аминам.
Эти яды влияют через мозг на процессы окисления молочной, глутаминовой, пировиноградной кислот и глюкозы. За психозы, вызываемые мескалином н, по всей вероятности, за шизофрению вообще ответственными являются также изменения в процессе обмена веществ в печени. Мескалин связывается специфичными аминокислотами белка печени. По изменениям в распаде аминокислот делают вывод о характерном психическом действии мескалина.
То, что изменения в процессе обмена'веществ, в печени вызываются фармакологическими воздействиями, было неоднократно показано на примере психоактивных веществ. За психические воздействия ответственным является не только одни этот эффект, речь идет скорее о сложных н взаимообусловленных явлениях, исследование которых представляет большие трудности ввиду сложности таких биохимических процессов.
Мескалин (3,4,5-трнметокснфенэтнламнн) был выделен нз мексиканской разновидности кактусов семейства Anhalonium, которую в народе называли «пейотл». Из этого растения местные жители приготовляли наркотики.
В 1919 г. Шпету удалось синтезировать мескалин конденсацией альдегида трнметнлгалловой кислоты с нитрометаном с последующим восстановлением образовавшегося трнметоксн-о-ннтростнрола:
Общие свойства. В чистом виде мескалин — кристаллическое вещество с т. пл. 35—36 °C. Обычно его получают лишь в виде
365
бесцветной маслянистой жидкости, которая растворяется в воде, этаноле н галогеналканах, но нерастворима в эфире и петролейном эфире.
•Мескалин кнпнт прн 180°C, дает щелочную реакцию и поэтому образует с кислотами растворимые в воде бесцветные кристаллические соли (сульфат, гидрохлорид); с углекислотой воздуха образуется карбонат.
Психотропные свойства. Одноразовые дозы в 50—200 мг вызывают у людей психозы через 1—2 ч. Как правило, онн начинаются с патологической болтливости и повышенного двигательного возбуждения. Затем наступают головные болн, головокружение н легкое недомогание. На некоторое время расширяются зрачки и выступает пот. Пораженный злится, иногда пугается. На следующей стадии появляются зрительные, реже слуховые галлюцинации прн полном сознании, обычно приятные и очень красочные видения. Ощущение времени и пространства теряется. Отравленный проявляет чрезмерную веселость и уже не ощущает себя.
Характер и степень иллюзий зависят от принятой дозы мескалина. Психоз длится от 6 до 24 ч. Как последствия — жалобы на головные болн и боли в мышцах, а также на тошноту.
Эффективная доза мескалина близка по величине к смертельной, поэтому всегда существует опасность превышения дозы, что может привести к смерти от царалнча дыхания.
Средняя летальная доза (ЛДйо)_Для крыс (внутривенно) составляет 157 мг/кг, прн подкожном введении — 534 мг/кг.
8.2.2. 3,4,5-Триметоисифенизопропиламин (ТМА)
С Н з С Н 2 С Н N Н 2
сн3.
СН3
•СН3
Родственный мескалину ТМА вызывает галлюцинации прн приеме меньших доз, чем мескалин. Психоз очень близок к «мескалиновому опьянению». Характерным для зрительных представлений и видений является преобладание голубого цвета. Психоз начинается через 45—60 мин после приема и длится около 6—7 ч.
8.3. ПРОИЗВОДНЫЕ ПИПЕРИДИНА
Пнпернднн XI н различные его производные, например алкалоиды кониин ХП, конгидрин н седрнднн, встречаются во многих растениях, из которых онн были выделены.
NH
NH СНеСН2СНз
366
В последние годы пнпернднн как психотропное вещество нашел терапевтическое применение для лечения особых форм заболевания шизофренией.
Были синтезированы и исследованы различные психоактивные соединения, содержащие пиперидиновое кольцо в качестве основного элемента структуры.
< 8.3.1. Сериил
о.
£ /—х
' ^СбНа
Синоним: гидрохлорид или гидробромид 1-(1'-фенилциклогексил)-пипери-дина.
< Серннл — бесцветное, хорошо растворимое в воде вещество. За * последнее время он нашел клиническое применение как анестезирующее средство. Он» действует успокаивающе при болях кожи, f. мышц, костей и суставов. Из-за высокой пснхоактнвностн сернила его клиническое применение, пожалуй, ограничено.
Продолжительность психоза очень различна, она составляет от нескольких часов до двух дней. У разных подопытных действие сернила проявлялось очень по-разному.
Кроме обезболивающего действия серннл вызывает сильное воз-V бужденне либо утомление, депрессию, чувство одиночества и нзо-ляцнн, враждебность к окружающему миру и негативизм. После инъекций сернила кошкам в дозах 2 ма/ка. как и прн шизофрении, наступает каталепсия, которая может продолжаться до 3 суток. Она выражается в так называемом «восковом оцепенении». Тело сохраняет приданные ему положения, даже неудобные позы. Пораженный является, так сказать, пластичным и гибким, он ведет себя, как статуя.
Гу;
8.3.2. Пипериднлбеизилаты и пиперидилгликоляты
Пнпернднлбензнлаты и пиперидилгликоляты можно представить у общей формулой:
NR
Если R = СНз, С2Н5; I?' — I?" = CfiHs, то такие вещества псн-- хоактивны. Такими же свойствами обладают производные, у ко- торых R" _= С6Нц, С5Н9.
367
Наряду с LSD, эти вещества возможно будут иметь значение как пснхохимнческие ОВ. Их можно сравнительно легко синтезировать. Эффективные дозы этих веществ малы. Прн замещении фенильных групп цнклогексильнымн нлн цнклопентнльными радикалами психоактнвность увеличивается.
8.3.2.1. N-Алкил-З-пиперидилбеизилаты
/О—С—C(CgHs)2
J О ОН
NR
где R = СН3, С2Н6
Как N-метнл-, так и N-этил-З-пнперндилбензнлаты хранят в виде гидрохлоридов. Оба соединения Получают конденсацией метилового эфира бензиловой кислоты с соответствующими N-алкил-3-окснпнперндннами при действии метилата натрия в кипящем гептане: .
J + СН3О—С—С(СеН5)2
NR О ОН .
CH3ONa
/ О—С—С (С 6Н 5) 2
0 1 он +сн3он
nr
Пипериднлбензнлаты действуют подобно серннлу. Пороговая доза для людей перорально составляет примерно 5 мг на 70 кг массы.
Психозы, продолжающиеся несколько часов (5—6 ч), выражаются в слуховых и зрительных галлюцинациях и в состоянии страха. Прием повышенных доз приводит к полной потере личности (деперсонализации). Отравленный уже не воспринимает окружающее, не реагирует на него и целиком находится во власти психоза. * .
8.3.2.2. Дитраи. Препарат представляет собой смесь гидрохло-. рндов N-этнл-З-пиперндилфенилциклопентнлглнколята и М-этнл-2-пнрролнднлметнлфеинлциклопеитилглнколята:
4 Наиболее эффективным соединением в ряду пнпернднлглнколя-тов является N-этнл-З-пнпериднлфенилцнклопентнжликолят. Это соединение и его аналоги были получены и исследованы в лаборатории Лайксайд.
363
Психозы, вызванные днтраном, продолжаются примерно 20— 24 ч: Они почти не отличаются от истинных психических заболеваний.
Пороговая доза для людей (перорально) составляет 5—15 мг' на 70 кг массы. Отравление сначала проявляется в расширении зрачков, мышечной слабости, сухости рта н учащенном пульсе.
Психические расстройства наступают примерно через I ч. Интенсивным галлюцинациям предшествуют нарушения речи, утрата способности к сосредоточению, снижение и потеря чувства ориентировки. Сильные галлюцинации (зрительные, слуховые, осязательные) полностью отключают отравленного от его окружения и создают почву для появления у него бредовых идей и представлений. Поза и поведение отравленного определяются этими галлюцинациями, например поднятие несуществующих предметов, танец с несуществующим партнером без музыки. С другой стороны,' у некоторых пораженных наступает полная потеря реакций, они не отвечают на раздражения, хотя ц воспринимают их, появляется скованность, которая заходит столь далеко, что человек отказывается от приема пищи и т. Д. Психозы от Днтрана более интенсивны, чем от мескалина илн LSD.
8.4. БОЕВОЕ ПРИМЕНЕНИЕ И ЗАЩИТА
^Психояды могут быть использованы как яды для диверсий, а также : в бою. Существенным недостатком для их боевого применения являются их физико-химические свойства, отсутствие или недостаточное действие через кожу, а также отчасти дороговизна промышленного производства по сравнению с другими ОВ.
С тактической точки зрения они относятся к короткодейстВую-гщим ОВ. Возможно их применение в виде аэрозолей с помощью >бомб, мин, зараженных осколочных снарядов и прежде всего спе-циальных аэрозольных генераторов. Хотя многие из рассмотренных соединений легко разлагаются при нагревании, все же нужно принимать в расчет их возможное применение в дымовых смесях.
Псйхохимическне ОВ могут быть использованы для поражения разных целей и объектов. Прн этом стремятся к тому, чтобы ослабить волю и дух противника, вызвать сопротивление к выполнению ^боевых н трудовых задач и на время привести к полной потере боеспособности и работоспособности.
Особое внимание уделяют применению пснхоядов как ядов для диверсий.
; Своевременное надевание противогаза полностью предохраняет i от воздействия пснхоядов в виде аэрозолей. Трудность распознавания пснхоядов современными полевыми средствами и методами индикации может привести к запоздалому объявлению тревоги. Как закон, в современном бою каждое нападение следует считать химическим нападением н поступать в соответствии
Для своевременного распознавания отравления н принятия мер некоторые западные военные специалисты рекомендуют иметь в подразделениях наблюдателей с постоянно надетыми противогазами, которые должны непрерывно следить за поведением личного состава.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1, Чем отличаются по своему действию и'эффективности фосфорорганические ОВ от психоядов?
2, Что такое галлюциногены?
3. Напишите структурные формулы для трех важнейших групп психоядов.
4. Как объясняют психотропное действие LSD?
5. Опишите психотропное действие LSD.
6. Напишите схему получения псилоцибина.
7. Какой состав имеет сернил?
8. В каких случаях пиперидилбензилаты- и пиперидилгликоляты психоактивны?
9, Какой состав дитрана и Как он действует на психику?
ЛИТЕРАТУРА
L Science (Washington), 136, 151 (1962).
2. Hoffmann Ai, Planta med. (Stuttgart), 9, 354 (1961); Dtsch. med. Wschr, 86 , 885 (1961); C., 1963, 215.
3. Murphreen H. B„ Clin. Pharm. Therap., 3, 314 (1962); C., 1963, 14403.
4. Hoffmann A., Heim R„ Chimia, 14, 309 (1960); Experlmenta 14 397 (1958).
5, Kunz M., Vojenstri, 1962, 392.
5 Horita A., Weber L. J,, Toxicol, appl. Pharmacol., 4, 730 (1962); C., 1964, 16-1440.
9. ФИТОТОКСИЧЕСКИЕ ОВ (ЯДЫ РАСТЕНИЙ)
Во время второй мировой войны в США были заготовлены1 фито-токсические ОВ. Применение их планировалось иа конец войны, особенно для нападения иа Японию.
Среди тысяч соединений, полученных и прошедших испытания в биологическом исследовательском центре — американском армейском лагере Детрик и отобранных в качестве регуляторов роста растений, был найден так называемый препарат 2,4-D- (2,4-дихлор-феноксиуксусная кислота) I, соединение, пригодное для военных целей.
СНгСООН
NH
II
Его производство в США в 1945 г. составило уже 450 т в год. Летом 1945 г. находились в пути первые суда с препаратом, предназначенным для уничтожения японских рисовых полей. Одиако до военного применения тогда дело ие дошло.
Представления о том, что для своего развития растение иуж-дается наряду с питательными веществами в соответствующих гор-монах (фитогормоиах) и само их вырабатывает, восходят к трудо-Д? емким исследованиям ученых Бойзен-Йенсена (1907 г.), Н. Г. Хо-лодного и Вента (1924 г.), Кегля (1934 г.) и С. С. Наметкина \ (1940 г.) —специалистов по физиологии растений. Фитогормоны
регулируют процесс обмена веществ в растении и тем самым — его рост.
V После того, как стало известно, что к числу таких фитогор-монов относится гетероауксин (3-индолилуксусиая кислота) П — синтетически легко доступное соединение, появились надежные ос-(„ нования для проведения дальнейших исследований в этой области, л которые были направлены как во благо, так и во вред челове-
честву.
Применяемые в малых количествах синтетические стимуляторы
>.? растений играют большую и вполне заслуженную роль в сельском у;; хозяйстве и лесоводстве для повышения урожайности, борьбы с сорняками, для удаления листвы с растительных культур
371
(в результате чего возможно применение уборочных машин), выращивания плодов без семян, уничтожения порослей и т. д.
Уже во время второй мировой войны Химическая служба армии США провела успешные опыты по уничтожению посевов, урожая и по обеспложиванию цветущих растительных культур. При этом применялись те же вещества, что и в сельском хозяйстве н лесоводстве;
Фитогормоны интенсифицируют обмен веществ в растении. Связанное с этим ускорение физиологических процессов является причиной стимулирующего действия. Повышенная добавка таких стимуляторов приводит к ускоренному, чрезмерному обмену веществ в растении, который, в зависимости от примененного количества, приносит растению пользу либо вред.
Передозировки действуют на растения токсично. Обмен веществ претерпевает глубокие нарушения, поскольку в этих условиях растение не может столь быстро избавляться от больших количеств ненужных веществ. Его рост подавляется — клетчатка начинает злокачественно разрастаться, и растение погибает. Таким образом фитогормоны становятся фитоядамн. После выведения вредных веществ из растения оно продолжает нормально развиваться.
Под фитотокснчными ОВ подразумевают химические соединения, которые, будучи применёнными в определенных количествах, могут принести вред или вызвать гибель растения. Вред означает обеспложивание, осыпание листвы, аномальное развитие и т. д.
Наряду с неорганическими химикатами — цианамидом кальция и цианатами, которые могут применяться в качестве фитотоксиче-ского химического оружия, особого внимания заслуживают органические соединения типа фенил карбоновый или феноксикарбоновых кислот, гнбберелннов, триазииов и др. ' - ’
Впервые фнтояды были применены в больших количествах в 1952—1954 гг. в Малайе, а затем в 1961 —1966 гг. — во Вьетнаме. Во Вьетнаме американские войска сначала применяли фнтояды с целью уничтожения джунгле§ вдоль стратегически важных дорог, объектов, каналов и т. д,, чтобы затруднить силам освобождения Народного Вьетнама контроль за этими важными пунктами. Затем стали уничтожать возделанные поля, плантации и даже заражали ценныё пастбища. Десятки тысяч га риса и других сельскохозяйственных культур были уничтожены в последние годы, что сказалось на продовольственном снабжении Южного Вьетнама. Сотни людей заболели, многие умерли в результате отравлений, люди слепли нлн покрывались ранами.
Фитотокснчные ОВ могут применяться в следующих целях:
а) в качестве так называемых дефолиантов, чтобы вызвать опадение листвы в лесах с целью затруднения противнику естественной маскировки;
б) в качестве так называемой потравы для посевов для уничтожения н заражения полезных растений с целью подрыва продовольственной базы противника.
372
Из множества возможных химикатов с фнтотокенческнми свойствами было отобрано несколько важных представителей.
По фитотоксическому действию их подразделяют на следующие группы: сжигающие (едкие) гербициды, которые вызывают на поверхности листьев химические ожоги, например цианамид кальция; гербициды роста, которые воздействуют как на рост растения, так и на эмбриональное или плазменное развитие клеток, например, препарат 2,4-D.
Для-оценки фитотоксичности соединения не существует критериев сравнения, как в токсикологии; точная характеристика и оценка невозможны. Имеются чисто эмпирические наблюдения, которые пока не получили количественного выражения.
Если не рассматривать неорганические химикаты, действующие сжигающе, и некоторые другие исключения, то необходимыми условиями фитотоксического или гербицидного действия являются, по-видимому, следующие:
Г) наличие в молекуле гербицида циклического ядра с двойной связью;.
2) в боковой цепи ядра должен быть хотя бы один атом углерода и одна СООН-группа либо другая функциональная группа, легко превращающаяся в карбоксильную группу;
3) для соединений некоторых классов условием фнтотоксиче-ского действия является, по-видимому, определенное пространственное расположение ядра и карбоксильной группы, что можно наблюдать на примере некоторых изомерных производных фенилкарбоновых кислот. Так, цмс-коричная кислота (III) эффективна, а гранс-коричная кислота (IV) — нет.
Нх >н к zcooh
/С=С\ /С=С\
C6HZ ^СООН С6Н5/ . >н
III IV
9.1. ПРОИЗВОДНЫЕ ФЕНОКСИКАРБОНОВЫХ КИСЛОТ
RO(CH2)„COOH
Г где R — ароматическое ядро с различными заместителями.
Производные ф,еноксикарбоновых кислот по своему строению удовлетворяют условиям, предъявляемым к фитотоксичным препаратам. Наиболее известными в настоящее время и чаще всего применяемыми представителями этой группы являются производные феиоксиуксусной кислоты, из которых во Вьетнаме применялась 2,4-дихлорфеноксиуксусная кислота Ч
Соединения этой группы усваиваются всеми частями растения— листвой, стеблями и корнями. Оии во много сот раз эффективнее, чем природные фитогормоны типа гетероауксина (стр. 371).
Действие этих соединений заключается в стимулировании непропорционального развития, особенно за счет роста клеток в едином направлении. Онн действуют не только на молодые растения
373
или на семена, но и на вполне развившиеся растения, как однолетние, так и многолетние. Они пригодны для уничтожения растительных культур, обеспложивания растений н уничтожения лесных массивов.
9.1.1. 2,4-Дйхлорфеиоксиуксусиая кислота
С1
осн2соон
С1
Военные обозначения: 2,4-D, U-46.
Во время второй мировой войны в форте Детрик и в нью-йоркском институте Бойс-Томсона соединение 2,4-D было оценено как наиболее эффективное вещество этого типа.
Получают 2,4-D из 2,4-дихлорфенолята натрия и натриевой соли хлоруксусной кислоты с последующей обработкой полученного продукта соляной кислотой:
Приводим следующую методику получения 2,4-дихлорфеноксиуксусной кислоты г. Сплавляют 70 г технической хлоруксусной кислоты с 80 г 2,4-дихлорфенола и к плаву, охлажденному до ~20’С, прибавляют 156 г 40%-ного водного раствора гидроокиси йатрия, поддерживая температуру 40—60 °C. Реакционную смесь затем перемешивают 2 ч при 60—65 °C и нагревают еще 3—4 ч при 100 °C, после чего прибавляют ~200 мл горячей воды и нагревают смесь еще I ч. Натриевую соль 2,4-дихлорфеноксиуксусной кислоты отсасывают и промывают разбавленным раствором едкого натра и небольшим количеством воды.
2,4- Дихлорфеноксиуксусная кислота представляет собой бесцветные кристаллы, т. пл. 139—140°C (из 35%-ной уксусной кислоты). Растворимость в воде невелика.
Применяют свободную 2,4-дихлоруксусиую кислоту, либо ее натриевую соль.
9.1.2. 2,4,5-Трихлорфеиоксиуксусиая кислота и ее производные
Cl^^^^OCH.COOH
Военные обозначения: 2,4,5-Т, TCP, Weedone.
Препарат 2,4,5-Т и его производные в фитотоксическом отношении эффективнее, чем 2,4-D и более пригодны дли уничтожения
374
древесных растений. Его применяют в смесн с химикатами сжигающего действия, в результате чего удешевляется применение препарата. Такая смесь получила в США название «смерть кустам» («bush-killer»).
Кислота 2,4,5-Т представляет собой бесцветные кристаллы, т. пл. 153 ЧС, плохо растворима в воде.
Применяют свободную кислоту либо ее натриевую соль, обычно в смесн с более дешевыми гербицидами.
Из других производных фенокснкарбоновых кислот применяют: 2-метнл-4-хлорфенокснуксусную кислоту (МСРА) V, (2,4-днхлор-феноксн).-масляную кислоту (2,4-DB) VI, хлорбутеннловый эфир 2,4-днхлорфенокснуксусной кислоты (кротилнн) VII н некоторые сильно фнтотокснчные фторпронзводные, например 2,4-днхлор-6-(илн 5)фторфенокснуксусную кислоту (VIII н IX):
VIII . IX
Производные феноксикарбоновых кислот можно применять как опылением, так н опрыскиванием. Вследствие растворимости в воде _ солей щелочных металлов этих кислот, онн пригодны для прнме-* нення опрыскиванием. Однако чаще всего применяют свободные : кислоты либо в смесн с фракциями дизельного топлива либо в вод-иых эмульсиях с добавкой соответствующих смачивающих агентов V и эмульгаторов (продукты конденсации жирных кислот, сульфопро-‘изводные нафталина н др.).
Лучше применять алкиловые (метиловый, изопропиловый, нзо-- бутиловый) нли алкениловые эфиры (например кротнлнн VII). Эти ' вещества предпочтительно применять опрыскиванием, так как прн этом растения легче нх усваивают н онн быстрее в него проникают. < - Такне препараты более устойчивы н к метеорологическим воздей-ствням.
В последнее время налажено промышленное производство раз-( личных амннопронзводных фенокснкарбоновых кислот, например,
375
диметил- н диэтиламинопроизводиых кислот 2,4-D н 2,4-Т. Они обладают тем преимуществом, что будучи жидкостями лучше растворимы, и поэтому более удобны в применении.
В зависимости от примененных количеств, фнтояды могут сохранять свою эффективность в почве несколько недель и месяцев, что затрудняет или делает невозможным произрастание специфических видов растений. В проведенных в крупном масштабе экспериментах по прореживанию лесных массивов использовали3 натриевую соль 2,4-D в количестве до 6 кг/га, аммониевую соль 2,4,5-Т в количестве до 3,5 кг/га и изобутиловый эфир 2,4-D—-до 4,2 кг!га.
Количество препарата, необходимое для заражения лесных массивов, зависит как от способа применения (опрыскивание, опыление), так и От породы деревьев и их возраста.
Свекла, картофель, виноградники, клевер и плодовые деревья очень восприимчивы к этим неществам и даже небольшие концентрации препаратов причиняют им вред или уничтожают их.
Как показали опыты на животных4, при внутрибрюшинных инъекциях 2,4-D крысам наступают тяжелые поражения центральной нервной системы.
9.2. ДРУГИЕ КАРБОЦИКЛИЧЕСКИЕ ФИТОЯДЫ
Кроме производных феноксикарбоновых кислот было обнаружено много других фитотоксичных веществ, которые по специфичности своего действия и по эффективности во много раз отличаются друг от друга. Мы приводим здесь произвольно выбранные некоторые примеры из всей совокупности исследованных веществ.
9.2.1. Феиилалкиловые эфиры
К началу пятидесятых годов в американских военных лабораториях были получены и испытаны некоторые феиилалкиловые эфиры. Опыты проводились с хлор- и фторфенилалкнловымн эфирами, из которых особенно предпочтительными оказались эфиры с такими алкильными группами, как w-октил, н-децил, ундецил, тридецил и гексадецил.
Эти соединения получают взаимодействием соответствующих производных фенола с соответствующими алкнлгалогенндамн (хлоридами или бромидами) в этанольных растворах в присутствии иодида щелочного металла. Приводим реакцию получения 2,4,5-трихлорфенилгексадецнлового эфира:
С 1 в Н33
Y Y 4- НВг
376
Некоторые из этих эфиров являются маслянистыми жидкостями, легко растворимыми в жирах и быстро проникающими в растение, Их применяют опрыскиванием для замедления роста растений в количествах от 0,1 до 0,5 кг/га н более.
9.2.2. Производные фенилмочевины
Наиболее известными соединениями этого типа являются Ы-(3,4-дн-хлорфеннл) -№-н-пропнл-ЬГ-метнлмочевина X и N- (4-хлорфеннл) -N',N -днметилмочевнна XI:
XN(CH3)2
XI
В малых количествах оба соединения способствуют росту, но в повышенных количествах (10 кг(га) приводят к сильным поражениям, особенно в случае овощных культур н кормовых трав. Эти соединения могут применяться для уничтожения пастбищных угодий н овощных полей.
Для тех же целей пригодны н некоторые уретаны, например, изопропиловый эфир Ы-(З-хлорфеннл) -карбаминовой кислоты (CIPC) (XII) и не содержащий хлора аналог IPC (XIII). Онн за-- трудияют прорастание семян зерновых культур. Применяют нх по-< лнвкой в количестве около 2 ка/ап.
\ЭСН(СН3)2
XIII
Другие карбоциклические соединения с фнтотоксическнмн свойствами находят в ряду производных феннлкарбоновых, как насы-* щенных, так н ненасыщенных кислот, например цмс-коричной кнс-лоты. Даже низший член этого ряда — феннлуксусная кислота, СбН5СН2СООН (XIV), проявляет свойства фнтогормона,
9.2.3. Производные фенола
' Некоторые производные фенола, допускающие военное применение в качестве фнтотокснчных ОВ, давно применяются в качестве гербицидов, Онн являются важными исходными продуктами для хн-, мнческой промышленности, н нх получают в больших количествах.
377
2,4-Днннтрофенол (DNP) XV и 4,6-динитро-2-метилфенол (4,6-динитро-о-крезол, DNC, DNOC, DOK) XVI применялись во Вьетнаме.
Они являются типичными сжигающими препаратами, которые разъедают части растения. Этн соединения оказывают токсическое действие на человека и животных как при вдыхании, так и через кожу. Причина их токсичности для теплокровных заключается в сильной интенсификации процессов окисления, что ведет к увеличению потребления кислорода и к снижению запасов углеводов.
Симптомы отравления проявляются в головных болях, потливости, потере аппетита, повышении температуры тела до 40 °C, увеличении частоты дыхания, удушьи и отёке легких. Летальный исход отравлений наступает обычно в результате отёка мозга или теплового удара.
При их применении в мирных целях рекомендуется очень строгое соблюдение техники безопасности — защита тела и органов дыхания, смена одежды и тщательная гигиена.
2,4-Динитрофенол представляет собой желтые игольчатые кристаллы, т. пл, 113—114 °C; 4,6-динитро-2-метилфенол— желтые призматические кристаллы, т. пл. 85,8 °C.
Оба соединения мало растворимы в воде; используют их натриевые или аммониевые соли, растворимые в воде. Соединения применяют распылением и опрыскиванием. Кроме этих соединений, американцы часто употребляют 2,4-динитро-6-бутилфенол (DNBP) XVII:
ОН
no2
XVil
Этн динитрофенолы пригодны для уничтожения широколиственных растительных культур, пастбищных угодий н для заражения лесных массивов. Кроме того, их применяют в качестве инсектицидов и средств защиты древесины.
378
9.2.4. Гибберелииы
По своей природе гибберелииы являются своеобразными веществами роста, которые, в противоположность другим фитогормонам, в сравнительно малых количествах стимулируют рост. Они увеличивают интенсивность физиологических процессов в растении.
Гнбберелин был выделен японскими фитофизиологамн из гриба Gibberela fuikowi. Рисовые посевы, зараженные этим грибом, погибают от так называемого рисового бешенства (Вакапае).
К настоящему времени выделены и отчасти синтезированы восемь разновидностей гибберелниа. Основным фрагментом структуры гибберелинов является гибберелиновая кислота, которой приписывают следующее строение:
Как и все фитогормоиы, гибберелииы в больших количествах приносят вред растениям. Пораженные растения растут неестественно длинными, плоды у них не развиваются и впоследствии погибают, как и само растение.
В некоторых странах организовано промышленное производство гибберелинов для покрытия нужд сельского хозяйства. Из-за их сильного фитотоксического действия гибберелииы нужно считать возможными фитотоксичными ОВ. Для человека и животных гибберелииы безвредны.
9.3, ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ С ФИТОТОКСИЧЕСКИ МИ СВОЙСТВАМИ
9.3.1. Иидолил-З-карбоиовые кислоты
Как упоминалось, развитие области фитогормонов началось С открытия гетероауксина — индолил-3-уксусной кислоты (стр. 371).
Гетероауксин обладает меньшей фитотоксичностью, чем, например препарат 2,4-D и ему подобные. Высшие члены ряда более -.эффективны. Их получают взаимодействием, индола с эквнмоль-нымн количествами соответствующих лактонов при многочасовом нагревании. Например, нз индола и бутиролактана в присутствии 'основания получается индолнл-3-масляная кислота:
Индолнльные производные вряд ли могут иметь военное значение.
379
9.3.2. Производные 1,3,5-триазииа
Различные производные трназнна известны как гербициды. Из-за своего сильного фитотоксического действия при применении в больших количествах, чем это необходимо для подавления сорняков, они приводят к уменьшению числа плодовых ^завязей, обеспложи- -вают посевные площади, а также действуют как дефолианты.
Известный под названием симазин 2-хлор-4,6-бис-(этнленами-но)-1,3,5-триазнн получают нз хлораигндрида циануровой кислоты и этиламнна:
С1
С 4N
Cl—Сх ^С— С!
N
+2C2H6NH2
-
- 2НС1
С1
I
С Nx 4N 11 I
с2н5мн—С\ ^с—мнс2нб N
Другими фитотокснчными веществами из ряда производных 1,3,5-триазина являются полученные Хюмером5 перхлорметнлмер' каптоамниотрназины, например 2-(ЬТтрихлорметнлмеркапто-М-ме' тиламино)-4-Ь1-алкнламино-6-хлор-1,3,5-триазнн следующей общей формулы:
С1
л I
С
н\ II I ,
чкт г* кт ли
В настоящее время уже известны соединения этого типа, у которых R=CH3, С2Н5, пзо-С3Н7, н-С3Н7, н-С4Н9, трет-С4Н9.
Производные трназнна не растворимы или лишь малорастворимы в воде. Онн растворяются в алифатических кетоиах, бензоле, в растительных (1) и минеральных маслах. Их можно применять как опрыскиванием в смеси с минеральными маслами, так и распылением смесей с твердыми носителями.
9.4. ПРОЧИЕ ФИТОТОКСЯЧЕСКИЕ ВЕЩЕСТВА
Подразделения армии США во Вьетнаме применяли наряду с препаратами 2,4-D и 2,4,5-Т большие количества цианамида кальция, -гербицидные свойства которого известны уже давно.
Цианамид кальция, Са — N—C«N, бесцветное вещество; промышленный продукт (серый или черный порошок) применяют для удобрения как гербицид. Примерный состав промышленного продукта следующий: 20% N, 65% Са, 10% С и до 2% дегтя.
Цианамид кальция — ёдкнй гербицид. Части растений разъедаются и отмирают. Он эффективен как для высоких, так и дли низких растений. С деревьев осыпаются листья, цветы и плоды. Ус-380
§Ловиями дефолиантного действия являются определенная относительная влажность н температура воздуха.
й Водой и влагой воздуха цианамид кальция разлагается:
' CaCNa ------> CaCO3+2NH3
>' Цианамид кальция токсичен для человека и животных. Токсическое Действие вызывает дициаиамид (CNNHaJz, образующийся «иа слизистой оболочке в результате частичного гидролиза/ | При отравлениях кожа, лица сначала становится сине-красной, г Головокружение, головные боли, озноб и приливы жара, удушье и ?общая слабость являются симптомами, которые появляются при < вдыхании пыли цианамида кальция или при употреблении в пищу запыленных растений.
Г Цианамидные отравления почти не смертельны, через 1—2 су-ток оин проходят. Отравляющее действие значительно усиливается
Ч результате употребления алкоголя до, в момент или после отравления.
J На коже цианамид кальция вызывает химические ожоги, кото- грые принимают злокачественный характер, если цианамид попа--дает в открытые раны. Это приводит к общей интоксикации.
Цианамид кальция пригоден как дефолиант для низких порос-’ лей, плодовых деревьев и др. Он может служить средством для за-. ражеиия посевов и.пастбищ.
Как фитотоксичные вещества имеют значение еще хлораты, ар-“сениты натрия н кальция (также применявшиеся во Вьетнаме), ; трихлоруксусная кислота, а-нафтилуксусная кислота, 2,6-дихлор-Тбензонитрил, иодметнлат фенилгидразона пиридин-2-альдегида, аддукты триалкиламинов с трехокисью серы' общей формулы ORjN-SOs с числом атомов углерода в алкильной группе от 1 до 4 |И др- z
£’ 9.5. ВОЕННОЕ ПРИМЕНЕНИЕ ФИТОТОКСИЧрСКИХ ОВ
Из-за нх специфических свойств фитояды могут быть применены 1 для уничтожения урожаев, уничтожения и заражения пастбищ, для истощения плодородия почвы и для удаления листвы, нужного для того, чтобы сделать невозможной или затруднить- естественную маскировку.
t Их применение в больших количествах осуществимо путем распыления илн опрыскивания. Особая степень чистоты продукта не ; требуется, поэтому могут использоваться промышленные продукты ? Как таковые илн в соответствующих смесях, так как фитояды не ^.обладают достаточно широким характером действия. За счет нс-|пользоваиия смесей можно расширить область применения фито-токсичиых ОВ для военных целей.
£ Во Вьетнаме американская авиация сбрасывала их с самолетов с помощью выливных и распыляющих авиационных приборов, подобно тому, как эго принято ,в сельском хозяйстве. Использовались
381
также специальные выливные контейнеры, которые забрасывали на парашютах.
Применение фитоядов с помощью боеприпасов невероятно, так как ОВ оказывается эффективным не сразу, а лишь через несколько дней н немедленного эффекта при боевых действиях ожидать нельзя.
Фитотокснчные вещества могут применяться в глубоком тылу нлн прщпланомерном отходе для осуществления «тактики выжженной земли». Применение фитоядов в глубоком тылу возможно при наличии самолетов, соответствующих целей и вообще тогда, когда можно ожидать достаточно серьезных последствий.
Прн применении фитоядов нужно прибегать к тем же мерам защиты, что и в случае всех других химических ОВ.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие цели преследуют при применении фитоядов?
2. Каковы предпосылки фитотОксической эффективности соединения?
3. Напишите схему получения препарата 2,4-D.
4. Назовите способы применения фитоядов.
5. Какие производные триазина являются фитотоксичными?
6. Какое токсическое действие оказывает цианамид, кальция на человека и на растения? f
ЛИТЕРАТУРА
1. Beltz W., Militarwesen, 8, 1167 (1964).
2. Авт. свид. 118508; Бюлл. изобр., № 6, 11 (1959).
3. Шима некий П. С., Иав. АН БССР, 1962, 35
4. D е s 1 п I., J. S о с, Acta med. Acad. Sci. Hung., 18, 429 (1962).
5. Пат. ФРГ 1138978 KI. 451 (1960); С., 1964, 18-2153.
у?
10. ДИВЕРСИОННЫЕ яды
(ЯДЫ И ТОКСИНЫ, КОТОРЫЕ МОГУТ БЫТЬ ИСПОЛЬЗОВАНЫ ДЛЯ ЗАРАЖЕНИЯ ПРОДОВОЛЬСТВИЯ, воды И ДР.)
Одной нз доктрин Генеральных штабов империалистических ар-Йж^ЯИЙ является подавление глубокого тыла противника во время войны. Прн современных способах ведения войны нужно предви-уЖ деть постоянные н массированные налеты бомбардировщиков, как это было во время второй мировой войны, н постоянные ракетные ^\удары. Поэтому средствам массового уничтожения, будь то хнмн-^ческое, бактериологическое нлн ядерное оружие, придают особое f ; значение. Определенное место в этих замыслах занимает возможное применение так называемых диверсионных ядов.
Идея о том, чтобы во время войны заражать продовольствие, предметы широкого потребления, системы водоснабжения н т. п. какнмн-лнбо веществами — очень стара. Однако за немногими исключениями «прежние» яды почти не давали возможности массовых отравлении.
За последние десятилетия был выделен нз природных продук-Жг»'тов либ° синтезирован целый ряд высокотоксичных органических J н неорганических веществ. Многие нз них считаются пригодными в качестве диверсионных ядов.
Такне яды используют в «тайной войне» агенты разведок, диверсионные группы, диверсионно-десантные и воздушно-десантные v подразделения. Этн вещества даже в малых количествах могут оказаться весьма эффективными.
У Главным направлением в этой области являются широкие нс-SСледования, в первую очередь, природных ядов, проводимые в по-средние годы в военно-хнмнческнх исследовательских центрах нм-периалнстнческнх государств, а также в гражданских институтах. ужУЗ настоящее время прилагают усилия, чтобы синтезировать прн-уЖррдные яды, токсичность которых часто во много раз превышает :®СоКснчность самых ядовитых фосфорорганических соединений. Изу-||Ь.ченне строения ядовитых начал ядов животного н растительного ^происхождения привело к разработке ряда методов синтеза некоторых аналогов подобных соединений.
Ассортимент соединений, рассматриваемых как диверсионные .-’яды, велнк и никоим образом не исчерпывается соединениями, описанными в следующих разделах. Кроме них, для заражения предметов снабжения, продовольствия, одежды, воды, растительных культур на полях могут применяться и фосфорорганические
383
вещества, некоторые азотистые иприты, мышьяк- и свннецоргани-ческне соединения, психояды и многие другие.
К диверсионным ядам предъявляют следующие общие требования: максимальная токсичность в сочетании с достаточной, способностью к резорбции через слизистую оболочку пищеварительного тракта; отсутствие цвета, запаха н вкуса; отличная растворимость в воде н жирах; устойчивость к нагреванию н гидролизу; скрытый период действия; трудность обнаружения в организме и в зара-жеином материале; отравление не должно вызывать в организме каких-либо специфических патологических изменений, позволяющих распознать эти яды; необычный характер токсичности, отсутствие противоядий, либо Слабая их эффективность.
Этим требованиям удовлетворяют не все соединения, описанные ниже. Они почти выполняются для некоторых фтороргаийческих соединений, н благодаря дешевизне производства, эти соединения занимают преимущественное положение средн ядов для отравления воды, продовольствия, а также домашнего скота.
Какие размеры может принять действие ядов — показывают многочисленные примеры отравления больших продовольственных запасов, взятые из истории .или имевшие место в недавнее время.
В 1959 г. в Марокко поступило в продажу оливковое масло с примесью масла для авиационных двигателей, в результате чего отравилось более 10 000 человек; в 1960 г. в Голландии в течение 10 дней заболели почти 100 000 человек в результате употребления в пищу маргарина, к которому был добавлен вредный для здоровья эмульгатор.
Можно еще вспомнить о ставших уже историческими катастрофах — массовых случаях гангрены, вызванной употреблением спорыньи. Случилось так, что мука из ржи, зараженной спорыньей, на 1/3 состояла из спорыньи. Массовые заболевания эпидемического характера в 994 г. во Франции привели к гибели 4.0 000 человек. По историческим данным, до XIX века подобные катастрофы случались около 300 раз.
10.1. ФТОРОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ
К началу второй мировой войны во многих капиталистических странах исследовалась возможность использования в качестве ОВ фторорганическнх соединений. Впервые фторорганнческне соединения были получены свыше 100 лет назад. Систематические исследования в области химии фтора начались около 40 лет назад. После второй мировой войны эта область претерпела большой подъем. Организация крупнотоннажного производства фторорганическнх соединений стала необходимой вследствие использования их для производства пластмасс, хладоагентов, средств борьбы с вредителями сельского хозяйства, топлив, красителей, в текстильной промышленности, металлургии, производстве смазочных мате
384
риалов и других чрезвычайно ценных веществ, которым прииад--лежит большое будущее.
Высокая токсичность некоторых фторогранических соединений послужила поводом к военному исследованию соответствующих соединений с целью их получения и испытания. Одновременно еще большее значение в качестве ОВ (ср. раздел 7) приобрели фтор-. фосфорорганические соединения.
Из фторорганических соединений значение как ОВ могут иметь: метиловый эфир фторуксусной кислоты I (МФА), 2-фторэтанол II и некоторые их гомологи:
FCHt—СООСНз FCH2CH2OH
I и
По мнению военных специалистов капиталистических государств, эти соединения почти идеально отвечают требованиям, предъявляемым к диверсионным ядам. Они устойчивы, органолептически не обнаруживаются, довольно хорошо смешиваются с водой, сильно ядовиты, их действие проявляется только после латентного периода. Кроме того, затруднено их аналитическое н гистологическое определение, а также лечение отравлений. Поэтому исследовалось возможное применение этих соединений для отравления питьевой воды, продовольствия и фуража.
10.1.Г. Химическое строение и биологическая активность
Фтороргаиические соединения значительно различаются по токсичности. По Саундерсу, решающим для токсичности какого-либо соединения является его отношение к гидролизу и к окислению. Любое фторорганическое соединение ядовито, если, в результате биохимических реакций оно может образовать фторуксусиую кислоту <или фторацетат-иои. Это могут быть кислоты, простые и сложные ’эфиры, амиды, галогеиангидриды, спирты, альдегиды и т. д.
Саундерс и сотр. установили, что в ряду производных фторкар-боновых кислот III ядовиты лишь те соединения, у которых п — нечетное число. Соединения, у которых п — четное, менее или совсем :пе ядовиты (ср. табл. 39, стр. 388). Это относится почти ко всем Сфтороргаиическим соединениям приведенного типа.
Р(СН2)ЛСОХ FCH2CH2C(CH3)2COOH
ni IV
Чередование токсичности в пределах гомологического ряда сопоставляют с теорией Кноопа о р-окислении в организме жирных ию-кислот, которые ступенчато расщепляются до уксусной кислоты;
л +Н2О
RCH2CH2COOH -----> RCH=CHCOOH ------->
-Н2 -На
: +Н2о
—> RCOCH2COOH --------> RCOOH 4- СНзСООН
7213 Зак. 448
385
Расщепление фторзамещениых карбоновых кислот происходит аналогично. Если п — четное число, то такие соединения расщепляются только до неядовитой 3-фторпропиоиовой кислоты, а прн нечетном гг образуется ядовитая фторуксусиая кислота. Например:
FCH2CH2CH2COCHaCOOH FCH2CH2CH2COOH + СНзСООН
FCH2CH2CH2COOH—> FCH2COOH 4- СНзСООН
Соединения с разветвленной цепью не превращаются во фторуксусную Кислоту и поэтому неядовиты, например 4-фтор-2,2-диме-тилмасляная кислота IV (стр. 385).
Кроме р-окисления определенную роль играют и Другие механизмы, поскольку ядовитыми оказались некоторые фторорганиче-ские соединения этого ряда, ие удовлетворяющие правилу p-окисления.
В общем, условием токсичности этого типа фторорганических соединений, так называемых фторацетатов, является образование радикала РСНгСО—. Всякое замещение у с^углерода в этом радикале ведет к уменьшению или потере токсичности, в том числе и введение второго или третьего атома фтора.
В настоящее время известны следующие токсичные соединения: фторкарбоновые кислоты и их эфиры; фторированные спирты и альдегиды; амиды и гидразиды фторкарбоновых кислот; фторке-тоиы и простые фторалкиловые эфиры; фторалкиламниы и фтор-алкилнитрилы; фторированные алканы, алкены и алкины.
Фтороргаиические соединения этого типа эффективны при приеме через рот, при вдыхании паров, а некоторые также в результате резорбции через кожу.
Их действие наступает после латентного периода от 0,5 до 6 ч. Симптомы отравления следующие: двигательное возбуждение, тошнота, рвота, слюнотечение, повторяющиеся нерегулярно эпилептические судороги, иногда потеря речи, отделение мочи и кала, полная потеря сознания, расстройства дыхания вследствие сильного выделения жидкости в бронхиальные лимфоузлы, смерть от остановки сердца.
Со смертельным исходом протекает 75% случаев отравления. Смерть в результате остановки кровообращения наступает через 3 ч или через 4—5 суток, в зависимости от концентрации и дозы ОВ. Поскольку токсичность рассматриваемых соединений почти одинакова, то, независимо от типа соединения (кислота, эфир, альдегид и т. д.), смертельной дозой для людей считают от 2 до 10 мг/кг.
10.1.2. о)-Фторкарбоиовые кислоты
F(CH2)„COOH
Фторуксусная кислота и некоторые ее производные были получены Свартом в прошлом веке. Она практически является соединением»
386
йот которого берет начало ряд токсичных производных фторкарбо-^иовых кислот, которые имеют общее, но неточное название фтор-ацет а ты.
v . Известно несколько методов получения фторкарбоновых кислот. -.Их преимущественно получают гидролизом алкиловых эфиров соответствующих фторкарбоновых кислот. Свободную фторуксусную .дкислоту получают омылением ее этилового эфира слабой щелочью:
+Н2о
К' FCH2COOC2H5 ---------> FCH2COOH
-с2н5он
| Фторуксусная кислота — бесцветные игольчатые кристаллы, пл. 31 °C, т. кип. 167 °C. Соединение легко растворимо в воде и спирте. Чистую фторуксусную кислоту получают повторной перегонкой при атмосферном давлении..
Замещение атома водорода уксусной кислоты на фтор оказывает сильное влияние на кислотность; константы диссоциации со-^.ставляют:
t; СН3СООН .................1,76- i0-s
FCH2C00h........... . ? . 1,2-10~3
В качестве диверсионного яда опасен также фторацетат натрия £FCH2COONa, игольчатые кристаллы, т. пл. 76 °C.
С Наряду с другими это соединение служит для истребления ?*крыс. Оно растворимо в воде, в спирте растворяется лишь умеренно.
Калиевая соль этой кислоты была выделена из произрастающего в Западной и Южной Африке растения «гифблаар» (ядовитый лист, Dichapetalum cymosum). По опыту туземцев; нескольких ^листочков растения достаточно, чтобы умертвить мелкое или даже ^крупное животное. В плодах произрастающего там же растения Ш. toxicarium содержатся фторкарбоновые кислоты с длинной -цепью — (о-фторолеиновая н со-фторпальмнтиновая 2.. Семена этого растения, растертые в порошок, применяют как крысиный яд. Во Йфемя колонизации туземцы использовали их для отравления ко-|ж>Дцев, источников н ручьев, а также для изготовления яда для |£трел.
Ц ' Средняя летальная доза фторацетата натрия при приеме через Йют составляет3 для овец 0,7 мг/кг, для лошадей 1,0 мг/кг, для лю-Оей от 2 до 10 мг/кг.
Ж.-' Опыты на овцах показали, что меньшие дозы не вызывают за-|йетных симптомов отравления, но прн повторных приемах те же Орзы оказываются токсичными.
Лошади погибают уже при добавлении нескольких капель фтор-уксусной кислоты на ведро воды. Собаки, съевшие мясо такой ло-|Щади, — умирают.
В- В зависимости от принятой дозы, действие на людей начинается рчерез латентный период до 6 ч.
387
Некоторые свойства со-фтор карбоновых кйслот приведены в табл. 39.
Таблица 39. Свойства <о-фторкарбоновых кислот
В круглых скобках приведена давление в мм рт. ст.
п Т. кип., °C Т. пл., °C - ЛДао (для мышей, внутрибрюшинно), мг/кг
I 167-168,5 (760) 31-32 0,6
3 60-62 (2) — 0,65
5 193 (762) — 1,35
7 145-148 (10) — 0,64
9 <—‘ 49-49,5 1,5
11 59,5—61 1,25
17 —— 69 5,7
2 — 60
4 — >100
6 —. — 40
8 — <—, >100
10 — — 58
10.1.3. Алкиловые эфиры фторуксусиой кислоты
fch2c^
XOR
Из сложных эфиров фторуксусной кислоты был предложен в качестве ОВ метиловый эфир. Ои является эталонным соединением в ряду так называемых фторацетатов, с которым сравнивают токсичность и физиологическое действие других аналогичных соединений.
Наряду с другими методами \ алкиловые эфиры фторуксусиой кислоты получают реакцией соответствующих алкиловых эфиров хлоруксусиой кислоты с фтористым калием:
+ KF Z
C1CH2COOR ---->- FCH2COOR
-КС1
Алкиловые эфиры фторуксусной кислоты — бесцветные жидкости, некоторые без запаха. Свойства ряда алкиловых эфиров фторуксусиой кислоты приведены в табл. 40.
Метиловый эфир фторуксусиой кислоты (метил фтор ацет ат, МФА) растворим в обычных органических растворителях (спирт, петролейиый эфир и др,). Его растворимость в воде составляет 15%, ои смешивается во всех отношениях с ипритом и другими ОВ. Это лишенная запаха жидкость, т. кип. 104 °C, т. застыв, около —32 °C, давление пара при 20 °C составляет 15 мм рт, ст., а соответствующая летучесть — 92 мг/л.
388
Таблица 40. Свойства алкиловых эфиров фторуксусной кислоты
FCH3COOR
Радикал т. кип.. □с Относит, л20 плотность аЛ 4 Показатель преломления „20 п£> ЛК50 (на кроликах), мг/л
сн3 104,5 1,1744 1,3679 0,1
С2Н5 117-118 1,093 1,3767 о,1
Н'СзН? 135-137 '— — 0,1
изо- С3Н7 124 1,033 1,3804 0>1
Согласно 5, давление пара (в мм рт. ст.) этого и некоторых других производных фторкарбоиовых кислот можно рассчитать по формуле
, Л В
1g р=^Д —
а летучесть (мг/л) по формуле:
lgCsT^A'~^.
Постоянные А, А' и В приведены в табл. 41.
Таблица 41. Константы для расчета давлении ияра и летучести различных фтороргаиических соединений по Редеману и сотр.
Соединение 4 А' в
Эфиры фторуксусной кислоты 8,7860 12,0249 2229,4
метиловый
этиловый 8,4203 11,6510 2188,9
2-фторэтиловый 9,713 13,012 2876
изопропиловый 8,750 12,035 2316
Метиловый эфир 4-фтормасляной кислоты 9,0765 12,3611 2471,7
2-Фторэтанол 9,060 12,072 ‘ 2304
2-Фторэтиловый эфир хлоругольной кислоты 9,001 12,309 2435
Метиловый эфир фторуксусной кислоты химически устойчив; в водном растворе он гидролизуется с образованием столь же ядовитой фторуксусиой кислоты:
РСН2СООСНз
+Н2о
~СН3ОН
> FCHiCOOH
13 Зак, 448
389
Гидролиз по связи С—F происходит медленно. При 23 °C в 15%-ном водном растворе от МФА за 60ч отщепляется только 2,5% от общего количества фтора, через 14 суток —около 50%.
Малая скорость гидролиза по связи С—О объясняется очень сильно полярным характером связи С—F, что препятствует разрыву связи С—О.
Добавление щелочей способствует гидролизу. Так, прн действии известковой воды фтор отщепляется уже иа холоду. Превращение иа 50% с отщеплением фторид-иоиов достигается после 20 ч кипячения в 20 %-ном спиртовом растворе едкого кали. Кислоты мало способствуют гидролизу. Ни кислоты, ни щелочи не могут быть использованы в качестве дегазационных средств.
Водные, спиртовые или эфириые растворы аммиака реагируют с метиловым эфиром фторуксусной кислоты очень энергично с образованием фторацетамида:
FCH2COOCH3
+NH3
------> fch2C<
-СНзОН \NH2
Фторацетамид столь же ядовит, как и МФА. Это бесцветные кристаллы, т. пл. 108 °C. Его растворимость в воде составляет около 15%.
Аналогично иа МФА действуют первичные амииьц в результате реакции с которыми образуются соответствующие токсичные N-ал-килацетамиды:
+ch3nh2
FCH^COOCHs --------> FCH2CT
ЧЧНСНз
а-Фтор-Ы-метилацетамид — кристаллическое соединение, т. пл. 64 °C.
Полностью разрушить молекулу сложного эфира, способны тдлько сильные окислители. При действии хромовой смеси в качестве продуктов разложения образуются двуокись углерода и фтористый водород:
’ +60
fch2cooch3 ——-> HF + 3CO2
—2Н2О
Так же действуют перманганат калия и азотная кислота. Разбавленные растворы гипохлорита не реагируют с МФА, соединения с высоким содержанием активного хлора разрушают эфир.
Для кроликов (внутривенно) ЛД50 составляет 0,25 ма/яа. В опытах иа животных отравление начиналось через 30—60 лшн. Животное погибает в течение нескольких часов. Токсичность через кожу для кроликов составляет около 20 ?нг/кг. (
По наблюдениям Саундерса, эфир в разбавлении 1 : 10е отравления не вызывает; для людей опасны концентрации выше 1:105.
390
10.1,4. Алкиловые эфиры ш-фторкарбоновых кислот
р(сн2)„с:
.' Как уже упоминалось, токсичным является любое соединение, которое может при гидролизе или окислении дать фторуксусиую кис-лоту.’
Это утверждение верно и в случае сложных эфиров со-фторкар-боновых кислот. Как видно из данных табл. 42, токсичны лишь те ; соединения, у которых п-иечетиое число, т. е. это значит, что токсичная фторуксусиая кислота образуется только прн ₽-окисле-1 иии. Еслц «-четное число, то такие соединения нетоксичны, поскольку при окислении получается неядовитая р-фторпропиоиовая кислота.
Некоторые свойства этих эфиров приведены в табл. 42.
Таблица 42. Свойства алкиловых эфиров ©-фторкарбоновых кислот F(CH2)ftCOOR
В круглых скобках приведено давление в мм рт, ст.
я n T. кип., °C ЛД50, на мышах, мг[кг
подкожно вя у трибрюшинно
СНз 1 104,5 (760) 15
СН3 3 78,5 (100) ' Ядовит 0,7
СНа 5 70—71 (9) — 1,6
С2Н5 I 117-118 (760) -15 —
с2н5 2 >200 ——’
C2HS 3 Ядовит —
С2Н5 4 56-60 (16) >160 —
С2Н5 5 82-84(14) 4 4,0
C2H6 7 145-150(12). 9 , 1,75
с2н5 9 135—138(10) 10 1,65
с2н5 10 140—141' (11) >100 —
С2н5 II 152-153 (11) <20 —
fch2ch2 1 158,146 (760) 8,5 ——
fch2ch2. 5 103-105(14) 2,5
fch2ch2 7 128-130 (13) 7 —
fch2ch2 9 145—149(12) 10
кислоты, в частно-
Гомологи алкиловых эфиров фторуксусиой сти, метиловый н этиловый'эфиры, получают окислением соответствующих со-фторзамещеиных спиртов с последующей их этерификацией, либо их получают фторированием алкиловых эфиров co-бром- илн (о-йодкарбоиовых кислот.
Метиловые и этиловые эфиры о-фторкарбоиовых кислот — бесцветные, легколетучне жидкости, по своим химическим свойствам сравнимые с метиловым и этиловым эфиром фторуксусной кислоты.
13*
391
С возрастанием молекулярного веса уменьшается реакционная способность и растворимость в воде.
Примечательной в этом ряду является группа 2-фторэтиловых эфиров, которые обладают более высокой токсичностью по сравнению с незамещенными алкиловыми эфирами. Эти соединения были получены с Целью сочетать токсические свойства са-фторированиых спиртов со свойствами эфиров co-фторкарбоновых кислот.
Важнейшими представителями этой группы являются 2-фтор-этиловый эфир фторуксусной кислоты V н 2-фторэтиловый эфир w-фторвалериановой кислоты VI. Токсичность последнего соединения почти в 11 раз превосходит токсичность метилового эфира фторуксусиой кислоты.
FCH2C^ FCH2(CH2)3C^
>och2ch2f ^och2ch2f
V VI
2-Фторэтиловый эфир фторуксусной кислоты получают взаимодействием фтор а цетил хлор ид а с 2-фторэтанолом при ПО—120° С: уС1
fch2c; + hoch2ch2f —> fch2c—och2ch2f + на
So J
Это бесцветная жидкость со слабым запахом, обладающая следующими физическими константами:
• Т. кип., °C
при 760 лл рт. ст. . 158 (146)
при 48 мм рт. ст.................. 85
Т. пл., °C................................ -25,4
Относит, плотность d?°............... 1,2862 (1,2906)
Показатель преломления ..... 1,3900
Высшие члены ряда— легкоподвижные жидкости с запахом, напоминающим запах фруктов. Эти соединения1 сравнительно устойчивы, не гидролизуются и полностью разрушаются только сильными окислителями, например, хромовой кислотой. С алкоголятами щелочных металлов образуются продукты конденсации.
Средняя летальная концентрация 2-фторэтилового эфира фторуксусной кислоты ЛК50 для кроликов составляет 0,05 мг/л (в случае МФА 0,1 мг/л). .Повышенная токсичность по сравнению с метиловым эфиром фторуксусной кислоты и с 2-фторэтаиолом, объясняется не только возможным гидролизом в организме по следующей схеме
+нао fch2cooch2ch2f-------> fch2ch2oh + fch2cooh
но и наличием двух активных фтор содержащих групп в молекуле.
392
10.1.5. «-Фторированные спирты
£
(й-Фторировацные спирты былн нсследованы в Англии и в Германии перед второй мировой войной и во время ее. В последние годы стало известно множество методов их получения.
Низшие фторированные спирты — бесцветные, растворимые в воде Жидкости. Они растворяются в органических растворителях, но не в петролейном эфире. Это — химически очень устойчивые соединения, которые при окислении дают столь же ядовитые фтор-ал ьдег иды и фтор к ар боновые кислоты.
Некоторые свойства «-фторированных спиртов приведены в табл. 43.
Таблица 43, Свойства ©-фторированных спиртов F(CH2)„CH2OH
В круглых скобках приведено давление в ям рт. ст.
_ п Т. кип., °C ЛД50 (на мышах, внутрибрюшинно), мг/кг
1 102—104 (760) 10
2 126-128 (760) 46,5
3 52-53 (11) 0,9
5 85-86(14) 1,24
7 106-107(10) 0,6
9 136-137 (15) 1,0
г 2-Фторэтанол имеет запах этанола. Ои смешивается с водой £ во всех отношениях, т. кип. 102—104°C (103,5°С), т. пл. — 43°С, d?l,104; d?lt1002; >$1,3647.
f, Фторэтанол устойчив к гидролизу и по сравнению с этанолом ’Ч обладает более сильными кислотными свойствами. Он очень устойчив к действию окислителей, при окислении двуокисью марганца ? в серной кислоте при 100°C образуется только 6% фторацеталь-Гдегида:
+MnO2(H2SO4)
FCH.CHiOH > FCH2CHO
-н2
При действии перманганата калия получается в небольшом ко-' личестве фторуксусная кислота.
Взаимодействие фторэтанола с ’ при 60 °C приводит к образованию ? сульфоновой кислоты: - 4-SO9CI2
i 2FCH2CH2OH —>
избытком сульфурилхлорида
2-фторэтилового эфира хлор-
fch2ch2oso2ci
j Это соединение столь же ядовито, что и метиловый эфир фтор-уксусной кислоты. Оно действует как раздражающее ОВ на сли- зистые оболочки носа и глотки.
393
Если же вводить в реакцию избыток фторэтанола, то получается менее ядовитый бис-2-фторэтиловый эфир сериой кислоты, ие обладающий раздражающим действием;
+so2ci2
FCH2CH2OH --С1> - (FCH2CH2O)2SO2
Фосгеи с фторэтанолом при О °C образуют фторэтиловый эфир хлоругольной кислоты, сильное раздражающее ОВ (т. кнп. 128°С, iff 1,3995):
+COC1-J fch2ch2oh ——> о=с;
^och2ch2f
При реакции фторэтанола с треххлорнстым фосфором в присутствии пиридина в качестве связывающего кислоту агента обра-. зуется трис-(2-фторэтил)-фосфит PfOCHsChbF)*, соединение, которое должно оказывать на центральную нервную систему угнетающее действие.
Фторэтаиол действует подобно фторацетатам. Для мышей ЛКео составляет 1,1 мг/л. По данным Саундерса, при экспозиции 10 мин и Концентрации 0,29 мг/л погибало 62% подопытных животных (кролики, крысы, морские свинки), а при концентрации 0,14 мг/л — 38%. Смерть животных наступала через 12 ч.
По данным Дукельской6, сроки наступления смерти1 у белых крыс после проглатывания отравленной приманки распределялись следующим образом: прн дозах 0,5—0,6 мг/кг через 24 ч погибало 100% животных, при меньших дозах время наступления смерти достигало у отдельных животных до 4 суток. Сроки наступления смерти в зависимости от принятой дозы приведены в табл. 44.
Таблица 44. Сроки наступлении смерти у белых крыс в зависимости от дозы фторэтанола6
Число подопытных животных Доза, jH&Iks Число погибших животных Г Число ВЫЖИВ- ШИХ ЖИВОТ- НЫХ
через4 2 ч через 4 ч через 6 ч через 12 ч через 18 ч .через 24 ч через 48 ч через 72 ч через 96 ч
25 0,08 __ 2 5 1 2 6 2 1 1 5
20 0,5 2 6 4 — 8 ——- ——- ——-
10 1,0 1 1 1 3 3 1
5 2,0 1 -— 3 1 — — —
15 5,0 2 —. 10 — 1 2 — —— — -
5 10,0 — 4 I — — — — — .— —
10.2. ПРИРОДНЫЕ яды
Природные яды растительного и животного происхождения известны издавна н находили применение как в «военных» целях (отравленные стрелы и т. д.), так и в медицине еще до того, как
394
устало возможным их выделение и был установлен их состав. ^Строение многих природных ядов окончательно ие выяснено еще теперь. Очень часто ядовитые начала растений и животных являются смесью нескольких соединений. Природные яды обра-: зуются в различных частях растений и органах животных, причем в очень небольшом числе случаев имеется полная ясность о ха-рактере происходящих биохимических процессов.
£ Природные ядовитые вещества подразделяют на яды и токсины, с Токсины представляют собой ядовитые вещества животного, рас-I тительного или бактериального происхождения. Как правило, оии ‘ являются белковыми веществами.
Токсины, вызывающие отравление, а не инфекционное заболе-; вание как мы его обычно понимаем, можно отнести к химическим ядам. Токсины, подобные принятому впоследствии на вооружение ' империалистических армий столбнячному токсину (стр. 409), кото-- рые приводят к инфекционным заболеваниям, относят к бактериологическому оружию. Растительные яды составляют исключение, к иим относятся, например, ядовитые начала бледной погаики, которые являются типичными токсинами. Такие яды, как никотин, атропин, стрихнин и акоиитии вследствие, своей высокой токсич-’. ностн могут представлять опасность как диверсионные яды.
В последнее время военные химики капиталистических госу-: дарств уделяют особое внимание ядовитым продуктам жизнедея-* тельности животных, в том числе морских. Из них были выделены Ъ высокотоксичные соединения; некоторые из иих уже синтези-роваиы.
10.2.1. Яды растительного происхождения. Алкалоиды
10.2.1.1. Никотин (3-метнлпнроллидии-2'-пириди#)
I Никотин вырабатывается корнями табачного растения Nico-: tiana tabacum и оттуда поступает в другие части растения. Содержание никотина в растении Колеблется от 0,5 до 8%, в зависи-разводят табачных
в общем,
ржание никотина в растении Колеблется от 0,5 до 8%, Мости от сорта. Богатые никотином растения, которые
Специально для добывания никотина н для получения ^щелоков, могут содержать до 15% никотина.
t Никотин можно синтезировать. Потребность в нем, ^покрывается природным продуктом.
" Это — бесцветное масло, которое на воздухе становится коричневым. В очень чистом виде он не имеет запаха, т. кип. 246°C (разл.), т. пл. ниже —30°C, d 1,014—1,015 г/слР. Никотин
395
растворим в воде и в органических растворителях, летуч с водяным паром. С кислотами ои дает солц, при окислении образуется никотиновая (пиридин-3-карбоиовая) кислота.
Никотин быстро проникает в кожу и слизистые оболочки; особенно опасен никотин прн попадании в открытые раны. Летальная доза для кроликов (внутривенно) составляет 6 мг/кг. Некоторые животные, например, козы и косули — невосприимчивы к никотину. Для людей смертельную Дозу оценивают в 50—100 мг. Уже в количестве 3—5 мг никотин вызывает симптомы сильного отравления — одышку, спастическое состояние, обмороки, которые длятся 3 и более суток. Никотин действует на центральную нервную си-' стему. Благодаря растворимости в воде и жирах он может быть использован Для заражения продуктов питания и продовольствия,
10.2.1.2. Алкалоиды чилибухи. В индийском рвотном орехе Strychnos nux vomica и многих других видах чилибухи наряду с другими алкалоидами содержатся стрихнин и бруцин (от 2 до 3%). Оба этих индольных алкалоида превращаются друг в друга. Бруцин является метоксипроизводным стрихнина (стрихнни R==H, бруцнн R = ОСНз):
Оба алкалоида можно синтезировать. Стрихнин и бруцин — бесцветные кристаллические вещества, горькие на вкус; т. пл. стрихнина 268 °C, бруцина 178 °C. Они умеренно растворимы в воде и в органических растворителях- Несколько лучшей растворимостью обладает нитрат стрихнина. Водные растворы имеют нейтральную реакцию.
Стрихнин со щелочами в жестких условиях образует триптамин (стр. 361). При действии концентрированной серной и хромовой кислот стрихнин приобретает сине-фиолетовую окраску.
Оба ядовитых растительных основания по своему токсическому действию приближаются к токсину столбняка. Стрихнин увеличивает чувствительность органов чувств и рефлекторную возбудимость. При повышенных дозах появляются страх, дрожание конечностей, нарушения речи, мучительное окоченение мышц, ригидность затылка (голову тянет назад), искажение лица. В результате внешнего раздражения (шум, свет) мгновенно наступает первая судорога. Истощение и паралич дыхательного центра приводят отравленного к смерти. Судороги, предшествующие смерти, непрерывно усиливаются. Они чрезвычайно болезненны и мучительны.
На взрослых людей дозы в 15 мг могут действовать летально, доза для детей — 5 мг. В общем считают, что для взрослых дозы 396
Т 100—ЗОо мг являются смертельными, при uptium. —
рядка 1 г смерть наступает через 30 мин от паралича дыхательного центра.
Бруцин в 8—40 раз менее ядовит, чем стрихнин. Предполагают, что оба вещества могут быть пригодны для заражения подслащенных напитков и продуктов питания. Нитрат или сульфат стрихнина растворяются в воде. Водные растворы на вкус горькие.
10.2.1.3. Курарин. Кураре был известен еще в XVI веке. Индейцы использовалн его для отравления стрел. Это — коричневая, растворимая в воде, горькая масса, которую извлекают из коры лиановых растений Strychnos toxifera,, St. Castelniae экстракцией водой. Различают несколько видов кураре: трубочный, горшечный И тыквенный кураре. Самый эффективный из них — тыквенный (туземцы хранили его в сосудах из выдолбленной тыквы). Он содержит самые ядовитые растительные вещества из известных в настоящее время.
Алкалоиды кураре являются производными индола. Тыквенный кураре содержит две группы алкалондов, примерный состав одной группы C20N2, другой — C4oN2Алкалоиды последней группы, очевидно, обладают наибольшей токсичностью. В их число входят С-калебасснн, С-курарин и токсиферины (С-токсиферин-1, С-дигидротоксиферин). Токсиферины являются самыми ядовитыми компонентами курарина.
Впервые С-токсиферин-1 был выделен в 1941 г. Виландом и Пистором, позднее его удалось получить из одного из продуктов разложения стрихнина. Ему приписывают следующее строение:
С-Токсиферин-1 в 5—10 раз токсичнее, чем аконитин (ср. раздел 10.2.1.5). Дозы в 7,7 мкг(кг вызывают паралич у лягушек. Неочищенный тыквенный курарин примерно в 10 раз менее эффективен. Хлорид трубочного курарина в 10—100 раз менее ядовит. Его летальная доза для людей составляет 0,25—0,4 мг[кг.
При попадании через рот курарины во много раз менее ядовиты, чем прн других способах введения. Онн не разлагаются 397
в желудочно-кишечном тракте и лишь медленно резорбируются. В моче курарин остается неизменным и выводится с нею из организма. Поэтому мясо и другие продукты, отравленные кураре, можно употреблять в пищу, если только'он не содержится В слишком больших количествах. Кураре, попавший в организм через раневые поверхности илн другими путями, приводит к параличу или к деполяризации рецепторов моторных концевых пластин. Паралич обратим. Курариновый паралич поперечнополосатых мышц начинается с мышц лица (сначала с внешних глазных мышц, затем мышц шеи и затылка), распространяется на мышцы конечностей н живота (полная неподвижность) и переходит на дыхательные мышцы. Субъективные ощущения при отравлении кураре состоят, например, в двоении в глазах, затруднении глотания, опущении головы, затруднении дыхания. При внутривенных инъекциях отравление обычно наступает через 2—3 мин.
Самые сильные отравления курарин и его алкалоиды вызывают при попадании через маленькие ранки" непосредственно в кровь.
Курарин непригоден для заражения воды и продовольствия.
Даже в настоящее время туземцы еще применяют его в качестве яда для стрел. В качестве такового не исключена возможность и его военного применения в виде отравленных снарядов, специальных осколочных гранат и т. д.
10.2.1.4. Атропин н (—)-гносцнамин
- . СН2 СН—сня
сн3—n с^н-о-с—си—сн2он
' СН2 СИ—<^Н2 А сен5
Атропин (тропиновый эфир троповой кислоты) содержится
в красавке Atropa belladonna; (—)-гиосциамин, кроме того встречается в других растениях семейства пасленовых (Solanacea)—белене, скополии, мандрагоре. Атропин и (—)-гиосциамин изомерны н различаются лишь тем, что атропин содержит остаток рацемической троповой кислоты, а (—)-гиосциамин содержит оптически активную троповую кислоту. Основное действие атропина приписывают (—)-гиосциамину.
Атропин может быть получен синтетически. На заключительных стадиях синтеза полученный спирт тропин этерифипируют хлора нгидридом 1 -фенил-2-ацетоксипропионовой (ацетилтропо-вой) кислоты и затем гидролизом отщепляют ацетильный остаток. Конечный продукт представляет собой рацемическую (оптически недеятельную) смесь (+)- и (—)-гиосциаминов — атропин.
Тропин был синтезирован из янтарного диальдегида, метиламина и ацетоидикарбоновой кислоты через тропином, который при
39$
шислотиом восстановлении дает тропин:
Й' Н2С—COOH(R) СН2 СН—СН—СООН н ’ ~
ХНз-СНО \ III S1
1 + H2NCH3 + ;с=о
СНз—СНО /
H2C—COOH(R)
СН3—N
:Н2--С1
:н— С ООН
сн2—сн—сн2
СН2--СН—СН2
—► СНз—N С
СНз--СН— (!на СН2—СН—СНз
тропинон тропин
Оба соединения представляют собой игольчатые кристаллы, ^умеренно растворимые в 'воде. Они растворяются в органических растворителях (в спирте, эфире, хлороформе) и в жирных маслах. ^ Атропин н (—)-гиосциамий имеют т. пл. 115 и 165,5°C соответственно; сульфат атропина растворяется в воде.
f Будучи эфирами (±)-троповой кислоты, атропин и гиосциамин Смогут гидролитически расщепляться на тропин и троповую кислоту.
Особенно хорошо идет гидролиз в горячей воде:
СН2--СН—СН2
СНз--СН—СН2
СеНд
' : СНз—N СН—О—с—СН СНз--:Ан— СН2
восстановление
СН3—N
:СН—ОН
+ н2о
С6Й3
:н2он
СНз—]
:нон + ноос—СН
СН2 СН—сн2
СН2ОН
' Атропин и (—)-гиосциамин можно окислить.
В малых дозах атропин и гиосциамин возбуждают центральную нервную систему, а в больших дозах парализуют ее. При Пероральном приеме происходит быстрая резорбция алкалоидов Й пищеварительном тракте. При подкожных инъекциях летальная Доза для людей составляет 15 мг[кг.
В зависимости от введенной дозы у людей появляются следующие симптомы (данные Хаушилда):
0,5 мг
0,5-1
1-2
3-5
7 мг
10 мг
мг мг
мг
Сухость кожи, замедление пульса ...............
к - Сухость во рту, жажда • . ........................
Расширение зрачков, учащение пульса.............
Беспокойство, мышечная слабость, затруднение гло-такня, головные боли............................
Максимальное расширение зрачков, нарушение мы-щечной координации..............................
Апатия, галлюцинации, бред, потеря сознания ....
Г- Характерной для отравления атропином является горячая, баг-фовая кожа, повышение температуры тела, дрожание конечностей непрерывное их движение. Воздействие иа центральную нервную Систему выражается в патологической болтливости, танцах, приступах смеха, неистовстве и бешенстве. Дозы свыше 100 лег ведут параличу дыхация,
399
Атропин применяют для подавления действия мускарина и никотина на ацетилхолин (фосфорорганические отравления!).
10.2.1.5. Аконитин. Алкалоид аконитин содержится в различных видах аконита, например, в синем борце. Это — один из сильнейших растительных ядов, строение его не установлено.
Аконитин — пластинчатые кристаллы, лишенные цвета и запаха, т. пл. 197—198 °C. Ои мало растворим в воде, растворим в органических растворителях. Это соединение неустойчиво, разлагается щелочами, кислотами и при нагревании. При разложении наряду с другими продуктами образуются уксусная и бензойная кислоты. В результате гидролиза в водных растворах аконитин перестает быть ядовитым.
Действие аконитина проявляется сначала в учащении дыхания, но затем, при известных условиях, дыхание замедляется вплоть до полного прекращения. После резорбции яда все тело начинает чесаться (парестезия, ощущение ползания мурашек), затем появляется озиоб и выделение пота, паралич скелетных мышц (язык, лицо), рвота, усиление двигательного возбуждения. Отравленный вскакивает на ноги и снова бросается наземь. Сознание сохраняется до наступления смерти (остановка сердца, паралич дыхания). Смерть Может наступить примерно в течение 1 ч.
Как показали опыты на крысах \ при инъекциях аконитина (0,02 мл, в гипоталамус) развивается отёк легких, прн котором легкие увеличиваются в 2—3 раза.
Для крыс внутривенно ЛД50 составляет 0,11 м&!кг, для лошадей уже смертельны дозы в 0,004 мг]кг, тогда как для людей прн пероральном приеме смертельны дозы около 2—5 мг.
Фашистские войска СС в концентрационном лагере Бухенвальд проводили на узниках бесчеловечные опыты с нитратом аконитина. Заключенным стреляли в бедро пулями, отравленными аконитином. Первые симптомы отравления появлялись через 20—25 мин (двигательное возбуждение, легкое слюнотечение). Через 40 мин слюнотечение усиливалось и становилось столь интенсивным, что отравленные уже не могли сглатывать слюну (позыв на рвоту, рвота). Резко усиливалось двигательное возбуждение (вскакивание иа ноги, броски наземь, закатывание глаз). Затем возбуждение уменьшалось (расширение зрачков). Эти зверски замученные люди погибли через 121 —129 мин после ранения-
Этот пример показывает, что алкалоиды такого типа и другие яды могут найти военное применение посредством использования отравленных пуль. Аконитин и родственные ему алкалоиды (аконитины) могут быть также использованы и качестве диверсионных ядов, особенно для заражения продовольствия и напитков, не нуждающихся в подогреве.
В Японии было выделено одно из столь же ядовитых соединений— анизатин С^НаоОа, являющийся компонентом семян Illicium anisatum L. Опытами на мышах установлено, что ЛД50 (внутрибрюшинно) составляет 0,7 мг]кг. Смерть в судорогах наступала 400
в результате Паралича дыхания. Уже дозы в 0,04 ж/кг оказывают влияние на дыханне н ведут к судорогам.
Другими сильно ядовитыми алкалоидами являются, например, эритропленн, гоматропин, колхицин, кониин, физостигмин и вератрин.
10,2.2. Токсины грибов
Ядовитые начала большей части ядовитых грнбов были выделены н отчасти идентифицированы. Вследствие высокой токсичности военный интерес при известных условиях представляют токсины клубневидных мухоморов рода Amanita.
Яды бледной поганки (Amanita phalloides, A. virosa, A. verna) представляют собой полипептиды. Как вндио нз структуры фал-лоина VII, вокруг индольного ядра расположен ряд аминокислот, соединенных амндоподобнымн связями. В состав токсинов входят такие аминокислоты, как (—)-алаиин, (—)-треонии, (—)-цистеин, (—)-триптофан, (—)-алло-оксипролин, лейцин и др.
(СнДс-Он ^СНОН
1 н2с \:н2
СН2 |
CH-NH-CO-CH—NH-CO-CH—N
! ' I
С° ,СН2 СО
kJ-Д-----s—сн2 I
СН3-СН Y I СН-СН3'
I и ! I
СО —NH—СН—СО—NH—СН —СО—NH 1 снон-сн3
VII
Кроме фаллоина удалось выделить фаллоидин, C40H47O10N9S н аманитин, C33H45O12N7S. Основным ядом мухомора является, невидимому, аманитин, которой Виланд и сотр. выделили еще в 40-х годах этого столетия.
Фаллоидин и фаллоии на порядок менее токсичны, чем аманитин: для мышей их ЛД5о составляет 1,4 мг/кг, а аманитина 0,097 мг/кг.
Аманитин — кристаллическое вещество (т. пл. 245 °C), которое в грнбе находится в виде а- и р-амаинтинов. Летальная доза составляет: для «-аманнтина 2,5 мкг, для р-аманнтнна 8—10 мкг на 1 мышь.
Оба токсина содержат одинаковые аминокислоты, ио их пространственное расположение в молекуле иное.
Токсины грнбов не разлагаются в кишечнике, а также при Сушке и варке'грибов. В организме они активны в виде либо
401
целой молекулы, либо фрагментов с особым пространственноструктурным расположением.
Симптомы отравления появляются после латентного периода в несколько часов. Отравление начинается со рвоты, поноса, болей в животе (коликообразных), учащения дыхания и потери сознания. Смерть наступает в результате тяжелых поражений печени, сердца и сосудистого центра.
Токсины клубневидных мухоморов вследствие своей высокой' токсичности и устойчивости к гидролизу и нагреванию могут быть использованы как диверсионные яды.
Ядовитыми началами других грибов в основном являются алкалоиды. Из-за меньшей токсичности и недостаточной устойчивости они, пожалуй, почти не представляют военного интереса. Яд красного мухомора, мускарин — легко разлагающееся кристаллическое вещество. Из 100 г красных мухоморов можно выделить 16 мг алкалоида, его ЛД50 для мышей (внутривенно) составляет 0,23 мг!кг. В водном растворе мускарин дает щелочную реакцию.*
10.2.3. Яды животного происхождения
10.2.3.1. Кантаридйн (т. пл. 218°С)
СНз о
I J
СН2—СН—С—С"
I I \
СН2—СН—с— . СНз 0
Кантаридин является производным циклогекс ан ди кар боновой кислоты. В природе он содержится в так называемой шпанской мушке Lytta vesicatoria. Это кристаллическое соединение, растворимость которого в Холодиой воде составляет 0,003%; его можно синтезировать реакцией фурайа с пирохининовой кислотой.
При приеме через рот -кантаридин является эффективным ядом. Ои растворяется в жирах и маслах. Летальная доза кристаллического кантаридина составляет 40—80 мг. При приеме внутрь появляются слюнотечение, жжение, волдыри и образуются струпья в полости рта (тяжелые поражения слизистой оболочки), возникает сильная жажда, тошнота, рвота с кровью, кровь при дефекации и мочеиспускании (геморрагия), сильные боли в почках, мочеиспускательном канале н в мочевом пузыре, маточные кровотечения, патологическая эрекция (приапизм). Приостановка мочеотделения ведет к тяжелым поражениям почек. С судорогами в области почек и мочевого пузыря (уремические судороги) наступает смерть.
402
Растворённый в масле Кантаридин вызывает на поверхности кожи нарывы, на слизистых оболочках появляются болезненные и глубокопроиикающие некротические распады.
Из-за хорошей растворимости кантаридина в жирах ие исключено его использование в качестве диверсионного, яда, прежде всего для заражения содержащих жиры продуктов.
10.2.3.2. Яды змей. Известно около 300-400 видов ядовитых змей. Змеиные яды ие имеют постоянного состава. В общем оии состоят из множества ферментов (гидролазы и десмолазы) с необычной активностью. В составе ядов обнаружены также другие протеины, альбумины, пептоны, соли различных металлов, жиры и др.
Вследствие разнообразия своего состава змеиные яды ие дают единой картины отравления. Оии могут действовать как нейротоксины, как ингибиторы ферментов, вызывать распад крови и поражать кровеносные сосуды. .
Симптомами отравления являются как головные боли и головокружение, так и кровавые поносы, кровь в моче, тошиота, рвота, страх, потовыделение, паралич и смерть от паралича дыхательного центра.
Из яда кобры был выделен так называемый офиотоксин— гликозид состава С^НзбОы- Смертельная доза яда кобры для мышей составляет около 0,1 мг}кг.
Даже в малых дозах этот яд действует на людей смертельно через 8 ч (паралич дыхательного центра).
Кроталотоксин, С17Н2б01о-0,5Н20, был выделен из яда гремучей .змеи. Доза в 0,35лл/г/ке смертельна для кроликов.
Неочищенные змеиные яды представляют собой прозрачные илн молочно-мутные, иногда бесцветные, иногда золотисто-желтые жидкости. Оии свертываются при длительном стоянии и хорошо растворяются в воде. В высушенном виде оин образуют порошок, который, как правило без потери токсичности можно хранить хвыше 20 лет.
10.2.3.3. Токсин скорпиона. Особенно опасны яды тропических скорпионов. Эти токсины содержатся в хвостовом жале животного. ДЭии легко резорбируются и вызывают судороги шейных н жевательных мышц, слюнотечение и другие явления. Для мышей ЛД50 Доставляет 0,5 мг!кг. Одни скорпион прн каждом уколе выпускает ^коло 40 мкг яда. От одного животного можно получить от 0,1 до $ мг яда в высушенном виде. Ядом североафрикаиского толсто-Двостого скорпиона в количестве 0,5 мг можно убить около ^?00 морских свинок. Строение яда скорпиона пока выяснено недостаточно.
10.2.3.4. Яды жаб. Буфотоксин. В разделе 8.1.2.2 уже упоминалось об одном яде жаб, буфотенине, который наряду с обще-ядовитым действием вызывает психозы, сходные с психозами от XSD или мескалина. Кожные секреты жаб грубо делятся иа две Д ру п пы веществ: с ое динения, которые можно произ вести от
403
триптофана (к этой группе относится буфотенин и др.), и ие содержащие азота и действующие на сердце буфогенины, или буфагины и их производные с пробковой кислотой н 2-амнно-5-гуанидилва-лериаиовой кислотой (аргинином), так называемые буфотоксины И Др.
Буфотоксины имеют общий состав QoHeoOioNi. Строение бу-фотоксина земляной жабы следующее8:
СН3
Буфотоксин принадлежит к самым токсичным ядам жаб, он оказывает действие иа сердце и в этом отношении сравним с растительными сердечными ядами — строфантином (гликозидом из семян произрастающих в Африке видов Strophantus) и сцилларё-иом (гликозидом нз морского лука). Это игольчатые кристаллы, горькие на вкус, т. пл. 204°C (разл.). Летальная доза для кошек (внутривенно) составляет 0,3 мг!кг массы. Буфотоксин обладает сильным резорбтивным действием. При отравлении наблюдается повышение кровяного давления, учащение пульса, позыв к рвоте и судороги.- Смерть наступает от мерцания желудочков сердца.
Яд кокои. Одним из сильнейших ядов животного происхождения является яд кокои, высокоактивное вещество из кожи ядовито-жальной лягушки Phyllobates bicolor, которая водится в Колумбии. Токсичность очищенного яда для мышей составляет 2,7 ± 0,2 мкг}кг массы. Экстракт природного яда9, сконцентрированный в 60 раз, обладает ЛД50 около 5 мкг/кг массы.
Яд кокои представляет собой основание, растворимое в гало-геналканах, например в хлороформе.
Ои необратимо блокирует моторные концевые пластины, результатом являются судороги. Смерть наступает от паралича дыхания.
10.2.3.5. Яды морских животных 10. Мурексим— холиновый эфир имидазол-4-акриловой кислоты — производное имидазола, имеет следующее строение:
N---С—СН=СН—СО—OCH2CH2N(CH3)3
II
НСХ /СН
NH I
Ои был выделен из пурпурных улиток Muricidae. В организме этих морских животных он, по-видимому, образуется нз холина и
404
имидазол-4-акриловой кислоты, либо одного из продуктов метаболизма гистидина
N---С— СН2— СН—СООН
II ' tin НСХ /СН NH2
NH
Мурексни можно синтезировать. Ои обладает теми же фармакологическими свойствами, что и другие производные холина. Так, он возбуждающе действует на ганглии и блокирует передачу раздражения нервами к мышцам. Аналогично действию фосфорорганических соединений, мурексин поляризует область концевых пластин; таким образом, ои вызывает тот же эффект, что и при нарушении обмена ацетилхолина вследствие ингибирования холинэстеразы. Как холиновый эфир мурексин ие гидролизуется холинэстеразой (!).
Действие мурексина выражается в параличе скелетных мышц. Смерть наступает в результате паралича дыхательных мышц и последующей кислородной недостаточности (аноксемии). После остановки дыхания сердце продолжает еще несколько минут биться.
$,$-Диметилакрилоилхолин (сенецило'илхолин)
(СН3)2С=СН—СО—О—СН2СН2Й(СН3)3
был выделен из морских животных Thais floridiana. Вероятно этот эфир образуется в них из аминокислоты — валина (а-амниоизова-лериановой кислоты) и холина:
(снэ)2сн—сн—соон ———> (сн3)2с=сн—соон холин>
| —NH3
йн2
(CH3)2C=CH—СО—О— СН2СН2Й(СН3)з
Токсическое действе этого яда такое же, как у мурексина. Деполяризующее действие на моторные концевые пластины более продолжительное, чем в случае ацетилхолина. Этот токсин не разлагается или лишь мало разлагается холинэстеразой.
Сакситоксин (митилотоксин) является одним из самых сильных паралитических ядов, существующих в природе. Уже несколько лет он служит предметом исследований в воеино-химических лабораториях в Кэмп Детрик, США. Сакситоксин был выделен из некоторых морских моллюсков, в частности, из раковины Saxidomus giganteus и ракушечника Mytilus californianeus. В кристаллическом виде потучнть его пока не удалось. Токсин имеет щелочную реакцию и образует соли с минеральными кислотами, Молекулярный вес его 372, состав гидрохлорида CioHnNyO^HCl.
Сакситоксин растворим- в воде, спирте, метаноле и водном ацетоне, не растворяется в жидких углеводородах. При гидролизе и окислении'сакситоксин разлагается с образованием гуанидина,
405
мочевины, гуанидопропноновой кислоты, аммиака и двуокиси углерода.
Симптомы отравления сакситоксином или моллюсками проявляются в покалывании и онемении губ, языка и кончиков пальцев, расслаблении мышц затылка, рук, йог и в удушьи. Смерть наступает от паралича дыхания.
Хотя отравляющее действие сакситоксина сходно с действием кураре, противоядия от кураре в этом случае неэффективны. Этот токсин — ингибитор холинэстеразы. Токсичность для мышей составляет 0,2 мкг. Летальная активность белого аморфного порошка сакситоксина составляет 5,5-Ю6 ME* на 1 г токсина.
Нереистоксин— соединение, родственное липоновой (6,8-тнокто-вой) кислоте.
ГА
^^^СНаСНгСНэСНгСООЙ
В нереистокснне дисульфидное кольцо связано с третичным атомом азота. Он может иметь структуру I нлн П:
।---j j----pN(CH3)2-
Sxsx^N(CH3)2
I II
Нереистоксин был выделен в кристаллическом виде в 194! г. японским химиком Ннтта нз морского кольчатого червя Lumbrico-nereis heteropada. Это сильный нервный яд, приводящий к сужению зрачков, слюно- и слезотечению, к сокращению гладких мышц. Летальная доза для кроликов (подкожно) составляет 1,8 мг/кг.
Тетродотоксан (тарихатоксин). Вещество было выделено из многих морских животных. Например, он содержится в разных видах мурены, в печени и янчннках рыбы Tetraodontidae. В Японии, где эта рыба считается деликатесом, сотни человек ежегодно отравляются насмерть в результате небрежного приготовления блюда (так называемое отравление фугу).
Для выделения примерно 10 г очищенного кристаллического тетродотоксина требуется переработать около 1т яичников этой рыбы. ЛД50 препарата, полученного химиком Цуда, равна 0,01 мкг/г /для мышей). Позже был получен еще более эффективный препарат с токсичностью 0,008 мкг/г.
Тетродотокснн образует призматические кристаллы, т. пл. 220°С. Прн нагревании выше этой температуры он разлагается, дает соли с различными кислотами, например гндрогалогеннды, тартрат.
Явления, наблюдаемые11 в опытах с тетродотоксином на кроликах приведены в табл. 45.
* ME — «мышиная единица»; 1 ME — количество яда, которое при внутрибрюшинной инъекции за 15 мин убивает мышь весом 20 г.
406
Таблица 45. Результаты испытания отравляющего действия тетродотоксина иа кроликах
Доза, мк.г!кг Способ введения Действие
2 Внутривенно Падение кровяного давления, затруднение дыхания (в течение 30 мин)
3 Внутривенно Полное прекращение дыхания, смерть. Стимуляция дыхания, разгибание тела (в течение 2 ч)
6 Субвальвулярно
7 Субвальвулярно Нарушение координации движений (в течение 8 ч)
10 Субвальвулярно Прекращение дыхании (через 2 ч)
20 Перорально Тяжелое двигательное расстройство, прекращение дыхания (через 4 ч)
Дозы в 5 мкг}кг (подкожно) вызывают у собак рвоту и нарушение дыхания, а дозы в 6 мкг уже в течение 1 ч приводят к смерти в результате прекращения дыхания.
Тетродотоксин является одним из сильнейших животных ядов своего типа. У людей отравление выражается в зуде, потере чувствительности кожи, в параличе моторной нервной системы. Смерть наступает от паралича дыхательных мышц или дыхательного центра. Действие токсина примерно соответствует отравлению кураре.
Токсин рыбы-дракона. Встречающиеся в Северном море драконы Trachinus draco выделяют из своих кожных желез очень сильный яд. Уже уколы при прикосновении к рыбе (шип спинного плавника) ведут к болезненным поражениям кожи (некрозы).
Вследствие резорбции в другие части тела яд оказывает общеядовитое действие и вызывает удушье, головокружение, выделение пота и замедление, деятельности сердца12. Для мышей 100%-ная летальная доза неразбавленного токсина составляет 4* Ю-4 мл, для морских свннок 8-Ю-4 мл (смерть через 24 ч). В одной рыбе содержится около 250 ME токсина.
Чистый токсин с добавкой глицерина устойчив при хранении при —60 °C в течение 2 лет без потери токсичности.
10.2.4. Бактериальные токсины
10.2.4.1. Ботулинический токсин. Токсин выделяется микроорганизмами Clostridium botulinum. Микроорганизмы представляют собой палочковидные бактерии, которые размножаются в почве, воде, продуктах питания и в живых организмах. Они развиваются преимущественно в содержащей белок среде и являются причиной пищевых отравлений (ботулизма), особенно при употреблении испорченных консервов, например гороха, фасоли, консервированного сыра или плохо прокопченных, недостаточно просоленных колбасных изделий, Ботулинические микроорганизмы устойчивы к
407
кипячению н развиваются в отсутствие воздуха (анаэробно). Для своего развития и для образования токсина микроорганизмы нуждаются в триптофане (стр. 361).
4 Ботулизм не является инфекционным контагиозным заболеванием, это — отравление, вызываемое выделенным токсином, и поэтому не передается от человека к человеку, как в случае типичных инфекционных заболеваний.
В кристаллическом виде токсин был выделен13 во время второй мировой войны в американском военно-бнологнческом исследовательском центре Кэмп Детрик химиком Ламанна. Процесс этот чрезвычайно сложный, поэтому практическое использование кристаллического вещества, пожалуй, маловероятно; в военных целях, возможно, будут применяться сильно концентрированные субстраты.
Имеется пять типов ботулинических токсинов — типы А, В, С, D и Е, из которых чрезвычайно опасными, особенно для людей, являются типы А, В и Е. Речь идет о высокомолекулярных протеинах еще неизвестного строения.
Ботулинический токсин типа А представляет собой белые игольчатые кристаллы. Это не индивидуальное вещество, а смесь глобулярных протеинов, построенных из аминокислот. Средний молекулярный вес токсина равен 900 000. В водных растворах токсин частично гидролизуется, но ядовитость раствора прн этом не теряется. В результате гидролиза прн pH 6,5 образуются продукты с молекулярными весами между 100000 и 400000. К настоящему времени в продуктах гидролиза удалось идентифицировать 19 взвестных аминокислот. Считают поэтому, что токсичность определяется, во-первых, определенной Пространственной структурой, которая ие затрагивается при гидролизе, во-вторых, наличием триптофана, входящего в состав молекулы, ботулинического токсина.
При pH 9,2 токсичность очищенного токсина несколько уменьшается. По-видимому, при протеолизе отщепляется небольшое количество «носителей токсичности».'
Токсин флуоресцирует в ультрафиолетовом свете. Некоторые вещества, которые гасят флуоресценцию, влияют также и иа его токсичность14. Так, например, токсин обезвреживается N-бром-сукцинимидом с одновременным гашением флуоресценции. Вероятно, бромсукцинимнд реагирует с имеющимися в молекуле токсина триптофаном; 6 AI раствор мочевины оказывает такое же действие без гашения флуоресценции. По-виднмому, это происходит в результате изменения пространственных соотношений в молекуле токсина. Кроме того, обезвреживающее действие оказывает формальдегид (формалин).
Летальная активность кристаллического токсина составляет около 3-1010 ME на 1 г токсина. Обычно для мышей приводится величина токсичности от 3-10-5 до 10~4 мкг. Если принять, что мыши н люди одинаково восприимчивы к этому токсину, то, по
408
мнению Розбери, 1 г ботулинического токсина достаточно, чшиы г отравить 8 миллионов человек.
Д Вероятная летальная доза токсина для людей при внутривен-Ь ной инъекции составляет от 0,15 до 0,3 мкг, а пероральная, невидимому, 8—10 мкг.
Ботулинический токсин — одни из сильнейших нейротропных ядов. Ои препятствует образованию ацетилхолина и тем самым парализует мышцы, работа которых контролируется сознанием. Передача возбуждения к моторным концевым пластинам прекра-£. щается. Этот токсин — сильный яд при приеме через рот. Он ие разлагается или мало разлагается в пищеварительном тракте и оттуда проникает в круг кровообращения. Почками ботулинический токсин не выводится из организма.
Отравление (ботулизм) наступает после латентного периода в несколько часов ( -^36 ч). Первые симптомы выражаются в головных болях, тошноте, иногда слюнотечении, рвоте, головокружении, ломоте в конечностях и поносе. Действие на центральную нервную систему проявляется в диплопии (двоение в глазах), снижении степени аккомодации, иногда расширении зрачков, сухости в полости носоглотки. Функция слюнных и потовых желез прекращается, дело доходит до паралича пищевода и мышц глотки (паралич глотания), а также паралича других групп мышц. В тяжелых случаях смерть от паралича дыхания наступает на десятый день (иногда через 3 недели).
Смертельные исходы от отравления ботулиническим токсином составляют от 15 до 30%. При несвоевременном лечении анти-ботулиновой сывороткой (токсоидом, т. е. обезвреженным токсином) смертность может достигать 90%.
Ботулинический токсин или его природные концентраты из-за их чрезвычайно высокой токсичности опасны в первую очередь как яды для отравления пищевых продуктов, воды н т. п. Зараженную воду можно обезвредить кипячением в течение 1 ч.
По американским данным, этот токсин в боевой обстановке могут применять ВВС с помощью кассетных авиабомб. В настоящее время ботулинический токсин считают наиболее опасным прн военном применении.
10.2.4.2. Токсин столбняка. Представляет собой высокомолекулярный протеин, выделяемый бациллой столбняка Clostridium tetani. Этот токсин, как и ботулинический токсин, вырабатывается бактериями в соответствующих питательных средах. Из ядовитого фильтрата токсин выделяют и кристаллизуют; он относится к самым ядовитым веществам.
Кристаллический токсин столбняка неустойчив при нагревании. Порядок величины летальной активности составляет 1010 ME на 1 г токсина. Доза в 1,3-10~5мкг вызывает у мышей столбняк. Лошади менее восприимчивы к токсину, чем мыши и другие мелкие животные. Для людей смертельная одноразовая доза концентрата токсина составляет 0,2—0,3 мг.
14 Зак. 448 409
Действие токсина столбняка приближается к действию стрихнина. В отличие от последнего, столбнячный токсин, по-видимому, ие попадает по кровеносному руслу в исполнительные органы, а воздействует через нервные тракты на преобразовательные центры моторной нервной системы.
В противоположность ботулизму, столбняк (тетанус) является инфекционным заболеванием людей и животных. После весьма нехарактерных первых симптомов, как правило, дело доходит сначала до судорог жевательных мышц, а затем и всей скелетной мускулатуры. Смерть от удушья наступает в результате судорог дыхательных мышц.
Столбняк развивается после инкубационного периода от 4 до 21 суток. Столбнячная палочка широко распространена, она имеется в содержимом кишечника, в земле, в пыли и т. д.
Столбнячный токсин обезвреживают щелочными солями высших ненасыщенных жирных кислот, например, Олеатом натрия.
Со временем удалось выделить токсины соответствующих возбудителей почти всех инфекционных болезней, например, токсины дифтерии, чумы н скарлатины. Последний содержит 16 различных ймииокислот и имеет молекулярный вес 29000.
10.3. НЕОРГАНИЧЕСКИЕ ЯДЫ
Хотя почти все неорганические соединения, в зависимости от дозы и продолжительности воздействия, в известной мере обладают определенными физиологическими и фармакологическими свойствами и порою могут быть очень ядовитыми, все же обычно их токсические свойства выражены не столь сильно, как в случае органических ядов. Яд°витые соединения при условии достаточной растворимости в воде могут служить для заражения некоторых продуктов питания и воды. Более трудно растворимые соединений могут быть использованы для отравления муки и других продуктов. Так, трехокись мышьяка может быть использована для заражения муки, а различные соли свинца и ртути(П) — для заражения сахара или поваренной соли.
Токсическое действие подобного рода ядов начинает проявляться уже в пищеварительном тракте. Для этого необходимо, чтобы эти вещества в достаточных количествах резорбировались стенками желудка и кишечника. Вещества, не обладающие такими свойствами, при пероральном приеме очень быстро выводятся из организма в неизменном виде, как например соединения ртути(I) и стронция. В общем, существует известная корреляция между пероральной токсичностью неорганических соединений и их растворимостью в воде.
Другим условием пероральной токсичности неорганических соединений является их способность к ионизации. Вообще, для того чтобы быть эффективными, оии должны существовать в ионной форме. Носителями отравляющего действия на исполнительный орган являются как катионы, так и анионы.
410
Если зависимость токсичности катионов от порядковых номеров соответствующих элементов представить графически 1S, то с увеличением порядкового номера наблюдается периодичность изменения показателей токсичности (рнс. 20).
Сравнительно неядовитые катионы (максимумы на кривой) — это катионы металлов с сильно отрицательным окислительно-восстановительным потенциалом. Самые ядовитые представители на-
ходятся среди катионов
потенциалом.
металлов с положительным нормальным
Рис. 20. Зависимость ЛД50/,30 (ммоль/кг) катиона от порядкового номера элемента Z.
В табл. 46 приведены показатели токсичности некоторых катионов, которые были определены15 на самцах белых мышей. Они представляют собой средние летальные дозы ЛД50 при внутрибрюшинном введении (смерть через 30 суток).
Различна и токсичность анноиов. В табл. 46 приведены показатели токсичности некоторых анионов (ЛД50, смерть через 30 суток), установленные прн внутрибрюшинных инъекциях белым мышам. Для этого применяли натриевые соли соответствующих кислот16.
Согласно этим данным, анноны, которые при известных условиях могут представить военную опасность, располагаются по токсичности в следующий ряд:
no;, г, AsO43', cn", Aso;
Токсичность анионов в солях HgCl2 и РЬС12 почти не играет роли; главным носителем токсичности является катион. Применение хлоридов, сульфатов, нитратов, ацетатов н т. п. обусловлено исключительно хорошей растворимостью таких соединений в воде.
14* 411
Таблица 46. Токсичность катионов и анионов
ЛДяо («а Анионы (в виде натриевых солей)
Катионы (в виде хлоридов, нитратов и др.) мышах, внутрибрюшинно) ]S. мг)кг ЛД50 <на мышах, внутрибрюшинно} 10> мг!кг
Na+ (хлорид) 1024 SO2’ 2255
Sr2+ (хлорид) 503 2166
Th4 (хлорид) 206 NO'
NH* (хлорид) 163 СГ 1576
Li+ (хлорид) 99,0 сю; 467
РЬ3+ (ацетат) 76,6 сю; 448
Сг3+ (хлорид) 46,8
Мп2+ (сульфат) 43,9 no; 106
Fe2+. (сульфат) 39,1 23,2
Ва2+ (хлорид) 35,5 СЮ2’
Со2+ (сульфат) 20,6 F’ 19
Т1+ (хлорид) 20,4 ЗеОГ 7,15
Ag+ (нитрат) 13,9 С 2- 7\05
Zn2+ (сульфат) 11,7
Ni2+ (сульфат) 7,93 SeOg 4,44
Hg2+ (хлорид) 3,92 AsO2- 4,17
Cd2+ (сульфат) 3,71
Си2+ (сульфат) 2,86 CN" 2,6
Вег+ (хлорид) 1,35 Se2~ 2,37
Aso; 0,95
Свойства приведенных ниже токсичных неорганических соединений известны из курса неорганической химии, поэтому достаточно их перечислить. При известных обстоятельствах эти соединения могут представлять опасность в качестве диверсионных ядов.
Соединение элементов I группы периодической системы элементов. Хлористый литий. Хотя это соединение н не обладает высокой токсичностью, однако при употреблении вместо поваренной соли может вызвать тяжелые отравления. Чрезмерное потребление Li+ нарушает баланс между катионами Na+ и К+.
Соли двухвалентной меди. Сульфат меди CuSO4 смертельно ядовит для людей в дозах 10—20 г (перорально). Так же ядовит цианид меди, другие ее соли менее ядовиты. Соли меди лишь медленно резорбируются в кишечнике. Отравления обычно приводят к рвоте, в результате чего большая часть невсосавшейся соли выводится.
Соединения эле ментов II группы периодической системы. Соли бериллия. Катионы бериллия — самые токсичные из катионов. В качестве ядов пригодны хлорид и ацетат. Нитрату хотя и растворим в воде, из-за кислой реакции раствора, пожалуй, не стоит рассматривать. Ацетат бериллия уже в малых дозах опасен. Он вытесняет магний и кальций из их соединений с протеинами. Отравления смертельны в 20% случаев.
412
Соли бария. Вследствие хорошей растворимости в воде из солей бария отравления вызывает только хлорид, и иногда еще нитрат и нитрит- Растворимые соли бария быстро резорбируются в кишечнике. Смерть может наступить уже через несколько часов. Дозы в 0,2—0,5 г приводят к тяжелым отравлениям, а доза в 0,8 г может быть смертельной. Безусловно смертельной является доза в 2 г.
Менее ядовитый' карбонат бария по ошибке однажды был использован вместе с мукой для выпечки хлеба (!).
Соли кадмия. Раньше соли кадмия часто обнаруживали в виде примесей к пищевым продуктам, особенно в кислых фруктовых соках, хранившихся в консервных байках, содержащих кадмий. Иоиы кадмия действуют как сильные яды для ферментов. При пероральном приеме смертельны для людей количества в 50— 60 мг. Соли кадмия быстро резорбируются в кишечнике.
Соли ртути. Токсичны прежде всего соли ртути(II). При пероральном приеме сулема смертельна для люден в количестве от 0,2 до 1 г. Если HgCl2 принять на пустой желудок, то летальный исход отравления может наступить в течение 36 я, но отравление может продолжаться и до трех недель, после чего наступит смерть.
Соединения элементов III группы периодической системы элементов. Соли таллия;. Из солей таллия большой проникающей способностью (даже через кожу) отличаются хлорид, ацетат и сульфат. Соли таллия уже неоднократно применяли в преступных целях. Для сульфата таллия летальная доза при пероральном приеме составляет для людей около 1 г. Известны случаи, когда смертельными оказывались дозы в 8 мг}кг, а также в Ю—15 мг{кг. Соли таллия являются клеточными ядами. Отравление продолжается несколько недель (2—3 недели), причем через 3—4 суток после приема яда наступает мнимое хорошее самочувствие (!).
Соединения элементов IV группы периодической системы элементов. Соли свинца. Соли свинца легко резорбируются через слизистые оболочки полости рта, дыхательных путей, пищеварительного тракта и даже через кожу. С белком оии образуют нерастворимые комплексные соединения. Б больших дозах соли свинца безусловно токсичны. Для ацетата свинца летальная доза при пероральном приеме составляет 5—30 г; дозы в 2—3 г ведут к тяжёлым отравлениям.
Соединения элементов V группы периодической системы элементов. Соединения мышьяка. Хотя все соединения мышьяка ядовиты, в качестве диверсионных ядов наибольшую опасность представляют только трехокись мышьяка и мышьяковистая кислота, иногда еще мышьяковая кислота, последние в виде своих солей (арсенитов и арсенатов). Соединения мышьяка как яды были «в моде» долгое время. Для людей смертельное количество AS2O3 при приеме через рот составляет 60—120 мг. Отравляющее действие зависит от размеров частиц яда. Арсениты
413
более токсичны, чем арсенаты. Производные мышьяка сравнительно быстро резорбируются в пищеварительном тракте. Они воздействуют на определенные ферментные системы и поражают капилляры.
В зависимости от количества яда отравление развивается в течение от ЗОлшн до 3 ч. Смерть от остановки дыхания наступает в течение 3 суток.
Соединения сурьмы. Эти соединения действуют так же и почти так же ядовиты, как и соединения мышьяка. Доза рвотного камня в 100 мг — смертельна.
Неорганические соединения других элементов, за исключением . фторидов, цианидов н нитритов, пожалуй, не имеют значения в качестве диверсионных ядов. Хотя соли никеля и кобальта более ядовиты, чем соли бария и свинца, из-за своей бросающейся в глаза окраски они для отравлении непригодны.
Несомненно, что самыми ядовитыми из солей являются цианиды. Цианиды щелочных и некоторых других металлов хорошо растворимы в воде (щелочная реакция).-По своей токсичности онн соответствуют синильной кислоте. Для людей смертельны ПО мг NaCN или 115 мг KCN. При сильных отравлениях смерть наступает через 5 мнн. Фториды существенно нарушают кальциевый баланс (осаждение кальция). Уже 250 мг фтористого натрия при пероральном приеме приводят к тяжелым отравлениям, количества около 3—4 г должны быть смертельными. Примерно такие же летальные дозы приводятся н для нитрита натрия.
В литературе известно много случаев отравлений этими н другими неорганическими соединениями, отравления были порой сознательными, порой ' случайными. В специальной литературе по токсикологии описано много случаев отравления.
ЛИТЕРАТУРА
1. S a u n d е г s В. С., S t а с е у В. J., J. Chem. Sex:., 1948, 1773.
2. Nature, 201, 611 (1964).
3. С h е п о w е t h М. В., Pharm. Rev., 1, 383 (1949).
4. Bergmann Е. О., Blank S., J. Chem, Soc., 1953, 3786; Гудлнцкий M„ Химия органических соединений фтора, М., Госхимиздат, 1961.
5. J. Am. Chem. Soc., 70, 3604 (1948).
6. Вестник МГУ, 1959, 65.
7. Sea ger L. D., Wood Ch. D., Proc. Soc. exp. Biol. Med., Ill, 120 (1962); C 1963 15513
8. В e h r 1 n g ё r H., Angew. Chem., 56, 83 (1943).
9. M a r k i F., W i t k о p B„ Experiment, 19, 329 (1963).
10. Ghiretti F.. Angew. Chem., 76. 982 (1964).
11. Nuki B., Naguno M., Folia pharm. Japan. 55, 1305 (1959); C., 1964, 16-1413.
12. Skele E., Acta pathol. microblol. Scand., 55, 166 (1962),
13. R о s e b u г у T., Frieden oder Pest, New York, 1949.
14. Angew. Chem., 74,35 (1962).
15. C. r. hebd. Seance Acad. Sci.. 256, 1043 (1963).
16. C. r. hebd. Seance Acad. Sci., 257, 791 (1964).
414
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
О’Брайн, Токсичные эфиры кислот фосфора, пер, с англ., М., «Мир», 1964.
В ii s с h е г Н,, Griin- und Gelbkreuz, Leipzig, 1932,
F i n k Z, u. a„ Der Gesundheitschutz gegen chemische Kampfstoffe, Berlin, 1962, Hauschild F„ Pharmakologie und Grundlagen der Toxikologie, Leipzig, i960. Lohs K., Synthetische Gifte, Berlin, 1963,
Matoulek J„ Tomecek 1,, Analyse synfhefischer Gifte, Berlin, 1965. Moeschlin S., Klinik und Therapie der Vergiftungen, Stuttgart, 1963.
M u n t s c h O„ Leitfaden der Pathologic und Therapie der Kampfstofferkran-kungen, Leipzig, 1941.
S a r t о г i M, F,, Die Chemie der Kampfstoffe, Braunschweig, 1940,
Саундерс Б„ Химия и токсикология органических соединений фосфора и фтора, перев, с англ, с доп. Н, А. ТТошаткина и В, В, Смирнова, М,, «Мир», 1961.
Шрадер Г., Новые фосфорорганические инсектициды, перев, с нем,, М,, «Мир», 1965,
Schrader Gu Die Entwicklung neuer Insektizide auf Grundlage organischer Fluor- und Phosphorverbindungen, Weinheim, 1951.
S t a de K., Pharmakologie und Klinik synthefischer Gifte, Berlin, 1964, Stohr R,, Die chemischen Kampfstoffe, Berlin, 1961.
i
ПРИЛОЖЕНИЯ
ПРИЛОЖЕНИЕ t. МЕТОДЫ РАБОТЫ С ХИМИЧЕСКИМИ ОВ
Работу с химическими ОВ можно проводить только по соответствующему разрешению и в оборудованных для этой цели лабораториях, В установленном порядке надлежит ознакомиться и организовать соответствующие меры безопасности в лаборатории, К работам такого рода допускаются только лица, которые обладают необходимыми знаниями о токсических, химических и физических свойствах этих ОВ, имеют опыт препаративной и аналитической работы и владеют методами защиты и оказания первой помощи.
Перед началом работ практикантов нужно проэкзаменовать по указанным выше вопросам. Их следует практически обучить мерам безопасности. Число практикантов определяется, исходя из наличия свободных мест, С одним ответственным лицом должны работать не более пяти человек. Как правило, нужно привлекать в качестве наблюдателя еще одного человека, не занятого непосредственно практической работой. Наблюдатель должен обращать внимание на соблюдение техники безопасности и в случае аварий и отравлений принимать немедленные меры,
Все работы с химическими ОВ н веществами сходного действия следует проводить в вытяжном шкафу. При работах с фосфорорганическими, свинецоргани-ческими соединениями и кожно-нарывными ОВ следует обязательно одевать защитную одежду. Защитную одежду, перчатки и другие части одежды прн их загрязнении следует немедленно, а при длительных работах — время от времени сменять и дегазировать.
При препаративных работах применяют обычные меры предосторожности. При получении ингаляционных ядов следует надевать противогаз.
Перед началом работ нужно приготовить в подходящих сосудах соответствующие растворы, необходимы^ для дегазации защитной одежды, лабораторной посуды и т. д. Всегда нужно применять свежеприготовленные дегазирующие растворы.
Следует безусловно избегать заражения рабочего места, защитной одежды и занесения яда в помещение или наружу. В необходимых случаях при заражении нужно проводить соответствующие мероприятия по дегазации. Остатки ядов, зараженные материалы и т. п. следует собирать в соответственно обозначенные емкости и дегазировать.
После снятия защитного снаряжения все практиканты должны пройти санитарную обработку: рукя надо мыть соответствующими дегазирующими средствами и теплой водой с мылом; в некоторых случаях требуется обмывание всего тела (ванна, душ).
Нужно контролировать состояние здоровья практикантов перед, во время и после практикума. В зависимости от продолжительности и объема практикума и особенно при работах с высокотоксичными ядами (фосфорорганические, психохимические вещества и производные фторкарбоновых кислот) нужно через определенные промежутки-времени проходить врачебный осмотр,
ПРИЛОЖЕНИЕ 2. ВАЖНЕЙШИЕ ДЕГАЗИРУЮЩИЕ РАСТВОРЫ, ПРИМЕНЯЕМЫЕ В ЛАБОРАТОРНЫХ УСЛОВИЯХ
Название ОВ
Фосфорорганические соединения
Галогенированные тио-
эфиры
Галогенированные тре-
тичные амины
Галогенированные алифатические арсины
Галогенированные ароматические арсины
Галогенированные кетоны, нитроалканы, бромбензилцианид
Цианпроизводиые
Дегазирующий раствор
10%-ный раствор NaOH в 30%-ном метаноле нли . в.воде с добавкой 1% эмульгатора; 10%-ный водный или водно-спиртовой раствор аммиака
10%-ный водный или водно-спиртовой раствор монохлорамина; 10%-ный раствор дихлорамина в дихлорэтане
20%-ный раствор дихлорамияа в дихлорэтане; насыщенный раствор тиосульфита натрия
20%-иый водный или водно-спиртовой раствор NaOH
5%-ный раствор перманганата калия; 10%-ный раствор НгОг; концентрированная HNO3
20%-ный раствор сульфида натрия с добавкой спирта или 1% эмульгатора
10%-ный раствор NaOH с добавкой FeSO4 и эмульгатора
ПРИЛОЖЕНИЕ 3. МЕРЫ ПЕРВОЙ ДОВРАЧЕБНОЙ ПОМОЩИ ПРИ ОТРАВЛЕНИЯХ ХИМИЧЕСКИМИ ОВ
При всех авариях и отравлениях нужно тотчас вызывать врача! Действовать быстро, первую помощь оказывать немедленно! Не в каждом случае степень отравления можно оценить сразу. В случае высокотоксичных соединений речь идет о жизни пострадавших.
Все перечисленные ниже меры являются только первой помощью и никоим образом не заменяют врачебного лечения. Для ^уменьшения степени поражения нужно принимать следующие немедленные меры;
удаление из зараженного помещения, снятие зараженной одежды, удаление яда с поверхности тела, надевание средств защиты и т. п.
Отравления фосфорорганическими соединениями. Искусственное дыхание.
Кожу — промыть 5—10%-ным раствором NaHCO3 или аммиачной водой, обработать дегазирующей мазью или раствором, содержащими хелат меди.
Глаза — промыть 2—3%-ным раствором NaHCO3, затем впустить 1—2 капли "раствора гоматропина, содержащего 0,4% новезина.
Желудок, — промыть желудок, принимать в больших количествах активированный уголь.
Отравления фторорганическими соединениями. Вызвать рвоту, опорожнить желудок, полоскать горло и полость рта раствором перманганата калия, внутрь — крепкий раствор винного спирта.
Отравления цианидами. В течение 2 мин (до 8 раз) через 30 сек давать вдыхать амилнитрит, сделать искусственное дыхание. Внутрь — крепкий кофе. Грелка.
Кожу — промыть 5—10%-ным раствором NaHCOg.
Желудок — вызвать опорожнение желудка, напоить смесью из 2 г FeSO4 и 20 г MgO в 100 мл воды.
Отравления фосфористым и мышьяковистым водородом. Вдыхание кислорода. В случае отравления РН3 — первак помощь такая же, как при отравлениях цианидами; в случае отравления AsH3 — 3 раза в час принимать по 1,5—3 мг/кг массы дитиоглицерина.
417 .
Отравления удушающими ОВ. Полный покой в положении лежа на спине, грелка, можно давать горячее питье, внутрь — 3 г гексаметилентетрамина (уротропина). Вдыхание кислорода без давления, для снятия раздражения — вдыхание эфира, ментола и т. д.
Отравления тетраэтилсвинцом. Поверхность кожи протереть керосином. Большие дозы витамина Вь
Отравления кожно-нарывными ОВ. Кожу — обрабатывают растворами моно-хлорамина, перманганата калия или перекиси водорода (окунания, влажные повязки), в случае иприта также раствором гексаметилентетрамина. Для ванн применять 1%-ный раствор перманганата калия, затем вымыть тело теплой водой с мылом.
Глаза — обрабатывают 2%-ным раствором NaHCO3; 3%-ной борной кислотой; показаны щелочные глазные мази.
Органы дыхания — горло полоскать раствором гексаметилентетрамина или перманганата калия, вдыхать вещества, облегчающие дыхание.
Желудок — принимать в больших количествах активированный уголь.
Отравления раздражающими ОВ. Глаза — обработать 3—5%-ным раствором NaHCOs или 3%-ным раствором борной кислоты; показаны щелочные глазные мази.
Органы дыхания — полоскание горла щелочными растворами, ингаляция веществ, облегча’ющих дыхание (ментол, эвкалиптовое масло), полоскание горла небольшим количеством коньяка.
ПРИЛОЖЕНИЕ 4. ЛЕТАЛЬНЫЕ КОНЦЕНТРАЦИИ НЕКОТОРЫХ ОВ
Звездочкой отмечены данные для ЛКэо
Название ОВ Подопытные животные Концентрация Л Као. мг/л Экспозиция, мин Время до наступления смерти
Диизопропилфторфосфат Мыши 0,44* 10 2 <4
(ДФФ) Мыши 2,7* 2 2 Ч
Мыши 5* 1 2 ч
Крысы 0,36* 10 2 ч
Крысы 1,8* 2 2 «
Крысы 4,2* 1 2 ч
Диметилфторфосфат Мыши 0,3* 10 6 ч
Дициклогексилфторфосфат Мыши 0,11* — —
Крысы
Кролики 0,14* ——* —
Диэтнлфторфосфат Мыши 0.5* 10 1 ч
Зарии Мыши 0,005 30 —
Крысы 0,03* — —
Иприт Собаки 0,01 Непрерывно 8 ч
Собаки 0,5 Непрерывно 5 мин
Синильная кислота Кошки 0,2 Непрерывно 5—10 мин
Кошки 0,35 Непрерывно Мгновенно
Собаки 0,2 Непрерывно 5—10 мин
Собаки 0,35 Непрерывно Мгновенно
Обезьяны 0,2 Непрерывно 5—10 мин
Обезьяны 0,35 Непрерывно Мгновенно
Табун Мыши 0,02* 30 —
Фосген Крысы 0,2—0,3* 30 14 суток
Кошки’ 0,05 20 —
Кошки 0,1 20 —
Собаки 0,3-0,4 30 24 и
Хлорпикрин Кошки 0,8 20 14 суток
Кролики 0,8 20 3 суток
Кролики 5 Непрерывно 30 мин
Хлорциан Мыши 0,7 7,5 —
Крысы 1,4 10
Собаки 0,8 7,5 —
Козы 2,2 7-10 —
Козы 5,1 1-2
419
ПРИЛОЖЕНИЕ 5. ЛЕТАЛЬНЫЕ ДОЗЫ (ЛД50) НЕКОТОРЫХ ОВ И ЯДОВ
Название ОВ Подопытное животное Способ введения * мг!кг
Азотистый иприт Мыши ч/к 7
Мыши п/к 2
Крысы ч/к 4,9
Крысы я/в 0,7
Собаки ч/к 1
Аконитин Крысы в/в 0,11
Амитон [О, О-диэтЯЛ'Б-(2-этила- Крысы п/о 3,5
миноэтил)-тнофос фат] Мыши в/б 0,5
Атропин Мыши п/о 400-800
Мыши п/к 900
Мыши в/в 90
• Крысы п/о 750
Буфотоксин Кошки в/в 0,27-0,41
Диизопропилфторфосфат (ДФФ) Мыши п/к 5
Мыши в/в 0,4
Мыши п/о 36,8
Крысы в/м 1,8
Крысы п/о 7,7-13,5
Собаки в/в 3,4
Собаки п/к 3
Козы в/в 0,8
Козы п/к 1
Обезьяны в/в * 0,25±0,3
Обезьяны п/к 1
N, N-Диэтиламидо-О’Этилцианфос- Мыши в/б 4
фат
О, О-Диэтил-бис (М, N-диметил- Мыши в/б 4,7-4,9
амидо)-пирофосфат, асимметрия- Крысы в/б 2,4-2,7
ный Крысы п/о 3,8
То же, симметричней Мыши в/б “ 17
Крысы в/б 10-11,5
Крысы ( п/о 12,4
О, О-Диэтил-S- (2-триэтиламмоний)- Мыши в/б 0,17
тиофосфат ио ди ст ый
О, О-Диэтоксифосфорилтиохолин Мыши в/б 0,14
Мыши п/к 0,26
Кролики в/в 0,31
Зарин (О-изопропилметилфторфос- Мыши п/к; в/в, в/б п/к 0,05-0,2
фон ат) Мыши 0,2
Крысы 0,12
Крысы в/м 0,17
Крысы в/в 0,05
Крысы п/о 1 ОД
420
Продолжение
Название ОВ Подопытное животное Способ введения * марсе
Зоман (3,3-О-диметилизобутилме- Мыши п/к 0,04-0,1
тилфторфосфонат)
Иприт (бис-2-хлорэтиловый тио- Мыши ч/к 92 **
эфир) Мыши и/к 26 **
Мыши в/в 8,6 **
Крысы ч/к 18
Крысы в/в 0,7 **
КОКОН ЯД Мыши — 0,2-10-3(л(кг/«г)
М-Метил-бис-М, N (2-хлорэтил)-амин Мыши п/к 2,6 •
Мыши в/в 2
Мыши ч/к 29
Мыши п/о 20
Крысы п/к 1,9
Крысы в/в 1,1
Крысы ч/к 22
Крысы п/о 10
Метилфторфосфорилгомохолин Мыши, кролики Мыши в/в в/б 0,006 0,05
Метилфторфосф о рил- 2- метилхолин Мыши в/б 0,03 (0,07)
Мыши, кролики в/в 0,008
Метилфторфосфорилхолин Мыши, кролики в/в 0,01
Мыши в/б 0,1
Метокси-4-нитрофеноксиметилфос- Крысы п/о 1
финсульфид
Никотин Мыши в/в 7,1
Мыши п/о ***
Собаки в/в 5
Параоксон [дИэтил-(4-нитрофеннл)- Мыши п/к 0,6—0,8
фосфат] Крысы п/о 3
Кролики ч/к 5
Паратион [диэтил-(4-нитрофенил)- Мыши в/б 5,5
тионфосфат] Мыши п/о 25—26
Крысы в/б 4—7
Крысы п/о 4—13
Лошади п/о б
Табун (N, N-диметиламидо-О-этил- Мыши п/к; в/б 0,6
цианфосфат) Мыши в/в 0,15
Крысы п/к 0,35
Крысы в/в 0,06
Крысы п/о 3,7
Собаки в/в 0,08
Собаки п/о 0,2
421
Продолжение
Название ОВ Подопытное животное Способ введения * ма/ка
Т и мет [ О, О - ди этил- S - (этилтиоме- Крысы ч/к 2,5—6
тил)-тнонфосфат] Крысы п/о 1,1-2,3
Тиолсистокс [О, О-диэтил-S-2-(этил- Мыши п/к 6
тиоэтил)-тиофосфат] Крысы п/о 1,5
Тионсистакс [О, О-диэтил-3-2 (этил- Мыши п/к 15
тибэтил)-тионфосфат] Крысы п/о 30
Тетраэтилсвинец Крысы в/б 10 ***
Кролики в/в 22-47
Трехокись мышьяка Крысы п/о 138±13
Собаки п/о 30-70
Тетраметилпйрофосфат Мыши в/б 1,7
Тетраэтилпирофосфат Мыши в/б 0,85
Мыши п/о 7
Крысы п/о 1,2-2
Кошки п/к 2,5-3
Фторацетат натрия Крысы п/о 6,9
Србади п/о 0,97
Овцы п/о 0,7
Лошади п/о 1
Хлорпикрин Кошки п/к 10
Кролики в/в 104***
Хлорциан Мыши п/к 39
Крысы п/к 20
Хлористый триэтилсвинец Крысы в/б 5***
М-Этил«бис-Ы, N (2-хлорэтил)-амин Мыши ч/к 13
Мыши п/к 1,2
Крысы ч/к 17
Крысы п/к 1
Этоксиметилфосфорилтиохолин Мыши в/б 0,03
О-Этил-S-(2-этиламиноэтокси)" метил-тиофосфиноксйд Крысы п/о 0,25
0-Этил-3-(2-этиламиноэтокси)-этил-тиофосфиноксид Крысы п/о 0,25
в/в —внутривенно; в/б — внутрибрюшинно; в/м —внутримышечно; п/о — перорально; п/к — подкожно; ч/к —через кожу (кожнореэорбтивно),
** — с растворителем;
*** —минимальная летальная доза (МЛД),
422
ПРИЛОЖЕНИЕ 6. ФИЗИЧЕСКИЕ свойства некоторых ов
В круглых скобках приведено давление в ям рт. ст.
Наименование Бру тто-ф о рм у ла T. кип., °C T. пл„ °C я* d4 Давление пара, мм рт. ст. Макс, концентрация при 20 °C, мг/л 4
Бис-трихлорметиловый эфир С3С!бО3 , 205 (760) 78 1,6го 1 *
угольной кислоты
Бис-(2-хлорэтил)-сульфид C4H8C12S 217(760) 13,5-14,5 1,274го 0,115го 0,63 1,5313эа
(иприт)
2,2'-Бнс-(2-хлорэтилтио)-диэ-тнловый эфир 174 (2) от —30 до —38 1,2311го — — —
1,2- Бис- (2- хлорэтилти о)-этан CgHisClaSa 140 (2) 57 — — —
Бромацетофенон C8H?BrO 260 (760) 50-51 170915 —— —
Бромбензилцианид C8H6BrCN 242 (760) 25—26 1,516го 0,012го 0,13 —
Бромпропанон C3H6BrO 136(760) -54 1,634го 9го 75 ——
Бромциаи BrCN 61,3 (760) 52 2,02го 120го 1500 —
Диизопропилфторфосфат C6HBFO3P 183 (760) -82 1,0611го 0,97 (0,57го) 5,6 (9,7) 1,3814го
Дигидрофенарсазияхлорид Ci2H9C1NAs ' 410(760) 195 1,648го 2- 10-17 2-10-5 —
Дифенилхлорарсин Cl2Hi0ClNAs 333 (760) 39 1,422го 5- 10-6 7- 10“4 1,63325»
Дифени лци ана рсии C13HioNAs 346 (760) 32-35 1,45го 2- 10~4 1,5- 10-4 1,6092го
Дихлорформоксим (фосфоге-иоксин) Зоман CHClzON 129 (760) 39-43 — 20—25 —
CjHibFOsP 190 (760) —80 1,0131го 0,92 10 1,4080го
О-Изопропилметилфторфос- C4HiqFO2P 147,3 (760) -57 1,0941го 1,48 11,2 1,3830го
фонат (зарин)
О-Изопропилэтилфторфосфо-и ат М-Метил-бис-М,М-(2-хлорэтил)- амии C5HJ2FO2P CsHnCkN 67-68 (18) 71 (9) от —65 до —60 1,381726 1,130го 0,95го 8 1.381726
Метил дихло рар сии CH3CI2As 132,5 (760): -42,5 1,8359го 10,S325 74,4 1,5677й*5
Метилфторфосфорил- 2-метил-холин CeHjSFO2NP 4® (0,1) 84 1,0625 — 1,4150» 1 ..
Наименование Брутто-формула
Метилфторфосфорилхолин Монохлорформоксим Морфолид пеларгоновой кислоты Мышьяковистый водород Пятифтористая сера Синильная кислота Табун CsH^FOi-NP CH2C1ON Ci 3H25O2N АзНз sf5 HCN C8HnO2N2P
Тетраметилсвинец Тетраэтилсвинец Трис-(2-хлорэтил)-амин Трихлорметиловый эфир хлоругольной кислоты Т рихлорнитрометан Фенилдихлорарсин фосфористый водород Хлорангидрид угольной кис-. лоты Хлорацетофенон 2-Хлорвинилдихлорарсин (цис~) 2-Хлорвинилдихлорарсин (транс-) Хлортрифторид Хлорциан N-Этил-бис-Ы, N-(2-хлорэтил -амнн) Этнлдихлорарсин C4H12 C&HjflPb C6H12C!3N C2CI4O2 CC13O2N CeHsClaAs PH3 CC13O CaH7C10 C2H3C13As C2H2C13As C1F3 C1CN C6HJ3C12N CaHsClaAs
Продолжение
Т. пл,, °C 4 Давление пара, мм рт, ст. Макс, концентрация при 20 °C, лсг)л nD
152 1,1425 1,4130^
60 - — __ —
0,96525 — — — 1,468425
— 113,5 — ~ —
—92 2,08° — —
-14 0,696918 612 873 1,2675го
-48 1,0823го 0,073го 0,6 1,4250го
—30,3 1,995го —
— 130,2 U52920 0,261го 4,6 1,5195го
-4 1,2348го 0,007 0,07 1,4957го
-57 1.64415 10,3 1-20 1,4566га
-69,2 1,6579го 16,91 184 —
—20 1,654го 0,01415 0,4 —-
-133,8 —
-118 1,38 (жидк.) 1173го 6370 —
58,5 1,321го 0,013го 0,105 1,5492е6
-44,7 1,8598 23 1.56225 1,589825
—2,4 1.879323 0,425 — 1.607625
-80-83 1,840315 —
-6 1,222° 1002го 3300 —
-34,4 1,086123 — 1,4653гз
-65 1,6595го 2,2921'5 20 L558814,5
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Адамсит (Дигидрофенарсазинхлорид, Фенарсазин хлорид, 10-Хлор-9,10-ди-гидрофенарсазин) 24, 25, 29, 92 сл., 106
биологическая активность 83, 86
стойкость 40
токсичность 93, 97, 98
физические свойства 93 химические свойства 94 сл. хранение 45
Азотистый иприт [Трис-(2-хлорэтил) -амин, 2.2/.2//-'Грихлорэтиламнн, ₽,₽/,₽"-Трихлорэтиламин] 53, 62, 137, 170, 175 сл.
боевое применение 209 дегазация 183, 184 получение 175
признаки отравления 187, 188
свойства 176 сл., 178 сл, токсичность 53, 62, 177, 420 Аконитин 400, 420 Алкалоиды 395 сл.
кураре 397
спорыньи 358, 359
чилибухи 396
О-Алкилалкилфторфосфонаты 293 сл.
М-Ллкил-N.N -бис-(2-хлорэтил) -амины см. Иприты азотистые
Алкилдихлорарсины 190 сл.
биологическая активность 82 сл., 86 сл., 191 сл.
свойства 195 сл.
токсичность 203, 417
N-Алкил-З-пиперпдилбензилаты 368
Алкилфосфорилтиохолины 393 сл.
Алкилфторфосфорилхолины 318 сл. токсичность 323 сл.
Алкоксиалкилфосфорилхолины 318
Аманитины 401
Амидазол-4-акриловая кислота, холиновый эфир (Мурексин) 404, 405
Амины галогенированные 170 сл., 417
Амитон см. Тетрам
Анизатнн 400
Арсин см. Мышьяковистый водород Арсины галогенированные 82, 87 см. также Алкилдихлорарсины Атропин (Тропиновый эфир троповой кислоты) 398 сл., 420
Барий, соли 412, 413
Безопасная концентрация ОВ 49
Бензилбромид 67, 80
Бензилиодид 80
Бензилхлорид 80
Бериллий, соли 412
Бис-2-бромэтиловый тиоэфир 167
Бис- (N.N-диметиламидо) -фторфосфат см. Димефокс
Бис-2-иодэтиловый тиоэфир 167
Бис- (3-метилбутил) -фторфосфат 264
Бис- (4-м етил изопентил) -фторфосфат 264, 265
Бис- (2-метилииклогексил) -фторфосфат 265
Бис-трихлорметиловый эфир угольной кислоты см. Трифосген
Бис- (триэтилсвинец) -фторфосфат 102
Бис-2-фторэтиловый тиоэфир 166, 167
1,2-Бис-(2-фторэтнлтио)-этан 170
Бис- (2-хлорвннил) -хлорарсин 190
Бис-(хлорметил)-формоксим 205
Бис-2-хлорэтиловый тиоэфир см. Иприт
Бис-(2-хлорэтилтио)-алканы 168 сл.
2,2'-Бис- (2-хлорэтилтио) -диэтиловый эфир («Иприт кислородный») 154, 167, 168, 423
1,2- Бис- (2-хлорэтилтио) -этан (Сескви-ипрйт) 168, 170, 423
Бладан см. ТЭПФ
Боевая концентрация ОВ 37, 58
Боевое применение ОВ 40 азотистого иприта 209 арсина 244 ботулинического токсина 409 бромбензнлцианида 105, 106 V-газов 326, 327
425
Боевое применение ОВ галогеноксимов 211 галоген фторидов 133 зарина 326, 327 иприта 208 сл. кожно-нарывных 208 сл. люизита 209, 210 метнлдихлорарейна 208 обще ядовитых 243, 244 окиси углерода 244 . психоядов 369 раздражающих 105, 106 синильной кислоты 243 табуна 290, 291 тетраэтилсвинца 244 удушающих 133 сл. фитоядов 381, 382 фосгена 133, фосфористого водорода 244 фосфорорганических 325 сл. хлорпикрина 133 хлорциана 243 этилдихлорарсина 208
Ботулизм 407 сл.
Ботулинический токсин 407 сл.
Бромацетон (Бромпропаион) 67, 69 сл., 80
получение 69 токсичность 70, 423 Бромацетофенон 76, 80, 423 получение 69
Бромбензилцианид 77 сл. боевое применение 105, 106 дегазация 81, 107, 417 токсичность 67, 79, 80, 423 1-Бромбутанон-2 69 сл., 80 Бромкетоны 69 сл.
«Бромлост» 167
Бромпропаион см. Бромацетон Бромуксусная кислота, этиловый эфир 80
Бромциан 228 сл.
токсичность 80, 229, 231 сл., 423 Бруцин 396, 397 Буфагины 404 Буфотенины 404 Буфотенин 381, 362, 403 Буфотоксин 403, 404, 420
Вератрин 401 Винсенит 218
Габеров фактор смертельности ОВ «Ве»-Газ 69
V-Газы 42, 48, 62, 298, 315, 316 боевое применение 326, 327 защита 328
Галлюциногены 356, 357, 359
426
Галогенангидриды угольной кислоты 113 сл.
Галогенарсины 82, 87, 190 сл.
Галогенфториды 131 сл.
Галогенцианы 228 сл.
биологическая активность 218 СЛ.
признаки отравления 221, 231
свойства 229 сл.
токсичность 231, 232
Гексахлоран 52
Гексахлор дим етилкарбонат см. Трифосген
Гексоген 52
Гербициды 373, 377, 378, 380
Г етер оа уксин (Ин дол и л -3 -у ксусна я
кислота) 379
Гибберелииы 379
Гидразид лизергиновой кислоты (LSH) 360
(—)-Гиосциамин 398 сл.
Глазные яды 173
Гоматропин 401
Гомомартониты 69
Горчичный газ см. Иприт
Горшечный кураре 396
Горючие смеси 105
2,4-D (2,4-Дихлорфеноксиуксусная
кислота) 371, 374
Дегазация ОВ 25, 36, 37, 44, 46, 417
азотистого иприта 183, 184, 417
бромбензилцианида 81, 107, 417 бромкетонов 70
галогенированных аминов 183, 184, 417
арсинов 417
кетонов 417
нитроалканов 129, 417
оксимов 214
тиоэфиров 417
гексахлораминов 36
дифениларсинов 91
ДФФ 283
иприта 44, 160, 162 сл., 417
кожно-нарывных 213, 214
люизита 200
мышьякорганических 417 общеядовитых 245, 246 раздражающих 107 тетраэтилсвинца 242, 245,
246
удушающих 134
фосгена 117, 118
фосфорорганических 328, 417
хлорпикрина 129, 417
О,О-Диалкилалкилфосфонаты 251, 265 N,N -Диалкиламидо-О-алкилацилфо-сфаты 251, 284 сл.
М.М-Диалкиламидо-О-алкилцианфо-сфаты 252, 286 сл.
Диалкиламидофторфосфаты 286
Диалкиламидоцианфосфаты 286 Диалкилацилфрсфаты 251, 263 сл. Диалкилфторфосфаты 252, 264 сл. биологическая активность 264 сл.-получение 266, 267 свойства 267 сл., 423 токсичность 284
Диалкоксифосфорилхолины 317
Диамидофторфосфаты 284, 285
Дибромдиметиловый эфир 80
1,3- Дибром пропан он 68
Дибромформоксим 205
Диверсионные яды 383 сл., 413
С-Дигидротоксиферин 397
Дигидрофенарсазинхлорид см. Адамсит
Диизобутилфторфосфат 265
Диизопропилфторфосфат (ДФФ) 56, 57, 248 264* 265, 271 сл.
гидролиз 275 дегазация 283 получение 271 признаки отравления 284 свойства 273 сл.
смеси с ипритом 154 274 токсичность 273, 283, 419, 420, 423
Р,р-Диметилакр ил оил холин 405
М.М-Диметиламидо-О-этилфторфо-сфат 285
М.М-Диметиламидо-О-этилциаифо-сфат см. Табун
3-(р-ДиметиЛаминоэтил) -индол (N,N-Диметилтриптамид)
3,3-0-Дим етилизобутилм етилфтор -фосфонат см. Зоман
Диметил-(1-карбметокси-1-пропен-2-ил) -фосфат (Фосдрин) 349
М,М-Диметилтриптамид 361 Диметилфторфосфат 267, 419 Димефокс [Бис- (N.N-Диметилам и до) -фторфосфат, Пестокс 14, CR-409, Ханан] 285, 348, 349
4,6-Динитро-о-крезол (4,6-Динитро-2-метилфенол) 378
2,4-Динитрофенол 378
Ди-З-пентилфторфосфат 264
Ди -н-лр опил фтор фосф ат 264
Дисистон [Дитиосистокс, О,О-Диэтил-S-2- (этилтиоэтил) -тионфосфат, О.О-Д иэтил-5-2- (этилмеркаптоэтил)-дитиофосфат] 339 слГ
Дитион (Сульфо-ТЭПФ, Сульфотеп, О.О.О'.О'-Тетраэтилдитион-пирофосфат) 255
Дитиосистокс см, Дисистон
Дитран 62, 83, 368, 359
Дифен илам ин хлора ррин 423
Дифенилбром(фтор)арсин 92
Дифенилфторфосфат 265
Дифенилхлорарснн 40, 55, 63, 82, 83, 85, 87 сл., 97, 98, 106, ‘423
Дифенилхлорстибин 92
Дифенилцианарсин 24, 83, 87 сл., 97, 98, 423
Дифеннлцианстибин 92
Дифосген биологическая активность 110 сл. боевое применение 133 признаки отравления 112, 113 свойства ПО, 113 сл., 120 сл. стойкость 27 токсичность 121 сл., 424 хранение 121
Дифосфорная (пирофосфорная) кислота, производные 253, 254 смл-Дифтор дихлор динитроэтан 131 Дифторхлорнитрометан 130
Дихлорангидрид угольной кислоты см. Фосген
Дихлордиметиловый эфир 80
2,2'-Дихлордиэтиловый тиоэфир (2,2'-Дихлордиэтилсульфид) см. Иприт
N- (3,4-Дихлорфенил) -N'-н-пропил-N'-метилмочевина 377
2,4-Днхлорфеноксимасляная кислота 375
2,4-Дихлорфеноксиуксусная кислота (2,4-D) 371, 374
хлорбутениловый эфир (Кроти-лин) 375
Дихлорформальдоксим (Дихлорформоксим) см. Фосгеноксим 2,4-Дихлор-6(5) -фторфен оксиуксус-ная кислота 375
1,2-Дихлорэтан, допустимая концентрация 52
Дициан 219
Дициклогексилфторфосфат 265, 419
Диэтиламид лизергиновой кислоты (LSD, LSD-25) 62, 63, 359, 360
N,N-Диэтил амидо-О-этилциан фосфат 420
О.О-Диэтил-бис- (М,М-диметиламидо-пирофосфат) 420
Диэтилдив ин ил свинец 243
О.О-Диэтил-S- (2-диэтил аминоэтил) -тиофосфат см. Тетрам
симм-О, О-Диэтил-S, S-диэтилдитио-пирофосфат 254
Диэтил-4-нитрофенилтионфосфат 255 О,О-Диэтил- (4-нитрофенил) -тионфосфат см. Паратион
О.О-Диэтил- (4-нитрофенил)-фосфат см. Параоксон
427
О,О-Диэтилтионфосфорная кислота 255
Диэтилфторфосфат 419
О,О-Дяэтил-5- (этилмеркантометил) -дитиофосфат см. Тимет 0,0-Диэтил-5-2- (этил меркаптоэтил) -дитиофосфат [0,0-Диэтил-о-2-(этилтиоэтил)-тнонфосфат] см. Дней ст он
О,О-Диэтил;5-2- (этилтиоэтил) -тиофосфат (Тиолсистокс) 422 0,0-Диэтоксифосфорилтнохолин 420 ДФФ см. Диизопропилфторфосфат Дымовые шашки ОВ 40
Е 600 см. Параоксон
Е 605 см. Паратион
Зажигательные средства 131
Заражения плотность 37
Зарин (О-Изопропилметилфторфос-фонат) 24, 26, 42, 298 сл. боевое применение 326, 327 защита 328 летальная доза 419, 420 получение 295, 298 сл. признаки отравления 312 свойства 303 сл. стойкость 27, 40 токсичность 53, 55, 294, 303, 312 сл., 423
Защита от
V-газов 328 зарина 328 иприта 155, 212 кожно-нарывных 118, 211, 212 общеядовитых 244 сл. пснхоядов 369 раздражающих 106, 107 синильной кислоты 245 тетраэтилсвинца 245 удушающих 134 фосфорорганических 327, 328
Змеиные яды 402, 403
Зоман (3,3-О-Диметилизобутил-метилфторфосфонат, Метил-фторфосфоновой кислоты пи-наколиловый эфир, втор-О-Нео-гексилметилфторфосфонат) свойства 314 токсичность 55, 294, 315, 421, 423
О-Изопропялметилфторфосфонат ем. Зарин
0-Изопропилэтилфторфосфонат
(Этилзарин) 297, 298, 423
Изопропоксиметилфосфорилхолин 317
428
Ингаляционные яды 125, 132, 138, 164, 173, 194, 201, 219
Индол, производные 358 сл, 3-Ин долил-{}-диметил аминоэтан (М,М-Диметилтриптамид) 361
Индолил-З-карбоновые кислоты 379 Индолил-З-уксусная кислота (Гетероауксин) 379
Инсектициды фосфорорганические
военное значение 351 контактные 340 системные 329, 335 сл. Токсичность 336, 337
Иодуксусная кислота, этиловый эфир 80
Иприт [Бис-2-хлорэтиловый тиоэфир, Горчичный газ, 2,2'-Дихлор диэтиловый тиоэфир, 2,2'-Дихлор-диэтилсульфид, Бис- (2-хлорэтил)-сульфид, 1 -Хлор-2- (2'-хлорэтил-
тио)-этан] 136 сл., 144 сл, биологическая активность 140 боевое применение 208 сл. в смесях 31, 34, 35, 154, 277 гидролиз 41, 156 сл. дегазация 44, 160, 162 сл, защита 155, 212 «кислородный» [2,2'«Бис- (2-хлор-этилтио) -диэтиловый эфир 154 167, 168, 423 маркировка 144 окисление 44, 160 сл. получение 145 сл.
признаки отравления 165, 166 помощь при отравлениях 418
свойства 151 сл., 155 сл. стабильность 40, 42, 164 стойкость 27, 28, 152, 153 токсичность 51, 53, 55, 152 165 166, 193, 194, 419, 421, 423 хранение 45
Иприт азотистый см. Азотистый иприт
Иприт кислородный 154 167, 168, 423
Иприты азотистые [М-Алкил-М,Ы-бис-(2-хлорэтил)-амины] 43, 137, 170 сл., 188 сл.
Кадмий, соли 413
С-Калебассин 397
Кантаридин 402, 403
Карбонил хлорид см. Фосген
Карбонилы металлов 238 сл.
Кетоны галогенированные 69 сл.,
71 сл., 417
Классификация ОВ 57, 60 сл. военно-тактическая 63 по агрегатному состоянию 60 по быстроте наступления поражающего действия 63 по патологическим реакциям 62 по продолжительности заражающего действия 64 по способу заражения воздуха 61 по характеру действия 63 токсикологическая 62, 63 учебно-методическая 64 физическая 60 химическая 61
Кожно-нарывные ОВ 136 сл. биологическая активность 138 сл., 171 сл., 191 сл., 204, 205 боевое применение 208 сл. дегазация 213, 214 защита 118, 211, 212 маркировка 137 помощь при отравлениях 418 признаки отравления 141, 142, 173, 174, 192 сл.
Кокои яд 404, 421
Колхицин 401
Кониин 401
Контактные яды 165, 173
Концентрация ОВ 37, 38, 49 сл., 55 безопасная 49 боевая 25, 37, 58 летальная 56, 57, 419 сл. максимальная 24 сл., 52 сл. 423, . 424 массовая 37 незначительно опасная 55 опасная 49 очень опасная 49, 55 пороговая 50, 51 предельно допустимая 52 смертельная 49 средне опасная 55
Крапивные ОВ 170, 208 Кроталотоксин 403
Кротил ин (2,4-Дихлорфен оксиуксус -
ной кислоты хлор бу тениловый эфир) 375
Крысиный яд 387 Ксилилбромид 80 Кураре 396 сл. Курарин 397, 398
Лакриматоры (Слезоточивые ОВ) 66 сл.. 77 биологическая активность 66 сл., 219 признаки отравления 68, 125 строение 67 токсичность 80, 81
Летальная
доза ОВ 56, 420 сл.
концентрация ОВ 57, 419 Летучесть ОВ 24 сл.
Летучие (короткодействующие) ОВ 25, 61, 64
Лизергиновая кислота, производные 358 сл.
Липоиовая кислота 406
ЛСД 62, 63, 359, 360
Люизит (2-Хлорвинилдихлорарсин, p-Хлорвинилдихлорарсин) 190 сл., 196 сл.
биологическая активность 191 сл. боевое применение 209, 210 дегазация 200
маркировка 137 получение 196 сл. признаки отравления 192 сл. свойства 198 сл.
стойкость 27
токсичность 193, 201, 424
Максимальные концентрации ОВ
24 сл., 52 сл., 423, 424
Манганит 218
Мангвинит 218
Мартониты 69
Массовая концентрация ОВ 37
Медь, соли неорганические 412 хелаты 276
Мескалин (3,4,5-Триметоксифенил-этиламин) 63, 365, 366
Метасистокс 330, 332 сл.
летальная доза 337, 338 производные 338
системное действие 335, 338 токсичность 336, 337
N-Метил-М, N-бис- (2-хлорэтнл)-амин»
188 сл., 421, 423
Метилдихлорарсин 195, 201 сл.
боевое применение 203 токсичность 82, 97, 203, 424
3-Метилпирролидин-2'-п иридии (Ни-
котин) 395, 396, 421
Метилфосфорилхолины 318 сл.
Метилфторацетат (МФА) 388 сл.
Метилфторфосфоновая кислота, пи-наколиловый эфир см. Зомап
Метилфторфосфорилгомохолин 317 421
Метилфторфосфорилкарбохолин 318 Метилфторфосфорил-2-метилхолин 424
Метилфтор фосфор ил хол ин 252 317, 421, 424
2-Метил-4-хлорфеноксиуксусная кислота 375
Метокси-4-нитрофепоксиметил фосфин сульфид 421
429
Минтакол см. Параоксон Митилотоксин 405 Монохлор метиловый эфир хлоругольной кислоты 68
Монохлор фар моксим 204, 205, 208, 424 Морфолид
пеларгоновой кислоты 102, 103, 424
лизергиновой кислоты (LSM) 361 Мурексин 404, 405 Мускарин 402
МФА (Метил фтор ацетат) 388 «л.
Мышиная единица (ME) 406 Мышьяк 52
Трехокись' 422
Мышьяковистая кислота 413 Мышьяковистый водород (Арсин)
232, 233
боевое применение 244
помощь при отравлениях 417 токсичность 52, 233, 424
Мышьякорганические ОВ 81 сл., 190 сл., 413 ароматические 86 сл. дегазация 417 физиологическая активность 97 сл.
Нейротропные яды 409 етор-О-Неогексилметилфторфосфо-нат см. Зоман
Нервно-паралитические ОВ 292 Нереистоксин 406 НЕТ см. ТЭПФ
Никотин (3-Метил пиррол идин-2'-пи-
ридин) 395, 396, 421
Нитроалканы галогенированные
123 сл., 417 о-Нитробензилхлорид 80 Нитробензол 52 Нитроглицерин 52 4-Нитрофенол 255 Нитрохлороформ см. Хлорпикрин Нифос Т см. ТЭПФ
Общеядовитые ОВ 62, 217 сл. биологическая активность 218 сл. боевое применение 243, 244 дегазация 245, 246 защита 244 сл.
признаки отравления 221, 231 токсичность 231, 232
Окись углерода 235 сл.
боевое применение 244 токсичность 52, 236 сл.
5-Окси-З- (р-диметилам ин оЭтил) -индол см. Буфотенин
Оксимы галогенированные 45, 203 сл., 211, 214
Опасная концентрация ОВ 49
430
Отравляющие вещества (ОВ) агрегатные состояния 60 аэрозольные 24, 25, 29, 40, 61 боевое применение см. Боевое применение ОВ быстродействующие 63 гидролиз 41 сл.
давление паров 20 сл. 33 сл,, 423, 424
дегазация см. Дегазация ОВ действие на материалы 46 «желтого креста» 137, 144, 190, 201
защита см. Защита от ОВ «зеленого креста» 190, 201 классификация см. Классификация ОВ
кожно-нарывные см. Кожно-на-рывн ые 'ОВ
концентрация см. Концентрация ОВ
крапивные 170, 208
«красного креста» 170, 204 летучесть 24 сл.
летучие (короткодействующие) 25, 61, 64
медленнодействуюшие 63 мыщьяксо держащие 81 сл., 190сл., 413
нервно-паралитические 292 общеядовитые см. Общеядовитые ОВ
ограниченного значения 60 первая помощь при отравлениях 417, 418
плотность заражения 37 - паров 28
психохимические см. Психоя-ды
растворимость (смешиваемость) 30 сл., 36
резервные 60 .слезоточивые см. Лакриматоры смеси 31 стабилизация 45 степень токсичности 54 стойкие (долгодействующие) 26, 61, 64
стойкость 27, 28, 40 табельные 41, 60 температура застывания 32, 33 — кипения 23, 24, 26 — плавления 23, 24 токсические свойства 46 сл. удушающие см. Удушающие ОВ фактор смертельности 54, 55 физические свойства 20 сл 423 424
фитотоксические (Фитояды, Яды растений) 30, 370 сл.
Отравляющие вещества (ОВ) фосфорорганические см. Фосфорорганические ОВ фторорганические см. Фторорга-иические ОВ
хранение 45
Офиотоксин 403
Па р а оксон [0,0-Диэтил- (4-нитрофе-
нил)-фосфат, Е 600, Минтакол] 251, 344 сл.
токсичность 346, 347, 421
Паратион [О,О-Диэтил-(4-нитрофе-нил)-тионфосфат, Е 605, Парафос, Тиофос] 344 сл.
летальная доза 347, 421
максимальная концентрация 345 токсичность 346, 347
Пеларгоновая кислота, морфолид 102, 103, 424
Пентакарбонил железа 238 сл-
Пер хлор ацетальдоксим (Трихлорме-
тилформоксим) 205, 208
Перхлорметилмеркаптотриазины 380 Пестокс 14 см. Димефокс
Пиперидилбензилаты 367, 368
Пиперидилгликоляты 367, 368 Пиперидин, производные 366 сл. Пиридин, допустимые концентрации 52
производные 368 сл.
Пирофосфорная кислота, производ-' ные 253, 254
Плотность заражения ОВ 37
Порог раздражения О,В 50, 80 галогенированными арсинами 203 дифосгеном 123 раздражающими носоглотку 97 слезоточивыми 80 трибромнитрометаном 125 хлорпикрином 125 хлорцианом 232
Пороговая концентрация ОВ 50, 51
«Пороховая болезнь» 244
Предел переносимости ОВ 51 сл., 80
ароматических арсинов 83 галогенированных арсинов 203 раздражающих носоглотку 97 слезоточивых 80
Предельно допустимые концентрации ОВ 52
Природные яды 394 сл.
Произведение Ct 54
Прохлорит 150
Псилоцибин и псилоцин 382 сл.
Психодислептики 357
Психояды (Психохимические ОВ) 29, 40, 46, 62, 355 сл.
боевое применение 369 действие на организм 356, ЗЕГ . защита 369 пороговые концентрации 51 хранение 45
Пятифтористая сера, допустимые концентрации 133, 424
Раздражающие ОВ 60, 65 сл.
биологическая активность 66. с л., 81 сл.
боевое применение 105, 106 дегазация 107 защита 106, 107 маркировка 106 помощь при отравлениях 418 признаки отравления 68, 85 стерниты 62, 81 сл.
Рвотный камень 414
Резервные ОВ 60 «Роса смерти» 196 Ртуть, соли 413
Сакситоксин 405, 406
Салин-процесс получения зарина 302 Свинец, соли 413
Свинецорганические ОВ 83 сл., 98 сл.
Сенцилоилхолин 405
Сернил 367
Сероводород, допустимые концентрации 52
Сероуглерод, допустимые концентрации .52
Серы двуокись, допустимые концентрации 52
Сесквииприт 168, 170, 423
Симазин 380
Синильная кислота (Цианистый во-* дород) 217 сл., 221 сл., 226 сл. адсорбционная способность 225 биологическая активность 218сл. боевое применение 243 защита 245 как пестицид 226 получение 222 сл. признаки отравления 221, 231 свойства 224, 225 сл.
соли (Цианиды) 414 токсичность 52, 224, 231 232, 419 424
хранение 23, 28, 225 Систокс 330, 332 сл.
производные 330 сл., 338 свойства 331 сл.
сульфоксид 336 сульфон 336 токсичность 338, 337
431
Сй Эс (Хлорбензилиденмалонодинит-рил) 103 сл.
Скорпиона токсин 403
Слезоточивые ОВ см. Лакриматоры Смертельная концентрация ОВ 49 «Смерть кустам» 375
Стерниты (Раздражающие носоглотку ОВ) 62, 81 сл.
Столбняк (Тетанус) 410
токсин 396, 409, 410
Стрихнин 396, 397 »
Строфантин 404
Сулема 413
Сульфотеп (Сульфо-ТЭПФ) см. Ди-тион
Сурьма, производные 414
Сцилларен 404
Табельные ОВ 41, 60
Табун' (Диметиламидоцнанфосфорной кислоты этиловый эфир, N.N-Диметиламидо-О-этилцианфо-сфат) 288 сл.
биологическая активность 284, 285 боевое применение 290, 291 получение 288, 289 признаки отравления 292 свойства 290 стойкость 27
токсичность 290, 292 сл., 419, 421, 424
Тактические смеси ОВ 31, 32
Таллий, соли 413
Тарихатоксин (Тетродотоксин) 406, 407
ТЕРР см. ТЭПФ
Тетанус см. Столбняк
Тетраалкилпирофосфаты 349, 350
Тетра в ин ил свинец 243
Тетракарбонил никеля 52, 239
Тетракис-(2-хлорэтил)-свинец 243
Тетрам [Амитон, О,О-Диэтил-5-(2-ди-этиламиноэтил)-тиофосфат] 318, 342 сл„ 420
Тетраметилпирофосфат 422
Тетраметилсвинец 243, 424 сильи-Тетранитродихлорэтан 124 сп.и.и-Тетрафтординитроэтан 131 Тетрахлординитроэтан 124, 130 Тетрахлорэтан 52
действие па организм 124 смеси с ипритом 154
О.О.О^О'-Тетраэтилдитионпирофо-сфат см. Дитион
Тетраэтилпирофосфат см. ТЭПФ Тетраэтилсвинец 240 сл.
биологическая активность 240, 241
боевое применение 244 дегазация 242, 245, 246
Тетраэтилсвинец 240 сл.
защита 245
помощь при отравлениях 417
токсичность 52, 240 сл., 422,. 424
Тетродотоксин (Тарихотоксин) 406,
Тимет [О.О-Диэтил-S- (этилмеркапто-метил) -дитиофосфат, О,О-Ди-этил-S- (этилтиометил) -тионфо-сфат] 339 сл.
летальная доза 341, 422
Тиолсистокс [О,О-Диэтил-5-2- (этнл-тиоэтил)-тиофосфат] 422
Тиофос см. Паратион
ТМА (3,4,5-Триметоксифениламино-пропан) 366
Токсины 395
бактериальные 407 сл. ботулинический 407 сл.
грибов 401, 402 рыбы-дракона 406 скорпиона 403 столбняка 396, 409, 410 Токсиферины 397 Тониты 69
Транквилизаторы 357
1,3,5-Триазин, производные 380 Триалкилсвинец, соли 98 сл.
Трнбромнитрометан 80
М,М,М-Триметиламмонийалкоксиал-килфторфосфонаты см. Алкил-фторфосфорилхолины
3,4,5-Триметоксифенизопропиламин (ТМА) 366
3,4,5-Триметоксифенэтила мин (Мескалин) 63, 365, 366
Тринитрометан 52
Тринитротолуол 52
Триптамин 396
производные 361 сл.
Триптофан 361, 408
Трис-N.N- (диметиламидо) -О-Этил-пирофосфат 253
Трис-(2-фторэтил)-фосфит 394
Трис- (2-хлорвинил) -арсин 190
Трис-(2-хлорэтил )-амин см. Азотн-. стый иприт
Трифениларсин 82
Трифосген (Бис-трихлорметиловый эфир угольной кислоты, Гекса хлордиметилкарбонат) 113, 123, 423
Трифторнитрометая (Фторпикрип) 130
1,1,2-Трифтор-2-хлор-1,2-дин итроэтан 131
2-(М-Трихлорметилмеркапто-М-ме-тиламино) -4-М-алкиламино-6-хлор-1,3,5-триазин 380
432
Трихлорметиловый эфир хлормуравьиной (хлоругольной) кислоты, Трихлорметилхлоркар-
бонат, Трихлорметилхлорфор-миат) см. Дифосген
Трихлорметилформоксим (Перхлорацетальдоксим) 205, 208
Трихлорнитрозометан 129
Трихлорнитрометан см, Хлорпикрин 2,4,5-Три хлор уксусная кислота 374 сл. производные 375, 376
2,4,5-Трихлорфенилгексадециловый эфир 376
Трихлорэтан 52, 124
Р.Р'.Р^-Трихлорэтиламин см. Азотистый иприт
Трифосген 123
Триэтилсвинец, тиофенолят 101 — тиоэтилат 101 — фторацетат 101 — хлористый 422
Троповая кислота, пропиновый эфир (Атропин) 398 сл., 420
Трубочный кураре 396
Тыквенный кураре 396
ТЭПФ (Бладан, НЕТ, Нифос Т, ТЕРР, Тетраэтилпирофосфат) 253, 349, 350, 422
Угольная кислота бис-трихлорметиловый эфир (Трифосген) 113, 423 галогеипроизводные НО сл. хлорангидриды ПО сл., 424 эфиры 113, 123, 423
Удушающие ОВ 109 сл., 134, 417 биологическая активность ПОсл., 124, 125
боевое применение 133 сл. дегазация 134
' защита 134
помощь при отравлениях 417 признаки отравления 112 сл.
Фактор смертельности ОВ 54, 55 Фаллоидин 401
Фаллоин 401
Фармакологическая эффективность ОВ 46 сл.
Фенарсазинхлорид см. Адамсит Фенацилхлорид см. Хлорацетофенон Фен ил алкил а мины 364 сл.
Фенилалкиловые эфиры 376, 377
N-Фениламидо-О-этилфторфосфат 285 Фен ил дихлор арсин 83, 98, 195, 201 сл. токсичность 203, 424
Фенилкарбиламинхлорид 80, 105
‘ Фенилмочевина, производные 377
Фенилуксусная кислота 375
Фен илхлор метил кетон см. Хлорацетофенон
(1- (Г-Фенилциклогексил) -пиперидин, гидрогалогенид 367
Фен оксикарбоновые кислоты, производные 373 сл.
Фенол, производные 377, 378
Фенэтил а мин 364
Физостигмин 401
Фитотоксические ОВ (Фитояды) 30, 64, 370 сл.
военное применение 381, 382
Фосген (Дихлорангидрид угольной кислоты, Карбонилхлорид, Хлорокись углерода) 114 сл.
биологическая активность ПО сл.
, боевое применение 133 дегазация 117, 118 получение 114 сл. признаки отравления 112, 113 свойства 115, 116 сл.
токсичность 50, 52, 55, 116 119
120, 419 ’ ’
хранение 117
Фосген-карбонильные смеси 235
Фосгеноксим (Дихлорформальдоксим, Дихлорформоксим) 129, 204 сл„ 206 сл.
токсичность 207, 423
Фосдрин 349
Фосфин см. Фосфористый водород
Фосфорилтиохолины 29, 315 сл. биологическая активность 316 сл. токсичность 323 сл.
Фосфор ил холины 315 сл.
Фосфористый водород (Фосфин) 232 сл.
боевое применение 244
помощь при отравлениях 417 токсичность 52, 424
Фосфорорганические инсектициды 329 сл.
Фосфорорганические ОВ 247 сл., 329 сл. биологическая активность 254 сл., 264 сл., 294, 316 сл. боевое применение 325 сл.
гидролиз 43, 44 дегазация 328, 417 защита 327, 328 классификация 248—253 помощь при отравлениях 417 признаки отравления 261 сл„ 284 токсичность 48. 52, 57, 284
Фтор-М-алкилацетамиды 388
Фтор ацетамид 889
Фтор ацетаты •
калия 387 натрия 387, 422 Токсичность 386 триэтилсвинца 101
433
Фторацетофенон 76
ад-Фторвалериановая кислота, 2-фтор-этиловый эфир 392
ад-Фторированные спирты 393, 394
Фтористый водород, допустимая концентрация 52
ад-Фторкарбоновые кислоты 386 сл. алкиловые эфиры 391, 392 фторэтиловые эфиры 392
4-Фтормасляная кислота, эфиры 389 а-Фтор-1Ч-метнлацетамид 390 ад-Фторолеиновая кислота 387 Фторорганические ОВ 32, 384 сл.
биологическая активность 385, 386
помощь при отравлениях 417 признаки отравления 385, 386
о-Фторпальмитиновая кислота 387
Фторпикрин (Трифторнитрометан) 130
Фторуксусная кислота 386, 387, 391 алКиловые эфиры 388 сл.
метиловый эфир (МФА) 385, 388 сл., соли 387
2-фторэтиловый эфир 392 Фторфосфорилхолины 317, 318 Фторфосфоновая кислота, изопропиловый эфир см. Зарин
Фторфосфорная кислота, диэфиры см. Ди алкил фторфосфаты
Фторхлорформ оксим 205
Фторциан 229 ~
2-фторэтанол 385, 388, 392 сл.
Хаиан см. Димефокс
Хлорацетофенон (ФенилХлорметилке- » тон, Фенацилхлорид) 29, 40, ? 71 сл. j
биологическая активность 67 сл.
дегазация 107
стабильность 40
токсичность 73, 424 хранение 45
2-Хлорбензил ид ей мал оно динитрил (Си Эс) 103 сл.
Хлорбензол 52, 154
2-Хлор-4,6-бис- (этилен амин о) - 1,3,5-триазин (Симазин) 380
2-Хлорвинилдйхлбрарсин см. Люизит окись 199
10-Хлор-9,10-дигидрофенарсаэин хлорид см. Адамсит ,
Хлорметилхлорформоксим 205
Хлормуравьиная кислота, трихлорметиловый эфир см. Дифосген
Хлороформ 124
Хлорпикрин (Нитрохлороформ, Три-хлорнитрометан) 126 сл.
боевое применение 133
Хлорпикрин
дегазация 129
получение 126, 127
признаки отравления 123, 124, 130
свойства 127 сл.
смеси с ипритом 34, 35, 154
стабильность 40
стойкость 27
токсичность 55, 80; 127 130 419, 422, 424
Хлорпропанон 67, 80
Хлорсульфоновая кислота, 2-фторэтй-ловый эфир 393
ХлЪртрифторид 424
Хлоругольная кислота, эфиры 40, 68, 113, 114, 120 сл., 394
N- (4-Хлорфенил) -М'^'-диметилмоче-вина 375
1Ч-(3-Хлорфенилкарбаминовая кислота, изопропиловый эфир 375
1-Хлор-2- (2'-хлорэтилтио) -этан см.
Иприт
Хлорциан 228 сл.
боевое применение 243 -
токсичность 55, 80, 229, 232, 419, 422, 424
2-Хлорэтил-2'-бромэтиловый тиоэфир
2-Хлорэтилдихлорарсин 97
4- (2-Хлорэтил тцо)-фенилдихлорарсии
Цианамид кальция 380, 381
Цианиды 414
помощь при отравлениях 417
Цианистый водород см. Синильнай
кислота
Цианхлорформоксим 205
Циклозап ин 294
Циклон Б 226
Чилибухи алкалоиды 396, 397
Эргоалкалоиды 358
Эритроплеин 401
Этиламид . лизергиновой кислоты (LAE) 360
N-3nwi-N,N-(fac- (2-хлорэтил) -амин
188 сл., 422, 424
Этилдихлорарсии 201 сл.
боевое применение 208
токсичность 97, 195, 203, 424
Этилзарин . (О-Изопропилэтилфтор-фосфонат) 297, 298, 423
О-Этил-S- (2-этиламиноэтокси) -метил-тиофосфипоксид 422
О-Этил-S- (2-этиламиноэтокси) -этил-тиофосфиноксид 422
434
Этоксиметялфосфорилтиохолин 422
Яды
бледной поганки 401
глазные 173
диверсионные 383 сл., 412
жаб 403, 404
животного происхождения 402 сл.
змей 402, 403
ингаляционные 125, 132, 138 164, 173, 194, 201, 219
кобры 403
* кокои 404, 421
Яды
контактные 165, 173
крысиный 387
летальные дозы 420 сл.
морских животных 404 сл.
мухомора 401
нейротропные 409
неорганические 410 сл.
нервно-паралитические 84 природные 394 сл.
психохнмические см. Психояды растений см. Фитотоксические ОВ растительные 395 сл., 404