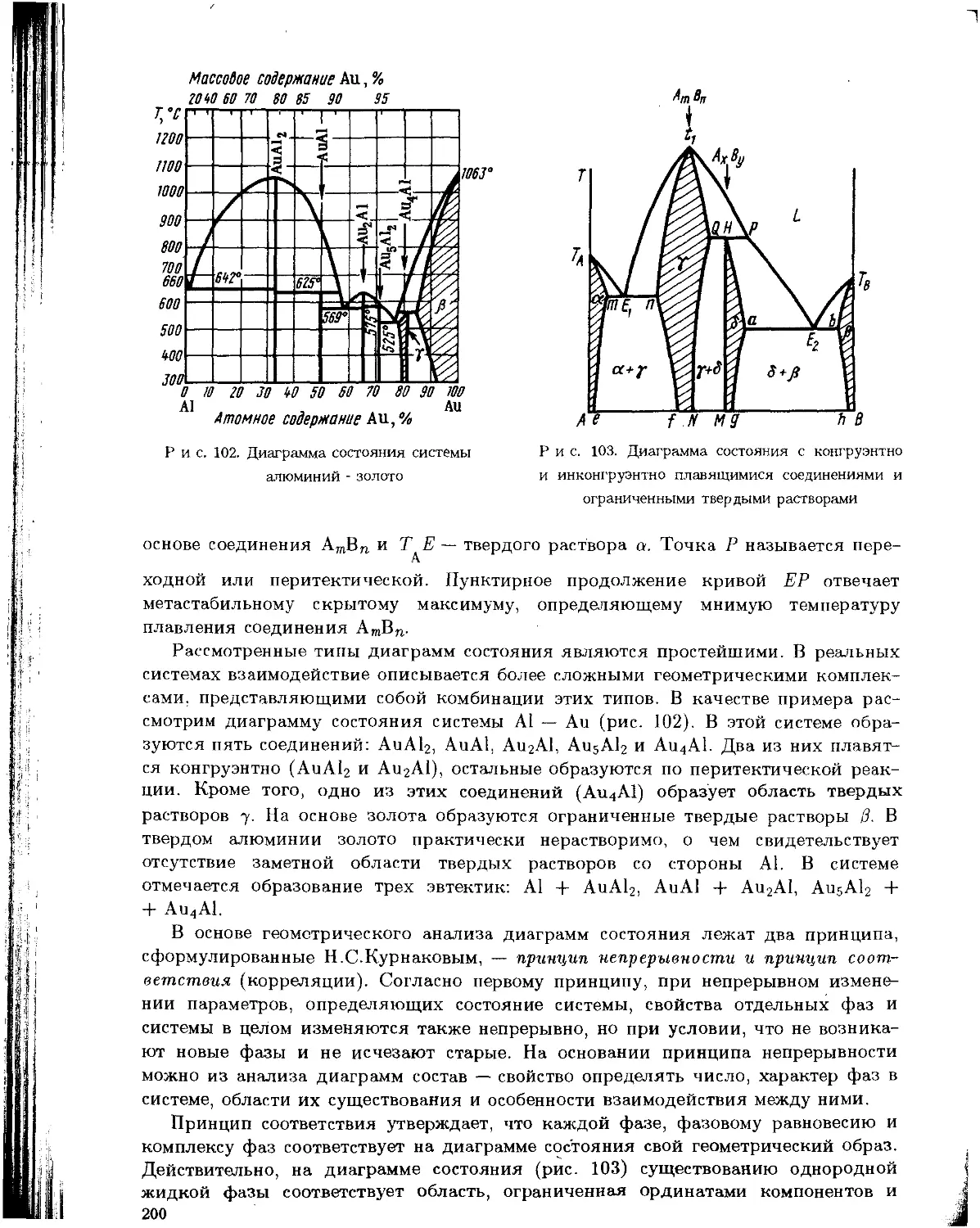
/

















































































































































































































































































































































































































































































































































Text
ЯЛ. Угай
ОБЩАЯ И НЕОРГАНИЧЕСКАЯ ХИМИЯ
М: Высш. ж.. 1997. — 527 с.
В учебнике по-новому излагаются вопросы химической атомистики и стехиометрические законы
химии. Даны современная трактовка фундаментальных законов химии, квантово-химическая
трактовка природы химической связи, учение о химических процессах, основы химии
межмолекулярного взаимодействия, включая комплексообразование. Наряду с жидкими растворами
большое внимание уделено химии твердых растворов.
Материал по неорганической химии излагается на базе современной интерпретации
Периодической системы элементов Д.И.Менделеева, учения о химическом и кристаллическом
строении вещества, а также элементов химической термодинамики.
ОГЛАВЛЕНИЕ
Предисловие 4
ЧАСТЬ I. ОБЩАЯ ХИМИЯ
Глава I. Фундаментальные законы к теории химии 5
1. Определение химии E). 2. Общая химия. Неорганическая химия F). 3. Фундаментальные теории
и законы химии G)
Глава II. Химическая атомистика. Основные законы и понятия 10
1. Атомистика Дальтона A0). 2. Газовые законы химии A1). 3. Атомная масса. Молекулярная
масса. Молярная масса A1). 4. Структура неорганических веществ A2).5. Неорганические
полимеры A5). 6. Фаза A5). 7. Постоянный и переменный состав. Формульная масса A6). 8.
Ограниченный характер и границы применимости стехиометрических законов химии. Современная
формулировка стехиомет-рических законов A8). 9. Закон постоянства свойств.
Кристаллохимическое строение и свойства A9). 10 Химический индивид. Химическое соединение
B1)
Глава III. Строение атома и Периодическая система ояементов 23
1. Модели строения атома B3). 2. Строение атома по Бору B4). 3. 0 квантовой механике B7). 4.
Основы квантово-механического рассмотрения атома водорода. Орбитали C0). 5. Квантовые числа
C4). 6. Многоэлектронные атомы C6). 7. Периодическая система элементов и атектронная
структура атомов C9). 8. Строение электронной оболочки и свойства элементов D5)
Глава IV. Химическая связь 56
1. Химическая связь и валентность E6). 2. Энергия химической связи E7). 3. Длина химической
связи E8). 4. Электрический момент диполя и направленность связи F0). 5. 0 ионной связи F2). 6.
Ковалентная связь F5). 7. Понятие о квантовой химии F6). 8. Метод валентных связей (МВС) F7).
9. Валентность и МВС G1). 10. Насыщаемость ковалентной связи G4). 11. Поляризация
химической связи G5). 12. Направленность ковалентной связи G9). 13. Кратность химической
связи (84). 14. О связях с избытком и дефицитом валентных электронов (86). 15. Понятие о методе
молекулярных орбиталей (88). 16. Сравнение МВС и ММО (93). 17. Металлическая связь (94). 18.
Химическая связь в твердых неорганических веществах (97)
Глава V. Межмолекулярное взаимодействие и комплексообразование 98
1. Силы Ван-дер-Ваальса (98). 2. Водородная связь A00). 3. Комплексные соединения.
Координационная теория Вернера A03). 4. Номенклатура комплексных соединений A04). 5.
Классификация комплексных соединений A06). 6. Устойчивость комплексных соединений.
Константа нестойкости. Двойные соли A07). 7. Хелаты и внутрикомплексные соединения A08). 8.
Изомерия комплексных соединений (НО). 9. Трансвлияние A11). 10. Природа химической связи в
комплексных соединениях A13)
Глава VI. Учение о химических процессах 121
1. Понятие о химической термодинамике A21). 2. Экзо- и эндотермические реакции. Основы
термохимии A24). 3. Направление химических процессов. Энтропия. Свободная энергия A27). 4.
Понятие о химической кинетике. Скорость химических реакций A29). 5. Основной закон
химической кинетики A30). 6. Параллельные, последовательные, сопряженные и цепные реакции
A31). 7. Зависимость скорости реакции от температуры. Энергия активации A34). 8. Обратимые
химические реакции. Химическое равновесие A36). 9. Смещение химического равновесия.
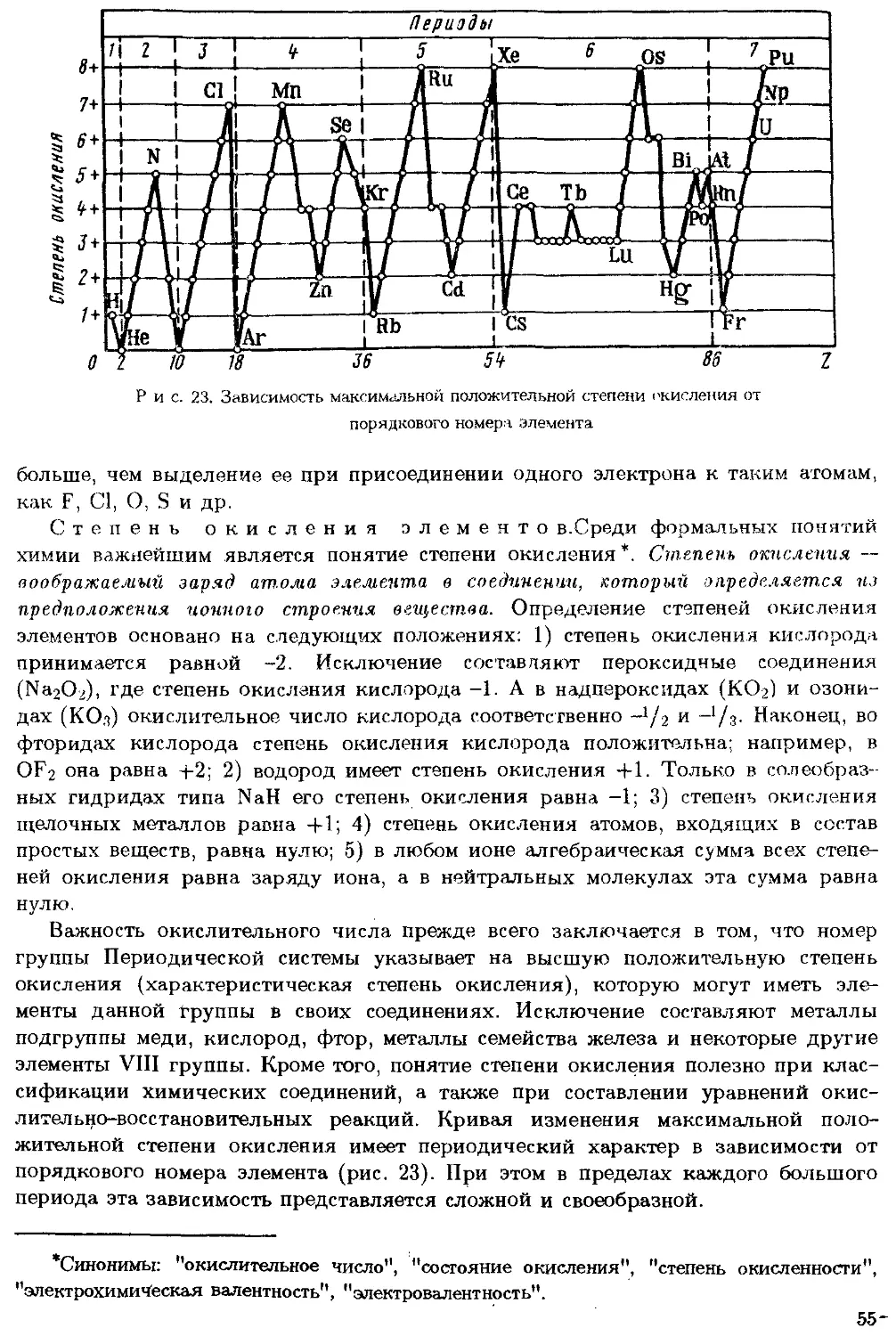
Принцип Ле Шателье A38). 10 Понятие о катализе. Гомогенный и гетерогенный катализ A41)
Глава VII. Жидкое состояние. Растворы 144
1. Жидкое состояние. Структура жидкости A44). 2. Дисперсные системы. Растворы A45). 3.
Процесс образования растворов A47). 4. Идеальный раствор. Законы разбавленных растворов A48).
5. Электролитическая ионизация. Степень и константа ионизации A52). 6. Понятие о теории
сильных электролитов. Активность A55). 7. Кислотно-основная ионизация A56). 8. Теории кислот
и оснований A58). 9. Водородный показатель. Индикаторы A59). 10. Обменные реакции между
ионами. Произведение растворимости A62). 11. Реакции нейтрализации и гидролиза A63). 12.
Окислительно-восстановительные реакции A70). 13. Гетерогенные реакции в растворах A74). 14.
Химические источники тока. Электролиз A81)
Глава VIII. Твердое состояние. Твердые растворы 185
1. Понятие о твердой фазе A85). 2. Кристаллическое, стеклообразное, аморфное состояния A86). 3.
Представление о зонной теории. Металлы, полупроводники, изоляторы A88). 4. Основы физико-
химического анализа A92). 5. Типы диаграмм состояния A94). 6. Твердые растворы B01). 7.
Соединения постоянного и пере-менног состава. Дальтониды и бертоллиды B04)
Глава IX. Металлохимия 208
1. Элементохимия B08). 2. Металлохимические свойства элементов B10). 3. Примитивные типы
химического взаимодействия в металлических системах B12). 4. Образование соединений в
металлических системах B14). 5. Металлохимические свойства и диаграммы состояния B21)
ЧАСТЬ П. НЕОРГАНИЧЕСКАЯ ХИМИЯ
Раздел 1. Периодический закон как основа химической систематики 225
Глава X. Структура Периодической системы 225
1. Этапы развития Периодического закона B25). 2 Групповая и типовая аналогии B27). 3.
Электронная аналогия. Кайносимметрия B28). 4. Переходные металлы. Контракционная аналогия
B32). 5. Орбитальные радиусы. Вторичная и внутренняя периодичность B33). 6. Горизонтальная и
диагональная аналогии B36)
Глава XI. Простые вещества как гомоатоыные соединения 239
1. Химическое и кристаллохимическое строение простых веществ B39). 2. Металлы и неметаллы в
Периодической системе B41). 3. Физические свойства простых веществ B44). 4. Химические
свойства простых веществ B49). 5. Нахождение в природе и общие принципы получения простых
веществ B50). 6. Особо чистые вещества B54)
Глава XII. Бинарные химические соединения 255
1. Классификация бинарных соединений B56). 2. Кристаллохимическое строение сиды B65). 5.
Водородные соединения B68). 6. Галогениды B71). 7. Халькогениды B73). 8. Пниктогениды B75).
9. Карбиды, силициды, бориды B77), 10. Интерметаллические соединения B79)
Глава XIII. Сложные химические соединения 279
1. Классификация сложных соединений B80). 2. Гидроксиды как характеристические соединения
B82). 3. Кислотно-основные свойства. Амфотерность гидроксидов B83). 4. Окислительно-
восстановительные свойства гидроксидов B87). 5. Соли кислородсодержащих кислот B89). 6.
Комплексные соединения B90)
Раздел 2. Химия элементов 292
Глава XIV. Водород 292
1. Уникальное положение водорода в Периодической системе B92). 2. Изотопы водорода B93). 3
Атомарный и молекулярный водород B94). 4. Физические и химические свойства водорода B95). 5.
Гидриды и летучие водородные соединения B96). 6. Получение водорода B99). 7. Вода B99). 8.
Пероксид водорода C01)
Глава XV. Элементы I группы 303
1. Литий C04). 2. Щелочные металлы C07). 3. Подгруппа меди C10)
Глава XVI. Элементы II группы 315
1. Бериллий C15). 2. Магний C18). 3. Щелочно-земельные металлы C20). 4. Подгруппа цинка C22)
Глава XVII. Элементы III группы 325
1. Бор C25). 2. Алюминий C31). 3. Подгруппа галлия C37). 4. Подгруппа скандия и РЗЭ C46)
Глава XVIII. Элементы IV группы 355
1. Углерод C56). 2. Кремний C69). 3. Подгруппа германия C79). 4. Подгруппа титана C90)
Глава XIX. Элементы V группы 396
1. Азот C97). 2. Фосфор D09). 3. Подгруппа мышьяка D17). 4. Подгруппа ванадия D26)
Глава XX. Элементы VI группы 432
1. Кислород D32). 2. Сера D35). 3. Подгруппа селена D43). 4. Подгруппа хрома D48)
Глава XXI. Элементы VII группы 456
1. Фтор D57). 2. Хлор DСЗ). 3. Подгруппа брома D68). 4. Подгруппа марганца D73)
Глава XXII. Элементы VIII группы 482
1. Элементы VIIIA-группы D83). 2. Металлы триады железа D88). 3. Платиновые металлы D95)
Глава XXIII. Радиоактивные и синтезированные элементы 501
1. Радиоактивные аналоги стабильных элементов Периодической системы E02). 2. Металлы
семейства актинидов E04). 3. Трансактиниды E16)
Литература 519
Предметный указатель 520
Оглавление 524
Важнейшие константы, единицы энергии
и некоторые другие физические вели чипы
Постоянная Авогадро N — 6,02* 1023 моль
А
Постоянная Больцмана к— 1,38-106 эргтрад = 1,38• 10~23 Джтрад
Постоянная Планка h = 6.62-107 эрг-г = 6,62-104 Дж-с
Постоянная Фарадея F— 96485 Кл-моль
Постоянная Ридберга Rx = 1,097 *105 см = 1,097 • 107 м
Элементарный электрический заряд е = 4,80 -100 эл.ст.ед. = 1,60 • 109 Кл
Атомная единица массы: 1 а.е.м. = 1,66-107 кг
Масса покоя электрона щ — 9,11 *101 кг
Скорость света в вакууме с = 3-Ю10 см*с'1 = 3-Ю8 м-с
Молярная газовая постоянная R = 8,31 Дж • моль • град
1 Дж = 6,25* 1018 эВ
1 эВ = 1,60* 109 Дж
1 мк = 1 мкм = 10 м
ПРЕДИСЛОВИЕ
В 1984 г. в издательстве "Высшая школа" вышел в свет учебник Я.А.Угая
"Общая химия" B-е изд.) для химических факультетов университетов, а в
1989 г. — 1-е издание учебника того же автора "Неорганическая химия" для
студентов вузов, обучающихся по специальности "Химия". Обе книги стали
библиографической редкостью.
Однако опыт работы со студентами Воронежского государственного универси-
университета показывает целесообразность объединения обоих учебников под названием
"Общая и неорганическая химия". В результате создан единый учебник, содер-
содержащий информацию разумной достаточности как для нынешних студентов, так и
для подготовки химиков-бакалавров.
Приношу искреннюю признательность рецензентам рукописи д-ру хим. наук,
проф. С.А. Симановой (С.-Петербургский технологический институт) и коллекти-
коллективу кафедры общей и неорганической химии Ростовского госуниверситета (зав.
кафедрой — д-р хим. наук, проф. Т.Г. Лупейко), чьи замечания позволили улуч-
улучшить качество учебника.
Автор благодарит также канд. хим. наук, доц. кафедры неорганической химии
В.Р. Пшестанчика и канд. хим. наук, асе. кафедры общей химии Т.П. Сушкову
за помощь при подготовке настоящего издания учебника.
Замечания и пожелания можно направлять по адресу. 394693, Воронеж, Уни-
Университетская площадь, 1, Государственный университет, кафедра неорганической
химии.
Автор
ЧАСТЬ I
ОБЩАЯ ХИМИЯ
Г Л А В А 1. ФУНДАМЕНТАЛЬНЫЕ ЗАКОНЫ
И ТЕОРИИ ХИМИИ
1. Определение химии. Современная химия является одной из естественных
наук и представляет собой систему отдельных дисциплин: общей и неорганиче-
неорганической химии, аналитической химии, органической химии, физической и коллоид-
коллоидной химии, геохимии, космохимии и т.п.
Химия — наука, изучающая процессы превращения веществ, сопровождаю-
сопровождающиеся изменением состава и структуры, а также взаимные переходы между этими
процессами и другими формами движения материи.
Таким образом, главным объектом химии как науки являются вещества и их
превращения.
Известны две формы существования материи как объективной реальности:
вещество и поле. Вещество — материальное образование, состоящее из элементар-
элементарных частиц, имеющих собственную массу, или массу покоя. К элементарным
частицам с конечной массой покоя* относятся электроны и позитроны (лептоны),
протоны, нейтроны (нуклоны), гипероны и другие тяжелые частицы (барионы).
Промежуточные по массе частицы между лептонами и нуклонами называют
мезонами. Мезоны и барионы вместе именуются адронами. Все вещества в
конечном итоге состоят из атомов, следовательно, из электронов, протонов и
нейтронов.
В отличие от вещества поле — материальная среда, в которой осуществляется
взаимодействие частиц. Так, в электромагнитном поле происходит взаимодейст-
взаимодействие между заряженными частицами, а ядерное поле осуществляет взаимодействие
между нуклонами и т.п. Полевая форма материи не является непосредственным
объектом химии и проявляется прежде всего энергетическими характеристиками,
хотя и обладает массой-
В определении химии подчеркивается взаимосвязь химической и других форм
движения материи. Движение есть способ существования материи, ее коренное и
неотъемлемое свойство. Многообразные явления Вселенной, несмотря на их каче-
качественное различие, представляют собой различные формы и виды движущейся
материи. Специфика химической формы движения материи — изменение состава
вещества. Химические процессы образования и разрушения веществ всегда сопро-
*Речь идет о покое относительном, так как абсолютного покоя не существует. Фотоны,
например, не обладают массой покоя.
5
вождаются изменением их состава и структуры. При этом разрываются, вновь
возникают или перераспределяются химические связи между атомами, входящи-
входящими в состав вещества.
Различные формы движения материи взаимосвязаны и взаимопревращаемы.
Хорошо известны химические реакции с выделением теплоты и светового излуче-
излучения. Теплота и световое излучение наряду с другими физическими явлениями
составляют предмет физики. Основатель научной химии М.В.Ломоносов говорил:
"Химик без знания физики подобен человеку! который всего искать должен
ощупом. И сии две науки так соединены между собой, что одна без другой в
совершенстве быть не могут". Свидетельством этому служит процветание в наше
время физической химии, основателем которой является Ломоносов, и химиче-
химической физики.
Еще один пример тесной связи между разными формами движения материи,
нашедшими отражение в соответствующих естественных науках, — взаимоотноше-
взаимоотношение химии и биологии. В последние годы биологическая наука сделала качест-
качественный скачок в сторону молекулярной биологии. Зарождение и бурное развитие
биохимии, биоорганической химии, биофизической химии, бионеорганической
химии и других наук на стыке между химией и биологией — яркое доказательст-
доказательство взаимосвязи между химическими и биологическими явлениями.
Процесс взаимопроникновения различных естественных наук — объективное
следствие существующей в природе всеобщей взаимосвязи и взаимообусловленно-
взаимообусловленности различных форм движения материи.
2. Общая химия. Неорганическая химия. Неорганическая химия — это химия
элементов Периодической системы и образованных ими простых и сложных ве-
веществ.
Неорганическая химия неотделима от общей химии. Исторически при изуче-
изучении химического взаимодействия элементов друг с другом были сформулированы
основные законы химии, общие закономерности протекания химических реакций,
теория химической связи, учение о растворах и многое другое, что составляет
предмет общей химии. Таким образом, общая химия изучает теоретические пред-
представления и концепции, составляющие фундамент всей системы химических
знаний.
Неорганическая химия давно перешагнула стадию описательной науки и в
настоящее время переживает свое "второе рождение" в результате широкого
привлечения квантово-химических методов, зонной модели энергетического
спектра электронов, открытия валентно-химических соединений благородных
газов, целенаправленного синтеза материалов с особыми физическими и химиче-
химическими свойствами. На основе глубокого изучения зависимости между химическим
строением и свойствами она успешно решает главную задачу — создание новых
неорганических веществ с заданными свойствами. Из экспериментальных методов
химии важнейшим является метод химических реакций. Химические реакции —
превращение одних веществ в другие путем изменения состава и химического
строения. Во-первых, химические реакции дают возможность исследовать хими-
химические свойства вещества. Кроме того, по химическим реакциям исследуемого
вещества можно косвенно судить о его химическом строении. Прямые же методы
установления химического строения в большинстве своем основаны на использо-
использовании физических явлений. Во-вторых, на основе химических реакций осущест-
осуществляется неорганический синтез. За последнее время неорганический синтез дос-
6
тиг большого успеха, особенно в получении особо чистых соединений в виде
монокристаллов. Этому способствовали применение высоких температур и давле-
давлений, глубокого вакуума, внедрение бесконтейнерных способов синтеза и т.п.
При проведении химических реакций, а также при выделении веществ из
смеси в чистом виде и поныне исключительно важную роль играют
препаративные методы: осаждение, кристаллизация, фильтрование,
сублимация, перегонка и т.п. В настоящее время многие из этих классических
препаративных методов получили дальнейшее развитие и являются ведущими в
технологии получения особо чистых веществ и монокристаллов. К ним относятся
методы направленной кристаллизации, зонной перекристаллизации, вакуумной
сублимации, фракционной перегонки. Одна из примечательных особенностей
современной неорганической химии — исследование особо чистых веществ на
монокристаллах.
Обычно препаративным методам противопоставляют методы физико-химичес-
физико-химического анализа. Последние широко применяются при изучении растворов и спла-
сплавов, когда образующиеся в них соединения трудно или практически невозможно
выделить в индивидуальном состоянии. Тогда вместо выделения отдельных ве-
веществ с последующим изучением их свойств исследуют физические свойства
систем в зависимости от изменения состава. В результате строят диаграмму сос-
состав — свойство, анализ которой позволяет делать заключение о характере хими-
химического взаимодействия компонентов, образовании соединений и их свойствах.
Совершенно очевидно, что физико-химический анализ не должен противопостав-
противопоставляться препаративной химии, так как его методы дополняют препаративные
методы исследования, а не исключают.
Однако для познания сущности явления одних экспериментальных методов
недостаточно, поэтому Ломоносов говорил, что истинный химик должен быть
теоретиком. Только через мышление, научную абстракцию и обобщение познают-
познаются законы природы, создаются гипотезы и теории, открывающие путь для пред-
предсказания новых фактов. А научное предвидение — главная черта любой истинной
науки. Теоретическое осмысливание опытного материала и создание стройной
системы химических знаний в современной общей и неорганической химии бази-
базируются на: 1) квантово-механической теории строения атомов и Периодической
системе элементов Д.И.Менделеева; 2) квантово-химической теории химического
строения и учении о зависимости свойств вещества от его химического строения;
3) учении о химическом равновесии, основанном на понятиях химической
термодинамики.
3. Фундаментальные теории и законы химии. К числу основополагающих
обобщений химии и естествознания относятся атомно-молекулярная теория,
закон сохранения массы и энергии, Периодическая система и теория химического
строения.
Атомно-молекулярная теория. Создатель атомно-молекуляр-
ного учения и первооткрыватель закона сохранения массы веществ М.В.Ломоно-
М.В.Ломоносов по праву считается основателем научной химии. Ломоносов четко различал
две ступени в строении вещества: элементы (в нашем понимании — атомы) и
корпускулы (молекулы). Согласно Ломоносову, молекулы простых веществ состоят
из одинаковых атомов, а молекулы сложных веществ — из разных атомов. Всеоб-
Всеобщее признание атомно-молекулярная теория получила в начале XIX в. после
утверждения в химии атомистики Дальтона. С тех пор главным объектом иссле-
дования химии стали молекулы. Химией молекул продолжает оставаться совре-
современная органическая химия, а большинство неорганических веществ не имеют
молекулярной структуры. В последнем случае макротела состоят либо из атомов
одного и того же химического элемента, либо из атомов разных элементов. Приз-
Признание немолекулярной формы существования твердого вещества приводит к
необходимости пересмотра некоторых положений химической атомистики, модер-
модернизации основных законов и понятий, справедливых для пневматической (газо-
(газовой) химии.
Закон сохранения массы и энергии. В 1760 г. Ломоносов,
по существу, сформулировал единый закон сохранения массы и энергии: "Все
перемены, в натуре случающиеся, такого суть состояния, что сколько чего у
одного тела отнимается, столько же присовокупится к другому. Так, ежели где
убудет несколько материи, то умножится в другом месте... Сей всеобщий естест-
естественный закон распространяется и в самые правила движения, ибо тело, движущее
своею силою другое, столько же оныя у себя теряет, сколько сообщает другому,
которое от него движение получает". Однако до начала XX в. эти законы обычно
рассматривались независимо друг от друга. Химия в основном имела дело с зако-
законом сохранения массы вещества, а физика — с законом сохранения энергии. В
1905 г. основоположник современной физики А.Эйнштейн показал, что между
массой и энергией существует взаимосвязь, выражаемая уравнением
Е = тс2, A.1)
где Е — энергия; т — масса; с — скорость света в вакууме (коэффициент пропор-
пропорциональности).
Ввиду исключительно большого значения квадрата скорости света в вакууме
очень малые изменения массы ведут к колоссальному изменению энергии. Так, из
формулы A.1) следует, что 1 кДж соответствует изменению массы 2-Ю0 г. Если
считать тепловые эффекты химических реакций порядка сотен килоджоулей,
соответствующие изменения массы должны быть порядка 10~8 — 10"9 г. Эта вели-
величина лежит за пределами чувствительности весов, применяемых в химии, вклю-
включая ультрамикровесы. Таким образом, из уравнения A.1) следует, что при хими-
химических реакциях, поскольку они сопровождаются энергетическими эффектами, в
действительности происходит изменение массы. Однако эти изменения настолько
малы, что ими можно пренебречь. Ощутимые же изменения массы наблюдаются в
ядерной химии и энергетике, где энергетические эффекты измеряются миллиона-
миллионами и миллиардами килоджоулей.
Периодический закон. Важнейшая задача неорганической химии
заключается в изучении свойств элементов, в выявлении общих закономерностей
их химического взаимодействия между собой. Самое крупное научное обобщение
в решении этой проблемы сделал Д.И.Менделеев, открывший Периодический
закон и графическое его выражение — Периодическую систему. Только вследст-
вследствие этого открытия стало возможным химическое предвидение, предсказание
новых фактов. Поэтому Менделеев является основателем современной химии.
Периодический закон Менделеева является основой естественной систематики
химических элементов. Химический элемент — совокупность атомов с одинако-
одинаковым зарядом ядра. Закономерности изменения свойств химических элементов
определяются Периодическим законом. Учение о строении атомов объяснило
физический смысл Периодического закона. Оказалось, что периодичность изме-
изменения свойств элементов и их соединений зависит от периодически повторя-
8
ющейся сходной структуры электронной оболочки их атомов. Химические и
некоторые физические свойства зависят от структуры электронной оболочки,
особенно ее наружных слоев. Поэтому Периодический закон является научной
основой изучения важнейших свойств элементов и их соединений: кислотно-ос-
кислотно-основных, окислительно-восстановительных, каталитических, комплексообразова-
тельных, полупроводниковых, металлохимических, кристаллохимических, радио-
радиохимических и т.п. Помимо теории строения атома Периодическая система элемен-
элементов сыграла колоссальную роль в учении о естественной и искусственной радио-
радиоактивности, освобождении внутриядерной энергии. В настоящее время синтез
заурановых и заплутониевых элементов немыслим без Периодической системы, не
говоря уже о том, что геохимия и космохимия полностью основываются на ней.
Периодический закон и Периодическая система беспрерывно развиваются и
уточняются. Доказатапьством тому служит современная формулировка Периоди-
Периодического закона: свойства элементов, а также формы и свойства их соединений
находятся в периодической зависимости от величины заряда ядра их атомов.
Таким образом, положитапьный заряд ядра (а не атомная масса) оказался более
точным аргументом, от которого зависят свойства элементов и их соединений.
Д.И.Менделеев в "Основах химии" писал: "Периодический закон ждет не
только новых приложений, но и усовершенствований, подробной разработки и
свежих сил... По-видимому, Периодическому закону будущее не грозит разруше-
разрушением, а только надстройка и развитие обещается". Эти пророческие слова творца
Периодического закона и Периодической системы целиком и полностью оправды-
оправдываются в наше время.
Теория химического строения. Фундаментальная задача
химии — изучение зависимости между химическим строением вещества %s его
свойствами. Свойства вещества являются функцией его химического строения.
До А.М.Бутлерова считали, что свойства вещества определяются его качествен-
качественным и количественным составом. Он впервые сформулировал основное положение
своей теории химического строения так: химическая натура сложной частицы
определяется натурой элементарных составных частиц, количеством их и химиче-
химическим строением. Это знаменитое положение может быть по праву названо законом
Бутлерова и приравнено к фундаментальным законам химии. В "переводе" на
современный язык закон Бутлерова утверждает, что свойства молекулы определя-
определяются природой составляющих ее атомов, их количеством и химическим строением
молекулы. Таким образом, первоначально теория химического строения относи-
относилась к химическим соединениям, имеющим молекулярную структуру. Это одна из
причин, почему она считалась теорией строения органических соединений. Меж-
Между тем сам Бутлеров считал созданную им теорию химического строения A861)
общехимической теорией и для ее обоснования пользовался примерами как орга-
органической, так и неорганической химии.
Химия твердых неорганических веществ установила, что важнейшие свойства
этих тел также зависят от их химического строения. Само понятие химического
строения применимо не только к молекулам, но и к веществам, не имеющим
молекулярной структуры. Это и правильно, поскольку более широкое понятие
химического строения включает в себя структуру, т.е. внутреннее строение веще-
вещества. А структурой обладает любое вещество независимо от того, образуют состав-
составляющие атомы дискретные молекулы или нет. Не случайно поэтому учение о
химическом строении пронизывает такие разделы неорганической химии, как
химия координационных соединений, химия неорганических полимеров, химия
полупроводников и др.
В настоящее время есть все основания считать теорию Бутлерова фундамен-
фундаментальной общехимической теорией строения химических соединений и зависимости
свойств их от химического строения. Эта теория — продолжение и развитие
атомно-молекулярного учения Ломоносова, являющегося фундаментом всей хи-
химии. Спустя столетие после создания теории Бутлерова в результате успешного
приложения к химическим объектам методов теоретической и экспериментальной
физики стали глубже и полнее понимать само химическое строение. Сегодня
химическое строение — это не только порядок валентной связи атомов и их вза-
взаимное влияние в веществе, но и направления и прочность связей, межатомные
расстояния, распределение плотности электронного облака, эффективные заряды
атомов и т.п. Химическое строение в первую очередь определяется характером
химической связи между атомами, связанными непосредственно друг с другом.
Поэтому основу теории химического строения составляет учение о химической
связи.
По вопросу дальнейшего развития теории химического строения Бутлеров
писал: "Само собой разумеется, что когда мы будем знать ближе натуру химиче-
химической энергии, самый род атомного движения, — когда законы механики получат
и здесь приложение, — тогда учение о химическом строении падет, как падали
прежние химические теории, но, подобно большинству этих теорий, оно падет не
для того, чтобы исчезнуть, а для того, чтобы войти в измененном виде в круг
новых и более широких воззрений". Итак, автор теории химического строения
предвидел приложение механики атомного мира (т.е. квантовой механики) к его
теории. Именно применение квантовой механики к проблемам структуры вещест-
вещества подняло теорию химического строения Бутлерова на новую, высшую ступень.
Только в одном не прав был Бутлеров: его теория не пала, а превратилась в
общехимическую теорию, являющуюся фундаментом современной химии.
Г Л А В А II. ХИМИЧЕСКАЯ АТОМИСТИКА.
ОСНОВНЫЕ ЗАКОНЫ И ПОНЯТИЯ
1. Атомистика Дальтона. После Ломоносова английский ученый Дальтон под-
подробно развил атомистические воззрения применительно к химии. Сущность уче-
учения Дальтона A803) сводится к следующему:
1. Вещества состоят из мельчайших неделимых частиц — атомов. Атомы при
химических реакциях не разрушаются и не возникают вновь. Все атомы простого
вещества одинаковы между собой по форме и массе.
2. Сложные вещества состоят из "сложных атомов", которые при химических
реакциях могут распадаться на атомы простых веществ. Простые атомы сочетают-
сочетаются в сложные атомы в простейших числовых пропорциях: атом на атом, атом на
два атома и т.д. Масса сложного атома равна сумме масс входящих в него прос-
простых атомов.
Дальтон не видел качественной разницы между простыми и сложными атома-
атомами, следовательно, не признавал две ступени (атомы и молекулы) в строении
вещества. В этом смысле атомистика Дальтона была шагом назад по сравнению с
элементно-корпускулярной концепцией Ломоносова. Однако "рациональным
зерном" атомистики Дальтона явилось его учение о массе атомов. Совершенно
правильно считая, что абсолютные массы атомов чрезвычайно малы, Дальтон
предложил определять относительные атомные массы. При этом масса атома
водорода, как самого легкого из всех атомов, была принята за единицу. Таким
образом, впервые Дальтон определил атомную массу элемента как отношение
массы атолю данного элемента к массе атома водорода. Он же составил первую
таблицу атомных масс 14 элементов. Учение Дальтона об атомных массах сыграло
неоценимую роль при превращении химии в количественную науку и открытии
Периодического закона.
2. Газовые законы химии. При определении атомной массы элемента Дальтон
исходил из понятия атомной массы и результатов химического анализа. Однако
для установления правильных атомных масс элементов оказались недостаточными
указанные исходные позиции Дальтона. Необходимо было атомистику Дальтона
дополнить ясными представлениями о молекулах. На этом пути важную роль
сыграли газовые законы и особенно закон объемных отношений Гей-Люссака и
закон Авогадро. Экспериментальные исследования но изучению химических
реакций между газообразными веществами привели Гей-Люссака к открытию
закона объемных отношений A808): при неизменных температуре и давлении
объемы вступающих в реакцию газов относятся друг к другу, а также к объемам
образующихся газообразных продуктов как небольшие целые числа. Так, при
образовании хлорида водорода из простых веществ объемы реагирующих и полу-
получающихся газов относятся друг к другу как 1:1:2. А при синтезе воды из прос-
простых веществ это отношение равно 2:1:2. Эти пропорции небольших и целых чисел
нельзя объяснить, исходя из атомистики Дальтона. Закон объемных отношений
нашел объяснение в гипотезах Авогадро A811):
1. В равных объемах разных юзов при одинаковых условиях (давлении и темпе-
температуре) содержится равное количество молекул.
2. Молекулы, простых газообразных веществ, таких, как водород, кислород,
азот, хлор и др., состоят из двух атомов.
Таким образом, обе гипотезы Авогадро получили опытное подтверждение в
газовой химии. Первая гипотеза впоследствии превратилась в один из основных
законов идеальных газов, имеющих первостепенное значение для химии. Из
закона Авогадро вытекают два очень важных следствия:
1. Молекулярная масса газа или пара равна произведению его плотности по
отношению к любому другому газу на люлекулярную массу последнего. При этом
под плотностью понимают отношение массы определенного объема данного газа к
массе такого же объема другого газа (при одинаковых температуре и давлении),
молекулярная масса которого известна.
2. Моль любого газа при нормальных условиях B73 К и 1,033-105 Па) занимает
объем 22,4 л. В условиях, отличных от нормальных, объем любого количества
газа может быть рассчитан из уравнения Клапейрона — Менделеева:
PV=(m/M)RT, A1.1)
где т — масса газа; М — молекулярная масса; R — молярная газовая постоянная;
р — давление; Т — абсолютная температура; V — объем газа, а m/М — п — число
молей.
3. Атомная масса. Молекулярная масса. Молярная масса. В настоящее время
атомная масса — одна из фундаментальных характеристик химического элемента.
Она служит основой для всевозможных стехиометрических расчетов по химиче-
химическим формулам и уравнениям, вычисления молекулярных масс химических со-
соединений. Понятие атомной массы приложимо не только к элементам (элемент-
(элементная масса), но и к отдельным изотопам (изотопная масса).
Все физико-химические методы определения атомных масс дают величину
элементной массы. Только для моноизотопных элементов, представленных
единственным природным изотопом, элементная масса совпадает с изотопной.
Современные точные физические методы установления атомных масс (например,
масс-спектроскопия) позволяют получать значения изотопных масс. Поэтому для
установления атомной (элементной) массы необходимо еще знать изотопный
состав элемента.
Долгое время в качестве единицы атомной массы была принята '/щ средней
массы атомов природного кислорода, состоящего из изотопов 1бО. 17О и 18О. Эта
единица составляла основу химической шкалы атомных масс. В основе же физиче-
физической шкалы лежала 1/\6 массы изотопа 160. Переходный множитель от одной
шкалы к другой 1,000275. Существование двух шкал атомных масс создавало
определенные трудности. Разница между ними намного превышает точность
определения атомных масс современными физическими и физико-химическими
методами. В 1916 г. Международный союз по теоретической и прикладной химии
(IUPAC) утвердил единую углеродную шкалу атомных масс. Основа ее — атомная
единица массы (а.е.м.), равная 'Дз массы изотопа углерода 12С. По углеродной
шкале относительные атомные массы водорода и кислорода соответственно равны
1,0079 и 15,9994. Таким образом, атомная (элементная) масса — среднее значение
массы атома химическою элемента, выраженное, в атомных единицах массы.
Молекулярная масса — масса молекулы, выраженная в атомных единицах массы*:
она равна сумме масс всех атомов, из которых состоит молекула.
В химии практически важной единицей количества вещества является моль. В
1 моль содержится столько структурных единиц (атомы, молекулы или др.;,
сколько атомов в 12 г изотопа углерода 12С. Число структурных единиц, содер-
содержащихся в 1 моль вещества, равно 6,022 *1023 моль (постоянная Авогадро). Мас-
Масса 1 моль данного вещества, выраженная в граммах, называется его .полярной
(мольной) массой. Так, молярная масса молекулярного кислорода равна 31,9988,
а атомарного водорода — 1.0079 г/моль. Молярная масса вещества в граммах
численно равна его молекулярной (атомной) массе, выраженной в атомных едини-
единицах массы.
4. Структура неорганических веществ. Подавляющее большинство неоргани-
неорганических веществ при комнатной температуре находится в твердом состоянии. А
для твердых тел обычным и устойчивым состоянием является кристаллическое**.
Кристаллы характеризуются упорядоченным расположением частиц в строго
определенных точках пространства. Если эти точки соединить пересекающимися
друг с другом прямыми линиями, получится пространственный каркас, называ-
называемый кристаллической решеткой. Для ее описания необходимо знать расположе-
расположение структурных единиц (атомов, молекул) в ее элементарной ячейке, параллель-
*Молекулярнал масса в действительности представляет собой среднюю массу молекулы
с учетом изотопного состава всех элементов, образующих данное соединение.
* "Даже микрообъемы стекла имеют кристаллическую структуру.
P n c. 1. Структура хлорила нат-
натрия
Р и с. 2. Структура метаплическо-
го натрия с выделением илелген-
тарной ячейки (заштриховано)
ными переносами которой (трансляциями) в трех измерениях можно построить
всю кристаллическую решетку. Под элементарной ячейкой понимают наимень-
наименьшую часть кристаллической решетки, которая отражает все особенности ее струк-
структуры. Узлы решетки — трансляционно идентичные точки, в которых находятся
частицы. В узлах решетки частицы — атомы, молекулы — совершают малые коле-
колебания *.
Рассмотрим структуру типичных неорганических веществ. На рис. 1 приведена
кристаллическая структура хлорида натрия. Принято считать, что в узлах решет-
решетки NaCl находятся ионы**. Атомы одного вида находятся в вершинах куба и в
центрах каждой грани, противоположно поляризованные атомы также расположе-
расположены по вершинам куба и в центрах каждой его грани. Только второй куб смещен
от первого на кратчайшее расстояние между атомами натрия и хлора. В целом
хлорид натрия образует гранецентрированную кубическую решетку. В этой ре-
решетке каждый атом натрия окружен ближайшими шестью атомами хлора, и
наоборот.
Существенно то, что в структуре поваренной соли нельзя очертить отдельные
молекулы NaCl, так как их нет. Атомы натрия и хлора в решетке хлорида нат-
натрия не связаны попарно между собой. Между тем в условиях повышенной темпе-
температуры в парах хлорида натрия существуют молекулы NaCl. При этом равновес-
равновесное расстояние между натрием и хлором в кристалле на 15% больше, чем в газо-
газообразной молекуле NaCl, т.е. последняя менее ионна.
В отличие от NaCl металлический натрий образует объемно центрированную
кубическую решетку. На рис. 2 приведена кристаллическая структура натрия с
выделением (штриховка) одной элементарной ячейки. Как это видно из рис. 2, в
структуре натрия также отсутствуют молекулы. В парах же натрия обнаружены
молекулы Na2 с межатомным расстоянием 0,308 нм против 0,372 нм в твердом
металлическом натрии.
Один из важнейших элементов неорганической химии — кремний — в виде
простого вещества имеет кристаллическую структуру типа алмаза (рис. 3). Атомы
'Колебательные движения в узлах решетки с малой амплитудой.
* *На самом деле в узлах решетки находятся поляризованные атомы Na и С1 с эффек-
эффективными зарядами + 0,87 и — 0,87 соответственно.
1.4
кремния располагаются по вершинам и в центрах каждой грани элементарной
кубической ячейки. Тремя перпендикулярными плоскостями, проходящими
через центр ячейки, ее можно мысленно разбить на 8 малых кубов (октантов).
Один из восьми октантов на рис. 3 показан пунктиром. По каждому координет-
ному направлению "заселенные" октанты, в центре которых находятся атомы
кремния, чередуются с пустыми. Таким образом, из восьми октантов заселенными
оказываются только четыре. При таком расположении каждый атом кремния
окружен четырьмя другими, которые в свою очередь окружены четырьмя следу-
следующими атомами, находящимися на тех же расстояниях 0,225 нм. Таким образом,
в кристаллической структуре кремния все атомы его тождественны друг другу,
т.е. отсутствуют молекулы.
Структуре кремния подобен сульфид цинка. На рис. 4 приведена элементар-
элементарная ячейка кристаллического сульфида цинка. Одни атомы (безразлично, цьька
или серы) занимают вершины и центры граней куба, а другие — центры четырех
(кз восьми) октантов. Пизтоку в структуре ZnS каждый атом цгкка окружен
четырьмя атомами серы и, наоборот, каждый атом серы соединен с четырьмя
атомами цинка. Значит, структура ZiiS аналогична структуре кремния с той
только рагш;цей, что центры малых кубов заняты атомг..ь и другого вида по срав-
сравнению с вершинами и центрами граней большого куба. В целом кристалл ZnS
можно рассматривать как совокупность бесконечно повторяющихся зъенъев
-Zn-
в пространстве. Опять приходим к выводу о том, что в структуре сульфида цинка
непьзя найти обособленные молекулы ZnS. В то же время измерение плотности
паров, а также масе-снектрометричеекие исследования сульфида цинка подтверж-
подтверждают существование молекул ZnS в газообразном состоянии.
Таким образом, во всех рассмотренных структурах нельзя выделить обособ-
обособленные молекулы. Такие кристаллические структуры, в которых отсутствуют
дискретные молекулы, а межатомные связи однотипны, называют координацион-
координационными. Другими словами, координационные структуры гомодесмичны, т.е. атомы в
кристалле соединены одинаковым типом химической связи. Для большинства
неорганических веществ (более 95%) характерны именно координационные струк-
структуры.
Р и с. 3. Структура кремния
Р и с. 4. Структура сульфида
цинка
На рис. 5 для сравнения приведены элемен-
элементарные ячейки молекулярных структур иода
(а) и диоксида углерода E). Их важнейшей
особенностью в отличие от предыдущих типов
кристаллов является то, что в узлах кристалли-
кристаллической решетки находятся не атомы, а молеку-
молекулы. При этом расстояния между атомами в
молекуле меньше, чем межмолекулярные рас-
расстояния в кристалле. В них атомы связаны в
молекулы прочными ковалентными связями, а
между молекулами действуют слабые силы Ван-
дер-Ваальса (см. § 1 гл. V). Это значит, что
структуры иода и диоксида углерода являются
гетеродесмичными. К рассмотренным выше так
называемым островным молекулярным структу-
структурам Aг и СОг) относится абсолютное большин-
большинство органических соединений. Однако некото-
некоторые неорганические вещества, не имеющие
молекулярной структуры (цепочечные, слоис-
слоистые, каркасные), также гетеродесмичны, так
как внутри цепей, слоев и каркасов межатом-
межатомные связи ковалентные, а между цепями, слоя- р и с. 5. Молекулярные структуры крис-
ми и каркасами функционируют силы Ван-дер-
Ваальса.
5. Неорганические полимеры. Вещества со структурой координационных решэ-
ток могут быть названы неорганическими полимерами, если под последними
понимать макротела, состоящие из большого числа повторяющихся звеньев.
По составу неорганические полимеры делятся на дзе основные труп 1ы: w.uo-
атомные, построенные из одинаковых атомов, и гетероатомнче, в которых
звенья образованы из двух и большего числа атомов различных химических
элементов.
В отличие от органических полимерных веществ, в которых преобладают
линейные гомоцепные (состоящие из одинаковых атомов) структуры, неорганиче-
неорганические полимеры характеризуются преимущественно гетероцепной пространствен-
пространственной структурой. Неорганические полимеры отличаются повышенной термостой-
термостойкостью, высокими температурами плавления, большой прочностью и твердостью.
Многие из них относятся к полупроводникам и сверхпроводникам. Большинство
неорганических полимеров характеризуется большой хрупкостью. Однако некото-
некоторые линейные гетероцепные полимеры обладают высокозластичными свойствами
и являются настоящими неорганическими эластомерами.
6. Фаза. В современной общей и неорганической химии принципиально важ-
важным является понятие фазы. Фазой называется гомогенная* часть гетерогенной*
системы, обладающая одинаковым химическим составом и термодинамическими
свойствами, ограниченная поверхностью раздела, при переходе через которую
свойства меняются скачком. Термодинамические свойства — это свойства, зави-
зависящие от состава вещества, температуры, давления и концентрации. К ним отно-
диоксида угяерода (б)
"Гомогенный (лат.) — однородный, гетерогенный — неоднородный.
сятся, например, теплоемкость и удельный объем. Существенно, что на границе
данной фазы с внешней средой или другими фазами термодинамические свойства
и химический состав меняются скачкообразно. Под системой понимают совокуп-
совокупность всех веществ, участвующих в химическом равновесии. Данное определение
фазы является термодинамическим. Это понятие применимо лишь для равновес-
равновесных систем. Система называется равновесной, если в ней не происходят измене-
изменения во времени и не имеет места перенос вещества или энергии через нее. Для
гомогенной системы вся совокупность свойств строго одинакова во всех ее частях.
Так, ненасыщенный раствор соли в воде представляет собой пример гомогенной
системы, если не считаться с паром над раствором. Гомогенная система однофаз-
однофазна, гетерогенные системы состоят более чем из одной фазы.
Количественный состав фаз может меняться в определенных пределах. На
примере раствора соли в воде предельный количественный состав жидкой фазы
совпадает с концентрацией насыщенного раствора при данной температуре. Если
содержание соли превышает концентрацию насыщенного раствора, возникает
вторая фаза (кристаллы избыточной соли) и система становится гетерогенной.
Очевидно, что газо- и парообразные системы всегда однофазны независимо от
качественного и количественного состава. Существенно, что количественный
состав твердой фазы также может изменяться в некоторых границах. Это касает-
касается не только твердых растворов, но и химических соединений, находящихся в
твердом состоянии. Таким образом, фазы могут обладать постоянным нли пере-
переменным составом. К фазам постоянного состава относится большинство химиче-
химических соединений в газообразном состоянии. Фазы переменного состава — это
газообразные, жидкие и твердые растворы, а также твердые соединения, состав
которых в зависимости от условий получения варьирует в некоторых пределах.
Для немолекулярных кристаллов понятие молекулы лишено смысла. Для них
формой существования химического соединения в твердом состоянии является
фаза. Поэтому фаза — носитель всех физических, физико-химических и химических
свойств вещества, кристаллизующегося в коордгшационной структуре, т.е. его
свойства зависят от состава и химического строения фазы. В этом заключается
фундаментальность понятия фазы в современной химической атомистике. Однако
понятие фазы здесь употребляется уже не в термодинамическом, а в несколько
ином смысле. Если с термодинамической точки зрения понятие фазы можно
применять только к равновесным системам, то фаза как носитель свойств вещест-
вещества с немолекулярной структурой — это однородная но составу и свойствам часть
системы. При этом подразумевается, что фаза может быть и метастабильной
(неравновесной), но тем не менее она вполне характеризует свойства объекта.
7. Постоянный и переменный состав. Формульная масса. Во времена Д.И.Мен-
Д.И.Менделеева химические соединения считались определенными, т.е. имеющими посто-
постоянный и неизменный состав. В качестве неопределенных соединений с перемен-
переменным химическим составом Менделеев приводил растворы и сплавы. В металличе-
металлических сплавах важнейшие структурные составляющие — соединения металлов
между собой (металлиды). Характерной особенностью металлидов оказалась
изменчивость их составов в определенных границах. Таким образом, металлиды
являются типичным примером соединений переменного состава.
В настоящее время установлено, что к соединениям переменного состава отно-
относятся не только металлиды, а вообще большинство немолекулярных соединений.
Так, многочисленные оксиды, сульфиды, селениды, теллуриды, нитриды, фосфи-
ды, карбиды, силициды и др., как правило, относятся к соединениям переменно-
переменного состава. Больше того, галогениды металлов в твердом состоянии также пред-
представляют собой фазы переменного состава (например, NaCl), хотя для доказа-
доказательства этого требуются более тонкие методы. Рассмотрим некоторые типичные
примеры соединений переменного состава.
Состав природного (пирротин) и искусственно полученного сульфида жапеза
FeS характеризуется "избыточным" содержанием серы против стехиометрии.
Согласно стехиометрическому составу, в FeS на один атом железа приходится
один атом серы, т.е. атомное содержание 50% Fe и 50% S. В действительности
оказалось, что сильфид железа содержит не избыток серы, а в нем недостает
атомов железа по сравнению со стехиометрическим составом. В синтетических
образцах FeS атомное содержание его меняется от 45 до 50% Fe. Таким образом,
формулу сульфида железа правильнее писать в виде Fei-^S, где х меняет значе-
значения от нуля E0% железа) до 0,05 D5% Fe). Для природных кристаллов сульфи-
сульфида железа х колеблется в пределах 0,1 — 0.2, т.е. наблюдается недостаток от 10
до 20% атомов железа против формульного состава. Это значит, что условия
формирования пирротина в земной коре были существенно иные по сравнению с
синтетическим сульфидом железа. В то же время природный сульфид железа
представляет собой пример истинного неспи-хиометрическою соединения, для
которого состав FeS A:1) является идеальным, а не реальным {х — 0,1 т 0,2).
Хорошо изученным соединением переменного состава является и оксид железа
FeO. Как и в моносульфиде, в оксиде железа (+2) наблюдается недостаток ато-
атомов железа по сравнению со стехиометрическим составом. Поэтому формулу окси-
оксида железа (+2) следует изображать так: Fei-^O. Нестехиометричность оксида
железа в сторону недостатка железа понятна, если учесть химическую аналогию
кислорода и серы. Для оксида железа (+2) впервые установлен факт повышения
температуры плавления с нарушением стехиометричеекого состава. Так, для
Fe0>3SO (х - 0,07) т.пл. 1378°С. Fe0,9iO (х = 0,09) и Fe0>89O (x = 0,11) плавятся
соответственно при 1382 и 1387°С. Для координационных кристаллов температу-
температура плавления характеризует прочность соединения. Таким образом, до опреде-
определенного предела устойчивость оксида железа растет вместе со степенью наруше-
нарушения стехиометрического состава. Кроме того, оксид железа (+2) как соединение
эквиатомарноо A атом Fe на 1 атом О) просто не существует, так как область
нестехиометрии на самом деле не включает стехиометрический состав.
Для оксида титана (+2), кристаллизующегося в структуре NaCl, нарушение
стехиометрического состава наблюдается относительно обоих сортов атомов. В
TiO в зависимости от условий получения (температура, давление кислорода)
стехиометрический индекс кислорода может меняться от 0,58 до 1,33. Это значит,
что все составы оксида титана (+2) от 0,58 до 1,00 будут характеризоваться недо-
недостатком атомов кислорода (соответственно избыток атомов титана) против стехио-
стехиометрии. А составы от 1,00 до 1,33 будут иметь избыток атомов кислорода (или
недостаток атомов титана) по сравнению со стехиометрическим составом. Таким
образом, формула оксида титана (+2) с учетом нарушения стехиометрического
состава может быть представлена как TiOo,53-i,33i те- на Ю0 атомов титана может
приходиться от 53 до 133 атомов кислорода.
Составы, укладывающиеся внутри граничных значений нарушения стехиомет-
стехиометрического состава, называют областью нестехиометрии или областью гомоген-
гомогенности. Такие соединения, стехиометрический состав которых лежит внутри обла-
17
сти гомогенности, называют двусторонними фазами. А сульфид и оксид железа
(+2) являются примерами односторонних фаз, так как область гомогенности для
них наблюдается по одну сторону от стехиометрического состава. Однако соеди-
соединения переменного состава не обязательно должны иметь очень широкие области
гомогенности, как это было в приведенных примерах. Ширина области гомоген-
гомогенности прежде всего зависит от физико-химической природы самого соединения.
Примером двусторонней фазы с узкой областью гомогенности может служить
сульфид свинца PbSo,9995-1,0005- Еще меньшую область гомогенности имеет суль-
сульфид кадмия CdS, представляющий собой одностороннюю фазу с недостатком
серы против стехиометрического состава.
Наибольшие области гомогенности наблюдаются у металлических соединений.
Для них обычные методы классического химического анализа, как правило,
более чем достаточны для установления области нарушения стехиометрическо-
стехиометрического состава. У условно ионных и ковалентных координационных кристаллов ко-
количественное определение области гомогенности требует привлечения современ-
современных прецизионных физико-химических и физических методов. Поэтому длитель-
длительное время объектами классической химии считались соединения постоянного
состава.
Для соединений переменного состава, не имеющих молекулярной структуры,
вместо молекулярной массы целесообразно ввести понятие формульной массы.
Формульная масса равна сумме атомных масс входящих в данное соединение
элементов, умноженных на фактические стехиомзтрическле индексы в химиче-
химической формуле соединения. К примеру, формульная масса оксида титана (-1-2)
состава ТЮо,з2 равна 47,9 + 16,00-0,82 = 61,02. Для молекулярных структур
формульная масса вещества совпадает с его молекулярной массой.
8. Ограниченный характер и границы применимости стехичметрических зако-
законов химии. Современная формулировка стехиометричесних з.шонов. При образо-
образовании подавляющего большинства неорганических соединений их состав можэт
быть переменным в пределах области гомогенности. Постоянный и неизменный
химический состав наблюдается только для молекул (например, NH3, SO2 и т.п.),
а также кристаллов с молекулярной структурой. А последних среди твердых
неорганических веществ очень мало, и они представляют исключения (менее 5%).
Таким образом, молекулы являются одной из форм существования химических
соединений, но не единственной. Для типичных твердых неорганических простых
веществ и соединений характерна немолекулярная форма существования веще-
вещества.
Стехиометрические законы химии — постоянства состава, эквивалентов и
кратных отношений — были в свое время сформулированы применительно к
молекулам, а потому справедливы для молекулярной формы вещества. Для немо-
немолекулярных структур постоянство состава и вытекающие из него следствия не
являются уже критерием образования химических соединений. Поэтому в насто-
настоящее время стехиометрические законы химии формулируются с учетом единства
молекулярной и немолекулярной форм существования вещества.
Закон постоянства состава. Состав молекулярного соединения
остается постоянным независимо от способа его получения. В отсутствие
молекулярной структуры его состав зависит от условий получения и предыдущей
обработки. Возьмем, к примеру, аммиак. Независимо от способов получения
(прямой синтез из простых веществ, разложение аммонийных солей, действие
кислот на нитриды активных металлов и т.п.) состав молекулы аммиака всегда
постоянен и неизменен: на атом азота приходится три атома водорода. А для
оксида титана (+2) состав соединения зависит от условий получения и предыду-
предыдущей обработки. В молекуле аммиака, состоящей лишь из четырех атомов, исклю-
исключается изменчивость состава. Оксид же титана (+2) представляет собой фазу,
состоящую из огромного числа атомов (порядка постоянной Авогадро), которая и
определяет свойства этого соединения. Это ярчайший пример перехода количест-
количества в качество: коллектив из колоссального числа частиц обладает уже новым
качеством — непостоянством состава.
Закон эквивалентов. Для молекулярных соединений массовые
количества составляющих элементов пропорциональны их химическим эквивален-
т.ал1; при отсутствии молекулярной структуры массовые количества составля-
составляющих элементов могут отклоняться от значений их химических эквивалентов. В
аммиаке на 1 масс.ч. водорода (его химический эквивалент) приходится точно
14/3 масс.ч. азота. Последняя величина и есть эквивалентная масса азота. Для
оксида титана (+2) стехиометрического состава TiO 47,90/2 масс.ч. Ti (эквива-
(эквивалентная масса титана в этом соединении) соединяются с 8 масс.ч. кислорода. В
оксиде титана состава TiOo,s2 то же количество титана соединяется с 8-0,82 =
= 6,56 масс.ч. кислорода, т.е. на 8 — 6,56 = 1,44 меньше его эквивалентной массы.
Итак, если валовой состав соединения содержит дробные индексы, то массовые
количества составляющих элементов отличаются от эквивалентных масс.
Закон кратных отношений. Если два элемента образуют меж-
между собой несколько молекулярных соединений, то массовые количества одного
элемента, приходягциеся на одно и то же массовое количество другого, относят-
относятся Л1ежду собой как небольшие целые числа. Для соединений, не имеюгцих молеку-
молекулярной структуры, массовые количества одною из них, приходящиеся на одно и
то же количество другою, могут относиться между собой как дробные числа.
Нетрудно подсчитать, что в оксидах углерода СО2, СО и С3О2 массовые части
углерода, приходящиеся на одну и ту же массовую часть кислорода, например на
16, относятся между собой как целые небольшие числа 1:2:3. Отношения массо-
массовых частей кислорода, приходящихся на одну и ту же массовую часть титана в
оксидах переменного состава TiOo^e-i зз, TiOi 45-1,56 и TiOi 9-2,0- выражаются
дробными числами. Только у оксидов стехиометрического состава TiO, T12O3
(TiOi 5) и TiO2 массовые количества кислорода на постоянную массовую часть
титана относятся между собой как 2:3:4.
9. Закон постоянства свойств. Кристаллохимическое строение и свойства.
Логическим следствием закона постоянства состава является закон постоянства
свойств (Пруст, 1806) — свойства вещества не зависят от способа его получения
и предыдущей обработки. Этот закон относится только к молекулярным соедине-
соединениям. Свойства химических соединений, не имеющих молекулярной структуры,
прямо зависят от способа получения и предыдущей обработки. Это прежде всего
связано с тем, что количественный состав соединения зависит от условий его
получения. А свойства вещества являются в первую очередь функцией состава.
Однако, по Бутлерову, свойства вещества зависят не только от качественного и
количественного состава, но и от химического строения. Но классическая теория
химического строения Бутлерова относится к молекулярной химии, поскольку
она рассматривает химическое строение именно молекул. Это и понятно, так как
IQ
во времена Бутлерова вся химия (органическая и неорганическая) развивалась на
уровне молекулярной химии.
Подавляющее большинство неорганических веществ в условиях комнатной
температуры и атмосферного давления — твердые вещества с немолекулярной
структурой. Поэтому на первый взгляд может показаться, что теория химическо-
химического строения Бутлерова неприменима для типичных неорганических соединений.
На самом же деле такой вывод является преждевременным. Дело в том, что ос-
основная идея Бутлерова о взаимосвязи между химическим строением и свойствами
остается в силе и для веществ, не имеющих молекулярной структуры. Только
для последних вместо химического строения вводится понятие кристаллохимиче-
ского строения.
Криста.глохимичсское строение — порядок расположения и природа связи агп.о-
люв в пределах элементарной ячейки, их взаимное влияние dpyi па друга, а также
распределение электронной плотности, величины эффективных зарядов. Как
видно из этого определения, понятие кристаллохимического строения представ-
представляет собой превращенную форму химического строения молекул применительно к
немолекулярным структурам. Вот почему теория химического строения Бутлеро-
Бутлерова — общехимическая теория, в одинаковой степени приложимая как к органи-
органическим, так и к неорганическим объектам. На рис. 6, а приведена кубическая
структура стехиометрического соединения АВ. Она показывает только порядок
размещения атомов в элементарной ячейке и не отображает природу межатомных
связей, а также их взаимное влияние. Вообще кристаллическая структура в той
мере отражает кристаллохимическое строение вещества, в какой структурная
формула — химическое строение молекулы. В действительности химическое и
кристаллохимическое строение — понятие динамическое, а не статическое.
Кристаллическая структура твердого хлорида моди (+1) аналогична структуре
сульфида цинка (см. рис. 4). Атомы меди располагаются ь вершинах и центрах
граней куба, а атомы хлора находятся в центрах четырех из восьми октантов.
Каждый атом хлора непосредственно связан с четырьмя атомами меди, и, наобо-
наоборот, каждый атом меди — с четырьмя атомами хлора. В результате в твердом
хлориде меди элементы проявляют валентность, равную четырем.
Таким образом, молекулы CuCl в паровой фазе и твердый хлорид меди — это
вещества, разные по структуре, а следовательно, и по свойствам. Количественный
состав твердого хлорида меди такой же, как и молекул: на атом меди приходится
атом хлора. Для хлорида меди в парах формула CuCl является истинной, а для
твердого состояния — только простейшей. Истинная формула кристаллического
хлорида меди может быть написана, как для полимерного вещества (CuCl)n. На
рис. 6, а представлена идеальная структура стехиометрического соединения АВ,
когда все атомы размещены по узлам решетки. На рис. 6, б один атом А находит-
находится в междоузлии, а узел (откуда ушел атом А) остается незанятым. Рис. 6 отра-
отражает различное кристаллохимическое строение стехиометрического соединения
одного и того же состава. Как показывают опыт и теория, реальные кристаллы
предпочтительнее образуют дефектную структуру (рис. 6, б). Концентрация же
дефектов (в данном примере атом А в междоузлии и вакансия в узле) находится
в зависимости от способа получения и предыдущей обработки вещества.
Итак, кристаллы одного и того же соединения, полученные разными метода-
методами, отличаются своей реальной структурой и свойствами. Поэтому при одинако-
одинаковом качественном и количественном составе веществ с координационной струк-
Р и с. 6. Кубическая структура стеююмегричеекого соединения
АВ: а - идеальная структура; б - атом А в междоузлии
турой их свойства зависят от. условий получения и предыдущий обработки (на-
(например, термической). Здесь в первую очередь речь идет об электрических и
оптических свойствах, которые несравненно более чувствительны к нарушениям
регулярности решетки и примесям, чем свойства химические. Химические свойст-
свойства (они основаны на химических реакциях) более чувствительны к тонкостям
химического строения молекул.
10. Химический индивид. Химическое соединение. В химии главными объек-
объектами изучения являются химические индивиды. Последние противопоставляются
механическим смесям и отличаются от растворов, состоящих из различных хими-
химических индивидов. Химический индивид представляет собой фазу, состоящую u.i
одною вида вещества — простого или сложного. Все простые вещества и химиче-
химические соединения в чистом виде являются химическими индивидами*.
Химическое соединепие — однородное вещество постоянного или персмепного
состава с качественно своеобразным химическим или кристалл охимическим
строением, образованное из атомов одного или нескольких химических элементов.
Химическое соединение прежде всего характеризуется однородностью. В
"Основах химии" Менделеев пишет: "Ближайший предмет химии составляет
изучение однородных веществ... Химия занимается только однородными телами...
Химическим соединением вообще называют такое соединение двух или более тел,
продукт которого представляется нам однородным, однообразным во всех своих
мельчайших частицах... Это есть единственное определение, какое можно дать
химическому соединению, и в этом отношении неопределенные соединения также
совершенно ему подчиняются".
Таким образом, основатель современной химии Д.И.Менделеев отмечал суще-
существование однородных неопределенных соединений, т.е. веществ переменного
состава в области гомогенности. К тому же современное учение о фазах не делает
принципиального различия между химическими соединениями постоянного и
переменного состава. А химическое соединение, по существу, представляет собой
фазу, ибо оно всегда однородно. Поэтому состав химического соединения может
быть как постоянным (молекулы), так и переменным (координационные кристал-
Согласно определению, химические индивиды должны быть абсолютно чистыми, что
практически не достигается. В этом смысле понятие "химический индивид" является
идеальным. Реальный химический индивид всегда содержит примеси.
21
лы). В этом также заключается характерная черта определения химического
соединения.
Однако к фазам переменного состава относятся и растворы. В газовых раство-
растворах, несмотря на их однородность, имеется смесь молекул (например, молекул
кислорода, азота, диоксида углерода и т.п. в воздухе). В жидких растворах
отсутствуют молекулы с качественно новым химическим строением по сравнению
с химическим строением исходных компонентов. Однако в растворах неорганиче-
неорганических веществ часто возникают новые структурные образования (неопределенные
сольваты, гидраты, гидратированные ионы и т.д.), не относящиеся к типическим
новообразованиям. Твердые растворы обладают кристаллохимическим строением
компонента-растворителя. В отличие от твердых растворов химическое соедине-
соединение переменного состава характеризуется присущим только ему кристаллохимиче-
кристаллохимическим строением, не свойственным строению компонентов. Поэтому в противопо-
противоположность твердым растворам свойства соединений переменного состава резко
отличаются от свойств составляющих веществ.
Наконец, классическая химия к химическим соединениям относила лишь
химические индивиды, состоящие из атомов различных элементов. Поэтому,
например, молекула кислорода с характерным для нее химическим строением и
специфическими свойствами не считалась химическим соединением. В действи-
действительности понятие химического соединения относится к соединению атомов друг
с другом. Соединяющиеся атомы могут принадлежать либо к одному, либо к
нескольким химическим элементам. Поэтому все простые вещества, по существу,
являются химическими соединениями, образованными из атомов одного и того
же химического элемента. Молекулы газов (водород, кислород, азот и т.д.) состо-
состоят из двух атомов, а простые твердые вещества — из огромного числа одинаковых
атомов, соизмеримого с постоянной Авогадро.
Химическому соединению присуще только ему свойственное химическое или
кристаллохимическое строение. В химическом или кристаллохимическом строе-
строении главное — это химическая связь, ее природа. Именно химические соединения
характеризуются наличием химической связи. С этой точки зрения молекулы и
кристаллы, построенные из одинаковых атомов, являются химическими соедине-
соединениями. Атомы в молекуле водорода связаны ковалентной связью. Все свойства
(физические, химические, спектральные и т.п.) молекулярного водорода отличны
от атомарного *. А по Менделееву, в результате химического взаимодействия
образуется тело, отличное от взаимодействующих веществ. Еще большее разли-
различие в свойствах, например, металлической меди (атомы связаны металлической
связью) от свойств составляющих атомов меди. Вообще кажется странным,
почему классическая химия считает, что в результате процесса Н + F —> H~~F
образуется химическое соединение, а в процессе Н + Н —> Н~~Н или F + F —>
—> F—F оно не возникает. Это по меньшей мере не логично. Естественно призна-
признание как гетероатомных (например, HF), так и гомоатомных химических соедине-
соединений (Нг, F2, металлы и т.п.).
"Свободные атомы имеют линейчатый спектр (например, атомарный водород); молеку-
молекулы же (например, молекулярный водород) характеризуются полосатым спектром, состоя-
состоящим из полос или групп густо расположенных линий; нагретые твердые тела испускают
сплошной спектр.
ГЛАВА III. СТРОЕНИЕ АТОМА И ПЕРИОДИЧЕСКАЯ
СИСТЕМА ЭЛЕМЕНТОВ
1. Модели строения атома. В химии своеобразными элементарными частицами
являются атомы, из которых построены все химические индивиды. Громадное
разнообразие химических соединений обусловлено различным сочетанием атомов
химических элементов в молекулы и немолекулярные вещества. Способность же
атома вступать в химические соединения, его химические и физические свойства
определяются структурой атома. Отсюда для химии первостепенное значение
имеет строение атома, в первую очередь структура его электронной оболочки.
Согласно модели первооткрывателя электрона Томсона A904), атом представ-
представляет собой "сферу положительного электричества" одинаковой плотности по
всему объему диаметром порядка 0,1 нм. Электроны как бы "плавают" в этой
сфере, нейтрализуя положительный заряд. Экспериментальную проверку этих
наглядных представлений предпринял английский физик Эрнест Резерфорд в
своих знаменитых опытах по рассеянию а-частиц (ядра атома гелия). Схема
установки Резерфорда A907) приведена на рис. 7. Радиоактивный препарат Р
излучает о-частицы ("снаряды") в виде узкого пучка, на пути которого ставится
тонкая золотая фольга Ф. Регистрация а-частиц, прошедших через фольгу,
производится микроскопом М на люминесцирующем экране Э по вспышкам
световых точек [сцинтилляция). Если модель Томсона верна, а-частицы не могут
пройти даже через очень тонкую фольгу*, так как атомы заполняют все про-
пространство: летящие снаряды должны остановиться, передач свою энергию и им-
импульс фольге.
Однако результаты опыта Резерфорда (рис. 8) показали, что: 1) для подав-
подавляющего большинства а-частиц фольга прозрачна и они проходят сквозь нее, не
изменяя своего первоначального направления; 2) наблюдаются с^-частицы, рас-
рассеянные под разными углами в\ 3) с увеличением угла рассеяния в число рас-
рассеянных а-частиц убывает; 4) ничтожная часть о^частиц (примерно 1 частица на
10000) отбрасывается в обратном направлении, т.е. В = 180°. Летящая а-части-
ца, имеющая энергию несколько МэВ, может быть отброшена назад только при
* К насосу
Рис. 7. Схема установки Ре-
Резерфорда
III
—0—»~ДУ 90
Угол рассеяния, град
Р и с, 8. Зависимость относительного чис-'
ла а-частиц, прошедших через фольгу
в 1 мин, от угла рассеяния
В опытах Резерфорда золотая фольга имела толщину 0,001 мм, поперек которой ук-
укладывалось около 3300 слоев атомов золота.
23
столкновении с положительно заряженной части-
частицей большой массы. Подсчет числа таких частиц
позволил определить размер этой положительно
заряженной массы порядка 10~13 см. Основываясь
на этом, Резерфорд в 1911 г. предложил так
р называемую планетарную модель строения атома.
Р и с. 9. Интерпретация опыта Ре- Согласно этой модели, атом уподобляется Сол-
зерфорда нечной системе в микромасштабе: а) в центре
атома имеется положительно заряженное ядро с зарядом Ze, в котором
сосредоточена почти вся масса атома; 6) вокруг ядра по орбитам двиисутся Z
отрицательно заряженных электронов: в) размеры атомного ядра на много
порядков меньше размеров самого атома (соответственно 10'13 и 10"8 см). Этим и
объясняется "прозрачность" фольги — снаряды пролетают через огромное (в
масштабах атома) пространство между ядром и электронами.
На базе планетарной модели рассеяние а-частиц объясняется следующим
образом. Если бы «-частица не взаимодействовала с ядром, она пролетела бы от
него иа некотором расстоянии П, называемом прицельным расстоянием (пунктир-
(пунктирная прямая на рис. 9). Однако в результате одноименности зарядов ядро от-
отталкивает «-частицу, которая начинает двигаться по гиперболе, отклонившись на
угол в от первоначального направления. При этом влиянием электронов на тра-
траекторию а-частицы можно пренебречь, так как масса электрона очень мала по
сравнению с ядрами атома гелия. Величина угла тем больше, чем больше Z и чем
меньше П и кинетическая энергия летящей а-частицы. Из опытов по рассеянию
а-частиц Резерфордом была определена величина положительного заряда ядер Z
различных химических элементов. Оказалось, что положительный заряд ядра
равен приблизительно половине атомной массы рассматриваемого элемента (ма-
(материал фольги). Впоследствии Чэдвик A920) усовершенствовал опыты по рас-
рассеянию а-частиц ядрами атомов различных химических элементов. На примере
атомов меди, серебра и платины он показал, что заряд ядра Z численно равен
порядковому номеру элемента в Периодической системе элементов Д.И.Менде-
Д.И.Менделеева.
2. Строение атома по Бору. Планетарная модель Резерфорда противоречила
факту устойчивого существования самих атомов с точки зрения законов классиче-
классической физики. Дело в том, что движение электрона по орбите есть электрический
ток, который индуцирует в пространстве электромагнитное поле. На создание
последнего расходуется энергия электростатического взаимодействия электрона с
ядром, в результате чего электрон должен двигаться по спирали (а не по замкну-
замкнутой орбите) и упасть на ядро, что равносильно ликвидации атома.
Расчеты показывают, что продолжительность жизни атома в таком случае
должна быть порядка 10~8 с. В действительности же атомы — исключительно
устойчивые образования. Кроме того, при движении электрона по спирали энер-
энергия его должна уменьшаться непрерывно и атомный спектр должен быть также
непрерывным. А опыт показывает, что все атомные спектры имеют дискретный
(линейчатый) характер.
Выход из создавшегося положения был найден великим датским ученым
Нильсом Бором в 1913 г. Он исходил из модели Резерфорда, опирался на учение
Эйнштейна о световых квантах A905) и на квантовую теорию излучения Планка
A900). Согласно последней, вещества поглощают и излучают энергию отдельны-
ми порциями — квантами. При этом энергия кванта электромагнитного излуче-
излучения пропорциональна частоте этого излучения и:
Е = hv, (III Л)
где h — постоянная Планка. Наравне со скоростью света и зарядом электрона
постоянная Планка относится к числу фундаментальных констант природы.
Основные положения своей теории строения атома Бор сформулировал в виде
постулатов. Эти постулаты накладывают определенные ограничения на разрешен-
разрешенные классической физикой формы движения. Первый постулат Б о -
р а: электрон в атоме может находиться только в стационарных или квантовых
состояниях с дискретными значениями энергии _?', в которых атом не излучает.
Для стационарных состояний момент количества движения электрона М равен
целому кратному постоянной Планка fi — Л/Bтг), т.е.
М = nt= nh/Bx) *, (III.2)
где п — целое число. Первый постулат Бора, называемый условием квантования
орбит, находится в явном противоречии с классической физикой, согласно кото-
которой энергия движущегося электрона может принимать любые значения.
Второй пост}'лат Бора (условие частот): при переходе из одного
стационарною состояния о друюе атом испускает или поглощает квант элек-
тромагнитноло излучения, частота которою определяется соотношениелг.
Еп- Ет= hv. (III.3)
Если п > т, то атом переходит из стационарного состояния с более высокой
энергией на орбиту с меньшей энергией с выделением кванта лучистой энергии
При п < т наблюдается обратная картина с поглощением фотона. Атомы в ос-
основном (нормальном) состоянии могут только поглощать кванты света, переходя в
возбужденное состояние. Возбужденный же атом может как поглощать, так и
испускать фотоны. Продолжительность пребывания атома в возбужденном состоя-
состоянии порядка 10~8 с.
Модели стационарных состояний соответствуют боровским или дозволенным
орбитам, при движении по которым электрон не теряет энергии. Бор предполо-
предположил, что электроны движутся вокруг ядра по круговым дозволенным орбитам.
Тогда, согласно (III.2), можно написать
movr = nt, (HI-4)
где mo — масса электрона; v — скорость электрона; г — радиус орбиты. Для дви-
движения электрона по окружности ему необходимо сообщать центростремительное
ускорение. Это ускорение v2/r электрону сообщает сила кулоновского взаимодей-
взаимодействия электрона с ядром, а потому справедливо равенство
или
*Л = А/Bтг) называется также постоянной Дирака и равна il = 1,054-10~34 Дж-с.
25
Разделив почленно (III.5) на (Ш.4), получим
v =
т.е. скорость и, следовательно, кинетическая энергия электрона обратно пропор-
пропорциональны п. Целое число п называют главным квантовым числом, и в модели
атома Бора оно означает порядковый номер дозволенной орбиты от ядра. Из
(III.5) и (III.6) получаем
г = ЪЧ.2/(т0еЩ. (III.7)
Таким образом, радиусы дозволенных орбит относятся как 12:22:32 и т.д. Для
атома водорода (п = 1 и Z — 1) расчеты по (III.7) приводят к г = 0,053 нм. Эта
величина служит характеристической длиной и называется боровским радиусом.
Для элемента с порядковым номером Z радиус первой орбиты будет в Z раз мень-
меньше.
Важнейшей заслугой теории Бора явилось количественное обоснование спект-
спектра атома водорода и водородоподобных атомов. Полная энергия электрона в
атоме складывается из кинетической (mov2/2) и потенциальной — (e2Z/r), или с
учетом (III.5) получаем
Е = mov2/2 - e2Z/r = m0v?/2 - m0v2 - -moi?/2. (III.8)
Итак, полная энергия электрона равна его кинетической энергии, взятой с обрат-
обратным знаком. Теперь в (III.8) подставим выражение скорости электрона из (III.6) и
получим
Е = -m0i,2/2 = -moe«27Bn2»2). (Ш.9)
Из условия частот Бора (III.3) с учетом (Ш.9) имеем
ИЛИ
hu =
откуда
Коэффициент перед скобками есть теоретическое выражение константы Ридберга
R в его формуле для описания линий спектра атома водорода в видимой области
(серия Бальмера):
J/= ДA/22 - 1/п2). (III.11)
Сравнение (ШЛО) и (III.11) для атома водорода (Z = 1) дает
R = т0е*/DжП). (III.12)
Рассчитанная по формуле (III.12) константа Ридберга хорошо совпадает с опыт-
опытной величиной.
3. О квантовой механике. Доминирующей современной теорией поведения
электронов и других микрообъектов, обладающих очень малой массой, является
квантовая механика. Квантовая волновая механика изучает законы движения
микрообъектов в силовых полях. Главной особенностью квантовой механики
является ее вероятностный статистический характер: она даст возможность нахо-
находить вероятность того или иного значения некоторой физической величины. В
отличие от классической физики в квантовой механике все объекты микромира
(электроны, атомы, молекулы и др.) выступают как носители и корпускулярных
и волновых свойств (волново-корпускулярный дуализм), которые не исключают, а
дополняют друг друга. Не представляет труда обосновать объективность волново-
корпускулярного дуализма для световых квантов — фотонов. Так, фотоэффект
Столетова и эффект Комптона доказывают корпускулярную природу видимого и
рентгеновского излучений, а интерференция и дифракция — волновую природу
света. Поэтому для фотонов легко показать единство волны и корпускулы. Дейст-
Действительно, из формул A.1) и (III.1) следует Е = с2т. = hi/, откуда с учетом v — с/А
получаем
с2т = hc/X
или
А = h/(mc) = h/p, A11.13)
где А — длина волны; р — количество движения, или импульс фотона.
В 1924 г. французский ученый де Бройль высказал идею, что волново-корпус-
кулярный дуализм присущ не только фотонам, но и всем микрообъектам. Поэто-
Поэтому, по де Бройлю, их движение связано с распространением волны, т.е. движение
микрообъекта можно рассматривать как волновой процесс, при котором справед-
справедливо соотношение
А = h/(mov), (III.14)
аналогичное (III.13) для фотона. Формула (III.14) связывает важнейшую характе-
характеристику вещественной формы существования материи (количество движения mov)
с характеристикой материального поля (длина волны А) через постоянную План-
Планка.
Однако идея де Бройля послужила только началом создания квантовой меха-
механики. Она рассматривала поведение микрообъекта, свободного от силового поля.
В действительности же материальные частицы, например электроны, всегда нахо-
находятся в поле действия определенных сил. С этой точки зрения электроны в атоме
движутся в центрально-симметричном поле, для которого потенциальная энергия
зависит только от расстояния до ядра. Законы движения в поле центральных сил
образуют основу атомной механики: решение общей задачи о движении электро-
электронов в атоме опирается на результаты, относящиеся к движению одной частицы в
поле центральных сил. На основании гипотезы де Бройля австрийский ученый
27
r\
\x
P и с. 10. Распространение волны
ВДОЛЬ ОСИ X
т
\
Рис. 11. Стоячая волна
Шрёдингер A925 — 1926) интуитивно использовал волновое уравнение классиче-
классической механики в качестве модели для описания поведения электрона в атоме. Из
учения о колебаниях и волнах известно, что распространение волны вдоль коор-
координатной оси х (рис. 10) описывается дифференциальным уравнением в частных
производных второго порядка
д2А _ 1 &А
!h& ~~ 'с2 dt2"
(III.15)
где А — амплитуда волны; с — скорость перемещения волны; t — время перемеще-
перемещения волны.
Но, по Шрёдингеру, атомная система замкнутая, а потому поведение электро-
электрона, его движение подобно стоячей волне (рис. 11). А математическое уравнение,
описывающее стоячую волну, значительно проще, так как не содержит скорости
и времени. Только атомная система является трехмерной, а потому в уравнение
для описания модачи атомной стоячей волны Шрёдингер вводит все три аргумен-
аргумента — кооординаты х, у и г.
ду2 + dz2 + Л2
(III.16)
где ф — трехмерный аналог величины А в (III. 15).
Если в выражении (III.16) вместо А подставим значение длины волны де Брой-
ля из (III. 14), то получим
дх2 + ~ду2 + dz2
^- I - П
t2 ~
(III.17)
Полная энергия системы Е равна сумме потенциальной U и кинетической m0v2/2
энергий, т.е.
Е= U+ mov2/2,
откуда v2 — 2(Е - [1)/т0. Подставив значение v2 в (III.17), получим
A11.18)
A11.19)
или, введя оператор Лапласа* V2 (набла в квадрате), запишем
~(Е- Ц)ф = 0, A11.20)
где
\ ^ — -f- ~ь —'—¦
дх2 ду2 az2
Выражения (III.19) и A11.20) есть волновое уравнение Шрёдингера для ста-
стационарного состояния, когда энергия системы не зависит от времени. В большин-
большинстве случаев задачи сводятся именно к нахождению стационарных состояний.
Уравнения (III.19) и (III.20) не выводятся из более общих законов, а являются
следствием эмпирического выбора уравнения стоячей волны в качестве модели
для описания поведения электрона в атоме с учетом волны де Бройля. Правомер-
Правомерность такого вывода уравнения Шрёдингера доказывается тем, что при его реше-
решении получают значения энергии Е, точно соответствующие опытным данным из
атомных спектров.
Функция ф в уравнении Шрёдингера называется волновой функцией и опреде-
определяет амплитуду стоячей электронной волны. Физический смысл имеет величина
y2dv**, равная вероятности нахождения электрона в элементарном объеме dv —
= dxdydz. Таким образом, квантовая механика дает лишь вероятность нахождения
электрона в том или ином месте атомной системы. Поэтому такие понятия, как
траектория частицы (например, электронная орбита), в квантовой механике не
имеют смысла. В соответствии с физическим смыслом ф2 сама волновая функция
должна удовлетворять определенным условиям, которые называются стандартны-
стандартными. Согласно последним, волновая функция должна быть: 1) непрерывной, так
как состояние квантовой системы в пространстве меняется непрерывно; 2) конеч-
конечной, т.е. она не должна обращаться в бесконечность ни при каких значениях
аргументов; 3) однозначной, ибо по смыслу ф есть амплитуда вероятности, а пото-
потому для любой данной точки она может иметь только одно значение; 4) обращать-
обращаться в нуль на бесконечности. Кроме того, функция ф должна быть нормированного.
Это означает, что суммарная вероятность нахождения электрона в околоядерном
пространстве должна быть равна единице, т.е. результат проявления волново-
корпускулярного дуализма не ведет к исчезновению электрона. Математически
условие нормировки записывается как $ф>2йю — 1, т.е. суммирование (точнее,
интегрирование) ведется по всему объему значений каждой из координат от — оо
до + оо. Из статистической интерпретации волновой функции возникает вопрос,
обладает ли волновыми свойствами отдельная микрочастица или они присущи
коллективу их. В опытах по дифракции электронных пучков очень малой интен-
*Оператор есть символическая запись тех действий, которые необходимо проделать над
произвольной функцией для получения некоторой другой функции.
В общем случае вероятность нахождения частицы равна квадрату волновой функции
по модулю, т.е. |i/>|2<fo, так как волновая функция может выражаться и комплексной
величиной.
29
Р и с. 12. Связь между поляр-
полярными и декартовыми коорди-
координатами
сивности было показано, что волновыми свойст-
свойствами обладает каждая микрочастица.
4. Основы квантово-механяческого рассмотре-
рассмотрения атома водорода. Орбятали. Решения уравне-
уравнения Шрёдингера даже для атома водорода весь-
весьма сложны. В то же время результаты, получен-
полученные при приложении квантовой механики к
задаче атома водорода, имеют принципиальное
значение для современной теории строения ато-
атомов вообще. Поэтому рассмотрим лишь узловые
вопросы квантово-механического представления
атома водорода, опуская математические подроб-
подробности. Уравнение Шрёдингера (III. 19) примени-
применительно к атому водорода запишется так:
дх2 ду2 dz2
+
2ю0
е2]
(III.21)
где — е2/г — потенциальная энергия; г — расстояние электрона от ядра. Задачу
движения электрона в атоме удобнее рассматривать, предварительно преобразо-
преобразовав уравнение (III.21) от декартовых координат (х, у и z) к сферическим поляр-
полярным координатам. Положение точки р в полярных координатах, центр которых
совпадает с ядром атома, показано на рис. 12. Линия, соединяющая точку р с
началом координат, представляет собой радиус-вектор г, а д — угол (широта),
который эта линия образует с осью z. Проекция радиуса-вектора на плоскость ху
составляет угол <р (долгота) с осью х. Полярные координаты г, В и <р однозначно
определяют р. Они связаны с декартовыми координатами следующими соотноше-
соотношениями:
х — rsm У cos р, у = rsm t) sin <p, z = rcos
С учетом формул (III.22) уравнение (III.21) преобразуется в
(III.22)
д
7г!
1
д2ф
2sin 20 д<р2
д Г . дф
a T7i SIn " Т7Г
в дв \ дд
(III.23)
Решение уравнения Шрёдингера для атома водорода в полярных координатах
(III.23) после разделения переменных удается представить в виде произведения
трех отдельных функций, каждая из которых зависит только от одного аргумен-
аргумента. В самом общем виде это приводит к волновой функции вида
ф(г,
(III.24)
в которой R (г) называется радиальной составляющей волновой функции, а про-
произведение &@)Ф(<р) представляет собой ее угловую часть*. Уравнение (III.23)
'Функции 0(#) и Ф(^) называют также сферическими.
имеет бесконечное множество решений. Но чтобы они имели смысл для описания
движения электрона в атоме, они должны удовлетворять стандартным условиям.
При этом в соответствии с тремя степенями свободы автоматически появляются
три величины, которые могут принимать только целочисленные значения. Ради-
Радиальная часть волновой функции содержит пи/, угловая функция — / и пц, а
другая часть волновой функции Ф (у) включает число пц. Таким образом, общим
решением уравнения (III.23) является функция
^nXmi.r, 9, <р) = Д„,г(гHи^)Фж/^. (Ш.25)
Безразмерные величины п, I и т/, входящие в уравнение (III.25), называются
квантовыми числами. При решении уравнения (III.23) на квантовые числа накла-
накладываются строгие граничные условия и они могут принимать следующие цело-
целочисленные значения:
п - 1, 2, 3, 4, ..., оо;
/=0, 1,2,3, ..., (п-1);
т/=0, ± 1,±2, ±3, ..., ± /. A11.26)
Дозволенные решения уравнения (III.23) с соблюдением стандартных условий и
требований (III.26) называют собственными функциями. Выражение (Ш.25) явля-
является полной собственной функцией уравнения Шрёдингера для атома водорода.
Собственная функция характеризует состояние электрона в атоме и называется
атомной орбиталъю. Состояние электрона, или орбиталь, однозначно определя-
определяется набором квантовых чисел п, / и пц. Этот вывод имеет общее значение, так
как эти квантовые числа определяют состояние электрона не только в атоме
водорода, но и в любом другом атоме.
В табл. 1 приведены собственные функции или орбитали водородного атома.
В ней представлены в явном виде выражения для радиальной и угловой частей.
Поэтому полная собственная функция получается как произведение радиальной
и угловой функций. Кроме того, для обозначения орбитали (символ орбитали)
квантовое число п выражается цифрами, а значения / — строчными латинскими
буквами:
/ О 1 2 3 4 ...
Обозначение а р d f g\\ т.д.
А различия в значениях квантового числа т\ при одних и тех же п и / обозначе-
обозначены нижними индексами справа от букв. Для графического представления атом-
атомных орбиталей (зависимость Ф от г, 9 и ip) требуется четырехмерное пространст-
пространство, что практически невозможно. Поэтому в соответствии с табл. 1 разобьем пол-
полную собственную функцию на радиальную и угловую части и воспользуемся
двумя типами графической зависимости. Вероятность нахождения электрона на
различных расстояниях от ядра можно наглядно выразить при помощи так назы-
называемого графика радиального распределения. Это мера нахождения электрона в
сферическом слое между расстояниями г и г + froT ядра вдоль линии с задан-
заданными значениями углов В и ip. Объем, лежащий между двумя сферами, имеющи-
имеющими радиусы г и г + dr, равен Arr^dr, а вероятность пребывания электрона в этом
элементарном шаровом слое пропорциональна 4ят2[Дп/(г)]2. На рис. 13 приведено
радиальное распределение величины 4irr2[Rnj(r)]2, которая характеризует плот-
плотность вероятности нахождения электрона на различных расстояниях от ядра.
31
Таблица 1. Орбитали атома водорода
Квантовые числа
п
1
2
2
2
2
3
3
3
3
со со со со со
1
0
0
1
1
1
0
1
1
1
2
2
2
2
2
0
0
0
1
-1
0
0
1
-1
0
1
-1
2
-2
Символ
орбитали
Is
2s
3s
3Pz
3dxz
Радиальная часть
2B
1 ..,,
2
B7- 18г +
81J3
8l(i
4
81C0
Угловая часть
Г~з~ л
COS Р
1 -— sm 0 cos 'f
-— sin в sin ifi
-— cos 9
V^ ¦ a
-— sin У cos ш
^ 4л- r
Г~з~
-;— sin в sin <p
^ 4л-
fl5 . . .
-— sm в cos w cos и
\Av
Щ . a .
I j— sm ip cos p sm ip
W -га о
¦гт— Sill' в COS2 M
^ IDT
7-r— sin2 ^ sin2 ip
\ 16Я"
Рис. 13 показывает, что в отли-
отличие от теории Резерфорда — Бора,
согласно которой электрон движет-
движется по круговым орбитам *, по кван-
тово-механической модели элект-
электрон может пребывать в любой
области атомного пространства,
только с различной вероятностью.
Если бы удалось через малые
промежутки времени сфотографи-
сфотографировать положение электрона, то
при наложении тысяч таких фото-
фотографий получили бы картину
электронного облака. Совершенно
очевидно, что плотность электрон-
электронного облака в различных точках
п=3
В 0,2
о oj. ojt ф
Г, ИМ
Рис. 13. Функции радиального распределения пере-
передающие вероятность нахождения электронов на рас-
расстояниях г от ядра
определяется вероятностью пребывания электрона. Поэтому электронное облако—
это облако вероятности нахождения электрона. Эти облака состоят из сгущений
и разрежений, придающих им специфическую форму. На рис. 14 приведено
поперечное сечение электронного облака 2*-орбитали возбужденного атома водо-
водорода, которое соответствует кривой 2s (п = 2, / = 0) на рис. 13. В дальнейшем
изложении будем попеременно пользоваться взаимозаменяющими терминами
"вероятность нахождения электрона", "электронная плотность" и "плотность
электронного облака".
Теперь сравним радиусы боровских стационарных орбит с абсциссами макси-
максимума электронной плотности на рис. 13. Радиусы боровских орбит меняются
пропорционально квадрату главного квантового числа и равны соответственно
0,053-I2, 0,053-22; 0,053-З2 нм и т.д. На рис. 13 для орбиталей Is, 2s и 3s эти
расстояния показаны пунктирными линиями. Для всех орбиталей ns только
радиус первой боровской орбиты совпадает с местами максимальной плотности
нахождения электрона. Из других состояний наблюдается совпадение для 2р,
3d, ... орбиталей.
Для химии первостепенное значение имеет угловое распределение плотности
электронного облака. Как это видно из табл. 1, ns-орбитали не зависят от сфери-
сферических углов: их собственные функции не содержат членов, зависящих от углов в
и <р. Поэтому все атомные ns-орбитали обладают сферической симметрией. На
рис. 15 показаны формы электронных облаков, соответствующие различным
атомным орбиталям. Рис. 15 представляет собой геометрическое выражение квад-
квадрата угловой части собственной функции О/,т@)Фте (^)i выборочно взятой из
табл. 1. Для правильного понимания рис. 15 необходимо заметить, что он дает
наглядное представление о граничной поверхности, внутри которой электронная
плотность велика (90%), а вне ее мала. Граничная поверхность характеризуется
изоэлектронной плотностью, т.е. она является поверхностью равной электронной
плотности. Все орбитали изображаются трехмерными фигурами. Возникновение,
например, р-орбиталей можно представить вращением их двумерных изображений
*Следовательно, между орбитами электрону быть запрещено.
IS
Рис. 14. Вид поперечного
сечения облака 2.9-орбитали
атома водорода
Рис. 15. Формы электронных об-
облаков, соответствующие различ-
различным атомным орбиталям
на 180° вокруг осей. Орбитали 3d 2 и 3d 2 2 располагаются вдоль осей, a 3dXy,
z х у
ZdyZ и 3dxz — между осями.
Наиболее характерная особенность атомных орбиталей — их зависимость от
углов в и <р (за исключением s-орбиталей). Отсюда их граничные поверхности не
являются сферически-симметричными и при некоторых значениях углов элект-
электронная плотность практически обращается в нуль, т.е. электронные облака ато-
атомов, как правило, пространственно направлены. Это служит теоретическим обос-
обоснованием учения о направленной химической связи.
5. Квантовые числа. Итак, набор квантовых чисел п, I, mi и их вариация —
следствие решения уравнения Шрёдингера для состояния электрона в атоме
водорода. Эти же квантовые числа однозначно характеризуют состояние электро-
электронов любого другого атома Периодической системы*. В этом заключается прин-
принципиальная значимость квантовых чисел в теории атома и в раскрытии физиче-
физического смысла Периодического закона. Однако квантовая механика не отличается
наглядностью и не дает физической интерпретации квантовых чисел п, I и пц.
Для придания этим квантовым числам физического смысла обратимся к модели
атома Бора, в которой уже фигурировало главное квантовое число п. Оно опреде-
*При этом допускают: 1) многоэлектронный атом характеризуется набором атомных
орбиталей, которые соответствуют водородным орбиталям, т.е. является "водородопо-
добным"; 2) отдельный электрон многоэлектронного атома не взаимодействует с другими
электгюнами и ведет себя так, как если бы он был один. ' :
ляет основное свойство электрона — его энергию в поле ядра или атомного осто-
остова. ЕГ модельной картине главное квантовое число определяет порядок (номер)
электронного слоя от ядра в атомах. Главное квантовое число п не может быть
равно нулю, так как, согласно (III.9) и (III.27), энергия электрона становится
равной бесконечности, что не имеет физического смысла. Энергия электрона в
атоме водорода и водородоподобных ионах (например, Не+, Li2+ и др.) зависит
только от главного квантового числа. Тогда решение уравнения Шрёдингера
приводит к выражению
Е = -[m0e427Bn2fi2)]. A11.27)
Уравнение (III.27), полученное из квантовой механики, идентично выражению
(III.9) из теории Бора. Но в отличие от последней квантовая механика приходит
к (III.27), не прибегая к постулату о существовании стационарных орбит. В соот-
соответствии с (III.27) все атомные орбитали с постоянным значением главного кван-
квантового числа должны иметь одну и ту же энергию. Такие состояния с одинаковой
энергией называют вырожденными. Таким образом, например, np-орбитали трех-
трехкратно вырождены, nrf-орбитали — пятикратно.
Квантовое число / называется орбитальным или побочным и определяет меха-
механический момент электрона, обусловленный его движением вокруг ядра. Орби-
Орбитальный момент квантуется и связан с побочным квантовым числом соотноше-
соотношением
\Mi\ =\l{l+ 1). A11.28)
С ним связан орбитальный магнитный момент
M =^/(/+1), A11.29)
где
ltB - ей/Bтос); (Ш.ЗО)
(I — магнетон Бора, являющийся единицей измерения магнитного момента
В
электрона и атома.
Механический и магнитный моменты являются векторными величинами.
Магнитное квантовое число пц определяет проекцию орбитальных механического
М и магнитного \i моментов электрона на некоторое произвольное направление z
или на направление магнитного поля:
Mz = пцИ; ц2 - тш. (III.31)
в
Значения соответствующих проекций орбитальных моментов квантованы. На рис.
16 показана ориентация вектора орбитального механического момента для пяти
rf-орбиталей относительно оси г. Из рис. 16 видно, что квантовое число т/ харак-
характеризует расположение вектора орбитального момента в пространстве. При задан-
заданном значении / магнитное квантовое число может иметь 2/ + 1 значений. Тогда
для заданного значения главного квантового числа п всего различных состояний
будет
n-i
B/ + 1) = п2. A11.32)
1*0
2* 35
I
Интерпретация тонкой структуры спектров атомов
показала, что три квантовых числа п, I и mi, фигури-
фигурирующие в решении уравнения Шрёдингера, не явля-
являются исчерпывающими характеристиками состояния
электронов в атомах. Английский ученый Дирак
A928) предложил обобщенное уравнение Шрёдингера
с учетом теории относительности (уравнение Дирака).
Оказалось, что у электронов, протонов, нейтронов
существует специфическая внутренняя степень свобо-
Р и с. 16. Ориентация вектораДы. С ней ассоциируется собственный механический
орбитального момента для момент частицы, не связанный с ее орбитальным
B-орбиталей движением.. Этот механический момент называется
спином. Электронный спин вытекает из решения уравнения Дирака. Спин
электрона — такое же фундаментальное его свойство, как масса и заряд. По
аналогии с формулой (III.28) для спина электрона можно написать
Т~Щ (Ш.ЗЗ)
-2..
\MS\ =Щ7+Щ,
где s = ^11- Проекция спина на направление z будет
sz = тД (III. 34)
в которой спиновое квантовое число ms = ± '/г- Итак, спиновое квантовое число
может иметь только два значения: Ч-'/г и ~ '/г- Они, как и другие квантовые
числа, отличаются на единицу. Положительное и отрицательное значения спина
связаны с его направлением. Разность в энергиях, связанная с противоположно
направленными спинами, снимает вырождение и приводит к наблюдаемой экспе-
экспериментально тонкой структуре спектра.
Таким образом, с учетом спинового квантового числа состояние электрона в
поле центральных сил полностью определяется заданием четырех квантовых
чисел: п, /, пц и ms. Никаких других, независимых от указанных квантовых чи-
чисел, характеристик для этого не требуется. Теперь в развитие (III.32) данному
значению главного квантового числа п будет отвечать 2п2 отдельных состояний
Фп,1,т,т (г, 0, <р), Т.е.
I s
п-\
2B/+ 1) =
A11.35)
6. Многоэлектронные атомы. В сложных атомах на данный электрон влияет не
только ядро, но и все имеющиеся электроны. Каждый электрон отталкивается от
всех остальных электронов в соответствии с законом Кулона, а потому все волно-
волновые функции взаимозависимы. Расчет энергии уровней и распределения элект-
электронной плотности в принципе может быть проведен на основе решения уравнения
Шрёдингера для многих частиц. Однако точное решение подобных уравнений
неизвестно.
Приближенный метод Томаса и Ферми исходит из статистической модели
атома и применим к атомам, содержащим достаточно большое число электронов
(начиная примерно с середины Периодической системы). При помощи этого
метода приближенно определяют радиальное распределение плотности электрон-
He Li Be N № Ca Zn Zr Nd Hg-
Рис. 17. Зависимость энергии электрона от порядкового
номера элемента
ного облака. Аналогичную задачу для легких атомов можно решить и методом
самосогласованного поля (метод ССП), предложенным Хартри и развитым
Фоком. В этом методе рассматриваются одноэлектронные волновые функции,
электронов, движущихся в квазицентральном поле, создаваемом ядром и
усредненным полем всех остальных электронов (одноэлектронное приближение).
Эти одноэлектронные волновые функции и представляют собой атомные
орбитали (АО). Наконец, в методе Слейтера предполагается движение электрона
многоэлектронного атома в центрально-симметричном поле, создаваемом
эффективным зарядом ядра Z — S, где 5 — постоянная экранирования ядра
всеми остальными электронами. Постоянная экранирования — количественная
характеристика того, насколько внутренние электроны экранируют внешние
электроны от действия заряда атомного ядра. Принципиальные результаты, к
которым приводят расчеты многоэлектронных атомов указанными
приближенными методами, следующие:
1. Многоэлектронность сказывается только ца радиальной части волновой
функции и не влияет на угловое распределение электронной плотности.
' . ' . 37
2. В многоэлектронном атоме состояние электрона, так же как и в атоме водо-
водорода, однозначшо определяется значениями квантовых чисел п, /, пц и ms, кото-
которые принимают те же значения, что и в атоме водорода.
3. Энергия электронов в многоэлектронных атомах определяется (в отличие от
атома водорода) значениями главного и побочного квантовых чисел. При этом
энергия возрастает с увеличением обоих квантовых чисел, так что орбитали ns,
(п — \)d и (и — 2)/ сравнительно мало отличаются по энергии и всегда имеют
более низкую энергию, чем пр.
4. Энергия орбиталей меняется от атома к атому в зависимости от порядково-
порядкового номера элемента. На рис. 17 показана зависимость энергии электрона от по-
порядкового номера элемента. За единицу взята энергия электрона атома водорода
в основном состоянии, равная 13,6 эВ *.
Электроны в многоэлектронных системах (атомы, молекулы, кристаллы) под-
подчиняются квантово-механической закономерности, называемой принципом Паули.
Согласно этому принципу, в любой миоюэлектронной системе в каждом состоя-
состоянии, определяемом полным набором четырех квантовых чисел, не может быть
больше одною электрона. Следовательно, все наборы значений квантовых чисел
должны отличаться друг от друга хотя бы одним квантовым числом. Принцип
Паули применим к любым частицам, имеющим полуцелый спин (электрон, про-
протон, нейтрон, позитрон и др.). Принцип Паули** ограничивает число электронов
в атоме для каждого значения главного квантового числа п. Согласно (III.35),
для данного квантового числа п возможно всего 2п2 состояний и, следовательно,
по запрету Паули в этом электронном слое может быть не больше 2п2 электро-
электронов. Рассмотрим вариации квантовых чисел, определяющих 2п2 состояний элект-
электронов.
Пусть главное квантовое число п = 1. Тогда, согласно (III.26), / и пц могут
принимать только нулевые значения. Отсюда электроны с п = 1 могут отличать-
отличаться друг от друга только значениями спиновых квантовых чисел:
1-й электрон
2-й >
1
1
i
0
0
тщ
0
0
77ls
+ 72
-72
Таким же образом можно показать, что в слое с п — 2 максимально могут
находиться 2п2 = 8 электронов с квантовыми числами:
п I пц гщ
1-й электрон 2 0 0 +7г
2-й > 2
3-й > 2
4-й > 2
5-й > 2
6-й > 2
7-й > 2
8-й > 2
0
1
1
1
1
1
1
0
-1
-1
0
0
+1
+1
-72
+ 72
2
+ 72
-72
+72
-7г
*Эта величина получается Из формулы (III. 9) при Z = 1 и в = 1.
* 'Ввиду негативного характера его также называют запретом Паули.
Совокупность электронов с одинаковым значением побочного квантового чис-
числа / удобно называть электронной оболочкой *. Независимо от значения главного
квантового числа электронная емкость «-оболочки равна 2, р-оболочки — 6, <?-
оболочки — 10, а /оболочка вмещает 14 электронов.
Существует два условных способа изображения заселенности электронных
оболочек атомов: в виде электронных формул и в форме квантовых ячеек. В
первом способе сначала пишется символ соответствующей электронной оболочки,
а в виде показателя степени изображается число электронов на данной оболочке.
Например, электронная формула атома водорода в нормальном невозбужденном
состоянии будет Is1, а натрия — Is22s22p63s1. Сумма верхних индексов должна
быть равна общему числу электронов в атоме, т.е. порядковому номеру элемента.
Недостатком электронных формул является использование только двух кванто-
квантовых чисел: пи/. Более полно описывает состояние электронов в атоме метод
квантовых ячеек, использующий все четыре квантовых числа. Каждой ячейке
отвечает определенная орбиталь, электрон изображается в виде стрелки, а на-
направление последней олицетворяет спиновое квантовое число. На каждой орбита-
ли (в квантовой ячейке) могут находиться или один электрон, или два электрона
с противоположными спинами **. Свободная ячейка означает свободную орбиталь,
которую может занимать электрон при возбуждении атома.
На рис. 18 приведена электронная структура атома кислорода в виде кванто-
квантовых ячеек. Подразумевается, что энергия орбиталей растет по вертикали, а пото-
потому при одном и том же значении главного квантового числа р-ячейки должны
располагаться выше, чем «-ячейки. В многоэлектронных атомах энергия электро-
электронов зависит от квантовых чисел пи/, что является следствием электрон-элект-
электрон-электронного взаимодействия (в первом приближении отталкивания). При этом с
ростом этих квантовых чисел энергия увеличивается. Однако на рис. 18 кванто-
квантовые ячейки для данного слоя (квантовое число п постоянно) изображены на
одной горизонтали, хотя в действительности существуют небольшие различия в
энергиях разных орбиталей. У атома кислорода первый электронный слой (п =
= 1) состоит из двух спаренных ls-электронов (/ = 0, mi = 0) с антипараллельны-
антипараллельными спинами (ms = ± Уг)- Второй слой (п = 2)
включает два спаренных электрона в 2*-ячейке, а
2р-ячейки заселены четырьмя электронами. Поэто-
Поэтому у двух спаренных р-электронов магнитные
квантовые числа mi одинаковы и отличаются
только противоположными спинами. Два же дру-
других неспаренных (одиночных) р-электрона харак-
характеризуются разными величинами mi
7. Периодическая система элементов и элект-
электронная структура атомов. Для каждого атома в
принципе возможно неограниченное число отдель- Рис. 18. Электронная структура
ных состояний, различающихся по своей энергии, атома кислорода в виде квантовых
Среди них одно-единственное состояние с наи- ячеек
п=2
*Вместо терминов "сло#' и "оболочка" часто употребляются термины "уровень" и
"подуровень", а также "оболочка" и "подоболочка".
"Противоположная ориентация спинов называется антипараллельностью.
меньшей энергией называется нормальным или невозбужденным. Все остальные
энергетические состояния с большим запасом энергии называются возбужденны-
возбужденными. Для перевода атома из нормального состояния в возбужденное необходимо
сообщить ему некоторую энергию — энергию возбуждения. Когда речь идет об
электронной структуре атомов, имеют в виду прежде всего их нормальное состоя-
состояние.
При переходе атома из возбужденного состояния в нормальное полностью
восстанавливается структура электронной оболочки. Если строение атомов можно
было бы описать классической физикой, отражающей непрерывность изменения
свойств, атомы одного и того же химического элемента в нормальном состоянии
имели бы различные электронные структуры и утратили бы свою индивидуаль-
индивидуальность. При этом исчезло бы различие между элементами и само понятие химиче-
химического элемента утратило бы свой смысл. Существование химических элементов с
их специфическими свойствами связано с квантовыми закономерностями, соглас-
согласно которым структура электронной оболочки атома в нормальном состоянии
однозначно определяется зарядом ядра или порядковым номером элемента в
Периодической системе.
При заполнении электронных слоев и оболочек атомы подчиняются: 1) прин-
принципу наименьшей энергии, согласно которому электроны сначала заполняют
вакантные орбитали с минимальной энергией; 2) принципу Паули; 3) правилу
Гунда — на вырожденных орбиталях суммарное спиновое число электронов долж-
должно быть максимальным. В квантовых ячейках с одинаковой энергией заселение
электронами происходит так, чтобы атом имел наибольшее число неспаренных
электронов. Это отвечает нормальному состоянию атома (минимум энергии).
Рассмотрим связь между электронным строением атомов и положением элементов
в короткой 8-клеточной Периодической системе (см. форзац). У каждого следу-
следующего элемента Периодической системы по сравнению с предыдущим на один
электрон больше. Наиболее прост первый период системы, состоящий
лишь из двух элементов. У водорода единственный электрон заселяет наинизшую
по энергии орбиталь Is, а у гелия на этой орбитали два электрона с антипарал-
антипараллельными спинами. Гелием заканчивается первый период системы и исчерпаны
все вариации квантовых чисел при п = 1. Таким образом, у атома гелия полно-
полностью формируется наиболее близкий к ядру .ЙГ-слой.
Формирование ?-слоя (и = 2) начинается с лития, у которого имеется три
электрона. Два электрона, как у гелия, заполняют А'-слой. Третий электрон
лития не может находиться в этом слое, так как на ls-орбитали электронных
вакансий нет. Помещение третьего электрона на s-орбиталь, максимальная элект-
электронная емкость которой равна двум, противоречило бы принципу Паули. У пос-
последнего элемента второго периода — неона — все s- и р-орбитали при п = 2 за-
заполнены. Электронное строение атомов элементов в нормальном состоянии при-
приведено в табл. 2. В ней квадратные скобки символизируют электронные структу-
структуры благородных газов, которые органически входят в строение атомов последу-
последующих элементов.
Третий период (п = 3) начинается с натрия, электронная формула
которого Is22s22p63s1, или, согласно табл. 2, [NejSs1. У аргона заполняются
полностью все в- и р-оболочки при п — Z. Аргоном заканчивается третий период
Периодической системы. Однако не исчерпаны все возможности вариации кван-
квантовых чисел при главном квантовом числе, равном трем (см. табл. 2). При п = 3
а б л и ц
Символ
элемента
Н
Не
Li
Be
В
С
N
О
F
Ne
Na
Mg
Al
Si
P
S
Cl
Ar
К
Ca
Sc
Ti
V
Cr
Mn
Fe
Co
Ni
Cu
Zn
Ga
Ge
As
Se
Br
Kr
Rb
Sr
Y
Zr
Kb
Mo
a 2. Электронное строение атомов элементов в нормальном состоянии
Электронное
строение
и
1*2
[Не] 2s1
2s2
2«22р2
2s22p3
2s22p4
2?2p>
2s22p6
[Ne] 3 s1
3s2
Зв2^
3s2p2
З^р3
3s2p4
3s2p5
3s2p6
[Ar] 4s1
4s2
4s23<P
4^3<Р
4513 d5
4s23rf5
4s23rfS
4s23(f
4s23<f
4^3^10
4^23^0
4s23iiO4pi
4s23rflO4p2
4s24rf104p3
4s23<P°4p4
4«23 (РЧр*
4s23#°4p6
[Kr] 5sl
5*2
5^4сР
5s4d*
Поряд-
Порядковый
номер
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67 .
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
Символ
элемента
Тс
Ru
Rh
Pd
Ag
Cd
In
Sn
Sb
Те
I
Xe
Cs
Ba
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Hf
Та
W
Re
Os
Ir
Pt
Au
Hg
TI
Pb
Bi
Po
Электронное
строение
5*24 (P
ЬМсР
bs4<P
4<f°
5 s 4 ur
5s^4 ur
5s24<?05p'
5s24<?°5p2
5s24<?('5p3
5s24 ifi^bp^
5s24<?^5p5
5s^4if^5p^
[Xe] 6s1
6*2
6s24/2
6s24/3
6s24/4
6s24/5
6s24/6
6s24/7
6s25<?4/7
6s24/9
6s24/10
6s24fH
6*24/• 12
6s24/13
6s24/H
6*25 <?4/14
6s25rf24yl4
6в25й34/14
6s25rf44/14
6s25rf>4/H
6^5tf4fu
6^5 #4/14
6s25<?°4/4
6s25d104/46p1
ъ&ьёчрчр1
6s25#°4/46p3
6s25d104/46p4
Продолжение табл. 2
Поряд-
Порядковый
номер
85
86
87
88
89
90
91
92
93
94
95
Символ
элемента
At
Rn
Fr
Ra
Ac
Th
Pa
U
Np
Pu
Am
Электронное
строение
6s25<?°4/46p5
6s25<?°4/46p6
[Rn] 7s1
7s2
7s4<?
7s26 cP
7s26<?5/2
7^6 <fibfs
7^6 cP5/5
7^6 d°5/6
7Mtf5f
Поряд-
Порядковый
номер
96
97
98
%
100
101
102
103
104
105
Символ
элемента
Cm
Bk
a
Es
Fm
Md
(No)
Lr
Ku
Ns
Электронное
строение
7^6 <P5f
7s4<f>bf
7^6 (P5fil
7s26 <fbfi2
7^6 <P5f3
7s4<Pbf4
7^6(tof4
7a26(P5/4
7^6 #5/4
существует пять З^-орбиталей (/ = 2), каждая из которых вмещает по два элект-
электрона. Пока все эти орбитали вакантны, а следующим после аргона элементом яв-
является калий — первый представитель четвертого периода системы.
Хотя в третьем слое остается незаполненной вся rf-оболочка A0 вакансий), у
калия и кальция начинает заполняться четвертый слой (п — 4). Об этом свиде-
свидетельствуют спектры и химические свойства этих элементов, являющихся химиче-
химическими аналогами соответственно натрия и магния. Если у последних наружными
являются 3«-электроны, то у калия и кальция наиболее удалены от ядра перифе-
периферические электроны 4s.
Помимо чисто "химического" обоснования очередности заселения, основанного
на аналогии в химических свойствах элементов, из рис. 17 видно, что в области
Z = 19 ? 21 энергии As- и З^-орбиталей близки. Поэтому у калия (Z = 19) и
кальция (Z — 20) электроны заселяют 4«-орбиталь. На основе анализа зависимо-
зависимости энергии электрона от порядкового номера (см. рис. 17) В.М.Клечковский
A954) сформулировал правило последовательности заселения электронных оболо-
оболочек атомов химических элементов. Это правило, именуемое (п + /)-правилом,
детализирует условие минимума энергии при заполнении электронных оболочек
и слоев. По Клечковскому, сначала заселяются орбитали с меньшим значением
суммы главного и побочного квантовых чисел (п + 0- При равенстве этой суммы
для нескольких состояний сохраняется ведущая роль главного квантового числа,
т.е. заполнение орбиталей происходит последовательно от меньшего его значения
к большему. Рассмотренное электронное строение атомов калия и кальция соот-
соответствует правилу Клечковского. Действительно, для 4в-орбитали (п = 4, / = 0)
сумма (п + /) = 4, а для З^-орбитали (п — 3, / = 2) эта сумма равна 5. Поэтому
4#-оболочка заполняется раньше, чем З^-оболочка.
Начиная с 21-го элемента скандия заполняется З^-оболочка*, которая фор-
*Для скандия два выбора: начать заполнение 4р- (п + / = 5) или 34-орбитали (п +
+ / = 5). Так как сумма п + / для этих орбиталей одинакова, то, согласно правилу Клеч-
Клечковского, сначала заполняется ЗЙ-оболочка.
малыш принадлежит предыдущему слою при п = 3. Поэтому в четвертом ряду
Периодической системы слева направо не наблюдается заметного убывания ме-
металлических свойств, так как на внешнем электронном слое (п = 4) имеется всего
два электрона Ds2). Исключение составляют хром и медь, для которых наблюда-
наблюдается "провал" одного электрона с 4s- на 3</-орбиталь. Провалы электронов наб-
наблюдаются и для других элементов (см. табл. 2). Они оправданы энергетически,
т.е. подчиняются принципу наименьшей энергии, и находят экспериментальное
подтверждение при изучении тонкой структуры спектров *. Полностью 3</-орби-
таль заполнена у цинка. У галлия, подобно алюминию, появляется один элект-
электрон на ;р-оболочке D/I1). Четвертый период заканчивается также благородным
газом криптоном с полностью заполненной 4р6-оболочкой. Между кальцием Ds2)
и галлием D/»1) "вклиниваются" десять элементов от скандия до цинка, для
которых характерно заселение электронами 3</-орбиталей. Эти металлы Sc — Zn
образуют первую десятку элементов вставных декад.
Пятый период аналогичен четвертому. Здесь вторую десятку элемен-
элементов вставной декады составляют металлы Y — Cd, для которых свойственно
заполнение 4</-орбиталей. С индия начинается заполнение 5;р-орбитали, которое
заканчивается у атома ксенона. При этом 4/-, 5d- и 5/-орбитали вакантны, а
пятый период полностью завершен. Объясняется это тем, что периоды формиру-
формируются быстрее, чем квантовые слои. Эта закономерность четко прослеживается
начиная с третьего периода.
В шестом периоде после лантана, у которого на 5</-орбитали появ-
появляется один электрон, следуют 14 лантаноидов. Для них характерно заполнение
в основном 4/орбитали. Один электрон на 5</-орбитали (как у лантана) появляет-
появляется только у гадолиния {Z = 64) и лютеция (Z = 71). Поскольку у лантаноидов,
представителей шестого периода, происходит заполнение глубоколежащего внут-
внутреннего (га — 2)-слоя, структура внешнего и второго снаружи слоев у них совер-
совершенно идентична. Это является определяющим в химическом поведении ланта-
лантаноидов и объясняет чрезвычайно сильно выраженную аналогию в химических
свойствах этих элементов.
Второй электрон на 5</-орбитали появляется только у гафния (Z = 72), а
полностью 5</-орбитали заполняются у атома ртути. Таким образом, десять ме-
металлов от лантана до ртути (без лантаноидов) формируют третью вставную дека-
декаду элементов. Тогда лантаноиды, у которых происходит заселение 4/орбиталей,
рассматриваются как вставка во вставку, так как они вклиниваются между ланта-
лантаном и гафнием. У таллия начинает заполняться бр-оболочка, которая завершает-
завершается у атома радона.
В незаконченном седьмом периоде у франция начинается, а у ра-
радия заканчивается заполнение 7в-орбитали. Атом актиния, как и лантана, начи-
начинает заполнение <?-орбитали. Для актиния это будут 6</-орбитали. Актиноиды
(90 — 103) "застраивают" 5/юрбиталь. Так как с ростом порядкового номера раз-
разница в энергиях соответствующих орбиталей делается все меньше (см. рис. 17), в
*В тонкой структуре спектров различают одиночные, двойные, тройные и т.д. спект-
спектральные линии, называемые соответственно синглетами, дублетами, триплетами и т.д.
— мультиплетами. Мультиплетность связана с числом неспаренных электронов t] равен-
равенством М = 1/ + 1. Экспериментально определяя М, находят число неспаренных электронов
в атоме.
атомах актиноидов происходит своеобразное "соревнование" в заполнении 5/- и
6</-орбиталей (см. табл. 2), энергии которых очень близки. У 104-го элемента —
курчатовия — очередной электрон заселяет 6<йэболочку, доводя ее до 6(Р. Поэто-
Поэтому курчатовий является химическим аналогом гафния, что доказано эксперимен-
экспериментально. По-видимому, у 105-го элемента* бсйэболочка состоит из трех электро-
электронов, т.е. 105-й элемент должен быть химическим аналогом тантала — экатанталом.
Рассмотрим особенности заполнения электронных слоев и оболочек атомов
Периодической системы.
1. Номер периода совпадает с максимальным значением главного квантового
числа для элементов данного периода, т.е. начало каждого периода совпадает с
началом нового электронного слоя.
2. Каждый период начинается с элементов (водорода и щелочных металлов),
для которых наружная оболочка состоит из одного электрона ns1. Завершается
любой период благородным газом с октетной (кроме гелия) внешней электронной
оболочкой ns2np6 при п > 1.
3. У типических элементов** и элементов главных подгрупп (группы А), непо-
непосредственно следующих за типическими по вертикали, заполняются либо внеш-
внешние ns-орбитали (IA- и ИА-группы), либо внешние п^мэрбитали (IIIA — VIIIА-
группы). Элементы с заселяющимися ns-орбиталями называются «-элементами, а
те, для которых характерно заполнение п^мэрбиталей, именуются р-элементами. У
элементов побочных подгрупп (группы В), включая побочную подгруппу VIII
группы***, происходит заполнение внутренних (и — 1)rf-орбиталей (если не счи-
считаться с отдельными провалами электронов). Они называются (/-элементами или
переходными металлами в отличие от s- и р-металлов, которые называются прос-
простыми металлами.
4. Для лантаноидов и актиноидов характерно заполнение глубинных (п — 2)/-
орбиталей (соответственно 4/- и 5/орбиталей). Поэтому лантаноиды и актиноиды
относят к /-элементам.
5. Для элементов-аналогов наблюдается одинаковое число электронов на одно-
одноименных орбиталях при разных значениях главного квантового числа. Поэтому
физический смысл Периодического закона заключается в периодическом изменении
свойств элементов в результате периодически возобновляющихся сходных элект-
электронных оболочек атомов при последовательном возрастании значений главного
квантового числа.
Таким образом, с ростом порядкового номера заполнение орбиталей происхо-
происходит в следующем порядке:
Is < 2s < 2р < 3s < Зр < 4s < 3</ < Ар < 5s »
« Ad < Ьр < 6s и 4/й Ы <6р < 7s » 5/« 6rf < 1р
не завершены
*Открытие 105-го элемента зарегистрировано Государственным комитетом по делам
изобретений и открытий Совета Министров СССР 16 мая 1972 г. Авторы открытия пред-
предложили назвать его Нильсборий (Ns) в честь Нильса Бора.
* *Типическими Менделеев назьшал элементы второго и третьего периодов.
. * * *Необычная побочная подгруппа VIII группы состоит из триады металлов семейства
железа и шести элементов семейства платины.
I
В полудлинной 18-клеточной Периодической
системе (табл. 3) элементы вставных декад за-
занимают клетки между а- и р-элементами. Такую
систему легко разбить на отдельные секции (рис.
19) по расположению в ней э-, 'р-, d- и /-элементов.
Секция, обозначенная s, содержит по два элемента
каждого периода, секция р — по шесть, секция d —
по десять и т.д. в соответствии с максимальной р и с- 19- Схема заполнения *-, «*-.
емкостью той или иной оболочки. Такое естест- Р" и /орбитами полудлинной
венное разделение Периодической системы на формы Периодической системы
отдельные секции еще раз демонстрирует ее неразрывную связь со строением
электронных оболочек атомов химических элементов.
Однако в полудлинном варианте Периодической системы ^элементы продол-
продолжают оставаться за ее пределами. От этого недостатка свободна длиннопериодная
32-клеточная Периодическая система, которая приводится в табл. 4. В ней ланта-
лантаноиды и актиноиды вставлены между ШВ- и IVB-группами rf-элементов. В длин-
нопериодном варианте системы в'идно, что ^элементы (лантаноиды и актиноиды)
являются вставкой во вставку из (/-элементов.
8. Строение электронной оболочки и свойства элементов. Структура электрон-
электронной оболочки атомов химических элементов изменяется периодически с ростом
порядкового номера элемента. Поскольку свойства есть функция строения элект-
электронной оболочки, они должны находиться в периодической зависимости от заря-
заряда ядра атома. И действительно, для самых разнообразных характеристик эле-
элементов указанная зависимость выражается периодическими кривыми, имеющими
ряд максимумов и минимумов. Даже такие на первый взгляд непериодические
свойства, как удельная теплоемкость простых веществ, частота линий рентгенов-
рентгеновского спектра элементов и т.д., при внимательном анализе оказываются периоди-
периодическими. Объясняется это тем, что периодичность присуща всей электронной
оболочке атомов, а не только ее внешним слоям. Рассмотрим кратко наиболее
важные периодические свойства элементов.
Потенциал ионизации атомов и сродство к
электрону. Одним из важнейших свойств химического элемента,
непосредственно связанным со структурой электронной оболочки, является
ионизационный потенциал. Последний представляет собой энергию, необходимую
для отрыва наиболее слабо связанного электрона из атома в его нормальном
состоянии. Это есть потенциал ионизации первого порядка, который отвечает
процессу Э = Э+ + е~. Энергию ионизации можно выражать в любых единицах,
имеющих размерность энергии, например в килоджоулях, но чаще ее выражают в
электрон-вольтах. Для многоэлектронных атомов в принципе существует столько
энергий ионизации *, сколько электронов в атомах. От атомов химических
"Потенциалы ионизации, выраженные в вольтах, численно равны величинам энергии
ионизации в электрон-вольтах, поскольку работа отрыва электрона под действием элект-
электрического поля равна А — QU — el, где е — заряд электрона; / — ионизационный потен-
потенциал. Величины 7i, 12, 1$, ..., опознающие отрыву первого, второго и т.д. электронов,
называются ионизационными потенциалами соответственно первого, второго и т.д. поряд-
порядков.
Таблица 3. Полудлинная 18-клеточная Периодическая система
^ГРУППЫ
1
2
3
4
5
6
7
IA
(Н)
3
Li
1.57
11
Na
1,80
К
2,16
37
Rb
2,29
55
Cs
2.52
17
Fr
ПА
4
Be
A,08)
12
Mg
A,37)
2a
Ca
A,68)
31
Sr
0,84)
M
Ba
•t
Ra
1ПВ
IVB
VB
21
Sc
1,57
3»
Y
1,69
La*"
1,92
A?"
1,90
22
Ti
1.4»
4a
Zr
72
Hf
1,4»
И4
Ко
23
1,40
41
Nb
1,59
73
Та
M5
(Ns)
VIB
YOB
-v"
1,40-^
24
Cr
1.45
42
Mo
1,52
74
w
1,3»
—
25
Mn
1,28
43
Tc
1,39
75
Re
1,36
vine
IB
Орбитальный радиус атома, А*
24
Fe
из
Rn
1.41
74
Os
U7
27
Co
Rb
1,36
77
Ir
us
21
Ni
1,14
Pd
@Л)
78
Pt
U2
2»
Cn
A.19)
Ag
U9
7»
An
1,18
ПВ
за
Zn
A.07)
Cd
1.18
Hg"
1.13
ША
В
0.84
13
Al
1,43
31
Ga
1.25
In
1,38
И
TI
1,32
IVA
С
0,65
|4
Si
32
Ge
1,09
50
Sn
U4
«1
Pb
1,22
VA
N
0,52
is
P
0,96
33
As
0,98
51
Sb
1.14
Ю
Bi
1.12
VIA
0
0,45
s
0:85
34
8e
0.92
52
Те
i.n
14
Po
1,21
VDA
H
0.53
F
0,39
17
Cl
0,73
35
Br
0.87
S3
I
1.07
15
At
1,14
УША
2
He
0,29
И
Ne
0.3J
и
Ar
0,69
34
Kj
0,80
54
Xe
0,99
M
Rn
1,10
• ЛАНТАНОИДЫ
Се
Я
Pr
1.94
И
Nd
1.91
<1
Pm
l.M
«2
Sm
l,«5
a
Ев
1,12
M
Gd
1,71
<5
Tb
1,77
M
Dy
1,75
G
Ho
1,72
<•
Er
1.70
И
Tm
i.6i
70
Yb
1,65
71
Ln
US
•• АКТИНОИДЫ
я
Th
1,79
N
Pa
1,80
•2
U
1,71
«3
Np
1,52
M
Pn
1,78
U
Am
M
Cm
n
Bk
я
Cf
»
Es
Fm
m
Md
И2
(No)
M3
Lr
Таблица 4. Длиннопериодная 32-клеточная Периодическая система
юды|
§
i
2
3
4
5
6
7
IA
(H)
3
LI
US
11
Nt
1,89
1»
К
3,3.
J7
Rb
2,4»
Я
Cs
2.48
87
Fr
240
ПА
4
Be
1,13
n
Me
l.ao
21
Ca
МП
38
Sr
2,H
M
Ba
2,21
81
Ra
W2
ШВ
]
Г Р
У П 1
1
ы
э
ЛАНТАНОИДЫ И АКТИНОИДЫ
Химичершй снкм | M
21
Sc
1,64
99
Y
i,ti
57
La
1,87
a*
Ac
2,03
©
58
Се
1,83
Я
Th
l.ao
я
Vr
i*i
N
Pa
1,63
и
Nt
1,81
•2
и
1,54
—rv
и
Pm
-
Np
1,50
а
Sm
1,81
Рв
1,60
,
ч
<3
Ев
2,02
Am
1,82
Порядковый но
x<pi
ДСМСМТ»
ЭффсжтатД рмшус «сма , A
1 «4
Gd
1.79
Cm
1,74
и
Tb
1,77
Bk
6t
Dy
1,77
Cf
«7
He
1,76
Es
•9
Er
1,75
IN
Fm
•9
Tn
1,74
Ш
Md
78
Yb
1,93
102
(Ne)
71
Lb
1,74
103
Lr
©
Л
IVB
22
Ti
1,46
Zr
1,60
Г 71
Hf
1,59
IM
Kb
?
VB
23
V
1,34
41
Nb
1,45
73
Tb
1.46
105
(Ns)
VIB
24
Cr
1,27
42
Me
1.39
74
w
1,40
M
VIIB
25
Mb
1,30
43
Tc
1,37
75
Re
1,37
?
.
2*
Fe
1,26
Rb
1,34
7t
Os
1,35
H
ШВ
27
Ce
1,25
Rh
1,34
77
lr
1,35
21
Ni
1.24
Pd
1,37
78
Pt
1,38
T
IB
2>
Co
1,28
Ac
1,44
79
An
1,44
О
ПВ
за
Zn
1,39
Cd
1,56
80
Hg
1,60
©
ША
В
0,88
13
Al
1,43
31
Ga
1,39
Ib
i,66
81
Tl
1,71
в
IVA
С
0,77
14
Si
1,18
32
Ge
1,3»
50
Sn
1,58
82
Pb
1.75
VA
N
0,70
15
P
1.10
33
As
1,21
51
Sb
1,41
83
Bi
1,46
VIA
О
0,66
к
S
1,02
34
8e
Мб
52
Те
1,35
84
Ре
1.40
УПА
H
0,37
F
0,64
17
CI
0,99
35
Br
1,14
S3
I
1,33
85
At
1,40
УША
2
He
0.93
Щ
Ne
1,12
И
Ar
1.54
Kr
1.»
54
Xe
1,90
К
Rn
1,20
©
Т а б л и ц а 5. Потенциалы ионизации некоторых элементов (В)
Эле-
Элементы
Н
. Не
Li
Be
В
С
N
О
F
Ne
13,60
24,58
5,39
9,32
8,31
Щ6
14,53
13,61
17,42
21,56
54,40
75,62
18,21
25,15
2438
25,59
35,15
34,98
41,07
122,42
153,85
37,92
47,86
47,43
54,93
62,65
63^0
217,66
259,30
64,48
77,45
77,39
87,23
97,16
340,13
391,99
97,86
Ш,87
114,21
126,40
489,84
55У»3
138,08
157,11
157,90
666,83
739,11
185,14
207,00
871,12
953,60
239,00
1196
элементов можно последовательно удалить все электроны, сообщив дискретные
значения потенциалов /ь /2, /3 и т.д. При этом h < h < h < ¦¦¦ ¦ В табл. 5
приведены потенциалы ионизации различных порядков для элементов первых
двух периодов Периодической системы. При сравнении величин ионизационных
потенциалов разных порядков для атомов одного и того же элемента обращает на
себя внимание сравнительная легкость отрыва электронов наружных слоев. Так,
для атома лития удаление одиночного электрона наружного слоя 2s1(/1 = 5,39 В)
происходит несравненно легче, чем двух электронов внутреннего слоя (/2 = 75,62
и 7з = 122,4 В), т.е. /х < 12 < /3- У азота для отрыва первых пяти электронов
второго от ядра слоя (п = 2) требуются лишь десятки электрон-вольт, а удаление
двух электронов внутреннего слоя с главным квантовым числом п = 1
сопровождается затратой энергии в сотни электрон-вольт. Для элементов второго
периода в табл. 5 границы резкого возрастания энергий ионизации обозначены
жирной линией. Таким образом, энергии ионизации отражают дискретность
структуры электронных слоев и оболочек атомов химических элементов.
Данные табл. 5 позволяют связать более тонкие изменения энергии ионизации
с характером заполнения электронных оболочек. Для элементов второго периода
при переходе от лития к неону наблюдается возрастание энергии ионизации. Это
объясняется увеличением заряда ядра при постоянстве числа электронных слоев.
В то же время возрастание энергий ионизации первого порядка происходит внут-
внутри периода немонотонно. Так, например, у бора и кислорода наблюдается замет-
заметное уменьшение 1\ по сравнению, с предыдущими элементами — бериллием и
азотом. Аналогичное нарушение монотонности в изменении числовых значений
первых ионизационных потенциалов характерно и для других периодов системы.
Объясняется это тем, что повышенной стабильностью отличаются атомы, у кото-
которых внешняя электронная орбиталь*, либо совсем не заселена электронами (бе-
(бериллий), либа заполнена наполовину (азот):
*У всех обсуждаемых атомов (Be, N, В, О) внешняя орбиталь 2р.
n-2
n-1
II
II
Is
Be (932эВ)
ЩТ
III
||| NA4,53эВ)
Is
Для бора и кислорода это условие нарушается, в связи с чем имеет место соответ-
соответствующее уменьшение потенциала ионизации первого порядка (В — 8,31; О —
13,61 эВ). Повышенная устойчивость электронных структур с полностью вакант-
вакантными внешними квантовыми ячейками, а также наполовину заполненными элект-
электронами является следствием взаимного отталкивания друг от друга электронов,
находящихся на одной и той же орбитали.
Поскольку ионизационные потенциалы — функция строения электронной
оболочки атомов, они обнаруживают периодическую зависимость от порядкового
номера элементов (рис. 20). Периодическую зависимость можно проследить и в
характере изменения ионизационных потенциалов второго, третьего и т.д. поряд-
порядков. Наименьшими величинами потенциалов ионизации первого порядка облада-
обладают атомы щелочных металлов. Это объясняется сильным экранированием заряда
ядра полностью завершенными электронными оболочками с конфигурацией,
свойственной благородному газу, которые предшествуют внешнему п*]-электрону
атомов щелочных металлов.
Эффект экранирования заключается в уменьшении воздействия на данный
электрон положительного заряда ядра из-за наличия между ним и ядром других
электронов. Экранирование растет с увеличением числа электронных слоев в
атомах и уменьшает притяжение внешних электронов к атомному ядру. Экрани-
Экранированию противоположен эффект проникновения, обусловленный тем, что, сог-
согласно квантовой механике, электрон может находиться в любой точке атомного
' порядка,
1
1
1
1
21
20
18
№
74
n
W
6
г
0
¦He
II
Hl
"Li
If.
Ne
о
I
oil
F\
I Г
8 12
At
I
Cl/l
P /I
oil
JS 1
9S1 1
Ll Vе"
n.
IS 20
КГ
ASn#
WJ/se
ЛееЬ
On
It
i i i 'i i i i i i
2k 28 32 36
Xe
ir ° In 1
b <
W 44 +J 52
Hg- Rn
w/A/
^7.
SB П 78 82 SB Z
Рис. 20. Зависимость ионизационных потенциалов атомов от порядкового номера
элемента
пространства. Поэтому во внутренних областях
атома, близких к ядру, вероятность нахождения
даже внешних электронов достигает конечной
величины. Радиальное распределение плотности
вероятности 3«-электрона натрия указывает на
проникновение этого электрона во внутренние К-
и i-слои атома (рис. 21). Эффект проникновения
увеличивает прочность связи электрона с ядром.
Эффекты экранирования и проникновения мож-
можно рассматривать с единой точки зрения, так как
г формально они являются способом учета взаим-
Р и с. 21. Радиальное распределе- ного влияния электронов друг на друга. В отсут-
ние вероятности нахождения ствие других электронов, согласно уравнению
^электрона в атоме натрия (III.27), энергия рассматриваемого электрона
зависит только от заряда ядра Z и главного квантового числа п. Влияние других
электронов на данный электрон уменьшает Z и п:
Е = -т.е
A11.36)
где ?;>фф — эффективный заряд ядра; Пзфф — эффективное главное квантовое
число. При этом экранирование ведет к ?эфф < Z, а. эффект проникновения дела-
делает п3фф < п. Поэтому первый эффект уменьшает энергию связи данного электро-
электрона с ядром, а второй — увеличивает. Это происходит потому, что, согласно
(III.27), чем больше Z и чем меньше п, тем ниже лежит энергетический уровень в
одноэлектронной системе, тем прочнее связан электрон с ядром. Сильное умень-
уменьшение эффективного заряда наблюдается у ядер атомов щелочных металлов по
сравнению с другими атомами. Так, для атома фтора 2эф<Ь — ^,20 (Z — 9), а для
атома натрия ?эфф = 2,2 при Z = 11. В табл. 6 приведены значения пэфф для
низших s-, р-, (I- и /чзрбиталей щелочных металлов и металлов подгруппы меди.
В скобках приведены значения главного квантового числа п, для которого вы-
вычислены соответствующие иэфф, учитывающие эффект проникновения.
Таблица 6. Значения Ядфф для некоторых металлов
Li
Na
К
Pb
Cs
Cu
Ag -
Au
s
1,59 B)
1,63 C)
1,77 D)
1,80 E)
1,87 F)
1,33 D)
1,34 E)
1,27 F)
Орбитали
P
1,96 B)
2,12C)
2,23 D)
2,28 E)
2,33 F)
1,86 D)
1,87 E)
1,72 F)
d ¦
3,00 C)
2,99 C)
2,85 C)
2,77 D)
2,55 E)
2,98 E)
2,98 E)
2,98 F)
/
4,00 D)
4,00 D)
3,99 D)
3,99 D)
3,98 D)
4,00 D)
3,99 D)
>
50
Из данных табл. 6 видно, что наибольшим проникающим эффектом обладают
s-электроны, меньшим — р-электроны, еще меньшим — d- и /-электроны. Послед-
Последние (точнее, 4/-электроны) практически не характеризуются эффектом проникно-
проникновения. Кроме того, эффект проникновения более характерен для тяжелых атомов
с большим числом электронов во внутренних слоях, сквозь которые и проникает
внешний электрон. Наконец, проникновение внешних э- и р-электронов внутрь
атома исключительно сильно выражено для rf-элементов Периодической системы.
Однако с увеличением числа электронных слоев сильно возрастает расстояние
внешнего электрона от ядра, что уменьшает энергию ионизации. Например, для
щелочных металлов это играет доминирующую роль по сравнению с увеличением
эффекта проникновения. Поэтому в направлении сверху вниз для щелочных
металлов наблюдается слабое уменьшение (из-за проникновения внешнего элект-
электрона) энергий ионизации первого порядка. Вследствие ярко выраженного эффек-
эффекта проникновения s-злектронов энергии ионизации для металлов вставных декад
выше, чем у металлов главных подгрупп. В самих же вставных декадах ионизаци-
ионизационные потенциалы сравнительно мало изменяются при переходе от одного эле-
элемента к другому. Среди (/-элементов сравнительно большими значениями энергий
ионизации характеризуются, металлы, следующие за лантаноидами. Объясняется
это проникновением бе-электронов под двойной "экран" из 5rf- и 4/-электронов.
Периодически изменяется и сродство к электрону. Под последним понимают
энергию, которая выделяется при присоединении электрона к нейтральному ато-
атому, т.е. энергию процесса Э + е" = Э". Наибольшим сродством к электрону
характеризуются р-элементы VII группы. Наименьшие (и даже отрицательные)
величины сродства к электрону имеют атомы с конфигурацией внешних элект-
электронов ns2 и благородные газы. Ниже приведены величины сродства к электрону
для некоторых элементов:
Атом Н Не Li Be В С N О F
Е, эВ 0,75 0,19 0,82 -0,19 0,33 1,24 0,05 1,47 3,50
Атом Ne Na Mg Al Si P S Cl Ar
E, эВ -0,57 0,47 -0,32 0,52 1,46 0,77 2,15 3,70 -1,0
Атом К Br
E, эВ 0,82 3,51
Сродство к электрону определено далеко не для всех элементов. Даже для
типичных неметаллов квантово-механические расчеты показывают, что сродство
их атомов к двум и более электронам всегда отрицательно. Так, электронное
сродство второго порядка для атома кислорода равно -7,6, а для атома серы
-3,5 эВ. Поэтому многозарядные одноатомные отрицательные ионы типа О2", S2",
N3" и т.д. не могут существовать ни в свободном состоянии, ни в молекулах, ни в
кристаллах.
К тому же существование твердых электролитов и ионных проводников не
является доказательством наличия в них самостоятельных ионов. Под воздейст-
воздействием прилагаемого электрического поля происходит дополнительная поляриза-
поляризация, приводящая к возникновению ионов в твердом состоянии, в результате чего
наблюдается ионная проводимость. При растворении в воде солей, кислот и
оснований (также не имеющих готовых ионов) под воздействием электрического
поля полярных молекул воды протекает процесс электролитической диссоциации
растворенных электролитов с образованием гидратированных ионов.
Радиусы атомов. С точки зрения квантовой механики изолированный
атом не имеет строго определенного размера, так как электронная плотность
теоретически обращается в нуль лишь на бесконечно большом расстоянии от
ядра. В то же время электронное облако становится очень размытым уже на рас-
расстоянии в несколько десятков нанометров от ядра. Поэтому определить абсолют-
абсолютные размеры атомов невозможно.
Первоначально сложилось представление об эффективных радиусах атомов,
проявляющихся в их действиях, т.е. в химических соединениях. Эффективные
радиусы * определяли из экспериментальных данных по межъядерным расстоя-
расстояниям в молекулах и кристаллах. При этом предполагалось, что атомы представ-
представляют собой несжимаемые шары, которые соприкасаются своими поверхностями в
соединениях. При определении значения эффективного радиуса из межъядерных
расстояний в ковалентных молекулах подразумевали ковалентные радиусы, при
вычислении их из данных для металлических кристаллов — металлические ра-
радиусы. Наконец, эффективные радиусы, рассчитанные для кристаллов с преиму-
преимущественно ионной связью, назывались ионными радиусами. Для этого определя-
определяли радиус какого-нибудь иона, а затем вычисляли ионные радиусы других эле-
элементов из экспериментальных данных по межъядерным расстояниям в кристал-
кристаллических решетках. Так, с помощью оптических методов, а затем расчетом был
определен радиус аниона фтора, равный 0,113 нм. А расстояние между атомами
Na и F в решетке NaF было установлено равным 0,231 нм. Отсюда радиус иона
Na+ равен 0,231 — 0,113 = 0,118 нм. Металлические радиусы получены делением
пополам расстояния между центрами двух смежных атомов в кристаллических
решетках металлов. Ковалентные радиусы неметаллов также вычислены как
половина межъядерного расстояния в молекулах или кристаллах соответству-
соответствующих простых веществ. Для одного и того же элемента эффективные радиусы
(ковалентный, ионный, металлический) не совпадают между собой. Это свиде-
свидетельствует о зависимости эффективных радиусов не только от природы атомов,
но и от характера химической связи, координационного числа** и других факто-
факторов (см. табл. 4). Изменение эффективных радиусов атомов носит периодический
характер (рис. 22). В периодах по мере роста заряда ядра эффективные радиусы
атомов уменьшаются, так как происходит стягивание электронных слоев к ядру
(при постоянстве их числа для данного периода). Наибольшее уменьшение харак-
характерно для s- и р-элементов. В больших периодах для d- и /элементов наблюдает-
наблюдается более плавное уменьшение эффективных радиусов, называемое соответственно
d- и /сжатием. Эффективные радиусы атомов благородных газов, которыми
заканчиваются периоды системы, значительно больше эффективных радиусов
предшествующих им /(-элементов. Значения эффективных радиусов благородных
газов (см. табл. 4) получены из межъядерных расстояний в кристаллах этих
веществ, существующих при низких температурах. А в кристаллах благородных
газов действуют слабые силы Ван-дер-Ваальса в отличие, например, от молекул
галогенов, в которых имеются прочные ковалентные связи.
В подгруппах Периодической системы эффективные радиусы атомов увеличи-
*Эффективные радиусы часто называют кажущимися.
* 'Координационное чяаю (к.ч.) — число ближайших соседей, находящихся в молекуле
или кристалле на одинаковых расстояниях от данного атома.
10 ZO JO W SO 60 70 80 90 Z
Рис. 22. Зависимость эффективных радиусов атомов от порядкового
номера элемента
ваются из-за роста числа электронных слоев. При этом в подгруппах s- и />-эле-
ментов рост эффективных радиусов происходит в большей мере по сравнению с
подгруппами из d-элементов.
За последние десятилетия (начиная с 1965 г.) в связи с бурным развитием
электронно-вычислительной техники получило определенное распространение
понятие об орбитальных радиусах атомов. Орбитальные радиусы — это рассто-
расстояния от ядра до наиболее удаленною от- него максимума функции радиального
распределения электронной плотности (см. рис. 13). Для любого атома может
быть только один орбитальный радиус в нормальном состоянии (см. табл. 3) и
сколько угодно значений орбитального радиуса в возбужденных состояниях.
Подобно эффективным радиусам, орбитальные радиусы атомов также обнару-
обнаруживают явную периодичность в зависимости от порядкового номера элемента. В
пределах каждого периода наибольшим орбитальным радиусом обладает щелоч-
щелочной металл, а наименьшим — атомы благородных газов. В отличие от эффектив-
эффективных радиусов орбитальные радиусы благородных газов хорошо укладываются в
общую закономерность уменьшения размеров атома по мере увеличения заряда
ядра внутри данного периода.
Для металлических элементов характерно удовлетворительное совпадение
значений эффективных и орбитальных радиусов, чего нельзя сказать относитель-
относительно неметаллов. Ниже приведены эффективные и орбитальные радиусы некоторых
элементов Периодической системы:
Элемент.
Гэфф, НМ.
ГОрб, НМ..
Li
0,155
0,157
Na
0,189
0,180
0
0
К
,236
,216
0
0
Ti
,146
,148
Zr
0,160
0,159
Hf
0,159
0,158
Элемент О S , Se
гэфф, нм 0,066 0,102 0,116
горE, нм 0,045 0,085 0,092
Из самого понятия орбитального радиуса следует, что он ближе к истинному
размеру атома, чем эффективный радиус *. В отличие от эффективного орбиталь-
орбитальный радиус является характеристикой свободного атома или иона и не зависит
от природы химической связи и других факторов. Для предсказания межатом-
межатомных расстояний в молекулах и кристаллах необходимо знание орбитальных ра-
радиусов атомов не только в нормальном, но и в возбужденных состояниях. Однако
даже с помощью мощных компьютеров задача вычисления орбитальных радиусов
для возбужденных состояний атомов еще не решена.
По сравнению с возбужденными состояниями легче производится расчет орби-
орбитальных радиусов ионов. Для катиона натрия, например, его орбитальный радиус
определяется расстоянием от ядра до максимума электронной плотности 2р-
электронов (i-слой на рис. 21), так как у Na отсутствует Зз-электрон.
Теоретический расчет орбитальных радиусов анионов аналогичен расчету
соответствующих радиусов нормальных состояний. В табл. 7 приведены
орбитальные и эффективные радиусы некоторых ионов и нейтральных атомов.
Таблица 7. Орбитальные и эффективные радиусы
некоторых атомов и ионов
Атом
Li
Na
К
Rb
rOp6i
нм
0,157
0,180
0,216
0,229
Катион
Li+
Na+
К+
Rb'
горб>
нм
0,019
0,028
0,059
0,073
гэфф,
нм
0,068
0,098
0,133
0,149
Атом
F
С1
Вг
I
Горб,
нм
0,039
0,073
0,087
0,107
Анион
F"
а-
Вг"
I-
горб>
нм
0,040
0,074
0,089
0,109
*эфф,
нм
0,133
0,181
0,196
0,220
Переход нейтрального атома в катион (например, Na -> Na+ с упразднением;
внешнего электронного слоя) сопровождается резким уменьшением орбитального
радиуса. Этот факт согласуется как с теорией Бора [см. формулу (III.7)], так и с
выводами квантовой механики (см. рис. 13). В то же время анионизация (F -> F" и
т.д.) почти не изменяет орбитального радиуса нейтрального атома. Это и понят-
понятно, поскольку образование аниона, как правило, не связано с возникновением
новых электронных слоев и оболочек. Например, при образовании иона С1" лиш-
лишний электрон заполняет внешнюю З^мэболочку, на которой у атома хлора было 5
электронов. Поэтому орбитальные атомный и ионный радиусы хлора практически
не отличаются друг от друга и соответственно равны 0,073 и 0,074 нм. Таким
образом, эффективные радиусы катионов и анионов оказываются в несколько раз
превосходящими их орбитальные радиусы. Это указывает на отсутствие в молеку-
молекулах и кристаллах самостоятельных ионов вообще. Об этом же свидетельствует тот
факт, что затрата энергии на отрыв одного электрона от атомов металлов всегда
"¦Относительность и субъективность эффективных радиусов видны хотя бы из того
факта, что для атома кислорода он определен по Брэггу 0,066, по Гольдшмидту — 0,132,
по Полингу — 0,140, по Слетеру — 0,060 нм.
Ю 18 36 5? 86
Рис. 23. Зависимость максимальной положительной степени окисления от
порядкового номера элемента
больше, чем выделение ее при присоединении одного электрона к таким атомам,
как F, C1, О, S и др.
Степень окисления элементе в.Среди формальных понятий
химии важнейшим является понятие степени окисления*. Степень окисления —
воображаемый заряд атома элемента в соединении, который определяется из
предположения ионного строения вещества. Определение степеней окисления
элементов основано на следующих положениях: 1) степень окисления кислорода
принимается равной -2. Исключение составяяют пероксидные соединения
(Na2O2), где степень окисления кислорода -1. А в надпероксидах (КО2) и озони-
дах (КО.ч) окислительное число кислорода соответственно -'/2 и -'/з- Наконец, во
фторидах кислорода степень окисления кислорода положительна; например, в
OF2 она равна +2; 2) водород имеет степень окисления +1. Только в солеобраз-
ных гидридах типа NaH его степень окисления равна -1; 3) степень окисления
щелочных металлов равна +1; 4) степень окисления атомов, входящих в состав
простых веществ, равна нулю; 5) в любом ионе алгебраическая сумма всех степе-
степеней окисления равна заряду иона, а в нейтральных молекулах эта сумма равна
нулю.
Важность окислительного числа прежде всего заключается в том, что номер
группы Периодической системы указывает на высшую положительную степень
окисления (характеристическая степень окисления), которую могут иметь эле-
элементы данной Группы В своих соединениях. Исключение составляют металлы
подгруппы меди, кислород, фтор, металлы семейства железа и некоторые другие
элементы VIII группы. Кроме того, понятие степени окисления полезно при клас-
классификации химических соединений, а также при составлении уравнений окис-
окислительно-восстановительных реакций. Кривая изменения максимальной поло-
положительной степени окисления имеет периодический характер в зависимости от
порядкового номера элемента (рис. 23). При этом в пределах каждого большого
периода эта зависимость представляется сложной и своеобразной.
*Синонимы: "окислительное число", "состояние окисления", "степень окисленности",
"электрохимическая валентность", "электровалентность".
55"
Несмотря на широкое применение в химии понятия степени окисления, оно
является сугубо формальным. В настоящее время экспериментально определя-
определяемые истинные заряды атомов в соединениях не имеют ничего общего со степеня-
степенями окисления этих элементов. Так, действительные заряды атомов водорода и
хлора в молекуле НС1 соответственно равны +0,17 и —0,17 (а степени окисления
+1 и -1). В кристаллах сульфида цинка ZnS заряды атомов цинка и серы равны
+0,86 и -0,86 вместо формальных степеней окисления +2 и -2.
Нельзя отождествлять степень окисления с валентностью элемента, если даже
их абсолютные значения совпадают. Валентность атома, определяемая как число
химических связей, которыми данный атом соединен с другими атомами, не
может иметь знака (+ или -) и быть равен нулю. Поэтому особенно неудачны
выражения "положительная и отрицательная валентность", тем более "нулевая
валентность", бытующие поныне в химической литературе. Например, у метана
СЩ, метилового спирта СНзОН, формальдегида НСОН, муравьиной кислоты
НСООН и диоксида углерода СОг валентность углерода равна четырем, а степени
окисления его равны соответственно -4, -2, 0, +2 и +4. Кроме того, для установ-
установления валентности атома требуется знание химического строения, а определение
степени окисления производится в отрыве от структуры вещества, т.е. формально.
ГЛАВА IV. ХИМИЧЕСКАЯ СВЯЗЬ
1. Химическая связь и валентность. Понятие о химической связи является
одним из основополагающих в современной химической науке. Физико-химиче-
Физико-химическая природа вещества целиком определяется его химическим или кристаллохи-
мическим строением. В настоящее время под химическим или кристаллохимиче-
ским строением понимают совокупность энергетических, геометрических и кван-
тово-химических характеристик вещества: порядка, длины, кратности и энергии
связи, распределения и пространственной направленности электронного облака,
эффективных зарядов атомов и т.п. Но главное в учении о химическом и крис-
таллохимическом строении вещества — химическая связь. Химическое и кристал-
лохимическое строение в первую очередь определяется характером межатомных
связей всех атомов, входящих в состав данного вещества.
Химическая связь — явление взаимодействия атомов, обусловленное перекры-
перекрытием электронных облаков связывающихся частиц, которое сопровождается
уменьшением полной энергии системы (молекула, комплекс, кристалл и т.п.).
Химическая связь характеризуется энергетическими и геометрическими парамет-
параметрами. Важнейшей энергетической характеристикой служит энергия химической
связи, определяющая ее прочность. К геометрическим параметрам относятся
длина химической связи, углы между связями в молекулах, комплексах, кристал-
кристаллах и т.п.
Валентность зависит от состояния атомов рассматриваемого элемента, природы
партнера, с которым реагирует данный элемент, условий взаимодействия. Так,
углерод с одним и тем же партнером — кислородом — в зависимости от условий
взаимодействия образует СОг и СО, в которых состояния атомов углерода раз-
различны. На основе валентности элементов легко определить формульный состав
химического соединения. Поэтому величину валентности часто называют стехио-
метрической валентностью.
56
Учение о химической связи неразрывно связано с понятием валентности.
Однако для правильного понимания рассматриваемого вопроса необходимо четко
разграничить понятия валентности и химической связи. Валентность возникает
как формальная числовая характеристика элемента, а химическая связь пред-
представляет собой физико-химическое явление. Валентность отражает форму химиче-
химического взаимодействия элементов, а химическая связь — его содержание. Поэтому
между валентностью и химической связью существует различие в той мере, в
какой разграничиваются понятия формы и содержания предмета. В диалектиче-
диалектическом единстве формы и содержания (валентности и химической связи) определя-
определяющим является содержание, т.е. химическая связь.
2. Энергия химической связи. Мерой прочности химической связи служит
энергия связи. Ее величина определяется работой, необходимой для разрушения
связи, или выигрышем в энергии при образовании вещества из отдельных атомов.
Например, энергия связи Н—Н в молекуле водорода равна 435 кДж/моль. Это
значит, что при образовании 1 моль газообразного водорода из изолированных
атомов по уравнению
Н + Н = Н2 + 435 кДж/моль
выделяется 435 кДж. Такое же количество энергии должно быть затрачено на
распад 1 моль Н2 до атомарного состояния (энергия атомизации молекулы). При
образовании многоатомных молекул, содержащих одинаковые связи (например,
молекул метана СЩ или воды), средняя энергия связи в пересчете на 1 моль
вещества определяется делением энергии образования этого вещества из изолиро-
изолированных атомов на число связей. В приведенных примерах энергии образования
определяются уравнениями
С + 4Н = СН4 + 1647 кДж/моль,
2Н + О = Н20 + 924 кДж/моль.
Отсюда средние энергии связей С—Н и О—Н равны соответственно 1647:4 = 412
кДж/моль и 924:2 = 462 кДж/моль. Для определения энергии единичной связи
необходимо средние энергии разделить на число Авогадро. Все связи в каждой
из рассмотренных молекул равноценны.
Представление об энергии связи является универсальным и в равной мере
приложимо как к молекулам, так и к кристаллическому состоянию. Однако
величина энергии связи при переходе от молекул к кристаллу изменяется, по-
поскольку при этом изменяются координационное число и энергетическое состо-
состояние атомов. При образовании кристалла из газообразных молекул наблюдается
выигрыш в энергии, обусловленный упорядоченным расположением атомов в
кристаллической решетке. Чтобы оценить этот выигрыш, нужно сравнить между
собой энергии разрыва связи в кристалле и газообразной молекуле. Разрыв связи
в молекуле может быть осуществлен юмолитгсчески (с образованием нейтральных
атомов) и гетеролитически (с образованием ионов). Для молекулы NaCl в первом
случае, согласно уравнению
NaCl (г) = Na (г) + С1 (г) - 414 кДж/моль,
необходимо затратить 414 кДж/моль. При гетеролитическом распаде
NaCl (г) = Na+ (г) + С1" (г) - 548 кДж/моль
эта энергия возрастает на 134 кДж/моль. Связи в кристалле можно также разор-
разорвать по гемолитическому и гетеролитическому механизмам:
NaCl (тв) = Na (г) + С1 (г) - 644 кДж/моль, (I)
NaCl (тв) = Na+ (r)v+ C1" (г) - 778 кДж/моль. (II)
Уравнение (I) отражает процесс сублимации кристаллического NaCl с образова-
образованием моноатомных паров компонентов. Соответствующая энергия F44 кДж/моль)
называется энергией атомиэации кристаллического соединения. При образовании
свободных газообразных ионов в соответствии с уравнением (II) энергия, необхо-
необходимая для осуществления этого гипотетического процесса, равна по величине и
противоположна по знаку так называемой энергии кристаллической региетки.
Энергия связи в кристаллическом NaCl, представляющая гобой энергию раз-
разрыва, взятую с противоположным знаком, на 230 кДж/моль больше, чем для
газообразной молекулы F44 — 414 = 778 — 548). Энергия разрыва связи по гомо-
литическому и гетеролитическому типам как в молекуле, так и в кристалле отли-
отличается на одну и ту же величину 134 кДж/моль. Она представляет собой раз-
разность между энергией ионизации Натрия (-495,3 кДж/моль) и энергией сродства
к электрону для хлора C61,5 кДж/моль) и определяет энергию, необходимую
для образования газообразных ионов Na* и С1" из изолированных атомов.
Таким образом, необходимо подчеркнуть, что характер химического взаимо-
взаимодействия коренным образом меняется при переходе от газообразных молекул к
кристаллическому твердому телу, что прежде всего отражается на энергии связи
в различных агрегатных состояниях. Энергии связи в рядах однотипных соедине-
соединений как для газообразных молекул, так и для кристаллов изменяются законо-
закономерно. На рис. 24, в, б показано изменение энергии связи в рядах галогенидов
щелочных металлов для газообразных молекул и кристаллов. Отметим три основ-
основные закономерности: во-первых, энергия связи уменьшается при увеличении
атомного номера элементов; во-вторых, энергия связи в кристалле всегда выше,
чем в соответствующей молекуле; в-третьих, энергия диссоциации по гемолитиче-
гемолитическому механизму и для молекул и для кристаллов ниже, чем по гетеролитическо-
гетеролитическому. Следовательно, при нагревании эти соединения распадаются на атомы, а не
на ионы. Отмеченные закономерности универсальны и сохраняются для соедине-
соединений с различным характером взаимодействия и типом связи.
3. Длина химической связи. Под длиной связи понимают расстояние между
центрами ядер атомов в молекуле (и кристалле), когда силы притяжения урав-
уравновешены силами отталкивания и энергия системы минимальна. Длины связей
определяют экспериментально по рентгеноструктурным и спектральным данным.
Длины связей в рядах однотипных соединений также подчиняются общей
закономерности (рис. 25). Длина связи увеличивается с возрастанием атомного
номера элемента, что хорошо коррелирует с уменьшением энергии связи как в
ряду молекул, так и в ряду кристаллов. Однако при переходе от газообразных
молекул к кристаллам наблюдается заметное увеличение длины связи, которое
тем не менее сопровождается ее упрочнением. Это кажущееся противоречие легко
объяснимо. В самом деле, несмотря на то что каждая отдельная связь в кристал-
кристалле слабее, чем в соответствующей молекуле, число таких связей намного больше
F для структуры типа NaCl и 8 для структуры типа CsCl), что и увеличивает
общую энергию взаимодействия. Таким образом, кристаллическое состояние
LI Na К Rb Cs
a
ввв
500
too
200
—* ^^_——•
~° 0 o/
-o~—-o-— — -o I
.O- —— —O- — — ~oj
Li Na
К Rb CS
6
Li Na К Rb CS
Рис. 24. Изменение энергии связи в ря-
ряду галогенидов щелочных металлов для
газообразных молекул и кристаллов:
а - гетеролитаческий распад; б - гемоли-
гемолитический распад; - - - кристаллическое
состояние вещества; — - парообразное
состояние вещества; I - фториды; 2 -
хлориды; 3 - бромиды; 4 ' иодиды
Рис. 25. Изменение длины химичес-
химической- связи для галогенидов щелочных
металлов:
---кристаллическое состояние вещества;
парообразное состояние вещества;
1 - фториды; 2 - хлориды; 3 - броми-
бромиды; 4 - иодиды
вещества отличается от газообразного энергией связей и механизмом их образова-
образования.
Атомы в молекулах и кристаллах совершают колебания около положения
равновесия, причем частота колебаний характерна для каждой связи и не зави-
зависит от температуры, в то время как амплитуда колебаний растет с увеличением
температуры. Исследование колебательных спектров молекул и кристаллов позво-
позволяет оценить жесткость связи, т.е. ее сопротивляемость внешним воздействиям,
вызывающим изменение ее длины.
При изучении колебательных спектров часто вместо характеристических час-
частот колебаний v — с/А [с] используют волновые числа а; = 1/А [см'1], которые
определяют число длин волн, укладывающихся в 1 см. На основании эксперимен-
экспериментально определенных волновых чисел, характерных для данной связи, может
быть рассчитана так называемая силовая константа связи к, характеризующая ее
жесткость. Соотношение между к и и> имеет вид
О- (IV.1)
59
где с — скорость света; ц — приведенная масса колеблющейся системы, которая
определяется из формулы
= 1/mi + \/ni2 или /I = mim2/(mi + m?),
где mj и пг2 — абсолютные массы * связанных атомов.
Если использовать только массовые числа, то расчетная формула для опреде-
определения силовой константы (Н/нм) принимает вид
*=5,8-10-17/«<>2- (IV.2)
Так, для связи Н—Вг характеристическое волновое число w = 2558 см'1. Тогда
силовая константа к = 3,8 -10 Н/нм. Как следует из размерности силовой кон-
константы, она по своему физическому смыслу определяет силу, которую надо при-
приложить к системе двух связанных атомов в направлении связи, чтобы изменить ее
длину на 1 нм. Преобразуя (III.1), можно написать Е = hv = hew, что устанавли-
устанавливает прямую связь между волновым числом и соответствующей энергией. Следо-
Следовательно, силовая константа характеризует энергию, необходимую для возбужде-
возбуждения собственных (резонансных) колебаний данной связи. Чем больше величина Jk,
тем большую энергию необходимо затратить для возбуждения колебаний, тем
более жесткой является связь. Если энергия связи определяет ее прочность, то
силовая константа — ее жесткость. Несмотря на отсутствие однозначного соответ-
соответствия между энергией связи и силовой константой, эти величины в рядах одно-
однотипных соединений меняются симбатно (табл. 8).
Таблица 8. Характеристики связей Н—Г
Связь
H-F
н-ci
H-Br
H-I
Силовая константа также зависит от кратности связи и определяет степень
возрастания прочности и жесткости при переходе от одинарных связей к двой-
двойным и тройным. Силовые константы кратных связей приведены ниже:
Связь.... С~С
Jfc-107, Н/нм 4,6
4. Электрический момент диполя и направленность связи. Химическая связь
характеризуется определенной пространственной направленностью. Если двух-
двухатомные молекулы всегда линейны, то формы многоатомных молекул могут быть
Энергия
Е, кДж/моль
564,3
430,5
364
297
Волновое
число и, см
3935
2886
2553
2233
Силовая константа
к-107, Н/нм
8,65
4,74
3,78
2 о
с=с
9,5
СеС
15,8
с-о
4,9
С=О
12,3
СТО
18,6
'Абсолютная масса — массовое число, умноженное на атомную единицу массы
1,6605655-107 кг.
различными. Так, трехатомные молекулы типа AB2 бывают как линейными
HgCl2), так и угловыми (Н2О, SO2) H2S). Пространственное строение молекул
может быть выявлено различными методами. К их числу относятся, например,
исследование вращательных спектров молекул в дальней инфракрасной области,
определение электрических моментов диполей и некоторые другие.
Электрический момент* диполя является мерой полярности молекулы. Меж-
Между взаимодействующими атомами, которые различаются по электроотрицательно-
электроотрицательности, возникают полярные связи. В результате смещения электронной плотности в
сторону более электроотрицательного партнера происходит разделение "центров
тяжести" положительного и отрицательного зарядов и возникает диполь, пред-
представляющий собой систему из двух равных и противоположных по знаку зарядов
6+ и 6-, находящихся на определенном расстоянии / (длина диполя) друг от
друга. Длину диполя не следует отождествлять с длиной связи, поскольку "цент-
"центры тяжести" зарядов не совпадают с центрами ядер взаимодействующих атомов.
Длина диполя для отдельной связи всегда меньше длины связи и изменяется от
нуля для гомоядерных молекул (типа А2) до 0,17 нм для одной из наиболее
полярных молекул LiF (длина свйзи Li~~F равна 0,21 нм).
Диполь выражают через электрический момент диполя ц, который представ-
представляет собой произведение заряда 6 на длину диполя /:
ц = 16.
В отличие от длины диполя электрический момент диполя является векторной
величиной. Направление электрического момента диполя условно принимают от
отрицательного к положительному полюсу диполя.
Для многоатомных молекул следует различать понятие об электрических мо-
моментах диполя отдельных связей и молекулы в целом. При наличии нескольких
связей в молекуле их электрические моменты (векторы) складываются по прави-
правилу параллелограмма. В зависимости от формы молекулы, определяемой направ-
направленностью связей, результирующий электрический момент диполя отличается от
электрических моментов диполя отдельных связей и для высокосимметричных
молекул может быть равен нулю, несмотря на значительную полярность отдель-
отдельных связей. Например, линейная молекула СО2 неполярна (ц = 0), хотя каждая
связь С=О имеет значительный электрический момент диполя (fi = 8,9-109
Кл-м). Это объясняется тем, что равные по величине электрические моменты ди-
диполя связей направлены навстречу друг другу:
8- + 2 * 8-
8,9-КГ29 * i 8,9- 1(Г29
Также это относится к неполярным плоской треугольной молекуле BF3 и окта-
эдрической молекуле SF6 (рис. 26). Для несимметричных молекул электрические
моменты диполя отдельных связей не компенсируют друг друга и суммарный
электрический момент диполя будет отличным от нуля. Классическим примером
могут служить угловые молекулы типа АВ2(Н2О, H2S и т.п.). Так, в молекуле
*Речь идет о собственном, а не наведенном электрическом моменте диполя.
61
/ \
Р и с. 26. Гомеополярность симметричных мо-
молекул BF3 и SFg
воды электрические моменты диполя
каждой связи О"~Н равны 5,2 -109
Кл'М, однако электрический момент
диполя молекулы в целом составляет
6,07 -Ю9 Кл*м. Следовательно, элект-
электрические моменты диполя связей скла-
складываются геометрически и связи О"~Н
должны быть направлены под углом
105°- друг к другу (рис. 27). Строение
молекулы аммиака можно гипотетиче-
гипотетически представить двояко: в виде плоско-
плоского треугольника или тригональной пирамиды (рис. 28). В первом случае резуль-
результирующий электрический момент диполя молекулы NH3 должен быть равен
нулю. Однако экспериментально найденная величина D,8-10~29 Кл-м) свидетель-
свидетельствует о пирамидальной структуре.
Таким образом, существование электрических моментов диполей в молекулах
и их величина определяются пространственной направленностью химической
связи.
В кристаллах направленность химической связи также существует, что особен-
особенно ярко проявляется, например, в веществах с преимущественно ковалентной
связью (кремний, германий, ZnS, InP и т.п.). Связи в таких кристаллах направ-
направлены к вершинам тетраэдра (см. рис. 3 и 4), поэтому подобные вещества часто
называют тетраэдрическими фазами.
5. О ионной связи. Первоначально Коссель A916) считал, что при химическом
взаимодействии разнородные атомы стремятся приобрести конфигурацию внеш-
внешней оболочки благородных газов. Это достигается отдачей и присоединением
электронов нейтральными атомами химических элементов. Атомы, отдающие свои
электроны, превращаются в положительно заряженные ионы (катионы). Атомы,
присоединяющие электроны, превращаются в отрицательно заряженные ионы
(анионы). Химическая связь осуществляется за счет электростатического при-
притяжения образовавшихся разноименных ионов. В этом заключается сущность
теории ионной связи.
Согласно теории ионной связи, в решетке ионного кристалла (например,
NaCl) происходит не только притяжение между разноименными ионами, но и
отталкивание одноименных ионов. В этих условиях устойчивость подобных крис-
кристаллов объясняется тем, что расстояния между разноименными ионами меньше,
36-
Рис. 27. Электрический момент
диполя в молекуле воды
Р и с. 28. Обоснование конечного значения электричес-
электрического момента диполя молекулы аммиака
чем между одноименными. Поэтому кулоновские силы притяжения превалируют
над силами отталкивания, что и обеспечивает ионную связь.
Однако идеально ионных соединений вообще не существует, а следовательно,
истинной ионной связи тоже. Даже при химическом взаимодействии наиболее
электроположительных и электроотрицательных элементов образуются соедине-
соединения, в которых химическая связь не на 100% ионная. Поэтому в молекулах и
кристаллах ионная связь должна рассматриваться как предельный случай час-
частично ионной связи. Прежде всего об этом свидетельствуют экспериментальные
данные по эффективным зарядам атомов, входящих в состав соединений.
Таблица 9. Эффективные заряды * атомов в некоторых соединениях
Соеди-
Соединение
NaCl
NaBr
Nal
MgCl2
MgBr2
A1N
A1P
AlSb
GaSb
InSb
Na2S
Эффективный
заряд
+0,87
+0,83
+0,75
+ 1,50
+ 1,38
+ 1,32
+0,87
+0,57
+0,33
0
+0,75 -0,96
Соединение
SiC-2
А12О3
A12S3
MgO
ZnS
K2S
Na2SO4
Na3PO4
MgSO4
A1PO4
Эффективный
заряд
+ 1,97 -0,99
+ 1,77 -1,02
+ 1,26 -1,00
-1,01
-0,86
-1,06
-1,09
-1,15
-0,88
-0,94
*Для тройных соединений заряд относится к атому кислорода.
Эффективный заряд атома, входящею в состав соединения, определяется как
алгебраическая сумма ею отрицательного электронного заряда и положительною
заряда ядра. В настоящее время известно более десятка экспериментальных мето-
методов определения значений эффективных зарядов в большинстве своем с точ-
точностью @,1 — 0,3)е, что соизмеримо с точностью вычисления этих зарядов в
квантовой химии и теории твердого тела. В табл. 9 приведены данные по эффек-
эффективным зарядам атомов, которые получены рентгеноспектральным методом для
ряда типичных неорганических веществ. Знаком "+" отмечены эффективные
заряды на металлических элементах, знаком "—" — на электроотрицательных
атомах. К чисто ионным соединениям близки только галогениды щелочных ме-
металлов, хотя и для них эффективные заряды не достигают единицы. Все осталь-
остальные соединения,^ том числе галогениды, оксиды, сульфиды кальция и магния,
являются только частично ионными. Кроме того, эффективные заряды на типи-
типических электроотрицательных атомах (кислород, сера) почти не превосходят 1, в
то время как заряды металлических элементов (кальций, алюминий) могут быть
заметно больше единицы. Это объясняется тем, что энергия присоединения двух
электронов к кислороду и сере (сродство к электрону второго порядка) отрица-
тельйа. Расчеты показывают, что сродство к электрону второго порядка для
кислорода равно -732, а для серы составляет -334 кДж/моль. Значит, ионы типа
О2", S2" не существуют* и все оксиды, сульфиды независимо от активности
металлов не относятся к ионным соединениям. Если двухзарядные ионы в дейст-
действительности не существуют, тем более нереальны многозарядные одноатомные
отрицательные ионы.
Самопроизвольная передача электрона от металлического атома к атому неме-
неметалла в действительности вряд ли осуществляется. Дело в том, что потенциал
ионизации первого порядка даже для наиболее активных щелочных металлов
больше, чем сродство к электрону типичных электроотрицательных элементов. С
этой точки зрения оказывается энергетически невыгодным образование ионной
молекулы NaCl из элементов, так как первый ионизационный потенциал натрия
равен 5,14 В, а сродство к электрону атома хлора — 3,7 эВ (ионизационный по-
потенциал, выраженный в вольтах, численно равен энергии ионизации в электрон-
вольтах). Из квантовой механики также следует, что полное разделение зарядов с
возникновением идеальной ионной связи А1+В1- никогда не может осуществиться,
так как из-за волновых свойств электрона вероятность его нахождения вблизи
ядра атома А может быть мала, но отлична от нуля.
Таким образом, вопреки довольно распространенному мнению чисто ионных
соединений с идеальной ионной связью** на самом деле не существует***. Меж-
Между тем принято считать, что химическая связь у подавляющего большинства не-
неорганических соединений имеет ионный характер. Объясняется это двумя исто-
исторически сложившимися причинами. Во-первых, почти все химические реакции
исследовались в водной среде и представляли, по существу, ионные реакции. В
то же время поведение вещества в водных растворах коренным образом отличает-
отличается от его свойств в отсутствие воды. Так, соляная кислота относится к числу
сильнейших электролитов: растворенный в воде хлорид водорода полностью дис-
диссоциирует на ионы водорода и хлора. Основываясь на этом факте, можно было
бы допустить ионную связь в молекуле НС1. Однако безводный хлорид водорода
представляет собой почти неионное соединение, в котором эффективные заряды
водорода и хлора соответственно равны +0,17 и -0,17. Во-вторых, в свете учения
об ионной связи в неорганической химии укоренились представления о положи-
положительной и отрицательной валентности (электровалентности). Даже если невозмож-
невозможны отдача и присоединение электронов, нередко подразумевали электровалент-
электровалентность, т.е. ионную связь. Это усугублялось еще и тем, что в неорганической хи-
химии исключительно важную роль играет электронная теория окислительно-вос-
окислительно-восстановительных реакций, постулирующая переход электронов от восстановителей
к окислителям. При этом степень окисления полностью отождествлялась с элект-
электровалентностью и для удобства подсчета числа отдаваемых и присоединяемых
электронов заведомо неионные соединения рассматривались как вещества с ион-
ионной связью. Между тем понятие степени окисления не имеет ничего общего
*Согласно данным опытов по рассеянию молекулярных пучков, эти ионы могут суще-
существовать как чрезвычайно неустойчивые частицы с временем жизни порядка 100 с.
* *Чтобы ионы характеризовались целочисленными дискретными электрическими заря-
зарядами, например А1+, А2+, А3+, В1", В2" и т.д.
* * *Такие соединения лучше называть преимущественно ионными.
Ad
ни с эффективными зарядами атомов, ни с фактическим числом связей, которые
образует данный атом (валентность).
Таким образом, нужно говорить о большей или меньшей степени ионности
(доля ионности). При этом чем выше степень ионности связи, тем больше величи-
величины эффективных зарядов атомов, входящих в состав соединения. Термин "эф-
"эффективные заряды" неудачен, так как в действительности опытные и расчетные
их значения представляют собой фактические заряды атомов в соединениях.
Идеальная же ионная связь наравне с идеальными газами и идеальными раство-
растворами представляет собой типичный пример научной абстракции.
6. Ковалентная связь. Начало учению о парно-электронной (ковалентной)
связи положил Льюис A916). Подобно Косселю, он считал, что при химическом
взаимодействии атомы стремятся приобрести конфигурацию внешней электрон-
электронной оболочки последующего благородного газа. Только, по Льюису, это достига-
достигается не отдачей и присоединением электронов, а образованием общей электрон-
электронной пары. Каждый из взаимодействующих атомов отдает по одному электрону на
образование указанной электронной пары, если возникает одинарная связь. Так,
образование молекулы водорода происходит за счет неспаренных электронов
атомов *: Н:Н.
Главное в учении о ковалентной связи — обобщение валентных электронов. В
молекуле водорода обобществляются оба электрона — по одному от каждого ато-
атома водорода, которые и являются валентными. При этом одна обобществленная
электронная пара соответствует одной "единице валентности" в теории химичес-
химического строения Бутлерова. Общая электронная пара, ответственная за химическую
связь, иначе называется поделенной парой электронов. Возникновение кратной —
двойной и тройной — связи сопровождается образованием соответственно двух и
трех поделенных электронных пар. Соединение атомов азота с возникновением
трех ковалентных связей (тройная связь), по Льюису, можно представить следую-
следующим образом * *:
:Ni + -N: —>- :N| IN:
\/
неподеленные
пары электронов
Каждый атом азота имеет по три неспаренных электрона, которые и образуют
три парно-электронные связи. При этом у каждого атома остается по одной непо-
деленной паре электронов, т.е. два электрона с антипараллельными спинами на
одной атомной орбитали.
Таким образом, ковалентная связь осуществляется электронной парой, нахо-
находящейся в общем владении двух атомов, образующих химическую связь. Ковалент-
ную связь между одинаковыми атомами (например, в Нг, N2) называют также
атомной или гомеополярной***. Молекулы или соединения, образованные на ос-
*В теории Льюиса электроны обозначают точками.
* "Около символа элемента принято обозначать электроны только наружного электрон-
электронного слоя.
* * * Атомную или гомеополярную связь иногда называют неионной.
3 я д v™« - - вй
нове этих связей, называются неполярными или юмеополярными. Их электричес-
электрический момент диполя равен нулю. Ковалентная связь возникает и при химическом
взаимодействии атомов разных химических элементов. Тогда обобществленная
электронная пара (или электронные пары) несколько смещается в сторону более
электроотрицательного партнера. Несмотря на такое смещение, электронная пара
продолжает быть коллективной собственностью обоих взаимодействующих ато-
атомов. Такая ковалентная связь называется полярной и показана на примере обра-
образования молекулы HF:
Н' + .F: —*~ Н :F:
Здесь 6 < \. Таким образом, полярная связь не является самостоятельным типом
химической связи, а представляет собой результат поляризации * ковалентной
связи.
В широком смысле слова ковалентная связь — химическая связь между атома-
атомами, осуществляемая обобществленными электронами. Ковалентная связь являет-
является универсальным типом химической связи. Ионная же связь, по Косселю, может
быть рассмотрена как предельный случай полярной ковалентной связи. В самом
деле, общая электронная пара Льюиса в пределе может быть смещена (теоретиче-
(теоретически) полностью в сторону одного из взаимодействующих атомов. Это равносильно
тому, что один из партнеров безвозмездно отдает принадлежавший ему ранее
электрон (входивший в состав электронной пары) другому атому.
Ковалентная связь — самый распространенный тип химической связи. Меж-
Межатомная связь абсолютного большинства неорганических и органических соедине-
соединений ковалентна. По механизму образования ковалентных связей нет никакой раз-
разницы между неорганическим соединением аммиаком NH3 и органическим соеди-
соединением метаном СН4. Для неорганических соединений типа кислот, оснований и
солей наблюдаются межатомные связи с несколько большей долей ионности, т.е.
более полярные ковалеитные связи по сравнению с органическими соединениями.
Следовательно, по фундаментальной характеристике молекул — природе меж-
межатомной химической связи — нет принципиальной разницы между неорганичес-
неорганической и органической химией. Отличие состоит в том, что в твердых органических
веществах действуют слабые межмолекулярные силы, а в типичных неорганичес-
неорганических кристаллах отсутствуют молекулы и доминирует ковалентная связь между
атомами.
7. Понятие о квантовой химии. В общем виде квантовая химия — это прило-
приложение современной квантовой теории для решения химических проблем. Она
изучает строение и физико-химические свойства молекул, радикалов, комплексов
и кристаллов на основе представлений современных квантовых теорий, в частно-
частности квантовой механики. Квантовая химия охватывает учение о природе химиче-
химической связи, об электронной структуре молекул и других объектов исследования
химии, а также вскрывает взаимосвязь между структурой и свойствами, включая
реакционную способность веществ. Квантовая химия — современное учение о
химическом и кристаллохимическом строении вещества, а также о взаимосвязи
между строением и свойствами на основе представлений и методов квантовой
*Поляризация — явление смещения электрических зарядов под действием каких-то
сил.
66
механики. Таким образом, квантовая химия представляет собой дисциплину на
стыке физики и химии и имеет первостепенное значение для всей современной
химии.
В настоящее время главная заслуга квантовой химии заключается в раскры-
раскрытии природы химической связи. Наибольшее распространение получили два
квантово-химических способа приближенного расчета систем из ядер и электро-
электронов, отвечающих химическим объектам, — метод валентных связей и метод
молекулярных орбиталей. В обоих методах приближенные волновые функции
сложной системы конструируются по определенным правилам (специфичным для
каждого метода) из одноэлектронных атомных волновых функций, т.е. атомных
орбиталей.
8. Метод валентных связей (МВС). Начало квантовой химии было положено
немецкими учеными Гейтлером и Лондоном, которые в 1927 г. опубликовали
квантово-механический расчет молекулы водорода. Впервые было дано научное
обоснование электронной пары Льюиса, ответственной за ковалентную связь. В
дальнейшем метод Гейтлера — Лондона получил развитие в трудах Слейтера,
Ван Флека и особенно Полинга и был назван методом валентных связей (МВС)
или локализованных электронных пар.
Волновое уравнение Шрёдингера для стационарного состояния (III.20) легко
преобразовать в
Нф = Еф,
(IV.3)
л
где Я - квантово-механический оператор полной энергии Гамильтона, равный
Я = -
+ U (х, у, г),
(IV.4)
в котором U — потенциальная энергия электрона. Умножив обе части уравнения
(IV.3) на 4>dv (где dv — бесконечно малый элемент объема внутримолекулярного
пространства), имеем
(IV.5)
v - f'Hipdv.
Интеграл выражения (IV.5) по всему объему молекулы запишется как
Efip2dv =
Сомножитель fijpdv, согласно условию нормировки волновой функции (см. § 3
гл. III), равен единице, а потому энергия равна
Е = Ji)H4>dv.
(IV.6)
Молекула водорода представляет собой систему из двух электронов и двух
протонов (рис. 29). Потенциальная энергия* такой системы запишется в виде
Rah
Га1 га2
С1
П>2
*Выражение для потенциальной энергии U необходимо знать, так как она входит в
(IV.4).
Пг
IP.
Рис. 29. Расстояния между части-
частицами в молекуле водорода
Рис. 30. Зависимость энергии
молекулы водорода от межъя-
межъядерного расстояния
В МВС при построении волновой функции электронов молекулы исходят из
волновых функций электронов составляющих атомов. Пусть фа A) и фь B) озна-
означают собственные функции электронов изолированных атомов водорода На и Нь,
где A) и B) — символы пространственных координат первого и второго электро-
электронов, т.е. фа A) = фа (х{, уи zx) и фь B) = фЬ (х2, у2, ъ).
Вероятность одновременного нахождения электрона A) у ядра я, а электрона
B) — у ядра Ь равна произведению частных вероятностей фа A) и фь B):
Ф = Фа A) ФЬ B).
(IV.7)
Ввиду абсолютной неразличимости электронов выражению (IV.7) эквивалентно
уравнение
Ф = Фа B) фь A). (IV.8)
По Гейтлеру и Лондону, волновая функция молекулы водорода представляется
как линейная комбинация функций (IV.7) и (IV.8):
Ф = Фа A) ФЬ B) ± фа B) фЬ A). (IV.9)
Подстановка волновой функции (IV.9) в (IV.6) и учет (IV.4) приводит к выра-
выражению полной энергии системы общего вида
Е = (К± 0)/A ± IP), (IV.10)
где К, О и П — кулоновский, обменный интегралы и интеграл перекрывания
соответственно.
Интеграл перекрывания показывает степень перекрытия волновых функций
атомов водорода и изменяется от нуля при межъядерном расстоянии R = оо до
единицы (R — 0). При равновесном расстоянии между атомами водорода в
молекуле он равен 0,75. Поэтому без большой ошибки можно принять, что
полная энергия системы равна алгебраической сумме кулоновского и обменного
интегралов:
Е= R± О.
(IV.11)
68
Кулоновский интеграл характеризует электростатическое взаимодействие заря-
заряженных частиц в рассматриваемой системе. По абсолютной величине он неизме-
неизмеримо меньше обменного интеграла, обусловленного движением каждого электрона
около обоих ядер (возникновение электронной пары). Кроме того, обменный
интеграл имеет отрицательный знак. Поэтому он вносит основной вклад в энер-
энергетику химической связи, т.е. уменьшение энергии молекулярной системы по
сравнению с изолированными атомами в основном обусловлено величиной обмен-
обменного интеграла.
На рис. 30 показана зависимость энергии молекулы водорода от межъядерного
расстояния, образование молекулы водорода представлено сплошной кривой. Она
состоит из двух ветвей: притяжения аЬ и отталкивания be атомов. В точке мини-
минимума силы притяжения уравновешиваются силами отталкивания. Равновесное
расстояние Го, т.е. расстояние от минимальной точки Ь до оси ординат, представ-
представляет собой длину химической связи, а отрезок от минимума кривой до оси аб-
абсцисс характеризует энергию связи или энергию диссоциации Ед молекулы водо-
водорода на атомы*. При образовании молекулы водорода (рис. 30, сплошная кри-
кривая) спины электронов антипараллельны, а отсутствие химического взаимодейст-
взаимодействия (пунктирная кривая) характеризуется параллельностью электронных спинов.
Это вытекает из анализа уравнения (IV.9) при перемене координат электронов с
соблюдением принципа Паули. Уравнение (IV.9) можно записать в виде двух
самостоятельных выражений:
Ф+ = Фа A) Фъ B) + Фа B) фЪ A) (IV. 12)
И
Ф= Фа A) ФЬ B) - фа B) фЪ A). (IV.13)
Перемена электронных координат в уравнении (IV.12), т.е. перестановка коорди-
координат A) и B), не изменяет знака функции ф+. Такую функцию называют симмет-
симметричной. Наоборот, подобная инверсия пространственных координат электронов в
(IV.13) сопряжена с изменением знака функции ф-. Поэтому функция ф- называ-
называется антисимметричной. Однако функции (IV.7) и (IV.8), из которых составлено
уравнение (IV.9), не учитывают спина электрона (как и вся нерелятивистская
квантовая механика Шрёдингера). Поэтому принцип
Паули требует, чтобы для антисимметричной функ-
функции (IV. 13) электронные спины были параллельны,
т.е. оба электрона должны иметь одинаковые спино-
спиновые квантовые числа. Только в этом случае при пере-
перемене местами электронов ф. изменит свой знак. Нао-
Наоборот, ф+ отвечает такому состоянию, когда электро-
электроны в молекуле Характеризуются различными спино- Рис. 31. Вид электронного об-
выми квантовыми числами, т.е. имеют противополож- лака в системе из двух атомов
но направленные, или антипараллельные, спины. водорода для симметричной и
Симметричной и антисимметричной волновым антисимметричной волновых
функциям отвечают картины распределения функций
За вычетом небольшой по величине так называемой нулевой колебательной энергии
атомных ядер. ' .
электронного облака в системе из двух атомов водорода (рис. 31). Вероятность
нахождения электронов, или плотность электронного облака, определяется
квадратом волновой функции (см. гл. III). Возведя в квадрат уравнения (IV.12) и
(IV.13), получим
= ф1 A) ф\ B) + ф\ B) фь A) + 2фа A) фъ B)фа B) фь A) (IV.14)
\ф.\2 = ф2а A) ф\ B) + ф\ B) ф\ A) - 2фа A) фь B)фа B) фь A). (IV.15)
Для симметричной волновой функции, когда электронные спины антипарал-
лельны, их волновые функции складываются. Поэтому симметричной функции
отвечает увеличение плотности электронного облака между ядрами (IV. 14). Тогда
говорят, что электронные облака перекрываются *. Это соответствует соединению
атомов друг с другом с образованием молекулы (рис. 31). Как это видно из
(IV. 14), при перекрывании электронных облаков электронная плотность между
атомами делается больше суммы плотностей электронных облаков изолированных
атомов**. Перекрывание электронных облаков нельзя рассматривать как простое
наложение друг на друга электронных облаков, существовавших до взаимодейст-
взаимодействия изолированных атомов.
Для антисимметричной волновой функции, характеризующейся парал-
параллельностью электронных спинов, наблюдается уменьшение электронной плотно-
плотности между атомами [см. (IV. 15)] и, следовательно, химическая связь не возникает,
т.е. соединение не образуется***. При этом плотность электронного облака между
ядрами падает до нуля и в результате электроны выталкиваются из этого прост-
пространства. Наоборот, при возникновении химической связи и образовании соедине-
соединения электронные облака стремятся вытянуться навстречу друг другу.
Вычисленная по методу Гейтлера — Лондона энергия ковалентной связи в
молекуле водорода была равна 414,0 кДж/моль при равновесной длине связи
0,086 нм. Опытные значения энергии и длины связи**** в Нг соответственно рав-
равны 457,67 кДж/моль и 0,074 нм. Расхождение между расчетными и эксперимен-
экспериментальными данными, равное 10%, можно считать небольшим, если принять во
внимание приближенный характер волновых функций (IV.7) и (IV.8), составлен-
составленных из неизменных волновых функций атомов.
В целом расчет молекулы водорода Гейтлером и Лондоном явился убедитель-
убедительным доказательством применимости квантовой механики для решения проблемы
химической связи и положил начало методу валентных связей. Согласно МВС,
приближенная волновая функция молекулы строится в виде линейной комбина-
комбинации выбранных исходных волновых функций фг:
N
ф = ?
г=1
(IV.16)
"Точнее, перекрываются волновые функции электронов.
* *Для тех же расстояний от ядра.
* * "Такое состояние системы называют репульсивным.
****Из анализа молекулярных спектров водорода.
7П
Результаты квантово-механического расчета молекулы водорода методом ва-
валентных связей с использованием различного числа волновых функций фг пока-
показывают, что точность повышается при увеличении числа членов в сумме (IV. 16).
Главные положения МВС можно сформулировать так: 1) ковалентную связь
образуют два электрона с антипараллельными спинами; 2) при образовании
ковалентной связи происходит перекрывание волновых функций электронов и
между взаимодействующими атомами увеличивается плотность электронного
облака A5—20%), что приводит к уменьшению энергии системы; 3) ковалентная
связь направлена в сторону максимального перекрытия электронных облаков
взаимодействующих атомов (критерий наибольшего перекрывания).
Укажем на две часто встречающиеся ошибки при популярном изложении
МВС. Во-первых, на основе того, что ковалентная связь образуется электронами
с антипараллельными спинами, часто неправильно считают, будто причиной
химической связи является взаимное притяжение противоположно направленных
магнитных моментов электронов. В действительности же магнитное взаимодейст-
взаимодействие крайне незначительно и не оно определяет химическую связь. Во-вторых,
воздавая должное роли обменных интегралов в (IV.10) и (IV.11), иногда говорят
об обменной природе сил химической связи. На самом же деле "обмен" характе-
характеризует не природу связи, а является лишь следствием метода расчета, называемо-
называемого МВС. Поскольку электроны неразличимы, нельзя говорить об их обмене мес-
местами как о физическом явлении.
9. Валентность и МВС. Согласно МВС, пребывание двух электронов с анти-
антипараллельными спинами в поле двух ядер энергетически более выгодно, чем
нахождение каждого электрона в поле своего ядра. Тогда объяснение валентно-
валентности состоит в том, что каждый атом для образования химической связи предо-
предоставляет один неспаренный электрон. Поскольку у атома водорода всего лишь
один электрон в нормальном и возбужденном состояниях, он функционирует как
одновалентный элемент.
У атома гелия в нормальном состоянии нет неспаренных электронов — оба его
электрона находятся на орбитали Is. Возбуждение же атома с переходом электро-
электронов из состояния с одним главным квантовым числом п на орбиталь с другим
главным квантовым числом, особенно для низких их значений (п — 1; 2), требует
большой затраты энергии. Для атома гелия энергия возбуждения электрона из
состояния Is на орбиталь 2s равна 1672 кДж/моль. Такие высокие энергии воз-
возбуждения в условиях обычных химических реакций не наблюдаются. Поэтому
гелий не образует валентно-химических соединений.
Атом лития в нормальном состоянии имеет один неспаренный электрон. Пере-
Переход Is -» '2р требует большой энергии. Поэтому литий одновалентен. Для берил-
бериллия перевод его в возбужденное состояние требует всего 259 кДж/моль:
[Be *]П^2рК
Таким образом, в возбужденном состоянии у атома бериллия два неспаренных
электрона, благодаря которым он проявляет валентность, равную двум. При этом
энергия возбуждения с избытком компенсируется энергией образования двух
химических связей. Переход электрона в процессе возбуждения атома на другие
орбитали с более высокой энергией называется промотированием.
Атом бора в нормальном состоянии (Is22s22pl) имеет один неспаренный элект-
электрон, а потому должен быть одновалентным. Однако для бора одновалентное
71
II. ii
состояние не характерно, так как происходит промотирование с переходом атома
в возбужденное состояние:
531 кДж
[В *}\s22s2p2.
Именно в возбужденном состоянии бор проявляет валентность, равную трем, из-
за наличия в атоме трех неспаренных электронов. Как и у других атомов, энер-
энергия возбуждения компенсируется возникновением большего числа химических
связей. У атома углерода имеется два одиночных электрона, но двухвалентное
состояние для него не характерно. Возбуждение атома углерода с возникновением
четырехвалентного состояния показано схемой
401 кДж
n = 2
n = 1
s
и
s
j
lit
1
}
II
Приведенных примеров достаточно для подтверждения так называемой спино-
спиновой теории валентности, согласно которой валентность элемента определяется
общим числом неспаренных электронов как в нормальном, так и в возбужденном
состоянии. Способ образования ковалентной связи, когда каждый из взаимодей-
взаимодействующих атомов отдает по одному электрону для образования общей электрон-
электронной пары, называется обменным. Но нередко валентность элемента превосходит
число неспаренных электронов в его атомах. Происходит это потому, что помимо
обменного механизма образования ковалентной связи существует и другой, зак-
заключающийся в том, что один атом отдает в общее пользование неподеленную
пару электронов, а партнер предоставляет свободную орбиталь. Первый называет-
называется донором, а второй — акцептором. Ковалентную связь, образующуюся за счет
неподеленной пары электронов донора и свободной орбитали акцептора, называ-
называют донорно-акцепторной связью. Схематически ее образование может быть пока-
показано так:
д: +
Донорно-акцепторный механизм возникновения ковалентной связи отличается
от обменного только происхождением общей электронной пары, ответственной за
химическую связь. При донорно-акцепторном механизме связь осуществляется за
счет неподеленной электронной пары одного из атомов, а при обменном механиз-
механизме — поделенной пары электронов. Во всем остальном оба вида ковалентной
связи тождественны: понижение общей энергии системы (см. рис. 30), антипарал-
антипараллельность спинов электронов, перекрытие электронных облаков (рис. 31).
Рассмотрим химическое строение молекулы монооксида углерода. У атомов
углерода и кислорода имеется по два неспаренных электрона. Поэтому можно
было предположить, что между этими атомами возникает двойная связь по об-
обменному механизму. Однако монооксид углерода представляет собой очень проч-
прочную молекулу с энергией связи, близкой к энергии связи для молекулы азота, у
которой тройная связь. Кроме того, молекулы монооксида углерода и азота изо-
электронны, т.е. имеют одинаковое число электронов, равное 14. Современная
72 t
трактовка химического строения молекулы СО, согласно МВС, базируется на
признании тройной связи между атомами углерода и кислорода, одна из которых
донорно-акцепторная:
Ifl
[ill
It
It
III 1
1
Таким образом, атом углерода функционирует как акцептор за счет одной ва-
вакантной 2т>-орбитали, а атом кислорода является донором двух спаренных 2р-
электронов. Для обозначения донорно-акцепторной связи * применяют стрелку (в
отличие от обычной валентной черточки) от донора к акцептору:
:С = О:
Монооксид углерода представляет собой пример химического соединения,
когда валентность элементов превышает число неспаренных электронов. Углерод
и кислород трехвалентны, хотя атомы этих элементов имеют по два неспаренных
электрона. Не следует думать, что монооксид углерода — исключение. Наоборот,
подавляющее большинство неорганических соединений образуется или на основе
донорно-акцепторного механизма ковалентной связи, или одновременно сочетает
в себе обменный и донорно-акцепторный механизмы. Рассмотрим сульфид цинка,
кристаллохимическое строение которого показано на рис. 4. Каждый атом цинка
связан с четырьмя атомами серы, и, наоборот, каждый атом серы — с четырьмя
атомами цинка. Поэтому атомы цинка и серы проявляют одинаковую валент-
валентность, равную четырем. Между тем атом цинка в нормальном состоянии не имеет
ни одного неспаренного электрона, а атом серы характеризуется двумя одиноч-
одиночными электронами. При возбуждении атома цинка происходит промотирование
4в-электрона на 4р-уровень и появляются два неспаренных электрона:
Zn n =41И1 I 1 I — МИ I I
В то же время у возбужденного атома цинка вакантны две 4р-орбитали, а атом
серы имеет две неподеленные пары электронов при главном квантовом числе
п = 3:
Р
Эту связь часто называют координационной или семиполярной, что означает полупо-
полуполярная. Последний термин несколько неудачен, так как никакой "полуполярности" не
существует. ' . /
Zn:
.. г
S j:
Znj:
S 1:
Zn :
S :
Zn:
S :
Zn :
• *
~S :
Zn
s
Zn
s
Zn
: s
: Zn
: s
: Zn
: s
: Zn
:j s
:jZn
:j s
: Zn
В результате четырехвалентность атомов цинка и
серы достигается тем, что две ковалентные связи
образуются по обменному механизму, а две другие —
по донорно-акцепторному. На рис. 32 показано воз-
возникновение химических связей в сульфиде цинка, где
более крупными точками обозначены неподеленные
пары электронов от атомов серы, осуществляющие
донорно-акцепторную связь. Следовательно, в суль-
Р и с. 32. Схема возникновения фиде цинка атомы цинка функционируют как акцеп-
химических связей в сульфиде торы, а атомы серы — как доноры.
цинка Таким образом, сульфид цинка — соединение, в
котором валентность элемента больше числа неспа-
ренных электронов не только в нормальном, но и в возбужденном состоянии
атома. Это свидетельствует о неточности и неправомерности спиновой теории
валентности. Объясняется это тем, что спиновая теория валентности, по существу,
является формалистической трактовкой метода валентных связей, в которой ос-
основной упор делается на внешний признак образования ковалентной связи. МВС
в действительности гораздо глубже и фундаментальнее, чем спиновая теория
валентности.
Обобщение огромного теоретического и экспериментального материала совре-
современной химии приводит к выводу, что валентность элемента (число ковалент-
ности) равна числу электронных орбиталей его атома, участвующих в образова-
образовании химических связей как по обменному, так и по донорно-акцепторному меха-
механизму.
10. Насыщаемость ковалентной связи. Среди многих свойств ковалентной свя-
связи наиболее важны насыщаемость, поляризация и направленность. Насыщаемость
химической связи — это то, что отличает ее от всех других видов взаимодействия
частиц. Собственно, вся изложенная квантово-химическая теория ковалентной
связи — основные положения МВС, обменный и донорно-акцепторный механизмы
ее образования — служат обоснованием насыщаемости химической связи.
Гейтлер и Лондон уже в своей первой работе рассмотрели возможности хими-
химического взаимодействия молекулы водорода с третьим атомом водорода.
Квантово-механический расчет энергии взаимодействия в системе Нг + Н
показал, что третий атом водорода не будет притягиваться, т.е. образование
молекулы Н3 энергетически невыгодно, а следовательно, невозможно. Это явилось
теоретическим обоснованием насыщаемости ковалентной связи. Формальная
интерпретация указанного расчета Гейтлера и Лондона и, следовательно,
насыщаемости ковалентной связи сводится к тому, что присоединение третьего
атома водорода к молекуле Н2 невозможно, так как спин его электрона будет
обязательно совпадать со спином одного из двух электронов молекулы. Поэтому
между третьим атомом водорода и Нг будут действовать силы отталкивания и в
результате никакого перекрывания электронных облаков не может быть.
Таким образом, именно насыщаемость ковалентной связи определяет стехио-
стехиометрию молекулярных химических соединений. В этом заключается колоссальная
роль насыщаемости ковалентной связи, так как от стехиометрии зависят фор-
формульный состав, массовые соотношения элементов, расчеты по формулам и урав-
уравнениям и т.п.
В то же время электростатическое взаимодействие разноименных зарядов не
знает насыщаемости. Если положительный заряд притягивается к одному отрица-
отрицательному, то это ему отнюдь не мешает одновременно притягиваться к другим
отрицательным зарядам.
11. Поляризация химической связи. Ковалентная связь гомеополярна только
для молекул и соединений, состоящих из одинаковых атомов*. А таких веществ
не может быть больше (с учетом аллотропии) числа элементов в Периодической
системе. В настоящее время металлов и металлидов (соединений с преимущест-
преимущественно металлической связью) насчитывается лишь свыше 10 000. Все остальные
химические соединения характеризуются полярной ковалентной связью. Это
происходит потому, что абсолютное большинство соединений образуется сочета-
сочетанием неодинаковых атомов. При этом происходит смещение связывающего элект-
электронного облака под влиянием одного из атомов — поляризация, результатом чего
является полярная связь. Смещение связующего электронного облака происходит
в сторону более электроотрицательного атома. И потенциал ионизации, и сродст-
сродство к электрону порознь не могут служить достаточной мерой электроотрицатель-
электроотрицательности элемента. Малликен предложил количественную меру электроотрицатель-
ности атома в виде полусуммы первого ионизационного потенциала и сродства к
электрону
ЭО = (/+ Е)/2, (IV.17)
где / — первый ионизационный потенциал; Е — сродство к электрону.
Однако в отличие от ионизационного потенциала, определяемого с большой
точностью для любого элемента по спектроскопическим данным, для электронно-
электронного сродства пока нет надежных методов количественного определения. Поэтому
Полинг пошел по другому пути и выразил значения электроотрицательностей
элементов в условных относительных единицах. По Полингу, энергия диссоциа-
диссоциации соединения АВ D(A~B) должна быть больше полусуммы энергий диссоциа-
диссоциации молекул А2 и В2 в отдельности и разность между D(A~~B) и полусуммой
обозначается через Д:
Д = ?>(А-В) - У2[?(А-А) + Я(В-В)],
(IV.18)
где D(A~A) и D(B~B) — соответствующие энергии диссоциации молекул Аг и В2.
Если атомы А и В химически реагируют между собой, Д не может быть отрица-
отрицательной величиной. Так, при взаимодействии Нг и Вгг с образованием НВг будет
ДН-Вг) = 364,9, ?>(Н-Н) = 432,2 и D(Br-Br) = 192,7 кДж/моль. Тогда
Д = 364,9 - 1/2D32,2 + 192,7) = 52,45 кДж.
Анализируя экспериментальные данные, Полинг пришел к выводу, что химиче-
химическим элементам можно приписать такие относительные значения электроотрица-
электроотрицательностей (ОЭО), разность которых для определенного соединения будет равна
0,102 |Д. Ниже приведены значения 0,102 [А для некоторых химических связей:
Согласно квантовой механике, в силу делокализации электронов и для этих веществ,
вероятно, существуют мгновенные диполи. Однако средняя статистическая картина такова,
что соединения, состоящие из одинаковых атомов, бездйпольны и гомеополярны.
Связь Si-H
Д 8,78
0,102 [А* 0,30
Разность ОЭО 0,3
Вг-Н
52,5
0,74
0,7
с-о
96,9
1,00
1,0
As-Cl
99,5
1,01
1,0
s-ci
22,1
0,58
0,5
F-C1
65,6
0,82
0,9
Вг-1
7,1
0,27
0,3
*0,102 [А = ,| Д/96,485 эВ.
В шкале электроотрицательностеи каждому химическому элементу приписыва-
приписывается вполне определенное значение ОЭО. В табл. 10 приведены данные по ОЭО.
На рис. 33 представлено изменение характера межатомной связи в химических
соединениях. Начало координат характеризует гомеополярную связь. В качестве
"идеально ионного" вещества возьмем фторид цезия — соединение атомов, наибо-
наиболее сильно различающихся по ОЭО. В этом соединении (разность ОЭО = 3,2)
степень ионности пусть равна единице. Начало координат соединим прямой с
фигуративной точкой для CsF. Такая аппроксимация не является грубой, по-
поскольку зависимость степени ионности связи от разности электроотрицательно-
электроотрицательностеи выражается кривой, мало отличающейся от прямой. Значения разности
ОЭО отдельных соединений наносятся на прямую. Тогда степень ионности, или
полярность, любого соединения на прямой определится по перпендикулярам к
оси ординат.
Таким образом, из рис. 33 видно, что чем больше разность ОЭО компонентов
соединения, тем более полярна ковалентная связь. Для галогенидов и оксидов
металлов межатомная связь наиболее полярна потому, что галогены и кислород
имеют высокие значения ОЭО. Однако, привлекая концепцию электроотрица-
электроотрицательности как условной величины, характеризующей относительную способность
атома в соединении притягивать к себе связующее электронное облако, необходи-
необходимо учитывать следующее:
1,0
0,5 1,0 1,5 1,0 7,5
Разность ОЭО
Рис. 33. Зависимость степени ионности от ОЭО
Таблица 10. Относительная электроотрицательность элементов (ОЭО)
Порядко-
Порядковый номер
элемента
1
3
4
5
6
7
8
9
11
12
13
14
15
16
17
19
20
21
22
B2)
23
B3)
B3)
24
B4)
B4)
25
B5)
B5)
26
B6)
27
28
29
B9)
30
31
32
33
Элемент
Н
Li
Be
В
С
N
0
F
Na
Mg
Al
Si
P
S
Cl
к
Ca
Sc
TiB) *
TiD)
VC)
VD)
VE)
CrB)
CrC)
CrF)
MnB)
MnD)
MnG)
FeB)
FeC) '
Co
Ni
Cu(l)
CuB)
Zn
Ga
Ge
As
ОЭО
2,1
0,95
1,5
2,0
2,6
3,0
3,5
3,9
0,9
1,2
1,5
1,9
2,1
2,6
3,1
0,8
1,0
1,3
1,1
1,6
1,4
1,7
1,9
1,4
1,6
2,4
1,4
2,1
2,5
1,7
1,8
1,7
1,8
1,8
2,0
1,5
1,6
2,0
2,0
1орядко-
вый номер
элемента
34
35
37
38
39
40
41
42
D2)
43
D3)
44
45
46
47
48
49
50
E0)
51
E1)
52
53
55
56
57
58
59
60
61
62
63
64
65
66
67
¦68
69
70
Элемент
Se
Вг
Rb
Sr
Y
Zr
Nb
MoD)
MoF)
TcE)
TcG)
Ru
Rh
Pd
Ag
Cd
In
SnB)
SnD)
SbC)
SbE)
Те
I
Cs
Ba
La
Ce
Pr
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tu
Yb
ОЭО
2,4
2,9
0,8
1,0
1,2
1,5
1,7
1,6
2,1
1,9
2,3
2,0
2,1
2,1
1,9
1,7
1,7
1,7
1,9
1,8
2,1
2,1
2,6
0,75
0,9
1,2
1,2
1,2
1,3
1,3
1,3
1,2
1,3
1,3
1,3
1,3
1,3
1,3
1,2
Порядко-
Порядковый номер
элемента
71
72
73
G3)
74
G4)
75
G5)
G5)
76
77
78
79
80
81
(81)
82
(82)
83
84
85
87
88
89
90
(90)
91
(91)
92
(92)
93
94
95
96
97
98
99
100
101
Элемент
Lu
Hf
TaC)
TaE)
WD)
WF)
ReE)
ReF)
ReG)
Os
Ir
Pt
Au(l)
Hg
Tl(l)
TlC)
PbB)
PbD)
Bi
Po
At
Fr
Ra
Ac
ThB)
ThD)
PaC)
PaE)
UD)
UF)
. NpD)
Pu
Am
Cm
Bk
a
En
Fm
Md
ОЭО
1,3
1,4
1,3
1,7
1,6
2,0
1,8
2Д
2,2
2,1
2,1
2,2
2,3
1,8
1,4
1,9
1,6
1,8
1,8
2,3
2,2
0,7
0,9
1Д
1,0
1,4
1,3
1,7
1,4
1,9
1,4
1,3
1,3
1,3
1,3
1,3
1,3
1,3
1,3
В скобках указана положительная степень окисления элемента.
77
1) электроотрицательность — не строгая физическая величина, которую можно
непосредственно определить;
2) величина электроотрицательности не постоянна и зависит от природы
другого атома, с которым химически связан данный атом;
3) один и тот же атом в данной химической связи иногда одновременно может
функционировать и как электроположительный (донор), и как электроотрица-
электроотрицательный (акцептор).
Обычно полярная ковалентная связь трактуется исходя из представлений об
электроотрицательности. Чем больше 0Э0 второго компонента, тем большей
поляризации подвергается ковалентная связь, а следовательно, растет доля ион-
ности связи. В то же время к проблеме степени ионности или полярности связи в
соединениях можно подойти с диаметрально противоположных позиций, а именно
с точки зрения поляризации ионов. Постулируется при этом, что молекулы сое-
соединения образуются путем полной передачи электронов и состоят из обособлен-
обособленных и самостоятапьных ионов. Затем происходит смещение электронов под дейст-
действием электрического поля, создаваемого ионами — поляризация ионов.
Поляризация — процесс двусторонний, в котором сочетается поляризующее
действие ионов с их поляризуемостью. Поляризуемость — способность электрон-
электронного облака частицы к деформации под действием электростатического поля дру-
другого иона, а напряженность этого поля опредачяет поляризующее действие иона.
Практически можно считаться только с поляризующим действием катионов и
поляризуемостью анионов. Поляризующее действие катиона в первую очередь
зависит от его электронной структуры, величины заряда (степени окисления) и
радиуса. Чем меньше радиус и главное квантовое число внешних электронных
орбиталей иона и больше его заряд, тем значительнее его поляризующее дейст-
действие. Отсюда сильным поляризующим действием обладают небольшие катионы
первых рядов Периодической системы, особенно при передвижении слева на-
направо*. Поляризуемость анионов зависит от тех же факторов, что и поляризу-
поляризующее действие катионов. Анионы с большим радиусом (размером) и зарядом
сильнее поляризуются. Чем больше главное квантовое число внешних электрон-
электронных орбиталей аниона, тем выше его поляризуемость. При одинаковом главном
квантовом числе р-электронные облака поляризуются в большей степени, чем *-
облако. Поляризующее действие катиона сводится к оттягиванию на себя элект-
электронного облака от аниона. В результате ионность химической связи уменьшается,
а степень ковалентности увеличивается, т.е. связь становится полярной ковалент-
ной. Таким образом, поляризация ионов уменьшает степень ионности химической
связи и по своему эффекту противоположна поляризации ковалентной связи.
На основании концепции поляризации ионов рассмотрим хлориды элементов
третьего периода Периодической системы:
NaCl
MgCl2
з
А1С13
4+
SiCL,
5 +
PCI5
Слева направо увеличивается заряд и уменьшается радиус поляризующего агента
— катиона — при одном и том же хлорид-анионе. В результате в этом же направ-
направлении увеличивается поляризующее действие катионов и в соответствии с этим
"Например, воображаемый катион Р5+.
**В действительности соединение SCle не существует.
7Я
закономерно уменьшается ионность межатомной связи. Если хлорид натрия пред-
представляет собой почти ионное соединение, тетрахлорид кремния характеризуется
сравнительно малой полярностью, а гипотетический хлорид SCle был бы почти
гомеополярным. К такому же выводу относительно характера межатомной связи
можно прийти на основе концепции электроотрицательности — поляризации
первоначально гомеополярной связи. Но гомеополярная связь в действительности
существует, а ионная связь является абстрактной и на самом деле ее нет. Поэтому
методологически более правильной и научно обоснованной следует считать поля-
поляризацию ковалентной связи, а не поляризацию ионов.
12. Направленность ковалентной связи. Направленность ковалентной связи
является тем главным свойством, от которого зависит структура молекул и немо-
немолекулярных химических соединений. Пространственная направленность ковалент-
ковалентной связи определяет химическое и кристаллохимическое строение вещества.
Поэтому нередко МВС называют методом направленной валентности.
Согласно МВС, ковалентная связь направлена в сторону максимального пере-
перекрывания электронных облаков взаимодействующих атомов. Поэтому атомы с ns-
валентными электронами способны образовывать одинаково прочные связи в
любом направлении и все направления в этом смысле равнозначны (рис. 34). Для
np-электронов максимальное перекрывание облаков связывающих электронов
происходит по направлению "восьмерок" (рис. 35). Поэтому угол между двумя
связями, образованными р-электронами одного атома с s- и р-электронами двух
других, теоретически должен быть равен 90°. Это происходит потому, что две р-
орбитали одного атома обязательно располагаются под прямым углом. Такое
расположение облаков наиболее выгодно энергетически, так как неспаренные
электроны отталкиваются друг от друга. Следовательно, причина направленности
химических связей кроется в зависимости атомных волновых функций от сфери-
сферических углов в и <р.
Нередко электроны, участвующие в образовании химической связи, находятся
в различных состояниях, например один валентный электрон на s-орбитали,
другой — в р-состоянии и т.д. Так, возбужденный атом бериллия имеет один
неспаренный электрон на 2в-орбитали, а другой — на 2р. Тогда в соединениях
бериллия с одновалентными элементами, например в ВеС12, две связи не должны
быть равноценными. Одна связь Be—C1 должна быть s—р-связью, а другая р—р-
связью. Последняя характеризуется большей прочностью, так как р-орбитали
более вытянуты от ядра по сравнению с s-орбиталями, а потому сильнее перекры-
перекрываются с орбиталями других атомов. В то же время совокупность всех свойств
молекулы ВеСЬ (пар хлорида бериллия) свидетельствует, что обе связи Be—C1
одинаково прочны и расположены под углом 180°, т.е. молекула ВеС^ линейна.
Рис. 34. Образование
химической связи s-электроиами
Рис. 35. Образование химических
¦ связей *• и р-электронами
В возбужденном атоме бора имеются один 2в-злектрон и два электрона на двух
2р-орбиталях. А возбужденный атом углерода характеризуется одним s-электро-
ном и тремя неспаренными р-электронами. Однако в соединениях бора и углеро-
углерода (например, ВС1з, СЩ, ССЦ и др.) соответственно все три или четыре связи
эквивалентны,
Для объяснения подобных фактов в рамках МВС Полинг ввел представление
о гибридизации электронных орбиталей. При этом Полинг исходил из того, что
значения энергии Е для s- и р-орбиталей либо совпадают между собой *, либо
различаются не намного. Тогда состояния валентных электронов описываются не
чистыми s-, р-, rf-функциями, а смешанными или гибридными волновыми
функциями, которые представляют собой линейную комбинацию собственных
функций, описывающих состояние исходных электронов. Например, для возбуж-
возбужденного атома углерода вместо одного 2s- и трех 2р-состояний в результате гиб-
гибридизации получаются четыре вырожденные гибридные орбитали, энергия кото-
которых промежуточна между энергиями 2s- и 2р-орбиталей. Такая гибридизация
называется тетраэдрической или sp^-гибридизацией.
Таким образом, число гибридных орбиталей всегда равно суммарному числу
исходных орбиталей. Кроме того, при возникновении гибридных орбиталей необ-
необходимо соблюдение следующих условий: 1) хорошее перекрывание гибридизу-
емых электронных орбиталей; 2) небольшая разница в энергиях атомных орбита-
лей, участвующих в гибридизации. Например, Ь-орбитали не могут гибридизо-
ваться с 2р-орбиталями, так как у них различные значения главного квантового
числа, а потому их энергии сильно различаются. Гибридизация всегда сопровож-
сопровождается изменением формы электронного облака. При этом гибридное электронное
облако асимметрично: имеет большую вытянутость по одну сторону от ядра, чем
по другую. Поэтому химические связи, образованные с участием гибридных
орбиталей, обладают большей прочностью, чем связи за счет чистых негибрид-
негибридных электронных облаков. Гибридизация одной s-орбитали и одной р-орбитали
приводит к возникновению двух гибридных облаков, расположенных под углом
180° (рис. 36). Это так называемая «^-гибридизация, в результате которой гиб-
гибридные облака располагаются по прямой. Отсюда легко объяснить прямолиней-
прямолинейность молекулы ВеС12: s- и р-орбитали атома бериллия подвергаются sp-гибриди-
зации и образуют две гибридные связи с двумя атомами хлора (рис. 37). У каж-
каждого атома хлора имеется по одному неспаренному р-электрону, которые и явля-
являются валентными.
sp- гибридизация
Рис. 36. Схема sp-гибридизации
Рис. 3?. Линейная молекула ВеС1г
Комбинация одной s- и двух р-орбиталей приводит к возникновению трех
асимметричных гибридных орбиталей, расположенных под углом 120°. Это sp2-
гибридизация. Например, возбужденный атом бора подвергается «р2-гибридиза-
ции, в результате ВС1з представляет равносторонний треугольник (рис. 38).
*Как это наблюдается в атоме водорода и водородоподобных ионах.
f
Рис. 38. Треугольная молекула
ВС13
н
Рис. 39. Тетраэдрическая
молекула СЩ
При 8р3-гибридизации четыре гибридных облака располагаются под тетраэд-
рическим углом 109°28'. Этот угол является оптимальным, обеспечивающим
максимальное взаимное удаление и минимальную энергию отталкивания асим-
асимметричных гибридных облаков своими утолщенными частями, что обеспечивает
минимум энергии системы. Поэтому молекула метана представляет собой пра-
правильный тетраэдр, в центре которого находится атом углерода с четырьмя тетра-
эдрически направленными гибридными облаками. Четыре атома водорода зани-
занимают вершины тетраэдра, химические связи направлены к вершинам тетраэдра
(рис. 39). Угол между связями равен точно тетраэдрическому.
В атомах с rf-электронными орбиталями гибридизация приводит к образова-
образованию более сложных конфигураций электронных облаков. Гибридизация с участи-
участием ^электронных состояний пока еще почти не разработана. В табл. 11 приведена
геометрия гибридных орбиталей в зависимости от типа гибридизации орбиталей
Таблица 11. Пространственное расположение для различных типов гибридизации
с участием s-, р-, d-орбиталей
Тип гибридизации орбиталей
центрального атома
sp или dp
sp2, dp2 или sd2
pd2
sp3 или sd3
dsp2
sp^d- или spd?
S^d?-y2 (Sp3<f)
sp9W*(sp3rf2)
sp*d & d (sp^d?)
spPd4
Геометрическая модель
гибридных орбиталей
Прямая линия
Плоский треугольник
Тригональная пирамида
Тетраэдр
Квадрат
Тригональная бипирамида
Квадратная пирамида
Октаэдр
Тригональная призма
Пентагональная бипирамида
Додекаэдр
«1
Рис. 40. Искаженный тетраэдр
молекулы
Рис. 41. Пространственная
ориентация трех р-орбитапей
центрального атома. Данные табл. 11 показывают, что геометрическая модель
соединения определяется состоянием d-орбиталей центрального атома, участвую-
участвующих в гибридизации. Так, при sp3(^-гибридизации с участием d 2 п- и d „-орби-
талей получим октаэдр, а с участием dxy- и «^^горбиталей — тригональную
призму.
Рассмотрим реальные примеры химического и кристаллохимического строения
типичных неорганических веществ на основе концепции гибридизации элект-
электронных орбиталей атомов.
В молекуле аммиака орбитали атома азота подвергаются $р3-тибридизации *.
При этом атомы водорода занимают три вершины тетраэдра и образуются три
s — «р3-гибридные связи. А четвертая вершина тетраэдра занята гибридным
электронным облаком, не участвующим в образовании химической связи**. Одна-
Однако разница в химическом строении метана и аммиака заключается и в том, что
угол между связями в аммиаке меньше тетраэдрического и равен 107° (рис. 40).
Таким образом, геометрическая модель молекулы аммиака представляет собой
чуть искаженный тетраэдр. Дело заключается в том, что увеличение "примеси"
s-состояния в гибридной орбитали приводит к увеличению угла между связя-
связями, тогда как возрастание доли /^-состояний уменьшает значение валентного
угла:
Гибридизация
Угол, град
sp
180
120
sp3 р3
109° 28' 90
Три чистые р-орбитали в пространстве будут ориентированы друг относитель-
относительно друга под углом 90°. При этом две "восьмерки" р-облаков расположатся под
прямым углом на плоскости, а третья /ьорбиталь — перпендикулярно этой плос-
плоскости. Именно такой пространственной ориентации р-облаков отвечает минимум
энергии системы (рис. 41). Значит, в молекуле аммиака степень ^-гибридизации
*Гибридизация — смешение орбиталей, а потому может происходить и в отсутствие
электронов на них и при наличии спаренных электронов на одной или нескольких ор-
биталях.
**Орбиталь, на которой находится неподеленная электронная пара, называется неэкви-
валентной. ,
не достигает 100%: примесь s-состояния несколько меньше, в результате чего угол
между связями меньше тетраэдрического.
Для гомологов аммиака — фосфина РН3, арсина AsH3 и стибина SbH3 — углы
между химическими связями почти не отличаются от прямого и соответственно
равны 93,5, 92 и 91°. У более тяжелых элементов сверху вниз по Периодической
системе орбитали имеют большие "размеры", более диффузны, а потому гибри-
гибридизация почти не происходит.
В структуре молекулы воды угол между связями О—Н равен 104,5°, т.е. бли-
близок к тетраэдрическому. Объясняется это тем, что атом кислорода также подвер-
подвергается неполной ятK-гибридизации. При этом примесь s-состояния еще меньше,
чем для азота в аммиаке. Отсюда геометрическая модель молекулы воды пред-
представляет также несколько искаженный тетраэдр, в котором две вершины заняты
двумя атомами водорода, а две другие — неэквивалентными электронными обла-
облаками, не участвующими в образовании химических связей (рис. 42).
В настоящее время принято считать, что в галогеноводородах орбитали атомов
галогенов подвержены я^-гибридизации. Тогда только одна вершина тетраэдра
занята атомом водорода, а три другие — совершенно одинаковыми гибридными
электронными облаками (рис. 43). На первый взгляд может показаться излишним
постулирование яр3-гибридизации для объяснения формы двухатомной (линей-
(линейной) молекулы НГ. Однако ковалентная связь, образованная в результате гибри-
гибридизации, обладает большей прочностью и, самое главное, неподеленные пары
электронов становятся совершенно идентичными. Если же допустить образование
НГ чистой /мэрбиталью (один неспаренный электрон) атома галогена, то остав-
оставшиеся неподеленные электронные пары будут разной симметрии: пара электро-
электронов s-состояния, а две пары — р-состояния. Между тем совокупность физических
и химических свойств галогеноводородов свидетельствует о тождественности всех
неподеленных электронных пар. Это же положение характерно и для воды, т.е.
оставшиеся неподеленными две пары электронов центрального атома обладают
одинаковой симметрией (гибридной).
Итак, материал по гибридизации электронных орбиталей атомов при образо-
образовании химических связей подтверждает исключительную плодотворность и важ-
важность самой идеи гибридизации в МВС. Прежде всего гибридизацией определя-
определяется химическое и кристаллохимическое строение веществ. А свойства веществ в
первую очередь зависят от их химического и кристаллохимического строения.
Кроме того, гибридизация делает тождественными неподеленные электронные
Н
Рис. 42. Искаженный тет-
тетраэдр молекулы воды
Рис. 43. Строение молекул
галогеноводородов
пары атомов. Наконец, гибридные связи обладают большей прочностью (энер-
(энергетически более выгодны) по сравнению со связями, образованными чистыми
электронными облаками. Относительная прочность гибридных связей (прочность
s-связей принята за единицу) приведена ниже:
Тип орбиталей, образу-
образующих связь s
Относительная проч-
прочность 1
р sp sp2 sp3 sjp-d s<P
1,73 1,93 1,99 2,00 2,69 2,95 2,93
13. Кратность химической связи. Кратные связи — ковалентные связи, осу-
осуществляемые более чем одной парой электронов. В молекуле этилена C2H<j каж-
каждый из возбужденных атомов углерода подвержен ^-гибридизации. Две гибрид-
гибридные орбитали используются на образование связи с двумя атомами водорода, а
третья гибридная орбиталь — для связи с другим атомом углерода. Таким обра-
образом, у каждого атома углерода в запасе остается еще по одному неспаренному 2р-
электрону. При «р2-гибридизации электронные облака располагаются в одной
плоскости под углами 120° друг относительно друга. Из экспериментальных
данных действительно следует, что молекула этилена имеет плоское строение
(рис. 44).
Химическая связь, для которой линия, соединяющая атомные ядра, являет-
является осью симметрии связывающею электронного облака, называется а-связъю.
(Т-Связь возникает при "лобовом" перекрывании атомных орбиталей. В молекуле
этилена каждый атом углерода образует по три <т-связи: одну — друг с другом, а
две другие — с двумя атомами водорода. Имеющиеся у атомов углерода негиб-
негибридные орбитали образуют одну так называемую тг-связь. Химическая связь, для
которой связывающее электронное облако имеет только плоскость симметрии,
проходящую через атомные ядра, называется ж-связью. На рис. 44 тг-связь пока-
показана пунктиром. В действительности при образовании тг-евязей происходит "бо-
"боковое" перекрывание атомных орбиталей (рис. 45).
Таким образом, две связи между атомами углерода в этилене неравноценны:
одна из них — it-связь, а другая тг-связь. Перекрывание электронных облаков при
образовании тг-связи меньше. Кроме того, области перекрывания лежат дальше от
ядер, чем при образовании <т-связи. Вследствие этих причин тг-связь обладает
меньшей прочностью по сравнению с сг-связью. Поэтому энергия двойной связи
меньше удвоенной энергии одинарной связи, которая всегда является сг-связью.
Кроме того, it-связь имеет осевую цилиндрическую симметрию и представляет
собой тело вращения вокруг линии, соединя-
соединяющей атомные ядра. тг-Связь, наоборот, не
обладает цилиндрической симметрией. Это
является главной причиной, почему невоз-
невозможно свободное вращение одной группы СЩ
относительно другой по линии связи между
углеродными атомами в молекуле этилена.
Таким образом, нельзя осуществить свободное
вращение фрагментов молекул вокруг двой-
двойной и тройной связи;
В молекуле этилена различаются два типа
G-связей. G-Связь между атомами углерода,
Н
Рис. 44. Строение молекулы этилена
Рис. 45. Различные типы тг-связей и (Т-связь
образованная перекрытием двух s/ьэлектронных облаков, обозначается а 2 ,.
В образовании ст-связей между углеродом и атомами водорода со стороны угле-
углеродных атомов участвуют в^2-орбитали, а водородных атомов — s-орбитали. Поэ-
Поэтому it-связи такого типа обозначаются а „. Существуют химические связи
s—spz
и т.п. На рис. 45 показаны типы тг-связей и так называемая й-связь. тг-Свя-
. 5-Связи образуются при перекрывании
s—d
зи подразделяют на it
, ж „ ж
р—р р—а а—а
rf-орбиталей всеми своими "восьмерками".
Рассмотрим молекулу азота, энергия диссоциации которой на атомы равна
945 кДж/моль. Раньше считали, что, поскольку в атоме азота уже в нормальном
состоянии имеется три неспаренных электрона, между атомами азота осуществля-
осуществляется тройная связь: одна а и две ж . По экспериментальным данным, энер-
р—р р—р
гия разрыва сг-связи довольно значительна и равна 543,4 кДж/моль. Простая
(негибридная) а -связь не может характеризоваться такой большой энергией
р—р
диссоциации. При образовании молекулы азота каждый атом подвергается sp-
гибридизации и два /ьэлектрона остаются негибридными. Разрыв гибридной
и -связи требует значительно большей энергии. Сказывается эффект наи-
наибольшего перекрывания электронного облака. Таким образом, в молекуле азота
осуществляются две связи л- за счет двух чистых негибридных орбиталей,
р—р
имеющихся у каждого атома, и одна гибридная и -связь, которая вносит
sp—sp
значительный вклад в энергетику молекулы. Естественно, две связи ж ориен-
Р—Р
тированы взаимно перпендикулярно. Кроме того, у атомов азота имеется по од-
одной неэквивалентной орбитали (рис. 46).
Приведенные примеры дают основание утверждать, что одинарная связь всег-
всегда является чистой или гибридной сг-связью. Двойная связь состоит из одной <г-
и одной it-связи. Наконец, тройная связь слагается из одной а- и двух тг-связей,
расположенных перпендикулярно друг другу.
В диоксиде углерода также наблюдается я/ьгибридизация центрального атома.
Что касается атомов кислорода, у них происходит в^-гибридизация, в результате
чего у каждого атома остается по одному чистому /ьэлектрону, которые и исполь-
используются для образования тг -связей. Таким образом, двойная связь между ато-
атомами углерода и кислорода опять же слагается из одной гибридной <т - и
sp^—sp
одной чистой тг -связей» Центральный атом в диоксиде углерода имеет четыре
электронных облака, два из которых гибридные. Два последних и чистое р-обла,-
85
Рис. 46. Строение молекулы азота
Р и с. 47. Строение молекулы диок-
диоксида углерода
ко одного атома кислорода расположены на одной плоскости (рис. 47), а негиб-
негибридная р-орбиталь второго атома кислорода ориентирована перпендикулярно
плоскости рисунка. Поэтому плоскости симметрии /мэблаков, являющихся состав-
составляющими двух разных кратных связей, ориентированы друг относительно друга
перпендикулярно. Наконец, каждый атом кислорода характеризуется двумя
неэквивалентными «т>2-орбиталями, расположенными под углом 120°.
14. О связях с избытком и дефицитом валентных электронов. По своей сути
МВС приложим к двухэлектронным и двухцентровым связям. В этом методе
рассматриваются химические связи, осуществляемые парой электронов (независи-
(независимо от их происхождения), обслуживающих два атомных центра. В то же время
встречаются соединения, в которых число электронов, приходящихся на одну
связь, больше или меньше двух. Рассмотрим связи с избытком электронов против
двухэлектронной связи. Раньше считали, что в молекуле кислорода осуществля-
осуществляется двойная связь между атомами: одна <тр-р и другая 1гр-р. Это было вполне
логично, так как каждый атом кислорода имеет два неспаренных электрона.
Однако кислород обладает парамагнетизмом, доказательством чему служит при-
притягивание жидкого и твердого кислорода к магниту *. А парамагнетизм вещества
обусловлен наличием в нем хотя бы одного неспаренного электрона. Но двойная
связь в молекуле кислорода исключает его парамагнитные свойства, так как в
структуре не оказывается ни одного неспаренного электрона.
Для обоснования парамагнетизма кислорода была предложена так называемая
трехэлектроппая связь: два неподеленных электрона от одного атома и один
неспаренный электрон от его партнера. Считается, что последний и ответствен за
парамагнетизм вещества. Ниже приводим строение молекулы кислорода с трех-
электронными связями, где для удобства крестиками обозначены электроны от
одного атома кислорода, а точками — от другого. Валентная черточка означает
простую (Тр-р-связь за счет одного из двух неспаренных электронов от каждого
атома кислорода. Другой неспаренный электрон, входящий в состав трехэлект-
ронной связи, обозначен стрелкой:
f
|)| ,i| *Парамагнитные вещества имеют положительную магнитную восприимчивость и втя-
втягиваются в магнитное поле. Диамагнитные характеризуются отрицательной магнитной
восприимчивостью и выталкиваются из магнитного поля.
86
Рис. 48. Строение молекулы диборана
в2н6
В
Рис. 49. Мостиковые орби-
тали в BoHg
Принимается, что прочность трехэлектронной связи небольшая: энергия ее мень-
меньше it-связи.
Существуют также соединения, в которых на каждую связь приходится мень-
меньше двух электронов. Для молекулярного иона водорода Н* энергия связи состав-
составляет 267 кДж/моль при длине ее 0,106 нм. Это стабильно существующее образо-
образование, связь между протонами в котором осуществляет единственный электрон.
Другим примером вещества с дефицитом валентных электронов может служить
молекула диборана (борэтан) E^Hg. В отличие от этана СгНб в молекуле диборана
всего 12 валентных электронов F от двух атомов бора и 6 от атомов водорода).
Изучение свойств диборана позволило установить строение его молекулы (рис.
48). Атомы водорода, через которые связываются два атома бора, называются
мостиковыми. На рис. 48 мостиковые атомы водорода связаны с бором пунк-
пунктирными линиями. Кроме того, мостиковые атомы водорода лежат на плоскости,
перпендикулярной плоскости расположения атомов бора и остальных четырех
атомов водорода. По своей геометрии диборан представляет собой два тетраэдра
с общим ребром из мостиковых атомов водорода. Каждый мостиковый атом во-
водорода образует две мостиковые связи*. Как видно из рис. 48, в молекуле ди-
диборана восемь межатомных связей, которые обслуживаются всего лишь 12 элект-
электронами (вместо 16). Это возможно потому, что каждый мостиковый атом водоро-
водорода может образовать с двумя атомами бора двухэлектронпую трехцеитровую
связь В—Н В. При образовании последней возможно перекрывание двух гиб-
гибридных зу3-орбиталей от двух атомов бора и s-орбитали атома водорода (рис. 49).
Ввиду изогнутости мостиковой связи В Н—В и ее иногда называют "бана-
Мостиковая связь — участие атомов в одновалентном состоянии одновременно в двух
связях.
87
I i
В ней два электрона (один — от водорода, а другой — от одного атома бора)
обслуживают три центра: два атома бора и один атом водорода. Именно в банано-
банановых связях наблюдается дефицит электронов: на четыре связи приходится столь-
столько же электронов.
Наиболее известными и распространенными электронодефицитными вещества-
веществами являются металлы и металлические соединения — металлиды. Речь идет о
металлах в конденсированном состоянии. В газообразном состоянии металличе-
металлические молекулы ничем не отличаются от других типичных молекул по природе
химической связи. Например, молекулы щелочных металлов Ы2, Na2, K2, CS2, как
и молекула водорода Нг, характеризуются парно-электронной <т5-5-связью. Одна-
Однако металлы и металлиды в их обычном твердом состоянии коренным образом
отличаются от их пара. Возьмем, к примеру, кристаллический литий, объемно
центрированная решетка которого показана на рис. 50. Каждый атом лития окру-
окружен восемью другими, и один 2«-электрон атома лития должен обеспечивать его
связи с 8 ближайшими соседями. Следовательно, в металлическом литии сущест-
существует большой дефицит валентных электронов против парно-электронной двух-
центровой ковалентной связи. Это означает, что металлы и металлиды нельзя
интерпретировать, оставаясь в рамках МВС. Кроме того, метод локализованных
электронных пар не может объяснить такое ярко выраженное свойство металлов и
металлидов, как их электрическая проводимость.
Таким образом, и вещества с дефицитом валентных электронов, по существу,
выходят за границы применимости МВС. "Факты, не объяснимые существующи-
существующими теориями, — писал А.М.Бутлеров, — наиболее дороги для науки, от их разра-
разработки следует по преимуществу ожидать ее развития в ближайшем будущем".
Метод молекулярных орбиталей (ММО) объясняет химическую связь в ковалент-
ных веществах, а также в соединениях с избытком и с дефицитом валентных
электронов.
15. Понятие о методе молекулярных орбиталей. Более универсальным кванто-
во-химическим методом описания химической связи является метод молекуляр-
молекулярных орбиталей (ММО), развитый в трудах Леннарда—Джонса, Гунда и особенно
Малликена. В этом методе состояние электронов в многоатомной системе описы-
описывается молекулярными орбиталями (МО), подобно тому, как электроны в атомах
характеризуются атомными орбиталями (АО). При этом и АО и МО представля-
представляют собой одноэлектронные волновые функции атома или молекулы соответствен-
соответственно. Разница заключается в том, что АО — одноцентровые, а МО — многоцентро-
многоцентровые орбитали. Итак, ММО — квантово-химический метод описания химической
связи, рассматривающий молекулу и любую многоатомную
систему как "лногоядерный атом", в котором электроны
заселяются по молекулярным орбиталям.
Одноэлектронное приближение в ММО обычно принимает
форму МО ЛКАО, что означает "молекулярная орбиталь
как линейная комбинация атомных орбиталей". Рассмотрим
^^0 \ I J^b принципы метода МО ЛКАО на примере молекулы водоро-
водороду Сг да. Как и прежде, фа и фь — волновые функции электронов
атомов водорода На и Щ. Согласно основной идее метода
МО ЛКАО, волновые функции электрона в молекуле Н2
записываются так:
t \2Фъ- (IV.19)
Рис. 50. Объемно це-
центрированная решетка,
металлического лития
МО
88
Коэффициенты Ai и А2 показывают долю участия соответствующей АО при
конструировании МО. Если А] > А2, то АО фа вносит больший вклад в образова-
образование МО по сравнению с КОфь. При взаимодействии разных атомов \\ и А2 служат
мерой полярности связи. Действительно, при \\ > А2 вероятность пребывания
электрона у атома с индексом 1 больше, чем у атома с индексом 2. Это свидетель-
свидетельствует о большей электроотрицательности первого атома по сравнению со вторым.
Таким путем метод МО ЛКАО автоматически учитывает поляризацию химичес-
химической связи. Энергию образования МО рассчитывают по формуле (IV.6). Выраже-
Выражение для потенциальной энергии системы аналогично выражению для нее в МВС.
Подставляя (IV.19) в (IV.6), вычисляют энергию системы Е, которая может быть
представлена как алгебраическая сумма
Е= Q±
(IV.20)
где Q — кулоновский интеграл; /? — обменный интеграл.
Как и в МВС, обменный интеграл имеет отрицательный знак и Q € |/?|. Если
Е = Q + /?, то возникает связывающая молекулярная орбиталь (СМО), т.е. энер-
энергия системы уменьшается (рис. 51). СМО отвечает симметричная волновая функ-
функция
Х\фа
(IV.21)
для которой характерны перекрытие электронных облаков и, следовательно,
повышенное значение плотности электронного облака (рис. 52).
Антисимметричной волновой функции
Ф- =
(IV.22)
отвечает Е — Q — /?, что соответствует разрыхляющей молекулярной орбитали
(РМО). Для последней энергия выше исходных АО (рис. 51) и происходит
уменьшение электронной плотности в пространстве между ядрами (рис. 52), чем и
объясняется отталкивание атомов. Поэтому в молекуле водорода оба электрона
находятся на СМО, энергетический уровень которой лежит ниже уровня АО. По
аналогии с атомными орбиталями (s, p, d, f) молекулярные орбитали также обоз-
Р и с. 51. Энергетическая схема
АО и МО в системе из двух ато-
атомов водорода
¦СМО
РМО
Рис. 52. Распределение электронной плотности в системе из
двух атомов водорода
89
начаются соответствующими буквами греческого алфавита а, тг, 8 и <р. Разрыхля-
Разрыхляющие молекулярные орбитали отмечают звездочками. Заселение их электронами
уменьшает энергию и прочность химической связи. Сами электроны на РМО
называются разрыхляющими. Для СМО добавление электронов увеличивает
энергию связи. Электроны на СМО также называются связывающими. Число МО
равно суммарному числу исходных АО. При образовании молекулы водорода из
двух АО получили две МО, одна из которых СМО, а другая — РМО (рис. 51).
При расчетах МО многоатомных систем (начиная с трехатомных молекул) могут
появиться энергетические уровни, лежащие посередине между СМО и РМО.
Такие МО называются несвязывающими молекулярнылш орбиталями (НМО). В
какой-то мере НМО характеризуют энергию электрона на АО. В целом имеется
четыре типа молекулярных орбиталей: СМО, РМО, НМО внутренних электронов
и НМО вачентных электронов *. Возникновение МО в методе МО ЛКАО требует
определенных условий: 1) энергии АО не должны сильно отличаться друг от
друга — они соизмеримы по величине; 2) максимальное перекрытие электронных
облаков взаимодействующих АО; 3) АО должны обладать одинаковыми свойства-
свойствами симметрии относительно оси молекулы (например, могут взаимодействовать
АО рх с рх, но не могут комбинировать рх и ру или рх и pz). Кроме того, заполне-
заполнение электронами МО происходит в соответствии: а) с принципом наименьшей
энергии системы; б) с принципом Паули: в) с правилом Гунда.
Метод МО не нуждается в понятии валентности, но зато вводит понятие "по-
"порядок гвяги". Порядок связи равен частному от деления разности числа электро-
электронов на СМО и РМО на число взаимодействующих атомов. Порядок связи может
принимать и целочисленные, и дробные значения, но только положительные.
Если порядок связи равен нулю, система неустойчива и связь не возникает. Это
значит, что для возникновения молекулы или любой многоатомной системы
необходимо, чтобы заселенность электронами СМО была всегда больше, чем
заселенность РМО. Тогда образующаяся система энергетически выгодна и ста-
стабильно существует. Порядок связи в ММО есть, по существу, превращенная
форма понятия валентности классической химии, кратности связи в МВС. Неце-
Нецелочисленные же значения порядка связи — прямое следствие многоцентровости и
делокализации связей в методе МО.
Рассмотрим некоторые химические соединения с точки зрения ММО. Прежде
всего молекулярный ион водорода Щ, который в принципе не интерпретируется
в рамках МВС, является самым простым и понятным в методе МО. Отличие
между молекулой водорода и ионом В* заключается в заселенности СМО c\s\ в
молекуле на этой орбитали находятся два электрона (рис. 51), в ионе — один.
Порядок связи в молекуле водорода равен 1, а в молекулярном ионе — 0,5.
Поскольку удаление электрона из СМО уменьшает энергию связи, ион.Н^ менее
прочен, чем молекула Нг- Известен также молекулярный ион Н~. Третий (по
сравнению с Н2) электрон может быть помещен только на разрыхляющей мо-
молекулярной орбитали <r*s (рис. 51). Порядок связи в отрицательном молеку-
молекулярном ионе водорода также будет равен 0,5. Но прочность связи в Н~ очень
*Понятия СМО и РМО относятся только к валентным электронам.
90
С МО
Рис. 53. Схема образования из
АО o-j-CMO и <т*51-РМО
GMO
Р и с. 54. Схема образования из
АО ар -СМО и <Т *р -РМО
мала A6,7 кДж/моль), так как заселение электронами РМО уменьшает анергию
связи.
Очевидно, молекула гелия Не2 возникнуть не может, так как в этом гипотети-
гипотетическом образовании два электрона должны находиться на СМО и два других —
на РМО. Таким образом, порядок связи равен нулю. В то же время известен
молекулярный ион Не*, энергия диссоциации которого близка к энергии диссо-
диссоциации Н^. В молекулярном ионе гелия на СМО находятся два электрона, а на
РМО — один. Это и определяет его стабильное существование. Подобно Не2, не
могут образоваться молекулы состава Ве2 Са2, Ва2, Mg2 и др.
Наряду с энергетической диаграммой образования МО (рис. 51) можно пока-
показать вид молекулярных электронных облаков, полученных из исходных атомных
орбиталей путем перекрытия их (СМО) или, наоборот, отталкивания (РМО). На
рис. 53—55 приведены схемы образования СМО и .РМО различной симметрии*.
На рис. 53 представлено взаимодействие электронных облаков атомов водорода с
образованием связывающих и разрыхляющих электронных облаков молекулы
водорода. На рис. 54 видно, что при взаимодействии р^-облаков атомоь возника-
возникают и -МО, а не ж -МО. Образующаяся СМО обладает осевой цилиндрической
Рх Рх
симметрией, а потому ее нельзя обозначать тг -МО. В этом факте, в частности,
Рх
сказывается разница между АО и МО. Только при взаимодействии ру- и р^-орби-
талей атомов образуются соответственно ж - и ж -МО. Последние две МО энер-
Ру Vz
гетически вырождены.
Для сравнения ММО и МВС рассмотрим молекулу азота, обсужденную ранее
с позиций МВС. На рис. 56 показаны энергетическая схема и заселенность орби-
орбиталей в молекуле азота. Заполненные полностью ls-орбитали атомов азота прак-
практически не участвуют в образовании связей. Поэтому на два атома азота прихо-
приходится всего 10 валентных электронов при п = 2 Bs22p3). 2я-Электроны полностью
заселяют <г25- и <г*^-МО. Они не вносят вклада в образование связи, так как
число электронов на СМО точно равно числу электронов на РМО. Шесть р-
электронов размещаются на МО так: два электрона — на <т2р-МО, а четыре — на
двух вырожденных тг2р~МО. Следовательно, в молекуле азота имеется восемь
связывающих на два разрыхляющих электрона, т.е. порядок связи равен трем. В
действительности собственно валентных электронов у атома азота всего три Bр3),
МО обозначают указащ1ем в виде индексов исходных атомных орбиталей.
ll! i'l
PMO
С MO
Рис. 55. Схема образования из
АО жр -СМО и 1Г *р -РМО
АО
МО
Рис. 56. Энергетическая диаграмма и
заселенность орбиталей в молекуле азота
так как 2,9-электроны, по существу, не участвуют в образовании химических
связей. Поэтому можно считать, что в молекуле азота на СМО находится шесть
электронов, а на РМО нет ни одного. Этим и объясняются большая энергия
диссоциации N2 на атомы и диамагнетизм молекулы азота.
Из гетероатомных молекул на молекулу азота похожа по химическому строе-
строению и свойствам молекула монооксида углерода. Заселенность молекулярных
орбиталей в СО точно такая же, как и в молекуле азота (рис. 56). Разница за-
заключается только в структуре АО: со стороны углерода участвуют в образовании
МО четыре электрона, а со стороны кислорода — шесть. Порядок связи равен
трем, т.е. соответствует кратности связи в СО согласно МВС.
По сравнению с мрлекулой азота в молекуле кислорода имеется на два элект-
электрона больше. Энергетическая диаграмма и заселенность МО молекулы кислорода
показаны на рис. 57. Во-первых, порядок связи в О2 равен двум. Во-вторых, на
двух вырожденных ж* -РМО находится по одному неспаренному электрону (сог-
(согласно правилу Гунда). Они-^го и являются "виновниками" парамагнетизма моле-
молекулы кислорода. Таким образом, в ММО парамагнетизм О2 обоснован строго
научно и нет необходимости в постулировании трехэлектронной связи.
Молекулярный ион кислорода О* (диоксигенил-катион) по сравнению с моле-
молекулой кислорода имеет на один электрон меньше. Это значит, что на ж* -РМО
будет находиться всего один электрон (рис. 57). Порядок связи в О* равен 2,5. И
большая величина порядка связи, и меньшее число электронов на РМО у О*
приводят к тому, что он прочнее молекулы кислорода. Энергия диссоциации
молекулы кислорода на атомы равна 497 кДж/моль, а для О^ она достигает 643
кДж/моль. Диоксигенил-катиону изоэлектронна гетероатомная молекула оксида
азота (+2) с энергией диссоциации на атомы 627 кДж/моль. Следовательно,
высокую прочность молекулы N0 и ее парамагнетизм можно предсказать на
основе ММО (рис. 57). Это еще одно убедительное доказательство того, что такие
молекулы, как Ог, N0 и др., в принципе нельзя и, самое главное, не нужно пы-
пытаться интерпретировать с точки зрения МВС. Тогда отпадает необходимость
АО
МО
Рис. 57. Энергетическая диаграмма и заселенность орбиталей электронами в молекуле
кислорода (а) и оксида азота B+) (б)
прибегать к формальным представлениям о трехэлектронной связи. Таким обра-
образом, в ММО отчетливо проявляется принцип изоэлектронности, согласно которо-
которому молекулы с одинаковым числом электронов обладают аналогичной картиной
заселенности МО и, следовательно, имеют сходную электронную структуру и
близкие физико-химические характеристики.
16. Сравнение МВС и ММО. МВС и ММО — это квантово-химические мето-
методы. Для них характерны следующие общие черты в подходе к описанию химиче-
химической связи: 1) оба метода являются приближенными; 2) для данной молекулы
они приводят к сходному результирующему распределению электронов *; 3) в
обоих методах наиболее существенны обобществление электронной плотности
связывающих электронов между ядрами и концентрирование плотности электрон-
электронного облака между ними; 4) а- и тг-связи отличаются друг от друга как по харак-
характеру перекрывания электронных облаков, так и по свойствам симметрии.
Эта общность методов наблюдается лишь для двухцентровых связей, так как
МВС в отличие от ММО не может в принципе рассматривать многоцентровые
связи. Каждый из обсуждаемых методов обладает своими преимуществами и
недостатками. Метод МО — более общий и универсальный. Его представления о
полностью делокализованных молекулярных орбиталях, охватывающих все ядра
системы, прогрессивны и физически адекватны.
Однако эти представления, а также основная идея ММО о делокализованных
орбиталях непривычны обычному химическому мышлению о локализации хими-
химической связи, т.е. об одной, двух или трех парах электронов, связывающих толь-
только два атома и не участвующих в связывании других атомов той же молекулы.
Кроме того, ММО трактует молекулу в целом, тогда как для химии более важны
характеристики ее отдельных атомных сочетаний — валентных связей и фрагмен-
фрагментов молекулы. В то же4 время ММО автоматически учитывает возбужденные
состояния молекул, чего нельзя сказать относительно МВС.
Обсуждаемые методы (МВС и ММО) не исключают один другого, а взаимно,
дополняют. Основные идеи ММО были высказаны почти одновременно с концеп-
Для тех соединений, которые описываются обоими методами.
циями МВС в 20-х годах нашего века. Однако долгое время ММО находился в
забвении только потому, что он далек от языка химических структурных формул.
В целом и МВС и ММО — квантово-химическое обоснование и дальнейшее раз-
развитие теории химического строения А.М.Бутлерова.
17. Металлическая связь. Металлы отличаются от других веществ высокими
значениями электро- и теплопроводности, а их структуры характеризуются высо-
высокими координационными числами. О существовании межатомной связи в метал-
металлах свидетельствуют энергии атомизации металлов, которые меняются в широких
пределах (табл. 12). Для сравнения здесь помещены данные по энергии атомиза-
атомизации таких типичных ковалентных кристаллов, как кремний и германий.
Т а б л и ц а 12. Энергии атомизации металлов нри 298 К
Металл
Li
К
Na ¦
Rb
Cs
Си
Ag
Аи
Энергия
атомизации,
кДж./моль
263,8
225,7
181
128,3
125
539
458
383,5
Металл
AJ
Ga
In
Tl
Si
Ge
Sn
Pb
Энергия
атомизации,
кДж/моль
514
418
332
239
763
643,3
493,7
291,8
Металл
V
Nb
Та
Cr
Mo
W
Ru .
Os
Энергия
атомизации,
кДж/моль
510,8
719
781,7
395
654
855
654
788,3
Сравнительно небольшими энергиями атомизации характеризуются щелочные
и простые sp-металлы. Для этих металлов сверху вниз по Периодической системе
происходит закономерное уменьшение значений энергий атомизации. Для пере-
переходных металлов наблюдаются большие величины энергий атомизации, соизме-
соизмеримые с энергией ковалентных связей.
Между ковалентной и металлической связями имеется большое сходство — оба
типа химической связи основаны на обобществлении валентных электронов.
Только в металлах обобществленные электроны обслуживают весь кристалл, т.е.
они полностью делокализованы. Этим объясняются отсутствие пространственной
направленности металлической связи и высокие координационные числа метал-
металлических структур. Это означает, что металлическая связь не проявляет свойств
насыщаемости, столь характерных для ковалентной связи. Делокализация же
валентных электронов в металлах является следствием многоцентрового характе-
характера металлической связи. Многоцентровость металлической связи обеспечивает
высокую электрическую проводимость и теплопроводность металлов.
f Металлическая связь — многоцентровая химическая связь с дъфицитом элект-
•, ронов в твердом или жидком веществе, основанная на обобществлении внешних
электронов атомов. Таким образом, металлическая связь характерна только для
конденсированного состояния вещества. В паро- и газообразном состоянии атомы
всех веществ, в том числе и металлов, связаны между собой только ковалентной
связью или не связаны вообще.
Аналогия между ковалентной и металлической связями подтверждается рас-
94
пределением электронной плотности между атомными остовами. Для обоих типов
связей в пространстве между атомными остовами сохраняется значительная
электронная плотность, образуя электронные мостики между взаимодействующи-
взаимодействующими атомами (рис. 58). Разница заключается в том. что в ковалентной связи элект-
электронные мостики имеют строго определенные пространственные направления, а у
металлической связи электронная плотность равномерно распределена по всем
направлениям.
Метод ВС не может интерпретировать металлическую связь. В металлах с их
высокими координационными числами наблюдается сильный недостаток валент-
валентных электронов по сравнению с двухэлектронной и двухцентровой ковалентной
связью. С точки зрения ММО металлическая связь характеризуется дефицитом
электронов против нормальной ковалентной связи. Поэтому порядок связи в
металлах и истинных металлидах может быть любым дробным числом. Отсюда
металлиды, как правило, не подчиняются правилам классической валентности,
т.е. ковалентности. Из-за этого для истинных металлидов невозможно предска-
предсказать их формульный состав на основе классических представлений о валентности,
а потому здесь нужны другие концепции.
Металлическая связь не исключает некоторой доли ковалентности. Металли-
Металлическая связь в чистом виде характерна только для щелочных и щелочно-земать-
ных металлов. Ряд физических свойств других металлов, особенно переходных
(температуры плавления и кипения, энергия атомизации, твердость, межатомные
расстояния), свидетельствуют о несводимости химической связи в них только к
металлической. Современными физическими методами исследования установлено,
что в переходных металлах лишь небольшая часть валентных электронов нахо-
находится в состоянии обобществления. Число электронов, принадлежащих всему
кристаллу, невелико — - 1 электрон/атом. Например, такой типичный переход-
переходный металл, как ниобий, имеет концетрацию обобществленных электронов всего
лишь 1,2 на один атом Nb. Остальные же электроны осуществляют направлен-
t,r
6000
5000
то
зооо
2000
woo
too
BOO
ш
CS
W
Рис. 58. Распределение элек-
электронной плотности при обра-
образовании ковалентной (а) и ме-
металлической (б) связей
Рис. 59. Температуры плавления
(/), кипения C) и энергии атомиза-
атомизации B) для металлов 6-го периода
Периодической системы
95
ные ковалентные связи между соседними атомами. Перекрывание направленных
электронных облаков наблюдается уже в простых sp-металлах, но особенного
развития достигает в переходных .sJ-металлах. Это объясняется большими
размерами и сложной геометрией (/-электронных облаков.
В переходных металлах, характеризующихся высокими температурами плавле-
плавления и кипения, а также высокими значениями энергии атомизации, основной
вклад в энергетику связи вносит перекрывание rf-орбиталей с образованием кова-
лентных связей. Это особенно заметно для элементов середины вставных декад,
атомы которых характеризуются максимальным числом неспаренных электронов
(рис. 59).
В настоящее время принято считать, что все основные свойства металлов
определяются природой металлической связи. Но наиболее специфическим свой-
свойством металлов, качественно отличающим их от других веществ в конденсирован-
конденсированном состоянии, является отрицательный температурный коэффициент электриче-
электрической проводимости. Это означает, что металлы с ростом температуры уменьшают
электрическую проводимость. А носителями электрического тока в металлах как
раз выступают обобществленные электроны.
В начале XX в. Друде и Лоренц применили к электронам проводимости
металлов кинетическую теорию газов и ввели представления об электронном газе.
Эта теория свободных электронов хорошо объяснила закон Ома и связь
электрической проводимости с теплопроводностью (закон Видемана—Франца), но
не объяснила главного отличия металлов от других твердых тел, а именно
температурную зависимость электрической проводимости. Действительно, в
теории свободных электронов Друде и Лоренца кинетическая энергия электронов
равна
кТ,
(IV.23)
где к — постоянная Больцмана. Из (IV.23) следует, что с повышением температу-
температуры скорость движения электронов v должна расти, а вместе с ней и проводи-
проводимость. Однако с ростом температуры электрическая проводимость падает и в этом
заключается характерная особенность металлов.
Поведение электронов проводимости правильно описывается квантовой теори-
теорией металлов, которая представляет собой приложение квантовой статистики к
металлам. Ее исходные представления: 1) электроны системы неразличимы; 2)
обязательное выполнение принципа Паули, т.е. в любой системе в данном состоя-
состоянии не может находиться более одного электрона с данной ориентацией спина; 3)
изменение состояния электронов определяется изменением хотя бы одного из
четырех квантовых чисел. Расчеты, проведенные с учетом основных положений
квантовой статистики применительно к металлам, позволяют вывести уравнение
Ео = 36,1 (п0/
(IV.24)
где Eq — энергия Ферми *; »о — число электронов проводимости на один атом;
Vq — молярный объем металла.
*Энергия Ферми представляет собой максимальную энергию, которую имеют электроны
в металлах при Т = О К.
Na
3,16
К
2,06
Rb
1,76
Cs
1,53
Cu
7,10
Ag
5,52
Au
5,56
Ниже приведены значения энергии Ферми для металлов I группы Периодиче-
Периодической системы (при % = 1):
Металл Li
Ео, эВ 4,74
Согласно квантовой теории, уже при Т = 0 К энергия электронов в металлах
измеряется несколькими электрон-вольтами. По классической теории электронно-
электронного газа, средняя энергия электронов при комнатной температуре B93,2 К) долж-
должна быть равна ~ 0,03 эВ. Таким образом, в действительности (квантовая теория)
электроны в металлах уже при 0 К имеют энергию на два порядка выше энергии,
рассчитанной по кинетической теории газов при комнатной температуре. Отсюда
повышение температуры практически не влияет на скорость электронов. Такое
состояние системы называется вырожденным. Следовательно, в металлическом
состоянии вещества электроны проводимости энергетически вырождены.
Вырождение электронов служит главной причиной, в результате которой
металлы с повышением температуры уменьшают свою проводимость. С ростом
температуры увеличивается амплитуда колебаний атомов в узлах кристалличес-
кристаллической решетки, что ведет к более интенсивному рассеянию электронов. Из-за этого
длина свободного пробега электронов падает, что уменьшает их подвижность.'
Колебания атомных остовов решетки в современной физике уподобляются стоя-
стоячим звуковым волнам. Кванты звуковых волн называют фононами. С повышением
температуры энергия фононов растет и вместе с ней увеличивается рассеяние
электронов на фононах. Таким образом, падение электрической проводимости с
ростом температуры (металлический ход проводимости) обусловлено уменьшени-
уменьшением подвижности при практически неизменной концентрации электронов проводи-
проводимости.
18. Химическая связь в твердых неорганических веществах. В металлах и
металлидах доминирует металлическая связь, хотя и в них немаловажную роль
играет ковалентная составляющая связи. В твердых неорганических веществах,
состоящих из одинаковых неметаллических атомов, господствующей межатомной
связью является ковалентная. При взаимодействии различных атомов с образова-
образованием твердого вещества природа межатомной связи имеет более сложный харак-
характер. Именно физико-химическая природа связи между неодинаковыми атомами
представляет наибольший интерес, так как подавляющее большинство неоргани-
неорганических соединений образовано сочетанием разнородных атомов.
Исследования химической связи в твердых телах современными физическими
и физико-химическими методами приводят к выводу о том, что межатомная связь
в твердых неорганических веществах неоднозначна. Как и для молекул, межатом-
межатомная связь в координационных кристаллах, за исключением металлов и металли-
дов, имеет ковалентный характер. Однако вследствие различных значений ОЭО
партнеров ковалентная связь подвергается поляризации, т.е. электронное облако
смещается в сторону более электроотрицательного атома. В результате на кова-
лентность накладывается определенная доля ионности. Поляризация приводит к
полярной ковалентной связи. Кроме того, уже при температуре, немного отлич-
отличной от абсолютного нуля, существует вероятность распада электронной пары,
ответственной за ковалентную связь. Эта вероятность растет пропорционально
температуре. А распад электронной Пары означает начало металлизации связи,
4 Я. А. Упй 1 ' , ¦ . 97
П-t
так как эти электроны принадлежат- теперь всему кристаллу. Таким образом,
металлизация связи — второй этап обобществления электронов, если считать за
первый этап образование ковалентной связи.
В целом химическая связь между разнородными атомами в твердых неоргани-
неорганических веществах имеет ковалентно-ионно-металлическиЛ или полярно-металли-
полярно-металлический характер. Тогда состояние электронов, участвующих в межатомной связи,
может быть описано функцией,
Ф = С1^ков + C2iW + CgV-мет, (IV.25)
где коэффициенты с\, c<i, C3 определяют долю ковалентной, ионной и металличе-
металлической составляющих химической связи и в сумме равны единице.
В зависимости от того, каким из трех коэффициентов в реально существую-
существующем соединении можно пренебречь, говорят о двойственном характере химиче-
химической связи. Например, в сульфиде цинка коэффициент с3 пренебрежимо мал
поэтому межатомная связь в нем является ковалентно-ионной. В антимониде
(стибиде) индия InSb, наоборот, практически отсутствует ионная доля связи
(С2 ¦* 0), поэтому в этом веществе химическая связь преимущественно ковалентно-
металлическая. В соединении NaSb химическая связь имеет ионно-металлический
характер, т.е. С\ -» 0.
Г Л А В А V. МЕЖМОЛЕКУЛЯРНОЕ ВЗАИМОДЕЙСТВИЕ
И КОМПЛЕКСООБРАЗОВАНИЕ
Электрически нейтральные атомы и молекулы, валентно-насыщенные в обыч-
обычном понимании, способны к дополнительному взаимодействию друг с другом.
Степень проявляемого при этом взаимодействия может меняться в весьма широ-
широких пределах. Так, при процессах комплексообразования, когда в координацион-
координационной связи участвуют электронные орбитали частиц и образуются достаточно
устойчивые сложные продукты, эффект взаимодействия проявляется очень ярко.
С меньшим энергетическим эффектом и более слабым участием электронных
орбиталей, а следовательно, и с менее выраженным химизмом происходит образо-
образование водородных связей. И наконец, совсем слабыми силами с очень незначи-
незначительным энергетическим эффектом характеризуется ван-дер-ваальсово или меж-
межмолекулярное взаимодействие, проявляющееся между любыми частицами на
расстояниях, исключающих возможность перекрывания электронных орбиталей.
1. Силы Ван-дер-Ваальса. Очень слабые силы притяжения между нейтраль-
нейтральными атомами или молекулами, проявляющиеся на расстояниях, превосходящих
размеры частиц, называют межмолекулярным притяжением или силами Ван-дер-
Ваальса. Они действуют в веществах, находящихся в газообразном или жидком
состоянии, а также между молекулами в молекулярных кристаллах, Ван-дер-
ваальсово притяжение имеет электрическую природу и рассматривается как
результат действия трех эффектов — ориентационного, индукционного и диспер-
дисперсионного:
Е —
Ориентационный эффект возникает только в полярных вещест-
веществах, молекулы которых представляют собой диполи. При этом молекулы вещест-
вещества поворачиваются друг к другу разноименными полюсами и в результате такого
диполь-дипольного взаимодействия определенным образом ориентируются в
пространстве.
Величина ориентационного эффекта тем больше, чем выше электрический
момент диполя молекул и чем меньше расстояние между ними. С ростом темпера-
температуры эффект уменьшается, так как усиливающееся тепловое движение нарушает
взаимную ориентацию диполей.
Индукционный эффект связан с процессами поляризации моле-
молекул диполями окружающей среды. При этом в неполярной молекуле центры
тяжести положительных и отрицательных зарядов перестают совпадать. Возника-
Возникает наведенный, или индуцированный, диполь. Подобное явление может наблю-
наблюдаться и для полярных частиц. Тогда индукционный эффект накладывается на
диполь-дипольное взаимодействие, в результате чего увеличивается взаимное
притяжение. Индукционное взаимодействие возрастает с ростом электрического
момента диполя и поляризуемости, быстро уменьшается при увеличении расстоя-
расстояния. В то же время Еиня от температуры не зависит, так как наведение диполей
происходит при любом пространственном расположении молекул.Более или менее
ощутимое влияние индукционного взаимодействия наблюдается для частиц,
обладающих сравнительно большой поляризуемостью.
Дисперсионный эффект возникает при взаимодействии любых
атомов и молекул независимо от их строения и полярности. Характерной особен-
особенностью дисперсионных сил является их универсальность. В основе квантово-
механической теории, объясняющей природу дисперсионного эффекта (Дебай,
1930), лежит представление о синхронизации движения мгновенных диполей
взаимодействующих частиц.
Рассмотрим систему, состоящую из ядра и движущихся электронов. В любой
момент времени вследствие несовпадения центров тяжести зарядов электронного
облака и ядра такая система представляет собой мгновенный диполь. Электриче-
Электрическое поле мгновенных диполей атомов и молекул индуцирует мгновенные диполи
в соседних частицах. Каждый из диполей будет влиять на ориентацию подобных
мгновенных диполей, возникающих в близлежащих молекулах, Движение всех
мгновенных диполей системы перестает быт? независимым и становится синх-
синхронным. Их появление и исчезновение в разных молекулах происходит в такт
друг другу. В результате соседние частицы испытывают взаимное притяжение и
энергия системы понижается.
Общим для рассмотренных эффектов является то, что все они уменьшаются
обратно пропорционально шестой степени расстояния между взаимодействующи-
взаимодействующими частицами, т.е. очень быстро падают с увеличением расстояния. Величина
вклада отдельных составляющих в суммарный эффект зависит от свойств моле-
молекул. Наиболее существенны такие их свойства, как полярность и поляризуемость. .
Чем выше полярность, тем значительнее роль ориентационных сил. С увеличени-
увеличением поляризуемости молекулы возрастает дисперсионный эффект. Индукционный
эффект зависит от обоих факторов, но сам имеет лишь второстепенное значение
(табл. 13).
Число молекулярных кристаллов, где ван-дер-ваальсова связь обусловливает
взаимодействие отдельных молекул, довольно велико. К кристаллам этого типа
Т а б л и ц а 13. Вклад отдельных составляющих в полную энергию
межмолекулярного взаимодействия
Вещество
н2
Аг
Хе
НС1
НВг
HI
NH3
Н20
Электри-
Электрический мо-
момент дипо-
диполя, 1029,
Кл-м
0
0
0
3,4
2,57
1,25
4,95
6,07
Поляри-
Поляризуемость
0,20
1,63
4,00
2,63
3,58
5,4
2,21
1,48
Эффект, кДж/моль
ориента-
ционный
0
0
0
3,34
1,09
0,58
13,28
36,32
индукци-
индукционный
0
0
0
1,003
0,71
0,295
1,55
1,92
дисперси-
дисперсионный
0,17
8,48
18,4
16,72
28,42
60,47
14,72
8,98
Суммарная
энергия,
кДж/моль
0,17
8,48
18,4
21,05
30,22
61,36
29,55
47,22
Темпера-
Температура плав-
плавления, К
20,21
76
167
188
206
^38
239,6
373
относятся твердые водород, кислород, азот, галогены, НГ, СОг, белый фосфор,
ромбическая и моноклинная сера и др., а также кристаллы большинства органи-
органических соединений. Все благородные газы и многие молекулярные вещества
кристаллизуются в молекулярных решетках с плотнейшей упаковкой. Это указы-
указывает на то, что для межмолекулярных связей характерны ненаправленность и
ненасыщенность. Структуры молекулярных кристаллов относятся к гетеродесми-
ческиш: в них сосуществуют два типа связи — внутри молекул и между молекула-
молекулами. Связи, действующие между молекулами, намного слабее, чем межатомные
внутри молекул. Поэтому именно межмолекулярные силы в первую очередь опре-
определяют многие физические свойства веществ (температуры плавления, твердость,
плотность, тепловое расширение и др.). Низкие температуры плавления, высокая
летучесть, малая твердость, незначительная плотность и высокий коэффициент
теплового расширения — все это свидетельствует о слабости ван-дер-ваальсовой
связи. На рис. 60 представлена типичная кривая изменения энергии для межмо-
межмолекулярного взаимодействия. Она характеризуется неглубоким минимумом, кото-
который расположен на значительном расстоянии от начала координат. Длина ван-
дер-ваальсовой связи больше, а прочность меньше,
чем те же параметры для ковалентной связи.
2. Водородная связь. Промежуточный характер
между валентным и межмолекулярным взаимодей-
взаимодействием имеет так называемая водородная связь.
Она осуществляется между положительно поляри-
поляризованным атомом водорода, химически связанным
в одной молекуле, и отрицательно поляризован-
поляризованным атомом фтора, кислорода и азота (реже хлора,
серы и др.), принадлежащим другой (или той же)
Рис. 60. Зависимость энергии молекуле. То, что подобное взаимодействие не
взаимодействия от межмолекуляр- обнаруживается у других атомов, обусловлено
ного расстояния • уникальными свойствами поляризованного атома
160' .
водорода — его малым размером и отсутствием внутренних электронных слоев.
Ьч Приводим примеры образования водородной связи (пунктир):
н-с/
о
\
с—н -
о
о—н о
н—cf Nc—н
\ 0Л07нм 0,163нм /г
о н о
Сравнение длин связей, например, для муравьиной кислоты показывает, что
ковалентная связь в исходной молекуле мономера испытала деформацию. Ее дли-
длина увеличилась от 0,097 в мономере до 0,107 нм в димере. Энергия- водородной
связи невелика и лежит в пределах 8 — 80 кДж/моль. Так, энергия водородной
связи H...F равна 82, Н...0 — 21, H...N — 8 кДж/моль. Водородная связь прояв-
проявляется тем сильнее, чем больше относительная электроотрицательность и меньше
размер атома-партнера. Поэтому она легко возникает с атомами неметаллических
элементов второго периода системы и в меньшей степени характерна для хлора и
серы. Благодаря наличию водородной связи молекулы объединяются в димеры и
более сложные ассоциаты, устойчивые при достаточно низких температурах. Ас-
социаты могут представлять собой одномерные образования (цепи, кольца), дву-
двумерные плоские сетки и трехмерные пространственные структуры.
В кристалле льда молекулы воды расположены тетраэдрически (рис. 61).
Каждый атом кислорода тетраэдрически связан с четырьмя атомами водорода,
Рис. 61. Структура льда с водородными связями
101
I I
причем с двумя из них — полярной ковалентной связью (d _^ = 0,109 нм), а
с двумя другими — водородной связью (d _ = 0,176 нм). В свою очередь,
Н—О
каждый атом водорода связан одновременно с двумя атомами кислорода: с од-
одним — ковалентной, а с другим — водородной связью. Создается ажурная струк-
структура, далекая от плотной упаковки. При плавлении льда водородные связи час-
частично разрушаются (примерно на 15%). Это сближает молекулы, поэтому вода
плотнее льда. Одновременно с уплотнением структуры при нагревании происхо-
происходит расширение воды. Результатом наложения обоих процессов оказывается
аномальный ход изменения плотности воды: наличие максимума плотности при
4°С. При переходе воды в парообразное состояние разрушаются уже все водород-
водородные связи. В парах фтороводорода и некоторых карбоновых кислот водородные
связи сохраняются.
Способность к ассоциации проявляют аммиак, спирты, пероксид водорода,
гидразин, серная кислота и многие другие вещества. Многие физические свойст-
свойства веществ с водородной связью выпадают из общего хода их изменения в ряду
аналогов. Так, летучесть ассоциированных жидкостей аномально мала, а вяз-
вязкость, диэлектрическая постоянная, теплота парообразования, температура кипе-
кипения аномально повышены. Ассоциация приводит к изменению растворяющей
способности. Часто возможность растворения вещества связывают с его способ-
способностью образовывать водородные связи.
Наряду с межмолекулярной водородной связью существует внутримолекуляр-
внутримолекулярная связь, которая широко распространена и также оказывает заметное влияние
на химические и физические свойства веществ. Вот примеры образования внутри-
внутримолекулярной водородной связи:
салициловый
альдегид
Обычно водород входит в плоское шести- или пят,ичленное кольцо.
Трудность квантово-механического расчета водородной связи обусловлена тем,
что погрешность вычисления значительно больше величины энергии этой связи.
По-видимому, наиболее надежных результатов можно ожидать от ММО. Расчеты
для иона HF' дают три трехцентровые молекулярные орбитали: СМО, РМО и
НМО. Две электронные пары, вначале заселявшие орбитали фтора и водорода,
располагаются на СМО и НМО, образуя две эквивалентные связи H~F: -
РМО
F м F~ 2p
АО
НМО
СМО
HF,"
If Н
АО
102
Водородная связь играет важную роль в неорганической и органической
химии. Ее универсальность связана с распространенностью в природе воды и
соединений со связями О~Н. Низкая энергия водородной связи, способность
легко разрушаться и восстанавливаться при комнатной температуре вместе с ее
огромной распространенностью обусловливает значение водородной связи в био-
биологических системах. Упорядоченное расположение полипептидных цепей в
структуре белка, поперечные связи в структуре целлюлозы и в двойной спирали
дезоксирибонуклеиновой кислоты (ДНК) объясняются наличием водородной
связи. Кроме того, доказано образование водородной связи на некоторых стадиях
почти всех биохимических процессов.
3. Комплексные соединения. Координационная теория Вернера. К началу 90-х
годов прошлого столетия был накоплен материал по особой группе соединений,
состав которых не находил объяснений с позиций классической теории валентно-
валентности. Соединения типа BF3, CH4, NH3, НгО, СОг и др., в которых элемент прояв-
проявляет свою обычную максимальную валентность, называются валентно-
насыщенными соединениями или соединениялш первого порядка. При
взаимодействии соединений первого порядка друг с другом образуются
соединения высшею порядка. К соединениям высшего порядка относятся гидраты,
аммиакаты, продукты присоединения кислот, органических молекул, двойные
соли и многие другие. Вот примеры образования соединений высшего порядка:
СоС13 + 6NH3 = CoCl3-6NH3
Fe(CNK + 3KCN = Fe(CNK-3KCN
Швейцарский химик Альфред Вернер ввел в химию представления о соеди-
соединениях высшего порядка и дал первое определение понятию комплексного
соединения. К комплексным соединениям он относил наиболее устойчивые со-
соединения высшего порядка, которые в водном растворе либо вообще не рас-
распадаются на составные части, либо распадаются в самой незначительной степени.
Тот факт, что в водных растворах комплексных соединений не наблюдались
реакции, свойственные отдельным составляющим, он объяснил особой устой-
устойчивостью этих соединений. Вернер в 1893 г. высказал предположение о том, что
любой элемент после насыщения его обычных валентностей способен проявлять
еще и дополнительную валентность, — координационную. Именно за счет коор-
координационной валентности и происходит образование соединений высшего по-
Рядка.
По координационной теории Вернера, в каждом комплексном соединении
различают внутреннюю и внешнюю сферы. Более тесно связанные частицы внут-
внутренней сферы называют комплексным ионом или комплексом. При написании
координационной формулы эту часть комплексного соединения заключают в
квадратные скобки. Координационные формулы соединений высшего порядка
[ могут быть записаны так: CoCl3-6NH3 или [Co(NH3N]Cl3; Fe(CN3)-3KCN или
| K3[Fe(CNN].
В химических реакциях, в процессах растворения,' в структуре кристалла
комплекс выступает как самостоятельная единица. Частицы внешней сферы,
Связанные в соединении менее прочно, в пррцессах растворения или в реакциях
б
отщепляются от соединения, образуя самостоятельные ионы:
103
[Co(NH3N]Cl3 = [Co(NH3N]3+ + ЗСГ
K3[Fe(CNN] = 3K+ + [Fe(CNN]3-
Центральный атом внутренней сферы, вокруг которого группируются ионы
или молекулы, называется комплексообразователем или ядром комплекса. Роль
комплексообразователей чаще всего выполняют катионы металлов, реже нейт-
нейтральные атомы или анионы. Ионы или молекулы, координирующиеся вокруг
центрального атома во внутренней сфере, называются лигандами. Лигандами
могут служить анионы Г", ОН", CN", CNS", N0', С0|~, С20^" и др., нейтральные
молекулы Н20, NH3, CO, N0, Г2, N2H4, этилендиамин NH2—CH2—CH2—NH2, ами-
ноуксусная кислота NH2—CH2~Cv,riTi и т.д.
Координационное число, или координационная валентность (KB), — число мест
во внутренней сфере комплекса, которые могут быть заняты лигандами. Коор-
Координационное число обычно больше степени окисления комплексообразователя.
Известны координационные числа 1, 2, 3, 4, 5, 6, 7, 8, 9, 12. Чаще встреча-
встречаются комплексные соединения с координационной валентностью 4, 6 и 2. Эти
числа соответствуют наиболее симметричной геометрической конфигурации комп-
комплекса — октаэдрической F), тетраэдрической или квадратной D) и линейной B).
Координационная валентность зависит от природы комплексообразователя и
лигандов. Незаряженные лиганды обычно могут присоединяться к комплексооб-
разователю в большем числе, чем заряженные: [Со(Н20N]2+ и [СоСЦ]2"- Коорди-
Координационная валентность зависит также от размеров комплексообразователя и
лигандов. Например, с ионами О", Вг", Г алюминий проявляет координационное
число 4, а с меньшим ионом F" — число 6: К[А1С14] и K3[A1F6].
Координационная емкость лиганда — число мест во внутренней сфере комплек-
комплекса, занимаемых каждым лигандом. Для большинства лигандов координационная
емкость равна единице, реже двум. Анионы галогенов, CN", NH3 занимают в
комплексах одно координационное место и называются монодентатными лиган-
лигандами. Гидразин, аминоуксусная кислота, этилендиамин, а также ионы С2О2",
S04", СО^" занимают по два координационных места и являются бидентатными.
Существуют лиганды с большей емкостью — 3, 4, 6. Их называют полидентатны-
ми. Они способны осуществлять одновременно несколько связей с комплексообра-
комплексообразователем. Одним из наиболее важных полидентатных лигандов (шести дентат-
ным) является анион этилендиаминтетраацетат — ЭДТА (см. с. 109).
Заряд комплекса должен быть численно равен суммарному заряду внешней
сферы и противоположным ему по знаку. Во внешней сфере K3[Fe(CNN] находят-
находятся три положительно заряженных иона калия. Следовательно, сам комплексный
ион имеет три отрицательных заряда. Степень окисления комплексообразователя
равна и противоположна по знаку алгебраической сумме зарядов всех остальных
ионов. Отсюда в K3[Fe(CNN] степень окисления железа равна +3. Однако комп-
комплексы [Ca(NH3N], [Fe(COM], [Хе(Н2О)б] и другие нейтральны. Они не имеют
внешней сферы, а их внутренняя сфера состоит из нейтральных молекул-лиган-
дов и комплексообразователя в степени окисления 0. v
4. Номенклатура комплексных соединений. Многие комплексные соединения
до сих пор сохранили свои исторические названия, связанные с их одетом или
\ол .,.¦.¦ . ' .:.;-'-. •: ¦ .. - >,;¦;; ¦¦-.-.-• \ .. /¦'.,-.¦:.% ¦.'¦
именем ученого, их синтезировавшего. В настоящее время принципы наимено-
наименования комплексных соединений утверждены комиссией по номенклатуре ШРАС.
Ниже приведены основные правила, заимствованные из материалов этой комис-
комиссии, с учетом особенностей номенклатуры соединений, принятой в русском
языке.
Порядок перечисления ионов. Первым называют анион,
затем катион:
[Ag(NH3J]Cl — хлорид диамминсеребра (+1),
К2[СиС1з] — трихлорокупрат (+1) калия.
*
При этом в названии аниона употребляется корень латинского наименования
комплексообразователя, а в названии катиона — его русское наименование в
родительном падеже:
К3(Те(С]Ч)б] — гексацианоферрат (+3) калия,
но [Ре(КНз)в]С1з — хлорид гексаамминжелеза (+3).
Порядок перечисления лигандов. Лиганды в комплексе
перечисляются в следующем порядке: а) анионные; б) нейтральные; в) катион-
ные — без разделения друг от друга дефисом. Анионы перечисляются в порядке
Н", О2", ОН", простые анионы, полиатомные анионы и, наконец, органические
анионы — в алфавитном порядке:
[PtEn(NH3J(NO2)Cl]SO4 -
сульфат хлоронитродиамминэтилендиаминплатины (+4)
Окончания координационных групп. Нейтральные груп-
группы называются так же, как молекулы. Исключениями являются аква (Н20) и
аммин (NH3). К отрицательно заряженным ионам прибавляют соединительную
гласную "о":
[Co(NH3N][Fe(CNN] — гексацианоферрат (+3) гексгаамминкобальта (+3)
Приставки, указывающие число лигандов. Ди-, три-,
тетра-, пента-, гекса- и т.д. используются перед простыми лигандами, например
Дихлоро-, триоксалато-, тетраоксо-, гексагидроксо-:
K^SnFs] — октафторостаннат (+4) калия.
Приставки "бис", "трис" используют перед лигандами со сложным названием,
где в самом названии лиганда уже имеются приставки (моно-, ди- и т.д.). Назва-
Названия таких лигандов заключают в скобки:
[Ге(Еп)з]С1з-— хлорид трмс(этилендиамин)железа (+3)..
105
! ;
Окончания названий комплексов. Характерным окон-
окончанием для анионных комплексов является "ат" или "овая", если называется
кислота. Для катионных и нейтральных комплексов типичных окончаний нет:
K3[Co(CNN] — гексацианокобальтат (+3) калия, H2[Pt(CNN] — гексацианоплати-
новая (+4) кислота, или гексацианоплатинат (+4) водорода.
Состояние окисления. Окислительные числа комплексообразова-
теля обозначаются арабскими цифрами в скобках после его названия. Знак ста-
ставится перед цифрой:
[Cu(NH3J]OH — гидроксид диамминмеди (+1).
Названия мостиковых групп. Группы, которые являются
мостиком между двумя центрами координации, обозначают греческой буквой ц,
которая повторяется перед названием каждой мостиковой группы:
0Н
4-
г^ р
ди-//-гидроксотетраоксалато дихромат (+3)
5. Классификация комплексных соединений. Применяющиеся системы класси-
классификации комплексных соединений основываются на различных принципах.
По принадлежности к определенному классу со-
соединений различают:
комплексные кислоты — H2[SiFe], H[AuCl4];
комплексные основания — [Ag(NH3J]OH, [Со(ЕпK](ОНK;
комплексные соли — K2[HgI4], [Cr(H2ON]Cl3.
По природе лигандов. Если лигандом является вода, комплексы
называются аквакомплексами: [Co(H2ON]SO4, [Cu(H2OL](NO3J. Комплексы,
образованные аммиаком, — аммиакаты: [Ag(NH3J]Cl, [Cu(NH3L]SO4,
[Co(NH3N]Cl2. Оксалатные, карбонатные, цианидные, галогенидные и другие
комплексы, содержащие в качестве лигандов анионы различных кислот, называ-
называют ацидокомплексами. Например, K4[Fe(CNN] и K>[HgI4] — цианидный и иодид- '
ный ацидокомплексы. Соединения с ОН-группами в виде лигандов называют
гидроксокомплексами, например: К3[А1(ОНN].
По знаку заряда комплекса различают:
катионные комплексы — [Co(NH3N]Cl3, [Zn(NH3L]Cl2;
анионные комплексы — Li[AlH4], K2[Be(CO3J];
нейтральные комплексы — [Pt(NH3JCl2], [Co(NHsKCl3].
Нейтральные комплексы не имеют внешней сферы. Более сложными являются
бикомплексы, состоящие из комплексных катионов и анионов, например-J
[Co(NH3N][Fe(CN)«].
106
\
По внутренней структуре комплексного соеди-
соединения, а) По числу ядер, составляющих комплекс, различают моно- и поли-
полиядерные комплексные' соединения. Пример двухъядерного комплекса — это
[(NH3MCr—ОН—Cr(NH3M]Cl5, в котором два иона хрома (комплексообразователя)
связаны посредством мостиковой группы ОН. В качестве мостиковых могут функ-
функционировать частицы, обладающие неподеленными электронными парами: ионы
F", С1~, О2", S2", S02-, NH2", NH^ и др. Полиядерные комплексы, в которых мости-
мостики образованы гидроксильными группами, называются оловыми соединениями.
Структурно мостиковая группа ОН отличается от гидроксильной группы в одно-
одноядерных комплексах. Координационное число кислорода в оловом мостике равно
трем, а в ОН-группе одноядерных комплексов — двум.
б) По отсутствию или наличию циклов в составе комплексных соединений
различают простые и циклические комплексные соединения. Комплексы, которые
содержат лиганды, занимающие одно координационное место, называются прос-
простыми. Полидентатные лиганды, связывающиеся с одним и тем же центральным
атомом несколькими связями, дают циклические комплексы.
Особую группу составляют сверхкодтлексные соедгшепия. В них число лиган-
дов превышает координационную валентность комплексообразователя. Примером
может служить CuSO4*5H2O. У меди координационная валентность равна четы-
четырем и во внутренней сфере координированы четыре молекулы воды. Пятая моле-
молекула присоединяется к комплексу при помощи водородных связей. Она связана
менее прочно и в координационной формуле записывается после внутренней и
внешней координационных сфер: [Cu(H2OL]SO4"H2O.
Комплексные соединения с мостиковыми атомами кислорода называются
оксоловыми. Одной из групп многоядерных оксоловых комплексов являются
поликислоты. Если кислота включает ядра одного и того же элемента, она отно-
относится к изополикислотам:
-]Н2 — полиметакремниевая кислота.
В гетерополикислотах мостиковый кислород соединяет атомы разных элемен-
элементов:
Нз[0зР~О—МоОз] — фосфорно-молибденовая кислота.
В гетерополикислотах сочетаются кислотные остатки металлических и неметал-
неметаллических элементов. Твердо установлено, что все они представляют собой много-
многоядерные комплексы, в которых связь между ядрами осуществляется посредством
кислородных мостиков.
6. Устойчивость комплексных соединений. Константа нестойкости. Двойные
соли. При растворении в воде комплексные соединения обычно распадаются так,
что внутренняя сфера ведет себя как единое целое:
K[Ag(CNJ] ^КЧ [Ag(CNJ]-
Наряду с этим процессом, хоть и в незначительной степени, происходит диссо-
диссоциация внутренней сферы комплекса;
[Ag(CNJ]*
Ag+ + 2CN-
Применив к последнему равновесию выражение для константы ионизации (см.
с. 154), получим соотношение, называемое константой нестойкости комплекса:
2
1
К
CN"
HecT,[Aft(CNJ]"
C[Ag(CN):
= ~ Ю-22.
Константа нестойкости есть мера прочности комплекса. Вместо константы нестой-
нестойкости иногда пользуются обратной величиной, называемой константой устойчи-
устойчивости: Куст = 1/йнест •
У ряда соединений, которые рассматриваются как комплексные, константы
нестойкости настолько велики, что концентрации составляющих частиц оказыва-
оказываются больше концентрации комплексного иона. К таким соединениям относятся
двойные соли, которые в твердом состоянии имеют координационную структуру,
а в растворе в значительной мере распадаются на составные ионы, например
К2[СиС14]
2К* +
2К+
Cu2+ + 4C1-
В умеренно разбавленных растворах этой соли существуют как комплексные, так
и простые ионы. Дальнейшее разбавление приводит к полному распаду комплекс-
комплексных ионов [CuClJ2".
Известны соли, которые занимают промежуточное положение между типичны-
типичными двойными и типичными комплексными солями, например КРЫз- Эта соль в
концентрированном водном растворе существует в виде простого иона К+ и комп-
комплексного иона [РЫз]-. При разбавлении раствора этой соли комплексный ион
распадается на Г и РЫг, который выпадает в осадок.
7. Халаты и внутрикомилекспые соединения. К очень важным циклическим
соединениям относятся так называемые хелаты или клешнеобразные соединения,
в которых центральный атом и полидентатный лиганд образуют цикл. Например,
при взаимодействии гидроксида меди с аминоуксусной кислотой (гликокол)
образуется нейтральный комплекс»:
СН2—NH2
+ _ Си
с—о—Гн~~ноТ
нно
. .
O
NH2—CH2
О
-2Н,0
CH2—NH2
0
NH,—CH,
ч
0-
40
Каждая молекула гликокола использует обе функциональные группы. В одном
случае она соединяется с центральным атомом через две аминогруппы по донор-
но-акцепторному механизму, а во втором — через кислород карбоксильной
группы обычной ковалентной связью. Комплексообразователь при этом
оказывается как бы втянутым внутрь лиганда, охвачен связям» наподобие
^клешни рака. Отсюда и происходит название хелат *. Примерами других хелатов
могут служить комплексы с этилендиамином NH2~CH2~~CH2—NH2, этилендиамин-
тетрауксусной кислотой (ЭДТА), щавелевой кислотой:
*Хелат — клешня рака.
108. ¦- ¦ •• '. ,. -..- '.-V ¦ ¦.
Na3
трноксалатоферрат (+3) натрия
О.,
Na,
^С СН, С»х, \"*»у
CH,—NH, NH,—CH,'
Cu
CH,—NH, NH,—CH,
Cl,
хяорнд 5ас(
\
NC—0 O-
этнленднамннтетраацетатокупрат (+2) натрия
нлн
ЭДТА-купрат (+2) яатряя
Наличие в хелатах циклических группировок сильно увеличивает их устойчи-
устойчивость по сравнению с соединениями подобного состава, но не имеющими циклов.
Такое повышение устойчивости называют хелатпным эффектом. Известны хела-
хелаты, которые не разлагаются даже при 500°С. Константы нестойкости двух комп-
комплексов кобальта (в водном растворе), в которых лиганды координированы через
азот, различаются на 10 порядков:
3 +
NH
NH,
вон
гексааииянкобальта (+3)
К».™ =7-1(Г39
сн2
CH,
4NH, NH.
<CH2
NH,
— С"
NH,
/ \
NH2
сн2—сн2
вон
д»рис(этялендяамнн)кобальта (+3)
Особенно следует выделить такую разновидность хелатов, как внутрикомп-
лексные соединения. Впутрикомплекспыми соединениями называют такие хелаты,
в которых один и тот же лиганд связан с комплексообразователем как парно-
электронной, так и донорно-акцепторной связью, например гликоколат меди,
этилендиаминтетраацетатокупрат натрия. Соединения триоксалатоферрат (+3)
натрия и хлорид ?ис(этилендиамин)меди (+2) являются обыкновенными хела-
тами. 1
^ К хелатам отнбсятся такие важные для жизни соединения, как хлорофилл и
^гемоглобин: ,
109
Ill I
хлорофилл
гемоглобин
Комплексообразователем в хлорофилле выступает магний, а в гемоглобине —
железо. В одной плоскости с металлом располагаются четыре атома азота органи-
органического лиганда. По одну сторону от плоскости железо присоединяет молекулу
белка (глобина), а по другую — молекулу кислорода. Такой продукт называется
оксигемоглобином. Он образуется в легких, где гемоглобин присоединяет кисло-
кислород воздуха и далее в виде оксигемОглобина разносится по всему организму.
Хлорофилл играет важнейшую роль в процессах фотосинтеза, протекающих во
всех зеленых растениях.
8. Изомерия комплексных соединений. В химии комплексных соединений
изомерия очень распространена.
Сольватная (в частности, гидратная) изомерия обнаруживается в
отдельных изомерах, когда распределение молекул воды между внутренней и
внешней сферами оказывается неодинаковым. Например, для хлоридов гекса-
аквахрома (+3) известны три изомера:
[Сг(Н2ОN]С13
фиолетовый
светло-зеленый
[Сг(Н2ОLС12]С1-2Н2О
темно-зеленый
Валовой состав всех изомеров одинаковый. Но в первом случае все шесть молекул
воды находятся во внутренней сфере, во втором — пять, а в третьем — только
четыре. Так как структура всех комплексов различна, различны и свойства.
Ионизационная изомерия связана с различной легкостью
диссоциации ионов из внутренней и внешней сферы комплекса. Примерами иони-
ионизационных изомеров могут служить
[Pt(NH3LCl2]Br2 и [Pt(NH3LBr2]Cl2
Первая соль дает желтый осадок с раствором AgNO3- Вторая реагирует с раство-
раствором нитрата серебра с образованием белого творожистого осадка AgCl.
Координационная изомерия встречается только у бикомп-
лексных соединений, например
[Co(NH3N][Fe(CNN] и [Fe(NH3N][Co(CNN].
Изомерия связи возникает тогда, когда монодентатные лиганды
могут координироваться через два разных атома. Например, ион NOj может
110
присоединяться к центральному атому через азот или через кислород. Это обус-
обусловливает существование у иридия, кобальта и некоторых других металлов двух
изомеров:
[(NH3M-Ir-NO2]Cl2 и [(NH3M-Ir-ONO]C12
Таким образом, изомерия связи может наблюдаться у амбидентпатных лигандов,
которые содержат по крайней мере два разных атома с неподеленными электрон-
электронными парами.
Пространственная (геометрическая) изомерия
обусловлена тем, что одинаковые лиганды располагаются вокруг комплексообра-
зователя либо рядом (чис-положение), либо напротив (m/хшс-положение). Этот
тип изомерии часто называется ?{ис-транс-изомерией. Примером может служить
дихлородиамминплатина:
CL NH3
Pt
СГ NH3
yuc-изомер
(оранжевые кристаллы)
NH, С1
Pt
С Г
транс-изомер
(желтые, менее растворимые
в воде кристаллы)
Цис- и транс-изомеры отличаются друг от друга физическими и химическими
свойствами. Изучение геометрической изомерии имело большое значение для
установления пространственного строения комплексных соединений. На основа-
основании того, что для некоторых комплексов МА2В2 и МА4В2 удавалось синтезиро-
синтезировать по два изомера, Вернер приписал им квадратное и октаэдрическое строение.
Подавляющее большинство комплексных соединений МА2В2 изомеров не имеет.
Для них Вернер постулировал тетраэдрическую структуру. Все предположения
были позднее подтверждены современными методами исследования строения
вещества.
9. Трансвлияние. Важнейшая закономерность, которой подчиняется реакцион-
реакционная способность комплексных соединений, была открыта И.И.Черняевым A!J(>) 11
названа им трансвлиянием. Работая с комплексами платины Pt2+, он установи.!,
что неоднородные лиганды в трамс-положении оказывают друг на друга в.шя-
ние, проявляющееся в большей или меньшей способности этих лигандов вступать
в реакции замещения (обмена). Впоследствии оказалось, что трансвлияние явля-
является общей закономерностью для неоднородных комплексов квадратной или
октаэдрической структуры. По силе своего трансвлияния лиганды располагаются
в ряд:
СО, NO, NO", CNS", I", Вг , С1", ОН", NH3, H20
Активность членов ряда уменьшается слева направо. Каждый предыдущий член
более активен в отношении трансвлияния по сравнению с последующими. Лиган-
Лиганды, расположенные в ряду слева, способствуют удалению лигандов, стоящих
справа. И наоборот, лиганды конца ряда закрепляют во внутренней сфере-лиган-
сфере-лиганды начала ряда. Например, для комплекса.
111
12-
Pt
(транс-лабилизирующее влияние)
из трех ионов хлора легче других замещается тот, который находится в транс-
положении к сильно mpawc-активному лиганду NOj. И наоборот, в комплексе
С1
С1
Pt
Cl
(транс-стабилизирующее влияние)
хлор, стоящий в гпранс-положении к менее активному лиганду NH3, закреплен
прочнее других атомов хлора и потому труднее вступает в реакции замещения.
Закономерность трансвлияния объясняет, почему при реакциях замещаются
только определенные лиганды, и открывает пути сознательного синтеза веществ.
Рассмотрим процесс образования чмс- и транс-изомеров дихлородиамминплатины
[Pt(NH3JCl2]. При действии аммиака на K^PtCl^ образуется умс-изомер. Следо-
Следовательно, реакция идет по схеме
С1.
сг
Pt
Cl
'Cl
+ 2 NH3 =
Cl
Cl
Pt
+ 2 KC1
С другой стороны, при обработке [Pt(NH3L]Cl2 соляной кислотой образуется
транс-изомер по уравнению реакции
NH3 NH;
Pt
NH;
NHc
Cl2 + 2 HC1 =
Cl
NH.
Pt
NHc
'Cl
+ 2 NH4CI
Таким образом, исходя из иона [PtC^]2" можно получить только цис-, а из
[Pt(NH3L]2+ — транс-изомер. С точки зрения принципа трансвлияния эти резуль-
результаты объясняются просто. Замена иона С1" на аммиак в [PtC^]2" ведет к закреп-
закреплению хлора, расположенного в mpawc-положении к замещенному. Поэтому вто-
вторая молекула аммиака замещает один из менее прочно связанных ионов С1~,
находящихся в уис-положении по отношению к первой. Замена же одной молеку-
молекулы аммиака в [Pt(NHaL]2+ хлором ведет к ослаблению связи молекулы аммиака,
занимающей трам<?-положение. Поэтому второй ион СГ легко замещает ее и
входит в трамс-положение к первому.
Согласно поляризационной теории трансвлияния, комплексообразователь
полязирует лиганды и создает в них индуцированные диполи. Когда ядро комп-
комплекса окружено одинаковыми лигандами, оно находится в симметричном поле и
все индуцированные диполи компенсируют друг друга. При замещении лиганда
112
Рис. 62. Обоснование трансвлияния с
точки зрения поляризационных пред-
представлений
Рис. бЗ.Образование тг-связи при перекрывании
битали комплексообразователя и р^-орбитали лиганда
на более отрицательную или более легко поляризуемую группу симметрия поля
вокруг комплексообразователя нарушается и в нем индуцируется некомпенсиру-
емый диполь. Так, первоначальный заряд на Pt2+ индуцирует диполь на лиганде
L, который в свою очередь поляризует ион металла. А поляризация электронной
системы комплексообразователя ведет к отталкиванию отрицательного заряда на
лиганде X. Следовательно, связь Pt~X ослабляется и удлиняется. Поэтому в
реакциях лиганд X наиболее подвижен (рис. 62). Таким образом, в поляриза-
поляризационной модели трансвлияние сводится к сравнению прочности связей между
комплексообразователем и лигандами. Большей реакционной способностью обла-
обладает тот лиганд, связь которого с комплексообразователем наиболее слабая. Од-
Однако иногда это положение не соблюдается.
Теория тг-связей объясняет трансвлияние способностью тромс-лиганда образо-
образовывать двойные связи с комплексообразователем. На рис. 63 представлено обра-
образование двойной а- и тг-связи в комплексе. Для простоты даны только орбитали,
лежащие в плоскости yz. тг-Связывание может осуществляться в тех комплексах,
в которых комплексообразователь имеет неподеленные электронные пары, а ли-
лиганд — свободные орбитали. В результате донорно-акцепторного взаимодействия
электронная плотность с (/у^^орбитали центрального атома частично переходит на
орбиталь лиганда. Возникающая асимметрия облака dyZ открывает доступ к
игранс-лиганду. Чем интенсивнее тг-связывание с левым лигандом (рис. 63), тем
больше правый лиганд открыт для атаки, т.е. легче происходит замена Li на L.
Таким образом, по этой концепции, реакционная способность лиганда не связы-
связывается с ослаблением связи между ним и комплексообразователем, а зависит от
способности игранс-лиганда к взаимодействию с комплексообразователем. Поэто-
Поэтому нет ничего удивительного в том, что часто скорость обмена более прочно
связанных лигандов оказывается большей, чем менее прочно связанных.
10. Природа химической связи в комплексных соединениях. Электро-
Электростатические представления. По простой электростатической
модели (Коссель и Магнус, 1916—1922), взаимодействие между комплексообразо-
комплексообразователем и ионными (или полярными) лигандами подчиняется закону Кулона.
При этом предполагается, что образующие комплекс частицы^представляют
собоД недеформируемые шары с определенным зарядом и радиусом. Устойчивый
113
I I
комплекс получается, когда силы притяжения к ядру комплекса уравновешивают
силы отталкивания между лигандами. Согласно закону Кулона, прочность комп-
комплекса увеличивается с ростом заряда и уменьшением радиусов комплексообразо-
вателя и лигандов. Например, для галогенидных комплексов алюминия [AIFJ"
устойчивость убывает в направлении от фторокомплекса к иодокомплексу, так
как в этом ряду закономерно увеличивается размер лигандов. Устойчивость
комплексов с одними и теми же лигандами в более высокой степени окисления
комплексообразователя всегда выше, например К. +/мц \ i+ = 101 и
^го 9+/хти т>+ = Ю3- Так как силы отталкивания между ионами больше, чем
[Cu^ (JNH3LJ'
между нейтральными лигандами, координационное число в комплексе с ионными
лигандами меньше, чем в комплексе с дипольными молекулами. С ионными ли-
лигандами Со2+ дает комплексы с KB = 4, тогда как с дипольными молекулами
[Co(CNSL]
2-
но
координационное число возрастает до 6: [Со^гО^г]2'
[Со(Н20N]2+ и [Co(NH3N]2+.
Электростатическая теория очень наглядна, и потому для качественных выво-
выводов ею широко пользуются и теперь. Однако она не в состоянии объяснить це-
целый ряд фактов: 1) почему существуют комплексы с неполярными лигандами и
комплексообразователем в нулевой степени окисления, например [Fe(CO)s],
[Ca(NH3)e] и др.; 2) чем обусловлены магнитные и оптические свойства соеди-
соединений.
Метод валентных связей.. Весьма наглядным способом описания
комплексных соединений является МВС, предложенный и разработанный Полин-
гом в 30-х годах. В основе метода лежат следующие положения.
1. Связь между комплексообразователем и лигандами донорно-акцепторная.
Лиганды предоставляют электронные пары, а ядро комплекса — свободные орби-
тали. Мерой прочности связи служит степень перекрывания орбиталей.
2. Орбитали центрального атома, участвующие в образовании связей, подвер-
подвергаются гибридизации. Тип гибридизации определяется числом, природой и
электронной структурой лигандов. Гибридизация электронных орбиталей комп-
комплексообразователя определяет геометрию комплекса.
3. Дополнительное упрочнение комплекса обусловлено тем, что наряду с а-
связями могут возникать и тг-связи. Это происходит, если занятая электронами
орбиталь центрального атома перекрывается с вакантной орбиталью лиганда.
Перераспределение электронной плотности в результате а- и тг-связывания проис-
происходит в противоположных направлениях: при возникновении <т-связи идет пере-
перенос на комплексообразователь, при тг-связывании — от него к лйгандам.
4. Магнитные свойства, проявляемые комплексом, объясняются исходя из
заселенности орбиталей. При наличии неспаренных электронов комплекс пара-
парамагнитен. Спаренность электронов обусловливает диамагнетизм комплексного
соединения.
Рассмотрим, как МВС описывает электронную структуру и свойства некото-
некоторых комплексов, образованных металлами первого переходного ряда: медью,
цинком, никелем и кобальтом.
При образовании комплекса распределение электронов на rf-орбиталях комп-
комплексообразователя может оставаться таким же, как у ¦изолированного иона, или
Испытывать изменения {табл! 14). В приведенных примерах медь, никель в
[NiClJ2" и кобальт в [CoFe]3" сохранили электронную структуру катионов, в то
Ш
Комплекс
[Cu(NH,J]+
ZnfNH,),]2*
[Ni(CN)J2'
[NiClJ2"
[CotNH^J84
[CoF6]3"
I*
|l
|i
T
а б л и ц а 14.
Некоторые
комплексы
Заселенность орбяталей
комплексообравователя
3d As Ар
н
TUtlltlltlUtllltl 1 II
tlltlJtUtlltlMtlUtUtUtll
¦1Ш1Ш11М1Ш1 II
щи
' lltlllllllflUtlitlitll
iiti
ii
t||tli;li:lli!ll
tlitlitl 1
[11ШШПШ1,
MW
|
|
I
I
1
lit
A
|
|
1
I
1
1
d
|
|
1
1
1
1
|
1
1
1
1
металлов в МВС
|
I
1
I
1
J
Тип гиб-
ридиаацин
орбиталей
ядра
комплекса
dtp*
Структура
комплекса
Линейная
Тетраэдрическая
Квадратная
Тетраэдрическая
Октаэдрическая
Октаэдрическая
время как в остальных комплексах произошло спаривание электронов. Освобож-
Освобождающиеся электронные орбитали участвуют в образовании <т-связей с лигандами.
Как видно из табл. 14, при образовании октаэдрических комплексов гибридиза-
гибридизация может осуществляться либо с использованием внутренних (п — 1)<йэрбиталей
([Со(КНз)б]3+), либо внешних nrf-орбиталей ([CoFg]3"). В табл. 14 собственные
электронные пары комплексообразователя изображены сплошными стрелками, а
электронные пары лигандов, ответственные за донорно-акцепторные <т-связи, —
пунктирными стрелками. При внешнеорбитальной гибридизации sp3 (Р-связи
образуют более удаленные и менее плотные 4Л-орбитали. Степень перекрывания
электронных облаков при этом меньше и связь лигандов с комплексообразовате-
лем слабее, чем при внутренней гибридизации. Поэтому в комплексе [CoFg]3"
замещение ионов фтора идет легко и он более реакционноспособен, чем
[Со(КНз)б]3+, в котором гибридизация внутриорбитальная. Приведенные в табл.
14 электронные структуры комплексов правильно отражают их магнитные свойст-
свойства. Так, [Cu(NH3J]+, [Zn(NH3L]2+, [Ni(CNL]2-, [Co(NH3N]3+ диамагнитны: у них
нет неспаренных электронов. В противоположность им [NiClJ2" и [CoF6]3" пара-
парамагнитны. При этом парамагнетизм этих соединений пропорционален числу
неспаренных электронов.
МВС дает возможность трактовать комплексы с нейтральными лигандами и
комплексообразователями в нулевой степени окисления, например карбонилы
металлов.
Недостатки МВС: 1) пригоден для описания только ограниченного круга
веществ. Комплексные соединения с многоцентровыми связями МВС совсем не
рассматривает; 2) не объясняет и не предсказывает оптические свойства комп-
комплексных соединений, так как не учитывает возбужденные состояния.
Теория кристаллического поля. Теория кристаллического
г
'¦ i r
* Я) 1-
P и с. 64. Ориентация d-орбиталей комплексообразователя в октаэдрическом поле
лигандов
поля (ТКП) является дальнейшим развитием на квантово-механической основе
электростатической теории Косселя и Магнуса. Согласно ТКП, связь между
ядром комплекса и лигандами ионная * или ион-дипольная. При этом комплексо-
образователЬ' рассматривается с детальным учетом его электронной структуры, а
лиганды — как бесструктурные заряженные точки, создающие электростатиче-
электростатическое поле. Основное внимание ТКП уделяет рассмотрению тех изменений, кото-
которые происходят в комплексообразователе под влиянием поля лигандов. Вырожде-
Вырождение орбиталей, характерное для изолированного атома или иона металла, в не-
несферическом поле лигандов частично снимается. Причина снятия вырождения —
различие в форме rf-орбиталей и их ориентации в пространстве. На рис. 64 пока-
показано расположение d-орбиталей комплексообразователя для октаэдрического
комплекса.
Электронная плотность орбиталей d „ и d „ „ сконцентрирована вдоль коор-
динатных осей, тогда как орбитали d . d , d расположены по биссектрисам
ху х* yz г
ху
между осями. Поэтому электроны d „- и d
»¦
-орбиталей (обозначаемых
испытывают со стороны отрицательно заряженных лигандов большее отталкива-
отталкивание, чем электроны трех других орбиталей, называемых de. В результате энергия
^7-орбиталей повышается, а энергия rfe-орбиталей понижается, т.е. происходит
энергетическое расщепление (рис. 65). При этом </7"~°Рбитали дважды вырожде-
вырождены, а rfe-орбитали — трижды. Энергетическое расстояние между dt- и
лями называется энергией расщепления и обозначается D — Д . Так как сред-
средняя энергия орбиталей должна быть неизменной, то понижение энергии трех-
трехкратно вырожденных rfe-орбиталей должно быть скомпенсировано повышением
энергии двукратно вырожденных </7~°рбиталей. Поэтому </7~°рбитали располага-
располагаются на 0,QD выше, а «fe-орбитали — на 0,4D ниже средней энергии вырожден-
q q
ных rf-орбиталей в поле лигандов. В тетраэдрическом комплексе (рис. 66) орбита-
орбитали d „ и d ~ „ испытывают меньшее отталкивание от лигандов и потому облада-
2? Я2—у2
*Ионная в смысле электростатического взаимодействия катиона-1сомплексообразователя
и анионов-лигандов. ^
Средняя энергия
Вырожденных d-op- 0,<fDe
биталей изо/iupo- '
ванного атома
,dT
de
, Расщепление
в попе лиганооб
Рис. 65. Диаграмма расщепления
энергетических уровней d-орбиталей
комплексообразователя в октаэдричес-
ком поле
Рис. 66. Расположение d-орбиталей
комплексообразователя в тетраэд-
рическом поле лигандов
ют более низкой энергией, чем орбитали d , d , d . Энергетическое расщепле-
ху у? zx
ние для тетраэдрического поля лигандов представлено на рис. 67.
Поля другой симметрии дают более сложную картину расщепления. Величина
энергии расщепления Д, являющаяся мерой силы кристаллического поля, зави-
зависит от природы образующих комплекс частиц и от симметрии поля. Расщепление
увеличивается с ростом заряда комплексообразователя. При переходе от комплек-
комплексов Зй-металлов к комплексам Ad- и 5</-металлов энергия расщепления при одина-
одинаковых лигандах возрастает на 30—80% — более тяжелые ионы имеют больший
радиус и большую поляризуемость. Расщепление, получаемое в комплексах одно-
одного и того же комплексообразователя с различными лигандами, убывает в следу-
следующем порядке:
CN-*>NO">9ATA> NH3>CNS"> H2O>F->OH->CNS->Cl->Br->I-
Эта последовательность лигандов по созда-
создаваемому ими кристаллическому полю назы-
называется спектрохимическим рядом, так как
он получен из данных спектров поглощения
комплексов.
Распределение электронов комплексо-
комплексообразователя по расщепленным энергетиче-
энергетическим уровням в слабом поле подчиняется
общим правилам и принципам: а) перво-
изолированного ращеШяис
атома. s тле лигандов
Рис. 67. Расщепление энергетических
уровней d-орбиталей в тетраэдричес-
ком поле
"Подчеркнут координирующий с ядром комплекса атом (изомерия связи).
I
очередного заполнения наиболее низких уровней; б) Гунда; в) Паули. Однако в
сильных полях при достаточном числе электронов происходит полное заполнение
орбиталей с низкой энергией сначала по одному, а затем по два электрона на
каждой орбитали (спаривание). Лишь после этого начинают заполняться элект-
электронные орбитали с высокой энергией. Спаривание требует затраты энергии Есп,
так как оно принуждает электроны находиться в одной области околоядерного
пространства и тем самым увеличивает отталкивание между ними. Величина Есп
рассчитывается методами квантовой механики и может быть определена экспери-
экспериментально из спектральных данных. Энергия спаривания электронов уменьшает-
уменьшается в группах по мере увеличения атомной массы комплексообразователя, так как
орбитали с увеличением главного квантового числа становятся все более диффуз-
диффузными и поэтому уменьшается отталкивание спариваемых электронов.
Заселение электронами орбиталей в каждом конкретном случае зависит от
соотношения между величинами энергий расщепления и спаривания Есп. При
Д < Есп (слабое поле) электроны будут занимать разные орбитали и спины их
параллельны. В этом случае комплекс называется высокоспиновым. При Д > Есп
(сильное поле) электроны спариваются на (/^-уровнях *, в результате образуются
низкоспиновые комплексы. Наконец, при Д = Есп оба состояния (высоко- и низ-
низкоспиновое) равновероятны. Средняя энергия спаривания для ионов первого
ряда переходных металлов в аквакомплексах значительно превышает энергию
расщепления, т.е. они должны быть высокоспиновыми и парамагнитными, что и
наблюдается в действительности.
Рассмотрим распределение rf-электронов иона Со3+ при образовании октаэдри-
ческих комплексов [CoFg]3" и [Co(NH3N]3+. В изолированном ионе Со3+ внешние
электроны располагаются следующим образом:
Co*
3d 4* 4p id
НЖ1Н ? ГТП МММ
В слабом поле лигандов F" энергия расщепления мала: Д < Есп и заселение
электронов по орбиталям Со3+ такое же, как и в свободном ионе. В сильном же
поле, создаваемом молекулами аммиака (спектрохимический ряд), Д > Есп и
энергетически более выгодно, когда электроны иона Ср3+ располагаются только
на rfe-орбиталях. В соответствии с этим комплекс [CoF6]3" является высокоспино-
высокоспиновым, а [Со(МНз)б]3+ — низкоспиновым. При этом в первом комплексе осуществля-
осуществляется 5р3<#-гибридизация, а во втором — <Psps. В результате комплекс [СоЕб]3"
парамагнитен, a [Co(NHaN]3+ диамагнитен.
Наряду с магнитными свойствами представление о расщеплении энергетиче-
энергетических уровней комплексообразователя может быть использовано для объяснения
окраски комплексных соединений. Предположим, что у комплексообразователя
имеется электрон, который в основном состоянии находится на орбитали de (при
октаэдрической координации). Если сообщить комплексу квант энергии, отвеча-
отвечающий разности энергий между уровнями de и df, то он поглотится, а электрон
перейдет на уровень dу. Состояние возбуждения существует недолго и система
возвратится в исходное состояние. Этот процесс происходит непрерывно, а так
"Для октаэдрического расположения лигандЪв.
118
как он связан с поглощением квантов определенной энергии, излучение будет
поглощаться избирательно. Значит, комплексное соединение будет окрашено.
Эти закономерности наблюдаются, например, в водных растворах солей тита-
титана Ti3+. Единственный rf-электрон, например, в ионе [Ti(H2O)e]3+ переходит с de
на dj (поглощая свет), что и обусловливает фиолетовую окраску комплекса.
Таким образом, цвет комплекса обусловлен величиной энергии расщепления Д
(см. рис. 65). Постепенное возрастание энергии расщепления соответствует изме-
изменению цветности комплекса от инфракрасной части спектра до ультрафиоле-
ультрафиолетовой .
ТКП, так же как и МВС, качественно объясняет основные факты химии комп-
комплексных соединений: координационные числа, пространственные структуры,
магнитные и, кроме того, оптические свойства. Тем не менее ТКП несовершенна.
Исходя из предположения о ионном характере связи, она хорошо объясняет и
дает близкие к опыту расчетные величины для комплексов с недеформируемыми
или малополяризующимися лигандами, т.е. если доля ковалентной составляющей
в связи невелика. При значительной доле ковалентности расчетные величины,
характеризующие комплекс, не согласуются с опытными данными.
ТКП приложима лишь к комплексным соединениям, в которых комплексооб-
разователь (rf-элемент) имеет свободные электроны. Поэтому она не позволяет
изучать не только все производные непереходных элементов, но и многие комп-
комплексы переходных металлов (Sc3+, Ti4+, Nb5+ и др.). Кроме того, детально разби-
разбирая изменения центрального атома, ТКП вовсе не учитывает ни структурных
особенностей лигандов, ни склонности некоторых из них образовывать тг-связи.
Основным затруднением ТКП является то, что она не учитывает частично
ковалентный характер связи комплексообразователь — лиганд. Поэтому все эф-
эффекты, обусловленные ковалентным вкладом в связь, в простом методе ТКП
остаются необъясненными.
Усовершенствованная модель ТКП, в которой электростатическое взаимодей-
взаимодействие дополнено идеей перекрывания орбиталей, называется теорией поля лиган-
лигандов (ТПЛ). Она с успехом применяется к большому числу комплексов переход-
переходных металлов в обычных степенях окисления, где величины перекрывания элект-
электронных облаков не слишком велики. В тех же комплексах, где перекрывание
существенно, методы ТКП и ТПЛ непригодны. Для описания подобных комплек-
комплексов надо пользоваться ММО.
В ММО учитывается детальная электронная структура не только комплексо-
образователя, но и лигандов. Здесь комплекс рассматривается как единая кванто-
во-механическая система, в которой отдельные атомы и молекулы теряют свои
индивидуальные черты. Валентные электроны системы располагаются на много-
многоцентровых МО, охватывающих ядра комплексообразователя и всех лигандов,
входящих в состав комплекса. Движение каждого электрона определяется поло-
положением ядер и характером движения всех остальных электронов. Главные резуль-
результаты получены в рамках так называемого одноэлектронного приближения.
Рассмотрим октаэдрический комплекс переходного металла, лиганды которого
не проявляют склонности к образованию т-связей. Из шести АО лигандов и
девяти АО комплексообразователя возникают 15 МО. Для образования <т-связей
пригодны шесть орбиталей: d „, d~_ ,, s, р , р р , так как их ветви лежат вдоль
прямых, соединяющих атомы металла и лиганда. Остальные три орбитали (d ,
v ху'
< 119
d , d ) являются несвязывающими, так как их электронные облака направлены
между лигандами, и энергия этих орбиталей практически не меняется. Если
использовать изображение орбиталей, принятое в МВС, и дополнить его изоб-
изображением разрыхляющих орбиталей, схемы комплексов [Co(NH3N]3+ и [CoF6]3"
будут иметь вид
d d d
axy uyz axi
HJtumt
HMO
ttz2 <
[ 9
Z 8
Px
Py
Рг
PMO
С MO
d d d
axy ayi axz
\\\ \\ \
HMO
\
w
лх2-у
\
if
T 1
2 S
i'.
Px
it
Py
it
it
PMO
CMO
В первом комплексе шесть электронов занимают три НМО иона Со3+. Шесть
СМО, соответствующие (^«^-гибридизации, заселяются электронами лигандов
(по два от каждого). И наконец, шесть РМО в диамагнитном комплексе
[Со(МНз)б]3+ остаются вакантными. Для высокоспинового комплекса [CoFg]3" в
МВС постулировалась внешнеорбитальная «рЗ^-гибридизация, так как все 3rf-
орбитали заселены. В ММО нет необходимости использовать 4</-орбитали, так
как электроны комплексообразователя имеют возможность расположиться на
РМО.
На рис. 68 представлена энергетическая диаграмма октаэдрического комплек-
комплекса. Такая диаграмма объясняет магнитные и оптические свойства, а также целый
ряд других особенностей комплексных соединений. Как видно из рис. 68, карти-
картина комплекса в ММО значительно полнее, чем в ТКП. Так, для октаэдрического
комплекса ММО учитывает 15 МО, в то время как ТКП оперирует только вкла-
вкладом пяти (/-орбиталей. rfe-Орбиталям ТКП на энергетической диаграмме ММО
отвечают трижды вырожденные НМО, а ^7~°рбиталям — дважды вырожденные
РМО. В ММО энергетическое расщепление происходит в результате образования
ковалентной связи и перекрывания орбиталей центрального атома и лигандов.
Чем больше перекрывание электронных облаков, тем больше значение энергии
расщепления Д. Так же как и в ТКП, в ММО распределение электронов по
орбиталям de и dy рассматривается в зависимости от соотношения энергий рас-
расщепления и спаривания. Энергия связи может быть оценена на основании вели-
величин энергий отрыва электронов с соответствующей СМО. Выигрыш в энергии,
например, для [Со(МНз)б]3+ выше, чем для [CoFe]3", так как в первом случае
на СМО содержится 12 некомпенсированных электронов, а во втором — только
10.
Если электрон, поглощая квант света, переходит с rfe-уровня на df, возникают
полосы поглощения, называемые в ТКП d — rf-спектрами. Спектры переноса
заряда в ММО интерпретируются как результат поглощения световой энергии,
связанного с переносом электрона со СМО, локализованных у лигандов, на НМО
или РМО комплексообразователя. Эти полосы обычно располагаются в ультра-
ультрафиолетовой части спектра и характеризуются большой интенсивностью. Для
120
> PMO
АО
коыплексообраэо&ателя
Р и с. 68. Диаграмма энергетических уровней октаэдрического комплекса [Со(МНз)б]3+
комплексообразователей rf-подгрупп Периодической системы с ростом порядково-
порядкового номера максимум полосы поглощения смещается в коротковолновую сторону.
Это находится в соответствии с ростом устойчивости высших степеней окисления
для d-элементов системы сверху вниз.
Важная особенность ММО — возможность учета эффекта тг-связывания и
упрочняющего влияния его на состояние комплекса. Взаимодействие не занятых
в G-связях тг-орбиталей лигандов и несвязывающих rfe-орбиталей комплексообра-
зователя ведет к росту параметра расщепления Д. Часто эффективное тг-связыва-
ние настолько увеличивает Д, что становится причиной образования низкоспино-
низкоспиновых комплексов. Таким образом, согласно ММО, рост энергии расщепления
обусловлен дополнительным упрочнением ковалентной связи за счет тг-связыва-
ния, а не увеличением силы кристаллического поля, как это утверждает ТКП.
Г Л А В А VI. УЧЕНИЕ О ХИМИЧЕСКИХ ПРОЦЕССАХ
Изучение характера протекания и динамики химического процесса следует
считать одной из основных задач химии, поскольку ее успешное решение позво-
позволяет найти оптимальные пути создания новых материалов с заданными, "запро-
"запрограммированными", свойствами. Это имеет первостепенное значение не только
для развития и углубления основных представлений химии как науки, но и для
непосредственного практического использования ее достижений.
1. Понятие о химической термодинамике. К изучению химических процессов
следует подходить через ряд последовательных приближений. На первом этапе
целесообразно рассмотреть лишь начальное и конечное состояния взаимодейству-
взаимодействующих веществ, не учитывая путь, по которому протекает процесс, и развитие
процесса во времени. В этом и заключается термодинамический' подход. Химиче-
Химическая термодинамика использует законы общей термодинамики для исследования
химических и физико-химических процессов. Она включает термохимию, учение
о химическом равновесии, жидких и твердых растворах, фазовых переходах и о
возможности самопроизвольного протекания процессов.
121
I 1
Для удобства изучения необходимо изолировать объекты исследования от
окружающего пространства. Такая совокупность тел, выделенная из пространст-
пространства, образует систему. Если в системе возможен массо- и теплообмен между всеми
ее составными частями, то такая система называется термодинамической. Хими-
Химическая система, в которой возможно протекание реакций, представляет собой
частный случай термодинамической. Если между системой и окружающей средой
отсутствует массо- и теплообмен, то такая система называется изолированной.
Если отсутствует массообмен, но возможен теплообмен, то система называется
закрытой. Если же между системой и окружающей средой возможен и массо-, и
теплообмен, то система открытая. Система, состоящая из нескольких фаз, назы-
называется гетерогенной, однофазная система — гомогенной. Реакции, протекающие в
гомогенной системе, развиваются во всем ее объеме и называются гомогенными.
Реакции, происходящие на границе раздела фаз, называются гетерогенными.
Состояние системы определяется ее свойствами. Свойства подразделяются на
интенсивные (температура, давление, концентрация и др.), которыми характери-
характеризуется каждая точка системы, и экстенсивные, зависящие от количества вещества
или массы. К последним относятся, например, объем, энергия, значения которых
в заданной точке системы не имеют смысла. И для интенсивных, и для экстен-
экстенсивных свойств в размерности физических величин не входит время. Объясняет-
Объясняется это тем, что термодинамика изучает равновесные процессы. Состояние системы
аналитически можно представить в виде так называемого уравнения состояния
f(P, V,T) = 0,
связывающего все параметры системы. Конкретный вид уравнения состояния
известен лишь для ограниченного числа наиболее простых объектов. Например,
уравнение Клапейрона—Менделеева (II.1) является уравнением состояния идеаль-
идеального газа.
Поскольку для большинства систем уравнение состояния в явном виде неиз-
неизвестно, для термодинамического описания системы пользуются так называемыми
функциями состояния. Под последними понимают любую' физическую величину,
значения которой однозначно определяются термодинамическими свойствами
системы. Особую роль играют функции состояния, с помощью которых можно в
явном виде выразить все термодинамические свойства системы. Такие функции
состояния называются характеристическими.
Одна из основных фундаментальных функций состояния — полная энергия
системы Е, которая представляет собой сумму трех составляющих: кинетической
энергии -Ё^ин движущейся системы, потенциальной энергии E^q^, обусловленной
воздействием на систему внешних силовых полей, и внутренней энергии системы
U:
Е — Е
К1ЛН
При термодинамическом описании предполагают, что система находится в
состоянии относительного покоя (#кин = 0) и воздействие внешних полей прене-
пренебрежимо мало (.Енот = 0)- Тогда полная энергия системы определяется запасом ее
внутренней энергии (Е — Щ. Количественный учет всех составляющих внутрен-
внутренней энергии невозможен, но для термодинамического анализа систем в этом нет
необходимости, так как достаточно знать лишь изменение внутренней энергии
при переходе системы из одного состояния в другое, а
не ее абсолютные значения в этих состояниях. В соот-
соответствии с законом сохранения энергии, выражающим
первое начало термодинамики, общий запас внутренней
энергии остается постоянным, если отсутствует тепло-
тепловой обмен с окружающей средой. В ходе процессов,
протекающих в изолированной системе, возможно лишь
перераспределение внутренней энергии между отдель-
отдельными составляющими системы.
Сообщенная системе теплота расходуется на прира- Р и с . 69. К пояснению физи-
щение внутренней энергии и на совершение работы ческого смысла работы расши-
против внешних сил: рения
S
\р
h
Q= AU+ А.
(VI.1)
Уравнение (VI. 1) представляет собой математическое выражение первого начала
термодинамики •— закона сохранения энергии. Для наглядного представления
физического смысла работы против внешних сил рассмотрим систему, представ-
представляющую собой газ, заключенный в цилиндр, который отделен от внешней среды
перемещающимся без трения поршнем (рис. 69). Если поршень закреплен непод-
неподвижно (V = const), то сообщенная системе теплота полностью идет На увеличение
запаса внутренней энергии
Qv = A U,
(VI.2)
поскольку работа расширения при V = const равна нулю. Если теперь дать воз-
возможность поршню свободно перемещаться, то газ, расширяясь, совершит работу
А = Fh = pSh,
где F — сила, действующая на поршень; h — высота перемещения поршня; р —
давление; S — площадь поршня. Так как Sh = V — изменение объема, то
А = PAV= p(V2- Vx).
Это соответствует условию постоянства давления или изобарному условию (р =
— const). Тогда
Qp= AU+P(V2- Vx).
Если через Ui и G2 обозначить запас внутренней энергии в исходном и конечном
состояниях, то Д{/.= Ц2 — U\. Тогда
- К,)
или
Введем новую функцию
U+pV=H,
(VI.3)
123
которая больше величины внутренней энергии на величину работы расширения.
Эта функция называется энтальпией. Энтальпия, как и внутренняя энергия,
является функцией состояния. Таким образом, при условии постоянства давле-
давления подводимая к системе теплота Qp идет на увеличение энтальпии системы
ДЯ:
Qp= Н2-Щ = АН. (VIA)
Из сравнения выражений (VI.2) и (VI.4) следует, что энтальпия как функция
состояния эквивалентна внутренней энергии при термодинамическом описании
процессов, протекающих при постоянном давлении.
2. Экзо- и эндотермические реакции. Основы термохимии. Химическое взаи-
взаимодействие, как правило, сопровождается тепловым эффектом. При этом теплота
может как выделяться, так и поглощаться. Процессы, протекающие с выделением
теплоты, называются экзотермическими, а идущие с поглощением теплоты —
эндотермическими. Уравнения реакций, учитывающие тепловые эффекты, назы-
называются термохимическими, например
2Н2(г) + О2(г) ^=* 2Н2О(г) + 476 кДж.
Тепловой эффект реакции необходимо характеризовать не только абсолютной
величиной, но и знаком. Исторически сложились две системы отсчета: термохи-
термохимическая и термодинамическая. В первой из них знак теплового эффекта экзо-
экзотермической реакции считается положительным (экзо — наружу). Эндотермиче-
Эндотермические реакции сопровождаются отрицательным тепловым эффектом, теплота пог-
поглощается системой (эндо — внутрь). В термодинамике принята обратная система
знаков, т.е. теплота, поглощенная системой, считается положительной, а теплота,
отданная системой в окружающую среду, — отрицательной. В термодинамической
системе знаков тепловой эффект реакции отождествляется с изменением энталь-
энтальпии системы (если процесс протекает при постоянном давлении). При записи
термохимического уравнения в этой системе тепловой эффект не включается в
уравнение, а записывается рядом. Так, приведенное уравнение реакции получе-
получения воды из простых веществ запишется следующим образом
2Н2(г) + О2(г)
2 Н2О(г), ДЯ = -476 кДж.
В дальнейшем будем придерживаться термодинамической системы знаков и
полагать, что тепловой эффект эндотермической реакции связан с возрастанием
энтальпии системы (ДЯ > 0), а экзотермической — с ее уменьшением (ДЯ < 0).
Обычно химические реакции протекают при постоянном давлении (в открытых
системах). Однако иногда необходимо проводить реакции в закрьггых герметизи-
герметизированных аппаратах, когда соблюдается условие постоянства объема. В этом
случае, согласно (VI.2), тепловой эффект реакции связывается с изменением
внутренней энергии системы. При рассмотрении энергетического баланса химиче-
химических процессов в изобарных условиях тепловой эффект реакции определяется
изменением энтальпии (VI.4), т.е. разницей энтальпий конечного и исходного
состояний. Рассмотрим обратимую реакцию:
А + В
прямая
обратная
АВ, ДН < 0.
124
Прямая реакция идет с уменьшением энталь-
энтальпии и представляет собой экзотермический
процесс (рис. 70, а). Обратная реакция проте-
протекает с увеличением энтальпии и является _^
эндотермическим процессом (рис. 70, б). Теп-
Теплота, выделяющаяся при образовании вещест-
вещества, равна теплоте, поглощаемой при разложе-
разложении такого же его количества на исходные
составные части. Это положение следует
рассматривать как частный случай закона
сохранения материи и энергии. Оно называет-
называется законом Лавуазье—Лапласа.
"на
+АН
"кт
пК1Н
"на
д
Рис. 70. Изменение энтальпии
системы: а-экзотермический про-
процесс (ДЯ < 0); б-эндотермичес-
кий процесс (ДЯ > О)
Более глубокие обобщения термохимических закономерностей дает основной
закон термохимии, сформулированный Г.И.Гессом A840): тепловой эффект
химических реакций, протекающих или при постоянном давлении, или при по-
постоянном объеме, не зависит от числа промежуточных стадий, а определяется
лишь начальным и конечным состояниями системы.
Закон Гесса можно проиллюстрировать схемой
а + в=Хв (дя)
А Г-*1АВ
Здесь образование соединения АВ представлено двумя путями: непосредственным
синтезом из компонентов (ДЯ) или через стадию образования промежуточного
соединения AC (ДЯ1), которое, реагируя с В (ДЯг), дает тот же конечный про-
продукт. В соответствии с законом Гесса тепловой эффект прямого синтеза АВ равен
сумме тепловых эффектов реакций с участием промежуточного продукта АС, т.е.
ДЯ = ДЯ1 + ДЯг- Как следует из закона Гесса, тепловой эффект реакции обра-
образования одного моля соединения из простых веществ в стандартном состоянии
при заданных Тир — теплота образования — не зависит от способа его пблуче-
ния. В термодинамике в качестве стандартных условий принимаются температура
25°С = 298 К и давление 1,013-105 Па. Теплоты образования соединений в этих
условиях называются стандартными теплотами образования (ДЯ^2д8)* и приво-
приводятся в таблицах термодинамических величин. В качестве примера рассмотрим
гидриды sp-элементов V группы. Энтальпия образования гидридов элементов VA
группы:
Соединение NH3(r) РН3(г)
ДЯд298 -46,15 +12,96
AsH3(r) SbH3(r)
+66,38 +145,0
В этом ряду образование аммиака сопровождается экзотермическим эффектом
(ДД < 0). Три следующих гидрида образованы" в результате эндотермической
*Индекс / происходит от англ. formation — образование.
1
реакции (ДЯ > 0), причем величина эндотермического эффекта увеличивается в
пределах группы сверху вниз. Таким образом, Газообразный аммиак является
устойчивым соединением, в то время как РНз и AsHs неустойчивы (нестабильны),
a SbHs разлагается в момент получения и теплота его образования может быть
вычислена лишь косвенным путем.
Рассмотрим еще один пример применения закона Гесса для расчета энергии
кристаллической решетки. Энергия кристаллической решетки — это энергия,
которую надо затратить для разрушения кристалла по гетеролитическому меха-
механизму. Образование 1 моль кристаллического хлорида натрия может быть пред-
представлено двумя путями:
I. Непосредственный синтез из элементов:
Na(K) + Чг С12(г) = NaCl(K) (ДЯ[)
ДЯ] = —411,7 к.Дж/моль — стандартная теплота образования NaCl.
II. Гипотетический многостадийный процесс, приводящий к образованию
кристалла из газообразных ионов:
а) атомизация натрия
Na(K) ^=^ Na(r) (ДЯ2)
ДЯг = 107,8 кДж/моль — стандартная теплота возгонки металлического нат-
натрия;
б) атомизация хлора
Чг С12(г) *=^ С1(г) (ДЯ3)
ДЯз = 120,8 кДж/моль — стандартная теплота атомизации газообразного
хлора;
в) ионизация газообразного натрия
Na(r) *
Na+(r) + е" (Г)
I = 501,6 кДж/моль — энергия ионизации газообразного натрия;
г) ионизация газообразного хлора
+ С1(г)
(Е)
Е = —369,8 кДж/моль — энергия сродства к электрону для хлора;
д) взаимодействие ионов в газе с одновременной конденсацией
Na*(r)
= NaCl(K) ( U)
U — искомая энергия кристаллической решетки.
Схематически этот круговой процесс можно представить следующим образом
(цикл Борна—Габера):
126 •
. ДН„, 4Н,
Маю, та>(г) «то»«8«цм
ковдевс*цвя
N«(D, Cl (r)
Na'r),
На основании закона Гесса можно записать
A Hi = АН2 + АН3 + /+ Е + I/,
-411,7 = 107,8 + 120,8 + 501,6 - 369,5 + U.
Отсюда U = —772,4 кДж/моль. Большая отрицательная величина энергии крис-
кристаллической решетки хлорида натрия указывает на экзотермичность процесса
образования и значительную стабильность кристаллического NaCl.
3. Направление химических процессов. Энтропия. Свободная энергия. Закон
сохранения энергии и следствия из него позволяют представить энергетический
баланс реакций, однако они не указывают, в каком направлении должен самопро-
самопроизвольно (без действия внешних сил) протекать процесс. Функции состояния АН
и Д U также не дают возможности судить о направлении процесса. Они указыва-
указывают лишь, что энергия изолированной системы постоянна.
Одной из наиболее известных попыток воспользоваться первым началом тер-
термодинамики для оценки направленности процессов является так называемый
принцип Бертло A867), согласно которому химические реакции самопроизвольно
протекают в сторону выделения теплоты (по современной терминологии, в сторо-
сторону уменьшения энтальпии, ДЯ < 0).
Для суждения о направлении процесса необходимо ввести новую функцию
состояния. Физический смысл этой новой функции можно представить на приме-
примере плавления индивидуального кристаллического вещества. Плавление
происходит при постоянной температуре и сопровождается поглощением так
называемой скрытой теплоты плавления Д#пл (энтальпии плавления).
Поглощение теплоты должно было бы способствовать увеличению внутренней
энергии системы, что выразилось бы в увеличении ее температуры. Однако это не
наблюдается. Следовательно, в процессе плавления действует другой фактор,
способствующий сохранению постоянной температуры. Аналитически это
положение можно представить в виде
= TnjlAS}
Jroi>
где Д5пл — величина, характеризующая какой-то процесс в системе, на который
расходуется поглощаемая системой теплота, Дж/(моль-К). Так как энтальпия
плавления является функцией состояния, то и величина Д5пл должна быть функ-
функцией состояния.
При плавлении кристалла происходят разрушение упорядоченной кристалли-
кристаллической структуры и образование расплава, который характеризуется неупорядо-
неупорядоченным расположением частиц, т.е. наблюдается увеличение беспорядка в систе-
127
ме. Таким же образом происходит увеличение беспорядка при процессах возгон-
возгонки, испарения, диссоциации и т.д. Критерием направленности процесса может
служить степень неупорядоченности системы. Мерой этой неупорядоченности
является функция 5, которую называют энтропией.
В соответствии со смыслом функции 5 можно дать следующее определение: в
изолированной системе самопроизвольные процессы протекают в сторону увели-
увеличения энтропии. Таким образом, если в результате процесса AS > О, то процесс
термодинамически возможен, если же AS < О, то его самопроизвольное протека-
протекание в изолированной системе исключается. Приведенная формулировка составля-
составляет смысл второго начала термодинамики для изолированных систем.
В неизолированных системах возможны процессы, сопровождающиеся умень-
уменьшением энтропии (например, кристаллизация расплава, конденсация пара и т.п.,
протекающие в условиях отвода теплоты в окружающую среду). Целесообразно
ввести такую новую функцию состояния, которая учитывала бы совместное влия-
влияние энтальпийного и энтропийного факторов. Такая функция представляет собой
разность
AG= AH-TAS. (VI.5)
Функция G называется свободной энергией Гиббса и при независимых перемен-
переменных Тар является характеристической функцией. Она является мерой устойчи-
устойчивости системы при постоянном давлении. При условии постоянства объема со-
состояние системы может быть описано аналогичной характеристической функцией,
которая называется свободной энергией Гельмгольца:
AF= AU- TAS. (VI.6)
Соотношение между AG и AF такое же, как между АН и Д U, т.е. они различа-
различаются на величину работы расширения:
AG- AF= pAV.
Судить о возможности самопроизвольного протекания процесса можно по
знаку изменения функции свободной энергии: если Д G < О, т.е. в процессе вза-
взаимодействия происходит уменьшение свободной энергии, то процесс термодина-
термодинамически возможен. Если AG > О, то протекание процесса невозможно. Таким
образом, все процессы могут самопроизвольно протекать в сторону уменьшения
свободной энергии. Эта формулировка справедлива как для изолированных, так и
для открытых систем. Самопроизвольное осуществление реакции (Д G < 0) воз-
возможно при следующих условиях:
1) АН < 0 (экзотермический процесс) и в то же время |Д#| > |ГД5|, т.е.
при экзотермических процессах знаки ДЯ и AG в выражении (VI.5) совпадают,
что означает возможность протекания реакции независимо от знака AS;
2) ДЯ > 0 (эндотермический процесс) и |ДЯ| < TAS, тогда возрастание
энтальпии компенсируется значительно большим ростом энтропийного члена, что
осуществимо при высоких температурах или при реакциях с участием газовой
фазы, когда наблюдаются значительные изменения энтропии. Этим и объясняет-
объясняется возможность протекания эндотермических реакций, что не согласуется с прин-
128 V
ципом Бертло. Судить о направлении процесса по знаку изменения энтальпии в
соответствии с этим принципом можно лишь: а) при низких температурах (при
Т ¦* О, TAS -» 0 и TAS < АН), когда знаки изменения свободной энергии и
энтальпии совпадают; 6) в конденсированных системах, в которых в процессе
взаимодействия энтропия меняется незначительно (беспорядок не может
существенно возрасти, если, например, одно кристаллическое вещество
превращается в другое кристаллическое вещество). Поэтому при низких
температурах и в конденсированных системах возможно протекание лишь
экзотермических реакций (AG < 0, когда АН < 0).
Самопроизвольное протекание процесса невозможно, если АН > 0 и |Д#| >
> | TAS\. Тогда AG = АН — TAS > 0. Положительное значение AG определяет
принципиальную невозможность самопроизвольного осуществления процесса,
поскольку это противоречило бы первому и второму началам термодинамики. В
то же время отрицательный знак AG свидетельствует лишь о возможности проте-
протекания данного процесса. Будет ли наблюдаться процесс в действительности,
зависит от конкретных условий и кинетических факторов.
4. Понятие о химической кинетике. Скорость химических реакций. С термоди-
термодинамических позиций невозможно анализировать развитие процесса во времени,
поскольку время (как переменная) не учитывается при термодинамическом описа-
описании. Поэтому вторым этапом в изучении закономерностей протекания химических
процессов является рассмотрение их развития во времени, что представляет собой
основную задачу химической кинетики. В реальных условиях протекание химиче-
химических реакций связано с преодолением энергетических барьеров, которые иногда
могут быть весьма значительными. Именно поэтому термодинамическая возмож-
возможность осуществления данной реакции (AG < 0) является необходимым, но недо-
недостаточным условием реализации процесса в действительности. Химическая кине-
кинетика кроме выяснения особенностей развития процесса во времени (формально-
кинетическое описание) изучает также механизм взаимодействия реагентов на
атомно-молекулярном уровне (молекулярно-кинетическое описание). Оба метода
описания кинетических закономерностей взаимно дополняют друг друга.
Исходным и основополагающим понятием в химической кинетике является
понятие о скорости химической реакции. Скорость химической реакции — это
количество элементарных актов взаимодействия в единицу времени. Так как при
взаимодействии изменяются концентрации реагирующих веществ, то скорость
реакции обычно измеряют изменением концентрации реагентов или продуктов
реакции в единицу времени. При этом нет необходимости следить за изменением
концентрации всех веществ, участвующих в реакции, поскольку ее стехиометриче-
ское уравнение устанавливает соотношение между концентрациями реагентов.
Рассмотрим уравнение гомогенной реакции общего вида А + В = АВ. Пусть
эта реакция протекает в изобарно-изотермических условиях (р — const, T =
= const). Пусть концентрация (моль/л) вещества А в момент времени /0 равна с0.
По истечении времени в некоторый момент /j (<j > <0) концентрация реагента А
уменьшается за счет образования продукта АВ и равна с\ {с\ < Со). За проме-
промежуток времени At == Ц — t0 изменение концентрации реагента А равно —Дс =
= С1 — с0 и в соответствии с определением средняя скорость изменения
концентрации А выразится уравнением
Дс
- _ q - со _ а
\~ ti-t0 - -кг: , <VL7)
. . ' 129
a.
7
Рис. 71. Зависимость концентрации реагирующих веществ от времени для ли-
линейного (а) и нелинейного (б) процессов *
Знак "—" в выражении (VI.7) свидетельствует о том, что в ходе реакции
концентрация вещества А уменьшается. Аналогичное выражение для скорости
изменения концентрации продукта реакции АВ имеет вид
т.е. концентрация продукта реакции со временем возрастает.
Концентрация вещества А в зависимости от химической природы
взаимодействия может изменяться со временем как линейно, так и в соответствии
с более сложными закономерностями (параболическими, гиперболическими,
экспоненциальными и т.п.). Для линейной зависимости средняя скорость
однозначно характеризует течение процесса (рис. 71, а), поскольку она постоянна
и определяется тангенсом угла наклона прямой к оси времени (Ас /At = tga).
А
Для нелинейной зависимости концентрации от времени (рис. 71, б) средняя
скорость является грубым приближением, поскольку в интервале времени Ц — t0
она не остается постоянной. Чем меньше интервал времени At, тем ближе
значение средней скорости процесса к ее истинному значению в момент времени
t. В пределе при At -> 0 получим
v = lim (-Ac /At) = -(dc /dt).
А д^о А А
(VI.8)
Таким образом, истинная скорость v равна первой производной от концентрации
по времени и определяется тангенсом угла наклона касательной к кривой с =
• А
= Д<) в точке, соответствующей данному моменту времени (dc /dt — tga]).
5. Основной закон химической кинетики. Взаимодействие между частицами
возможно лишь в момент их столкновения. Вследствие этого скорость реакции
пропорциональна вероятности столкновений. А вероятность столкновений частиц
определяется их концентрацией. Пусть вероятность w нахождения частицы А в
некоторой точке пространства определяется выражением '
w = <pc (VI.9)
A A A
где с — концентрация вещества А; <р — коэффициент пропорциональности, а
А А
вероятность нахождения частицы В в той же точке пространства равна
w — ф с .
В ГВ В
Тогда вероятность одновременного нахождения А и В в данной точке
пространства — вероятность их столкновения — определяется произведением
частных вероятностей w им:
W = 11} И) = ip СС,
АВ А В ГАВ А В
где if — if ip . Так как скорость реакции пропорциональна вероятности
столкновений, то можно записать
v = А то =А<? с с ,
АВ ГАВ А В
где Л — коэффициент пропорциональности. Объединяя коэффициенты
пропорциональности \<р —к, получаем
v= kc с , (VI.10)
А в' v '
т.е. истинная скорость химической реакции пропорциональна произведению
концентраций реагирующих веществ. Этот основной закон химической кинетики
называется законоль действующих масс.
Коэффициент пропорциональности к называется константой скорости. Смысл
ее становится ясным, если принять с = с = 1 моль/л, тогда v — к. Константа
A D
скорости — это скорость реакции при концентрациях реагирующих веществ,
равных единице. Таким образом, константу скорости можно рассматривать как
удельную скорость. По определению, константа скорости не зависит от
концентрации реагирующих веществ.
6. Параллельные, последовательные, сопряженные и цепные реакции.
Реальные химические процессы лишь редко могут быть описаны простым
механизмом и, как правило, представляют собой сложные реакции, в которых
помимо молекул могут участвовать и такие нестойкие промежуточные
образования, как ионы, свободные радикалы, активные комплексы и т.п.
Сложные реакции подразделяются на параллельные, последовательные и
сопряженные. Особо следует выделить цепные реакции, которые можно
рассматривать как комбинацию трех основных типов.
Параллельными реакциями называют связанную систему реакций, имеющих
одни и те же исходные вещества, но различные продукты реакции. Например,
термическое разложение хлората калия может одновременно протекать в двух
направлениях:
¦+4КС1 + 6О2
4 КСЮз
•+ ЗКС1О 4 + KCL
В каждой реакции в единичном акте разложения участвует одна формульная
единица КСЮз. Скорости этих параллельных реакций определяются выраже-
выражениями
' "' = *1Сксю3'
Суммарная скорость процесса равна
= *2С
ксю3-
т.е. скорость системы параллельных реакций равна сумме скоростей отдельных
стадий.
Последовательными реакциями называют связанную систему реакций, в кото-
которых продукты предыдущих стадий являются исходными веществами для последу-
последующих. Рассмотрим наиболее простой случай двух последовательных реакций,
которые в общем виде могут быть представлены схемами
В (I); В
С (И).
Продукт В, образующийся по первой стадии, является промежуточным. Ско-
Скорость расходования вещества А определяется скоростью реакции (I), и его коли-
количество непрерывно убывает со временем:
Скорость накопления вещества В в соответствии с реакцией (I)
v - +(dc /dt) = kxc .
IB v в' ' В
Однако это вещество одновременно расходуется в соответствии с реакцией (II):
Так как вещество В одновременно участвует в двух реакциях — в одной как
продукт, а в другой как исходное вещество, — то общее изменение его концентра-
концентрации определяется разностью скоростей его накопления и расходования:
Накопление конечного продукта С определяется общей концентрацией В,
которая сложным образом меняется со временем. Поскольку обычно различные
стадии серии последовательных реакций протекают с различными скоростями,
общая скорость этого сложного процесса (скорость превращения А в С) опреде-
определяется стадией, протекающей с наименьшей скоростью. Эта стадия процесса
называется контролирующей или лимитирующей.
Сложное химическое взаимодействие возможно и тогда, когда протекание
одной химической реакции вызывает (индуцирует) протекание другой реакции в
той же системе, которая в отсутствие первой не протекает. Это явление называет-
ся химической индукцией. Например, непосредственного взаимодействия иодо-
водородной кислоты с хромовой не происходит. Однако при введении в систему
оксида железа (+2) наряду с его окислением по уравнению
6FeO + 2Н2Сг04 = 3Fe2O3 + Сг2О3 + 2Н2О
одновременно происходит окисление HI:
12HI + 2Н2Сг04 = 312 + 2Сг13 + 8Н2О
Две реакции, одна из которых индуг^ирует протекание другой, называются
сопряжеппыми. С формальной точки зрения систему сопряженных реакций можно
рассматривать как частный случай параллельных реакций, поскольку один из
исходных компонентов — общий для обоих процессов (в приведенном примере
Н2СЮ4). Однако сопряженные реакции могут протекать только в сложных систе-
системах, когда взаимодействие осуществляется через ряд последовательных промежу-
промежуточных стадий.
Детальное рассмотрение химических процессов с молекулярно-кинетической
точки зрения показывает, что многие из них протекают по так называемому
радикально-цепному механизму. Особенность цепных реакций заключается в
образовании на промежуточных этапах свободных радикалов — нестабильных
фрагментов молекул с малым временем жизни, имеющих свободные связи: •СНз,
•С2Н5, C1-, НО^, N: и т.п. Связанная система сложных реакций, протекающих
последовательно, параллельно и сопряженно с участием свободных радикалов,
называется цепной реакцией. Основные стадии цепных реакций — зарождение
цепи, продолжение цепи, разветвление цепи и обрыв цепи.
Зарождение цепи — стадия цепной реакции, в результате которой возникают
свободные радикалы из валентно-насыщенных молекул. Эта стадия осуществля-
осуществляется разными путями. Так, при синтезе хлороводорода из водорода и хлора
образование радикалов осуществляется за счет разрыва связи С1—С1 под воз-
воздействием кванта света: С12 + hv —> С1- + С1-. Образование свободных радика-
радикалов можно инициировать введением посторонних веществ, обладающих специфи-
специфическим действием (инициаторов). В качестве инициаторов часто используют
малостабильные пероксидные и гидропероксидные соединения.
Продолжение цепи — стадия цепной реакции, протекающая с сохранением
общего числа свободных связей. В процессе синтеза НС1 продолжение цепи
обеспечивается следующими элементарными стадиями:
Н2 + С1- —+ Н- + НС1
Н- + С12—> НС1 + С1- и т.д.
Разветвление цепи — элементарная стадия цепного процесса, протекающая с
увеличением числа свободных связей. Например, при окислении водорода это
может происходить за счет взаимодействия Н- + О2 = -ОН + -О- — вместо
одной свободной связи появляются три.
Обрыв цепи — элементарная стадия цепного процесса, приводящая к исчезно-
исчезновению свободных связей. Например, обрыв цепи может происходить в результате
взаимодействий:
Н- + СЬ = НС1
СЬ + стенка = С1 (адсорбирован)
133
11 i-l
Одним из наиболее важных понятий в теории цепных реакций является длина
цепи. Под длиной цепи понимают среднее число элементарных стадий продол-
продолжения цепи после возникновения свободного радикала до его исчезновения (об-
(обрыва цепи). Пусть вероятность обрыва цепи на данном звене равна а (а < 1).
Тогда вероятность ее продолжения равна 1 — а, а длина цепи представляется
соотношением вероятностей ее продолжения и обрыва: п — A — а)/а. Отношение
вероятностей продолжения и обрыва цепи равно отношению соответствующих
скоростей, т.е. п = %род/^обр- Длина цепи может составлять от нескольких десят-
ков до миллионов звеньев, как при синтезе НС1. Общая скорость неразветвленной
цепной реакции равна произведению скорости зарождения цопи Vq (количество
свободных радикалов, возникающих в единице объема в единицу времени) на
длину цепи:
v — von =
7. Зависимость скорости реакции от температуры. Энергия активации. Ско-
Скорость химических реакций сильно зависит от температуры. В соответствии с
эмпирическим правилом Вант-Гоффа A884) при повышении температуры на 10°
скорость большинства реакций возрастает в 2—4 раза. Отношение константы
скорости при температуре t + 10° к константе при температуре t называется
температурным коэффициентом скорости. Как следует из правила Вант-Гоффа,
1
На первый взгляд может показаться, что зависимость скорости реакции от
температуры обусловлена увеличением числа столкновений реагирующих частиц.
Однако, как показывает расчет, число столкновений с ростом температуры увели-
увеличивается незначительно * и не может сравниться со значением температурного
коэффициента реакции. С другой стороны, число столкновений частиц в 1 л
газовой смеси при нормальных условиях достигает порядка 1028 соударений в
секунду. Если бы каждое соударение приводило к химическому взаимодействию,
скорости реакций были бы огромны. Так, например, реакция синтеза иодоводо-
рода (одна из наиболее изученных простых реакций) заканчивалась бы за время
порядка 100 с. В действительности же скорость взаимодействия молекул Нг и 1г
в 1014 раз меньше. Отсюда можно сделать вывод, что далеко не каждое соударе-
соударение приводит к химическому взаимодействию. Для осуществления взаимодейст-
взаимодействия молекулы должны обладать определенным запасом энергии. Энергия Еа,
представляющая собой минимальную энергию, достаточную для осуществления
акта химического взаимодействия, называется энергией активации. Большинство
молекул в данных условиях этой энергией не обладает.
Согласно кинетической теории газов, средняя энергия молекулы при темпера-
температуре Т равна Еср = s/^kT). В то же время энергия распределена между отдель-
отдельными молекулами неравномерно. Это обусловлено передачей энергии от одних
молекул другим при их столкновениях в процессе хаотического движения. При
данной температуре энергия отдельных молекул в системе распределена по опре-
определенному закону, который называется распределением Максвелла—Больцмана..
*При увеличении температуры на 10° число столкновений растет в среднем всего лишь
на 1—2%.
134
Рис. 72. Распределение молекул в системе
по энергиям при разных температурах
На рис. 72 представлено распределение
молекул в системе по энергиям при раз-
ных температурах. Координаты выбраны
таким образом, чтобы площадь фигуры
под кривой распределения была пропор-
пропорциональна общему числу частиц в систе-
системе. Площади под кривыми при Т\, Т2, Тз
равны друг другу, так как общее число
частиц в системе постоянно. Максимум
на каждой кривой отвечает наиболее
вероятной при данной температуре энер-
энергии частиц Е'. Этой энергией обладает наибольшее число частиц. Однако, как
следует из кривых распределения частиц по энергии, некоторая их доля
обладает энергией, значитачьно превышающей Е'. Доля частиц, обладающих
энергией Е ^ Еа, с ростом температуры сильно возрастает. На рис. 72 этому
отвечают заштрихованные площади под кривыми, соответствующие различным
температурам. Таким образом, скорость реакции определяется не общим числом
частиц, а числом частиц с достаточно высокой энергией (Е > Еа), так называемых
активных частиц. С повышением температуры резко возрастает именно число
активных частиц.
Все выводы относительно зависимости скорости реакции от числа соударений
не теряют смысла, гак как число активных столкновений пропорционачьно обще-
общему числу столкновений. Число активных частиц, а следовательно, и скорость
реакции возрастают с температурой по экспоненциальному закону:
или
* = Aexp[-Ea/(RT)]
Ink = In A - Eal(RT).
(VI.11)
(VI.12)
Уравнение (VI.11), отражающее зависимость константы скорости от температуры,
называется уравнением Аррениуса. Предэкспоненциальный множитель А фор-
формально определяет константу скорости при нулевой энергии активации (или при
бесконечной температуре). Действительно, зависимость (VI. 12) можно представить
в виде
\пк=В-с/Т,
(VI. 13)
где В — 1пЛ = const, с = Еа/R = const. Выражение (VI.13) представляет собой
уравнение прямой в координатах \пк — \/Т. Тангенс угла наклона этой кривой к
оси абсцисс определяет энергию активации: tga — Ea/R. В настоящее время
показано, что энергии активации химических реакций лежат в пределах 40 —
400 кДж/моль.
Фундаментальным представлением в теории элементарных химических процес-
процессов является понятие об активном комплексе. Схема химической реакции может
быть представлена в виде
исходные вещества
активные частицы +
+ инертные молекулы
продукты реакции
!
т,п
Координата реакции
Рис. 73. Энергетическая диаграмма
взаимодействия H2+I2 ^=^ 2HI: Е^-
энергия активации образования HI;
?а-энергия активации диссоциации
HI; E'-энергия активации, необходи-
необходимая для полной диссоциации Н2 и
12', Е -энергия активации диссоциации
HI с образованием атомов Н и I
При химическом взаимодействии, например,
Н2 и 12 должны разорваться связи Н—Н и I—I
и образоваться связи Н—I. В некоторый мо-
момент времени при сближении активных моле-
молекул компонентов, обладающих достаточной
энергией для преодоления взаимного оттал-
отталкивания электронных облаков, возникает
переходное состояние, когда одни связи не
полностью разорвались, а другие уже начали
формироваться. Такой возбужденный неста-
нестабильный ассоциат называется активным
комплексом. Образование активного комплек-
комплекса можно представить схемой
н-н
+
н-
I
• н
I
н
н
+ I
I I
На рис. 73 представлена энергетическая
диаграмма взаимодействия Н2 + Ь ^^ 2HI.
Разность энергий активации прямой и обрат-
обратной реакций равна изменению энтальпии
реакции: Е'а — Е"а — ДН. Точно так же разность "энергий активации" при пол-
полной диссоциации* дает изменение энтальпии реакции Е' — Е" = АН, что и
следовало ожидать на основании закона Гесса, так как тепловой эффект реакции
не зависит от пути процесса. С термодинамической точки зрения оба процесса
эквивалентны, однако кинетически более выгоден процесс, идущий с образовани-
образованием активного комплекса, так как он требует меньшей затраты энергии.
8. Обратимые химические реакции. Химическое равновесие. Большинство
химических реакций не протекает до конца. Реакции, которые могут одновремен-
одновременно протекать в прямом и обратном направлениях, называются обратимыми. В
реакции
h
Н2 + 12 . ' 2HI
в начальный момент времени концентрация HI равна нулю, а концентрации Н2 и
12 равны исходным (максимальны). В этих условиях реакция протекает слева
направо и скорость ее определяется выражением
v = к] с с .
Н2 12
В ходе реакции, которую будем называть прямым прогрессом, концентрации
исходных веществ непрерывно падают и в соответствии с этим скорость взаимо-
взаимодействия водорода с иодом уменьшается. Одновременно начинает возрастать
концентрация продукта прямой реакции (HI) и, следовательно, появляет-
*3десь это просто энергия диссоциации.
ся возможность протекания обратной реакции (справа налево). Скорость ее
равна
По мере увеличения концентрации HI скорость его диссоциации возрастает. На-
Наступит такой момент, когда скорости прямой и обратной реакций станут равны-
равными: v = v. Такое состояние системы, когда в ней протекают два противополож-
противоположно направленных химических прогресса с одинакового скоростью, называется состо-
состоянием химического равновесия.
Условие равенства скоростей прямого и обратного процессов для реакции
Н2 + Ь '==i 2HI можно записать в виде
откуда следует
к\с с = к2с2 ,
н2 i2 hi
%/%2 = с2 /(с с ).
11 1 т'К н2 \г
Соотношение констант скоростей к\/к2 является также константой: Xi/k2 = К =
= const. Тогда
К = с2 /(с с ).
HI/V Н2 I/
(VI.14)
Константа К, отражающая соотношение концентраций компонентов обратимой
реакции в состоянии химического равновесия, называется константой равно-
равновесия.
Константа равновесия — важнейшая характеристика химического взаимодейст-
взаимодействия. Ее величина позволяет судить о полноте протекания реакции. Для необрати-
необратимых реакций (протекающих до конца) К -» оо, поскольку равновесная концентра-
концентрация продуктов реакции намного превышает концентрацию исходных веществ.
Если к -> О, то это свидетельствует о практически полном отсутствии взаимодейст-
взаимодействия в прямом направлении.
Достижению химического равновесия часто препятствуют факторы, связанные
с особенностями кинетики процессов, т.е. преодолением активационных барьеров.
Значительная величина энергетического барьера может практически полностью
воспрепятствовать достижению химического равновесия, в результате чего можно
прийти к неправильному выводу о том, что система уже находится в состоянии
химического равновесия. Так, смесь Н2 + Ог при комнатной температуре может
неограниченно долго находиться в неизменном состоянии. Однако это состояние
не является равновесным, поскольку внешнее воздействие (например, нагревание)
приводит к мгновенному взаимодействию с образованием воды. Если эту систему
затем охладить до прежней температуры, то она не вернется в исходное состоя-
состояние, так как новое состояние является термодинамически более устойчивым.
Если состояние системы неизменно во времени, но при изменении внешних
условий в системе происходит необратимый процесс, то такое состояние назы-
называется ложным (заторможенным) равновесием. Особенно характерны ложные
равновесия для твердофазных систем, в которых осуществление химических
процессов сильно затруднено за счет малой подвижности частиц и высоких энер-
энергетических барьеров.
В отличие от ложного истинное химическое равновесие не только неизменно во
времени в отсутствие внешних воздействий, но и после воздействия система
может вернуться в прежнее состояние. Классическим примером истинного равно-
равновесия является равновесие в системе N2O4 '=i 2NC>2- При нагревании в замкнутом
объеме димер N2O4 диссоциирует на две молекулы NO2. При охлаждении же
система возвращается в исходное состояние: из двух молекул NO2 вновь образует-
образуется N2O4. Истинное химическое равновесие характеризуется минимальным значе-
значением свободной энергии (AG = 0). При ложном равновесии AG < 0 т.е. не ис-
исключена возможность протекания химического процесса.
9. Смещение химического равновесия. Принцип Ле Шателье. Состояние хими-
химического равновесия зависит от ряда факторов, основные из которых — температу-
температура, давление и концентрация реагентов. Изменение хотя бы одного из этих фак-
факторов приводит к смещению равновесия.
Рассмотрим влияние температуры на химическое равновесие. Из сравнения
выражений (VI.14) и (VI.11) следует
К =
k2
А2ехр[-Е"а/(Щ)]
= А ехр [-(Еа - E'a)/(RI)}.
Как видно из рис. 74, разность энергий активации прямой и обратной реак-
реакций равна изменению энтальпии в результате взаимодействия:^ - Е"а = ДЯ.
Тогда
или
К= A exp[-AH/(RT)]
\пК= \nA-AH/(RT).
(VI.15)
(VI.16)
Таким образом, изменение температуры влияет на значение константы равнове-
равновесия. Уравнение (VI.15) по форме аналогично уравнению Аррениуса, только вместо
константы скорости и энергии активации здесь используют равновесные характе-
характеристики: изменение энтальпии в результате реакции и константу равновесия.
Уравнение (VI. 15), отражающее зависимость константы равновесия от температу-
температуры, называется уравнением Вант-Гоффа*. Характер изменения константы равно-
равновесия от температуры определяется знаком изменения энтальпии АН. При
АН > 0 (эндотермический процесс) с ростом температуры константа равновесия
увеличивается. При АН < 0 (экзотермический процесс) увеличение температуры
приводит к уменьшению К. При повышении температуры в соответствии с
уравнениями (VI.11) и (VI.12) увеличиваются скорости и прямой и обратной
реакций. При этом константа равновесия может и возрастать и уменьшаться в
Так как в уравнении (VI. 15) используется энтальпия, то процесс рассматривается в
изобарных условиях. При V = const (изохорные условия) вместо ДЯ в уравнение Вант-
Гоффа, подставляют изменение внутренней энергии Д U. ,
Активный комплекс
Г\ *
1 ч
/ Продукты ,
/ реакции
• л
1 / *,
/
исходные
бещестда
Координата реакции
а
V
Активный
/
з /
i /
Исходные
Вещества
комплекс
\
\
\ *
\
\
\
Продукты
реакции
координата реакции
5
Рис. 74. Энергетическая диаграмма соотношения энергий
активации прямой и обратной реакций
зависимости от того, скорость какой реакции быстрее растет с температурой.
Если скорость прямой реакции сильнее зависит от температуры (ее энергия акти-
активации больше, чем для обратной), то, как следует из рис. 74, а, прямой процесс
эндотермичен и константа равновесия возрастает с температурой. Если же энер-
энергия активации обратной реакции выше (Е"а > Е'а , рис. 74, б), то прямой процесс
экзотермичен и константа равновесия с ростом температуры уменьшается. Напри-
Например, при повышении температуры равновесной системы Н2 + Ь ^^ 2HI константа
скорости прямой реакции Xj растет медленнее, чем величина к2 (для реакции
2HI -* Нг + Ь)- Поэтому константа равновесия обратимой реакции при нагревании
будет уменьшаться.
Построив график в координатах 1пА' — \/Т (рис. 75), получим возможность
количественной оценки энтальпии реакции на основании экспериментальных
данных по температурной зависимости константы равновесия [см. уравнение
(VI.16)]. Отрезок, отсекаемый прямой на оси ординат, равен 1пЛ. В первом приб-
приближении коэффициент пропорциональности А можно трактовать как константу
равновесия при бесконечно большой температуре.
Изменение концентрации компонентов равновесной химической системы при
постоянной температуре смещает равнове-
равновесие, однако константа равновесия при
этом остается неизменной. В самом деле,
если в систему, состоящую из Н2, h и HI,
в которой установилось химическое рав-
равновесие, определяемое константой равно-
равновесия ш
In К
Ш К
'/г
а
ввести некоторое количество водорода
Рис. 75. Определение энтальпии реакции по
тангенсу угла наклона прямой:
(или иода), то должно образоваться а - эндотермический процесс; б - экзотерми-
дсгполнительное количество HI, посколь-
ческий процесс
ку константа равновесия (при Т = const) — величина постоянная. При этом рав-
равновесие сместится в сторону увеличения продукта (HI). При увеличении концент-
концентрации иодоводорода равновесие смещается в сторону возрастания концентраций
исходных веществ. Таким образом, изменяя концентрации компонентов, можно
сместить равновесие в нужную сторону, увеличив выход продукта реакции и
степень использования дефицитных реагентов.
Влияние различных факторов на состояние химического равновесия качест-
качественно описывается принципом смещения равновесия Ле Шателье A884). Согласно
этому принципу, при всяком внешнем воздействии на систему, находящуюся в
состоянии химического равновесия, в ней протекают процессы, приводящие к
уменьшению этого воздействия. Действительно, повышение температуры увеличи-
увеличивает выход продуктов эндотермических реакций, в ходе которых подводимая
извне теплота поглощается. Реакции, сопровождающиеся выделением теплоты,
протекают более полно при охлаждении. Аналогичным образом при увеличении
давления стимулируется реакция, сопровождающаяся уменьшением объема, так
как этот процесс способствует уменьшению влияния давления. Добавление в
реакционнуй смесь, находящуюся в равновесии, одного из компонентов благо-
припятствует протеканию той реакции, в ходе которой этот компонент расходует-
расходуется. Практическое использование принципа смещения равновесия можно показать
на примере синтеза аммиака. Эта реакция экзотермична и протекает с уменьше-
уменьшением объема:
ЗН2 + N2 *=± 2NHS (АН < 0).
Синтез аммиака идет тем полнее, чем ниже температура (ДЯ < 0). Однако при
низких температурах процесс достижения равновесия протекает очень медленно и
для его ускорения необходим подогрев. Нагревание же способствует уменьшению
выхода аммиака вследствие ускорения эндотермической реакции диссоциации
NH3. Однако, поскольку образование аммиака сопровождается уменьшением
объема, снижение выхода аммиака за счет нагревания можно компенсировать
увеличением давления в системе. Таким образом, одновременное повышение
температуры и увеличение давления ускоряет процесс достижения равновесия и
увеличивает выход аммиака (табл. 15).
Т а б л и ц а 15. Зависимость выхода аммиака от температуры и давления
Температура, °С
400
450
500
550
600
0,1
0,405
0,214
0,1216
0,0736
0,0470
Объемное содержание
10
25,37
16,10
14,87
6,82
4,53
аммиака
30
48,18
35,87
25,80
18,23
12,84
(%) при давлении, МПа
80
79,34
62,70
51,05
40,19
30,92
Из табл. 15 видно, что при р = const с ростом температуры выход аммиака
уменьшается, однако при Т — const с ростом давления содержание аммиака в
газовой смеси резко растет. ¦ -
ЛАП
Рис. 76. Кинетические кривые изменения
скорости (а) и накопления продуктов (б) ав-
автокаталитической реакции
Координата реакции
Рис. 77. Энергетическая диаграмма дейст-
действия катализатора
Принцип смещения равновесия применим только к системам, находящимся в
состоянии истинного химического равновесия. Поэтому в конденсированных
(особенно твердофазных) системах, процессы в которых часто заторможены, при-
применение этого принципа ограниченно.
10. Понятие о катализе. Гомогенный и гетерогенный катализ. Катализом
называется явление изменения скорости peaKV,uu под воздействием небольших
добавок специфических веществ, количество которых в ходе реакции не изменяет-
изменяется. В каталитических процессах скорость основной реакции может и увеличивать-
увеличиваться, и уменьшаться. В соответствии с этим каталитическое действие может быть
положительным и отрицательным. Вещества, ускоряющие реакцию, называются
катализаторами, а замедляющие — ингибиторами.
Для каталитических реакций характерны некоторые особенности. Как прави-
правило, катализатор вводится в систему в очень небольших количествах по сравнению
с массой реагентов. Тем не менее эффективность действия этих малых добавок
необыкновенно высока. В результате реакции катализатор остается в химически
неизменном состоянии и не расходуется, т.е. участие катализатора в реакции не
отражается общим стехиометрическим уравнением. Однако физически катализа-
катализатор изменяется. Например, кристаллический МпОг в процессе каталитического
разложения хлората калия КСЮз превращается в мелкодисперсный порошок.
Эти изменения свидетельствуют о том, что в ходе реакции на определенных
стадиях катализатор вступает во взаимодействие с реагентами, а по окончании ее
вновь выделяется.
Нередко один из продуктов реакции служит катализатором, ускоряющим эту
реакцию. Например, сухой фтороводород практически не действует на металлы и
оксиды. Но в процессах с оксидами типа МеО + 2HF = MeF2 + Н2О появление
молекул воды (катализатор) резко ускоряет реакцию слева направо. Такого рода
реакции, когда катализатор не вводится в систему извне, а является продук-
продуктом самой реакции, называются автокаталитическими. Типичные кинетические
кривые изменения скорости и накопления продуктов реакции приведены на
рис. 76.
Для обратимой реакции катализатор не смещает равновесие и не влияет на
константу равновесия, а лишь ускоряет процесс достижения равновесного состоя-
состояния. Вблизи этого состояния катализатор в равной мере влияет на скорости
141
прямой и обратной реакций. Действие катализатора сводится к снижению энер-
энергии активации за счет образования промежуточных нестойких ассоциатов, кото-
которые в дальнейшем распадаются на продукты реакции с выделением катализатора
в химически неизменном виде. На рис. 77 представлено изменение энергии систе-
системы без катализатора К (А + В ^^ АВ) и с его участием (А + К ^^ АК, АК +
+ В ^^ АВ + К). Уровень 1 соответствует энергии смеси исходных компонентов
А + В, уровень /' — энергии активного комплекса при взаимодействии без ката-
катализатора.
Разность энергий этих уровней представляет собой энергию активации прямо-
прямого взаимодействия Еа. Уровень 2 соответствует энергии активного комплекса при
взаимодействии А + К ?=^ АК. Уровень 3 отражает энергию промежуточного
соединения АК, причем 2 — 1 — Е^. Обменное разложение промежуточного сое-
соединения с компонентом В, протекающее через активный комплекс 4 с энергией
активации 4 ~~ 3 — Е'^, приводит к образованию продукта реакции и выделению
катализатора. Если энергии активации Е^ и Е^ меньше энергии Еа, то косвенный
путь оказывается кинетически более выгодным, чем прямое взаимодействие.
Поскольку тепловой эффект реакции не зависит от пути процесса (Д# =
= const), константа равновесия остается постоянной согласно выражению (VI.15).
Отсюда и следует, что катализатор не смещает равновесие, а лишь ускоряет его
достижение.
Активность катализатора сильно зависит от присутствия посторонних веществ,
действие которых может быть двояким: увеличивать активность катализатора или
снижать ее. Вещества, не обладающие каталитической активностью, но увеличи-
увеличивающие активность катализатора, называются промоторами или активаторами.
Так, например, катализаторы синтеза аммиака (Fe, Mo, W, Ni, Co) значительно
активируются в присутствии тугоплавких оксидов AI2O3, MgO, Сг2Оз- Вещества,
способствующие снижению активности катализатора, вплоть до полной ее потери,
называются каталитическими ядами. При взаимодействии ядов с катализатором
образуются малоактивные продукты, не обладающие каталитическим действием.
Это явление называется отравлением катализатора. В зависимости от стабильно-
стабильности образовавшихся продуктов отравление бывает обратимым и необратимым.
При обратимом отравлении активность катализатора может быть восстановлена
пропусканием свежих порций реакционной смеси. При необратимом отравлении
требуется полная регенерация катализатора или его замена.
Механизмы каталитического взаимодействия очень сложны, разнообразны и
очень редко твердо установлены.
При гомогенном катализе все взаимодействия, включая и промежуточные
стадии, протекают в однородной среде (в газовой или жидкой фазе). При ге-
гетерогенном катализе катализатор и реагирующие вещества находятся в разных
фазах и каталитическая реакция протекает на границе раздела фаз. При этом
катализатор обычно бывает твердым и на его поверхности происходят все про-
промежуточные взаимодействия. Реагирующие вещества находятся в жидкой или
газовой фазе.
Примером достаточно хорошо изученной гомогенной каталитической реакции
может служить нитрозный способ получения серной кислоты. В процессе ее полу-
получения диоксид серы поглощается раствором оксидов азота в концентрированной
серной кислоте (нитрозой) и окисляется в этом растворе по схеме
142 .
S02 + N2O3 + H20 *=* H2SO4 + 2N0
Процесс окисления при этом идет значительно быстрее, чем прямое окисление
диоксида серы. Это связано с каталитической активностью нитрозы, обусловлен-
обусловленной образованием нестабильного промежуточного соединения — нитрозилсерной
кислоты — по уравнению
N2O3 + 2H2SO4 *=* 2SO2(OH)ONO + Н2О
Это равновесие при высоких температурах смещено влево, а при охлаждении —
вправо. В процессе окисления диоксида серы нитрозой образуется оксид азота,
выделяющийся из нитрозы (в которой он плохо растворим), и частично окисля-
окисляется кислородом воздуха. Регенерированная таким образом смесь оксидов азота
вновь возвращается в процесс. В приведенном примере медленно протекающий
процесс прямого окисления
О2 + 2H2SO3 «=* 2H2SO4 (A + В = АВ)
заменяется двумя быстро протекающими процессами с участием катализатора:
О2 + 2NO *=± 2NO2 (А + К = АК)
и
2NO2 + 2H2SO3 = 2H2SO4 + 2NO (АК + В = АВ + К)
Для гомогенного катализа разработана количественная теория промежуточных
соединений, основные положения которой: 1) каталитическое взаимодействие
протекает через образование метастабильного промежуточного соединения ката-
катализатора с реагентами; 2) промежуточное соединение образуется по обратимой
реакции, которая протекает с большой скоростью; 3) разложение промежуточного
соединения является лимитирующей стадией каталитического процесса.
В отличие от гомогенного для гетерогенного катализа нет единой теории,
позволяющей описать все наблюдаемые явления. Особенность гетерогенных
каталитических реакций заключается в образовании на твердой поверхности
катализатора хемосорбированных (на активных центрах) комплексов, которые не
способны существовать индивидуально и не могут быть названы промежуточными
соединениями. Хемосорбционные комплексы одного из реагентов в дальнейшем
вступают во взаимодействие с компонентами реакционной смеси, образуя продук-
продукты реакции и освобождая активные центры поверхности. Характер взаимодейст-
взаимодействия в значительной мере зависит от электронной структуры твердого катализато-
катализатора. С этой точки зрения, активные металлы с их легкоподвижными электронами
обычно склонны к образованию прочных поверхностных комплексов и поэтому
каталитически малоактивны. Диэлектрики с ничтожно малой концентрацией
свободных электронов плохо образуют поверхностные комплексы и поэтому также
не отличаются каталитической активностью. А на поверхности полупроводников
и малоактивных металлов, которые характеризуются промежуточными значени-
значениями электронной концентрации, хорошо образуются метастабильные ассоциаты,
чем и определяется их высокая каталитическая активность. Так, например, про-
йотирование V2O5 сульфатами щелочных металлов при каталитическом окисле-
окислении объясняется резким увеличением электронной концентрации, а следователь-
следовательно, и электрической проводимости оксида ванадия (V), который сам по себе
обладает малой концентрацией свободных электронов.
Г Л А В А VII. ЖИДКОЕ СОСТОЯНИЕ. РАСТВОРЫ
В твердых телах и жидкостях наблюдается ближний порядок, под которым
понимают упорядоченное расположение частиц на расстояниях десятых долей
нанометров (первая координационная сфера). В пределах первой координа-
координационной сферы наблюдается более сильное взаимодействие частиц. Кроме того,
при переходе от пара к конденсированным системам вследствие агрегации (объ-
(объединения) большого количества частиц вступают в силу законы больших чисел.
Поэтому многие законы, строго соблюдаемые в пределах химии газообразного
состояния, становятся приближенными и ограниченно применимыми к конденси-
конденсированному состоянию. Так, например, один из основных стехиометрических
законов — закон постоянства состава — применительно к конденсированным
системам может быть использован с известными ограничениями.
1. Жидкое состояние. Структура жидкости. Жидкость имеет много общего с
твердым состоянием. Компактное расположение частиц обусловливает высокую
плотность и малую сжимаемость по сравнению с газами. Структура и внутреннее
строение жидкостей и твердых тел во многом схожи и характеризуются упорядо-
упорядоченным расположением частиц. В кристаллических твердых телах упорядочение
распространяется на огромное количество межатомных расстояний, т.е. ближний
порядок переходит в дальний. В жидкости вследствие относительно высокой
подвижности частиц упорядоченность ограничивается небольшими островками
(агрегатами или кластерами), причем последние ориентированы друг относитель-
относительно друга беспорядочно и часть пространства между ними остается не заполнен-
заполненной веществом. Эти образования нестабильны, связи в них постоянно разрушают-
разрушаются и вновь возникают. При этом происходит обмен частиц между соседними
кластерами. Таким образом, в структурном отношении для жидкости характерно
наличие лабильного (подвижного) равновесия, обусловленного относительной
свободой перемещения частиц. Образование .лабильных агрегатов в жидкости
наблюдается даже при температурах, намного превышающих температуру крис-
кристаллизации. С понижением температуры стабильность таких агрегатов увеличива-
увеличивается и вблизи температуры кристаллизации жидкости имеют квазикристалличе-
квазикристаллическое строение, т.е. возрастает количество агрегатов, они становятся больше по
размерам и начинают определенным образом ориентироваться друг относительно
друга.
В то же время между жидким и твердым состояниями, естественно, наблюда-
наблюдаются и различия. Прежде всего в отличие от кристаллов жидкости изотропны,
т.е. их физические свойства одинаковы в различных направлениях. Для кристал-
кристаллического же состояния характерно явление анизотропии, проявляющееся в том,
что физические свойства зависят от направления, в котором они измерены.
Различаются твердые тела и жидкости также по их отношению к деформиру-
деформирующим нагрузкам. Если твердое тело под действием нагрузки обычно испытывает
144
упругие деформации (до определенного предела), то жидкость при любых, сколь
угодно малых усилиях легко изменяет свою форму, что проявляется в текучести.
Естественно, что текучесть (или обратная ей величина — вязкость) для различ-
различных жидкостей меняется в широких пределах. Существуют жидкости, облада-
обладающие весьма высокой вязкостью (например, некоторые битумы), вследствие чего
при резком приложении нагрузки — ударе — они разрушаются подобно твердым
телам. В то же время постепенное и непрерывное увеличение нагрузки позволяет
обнаружить у них текучесть.
С этой точки зрения стеклообразное состояние можно рассматривать как
переохлажденную жидкость с очень высокой вязкостью. Действительно, для
стекла, как и для жидкости, характерны изотропность свойств и сохранение
структуры ближнего порядка при отсутствии дальнего порядка. В отличие от
кристаллических тел стекло не имеет фиксированной температуры плавления, а
при нагревании постепенно размягчается. В термодинамическом отношении стек-
стеклообразное состояние, будучи переохлажденным, является неустойчивым (мета-
стабильным), несмотря на то что может сохраняться сколь угодно долго. При
определенных условиях (при нагревании) наблюдается самопроизвольный пере-
переход в кристаллическое состояние (кристаллизация или расстекловывание), кото-
который, однако, сильно заторможен вследствие высокой вязкости стекла.
Таким образом, в структурном отношении жидкость занимает промежуточное
положение между твердыми телами и газами. Жидкое состояние существует в
определенном температурном интервале, ограниченном, с одной стороны, темпера-
температурой плавления, а с другой — критической температурой. Вблизи температуры
плавления жидкость имеет квазикристаллическое строение, т.е. имеет много
общих черт с твердым телом. При температурах, близких к критической, строе-
строение и свойства жидкости напоминают газообразное состояние. В связи с проме-
промежуточным характером жидкого состояния теоретическое рассмотрение структуры
и свойств жидкости представляет собой сложную задачу. Если для твердого и
газообразного состояний в первом приближении существуют идеализированные
модели (идеальный кристалл и идеальный газ), то для жидкого состояния подоб-
подобная относительно простая модель отсутствует.
2. Дисперсные системы. Растворы. В течение длительного периода развития
химии основными объектами исследования были вещества постоянного состава,
образующиеся при некоторых рациональных и строго фиксированных стехиомет-
рических соотношениях компонентов, что представляло собой так называемую
"привилегию дискретности" в химии. Фазы, не подчиняющиеся стехиометриче-
ским законам и обладающие переменным составом (в частности, растворы), иск-
исключались из рассмотрения в рамках классической химии. Бурное развитие химии
в конце XIX и начале XX в., особенно учения о химическом равновесии, теории
растворов (Гиббс, Вант-Гофф, Ле Шателье, Д.И.Менделеев, Д.П.Коновалов,
Н.С.Курнаков); показало, что наиболее общий случай химического взаимодейст-
взаимодействия — именно непрерывность изменения состава в зависимости от условий полу-
получения. А дискретность взаимодействия, проявляющаяся в образовании фаз пос-
постоянного состава, представляет собой частный случай, хотя и очень распростра-
распространенный. В связи с этим в настоящее время изучение растворов приобретает со-
совершенно особый смысл, так как закономерности их образования и свойства в
определенном смысле применимы для изучения характера химического взаимо-
взаимодействия с общих позиций.
1 145
Растворы — гомогенные системы переменного состава, находящиеся в состоя-
состоянии химического равновесия. Растворы представляют собой дисперсные системы,
в которых частицы одного вещества равномерно распределены в другом. Дисперс-
Дисперсные системы по характеру агрегатного состояния могут быть газообразными,
жидкими и твердыми, а по степени дисперсности — взвесями, коллоидными и
истинными растворами. Частицы взвесей обычно имеют размер порядка 1 мкм
и более. Такие частицы сохраняют все свойства фазы. Поэтому взвеси следует
рассматривать как гетерогенные системы. Характерным признаком взвесей слу-
служит их нестабильность во времени. Они расслаиваются, причем диспергирован-
диспергированная фаза (т.е. вещество, распределенное в среде) выпадает в виде осадка
или всплывает в зависимости от соотношения плотностей. Примерами взвесей
могут служить туман (жидкость распределена в газе), дым (твердое + газ), сус-
суспензии (твердое + жидкость), эмульсии (жидкость + жидкость), пены (газ +
жидкость).
При образовании истинного раствора (или просто раствора) распределенное в
среде вещество диспергировано до атомного или молекулярного уровня. Примеры
гаких систем многочисленны: воздух (газообразный раствор, содержащий кисло-
кислород, азот и т.д.), жидкие водно-солевые системы, сплавы меди с золотом, пред-
представляющие собой пример твердых растворов, и многие другие. Для истинных
растворов — термодинамически равновесных систем — в противоположность взве-
взвесям характерна неограниченная стабильность во времени. Наибольшее значение
имеют жидкие, а в последнее время и твердые растворы, находящие широкое
применение в самых различных областях науки и техники. Промежуточное поло-
положение по степени дисперсности и свойствам занимают коллоидные растворы. В
них частицы диспергированного вещества представляют собой относительно
простые агрегаты с размерами, промежуточными между истинными растворами и
взвесями. С этой точки зрения коллоидные растворы можно рассматривать как
микрогетерогенные системы.
Растворы — это как минимум двухкомпонентные системы. Обычно растворите-
растворителем считают компонент, который в данных условиях находится в том же агрегат-
агрегатном состоянии, что и образующийся раствор. Это определение существенно лишь
тогда, когда растворяемые вещества находятся в другом агрегатном состоянии
(например, растворение солей и газов в воде с образованием жидких растворов).
Если же компоненты, образующие раствор, находятся в одном и том же агрегат-
агрегатном состоянии, то понятия растворителя и растворенного вещества становятся в
известной мере условными.
Многие свойства растворов зависят от их концентрации. Концентрация вы-
выражается через число частиц в единице объема или как отношение числа час-
частиц данного вида к общему числу частиц в растворе (в долях единицы или
в процентах). Существует несколько способов выражения концентрации раст-
растворов.
В химической практике концентрацию чаще всего выражают через моляр-
ность. Молярностью раствора называется количество молей вещества, содер-
содержащееся в 1 л раствора. При расчетах, связанных с использованием закона экви-
эквивалентов (например, в аналитической химии при объемном анализе), удобно
пользоваться эквивалентной или нормальной концентрацией. Нормальная кон-
концентрация раствора определяется количеством эквивалентов растворенного
вещества в 1 л раствора. При физико-химических исследованиях растворов, в
146 .
частности при криоскопических и эбуллиоскопических определениях (см. ниже),
используют моляльный способ выражения концентрации. Молялъностъ раствора
определяется числом молей растворенною вещества в 1 кг растворителя; раз-
размерность — моль/кг. Основная особенность этого способа выражения концентра-
концентрации заключается в том, что моляльная концентрация раствора не зависит от
температуры, поскольку для определения моляльности не привлекается объем.
Этим же свойством обладает способ выражения концентрации через молярные
(атомные) доли. При этом концентрация выражается отношением числа молей
данного компонента к общему числу молей всех компонентов в растворе.
3. Процесс образования растворов. Растворение обычно сопровождается замет-
заметным тепловым эффектом (эндо- или экзотермическим), изменением объема (об-
(общий объем раствора не равен сумме объемов компонентов), иногда изменением
окраски и т.п. Например, при растворении гидроксида калия в воде наблюдается
сильное разогревание раствора:
КОН + aq = KOH-aq (АН = -54 кДж),
а при растворении нитрата аммония — охлаждение:
NH4NO3 + aq = NH4NO3-aq (АН = +25 кДж).
При смешении 100 мл воды с равным объемом этилового спирта вместо ожида-
ожидаемых 200 мл раствора получается лишь около 180 мл, т.е. в результате растворе-
растворения происходит уменьшение объема. Растворение безводного сульфата меди, не
имеющего окраски, сопровождается появлением интенсивной голубой окраски.
Все эти явления свидетельствуют об изменении химической природы компонен-
компонентов раствора в процессе его образования. В связи с этим Д.И.Менделеев, осново-
основоположник современной теории растворов, в 1887 г. выдвинул положение, отража-
отражающее химический аспект образования растворов: "Растворы представляют жид-
жидкие диссоциационные системы, образованные частицами растворителя, растворен-
растворенного вещества и тех определенных нестойких, но экзотермических соединений,
которые между ними происходят, одного или нескольких, смотря по природе
составляющих начал". Основное содержание современной химической теории
растворов отражается в этом определении, хотя в свете новых данных необходимо
уточнить, что промежуточные соединения могут и не быть определенными, т.е.
соединениями постоянного состава.
Промежуточные соединения, образующиеся в растворах между молекулами
растворителя и частицами (молекулами, ионами, атомами) растворенного вещест-
вещества, называются сольватами. Для водных растворов такие соединения называются
гидратами. Иногда гидраты могут быть настолько прочными, что при выделении
растворенного вещества из раствора вода входит в состав растущего кристалл а. в
химически связанном виде. Такие кристаллы называются кристаллогидратами, а
входящая в их состав вода — кристаллизационной. Примеры кристаллогидратов
многочисленны: CuSO4-5H2O, Na2SO4-10H2O, СгС13'6Н2О и т.п. Кристаллогидра-
Кристаллогидраты сохраняют окраску, характерную для соответствующих растворов. Это служит
доказательством существования в растворе аналогичных аквакомплексов.
С термодинамической точки зрения растворение всегда сопровождается
убылью энергии Гиббса. При этом независимо от знака изменения энтальпии при
; . '..-¦- '147
о
180
ISO
ПО
I 120
I 100
I 80
I 60
to
a?
/7
Температура, 'С
Рис. 78. Зависимость растворимости раз-
различных веществ в воде от температуры
/
и
J
А
!
щ
'"ft
? 5L
—¦
Nac
V
ч
/
А"
7 S,
1
*«¦
— —¦
растворении всегда AG < 0, так как
переход вещества в раствор сопровожда-
сопровождается значительным возрастанием энтро-
энтропии вследствие стремления системы к
разупорядочению. Раствор, в котором
при данных условиях невозможно даль-
дальнейшее растворение вещества, называет-
называется насыщенным относительно данного
вещества. Таким образом, насыщенный
раствор можно определить как раствор,
находящийся в динамическом равнове-
равновесии с осадком растворяемого вещества
(или с растворяемым газом). Концент-
Концентрация насыщенного раствора определя-
определяет растворимость вещества при данной
температуре. Растворы с меньшей кон-
концентрацией называются ненасыщенными. Растворимость вещества сильно зависит
от температуры. С повышением температуры растворимость большинства твердых
веществ растет (рис. 78). Однако известны вещества, растворимость которых с
ростом температуры понижается. К их числу относятся, например, карбонат и
сульфат кальция, гидроксиды кальция и магния и др. Понижение растворимости
с температурой иногда связано с переходом в менее гидратированную форму.
Если насыщенный раствор, приготовленный при повышенной температуре,
охлаждать, то вследствие уменьшения растворимости часть растворенного вещест-
вещества будет выпадать в осадок. Это явление часто используют для очистки веществ
перекристаллизацией. При медленном и осторожном охлаждении в отсутствие
посторонних частиц, которые могут стать центрами кристаллизации, иногда
удается предотвратить выделение вещества из раствора. Такой раствор, содержа-
содержащий больше растворенного вещества, чем это определяется его растворимостью,
называется пересыщенным. Пересыщенный раствор представляет собой неустой-
неустойчивую, метастабильную систему. При любом внешнем воздействии (например,
встряхивании, внесении кристалла затравки и т.д.) в системе происходят необ-
необратимые изменения, сопровождающиеся осаждением избыточного количества
растворенного вещества.
4. Идеальный раствор. Законы разбавленных растворов. При образовании
растворов характер взаимодействия компонентов определяется их химической
природой, что затрудняет выявление общих закономерностей. Поэтому удобно
прибегнуть к некоторой идеализированной модели раствора, в которой исключа-
исключаются конкретные особенности процесса растворения, однако сохраняются наибо-
наиболее существенные черты всех растворов.
В первом приближении полезно рассмотреть образование раствора в результа-
результате простого "физического" смешения компонентов, не сопровождающегося тепло-
тепловым эффектом и изменением объема. При этом процесс растворения не сопровож-
сопровождается специфическим химическим взаимодействием. Такой раствор, образование
которого не связано с изменением объема и тепловым эффектом (Д Vpacre = 0>
Д~йраств = 0), называют идеальным раствором. Это понятие имеет большое теоре-
теоретическое и практическое значение. Хотя большинство растворов и не обладает в
148
полной мере свойствами идеальных, однако поведение многих из них достаточно
удовлетворительно может быть описано при помощи этой модели, например так
называемых разбавленных растворов, т.е. таких, в которых содержание растворен-
растворенного вещества очень мало по сравнению с содержанием растворителя.
Рассмотрим разбавленный раствор нелетучего (твердого) вещества А в летучем
жидком растворителе В, например раствор сахара в воде. При этом общее давле-
давление насыщенного пара над раствором определяется парциальным давлением пара
растворителя, поскольку давлением пара растворенного вещества можно прене-
пренебречь. Рауль A886) показал, что давление насыщенного пара растворителя над
раствором рв меньше, чем над чистым растворителем р" . Разность р" — в = Ар
называется абсолютным понижением! давления пара над раствором. Эта величина,
отнесенная к давлению пара чистого растворителя, те (р° ~ р )/р° = Д» /»°
в w' В в в'
называется относительным понижением давления пара.
Согласно закону Рауля, относительное понижение давления насыщенного пара
растворителя над раствором равно молярной доле растворенною нелетучею
вещества:
где хд = "A/(«A 4- пв); хд — молярная доля растворенного вещества; и — число
молей растворенного вещества; п^ — число молей растворителя.
Понижение давления пара над раствором нелетучего вещества, например в
воде, можно пояснить с привлечением принципа смещения равновесия Ле Ша-
телье. Действительно, при увеличении концентрации нелетучего компонента в.
растворе равновесие в системе вода - насыщенный пар сдвигается в сторону
конденсации части пара (реакция системы на уменьшение концентрации воды
при растворении вещества), что и вызывает уменьшение давления пара.
Понижение давления пара над раствором влияет на температуры замерзания и
кипения. На рис. 79 представлены температурные зависимости давления пара над
чистым растворителем и двумя растворами различной концентрации. Кривая ао
ЮЯгПа
Рис. 79. Температурная зависимость давления пара для твердого и
жидкого растворителей и растворов различной концентрации
149
I >
представляет собой температурную зависимость давления насыщенного пара для
твердого растворителя, а кривая об — аналогичную зависимость для чистого
жидкого растворителя. Кривые о'Ь' и о"Ь" отражают температурную зависимость
давления пара растворителя над растворами, причем концентрация второго раст-
раствора выше концентрации первого. Точка о, в которой пересекаются кривые дав-
давления пара над твердым и жидким растворителями и в которой, следовательно,
эти давления равны, является точкой плавления (замерзания) чистого раствори-
растворителя. Соответственно точки о' и о"- точки замерзания растворителя в растворах I
и II, если из растворов кристаллизуется чистый растворитель. Точки 6, .6', Ь"
соответствуют температурам кипения растворителя, раствора I и раствора II,
поскольку при этих температурах достигается давление пара растворителя, рав-
равное внешнему (атмосферному) давлению. Как следует из рис. 79, растворы замер-
замерзают при более низкой температуре {Т'3, Т^'), а кипят при более высокой (Т^,
TQ, чем чистый растворитель (соответственно Т°3, и Т^). Это объясняется тем,
что р — Т-кривые для растворов проходят ниже аналогичной кривой для раство-
растворителя (поскольку, согласно закону Рауля, р° > р' > р") и пересекают р -<- Т-
кривую твердого растворителя при более низких температурах, а изобару р —
— Рвнешн — 1,013 -105 Па — при более высоких.
И повышение температуры кипения, и понижение температуры замерзания
растворов по сравнению с чистым растворителем (AT), согласно закону Рауля,
пропорциональны моляльной концентрации растворенного вещества — неэлектро-
неэлектролита, т.е. Д Г = Кст, где ст — моляльность раствора. Коэффициент пропорцио-
пропорциональности К в случае повышения температуры кипения называется эбулиоскопи-
ческой константой для данного растворителя, а для понижения температуры
замерзания — криоскопической константой. Эти константы, численно различные
для одного и того же растворителя, характеризуют повышение температуры
кипения и понижение температуры замерзания одномоляльного раствора, т.е. при
растворении 1 моль нелетучего неэлектролита в 1000 г растворителя. Поэтому их
часто называют моляльным повышением температуры кипения и моляльным
понижением температуры замерзания раствора. Криоскопическая и эбулиоскопи-
ческая постоянные не зависят от концентрации и природы растворенного вещест-
вещества, а зависят лишь от природы растворителя и характеризуются размерностью
кг-град/моль. Ниже приведены криоскопические Кк и эбулиоскопические Кэ
константы для некоторых растворителей:
Растворитель
К*
H20
0
1,86
100
0,52
CeHfi
5,5
5,12
80,1
2,53
ССЦ
Г22
30,0
76,5
5,03
СНС13
-63,5
4,7.
61,7
3,63
Криоскопия и эбулиоскопия — методы определения молекулярных масс раст-
растворенных веществ. Методы криоскопии и эбулиоскопии позволяют определить
молекулярную массу не диссоциирующих при растворении веществ (обычно
органических) по понижению температуры замерзания и повышению температу-
температуры кипения растворов известной концентрации. Пусть в Г граммах растворителя
растворена навеска Н вещества с молекулярной массой М. Тогда
150
Д Г = Кст- KB 1000/(М-/)-
^Экспериментально определяя AT, по этой формуле вычисляют М.
Закон Рауля и следствия из него в полной мере приложимы лишь для описа-
описания свойств идеальных растворов. Не случайно поэтому методы криоскопии и
эбулиоскопии дают удовлетворительные результаты для не диссоциирующих в
растворителе веществ и к тому же в малых концентрациях, когда взаимодействи-
взаимодействием частиц можно пренебречь. Очевидно, что понятие идеального раствора не
определяется его концентрацией, достаточно лишь отсутствие взаимодействия
компонентов. Однако на практике встречается очень мало систем, которые в
широком интервале концентраций удовлетворяют условию идеальности. К таким
системам относятся, в частности, смеси газов при низких давлениях и некоторые
растворы неэлектролитов и металлические расплавы. В то же время существует
категория растворов, для которых законы идеальных растворов могут быть ис-
использованы с достаточной точностью. Это так называемые разбавленные раство-
растворы, в которых концентрация растворенного вещества мала, вследствие чего можно
пренебречь взаимодействием растворенных частиц. Термодинамика не устанавли-
устанавливает количественного критерия разбавленного раствора. Единственным критерием
является применимость в данном интервале концентраций законов идеальных
растворов.
Помимо явлений, связанных с зависимостью давления пара над раствором от
концентрации раствора, к общим свойствам растворов относят также осмос. При
исследовании явления осмоса широко используются полупроницаемые перегород-
перегородки (мембраны и диафрагмы). Характерная их особенность заключается в способ-
способности пропускать молекулы растворителя, но задерживать частицы растворенного
вещества. Исключительно важную роль полупроницаемые перегородки играют в
процессах жизнедеятельности организмов.
Рассмотрим замкнутый цилиндр, разделенный на две части поршнем, который
представляет собой полупроницаемую перегородку, пропускающую только раст-
растворитель (рис. 80). В нижней части помещается растворитель, а в верхней —
раствор. Вследствие различия концентраций растворителя по обе стороны перего-
перегородки он самопроизвольно (в соответствии с принципом Ле Шателье) поступает
через полупроницаемую перегородку в раствор, разбавляя его. Движущей силой
преимущественной диффузии растворителя в раствор является разность свобод-
свободных энергий чистого растворителя и растворителя в растворе. При разбавлении
раствора за счет самопроизвольной диффузии растворителя
объем раствора увеличивается и поршень перемещается из
положения I в положение II.
Явление селективной диффузии определенною сорта
частиц в растворе через полупроницаемую перегородку
называется осмосом. А сила, обусловливающая осмос, .Л
отнесенная к единице поверхности полупроницаемой мем- I—t-
браны называется осмотическим давлением. Вант-Гофф
показал, что осмотическое давление в растворе неэлектро-
неэлектролита пропорционально молярной концентрации растворен-
растворенного вещества:
р = cRT,
Рис. 80. К понятию
(VII. 1) I явления осмоса
151
где с = n/V — молярная концентрация, моль/л. Выражение (VII. 1) по форме
аналогично уравнению Клапейрона—Менделеева для идеальных газов (II.1),
однако эти уравнения описывают разные процессы. Осмотическое давление воз-
возникает в растворе при проникновении в него дополнительного количества раство-
растворителя через полупроницаемую перегородку. Это давление — сила, препятствую-
препятствующая дальнейшему выравниванию концентраций.
Формальная аналогия позволила Вант-Тоффу A887) сформулировать закон
осмотического давления: осмотическое давление равно тому давлению, которое
производило бы растворенное вещество, если бы оно в виде идеального газа зани-
занимало тот же объем, который занимает раствор, при той же температуре.
Все описанные законы относятся к бесконечно разбавленным идеальным раст-
растворам. Применение их к реальным растворам ограничено тем в большей степени,
чем выше концентрация раствора.
5. Электролитическая ионизация. Степень и константа ионизации. Изучение
разбавленных растворов показало, что все их общие свойства (понижение давле-
давления пара, изменение температур замерзания и кипения, величина осмотического
давления) изменяются пропорционально числу частиц растворенного вещества.
Такие свойства называются коллтативными. Эта общая закономерность оказа-
оказалась справедливой для растворов органических веществ в воде и для растворов в
органических растворителях. При исследовании водных растворов солей, кислот,
оснований было обнаружено, что изменение соответствующего свойства в зависи-
зависимости от концентрации раствора значительно превышает ожидаемую величину.
Например, понижение температуры замерзания моляльного раствора NaCl превы-
превышает почти в два раза криоскопическую постоянную для воды C,36° вместо
1,86°). Это свидетельствует о том, что число частиц в водных растворах кислот,
оснований и солей не соответствует молярной концентрации раствора.
Кроме того, растворы, для которых характерны отклонения от законов разбав-
разбавленных растворов, обладают значительной электрической проводимостью в отли-
отличие от водных растворов некоторых органических веществ. Это моз&но было
объяснить наличием в растворе заряженных частиц. Вещества, растворы (или
расплавы) которых проводят электрический ток, были названы электролитами.
Свойства растворов электролитов были рассмотрены и обобщены основополож-
основоположником теории электролитической ионизации Аррениусом A887) и развиты в
трудах И.А.Каблукова, В.А.Кистяковского на основе химической теории раствора
Д.И.Менделеева. Основные положения теории электролитической ионизации:
1) при растворении солей, кислот и оснований в воде происходит диссоциация
этих веществ с образованием электрически заряженных частиц — катионов и
анионов;
2) электрическая проводимость водных растворов солей, кислот и оснований
пропорциональна общей концентрации ионов в растворе.
Электролитическая ионизация вызывается взаимодействием полярных моле-
молекул растворителя с частицами растворяемого вещества. Это взаимодействие при-
приводит к поляризации даже преимущественно ковалентных связей, как, например,
в хлороводороде. При растворении этого газа в воде происходит образование
ионов водорода и хлора за счет ослабления связи Н—С1 в среде с большой ди-
диэлектрической постоянной. Переход ионов в раствор сопровождается их гидрата-
гидратацией:
НС1 + иН2О *=* Н^НгО)* + С1-(Н2О)П-*
152
Такой же процесс наблюдается и при растворении преимущественно ионных
кристаллов (например, NaCl) в воде. Хотя в кристаллической решетке нет ионов
Na+ и СГ, однако взаимодействие с полярными молекулами растворителя способ-
способствует поляризации связей в кристалле, их ослаблению и обеспечивает возмож-
возможность перехода частиц в раствор с образованием гидратированных ионов:
NaCl + иН2О *=* Na+(H2O)X + С1-(Н2О)„-Я
Процесс гидратации сильно экзотермичен и идет самопроизвольно с уменьше-
уменьшением энтальпии. Обычно степень гидратации, т.е. число молекул растворителя,
окружающих каждый ион, очень велико (п и х — целые числа, п > 1, х > 1); лишь
при ионизации кислоты (х = 1):
НА + яН2О ?=± Н3О+ + А-(Н2О)„-,
Это объясняется малым размером иона водорода (протона), который составляет
~ 10~4 от размера атома. При гидратации протон внедряется в сферу молекулы
Н2О с образованием иона гидроксония ЩО*. Новая ковалентная связь образуется
по донорно-акцепторному механизму за счет свободной электронной пары кисло-
кислорода и является насыщенной.
Одной из количественных характеристик электролитической ионизации явля-
является степень ионизации, которая определяется как отношение ионизированных
молекул * к общему числу растворенных молекул. Обычно степень ионизации
выражают в долях единицы или в процентах:
а = (я/яоI00, (VII.2)
где я — число частиц, подвергшихся электролитической ионизации; Яо — число
растворенных частиц.
По степени ионизации электролиты условно подразделяются на сильные
(а > 30%) и слабые (а < 3%). Степень ионизации зависит от природы раствори-
растворителя. Чем более полярна молекула растворителя, тем при прочих равных услови-
условиях выше степень ионизации растворенного вещества. Так как электролитическая
ионизация сопровождается тепловым эффектом, то степень ионизации зависит от
температуры, причем влияние температуры можно оценить по принципу Ле Ша-
телье: если электролитическая ионизация представляет собой эндотермический
процесс, то с повышением температуры степень ионизации растет, а с понижением
температуры уменьшается.
Сильно влияет на степень электролитической ионизации концентрация раст-
раствора. Если рассматривать электролитическую ионизацию как равновесный обра-
обратимый химический процесс
КА + иН2О ?=ь К^НгО)* + А-(Н2О)„-Я
то в соответствии с принципом смещения равновесия разбавление водой увеличи-
увеличивает количество диссоциированных** молекул, т.е. степень ионизации при разбав-
разбавлении возрастает.
*Для веществ с немолекулярной структурой — формульных единиц.
**Следует различать термическую диссоциацию, протекающую по гемолитическому
механизму (например, NH4CI ^=^ NH3 + НО), и электролитическую диссоциацию (иони-
(ионизацию), сопровождающуюся гетеролитическим распадом на ионы (NH4C1 =?=*¦ NH* + СГ).
.153
Процесс электролитической ионизации удобнее характеризовать константой
ионизации, применив к нему законы химического равновесия. Так, для реакции
КА '^ К+ + А" константа ионизации
КИ =. [К*][А-]/[КА].
Здесь и далее в квадратных скобках обозначаются молярные концентрации ком-
компонентов. В отличие от степени ионизации константа электролитической иониза-
ионизации зависит лишь от природы электролита и температуры. Чем больше величина
Аги, тем сильнее электролит. Между константой и степенью электролитической
ионизации существует количественная связь. Действительно, пусть в рассмотрен-
рассмотренном процессе общая концентрация растворенного вещества КА равна с, а степень
ионизации равна а. Тогда [К+] = [А"] = ас и соответственно концентрация недис-
социированных частиц [КА] = A — а)с. Подставив значения ас и A — а)с в
выражение для константы ионизации, получим
г, _
асах
(VII.3)
Соотношение (VII.3) называется законом разбавления Оствальда. Для слабых
электролитов, когда а < 1, Кк « а2с. Отсюда
или
(VII.4)
где V — 1/с — разбавление. Из формулы (VII.4) следует, что если разбавить
раствор в 100 раз, то степень ионизации возрастет в 10 раз.
С учетом степени электролитической ионизации можно применить законы
разбавленных растворов и к растворам электролитов введением поправочного
множителя «', называемого изотоническим коэффициентом Вант-Гоффа. Тогда
отношение коллигативного свойства для электролита к аналогичному свойству
раствора неэлектролита той же концентрации равно коэффициенту Вант-Гоффа,
т.е.
Ар'/Ар = А^з/ДТз = ATJATK = piJVocu = I'. (VII.5)
Очевидно, что для растворов электролитов всегда г > 1, а для растворов неэлект-
неэлектролитов 1 = 1.
Изотонический коэффициент можно связать со степенью ионизации. Пусть
степень ионизации некоторого электролита с общим числом молекул в растворе с
равна а. Предположим, что при диссоциации каждая молекула электролита
распадается на п ионов. Тогда число молекул электролита, распавшихся на ионы,
равно ас, число ионов в растворе — пас, а число молекул, не распавшихся на
ионы, — A — а)с. Общее число частиц в растворе составляет A — а)с + пас.
Отношение общего числа частиц в растворе к числу растворенных молекул пред-
представляет собой изотонический коэффициент
с A - а + па) • 1 /, ч
t = —' : , » = 1 - аA — п).
154
Отсюда
а = (,' - 1)/(я - 1). (VII.6)
Соотношение (VII.6) позволяет определить степень ионизации электролита по
отклонению его свойств от законов разбавленных растворов.
6. Понятие о теории сильных электролитов. Активность. Для сильных элект-
электролитов, когда степень ионизации велика, константа ионизации зависит от кон-
центрации^так как при накоплении в растворе большого числа ионов сказывает-
сказывается их взаимное влияние.
При определенных условиях, например когда растворитель обладает малой
диэлектрической проницаемостью, создаются условия для электростатического
взаимодействия сольватированных ионов противоположного знака. При этом
последние подходят друг к другу на близкое расстояние и образуют так называ-
называемую ионную пару — сложный агрегат, состоящий из двух противоположно заря-
заряженных ионов, окруженных молекулами растворителя, в котором электрические
заряды взаимно компенсированы.
Представление об образовании ионных пар в растворах сильных электролитов
было введено Бьёррумом и Семенченко. В соответствии с этой концепцией для
каждого растворителя существует определенный параметр q (параметр Бьёррума),
представляющий собой расстояние, на которое подходят друг к другу ионы в
процессе образования ионной пары. Этот параметр определяется из соотношения
q=
где 2+) z_ — заряды катиона и аниона; е = 4,8-100 эл.ст.ед. — заряд электрона;
D — диэлектрическая проницаемость растворителя; к — постоянная Больцмана;
Т — абсолютная температура.
Из соотношения (VII.7) следует, что при увеличении заряда ионов расстояние,
на котором они начинают взаимодействовать, увеличивается. Наоборот, при уве-
увеличении диэлектрической проницаемости растворителя сила электростатического
взаимодействия между ионами уменьшается в D раз. Поэтому полярные раствори-
растворители, характеризующиеся большим значением диэлектрической проницаемости,
способствуют образованию растворов с малой склонностью к возникновению
ионных пар. Даже на сравнительно малых расстояниях взаимодействием ионов
можно пренебречь (q мал по величине), поэтому ионы можно считать практически
изолированными. Параметр Бьёррума имеет вполне определенное значение для
каждого растворителя при заданных температуре и заряде ионов. Например, для
однозарядных ионов в воде (г+ = z_ = 1) при 25°С = 298 К
Если расстояние между ионами меньше этой величины, то растворенную частицу
можно считать недиссоциированной. Если же q > 0,357 нм, то ионы рассматрива-
рассматриваются как изолированные.
При повышении концентрации раствора расстояния между ионами сокраща-
сокращаются, что усиливает межионное взаимодействие. Вследствие этого зксперимен-
1RS
тально определяемые свойства растворов сильных электролитов (Ар, ДТКИП, ДТ3
и т.п.), зависящие от общего количества частиц в растворе, оказываются меньше
рассчитанных в предположении полной ионизации.
Чтобы можно было пользоваться простыми соотношениями идеальных раство-
растворов для описания поведения реальных растворов, Льюис A907) ввел формальное
представление об эффективной концентрации — активности. Активность связана
с истинной концентрацией растворенного вещества соотношением
= Тс,
(VII.8)
где а — активность; -у — коэффициент активности; с — концентрация.
Активность выражается в тех же единицах, что и концентрация, поскольку
коэффициент активности — величина безразмерная. Он характеризует степень
отклонения свойств данного раствора от свойств идеального раствора. Для бес-
бесконечно разбавленных растворов электролитов, где практически отсутствует
взаимодействие ионов, активность становится равной концентрации и коэффици-
коэффициент активности равен единице. Введение понятия об активности позволяет, не
выясняя сложной картины взаимодействия частиц в реальном растворе, оценить
суммарный эффект этого взаимодействия, проявляющийся в отклонении свойств
системы от идеальной и применять законы идеальных растворов для анализа
реальных систем.
7. Кислотно-основная ионизация. Характер электролитической ионизации
гидроксидов общей формулы ЭОН зависит от сравнительной прочности и поляр-
полярности связей Э~О и О~Н и может протекать по двум типам:
I
II
i
i
II
эо- + н*
Полярность связей, как известно, определяется разностью электроотрицатель-
ностей компонентов, размерами и эффективным зарядом атомов. Щелочные и
щелочно-земельные металлы, а также переходные элементы в низших степенях
окисления образуют ионы относительно большого размера и с малым эффектив-
эффективным зарядом. Поэтому ионный потенциал* таких ионов невелик и их поляризую-
поляризующее действие выражено слабо. При этом связь Э~О обладает сравнительно малой
прочностью и диссоциация ЭОН идет преимущественно за счет отщепления гид-
роксид-ионов, т.е. по основному типу.
С ростом степени окисления увеличивается ионный потенциал элемента Э и
преобладает диссоциация по кислотному типу с отщеплением иона водорода, так
как связь Э~О упрочняется, а за счет перераспределения электронной плотности
у кислорода связь О—Н ослабевает. Таким образом, диссоциация по кислотному
типу протекает, если Е (О—Н) < Е (Э~О), а по основному типу — если Е (О—Н) >
> Е (Э~О). При сравнимой прочности связей О~Н и Э"~О диссоциация гидрок-
сида может одновременно протекать и по I, и по II типам с отщеплением как
ионов водорода, так и гидроксид-ионов. Электролиты, которые в растворе иони-
*Ионный потенциал — отношение заряда иона к его радиусу.
Mg
+2
0,078
Al
+3
0,057
Si
+4
0,039
P
+5
0,034
S
+6
0,029
Cl
+7
0,026
зируются одновременно по кислотному и основному типам, называются амфотер-
ными (амфолиты). Например, для гидроксида галлия Ga(OHK константы иони-
ионизации, соответствующие уравнениям
^осн
Ga(OHK > Ga3+ + ЗОН"
и
-Ккисл
Ga(OHK i 3H+ + GaOf
равны приблизительно 102 (т.е. Кжн и Ккисл). Следовательно, гидроксид галлия
служит примером идеального амфолита. Совершенно очевидно, что в кислой
среде амфолит проявляет основный, а в щелочной — кислотный характер.
Рассмотрим основные закономерности изменения характера ионизации гид-
роксидов в растворе в зависимости от положения элемента в Периодической
системе. В ряду элементов III периода от натрия к хлору степень окисления
растет, а эффективные ионные радиусы уменьшаются. Ниже приведены ионные
радиусы элементов III периода в высшей степени окисления:
Элемент Na
Степень окисления +1
Ионный радиус, нм 0,098
В этом ряду резко возрастает объемная плотность формального заряда. Поэто-
Поэтому поляризующее действие ионов элементов, приводящее к перераспределению
электронной плотности между связями Э~О и О—Н, возрастает в том же направ-
направлении. Гидроксиды крайних элементов рассматриваемого ряда обладают резко
выраженными основными (NaOH) и кислотными (НСЮ4) свойствами. Гидроксид
натрия принадлежит к хорошо растворимым основаниям и является одной из
самых сильных щелочей. Хлорная кислота по силе превышает такие кислоты, как
НС1, H2SO4, HNO3 и др. При переходе от натрия к магнию наблюдается ослабле-
ослабление основных свойств, уже Mg(OHJ представляет собой основание средней силы.
Гидроксид алюминия А1(ОН)з уже амфолит с некоторым преобладанием основ-
основных свойств, а гидроксид кремния Si(OHL — обладает только кислотными свой-
свойствами. В последующих членах ряда ярко выражена склонность к образованию в
растворе сложных ионов и соответствующие гидроксиды относятся к кислотам,
причем их сила в ряду высших кислот Н3РО4 — H2SO4 — HCIO4 возрастает.
Для переходных металлов, образующих гидроксиды с переменными степенями
окисления, характерны те же закономерности в изменении свойств. С возрастани-
возрастанием степени окисления и уменьшением при этом эффективного радиуса ионов
ослабевают основные и усиливаются кислотные свойства. В качестве примера
рассмотрим ряд гидроксидов марганца Мп(ОНJ, Мп(ОНK, Мп(ОНL, Н2Мп04,
НМпО4, в котором степень окисления марганца меняется в последовательности
+2, +3, +4, +6, +7. Первые два гидроксида — основания, последние два — кис-
кислоты, а гидроксид Мп(ОНL — амфолит с некоторым преобладанием кислотных
свойств. Таким образом, если элемент образует гидроксиды в нескольких степе-
степенях.окисления, то его гидроксиды в низших степенях окисления обладают более
основным характером, а гидроксиды в высших степенях окисления — более кис-
кислотным (или менее основным) характером.
157
8. Теории кислот и оснований. С признанием теории Аррениуса возникла
возможность классификации кислот и оснований по характеру их диссоциации в
растворах. С этой точки зрения кислотой следует считать электролит, в водном
растворе которого присутствуют гидратированные ионы водорода (ионы гидро-
ксония). Основания в водных растворах отщепляют гидроксид-ион.
Характер кислотно-основного равновесия в неводных растворах имеет свои
особенности. В соответствии с теорией солъвосистем это равновесие должно
удовлетворять условию
кислота + основание ^^ соль + растворитель.
Так, для растворов в жидком аммиаке, который подвергается самоионизации по
схеме
2NH3 ^=* NH; + NH-
взаимодействие хлорида аммония с амидом калия протекает в соответствии с
уравнением
NH4C1 + KNH2 ^ KC1 + 2NH3
Хлорид аммония здесь выступает в качестве кислоты, а амид калия — в качестве
основания. Эти представления позволили дать определенную классификацию
взаимодействий в неводных растворителях, но такой подход был формальным и
теория сольвосистем не получила широкого распространения.
В основу современных теорий кислот и оснований положены представления
Вренктеда и Льюиса. По протонной теории кислот и оснований Вренстеда, кис-
кислота является донором протона, а основание — акцептором протона. Сила кисло-
кислоты НА определяется константой кислотности, соответствующей равновесию
НА 5=i Н+ + А":
Однако для реализации этого равновесия необходимо присутствие вещества —
акцептора протона. Кислотно-основное равновесие заключается в переходе про-
протона от кислоты к основанию:
НА + Э" ^ НЭ + А-
где НА — кислота; А" — основание, сопряженное с этой кислотой. Точно так же
НЭ и Э" являются сопряженной парой кислота — основание. Реакции такого типа
называются протолитичёскими. С количественной точки зрения протолитическая
реакция характеризуется константой равновесия, которая называется константой
протолиза:
[НЭ][А']
_
п~
[Э-][НА]
Так как Кг = ([Н+][А-])/[НА], а К2 = ([Н+][Э-])/[НЭ], то Ка - Кх/Кг. Например, в
реакции '
158
HSO; + П7О ^ SO2- + H3O+
константа протолиза равна
н2о
т.е. она выражается через соотношение констант кислотности соответствующих
кислот по Вренстеду. Если какую-нибудь кислоту взять в качестве эталона, то
силу других кислот можно выразить через константу протолиза. Для водных
растворов в качестве эталонной кислоты выбран моногидрат протона — гидроксо-
ний Н3О+.
Протонная теория кислот и оснований Бренстеда позволила достаточно четко
разграничить эти два класса веществ и сделать целый ряд количественных выво-
выводов, касающихся характера кислотно-основного равновесия. Однако существуют
вещества, которые обладают сильно выраженными кислотными и основными
свойствами, но не могут быть отнесены к кислотам и основаниям Бренстеда из-за
отсутствия протона. К числу таких веществ относятся, в частности, галогениды
бора, алюминия, кремния, олова, обладающие кислотными свойствами.
Согласно электронной теории кислот и оснований Льюиса, кислота — акцеп-
акцептор электронной пары, а основание — донор. Взаимодействие между кислотой и
основанием с этой точки зрения заключается в возникновении ковалентной связи
по донорно-акцепторному механизму:
F H FH
F:B + :N:H ^ F:B:N:H
F H F H
Здесь донором электронной пары, т.е. основанием по Льюису, является аммиак, а
в качестве льюисовской кислоты выступает акцептор электронной пары BF3.
Протонная теория кислот и оснований Бренстеда и электронная теория Льюиса
дополняют друг друга и имеют глубокую внутреннюю связь. В определенном
смысле кислоты, по Бренстеду, представляют собой частный случай льюисовских,
поскольку протон характеризуется большим сродством к электронной паре и, по
Льюису, может рассматриваться как сильная кислота.
Таким образом, общее свойство кислот и оснований, по Льюису и Бренсте-
Бренстеду, — способность к взаимной нейтрализации с образованием стабильных кова-
лентно-насыщенных соединений.
9. Водородный показатель. Индикаторы. Вода служит не только наиболее
распространенным растворителем для многих электролитов, но и сама является
идеальным амфолитом. В соответствии с равновесием
Н2О 5=± Н+ + ОН-
в воде присутствуют катионы водорода и гидроксид-анионы в строго эквива-
эквивалентных количествах. Константа ионизации воды
Кк = [Н*][ОН-]/[Н2О]
-. -¦,!.... " 159
может быть определена по электрической проводимости. Измерения показывают,
что при 22°С Ки = 1,8-10~16, т.е. вода диссоциирована в очень малой степени.
Так как вода — очень слабый электролит, то концентрация недиссоциированных
молекул может быть принята равной общему количеству молей в 1 л воды, т.е.
[Н2О] = 1000/18 = 55,56 моль/л. Тогда КИ[Е2О] = [Н+][ОН"], или
[Н+][ОН-] = 1,8-10-16-55,56 = Ю-14.
Величина [Н+][ОН~] = 10~14 = Kw называется ионным произведением воды. Так
как в воде концентрации гидратированных ионов равны, то
[Н+] = [ОН"] =
= 10 моль/л.
При добавлении кислоты концентрация ионов водорода увеличивается и соответ-
соответственно уменьшается концентрация гидроксид-ионов, поскольку при данной
температуре ионное произведение воды — величина постоянная. При добавлении
щелочи наблюдается обратная картина. Таким образом, концентрация ионов
водорода в растворе может служить мерой кислотности или щелочности среды. В
кислых растворах [Н+] > 10, в щелочных [Н+] < 10. Вводится значение отри-
отрицательного десятичного логарифма концентрации водородных ионов, которое
называют водородным показателем рН:
рН = -lg[H+] *.
(VII.11)
Тогда для нейтральной среды рН = —lglO~7 = 7, для кислых растворов рН < 7, а
для щелочных рН > 7. Аналогичным образом реакция среды может быть охарак-
охарактеризована так называемым гидроксильным показателем:
pOH = -
(VII.12)
Для воды рН = рОН = 7, а изменение рОН в кислых и щелочных растворах
противоположно изменению рН. Прологарифмировав ионное произведение воды,
получим
(VII.13)
+ lg[OH-] = -14.
Взяв отрицательные логарифмы, получим соотношение
рН + рОН = 14.
В растворах величина рН может меняться в широких пределах. Так, при
рН1(рОН 13) имеем [Н+] = 10 моль/л, а [ОН"] = 103 моль/л, что соответствует
0,1 н. раствору кислоты. Для 1 М раствора кислоты, когда [Н+] = 1 моль/л, рН 0
(рОН 14), т.е. [ОН"] = 104 моль/л, а для 10 н. раствора кислоты рН —1, а
рОН 15, т.е. [ОН"] = 105 моль/л. Отсюда следует, что даже в концентрирован-
концентрированных растворах кислот всегда в ничтожно малых количествах присутствуют гид-
роксид-ионы.
*Точнее рН = —lea +, где о — активность.
н
Аналогично, для щелочных растворов рН 13 в 0,1 н. растворе NaOH, т.е.
рОН 1, и соответственно [Н+] = 103 моль/л, [ОН"] = 10 моль/л. Для 1 н.
раствора NaOH рН 14 и рОН 0, т.е. [Н+] = 10"и моль/л, а [ОД'] = 1 моль/л.
Водородный показатель Юн. раствора щелочи должен быть равен 15, поскольку
для [ОН"] = = 10 мель/л рОН —1. Практически растворы со значениями рН < 0
и рН > 14 встречаются редко.
Для определения рН используют так называемые кислотно-щелочные индика-
индикаторы, которые представляют собой слабые органические кислоты и основания,
имеющие различную окраску в диссоциированной и молекулярной формах. Так,
индикатор фенолфталеин представляет собой слабую органическую кислоту,
формулу которой в общем виде можно представить как Hind. В водном растворе
фенолфталеин диссоциирует по схеме
Hind ^
бесцветный
Н+
Ind"
мали новый
причем окраска остается бесцветной, поскольку концентрация окрашенного иона
ничтожно мала. При добавлении щелочи равновесие сильно смещается вправо,
так как ионы водорода связываются гидроксид-ионами в малодиссоциированные
молекулы воды. Поэтому в щелочной среде раствор приобретает малиновую ок-
окраску. При работе в кислых средах в качестве индикатора используют слабые
органические основания, диссоциирующие по схеме
IndOH ?=* Ind+ + ОН"
окраска I окраска II
Увеличение концентрации ионов водорода (повышение кислотности среды) сдви-
сдвигает равновесие вправо за счет связывания ионов ОН", и наблюдается изменение
цветности раствора. В табл. 16 приведены наиболее употребительные кислотно-
основные индикаторы и указан интервал рН, в котором происходит изменение
окраски.
Т а б л и д а 16. Свойства кислотно-основных индикаторов
Индикатор
Пикриновая кислота
га-Метиловый красный
Метиловый оранжевый
2,5-Динитрофенол
Метиловый красный
Бромтимоловый синий
Лакмус
Феноловый красный
Тимоловый синий
Фенолфталеин
Тимолфталеин
Ализариновый желтый
вял v™o
Интервал перехода рН
0-2,2
1,0-3,0
3,1-4,5
4,0 - 5,8
4,2-6,2
6,0-7,8
5,8 - 8,0
6,9 - 8,5
8,0 - 9,3
8,3 - 10,0
9,5 - 10,6
10,0-12,0
Изменение цвета
Бесцветный - желтый
Красный - желтый
То же
Бесцветный - желтый
Красный - желтый
Желтый - синий
Красный - синий
Желтый - красный
Желтый - синий
Бесцветный - красный
Бесцветный - синий
Желтый - фиолетовый
161
10. Обменные реакции между ионами. Произведение растворимости. Сильные
электролиты в достаточно разбавленных растворах практически полностью иони-
ионизированы. Поэтому уравнения реакций, записанные в молекулярной форме, на-
например
КС1 + NaNO3 ^ NaCl + KNO3
для реакций в растворах неприменимы, так как они не отражают истинного
состояния веществ в растворе (недиссоциированные молекулы в растворе отсутст-
отсутствуют) и вследствие этого не дают возможности судить о направлении процесса. В
рассмотренной системе в растворе присутствуют ионы К+, Na+, С1~ и NOj. Если
записать уравнение реакции в ионной форме
К* + С1- + Na+
NO"
К+ + NO-
Na+ + СГ
то становится очевидным, что в данной системе никакого взаимодействия не
происходит, так как и справа, и слева в уравнении реакции стоят одни и те же
ионы в равных концентрациях. Если вместо раствора NaNOe в приведенной реак-
реакции взять AgNC>3, то в ионной форме уравнение запишется следующим образом:
К+ + С1- + Ag+ + NO: «=* AgCll + К+ + N0-
т.е. в результате реакции образуется малорастворимый AgCl и равновесие смеща-
смещается вправо. Исключив из записи ионы К+ и NOj, присутствующие и в левой, и в
правой частях уравнения, получим так называемое сокращенное ионное уравне-
уравнение реакции:
Ag+ + С1"^ AgCll
Поскольку реакции между ионами в растворе представляют собой пример
химического равновесия, к ним приложим принцип смещения равновесий Ле
Шателье. В соответствии с этим принципом равновесие можно сместить в одну
сторону, если какое-либо вещество будет удаляться из сферы реакции по мере ее
протекания. Удаление вещества может быть осуществлено следующим путем:
1) образованием малорастворимого осадка; 2) выделением газообразного продукта
реакции; 3) образованием малодиссоциированного соединения; 4) образованием
прочного комплекса.
Практически важны обменные реакции в растворах с образованием нераство-
нерастворимых осадков. Осаждение веществ в результате химической реакции является
одной из основных операций в качественном и количественном химическом ана-
анализе, когда реакцию необходимо довести до конца и обеспечить полноту осажде-
осаждения.
Твердые вещества характеризуются самой различной растворимостью. Наряду
с хорошо растворимыми веществами существуют малорастворимые и практически
нерастворимые (в воде). Однако абсолютно нерастворимых веществ в природе
нет. Любое вещество, хотя бы в ничтожной степени, все же обладает раствори-
растворимостью. Рассмотрим равновесие между твердым осадком труднорастворимой соли
AgCl и ее ионами в водном растворе:
) ч=± Ag+
Константа равновесия имеет вид
При этом концентрация конденсированной фазы [AgCl(Te)] как постоянная
входит в величину К. Тогда константа равновесия определяется только
произведением концентраций ионов [Ag+] и [С]"] в растворе и называется
произведением растворимости:
ПР = [Ag+][C1-] *.
Для соединения AmBn
ПР = [A+]m[B-]n (VII.14)
Величина ПР характеризует растворимость труднорастворимого сильного
электролита при постоянной температуре. Значения произведения растворимости
изменяются в широких пределах, например ПР = 6,1-10'5, а ПР
CabU4
Л =
ВГ203
= ~ 10~96 при 20°С. В рассмотренном примере ПР = 1,6* 100. При отсутствии
AgLl
посторонних веществ произведение растворимости позволяет вычислить концент-
концентрацию насыщенного раствора труднорастворимого сильного электролита. Дейст-
Действительно, так как [Ag+] = [Cl"], то
[Ag+] = ЩР~= 41,6-Ю0 = 1,27-Ю-5 моль/л
и растворимость AgCl равна 1,27-10 моль/л.
Присутствие посторонних электролитов в растворе вызывает так называемый
солевой эффект, выражающийся в увеличении растворимости труднорастворимо-
труднорастворимого осадка. Причиной этого эффекта является усиление электростатического вза-
взаимодействия между ионами, в результате чего эффективная концентрация (актив-
(активность) ионов растворенного вещества уменьшается и новые их порции переходят в
раствор из осадка.
При введении в раствор вещества, способного.образовывать комплексы с иона-
ионами малорастворимого соединения, растворимость последнего также увеличивает-
увеличивается. Например, в аммиачных растворах осадок AgCl легко растворяется вследствие
образования аммиачного комплекса серебра [Ag(NH3J]Cl.
Таким образом, произведение растворимости характеризует гетерогенное рав-
равновесие осадок ^*=ь раствор для труднорастворимых сильных электролитов. В
данных термодинамических условиях величина ПР постоянна и не зависит от
присутствия в растворе посторонних веществ. В то же время растворимость веще-
вещества в значительной мере определяется влиянием одноименных ионов, солевым
эффектом и присутствием комплексообразователей.
11. Реакции нейтрализации и гидролиза. К обменным реакциям в растворах
относится, в частности, взаимодействие между кислотами и основаниями, в ре-
*Т#чнее, ПР — а. .я , где а — активность.
Ag* CT
*» * 164
зультате которого образуются соль и вода. Такие реакции называются реакциями
нейтрализации. В соответствии с закономерностями протекания ионных реакций
нейтрализация доходит до конца только тогда, когда единственным малодиссо-
циированным веществом в системе является вода, например
НС1 + КОН = КС1 + Н20
A)
В этой системе из четырех веществ лишь одно (Н20) малодиссоциировано, а все
остальные — сильные электролиты. Сокращенное ионное уравнение этой реакции
Н+ + ОН" = Н2О
показывает, что равновесие полностью смещено в сторону образования воды.
Доказательством того, что реакции нейтрализации сильных кислот сильными
основаниями сводятся к образованию воды из ионов гидроксония и гидроксид-
ионов, служит одинаковый тепловой эффект этих реакций в пересчете на 1 моль
образующейся воды: ДЯнейтр = —55,76 кДж/моль. Взаимная нейтрализация кис-
кислот и оснований, отличающихся по силе, до конца не протекает. Так, при вза-
взаимодействии слабой уксусной кислоты СН3СООН (НАс) с гидроксидом калия из
четырех веществ, присутствующих в системе, два слабо диссоциированы:
НАс + КОН ^ HjO + КАс B)
или в сокращенном ионном виде
НАс + ОН" ^ Н20 + Ас"
Эта реакция нейтрализации обратима. Обратная реакция (взаимодействие
соли с водой с образованием — в простейшем случае — кислоты и основания)
называется реакцией гидролиза соли.
Реакция нейтрализации будет обратимой и при взаимодействии сильной кис-
кислоты со слабым основанием:
НС1 + NH4OH ^ NH4C1 + Н20
а также при взаимодействии слабой кислоты и слабого основания:
НАс + NH4OH *=* NH4Ac + Н2О
C)
D)
Наиболее общим случаем реакций нейтрализации является взаимодействие
кислот и оснований, различающихся по силе, когда реакция не доходит до конца
в силу протекания обратной реакции — гидролиза образующейся соли. Если соль
образована сильной кислотой и сильным основанием [реакция A)], гидролиз ее не
протекает, так как в системе присутствуют три сильных электролита и лишь
один слабый (вода). Поэтому, согласно принципу смещения равновесия, примени-
применительно к ионным реакциям в растворе это равновесие целиком смещено вправо. В
реакциях B), C) и D) гидролиз протекает в заметной степени и реакция нейтра-,
лизации обратима, поскольку слабые электролиты присутствуют и в левой, "'и в
правой частях уравнения реакции. Таким образом, причина гидролиза заюцючает-
ся не только в участии в реакции слабой кислоты или основания, но и в диссо-
диссоциации самой воды. Сущность гидролиза с этой точки зрения состоит в том, что
катион соли (слабое основание) либо ее анион (слабая кислота) преимущественно
связывает соответственно ионы ОН" или Н+ с образованием слабого электролита
(соответственно основания или кислоты).
Рассмотрим гидролиз соли, образованной сильным основанием и слабой кис-
кислотой. К таким солям относятся NaAc, KCN и т.п. В частности, гидролиз ацетата
натрия протекает по уравнению
NaAc + Н20 ?=* НАс + NaOH
или в сокращенной ионной форме
Ас" + Н20 ^ НАс + 0Н-
(слабые электролиты представлены в недиссоциированной форме). Так как обра-
образующееся в результате гидролиза основание представляет собой сильный элект-
электролит, то концентрация гидроксид-ионов в растворе превалирует и реакция
среды щелочная. Таким образом, соль сильною основания и слабой кислоты гид-
ролизуется с увеличением концентрации гидроксид-ионов в растворе.
Если соль образована слабым основанием и сильной кислотой (NH4CI,
NH4NO3, А1С1з и т.п.), в процессе гидролиза главную роль играет катион соли,
связывающий гидроксид-ионы воды в слабое основание. При этом в растворе
накапливается избыток катионов водорода и реакция среды становится кислой:
NH4C1 + Н20 5=* NH4OH + HC1
или в сокращенной ионной форме
NH; + Н20 ^ NH4OH + Н+
Соль слабою основания и сильной кислоты гидролизуется с увеличением кон-
11,ентрации ионов водорода в растворе. Из приведенных примеров следует, что
реакция среды в результате гидролиза определяется тем продуктом гидролиза,
который является сильным электролитом.
Сложнее протекает гидролиз солей, образованных слабой кислотой и слабым
основанием (NH4CN, (NH4JCO3, NH4Ac). При этом в реакции гидролиза участву-
участвуют и катион, и анион соли, связывающие соответственно гидроксид-ионы и ионы
водорода воды. Поэтому реакция среды в результате гидролиза определяется
относительной силой образующихся слабой кислоты и слабого основания и, в
частности, может быть близка к нейтральной, хотя гидролиз протекает практи-
практически полностью. Например, ацетат аммония гидролизуется по уравнению
NH4Ac + Н20 ^ НАс + NH4OH
Константы ионизации уксусной кислоты и гидроксида аммония равны
tf(HAc) = 1,76-Ю и tf(NH4OH) = 1,79-10-s. Этим и объясняется нейтральная
реакция среды при гидролизе NH4Ac. При гидролизе цианида аммония
NH4CN + Н2О ч=± NH4OH + HCN
165
сравнение констант ионизации кислоты и основания [A'(HCN) = 7,2-100] позво-
позволяет сделать вывод, что реакция среды будет щелочной. Напротив, гидролиз
формиата аммония HCOONH4, протекающий по уравнению
HCOONH4 + Н20 ?=* НСООН + NH4OH
характеризуется слабокислой реакцией среды, что подтверждается сравнением
констант ионизации муравьиной кислоты (К — 1,8* КГ4) и гидроксида аммония.
Итак, при гидролизе солей, образованных одноосновными кислотами и одно-
кислотными основаниями, единственными продуктами гидролиза будут кислота и
основание. Соли многоосновных кислот и многокислотных оснований подвергают-
подвергаются ступенчатому гидролизу и в числе продуктов могут образоваться соответствен-
соответственно кислые и основные соли.
I. При гидролизе солей, образованных многоосновными кислотами и одноки-
слотными основаниями, возможны четыре варианта, аналогичные рассмотренным:
1) соли, образованные сильной кислотой и сильным основанием (Na2SO4),
гидролизу не подвергаются;
2) соли сильной кислоты и слабого основания гидролизуются ступенчато с
образованием кислых солей:
(NH4JSO4 + НОН s=± NH4HSO4 + NH4OH
кислая соль
или
NHJ + НОН ?=* NH4OH + Н+ (рН < 7)
Накапливающиеся в растворе ионы водорода препятствуют протеканию вто-
второй ступени гидролиза (до свободной кислоты);
3) при гидролизе солей слабых кислот и сильных оснований образуются кис-
кислые соли, но в растворе накапливаются свободные гидроксид-ионы:
Na3PO4 + НОН =?=* Na2HPO4 + NaOH
кислая соль
f + НОН ч=* HPOf + ОН" (РН > 7)
При избытке воды (в сильно разбавленном растворе) гидролиз протекает дальше:
Na2HPO4 + НОН *=± NaH2PO4 + NaOH
кислая соль
НРО2- + НОН *=* Н2РО- + 0Н-
При этом гидролиз также не доходит до конца, так как накопление в растворе
гидроксид-ионов препятствует образованию ортофосфорной кислоты Н3РО4;
4) гидролиз соли слабого основания и слабой кислоты протекает практически
до конца с образованием в промежуточной стадии кислых солей:
(NH4JS.+ НОН ?=* NH4HS + NH4OH
NH4HS + НОН ?=* NH4OH + ff2S
Таким образом, при ступенчатом гидролизе солей многоосновных кислот и
однокислотных оснований всегда образуются кислые соли, а полнота гидролиза
определяется относительной силой кислоты и основания.
II. Гидролиз солей многокислотных оснований и одноосновных кислот также
протекает ступенчато, но при этом образуются основные соли:
1) соли сильных кислот и сильных оснований (ВаС12) практически не гидро-
лизуются. Однако сильных многокислотных оснований сравнительно немного
(гидроксиды щелочно-земельных металлов);
2) соли сильной кислоты и слабого основания, например хлорид алюминия
AlClg, гидролизуются по следующей схеме:
А1С13 + НОН ^ А1 (ОН)С12 + НС1
основная соль
А13+ + НОН *=± А1(ОНJ+ + Н+ (рН < 7)
и далее
А1(ОН)С12 + НОН ?=* А1(ОНJС1 + НС1
А1(ОНJ+ + НОН s^ Al(OH); + Н+
До конца гидролиз не протекает, так как накапливающиеся в растворе ионы Н+
препятствуют образованию А1(ОН)з;
3) соли, образованные сильным многокислотным основанием и слабой кисло-
кислотой, также немногочисленны. Гидролиз цианида бария Ba(CNJ протекает лишь
до образования основной соли, но в растворе накапливаются гидроксид-ионы,
сообщая ему щелочную реакцию:
Ba(CNJ + НОН *=* Ba(OH)CN + HCN
CN" + НОН ?=^ HCN + ОН" (рН > 7);
4) соли слабой кислоты и слабого основания при гидролизе образуют основ-
основные соли, а реакция среды зависит от сравнительной силы кислоты и основания:
А1(АсK + НОН 5=^ А1(ОН)(АсJ + НАс
А1(ОН)(АсJ + НОН ?=± А1(ОНJАс + НАс
III. Гидролиз солей многоосновных кислот и многокислотных оснований также
может быть представлен четырьмя вариантами:
1) соли сильных кислот и сильных оснований встречаются сравнительно редко
и, как правило, нерастворимы в воде (BaSO4);
2) соли сильной кислоты и слабого основания образуют при гидролизе основ-
основные соли и свободную кислоту. При этом гидролиз до конца не протекает:
A12(SO4K + 2НОН =?=^ 2A1(OH)SO4 + H2SO4
Al3+ + НОН *=± А1(ОНJ+ + Н+ (рН < 7)^
IR7
3) соли, образованные слабой кислотой и сильным основанием, немногочислен-
немногочисленны и гидролизуются с образованием кислой соли и свободного основания
2BaS + 2НОН ?=* Ba(HSJ + Ba(OHJ
S?" + НОН ?=* HS- + OH" (рН > 7);
4) соли слабых кислот и слабых оснований полностью гидролизованы в раст-
растворе до свободной кислоты и свободного основания:
A12S3 + 6НОН = 2А1(ОНK + 3H2S
Такие соли в водных растворах существовать не могут.
Общие закономерности протекания гидролитических реакций представлены в
табл. 17.
С количественной точки зрения гидролиз в разбавленных растворах можно
охарактеризовать константой гидролиза. Например, при гидролизе ацетат-иона
Ас" + НОН ?=* НАс + ОН' '
константа равновесия имеет вид
[НАс] [ОН-]
Л-[Ас-][Н2О]'
Концентрация недиссоциированных молекул воды постоянна. Тогда
к [но] = [НАсДШП = к
2 [Ас"] г
(VII.15)
есть также величина постоянная и представляет собой константу гидролиза. Так
как гидролиз определяется диссоциацией слабой кислоты и воды, то константу
гидролиза удобно связать с константой ионизации кислоты и ионным произ-
произведением воды. Действительно, умножив числитель и знаменатель выражения
(VII.15) на [Н+], получим
_ [НАс][ОН-][Н+]
Так как
= Kw, а [Ас-][Н*]/[НАс] = ЛГв(НАс),
то
Кг = КШ/КЯ(Е Ас).
(VII.16)
Аналогично, константа гидролиза соли слабого основания и сильной кислоты
выражается отношением ионного произведения воды и константы ионизации
соответствующего основания. Например, при гидролизе NH4CI
Таблица 17. Гидролиз солей
Основание
Многокислотное
слабое [А](ОН)з]
Многокислотное
сильное [Ва(ОНJ]
Однокислотное слабое
(NH^OH)
Однокислотное силь-
сильное (NaOH)
Кислота
одноосновная
сальная
(НС1)
А1С13
Продукты основные со-
соли АК>на2> А1(онJа.
Гидролиз до конца не идет;
рН-<7
Bad2
Гидролиз практически
нейдет
NH4C1 .
Продукты: NH4OH, HQ.
Гидролиз идет до свобод-
свободных основания и кислоты;
рН<7
NaCl
Гидролиз не идет
слабая
(HCN)
A1(CNK
Продукты: основные со-
соли AHOHXCNJ, A1(OHJCN,
А1(ОНK и HCN. Гидролиз
протекает полностью; рН
зависит от силы кислоты и
основания
Ba(CNJ
Продукты: Ba(OH)CN,
HCN. Гидролиз до конца
не идет; рН>7 (редкий слу-
случай)
NH4CN
Продукты: NH4OH,HCN.
Гидролиз идет до конца;
рН зависит от силы кисло-
кислоты и основания
NaCN
Продукты: NaOH, HCN.
Гидролиз идет до свобод-
свободных кислоты и основания;
рН>7
многоосновная
сильная
<H2SO4)
A12(SO4K
Продукты: основные со-
соли A1(OH)S04 и H2SO4.
Гидролиз до конца не идет;
рН<7
BaSO4
Соли, практически не-
нерастворимые в воде
(NH4JSO4
Продукты: кислая соль
NH4HSO4 и Нг8О4. Гидро-
Гидролиз до конца не идет; рН < 7
Na2SO4
Гидролиз не идет
слабая
<H2S)
A12S3
Продукты: A1(OH)S,
А1(ОНK и H2S. Гидролиз
идет до конца; рН зависит
от силы кислоты и основа-
основания
BaS
Продукты: BaCHS^,
Ва(ОН>2. Гидролиз до кон-
конца не идет; рН >7 (редкий
случай)
(NH4bS
Продукты: NH4OH, H^.
Гидролиз идет до конца;
рН зависит от силы кисло-
кислоты и основания
Na2S
Продукты: NaOH, NaHS.
Гидролиз до конца не идет;
рН>7
Кг = A
(VII.17)
Константа гидролиза соли слабого основания и слабой кислоты NH4AC опре-
определяется соотношением
Кг = Kw/[Ka(EАс)КЪ(ХЕ4ОЩ] *.
(VII.18)
Воспользовавшись принципом Ле Шателье, рассмотрим влияние различных
факторов на положение гидролитического равновесия. Разбавление раствора
соли, что равносильно увеличению концентрации одного из реагентов (воды),
усиливает гидролиз. Напротив, гидролиз концентрированных растворов солей
протекает в меньшей степени. Повышение температуры влияет на гидролиз
главным образом вследствие резкой температурной зависимости степени
диссоциации воды. Поэтому при нагревании концентрация ионов Н+ и ОН" в
растворе сильно возрастает, что увеличивает вероятность связывания их с
образованием малодиссоциированной кислоты или основания, и при высоких
температурах гидролиз протекает полнее.
При обратимом гидролизе соли сильной кислоты и слабого основания, в ре-
результате которого увеличивается кислотность среды, добавление кислоты подав-
подавляет гидролиз. Если же в результате гидролиза образуется свободное сильное
основание, то гидролиз можно подавить, вводя в раствор дополнительное коли-
количество щелочи.
12. Окислительно-восстановительные реакции. Ионные реакции в растворах
можно подразделить на два типа. Реакции первого типа, примеры которых были
рассмотрены, не связаны с изменением заряда ионов и представляют собой ион-
но-обменные взаимодействия. Ко второму типу относятся реакции, в ходе
которых заряды ионов изменяются, что обусловлено передачей электронов от
одного иона другому. Так, при взаимодействии
2HI + Вг2 -> 2НВг + 12 или 2Г + Вг2 -» 2Вг" + 12
формальный заряд иода изменяется от —1 до 0, а заряд брома — от 0 до —1.
Взаимодействия, связанные с передачей электронов от одних атомов другим,
называются окислительно-восстановительными. Это название сложилось истори-
исторически в связи с тем, что процессы окисления металлов (Си + У2О2 -» СиО) и их
восстановления из оксидов (СиО + Н2 -* Си + Н2О) были давно известны. В
настоящее время к окислительно-восстановительным относят не только реакции с
участием кислорода, но и -все процессы, сопровождающиеся передачей электро-
электронов. Окислителем называется ион**, который при взаимодействии принимает
электроны, восстановителем — ион, отдающий электроны.
Окисление и восстановление представляют единый процесс, в ходе реакции
окислитель восстанавливается, а восстановитель окисляется. Окислитель может
проявить свою функцию только тогда, когда в растворе одновременно присутст-
присутствует и восстановитель, и наоборот. Так, FeClg является довольно сильным окис-
*К& и Кь — константы ионизации кислоты и основания.
* *В частном случае — нейтральный атом или молекула.
17П
лителем, но присоединение электрона может осуществиться только при наличии в
растворе какого-либо восстановителя, способного этот электрон отдать (например,
SnCl2):
2FeCl3 + SnCl2 ^ 2FeCl2 + SnCl4
или
3+ 2 + 2+ 4 +
2Fe + Sn ?=* 2Fe + Sn
Используя понятие о степени окисления, можно дать более общее определение
процессов окисления и восстановления. При окислении степень окисления увели-
увеличивается, при восстановлении — уменьшается. Коэффициенты в уравнении окис-
окислительно-восстановительной реакции опредачяют двумя методами: электронного
баланса и полуреакций. Первый метод можно рассмотреть на примере окисления
концентрированной соляной кислоты перманганатом калия. Эта реакция исполь-
используется в лабораториях для получения хлора:
КМпО4 + НС1 -> С12 + КС1 + МпС12 + Н20
Для определения стехиометрических коэффициентов в соответствии с приведен-
приведенными правилами необходимо знать степень окисления каждого атома:
+ 1+7-2 +1-1 0 +1-1 +2-1 +1-2
КМпО4 + НС1 -> С12 + КС1 + МпС12 + Н2О
В ходе взаимодействия только марганец и хлор изменяют степень окисления.
При этом степень окисления марганца уменьшается (Мп7+ — окислитель), а хло-
хлора — увеличивается (СГ — восстановитель). Затем составляют схемы, отражающие
процесс передачи электронов:
Мп7+ + 5 е" = Мп2+
2С1- -if = С1°
Поскольку число отданных и принятых электронов должно быть равным,
вводят дополнительные множители. Эти множители подбираются по правилу
нахождения наименьшего общего кратного и представляют собой стехиометриче-
ские коэффициенты при окислителе и восстановителе:
2КМпО4 + ЮНС1 -> 5С12 + КС1 + 2МпС12 + Н20
Далее уравнивают число атомов, не участвующих в окислении — восстановлении,
в следующем порядке: 1) число атомов металлов, не изменивших степень окисле-
окисления (калий); 2) ионы кислотных остатков, не изменивших степень окисления (в
приведенном примере ионы хлора, которые использованы для связывания К+ и
Мп2+):
2КМпО4 + 10НС1 + 6НС1 -+ 5С12 + 2КС1 + 2МпС12 + Н20
на окис- на свя-
ление "зывание
• ¦ ¦ - 171
3) число атомов водорода:
2КМпО4 + 16НС1 = 5С12 + 2КС1 + 2МпС12 + 8Н2О
4) проверка правильности расстановки коэффициентов осуществляется подсчетом
общего числа атомов кислорода слева и справа.
Полезно рассмотреть также порядок составления уравнений для реакций
самоокисления — самовосстановления, часто называемых реакциями диспропор-
ционирования *, например
КСЮз -> КСЮ4 + КС1
+ 5
В таких процессах один и тот же элемент (С1) выступает в качестве и окислителя
-1
+ 7
(восстанавливаясь до С1), и восстановителя (окисляясь до С1). Для облегчения
подбора коэффициентов удобно мысленно рассматривать обратные реакции:
КС! + КСЮ4 -> КС1О3 + КСЮз
Тогда получим
или окончательно
С1- - бе" -> С15+
С17++ 2 е- -+ С15+
КС1 + ЗКСЮ4 ^ ЗКСЮз + КСЮз
КС1
4КСЮ3 = ЗКСЮ4
В методе полуреакций (ионно-электронного баланса) коэффициенты в уравне-
уравнении окислительно-восстановительной реакции определяют с учетом конкретной
формы ионов, участвующих во взаимодействии. Преимущество метода состоит в
том, что нет необходимости пользоваться формальным представлением о степени
окисления. Кроме того, этот метод позволяет учесть влияние реакции среды на
характер окислительно-восстановительного процесса.
Восстановление перманганат-иона МпО^ в зависимости от реакции среды
может протекать до Мп2+ (в кислой среде), МпО2 (в нейтральной) и МпО^ (в
щелочной). Эти-взаимодействия описываются следующими схемами:
K2SO3 + КМпО4 + H2SO4 -» K2SO4 + MnSO4 + Н20 (рН < 7)
K2SO3 + KMnO4 + Н20 -» K2SO4 + MnO2 + КОН (рН 7)
K2SO3 + KMnO4 + КОН -* K2SO4 + K2MnO4 + H20 (рН > 12)
В соответствии со схемой A) в ходе взаимодействия идет восстановление:
МпО" + 8Н+ -> Мп2+ + 4Н2О
(полуреакция восстановления окислителя)
A)
B)
C)
*В результате диспропорционирования степень окисления одного и того же элемента и
повышается, и понижается.
172 '
Для соблюдения условия электронейтральности в левую часть выражения необ-
необходимо добавить пять электронов (это следует из подсчета положительных и
отрицательных зарядов слева и справа):
Ье- + МпО" + 8Н+ = Мп2+ + 4Н2О
Одновременно протекает процесс окисления сульфит-иона SO2" в сульфат-ион
по уравнению
5e + МпО- 4- 8H+ = Mn2+ 4- 4H2O
4- H2O = SQ2- + 2H* + 2e"
4- H2O = SOf + 2H+ 4- 2e"
(полуреакция окисления восстановителя)
Так как в соответствии с первой полуреакцией потребляется пять электронов, а
согласно второй освобождается два электрона, необходимо уравнять общее число
участвующих во взаимодействии электронов подбором дополнительных множите-
множителей по правилу наименьшего общего кратного
2
5
О - - ч
Суммируя, получаем
10е 4- 2МпО" + 16Н+ 4- 5SO2- + 5Н2О = 2Мп2+ + 8Н2О + 5SO2" + ЮН+ + Юе"
После приведения подобных членов
2МпО" + 6Н+ + 5SOf = 2Мп2+ + ЗН2О + 5SO2-
или в молекулярной форме с учетом одной дополнительной формульной единицы
K2SO4 (за счет связывания серной кислотой двух ионов К+ из КМпО4) получим
2КМпО4 + 5K2SO3 + 3H2SO4 = 2MnSO4 + 6K2SO4 + 3H2O
Используя тот же подход для нахождения стехиометрических коэффициентов
при взаимодействии в нейтральной среде [по схеме B)], запишем:
Зе" + МпО" 4- 2Н2О = МпО2 4- 4ОН"
SO|- 4- 2 ОН- = SO|" + Н 2О 4- 2е "
Суммируя, получаем
бе" 4- 2MnQ" 4- 4Н2О 4- 3SO2" 4- 6ОН" = 2МпО2 4- 8ОН" 4- 3SO|- 4- ЗН2О 4- 6е"
или после приведения подобных членов
173
2MnO"
H20
= 2MnO2 + 20H"
3SO2/
B молекулярной форме это взаимодействие описывается уравнением
' 2КМпО4 + Н2О + 3K2SO3 = 2МпО2 + 2КОН + 3K2SO4
Аналогичным образом применение метода полуреакций к взаимодействию в
щелочной среде [см. схему C)] дает следующие уравнения ионно-электронного
баланса:
е- +МпО" = МпО2 "
SOf + 2ОН- = SQ2- + Н2О+ 2е"
2 е" + 2MnO;
SO2
+ 2ОН" = 2MnQ2- +
+ H20 + 2 е-
или
2МпО" +
В молекулярной форме
+ 2ОН- = 2МпО2" +
+ Н2О
2KMnO4 + K2SO3 + 2K0H = 2K2Mn04 + K2SO4 + Н20
Таким образом, использование метода полуреакций для составления урав-
уравнений окислительно-восстановительных взаиКЬдействий позволяет, во-первых,
избежать формального представления о степени окисления, во-вторых, соста-
составить сокращенное ионное уравнение окислительно-восстановительной реакции и,
в-третьих, выявить влияние среды на характер процесса. Из рассмотренных при-
примеров следует, что в процессе восстановления МпО^ -» Мп2+ участвуют ионы водо-
водорода (необходима кислая среда), восстановление МпО^ -» МпО2 протекает в ней-
нейтральной среде с участием молекул Н2О, а превращение МпО^ -> МпО|" происхо-
происходит только в щелочной среде (во взаимодействии участвуют ионы ОН").
Метод полуреакций применим лишь для описания и подбора коэффициентов
окислительно-восстановительных процессов в растворах. Метод электронного
баланса более универсален, поскольку позволяет на формальной основе устанав-
устанавливать стехиометрические отношения в процессах окисления — восстановления в
любых гомогенных и гетерогенных системах.
13. Гетерогенные реакции в растворах. В отличие от
гомогенных реакций, протекающих во всем объеме
системы, гетерогенные взаимодействия представляют
собой реакции, осуществляемые на границе раздела
фаз.
Рассмотрим металлический стержень, погруженный в
воду (рис. 81). Поскольку совершенно нерастворимых
веществ не существует, любой металл хотя бы в нич-
ничтожной степени растворяется в воде, что обусловлено
Рис. 81. Образование двойно- возрастанием энтропии при растворении. При этом в
го электрического слоя при раствор переходят положительно заряженные ионы
погружении металлического металла, а не нейтральные атомы. В металле же оста-
электрода в воду ются избыточные свободные электроны, т.е. на границе
174 "
е
Me
в в
в в
в в
е в
в в
в в
в е
%1.1
©
е—
раздела двух фаз возникает двойной электрический слой, электрическое поле в
котором направлено так, что препятствует дальнейшему растворению металла в
воде. В результате нарушения условия электронейтральности и возникновения на
границе раздела разности потенциалов ионы металла в растворе не диффундиру-
диффундируют в его объем, а скапливаются в тонком приповерхностном слое раствора. Обра-
Образующийся раствор, состоящий из гидратированных катионов металла (поверхно-
(поверхностный раствор), является практически двумерным образованием и концентрация
этого раствора может достигать значительной величины при общей малой раство-
растворимости металлов в воде. Для всех металлов, погруженных в воду, характерен
одинаковый механизм образования двойного электрического слоя: металлический
электрод заряжается отрицательно, а в поверхностном растворе концентрируются
гидратированные положительные ионы металла. Хотя качественно картина обра-
образования двойного электрического слоя одинакова для всех металлов, погружен-
погруженных в воду, количественно концентрация поверхностного раствора (с) может
очень сильно отличаться для разных металлов.
Если металлический электрод погрузить в раствор его соли, то процессы,
протекающие на границе металл — раствор, будут аналогичны рассмотренным
выше. Отличие состоит лишь в том, что для достижения равновесия при образо-
образовании двойного электрического слоя требуется меньшее растворение металла, так
как частично ионы металла уже присутствуют в растворе его соли.
Очевидно, что чем выше концентрация соли в растворе, тем меньшей должна
быть величина скачка потенциала на границе металл — раствор. Таким образом,
она зависит от концентрации раствора. Кроме того, эта величина зависит от
температуры и ряда других факторов. Но прежде всего она определяется приро-
природой металла. Поэтому для сравнения электродных потенциалов необходимо выб-
выбрать некоторые стандартные условия. Обычно сравнение производят при стан-
стандартной температуре 25°С B98 К), давлении 1,013-105 Па и в растворе с актив-
активностью одноименного иона, равной единице (в 1М растворе). Абсолютное значе-
значение электродного потенциала измерить невозможно, поскольку введение любых
измерительных зондов неизбежно приводит к появлению новой контактной раз-
разности потенциалов. В связи с этим измеряют разность потенциалов между дан-
данным электродом и некоторым электродом сравнения, потенциал которого условно
принимают равным нулю. В качестве стандартного электрода сравнения исполь-
используют так называемый стандартный водородный электрод *. Электрод изготовля-
изготовляют из губчатой платины с сильно развитой поверхностью (платиновая чернь) и
погружают в раствор кислоты с активностью ионов водорода, равной 1 моль/л.
Через раствор пропускают газообразный водород под давлением 1,013 -105 Па,
который адсорбируется платиной**. Электродные потенциалы, измеренные по
отношению к водородному электроду в стандартных условиях, называются стан-
стандартными электродными потенциалами. В зависимости от величины и знака
Поверхность электрода сложного состава приобретает потенциал, свойственный менее
благородному компоненту. Для платинового электрода, насыщенного водородом, потенциал
определяется равновесием водорода с собственными катионами:
Н2(г) т=* 2Н(адс) ^ 2Н+ (р-р) + 2е"
* * Аналогичным образом могут быть созданы и другие газовые электроды, например
кислородный, хлорный и т.п.
175
стандартного электродного потенциала все металлы можно расположить в ряд
стандартных электродных потенциалов, который фактически представляет собой
ряд активности, эмпирически установленный Н.Н.Бекетовым по взаимному вытес-
вытеснению металлов (табл. 18). Аналогичный ряд стандартных электродных потен-
потенциалов для неметаллов не установлен столь полно, так как механизм электрод-
электродных реакций гораздо сложнее.
Т а б л
Элемент
Цезий
Литий
Рубидий
Калий
Барий
Стронций
Кальций
Натрий
Лантан
Магний
Бериллий
Алюминий
Марганец
Цинк
и ц а 18. Стандартные электродные потенциалы ыеталлов
Электродная
реакция
Cs+ +
Li+ +
Rb+ +
К+ +
Ba2t -
Sr2+ 4
Ca2+ -
Na+ +
е" ^^
е" ^=^
е" ;!=:й
t ^^
f 2e-<?=
- 2е"^=
<г 2е ^
е" ^^
La3+ + Зе" ^
Mg2+
Be2+ -
Al3+ H
Mn2+
Zn2t -,
+ 2е'^
f- 2e"^=
- Ъе*=
+ 2е"^
h 2e" *=
Cs
Li
Rb
К
=*Ва
^Sr
— Са
Na
-La
— Mg
— Be
- Al
— Mn
-Zn
E°, В
-3,08
-3,02
-2,99
-2,92
-2,90
-2,89
-2,87
-2,71
-2,37
-2,34
-1,70
-1,67
-1,05
-0,76
Элемент
Хром
Железо
Кадмий
Кобальт
Никель
Олово
Свинец
Водород
Медь
Серебро
Палладий
Ртуть
Платина
Золото
Электродная
Сг3+
Fe2+
Cd2'
Со2+
Ni2+
Sn2+
Pb2+
2H+
Cu2+
Ag+
Pd2+
Hg2+
Pt2+
Au+
реакция
+ 3e" :?=± Cr
+ 2e" :?=± Fe
+ 2e" ^^ Cd
+ 2e" :?=± Co
+ 2e" ^ Ni
+ 2e' 5=^ Sn
+ 2e" ^=^ Pb
+ 2e" ^^ H2
+ 2e" ?=^ Cu
+ t ^^Ag
+ 2e' ^^ Pd
+ 2e" ^=i Hg
+ 2e" ^^ Pt
+ e" т=* Аи
E\ В
-0,71
-0,44
-0,40
-0,28
-0,25
-0,14
-0,13
0,00
+0,34
+0,80
+0,83
+0,85
+ 1,20
+ 1,68
На основании ряда стандартных электродных потенциалов можно сделать два
практически важных заключения: 1) металлы, обладающие более отрицательным
электродным потенциалом, способны вытеснять менее активные металлы (с более
положительным потенциалом) из водных растворов их солей. Например, взаимо-
взаимодействие Zn + Cu2+ -» Cu + Zn2+ протекает только слева направо. Обратная реак-
реакция практически невозможна; 2) металлы, выступающие в качестве отрицательно-
отрицательного электрода по отношению к водородному, вытесняют водород из кислот, а
металлы с более положительным элект-
электродным потенциалом не обладают этим
свойством.
Ячейка для измерения электродного
потенциала (рис. 82) — простейший при-
пример электрохимического (гальваническо-
(гальванического) элемента. Э.д.с. этого элемента возни-
возникает за счет протекания окислительно-
восстановительной реакции. Движущей
силой химической реакции является
убыль свободной энергии Гиббса АО. С
другой стороны, убыль свободной энер-
энергии Гиббса определяет максимальную
работу химической реакции. Для
реакции, осуществляемой в условиях
ZnS04
Рис. 82. Схема измерения стандартного
электродного потенциала относительно во-
водородного электрода
176 . :
гальванического элемента, работа А, производимая системой, равна А — iUt =;
= QU, где / — сила тока в цепи; {/—падение напряжения; t — время;
Q — количество электричества.
Если в системе протекает ток, то часть энергии системы расходуется на выде-
выделение джоулевой теплоты и процесс протекает необратимо. Максимальную работу
гальванический элемент совершает, если реакция протекает обратимо в равновес-
равновесных условиях. Это возможно при / = 0, когда э.д.с. элемента скомпенсирована
равной по величине и противоположной по знаку внешней э.д.с. (или элемент
замкнут на бесконечно большое сопротивление). При этом условии U — Е (напря-
(напряжение элемента равно его э.д.с.) и максимальная работа
Лтах = QE.
(VII.19)
Для 1 моль вещества, вступившего в реакцию, Q = nF, где п — число электро-
электронов, принимаемых или отдаваемых веществом (заряд иона); F = 96485 Кл/моль —
постоянная Фарадея, т.е. количество электричества, необходимое для выделения
одного эквивалента вещества. Тогда максимальная работа химической реакции
равна убыли энергии Гиббса:
-ДС = Атах = nFE.
При стандартных условиях
-AG° = nFE°,
(VII.20)
(VII.21)
где Е° — э.д.с. гальванического элемента при стандартных условиях.
Взаимосвязь между э.д.с. гальванического элемента и изменением свободной
энергии Гиббса дает возможность непосредственного определения величины Д G.
Этот метод — один из наиболее чувствительных, так как реакцию в гальваниче-
гальваническом элементе можно осуществить в условиях, максимально приближенных к
равновесным, а измерение э.д.с. производится с высокой точностью.
Согласно принятой в уравнениях (VII.20) и (VII.21) системе знаков, процесс
самопроизвольно протекает тогда, когда э.д.с. соответствующего элемента поло-
положительная. В качестве примера рассмотрим гальванический элемент (элемент Да-
Даниэля—Якоби), состоящий из медного и цинкового электродов в растворах их
солей (рис. 83). Для определения э.д.с. этого
элемента сравнивают стандартные электродные
потенциалы цинкового и медного полуэлементов.
При записи электродных реакций принято, что
окисленная форма находится в левой части
уравнения, а восстановленная — в правой:
Zn2+ -(- 2е" = Zn°; E°(Zn2+/Zn°) = -0,76 В
и
Cu2+ + 2е" = Cu°; ?°(Cu2+/CV) = +0,34 В.
Отрицательный знак E°(Zn2+/Zn) свидетельству-
свидетельствует о том, что переход цинка из окисленной фор- Рис. 83. „Схема медно-щшкового
мы (Zn2+) в восстановленную (Zn°) в паре с водо- гальванического элемента Даниэля -
родным полуэлементом самопроизвольно проте- Якоби
177
¦lii
кать •не может. Термодинамически возможен обратный процесс Zn° =. Zn2t +
+ 2е", для которого электродный потенциал положителен [?"°(Zn°/Zn2+) =
= +0,76 В]. Для медного полуэлемента в комбинации с водородным термодина-
термодинамически осуществим процесс восстановления, о чем свидетельствует положитель-
положительный знак электродного потенциала. Таким образом, в элементе Даниэля—Якоби
осуществляются следующие полуреакции:
анодный процесс: Zn° = Zn2+ + 2е~ (окисление цинка), Е° = +0,76 В;
катодный процесс: Си2+ + 2е~ = Си° (восстановление меди), Е° = +0,34 В.
После суммирования получим
Zn° + Cu2+
I 2e- T
= Zn2+
Cu°
Э.д.с. элемента равна сумме +0,76 + 0,34 = 1,10 В. Положительный знак э.д.с.
определяет направление самопроизвольного протекания реакции слева направо. В
соответствии с (VII.21) изменение энергии Гиббса рассчитывают по формуле
Д G" = -96485 пЕ°.
Для элемента Даниэля &G° = —96485-2-1,1 = —212267 Дж/моль « —212
кДж/моль. Большая отрицательная величина AG° свидетельствует о самопроиз-
самопроизвольном восстановлении меди цинком.
Для вычисления стандартной э.д.с. элемента на основании стандартных элект-
электродных потенциалов следует из величины потенциала более благородного компо-
компонента (более положительного) вычесть значение потенциала менее благородного
компонента. Например, элемент, составленный из натриевого и цинкового полу-
полуэлементов, обладает э.д.с.
?°(Na - Zn) = F(Zn2+/Zn) - ?°(Na+/Na) = -0,76 - (-2,71) = +1,95 В,
а элемент, состоящий из медного и золотого полуэлементов, имеет э.д.с.
Е°(Си - Аи) = ?°(Аи7Аи) _ ?"(Си2+/Си) = 1,68 — 0,34 = +1,34 В.
т.е. в первом случае в качестве анода выступает натриевый полуэлемент (натрий
окисляется Na° -+ Na+ + e"), а катодом служит цинковый полуэлемент (цинк
восстанавливается Zn2+ + 2е~ -> Zn°). Во втором элементе происходит окисление
меди и восстановление золота (на аноде Си0 -+ Си2+ + 2е~, на катоде Аи+ + е" ->
До сих пор в расчетах использовались стандартные электродные потенциалы.
В то же время на величину электродного потенциала существенно влияют темпе-
температура и концентрация раствора соли. Эта зависимость выражается так называ-
называемым уравнением Нернста:
Ej = tt H — Ш — . yvVV.LL)
178 ' - .
Из уравнения (VII.22) следует, что при с = с0 электродный потенциал Е = Е°,
т.е. в стандартных условиях (с = с0 = 1 моль/л) электродный потенциал равен
стандартному потенциалу. При росте концентрации по сравнению со стандартной
(с > со) электродный потенциал увеличивается (становится более положитель-
положительным). Так же действует и увеличение температуры. При с < cq ln(c/co) < 0 и
электродный потенциал становится меньше стандартного значения.
Таким образом, составив гальванический элемент из одинаковых электродов,
погруженных в растворы их солей с различными концентрациями, получим так
называемый концентрационный элемент, э.д.с. которого определяется по уравне-
уравнению (VII.22) соотношением концентраций растворов, так как величина Е° для
обоих полуэлементов будет одинаковой.
Каждый элементарный акт окисления — восстановления связан с передачей
электронов, что может быть записано в виде
Ох + е" = Red (a)
где Ох — окисленная форма (oxydation — окисление); Red — восстановленная
форма (reduction — восстановление). Окислительно-восстановительный процесс
(или редокс-процесс) состоит из двух полуреакций: катодной (восстановление)
и анодной (окисление). Катодный процесс описывается уравнением (а), а анод-
анодный —
Red ' = Ох' + е" (б)
Общее уравнение редокс-реакции получим суммированием полуреакций (а) и (б):
Ох + Red' = Ох' + Red
Если окислительно-восстановительное взаимодействие протекает в одном раст-
растворе, то система не производит работы, так как суммарный ток, обусловленный
передачей электронов от восстановителя к окислителю, равен нулю. Это связано
с беспорядочным перемещением ионов и электронов. Если же катодный и анод-
анодный процессы пространственно разделить (как это сделано в гальваническом
элементе), то при замыкании внешней цепи наблюдается направленное перемеще-
перемещение электронов от анода к катоду, а в растворе осуществляется движение анионов
от катода к аноду (см. рис. 83).
В гальванических элементах могут реализоваться два принципиально различ-
различных типа электрохимических редокс-взаимодействий. В первом случае сами
электроды участвуют в окислительно-восстановительной реакции, как, например,
в элементе Даниэля—Якоби. Гальванические цепи такого типа можно назвать
редокс-цепями с расходуемыми или активными электродами. Во втором случае
вещество электродов инертно по отношению к реакции, протекающей в растворе.
Рассмотрим элемент, схема которого приведена на рис. 84. В отличие от элемента
Даниэля—Якоби здесь электроды не участвуют во взаимодействии, а являются
лишь передатчиками электронов между ионами, находящимися в растворах.
Левый полуэлемент представляет собой раствор, состоящий из смеси солей Sn2+ и
Sn4+, в который погружен платиновый электрод. На поверхности электрода уста-
устанавливается равновесие Sn4t + 2e~ ^=^ Sn2+, которое и определяет потенциал
179
электрода. В правом полуэлементе платиновый электрод погружен в раствор
смеси солей Fe2+ и Fe3+. На его поверхности осуществляется равновесие Fe3+ +
+ е" 5=^ Fe2+, определяющее потенциал электрода.
Эти потенциалы, измеренные по отношению к стандартному водородному
электроду, называются стандартными редокс-потенциалами, если активность
окисленной и восстановленной форм в растворе равна единице A моль/л). Подоб-
Подобно ряду стандартных электродных потенциалов для металлов, существует ряд
редокс-потенциалов окислительно-восстановительных пар. Значения стандартных
редокс-потенциалов для рассматриваемых полуреакций:
Sn4+ + 2е" = Sn2+; ?°(Sn4+/Sn2+) = +0,15 В, (а)
Fe3+ + е" = Fe2+; ?°(Fe3+/Fe2+) - +0,77 В. (б)
Э.д.с. элемента равна
Е° - ?°(Fe3+/Fe2+) - ?°(Sn4+/Sn2+) = 0,77 - 0,15 = 0,62 В.
Таким образом, полуреакция (а) протекает в обратном направлении (как процесс
окисления) и соответствующий полуэлемент является анодом:
Sn2+ =
2e"
А полуэлемент Fe3+/Fe2+ представляет собой катод, на котором происходит вос-
восстановление:
Fe3+ + е- = Fe2+
Общее уравнение реакции имеет вид
Sn2+ + 2Fe3t = Sn4+ + 2Fe2+
Э.д.с. не зависит от стехиометрических коэффициентов уравнения, поэтому в
расчетах, связанных с использованием стандартных редокс-потенциалов, они не
учитываются. Однако изменение свободной энергии Гиббса AG [см. уравнение
(VII.21)] зависит от числа электронов, участвующих во взаимодействии, а следо-
следовательно, и от стехиометрических коэффициентов в уравнении реакции. Гальва-
Гальванические цепи подобного типа (рис. 84) называ-
называются редокс-цепями с неактивными (инертными)
электродами. • Величины редокс-потенциалов
определяются природой раствора, соотношением
концентраций окисленной и восстановленной
форм и температурой. Эта зависимость количест-
количественно выражается уравнением Нернста, которое
для редокс-систем имеет вид
Е=Е° + [flT/(nl9]ln([Ox]/[Red]). (VII.23)
А-
Рис. 84. Окислительно-восстано-
витальный (редокс) элемент с
инертными электродами
,180-
При [Red] = [Ох] = 1 МОЛь/л Е — Е°. Если
[Ох] > [R*d], редокст-потенциал становится более
положительным по сравнению с Е°. Если [Ох] <
< [Red], то потенциал смещается в отрицательную сторону. В ряду редокс-
потенциалов (табл. 19) отмечают следующие закономерности.
1. Если стандартный редокс-потенциал отрицателен, то полуэлемент по отно-
отношению к водородному выступает в качестве восстановителя (окисленная форма
устойчивее). Восстановительная активность тем выше, чем более отрицателен
редок с-п оте н ци ал.
2. Если стандартный редокс-потенциал положителен, то полуэлемент по отно-
отношению к водородному является окислителем (устойчивее восстановленная фор-
форма). Чем положительнее величина редокс-потенциала, тем выше окислительная
активность полуэлемента.
3. С увеличением э.д.с. редокс^епи окислительно-восстановительные реакции
протекают энергичнее.
Таким образом, ряд редокс-потенциалов позволяет количественно оценить
активность окислителя и восстановителя, направление и интенсивность протека-
протекания окислительно-восстановительной реакции.
14. Химические источники тока. Электролиз. Редокс-элементы с активными
или инертными электродами могут служить химическими источниками тока. При
работе этих источников энергия химической реакции непосредственно превраща-
превращается в электрическую в соответствии с (VII.23).. Химические источники тока
подразделяются на аккумуляторы и гальванические элементы. Последние допус-
допускают лишь однократное использование, поскольку один из электродов (напри-
(например, цинк в элементе Даниэля—Якоби) необратимо расходуется. Аккумуляторы
можно использовать многократно, так как их работоспособность может быть
восстановлена при пропускании тока в обратном направлении от внешнего источ-
источника.
В качестве примера обратимого химического источника тока рассмотрим
принцип действия так называемого кислотного аккумулятора. Электроды свинцо-
свинцового кислотного аккумулятора изготавливают заполнением ячеек свинцовой ре-
решетки пастой из оксида свинца РЬО. Электролит — 30%-ная (d = 1,2 г/см3)
серная кислота. При погружении электродов в серную кислоту на поверхности
пластин за счет реакции
РЬО + H2SO4 = PbSO4 + Н2О
образуется труднорастворимая соль — сульфат свинца. В этом состоянии оба
электрода имеют один и тот же состав, окислительно-восстановительное взаимо-
взаимодействие невозможно и аккумулятор разряжен (рис. 85). При зарядке через акку-
аккумулятор пропускают постоянный электрический е-
ток. При этом на отрицательном электроде про- _"pl|LJ_. +
исходит процесс восстановления ' '
Л* i-l-i
PbSO4 + 2 е- + 2Н+ = Pb + H2SO4
или
РЬ2+ + 2 е" = Pb
На положительном электроде
PbSO4 - 2 с- + SOf = Pb(SO4J
(Pb2+ - 2 е- = Pb4+)
1
Oh
Рис. 85. Схема работы свинцового
кислотного аккумулятора при заря-
заряде (а) и разряде (б)
181
Таблица 19. Окислительно-восстановительные
потенциалы (инертный электрод — платина)
Электродная
система
н2>н-
н2, он-
Сг3+, Сг2+
уЗ+^ у2+
Н+, SOf, S2O2"
Sn4+, Sn2+
Cu2+, Cu+
CIO", CIO", OH-
Fe(CN)|-, Fe(CNL"
H+, H2O2, O2
Fe3+, Fe2+
H+, NO", HNO2
H+, NO", NO
H+, N2O4, HNO2
H+, СЮ", СЮ-
H+, 10", I2
H+, O2
O3, O2, OH-
Tl3+, Tl+
H\ Cr2O2;, Cr3+
H+, PbO2, Pb2+
H+, MnO;, Mn2+
Ce4+, Ce3+
H+, HC1O, Cl2
H+, MnO', MnO2
•H+, H2O2
Co3+, Co2+
S2O2-, SO2-
H+, O3, O2
H+, О
Электродная
реакция
H2 + 2e ^=* 2H-
2Н2О + 2е *=± Н2 + 2ОН"
Сг3+ + е" ?=± Сг2+
V3+ + е 5=^ V2+
2SO2- + 4Н+ + 2е" ^=^ S2O2" + 2Н2О
Sn4+ + 2е 5=i Sn2+
Cu2+ + е *=* Cu+
СЮ" + Н2О + 2е 5=i СЮ" + 2ОН"
Fe(CN)|- + e 5=± Fe(CNL"
I' + 2е 5=* ЗГ
О2 + 2Н+ + 2е ?=± Н2О2
Fe3+ + е ?=* Fe2+
NO" + ЗН+ + 2е 5=* HNO2 + Н2О
NO' + 4Н* + Зе" 5=^ N0 + 2Н2О
N2O4 + 2Н+ + 2е ?=± 2HNO2
СЮ" + 2Н+ + 2е ^=^ СЮ" + Н20
10" + 6Н+ + Ье *=± 1/2 12 + ЗН2О
02 + 4Н+ + 4е" 5=^ 2Н2О
Оз + Н20 + 2е ^02 + 20Н"
Т13+ + 2е 5=* Т1+
Сг2О2- + 14Н+ + бе" ^=i 2Cr3+ + 7Н2О
РЬО2 + 4Н+ + 2е" ^=* РЬ2+ + 2Н2О
МпО; + 8Н+ + 5е" ^^ Мп2+ + 4Н2О
Се4+ + е" ^=* Се3*
НСЮ + Н+ + е" ^=* 1/2С12 + Н20
МпО; + 4Н+ + Зе" ^=^ МпО2 + 2Н2О
Н2О2 + 2Н+ + 2е" ?=^ 2Н2О
СоЗ* + е- ^=i Co2+
S2O|- + 2е *=*¦ 2SO2-
О3 + 2Н+ + 2е 5=*О2+ Н2О
О + 2Н+ + 2е' 5=* Н2О
Г, В
-2,23
-0,82
-0,41
-0,23
-0,22
+0,15
+0,15
+0,33
+0,36
+0,54
+0,68
+0,77
+0,94
+0,95
+ 1,07
+ 1,20
+1,22
+ 1,23
+ 1,24
+ 1,25
+ 1,33
+ 1,50
+ 1,51
+ 1,61
+ 1,63
+ 1,70 '
+ 1,78
+ 1,81
+2,01
v +2,07
+2,42
и далее
Pb(SO4J + 2Н2О = РЬО2 + 2H2SO4
Таким образом, после зарядки один электрод аккумулятора представляет
собой губчатый металлический свинец, а другой — диоксид свинца (рис. 85, б).
При работе аккумулятора (разрядке) процессы на электродах протекают в обрат-
обратном направлении. Окисление на аноде:
РЪ + SOf =PbSO4 + 2e"
(РЬ° - 2 е" = РЬ2+)
Восстановление на катоде:
РЬО2 + 2H2SO4 = Pb(SO4J + 2Н2О
и
Pb(SO4J + 2 е" + 2Н+ = PbSO4 + H2SO4
(Pb4+ + 2 е" = Pb2+)
Общее уравнение реакции, отражающее работу свинцового аккумулятора, имеет
вид
разрядка
РЬО2 + Pb + 2H2SO4 *=* 2PbSO4 + 2H2O
4
зарядка
Из уравнения видно, что в процессе зарядки увеличивается концентрация серной
кислоты. При разрядке плотность электролита уменьшается, поскольку образу-
образующаяся вода разбавляет электролит.
На примере работы свинцового аккумулятора показана возможность изменения
направленности химического процесса за счет приложения к ячейке внешней
э.д.с. В принципе любая электрохимическая реакция, протекающая в гальваниче-
гальваническом элементе, может быть осуществлена в противоположном направлении, если
приложить встречную э.д.с, превышающую э.д.с. элемента. При этом элемент
работает как электролизер.
Электролиз — процесс раздельного окисления и восстановления на электродах,
осуществляемый за счет протекания тока от внешнего источника э.д.с. Если в
гальваническом элементе энергия химической реакции преобразуется в электри-
электрический ток, то в электролизере идет обратный процесс — преобразование элект-
электрической энергии в химическую. При электролизе, как и в гальваническом эле-
элементе, на аноде происходит окисление, а на катоде — восстановление. Однако
при этом анодом служит положительный электрод, а катодом — отрицательный.
Как и в гальваническом элементе, при электролизе могут быть использованы
активные (расходуемые) и инертные (нерасходуемые) аноды. Активный анод,
окисляясь, посылает в раствор собственные ионы. Инертный анод является лишь
передатчиком электронов, а сам химически не изменяется. В качестве инертных
анодов обычно используют графит и платину. Рассмотрим простейший пример
электролиза расплава хлорида натрия с применением угольных электродов. Рас-
Расплав NaCl диссоциирует с образованием ионов Na* и С1~:
NaCl *=ь Na+ + С1"
На катоде идет восстановление:
Na* + е" = Na°
На графитовом аноде — окисление ионов хлора:
СГ - е- = СГ и 2СГ = C12J
Этот процесс используется в промышленности для получения активных металлов:
щелочных, щелочно-земельных, бериллия, магния, алюминия. Все эти металлы —
энергичные восстановители, и для их выделения из соединений требуется
еще более активный восстановитель, роль которого и выполняет катод электроли-
электролизера.
Примером электролиза с активным анодом может служить электрохимическое
рафинирование меди. При этом анод представляет собой пластину черновой
меди, подлежащую очистке, а катод — пластину из химически чистой меди.
Электролитом служит водный раствор сульфата меди. При рафинировании медь
окисляется на аноде с переходом ионов Си2+ в раствор:
Си° - 2е" = Си2+
а на катоде выделяется чистая медь из раствора:
+ 2 е" = Си°
Примеси, содержащиеся в медном аноде, оседают в виде так называемого
анодного шлама (неметаллы) или остаются в растворе (металлы).
Количественно электролиз описывается двумя законами Фарадея.
1. Масса выделяющегося на электроде вещества пропорциональна количеству
электричества, протекшего через электролизер:
т = kit- kQ,
где / — сила тока; t — время протекания тока; Q = It — количество электричест-
электричества; к — коэффициент пропорциональности, значение которого зависит от выбран-
выбранной системы единиц. Если Q = 1 Кл, то т = к.
Масса вещества, выделяющаяся при прохождении 1 Кл электричества, называ-
называется электрохимическим эквивалентом.
2. Для выделения на электроде одного эквивалента любого вещества необхо-
необходимо затратить одно и то же количество электричества, равное постоянной Фара-
Фарадея F — 96485 Кл/моль. Действительно, один эквивалент вещества содержит
N = 6,02322 *1023 частиц. Чтобы восстановить такое число однозарядных ионов
А
на катоде, необходимо затратить количество электричества
F= N е= 6,02322¦ 1023 моль"»-1,6021 • 109 Кл = 96485 Кл/моль,
А
где заряд электрона е = 1,6021 *109 Кл.
Обобщая оба закона Фарадея, можно записать
r m = 9(It/F) = Э(Л/96485),
(VII.24)
Где m — масса вещества, г; Э — эквивалент вещества, г/моль; I — сила тока, А;
t — продолжительность электролиза, с; F — постоянная Фарадея, Кл/моль.
ГЛАВА VIII. ТВЕРДОЕ СОСТОЯНИЕ. ТВЕРДЫЕ РАСТВОРЫ
Большинство известных простых и сложных веществ в обычных условиях
представляют собой твердые тела. Одной из важнейших задач современной неор-
неорганической химии является исследование свойств твердых тел в зависимости от
их состава и структуры. Изучение твердых тел, которое интенсивно развивается в
течение последних десятилетий и обусловлено растущими потребностями различ-
различных областей новой техники, заставляет с новых позиций подойти к пониманию
фундаментальных законов общей химии (представления о валентности, стехио-
метрические законы и др.). Успехи химии металлов, химии полупроводников и
вообще химии твердого состояния оказывают в настоящее время определяющее
влияние на развитие химической науки в целом и неорганической химии в част-
частности.
1. Понятие о твердой фазе. Термодинамическое определение фазы (см.. § 9
гл. II) включает следующие основные положения. Во-первых, подразумевается,
что система находится в состоянии термодинамического равновесия. Во-вторых,
каждая фаза, составляющая систему, должна быть физически однородной ее
частью. При этом химическая однородность фазы не обязательна. Примерами
физически однородных (однофазных), но химически неоднородных систем яв-
являются воздух — молекулярный раствор газов, не взаимодействующих друг с
другом, молекулярные водные растворы неэлектролитов и т.п. В-третьих, термо-
термодинамическое определение фазы предполагает наличие межфазной границы
раздела — поверхности, отделяющей данную фазу от всех остальных фаз в систе-
системе и от окружающего пространства. Поверхностный слой фазы находится в иных
условиях по сравнению с объемом и обладает избыточной свободной энергией.
Вследствие этого свойства поверхности отличаются от свойств вещества в целом.
Поэтому понятие фазы применимо к макроскопическим объектам, для которых
объемные свойства являются определяющими. Если поверхностными свойствами
по сравнению с объемными пренебречь нельзя (что наблюдается, например, в
тонких пленках), то классическое понятие фазы становится неприменимым. При
этом не имеет значения абсолютное количество вещества в объеме данной фазы,
важно лишь соотношение между поверхностью и объемом. Например, фазой в
термодинамическом смысле нельзя считать тонкую масляную пленку на поверх-
поверхности воды, хотя общая масса этой пленки может быть значительной.
Важность применения понятия фазы к твердому состоянию заключается в
том, что, как правило (за исключением молекулярных кристаллов), носителем
всех свойств твердого вещества является фаза. В жидком и газообразном состоя-
состояниях, а также в молекулярных кристаллах носитель химических свойств — моле-
молекула, хотя представление о фазе к ним приложимо. В связи с этим твёрдая фаза
представляет собой высшую ступень химической организации вещества. Рассмот-
Рассмотрим взаимосвязь и характерные особенности различных форм организации веще-
вещества на примерах иода, кремния и диоксида кремния (рис. 86). Изолированный
атом не является конкретным носителем химических свойств вещества в обычных
условиях, а у SiC>2 (сложное вещество) организация на атомном уровне отсутству-
отсутствует вообще. Для. иода первичным носителем химических свойств выступает моле-
молекула. При образовании молекулярного кристалла 12) в котором молекулы связаны
слабыми ван-дер-ваальсовыми силами, возникающая твердая фаза не будет спе-
специфическим носителем свойств йода, так как последние целиком определяются
Лтвм
©
Молекула
*'-*
Рис. 86. Взаимосвязь атом - молекула -
фаза
тпл ткрт
Рис. 87. Диаграмма возгонка - ис-
испарение
свойствами самих молекул 12. Иная картина наблюдается для кремния и SiC>2,
которые образуют координационные решетки. Здесь отсутствует молекулярная
организация вещества и носителем всех свойств этих объектов служит фаза. Ди-
Диоксид кремния существует как в виде координационных кристаллов (кварц, три-
димит, кристобалит и т.п.), так и в стеклообразном состоянии. В обоих состояни-
состояниях для характеристики свойств объекта используют понятие фазы. Однако при-
применение этого понятия к стеклообразному состоянию условно, так как не выпол-
выполняется один из основных критериев термодинамического определения фазы —
равновесность системы (поскольку стеклообразное состояние метастабильное).
Для твердого состояния вообще характерна заторможенность процессов и вслед-
вследствие этого реализация метастабильных (ложных) равновесий. Поэтому примене-
применение понятия фазы к твердому состоянию иногда затруднительно. Отсюда следу-
следует, что необходимо различать понятие о фазе как носителе свойств твердого ве-
вещества (что эквивалентно понятию о химическом индивиде) и представление о
фазе как гомогенной части термодинамически равновесной системы.
2. Кристаллическое, стеклообразное, аморфное состояния. В подавляющем
большинстве случаев твердые тела представляют собой кристаллы. Если в струк-
структурном отношении жидкость характеризуется наличием только ближнего поряд-
порядка, то в кристаллах ближний порядок переходит в дальний, т.е. упорядоченное
расположение атомов распространяется на весь объем твердой фазы. С термоди-
термодинамической точки зрения образование упорядоченной кристаллической структу-
структуры энергетически выгодно (ниже температуры плавления), т.е. в этих условиях
кристаллическому состоянию отвечает минимум свободной энергии Гиббса. Хотя
при понижении температуры энтропия уменьшается (упорядоченность возраста-
возрастает), но при этом наблюдается значительное уменьшение внутренней энергии (или
энтальпии). В результате, как следует из уравнения (VI.5), при образовании
кристалла происходит уменьшение свободной энергии (AG < 0).
Одной из особенностей кристаллического вещества в термодинамическом
отношении является невозможность сколько-нибудь заметного перегрева выше
температуры плавления. Если жидкость можно легко переохладить на десятки и
даже сотни градусов ниже температуры кристаллизации, то кристаллы практиче-
практически всегда расплавляются по достижении температуры плавления. Плавление
(кристаллизация) наступает при равенстве давления пара над твердой и жидкой
фазами (рис. 87), т.е. температура плавления определяется как точка пересечения
кривых давления пара для твердого тела и жидкости. Поскольку жидкости свой-
свойственно явление переохлаждения, кривая be может быть продолжена в область
метастабильных состояний, лежащих ниже температуры плавления F6'). В то же
время кривая давления пара над твердой фазой (кривая возгонки ab) заканчива-
заканчивается в точке плавления и не может быть продолжена выше. Следовательно, тем-
температура плавления — последняя точка на кривой возгонки и принадлежит толь-
только этой кривой. Отсюда следует, что температура плавления — истинная верхняя
граница существования кристаллического твердого тела. Для жидкости нижняя
граница ее существования условна (вследствие склонности к переохлаждению), а
верхняя граница — критическая температура ТКр-, так же как и для твердого
состояния, будет истинной. Эти особенности поведения твердого тела и жидкости
вблизи температуры плавления связаны с исчезновением (при плавлении) или
возникновением (при кристаллизации) межфазной границы. Так как поверхность
обладает избытком свободной энергии по сравнению с объемом, то при достиже-
достижении температуры плавления разрушение кристалла начинается именно с поверх-
поверхности. Таким образом, исчезновение фазовой границы не требует затраты допол-
дополнительной энергии и осуществляется самопроизвольно. Именно поэтому перегрев
твердого тела выше температуры плавления практически невозможен.
Процесс кристаллизации жидкости начинается с возникновения зародышей
твердой фазы. При этом не любой возникший зародыш способен к самостоятель-
самостоятельному существованию и дальнейшему росту. Образование межфазной границы
связано с затратой дополнительной энергии*. Поэтому зародыш станет стабиль-
стабильным, когда он достигнет определенного размера, при котором величина его объ-
объемной свободной энергии превысит запас поверхностной энергии. При некотором
критическом размере зародыша его объемная и поверхностная свободные энергии
равны друг другу и в этом состоянии система будет находиться в неустойчивом
равновесии. Для осуществления процесса кристаллизации с заметной скоростью
необходимо обязательно переохладить расплав, чтобы скомпенсировать затрату
энергии на возникновение фазовой границы. При охлаждении расплава только до
равновесной температуры кристаллизации образование кристаллов будет проис-
происходить с бесконечно малой скоростью.
Существование энергетического барьера при образовании фазовой границы и
служит основной причиной переохлаждения жидкости. При этом иногда переох-
переохлаждение можно осуществить в таких условиях, когда упорядочение частиц,
сопровождающее процесс кристаллизации, затруднено вследствие резкого воз-
возрастания вязкости жидкости. Таким образом можно получить некристаллическую
твердую фазу, находящуюся в метастабильном состоянии и фактически представ-
представляющую собой сверхвязкую жидкость. Это состояние вещества называется стек-
стеклообразным.
Условия стеклообразования характеризуются кривой давления пара над пере-
переохлажденной жидкостью (см. рис. 87, кривая 66'). Однако даже глубокое переох-
переохлаждение жидкости не всегда приводит к образованию стекла. Возможность
С термодинамической точки зрения, равновесие при температуре плавления должно
быть обратимым, т.е. затрата энергии при плавлении (теплота плавления) равна скрытой
теплоте кристаллизации — энергии, выделяющейся при образовании твердой фазы из
жидкости. Однако, чтобы кристаллизация протекала с конечной скоростью, необходимо
переохлаждение, компенсирующее затрату энергии на возникновение фазовой границы.
Следовательно, кристаллизация в отличие от плавления является принципиально нерав-
неравновесным процессом.
187
стеклообразования при затвердевании жидкости определяется характером хими-
химической связи и особенностями структуры твердой и жидкой фаз. Жидкости,
обладающие преимущественно металлической связью или жидкости с ионной
природой, как правило, мало склонны к стеклообразованию вследствие ненаправ-
ненаправленности и ненасыщенности этих типов связи. Поэтому возникновение дальнего
порядка при затвердевании происходит достаточно легко и быстро, если не соз-
созданы специальные условия, воспрепятствующие этому процессу. Затвердевание
жидкостей, в которых преобладает ковалентная связь, приводит к образованию
кристаллов с сохранением того же типа связи. Процессы упорядочения при обра-
образовании кристаллов с ковалентной связью из-за ее направленности и насыщаемо-
насыщаемости затруднены и протекают сравнительно медленно. В условиях достаточного
переохлаждения при возрастании вязкости жидкости образование упорядоченной
(кристаллической) фазы не происходит. Это и приводит к возникновению стекол.
Стекло представляет собой типичный пример так называемого аморфного
состояния вещества, которое в отличие от кристаллического характеризуется
двумя признаками: изотропностью свойств и отсутствием температуры плавления.
Аморфные тела встречаются обычно в виде двух форм: компактной и дисперс-
дисперсной.
Для аморфного состояния характерно наличие только ближнего порядка в
расположении структурных единиц. Дальний порядок, свойственный кристаллам,
отсутствует. Компактное аморфное состояние представляет собой сильно переох-
переохлажденную жидкость и отличается от последней только отсутствием лабильного
обмена местами между отдельными структурными фрагментами. В дисперсном
аморфном состоянии, представляющем собой тонкий порошок, состоящий из
агрегатов, не имеющих упорядоченного строения, химическое взаимодействие
между отдельными частицами полностью отсутствует. В стекле отдельные
ассоциаты связаны друг с другом силами химического взаимодействия, но эти
связи не имеют пространственно упорядоченного характера, как в кристалле. Обе
формы аморфного состояния вещества в термодинамическом отношении
метастабильны и при благоприятных условиях способны кристаллизоваться с
выделением теплоты.
3. Представление о зонной теории. Металлы, полупроводники, изоляторы.
Валентные отношения в твердом теле с координационной структурой * определя-
определяются иными законами, чем в молекулах. Само представление о валентности как о
способности атома присоединять определенное число партнеров в применении к
твердому телу теряет смысл, так как здесь реализуется возможность коллективно-
коллективного взаимодействия. Так, валентности натрия и хлора в молекуле NaCl равны
единице, а в твердом состоянии каждый атом натрия окружен шестью атомами
хлора, и наоборот.
Химическая связь в твердом теле с координационной структурой может быть
хорошо описана с позиций ММО. Если при описании простых молекул методы
ВС и МО могут быть использованы одинаково широко, то образование твердых
тел * нельзя интерпретировать методом ВС. Здесь наиболее очевидны преиму-
преимущества ММО. В рамках этого метода химическая связь между партнерами может
осуществляться не только при парно-электронных (валентных) взаимодействиях,
но и при образовании "невалентных" орбитальных связей. В кристаллах, образо-
*Речь идет о металлах и структурах с преимущественно ионной связью.
1ЯЯ
ванных с участием таких связей, электроны делокализованы или в части систе-
системы, охватывающей несколько атомов, или во всем кристалле. Например, при
образовании металлических кристаллов наблюдаются большие координационные
числа (как правило, 8 и 12). В то же время количества валентных электронов в
металлах явно недостаточно для образования такого числа парно-электронных
связей. При этом химическая связь осуществляется за счет обслуживания элект-
электроном большого числа структурных единиц (атомов) и является многоцентровой
связью с дефицитом электронов. Таким образом, в отличие от валентных соеди-
соединений здесь нельзя выделить отдельные связи, попарно соединяющие между
собой соседние атомы. Хотя атомы связаны в устойчивую систему, между ними не
существует классически понимаемых химических связей. Специфика взаимодей-
взаимодействия большого количества частиц состоит в том, что при образовании ансамбля
при сближении частиц и их взаимном влиянии друг на друга происходит "рас-
"расщепление" атомных орбиталей. На рис. 88 показано расщепление орбиталей
щелочного металла, валентный электрон которого находится на ns-уровне.
Энергия валентного уровня в изолированном атоме (N = 1) равна Eq (рис. 88,
а). При сближении двух атомов происходит квантово-механическое взаимодейст-
взаимодействие между валентными электронными орбиталями, в результате чего из двух
атомных орбиталей с одинаковой энергией образуются две молекулярные орбита-
ли. Энергия одной из них больше (РМО), а другой меньше (СМО), чем для
изолированного атома (рис. 88, б). Взаимодействие четырех, шести и т.д. атомов
приводит к расщеплению валентных АО на такое же число МО (рис. 88, в, г).
При этом кроме СМО и РМО при достаточно большом числе взаимодействую-
взаимодействующих атомов появляются молекулярные орбитали, энергия которых примерно
равна энергии изолированной атомной орбитали (НМО). Это равносильно пред-
предположению, что в металлах могут существовать (статистически) неионизирован-
ные атомы, не отдавшие электрон в общее пользование. Образование кристалла в
результате взаимодействия большого числа атомов (порядка N ~ 1023) приводит
к появлению практически сплошного спектра энергий МО (рис. 88, д), количест-
количество которых равно числу взаимодействующих частиц. Общая ширина полученной
таким образом энергетической зоны, т.е. разность энергий между самым низким и
самым высоким уровнями, не зависит от числа атомов, участвующих во взаимо-
взаимодействии (при их достаточно большом количестве). Если принять ширину зоны
равной 1 эВ, то при наличии в ней 1023 дискретных уровней энергетический
зазор между соседними состояниями равен 103эВ *. Столь ничтожная разница
между энергиями отдельных уровней позволяет утверждать, что энергия внутри
зоны меняется практически непрерывно, хотя эта непрерывность в действитель-
действительности складывается из отдельных очень близко лежащих дискретных энергетиче-
энергетических состояний.
Таким образом, применение ММО к системам, состоящим из многих частиц,
приводит к представлению о наличии энергетических зон, характеризующих
состояние валентных электронов в кристаллическом твердом теле. Понятие об
энергетической зоне является основополагающим в зонной теории твердого тела.
Подобно тому, как в изолированном атоме существуют разрешенные и запрещен-
*Для сравнения укажем, что средняя тепловая энергия электронов при 300 К составля-
составляет величину порядка 10~2 эВ.
189
f'¦¦
a
Рис. 88. Расщепление валентного энергетичес-
энергетического уровня щелочного металла при различном
числе взаимодействующих атомов
1S
г" г'" г" г' г», г
Рис. 89. Возникновение и перекрывание
энергетических зон в кристалле натрия в
зависимости от межатомного расстояния
ные уровни энергии, в кристалле существуют зоны разрешенных и запрещенных
энергий. На рис. 89 представлена схема возникновения энергетических зон в
кристалле натрия в зависимости от межатомного расстояния г.
В изолированном атоме Na (гте) все уровни разрешенных энергий (Is22s22p63sl)
дискретны. Такое состояние характерно для одноатомного пара натрия. При
химическом взаимодействии атомов с образованием кристалла по мере их сближе-
сближения происходит расщепление уровней энергии в энергетические зоны. В первую
очередь начинают расщепляться внешние уровни (вакантный уровень Зр начина-
начинает расщепляться на расстоянии г', затем наполовину заполненный 3*-уровень —
на расстоянии г"). При дальнейшем сближении атомов на расстоянии г'" происхо-
происходит перекрытие 3*- и Зр-зон разрешенных энергий. На расстоянии г° (межатом-
(межатомное расстояние в кристалле натрия) сближение атомов заканчивается и в этом
состоянии валентная 3*- и вакантная Зр-зоны перекрыты. Таким образом, валент-
валентные Зя-электроны натрия могут занимать любое энергетическое состояние в пре-
пределах общей зоны, образующейся в результате перекрывания. Внутренние энерге-
энергетические уровни Is, 2s, 2p могут расщепиться в зоны только на расстояниях
меньше г°, и в реальном кристалле натрия эти состояния в химической связи не
участвуют, а связь осуществляется коллективом валентных электронов, энергети-
энергетическое состояние которых характеризуется
общей зоной, полученной в результате
перекрывания.
В кристалле кремния особенности рас-
расщепления энергетических уровней в зоны
и их перекрывания отличаются от зонной
структуры металлов (рис. 90). При образо-
образовании кристаллической решетки начиная с
некоторого межатомного расстояния
г'(г' > Го) наблюдается вр3-гибридизация
электронных состояний атомов, что при-
приводит не просто к перекрыванию 3 s- и Зр-
зон, а к их полному слиянию с возникно-
возникновением единой «р3-гибридной валентной
зоны. В кристаллическом кремнии каждый
a S
Р и с. 90. Возникновение энергетических
зон в кремнии (а) и зонная структура
кремния (б)
190
J
атом образует тетраэдрические парно-электронные насыщенные ковалентные
связи, достраивая свою валентную оболочку до октета. Таким образом, в валент-
валентной зоне все 8N состояний оказываются занятыми.
Другой характерной особенностью зонной структуры кремния является то, что
следующая вакантная 4«-зона не перекрывается с валентной на межатомных рас-
расстояниях г = г0, а отделена от последней зоной запрещенных энергий АЕ*.
Электроны, находящиеся в валентной зоне, участвовать в электрической прово-
проводимости не могут, так как в этой зоне все состояния заняты. Для возбуждения
электрической проводимости необходимо любым путем (нагревание, облучение)
сообщить электронам энергию, равную Д2? (рис. 90, б). Тогда возбужденные
электроны попадают в свободную 4«-зону, которая называется зоной проводимо-
проводимости, и становятся способными участвовать в электрической проводимости. Энер-
Энергетический промежуток между верхним краем (потолком) валентной зоны и ниж-
нижним краем (дном) зоны проводимости (Д-Е) называется шириной запрещенной зо-
зоны. Эта величина представляет собой важнейшую характеристику кристалличес-
кристаллического вещества. В зависимости от ширины запрещенной зоны все кристаллические
вещества подразделяются на три класса: металлы, полупроводники и изоляторы
(диэлектрики). В металлах ширина запрещенной зоны равна нулю, так как за-
заполненная и свободная зоны перекрываются между собой и, в сущности, ватент-
ная зона одновременно будет и зоной проводимости. Именно способность валент-
валентных электронов в металлах к свободному перемещению по всему объему кристал-
кристалла и обусловливает их высокие электрическую проводимость и теплопроводность.
Если ширина запрещенной зоны очень велика (больше ~ 4 эВ), то электричес-
электрическую проводимость в веществе (нагреванием или облучением) возбудить практи-
практически невозможно. Это объясняется тем, что энергия теплового возбуждения
электронов при нагревании даже до температуры плавления (Е — 3/2 кТПЛ) недо-
недостаточна для преодоления зоны запрещенных энергий. Следовательно, при нагре-
нагревании кристалл расплавится прежде, чем возникнет электронная проводимость.
Такие вещества называются изоляторами. К их числу относятся, например, алмаз
(Д2? = 5,1 эВ), кварц (АЕ = 5,2 эВ), многие типичные соли и т.п.
Промежуточные значения АЕ (в пределах ~ 0,1 — 4,0 эВ) характерны для
полупроводников. Наиболее типичными примерами веществ с полупроводниковы-
полупроводниковыми свойствами являются германий (АЕ = 0,78 эВ), кремний (АЕ = 1,14 эВ), би-
Ш V П VI
нарные соединения элементов III и V групп (А В ), II и VI групп (А В ) и т.п.
Электрическая проводимость этих кристаллов при комнатной температуре, как
правило, невелика, а при 0 К они ведут себя подобно изоляторам. Однако при
повышении температуры часть электронов приобретает энергию, достаточную для
преодоления запрещенной зоны, в результате чего электрическая проводимость
резко возрастает. Таким образом, принципиальное отличие полупроводников от
металлов заключается в различной зависимости электрической проводимости от
температуры. Если у металлов с ростом температуры электрическая проводимость
снижается (отрицательный температурный коэффициент электрической проводи-
проводимости), то у полупроводников при повышении температуры наблюдается значи-
значительное увеличение электрической проводимости.
*3десь зонная структура кремния трактуется упрощенно. В действительности структура
валентной зоны и зоны проводимости более сложная.- ,
4. Основы физико-химического анализа. В конце XIX и начале XX в. форми-
формируется новый раздел химии — физико-химический анализ. Основоположниками
нового направления были Гиббс, Ван-дер-Ваальс, Розебом; Тамман, Д.И.Менде-
Д.И.Менделеев, Д.П.Коновалов, Н.С.Курнаков. Особая заслуга акад. Курнакова состоит в
том, что он разработал основы геометрического анализа диаграмм состояния и
создал крупнейшую в мире школу физико-химического анализа. Основная задача
этого раздела химии состоит в измерении физических свойств системы, находя-
находящейся в состоянии равновесия, при последовательном изменении ее состава.
Результатом такого исследования является диаграмма состав — свойство, пред-
представляющая собой геометрическое отражение процессов, которые протекают в
системе. Геометрический анализ диаграмм состав — свойство, сочетая в себе на-
наглядность и универсальность, позволяет определить число образующихся в систе-
системе фаз, их природу, области их существования и особенности взаимодействия
между ними. Это обусловлено возможностью наблюдения за изменениями в систе-
системе в процессе химического взаимодействия, не выделяя образующиеся фазы для
исследования. Таким образом, химия получила метод, при помощи которого
открываются пути непосредственного изучения химических процессов, что пред-
представляет собой качественно новую ступень в познании природы вещества.
В качестве экспериментально измеряемого свойства (функции состава) могут
быть использованы температура фазовых превращений, твердость, электрическая
проводимость, плотность, вязкость и т.п. Если функцией состава служит термоди-
термодинамическое свойство, определяющее состояние системы, например температура
фазовых превращений, то получающийся геометрический образ называется диаг-
диаграммой состояния или фазовой диаграммой. Диаграммы, отражающие зависи-
зависимость нетермодинамических физических свойств (электрических, магнитных и
пр.) от состава, называются диаграммами состав — свойство.
Фундаментальным принципом построения диаграмм состояния является пра-
правило фаз Гиббса. Введем предварительно понятие о независимом компоненте и
степени свободы. Независимые компоненты — химически индивидуальные вещест-
вещества, наименьшее число которых достаточно для образования всех фаз в системе.
Следует различать термодинамическое понятие о независимом компоненте и
тривиальное понятие о компоненте как составной части системы *. Если система
физическая, то оба понятия совпадают. Например, система, состоящая из воды,
льда и водяного пара при 0,01°С и 6,12 гПа, однокомпонентная, поскольку для
формирования всех трех фаз в системе достаточно одного индивидуального веще-
вещества — воды. Система, состоящая из насыщенного раствора сахара в воде и
водяного пара над раствором, образует три фазы (кристаллы сахара, раствор и
пар), но является двухкомпонентной (сахар + вода).
В химических системах, в которых возможно протекание реакции, понятия
независимых компонентов и веществ, слагающих систему, не тождественны. Число
независимых компонентов в таких системах определяется числом индивидуаль-
индивидуальных веществ в системе за вычетом возможных между ними обратимых взаимодей-
взаимодействий. Например, система из трех индивидуальных веществ СаСОз, СаО и СОг
будет двухкомпонентной, так как для образования всех фаз достаточно любых
*В литературе часто смешивают эти понятия. В дальнейшем под компонентом, будем
понимать именно независимый компонент, а не составляющие систему индивидуальные
вещества. .
двух веществ, поскольку они связаны между
собой стехиометрическим уравнением реакции
СаСОз <р=* СаО + СО2.
Степени свободы — число внешних парамет-
параметров равновесия (р, Т), которые можно произволь-
произвольно изменять без изменения числа фаз в системе.
Рассмотрим диаграмму состояния однокомпо-
нентной системы — воды (рис. 91). Тройную
точку, координаты которой определяют условия о,О1 "С tKp t
сосуществования трех фаз, можно рассматривать
г f ' r f P и с. 91. Диаграмма состояния
как геометрический образ с нулевым числом
ВОДЫ
измерений. При этом число степеней свободы
равно нулю. Действительно, если изменить хотя бы один из параметров (давле-
(давление или температуру), неизбежно изменится число сосуществующих фаз. Линии,
описывающие условия равновесного сосуществования двух фаз (лед :5=± пар,
лед ^* вода, вода ^^^ пар), представляют собой одномерный геометрический
образ (число степеней свободы равно единице). В самом деле, можно произвольно
менять любой параметр, сохраняя равновесие двух фаз, но значение второго
параметра будет при этом строго определено. Таким образом, линия двухфазного
равновесия представляет собой функциональную зависимость одного параметра
от другого: р = /(Т) или Т = <р(р).
Поля диаграммы состояния, отвечающие областям существования льда, воды
и пара, которые ограничены соответствующими линиями двухфазных равновесий,
представляют собой двумерные геометрические комплексы (имеют две степени
свободы). В пределах этих полей можно произвольно менять оба параметра (и
температуру, и давление), а система при этом будет оставаться однофазной. Итак,
тройная точка, в которой сосуществуют в однокомпонентной системе три фазы,
представляет равновесие с нулевой степенью свободы, или нонвариантное равно-
равновесие. Аналогично двух- и однофазные состояния, отвечающие линиям и полям
диаграммы, принадлежат равновесиям с одной или двумя степенями свободы или
представляют моновариантные или дивариантные (бивариантные) равновесия.
Правило фаз Гиббса определяет связь между числом внешних и внутренних
факторов равновесия и числом сосуществующих фаз. Из внешних факторов рав-
равновесия для химических систем наибольшее значение имеют давление и темпера-
температура. Влиянием остальных факторов (гравитационных, электромагнитных полей,
капиллярных сил и т.п.) пренебрегают. Под внутренними факторами равновесия
понимают число К независимых компонентов системы. Тогда общее число факто-
факторов, определяющих фазовое равновесие, равно К + 2, где 2 соответствует внеш-
внешним факторам равновесия. Число степеней свободы С определяется как разность
между максимально возможным числом фаз К + 2 и действительно существу-
существующим числом фаз Ф в системе;
С= К + 2-Ф,
или
ф + С = К+2, (VIII.1)
т.е число фаз плюс число степеней свободы равно числу компонентов плюс два.
7 Я. А. Угай 193
1,0 Oft 0,8 0,7< 0,Б 0? 0,<t 0,3 0,1 0,1 0
/)• 1 \ MM—+—I 1 1 1—•*
0 0,1 0Л OJ 0Л 0,5 0,e 0,7 0,8 0,9 1,0
Атомное содержание,доля
В "
Р и с. 92. Способ изображения составов в
бинарной системе
}М
А хм В
Атомное содержание
Рис. 93. Координаты диаграмм
состояния при постоянном дав-
давлении
Если один из внешних параметров сохраняет постоянное значение, то правило
фаз принимает вид
= К+
(VIII.2)
В этой форме (постоянным принимается давление) правило фаз применяется к
системам с практически нелетучими компонентами (металлические сплавы и
солевые системы).
5. Типы диаграмм состояния. Построение диаграмм состояния и их геометри-
геометрический анализ при изучении химических превращений в системе составляют
основную задачу физико-химического анализа — одного из наиболее мощных и
универсальных методов современной химии. Для графического изображения
состояния однокомпонентной системы достаточно двух координат р и Т (плос-
(плоскость), поскольку состав ее фиксирован. Для определения состояния двухкомпо-
нентной системы, зависящего от четырех параметров в соответствии с уравнением
f(p, Т, х\, хг) — 0, достаточно знать три из них: температуру, давление и концен-
концентрацию одного из компонентов. Учитывая связь молярных долей компонентов
(х\ 4- ж2 = 1), удобно состав системы представить в виде отрезка, длину которого
принимают за единицу. При этом один его конец соответствует чистому компо-
компоненту A(^ = 1, ж2 = 0), другой — чистому компоненту В(х\ = 0, х2 = 1). Тогда
любой промежуточный состав характеризуется точкой на этом отрезке. Так, для
точки М на рис. 92 доля В равна 0,35, доля А — 0,65 (или 35% В и 65% А).
Значения внешних параметров р и Т в этой координатной системе удобно откла-
откладывать по двум взаимно перпендикулярным направлениям.
Для описания конденсированных систем в ряде случаев можно полагать, что
давление не влияет на положение равновесия. Тогда, приняв р = const, получим
возможность изобразить состояние бинарной системы на плоскости (в координа-
координатах Т — х) (рис. 93). Любая фигуративная точка* М в этой координатной сетке
однозначно характеризует состояние системы при р = const. Для построения
Т — гс-диаграмм исследуют зависимость температуры от времени охлаждения для
расплавов различного состава. Полученные таким образом кривые температура —
*Точка, которая отражает состояние системы, называется фигуративной, и ее
координаты равны значениям параметров.
1Й4
Время
Р и с. 94. Кривые охлаждения
время называются кривыми охлаждения. Вид этих Т 1 г
кривых свидетельствует о наличии или отсутствии
фазовых превращений при некоторых определенных
температурах или в интервале температур (рис. 94).
В соответствии с возможностью осуществления трех
типов равновесия (ди-, моно- и нонвариантных)
наблюдается три вида кривых охлаждения. Кривая
1 отвечает охлаждению образца, не претерпевающе-
претерпевающего фазовых превращений. Кривая 2 отражает моно-
моновариантный процесс, протекающий в интервале
температур Тн — Тк. Кривая 3 соответствует нонва-
риантному процессу, при котором температура должна оставаться постоянной.
Температурная остановка на кривых охлаждения наблюдается при кристал-
кристаллизации чистого компонента, химического соединения постоянного состава из
стехиометрического расплава (р = const), когда с точки зрения правила фаз
система также однокомпонентна (С = 1 + 1 — 2 = 0). Кроме того, при одновре-
одновременной кристаллизации двух компонентов в бинарной системе число степеней
свободы также равно нулю: К = 2, Ф = 3 (две твердые и жидкая фазы) и С =
= 2+1—3 = 0). Нонвариантными должны быть и любые другие равновесия,
когда в бинарной системе присутствуют три фазы. По характерным изломам и
остановкам на кривых охлаждения сплавов различного состава строят Т — з^диа-
грамму состояния. В зависимости от особенностей взаимодействия компонентов
существуют различные типы диаграмм состояния.
Наиболее часто встречаются системы, в которых компоненты неограниченно
смешиваются в жидком состоянии (полная взаимная растворимость). Если при
этом компоненты совершенно нерастворимы друг в друге в твердом состоянии и
не образуют химических соединений, то это отражается диаграммой состояния
эвтектического типа (рис. 95). Как следует из законов идеальных растворов (кри-
(криоскопия), добавление второго компонента понижает температуру плавления пер-
первого. Рассмотрим ход кристаллизации сплава, составу которого на рис. 95 соот-
соответствует фигуративная точка 1. До тех пор, пока фигуративная точка находится
в области жидкой фазы L, никаких фазовых превращений в системе не происхо-
происходит. Равновесие будет дивариантным * (С = 2 + 1 — 1 = 2). Падение температу-
температуры при охлаждении расплава происходит по плавной кривой. При достижении
фигуративной точкой кривой Т Е при температуре Т% расплав насыщается отно-
сительно компонента А, кристаллы которого начинают выпадать из расплава при
дальнейшем понижении температуры. Ввиду того что при этом происходит выде-
выделение теплоты кристаллизации, на кривой охлаждения появляется излом. Так
как из расплава выделяется первоначально только компонент А, жидкая фаза
обогащается компонентом В. По мере снижения температуры в равновесии с
кристаллами компонента А будет находиться жидкая фаза иного состава, переме-
перемещающегося по кривой Т Е в сторону точки Е. Число степеней свободы при этом
А. '
равно единице (С = 2 + 1 — 2 = 1).
*Вариантность системы при р — const условная. Истинная вариантность на единицу
больше.
7* 195
Рис. 95. Диаграмма состояния с эвтектикой (а) и кривые ох-
охлаждения (б)
Как только система охладится до температуры ТЕ, расплав окажется насы-
насыщенным и относительно компонента В. Поэтому в дальнейшем одновременно
кристаллизуются оба компонента:
где L — жидкость; S и S — кристаллы компонентов. Состав расплава при этом
не изменяется и отвечает на диаграмме точке Ё. Так как в равновесии находится
максимальное число фаз, число степеней свободы С = 2+1—3 = 0и равнове-
равновесие является нонвариантным. На кривой охлаждения ему отвечает горизонталь-
горизонтальный участок. По окончании кристаллизации кривая охлаждения вновь плавно
понижается (С =2 + 1— 2=1). Аналогично будет протекать кристаллизация
всех сплавов, составы которых на диаграмме состояния лежат левее точки Е.
Затвердевание сплавов, составы которых лежат правее точки Е (точка 3), начина-
начинается с выделения кристаллов компонента В. Жидкая фаза при этом обогащается
компонентом А. Процесс также заканчивается совместной кристаллизацией обоих
компонентов. На кривой охлаждения 2 отмечается лишь горизонтальная площад-
площадка при температуре Т?. При этой температуре расплав оказывается сразу же
насыщенным относительно двух компонентов, кристаллизующихся совместно
(С = 2 + 1 — 3 = 0). На диаграмме состояния кривые Т Е и Т Е называются
линиями ликвидуса. Они представляют собой геометрическое место точек, отвеча-
отвечающих температурам начала кристаллизации. Обе линии сходятся в точке Е,
называемой эвтектической. Сплав, состав которого отвечает этой точке, кристал-
кристаллизуется (плавится) при постоянной и самой низкой для данной системы темпе-
температуре и называется эвтектикой*. Горизонтальная линия тп, соответствующая
температуре Tg, называется эвтектической горизонталью. Она является геометри-
196
*Эвтектика в переводе с греческого означает "легко плавящийся".
^4 +-4- —
Состав
Время
а 6
Рис. 96. Диаграмма состояния с неограниченными твердыми раст-
растворами (а) и кривые охлаждения (б)
ческим местом точек, отвечающих окончанию кристаллизации всех промежуточ-
промежуточных сплавов в системе. Так как чистые компоненты А и В кристаллизуются
выше эвтектической горизонтали (при температурах Т , Т соответственно), то
линия солидуса, ниже которой существуют только твердые фазы, представляет
собой ломаную Т тппТ , замыкающуюся с линией ликвидуса в точках плавления
компонентов.
Рассмотрим случай, когда компоненты неограниченно растворимы друг в
друге как в жидком, так и в твердом состоянии. На рис. 96, а представлена ди-
диаграмма состояния такой системы. Поскольку при охлаждении образуется лишь
одна твердая фаза переменного состава, диаграмма состоит из двух непрерывных
линий (ликвидуса и солидуса), отвечающих температурам начала и конца крис-
кристаллизации твердого раствора а(Р) *. Ни в одной из точек (за исключением сос-
составов, отвечающих чистым компонентам) система не может быть нонвариантной,
так как минимальное число степеней свободы С=2+ 1—2= 1. Следовательно,
кристаллизация всех промежуточных расплавов будет происходить не при посто-
постоянной температуре, а в некотором интервале температур. На кривых охлаждения
отмечаются два перегиба, отвечающих началу и концу превращения (рис. 96, 6).
При образовании непрерывных твердых растворов встречаются системы, когда
линии ликвидуса и солидуса имеют общую точку касания и проходят через
экстремум — минимум или максимум (рис. 97, а, б). Составы, отвечающие экстре-
экстремальным точкам, имеют на кривой охлаждения нонвариантную обстановку, что,
казалось бы, противоречит правилу фаз**.На самом деле для этих составов в
уравнении правила фаз следует учесть дополнительное условие х^ = Xg, что сни-
Обычно символом а обозначаются твердые растворы на основе компонента А, симво-
символом Р — твердые растворы на основе компонента В. При образовании непрерывного ряда
твердых растворов безразлично, какой компонент считать растворителем, что и отражается
обозначением а(/7).
**Равновесие двух фаз в бинарной системе должно быть моновариантным: С = 2 +
+ 1—2=1.
197
Pi1
fL+S
В
P и с. 97. Образование непрерывных растворов с экстремумами:
а - с максимумом; б - с минимумом
Ар
Рис.
с ограниченными твердыми раст-
растворами (эвтектический тип)
q В
. Диаграмма состояния
жает вариантность системы на единицу. Условно такие системы можно считать
однокомпонентными и тогда С = 1 + 1 — 2 = 0.
Рассмотренные типы диаграмм состояния являются предельными. Так, диа-
диаграмма на рис. 95 представляет идеальный вариант, поскольку абсолютно нераст-
нерастворимых веществ в природе не существует. С учетом ограниченной растворимости
компонентов друг в друге диаграмма состояния эвтектического типа видоизменя-
видоизменяется (рис. 98). Отличие ее от рис. 95 состоит в том, что при охлаждении расплава
L из него кристаллизуются не чистые компоненты А и В, а твердые растворы а
(твердый раствор В в А) и /? (твердый раствор А в В). Первый выделяется при
кристаллизации доэвтектических сплавов, второй — при кристаллизации заэвтек-
тических сплавов. Новым фазам (о и /)) отвечают геометрические образы — участ-
участки плоскости, примыкающие к ординатам компонентов (ApntT , BqnT ). Линии
ликвидуса Т Е и Т Е отвечают началу кристаллизации твердых растворов аи/)
А В
соответственно. Они пересекаются в эвтектической точке Е. Состав твердых раст-
растворов, сосуществующих с расплавами, определяется участками линии солидуса
Т m и Т п. Обе линии в точках тип пересе-
А В
каются с кривыми рт и qn, указывающими на
изменение взаимной растворимости компонен-
компонентов в твердом состоянии в зависимости от тем-
температуры. Точки тип отвечают составам
твердых растворов аи/?, насыщенных при
эвтектической температуре. Участок плоскости,
заключенный между линиями тр, nq и эвтекти-
эвтектической горизонталью тп, соответствует гетеро-
гетерогенной смеси а + /? твердых растворов. Кривые
охлаждения сплавов, составы которых лежат
между точками тип, имеют такой же вид, как
и на диаграммах эвтектического типа. На кри-
А с d в вых охлаждения сплавов, составы которых
Рис. 99. Диаграмма образования лежат левее точки m и правее точки п, гори-
ограниченных твердых растворов зонтальные участки, отвечающие кристаллиза-
(перитектический тип) ции эвтектики, отсутствуют.
1Q8
Эвтектическая горизонталь, отвечающая нонвариантному превращению, лежит
ниже температур кристаллизации чистых компонентов А и В. Однако встречают-
встречаются системы, в которых нонвариантная горизонталь расположена между температу-
температурами кристаллизации компонентов. Такая горизонталь называется перитектиче-
перитектической. Диаграмма состояния этого типа представлена на рис. 99. На этой диаграм-
диаграмме вместо эвтектического происходит нонвариантное перитектическое превраще-
превращение. Оно заключается в растворении ранее выпавших кристаллов твердого раст-
раствора /3 в расплаве определенного состава, в результате чего образуются кристал-
кристаллы твердого раствора a: L(p) -f Дп) ^^ а{т). Подобное превращение претерпева-
претерпевают в ходе охлаждения сплавы, составы которых лежат в пределах протяженности
перитектической горизонтали рп.
Если в системе образуется химическое соединение AmBn, то это отражается на
диаграмме состояния (рис. 100). Эта диаграмма формально может быть представ-
представлена как совмещение двух диаграмм состояния эвтектического типа А — АШВ„ и
АГОВП — В (см. рис. 98). Линия ликвидуса состоит из трех участков, соответству-
соответствующих началу кристаллизации компонентов и соединения. Солидус представлен
ломаной линией Т mnT pqT . Если на диаграмме состояния соединению
А АтВп В
отвечает явный максимум, соединение существует вплоть до температуры плавле-
плавления. При этом составы твердой и жидкой фаз в точке плавления совпадают.
Такое плавление называется кошруэнтныл! (от лат. congruens — совпадающий), а
температурный максимум на кривой ликвидуса называется дистпектпикой (от греч.
"трудноплавящийся"). Если соединение разлагается, не достигая температуры
плавления, явный максимум на диаграмме состояния отсутствует (рис. 101). По-
Появляется новая линия PQ (перитектическая горизонталь), отвечающая нонва-
нонвариантному равновесию:
АЯВ„ 5=* L(P) + S@); C = 2+l-3 = 0.
По смыслу она аналогична перитектической горизонтали рп (см. рис. 99). При
температуре Тр кристаллы AmBn разлагаются в соответствии с приведенным
уравнением на жидкость состава L(P) и твердый компонент S@) (рис. 101). По-
Поскольку составы твердой и жидкой фаз не совпадают, такое плавление называет-
называется инкомруэнтпным (incongrue.ns — несовпадающий). Ликвидус состоит из трех
ветвей: Т Р — кристаллизация твердого раствора /?, РЕ — твердого раствора у на
Г
Т
Рас. 100. Диаграмма состояния
конгруэнтно плавящегося соедине-
соединения с ограниченными твердыми
растворами
Рис. 101. Диаграмма состояния
инконгруэнтно плавящегося сое-
соединения с ограниченными тверды-
твердыми растворами
199
Массодое содержание Аи, %
ТОМ 60 70 80 85 90 95
( I
1,4 i
г, г
то
поп
1000
900
800
700
660
600
500
т
О 10 20 JO W 50 50 70 SO 90 100
Al Au
Атомное содержание Аи, %
Рис. 102. Диаграмма состояния системы
алюминий - золото
1
1
1—
/
1-
1
\
625°
\
\
Л
S69°
f
f
b
•—
$
и
щ
\
R
1"
A
P
ТУ,
1063°
f n мд Тв
Рис. 103. Диаграмма состояния с конгруэнтно
и инконгруэнтно плавящимися соединениями и
ограниченными твердыми растворами
основе соединения AmBn и Т Е — твердого раствора а. Точка Р называется пере-
/л.
ходной или перитектической. Пунктирное продолжение кривой ЕР отвечает
метастабильному скрытому максимуму, определяющему мнимую температуру
плавления соединения AmBn.
Рассмотренные типы диаграмм состояния являются простейшими. В реальных
системах взаимодействие описывается более сложными геометрическими комплек-
комплексами, представляющими собой комбинации этих типов. В качестве примера рас-
рассмотрим диаграмму состояния системы А1 — Аи (рис. 102). В этой системе обра-
образуются пять соединений: AuAl2, AuAl, Au2Al, Au5Al2 и Au4Al. Два из них плавят-
плавятся конгруэнтно (AuAI2 и Au2AI), остальные образуются по перитектической реак-
реакции. Кроме того, одно из этих соединений (Аи4А1) образует область твердых
растворов 7- На основе золота образуются ограниченные твердые растворы в. В
твердом алюминии золото практически нерастворимо, о чем свидетельствует
отсутствие заметной области твердых растворов со стороны А1. В системе
отмечается образование трех эвтектик: Al + AuAl2, AuAl + Au2AI, Au5Al2 +
+ Au4Al.
В основе геометрического анализа диаграмм состояния лежат два принципа,
сформулированные Н.С.Курнаковым, — принцип непрерывности и принцип соот-
соответствия (корреляции). Согласно первому принципу, при непрерывном измене-
изменении параметров, определяющих состояние системы, свойства отдельных фаз и
системы в целом изменяются также непрерывно, но при условии, что не возника-
возникают новые фазы и не исчезают старые. На основании принципа непрерывности
можно из анализа диаграмм состав — свойство определять число, характер фаз в
системе, области их существования и особенности взаимодействия между ними.
Принцип соответствия утверждает, что каждой фазе, фазовому равновесию и
комплексу фаз соответствует на диаграмме состояния свой геометрический образ.
Действительно, на диаграмме состояния (рис. 103) существованию однородной
жидкой фазы соответствует область, ограниченная ординатами компонентов и
200
линией ликвидуса (снизу). Областям твердых растворов на основе компонентов
А, В и соединений AmBn и АжВу отвечают замкнутые геометрические комплексы
а, р, 7, Ь, ограниченные соответствующими линиями моновариантных равновесий
и ординатами компонентов. Кристаллизации каждого химического индивида
отвечает свой участок линии ликвидуса (компоненту А — Т Ev соединению
АтВ„ — E\t\P, соединению АжВу — РЕ2, компоненту В — Г Е2). Области, за-
заключенные между линиями ликвидуса и солидуса (Т тпЕ., Einti, t\QP и т.д.),
А '
отвечают равновесным комплексам жидкой и соответствующих твердых фаз.
Области emnf, MNQH и gabh отвечают существованию гетерогенной смеси твер-
твердых фаз.
6. Твердые растворы. В термодинамическом отношении можно провести пол-
полную аналогию между жидкими и твердыми растворами. К обоим типам систем
применимы основные законы растворов. Отсюда следует, что твердые растворы,
так же как и жидкие, можно определить как фазы переменною состава, находя-
находящиеся в состоянии химическою (термодинамическою) равновесия.
Вследствие специфики взаимодействия в твердом состоянии между жидкими и
твердыми растворами существуют и различия. Последние определяются наличи-
наличием дальнего порядка и более сильным взаимодействием частиц в твердом состо-
состоянии. Поэтому для твердых растворов в еще большей степени характерны откло-
отклонения от идеальности и применимо понятие активности. Кроме того, по этим же
причинам состояние равновесия в твердых растворах достигается медленнее, со
значительными кинетическими затруднениями.
При образовании твердых растворов обычно происходит деформационное
искажение кристаллической решетки, обусловленное различием в размерах ато-
атомов растворителя и растворенного вещества. Это должно приводить к возраста-
возрастанию внутренней энергии системы U (или энтальпии Я = U + pV), т.е. процесс
образования твердых растворов должен быть эндотермическим (ДЯдефорМ > 0). С
другой стороны, перекрывание электронных орбиталей компонентов при образо-
образовании твердых растворов приводит к возникновению химических связей между
ними, что связано с уменьшением энтальпии (ДЯСВ < 0). Последнее явление до
некоторой степени аналогично уменьшению энтальпии в жидких растворах за
счет сольватации. В результате суммарное изменение энтальпии при образовании
твердых растворов АН = —ДЯСВ + ДЯдеформ может быть как положительным, так
и отрицательным. Термодинамически образование твердого раствора будет воз-
возможно, если изменение свободной энергии AG = АН — TAS будет отрицатель-
отрицательным. Если ДЯ < 0, т.е. энергия химического взаимодействия преобладает над
энергией деформации решетки, то всегда AG < 0 (так как AS при образовании
всегда положительно вследствие возрастания неупорядоченности в системе). При
ДЯ > 0 (преобладание деформационного эффекта над химическим) возможность
образования твердых растворов будет определяться соотношением между АН и
TAS. Изменение свободной энергии здесь будет отрицательным только тогда,
когда TAS > АН. Вблизи чистых компонентов А и В наблюдается очень резкое
возрастание энтропии смешения (AS > 0, рис. 104). Таким образом, при малой
концентрации растворенного вещества образование твердого раствора всегда
термодинамически выгодно, поскольку в этих условиях TAS > ДЯ независимо от
абсолютной величины АН. Отсюда и следует, что абсолютно нерастворимых в
твердом состоянии веществ в природе не существует.
Afcr
Рис, 104. Изменение энтропии
смешения в зависимости от сос-
Растворимость в твердом состоянии в первую
очередь определяется характером химической
связи. Важную роль при этом играют близость
кристаллохимического строения и размерный
фактор — соотношение атомных радиусов взаимо-
взаимодействующих компонентов. В соответствии с эмпи-
эмпирическим правилом Руайе различие в размерах
атомов компонентов при образовании неограничен-
неограниченных твердых растворов не должно превышать
8 — 15%. Последний признак служит необходи-
необходимым, но не достаточным условием образования
тава твердых растворов. Действительно, медь и железо
имеют почти одинаковые радиусы атомов (для координационного числа 12):
г (Си) = 0,127 нм, г (Fe) = 0,126 нм, однако их взаимная растворимость в
твердом состоянии мала @,35% Fe в Си и 1% Си в Fe). С другой стороны, хотя
радиусы атомов меди и золота различаются значительно больше [г (Аи) =
= 0,144 нм, т.е. различие составляет 11%], тем не менее эти два металла образуют
непрерывный ряд твердых растворов. Это и свидетельствует об определяющем
влиянии характера химической связи, близости физико-химических свойств
компонентов при образовании твердых растворов. По механизму образования
твердые растворы можно подразделить на следующие основные типы: замещения,
внедрения и вычитания.
I. Твердые растворы замещения возникают в результате статистического заме-
замещения одних частиц другими в узлах кристаллической решетки растворителя.
Все рассмотренные общие закономерности образования твердых растворов отно-
относятся именно к этому типу. Это единственный случай при образовании твердых
растворов, когда возможна полная взаимная растворимость. Например, при посте-
постепенном замещении атомов натрия в кристаллической решетке NaCl атомами ка-
калия можно непрерывно перейти от одного чистого компонента (NaCl) к другому
(KCI). Непрерывный ряд твердых растворов замещения может образоваться при
значительном сходстве физико-химического характера компонентов. Если же со-
сочетание факторов, определяющих растворимость, не очень благоприятно, то наб-
наблюдается ограниченная взаимная растворимость. Обычно различают твердые
растворы с катионным (Na^Ki-^Cl) и анионным (KClxBri-x) замещением. Кроме
того, следует различать изовалентное и гетеровалентное замещение. При изова-
лентном замещении атомы растворяемого вещества и растворителя имеют одина-
одинаковую валентность* (AlSb — GaSb, KCI — КВг и т.п.). Гетеровалентное замеще-
замещение наблюдается при разной валентности атомов растворителя и растворенного
вещества. Так, твердые растворы Ga2Se3 — GaAs представляют собой пример
твердых растворов с анионным гетеровалентным замещением. Встречаются более
сложные. варианты образования растворов замещения, когда одновременно наб-
наблюдается и катионно-анионное, и гетеровалентное замещение. Например, в
системе KMnO^ — BaSC>4 в катионной и в анионной подрешетках осуществляется
гетеровалентное замещение.
II. Твердые растворы внедрения образуются в результате встраивания атомов
*Следует помнить, что понятие валентности применительно к твердому состоянию яв-
является формальным и условным.
растворяемого вещества в междоузлия кристаллической решетки растворителя.
Растворы такого типа могут быть только ограниченными, так как общий объем
пустот в кристаллической структуре растворителя не беспределен, и при опреде-
определенной концентрации растворенного вещества напряжения в решетке (ДЯдефорМ)
становятся столь значительными, что само существование твердого раствора
маловероятно. Размер внедряемого атома должен быть небольшим и оптимально
соответствовать объему пустот в междоузлиях кристаллической решетки раство-
растворителя. В соответствии с эмпирическим правилом Хэгга г (Э): г (Me) ^ 0,59, где
г (Э) — радиус внедренного атома; г (Me) — радиус металла-растворителя. В
частности, для металлов, образующих плотноупакованные гранецентрированные
кубические (ГЦК) и гексагональные плотноупакованные (ГПУ) решетки, воз-
возможны два типа пустот: тетраэдрические и октаэдрические. Первые окружены
четырьмя атомами, вторые — шестью. Соотношение размеров пустот и внедряе-
внедряемых атомов определяет их положение в решетке и ближайшую координацию.
При образовании металлических твердых растворов внедрения в качестве внед-
внедряемых атомов обычно выступают легкие неметаллы, обладающие малым радиу-
радиусом: Н, В, С, N, О. При этом встраивание их в пустоты решетки существенным
образом не влияет на характер взаимодействия атомов и не изменяет типа крис-
кристаллической решетки (Д-Ндеформ незначительно). Например, твердые растворы
водорода в титане и палладии, бора в переходных металлах обладают металли-
металлическими свойствами, поскольку внедрение атомов водорода и бора в кристалли-
кристаллические, структуры соответствующих металлов, во-первых, не нарушает общего
металлического характера взаимодействия атомов, а во-вторых, электроны внед-
внедряемых атомов также обобществляются.
III. Твердые растворы вычитания образуются только на основе химических
соединений при недостатке одного из компонентов в соответствующей подрешет-
ке. Типичный пример твердого раствора вычитания представляет собой фаза
ТЮ, состав которой характеризуется формулой Tio^s-i.ooO^oo-o^R. Реально эта
фаза существует как в виде твердого раствора вычитания по титану Tiojs-i.ooO
(вакантна часть мест в подрешетке титана), так и в виде твердого раствора
вычитания по кислороду TiOi>0o-o,58 (все места в подрешетке титана заняты, а
часть кислородных вакантна). Следует отличать фазы с избытком одного из
компонентов против стехиометрии, который внедрен в междоузлия (твердые
растворы внедрения), от фаз с недостатком другого компонента в узлах
кристаллической решетки. Хотя валовой состав при этом может оставаться одним
и тем же (например, запись АВ1I2 формально эквивалентна записи А1)Оод,12В =
= Ao^gB), однако в первом случае будет твердый раствор внедрения по В
(подрешетка А полностью заполнена, а избыток В находится в междоузлиях), а
во втором — раствор вычитания по А (подрешетка В полностью заполнена, а
часть узлов А вакантна). Твердые растворы вычитания (как и растворы
внедрения) могут быть только ограниченными.
Одним из объективных критериев образования твердых растворов служит ха-
характер изменения физических свойств системы в зависимости от состава, в ре-
результате изучения которого осуществляется построение диаграммы состав — свой-
свойство. Согласно Курнакову, при непрерывном изменении состава твердого раствора
свойства системы должны меняться также непрерывно и графически отражаться
плавными кривыми, которые иногда имеют экстремум, а в некоторых случаях
переходят в прямые линии, соединяющие ординаты свойств чистых компонентов.
Наиболее характерными свойствами, подтверждаю-
подтверждающими образование твердых растворов, являются
изотермы электрической проводимости и твердости
в зависимости от состава, которые впервые были
получены для металлических растворов Курнако-
вым. На рис. 105 представлены типичные диаграм-
диаграммы состав — свойство для непрерывного ряда твер-
твердых растворов. Зависимость электрической прово-
проводимости от состава выражается плавной кривой,
проходящей через минимум около 50%, а кривая
твердости имеет максимум вблизи того же состава.
Наблюдаемые закономерности объясняются возрас-
возрастанием деформации решетки растворителя по мере
увеличения концентрации раствора. При этом мак-
максимальные искажения в решетке наблюдаются при
эквиатомном соотношении компонентов. Для
металлов, изменение электрической проводимости
которых определяется главным образом подвижно-
подвижностью носителей (поскольку их концентрация
А Состав В
Рис. 105. Изотермы электри-
электрической проводимости S и твер-
твердости Я для непрерывных твер-
твердых растворов
остается практически постоянной), искажения решетки приводят к увеличению
рассеяния носителей тока, снижению их подвижности и, следовательно,
электрической проводимости. Максимум на кривой твердости обусловлен напря-
напряжениями, возникающими вследствие упругой деформации решетки при образова-
образовании твердых растворов. Эти закономерности, называемые иногда законами Курна-
кова, в полной мере справедливы только для металлических твердых растворов.
В применении к твердым растворам с преимущественно ковалентной связью,
которые обладают полупроводниковым характером проводимости, эти законы
могут не соблюдаться. Это, в частности, обусловлено тем, что, например, электри-
электрическая проводимость полупроводников зависит не только от подвижности, но и
главным образом от концентрации носителей тока.
7. Соединения постоянного и переменного состава. Дальтониды и бертоллиды.
Стехиометрические соотношения компонентов, образующих соединение, соблюда-
соблюдаются только в парообразном состоянии, в молекулярных кристаллах и жидко-
жидкостях. При образовании твердых фаз с координационной структурой эти соотно-
соотношения не соблюдаются. В настоящее время доказано, что большинство твердых
веществ с немолекулярной структурой могут образовывать твердые растворы со
своими компонентами, т.е. существовать в некотором интервале составов. Так, на
диаграмме состояния (см. рис. 103) промежуточная фаза AmBn образует твердые
растворы как с одним, так и с другим компонентом. Аналогично этому существу-
существуют области гомогенности (области твердых растворов а и (?) на основе компонен-
компонентов А и В. С термодинамической точки зрения, образование ограниченных твер-
твердых растворов всегда энергетически выгодно. Поэтому отсутствие эксперимен-
экспериментально установленной области гомогенности у определенного ряда соединений с
координационной структурой (так называемые линейные фазы, которые на диаг-
диаграмме состояния отображаются вертикальной линией — ординатой соответству-
соответствующего состава) свидетельствует лишь о недостаточной чувствительности совре-
современных методов физико-химического исследования. Очевидно, истинно линей-
линейными могут быть только твердые фазы с молекулярной структурой.
Возникновение области гомогенности на базе химического соединения связано
с возникновением твердых растворов -до одному из рассмотренных выше трех
типов. Возможно замещение атомов одного из компонентов соединения в его
подрешетке атомами другого. Может возникнуть и твердый раствор внедрения в
результате встраивания избыточных атомов одного из компонентов в междоузлия
кристаллической решетки при благоприятном соотношении размерных факторов.
Кроме того, для промежуточных фаз* характерно образование твердых растворов
вычитания с возникновением недоукомплектованной подрешетки на базе одного
из компонентов. Во всех этих случаях в решетке возникают так называемые
точечные дефекты: чужеродный атом в узле подрешетки одного из компонентов
(твердый раствор замещения), атом в междоузлии (твердый раствор внедрения)
или вакансия в узле решетки (твердый раствор вычитания). Эти типы дефектов
могут встречаться как порознь, так и в комбинации друг с другом. Например,
при перемещении атома из узла в междоузлие (дефект по Френкелю) одновре-
одновременно возникают и вакансия в узле кристаллической решетки, и атом в междоуз-
междоузлии, что равносильно одновременному сосуществованию твердых растворов вычи-
вычитания и внедрения. Реальный кристалл всегда содержит термодинамически рав-
равновесное количество дефектов, концентрация которых однозначно определяется
внешними параметрами равновесия — температурой и давлением.
Экспериментально область гомогенности промежуточных фаз можно обнару-
обнаружить при исследовании диаграмм состав — свойство. На рис. 106 представлен
общий вид изотерм электрической проводимости и твердости в системе с образо-
образованием одного промежуточного соединения, причем
вблизи ординат компонентов и соединения сущест-
существуют области гомогенности. В гетерогенной области
изотермы свойств имеют вид аддитивных прямых, а
в области твердых растворов они подчиняются
законам Курнакова. Характерной особенностью
таких диаграмм состав — свойство является наличие
особой точки на изотермах свойств, которая отвеча-
отвечает некоторому составу промежуточной фазы. При
этом для любого измеряемого при данных условиях
физического свойства экстремальная точка на изо-
изотермах состав — свойство соответствует одному и
тому же составу. Согласно Курнакову, такие особые
точки на изотермах состав — свойство называются
сингулярными. Данное понятие привлечено из гео-
геометрической топологии и характеризует точки,
инвариантные относительно преобразования коорди-
координат. В рамках физикО-ХИМИЧескОГО анализа ЭТО Рис. 106. Диаграмма образования
означает, что при замене координат физических дальтонида и характер изотерм
свойств на диаграммах состав — свойство [например, электрической проводимости б и
° —/(О на Я = /(я )] положение сигнулярной твердости Я
Под промежуточными фазами понимают все многообразие химических объектов, ото-
отображаемое на диаграмме состояния между ординатами чистых компонентов (от ограни-
ограниченных твердых растворов до соединений дальтоновского типа включительно).
205
*^^Ш
точки остается постоянным по составу. В соответствии с этими представлениями
сформулировано понятие о дальтониде. Далътонидом называется промежуточ-
промежуточная фаза, которая на диаграммах состав — свойство характеризуется сингуляр-
сингулярной точкой.
В соответствии с современными представлениями класс дальтонидов не
ограничивается соединениями постоянного состава, но включает в себя большое
число фаз переменного состава. Необходимым условием существования
дальтонидной фазы является лишь наличие внутри области ее гомогенности
некоторого предпочтительного состава, которому отвечает сингулярная точка на
диаграммах состав — свойство. При изменении внешних условий состав
дальтонида может изменяться. Однако при заданных температуре и давлении
состав, отвечающий сингулярной дальтоновской точке, отражает "привилегию
дискретности" в химии. Таким образом, по Курнакову, не состав твердой фазы
характеризует дискретное химическое соединение, поскольку он может быть
переменным в пределах области гомогенности, а состав, отвечающий
дальтоновской точке на диаграммах состав — свойство. С этой точки зрения,
далътонидами в широком смысле являются фазы переменного состава, внутри
области гомогенности которых существует, некоторый состав (не обязательно с
целочисленными и небольшими undtKcaMii), отвечающий инвариантности свойств
в данных условиях.
Экспериментальный материал по исследованию диаграмм состояния и диаг-
диаграмм состав — свойство свидетельствует о широком распространении промежуточ-
промежуточных фаз переменного состава с иным характером изменения свойств в пределах
области гомогенности. Отличительным признаком диаграмм состав — свойство
таких систем является отсутствие инвариантной дальтоновской точки на изотер-
изотермах физических свойств, например, твердости Н, в пределах области гомогенно-
гомогенности промежуточной фазы (рис. 107). При этом, очевидно, ни один из составов в
области существования фазы не будет предпочтительным и не характеризует
состав соединения, на основе которого эта фаза существует. Такие фазы перемен-
переменного состава были названы Курнаковым бертоллидами. По своей природе бертол-
лиды занимают промежуточное положение между твердыми растворами и соеди-
соединениями дальтонидного типа. С химическими соединениями подобные фазы
сходны в том отношении, что они обладают своеобразным, отличным от компо-
компонентов кристагыюхимичееким строением. С твердыми растворами бертоллиды
роднит отсутствие предпочтительного состава в пределах области гомогенности,
на базе которого сформирована фаза.
В настоящее время взгляды на природу бертоллидных фаз расширены. Часто
рациональный состав с целочисленными индексами,
отвечающими закону кратных отношений Дальтона,
может оказаться внутри области гомогенности бер-
толлида. Однако этот состав не является предпоч-
предпочтительным и на изотермах свойств ему не отвечает
сингулярный экстремум. Классическим примером
такого типа может служить соединение TiO, сущест-
существующее в интервале концентраций ТЮо^в-л.зз, при-
о ,._ _ чем рациональный состав TiO находится внутри
Рис. 107. Диаграмма состоя- к
ния с бертоллидом и характер этой области. Однако характер изотерм состав
изотермы твердости Я
свойство, типичный для бертоллидных фаз, не
Соединения
постоянного
состава
(дальтоннды
в узком
смысле)
Фазы
переменного
состава-
растворы
на основе
соединения
(дальтониды
» широком
смысле)
Фазы Курнакова
(растворы между
дальтонидами и берголлндамн)
Фазы
переменного
состава -
бертоллнды
позволяет выделить этот состав среди всех остальных. Соединения такого типа
распространены очень широко (VO0)90-i,30> UO^-s.o и т.п.). С этой точки зрения,
бертоллидалш в широком смысле слова следует считать промежуточные фазы,
не обнаруживающие на изотермах свойств дальтоновской точки в пределах облас-
области гомогенности. При этом совершенно безразлично, находится ли рациональ-
рациональный состав внутри этой области или за ее пределами. Таким образом, следует
различать фазы переменного состава, представляющие собой твердые растворы
на основе химического соединения дальтоновского типа, и фазы переменною
состава — бертоллиды, которым в принципе невозможно приписать какой-то
определенный "предпочтительный" состав в пределах их области гомогенности.
Рассмотрим взаимосвязь между различными твердыми фазами. В основу пред-
предлагаемой классификации положены два признака: кристаллохимическая инди-
индивидуальность и постоянство или переменность состава фазы. Предельно общим в
данной классификации является понятие о твердой фазе. Твердые фазы подраз-
подразделяются на химические индивиды — фазы постоянного или переменного соста-
состава, характеризующиеся качественно своеобразной кристаллической структурой, и
твердые растворы — фазы переменного состава, структура которых идентична
структуре компонента-растворителя и свойства которых, следовательно, качест-
качественно не отличаются от свойств компонентов.
Химические индивиды представляют собой как простые вещества, так и слож-
сложные (соединения). Простые вещества можно рассматривать как частный случай
соединений постоянного состава, называемых дальтонидами в узком смысле. К
этому же классу следует отнести и сложные вещества, обладающие молекулярной
кристаллической структурой (например, твердый ССЬ). Отличительный признак
молекулярных кристаллов — образование фаз постоянного состава. Таким обра-
образом, на диаграмме состояния эти соединения представляют собой линейные фазы
с нулевой областью гомогенности.
Твердые растворы, являющиеся фазами переменного состава, представляют
собой или растворы на основе простых веществ, не обладающие качественной
кристаллохимической индивидуальностью по сравнению с чистым веществом, или
растворы на основе соединения. Последние представляют собой фазы переменно-
переменного состава, характеризующиеся наличием дальтоновской точки на кривых сос-
состав — свойство в пределах области гомогенности. Эта дальтоновская точка отве-
отвечает составу соединения, и твердые растворы этого типа целесообразно назвать
дальтонидами в широком смысле и рассматривать не только как растворы, но и
как химические индивиды, причем последний признак следует считать домини-
доминирующим. Соединения переменного состава — бертоллиды — при своеобразии
кристаллохимического строения не характеризуются сингулярной точкой на
изотермах свойств, что роднит их с твердыми растворами.
Деление химических индивидов на дальтониды и бертоллиды относительно,
поскольку экспериментально установлено существование так называемых фаз
Курнакова — твердых растворов между дальтонидами и бертоллидами, образу-
образующихся в некоторых тройных системах и представляющих собой, таким образом,
непрерывный переход между этими типами соединений.
Г Л А В А IX. МЕТАЛЛОХИМИЯ
С глубокой древности металлы и сплавы на их основе играют колоссальную
роль в развитии человеческого общества. Однако систематические научные иссле-
исследования природы химического взаимодействия металлов друг с другом были
начаты сравнительно поздно — на рубеже XIX и XX вв. Это было связано с тем,
что препаративный метод исследования веществ, используемый классической
неорганической химией, оказался бессильным для изучения металлических спла-
сплавов вследствие своеобразия их свойств.
Необходим был новый метод исследования, позволяющий установить природу,
состав и число образующихся фаз в системах, не прибегая к их выделению и
анализу. В 1889 г. была опубликована работа Курнакова "О взаимных соединени-
соединениях металлов", в которой на основе исследования температуры плавления и
микроструктуры некоторых сплавов натрия ученый приходит к выводу о сущест-
существовании металлических соединений, которым на кривых зависимости свойств от
состава отвечают характерные точки, названные впоследствии сингулярными или
дальтоновскими.
Таким образом, наметились новые пути исследований, в основе которых лежа-
лежало изучение свойств сплавов в зависимости от изменения их состава, что стало
содержанием нового метода исследования — физико-химического анализа. По
мере накопления материала в области изучения металлических сплавов развилась
новая область общей и неорганической химии — химия металлических сплавов.
Эта область теснейшим образом связана с физической химией, физикой и химией
твердого тела, кристаллохимией, металловедением.
1. Элемептохимия. Часть общей и неорганической химии, рассматривающую
взаимодействия элементов Периодической системы друг с другом, целесообразно
назвать элементохимией. Это понятие не адекватно определению химии элемен-
элементов. Последний термин более широк и охватывает все многообразие соединений
рассматриваемого элемента, в том числе комплексные соединения. Главная же
задача элементохимии состоит в выяснении общих закономерностей образования
фаз при взаимодействии простых веществ друг с другом, в изучении кристалле»-
химических превращений этих фаз (распад твердых растворов, образование
сверхструктур и т.п.). Кроме того, элементохимия изучает природу химической
2ПЙ
связи, химические, физико-химические, электрофизические и иные свойства та-
таких систем в зависимости от состава и внешних условий (давления и темпера-
температуры).
В соответствии с разделением элементов на катионо- и анионообразователи —
металлы и неметаллы — в рамках элементохимии возможны три типа взаимодей-
взаимодействия: неметалл + неметалл, неметалл + металл, металл + металл. В зависимости
от физико-химической природы промежуточных фаз эти взаимодействия приво-
приводят к образованию двух групп объектов. Химия неметаллических фаз изучает
объекты, возникающие при взаимодействии неметалл + неметалл и неметалл +
металл. А предмет химии металлических фаз, или металлохимии, составляет
обширный класс разнообразных фаз, образованных в результате взаимодействия
катионообразователей друг с другом. Правомерность такого разделения под-
подтверждается различием природы образующихся соединений. Ниже представлена
классификация взаимодействий в рамках злементохимии, в которой отмечены
характерные особенности и признаки промежуточных фаз различного типа. При-
Приведенная классификация относительна уже хотя бы потому, что нет четкой грани
между металлами и неметаллами. В соответствии с этим по ряду признаков объ-
объекты химии неметаллических фаз обладают сходными свойствами. Разделение их
по свойствам возможно провести только для фаз, подчиняющихся правилу фор-
формальной валентности, — так называемых нормально-валентных соединений. Ха-
Характерной особенностью нормально-валентных продуктов взаимодействия в рам-
рамках химии неметаллических фаз является наличие только "катион-анионных"
связей.
Ни одна классификация не в состоянии отразить всего многообразия взаимо-
взаимодействий. Например, многие нитриды переходных металлов, представляющие
собой так называемые фазы внедрения, обладают ярко выраженными металличе-
металлическими свойствами, что позволяет отнести их к объектам металлохимии.
Элементохимия
У
Химия неметаллических фаз
, неметалл+неметалл
неметалл+металл
t т
Связь:
ковалентно-нонная (с преоб-
преобладанием ковапентной сос-
составляющей и незначительной
долей металличности)
Связь:
ионно-ковалентиая (с преоб-
преобладанием ионной составля-
составляющей и незначительной
долей металличности)
Структура:
рыхлая
ьч.< 4
Структура:
рыхлая и плотная
4 < к.ч.< 8
Состав:
небольшие целочисленные стехиометрические индексы,
рациональный состав, узкие области гомогенности
Свойства:
диэлектрики,
полупроводники
Свойства:
полупроводники,
диэлектрики
Химия металлических фаз (металлохимия) [
металл+мегалл
Связь:
металлическая, ковалентно-металличе-
ская (с преобладанием металлической
составляющей и незначительной
долей ионности)
Структура:
плотная и шютнейшая
к.ч.> 8
Состав:
произвольные стехиометричесхие ин-
индексы, широкие области гомогенности
Свойства:
металлы
209
Соединения между металлами часто называют интерметаллическими соедине-
соединениями (например, NaZn13, NaK2, Na4Pb). Этот термин подчеркивает лишь проис-
происхождение фазы, но не ее природу. Если же хотят отметить металлический харак-
характер промежуточной фазы со всем комплексом присущих металлу свойств, то упот-
употребляется термин металлид или металлическое соединение. Это понятие более
широкое, так как металлической природой могут обладать не только интерметал-
интерметаллические соединения, но и некоторые фазы, образованные при взаимодействии
металлов с неметаллами (например, некоторые оксиды, карбиды, нитриды пере-
переходных металлов и т.п.). В то же время не могут существовать металлические
соединения с неметаллическим характером, поскольку делокализация электронов
при наличии вакантных мест в зоне проводимости (характерная для метал-
металлических компонентов) сохраняется и после взаимодействия.
Строго говоря, в металлохимии следует рассматривать лишь интерметалличе-
интерметаллические фазы (соединения, растворы и т.п.), которые всегда обладают металличе-
металлическим характером. Однако сюда целесообразно включать и металлидные фазы,
образованные в результате взаимодействия металлов с неметаллами (некоторые
карбиды, бориды, нитриды, субоксиды и т.п.). Свойства этих фаз в значительной
мере определяются именно металлическим компонентом.
2. Металлохимические свойства элементов. Характер взаимодействия элемен-
элементов друг с другом определяется в основном разностью их электроотрицательно-
стей, электронной концентрацией (количеством валентных электронов, приходя-
приходящихся на каждый атом в формульной единице вещества, а также типом валент-
валентных электронных орбиталей) и соотношением размеров атомов взаимодейству-
взаимодействующих компонентов.
При образовании интерметаллических фаз роль разности электроотрицатель-
ностей не может быть определяющей, поскольку элементы, расположенные слева
от границы Цинтля *, характеризуются сравнимыми величинами электроотрица-
тельностей и разность их не превышает 0,8. При оценке разности электроотрица-
тельностей в металлохимических реакциях необходимо использовать значения,
характеризующие элементы в низших степенях окисления, поскольку в интерме-
интерметаллических фазах высшие степени окисления реализоваться не могут. Опреде-
Определенную роль при взаимодействии металлов друг с другом играют факторы элект-
электронной концентрации и размерный.
Различие между простыми и переходными металлами проявляется уже при
сравнении атомных радиусов, d-, /-Элементы характеризуются меньшими значе-
значениями радиусов, чем sp-металлы. Кроме того, различие атомных радиусов у sp-
элементов-аналогов значительно больше, чем у элементов вставных декад. Так, у
металлов IA-группы радиусы изменяются от 0,250 для Rb до 0,155 нм у Li, a
атомные радиусы всех rf-элементов — в интервале 0,124 — 0,181 нм. Еще более
близки атомные радиусы у /-металлов, что объясняется заполнением третьего
снаружи энергетического уровня. Так, все элементы семейства лантаноидов имеют
атомные радиусы в пределах 0,174 — 0,183 нм.
Различие между простыми и переходными металлами проявляется и в отно-
отношении электронной концентрации. Электронная концентрация представляет
собой общее количество валентных электронов на один атом, причем часть этих
"Условная граница, проходящая по элементам IVA группы в полудлинной, форме Пе-
Периодической системы Менделеева (см. табл. 4).
электронов обобществляется с образованием металлической связи, а некоторая их
доля может участвовать в образовании ковалентной связи. Поэтому понятия
электронной концентрации и концентрации электронов проводимости не всегда
совпадают. У щелочных и щелочно-земачьных металлов валентные электроны
полностью отданы в коллективное пользование. У переходных элементов общее
число валентных электронов возрастает, но число электронов, участвующих в
образовании металлической связи, колеблется в тех же пределах (в среднем 1—2
электрона на атом). Однако прочность связи в кристаллах d-элементов значитель-
значительно выше за счет усиления ковалентного взаимодействия, обусловленного возник-
возникновением d — rf-связей. Таким образом, эти элементы обладают в кристалличе-
кристаллическом состоянии не чисто металлической, а ковалентно-металлической связью.
На основании рассмотренных металлохимических свойств элементов можно
проанализировать общие закономерности взаимодействий металлов друг с дру-
другом. Интерметаллические взаимодействия сводятся к следующим основным ти-
типам: образование ограниченных твердых растворов, полная взаимная раствори-
растворимость, возникновение интерметаллических соединений.
Простые металлы являются плохими растворителями, поскольку для них
характерно отсутствие внутренних вакантных орбиталей. Особенно плохие раство-
растворители — щелочные металлы. Вследствие значительных размеров атомов они и
сами довольно плохо растворяются в других металлах. Для систем с участием 9-
и sp-металлов часто наблюдается расслоение в жидкой фазе (Na — А1, К — Fe и
ДР-)-
От щелочно-земельных металлов бериллий и магний существенно отличаются
по металлохимическим свойствам, хотя магний примыкает к щетючно-земельным
металлам несколько ближе. В частности, бериллий уже способен образовывать
химические соединения с дефектными (/-металлами, в то время как магний обла-
обладает этой способностью в меньшей степени. sp-Металлы ША-группы непрерыв-
непрерывных твердых растворов не образуют ни с одним элементом Периодической
системы (даже друг с другом), но образуют химические соединения с элементами
IA- и ПА-групп, большинством переходных металлов, полуметаллами и неметал-
неметаллами.
Переходные металлы в силу дефектности d- и /оболочек, близости значений
ионизационных потенциалов и атомных радиусов обладают обширными металло-
химическими возможностями. Как правило, они являются прекрасными раствори-
растворителями для других элементов (за исключением щелочных и щелочно-земельных),
однако образуют непрерывные твердые растворы лишь между собой. Переходные
металлы способны давать большое количество интерметаллических фаз разнооб-
разнообразного состава как при взаимодействии друг с другом, так и с элементами ША-
группы, бериллием и магнием. Кроме того, они хорошо взаимодействуют с эле-
элементами, расположенными справа от границы Цинтля.
При общей оценке типа взаимодействия в металлических системах необходимо
обращать внимание на разницу температур плавления и разность электроотрица-
тельностей. Хотя последний фактор в металлохимии и не играет столь существен-
существенной роли, как в химии металлов и неметаллов в целом, тем не менее он может
указывать на возможность образования интерметаллического соединения. Если
простые вещества обладают значительной разницей температур плавления, то
возможно расслоение в жидкой фазе. Этот вывод приобретает еще большую опре-
определенность, если элементы обладают различным строением валентных электрон-
211
ных оболочек и различаются по ионизационным потенциалам. Так, щелочные
металлы подгруппы калия, несмотря на их высокую активность, не взаимодейст-
взаимодействуют с алюминием, что обусловлено существенной разницей температур плавле-
плавления, ионизационных потенциалов и электронного строения. При этом даже замет-
заметная разность электроотрицательностей не способствует взаимодействию. Если
температуры плавления компонентов относительно близки, а разность электроот-
электроотрицательностей значительна, то в системе возможно образование соединений.
Например, в системах с участием алюминия и щелочно-земельных металлов суще-
существует целая серия промежуточных фаз: МеАЦ, MeAl2, MeAl. Если оба фактора
(т.пл. и ОЭО) близки, то определяющее влияние на характер взаимодействия
оказывает соотношение атомных факторов. При близости атомных радиусов воз-
возможно образование непрерывных твердых растворов (Са — Ва, К — Rb, К — Cs).
Если размеры атомов отличаются более значительно, то возможно образование
ограниченных твердых растворов, а при существенной разности ОЭО — и соеди-
соединений. Так, натрий со своими аналогами (К, Rb, Cs) дает лишь ограниченные
твердые растворы. Кроме того, поскольку электроотрицательность натрия заметно
выше, чем у калия, рубидия и цезия, то возможно образование интерметалличе-
интерметаллических соединений. В самом деле, в системах Na — К, Na — Cs существуют фазы
KNa2) CsNa2.
Попытаемся оценить характер взаимодействия в неизученных системах бария
с рубидием и цезием на основании металлохимических свойств компонентов (рис.
108). Для данных металлов характерно существенное различие в температурах
плавления. Это могло бы свидетельствовать о тенденции к расслоению. С другой
стороны, для компонентов отмечается значительная разность ОЭО, что определя-
определяет вероятность образования соединений. В то же время различие атомных ра-
радиусов B0%) исключает возможность возникновения широкой области гомогенно-
гомогенности в твердом состоянии. Если же учесть близость ионизационных потенциалов
компонентов, благоприятствующую взаимному обобществлению электронов, то из
030
0,5
г,нм
0,3
Т.пл.,
Li Na К RbCs BeMgCaSrBa Al Ga in Tl
as1 HP п$г np'
Рис. 108. Изменение основных металлохимических
факторов s- и sp-элементов I - III групп:
/ - температура плавления; 2 - ОЭО; 3 - атомные ра-
радиусы; 4 ' ионизационные потенциалы
двух возможностей (расслоение —
соединение) более вероятным пред-
представляется образование промежу-
промежуточных фаз. Итак, в системах Ва —
Rb, Ва — Cs возможно образование
весьма ограниченных твердых
растворов на основе компонентов, а
также металлических соединений.
3. Примитивные типы химическо-
химического взаимодействия в металлических
системах. К примитивным типам
взаимодействия относят в порядке
усложнения расслоение (отсутствие
взаимодействия), образование эв-
эвтектической смеси, образование
ограниченных и непрерывных твер-
твердых растворов. Характерной осо-
особенностью рассматриваемых типов,
которая позволяет их объединить,
является то, что в результате обра-
212
зуются фазы, физические, химические и кристаллохимические свойства которых
качественно не отличаются от свойств компонентов. Так, на диаграммах состав —
свойство при образовании эвтектических смесей наблюдаются аддитивные пря-
прямые, соединяющие величины свойств чистых компонентов. На рис. 109 представ-
представлены диаграмма состояния с эвтектикой и изотермы интегральных свойств систе-
системы — твердости (Я) и электрической проводимости (<т) в зависимости от состава.
Так как в твердом состоянии существует лишь смесь кристаллов чистых компо-
компонентов S + S , то свойства отдельных кристаллов каждой из фаз остаются по-
постоянными (штриховые линии), а интегральное свойство сплавов изменяется про-
пропорционально концентрации:
Н= х Н + A -х )Н ,
В В V В7 А
где Н и Н — твердость компонентов; Н — интегральная твердость сплава; х —
А В В
молярная доля компонента В в сплаве.
При образовании непрерывных твердых растворов в соответствии с законами
Курнакова изотермы состав — свойство представляют собой плавные кривые, на
которых иногда наблюдаются экстремумы вблизи 50% одного из компонентов. В
частности, такие экстремумы характерны для изотерм а = f (х )и Я = / (х ) (см.
В В
рис. 105), что объясняется максимальными искажениями кристаллической решет-
решетки для средних составов, а не свидетельствует о качественном своеобразии экви-
атомного сплава. Изотерма молярного объема для идеального раствора представ-
L+S
В
Рис. 109. Изотермы твердости Я
и удельной электрической прово-
проводимости (Т для диаграммы состоя-
состояния системы с эвтектикой
в
Рис. 110. Изотермы твердости Я и
удельной электрической проводимости (Т
для диаграммы состояния системы "с
ограниченными твердыми растворами
213
ляет собой аддитивную прямую. Для реальных растворов, которые образуются с
изменением объема вследствие заметного химического вклада во взаимодействие
компонентов, наблюдаются отклонения от прямой, однако экстремумы отсутст-
отсутствуют.
При образовании ограниченных твердых растворов изотермы состав — свойст-
свойство в пределах области гомогенности имеют вид плавных кривых, в гетерогенной
области — аддитивных прямых (рис. 110). При отсутствии взаимодействия (рас-
(расслоение) свойства каждой фазы в твердом состоянии остаются постоянными. От
расслоения к непрерывным твердым растворам возрастает химический вклад во
взаимодействие. Этот вклад, однако, определяется фактором низшего порядка —
размерным, поэтому взаимодействие не приводит, как правило, к образованию
химических соединений. В самом деле, основным критерием образования непре-
непрерывных твердых растворов является сходство физико-химического характера
взаимодействующих компонентов, что определяется близостью значений ОЭО,
электронного строения и типа химической связи. Кроме того, в соответствии с
правилом Руайте размеры атомов при этом не должны различаться более чем на
8 — 15%, что с учетом подобия остальных факторов предопределяет одинаковый
тип кристаллической решетки. Если при сходстве электронных конфигураций и
значений ОЭО атомные размеры компонентов отличаются более значительно, то
вместо непрерывных образуются ограниченные твердые растворы с возникновени-
возникновением между ними гетерогенной области — эвтектической смеси. Образование эвтект
тики возможно и тогда, когда атомные размеры компонентов близки, а электрон-
электронное строение различно. Это различие не должно возрастать настолько, чтобы
существенным становился вклад электроотрицательности, поскольку тогда воз-
возможно образование химических соединений.
Рассмотренным условиям образования твердых растворов в особой степени
удовлетворяют переходные металлы, способные давать непрерывные твердые
растворы как в пределах групп (Ti — Zr — Hf, V — Nb — Та, Сг — Mo — W
и т.д.), так и при взаимодействии металлов близко расположенных групп
(Ti - V, Ti - Cr, Zr - Nb, Nb - Mo, Nb - W и т.д.). По мере увеличения
различия металлохимических свойств растворимость металлов друг в друге будет
падать. Для примера приведем значения растворимости металлов IV периода в
титане:
Металл
Растворимость в /?-Ti, %
V
100
Сг
100
Мп
28
Fe
20
Со
13
Ni
10
4. Образование соединений в металлических системах. В противоположность
примитивным типам взаимодействий металлохимические реакции, приводящие к
образованию соединений, можно условно отнести к сложным типам. Основное
отличие этих процессов заключается в возникновении при взаимодействии каче-
качественно нового химического индивида, характеризующегося своеобразными
структурой и свойствами по сравнению с исходными компонентами. По мере
нарастания взаимного химического сродства металлов образующиеся соединения
приобретают все более ярко выраженную индивидуальность. В зависимости от
того, какой из металлохимических факторов преобладает при взаимодействии,
возникают фазы различного типа: соединения Курнакова, фазы Лавеса, фазы
внедрения, электронные соединения Юм-Розери и, наконец, соединения, отвеча-
214
ющие правилу формальной валентности. Последние соединения возникают при
взаимодействии металлов с неметаллами, когда преобладает фактор электроотри-
электроотрицательности, и в рамках металлохимии обычно не рассматриваются. Тем не менее
для получения полной картины взаимодействия металлов этот случай целесо-
целесообразно рассмотреть в общей связи.
Переходным этапом от примитивных типов взаимодействия к более сложным
является образование соединений Курникова. В 1914 г. Курнаков с сотрудниками,
исследуя систему Си — Аи, показал, что непрерывные твердые растворы при
медленном охлаждении претерпевают превращения с образованием металличе-
металлических соединений Cu3Au и CuAu, дающих твердые растворы с избытком своих
компонентов. Образование этих соединений из непрерывных твердых растворов
можно сравнить с выделением кристаллогидратов из водных растворов. Это явле-
явление было подтверждено как методом термического анализа, так. и изучением
твердости, микроструктуры и электрофизических свойств исследуемых образцов.
В этом отношении работа Курнакова представляет собой классический пример
исследования твердофазных превращений методами физико-химического анали-
анализа. Было отмечено, что изотермы свойств быстро охлажденных (закаленных)
сплавов представляют собой плавные кривые, свойственные непрерывным твер-
твердым растворам во всем интервале концентраций. Напротив, отличительным приз-
признаком изотерм свойств сплавов, подвергнутых длительной термообработке (отжи-
(отжигу) при температуре ниже линии голидуса, служит наличие сингулярных точек,
отвечающих соединениям Cu3Au и CuAu (рис. 111). Эти соединения имеют до-
довольно широкие области гомогенности: 22 — 40% для Cu3Au и 42,5 — 70% для
CuAu. Полученные данные позволили объяснить причину хрупкости отожженных
сплавов в области существования данных соединений. По сравнению с твердыми
растворами металлиды менее способны к пластической деформации. Последу-
Последующие исследования системы Си — Аи при помощи рентгеноструктурного анализа
подтвердили факт существования твердофазных превращений в системе и в зна-
значительной степени способствовали раскрытию их природы.
Медь и золото, кристаллизующиеся в кубической гранецентрированной ре-
решетке, образуют между собой при повышенных
температурах и закалке непрерывный ряд твердых
растворов. При этом атомы металлов статистически
неупорядоченно распределены по узлам решетки
(рис. 112, а). При отжиге происходит процесс упо-
упорядочения в распределении атомов золота и меди в
кристаллической структуре, причем степень упоря-
упорядочения будет наибольшей для атомных соотношений
Си : Аи = 3:1 и Си : Аи = 1:1, отвечающих
соединениям Cu3Au и CuAu. В Cu^Au атомы золота
будут располагаться в вершинах кубической элемен-
элементарной ячейки, а атомы меди займут центры всех
граней (рис. 112, б). Поскольку каждый атом в вер-
вершине куба принадлежит одновременно восьми сосед-
соседним ячейкам, на данную ячейку приходится !Д атома
золота. Так как в вершинах куба находится 8 атомов
Рис. 111. Диаграмма состоя-
состояния системы Си - Аи и изотер-
Аи, то данной ячейке соответствует 8-1/8 = 1 атом мы электрической проводимос-
Аи, Атомы меди, располагающиеся в центрах граней, ™ а и твердости Я
215
Рис. 112. Кристаллическая структура сплавов системы медь - золото:
а - неупорядоченный твердый раствор; б - соединение Курнакова состава
CugAu; в - соединение Курнакова состава CuAu
принадлежат каждый двум соседним ячейкам. Поэтому на данную ячейку при-
приходится бл/г = 3 атома, меди, что и отвечает составу Cu3Au. В CuAu (рис. 112, в)
атомы золота расположены в вершинах куба и в центрах двух граней, а атомы
меди — в центрах четырех граней. Подсчет числа атомов на ячейку дает для
золота 8-1/8 + 2-1/2 = 2 атома, для меди АЛ/2 = 2 атома, что соответствует
формульному составу CuAu. На практике полной упорядоченности в расположе-
расположении атомов получить не удается. Степень упорядоченности составляет обычно
80 - 90%.
Металлические соединения, образующиеся из твердых растворов замещения
при отжиге или медленном охлаждении и характеризующиеся упорядоченным
расположением атомов в узлах кристаллической решетки, называются соединени-
соединениями Курнакова*. В настоящее время соединения Курнакова обнаружены в много-
многочисленных системах, образующих твердые растворы. Даже в такой системе, как
Ag — Аи, образованной металлами, весьма близкими по металлохимическим свой-
свойствам, в результате длительного отжига (более 1300 ч) отмечено образование
соединений Курнакова AuAg3 и AuAg. Соединения Курнакова образуются не
только в результате упорядочения непрерывных твердых растворов, но и в систе-
системах с ограниченной растворимостью компонентов. Примером могут служить со-
соединения MgAg3, F3A1, VNi3, PtCu, VCo3, MnAu3, Ni3Au, Ti3Al.
В соединениях Курнакова сохраняется тип кристаллической структуры компо-
компонентов, однако свойства их качественно отличны от свойств компонентов. Именно
по этой причине данные фазы представляют собой переходную ступень от твер-
твердых растворов к соединениям. Образование соединений Курнакова регламентиру-
регламентируется фактором низшего порядка — размерным. Ни электронная концентрация, ни
тем более электроотрицательность не играют здесь существенной роли, поскольку
взаимодействующие компоненты близки по физико-химической природе. С тер-
термодинамической точки зрения, существование соединений Курнакова (упорядо-
(упорядоченных твердых растворов) возможно при низких температурах, когда энтропий-
энтропийным членом в выражении AG — АН — TAS можно пренебречь. С повышением
*Эти соединения иногда называют также упорядоченными твердыми растворами или
фазами со сверхструктурой.
216
• Си. OMJ
Рис. 113. Кристаллическая струк-
структура MgCu2
температуры энтропия системы возрастает вследст-
вследствие увеличения разупорядочения. Это приводит к
исчезновению соединений Курнакова и образова-
образованию статистически неупорядоченного твердого
раствора. По своей природе все известные соедине-
соединения Курнакова являются дальтонидами.
Ряд интерметаллических соединений объединя-
объединяется под общим названием фаз Лавеса. Общими
для этих фаз служат следующие признаки: им
отвечает состав АВг, все они принадлежат к
плотноупакованным структурам с высокими коор-
координационными числами и, так же как и соедине-
соединения Курнакова, образуются с преобладанием раз-
размерного фактора. Однако роль размерного фактора для этих групп соединений
различна. Если условием образования соединений Курнакова является близость
атомных размеров компонентов, обеспечивающая их изоморфное замещение в
узлах кристаллической решетки, то для фаз Лавеса важно определенное
различие (в идеальном случае — в два раза) объемов атомов компонентов. Если
соблюдается условие V и 2V (или г : г и 1,26), то атом компонента А занимает
в структуре соединения два места плотнейшей упаковки, а атом компонента В —
одно, что и отвечает формульному составу АВ2. Типичным и характерным
примером фаз Лавеса является соединение MgCii2. Действительно, атомные
радиусы магния и меди равны соответственно 0,16 и 0,128 нм, а объемы атомов
V (Mg) и 4,1-10 нм, V (Си) «' 2,1-10 нм. Отношение атомных объемов
V (Mg) : V(Cu) и 2, что соответствует условию образования фаз Лавеса. Объемы
атомов меди и золота [V^(Au) и 3-Ю нм] различаются меньше, поэтому здесь
имеет место изоморфное замещение.
Структура MgCu2 представлена на рис. 113. Основу ее формально составляет
структура типа алмаза, сформированная атомами магния. Это гранецентрирован-
ная кубическая решетка, в которой центры четырех из восьми октантов также
заняты атомами магния *. Оставшиеся свободными октанты заняты тетраэдриче-
скими комплексами Си^ таким образом, что центр тетраэдра совпадает с центром
октанта. Такую структуру (типа MgCii2) образуют многие фазы Лавеса. Помимо
этого для них характерны еще два структурных типа: MgZii2 и MgNi2, не отлича-
отличающиеся существенно от описанного. Во всех этих соединениях в кристалле суще-
существуют разнотипные связи А~В, А~А, В~В. Фазы Лавеса, являющиеся следу-
следующей ступенью на пути усложнения химической организации вещества, фактиче-
фактически выступают как первые представители собственно химических соединений.
Несмотря на то что их образование также определяется размерным фактором,
они уже отличаются от компонентов не только по свойствам, но и в структурном
отношении. Как и для соединений Курнакова, для многих фаз Лавеса характерна
сверхпроводимость.
К числу фаз, возникновение которых определяется соотношением атомных
размеров компонентов, относятся и так называемые фазы внедрения. Как и
*Октант — 1/8 куба элементарной ячейки, полученная рассечением его тремя взаимно
перпендикулярными плоскостями, проходящими через центр.
217
твердые растворы внедрения, они образуются при взаимодействии металлов и
легких неметаллов (водород, бор, углерод, азот, кислород)*. Образование ме-
таллидных фаз внедрения (карбидов, нитридов и т.п.) характерно только для
переходных металлов с дефектными rf- и /оболочками. При низких концент-
концентрациях неметалла наблюдается простое растворение с образованием твердых
растворов внедрения, которое сопровождается поглощением теплоты. Вследствие
итого в соответствии с правилом Ле Шателье растворимость неметалла с по-
повышением температуры растет. Растворение сопровождается возрастанием упру-
упругих напряжений и увеличением межплоскостных расстояний, хотя кристал-
кристаллическая структура металла-растворителя при этом сохраняется. По мере уве-
увеличения концентрации внедряемого неметалла развивается значительное внут-
внутреннее давление, происходит изменение структуры и возникает новый хими-
химический индивид — фаза внедрения. Например, тантал имеет структуру ОЦК, а в
фазе внедрения ТаС атомы тантала располагаются по позициям кубической
плотнейшей упаковки (ГЦК).
Образование фаз внедрения в отличие от твердых растворов внедрения сопро-
сопровождается экзотермическим эффектом, иногда довольно значительным
(например, для ZrH АН - —169,3 кДж/моль, для TiC АН = —183,5 кДж/моль).
Это обусловлено уменьшением свободной энергии системы в процессе пере-
перестройки структуры. Возможность образования фаз внедрения регламентируется
правилом Хэгга. Но здесь существенно сказывается и влияние электронной кон-
концентрации, так как электроны внедренного неметалла обобществляются, попадая
на вакантные d- и /-орбитали металла. Это приводит к образованию новой струк-
структуры с металлическими свойствами, в которой атомы неметалла тоже металлизо-
ваны.
Металлизация атомов неметалла способствует увеличению электронной кон-
концентрации в решетке переходного металла, деформированной в процессе внедре-
внедрения, что приводит к заполнению вакантных состояний в (/-зоне металла и усиле-
усилению ковалентности связи. При этом образуются прочные гиб>ридкые rfs/h-связи с
участием rf-электронов переходного металла и sp-электронов внедряющихся ато-
атомов. Именно поэтому максимальной тугоплавкостью обладают карбиды и нитри-
нитриды начальных элементов (/-рядов (метаплов IVB — VB-групп). Сами же эти ме-
металлы не являются наиболее тугоплавкими в своих рядах. В то же время карби-
карбиды и нитриды хрома, молибдена и вольфрама, обладающих максимальными
температурами плавления, относительно менее тугоплавки. Это можно объяснить
тем, что в самих металлах VIB-группы ковалентность максимальна, дефицит
электронов ощущается не столь остро и электроны внедряемых атомов способст-
способствуют главным образом металлизации связи.
Состав фаз внедрения определяется не взаимным сродством компонентов, а
геометрическими соображениями. Число октаэдрических пустот на одну элемен-
элементарную ячейку равно числу атомов в этой ячеке, а число тетраэдрических пустот
в два раза больше. Если внедряемые атомы занимают октаэдрические пустоты, то
ожидаемый состав фазы внедрения будет отвечать формуле АВ, если же зани-
*Металлохимия не изучает взаимодействия между металлами и неметаллами. Однако
здесь рассматриваются только те, которые приводят к образованию соединений, облада-
обладающих металлидным характером.
маются тетраэдрические пустоты — АВг (А — металл, В — неметалл) *. Так как
размер тетраэдрических пустот меньше, то фазы типа АВг могут образоваться
только при внедрении малых атомов водорода. Действительно, существуют гид-
гидриды TiH2, ZrH2 и т.д. Для карбидов, нитридов и боридов более характерны
фазы внедрения состава АВ (TiC, TaN, HfH, ZrB и т.п.), что указывает на внед-
внедрение атомов неметалла в октаэдрические пустоты * *.
В химическом отношении многие фазы внедрения (особенно некоторые карби-
карбиды и нитриды) представляют собой инертные материалы, безразличные к дейст-
действию сильных минеральных кислот, что открывает возможности их использования
для изготовления деталей химической аппаратуры. Помимо этого, обработка
поверхности металлов, позволяющая создать на ней карбидный, боридный или
нитридный слой, не только улучшает механические свойства, но и повышает
коррозионную стойкость. Фазы внедрения обладают высокой твердостью, туго-
тугоплавкостью и жаропрочностью, а потому являются прекрасными конструкцион-
конструкционными материалами в современной технике.
В определенном смысле фазы внедрения представляют собой новый этап в
химической организации вещества. Их образование контролируется не только
соотношением размеров атомов, но и фактором более высокого порядка — элект-
электронной концентрацией. Это приводит к еще большему качественному своеобра-
своеобразию продуктов взаимодействия по сравнению с исходными компонентами.
Существование большой группы интерметаллических соединений разнообраз-
разнообразного качественного и количественного состава, но сходных по физико-химической
природе, обусловлено влиянием фактора электронной концентрации. Все эти
фазы обладают металлическим характером и кристаллизуются в структурах трех
типов: /?-латуни (ОЦК), 7~латУни (сложная кубическая структура с 52 атомами в
элементарной ячейке) и е-латуни (ГПУ). Тип кристаллической структуры опре-
определяется не свойствами взаимодействующих компонентов, а так называемой фор-
формальной электронной концентрацией (ФЭК), т.е. отношением общего числа ва-
валентных электронов (соответствующих номеру группы) к числу взаимодейству-
взаимодействующих атомов в формульной единице. Эти фазы называются электронными соеди-
соединениями Юм-Розери. Обычно электронные соединения образуются в системах,
содержащих, с одной стороны, элементы IB- и VIIIB-групп, а с другой — металлы
IIB-, ША-и IVA-групп. Эти соединения не подчиняются классическим правилам
валентности, и их состав определяется лишь формальной электронной концентра-
концентрацией. Трем видам электронных соединений соответствует определенная формаль-
формальная электронная концентрация. Так, для ОЦК-структуры /?-латуни ФЭК =
= 21/14 = 3/2 (числитель — общее число валентных электронов, знаменатель —
число атомов в формульной единице соединения). Сложная структура 7~латуни
определяется величиной ФЭК, равной 21/13, а структуре е-латуни (ГПУ) отвеча-
отвечает ФЭК = 21/12 = 7/4. Примеры типичных электронных соединений в различ-
различных системах приведены в табл. 20. Обращает на себя внимание существенно
различный состав соединений Юм-Розери, кристаллизующихся в одинаковом
*В данном случае рассматривается внедрение легких атомов в искаженную решетку, не
отвечающую исходному металлу. Если же атомы неметалла внедряются в исходную метал-
металлическую кристаллическую решетку, то образуется просто твердый раствор внедрения.
* *Существуют и гидриды такого же типа, например PdH.
219
Таблица 20. Характеристика некоторых электронных соединений Юм-Роэери
Соединение
CuZn
AgMg
Cu3Al
InNi*
Cu5Zn8
Cu9Al4
Cu31Sn8
Au5Zn8
C03Z1121 *
AgCd3
CuZn3
Ag5Al3
CuaSn
Число валентных
электронов
1 + 2
1 + 2
3+3
3 + 0
5 + 16
9+12
31 + 32
5 + 6
0 + 42
1 + 16
1 + 6
5 + 9
3 + 4
Число атомов
в соединении
2
2
4
2
13
13
39
13
26
4
4
8
4
ФЭК, элект-
рон/атом
3/2
3/2
3/2
3/2
21/13
21/13
21/13
21/13
21/13
7/4
7/4
7/4
7/4
Тип структуры
0-Фааа.
м
м
It
7-фаза
11
и
и
II
С-Фаза
м
1)
II
t;c
1100
Массовое содержание, %
10 20 30 W 50 60 70 80 90
* В соединениях, содержащих элементы VIIIB-группы, число валентных электронов у
последних принимается равным нулю.
структурном типе. Так, соединение CuZn содержит 50 ат. % цинка, a CuaAl —
только 25 ат. % А1. Но именно при этом составе и достигается величина ФЭК,
необходимая для возникновения структуры /?-фазы.
Смысл определяющего влияния ФЭК на состав и структуру электронных
соединений можно понять с привлечением представлений зонной теории. Каждой
кристаллической структуре отвечает характерный для нее зонный энергетиче-
энергетический спектр электронов. Валентная зона заполняется электронами не беспредель-
беспредельно и вмещает только определенное их число.
По заполнении зоны наступает такой момент,
когда энергия электронов так резко повышает-
повышается, что данная структура оказывается неста-
нестабильной и происходит изменение кристалли-
кристаллического строения сплава. Возникающая при
этом новая структура будет соответствовать
большей электронной концентрации. В качестве
примера рассмотрим систему медь — цинк (рис.
114). Чистая медь имеет ГЦК-структуру (куби-
(кубическая плотнейшая упаковка). При плавлении
меди с возрастающим количеством цинка (до
37%) атомы цинка замещают часть атомов меди
статистически без изменения типа кристалли-
кристаллической структуры матрицы. Образуется а-твер-
дый раствор, которому отвечает вполне опреде-
определенная область электронной концентрации. Эта
ю го so w 50 so w so so ко
cu zn
Атомное содержание, %
Рис. 114. Диаграмма состояния сис-
системы медь - цинк
фаза стабильна в интервале электронных концентраций от 1 до 1,4 элект-
электрон/атом. Превышение предельной электронной концентрации приводит к пере-
перестройке кристаллической структуры. Вместо «-фазы появляется /3-фаза, которой
отвечает объемно центрированная кубическая структура с электронной концент-
концентрацией 1,5 электрон/атом. В область гомогенности данной фазы как раз и укла-
укладывается состав, отвечающий электронному соединению CuZn. /2-Фаза сменяется
7-фазой, стабильность которой сохраняется в пределах содержания 58 — 67 ат. %
Zn. Это происходит, если электронная концентрация превышает 1,5 элект-
электрон/атом. Идеальный состав 7"~Фазы' отвечающий электронному соединению, бу-
будет CusZns- Он соответствует содержанию 61,5 ат. % Zn и укладывается в область
гомогенности 7~Фазы- 7~*^>аза устойчива в довольно широкой области вблизи
ФЭК = 21/13 = 1,615 электрон/атом. Дальнейшее увеличение содержания цинка
в сплавах G8 — 86 ат. %) приводит к возникновению е-фазы с ФЭК. = 7/4 =
= 1,75 электрон/атом, вблизи области существования которой укладывается
состав, отвечающий электронному соединению CuZn G5 ат. % Zn).
Из всего этого следует, что основой устойчивости рассмотренных фаз служат
значения электронной концентрации, которые определяют собой предельный
состав, при котором данная фаза стабильна. Большинство электронных соедине-
соединений Юм-Розери представляют соединения переменного состава, относящиеся к
классу бертоллидов. На диаграммах состояния им отвечают обычно широкие
области гомогенности. На кривых зависимости свойств от состава в области суще-
существования соединений сингулярные точки отсутствуют.
5. Металлохимические свойства и диаграммы состояния. По мере усложнения
химической организации вещества в ряду соединения Курнакова — фазы Лаве-
са — фазы внедрения — электронные соединения Юм-Розери происходит нарас-
нарастание качественного отличия промежуточных фаз от компонентов, их образую-
образующих. При этом происходит ослабление влияния объемно-геометрических и усили-
усиливается роль физико-химических и химических факторов. Так, фактор электрон-
электронной концентрации, проявляющийся уже при образовании фаз внедрения и наибо-
наиболее ярко выступающий при возникновении электронных соединений, является
преимущественно химическим, поскольку его действие связано с перераспределе-
Неблагопрнятиое
I сочетание I Размерный фактор I
. цгак гирип ¦
1 • г-г—q__*L_
химической I химической
активности I активности
Рис. 115. Влияние металлохимических факторов на увеличение химическо-
химического сродства:
рсл - расслоение; эвт - эвтектика; огр - ограниченные твердые растворы;
нтр - непрерывные твердые растворы; ск - соединения Курнакова; фл - фа-
фазы Левеса; фв - фазы внедрения; эс - электронные соединения; вс - валент-
валентные соединения; ФЭК - формальная электронная концентрация; ЭО - элек-
электроотрицательность
221
CU AS
Содержание, %
Си Se
Содержанием/о
Си Вг
Содержатели
Рис. 116. Изменение диаграмм состояния с участием меди при уве-
увеличении электроотрицательности второго компонента
нием валентных электронов. Фактором более высокого порядка выступает
разность электроотрицательностей, в наиболее сильной форме отражающая
химический аспект взаимодействия.
Все рассмотренные типы взаимодействия (примитивные и сложные) по мере
нарастания химического сродства представлены в виде ряда (рис. 115). Своеоб-
Своеобразной границей в этом ряду служат непрерывные твердые растворы, которые
разделяют примитивные и сложные типы взаимодействия. С одной стороны,
образование твердых растворов можно рассматривать как наиболее общий случай
физико-химического взаимодействия, а с другой стороны, это начальный этап
химической организации вещества в твердом состоянии.
Глубокую внутреннюю генетическую взаимосвязь различных типов взаимодей-
взаимодействия в бинарных системах в зависимости от соотношения металлохимических
свойств компонентов можно выявить, проследив развитие геометрических элемен-
элементов диаграмм состояния. В качестве примера рассмотрим изменения диаграмм
состояния в системах с участием меди: Си — Аи, Си — Zn, Си — Ga, Си — Ge,
Си — As, Си — Se, Си — Вг (рис. 116). Система Си — Аи (см. рис. 111), компо-
компоненты которой являются элементами-аналогами с близкими металлохимическими
характеристиками, представляет собой непрерывный ряд твердых растворов. Од-
Однако уже здесь проявляется склонность'к химическому взаимодействию, что под-
подтверждается возникновением в твердом состоянии упорядоченных твердых раст-
растворов — соединений Курнакова Cu3Au, CuAu. При сравнении системы Си — Аи с
системой Си — Zn (см. рис. 114) можно видеть, что в последней более существен-
существенную роль играет фактор электронной концентрации. Это приводит, с одной сто-
стороны, к ограничению поля твердых растворов со стороны меди (вследствие ос-
ослабления влияния размерного фактора), а с другой стороны, к возникновению
типичных электронных соединений Юм-Розери: /?-фаза (CuZn, ФЭК = 21/14 =
= 3/2), 7-фаза (Cu5Zn8, ФЭК = 21/13), е-фаза (CuZn3, ФЭК = 21/12 = 7/4).
В системе Си — Ga фактор электронной концентрации также играет домини-
доминирующую роль. Хотя базовые составы промежуточных фаз и отличаются от соот-
соответствующих аналогов в системе Си — Zn (Д-фаза СизСа, ?-фаза CugGa,}, е-фаза
Cu5Ga3), однако характерные величины ФЭК, определяющие границы устойчиво-
устойчивости фаз, остаются теми же (соответственно 6/4, 21/13 и 14/8).
Переходя к системе Си — Ge, можно отметить, что здесь становится заметным
вклад разности электроотрицательностей. Это приводит к возникновению фазы
Cu3Ge, плавящейся с открытым максимумом, который, однако, выражен очень
слабо. Кроме того, в этой системе отмечена фаза Cu5Ge2, существующая в интер-
вапе 612 — 700 °С и разлагающаяся по пёритектической схеме.
В системе Си — As происходит дальнейшее нарастание влияния разности
электроотрицательностей. Температура плавления соединения Cu3As (830 °С)
выше, чем у Cu3Ge G49 °С), температурный интервал устойчивости фазы Cu5As2
C00 — 710 °С) также выше, чем у Cu5Ge2. Все это свидетельствует об усилении
химического взаимодействия в последней системе, хотя общий вид диаграмм
состояния Си — Ge и Си — As аналогичен. Все промежуточные фазы в рассмот-
рассмотренных системах обладают металлическим характером.
И наконец, в системе Си — Se и особенно в системе Си — Вг фактор элект-
роотрицательности играет доминирующую роль, предопределяя возникновение
прочных ионно-ковалентных соединений Cu2Se, CuSe, CuBr и CuBr2, причем
последние две фазы представляют собой типичные соли. Хотя в системе Си — Se
и наблюдается область расслоения в расплаве, однако сильное взаимное сродство
компонентов обеспечивает возможность их химического взаимодействия.
Генетическую связь между различными типами взаимодействия и формами
химической организации вещества можно проследить также на отдельной диаг-
диаграмме состояния с несколькими промежуточными фазами. В качестве примера
рассмотрим диаграмму состояния Ti — О (рис. 117). В этой системе отмечается
образование как твердых растворов внедрения на основе о- и /^-титана, так и
соединений Курнакова Ti6O [(Ti3JO] и Ti3O,
формирующихся в результате упорядочения
внутри обширной области твердых раство-
растворов кислорода в cf-Ti. Кроме того, в системе
отмечено существование соединения Ti2O,
которое можно представить как фазу внед-
внедрения с наполовину заполненными октаэд-
рическими пустотами в искаженной решетке
o-Ti (TiOo,5). Данные фазы, как и следовало
ожидать, обладают металлическими свойст-
свойствами. При дальнейшем увеличении относи-
относительного содержания кислород а. формируют-
формируются фазы с полупроводниковыми и диэлект-
диэлектрическими свойствами. Сначала фиксирует-
фиксируется бертоллид в широком смысле "TiO"
(TiOo,58-i,2o)> а затем полуторный оксид
Ti2O3 с узкой областью гомогенности
(TiO1L9-i,5i)- Наконец, образуется ковалент-
шяионный высший оксид титана ТЮ2 даль-
тонидного типа с узкой областью гомогенно-
10 20 30 M SO SO 70 SO SO WO
Л томное содержание, %
Рис. 117. Диаграмма состояния систе-
системы титан - кислород
223
сти и диэлектрическими свойствами, очень инертный и тугоплавкий; он относит-
относится к типичным валентно-химическим соединениям.
Итак, в системе Ti — О постепенно нарастает стремление к химическому взаи-
взаимодействию через ряд промежуточных фаз; от твердого раствора с соединениями
Курнакова через фазу внедрения к химическим соединениям:
Ti«-0
твердый
раствор
Ti6O, Ti3O
соединения
Курнакова
> Ti2O -
фаза
внедре-
внедрения
бертол-
лид
Ti2O3, ТЮ2
дальтониды
Этот переход сопровождается постепенным изменением структуры и нараста-
нарастанием качественных отличий промежуточных фаз от исходных компонентов. По
мере увеличения относительного содержания кислорода возрастает роль фактора
электроотрицательности, в результате чего увеличивается тепловой эффект обра-
образования оксидных фаз от 40 — 80 кДж/моль для твердых растворов и соедине-
соединений Курнакова до 250 — 310 кДж/моль для Т12Оз, TiO2.
Таким образом, взаимосвязь между различными типами промежуточных фаз в
бинарных системах прослеживается не только для различных систем, но и в
пределах одной и той же диаграммы состояния. Развитие металлохимии с ее
специфическими методами исследования открывает возможность детально просле-
проследить эту зависимость, что способствует расширению и углублению фундаменталь-
фундаментальных представлений о характере и природе химического взаимодействия.
lii
ЧАСТЫ1
НЕОРГАНИЧЕСКАЯ ХИМИЯ
РАЗ ДЕЛ 1
ПЕРИОДИЧЕСКИЙ ЗАКОН КАК ОСНОВА
ХИМИЧЕСКОЙ СИСТЕМАТИКИ
ГЛАВАХ. СТРУКТУРА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
Систематическое, целенаправленное и осознанное изучение огромного факти-
фактического материала современной неорганической химии невозможно без руководя-
руководящего принципа, роль которого играют Периодический закон и Периодическая
система элементов как его графическое выражение. Без преувеличения можно
сказать, что уровень квалификации химика определяется тем, насколько он спо-
способен творчески и свободно использовать те общие закономерности в изменении
характера химической связи, химического и кристаллохимического строения,
свойств веществ, которые диктуются явлением периодичности. Физическая
сущность этого явления заключается в особенностях электронного строения ато-
атомов.
На основе современных квантово-механических представлений об электронном
строении атомов можно детально проанализировать структуру Периодической
системы. При этом выявляются не только общие закономерности в изменении
свойств элементов (расположение их по группам и подгруппам), но и более тон-
тонкие детали, позволяющие объяснить вторичную и внутреннюю периодичность,
горизонтальную и диагональную аналогии. Одним из важных представлений,
объясняющих немонотонный характер изменения свойств элементов в пределах
группы, является представление о кайносимметричных орбиталях и кайносиммет-
ричных элементах.
1. Этапы развития Периодического закона. Периодический закон, открытый
Д.И.Менделеевым в 1869 г., представляет собой один из фундаментальных зако-
законов в современном естествознании. Он отражает материальное единство мира и
именно поэтому имеет непреходящее значение не только для химии, но и для
всего естествознания в целом. В нем сконцентрирована сущность химии как науки
О качественных изменениях тел, происходящих под влиянием изменения коли-
количественного состава.
Трудно переоценить роль Периодического закона и в развитии других естест-
естественных наук: физики, геохимии, космохимии и т.д. Значение его не ограничива-
ограничивается только возможностью классификации элементов на единой основе. Он позво-
позволяет предсказать свойства каждого элемента на основании его расположения в
системе. Это распространяется не только на физические свойства простых ве-
веществ, но и на весь комплекс их химических свойств: способность к взаимодейст^
« „ ¦ „„* 225
I.
вию с другими элементами, строение, состав и свойства образующихся бинарных
и более сложных соединений, кислотно-основные и окислительно-восстановитель-
окислительно-восстановительные свойства элементов и т.д. Известно, что Д.И.Менделеев, пользуясь Периоди-
Периодическим законом, предсказал свойства еще не известных в то время элементов. А
сила подлинно научной теории определяется не только и не столько возмож-
возможностью объяснения на ее основе известных фактов, но главным образом предви-
предвидением новых фактов.
Однозначное описание свойств элементов предполагает, что каждый элемент
должен находиться в Периодической системе на строго определенном постоянном
месте. Это называется инвариантностью (неизменностью) положения. Положение
элемента в системе Д.И.Менделеева определяется не только его порядковым
номером, но также номером периода (строки) и группы (столбца), в которых он
находится. Однако даже в современной наиболее распространенной форме Перио-
Периодической системы принцип инвариантности положения элемента не всегда соблю-
соблюдается. В качестве примера можно привести неопределенное положение в ней во-
водорода. Очевидно, необходим общий критерий, позволяющий однозначно опре-
определять положение элемента. Д.И.Менделеев в качестве такого критерия выбрал
химические свойства элементов, которые он считал более фундаментальной ха-
характеристикой, чем значения атомных масс, несмотря на то что именно последние
были им положены в основу классификации элементов. Поэтому он допускал пе-
перестановки элементов (Аг — К, Те — I и т.д.), с тем чтобы привести в соответст-
соответствие положение элемента в Периодической системе с его химическими свойствами,
отражаемыми групповой аналогией. В дальнейшем разными исследователями бы-
были предложены различные варианты системы (в настоящее время их известно
более четырехсот), в основу которых взяты разные, нередко частные критерии.
С развитием электронной теории строения атомов стало ясно, что химические
свойства элементов являются функцией электронной структуры атомов. Отсюда
следует, что в качестве объективного критерия, однозначно определяющего поло-
положение элемента в Периодической системе, целесообразно выбрать именно элект-
электронное строение атома. Поэтому в развитии Периодического закона выделяют
три этапа. На первом этапе в качестве аргумента, определяющего свойства эле-
элементов, была выбрана атомная масса и закон был сформулирован Д.И.Менделе-
Д.И.Менделеевым следующим образом: "Свойства элементов, а также формы и свойства их
соединений находятся в периодической зависимости от их атомного веса". На
втором этапе было выяснено значение атомного номера, который, как оказалось,
определяет заряд ядра атома. Открытие изотопов и изобаров показало, что ис-
истинным аргументом, определяющим природу элемента, является именно заряд
ядра, а не атомная масса. Действительно, атомы с одинаковой атомной массой —
изобары (например, ^Аг, |°К, ^Са) — принадлежат разным элементам, в то вре-
время как атомы с одинаковым зарядом ядра принадлежат одному элементу, не-
несмотря на то что у них могут быть различные атомные массы (например, изотопы
!|О, ^О, Х|О). В связи с этим по-новому стала звучать и формулировка закона,
отражающая периодическую зависимость свойств элементов от атомного номера
(заряда ядра), а не от атомной массы. Это изменение носит принципиальный
характер и свидетельствует о качественно новом уровне понимания природы
элементов.
Тем не менее даже на этом этапе развития Периодического закона оставался
неясным физический смысл периодичности, т.е. констатировался сам факт перио-
периодического изменения свойств элементов, но не было понятно, почему при моно-
монотонном возрастании атомного номера свойства элементов меняются не монотонно,
а именно периодически. И только на третьем этапе, с развитием квантово-механи-
ческой теории электронного строения атома, стало возможным вскрыть физиче-
физический смысл Периодического закона. Выяснилось, что сущность периодичности
заключается в существовании предельной емкости электронных слоев и в перио-
периодическом воспроизведении сходных валентных электронных конфигураций на все
более высоком энергетическом уровне в результате наложения квантово-механиче-
ского принципа Паули на классический принцип наименьшей энергии в атомной
системе.
В дальнейшем, по мере углубления теоретических представлений о свойствах
атомов (эффекты проникновения и экранирования, р-, d-, /-контракция, учение о
кайносимметричных и некайносимметричных орбиталях и др.), появилась воз-
возможность обосновать наряду с групповой, типовой и другими вертикальными
аналогиями вторичную, внутреннюю и горизонтальную аналогии. Кроме того,
были объяснены специфические особенности химии первых типических элемен-
элементов, а также первого ряда элементов вставных декад. Таким образом, по мере
углубления представлений о строении вещества открываются новые возможности
в понимании Периодического закона, который находится в постоянном развитии.
2. Групповая и типовая аналогии. Периодический закон является фундамен-
фундаментальным законом природы, отражающим единство количественной (заряд ядра,
число электронов, атомная масса) и качественной (распредачение электронов,
совокупность свойств) характеристик элементов.
Совокупность этих характеристик должна обеспечивать инвариантность поло-
положения элемента в таблице. В свете современных представлений о строении атома
принадлежность элемента к конкретному периоду определяется числом электрон-
электронных слоев атома в нормальном, невозбужденном состоянии. Номер периода отве-
отвечает номеру внешнего слоя, который не завершен и заполняется электронами. А
принадлежность элемента к той или иной группе определяется общим числом
валентных электронов, т.е. электронов, находящихся на внешней и недостроен-
недостроенных внутренних оболочках*. Например, хром [Сг]24 — [Ar]183rf54s1 и сера [S]16 —
[Ne]103s23p4 являются элементами одной и той же VI группы, поскольку оба атома
имеют по 6 валентных электронов. Отметим, что деление на периоды и группы
введено Д.И.Мендачеевым, который определял принадлежность элемента к кон-
конкретной группе, ориентируясь на химические свойства, в частности на форму и
характер высших оксидов и гидроксидов. Действительно, такие непохожие друг
на друга металлический хром и неметаллическая сера в высшей степени окисле-
окисления, соответствующей номеру группы, образуют оксиды одинакового состава ЭОз
(СгО3 и SO3), которые к тому же обладают сходными (кислотными) свойствами.
Им отвечают гидроксиды, имеющие ярко выраженный кислотный характер, —
хромовая Н2СЮ4 и серная H2SO4 кислоты. Таким образом, в группы Периоди-
Периодической системы объединяются элементы с одинаковым общим числом электронов
на достраивающихся оболочках независимо от их типа. Подобное объединение
позволяет выделить наиболее общий вид аналогии, который называется группо-
*Исключениями являются гелий и вертикальные триады элементов VHIB-группы
Со — Rh - Ir, Ni — Pd - Pt.
a* 2517
j ¦
вой аналогией и просматривается только в высшей степени окисления, отвеча-
отвечающей номеру группы. Именно по этому признаку главные и побочные подгруппы
(группы А и группы В) объединяются в одну группу. Так, в III группу объедине-
объединены элементы В, Al, Ga, In, Tl (ns2npl) и элементы подгруппы скандия — Sc, Y,
La[ns2(n — 1)<Р], имеющие одинаковое число валентных электронов, равное трем.
Такое же положение характерно и для других групп системы.
Групповая аналогия далеко не отражает всех особенностей элементов, входя-
входящих в данную группу, поскольку формируется она по наиболее общему призна-
признаку — числу валентных электронов — без учета типа валентных орбиталей. Эта
аналогия пропадает для элементов в низших степенях окисления и тем более в
свободном состоянии. Однако в пределах каждой группы можно выделить эле-
элементы, которые обладают более глубоким сходством между собой. Это сходство
проявляется не только в высшей, но и во всех промежуточных степенях окисле-
окисления, и обусловлено не только одинаковым числом валентных электронов, но и
одинаковым типом орбиталей, на которых эти электроны расположены. По этому
признаку и выделяются подгруппы элементов в пределах одной группы. Элемен-
Элементы, принадлежащие к одной подгруппе, обладают более близким сходством в
свойствах, в основе которого лежит одинаковый тип валентных орбиталей, запол-
заполняющихся электронами. Эта более глубокая аналогия называется типовой анало-
аналогией. Таким образом, элементы, принадлежащие одной подгруппе, являются тип-
аналогами. Так, в рассмотренном выше примере III группы бор, алюминий и
подгруппа галлия, образующие главную подгруппу (или ША-группу), являются
тип-аналогами, поскольку для всех этих элементов характерен одинаковый тип
валентных орбиталей (ns2npl). Элементы подгруппы скандия, образующие побоч-
побочную подгруппу III группы (или ШВ-группу), также являются между собой тип-
аналогами (валентная электронная конфигурация (п — 1)(#пя2).
Типовая аналогия (аналогия между элементами в подгруппах), несомненно,
характеризует более глубокое сходство между элементами по сравнению с группо-
групповой, что находит свое отражение в закономерностях изменения свойств как самих
элементов, так и их соединений. Тем не менее и этот вид аналогии не полностью
охватывает все особенности физико-химической природы отдельных элементов и
их взаимосвязь с соседями по группе.
3. Электронная аналогия. Кайносимметрия. Элементы малых периодов B-го и
3-го) с точки зрения электронного строения характеризуются качественным
своеобразием по сравнению со всеми остальными элементами. Это своеобразие
заключается в том, что у элементов 2-го и 3-го периодов под валентной электрон-
электронной оболочкой находится атомный остов предыдущего инертного газа. Что каса-
касается «-элементов, то аналогичная ситуация наблюдается и для элементов боль-
больших периодов. Например, электронные конфигурации атомов Na и Cs соответст-
соответственно [Ne]11^1 и [Xe]546s1, а электронные конфигурации ионов Na+ и Cs* совпада-
совпадают с электронными структурами неона и ксенона, т.е. аналогичны. Для р-элемен-
тов наблюдается другая картина. Если в малых периодах под валентной оболоч-
оболочкой, как и у s-элементов, располагается остов благородного газа, то в атомах р-
элементов больших периодов валентным nsnp-оболочкам предшествуют заполнен-
заполненные (п — 1)</-орбитали сверх оболочки предыдущего благородного газа. Так,
электронные конфигурации серы и селена соответственно [Ne]103s23p4 и
[Ar]183tf°4s24p4, а конфигурации "ионов" S6+ и Se6+, формально отвечающих выс-
высшим степеням окисления элементов, таковы: [S6+] — [Ne]10; [Se6+] — [Аг]183<#°.
Таким образом, трудно ожидать полной аналогии в свойствах серы и селена в
высших степенях окисления. В то же время в промежуточных положительных, а
также в отрицательных степенях окисления у серы и селена наблюдается полная
электронная аналогия, т.е. подобие валентных электронных конфигураций. С.
другой стороны, в промежуточных степенях окисления отсутствует какая-либо
аналогия между элементом VIA-группы — серой — и элементами VIB-группы, на-
например хромом. Однако эта аналогия появляется именно в высшей степени окис-
окисления, когда у атомов этих элементов "обнажается" электронная оболочка преды-
предыдущего благородного газа: [S6+] — [Ne]10 и [Сг6+] — [Аг]18.
Подобные закономерности в электронном строении наблюдаются и в осталь-
остальных группах. Таким образом, элементы малых периодов проявляют электронную
аналогию как с элементами главных подгрупп, так и с представителями побоч-
побочных подгрупп (в различных степенях окисления) и, следовательно, определяют
облик группы в целом. Поэтому элементы малых периодов называют типически-
типическими. Этот термин был введен Д.И.Менделеевым.
Таким образом, можно ввести представление о полной и неполной электронной
аналогии. Полными электронными аналогами называются элементы, которые
имеют сходное электронное строение во всех степенях окисления, чем и объясня-
объясняется близкое подобие их химических свойств. Например, в рассматриваемой VI
группе Периодической системы полными электронными аналогами являются
кислород и сера [О]8 — [He]22s22/>4; [S]16 — [Ne]103s23p4, селен, теллур и полоний
[Se]34 _ [Ai]^3^4s4p*; [Те]52 - [Kr]364<P°5s25p4; [Ро]84 - [Xe]544/45<P°6s26p, a
также хром, молибден и вольфрам [Сг]24 — [Arp^dMs1; [Mo]42 — [Kr]364rf55s!;
[W]74 — [Xe]544/45d46s2. У полония и вольфрама в отличие от остальных элемен-
элементов присутствует внутренняя завершенная 4/оболочка, наличие которой проявля-
проявляется в лантаноидном сжатии. Поскольку 4/-оболочка располагается глубоко, она
мало влияет на свойства и не нарушает характер электронной аналогии. Атомы
типических элементов — кислорода и серы -— по электронному строению отлича-
отличаются как от атомов элементов подгруппы селена (в высшей степени окисления),
так и от атомов элементов подгруппы хрома (во всех степенях окисления, кроме
высшей). Это значит, что кислород, и сера по отношению к остальным элементам
VI группы являются неполными электронными аналогами. В то же время анало-
аналогия в электронном строении между типическими элементами и подгруппой селена
более близкая. Они являются, как отмечено выше, не только групповыми, но и
типовыми аналогами. Характер электронной аналогии в VI группе можно проил-
проиллюстрировать следующей схемой:
О
С г 4Se
I I
Mo Те
I I
W Ро
Здесь сплошной линией показана полная электронная аналогия, штриховой
линией — неполная аналогия, а точками — аналогия только в высшей степени
окисления (если она существует).
Подобным же образом можно представить характер электронной аналогии во
всех группах Периодической системы *. Отметим некоторые особенности характе-
характера электронной аналогии, вытекающие из приведенной схемы. «-Элементы (IA—и
ПА-группы) являются полными электронными аналогами и в то же время прояв-
проявляют групповую и типовую аналогию. Это обусловлено аналогичным строением
электронных орбиталей (ns1'2) у всех представителей одной группы. Заполненные
предвнешние rf-оболочки у этих элементов отсутствуют. В то же время типиче-
типические элементы обеих групп (Li и Na, Be и Mg) являются неполными электронны-
электронными аналогами с элементами побочных подгрупп (Си, Ag, Аи и Zn, Cd, Hg). Эти
последние представляют собой семейства типовых аналогов и характеризуются
полной электронной аналогией между собой. У элементов III — VII групп сквоз-
сквозная полная аналогия отсутствует. В каждой из этих групп выделяются три се-
семейства полных электронных аналогов: 1 — типические элементы; 2 — остальные
элементы главной подгруппы; 3 — элементы побочной подгруппы. Между собой
эти семейства связаны через типические элементы неполной электронной анало-
аналогией, причем аналогия между элементами главных подгрупп проявляется более
ярко, поскольку они являются типовыми аналогами. Отличие характера взаимо-
взаимосвязи между электронными аналогами в этих группах от I и II объясняется вкли-
вклиниванием между ПА- и ША-группами вставных декад переходных элементов.
Сквозная полная аналогия не просматривается и среди элементов VIIIA-rpyn-
пы, хотя все они относятся к благородным газам. Однако и здесь можно выде-
выделить типические элементы (неон и аргон) и элементы подгруппы криптона, у
которых в отличие от типических присутствует заполненная предвнешняя (и —
— 1)<?-оболочка. Элементы подгруппы криптона характеризуются заметной хими-
химической активностью и, как известно, в своих высших оксидах и фторидах могут
проявлять степень окисления, отвечающую номеру группы (ЭО4, ЭРв). Следова-
Следовательно, и в этой группе, как и у всех р-элементов, существует неполная электрон-
электронная аналогия между типическими элементами и остальными ^элементами VIII
группы. Следует отметить, что здесь типические элементы вообще не определяют
облик группы в целом в силу своей химической инертности.
Сравнивая между собой типические элементы 2-го и 3-го периодов, отметим,
что с точки зрения электронного строения они также различны. Дело в том, что
у элементов 3-го периода существует вакантная Зсйэболочка, которая при опреде-
определенных условиях может принимать участие в химическом взаимодействии. Это
реализуется вследствие промотирования валентных электронов с 3s- и Зр-оболо-
чек на близлежащую по энергии Згйэболочку при одном и том же значении глав-
главного квантового числа. А у элементов 2-го периода возможность промотирования
на следующую вакантную Зз-оболочку исключается по энергетическим соображе-
соображениям: затраты энергии на промотирование не могут быть компенсированы выиг-
выигрышем энергии за счет образования дополнительных связей. Именно по этой
причине не реализуется, например, пятивалентное состояние азота, хотя для
фосфора оно является обычным. Так, галогениды азота имеют формулу NT3, a
для фосфора известны и пентагалогениды, например PCU- В самом деле, струк-
структуру валентных орбиталей азота (п = 2) и фосфора (и = 3) можно представить
следующим образом:
*3а исключением VIII группы, где побочная подгруппа состоит из элементов семейства
железа и платиновых металлов.
пш
я-З
II
1 1
Следовательно, с точки зрения спиновой теории валентности ковалентность
азота и фосфора равна трем (три неспаренных электрона), а с учетом донорно-
акцепторного взаимодействия за счет неподеленной s-пары ковалентность азота и
фосфора достигает четырех, что реализуется, например, в катионах аммония и
фосфония:
Н
Н—;
Н
н
I
н—р—>н
I
н
+
Но в отличие от азота у фосфора возможно и пятиковалентное состояние:
р [Н
S
*
промотирование
Этим и объясняется существование пентагаяогенидов фосфора.
С другой стороны, своеобразие первого ряда типических элементов заключает-
заключается в том, что у них jj-орбитали появляются впервые. Функция радиального рас-
распределения электронной плотности для этих орбиталей имеет один максимум.
Орбитали, которые появляются впервые, называются к айн о симметричными (от
греч. "кайнос" — новый, т.е. новый тип симметрии орбиталей). К таким орбита-
лям относятся Is, 2p, 3d, 4/и т.д. Для всех таких орбиталей и характерно нали-
наличие единственного максимума на кривой радиального распределения электронной
плотности.
В отличие от перечисленных все остальные орбитали той же симметрии имеют
дополнительные максимумы на кривых радиального распределения электронной
плотности. Таким образом, для кайносимметричных орбиталей характерно отсут-
отсутствие внутренних заполненных орбиталей той же симметрии. Это при-
приводит к усилению связи кайносимметричных электронов с ядром за счет сущест-
существенного ослабления эффекта экранирования, уменьшению орбитальных атомных
радиусов, повышению потенциалов ионизации, а следовательно, к ослаблению
металлических свойств кайносимметричных элементов по сравнению с некайно-
симметричными.
В качестве иллюстрации можно привести пары элементов, различие в свойст-
свойствах которых общеизвестно и может быть объяснено с точки зрения представления
231
о кайносимметричных орбиталях. В самом деле, водород (Is1) значительно менее
"металличен", чем литий Bs1), а бор Bs22pl) и углерод Bs22p2) менее "металлич-
ны", чем алюминий Cs2, Зр1) и кремний Cs23p2). Эти особенности кайносиммет-
кайносимметричных элементов обусловлены меньшим экранированием валентных электронов.
Внутренние максимумы радиального распределения электронной плотности для
некайносимметричных валентных орбиталей совпадают с аналогичными максиму-
максимумами заполненных внутренних орбиталей той же симметрии. Вследствие этого
некайносимметричные электроны испытывают значительно больший эффект
экранирования, из-за чего их связь с ядром существенно слабее по сравнению с
кайносимметричными электронами.
Таким образом, представление о кайносимметрии позволяет понять, например,
своеобразие свойств элементов 1-го периода (Н, Не) и особые свойства р-элемен-
тов 2-го периода (первый ряд типических элементов) по сравнению с другими
типическими элементами C-й период).
Действительно, водород и гелий, обладающие кайносимметричными ls-орби-
талями, характеризуются весьма высоким потенциалом ионизации A3,6 и 24,6 В
соответственно). Бор (первый типический элемент III группы), у которого налицо
один кайносимметричный 2р-электрон, имеет первый ионизационный потенциал
8,3 В. У второго типического элемента той же группы — алюминия — 1\ = 5,9 В,
т.е. намного меньше, чем у бора, из-за некайносимметричности З/мэрбитали алю-
алюминия.
Используя представление о кайносимметрии, можно выделить более тонкий
вид электронной аналогии, так называемую слоевую аналогию (в дополнение
к групповой и типовой аналогии). Слоевыми аналогами называют элементы,
которые являются типовыми аналогами, но не имеют внешних или предвнешних
кайносимметричных электронов. К таким аналогам относятся, например, в IA-
группе К, Rb, Cs и Fr, a Li и Na не являются слоевыми аналогами с остальны-
остальными щелочными металлами, поскольку у Li присутствует внешняя кайно-
симметричная 2р-оболочка (вакантная), а у Na кайносимметричная заполненная
2р-оболочка является предвнешней. Во НА-группе слоевыми аналогами являются
щелочно-земельные металлы (подгруппа кальция). С точки зрения электронного
строения, слоевые аналоги являются между собой полными электронными анало-
аналогами. Поэтому рассматривать химические свойства элементов группы мы будем в
такой последовательности: первый * типический элемент, второй типический эле-
элемент, остальные элементы главной подгруппы, элементы побочной подгруппы.
Например, в III группе отдельно рассматриваются бор, алюминий, подгруппа
галлия, подгруппа скандия; в V группе — азот, фосфор, подгруппа мышьяка,
подгруппа ванадия и т.д.
4. Переходные металлы. Контракционная аналогия. Для атомов переходных
элементов отмечаются две тенденции, определяющие в конечном итоге их хими-
химические свойства. С одной стороны, заполняющиеся внутренние d- или /орбитали
по мере увеличения в них числа электронов способствуют экранированию внеш-
внешних ns-электронов, в силу чего их связь с ядром должна ослабляться. Но, с дру-
другой стороны, рост числа электронов на внутренних уровнях приводит к уменьше-
уменьшению атомного радиуса. Наиболее ярко это явление выражено у лантаноидов и
получило, как известно, название лантаноидной контракции. Но оно характерно
и для rf-элементов (d-сжатие). В результате контракции связь внешних электро-
электронов с ядром должна усиливаться. Наложение этих двух противоположных тенден-
232 . .% " >¦ ¦ ' . ,-
ций приводит к тому, что хотя d- и /-элементы обладают металлическими свойст-
свойствами, эти свойства выражены у них менее ярко, чем у s- и р-металлов.
Отметим еще одно важное обстоятельство, существенно влияющее на зако-
закономерности изменения свойств переходных металлов. Дело в том, что у элементов
ряда Sc — Zn (вставная декада 4-го периода) заполняющаяся 3</-оболочка
появляется впервые и является кайносимметричной. Вследствие этого наблю-
наблюдается более прочная связь 3i-электронов с ядром, чем у \d- и бсйэлементов. Это
наиболее наглядно иллюстрируется значениями 3-го потенциала ионизации,
отвечающего отрыву первого ^-электрона. В самом деле, сравнивая эти величины
для элементов начала декад (Sc — Y — La), можно видеть, что хотя сверху вниз
потенциалы ионизации и уменьшаются, это уменьшение немонотонно: при пе-
переходе от Sc к Y 3-й потенциал ионизации уменьшается на 4 В, а от Y к La —
всего на 1 В. В дальнейшем в рядах (/-металлов эта закономерность так четко не
прослеживается, потому что на нее накладывается лантаноидное сжатие. По-
Подобное же положение характерно и для 4/^элементов по сравнению с остальными
/^элементами, поскольку 4/оболочка также является кайносимметричной.
Таким образом, первый ряд rf-элементов и первый ряд ^элементов также об-
обладают своеобразием по сравнению со своими более тяжелыми аналогами, по-
подобно элементам 2-го периода по сравнению с типическими р-элементами 3-го
периода.
Совместное влияние кайносимметрии 3 (/-оболочки и лантаноидной контракции
для (/-элементов 6-го периода (Hf — Hg) приводит к существованию более тонкого
вида химической аналогии, чем рассмотренные ранее групповая, типовая и сло-
слоевая. Этот вид аналогии целесообразно назвать конг/гращионной аналогией. Сущ-
Сущность его состоит в том, что пары Zr — Hf, Nb — Та, Mo — W и т.д. обладают
особенно близкими свойствами, а их более легкие аналоги — Ti, V, Сг и др. —
отличаются от них. Эта закономерность хорошо иллюстрируется значениями
металлических атомных радиусов, которые очень близки для элементов 5- и 6-го
периодов. Именно контракционной аналогией объясняется тот факт, что элемен-
элементы Zt. — Hf, Nb — Та часто называют элементами-близнецами.
5. Орбитальные радиусы. Вторичная и внутренняя периодичность. При рас-
рассмотрении характера изменения атомных орбитальных радиусов следует отметить
ряд важных принципиальных особенностей (рис. 118).
1. При переходе от nsl- к п«2-элементам (Н — Не, Li — Be, Na — Mg и т.д.)
наблюдается резкое уменьшение радиуса, что обусловлено формированием полно-
полностью завершенной п«2-оболочки. При этом изменение радиуса (угол наклона
кривой зависимости горб от Z) сохраняется практически постоянным во всех
периодах. Однако величина орбитальных радиусов для s-элементов различных
периодов меняется нелинейно. Радиусы 1 s-элементов — водорода и гелия — много
меньше, чем у всех остальных s-элементов, что объясняется кайносимметрич-
ностью ls-орбиталей и, как следствие этого, более сильным взаимодействием этих
электронов с ядром. Радиусы типических элементов (Li — Na, Be, — Mg) увеличи-
увеличиваются более плавно при переходе от 2-го к 3-му периоду. Радиусы s-элементов
больших периодов заметно больше, чем у соответствующих им типических, одна-
однако их изменение в рядах аналогов К — Ш> — Cs, Са — Sr — Ва также довольно
плавное. Это хорошо иллюстрирует отмеченную ранее слоевую аналогию между
этими элементами и неполную электронную аналогию между ними и типически-
типическими элементами. Наконец, отметим, что орбитальные радиусы последних s-элемен-*
1
5 10
2
15
3
W 15 30 35
h
W tf 50
5
55 60 65 70 75 80 85
6
90 "95 100 Z
7 периоды
Рис. 118. Атомные орбитальные радиусы элементов
тов — франция и радия — несколько меньше, чем у предыдущих (Cs, Ba), что
обусловлено проявлением контракционной аналогии.
2. Орбитальные радиусы р-элементов в пределах каждого периода также зако-
закономерно и монотонно уменьшаются, однако это уменьшение более плавное, чем у
s-элементов. Если рассматривать изменение радиусов р-элементов в каждой груп-
группе с ростом числа электронных слоев, то обращает на себя внимание немонотон-
немонотонность этого изменения. Радиусы кайносимметричных 2р-элементов заметно мень-
меньше, чем у их более тяжелых некайносимметричных аналогов. Вследствие этого,
например, во 2-м периоде радиус бора меньше, чем радиус предшествующего
бериллия, а в 3-м периоде орбитальный радиус алюминия оказывается несколько
большим, чем у магния. При переходе от р-элементов 3-го периода к р-элементам
4-го периода в пределах каждой группы наблюдается очень незначительное уве-
увеличение орбитального радиуса (Si — Ge, P — As, S — Se, Cl — Br, Ar — Кг), а для
элементов III группы — даже его уменьшение от А1 к Ga, что объясняется rf-сжа-
тием. При переходе в пределах одной группы от р-элементов 4-го периода к 5-му,
а затем к 6-му (Ge — Sn — Pb, As — Sb — Bi и т.д.) наблюдается увеличение
орбитальных радиусов. Однако в ША-группе орбитальный радиус изменяется
немонотонно: от Ga к In увеличивается, а затем уменьшается (Т1). Последнее
также обусловлено влиянием лантаноидного сжатия, которое уже не проявляется
в явном виде у следующих за таллием р-элементов 6-го периода.
3. В рядах rf-элементов орбитальный радиус в пределах каждого периода
уменьшается еще более плавно, чем у s-и р-элементов. В пределах каждой В-
группы, как и для s- и р-элементов, наблюдается немонотонное изменение орби-
орбитального радиуса: увеличение при переходе от 3d- к 4<?-эл§ментам и уменьшение
от 4d- к 5с?-элементам. Следствием этого является немонотонное изменение и
других характеристик атомов, определяющих их свойства (потенциал ионизации,
электроотрицательность и т.п.). Это явление получило название вторичной перио-
периодичности. Для (f-элементов такая немонотонность объясняется тем, что впервые
появляющаяся Зй-оболочка является кайносимметричной и обусловливает мень-
ший орбитальный радиус элементов первой вставной декады. Уменьшение орби-
орбитального радиуса 5 rf-элементов обусловлено, как и в предыдущих случаях, ланта-
лантаноидной контракцией.
4. Наконец, в семействе ^элементов изменение орбитальных радиусов еще
более плавное, за исключением некоторых отклонений (Ей — Gd, Np — Pu, Am —
Cm и т.п.), причина которых будет рассмотрена ниже.
Таким образом, анализ рис. 118 позволяет сделать вывод, что наиболее резкое
изменение орбитальных радиусов в периодах наблюдается тогда, когда электро-
электроны попадают на самую внешнюю оболочку. Когда же заполняются предвнешние
оболочки, орбитальные радиусы в пределах каждого периода изменяются более
плавно. Немонотонное, в общем, изменение орбитальных радиусов в пределах
групп объясняется главным образом двумя причинами: кайносимметричностью и
контракцией. Обращение к анализу изменения орбитальных радиусов в периодах
и группах — чисто геометрического фактора, который сам по себе еще не опреде-
определяет химические свойства элементов, — не является самоцелью. Следует иметь в
виду, что атомные радиусы существенно влияют на энергетические характеристи-
характеристики элементов (потенциалы ионизации, сродство к электрону, электроотрицатель-
электроотрицательность), которые непосредственно определяют устойчивость валентных электрон-
электронных оболочек, способность их к деформации и перестройке, т.е. способность
атомов к химическому взаимодействию.
Наряду с упомянутой выше вторичной периодичностью, являющейся следст-
следствием d- и /сжатия и появления кайносимметричных 2р- и З^-орбиталей у первых
представителей тип-аналогов, в Периодической системе элементов существует еще
один вид периодичности в горизонтальных рядах р-, d- и ^элементов. Это так
называемая внутренняя периодичность, сущность которой состоит в немонотон-
немонотонном изменении в этих рядах характеристик атомов, отражающих устойчивость
заполняющихся валентных электронных орбиталей. Наиболее наглядно внутрен-
внутреннюю периодичность можно проиллюстрировать изменением соответствующих
потенциалов ионизации A-го для р-элементов, 3-го для d- и ^элементов) в гори-
горизонтальных рядах. В самом деле, например, в ряду 2|>-элементов (В — Ne) первый
потенциал ионизации, характеризующий отрыв р-электрона от нейтрального
атома, у первых трех элементов (В — С — N) растет монотонно (8,3; 11,3: 14,5 В
соответственно), а у следующего элемента — кислорода — его значение меньше
A3,6 В). В ряду же последующих трех элементов 2-го периода (О — F — Ne)
вновь наблюдается монотонный рост ионизационного потенциала A3,6; 17,4;
21,6 В соответственно). Таким образом, шесть ^элементов отчетливо разделяются
на две тройки.
Аналогичная картина наблюдается и в горизонтальных рядах d- и /-элемен-
/-элементов, но здесь для выявления этой закономерности необходимо рассматривать 3-й
ионизационный потенциал, характеризующий отрыв соответственно d- или ^элек-
^электрона. Действительно, для id- и 4rf-элементов отчетливо просматриваются две
пятерки элементов с монотонным изменением 3-го ионизационного потенциала,
причем при переходе от 5-го к 6-му элементу (Мп — Fe, Тс — Ru) ионизационный
потенциал скачкообразно уменьшается C3,7 — 30,6; 29,5 — 28,5 В). В рядах /-эле-
/-элементов также четко просматривается внутренняя периодичность, а весь ряд из 14
элементов разделяется на две семерки, для которых характерно монотонное изме-
изменение 3-го ионизационного потенциала. Так, в ряду лантаноидов в первую семер-
семерку входят элементы от лантана (La — № 57) до европия (Ей — № 63), 3-й иониза-
235
ционный потенциал у которых меняется соответственно от 19,2 до 24,0 В. При
переходе к следующему элементу — гадолинию (Gd — № 64) — ионизационный
потенциал уменьшается до 20,6 В и вновь монотонно растет, достигая 25,5 В у
иттербия (Yb — № 70). Следует подчеркнуть, что последний из лантаноидов —
лютеций (Lu — № 71) — явно выпадает из этой общей закономерности, в то время
как сам лантан в отмеченную закономерность хорошо вписывается.
Внутренняя периодичность находит естественное объяснение, если учесть, что
энергетически вырожденные р~, d- и /орбитали заполняются в соответствии с
правилом Гунда, причем повышенной стабильностью обладают вакантные, полно-
полностью завершенные (р6, d10, /4), а также наполовину завершенные (р3, &, f) вы-
вырожденные орбитали. В соответствии с этим, например, валентная электронная
конфигурация кислорода Bs22p4) оказывается несколько менее стабильной, чем у
азота Bs22p3). У rf-элементов при переходе, например, от Mn Ds23<P) к Fe Ds23rf6)
наблюдается та же закономерность. Для 4^элементов следует иметь в виду кон-
конкуренцию между 4/- и 5с?-оболочками. У европия Fs24/) 5^-оболочка вакантна, а
4/-оболочка заполнена наполовину. Поэтому 3-й ионизационный потенциал харак-
характеризует энергию отрыва одного из электронов с 4/-уровня, обладающего ста-
стабильной 4/-конфигурацией, а потому относительно высок. У следующего элемен-
элемента — гадолиния Fs24/5</1) при сохранении стабильной /"-конфигурации очеред-
очередной электрон попадает на 5 (/-оболочку и 3-й ионизационный потенциал отвечает
отрыву именно этого электрона, который, естественно, значительно слабее связан
с ядром. Аналогичная ситуация наблюдается при переходе от иттербия Fs24/4) к
лютецию F«24/45</1). С этой точки зрения лютеций целесообразно рассматривать
не как последний элемент в семействе лантаноидов, а как первый элемент в ряду
5rf-элементов, а лантан, который хорошо вписывается в общую закономерность
изменения ионизационного потенциала, должен возглавлять семейство лантано-
лантаноидов.
6. Горизонтальная и диагональная аналогии. Своеобразным следствием внут-
внутренней периодичности является так называемая горизонтальная аналогия, смысл
которой заключается в том, что в ряде случаев соседи в горизонтальных рядах
обладают заметным химическим сходством. Наиболее известна горизонтальная
аналогия в триадах VIIIB-группы (Fe — Со — Ni; Ru — Rh — Pd; Os — Ir — Pt).
Однако не следует думать, что это исключение. Еще Д.И.Менделеев, предсказы-
предсказывая свойства неизвестных элементов, ориентировался не только на вертикальную
(групповую) аналогию, но и на сходство по горизонтали, находя, например, атом-
атомную массу как среднее арифметическое из атомных масс соседей сверху, снизу,
слева и справа. Причиной горизонтальной аналогии можно считать некоторое
отличие первой и второй пятерок rf-элементов, первой и второй семеррк /-элемен-
/-элементов, обусловленное тем, что у перых 5 или 7 элементов заполнение оболочек
происходит в соответствии с правилом Гунда, а у последних правило Гунда не
выполняется.
Отсюда следует сравнительно малое изменение энергетических и размерных
характеристик атомов в середине вторых пятерок и семерок, что и проявляется в
их химических свойствах. Хорошо известно, что элементы второй семерки ланта-
лантаноидов (Tb — Lu) в меньшей мере отличаются друг от друга по свойствам, чем
элементы первой семерки (Ge — Gd), особенно близка эта аналогия у средних
элементов второй семерки (Но — Ег — Тт). Известно также, что в семействе
актинидов наиболее близкими свойствами обладают так называемые кюриды, т.е.
°'1 10 20 30 W 50 60 70 SO Z
P и с. 119. Зависимость металлических радиусов элементов от их поряд-
порядкового номера
элементы, следующие за кюрием (Bk — Lr), которые и являются представителями
второй семерки.
Для rf-элементов горизонтальная аналогия в пределах всего ряда менее замет-
заметна, что обусловлено заполнением предвнешней (п — 1) (/-оболочки. Эта аналогия в
rf-рядах не может быть обнаружена при сравнении потенциалов ионизации, пото-
потому что для этих элементов свойственны разные степени окисления. Однако изме-
изменение металлических радиусов (рис. 119) у (i-элементов обнаруживает неглубокий
минимум, который приходится как раз на элементы триад VIHB-группы. А хоро-
хорошо известно, что вблизи минимума любая функция изменяется слабо (первая
производная, определяющая скорость изменения функции, близка к нулю).
Именно по этой причине у элементов триад горизонтальная аналогия выражена
наиболее ярко. Что касается р-элементов, особенно принадлежащих малым перио-
периодам, то здесь горизонтальная аналогия не наблюдается, поскольку электронами
заполняется самый внешний уровень. Однако у тяжелых представителей р-эле-
р-элементов горизонтальное сходство более заметно (Ge — As, Pb — Bi и т.п.), что
обусловлено существованием предвнешней заполненной (п — ^(/^-оболочки,
сближением энергетических характеристик заполняющихся оболочек, усилением
металличности сверху вниз в пределах каждой группы р-элементов.
Помимо отмеченных выше видов аналогии (групповая, типовая, слоевая, конт-
ракционная и горизонтальная) в Периодической системе существует определенное
сходство элементов, расположенных по диагонали, — так называемая диагональная
аналогия. Наиболее известна аналогия в диагональных парах Li — Mg, Be — Al,
В — Si. Диагональная аналогия может проявляться в двух формах: сходстве
общего химического характера элементов, проявляющемся во всех однотипных
соединениях (диагональная аналогия в широком смысле), и в возможности изо-
изоморфного замещения диагональных аналогов в сложных соединениях (диагональ-
(диагональная аналогия в узком смысле). Последний тип аналогии широко известен в гео-
геохимии. Диагональная аналогия в широком смысле обусловлена близостью энерге-
энергетических (Д/, Д?, ДОЭО) и размерных (ДОЭО/Дг) характеристик* элементов-
аналогов. В свою очередь, это определяется немонотонным изменением, напри-
например, электроотрицательности и орбитальных радиусов элементов по горизонтали
(в периоде) и по вертикали (в группе). Причинами немонотонного изменения
энергетических и силовых характеристик элементов, как обсуждалось выше,
являются эффекты кайносимметрии, экранирования, проникновения внешних
Например, энергия кулоновского взаимодействия обратно пропорциональна расстоя-
расстоянию между зарядами U = йй/г. Если эту энергию разделить на расстояние между за-
зарядами, то получим силу взаимодействия, отражаемую законом Кулона F — U/r= gift/r1.
237
1,0
о?
3 <t 5 6 7 8 9 10111213П1516ПШ1 J ¦ 5 6 7 8 3 10 11 1213П 151617 IS Z
a S
Рис. 120. Зависимость электроотрицательности (а) и орбитальных радиусов (б)
элементов 2-го и 3-го периодов от порядкового номера
электронов к ядру и т.д. В силу этой немонотонности и оказывается возможной
такая ситуация, когда различие между характеристиками элементов по диагона-
диагонали оказывается меньше, чем по горизонтали и по вертикали, что и приводит к
большему химическому сходству диагонально расположенных элементов в согед-
них группах по сравнению с групповой аналогией. Это положение хорошо иллю-
иллюстрирует рис. 120, где сопоставлены изменения электроотрицательностей ОЭО
(энергетическая характеристика) и орбитальных радиусов горб (размерный фак-
фактор) для ряда элементов 2-го и 3-го периодов. Из рис. 120, а, видно, что разность
электроотрицательностей в парах Li — Mg, Be — А1, В — Si (диагональ) значи-
значительно меньше, чем между групповыми аналогами Li — Na, Be — Mg, В — Al,
С — Si и между соседними элементами 2-го (Li ~ Be — В — С) и 3-го (Na — Mg —
— Al — Si) периодов, хотя изменение орбитальных радиусов по диагонали доволь-
довольно значительно (рис. 120, 5).
Отметим, что диагональная аналогия в широком смысле возможна только для
элементов начала малых периодов и не наблюдается как при переходе к более
тяжелым групповым аналогам, так и при дальнейшем продвижении вправо. На-
Например, изменение характеристик диагональной пары С — Р уже одного порядка
с вертикальными парами С — Si, N — Р и горизонтальными парами С — N,
Si-Р.
Диагональная аналогия, обусловленная изоморфным замещением элементов,
распространена значительно шире. Так как возможность изоморфного замещения
объясняется преимущественно размерным фактором, особенно при замещении
катионообразователей, то причиной этого вида диагональной аналогии является
лишь малое различие атомных радиусов. Так, можно отметить близость орби-
орбитальных радиусов в диагональной паре Na — Ca, что проявляется в широком
изоморфизме этих элементов в силикатных и сложных оксидных системах. В то
же время о глубокой химической аналогии между ними говорить нельзя, так как
их силовые характеристики сильно различаются. Такие диагональные пары,
аналогия между которыми проявляется в изоморфизме, достаточно многочислен-
многочисленны: Sc — Zr, Ti — Nb, V — Mo, Cu — Cd, Ag — Hg. Этот вид диагональной анало-
аналогии можно назвать прямой или нисходящей (от более легкого элемента вниз и
238
вправо к более тяжелому). Существует, однако, и обратная или восходящая,
диагональная аналогия такого типа (от более тяжелого элемента вверх и вправо к
более легкому), наблюдающаяся у некоторых р-элементов-анионообразователей.
Наиболее известны такие изоморфные пары, как As — S, Sb — Se, Bi — Те, что
проявляется во многих природных минералах. Так, наряду с диарсенидом железа
FeAs2 (леллингит) и дисульфидом железа FeS2 (пирит) широко распространен
минерал арсенопирит FeAsS, который можно рассматривать как продукт изо-
изоморфного замещения серы или мышьяка в любом из предыдущих соединений.
Г Л А В А XI. ПРОСТЫЕ ВЕЩЕСТВА КАК ГОМОАТОМНЫЕ
СОЕДИНЕНИЯ
Изучение свойств простых веществ имеет фундаментальное значение в неорга-
неорганической химии. Оно является первым этапом в описательной химии элементов.
Последовательное и аналитическое восприятие фактического материала о свойст-
свойствах простых веществ (физических, физико-химических, химических) позволяет
составить общее представление о химическом облике элемента, предвидеть приро-
природу химической связи, состав и свойства его характеристических соединений, их»
кислотно-основные и окислительно-восстановительные характеристики и т.п.
Принципиальная особенность простых веществ состоит в том, что при рассмотре-
рассмотрении их свойств нет необходимости учитывать вопросы, связанные с постоянством
или переменностью состава, поскольку состав простых веществ, естественно, всег-
всегда постоянен. Однако даже у простых веществ следует учитывать явление алло-
аллотропии и наличие собственных дефектов в реальном кристалле, что позволяет
выявить зависимость свойств простых веществ от их химического и кристаллохи-
ми чес кого строения.
Еще одна проблема, которая тесно связана с постоянством состава и выпукло
вырисовывается при изучении простых веществ, — это проблема получения и
изучения свойств вещества в особо чистом состоянии. Часто даже ничтожное
содержание примесей коренным образом меняет физические и химические свойст-
свойства вещества. Такие характеристики, как, например, твердость, электрическая
проводимость, коррозионная стойкость и т.п., радикально изменяются у особо
чистых веществ по сравнению с веществами обычной технической чистоты.
1. Химическое и кристаллохимическое строение простых веществ. Основопола-
Основополагающим понятием современной химии является понятие о химическом элементе,'
т.е. виде атомов с определенной совокупностью свойств. Под свойствами изолиро-
изолированных атомов подразумеваются заряд ядра и атомная масса, особенности элект-
электронного строения, потенциалы ионизации, сродство к электрону и электроотри-
электроотрицательность, атомные, орбитальные и ионные радиусы и т.д. Однако необходимо
иметь в виду, что изолированные атомы как форма организации вещества могут
существовать в природе лишь при достаточно высоких температурах в виде моно-
моноатомного пара. Единственным исключением являются благородные газы, для
которых при любых условиях и в любом агрегатном состоянии структурной еди-
единицей является атом. Все остальные элементы существуют в природе в виде более
сложных агрегатов: молекул и кристаллов. Таким образом, следует строго разли-
различать понятия элемента как вида изолированных атомов и простого
вещества как формы существования элемента в свободном состоянии. Сле-
239
дует особо подчеркнуть нетождественность этих понятий хотя бы потому, что
один элемент может существовать в виде нескольких простых веществ (аллотро-
(аллотропия). К тому же сама Периодическая система является системой эле-
элементов, а не простых веществ.
Несмотря на очевидность рааличия между этими понятиями, даже в учебной и
научной литературе допускают их смешение, употребляя, например, такие выра-
выражения, как "элементарный азот", "взаимодействие элементарного цинка с кисло-
кислотой" и т.п., хотя речь идет о простых веществах, а не об элементах. При образо-
образовании простых веществ из элементов возникают объекты, характеризующиеся
качественно иным набором свойств, чем изолированные атомы. Даже в тех случа-
случаях, когда в результате взаимодействия атомов образуются газообразные молеку-
молекулы, их свойства существенно иные. Например, хорошо известно, что атомарный
азот принадлежит к числу наиболее активных неметаллов, в то время как в моле-
молекулярной форме простое вещество — азот — характеризуется малой химической
активностью. Это обусловлено большим значением энергии химической связи в
молекуле азота. По той же причине все газы в атомарном состоянии существенно
более активны химически, чем их молекулы. Еще резче качественное отличие
простого вещества от соответствующего химического элемента при образовании
•конденсированной фазы с немолекулярной структурой. Конденсированное состоя-
состояние характеризуется свойствами, которые принципиально неприменимы к атомам,
например твердость и температура плавления (для кристаллов), вязкость и тем-
температура кипения (для жидкостей), электрическая проводимость и т.п.
Появление нового комплекса свойств у молекул и подавляющего большинства
кристаллов по сравнению с исходными атомами обусловлено возникновением
химических связей в процессе агрегации. Основополагающей идеей, проходящей
красной нитью через всю современную химию, является тезис о том, что все
свойства веществ обусловлены их химическим и кристаллохимическим строением.
А химическое и кристаллохимическое строение вещества определяется в первую
очередь природой химических связей взаимодействующих атомов. С этой точки
зрения переход от изолированных атомов к молекулам и кристаллам, сопровож-
сопровождающийся перераспределением электронной плотности и возникновением химиче-
химических связей, представляет собой процесс химический.
Большинство простых веществ существует не в виде молекул, а представляет
собой более сложные макроскопические образования с немолекулярной структу-
структурой. Характерной особенностью этого состояния является агрегация большого
числа атомов (порядка постоянной Авогадро) в едином ансамбле, в результате
чего и возникают новые свойства, о которых нельзя говорить применительно к
молекулам. Так, молекулярный пар натрия Na2 (г), существующий при высоких
температурах, принципиально отличается от одноатомного пара тем, что здесь
возникает ковалентная сг^-связь (как в молекуле Нг). В силу насыщенности
ковалентной связи в молекуле Na2 (г) и отсутствия межмолекулярного взаимодей-
взаимодействия натрий в парообразном состоянии обладает диамагнетизмом (в отличие от
одноатомного пара) и является диэлектриком. В то же время при конденсации
пара натрия в жидкость и ее кристаллизации возникает простое вещество с ме-
металлической связью и всеми характерными для металла свойствами: парамагне-
парамагнетизмом, высокой электрической проводимостью, пластичностью и т.п.
Таким образом, при образовании простых веществ из элементов в общем слу-
случае выделяются две стадии химического превращения: атом- — молекула и моле-
240
кула — координационный кристалл *. Уже на первой стадии из одного элемента
может образоваться несколько простых веществ. Например, элементу кислороду
отвечают два простых вещества: 02 и 03, — различающихся составом, строением,
а следовательно, и свойствами. На второй стадии образования простых веществ
возникающие координационные кристаллы в зависимости от температуры и
давления могут существовать в нескольких полиморфных модификациях, напри-
например ромбическая и моноклинная сера, белый, красный и черный фосфор, ГПК- и
ОПК-модификации железа и т.п.
Иногда полиморфные разности могут иметь одну и ту же решетку, но разные
структуры. Так, например, а- и /^-модификации марганца ОЦК-решетки, но
имеют разные структуры: с 58 и 2 атомами в элементарной ячейке.
В соответствии с двумя стадиями образования простых веществ из элементов
вводятся представления об их химическом и кристаллохимическом строении.
При этом термин "химическое строение" относится к молекулярной форме хими-
химической организации вещества. Итак, процесс образования простого вещества из
атомов химического элемента представляет собой химическую реакцию. Образо-
Образовавшийся продукт обладает качественно своеобразными свойствами. За счет
возникновения химических связей энергия системы уменьшается. Следовательно,
простое вещество представляет собой продукт химического взаимодействия оди-
одинаковых атомов и его необходимо рассматривать как юмоатомное химическое
соединение.
2. Металлы и неметаллы в Периодической системе. Периодический закон как
основа химической систематики дает возможность проанализировать положение
простых веществ с учетом особенностей их свойств. Все простые вещества, как
известно, подразделяются на две категории: металлы и неметаллы. Эта класси-
классификация основана на существенно различном характере физических и химиче-
химических свойств веществ, принадлежащих к разным классам. Принимая во внимание,
что свойства веществ являются функцией химической связи, следует сделать
вывод, что причины различий между металлами и неметаллами кроются в разном
типе межатомного взаимодействия при образовании простых веществ.
При образовании гомоатомных соединений (простых веществ) все эффекты,
связанные с разностью электроотрицательностей взаимодействующих атомов,
исключаются. Поэтому в простых веществах не реализуются полярные, а тем
более преимущественно ионные связи. Следовательно, в простых веществах осу-
осуществляется лишь металлическая и ковалентная связь. Следует при этом учесть
и возможность возникновения дополнительного ван-дер-ваальсова взаимодейст-
взаимодействия. Преобладание вклада металлической связи приводит к металлическим свой-
свойствам простого вещества, а неметаллические свойства обусловлены преимущест-
преимущественно ковалентным взаимодействием. Для образования ковалентной связи взаи-
взаимодействующие атомы должны обладать достаточным числом валентных электро-
электронов. При дефиците валентных электронов осуществляется коллективное элект-
электронно-атомное взаимодействие, приводящее к возникновению металлической
связи. На этой основе в Периодической системе можно провести вертикальную
*В частном случае при образовании молекулярных кристаллов (например, иода) отсут-
отсутствует вторая стадия превращения. Но возможно образование координационных кристал-
кристаллов из атомов (например, конденсация переходных металлов, выделение металла на катоде
при электролизе). В этом случае отсутствует первая стадия.
241
границу по элементам IVA-группы (граница Цинтля), слева от которой распола-
располагаются элементы с дефицитом валентных электронов, а справа — с избытком. Ее
положение в Периодической системе обусловлено тем, что в соответствии с совре-
современными представлениями о механизме образования ковалентной связи особой
устойчивостью обладает полностью завершенная октетная электронная ns2np6-
конфигурация, свойственная благородным газам. Поэтому для реализации кова-
лентного взаимодействия при образовании простых веществ необходимо, чтобы
каждый атом имел не менее четырех электронов в s- и р-еостояниях. В этом
случае возможно образование четырех ковалентных связей («ря-гибридизация),
что и реализуется у элементов IVA-группы (решетка типа алмаза у углерода,
кремния, германия и а-олова с координационным числом 4). Если атом имеет
пять валентных электронов (VA-группа), то до завершения октета ему необходи-
необходимо три электрона. Поэтому он может иметь лишь три ковалентные связи с парт-
партнерами (к.ч. 3). В этом случае кристалл образован гофрированными сетками,
которые связаны между собой более слабыми силами. Получается слоистая струк-
структура, в которой расстояние между атомами, принадлежащими одному слою, на-
намного меньше, чем между атомами различных слоев (черный фосфор мышьяк,
сурьма):
Элемент Р (черн) As Sb
Расстояние между атомами в слое, нм 0,217 0,251 0,287
Расстояние между атомами различных
слоев, нм 0,387 0,315 0,337
На этом основании было сформулировано кристаллохимическое правило Юм-
Розери, согласно которому координационное число в кристаллических структурах
простых веществ, расположенных справа от границы Цинтля, равно 8 — N, где
Л' — номер группы Периодической системы. Для элементов VIA-группы (S, Se,
Те), у которых до октета недостает двух электронов, структурными элементами в
кристаллах простых веществ являются линейные зигзагообразные цепочки (или
замкнутые кольца) с к.ч. 2, которые между собой связаны слабыми силами Ван-
дер-Ваальса. При этом расстояния между ближайшими соседями в пределах
одной цепочки также значительно меньше, чем между цепочками:
Элемент S (ромб)
Расстояние между атомами в цепочке, нм 0,210
Расстояние между цепочками, нм 0,330
Se (гекс) Те (гекс)
0,232 0,286
0,346 0,374
Элементы VHA-группы (галогены) в соответствии с правилом Юм-Розери
должны иметь в кристаллах простых веществ координационное число, равное
единице, т.е. каждый атом может иметь лишь одного соседа. Действительно, все
галогены (иод при обычных условиях, а остальные при низких температурах)
образуют молекулярные кристаллические структуры, в которых расстояния меж-
между атомами в молекулах значительно меньше, чем расстояния между молекулами
в кристалле. Так, для хлора длина связи в молекуле 0,202 нм, а расстояние
между молекулами в кристалле составляет 0,334 нм. Наконец, правило Юм-Розе-
Юм-Розери можно применить и к элементам VHIA-группы (благородные газы). В соот-
соответствии с этим правилом при образовании кристаллов простых веществ коор-
координационное число должно быть равно нулю. Действительно, кристаллы благо-
благородных газов состоят из одноатомных молекул, объединенных невалентными
силами Ван-дер-Ваальса.
242
Все элементы, располагающиеся слева от границы
Цинтля, характеризуются дефицитом валентных элект-
электронов, в силу чего в плотноупакованных кристалличес-
кристаллических структурах соответствующих простых веществ
доминирует металлическая связь. При этом граница
Цинтля не является границей между металлами и
неметаллами, а лишь разграничивает элементы с дефи-
дефицитом и избытком валентных электронов, что опреде-
определяет особенности кристаллохимического строения р и с 121 икосаэдр - элемент
простых веществ. Обращает на себя внимание ряд крисгалличеСкой структуры
исключений из правила 8 - N Так, свинец, располо- полиморфных модификаций
женный на границе Цинтля, обладает плотноупакован- ^л
ной кристаллической структурой с металлическим
типом связи. Для последнего представителя VA-группы — висмута — характерно
малое различие в межатомных расстояниях внутри слоя и между слоями @,310 и
0,347 нм), что фактически приводит к координационному числу 6. Ни одна из
двух известных структур полония также не отвечает правилу Юм-Розери.
Объясняется это тем, что с увеличением атомного номера элемента в пределах
каждой группы возрастает число электронных слоев и уменьшаются различия в
энергиях соседних орбиталей. Вследствие этого у тяжелых аналогов при
образовании простых веществ валентные электроны могут занимать более высокие
вакантные орбитали. У свинца, висмута и полония валентные электроны могут
попадать на вакантные (в изолированных невозбужденных атомах) 6^-орбитали.
При этом число возможных состояний превышает число неспаренных электронов,
т.е. наблюдается дефицит электронов, что и приводит к возникновению
плотноупакованных структур с металлическим типом связи.
С другой стороны, бор, находящийся слева от границы Цинтля и обладающий
дефицитом электронов, в виде простого вещества характеризуется неметалличес-
неметаллическими свойствами. Из-за кайносимметричности 2р-орбитали и вследствие этого
высоких значений потенциала ионизации и электроотрицательности бора имеет
место затруднение к обобществлению электронов в пределах всего кристалла. В
силу дефицита валентных электронов в кристаллических модификациях бора
наблюдается их обобществление, которое ограничено, однако, локальными атом-
атомными группами. Поэтому для бора характерны сложные кристаллические решет-
решетки, структурным элементом которых служит икосаэдр (рис. 121), который являет-
является своеобразным кластером*, состоящим из 12 атомов бора.
Таким образом, граница между металлами и неметаллами не совпадает с гра-
границей Цинтля, а проходит по диагонали в общем направлении от бериллия к
астату между элементами В — Al, Si — Ge, As — Sb, Те — Ро. Обоснованность
диагональной границы между металлами и неметаллами наглядно проявляется в
18- и 32-клеточной формах таблицы Менделеева, в которых элементы В-трупп
(переходные металлы), а также лантаноиды и актиниды естественным образом
располагаются слева от этой границы. Все в.- и /-элементы в виде простых ве-
веществ образуют плотноупакованные кристаллические структуры с доминиру-
доминирующим металлическим типом связи, хотя здесь проявляется и ковалентный вклад,
обусловленный наличием дефектных внутренних электронных орбиталей.
*От англ. cluster — группировка, рой, гроздь. .
24.4
Деление элементов и простых веществ на металлы и неметаллы в известной
степени неоднозначно. С одной стороны, металлы и неметаллы различают по их
физическим свойствам, которые проявляются у соответствующих простых ве-
веществ. Так, для металлов характерны высокая теплопроводность и электрическая
проводимость, отрицательный температурный коэффициент проводимости, специ-
специфический металлический блеск, ковкость, пластичность и т.п. Физические свой-
свойства неметаллов существенно иные: они хрупки, обладают низкой теплопровод-
теплопроводностью и электрической проводимостью с положительным температурным коэф-
коэффициентом (возрастание с температурой) и т.п. С другой стороны, различие
между металлами и неметаллами проявляется в их химических свойствах: для
первых характерны основные свойства оксидов и гидроксидов и восстановитель-
восстановительное действие, для вторых — кислотный характер оксидов и гидроксидов и
окислительная активность. Ориентируясь на физические свойства, к типичным
металлам следует отнести, например, медь, серебро и золото, обладающие
наиболее высокой электрической проводимостью и пластичностью. Однако по
химическим свойствам эти вещества вовсе не относятся к типичным металлам,
поскольку стоят в ряду стандартных электродных потенциалов (ряд напряжений)
после водорода. В то же время для элементов IA-группы, являющихся по
химическим свойствам самыми активными металлами, некоторые физические
характеристики (например, электрическая проводимость) выражены не так ярко.
Таким образом, подразделяя элементы на металлы и неметаллы, всегда следует
иметь в виду, по каким свойствам это деление осуществляется: по химическим
или по физическим.
Деление на металлы и неметаллы относительно, поскольку существуют так
называемые амфотерные элементы. Последние, как и следует ожидать, группиру-
группируются вблизи диагональной границы, разделяющей металлы и неметаллы.
3. Физические свойства простых веществ. При рассмотрении физических
свойств и характера их изменения в Периодической системе следует различать
атомные свойства (свойства элементов) и свойства простых веществ (гомоатомных
соединений). Кроме того, физические свойства простых веществ могут характери-
характеризовать обе формы химической организации вещества (молекула и кристалл) или
только одну из них. Очевидно, такие свойства, как температура плавления и
кипения, твердость и вязкость, электрическая проводимость и т.п., относятся
только к конденсированному состоянию вещества. С другой стороны, например,
магнитные свойства (диа- или парамагнетизм) характерны как для кристаллов,
так и для молекул.
Атомы элементов характеризуются сравнительно небольшим набором физиче-
физических свойств: заряд ядра, атомная масса, орбитальный радиус, потенциал иониза-
ионизации, сродство к электрону. Для простых веществ, особенно в конденсированном
состоянии, набор физических свойств, т.е. существенных признаков, отличающих
одно вещество от другого, весьма обширен. В качестве примера можно перечис-
перечислить классы таких характеристик: термодинамические, кристаллохимические,
физико-механические, электрофизические, оптические, магнитные и иные свойст-
свойства. Рассматривая закономерности изменения физических свойств простых ве-
веществ, целесообразно ограничиться сравнительно небольшим набором характе-
характеристик, которые обусловлены в первую очередь особенностями химической связи
(молярные объемы, энтальпии атомизации, энергии диссоциации двухатомных
молекул, температуры плавления, магнитная восприимчивость).
¦¦Q
S
«a
•a
1
8-
Мол
p
70
60
50
40
30
20
10
0
и с.
Ra
го
20 3d W '"Sb W Id ' W M Z
122.
Зависимость молярного объема гомоатомных соединений
от порядкового номера элементов
Фундаментальной характеристикой простого вещества является молярный
объем, представляющий собой отношение молярной массы вещества (М, г/моль)
к его плотности (d, г/см3): Vm = M/d (см3/моль). Молярный объем простых ве-
веществ — одно из первых свойств, подтвердивших явление периодичности (рис.
122). В пределах каждого периода наибольшим молярным объемом обладают
литий и щелочные металлы. К середине периода молярные объемы уменьшаются,
а затем вновь возрастают начиная с IVA-группы. Эта закономерность особенно
ярко выражена в малых периодах. В больших периодах, где вклиниваются d- и f-
элементы, в этих пределах молярные объемы меняются незначительно. Такой
характер зависимости определяется как атомными свойствами элементов (значе-
(значением атомной массы), так и характером химической связи и особенностями кри-
кристаллической структуры простых веществ.
Уменьшение молярного объема до середины малого периода, несмотря на
монотонное возрастание молярной массы, обусловлено более резким возрастанием
плотности. Действительно, в IA — ША-группах располагаются металлы, облада-
обладающие плотноупакованными структурами. Вследствие уменьшения атомных радиу-
радиусов по периоду слева направо наблюдается уменьшение межатомных расстояний,
что в совокупности с увеличением атомной массы и приводит к возрастанию
плотности, а следовательно, к уменьшению молярного объема. У простых веществ
второй половины малых периодов начиная с IVA-группы в соответствии с прави-
правилом 8—N реализуются "рыхлые" структуры с малыми координационными числа-
числами, что и приводит к резкому уменьшению плотности, несмотря на возрастание
атомной массы. Поэтому молярные объемы во второй половине периода возраста-
возрастают. Следуя этой закономерности, можно было бы ожидать, что наибольшими
молярными объемами в пределах каждого периода должны обладать благородные
газы (в кристаллическом состоянии). Однако вследствие образования плотноупа-
кованных структур (хотя и обусловленных силами Ван-дер-Ваальса) плотность их
кристаллов оказывается несколько выше ожидаемой, что и приводит к некоторо-
некоторому уменьшению молярного объема. У переходных «йяеталлов с близкими по
характеру упаковки кристаллическими структурами в пределах одного периода
245
I 800
v? 720
hOO
320
160
80
I
- С
W М- т <iO ..??•¦ SO. . 70 80 I
Рис. 123. Зависимость энтальпии атомизации гомоатомных соеди-
соединений от порядкового номера элементов
плотность варьирует в сравнительно небольших пределах с общей тенденцией
увеличения от начала вставных декад к элементам VIIIB-группы (триады). С
учетом монотонного возрастания атомных масс это приводит к относительному
постоянству молярного объема. В ряду лантаноидов наблюдается монотонное
уменьшение молярного объема, которое обусловлено возрастанием плотности
вследствие уменьшения межатомных расстояний за счет лантаноидной контрак-
контракции.
Явно выраженная периодичность характерна для энтальпий атомизации прос-
простых веществ (рис. 123). Для элементов малых периодов кривая зависимости эн-
энтальпии атомизации от атомного номера проходит через четко выраженный мак-
максимум, приходящийся на элементы IVA-группы (алмаз, кремний). Это обуслов-
обусловлено, с одной стороны, упрочнением связей в кристаллах по мере увеличения
числа валентных электронов от одного до четырех, а с другой — уменьшением
прочности кристаллической решетки за счет уменьшения координационного
числа ковалентных структур по правилу 8 — N после IVA-группы. Минимумы на
кривой соответствуют кристаллам благородных газов, образованным за счет
слабых сил межмолекулярного взаимодействия. В больших периодах для s- и р-
элементов (главные подгруппы) эта закономерность также просматривается. Од-
Однако на нее накладывается изменение энтальпий в рядах переходных металлов.
При этом для металлов первой вставной декады, обладающих кайносимметрич-
ными Зсйэлектронами, наблюдается четко выраженная внутренняя периодич-
периодичность, обусловленная особой стабильностью rf5- и <#°-конфигурации и меньшей
способностью электронов в этом состоянии к валентному взаимодействию. Имен-
Именно по этой причине кристаллические структуры марганца (Sd^As2) и цинка
C<flo4s2) менее прочны. Эта тенденция менее ярко выражена у остальных rf-эле-
ментов, особенно в середине вставных декад, где более явно проявляется гори-
горизонтальная аналогия. Тем не менее, для простых веществ, замыкающих d-ряды
(Cd, Hg), отмечаются минимумы энтальпии атомизации. Принципиально важным
является то, что в главных подгруппах максимальное значение энтальпии атоми-
атомизации в ряду С — Si — Ge — Sn — Pb уменьшается, а для rf-элементов — возраста-
возрастает в направлении 3d — Ad — bd. Первый факт обусловлен ростом доли металличе-
с
¦ I
ЩкАгь
Mo
1 Л
w
л
Sm ?Hf\p
3D \fi
CS HgW
3775
3275
2775
2275
1775
1275
775
275
2 IB 20 30 kO 50 60 70' 80 90 Z
P и с. 124. Периодичность в изменении температур плавле-
плавления гомоатомных соединений
Порядкобый номер
Р и с. 12а, Энергии диссоциации двух-
двухатомных молекул гомоатомных соедине-
соединений
ской связи при образовании кристаллов простых веществ от С к РЬ, а второй —
увеличением доли ковалентного взаимодействия в кристаллах переходных метал-
металлов по мере увеличения атомной массы в В-группах.
Качественная корреляция с характером изменения энтальпий атомизации
наблюдается и в изменении температур плавления простых веществ (рис. 124).
которые также в определенной мере обусловлены сравнительной прочностью
связей в кристаллах. При этом надо иметь в виду, что полная корреляция была
бы возможна, если бы простые вещества обладали одинаковой структурой и
одинаковыми значениями энтропии. Дело в том, что плавление как фазовый
переход характеризуется равенством свободных энергий Гиббса сосуществующих
фаз, т.е. одновременно надо учитывать и энтальпийный и энтропийный факторы.
Значения же энтальпии атомизации сопоставимы только с одним из них. Тем не
менее наинизшие температуры плавления в пределах каждого периода свойствен-
свойственны благородным газам, в пределах главных подгрупп температуры плавления
понижаются, а для rf-элементов наблюдается более сложная зависимость. В нача-
начале и конце вставных деках (Sc — Y — La и Zn — Cd — Hg) сверху вниз температу-
температуры плавления уменьшаются, а в середине (Сг — Mo — VV) возрастают.
Периодичность проявляется и в энергиях диссоциации двухатомных молекул
(рис. 125). Если энергия атомизации характеризует прочность связей в кристалле
как высшей форме организации вещества, то энергия диссоциации является
аналогичной характеристикой молекулярной формы. Молекулярная форма орга-
организации у простых веществ встречается сравнительно редко: в стандартных усло-
условиях двухатомные молекулы образуют водород, азот, кислород и галогены, а при
высоких температурах в этой форме существуют пары щелочных металлов, угле-
углерода (выше 3600°С), халькогенов (кроме полония) и пниктогенов* (кроме висму-
висмута). Таким образом, молекулярная форма в парообразном состоянии наиболее
характерна для неметаллов. Большинство же металлов (за исключением щелоч-
Термин "пниктогены", объединяющий элементы VA-труппы, введен по аналогии с
терминами "галогены" и "халькогены" и образован от символов химических элементов:
фосфора Р и азота N.
247
Tm
ных металлов и меди) при высо-
высоких температурах существуют в
виде одноатомного пара. Мини-
Минимальными значениями энергии
диссоциации характеризуются
щелочные металлы и литий, мак-
максимальными — молекулы пникто-
генов (N2, Р2, As2, Sb2), где реа-
реализуется тройная связь. Молеку-
Молекула азота вообще является самой
прочной из двухатомных молекул
(Е = 941,4 кДж/моль). Для халь-
когенов и галогенов энергии дис-
О 10 20 30 *Г'5О 60 70 ~8п Z с0^иаЦ"и закономерно уменьша-
ются в пределах каждого перио-
Р и с. 126. Зависимость магнитной восприимчивости
да, что соответствует образова-
гомоатомных соединений от их порядкового номера
нию двойных и одинарных
связей. В молекулах галогенов, кроме фтора, может осуществляться дативная
связь*, поэтому в группе галогенов энергия диссоциации изменяется немонотон-
немонотонно: возрастает от фтора к хлору, а затем уменьшается к брому и иоду. Меньшие
по сравнению с фтором энергии диссоциации одинарных связей в молекулах
щелочных металлов объясняются тем, что их атомы образуют (Т^-связи, в то
время как атомы фтора в его молекуле — <гр_р-связи. Однако в молекуле Н2, где
также существует <7,-,гСвязь, энергия диссоциации существенно выше, что объяс-
объясняется кайносимметричностью ls-орбиталей.
Магнитные свойства простых веществ также обнаруживают периодическую
зависимость от порядкового номера элемента (рис. 126), но закономерности, кото-
которым подчиняется эта зависимость, требуют пояснения. В стандартных условиях
простые вещества находятся в разном агрегатном состоянии. Все газообразные и
жидкие простые вещества являются диамагнитными. Единственным исключением
является кислород, парамагнетизм двухатомной молекулы которого объясняется с
позиций метода МО. Сложнее обстоит дело с кристаллическими веществами.
Магнитные свойства кристаллов определяются главным образом тремя вкладами:
диамагнетизмом атомного остова, орбитальным диамагнетизмом валентных элект-
электронов и спиновым парамагнетизмом. У неметаллов, в кристаллах которых доми-
доминирует ковалентная связь, вклад спинового парамагнетизма пренебрежимо мал,
поэтому все они диамагнитны. Парамагнитными свойствами обладают все пере-
переходные металлы с недостроенными d- и /оболочками, щелочные, щелочно-зе-
мельные металлы и магний, а также алюминий. rf-Металлы с заполненными
внутренними оболочками (подгруппы меди и цинка) диамагнитны, так как у них
спиновый парамагнетизм не перекрывает двух диамагнитных составляющих (ор-
(орбитального диамагнетизма валентных электронов и диамагнетизма атомного
остова). По той же причине диамагнитными свойствами обладают металлы под-
подгруппы галлия, олово и свинец.
*Разновидность донорно-акцепторного взаимодействия, при котором атом одновременно
выступает в качестве донора электронной пары и в качестве акцептора.
оля
Электрические свойства простых веществ, как известно, являются одним из
признаков, пс которым их делят на металлы и неметаллы. С электрической про-
проводимостью тесно связана теплопроводность кристаллов, обусловленная переда-
передачей теплоты за счет колебаний атомов в узлах кристаллической решетки (фоно-
ны) и передачей теплоты электронами. В кристаллах неметаллов концентрация
свободных электронов незначительна. Поэтому все они являются полупроводни-
полупроводниками и диэлектриками и обладают низкой теплопроводностью, обусловленной
колебаниями решетки. В противоположность этому для металлов характерны
высокие значения электрической проводимости (порядка 105 — 106 Ом-см) и
теплопроводности, поскольку в этом случае вклад свободных электронов в тепло-
теплопроводность является определяющим. Наиболее высокой электрической проводи-
проводимостью и теплопроводностью обладают металлы подгруппы меди и алюминий.
Для переходных металлов характерны достаточно высокие, но несколько мень-
меньшие значения электрической проводимости.
4. Химические свойства простых веществ. При рассмотрении физических
свойств простых веществ подчеркивалось, что они в основном присущи макроско-
макроскопическим количествам вещества (особенно в конденсированном состоянии). Что
же касается химических свойств, то они главным образом определяются свойства-
свойствами атомов или молекул, поскольку химическое взаимодействие всегда протекает
на атомном или молекулярном уровне. Однако реально наблюдаемая химическая
активность твердых простых веществ в заметной мере зависит, например, от
величины поверхности соприкосновения, ее состояния, структуры кристалла и
т.п., т.е. опять-^гаки от макроскопических характеристик. Так, мелкодисперсный
цинк (цинковая пыль) значительно энергичнее взаимодействует с кислотами, чем
гранулированный. Например, цинковая пыль восстанавливает азотную кислоту до
аммиака, а гранулированный цинк — только до низших оксидов азота. Хорошо
известна также способность многих металлов (Al, Fe, Ti, Cr и др.) к пассивации в
агрессивных окисляющих средах, хотя сами эти металлы достаточно активны.
Кроме того, различные модификации одного и того же простого вещества могут
заметно различаться по химической активности (например, белый и красный
фосфор). Таким образом, химические свойства простых веществ представляют
собой единство атомной, молекулярной и кристаллической форм химической
организации со всеми характерными для них особенностями.
По химическим свойствам простые вещества, как известно, также подразделя-
подразделяются на металлы и неметаллы. С этими двумя классами генетически связаны
соответствующие ряды характеристических соединений: оксидов (основных и
кислотных), гидроксидов (оснований и кислот). Отличительной особенностью
этих рядов является способность к взаимодействию с образованием солей, т.е. к
взаимной нейтрализации в широком смысле слова. Чем ярче выражены металли-
металлические и неметаллические свойства простых веществ, тем активнее взаимодейст-
взаимодействие между ними и их характеристическими соединениями. Таким образом, в
химии ярко проявляется симметричность относительно кислотно-основного взаи-
взаимодействия, причем каждый из генетических типов базируется на одном из двух
классов простых веществ.
С этих позиций становится более понятным явление амфотерности в широком
смысле как общего свойства простых веществ и их характеристических производ-
производных. Осложняющим обстоятельством здесь является то, что кислотно-основное
взаимодействие между характеристическими соединениями происходит без изме-
249
нения степени окисления, а взаимодействия с участием простых веществ непре-
непременно являются окислительно-восстановительными.
Таким образом, для простых веществ два вида взаимодействия (кислотно-
основное и окислительно-восстановительное) сливаются воедино. Эта особенность
наиболее наглядно прослеживается при взаимодействии металлов с неметаллами,
причем первые — восстановители и в соединениях присутствуют в окисленной
форме (катионообразователи), а вторые — окислители и выполняют роль анионо-
образователя, выступающего в соединении в восстановительной форме. Таким
образом, окислительно-восстановительная сущность при взаимодействии металлов
с неметаллами выражена весьма выпукло. Но при этом образуются соли бескисло-
бескислородных кислот, т.е. происходит взаимная нейтрализация в широком смысле.
5. Нахождение в природе и общие принципы получения простых веществ.
Подавляющее большинство элементов в природе находится в связанном состо-
состоянии — в виде соединений. При этом преобладающие формы соединений специ-
специфичны и определяются главным образом активностью элемента. Природные
соединения металлов зависят от их активности: чем более активен металл, тем
более ярко выраженный солеобразный характер имеют его соединения в природе.
Так, щелочные металлы находятся в природе в виде галогенидов, нитратов,
сульфатов, изредка карбонатов (содовые озера). Все эти соединения хорошо
растворимы в воде, что определяет специфику распределения месторождений на
Земле: в морях и океанах, соленых озерах, подземных минеральных источниках и
т.п. Месторождения, сконцентрированные на суше (залежи каменной соли NaCl,
сильвинита NaCl-KCl и др.), образуются в результате испарения водоемов и
имеют осадочное происхождение. Для несколько менее активных щелочно-зе-
мельных металлов и магния галогенидные и нитратные месторождения мало
характерны, хотя изредка и встречаются [карналлит KCbMgCl2<6H2O, норвеж-
норвежская селитра Са(МОзJ]- Наиболее типичны для них малорастворимые в воде
сульфаты и карбонаты (мел, известняк, мрамор СаСО3, гипс CaSO4-2H2O, доло-
доломит СаСОз• MgCO3 и т.д.). Многие из этих минералов являются породообразу-
породообразующими и образуют горные массивы (например, Доломитовые Альпы).
Нахождение в природе алюминия обусловлено главным образом высоким
сродством к кислороду, амфотерностью его соединений и способностью к образо-
образованию комплексов (например, криолит Na3[AlFg]). Вследствие этого для алюми-
алюминия характерно образование алюмосиликатов разнообразного строения. Продук-
Продуктами их выветривания в природе являются оксидно-гидроксидные месторожде-
месторождения [бёмит АЮ(ОН), каолин Al2Cv2SiO2-2H2O, боксит А12О3-хН2О, гидраргил-
лит А1(ОНK и т.п.]. Галогенидные (кроме криолита), нитратные, сульфатные и
карбонатные природные соединения для алюминия не характерны. Для металлов
IVA-группы (Sn, Pb) и большинства переходных металлов наиболее характерны
многочисленные и разнообразные оксидно-сульфидные месторождения. Следует
подчеркнуть, что в геохимическом отношении сера и кислород в соединениях с
металлами конкурируют между собой. Многочисленные сульфиды (а также суль-
фоарсениды) образуют так называемые полиметаллические руды, которые пред-
представляют собой конгломерат различных соединений и могут иметь осадочное,
гидротермальное и магматическое происхождение.
Наконец, некоторые металлы (Си, Ag, Аи, Hg и др.) встречаются в природе не
только в виде оксидов и сульфидов (киноварь HgS, халькозин Cu2S, куприт СиО2
и т.п.), но и в самородном состоянии.
Что касается природных соединений неметаллов, то здесь соблюдаются те же
общие принципы. Наиболее активные неметаллы — галогены — встречаются в
природе исключительно в виде соединений, главным образом со щелочными
металлами. Кислород и сера также активные минералообразователи и ассоцииро-
ассоциированы преимущественно с переходными металлами. Кислород, кроме того, образует
многочисленные силикаты и алюмосиликаты, а сера — сульфаты. Хотя эти два
элемента встречаются в природе и в состоянии простых веществ (атмосферный
кислород, озонный слой в стратосфере, самородная сера), но это обусловлено
вторичными процессами, связанными преимущественно с фотосинтезом растений
и вулканической деятельностью.
Основной формой азота в природе является атмосферный азот, что обусловле-
обусловлено исключительной прочностью молекулы N2. Главные природные соединения
фосфора — фосфаты, в основном фосфорит Са^РО^г и апатиты Са.5Х(РО4)з- где
X — F", С1~, ОН". Эти соединения нерастворимы в воде и образуют мощные зале-
залежи. Остальные пниктогены — As, Sb, Bi — встречаются обычно в виде сульфид-
сульфидных минералов (например, реальгар AS4S4, сурьмяный и висмутовый блеск ЭгБз).
Мышьяк, кроме того, выступает и в роли анионообразователя в минералах (на-
(например, сульфоарсенид железа — арсенопирит FeAsS). Эти элементы часто ассо-
ассоциированы с полиметаллическими рудами. Природный углерод встречается в
двух формах — органического и минерального происхождения. К первой относят-
относятся залежи каменного угля, нефти, природного газа, вторая представлена главным
образом карбонатными породами и углекислым газом. Кремний — основной эле-
элемент твердой земной коры (литосферы) — представлен многочисленными силика-
силикатами и алюмосиликатами.
Таким образом, все элементы с точки зрения их локализации в природе под-
подразделяются на четыре группы: атмофильные, литофильные или оксифильные,
халькофильные и сидерофильные *. К первой группе относятся азот, водород,
кислород и благородные газы, концентрирующиеся в атмосфере (водород — в
виде водяного пара). Литофильные элементы концентрируются в самой внешней
оболочке Земли — литосфере. Их соединения характеризуются сравнительно не-
невысокой плотностью и в процессе дифференциации вещества по мере остывания
планеты формируют ее внешнюю твердую оболочку. Халькофильные элементы,
имеющие повышенное сродство к халькогенам, входят в состав так называемой
халькосферы, "подстилающей" литосферу. Наконец, сидерофильные элементы —
элементы триад VIIIB-группы — образуют наиболее плотную часть Земли — ее
ядро.
Различные элементы представлены и распространены на Земле неравномерно.
Большинство легких элементов с массовыми числами до ~ 50 составляют в сумме
~ 99,4% трех оболочек: атмосферы, гидросферы и литосферы. На долю остальных
элементов приходится всего ~ 0,6%. В соответствии с этим выделяют так называе-
называемые редкие элементы, содержание которых на Земле мало. Так, для цезия оно
составляет 9-10%, для рения — 9<10"9%, для церия — 5-10%, а содержание
других лантаноидов значительно меньше. Другой характеристикой, отражающей
распространенность элементов в природе, является способность концентрировать-
концентрироваться, образуя месторождения. Так, общее содержание меди на Земле оценивается в
3-10~3%, т.е. сравнительно невелико. Однако медь — металл, известный челове-
*От греч. sideros — железо, philes — люблю.
честву с глубокой древности. Это обусловлено способностью меди концентриро-
концентрироваться в отдельных участках, формируя собственные месторождения. С другой
стороны, германий B-10%) самостоятельных месторождений не образует, встре-
встречается в виде примеси в полиметаллических рудах и в некоторых сортах углей.
Он относится к рассеянным элементам. Многие редкие элементы являются и
рассеянными, однако к числу рассеянных относятся и некоторые такие элементы,
содержание которых на Земле сравнительно велико.
Получение простых веществ из их природных соединений — это всегда окис-
окислительно-восстановительный процесс, кроме тех случаев, когда простые вещества
встречаются в самородном состоянии. В последнем случае их обычно выделяют
из смесей физическими методами (например, разгонка сжиженного воздуха при
получении N2, O2, благородных газов). Все металлы (кроме самородных) нахо-
находятся в природе в окисленном состоянии, и их выделение из природных соедине-
соединений сводится к восстановлению. Неметаллы в природных соединениях могут
находиться как в окисленном, так и в восстановленном состоянии. При этом
наиболее активные неметаллы (галогены, кислород) находятся в природных
соединениях исключительно в восстановленном состоянии. Халькогены также
преимущественно находятся в восстановленном состоянии, хотя, например, в
сульфатах сера окислена. Азот, фосфор, кремний, бор, сурьма, висмут в природе
встречаются всегда в окисленной форме (нитраты, фосфаты, силикаты, сульфиды
сурьмы и висмута и т.п.).
Общий принцип получения металлов из природных соединений заключается в
следующем: чем более активен данный металл, тем более энергичный восстанови-
восстановитель необходимо использовать для его выделения. Типичными восстановителями
в металлургии являются водород, углерод, активные металлы (Al, Zn, Mg, Ca,
щелочные металлы). Выбор подходящего восстановителя определяется не только
возможностью протекания самой окислительно-восстановительной реакции (отри-
(отрицательно значение ДG), но и протеканием побочных реакций избытка восстано-
восстановителя с восстановленным металлом. Многие переходные металлы можно восста-
восстанавливать из оксидов углеродом. Однако он образует с рядом металлов хрупкие
и тугоплавкие фазы внедрения. Иногда этот эффект используют сознательно,
например при карботермическом восстановлении железной руды в доменных
печах с образованием чугуна.
Для многих металлов формой, подлежащей восстановлению, является оксид.
Поэтому сульфидные руды для перевода в оксидную форму подвергают обжигу.
Водородным восстановлением оксидов получают такие металлы, как Mo, W, Re и
т.п. Водород — сравнительно мягкий восстановитель. Карботермическое восста-
восстановление используют для получения Fe, Co, Ni, Pb, Sn, Си, Zn, Mn и др. Более
энергичным восстановителем является металлический алюминий. Алюминотер-
Алюминотермия широко используется для получения таких металлов, как Сг, Mn, Fe (алю-
минотермическая сварка), щелочно-земельных металлов. Восстановление оксидов
металлов алюминием протекает с большим выделением теплоты, что обусловлено
высоким сродством алюминия к кислороду. Еще энергичнее как восстановитель
действует магний, который используют для восстановления как оксидов (на-
(например, ВгО3), так и галогенидов (например, при получении титана и его ана-
аналогов). Наконец, самые активные металлы — алюминий, магний, щелочно-земель-
ные и щелочные — получают электролизом расплавов солей (как правило, хлори-
Т а б л и ц а 21. Основные способы получения простых веществ
Способы проведения
процесса
Исходная форма
Получаемые вещества
Разделение смесей
Фракционная перегонка
Водородное
Карботермия
Алюминотермия
Цинктермия
Магнийтермия
Кальцийтермия
Щелочными металлами
Катод электролизера
Вытеснение хлором
Анод электролизера
Физические методы
Самородные простые ве-
вещества
Воздух (жидкий)
Химические методы
А. Восстановление
Оксиды, халькогениды,
соли
Оксиды, соли
S, Аи, Pt, Hg, платиновые
металлы
Ог, N2, благородные газы
Оксиды
Галогениды, соли
Оксиды, галогениды
То же
Оксиды
Галогениды, соли, вода
Б. Окисление
Бромиды, иодиды
Расплавы фторидов, рас-
растворы, расплавы хлоридов,
вода
В. Разложение
и диспропорционирование
Галогениды, субгалогениды
(кроме фторидов)
Карбонилы
Ga, In, Si, Ge, Mo, W,
Re, В
Fe, Co, Ni, Cr, Mn, Si,
Pb, Sn, Zn, Cd, Си, Р, As,
Sb, Bi, Mo, W
Cr, Fe, Co, Ni, Mn, ще-
лочно-земельные металлы
Si, Ag, Au
Be, Ti, Zr, Hf, Si, В
U, V, Nb, Та, Sc, Y, La,
лантаноиды
Nb, Та, Ti, Zr, Hf
Щелочные, щелочно-
щелочноземельные металлы, AJ, Ga,
In, TI, Be, Mg, Zn, ланта-
лантаноиды, Pb, Nb, Та, Mn, Cu,
H
Br2, I2
Cl2, F2, O2
Si, Ge, Ti, Zr, Hf, Mo,
W, V, Nb, Та, Al, В
Fe, Co, Ni, Mn
дов или фторидов). Катод электролизера можно рассматривать как наиболее
энергичный восстановитель — непосредственный донор электронов.
Сравнительно малоактивные неметаллы, которые находятся в природе в окис-
окисленном состоянии (В, Si, Ge, P, As, Sb), выделяют в свободном состоянии также
действием соответствующих восстановителей. Так, бор и кремний восстанавлива-
ют магнийтермически из оксидов, водородом из хлоридов (BCI3, SiCl4), а крем-
кремний — еще и цинком из SiCl4. Германий выделяют в свободном состоянии водо-
водородным восстановлением веОг- Пниктогены (кроме азота и фосфора) получают
карботермическим восстановлением оксидов Э2О3, а фосфор — карботермическим
восстановлением Саз(РО4J- Наиболее активные неметаллы — галогены — можно
выделить из природных соединений только окислением. Бром и иод можно вы-
вытеснить из растворов бромидов и иодидов более энергичным окислителем —
хлором. Хлор и фтор получают электролизом, причем в случае хлора используют
растворы хлоридов, а фтора — только расплавы, поскольку фтор, как наиболее
энергичный окислитель, активно разлагает воду с выделением ряда побочных
продуктов. Во всех этих процессах анод электролизера, является самым активным
окислителем как непосредственный акцептор электронов.
Своеобразную группу методов получения простых веществ (металлов и неме-
неметаллов) составляют методы, основанные на термическом разложении (пиролиз)
или диспропорционировании летучих галогенидов:
Sil4
Si + 2I2; 2TiCl2
Ti + TiCl4; Til4
; Ti + 2I2
Таким путем получают бор (разложение ВВгз, В1з). кремний, металлы подгруппы
титана и т.п. Особенностью этих методов является то, что в результате реакций
пиролиза, а также диспропорционирования получаются вещества очень высокой
степени чистоты. Поэтому данная группа методов находит все большее примене-
применение в современной технологии. В табл. 21 представлены основные способы полу-
получения простых веществ.
6. Особо чистые вещества. Понятие о чистоте вещества имеет принципиальное
значение в современной неорганической химии. Абсолютно чистые вещества в
природе не существуют, поскольку загрязнение примесями происходит самопро-
самопроизвольно вследствие резкого возрастания энтропии. Поэтому нет абсолютно не-
нерастворимых веществ и, следовательно, любое вещество загрязнено примесями.
Даже в тех случаях, когда вещество очищено до очень высокой степени, абсолют-
абсолютное число атомов примеси в единице массы или объема все еще остается огром-
огромным. Так, в германии полупроводниковой чистоты 99,9999999% содержание ато-
атомов примеси не превышает 10~7 ат.доли, %,т.е. один атом примеси приходится на
миллиард атомов основного вещества. Тем не менее 1 см3 этого особо чистого
германия содержит около 1013 атомов примеси. Примеси коренным образом вли-
влияют на свойства вещества. Например, хорошо известная хрупкость и исключи-
исключительная твердость металлического хрома, как выяснилось, являются следствием
наличия небольшого количества примесей, в основном кислорода. Хром, получен-
полученный в условиях глубокого вакуума, оказался мягким и пластичным.
Проблема получения чистых веществ имеет три основных аспекта. Первый из
них состоит в том, что свойства вещества можно определять только получив его в
достаточно чистом состоянии. Более того, сравнение одноименных свойств раз-
различных веществ допустимо только при их одинаковой чистоте. Второй аспект
заключается в выборе подходящих методов, позволяющих очистить вещество до
необходимой чистоты. И наконец, третий аспект проблемы — обеспечение доста-
достаточно чувствительных и селективных методов контроля чистоты.
По мере развития науки и техники возникает проблема получения все более
чистых веществ. До сравнительно недавнего времени использовались вещества,
которые в настоящее время можно считать "грязными". Очистка таких веществ
обеспечивалась главным образом химическими методами, контроль чистоты —
химическим анализом. Такие вещества в ряде случаев и сейчас используются в
химической практике. Для них разработана градация оценки чистоты на четырех
уровнях: "технический" (т), "чистый" (ч), "чистый для анализа" (ч.д.а) и "хи-
"химически чистый" (х.ч). Так, серная кислота квалификации "ч" содержит 1-10
масс, доли, % примесей, "ч.д.а" — 2-Ю масс.доли, %, а "х.ч" — 1-Ю масс.
доли, %.
В современных отраслях техники — ядерной энергетике, квантовой электрони-
электронике, полупроводниковом приборостроении и т.п. — требуются материалы значи-
значительно более высокой степени чистоты. Такие особо чистые вещества можно
получить только с помощью специальных физико-химических методов очистки,
основанных на различном содержании примесей в сосуществующих фазах. Мето-
Методами сублимации, экстракции, хроматографии, направленной кристаллизации,
зонной плавки удается получить вещества, которым присваивается квалификация
"особо чистый" (ос.ч). Для характеристики таких материалов используется не
общее содержание примесей, а содержание так называемых анализируемых при-
примесей. При этом число анализируемых примесей достаточно велико: 10 — 20, а
иногда и более. В маркировке ОСЧ-материала указывается количество анализи-
анализируемых примесей и их общее содержание. Например, мышьяк маркировки
ОСЧ10-5 означает, что в материале определено содержание 10 примесей, а их
суммарное количество — 10~5 масс.доли, %, т.е. содержание основного вещества
99,99999%.
В ряде случаев вводится понятие о так называемой целевой чистоте и вещест-
веществах эталонной чистоты (ВЭЧ). В маркировке таких веществ три сорта значащих
цифр, например 003ВЭЧ4-7. Первые три цифры означают общее содержание всех
примесей A0~3 масс.доли, %), вторая — число анализируемых примесей D), а
третья обозначает их содержание A0~7 масс.доли, %). В веществах эталонной
чистоты в отличие от особо чистых веществ количество анализируемых примесей
невелико, поэтому их суммарное содержание не совпадает с общим содержанием
примесей.
ГЛАВА XII. БИНАРНЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ
Бинарные соединения, несмотря на кажущуюся простоту их химического
состава, представляют собой следующий после простых веществ принципиально
важный объект изучения природы вещества. С химической точки зрения, этот
класс веществ обладает и качественно иными характеристиками, с которыми не
приходится сталкиваться при изучении простых веществ. Во-первых, помимо
внешних факторов, влияющих на состояние и свойства вещества (температура и
давление), здесь появляется и внутренний фактор — состав, и связанная с ним
проблема постоянства и переменности состава, имеющая фундаментальное значе-
значение в химии. Во-вторых, при описании бинарных соединений впервые формиру-
формируются такие базисные понятия, как валентность, степень окисления, поляризация
химической связи. Здесь в отличие от простых веществ появляются гетерополяр-
ная составляющая химической связи и все эффекты, связанные с разностью
электроотрицательностей компонентов.
Исключительно важную роль играют бинарные соединения с классификаци-
классификационной точки зрения. Многие из них относятся к так называемым характеристи-
характеристическим соединениям, отражающим типичные степени окисления и их сравнитель-
сравнительную стабильность. К таким соединениям относятся прежде всего оксиды, летучие
водородные соединения, а также галогениды.
1. Классификация бинарных соединений. Бинарные соединения — это проме-
промежуточные фазы в двухкомпонентных системах, образованных простыми вещества-
веществами. Иными словами, бинарными соединениями являются продукты взаимодейст-
взаимодействия атомов различных элементов Периодической системы друг с другом. Нередко
при таком взаимодействии может образоваться несколько бинарных соединений.
Так, в системе N2 — 02 существует пять соединений: N20, NO, N2O3, N02, N2O5; в
системе Fe — Si образуются Fe3Si, Fe3Si2, FeSi, FeSi2, FeSi2+^ и т.д. Среди них
встречаются соединения как постоянного, так и переменного состава. Соединения
с координационной структурой всегда характеризуются переменностью состава,
хотя в ряде случаев область гомогенности еще экспериментально не определена.
Истинно постоянным составом обладают лишь бинарные соединения с молеку-
молекулярной структурой.
Разнообразие состава бинарных соединений обусловлено еще и тем, что
среди них широко распространены не только соединения, подчиняющиеся так
называемому правилу формальной валентности, но и соединения, не подчиня-
подчиняющиеся этому правилу. При этом под формальной (стехиометрической) валент-
валентностью понимают величину, определяемую положением элемента в Периодиче-
Периодической системе и возможными промежуточными степенями окисления. Если прави-
правило формальной валентности не соблюдается, то это однозначно свидетельствует о
наличии в структуре соединения "катион-катионных" или "анион-анионных"
связей.
Даже в таком простом молекулярном соединении, как Н2О2, несоблюдение
формальной групповой валентности кислорода объясняется наличием "анион-
анионной" связи —0~~0~. В том случае, когда правило стехиометрической ва-
валентности соблюдается, в структуре соединения присутствуют только "катион-
анионные" связи.
Разнообразие состава и структуры приводит к многообразию свойств бинар-
бинарных соединений. Среди них существуют и солеобразные, и металлоподобные, и
летучие, и тугоплавкие и т.п. Среди бинарных индивидов переменного состава
встречаются дальтониды и бертоллиды, свойства которых в пределах области
гомогенности меняются различным образом. Такая широкая вариация состава,
структуры и свойств бинарных соединений затрудняет их систематику. Класси-
Классификация и номенклатура бинарных соединений общеприняты. В их названиях
употребляется корень латинского наименования анионообразователя с окончани-
окончанием -ид, например MgCl2 — хлорид магния, TiC — карбид титана, SFg — гексафто-
рид серы и т.п.*. Так формируются классы бинарных соединений: гидриды,
оксиды, галогениды (фториды, хлориды, бромиды, иодиды), халькогениды (суль-
(сульфиды, селениды, теллуриды), пниктогениды [нитриды, фосфиды, арсениды,
антимониды (стибиды), висмутиды], карбиды, силициды, германиды, бориды.
Неуниверсальность этой классификации видна хотя бы из того, что она не вклю-
включает бинарные интерметаллические соединения. Например, соединения CuZn
Такие химические объекты называют идо-соединениями. -,
невозможно назвать ни "куприд цинка", ни "цинкид меди", так как терминоло-
терминология идо-соединений правомерна лишь в том случае, когда в роли анионообразо-
вателя выступает неметалл.
В соответствии с рекомендациями IUPAC в систематических названиях интер-
интерметаллических соединений не следует употреблять окончание -ид. Например,
соединение СизАи рекомендуется называть тримедь-золото.
С другой стороны, многие гидриды, оксиды, карбиды и т.п. обладают метал-
металлическими свойствами и относятся к металлидам. Следовательно, в этом случае
неметаллический компонент не выступает в роли анионообразователя и номенкла-
номенклатура идо-соединений становится условной. Фундаментальной характеристикой
химического соединения, определяющей все его особенности — структуру, состав
и свойства, является доминирующий тип химической связи. Только на этом
основании можно осуществить систематику бинарных соединений. По этому приз-
признаку все бинарные соединения следует подразделить на три типа: преимущест-
преимущественно ионные (солеобразные), ковалентные и металлоподобные. Следует также
различать координационные ковалентные и молекулярные ковалентные соедине-
соединения. А преимущественно ионные и металлические бинарные соединения могут
быть только координационными в силу ненасыщенного и ненаправленного харак-
характера химических связей в них.
2. Кристаллохимическое строение бинарных соединений. Систематика бинар-
бинарных соединений по характеру химической связи позволяет на основании положе-
положения компонентов в Периодической системе прогнозировать особенности кристал-
лохимического строения этих соединений. Руководящим принципом при этом
является расположение компонентов относительно границы Цинтля. Если оба
компонента располагаются слева от границы Цинтля, т.е. у обоих существует
дефицит валентных электронов, то образующиеся промежуточные фазы обладают
металлическими свойствами (исключение составляют некоторые бориды). Когда
оба компонента размещены справа от этой границы, т.е. обладают достаточным
числом валентных электронов для образования ковалентных связей, образующи-
образующиеся бинарные соединения характеризуются ковалентным типом взаимодействия.
В случае нахождения компонентов по разные стороны от границы Цинтля воз-
возможно образование соединений с различным доминирующим типом химической
связи — ионным *, ковалентным и металлическим. При этом существенную роль
играют три фактора. Во-первых, это разность электроотрицательностей. При
значительной разности ОЭО образуются ионные солеобразные соединения (на-
(например, галогениды щелочных металлов). При небольшой разности ОЭО взаимо-
взаимодействие компонентов приводит к образованию бинарных соединений с преиму-
, , Ш„У , П„У1 . „
щественно ковалентным типом связи (например, соединения А В , А В — InP,
GaAs, ZnS, CdTe и т.п.). Во-вторых, это размерный фактор, который определяет
возможность образования фаз внедрения с ковалентно-металлическим типом
связи В-третьих, это относительное содержание анионообразователя в формуль-
формульной единице соединения в системе с несколькими промежуточными фазами. Так,
например, в системах с участием переходных металлов, с одной стороны, и кисло-
кислорода, фосфора, кремния и т.п. — с другой, существует большое число промежу-
промежуточных фаз (Ti6O, Ti3O, TiO, Ti2O3, TiO2 в системе Ti~O; CO2P, CoP, CoP3 в
"Далее под термином "ионные" подразумеваются ''преимущественно ионные".
9 Я. А. Угай 257
системе Со—Р; Ni3Si, Ni5Si2, Ni2Si, Ni3Si2, NiSi, NiSi2 в системе Ni—Si). Характер
химической связи в них изменяется от преимущественно металлического до преи-
преимущественно ковалентного по мере увеличения относительного содержания неме-
неметаллического компонента.
В соответствии с преобладающим типом химической связи в бинарных соеди-
соединениях реализуются различные кристаллические структуры: плотно упакованные
ОЦК и другие для металлидов (к.ч. 8, 12 и более), менее плотно упакованные
(к.ч. 6, 8) для солеобразных ионных кристаллов и "рыхлые" структуры с невысо-
невысокими координационными числами (к.ч. < 4) для ковалентных соединений. В
последнем случае возможно также образование слоистых, цепочечных и молеку-
молекулярных кристаллических структур. Изменение типа кристаллической структуры
в зависимости от характера химической связи в бинарных соединениях можно
проследить в так называемых изоэлектронных рядах. Изоэлектронным рядом
называют последовательность соединений с одинаковым средним числом валент-
валентных электронов на атом. Наиболее известны и показательны в этом отношении
изоэлектронные ряды соединений, компоненты которых расположены симметрич-
симметрично относительно элементов IVA-группы. Четыре валентных электрона на атом
обеспечивают возникновение пространственных тетраэдрических структур с кова-
лентным типом связи у простых веществ этой группы.
Рассмотрим изоэлектронные ряды углерода и кремния. Первый из них вклю-
включает в себя С—BN—BeO—LiF, а второй — Si—А1Р — MgS~NaCl. Принцип форми-
формирования изоэлектронных рядов состоит в следующем. Возглавляет ряд простое
вещество IVA-группы (четыре электрона на атом). Остальные члены ряда — это
соединения, компоненты которых равно отстоят от IVA-группы. Число электро-
электронов у катионообразователя (ША — IA) уменьшается, а у анионообразователя (VA
— VIIA) увеличивается. При этом среднее число валентных электронов на атом в
формульной единице остается постоянным. Разность ОЭО компонентов соедине-
соединений в изоэлектронных рядах растет; следовательно, нарастает ионный вклад в
химическую связь и закономерно изменяется характер кристаллохимического
строения фаз.
Сам углерод известен главным образом в двух полиморфных модификациях:
алмаз и графит. В первой из них реализуется пространственная тетраэдрическая
структура Eр3-гибридизация), а во втором — слоистая гексагональная структура
(вр2-гибридизация) с более слабыми связями между слоями. Первый изоэлектрон-
ный аналог углерода — нитрид бора BN — также образует "алмазоподобную"
кубическую (сфалеритную) и "графитоподобную" слоистую структуры. Однако
появление некоторой доли ионности химической связи обусловливает появление
третьей полиморфной модификации BN — гексагональной структуры типа вюрт-
цита. Таким образом, в бинарных соединениях с тетраэдрической структурой и
преимущественно ковалентным типом связи вюрцитоподобная модификация
стабилизируется при наличии заметного ионного вклада. Это положение особенно
наглядно проявляется у следующего изоэлектронного аналога углерода — ВеО, в
котором стабильной модификацией является гексагональная типа вюрцита, что
обусловлено еще большей разностью ОЭО компонентов. И наконец, преоблада-
преобладающий ионный вклад в химическую связь последнего члена этого изоэлектронно-
изоэлектронного ряда — LiF — обеспечивает образование кристаллов с решеткой типа NaCl
(К.Ч. 6).
Подобная тенденция прослеживается и в изоэлектронном ряду кремния: у
самого кремния кристаллическая структура типа алмаза, а у А1Р — кубическая
(сфалерит), а уже у MgS и тем более у NaCl реализуется ионная решетка с к.ч. 6.
Различие между структурами вертикальных изоэлектронных аналогов ВеО и MgS
определяется большей ионностью связи в последнем.
В изоэлектронных рядах германия и оюлова, компоненты которых принадле-
принадлежат большим периодам, в целом наблюдаются те же закономерности. Однако в
отличие от изоэлектронных рядов в малых периодах здесь возможно разветвле-
разветвление, обусловленное тем, что в качестве катионообразователя могут выступать как
элементы главных, так и побочных подгрупп I — III групп:
, GaAs—> ZnSe —> CuBr
Ge '
ч
(ScAs) —> CaSe —> KBr
InSb —> CdTe —> Agl
^•(YSb) —> SrTe —> Rbl
Кристаллохимия арсенидов и антимонидов (стибидов) скандия и иттрия почти
не изучена. Во всех остальных случаях реализуются те же кристаллические
структуры. В рядах, где катионообразователи — rf-элементы, сохраняются тетра-
эдрические структуры. Если катионообразователи — .s-элементы I и II групп (Са,
Sr, К, Rb), то реализуются структуры типа NaCl.
Рассмотренные изоэлектронные ряды сформированы по горизонтальному
принципу — в пределах одного периода. Но для каждого члена изоэлектронного
ряда существует ряд аналогов, построенный путем "катионного" или "анионного"
замещения. Так, для А1Р при "анионном" замещении фосфора на другие пникто-
гены получаются A1N, AlAs, AlSb, а при "катионном" замещении — ВР. GaP,
InP. Таким образом и формируются изоэлектронные семейства бинарных соеди-
соединений: А В , А В и А В . Все эти соединения являются полупроводниками
или диэлектриками, что обусловлено ионно-ковалентным взаимодействием. При
этом поперечное "анионное" замещение (например, A1N —> А1Р —> AlAs —>
—> AlSb) приводит к уменьшению ионного вклада в силу уменьшения ОЭО
анионообразователя. При этом ослабляется энергия ковалентного взаимодейст-
взаимодействия, что приводит к металлизации химической связи. В связи с этим понижают-
понижаются температуры плавления и уменьшается ширина запрещенной зоны в ряду:
Соединение A1N А1Р AlAs AlSb
Гпл, °С 2200 2000 1700 1070
?,эВC00К) 3,8 2,42 2,16 1,6
Необходимо иметь в виду, что, несмотря на некоторую металлизацию связи,
преимущественным типом химической связи в кристаллах всех изоэлектронных
соединений остается ионно-ковалентный или ковалентно-ионный.
Рассмотренные изоэлектронные ряды включают соединения эквиатомного
состава (типа АВ). Все они подчиняются правилу октета (содержат восемь ва-
9* 259
лентных электронов на формульную единицу). Выполнение этого правила обеспе-
обеспечивает неметаллические свойства соответствующих соединений. Кроме того, сос-
состав всех этих соединений удовлетворяет правилу формальной валентности.
Кристаллохимические закономерности, характерные для изоэлектронных рядов,
IV II V
можно обобщить для бинарных соединений более сложного состава (АдВ , А3В2.
А В2 и т.п.). Общий принцип составления стехиометрических формул этих
соединений состоит в том, чтобы на формульную единицу приходилось восемь
или кратное восьми число валентных электронов. Например, если в формуле A1N
один атом алюминия заменить на три атома лития (сохраняется общее число
валентных электронов катионообразователя), получим соединение LiaN (число
валентных электронов на формульную единицу равно восьми). Если в том же A1N
атом анионообразователя заменить на три атома галогена, то получим А1Гз с
общим числом валентных электронов 24 C х 8). Подобным образом, если в фор-
формуле 2GaP два атома галлия заменить на три атома кадмия с тем же общим
числом валентных электронов, а два атома фосфора сохранить в формуле, то
получим Cd3P2 A6 валентных электронов, или 2 * 8). Если при этом общее число
валентных электронов пе разделить на число атомов анионообразователя JVa, то
во всех этих случаях получим 8, т.е. соблюдение правила октета:
ne/Na = 8.
(ХИЛ)
Поскольку при таком замещении разность электроотрицательностей компонен-
компонентов возрастает, соответствующие бинарные соединения обладают солеобразным
характером и структурами, свойственными преимущественно ионной связи. Одна-
Однако в этих случаях координационные числа катионо- и анионообразователей
должны быть разными — обратно пропорциональными числу атомов в формуль-
формульной единице. Например, в кристаллической решетке флюорита CaF2 (рис. 127)
к.ч. (Са) 8, а к.ч. (F) 4; в структуре 1лгО, наоборот, к.ч. (Li) 4, а к.ч. @) 8
(структура антифлюорита). В структуре A1F3 (рис. 128) к.ч. (А1) б, а к.ч. (F) 2.
Некоторые из этих соединений обладают более сложными структурами псевдомо-
псевдомолекулярного типа, когда разность ОЭО компонентов недостаточно велика для
образования ионных кристаллов.
Приведенное правило октета в форме (ХИЛ) справедливо лишь для соедине-
соединений, подчиняющихся правилу формальной валентности и обладающих неметал-
O-F т-Са
Рис. 127. Элементарная ячейка
кристалла фторида кальция
(структурный тип флюорита
• -А1 O-F
Рис. 128. Кристаллическая
структура фторида алюминия
лическими свойствами (полупроводники или диэлектрики). Его можно обобщить
на бинарные соединения, содержащие в структуре анион-анионные связи. Такие
соединения, не подчиняющиеся правилу формальной валентности, называются
анионоизбыточными. В этом случае валентная насыщенность связей, Обеспечива-
Обеспечивающая проявление неметаллических свойств, обусловлена именно возникновением
анион-анионных связей. Правило октета с учетом числа таких связей Ьа принима-
принимает вид (правило Музера — Пирсона)
Например, в соединениях CCI3P2 и CdP2 общее число валентных электронов на
формульную единицу (яе) равно соответственно 16 и 12, а число атомов анионооб-
разователя (Na) — 2 и 2. Для Сс1зР2 получаем 16/2 + ba — 8, т.е. ba = 0 (анион-
анионные связи отсутствуют). В случае CdP 12/2 + ba = 2, т.е. ba = 2 (присутст-
(присутствуют две анион-анионные связи на формульную единицу). Это правило примени-
применимо лишь для бинарных соединений э- и р-металлов, а также rf-элементов с пол-
полностью завершенной rf-оболочкой (подгруппы меди и цинка). Для переходных
металлов с дефектной rf-оболочкой это правило трудно использовать, поскольку
заранее невозможно оценить число валентных электронов металла, участвующих в
образовании связей. Кристаллохимическое строение анионоизбыточных фаз
достаточно сложно. Часто в структурах существуют слои, цепочки или изолиро-
изолированные группы из нескольких атомов анионообразователя.
Среди бинарных соединений, компоненты которых расположены по разные
стороны от границы Цинтля, особое место занимают фазы внедрения. Они обра-
образуются в системах переходных металлов с углеродом, азотом, кислородом. Сюда
же примыкают гидриды и некоторые бориды, хотя положение водорода в Перио-
Периодической системе неоднозначно, а бор расположен слева от границы Цинтля.
Определяющим фактором при образовании фаз внедрения являются не индиви-
индивидуальные химические особенности неметалла, а лишь соотношение атомных раз-
размеров (размерный фактор). Все фазы внедрения образуют плотноупакованные
структуры и обладают металлическими свойствами.
Бинарные соединения, оба компонента которых расположены справа от грани-
границы Цинтля, как отмечено выше, характеризуются преимущественно ковалентным
типом взаимодействия в силу незначительной разности 0Э0. Правило октета
здесь соблюдается, поскольку число валентных электронов у обоих компонентов
достаточно для реализации ковалентного взаимодействия. Для соединений, под-
подчиняющихся правилу формальной валентности, у катионообразователя необходи-
необходимо учитывать лишь число электронов, участвующих в образовании связи. Так, в
оксидах мышьяка As2O3 и AS2O5 у кислорода в обоих случаях учитывается шесть
электронов, а у мышьяка в первом случае три, а втором — пять электронов. Тог-
Тогда для As2O3 получим C-2 + 6-3)/3 = 8, а для As2O5 — E-2 + 6-5)/5 = 8. Если
соединения не подчиняются правилу формальной валентности, то применимо
правило Музера — Пирсона. Особенности кристаллохимического строения бинар-
бинарных соединений с компонентами, расположенными справа от границы Цинтля,
состоят в том, что в их структуре присутствуют группировки атомов в виде цепо-
цепочек, сеток и молекул. Следовательно, кроме ковалентной связи здесь реализуется
и межмолекулярное взаимодействие.
Когда оба компонента бинарного соединения располагаются слева от границы
261
Цинтля, доминирующей является металлическая связь. При этом возникают
металлиды с плотноупакованными кристаллическими структурами. Формальные
стехиометрические соотношения при этом не соблюдаются в силу коллективного
электронно-атомного взаимодействия из-за дефицита валентных электронов.
Формульный состав этих соединений определяется размерным фактором и элект-
электронной концентрацией. В этом случае правило октета не выполняется, а разнооб-
разнообразие состава при сохранении плотной упаковки атомов в кристаллических
структурах приводит к образованию соединений Курнакова, фаз Лавеса, элект-
электронных соединений Юм-Розери и т.п.
Таким образом, на основании положения компонентов бинарных соединений в
Периодической системе можно предвидеть характер химической связи, а следова-
следовательно, особенности кристаллохимического строения и свойства этих соединений.
3. Постоянство и переменность состава. Вопрос о постоянстве и переменности
состава химических соединений является одним из центральных в современной
неорганической химии. В настоящее время признано, что твердые соединения с
координационной структурой в принципе являются фазами переменного состава,
хотя ширина области гомогенности при этом может быть весьма различной. Пос-
Постоянство же состава свойственно лишь соединениям с молекулярной структурой.
Отклонения от стехиометрии, обусловленные переменностью состава, в наиболь-
наибольшей степени изучено для бинарных соединений. В связи с этим роль бинарных
соединений в химии приобретает особый смысл.
Само существование соединений постоянного и неременного состава, служит
отражением общей идеи о единстве непрерывности и дискретности при химиче-
химических превращениях. Соединения постоянного состава символизируют так называ-
называемую "привилегию дискретности" в химии, поскольку для химического взаимо-
взаимодействия характерно скачкообразное изменение состава и свойств продуктов при
некоторых определенных соотношениях компонентов. Эти соотношения регламен-
регламентируются основными стехиометрическими законами: кратных отношений, эквива-
эквивалентов и т.п. Для соединений переменного состава в пределах области гомогенно-
гомогенности соотношения компонентов изменяются непрерывно при сохранении кристал-
кристаллохимического строения фазы. В соответствии с этим непрерывно изменяются и
свойства фазы. При этом для фаз переменного состава, которые относятся
к дальтонидам в широком смысле, внутри области гомогенности существует
"предпочтительный" состав, которому отвечает сингулярная точка на диаграмме
состав — свойство. Для бертоллидных фаз, которые также обладают качественно
своеобразным кристаллохимическим строением (по этому признаку относятся к
соединениям), характерно монотонное изменение свойств в пределах области
гомогенности, что роднит их с твердыми растворами. Для бертоллидов (в отли-
отличие от дальтонидов) внутри области гомогенности ни один из составов не облада-
обладает особыми свойствами, т.е. не является предпочтительным. Таким образом,
бертоллиды представляют собой промежуточную ступень между твердыми раство-
растворами и химическими соединениями дальтонидного типа. Если учесть, что сами
твердые растворы являются промежуточной ступенью между механическими
смесями и химическими соединениями, то прослеживается взаимосвязь:
смесь —> раствор —> бертоллид —> дальтонид,
где нарастает степень химического взаимодействия и наблюдается постепенный
переход к все более высоким формам химической организации вещества.
Постоянство или переменность состава бинарных, соединений непосредственно
обусловлены характером химической связи и вытекающими отсюда особенностя-
особенностями кристаллической структуры этих соединений. Из общих соображений следует
ожидать, что в наибольшей степени переменность состава должна быть свойствен-
свойственна металлидным фазам, в которых доминирующим типом химической связи
является металлическая. Это обусловлено не только ненаправленностью и нена-
ненасыщенностью металлической связи, но и делокализацией валентных электронов.
В преимущественно ионных кристаллах области гомогенности сравнительно неве-
невелики по сравнению с металлидами, что объясняется особенностями электростати-
электростатического взаимодействия (соблюдение принципа электронейтральности). По этой
причине, несмотря на ненасыщенность и ненаправленность ионной связи, первая
координационная сфера каждого иона в соответствии с размерным фактором
насыщается практически одинаковым числом ионов противоположного знака. В
соединениях с преимущественно ковалентной связью отклонения от стехиометрии
возможны лишь для координационных структур. Однако в отличие от металлопо-
добных и солеобразных соединений, которые образуют только координационные
кристаллы, ковалентные соединения существуют и в молекулярной форме. В
последнем случае, естественно, переменность состава исключается и такие соеди-
соединения представляют собой дальтониды в узком смысле. В координационных же
ковалентных кристаллах области гомогенности невелики, что обусловлено насы-
насыщенным и направленным характером ковалентной связи.
Природа отклонений от стехиометрии в соединениях переменного состава
состоит в том, что при любых температурах, отличных от абсолютного нуля, в
реальном кристалле существуют дефекты структуры. С повышением температуры
концентрация этих дефектов возрастает в силу увеличения энтропии системы,
Наиболее упорядоченной структурой должен обладать идеальный кристалл, в
котором каждый атом занимает предназначенный ему узел в кристаллической
решетке. При этом все узлы заняты, а все междоузлия свободны. Такая структу-
структура обладает полным порядком (энтропия равна нулю) и может быть реализована
только при абсолютном нуле. При повышении температуры нарушения идеальной
структуры возможны за счет возник-
возникновения незанятых узлов в кристал-
кристаллической решетке, появления агомов
в междоузлиях или существования в
узлах решетки чужеродных атомов.
Эти типы дефектов в кристалле
являются простейшими. В реальных
случаях возможно появление комби-
комбинаций этих дефектов. Возникновение
таких дефектов в реальных кристал-
кристаллах приводит к образованию ограни-
ограниченных твердых растворов и появле-
появлению области гомогенности. Основные
типы дефектов представлены на рис.
о»о»о
•о*о»
о»о»о
•о»о*
а
O»Q*O
• ооо»
о»о»о
•о»о»
S
O»O»O O»Q»Q
о#чо»о
< >о»
о»о»о
• о» о»
д
о»о«о
*о
оЪ
•о»о»
г
Ж8
•о»о»
д
О-А т-в
Рис. 129. Точечные дефекты в реальном кристал-
кристалле бинарного соединения АВ:
а - идеальный кристалл; 6 - вакансия в подрешет-
129. Рис. 129, а представляет схему ке компонента В; в - вакансия в подрешетке ком-
идеальной кристаллической структу- понента A; i - сверхстехиометрический атом В в
рЫ бинарного соединения АВ. Рис. междоузлии; д-антиструктурное разупорядочение;
129, б, в Отражает существование е - дефект Френкеля
263
незанятых узлов в подрешетках компонентов А и В. Такие незанятые узлы назы-
называются вакансиями или дефектами Шотки. Это соответствует образованию твер-
твердого раствора вычитания по соответствующему компоненту, т.е. подрешетка одно-
одного из компонентов полностью укомплектована, а в подрешетке другого существу-
существуют вакансии. Состав соединений должен выражаться формулами ABi-^ (рис. 129,
б) или Ai-y (рис. 129, в). Это один из примеров односторонних фаз, в которых:
область гомогенности на диаграмме состояния расположена по одну сторону от
ординаты соединения. Отклонение от стехиометрии возможно и за счет внедре-
внедрения избыточных атомов одного из компонентов в междоузлия укомплектованной
(не содержащей вакансий) кристаллической решетки (рис. 129, г). При этом
образуются твердые растворы внедрения, а состав фазы описывается формулой
Ai+IB (или ABj+y). Такие фазы также могут быть только односторонними, а об-
область гомогенности — ограниченной.
Образование дефектов не всегда может приводить к нарушению стехиометрии
На рис. 129, д приведен пример так называемого антиструктурного разупорядоче-
ния, при котором атомы А и В попадают в узлы "чужой" подрешетки. Это воз-
возможно в том случае, если компоненты соединения близки между собой по метал-
лохимическим свойствам. Кристаллы с антиструктурными дефектами представля-
представляют собой твердые растворы взаимозамещения. Комбинации простых дефектов
могут привести к взаимной компенсации, в результате чего нарушений стехиомет-
стехиометрии может и не быть. Так, смещение атома из его равновесного положения в
междоузлие (рис. 129, е) приводит к появлению комбинированного дефекта ва-
вакансия — атом в междоузлии. Такая комбинация называется дефектом Френкеля.
Очевидно, что при этом изменение состава не наблюдается. В принципе такая же
ситуация возможна и при возникновении равного количества вакансий в обеих
подрешетках бинарного соединения. Однако в реальном случае одинаковые кон-
концентрации вакансий в обеих подрешетках невозможны, поскольку компоненты
различаются по свойствам. В этом случае валовое отклонение от стехиометрии
определяется разностью концентраций вакансий в двух подрешетках и определя-
определяемая экспериментально область гомогенности будет меньше той, которая следует
из действительной концентрации вакансий.
Существование двусторонних фаз возможно потому, что в зависимости от
внешних условий наблюдается преимущественная генерация вакансий, антиструк-
антиструктурных дефектов или междоузельных атомов либо одной, либо другой подрешет-
подрешетки. Здесь речь идет о так называемых собственных дефектах, обусловленных
различным положением атомов компонентов, а не примесных дефектах, которые
определяются чужеродными посторонними атомами.
В тесной связи с постоянством и переменностью состава находятся и свойства
химических соединений. Очевидно, что для соединений постоянного состава
(дальтониды в узком смысле) свойства являются константами. Для соединений
переменного состава наблюдается зависимость свойств от соотношения компонен-
компонентов в пределах области гомогенности. Если фаза односторонняя, а стехиометриче-
ский состав не достигается, то в соответствии с законами Курнакова для твердых
растворов должно наблюдаться плавное изменение свойств в пределах области
гомогенности. Такие фазы всегда обладают бертоллидным характером. Если
односторонняя фаза включает в качестве предельного стехиометрический состав,
то сделать вывод о принадлежности фазы к бертоллидам или дальтонидам не
представляется возможным. Для двусторонних фаз возможны два принципиально
различных варианта. При дальтонидном характере фазы (дальтонид в широком
смысле) внутри области гомогенности существует предпочтительный состав, кото-
которому отвечает экстремальное значение свойств. Важно подчеркнуть, что в этом
случае экстремальное значение различных свойств соответствует одному и тому
же составу (инвариантность). Когда двусторонняя фаза обладает бертоллидным
характером, внутри области гомогенности наблюдается плавное изменение
свойств, т.е. ни один состав не обладает особыми свойствами.
4. Оксиды. Оксиды занимают особое положение среди всех бинарных соедине-
соединений. Еще Д.И.Менделеев относил "высшие солеобразующие окислы" к характе-
характеристическим соединениям. Состав высшего оксида давал возможность определить
групповую принадлежность элемента. Свойства оксидов позволяли характеризо-
характеризовать сам элемент как металл или неметалл. Кроме того, с учетом кислотно-основ-
кислотно-основных свойств оксидов делались выводы о характере соответствующих гидроксидов,
а также о составе и свойствах соответствующих солей. На первом этапе становле-
становления и развития Периодического закона роль оксидов как характеристических
соединений была исключительно велика. С развитием теории строения атома и в
результате выявления физического смысла Периодического закона, казалось бы,
роль характеристических соединений утрачивается. Но периодически изменяются
не только свойства элементов, но также формы и свойства их соединений. Поэто-
Поэтому для описания химического облика элементов характеристические соединения
по-прежнему играют исключительно важную роль.
Кислород по ОЭО уступает лишь единственному элементу — фтору, а потому в
бинарных соединениях со всеми остальными элементами он выступает в качестве
анионообразователя. Соединения элементов (кроме фтора) с кислородом, которые
подчиняются правилу формальной валентности, называются оксидами. Во всех
таких соединениях кислород имеет степень окисления —2. Кроме оксидов, соот-
соответствующих высшим степеням окисления элементов, сюда относятся и оксиды,
отвечающие промежуточным степеням окисления (например, SnO, SO2 и др.).
Помимо нормальных оксидов существуют и другие соединения элементов с кис-
кислородом: пероксиды (перекиси), в которых степень окисления кислорода равна
—1 (например, Н2О2 — пероксид водорода); супероксиды (надперекиси) со сте-
степенью окисления кислорода —!/2 (КО2 — супероксид калия); озониды, где сте-
степень окисления кислорода равна —1/з (КОз — озонид калия). Все эти индивиды
не подчиняются правилу формальной валентности, являясь анионоизбыточными
соединениями, следовательно, в их структуре присутствуют связи кислород —
кислород. Вместе с тем существуют кислородные соединения, главным образом
переходных металлов, которые являются катионоизбыточными и также не подчи-
подчиняются правилу формальной валентности (например, TigO и Т1зО). По традиции
их также называют оксидами, хотя кислород здесь не выступает в роли анионо-
анионообразователя. Подобные соединения относятся к металлидам, поскольку в них
доминирует металлическая связь, и говорить о степени окисления кислорода в
этом случае не имеет смысла. Известна небольшая группа несолеобразующих
оксидов (СО, N2O, NO), которые не относятся к характеристическим соединени-
соединениям, поскольку им не соответствуют гидроксиды. В соединениях фтора с кислоро-
кислородом анйонообразователем выступает более электроотрицательный фтор. Поэтому,
например 0F2 — это не оксид фтора, а фторид кислорода, в котором кислород
имеет степень окисления +2.
О-о •si
Рис. 130. Кристаллическая струк-
структура кубической модификации SiC>2
(/?-кристобалит)
О-о •-п
Рис. 131. Кристаллическая структура тетра-
тетрагональной модификации TiC>2 (структурный
тип рутила)
Систематизируя кислородные соединения элементов по доминирующему типу
химической связи, можно выделить три основных типа соединений: с металличе-
металлической, преимущественно ионной и ковалентной связью. К характеристическим
соединениям относятся только оксиды, подчиняющиеся правилу формальной
валентности. В характеристических оксидах доминирующим типом связи являет-
является ионно-ковалентная, поэтому их можно подразделить на два типа: с преимуще-
преимущественно ионной и преимущественно ковалентной связью. Последние, в свою оче-
очередь, по структурному признаку подразделяются на координационные и молеку-
молекулярные (например, ЯЮг и СОг). Ионные оксиды всегда имеют координационную
структуру. Ионно-ковалентное взаимодействие характерно и для анионоизбыточ-
ных кислородных соединений, однако они обладают особыми свойствами и обыч-
обычно рассматриваются отдельно. Такую же специфическую группу составляют и
металлоподобные оксиды. Принимая во внимание зависимость типа кристалличе-
кристаллической структуры оксидов от характера химической связи, можно сделать вывод,
что в немолекулярных структурах с ковалентной связью координационные числа
не должны превышать 4, а в ионных кристаллических решетках реализуются
более высокие координационные числа. Так, в кубической структуре SiCh {Р~
кристобалит) к.ч. (Si) 4, а к.ч. (О) 2 (рис. 130), в структуре TiO2 (рутил) к.ч. (Ti)
6, а к.ч. (О) 3 (рис. 131), т.е. здесь уже реализуется преимущественно ионный тип
взаимодействия. А в еще более ионном соединении MgO (структурный тип NaCi)
к.ч. (Mg, О) 6.
Рассматривая структуры оксидов неметаллов, обратим внимание на то, что
для реализации координационной структуры при сохранении преимущественно
ковалентного взаимодействия необходима заметная доля ионности связи. В про1-
тивном случае образуются молекулярные структуры. Так, сравнивая между собой
структуры высших оксидов углерода и кремния, отметим, что СОг обладает моле-
молекулярной структурой, a SiCb — координационной структурой ковалентного типа.
Это обусловлено возрастанием разности ОЭО элементов в оксидах при переходе
от углерода к кремнию. Остальные высшие оксиды элементов IVA-группы (GeO2,
ЭпОг, РЬОг) кристаллизуются в структурном типе рутила ТЮг, свойственном бо-
более ионным соединениям. При переходе к оксидам элементов VA — VHA-rpynn
наблюдается увеличение числа молекулярных структур и уменьшение числа ко-
ординационных, причем первые более характерны для легких элементов, а вто-
вторые для тяжелых. Так, в VA-группе для оксидов азота (от N2O до N2O5),
фосфора (Р2Оз и Р2О5) и мышьяка (As2O3 и As2Os) характерны молекулярные
структуры,- для оксида сурьмы — псевдомолекулярная цепная структура ромбиче-
ромбической сингонии, а для Bi2O3 — координационная с к.ч. (Bi) 6 и к.ч. (О) 4. В VTA-
группе оба оксида серы (SO2, SO3) молекулярны. У селена низший оксид SeO2
обладает псевдомолекулярной, а высший БЮз — молекулярной структурой. Низ-
Низший оксид теллура ТеО2 имеет уже координационную структуру (типа рутила
TiO2), а ТеОз — молекулярную. Наконец, все оксиды элементов VIIA-группы (С1,
Вг, I) имеют молекулярные кристаллические структуры.
Все характеристические оксиды, как известно, относятся к основным и кис-
кислотным. Первые являются оксидами металлов, вторые генетически связаны с
неметаллами. Поскольку нет четкой границы между металлами и неметаллами,
существует большая группа амфотерных оксидов. Амфотерность определяется не
только положением элемента в Периодической системе, но и зависит от его степе-
степени окисления. Ориентируясь на разность ОЭО, можно утверждать, что оксиды
металлов должны быть преимущественно ионными, а оксиды неметаллов — пре-
преимущественно ковалентными. Поскольку для одного и того же элемента с увели-
увеличением степени окисления его электроотрицательность растет, то в этом направ-
направлении — от низших к высшим оксидам — растет ковалентный вклад. Вследствие
этого наблюдается изменение свойств оксидов от основных к кислотным, напри-
например ОЭО (Сг2+) = 1,4, ОЭО (СгЗ*) = 1,6, ОЭО (Сг6+) = 2,4, и свойства оксидов
закономерно изменяются:
СгО Сг2О3 СгО3
основный амфотерный кислотный
, Деление оксидов на основные и кислотные базируется на их собственном
отношении к кислотам и щелочам, а также на свойствах соответствующих им
гидроксидов. Большая группа оксидов по этим признакам относится к амфотер-
ным. Элементы, образующие амфотерные оксиды, характеризуются средними
значениями ОЭО в пределах 1,4 — 1,8 и степенями окисления (+2) — D-4). Если
при степени окисления 4-2, 4-3 электроотрицательность менее 1,4, то оксиды (и
отвечающие им гидроксиды) обладают основными свойствами. Так, ОЭО [СаD-2)]
составляет 1,0, ОЭО лантаноидов [Ln (+3)] равна 1,2 — 1,3. Бели при степени
окисления 4-4 электроотрицательность элемента больше 1,8, оксид обладает
кислотными свойствами. Например, ОЭО СD-4), SiD-4), GeD-4) равны соот-
соответственно 2,6, 1,9 и 2,0. Если электроотрицательность элемента находится в
пределах 1,4 — 1,8 или даже несколько превышает этот интервал, а степень окис-
окисления +1, оксид принадлежит к основным (у Ag+ ОЭО 1,9). Когда же степени
окисления элементов превышают 4-4 и значения ОЭО высоки, соответствующие
оксиды кислотные.
По отношению к воде характеристические оксиды ведут себя различным
образом и по этому признаку их можно подразделить на четыре группы. Доволь-
Довольно редки оксиды, растворяющиеся в воде без заметного химического взаимодей-
взаимодействия (высшие, оксиды рутения и осмия): Большинство оксидов химически не
взаимодействует с водой и не растворяется в ней — соответствующие гидроксиды
получаются лишь косвенным путем (в частности, амфотерные оксиды А12О3,
Сг2Оз, Fe20j, ZnO и т.п.). Взаимодействующие с водой оксиды составляют две
группы: одни при взаимодействии образуют растворимые в воде гидроксиды
основного или кислотного характера (оксиды бора, углерода, азота, фосфора,
серы, щелочных и щелочно-земельных металлов), а вторые — нерастворимые в
воде гидроксиды (оксиды бериллия, магния, редкоземельных элементов) основно-
основного характера. Учитывая, что сама вода является идеальным амфолитом, индиф-
индифферентность оксидов по отношению к ней вовсе не связана с их индифферент-
индифферентностью по отношению к кислотам и щелочам. Все кислотные оксиды, независимо
от их отношения к воде, реагируют со щелочами, а все основные с кислотами.
Так, нерастворимые в воде СиО и S1O2 хорошо взаимодействуют с кислотами и
щелочами соответственно. В то же время амфотерные оксиды, как правило, ус-
устойчивы не только по отношению к воде, но и к кислотам и щелочам. Их двойст-
двойственная природа проявляется в свойствах соответствующих гидроксидов, которые
более реакционноспособны.
5. Водородные соединения. Водород занимает в Периодической системе особое
место. Двойственная роль водорода обусловлена тем, что, с одной стороны, у него
на валентном уровне находится единственный электрон (как у щелочных метал-
металлов), а с другой стороны, в силу специфики 1-го периода ему недостает всего
одного электрона до устойчивой электронной оболочки благородного газа (как у
галогенов). По значению ОЭО B,1) он занимает среднее положение среди элемен-
элементов [ОЭО (F) = 3,9, ОЭО (Cs) = 0,7]. Поэтому с менее электроотрицательными
элементами он выступает в роли анионообразователя, а с более электроотрица-
электроотрицательными является катионообразователем. С учетом общих правил номенклатуры
бинарных соединений к гидридам относятся только соединения водорода, в
которых он отрицательно поляризован, т.е. в основном его соединения с металла-
металлами. Соединения водорода с неметаллами с этой точки зрения не являются гидри-
гидридами. Так, существуют галогениды водорода (НС1, НВг и т.п.), халькогениды
водорода (H2S, НгБе, НгТе), пниктогениды водорода (NH3, РЩ). Имея в виду,
что в формулах бинарных соединений более электроотрицательный компонент
пишется на втором месте, формулу аммака NH3 иногда записывают в виде H^N
(нитрид водорода). Однако в силу сложившихся традиций этот номенклатурный
принцип не всегда соблюдается. Так, с точки зрения формальной номенклатуры
метан СН4 представляет собой карбид водорода [ОЭО (С) = 2,6] и его формулу
следовало бы записывать в виде Н4С. Но уже для силана SiH4 [ОЭО (Si) = 1,9]
номенклатурная формула оказывается верной. Подобное же положение характер-
характерно для элементов подгруппы мышьяка: AsH3, SbH3, ВШ3 — это гидриды [ОЭО
(As) = 2,0, ОЭО (Sb) = 1,8, ОЭО (Bi) = 1,8]. Такая неоднозначность в номенкла-
номенклатуре обусловлена небольшими различиями электроотрицательностей компонентов
в рассматриваемых случаях.
Водородные соединения наряду с кислородными играют особую роль в хими-
химической характеристике элементов. Д.И.Менделеев кроме "высших солеобразу-
ющих окислов" к характеристическим относил и "летучие водородные соедине-
соединения". Оказалось, что сумма валентности элементов по водороду и высшей валент- .
ности по кислороду всегда равна 8. Это положение было и остается справедли-
справедливым для элементов, расположенных справа от границы Цинтля. Именно для этих
элементов характерно образование летучих (главным образом газообразных)
водородных соединений.
Следуя принятой систематике на основании преимущественного типа химиче-
ской связи, все бинарные водородные соединения можно разделить на три основ-
основных класса: солеобразные (ионные), металлоподобные и летучие (ковалентные).
Солеобразные гидриды образуются при непосредственном соединении с водоро-
водородом щелочных и щелочно-земельных металлов. Водород в солеобразных гидри-
гидридах формально функционирует как галогены, однако связь здесь имеет менее
ионный характер. Тем не менее гидриды щелочных металлов образуют кристал-
кристаллические структуры типа NaCl, а гидриды щелочно-земельных металлов — более
сложные слоистые структуры. Состав солеобразных гидридов отвечает правилам
формальной валентности, причем водород здесь имеет степень окисления —1.
Характерной особенностью солеобразных гидридов в отличие от галогенидов
является способность энергично взаимодействовать с водой с выделением водо-
водорода:
эн + н2о = эон + н2
ЭН2 + 2Н2О = Э(ОН2) + 2Н2
Это указывает на нестабильность степени окисления —1 для водорода, в силу
чего Н" обладает значительной восстановительной активностью (значительно
большей, чем у I") и способен восстанавливать Н+ из воды (как активные ме-
металлы):
Н' (из гидрида) + Н+ (из воды) = Н°
При нагревании все солеобразные гидриды (кроме LiH) начинают диссоцииро-
диссоциировать с отщеплением водорода до достижения температуры плавления. Выделя-
Выделяющийся при этом водород способен частично растворяться в расплавленном
металле.
Большая группа элементов (многие переходные металлы) образует гидриды с
преимущественно металлическим характером связи. Все они являются фазами
внедрения. Состав большинства металлоподобных гидридов отвечает формулам
ЭН, ЭН2. Иногда встречаются и гидриды состава ЭН3. Соотношение элементов в
формульных единицах не зависит от природы металла, правило формальной
валентности здесь не соблюдается, а состав определяется общими закономернос-
закономерностями образования фаз внедрения. Водород способен внедряться не только в
октаэдрические пустоты плотноупакованных структур, что отвечает составу АВ,
но и в тетраэдрические (состав АВ2). Бели же атомы водорода занимают и окта-
октаэдрические, и тетраэдрические пустоты, реализуется состав АВ3. Поскольку в
реальных условиях водород может занимать лишь часть пустот соответствующего
типа, указанные составы являются предельными и возможно отклонение от них в
сторону недостатка водорода. Поэтому все металлоподобные гидриды являются
односторонними фазами переменного состава 3Hi-x, ЭН2-Ж, ЭНз-^. Переходные
металлы 4-го периода с кайносимметричной 3 d-оболочкой, во-первых, растворяют
водород, а во-вторых, образуют фазы внедрения. При этом первая четверка 3d-
металлов (Ti — Mn, взаимодействие скандия с водородом не изучено) хорошо
растворяет водород в твердом состоянии, но образуют лишь по одному гидриду.
Металлы VIIIB-группы (Fe, Co, Ni), напротив, плохо растворяют водород, но
образуют по нескольку гидридов. Взаимодействие с водородом первых пяти
элементов 5-го и б-го периодов подчиняется тем же закономерностям — образова-
образование ограниченных твердых растворов и гидридов. Исключением является молиб-
ден, который с водородом не взаимодействует. Платиновые металлы, за исключе-
исключением палладия D<Р°55°) и платины (Б^бв1), гидридов не образуют, растворимость
водорода в них, как правило, мала.
Металлы подгрупп меди и цинка с водородом практически не взаимодейству-
взаимодействуют, хотя имеются указания на незначительную растворимость водорода в меди и
серебре и на существование м&яостабильного гидрида СиН. Таким образом выяв-
выявляется общая закономерность, согласно которой повышенная растворимость водо-
водорода и способность к образованию металлоподобных фаз внедрения наблюдается
у rf-элементов с сильно дефектными rf-оболочками. А элементы конца вставных
декад обладают малым сродством к водороду. Это объясняется повышенной
возможностью обобществления электрона внедренного атома водорода в случае,
когда не все электронные уровни в соответствующей энергетической зоне запол-
заполнены.
Бинарные водородные соединения с преимущественно ковалектным типом
связи образз'ют элементы 1VA — VlIA-групп. Именно эти соединения относятся к
характеристическим. В них водород выступает, как правило, в качестве катионо-
образователя. Считается, что соответствующие элементы имеют отрицательную
степень окисления по отношению к водороду (ЭН4, ЭНд, ЭНг, ЭН), хотя в ряде
случаев это не согласуется с принятыми значениями ОЭО. Особенно это заметно
у элементов IVA- VA-групп. Так, степень окисления кремния, германия, олова в
SiH4, GeH4, SnH4 равна —4, хотя их злектроотрицательности составляют соответ-
соответственно 1,9; 2,0; 1,8. Фосфор, мышьяк, сурьма и висмут в PHg, AsHg, SbH3, ВШЙ
имеют степень окисления —3 (ОЭО составляют 2,1; 2,0; 1,8; 1,8). Термическая
устойчивость летучих водородных соединений уменьшается в группах сверху вниз
вплоть до того, что PbH4, P0H2 и AtH не получены, a BiH3 разлагается в момент
выделения. Полярность связей в молекулах летучих гидридов возрастает при
переходе от элементов IVA-группы к элементам VlIA-группы и, естественно,
уменьшается в пределах каждой группы сверху вниз. С изменением прочности и
полярности связей в летучих водородных соединениях соответственно изменяется
их отношение к воде, а также кислотно-основные свойства образующихся продук-
продуктов. Чем менее прочна или более полярна связь, тем легче происходит электроли-
электролитическая ионизация по этой связи в водных растворах. Преимущественно кова-
лентные водородные соединения элементов IVA-группы плохо растворимы в воде
(но хорошо растворимы в неполярных органических растворителях). В растворе
они медленно гидролизуются с образованием гидроксокомплексов и выделением
водорода. Соединения элементов VA-группы хорошо растворимы в воде и про-
продукты взаимодействия диссоциированы по основному типу. Более полярные H2S,
H2Se и НгТе при растворении в воде диссоциируют уже по кислотному типу.
Галогеноводороды, как более полярные соединения, еще лучше растворимы в
воде, причем их водные растворы уже являются сильными кислотами (кроме
HF).
Водородные соединения первых элементов VA — VIIA-групп (NH3, H2O, HF)
заметно отличаются по целому ряду свойств от своих более тяжелых аналогов.
Это обусловлено их сильной ассоциацией за счет образования водородных связей.
Их температуры плавления, кипения, значения констант ионизации в водных
растворах определяются именно этим обстоятельством. Вода является идеальным
амфолитом, и слева направо в ряду NH3—H2O—"HF закономерно изменяется ха-
характер диссоциации:
NH3 + H20 «=± NHj + OH-
H2O + Н2О *=* Н3О+ + ОН-
Н2О + HF ^НзО + F"
Из двух связей N—Н и Н~О более полярна последняя, поэтому неподеленная
электронная пара азота легче атакует положительно поляризованный атом водо-
водорода в воде, образуя донорно-акцепторную связь. В случае HF связь водорода с
фтором более полярна, чем связь О—Н, поэтому здесь атаке подвергается протон
в HF.
Несколько отличается от остальных водородных соединений группа так назы-
называемых полимерных гидридов. К ним относятся гидриды бериллия, магния,
алюминия (BeH2)j;, (MgH2)x, (A1H3)X. Это твердые вещества, термически распа-
распадающиеся на элементы соответственно при 100, 300 и 100°С. Близки к ним по
свойствам гидриды меди, серебра, цинка и кадмия, а также твердые гидриды
фосфора (РН)^. Гидриды бора В2Н6 и галлия Ga2He представляют собой летучие
димеры, в обычных условиях газообразные или жидкие.
Таким образом, намечается постепенный переход от металлоподобных гидри-
гидридов через гидриды меди, цинка и их аналогов к полимерным гидридам, а от
последних, в свою очередь, через летучие димерные гидриды бора и галлия к
летучим характеристическим водородным соединениям. В то же время полимер-
полимерные гидриды бериллия, магния и алюминия генетически связаны и с солеобраз-
ными гидридами щелочных и щелочно-земельных металлов.
6. Галогениды. Классификация галогенидов с позиций особенностей химичес-
химической связи в них проще, чем в предыдущих случаях, поскольку ни один из гало-
галогенов ни с одним из элементов Периодической системы не образует металлопо-
металлоподобных фаз. По отношению к галогенам все элементы выступают в качестве ка-
тионообразователей. Поэтому максимальное число атомов галогена на формуль-
формульную единицу часто соответствует номеру группы, в которой расположен элемент,
т.е. максимальной валентности последнего. Это безусловно справедливо для боль-
большинства s- и /j-элементов, а также наблюдается для ряда d- и /-металлов. Особен-
Особенно характерно это положение для фторидов, поскольку фтор — наиболее электро-
электроотрицательный элемент и наиболее энергичный окислитель. В ряду F — С1 —
— Вг — I максимальная валентность элемента по галогену уменьшается. Так,
существует гексафторид серы SFg, но не существует гексахлорид, существуют
PF5, РС15, но лишь РВга и Р!3.
Для галогенов характерно образование соединений, отвечающих промежуточ-
промежуточным степеням окисления элементов. При этом повышение температуры, как пра-
правило, способствует термической диссоциации высших галогенидов, что приводит
к понижению степени окисления катионообразователя. Например, хлориды пла-
платины в зависимости от температуры существуют в следующих формах (под давле-
давлением пара галогена 1,013-105 Па):
PtCl3 ^L^ PtCl2 ЪЖ±> PtCl
Если элемент способен проявлять переменные степени окисления, все эти степе-
степени окисления могут быть реализованы в галогенидах в зависимости от условий
271
(температура, давление пара галогена и его окислительная активность). Таким
образом, галогениды также следует отнести к характеристическим соединениям
элементов, поскольку в них четко прослеживается взаимосвязь между положе-
положением элемента в Периодической системе, характерными для него степенями окис-
окисления и их сравнительной устойчивостью в различных условиях. В известном
смысле галогениды более универсальны и дают больше информации о химиче-
химической природе элемента, чем оксиды и водородные соединения, так как все они
подчиняются правилу формальной валентности. Среди них полностью отсутст-
отсутствуют анионоизбыточные фазы *, а немногочисленные катионоизбыточные соеди-
соединения (например, каломель Hg2Cl2) при ближайшем рассмотрении оказываются
координационными кристаллами солеобразного типа с существенно ионной
связью.
.Структуры галогенидов можно подразделить на два основных типа: ионные и
ковалентные, а последние, в свою очередь, на координационные и псевдомолеку-
псевдомолекулярные — цепные, слоистые. Условной границей между этими двумя типами
структур является диагональная граница между металлами и неметаллами. Ион-
Ионные структуры, как правило, реализуются, если степень окисления элемента не
превышает +2. При этом элемент должен быть расположен слева от диагональной
границы. В этом случае типичными являются структуры типа NaCl, CsCl, CaF2.
Когда разность электроотрицательностей недостаточна для возникновения ион-
ионных структур, наблюдаются координационные структуры сфалерита (CuBr, Cul),
вюрцита (например, Agl) и рутила (например, Z11F2). реже слоистые (HgBr2,
Hg^). Если степень окисления +1 или +2 существует для неметаллического
элемента, галогениды образуют молекулярные структуры (S2CI2, SCI2). При более
высоких степенях окисления катионообразователя существуют псевдомолекуляр-
псевдомолекулярные и молекулярные структуры.
Ионные кристаллы галогенидов обнаруживают тенденцию к уменьшению
температур плавления при переходе от фторидов к иодидам, что связано с умень-
уменьшением прочности связи за счет увеличения радиуса анионообразователя:
Соединение
*пл> С
NaF
995
NaCl
800
NaBr
750
Nal
662
Для молекулярных галогенидов характерна обратная закономерность, т.е.
повышение температур плавления (и кипения). Это объясняется усилением меж-
межмолекулярного взаимодействия вследствие роста поляризуемости анионообразова-
анионообразователя в ряду F — С1 — Вг — I. При этом сами температуры плавления и кипения
существенно ниже, чем у ионных галогенидов:
Соединение SiF4 S1CI4 SiBr4 Sil4
^, "С -90 (под -68 +5 +122
давл.)
<к„п> °С -95 +57 +153 +290
По отношению к воде галогениды можно подразделить- на две группы. К пер-
первой относятся преимущественно ионные галогениды, которые хорошо дисеоци-
"Известный К1з является комплексом К+р'Aг)].
272
ируют в водных растворах и не подвергаются заметному гидролизу, хотя по
растворимости они могут сильно различаться (например, NaCl и AgCl). Ко вто-
второй группе относятся преимущественно ковалентные галогениды неметаллов и
некоторых переходных металлов в высших степенях окисления, в сильной степе-
степени склонные к гидролизу. Многие из них настолько энергично реагируют с во-
водой, что реакции носят взрывной характер. Например, активно разлагается водой
ВВг3:
ВВг3 + ЗН2О = ЗНВг + Н3ВО3
Такие галогениды дымят на воздухе вследствие интенсивного гидролиза в паро-
паровой фазе за счет взаимодействия с тем небольшим количеством водяного пара,
которое обычно содержится в воздухе. Высшие галогениды наиболее типичных
неметаллов (углерод, азот, сера) малорастворимы в воде и практически не гидро-
лизуются (CCI4, SFe, NI3 и т.п.). Существует сравнительно небольшая группа га-
галогенидов (HgCl2, Cdl2, PtCl4, АиС1з и т.п.), которые, растворяясь в воде, прак-
практически не диссоциируют и вследствие этого не подвергаются гидролизу, хотя
формально их можно рассматривать как соли слабого основания и сильной кис-
кислоты.
Галогениды водорода рассмотрены выше как характеристические водородные
соединения. Солеобразные галогениды являются солями соответствующих галоге-
новодородных кислот. Так как окислительная активность галогенов уменьшается
от фтора к иоду, то в том же направлении увеличивается восстановительная
активность галогенид-ионов Г". Изменение восстановительных свойств этих ионов
видно из сопоставления следующих реакций:
NaF(K) + H2SO4(koh4) = NaHSO4 + HFf
NaCl(K) + H2SO4(koh4) = NaHSO4 + HC1|
2NaBr(K) + 2Н28О4(конц) = Na2SO4 + SO2f + Br2 + 2H2O
8NaI(K) + 5H2SO4(koh4) = 4Na2SO4 + H2Sf + 4I2 + 4H2O
Если F" и СГ не проявляют в данных условиях восстановительных свойств, то Вг~
восстанавливает H2SO4 до SO2, а Г — даже до H2S.
Большинство галогенидов (кроме галогенидов типичных неметаллов) склонны
к реакциям комплексообразования. Галогенсодержащие ацидокомплексы харак-
характерны не только для переходных металлов, но и для ряда *- и р-элементов. Так,
фторидные комплексы очень характерны для бериллия, алюминия и кремния:
Me2[BeF4], MejAlFe], Me2[SiFe]. Обычно устойчивость галогенидных ацидокомп-
лексов уменьшается в ряду F — С1 — Вг — I. Если же комплексообразователь об-
обладает значительной деформируемостью, может наблюдаться обратная картина.
Так, для HgF2 комплексные соединения неизвестны, в то время как для осталь-
ных галогенидов ртути комплексы типа Me2[HgF4] существуют, например
K2[HgI4].
7. Халькогениды. Сера, селен и теллур, образующие группу халькогенов, по
электроотрицательности уступают галогенам и кислороду. В то же время они
273
7 \
являются групповыми и типовыми аналогами кислорода, хотя последний и обла-
обладает особыми свойствами из-за наличия кайносимметричных 2р-электронов. По
этой причине кислород обладает значительно большей электроотрицательностью
C,5), чем его аналоги (S — 2,6; Se — 2,4; Те — 2,1). Следовательно, химическая
связь в халькогенидах должна иметь менее полярный характер , чем в оксидах и
галогенидах. Халькогенидами называют соединения серы, селена и теллура с
менее электроотрицательными элементами. С этой точки зрения, H2S, HjSe и
Н2Те также являются халькогенидами, которые были рассмотрены выше как
характеристические водородные соединения.
Несмотря на значительно меньшую электроотрицательность, сера в соединени-
соединениях часто выступает как "конкурент" кислорода. Это наглядно видно в сложных
соединениях (например, существуют кислоты НтЭОи и тиокислоты HOT3Sn, спир-
спирты R—ОН и тиоспирты R—SH и т.п.), но проявляется и в бинарных соединениях
(С02 и CS2, C112O и Cu2S и т.п.). Для более тяжелых аналогов — селена и теллу-
теллура — эта тенденция менее характерна. Другой особенностью халькогенов, отли-
отличающей их от кислорода, является повышенная склонность к образованию гомо-
атомных цепей. Если для кислорода характерны лишь двухатомные цепочки
—О О—, то у халькогенов, особенно у серы, устойчивы более длинные цепи.
Эта особенность реализуется в полисульфидах, полиселенидах и полителлури-
дах, хотя устойчивость таких связей и разнообразие нолисоединений в ряду
S~Se — Те уменьшается. Отсюда следует, что для халькогенидов должны быть
характерными анионоизбыточные фазы, не подчиняющиеся прааилу формальной
валентности. По аналогии с кислородом возможно существование и катионоизбы-
точных фаз, особенно среди халькогенидов переходных металлов.
По характеру химической связи халькогениды (как и оксиды) подразделяют-
подразделяются на ионные, ковалентные и металлические. Преимущественно ионным характе-
характером обладают лишь халькогениды наиболее активных металлов, подчиняющиеся
правилу формальной валентности. Так, все халькогениды щелочных металлов и
лития состава Ме2Х имеют кристаллические структуры антифлюоритного типа,
халькогениды кальция, стронция, бария, а также сульфид и селенид магния
состава Me X обладают структурами типа NaCl. В то же время теллурид магния
уже кристаллизуется в структурном типе вюрцита, а еще менее ионные халькоге-
халькогениды бериллия (BeS, BeSe, BeTe) образуют структуры типа сфалерита.
Наименее электроотрицательные элементы IA- и НА-групп склонны к образо-
образованию полихалькогенидов. Так, среди элементов IA-группы литий образует толь-
только нормально-валентные халькогениды, натрий помимо них ещё и Na2Sn и Na2Sen
(п = 2 — 9 для серы и 2 — 6 для селена), а калий — K2Sn, K2Sen и К2Те„ (п =
= 2 — 6,2 — 5и2 — 3 соответственно). Среди элементов ПА-группы лишь строн-
стронций и барий образуют полисульфиды SrSn и ВаБг-з (но не селениды и теллури-
ды). Халькогениды р-элементов имеют более разнообразный состав. В большинст-
большинстве случаев в них наблюдается преимущественно ковалентное взаимодействие. При
этом для халькогенидов легких р-элементов, а также неметаллов VA-группы реа-
реализуются молекулярно-ковалентные структуры. Так, B2S3l B2Se3 — легкоплавкие
твердые вещества, CS2 и CSe2 — летучие жидкости. Молекулярные кристаллы
образуют и халькогениды фосфора P4S3, P4S7, P4S10 (в паровой фазе P2S5), P4Se3-
Псевдомолекулярным строением обладают халькогениды мышьяка, сурьмы и вис-
висмута (слоистые и цепные структуры). При этом существуют и соединения, подчи-
274
няющиеся правилу формальной валентности (AS2X3, As2Ss, 8Ь2Хз), и катионо-
избыточные фазы (AsS, BiSe, BiTe) с цепочками из атомов пниктогена. Менее
электроотрицательные р-элементы ША- и IVA-групп образуют координационно-
ковалентные структуры. Для алюминия характерны только нормально-валентные
соединения А12Хз, а для галлия, индия и таллия помимо них еще и эквиатомные
соединения состава ЭХ. Кремний, германий и олово образуют монохалькогениды
ЭХ и дихалькогениды ЭХ2. Эти соединения обладают координационными струк-
структурами, причем у кремния они имеют выраженную слоистость. Наконец, халько-
гениды свинца имеют лишь состав РЬХ и кристаллизуются в структурном типе
NaCl, что свидетельствует об увеличении ионного вклада в химическую связь.
Для переходных металлов характерно большое разнообразие халькогенидов
различного состава. Однако при этом можно выделить ряд общих закономернос-
закономерностей. Во-первых, в большинстве случаев разнообразие соединений уменьшается от
сульфидов к теллур идам. Так, для молибдена известны 5 сульфидов, 4 селенида
и 3 теллурида, для никеля — б сульфидов (Ni3S2, Ni6S5, Ni7S6, NiS, Ni3S4, NiS2), 3
селенида (№з8е2, NiSe, NiSe2) и 2 теллурида (NiTe, NiTe2). Во-вторых, для боль-
большинства переходных металлов характерны халькогениды МеХ и МеХ2. В-^гретьих,
наименьшее число халькогенидов характерно для переходных металлов с относи-
относительно устойчивыми сР- и (/^-конфигурациями. Так, у хрома, вольфрама, марган-
марганца и рения — не более двух халькогенидов, у серебра, золота — по 1 — 2 халько-
генида (Ag2X, Au2S, Au2Te, AuTe2), a y Zn, Cd, Hg — по одному (ЭХ). В-четвер-
В-четвертых, по мере увеличения содержания халькогена в формульной единице соедине-
соединения закономерно меняется характер химической связи от металлоковалентной к
ковалентной вплоть до образования слоистых псевдомолекулярных структур. При
этом в силу сравнительно высоких значений электроотрицательности (/-элементов
ионный вклад в химическую связь не является доминирующим. В то же время
MnS обладает полиморфизмом: низкотемпературная «-модификация имеет струк-
структуру типа NaCl (преимущественно ионный тип связи), а /^-модификация, сущест-
существующая при более высоких температурах, образует кристаллы типа сфалерита
(преобладающая ковалентная связь). Халькогениды переходных металлов имеют
довольно сложные кристаллические структуры, в которых обычные валентные
соотношения, как правило, не соблюдаются. Они представляют собой часто ка-
тионо- и анионоизбыточные фазы. Все халькогениды относятся к бинарным
соединениям с летучим компонентом и склонны к термической диссоциации. При
этом координационным халькогенидам свойственна более или менее ярко выра-
выраженная переменность состава, особенно заметная у низших халькогенидов пере-
переходных металлов.
8. Пниктогениды. Пниктогены (элементы VA-группы) по .электроотрицатель-
.электроотрицательности уступают халькогенам. Так, наиболее тяжелый представитель этой груп-
группы — висмут (ОЭО = 1,8) — приближается уже к переходным металлам. Однако
азот (ОЭО = 3,0) принадлежит к числу наиболее электроотрицательных элемен-
элементов. Этому способствует кайносимметричность 2р-орбиталей, которые к тому же
заселены электронами наполовину Bр3). В силу этого потенциал ионизации азота
выше, чем у следующего за ним кислорода. Эта особенность (повышенные потен-
потенциалы ионизации) в известной мере свойственна всем пниктогенам, однако для
более тяжелых аналогов она выражена менее ярко.
В силу отмеченных особенностей азота, а также учитывая его малый атом-
атомный радиус, можно ожидать, что нитриды должны отличаться от остальных
пниктогенидов. В самом деле, с $- и р-элементами азот образует соедине-
соединения, подчиняющиеся правилу формальной валентности. Нитриды активных
металлов имеют солеобразный характер, но обладают повышенной склонностью
к гидролизу, поскольку кислотные свойства аммиака, солями которого они
формально могут считаться, выражены слабо. Нитриды элементов IIIА- и особен-
особенно IVA-группы являются преимущественно ковалентными соединениями (A1N,
GaN, Ge3N4, Ge3N2, SisN,}), а углерод, олово и свинец с азотом не взаимодейст-
взаимодействуют.
Особенностью взаимодействия переходных металлов с азотом является склон-
склонность к образованию металлидных фаз внедрения, для которых типичен состав
АВ (ScN, TiN, VN, CrN и т.п.). Все эти соединения представляют собой односто-
односторонние фазы переменного состава 3Ni-# с широкими областями гомогенности
(состав 3N является предельным). В ряде случаев в системе Э — N существует
несколько нитридов, например V3N, V2N, VN в системе V — N; Mn4N, Mn2N,
Mn5N2, Mn3N2, MneNs в системе Мп — N. Однако их можно рассматривать как
составы, отвечающие дефициту азота в фазе внедрения 3Ni-x. В противополож-
противоположность солеобразным нитридам щелочных и щелочно-земельных металлов все они
тугоплавки, инертны даже в большей мере, чем ковалентные нитриды кремния и
германия.
Для остальных пниктогенов в системах с активными металлами также харак-
характерно образование соединений, подчиняющихся правилу формальной валентнос-
валентности Ме3Э и Ме3 Э2. Однако помимо этих фаз существуют и анионоизбыточные
соединения, например LiAs5, LiBi, Na2P5, NaSb, KBiz, СаВ1з и т.п. Тем не менее
для бериллия и магния (типические элементы) характерны только нормально-
валентные соединения. Анионоизбыточные пниктогениды в силу наличия связей
П—П являются полупроводниками и обладают сложными структурами. Пниктоге-
Пниктогениды элементов ША-группы относятся к классу алмазоподобных полупроводни-
ков (соединения А В ) и кристаллизуются в структурном типе сфалерита.
Висмут подобных соединений не образует.
Из пниктогенидов кремния и германия известны только фосфиды и арсениды.
Их состав отвечает формулам АВ и АВ2 (SiP, SiP2, GeAs, GeAs2 и т.н.). Хотя,
согласно обобщенному правилу, 8 — N первые из них являются катионоизбыточ-
ными, а вторые — анионоизбыточными, все они обладают неметаллическими
свойствами, поскольку катионообразователи расположены справа от границы
Цинтля. Олово, обладающее более металлическими свойствами, с фосфором
образует три соединения (Sn4P3, Sn3P<b S11P3), с мышьяком — два (Sn3As2, SnAs),
а с сурьмой — только SnSb. Свинец с пниктогенами не реагирует, но в расплав-
расплавленном состоянии хорошо растворяет фосфор, который при кристаллизации
выделяется в виде так называемого фосфора Гитторфа.
Все переходные металлы с пниктогенами образуют большое число соединений,
состав которых весьма разнообразен. В системе Me — П может существовать до
4 — 6 соединений с постепенно увеличивающимся содержанием анионообразова-
теля. Так, никель с фосфором образует шесть соединений: №зР, NisP2, Ni2P,
Ni6P5, NiP2, NiP3; палладий с мышьяком — также шесть соединений: Pd7As,
PdgAs, PdsAs2, Pd3As2, PdAs, PdAsj. Все эти соединения не подчиняются правилу
формальной валентности, являются фазами переменного состава со сложными
кристаллическими структурами. При этом ширина области гомогенности по мере
276
нарастания ковалентной доли связи- при переходе от низших пниктогенидов к
высшим уменьшается. В этом же направлении ослабляется металлический и
усиливается неметаллический характер фаз. Все эти пниктогениды устойчивы к
воде и неокисляющим минеральным кислотам. Поскольку они содержат летучий
компонент, высшие пниктогениды (аналогично халькогенидам) при нагревании
подвергаются термической диссоциации, отщепляя летучий компонент и пос-
последовательно переходя в фазы, более бедные пниктогеном. Термическая устой-
устойчивость сильно повышается в направлении от фосфидов к арсенидам, антимо-
нидам и висмутидам в соответствии с уменьшением летучести анионообразова-
теля.
Особенно следует выделить пниктогениды элементов ПВ-группы (Zn, Cd, Hg),
у которых rf-уровни полностью завершены. С азотом эти элементы не взаимодей-
взаимодействуют, а с остальными пниктогенами образуют нормально-валентные соединения
Ме3Пг, которые являются полупроводниками. Цинк и кадмий, помимо того,
образуют и анионоизбыточные фазы (ZnP2) ZnAs2, ZnSb, CdP2, CdP4, CdAs2,
CdSb), которые тем более проявляют полупроводниковые свойства. Таким обра-
образом, элементы ИВ-группы по отношению к пниктогенам ведут себя как s- и р-
элементы.
9. Карбиды, ^силициды, бориды. Сопоставляя особенности строения и свойств
рассмотренных выше классов бинарных соединений, можно прийти к выводу, что
при переходе от галогенидов к халькогенидам и далее к пниктогенидам наблю-
наблюдается постепенное уменьшение ионного вклада в химическую связь, что сопро-
сопровождается изменением преобладающих типов кристаллических структур. Уже
среди пниктогенидов встречаются фазы с ковалентио-металлическим характером
взаимодействия компонентов. Еще более эта тенденция усиливается у карбидов,
силицидов и боридов. Возрастающее число металлоподобных фаз среди этих
соединений позволяет заключить, что они являются связующим звеном между
бинарными соединениями металлов с неметаллами и интерметаллическими
соединениями.
Углерод и кремний располагаются на границе Цинтля, в то время как бор —
единственный неметаллический элемент, расположенный слева от этой границы.
В силу дефицита валентных электронов у бора среди боридов принципиально
невозможны соединения, подчиняющиеся правилу формальной валентности. По
этому признаку бориды также близки к интерметаллическим соединениям. Из
металлов IA-группы с бором взаимодействует только литий, образуя гексаборид
LiBe. Щелочные металлы с бором не взаимодействуют. Щелочно-земельные ме-
металлы (Са, Sr, Ba), подобно литию, образуют с бором по одному соединению
МеВб. Но уже для магния и бериллия известны по три борида: MgB2, MgBe,
MgBi2, ВегВ, ВеВг, ВеВб- Из р-элементов бориды образует только алюминий
(AIB2, AlBi2, А1В70). Все эти соединения относятся к полиборидам. Остальные р-
металлы с бором не взаимодействуют.
Среди переходных металлов с бором не взаимодействуют элементы IB- и НВ-
групп (подгруппы меди и цинка), что подтверждает двойственный характер этих
элементов. Остальные rf-элементы образуют бориды двух типов: фазы внедрения
(МеВ!-х) и полибориды МеВ„, где п может достигать 66 — 70. Все бориды туго-
тугоплавки, особенно бориды переходных металлов. Так, ТаВ2 плавится при 3200° С,
a HfB2 — при 3500°С. Бориды активных металлов разлагаются водой и минераль-
минеральными кислотами с выделением смеси бороводородов. Бориды переходных метал-
лов стойки по отношению к неокисляющим минеральным кислотам и кислороду
воздуха даже при высоких температурах. А некоторые из них (например, ТаВг)
устойчивы к действию кипящей царской водки.
У карбидов единственной фазой, в которой соблюдаются обычные валент-
валентные отношения, является карбид кремния SiC — типичное ковалентное соеди-
соединение, в котором атомы Si и С находятся в состоянии «р3-гибридизации. Кро-
Кроме того, правилу формальной валентности подчиняются карбиды алюминия
AI4CI3 и бериллия Ве2С, которые можно рассматривать как производные метана.
Некоторые карбиды трактуют как производные ацетилена (ацетилениды). Наи-
Наиболее известен среди них карбид кальция СаС2. Карбиды такого же состава
образуют и другие элементы ПА-группы, кроме бериллия (MgC2, SrC2, BaC2).
Ацетилениды состава Ме2С2 образуют литий, натрий и, вероятно, собственно
щелочные металлы. Все карбиды активных металлов разлагаются водой, причем
в качестве газообразных продуктов гидролиза выделяются либо метан, либо
ацетилен:
А14С3 + 12Н2О = 4А1(ОНK + ЗСН4
СаС2 + 2Н2О = Са(ОНJ + С2Н2
Таким образом, карбиды активных металлов можно рассматривать как солеобраз-
ные производные слабых кислот — "метановой" СН4 и "ацетиленовой" С2Н2. Для
щелочных металлов помимо ацетиленидов характерны соединения включения
(аддукды) с графитом (NaCs, NaCi6, NaC24 и т.п.). Энтальпии образования одно-
однотипных аддуктов, например Me C8, возрастают в ряду К — Rb — Cs от 365 до 670
кДж/моль графита. Натрий образует такие соединения с трудом, а литий не
образует вовсе. Такие аддукты иногда называют графитидами. Графитиды актив-
активно реагируют с водой, как и щелочные металлы. Механизм образования графити-
дов состоит в регулярном заполнении пустот в графитовой сетке атомами метал-
металла. Поскольку графитиды обладают большей электрической проводимостью, чем
сам графит, можно предположить, что при внедрении электроны щелочных ме-
металлов делокализуются.
Карбиды переходных металлов, в которых, на первый взгляд, соблюдается
правило формальной валентности (TiC, HfC и т.п.), относятся в действительности
к фазам внедрения типа ABi-^, а потому обычные валентные отношения здесь
неприменимы. Фазы внедрения являются наиболее обычными соединениями
переходных металлов с углеродом. Несмотря на разнообразие карбидных фаз в
некоторых системах, содержание углерода в них никогда не превышает 50 ат.
долей, % (состав МеС). Так, в системе Fe — С существуют три карбида (ГезС,
Fe2C, FeC), в системе Мп — С — также три карбида (Мп3С, Мп5С2, Мп7Сз) и т.п.
Более бедные углеродом карбиды можно рассматривать как продукты частичного
замещения октаэдрических пустот в плотноупакованных структурах. Все эти
карбиды являются фазами переменного состава. Карбидные фазы внедрения
образуют rf-элементы с дефектной rf-оболочкой. Элементы подгрупп меди (dlos1) и
цинка (d10*2) с*'углеродом не взаимодействуют. Не образуют карбидов и платино-
платиновые металлы, хотя некоторые из них, например платина, растворяют углерод в
твердом состоянии (твердые растворы внедрения).
Кремний заметно уступает углероду по электроотрицательности. Поэтому
i
солеобразные силициды с преимущественно ионным типом связи не существуют.
Лишь некоторые силициды наиболее активных металлов подчиняются правилу
формальной валентности (L^Si, Mg2Si). Однако даже в этих системах существуют
соединения, в которых это правило не соблюдается (Li2Si, LiSio, NaSi2, KSig, CaSi,
CaSi2, BaSi3, BaSi4 и т.п.). Металлы ША-группы (Al, Ga, In, Tl), а также металлы
IVA-группы (Sn, Pb), т.е. все р-металлы, с кремнием соединений не образуют.
Химически не взаимодействуют с кремнием и металлы, завершающие вставные
декады (подгруппы меди и цинка). Напротив, rf-металлы с дефектной d-оболоч-
кой образуют большое количество силицидных фаз с преобладанием составов
ЭзБ!, 32Si, 3Si и 3Si2. Доминирующим типом связи здесь является металлокова-
лентный, хотя некоторые высшие силициды (состава 3Si2) обладают полупровод-
полупроводниковыми свойствами (CrSi2, FeSi2, MnSi2 и т.п.). Напротив, низшие силициды
переходных металлов являются металлидами. В этом проявляется общая тенден-
тенденция, отмеченная выше, — нарастание ковалентного вклада по мере увеличения
содержания анионообразователя в формульной единице.
Силициды переходных металлов тугоплавки, устойчивы к окислению на воз-
воздухе даже при высоких температурах и не взаимодействуют с неокисляющими
кислотами. Например, TaSi2 плавится при 2200°С, MoSi2 устойчив к окислению
до 1800° С, в силу чего его используют в качестве материала нагревательных
элементов, работающих в окислительной атмосфере. Многие силициды переход-
переходных металлов, особенно с большим содержание кремния, устойчивые в кислых
средах, разлагаются щелочами.
10. Интерметаллические соединения. Закономерности образования этих соеди-
соединений обычно обусловлены металлохимическими факторами низшего порядка —
размерным фактором и электронной концентрацией. Типичными представителя-
представителями интерметаллических соединений являются электронные соединения Юм-Розе-
ри, фазы Лавеса, соединения Курнакова. Последние со структурной точки зрения
близко примыкают к твердым растворам, в чем проявляется единство непрерыв-
непрерывности и дискретности при химическом взаимодействии.
Обычно интерметаллическим соединениям приписывают некий базовый сос-
состав, несмотря на наличие широкой области гомогенности. Так, в системе Си — Zn
существует соединение CusZn8 F1,5% Zn), хотя его область гомогенности нахо-
находится в интервале 58—67% Zn. Однако часто не прибегают к такой индивидуали-
индивидуализации и не приписывают интерметаллическому соединению определенной форму-
формулы, обозначая промежуточные фазы в бинарных металлических системах буквами
греческого или латинского алфавита с указанием интервала составов, в котором
они существуют. Например, в системе Си — Be отмечено существование двух
интерметаллических соединений; /3-фазы B7—39% Be) и /?'-фазы D7,5—48,5%
Be), в системе Та — Ru существуют зьфаза B7,5—38,5% Ru), «/-фаза C9—45% Ru),
г-фаза D5,5-54% Ru), о-фаза E4,5-57% Ru).
ГЛАВА XIII. СЛОЖНЫЕ ХИМИЧЕСКИЕ СОЕДИНЕНИЯ
Основной массив объектов неорганической химии составляют многоэле-
многоэлементные соединения (с числом элементов три и более), которые можно назвать
сложными. Если бинарные соединения являются продуктами взаимодействия
простых веществ, то сложные, в свою очередь, можно рассматривать как прЪ-
27»
дукты взаимодействия бинарных соединений. Руководящим принципом при
изучении этих объектов, как и ранее, являются природа химической связи,
химическое и кристаллохимическое строение и, как следствие этого, свойства
соединений.
Анализ свойств сложных соединений, образуемых тем или иным элементом,
позволяет еще более детально и подробно охарактеризовать его химическую
природу. При этом наиболее рельефно выявляются особенности кислотно-основ-
кислотно-основного и окислительно-восстановительного взаимодействия в зависимости от степе-
степени окисления, сравнительная стабильность соединений в разных степенях окисле-
окисления элемента, их способность к термической и электролитической диссоциации,
гидролизу, комплексообразованию и т.п.
Изучение закономерностей образования и свойств сложных соединений завер-
завершает химическую характеристику элемента и служит основой для прогнозирова-
прогнозирования областей практического применения как самого простого вещества, так и его
соединений.
1. Классификация сложных соединений. Термином сложные химические
соединения определяют химические индивиды, содержащие три элемента и более.
Если простых веществ (с учетом аллотропии и полиморфизма) насчитывается
около 200, а бинарных соединений — порядка 10 000, то сложных химических
соединений значительно больше. Традиционно эти объекты подразделяются на
три класса: основания, кислоты и соли. В эту же классификацию обычно вклю-
включают и комплексные соединения, поскольку существуют комплексные кислоты,
комплексные основания и комплексные соли. Однако уже среди комплексных
соединений встречаются такие, которые невозможно отнести ни к одному из
перечисленных классов. Таковы, например, карбонилы металлов, многие хелаты
и внутрикомплексные соединения. Таким образом, уже применительно к комп-
комплексным соединениям приведенная классификация не является полной. Но
существуют сложные соединения, которые не относятся и к комплексным, хотя
их также нельзя рассматривать в рамках данной классификации. В частности,
такие соединения, как CdSnAs2, ZnGeP2, CuFeS2 и т.п., невозможно отнести к
солям, в том числе и комплексным. Причиной неуниверсальности этой класси-
классификации служит то, что она охватывает только объекты, в которых существенная
роль принадлежит преимущественно ионной связи между структурными эле-
элементами. Отсюда, в частности, вытекает принципиальная возможность электро-
электролитической диссоциации в водных растворах с разрывом преимущественно
ионной связи по одному из трех типов: кислотному, основному или "солевому".
Очевидно, если в веществе отсутствует ионная
связь, то оно не подпадает под данную клас-
классификацию.
Таким образом, универсальным признаком,
позволяющим провести единую классифика-
классификацию сложных соединений, как и в случае
бинарных, является доминирующий тип
химической связи. В соответствии с этим
признаком можно выделить три типа сложных
соединений. Так, к первому типу
Рис. 132. Кристаллическая структура соединений относятся те сложные соли, а
сульфата калия также кислоты и основания, в которых ионное
280
взаимодействие является существенным, преимущественным, по крайней мере
между отдельными фрагментами структуры (в молекуле или кристалле). Напри-
Например, сульфат калия (рис. 132) кристаллизуется в ромбической структуре, в узлах
его решетки находятся тетраэдры [SOJ (S и О связаны ковалентной связью) и
атомы калия. Между последними и тетраэдрами [SO4] имеет место преимущест-
преимущественно ионная связь. Комплексные группировки [SOJ представляют собой единые
структурные фрагменты и вместе с атомами калия формируют плотноупакован-
ную структуру с координационными числами, характерными для ионных крис-
кристаллов: к.ч. (К) = 4, к.ч. [SOJ = 8 (подобно структуре антифлюорита). Наличие
преимущественно ионной связи в структуре предопределяет возможность диссо-
диссоциации в полярных растворителях по "солевому" механизму. При этом ковалент-
но-связанные фрагменты [SOJ в растворе сохраняют свою индивидуальность.
Аналогичная ситуация наблюдается в кристаллах NaOH, КОН и т.п., в кото-
которых атомы щелочного металла и ковалентно-связанные группы ОН совместно
образуют структуру типа NaCl, типичную для преимущественно ионных соедине-
соединений. Отличительной особенностью оснований, кислот и солей является гетеродес-
мический характер соответствующих структур, в которых сочетаются преиму-
преимущественно ковалентное взаимодействие внутри фрагментов и преимущественно
ионное — между ними.
Ко второму типу сложных соединений относятся вещества, в которых
наблюдается только ковалентное взаимодействие. В функциональном отношении
такие соединения не обладают ни кислотным, ни основным, ни солеобразным
характером. Так, халькопирит CuFeS2 лишь формально можно рассматривать как
солеобразное производное сероводородной кислоты, поскольку его структура
типична для ковалентных соединений (тетраэдрические ковалентные яр3-гибрид-
ные связи). Такие соединения, как CdSnP2, ZnGeAs2 и другие тройные алмазопо-
добные полупроводники, относящиеся к изоэлектронным аналогам А В , тем
более нельзя рассматривать как соли. Здесь реализуются пространственные трех-
трехмерные ковалентные структуры с к.ч. 4. Среди преимущественно ковалентных
сложных соединений кроме гомодесмичных тетраэдрических структур могут
реализоваться слоистые, островные, цепные и, наконец, молекулярные структуры.
Таковы, например, карбонилы переходных металлов, внутрикомплексные соеди-
соединения и хелаты. Существование таких структур обусловлено наличием ван-дер-
ваальсова взаимодействия между фрагментами.
Третий тип сложных соединений — это интерметаллические многоком-
многокомпонентные фазы с доминирующим металлическим типом связи. Наличие коллек-
коллективного электронно-атомного взаимодействия и, как следствие этого, реализация
плотноупакованных структур с широкими областями гомогенности приближают
многокомпонентные металлические фазы к твердым растворам.
Для того чтобы сложные соединения генетически связать с более простыми
бинарными, можно рассматривать многокомпонентные фазы как продукт взаимо-
взаимодействия бинарных соединений друг с другом. Так, H2SO4 и Са(ОНJ получают
непосредственным взаимодействием SO3 и СаО с водой. Сульфат кальция CaSO4
может быть синтезирован из соответствующих оксидов СаО и SO3. Нередко пря-
прямая реакция между соответствующими бинарными соединениями неосуществима
по кинетическим причинам. Тем не менее такие соединения, как А1(ОНK,
Cu(OHJ, Na2SiO3 и т.п., можно рассматривать как продукты взаимодействия
А12О3 + ЗН2О; CuO + H2O; Na2O + SiO2 и.т.п. Аналогично, CuFeS2 является
281
продуктом взаимодействия CuS и FeS, a CdSnP2 — Cd3P2 + S113P4 и т.д. Этот
подход справедлив и для комплексных соединений:
4KCN + Fe(CNJ = K4[Fe(CNN], 3NaF + A1F3 = Nas[AlF6]
Часто формулы сложных минералов записывают в виде комбинации соответ-
соответствующих бинарных соединений, например асбест MgsHUSiaOg — 3MgO-2SiO2x
х2Н2О.
Очевидно, что при таком подходе выявляются особенности химической связи
в образующихся сложных соединениях. Чем резче бинарные соединения отлича-
отличаются друг от друга по свойствам, тем более вероятно возникновение сильнопо-
сильнополярного взаимодействия между структурными фрагментами сложного соединения.
Так, взаимодействие основных оксидов с кислотными приводит к образованию
солей, основных оксидов с водой — к образованию оснований, а кислотных с во-
водой — кислот. Если же бинарные соединения, из которых можно образовать
сложные, различаются по характеру не сильно, то преимущественно ионное взаи-
взаимодействие не реализуется (CuFeS2)- В случае образования сложного вещества
несколькими бинарными соединениями можно предвидеть неравноценность функ-
функций его составляющих. Например, в ортоклазе b^Al-SihOx; или К2О-Al2Og-6SiO2
амфотерный оксид AljOs и слабокислотный оксид SiC>2 образуют прочный алю-
мосиликатный каркас, а калий, входящий в состав сильно основного оксида К2О,
оказывается связанным с этим каркасом преимущественно ионной связью. В силу
этого наблюдается, например, выщелачивание его при длительном контакте с
водой.
Подобный подход продуктивен тогда, когда при образовании сложного сое-
соединения из бинарных не происходит изменения степени окисления элементов.
При этом условии таким образом можно рассмотреть и взаимодействие бинарных
соединений с простыми веществами, приводящее к образованию кои&лентных
комплексов, например карбонилов. Так, металлическое железо при определенных
условиях присоединяет СО: Fe + 5С0 = [Fe(COM], причем в карбониле степень
окисления железа остается раьной нулю. Во всех остальных случаях взаимодей-
взаимодействие бинарного соединения с простым веществом происходит с изменением сте-
степени окисления и полученные таким образом продукты не могут рассматриваться
как комбинации бинарных соединений. Например, с этих позиций нельзя рас-
рассматривать SO2CI2, СаОСЬ, H2S2O3 и т.п., поскольку при взаимодействии меняет-
меняется степень окисления:
+SO2 + Cl =+SO2Cl2; СаО + С12 = CaC'loCl; H2SO3 + S = H2S0J
Тем не менее рассмотрение сложного соединения как продукта взаимодействия
более простых позволяет не только установить генетическую связь между ними,
но и прогнозировать свойства сложных соединений с учетом особенностей хими-
химической связи в них.
2. Гидроксиды как характеристические соединения. Как известно, гидрокси-
ды обычно рассматриваются как продукты взаимодействия оксидов с водой
независимо от того, наблюдается это взаимодействие в действительности или
гидроксид может быть получен только косвенным путем. Гидроксиды являются
характеристическими соединениями, поскольку свойства гидроксида определяют
в конечном итоге принадлежность элемента к металлам или неметаллам. Так,
282
оксиды металлов, взаимодействуя с водой, дают основные гидроксиды (основа-
(основания), а оксиды неметаллов — кислотные гидроксиды (кислоты)*. Следует, одна-
однако, подчеркнуть, что характеристическими в этом смысле свойствами гидроксиды
обладают только в водных растворах. В других растворителях их кислотно-основ-
кислотно-основные функции могут резко измениться. Например, оксид азота N^Oj с водой обра-
образует гидроксид HNOa, который в водном растворе является сильной кислотой.
Это характеризует азот как типичный неметалл, a N2O5 — как кислотный оксид.
В то же время в среде сжиженного фтористого водорода HNO3 ведет себя как
типичное основание, присоединяя протон:
HNO3 + HF 5=* H2NO; 4- F"
В общем случае многие основания и кислоты в неводных средах могут сильно
изменить степень диссоциации. Например, слабая в водном растворе сернистая
кислота HjSOg в жидком аммиаке диссоциирована примерно в такой же степени,
как HNO3 в водном растворе, т.е. является сильной кислотой. Таким образом,
характер диссоциации гидроксидов (по кислотному или основному типу), а также
степень диссоциации зависит от природы растворителя, в частности от его донор-
но-акцепторной активности по отношению к протону. Так, в ряду
HF: -+ Н2б: > : NH3
увеличивается сродство к протону вследствие уменьшения числа неподеленных
электронных пар у анионообразователя. В силу этого слабые в водных растворах
кислоты становятся сильными кислотами в среде жидкого аммиака и основания-
основаниями в среде жидкого фтороводорода. Отсюда следует, что сравнивать кислотно-
основные свойства гидроксидов можно лишь по отношению к определенному
растворителю. И в настоящее время кислотно-основные свойства гидроксидов как
характеристических соединений рассматриваются по отношению к водной среде.
3. Кислотно-основные свойства. Амфотерность гидроксидов. Общая формула
гидроксидов может быть представлена в виде Э—(OH)n, где п равно степени
окисления элемента. Максимальное значение п может быть равно 8 (наибольшее
значение формальной валентности, определяемое номером группы Периодической
системы). Однако координационные сферы, содержащие большое число гидрок-
согрупп, оказываются неустойчивыми. Повышение устойчивости в этом случае
достигается путем уменьшения координационного числа за счет отщепления
одной или нескольких молекул воды. Чем выше степень окисления элемента, а
следовательно, меньше размер ион-атома, тем большее число молекул воды
отщепляется для обеспечения устойчивости координационной сферы. Это
положение характерно для гидроксидов с кислотными свойствами, т.е. для
гидроксидов неметаллов и металлов со степенью окисления, превышающей +3.
Ряд кислотных гидроксидов, отвечающих высшим степеням окисления элементов
III—VIII групп Периодической системы, представлен в табл. 22.
Такое утверждение в действительности не может считаться категоричным, посколь-
поскольку, как указывалось выше, основность и кислотность оксида зависят от степени окисления
элемента. Так, Мп2О7 является кислотным оксидом, а МпО — основным. К тому же пос-
последний не растворяется и не взаимодействует с водой и Мп(ОНJ получают косвенным
путем.
, . 283
Таблица 22. Кислотные гидроксиды, номинально отвечающие высшим степеням
окисления элементов Ш—VIII ipyim Периодической системы
Период
2
3
4
5
6
III
В(ОН)з
-
-
-
-
-
-
IV
С(ОНL
Si(OHL
Ti(OHL
Ge(OHL
Zr(OHL
Sn(OHL
Hf(OHL
V
N(OHM
P(OHM
V(OHM
As(OHM
Nb(OHM
Sb(OHM
Ta(OHM
Группа
VI
_
S(OHN
Cr(OHN
Se(OHN
Mo(OHN
Te(OHN
W(OHN
VII
_
C1(OHO
Mn(OHO
Br(OHO
-
I(OHO
Re(OHO
VIII
-
-
-
-
Ru(OH)8
-
Os(OH)8
Во втором периоде при переходе В(+3) — С(+4) — N(+5) атомный радиус
уменьшается. Поэтому для бора характерно сохранение координационного окру-
окружения из трех гидроксогрупп в ортоборной кислоте Н3ВО3 (хотя известны произ-
производные и метаборной кислоты HBO2). Для углерода повышение координационной
устойчивости достигается за счет удаления одной молекулы воды и замены двух
лигандов —ОН на один лиганд =О:
I г
i i
ОН
, Н'О-С-ОН - Н2О
( I
он
он
+ о=с
он
СО(ОНJ или Н2СО3
Для азота более устойчивой оказывается структура, образованная удалением двух
молекул воды:
N(OHM - 2Н2О —> N0 2 ( ОН)
HNO3
Устойчивые формы кислотных гидроксидов элементов 3-го периода подчиняются
той же закономерности: с увеличением номера группы возрастает число отщеп-
отщепленных молекул воды, поскольку в этом направлении размеры атомов уменьша-
уменьшаются. Однако в пределах групп [С(+4) —> Si(+4), N(+5) —> Р(+5)] атомные
радиусы возрастают. Поэтому для кремния и фосфора, помимо мета-форм кис-
кислот (H2Si03 и НРО3), известны и opmo-формы (H4SiO4 и Н3РО4). С учетом су-
существования ортокремниевой и ортофосфорной кислот для элементов 3-го пе-
периода наблюдается общая закономерность:
Si(OHL=H4SiO4
Р(ОНM - Н20 —> РО(ОНK
Н3РО4
284
S(OHN - 2H2O —¦ SO2(OHJ
H2SO4
C1(OHO - 3H2O —> С 1 03 (ОН)
HC1O4
т.е. с ростом степени окисления и уменьшением размера центрального атома
число отщепленных молекул воды увеличивается. Для гидроксидов более тя-
тяжелых р-элементов отмечается тенденция к увеличению количества связанных
гидроксоанионов, что также обусловлено возрастанием атомного радиуса эле-
элемента при переходе сверху вниз в пределах каждой группы. При этом у элемен-
элементов 4-го периода (Ge, As, Se, Вг) сохраняется та же тенденция, что и у их анало-
аналогов из 3-го периода (Si, P, S, C1), поскольку атомные радиусы в направлении
Si —> Ge, P —> As, S —> Se, Cl —> Вг возрастают незначительно. Это обусловле-
обусловлено rf-сжатием в силу наличия у ^элементов 4-го периода завершенной кайиосим-
метричной Зй-оболочки. Вследствие этого формулы соответствующих высших
кислот имеют вид H4Ge04, H3As04 (-1Н20), H2SeO4(-2H2O) и HBrO4(-3H2O).
Но для р-элементов 5-го периода характерны более богатые водой гидратные
формы. Наряду с Н2Те04 известна и ортотеллуровая кислота НбТеОе [Те(ОН)б
без потери молекул воды]. А для иода кроме НЮ4 известна и ЩЮб с потерей
всего одной молекулы воды от гидроксида номинального состава 1@Н)у-
Таким образом, наиболее распространенными гидратными формами р-элемен-
тов являются те, формулы которых можно получить путем вычитания из пре-
предельной формы 9@H)n четного числа молекул воды для элементов четных групп
и нечетного — для элементов нечетных групп. Исключение составляют бор, угле-
углерод и азот с кайносимметричными 2р-орбиталями.
Кислотные гидроксиды переходных металлов в высших степенях окисления
подчиняются той же закономерности: с увеличением номера группы число отщеп-
отщепляемых молекул воды возрастает. Для гидроксидов кайносимметричных Srf-эле-
ментов наиболее характерны лета-формы: Н2ТЮз(—IH2O), HVOg(—2Н2О),
Н2Сг04(—2Н2О) и НМпО4(—ЗН2О). Элементы триады железа не проявляют степе-
степеней окисления, отвечающих номеру группы. У (/-элементов 5-го и 6-го периодов
помимо мета-форм известны и opmo-производные: H4Zr04, H3NbO4(—ШгО).
Гидроксид рутения в степени окисления +8 не существует, поскольку в соответ-
соответствии с общей закономерностью от Ru(OH)s должны отщепиться четыре молеку-
молекулы воды, что приводит к составу RuO4. Особенно наглядно эта тенденция прос-
прослеживается у rf-элементов 6-го периода (у Н4НЮ4 вода не отщепляется):
Та(ОНM - Н20 —> H3Ta04; W(OHN - 2Н2О —> H2WO4
Re(OHO - ЗН2О —> HReO4; Os(OH)8 - 4Н2О —> OsO4
Оксиды рутения и осмия в высшей степени окисления не могут химически взаи-
взаимодействовать с водой с образованием устойчивых гидратных форм.
Таким образом, общую формулу гидроксидов правильнее представлять не в
виде Э(ОН)„, а в виде ЭОя^ОН)^, где т — 0f4, a n — Отб в реально существую-
существующих гидроксидах. При т - О гидроксиды Э(ОН)„ могут обладать как основным,
так и кислотным характером, причем с увеличением числа гидроксогрупп в фор-
формульной единице (т.е. с увеличением степени окисления Э) оснбвные свойства
285
ослабевают, а кислотные нарастают. Это можно объяснить ростом поляризующего
действия катионообразователя в пределах каждого периода в связи с уменьшени-
уменьшением его радиуса и увеличением степени окисления, например в ряду NaOH,
Mg(OHJ, A1(OHK, Si(OHL- В общем случае для гидроксидов типа Э(ОН)„ (т =
— 0) при переходе сверху вниз в пределах каждой группы основный характер
гидроксида усиливается, что связано с ослаблением поляризующего действия
катионообразователя за счет увеличения его радиуса.
Гидроксиды ЭОт(ОН)п (т Ф 0) обладают исключительно кислотными свой-
свойствами. Это объясняется тем, что атомы кислорода, ковалэнтно связанные с эле-
элементом, способствуют поляризации связи О—Н, в силу чего и облегчается диссо-
диссоциация по кислотному типу с отщеплением протона. Очевидно, г. ростом т (числа
ковалентно связанных с элементом кислородных лигандов) сила кислот должна
возрастать. От величины п сила кислоты практически не зависит, поскольку для
многоосновных кислот концентрация ионов водорода в растворе определяется в
основном первой константой диссоциации, отвечающей схеме
ЭОт(ОН)п ^ Н+ + [ЭОт(ОН)п-,О]-
(Н3РО4 ^ Н+ + Н2РО4; H4Si04 ^ Н+ + H3Si04 и т.п.)
Кислоты типа Э(ОН)П (т — 0) принадлежат к очень слабым электролитам
(Н3ВО3, НОС1, ЩТеОб имеют константы диссоциации по первой ступени, равные
соответственно 6-Ю0, 5-Ю"8, 2-Ю"8). Кислоты типа ЭО(ОН)П (т — 1) слабые
(для НС1О2, HNO2, Н2СО3 константы диссоциации равны МО, 4-Ю, ЬН)).
Кислоты типа ЭО2(ОН)П (т = 2} 0 сильные (HNO3, H2SO4 имеют первые констан-
константы диссоциации, равные 4-Ю1 и 1-10' соответственно); ЭО3(ОН) (т = 3, п — 1) —
очень сильные кислоты (НС1О4 — Кл = МО10).
Этот подход позволяет объяснить изменение кислотно-основного характера
гидроксидов в зависимости от степени окисления элементов. Так, азотистая кис-
кислота HNO2 или NO(OH) слабее азотной HNO3 или NO2(OH), сернистая H2SO3
или SO(OHJ слабее серной H2SO4 или SO2(OHJ. В ряду Мп(ОНJ, Мп(ОНK,
Мп(ОНL, (НО)зМпО (или Н3Мп04), (НОJМпО2 (или Н2Мп04), (НО)МпО3 (или
НМпО4) для гидроксидов Э(ОН)П с увеличением п ослабевает основный характер,
а для MnOm(OH)n с возрастанием т усиливаются кислотные свойства. При этом
гидроксиды Мп(ОНK и Мп(ОНL проявляют амфотерность. Хотя оба гидроксида
принадлежат к очень слабым электролитам, у первого ярче выражены основные,
а у второго — кислотные свойства.
С общей точки зрения кислотно-основные свойства гидроксидов, как и кис-
кислотно-основные свойства других соединений, могут быть выявлены лишь при
химическом взаимодействии. Эти свойства являются проявлением единого про-
процесса кислотНо-основного взаимодействия. Кислотный характер соединения ста-
становится очевидным лишь при взаимодействии с соединениями, обладающими
основной функцией, и наоборот. Поэтому амфотерность в известном смысле мож-
можно рассматривать как универсальное свойство гидроксидов. Например, Мп(ОН)г и
Си(ОНJ обладают преимущественно основным характером. Однако в жестких
условиях — при кипячении с концентрированными растворами щелочей, а тем
более при сплавлении со щелочами — образуются соединения, отвечающие кис-
кислотной функции этих гидроксидов. С другой стороны, HNO3 в большинстве
случаев проявляет кислотные свойства. Однако для HNO3 можно обнаружить и
основную функцию, например при взаимодействии с НС1О4 и H2SO4:
286
hno3 + 2hcio4 = н3о+ + no;
HNO3 + 2H2so4 = н3о+ + no; + 2hso;
При этом образуются перхлорат и гидросульфат нитроила: NO2CIO4 и NO2HSO4.
Таким образом, в зависимости от степени проявления кислотно-основных
свойств характер диссоциации гидроксидов в водной среде, а также и особен-
особенности взаимодействия оксидов с водой закономерно изменяются. При этом можно
выделить основные типы такого взаимодействия. Оксиды типичных неметаллов с
водой дают сильные кислоты, диссоциирующие с образованием оксоанионов:
SO3 + Н2О = SOf + 2Н+
Слабокислотные оксиды образуют при взаимодействии с водой соответствующие
кислоты, которые диссоциируют преимущественно до кислых анионов, причем
последние в малой степени склонны к дальнейшей диссоциации:
SO2 + Н20 ?=* HSOg + Н-
Амфотерные оксиды с преобладанием кислотных свойств образуют с водой
гидроксоанионы в щелочной среде:
GeO2 + 2Н2О + 2ОН- = [Ge(OH)fip-
Напротив, диссоциация амфотерных гидроксидов с преобладающей основной
функцией приводит к образованию гидроксокатионов (основных солей) в кислой
среде:
Zn(OHJ + Н+ = Zn(OH)+ + Н2О
Если кислотные и основные свойства амфотерного гидроксида выражены пример-
примерно в одинаковой степени, то возможны взаимодействия по обеим схемам:
1Г+ ЧОН"
Ga(OH)^ + Н2О < Ga(OHK > [Ga(OHN]3"
Диссоциация слабоосновных гидроксидов протекает с образованием гидроксо-
гидроксокатионов и гидроксидионов:
Fe(OHJ *=* Fe(OH)+ + ОН"
Гидроксиды типичных металлов, обладающие сильноосновным характером, в
водных растворах образуют гидратированные простые катионы (аквакомплексы)
и анионы ОН":
NaOH + aq 5=± Na+-aq + ОН"
4. Окислительно-восстановительные свойства гидроксидов. Характеристич-
Характеристичность гидроксидов проявляется не только в закономерностях изменения кислот-
кислотно-основных функций, но и в том, что на их примере можно оценить
относительную стабильность различных степеней окисления элемента, взаимные
переходы между ними, т.е. рассмотреть окислительно-восстановительную
активность элементов в различных степенях окисления. Специфика
окислительно-восстановительного поведения гидроксидов обусловлена тем, что
взаимодействия в данном случае протекают в. водных растворах. При этом вода
287
является активным участником процесса. В большинстве случаев окислительно-
восстановительные реакции с участием гидроксидов зависят от реакции среды.
Дело в том, что потенциал водородного электрода равен нулю только в
стандартных условиях, т.е. при концентрации (активности) ионов водорода в
растворе, равной 1 моль/л (рН 0). В соответствии с уравнением Нернста
потенциал водородного электрода определяется соотношением
Е = 0,059 lg[H+] = -0,059рН.
С увеличением рН электродный потенциал смещается в отрицательную сторону.
Так, при рН 7 потенциал водородного электрода Е (Н+/Н) = -0,413 В, а при
рН 14 Е (Н+/Н) = -0,826 В. Устойчивость окислителей и восстановителей в вод-
водной среде зависит от их редокс-потенциала в конкретных условиях (температура,
концентрация окисленной и восстановленной форм и рН среды).
Окислительно-восстановительная активность гидроксидов зависит от степени
окисления элемента. В общем случае с увеличением степени окисления растет
способность к окислительному действию (редокс-потенциал становится более
положительным). Так, цепь окислительно-восстановительных превращений мар-
марганца и его гидроксидов, представленная ниже, демонстрирует зависимость ре-
редокс-потенциала системы от степени окисления марганца и рН среды:
МпОг+ 2Н;О+2«-~-Мп(ОНJ+2ОН
МпО7+ 8Н++5<Г- Мп2++ 4НгО
1,51В
Из схемы видно, что в кислой среде окислительная активность оксо- и гид-
роксопроизводных марганца в степени окисления +4, +6, +7 выше (более по-
положительные значения редокс-потенциала), чем в нейтральной и щелочной. На-
Напротив, восстановительные свойства Мп ярче проявляются именно в щелочной
среде.
Характерной особенностью некоторых соединений в промежуточных степенях
окисления элемента является способность к реакциям диспропорционирования:
ЗНОС1
2НС1
НС1О3; 4НС1Оз
3HC1O4 + НС! и т.п.
.1'
Этот общий признак касается не только гидроксидов, но и в еще большей степе-
степени распространен среди их солеобразных производных, которые более стабильны,
чем соответствующие кислоты (например, NaOCl и НОС1, КСЮ3 и НСЮз)- Тем
не менее соли также подвержены диспропорционированию. Склонность к реакци-
288
ям диспропорционирования служит важным классификационным признаком,
поскольку свидетельствует о нестабильности соответствующих степеней окисления
и повышенной склонности их к окислительно-восстановительному распаду.
5. Соли кислородсодержащих кислот. Выше было отмечено, что тройные и
более сложные соединения можно рассматривать как продукт взаимодействия
соответствующих бинарных. Так если взаимодействуют (в действительности или
мысленно) оксиды, один из которых — оксид водорода (вода), то образуются
гидроксиды. Если взаимодействуют основные и кислотные оксиды, то образуются
соли кислородсодержащих кислот. Таким образом, гидроксиды (основания и
кислоты) являются своеобразным частным случаем солей. При этом в результате
электролитической диссоциации (например, в водных растворах) единственным
типом катионов являются катионы НзО+ в случае кислот и единственным типом
анионов — ОН" в случае оснований. Если же в растворе присутствуют какие-либо
иные катионы или анионы, то объект является солью. Так, при диссоциации
NaHSO4 ;г=? Na+ + Н+ + SO?"
кроме катионов Н+ (НзО+) присутствуют катионы Na+, а при диссоциации
Ba(OH)CN ^ Ва2+ + ОН"+ CN"
кроме анионов ОН" присутствуют анионы CN". Не случайно кислотно-основное
взаимодействие сводится к образованию солей. С этой точки зрения к данному
типу взаимодействия относятся не только реакции между кислотами и основания-
основаниями, но и между основными и кислотными оксидами. Нейтрализация в широком
смысле слова сводится не к возникновению нейтральной реакции среды в раство-
растворе, а к взаимной компенсации кислотных и основных свойств реагентов. Строго
говоря, полная взаимная компенсация этих свойств возможна лишь в том случае,
если они выражены у обоих реагентов сильно и в равной мере. Если же кислота и
основание, образующие соль, различаются по силе, что чаще всего и наблюдается
на практике, то полной взаимной нейтрализации не происходит и кислотно-ос-
кислотно-основное равновесие фиксируется на некоторой промежуточной стадии. В водных
растворах это проявляется как гидролиз солей. Если соль рассматривать как
продукт взаимодействия основного и кислотного оксидов, в результате могут
образоваться лишь средние соли. Кислые и основные соли можно рассматривать
как продукт частичной нейтрализации многоосновных кислот основаниями, или
основными оксидами, или многокислотных оснований кислотами, или кислотны-
кислотными оксидами.
Соли часто являются характеристическими объектами, так как по ним можно
судить о свойствах соответствующих кислот. Это особенно важно, если кислоты в
свободном состоянии не существуют, в то время как их соли устойчивы. Примеры
такого рода многочисленны (Н2СО3 и Na2CO3, HOC1 и КОС1 и т.п.). Существует
общая закономерность: соли устойчивее кислот, что обусловлено повышением
прочности за счет поляризации связи Me—О.
Одним из существенных свойств солей вообще и солей кислородсодержащих
кислот в частности является растворимость. В настоящее время строгие коли-
количественные критерии, позволяющие связать растворимость с физико-химической
природой катиона и аниона, образующих соль, отсутствуют. Ясно лишь, что
растворимость является сложной функцией энергии кристаллической решетки,
энергии сольватации катиона и аниона, их поляризационных характеристик,
10 Я. А. Угай 289
энтальпии образования соли и т.д. Однако можно привести некоторые качествен-
качественные закономерности, характеризующие растворимость солей кислородсодержа-
кислородсодержащих кислот в воде. Так, практически все соли одноосновных кислот (нитраты,
ацетаты-, перхлораты и т.п.) растворимы в воде. Хорошей растворимостью облада-
обладают все кислые соли независимо от природы катиона и аниона (гидросульфаты,
гидрокарбонаты, моно- и дигидрофосфаты и т.п.). Напротив, многие основные
соли малорастворимы в воде. Исключения составляют некоторые основные соли
сильных многокислотных оснований, например Ba(OH)CN. Закономерности раст-
растворимости средних солей многоосновных кислот более сложны. Так, соли кислот
HXY4 и цезия сравнительно малорастворимы независимо от природы X и Y.
Растворимость CSCIO4, CsMnO4, CsReO4, CSBF4, CSIO4 и т.п. составляет около
0,01—0,07 моль/л. Растворимость солей бария крайне мала для всех кислот типа
Н2ЭО4 (Э — S, Se, Сг, Mo, W, Mn, Re, Fe, Ru, Os). Труднорастворимы также
сульфаты кальция, стронция,радия, свинца. С другой стороны, многие сульфаты
р- и особенно rf-металлов (Al, Ga, In, Сг, Mn, Fe, Zn и т.п.) хорошо растворимы в
воде. Важно подчеркнуть, что именно эти сульфаты весьма склонны к образова-
образованию кристаллогидратов, в то время как малорастворимые сульфаты образуют
кристаллогидраты редко (например, CaSO4-2H2O). Это услужит указанием на
существенную зависимость растворимости солей от способности катионов образо-
образовывать прочные аквакомплексы, т.е. на значительную роль гольватациоиных
эффектов в процессе растворения.
6. Комплексные соединения. Строго говоря, кислородсодержащие кислоты и
их соли уже являются комплексными соединениями, хотя обычно комплексными
считают более сложные объекты. В большинстве случаев комплексные соедине-
соединения можно рассматривать как продукты взаимодействия соединений первого
порядка. Помимо рассмотренных выше вариантов образования комплексных
соединений с участием бинарных сюда следует отнести и более сложные случаи
взаимодействия солей, солей и оснований, солей и кислот и т.п. В результате
возникают соединения с существенно ионной связью между внутренней и внеш-
внешней сферами. В силу этого такие соединения в водных растворах могут быть
только солями, кислотами и основаниями. Если при взаимодействии образуются
нейтральные комплексы, они не принадлежат ни к одному из этих классов.
Комплексные соединения очень разнообразны по строению и составу и широ-
широко распространены. Поэтому они сами по себе являются интересными объектами
неорганической химии. Однако не менее важно их общехимическое значение,
поскольку они в определенной мере характеризуют природу элемента-комплексо-
образователя. Так, уже сама способность к образованию комплексных соединений
является важной "паспортной" характеристикой элемента. Известно, например,
что все s- и ^элементы в меньшей степени склонны к комплексообразованию, чем
d- и /металлы. При этом выявляются и более тонкие особенности элементов.
Например, для бериллия свойственны комплексные соединения, в то время как
для магния они не характерны, что объясняется кайносимметричностью 2|>-обо-
лочки бериллия в отличие от Зр-оболочки магния. Поэтому четыре вакант-
вакантные орбитали Bs и 2р) иона Ве2+ могут быть акцепторами электронных пар от
четырех лигандов (тетраэдрические «у3-гибридные комплексы [Ве(ОНLр~)
[Be(NH3Lp+ и т.п.). Аналогичные комплексы магния неустойчивы, поскольку для
некайносимметричной Зр-оболочки в существенно большей степени выражен,-
эффект экранирования, ослабляющий связь лигандов с комплексообразователем.
290
F
В то же время алюминий, являющийся соседом магния по периоду, проявляет
склонность к комплексообразованию, но уже при к.ч. 6. Здесь наблюдается sp^d1-
гибридизация с октаэдрической координацией, т.е. в образовании связей прини-
принимают участие вакантные кайносимметричные Зсйзрбитали алюминия.
При образовании комплексных соединений валентные возможности элемента
расширяются за счет донорно-акцепторного взаимодействия с лигандами. Устой-
Устойчивые комплексы образуются в тех случаях, когда у комплексообразователя
имеются вакантные орбитали, близкие по энергии к заполненным или заполняю-
заполняющимся. Поэтому-то более прочные связи и наблюдаются, когда в их образовании
участвуют вакантные кайносимметричные 2р-, 3rf-, 4/юрбитали. Кроме того, ус-
устойчивость многих комплексов повышается при возможности тг-взаимодействия
между комплексообразователем и лигандами. Это возможно в тех случаях, когда
лиганды имеют не только неподеленные электронные пары, обусловливающие их
донорную активность, но и вакантные орбитали.
За счет комплоксообразования часто стабилизируются такие степени окисле-
окисления элементов, которые в соединениях первого порядка неустойчивы или вовсе
не характерны. Так, хорошо известно, что степень окисления +4 для свинца не-
нестабильна и соответствующие соединения являются сильными окислителями.
Кроме того, многие из них, например РЬСЦ, неустойчивы даже при комнатной
температуре, а РЬВг4 и РЫ4 вообще не существуют. Однако комплексные кисло-
кислоты Н2[РЬГ6] и их солеобразные производные весьма устойчивы. Это тем более
интересно, если учесть, что, например, HI — сильный восстановитель, а РЫ4
должен был бы быть сильным окислителем. Таким образом, комплексообразова-
ние в этой системе подавляет даже окислительно-восстановительную активность
реагентов. Повышение устойчивости малостабильных степеней окисления элемен-
элементов возможно даже за счет образования двойных солей. Например, производные
Fe(+2) неустойчивы в водных растворах по отношению к окислению кислородом
воздуха, что создает известные трудности при их изучении. Однако уже в крис-
кристаллогидрате FeSO4-7H2O Fe(+2) более стабильно. Особенно заметно повышение
стабильности Fe(+2) в двойной соли (NH4JSO4-FeSO4-6H2O (соль Мора). При
этом в растворе могут протекать все реакции, характерные для Fe(+2), а устойчи-
устойчивость по отношению к кислороду воздуха существенно выше.
За счет комплексообразования изменяются и кислотно-основные свойства
соединений. Так, кислота средней силы HF при взаимодействии с BFj или SiF4
образует сильные комплексные кислоты H[BF4] и H2[SiF6], причем последняя в
водных растворах по силе соответствует серной кислоте. Слабая синильная кис-
кислота HCN при взаимодействии с цианидом серебра AgCN дает сильную кислоту
H[Ag(CNJ], соли которой, например Na[Ag(CNJ], в водных растворах не гидро-
лизуются. Существует закономерность и в увеличении силы комплексных кислот
при усложнении структуры комплексного аниона за счет образования изо- и
гетерополисоединений. Известно, что, например, дихромовая кислота Н2Сг207
значительно сильнее хромовой НгСгО4. В еще большей степени это заметно на
примерах поликислот молибдена и вольфрама, которые помимо дикислот образу-
образуют три-, тетра- и более сложные кислоты, сила которых возрастает с увеличением
числа ядер в комплексном ионе.
Таким образом, химия комплексных соединений представляет собой важную
область современной общей и неорганической химии и существенно расширяет
представления о химических свойствах элементов.
10* 291
РАЗДЕЛ 2
ХИМИЯ ЭЛЕМЕНТОВ
ГЛАВА X Г V. ВОДОРОД
Атом первого элемента в Периодической системе — водорода — обладает наи-
наипростейшим строением. Он состоит всего из двух частиц: протона и электрона, —
между которыми существуют лишь силы притяжения. Не случайно именно для
атома водорода оказалась успешно применимой первая квантовая теория — тео-
теория Бора, и только для этого атома волновое уравнение Шредингера имеет точ-
точное решение.
Исключительна роль водорода и в химическом отношении. Если атомы всех
остальных элементов (кроме химически инертного гелия) под валентной оболоч-
оболочкой имеют электронный остов предыдущего благородного газа и размеры их
положительных ионов не намного меньше размеров нейтральных атомов, то ион
Н+ представляет собой просто протон, размеры которого примерно в 104 раз
меньше размеров атома. Поэтому положительно поляризованный атом водорода
обладает исключительно сильно выраженным поляризующим действием, что
является одним из основных мотивов в химии этого элемента. С этим связаны
такие особые свойства элемента, как образование водородных связей, "ониевых"
соединений (оксоний, аммоний и т.п.), протолитические реакции, протонная
(бренстедовская) концепция кислот и оснований и пр.
1. Уникальное положение водорода в Периодической системе. Водород —
первый элемент и один из двух представителей первого периода системы. По
электронной формуле Ls1 он формально относится к s-элементам и является
аналогом типических элементов I группы (лития и натрия) и собственно щелоч-
щелочных металлов (подгруппа калия). Это обусловливает сходство оптических спект-
спектров водорода и щелочных металлов. Водород и металлы IA-группы проявляют
степень окисления +1 и являются типичными восстановителями. Однако в сос-
состоянии однозарядного катиона Н+ (протона) водород не имеет аналогов. В метал-
металлах IA-группы валентный электрон экранирован электронами внутренних орбита-
лей. У атома водорода отсутствует эффект экранирования, чем и объясняется
уникальность его свойств. Кроме того, единственный электрон атома водорода
является кайносимметричным, а потому исключительно прочно связан с ядром
(/i = 13,6 В или 1312 кДж/моль).
С другой стороны, как у водорода, так и у галогенов не хватает одного элект-
электрона до электронной структуры последующего благородного газа. Действительно,
водород, подобно галогенам, проявляет степень окисления -1 и окислительные
свойства. Сходен водород с галогенами и по агрегатному состоянию, и по составу
молекул Э2- Но МО водорода не имеют ничего общего с таковыми молекул
галогенов. В то же время МО Нг имеют определенное сходство с МО двухатом-
двухатомных молекул щелочных металлов, существующих в парообразном состоянии.
Таким образом, в отличие от других элементов Периодической системы у
водорода нет истинных элементов-аналогов вследствие исключительности строе-
строения его атома. Не случайно только для соединений водорода в степени окисле-
окисления + 1 имеет место специфический вид связи — водородная связь. Все это свиде-
свидетельствует о том, что в Периодической системе водороду должно быть отведено
необычное место (см. первый форзац книги). Водород по праву занимает одну
протяженную клетку над элементами 2-го периода системы (исключая неон).
Такое расположение водорода в системе вполне логично, так как 1-й период
содержит всего два элемента. Особое положение водорода в системе заслуживает
отдельного рассмотрения его свойств. Кроме того, водород (а не углерод!) образу-
образует наибольшее число химических соединений.
Водород — самый распространенный элемент Вселенной. Он составляет основ-
основную массу Солнца, звезд и других космических тел. В недрах звезд на опреде-
определенной стадии их эволюции протекают разнообразные термоядерные реакции с
участием водорода. Они и являются источником неисчислимого количества энер-
энергии, излучаемого звездами в космическое пространство. Распространенность
водорода на Земле существенно иная. В свободном состоянии на Земле он встре-
встречается сравнительно редко — содержится в нефтяных и горючих газах, присут-
присутствует в виде включений в некоторых минералах. Некоторое количество водорода
появляется постоянно в атмосфере в результате разложения органических ве-
веществ микроорганизмами, но затем водород быстро перемещается в стратосферу
вследствие его легкости. Основная масса водорода в земной коре находится в
виде химических соединений с другими элементами: большая часть его связана в
форме воды, глин и углеводородов; последние составляют основу нефти и входят
составной частью в природные горючие газы. Кроме того, растительные и живот-
животные организмы содержат сложные вещества, в состав которых обязательно входит
водород. Общее содержание водорода составляет 0,88% массы земной коры, и по
распространенности на Земле он занимает 9-е место.
2. Изотопы водорода. Природный водород представляет собой смесь изотопов:
протия ]Н, дейтерия fH (или JD) и трития ]Н (или fT). Искусственно получены
еще два изотопа водорода: f H и ]Н, но они весьма неустойчивы. Ядра протония и
дейтерия стабильны. Тритий же радиоактивен: выбрасывая электрон, он превра-
превращается в легкий изотоп гелия \\\е с периодом полураспада 12,26 года. В естест-
естественной смеси изотопов преобладает протий (99,985%), массовое содержание дейте-
дейтерия меньше (порядка 0,015%), содержание трития ничтожно. Количественные
соотношения между изотопами водорода H:D:T могут быть представлены как
1:1,46-10-5:4,0-10-15.
Для водорода, как ни для какого другого элемента, относительное различие
изотопных масс достигает значительной величины. Поэтому, хотя все изотопы
характеризуются одинаковой электронной структурой, они заметно различаются
не только физическими, но и химическими свойствами. Вследствие резкого пре-
преобладания протия влияние тяжелых изотопов сказывается незначительно и может
быть зафиксировано лишь в очень точных экспериментах. Поэтому без большой
погрешности можно считать, что свойства природного водорода соответствуют
свойствам системы, состоящей из чистого протия.
Небольшие различия свойств, именуемые изотопным эффектом, обусловлены
различием масс изотопных атомов, которое в первую очередь сказывается на
частоте колебаний изотопов в молекулах и твердых телах. Так, колебательная
энергия молекул трития и дейтерия меньше, чем протия. А это, в свою очередь,
сказывается на термодинамических свойствах: теплоемкости, температуре плавле-
плавления и кипения, энтальпии плавления и испарения, давлении насыщенного пара и
т.д. Так, дейтерий по сравнению с обычным водородом обладает меньшей
293
I
теплоемкостью, теплопроводностью и скоростью диффузии. Таким образом, для
изотопных соединений характерна термодинамическая неравноценность. Послед-
Последняя ведет к неравноценности активных комплексов при химических реакциях, в
результате чего имеет место различие в скоростях протекания реакций, т.е. наб-
наблюдается кинетический изотопный эффект. Он выражается отношением констант
скоростей химических реакций для различных изотопных соединений. Например,
отношение констант скоростей синтеза бромидов протия и дейтерия равно 5.
Такие значительные отличия физических и химических свойств изотопов одного
и того же элемента уникальны и не имеют аналогов в Периодической системе.
Все это в какой-то мере оправдывает применение для каждого изотопа водорода
собственного названия (особенно для протия и дейтерия).
3. Атомарный и молекулярный водород. Давно известно, что реакционная
способность водорода резко повышается, если использовать его в момент выделе-
выделения. В этом случае химически реагируют не молекулы, а атомы водорода. Ато-
Атомарный водород уже при комнатной температуре восстанавливает перманганат
калия, реагирует с кислородом, многими металлами и неметаллами. Атомарный
водород можно получить не только термической диссоциацией молекулярного
или при химических реакциях, но также действием тихого электрического разря-
разряда или ультрафиолетового излучения на обычный водород. Атомарный водород
может сохраняться неограниченное время в условиях малой вероятности столкно-
столкновений атомов со стенками сосуда, в отсутствие примесей. При столкновении двух
атомов водорода возникают неустойчивые частицы, имеющие избыточную энер-
энергию, выделившуюся при образовании химической связи. Эти неустойчивые час-
частицы мгновенно распадаются вновь с образованием атомов водорода. Молекулы
водорода образуются из атомов при так называемых тройных соударениях, когда
третья частица уносит с собой избыток энергии. Роль такой третьей чагтицы
могут играть молекулы водорода, примеси и стенки сосуда. Практически проме-
промежуток времени, в течение которого половинное число атомов соединяется в моле-
молекулы, равен ~ '/з с. При образовании молекул водорода из атомов (рекомбина-
(рекомбинация) выделяется столько энергии, сколько поглощается при диссоциации, т.е.
436 кДж/моль.
Молекула водорода представляет собой пример простейшей молекулы, состоя-
состоящей из двух атомов, связанных ковалентной связью. Вследствие большой проч-
прочности и высокой энергии диссоциации распад молекул водорода на атомы проис-
происходит в заметной степени лишь при 2500°С (степень термической диссоциации
0,0013). А при 5000°С молекулярный водород почти полностью диссоциирован на
атомы (степень диссоциации равна 0,95). Интересно, что для молекулы дейтерия
Бг энергия диссоциации несколько больше и равна 439,56 кДж/моль при практи-
практически равных межатомных расстояниях в Нг и D2 @,07414 и 0,07417 нм соответст-
соответственно).
Интересной особенностью молекулярного водорода является наличие в смеси
двух сортов молекул. Обе модификации отличаются друг от друга направлением
собственного момента вращения протонов. В opmo-форме о-Нг оба протона враща-
вращаются вокруг своей оси в одинаковых направлениях, т.е. спины ядер параллельны
A1). У пара-водорода р-Щ яДРа вращаются в противоположных направлениях и
ядерные спины антипараллельны (|1). Обе модификации связаны друг с другом
взаимными переходами, которые протекают очень медленно, но могут быть уско-
ускорены введением парамагнитных катализаторов (Ог, NO2 и др-)- При комнатной
294
температуре в равновесной смеси находится 75% о-Нг- При температуре, близкой
к абсолютному нулю, смесь практически содержит только р-Нг- Обе формы моле-
молекулярного водорода различаются по термодинамическим свойствам (теплоем-
(теплоемкость, энтропия и т.п.). В химическом отношении поведение обеих модификаций
практически тождественно. Существование орта- и пара-водорода — пример
новой разновидности аллотропии. Орта- и пара-модификации характерны не
только для протия, но также для молекул дейтерия и трития. Кроме того, име-
имеются указания, что подобная аллотропия наблюдается для молекул азота.
4. Физические и химические свойства водорода. При нормальных условиях
водород представляет собой очень легкий (в 14,32 раза легче воздуха) бесцвет-
бесцветный газ без запаха и вкуса. Плотность его при 0°С и давлении 1,01325• 105 Па
равна 8,99 -10 кг/л. Из всех газов водород обладает наибольшей теплопровод-
теплопроводностью (в 7 раз больше теплопроводности воздуха). Из-за малой поляризуемости
водород очень трудно сжижается. Точки кипения (-252,6° С) и плавления
(-259.1 °С) отстоят друг от друга всего на 6,5Г. Жидкий водород — прозрачная
бесцветная неэлектропроводная жидкость, поверхностное натяжение которой в 35
раз меньше, чем у воды. Плотность жидкого водорода (-253°С) равна 0,0708 кг/л.
Критическая точка характеризуется температурой -239°С и давлением 12,969 -105
Па. Твердый водород имеет малоплотную гексагональную решетку. Сжимаемость
твердого водорода наибольшая по сравнению с другими твердыми телами. Кон-
Конденсированное состояние характеризуется малыми значениями энтальпий плавле-
плавления @,116 кДж/моль) и кипения @,882 кДж/моль). Таким образом, теплота
кипения жидкого водорода во много раз превосходит теплоту плавления твердого
водорода.
Водород плохо растворяется в воде, еще хуже в органических растворителях.
Небольшие количества водорода растворяются во всех расплавленных металлах.
Хорошо растворим водород во многих твердых металлах, особенно с дефектными
d- и /орбиталями. Например, один объем палладия растворяет до 900 объемов
водорода. При этом гранецентрированная кубическая решетка металлического
палладия сохраняется, но несколько изменяются параметры элементарной ячей-
ячейки. Растворение водорода приводит к ликвидации парамагнетизма палладия, что
свидетельствует о переходе электронов от атомов водорода на вакантные d-орби-
тали атомов палладия. Это иллюстрация того, что растворение представляет
собой сложный физико-химический процесс.
Температурная зависимость процесса растворения водорода в металлах опре-
определяется знаком теплового эффекта. Для многих металлов (хром, железо, ко-
кобальт, никель, медь, серебро, платина, молибден и др.) АН > 0 и с повышением
температуры растворимость растет. Экзотермически поглощают водород (АН <
< 0) титан, цирконий, гафний, ванадий, ниобий, тантал, торий, уран и РЗЭ за
счет образования металлидных фаз внедрения. В то же время есть металлы, в
которых водород практически не растворяется. Это вольфрам, золото, цинк,
кадмий, ртуть, индий. Если при растворении водорода кристаллохимическое
строение металла не изменяется, в результате возникают твердые растворы внед-
внедрения. При растворении значительного количества водорода, как правило, крис-
кристаллохимическое строение металла-растворителя претерпевает изменения. Тогда
образуются фазы внедрения.
Исключительная прочность молекул водорода (например, прочнее молекул
фтора-в 2,7 раза) обусловливает высокие энергии активации химических реакций
295
с участием молекулярного водорода. При обычных условиях в газообразном
водороде активных молекул немного и молекулярный водород малоактивен. Он
способен непосредственно соединяться лишь с наиболее активными из неметаллов
— с фтором и на свету с хлором. Для инициирования реакций молекулярного
водорода с другими веществами требуется нагрев или другие способы активации.
При нагревании же молекулярный водород вступает в химическое взаимодействие
со многими металлами, неметаллами и сложными веществами.
Значение электроотрицательности водорода промежуточное между ОЭО метал-
металлов и неметаллов и равно 2,1. Поэтому для химии водорода характерны реакции
с понижением степени окисления, в которых он функционирует как окислитель,
и процессы с повышением окислительного числа, где он играет роль восстанови-
восстановителя. И окислительные, и восстановительные функции может выполнять и ато-
атомарный, и молекулярный водород. Однако способность быть окислителем у водо-
водорода выражена менее ярко, чем его восстановительные свойства. Это обусловлено
сравнительно небольшим значением сродства к электрону для атома водорода.
Окислительные свойства водорода проявляются, например, в реакциях со щелоч-
щелочными и щелочно-земельными металлами с образованием их гидридов. По восста-
восстановительной активности водород также уступает таким широко распространен-
распространенным в технике восстановителям, как уголь, алюминий, кальций и др.
5. Гидриды и летучие водородные соединения. Общие вопросы химии водо-
водородных соединений были рассмотрены в § 5, гл. XII. Здесь же рассмотрим неко-
некоторые вопросы, не затронутые выше. Считается, что в простых солеобразных
гидридах существует анион Н". Однако процесс его образования из молекул Н2
эндотермичен:
7гН2 (г) = Н (г), АН = 218,0 кДж/моль, A)
Н (г) + е" = И" (г), АН - -€6,9 кДж/моль B)
V2H2 (г) + е" = Н- (г), АН - 151,1 кДж/моль
Поэтому наиболее полярная связь наблюдается в солеобразных гидридах самых
активных щелочных и щелочно-земельных металлов. Ниже приведены энтальпии
образования и межатомные расстояния солеобразных гидридов щелочных метал-
металлов:
Соединение КН RbH CsH
ДЯ}|298, кДж/моль -57,82 -52,30 -54,04
г, нм : 0,285 0,302 0,319
При закономерном возрастании межатомных расстояний энтальпии образова-
образования гидридов щелочных металлов мало отличаются друг от друга. В то же время
эти величины несравненно меньше энтальпий образования галогенидов этих
металлов (АН°*2д8 и -400 кДж/моль). Это обусловлено малым сродством к элект-
электрону атома водорода, меньшей величиной ОЭО по сравнению с галогенами.
Солеобразные гидриды не только выделяют водород из воды, но и окисляют-
окисляются кислородом воздуха:
СаН2 + О2 = Са(ОНJ
Здесь каждый гидрид-ион отдает по два электрона двум атомам кислорода, при-
приобретая степень окисления +1.0 ярко выраженных восстановительных функциях
гидрид-иона свидетельствует большая отрицательная величина стандартного
электродного потенциала системы УгНг/Н" (?" = —2,23 В).
Формально к соединениям водорода со степенью окисления —1 относятся и
комплексные гидриды, например боро- и алюмогидриды лития LijBHj и LiJAlHJ
(тетрагидроборат и тетрагидроалюминат лития). Способность образовывать комп-
комплексные анионы характерна для координационно ненасыщенных простых гидри-
гидридов бора, алюминия и других sp-металлов III группы Периодической системы.
Комплексные гидриды термодинамически более стабильны по сравнению с прос-
простыми. Боро- и алюмогидриды щелочных и щелочно-земельных металлов плавятся
без заметного разложения, хорошо растворяются во многих органических раство-
растворителях. В воде они также разлагаются с выделением водорода. Комплексные
гидриды активных металлов получают либо прямым синтезом из простых ве-
веществ при повышенных температуре и давлении водорода, либо взаимодействием
простых гидридов с галогенидами. Комплексные гидриды других металлов полу-
получают обменным разложением их галогенидов с боро- и алюмогидридами щелоч-
щелочных металлов, например
ScCl3 = Li[ScH4] + А1С13
Водород в соединениях с неметаллами поляризован положительно. Поскольку
он сам является неметаллом, эти соединения сравнительно малополярны. Даже
соединения с галогенами, например НС1, представляют собой почти идеально
ковалентную молекулу. Если допустить образование положительного иона водо-
водорода при взаимодействии с сильно электроотрицательными элементами (что
маловероятно из-за большого потенциала ионизации), образующиеся соединения
должны быть малополярными в результате исключительно высокого поляризу-
поляризующего действия Н+. Таким образом, соединения водорода со степенью окисления
+1 — малополярные ковалентные вещества. Они летучи по той простой причине,
что между молекулами действуют слабые ван-дер-ваальсовы силы или водород-
водородная связь. Прочность межатомных связей и термическая устойчивость летучих
гидридов зависят в первую очередь от ОЭО и размера атома второго элемента, с
которым связан водород. Как видно из рис. 133, внутри группы прочность связей
Н—Э уменьшается сверху вниз. В этом же направлении возрастает атомный раз-
размер второго элемента и уменьшается его
ОЭО. Оба фактора действуют в направле-
направлении уменьшения прочности связи Н—Э. За
небольшими исключениями внутри пери-
периода с ростом порядкового номера Э проч-
прочность связи Н—Э возрастает из-за увеличе-
увеличения ОЭО и уменьшения размера Э. Если
же взять два элемента с одинаковой ОЭО,
более тяжелый образует менее устойчивый
летучий гидрид. Так, например, устойчи-
устойчивость метана выше, чем сероводорода, хотя
углерод И сера характеризуются Одйнако-
ВОЙ ОЭО.
Группы системы
Ж I И
ж
Рис. 133. Энергия связи в летучих водо-
родных соединениях
Периоды системы
г ' з
Рис. 134. Температуры кипения
соединений
летучих водородных
Интересно, что косвенные экспериментальные доказательства реальности
водородных связей впервые были получены сравнительным анализом некоторых
физических свойств летучих гидридов. Хорошо известен факт аномально высоких
температур кипения Н2О, HF, NH3 и отчасти НС1 и H2S (рис. 134), который обус-
обусловлен ассоциацией их молекул за счет водородных связей с образованием ди-,
три- и полимеров в жидкой фазе. Вода, например, имела бы температуру кипе-
кипения —80, а не +100°С, если бы в жидкой фазе не было водородных связей между
молекулами. Аномалия наблюдается при сравнении энтальпий испарения и тем-
температур плавления (водородная связь в твердом состоянии) обсуждаемых водо-
водородных соединений.
В газовой фазе образование протона затруднено из-за высокой энергии иони-
ионизации атомарного водорода A312 кДж/моль). В растворе же протон подвергается
сольватации с выделением значительного количества энергии, которая компенси-
компенсирует энергетические затраты на образование протона. Эти соображения делают
понятным тот факт, что в растворах и других конденсированных средах протон
существует не в свободном состоянии, а в виде ассоциатов с растворителем, т.е.
сольватов. Стремление протона к ассоциации с другими атомами и молекулами
объясняется его малым размером (~ 10~14 м) по сравнению с размерами атомов и
молекул (-» 100 м), а также его уникальной способностью поляризовать элект-
электронные оболочки взаимодействующих с ним атомов и молекул. В случае воды
протон с молекулой воды образует ион гидроксония Н3О+. Аммиак и фторид
водорода с водой образуют соответственно ионы аммония NH4 и фторония H2F+-
Энтальпии образования ониевых ионов закономерно уменьшаются от аммония к
фторонию. Так, энтальпия образования аммония -862,5, оксония — 692,8, а фто-
фторония еще меньше и равна -550,5 кДж/моль. Поэтому соли фторония в индиви-
индивидуальном состоянии не выделены и существуют лишь в жидком фториде водоро-
водорода. Соли же аммония очень распространены, в то время как производные оксония
выделены в свободном состоянии в виде кристаллических моногидратов только
сильных кислот, например OH3NO3 и ОН3СЮ4 (HNO3-H2O и НСЮ4-Н2О).
Все ме^таллоподобные гидриды обладают собственным крис-
таллохимическим строением (в отличие от твердых растворов водорода в метал-
металлах) и свойствами, типичными для металлов: металлическим блеском, значитель-
значительной твердостью. Многие из них являются жаропрочными и коррозионностойкимй
веществами. По механическим свойствам металлоподобные гидриды уступают
металлам, так как они более хрупки. Плотность этих гидридов меньше плотности
исходных металлов, а энтальпии образования больше, чем у солеобразных гидри-
гидридов, например для ZrH2 ДН^298 = -169,6 кДж/моль. В металлоподобных гидри-
гидридах часть атомов водорода отдает электроны в зону проводимости металла, а
электроны остальных атомов образуют с неспаренными электронами металла
ковалентные связи. Последние и являются причиной увеличения твердости при
образовании металлоподобных гидридов по сравнению с исходными металлами.
Эти представления хорошо согласуются с фактом миграции водорода к катоду
при длительном пропускании постоянного электрического тока, а также с умень-
уменьшением магнитной восприимчивости гидридных фаз из парамагнитных металлов.
Полимерные гидриды существуют в виде сложных структур с
цепями и полиэдрами. Сюда же относятся и твердые бороводороды. Наиболее
изучен полимерный гидрид алюминия (АШз)п- Полагают, что полимеризация
происходит за счет возникновения трехцентровых связей. Наибольшей устойчи-
устойчивостью отличаются полимерные гидриды легких элементов.
6. Получение водорода. Конверсионный метод получения водорода основан на
каталитических реакциях взаимодействия водяного пара с метаном (главный
компонент природного газа), а затем с монооксидом углерода (продукт реакции):
р
СН4 + Н2О " > СО + ЗН2, ДЯ°298 = 206,2 кДж
р
СО + Н2О %Д > СО2 + Н2, Д/Г298 = -41,2 кДж
Эндотермичность процесса конверсии метана можно частично восполнить
энергией, выделяющейся при неполном его окислении. Для проведения такого
процесса природный газ смешивается с водяным паром и кислородом, а реакция
протекает по схеме
ЗСН4 + О2 + Н2О **°С > ЗСО + 7Н2
Важным способом получения водорода является выделение его из коксового газа
и газов нефтепереработки путем глубокого охлаждения. При этом в газообразном
состоянии остается только водород, а все остальные компоненты исходной газо-
газовой смеси конденсируются. Электролиз воды обеспечивает получение наиболее
чистого водорода. Электролитом обычно служит водный раствор щелочи, приме-
применение же серной кислоты нерационально из-за быстрого коррозионного разруше-
разрушения стальной аппаратуры. Этим способом целесообразно получать водород в
районах с дешевой электроэнергией.
В лабораторных условиях водород обычно получают действием цинка на
соляную или серную кислоту.
7. Вода. Вода является одним из наиболее распространенных и важных хими-
химических соединений на Земле. Поверхность земного шара на 3/4 покрыта жидкой
(океаны, моря, озера, реки) и твердой (ледники) водой. В больших количествах
вода содержится также в атмосфере и земной коре, в связанном виде входит в
состав различных минералов и пород (глина, гипс, кристаллогидраты солей).
Вода составляет больше половины массы живых организмов. Она играет особую
роль в самых разнообразных процессах и явлениях живой и неживой природы, а
также в практической деятельности человека. Несмотря на большую распростра-
распространенность воды, запасы пресной воды, пригодной для питья и промышленных
нужд, весьма ограничены. Непрерывный рост потребления воды как в быту, так и
для нужд народного хозяйства требует рационализации ее использования, охра-
охраны водных ресурсов, очистки сточных вод, борьбы с загрязнениями рек и озер
промышленными отбросами. В настоящее время пресная вода рассматривается
как один из наиболее важных минеральных ресурсов. Широким фронтом ведутся
работы по обессоливанию морской воды.
Чистая вода прозрачна и бесцветна. Она не имеет ни запаха, ни вкуса. Вкус и
запах воде придают растворенные в ней примесные вещества. Многие физические
свойства и характер их изменения у чистой воды аномальны. Это относится к
температурам плавления и кипения, энтальпиям и энтропиям этих процессов.
Аномален и температурный ход изменения плотности воды. Вода имеет макси-
максимальную плотность при +4° С. Выше и ниже этой температуры плотность воды
уменьшается. При отвердевании происходит дальнейшее резкое уменьшение
плотности, поэтому объем льда на 10% больше равного по массе объема воды при
той же температуре. Все указанные аномалии объясняются структурными измене-
изменениями воды, связанными с возникновением и разрушением межмолекулярных
водородных связей при изменении температуры и фазовых переходах. Аномалия
плотности воды имеет огромное значение для жизни живых существ, населяющих
замерзающие водоемы. Поверхностные слои воды при температуре ниже +4° С не
опускаются на дно, поскольку при охлаждении они становятся более легкими:
Поэтому верхние слои воды могут затвердевать, в то время как в глубинах водое-
водоемов сохраняется температура +4°С. В этих условиях жизнь продолжается. Если
бы плотность льда была больше плотности воды (как у большинства других
веществ), все водоемы на Земле постепенно промерзли бы до дна и живые орга-
организмы в них погибли бы. Кроме того, получаемой от Солнца теплоты (включая
теплое время года) недостаточно для оттаивания всей массы воды, если бы она
превратилась в лед.
Химические свойства воды во многом определяются ее химическим строением,
в частности наличием двух неподеленных электронных пар и значительной по-
полярностью молекулы. Вода является весьма реакционноспособным веществом.
Она может быть окислителем, восстановителем, вступать в реакции без изменения
степени окисления, функционировать в качестве лиганда в комплексных соедине-
соединениях. Конкретно подобные реакции будут рассмотрены при описании химии
отдельных элементов.
В воде растворяется большинство неорганических кислот, оснований и солей.
Из ковалентных водородных соединений в воде хорошо растворяются те, которые
подвергаются электролитической ионизации с образованием гидратированных
ионов (например, НС1) и способны давать межмолекулярные водородные связи с
молекулами воды (например, РШз). Из органических веществ растворимы в воде
те, молекулы которых содержат полярные функциональные группы: многие кис-
кислоты, спирты, амины, сахара и т.д. С другой стороны, практически все вещества,
с которыми мы имеем дело, содержат следы воды. Например, температуры кипе-
кипения ртути, брома, этилового спирта и т.п. после тщательного высушивания повы-
повышаются на десятки градусов.
Вода является катализатором целого ряда химических процессов. В ее отсут-
1
г
ствие многие вещества почти не взаимодействуют химически. Например, после
длительного высушивания гремучая смесь не взрывается даже при высокой тем-
температуре, угарный газ не горит в кислороде, хлор не действует на металлы, фто-
фторид водорода не разъедает стекло, натрий и фосфор не окисляются на воздухе и
не реагируют с хлором. Следы воды катализируют также некоторые реакции
разложения. В то же время вода иногда затрудняет протекание реакций, а также
может выступать как каталитический яд. Так, при синтезе аммиака из элементов
на железном катализаторе присутствие в реакционной смеси следов водяного
пара отравляет катализатор.
Вода, молекулы которой включают тяжелые изотопы водорода и кислорода,
обобщенно называется тяжелой водой. Однако под тяжелой водой прежде всего
имеют в виду дейтериевую воду D2O16. В природной воде 99,73% приходится на
обычную воду Н2О16. Из тяжелых разновидностей в природной воде больше
других содержится Н2О18 @,2 мол. доли, %), Н2О17 @,04 мол. доли, %) и HDO
@,03 мол. доли, %). Содержание остальных разновидностей тяжелой воды, в том
числе и тритиевой Т2О, составляет не более 10~5 мол. доли, %. Химическое строе-
строение молекул тяжелой воды такое же, как у обычной, с очень малыми различиями
в длинах связей и углах между ними. Однако частоты колебаний в молекулах с
тяжелыми изотопами заметно ниже, а энтропия выше, чем в протиевой воде.
Химические связи D—О и Т—О прочнее связи Н—О, числовые значения измене-
изменения энергии Гиббса реакций образования D2O и Т2О более отрицательны, чем
для Н2О (-190,10. -191,48 и -185,56 кДж/моль соответственно). Следовательно,
прочность молекул в ряду Н2О, D2O, T2O растет. Для конденсированного состоя-
состояния разновидностей тяжелой воды также характерна водородная связь. Лучше
других исследованы свойства дейтериевой воды D2O, которую обычно и называ-
называют тяжелой водой. По сравнению с Н2О она характеризуется большими значени-
значениями плотности, теплоемкости, вязкости, температур плавления и кипения. Раст-
Растворимость большинства веществ в тяжелой воде значительно меньше, чем в про-
протиевой. Более прочные связи D—О приводят к определенным различиям в кине-
кинетических характеристиках реакций, протекающих в тяжелой воде. В частности,
протолитические реакции и биохимические процессы в ней значительно замед-
замедлены. Вследствие этого тяжелая вода является биологическим ядом. Получают
тяжелую воду многоступенчатым электролизом воды, окислением обогащенного
дейтерием протия, изотопным обменом между молекулами воды и сероводорода с
последующей ректификацией обогащенной дейтерием воды.
8. Пероксид водорода. Помимо воды к водородным соединениям кислорода с
полярной ковалентной связью относится также пероксид водорода Н2О2. Молеку-
Молекула еср нелинейна, две связи О—Н расположе-
расположены не симметрично, а в двух плоскостях под
углом 120° (рис. 135). Вследствие этого по-
полярность и электрический момент диполя
молекулы Н2О2 больше, чем у воды. Связь
между атомами кислорода в пероксидной
группе —О~О— почти в 3 раза слабее связи
О—Н. Жидкий и твердый пероксид водорода,
а также его водные растворы характеризуются
УСТОЙЧИВЫМИ ВОДОРОДНЫМИ СВЯЗЯМИ. Водород- Рис. 135. Строение молекулы перок-
ная связь приводит к ассоциации молекул сида водорода
120°
Н2О2, вследствие чего пероксид водорода представляет собой вязкую светло-
голубую жидкость без запаха. Пероксид водорода почти в 1,5 раза тяжелее воды,
его температура кипения выше, а поверхностное натяжение больше, чем у воды.
Получают пероксид водорода окислением алкильных производных гидрохино-
гидрохинона воздухом (80% от общего количества), а также электролизом растворов суль-
сульфата аммония или серной кислоты. В последнем случае гидрогульфат-анионы
окисляются на аноде в пероксодисерную (надсерную) кислоту, при взаимодей-
взаимодействии которой с водой получается перокгид водорода:
2HSO4 - 2с- = H2S2OS; H2S2Og + 2Н2О = 2H2SO4 + Н2О2
Небольшие количества пероксида водорода постоянно образуются в природе при
грозовых разрядах, атмосферной коррозии металлов, медленном окислении орга-
органических и неорганических веществ. Согласно теории Баха — Энглера, окисление
многих веществ протекает через стадию образования неустойчивых промежуточ-
промежуточных пероксидных соединений, при превращении которых в устойчивые оксиды
часто выделяется пероксид водорода.
Пероксид водорода высокой чистоты и разбавленные водные растворы его при
комнатной температуре вполне устойчивы, но в присутствии ионов переходных
металлов и серебра, а также под действием УФ-излучения разлагаются с выделе-
выделением кислорода. Неустойчивость молекулы Н2О2 и легкость отщепления кислоро-
кислорода связаны с малой прочностью пероксидной цепочки —О—О—. Во избежание
разложения пероксид водорода хранят в темных скляьках в прохладном месте.
Для этой же цели используют ингибиторы, например фосфат натрия. В продажу
поступает высококонцентрированный раствор пероксида водорода с 85—90%-ным
содержанием Н2О2, а также пергидроль, представляющий собой 30%-ный водный
раствор.
Подобно воде, пероксид водорода хорошо растворяет многие вещества с по-
полярными связями. Высокая диэлектрическая проницаемость, наличие водород-
водородных связей делают Н2О2 и ее водные растворы превосходными ионизирующими
растворителями.
В водных растворах пероксид водорода проявляет слабые кислотные свойства:
Н2О2 + Н2О = Н3О+ + НО2, рА'а = 11,65 *
Как кислота Н2О2 может реагировать со щелочами, образуя пероксиды металлов:
Н2О2 + Ва(ОНJ = ВаО2 + 2Н2О
ВаО2 представляет собой соль пероксида водорода, а не оксид бария. Аналогич-
Аналогичные по составу кислородные соединения, например SnO2, PbO2, MnO2, в структу-
структуре которых отсутствуют пероксидные группы —О—О—, являются нормальными
оксидами, а не пероксидами. При взаимодействии последних с кислотами выде-
выделяется пероксид водорода, а не вода:
ВаО2 + H2SO4 = BaSO4 + Н2О2
SnO2 + 2H2SO4 = Sn(SO4J + 2H2O
S' ¦
* Для удобства вместо К (константа диссоциации) используется отрицательный деся-
десятичный логарифм этой величины рК (подобно рЯ). Такой же смысл имеют pJ^,ecr Для
комплексов, рПР для труднорастворимых солей (ПР — произведение растворимости).
Производными пероксида водорода являются надкислоты и надоснования (пе-
роксосоединения). В их химическом строении обязательно наличие пероксидной
цепочки. Формально формулы надкислот получают при замещении водорода в
молекуле Н2О2 на кислотные радикалы:
О О
Н-О-О О || || Н-О-О^ О
s h-0-s-o-o-s-o-h р^
н-о ^о || || н-о о-н
о о
мононадсерная динадсерная надфосфорная
(пероксомоносерная) (пероксодисерная) (пероксомонофосфорная)
При замене одного атома водорода в молекуле Н2О2 на атом металла получаем
надоснования, например К—О—О—ОН. Поскольку подобные соединения можно
рассматривать как кислые соли пероксида водорода, их часто именуют гидро-
пероксидами. Характерное свойство пероксидов металлов, пероксокислот и гид-
ропероксидов — способность подвергаться гидролизу с образованием пероксида
водорода и разлагаться, выделяя кислород, т.е. быть окислителями. Сам перок-
сид водорода может функционировать и как окислитель, и как восстановитель:
Окислитель: Н2О2 + If + 2Н+ = 2Н2О, Е° = +1,776 В,
Восстановитель: О2 + 2Н+ + 2е" = Н2О2, Е° = +0,682 В.
Как видно из значений стандартных редокс-потенциалов, окислительные свой-
свойства пероксида водорода выражены сильнее, чем восстановительные. Пероксид
водорода легко окисляет иодиды в свободный иод, нитриты в нитраты, хроматы
в пероксохроматы и т.д. Восстановительные свойства проявляет реже, только в
реакциях с сильными окислителями, например в кислой среде Н2О2 восстанавли-
восстанавливает М11О4 до Мп2+. Кроме того, пероксид водорода подвержен реакции диспро-
порционирования, примером которой может служить процесс его разложения на
свету, в присутствии примесей и при нагревании:
Н262 + Н262 = 62 + 2Н262
Г Л А В А XV. ЭЛЕМЕНТЫ I ГРУППЫ
Первая группа Периодической системы характеризуется тем, что в ней разме-
размещаются элементы с резко отличными свойствами. С одной стороны, это литий и
натрий, а также исключительно химически активные собственно щелочные метал-
металлы, а с другой — медь и такие благородные металлы, как серебро и золото. Все
они объединяются групповой аналогией. Как и в других группах, между типичес-
типическими элементами, а также элементами подгрупп калия и меди соответственно
наблюдается типовая аналогия. Кроме того, металлы подгруппы калия являются
слоевыми аналогами. Несколько отличается химия лития как первого типическо-
типического и кайносимметричного элемента IA-группы. Кроме того, имеет место диаго-
диагональная аналогия между литием и магнием. Диагональными аналогами в узком
смысле являются также натрий и кальций. С металлохимической точки зрения,
между элементами IA- и IB-групп также имеется существенное различие. Для
металлов IA-группы вовсе не характерно образование широких областей твердых
растворов с металлами других групп, а элементы подгруппы меди, наоборот,
дают непрерывные или ограниченные твердые растворы с широкими областями
гомогенности. В то же время и те и другие металлы не образуют фаз внедрения.
1. Литий. Особенности лития. В отличие от водорода, у которого
единственный электрон на кайносимметричной ls-орбитали, у лития на кайно-
симметричной 2р-орбитали нет еще ни одного электрона. Имеющийся один элек-
электрон 2в-орбитали не является кайносимметричным. Это один из главных аргу-
аргументов, почему водород не может быть предшественником лития по группе IA.
Как и другие металлы IA-группы, литий стабильно проявляет степень окисления
+1. Тем йе менее из-за наличия кайносимметричной 2р-оболочки химия лития
существенно отличается от химии его аналогов по группе. В степени окисления
+1 литий по сравнению с другими элементами IA-группы является лучшим комп-
лексообразователем. Этим, в частности, объясняется большая отрицательная
величина стандартного электродного потенциала лития (-3,05 В). Дело в том, что
энтальпия гидратации катионов лития сравнительно велика (ДЯ°д8 = -468,6
кДж/моль), т.е. прочность аквакомплекса лития значительно выше, чем натрия и
щелочных металлов. При окислении кислородом только литий образует характе-
характеристический оксид Li2O, а остальные металлы обсуждаемой группы дают перок-
сиды, супероксиды. При окислении на воздухе только литий наряду с нормаль-
нормальным оксидом образует нитрид LisN, отвечающий правилам формальной валент-
валентности. Фторид, карбонат и фосфат лития плохо растворимы в воде в противопо-
противоположность аналогичным соединениям других металлов IA-группы. Наконец, литий
хорошо образует литийорганические соединения аналогично магнию. Металлохи-
Металлохимия лития также во многом отличается от таковой натрия и металлов подгруппы
калия.
Природные соединения и получение лития. Суммар-
Суммарное содержание лития в земной коре 3,4-10%. Он входит в состав многих мине-
минералов, содержится в каменных углях, почвах, морской воде, а также в живых
организмах и растениях. Промышленным минералом лития является сложный
полисиликат сподумен LiAlj^Oe]. При вакуум^гермическом восстановлении спо-
сподумена или оксида лития в технике в качестве восстановителя применяют крем-
кремний или алюминий. При электролитическом восстановлении используют эвтекти-
эвтектическую смесь (для понижения температуры) хлоридов лития и калия. Содержа-
Содержание основного металла 99,4%. Электролиз расплавов с применением эвтектики из
хлорида и бромида лития дает особо чистый металл.
Физические и химические свойства лития. Литий —
серебристо-белый металл, на воздухе покрывается темно-серым налетом (смесь
1Л2О и LJ3N). Литий является самым легким из всех металлов, его плотность при
0°С равна 0,539 г/см3. Литий относительно мягок, но режется ножом значительно
труднее натрия, хотя отличается высокой пластичностью. Литий имеет самые
высокие температуры плавления и кипения A80,5 и 1327°С) среди металлов IA-
группы.
По сравнению с натрием и собственно щелочными металлами литий является
химически менее активным элементом. Объясняется это тем, что валентный 2s-
электрон лития значительно ближе к.ядру и более прочно с вим связан. Взаимо-
действие лития с водой протекает более спокойно, чем у других металлов IA-
группы. С кислородом и азотом литий реагирует уже при комнатной температуре,
.а выше 200°С воспламеняется. В обычных условиях он бурно реагирует со всеми
галогенами. При небольшом нагревании литий взаимодействует с водородом,
серой и другими неметаллами.
Характеристические соединения. Оксид лития Li2O полу-
получается непосредственным взаимодействием металла и кислорода. Он представляет
собой бесцветное кристаллическое вещество с преимущественно ионной связью
(*пл = 1570°С; ДЯу298 = -595,8 кДж/моль). По химической природе Li2O — ос-
основный бксид, а потому при взаимодействии с кислотными оксидами и кислота-
кислотами образует соли. Так, Li2O легко поглощает СО2 с образованием карбоната
лития Термическим разложением карбоната, а также гидроксида и нитрата в
токе сухого водорода также можно получить оксид лития. Оксид лития легко
соединяется с водой, образуя гидроксид. LiOH — бесцветные кристаллы с tun =
= 462°С и АН", 298 = -487,2 кДж/моль. LiOH является сильным основанием. В
отличие от гидроксидов других элементов IA-группы LiOH при 600°С разлагает-
разлагается по реакции 2LiOH = Li2O + Н2О. В этом отношении гидроксид лития похож
на ЭОН металлов подгруппы меди. Из водных растворов кристаллизуется моно-
моногидрат, который полностью обезвоживается при температуре выше 600°С. Полу-
Получают LiOH электролизом раствора хлорида лития с ртутным катодом.
Характеристическими соединениями лития являются также его галогениды. В
ряду галогенидов лития закономерно изменяются температуры плавления и
энтальпии образования:
Соединение LiF LiCl LiBr Lil
ion, °C 870 614 549 450
-AH°f 2g8, кДж/моль 612,1 408,8 350,3 271,1
Кристаллохимическое строение всех галогенидов одинаково: все они кристал-
кристаллизуются в ГЦК-структуре типа NaCl. Поэтому температура плавления характе-
характеризует прочность химической связи. Для фторида лития наблюдаются резкое
увеличение как температуры плавления, так и энтальпии образования. Здесь
сказывается большая ОЭО атома фтора и малые размеры взаимодействующих
атомов. Кроме того, фторид лития обладает малым сродством к воде: плохо раст-
растворяется в ней и не образует кристаллогидратов. Все остальные галогениды
лития гигроскопичны, хорошо растворяются в воде и образуют множество крис-
кристаллогидратов.
Соединения лития с другими неметаллами. Гидрид
лития получается гидрированием расплавленного лития водородом. LiH устой-
устойчивее гидридов других элементов IA-группы (Д#°*298 = -90,41 кДж/моль). В
отсутствие воздуха плавится при 690°С без разложения. С водой реагирует, как и
все солеобразные гидриды, с выделением водорода и образованием гидроксида
лития, а кислородом окисляется только при температуре красного каления. Уже
в обычных условиях литий взаимодействует с азотом, образуя нитрид Li3N
(ДЯ^298 = -90,41 кДж/моль). Скорость образования нитрида резко возрастает
при 250°С. Он плавится без разложения при 845°С, а при растворении в воде
305
выделяет аммиак и LiOH. Восстановлением карбоната лития углем, а также не-
непосредственным взаимодействием простых веществ получают карбид Li2C2. Кар-
Карбид лития является сильным восстановителем, а при нагревании термически
диссоциирует на литий и графит. В отличие от углерода кремний образует с
литием силициды Li4Si и Li2Si, отвечающие правилам формальной валентности.
При сплавлении серы и лития образуется сульфид Li2S (*пл = 950°С, Д#у298 =
= -448 кДж/моль). Он представляет собой бесцветные кристаллы, в водных
растворах легко гидролизуется:
Li2S + Н20 = LiHS + LiOH
При нагревании сухой сульфид окисляется до сульфата, присоединяя две
молекулы кислорода. В отличие от других элементов IA-группы литий не образу-
образует полисульфидов. С фосфором литий образует 1лзР, который нацело разлагается
водой:
L3P + ЗН2О = ЗЫОН + РН3
Соли кислородсодержащих кислот. Для сравнения приве-
приведем температуры плавления и энтальпии образования литиевых солей трех кис-
кислородсодержащих кислот:
Соединение LJNO3 U2CO3 U2SO4
1шь °С 261 732 860
-AH°f 29g, кДж/моль 482,3 1215,6 1434,4
Наименее прочный нитрат лития при 600°С разлагается:
2LiNO3 = Li2O + 2NO2 + V2O2
Нитраты других элементов IA-группы при термическом разложении превращают-
превращаются в нитриты. Получают 1ЛМОз действием на LiOH или Li2CO3 разбавленной
азотной кислотой. Карбонат лития получают нейтрализацией раствора LiOH
диоксидом углерода. В отличие от нитрата и сульфата карбонат лития плохо
растворим в воде. Термическая диссоциация на Li2O и СО2 начинается выше
температуры плавления. Сульфат лития — термически наиболее устойчивая из
солей кислородсодержащих кислот, характеризуется многими полиморфными
модификациями, получается разложением карбоната лития разбавленной серной
кислотой с последующим упариванием раствора.
Металлохимия лития. По металлохимическим свойствам литий
также отличен от других элементов IA-группы. Объясняется это аномально малой
плотностью, резким увеличением температуры плавления в направлении от нат-
натрия к литию, а также размерными факторами. Так, литий при сплавлении со
своими групповыми аналогами (IA-группа) дает расслоение. В противоположность
другим металлам IA-группы литий не образует металлидов с металлами подгруп-
подгруппы меди. Литий с алюминием образует интерметаллические соединения, тогда
как остальные металлы IA-группы не смешиваются с алюминием в расплавленном
состоянии. В то же время все металлы IA-группы, включая литий, хорошо обра-
образуют амальгамы. Кроме того, однотипный характер имеет взаимодействие метал-
металлов IA-группы с Ga, In, Pb и Sn.
306
2. Щелочные металлы. Характеристика элементов I A -
группы. Сопоставление некоторых физических и химических свойств натрия
и лития, с одной стороны, и элементов подгруппы калия — с другой, свидетель-
свидетельствует о том, что натрий ближе к собственно щелочным металлам (подгруппа
калия). Поэтому второй типический элемент не выделен в отдельный параграф,
чтобы не создавалось впечатление искусственного отделения его от собственно
щелочных металлов. В ряду Na—Cs наблюдается плавное изменение плотности,
температур плавления и кипения, а также энергий диссоциации двухатомных
молекул Э2 и стандартных электродных потенциалов в водных растворах. Общим
для всех щелочных металлов является ярко выраженная электроположительность
и химическая активность вследствие больших величин радиусов, малых значений
ионизационных потенциалов и ОЭО. Ниже приведены некоторые свойства эле-
элементов и простых веществ 1А-группы:
Li Na К Rb Cs
Содержание в земной коре,
масс, доли, % 3,4-10 2,64 2,6 1,5-КГ2 3,7-10
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Ме1+, им
Потенциал ионизации, В
Д: Me —> Ме1+ + е
0Э0
Температура плавления, "С
Температура кипения, ° С
Платность, г/см3
Я". (Ме*.р/Ме), В
Природные соединения и получение щелочных
металлов. По содержанию в земной коре B,6 масс, доли, %) натрий и калий
являются одними из самых распространенных. Содержание рубидия меньше, а
цезия еще меньше. Натрий и калий входят в состав всех силикатных пород. Из
отдельных минералов натрия наиболее важны каменная соль, мирабилит
(Na2SCv 10H2O), а для калия — сильвинит, карналлит. Рубидий часто изоморфно
замещает калий в сильвините и карналлите. Для цезия известен минерал поллу-
цит состава CsAl[Si2O6J. Все эти металлы могут быть выделены в свободном состо-
состоянии электролизом расплавов соответствующих хлоридов. Для натрия и калия
это промышленный способ получения. Рубидий и цезий чаще получают вакуум-
термическим восстановлением их хлоридов металлическим кальцием. Все щелоч-
щелочные металлы хорошо очищаются возгонкой в вакууме.
Физические и химические свойства. Все щелочные ме-
металлы серебристо-белого цвета, а ничтожные примеси кислорода придают цезию
золотисто-желтую окраску. Натрий и калий легче воды, а цезий почти в два раза
тяжелее. Все щелочные металлы мягки, пластичны, в атмосфере сухого воздуха
быстро тускнеют. При этом Na и К образуют оксиды Э2О, a Rb и Cs — перокси-
ды Э2О2. С водой натрий реагирует бурно, калий — со взрывом, a Rb и Cs вос-
воспламеняются даже при соприкосновении со льдом. При взаимодействии с влаж-
307
[Heps1
0,155
0,068
5,39
0,95
180,0
1340,0
0,53
-3,045
[Ne]3s!
0,189
0,098
5,14
0,90
97,8
882,5
0,97
-2,714
[Ar]4s!
0,236
0,133
4,34
0,80
63,5
758,5
0,86
-2,924
[Кфв1
0,248
0,149
4,18
0,8
38,7
696,0
1,52
-2,925
[Хф*1
0,267
0,165
3,89
0,75
28,5
706,0
1,89
-2,923
!*• ¦
ным воздухом все щелочные металлы в конечном итоге превращаются в карбона-
карбонаты. Поэтому натрий и калий обычно хранят под керосином, а рубидий и цезий —
в эвакуированных ампулах. В атмосфере кислорода щелочные металлы сгорают.
При этом натрий образует пероксид Na2O2, а остальные — надпероксиды (Э02).
Для калия, рубидия и цезия известны озониды ЭО3. При обычных условиях
щелочные металлы горят в атмосфере фтора и хлора, а при небольшом нагрева-
нагревании легко взаимодействуют с серой, водородом, бромом и другими неметаллами.
Характеристические соединения. В силу большого срод-
сродства щелочных металлов к кислороду их характеристические оксиды Э20 непроч-
непрочны и обладают тенденцией к дальнейшему присоединению кислорода. В отличие
от них пероксиды Э2О2 не разлагаются. Нормальные оксиды могут быть получе-
получены восстановлением пероксидов одноименными металлами. Для сравнения приво-
приводим энтальпии образования оксидов и пероксидов щелочных металлов
(кДж/моль):
Элемент Na К Rb Cs
Оксид Э20 -418 -364 -331 -318
Пероксид Э2О2 -510 -494 -422 -402
Оксид натрия бесцветен, а остальные оксиды желтого и оранжевого (Cs2O)
цвета. Все они очень гигроскопичны, с присоединением воды переходят в харак-
характеристические гидроксиды.
Гидроксиды щелочных металлов МеОН — кристаллические вещества, раство-
растворимые в воде и спиртах. Их водные растворы — едкие щелочи — самые сильные
основания. Гидроксиды получают электролизом водных растворов хлоридов. При
этом в катодном пространстве выделяется водород и образуется гидроксид ще-
щелочного металла. Побочными продуктами производства являются водород и хлор
(на аноде). При нейтрализации растворов гидроксидов щелочных металлов гало-
геноводородными кислотами образуются их галогениды, которые являются ха-
характеристическими соединениями. Они также получаются непосредственным
взаимодействием щелочных металлов с галогенами. Галогениды щелочных метал-
металлов характеризуются высокими температурами плавления и кипения, по природе
химической связи они — самые ионные соединения.
Соединения с другими неметаллами. Гидриды щелочных
металлов МеН в отличие от гидрида лития разлагаются на элементы до плавле-
плавления. Все они кристаллизуются в структуре NaCl и являются сильными восстано-
восстановителями. При непосредственном взаимодействии компонентов в вакууме образу-
образуются сульфиды 92S. Они представляют собой бесцветные кристаллы, хорошо
растворимые в воде и спирте. В водном растворе они, как и сульфид лития,
подвергаются гидролизу с образованием гидроксидов и гидросульфидов щелоч-
щелочных металлов. Под действием кислорода воздуха Na2S медленно окисляется до
тиосульфата:
2Na2S + 2О2 + Н2О = Na2S2O3 + 2NaOH
Кроме того, щелочные металлы образуют полисульфиды. Селениды и теллуриды
щелочных металлов получают сплавлением компонентов в вакууме. В воде они
разлагаются с выделением селена и теллура, например
Na2Se + 2Н2О = 2NaOH + Se + Н2
ЗОЯ
Нитриды щелочных металлов образуются при взаимодействии паров металлов с
возбужденными атомами азота (тлеющий разряд). Они гигроскопичны и энергич-
энергично взаимодействуют с водой:
Na3N + ЗН2О = 3NaOH + NH3
Фосфиды щелочных металлов получают прямым синтезом из компонентов или
действием белого фосфора на раствор металла в жидком аммиаке. Натрий и
калий образуют фосфиды Э3Р и 32Ps- Последний формульный состав характерен
также для рубидия и цезия. Водой фосфиды нацело гидролизуются с выделени-
выделением фосфина и гидроксида. Арсениды щелочных металлов ЭзАв менее стабильны,
чем фосфиды, и совершенно неустойчивы к действию влаги. Действием ацетилена
на нагретые металлы получают карбиды щелочных металлов Э2С2 (ацетилиды).
При нагревании они разлагаются на элементы, причем термическая стойкость
растет в ряду Li2C2 —> Na2C2 —> Cs2C2 —> Rb2C2 —> K2C2. В этом отношении
натрий ближе к литию, чем к остальным щелочным металлам. Водой все ацети-
ацетилиды разлагаются с выделением ацетилена и образованием гидроксида.
Соли кислородсодержащих кислот. Физические свойства
солей кислородсодержащих кислот щелочных металлов закономерно изменяются
в зависимости от положения элемента внутри группы Периодической системы.
Ниже приводятся температуры плавления и энтальпии образования нитратов,
карбонатов и сульфатов натрия и калия. Для каждого из соединений первая
цифра означает температуру плавления (°С), а вторая — энтальпию образования
(кДж/моль):
Нитрат Карбонат Сульфат
Натрий 308; -466,7 853; -1130,93 884; -1384,5
Калий 336; -492,7 891; -1146,1 1074; -1433,7
Как и в случае соответствующих соединений лития, слева направо возрастают
и температура плавления, и энтальпия образования, что и следовало ожидать. Но
при переходе от лития к аналогичным соединениям натрия и калия практически
не увеличивается энтальпия образования, хотя наблюдается заметное увеличение
температур плавления соответствующих соединений. Больше того, при переходе
от соединений лития к соединениям натрия имеет место уменьшение теплот обра-
образования, что свидетельствует о большей прочности рассматриваемых соединений
для лития. Рост температур плавления от литиевых к калиевым соединениям
объясняется увеличением доли ионной" связи между атомами металла и остальной
частью фрагмента структуры внутри каждой группы обсуждаемых веществ.
Из солей кислородсодержащих кислот наиболее важны карбонаты. Сода
Na2CO3 относится к числу продуктов основной химической индустрии. Получают
ее аммиачным способом, при котором концентрированный раствор NaCl насыща-
насыщается аммиаком, а затем обрабатывается диоксидом углерода. В этом процессе
осаждается гидрокарбонат, который прокаливанием переводят в карбонат:
NaCi + NH3 + CO2 + Н2О = NH4C1 + NaHCO3
2NaHCO3 = Na2CO3 + CO2 + H2O
309
Поташ К2СОз получают при переработке природного КС1, а также выщела-
выщелачиванием золы лиственных пород деревьев и подсолнечника. И сульфат, и
нитрат натрия, а также сульфат калия в больших количествах встречаются в
природе.
Металлохимия. Металлы подгруппы калия между собой образуют
непрерывные твердые растворы. Натрий не дает непрерывных твердых растворов
с другими щелочными металлами и согласно этому металлохимическому крите-
критерию стоит ближе к литию. Для щелочных металлов наиболее характерно образо-
образование металлидов с s- и sp-металлами, а также с элементами с полностью запол-
заполненными (и — 1)<йэрбиталями (металлы подгрупп меди и цинка). Так как щелоч-
щелочные металлы не смешиваются с жидким алюминием, они с ним не образуют ни
твердых растворов, ни металлидов. В то же время литий и натрий дают металли-
ды с галлием и индием. С переходными металлами с дефектной (п — ^(/-оболоч-
^(/-оболочкой щелочные металлы не взаимодействуют, а при высоких температурах наблю-
наблюдается расслоение в широком диапазоне концентраций.
3. Подгруппа меди. Характеристика элементов I В -г р у п -
п ы. Сравнительно малая химическая активность элементов подгруппы меди
объясняется двумя причинами. Во-первых, ярко выраженным в их атомах эффек-
эффектом проникновения s-электронов внутрь непосредственно подстилающих (п — Y)d-
оболочек [в случае золота и (и — 2)/оболочки]. Во-вторых, в результате (/-конт-
(/-контракции и совместной d- и /-контракции (золото) радиусы их атомов значительно
меньше радиусов атомов щелочных металлов [г (К) > г (Си), г (Rb) > г (Ag),
г (Cs) > г (Аи)]. В результате металлы подгруппы меди характеризуются несрав-
несравненно большими значениями первого ионизационного потенциала, ОЭО, сродства
к электрону, чем щелочные металлы.
У элементов IB-группы реализуются степени окисления +1, +2 и +3. Для
меди наиболее характерны соединения со степенями окисления +1 и +2, для
золота +1 и +3, а для серебра +1. Все они обладают ярко выраженной склоннос-
склонностью к комплексообразованию. Серебро несколько выпадает из этого ряда элемен-
элементов не только по степени окисления. Даже в форме простого вещества серебро
отличается от меди и золота (цвет, температура плавления, энергия атомизации и
т.д.). Эта вторичная периодичность элементов IB-группы еще ярче проявляется в
их металлохимии.
Природные соединения и получение металлов, Все
элементы IB-группы относятся к сравнительно мало распространенным в земной
коре. Медь чаще встречается в виде сульфидов (CU2S — медный блеск, CuFeS2 —
медный колчедан) и кислородсодержащих соединений [Си2О — куприт,
Си2(ОНJСОз — малахит]. Кроме аргентита Ag2S серебро содержится в полиметал-
полиметаллических рудах. Для золота известен минерал калаверит (АиТе2), но обычно он
находится в самородном состоянии.
Медь получают пирометаллургическим восстановлением окисленных сульфид-
сульфидных концентратов. Выделяющийся при обжиге сульфидов диоксид серы SO2 идет
на производство серной кислоты. Восстановленную черновую медь очищают
электрохимическим рафинированием. Из анодного шлама извлекают благород-
благородные металлы, селен, теллур и др. В целом в производстве меди намечаются кон-
контуры безотходной технологии. Серебро получают при переработке полиметалли-
полиметаллических (серебряно-свинцово-цинковых) сульфидных руд. После окислительного
обжига концентрата плавку ведут так, что серебром обогащается расплав цинка.
310
В дальнейшем цинк отгоняют, примесь свинца окисляют, а черновое серебро
подвергают электрохимическому рафинированию. При цианидном способе добы-
добычи золота сначала золотоносную породу отмывают водой, затем обрабатывают
раствором NaCN на воздухе. При этом золото образует комплекс Na[Au(CNJ], из
которого его осаждают цинком:
2Na[Au(CN2)] + Zn = Na2[Zn(CNL] + 2Au
Этим способом можно выделять и серебро из бедных руд. При ртутном способе
золотоносную породу обрабатывают ртутью с целью получения амальгамы золота.
Затем ртуть отгоняется и остается металлическое золото.
Физические и химические свойства. Медь и золото в
отличие от большинства металлов обладают ярко выраженной цветностью. Медь
красного, а золото желтого цвета. Серебро не имеет яркой окраски; оно, как и
многие металлы, — белый блестящий металл. Это обусловлено хорошо выражен-
выраженной отражательной способностью металлов. Ниже для сравнения приведены неко-
некоторые константы представителей IB-группы:
Си Ag Аи
Содержание в земной коре,
масс, доли, % 3-Ю 6-Ю 4-Ю
Валентная электронная конфи-
конфигурация [Ar]3rflo4s1
Атомный радиус, нм 0,128
Ионный радиус, нм 0,093
Потенциал ионизации, В
h: Э -> Э+ + е 7,726
ОЭО 2,0
Температура плавления, °С 1083
Температура кипения, °С 2600
Плотность, г/см3 8,96
ЕГ (Эр-р/Э), В 0,521 0,799 1,691
Е° (Эр!р/Э), В ¦ 0,337
ЕГ (Э?р/Э), В - - 1,50
Для серебра наблюдаются заниженные (по сравнению с медью и золотом)
значения температур плавления и кипения, энергии атомизации и первого иони-
ионизационного потенциала. В то же время серебро обладает наивысшими (среди
металлов одинаковой чистоты) теплопроводностью и электрической проводимос-
проводимостью. Объясняется все это тем, что валентный электрон серебра меньше подвержен
эффекту проникновения из-за особой стабильности полностью заселенной 4rf-
оболочки (уже у Pd). В результате именно для серебра наблюдается устойчивая
степень окисления +1.
На воздухе медь покрывается рыхлым слоем основных карбонатов, серебро
чернеет (Ag2S) от наличия сероводорода, в то время как золото не изменяется.
Химическая активность элементов в подгруппе меди сверху вниз падает. Об этом
свидетельствуют также закономерно возрастающие положительные значения
стандартных электродных потенциалов.
311
[Kr]4rflo5.s1
0,144
0,113
7,576
1,9
960,5
2212
10,5
[Хе]4Я
0,144
0,137
9,226
2,3
1063
2947
19,3
Характеристические соединения. Характеристические
оксиды Э2О известны для всех элементов. Си2О получают прямым взаимодей-
взаимодействием меди с кислородом, Ag2O и Аи2О — осторожным нагреванием соответству-
соответствующих гидроксидов. Си2О плавится без разложения при 1235° С, Ag2O и АигО
при нагревании до 200°С распадаются на компоненты.
Из гидроксидов ЭОН устойчив AgOH, два других распадаются на воду и Э2О.
Гидроксид серебра — амфолит — с сильнее выраженными основными свойствами.
Так, рК\) = 3,6, a p/ifa = 11,4. Поскольку для элементов подгруппы меди в обра-
образовании химических связей помимо пв'-электронов принимают участие сравни-
сравнительно близкие по энергии электроны (к — 1)с/-оболочки, они проявляют степени
окисления выше характеристической (так называемые .экстравалентные состоя-
состояния). Из оксидов в степени окисления +2 и +3 устойчивы СиО и Аи2Оз- Первый
получается непосредственным взаимодействием компонентов, а второй — осторож-
осторожным обезвоживанием Аи(ОН)з- Гидроксиды Си(ОНJ и Аи(ОН)з получают дей-
действием щелочи на растворимые соли Си2+ и Аи3+. Гидроксиды Си(ОНJ и
Аи(ОНK амфотерны:
Cu(OHJ + 2NaOH = Na2[Cu(OHL]
Au(OHK + КОН = K[Au(OHL]
Известен оксид черного цвета AgO (Ag2O — темно-бурый), который разлагается
при 100°С (ДЯу 2д8 = -13,4 кДж/моль), при действии серной кислоты образует
Ag2SO4 и выделяет кислород. Анодное окисление AgO, растворенного в НСЮ4,
ведет к образованию Ag2O3, который моментально разлагается при выделении из
раствора. Таким образом, бинарные соединения в степенях окисления +2 и +3
для серебра нестабильны. Действием пероксида водорода на сильнощелочной
раствор растворимой соли Си2+ получают гранатово-красный порошок Си2Оз- Он
выделяет кислород уже при 100°С и является сильнейшим окислителем, напри-
например окисляет соляную кислоту до хлора.
Моногалогениды характерны прежде всего для серебра, так как только сереб-
серебро проявляет устойчивую степень окисления +1. Для серебра хорошо известны
все моногалогениды, плавящиеся без разложения. В то же время для золота
моногалогениды почти не характерны. Так, монофторид золота неизвестен, а
остальные неустойчивы, даже монохлорид. Энтальпия образования Aul равна
+4,0 кДж/моль. Галогениды меди занимают промежуточное положение, монофто-
монофторид также не существует. Из галогенидов меди в степени окисления +2 наиболее
устойчив CuF2, а иодид не получен. Для золота известны все галогениды в степе-
степени окисления +3.
Соединения с другими неметаллами. Для меди получен
неустойчивый гидрид СиН, который разлагается при комнатной температуре
(AH°f2gs = +21 кДж/моль). Серебро и золото с водородом не взаимодействуют.
Прямым синтезом из элементов получают сульфиды Cu2S и CuS. Оба соедине-
соединения распространены в природе. При нагревании в вакууме CuS распадается на
Cu2S и серу. Медь образует с селеном и теллуром целый ряд соединений, из
которых наиболее устойчивы Cu2Se (^щ, = 1113°С) и Си2Те (<пл = 855°С). Осаж-
Осаждением из водных растворов растворимых солей Си2+ может быть выделен CuSe.
Известны все халькогениды серебра: Ag2S, Ag2Se и Ag2Te. Непрямым синтезом
312
получен сульфид AgS, устойчивый при комнатной температуре. Золото с серой
непосредственно не взаимодействует. Действием сероводорода на раствор
K[Au(CN2)] получают Au2S. При реакции сухого Li[AuCl4] с H2S образуется
A112S3. Сульфиды золота разлагаются при Т = ~200°С. Известен A\i2Se3, а с
теллуром золото образует соединение АиТег, встречающееся в природе.
С пниктогенами только медь образует устойчивые соединения. Нитрид C113N
получается при пропускании аммиака над нагретой медью. С фосфором медь
образует два конгруэнтно плавящихся соединения Си3Р и СиРг, из которых
первое обладает металлической проводимостью. Сплавлением компонентов полу-
получают Cu3As и CU5AS2. Серебро, как и медь, непосредственно не взаимодействует с
азотом. Взрывчатый Ag3N получен косвенным путем. Нитриды для золота неиз-
неизвестны. Длительным нагреванием золота с фосфором в ампулах получен Аи2Р3. С
мышьяком золото химически не реагирует, но образует эвтектику.
Для элементов IB-группы получены карбиды состава Э2С2 (как и для щелоч-
щелочных металлов), взрывающиеся при нагревании.
Комплексные соединения. Одним из основных свойств элемен-
элементов IB-группы в любых степенях окисления является способность образовывать
комплексные соединения. Большинство растворимых соединений меди, серебра и
золота являются комплексными соединениями. Комплексообразовательная спо-
способность элементов IB-группы объясняется дефектностью (и — 1)сйзболочек (при
степенях окисления +2 и больше), а также тг-связыванием спаренных электронов
тех же орбиталей с лигандами. Поскольку последний фактор играет домини-
доминирующую роль, при переходе от меди к золоту комплексообразовательная способ-
способность возрастает вследствие лабильности ^-электронных пар у более тяжелых
элементов.
Из ацидокомплексов Э(+) наиболее устойчивы цианидные. Так, для [СиСЬ]"
рА'нест 5,96, а для [Cu(CNJ] рА'неСт 16,0. Как указывалось выше, у более тяжелых
элементов возрастает устойчивость однотипных комплексов: [Ag(CNJJ"
(рКнест 21,1) и [Au(CNJ]~ (рА'нест 38,8). Из комплексов Ag+ практически важен
[Ag(S2O3J]3~, который образуется при закреплении фотоматериалов тиосульфатом
натрия Na2S2O3. При этом не разложившийся под действием света AgBr связыва-
связывается в прочный комплекс и переходит в раствор:
AgBr + 2Na2S2O3 = Na3[Ag(S2O3J] + NaBr
Аквакомплексы катионов металлов подгруппы меди нестойки. Для Си(+1) и
Ag(-)-l) гораздо устойчивее аммиакаты.
Золото в степени окисления +2 не образует комплексов. Серебро, для которо-
которого бинарные соединения Ag(+2) малостойки, образует более устойчивые фторо-
комплексы [AgF3]" и [AgF4]2". Производные Си(-)-2) в равной мере характерны и
для соединений первого порядка, и для комплексов. Хорошо известны голубые
аквакомплексы в водных растворах [Си(Н2ОL]2+ и кристаллический медный
купорос CuSCv5H2O, который является примером сверхкомплексного соедине-
соединения. Устойчивый аммиакат характеризуется к.ч. 4 ([Cu(NH3L]2+, рА^ест 12,7), а
комплекс [Cu(NH3N]2+ можно получить только в жидком аммиаке. Из комплек-
комплексных анионов Си(-)-2) отметим гидроксокомплекс [Си(ОНL]2" (рА'нест 16,1) синего
цвета, благодаря образованию которого Си(ОНJ растворяется в концентрирован-
концентрированных растворах щелочей. Купраты состава КгСиОг не существуют в растворах. В
Q1Q
твердом состоянии их можно синтезировать спеканием оксидов (или солей) ще-
щелочного металла и СиО:
К2СО3 + СиО = К2Си02 + СО2
Для Аи(+3) характерны очень устойчивые цианидный [Au(CNL]~ (р^нест 56,0)
и роданидный [Au(CNSL]" (р-Кцест 42>°) комплексы. Устойчивость цианидных
комплексов определяется наложением двух факторов: увеличением степени окис-
окисления комплексообразователя и мощным кристаллическим полем лигандов CN",
которыми и открывается спектрохимический ряд. В химической практике важны
гидроксо- и хлорокомплексы Аи(+3). Аи(ОН)з по сравнению с Си(ОНJ легко
растворяется в щелочах с образованием [Аи(ОНL]~. Объясняется это большей
прочностью последнего по сравнению с [Си(ОНL]2~ и более кислым характером
Аи(ОНK, чем Си(ОНJ. При растворении золота в царской водке образуется не
A11CI3. а стойкий комплекс Н[АиС14] (рА'нест 21,3) — золотохлористоводородная
кислота:
Аи + HNOS + 4НС1 = Н[АиС14] + N0 + 2Н2О
Медь и серебро в степени окисления +3 значительно стабилизируются за счет
комплексообразования. Для меди известен светло-зеленый высокоспиновый окта-
эдрический комплекс [CuFg]3 , а серебро образует квадратный комплекс [AgF4]~, в
котором в качестве внешнесферных катионов функционируют элементы подгруп-
подгруппы калия. Все эти соединения являются сильными окислителями.
Металлохимия. Роль и значение элементов подгруппы меди в метал-
металлохимии трудно переоценить. Достаточно сказать, что впервые соединения Кур-
накова были открыты в системе Си—Аи. фазы Лавеса — в системе Си—Mg, a
электронные соединения Юм-Розери изучены в системе Си—Zn. Ниже приведена
характеристика взаимодействия этих металлов друг с другом:
Система
Характер взаимодейст-
взаимодействия
Cu-Ag
ограниченные
твердые растворы
Си—Аи-
непрерывные
твердые растворы
Ag—Аи
непрерывные
твердые растворы
Обращает на себя внимание тот факт, что расположенные друг под другом
в Периодической системе медь и серебро образуют ограниченные твердые раст-
растворы. В то же время медь с золотом дают непрерывные твердые растворы. По
способности давать непрерывные твердые растворы с другими переходными
металлами серебро также отличается (плюс — образуется, а минус — не образу-
образуется):
Ми
Ni
Pd
Pt
Медь + + + +
Серебро — — + -
Золото — + + +
Зато по склонности образовывать ограниченные твердые растворы типа латуни
серебро стоит на первом месте:
I
Li Zn Cd Al Ga In Sn Pb
Медь - + - + + ++-
Серебро + + ++ + ++ +
Золото _-)-______
По способности образовывать металлиды с другими металлами системы
Д.И. Менделеева выделяются медь и золото. Эти металлы образуют с другими
элементами, как правило, по нескольку конгруэнтно и инконгруэнтно плавящих-
плавящихся металлидов. Таким образом, и по металлохимическим свойствам в подгруппе
IB обнаруживается четкая вторичная периодичность, В то же время все обсуж-
обсуждаемые металлы не образуют фаз внедрения из-за полной заселенности (п — l)d-
орбиталей.
Г Л ABA X V I. ЭЛЕМЕНТЫ II ГРУППЫ
Вторая группа Периодической системы включает типические элементы берил-
бериллий и магний, а также металлы подгрупп кальция и цинка. В этой группе отли-
отличие в свойствах между элементами ПА- и (Ш-грулп намного меньше. Все элемен-
элементы проявляют характеристическую степень окисления -f-2. Даже в соединениях
ртути (например, Hg^C^) атомы Нв; связаны между собой ковалентно, а потому
функционируют катионообразователи Hgo+. Кроме того, в обсуждаемой группе
типические элементы проявляют гораздо большую аналогию с элементами под-
подгруппы цинка. Бериллий, магний, как и цинк и кадмий, имеют ГПУ-структуру, а
высокотемпературные модификации Са и Sr характеризуются ОЦК-решеткой *.
Металлический магний образует непрерывные твердые растворы с единственным
металлом — кадмием. Кроме того, сульфаты типических элементов, цинка и кад-
кадмия хорошо растворяются в воде в противоположность сульфатам щелочно-зе-
мельных металлов. Магний и металлы подгруппы цинка образуют двойные соли
типа шенитов.
1. Бериллий. Особенности бериллия. В нормальном состоянии
оба валентных электрона бериллия находятся в состоянии 2s. При химическом
взаимодействии атом бериллия возбуждается и один из 2«-электронов промотиру-
ет на 2]п-орбиталь. Появление одного электрона на кайносиммотричной 2р-орби-
тали определяет специфические особенности химии бериллия. Бериллий может
проявлять максимальную ковалентность, равную 4: две связи по обменному меха-
механизму и две — по донорно-акцепторному. Первый потенциал ионизации бериллия
наибольший не только среди элементов ПА-группы, но больше 1\ лития и бора.
Для химии водных растворов бериллия аномально большое значение ионного
потенциала играет особую роль: Ве2+ — 58,5; Mg2+ — 27,3; Са2+ — 19,2; Sr2+ — 16,6;
Ва2+ — 15,0. Наконец, бериллий проявляет диагональную аналогию с алюминием
в большей мере, чем литий с магнием.
Природные соединения и получение бериллия.
Содержание бериллия в земной коре сравнительно невелико. Важнейшие минера-
Барий и низкотемпературные модификации кальция и стронция кристаллизуются в
ГЦК-решегке.
SIR
лы бериллия ВеО-А^Оз (хризоберилл) и BeaA^SieOis (берилл). Природное сырье
после технологических операций переводят в BeF2 или ВеС12. Бериллий получа-
получают либо магнийтермически из BeF2 при 1000—1200°С, либо катодным восстанов-
восстановлением расплава смеси ВеС1г с NaCl (для понижения температуры и увеличения
электропроводности). Особо чистый бериллий получают зонной плавкой.
Физические и химические свойства. Бериллий — сереб-
серебристо-белого цвета, отличается твердостью и хрупкостью. В отличие от многих
металлов он диамагнетик. На воздухе бериллий покрывается тонким слоем окси-
оксида, предохраняющим от коррозии (как и алюминий). Из элементов ИА-группы
бериллий наименее активен, а потому его стандартный электродный потенциал
наименее отрицателен. Следует также отметить близость этой характеристики
для бериллия (Е° = -1,7 В) и алюминия (Е° — -1,67 В), т.е. по химической
активности эти металлы очень близки. Бериллий растворяется в разбавленных
щелочах и кислотах, в том числе и в HF. С водородом бериллий непосредственно
не взаимодействует, при нагревании реагирует с галогенами, в атмосфере кисло-
кислорода сгорает, при повышении температуры взаимодействует с азотом и серой.
Характеристические соединения. ВеО получают терми-
термическим разложением гидроксида, сульфата, нитрата или основного карбоната
бериллия. Он бесцветен, плавится при 2580°С (Д#°,2д8 = -611 кДж/моль), а
охлаждение расплава ведет к образованию стекла. Кристаллы ВеО имеют струк-
структуру вюрцита. И стеклообразование, и вюрцитная структура ВеО свидетельству-
свидетельствуют о его малой полярности. В воде он растворяется, водородом не восстанавлива-
восстанавливается, взаимодействует с кислотами и основаниями. Однако сплавленный оксид
бериллия (т.пл. 2580°С) химически стоек к действию кислот (кроме HF) и раст-
растворов щелочей.
Гидроксид Ве(ОНJ является полимерным соединением, поэтому не растворя-
растворяется в воде (рПР 22). При осаждении из растворов растворимых солей Ве(+2) он
образует белую студенистую массу. Аналогично оксиду гидроксид Ве(ОНJ —
амфолит. При растворении его в кислотах образуются аквакомплекс [Ве(Н2ОL]2+,
в щелочах — гидроксокомплекс [Ве(ОНL]2~. Однако для гидроксида бериллия
основные свойства более характерны. Бериллаты типа Na2BeO2 существуют толь-
только в твердом состоянии.
Из галогенидов бериллия наиболее характерны BeF2 (<пл = 595°С, Д#°. 2д8 =
= -1008,3) и ВеС12 (W = 440°С, Д#}298 = -471-1 кДж/моль). BeF2 получают тер-
термическим разложением фторобериллата (NH4J[BeF4]. BeF2 изоморфен с SiO2-
При охлаждении расплава как SiO2, так и BeF2 образуется стекло. BeF2 хорошо
растворяется в воде, из водных растворов выделяется в виде 5BeF2-2Be0, что
свидетельствует о его гидролизе. Хлорид бериллия ВеСЬ получают действием
хлора на смесь ВеО с углем при 800°С. Это гигроскопичное, легко гидролизу-
ющееся соединение. Волокнистая орторомбическая модификация ВеС1г представ-
представляет собой типичный неорганический полимер с мостиковыми связями:
-О-
"С1-
Ве
С1
С1-
Ве
.41 й
Атом бериллия подвергается з/>3-гибридизации, поэтому чередующиеся фраг-
фрагменты структуры лежат в разных плоскостях. Кроме того, ковалентность берил-
бериллия в этом соединении равна 4. Две связи образованы по обменному механизму
за счет двух неспаренных электронов атома бериллия (после промотирования) и
двух электронов от двух атомов хлора. Две другие связи осуществляются по
донорно-акцеп торному механизму за счет двух вакантных 2р-орбиталей бериллия
(акцептор) и двух неподеленных электронных пар атомов хлора. В качестве мос-
тиковых функционируют атомы хлора.
Соединения с другими неметаллами. С водородом берил-
бериллий непосредственно не взаимодействует. Полимерный гидрид (ВеН2)„ может
быть получен разложением бериллийорганических соединений. По свойствам
(ВеН2)п похож на (АШз)п- Сульфид BeS синтезируют из простых веществ при
высокой температуре или получают действием сероводорода на бериллий. BeS
д8 = -235,6 кДж/моль) кристаллизуется в структуре сфалерита, легко
гидролизуется. Образует при взаимодействии с основными сульфидами тиоберил-
латы (например, К2Ве8г), с кислотными сульфидами — бериллиевые соли тиокис-
лот (например, ВеСЯз). Таким образом, подобно оксиду, сульфид бериллия про-
проявляет амфотерные свойства.
Из других халькогенидов бериллия следует отметить селенид, который также
обладает кристаллохимическим строением сфалерита. В то же время селениды
магния и щелочно-земельных металлов кристаллизуются по типу NaCl. Этот
факт указывает, что в селениде бериллия доля ионной связи мала.
Из металлов ИА-группы бериллий обладает наименьшим сродством к пникто-
генам. Нитрид Be3N2 синтезируют из простых веществ при 1000°С (Д#^ 298 =
= -192,8 кДж/моль). Кристаллический нитрид бериллия — твердое тугоплавкое
вещество (^ ~ 2200°С), химически инертное и в целом похожее на A1N. Однако
порошкообразный нитрид бериллия медленно разлагается водой и разбавленны-
разбавленными кислотами:
Be3N2 + 8HC1 = ЗВеС12 + 2NH4C1
С углеродом бериллий дает метанид Ве2С (как А14Сз) и, подобно щелочно-
щелочноземельным металлам, ацетиленид ВеС2. В отличие от магния и щелочно-земель-
щелочно-земельных металлов бериллий не дает соединений с кремнием и германием.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Сульфат бериллия из водных растворов всегда выделя-
выделяется в виде кристаллогидрата BeSO4-4H2O, а последний представляет собой
аквакомплекс [Be(H2OL]SO4 с тетраэдрическим строением. Из элементов ИА-
группы Ве(+2) характеризуется наибольшей комплексообразовательной способ-
способностью. Объяснение этому — и наличие четырех вакантных орбиталей (одна
орбиталь 2s и три вырожденные орбитали 2р), и аномально высокий ионный
потенциал Ве(+2). В результате этого катион бериллия обладает сильным поля-'
ризующим действием, что ведет к усилению его комплексообразовательной спо-
способности. Сульфаты щелочных металлов с BeSO4 образуют двойные соли типа
Me2SO4-BeSO4-2H2O, которые можно представить в виде малостойких комплек-
комплексов Me2[Be(SO4J]-2H2O. Вследствие наличия четырех вакантных орбиталей комп-
комплексы Ве(+2) характеризуются к.ч. 4:
.417
p*, [3e(NH3)j2*, [BeF4]2-, [Be(CNLp-
[Be(CNSL]2-, [Be(OHLp-, [Be(NO3LI2-, [Be(CO3J]2- и т.д.
Об этом же свидетельствует легкорастворимый в воде нитрат бериллия, кото-
который кристаллизуется из воды в виде Ве(гЮзJ*4Н2О. Для карбоната также ха-
характерен кристаллогидрат ВеСОз-4Н2О. Для магния и щелочно-земельных ме-
металлов в отличие от бериллия характерно к.ч. 6.
Металлохимия бериллия. На металлохимию бериллия опреде-
определяющее влияние оказывают малый размер атома и относительно большая по
сравнению с магнием и щелочно-земельными металлами ОЭО. Бериллий не сме-
смешивается с магнием даже в жидком состоянии и ни с одним элементом Периоди-
Периодической системы не образует непрерывных твердых растворов. Он плохо растворя-
растворяет другие металлы, сам несколько больше растворяясь в них. Со многими элемен-
элементами образует большое число металлидов. Бериллий не дает эвтектических сме-
смесей и не взаимодействует с индием и сурьмой.
2. Магний. Особенности химии магния. В отличие от берил-
бериллия магний не является кайносимметричным элементом. В невозбужденном состо-
состоянии два его валентных электрона находятся на 3*-орбитали. В силу этого иони-
ионизационные потенциалы магния меньше, чем бериллия, а потому соединения ммг-
ния характеризуются большей долей ценности связи. Не случайно многие авторы
относят магний к щелочно-земельным металлам. По комплексообразовательной
способности магний также уступает бериллию. Комплексы магния с органически-
органическими лигандами очень важны для жизнедеятельности живых организмов (например,
хлорофилл). Поэтому магний является одним из главных элементов бионеоргани-
бионеорганической химии. По металлохимическим свойствам магний также более близок к
щелочно-земельным металлам.
Природные соединения и получение магния. Маг-
Магний относится к числу распространенных элементов. Встречается в виде силика-
силикатов, хлоридов, карбонатов и сульфатов. В основном в производстве металлическо-
металлического магния используют доломит, магнезит MgCO3 и карналлит. Получают его
либо электролизом хлорида, либо карботермией из обожженных магнезита или
доломита.
Физические и химические свойства. Магний — блестя-
блестящий серебристо-белый металл, тускнеющий на воздухе в результате окисления,
сравнительно мягкий и пластичный. В отличие от бериллия парамагнитен. Для
сравнения магния с бериллием и щелочно-земельными металлами ниже приведе-
приведены некоторые свойства элементов ПА-группы:
Содержание в земной коре,
масс. %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э2+, нм
Потенциалы ионизации
1\: Э -> Э+ + е
h: Э* -? Э2+ + е
Ве
Mg
3,8-Ю 1,9
0,160
0,074
7,645
15,03
Ca Sr Ba
3,3 3,4-10-2 6,5-10-2
0,113
0,034
9,323
18,21
0,197
0,104
6,133
11,87
0,215
0,120
5,695
11,03
0,221
0,138
5,212
10,00
41S
hli 27,53 22,69 18,0 16,73 15,21
t=l
ОЭО
Температура плавления, °С
Температура кипения, ° С
Плотность, г/см3
ЕГ (ЭгуЭ), В
1,5
1283
2970
1,85
-1,847
1,2
650
1104
1,74
-2,363
1,0
847
1470
1,54
-2,866
1,0
770
1375
2,63
-2,888
0,9
718
1687
3,76
-2,906
Магний — химически активный металл. На холодную воду не действует, а
кипящую энергично разлагает. Растворяется в минеральных кислотах (кроме
HF), но не растворяется в щелочах. На воздухе сгорает с образованием смеси
оксида и нитрида магния. При нагревании магний соединяется с галогенами,
халькогенами и пниктогенами.
Характеристические соединения. Характеристический
оксид MgO — тугоплавкое вещество (^ = 285СГС, Д#^2д8= -598,4 кДж/моль)
кристаллохимического строения типа NaCl. MgO в отличие от ВеО не растворя-
растворяется в щелочах, что доказывает его основность. При хранении на воздухе посте-
постепенно превращается в смесь Mg(OHJ и MgCOa. Гидроксид Mg(OHJ плохо раст-
растворяется в воде (рПР 11). является основанием средней силы (рА'г 3), не раство-
растворяется в щелочах. Характеристические галогениды Mg^ — бесцветные вещества,
очень гигроскопические (кроме MgF2), их температуры плавления и энтальпии
образования закономерно снижаются от фторида к иодиду. За исключением
фторида (рПР 9), они хорошо растворяются в воде, из водных растворов выделя-
выделяются в виде кристаллогидратов.
Соединения с другими неметаллами. Косвенным путем
(из магнийорганических производных) можно получить белый твердый гидрид
магния, который представляет собой полимер (MgH2)n. Однако при повышенных
температурах и под избыточным давлением водорода MgH2 может быть синтези-
синтезирован из компонентов. Около 300"С он распадается на элементы, медленно разла-
разлагается водой, является сильным восстановителем. Все халькогениды MgX синте-
синтезируют из простых веществ. В той или иной степени они подвержены гидролизу.
Пниктогениды Mg3n2 также получаются непосредственным взаимодействием
компонентов. Они еще более неустойчивы к воде. С углеродом магний дает эндо-
эндотермические карбиды MgC2 и М^Сз, которые при действии воды выделяют
углеводороды. В отличие от бериллия магний с кремнием и германием образуют
силицид MgoSi и германид Mg2Ge.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Сульфат MgSO4'7H2O может быть представлен как
сверхкомплексное соединение [Mg(H2O)B]SO4*H2O. С сульфатами щелочных ме-
металлов он образует двойные соли типа шёнита Me2SO4-MgSO446H2O. В отличие
от бериллия для Mg(+2) характерно к.ч. 6, а сами комплексы магния менее
стойки и разнообразны. Зато магний дает значительно больше комплексов с
органическими лигандами.
Металлохимия магния. Расплавленный и твердый магний хоро-
хорошо растворяет водород. С металлами В-групп образует ограниченные твердые
растворы. Незначительно растворяется в переходных металлах и не образует с
ними металлиДов. Щелочные, щелочно-земельные, благородные металлы, а также
металлы подгруппы цинка с магнием образуют металлиды. Магний не взаимодей-
ствует с молибденом, вольфрамом, ураном и железом. В целом по металлохими-
ческим свойствам магний близок к щелочно-земельным металлам.
3. Щелочно-земельные металлы. Характеристика элементов
подгруппы кальция. Элементы подгруппы кальция (щелочно-земель-
(щелочно-земельные металлы) характеризуются наибольшим сходством между собой, поскольку
для них имеет место не только групповая и типовая аналогия, но и слоевая. При
наличии в атоме заполненной ns-орбитали пр- и (п — 1)^-оболочки вакантны.
0Э0 обсуждаемых элементов практически одинакова, равно как и значения
стандартных электродных потенциалов. В целом от Са к В а незначительно воз-
возрастает химическая активность элементов. Во многих отношениях щелочно-зе-
щелочно-земельные металлы напоминают щелочные. Те и другие образуют солеобразные
гидриды, их гидроксиды представляют собой сильные основания, они являются
плохими комплексообразователями и т.д.
Природные соединения и получение. По распространен-
распространенности в земной коре Са занимает пятое место. Содержание стронция и бария
намного меньше. Помимо силикатных пород эти элементы встречаются в виде
карбонатов и сульфатов: СаСОз (кальцит), SrSO4 (целестин), BaSO4 (тяжелый
шпат) и т.д. Общий способ их получения в свободном состоянии — алюмотермия
в вакууме. Кроме того, кальций еще получают катодным восстановлением распла-
расплава его хлорида.
Физические и химические свойства. В свободном состоя-
состоянии Са, Sr и Ва — белые блестящие металлы, на воздухе окисляются. Са облада-
обладает наибольшей электрической проводимостью и твердостью. Все эти металлы
активнее бериллия и магния, вытесняют водород из воды и разбавленных кис-
кислот. Металлы подгруппы кальция при обычных условиях взаимодействуют с
кислородом и галогенами. С менее активными неметаллами (азот, халькогены,
водород и др.) — при умеренном нагревании.
Характеристические соединения. В отличие от щелочных
металлов элементы подгруппы кальция образуют прочные характеристические
оксиды ЭО. Их получают термическим разложением карбонатов или нитратов.
Все оксиды — тугоплавкие бесцветные гигроскопические вещества. Они бурно
взаимодействуют с водой с выделением большого количества теплоты и образова-
образованием гидроксидов. Все гидроксиды Э(ОН)г являются сильными основаниями. Их
растворимость в воде и сила основания растут от Са(ОН)г к Ва(ОН)г. Помимо
характеристических оксидов металлы подгруппы кальция (в отличие от Be и Mg)
образуют пероксиды ЭОг- Они намного менее стабильны в сравнении с оксидами
(например, СаОг взрывается при 275°С) и сильные окислители.
Среди характеристических галогенидов резко отличаются фториды. Ниже
приведены стандартные свободные энергии образования
Соединение
AG^298, кДж/моль -1168,6
-749,6
СаВг2
-666,5
Са12
-532,5
Фториды почти нерастворимы в воде (например, для CaF2 рПР 11), а осталь-
остальные галогениды хорошо растворяются и выделяются из воды в виде кристалло-
кристаллогидратов. Безводные хлориды, бромиды и иодиды могут быть получены медлен-
медленным нагреванием кристаллогидратов.
Соединения с другими неметаллами. Гидриды щелочно-
щелочноземельных металлов синтезируют из простых веществ при нагревании. Подобно
г
гидридам щелочных металлов, они относятся к солеобразным и энергично взаи-
взаимодействуют с водой. Этот процесс является удобным методом получения водоро-
водорода. От Са к Ва термическая устойчивость гидридов несколько растет, но при
600°С все они заметно разлагаются. С халькогенами синтез из элементов дает
халькогениды ЭХ. Сульфид кальция также можно получить восстановлением
сульфата углем. ЭХ — бесцветные кристаллические вещества со структурой типа
NaCl. Меньше других растворимы в воде теллуриды. Из сульфидов наименьшей
растворимостью обладает CaS. В воде все ЭХ гидролизуются. Степень гидролиза
увеличивается от теллуридов к сульфидам- Пниктогениды щелочно-земельных
металлов — бесцветные тугоплавкие вещества обобщенной формулы Э3П2. Ввиду
большого сродства обсуждаемых элементов к азоту были синтезированы нитриды,
более богатые азотом, например 33N4. Для бария известен даже BaN2. Все пник-
пниктогениды Э3П2 разлагаются водой с выделением летучих гидридов пниктогенов и
образованием гидроксидов. С углеродом щелочно-земельные металлы образуют
только ацетилениды ЭС2. Практически важен СаС2, получаемый карботермичес-
ки из оксида кальция. В отличие от магния для щелочно-земельных металлов
известны силициды 3Si и 3Si2.
Слои кислородсодержащих кислот и комплексы.
3SO4 плохо растворяются в воде, причем растворимость уменьшается от сульфата
кальция к BaSO<j. Сульфаты стронция и бария кристаллизуются без воды, а
CaSO4-2H2O встречается в природе (гипс). Карбонаты ЭСО3 также плохо раст-
растворимы в воде (например, для СаСО3 рПР 9). При прокаливании все выделяют
СО2. Нитраты кальция и стронция дают кристаллогидраты с четырьмя молекула-
молекулами воды. Ba(NO3J кристаллизуется без воды и плохо растворяется в ней. В
отличие от средних кислые соли хорошо растворимы в воде. Присутствие в при-
природной воде солей кальция и магния придает ей "жесткость". Временная жест-
жесткость обусловлена в основном содержанием растворимого гидрокарбоната каль-
кальция Са(НСО3J, который при кипячении разрушается, переходит в СаСО3 и
образует накипь на стенках сосудов. Постоянная жесткость обусловлена присутст-
присутствием сульфатов и хлоридов кальция и магния, не дающих осадка при кипяче-
кипячении. Поэтому от нее можно избавиться только химическим путем, например до-
добавлением соды.
Комплексообразовательная способность элементов подгруппы кальция усту-
уступает таковой типических элементов II группы, но выше, чем у щелочных метал-
металлов. При переходе от последних к щелочнонземельным металлам уменьшаются
ионные радиусы, а заряды увеличиваются в два раза. В результате поляризую-
поляризующая сила Э(+2) намного больше, чем Э(+1), что и ведет к лучшей комплексооб-
разовательной способности элементов подгруппы кальция. Так, при растворении
в жидком аммиаке щелочных металлов образуются коллоидные растворы, т.е. не
возникают комплексы, тогда как растворение в нем щелочно-земельных металлов
ведет к образованию нейтральных комплексов [9(NH3N], которые полностью
разлагаются водой. Неустойчивы аммиакаты [9(NH3N]2+. Кальций и стронций
дают аквакомплексы [Э(Н2О)б]Г2, где Г — хлорид- и бромид-анионы.
Металлохимия. Металлы подгруппы кальция вследствие близости
металлохимических факторов образуют между собой непрерывные твердые раст-
растворы. Области однородности ограниченных твердых растворов на основе Са, Sr,
Ва растут слева направо с увеличением металлических радиусов. Вследствие
большого различия физико-химических свойств переходных и щелочно-земель-
11 я а у™а Я91
ных металлов между ними имеет место полное или частичное расслоение в жид-
жидкой фазе. Cm р-металлами, а также цинком и его аналогами элементы под-
подгруппы кальция образуют металлиды.
4. Подгруппа цинка. Характеристика элементов ИВ-г р у п -
п ы. Элементы подгруппы цинка характеризуются полностью заполненными (п —
— \)d- и ns-орбиталями. Поэтому для них в принципе невозможна дефектность
(п — 1)</-оболочки (в отличие от элементов IB-группы). Другими словами, элемен-
элементами ИВ-группы заканчиваются три ряда переходных rf-металлов. За этими эле-
элементами следуют sp-металлы. В результате этого с учетом стабильности заполнен-
заполненной rf-оболочки металлы ИВ-группы являются связующим звеном между элемен-
элементами подгрупп меди, с одной стороны, и галлия — с другой. Так, комплексообра-
зовательная способность обсуждаемых элементов является промежуточной между
таковой металлов подгрупп меди и галлия. Гидроксид цинка является амфоли-
том, как и гидроксид галлия. Таллий также проявляет нехарактеристическую
низшую степень окисления, подобно ртути. Металлы подгрупп цинка и галлия
относятся к самым легкоплавким и т.д. В то же время элементы ИВ-группы во
многом сходны по свойствам с металлами подгруппы меди. В подгруппе цинка
сверху вниз также уменьшается химическая активность элементов, а ртуть можно
отнести к благородным металлам (Е° = +0,85 В). Химия и металлохимия кадмия
(как и серебра) и его гомологов по группе несколько различаются. Эта вторичная
периодичность объясняется двумя причинами: во-первых, кайносимметричностью
Згйэлектронов атомов меди и цинка и ее последствиями; во-вторых, на химию
золота и ртути оказывает большое влияние совместное действие d- и /-контрак-
/-контракции.
Природные соединения и получение металлов.
Содержание элементов в земной коре относительно невелико и уменьшается от Zn
к Cd. Важнейшие цинковые руды — ZnS (сфалерит, вюрцит) и ZnCOe (галмей).
Кадмий сопутствует цинку в полиметаллических сульфидных рудах и редко
образует самостоятельное месторождение CdS (гринокит). Киноварь HgS является
главной рудой в производстве ртути.
Для получения цинка и кадмия их сульфидные концентраты подвергают
окислительному обжигу, а затем проводят карботермическое восстановление.
Наряду с пирометаллургическим цинк и кадмий получают и гидрометаллурги-
гидрометаллургическим способом. В этом методе обожженную руду в виде оксидов растворяют в
разбавленной серной кислоте, а раствор подвергают катодному восстановлению.
Физические и химические свойства. Цинк и кадмий —
серебристо-белые мягкие металлы, а ртуть — единственный металл, жидкий в
обычных условиях. Кристаллизуется ртуть в ромбоэдрической структуре. В отли-
отличие от щелочно-земельных металлов цинк, кадмий и ртуть принадлежат к тяже-
тяжелым металлам. Ниже приведены некоторые характеристики элементов и простых
веществ НВ-группы:
1
1
It
If
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э2+, нм
Zn
8,3-Ю-з
[Ar]3dl04s2
0,139
0,083
Cd
1,3-10
[Kr]4«P°5s2
0,156
0,099
Hg
7-Ю-6
[Xe]4/45
0,160
0,112
9,304
17,964
1.5
419,7
906,4
7,14
-0,763
8,994
16,908
1,7
321,3
766,7
8,65
-0,403
10,438
18,756
1,8
-38,7
356,8
13,55(ж)
+0,854
Потенциал ионизации, В
к Э -> Э+ + е
/2: Э -> Э2+ + €
ОЭО
Температура плавления, °С
Температура кипения, °С
Плотность, г/см3
ЕГ (Э2!р/Э), В
Обращают на себя внимание высокие значения ионизационных потенциалов
ртути (из-за чего не укладывается в ряд кадмий), что объясняется не только
результирующим действием d- и /-контракции, но и сильно выраженным эффек-
эффектом проникновения 6 s-электронов под двойной экран из d- и /-электронных обла-
облаков.
На воздухе металлы НВ-группы теряют блеск, так как покрываются оксидной
пленкой. Ртуть окисляется медленно, а цинк и кадмий при нагревании сгорают
до оксидов ЭО. При нагревании цинк и кадмий реагируют с галогенами и халь-
когенами. При растирании в ступке серы с ртутью на холоду образуется HgS.
Этот пример иллюстрирует, как жидкое состояние облегчает химическое взаимо-
взаимодействие.
Характеристические соединения. Характеристические
оксиды ЭО получают из компонентов. Оксиды разлагаются до плавления. От
цинка к ртути термическая стойкость уменьшается. В отличие от ZnO (структура
вюрцита) и HgO (ромбическая структура) оксид кадмия имеет кристаллохими-
ческое строение NaCl, что свидетельствует о большей ионности CdO. Оксид цин-
цинка амфотерен, a CdO и HgO — основные оксиды. Гидроксиды Э(ОНJ практичес-
практически не растворяются в воде: Zn(OHJ - рПР 11; Cd(OHJ - рПР 14; Hg(OHJ -
рПР 16. Гидроксид ртути химически малостоек. Гидроксид цинка — амфолит с
преобладанием основных свойств. При растворении в щелочах образуются гид-
роксокомплексы Me2[Zn(OHL], а не цинкаты типа Na2Zn02.
Все характеристические галогениды цинка, кадмия и ртути — бесцветные
кристаллические вещества. Температуры плавления фторида, хлорида и бромида
кадмия намного выше, чем у соответствующих производных цинка и ртути. Кро-
Кроме того, CdF2 кристаллизуется в структуре CaF2. Все эти факты говорят о боль-
большей ионности обсуждаемых производных кадмия и о вторичной периодичности в
ПВ-группе.
Соединения с другими неметаллами. Как и в случае
элементов IB-группы, для металлов подгруппы цинка не характерны соединения
с водородом. Синтезированный косвенным путем ZnH2 разлагается около 90°С,
CdH2 неустойчив выше -20°С, a HgH2 — выше -125°С.
Халькогениды синтезируют сплавлением компонентов. ZnS (рПР 24) и CdS
(рПР 27) можно также получить осаждением из растворов соединений цинка и
кадмия сульфид-ионом. Для всей группы обсуждаемых халькогенидов сфалерит
является низкотемпературной модификацией, а вюрцитная структура — высоко-
высокотемпературной. С увеличением порядкового номера катион о- и анион ообразовате-
ля на ковалентюмюнную связь накладывается определенная доля металлической
связи. Поэтому ширина запрещенной зоны у халькогенидов закономерно умень-
уменьшается. Сульфиды цинка и кадмия являются основой лучших неорганических
i ¦'
люминофоров*. Селениды и теллуриды кадмия и ртути — важнейшие полупро-
. II_VI _
водниковые соединения группы А В . С азотом элементы подгруппы цинка
непосредственно не взаимодействуют. Нитриды ЭзЩ неустойчивы и разлагаются
водой. Остальные пниктогениды получают синтезом из элементов. Кроме нор-
нормально-валентных Э3П2 известны дифосфиды и диарсениды цинка и кадмия
(например, Z11P2, CdAs2), а также CdP4. Все пниктогениды цинка и кадмия,
вплоть до антимонидов, являются полупроводниковыми соединениями группы
aV.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Соли сильных кислот, образованные элементами под-
подгруппы цинка (нитраты, сульфаты и др.), хорошо растворяются в воде и выделя-
выделяются из растворов в виде кристаллогидратов. Карбонаты ЭСОз известны только
для цинка и кадмия. Для ртути помимо солей Hg(+2) существуют производные
ртути Hg2(+2). Разбавленная азотная кислота A:1) с избытком ртути взаимодей-
взаимодействует согласно уравнению реакции
8HNO3 + 6Hg = 3Hg2(NO3J + 4H2O + 2NO
В ионе Hg2+ атомы ртути связаны между собой ковалентной связью, в результате
чего все соединения Hg2(+2) диамагнитны. Все производные Hg2+ постепенно
диспропорционируют: Hgf+ = Hg2+ + Hg°. Еще менее стабильны описанные в
литературе Hg!+ и Hg|+.
Цинк и кадмий в отличие от щелочно-земельных металлов образуют двойные
соли типа шёнитов. Цинк вследствие амфотерности образует наиболее устойчи-
устойчивый гидроксокомплекс [Zn(OHL]2~ (рА'нест 15,5). Вторичная периодичность имеет
место и в химии комплексных соединений. Это видно, например, из сравнения
рА'нест Для аммиакатов [3(NH3LJ: S,46; 7,12; 19,28 соответственно для Zn(+2),
Cd(+2), Hg(+2). Такая же картина наименьшей устойчивости координационных
соединений кадмия наблюдается и для комплексов с тиомочевиной. Не надо
думать, что такое положение фиксируется только для комплексных катионов.
Так, рА'нест для ацидокомплексов с тиосульфат-анионом [Э^Оз^]2" от цинка к
ртути принимает значения 8,2; 6,4 и 24,4. Кроме того, Cd2+ чаще других имеет
к.ч. 6, например в [Cd(NH3N]2+, [Cdle]4', [Cd(CNSN]4- и др.
Металлохимия. За исключением кадмия (Cd — Mg), металлы подгруп-
подгруппы цинка не дают непрерывных твердых растворов. Между собой образуют эв-
эвтектику. Для металлов НВ-группы наиболее характерно образование металлидов.
Они образуются с щелочными, щелочно-земельными, sp-, а также с переходными
и благородными металлами. Наибольшим числом металлидов характеризуется
кадмий. Таким образом, вторичная периодичность проявляется в металлохимии
элементов ПВ-группы. Металлохимической особенностью ртути является сущест-
существование амальгам — металлидов ртути с щелочными, щелочнц-земельными метал-
металлами и элементами подгруппы меди.
* Люминофоры — вещества, способные к люминесценции. Неорганические кристалли-
кристаллические люминофоры называются также кристаллофосфорами.
ГЛАВА XVII. ЭЛЕМЕНТЫ Ш ГРУППЫ
Третья группа Периодической системы — самая элементоемкая. Она содержит
37 элементов, включая лантаноиды и актиноиды. Все элементы III группы, за
исключением бора, являются металлами. Первый типический элемент бор —
неметалл. В какой-то мере бор выполняет роль переходного элемента от металли-
металлического бериллия к углероду. Но, поскольку у атома бора уже в нормальном
состоянии на кайносимметричной 2р-орбитали имеется один электрон (а в воз-
возбужденном состоянии два электрона), он функционирует как неметалл. Наконец,
в III группе наблюдается наименьшая разница в свойствах элементов ША- и ШВ-
групп. Элементы подгруппы галлия, как и А1, являются «^металлами. В отличие
от них элементы подгруппы скандия принадлежат к srf-металлам. Но в характе-
характеристической степени окисления +3 элементы подгруппы галлия имеют внешнюю
электронную конфигурацию (п — l)^10, а типовые аналоги скандия, как и А1(+3),
— электронную структуру благородных газов s2pe. Поэтому некоторые авторы
располагают элементы подгруппы скандия непосредственно под А1, т.е. относят
их к ША-группе. Тогда подгруппа галлия должна составлять ШВ-группу. Все
это свидетельствует о близости химии элементов ША-и ШВ-групп между собой, с
одной стороны, и с химией алюминия — с другой.
1. Бор. Особенности бора. Электронная формула атома бора
Is22s22p1. Наличие одного неспаренного электрона могло бы обусловить существо-
существование одновалентных соединений, что мало характерно для бора. Объясняется
это тем, что один из спаренных 2 s-электронов сравнительно легко промотирует
C43,0 кДж/моль) на 2р-орбиталь и тогда бор функционирует как трехвалентный:
дополнительно образующиеся две ковалентные связи дают больший выигрыш в
энергии, чем ее затрачивается на промотирование. Реже бор проявляет валент-
валентность 4 с привлечением вакантной 2р-орбитали по донорно-акцепторному меха-
механизму. В соединениях бора химические связи малополярны. Вследствие малого
размера атома бора и кайносимметричности 2/ьорбитали ионизационные потенци-
потенциалы бора намного больше, чем у его аналогов по группе. Кроме того, значение
ОЭО бора сильно превышает значения ОЭО других элементов III группы. Все это
вместе взятое определяет неметаллическую природу бора. В то же время по хи-
химической активности бор уступает следующим за ним элементам 2-го периода
(кроме неона). Как известно, бор обнаруживает диагональную аналогию с крем-
кремнием. Для бора и кремния наиболее характерны производные, в которых эти
элементы поляризованы положительно. Для обоих элементов их низшие гидриды
малоустойчивы и газообразны. Много общего имеет химия кислородных соедине-
соединений бора и кремния: кислотная природа оксидов и гидроксидов, стеклообразова-
ние оксидов, способность образовывать многочисленные полимерные структуры и
т.д.
Природные соединения и получение бора. Содержа-
Содержание бора в земной коре 3-10%. В природе бор встречается исключительно в
виде кислородных соединений: борной Кислоты Н3ВО3, буры Na2B4O7-10H2O и
минерала ашарита MgHBOa- Для получения из природных соединений бор пере-
переводят в оксид В2О3, в галогениды или в бороводороды. Чаще других пользуются
методами металлотермии, например
В2О3 + 3Mg = 3MgO + 2В, AG298 = -532,2 кДж,
: 2ВС13 + 3Zn = 3ZnCl2 4- 2В, &G°2ga = -504,5 кДж.
Я9К
\i
P и с. 136. Проекция кристаллической структуры ром-
ромбоэдрического бора
более
В этих процессах получается амор-
аморфный бор, загрязненный примеся-
примесями. Более чистый бор получают
восстановлением его хлорида водо-
водородом или осаждением кристалли-
кристаллического бора на раскаленной про-
проволоке из тантала или вольфрама.
Наиболее перспективны методы,
основанные на пиролизе боранов,
например В2Н6 = 2В + ЗН3. Они
сравнительно меньшей стоимости исходного
экономичны вследствие
продукта и низких температур проведения процесса.
Физические и химические свойства бора. Наиболее
устойчивой кристаллической формой бора является /^-ромбоэдрическая. Сущест-
Существуют также о-ромбоэдрическая и тетрагональная модификации бора. Все эти
кристаллические структуры слагаются из икосаэдров (см. рис. 121). Каждый из
атомов бора внутри икосаэдра связан с пятью соседями, а атомы в вершинах
осуществляют сочленение икосаэдров друг с другом непосредственно или через
промежуточные атомы бора. На рис. 136 представлен фрагмент кристаллохими-
ческого строения ромбоэдрического бора, в котором икосаэдры связаны между
собой мостиковыми атомами бора. В целом кристаллохимия бора необычна и
характеризует его как переходный элемент между металлами и неметаллами.
Кристаллы чистого бора серовато-черного цвета, отличаются тугоплавкостью
и хрупкостью. По твердости он уступает только алмазу и кубическому нитриду
бора. Бор — полупроводник с шириной запрещенной зоны 1,53 эВ. Бор является
диамагнетиком.
При обычных условиях бор химически инертен. При высоких температурах он
взаимодействует с кислородом, хлором, серой, азотом. Кипящие соляная и
плавиковая кислоты на него не действуют. Он медленно реагирует с такими
сильными окислителями, как фтор, горячая концентрированная азотная кислота
и царская водка. Во всех случаях кристаллический бор химически менее активен,
чем аморфный. Так, концентрированные щелочи не действуют на кристалличес-
кристаллический бор даже при кипячении, а аморфный постепенно растворяют:
2В + 2NaOH + 2Н2О = 2NaBO2 + ЗН2
В химических реакциях бор чаще выступает как восстановитель. Тем не менее
он, например, не может вытеснить кремний из его диоксида. Расчеты показыва-
показывают, что для реакции
4В + 3SiO2 - 2В2О3 + 3Si
слева направо AG°gg = +183,3 кДж/моль. Другими словами, реакция идет справа
налево, т.е. кремний вытесняет бор из В2О3.
Характеристические соединения. Бор, подобно кремнию,
обладает большим сродством к кислороду, а в кислородных соединениях чаще
всего имеет к.ч. 3. Реже вокруг атома бора координируется четыре атома кисло-
рода. В первом случае структура содержит плоские группировки [ВО3] с атомом
бора в *р2-тибридном состоянии, во втором — тетраэдрические образования [BOJ
с «р3-гибридизацией атома бора.
Характеристический оксид В2О3 существует в виде нескольких модификаций.
Кристаллические формы характеризуются дальним порядком в кристаллохими-
ческой связи отдельных группировок [ВО3] или [BOJ- В стеклообразном В2Оз
существует сетка из беспорядочно ориентированных групп [ВОз]. На воздухе при
температуре около 700°С бор сгорает и образует ВгОз- Последний удобнее полу-
получать обезвоживанием гидроксида бора В(ОН)з:
2ЩВО3 ^ 2Н2О + 2НВО2; 2НВО2 ^ Н2О + В2О3
Сначала образуется метаборная кислота НВО2 (рК& ~ 10). Обе реакции обратимы,
вода растворяет В2Оз и переводит его в борную кислоту Н3РОз- Ортоборная (или
просто борная) кислота — белое кристаллическое вещество, легко расслаивающе-
расслаивающееся на очень тонкие перламутровые чешуйки. Обусловлено это слоистым строени-
строением кристаллов НйВОз- Молекулы Н3ВО3, расположенные в плоских параллельных
слоях, связаны между собой водородными связями, а связь между слоями осу-
осуществляется слабыми силами Вэн-дер-Ваальса.
Борная кислота хорошо растворима в воде, а также в некоторых органических
растворителях. Несмотря на наличие трех гидроксогрупп, в водных растворах она
проявляет себя как одноосновная кислота вследствие образования гидроксокомп-
лекса:
В(ОНK + Н2О = Н[В(ОНL]
Тетрагидроксоборат водорода представляет собой слабую кислоту, близкую по
силе синильной и угольной кислотам. Необычным в поведении борной кислоты
является и то, что с ростом концентрации кислотность ее растворов не падает,
как у других кислот, а увеличивается. Причиной этого является возникновение в
крепких растворах Н3ВО3 ряда полиборных кислот пВ2Оз-игН2О, не выделенных
в свободном состоянии. В растворах умеренной концентрации при комнатной
температуре преобладающей формой является циклическая триборная кислота
(ЗВ2О3-ЗН2О = H6B6Oi2 = 2Н3В3О6):
НО
но
Циклизация ведет к перераспределению электронной плотности, и связь О—Н
делается более полярной, чем и объясняется увеличение кислотности растворов
борной кислоты.
Ортоборная кислота Н3ВО3 при 100°С с отщеплением молекулы воды перехо-
переходит в метаборную HBO2. И ортобораты, и метабораты активных металлов в воде
подвержены гидролизу и имеют щелочную реакцию. Строение неизвестной в
свободном состоянии тетраборной кислоты Н2В4О7 может быть представлено
следующим образом:
327
н-о-в
в-о-в
в-о-н
I
= 4, рА'г ~ 5). Соли ее — тетрабораты — хорошо известны и встречаются в
природе. Наиболее известна натриевая соль — тетраборат натрия (бура)
Na2B4O7*10H2O. Тетраборат натрия образуется при нейтрализации водными
растворами щелочей тетрагидроксобората водорода:
4Н[В(ОНL] + 2NaOH = Na2B4O7 + 11Н2О
С другой стороны, при действии на тетрабораты сильных кислот выделяется
тетрагидроксоборат водорода:
Na2B4O7 + H2SO4 + 9H2O = 4Н[В(ОНL] + Na2SO4
Многие полибораты могут быть получены синтетически. В целом соли борных
кислот многочисленны и разнообразны, как и силикаты.
Известны все характеристические галогениды BF3. Все они могут быть полу-
получены синтезом из компонентов при нагревании. BF3 представляет собой газ, ВСЦ
и ВВг3 при обычных условиях — жидкости, а В1з — летучее твердое вещество
(как и SiF4!). Вследствие наличия вакантной 2;мэрбитали у атома бора его гало-
галогениды склонны к реакциям присоединения. В качестве доноров неподеленной
электронной пары могут выступать молекулы аммиака, воды, фосфина, спиртов,
эфиров и др. В продуктах присоединения координационное число бора равно 4,
а геометрия первой координационной сферы отвечает вр3-гибридизации атома
бора. Из производных BF3 наибольшее значение имеет комплексная тетрафторо-
борная кислота H[BF4] и ее соли — тетрафторобораты. В водном растворе H[BF4]
практически полностью ионизирована и поэтому является сильной одноосновной
кислотой. Большинство солей H[BF4] бесцветны и хорошо растворимы в воде.
Тетрагалогенобораты для хлора и брома малоустойчивы и известны только для
тяжелых щелочных металлов.
Соединения с другими металлами. Простейший бороводо-
род — боран ВН3 — в обычных условиях не существует вследствие координацион-
координационной ненасыщенности и димеризуется в диборан В2Н6. В последнем бор имеет к.ч.
4. Во многих отношениях бораны являются необычными соединениями; для
понимания их природы представления классической валентности недостаточно.
Все они, как диборан, электронодефицитны и у них наблюдаются многоцентро-
многоцентровые химические связи. По свойствам бораны- имеют много общего с кремневодо-
родами и углеводородами. Диборан — газ, остальные — твердые летучие вещества
или жидкости. Все бораны имеют характерный неприятный запах, очень токсич-
токсичны: вдыхание их паров вызывает головную боль и рвоту. Химически бораны
весьма реакционноспособны и по отношению к большинству реагентов менее
устойчивы, чем кремневодороды. Водой, щелочами, окислителями бораны разру-
разрушаются, например
В2Н6 + 6Н2О = 2В(ОНK + 6Н2
При нагревании бораны разлагаются; наименее устойчив тетраборан. Ниже при-
приведена схема его разложения в зависимости от температуры:
328
Нагревание боранов выше 250° С заканчивается, как правило, полным их разло-
разложением на водород и бор. Регулируя температуру и давление, удается целенап-
целенаправленно проводить процесс в сторону образования желаемого продукта.
Для боранов, в частности диборана, наиболее характерны реакции, протекаю-
протекающие с разрывом мостиковых связей. К подобным реакциям относится приведен-
приведенная выше реакция гидролиза, а также окисление и т.д. Реакции с солеобразными
гидридами формально можно отнести к реакциям присоединения:
В2Н6 + 2NaH = 2Na[BH4]
Однако в действительности в этих реакциях также имеет место разрушение бана-
банановых связей, превращение их в нормальные электрононедефицитные связи.
Соединения типа Me [BH4] называются боранатами или гидроборатами. Гидробо-
Гидробораты известны для щелочных, щелочно-земельных и некоторых других металлов.
В практике чаще других используется NafBHj — белое нелетучее вещество, ус-
устойчивое в сухом воздухе. Боранаты ИА-группы по сравнению с таковыми эле-
элементов IA-группы термически менее стойки и более активны как восстановители.
Только Ве[ВН4]г мало похож на своих гомологов и по свойствам стоит ближе к
А1[ВН4]3.
Для бинарного соединения бора с азотом — нитрида бора — известны в основ-
основном две модификации. Одна из них может быть получена синтезом из элементов.
Однако синтез осуществляется трудно, требуется температура выше 1200°С.
Поэтому так называемый гексагональный нитрид бора получают из соединений,
например
400° О
В2О3 + 2NH3 = BN + ЗН2О или 4H3NBF3 > BN + 3NH4[BF4]
Полученный таким образом нитрид бора представляет собой белый, похожий на
тальк порошок. Его часто называют "белым графитом". Во-первых, его кристал-
лохимическое строение аналогично таковому графита. Атомы бора и азота связа-
связаны между собой 5р2-гибридными связями. Кроме того, в плоскости слоев осущест-
осуществляется дополнительное тг-связывание за счет пустой ^мэрбитали атома бора и
неподеленной электронной пары атома азота. Отдельные слои между собой связа-
связаны силами Ван-дер-Ваальса. Во-вторых, "белый графит" обладает высокой огне-
огнеупорностью (tfjm ~ 3000°С), химически инертен даже при высоких температурах,
расслаивается на чешуйки, как графит.
Другая модификация нитрида бора имеет кубическую алмазоподобную струк-
структуру. В ней атомы бора и азота находятся в яр3-гибридном состоянии. При к.ч. 4
три связи образованы по обменному механизму, а одна — по донорно-акцепторно-
Му, причем атом бора является акцептором электронной пары, а атом азота —
Донором. Алмазоподобная форма нитрида бора называется боразоном или эльбо-
ром. В условиях высокой температуры и давления эльбор можно получить из
белого графита подобно тому, как алмаз получается из черного графита:
Другой способ получения боразона — азотирование фосфида бора
ВР + NH3 = BN + РН3
По химической инертности и твердости боразон не уступает алмазу, но в отли-
отличие от алмаза эти ценные качества сохраняет и при высоких температурах (до
2000°С). Подобно алмазу, боразон является изолятором. Таким образом, нитрид
бора удивительно похож на углерод: гексагональная модификация — на графит,
а кубическая — на алмаз. Это еще один пример аналогии изоэлектронных образо-
образований. Одна формульная единица BN и два атома углерода содержат одинаковое
число валентных электронов — по 8.
При взаимодействии гидробората лития с хлоридом аммония
3Li[BH4] + 3NH4C1 = B3N3H6 + 3LiCl + 9H2
образуется боразол В3КзН6, называемый также неорганическим бензолом. Анало-
Аналогично бензолу, в молекуле боразола атомы азота и бора находятся в состоянии
«р2-гибридизации, образуя шестичленный цикл
Атомы бора и азота связаны друг с другом а- и делокализованными тг-связями
(пунктир) за счет шести электронов от трех атомов азота по донорно-акцепторно-
му механизму; атомы бора — акцепторы, Боразол, как и бензол, представляет
собой жидкость, которая химически значительно более устойчива, чем гидриды
бора и их производные. Однако по сравнению с бензолом боразол более реакци-
онноспособен: медленно окисляется на воздухе, склонен к реакциям замещения и
присоединения. С серой бор образует сульфид B2S. Реакцию проводят между
твердым бором и парами серы при 600°С. Сульфид бора легко образует стекло,
разлагается водой с выделением сероводорода.
Соединение бора с углеродом (В4С)П — карбид бора — по структуре является
пространственным полимером. Получается он сплавлением оксида бора с углем.
Представляет собой черные блестящие кристаллы, обладающие очень высокой
твердостью, термически очень стойкие и устойчивые к различным химическим
воздействиям.
Соединения бора с металлами. Бор ни с одним из металлов
не образует непрерывных твердых растворов. Он не взаимодействует с металлами
группы цинка, индием, таллием, оловом, свинцом и висмутом. С металлическим
галлием бор образует эвтектическую смесь. С активными металлами бор дает
бориды, образованные в соответствии с правилами валентности, например 3Mg +
+ 2В = Mg3B2. Эти бориды химически активны, легко разлагаются
минеральными кислотами:
Mg3B2 + 6HC1 = В2Н6 + 3MgCl2
Наибольшее число соединений бор образует с переходными металлами. Мно-
Многие из них представляют собой фазы внедрения, являются металлоподобными
бооидами. Эти металлоподобные бориды исключительно тугоплавки, жаростойки,
жаропрочны и коррозионностойки. Кроме того, они отличаются высокой твердос-
твердостью, электрической проводимостью и теплопроводностью. Так, теплопроводность
и электрическая проводимость ZrB2 и TiB2 на порядок выше, чем эти же харак-
характеристики для металлических циркония и титана.
В зависимости от условий синтеза и соотношения исходных компонентов
состав боридов #-, sp- и переходных металлов может быть самым различным:
Ме4В, Ме3В, Ме2В, Ме3В2, МеВ, Ме3В4, МеВ2, Ме2В5, МеВ4, МеВ6, МеВ12. По
мере увеличения содержания бора в формульной единице структура боридов
усложняется. В соединениях с относительно большим содержанием металла отсут-
отсутствуют связи между атомами бора. Наоборот, при избытке атомов бора последние
образуют пары, цепочки, сетки, каркасы, в которых атомы бора группируются в
октаэдрические и икосаэдрические кластеры. Для примера на рис. 137 показана
структура А1В2, в которой атомы бора, расположенные в пустотах между атомами
А1, образуют графитоподобные слои из шестичленных колец. Изменение структу-
структуры влечет за собой изменение характера химической связи. У начальных членов
указанного ряда на полярную связь накладывается значительная доля металли-
металлической связи. С возрастанием содержания бора на первый план выдвигаются
неполярные ковалентные связи с частичной делокализацией электронов. В целом
между атомами реализуется химическая связь, представляющая собой наложение
ковалентной, металлической и ионной.
2. Алюминий. Особенности химии алюминия. Второй типи-
типический элемент III группы Периодической системы — алюминий — является
первым и самым легким sp-металлом с электронной формулой Is22s22p63s23p'. У
алюминия по сравнению с бором атомный радиус больше, а потенциалы иониза-
ионизации меньше; следовательно, возрастают металлические свойства. В отличие от
неметалла бора алюминий является амфотерным элементом в широком смысле
слова. Так, металлический алюминий и его гидроксид растворяются и в кисло-
кислотах, и в щелочах, а А1(+3) образует и комплексные катионы, и ацидокомплексы.
Алюминий по праву можно считать родоначальником как элементов подгруппы
галлия, так и элементов подгруппы скандия. Это видно из рис. 138, на котором
показан характер изменения энтальпий образования оксидов и галогенидов алю-
алюминия и элементов подгрупп галлия и скандия.
Рис. 137. Схема упаковки атомов А1 и В
в кристаллической структуре борида
алюминия А1В2
Рис. 138. Энтальпии образования окси-
оксидов и галогенидов алюминия и элементов
подгрупп галлия и скандия
20 И *0 SO 80 70 W 90 %
Для химии алюминия исключительно важное значение, имеет его большое
сродство к кислороду. Ниже для сравнения приведены энтальпии и энергии
Гиббса образования характеристических оксидов алюминия и элементов подгруп-
подгруппы галлия:
Соединения А12О3 Ga2O3 In2O3 Т12О3
Alff 298> кДж/моль -1675,7 -1089,1 -925,9 -390,4
AG°/298, кДж/моль -1582,4 -998,3 -831,8 -321,3
И энтальпия, и энергия Гиббса образования алюминия резко отличаются от
таковых для галлия и его аналогов. Только металлы подгруппы скандия и ланта-
лантаноиды имеют еще большие значения указанных характеристик, чем алюминий.
Наконец, для алюминия наблюдается горизонтальная аналогия со вторым
типическим элементом IV группы — кремнием. Отчетливо проявляется она в
алюмосиликатах, которые являются наиболее распространенными в земной коре
химическими соединениями. К ним относятся полевые шпаты, составляющие
более половины массы земной коры, глинистые минералы, слюды и др. В алюмо-
алюмосиликатах часть кремнекислородных тетраэдров [SiO^4" замещена алюмокисло-
родными тетраэдрами [AlOJ5"- Возникающие при таком замещении избыточные
отрицательные заряды компенсируются ионами К+ и Na+, так что электроней-
электронейтральность алюмосиликатов сохраняется.
Природные соединения и получение алюминия.
Алюминий — один из самых распространенных на Земле элементов (8,8%), зани-
занимает третье место по распространенности после кислорода и кремния. Таким
образом, алюминий является самым распространенным из металлов. Помимо
алюмосиликатов он встречается в природе в виде KA1(SO4J'2A1(OHK (алунит),
Na3[AlF6] (криолит), Mg[Al2O4] (шпинель), а также в виде оксида и его гидрат-
ных форм. Это прежде всего корунд о^-А12О3. В зависимости от содержания при-
примесей корунд имеет различную окраску и в соответствии с ней названия: бесцвет-
бесцветный — лейкосапфир, голубой — сапфир, рубиново-красный — рубин. Из гидрат-
ных форм оксида алюминия отметим боксит А12О3-пН2О с примесями оксидов
кремния, железа, титана и т.д. В зависимости от минералогической формы бокси-
боксита и содержания связанной воды различают гидраргиллит А12Оз-ЗН2О и диаспор
А12Оз'2Н2О. Боксит является наиболее ценной алюминиевой рудой, содержащей
более 50% А12О3.
Первый этап производства алюминия заключается в извлечении А12О3 (глино-
(глинозема) из алюминиевых руд щелочным, кислотным, электротермическим или
комбинированными способами. На второй стадии проводят электролиз глинозема
в расплаве криолита при температуре около 950° С. Хотя криолит встречается в
природе, чаще всего его получают искусственно. Не принимая во внимание взаи-
взаимодействие глинозема с растворителем, его ионизацию в расплаве можно предста-
представить уравнением
А12О3 *=* АЮ+ + АЮг
Катодом служит подина железной ванны, анодом — угольные электроды. На
катоде выделяется алюминий:
ЗАЮ* + Зе" = А1 + А12О3
На аноде разряжаются алюминат-ионы:
2АЮг - 2е- = А12О3 + 7гО2
Поскольку на аноде выделяется кислород, уголь постепенно сгорает и анод не-
непрерывно приходится доращивать. Несмотря на большую энергоемкость электро-
электролизного процесса, в настоящее время он является единственным промышленным
способом получения алюминия.
Полученный электролитический алюминий содержит 98,5—99,8% основного
вещества. Примесями являются железо, медь, титан, кремний, механически захва-
захваченные при кристаллизации криолит, глинозем, уголь. Сырой металл сначала
переплавляют, а затем подвергают электрохимическому рафинированию в расп-
расплаве из фторидов алюминия и натрия и хлорида бария. При рафинировании
чистота алюминия достигает 99,9%. Особо чистый алюминий, необходимый,
например, в электронной технике, получают специальными методами: вакуумной
дистилляцией и зонной плавкой.
Физические и химические свойства алюминия.
Алюминий — серебристо-белый металл. Кристаллизуется в ГЦК-структуре. Отли-
Отличается высокой пластичностью, легко поддается прокату, штампованию, волоче-
волочению. По электрической проводимости алюминий занимает четвертое место после
меди, серебра и золота. Металлический алюминий слабо парамагнитен.
Алюминий химически активен, но на воздухе покрывается тончайшей E—10
нм) оксидной пленкой, надежно защищающей металл от дальнейшего окисления.
Именно вследствие электрически и механически прочной защитной пленки при
обычных условиях А1 ведет себя довольно инертно, хотя Е° (А13+/А1) = -1,67 В.
При температуре плавления алюминия 660"С гранулированный А1 не сплавляется
в слиток даже при нагреве до 1200° С, так как каждая капля расплава металла
оказывается как бы в мешке из оксида. Поэтому почти все реакции с участием
алюминия идут с латентным (скрытым) периодом, необходимым для разрушения
оксидной пленки или диффузии реагента через нее.
С пниктогенами и халькогенами алюминий взаимодействует при высокой
температуре. С галогенами, за исключением иода, А1 реагирует при комнатной
температуре. В присутствии воды как катализатора алюминий легко взаимодей-
взаимодействует и с иодом. С водородом алюминий непосредственно не взаимодействует,
хотя водород растворяется в нем. При обычных условиях А1 не взаимодействует с
водой, но легко растворяется в растворах кислот и щелочей. Концентрированная
азотная кислота пассивирует алюминий, поэтому в ней он не растворяется.
Характеристические соединения. Характеристический
оксид алюминия AI2O3 является важнейшим химическим соединением. Это белое
очень тугоплавкое вещество (<пл = 2053 и <кип > 3000°С). Чистый AI2O3 получают
термическим разложением квасцов:
2NH4A1(SO4J = А12Оз + 2NH3 + 4SO3 + Н2О
или гидроксида А1(ОНK при 600—800°С. В последнем случае получается 7-AI2O3,
который при температуре выше 1000°С монотропно* переходит в №-А12О3. f-
А12Оз химически более активен и гигроскопичен. Водородом оксид алюминия не
* Монотропное превращение — необратимый фазовый переход из одной модификации
в другую.
восстанавливается. Прокаленный AI2O3 в воде не растворяется. В растворимое
состояние его можно перевести сплавлением со щелочами или пиросульфатами:
А12О3 + 2КОН = 2КАЮ2 + Н20
А12О3 + 3K2S2O7 = A12(SO4K + 3K2SO4
При комнатной температуре оксид алюминия практически не проводит электри-
электрического тока и является диэлектриком. При температурах выше 1000" С начинает
заметно проводить электрический ток и проявляет полупроводниковые свойства.
С повышением температуры электрическая проводимость растет экспоненциаль-
экспоненциально. В этих условиях в А12Оз происходит отщепление кислорода, в результате чего
нарушается стехиометрический состав, что и приводит к электронной проводи-
проводимости.
Гидроксид алюминия получают осаждением основаниями из растворов раство-
растворимых солей (А1(+3). Он представляет собой белый студенистый осадок, нераст-
нерастворимый в воде. Этот осадок в действительности является продуктом полимери-
полимеризации тригидрата гидроксида алюминия, который и растворяется в кислотах и
щелочах:
[А1(Н2ОN]
з+
ЗОН
AKOHV
ЗН2О
[А1(Н2О)з(ОН)з1
|
V
осаж/
i
1
i
зон
[Al (ОН)*!3
[А1(Н2О)з(ОН)з)„
Как у любого амфолита, кислотные и основные свойства гидроксида алюминия
выражены слабо. Метаформа от Н3АЮ3 — НАЮ2- Соли кислоты НАЮ2 называют-
называются метаалюминатами или алюмошпинелями, так как некоторые из них встречают-
встречаются в природе (MgAl2O4, ZnAl2O4) и входят в группу минералов, именуемых
шпинелями. Эти соли могут быть получены спеканием оксида алюминия с
оксидами металлов или карбонатами. Шпинели имеют кубическую решетку.
Атомы кислорода образуют плотноупако-
ванную структуру с тетра- и октаэдричес-
кими пустотами. В так называемых нор-
нормальных шпинелях, таких, как MgAl2O4,
в тетраэдрических пустотах находятся
атомы Mg, а в октаэдрических — атомы
А1 (рис. 139).
При обезвоживании студнеобразного
гидроксида алюминия получается актив-
активная форма А12Оз — алюмогель. Это по-
пористое вещество, сильно отличающееся
по виду от обычного мелкокристалличес-
мелкокристаллического порошка А12Оз- Алюмогель не раст-
растворяется в воде и не набухает, является
эффективным осушителем. Кроме того,
алюмогель — хороший носитель катали-
катализаторов (в основном никеля и платины).
Ш-А1 т-Мд О-В
Рис. 139. Кристаллическая структура нор-
нормальной шпинели
Из характеристических галогенидов А1Р3 фторид по свойствам резко отлича-
отличается от своих гомологов, в том числе по энтальпии образования (см. рис. 138),
температурам плавления и кипения. Аномально высокие значения указанных
констант для AlFg по сравнению с другими галогенидами объясняются большей
ионностью этого вещества вследствие наибольшей ОЭО фтора. В отличие от
других галогенидов алюминия его фторид в воде практически нерастворим. В
результате гидролиза хлорид, бромид и Иодид алюминия дымят на воздухе; в
парах они существуют в виде димеров А^Гд с мостиковыми связями:
Al A1
В димерных галогенидах алюминия его к.ч. 4. При этом из четырех ковалентных
связей три образованы по обменному механизму, а одна — по донорно-акцептор-
ному. В качестве акцептора электронной Пары выступает А1, а в качестве доно-
донора — атом галогена. Здесь еще раз дает себя знать диагональная аналогия, су-
существующая между алюминием и бериллием.
Фторид алюминия получают синтезом из элементов или растворением гидрок-
сида алюминия в плавиковой кислоте. Безбедный А1С1з можно получить нагрева-
нагреванием алюминия в токе хлора или НС1, а также пропусканием хлора над нагретой
смесью А12Оз с углем. Бромид и иодид алюминия синтезируют из элементов при
нагревании.
Соединения с другими неметаллами. В эфирном раство-
растворе между алюмогидридом (тетрагидроалюминатом) лития и хлоридом алюминия
протекает реакция 3Li[AiH4] + AICI3 = 4А1Н3 + 3LiCl с образованием белого
осадка — полимерного гидрида (А1Нз)га. Структура (АШз)п — трехмерная сетка с
мостиковыми связями А1 Н А1. Выше 300°С он разлагается на элемен-
элементы, водой гидролизуется с выделением водорода, является сильным восстановите-
восстановителем. Поскольку в гидриде алюминия имеется свободная Зр-орбиталь, он склонен
к реакциям присоединения, выступая как акцептор электронной пары. Так, он
присоединяет аммиак с образованием гидрида HsN-АШз, который при нагрева-
нагревании до 150°С переходит в нитрид алюминия A1N.
С халькогенами алюминий образует соединения типа AI2X3. Все они синтези-
синтезируются из компонентов в откачанных кварцевых ампулах. Энтальпия их образо-
образования закономерно уменьшается от сульфида к теллуриду в результате уменьше-
уменьшения доли ионности химической связи между разнородными атомами. Поскольку
они являются солями слабого основания и несильных кислот, в водном растворе
нацело подвергаются гидролизу. Этими реакциями удобно пользоваться для
получения соответствующих халькогеноводородов.
Нитрид алюминия частично образуется при сгорании металла на воздухе,
хотя и в очень малых пропорциях по отношению к оксиду. Существует несколько
способов получения A1N: действием на нагретый металл аммиаком, взаимодей-
взаимодействием азота и сульфида алюминия. Наконец, нитрид алюминия можно получить
по реакции
А12О3 + ЗС + N2 5= 2A1N + ЗСО
335
В начале нашего века эта реакция была предложена для связывания атмосферно-
атмосферного азота. Она протекает интенсивно при 1500—1600°С, для поддержания высокой
температуры достаточно теплоты сгорания выделяющегося оксида углерода.
Нитрид алюминия обладает структурой вюрцита. При нагревании он начинает
разлагаться при 1900 и плавиться при 1200°С под давлением 4,04• 10s Па. Непе-
реплавленный A1N гидролизуется даже во влажном воздухе. После переплавки
нитрид алюминия приобретает значительную химическую стойкость. С остальны-
остальными пниктогенами (кроме висмута) алюминий также дает эквиатомные соединения,
температуры плавления и склонность к гидролизу которых уменьшается от фос-
фосфида к антимониду. Все они являются перспективными полупроводниковыми
соединениями.
При взаимодействии алюминия с углем при 1800—2000°С в атмосфере водоро-
водорода происходит реакция синтеза карбида алюминия AI4C3 в виде бесцветных
прозрачных ромбоэдрических кристаллов. Примеси окрашивают его в желтый
цвет. Карбид алюминия — один из немногих карбидов, отвечающих правилам
формальной валентности. Галогены и халькогены вытесняют из карбида алюми-
алюминия углерод. Получен также карбид состава А12Сб (ацетиленид).
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Нерастворимый в воде AIPO4 интересен тем, что он
имеет структуру кварца. Обусловлено это горизонтальной аналогией, близостью
атомных радиусов Al, Si, P, изоэлектронностью. Если представить фосфат алю-
алюминия в виде АЮг-РОг, а диоксид кремния как SiO2-SiO2, то сумма валентных
электронов у атома алюминия и фосфора равна 8 и число валентных электронов
двух атомов кремния также равно 8, т.е. точно соответствует сумме валентных
электронов алюминия и фосфора.
Хорошо растворимый в воде нитрат А1(ГТОз)з кристаллизуется из раствора с 9
молекулами воды. Безводный нитрат алюминия в концентрированной HNO3 обра-
образует ацидокомплекс [А1(ГТОзL]~- Однако он полностью разрушается в воде. Суль-
Сульфат A12(SO4K кристаллизуется из водного раствора в форме Al2(SO4)s- I8H2O. С
сульфатами металлов в степени окисления +1 сульфат алюминия образует двой-
двойные соли типа Me2SO4" A12(SO4K'24H2O. Эти соединения получили название алю-
алюминиевых квасцов. Квасцы в твердом состоянии вполне устойчивы, а в растворах
сильно диссоциированны на составляющие ионы. Квасцы растворяются в воде
намного хуже, чем составляющие сульфаты, а потому меньше подвергаются
гидролизу, хотя растворы имеют кислую реакцию. Квасцы образуют не только
А1(+3), но и другие элементы в степени окисления +3, в том числе галлий и
индий. В табл. 23 порядок следования обоих типов ионов слева направо и сверху
вниз соответствует убыванию способности к образованию квасцов.
Т а б л и ц.а 23. Способность элементов к образованию квасцов
Rb+
Cs+
NH4
Na+
A13+
Ga3+
Fe3-
Rh3*
V3+
In»*
Co3+
+
:
Ti3+
- ¦
Mn3+
+
-
—
336
По вполне понятным причинам среди s- и sp-металлов алюминий является
лучшим комплексообразователем. Это видно хотя бы из того, что А1(+3) лучше
других катионов образует квасцы, в природе встречается такой устойчивый комп-
комплекс, как криолит Na3[AlF6] (рА'неСт 19,8). Дело в том, что ион А13+ характеризу-
характеризуется большим зарядом, малым размером и вакантными 3s-, Зр- и даже 3</-орбита-
лями. По комплексообразовательной способности из s- и sp-элементов с алюмини-
алюминием может сравниться только его диагональный аналог — бериллий. В своих коор-
координационных соединениях А13+ показывает тенденцию к октаэдрической и тетра-
эдрической координации. Помимо вышеупомянутых аква-, гидроксо- и фторо-
комплексов с к.ч. 6 следует отметить устойчивый оксалатокомплекс [А^СгС^Ь]" с
рА'нест 13,0 и к.ч. 4. Со всеми галогенид-ионами А13+ образует комплексы с к.ч. 4,
среди которых наибольшей устойчивостью отличается [AIF4]" (рАцест 17,8). Ус-
Устойчивость тетрагалогенидных комплексов [AIFJ от фтора к иоду закономерно
снижается. Известны также цианидные и роданидные комплексы с к.ч. 4, разла-
разлагающиеся при долгом хранении, гигроскопические и легко гидролизующиеся.
Металлохимия. Ни с одним металлом Периодической системы алюми-
алюминий не дает непрерывных твердых растворов. Алюминий является плохим раство-
растворителем для других металлов, хотя сам хорошо растворяется в них, особенно в
переходных. Для алюминия исключительно характерно образование большого
числа металлидов: с литием, щелочно-земельными и со всеми переходными ме-
металлами. Как видно из диаграммы состояния А1—Ni (рис. 140), алюминий с нике-
никелем образует широкую область твердых а-растворов, тогда как со стороны алю-
алюминия отсутствует область твердых растворов. Кроме того, эквиатомный металлид
AINi плавится при значительно более высокой температуре, чем тугоплавкий
компонент — никель, тем более алюминий. Это соединение является истинным
металлидом, ибо на его основе существует широкая область однородности. Нако-
Наконец, алюминий с никелем образуют ряд инконгруэнтно плавящихся металлидов.
Только с оловом (тоже sp-металл) алюминий дает на диаграмме состояния
простую эвтектику. Алюминий не взаи-
взаимодействует с натрием и собственно щелоч- f°?
ными металлами, а также с таллием,
свинцом и висмутом.
3. Подгруппа галлия. Характе-
Характеристика элементов под-
подгруппы галлия. Подобно типи-
типическим элементам, металлы подгруппы
галлия являются sp-элементами. Несмотря
на то что элементы подгруппы галлия —
типовые аналоги, наблюдаются особен-
особенности в свойствах отдельных ее предста-
представителей. Элемент галлий непосредственно
следует за первой десяткой кайносиммет-
ричных переходных 3</-металлов, для
которых особенно сильна rf-контракция.
Поэтому атомный радиус галлия меньше """ ° 10 20 30 ^ 50_ 6" 70 80 90 т
таковых не только его более тяжелых Al Am- вШ' % Ni Ni
аналогов, НО И аЛЮМИНИЯ. Вследствие Рис. 140. Диаграмма состояния системы
этого ионизационное потенциалы галлия алюминий - никель
337
более высокие и связанные с ними энергетические характеристики отличаются от
его аналогов. Уже у элементов НВ-группы заметна тенденция к уменьшению
степени окисления сверху вниз, в частности для ртути. Такое понижение положи-
положительной степени окисления еще более заметно в подгруппе галлия. В этом в
определенной мере проявляется горизонтальная аналогия. Уже для таллия сте-
степень окисления +1 более стабильна, чем характеристическая степень окисления
+3. Вследствие d- и особенно /-контракции переход от индия к таллию сопровож-
сопровождается только незначительным увеличением атомного радиуса. В то же время
ионизационные потенциалы таллия заметно больше, чем индия. Дело в том, что
оба 6в2-электрона атома таллия подвержены сильному эффекту проникновения
через двойной экран d- и /-электронных облаков В результате «-электроны с
трудом участвуют в образовании химических связей. Этот факт получил наиме-
наименование концепции инертной электронной пары. Поэтому у таллия часто валент-
валентным является бр-электрон, который, переходя к окислителю, превращает таллий
в устойчивый ион Т!(+1). По этой причине производные Tl(-fl) почти не прояв-
проявляют восстановительных свойств и, наоборот, производные Т1(+3) являются
сильными окислителями.
Природные соединения и получение. В противовес алю-
алюминию металлы подгруппы галлия относятся к малораспространенным и рассеян-
рассеянным элементам. Практически существует один минерал галлия — галлит CuGaS2,
редко встречающийся (Южная Америка). Ввиду близости значений ионных
радиусов А1(+3) и Ga(+3) галлий частично замещает алюминий в бокситах.
Кроме тоге, параметры решеток GaS и ZnS почти одинаковы, а потому галлий
способен входить в виде примеси в сфалерит. Все это приводит к тому, что гал-
галлий присутствует в бокситах, сфалерите и полиметаллических рудах. Из отходов
переработки боксита на глинозем или из полиметаллических руд галлий осажда-
осаждают в виде гидроксида Са(ОН)з- Затем выделяют галлий электролизом сильноще-
сильнощелочных растворов гидроксида. Полученный продукт содержит не более 99,5%
основного металла. Галлий высокой чистоты получают переплавкой в вакууме.
При этом примеси улетучиваются, а сам галлий практически не испаряется
вследствие колоссальной разницы между температурами плавления и кипения
B9,8 и 2070°С).
Индиевые руды рокезит CuInS2 и индит FeInS3 также встречаются очень
редко. В основном индий получают при переработке цинковых, кадмиевых и
оловянных руд. Нахождение его в последних подчеркивает его горизонтальную
аналогию с кадмием и оловом. Извлечение индия сводится к обогащению им
исходного продукта и действию на концентрат серной кислотой. Черновой индий
извлекают из растворов реакцией замещения металлическим цинком или алюми-
алюминием. Особо чистый индий получают зонной плавкой.
Хотя таллий и образует несколько минералов, например лорандит T!AsS2 и
крукезит (Tl,Cu,AgJSe, они также весьма редки. Интересно отметить, что в
природе таллий встречается в степени окисления +1. Это подтверждается вало-
валовым составом указанных минералов. Основным сырьем для получения таллия
служат полиметаллические руды, в которых он присутствует в виде примеси.
Извлечение таллия из пыли, получающейся при окислительном обжиге этих руд,
основано на растворимости оксида таллия в горячей воде. Полученный гидрок-
сид переводят в сульфат таллия, который и подвергают электролизу. Существу-
Существуют способы, по которым сначала получают плохо растворимый Т1С1, который
338
восстанавливают до металла цинка. Возможно также восстановление оксида тал-
таллия углем или водородом.
Физические и химические свойства. Существование
галлия — экаалюминия — было предсказано Д.И. Менделеевым. На основе Пе-
Периодической системы им были в 1870 г. "вычислены" основные свойства этого
элемента. В 1875 г. французский химик Лекок де Буабодран открыл и выделил
этот элемент. Научный мир был поражен совпадением предсказанных свойств
экаалюминия со свойствами вновь открытого элемента галлия. Открытие галлия
явилось первым триумфом на пути становления и укрепления Периодического
закона и Периодической системы Д.И. Менделеева.
В форме простых веществ галлий, индий и таллий представляют собой сереб-
серебристо-белые металлы, при этом галлий хрупок, а индий и таллий очень мягкие.
Индий и таллий кристаллизуются в плотноупакованной кубической или близкой
к ней решетке. Кристаллохимическое строение галлия оригинально и необычно
для металлов. Структуру галлия лишь условно можно назвать псевдотетрагональ-
псевдотетрагональной. Каждый атом галлия имеет ближайшего соседа на расстоянии 0,243 нм,
шесть других находятся на расстоянии от 0,270 до 0,279 нм. Другими словами,
металлический галлий как бы состоит из двухатомных молекул, образующих
слои, связь между которыми слабая, чем и объясняется его аномально низкая
температура плавления. Характеристики элементов и простых веществ ША-груп-
пы приведены ниже.
Обращают на себя внимание довольно низкие температуры плавления метал-
металлов подгруппы галлия, чем эти металлы также похожи на металлы подгруппы
цинка. Только в последней самый легкоплавкий металл — ртуть — замыкает
подгруппу, а обсуждаемая подгруппа, наоборот, легкоплавким галлием начинает-
начинается. Это обстоятельство еще раз доказывает переходный характер * металлов
подгруппы цинка, если иметь в виду, что все sp-металлы сравнительно легко-
легкоплавки.
В Al Ga
Содержание в земной коре,
масс.доли, % 3-Ю 8,8 1,5-КГ3
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э3+, нм
Потенциалы ионизации, В
lv Э -+ Э+ + е
12: Э+ -+ Э2+ + е
h: Э2+ -> Э3+ + е
ОЭО
Температура плавления, °С
Температура кипения, ° С
Плотность, г/см3
), В - -1,67 -0,65
0,091
0,023
8,298
25,155
37,930
2,0
2040
2550
2,46
0,143
0,057
5,986
18,828
28,447
1,5
660,2
2270
2,70
0,139
0,062
5,998
20,514
30,710
1,6
29,8
2070
5,90
Здесь речь идет о переходном характере от IB- к ША-группе.
339
i'
Содержание в земной коре,
масс.доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э+, нм
Ионный радиус Э3+, нм
Потенциалы ионизации, В
h: Э -+ Э+ + е
12: Э+ -+ Э2+ + е
13: Э2+ -? Э3 + + е
ОЭО
Температура плавления, °С
Температура кипения, ° С
Плотность, г/см3
), В
In
1,5-10
?"(э;_р/э), в
0,166
0,130
0,092
5,786
18,869
28,03
1,7
156,2
2075
7,31
-0,343
-0,25
Tl
4,5-10
0,171
0,136
0,105
6,108
20,428
29,83
1,9
302,5
1457
11,85
+0,71
-0,336
Оксидные пленки на галлии, индии и особенно на таллии не столь прочны по
сравнению с алюминием и в гораздо меньшей степени пассивируют поверхность
металлов. При нагревании в кислороде или на воздухе галлий, индий и таллий
сгорают, но продукты окисления различны. Галлий преимущественно образует
монооксид GaO, индий дает 1п2Оз, а таллий — смесь Т12Оз и Т12О с тем большим
содержанием последнего, чем выше температура процесса окисления. Интенсивно
реагируют металлы подгруппы галлия с галогенами. Только взаимодействие
таллия с иодом требует предварительного подогрева. К водороду все эти металлы
инертны, хотя и образуют гидриды косвенным путем. Реакции с халькогенами и
пниктогенами требуют сильного нагревания, а таллий при сплавлении с фосфо-
фосфором, мышьяком и сурьмой образует простую эвтектику. Галлий, индий и таллий
не взаимодействуют с водой, хотя последний медленно растворяется в воде, если
она содержит большое количество растворенного кислорода. В кислотах все три
металла хорошо растворимы. При этом галлий и индий дают производные степе-
степени окисления +3, а таллий окисляется до Т1(+1). Лишь кислоты, обладающие
большой окислительной способностью, переводят таллий в Т1(+3). Так, концент-
концентрированная азотная кислота дает смесь TINO3 и Т1(КОз)з и только царская водка
переводит таллий в Т1С1з- Галлий растворим в концентрированных растворах
щелочей, а индий и таллий инертны к ним. Однако индий (как алюминий и
галлий) взаимодействует с расплавленной щелочью в присутствии кислорода
воздуха:
2In + 2NaOH + 3/2О2 = 2NaInO2 + Н2О
Характеристические соединения. Характеристические
оксиды элементов подгруппы галлия менее прочны и тугоплавки по сравнению с
оксидом алюминия. Без термической диссоциации плавится Ga2O3 A725°С), а
1п2Оз выше 850°С отщепляет кислород и переходит в I113O4. 1п2Оз и Т12Оз могут
быть расплавлены под избыточным давлением кислорода, причем TI2O3 начинает
340
заметно разлагаться при 90° С, переходя в TI2O. С увеличением атомного номера
возрастает интенсивность окраски: Ga2O3 — белый, I112O3 — светло-желтый,
TI2O3 — коричневый. Этот факт также указывает на уменьшение доли ионности в
оксидах с ростом атомной массы элемента. вагОз изоструктурен корунду, a I112O3
и Т12Оз кристаллизуются в решетке Мп2О3 (ОЦК).
В отличие от AI2O3 оксид галлия начинает восстанавливаться водородом уже
при 600°С. Как и оксид алюминия, свежеполученный непрокаленный Ga2O3
легко растворяется в сильных кислотах и утрачивает эту способность после про-
прокаливания. В расплавах щелочей легко растворяется даже прокаленный Ga2O3,
образуя растворимые в воде галлаты типа Me GaO2. Галлаты металлов Ме(+2),
например MgGa2O4, имеют структуру шпинели. Высший оксид индия еще легче
восстанавливается водородом, чем Са2Оз, а с порошком магния 1п2Оз образует
взрывчатую смесь. Т12Оз представляет собой нестехиометрическое соединение с
избытком атомов таллия (односторонняя фаза), а потому обладает аномально
высокой проводимостью среди оксидов Э2Оз.
Характеристические гидроксиды Э(ОН)з — почти нерастворимые в воде
вещества. Так, для Ga(OHK и 1п(ОНK рПР 37, а для Т1(ОНK рПР 45. Гид-
Гидроксид галлия растворяется в сильных кислотах и основаниях. Он является
редким примером идеального амфолита, для которого кислотные и основные
свойства выражены практически в равной мере. Ниже приведены последова-
последовательные константы электролитической ионизации по кислотному и основному
типам:
К\ К^ Кз
H3Ga03
Ga(OHK
Гидроксид индия 1п(ОН)з выделен в свободном состоянии; выше 300° С час-
частично теряет воду и переходит в InO(OH), который при дальнейшем нагревании
превращается в оксид. Гидроксид индия с трудом растворяется в крепких раст-
растворах щелочей, т.е. тенденция к образованию индатов выражена слабо. Достовер-
Достоверно установлено существование индатов щелочных и щелочно-земельных металлов.
Кроме того, твердофазным путем получена индиево-магниевая шпинель MgIn2O4-
Гидроксид Т1(ОН)з неустойчив и при хранении в эксикаторе с серной кислотой
превращается в оксид. Гидроксид таллия в отличие от его более легких аналогов
и гидроксида алюминия обладает только основными свойствами.
Характеристические тригалогениды (кроме фторидов) сравнительно легко-
легкоплавки и сильно летучи. Ниже приведены температуры плавления и кипения
этих соединений (верхняя строка означает tnJl, а нижняя — tKim или <Субл, °С):
F Cl Br I
Ga
In
Tl
930 - - -
341
1,6-
1,2-
Ю-7
lO
5-
2-
10-u
10-u
2-
4-
102
102
1000
950
1172
1200
550
930
78
215
586
546
155
122
279
436
372
-
_
212
,346
200
500
—
В трифторидах доля ионной связи наибольшая, а потому они отличаются
более высокой температурой плавления и кристаллизуются в координационных
структурах. Трихлориды, за исключением GaCb, имеют слоистую решетку. Три-
хлорид галлия характеризуется молекулярной структурой, состоящей из димер-
ных молекул, между которыми действуют слабые силы Ван-дер-Ваальса. Этим и
объясняется аномально низкая температура плавления GaCl3- В парообразном
состоянии все ЭГз, как и А1Гз, димеризованы с образованием мостиковых связей.
При этом все элементы подгруппы галлия проявляют ковалентность, равную
четырем. Из-за склонности к димеризации в парообразном состоянии многие
галогениды ЭГз возгоняются (сублимируют) до плавления, например GaF3, 1пС1,ч
и 1пВг3. Все тригалогениды легко взаимодействуют с водой. Фториды галлия и
индия образуют кристаллогидраты ЭРз'ЗН2О. Для остальных ЭГ3 характерны
реакции гидролиза с образованием оксида и галогеноводорода. При гидролизе
выделяется большое количество теплоты, особенно в случае хлоридов и бромидов
галлия и индия. Поэтому эти галогениды сильно гигроскопичны. Фториды, не
гидролизующиеся при обычных условиях, гидролизуются горячим водяным
паром.
Соединения с другими неметаллами. Подобно аланату
лития, в эфирном растворе при низких температурах образуются галланат, инда-
нат и талланат лития: Li[GaH4], Li[InH4] и Li[TlH4]. В ряду [ВН4]~, [А1Н4]~,
[GaH4]~, [InH4]~, [Т1Н4]~ устойчивость снижается. Взаимодействием указанных
соединений с соответствующими хлоридами получены полимерные гидриды:
(GaH3)n, (InH3)n, (Т1Нз)п. Из них (GaII3)n — стабильное соединение, (Т1Н3)П
устойчив только ниже -90°С, полимерный гидрид индия при 0° разлагается до
моноформы 1пНз, которая устойчива до 380°С. Интересно, что в низкотемпера-
низкотемпературной плазме водорода при введении паров триметилгаллия Ga(CH3K получен
жидкий дигаллан Ga2H6, стабильный до 130°С. Это соединение аналогично дибо-
рану и в нем также установлено существование банановых связей. В целом химия
галлия (как и химия бора) несколько отличается от химии других представите-
представителей Ш-А группы, что является проявлением вторичной периодичности.
Халькогениды ЭгХж известны для галлия и индия. Энтальпии образования
соединений закономерно уменьшаются в направлении от сульфидов к селенидам.
Для таллия известен только сульфид TI2S3, который может быть получен сухим
путем. Халькогениды Э2Х3 являются полупроводниками типа А2 Вз • Они ха-
характеризуются большим числом полиморфных модификаций; из них наиболее
стабильна гексагональная вюрцитоподобная, в которой треть узлов в подрешетке
металла вакантна. Таким образом, эти соединения по своей генетической природе
являются дефектными. Такие дефекты получили название биографических.
Сверхстехиометрические и примесные атомы в этих халькогенидах занимают
места биографических вакансий и потому мало влияют на свойства соединения.
Исключительно важны пниктогениды элементов подгруппы галлия — самые
ш V
важные полупроводниковые соединения А В . Висмут такие соединения не
образует. Желтый нитрид галлия получается при пропускании аммиака над
нагретым до 1000°С галлием, а также в результате разложения (NH4)a[GaF6] в
атмосфере аммиака. Нитрид индия получен аналогичным"образом из (NH4K[InFe]
при более низкой температуре. Остальные пниктогениды получают синтезом из
компонентов. От^фосфидов к антимонидам (стибидам) закономерно уменьшается
342
температура плавления вследствие уменьшения доли ионной связи. Все соедине-
а И1„У ,
ния типа А В , кроме нитридов, имеют кристаллохимическое строение сфале-
„ . III „V .
рита. И атомы А , и атомы В проявляют ковалентность, равную 4, причем три
связи образуются по обменному механизму, одна — по донорно-акцепторному.
III V
Соединения А В являются изоэлектронными аналогами алмаза, кремния,
германия и о-олова. Однако химическая связь в А В имеет и существенное
отличие, связанное с тем, что атомы элементов V группы имеют гораздо большее
сродство к электрону, чем атомы металлов. Это приводит к смещению электрон-
электронной плотности и появлению третьего вклада в связь — ионного, в результате чего
ширина запрещенной зоны заметно увеличивается.
В химическом отношении наиболее устойчивы нитриды. Так, GaN не раство-
растворяется даже в кипящей царской водке, но горячая концентрированная серная
кислота и концентрированные растворы щелочей медленно переводят его в раст-
,. . Шг> V
вор. Остальные соединения А В ведут себя как металлопроизводные пниктоге-
новодородов. Соединения галлия и индия растворяются 8 кислотах. Некоторые
из них, например InP, пассивируются в азотной кислоте. Растворы щелочей на
эти вещества, кроме антимонида галлия, не действуют.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Сульфаты элементов подгруппы галлия бесцветны и
легко растворяются в воде. Кристаллизуются из растворов с различным содержа-
содержанием молекул воды. С сульфатами металлов в степени окисления +1 сульфат
галлия и сульфат индия образуют квасцы. Бесцветные нитраты Ga(+3), 1п(+3) и
Т1(+3) также выделяются из воды в виде кристаллогидратов. Все это свидетель-
свидетельствует об определенной комплексообразовательной способности элементов под-
подгруппы галлия.
При растворении гидроксида галлия в избытка кислоты и щелочи имеют
место те же процессы, что и в случае А1(ОН)з'.
[Ga(H2ONp*
Ga(OHK
[Ga(OHN
-
Таким образом, в аква- и гидроксокомплексах Ga(+3) имеет к.ч. б. Однако для
всех трех элементов существуют комплексы с координационной валентностью и 4,
и 6. Ниже приводим примеры некоторых из этих комплексов (в скобках рКнесг):
[GaF6p- A6,8), [In(OHL]" B9,6), [In(CH3COONp- A8,3), [TlBrJ" B6,1), [TlBr6p-
C1,6). Для Ga(+3), как и для А1(+3), наиболее характерны фторокомплексы, а
1п(+3) и Т1(+3) обладают большим сродством к другим галогенид-ионам. Здесь
сказывается размерный фактор: увеличение ионных радиусов комплексообразова-
телей в ряду от галлия к таллию.
Соединения низших степеней окисления*. Устойчи-
Устойчивость соединений металлов ША-группы, где они проявляют степень окисления
+1, растет от алюминия к таллию. Для всех этих металлов существуют оксиды
типа Э2О. AI2O получают восстановлением высшего оксида алюминием или крем-
* Для сравнения здесь обсуждаются и соединения А1, которые не рассматривались в
§ 2 Данной главы.
343
нием при 1800—2000° С. Он представляет собой светло-голубые кристаллы, устой-
устойчивые к действию растворов кислот и медленно растворяющиеся в щелочах. На
воздухе при 1000—1100°С А12О окисляется с поверхности до А12Оз- Ga2O получа-
получается по реакции Ga2O3 с металлическим галлием уже при 500°С. Это темно-ко-
темно-коричневый порошок, легко окисляющийся при нагревании и восстанавливающий
серную кислоту до сероводорода. Восстановлением оксида индия водородом
получают 1п2О — мелкокристаллическое вещество черного цвета. На воздухе оно
окисляется, растворяется в соляной кислоте, но стойко к действию воды. Оксиды
Э2О сравнительно легкоплавки (Ga2O 660°С, 1п2О 327° С) и легколетучи. Т12О
получают прямым окислением металла на воздухе при небольшом нагревании
или прокаливанием ТЮН в инертной атмосфере. Т12О — порошок черного цвета,
в расплавленном состоянии {tnjl 300°С) желтый, устойчив вплоть до температуры
кипения 1080° С. Т12О легко растворяется в воде с образованием щелочи, т.е.
ведет себя как оксид щелочного металла. При нагревании в кислороде Т12О
переходит в Т12Оз- Расплав Т12О разрушает силикаты — стекло, фарфор. Такое
же действие оказывает и раствор ТЮН — сильной щелочи, растворяющей, в
частности, гидроксид алюминия. Энтальпия образования субоксидов * резко
уменьшается от А12О к 1п2О, а для Т12О несколько растет:
Соединение
2gg, кДж/моль
А12О
-1040
Ga2O
-344
In2O
-168
Т12О
-177
Подобно субоксидам, для всех этих металлов существуют субсульфиды. A12S
получается нагреванием А12Бз с алюминием при 1300° С. Аналогичным путем
можно получить и Al2Se. Ga2S получают взаимодействием галлия с сероводоро-
сероводородом при высокой температуре. Субсульфиды индия и таллия получаются прямым
сплавлением компонентов или восстановлением сульфида водородом:
In2S3 + 2Н2 = In2S + 2H2S
Сплавлением стехиометрических количеств Ga и In с Se и Те в откачанных квар-
кварцевых ампулах синтезируют эквиатомные халькогениды (ЭХ), например GaSe и
InTe. Они весьма стойки, не гидролизуются и плавятся без разложения, облада-
обладают полупроводниковыми свойствами. Все эти элементы образуют моногалогениды
ЭГ. Для алюминия эти соединения, существующие только при высоких темпера-
температурах, получаются по реакции тригалогенидов с металлом. Реакция
А1С13
600° С
ЗА1С1
при 1000°С смещена в сторону образования монохлорида, который при низких
температурах диспропорционирует. Так как монохлорид более летуч, чем трихло-
рид, этот процесс является типичной транспортной реакцией и используется для
получения особо чистого алюминия. Моногалогениды галлия также устойчивы
лишь при высоких температурах, могут быть получены при нагревании металла в
атмосфере галогеноводорода. Для индия монохлорид, монобромид и моноиодид
уже устойчивы при нормальных условиях.
* Префикс а/б- используется для обозначения соединений в низших малохарактерных
степенях окисления.
344
Наиболее типичны моногалогециды для таллия. Монофторид легко получить,
нейтрализуя гидроксид таллия Т1ОН плавиковой кислотой. Он хорошо раство-
растворим в воде и может быть обезвожен переплавкой. Растворимость остальных моно-
галогенидов (получаются прямым синтезом) мала и уменьшается от хлорида к
иодмду. В этом отношении моногалогениды таллия напоминают галогениды
серебра. Сходство еще более усиливается фоточувствительностью галогенидов
таллия и окраской их кристаллов: Т1С1 — бесцветный, Т1Вг — бледно-желтый, ТН
имеет две модификации — желтую и красную. Карбонат таллия (+1) хорошо
растворяется в воде, подобно карбонатам щелочных металлов, и дает щелочную
реакцию. Таким образом, в степени окисления +1 таллий ведет себя и как сереб-
серебро, и как щелочной металл.
Степень окисления +2 соответствует неустойчивым соединениям. Поэтому она
для алюминия и элементов ША-группы мало характерна. Их образование связано
с распариванием s-электронной пары, что особенно затруднено для таллия (эф-
(эффект инертной электронной пары). При этом у атома металла остается неспарен-
ный электрон, что уменьшает стабильность этой степени окисления. Очень неус-
неустойчивые оксиды типа ЭО обнаружены для всех этих элементов, кроме таллия.
А10 обнаружен в парах при нагревании смеси металла с характеристическим
оксидом выше 3000°С. GaO и InO получаются при восстановлении Э2О3 водоро-
водородом. Для галлия и индия известны также дихлориды и дибромиды. Так как эти
соединения диамагнитны, их структура отвечает формуле Э2Г4:
т.е. они являются дигалогенидами лишь формально (как и в случае ртути). Дига-
логениды индия получаются прямым синтезом при осторожном нагревании ком-
компонентов, дихлорид и дибромид галлия — восстановлением тригалогенидов ме-
металлическим галлием. При неполном сгорании галлия во фторе или при его
обработке плавиковой кислотой получают белый кристаллический GaF2, плохо
растворимый в воде. Легко гидролизующиеся дихлорид и дибромид таллия
представляют собой автокомплексы * Т1[Т1С14] и Т1[Т1Вг4]. Прямым синтезом
получены также GaS и InS. Первый из них может быть получен также восстанов-
восстановлением Ga2S3 водородом.
Металлохимия. Кристаллохимическое строение всех трех металлов
различно. Галлий имеет орторомбическую решетку, индий — тетрагональную, а
таллий обладает диморфизмом: о-модификация ГПУ и /?-форма ОЦК. Ни один
из обсуждаемых металлов не образует непрерывных твердых растворов с другими
элементами Периодической системы. Между собой галлий с таллием дают рас-
расслоение в жидком состоянии, галлий с индием — ограниченные твердые растворы
со стороны индия с эвтектикой, а индий с таллием — ограниченные твердые
растворы с перитектикой. Из-за низких температур плавления области гомоген-
гомогенности со стороны металлов подгруппы галлия очень малы. Металлидов они обра-
образуют также сравнительно немного: главным образом с щелочными, щелочно-зе-
* Автокомплексы — комплексные соединения, в которых один и тот же атом функ-
функционирует и как комплексообразователь, и как внешнесферный катион.
345
20 НО № SO 100
In Am. Воли, % Те Те
Рис. 141. Диаграмма состояния
системы индий - теллур
мельными и некоторыми переходными металлами.
Интересно отметить, что в случае галлия и индия
монохалькогениды на диаграммах состояния пред-
представлены более высокими дистектическими точка-
точками по сравнению с халькогенидами этих металлов
в степени окисления +3. Для иллюстрации на рис.
141 приведена диаграмма состояния системы In —
Те (ср. InTe и Iri2Tes). Аналогичный характер
имеет и фазовая диаграмма системы Ga — Те.
Другими словами, отсутствие эффекта инертной
электронной пары для галлия и индия четко
проявляется и на фазовых диаграммах состояния.
А соединения Ir^Te, Ii^Tes представлены перитек-
перитектикой, т.е. они разлагаются до плавления.
4. Подгруппа скандия и РЗЭ. Характе-
Характеристика элементов подгруппы скандия и РЗЭ. Эле-
Элементы подгруппы скандия объединяет с типическими элементами и элементами
подгруппы галлия наличие трех валентных электронов. Но у элементов ША-
группы ватентные электроны — это два электрона на s-орбитали и один на р-
орбитали, а у элементов подгруппы скандия третий электрон находится на (п —
1)о'-орбитали. Таким образом, типические элементы и элементы подгруппы гал-
галлия являются sp-элгментами. а элементы подгруппы скандия — sa'-элементами. В
состоянии трехзарядных ионов Э3+ элементы ШВ-группы имеют электронную
конфигурацию благородных газов, а потому наблюдается горизонтальная анало-
аналогия с щелочно-земельными элементами, например в характере оксидообразова-
ния, в поведении гидроксидов и т.д. Несколько отличается химия скандия —
первого кайносимметричного Згйэлемента, открывающего первый ряд переходных
металлов.
К обсуждаемой подгруппе относятся и лантаноиды — 14 элементов, следую-
следующих за лантаном, для которых характерно заселение (п — 2)/орбиталей. Все
лантаноиды вместе с иттрием и лантаном именуются редкоземельными элемента-
элементами (РЗЭ). Название происходит от средневекового наименования природных
оксидов — "земли" (как и щелочно-земельных металлов). К ним обычно не отно-
относят скандий. Элементы подгруппы скандия (Sc, Y, La) проявляют характеристи-
характеристическую степень окисления +3, а некоторые лантаноиды помимо указанной глав-
главной степени окисления еще проявляют степени окисления +2 и +4.
Природные соединения и получение металлов.
Содержание скандия и редкоземельных элементов в земной коре не так уж мало:
для скандия оно примерно такое же, как для мышьяка, и в два раза больше, чем
бора. Суммарное содержание одного из самых редких редкоземельных элемен-
элементов — тулия — больше, чем серебра, кадмия, ртути, сепена. С точки зрения на-
нахождения в природе РЗЭ делятся на две группы: цериевую (La, Се, Pr, Nd, Pm,
Sm) и иттриевую (Y, Eu, Gd, Tb, Dy, Но, Er, Tm, Yb, Lu). Это деление основано
на том, что в одних природных минералах встречаются преимущественно церие-
вые земли, а в других — иттрий вместе с остальными элементами. Скандий обра-
образует собственные минералы ScPCV2H2O (стереттит), Sc2Si207 (тортвейтит), встре-
встречающиеся крайне редко. Кроме того, скандий содержится в касситеритах (SnC^),
вольфрамитах (Mn, Fe)WO4, урановых рудах и др. Существует более 170 минера-
346
лов, в которых в тех или иных пропорциях содержатся РЗЭ. Относительное
содержание каждого из них также неодинаково. Меньше других содержится РЗЭ
с большей атомной массой и нечетным порядковым номером. Из минералов це-
риевой группы укажем на монацит (Се, La, Nd, ...)PO4- Он образует россыпи
монацитового песка, куда кроме него входят кварц, рутил, диоксид тория. В
монацитовом песке содержатся все минералы цериевой группы. Элементы этой же
группы содержатся в изоморфных фторокарбонатах (Се, La,...)FCO3 (бастнезит),
а также в собственном силикате Ce2Si2O7 (церит). Из минералов иттриевой
группы отметим ксенотим (Y, Eu, Gd, ...)P04, в котором РЗЭ изоморфно замеща-
замещают друг друга (как в монаците).
Для получения скандия его концентраты переводят в трифторид ScF3, кото-
который кальцийтермически восстанавливают до металла. Рафинируют перегонкой в
высоком вакууме. Разделение РЗЭ в свое время представляло трудную химико-
технологическую задачу. Во-первых, они но химическим свойствам настолько
похожи между собой, что нет аналогичных примеров в химии. Это особенно отно-
относится к химии лантаноидов, что обусловлено заселением электронами третьего
глубинного электронного слоя (п — 4) атома, считая от наружного со значением
главного квантового числа 6. Во-вторых, все РЗЭ геохимически очень тесно свя-
связаны между собой. Наконец, в-третьих, не было эффективных методов разделе-
разделения РЗЭ. Пользовались в основном малоэффективной и малопроизводительной
дробной кристаллизацией* через двойные сульфаты и нитраты РЗЭ. В результа-
результате получали мишметалл, содержащий 40—45% Се, 20—25% La, ~15% Nd, ~10%
других редких земель и до 5% Fe. Современные высокоэффективные методы
хроматографии * * и экстракции * * * дают возможность получать каждый из РЗЭ
чистотой 99,7—99,9%. Конечной стадией процесса получения РЗЭ является либо
кальций- или магнийтермия их фторидов, либо электролиз расплавов хлоридов.
Погле переплавки и дистилляции в глубоком вакууме чистота получаемых РЗЭ
достигает 99,99%.
Физические и химические свойства. Скандий и все РЗЭ
в виде простых вещоств — серебристо-болые металлы, тускнеют во влажном воз-
воздухе. Скандий обладает диморфизмом: ниже 1334°С устойчича ГПУ-структура, а
выше этой температуры — ОЦК-решетка. Все РЗЭ в основном имеют структуру
ГПУ, за исключением европия (ОЦК), иттербия (ГЦК) и самария, который крис-
кристаллизуется в ромбоэдрической структуре. Однако последнюю можно рассматри-
рассматривать как слегка искаженную ГПУ. Металлы цериевой группы пластичны, сравни-
сравнительно мягки, причем их твердость возрастает с увеличением атомного номера.
Скандий и металлы иттриевой группы несколько тверже; исключением является
довольно ковкий иттербий. Он же имеет аномально высокую электрическую
проводимость: она в три раза больше, чем у других РЗЭ, которые по этому пара-
Дробная кристаллизация — способ разделения и очистки смеси близких по свой-
свойствам веществ с помощью многократной кристаллизации одного.
Хроматография — метод разделения и анализа смесей, основанный на различном
распределении их компонентов между двумя фазами: неподвижной (например, ионитом) и
подвижной (элюентом).
* * * Экстращия — разделение смеси веществ с помощью растворителя (экстрагента), в
котором компоненты смеси неодинаково растворимы.
метру приближаются к ртути. Скандий и редкоземельные металлы — парамагне-
парамагнетики, но Sc, Y и Lu обладают очень слабой магнитной восприимчивостью. В то
же время Eu, Gd, Dy, Er при температурах ниже комнатной обладают ферромаг-
ферромагнетизмом. Только Gd имеет наивысшую точку Кюри A6°С). Интересными маг-
магнитными свойствами обладает Dy, который в зависимости от температуры прояв-
проявляет свойства парамагнетика, ферромагнетика и антиферромагнетика*. В табл.
24 даны некоторые характеристики скандия и представителей РЗЭ как в форме
простых веществ, так и в качестве элементов ШВ-группы. Распространенность
элементов подгруппы скандия и лантаноидов подчиняется общей закономернос-
закономерности: элементов с четными порядковыми номерами содержится больше, чем с нечет-
нечетными Z. Скандий и иттрий являются самыми легкими из обсуждаемых простых
веществ и одновременно наиболее тугоплавкими. По температуре плавления их
превосходят лишь лантаноиды тулий и лютеций. В целом в характере изменения
температур плавления лантаноидов четко проявляется внутренняя периодич-
периодичность. Минимальными температурами плавления обладают европий и иттербий, у
которых реализуются устойчивые 4fbd°6s2 и 4/45i°6s2 электронные конфигура-
конфигурации. Для этих же элементов наблюдаются повышенные значения атомных ради-
радиусов. Иными словами, атомы европия и иттербия в простых телах между собой
взаимодействуют слабее из-за устойчивости их электронных конфигураций и
больших размеров атомов по сравнению с другими гомоатомными соединениями
лантаноидов. Интересно также отметить, что сравнительно легкоплавкие лантан,
церий и празеодим характеризуются высокими температурами кипения, т.е. явля-
являются трудноиспаряемыми (аналогия с галлием). В то же время те же европий и
иттербий в ряду лантаноидов имеют наинизшие температуры кипения — наибо-
наиболее легколетучи.
Скандий и РЗЭ по химической активности сравнимы с щелочно-земельными
металлами. Стандартные электродные потенциалы меняются от -2,08 (Sc) до
-2,52 В (La). Скандий не растворяется в воде вследствие наличия пленки проч-
прочного оксида, образующегося на воздухе. Остальные металлы подгруппы скандия
и лантаноиды энергично разлагают воду. Sc и РЗЭ растворяются в разбавленных
кислотах, за исключением плавиковой и фосфорной (из-за образования нераство-
нерастворимых фторидов и фосфатов). ОЭО скандия и РЗЭ практически одинакова и
меняется от 1,2 до 1,3. Эти элементы взаимодействуют с большинством металлов
и неметаллов.
Характеристические соединения. Характеристические
оксиды скандия и РЗЭ являются самыми прочными кислородными соединения-
соединениями. Отрицательные величины энтальпий их образования значительно больше,
чем для оксида алюминия (АН°,2д& = -1675,7 кДж/моль):
Соединение S02O3
АН°, 298, кДж/моль -1908,3
Соединение Сс^Оз
АН°,2д&, кДж/моль -1820,5
Y2O3
-1904,1
Dy2O3
-1862,7
La2O3
-1794,9
Er2O3
-1893,7
Nd2O3
-1808,3
Yb2O3
-1814,6
Eu2O3
-1661,1
Lu2O3
-1878,2
* Антиферромагнетики — твердые вещества, в которых магнитные моменты атомов в
соседних узлах кристаллической решетки ориентированы антипараллеЯьно и поэтому
интегральная намагниченность мала.
зля
Таблица 24. Некоторые свойства элементов подгруппы скандия и РЗЭ
Элемен-
ф* т гг
простые
вещест-
вещества
Sc
Y
La
Се
Рг
Nd
Pm
Sm
Eu
Gd
Tb
Dy
Ho
Er
Tm
Yb
Lu
Содержание
с шнилА
D JcMnUH
коре,
масс.доли,
%
6,0-10-4
2,8-Ю-3
1,8-10-3
4,5-10-3
7,0-10-4
2,5-Ю-з
_
7,0-10-4
1,2-10-4
1,0-10-з
1,5-10-4
4,5-10-4
4,3-10-4
4,0-10-4
8,0-Ю
3,0-10-4
1,0-10-4
Плот—
илсп
nOClb,
г/см3
2,99
4,72
6,17
6,66
6,78
7,00
7,22
7,54
5,26
7,90
8,27
8,54
8,80
9,05
9,33
6,98
9,84
Темпе—
пятлтя
paiypd.
плавле—
ния, С
1539
1509
920
795
935
1024
1027
1072
826
1312
1356
1406
1461
1497
1545
824
1652
Темпе-
ПЯ ТЛ7Т*Я
кипе-
кипения,
°с
2832
3357
3454
3257
3212
3127
2730
1752
1597
3233
3041
2335
2572
2510
1732
1193
3315
Атомный
радиус,
НМ
0,161
0,181
0,187
0,183
0,182
0,182
0,181
0,181
0,202
0,179
0,177
0,177
0,176
0,175
0,174
0,193
0,174
Ионный
радиус
Э3*,нм
0,068
0,088
0,106
0,103
0,101
0,099
0,098
0,096
0,095
0,094
0,092
0,091
0,089
0,088
0,087
0,086
0,085
Потенциал ионизации, В
Э—КЭ++е'
6,54
6,38
5,61
6,54
5,76
6,31
5,90
5,60
5,67
6,16
6,74
6,82
6,99
6,70
6,60
6,22
6,15
Э+—>Э2++е-
12,80
12,23
11,43
12,31
11,54
12,09
11,70
11,40
11,24
12,00
12,52
12,60
12,70
12,50
12,40
12,10
12,00
Э2+—»Э3++е-
24,75
20,51
19,17
19,87
20,96
21,51
22,00
24,00
24,56
20,60
21,02
21,83
22,20
22,40
23,90
25,50
23,70
3
S /?
1=1
44,09
39,12
36,21
38,72
38,26
39,91
39,60
41,00
41,43
38,76
40,28
42,25
41,89
41,60
42,90
43,88
41,85
?"(Э?Р/Э), В
-2,077
-2,372
-2,522
-2,483
-2,462
-2,462
-2,431
-2,414
-2,407
-2,397
-2,391
-2,357
-2,319
-2,296
-2,278
-2,267
-2,255
Таблица 25. Внутренняя периодичность
Элемент
Валентные электроны
Цвет гидратированных
ионов Э!+
Степень окисления
AG}j29fjCF3), кДж/моль
La
4/5 #6^
Бесцвет-
Бесцветный
+ 3
-1653,9
Се
4/5 <Р№
Бесцвет-
Бесцветный
+3, +4
-1642,2
Рг
4/5 «Рвя2
Желто-зе-
Желто-зеленый
+3 (+4)
-1634,2
Nd
4/5 «Рбя2
Красно-фи-
Красно-фиолетовый
+3
-1637,2
Рт
4/5 ^б*2
Розовый
+3
-1611,2
Sm
4/5 «Рб*2
Желтый
+3 (+2)
-1629,2
Сравнение теплот образования Э^Оз показывает, что наименьшими их значе-
значениями характеризуются оксиды лантана и европия. В то же время энтальпии
образования оксида европия практически равна таковой для А!2Оз, а оксида
лантана более чем на 100 Дж/моль ее превосходит. Таким образом, среди харак-
характеристических оксидов лантаноидов наименьшей прочностью отличается оксид
европия, хотя он сам по себе относится к категории таких прочных соединений,
как А12Оз. Кроме того, энергии активации оксидирования, как правило, столь
малы, что многие из РЗЭ неустойчивы уже при обычных условиях. Лантан, на-
например, хранят в бензоле во избежание окисления на воздухе.
Оксиды скандия и РЗЭ — бесцветные (большинство), тугоплавкие и трудно-
труднорастворимые в воде вещества, хотя интенсивно (с выделением теплоты) взаимо-
взаимодействуют с ней с образованием характеристических гидроксидов Э(ОН)з. Полу-
Получают оксиды прокаливанием соответствующих гидроксидов, нитратов и карбона-
карбонатов. Гидроксиды получают действием растворов щелочей на растворимые соли
скандия и РЗЭ. Гидроксиды также труднорастворимы в воде. В подгруппе скан-
скандия растворимость гидроксидов растет: Sc(OHK — рПР 28, Y(OHK — рПР 22,8,
Ьа(ОН)з — рПР 18,9. А все гидроксиды лантаноидов характеризуются примерно
такой же растворимостью, как Y(OHK (порядок величины рПР 22—23). Гидрок-
сид скандия — амфолит с более сильно выраженными основными свойствами, а
гидроксиды РЗЭ представляют собой довольно сильные основания. В ряду лан-
лантаноидов основная сила гидроксидов постепенно уменьшается с уменьшением
радиусов 3i+ в результате лантаноидной контракции. С уменьшением ионных
радиусов растет их ионный потенциал и связь с кислородом становится более
прочной. Поэтому гидроксиды иттербия и лютеция проявляют слабую
амфотерность и примыкают в этом отношении к Бс(ОН)з.
Характеристические тригалогениды скандия и РЗЭ в отличие от аналогичных
соединений элементов подгруппы галлия тугоплавки и труднолетучи. Трифтори-
ды практически нерастворимы в воде, а остальные ЭГ растворяются не только в
воде, но и в низших спиртах. Ниже приводим энтальпии образования трифтори-
трифторидов элементов подгруппы скандия и тех РЗЭ, для оксидов которых выше были
представлены эти характеристики:
Соединение
ДЯ\ кДж/моль -648,9
Соединение GdF3
Д#\ кДж/моль -1713,3
YF3
1718,3
DyF3
1719,6
LaF3
-1731,8
ErF3
-1722,5
NdF3
-1712,9
YbF3
-1656,9
EuF3
-1619,2
LuF3
-1700,8
Теплоты образования трифторидов несколько уступают таковым для оксидов,
но почти в два раза больше, чем для хлоридов и бромидов рассматриваемых
лантаноидов
Eu
4/5d°6s2
Почти бес-
бесцветный
+3, +2
-1537,6
Gd
4/5</16s2
Бесцвет-
Бесцветный
+3
-1637,2
Tb
4/5 «PCs2
Бесцвет-
Бесцветный
+3, +4
-1631,3
Dy
4/05<P6s2
Желто-зе-
Желто-зеленый
+3 (+4)
-1642,6
Ho
4/15<f6s2
Коричне-
Коричнево-желтый
+ 3
-1637,2
Er
4/25d°6s2
Розовый
+ 3
-1621,3
Tm
4/35<f6s2
Бледно-зе-
Бледно-зеленый
+ 3 (+2)
-1617,9
Tb
4/45<f6s2
Бесцвет-
Бесцветный
+ 3, +2
-1581,1
элементов. В подгруппе скандия энтачьпии образования ЭГз закономерно возрас-
возрастают, а в ряду лантаноидов имеет место довольно слабое последовательное умень-
уменьшение теплот образования для всех галогенидов ЭГ3. Естественно, от фторидов к
иодидам теплоты образования убывают для всех элементов. При сильном нагре-
нагревании на воздухе трифториды сначала превращаются в оксофториды 90F, а
затем в оксиды. Для хлоридов подобное же превращение наблюдается при нагре-
нагревании кристаллогидратов.
В химии редкоземельных элементов наиболее ярко проявляется внутренняя
периодичность, особенно для производных в характеристической степени окисле-
окисления. Объяснение этому факту было дано в § 5 гл. X. Для иллюстрации внутрен-
внутренней периодичности в табл. 25 приведены цвет гидратированных ионов Э3+, стан-
стандартные энергии Гиббса образования трифторидов и проявляемые степени окис-
окисления. Наблюдается удивительная аналогия в свойствах элементов, находящихся
друг под другом. В каждой семерке, составляющей внутренний период, иониза-
ионизационные потенциалы третьего порядка монотонно растут (см. табл. 24) с умень-
уменьшением атомных радиусов вследствие лантаноидной контракции. Но начало
нового внутреннего периода (переход от Ей к Gd) сопровождается уменьшением
третьего ионизационного потенциала на 4 В. У европия впервые в первой семерке
достигается устойчивая наполовину заполненная 4/-оболочка. У гадолиния же
при той же устойчивой 4/-оболочке появляется один электрон на 5(/-оболочке,
который намного легче удаляется, потому что этот электрон делает стабильную
5(/°-оболочку неустойчивой. Для элементов, следующих за Gd, вновь наблюдается
монотонное возрастание третьего ионизационного потенциала вследствие лантано-
лантаноидного сжатия. Вследствие стабильности 4/-оболочки европий часто функциони-
функционирует в степени окисления +2 за счет 6в2-электронов, а один из семи неспаренных
электронов на 4/оболочке участвует в образовании связей в более жестких усло-
условиях. Для его аналога иттербия картина схожая, только в качестве устойчивой
выступает уже полностью заселенная 4/4-оболочка. В случае самария и тулия.
находящихся левее указанных выше Ей и Yb, Ар- и 4/3-оболочки близки к дос-
достижению стабильного состояния, а потому в основном проявляют характеристи-
характеристические степени окисления. Но эти же элементы в более мягких условиях могут
быть в степени окисления +2 за счет 6з2-электронов при квазистабильных Ар- и
4/3-оболочках. Для элементов начала внутренних периодов — La и Gd — наблю-
наблюдается только степень окисления +3 вследствие устойчивости Af- и 4/г-оболочек,
полностью вакантной или наполовину заполненной. А электронами, участвующи-
участвующими в химическом взаимодействии, у них являются 5<Рбя2-электроны, т.е. по три
электрона. Следует отметить, что заполненные бв-орбитали также должны быть
стабильны, но для лантана и лантаноидов электроны на них являются внешними,
а потому слабее связанными с ядром и вследствие этого наиболее подвижными. У
351
!'¦<
следующих за лантаном и гадолинием церия и тербия против стабильных 4/3- и
4/г-оболочек появляются сразу по два "лишних" электрона в результате перехода
одного электрона с 5</-орбитали и появления очередного. Эти два электрона и
плюс два на 6*-орбитали и определяют степень окисления +4, причем церий
больше проявляет в своих производных именно высшую степень окисления +4, а
не характеристическую вследствие наибольшей стабильности полностью вакант-
вакантной 4/*-оболочки. Для Рг и Dy степень окисления +4 уже менее характерна по
сравнению с предшествующими элементами, так как все большая заселенность 4/-
орбиталей неспаренными электронами делает их более устойчивыми.
Обращает на себя внимание почти полное совпадение цвета гидратированных
ионов элементов-аналогов (диад). В действительности речь здесь идет об окра-
окрашенности аквакомплексов Э3+. Аналогия в их окраске свидетельствует о том, что
механизм комплексообразования с молекулами воды и характер участия элект-
электронных /- и d-орбиталей элементов диад одинаков. Конечно, трудно говорить о
полном совпадении значений энергий Гиббса образования трифторидов, так как
они велики. Кроме того, от элемента к элементу энергия Гиббса образования три-
трифторидов практически мало меняется, так как лантаноиды характеризуются при-
примерно одинаковой химической активностью. Но нельзя не отметить факт явно
заниженных значений указанных характеристик для замыкающих каждый внут-
внутренний период элементов — европия и иттербия. Интерпретация этого факта та
же — стабильность наполовину и полностью заселенных 4/юрбиталей, что и дела-
делает эти элементы менее активными по отношению к акцепторам электронов. Устой-
Устойчивые 4f- и 4/-оболочки имеют также La и Gd, которыми начинаются семерки
элементов. Но у этих элементов в отличие от Ей и Yb имеется по одному электро-
электрону на 5(йэрбитали, который вместе с 6в2-электронами определяет их электрополо-
электроположительную активность.
Таким образом, последний лантаноид — лютеций — не вписывается в семерки
элементов и не входит в состав диад. У него при полностью заполненных 4/4- и
6в2-орбиталях имеется один электрон на 5 i-оболочке. Поэтому лютеций можно
поместить под иттрием, вынеся за систему элементы с порядковыми номерами от
57 (La) до 70 (Yb). В этом случае внутренняя периодичность лантаноидов полу-
получает более строгую формальную основу.
Соединения с другими неметаллами. Скандий, европий и
иттербий образуют гидриды состава ЭНг — черные порошки, обладающие высо-
высокой электрической проводимостью. По своей физико-химической природе они
являются металлоподобными гидридами и, следовательно, только формально
похожи на солеобразные гидриды щелочно-земельных металлов. Остальные РЗЭ
образуют гидриды ЭНг и ЭНз- Последние также представляют собой фазы внед-
внедрения, т.е. металлоподобны. Для лантана наиболее устойчивым является гидрид
состава LaH2r5, который можно рассматривать как эквиформульный раствор ЬаЩ
и ЬаНз. Гидриды ЭНз легко гидролизуются.
Сульфиды скандия и РЗЭ отвечают валовым составам 3S, 32Sy, Э^Б^, Эг8з,
3S2. Большинство сульфидов — фазы переменного состава с преобладающим
металлическим типом связи. Для многих РЗЭ характерны тугоплавкие моносуль-
моносульфиды, кристаллизующиеся в кубической структуре. В сульфидах 9S степень
окисления РЗЭ +2 чисто формальная, так как при растворении в кислотах они
выделяют сероводород и водород. Правильнее считать их фазами внедрения
металлов в 32S3. Состав селенидов и теллуридов чаще всего отвечает формулам
352
3Se(Te) или Э28е(Те)з. Только при сплавлении церия с теллуром получается
CeTeg. В отличие от сульфидов селениды устойчивы к воде и разлагаются только
кислотами. При нагревании селенидов типа 32Se3 до 1200—1700°С они выделяют
селен и переходят в селениды Эз8е4 с металлическим блеском.
В кубической структуре кристаллизуются и нитриды 3N. Они очень тугоплав-
тугоплавки, например ScN плавится при 2900°С. Нитриды гидролизуются уже во влажном
воздухе с выделением аммиака. Известны также фосфиды типа ЭР, имеющие
структуру NaCl. При сплавлении компонентов легко образуются изоморфные
фосфидам арсениды, стибиды и висмутиды. Напомним, что элементы подгруппы
галлия висмутидов не образуют.
Карбиды желтого цвета имеют состав ЭС2. При их гидролизе выделяются
углеводороды, среди которых доминирует ацетилен. Скандий наряду с карбидом
ScC2 образует темно-серый SC4C3, аналогичный карбиду алюминия AI4C3.
Сплавлением компонентов при высоких температурах для скандия и большин-
большинства РЗЭ получены моно- и дисилициды 9Si и 3Si2- Известны также силициды с
меньшим содержанием кремния.
Для скандия, иттрия и лютеция получены дибориды ЭВг- Для всех РЗЭ
получены наиболее устойчивые гексабориды ЭВе. Кроме того, для многих РЗЭ
известны ЭВ4 и ЭВ12, а также более богатые бором фазы. Все они
металлоподобны, тугоплавки B000—2500°С), обладают высокой электрической
проводимостью и твердостью.
Соли кислородных кислот и комплексные соеди-
соединения. Для скандия и РЗЭ наиболее характерны соли кислородсодержащих
кислот и комплексные соединения на их основе. Нитраты и сульфаты растворимы
в воде лучше, чем карбонаты и фосфаты. Интересно отметить, что растворимость
нитратов с увеличением порядкового номера РЗЭ сначала уменьшается, а затем
вновь растет. В результате минимум растворимости приходится на Сс1AЧОз)з-
Аналогично растворимости сульфатов щелочно-земельных элементов раствори-
растворимость сульфатов в подгруппе скандия снижается сверху вниз. Соли скандия,
иттербия и лютеция подвержены гидролизу вследствие амфотерности их гидрок-
сидов.
Скандий и иттрий, подобно алюминию, образуют комплексные фториды
[9F6]3~, а также двойные соли с сульфатами, нитратами и карбонатами щелочных
металлов типа МегЗС^-Эг^О^з и т.п. Комплексообразовательная способность
лантаноидов невелика. Это связано с неблагоприятной для орбитальной гибриди-
гибридизации электронной структурой, так как достраивающиеся 4/-оболочки расположе-
расположены слишком глубоко. Кроме того, 5rf-, 6s- и бр-орбитали (для ионов Э3+) прини-
принимают мало участия в комплексообразовании. Таким образом, доминирующим
фактором в образовании комплексных соединений для РЗЭ остается электроста-
электростатическое притяжение между ионами Э3+ и лигандами. Но устойчивость комплек-
комплексов резко повышается при взаимодействии Э3+ с анионами поликарбоновых кис-
кислот с образованием ацидокомплексов. Так, оксалатный комплекс диспрозия
[Оу(С2О4)г]" имеет рА'нест 10,4, образование циклических комплексов хелатного
типа на много порядков увеличивает их устойчивость, например для [Т)у(ЭДТА)]~
рА'нест 17,98.
Соединения степеней окисления +4 и +2. Некоторые
лантаноиды имеют помимо характеристической еще степени Окисления +4 и +2.
Среди лантаноидов, проявляющих степень окисления +4, выделяется церий.
ю „ . „_.» 353
Относительно более стабильные соединения в степени окисления +2 дает евро-
европий.
Диоксид СеОг образуется при непосредственном взаимодействии компонентов
и кристаллизуется в структуре флюорита. Он плавится при 2600°С под давлени-
давлением кислорода, начинает отщеплять кислород только при 2300° С. При 1250° С
СеОг восстанавливается водородом до Се2О;). Диоксид церия не растворяется в
воде, а после прокаливания — и в кислотах, и в щелочах. -СеОг является сильным
окислителем, например выделяет хлор из соляной кислоты:
2СеО, + 8НС1 = 2СеС13 + С12 + 4Н2О
Сплавление диоксида церия с оксидами или карбонатами активных металлов
приводит к образованию цератов типа Ме2Се0з- Гидроксид Се(ОНL не растворя-
растворяется в воде (рПР 50). в щелочах, а при растворении в водных растворах кислот
возникают растворы розового цвета из-за образования аквакомплекса
[Се(Н2О)8]4*.
Нагревание металлических тербия и празеодима на воздухе впдет к образова-
образованию смешанных оксидов ТЬ^тСГЬгО.з'ЗТЬОг) и РГ7О12 ^РггОз'ЗРгО?). Диоксид
тербия неустойчив. Диоксид РгОг — черно-коричневые кристаллы структурного
типа флюорита, разлагается уже при 400° С. Он сильный окислитель: окисляет
соли Мп(+2) до перманганата.
Из тетрагалогенидов известны только 3F4:CeF4 получается растворением СеОг
в плавиковой кислоте и представляет собой бесцветный порошок, разлагающийся
при 390°С; TbF\j можно получить окислением трифторида фтором. Желтые крис-
кристаллы TbF4 разлагаются при 180"С; известен бесцветный PrF4 с температурой
разложения 90°С.
Из солей кислородсодержащих кислот известны только производные церия.
Сульфат Ce(SO4J получается нагреванием СеОг с горячей концентрированной
серной кислотой. Ce(SO4J — порошок желтого цвета, хорошо растворяется в
воде. Из водных растворов он выделяется в виде розовых кристаллов с 8 молеку-
молекулами воды. Известны только основные нитраты и карбонаты: Се(ОН)(ГЮз)з и
Се2(ОН)г(СОз)я- В то же время Се(+4) образует устойчивые ацетат и перхлорат:
Се(СН3СОО),, Се(СЮ4L.
Поскольку катион Се4+ обладает большим значением ионного потенциала, его
комилексообразовательная способность несколько выше других лантаноидов в
степени окисления +4. При этом высшая степень окисления более стабильна по
сравнению с соединениями первого порядка. Так, для Се(+4) известны довольно
устойчивые комплексы [Се(СгО4)з]2~ и [Ce(NOsN]2\ Из галогенидных комплексов
наиболее устойчивы фторидные: [3Fg]2" (Се и Pr), [3F7]2" (Се, Pr, Tb), а для
диспрозия известен только Csa[DyF7].
Степень окисления +2 наиболее характерна для европия. Нагреванием на
воздухе Еи2О3 с графитом до 1700°С получен темно-коричневый оксид ЕиО-
Монооксид европия — тугоплавкие кубические кристаллы-— медленно разлагают-
разлагаются водой с выделением водорода, т.е. является сильным восстановителем. Извест-
Известны также монооксиды самария и иттербия структурного типа NaCl. Восстановле-
Восстановлением EuFg водородом при 1000°С можно получить дифторид EuF-2, который
изоморфен CaF2. Известны дихлориды, дибромиды, дииодиды Sm, Eu, Tm и Yb.
Их устойчивость в указанном ряду лантаноидов снижается слева направо и, ес-
естественно, от хлоридов к иодидам.
354
МО'
Катодным восстановлением сульфатов
Э(+3) получены белый EuSO4, светло-
зеленый YbSO^ и красный SmSO4. Суль-
Сульфаты Э(+2) плохо растворяются в воде;
например, для EuSO4 рПР 10. EuSO4
изоморфен с BaSO4. Вообще химия Ей в
степени окисления +2 во многом схожа с
химией щелочио-земелььых элементов. Все
производные РЗЭ в степени окисления +2
являются восстановителями.
\1 е т а л л о х и м и я. В металлохи-
металлохимии отличия между элементами подгрупп
галлия и скандия проявляются более от-
отчетливо. Если металлы подгруппы галлия
(.SjP-металлы) не образуют непрерывных
твердых растворов, элементы подгруппы
скандия дают неограниченную раствори-
растворимость в твердом состоянии со многими
металлами. Так, иттрий образует непре-
непрерывные твердые растворы со скандием, лантаном, титаном, торием, гадолинием и
др. Кроме того, металлы подгруппы "кандия (в отличие от галлия и его анало-
аналогов) на диаграммах состояния дают широкие области ограниченных твердых
растворов. Необходимо подчеркнуть, что именно в металлохимии элементов под-
подгруппы скандия ярко проявляются их свойства как первых представителей каж-
каждого ряда переходных rf-злементов. В обычной химии особенность поведения </-•
элементов сказывается, как правило, начиная с подгруппы титана.
Скандий и РЗЭ взаимодействуют с щелочными металлами. При сплавлении с
металлами вставных декад нередко наблюдается расслоение в жидком состоянии.
Тем не менее со многими переходными металлами скандий и РЗЭ дают многочис-
многочисленные металлиды. Наконец,, интересно отметить, что при взаимодействии РЗЭ с
металлами подгруппы галлия возникают металлиды, часто очень прочные. Для
иллюстрации на рис. 142 приведена диаграмма состояния системы Pr — Ga, в
которой налицо конгруэнтно плавящийся при высокой температуре PrGa2 и ряд
металлидов, представленных перитектическими горизонталями.
'в 1A IS 30 ^ SO $5 W SS S3 WO
Pr Am. Ый, % 9s. 5п
F и с, 142. Дмагуалша состояния системы
празеодим - гяллий
ГЛАВА XVIII ЭЛЕМЕНТЫ IV ГРУППЫ
Четвертая группа Периодической системы включает два типических элемен-
элемента — углерод и кремний — и подгруппы германия и титана. По значимости тех
элементов, которые входят в IV группу, с ней не может сравниться никакая дру-
другая группа системы. Углерод является основой органической химии, главным
органогенным элементом; следовательно, необходимым компонентом организма
всех живых существ. Второй типический элемент группы — кремний — главный
элемент неорганической химии и всей нежирой природы. По целому ряду экстре-
экстремальных свойств титан и сплавы на его основе являются уникальными конструк-
конструкционными материалами, которые широко применяются в авиа- и судостроении,
космической технике. Еще в большей мере титан — металл будущего. Со времени
создания первого твердотельного транзистора на основе германия A948), произ-
произведшего целую революцию в радиоэлектронике, в течение 10 лет германий оста-
оставался доминирующим полупроводниковым материалом, уступив затем первое
место опять же представителю IV группы — кремнию. В настоящее время интег-
интегральные схемы на основе кремния являются основой компьютеров, микропроцес-
микропроцессоров, логических устройств и т.п., без чего нельзя представить себе современную
научно-техническую революцию.
Важность названных выше элементов IV группы для современной науки и
техники не случайна. Она обусловлена специфическими особенностями структуры
и заселенности электронных орбиталей их атомов и, как следствие этого, уни-
уникальными свойствами образуемых ими гомо- и гетероатомных соединений. Все
элементы содержат по четыре валентных электрона независимо от их орбитально-
орбитального происхождения. Это число валентных электронов является оптимальным,
например, для возникновения особо важных тетраэдрических связей по обменно-
обменному механизму. И вообще число валентных электронов, равное четырем, определя-
определяет IV группу как середину Периодической системы, если не считать VIII груп-
группу — благородных элементов. Другими словами, для элементов I, II и III групп до
четырех валентных электронов не хватает соответственно трех, двух и одного
электрона, а для элементов V, VI и VII групп, наоборот, отмечается избыток
электронов против четырех. Все это приводит к тому, что химию остальных
элементов системы целесообразно рассматривать в сравнении с химией элементов
IV группы, особенно ее типических элементов и подгруппы германия. И не слу-
случайно для элементов IVA-группы одновременно так типичны и характеристичес-
характеристические оксиды, и характеристические летучие водородные соединения по Д.И. Мен-
Менделееву.
1. Углерод. Особенности химии углерода. В невозбужден-
невозбужденном состоянии атом углерода имеет электронную формулу Is22s22p2. При этом два
валентных электрона спарены, а два других находятся в неспаренном состоянии:
я-2
гтп
г
401 кДж
s
1
В этом состоянии углерод может образовать три ковалентные связи, две из кото-
которых осуществляются по обменному механизму, а одна — по донорно-акцепторно-
му. Первые две связи возникают за счет двух неспаренных электронов, а третья —
с использованием одной вакантной 2р-орбитали. Такова картина ковалентных
связей в монооксиде углерода СО. При этом у атома углерода остается неисполь-
неиспользованной одна неподеленная пара валентных электронов на 2я-орбитали. Как раз
эти электроны используются в реакциях присоединения оксида углерода, напри-
например при образовании карбонилов металлов.
Возбуждение атома углерода сопровождается промотированием одного из 2s-
электронов на 2р-орбиталь. В результате возбуждения атом характеризуется
четырьмя холостыми электронами, что обеспечивает возможность возникновения
четырех химических связей. Энергия, необходимая для возбуждения, окупается
энергией, которая выделяется при образовании двух дополнительных ковалент-
ковалентных связей. Различие между е- и р-электронами снимается sp-, sp2- или «р3-гиб-
ридизацией, которым подвергается атом углерода в зависимости от партнера по
химическим связям. Таким образом, углерод функционирует в своих гомо- и
гетероатомных производных прежде всего как четырехвалентный элемент. Ввиду
отсутствия (f-орбиталей максимачьная валентность углерода может быть равна
четырем. Следует при этом отметить, что только у двух элементов Периодической
системы — водорода и углерода — имеет место одинаковое число валентных элек-
электронов и валентных орбиталей. Поэтому водород и углерод образуют наибольшее
число химических соединений как с другими элементами системы, так и между
собой.
it-Связи между атомами углерода отличаются наибольшей прочностью
(кДж/моль) по сравнению с гомоатомными связями других элементов, способных
образовывать цепочки из одинаковых атомов:
Этим и объясняются большая распространенность и исключительное разнообра-
разнообразие гомоцепных производных углерода. Гомоцепные структуры, содержащие
связь С—С, бывают самых различных типов: линейные, разветвленные, цикличес-
циклические и др. Естественно, эти образования включают и атомы других элементов. Из
гетеросвязей атомов углерода наиболее распространены связи с атомом водорода
С—Н вследствие сравнительно высокой их прочности D41,2 кДж/моль). Помимо
одинарных связей углерод легко образует кратные связи. При образовании крат-
кратных связей для углерода характерны только тгр.р-связи из-за отсутствия в его
атоме rf-электронов. Наконец, значение ОЭО углерода B,6) промежуточное между
таковыми электроположительных и электроотрицательных элементов, хотя ближе
к последним. Поэтому даже в случае максимальной поляризации атомов углерода
в его соединениях не возникает самостоятельных ионов С4+ и С4". Эффективные
заряды на атоме углерода во всех исследованных соединениях значительно мень-
меньше 1, т.е. химические соединения углерода малополярны.
Углерод в природе. По распространенности в земной коре углерод
уступает многим другим элементам. На его долю приходится лищь 0,1 масс, доли,
%. Несмотря на это, роль углерода в живой и неживой природе огромна. Ткани
животных и растений состоят из соединений углерода. Из продуктов разложения
органических веществ образовались каменные угли, торф, нефть, природные
газы. Формирование залежей этих горючих материалов происходило в условиях
отсутствия атмосферного кислорода в течение геологических эпох. Углерод вхо-
входит в состав многих солеобразных минералов, имеющих обобщенную формулу
МСОз, где М — металл в степени окисления +2. Из этих карбонатных пород
наиболее распространены: известняк и мрамор СаСОз, магнезит MgCC>3, доломит
MgCO3-CaCO3, сидерит FeCOs и др. Некоторые карбонаты имеют органическое
происхождение. Например, мел и известняк состоят из остатков микроскопичес-
микроскопических раковин, а коралловые рифы являются продуктами жизнедеятельности мик-
микроскопических организмов — кораллов. В районах с засушливым климатом встре-
встречаются водоемы с повышенным содержанием соды Na2CO3- Углерод в форме
углекислого газа содержится в атмосфере и в растворенном состоянии — во всех
природных водах. Интересно отметить, что в земной коре большая часть углеро-
углерода находится не в виде органических соединений, а в составе двух минералов —
известняка и доломита. ,
357
I Г
Физические и химические свойства углерода1. В
виде простых веществ углерод встречается в природе в трех аллотропных моди-
модификациях: алмаза, графита и карбина. Все они представляют собой гомоатомные
соединения углерода с различным кристаллохимическим строением. В отличие от
алмаза и графита карбин был вначале получен синтетически, а потом найден в
природе (минерал чаоит — вкрапления карбина в природном графите). Так назы-
называемый аморфный углерод (сажа, древесный и костяной уголь и др.) не является
самостоятельным аллотропным видоизменением углерода, а состоит из мельчай-
мельчайших разноориентированных кристалликов графита.
.Алмаз — бесцветное, прозрачное, сильно преломляющее свет вещество. Он
тверже всех найденных в природе веществ, но довольно хрупок. Кристаллы алма-
алмаза имеют координационную структуру, в которой атомы углерода связаны друг с
другом направленными «р3-гибридными связями. ГЦК-структура алмаза отлича-
отличается от ГЦК-структуры меди тем, что углеродные атомы располагаются не только
на гранях куба, но и в центрах малых кубов (октантов), чередующихся с пустыми
малыми кубами. Каждый атом углерода имеет четыре ближайших соседа (валент-
(валентность и к.ч. 4), расстояние между которыми 0,154 нм. По отношению к любому
атому углерода четыре ближайших соседних атома расположены в вершинах
правильного тетраэдра. Поэтому структуру алмаза можно представить в виде
комбинации тетраэдров, у которых в центре находится пятый атом углерода.
Каждая вершина тетраэдра является общей для четырех смежных тетраэдров.
Непрерывная трехмерная сетка к.овалентпых связей, которая в алмазе характери-
характеризуется наибольшей прочностью, определяет его важнейшие свойства: низков
значение энтропии, тугоплавкость, высокую твердость, плохую теплопроводность
и электрическую проводимость, а также химическую инертность.
Как видно из диаграммы состояния углерода (рис. 143), алмаз является его
модификацией, стабильной только при высоких давлениях. Область термодина-
термодинамической стабильности алмаза отвечает давлениям больше 1000 МПа. При мень-
меньших давлениях и температурах устойчивой модификацией углерода является
графит, а алмаз метастабилен. Однако переход алмаза в графит кинетически
заторможен и практически не происходит в обычных условиях. В отсутствие
катализатора даже в условиях высоких температур C000°С) и давлений "A2 500
МПа) скорость превращения графита в алмаз очень мала. Поэтому синтез алмаза
Р,МПа
10000
то
too
10
1
0.1
* о im ж зооо wo sm %°c
Рис. 143. Диаграмма состояния угле-
углерода
Алмаз
Графит
уКиВтсть
ж ^
1 Пар
I ! 1
Рис. 144. Кристаллическая струк-
структура графита
358
проводят в присутствии катализатора (железо, никель, платиновые металлы).
Алмазы образуются на поверхности раздела между графитом и расплавленным
металлом — катализатором. Для извлечения алмазов охлажденную массу дро-
дробят и обрабатывают смесью кислот. В отсутствие катализатора алмазы могут
быть получены при 5000°С и давлении 2100 МПа действием ударной волны на
графит.
Графит — устойчивая при нормальных условиях аллотропная форма углерода.
Он имеет серо-черный цвет и металлический блеск, кажется жирным на ощупь,
очень мягок, оставляет черные следы на бумаге. Графит хорошо проводит тепло-
теплоту и электрический ток, но его свойства резко анизотропны. Кристаллохимичес-
кое строение графита существенно отличается от структуры штааза. Он имеет
гексагональную структуру (рис. 144). Атомы углерода в графите расположены
отдельными слоями, образованными из плоских шестиугольников. Каждый атом
углерода на плоскости окружен тремя соседями (в^-гибридизация), расположен-
расположенными вокруг него в виде правильного треугольника на расстоянии 0,412 нм. А
расстояние между ближайшими атомами соседних слоев равно 0,340 нм и более
чем в два раза превышает кратчайшее расстояние между атомами углерода в
плоском слое. Поэтому графит имеет меньшую плотность по сравнению с алма-
алмазом, легко расщепляется на тонкие чешуйки. Химическая связь между атомами
углерода внутри слоя имеет ковалентный характер с ярко выраженной склоннос-
склонностью к металлизации. Последняя обусловлена возникновением делокализованных
¦Лр-р-связей в пределах шестиугольников (как в молекуле бензола) и всего макро-
глоя. Этим и объясняются хорошая электрическая проводимость и металличес-
металлический блеск графита. Углеродные атомы различных слоев связаны слабыми силами
Ван-дер-Ваальса. Преимущественно ковалентная связь между атомами углерода
внутри слоя сближает графит с алмазом: и тот и другой необычайно тугоплавки
и обладают малой упругостью паров при нагревании.
Карбин был получен окислительной конденсацией ацетилена. Представляет
собой белое вещество с плотностью, промежуточной между таковыми алмаза и
графита. Гексагональная решетка карбина построена из слабо связанных прямо-
прямолинейных цепей —С—С—С—-С—", в которых каждый атом углерода находится в
состоянии sp-гибридизации и образует по две <rSp-sp- и Яр-р-связи. При нагрева-
нагревании до 800° С карбин превращается в графит. В 1990 г, была получена четвертая
модификация углерода — фуллерит.
В химическом отношении алмаз и графит при обычных условиях инертны.
Сгорают они лишь в чистом кислороде при температуре около 800° С с образова-
образованием СОг- Обе модификации углерода устойчивы к действиям кислот и щелочей.
Реакционная способность карбина выше, чем алмаза и графита. Углерод в "амор-
"аморфном" состоянии (уголь, кокс, сажа) легко сгорает на воздухе. Углерод непосред-
непосредственно реагирует из галогенов только с фтором. При высоких температурах он
соединяется с серой и азотом.
Характеристические соединения. Простейшими соедине-
соединениями углерода с кислородом являются диоксид СОг (углекислый газ), оксид
СО (угарный газ) и диоксид триуглерода СзО2 (недокись). Диоксид углерода
играет исключительно важную роль в разнообразных процессах живой и неживой
природы. Оксид С3О2 неустойчив и практического применения не имеет. Диок-
Диоксид СОг является постоянной составной частью воздуха, образуется при всевоз-
всевозможных процессах окисления органических веществ, например при дыхании
л ' ' 359
t f
живых организмов, брожении, горении топлива, выбрасывается при вулканичес-
вулканических извержениях и выделяется из вод многих минеральных источников, а также
в процессе обжига известняка и других карбонатных пород.
Диоксид СО2 — газ, бесцветный, слегка кисловатый на вкус, в 1,5 раза тяже-
тяжелее воздуха. При комнатной температуре под давлением около 60,6 • 105 Па пере-
переходит в бесцветную жидкость. При ее охлаждении в результате испарения СО2
частично превращается в твердую снегоподобную массу ("сухой лед"). Во всех
агрегатных состояниях диоксид углерода состоит из линейных, бездипольных,
неассоциированных молекул СО2. Химическое строение диоксида углерода опре-
определяется sp-гибридизацией центрального атома углерода и образованием допол-
дополнительных тгр-р-связей.
Химически и термически диоксид углерода очень устойчив. Около 2000°С
начинается его диссоциация:
2СО2 ^ 2СО + О2
При 5000°С доля продиссоциированных молекул СО2 достигает 100%. Разложе-
Разложению СО2 способствуют ультрафиолетовое излучение и электрический разряд.
Степень окисления углерода в СО2 максимальная (+4), а потому диоксид не
может быть восстановителем, не горит и не поддерживает горения обычного
топлива. Но в его атмосфере горят простые вещества, атомы которых обладают
большим сродством к кислороду, чем углерод. Так, зажженная на воздухе лента
магния продолжает гореть в атмосфере СО2:
2Mg + СО2 = 2MgO + С,
= -349,8 кДж.
По своей химической природе диоксид углерода является кислотообразующим
оксидом.
Оксид углерода СО — бесцветный, очень ядовитый газ, без запаха. В воде он
растворяется плохо, намного лучше — в спирте. Вследствие малой деформиру-
деформируемости электронных оболочек оксид углерода имеет низкие температуры плавле-
плавления (-205°С) и кипения (-191,5°С). В лаборатории его легко получают, приливая
по каплям муравьиную кислоту к концентрированной серной кислоте. Последняя
служит водоотнимающим средством:
НСООН = СО + Н2О
А щавелевая кислота при нагревании с серной кислотой дает смесь СО и СО2:
Н2С2О4 = СО + СО2 + Н2О
Для отделения оксида углерода смесь газообразных продуктов пропускают через
раствор гидроксида бария, поглощающего только СО2. В промышленности СО
получают газификацией твердого топлива.
По химическому строению молекулы, химическим и физическим свойствам
оксид углерода проявляет большое сходство с молекулярным азотом. Молекулы
СО и N2 изоэлектронны, имеют равные молекулярные массы, высокий порядок
связи и относятся к самым прочным двухатомным частицам. В отличие от СО2
оксид углерода не обладает кислотной природой. Для него наиболее характерны
реакции окисления и присоединения. Первые обусловлены степенью окисления
углерода в СО(+2), а вторые — неподеленными электронными парами атомов
углерода и кислорода.
При комнатной температуре оксид углерода восстанавливает некоторые благо-
благородные металлы и выделяет их из водных растворов солей, например
PdCl2 + СО + Н20 = Pd + 2HC1 + С02
В условиях повышенной температуры химическая активность оксида углерода
как восстановителя возрастает. Он реагирует с такими неметаллами, как кисло-
кислород, хлор, сера:
8 = -256,9 кДж
2СО + О2 = 2СО2, ;
СО + С12 = СОСЬ (фосген)
СО + S = COS (тиооксид углерода)
Реакция образования фосгена протекает при наличии катализатора (активиро-
(активированного угля) и при комнатной температуре. Способность оксида углерода восста-
восстанавливать многие оксиды металлов широко используется в пирометаллургии. Эти
реакции лежат в основе промышленного производства таких металлов, как Fe,
Со, Ni, Cu, Ag, Mn, Мо и др. Условия восстановления определяются природой
оксида металла. Температуры восстановления металлических оксидов варьируют
от 300 до 1500°С. В доменном процессе суммарная реакция представлена уравне-
уравнением
Fe3O4 + 4СО = 3Fe + 4СО2
Из реакций присоединения интересна реакция взаимодействия СО с NaOH
B00°С, 15,2-105 Па): СО + NaOH = HCOONa. В результате образуется формиат
натрия, в котором степень окисления углерода +2, как и в оксиде. В этой реак-
реакции, равно как и вышеприведенной реакции получения СО из муравьиной кисло-
кислоты, формально проявляется кислотная функция оксида углерода.
Важными продуктами присоединения оксида углерода являются карбонилы
металлов, обобщенная формула которых Меж(СО)у, например Сг(СО)б,
Мп2(СО)ю, Fe(COM, Fe2(CO)g, Co2(CO)8, Ni(COL. Карбонилы переходных метал-
металлов — жидкости или летучие твердые вещества. Они хорошо растворяются в
органических растворителях, отличаются химической устойчивостью. Все они
ядовиты, но их токсическое действие не кумулятивно. При нагревании выше
определенной температуры карбонилы разлагаются с выделением оксида углеро-
углерода и металла в мелкодисперсном состоянии. В химическом отношении карбонилы
представляют собой комплексные соединения, в которых металлический элемент
функционирует в нулевой степени окисления, а в качестве лигандов выступают
молекулы оксида углерода. Их донорная активность обусловлена наличием непо-
деленной электронной пары атома углерода.
Соединяясь с серой, оксид углерода превращается в тиооксид — бесцветный
газ без запаха. В воде он довольно хорошо растворяется, но при этом медленно
гидролизуется:
COS + 2Н2О = H2S + Н2СО3
Присоединение хлора к оксиду углеррда приводит к образованию хлороокси-
да углерода — фосгена. Он представляет собой бесцветный легко сжижаемый газ
со слабым запахом влажного сена. Фосген медленно гидролизуется водой:
сось + 2н2о = 2НС1 + riica3
361
Фосген очень ядовит. Процессы присоединения СО к сере и хлору одновре-
одновременно можно рассматривать как реакции окисления оксида углерода.
Оксид углерода С3Ог получают отнятием воды от малоновой кислоты:
НООС-СН2-СООН = С3О2 + 2Н2О
Оксид С3О2 — бесцветный и удушливый, легко сжижаемый газ. Линейная моле-
молекула О—-С—С—С—-О неполярна, атомы углерода находятся в состоянии sjs-гибри-
дизации. Помимо crSp-5р-связей возникают я^-р-связи, плоскости симметрии р-
облаков которых расположены взаимно перпендикулярно. При нагревании или
длительном хранении С3О2 полимеризуется с образованием полимера (СзО2)п
красного цвета. Одновременно с полимеризацией повышение температуры вызы-
вызывает реакцию диспропорционирования:
С3О2 = СО2 + 2С
Характеристическим летучим водородным соединением углерода является
метан. В обычных условиях водород с углеродом не реагирует. Синтез метана
идет только при достаточно высокой температуре и в присутствии катализатора
(мелкораздробленный никель). Применяются также и другие способы получения
метана из сложных органических веществ. В лаборатории метан можно получить
разложением карбида алюминия водой. В природе метан постоянно образуется
при разложении органических веществ без доступа воздуха. Химическое строение
метана определяется «р3-гибридизацией атома углерода. Молекула метана пред-
представляет собой правильный тетраэдр, в центре которого находится атом углерода,
а по вершинам — атомы водорода. Метай — газ легче воздуха, почти нерастворим
в воде, устойчив вплоть до 1000° С. Выше этой температуры разлагается с образо-
образованием ацетилена и водорода:
— C2H2 + ЗН2
В химическом отношении метан инертен, не взаимодействует ни с кислотами, ни
с щелочами. С кислородом вступает в реакцию только при нагревании:
СН4 + 2О2 = СО2 + 2Н2О,
= -893,7 кДж
Метансодержащим топливом являются природные газы, в которых содержание
СН4 доходит до 99%.
Простейшие галогениды углерода СГ4, являющиеся также характеристически-
характеристическими соединениями, можно рассматривать как производные метана. В ряду от
фторида к иодиду устойчивость галогенидов углерода уменьшается, а химическая
активность их возрастает. В химическом отношении все тетрагалогениды похожи
на СС14- Молекулы СГ4 неполярны и имеют форму тетраэдров. Наибольшее
практическое значение имеет тетрахлорид углерода. Его можно получить хлори-
хлорированием метана, но большее распространение получил способ хлорирования
сероуглерода: ' .
CS2
2CI2 = СС14 + 2S
Для получения более тяжелых галогенидов применяют реакцию обмена CCI4 с
соответствующим галогенидом алюминия, например
ЗСС14 + 4А1Вг3 = ЗСВг4 + 4А1С13
Тетрахлорид СС14 представляет собой тяжелую, бесцветную и негорючую жид-
жидкость. Он не растворяется в воде, но хорошо растворяется в органических раство-
растворителях. Химически ССЦ весьма инертен. В обычных условиях он не горит, не
гидролизуется, не взаимодействует с кислотами и щелочами. Однако при повы-
повышенной температуре во влажной атмосфере тетрахлорид углерода разлагается
водой:
СС14 + 2Б2О = С02 + 4НС1
Эта реакция СС14 (или других галогенидов углерода) с водой свидетельствует о
его кислотной природе.
Соединения с другими неметаллами. При высоких темпе-
температурах углерод соединяется непосредственно с серой:
С + 2S = CS2
Дисульфид углерода (сероуглерод) CS2 — низкокииящая горючая жидкость.
Пары ого очень токсичны и легко воспламеняются. Он почти нерастворим в воде,
является хорошим растворителем многих органических веществ, например, жи-
жиров, красок и др., а также некоторых неорганических веществ (бром, иод. сера,
белый фосфор). Во всех агрегатных состояниях сероуглерод представляет собой
линейные неассоциирсванные молекулы CS2. Его химическое строение аналогич-
аналогично структуре СОг- Поэтому молекула CS; также не имеет электрического момента
диполя. Реакционная способность сероуглерода в обычных условиях невысока.
Он горит синим пламенем с образованием диоксидов серы и углерода:
CS2 4 30-> = СО2 + 2SO2
Вода гидролизует его лишь при температуре выше 150°С:
CS2 + 2Н2О = СО2 + 2H2S
В условиях высоких температур углерод также непосредственно взаимодей-
взаимодействует с азотом, образуя бесцветный ядовитый газ — циан (дициан):
2С + N2 = (CNJ
Реакция протекает при прохождении электрического тока между угольными
электродами в атмосфере азота. Проведение этого процесса в присутствии водоро-
водорода приводит к образованию циановодорода:
2С + N2 + Н2 = 2HCN
При охлаждении до —20,7°С циан сжижается в бесг&етную сильно ассоциирован-
ассоциированную жидкость. Молекулы циана очень устойчивы. Разложение на свободные
Для получения более тяжелых галогенидов применяют реакцию обмена СС14 с
соответствующим галогенидом алюминия, например
ЗССЦ + 4А1Вг3 = ЗСВг4 + 4А1С13
Тетрахлорид СС14 представляет собой тяжелую, бесцветную и негорючую жид-
жидкость. Он не растворяется в воде, но хорошо растворяется в органических раство-
растворителях. Химически ССЦ весьма инертен. В обычных условиях он не горит, не
гидролизуется, но взаимодействует с кислотами и щелочами. Однако при повы-
повышенной температуре во влажной атмосфере тетрахлорид углерода разлагается
водой:
СС14 + 2Н2О = С02 4- 4НС1
Эта реакция СС14 (или других галогенидов углерода) с водой свидетельствует о
его кислотной природе.
Соединения с другими неметаллами. При высоких темпе-
температурах углерод соединяется непосредственно с серой:
С + 2S = CS,
Дпсулъфил углерода (сероуглерод) GS2 — ниакокииящая горючая жидкость.
Пары ого очень токсичны и легко воспламеняются. Он почти нерастворим в воде,
является хорошим растворителем многих органических веществ, например, жи-
жиров, красок и др., а также некоторых неорганических веществ (бром, иод. сера,
белый фосфор), Во всех агрегатных состояниях сероуглерод представляет собой
линейные ноассоциирсванные молекулы CS^. Его химическое строение аналогич-
аналогично структуре СО2- Поэтому молекула CS2 также не имеет электрического момента
диполя. Реакционная способность сероуглерода в обычных условиях невысока.
Он горит синим пламенем с образованием диоксидов серы и углерода:
CS2 4- 30, - С02 4- 2SO2
Вода гидролизует его лишь при температуре выше 150°С:
CS2 4- 2Н2О = СО2 4 2H2S
В условиях высоких температур углерод также непосредственно взаимодей-
взаимодействует с азотом, образуя бесцветный ядовитый газ — циан (дициан):
2С 4 N2 = (CNJ
Реакция протекает при прохождении электрического тока между угольными
электродами в атмосфере азота. Проведение этого процесса в присутствии водоро-
водорода приводит к образованию циановодорода:
2С + N2 + Н2 = 2HCN
При охлаждении до —20,7°С циан сжижается в бесг&етную сильно ассоциирован-
ассоциированную жидкость. Молекулы циана очень устойчивы. Разложение на свободные
ГТ" '
радикалы (CNJ = 2CN начинается лишь при температуре выше 1000° С. Терми-
Термическая стабильность циана обусловлена прочностью межатомных связей в нем. В
молекуле циана центральные атомы углерода линейной молекулы находятся в
состоянии sp-гибридизации и образуют по три связи: одну с другим атомом
углерода и две с атомом азота. При этом у всех атомов остается по одному р-
электрону, которые образуют делокализованные 7гр-р-связи, упрочняющие моле-
молекулу: :N=C—¦C=N:. По химическим свойствам циан сходен с галогенами (кроме
фтора). Например, гидролиз циана на первой стадии протекает по уравнению
(CNJ + Н2О = HCN + HCNO
Более глубокий гидролиз циана приводит к образованию оксалата аммония:
(CNJ + 4Н2О = (NH4JC2O4
Циановодород HCN при обычных условиях представляет собой бесцветную лег-
легколетучую жидкость (^кип 26,5°С). Он имеет резкий запах горького миндаля. В
твердом, жидком и отчасти в парообразном состоянии между молекулами циано-
водорода действуют водородные связи вследствие большой полярности HCN.
Жидкий циановодород является неэлектролитом: вследствие наличия водород-
водородных связей его собственная ионизация ничтожно мала. В жидком состоянии HCN
представляет смесь двух изомерных форм — нормальной и изоформы:
H-C=N:
H-N=EC:
изоформа
В нормальной форме атом водорода связан с четырехвалентным атомом
углерода, а в изоформе водород соединен с азотом, а углерод имеет
ковалентность, равную трем. Обе формы находятся в динамическом равновесии и
легко переходят друг в друга. Высокую токсичность циановодорода связывают с
изоформой. Она соединяется с окислительными ферментами клеток через
ненасыщенный трехвалентный углерод с неподеленной электронной парой (как в
ядовитом угарном газе СО), прекращает окисление на уровне клеток, вызывает
удушье и паралич дыхания.
С водой HCN смешивается во всех отношениях. Водные растворы циановодо-
циановодорода называются циановодородной или синильной кислотой. Она является сла-
слабой кислотой (рА'а 9,3). Из ее солей — цианидов — она вытесняется даже такими
слабыми кислотами, как борная и угольная. Как и циан, синильная кислота
подвержена гидролизу, причем атаке подвергаются связи между углеродом и
азотом: водород присоединяется к азоту, а оксо- и гидроксогруппы — к углероду:
SN: + 2H2O =
NHa
ОН
Одним из наиболее распространенных способов получения синильной кислоты
является окисление смеси метана и аммиака. Реакцию проводят при 900°С над
платиновым катализатором:
2СН4 + 2NH3 + ЗО2 = 2HCN + 6Н2О
Цианиды щелочных и щелочно-земельных металлов легко растворимы в воде,
в результате гидролиза имеют щелочную реакцию:
KCN + НОН ?=* КОН + HCN
Цианиды активных металлов плавятся без разложения. Цианиды тяжелых метал-
металлов термически нестабильны, особенно те из них, в которых металлический эле-
элемент находится в высокой степени окисления.
Выше уже отмечалась аналогия в свойствах циана и галогенов. К этому доба-
добавим: их молекулы димерны [Г2 и (CNJ], легко превращаются в анионы Г" и CN",
подвергаются диспропорционированию при взаимодействии с водой и растворами
щелочей. Поэтому закономерно существование галоген цианидов FCN. Они могут
быть получены действием галогена на синильную кислоту:
Г2 + HCN = rCN + НГ
Галогенцианиды — бесцветные, летучие и токсичные вещества, по химическим
свойствам очень близкие к галогенам. С аммиаком галогенцианиды образуют
цианамид:
2NH3 + C1CN = NH4CI + NH2-C=N
Цианамид — бесцветные кристаллы, хорошо растворимые в воде. При взаимодей-
взаимодействии с водой в кислых и щелочных средах он образует мочевину (карбамид):
NH2CN + Н20 = CO(NH2J
Водород аминогруппы в цианамиде способен замещаться на металлы. Из солей
цианамида практически важен цианамид кальция CaCN2.
Циановодород и цианиды, являясь производными углерода (+2), сравнитель-
сравнительно легко окисляются. В мягких условиях происходит присоединение кислорода.
Уже на воздухе цианиды превращаются в цианаты — соли циановой кислоты
HCNO: 2KCN + 02 = 2KCN0. Цианаты щелочных металлов и аммония легко
растворяются в воде и медленно разлагаются ею:
2NaCNO -Ь 4Н2О = 2NaHCO3 + 2NH3
Свободная циановая кислота —летучая (tKim = 25°С), неустойчивая жидкость. Ее
водные растворы имеют ярко выраженный кислотный характер, представляют
собой кислоту средней силы (рКа 3,53).
Химическое строение HCNO отвечает трем структурам:
H-O-C^N: H-N=C=O: H-O-N^C
циановая изоциановая гремучая
кислота кислота кислота
Первые две формы связаны таутомерным* переходом. При комнатной температу-
температуре молекул изоциановой кислоты в равновесной смеси совсем немного. С пониже-
Таутомерия — динамическое равновесие между структурными изомерами.
365
кубическую и гексагональную. Поскольку кристаллохимическое строение SiC
обеспечивается прочными ковалентными связями, карборунд обладает высокой
твердостью, износостойкостью (кубическая модификация) и тугоплавкостью.
Химически и термически SiC очень устойчив. Термическое разложение на эле-
элементы заметно лишь при температуре выше 2300"С. На карбид кремния не дей-
действуют индивидуальные минеральные кислоты, но он растворяется в смеси HF +
+ HNO3. Хлор окисляет SiC при 600°С, а при 800°С наблюдается заметное окси-
оксидирование на воздухе:
SiC + 2С12 =¦ SiCl4 + С; SiC + О2 =¦ SiO2 + С
Карбид бора В4С получают, аналогично карбиду кремния, в электропечах
сопротивления при высокой температуре:
2В2Оз + 7С = В4С + 6СО
Карбид бора характеризуется тугоплавкостью (/пп — 2360°С), высокой твердос-
твердостью, близкой к твердости алмаза. Он устойчив по отношению к различным хими-
химическим реагентам при обычных условиях. Нл него не действуют кислогы, щелочи
и окислители.
У глеродсо держащие кислоты и их производные.
В водном растворе диоксида углерода существует равновесие:
СО2 + Н2О *=± Н2СО3 ^ Н+ + НСОз ^ 2Н* + СО|"
Оно сильно сдвинуто влево, поэтому концентрация ионов СО| мала, а большая
часть диоксида углерода находится в растворенной форме, не вступая во взаимо-
взаимодействие с водой (более 60% при 25°С). Нагревание еще больше смещает равнове-
равновесие влево, а при длительном кипячении диоксид углерода полностью удаляется
из воды. Наличие угольной кислоты Н2СО3 придает раствору кислый вкус. Она
является кислотой средней силы (рА[ 3,93). Угольная кислота не выделена в
свободном состоянии и существует только в растворах.
За исключением карбонатов щелочных металлов, большинство средних угле-
углекислых солей плохо растворимо в воде. Напротив, гидрокарбонаты характеризу-
характеризуются значительно большей растворимостью В водном растворе карбонаты под-
подвергаются гидролизу, в результате чего рН раствора больше 7:
Na2CO3 + Н2О ^ NaHCO3 + NaOH
Гидрокарбонаты гидролизуются слабее, и реакция раствора, например гидрокар-
гидрокарбоната натрия, близка к нейтральной. Методом обменных реакций можно полу-
получить карбонаты тех металлических элементов, для которых растворимость других
солей больше растворимости карбоната. Получить соли угольной кислоты можно
также растворением диоксида.углерода в щелочах, а при избытке СО2 карбонат
переходит в гидрокарбонат:
СО2 + Са(ОНJ = СаСОз + Н2О; СаСО3 + СО2 + Н2О = Са(НСО8J
В угольной кислоте и карбонатах анион угольной кислоты СОз~, по существу,
представляет собой ацидокомплекс, в котором центральный атом углерода под-
367
кубическую и гексагональную. Поскольку кристаллохимическое строение SiC
обеспечивается прочными ковалентными связями, карборунд обладает высокой
твердостью, износостойкостью (кубическая модификация) и тугоплавкостью.
Химически и термически SiC очень устойчив. Термическое разложение на эле-
элементы заметно лишь при температуре выше 2300'С. На карбид кремния не дей-
действуют индивидуальные минеральные кислоты, но он растворяется в смеси HF +
+ IINO3. Хлор окисляет SiC при 600°С, а при 800°С наблюдается заметное окси-
оксидирование на воздухе:
SiC + 2С12 - SiCL, + С; SiC -j- O2 = SiO2 + С
Карбид бора В4С получают, аналогично карбиду кремния, в электропечах
сопротивления при высокой температуре:
2В.С-3 + 7С - В4С + 6СО
Карбид бора характеризуется тугоплавкостью Aпл — 2360°С), высокой твердос-
твердостью, близкой к твердости алмаза. Он устойчив по отношению к различным хими-
химическим реагентам при обычных условиях. На него не действуют кислоты, щелочи
и окислители.
Углерод с о держащие кислоты и их производи ы е.
В водном растворе диоксида углерода существует равновесие:
СО2 + Н2О ^ Н2СО3 ^ Н+ + НСОз ^ 2Н+ ч- СОя'
Оно сильно сдвинуто влево, поэтому концентрация ионов ССI~ мала, а большая
часть диоксида углерода находится в растворенной форме, не вступая во взаимо-
взаимодействие с водой (более 60% при 25°С). Нагревание еще больше смещает равнове-
равновесие- влево, а при длительном кипячении диоксид углерода полностью удаляется
из воды. Наличие угольной кислоты Н2СОз придает раствору кислый вкус. Она
является кислотой средней силы (pA'i 3,93). Угольная кислота не выделена в
свободном состоянии и существует только в растворах.
За исключением карбонатов щелочных металлов, большинство средних угле-
углекислых солей плохо растворимо в воде. Напротив, гидрокарбонаты характеризу-
характеризуются значительно большей растворимостью. В водном растворе карбонаты под-
подвергаются гидролизу, в результате чего рН раствора больше 7:
Na2CO3 + Н2О ?=* NaHCO3 + NaOH
Гидрокарбонаты гидролизуются слабее, и реакция раствора, например гидрокар-
гидрокарбоната натрия, близка к нейтральной. Методом обменных реакций можно полу-
получить карбонаты тех металлических элементов, для которых растворимость других
солей больше растворимости карбоната. Получить соли угольной кислоты можно
также растворенном диоксида.углерода в щелочах, а при избытке СО2 карбонат
переходит в гидрокарбонат:
СО2 + Са(ОНJ = СаСОз + Н2О; СаСО3 + СО2 + Н2О = Са(НСО3J
В угольной кислоте и карбонатах анион угольной кислоты СО3", по существу,
представляет собой ацидокомплекс, в котором центральный атом углерода под-
367
вержен в^-гибридизации. Остающиеся у всех атомов по одному р-электрону
образуют делокализованные 1гр-р-связи, сильно увеличивающие прочность комп-
комплекса. При этом атом углерода находится в центре равностороннего треугольни-
треугольника, а атомы кислорода — по его вершинам:
2-
По отношению к нагреванию карбонаты ведут себя неодинаково. Карбонаты
металлов IA-группы, кроме 1л2СОз, плавятся без разложения, а остальные при
более или менее сильном нагревании разлагаются, например
СаСОз = СаО + СО2
Гидрокарбонаты менее термически стойки и разлагаются при более низких тем-
температурах, превращаясь в карбонаты:
Са(НСО3J = СаСОз + Н2О + СО2
Сильные кислоты разрушают карбонаты и гидрокарбонаты, вытесняя из них
более слабую угольную кислоту:
СаСОз + 2НС1 = СаС12 + Н2О + СО2
Са(НСО3J + НС1 = СаС12 + Н2О + СО2
При взаимодействии сероуглерода с сульфидом щелочного металла, например
CS2 -f K2S = K2CS3, образуется соль тиоугольной кислоты H2CS3- Тиоугольную
кислоту выделяют из ее солей — тиокарбонатов — взаимодействием с сильными
кислотами:
K2CS3 + 2НС1 = 2КС1 + H2CS8
Свободная тиоугольная кислота — маслянистая жидкость, легко разлагающаяся
на H2S и CS2. По термической стойкости и кислотным свойствам она превосходит
угольную кислоту. Тиоугольная кислота относится к сильным кислотам (рК\ 2,7).
В ионе CS3" атом углерода находится в вр2-гибридном состоянии. Его структура
аналогична структуре карбонат-иона.
Пероксопроизводными угольной кислоты являются соли пероксомоноугольной
(мононадугольной) Н2СО4 и пероксодиугольной (надугольной) ЩСгОб кислот,
называемые пероксокарбонатами. Сами кислоты в свободном состоянии не выде-
выделены, но в их структуре обязательно наличие пероксидной цепочки. Пероксокар-
бонаты получают анодным окислением концентрированных растворов карбонатов:
2СО!~ — 2е~ = С2О|". При нагревании пероксокарбонаты разлагаются, в воде
гидролизуются, а кислотами разрушаются.
368
Для пероксомоноугольной кислоты известны и средние, и кислые соли. Для
их получения углекислый газ пропускают над пероксидами и гидропероксидами:
Na2O2 + СО2 = Na2CO4; NaOOH + CO2 = NaHCO4
Все пероксокарбонаты — сильные окислители.
К производным угольной кислоты относится карбаминовая кислота
H2N—COOH. Она в свободном состоянии неизвестна, а ее соль H2NCOONH4 обра-
образуется присоединением аммиака к диоксиду углерода:
2NH3 + СО2 = H2NCOONH4
Нагревание карбамината аммония при 130°С и под давлением 202- Ю5 Па сопро-
сопровождается выделением воды и образованием мочевины (карбамида):
H2NCOONH4 = Н2О + H2N-CO-NH2
Мочевина представляет собой хорошо растворимое в воде белое кристаллическое
вещество. Ее молекула имеет плоское треугольное строение с атомом углерода в
вр2-гибридном состоянии:
Вследствие гидролиза растворы мочевины в воде пахнут аммиаком:
CO(NH2J + Н2О = 2NH3 + СО2
Карбамид является слабым основанием (рА'ь 13,7).
2. Кремний. Особенности химии кремния. Второй типичес-
типический элемент IV группы — кремний — является типовым аналогом углерода. Как и
у углерода, у атома кремния в невозбужденном состоянии на s-орбитали находят-
находятся два спаренных электрона, а ?>-орбитали имеют два неспаренных электрона.
Разница в том, что атом углерода располагает валентными электронами при
главном квантовом числе 2, а атом кремния характеризуется тем же числом ва-
валентных электронов D) при п — 3. В связи с увеличением числа электронных
слоев по сравнению с углеродом у кремния наблюдаются рост атомного радиуса,
понижение потенциала ионизации, уменьшение сродства к электрону и ОЭО.
Возрастание радиуса ведет к увеличению длины и уменьшению прочности меж-
межатомных связей, особенно в гомоатомных соединениях, вследствие чего растет
электрическая проводимость и сужается ширина- запрещенной зоны. Поэтому
углерод в виде алмаза представляет собой изолятор, а кремний — полупровод-
полупроводник. В целом переход от первого типического элемента ко второму свидетельству-
свидетельствует о нарастании металличности и ослаблении неметаллических свойств. Однако
вследствие наличия большого числа валентных электронов этот переход более
плавный, чем в Ш группе от бора к алюминию.
369
Существенной особенностью химии кремния сравнительно с химией углерода
является возможность вовлечения в связеобразование 3</-орбиталей. Это приводит
к увеличению валентных возможностей атома кремния. Теоретически максималь-
максимальная ковалентность кремния может быть равна 9 против 4 у углерода. На практике
помимо валентности 4 встречаются шестиковалентные производные, в которых
атом кремния находится в sp:i(^-гибридном состоянии. Однако для кремния
наиболее характерны структуры, где атомы кремния имеют к.ч. 4 и находятся в
.«^-гибридном состоянии. Производные с зр- и ар2-гибридизацией атома кремния
редки и. как правило, мало устойчивы. Кремний в отличие от углерода менее
склонен образовывать кратные связи. Для кремния наиболее характерно допол-
дополнительное 7Гр-(;-связывание в отличие от Яр-р-взаимодейгтвия для углерода. Та-
Таким образом, в случае кремния связывание часто возникает за счет участия ва-
вакантных Зй-орбитален и пеподеленных электронных пар атомов партнеров. Так
обстоит дело в соединениях кремния с азотом, кислородом, фтором и хлором.
Прочность связей кремния с кислородом, азотом и галогенами из-за дополни-
дополнительного ^-связывания выше, чем соответствующих связей для углерода. Наобо-
Наоборот, связь атома углерода, например, с водородом прочнее, чем у кремния, так
как водород не располагает нэподеленной электронной парой. Ниже для сравне-
сравнения приводим энергии различных связей углерода и крел'ния (кДж/моль):
Элемент
Углерод
Кремний
т
347,7
328,3
Si I Н
328,3 414.2
176,6
319,5
О
359,3
415,1
N
206.5
330,8
F
487,7
567.7
С1
340,1
382/1
Вг
285,6
310.3
I
214,4
235."
В'-ледствие малой прочности гомоатомных связей кремний в отличие от угле-
углерода не образует устойчивых цепей из многих атомов Кроме того, производные
кремния с типическими неметаллами более полярны по сравнению г, аналогичны-
аналогичными соединениями углерода. Это обусловлено большей моталлпчностыо кремния и
меньшей ОЭО. В наиболее стабильных соединениях кремний проявляет степени
окисления +4, 0 и —4. Окислительное числе +2 мало характерно для кремния.
Наконец,, для химии кремния имеет принципиальное значение большое срод-
сродство к кислороду, что обусловлено энергией связи атомов кремния с кислородом,
которая превосходит энергию связи между атомами кремния в 2.5 раза. И не
случайно земная кора более чем наполовину состоит из кремнезема SiO2, его
гидратпых форм xSiO^r jjllzO, различных силикатных и алюмосиликатных пород.
Природные соединения и получение кремния.
Содержание кремния в земной коре составляет 27,6 масс.доли, %, и по распрост-
распространенности он уступает только кислороду. При оценке распространенности эле-
элементов в ат.долях, % кремний смещается на третье место, располагаясь после
кислорода и водорода. В природе кремний находится только в связанном состоя-
состоянии. Среди важнейших минералов кремнезем SiCb, каолинит AJ^Si^OioXOH^,
полевые шпаты MeAl(Si3O8), где Me — Na, К, слюды Me32Al(SisOio)(OH,FJ, где
Me - Li, NTa, К, а Э - Al, Fe.
Технический кремний получают восстановлением кремнезема коксом в элект-
электропечах при 1500-1750°С:
SiO2 + 2С = Si + 2CO
370
Чистота технического продукта 93—99%. Для получения особо чистого кремния
технический кремний переводят в летучие производные, которые легко очищают-
очищаются перегонкой, ректификацией, глубоким охлаждением. Такими соединениями
кремния оказались тетрахлорид SiCl4, трихлорсилан SiHCl3 и моносилан SiH4- На
втором этапе очищенные соединения кремния восстанавливают водородом или
используют термический распад:
SiCi4 + 2Н2 = Si + 4НС1; SiHCl3 + Н2 = Si + ЗНС1; SiII4 = Si + 2Н2
Конечным продуктом приведенных выше реакций является поликристаллический
кремний. Для получения монокристаллов кремния и дальнейшей очистки приме-
применяют бесконтейнерную зонную плавку. 13 вакууме или инертной атмосфере с
помощью высокочастотного индуктора в вертикально установленном стержне
кремния создастся расплавленная зона, которая не растекается благодаря силам
поверхностного натяжения жидкого кремния. Расплавленная зона с определенной
скоростью многократно перемещается в одном и том же направлении. В результа-
результате получаются совершенные монокристаллы кремния с суммарным содержанием
примесей не более 10'7—10~8 масс.доли, %. Только бестигельная зонная очистка
A958) дала возможность кремнию стать ведущим современным полупроводнико-
полупроводниковым материалом. Дело в том. что из-за высокой температуры плавления A414'С)
жидкий кремний реагирует с материалом контейнера. Поэтому для очистки и
получения монокристаллов кремния в принципе непригодны все контейнерные
методы, например горизонтальная направленная кристаллизация.
Впервые кремний был получен металлотермическим восстановлением кремне-
кремнезема по реакции
SiOj + 2Mg -- Si + 2MgO
Продукт восстановления представляет собой мягкий коричневый порошок, назы-
называемый аморфным кремнием. Таким образом, путь от первого порошкообразного
кремния до современного особо чистого монокристалла с ничтожной концентра-
концентрацией дефектов — это революция в химии гомоатомных соединений кремния,
стимулированная развитием техники.
Физические и химические свойства кремния. Тер-
Термодинамически стабильная при нормальных условиях кубическая алмазоподоб-
ная модификация кремния серо-стального цвета со смолистым блеском, характе-
характеризуется значительной твердостью и хрупкостью. Вы-
Высокотемпературная гексагональная разновидность крем-
кремния неустойчива и практического значения не имеет.
Как показали исследования последних десятилетий,
аморфный кремний в действительности состоит из
мелких кристалликов кубической формы и некоторых
примесей. В алмазоподобной кубической структуре
кремния его атомы находятся в ярЗ-гибридном состоя-
состоянии. Трехмерную сетку тетраэдрических связей в крис-
кристалле- кремния МОЖНО изобразить на ПЛОСКОСТИ (рис. Рис. 145. Схема тетраэдри-
145). Однако вследствие меньшего перекрытия элвкт- ческих связей в кристалле
ронных облаков при образовании тетраэдрических кремния
371
Диоксид S1O2 — наиболее характерное и устойчивое кислородное соединение
кремния. Он образует три кристаллические модификации: кварц, тридимит и
кристобалит. Недавно были получены новые модификации S1O2 — стишовит и
коусит. Последние получаются только под высоким давлением, а при нормальных
условиях в метастабильном состоянии могут существовать неограниченно долго
(как алмаз). Часто встречающаяся разновидность кварца в природе -— горный
хрусталь. Окрашенные разновидности кварца: марион (черный), топаз (дымча-
(дымчатый), аметист (фиолетовый), цитрин (желтый). Описаны также волокнистые
модификации SiO2 (халцедон и кварцин). Кроме того, на дне морей и океанов из
водорослей и инфузорий образуется аморфный S1O2. В целом диоксид кремния —
самый распространенный оксид в земной коре. Кварц, тридимит и кристобалит
могут превращаться друг в друга, однако эти переходы сильно заторможены.
Вследствие этого тридимит и кристобалит, несмотря на свою термодинамическую
нестабильность, могут неограниченное время сохраняться при комнатной темпера-
температуре и существовать в природе в виде самостоятельных минералов. Каждая из
этих кристаллических модификаций, в свою очередь, может находиться в виде
двух или большего числа взаимно превращающихся форм, из которых №-форма
устойчива при комнатной, а /3-форма — при более высокой температуре. Ниже
приводим схему взаимных переходов кристаллических модификаций диоксида
кремния:
870° . 1470° _ , 1723°
fi— кварц _« ** р- тридимит « "* р—кристобалит;
а-кварц в—тридимит в-кристобалит стекло
Устойчивая при высокой температуре модификация — iS-кристобалит — плавится
при 1723° С. При быстром охлаждении расплавленного кремнезема образуется
стекло.
Различные кристаллические модификации SiO2, как и безводный аморфный
кремнезем, представляют собой неорганические гетероцепные полимеры. Во всех
формах (кроме стишовита) структурным мотивом является кремнекислородный
тетраэдр: в центре правильного тетраэдра находится атом кремния, а по верши-
вершинам — атомы кислорода. Кремнекислородные тетраэдры соединены друг с другом
своими вершинами, т.е. каждый атом кислорода связан с двумя атомами кремния,
что можно изобразить на плоскости:
I I I
— Si—О — Si — О—Si— О —
I I I
0 о о
I I I
— Si — O-^-Si—О—Si—О —
1 I I
Несмотря на одинаковый способ сочленения структурных мотивов [SiOJ, их
пространственное расположение для различных модификаций различно. Поэтому,
например, /?-кристобалит (см. рис. 130) имеет кубц^ескую решетку, а /3-тридимит
(рис. 146) — гексагональную. Между этими структурами такая же разница, как
между сфалеритом и вюрцитом. Наиболее плотная модификация SiO2 (стишовит)
373
O-Si •-#
Рис. 146. Кристаллическая
структура /?тридимита
характеризуется необычной для кислородных
соединений кремния координацией атомов. Здесь
каждый атом кремния окружен не четырьмя, а
шестью атомами кислорода. Поэтому структура
стишовита образована сочетанием кремнекисло-
родных октаэдров [SiOe].
Химическая активность модификаций SiO2
возрастает от кварца к кристобалиту и особенно
кремнезему, полученному обезвоживанием геля
кремниевой кислоты. Фтор, газообразный 1IF и
плавиковая кислота энергично взаимодействуют
с SiO2:
SiO2 + 2F2 = SiF4 + O2;
SiO2 + 4HF = SiF4 + 2H2O
В первой реакции фтор вытесняет кислород из оксида кремния. Обе реакции
протекают потому, что тетрафторид кремния — более прочное соединение, чем
диоксид. Энтальпия образования последнего -910,9, а для SiF4 AH°f Oflg = -1614,9
кДж/моль. Кроме того, эти процессы сопровождаются возрастанием энтропии
(слева — твердое вещество и газ, а справа — два газа). Поэтому свободная энер-
энергия Гиббса в результате этих взаимодействий сильно уменьшается.
В воде SiO? практически нерастворим. Не действуют на него кислоты и царс-
царская водка. В щелочных растворах, особенно при нагревании, SiO2 легко раство-
растворяете я:
SiO2 + 2NaOH = Na2Si03 + H2O
Обычно реакцию получения силикатов проводят не в растворе, а путем спекания
SiO^ со щелочами, карбонатами и оксидами металлов:
SiO2 + Na2CO3 = Na2Si03 + СО2; SiO2 4 PbO = PbSiO3
Все эти реакции доказывают кислотную природу диоксида кремния. Химичес-
Химические свойства кварцевого стекла практически такие же, как и кристалллическо-
го SiO2.
Поскольку S1O2 нерастворим в воде, кремниевую кислоту получают косвенным
путем:
Na2Si03 + 2HC1 = H2Si03 + 2NaCl
Образующаяся кремниевая кислота выделяется из раствора в виде студенистого
осадка или остается в растворе в коллоидном состоянии. Состав ее отвечает фор-
формуле xSiO^yH^O со значениями х и у, меняющимися в зависимости от условий.
Кислота с i= 1и }- 1 называется метакремниевой H2S1O3, а у ортокремниевой
H4Si04 у — 2. Все кислоты, для которых х > 1, называются поликремниевыми. В
свободном состоянии эти кислоты выделить не удается. Их состав определяется
374
по солям — силикатам. Все кремниевые кислоты очень слабые. Так, H4S1O4 имеет
pA'i 10. Поэтому растворимые в воде силикаты сильно гидролизованы:
Na2Si03 + 2Н2О = 2NaOH + H2Si03
Частично обезвоженная студнеобразная кремниевая кислота представляет собой
твердую белую очень пористую массу, называемую силикагелем. Он обладает
высокой адсорбционной способностью и энергично поглощает воду, масла, зфиры
и т.д.
Водородные соединения кремния — кремневодороды или силаны — получают
действием кислот на силициды активных металлов, например
Mg2Si 4 4НС1 - 2MgCi2 + SiH4
Наряду с моносиланом SiH.j выделяются водор(>д и птисиланы, вплоть до гекса-
силана Si6Hi.j. Содержание других кремноводородов в продуктах разложения
силицида магния закономерно увеличивается по мере уменьшения их молекуляр-
молекулярной массы. По структуре и физическим свойствам силаны похожи на углеводоро-
углеводороды гомологического ряда метана. Известны все гомологи моносилана, вплоть до
октасилана SigHis- Для получения практически наиболее важного моносилана
используют реакции восстановления галогеиидов кремния водородом или алюмо-
гидридом лития:
Sir., + 4Н2 = SiH4 + 4НГ
SiCl4 + Li[AlH4] = SiH4 4 LiCl + A1C!3
Все силаны имеют характерный неприятный запах и токсичны. По сравнению
с углеводородами силаны характеризуются большей плотностью и более высоки-
высокими температурами плавления и кипения, но термически менее стойки. По хими-
химическим свойствам силаны резко отличаются от представителей гомологического
ряда метана и напоминают бораны (диагонатьное сходство с бором). Они легко
окисляются на воздухе, т.е. являются восстановителями:
SiH4 4- 2О2 = SiO2 4- 2Н2О
Силаны восстанавливают КМпО.) до МпО2, производные FeD-3) до FeD-2). Кроме
того, для силанов характерен гидролиз. В присутствии следов кислот и особенно
щелочей силаны разрушаются:
SiH4 4- 2Н2О = SiO2 4- 4Н2
SiH4 4- 2NaOH 4- Н2О = Na2Si03 4- 4Н2
Образование в процессе гидролиза силанов кремнезема или силикатов указывает
на кислотную природу силанов.
Для кремния известны также немногие представители непредельных кремне-
водородов типа полисиленов (SiH2)n и полисилинов (SiH)n. Все они — твердые
вещества, неустойчивые к нагреванию и исключит^ьно реакционноспособныс
Они самовоспламеняются на воздухе и нацело разлагаются водой.
375
Галогениды кремния SiF4 могут быть получены синтезом из простых веществ.
Все они энергично взаимодействуют с водой:
Sir4 + 2Н2О = SiO2 + 4НГ
Для фторида реакция обратима (поэтому SiO2 растворяется в HF), а для осталь-
остальных галогенидов — практически полностью смещена вправо. При нагревании
галогенидов SiF4 с кремнием выше 1000°С протекает реакция образования дига-
логенидов: SiF4 + Si 15=i 2SiF2 *, которые при охлаждении диспропорционируют с
выделением кремния. Эту реакцию можно использовать как транспортную для
получения кремния высокой чистоты.
Из галогенидов кремния наибольшее значение имеют SiCU, SiF4 и SiHCb-
Тетрахлорид кремния получают при хлорировании смеси угля и кварцевого
песка F00-700°С):
2С12 + SiO2 + 2С = SiCl4 + 2CO
Значительные количества SiF4 в качестве побочного продукта улавливаются на
суперфосфатных производствах, работающих на апатитовом сырье. Кроме того,
он может быть получен нагреванием смеси кварцевого песка, фторида кальция и
серной кислоты:
SiO2 + 2H2SO4 + 2CaF2 = 2CaSO4 + SiF4 + 2H2O
Тетрафторид кремния, присоединяя две формульные единицы HF, переходит в
кремнефтористоводородную (гексафторокремниевую) кислоту:
SiF4 + 2HF = H2[SiF6]
В индивидуальном состоянии H2[SiF6] не выделена, по силе близка к серной
кислоте. Соли ее — гексафторосиликаты — при нагревании разлагаются на SiF4 и
фториды металлов. В октаэдрической структуре ионов [SiF6]2" кремний находится
в состоянии зр^-гибридизации и его к.ч. 6. Для других галогенов соединения
аналогичного состава неизвестны.
Трихлорсилан (или силикохлороформ) получают пропусканием тока сухого
хлороводорода над кремнием D00—500°С). На воздухе он не воспламеняется, но
горит при поджигании. Аналогичные трихлорсилану соединения кремния извест-
известны и для других галогенов. При восстановлении трихлорсилана получают крем-
кремний высокой чистоты.
Соединения с другими неметаллами. Дисульфид крем-
кремния SiS2 получается при непосредственном взаимодействии компонентов. Образу-
Образуется дисульфид также вытеснением кремнием водорода из H2S в отсутствие воз-
воздуха при 1300°С:
2H2S + Si = SiS2 + 2Н2
* Подобные процессы, обратные диспропорционированию, называются конпропорцио-
нированием. В результате этих реакций разные степени окисления одного и того же эле-
элемента принимают одно промежуточное значение.
376
Дисульфид кремния — белые шелковистые кристаллы. Водой дисульфид крем-
кремния разлагается на H2S и SiO2. Известен также моносульфид кремния SiS. Он
получается восстановлением дисульфида в вакууме при 900° С. Моносульфид
представляет собой полимерные игольчатые кристаллы, разлагающиеся водой:
SiS + 2Н2О = H2S + SiO2 + Н2
Нитрид кремния S13N4 получают либо взаимодействием компонентов (при
температуре выше 1300°С), либо из NH3 и SiCl4. В последнем случае в качестве
промежуточного продукта образуется имид кремния Si(NHJ, который в процессе
термического разложения превращается в нитрид:
SiCl4 + 2NH3 = Si(NHJ + 4НС1; 3Si(NHJ = Si3N4 + 2NH3
Бесцветные кристаллы SigN4 отличаются большой химической стойкостью. До
1000°С на него не действуют кислород, водород и водяной пар. Он не растворя-
растворяется в кислотах и растворах щелочей. Только расплавы щелочей и горячая кон-
концентрированная плавиковая кислота медленно его разлагают.
Из соединений кремния с фосфором наиболее известны моно- и дифосфид:
SiP и SiP2. Они получаются непосредственным взаимодействием компонентов в
нужных стехиометрических количествах, отличаются химической стойкостью.
Аналогичный состав имеют и арсениды кремния.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Кремниевую кислоту и силикаты уже можно рассмат-
рассматривать как силикатные оксокомплексы водорода и металлов. Большинство сили-
силикатов не окрашены, отличаются тугоплавкостью и практически нерастворимы в
воде. К числу немногих растворимых производных кремниевой кислоты относят-
относятся силикаты щелочных металлов. Из растворимых силикатов наибольшее значе-
значение имеет Na2Si03- Эту соль часто называют растворимым стеклом, а ее водные
растворы — жидким стеклом. Водные растворы силикатов химически активны,
обладают клейкостью и вяжущими свойствами. В закрытой посуде эти растворы
хорошо сохраняются, а на воздухе разлагаются под действием СО2: углекислота
вытесняет кремниевую кислоту.
Нерастворимые силикаты исключительно распространены в природе. Природ-
Природные силикаты отличаются разнообразием составов и структур. Как и в кремнезе-
кремнеземе и кремниевых кислотах, основной структурной единицей силикатов является
тетраэдрическая группировка [SiO4]4~. Тетраэдры могут объединяться попарно
[S12O7]6", образовывать кольца из трех [SisOg]6", четырех [Si4Oi2]8~, шести [SieOis]12"
тетраэдров. Эти сравнительно простые структурные образования могут объеди-
объединяться в цепи, ленты, слои, островки и координационные структуры.
Часть атомов кремния в структуре силикатов может быть заменена атомами
алюминия (алюмосиликаты), а оксо-ионы — на ионы фтора, хлора, гидроксид-
ионы, борат-ионы и др. В алюмосиликатах вместо части тетраэдров [SiO4] встраи-
встраиваются тетраэдры [A1OJ. Если тетраэдры [SiO4]4~ связаны в трех точках, они
образуют структурную единицу [Si2O5]2". К таким структурам относится каолин
Al2[(Si205)@HL]. Его, как и другие алюмосиликаты, можно представить форму-
формулой H4Al2Si20g или Al2Os-2SiO2-2H2O. При связывании кремнекислородных
тетраэдров всеми четырьмя вершинами образуются пространственные структуры.
Замещение кремния на гшюминий дает избыточные отрицательные заряды, кото-
которые нейтрализуются катионами Na+, К+, Са2+и Ва2+. К таким структурам и отно-
Р и с. !47. Схема типичной структуры
цеолита (налролкт)
сятся чрезвычайно распространенные
алюмосиликаты: полевые шпаты, цеолиты
и ультрамарины. Калиевый полевой шпат
ортоклаз K[AlSi3Os] может быть пред-
представлен формулами К2О-Al2O,v6SiO2 или
K2Al2Si6Oi6- В цеолитах встречаются
кольца, образованные из трех тетраэдров
[S1O4] и двух [AIO4], расположенных так,
что в пространстве образуются пустоты в
виде каналов, в которых винтообразно
располагаются катионы щелочного метал-
металла и молекулы воды, слабо связанные с
остальной решеткой. Такие структуры
легко теряют воду при нагревании и
вновь поглощают ее из атмосферы после
охлаждения. На рис. 147 приведена
проекция фрагмента структуры одной из
разновидностей цеолитов — натролита, в которой хорошо видно винтообразное
расположение молекул воды и катионов натрия. Под действием природных
факторов алюмосиликаты претерпевают деструкцию. Этот процесс называют
выветриванием. Конечным продуктом выветривания большинства силикатов
является глина (каолин).
Силикаты, как и кварц, при быстром охлаждении не кристаллизуются, а
образуют стекла. Обыкновенное оконное и бутылочное стекло имеет состав, близ-
близкий к Na20-Ca0-6Si0i. Получают его сплавлением смеси кварцевого песка, из-
известняка и соды:
Na2CO,4 + СаСО3 + 6SiO2 = Na2OCa0-6Si02 + 2СО2
Вместо соды можно использовать смесь сульфата натрия и угля:
Na?SO4 + С + СаСО3 4- 6SiO2 = Na2O-CaO-6SiO2 4- SO2 + СО2 4- СО
Примеси в исходных материалах придают стеклу определенную окраску. В част-
частности, зеленый цвет бутылочного стекла связан с железом (+2). При получении
специальных сортов стекла в шихту вводят легирующие добавки. Чаще других
используются для этих целей соединения Pb, Sb, Zn и Ва. Заменяются также
(частично или полностью) карбонаты натрия и кальция карбонатами других
мет&пл'ов, a SiO2 — оксидами неметаллов, например В2О3. Так, калиевое стекло,
отличающееся более высокой температурой размягчения, получают при замене
соды поташем. Свинцовое стекло (хрусталь) содержит вместо кальция свинец, а
вместо натрия — калий. Хрусталь обладает большой плотностью и высоким пока-
показателем преломления света. В боросиликатных стеклах (легирование борным
ангидридом) появляются группировки [ВО4] («р3-гибридизация атома бора),
включенные в беспорядочно ориентированные цепи кремнекислородных тетраэд-
тетраэдров [SiO4]. Эти стекла отличаются повышенной прочностью и термостойкостью,
устойчивостью к действию кислот и воды.
В настоящее время на основе стекла получают и используют новый класс не-
неорганических материалов — ситаллы, т.е. стеклокристаллы. Если в стекле имеют-
имеются лишь области микрокристаллитов, то ситаллы — продукты почти полной кри-
Я7Я
сталлизации стекла с тонкими прослойками остаточного стекла. Для массовой
кристаллизации стекла добавляют минерализаторы (чаще всего оксиды метал-
металлов), которые являются своеобразными катализаторами кристаллизации стекла.
Размеры отдельных кристаллитов в ситалле около 30 нм, а содержание прослоек
стекла не более 5%. Ситаллы обладают высокой механической прочностью; они
химически и термически устойчивы, их температура размягчения 1400—1500"С и
температурный коэффициент расширения невелик. Например, магниевые ситаллы
(MgO — А12Оз — SiOi) в 10 раз прочнее прокатанного стекла, легче алюминия,,
тверже высокоуглеродистой стали, а по термостойкости не отличаются от кварца.
М е т а л 1 о х и м и я к р е м н и я. При взаимодействии кремния с метал-
металлами различие между ним и углеродом сказывается гораздо сильнее. Дело в том,
что нарастание некоторой моталличности от углерода к кремнию не так резке
алияет на, их отношение к типическим неметаллам, хотя бы формально. Сравни-
Сравните, например. СО2 и SiO2. CO и SiO, CS2 и SiS?. СГ4 и SiT.h CH4 и SiH4, K2CO3 и
H2S1O3 и т.д. В металлохимии углерода важно его взаимодействие с железом и
образование металлоподобных карбидов. На диаграммах состояния углерода с
переходными металлами, как правило, имеется единственный тугоплавкий моно-
монокарбид металла. Для кремния металлоподобные силициды менее характерны, и
они не отличаются такими экстремальными свойствами, как металлоподобные
карбиды. На диаграммах состояния для кремния с переходными металлами су-
существует множество силицидных фаз. Все оьи в основном образованы не sin пра-
правилам валентности, т.е. являются истинными металлидами. Ниже приводим число
силицидов, фиксируемых на диаграммах состояния кремний — переходный ме-
металл: Ы - Zr 7, Si - Ni 6, Si - Та 4, Si - Mn 4, Si - Fe 4; Si - Th 4 Si - V 3.
Si - Pt 3. Si - Mo 3 и т.д.
Кремний образует непрерывные твердые растворы только с германием. С ме-
металлами IA- и ПА-групп кремний образует силициды, подчиняющиеся правилам
формальной валентности. Взаимодействие с s/ьметаллами, как правило, приводит
к образованию эвтектики. В силу неметаллического характера кремния он почти
не образует широкие области твердых растворов, хотя температура плавления его
высока. Кремний не взаимодействует с цинком, ртутью, таллием и висмутом.
3. Подгруппа германия. Характеристика элементов I V А-
группы. К. lVA-rpynne элементов помимо типических относятся элементы
подгруппы германия: Ge, Sn и Pb. Их валентная электронная конфигурация
(ns2np2 в невозбужденном состоянии) обусловливает возможность проявления
свойств и катионо- и анионообразователей. Кроме того, эти элементы расположе-
расположены на границе Цинтля и число валентных электронов достаточно для образова-
образования структур с ковалентной связью у соответствующих простых веществ с коор-
координационными числами согласно правилу Юм~Розери (8 — N). Действительно,
для гомоатомных соединений (кроме свинца и /3-олова) характерна кристалличес-
кристаллическая решетка типа алмаза. Однако преимущественная ковалентная связь в крис-
кристаллах соединений реализуется далеко не всегда. Причиной этого является вто-
вторая особенность IVA-группы, заключающаяся в том, что здесь наиболее рельефно
прослеживается изменение свойств от типично неметаллических 1С) до металли-
металлических (РЬ). Поэтому тяжелые представители этой группы (Sn, Pb), т.е. элементы
с большой атомной массой, характеризуются плотноупакованными структурами в
свободном состоянии.
Иногда элементы подгруппы германия называют металлами IVA-группы.
379
Формально в их число входит и сам германий. Однако германий является типич-
типичным полупроводником с преимущественно ковалентной связью, а следовательно,
металлом в свободном состоянии быть не может. Тем не менее в большом числе
соединений с более электроотрицательными элементами германий выступает в
роли катионообразователя, что с химической точки зрения отражает металличес-
металлическую природу элемента. В бинарных соединениях с металлами германий — ани-
онообразователь, однако все эти соединения обладают металлическими свойства-
свойствами, что характеризует германий как плохой анионообразователь. Точно так же он
не обладает ярко выраженной способностью к образованию анионных комплексов
типа [GeOsJ2".
Олово существует в двух полиморфных модификациях, причем низкотемпера-
низкотемпературная (a-Sn — серое олово) обладает кристаллической решеткой типа алмаза и
полупроводниковыми свойствами, а высокотемпературная (fi-Sn — белое олово),
хотя и представляет собой металл по физическим свойствам, тем не менее крис-
кристаллизуется в малохарактерной для металлов тетрагональной структуре. С хими-
химической точки зрения, олово ближе примыкает к германию, чем к свинцу, но ме-
металлический характер этого элемента выражен более ярко, чем у германия. Един-
Единственным типичным металлом в этой подгруппе является свинец. В виде простого
вещества он кристаллизуется в плотноупакованной ГЦК структуре с к.ч. 12. В
своих соединениях он выступает в основном в качестве катионообразователя.
Все три элемента проявляют две характерные степени окисления: +4 и 4-2,
причем сверху вниз в группе устойчивость высшей степени окисления уменьшает-
уменьшается, а низшей — возрастает. Германий и олово в степени окисления +2 являются
сильными восстановителями. У соединений Ge(+2) эта особенность настолько
ярко выражена, что в отсутствие окислителя они диспропорционируют. Для
Sn(+2) реакции диспропорционирования уже менее характерны. Что же касается
свинца, то для него степень окисления +2 является наиболее устойчивой, а сое-
соединения РЬ(+4) — сильные окислители.
В пределах IVA-группы наблюдается немонотонная зависимость свойств от
положения элемента в группе. Так, ОЭО германия оказывается больше, чем
кремния, хотя первый потенциал ионизации германия меньше. Это объясняется
существованием у атома германия в отличие от кремния заполненного внутренне-
внутреннего 3<#°-уровня, который служит экраном для jp-электронов. Если же сравнить 3-й
и 4-й потенциалы ионизации, характеризующие прочность связи с ядром s-элект-
ронов, можно сделать вывод о преобладании эффекта проникновения для s-элек-
тронов у германия под слой 3</-электронов. С учетом четырех потенциалов иони-
ионизации оказывается, что прочность связи валентных электронов с ядром выше у
атома германия. Этим и объясняется более высокое значение ОЭО германия по
сравнению с кремнием. Радиусы элементов также изменяются немонотонно. При
переходе от С к Si наблюдается резкое возрастание атомного радиуса, а затем
радиус меняется незначительно. Это можно объяснить влиянием (/-контракции у
германия и олова.
Природные соединения и получение германия,
олова и свинца. Содержание элементов подгруппы германия в природе
сравнительно невелико. Олово и свинец образуют самостоятельные месторожде-
месторождения (полиметаллические руды) и концентрируются в отдельных регионах/ Герма-
Германий не образует больших скоплений и относится к числу рассеянных элементов.
Известны такие германийсодержащие минералы, как аргиродит, 4Ag2S-GeS2,
380
германит 3Cu2S-FeS-2GeS2, которые сокристаллизуются в процессе остывания
магмы и горячих водных растворов с другими сульфидными минералами. Самы-
Самыми широко распространенными минералами олова являются касситерит (оловян-
(оловянный камень) SnO2 и станнин (оловянный колчедан) Cu2S-FeS-SnS2. Для свинца
наиболее известен свинцовый блеск, или галенит, PbS. Галенит нередко составля-
составляет основную минеральную фракцию полиметаллических руд. Помимо того, в
природе встречаются и кислородсодержащие минералы свинца: англезит PbSO4,
церуссит РЬСОз и др.
В настоящее время основным источником получения германия являются отхо-
отходы цинкового производства и коксования каменных углей. Германий получают
обработкой оксидного германийсодержащего сырья крепким раствором соляной
кислоты:
GeO2 + 4НС1 = GeCl4 + 2Н2О
В дальнейшем тетрахлорид германия (легкокипящая жидкость) подвергают
ректификационной очистке, гидролизу, а образующийся в результате чистый
GeO2 восстанавливают водородом:
GeO2 + 2Н2 = Ge + 2Н2О
Поскольку германий используется в основном как полупроводниковый материал в
виде монокристаллов, полученный после восстановления порошок сплавляют в
атмосфере водорода и подвергают направленной кристаллизации или зонной
плавке. При этом происходит дополнительная глубокая очистка германия.
Олово и свинец из кислородсодержащих соединений восстанавливают углем
или СО. Сульфидные минералы предварительно подвергаются окислительному
обжигу. Олово и свинец можно получить электролизом. Этот же процесс исполь-
используют для рафинирования этих металлов, полученных другими способами. Особо
чистые Sn и РЬ получают методом зонной плавки.
Физические и химические свойства. В компактном сос-
состоянии германий представляет собой хрупкое вещество серебристо-серого цвета с
металлическим блеском. При обычных условиях германий кристаллизуется в
структуре типа алмаза и обладает ярко выраженными полупроводниковыми свой-
свойствами. Однако при высоких давлениях германий претерпевает полиморфные
превращения, образуя сначала тетрагональную структуру /?-олова, а затем более
плотноупакованную О ЦК-структуру. Это сопровождается повышением координа-
координационного числа и появлением металлических свойств.
Олово — серебристо-белый легкоплавкий металл при обычных условиях. Ус-
Устойчивая при комнатной температуре тетрагональная /^-модификация олова при
13,2°С в равновесных условиях переходит в алмазоподобную а-модификацию.
Однако с заметной скоростью это превращение происходит при более низких
температурах порядка -ЗО...-4О°С. В ходе этого превращения происходит значи-
значительное увеличение удельного объема (на 25,6%), что обусловлено значительным
уменьшением координационного числа при переходе от плотноупакованной к
рыхлой алмазоподобной структуре. Этот фазовый переход инициируется и уско-
ускоряется при внесении затравки оюлова. При соприкосновении^белого олова с
серым при низких температурах процесс полиморфного превращения протекает
чрезвычайно быстро. Оловянные предметы при этом рассыпаются в порошок. Это
явление получило название "оловянной чумы".
381
Третий представитель этой подгруппы — свинец — представляет собой сереб-
серебристо-серый металл с синеватым отливом. Свинец в отличие от Ge и Sn не имеет
полиморфных модификаций и всегда кристаллизуется в плотноупакованной
ГЦК-структуре. Ниже приведены свойства элементов IVA-группы н простых
веществ на их основе:
Содержание в земной коре.
масс, доли, %
Валентная электронная кон-
конфигурация
Атомный ргциус. нм
Ионный радиу 34~, нм
Ионный ралнус Э'1+, нм
Потенциалы ионизации, В
1\. Э —> 3+ ¦•г г
h з+ —> з2+ + е-
/3: 32+ -> Э?* + е
74: 33t -> 3!+ + е
озо
Температура, плавления, "С
Температура кипения, °С
Плотность, г/см*
Твердость, кг/мм2
Ширина запрещенной зоны, эВ
FC?p/3), В
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конфигурация
Атомчый радиус, нм
Ионный радиус Э4", нм
Ионный радиус Э4+, нм
Потенциалы ионизации, В
А: 3 -+ Э+ + е
/2: Э+ —> Э24 + е
13: Э2+ —0 Э3+ + е
[ ¦ з3+ —+ Э4+ + е"
ОЭО
Температура плавления, °С
Температура кипения, ° С
Плотность, г/см3
Твердость, кг/мм2
Ширина запрещенной зоны, эВ
V n-tv /'
го»уэ). в
с
1-КГ1
\Ие}2^2р2
0,077
0.25С
0.015
П,2(Ю
24.382
47,883
•-•4,4«2
2.6
3747
3927
3.51 (алмаз)
Ю1 (алмаз)
5,2 (ачмаз)
-
Sri
4-Ю
[Kr]4rflo56-M]
0,158
0,294
0,071
7,344
14,632
30,502
40,73
1,8
231,9
2337
5,8
30,2
0,08 (a-Sn) «
0,009
-0,136
Si
27,6
fNe]3523^
0.118
0,271
0,041
8,151
16,342
33,530
45,141
1,9
1414
3249
2.33
980
1,21
-
Ge
7-10'4
[Ar]3fPCl4s
0,139
0,272
0,053
7,899
15.934
34,230
45,141
2,0
937,4
2847
5,32
385
0,78
0,2
Pb
l,6-10-J
;Xe]4/45rf106s2
0.175
0,313
0,084
7,417
15,032
31,981
42,32
1,7
327,4
1751
11*34
3,9
-
0,8
-0,126
382
Сравнение твердости германия, кремния и алмаза позволяет сделать вывод,
что в германии уже наблюдается некоторая металлизация связи. Это подтвержда-
подтверждается и более низкой температурой плавления германия по сравнению с кремнием
и алмазом, а также меньшей шириной запрещенной зоны и большей электричес-
электрической проводимостью. Тем не менее германий является типичным полупроводником
вплоть до температуры плавления.
Химические свойства простых веществ также подтверждают неуклонное нарас-
нарастание металлических свойств в ряду Ge—Sn—РЬ. При обычных условиях все три
вещества устойчивы по отношению к воде и воздуху. Однако свинец на воздухе
быстро покрывается матовой оксидной пленкой, предохраняющей его от дальней-
дальнейшего окисления. Очень тонкие пассивирующие пленки оксидов всегда присутст-
присутствуют и на поверхности германия и олова. При нагревании все эти вещества сое-
соединяются с кислородом воздуха, образуя оксиды Ge(+4), Sn(-H), но Pb(-f 2), что
опять-таки указывает на большую близость олова к германию, чем к свинцу. В
ряду стандартных электродных потенциалов германий стоит после водорода,
между медью и серебром. Поэтому с разбавленными if концентрированными раст-
растворами кислот, не являющихся одновременно окислителями, он ко реагирует.
Олово и свинец стоят непосредственно перед водородом. Олово медленно раство-
растворяется в разбавленной IIG1 и легко в концентрированной с выделением водорода.
При этом в концентрированной НС1 образуется анионный комплекс:
Sii f 4HC1 = H2[SnCl,,] + Н2
При растворении свинца в концентрированной IICI также образуется комплексная
кислота H2[PbClj- В концентрированной HNO3 германий и олово образуют гер-
германиевые и оловянные кислоты яЭО^уНгО. Напротив, в разбавленной 1ЩОз
олово проявляет металлические свойства, выступая в качестве катионообразова-
теля:
3Sn + 8HNO3 = 3Sn(NO3J + 2NO + 4Н2О
Свинец же подобным образом взаимодействует с азотной кислотой любой кон-
концентрации, образуя РЬ(ГЮзJ. Таким образом, олово здесь также занимает проме-
промежуточное положение, реагируя с концентрированной HNO3 подобно германию, а с
разбавленной — подобно свинцу.
С растворами щелочей олово и свинец реагируют с выделением водорода
лишь при нагревании, образуя соответствующие гидроксокомплексы, например
Э + 2КОН + 2Н2О = К2[Э(ОНL] + Н2
Это подтверждает амфотеряую природу и свинца и особенно олова, поскольку
для него эта реакция более характерна. Германий взаимодействует со щелочами
только в присутствии окислителей. Лучшими реагентами для перевода германия
в раствор является смесь HF + HNO3: ^
3Ge + 4HNO3 + 18HF = 3,H2[GeF6] + 4NO + 8Н2О
Образующийся гексафторидный комплекс аналогичен таковому для кремния.
Все три вещества непосредственно взаимодействуют с галогенами и с халькоге-
нами, а германий и олово, кроме того, с фосфором и мышьяком.
Характеристические соединения. Гидриды элементов
подгруппы германия общей формулы ЭН2 не характерны. Напротив, высшие
гидриды элементов ЭН4 (кроме РЬН4) хорошо изучены. Хотя непосредственно с
водородом эти элементы не взаимодействуют, косвенным путем получены гидри-
гидриды германия — германы — вплоть до GegH^, а для олова известны гидриды SnH4
(станнан), Sn2H6 (дистаинан). Существование гидрида свинца сомнительно. Кос-
Косвенным признаком возможности образования РЬН4 (плюмбана) является заметная
летучесть свинца в токе водорода. Способность к образованию гомоатомных цепей
Э—Э быстро снижается от германия к свинцу, термическая устойчивость умень-
уменьшается в этом же направлении. Все гидриды элементов подгруппы германия
получают при разложении кислотами их соединений с активными металлами,
например
Mg2Ge + 4HCI = 2MgCI2 + GeH4
Водой эти гидриды быстро гидролизуются:
GeH4 + 6Н2О = H2[Ge(OHN] + 4Н2
Кроме того, они энергично окисляются кислородом воздуха (самовоспламенение):
GeH4 + 20, = GeO2 + 2Н2О
Известны два ряда характеристических оксидов и гидроксидов, отвечающих
степеням окисления Э(+2) и Э(-Н). При нагревании простых веществ на воздухе
образуются диоксиды германия и олова — GeO2 и SnO2, но монооксид свинца
РЬО. Оксиды GeO и SnO менее стабильны. GeO является полупродуктом терми-
термического окисления германия при сравнитачьно низких температурах (порядка
600°С) и обладает повышенной летучестью (сублимирует при 710°С). Тем не
менее низший оксид германия существенно более стабилен, чем оксид кремния
SiO. Оксид олова SnO при 400°С начинает диспропорционировать:
4SnO = Sn + 2SnO-SnO2(Sn3O4)
Диоксид свинца можно получить лишь косвенным путем: при нагревании он
диссоциирует ступенчато с образованием низших оксидов:
РЬО2
РЬ2О3
РЬ3О4
>РЬО
Промежуточные оксиды РЬ2О3 и РЬ3О4 следует рассматривать как смешанные
оксиды свинца РЬО-РЬО2 и 2РЬО-РЬ02.
Стабильность оксидов элементов подгруппы германия можно проиллюстриро-
проиллюстрировать схемой
усиление окислительных свойств
GeO2
SnO2
окисление
GeO
PbO2
I восстановление
РЬО
усиление восстановительных свойств
Таким образом, в ряду Ge — Sn — Pb происходит увеличение стабильности низ-
низшей степени окисления в полном соответствии с нарастанием металлических
свойств.
Все оксиды элементов подгруппы германия мало растворимы в воде, поэтому
соответствующие им гидроксиды могут быть получены косвенным путем. Гидрок-
сиды Э(ОНJ на самом деле представляют собой гидратные формы с переменным
содержанием воды ЭО-гЩО. Высшие оксиды ЭОг в кислотах трудно раствори-
растворимы. Напротив, со щелочами они взаимодействуют лучше. При этом образуются
соответственно германаты, станнаты и плюмбаты, которые в водных растворах
представляют собой гидроксокомплексы, например
GeO2 + 2NaOH + 2H2O = Na2[Ge(OHN]
Эти особенности кислотно-основного взаимодействия ярче выражены для гидрок-
сидов, поскольку гидратированные формы оксидов более реакционноспособны.
Все гидроксиды Э(ОНJ и Э(ОНL амфотерны. В ряду низших гидроксидов при
переходе от Ge к РЬ усиливаются основные свойства, а у высших гидроксидов
нарастают кислотные свойства в обратном направлении.
Аналогично Э(ОНL, низшие гидроксиды при взаимодействии со щелочами
также образуют в растворе гидроксокомплексы [Э(ОНL]2~ и соответствующие
гидроксосоли — германиты, станниты, плюмбиты, например Na2[Sn(OHL]. В
твердом состоянии для гидроксосолей Ме2[Э(ОН)б] и Ме2[Э(ОНL] характерны
безводные метаформы МегЭОз и Ме2ЭС>2.
Германиты и станниты как производные низших степеней окисления явля-
являются сильными восстановителями:
3Na2[Sn(OHL] + 2Bi(OHK - 2Bi + 3Na2[Sn(OHN]
Для плюмбитов такие реакции не характерны, так как степень окисления +2 для
свинца является устойчивой. Напротив, плюмбаты должны были бы выступать
как сильные окислители. Однако они существуют в щелочной среде, а, как из-
известно, в щелочных средах окислительные свойства кислородсодержащих соеди-
соединений выражены слабо, в то время как восстановительные усиливаются. Именно
поэтому восстановительные свойства ярче проявляются у германитов и станнитов,
а окислительные свойства РЬ(+4) — в кислой среде.
Поскольку высшие гидроксиды германия и олова, как правило, не отвечают
простым формам Н2ЭО3 или Н4ЭО4, а являются гидратами переменного состава
9O2*iH2O, их свойства сильно зависят от количества присоединенной воды.
Особенно это характерно для оловянных кислот. В зависимости от условий полу-
получения их свойства могут сильно различаться. Так, свежеосажденный гидроксид
олова (+4) (а-оловянная кислота) растворяется в концентрированной НС1 с обра-
образованием Н2[БпС1б], со щелочами образует станнат Na2[Sn(OH)e]. С течением
времени гелеобразный осадок о-оловянной кислоты теряет способность взаимо-
взаимодействовать с НС1 и не реагирует со щелочами. Такой гидроксиД*8п(+4), поте-
потерявший часть воды, называется /^-оловянной кислотой. Причиной различия
свойств-»- и /^-оловянных кислот является именно дегидратация геля, сопровож-
сопровождающаяся частичной кристаллизацией SnO2, устойчивого к действию кислот и
щелочей.
\\\
Элементы подгруппы германия непосредственно взаимодействуют с гапогена-
ми. При этом образуются галогениды германия и олова ЭГ4, но РЬГ2. Косвенным
путем могут быть получены и менее характерные GeF2 и SnF2, а также РЬГ4
(кроме РЬВг4 и РЫ4).
Все дигалогениды германия очень неустойчивы, являются сильными восстано-
восстановителями и дигпропорционируют:
2Ger2 *=±
GeF4
Устойчивость дигалогенидов олова несколько выше, хотя они также являются
сильными восстановителями. Напротив, РЬГг являются устойчивыми соединения-
соединениями и восстановительной активностью не обладают.
Важно отметить, что все без исключения дигалогениды ЭГ2 представляют
собой твердые вещества, что говорит о преобладании свойств катионообразовате-
лей у Э(+2). Однако дигалогениды олова и особенно германия сравнительно
легкоплавки, т.е. представляют собой своеобразные молекулярные кристаллы,
состоящие из полимерных агрегатов, слабо связанных друг с другом. В противо-
противоположность им дигалогениды свинца являются тугоплавкими координационными
кристаллами со значительной долей ионности, т.е., по существу, представляют
собой типичные соли.
В противоположность дигалогенидам тетрагалогениды элементов подгруппы
германия представляют собой преимущественно ковалентные соединения. Среди
них встречаются и газы, и жидкости, и молекулярные кристаллы с невысокими
температурами плавления. Лишь PbF4 и в какой-то мере SnF4 по свойствам напо-
напоминают соли. Из-за неустойчивости степени окисления +4 для свинца тетрагало-
тетрагалогениды PbF4 и РЬСЦ являются окислителями.
Для элементов подгруппы германия помимо вр3-гибридизации возможно spz<P~
гибридное состояние с участием вакантных nrf-орбиталей. Это предопределяет
возможность образования дополнительных донорно-акцепторных связей и склон-
склонность к комплексообразованию:
2KF + 9F4 = K2[9F6]; 2HCI + SnCl4 = H2[SnCl6]
Отметим, что как для Sn(+4), так и для РЬ(+4) известны комплексы типа [ЭГ6]2"
для всех галогенид-ионов, хотя в свободном состоянии РЬВг4 и РЫ4 не существу-
существуют. Это обусловлено стабилизацией высшей степени окисления за счет комплек-
сообразования.
Все ЭГ4 склонны к глубокому гидролизу, поскольку для Э(+4) свойства ка-
тионообразователей, т.е. оснбвные свойства, выражены очень слабо. Формально их
можно рассматривать как соли очень слабого основания и сильной галогеноводо-
родной кислоты. Реакция гидролиза сильно экзотермична, и в итоге образуются
диоксиды элементов:
ЭС14 + 2Н2О = ЭО2 + 4НС1
Как продукты частичного гидролиза можно рассматривать и такие соединения,
характерные для химии элементов подгруппы германия, как оксогалогениды
ЭОГ2.
Соединения с другими неметаллами. Халькогениды
элементов подгруппы германия, как и оксиды, образуют два ряда: монохалькоге-
ниды ЭХ и дихалькогениды ЭХ2. Низшие халькогениды известны для всех эле-
элементов и всех халькогенов. Все монохалькогениды элементов можно получить как
непосредственным взаимодействием компонентов при нагревании, так и пропуска-
пропусканием сероводорода через водные растворы, содержащие ионы Э2+. Дисульфиды
германия и олова получают непосредственным взаимодействием компонентов при
повышенном давлении пара серы. Все монохалькогениды являются фазами пере-
переменного состава общей формулы ЭХця, хотя область гомогенности их невелика
(х < 1). Поэтому их свойства изменяются с составом и зависят от условий получе-
получения. Поскольку области однородности узки, небольшое изменение состава в пре-
пределах существования фазы приводит к резкому изменению свойств, особенно
электрических и оптических.
Дихалькогениды ЭХ2 известны только для Ge и Sn, с одной стороны, и для S,
Se — с другой. Дихалькогениды при повышенных температурах диссоциируют по
схеме
2ЭХ2 5=* 2ЭХ + Х2(г)
Все халькогениды ЭХ и ЭХ2 устойчивы при обычных условиях по отношению к
воде и кислороду воздуха. В концентрированных минеральных неокисляющих
кислотах ЭХ растворяются:
ЭХ + 2НС1 = ЭС12 + Н2Х
При этом олово и свинец образуют в растворе ацидокомплекс [ЭСЦ]2", который
для германия нехарактерен.
Моносульфиды германия и олова взаимодействуют с дисульфидом аммония
(NH4JS2 с образованием тиосолей (тиогерманаты и тиостаннаты):
9S + (NH4JS2 = (NH4h9S3
Дисульфиды GeS2 и SnS2 подобным же образом реагируют с моносульфидом
натрия или аммония:
9S2 + Na2S = Na29S3
Для олова известны также сульфиды состава Sn2S3 и Sn3S4, которые можно
рассматривать как смешанные сульфиды SnS-SnS2 и 2SnS-SnS2.
Соединения элементов подгруппы германия с пниктогенами известны далеко
не для всех элементов. Нитрид известен лишь для германия, причем его получа-
получают косвенным путем — нагреванием германия в токе аммиака:
3Ge + 4NH3 = Ge3N4 + 6Н2
Нитрид Ge3N4 представляет собой довольно устойчивое соединение. С водой,
разбавленными кислотами и щелочами не взаимодействует. При нагревании
начинает распадаться на элементы лишь при 1000°С. Кроме того, известен и
низший нитрид Ge3N2, который значительно менее стабилен (заметно диссоци-
диссоциирует при 5Q0°C) и легко гидролизуется. Нитриды олова такого же состава
(Sn3N4 и S113N2) отличаются меньшей устойчивостью. Так, Sn3N4 диссоциирует
уже при 360° С.
13*
387
в воде. Вследствие малой стабильности степени окисления +4 высший сульфат
Pb(SO4J можно получить в жестких условиях, например анодным окислением
PbSC>4 в сернокислотном растворе (этот процесс протекает при зарядке свинцово-
свинцового аккумулятора). Это соединение очень неустойчиво и легко гидролизуется:
Pb(SO4J + 2Н2О = РЬО2 + 2H2SO4
Нитраты германия неизвестны. При взаимодействии олова с HNO3 в зависи-
зависимости от ее концентрации и температуры можно получить Sn(NO3J и Sn(NO3).i:
4Sn + 10HNO3(pa36.) = 4Sn(NO3J + NH4NO3 + 3H2O @°C)
3Sn + 16HNO3(koh4.) = 3Sn(NO3L + 4NO + 8H2O @°C)
Нитрат свинца (+2) получают обычными способами, известными для солей.
Трехзамещенный фосфат РЬ3(РО4J образует лишь РЬ(+2). Для олова они
менее характерны, а для германия неизвестны.
Практически важным соединением свинца является его ацетат РЬ(СН3СООJ
(свинцовый сахар). Это одна из наиболее хорошо растворимых его солей, которая
используется при получении многих других производных свинца. Ацетат свинца
образуется при взаимодействии металлического свинца или РЬО с уксусной кис-
кислотой, например
РЬ + 2СН3СООН = РЬ(СН3СООJ + Н2
Таким образом, в подгруппе германия наиболее разнообразные соли образует
свинец (+2), что является следствием наиболее металличного характера этого
элемента в ряду Ge — Sn — РЬ.
Металлохимия элементов подгруппы германия.
Наиболее обычным для обсуждаемых элементов является образование эвтектичес-
эвтектических смесей и ограниченных твердых растворов, а также металлидов. Германий
образует непрерывный ряд твердых растворов только со своим ближайшим ана-
аналогом — кремнием, что обусловлено сходством кристаллохимического строения и
прежде всего характера химической связи в кристаллах этих веществ. Свинец и
олово не образуют непрерывных твердых растворов ни с одним из элементов
Периодической системы.
Все три элемента образуют разнообразные соединения с щелочными, щелочно-
щелочноземельными металлами и магнием. Как правило, в каждой из бинарных систем
образуется по нескольку соединений самого разнообразного состава, большинство
из которых — металлиды, за исключением германидов щелочных и щелочно-
щелочноземельных металлов, которые являются полупроводниками. Для всех элементов
подгруппы германия характерны и многочисленные соединения с d-металлами.
Здесь разнообразие металлидов обусловлено не столько свойствами элементов
подгруппы германия, сколько обширными металлохимическими возможностями
d-элементов.
Сравнивая химические и металлохимические свойства элементов, важно под-
подчеркнуть, что металлохимия фактически представляет собой часть химии элемен-
элементов и взаимодействие элементов подгруппы германия с металлами обусловлено
теми же их особенностями, что и взаимодействие с остальными элементами систе-
389
в воде. Вследствие малой стабильности степени окисления +4 высший сульфат
Pb(SO4J можно получить в жестких условиях, например анодным окислением
PbSO4 в сернокислотном растворе (этот процесс протекает при зарядке свинцово-
свинцового аккумулятора). Это соединение очень неустойчиво и легко гидролизуется:
Pb(SO4J + 2Н2О = РЬО2 + 2H2SO4
Нитраты германия неизвестны. При взаимодействии олова с HNO3 в зависи-
зависимости от ее концентрации и температуры можно получить Sn(NO3J и Sn(NO3L:
4Sn + 10HNO3(pa36.) = 4Sn(NO3J + NH4NO3 + 3H2O @°C)
3Sn + 16HNO3(koh4.) = 3Sn(NO3L + 4NO + 8H2O @"C)
Нитрат свинца (+2) получают обычными способами, известными для солей.
Трехзамещенный фосфат РЬ3(РО4J образует лишь Pb(+2). Для олова они
менее характерны, а для германия неизвестны.
Практически важным соединением свинца является его ацетат РЬ(СН3СООJ
(свинцовый сахар). Это одна из наиболее хорошо растворимых его солей, которая
используется при получении многих других производных свинца. Ацетат свинца
образуется при взаимодействии металлического свинца или РЬО с уксусной кис-
кислотой, например
РЬ + 2СН3СООН = РЬ(СН3СООJ + Н2
Таким образом, в подгруппе германия наиболее разнообразные соли образует
свинец (+2), что является следствием наиболее металличного характера этого
элемента в ряду Ge — Sn — Pb.
Металлохимия элементов подгруппы германия.
Наиболее обычным для обсуждаемых элементов является образование эвтектичес-
эвтектических смесей и ограниченных твердых растворов, а также металлидов. Германий
образует непрерывный ряд твердых растворов только со своим ближайшим ана-
аналогом — кремнием, что обусловлено сходством кристаллохимического строения и
прежде всего характера химической связи в кристаллах этих веществ. Свинец и
олово не образуют непрерывных твердых растворов ни с одним из элементов
Периодической системы.
Все три элемента образуют разнообразные соединения с щелочными, щелочно-
щелочноземельными металлами и магнием. Как правило, в каждой из бинарных систем
образуется по нескольку соединений самого разнообразного состава, большинство
из которых — металлиды, за исключением германидов щелочных и щелочно-
щелочноземельных металлов, которые являются полупроводниками. Для всех элементов
подгруппы германия характерны и многочисленные соединения с rf-металлами.
Здесь разнообразие металлидов обусловлено не столько свойствами элементов
подгруппы германия, сколько обширными металлохимическими возможностями
(^элементов.
Сравнивая химические и металлохимические свойства элементов, важно под-
подчеркнуть, что металлохимия фактически представляет собой часть химии элемен-
элементов и взаимодействие элементов подгруппы германия с металлами обусловлено
теми оке их особенностями, что и взаимодействие с остальными элементами систе-
389
мы. Амфотерная в целом природа Ge, Sn и Pb обусловливает сравнительно огра-
ограниченные металлохимические возможности, что в особенности относится к герма-
германию. Относительно высокое значение ОЭО, преимущественно ковалентный харак-
характер связей этого элемента, обусловленный склонностью к *р3-гибридизации, не
позволяют ему служить хорошим растворителем, и по той же причине он сам
плохо растворяется в металлах в твердом состоянии. Для олова и в особенности
для свинца это характерно в несколько меньшей степени.
4. Подгруппа титана. Общая характеристика элементов
подгруппы тит а,н а. Валентная электронная конфигурация элементов
IVB-группы (и — l)<Pns2. Наличие четырех валентных электронов предопределяет
возможность реализации высшей степени окисления +4, а энергетическая нерав-
неравноценность этих электронных состояний служит причиной проявления перемен-
переменных низших степеней окисления (+3 и +2), что характерно для титана. В бинар-
бинарных соединениях элементы подгруппы титана выступают исключительно в ка-
качестве катионообразователей. В то же время эти элементы образуют и комплекс-
комплексные катионы, и ацидокомплексы, что свидетельствует об их амфотерности в ши-
широком смысле слова.
Поскольку титан является кайносимметричным элементом, его 3(/-электроны
значительно прочнее связаны с атомным ядром. Поэтому 3-й и 4-й потенциалы
ионизации титана намного превосходят таковые его гомологов. Это обстоятельст-
обстоятельство является главной причиной проявления титаном и низших степеней окисле-
окисления. Цирконий и гафний преимущественно функционируют в характеристичес-
характеристической степени окисления +4, а производные низших степеней окисления неста-
нестабильны.
Ниже приведены некоторые свойства элементов подгруппы титана и их гомо-
атомных соединений:
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э4+, нм
Потенциалы ионизации, В
1\: Э -> Э+ + е
Н Э+ -> Э2+ + е
/3: Э2+ —> Э3+ + е
h\ Э3+ -> Э4+ + е
4
t = l '
ОЭО
Температура плавления, °С
Температура кипения, °С
Плотность, г/см3
Ti
6-Ю
Zr
2-Ю
Hf
3,2-10'4
В
Г(Э?р/Э), В
В
0,146
0,068
6,82
13,58
27,5
43,2
91,1
1,6
1668
3169
4,5
-1,17
-1,628
-1,21
[Кг]4<Р5«2
0,160
0,082
6,84
13,13
23,0
34,3
77,27
1,5
1855
4325
6,5
-1,529
-
_
[Хе]4/45
0,159
0,082
7,5
14,9
21,0
31,0
74,4
1,4
1949
5227
13,2
-1,70
-
300
Здесь уместно отметить одну важную особенность, свойственную всем элемен-
элементам побочных подгрупп, кроме ШВ-группы (подгруппа скандия): усиление хими-
химической благородности металлов в пределах группы с увеличением атомного номе-
номера элемента. В главных подгруппах и в подгруппе скандия сверху вниз нараста-
нарастают металлические свойства, а начиная именно с подгруппы титана наблюдается
обратная закономерность. С этой точки зрения, элементы IVB-группы, так же
как и элементы IVA-группы, являются своеобразной границей, разделяющей две
противоположные тенденции, Отмеченное обстоятельство связано с тем, что меж-
между ШВ- и IVB-группами вклиниваются семейства /-элементов, что наглядно отра-
отражается в развернутой C2-клеточной) форме системы. При этом валентные бе-
электроны тяжелых элементов подгрупп титана, ванадия и т.д., следующих за
лантаноидами, обнаруживают эффект проникновения сквозь двойной слой из bd-
и 4/-электронов. Этим и обусловлено ослабление металлических свойств гафния,
тантала, вольфрама и т.д. На этой особенности основана интерпретация законо-
закономерностей изменения степеней окисления, кислотно-основных и окислительно-
восстановительных свойств в группах (/-элементов.
Природные соединения и получение металлов
подгруппы титана. Титан и его аналоги достаточно широко распрост-
распространены в природе. Важнейшим природным минералом титана является его диок-
диоксид ТЮг, встречающийся в виде трех полиморфных модификаций: рутила, ана-
таза и брукита. Кроме того, известны и соединения FeTiO3 и СаТЮз — минералы
ильменит и перовскит. Циркониевая земля, или баддалеит, ZrC>2, а также циркон
ZrSiO4 встречаются достаточно часто. Гафний как элемент-близнец циркония
всегда сопутствует ему в цирконе, баддалеите и других минералах (например, до
5,5% НЮ2 в ZrO2).
Титан и цирконий получают восстановлением их тетрахлоридов расплавлен-
расплавленным магнием. В последнее время широко развивается метод иодидного рафини-
рафинирования титана и циркония. Метод основан на термической диссоциации летучих
тетраиодидов металлов на раскаленной до 1800°С вольфрамовой нити. При этом
нить обрастает кристаллами металла высокой степени чистоты. Полученные грут-
ки обладают хорошими механическими свойствами, ковкостью в холодном сос-
состоянии, высокой пластичностью. Гафний получают аналогичным способом.
Физические и химические свойства. Гомоатомные соеди-
соединения всех трех элементов представляют собой тугоплавкие серебристо-белые
металлы, обладающие высокой пластичностью, ковкостью, износоустойчивостью.
Характерной особенностью всех трех металлов является полиморфизм. При обыч-
обычных условиях они кристаллизуются в ГПУ-структуре. С повышением температу-
температуры увеличивается энтропия и происходит перестройка в более рыхлую структуру
ОЦК. Эта закономерность является общей для металлов: высокотемпературные
модификации являются, как правило, менее плотноупакованными.
Следует также отметить высокие температуры плавления титана и его анало-
аналогов, что свидетельствует о металлоковалентном (а не чисто металлическом) харак-
характере связей в кристаллах. При этом температура плавления в ряду Ti — Zr — Hf
возрастает в противоположность закономерности, наблюдающейся в главной
подгруппе (в ряду С — Si — Ge — Sn — Pb).
В ряду стандартных электродных потенциалов все три элемента расположены
до водорода. Однако они вполне устойчивы по отношению к воде и минеральным
кислотам, за исключением HF и горячей Н3РО4- При нагревании вещества прак-
391
тически не взаимодействуют"с концентрированными H2SO4 и HNC3. Эта особен-
особенность поведения титана и его аналогов обусловлена наличием на их поверхности
тонкой пассивирующей пленки оксидов. Поэтому энергичные окислители
[НгБО^конц.), 1ШОз(конц.)] способствуют лишь уплотнению этой пленки, кото-
которая предохраняет металл от воздействия реагента.
Такие кислоты, как HF, Н2С2О4 и т.п., являющиеся источниками кислотных
остатков для образования прочных ацидокомплексов, растворяют пассивирую-
пассивирующую пленку. Схема взаимодействия выглядит так:
Э02 + 6HF = H2[3F6] + 2Н2О или Э02 + ЗН2С2О4 = Н2[Э(С2О4)з] + 2Н2О
Далее происходит растворение металлов:
Э + 6HF = H2[3F6] + 2Н2 или Э + ЗН2С2О4 = Н2[Э(С2О4)з] + 2Н2
В смесях кислот, одна из которых является окислителем, а другая — источником
лигандов (HNO3 + НС1, HNO3 + HF), процесс растворения суммарно выглядит
следующим образом:
ЗЭ + 4HNO3 + 18НГ = ЗН2[ЭГ6] + 4N0 + 8Н2О
По отношению к растворам щелочей компактные металлы подгруппы титана
устойчивы. Это объясняется слабо выраженными кислотными свойствами оксидов
Э02. Поэтому гидроксокомплексы для них не характерны. Однако в расплавах
щелочей и хлоридов щелочных металлов на воздухе (в присутствии кислорода)
металлы сильно корродируют за счет образования титанатов, цирконатов и гаф-
натов (оксокомплексов).
Характеристические соединения. При нагревании в атмо-
атмосфере кислорода титан, цирконий и гафний сгорают с образованием диоксидов
ЭО2. Стандартные энтальпии образования высших оксидов из простых веществ
возрастают в ряду Ti — Zr — Hf, что свидетельствует о стабилизации высшей
степени окисления при переходе от Ti к Hf:
Соединения
ff 298,
ТЮ2
Alff 298, кДж/моль -943,5
ZrO2
-1080,3
НГО2
-1135,9
Кроме того, для титана известны и низшие оксиды Ti2O3 и TiO. Первый из
них может быть получен восстановлением диоксида водородом:
2ТЮ2 + Н2 = Ti2O3 + Н2О
Второй получают реакцией конпропорционирования:
TiO2 + Ti = 2TiO
Оксид Ti2O3 напоминает по свойствам А12Оз (диагональное сходство). Так, про-
прокаленный Ti2O3 индифферентен по отношению к кислотам и щелочам. TiO —
типичный бертоллид, его состав и свойства сильно зависят от условий получения.
Все три оксида могут быть получены препаративным путем. Низшие оксиды для
циркония мало характерны, а для гафния неизвестны.
Диоксиды титана и его аналогов обладают высокой инертностью и тугоплав-
тугоплавки. Их температуры плавления: ТЮ2 - 1855°С, ZrO2 - 2687°С, НЮ2 - 2790°С.
Поскольку эти оксиды характеризуются высокими значениями энтальпий образо-
образования, они стабильны по отношению к многим расплавленным металлам. Отвеча-
Отвечающие им гидратные формы могут быть получены только косвенным путем, на-
например
ЭС14 + 4NaOH = Э(ОНL + 4NaCl
Эти гидроксиды амфотерны. Однако для титана и основные, и особенно кислот-
кислотные свойства гидроксида выражены очень слабо. При переходе к Zr(OHL и
Hf(OHL основные свойства несколько усиливаются. Таким образом, несмотря на
повышение устойчивости высшей степени окисления в ряду Ti — Zr — Hf, основ-
основный характер высших гидроксидов в группе сверху вниз нарастает. При этом
стехиометрический состав Э(ОНL для гидроксидов титана и его аналогов являет-
является предельным. Фактически эти соединения имеют переменный состав ЭОг'^НгО,
зависящий от условий получения, и склонны к образованию коллоидных раст-
растворов.
Поскольку кислотные свойства для Э(ОНL выражены очень слабо, все они, в
том числе и Ti(OHL, не взаимодействуют с концентрированными растворами
щелочей. Эти гидроксиды при сплавлении со щелочами образуют два ряда со-
солей: орто- и мета-формы. Первые являются производными Н4ЭО4 (т.е.
ЭОг^НгО), например К4ТЮ4 — ортотитанат калия, Na4Zr04 — ортоцирконат
натрия. Вторые происходят от частично обезвоженной мета-формы гидроксида
Н2ЭО3 (т.е. ЭО2*Н2О), например Na2Zr03 — метацирконат натрия, BaTiOs —
метатитанат бария. Обычно производные мета-форм называются просто титана-
тами, цирконатами, гафнатами.
Так как для всех Э(ОНL основные свойства выражены сильнее кислотных, эти
гидроксиды (в свежеосажденном состоянии) растворяются в кислотах с образова-
образованием *олей, например Ti(SO4J, Zr(NO3L и т.п.
Для титана в отличие от его аналогов известен гидроксид Ti(OHK, обладаю-
обладающий только основными свойствами. Титан в степени окисления +3 неустойчив и
является сильным восстановителем, окисляясь в растворах кислородом воздуха:
2Ti(OHK + О2 + 2Н2О = 2Ti(OHL + Н2О2
Еще менее устойчива для титана степень окисления +2. Гидроксид такого соста-
состава неизвестен, а оксид TiO обладает настолько сильными восстановительными
свойствами, что, подобно металлам, вытесняет водород из разбавленных раство-
растворов кислот:
2ТЮ + 3H2SO4 = Ti2(SO4K + Н2 + 2Н2О-
Наиболее типичными галогенидами элементов подгруппы титана являются
соединения ЭГ4> отвечающие высшей степени окисления. Они известны для всех
металлов и всех галогенов. Обычно их получают либо непосредственным взаимо-
взаимодействием компонентов при нагревании, например
Ti + 2Вг2 = TiBr4
либо прокаливанием диоксидов углем в атмосфере галогена:
ЭО2 + 2С + 2Г2 = ЭГ4 + 2СО
Активность прямого взаимодействия увеличивается при переходе от титана к
цирконию и гафнию и уменьшается при переходе от фтора к иоду.
Все тетрагалогениды элементов IVB-группы бесцветны (кроме тетраиодидов и
тетрабромидов) и в обычных условиях представляют собой твердые вещества,
кроме TiCl4 (жидкость). Несмотря на это, их нельзя рассматривать как соли. Все
твердые галогениды при нагревании сублимируют и их можно расплавить только
под давлением собственного пара. В ряду TiF4 ~ ZrF4 — HIT4 температуры плав-
плавления однотипных галогенидов возрастают. Все это указывает на молекулярный
характер кристаллической решетки этих соединений. В молекулах ЭГ4 все четы-
четыре тетраэдрические связи имеют повышенный порядок за счет дополнительного
тг-связывания по донорно-акцепторному механизму. При этом тг-электронная
плотность распределена в пределах всей молекулы. Наличие вакантных d-
орбиталей и делокализованных тг-электронов в молекулах ЭГ4 способствует
проявлению тенденции к дополнительному координационному насыщению с
образованием прочных октаэдрических комплексов [ЭГб]2".
Все тетрагалогениды ЭГ4 гигроскопичны, а большинство из них (кроме поли-
полимерных фторидов) легко гидролизуются. Так как основный характер гидрокси-
дов Э(ОНL при переходе от титана к цирконию и гафнию несколько усиливает-
усиливается, степень гидролиза галогенидов в этом же ряду уменьшается. При гидролизе
производных Э(+4) образуются устойчивые оксокатионы:
Э4+ + Н20 = ЭО2+ + 2Н+
Эти ионы TiO2+, ZrO2+, HfO2+ называются соответственно титанил, цирконил,
гафнил. Они настолько устойчивы, что соответствующие галогенопроизводные,
например ТЮС1г (хлорид титанила), ZrOBr2 (бромид цирконила) и т.п., сущест-
существуют в твердом состоянии и обладают солеобразным характером. Образование
"ил"-катионов характерно для многих переходных и непереходных металлов
(например, хромил СгО2+, уранил UO|+, алюминил А1О+, висмутил BiO+ и т.д.), а
также для неметаллов (например, нитроил NO2, сульфурил SO2, тионил SO2+ и
т.д.). Объясняется это тем, что многозарядные положительные ионы нестабильны
и с присоединением кислорода самопроизвольно переходят в стабильные оксока-
оксокатионы.
Как уже отмечалось, низшие степени окисления (+2, +3) характерны в основ-
основном для титана. Низшие галогениды титана Т1Г2 и Т1Г3 известны для всех гало-
галогенов. Не существуют лишь TiF2. Т1Гг и ТЧГз обычно получают восстановлением
TiF4 водородом:
2TiCl4 + Н2 = 2TiCl3 + 2HC1
или реакцией конпропорционирования:
TiCl4 + Ti = 2TiCl2
TiBr2 и Til2 можно получить непосредственным синтезом из элементов. Бром и
иод в виде простых веществ не являются достаточно сильными окислителями,
чтобы перевести титан в более высокие степени окисления, если реакцию прово-
проводить в мягких условиях. В более жестких условиях получаются TiBr4 и ТИ4.
Низшие галогениды титана нестабильны и являются сильными восстановите-
лями. Это особенно относится к дигалогенидам, напоминающим субгалогениды
кремния и германия и также склонным к диспропорционированию.
Соединения с другими неметаллами. Элементы подгруп-
подгруппы титана при нагревании непосредственно взаимодействуют с халькогенами.
Наиболее типичны соединения состава ЭХ и ЭХг. Так, для титана известны все
шесть соединений, для циркония обнаружены ZrS, ZrS2, ZrSe2. Имеются сведения
о существовании анионоизбыточных сульфидов T1S3 и ZrS3 и соединений ТлгЗез и
Zr2S3- Свойства халькогенидов титана и циркония изучены недостаточно.
Еще меньше сведений о взаимодействии элементов подгруппы титана с пник-
тогенами, за исключением металлоподобных нитридов. Известны монофосфиды и
дифосфиды титана и циркония (TiP, T1P2, ZrP, ZrP2), характеризующиеся метал-
локовалентным типом связи. Монофосфиды примыкают к металлидам, а дифос-
дифосфиды — к полупроводникам. Фосфиды титана и циркония — тугоплавкие вещест-
вещества, устойчивые по отношению к воде и неокисляющим кислотам. Они обладают
высокой твердостью, вследствие чего насыщение поверхности металла фосфором
(фосфидизация) может быть использовано для повышения ее износостойкости.
Соли кислородсодержащих кислот. Соли кислородсодер-
кислородсодержащих кислот, в которых металлы подгруппы титана выполняют роль катионов,
мало характерны для этих элементов. Тем не менее сульфат титана (+4) в виде
нейтрального комплекса [Ti(SO4J] может быть получен насыщением раствора
TiCU триоксидом серы:
TiCL, + 4SO3 = [Ti(SO4J] + 2SO2C12
Эта реакция аналогична получению Ge(SO4J- Сульфат титана (+4) — бесцветное
гигроскопичное, вещество, весьма склонное к гидролизу с образованием сульфата
титанила TiOSCv2H2O. Последняя соль более характерна, чем собственно
Ti(SO4J- Сульфат титана (+3) Ti2(SO4K является сильным восстановителем.
Характерной его особенностью является образование двойных солей типа квас-
квасцов, например Rb2SCvTi2(SO4K-24H2O, Cs2SO4-Ti2(SO4K-24H2O, по свойствам
Е1апоминающих алюминиевые квасцы.
Нитраты для титана не характерны. Для циркония же известны нитрат
цирконила ZrO(NO3J-2H2O и особенно характерен нитрат дицирконила
Zr2O3(NO3J-5H2O — производное радикала Zr2Ojj* (первый представитель в ряду
изополиоксоловых радикалов циркония).
Для циркония и гафния в противополож-
противоположность титану особенно характерны фосфаты
типа Эз(РС*4L- Кроме того, существуют пиро-
фосфаты и ортосиликаты: ZrP2O7, HfP2O7,
ZrSiO4.
Металлохимия элементов
подгруппы титана. Титан, цирконий
и гафний образуют непрерывные твердые раст-
растворы друг с другом в обеих модификациях.
Тройная система Ti — Zr — Hf является единст-
единственным примером системы, в которой реализу-
реализуются два вида непрерывных твердых трехком-
понентных растворов в двух модификациях
Zr
Ti
Рис. 148. Непрерывная взаимная
растворимость в тройной системе
, титан - цирконий - гафний
(кислород), как и следовало ожидать, намного химически активнее серы. В V
группе, наоборот, второй типический элемент (фосфор, особенно белый) более
активен как гомоатомное соединение, чем азот. Дело в том, что образование сое-
соединений первого порядка — это процесс взаимодействия между атомами, а не
молекулами. Поэтому на химическую активность элемента (атома) решающее
влияние оказывает энергия диссоциации гомоатомных соединений на атомы. А
энтальпия диссоциации молекулы N2 на атомы в 1,5 раза больше этой величины
для молекул фосфора Р4 (с учетом энергии сублимации менее активного красного
фосфора). Это обстоятельство является основной причиной большей химической
активности фосфора по сравнению с азотом. В то же время атомы азота, естест-
естественно, химически гораздо активнее атомов фосфора. Так, ОЭО азота 3,0, а фос-
фосфора 2,1. Таким образом, когда речь идет о большей химической активности
фосфора по сравнению с азотом, нужно иметь в виду активность гомоатомных
соединений. Несмотря на имеющиеся различия между азотом и фосфором, оба
типических элемента и их производные — важнейшие составные части раститель-
растительных и животных организмов.
Как отмечалось выше, наименьшая разница в свойствах элементов двух под-
подгрупп, составляющих одну группу в Периодической системе, имеет место в III
группе. В IV группе подгруппа германия уже заметно отличается от подгруппы
титана. В V группе подгруппа мышьяка еще более отлична от подгруппы вана-
ванадия. С увеличением номера группы металлический характер элементов В-под-
групп сравнительно мало изменяется, так как электронами заселяются (п — l)d-
орбитали, т.е. второй слой от внешнего. А у элементов А-подгрупп электроны
заполняют внешние nsnp-орбитали, вследствие чего с увеличением номера группы
быстро нарастают неметаллические свойства.
1. Азот. Особенности азота. У атома азота на один электрон боль-
больше, чем у атома углерода; согласно правилу Гунда, этот электрон занимает пос-
последнюю вакантную 2р-орбиталь. Атом азота в невозбужденном состоянии харак-
характеризуется тремя вырожденными 2р-электронами при наличии двух спаренных
электронов на 2*-орбитали. Три неспаренных электрона на 2р-орбитали ответст-
ответственны прежде всего за трехковалентность азота. Именно поэтому характеристи-
характеристическим летучим водородным соединением азота является аммиак, в котором атом
азота образует три ковалентные связи по обменному механизму с тремя атомами
водорода. У азота нет возможности промотирования электронов с переходом в
возбужденное состояние, так как ближайшие орбитали при п = 3 слишком высо-
высоки по энергии. Поэтому максимальная валентность азота равна четырем. При
этом три ковалентные связи могут быть образованы по обменному механизму, а
одна — по донорно-акцепторному. Однако азот в состоянии N+ может образовать
все четыре связи по обменному механизму. Азот проявляет большое разнообразие
степеней окисления: -3, -2, -1, 0, +1, +2, +3, +4 и +5. Наиболее часто встреча-
встречаются производные от степеней окисления -3, +5 и +3.
Азот в природе и его получение. Содержание азота в зем-
земной коре в виде соединений составляет 0,01 масс, доли, %. Атмосфера более чем
на 75 масс, долей, % состоит из газообразного азота, что равно ~ 4-1015 т. Свя-
Связанный азот образует минералы в форме нитратов: чилийская NaNO3, индийская
КГТОз-и норвежская Ca(NO3J селитры. Азот в форме сложных органических
производных входит в состав белков, в связанном виде содержится в нефти (до
1,5 масс, доли, %), каменных углях (до 2,5 масс, доли, %).
407
(кислород), как и следовало ожидать, намного химически активнее серы. В V
группе, наоборот, второй типический элемент (фосфор, особенно белый) более
активен как гомоатомное соединение, чем азот. Дело в том, что образование сое-
соединений первого порядка — это процесс взаимодействия между атомами, а не
молекулами. Поэтому на химическую активность элемента (атома) решающее
влияние оказывает энергия диссоциации гомоатомных соединений на атомы. А
энтальпия диссоциации молекулы N2 на атомы в 1,5 раза больше этой величины
для молекул фосфора Р4 (с учетом энергии сублимации менее активного красного
фосфора). Это обстоятельство является основной причиной большей химической
активности фосфора по сравнению с азотом. В то же время атомы азота, естест-
естественно, химически гораздо активнее атомов фосфора. Так, ОЭО азота 3,0, а фос-
фосфора 2,1. Таким образом, когда речь идет о большей химической активности
фосфора по сравнению с азотом, нужно иметь в виду активность гомоатомных
соединений. Несмотря на имеющиеся различия между азотом и фосфором, оба
типических элемента и их производные — важнейшие составные части раститель-
растительных и животных организмов.
Как отмечалось выше, наименьшая разница в свойствах элементов двух под-
подгрупп, составляющих одну группу в Периодической системе, имеет место в III
группе. В IV группе подгруппа германия уже заметно отличается от подгруппы
титана. В V группе подгруппа мышьяка еще более отлична от подгруппы вана-
ванадия. С увеличением номера группы металлический характер элементов В-под-
групп сравнительно мало изменяется, так как электронами заселяются (п — \)d-
орбитали, т.е. второй слой от внешнего. А у элементов А-подгрупп электроны
заполняют внешние nsnp-орбитали, вследствие чего с увеличением номера группы
быстро нарастают неметаллические свойства.
1. Азот. Особенности азота. У атома азота на один электрон боль-
больше, чем у атома углерода; согласно правилу Гунда, этот электрон занимает пос-
последнюю вакантную 2р-орбиталь. Атом азота в невозбужденном состоянии харак-
характеризуется тремя вырожденными 2р-электронами при наличии двух спаренных
электронов на 2«-орбитали. Три неспаренных электрона на 2р-орбитали ответст-
ответственны прежде всего за трехковалентность азота. Именно поэтому характеристи-
характеристическим летучим водородным соединением азота является аммиак, в котором атом
азота образует три ковалентные связи по обменному механизму с тремя атомами
водорода. У азота нет возможности промотирования электронов с переходом в
возбужденное состояние, так как ближайшие орбитали при п — 3 слишком высо-
высоки по энергии. Поэтому максимальная валентность азота равна четырем. При
этом три ковалентные связи могут быть образованы по обменному механизму, а
одна — по донорно-акцепторному. Однако азот в состоянии N+ может образовать
все четыре связи по обменному механизму. Азот проявляет большое разнообразие
степеней окисления: -3, -2, -1, 0, +1, +2, +3, +4 и +5. Наиболее часто встреча-
встречаются производные от степеней окисления -3, +5 и +3.
Азот в природе и его получение. Содержание азота в зем-
земной коре в виде соединений составляет 0,01 масс, доли, %. Атмосфера более чем
на 75 масс, долей, % состоит из газообразного азота, что равно ~ 4-Ю15 т. Свя-
Связанный азот образует минералы в форме нитратов: чилийская NaNOg, индийская
KNO3-h норвежская СаA*ЮзJ селитры. Азот в форме сложных органических
производных входит в состав белков, в связанном виде содержится в нефти (до
1,5 масс, доли, %), каменных углях (до 2,5 масс, доли, %).
307
водородное соединение азота. По своей химической природе он представляет
собой нитрид водорода H3N. В химическом строении аммиака в^нгибридные
орбитали атома азота образуют три <г-связя с тремя атомами водорода, которые
занимают три вершины чуть искаженного тетраэдра. Четвертая вершина тетраэд-
тетраэдра занята неподеленной электронной парой азота, что обеспечивает химическую
ненасыщенность и реакционноспособность молекул аммиака, а также большую
величину электрического момента диполя (fi = 0,5-10~29 Кл-м).
При обычных условиях аммиак — бесцветный газ с резким запахом. Он токси-
токсичен: раздражает слизистые оболочки, а острое отравление вызывает поражение
глаз и воспаление легких. При охлаждении до -33°С аммиак сжижается, а при
-78"С затвердевает. В жидком и твердом аммиаке между молекулами действуют
водородные связи, вследствие чего аммиак обладает рядом экстремальных
свойств по сравнению с другими водородными соединениями элементов VA-rpyn-
пы. Энтальпия испарения сжиженного аммиака заметно превышает таковую боль-
большинства других сжиженных газов.
Вследствие полярности молекул и достаточно высокой диэлектрической про-
проницаемости жидкий аммиак является хорошим неводным растворителем. Жидкий
аммиак положил начало химии неводных растворов. Результаты исследования
поведения веществ в жидком аммиаке дали возможность построить обобщенную
теорию кислот и оснований, открыли перед химией новые пути проведения реак-
реакций синтеза ранее неизвестных веществ и т.д. В жидком аммиаке хорошо раство-
растворяются щелочные и щелочно-земельные металлы, сера, фосфор, иод, многие соли
и кислоты. Вещества с функциональными полярными группами в жидком амми-
аммиаке подвергаются электролитической диссоциации. Однако собственная иониза-
ионизация аммиака 21ЧН3(ж) ;f=± NH4 + NH2 ничтожно мала и ионное произведение
[NHJ][NH2 = 103 при -50°С.
По растворимости в воде аммиак превосходит любой другой газ: при 0°С один
объем воды поглощает 1200 объемов газообразного аммиака. Прекрасная раство-
растворимость аммиака в воде обусловлена возникновением межмолекулярных водород-
водородных связей. При этом возможны два механизма возникновения водородных свя-
связей между молекулами аммиака и воды:
H^i'F^-H-O: и
н н
Поскольку донорная способность молекулы аммиака выражена сильней, чем у
воды, а связь О—Н более полярна по сравнению с полярностью связи N—Н в
аммиаке, межмолекул яри ая водородная связь образуется по первому механизму.
Таким образом, физико-химические процессы в водном растворе аммиака можно
представить следующим образом:
NH3 + Н2О ¦ К= °'2 ' NH3 Н2О . К=
Возникновение гидроксид-ионов создает щелочную реакцию раствора аммиака в
воде. Константа ионизации невелика (рК 5)- В условиях пониженных температур
.чад
водородное соединение азота. По своей химической природе он представляет
собой нитрид водорода H3N. В химическом строении аммиака в^-гибридные
орбитали атома азота образуют три <г-связи с тремя атомами водорода, которые
занимают три вершины чуть искаженного тетраэдра. Четвертая вершина тетраэд-
тетраэдра занята неподеленной электронной парой азота, что обеспечивает химическую
ненасыщенность и реакционноспособность молекул аммиака, а также большую
величину электрического момента диполя (/* = 0,5-109 Кл-м).
При обычных условиях аммиак — бесцветный газ с резким запахом. Он токси-
токсичен: раздражает слизистые оболочки, а острое отравление вызывает поражение
глаз и воспаление легких. При охлаждений до -33° С аммиак сжижается, а при
-78°С затвердевает. В жидком и твердом аммиаке между молекулами действуют
водородные связи, вследствие чего аммиак обладает рядом экстремальных
свойств по сравнению с другими водородными соединениями элементов VA-rpyn-
пы. Энтальпия испарения сжиженного аммиака заметно превышает таковую боль-
большинства других сжиженных гадов.
Вследствие полярности молекул и достаточно высокой диэлектрической про-
проницаемости жидкий аммиак является хорошим неводным растворителем. Жидкий
аммиак положил начало химии неводных растворов. Результаты исследования
поведения веществ в жидком аммиаке дали возможность построить обобщенную
теорию кислот и оснований, открыли перед химией новые пути проведения реак-
реакций синтеза ранее неизвестных веществ и т.д. В жидком аммиаке хорошо раство-
растворяются щелочные и щелочно-земельные металлы, сера, фосфор, иод, многие соли
и кислоты. Вещества с функциональными полярными группами в жидком амми-
аммиаке подвергаются электролитической диссоциации. Однако собственная иониза-
ионизация аммиака 2NH3(«) ;f=± NH4 + NH2 ничтожно мала и ионное произгедение
= КГ33 при -50эС.
По растворимости в воде аммиак превосходит любой другой газ: при 0°С один
объем воды поглощает 1200 объемов газообразного аммиака. Прекрасная раство-
растворимость аммиака в воде обусловлена возникновением межмолекулярных водород-
водородных связей. При этом возможны два механизма возникновения водородных свя-
связей между молекулами аммиака и воды:
тт тт
н н
Поскольку донорная способность молекулы аммиака выражена сильней, чем у
воды, а связь О—Н более полярна по сравнению с полярностью связи N—Н в
аммиаке, межмолекулярная водородная связь образуется по первому механизму.
Таким образом, физико-химические процессы в водном растворе аммиака можно
представить следующим образом:
NH3-H2O
NH3 + Н2О . К= °'2 ' NH3^^H2O ¦. К== 1Г5 ' ш; + ОН-
Возникновение гидроксид-ионов создает щелочную реакцию раствора аммиака в
воде. Константа ионизации невелика (рА)- В условиях пониженных температур
чад
.чад
На воздухе аммиак не горит, но в атмосфере кислорода он окисляется до
свободного азота:
4NH3 + ЗО2 = 2N2 + 6Н26
Аммиак функционирует как восстановитель и в реакциях с другими окислителя-
окислителями. Реже аммиак выступает как окислитель, например
Na + NH3 = NaNH2 + У2Н2
В этой реакции металлический натрий вытесняет водород из жидкого аммиака.
При этом водород аммиака понижает свою степень окисления и аммиак играет
роль окислителя. С другой стороны, подобные реакции иллюстрируют проявле-
проявление аммиаком кислотных свойств. Амиды металлов, например NaNH2, являются
солями аммиака, отвечающими его кислотной функции. Совершенно очевидно,
что кислотная природа у аммиака выражена значительно слабее, чем у Н2О и HF.
Константа кислотной ионизации ничтожно мала (рА'а 35), а потому соли аммиака
как кислоты в воде нацело гидролизуются:
NaNH2 + Н2О = NaOH + NH3
Кислотной функции аммиака отвечают не только амиды, но и имиды и нитриды
металлов. Если в амидах замещен один атом водорода (NaNH2), в имидах — два
(L^NH), то в нитридах — все три (A1N).
При осторожном окислении аммиака мягким окислителем, например гипохло-
ритом натрия, получают другое водородное соединение аммиака — гидразин или
диамид:
2NH3 + NaOCl = N2H4 + Nad + H2O
Диамид представляет собой бесцветную, легко испаряемую токсичную жидкость с
высокой диэлектрической проницаемостью (е = 52 при 25°С). Гидразин, подобно
аммиаку, является хорошим ионизирующим растворителем.
По химическим свойствам гидразин во многом похож на аммиак. В водных
растворах гидразина также возникают водородные связи, как и в случае ам-
аммиака.
При взаимодействии гидразина с одной молекулой воды с участием водород-
водородной связи образуется катион [N2H5]+, а с двумя — [N2H6]2t. Существование гид-
роксидов катионов N2H6 и N2H5 как индивидуальных веществ не установлено
(аналогично NH4OH). Это понятно, ибо основная функция гидразина выражена
слабее (pA'i 7, рА'2 15), чем у аммиака (рА'5). Тем не менее известны два типа
солей гидразина, например N2H5C1 и N2HfiCl2.
При восстановлении раствора азотной кислоты атомарным водородом получа-
получается гидроксиламин:
HNO3 + 6Н = NH2OH + 2Н2О
Гидроксиламин — бесцветные кристаллы (^ = 33°С), термически нестойкие,
вышеЛ00°С взрываются. Водные растворы гидроксиламина более устойчивы. В
растворе также возникают межмолекулярные водородные связи и устанавливает-
устанавливается динамическое равновесие:
401
На воздухе аммиак не горит, но в атмосфере кислорода он окисляется до
свободного азота:
4NH3 + ЗО2 = 2N2 + 6Н26
Аммиак функционирует как восстановитель и в реакциях с другими окислителя-
окислителями. Реже аммиак выступает как окислитель, например
Na + NH3 = NaNH2 + V2H2
В этой реакции металлический натрий вытесняет водород из жидкого аммиака.
При этом водород аммиака понижает свою степень окисления и аммиак играет
роль окислителя. С другой стороны, подобные реакции иллюстрируют проявле-
проявление аммиаком кислотных свойств. Амиды металлов, например NaNH2, являются
солями аммиака, отвечающими его кислотной функции. Совершенно очевидно,
что кислотная природа у аммиака выражена значительно слабее, чем у Н2О и HF.
Константа кислотной ионизации ничтожно мала (рА'а 35), а потому соли аммиака
как кислоты в воде нацело гидролизуются:
NaNH2 + Н20 = NaOH + NH3
Кислотной функции аммиака отвечают не только амиды, но и имиды и нитриды
металлов. Если в амидах замещен один атом водорода (NaNH2), в имидах — два
(Li2NH), то в нитридах — все три (A1N).
При осторожном окислении аммиака мягким окислителем, например гипохло-
ритом натрия, получают другое водородное соединение аммиака — гидразин или
диамид:
2NH3 + NaOcl = N2H4 + Nad + H2O
Диамид представляет собой бесцветную, легко испаряемую токсичную жидкость с
высокой диэлектрической проницаемостью (е = 52 при 25°С). Гидразин, подобно
аммиаку, является хорошим ионизирующим растворителем.
По химическим свойствам гидразин во многом похож на аммиак. В водных
растворах гидразина также возникают водородные связи, как и в случае ам-
аммиака.
При взаимодействии гидразина с одной молекулой воды с участием водород-
водородной связи образуется катион [N2H5]+, а с двумя — [N2H6]2t- Существование гид-
роксидов катионов N2He+ и N2Hs как индивидуальных веществ не установлено
(аналогично NH4OH). Это понятно, ибо основная функция гидразина выражена
слабее (pA'i 7, рА*2 15), чем у аммиака (рА'5). Тем не менее известны два типа
солей гидразина, например N2HsCl и N2H6C12.
При восстановлении раствора азотной кислоты атомарным водородом получа-
получается гидроксиламин:
HNO3 + 6Н = NH2OH + 2Н2О
Гидроксиламин — бесцветные кристаллы (^ = 33°С), термически нестойкие,
вышеЛ00°С взрываются. Водные растворы гидроксиламина более устойчивы. В
растворе также возникают межмолекулярные водородные связи и устанавливает-
устанавливается динамическое равновесие:
401
н
NH2OH + Н20 *=± H-N : Н-б: ^^ [NH3OH]+ + ОН"
ОН
Н
Однако основная функция гидроксиламина выражена еще слабее (pA'j 8), чем у
аммиака и гидразина. С кислотами гидроксиламин дает соли гидроксиламмония.
Наиболее известным препаратом является хлорид гидроксиламмония [NH3OH]C1.
Растворы солей гидроксиламмония более устойчивы, чем твердые вещества, и
имеют кислую реакцию вследствие гидролиза.
Поскольку атом азота в гидроксиламине имеет степень окисления —1, он может
функционировать и как окислитель, и как восстановитель. Но для него более
характерны восстановительные свойства, особенно в щелочной среде.
Среди водородных соединений азота наименьшая отрицательная степень окис-
окисления а.юта представлена в азиде водорода HN3. В этом соединении степень
окисления азота равна —'/j. Необычная степэнь окисления обусловлена структур-
структурной неравноценностью атомов азота в этом веществе. С позиций МВС эта струк-
структурная неравноценность может быть представлена схемой
120-
0,125 нм О.Шнм
n
1 2 з
Главное в этой схеме — долокализация тг-сзязей вдоль прямой, соединяющей
атомы азота. Правомерность схемы доказывается расстояниями между атомами
азота 1 — 2 и 2 — 3, являющимися промежуточными между длинами связей
-N-N-и №=N.
Водный раствор HNg называется азотистоводородной кислотой. Она получает-
получается окислением гидразинч азотистой кислотой:
N2H4 + Н№2 = HN3 + 2Н2О
По силе она приближается к уксусной. В разбавленных растворах азотистоводо-
родная кислота медленно диспропорционирует:
HN3 + Н2О = N2 + NH2OH
В безводном состоянии она может взорваться не только при нагревании, но и от
сотрясения:
2HN3 - Н2 + 3N2
Смесь азотистоводородной и концентрированной соляной кислот способна раство-
растворять даже благородные металлы. Соли азотистоводородной кислоты — азиды —
по растворимости в воде похожи на галогениды. Так, азиды щелочных металлов
хорошо'растворяются в воде, AgN3, Pb(N3J и Hg(N3J — плохо. Азиды щелочных
и щелочно-земельных металлов при медленном нагревании устойчивы вплоть до
плавления. Азиды тяжелых металлов легко взрываются при ударе:
Pb(N3J = РЬ + 3N2
Кислородные соединения азота. С кислородом азот образу-
образует ряд оксидов. Исходные сведения о них приведены в табл. 26.
402
Таблица 26. Оксиды азота,
Формульны
состав
Степень окис-
окисления азота
Химическое строение
молекулы
Агрегатное
состояние
N,0
N0
N02
N2O4
N2O5
+1
+2
+3
+4
+4
+5
NO
0,113 нм 0,118 нм "
0,115 нм "
0,186 нм
о-
120»
о
,0:
.м 0,175 им ft
N N. J126»
# \
¦=>" О:
Бесцветный газ
To же
Голубое твердое ве-
вещество (ниже -100° С)
Бурый газ
Бесцветный газ
Бесцветные кристаллы
403
Оксид N2O (закись аздта, "веселящий газ", поскольку он обладает наркоти-
наркотическим действием) получают термическим разложением нитрата аммония или
нитрита гидроксиламмония:
-1+3 +1
[1ЧНзОН]1ЧО2 = N2O + 2Н2О (внутримолекулярное конпропорционирование)
1 2с- Т
Оксид азота (+1) — эндотермическое соединение. Однако при комнатной темпе-
температуре химически мало активен. При нагревании его реакционная способность
сильно возрастает. Он окисляет водород, металлы, фосфор, серу, уголь, органи-
органические и другие вещества, например
Си + N20 = N2 + CuO
При нагревании N2O выше 700° С одновременно с реакцией разложения протекает
его диспропорционирование:
2N2O = 2N2 + O2; 2N2O = 2NO + N2
С водой оксид азота (+1) не взаимодействует, хотя известна кислота H2N2O2, в
которой азот также имеет степень окисления +1. Эта кислота называется азотно-
азотноватистой, и ей приписывается структура с двумя равноценными атомами азота:
H-O-N=N-O-H
Свободную азотноватистую кислоту можно получить так:
Н2О
NH2OH
I 2e-
HNO2 = H2N2O2
Она хорошо растворяется в воде, но кислота слабая (рК\ 8, а рА'2 12). Азотнова-
Азотноватистая кислота весьма неустойчива, при незначительном нагреве взрывается:
H2N2O2 = N2O + Н2О
Соли H2N2O2 — гипонитриты и гидрогипонитриты — в воде сильно подверже-
подвержены гидролизу. Большинство гипонитритов мало растворимо в воде, намного луч-
лучше растворяются гидрогипонитриты.
Четные степени окисления для азота сравнительно мало характерны. К числу
таких соединений относится оксид азота (+2). Молекула N0 содержит нечетное
число электронов и, по существу, представляет собой обладающий малой актив-
активностью радикал. В молекуле одна ковалентная связь по донорно-акцепторному
механизму и две тг-связи. Несмотря на эндотермичность и положительную вели-
величину энергии Гиббса образования N0 из простых веществ, оксид азота (+2) не
распадается на элементы. Дело в том, что, согласно ММО, порядок связи в N0
довольно высок и равен 2,5. Молекула N0 прочнее молекулы О2, так как у пер-
первой на разрыхляющей МО ж* всего один электрон, а у второй — два электрона.
404
В лаборатории оксид азота (+2) чаще всего получают действием разбавленной
азотной кислоты на медные стружки:
ЗСи + 8HNO3(pas6.) = 3Cu(NO3J + 2NO + 4Н2О
На воздухе оксид азота (+2) мгновенно окисляется:
2NO + О2 = 2NO2 (АЯ;98 = -113,4 кДж/моль)
Окисляется N0 и галогенами, образуя нитрозилгалогениды:
2N0 + Г2 = 2N0r
При взаимодействии с восстановителями N0 восстанавливается до N20, N2)
NH2OH, NH3 в зависимости от восстановительной способности партнера и условий
проведения процессов.
Водный раствор ококда¦чжк'в.-D 2} • жжсраявн. -й'скаких .саздагишкй г-&едо& or
не образует, хотя известны соли (гипонитраты) не выделенной в свободном сос-
состоянии азотноватой кислоты H2N2O3, в которой азот также имеет степень окисле-
окисления +2.
Оксид азота N2O3 существует в твердом состоянии (ниже -100°С). В жидком и
парообразном состояниях оксид азота (+3) в значительной степени диссоцииро-
диссоциирован за счет диспропорционирования:
N2O3 ^ NO + N02 (АЯ;98 = -41,2 кДж/моль)
Получают N2O3 охлаждением эквимолярных количеств N0 и N02. А равномер-
равномерный ток смеси нужного состава получается при взаимодействии 50%-ной азотной
кислоты с оксидом мышьяка (+3):
2HNO3 + As2O3 = 2HAsO3 + NO + N02
Оксиду азота (+3) отвечает известная лишь в растворе неустойчивая азотис-
азотистая кислота HNO2. Получить ее можно растворением равных объемов N0 и N02 в
воде:
N0 + N02 + Н20 = 2HNO2
При хранении и нагревании HNO2 диспропорционирует:
3HNO2 = HNO3 + 2N0 + Н20
Наиболее характерны для нее окислительные свойства:
2HNO2 + 2HI = 12 + 2N0 + 2Н2О
Однако сильные окислители переводят азотистую кислоту в азотную:
5HNO2 + 2КМпО4 + 3H2SO4 = K2SO4 + 2MnSO4 + 5HNO3 + 3H2O
Для азотистой кислоты известны две таутомерные структуры:
• 0,117 нм "
405
Соли азотистой кислоты — нитриты — производятся от обеих форм. Нитриты
активных металлов генетически связаны с первой структурой (связь через кисло-
кислород), а нитриты тяжелых металлов — со второй, т.е. металл непосредственно
связан с азотом.
Азотистая кислота является амфолитом с более сильно выраженной кислотной
функцией (рА'а 4). В ее водных растворах существуют равновесия:
NO+ 4- ОН- ^=* HNO2 ^НЧ N02
Ион N0+ — нитрозил или нитрозоний — изоэлектронен молекуле азота и чрезвы-
чрезвычайно устойчив. Производные азотистой кислоты, отвечающие ее основной функ-
функции, содержащие группу [N0]+, называются нитрозильными соединениями. Так,
нитрозилсерная кислота NOHSO4 является важным промежуточным продуктом
при нитрозном способе получения серной кислоты.
Оксид азота (+4) получают растворением меди в концентрированной азотной
кислоте:
Си + 4HNO3 = Cu(NO3J + 2NO2 -f 2H2O
Он является хорошим окислителем, в нем горят фосфор, сера, уголь и некоторые
органические вещества. Выше 150°С диоксид азота начинает разлагаться:
2NO2 = 2N0 + О2.
Поскольку молекула диоксида азота с неспаренным электроном по существу
представляет собой радикал, она легко димеризуется:
2NO2
N2O
2O4
= -58 кДж)
Димер бесцветен и диамагнитен в отличие от окрашенного в красно-бурый цвет и
парамагнитного NO2.
Диоксид азота при взаимодействии с водой диспропорционирует:
2NO2 + Н2О = HNO3 4- HNO2
При растворении NO2 в горячей воде получается азотная кислота, ибо первона-
первоначально образующаяся азотистая кислота диспропорционирует с выделением
оксида азота (+2) и образованием HNO3.
Оксид азота (+5) имеет молекулярную структуру только в газовой фазе. В
твердом состоянии N2Os имеет структуру, образованную ионами NO2 и NO3. N2Os
— легко возгоняющиеся кристаллы, причем испаряются молекулы. Таким обра-
образом, при возгонке оксида азота (+5) ионы NO2 и NO3 объединяются в молекулы
N2O5- Получают оксид азота (+5) дегидратацией азотной кислоты с помощью
Р2О5 или окислением NO2 озоном:
2HNO3 + Р2О5 = 2НРО3 + N2O5; 6NO2 + О3 = 3N2OS
Оксид азота (+5) является энергичным окислителем, многие реакции с его учас-
участием протекают весьма бурно. При растворении в воде дает азотную кислоту:
N2O5 + Н2О = 2HNO3
406
Азотная кислота — одна из сильных кислот. Молекула HNO3 и нитрат-ион
имеют строение, представленное схемами
Н
.0: "
120°
О: J
Безводная азотная кислота представляет собой бесцветную летучую жидкость.
При хранении (особенно на свету) и при нагревании частично разлагается:
4HNO3 = 4NO2 + 2Н2О -f O2
Так называемая "дымящая" азотная кислота (красного цвета) представляет собой
раствор выделяющегося диоксида азота в концентрированной HNOy.
В лаборатории HNO3 получают нагреванием нитрата натрия с серной кис-
кислотой:
NaNO3 + H2SO4 = HNO3 + N.1HSO4
В промышленности азотную кислоту получают из аммиака. Сначала аммиак
каталитически окисляют до оксида азота (+2), который далее окисляется до
NO2. Затем оксид азота (+4) растворяют в горячей воде и получают азотную
кислоту.
Азотная кислота является сильным окислителем и окисляет почти все метал-
металлы и неметаллы. Последние, как правило, переводятся ею в производные высшей
степени окисления, например
S + 6HNO3 = H2SO4 + 6NO2 + 2Н2О
Из металлов только золото, платина, осмий, иридий, ниобий, тантял и вольфрам
устойчивы к действию азотной кислоты. Некоторые металлы (например, Fe, A1,
Сг) пассивируются концентрированной азотной кислотой. Окислительными свой-
свойствами обладают и водные растворы азотной кислоты.
Обычно процесс восстановления HNO3 протекает в
нескольких параллельных направлениях, и в резуль-
результате получается смесь различных продуктов восста-
восстановления. Природа этих продуктов, их относительное
содержание в смеси зависят от силы восстановителя,
концентрации азотной кислоты и температуры. Рис.
149 иллюстрирует относительное содержание продук-
продуктов восстановления азотной кислоты железом в зави-
зависимости от ее концентрации. Чем разбавленнее кисло-
кислота, тем больше в смеси продуктов более глубокого
восстановления, например NH3, который с азотной
кислотой дает соль NH4NO3. В разбавленных раство-
растворах концентрация нитрат-иона меньше и каждый
атом азота D-5) принимает больше электронов от
1
J
80
60
20
16 31 W 6k
Конц. НН0,,мас. доли,%
J
Рис. 149. Продукты восста-
восстановления азотной кислоты в
зависимости от ее концент-
концентрации при взаимодействии с
железом
407
восстановителя, а потому происходит более глубокое восстановление. В концент-
концентрированных растворах, наоборот, много атомов азота (+5), в результате чего
каждая молекула HNO3 может принять меньше электронов от восстановителя.
Отсюда главными продуктами восстановления в концентрированных растворах
азотной кислоты являются N0 и NO2.
Более сильным окислителем является смесь концентрированных азотной и
соляной кислот — "царская водка". Она растворяет даже золото и платину,
которые не растворяются в азотной, а тем более в соляной кислоте. Ее окисли-
окислительная активность обусловлена снижением редокс-потенциала растворяющихся
металлов, т.е. усилением их восстановительных свойств за счет образования
прочных хлоридных комплексов (см. реакцию растворения золота в царской
водке на с. 314).
Соли азотной кислоты — нитраты — известны почти для всех металлов. Боль-
Большинство из них бесцветны и хорошо растворяются в воде. В кислых водных
растворах нитраты являются более слабыми окислителями, чем азотная кислота,
а в нейтральной среде вообще не обладают окислительными свойствами. Сильны-
Сильными окислителями они являются в расплавах, когда происходит разложение с
выделением кислорода. Оксид азота (+5) при взаимодействии со 100%-ным пе-
роксидом водорода образует пероксоазотную (надазотную) кислоту:
N2O5 + 2Н2О2 = 2HNO4 + Н20
Пероксоазотная кислота нестойка, легко взрывается, водой полностью гидролизу-
ется:
H-O-O-N
Н20 = Н2О2 + HNO3
Соединения с другими неметаллами. Известны все гало-
гениды азота NT3. Трифторид NF3 (ДЯ^298 = -117,2 кДж/моль) получают взаи-
взаимодействием фтора с аммиаком:
3F2 + 4NH3 = 3NH4F + NF3
Трифторид азота — бесцветный токсичный газ, молекулы которого обладают
пирамидальным строением. У основания пирамиды дислоцированы атомы фтора,
а вершина занята атомом азота с неподеленной электронной парой. К различным
химическим реагентам и к нагреванию NF3 весьма устойчив.
Остальные тригалогениды азота эндотермичны, а потому неустойчивы и реак-
ционноспособны. NC13 образуется при пропускании газообразного хлора в креп-
крепкий раствор хлорида аммония:
ЗС12 + NH4C1 = 4HC1 + NC13
Трихлорид азота представляет собой легколетучую. (Wi = 71° С) жидкость с
резким запахом. Небольшой нагрев или удар сопровождается взрывом с выделе-
выделением большого количества теплоты. При этом NCI3 распадается на элементы.
Тригалогениды NBr3 и NI3 еще менее стабильны.
408
Производные азота с халькогенами очень неустойчивы вследствие их сильной
эндотермичности. Все они плохо изучены', при нагреве и ударе взрываются.
Соединения с. металлами. Солеобразные нитриды получают
прямым синтезом из металлов и азота. Водой и разбавленными кислотами соле-
солеобразные нитриды разлагаются:
Mg3N2 + 6Н2О = 3Mg(OHJ + 2NH3
Ca3N2 + 8НС1 = ЗСаС12 + 2NH4C1
Обе реакции доказывают основную природу нитридов активных металлов.
Металлоподобные нитриды получают нагреванием металлов в атмосфере азота
или аммиака. В качестве исходных веществ могут применяться оксиды, галогени-
ды и гидриды переходных металлов:
2Та + N2 = 2TaN; Mn2O3 + 2NH3 = 2MnN + ЗН2О
CrCl3 + NH3 = CrN + 3HC1; 2TiH2 + 2NH3 = 2TiN + 5H2
Ковалентные нитриды образуют в основном металлы III группы. Внутри груп-
группы элементов-аналогов при переходе к более тяжелым металлам на доминирую-
доминирующий ковалентный тип связи все в большей мере накладывается металлическая
связь. Так, в ряду A1N, GaN и InN увеличивается металлизация связи, что ска-
сказывается на уменьшении ширины запрещенной зоны и росте электрической про-
проводимости.
2. Фосфор. Особенности химии фосфора. Второй типический
элемент V группы — фосфор — является неметаллом. По величине ОЭО он усту-
уступает таким типичным неметаллам, как фтор, кислород, хлор, азот и сера. Увели-
Увеличение главного квантового числа и атомного радиуса в группе при переходе от
азота к фосфору обусловливает ряд особенностей химии фосфора.
Во-первых, сумма первых пяти ионизационных потенциалов резко падает от
азота B66,8 В) к фосфору A76,7 В). Это ведет к стабильности положительных
степеней окисления, включая высшую характеристическую степень окисления
+5. Именно поэтому все производные, содержащие фосфор в степенях окисления
меньше +5, проявляют себя как восстановители. В то же время соединения фос-
фосфора (+5) в растворах окислителями не являются. Этим же объясняется большая
устойчивость кислородных соединений фосфора по сравнению с таковыми азота.
Наоборот, водородные соединения фосфора менее стабильны, чем водородные
соединения азота.
Во-вторых, валентные возможности фосфора гораздо богаче, чем у азота,
вследствие наличия у первого вакантных Збйэрбиталей. При промотировании
электрона на одну из 3</-орбиталей у атома фосфора появляется пять неспарен-
ных электронов, которые только по обменному механизму обеспечивают ковалент-
ность, равную 5. Кроме того, свободные 3(/-орбитали могут участвовать в образо-
образовании ковалентных связей по донорно-акцепторному механизму. В этом случае
фосфор акцептирует на эти орбитали электронные пары партнеров-доноров. В
соответствии с бблыними валентными возможностями у фосфора появляются
новые типы гибридизации, например sp3d (к.ч. 5) и sp3(P (к.ч. 6), которые в
принципе не могут быть осуществлены в случае атома азота.
В-третьих, в химии фосфора ярче проявляется склонность к образованию
ало
Рис. 150. Кристаллическая
структура черного фосфора
(фрагмент одного слоя)
Наиболее стабильной модификацией фосфора
является черный фосфор. Его получают аллотроп-
аллотропным превращением белого фосфора при температуре
220°С и давлении 1200 МПа. По внешнему виду он
напоминает графит. Кристаллическая структура
черного фосфора слоистая, состоящая из гофриро-
гофрированных слоев (рис. 150). Как и в красном фосфоре,
здесь каждый атом фосфора связан ковалентными
связями с тремя соседями. Расстояние между атома-
атомами фосфора соседнего слоя 0,387 нм. Белый и красный фосфор —диэлектрики, а
черный фосфор — полупроводник с шириной запрещенной зоны 0,33 эВ. В
химическом отношении черный фосфор наименее реакционноспособен, воспламе-
воспламеняется лишь при нагревании выше 400°С.
Окислительную функцию проявляет фосфор при взаимодействии с металлами:
ЗСа + 2Р = Са3Р2
Как восстановитель фосфор выступает в реакциях с активными неметаллами —
галогенами, кислородом, серой, а также с сильными окислителями:
4Р + ЗО2 = 2Р2О3; 4Р + 5О2 = 2Р2О5; 2Р + ЗС12 = 2РС13
2Р + 5С12 = 2РС15; 2Р + 3S = P2S3; 2P + 5S = P2S5
Р + 5HNO3 = Н3РО4 + 5NO2 + Н2О
Наконец, в растворах щелочей при нагревании белый фосфор диснропорциони-
рует:
8Р + ЗВа(ОНJ + 6Н2О = 2РН3 + ЗВа(Н2РО2J
Характеристические соединения. Фосфорсодер-
Фосфорсодержащие кислоты и их соли. При взаимодействии фосфора с кисло-
кислородом недостаток последнего ведет к образованию оксида Р2Оз, а в избытке
кислорода образуется P2Os- Механизм процесса окисления цепной, а продуктами
окисления являются димеры или более сложные полимеры. Оксид фосфора (+3)
существует в виде нескольких полиморфных модификаций, одна из них имеет
молекулярную решетку, в узлах которой дислоцированы димерные молекулы
Р4Об- Строение (на плоскости) этой молекулы, а также структура полимерного
оксида Р2О3 таковы:
I
I
.p>
X
p
I
о
I
^p.
о
I
Р4О6
полимерный
Молекула РгОб состоит из четырех пирамид (незавершенных тетраэдров) [РОз],
соединенных . атомами кислорода. Модификация, состоящая из молекул Р4О6,
411
Рис. 150. Кристаллическая
структура черного фосфора
(фрагмент одного слоя)
Наиболее стабильной модификацией фосфора
является черный фосфор. Его получают аллотроп-
аллотропным превращением белого фосфора при температуре
220°С и давлении 1200 МПа. По внешнему виду он
напоминает графит. Кристаллическая структура
черного фосфора слоистая, состоящая из гофриро-
гофрированных слоев (рис. 150). Как и в красном фосфоре,
здесь каждый атом фосфора связан ковалентными
связями с тремя соседями. Расстояние между атома-
атомами фосфора соседнего слоя 0,387 нм. Белый и красный фосфор — диэлектрики, а
черный фосфор — полупроводник с шириной запрещенной зоны 0,33 эВ. В
химическом отношении черный фосфор наименее реакционноспособен, воспламе-
воспламеняется лишь при нагревании выше 400°С.
Окислительную функцию проявляет фосфор при взаимодействии с металлами:
ЗСа + 2Р = Са3Р2
Как восстановитель фосфор выступает в реакциях с активными неметаллами —
галогенами, кислородом, серой, а также с сильными окислителями:
4Р + ЗО2 = 2Р2О3; 4Р + 5О2 = 2Р2О5; 2Р + ЗС12 = 2РС13
2Р + 5С12 = 2РС15; 2Р + 3S = P2S3; 2P + 5S = P2S5
Р + 5HNO3 = Н3РО4 + 5NO2 + Н2О
Наконец, в растворах щелочей при нагревании белый фосфор диспропорциони-
рует:
8Р + ЗВа(ОНJ + 6Н2О = 2РН3
Характеристические соединения. Фосфорсодер-
Фосфорсодержащие кислоты и их соли. При взаимодействии фосфора с кисло-
кислородом недостаток последнего ведет к образованию оксида Р2О3, а в избытке
кислорода образуется Р2О5. Механизм процесса окисления цепной, а продуктами
окисления являются димеры или более сложные полимеры. Оксид фосфора (+3)
существует в виде нескольких полиморфных модификаций, одна из них имеет
молекулярную решетку, в узлах которой дислоцированы димерные молекулы
Р4О6. Строение (на плоскости) этой молекулы, а также структура полимерного
оксида Р2Оз таковы:
I
I
о
ЧР'
I
О
Р'
I
О
,Р^
.0
I
Р4О6
полимерный Р2О1
Молекула РгО6 состоит из четырех пирамид (незавершенных тетраэдров) [РО3],
соединенных атомами кислорода. Модификация, состоящая из молекул Р4О6,
411
легкоплавка, летуча, незначительно растворима в CS2. В полимерных структурах,
образующих трехмерные сетки, также повторяются пирамидальные группировки
[РОз], т.е. незавершенные тетраэдры. Одна из вершин тетраэдра занята неподе-
: i j ленной электронной парой, а пирамиды соединены через атомы кислорода. В
обоих случаях орбитали атома фосфора находятся в состоянии вр'-гибридизации.
Химически оксид фосфора (+3) проявляет кислотную природу:
Р2О3 + ЗН2О = 2Н3РО3
Фосфористая кислота — бесцветные легкоплавкие хорошо растворимые в воде
кристаллы. По химическому строению она представляет собой искаженный тет-
тетраэдр, в центре которого находится атом фосфора с 5р3-гибридными орбиталями,
а вершины заняты двумя гидроксогруппами и атомами водорода и кислорода:
ОН
Атом водорода, непосредственно соединенный с фосфором, не способен к замеще-
замещению, а потому фосфористая кислота максимум двухосновна и нередко ее изобра-
изображают формулой Н2[НРОз]. Н3РО3 — кислота средней силы: рК\ 2,8, а рА'2 6,2.
Соли ее — фосфиты — получают взаимодействием Р2О3 со щелочами:
Р2О3 + 4NaOH = 2Na2HPO3 + H20
Фосфиты щелочных металлов и кальция легко растворимы в воде.
При нагревании фосфористая кислота диспропорционирует:
4Н3РО3 = РН3 + ЗН3РО4
Фосфористая кислота окисляется многими окислителями, в том числе галогена-
галогенами, например
Н3РО3 + С12 + Н20 = Н3РО4 + 2НС1
Получают обычно фосфористую кислоту гидролизом тригалогенидов фосфора:
РГ3 + ЗН2О = Н3РО3 + ЗНГ
При нагревании однозамещенных фосфитов получаются соли пирофосфористой
(дифосфористой) кислоты — пирофосфиты:
2NaH2PO3 = Na2H2P2O5 + H20
Пирофосфиты при кипячении с водой гидролизуются:
Na2H2P2O5 + ЗН2О = 2NaOH + 2Н3РО3
412
Сама пирофосфористая кислота Н4Р2О5 (пентаоксодифосфорная), как и фосфо-
фосфористая, только двухосновна и сравнительно малоустойчива:
Известна еще одна кислота фосфора (+3) — плохо изученная полимерная мета-
фосфористая кислота (НРОг)п-
Наиболее характерен для фосфора оксид Р2О5 — пентаоксид дифосфора. Это
белое твердое вещество, которое легко может быть получено и в стеклообразном
состоянии. В парообразном состоянии молекулы оксида фосфора (+5) имеют
состав P^tOjo-
Твердый Р2О5 имеет несколько модификаций. Одна из форм оксида фосфора
(+5) имеет молекулярную структуру с молекулами Р4О10 в узлах решетки. По
внешнему виду эта модификация напоминает лед. Она обладает небольшой плот-
плотностью, легко переходит в пар, хорошо растворяется в воде и реакционноспо-
собна.
Р2О5 — сильнейший дегидратирующий реагент. По интенсивности осушающе-
осушающего действия он намного превосходит такие поглотители влаги, как СаС12, NaOH,
H2SO4 и др. При гидратации Р2О5 сначала образуется метафосфорная кислота:
Р2О5 + Н20 = 2НРО3
дальнейшая гидратация которой последовательно приводит к пирофосфорной и
ортофосфорной кислоте (конечный продукт):
2НРО3 + Н20 = Н4Р2О7 и Н4Р2О7 + Н20 = 2Н3РО4
Ортофосфорная, или просто фосфорная, кислота Н3РО4 — одно из наиболее
важных производных фосфора (+5). Это бесцветные, легкоплавкие, расплы-
расплывающиеся на воздухе кристаллы, смешивающиеся с водой в любых соотношени-
соотношениях. В твердой кислоте и концентрированных растворах действуют межмолекуляр-
межмолекулярные водородные связи. Поэтому крепкие растворы Н3РО4 отличаются высокой
вязкостью. В более разбавленных растворах (менее 50 масс, долей, % Н3РО4)
возникают водородные связи между молекулами воды и кислоты. По химическо-
химическому строению молекула Н3РО4 представляет собой искаженный тетраэдр, в кото-
котором (в отличие от фосфористой) не две, а три вершины заняты гидроксогруппа-
ми, а четвертая — атомом кислорода. В водной среде Н3РО4 — кислота средней
силы: pA'i 2,2; рК2 7,3 и рА'3 12,4.
Из ПОЛИфосфорНЫХ КИСЛОТ ОТМеТИМ ДИфосфорНуЮ (пирофосфорную) Н4Р2О7 И
полимерные метафосфорные кислоты: тетраметафосфорную (НРОзL и гексамета-
фосфорную (НРОз)б- Дифосфорная немного сильнее ортофосфорной кислоты —
pA"i 1,6; рА^ 2,4; рА"з 6,6; рА^ 10,0. Метафосфорные кислоты являются сильными
кислотами. Например, для (НРО3)з рК1,7, а для тетраметафосфорной кислоты
рА-42,6.
В водном растворе ортофосфаты — соли фосфорной кислоты — подвергаются
гидролизу, причем рН среды при переходе от средней соли к кислой закономер-
413
но снижается. Ниже приведены гидролитические реакции для 1%-ных растворов
с указанием рН:
Na3PO4 +
Na2HPO4
Н20 = NaOH +
+ H2O = NaOH
Na2HPO4, pH 12,1,
+ NaH2PO4, pH 8,9.
При окислении влажного фосфора наряду с P2Os и Р2О3 образуется фосфор-
новатая (гексаоксодифосфорная) кислота ЩРзОе, в которой степень окисления
фосфора +4. В ее структуре атомы фосфора связаны друг с другом непосред-
непосредственно в отличие от полифосфорных кислот:
Н4Р2Ое — кислота средней силы, все ее четыре атома водорода могут быть заме-
замещены на металл. При нагревании ее водных растворов кислота, присоединяя
воду, распадается:
H4P2Ofi + Н2О = Н3РО8 + Н3РО4
Растворы ее солей — гипофосфатов — в воде вполне устойчивы. Из гипофос-
фатов хорошо растворимы в воде лишь соли щелочных металлов.
Наименьшая положительная степень окисления фосфора в фосфорноватистой
(диоксофосфорной) кислоте НзРО2. Ее можно получить в свободном состоянии
вытеснением из солей — гинофосфитов, например
Ва(Н2РО2J + H2SO4 = BaSO4 + 2Н3РО2
Фосфорноватистая кислота — бесцветные кристаллы, хорошо растворимые в воде.
Как видно из структуры НаРО2, она одноосновна:
Таким образом, в фосфорноватистой кислоте степень окисления фосфора +1, а
его ковалентность равна 5. Н3РО2 — сильная кислота (рА' 1,1). Сама фосфорнова-
фосфорноватистая кислота и гипофосфиты являются сильнейшими восстановителями.
Из пероксидных производных фосфора отметим мононадфосфорную (пероксо-
монофосфорную) Н3РО5 и динадфосфорную (пероксодифосфорную) Н4Р2Оз
кислоты. Кислотные свойства ярче выражены у последней, являющейся сильной
кислотой. А окислительная активность выше у пероксомонофосфорной. Их соли
в твердом состоянии вполне устойчивы, но при нагревании выделяют кислород и
превращаются в фосфаты.
Соединения фосфора с неметаллами. Фосфор и водород
в виде простых веществ практически не взаимодействуют. Водородные производ-
производные фосфора получают косвенным путем, например
Са3Р2 + 6НС1 = ЗСаСЬ + 2РН3
414
Фосфин РН3 представляет собой бесцветный сильнотоксичный газ с запахом
гнилой рыбы.
Мапекулу фосфина можно трактовать как аналог молекулы аммиака. Однако
угол между связями Н—Р—Н значительно меньше, чем у аммиака (93,7° против
107е). Это означает уменьшение доли участия s-облаков в образовании гибрид-
гибридных связей в случае фосфина. Кроме того, связи фосфора с водородом менее
прочны, чем связи последнего с азотом, а электрический момент диполя аммиака
почти втрое превосходит момент диполя фосфина. Донорные свойства у РНз
выражены несравненно слабее, чем у аммиака. И малая полярность молекулы
РНз, и слабая активность акцептировать протон приводят к отсутствию водород-
водородных связей не только в жидком и твердом состояниях, но и с молекулами воды в
растворах, а также к малой стойкости иона фосфония РН4- Последний является
аналогом аммоний-иона и характеризуется тетраэдрическим расположением свя-
связей. Самая устойчивая в твердом состоянии соль фосфония — это его иодид
PH4I. Водой и особенно щелочными растворами соли фосфония энергично раз-
разлагаются:
РН41 + КОН = РНЯ + KI + Н2О
Фосфин и соли фосфония являются сильными восстановителями. На воздзхе
фосфин сгорает до фосфорной кислоты:
РН3 + 2О2 = Н3РО4
В щелочных растворах его редокс-потенциал отрицателен:
Н2РО2 + ЗН2О + 4е" = РН3 + 5ОН"; Е° = -1,12 В
Поэтому фосфин восстанавливает ионы многих металлов: золота, серебра, меди,
свинца и др.
При разложении фосфидов активных металлов кислотами одновременно с
фосфином образуется в качестве примеси дифосфин РгН4. Дифосфин — бесцвет-
бесцветная летучая жидкость, по структуре молекулы аналогичная гидразину. Однако
он не проявляет основных свойств, с кислотами не взаимодействует. На воздухе
самовоспламеняется (сильный восстановитель), при хранении на свету и при
нагревании разлагается. В продуктах его распада присутствуют фосфор, фосфин
и аморфное твердое вещество желтого цвета. Этот продукт получил название
твердого фосфористого водорода, и ему приписывается формула Р^Щ. Согласно
другой точке зрения, твердый фосфористый водород есть раствор фосфина РН3 в
твердом фосфоре.
С галогенами фосфор образует три- и пентагалогениды — РГ3 и РГ5. Эти
производные фосфора известны для всех аналогов, но практически важны соеди-
соединения хлора. Все они получаются непосредственным взаимодействием простых
веществ. РГ3 и РГ5 токсичны. Соединения РГ3 термически более стойки, чем РГ5,
причем устойчивость уменьшается с ростом атомной массы галогена. РГ5 — ве-
вещества кислотной природы. При действии небольшого количества воды на РС15
образуется оксихлорид РОС13, а с избытком воды — фосфорная кислота:
РС15 + Н2О = РОС13 + 2НС1; РС15 + 4Н2О = Н3РО4 + 5НС1
415
Соединения фосфора с
металлами. С активными металлами
фосфор, аналогично азоту, образует соле-г
образные фосфиды, подчиняющиеся npd?
вилам классической валентности. р-Метал-
лы, а также металлы подгруппы цинка
дают и нормальные, и анионоизбыточные
фосфиды. Большинство из этих соедине-
соединений проявляют полупроводниковые свой-
свойства, т.е. доминирующая связь в них —
ковалентная.
Отличие азота от фосфора, обуслов-
обусловленное размерным и энергетическим фак-
факторами, наиболее характерно проявляется
при взаимодействии этих элементов с
переходными металлами. Для азота при
взаимодействии с последними главным
является образование металлоподобных
noo
1300
1200
1100
woo
900
800
V" /
^\f?OCa JtffS°
\l960"
13,1
1
?7»1 ^
\ \
\ 1W
1B90\/^\
42,6
лпЛПВ
1002
¦ж"
О 10 20 30 кО 50 60
Мп Am. доли Р,% Р
Рис. 151. Диаграмма состояния систе-
системы марганец - фосфор
нитридов. Фосфор также образует металлоподобные фосфиды, но в системах
переходный металл — фосфор другие фосфиды (независимо от того, подчиняются
они правилам валентности или нет) получают большую физико-химическую
индивидуальность. Для иллюстрации на рис. 151 приведена часть диаграммы
состояния системы Мп — Р. В целом в поведении фосфора по отношению к ме-
металлам проявляется большая аналогия с кремнием, чем с азотом. Многие фос-
фосфиды, особенно с преимущественно ковалентной связью, тугоплавки. Так,
А1Р плавится при 2197°С, а фосфид галлия имеет температуру плавления
1577° С. Фосфиды щелочных и щелочно-земельных металлов легко разлагаются
водой с выделением фосфина. Многие фосфиды являются не только полу-
полупроводниками (А1Р, GaP, InP и др.), но и ферромагнетиками, например СоР и
Fe3P.
3. Подгруппа мышьяка. Характеристика элементов VA-
группы. Валентная электронная конфигурация всех элементов VA-группы —
ns2np3, т.е. на внешнем энергетическом (валентном) уровне они содержат два
спаренных ns-электрона и три неспаренных электрона на трехкратно вырожден-
вырожденном np-уровне. Однако между элементами этой группы существуют и различия в
электронном строении. У мышьяка, сурьмы и висмута к вакантному nd-уровню
добавляется еще в отличие от фосфора полностью завершенный внутренний
(п — 1)й-уровень, а у висмута, следующего за лантаноидами, кроме того, и 4/4-
уровень. В силу наличия внутренних d- и /оболочек, экранирующих внешние
электроны, в ряду As — Sb — Bi проявляется вторичная периодичность. В ре-
результате этого для среднего элемента ряда — сурьмы — степень окисления +5
оказывается более стабильной, чем для мышьяка и висмута.
Основные характеристики атомов элементов VA-группы и образованных ими
гомоатомных соединений представлены на с. 418.
В ряду N — Р — As — Sb — Bi происходит монотонное возрастание радиусов
атомов и уменьшение электроотрицательности в соответствии с общей тенденцией
изменения этих характеристик в главных подгруппах Периодической системы.
Сумма первых трех потенциалов ионизации также уменьшается в этом ряду, что
14 Я. А. Угай
417
Соединения фосфора с
металлами. С активными металлами
фосфор, аналогично азоту, образует соле-
образные фосфиды, подчиняющиеся npd?
вилам классической валентности. р-Метал-
лы, а также металлы подгруппы цинка
дают и нормальные, и анионоизбыточные
фосфиды. Большинство из этих соедине-
соединений проявляют полупроводниковые свой-
свойства, т.е. доминирующая связь в них —
ковалентная.
Отличие азота от фосфора, обуслов-
обусловленное размерным и энергетическим фак-
факторами, наиболее характерно проявляется
при взаимодействии этих элементов с
переходными металлами. Для азота при
взаимодействии с последними главным
является образование металлоподобных
О 10 20 30 hO 50 SO
Мп Am. доли Р,% Р
Рис. 151. Диаграмма состояния систе-
системы марганец - фосфор
нитридов. Фосфор также образует металлоподобные фосфиды, но в системах
переходный металл — фосфор другие фосфиды (независимо от того, подчиняются
они правилам валентности или нет) получают большую физико-химическую
индивидуальность. Для иллюстрации на рис. 151 приведена часть диаграммы
состояния системы Мп — Р. В целом в поведении фосфора по отношению к ме-
металлам проявляется большая аналогия с кремнием, чем с азотом. Многие фос-
фосфиды, особенно с преимущественно ковалентной связью, тугоплавки. Так,
А1Р плавится при 2197° С, а фосфид галлия имеет температуру плавления
1577° С. Фосфиды щелочных и щелочно-земельных металлов легко разлагаются
водой с выделением фосфина. Многие фосфиды являются не только полу-
полупроводниками (А1Р, GaP, InP и др), но и ферромагнетиками, например СоР и
Fe3P.
3. Подгруппа мышьяка. Характеристика элементов VA-
группы. Валентная электронная конфигурация всех элементов VA-группы —
ns2np3, т.е. на внешнем энергетическом (валентном) уровне они содержат два
спаренных ns-электрона и три неспаренных электрона на трехкратно вырожден-
вырожденном np-уровне. Однако между элементами этой группы существуют и различия в
электронном строении. У мышьяка, сурьмы и висмута к вакантному nrf-уровню
добавляется еще в отличие от фосфора полностью завершенный внутренний
(п — 1)</-уровень, а у висмута, следующего за лантаноидами, кроме того, и 4/4-
уровень. В силу наличия внутренних d- и /оболочек, экранирующих внешние
электроны, в ряду As — Sb — Bi проявляется вторичная периодичность. В ре-
результате этого для среднего элемента ряда — сурьмы — степень окисления +5
оказывается более стабильной, чем для мышьяка и висмута.
Основные характеристики атомов элементов VA-группы и образованных ими
гомоатомных соединений представлены на с. 418.
В ряду N — Р — As — Sb — Bi происходит монотонное возрастание радиусов
атомов и уменьшение электроотрицательности в соответствии с общей тенденцией
изменения этих характеристик в главных подгруппах Периодической системы.
Сумма первых трех потенциалов ионизации также уменьшается в этом ряду, что
14 Я. А. Угай
417
свидетельствует об увеличении стабильности степени окисления +3 при переходе
от фосфора к висмуту. Состояние со степенью окисления +5 для висмута заметно
менее устойчиво, чем для сурьмы. Объясняется это, как и в предыдущих случаях
(Т1, РЬ), наличием инертной пары бв-эяектронов.
Содержание в земной коре,
масс.доли, %
Валентная электронная конфи-
конфигурация
Атомный радиус, нм
? к в
t=i
? к в
t=i
оэо
N
1 • Ю-2
[Не]2«22р3
0,070
91,5
266,8
3,0
Ю
'2
As
5-Ю
Характерные степени окис-
окисления
Температура плавления, °С
Температура кипения, °С
Плотность, г/см3
0,110
60,4
176,7
2,1
-3, -2, -1, +1, -3, (-2), +1,
+2, +3, +4, +5 +3, (+4), +5
0,121
58,0
171,0
2,0
-3, +3, +5
Удельное сопротивление,
Ом-см
Содержание в земной коре,
масс.доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
? к в
? /., в
г=1
ОЭО
Характерные степени окис-
окисления
Температура плавления, "С
Температура кипения, °С
Плотность, г/см3
мЬ \*-'p~p/4^J, "
Удельное сопротивление,
Ом • см
Sb
4-Ю
[Kr]4rflo5s25p3
0,141
52,3
158,4
1,9
-3, +3, +5
630,5
1634
6,7
0,24
40-10
-210,0 593 (под давл.) 817 (под давл.)
-195,8 429 возг. (краен.) 615 возг.
0,808 (жидк.) 2,0-2,4 (краен.) 5,72
0,3
1013
35 -Ю-6
Bi
2-Ю
0,146
52,0
153,0
1,8
+3, (+5)
271,4
1552
9,8
0,2
106 -10
418
Для элементов подгруппы мышьяка стабильность отрицательной степени
окисления в ряду As — Sb — Bi уменьшается вплоть до того, что для висмута она
вообще неизвестна. Это обстоятельство, в частности, также характеризует висмут
как металл.
Природные соединения и получение. В земной коре
мышьяк сравнительно широко распространен и находится в основном в связан-
связанном состоянии, хотя изредка встречается и самородный мышьяк. Известно более
120 минералов, содержащих мышьяк. В природе он ассоциирован главным обра-
образом с серой, образуя минералы двух типов: собственно сульфиды мышьяка (ре-
(реальгар AsS и аурипигмент As2S3) и сульфоарсениды и арсениды металлов (арсе-
нопирит FeAsS, лёллингит или мышьяковистый колчедан FeAs2). Последняя
группа минералов является породообразующей (полиметаллические руды). В
состав этих образований входят такие металлы, как Аи, Ag, Pb, Си, Со, Sn и т.д.
Содержание сурьмы в земной коре примерно на порядок меньше. Однако она
тоже образует самостоятельные минералы, наиболее распространенными из кото-
которых являются сурьмяный блеск (антимонит) Sb2S3, валентинит Sb2Og, сервантит
Sb2O4, стибиоканит Sb2O4*H2O и кермезит 3Sb2S3-Sb203. Как и мышьяк, сурьма
ассоциируется с полиметаллическими рудами. В природе висмут образует суль-
сульфиды, тиосоли, оксидные формы, встречается и в самородном состоянии. Наибо-
Наиболее известны висмутит Bi2S, висмит Bi2O3, тетрадимит Bi2Te2S.
Мышьяк, сурьму и висмут в свободном состоянии получают обычно путем
карбо- или металлотермического восстановления оксидов. Поскольку мышьяк и
его аналоги обычно ассоциированы со многими металлами, в процессе восстанов-
восстановления образуются сплавы. Восстановленный полупродукт подвергают хлорирова-
хлорированию. Летучие хлориды мышьяка, сурьмы и висмута отгоняют, подвергают дис-
дистилляции, а затем восстанавливают, например водородом, цинком и т.п. Оконча-
Окончательная очистка мышьяка достигается вакуумной пересублимацией. Сурьму и
висмут подвергают глубокой очистке методами направленной кристаллизации
или зонной плавки. Такие методы очистки позволяют получить мышьяк, сурьму
и висмут с суммарным содержанием примесей, не превосходящим 10—10 масс,
долей, %.
Простые вещества. Физические и химические
свойства. В свободном состоянии мышьяк, сурьма и висмут представляют
собой кристаллические вещества с металлическим блеском серого цвета (As), с
голубоватым отливом (Sb) или красноватым оттенком на изломе (Bi). При обыч-
обычных условиях они образуют слоистые кристаллические решетки ромбоэдрическо-
ромбоэдрического типа ((^-модификации).
Для мышьяка и сурьмы кроме окрормы известны и другие полиморфные
модификации. Так, при конденсации пара мышьяка на охлаждаемой жидким
азотом поверхности образуются желтые, мягкие, как воск, кристаллы кубической
сингонии, подобные белому фосфору. Превращение желтого мышьяка в стабиль-
стабильную о^-ромбоэдрическую форму обычно протекает через стадию образования так
называемого черного мышьяка, также похожего на аналогичную модификацию
фосфора. При 290° С черный мышьяк превращается в обычный серый металли-
металлический мышьяк. Аналогичные превращения наблюдаются и у сурьмы.
Чистые мышьяк, сурьма и висмут в компактном состоянии по отношению к
кислороду воздуха ведут себя существенно по-разному. Если сурьма и висмут в
сухой атмосфере практически не окисляются при обычных условиях, то мышьяк
И* 419
очень быстро тускнеет на воздухе с образованием оксида As2O3. Он не пассивиру-
пассивирует поверхность мышьяка. Мелкозернистый мышьяк, таким образом, может окис-
окислиться полностью. Во влажной атмосфере поверхностному окислению подверга-
подвергаются также сурьма и висмут. При нагревании на воздухе все три вещества сгора-
сгорают с образованием оксидов Э2Оз. В атмосфере кислорода при этих условиях
мышьяк может образовывать и высший оксид As2O5, сурьма окисляется лишь до
Sb(+4) (Sb2O,(). Sb2C>5 может быть получен окислением сурьмы в атмосфере озо-
озонированного кислорода. Висмут во всех случаях при непосредственном окислении
образует только Bi2O3.
В ряду стандартных электродных потенциалов все три элемента стоят правее
водорода (между водородом и медью). Вследствие этого они не могут взаимодей-
взаимодействовать с минеральными кислотами, не являющимися окислителями. Обычно
перевести их в раствор можно действием концентрированных H2SO4 или HNOS.
При этом по отношению к азотной кислоте эти элементы ведут себя по-разному.
Так, мышьяк при взаимодействии с азотной кислотой переходит в ортомышьяко-
вую кислоту:
As + 5HNO (конц.) = H3As04 + 5NO2 + Н2О
Сурьма с концентрированной HNO3 образует метасурьмяную кислоту:
3Sb + 5HNO3 (конц.) = 3HSbO3 + 5NO + Н2О
Висмут в отличие от мышьяка и сурьмы при взаимодействии с азотной кислотой
всегда образует нитрат висмута:
Bi + 4HNO3 = Bi(NO3K + NO + 2H2O
чем также подтверждается металлическая природа этого элемента.
С растворами сильных щелочей мышьяк и его аналоги не взаимодействуют.
Сурьма и висмут устойчивы и по отношению к расплавам щелочей, в то время
как мышьяк при сплавлении со щелочами в присутствии кислорода воздуха
образует арсенаты.
При комнатной температуре As и Sb непосредственно взаимодействуют с гало-
галогенами, в то время как висмут реагирует с ними лишь при нагревании. При наг-
нагревании все три вещества реагируют с халькогенами, сероводородом.
Характеристические соединения. Гидриды элементов
подгруппы мышьяка общей формулы ЭН3 можно получить лишь косвенным
путем, например разложением кислотой соответствующих соединений с активны-
активными металлами:
Mg332 + 6НС1 = 3MgCl2 + 2ЭН3
В обычных условиях арсин AsH3, стибин ЭЬНз, висмутин BiH3 газообразны, силь-
сильно эндотермичны и в силу этого неустойчивы. Висмутин разлагается в момент
получения, и о его существовании можно косвенно судить лишь на основании
повышенной летучести висмута в токе водорода.
Молекулы гидридов элементов подгруппы мышьяка имеют пирамидальную
структуру с атомом элемента в вершине и атомами водорода в основании, причем
420
угол Н—Э—Н близок к 90°. Это свидетельствует об образовании связей за счет
чистых, практически не гибридизованных р-орбиталей элемента. Вследствие
слабости электронодонорных свойств гидриды элементов подгруппы мышьяка
основными свойствами не обладают.
Очень важным свойством гидридов мышьяка и его аналогов является их высо-
высокая восстановительная активность, обусловленная нестабильностью отрицатель-
отрицательной степени окисления. Так, арсин способен восстанавливать даже фосфористую
кислоту Н3РО3 до фосфорноватистой Н3РО2, хотя фосфористая кислота обычно
сама является довольно энергичным восстановителем:
2AsH3 + ЗНзРО3 = ЗН3РО2 + 2As + 3H2O (AG°2g& = -60 кДж)
Рассматриваемые гидриды, особенно АэНз, являются сильнейшими неоргани-
неорганическими ядами. Это относится и ко всем летучим, и к растворимым соединениям
мышьяка.
Подобно оксиду фосфора (+3), As2O3 в парообразном состоянии существует в
виде димерной молекулы Ав4Об с тетраэдрической структурой. В твердом состоя-
состоянии As2O3 образует три кристаллические формы и одну стеклообразную. Оксид
сурьмы (+3) существует в двух кристаллических формах. При обычных условиях
стабильна псевдомолекулярная структура, состоящия из двойных бесконечных
цепей —Sb—О—Sb—О—Sb—, сшитых между собой кислородными мостиками:
Sb SbX NSbX \ь
/ / / /
О О О О
\ \ \ \
Sb Sb Sb Sb
' V V V ч
В отличие от молекулярных структур оксидов As2O3 и Sb2O3 оксид висмута (+3)
образует координационные решетки моноклинной или тетрагональной структуры.
Поэтому Bi2O3 более тугоплавок (<пл = 820°С) и менее летуч.
Если Аэ2Оз довольно хорошо растворим в воде, то два других практически
нерастворимы. Все гидроксиды амфотерны, однако у гидроксида мышьяка (+3)
сильно преобладает кислотный характер, а у Bi(OHK — основный. Гидроксид
сурьмы занимает промежуточное положение. Мышьяковистая кислота НзАэОз
(opmo-форма) известна лишь в растворах и представляет собой слабую кислоту:
pA'i 9,4; рА'2 12,8 и рА"з 13,4. Гидроксид сурьмы (+3) также существует только в
растворе в виде орто- и мета-форм И$БЪОз и HSbO2. Кислотные свойства гид-
гидроксида сурьмы (+3) выражены значительно слабее, чем у H3ASO3 (pA'i 11),одна-
11),однако в растворах сильных щелочей существуют комплексные гидроксостибат (+3)-
ионы: [Sb(OHL]' — метастибат; [Sb(OHN]3- — ортостибат.
Соли Sb(+3) существуют только в сильнокислых растворах, а при разбавлении
водой гидролизуются с образованием основных солей, например
SbCl3 + Н2О ^=± Sb(OH)Cl2 + НС1; Sb(OH)Cl2 + Н2О *=* Sb(OHJCl + HC1
Кроме того, для производных Sb(+3) характерно отщепление воды в процессе
гидролиза с образованием солей антимонила (стибила) SbO+:
Sb(OHJCl = SbOCl + Н2О
v 421
Оксид висмута (+3) обладает преимущественно оснбвными свойствами и хоро-
хорошо растворим в кислотах:
Bi2O3 + 6HNO3 = Bi(NO3K + ЗН2О
При взаимодействии солей Bi(+3) со щелочами образуется осадок Bi(OHK, на-
например
Bi(NO3K + 3NaOH = 3NaNO3 + Bi(OHK
Этот осадок нерастворим в избытке щелочи, т.е. кислотные свойства для него не
характерны. Однако основные свойства для Bi(OHK выражены также слабо, что
проявляется в сильной склонности солей Bi(-f-3) к гидролизу с образованием
основных солей и производных висмутила BiO+.
При повышении степени окисления элементов основные свойства их оксидов и
гидроксидов ослабевают, а кислотные усиливаются. Действительно, оксиды AS2O5
и Sb2Os нерастворимы в кислотах, но легко взаимодействуют со щелочами с
образованием соответствующих солей — арсенатов и стибатов, например
As2O5 + 6NaOH = 2Na3As04 + ЗН2О
Оксид мышьяка (+5) хорошо растворим в воде с образованием ортомышьяковой
кислоты:
As2O5 + ЗН2О = 2H3As04
Ортомышьяковая кислота хорошо растворима в воде и по силе примерно равна
фосфорной (pA'i 2,2, рА'2 6,7, рА'3 11,5). Sb2Os в воде растворим плохо. Соответст-
Соответствующая кислота в свободном состоянии не выделена. Из растворов стибатов при
обработке сильной кислотой выпадает обычно осадок переменного состава
zSb2O3-2/H2O. Сами стибаты существуют не как производные opmo-формы, а
происходят от гексагидроксосурьмяной кислоты H[Sb(OH)g]. Степень окисления
+5 для висмута вообще малохарактерна. В12О5 неустойчив и при нагревании
превращается в Bi2O3.
Таким образом, изменение кислотно-основных свойств оксидов, их стабильнос-
стабильности для мышьяка и его аналогов можно представить схемой
увеличение
стабильности
нарастание кислотных свойств
As2Os Sb2Os Bi2O5
увеличение
стабильности
As2O} Sb2Q} Bi2O
иарастаиие основных свойств
С различием стабильности производных в степенях окисления +3 и +5 связа-
связаны и закономерности изменения окислительно-восстановительных свойств в этих
рядах. Так как в ряду As(+3) — Sb(+3) — Bi(+3) увеличивается стабильность
соединений, то в этом же направлении уменьшаются восстановительные свойства.
Так, производные As(+3) в нейтральной и щелочной среде являются довольно
сильными восстановителями, легко переходя в более стабильные производные
As(+5).
422
Производные Sb(+3) к типичным восстановителям не относятся, однако в
щелочной среде могут быть окислены до Sb(+5):
Sb2S3 + 2(NH4JS2 = Sb2S5 + 2(NH4JS
Для производных Bi(+3) восстановительная активность вообще не характерна.
В ряду As(+5) — Sb(+5) — Bi(+5), напротив, стабильность производных силь-
сильно уменьшается, в соответствии с чем в этом же направлении резко возрастает
окислительная активность. Если производные As(+5) к типичным окислителям
не относятся и окислительная способность H3AsC>4 выражена лишь в кислой
среде, то производные сурьмяной кислоты в этих же условиях способны окислить
НС1 до С12. Производные же Bi(+5) являются сильнейшими окислителями, при-
причем не только в кислой, но даже в щелочной среде.
Для элементов подгруппы мышьяка помимо характерных степеней окисления
+3 и +5 иногда проявляются и менее устойчивые промежуточные степени окис-
окисления. Так, для сурьмы известен оксид формального состава Sb2O,(, отвечающий
степени окисления +4, а фактически являющийся смешанным оксидом
БЬгОз-БЬгОб. Этот оксид может быть получен прокаливанием Sb2O3 в атмосфере
кислорода при 900°С. При этом летучесть оксида Sb2O3 за счет образования
Sb2O4 резко уменьшается.
Соединения с другими неметаллами. Все три элемента
подгруппы мышьяка непосредственно взаимодействуют с галогенами. При этом
мышьяк и сурьма образуют два ряда галогенидов: ЭГз и ЭГ5, а для висмута
характерны низшие галогениды BiFa. Известен лишь BiF5. Помимо галогенидов,
отвечающих характерным степеням окисления, известны тетрахлориды сурьмы и
висмута ЭСЦ- Для висмута, кроме того, известны и дигалогениды (кроме фтори-
да).
В основном все галогениды характеризуются молекулярной структурой и в
обычных условиях представляют собой газы, жидкости или легкоплавкие твер-
твердые вещества. Лишь BiF3 обладает координационной структурой и солеобразным
характером, на что указывает сравнительно высокая температура плавления
G25°С). Обращает на себя внимание монотонное увеличение температур плавле-
плавления у галогенидов АвГ3 (-20, -6, +31 и 144°С от фторида к иодиду) и заметно
более высокие температуры плавления SbF3 B90°С), особенно BiF3 по сравнению
с остальными тригалогенидами. Это обусловлено тем, что электроотрицательность
мышьяка в степени окисления +3 заметно превышает таковую для сурьмы и
висмута в той же степени окисления. Поэтому AsF3 менее полярны. Для сурьмы и
висмута ионный вклад, обусловленный разностью электроотрицательностей, наи-
наиболее заметен у фторидов, обладающих вследствие этого максимальными точками
плавления в соответствующих рядах. Остальные галогениды сурьмы обладают
молекулярно-ковалентным характером. Что же касается тригалогенидов висмута,
то ионный вклад заметен у всех его производных. Поскольку электроотрицатель-
электроотрицательность элементов подгруппы мышьяка в высшей степени окисления велика, пре-
преимущественно ковалентный характер взаимодействия отмечается у всех известных
пентагалогенидов. Даже BiF5 сублимирует без плавления, что указывает на моле-
молекулярный характер его кристаллической решетки.
Для всех галогенидов мышьяка и его аналогов характерны три основных типа
химических реакций: термическая диссоциация, гидролиз и комплексообразова-
423
ние. Низшим галогенидам, кроме того, свойственны реакции диспропорциониро-
вания. Наиболее характерна термическая диссоциация для пентагалогенидов,
протекающая по схеме ЭГ5 = ЭГз + Г2, где Г — F, C1. Диссоциация галогенидов
ЭГз менее заметна.
Все известные галогениды элементов подгруппы мышьяка способны образовы-
образовывать и ацидокомплексы, и катионные комплексы. Для тригалогенидов более
характерны катионные комплексы, которые можно рассматривать как продукты
присоединения к ЭГз нейтральных молекул, имеющих неподеленные электронные
пары, например [Аз(МНзL]С1з и т.д. Кроме того, они образуют и ацидокомплексы
при взаимодействии с галогенидами активных металлов:
МеГ + ЭГ3 = Ме[ЭГ4]; 2МеГ + ЭГ3 = Ме2[ЭГ5]; ЗМеГ + ЭГ3 = Ме3[ЭГ6]
Для пентагалогенидов более характерно образование ацидокомплексов. Так.
пентафториды образуют комплексы типа Ме[ЭГб] с фторидами щелочных метал-
металлов. При этом комплексообразующая способность SbFs выражена сильнее, чем у
соответствующих производных мышьяка и висмута (вторичная периодичность).
Например, пентабромид сурьмы в свободном состоянии неизвестен, однако соот-
соответствующий анионный комплекс [БЬВгб]" существует и довольно устойчив.
Общим для элементов подгруппы мышьяка является существование всех халь-
когенидов состава Э2Х3, отвечающих правилу формальной валентности. Халькоге-
ниды Э2Х3 можно получить как непосредственным сплавлением компонентов,
взятых в стехиометрических соотношениях, так и пропусканием сероводорода
через подкисленные (во избежание гидролиза) растворы их солей, Халькогениды
висмута представляют собой типичные координационные кристаллы, тогда как
для структур As2X3 и Sb2X3 (особенно с легкими халькогенами) характерны
"молекулярные" фрагменты. Например, кристалл As2S3 построен из гофрирован-
гофрированных сеток, связь между которыми осуществляется за счет ван-дер-ваальсова вза-
взаимодействия. Внутри же сеток связи преимущественно ковалентные. В рядах
БЬ2Хз и Bi2X3 слоистость структуры сохраняется, однако на межатомное взаимо-
взаимодействие накладывается металлическая составляющая связи, особенно заметная у
теллуридов.
Халькогениды Э2Хз устойчивы на воздухе и во влажной атмосфере, нераство-
нерастворимы в воде и разбавленных неокисляющих минеральных кислотах. Поскольку
халькогениды являются льюисовскими кислотами, для них характерны реакции
с образованием солей тиокислот, например
As2S3 + 3(NH4JS = 2(NH4KAsS3
Все тиосоли в твердом состоянии вполне устойчивы, а соответствующие тиокисло-
ты в свободном состоянии неизвестны.
Из высших халькогенидов 92Xs известны лишь сульфиды, причем все они
могут быть получены только косвенным путем и их устойчивость резко падает в
ряду As2S5 — Sb2S5 — Bi2S5- Пентасульфиды мышьяка и сурьмы также обладают
кислотными свойствами и легко образуют тиосоли (тиоарсенаты и тиостибаты)
при взаимодействии с основными сульфидами:
As2S5 + 3Na2S = 2Na3AsS4
424
В свободном состоянии тиокислоты, отвечающие высшей степени окисления
мышьяка и его аналогов, также не существуют.
Соли кислородсодержащих кислот. В высшей степени
окисления мышьяк и его аналоги вообще не образуют солей, в которых они выс-
выступали бы в качестве катиона. Этим они существенно отличаются от элементов
IVA-группы. Для мышьяка, как наиболее электроотрицательного элемента в этой
подгруппе, даже в степени окисления +3 соли кислородсодержащих кислот неиз-
неизвестны. Однако уже для сурьмы и особенно для висмута такие соли существуют.
Сурьма при нагревании растворяется в концентрированной серной кислоте с
образованием сульфата сурьмы:
2Sb + 6H2SO4 = Sb2(SO4K + 3SO2 + 6Н2О
т.е. ведет себя в этом отношении, подобно, например, меди, что подтверждается и
сравнением нормальных электродных потенциалов [?"°(Cu2+/Cu) = +0,34 В,
i?°(Sb3+/Sb) = +0,24 В]. Отметим, что при взаимодействии с азотной кислотой
(любой концентрации) нитрат сурьмы не образуется.
Для висмута способность образовывать соли более характерна. Наиболее рас-
распространенным препаратом висмута является его нитрат ВЦМОз)з- Все соли вис-
висмута в водных растворах сильно гидролизованы, поэтому для растворимых солей
весьма характерны продукты частичного гидролиза — производные висмутила.
Металлохимия элементов подгруппы мышьяка.
Большинство арсенидов и стибидов s- и sp-элементов являются полупроводника-
полупроводниками, т.е. их нельзя рассматривать как интерметаллические соединения, а мышьяк
и сурьму — как металлы. В то же время арсениды и стибиды подавляющего
большинства переходных металлов являются металлидами, в которых мышьяк и
сурьма проявляют себя как металлические компоненты. В этом и заключается
своеобразие элементов подгруппы мышьяка, их двойственный характер. Висмут
вследствие заметного металлического характера со всеми катионообразователями
дает металлидные фазы.
В системах As — Sb и Sb — Bi наблюдается полная взаимная растворимость, в
то время как As и Bi образуют диаграмму состояния эвтектического типа.
Элементы подгруппы мышьяка с менее электроотрицательными компонентами
образуют многочисленные соединения. При этом с «-металлами, как правило,
образуются соединения, подчиняющиеся правилу формальной валентности, ус-
устойчивые не только в твердом состоянии, но и не диссоциирующие при плавле-
плавлении. Такие соединения образуют группы, например А3В (NasSb, K3Bi),
II V
A3 В2 (Mg3As2, Саз8Ьг). С sp-элементами ША-группы мышьяк и сурьма также
образуют соединения, подчиняющиеся правилам формальной валентности
I А И1„УЧ Г. 1 П1 . III „.
(А а ). а системах А — As и А — ЬЬ существует единственное эквиатомное
соединение (InAs, InSb и т.п.), плавящееся конгруэнтно.
Для соединений рассматриваемых элементов с переходными металлами харак-
характерны соотношения компонентов, не подчиняющиеся правилу формальной ва-
валентности (PdaBi, PtBi, RhBi4 и т.п.), хотя и встречаются нормально-валентные
соединения (Cu3As, C113A42 и др.). Большинство этих соединений — металлиды.
Лишь арсениды и стибиды цинка и кадмия обладают неметаллическими свой-
свойствами независимо от их состава. Этот факт еще раз подтверждает, что Zn и Cd
не являются типичными переходными металлами. ,
N> l"(
4. Подгруппа ванадия. Общая характеристика элемен-
элементов подгруппы ванадия. Атомы V, Nb и Та характеризуются ва-
валентной электронной конфигурацией (л — l)<Pns2. При этом у атома тантала
валентным орбиталям предшествует заполненная (и — 2)/4-оболочка. Наличие
пяти валентных электронов способствует проявлению характеристической степени
окисления +5. Их энергетическая неравноценность обусловливает возможность
проявления низших степеней окисления +2, +3, +4.
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус, Э(+5), нм
Потенциалы ионизации, В
If. Э -+ Э+ + е
12: Э+ -¦ Э2+ 4- е
1$: Э2+ —> Э3+ + е
Ц: Э3+ -¦ Э4+ + е
Е
в
оэо
Температура плавления, "С
Температура кипения, °С
Плотность, г/см3
В
V
1,5-10-2
0,134
0,040
6,74
15,13
30,31
48,35
68,70
69,23
1,9
1900
3400
5,96
-0,835
Nb
1 - Ю-3
0,145
0,068
6,88
13,48
24,70
37,70
51,90
134,66
1,7
2470
4760
8,57
-1,099
Та
2-10'4
[Xe]4/45d
0,146
0,068
7,88
12,70
22,27
33,08
44,80
120,73
1,6
3000
5500
16,6
_
Атомный радиус ванадия заметно меньше, чем ниобия, а при переходе от
ниобия к танталу радиус атома практически не изменяется, несмотря на то что у
тантала появляется новый электронный слой. У ниобия и тантала в степени
окисления +5 к тому же совпадают и ионные радиусы, что обусловливает боль-
большое сходство химических свойств этих элементов.
Первый потенциал ионизации возрастает от ванадия к ниобию незначительно
(на 0,14 В), а от ниобия к танталу более резко (на 1,0 В). Это объясняется замет-
заметным уплотнением электронной оболочки тантала за счет ярко выраженного эф-
эффекта проникновения 6«-электронов под экран 4/4-электронов. Однако вторые
потенциалы ионизации в этом ряду монотонно уменьшаются, что можно объяс-
объяснить относительным уменьшением прочности связи оставшегося неспаренного s-
электрона с ядром. В целом оказывается, что сумма первых двух потенциалов
ионизации у ванадия заметно больше B1,87 В), чем у ниобия и тантала B0,36;
20,58 В соответственно), а у последних эти характеристики практически совпада-
совпадают. Сравнивая последующие потенциалы ионизации, отметим, что /з, /4, h умень-
уменьшаются в ряду V — Nb — Та. Это приводит к уменьшению в этом же направле-
направлении суммы пяти потенциалов ионизации. Последнее обстоятельство и объясняет,
с одной стороны, увеличение стабильности высшей степени окисления при пере-
переходе от ванадия к танталу, а с другой стороны, нарастание металлических
свойств в степени окисления +5 в том же направлении, что вообще характерно
для многих rf-элементов.
Природные соединения и получение. Несмотря на то что
ванадий довольно широко распространен и известно более 65 минералов, содер-
содержащих этот элемент, самостоятельных рудных месторождений он обычно не обра-
образует и часто сопутствует другим элементам в полиметаллических рудах. Наиболее
известны минералы ванадия, принадлежащие к группам алюмосиликатов [рос-
коэлит — K(V, AlJ[AlSi30io](OH, FJ], комплексных ванадатов [ванадинит —
Pb5(VO4KCl, десклоизит - PbZn[VO4]OH, карнотит - K(UO2)[VO4] и сульфид-
сульфидных руд (натронит VzSy). Ниобий и тантал входят в состав около 100 минералов,
изоморфно замещают друг друга, но также довольно рассеяны. Промышленное
значение имеет колумбит^ганталит (Fe, Mn)[(Ta, 1ЧЬH3]г, представляющий собой
танталат-ниобат железа и марганца.
Основные методы получения этих металлов в свободном состоянии сводятся к
восстановлению оксидов, галогенидов, комплексных галогенидов, электролизу
расплавов солей. Предварительно руды, содержащие ванадий и его аналоги,
обогащают, концентрируют, затем переводят в оксиды или галогениды и подвер-
подвергают восстановлению. Металлы высокой степени чистоты получают иодидным
методом, подобно металлам подгруппы титана.
Простые вещества. „Ф изические и химические
свойства. В компактном состоянии все три металла — V, Nb, Та — представ-
представляют собой вещества светло-серого цвета, хорошо поддающиеся механической
обработке в чистом состоянии. Все эти металлы характеризуются кристалличес-
кристаллическими структурами с координационным числом 8 (ОЦК).
Лучше всего металлы подгруппы ванадия растворяются в смесях кислот, одна
из которых является окислителем, а другая — источником лигандов. Универсаль-
Универсальным реагентом, растворяющим все три металла, является смесь азотной и плави-
плавиковой кислот:
ЗЭ + 21HF + 5HNO3 = 3H2[9F7] + 5NO + 10Н2О
Ванадий реагирует с плавиковой кислотой, с кислотами, являющимися одно-
одновременно окислителями, и с царской водкой. Ниобий и тантал вполне устойчивы
не только в индивидуальных окисляющих кислотах, но и в царской водке. Таким
образом, можно сделать вывод, что благородность металлов в кислых средах
возрастает от ванадия к ниобию и танталу. Но при этом следует иметь в виду,
что в этом же направлении растет 1\, что коррелирует с ростом благородности
этих металлов.
В растворах щелочей все три металла вполне устойчивы, однако с расплавами
щелочей, особенно в присутствии кислорода воздуха, они взаимодействуют весьма
активно:
4Э + 12КОН + 5О2 = 4К3ЭО4 + 6Н2О
Таким образом, по отношению к агрессивным средам металлы подгруппы вана-
ванадия ведут себя аналогично металлам подгруппы титана. Однако в целом их хими-
химическая устойчивость в агрессивных средах (благородность) несколько выше, чем
у элементов IVB-группы.
Высшие оксиды ниобия и тантала с водными растворами кислот и щелочей не
реагируют. Горячая концентрированная серная и плавиковая кислоты способны
перевести их в раствор. Взаимодействие с HF обусловлено образованием раство-
растворимых фторокомплексов, например
Nb2O5 + 14HF = 2H2[NbF7] + 5Н2О
Обычно Nb2Os и Та2С>5 можно перевести в раствор путем сплавления со щелоча-
щелочами, карбонатами и кислыми сульфатами щелочных металлов, например
Nb2O5 + 3Na2CO3 сплавление » 2Na3Nb04 + ЗСО2
Оксиды тантала (+5) взаимодействуют подобным же образом, но менее энергич-
энергично. Таким образом, при взаимодействии со щелочами высшие оксиды ванадия,
ниобия и тантала образуют соответствующие соли — ванадаты, ниобаты и танта-
латы. Характерной особенностью этих анионов является способность к образова-
образованию изополисоединений.
Метаванадиевая кислота НУОз по силе близка к уксусной. Вероятность ее
диссоциации по кислотному и основному типу примерно одинакова. В кислых
растворах (рН < 1,5) она существует в виде ванадила VO2, который обладает
окислительными свойствами. Ванадат аммония NH4VO3 является наиболее обыч-
обычным препаратом этого элемента. Желтый цвет его раствора обусловлен тримери-
зацией ванадат-иона: ЗУОз ^^ V3O9". В кислых растворах происходит дальней-
дальнейшее усложнение состава с образованием изополикислот — гексаванадат-ионов, что
сопровождается дальнейшим углублением окраски до рубиново-красной:
2V3Oi~ + 2Н+ ^ Н2О + V6Ofт
Ниобий и тантал в водном растворе также способны к образованию полимерных
оксоанионов, состав которых зависит от рН среды.
Как отмечено выше, для ванадия в противоположность ниобию и танталу
весьма характерна степень окисления +4. Оксид ванадия (+4) обладает более
ярко выраженными основными свойствами, чем V2Os- Так, VO2 растворяется в
кислотах с образованием производных ванадила VO2+:
VO2 + 2НС1 = VOC12 + Н2О
Производные ванадила VO2+ подвергаются в водном растворе дальнейшему гид-
гидролизу:
VOSO4 + Н2О ^ VO2 + H2SO4
Кислотные свойства VO2 выражены значительно слабее. При взаимодействии с
сильными щелочами образуются гипованадаты разнообразного состава, поскольку
гипованадат-анион VO3 также склонен к образованию изополисоединений. В
свободном состоянии H2VO3 неизвестна, однако, по оценкам, сила этой кислоты
соответствует примерно угольной.
Низшие оксиды ванадия относятся к соединениям переменного состава с
Высшие оксиды ниобия и тантала с водными растворами кислот и щелочей не
реагируют. Горячая концентрированная серная и плавиковая кислоты способны
перевести их в раствор. Взаимодействие с HF обусловлено образованием раство-
растворимых фторокомплексов, например
Nb2O5 + 14HF = 2H2[NbF7] + 5Н2О
Обычно Nb2O5 и Та2С>5 можно перевести в раствор путем сплавления со щелоча-
щелочами, карбонатами и кислыми сульфатами щелочных металлов, например
Nb2O5 + 3Na2CO3 сплавление » 2Na3Nb04 + ЗСО2
Оксиды тантала (+5) взаимодействуют подобным же образом, но менее энергич-
энергично. Таким образом, при взаимодействии со щелочами высшие оксиды ванадия,
ниобия и тантала образуют соответствующие соли — ванадаты, ниобаты и танта-
латы. Характерной особенностью этих анионов является способность к образова-
образованию изополисоединений.
Метаванадиевая кислота НУОз по силе близка к уксусной. Вероятность ее
диссоциации по кислотному и основному типу примерно одинакова. В кислых
растворах (рН < 1,5) она существует в виде ванадила VO2, который обладает
окислительными свойствами. Ванадат аммония NH4VO3 является наиболее обыч-
обычным препаратом этого элемента. Желтый цвет его раствора обусловлен тримери-
зацией ванадат-иона: ЗУОз 5=^ УзОд". В кислых растворах происходит дальней-
дальнейшее усложнение состава с образованием изополикислот — гексаванадат-ионов, что
сопровождается дальнейшим углублением окраски до рубиново-красной:
2V8O|" + 2Н+ s=^ Н2О + V6O?t
Ниобий и тантал в водном растворе также способны к образованию полимерных
оксоанионов, состав которых зависит от рН среды.
Как отмечено выше, для ванадия в противоположность ниобию и танталу
весьма характерна степень окисления +4. Оксид ванадия (+4) обладает более
ярко выраженными основными свойствами, чем V2Os. Так, VO2 растворяется в
кислотах с образованием производных ванадила VO2+:
VO2 + 2НС1 = VOC12 + Н2О
Производные ванадила VO2+ подвергаются в водном растворе дальнейшему гид-
гидролизу:
VOSO4 + Н2О ^ VO2 + H2SO4
Кислотные свойства VO2 выражены значительно слабее. При взаимодействии с
сильными щелочами образуются гипованадаты разнообразного состава, поскольку
гипованадат-анион VO|" также склонен к образованию изополисоединений. В
свободном состоянии Н2УОз неизвестна, однако, по оценкам, сила этой кислоты
соответствует примерно угольной.
Низшие оксиды ванадия относятся к соединениям переменного состава с
Ala
сильно выраженной нестехиометрией. Так, оксид ванадия (+2) в зависимости от
условий получения может существовать в интервале составов VOo,9-i,3! a оксид
ванадия (+3) — в интервале VOif6-i,8- Последняя фаза обладает избытком кисло-
кислорода против V2O3 (VOit5) и, следовательно, дефектами в подрешетке ванадия.
Характерной способностью V(+3) является способность к образованию квас-
квасцов, чем он напоминает титан и хром в этой степени окисления. Здесь проявляет-
проявляется горизонтальная аналогия между «/-элементами.
Соединения с другими неметаллами. Металлы подгруп-
подгруппы ванадия, непосредственно взаимодействуя с галогенами при повышенных
температурах, образуют галогениды разнообразного состава, отвечающие различ-
различным степеням окисления. Так, взаимодействие с газообразным фтором приводит
к образованию пентафторидов всех трех элементов (VF5, NbFs, TaFs). Поскольку
степень окисления +5 для ванадия менее стабильна, чем для ниобия и тантала,
хлор способен окислить его лишь до VC14, бром — до VBr3, а иод — только до
VI2. В то же время ниобий и тантал окисляются до пентагалогекидов хлором и
бромом, а тантал — даже иодом с образованием Tals.
С химической точки зрения, для нентагалогенидов характерны две особеннос-
особенности. Во-первых, они склонны к гидролизу. При этом промежуточными продуктами
при гидролизе являются оксогалогениды, которые особенно характерны для
самого ванадия. Второй особенностью галогенидов ЭГ5 является >=рко выражен-
выраженная склонность к комнлексообразованию, которая характерна не только для
самих галогенидов, но и для их оксопроизводных типа ЭОГз- В сильнокислых
средах (во избежание гидролиза) при взаимодействии ЭГ5 с ганогенидами щелоч-
щелочных металлов образуются комплексные галогениды, например
NbF5 + NaF = Na[NbF6] или TaF5 + 2NaF = Na2[TaF7]
Координационное число 6 (вр3(/2-ги6ридизация) характерно для всех элементов
подгруппы ванадия и для всех галогенидов (для существующих пентагалогени-
дов). А координационное число 7 («р3</3-гибридизация) характерно, с одной
стороны, для самого маленького лиганда (иона F"), а с другой стороны, для
сравнительно больших ионов Nb5+ и Та5+ (против иона V5+).
Для низших галогенидов ванадия и его аналогов характерны отклонения от
стехиометрии, которые впервые наблюдались для галогенопроизводных металлов
именно на примере этих соединений. Так, фаза NbCle существует в довольно
широком интервале составов NbCl2|67-3,i3) а для ТаС1з область гомогенности ле-
лежит в пределах ТаС12,9-з,1-
В системе V — S зарегистрировано существование трех сульфидов — VS, V2S3
и VS4, которые могут быть получены при непосредственном сплавлении компо-
компонентов. Косвенным путем можно получить и сульфид V2S5. Черный порошок V2S5
нерастворим в воде, но легко растворяется в щелочах с образованием тиосолей. В
инертной атмосфере диссоциирует по схеме V2S5 = V2S3 + S2- Способность к
образованию тиосолей в высшей степени окисления весьма характерна для эле-
элементов подгруппы ванадия (особенно для самого ванадия):
NH4VO3 + 4(NH4JS + ЗН2О = (NH4KVS4 + 6NH4OH
Это роднит их с элементами главной подгруппы V группы.
Ниобий и тантал с серой образуют моно- и дисульфиды Ээ и 3S2, а тантал,
кроме того, и
Все три металла, взаимодействуя с фосфором, образуют соединения двух
типов: монофосфиды ЭР и дифосфиды ЭР2, причем первые обладают металли-
металлическими свойствами, а вторые являются полупроводниками. Все известные арсе-
ниды и стибиды проявляют металлидный характер независимо от их состава.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Солеобразные производные, в которых элемент высту-
выступает в качестве катионообразователя, известны главным образом лишь для вана-
ванадия. Однако и они не очень устойчивы.
Низшие оксиды ванадия VO и V2O3 растворяются не только в концентриро-
концентрированных, но и в разбавленных кислотах, образуя гексааквакомплексы [У(Н2Об)]2+ и
[У(Н2Об)]3+, отвечающие соответствующим солям ванадия. Например, известен
кристаллогидрат VSO4'7H2O, представляющий собой сверхкомплексное соедине-
соединение [V(H2O)E]SO4'H2O. Стабилизация солеобразных производных V(-t-2) и V(-f3)
достигается также путем образования двойных солей типа квасцов или шёнитов,
например Me2SO4-V2(SO4K-24H2O и Me2SO4-VSO4-6H2O.
Поскольку солеобразные производные ванадия, ниобия и тантала в водных
растворах сильно гидролизованы по катиону, их выделение представляет извест-
известные трудности, особенно с учетом ярко выраженной восстановительной активнос-
активности низших производных. Однако сухим путем удается получить индивидуальные
соли даже для нлобия и тантала в высших степенях окисления. Существование
сульфатов и фосфатов 92(SO4M и Э3(РО4M ниобия и тантала и отсутствие подоб-
подобных соединений ванадия указывают, во-первых, на стабилизацию высшей степени
окисления в ряду V — Nb — Та, а во-вторых, на усиление основного характера в
ряду V(+5) - Nb(+5) - Ta(+5).
В целом комплексообразование для элементов VB-группы не очень характер-
характерно, за исключением галогенидных ацидокомплексов типа Ме3[\Тй], Me [ЭГЙ],
МегрГб], а также рассмотренных выше оксокомплексов.
Металлохимия элементов V В-г р у п п ы. Ванадий, ниоблй и
тантал в любых комбинациях образуют друг с другом непрерывные твердые
растворы, что отмечается также в системах, образованных этими металлами с
изоструктурными (ОЦК) полиморфными модификациями других переходных
металлов, не сильно отличающихся по электронному строению. Так, ванадий
образует непрерывные твердые растворы с /?-титаном, металлами подгруппы
хрома, 6-марганцем, а-железом; ниобий образует непрерывные растворы в твер-
твердом состоянии с /^-модификациями всех металлов подгруппы титана, молибденом,
вольфрамом и 7~УРаном> тантал водет себя в этом отношении аналогично ниобию.
В тех случаях, когда сочетание металлохимических факторов не благоприятству-
благоприятствует полной взаимной растворимости, при взаимодействии с «/-металлами образуют-
образуются ограниченные твердые растворы с широкими областями гомогенности.
Для элементов VB-группы характерно образование соединений Курнакова,
причем не только в системах с другими переходными металлами (VFe, VFe3,
VCo3, VNi, VPd2 и т.д., NbPd3, NbPt3 и т.д., TaPd2), но и между собой (TaV2).
Помимо характерных для соединений Курнакова составов А3В и АВ здесь встре-
встречается состав АВ2, что обусловлено особенностями формирования упорядоченных
твердых растворов в системах с ОЦК-решеткой. Многие из этих соединений при
низших температурах обладают сверхпроводимостью.
Особое значение в металлохимии элементов подгруппы ванадия имеют метал-
лоподобные фазы внедрения. Бориды образуются по механизму внедрения и в
октаэдрические, и в тетраэдрические пустоты, т.е. имеют формульные составы
ЭВ^я и ЭВ2-а;- Карбиды и нитриды образованы только за счет внедрения углеро-
углерода и азота в октаэдрические пустоты. По тому же механизму образуются и гидри-
гидриды. Отметим, что даже кислород, обладающий значительной электроотрицатель-
электроотрицательностью, при небольшом относительном содержании способен образовывать метал-
лидные фазы внедрения, такие, как ТабО, ТагО, NbgO, МЪгО и даже TaO, NbO и
VO.
Г Л А В А XX. ЭЛЕМЕНТЫ VI ГРУППЫ
Первый типический элемент VI группы — кислород — самый распространен-
распространенный элемент на Земле: его содержание составляет почти 50 масс, долей, %. А по
ОЭО кислород стоит на втором месте после фтора и поэтому образует огромное
число соединений с другими элементами Периодической системы. Не случайно
большая часть неорганической химии посвящена кислородным соединениям.
Первоначально классификация неорганических веществ, кислотно-основное взаи-
взаимодействие, окислительно-восстановительные процессы рассматривались в рамках
приоритетной роли кислорода и его самого важного соединения — воды.
По числу соединений и их производных, образуемых кислородом со вторым
типическим элементом — серой, VI группа не знает аналогов, что свидетельствует
о большом химическом сродстве между двумя типическими элементами. Об этом
же говорит и тенденция к взаимозаменяемости этих элементов в рядах оксид —
сульфид, оксокислоты — тиокислоты, оксосоли — тиосоли и т.д., проходящая
красной нитью через всю неорганическую химию.
Для водородных соединений элементов VIA-группы наблюдается увеличение
прочности и характеристичности по сравнению с элементами VA-группы. Причи-
Причина этому — большая электроотрицательность элементов VIA-группы по сравнению
с элементами VA-группы в пределах каждого периода. Стабильность кислород-
кислородных соединений элементов VIA-группы, наоборот, меньше, чем для элементов
предыдущих групп.
1. Кислород. Особая роль кислорода в химии. В становле-
становлении и развитии классической неорганической химии неоценимая роль принадле-
принадлежит кислороду. Еще Берцелиус утверждал, что кислород — это та ось, вокруг
которой вращается химия. Обусловлено это двумя причинами. Во-первых, чрез-
чрезвычайно большая распространенность и исключительная реакционноспособность
кислорода определяет многообразие форм его соединений. Во-вторых, классичес-
классическая неорганическая химия в основном — это химия водных растворов. Другими
словами, она представляет собой химию самого распространенного и самого глав-
главного соединения кислорода — оксида водорода. Поэтому многие основополагаю-
основополагающие понятия, такие, как валентность по кислороду, окислительное число, окис-
окисление, горение, кислоты и основания, соли и т.д., были сформулированы приме-
применительно к кислороду и его важнейшим соединениям. Больше того, до 1961 г.
применялась кислородная шкала атомной единицы массы.
Кислород в природе. Получение кислород; а. Извест-
Известно более 1400 минералов, содержащих кислород. Важнейшие кислородсодержа-
кислородсодержащие минералы — кварц и его модификации, полевые шпаты, слюды, глины,
известняки. Огромное количество кислорода находится в воде как в химически
связанном, так и в растворенном состоянии. В свободном состоянии кислород
находится в атмосфере (около 1015 т). Кислород.воздуха расходуется в-процессах
горения, гниения, ржавления, дыхания и непрерывно регенерируется за счет
фотосинтеза. Кроме того, кислород является обязательной составной частью
организмов животных и растений. Так, в человеческом теле содержится до 65
масс, долей, % кислорода.
В технике кислород получают фракционированной перегонкой жидкого возду-
воздуха и электролизом воды (как побочный продукт при получении водорода), а в
лаборатории — при термическом распаде оксидов (СгОз), пероксидов (ВаОг),
солей оксокислот (KNO3, КС1О3, КМпО^).
Физические и химические свойства кислорода.
Кислород — газ без запаха и цвета. Вследствие плохой деформируемости элект-
электронной оболочки кислород имеет низкие температуры плавления и кипения.
Жидкий кислород светло-голубого цвета, а твердый — кристаллы синего цвета.
Во всех агрегатных состояниях кислород парамагнитен. Он мало растворим в
воде: в 100 объемах воды при 20°С растворяется 3 объема кислорода. Но эта
небольшая растворимость имеет огромное значение для жизнедеятельности живу-
живущих в воде организмов.
Метод МО обосновывает химическое строение молекулы кислорода. Наличие
двух неспаренных электронов на разрыхляющей МО ж* делает его молекулу
бирадикалом и объясняет парамагнетизм кислорода. Энергия атомизации моле-
молекулы кислорода 498,4 кДж/моль несравненно меньше, чем молекулы азота. Это
одна из причин большей реакционноспособности кислорода по сравнению с азо-
азотом. Под действием УФ-излучения легко происходит фотолиз молекул кислоро-
кислорода, поэтому на высоте более 100 км от поверхности земли основной формой су-
существования кислорода является атомарный. Аллотропной модификацией кисло-
кислорода является озон О3. В химическом строении молекулы озона центральный
атом кислорода подвергается зр2-гибридизации, а его 2р;горбиталь с такими же
орбиталями крайних атомов образует 7Гр-р-связи вдоль всей молекулы:
Длина связи между атомами кислорода меньше длины одинарной @,149 нм) и
больше длины двойной связи @,121 нм). Озон — газ синего цвета, молекулы
которого диамагнитны. Цвет его обусловлен большей поляризуемостью молекулы
Оз по сравнению с кислородом.
Озон получается при действии тихого электрического разряда на кислород
(до 10 масс, долей, % Оз). В атмосфере озон образуется при грозовых разрядах и
в верхних слоях под действием УФ-излучения. Озон сильно эндотермичен
(АЩШ — +142,3 кДж/моль), а потому реакционноспособен. Его окислительные
свойства выражены несравненно сильнее, чем у кислорода. Это видно из сравне-
сравнения редокс-потенциалов процессов с образованием воды:
Оз + 2Н+ + 2е" = О2 + Н2О, Ё° - +2,07 В,
О2 + 4Н+ + 4е" = 2Н2О, Е° = +1,23 В.
433
По химической активности кислород уступает только фтору. С большинством
простых веществ он реагирует непосредственно, за исключением галогенов, благо-
благородных газов, платины и золота. Два неспаренных электрона в невозбужденном
состоянии атома кислорода определяют его двухвалентность. Однако максималь-
максимальная ковалентность его равна 4. Атом кислорода может находиться в sp-, sp2- и
*р3-гибридном состоянии. В соединениях с делокализованными связями
координационное число кислорода может быть больше 4 и достигать 6 и 8.
Например, в оксидах со структурным типом NaCl каждый атом кислорода
соединен тремя трехцентровыми связями с шестью соседними атомами металла
(к. ч. 6). А в оксидах типа Na2O координационное число достигает 8, и он
образует четыре трехцентровые связи с 8 атомами металла.
Оксиды неметаллов. Известны оксиды всех неметаллов, получен-
полученные непосредственно или косвенно, за исключением оксидов гелия, неона и арго-
аргона. Поскольку разность ОЭО кислорода и неметаллов относительно невелика,
природа химической связи в оксидах неметаллов преимущественно ковалентная.
Поэтому в подавляющем большинстве случаев оксиды неметаллов — газы, легко-
летуччз жидкости или легкоплавкие твердые вещества. В твердом состоянии, как
правило, образуются молекулярные структуры из-за насыщаемости и направлен-
направленности ковалентных связей. Однако при наличии заметной доли ионной составля-
составляющей связи возникают координационные структуры, например в случае диокси-
диоксида кремния. В оксидах неметаллов кислород чаще всего подвергается .^гибри-
.^гибридизации.
Оксиды неметаллов в большинстве случаев являются кислотообразующими
оксидами. Некоторые оксиды неметаллов, например СО и N0, индифферентны
по отношению к воде. Обусловлено это исключительной прочностью их молекул.
Например, гипотетическая реакция с образованием муравьиной кислоты
СО + НОН —+ НСООН
в действительности не имеет места потому, что ее энергетика не компенсирует
разрушения прочной тройной связи в оксиде углерода (+2).
С повышением степени окисления неметалла в оксидах растет их кислотный
характер:
Р4О10(к) + 6Н2О(ж) = 4НаРО4, AG°298 =-59,8 кДж/моль,
SO3(k) + Н20(ж) = H2SO4(k), AG°298 = -81,8 кДж/моль,
С12О7(г) + Н20(ж) = 2НС1О4(ж), &G°29s = -330,9 кДж/моль.
Оксиды металлов. Оксиды с преимущественно ионной связью (на-
(например, щелочных и щелочно-земельных металлов) характеризуются координа-
координационными структурами с к.ч. (кислорода) б или 8. С ростом степени окисления
металлического элемента возрастает ковалентный вклад в химическую связь.
Химическое строение таких оксидов молекулярное (например, СгОз, М112О7,
ИегОт и OsO4). Для таких оксидов нарушение стехиометрии невозможно. Для
оксидов металлов с преимущественно ионной связью (координационные решетки)
нарушение стехиометрии термодинамически обосновано, так как при этом растет
энтропия системы. Нестехиометричные оксидные фазы могут быть как односто-
односторонними (FeO), так и двусторонними (TiO).
лял
По отношению к воде оксиды металлов могут быть оснбвными, например
ВаО(к) + Н2О(ж) = Ва(ОНJ(к), Дб*98 = -97,9 кДж/моль,
или кислотными:
Re2O7(K) + Н2О(ж) = 2HReO4(K), AG°2gs = -19,6 кДж/моль.
Амфотерные оксиды, как правило, не взаимодействуют с водой.
Пероксиды, супероксиды и озониды. Энтальпия процесса
О3 + е~ — Оз равна Д#298 = -188,3 кДж/моль, а потому реакции типа К + О3 =
= КОз протекают самопроизвольно с образованием озонидов металлов. Озониды
обычно окрашены в красный цвет. Парамагнетизм и цвет озонидов обусловлены
холостым электроном озонид-иояа Ojj. Присоединение одного электрона к моле-
молекуле кислорода также сопровождается выделением энергии (Д#298 = -48,1
кДж/моль). Прибавление одного электрона к молекуле кислорода уменьшает
порядок связи до 1,5, но на разрыхляющей МО ж* вместо двух неспаренных
электронов остается один. Таким образом, образование супероксид-иона также
энергетически выгодно. Производные аниона О2 называются супероксидами. И не
случайно элементы подгруппы калия при взаимодействии с кислородом воздуха
образуют именно супероксиды, например КО2. Наличие неспаренного электрона
делает супероксиды парамагнитными веществами и обусловливает их окраску.
Прибавление двух электронов к молекуле кислорода требует затраты энергии,
и пероксид-ион О2" эндотермичен (Д#°д8 = 160 кДж/моль). При этом порядок
связи уменьшается до 1, энергия диссоциации падает, а расстояние между атома-
атомами кислорода возрастает по сравнению с супероксид-ионом. Поэтому пероксиды
менее стабильны по сравнению с супероксидами. Пероксиды не окрашены вслед-
вследствие отсутствия непарного электрона. Все вещества с избытком кислорода про-
против формальной валентности являются сильными окислителями.
2. Сера. Особенности химии серы. Химия второго типического
элемента VI группы — серы — отличается от химии кислорода по меньшей мере
двумя аспектами. Во-первых, наличие вакантных 3 rf-орбиталей увеличивает ва-
валентные возможности серы с учетом промотирования электронов на эти орбитали.
При этом возрастают и положительные степени окисления серы. Кроме того,
создаются условия для дополнительного тг-взаимодействия. Наконец, с участием
3rf-орбиталей резко возрастает возможное число типов гибридизации, что ведет к
увеличению координационного числа серы в ее производных.
Во-вторых, для химии серы очень важна прочность химической связи между
ее атомами, которая уступает только межатомным связям С—С и Si—О—Si. Поэто-
Поэтому в химии серы определенную роль играют гомоцепные производные, например
полисульфиды, политионовые кислоты и т.д. В то же время сера образует и
гетероцепные структуры с участием атомов кислорода. В этом проявляется гори-
горизонтальная аналогия в ряду кремний — фосфор — сера.
Природные соединения и получение серы. Сера отно-
относится к числу распространенных элементов; Формы нахождения серы МНОГСЮбраЗ-
ртутью реакция протекает даже при температуре жидкого воздуха. В свою оче-
очередь, сера хорошо окисляется кислородом и галогенами, а также кислотами-
окислителями. В горячих растворах щелочей она диспропорционирует:
3S + 6NaOH = 2Na2S + Na2SO3 + ЗН2О
Наиболее часто сера проявляет четные степени окисления: —2, 0, +2, +4 и +6.
Характеристические соединения. Кислоты, со-
содержащие серу, и их соли. При сжигании серы на воздухе образу-
образуется диоксид серы (сернистый газ), обладающий резким запахом. Он токсичен,
легко сгущается в неэлектропроводную жидкость. В технике диоксид серы полу-
получают при окислительном обжиге сульфидов металлов, а в лаборатории — дейст-
действием концентрированной серной кислоты на медь:
Си + 2H2SO4 = CuSO4 + SO2 + 2Н2О
Химическое строение молекулы диоксида серы аналогично молекуле озона.
Атом серы находится в состоянии «р2-гибридизации. Из-за большего размера
атома серы длина связи S—О @,143 нм) больше, чем О—О @,128 нм) в молекуле
озона. Но по энергии связь S—О D89 кДж/моль) в молекуле SO2 в несколько раз
превосходит связь О—О A14,2 кДж/моль) в озоне. Обусловлено это дополнитель-
дополнительным ^-взаимодействием за счет вакантных d-орбиталей атома серы и заметным
вкладом ионной доли связи в случае молекулы SO2.
Полярные молекулы SO2 хорошо растворяются в воде. Лишь небольшая часть
растворенных молекул взаимодействует с водой и устанавливается равновесие:
SO2 + Н2О *=* H2SO3 ^НЧ HSOs ^ 2Н+ + SO!'
Равновесие сильно смещено влево. Сернистая кислота H2SO3 не выделена в сво-
свободном состоянии; это кислота средней силы (pA'i 1,7 и рА'2 7,3). Соли сернистой
кислоты — сульфиты и гидросульфиты — получают взаимодействием SO2 с щело-
щелочами или растворимыми карбонатами металлов.
В силу нестабильности степени окисления +4 SO2, H2SO3 и сульфиты функци-
функционируют как восстановители. Даже твердые сульфиты при хранении медленно
окисляются до сульфатов:
2Na2SO3 + О2 = 2Na2SO4
Однако с сильными восстановителями производные серы (+4) проявляют окисли-
окислительные свойства, например SO2 + 2H2S = 3S + 2Н2О (конпропорционирование).
Гидросульфиты при нагревании превращаются в пиросульфиты — соли неизвест-
неизвестной в свободном состоянии пиросернистой (дисернистой) кислоты H2S2O5:
2KHSO3 = K2S2OS + Н2О
Производное серы (+4) — тионилхлорид SOC12 — получают по реакции
SO2 + PCls = РОС13 + SOC12
Тионилхлорид — бесцветная жидкость с резким запахом. Химическое строение
его молекулы пирамидальное, с атомом серы в «рЗ-гибридном состоянии. Тионил-
. - . 437
ртутью реакция протекает даже при температуре жидкого воздуха. В свою оче-
очередь, сера хорошо окисляется кислородом и галогенами, а также кислотами-
окислителями. В горячих растворах щелочей она диспропорционирует:
3S + 6NaOH = 2Na2S + Na2SO3 + ЗН2О
Наиболее часто сера проявляет четные степени окисления: -2, 0, +2, +4 и +6.
Характеристические соединения. Кислоты, со-
содержащие серу, и их соли. При сжигании серы на воздухе образу-
образуется диоксид серы (сернистый газ), обладающий резким запахом. Он токсичен,
легко сгущается в неэлектропроводную жидкость. В технике диоксид серы полу-
получают при окислительном обжиге сульфидов металлов, а в лаборатории — дейст-
действием концентрированной серной кислоты на медь:
Си + 2H2SO4 = CuSO4 + SO2 + 2Н2О
Химическое строение молекулы диоксида серы аналогично молекуле озона.
Атом серы находится в состоянии яр2-гибридизации. Из-за большего размера
атома серы длина связи S—О @,143 нм) больше, чем О—О @,128 нм) в молекуле
озона. Но по энергии связь S—О D89 кДж/моль) в молекуле SO2 в несколько раз
превосходит связь О—О A14,2 кДж/моль) в озоне. Обусловлено это дополнитель-
дополнительным ^-взаимодействием за счет вакантных rf-орбиталей атома серы и заметным
вкладом ионной доли связи в случае молекулы SO2.
Полярные молекулы SO2 хорошо растворяются в воде. Лишь небольшая часть
растворенных молекул взаимодействует с водой и устанавливается равновесие:
SO2 + Н2О *=* H2SO3 ^НЧ ШОз ^ 2Н+ + SO|"
Равновесие сильно смещено влево. Сернистая кислота H2SO3 не выделена в сво-
свободном состоянии; это кислота средней силы (pA'i 1,7 и pA'2 7,3). Соли сернистой
кислоты — сульфиты и гидросульфиты — получают взаимодействием SO2 с щело-
щелочами или растворимыми карбонатами металлов.
В силу нестабильности степени окисления +4 SO2, H2SO3 и сульфиты функци-
функционируют как восстановители. Даже твердые сульфиты при хранении медленно
окисляются до сульфатов:
2Na2SO3 + О2 = 2Na2SO4
Однако с сильными восстановителями производные серы (+4) проявляют окисли-
окислительные свойства, например SO2 + 2H2S = 3S + 2Н2О (конпропорционирование).
Гидросульфиты при нагревании превращаются в пиросульфиты — соли неизвест-
неизвестной в свободном состоянии пиросернистой (дисернистой) кислоты H2S2O5:
2KHSO3 = K2S2O5 + Н2О
Производное серы (+4) — тионилхлорид SOC12 — получают по реакции
SO2 + PCI5 = РОС13 + SOC12
Тионилхлорид — бесцветная жидкость с резким запахом. Химическое строение
его молекулы пирамидальное, с атомом серы в «рЗ-гибридном состоянии. Тионил-
437
хлорид проявляет преимущественно восстановительные свойства. Бурно реагирует
с водой, образуя сернистую кислоту:
SOC12 + 2Н2О = 2НС1 + H2SO3
Производным серы (+1) является оксид S2O или S(SO), имеющий такую же
структуру, как и SO2:
ё <?
120"
S2O — желтый газ, легко реагирует с водой:
S2O + Н2О = H2S2O2
Образующуюся кислоту H2S2O2 можно рассматривать как сернистую кислоту, в
которой один атом кислорода заменен на атом серы. Поэтому она называется
тиосернистой. Однако в свободном состоянии не выделены ни тиосернистая кис-
кислота, ни ее соли.
Пропускание сероводорода в разбавленный раствор SO2 в воде приводит к
образованию жидкости Вакенродера. В ее составе коллоидная сера и политионо-
политионовые кислоты обобщенной формулы H2Si06, где х принимает значения от 3 до 20
и более. Степень окисления серы зависит от числа ее атомов в структуре кислоты
и может принимать самые различные значения, включая и дробные. В политио-
новых кислотах осуществляется цепочка из атомов серы, соединенных между
собой яр3-гибридными ковалентными связями. Только крайние атомы серы соеди-
соединены еще с тремя атомами кислорода. Для примера приводим химическое строе-
строение пентатионовой кислоты:
Н-О.
о
'о
о
о-н
чо
Политионовые кислоты не выделены в свободном состоянии, но представляют
собой довольно сильные кислоты. Значительно более стабильны политионаты,
особенно щелочных металлов. Кислые политионаты неизвестны.
Дитионовая кислота H2S2O6 не относится к политионовым, так как две пира-
пирамидальные [8Оз]-группы соединены между собой непосредственно через атомы
серы. Дитионат марганца получают пропусканием SO2 через взвесь МпО2:
2SO2 + MnO2 = MnS2O6
А взвесь цинковой пыли в воде, наоборот, восстанавливает SO2:
Zn + 2SO2 = ZnS2O4
Образующаяся дитионистая кислота H2S2O4 (тетраоксодисерная) представляет
собой двухоснбвную кислоту средней силы. Химическое строение дитионистой
ЛЧй
кислоты симметричное, с двумя четырехвалентными атомами серы. Дитионистая
кислота и ее соли — дитиониты — легко окисляются кислородом до сульфатов.
Степень окисления +3, как в дитионистой кислоте, сера имеет в полуторном
оксиде БзОз- Получается он взаимодействием сухого триоксида SO3 с мелкоиз-
мельченной серой. Полуторный оксид не отличается стабильностью и диспропор-
ционирует:
2S2O3 = 3SO2 + S
Еще меньше степень окисления серы в сульфоксиловой (диоксосерной) кисло-
кислоте, получающейся при гидролизе дихлорида серы SC12:
SC12 + 2Н2О = 2НС1 + H2SO2
Достоверно установлено существование только одной ее соли CoSO2 — сульфокси-
лата кобальта. Формально такой же степенью окисления серы +2 характеризуют-
характеризуются атомы серы в тиосерной кислоте H2S2O3 и ее солях — тиосульфатах. Последние
образуются при кипячении крепких растворов сульфитов с тонкоизмельченной
серой:
Na2SO3 + S = Na2S2O3
Тиосерная кислота но силе приближается к серной. Тиосульфат-ион представля-
представляет собой чуть искаженный тетраэдр, так как связь S—S длиннее связи S—О:
;
Тиосульфаты окисляются даже слабыми окислителями, например
2Na2S2O3 + h = Na2S4O6 + 2NaI
Диоксид серы на свету легко окисляется хлором:
SO2 + С12 = SO2C12
Хлористый сульфурил SO2C12 — резко пахнущая бесцветная жидкость. Горячей
водой разлагается с образованием серной кислоты:
SO2C12 + 2Н2О = H2SO4 + 2HC1
По структуре его можно представить как продукт замещения обоих гидроксидов
в молекуле серной кислоты на атомы хлора. А при замене одного гидроксила на
хлор получается хлорсульфоновая кислота HSO3C1. Водой хлорсульфоновая
кислота разлагается, как и сульфурил хлор ид. По химическому строению сульфу-
рилхлорид представляет собой искаженный тетраэдр.
439
При повышенной температуре и в присутствии катализатора оксид серы (+4)
окисляется кислородом с образованием оксида серы (+6):
2SO2 + О2 = 2SO3
| В парообразном состоянии триоксид SO3 существует в виде молекул с атомом
серы в центре правильного треугольника (яр2-гибридизация). В жидком состоя-
состоянии триоксид серы представлен тримерами. В них атомы серы находятся в сос-
состоянии яр3-гибридизации и образуют тетраэдры. Тетраэдры между собой связаны
мостиковыми атомами кислорода. В твердом состоянии SO3 имеет гетероцепную
полимерную структуру.
Растворение SO3 в воде сопровождается выделением теплоты (Д#°98 = -62,7
кДж/моль) и образованием серной кислоты:
SO3 + Н2О = H2SO4
Безводная серная кислота, называемая также моногидратом, представляет собой
вязкую маслянистую жидкость без запаха и цвета. В водных растворах ведет себя
как сильнейшая двухоснбвная кислота (рК\ 3 и рА'2 2). Химическое строение
молекулы H2SO4 отвечает искаженному тетраэдру. Еще в большей степени иска-
искажение в гидросульфат-ионе HSO^- Сульфат-ион SO4" имеет форму правильного
тетраэдра; все расстояния и углы между связями в нем равноценны:
о
I
ho-"-/S=^o
но
но-
о
о
Концентрированная серная кислота — сильный окислитель. В зависимости от
силы восстановителя она может быть восстановлена до диоксида серы, серы и
сероводорода.
Большинство сульфатов бесцветны, хорошо кристаллизуются, из водных раст-
растворов выделяются в виде кристаллогидратов. Термически стойки сульфаты ще-
щелочных и щелочно-земельных металлов. Сульфаты менее активных металлов при
нагревании разлагаются, например:
ZnSO4 = ZnO + S03; Ag2SO4 = 2Ag + S02 + 02
Гидросульфаты легко плавятся, а после плавления переходят в пиросульфаты
(дисульфатыI:
2NaHSO4 = Na2S2O7 + Н20
Пиросерная (дисерная) кислота H2S2O7 представляет собой продукт присоеди-
присоединения SO3 к моногидрату. Вообще серная кислота способна присоединять боль-
большие количества SO3. Такие растворы являются очень вязкими и называются
олеумом (маслоподобный). Состав олеума можно представить формулой
440
H2SO4-zSO3. При х = 1 получается дисерная кислота. Ока выделяется в виде
бесцветных кристаллов при охлаждении до комнатной температуры эквимоляр-
ных смесей H2SO4 и SO3. Химическое строение дисерной кислоты характеризует-
характеризуется тем, что атомы серы связаны через кислородный мостик, что дает основание
считать ее изополикислотой. В олеуме содержатся также трисерная и тетрасерная
кислоты. Олеум по сравнению с концентрированной серной кислотой обладает
еще большим водоотнимающим и окислительным действием.
Взаимодействием хлорсульфоновой кислоты с пероксидом водорода получают
пероксосерные кислоты. Пероксомоносерная (мононадсерная) H2SO5 (или кислота
Каро) и пероксодисерная (динадсерная) H2S2O8 представляют собой бесцветные
гигроскопические кристаллы. Обе кислоты бурно реагируют с водой:
H2S2O8 + Н2О = H2SO5 + H2SO4
H2SO5 + Н2О = H2SO4 + Н2О2
Пероксодисерную кислоту в технике получают анодным окислением гидросуль-
гидросульфат-аниона. Сначала на аноде гидросульфат-анион превращается в
гидросульфат-радикал, который димеризуется:
2HSOi - 2е" = 2HSO4; 2HSO4 = H2S2O8
Пероксосерные кислоты и их соли — пероксосульфаты — являются сильнейшими
окислителями. Так, для процесса S2OJ3 + 2е~ = 2SO4" редокс-потенциал равен
+2,01 В.
Соединения серы с другими неметаллами. Важней-
Важнейшим характеристическим летучим водородным соединением серы является серо-
сероводород. Для его получения обычно используют реакцию вытеснения H2S из
сульфида железа соляной кислотой:
FeS + 2HC1 = FeCl2 + H2S
Сероводород — бесцветный газ тяжелее воздуха, обладающий неприятным запа-
запахом. Он очень токсичен. Отравляющее действие сероводорода объясняют его
взаимодействием с железом гемоглобина. При этом функция гемоглобина как
переносчика кислорода нарушается или вовсе парализуется. Химическое строение
H2S аналогично строению воды, если не учитывать малую степень гибридизации
орбиталей атома серы. Молекула H2S намного менее полярна, чем молекула воды,
вследствие того, что ОЭО серы меньше, чем кислорода. Поэтому в сероводороде
водородные связи практически отсутствуют в любом агрегатном состоянии. Соб-
Собственная ионизация сероводорода ничтожна, и его ионное произведение
[HgS+][HS"] = 10~33. В воде ионизация сероводорода
H2S + Н2О ^ Н3О+ + HS"
приводит к образованию слабой сероводородной кислоты H2S (pifj 7,3 и
рК2 13,8). Соли сероводородной кислоты (сульфиды) тяжелых металлов в воде
нерастворимы.
441
Водные растворы сульфидов щелочных металлов растворяют серу с образова-
образованием полисульфидов:
Na2S + S = Na2S2; Na2S + 2S = Na2Ss и т.д.
С увеличением "содержания серы в полисульфиде интенсивность окраски раство-
растворов меняется от желто-оранжевого до красного цвета (Na2Sg). Из полисульфидов
получают многосернистые водороды — полисульфаны. Для этого концентрирован-
концентрированный раствор полисульфида медленно вливают в соляную кислоту:
2НС1 + Na2Sx = 2NaCl + H2SX
На дне сосуда собирается тяжапая маслянистая жидкость, состоящая из смеси
полисульфанов H2SX. Идентифицированы все члены ряда, вплоть до H2Sg. Моле-
Молекулы полисульфанов, равно как и полисульфиды, представляют собой зигзагооб-
зигзагообразные цепи из атомов серы в вр3-ги6ридном состоянии:
А А А/
s. ,s, s.
Цепи замыкаются либо атомами водорода (полисульфаны), либо атомами щелоч-
щелочных металлов (полисульфиды). В воде полисульфаны являются кислотами, при-
причем сила кислоты растет с увеличением числа атомов серы в полисульфид-ионе.
Так, тетрасульфан H2S4 — более сильная кислота, чем уксусная и тем более серо-
сероводородная. Полисульфиды устойчивее полисульфанов, проявляют и окислитель-
окислительные, и восстановительные свойства.
Отношение серы к галогенам в первую очередь регламентируется фактором
ОЭО, хотя некоторую роль играют и размерные факторы. Иод, ОЭО которого
равна таковой для серы B,6), не образует соединений с серой. Наибольшее число
соединений с серой образует фтор. Разность ОЭО между этими элементами сос-
составляет 1,3. Известны хлориды серы SC12 и S2C12. Из бромидов серы известен
только S2Br2.
Прямое фторирование серы приводит к образованию SF6 с примесями S2Fio и
SF4. Гексафторид SF6 — газ, очень устойчивый к химическим воздействиям. Он
не реагирует с водой, щелочами, кислотами, водородом, металлами. Его молекула
неполярна, структура октаэдрическая (sp* (^-гибридизация атома серы). Таким
образом, в молекуле SF6 атом серы валентно и координационно насыщен и прост-
пространственно экранирован шестью атомами фтора. К тому же связь S—F характери-
характеризуется высокой прочностью C21,3 кДж/моль). В то же время расчеты показыва-
показывают, что гидролитическая реакция SF6 с образованием SO2 и HF характеризова-
характеризовалась бы ДС°д8 = -460 кДж. Поэтому в целом низкая химическая активность SF6
обусловлена кинетическими факторами, а не термодинамической стабильностью
этого вещества.
Тетрахлорид серы SC14 неустойчив. При комнатной температуре диссоциирует
на низшие хлориды серы и хлор. Хлорирование расплавленной серы ведет к
образованию смеси ди- и монохлорида серы. Дихлорид нестоек и медленно раз-
разлагается:
2SC22 = S2C22 + С22
442
Молекула SCI2 — угловая с атомом серы в состоянии вр2-гибридизации. Устойчи-
Устойчивость моногалогенидов S2r2 уменьшается от фтора к брому. Все они легко гидро-
лизуются водой.
Сульфиды металлов. В силу существенно меньшей величины ОЭО
серы по сравнению с кислородом B,6 и 3,5) сульфиды металлов более ковалент-
ны, чем оксиды. Менее ионный характер межатомной связи приводит к тому, что
сульфиды металлов являются более явно выраженными полупроводниками в
отличие от оксидов, большинство которых — изоляторы. Если оксиды бесцветны
или слабо окрашены, сульфиды нередко отличаются интенсивной окраской.
Объясняется это тем, что при переходе от кислорода к сере растет емкость элект-
электронной оболочки, а вместе с ней ее способность к деформации. Другими словами,
атомы серы в сульфидах легче поляризуются, чем атомы кислорода в оксидах.
При этом с ростом поляризующего действия катионообразователя интенсивность
окраски возрастает. Так, HgS — красного, CdS — оранжевого, ZnS — желтого
цвета.
Сульфиды щелочных металлов с нормальной стехиометрией по своей химичес-
химической природе являются основными. Поэтому при их гидролизе возникает щелоч-
щелочная среда, например
Na2S + Н2О ^ NaHS + NaOH, РЛ™др -1,
NaHS + Н2О ^ H2S + NaOH, рА'гидр 7.
Сульфиды щелочных и щелочно-земельных металлов окисляются кислородом
воздуха, хлором, а также подвергаются карботермии с образованием карбидов
металлов.
Ковалентные сульфиды образуют в основном sp-металлы, особенно с конфигу-
конфигурацией внешних электронов ns2npl (Al, Ga, In, T1). Большая часть этих сульфи-
сульфидов имеет сложное кристаллохимическое строение, образуя слоистые и координа-
координационные структуры. По химической природе эти сульфиды амфотерны, малоус-
малоустойчивы по отношению к химическим реагентам, активно взаимодействуют с
кислородом,галогенами.
Наиболее разнообразны сульфиды переходных металлов: Me2S, MeS, Me,-?S4,
Me2S3, MeS2, MeS3. Многие из этих сульфидов (особенно не подчиняющиеся
правилу формальной валентности) являются соединениями переменного состава.
При малом содержании серы в формульной единице связь преимущественно
металлическая, т.е. эти сульфиды являются металлоподобными. С увеличением
содержания серы и появлением связей между атомами серы сульфиды становятся
полупроводниками. Наибольшим сродством к сере обладают лантаноиды. Напри-
Например, энтальпия образования LaS (ДЯ}298 = -751,3) и Ce2S3 (ДЯ}298 = -1257,2
кДж/моль) на порядок превосходит таковую для многих ковалентных сульфидов.
Среди сульфидов лантаноидов наиболее изучены MeS и Me2Ss. Первые металло-
подобны, а вторые — полупроводники. Их химическая стойкость к действию
различных реагентов несравненно выше, чем у других типов сульфидов металлов.
Особенно ценным их качеством является высокая устойчивость к действию ра-
расплавов солей, металлов и сплавов.
3. Подгруппа селена. Характеристика элементов VI А-
группы. Ниже приведены некоторые свойства элементов VIA-группы и их
гомоатомных соединений:
443
ции. Гексагональный селен — темно-серый с коричневым отливом, а теллур —
блестящее серебристо-серое хрупкое вещество с металлическим блеском. Селен и
теллур являются полупроводниками. Вследствие металлизации связи переход от
селена к теллуру сопровождается уменьшением ширины запрещенной зоны A,8 и
0,35 эВ),
По химическому поведению селен, как и сера, — типичный неметалл, чего
нельзя сказать относительно теллура. Так, например, галогениды ТеГ2 и ТеГ4
имеют солеобразный характер. Селен и теллур взаимодействуют с кислородом,
галогенами и другими окислителями, переходя в состояния окисления +4 и +6.
С разбавленными кислотами-окислителями селен не реагирует. Теллур окисляет-
окисляется даже водой:
Те + 2Н2О = ТеО2 + 2Н2
При нагревании селен и теллур взаимодействуют с металлами, выступая в качест-
качестве окислителей и образуя селениды и теллуриды:
2Cu + Se = Cu2Se, 2Ag + Те = Ag2Te
Кипячение с растворами щелочей приводит селен и теллур, подобно сере, к дис-
пропорционированию:
нагревание
3Se + 6К0Н ' охлаждение ' 2K2Se + K2Se03 + ЗН2О
Характеристические соединения и соли селен- и
телдурсодержащих кислот. При сгорании селена и теллура на
воздухе или в токе кислорода образуются диоксиды. В отличие от сернистого
газа диоксиды селена и теллура — твердые полимерные вещества. SeO2 имеет
цепочечную структуру, в которой атомы селена в состоянии врЗ-гибридизации
окружены тремя атомами кислорода, находящимися в трех вершинах сильно
искаженного тетраэдра. Четвертая вершина занята неподеленной электронной
парой селена.
ТеО2 кристаллизуется в координационной структуре рутила, что свидетельст-
свидетельствует о существенно ионном характере межатомной связи. Оба диоксида прекрасно
растворяются в щелочах, например
SeO2 + 2NaOH = Na2SeOs + Н2О
Образующиеся соли селенистой и теллуристой кислот — селениты и теллуриты —
бесцветны и похожи на сульфиты.
При действии сильных кислот на селениты и теллуриты селенистая и теллу-
ристая кислоты выделяются в свободном состоянии. H2Se03 — белое гигроскопи-
гигроскопическое вещество. Н2Те0з склонна к полимеризации, состав ее переменен и может
быть в общем виде изображен формулой ТеО2*жН2О. Обе кислоты относятся к
слабым кислотам и проявляют амфотерность, особенно теллуристая. Сильные
окислители переводят эти соединения в производные селена и теллура в степени
окисления +6:
5H2Se03 + 2КМпО4 + 3H2SO4 = 5H2Se04 + 2MnSO4 + K2SO4 + 3H2O
445
II
III
Селеновая кислота существует в форме летонкислоты H2Se04, а теллуровая — в
виде opmo-кислоты Н6ТеО6. Обе кислоты — бесцветные твердые вещества. По
силе селеновая кислота близка к серной, а теллуровая слабая. В то же время в
ортотеллуровой кислоте все шесть атомов водорода могут быть замещены на
металл; например, известны ортотеллураты Ag6TeOe и Hg3TeOe. При нагревании
Н6Те0б в запаянных ампулах образуется сиропообразная масса, переходящая в
моноклинные кристаллы слоистой структуры, представляющие собой метателлу-
ровую кислоту Н2Те04. Она намного сильнее opmo-формы. Растворы Н2Те04
постепенно превращаются в opmo-кислоту. И селеновая и теллуровая кислоты
образуют, подобно сере, изополикислоты, а теллуровая кислота способна давать и
гетерополикисл оты.
Селеновая и теллуровая кислоты — медленно действующие, но сильные окис-
окислители. По окислительной активности они превосходят серную кислоту:
SeOf + 4Н+ + 2е" = H2Se03 + Н2О, Е° = 1,15 В,
Н6Те06 + 2Н* + 2е" = ТеО2 + 4Н2О, Е° = 1,02 В,
SOf + 4Н+ + 2е" = H2SO3 + Н2О, Е° = 0,17 В.
Поэтому селеновая и теллуровая кислоты в отличие от серной окисляют НС1
до хлора:
H2Se04 + 2HC1 = С12 + H2Se03 + Н2О
В селеновой кислоте растворяется металлическое золото, которое нерастворимо в
горячей серной кислоте:
2Au + 6H2Se04 = Au2(Se04K + 3SeO2 + 6Н2О
А смесь концентрированных селеновой и соляной кислот растворяет даже пла-
платину:
2H2Se04 + 4НС1 + Pt = PtCl4 + 2SeO2 + 4H2O
В отличие от теллуратов селенаты во многих отношениях похожи на сульфа-
сульфаты. Сульфаты и селенаты однотипных металлов близки по растворимости, крис-
таллохимическому строению, по способности образовывать двойные соли типа
квасцов. Среди сульфатов и селенатов много примеров изоморфизма.
При нагревании ортотеллуровой кислоты образуется триоксид:
Н6Те06 = ЗН2О + ТеО3
Триоксид теллура диморфен. Обе модификации химически' малоактивны, не
растворяются в воде, разбавленных кислотах и основаниях. Одна из модифика-
модификаций взаимодействует с щелочью лишь при сплавлении.
SeOg получают вытеснением из селен ата триоксид ом серы:
K2SeO4 + SOS = K2SO4 + SeOs
Триоксид селена также образует две модификации: асбестовидную и стекловид-
стекловидную. Обе формы имеют молекулярную структуру. Триоксид селена хорошо раст-
растворяется в воде. В концентрированных растворах SeOe существуют смеси полисе-
полиселеновых кислот. С разбавлением полиселеновые кислоты деполимеризуются и
446
Селеновая кислота существует в форме летанкислоты H2Se04, a теллуровая — в
виде opmo-кислоты HeTeOg- Обе кислоты — бесцветные твердые вещества. По
силе селеновая кислота близка к серной, а теллуровая слабая. В то же время в
ортотеллуровой кислоте все шесть атомов водорода могут быть замещены на
металл; например, известны ортотеллураты AgeTeOe и Hg^TeOg. При нагревании
НбТеОе в запаянных ампулах образуется сиропообразная масса, переходящая в
моноклинные кристаллы слоистой структуры, представляющие собой метателлу-
ровую кислоту Н2Те04. Она намного сильнее opmo-формы. Растворы Н2Те04
постепенно превращаются в opmo-кислоту. И селеновая и теллуровая кислоты
образуют, подобно сере, изополикислоты, а теллуровая кислота способна давать и
гетерополикислоты.
Селеновая и теллуровая кислоты — медленно действующие, но сильные окис-
окислители. По окислительной активности они превосходят серную кислоту:
+ 4Н+ + 2е" = H2Se03 + Н2О, Е° = 1,15 В,
Н6Те06 + 2Н+ + 2е" = ТеО2 4- 4Н2О, Е" = 1,02 В,
SOf + 4Н+ + 2е" = H2SO3 4- Н2О, Е° = 0,17 В.
Поэтому селеновая и теллуровая кислоты в отличие от серной окисляют НС1
до хлора:
H2Se04 + 2HC1 = С12 + H2Se03 + Н2О
В селеновой кислоте растворяется металлическое золото, которое нерастворимо в
горячей серной кислоте:
2Au + 6H2Se04 = Au2(Se04K + 3SeO2 4- 6Н2О
А смесь концентрированных селеновой и соляной кислот растворяет даже пла-
платину:
2H2Se04 4- 4НС1 + Pt = PtCl4 4- 2SeO2 + 4H2O
В отличие от теллуратов селенаты во многих отношениях похожи на сульфа-
сульфаты. Сульфаты и селенаты однотипных металлов близки по растворимости, крис-
таллохимическому строению, по способности образовывать двойные соли типа
квасцов. Среди сульфатов и селенатов много примеров изоморфизма.
При нагревании ортотеллуровой кислоты образуется триоксид:
Н6Те06 = ЗН2О + ТеО3
Триоксид теллура диморфен. Обе модификации химически малоактивны, не
растворяются в воде, разбавленных кислотах и основаниях. Одна из модифика-
модификаций взаимодействует с щелочью лишь при сплавлении.
SeO3 получают вытеснением из селената триоксидом серы:
K2Se04 + SO3 = K2SO4 + SeO3
Триоксид селена также образует две модификации: асбестовидную и стекловид-
стекловидную. Обе формы имеют молекулярную структуру. Триоксид селена хорошо раст-
растворяется в воде. В концентрированных растворах БеОз существуют смеси полисе-
полиселеновых кислот. С разбавлением полиселеновые кислоты деполимеризуются и
446
образуется конечный продукт — селеновая кислота. Триоксид селена обладает
сильными окислительными свойствами: окисляет даже охлажденную соляную
кислоту.
Соединения с другими неметаллами. Характеристичес-
Характеристические летучие водородные соединения селена и теллура H2Se и H2Te являются
гомологами сероводорода. Получают селеноводород и теллуроводород разложени-
разложением селенидов и теллуридов разбавленными кислотами. Селено- и теллуроводо-
теллуроводород — бесцветные газы с характерным неприятным запахом. Селеноводород более
токсичен, чем H2S. В отличие от сероводорода они эндотермичны и в отсутствие
воздуха разлагаются на компоненты при незначительном нагревании. Их хими-
химическое строение аналогично сероводороду, но с утяжелением хапькогена длина
связи с водородом растет, а энергия уменьшается. В водных растворах селено- и
теллуроводород представляют собой кислоты. От селеноводорода к теллуроводо-
роду сила кислоты возрастает вследствие уменьшения прочности связи халькоге-
на с водородом (рК{ 4,2 и рК{ 3,3 соответственно).
В ряду H2S — H2Se — Н2Те восстановительная активность возрастает. Окисле-
Окисление селено- и теллуроводорода в водных растворах приводит к выделению сво-
свободных селена и теллура. Горение газообразных H2Se и Н2Те сопровождается
выделением дыма вследствие образования диоксидов. При окислении селенидов
и теллуридов в растворах выделяющиеся селен и теллур реагируют с халькогени-
дами с образованием полиселенидов и полителлуридов (например, Na2Se4 и
Na2Te6). Однако соответствующие водородные соединения не получены.
Вследствие нарастания металличности от серы к селену и теллуру галогениды
последних более разнообразны и стабильны, чем галогенопроизводные серы. Все
галогениды селена и топлура получаются прямым синтезом из простых веществ:
+6 +4 +2 +1
SeF6 SeF4, SeCl4, SeBr4 SeF2 Se2Cl2, Se2Br2
TeF6 TeF4, TeCl4, TeBr4, Tel4 TeCl2, TeBr2
SeF6 и TeFe газообразны, геометрия молекул — октаэдр с атомом халькогена в
центре. TeF6 сравнительно легко гидролизуется:
TeF6 + 6Н2О = Н6Те06 + 6HF
Тетрагалогениды селена имеют кислотную природу, а теллура — солеобразны.
Структура молекул тетрагалогенидов соответствует искаженной тригональной
бипирамиде, один из углов которой занят неподеленной электронной парой.
Поскольку тетра- и гексагалогениды селена и теллура являются слабыми кисло-
кислотами Льюиса, легко образуются комплексы типа [SeF5]', fSeF6]2~, [TeFe]2", [TeF5]",
[TeF7]", [TeF8]2' и т.д. Ацидокомплексы [SeCl6]2" и [TeF6]2~ строго октаэдрические,
а типа [TeFs]" — пирамидальные, так как одна из октаэдрических орбиталей
теллура (+4) занята неподеленной электронной парой.
Дихлорид и дибромид теллура солеобразны. Они почти не гидролизуются,
при нагревании диспропорционируют. Se2Cl2 и Se2Br2 — жидкости, которые при
незначительном нагревании также диспропорционируют: 2Se2F2 = 3Se + SeF4.
Соединения с металлами. Селениды и теллуриды получают
синтезом из простых веществ в вакууме или инертной атмосфере, взаимодействи-
взаимодействием селено- и теллуроводорода с металлами, восстановлением производных селена
и теллура (+4) и (+.6), взаимодействием компонентов в паровой фазе и т.д. По
447
свойствам селениды — более близкие аналоги сульфидов. Щелочные металлы,
медь и серебро образуют селениды и теллуриды нормальной стехиометрии, кото-
которые можно рассматривать как соли селено- и теллуроводородной кислот. Они
солеобразны, хорошо растворяются в воде и легко гидролизуются. С щелочно-
щелочноземельными металлами и металлами подгруппы цинка селен и теллур образуют
монохалькогениды. Селениды и теллуриды щелочно-земельных металлов легко
окисляются и разлагаются водой. Монохалькогениды металлов подгруппы цинка
отличаются большей устойчивостью.
Непереходные металлы и полуметаллы III—V групп образуют селениды и
теллуриды типа Ме7Хх и МегХ (например, T^Se), а также аномальной стехиомет-
стехиометрии: GaSe, InTe и др. И те и другие являются полупроводниками. С переходны-
переходными металлами селен и теллур дают целый ряд селенидов и теллуридов: МеХ,
МегХз, МезХ4, МеХг и т.д.; многие из них представляют собой соединения пере-
переменного состава. Селениды и теллуриды переходных металлов характеризуются
ковалентно-металлическим типом связи с некоторой долей ионности. При малом
содержании неметаллических элементов в формульной единице связь между
атомами имеет преимущественно металлический характер. С увеличением содер-
содержания халькогена доля ковалентной связи возрастает и происходит переход от
металлической проводимости к полупроводниковой.
4. Подгруппа хрома. К элементам VIB-группы Периодической системы отно-
относятся хром, молибден и вольфрам. В силу стабильности конфигурации (Р у ато-
атомов первых двух элементов подгруппы наблюдается проскок одного электрона с
оболочки ns на оболочку (n — \)d. У вольфрама валентной электронной конфигу-
конфигурации предшествует завершенная 4/4-оболочка. Поэтому на его свойствах сказы-
сказывается влияние лантаноидной контракции. Ниже сопоставлены некоторые харак-
характеристики элементов и простых веществ VIB-группы:
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конСг
2-Ю
Мо
3-Ю
W
1-Ю
фигурация
Атомный радиус, нм
Ионный радиус Э6+, нм
Потенциалы ионизации, В
h: Э -+ Э+ + е
12: Э+ -+ Э2+ + е
/3: Э2+ —> Э3+ + е
Ц: Э3+ -* Э4+ + е
15: Э4+ -+ Э5+ + е
16: Э5 -+ Э6+ + е
S h В
ОЭО
Температура плавления, °С
1 Температура кипения, °С
| Плотность, г/см3
111 V Г1-ГУ /'
1 448 '
[Ar]3d54s1
0,127
0,035
6,8
16,5
31,0
49,6
73,0
90,6
267,5
1,6
1875
2680
7,19
-0,744
[Кг] 4 S 5 s1
0,139
0,065 ,
7,1
16,1
27,1
46,4
61,2
68,0
225,9
1,8
2620
4630
10,22
-0,20
[Хе]4/45
0,140
0,069
8,0
14,1
24,1
35,5
47,7
61,0
190,4
1,7
3395
5680
19,35
-
Обращает на себя внимание тот факт, что первые потенциалы ионизации в
отличив от последующих (и их сумм) возрастают в ряду Сг — Mo — W. Увеличе-
Увеличение 1-го потенциала ионизации обусловливает возрастание химической благород-
ности простых веществ при переходе от хрома к вольфраму. Вторые потенциалы
ионизации у хрома и молибдена различаются незначительно, в то время как /2
для вольфрама заметно меньше. Это обусловлено проскоком электрона у первых
двух элементов, вследствие чего 1% у них характеризует энергию, необходимую
для отрыва rf-электрона с предвнешнего уровня. У вольфрама 2-й потенциал
ионизации соответствует отрыву 6«-электрона. Подобная же закономерность
наблюдается и в отношении четвертых потенциалов ионизации. Напротив, шес-
шестые потенциалы ионизации и сумма шести потенциалов более близки для молиб-
молибдена и вольфрама, вследствие чего следует ожидать большого сходства этих
элементов в высшей степени окисления.
Значения ОЭО этих элементов свидетельствуют о том, что они являются
сравнительно малоактивными металлами. Поэтому для химии хрома и его анало-
аналогов не характерно образование преимущественно ионных соединений даже при
взаимодействии с сильноэлектроотрицательными элементами. Напротив, следует
ожидать большого разнообразия интерметаллических соединений. Ионные ра-
радиусы в высшей степени окисления у молибдена и вольфрама практически оди-
одинаковы и резко отличаются от радиуса иона Сг6+. Это хорошо коррелирует с
б
отмеченной выше закономерностью изменения /6 и Е U и объясняет подобие
i-г
свойств Мо и W в характеристической степени окисления.
Природные соединения и получение металлов
V I В-г р у п п ы. Самым распространенным минералом хрома является хромис-
хромистый железняк (хромит) FeO-СггОз. Вторая по значимости руда хрома — кро-
коит — представляет собой хромат свинца РЬСгО^ Наиболее распространенный
минерал молибдена — молибденит (молибденовый блеск) МоБг- Вольфрам пред-
представлен в природе главным образом в виде вольфраматов двухвалентных метал-
металлов. К ним относятся, например, вольфрамит — изоморфная смесь вольфраматов
железа и марганца переменного состава Fe^Mni-^WC^, шеелит CaWC>4, штольцит
PbWC>4 и т.д. Помимо того, встречается вольфрамовый блеск WS2 в смеси с мо-
молибденитом.
В чистом виде эти металлы получают с помощью алюмотермии для хрома и
водородного восстановления для молибдена и вольфрама:
Сг2О3 + 2А1 = 2Сг + А12О3; ЭО3 + ЗН2 = Э + ЗН2О
При водородном или металлотермическом восстановлении получаются либо
порошкообразные, либо губчатые металлы. Для получения компактных металлов
и их дополнительной очистки используют обычно вакуумную плавку с примене-
применением электронно-лучевого нагрева или плавку в электродуговых печах с расхо-
расходуемым электродом из чернового металла в водоохлаждаемых медных тиглях.
Для нужд черной металлургии обычно нет необходимости получать очень чистый
легирующий металл. Поэтому при карботермическом восстановлении совместно с
железными рудами получают обычно феррометаллы (феррохром, ферромолибден,
ферровольфрам).
Простые вещества. Физические и химические
свойства. В компактном состоянии Сг, Мо и W представляют собой плотные
15Н.А.Уга» ЛЛЛ
серебристо-белые металлы. Все три металла обладают высокими температурами
плавления и являются самыми тугоплавкими в своих rf-рядах. Вольфрам вообще
является самым тугоплавким из всех металлов.
Для всех трех металлов характерно отсутствие полиморфизма. Вплоть до
температуры плавления они обладают ОЦК-решеткой.
В химическом отношении хром, молибден и вольфрам довольно инертны. При
обычных условиях они устойчивы по отношению к воде и кислороду воздуха.
Хром реагирует с разбавленными растворами НС1 и H2SO4, поскольку в этих
условиях пассивирующая пленка поверхностного оксида (близка по составу к
СГ2О3) постепенно разрушается. В ряду стандартных электродных потенциалов
хром располагается между цинком и железом. У молибдена и вольфрама коррози-
коррозионная устойчивость в кислых средах резко возрастает, поскольку состав их пасси-
пассивирующих пленок близок к кислотообразующим оксидам соответствующих кис-
кислот (МоОз и \?0з). Лучшими растворителями для Мо и W являются расплавы
щелочей в присутствии окислителей и горячая смесь HNO3 + HF:
Э + 3NaNO3 + 2NaOH = Na23O4 + 3NaNO2 + H20
Э + 6HNO3 + 8HF = H2[3F8] + 6NO2 + 6H2O
Хром при высоких температурах растворяет водород, а молибден и особенно
вольфрам водород практически не поглощают.
Элементы подгруппы хрома при нагревании реагируют с галогенами, халько-
генами, пниктогенами (кроме висмута), неметаллами IVA-группы, бором. Кроме
того, они реагируют с большинством металлов с образованием твердых растворов
или интерметаллических соединений.
Характеристические соединения. В соединениях с кисло-
кислородом хром проявляет степени окисления +2, +3, +4, +6 и образует соответству-
соответствующие оксиды СгО, Сг2Оз, СгО2 и СгОз- Для молибдена известны оксиды МоО2,
Мо2С>5 и МоОз- Для вольфрама известны всего лишь два оксида: W02 и \УОз-
Таким образом, в ряду Сг — Мо — W наблюдается, как и в предшествующих В-
группах, повышение стабильности высших степеней окисления и уменьшение
разнообразия соединений с кислородом.
Оксид хрома (+3) представляет собой порошок темно-зеленого цвета, нераст-
нерастворимый в воде, кислотах и щелочах. Удобным лабораторным способом получе-
получения Сг2Оз является термическое разложение дихромата аммония:
= Cr2O3 + N2 + 4Н2О
Поскольку Сг2Оз нерастворим, его гидроксид Сг(ОН)з можно получить, напри-
например, действуя раствором аммиака на раствор соли Сг(-)-З):
Cr2(SO4K + 6NH4OH = 2Сг(ОНK + 3(NH4JSO4
Гидроксид хрома обладает амфотерными свойствами. Со структурной точки
зрения, гидроксид хрома (+3), имеющий переменный состав СггОз'^НгО, облада-
обладает пространственным строением многоядерного комплекса. Структурными едини-
единицами его являются октаэдры [Сг(ОН)б]3" и [Сг(Н2О)б]3+, связанные между собой
бловыми ОН-мостиками. Относительное количество лигандов ОН" и Н2О может
меняться в зависимости от условий получения гидроксида хрома. Равновесия
между различными формами гидроксо- и аквакомплексов для Сг(+3) представле-
представлены схемой
Сг(ОНK-хН2О
п[Сг(Н2ОNр* ^=* [Сг(ОН)з(Н2ОK]„ ^р=^ И[Сг(ОНNр-
кислая среда нейтральная среда щелочная среда
Как следует из этой схемы, для Сг(+3) характерным является координацион-
координационное число 6. Оксид хрома (+3) и соответствующий гидроксид по свойствам силь-
сильно напоминают соединения алюминия AI2O3 и А1(ОН)з. В частности, при сплав-
сплавлении Сг2Оз с оксидами или карбонатами щелочных металлов образуются мета-
производные Me СгО2- Подобное же взаимодействие с производными щелочно-
щелочноземельных металлов, магния, железа (+2) приводит к образованию хромовых
шпинелей Me СГ2О4, относящихся к тому же классу соединений, что и собствен-
собственно шпинель MgAl^O^ Свободная НСгО2 представляет собой синевато-серые крис-
кристаллы. Она теряет воду при 430°С с сильным саморазогреванием. В водных раст-
растворах щелочей метапроизводные (хромиты) не существуют вследствие образова-
образования гидроксокомплексов, например
Сг(ОНK + 3NaOH = Na3[Cr(OH)b]
Из характеристических соединений хрома в низшей положительной степени
окисления 4-2 известны черный оксид СгО и соответствующий гидроксид
Сг(ОНJ желтого цвета. Сг(ОНJ обладает только основными свойствами. Осто-
Осторожным обезвоживанием Сг(ОНJ в восстановительной атмосфере можно получить
оксид СгО, который при небольшом нагревании диспропорционирует.
В степени окисления +4 характеристические соединения существуют для всех
трех элементов VlB-группы. Известны черный оксид СгО2, коричневато-фиолето-
коричневато-фиолетовый Мо02 и коричневый WO2- Оксид СгО2 представляет собой фазу переменного
состава с дефектами в подрешетке кислорода (твердый раствор вычитания
СгОг-х). Кристаллизуется в решетке рутила ТЮ2. Диоксиды хрома и его аналогов
нерастворимы в воде, трудно растворимы в кислотах и щелочах. Отвечающие им
гидроксиды неизвестны.
Наиболее типичной степенью окисления для молибдена и вольфрама и одной
из важнейших для хрома является 4-6. МоОз и WO3 получаются непосредствен-
непосредственным термическим окислением металлов газообразным кислородом. СгОз кристал-
кристаллизуется в виде ярко-красных игл при действии на раствор дихромата калия
К2Сг207 концентрированной серной кислотой:
К2Сг207 + H2SO4 = K2SO4 + 2CrO3 + Н2О
СгОз обладает цепочечной структурой из тетраэдров [СгО4], соединенных
вершинами. МоОз и WO3 имеют координационные структуры, построенные из
октаэдров [ЭОб], связанных между собой вершинами. Низкая температура плавле-
15* 451
ния СгОз A87°С) свидетельствует о заметном ван-дер-ваальсовом вкладе между
цепочками, а высокие температуры плавления МоОз и WO3 G95 и 1473°С) подт-
подтверждают их координационную структуру.
С химической точки зрения, СгОз, МоОз и WO3 представляют собой типич-
типичные кислотообразующие оксиды соответствующих кислот: хромовой Н2СЮ4,
молибденовой Н2М0О4 и вольфрамовой H2WC>4-
Этим кислотам отвечают многочисленные соли: хроматы, молибдаты и воль-
фраматы. Обычно соли хромовой кислоты окрашены в желтый цвет, свойствен-
свойственный иону CrOf , соли молибденовой и вольфрамовой кислот бесцветны.
Одной из характерных особенностей элементов VIB-группы в высшей степени
окисления является способность к образованию полисоединений. Для Сг свой-
свойственно образование лишь производных изополикислот, для Мо и W существуют
и производные гетерополикислот. Если в разбавленных водных растворах для
хрома характерно образование иона CrOij", то по мере повышения концентрации
раствора происходит образование полихромат-анионов СгпОзп+ь Сами изополи-
хромовые кислоты известны только в растворах. Однако их соли довольно много-
многочисленны. Наибольшее значение имеют дихроматы; они в отличие от желтых
хроматов имеют красно-оранжевую окраску и лучше растворимы в воде. Раство-
Растворы дихроматов имеют кислую реакцию, что объясняется взаимодействием дихро-
дихромат-иона с водой по схеме
Н2О + Сг2О?" ^± 2Н
2CrOf
Отсюда следует, что в кислой среде равновесие смещено влево, а при разбавле-
разбавлении водой или в щелочной среде — вправо.
Все хроматы являются сильными окислителями. Так, на холоду они окисляют
HI, H2S, H2SO3 и их соли, а при нагревании — НВг и даже НС1:
К2Сг207 + 14НС1 = 2КС1 + 2СгС13 + ЗС12 + 7Н2О
Для соединений хрома (+6) характерно взаимодействие с пероксидом водорода.
При этом получается пероксид CrOs синего цвета:
Н2Сг04 + 2Н2О2 = ЗН2О + СгО5
Образуются и пероксокислоты хрома H2Cr20i2 и ЩСтОв. Все пероксохроматы
неустойчивы, являются сильнейшими окислителями.
Для Мо(+6) и W(+6) еще более характерно образование изо- и гетерополи-
соединений. Структурной единицей сложных анионов изо- и гетерополикислот
молибдена и вольфрама является октаэдр [ЭОб]. Эти октаэдры могут соединяться
между собой вершинами или ребрами, образуя сложные каркасы. По мере услож-
усложнения состава аниона связь с атомами водорода во внешней сфере комплекса
становится более полярной вследствие перераспределения электронной плотности
и сила поликислот возрастает. Однако в целом, как отмечено выше, в ряду Сг —
Mo — W кислотный характер высших оксидов и гидроксидов быстро ослабевает.
В частности, Н2М0О4 и H2WO4 проявляют заметные признаки амфотерности, что
выражается в их взаимодействии с сильными кислотами:
ЭО2(ОНJ + 2НС1 = ЭО2С12 + 2Н2О
452
Эта реакция может быть проведена и с Н2Сг04, однако в- этом случае равновесие
сильно смещено влево. Соединения ЭО2С12 в водных растворах диссоциируют по
схеме
ЭО2С12 «=* ЭОГ + 2С1-
Оксокатион ЭО2+ называют хромилом, молибденилом или вольфрамилом.
Действием пероксида водорода в щелочной среде на растворы молибдатов и
вольфраматов можно получить пероксопроизводные общей формулы Ме2ЭОп (где
я = 5 — 8). Со структурной точки зрения, они аналогичны соответствующим
производным хрома и содержат от одной до четырех пероксогрупп —О—О—,
замещающих атомы кислорода в анионах ЭО| . Это твердые вещества, окрашен-
окрашенные в яркие тона от красного до желтого. При нагревании разлагаются со взры-
взрывом, а при комнатной температуре медленно отщепляют кислород, вследствие
чего обладают сильными окислительными свойствами. В ряду Сг — Mo — W
устойчивость пероксосоединений в высшей степени окисления заметно возрастает.
Соединения с другими неметаллами. Фтор, как наиболее
энергичный окислитель, способен образовывать высшие галогениды ЭГб, из кото-
которых CrFg — твердое вещество, MoFe — легкокипящая жидкость, a WFg — газ, что
свидетельствует об увеличении тенденции к образованию молекулярных струк-
структур. Наибольшее число фторидов образует хром, для которого реализуются все
степени окисления от +2 до +6 (в том числе и нехарактерная +5, не наблюдае-
наблюдаемая в ряду оксидов). Низшие фториды CrF2 и CrF3 солеобразны и тугоплавки. У
молибдена и особенно у вольфрама число известных фторидов значительно мень-
меньше. Ни один из фторидов Мо и W не является солеобразным.
В ряду хлоридов хрома и его аналогов наиболее ярко видна стабилизация
высших степеней окисления от хрома к вольфраму. Хлор способен окислить хром
только до CrCl4, молибден — до M0CI5, а вольфрам — даже до высшего хлорида
WC16.
Низшие хлориды хрома СгС12 и СгС1з — типичные солеобразные соедине-
соединения. СгС12 — бесцветное кристаллическое вещество, гигроскопическое и хоро-
хорошо растворимое в воде. Из раствора выделяется в виде кристаллогидрата
СгС12-4Н2О, который следует рассматривать как аквакомплекс [Сг(Н2ОL]С12.
Хлорид хрома (+3) также образует кристаллогидрат СгС1з*6Н2О, представляю-
представляющий собой аквакомплекс [Сг(Н2О)б]С1з сине-фиолетового цвета.
В отличие от низших хлоридов хрома МоС12 и WC12
не являются солеобразными соединениями, а
представляют собой так называемые кластеры, т.е.
многоядерные комплексы с непосредственной связью
между атомами комплексообразователя. Молекула
МоС12 представляет собой агрегат Mo6Cli2, причем
восемь атомов хлора находятся во внутренней сфере
комплексного многоядерного кластера [МобС18]4+ (рис.
153), а четыре атома С1 — во внешней сфере. Непо-
Непосредственно связанные между собой атомы Мо обра- Г\-м Л
зуют правильный октаэдр. На всех ребрах располага- ^
ЮТСЯ МОсТИКОВЫе атомы ХЛОра, каждый ИЗ КОТОРЫХ Рис. 153. Структура октаэдри-
связан одновременно с тремя атомами Мо, причем ческого кластера
образуются тетраэдры с атомом С1 в вершине и атомами металла в основании.
Таким образом, ковалентность хлора равна трем, а ковалентность молибдена —
восьми (каждый атом Мо связан с четырьмя другими атомами металла и четырь-
четырьмя атомами С1). Аналогична структура и хлорида вольфрама (+2).
Большой интерес представляет сопоставление свойств и структуры таких
комплексных ионов, как [ЭгСЩ3". В противоположность парамагнитному [СггСЩ3'
аналогичные по составу ионы на основе молибдена и вольфрама диамагнитны и
значительно более прочны. Причина этого различия заключается в разной струк-
структуре и разном характере связей рассматриваемых ионов. Строение парамагнитно-
парамагнитного комплекса [СггСЩ3" представлено ниже:
-.3-
CL
С1
С1
/
Сг'
•ск
0,31 нм
--С1
ЧС1
Неспаренные электроны у каждого иона Сг3+ (по три электрона) обеспечивают
парамагнетизм комплексного иона в целом. Расстояние между атомами хрома
в этом комплексе довольно велико и составляет 0,31 нм. Таким образом, комп-
комплекс [O2CI9]3" не является кластером. Аналогичные по составу комплексы
[M02CI9]3" и [W2C19]3" отличаются от [Сг2С19]3~ значительно меньшим расстоянием
между атомами металла, несмотря на то что размеры самих атомов больше. Так,
расстояние W—W в таком комплексе равно всего лишь 0,24 нм. Это обусловлено
тем, что более удаленные от ядра и более рыхлые валентные Ad- или ба'-орбитали
соседних атомов металла, содержащие по одному электрону, перекрываются по
обменному механизму, образуя тройную ковалентную связь. Именно поэтому эти
комплексы диамагнитны. Таким образом, комплексы [M02CI9]3" и [W^Clg]3" в
отличие от [Сг2С1д]3" представляют собой кластеры с непосредственной связью
Ме=Ме:
з-
Бром с металлами рассматриваемой группы образует соединения, в общем
напоминающие аналогичные производные хлора. При этом, однако, в силу не-
несколько меньшей окислительной активности брома высший бромид молибдена
отвечает формуле МоВг^. Иод как наименее активный окислитель среди галоге-
галогенов образует соединения с еще меньшими степенями окисления катионообразова-
ния Сг1з и Сг1г, обладающие солеобразным характером, и соединения Э1г, Э1з и
Э14 для молибдена и вольфрама. Низшие бромиды и иодиды молибдена и воль-
вольфрама (ЭГг), как и хлориды этих элементов, представляют собой кластеры и в
этом отношении заметно отличаются от аналогичных производных хрома.
Все сульфиды металлов подгруппы хрома (CrS, Cr2S3, 9S2 и ЭБз для Мо и W)
достаточно термически устойчивы и обладают полупроводниковыми свойствами,
что подчеркивает их неметаллическую природу. Все они представляют собой
координационные кристаллы и обладают переменным составом, что особенно
характерно для низших сульфидов. В этом отношении они заметно отличаются
от галогенидов, которые нередко образуют или молекулярные структуры, или
кластеры. Взаимодействие хрома, молибдена и вольфрама с селеном и теллуром
протекает менее энергично, причем вольфрам с теллуром соединений не образует,
а в остальных случаях в системах образуется небольшое число соединений, отве-
отвечающих лишь наиболее стабильным степеням окисления (СгХ, СГ2Х3, WSe2,
WSe8, MoTe2, MoTe3).
Фосфиды, арсениды и стибиды хрома и его аналогов принадлежат к классу
аномально построенных дальтонидов, содержащих "анион-анионные" и "катион-
катионные" связи. Наиболее характерны для фосфидов соединения состава Э3Р,
ЭР и ЭРг- Образование моно- и дифосфидов вообще весьма характерно для пере-
переходных металлов.
Для всех трех элементов существуют дисилициды 9Si2, представляющие собой
тугоплавкие соединения, обладающие полупроводниковыми свойствами. Дисили-
Дисилициды устойчивы к агрессивным средам при повышенных температурах. В системе
Cr — Si установлено существование соединений CrSi, C^Si, Ci^Si, первое из кото-
которых является полупроводником, а два других — металлиды. Таким образом, в
ряду силицидов хрома наблюдается та же закономерность, что была отмечена
для фосфидов.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Относительной стабильностью обладает CrSO4 в раз-
разбавленной серной кислоте в отсутствие кислорода. В твердом состоянии сульфат
хрома (+2) изоморфен сульфату железа (+2) и они оба образуют кристаллогид-
кристаллогидраты 9SO4-7H2O. Химия хрома (+2) в определенном смысле подобна химии
железа (+2). Так, для Сг (+2) известно соединение К4[Сг(С1Ч)б], аналогичное по
составу желтой кровяной соли K4[Fe(CNN], а также существует роданидный
комплекс K4[Cr(CNSN].
Оксид СггОз при сплавлении с пиросульфатом калия дает сульфат Cr2(SO4K,
который образует с сульфатами щелочных металлов двойные соли типа квасцов.
Тенденция к образованию квасцов у хрома выражена еще ярче, чем у алюминия,
и эти двойные соли более устойчивы. Хром является лучшим комплексообразова-
телем, чем алюминий. У хрома в отличие от алюминия существует внутренняя 3d-
оболочка с частично свободными орбиталями, способными принять участие в
донорно-акцепторном взаимодействии с лигандами. У алюминия З^-оболочка
также существует, но она является внешней и полностью вакантной. Таким обра-
образом, при координационном числе 6 для хрома характерна внутренняя <Рвр3-гиб-
ридизация, а для алюминия — внешняя sp3(^-гибридизация с меньшей прочнос-
прочностью связей.
Все три металла подгруппы хрома образуют однотипные гексакарбонилы
Ме(СО)б, в которых степень окисления металла формально равна нулю. Все эти
карбонилы представляют собой бесцветные ромбические кристаллы, устойчивые
на воздухе до 180—200°С. В карбонильных комплексах атомы металлов выступа-
выступают в аномально низких степенях окисления и лиганды, подобные СО, способст-
способствуют стабилизации этого состояния. Такие лиганды помимо неподеленных элект-
455
металлов подгруппы марганца содержит всего два ns-электрона, а подстилающая
электронная оболочка — стабильная, наполовину заполненная (n — l)d. По этой
причине элементы подгруппы марганца в первую очередь выступают как типич-
типичные переходные металлы. В то же время химия элементов подгруппы марганца в
высшей степени окисления приобретает закономерную аналогию с химией второ-
второго типического элемента — хлора.
1. Фтор. Особенности химии фтора. Как и в других группах
системы, химия типических элементов — фтора и хлора — имеет целый ряд осо-
особенностей. Наиболее ярко это проявляется у фтора. Специфика поведения фтора
по сравнению с другими галогенами связана не только с наименьшим радиусом,
наибольшими потенциалом ионизации и ОЭО атомов фтора. Главное, что опреде-
определяет особенности химии фтора, — ограниченные валентные возможности и степе-
степени окисления фтора. Атом фтора не располагает rf-орбиталями, а промотирование
электронов на орбитали с главным квантовым числом 3 для него энергетически
невыгодно. В результате в химии фтора представлены только две степени окисле-
окисления: 0 и -1. Отсюда следует, что фтор только окислитель, а восстановителем
быть не может. Поэтому для фтора неизвестны соединения с положительной
степенью окисления. Ниже приведены некоторые характеристики элементов
VIIA^-руппы и их гомоатомных соединений.
F Cl Br I
Электронная конфи-
конфигурация
Атомный радиус, нм
Радиус иона Г", нм
Атомная поляризуе-
поляризуемость, нм3
Длина связи в Гг, нм
Силовая константа
связи Гг, Н/м
/ь В
Сродство к электрону,
кДж/моль
ОЭО
ДЯ(дисс) Г2,
кДж/моль 159,1 242,8 192,6 150,7
ДЯ(гидр.)Г\
кДж/моль -535,9 -405,7 -386,0 -301,7
Ъ? (Г°/П> в 2,87 1,36 1,07 0,54
Температура плавле-
плавления, "С -220,6 -100,9 -7,2 Ц3,5
Температура кипения,
°С -187,7 -34,2 58,8 184,5
ДЯ(пл.), кДж/моль 0,51 6,41 10,60 15,56
ДЯ(кип.), кДж/моль 6,55 20,42 30,31 41,81
Плотность твердого
вещества, г/см3' 1,3 1,9 3,4 4,4
рК(дисс.) Г2 2,4 6,8 4,5 2,6
0,064
0,133
0,4
0,142
718,5
17,4
-339,13
о,»
0,099
0,181
2,3
0,199
329,2
13,0
-355,88
3,1
0,114
0,196
3,3
0,228
246,3
11,8
-330,76
2,9
0,133
0,220
5,1
0,267
173,4
10,5
-301,67
2,6
металлов подгруппы марганца содержит всего два ns-электрона, а подстилающая
электронная оболочка — стабильная, наполовину заполненная (n — \)d. По этой
причине элементы подгруппы марганца в первую очередь выступают как типич-
типичные переходные металлы. В то же время химия элементов подгруппы марганца в
высшей степени окисления приобретает закономерную аналогию с химией второ-
второго типического элемента — хлора.
1. Фтор. Особенности химии фтора. Как и в других группах
системы, химия типических элементов — фтора и хлора — имеет целый ряд осо-
особенностей. Наиболее ярко это проявляется у фтора. Специфика поведения фтора
по сравнению с другими галогенами связана не только с наименьшим радиусом,
наибольшими потенциалом ионизации и ОЭО атомов фтора. Главное, что опреде-
определяет особенности химии фтора, — ограниченные валентные возможности и степе-
степени окисления фтора. Атом фтора не располагает rf-орбиталями, а промотирование
электронов на орбитали с главным квантовым числом 3 для него энергетически
невыгодно. В результате в химии фтора представлены только две степени окисле-
окисления: 0 и -1. Отсюда следует, что фтор только окислитель, а восстановителем
быть не может. Поэтому для фтора неизвестны соединения с положительной
степенью окисления. Ниже приведены некоторые характеристики элементов
VIIA-группы и их гомоатомных соединений.
Cl Br I
Электронная конфи-
конфигурация
Атомный радиус, нм
Радиус иона Г", нм
Атомная поляризуе-
поляризуемость, нм3
Длина связи в Гг, нм
Силовая константа
связи Гг, Н/м
Л, в
Сродство к электрону,
кДж/моль
ОЭО
ДЯ(дисс) Г2,
кДж/моль
ДЯ(гидр.)Г,
кДж/моль
ЕГ (Г°/Г). В
Температура плавле-
плавления, °С
Температура кипения,
°С
ДЯ(пл.), кДж/моль
ДЯ(кип.), кДж/моль
Плотность твердого
вещества, г/см3/
рК(дисс) Г2
0,064
0,133
0,4
0,142
718,5
17,4
-339,13
3,9
159,1
-535,9
2,87
-220,6
-187,7
0,51
6,55
1,3
2,4
0,099
0,181
2,3
0,199
329,2
13,0
-355,88
3,1
242,8
-405,7
1,36
-100,9
-34,2
6,41
20,42
1,9
6,8
0,114
0,196
3,3
0,228
246,3
11,8
-330,76
2,9
192,6
-386,0
1,07
-7,2
58,8
10,60
30,31
3,4
4,5
0,133
0,220
5Д
0,267
173,4
10,5
-301,67
2,6
150,7
-301,7
0,54
1.13,5
184,5
15,56
41,81
4,4
2,6
акт
3 4 5 6 7 в 9 1
Рис. 154. Эффект обратного экрани-
экранирования у атомов 2р-элементов:
сплошная линия - 2s; пунктир • 2р
Прочность молекулы фтора, несмотря на
наименьшее межъядерное расстояние относи-
относительно других газогенов, намного меньше по
сравнению с молекулами хлора и брома. По
величинам энтальпии и рК (дисс.) молекула
фтора сравнима с молекулой иода. В то же
время силовая константа связи в молекуле
фтора в два с лишним раза превосходит тако-
таковую молекулы хлора. Другими словами, хими-
химическая связь в молекуле фтора менее прочна,
но более жестка. Сравнительно невысокая проч-
прочность молекулы фтора, которая является одним
из факторов его высокой химической активнос-
активности, обусловлена отсутствием у фтора d-орбиталей. В молекулах остальных
галогенов имеет место дополнительное тг-связывание за счет р-электронов и d-
орбиталей.
Сродство к электрону у атома фтора также меньше, чем у хлора. Фтор являет-
является менее электрофильным элементом по сравнению с хлором. Это объясняется
кайносимметричностью 2р-электронов атома фтора и связанным с ней эффектом
обратного экранирования. Дело в том, что 2р-орбитали в атоме фтора сильнее
притянуты к ядру и лежат глубже полностью заполненной электронами некайно-
симметричной 2«-орбитали (рис. 154). Последняя, будучи полностью заселенной,
отталкивает присоединяемый атомом фтора электрон, уменьшая электронное
сродство и увеличивая ионизационные потенциалы. Все перечисленные выше
факты определяют специфические особенности химии фтора.
Природные соединения и получение фтора. Фтор
довольно распространен в природе. Наиболее важными минералами фтора явля-
являются флюорит CaF2, криолит Na^fAlFg] и фторапатит ЗСа.^РО^г'СаГг. Свобод-
Свободный фтор получают электролизом смеси HF + KF или легкоплавких ацидокомп-
лексов KH2F3, KHjF4. Катод и анод отделены друг от друга диафрагмой для
разделения водорода и фтора. Фактически электролизу подвергается HF. а фто-
фторид калия обеспечивает электрическую проводимость расплава*:
3HF = H2F+ + HF2
На катоде 2H2F+ + 2е' = Н2 + 2HF
На аноде 2HF2 - 2е" = F2 + 2HF
В отсутствие KF фтороводород практически неэлектропроьоден, так как его
собственная ионизация незначительна: \>К (иониз.)Ю. Фтор хранят и транспорти-
транспортируют как в газообразном состоянии под давлением, так и в сжиженном виде.
Контейнеры и изделия для работы с жидким и газообразным фтором изготавли-
изготавливают из нержавеющей стали, никеля, меди или алюминия. Во всех случаях обра-
образуются зацщтные пленки из фторидов соответствующих металлов, которые пре-
предохраняют материал контейнера от дальнейшей коррозии.
* Ионизация по этому типу протекает вследствие склонности HF к образованию устой-
устойчивого иона HF2.
458
Физические и химические свойства фтора. Фтор —
светло-желтый газ с резким специфическим запахом. Газообразный фтор раство-
растворим в жидком HF. Твердый фтор ниже температуры -228°С имеет моноклинную
структуру, а выше этой температуры — кубическую молекулярную решетку.
При растворении фтора в воде вблизи 0°С вначале образуется фторноватистая
кислота HOF. При комнатной температуре HOF нацело разлагается с выделением
HF и кислорода. Таким образом, под действием фтора вода разлагается:
Н2О + F2 = 2HF + 1/2О2
Это еще одно доказательство невозможности получения стабильных соединений
фтора с положительной степенью окисления.
Фтор химически взаимодействует почти со всеми простыми веществами, в том
числе с тяжапыми благородными газами, окисляя их:
Хе(г) + F2(r) = XeF2(K), AG^29g = -161,2 кДж/моль,
Хе(г) + 2F2(r) = XeF4(K), Д G°fam = -256,7 кДж/моль.
С кислородом фтор реагирует при низких температурах в электрических разря-
разрядах с образованием эндотермичных фторидов кислорода. Углерод, кремний,
фосфор, сера и другие неметаллы, а также большинство металлов в виде порош-
порошков воспламеняются в атмосфере фтора при 20—300°С с образованием соответст-
соответствующих фторидов. Кроме того, многие реакции прямого фторирования протека-
протекают по цепному механизму и часто имеют взрывной характер.
Фторид водорода и фториды металлов. При взаимодей-
взаимодействии с водородом и металлами наиболее ярко проявляются окислительные свой-
свойства фтора. Реакция между газообразными Н2 и F2 протекает со взрывом на
холоду по механизму цепных реакций. В промышленности HF получают реакцией
вытеснения из CaF2 концентрированной серной кислотой. Безводный HF в лабо-
лабораторных условиях получают нагреванием высушенного KHF2.
Фтороводород выше 19,5°С представляет собой бесцветный газ с резким разд-
раздражающим дыхательные пути действием, а ниже указанной температуры кипения
— легкоподвижную бесцветную жидкость. HF характеризуется высоким значени-
значением электрического момента диполя @,64-Ю9 Кл*м), превосходящим электричес-
электрический момент диполя воды, сернистого газа и аммиака. Жидкий фторид водорода
имеет большое значение диэлектрической постоянной, равное 83,6 при 0°С. HF
ассоциирован за счет водородных связей в (HF)n, где п изменяется от 1 до 4 в
парах, а в жидком фтороводороде п > 4.
Несмотря на значительную ионность молекул HF, водный раствор его — пла-
плавиковая кислота — представляет собой кислоту средней силы в отличие от ос-
остальных сильнейших галогеноводородных кислот. Аномальное поведение плави-
плавиковой кислоты объясняется большой склонностью полярных молекул HF к ассо-
ассоциации за счет водородных связей. Энергия водородной связи между молекулами
HF больше, чем между молекулами воды, и составляет 83 кДж/моль. Возни-
Возникающая система водородных связей затрудняет гетеролитический распад HF в
воде. Кроме того, первичный акт ионизации HF в воде
. HF + Н2О *=* Н3О+ + F-, рК 3,2
¦г- ' 4S9
осложняется образованием более сложных частиц вследствие возникновения
водородных связей:
[ F
-0-2- F ] - Н F2, А Я2°,8 64*3 кДж/моль, р* -0,7
FH + HF7
0J7HM
¦"
В результате в водных растворах HF образуются устойчивые гидрофторид-ани-
гидрофторид-анионы. Простейший из них HF2 имеет линейную структуру с равными расстоя-
расстояниями @,126 нм) между мостиковым атомом водорода и атомами фтора*. Пока
это единственный случай симметричного мостика с водородной связью. На осо-
особый характер и "выравнивание" ковалентной и водородной связей в ионе HF2
указывает также очень большая для протонного мостика энергия связи, равная
114,6 кДж/моль. Более сложные гидрофторид-ионы, например H2F3, характери-
характеризуются зигзагообразным строением. Вследствие прочности водородных связей в
водных растворах HF при нейтрализации растворов плавиковой кислоты образу-
образуются не только нормальные фториды, но и гидрофториды типа MeHF2, MeH2F3,
MeHaF4 и т.д. С увеличением формульной массы стабильность соответствующих
гидрофторидов металлов возрастает:
Соединение KHF2(k) KH2F3(k) KH3F4(k)
AG}-298' кДж/моль -858,3 -1149,7 -1429,4
В образовании устойчивых кислых солей плавиковой кислоты заключается
принципиальное отличие от других галогеноводородных кислот, которые вообще
не дают гидропроизводных. Гидрофториды разлагаются при нагревании:
KHF2 = KF + HF
что используется для получения чистого HF и фторида металла.
Химическая активность HF сильно зависит от присутствия воды. Сухой HF не
действует на металлы и оксиды. В последнем случае образующаяся в результате
реакции вода резко увеличивает скорость процесса, т.е. фтористый водород под-
подвержен реакциям автокатализа. Специфической особенностью плавиковой кисло-
кислоты является ее способность химически взаимодействовать с материалами, содер-
содержащими диоксид кремния:
SiC-2 + 4HF = SiF4 + 2Н2О
Эта реакция лежит в основе травления стекла плавиковой кислотой. Поэтому
плавиковую кислоту нельзя хранить в стеклянной посуде, а лучший контейнер
для нее — емкость из фторопласта.
Так называемая симметричная водородная связь.
Вследствие особенностей химии фтора фториды металлов во многих отноше-
отношениях отличны от других галогенидов. Наивысшая окислительная активность и
наибольшая 0Э0 фтора обосновывают тот факт, что фториды металлов наиболее
тугоплавки и термически стабильны по сравнению с другими галогенидами.
Ниже приведены энергии Гиббса образования галогенидов кальция:
Соединение ,.., CaF2(K) СаС12(к) СаВг2(к) Са12(к)
AG°f 298, кДж/моль -1168,5 -749,4 -666,6 -533,1
Обращает на себя внимание большое значение энергии Гиббса для фторида,
резко отличное от таковых для других галогенидов кальция. Это одна из причин
малой растворимости CaF2 в воде, в то время как остальные галогениды хорошо
растворимы. Вообще фториды металлов малорастворимы в воде, за исключением
фторидов натрия, калия и некоторых других. В отличие от CaF2 фторид серебра
хорошо растворим в воде, хотя другие галогениды серебра, как известно, мало-
малорастворимы. Таким образом, даже по растворимости фториды в какой-то мере
противостоят остальным галогенидам металлов.
Во фторидах металлы способны проявлять высшие степени окисления, напри-
например AgF2, AuF3, T1F3, CoF3, PbF4, RhF4, BiF5, MoF6, ReF7 и т.д. При этом обра-
образование низших фторидов металлов малохарактерно и они менее стабильны. Для
иллюстрации приводим значения энтальпий образования фторидов тантала не-
нечетной степени окисления:
Соединение TaF(K) TaF3(K) TaF5(K)
MTf 298, кДж/моль +259,4 -786,6 -1903,6
Из металлов IA-группы наибольшим сродством к фтору обладает литий, а для
натрия и металлов подгруппы калия значения энергии Гиббса образования фто-
фторидов практически одинаковы. Подобное наблюдается и для «-металлов II груп-
группы Периодической системы. Из sp-металлов наиболее прочный фторид образует
алюминий. В подгруппах «^-металлов сверху вниз стабильность фторидов не-
несколько уменьшается. Еще более устойчивые характеристические трифториды
образуют металлы подгруппы скандия и лантаноиды (см. табл. 25).
В подгруппах металлов с полностью заселенными электронами d-орбиталями в
соответствии с уменьшением металличности элементов сверху вниз уменьшается
энергия Гиббса образования их фторидов, например:
Соединение CuF(k) AgF(K) ZiiF2(k) Cc1F2(k)
AGy298, кДж/моль -235,6 -187,9 -713,4 -649,4
В то же время для подгрупп металлов с дефектными rf-юрбиталями, как и
следовало ожидать, наблюдается обратная картина, например:
Соединение У?ъ(ж) NbF5(i<) TaFs(K)
ДС/,298- кДж/моль -1378,4 -1698,3 -1790,7
Соединения фтора с неметаллами. Большинство известных
соединейий фтора с кислородом — малоустойчивые вещества, существующие
лишь при низких температурах. Кислород в этих соединениях поляризован поло-
461
жительно. Фториды кислорода — единственные эндотермические соединения
фтора:
Соединение OF2 O2F2 O3F2
AH°f 29g, кДж/моль +16,7 +21,2 +26,1
Из них при комнатной температуре существует только OF2 — бесцветный
токсичный газ с запахом фтора. Ковалентная молекула дифторида кислорода
имеет структуру равнобедренного треугольника: d (F О) = 0,142 нм, a ZFOF =
= 103°. OF2 образуется при пропускании фтора в разбавленный раствор щелочи:
2F2 + 2NaOH = OF2 + 2NaF + H2O
OF2 термически устойчив до 200°С, но с водой, щелочами конпропорционирует:
OF2 + Н2О = О2 + 2HF; OF2 + 2NaOH = 2NaF + О2 + Н2О
Другие фториды кислорода OnF2 (n меняется от 2 до 6) получают действием
высоковольтного электрического разряда на смесь кислорода и фтора под умень-
уменьшенным давлением и при низких температурах. Все они полностью разлагаются
при температурах намного ниже комнатной. Так, период полураспада желто-
оранжевых кристаллов O2F2 при -50°С около 3 ч. Все фториды кислорода, вклю-
включая OF2, — сильнейшие окислители и фторагенты.
Из производных фтора с другими неметаллами представляют интерес фтори-
фториды галогенов. Последние являются интергалогенидами. Все фториды галогенов —
экзотермические соединения с нечетной положительной степенью окисления
хлора, брома и иода. Известны гептафторид иода, все пентафториды, трифтори-
ды и монофториды. Только IF не получен в чистом виде, а обнаружен в следовых
количествах спектроскопически. Дело в том, что стабильность фторидов возрас-
возрастает с увеличением положительной степени окисления галогенов. Поэтому наиме-
наименее устойчивы монофториды. Фториды галогенов диамагнитны, так как неспарен-
ные электроны галогенов входят в состав обобществленных электронных пар при
образовании ковалентных связей с атомами фтора.
Получают фториды галогенов прямым синтезом из простых веществ или дей-
действием фтора на готовый фторид галогена:
F2 + С12 = 2C1F; I2 + 7F2 = 2IFT; C1F + F2 = C1F3; C1F3 + F2 = C1F5
Структура трифторидов — тригональная бипирамида, два угла в основании кото-
которой заняты неподеленными электронными парами галогена в состоянии sjfid-
гибридизации. Согласно методу МО, трифториды характеризуются неравноцен-
неравноценными связями Г—F: одной трехцентровой F—Г—F и одной двухцентровой Г—F.
Молекулы пентафторидов имеют форму тетрагональной бипирамиды, одну из
вершин которой занимает неподеленная электронная пара галогена. Метод МО
предполагает наличие в FFs двух трехцентровых связей и одной двухцентровой.
Молекула IF7 имеет геометрию пентагональной бипирамиды, что, согласно мето-
методу ВС, отвечает sp3«^-гибридизации орбиталей центрального атома.
Интергалогениды, в том числе фториды галогенов, — реакционноспособные
вещества, функционируют как сильные окислители. Фториды галогенов' являют-
являются кислотными веществами, например:
C1F5 + ЗН2О = НСЮ3 + 5HF
462
2. Хлор. Особенности химии хлора. Второй типический эле-
элемент VII группы — хлор — характеризуется меньшей неметаллической активнос-
активностью по сравнению с фтором. Обусловлено это уменьшением потенциала иониза-
ионизации и 0Э0, а также возрастанием атомного радиуса и энтальпии диссоциации
молекул на атомы. Ббльшая прочность молекул С1г по сравнению с молекулами
F2 объясняется не только эффектом обратного экранирования в атомах фтора,
приводящим к ослаблению связи в его молекулах. В молекулах хлора имеет
место дополнительное тг-связывание за счет р-электронов и rf-орбиталей. тг-Связы-
вание возникает по донорно-акцепторному механизму, когда каждый атом хлора
одновременно является и донором и акцептором электронной пары (дативная
связь). В рамках метода ВС дополнительное тг-связывание можно представить
схемой
3d Зр
ТП |и|и| t
3s V
3s
ГГП ci
J_L_L
Зр 3d
Здесь сплошной вертикальной линией показано возникновение основной сг-связи
по обменному механизму, а пунктиром со стрелкой — дополнительное тг-связыва-
ние. При этом происходит лишь частичный перенос электронной плотности на d-
орбитали атома-партнера и поэтому порядок связи меньше 1,5.
Наличие Зсйэрбиталей атома хлора резко увеличивает валентные возможности
и вариации его положительных степеней окисления. Теоретически максимальная
ковалентность хлора может достигать 9 (девять орбиталей при п = 3). Однако
практически наблюдаемая координационная валентность хлора не превышает 6.
При взаимодействии атомов хлора между собой и с другими элементами хлор
проявляет степени окисления —1, 0, +1, +3, +5, +6 и +7. Разнообразие валент-
валентных состояний и степеней окисления делают химию хлора во многих отношениях
отличной от химии фтора. В то же время оба элемента — типичные неметаллы с
ярко выраженными окислительными свойствами. Поэтому главное в химии этих
элементов — функционирование в качестве анионообразователей в бинарных и
более сложных соединениях.
Природные соединения и получение хлора. Содержа-
Содержание хлора в земной коре почти в два раза больше, чем фтора. Основные минера-
минералы — каменная соль, сильвин КС1, карналлит, каинит KCl-MgSO4'3H2O и др.
Кроме того, морская вода содержит около 2,5 масс, доли, % NaCl.
В технике хлор получают электролизом концентрированных водных растворов
хлорида натрия. Н_а угольном аноде выделяется хлор, а на стальном катоде —
водород. В лаборатории хлор обычно получают из концентрированной соляной
кислоты, действуя на нее сильными окислителями, например КМпО4:
2МпО; + 10С1- + 1бН+ = 2Мп2+ + 5С12 + 8Н2О
Физические и химические свойства хлора. Хлор —
желто-зеленый ядовитый газ с резким раздражающим запахом.
463
Хлор непосредственно взаимодействует с большинством неметаллов (кроме
углерода, азота и кислорода), а также с подавляющим большинством металлов. В
жестких условиях хлор окисляет ксенон и образует ХеС12 и ХеСЦ. Выше 500°С
по отношению к хлору нестойки все металлы и сплавы. При комнатной темпера-
температуре сухой хлор не вступает в химическое взаимодействие с железом. Поэтому
хлор хранят и транспортируют в стальных баллонах. Растворяется хлор в воде,
SiCLj, CCI4 и других веществах. При температурах ниже 10° С из насыщенного
хлором водного раствора выпадают зеленовато-желтые кристаллы состава
С12'8Н2О. Это вещество плавится инконгруэнтно при 9,б°С. Известен также крис-
кристаллогидрат С12*6Н2О.
Характеристические соединения и соли хлорсо-
держащих кислот. Характеристическим летучим водородным соедине-
соединением хлора является хлороводород. В лаборатории его обычно получают дей-
действием концентрированной серной кислоты на поваренную соль. Основываясь на
термической стойкости НС1, в технике его получают прямым синтезом из простых
веществ:
У2С12(г) = НС1(г),
= -189,7 кДж/моль.
Эта реакция протекает по цепному механизму.
НС1 — бесцветный газ с резким запахом, раздражающе действует на дыхатель-
дыхательные пути, дымит на воздухе из-за образования с парами воды мелких капелек
раствора. В отличие от HF молекула НС1 малополярна, поэтому водородные
связи не возникают. Вследствие этого растворимость НС1 в воде меньше A4 моль
на 1 л при 10°С), чем HF, который смешивается с водой во всех пропорциях. При
температурах ниже 0°С из водных растворов НС1 были выделены кристаллогид-
кристаллогидраты НС1-Н2О, НС1-2Н2О и НС1-ЗН2О (<пл = -25, -18 и -15°С соответственно).
Эти продукты присоединения можно трактовать как аквакомплексы протона.
Поскольку комплексообразовательная способность катиона водорода выражена
слабо, указанные кристаллогидраты существуют только при относительно низких
температурах. Кристаллогидрат НСЬН2О может быть представлен как хлорид
гидроксония [НзО]С1.
При растворении НС1 в воде имеет место протолитическая реакция
НС1 + Н2О = Н3О+ + С1-
и образуется сильная (рКа -7) соляная кислота. С водой она образует азеотроп-
ную смесь, которая характеризуется одинаковым составом равновесных жидкой и
паровой фаз. Так, при атмосферном давлении и при 110"С перегоняется при
кипячении 20,2%-ный раствор НС1- Растворение металлов в соляной кислоте
приводит к образованию ее солей — хлоридов, например
Fe + 2НС1 = FeCl2 + Н2
В подобных реакциях соляная кислота выступает как окислитель вследствие
присоединения электрона к положительно поляризованному атому водорода. В
отличие от HF хлороводород окисляется до свободного хлора сильными .окисли-
.окислителями: KMnO4, K2Cr207, HNO3 и др. Однако вследствие большой величины
Е° (С1/С1") = 1,36 В даже концентрированная серная кислота не окисляет хлоро-
хлороводород.
464
Растворение хлора в воде сопровождается гидролитическим диспропорциони-
рованием:
С12 + НОН <?=* НС1 + НОС1
Равновесие этой реакции сдвинуто влево (рА'3,4), поэтому концентрация хлор-
хлорноватистой кислоты HQC1 при 20° С достигает лишь 0,03 моль/л. Растворением
хлора в водном растворе щелочи
С12 + 2КОН = КС1 + КОС1 + Н2О
практически нацело смещают равновесие вправо (рА" ~ -14) и получают соли
НОС1 — гипохлориты. НОС1 не выделена в свободном состоянии, максимальная
концентрация ее в воде 20 масс.долей, % (желтые растворы), представляет собой
слабую кислоту (рА"а 7,4). Солеобразующим оксидом этой кислоты является С12О
— желто-бурый газ с запахом хлора (<КИп — +2,2°С). Жидкий С12О взрывается, а
при комнатной температуре разлагается на хлор и СЮ2. Оксид CI2O — очень
сильный окислитель. Хлорноватистая кислота и гипохлориты также являются
окислителями. Сравнение стандартных редокс-потенциалов показывает, что хлор-
хлорноватистая кислота — более сильный окислитель, чем свободный хлор и гипохло-
гипохлориты:
кислая среда: НОС1 + Н+ + е - 1/2С12 + Н2О, Е° = 1,63 В,
щелочная среда: СЮ" + Н2О + с - у2С12 + 2ОН", Е° - 0,89 В.
Большая окислительная сила хлорноватистой кислоты объясняется сильным
поляризующим действием протона на связь С1—О, в результате чего связь дефор-
деформируется и, следовательно, НОС1 является нестабильным образованием по срав-
сравнению с гипохлоритами (щелочная среда). Хорошо известным окислителем также
является белильная известь СаОС12, представляющая собой смешанную соль
соляной и хлорноватистой кислот.
Вследствие малой стабильности НОС1 диспропорционирует:
2НОС1 = НС1 + НСЮ2) рА'-7,
ЗНОС1 = 2НС1 + НС1О3 (при нагревании),
НС1О2 — хлористая кислота — не выделена в индивидуальном состоянии, в раз-
разбавленных растворах быстро разлагается. Ее соли (хлориты) значительно более
стабильны, чем сама кислота. По окислительной силе НС1О2 сравнима с хлорно-
хлорноватистой кислотой, а хлориты несколько уступают гипохлоритам:
НСЮ2 + 2Н+ + 2е" = НСЮ + Н2О, Е° = 1,64 В,
С1О2 + Н2О + 2е" = СЮ" + 2ОН", Е° = 0,66 В.
Разложение хлористой кислоты по реакции
4НСЮ2 = 2СЮ2 + НСЮ3 + НС1 + Н2О
приводит к образованию диоксида СЮ2. Диоксид хлора — желто-бурый взрьцвча-
тый газ (ДЯу-298 = 109,5 кДж/моль). Молекула С1О2 имеет треугольную структу-
16 Я. А. Угнй , # , 465
ру с атомом хлора в состоянии врЗ-нгибридизации. СЮг парамагнитен, поэтому не
удивительно, что в растворе СС14 имеет место его димеризация в CI2O4.
Хлорноватая кислота НС1О3 существует только в растворе (максимальная
концентрация 40 масс, долей, %), кислота сильная (рКа 0). Окислительная актив-
активность НСЮз и ее солей (хлоратов) несколько уступает таковой НСЮг и их произ-
производных:
СЮ3 + 6Н+ + бе" = С1- + ЗН2О, Е° = 1,45 В,
СЮз + ЗН2О + бе' = С1- + 6ОН", Е° = 0,63 В.
Диоксид хлора взаимодействует с озоном, давая триоксид СЮз:
СЮ2 + О3 = СЮ3 + О2
СЮ3 — темно-красная дымящая жидкость (<пл = 3,5°С), взрывается (ДЯ^298 =
= 156 кДж/моль). Молекула СЮз парамагнитна, поэтому почти на 100% димери-
зована и в жидком состоянии представляет собой С12Об. Паровая фаза состоит из
малостабильных "нечетных" молекул СЮз. Жидкий триоксид хлора бурно реаги-
реагирует с водой:
С12О6 + Н2О = НСЮз + НС1С-4
Таким образом, С120б является смешанным солеобразующим оксидом хлорнова-
хлорноватой и хлорной (НСЮ4) кислот. НСЮ4 — самая сильная (рА' 10) из известных
кислот, представляет собой бесцветную дымящую подвижную жидкость. 100%-
ная хлорная кислота малоустойчива, часто взрывается, тогда как ее водные раст-
растворы вполне стабильны. Окислительные свойства НСЮ4 и ее солей — перхлора-
перхлоратов — выражены в меньшей степени, чем у НСЮз и хлоратов:
; + 8Н+ + 8е" = С1- + 4Н2О, Е° = 1,38 В,
СЮ4 + 4Н2О + 8е" = СГ + 8ОН", Е° = 0,56 В.
Перхлораты получают анодным окислением хлоратов, а также их диспропор-
ционированием.
Отнятием воды от НСЮ4 с помощью Р2О5 получают оксид С12О7 — бесцветное
масло с /кип = 83°С. Вследствие сильной эндотермичности (Д#^,д8 = 262,8
кДж/моль) CI2O7 при ударе и нагреве взрывается, медленно взаимодействует с
водой с образованием хлорной кислоты. Структура CI2O7 представляет собой два
тетраэдра, соединенных общей вершиной с одним мостиковым атомом кислорода.
В ряду кислородных кислот хлора с увеличением числа атомов кислорода в
формульной единице возрастает сила кислоты и уменьшается окислительная
активность. Все это обусловлено особенностями химического строения соответст-
соответствующих оксоанионов хлора:
6
л
v
¦С.1.
6
/
•Я-
9
X
9
СЮ"
cior
сю;
СЮ7
466
Центральный атом хлора находится в состоянии вр3-гибридизации. С уменьше-
уменьшением числа неподеленных электронных пар искаженность тетраэдра уменьшается
и СЮ4, у которого отсутствуют неподеленные электронные пары центрального
атома хлора, уже представляет собой правильный тетраэдр. В ряду СЮ" —?
—> С102 —> СЮз —> СЮ4 возрастает роль тг-связывания, вследствие чего зако-
закономерно уменьшается rf и увеличивается прочность связи кислорода с хло-
ром. И структурные, и энергетические факторы приводят к стабилизации оксо-
анионов в указанном ряду и одновременному уменьшению их окислительной
активности. Увеличение силы кислоты в обсуждаемом ряду обусловлено тем, что
с ростом числа кислородных атомов и увеличением степени окисления хлора
электронные облака все больше стянуты к центральному атому и связь Н—О
делается вге более ионной.
Хлориды металлов. Для металлов I—IV группы Периодической
системы максимальная степень окисления в хлоридах совпадает с номером груп-
группы. Только металлы подгруппы меди, некоторые лантаноиды и актиноиды дают
наряду с характеристическими хлориды более высокой и низкой степеней окис-
окисления. Но обусловлено это не образованием катионо- или анионоизбыточных фаз,
а проявлением переменных степеней окисления указанных металлов. В случае
металлов V—VIII групп максимальная степень окисления по хлору, отвечающая
номеру группы, как правило, не достигается. Главная причина этому — недоста-
недостаточная окислительная активность хлора. Даже фтор не дает многих теоретически
возможных высших фторидов. Кроме того, атомы металла не могут координиро-
координировать вокруг себя много атомов хлора из-за пространственных затруднений (стери-
ческий фактор).
Лишь небольшая часть хлоридов металлов с преимущественно ионной связью
кристаллизуется в ГЦК- и ОЦК-решетках (NaCl, KC1, RbCl, CsCl, T1C1, CuCl и
др.). Даже хлориды магния и щелочно-земельных металлов образуют ромбичес-
ромбическую структуру, а большинство хлоридов металлов — псевдомолекулярные слоис-
слоистые и цепные структуры (А1С13, ZnCl2, HgCl2, MnCl2, FeCl2, CoCl2, NiCl2, PdCl2)
FeCl3, СгС1з и др.). Хлориды металлов с координационными структурами —
нестехиометрические соединения. Нагревание хлоридов натрия и калия в парах
соответствующего щелочного металла приводит к изменению их окраски. Атомы
натрия или калия из паров проникают в кристаллы хлоридов, создают в них
точечные дефекты в анионной подрешетке. Известно, что электрические и опти-
оптические свойства обоих хлоридов зависят от условий получения (в избытке паров
металлов или газообразного хлора). Это обусловлено ничтожно малыми областя-
областями гомогенности, существующими около составов NaCl и КС1. Таким образом,
оба хлорида представляют собой двусторонние фазы.
Как и в случае других идо-соединений, увеличение степени окисления метал-
металла сопровождается ростом ковалентности хлоридов. Так, для свинца и титана их
высшие хлориды представляют собой малополярные молекулярные жидкости
(для РЬС14 <пл — 8,2; для ТЮЦ <пл = 21,1°С). В то же время низшие хлориды
этих металлов имеют солеобразный характер и температуры плавления TiCl2
1035°C и РЬС12 495'С. Устойчивость хлоридов s-металлов возрастает в группе
сверху вниз, как это видно на примере хлоридов IIA-группы:
16* 467
Соединение ВеС12(к) MgCl2(K) СаС12(к) SrCl2(K) ВаС12(к)
298, кДж/моль -449,7 -595,8 -749,9 -785,7 -796,3
412
714
772
874
961
Сравнительно низкая температура плавления ВеС12 объясняется его слоистой
структурой. В побочных подгруппах Периодической системы наблюдается умень-
уменьшение энергии Гиббса образования низших хлоридов, например:
Соединение
98- КДЖ/МОЛЬ
СгС12(к)
-356,3
МоС12(к)
-274,4
WC12(k)
-216,5
В то же время для высших хлоридов молибдена и вольфрама, как и следовало
ожидать, наблюдается противоположная тенденция:
Соединение
f 298, кДж/моль
МоС16(к)
-388,6
WC16(k)
-469,0
В зависимости от химической активности металла хлориды могут быть основ-
основными, амфотерными и кислотными; между ними возможны кислотно-основные
взаимодействия по Льюису. При этом амфотерный хлорид, например ВеС12, мо-
может функционировать и как основание, и как кислота:
ВеС12 + SnCl4 = Be[SnCl6]; 2KC1 4- ВеС12 = К2[ВеСЦ]
Хлориды металлов хорошо растворяются в воде, за исключением монохлори-
монохлоридов металлов подгруппы меди, Т1С1 и РЬС1г- При этом хлориды активных метал-
металлов не подвергаются гидролизу, большинство же хлоридов гидролитически рас-
расщепляется водой. Только некоторые хлориды металлов — HgCb, A11CI3, PtCU и
др. — не гидролизуются вследствие слабой электролитической ионизации их в
воде.
Хлориды неметаллов. В отличие от хлоридов металлов хлориды
неметаллов, как правило, летучие ковалентные соединения. Это означает, что в
твердом, жидком и газообразном состояниях они состоят из дискретных молекул.
Отдельные молекулы в твердом и жидком веществе удерживаются слабыми ван-
дер-ваальсовыми силами.
Хлориды неметаллов в основном получают непосредственным взаимодействием
простых веществ. С химической точки зрения, все они характеризуются кислот-
кислотной природой, например х
SiCl4 + 4Н2О = H4Si04 + 4HC1
3. Подгруппа брома. Характеристика элементов VII А-
группы. Поскольку каждый галоген в Периодической системе предшествует
инертному или благородному газу, он является самым электроотрицательным
элементом соответствующего периода. Действительно, до достижения электрон-
электронной конфигурации атомов инертных и благородных газов ns2np6 атомам галогенов
не хватает лишь одного электрона, вследствие чего для них наиболее характерна
468
тенденция к присоединению электрона. Тем не менее с ростом электроноемкости
атомов галогенов имеет место ослабление неметалличности и соответственно на-
нарастание признаков металличности. Об этом свидетельствуют уменьшение потен-
потенциалов ионизации, стандартных редокс-потенциалов и 0Э0. Если бром еще
является довольно сильным окислителем, то иод уже относится к числу мягких
окислителей. К тому же иод представляет собой твердое вещество с металличес-
металлическим блеском и проявляет заметные признаки амфотерности.
В ряду элементов VIIA-группы наблюдается более или менее закономерное
изменение физических и физико-химических характеристик атомов, ионов и
гомоатомных соединений. От фтора к иоду возрастают температуры плавления, и
кипения, энтальпии этих процессов, а также плотность. С ростом числа электрон-
электронных слоев увеличиваются размеры атомов и молекул; следовательно, усиливаются
дисперсионные силы межмолекулярного притяжения, что ведет к возрастанию
указанных характеристик. Прочность молекул от хлора к иоду уменьшается в
соответствии с ростом межъядерных расстояний, степень перекрывания электрон-
электронных облаков падает. Все это приводит к тому, что от хлора к иоду возрастает
константа термической диссоциации молекул галогенов на атомы.
Вследствие того что у брома имеет место 18-электронный предвнешний слой,
он обнаруживает явную вторичную периодичность, особенно в своих кислород-
кислородных соединениях. При этом 10 электронов 3</-орбиталей являются кайносиммет-
ричными, поэтому сильнее притянуты к атомному ядру. Это главная причина,
почему только сравнительно недавно были получены производные брома в степе-
степени окисления +7, в частности бромная кислота НВгО4. Предвнешние ^-электро-
^-электроны атома иода не относятся к кайносимметричным и не удивительно, что соеди-
соединения иода (+7) стабильны и давно известны.
Природные соединения и получение брома и и о -
д а. Содержание брома и иода в земной коре на несколько порядков меньше
типических элементов. Собственные минералы обоих элементов редки, практичес-
практического значения не имеют. Бром и иод содержатся в морской воде, в водах буровых
скважин нефтяных месторождений, рапе соляных озер. Бром — постоянный спут-
спутник хлора. Так, в сильвине и карналлите содержится до 3 масс, долей,'% брома в
виде твердого раствора замещения. Некоторые морские водоросли содержат зна-
значительные количества иода. Получают бром из морской воды, рапы соляных озер
и подземных рассолов окислением бромидов хлором с последующей отгонкой
брома с водяным паром и воздухом. Иод получают из буровых вод окислением
иодидов хлором или нитратом натрия.
Простые вещества. Физические и химические
свойства. Бром — темно-красная жидкость, а его пар — желто-бурого цвета
с резким раздражающим запахом. Бром растворим в воде C,53 г в 100 г воды
при комнатной температуре), во многих органических растворителях, обладает
высокой упругостью паров. Иод представляет собой черно-серое твердое вещество
с металлическим блеском, характеризующееся ромбической молекулярной решет-
решеткой. Иод легко возгоняется, образуя фиолетовые пары, состоящие из молекул 1г.
Иод плохо растворяется в воде, хорошо — в растворах иодидов металлов и орга-
органических растворителях.
Химическая активность брома меньше, чем хлора, но еще достаточно высока.
Со многими металлами и неметаллами он химически^ взаимодействует при обыч-
обычных условиях. Непосредственно не реагирует с кислородом, азотом, углеродом и
1 , • 469
благородными газами. Химическая активность иода — наименьшая в ряду галоге-
галогенов. Со многими элементами иод непосредственно не взаимодействует, а с некото-
некоторыми реагирует только при повышенных температурах (водород, кремний, многие
металлы). Основная причина уменьшения химической активности галогенов в
подгруппе сверху вниз — уменьшение их окислительных свойств.
Характеристические соединения, б р о м- и и о д с о -
держащие кислоты и их соли. Характеристические НВг и HI —
бесцветные газы, раздражающе действующие на стенки дыхательных путей. Ниже
приведены некоторые свойства бромоводорода и иодоводорода в сопоставлении со
свойствами HF и НС1:
&Н°, 2qg, кДж/моль
d (Н-Г), нм
Длина диполя, нм
/<-1029, Кл-м
Ионность связи, %
^пл. . С
4гип> С-
рК(терм.дис)
а @,1 н. р-р в Н2О), %
Растворимость в воде A0° С),
моль/л
HF
-270,1
0,092
0,036
0,640
43
-83,2
19,5
3,2
9,6
НС!
-93,8
0,128
" 0,023
0,347
14
-114,2
-85,6
_7
92,6
14
НВг
-35,1
0,141
0,017
0,263
12
-87,2
-66,7
-9
93,5
15
HI
+25,;
0,161
0,009
0,127
5
-50,8
-35,6
-11
95,2
12
Степень протолиза галогеноводородов в воде растет в ряду HF —> НС1 —>
—> НВг —> HI и одновременно увеличивается степень электролитической иони-
ионизации. В табл. 27 приведены расчетные значения энергии Гиббса процесса элект-
электролитической ионизации НГ в воде.
Таблица 27. Расчет ДCу 29в (кДж/моль) процесса
ионизации НГ в воде
Процесс
НГ(г) = Н(г) + Г(г)
Н(г) = Н+(г) + е
Г(г) + е = Г(г)
Г(г) = Г(р-р)
Н+(г) + Н2О = Н3О+(р-р)
HF
565,7
1312,0
-339,1
-535,9
-1168,2
НГ(г) + Н2О = Н3О+(р-р) + Г(р-р) -166,0
НС1
431,5
1312,0
-355,9
-405,7
-1168,2
-186,3
НВг
364,0
1312,0
-330,8
-386,0
-1168,2
-209,0
HI
297,8
1312,0
-301,7
-359,2
-1168,2
-219,3
Нарастание силы кислоты в указанном ряду НГ обусловлено уменьшением
прочности химической связи молекул галогеноводородов A-я строка табл. 27).
Этот фактор является определяющим в закономерном возрастании отрица-
отрицательных значений энергии Гиббса ионизации НГ в воде от HF к HI (резуль-
(результирующая строка табл. 27). Отсюда гетеролитический распад электролитов в
470
водном растворе обусловлен не столько полярностью, сколько прочностью хими-
химических связей.
В том же направлении от HF к HI увеличивается восстановительная актив-
активность галогенид-ионов. НВг и HI восстанавливают концентрированную серную
кислоту, например
2Н1(г) + H2SO4Ck) = 12(к) + SO2(r) + 2Н2О(ж), AG°2m = -82 кДж.
Поэтому НВг и HI обычно получают гидролизом бромидов и иодидов фосфора:
РГ3 4- ЗН2О = Н3РО3 + ЗНГ
Растворение брома и иода в воде приводит к установлению равновесия гидро-
гидролитического диспропорционирования:
Г2 + НОН ^ НГ + НОГ
По сравнению с хлором в случае брома и иода равновесие еще больше смещено
влево (рК 8,2 и 12,7 соответственно для брома и иода). Образующиеся бромнова-
тистая НОВг и иодноватистая HOI кислоты не выделены в индивидуальном сос-
состоянии, а соли — гипобромиты и гипоиодиты — вполне стабильны в отсутствие
влаги. Вследствие уменьшения ОЭО галогенов в ряду НОС1, НОВг и HOI сила
кислот падает: рКа соответственно равны 7,6; 10,7 и 12,4. Очень слабая иоднова-
иодноватистая кислота проявляет амфотерность:
Г 4- ОН" ^ HOI ^ Н+4- Ю'
Существование катиона 1+ — проявление металлических свойств иода.
Бромно- и иодноватистая кислоты диспропорционируют без нагревания:
ЗНОГ = 2НГ 4- НГО3
НВгОз — бромноватая кислота — существует только в растворе, а йодноватая
кислота НЮз выделена в свободном состоянии и представляет собой бесцветные
кристаллы с tnjl = 110°С. НВгОз и ее соли — броматы — по окислительной актив-
активности приближаются к соответствующим производным хлора. Окислительные
свойства йодноватой кислоты и иодатов заметно меньше. Кислоты НВгОз
(pA'a 0,7) и НЮз (рКа 0,8) относятся к разряду сильных кислот. Йодноватой
кислоте отвечает оксид I2Os — бесцветные кристаллы, плавящиеся при 300°С с
разложением. Это самый устойчивый оксид среди оксидов галогенов (Д#^298 =
= -184,5 кДж/моль). Он обладает сильными окислительными свойствами, а с
водой образует йодноватую кислоту.
Соли бромной кислоты НВгО4 — перброматы — получают при окислении
броматов в щелочной среде, например фтором:
КВгОз + Fl + 2KOH = КВгО4 4- 2KF + Н2О
Бромная кислота не выделена в индивидуальном состоянии, по силе близка к
НС1О4, но значительно менее устойчива, а потому является более сильным окис-
окислителем, чем хлорная кислота. Перброматы по свойствам похожи на перхлораты.
Йодная кислота НЮ4 может быть получена анодным окислением НЮ3. По
силе НЮ4 значительно уступает хлорной кислоте, а по окислительной активности
471
йодная кислота превосходит хлорную. Из водных растворов йодная кислота
выделяется в виде кристаллогидрата НЮ4-2Н2О. Это соединение представляет
собой кислоту Н5Юб- Хорошо известны ее производные, например Ag5IO6. Орто-
иодная кислота Н5Ю6 относится к разряду кислот средней силы: рК\ 3,2 и
рА'г 6,7. Анион Ю|' — октаэдр с атомом иода в центре, находящимся в состоянии
^«Р-гибридизации. Кристаллы ЩЮб плавятся приЛЗЗ°С, разлагаясь:
2Н5Ю6 = 12О5 + 5Н2О + О2
При взаимодействии НЮ4 с олеумом образуется оранжевое твердое вещество I2O7
— оксид, образующий йодную кислоту. Его свойства плохо изучены. Кроме того,
существуют малоустойчивые и менее изученные оксиды I2O4 и I4O9 — желтые
твердые вещества.
Наименее стабильны оксиды брома. Вг2О — красно-коричневая жидкость,
разлагается на простые вещества при -40° С. Вг2С>4 — светло-желтые Кристаллы,
неустойчивые при температурах выше -50°С. ВгОз — бесцветные кристаллы,
устойчивые только в атмосфере озона, разлагаются уже при -70° С. Таким обра-
образом, в химии оксидов и кислородных кислот брома ярко проявляется вторичная
периодичность.
Соединения с металлами. Соли бромо- и иодо водородных кис-
кислот — бромиды и иодиды — являются аналогами хлоридов металлов. В степени
окисления -1 бром также проявляет вторичную периодичность. В ряду хлорид —
бромид — иодид имеет место немонотонное изменение энергии Гиббса их образо-
образования из простых веществ, например:
Соединение ,., КС1(к) КВг(к) К1(к)
AG°f 2д8> кДж/моль -408,6 -380,1 -322,8
Аналогичный характер изменения энергии Гиббса наблюдается и для элемен-
элементов побочных подгрупп:
Соединение ,.... ZnO2(K) ZnBr2(K) ZiiI2(k)
AG°f ш, кДж/моль -369,7 -312,4 -209,3
В приведенных примерах разница между энергиями Гиббса образования хло-
хлоридов и бромидов намного меньше, чем между бромидами и иодидами. Кайно-
симметричность 3<йэрбиталей сильнее стягивает всю электронную оболочку
атома брома к ядру, а потому бром обладает несколько повышенной ОЭО и окис-
окислительной активностью. Отсюда вполне закономерно проявление бромом вторич-
вторичной периодичности и в производных с положительными степенями окисления, и
в степени окисления -1.'
Бромиды и иодиды металлов по химической природе могут быть основными,
амфотерными и кислотными; взаимодействие между ними- приводит к образова-
образованию комплексных бромидов и иодидов, например
2NaBr + CdBr2 = Na2[CdBr4]; Bel2 + Hgl2 = Be[HgI4]
Отношение бромидов и иодидов к воде в общих чертах такое, как и .в случае
хлоридов металлов. В воде плохо растворимы бромиды и иодиды меди, серебра и
ртути. .
Соединения с неметаллами. В производных с неметаллами,
472 . - •
которые представляют собой преимущественно ковалентные соединения, также
имеет место немонотонное изменение энергии Гиббса, например:
Соединение SiCl2(r) SiBr2(r) SiI2(r) SiCl4(r) SiBr4(r) SU4(r)
ДС/298' кДж/моль .. -171,2 -82,1 +23,9 -617,6 -435,8 -189,5
(89,1) A06,0) A81,2) B46,3)
Как видно из приведенных примеров, переход от бромидов к иодидам крем-
кремния сопровождается более резкой разницей в значениях энергии Гиббса (цифры
в скобках). В целом химии брома независимо от степеней окисления присуща
вторичная периодичность. По химической природе бромиды и иодиды неметал-
неметаллов являются кислотными веществами, например
Р13 + ЗН2О = Н3РО3 + 3HI
Кроме того, эти вещества могут проявлять свойства кислоты по Льюису.
4. Подгруппа марганца. X ар актеристика элементов VII В-
группы. Элементы VIIB-группы — марганец, технеций, рений — завершают
первые пятерки вставных декад rf-элементов. Их валентная электронная конфигу-
конфигурация (га — 1)<?ns2 позволяет сделать вывод об относительной стабильности степе-
степени окисления +2, поскольку наполовину заполненная rf-оболочка отличается
повышенной устойчивостью. Это положение должно быть особенно характерным
для марганца в силу заметной энергетической неравноценности 4s- и 3rf-coc-
тояний. Для технеция и особенно для рения энергетическое различие между
(п — l)d- и ns-оболочками становится меньше и особая роль ras-электронов ут-
грачивается.
Ниже сопоставлены основные характеристики элементов и простых веществ
VIIB-группы:
Mn Tc Re
Содержание в земной коре,
масс.доли, %
Валентная электронная конфи-
конфигурация
Атомный радиус, нм
Потенциалы ионизации, В
1у. Э -+ Э+ + е-
Н Э+ -+ Э2+ + е
13: Э2+ -+ Э3+ + е
1А: Э3+ -> Э4+ + е"
/5: Э4+ -+ Э5+ + е
16: Э5+ —f Э6+ + е
17: Э6+ -> Э7+ + е
S 4 В ,
i-l
оэо
Температура плавления, °С
Температура кипения, °С
Плотность, г/см3
?*(Э27Э), В (для Re Э3+/Э)
9-10
[АфсРАв2
0,130
7,40
15,64
33,7
53,4
76,0
100,7
119,2
406
1,5
1244
2080
7,44
-1,18
-
[Kr]4(f5s2
0,136
7,28
15,26
29,3
43,5
59,2
76,2
94,1
325
1,9
2200
4600
11,49
0,4
1-Ю'
[Хе]4/45
0,137
7,87
13,20
26,0
37,7
50,6
64,5
79,0
279
1,9
3180
5600
21,04
0,3
Обращает на себя внимание то, что первый потенциал ионизации в ряду Мп —
Тс — Re изменяется немонотонно: от марганца к технецию уменьшается, ЧТо
связано с увеличением атомного радиуса, а от технеция к рению заметно возрас-
возрастает, хотя атомные радиусы последних двух элементов близки. Это связано с
заметным проникновением 6*-электронов рения под экран заполненной 4/-оболоч-
ки. Увеличение первого потенциала ионизации у рения приводит к возрастанию
химической благородности этого металла по сравнению с его более легкими ана-
аналогами. Все остальные потенциалы ионизации, а также сумма семи потенциалов
ионизации убывают от марганца к рению. При этом соответствующие потенциалы
ионизации более близки для технеция и рения и отличаются от таковых для
марганца, что и подтверждает большее сходство между двумя последними пред-
представителями VIIB-группы. Об этом же говорят и близкие значения атомных
радиусов, и одинаковые значения электроотрицательностей.
Учитывая монотонное возрастание последовательных потенциалов ионизации,
равновероятными представляются все положительные степени окисления от -f-1
до +7. Действительно, для марганца и рения известны производные, отвечающие
всем положительным степеням окисления и даже нулевой (карбонилы). В то же
время относительная стабильность этих производных несколько различается. Для
марганца наиболее характерны степени окисления +2, +4 и 4-7, относительно
легко получаются производные, отвечающие степеням окисления 0, +3, +5 и +6.
Для рения наиболее устойчивы производные в высшей степени окисления +7,
что вообще характерно для тяжелых аналогов в побочных подгруппах, но особен-
особенно ярко проявляется у рения, накладывая отпечаток на всю химию этого элемен-
элемента. Для технеция типичными являются степени окисления +4, +6 и +7.
Природные соединения и получение металлов*
Если марганец относится к числу наиболее распространенных элементов на Земле
и следует непосредственно за железом, то рений относится к числу довольно
редких элементов. Что же касается технеция, то в природе этот элемент встреча-
встречается в исчезающе малых количествах как один из нестабильных продуктов распа-
распада урана (порядка 1-Ю3 г на 1 г урановой смоляной руды).
Основным природным соединением марганца является минерал пиролюзит
МпОг-хНгО — гидратированный оксид марганца (+4) серо-черного цвета. Марга-
Марганец часто встречается совместно с железом в оксидных, карбонатных и сульфид-
сульфидных рудах: гаусманит МП3О4 совместно с магнитным железняком ГезО.}, биксбит
представляет собой смешанный оксид марганца и железа (Mn,FeJO3, родохрозит
МпСОз встречается совместно с сидеритом ГеСОз, марганцевая обманка MnS —
совместно с пирротином FeS, марганцевый колчедан M11S2 — совместно с желез-
железным колчеданом FeS2-
Рений самостоятельных месторождений не имеет и собственных минералов не
образует. Он содержится в качестве примеси в молибденовых, вольфрамовых и
платиновых рудах. #
Основная масса металлического марганца в настоящее время получается путем
алюмотермического восстановления пиролюзита, гаусманита или предварительно
обожженных карбонатных и сульфидных руд:
ЗМп3О4 + 8А1 = 9Мп + 4А12О3
Технический марганец при необходимости подвергают электрохимическому ра-
рафинированию и вакуумной переплавке.
Получают рений из отходов медного и молибдено-вольфрамового производст-
производства. Эти отходы вначале подвергают окислительному обжигу. При этом рений
переходит в высший оксид Re2O7, который путем выщелачивания переводят в
перренат калия KReO4. Последний очищают от солей тяжелых металлов и избыт-
избытка щелочи многократной перекристаллизацией и восстанавливают водородом при
нагревании:
2KReO4 + 7Н2 = 2КОН + 2Re + 6Н2О
Получаемый таким образом рениевый порошок горячим прессованием переводят
в компактное состояние. Рениевые штабики при дуговой или электронно-лучевой
вакуумной плавке превращают в слитки металлического рения. Рений можно
получить и путем термической диссоциации галогенидов. Разложение галогени-
дов осуществляют на раскаленной нити из чистого рения.
Основная масса технеция получается в ядерных реакторах при реакциях деле-
деления ядерного горючего. Выход технеция составляет около 25 г на 1 кг плутония.
Простые вещества. Физические и химические
свойства. В компактном состоянии все элементы подгруппы марганца пред-
представляют собой металлы серебристо-белого цвета. Прежде всего следует отметить,
что в отличие от технеция и рения, не имеющих полиморфных модификаций и
образующих кристаллы с ГПУ-структурой, для марганца характерен полимор-
полиморфизм:
а-Мп > - /?-Мп 5=======* 7~Мп ч 8-Мп
причем а- и ^-модификации обладают ОЦК-структурой. Плотность полиморфных
модификаций уменьшается в направлении а —> /? —> у —> 6 в соответствии с
уменьшением координационного числа, что говорит о преобладающем вкладе
энтропии, который возрастает с увеличением температуры.
По химической устойчивости марганец и его более тяжелые аналоги заметно
различаются. Если марганец в электрохимическом ряду напряжений располагает-
располагается между Магнием и цинком и является, таким образом, довольно активным
металлом, то технеций и рений относятся к благородным металлам. В мелкодис-
мелкодисперсном состоянии марганец при нагревании разлагает воду с выделением водо-
водорода. Все три металла устойчивы на воздухе в обычных условиях. При нагрева-
нагревании марганец сгорает на воздухе, образуя оксид М113О4. Технеций и рений при
нагревании до 300° С образуют высшие оксиды Э2О7; они летучи и не предохра-
предохраняют металл от дальнейшего окисления.
Марганец энергично взаимодействует с неокисляющими разбавленными кис-
кислотами. При этом образуются только производные Мп(+2):
Мп + 2НС1 = МпС12 + Н2
Подобным же образом действует на марганец и азотная кислота:
Мп + 4HNO3 = Mn(NO3J + 2NO2 + 2Н2О
Технеций и рений с неокисляющими кислотами не взаимодействуют, а с азотной
кислотой реагируют по схеме
ЗЭ + 7HNO3 = ЗНЭО4 + 7N0 + 2Н2О
образуя соответственно технециевую НТсО4 и рениевую HReO4 кислоты.
Марганец энергично взаимодействует с галогенамиг однако при этом образу-
образуются только солеобразные производные МпГ2. При нагревании марганец взаимо-
взаимодействует и со всеми остальными неметаллами.
Металлический рений взаимодействует с фтором и хлором при нагревании до
100°С, с бромом — выше 300°С, а с иодом рений в реакции не вступает. При
сильном нагревании рений реагирует и со всеми остальными неметаллами. Техне-
Технеций в этом отношении подобен рению.
Характеристические соединения. Для марганца могут
быть получены следующие оксиды: МпО, Мп2Оз, МпО2, Мп2О7- Нагреванием
пиролюзита в токе водорода получают МпО:
МпО2 + Н2 = МпО + Н2О
Кроме того, МпО получают термическим разложением оксалата или карбоната
марганца. При длительном нагревании МпО2 на воздухе образуется оксид мар-
марганца (+3):
4МпО2 = 2Мп2О3 + О2
Установлено, что в структуре этого оксида марганец находится в различных
степенях окисления: +2 и 4-4, что можно представить в виде валовой формулы
+ 2 + 4
МпМпОз- Именно это соединение встречается в природе в виде минерала бра-
унита.
Оксид МпО является односторонней фазой переменного состава, область
гомогенности которой лежит в интервале MnOi,0-1,2- Все рассмотренные оксиды
марганца представляют собой тугоплавкие кристаллы с координационной струк-
структурой.
Высший оксид марганца Мп2О7 можно получить только косвенным путем при
взаимодействии перманганата калия с концентрированной серной кислотой:
2KMnO4 + 2H2SO4 = Mn2O7 + 2KHSO4 + Н2О
Мп2О7 представляет собой жидкость черно-зеленого цвета. Следовательно, в
отличие от рассмотренных выше оксидов М-п2О7 обладает молекулярной структу-
структурой и характеризуется постоянством состава.
Оксиды технеция и рения, отвечающие низшим степеням окисления, получа-
получают лишь косвенным путем. Так, при мягком восстановлении Re2O7 с помощью
оксида углерода СО образуется красный кристаллический порошок 11еОз с ме-
металлическим блеском:
Re2O7 + СО = 2ReO3 + СО2
При нагревании в вакууме триоксид рения диспропорционирует:
3ReO3 = Re2O7 + ReO2
Диоксиды технеция и рения обычно получают термическим разложением пертех-
йатов и перренатов аммония:
2NH43O4 = 4H2O + N2 + 2ЭО2
Оба диоксида при нагревании в отсутствие кислорода диспропорционируют:
7ЭО2 = 2Э2О7 + ЗЭ, а при нагревании на воздухе окисляются до Э2О7.
Оксиды марганца и его аналогов, отвечающие всем степеням окисления, кроме
высшей, с водой не взаимодействуют, поэтому соответствующие гидратные формы
получаются косвенным путем. Высшие же оксиды Э2О7 энергично взаимодейству-
взаимодействуют с водой, образуя соответствующие кислоты — марганцевую НМпО^, техне-
циевую НТсО4 и рениевую HReO4. Марганцевая кислота наименее устойчива и
существует лишь в растворах с концентрацией не более 20%. При больших кон-
концентрациях раствора она разлагается с выделением кислорода:
4НМпО4 = 4МпО2 + 2Н2О + ЗО2
Технециевая и рениевая кислоты более устойчивы. При концентрировании раст-
раствора НТсО4 ее красно-коричневые кристаллы выпадают в осадок, в то время как
HReO4 отщепляет воду лишь при 160°С и образует Re2O7-
Сила кислот, отвечающих высшим степеням окисления, в ряду Мп — Тс — Re
закономерно уменьшается. Если НМиО4 По силе напоминает соляную кислоту, то
НТсО4 и HReO4 диссоциированы в меньшей степени. Соли этих кислот называ-
называются соответственно перманганатами, пертехнатами и перренатами.
Гидратные формы, отвечающие степени окисления 4-6, также обладают кис-
кислотными свойствами. Эта степень окисления сравнительно мало стабильна. Одна-
Однако она стабилизируется в анионе ЭО4 , который устойчив в сильнощелочной
среде. В нейтральной и кислой средах происходит диспропорционирование:
ЗЭО4" + 2Н2О = 2ЭО4 + ЭО2 + 4ОН"
Ион ЭО4 называют манганатом (MnOf), технатом (ТсО4~), ренатом (ReO4~). В
свободном состоянии соответствующие кислоты не выделены. Солеобразные про-
производные Ме2ЭО4 получают восстановлением перманганатов, пертехнатов и пер-
ренатов в щелочной среде, например
2KMnO4 + K2SO3 + 2KOH = 2K2Mn04 + K2SO4 + Н2О
В противоположность разноцветным ионам МпО4 (фиолетовый), ТсО4 (красно-
желтый) и ReO4 (бесцветный) все ионы ЭО4 имеют темно-зеленую окраску. Твер-
Твердый перманганат при нагревании разлагается с выделением кислорода, что ис-
используется в качестве лабораторного способа его получения:
2КМпО4 = К2Мп04 + МпО2 + О2
Соответствующие производные технеция и рения при нагревании устойчивы.
Так, КТсО4 и KReO4 перегоняются при 1000° С без разложения.
При сплавлении диоксида марганца с содой и селитрой на воздухе появляется
характерное голубое окрашивание, свойственное производным MnOf", в которых
марганец проявляет степень окисления +5:
МпО2 + Na2CO3 + NaNO3 = Na3MnO4 + СО2 + NO2
477
В растворе соли марганца (+5) — гипоманганаты — легко диспропорционируют: :
2Na3Mn04 + 2Н2О = MnO2 + Na2Mn04 + 4NaOH
Поскольку оксиды ЭО2 нерастворимы в воде, соответствующие гидроксиды
Э(ОНL получаются только косвенным путем и также нерастворимы в воде. Так,
темно-бурый осадок Мп(ОНL можно получить окислением на воздухе гидроксида
Мп(ОНJ:
2Мп(ОНJ + О2 + Н2О = 2Мп(ОНL
Гидроксиды Э(ОНL являются амфотерными соединениями, в ряду Мп — Тс —
Re кислотная функция ослабевает. Сани, отвечающие кислотным свойствам
Mii(OHL, называются манганитами, а сам гидроксид в этом случае рассматрива-
рассматривается как марганцоватистая кислота. Манганиты неустойчивы, и их трудно выде-
выделить в чистом виде. Интересно, что оксид Мп3О4 может быть получен при взаи-
взаимодействии Мп(ОНL как кислоты и Мп(ОНJ как основания:
2Мп(ОНJ + Мп(ОНL = Мп2Мп04 + 4 Н2О
Таким образом, с химической точки зрения, оксид Мп3О4 является манганитом
марганца (+2) и встречается в виде минерала гаусманита.
Кислотная функция Тс(ОНL и Re(OHL выражена еще менее ярко. Соответст-
Соответствующие производные — рениты — могут быть получены лишь при сплавлении
ReO2 со щелочами:
ReO2 + 2NaOH = Na2Re03 + H2O
Гидроксиды Мп(ОНJ и Мп(ОНK характеризуются преимущественно основ-
основным характером. В кислой среде они легко образуют аквакомплексы и соответст-
соответствующие соли Мп(+2) и Мп(+3), причем последние малохарактерны для мар-
марганца:
Мп(ОНJ + 2Н+ + 4Н2О = [Мп(Н2ОN]2+
Получают Мп(ОНJ в виде студнеобразного осадка розоватого цвета (рПР 17,4)
действием щелочей на водные растворы солей Мп(+2).
Ниже сопоставлены кислотно-основные свойства известных гидроксидов эле-
элементов VIIB-группы. Формулы гидроксидов, не выделенных в свободном сос-
состоянии, приведены в скобках:
Усиление кислотных свойств
Усиление основных свойств
Mn(OH)j Mn(OHK Мп(ОНL (Н3Мп0<) (Н2МпО4) НМпО4
(Тс(ОНL) (Н2Тс04) НТсО4
Re2O3-*H2O Re(OHL (HjReO4) (H2Re04) HReO4
Повышение степеней окисления
47R
Соединения с неметаллами. Марганец с галогенами образует
соединения, отвечающие низшим степеням окисления +2, +3, 4-4, причем с
уменьшением окислительной активности галогенов в ряду F — С1 — Вг — I наб-
наблюдается снижение степени окисления марганца в предельных галогенидах. Так,
с фтором и хлором марганец образует ди-, три- и тетрагалогениды, в то время
как с бромом и иодом лишь дигалогениды. В противоположность марганцу рений
склонен образовывать с галогенами соединения, отвечающие более высоким сте-
степеням окисления, причем с фтором образует ReF4, ReF5, ReF6 и ReF7, с хлором
максимально насыщенный галогенид имеет состав КеСЦ, с бромом — ReBrs, а с
иодом — Rel4. Низшие галогениды марганца МпГг представляют собой сравни-
сравнительно тугоплавкие солеобразные соединения с преимущественно ионным типом
связи. Все соли МпГз известны как в безводном состоянии, так и в форме крис-
кристаллогидратов.
Из галогенидов, отвечающих степеням окисления марганца 4-3 и 4-4, известны
фториды и хлориды, которые сильно различаются по устойчивости. Красные
кристаллы MnF3 начинают разлагаться на MnF2 и F2 лишь при температуре
более 600°С. В противоположность этому хлорид марганца (+3) в свободном
состоянии не выделен и известен в виде двойных солей МпС1з'ЗКС1 или
МпС1з*2КС1. Синие кристаллы MnF4 получают при взаимодействии МпОг с
концентрированной плавиковой кислотой при охлаждении. Фторид марганца
D-4) — реакционноспособное вещество, медленно разлагающееся на воздухе и в
водном растворе. Он стабилизируется путем образования комплексов типа
Хлорид марганца D-4) также стабилизируется в виде комплекса
р-.
В противоположность солеобразным галогенидам марганца практически все
галогениды рения легкоплавки, что свидетельствует о молекулярном типе крис-
кристаллической структуры. Из фторидов рения наиболее стабильны ReF4, ReF6 и
ReF7- Пентафторид ReF5 диспропорционирует по схеме 2ReF5 = ReF4 4- ReF6.
Хлорид рения D-3) существует в виде тримерного кластера Re3Clg:
[МпС16
Среднее расстояние между атомами рения здесь составляет 0,248 нм и короче
расстояния между атомами в кристалле металлического рения @,275 нм), что
свидетельствует о высокой прочности внутрикластерной связи. Группировка
ResClg сохраняется в паровой фазе до 600° С. Тримерным кластером является и
трибромид рения.
Малоустойчивые тетрагалогениды рения, которые можно получить действием
соответствующих галогеноводородов на HReO4, также легко образуют двойные
соли ReIV2Mer, в которых степень окисления рения 4-4 более устойчива. Анало-
Аналогичные производные известны и для технеция.
ReCis является основным продуктом взаимодействия рения с хлором при
нагревании. Пентабромид ReBr5, получаемый действием брома на рений, сущест-
479
вует только при наличии избытка брома, а в противном случае разлагается на
ReBrs и Вг2.
В соединениях марганца и его аналогов с халькогенами в еще большей степе-
степени проявляется тенденция технеция и рения к образованию высших халькогени-
дов в противоположность марганцу. Для марганца характерны соединения соста-
состава МпХ и МпХг (где X — S, Se, Те), формально отвечающие степеням окисления
марганца +2 и +4. Однако дихалькогениды M11S2, MnSe2 и МпТе2 относятся к
полисоединениям, кристаллизуются в структурном типе пирита FeS2, где струк-
структурными элементами анионообразователя являются гантели Х2.
Моносульфид марганца можно получить не только прямым синтезом, но и
путем осаждения из водных растворов солей Мп(+2) сульфидом аммония. При
этом образуется осадок телесного цвета, содержащий переменное количество воды
MnS-xH^O. Сульфид марганца относится к числу труднорастворимых соединений
(рПР 15,2).
Для технеция и рения характерно образование высших халькогенидов общей
формулы Э2Х7 *. Высшие сульфиды технеция и рения получают действием серо-
сероводорода на растворы технециевой и рениевой кислот:
2НЭО4 + 7H2S = 92S7 + 8Н2О
TC2S7 и Re2S7 представляют собой черные кристаллические вещества, раствори-
растворимые в окисляющих кислотах. Для рения известны халькогениды ReS2 и ReSe2,
кристаллизующиеся в структурном типе пирита.
Соединения металлов VIIB-группы с пниктогенами представляют собой ка-
тионо- или анионоизбыточные фазы. Для первых характерно проявление метал-
лидных свойств (например, Mn4N, Mn3P, Mn2P, Mn3As, Mn2Sb, ReNx, Re2P).
Вторые характеризуются наличием анион-анионных связей. При этом существен-
существенную роль играет природа анионообразователя. Для полуметаллов As, Sb, Bi в
соединениях трудно ожидать проявления неметаллических свойств даже при их
высоком содержании, в то время как высшие фосфиды (МпРз, ReP2, ReP3) обла-
обладают полупроводниковыми свойствами.
Достаточно разнообразны соединения марганца и рения с углеродом, кремни-
кремнием и бором. Если углерод и бор образуют главным образом фазы типа внедрения,
то кремний с марганцем и рением дает ряд соединений, свойства которых зависят
в основном от содержания анионообразователя, поскольку кремний сам по себе
является полупроводником. Для обоих металлов характерны силициды следую-
следующего состава: Me3Si, Me5Si2, MeSi (металлиды или полуметаллы). Высшие сили-
силициды Mn4Si7 и ReSi2 являются полупроводниками.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Наиболее разнообразны солеобразные производные
Мп(+2), который образует соли практически со всеми известными анионами. Эти
соли, как правило, устойчивы, и большинство из них растворимы в воде, кроме
карбоната МпСО3 и фосфата Мп3(РО4J- Из водных растворов они обычно крис-
кристаллизуются в виде кристаллогидратов, например Mn(NO3J*6H2O, MnSO4-7H2O,
МпБО+'бНгО. Перечисленные кристаллогидраты представляют собой аквакомп-
лексы, в которых координационное число марганца равно 4 или 6:
[Mn(H2ON](NO3Jj [Mn(H2ON]SO4-H2O, [Mn(H2OL]SO4-H2O.
* Кроме теллура, так как его производные не изучены.
480
Помимо аквакомплексов для Мп(+2) характерны комплексные соединения с
такими лигандами, как ОН", CN", CNS", С2О4~, NH3, этилендиамин. Во всех этих
соединениях координационное число марганца равно 6. Известны и галогенидные
ацидокомплексы с к.ч. 4 для С1", Вг", Гиб для F", например K4[MnF6] и
К2[МпС14].
Комплексы Мп(+2) содержат комплексообразователь с неспаренными электро-
электронами на 3</-орбиталях как в сильном, так и в слабом поле лигандов:
Сильное поле Слабое поле
IV1 II •* U 1 » 1 ¦ i л
и и гги н
Поэтому большинство комплексов Мп(+2) парамагнитно. Сильнопарамагнитные
комплексы, отвечающие высокоспиновому состоянию Мп(+2), характеризуются
малой устойчивостью. Напротив, низкоспиновые комплексы более устойчивы.
Например, для комплекса [МпС16]4" рА'нест 3, а комплекс [Мп(ЭДТА)]2" характе-
характеризуется piTHeCT 14. Существенное повышение прочности комплекса в последнем
случае обусловлено еще и хелатным эффектом.
Солеобразные соединения Мп(+3) характеризуются неустойчивостью. В вод-
водных растворах они легко гидролизуются, диспропорционируют по схеме
2Мп(+3) = Мп(+2) + Мп(+4)
Стабильность Мп(+3) повышается за счет комплексообразования. Если сульфат
Mri2(SO4K неустойчив, то комплекс [Mn(CNN]3" вполне стабилен.
Соли Мп(+4), отвечающие основной функции Мп(ОНL, немногочисленны.
Наиболее известен сульфат Mn(SO4J черного цвета, который устойчив при из-
избытке серной кислоты. При разбавлении водой разлагается с выделением
Мп(ОНL и продуктов его дегидратации вплоть до МпО2.
Своеобразную группу комплексных соединений марганца и его аналогов обра-
образуют кластеры, главным образом галогенидные. Образование кластеров более
характерно для тяжелых аналогов, главным образом для рения, в то время как
для марганца существуют солеобразные галогениды.
Столь же своеобразным классом комплексных соединений элементов VIIB-
группы являются карбонилы. Для Мп, Тс и Re известны биядерные кластерные
карбонилы Э2(С0)ю со связью Э—Э. В ряду Мп — Тс — Re температуры плавле-
плавления карбонилов и их устойчивостью увеличиваются:
Соединение Мп2(СОI0 Тс2(СО)ю Re2(CO)i0
U°C 155 160 177
В этих соединениях комплексообразователь выступает и в качестве донора, и
как акцептор, т.е. здесь реализуется дативное взаимодействие. Диамагнетизм
карбонилов Э2(СО)ю подтверждает образование связи Э—Э по обменному меха-
механизму и возникновение биядерного кластера типа (СОM—Мп—Мп—(СОM.
Металлохимия. Марганец и рений неизоструктурны, заметно различа-
различаются по элек/гроотрицательности и особенно по температурам плавления. Поэтому
между собой они образуют лишь ограниченные твердые растворы и металлидные
, 481
фазы переменного состава с содержанием около 50 ат. долей, % компонентов. В
то же время вследствие близости свойств технеция и рения эти элементы образу-
образуют между собой непрерывный ряд твердых растворов.
Непрерывные твердые растворы с другими металлами образует только 7~маР~
ганец (ГЦК), причем в качестве партнеров выступают изоструктурные модифика-
модификации металлов 3<?-ряда, которые имеют с марганцем близкие значения атомных
радиусов (V, j-Fe, /?-Со, Ni, Си). Рений образует непрерывные твердые растворы
с металлами, обладающими ГПУ-структурой (Ru, Os, a-Co).
С большинством металлов Периодической системы, включая s- и р-элементы,
марганец и рений образуют ограниченные твердые растворы и соединения. Ха-
Характерно, что рений не дает соединений Курнакова, в то время как марганец
образует подобные соединения с никелем, платиной, золотом (MnNi, MnNi3,
MnPt3, MnPt, Mn3Pt, Mn3Au, MnAu3), а также с железом, кобальтом, медью,
алюминием.
ГЛАВА XXII. ЭЛЕМЕНТЫ VIII ГРУППЫ
Восьмая группа системы играет исключительно важную роль в понимании
Периодического закона и в структурном отношении не имеет аналогов среди
других групп. Если в группах I —> II —>Ш различие между А- и В-подгруппами
постепенно уменьшается, то начиная с IV группы оно вновь возрастает и в VIII
группе достигает максимума: главную подгруппу составляют химически инертные
и благородные газы, а побочную — триады железа и платиновых металлов. Уже в
VI и VII группах первый типический элемент (кислород и фтор) не полностью
отражает химический облик группы в целом: кислород практически не имеет, а
фтор не имеет положительных степеней окисления, тем более отвечающих номеру
группы. В VIII группе элементы малых периодов вообще не являются типически-
типическими элементами в силу своей инертности.
В целом VIII группа состоит из инертных (Не, Ne) и благородных (Аг, Кг, Хе,
Rn) газов, объединяемых в VIIIA-группу, и металлов VIIIB-группы. VIIIB-rpynna
также резко отличается в структурном отношении от остальных групп Периоди-
Периодической системы. В ее состав входят 9 элементов, объединяемых обычно в семей-
семейство железа (Fe, Co, Ni), и платиновые металлы, включающие остальные эле-
элементы. Кроме того, платиновые металлы целесообразно подразделить на три
вертикальные диады (Ru — Os, Rh — Ir, Pd — Pt), в пределах которых свойства
элементов особенно близки, что обусловлено лантаноидной контракцией. Поэтому
характер аналогии среди элементов VHIB-группы можно выразить схемой
Fe ¦"
1
1
f
Ru *¦
1
Со
1
1
t
Rh
1
*¦ Ni
1
1
¦
*¦ Pd
1
Os
Ir ¦ Pt
где сплошными стрелками отражена близкая аналогия свойств, а пунктирными
более отдаленная.
482
1. Элементы УША-грушш. Характеристика элементов
VIII А-г р у п п ы. Особенности гелия и неона. Все периоды системы заверша-
завершаются элементами VIIIA-группы. Эти элементы имеют полностью укомплектован-
укомплектованные электронные оболочки ns2np6 (у гелия I*2). С точки зрения электронного
строения, неон и тяжелые благородные газы естественно поместить в VIIIA-rpyn-
пу, поскольку на внешней оболочке они содержат восемь электронов и являются
sp-элементами. Гелий с этой точки зрения относится к s-элементам (как и водо-
водород) и формально должен возглавлять ПА-группу. Однако у атома гелия отсутст-
отсутствует возможность промотирования электрона, что и определяет его химическую
инертность. По этой причине его и помещают вместе с представителями VIIIA-
группы. Особенности электронной конфигурации неона (Is22s22p6 и отсутствие
вакантной rf-оболочки при га = 2) также предопределяют его химическую инерт-
инертность. Дело в том, что промотирование электрона с изменением главного кванто-
квантового числа (для Не Is —> 2s, для Ne 2р —> 3s) требует такой высокой энергии,
которая не может быть компенсирована энергией химической связи при образова-
образовании соединения. Кроме того, орбитали 1* (Не) и 2р (Ne) кайносимметричны, что
является еще одной причиной инертности этих элементов. У остальных предста-
представителей VIIIA-группы начиная с аргона существуют вакантные rf-орбитали при
том же значении главного квантового числа, который соответствует внешней
электронной оболочке (ns2np6nd°). Поэтому они в принципе способны образовы-
образовывать валентно-химические соединения за счет распаривания электронов. Прини-
Принимая во внимание эти различия в электронном строении, гелий и неон целесооб-
целесообразно назвать инертными газами, а аргон и более тяжелые газы VIIIA-группы —
благородными. Характеристики атомов элементов VIIIA-группы представлены
ниже:
Элемент
Электронная конфи-
конфигурация
Атомный радиус, нм
/ь В
Не
Is2
0,122
24,58
Ne
2«22р6
0,160
21,56
Ar
З^Зр6
0,192
15,69
Кг
C<floLs24p6 (
0,198
13,996
Хе
0,218
12,13
Rn
D/45 3 0N
0,220
10,75
Атомы инертных и благородных газов характеризуются экстремальными свой-
свойствами в своих периодах: наименьшими значениями атомных радиусов и наивыс-
наивысшими потенциалами ионизации, причем эти характеристики в VIHA-группе меня-
меняются немонотонно. В силу кайносимметричности ls-орбитали у Не и 2р-орбитали
у Ne их атомные радиусы заметно меньше, а потенциалы ионизации существенно
выше, чем у благородных газов. Что касается аргона, то вакантная 3^-оболочка у
него является также кайносимметричной, что отличает аргон от других благород-
благородных газов. Это является одной из причин отсутствия у аргона валентных соеди-
соединений, в то время как для остальных благородных газов они известны.
Инертные и благородные газы в природе. В силу
химической инертности элементы VIIIA-группы в природе встречаются только в
свободном состоянии главным образом в атмосфере. Их содержание невелико и
оценивается следующими величинами:
Элемент Не Ne Ar Кг Хе Rn
Содержание в атмо-
атмосфере, об. доли, % 5,24- 1<Г4 1,8- 10э 0,93 З-КГ4 8,7-1<Г6 7-КГ17
483
Иными словами, в 1 м3 воздуха содержится 9,3 л Аг, 16 мл Ne, 5 мл Не, 1 мл
Кг, 0,08 мл Хе и лишь 1—2 атома Rn в 1 см3. Гелий, являющийся продуктом ра-
радиоактивного распада, встречается в некоторых природных газах, в водах мине-
минеральных источников, а также в окклюдированном виде в минерале клевеите. Все
эти элементы (кроме аргона) принадлежат к редким. Это обстоятельство, а также
их исключительная инертность послужили причиной их сравнительно позднего
открытия. В космосе гелий наряду с водородом является наиболее распростра-
распространенным элементом G6 масс, долей, % Н и 23 масс, доли, % Не от общей массы
вещества во Вселенной). Источником космического гелия являются термоядерные
реакции, протекающие на определенной стадии эволюции звезд. Не случайно
поэтому гелий впервые был открыт A868) методом спектрального анализа на
Солнце. На Земле он был обнаружен спустя почти 30 лет.
Источниками получения гелия в настоящее время являются природные газы и
воздух. В некоторых газах его содержится до 7—16%. Остальные благородные
газы получают главным образом фракционной перегонкой жидкого воздуха. В
первой, наиболее легкокипящей, фракции содержатся Не, Ne, N2, во второй — N2,
Аг, Ог- Дополнительная разгонка третьей фракции позволяет выделить тяжелые
газы — Кг и Хе. Разделение благородных газов осуществляют также многократ-
многократной адсорбцией на активированном угле и других адсорбентах.
Физические свойства благородных газов. При обыч-
обычных условиях все элементы VIIIA-группы представляют собой моноатомные газы,
которые лишь при значительном охлаждении могут быть переведены в жидкое и
твердое состояния. Их характеристики представлены ниже:
Не
Температура плавле-
плавления, "С
Температура кипе-
кипения, "С
Энтальпия плавле-
плавления, кДж/моль
Энтальпия испаре-
испарения, кДж/моль
Растворимость в во-
воде @° С), см3/л
Плотность в жидком
состоянии (при
^ип), г/см3
Кристаллическая
структура в твер-
твердом состоянии
-272
B5-105 Па)
-269
0,021
0,084
10
0,13
ГПУ
ГЦК
ОЦК
'Ne
-249
-246
0,330
1,732
14,0
1,2
ГЦК
Аг
-189
-186
1,180
6,514
52,4
1,4
Кг
-157
-153
1,631
9,313
99,1
Хе
-112
-108
-71
-62
2,295 2,887
12,68
203,2
18,1
510
2,413 3,157 4,4
ГЦК ГЦК
ГЦК ГЦК
От гелия к радону возрастают температурь^ и энтальпии плавления и' кипе-
кипения, что указывает на увеличение прочности межмолекулярных связей в конден-
конденсированном состоянии. Вместе с тем низкие значения термических и термодина-
термодинамических характеристик фазовых переходов указывают на слабость межмолеку-
484
лярных сил. Особенно это касается инертных газов — гелия и неона. Ван-дер-
ваальсово взаимодействие обусловлено исключительно дисперсионным эффектом,
который тем значительнее, чем больше поляризуемость частиц, т.е. он возрастает
с увеличением атомных радиусов.
. Особое место среди простых веществ VHIA-группы занимает гелий. Во-пер-
Во-первых, это наиболее трудно сжижаемый газ; во-вторых, это единственный элемент.
для которого твердое состояние достигается только при повышенном давлении;
в-третьих, в жидком состоянии гелий обладает особыми свойствами. Вплоть до
температуры 2,172 К гелий — это бесцветная, прозрачная, легкая жидкость Не-1
(примерно в 10 раз легче воды). При отмеченной температуре наблюдается так
называемый фазовый переход II рода (не сопровождаемый тепловым эффектом) и
вплоть до сколь угодно низких температур, приближающихся к абсолютному ну-
нулю, гелий существует в виде жидкого Не-И. Эта жидкость с особыми и уникаль-
уникальными свойствами: она практически не обладает вязкостью (сверхтекучесть), имеет
колоссальную теплопроводность (в 3• 10s раз больше гелия-1), а также проявляет
ряд других аномальных эффектов. Эти явления связаны с тем, что при темпера-
температуре 1—2 К длина волны де Бройля для атома гелия сравнима со средним меж-
межатомным расстоянием. Поэтому сверхтекучий Не-П называют квантовой жидкос-
жидкостью. Из-за сверхтекучести гелий можно перевести в твердое состояние только под
большим давлением. Существует глубокая аналогия между сверхтекучестью ге-
лия-П и сверхпроводимостью металлов. При низких температурах свободные
электроны в металлах также ведут себя как "электронная квантовая жидкость".
К л а т р а т ы. До сравнительно недавнего времени F0-е годы XX в.) эле-
элементы VIIIA-группы называли инертными газами, подчеркивая тем самым их
полную неспособность к химическому взаимодействию. Однако уже в конце XIX
в., вскоре после открытия инертных газов, Вийяр, сжимая аргон под водой при
0°С, получил кристаллогидрат примерного состава Аг-бНгО. Затем были получе-
получены аналогичные гидраты ксенона и криптона. Оказачось, что эти соединения
неустойчивы. Их малая прочность свидетельствует об отсутствии валентного
взаимодействия между компонентами. Позднее подобные же соединения были
получены с фенолом, толуолом, гидрохиноном и др. Известны соединения
Хе-2С2Н5ОН, Хе-2СйН5СНз. Эти соединения оказались более устойчивыми, чем
гидраты. Поскольку с самого начала было ясно, что это межмолекулярные соеди-
соединения и здесь существенная роль принадлежит силам Ван-дер-Ваальса, не было
удивительным, что они более устойчивы для ксенона, а для неона и гелия не
были обнаружены. Более поздние исследования структуры подобных соединений
показали, что они представляют собой особый класс соединений — так называ-
называемые соединения включения. Такие соединения образуются при внедрении моле-
молекул и атомов в полости цепочечного, слоистого или каркасного кристалла,
образованного вторым компонентом. Первые молекулы в соединениях включения
называются "гостями", вторые — "хозяевами". В каркасных структурах, образо-
образованных молекулами-"хозяевами", возникают полости, в которых заключены моле-
кулы-"гости". Соединения включения (аддукты) с каркасным клеточным скеле-
скелетом получили название клатратов. Их существование уже не позволяет отнести
Аг, Кг, Хе (и радон) к инертным газам, так как они все же проявляют определен-
определенную склонность к рзаимодействию.
Валентно-химические соединения благородных
газов. В 1962 г. канадский ученый Бартлетт обратил внимание на то, что
• . ' ^ 485
первый потенциал ионизации ксенона A2,13 В) близок к потенциалу ионизации
молекулярного кислорода с образованием диоксигенил-катиона: Ог —> Ог + е",
/ = 12,2 В. Катион Oj входит в состав известного к тому времени комплексного
соединения (^[PtFe]. На основании этой аналогии путем взаимодействия PtFe с
ксеноном Бартлетту удалось получить красные кристаллы Xe[PtFe]. Это открытие
и положило начало химии благородных газов. Вскоре были получены Xe[RuFe],
Xe[RhFg] и Xe[PuFe]. Изучение этих соединений показало, что атомы ксенона
поляризованы положительно в результате окисления ксенона элементом-комп-
лексообразователем в степени окисления +6:
о
Хе
I
+6 +1+5
PtF6 = Xe[PtF6]
Как выяснилось, ксенон непосредственно может реагировать с фтором, образуя
XeF2, XeF4, XeF6, XeFg. Фториды ксенона получают в жестких условиях (высокие
температуры и давление, УФ-излучение, электрический разряд и т.п.), необходи-
необходимых как для возбуждения атома ксенона, так и для диссоциации молекулы F2.
Все кристаллы фторидов ксенона имеют молекулярное строение: низкие темпера-
температуры плавления (XeF2 140°C; XeF4 135°C; XeF6 50°C; XeF8 — газ), склонность к
сублимации.
Трактовка природы химической связи во фторидах ксенона возможна с пози-
позиций метода МО. Согласно этим представлениям, в дифториде XeF2 образуется
одна общая четырехвалентная трехцентровая связь за счет обобществления ва-
валентных электронов на СМО и HMO, a PMO остается вакантной, что и обеспечи-
обеспечивает устойчивость молекулы (порядок связи 2/з):
СХ2ХЗОО
F Хе F
АО
Образование XeF4 и XeFe сопровождается возникновением соответственно двух и
трех подобных трехцентровых связей. Фториды ксенона являются характеристи-
характеристическими соединениями этого элемента и свидетельствуют о его способности про-
проявлять положительные степени окисления четного ряда: +2, +4, +6 и +8. При
этом высшая характеристическая степень окисления ксенона в XeFg отвечает
номеру группы, в которой расположен ксенон. Фториды являются исходными
веществами для получения других соединений ксенона. В химическом отношении
фториды ксенона — очень реакционноспособные вещества, функционирующие
главным образом в роли энергичных окислителей. Кроме того, они склонны к
диспропорционированию, что позволяет легко переходить от низших фторидов к
высшим:
2XeF2 = Хе + XeF4; 3XeF4 = Хе + 2XeF6
486
По отношению к воде помимо окислительного действия высшие фториды ксенона
проявляют акцепторную активность:
XeF6 + Н2О = XeOF4 + 2HF; XeOF4 + 2Н2О = ХеО3 + 4HF
При этом образуются кислородсодержащие производные ксенона. Молекула
XeOF4 представляет собой искаженный октаэдр, одна из вершин которого занята
неподеленной электронной парой. Это бесцветная прозрачная жидкость (tun =
= -28°С), довольно устойчивая при обычных условиях. ХеО3 — твердое вещество
белого цвета, самопроизвольно взрывающееся, гигроскопичное. Его молекула
представляет собой тетраэдр, одна из вершин которого также занята неподелен-
неподеленной электронной парой. Оксид ХеОз обладает кислотными свойствами, и, хотя в
свободном состоянии соответствующий гидроксид Н2Хе04 существует лишь ниже
-2О...-ЗО°С, его солеобразные производные — ксенаты (+6) — известны. Так,,
ХеОд взаимодействует со щелочами:
ХеО3 + 2NaOH = Na2Xe04 + Н2О; ХеО3 + Ва(ОНJ = ВаХеО4 + Н20
Ксенаты (+6) напоминают производные серной кислоты, в частности ВаХеО4 по
строению идентичен BaSO4. При диснропорционировании соединений Хе.(+6)
или при их окислении энергичными окислителями (например, озоном) получают
производные Хе(+8) — перксенаты или ксенаты (+8):
4XeF6 + 18Ва(ОНJ = ЗВа2Хе06 + Хе + 12BaF2 + 18Н2О
ХеО3 + 4NaOH + 03 = Na4Xe06 + О2 + 2Н2О
В растворе перксенаты сильно гидролизованы, так как ксеноновая (+8) кисло-
кислота Н4ХеО(; относительно слаба (pA'i 2, рА'2 б, рА'3 11).
При взаимодействии перксенатов с безводной серной кислотой получается
ХеО4 — газ желтоватого цвета, медленно отщепляющий кислород уже при обыч-
обычных условиях:
Ва2Хе06 + 2H2SO4 = 2BaSO4 + ХеО4 + 2Н2О
В твердом состоянии ХеО4 взрывается даже при -40°С. Молекула ХеО4 — тетра-
тетраэдр с атомом ксенона в центре и d (Хе — О) = 0,16 нм.
Фториды ксенона склонны к реакциям присоединения. Так, гексафторид XeF6
присоединяет основные фториды щелочных металлов, образуя комплексные анио-
анионы:
CsF + XeF6 = Cs[XeF7]; 2RbF + XeF6 = Rb2[XeF8]
Эти соединения солеобразны и устойчивы вплоть до 400°С.
Таким образом, даже для благородного газа — ксенона — можно видеть, что
при повышении степени окисления стабильность бинарных соединений уменьша-
уменьшается, их окислительная активность растет, а устойчивость ацидокомплексов в
этом направлении повышается. Производные Хе(+8), особенно в кислой среде,
являются сильнейшими окислителями:
Н4Хе06 + 8Н+ + 8е" = Хе + 6Н2О, Е° = 4,8 В,
Н4ХеО6 + 2Н+ + 2е" = ХеО3 + ЗН2О, Е° = 3,0 В.
. 487
Химия валентных соединений ксенона в настоящее время наиболее богата.
Помимо указанных соединений получены также ХеСЬ, ХеВгг, ХеС14, ХеВг4- По-
Поскольку впервые настоящие химические соединения благородных газов были
открыты для одного из самых тяжелых представителей, у которого вакантная d-
оболочка доступна для взаимодействия, можно ожидать, что подобные соедине-
соединения должны быть еще более характерны для радона. Однако в силу его радиоак-
радиоактивности и крайне малой распространенности химия радона изучена явно недос-
недостаточно. В то же время для более легкого криптона уже получены K1F2, крипто-
криптоновая кислота Н2КГО4 и ее соли, которые, как и следовало ожидать, менее ста-
стабильны, чем соответствующие соединения ксенона. Химия соединений благород-
благородных газов в настоящее время интенсивно развивается.
2. Металлы триады железа. Характеристика элементов
триады железа. В VHIB-группе наблюдается уникальное в Периодичес-
Периодической системе явление: число валентных электронов элемента (Со, Ni) превосходит
номер группы. По этому признаку лишь Fe по праву можно было бы считать
элементом VIIIB-группы. Однако существует и вторая особенность, свойственная
элементам триады железа: ни в одном из своих соединений они не проявляют
валентности (или степени окисления), отвечающей номеру группы. С этой точки
зрения, даже Fe можно отнести к VIII группе лишь формально. Третья особен-
особенность, как отмечено выше, состоит в том, что в рамках одной подгруппы объеди-
объединяется горизонтальное семейство из трех .элементов.
Причины такого своеобразия структуры VIIIB-группы в целом и триады желе-
железа в особенности многоплановы. Существенную роль здесь играет то обстоятель-
обстоятельство, что в триаде железа наиболее ярко проявляется известная горизонтальная
аналогия, характерная для rf-элементов в целом. Немаловажным фактором, опре-
определяющим сходство свойств Fe, Co, Ni и их отличие от платиновых металлов,
является кайносимметричность 3^-оболочки.
Некоторые свойства элементов и простых веществ триады железа приведены
ниже:
Содержание в земной коре,
масс, доли, %
Валентная электронная кон-
конфигурация
Атомный радиус, нм
Ионный радиус Э2+, нм
Ионный радиус Э3+, нм
Потенциал ионизации, В
h: Э -* Э+ + е
ОЭО
Температура плавления, °С
Температура кипения, °С
Плотность, г/см3
В
Fe
5,1
[Аг]3^
0,126
0,080
0,067
7,89
A,7) 1,8
1536
2870
7,87
-0,44
Co
3-Ю
[Аг]3<^
0,125
0,078
0,064
7,87
1,7-1,8
1493
2960
8,84
-0,277
Ni
8-10
[Ar]3<P4s2
0,124
0,074
0,062
' 7,63
1,8
1453
2900
8,91
-0,25
В силу кайносимметричности З^-оболочки высшие степени окисления для
элементов триады железа малохарактерны. Наиболее типичны для них степени
окисления +2, +3. При этом у железа степень окисления -f-З заметно устойчивее,
488 . ' - -
чем +2, поскольку на 3<й>болочке существует лишь один лишний электрон сверх
устойчивой 5^-конфигурации. С дальнейшим увеличением числа электронов на
3^-орбиталях тенденция к их участию в химическом взаимодействии уменьшает-
уменьшается. Поэтому у Со обе характерные степени окисления устойчивы примерно в рав-
равной мере, а у Ni более стабильна степень окисления +2. В жестких условиях под
действием энергичных окислителей могут проявляться и более высокие положи-
положительные степени окисления, вплоть до +6. С другой стороны, для элементов
триады железа особенно характерна отмеченная ранее для Сг и Мп склонность к
образованию карбонилов, в которых степень окисления элементов равна нулю.
Природные соединения и получение железа, ко-
кобальта и никеля. Железо по распространенности в природе находится
на четвертом месте после кислорода, кремния и алюминия. Кобальт и никель
содержатся (ча Земле в значительно меньшей степени, хотя относятся к довольно
распрос тным элементам. Основными формами рудоносных минералов железа
являются чдные и сульфидные соединения: магнетит ГезО4, гематит Fe2O3,
лимонит Ft^- -гаН2О, пирротин FeS, пирит FeS2-
В отличие i. т железа кобальт и никель чаще образуют сульфидные и арсенид-
ные минерал:и, чем оксидные. Известны CoAsS — кобальтин, NiAs — никелин,
СоАвз — скуттерудит, NiAsS — гередорфит и т.п. Совместные с железом минералы
также принадлежат к сульфидно-арсенидному классу: (Fe,Ni)gSg — пентландит,
(Co,Fe)As2 — саффлорит и т.п.
Восстановление оксидных железных руд осуществляется главным образом в
доменных печах и с химической точки зрения может быть представлено такой
последовательностью реакций:
3Fe2O3 + СО = СО2 + 2Fe3O4, Fe3O4 + CO = СО2 + 3FeO
FeO + CO = С02 + Fe или FeO + С = СО + Fe
Сульфидные и арсенидные руды никеля и кобальта путем окислительного обжи-
обжига вначале переводят в оксиды, которые восстанавливают до металла углем в
электропечах. Черновые металлы подвергают электрохимическому рафинирова-
рафинированию.
Очень чистые металлы триады железа (чистотой 99,99% и выше) получают
карбонильным способом. Смесь карбонилов Fe(COM, Co2(CO)s, Ni(COL подверга-
подвергается фракционной разгонке с последующей глубокой очисткой. Затем карбонилы
термически разлагают с получением порошков особо чистых металлов.
Простые вещества. Физические и химические
свойства. Железо, кобальт и никель представляют собой серебристо-белые
металлы с сероватым (Fe), розоватым (Со) и желтоватым (Ni) отливом. Чистые
металлы пластичны, однако даже незначительное количество примесей (главным
образом углерода) повышает их твердость и хрупкость, что особенно заметно у
кобальта. Все три металла ферромагнитны *. При нагревании до определенной
температуры (точка Кюри) ферромагнитные свойства исчезают и металлы стано-
становятся парамагнитными. Переход ферромагнетика в парамагнетик не сопровожда-
, В ферромагнитных веществах магнитные моменты атомов (ионов) одинаково ориен-
ориентированы, а потому самопроизвольно намагничиваются даже в отсутствие внешнего маг-
магнитного поля.
489
ется перестройкой кристаллической структуры и представляет собой фазовый
переход 2-го рода.
Для железа и кобальта характерен полиморфизм, в то время как никель моно-
морфен и вплоть до температуры плавления обладает ГЦК-структурой. Кобальт
имеет две полиморфные модификации: низкотемпературный а-Со (ГПУ) и высо-
высокотемпературный /З-Со (ГЦК), причем переход а ^^ /? наблюдается при 450°С. У
железа три полиморфные модификации: a-Fe (ОЦК), 7~Fe (ГЦК) и б-Fe (ОЦК).
Переход a-Fe ^=^ /З-Fe при 769°С — это точка Кюри. В структурном же отноше-
отношении а и /З-Fe, лишь слегка различаются по параметру ОЦК-решетки.
В химическом отношении железо, кобальт и никель относятся к металлам
средней активности. В ряду стандартных электродных потенциалов они распола-
располагаются левее водорода, между цинком и оловом. Серная кислота концентрации
более 70% пассивирует железо. Концентрированная HNO3 пассивирует все три
металла. Смесь НС1 + HNO3 энергично растворяет металлы триады железа. По
отношению к растворам щелочей Fe, Co и Ni устойчивы, но с расплавами реаги-
реагируют при высоких температурах.
Наличие примесей сильно снижает устойчивость металлов, особенно железа, к
агрессивной атмосфере в присутствии влаги. Это приводит к развитию коррозии
(ржавление железа) за счет образования на поверхности рыхлого слоя смеси
оксидов и гидроксидов переменного состава, не предохраняющего поверхность от
дальнейшего разрушения. Схематически процесс коррозии можно представить
суммарным уравнением:
4Fe + 6Н2О + ЗО2 = 4Fe(OHK
Кобальт и никель в коррозионном отношении заметно устойчивее железа, что
согласуется с их положением в ряду стандартных электродных потенциалов.
Мелкодисперсное железо в кислороде сгорает при нагревании до образования
Fe3C>4, который является самым устойчивым оксидом железа. Термическое окис-
окисление кобальта и никеля протекает при более высоких температурах. При этом
образуются в основном NiO и СоО, обладающие переменным составом, завися-
зависящим от условий окисления. При высоких температурах Fe, Co и Ni активны по
отношению ко всем неметаллам и взаимодействуют со многими металлами.
Характеристические соединения. Для Fe, Co, Ni известны
оксиды ЭО и ЭгОз- Эти оксиды нельзя получить в чистом виде прямым синте-
синтезом, поскольку при этом образуется набор оксидов, каждый из которых является
фазой переменного состава. Их получают косвенным путем — разложением неко-
некоторых солей или гидроксидов. Если оксиды ЭО можно легко получить для всех
трех элементов, то Э2О3 устойчив лишь для железа и получается при обезвожива-
обезвоживании гидроксида. Оксиды кобальта (+3) и никеля (+3) неустойчивы и склонны к
разложению с отщеплением кислорода. Для железа и кобальта характерны ок-
оксидные соединения состава Э3О4. Эти оксиды являются производными элементов
в степенях окисления +2 и +3 (ЭО-ЭгОз).
Оксиды ЭО образуют кристаллические структуры типа NaCl, a Fe2O3 крис-
кристаллизуется в ромбоэдрической структуре. Оксиды ЭО нерастворимы в воде и не
взаимодействуют с нею и растворами щелочей. С кислотами они легко взаимо-
взаимодействуют с образованием солей Э(+2). Это же характерно и для соответствую-'
щих гидроксидов Э(ОНJ. Белый Fe(OHJ) розово-красный Со(ОН)г и зеленый
49Q
Ni(OHJ легко растворимы в кислотах и при нагревании отщепляют воду с обра-
образованием соответствующих оксидов ЭО.
Хотя устойчивый оксид Э2Оз известен только для железа, гидроксиды Э(ОНK
могут быть получены для всех элементов триады железа. Их основные функции
выражены слабее, чем для производных Э(+2). Гидроксид Fe(OHK образуется
уже при окислении Fe(OH)> кислородом воздуха:
4Fe(OHJ + О2 + 2Н2О = 4Ге(ОНK
Аналогичная реакция, но в меньшей степени, характерна для кобальта. Гидрок-
Гидроксид никеля по отношению городу воздуха устойчив. Вследствие этого гид-
гидроксиды Э(ОНK различными ом ведут себя при взаимодействии с кислотами.
Если Fe(OHK образует соли жь. \ (+3), то реакция Со(ОНK и Ni(OH)a с кисло-
кислотами сопровождается их восстанови яшем до Э(+2).
Гидроксид Fe(OHK проявляем и кислотную функцию, реагируя с горячими
концентрированными растворами щелочей с образованием гидроксокомплексов,
например Na3jTe(OHN]. Производные железистой кислоты HFeO2 (ферриты)
получают при сплавлении щелочей или карбонатов с Fe2O3:
2NaOH + Fe2O3 = 2NaFeO2 + H2O; MgCO3 + Fe2O3 = MgFe2O4 + CO,
Ферриты Me Fe2O4 относятся к классу шпинелей. Рассмотренные выше окси-
оксиды Fe3O4 и Со304 формально представляют собой шпинели FeFe2O4 и С0С02О4.
В отличие от кобальта и никеля для железа известны соединения, в которых
его степень окисления равна +6. Так, при окислении бромом взвеси Fe(OHK в
горячем концентрированном растворе щелочи образуются соли железной кислоты
H2Fe04 — ферраты:
2Fe(OHK + ЮКОН + ЗВг2 = 2K2Fe04 + бКВг + 8Н2О
Ферраты термически нестабильны и при небольшом нагревании A00—200°С)
превращаются в ферриты с отщеплением кислорода:
4K2Fe04 —> 4KFeO2 + 2К2О + ЗО2
В свободном состоянии железная кислота и соответствующий ей оксид FeO3 не
выделены. По растворимости и в структурном отношении ферраты близки к
соответствующим хроматам и сульфатам.
Соединения с неметаллами. Галогениды железа, кобальта и
никеля сравнительно немногочисленны и отвечают наиболее характерным степе-
степеням окисления +2 и +3. Так, для железа известны галогениды FeF2 и FeF3 с
фтором, хлором и бромом. Взаимодействие с иодом приводит к образованию Fel2.
При непосредственном взаимодействии образуются FeF3, FeCl3 и FeBr3. Дигало-
гениды получают лишь косвенным путем — растворением металла (или его окси-
оксида) в соответствующей галогеноводородной кислоте. Для кобальта из талогенидов
СоГ3 получены лишь CoF3 и СоС13. Эти соединения очень неустойчивы, особенно
СоС13. С бромом и иодом кобальт образует только дигалогениды. Никель вообще
не образует тригалргенидов, в то время как дигалогениды известны для всех
галогенов.
491
Все дигалогениды металлов триады железа являются типичными солеобраз-
ными соединениями с заметным ионным вкладом в химическую связь. Об этом
свидетельствуют сравнительно высокие температуры плавления, а также законо-
закономерное понижение температур плавления в ряду 3F2 — ЭС1г — ЭВг2 — ЭЬ. Ди-
фториды в кристаллическом состоянии имеют структуру рутила ТЮг- Остальные
дигалогениды образуют более сложную ромбоэдрическую решетку. Все соедине-
соединения 3F2 малорастворимы в воде. Остальные же хорошо растворяются.
Тригалогениды железа образуют сложные структуры, причем FeCla и FeBrg
имеют ярко выраженный слоистый характер и невысокие температуры плавления,
что указывает на псевдомолекулярный характер этих соединений. В паровой фазе
FeCls димеризован и состоит из молекул Fe2Cl6.
Все три элемента энергично взаимодействуют с халькогенами при нагревании
и образуют халькогениды двух типов: ЭХ и ЭХ2. Монохалькогениды можно
получать не только непосредственным взаимодействием компонентов при нагрева-
нагревании, но и в растворах, например
СоС12 + (NH4JS = CoS + 2NH4C1
При этом халькогениды образуются в виде черных осадков, крайне мало раство-
растворимых в воде. Препаративным путем можно получить также сульфиды железа и
кобальта состава Э28з и 9sS4. Первые отвечают степени окисления железа и
кобальта +3, а вторые можно рассматривать как "смешанные" сульфиды ЭБ-ЭоЗз-
аналогичные по составу оксидам Fe3O4 и Со304-
Все халькогениды являются фазами переменного состава, причем область
гомогенности у монохалькогенидов шире, чем у дихалькогенидов. Их состав, а
следовательно, и свойства сильно зависят от условий получения.
Соединения металлов триады железа с остальными неметаллами (пниктогены,
углерод, кремний, бор) заметно отличаются от рассмотренных выше. Все они не
подчиняются правилам формальной валентности и в большинстве своем обладают
металлическими свойствами. Поэтому их можно рассматривать как своеобразный
переходный класс соединений к объектам металлохимии. Для всех элементов
характерны ЭзС, а для железа и кобальта, кроме того, и Э2С. Такой лее состав
известен и для боридов. Остальные соединения обладают более сложной структу-
структурой и природой.
Нитриды железа, кобальта и никеля в отличие от нитридов предшествующих
rf-элементов фазами внедрения не являются. Об этом свидетельствуют их низкая
термическая устойчивость и способность к последовательной диссоциации при
нагревании с отщеплением азота и образованием все более бедных азотом соеди-
соединений. Склонностью к термической диссоциации с последовательным отщеплени-
отщеплением летучего компонента обладают также фосфиды и арсениды, причем первые —
в большей степени. Фосфиды, арсениды и стибиды получают прямым синтезом
из компонентов в эвакуированных и запаянных ампулах. Эти соединения разно-
разнообразны по составу, однако наиболее типичные фазы — это ЭзП, ЭгП, ЭП и ЭП2.
Для кобальта и никеля известны фосфиды ЭРз. Высшие фосфиды ЭР2 и ЭРз, а
также арсенид FeAs2 — полупроводники, остальные пниктогениды обладают
полуметаллическими и металлическими свойствами.
Силициды по составу и электрофизическим свойствам напоминают фосфиды и
арсениды, хотя они более тугоплавки и термически стабильны. Низшие силици-
492 . .
ды 33Si, 32Si, 9Si — металлиды, а соедини. ~- 3Si2 — полуметаллы или полупро-
полупроводники.
Среди боридов наиболее многочисленны соединения никеля, что характерно
для этого металла и в соединениях с фосфором и кремнием. Помимо типичных
составов Э2В, ЭВ для кобальта и никеля известны Э3В, ЭВ2, а для никеля — и
некоторые промежуточные (Ni3B2 и Ni2B3). Бориды тугоплавки и обладают ме-
таллидным характером.
Металлы триады железа, особенно никель, поглощают значительное количест-
количество водорода. При нагревании растворимость водорода в металлах возрастает
(АН > 0). Растворенный водород находится в атомарном состоянии.
Соли кислородсодержащих кислот и комплекс-
комплексные соединения. Сульфаты рассматриваемых элементов получают при
растворении металлов в H2SO4 (разб.). Из растворов они обычно выделяются в
виде кристаллогидратов 9SCv7H2O (купоросов) бледно-зеленого (Fe2+), красного
(Со2+), темно-зеленого (Ni2+) цветов. Эти цвета характерны для гидратированных
ионов Э2+. Сульфат железа (+3) можно получить по реакции
Fe2O3 + 3H2SO4 (конц.) = Fe2(SO4K + ЗН2О
Сульфат кобальта (+3) неустойчив как в растворе, так и в твердом состоянии.
Сульфат никеля (+3) неизвестен.
Нитраты ЭAЧОзJ получают при взаимодействии металлов с разбавленной
HNOg на холоду. Они легко растворимы в воде и из раствора выделяются в виде
кристаллогидратов Э(]\ГОзJ'6Н2О, цвет которых определяется цветностью гидра-
тированного иона. Из нитратов Э(+3) известен лишь Ре(ГЮз)з> который образует-
образуется при растворении металла в 25%-ной НГЮз-
Карботаны известны для всех трех элементов только в степени окисления +2.
В противоположность FeCOg, который осаждается из раствора без кристаллиза-
кристаллизационной воды, карбонаты кобальта и никеля образуют кристаллогидраты
ЭСО3-6Н2О.
Способность солей железа, кобальта и никеля к образованию кристаллогидра-
кристаллогидратов свидетельствует о склонности этих элементов к комплексообразованию. Крис-
Кристаллогидраты представляют собой типичный пример аквакомплексов, среди
которых встречаются как нормальные комплексные соединения, например
[Э(Н2О)б](СЮ4J, [Э(Н2О)б](РЮзJ, так и сверхкомплексные соединения, например
купоросы 3SO4-7H2O или [3(H2ON]SO4-H2O.
Элементы триады железа образуют типичные двойные соли типа шенитов и
квасцов. Из первых наиболее известна соль Мора (NH4JSCvFeSCv6H2O. Квас-
Квасцы известны лишь для железа и кобальта, например железоаммонийные квасцы
(NH4JSO4-Fe2(SO4K-24H2O, кобальтокалиевые квасцы K2S04-Co2(S04K-24H20 и
т.п.
Многочисленны для элементов триады железа1 анионные комплексы: галоге-
нидные (Ме^ЭГз], Ме2[ЭГ4], Мез[ЭГ6] и т.п.), роданидные (Me2[3(CNSL],
Me4[3(CNSN], Me'[3(CNSN]), оксалатные (Ме2[Э(С2О4J], Мез[Э(С2О4K]). Но
особенно характерны и устойчивы цианидные комплексы, например K4[Fe(CNN],
K3[Fe(CNN]. Дело в том, что лиганды CN", возглавляющие спектрохимический
ряд, образуют низкоспиновые комплексы, устойчивость которых весьма высока.
493
4s
Ар
Со0
Со0
||н|н|н|и
Ж-связь
]|H|*t|*t|*t
t
i ? с
-л
С— связь
I spi- гибридизация j
i ? с
Металлохимия. Железо, кобальт и никапь образуют непрерывные
твердые растворы не только между собой, но и с другими переходными металла-
металлами. Так, а-железо образует неограниченные твердые растворы с V и Сг (ОЦК),
7-железо и /^-кобальт (ГЦК) с 7~маРганЦем (ГЦК), родием, иридием, палладием и
платиной. Последнее подтверждает определенную аналогию между всеми элемен-
элементами УШВ-групны. /Мчобальт (ГПУ) непрерывно взаимно растворим с изоморф-
изоморфными Re, Ru. Os. Никель обнаруживает металлохимическое сходство с 7~Fe и 0~
Со, но в отличие от них образует непрерывные твердые растворы с металлами IB-
группы — медью и золотом. Таким образом, элементы VIHB-группы служат свое-
своеобразным связующим звеном между металлами VIIB- и IB-групп.
Характерной особенностью взаимодействия Fe, Co, Ni с переходными металла-
металлами является образование соединений Курнакова. Такие соединения известны,
например, в системах Fe — Pt, Fe — Pd. Fe — Co, Fe — Mn, Fe — Ni, Co — Ni.
Co - Mn. Co — Cr, Ni - Си и др.
В силу различия металлохимических факторов элементы триады железа не
взаимодействуют с металлами IA-, ПА-групп, а также с серебром и легкоплавки-
легкоплавкими ртутью, таллием и свинцом. С другими р-металлами (Al, Ga, In, Sn) образуют
интерметаллические соединения, например FeA^, CoSn, CoSn2, Ni3Al, NiAl,
NigGa, Niln, Ni3Sn. Структура и формульный состав этих соединений определя-
определяются фактором низшего порядка — размерным.
3. Платиновые металлы. Общая характеристика плати-
платиноидов. Структуры валентных электронных оболочек платиновых элементов
отличаются значительным разнообразием вследствие возможности проскока ns-
электронов на (и — 1)</-орбитали. Нередко проскоки «-электронов не связаны с
достижением стабильной «/^-конфигурации. Нормальное заполнение валентных
орбиталей (без проскоков электрона) характерно лишь для осмия и иридия.
Палладий — единственный элемент в Периодической системе, который в нор-
нормальном состоянии не имеет электронов на s-оболочке. У платины стабильна <Р-
конфигурация, что также не наблюдается у других элементов Периодической
системы.
Обращают на себя внимание близость атомных радиусов всех шести плати-
платиноидов @,133 —> 0,138 нм) и особая близость ионных радиусов Э4+ @,062—0,065
нм). Относительно высокие значения ОЭО B,0—2,2) говорят о благородное™
этих металлов, а их близкие значения — о близости химических свойств. !
Для платиновых металлов в соединениях характерны практически все степени
окисления от 0 до +8. При этом отмечается тенденция к понижению максималь-
максимальных степеней окисления в горизонтальных рядах. В вертикальных диадах наблю-
наблюдается обычно соответствие степеней окисления. Так, элементы первой диады
(Ru — Os) могут проявлять максимальную степень окисления +8 (даже в соедине-
соединениях первого порядка), элементы второй диады (Rh — Ir) достигают степени'
495
Co0
Co0
3d
|н|и|и|н
Я-связь
I и
и|и|н
4s
Ар
ггп
О— связь
spi- гибридизация
п
Металлохимия. Железо, кобальт и никель образуют непрерывные
твердые растворы не только между собой, но и с другими переходными металла-
металлами. Так, «-железо образует неограниченные твердые растворы с V и Сг (ОЦК),
7-железо и /^-кобальт (ГЦК) с -/-марганцем (ГЦК), родием, иридием, палладием и
платиной. Последнее подтверждает определенную аналогию между всеми элемен-
элементами VIJIB-rpyniibi. /З-Кобальт (ГПУ) непрерывно взаимно растворим с изоморф-
изоморфными Re, Ru, Os. Никель обнаруживает металлохимическое сходство с 7~Fe и 0-
Со, но в отличие от них образует непрерывные твердые растворы с металлами IB-
группы — медью и золотом. Таким образом, элементы VIlIB-группы служат свое-
своеобразным связующим звеном между металлами VIIB- и Ш-групн.
Характерной особенностью взаимодействия Fe, Co, Ni с переходными металла-
металлами является образование соединений Курнакова. Такие соединения известны,
например, в системах Fe — Pt, Fe — Pd, Fe — Co, Fe — Mn, Fe — Ni, Co — Ni,
Co - Mn, Co - Cr, Ni - Си и др.
В силу различия металлохимических факторов элементы триады железа не
взаимодействуют с металлами IA-, ПА-групп, а также с серебром и легкоплавки-
легкоплавкими ртутью, таллием и свинцом. С другими р-металлами (Al, Ga, In, Sn) образуют
интерметаллические соединения, например FeA^, CoSn, CoS)i2, Ni3Al, NiAl,
NigGa, Niln, Ni3Sn. Структура и формульный состав этих соединений определя-
определяются фактором низшего порядка — размерным.
3. Платиновые металлы. Общая характеристика плати-
платиноидов. Структуры валентных электронных оболочек платиновых элементов
отличаются значительным разнообразием вследствие возможности проскока ns-
электронов на (га — 1)</-орбитали. Нередко проскоки s-электронов не связаны с
достижением стабильной «Реконфигурации. Нормальное заполнение вачентных
орбиталей (без проскоков электрона) характерно лишь для осмия и иридия.
Палладий — единственный элемент в Периодичес'кой системе, который в нор-
нормальном состоянии не имеет электронов на s-оболочке. У платины стабильна (Р-
конфигурация, что также не наблюдается у других элементов Периодической
системы.
Обращают на себя внимание близость атомных радиусов всех шести плати-
платиноидов @,133 —> 0,138 нм) и особая близость ионных радиусов Э4+ @,062—0,065
нм). Относительно высокие значения ОЭО B,0—2,2) говорят о благородности
этих металлов, а их близкие значения — о близости химических свойств.
Для платиновых металлов в соединениях характерны практически все степени
окисления от 0 до +8. При этом отмечается тенденция к понижению максималь-
максимальных степеней окисления в горизонтальных рядах. В вертикальных диадах наблю-
наблюдается обычно соответствие степеней окисления. Так, элементы первой диады
(Ru — Os) могут проявлять максимальную степень окисления +8 (даже в соедине-
нияху первого порядка), элементы второй диады (Rh — Ir) достигают степени
495
окисления +6 (в комплексных соединениях), а палладий и платина имеют типич-
типичные степени окисления +2 и +4. Элементы первой диады напоминают по свой-
свойствам элементы VIIB-группы — технеций и рений (подобно тому, как железо
напоминает марганец). Элементы же последней диады проявляют определенное
сходство с элементами Ш-группы,
Содержание в земной
коре, масс, доли, %
Валентная электронная
конфигурация
Атомный радиус, нм
Ионный радиус Э4+, нм
Потенциал ионизации /j:
Э —> Э+ + е, В
ОЭО
Температура плавле-
ния, С
Плотность, г/см3
в
Ru
5-Ю
0,133
0,062
7,366
2,0
2334
12,45
+0,45
Rh
1-Ю
0,134
0,065
7,46
2,1
1960
12,41
+0,6
Pd
0,137
0,064
8,336
2,1
1552
12,02 '
+0,987
Os
5-10
0,135
0,065
8,5
2,1
3050
22,61
+ 0,7
Ir
1-10
D/4M<F6s2
0,136
0,065
9,1
2,1
2443
22,50
+ 1,0
Pt
5-10'7
D/4M<?l
0,138
0,064
9,0
2,2
1769
21,45
+ 1,2
Природные соединения, получение и аффинаж
платиновых металлов. Все платиновые металлы относятся к редким
элементам. Платиноиды обычно встречаются вместе и, как правило, в самородном
состоянии, а также в виде примеси в железных, хромовых, никелевых и медных
рудах. Они являются рассеянными элементами, и собственные минералы для них
малохарактерны. В основном платиноиды ассоциированы с сульфидно-арсенид-
ными, реже — с оксидными рудами.
Выделение, разделение и очистка (аффинаж) платиновых металлов представ-
представляют собой одну из сложнейших задач препаративной неорганической химии.
Исходные руды подвергают обогащению и из полученных концентратов выплав-
выплавляют медь и никель. При электролитическом рафинировании никеля в анодных
шламах концентрируются платиновые металлы, а при рафинировании меди,
кроме того, серебро и золото. Шламы растворяют в царской водке. При этом все
платиноиды, кроме осмия, переходят в раствор в виде комплексных хлорометал-
лических кислот (H2[PtCl6], Н2[11иС1б] и т.п.), а осмий в виде нерастворимого
OsO4 остается в осадке. Далее платину осаждают хлоридом аммония в виде
(NH4J[PtCle], а затем последовательно выделяют остальные платиноиды. Из
маточного раствора упариванием в присутствии HNO3 выделяют хлороиридат
(NH4J[IrCl6]. После осаждения платины и иридия из раствора, подкисленного
серной кислотой, выделяют палладий, рутений и родий в виде металлов путем
восстановления цинком. При этом выделяются и остатки платины и иридия.
Смесь порошкообразных металлов растворяют в царской водке. При этом родий
и рутений остаются в осадке в виде черни (тонкодисперсный порошок). Остав-
Оставшийся в растворе палладий осаждают в виде нейтрального комплекса
[Pd(NH3J]Cb- Родий из черни выделяют растворением в серной кислоте с после-
последующим осаждением, например в виде '[11п(]ЧНз)з]С1з, а рутений отгоняют в виде
летучего R11O4 с последующим восстановлением.
496
Простые вещества. Физические и химические
свойства. В компактном состоянии все платиноиды представляют собой
серебристо-белые металлы, по внешнему виду напоминающие серебро. Эти метал-
металлы мономорфны и образуют плотноупакованные кристаллические структуры с
к.ч. 12. При этом элементы первой вертикальной диады (Ru, Os) кристаллизуют-
кристаллизуются в ГПУ-структуре, а остальные формируют ГЦК-решетку. Первая триада отно-
относится к так называемым легким платиноидам. Металлы второй триады называют-
называются тяжелыми платиноидами. Они являются самыми плотными из всех металлов.
Тяжелые платиноиды имеют более высокие температуры плавления, чем легкие.
Наиболее пластичны металлы последней вертикальной диады (Pd и Pt).
В химическом отношении платиноиды принадлежат к благородным металлам
и в ряду напряжений располагаются правее водорода. Все платиновые металлы в
компактном состоянии устойчивы по отношению к неокисляющим минеральным
кислотам. Не действуют на них и горячая азотная кислота (кроме палладия), и
даже царская водка (кроме платины). В противоположность этому устойчивость
платиноидов к щелочам сравнительно невелика. Все они взаимодействуют с рас-
расплавами щелочей в присутствии окислителей, переходя в растворимые соедине-
соединения.
Платиноиды различаются по отношению к кислороду (в обычных условиях и
при нагревании). Элементы первой диады (Ru, Os) сравнительно легко окисляют-
окисляются кислородом, особенно при нагревании. При сильном нагревании с кислородом
реагируют также Rh и 1г (вторая диада). Сродство к кислороду у элементов
третьей диады (Pd, Pt) наименьшее. Они практически не окисляются даже при
нагревании.
Реакционная способность платиновых металлов по отношению к неметаллам
при обычных условиях выражена слабо. Даже при нагревании они не реагируют
с азотом, галогены лишь вызывают их повышенную коррозию, водород с плати-
платиновыми металлами химически не взаимодействуют и плохо растворяется. Исклю-
Исключением является палладий, способный поглощать значительные количества водо-
водорода A объем палладия поглощает до 900 объемов Н2 при комнатной температу-
температуре). При сильном нагревании платиновые металлы способны вступать во взаимо-
взаимодействие с халькогенами. Они образуют соединения также с бором, кремнием.
Углерод с платиновыми металлами соединений не образует, но при повышенных
температурах способен растворяться в них в значительных количествах.
Характеристические соединения. Элементы первой диады
образуют летучие оксиды RuO4 и OsO4- Это единственные в своем роде примеры
соединений, в которых степень окисления элемента VIIIB-группы равна +8, т.е.
отвечает номеру группы. В силу координационной насыщенности эти оксиды не
присоединяют воду, поэтому им не отвечают гидроксиды. Они способны раство-
растворяться в воде, химически с ней не взаимодействуя. Кислотный характер этих
оксидов проявляется лишь в их способности образовывать комплексные соли с
основными гидроксидами, например K2[OsO4(OHJ]. Отвечающие подобным комп-
комплексным солям кислоты называются аквакиолотами, например H2[OsO4(OHJ].
Наиболее стабильным для рутения является оксид R11O2 черного цвета, кото-
который образуется при окислении металла в кислороде F00°С). Оксид такого же
состава известен и для осмия. Он образуется при осторожном восстановлении
OsO4- OsO2 диспропорционирует: 2ОвО2 = Os + OSO4, a RuO2 при высоких тем-
температурах диссоциирует с отщеплением кислорода. Оксиды OsO2 и RuO2 нераст-
17 Я. А. Угай 497
i i.,'
воримы в воде и кислотах. Соответствующие гидроксиды Э(ОНL могут быть
получены, например, гидролизом тетрахлоридов:
ЭС14 + 2Н2О = Э(ОНL+ 4НС1
— и обладают амфотерными свойствами.
Элементы второй вертикальной диады — родий и иридий — обнаруживают
определенное сходство с кобачьтом. Как и последний, эти элементы, особенно
родий, склонны к проявлению степени окисления +3. Иридий, помимо того,
проявляет степени окисления +6 и +4, которые для родия менее характерны.
При нагревании на воздухе металлического родия или при прокаливании его
нитрата образуется черно-серый порошок Rh^Og, изоморфный корунду гг-А^Оя-
Мелкодисперсный иридий реагирует с кислородом при 1000 С, образуя 1гОг,
который при более высоких температурах диссоциирует на элементы. Соответст-
Соответствующий ему гидроксид 1г(ОНL получают косвенным путем:
2Na3[IrCl6] + 6NaOH + H2O +
= 2Ir(OHL + 12NaCl
Гидроксид иридия (+4) почти нерастворим в щелочах, но легко растворяется в
кислотах. В противоположность родию оксид ЬгОз менее стабилен.
Единственным стабильным оксидом палладия является PdO. Он образуется
при нагревании дисперсного палладия в токе кислорода. Безводный PdO нераст-
нерастворим в кислотах. Однако его гидроксид, получаемый гидролизом Pd(NOftJ.
легко взаимодействует с кислотами.
Платина при умеренном нагревании в кислороде растворяет кислород и обра-
образует смесь оксидов переменного состава. При дальнейшем повышении температу-
температуры все они диссоциируют. Поэтому индивидуатьные оксиды и их гидратные
производные получают косвенным путем. Все эти соединения химически неста-
нестабильны.
Соединения с неметаллами. Несмотря на химическую благо-
родность платиноидов, при нагревании они способны образовывать соединения с
галогенами, халькогенами и пниктогенами (кроме азота), кремнием и бором.
Поскольку оксиды и гидроксиды платиновых металлов малостабильны, роль
галогенидов как характеристических соединений существенно возрастает. Извест-
Известны фториды 9Fg, 9F4, ЭР5 и 31*7, для всех платиноидов, кроме палладия (PrlFo.
PdF3, PdF4), а для рутения и осмия получены и 3F*. Для хлоридов высшая
степень окисления металлов равна +4, за исключением пачладия, для которого
известен единственный хлорид PdCl2. У галогенидов ЭГ2, ЭГ3 и ЭГ4 в определен-
определенной мере выражен солеобразиый характер, причем с увеличением степени окисле-
окисления возрастает склонность к гидролизу, а соединения ЭГ4 гидролизованы в вод-
водных растворах практически нацело.
Галогениды, отвечающие степеням окисления +5 и выше, известны только
для фтора CF.5, 9F6, 9F8). Эти фториды легкоплавки и летучи. Они представля-
представляют собой типичные молекулярные структуры, причем с повышением степени
окисления элемента температуры плавления закономерно уменьшаются. PtF6 яв-
является сильнейшим окислителем, в то время как OsF6 и даже OsFg окислитель-
окислительными свойствами не обладают. Октафторид осмия гидролизуется с образованием
OsO4:
OsF8 + 4H2O = OsO4 + 8HF
498
Среди халькогенидов наиболее типичны составы ЭХ и ЭХг. Однако отноше-
отношение элементов к халькогенам несколько различается по вертикальным диадам.
Элементы первой диады образуют только дихалькогениды R11X2 и ОвХг, элемен-
элементы последней диады (палладий и платина) — моно- и дихалькогениды. Для
элементов первой диады ярче выражено сродство к кислороду, чем к халькоге-
халькогенам, а для элементов последней диады — наоборот. Элементы средней диады —
родий и иридий — занимают в этом отношении промежуточное положение: число
известных халькогенидов для них больше, чем для первой диады, но меньше,
чем для последней.
Все дихалькогениды, за исключением производных платины и палладия,
кристаллизуются в структурном типе пирита, т.е. являются анионоизбыточными
фазами. Дихалькогениды платины и палладия образуют слоистую гексагональ-
гексагональную решетку только с "катион-анионными" связями. Монохалькогениды ЭХ
кристаллизуются в структурном типе куперита PIS, представляющем собой тет-
тетрагональную решетку с координационным числом 4 (для Э и для X). Все халько-
гениды можно получить непосредственным синтезом из компонентов. Они явля-
являются фазами переменного состава, однако области гомогенности невелики.
Соли кислородсодержащих кислот и комплекс-
н ы е соединения. Элементы первой вертикальной диады, у которых
стабильными являются высокие степени окисления, вовсе не образуют солей, где
они выступали бы в качестве катионообразователей. Для элементов второй диады
известны солеобразные производные, отвечающие степени окисления -f3. главным
образом сульфаты, а также двойные сульфаты типа квасцов. Более многочис-
многочисленны солеобразные соединения палладия и платины, отвечающие главным об-
образом их степени окисления +2. Так получены 3SO4-2H2O, Э^ОзЬ'ЗЩО,
Э(С1О4J-4Н2О. Известен также ацетат палладия Pd(CHaCOOJ-
В противоположность простым солям комплексные соединения элементов
семейства платиноидов чрезвычайно распространены. Эти элементы являются
наилучшими комплексообразователями в Периодической системе. В этом отноше-
отношении они превосходят элементы триады железа за счет большего удаления от ядра
валентных орбиталей, что облегчает донорно-акцепторное взаимодействие с ли-
гандами и увеличивает энергию расщепления в кристаллическом поле лигандов.
Поэтому большинство комплексов платиноидов (в отличие от элементов триады
железа) относится к низкоспиновым. Для платиноидов характерны ацидокомп-
лексы с лигандами — анионами слабых кислот: CN", CNS". СН3СОО" и др.. а
также гапогенидные комплексы. Широко распространены катионные комплексы с
нейтральными лигандами, особенно аква- и амминокомплексы. Комплексные сое-
соединения этих элементов в нулевой степени окисления — карбонилы — также
хорошо известны, хотя и не имеют такого значения, как у элементов триады
железа.
Комплексные соединения, отвечающие степени окисления +2, наиболее харак-
характерны для палладия и платины при к.ч. 4. Широко распространены комплексные
галогениды [Pdr4]2', [PtT4]2~, чрезвычайно устойчивые амминокомплексы
[9(NH3L]2+. Так, для [Pt(NH3L]2+ рА'нест 38, а для [Pd(NH3L]2+ рА'нест 30. Комп-
Комплексные галогениды платины и палладия типа Н2[ЭГ4] являются сильными кис-
кислотами и характеризуются высокой устойчивостью внутренней сферы, причем
устойчивость комплексов увеличивается в ряду С1~ — Вг" —> Г и для платины
выше, чем для палладия:
17* 4QQ
дах (Pd — Rh, Pt — Ir). Вообще для палладия известно наибольшее среди плати-
платиноидов число систем с непрерывной взаимной растворимостью компонентов.
Родий и иридий образуют непрерывные твердые растворы между собой, с эле-
элементами триады железа, но не дают систем такого типа с элементами VIIB- и IB-
групп. Более того, они в определенном интервале составов расслаиваются с сереб-
серебром и золотом даже в жидком состоянии.
Ограниченные твердые растворы с довольно широкими областями гомогеннос-
гомогенности — типичное явление в системах с участием платиноидов. Они растворяют не
только другие переходные металлы, но также и такие неметаллы, как Р, As, С,
Si, О и т.п. Хуже растворяют они sp-металлы и еще в меньшей степени — «-эле-
«-элементы НА-группы.
Интерметаллические соединения характерны для платиноидов и весьма разно-
разнообразны по составу. Это типичные нестехиометрические соединения бертоллидно-
го типа. Весьма характерно для платиноидов образование соединений Курнакова.
Наиболее ярко эта особенность выражена у палладия и платины.
Г ЛАВА XXII I. РАДИОАКТИВНЫЕ И СИНТЕЗИРОВАННЫЕ
ЭЛЕМЕНТЫ
В настоящее время для любого элемента искусственно получены радиоактив-
радиоактивные изотопы. Поэтому под радиоактивными элементами понимают такие, которые
не имеют ни одного стабильного изотопа. Радиоактивные элементы в свою оче-
очередь подразделяются на естественные (встречающиеся в природе) и синтезиро-
синтезированные, изотопы которых в природе не встречаются. В основном радиоактивными
являются тяжелые элементы, расположенные в конце Периодической системы
после висмута. Висмут является последним стабильным элементом в системе,
поскольку у него достигается предельное соотношение числа нейтронов и прото-
протонов (N/Z = 126/83 = 1,518), еще обеспечивающее стабильность ядра.
В химическом отношении радиоактивные элементы разнородны, поскольку
они принадлежат к разным группам системы. Строго говоря, их химические
свойства можно было бы рассматривать совместно с их типовыми аналогами по
группам. Однако при изучении свойств радиоактивных элементов приходится
сталкиваться со специфическими трудностями, общими для всех этих элементов.
Главные из них — наличие проникающих излучений, нестабильность, малые
количества и рассеянность элементов. По этой причине радиоактивные элементы
в курсе неорганической химии целесообразно рассматривать отдельно. Единствен-
Единственное исключение было сделано для технеция, поскольку он является средним
элементом подгруппы марганца и его удаление из последовательности Мп — Тс —
Re не позволяет проследить закономерности изменения свойств элементов в под-
подгруппе. Остальные радиоактивные элементы либо являются завершающими в
подгруппах (Ро, At, Rn, Fr, Ra), либо входят в большое семейство (Рт в семей-
семействе лантаноидов), либо образуют самостоятельное семейство актиноидов.
Изучение химических свойств радиоактивных элементов прежде всего натал-
наталкивается на проблему их концентрирования и разделения. Эта задача относится
к числу труднейших в препаративной химии, поскольку, например, тяжелые
актиноиды были получены в количествах, исчисляемых всего десятками атомов.
Концентрирование и разделение осуществляется методами соосаждения, экстрак-
дах (Pd — Rh, Pt — Ir). Вообще для палладия известно наибольшее среди плати-
платиноидов число систем с непрерывной взаимной растворимостью компонентов.
Родий и иридий образуют непрерывные твердые растворы между собой, с эле-
элементами триады железа, но не дают систем такого типа с элементами VIIB- и IB-
групп. Более того, они в определенном интервале составов расслаиваются с сереб-
серебром и золотом даже в жидком состоянии.
Ограниченные твердые растворы с довольно широкими областями гомогеннос-
гомогенности — типичное явление в системах с участием платиноидов. Они растворяют не
только другие переходные металлы, но также и такие неметаллы, как Р, As, С,
Si, О и т.п. Хуже растворяют они sp-металлы и еще в меньшей степени — «-эле-
«-элементы ПА-группы.
Интерметаллические соединения характерны для платиноидов и весьма разно-
разнообразны по составу. Это типичные нестехиометрические соединения бертоллидно-
го типа. Весьма характерно для платиноидов образование соединений Курнакова.
Наиболее ярко эта особенность выражена у палладия и платины.
ГЛАВА X X J J J. РАДИОАКТИВНЫЕ И СИНТЕЗИРОВАННЫЕ
ЭЛЕМЕНТЫ
В настоящее время для любого элемента искусственно получены радиоактив-
радиоактивные изотопы. Поэтому под радиоактивными элементами понимают такие, которые
не имеют ни одного стабильного изотопа. Радиоактивные элементы в свою оче-
очередь подразделяются на естественные (встречающиеся в природе) и синтезиро-
синтезированные, изотопы которых в природе не встречаются. В основном радиоактивными
являются тяжелые элементы, расположенные в конце Периодической системы
после висмута. Висмут является последним стабильным элементом в системе,
поскольку у него достигается предельное соотношение числа нейтронов и прото-
протонов (N/Z = 126/83 = 1,518), еще обеспечивающее стабильность ядра.
В химическом отношении радиоактивные элементы разнородны, поскольку
они принадлежат к разным группам системы. Строго говоря, их химические
свойства можно было бы рассматривать совместно с их типовыми аначогами по
группам. Однако при изучении свойств радиоактивных элементов приходится
сталкиваться со специфическими трудностями, общими для всех этих элементов.
Главные из них — наличие проникающих излучений, нестабильность, малые
количества и рассеянность элементов. По этой причине радиоактивные элементы
в курсе неорганической химии целесообразно рассматривать отдельно. Единствен-
Единственное исключение было сделано для технеция, поскольку он является средним
элементом подгруппы марганца и его удаление из последовательности Мп — Тс —
Re не позволяет проследить закономерности изменения свойств элементов в под-
подгруппе. Остальные радиоактивные элементы либо являются завершающими в
подгруппах (Ро, At, Rn, Fr, Ra), либо входят в большое семейство (Рш в семей-
семействе лантаноидов), либо образуют самостоятельное семейство актиноидов.
Изучение химических свойств радиоактивных элементов прежде всего натал-
наталкивается на проблему их концентрирования и разделения. Эта задача относится
к числу труднейших в препаративной химии, поскольку, например, тяжелые
актиноиды были получены в количествах, исчисляемых всего десятками атомов.
Концентрирование и разделение осуществляется методами соосаждения, экстрак-
N
ции, хроматографии и т.п. Например, для выделения радия можно бы использо-
использовать малую растворимость его сульфата, которая должна быть меньше раствори-
растворимости сульфата бария. Однако следовые количества этого элемента в растворе не
позволяют достичь произведения растворимости и осадок RaSO4 не образуется. В
то же время изоморфизм RaSO4 и его аналога BaSO4 позволяет осуществить
выделение радия из раствора совместным его осаждением с сульфатом бария
в виде твердого раствора замещения Raa;Bai-ISO4. В этом и состоит сущность
метода соосаждения. Адсорбционные методы, к которым относятся хромато-
хроматография и ионный обмен, позволяют осуществить не только выделение, но и раз-
разделение микроколичеств элементов. В качестве адсорбентов используют алюмо- и
силикагель, мелкодисперсный гипс, синтетические ионообменные смолы (ио-
ниты).
1. Радиоактивные аналоги стабильных элементов Периодической системы.
Полоний. Элемент № 84 — полоний Ро — является аналогом селена и теллу-
теллура. Его валентная электронная конфигурация — [Xe]4/45rf106s26p4. В соответствии
с общей закономерностью металлические свойства полония должны проявляться
ярче, чем у теллура. Его атомный радиус @,176 нм) и значение ОЭО B,0) зако-
закономерно вписываются в ряд этих характеристик элементов VIA-группы.
Полоний принадлежит к числу редких элементов B-105 масс, долей, %).
Общее число изотопов полония, как природных, так и полученных искусственно,
равно 29. Самый долгоживущий из них 209Ро (Т. . =103 года). Природный поло-
/2
ний испытывает а- и /^-распад, превращаясь соответственно в изотопы свинца или
астата. Самый стабильный из природных изотопов — 210Ро (Т. . =138 сут).
/2
Для выделения полония из урановой руды его переводят в раствор в виде
РоС1г и восстанавливают сероводородом или активными металлами на платино-
платиновых или цинковых пластинках. Полоний — серебристо-белый с желтоватым оттен-
оттенком мягкий металл с температурой плавления 254°С. Он обладает диморфизмом.
Низкотемпературная о-модификация — простая кубическая решетка — при 36°С
переходит в ромбоэдрическую /^-модификацию. Плотность полония при 20° С
равна 9,32 г/см3. В ряду стандартных электродных потенциалов полоний занима-
занимает место между Си и Hg [E°(Ро2+/Ро) == 0,65 В]. При горении на воздухе образует
оксид РоОг — самый стабильный из всех оксидов полония. Ему отвечает частич-
частично обезвоженный гидроксид РоО(ОНJ- Восстановлением РоОг можно получить
РоО и Р02О3. При анодном окислении полония получается РоОз- Производные
Ро(+6) характеризуются кислотной функцией. Так, методом соосаждения вместе
с теллуратом свинца РЬТеО4 выделяется полонат РЬРоО4-
В степени окисления +3 полоний обладает преимущественно основными свой-
свойствами, образуя соли, которые похожи на соответствующие производные лантана
и РЗЭ. Из солей Ро(+3) труднорастворимы фосфаты и оксалаты.
Взаимодействуя с галогенами, полоний образует два ряда производных: РоГг
и Р0Г4. Дигалогениды РоГг солеобразны. Для галогенидов РоГ4 характерны
ацидокомплексы Мег[РоГв].
С металлами полоний образует полониды (Na2Po, PbPo, HgPo и т.п.), в кото-
которых его степень окисления формально равна -2.
Помимо названных соединений известны сульфиды PoS, Po2S3, нитрат, суль-
сульфат, хромат, иодат полония (+3). Таким образом, химия полония укладывается в
одчн ряд с халькогенами, однако нарастание металлических свойств и появление
стабильной степени окисления +3 определяет дополнительную аналогию с ланта-
лантаноидами и его соседом — висмутом.
Астат. Элемент JV» 85 — астат (At) — имеет электронную конфигурацию
[Xe]4/45rflo6s26p5 и принадлежит к VIIA-группе Периодической системы, являясь
более тяжелым аналогом иода. Стабильных изотопов не имеет. Известны 20 изо-
изотопов с массовыми числами 200—219. Из них наиболее устойчив 2sj;At (Tj . = 8,3
ч). Все природные изотопы астата подвергаются «-распаду, превращаясь в изото-
изотопы висмута. В свою очередь, методы искусственного получения At основаны на
бомбардировке изотопов висмута а-частицами, например
2O9RJ 4- 4Не > 211 At 4- 2'п
83DI + 2ПС 85Л1 + V
Химические свойства астата изучены недостаточно. Однако имеющиеся сведе-
сведения позволяют заключить, что астат более металличен, чем иод (Е° . =
= 0.2 В).
Известные степени окисления астата -1, +1, +5. В отрицательной степени
окисления At" образует астатиды металлов, напоминающие соответствующие
иодиды (например, AgAt no растворимости в воде напоминает Agl, с которым он
и соосаждается из растворов). В то же время из растворов астат осаждается серо-
сероводородом в виде At2S. Окисление астата сравнительно мягкими окислителями в
растворе приводит к образованию производных гипоастатит-иояа AtO" (аналог
гипохлорит-иона СЮ"):
2At + Br2 + 2Н2О = 2НАЮ + 2НВг
Более энергичные окислители (пероксодисульфат, гипохлориты) переводят астат
в состояние со степенью окисления +5. В этой степени окисления астат образует
ион AtOs — астатат (аналогичен иодату Юз) — и дает соответствующие соли
(AgAtOa). Из растворов солей астата в любой степени окисления этот элемент
вытесняется цинком.
Радон. Радон Rn (№ 86) является тяжелым аналогом благородных газов с
полностью завершенной 6«26/>6-оболочкой. Общее число известных изотопов — 17
с массовыми числами 204 — 224. Наиболее устойчив природный изотоп 222Rn,
который образуется в результате «-распада Ra и сам подвергается а-распаду с
образованием 218Ро.
В химическом отношении радон подобен ксенону, но более реакционноспосо-
бсн. Взаимодействие со фтором уже при комнатной температуре приводит к обра-
образованию фторидов: RnF2, RnF4, RnF6 и RnFg (в зависимости от давления фтора).
Эти фториды более устойчивы, чем соответствующие соединения ксенона. Поми-
Помимо фторидов получен и идентифицирован RnCl4.
Франций. Существование франция Fr (№ 87) как элемента 1А-группы
было предсказано Д.И. Менделеевым в 1871 г. (экацезий). Элемент № 87 был
открыт в 1939 г. при изучении продуктов радиоактивного распада актиния 227Ас:
227Д(, k 223Pr i 4Но
83rtL 87ГГ + 2Пе
Природные изотопы франция являются «-активными, превращаясь в астат. Из-
Известно 18 изотопов с массовыми числами 206 — 223, среди которых наиболее
устойчив 223Fr (Г, . = 21 мин). Этот изотоп /3-активен, превращается в радий.
503
i- 3
Химия франция практически не изучена вследствие значительных экспери-
экспериментальных трудностей при исследовании свойств следовых количеств коротко-
живущих изотопов. Содержание Fr в природе оценивается величиной 105 масс,
долей, %. Франций — самый активный металл Периодической системы, по свой-
свойствам и растворимости солей напоминает цезий. Его можно соосадить в виде
перхлората FrC104 и гексахлороплатината Fr2[PtCl6] совместно с аналогичными
производными цезия. Гидроксид франция FrOH является сильнейшим основани-
основанием. К числу хорошо растворимых солей относятся FrCl, FrNO3, Fr2SO4 и т.д.
Радий. В настоящее время известны 14 изотопов с массовыми числами 213,
218 - 230. Наиболее долгоживущий - 226Ra (Т. . = 1620 лет).
72
Радий ([Rn]7«2) является гомологом щелочно-земельных металлов и ближай-
ближайшим аналогом бария. Металлический радий впервые был получен электролизом
расплава RaCl2. Радий — серебристо-белый металл с плотностью 6,0 г/см3 и с
температурой плавления около 960°С.
Радий принадлежит к наиболее активным металлам (Е° 9, . = —2.92 В),
Ra^ /Ra
проявляет единственную степень окисления +2. На воздухе, как и барий, покры-
покрывается слоем нитрида Ra3N2. При взаимодействии с водой радий образует гид-
гидроксид Ra(OHJ, являющийся более сильным основанием, чем Ва(ОНJ. Обезво-
Обезвоживанием гидроксида получают белый оксид RaO, также обладающий ярко
выраженными основными свойствами.
С галогенами радий образует соли R,aF2 белого цвета, которые при хранении
желтеют под действием собственного радиоактивного излучения. Наиболее рас-
распространенными солями радия являются RaSO4 и RaCO3, растворимость которых
в воде столь же мала, как и соответствующих соединений бария. Получены также
R.a(NO3J) RaSO3, RaCrO4 и др.
Прометий. Элемент № 61 — прометий Рга — принадлежит к семейству
лантаноидов. Известны 16 изотопов прометия с массовыми числами 141 — 154.
Самый устойчивый, но малодоступный — 145Рш (Т. . =18 лет).
72
В настоящее время прометий выделяют из осколков деления 235U в ядерных
реакторах. От других осколков его отделяют хроматографией на специальном
ионообменнике — амберлите. Металлический прометий получен литийтермичес-
ким восстановлением трифторида.
Прометий — типичный металл серебристого цвета с плотностью 7,26 г/см' и
*пл = 1080°С. Электронная конфигурация атома в невозбужденном состоянии
[Xe]4/6s2. Он является типичным лантаноидом, проявляя во всех соединениях
степень окисления +3, Е° ,. ._ = -2,43 В. Гидроксид Рт(ОНK — светло-корич-
Pm-5+/Pm
невый желатинообразный осадок, обладающий основными свойствами. При про-
прокаливании отщепляет воду, переходя в Рт2Оз. В чистом состоянии выделены
растворимые в воде соли: желтый хлорид РтС13, розовый нитрат Рт(ГЮз)з-
Нерастворим в воде PmFa.
2. Металлы семейства актинидов. Положение актинидов в
Периодической системе. До синтеза тяжелых элементов, следую-
следующих за ураном, актиний, торий, протактиний и сам уран относили к соответству-
соответствующим В-группам системы (IIIB — VIB). Действительно, каждому из них свой-
свойственны высшие степени окисления, отвечающие номеру группы (Ас2О3, ThO2,
Ра2О5, 1Юз). Синтезированные тяжелые элементы (Np, Pu и др.) называли тран-
504
сурановыми и выносили за пределы графической формы Периодической системы
(подобно лантаноидам). Это выглядело несколько искусственным, так как не
было обосновано с точки зрения электронного строения атома. Было очевидно,
что в 7-м периоде должно существовать семейство из 14 б/^элементов, подобное
семейству лантаноидов, однако не было ясно, с какого именно элемента происхо-
происходит заполнение 5/оболочки. В 1942 г. Г. Сиборг высказал актинидную гипотезу,
согласно которой заполнение 5/-оболочки возможно уже у элементов, следующих
за актинием (начиная с тория — № 90). С этой точки зрения актиний является
аналогом лантана, а более тяжелые элементы — аналогами лантаноидов и образо-
образовывали соответствующее 5 ./-семейство.
Первоначально актинидная гипотеза вызывала возражения, главным из кото-
которых было отмеченное выше химическое подобие Th, Pa, U (а также Np и Рц) с
элементами побочных подгрупп. Так, М. Гайсинский считал, что это семейство
начинается с кюрия (Cm, № 96), поскольку он сам и следующие за ним элементы
(кюриды) обнаруживают сходство друг с другом и с лантаноидами, проявляя в
соединениях степень окисления +3. Однако противоречие между точками зрения
Сиборга и Гайсинского лишь кажущееся. Хорошо известно, что для металлов
переходных d- и ^рядов существует внутренняя периодичность, причем элементы
первой пятерки или семерки сильнее отличаются по свойствам друг от друга, чем
элементы второй половины рядов, у которых ярче проявляется горизонтальная
аналогия. Другой причиной является конкуренция между bf- и 6<йэболочками у
торидов. Действительно, спектроскопические и магнитные исследования показа-
показали, что элементы первой семерки могут иметь различную электронную конфигу-
конфигурацию;
Структура оболочки
5/6<?7б3 или
от bfi%S7s2 до &<P7s2
от 5/6<?7s2 до btfls2
от 5fb$7s2 до 6<P7s2
5f7s2 или
5/7s2 или 5
5/6 $7s2
Элемент
Th (90)
Ра (91)
U (92)
Np (93)
Pu (94)
Am (95)
Cm (96)
Bk (97)
На рис. 155 представлена зависимость энергии 5/- и 6</-оболочек от порядко-
порядкового номера элемента, из которой следует, что у элементов с Z < 90 предпочти-
предпочтительно заполняются 6<1-орбитали (Ас). Для элементов с 90 < Z < 95 наблюдается
конкуренция подуровней и только после Z > 96 предпочтительно заполняется 5/-
оболочка. По этой причине актиний являет-
является типичным rf-элементом, торий, протакти-
протактиний, уран и нептуний, а также плутоний в
некоторых соединениях проявляют опреде-
определенное сходство с элементами соответствую-
соответствующих В-групп. Этим и объясняются высокие
характеристические степени окисления в 90 91 92 S3 9Ь 95 96 87 38 X Ш I
торидов. Рис. 155. Конкуренция 5/- и 6й-уровней
Синтез курчатовия (№ 104) И нильсбория энергий в семействе актиноидов
(№ 105) подтвердил справедливость актинидной теории Сиборга, поскольку кур-
чатовий оказался аналогом гафния,-а не лантаноидов. Следовательно, лоуренсий
(№ 103) является последним из актинидов. С учетом неравноценности первой и
второй семерок актинидов сам актиний и последующие элементы до америция
далее рассматриваются индивидуально (чем подчеркивается различие их
свойств), а кюрий и кюриды — совместно.
Актиний (№ 89). Известны 13 изотопов Ас с массовыми числами 213—231.
Наиболее устойчив 227Ас с Т. . = 21.7 года. Содержание актиния в земной коре
/2
оценивается в 6-Ю0 масс, долей, %. В свободном состоянии выделен лишь в
1947 г. Металлический актиний получают литий- или калийтермическим восста-
восстановлением галогенидов. Это серебристо-белый металл, кристаллизующийся в
ГЦК-структуре с температурой плавления 1050°С. Актиний — очень активный
металл (Е° 3+/ = -2,6 В), легко окисляется на воздухе и энергично взаимодей-
ствует не только с минеральными кислотами, но и с водой. Во всех своих соеди-
соединениях проявляет степень окисления 4-3, являясь аналогом лантана. Оксид АсгОя
можно получить как непосредственным синтезом, так и прокаливанием Ае(ОН)д,
разложением оксалата или карбоната. И оксид, и гидроксид актиния обладают
ярко выраженными основными свойствами. Ас(ОН)з принадлежит к числу силь-
сильнейших оснований. К растворимым в воде солям Ас(+3) относятся Ас(гЮя)з-
Ac2(SO4K, а также АсС1з и АсВг3. Нерастворимы карбонат, фосфат, оксалат и
AcF3.
Торий (№ 90). Общее число изотопов тория равно 13 с массовыми числами
от 223 до 235. Наиболее устойчив природный долгоживущий изотоп 232Th (Т. . =
/2
= 1.4-1010 лет). Торий сравнительно широко распространен в природе F,5-10*
масс, долей, %), в основном в виде изотопа 232Th. Промышленными минералами
тория являются монацит (Се, La, Th, ...)РО4, торит Th(SO4J> торианит (Th,U)O2-
Переработка торийсодержащих руд сводится к выделению ТпОг с последую-
последующим кальцийтермическим восстановлением или к электролизу растворов ThF4
или ТЬСЦ в расплавах хлоридов щелочных металлов. Для получения тория
высокой чистоты термически разлагают ТЫ4 на раскаленной нити.
Торий — пластичный серебристо-белый металл с плотностью 11,72 г/см3
и температурой плавления 1750°С. Он обладает диморфизмом: низкотемпера-
низкотемпературная ГЦК-модификация (o^-Th) при 1450°С переходит в ОЦК-модификацию
@-Th).
Торий — активный металл, стандартный электродный потенциал его
Е° — -1,9 В. На воздухе и в воде торий довольно устойчив вследствие
пассивации его поверхности пленкой ТЬОг- Металлический торий медленно раст-
растворяется в минеральных кислотах. Концентрированная азотная кислота его
пассивирует, как Zr и Hf. При накаливании на воздухе торий сгорает с боль-
большим выделением теплоты, образуя устойчивый оксид ТЬОг (Д#^298
~ -1200 кДж/моль). ТЬОг — один из самых тугоплавких оксидов (<пл = 3200°С).
Прокаленный ТЬОг нерастворим в кислотах и щелочах. При взаимодействии
солей Th(+4) со щелочами и аммиаком образуется осадок Th(OHL белого цвета,
обладающий основными свойствами. При комнатной температуре Th реагирует с
фтором, а при нагревании — и с остальными галогенами, образуя солеобразные
галогениды ThF4. Фторид ThF4 в воде нерастворим.
При нагревании торий реагирует и с другими неметаллами. Гидриды тория
полностью диссоциируют при 900°С. С серой торий дает сульфиды ThS, Th2S3,
Th4S7 и ThS2- Нагревание тория в атмосфере азота приводит к образованию
Th2N3> который при 1500°С отщепляет частично азот, переходя в фазу внедрения
ThN с <пл = 2630°С. В системе Th — Р известны Th4P3 и ТЪ3Р4- Карбиды тория
ThC и ThC2 (<пл = 2624 и 2655°С), как и нитрид ThN, являются фазами внедре-
внедрения. С кремнием торий образует Th3Si2, ThSi, ThSi2, а с бором — ThB4 и ThB6,
т.е. соединения, характерные для большинства переходных металлов.
Соли кислородсодержащих кислот тория отвечают степени окисления +4. К
растворимым солям относятся Th(NOgL-пН2О, Th(SO4J-п.Н2О. Труднораствори-
Труднорастворимыми являются оксокарбонат ThOCO3-8H2O, оксалат Th(C2O4J-6H2O, фосфаты
Th3(PO4L-6H2O, Th(HPO4J-H2O. Все труднорастворимые соли тория могут быть
переведены в раствор за счет комплексообразования. Все растворимые соли тория
гидролизованы, и для него известны основные соли и солеобразные производные
торила ThO2+.
В металлохимическом отношении торий обладает меньшей способностью к
образованию непрерывных твердых растворов, чем металлы IVB-группы, вследст-
вследствие сравнительно большого атомного радиуса. Непрерывная взаимная раствори-
растворимость отмечена лишь в системах тория с Y, La, Zr, Hf, что опять же подчеркивает
ого сходство с элементами как ШВ- так и IVB-группы. В то же время торий
образует ограниченные твердые растворы и интерметаллические соединения с А-
металлами и тяжелыми sp-элементами (Pd, Sb, Bi). Торий не смешивается в жид-
жидком состоянии не только с щелочными металлами, но и с ураном, а с хромом и
вольфрамом образует эвтектические смеси.
Протактиний (№ 91). Наиболее долгоживущий изотоп 231Ра (Т. =
/2
— 3,4-104 лет). Всего известно 14 изотопов Ра с массовыми числами 224—237.
Протактиний принадлежит к числу наиболее редких элементов (~ 1-Ю0 масс,
долей, %). Для получения протактиния его концентрат переводят в тетрафторид
PaF4, который подвергают барийтермическому восстановлению. В свободном
состоянии Ра — серебристо-серый металл с плотностью 15,37 г/см3 и температу-
температурой плавления 1575°С. Протактиний обладает диморфизмом: переход тетраго-
тетрагонального а-Ра в /?-Ра (ОЦК) наблюдается при 1172°С.
В своих соединениях протактиний проявляет степени окисления от 4-2 до 4-5-
Степень окисления +2 малохарактерна и реализуется, например, в субоксиде
РаО. В степени окисления +3 Ра напоминает актиний, Ра(+4) похож на Zr и Hf,
а РаD-5) — на Nb и Та. Наиболее типичны степени окисления +4 и 4-5.
На воздухе металлический Ра тускнеет вследствие образования на поверхности
оксидов. При нагревании металл сгорает до Ра20.5 белого цвета. Восстановление
Ра2О5 водородом дает черный РаО2. Высший оксид Ра2О5 устойчив по отноше-
отношению к воде, соляной и азотной кислотам. Диоксид РаО2 также плохо растворим в
кислотах, но реагирует с HF за счет комплексообразования. Несмотря на это, оба
оксида обладают преимущественно основными свойствами. Так, в отличие от
Та2О5, которому соответствует кислота Н3Та04) гидроксид Ра(+5) РаО(ОНK
растворяется только в кислотах и представляет собой основание — гидроксид
протактиния а РаО3+.
С водородом Ра реагирует при 250—300° С с образованием РаН2. Известны
пента- и тетрагалогениды и оксогалогениды протактиния, например PaF4, PaF5,
РаС14, РаС15, РаОС1з и т.п. Они образуют комплексы с галогенидами металлов
507
галогенами — при нагревании. С фтором и хлором уран образует по четыре сое-
соединения, в которых его степень окисления изменяется от +3 до +6. С бромом и
иодом уран образует только иГд и UF4. Тригалогениды UF3 на воздухе окисля-
окисляются до производных уранила UO2F3. UF3 труднорастворим в воде и довольно
инертен к кислотам, в то время как остальные UF3 энергично взаимодействуют с
водой и выступают в роли восстановителей. В расплавах галогенидов щелочных
металлов ГГ3 (кроме UF3) образуют комплексы типа K2[UC15]. K3[UClrJ.
Mes[UBrf,] и т.п. Галогениды состава 1Т4 также легко окисляются до соединений
ураннла и склонны к комплексообразованию с галогенидами щелочных, щелоч-
но-земельных металлов и аммония: Me [UF.J, Me2[UF6], Me3[UF7].
Наименее характерна для урана степень окисления +5. Поэтому UF^ и UC1;,
склонны к диспропорционированию: 2UF5 — UF4 + UF6. В то же время за счет
комплексообразования степень окисления +5 несколько стабилизируется:
Me'[Urfi]: Ме'[иГ7].
Наиболее типичными и практически важными галогенидами урана являются
VFr, и UClfi. В отличие от инертного SF6 гексафторид урана чрезвычайно реакци-
онноспособен. Он активно реагирует с большинством металлов, образуя соответст-
соответствующие фториды, энергично взаимодействует с водой:
UF6 + 2Н2О = UO2F2 + 4HF
При нагревании UF<j восстанавливается водородом до UF4. Различие активности
SF6 и UFfj объясняется тем, что значительно больший атом урана недостаточно
экранируется атомами фтора.
Взаимодействие урана с водородом протекает при 250—300"С г образованием
нестойкого гидрида UHS, который разлагается при 430°С. В этом отношении
уран напоминает лантаноиды и актиний.
Уран образует большое число хапькогенидов. которые обычно являются фаза-
фазами переменного состава. Известны I S. U5S3, W2S3, US2; USe, U,Se3, USe9; ГТе,
U3Te4, UTp2- Эти халькогениды можно получить непосредственным синтезом из
компонентов.
Известны три нитрида урана (UN, U2N3. UN2), получаемые при взаимодейст-
взаимодействии урана или его гидрида с азотом или аммиаком. Самым стабильным из них
является UN (AH°*2gs — -286 кДж/моль). К фазам внедрения относятся и туго-
тугоплавкие карбиды иГ B350°С), U2C3 A775°C) и UC2 B475°C), получаемые пря-
прямым синтезом. При нагревании уран взаимодействует со всеми пниктогенами (в
том числе и с висмутом). Типичными составами являются Un, U3n4 и иП2, ха-
характерные для большинства переходных {/-металлов. Аналогичные закономернос-
закономерности наблюдаются и при взаимодействии урана с кремнием и бором, что сближает
его с элементами VIB-группы.
Типичными солеобразными производными урана являются соли уранила.
Хорошо растворимы в воде нитрат, сульфат уранила, ацетат иО2(СН3СООJ. К
труднорастворимым относятся фосфат UO2HPO4, оксалат UO2C2O4- Соли уранила
имеют обычно характерную желтую окраску с желто-зеленой флуоресценцией.
Помимо аквакомплексов для уранила характерны и ацидокомплексы. При дей-
действии щелочей на соли уранила получают производные диурановой кислоты
509
галогенами — при нагревании. С фтором и хлором уран образует по четыре сое-
соединения, в которых его степень окисления изменяется от 4-3 до +6. С бромом и
иодом уран образует только UFa и 11Г4. Тригалогениды 1ТГз на воздухе окисля-
окисляются до производных уранила UO2F3. UF3 труднорастворим в воде и довольно
инертен к кислотам, в то время как остальные UFg энергично взаимодействуют с
водой и выступают в роли восстановителей. В расплавах галогенидов щелочных
металлов 11Гз (кроме UF3) образуют комплексы типа KofUCls]. K3[UClr,].
Me3[UBrb] и т.п. Галогениды состава UF4 также легко окисляются до соединений
уранила и склонны к комплексообразованню с галогенидамн щелочных, щелоч-
но-земельных металлов и аммония: Me [UT5], MeofUI'e], Mes[Ur7].
Наименее характерна для урана степень окисления +5. Поэтому UF$ и UCU
склонны к диспропорционированию: 2UF5 — UF4 + UFH. В то же время за счет
комплексообразования степень окисления +5 несколько стабилизируется:
Ме'[иГE]; Me2[UT7].
Наиболее типичными и практически важными галогенидами урана являются
Шч я UClfi. В отличие от инертного SF6 гексафторид урана чрезвычайно реакци-
онноспособен. Он активно реагирует с большинством металлов, образуя соответст-
соответствующие фториды, энергично взаимодействует с водой:
UF6 + 2Н2О = UO2F2 + 4HF
При нагревании UFr, восстанавливается водородом до UF4. Различие активности
SF6 и UFr, объясняется тем, что значительно больший атом урана недостаточно
экранируется атомами фтора.
Взаимодействие урана с водородом протекает при 250—300° С г образованием
нестойкого гидрида UH:5, который разлагается при 430"С. В этом отношении
уран напоминает лантаноиды и актиний.
Уран образует большое число халькогенидов. которые обычно являются фаза-
фазами переменного состава. Известны US, U6S3, U2S3, US2; USe, U2Se3, USe2; UTe,
U3Te4, UTe2. Эти халькогениды можно получить непосредственным синтезом из
компонентов.
Известны три нитрида урана (UN, U2N:i, UN2), получаемые при взаимодейст-
взаимодействии урана или его гидрида с азотом или аммиаком. Самым стабильным из них
является UN (Д/Г^2И = -286 кДж/моль). К фазам внедрения относятся и туго-
тугоплавкие карбиды UC B350°C). U2C:i A775'C) и UC2 B475°C), получаемые пря-
прямым синтезом. При нагревании уран взаимодействует со всеми пниктогенами (в
том числе и с висмутом). Типичными составами являются 1Ш, и3П4 и иПг, ха-
характерные для большинства переходных d-металлов. Аналогичные закономернос-
закономерности наблюдаются и при взаимодействии урана с кремнием и бором, что сближает
его с элементами VIB-группы.
Типичными солеобразными производными урана являются соли уранила.
Хорошо растворимы в воде нитрат, сульфат уранила, ацетат UO2(CH3COOJ- К
труднорастворимым относятся фосфат UO2HPO4, оксалат иО2С2О4- Соли уранила
имеют обычно характерную желтую окраску с желто-зеленой флуоресценцией.
Помимо аквакомплексов для уранила характерны и ацидокомплексы. При дей-
действии щелочей на соли уранила получают производные диурановой кислоты
509
H2U2O7 — диуранаты, а при сплавлении оксидов 1Юз и Ме2О — моноуранаты
(аналогично дихроматам и хроматам). Тенденция к образованию изополикислот у
урана выражена ярче, чем у хрома, что сближает его с молибденом и вольфра-
вольфрамом. Так получены соли типа Мр2ипОз+ь например Na2Ufi0i9-
Металлохимия урана достаточно хорошо изучена и характеризует его как
переходный металл. Из трех полиморфных модификаций урана наибольшей спо-
способностью к образованию твердых растворов характеризуется f-V. Непрерывные
твердые растворы 7~U обрарует с Np, Pu, Ti, Zr, Nb. Достаточно хорошо раство-
растворим в y-V и молибден (~ 40%). В то же время многие Зс/--металлы лишь ограни-
ограниченно растворимы в уране в силу заметного различия атомных радиусов. При
ограниченной взаимной растворимости уран обрарует значительное число интер-
интерметаллических соединений, особенно с металлами VIIlB-rf уппы.
Нептуний. Первым искусственно полученным тяжелым элементом, сле-
следующим после урана, является элемент .N"! 93. Синтез /^-радиоактивного изотопа
23pN
Np с
72
= 2,3 с-ут впервые был осуществлен но реакции
239JJ
9'Z^
/Г.
23,4
В настоящее время известны 15 изотопов нептуьия с массовыми числами
230—242. Самым долгоживущьм из них яьляется 237\"р (Т.. — 2,14• 10ь лет). В
/2
чистом виде нептуний был выделен в 1944 г. путем барийтермиче( кого восстанов-
восстановления Nj. F4. Нептуний — серебристо-бьлый металл с плотностью
температуре й плавления 64СГС. Обладает полиморфизмом:
20 г/см3
u-Np
ромби-
ромбический
/?-Np
тетраго-
тетрагональный
7-NP
куби-
кубический
В химическом отношении Np принадлежит к активным металлам. Его стандарт-
стандартный электродный потенциал (Е° _ .., = -1,83 В) показывает, что в ряду стан-
v Np3+/Np '
дартных электродных потенциалов нептуний располагается между магнием и
апюминием. В своих соединениях Np проявляет все степени окисления от +3 до
4-7. Наиболее характерны из них +4 и +6, а в растворах важное значение имеет
степень окисления +5.
При сгорании на воздухе нептуний образует наиболее устойчивый оксид
NpO2. Его восстановлением получают Np2O3- Кроме того, существует и оксид
Np3Os, аналогичный соответствующему оксиду урана. Оксидам Np2Og и NpO2
соответствуют гидроксиды Np(OHK и Np(OHL, обладающие основным характе-
характером. Np(+3) и Np(+4) существуют в растворе как простые катионы, а производ-
производные Np(-f 5) и Np(+6) обнаруживают двойственную природу и могут образовывать
как катионы нептунилов NpO2 и NpOf\ так и соответствующие нептунат-анионы.
Однако кислотная функция Np(-f5) и Np(-f6) выражена в очень слабой степени и
нептунаты неустойчивы.
В отличие от урана для нептуния возможна степень окисления +7, что и было
доказано при окислении нептуната K2NPO4 озоном в щелочной среде:
2NpOf + О3 + 2ОН- = 2NpOf" + О2 + Н2О
510
Это подтверждает особую природу первой семерки актинидов, связанную с
конкуренцией 5/- и бсйэрбиталей, и некоторую аналогию Np(+7) с элементами
VIIB-группы.
Нептуний энергично взаимодействует с галогенами. Тригалогениды NpFj
известны для всех галогенов, NpF4 характерен для фтора, хлора и брома. A NpF5
и NpFe существуют только для фтора и хлора в соответствии с изменением окис-
окислительной активности в ряду галогенов. С применением суперокислителей —
фторидов ксенона — получен и
NpF3 + XeF4 = NpF7 + Xe
Низшие галогениды имеют солеобразный характер, a NpF6 похож на лвгколету-
чий UF6. Все галогениды, особенно отвечающие высоким степеням окисления,
склонны к гидролизу и комплексообразованию.
Нептуний энергично поглощает водород даже при легком нагревании, образуя
гидрид переменного состава NpH4-x. При более сильном нагревании этот гидрид
разлагается аналогично соответствующему производному урана.
Из других соединений нептуния с неметаллами получены сульфид Np2S3 и
нитрид NpN. Непосредственным синтезом получены также фосфид NpP, карбид
NpC^. Известен силицид NpSi2, изоморфный ThSi2-
Наряду с солями нептунилов NpO^ и NpO5+ известны и соли катионов NpJ+ и
Np4+ с кислородсодержащими кислотами: перхлораты, нитраты, сульфаты. В
виде кристаллогидрата выделен, например, Np(S04J.-rH_>0. Все они склонны к
гидролизу, диспропорционированию и комплексообразованию. Наилучшим комп-
лексообразователем является Np(+4). Гидратированные катионы нептуния в
растворах имеют характерную окраску: Np3+ — голубую, Np4+ — желто-зеленую,
МрОг — зеленую, NpC>2+ — розовую.
Плутоний. Самым важным для современной техники и наиболее изучен-
изученным из заурановых элементов является плутоний — элемент № 94. И тестны 15
изотопов Ри с массовыми числами 232—246. Наибольшее значение имеет 2!ЯРи
(Т. = 2,44• 104 лет), который наряду с 235U и 233U способен к цепн ж реакции
/2
деления. Самый долгоживущий изотоп 244рц (Т.. = 7,5-107 лет). В настоящее
/2
время плутоний получают в ядерных реакторах по реакции
- > 234Nd —
2.WIJ + In > IJ > Nd
92u +оп— 92u 2i3 мш 931NP 2,33 сут
Разделение урана, нептуния и плутония основано на различии их химических
свойств и значительно легче осуществляется, чем разделение изотопов урана.
Поэтому роль плутония ядерной технике неуклонно возрастает. Металлический
плутоний, как уран и нептуний, получают путем восстановления PuFg или PuF4
барием, кальцием или литием при высокой температуре.
Плутоний — серебристо-белый металл с температурой плавления 639°С. Имеет
шесть полиморфных модификаций, превосходя в этом отношении все металлы.
Плотность низкотемпературной о-модификации (орторомбической) равна 19,82
г/см3.
Плутоний — активный металл, располагается в ряду стандартных электродных
511
потенциалов, как и нептуний, между магнием и алюминием, но ближе к магнию
(Е° 3+. = -2,03 В). Из оксидов плутония наиболее устойчив РиО2 (как и для
нептуния) — коричнево-желтый кристаллический порошок. Известен также оксид
Ри2О3, но в отличие от U и Np оксид типа Ме3О8 для плутония уже не существу-
существует. Оксидам Ри(+3) и Ри(+4) отвечают гидроксиды Ри(ОНK и Ри(ОНL, причем
первый из них самопроизвольно окисляется кислородом воздуха, переходя во
второй.
Для плутония, как и для урана и нептуния, известны разнообразные степени
окисления: от +2 до +8. Однако высшие степени окисления для Pu менее харак-
характерны. Отличительной особенностью химии плутония является то, что его произ-
производные в различных степенях окисления, кроме +7, могут одновременно нахо-
находиться в растворе в равновесии вследствие легкости взаимных переходов (ЕГ, В):
в кислой среде A М НС1)
0 +2,03 . -0,97 L
Pu > Pu3+ > Pu4+
в щелочной среде A М NaOH)
Pu°-J^
Pu(OHK +°'95 > Pu(OHL °'76 > PuO2(OH) °'2S PuO2(OHJ
Действительно, особенно в кислой среде потенциалы взаимных переходов от
Ри(+3) до Ри(+б) различаются очень мало (порядка 1 В). В щелочной среде
индивидуальность каждой степени окисления несколько выше. В целом в водных
растворах плутоний — более активный металл, чем нептуний. В кислой среде для
Pu и Np более устойчива степень окисления +3, а в щелочной +4.
Плутоний активно взаимодействует с неметаллами. Галогениды плутония
напоминают галогениды нептуния: РиГз известен даже для иода. В отличие от
нептуния тетра- и гексагалогениды известны только для фтора, но и они при
нагревании легко переходят в P11F3, отщепляя фтор. PuF6 напоминает UF6 и
NpF6, но менее летуч. Трифторид P11F3 — солеобразное вещество. Он изоморфен
NpF3, UF3, LaF3, что подтверждает принадлежность Ри(+3) к актинидам и анало-
аналогию с лантаноидами.
Среди солей кислородных кислот преобладают производные Ри(+4): сульфа-
сульфаты, нитраты, перхлораты и др. Помимо комплексов с типичными лигандами для
Ри(+4) характерен нитратный комплекс Кг[Ри^Оз)б]. В степени окисления +5 и
+6 существуют соли плутонилов РиО2 и РиО2+, которые менее устойчивы, чем
производные нептунилов, и в растворах постепенно диспропорционируют. Дей-
Действием энергичных окислителей (озон в щелочной среде) получены производные
Ри(+7) и даже Ри(+8), которые еще более неустойчивы.
В металлохимическом отношении плутоний также активен. Он образует боль-
большое число интерметаллических соединений как с металлами главных подгрупп
(Be, Mg, A1), так и с переходными металлами (Fe, Co, Ni, Ag и др.). В то же
время возможности образования непрерывных твердых растворов у плутония
ограничены из-за его уникальной способности к полиморфизму.
В ряду Th — Ра — U — Np — Pu наблюдается повышение максимально воз-
возможной степени окисления элемента от +4 до +8. Это связано с конкуренцией 5/-
и erf-уровней. В образовании производных высших степеней окисления участвуют
512
все электроны незавершенных d- и /оболочек, а также 7в-электроны. Устойчи-
Устойчивость высших степеней окисления от Th к U возрастает, а для Np и Ри уменьша-
уменьшается.
На различной стабильности степеней окисления +6 и +4 для U, Np и Ри и
основано разделение этих элементов. В ядерных превращениях эти элементы
генетически связаны. Поэтому в продуктах ядерных реакторов они всегда при-
присутствуют совместно. В кислой среде при переработке вторичного сырья уран,
нептуний и плутоний существуют в виде UOf\ NpO'2+, РиОг*. Мягкое восстанов-
восстановление (например, с помощью SO2) переводит Np(+6) и Ри(+6) в производные
степени окисления +4, в то время как уран при этом остается в более характер-
характерной для него форме UO?*- Добавление в эту смесь ацетата натрия позволяет
осадить уран в виде желтого кристаллического натрийуранилацетата
КаиО2(СНзСОО)з, а нептуний и плутоний остаются в растворе. Затем их смесь
подвергают окислению (например, броматом калия КВгСЬ). Нептуний вновь
окисляется до NpO2+, а плутоний при этом сохраняет степень окисления +4.
Добавляя опять ацетат натрия, осаждают теперь нептуний в виде аналогичной
двойной соли NaNpO2(CH3COOK.
Америций. В 1944—1945 гг. при облучении нейтронами ->зуРи был полу-
получен изотоп нового элемента с массовым числом 241. Америций можно получить и
облучением 238U о-частицами:
§U + «Не —» 2!'Pu*+
Известны изотопы америция с массовыми числами 232, 234, 237—247. Наиболее
долгоживущий — 243Ат (Т. , = 7,6-103 лет).
/2
Металлический америций получают высокотемпературным восстановлением
AniFj или АтО2 активными металлами (Ва, La). Это серебристо-белый металл с
плотностью 11,9 г/см3 и температурой плавления 1176еС. Он диморфен: низко-
низкотемпературная «-модификация (гексагональная) при 1079°С переходит в ГЦК-
структуру (/З-Ат).
В соединениях америций проявляет степени окисления +2, +3, +4. Его стан-
стандартный электродный потенциал в растворе Е" 3+/ = -2,36 В. Оп легко раст-
воряется в разбавленных минеральных кислотах с образованием солей Аш(+3).
При сгорании на воздухе америций образует темно-коричневый оксид АшО2. При
дальнейшем нагревании АтО2 отщепляет кислород, переходя в Ат2Оз. При
окислении на воздухе получается черный АтО. Все оксиды америция и извест-
известный гидроксид Ат(ОН)з обладают основными свойствами. Америций энергично
взаимодействует с галогенами, образуя при этом AmF4, AmFg и для остальных
галогенов только АтГз- Галогениды америция солеобразны и напоминают соот-
соответствующие производные лантаноидов.
Кюрий и кюриды — элементы второй семерки ак-
актинидов. Получение новых тяжелых элементов представляет собой сложную
задачу, причем сложности возрастают по мере увеличения атомного номера эле-
элемента. Это объясняется тремя основными причинами. Во-первых, концентрация
исходных элементов, ядра которых необходимо подвергать бомбардировке, очень
невелика и соответственно вероятность попадания частицы-снаряда в ядро-ми-
R14
ная) и +4. Прокачиванием на воздухе ионита, содержащего сорбированный Вк,
получен ВкОг, который легко восстанавливается водородом до ВкгОд. Известны
галогениды ВкГз (для всех галогенов) и BkF^ По своим химическим свойствам
Вк является аналогом тербия и в то же время горизонтальным аналогом кюрия.
Элемент № 98 — калифорний Cf — также был получен путем бомбардировки
о-частицами. В качестве мишени служил 242Ст:
242fm 4- 4Не > 245Cf 4- 'п
S6UI1 ' 2 98^* + О
Известны изотопы калифорния с массовыми числами 244—254, а самый устойчи-
устойчивый — 249Cf (Т. . = 400 лет). По немногим существующим данным можно судить,
/2
что Cf — активный металл (E°3+/nf = -2,32 В), проявляющий степень окисления
\-Л' /*-Л
+3 и являющийся аналогом лантаноидов и предшествующих кюридов. Для него
известны оксид Cf20s, все галогениды CfFg, оксогалогениды СЮГ, сульфид
Cf2S3.
Следующие за калифорнием элементы № 99 (эйнштейний Es) и № 100 (фермий
Fm) впервые были открыты в продуктах термоядерного взрыва A952). Уже одно
это говорит о сложности синтеза ядер этих элементов. В самом деле, например,
синтез ядра эйнштейния путем последовательного захвата нейтронов ядрами 238U
возможен в результате захвата 15 нейтронов и семикратного /3-распада образую-
образующихся продуктов. Поэтому в дальнейшем изотопы эйнштейния и фермия получа-
получали бомбардировкой урана ядрами 14N и 16О соответственно:
2381] _L 14TVT » 24бр\, -4- 6!n- 238U 4- I6O > 248Fm 4- 6*П
Я2 ^ 71 S3 ^ Uo"> 92 ^ 8^ 100 + U0u
Наиболее долгоживущими изотопами этих элементов являются 254Es (Г, = 280
/2
сут) и 252Fm (T = 8 лет). Поскольку эти ядра склонны к спонтанному делению.
/2
эффективное время жизни Es и Fm много меньше отмеченного. Химические свой-
свойства этих элементов изучены недостаточно. Однако по поведению ионов Es(+3) и
Frn(+3) в процессе хроматографического разделения можно судить об их значи-
значительном сходстве с лантаноидами гольмием и эрбием. Кроме того, известно, что
эйнштейний и фермий соосаждается с LaFg и La(OH)g.
В 1955 г. получили 17 атомов элемента № 101 в ядерной реакции
4Не -> 256Md + ,п
Элемент был назван менделевием (Md) в честь Д.И. Менделеева. Несмотря на
исчезающе малое количество нового элемента, удалось хроматографически отде-
отделить его от других продуктов. Известны шесть изотопов менделевия с массовыми
числами 252, 264—258. Самый долгоживущий 258Md (Т. = 56 сут). Для менделе-
/2
вия характерна степень окисления +3, но под действием восстановителей Md
довольно легко переходит в состояние Md(+2), которое для него более характер-
характерно, чем для европия.
В 1957 г. интернациональная группа ученых, работавшия в Нобелевском инс-
институте (Стокгольм), объявила об открытии элемента № 102 по реакции
24|Ст
515
ная) и +4. Прокачиванием на воздухе ионита, содержащего сорбированный Вк,
получен ВкОг, который легко восстанавливается водородом до ВкгОд. Известны
галогениды ВкГз (для всех галогенов) и BkF4. По своим химическим свойствам
Вк является аналогом тербия и в то же время горизонтальным аналогом кюрия.
Элемент № 98 — калифорний Cf — также был получен путем бомбардировки
(^-частицами. В качестве мишени служил 242Ст:
242fm 4- 4Не > 245Of 4- 'п
Известны изотопы калифорния с массовыми числами 244—254, а самый устойчи-
устойчивый — 249Cf (Т. . = 400 лет). По немногим существующим данным можно судить,
/2
что Cf — активный металл (E°i+/nf = -2,32 В), проявляющий степень окисления
+3 и являющийся аналогом лантаноидов и предшествующих кюридов. Для него
известны оксид СГгОз, все галогениды CiTg, оксогалогениды СЮГ, сульфид
Cf2S3.
Следующие за калифорнием элементы № 99 (эйнштейний Es) и № 100 (фермий
Fm) впервые были открыты в продуктах термоядерного взрыва A952). Уже одно
это говорит о сложности синтеза ядер этих элементов. В самом деле, например,
синтез ядра эйнштейния путем последовательного захвата нейтронов ядрами 238U
возможен в результате захвата 15 нейтронов и семикратного /3-распада образую-
образующихся продуктов. Поэтому в дальнейшем изотопы эйнштейния и фермия получа-
получали бомбардировкой урана ядрами 14N и 1бО соответственно:
238U + I4N —> 246Es + 61П; 238U + ,60 —> ?48Fm + 61П
Наиболее долгоживущими изотопами этих элементов являются 254Es (Т.. — 80
/2
сут) и 252Fm (Tj . =8 лет). Поскольку эти ядра склонны к спонтанному делению,
эффективное время жизни Es и Fm много меньше отмеченного. Химические свой-
свойства этих элементов изучены недостаточно. Однако по поведению ионов Es(+3) и
Fm(+3) в процессе хроматографического разделения можно судить об их значи-
значительном сходстве с лантаноидами гольмием и эрбием. Кроме того, известно, что
эйнштейний и фермий соосаждается с LaFs и Ьа(ОН)з-
В 1955 г. получили 17 атомов элемента № 101 в ядерной реакции
253fV i 4Не к 256МЛ 4- >п
39 ^ 2 1011V1U ' 0
Элемент был назван менделевием (Md) в честь Д.И. Менделеева. Несмотря на
исчезающе малое количество нового элемента, удалось хроматографически отде-
отделить его от других продуктов. Известны шесть изотопов менделевия с массовыми
числами 252, 264—258. Самый долгоживущий 258Md {Т{ . = 56 сут). Для менделе-
менделевия характерна степень окисления +3, но под действием восстановителей Md
довольно легко переходит в состояние Md(+2), которое для него более характер-
характерно, чем для европия.
В 1957 г. интернациональная группа ученых, работавшия в Нобелевском инс-
институте (Стокгольм), объявила об открытии элемента № 102 по реакции
244Cm + I3C > 253Э + 41!!
515
Этот элемент был назван нобелием A02N0) в честь Нобеля. В том же 1957 г. совет-
советские ученые в Дубне осуществили реакцию
241 P.,
94
160
FoP
Подобная же реакция была воспроизведена в Беркли в 1958 г. Позже, в 1961 г.,
там же был получен еще один изотоп элемента № 102:
246Г)т I 12П
254'^
102
В 1963 г. иа ускорителе в Дубне была осуществлена реакция
25Off
9В
5
1* ' °0П
Там же путем бомбардировки урана ядрами неона был получен изотоп элемента
№ 102 с массовым числом 256. Таким образом, в настоящее время вопрос о при-
приоритете в открытии элемента №- 102 окончательно не решен. Для исследования
химических свойств этого элемента используют два наиболее доступных изотопа:
( Т, . =. 3 мин) и 259No
=1,5 ч). На стедовых количествах элемента
установлено, что наиболее характерной его степенью окисления является +2 и в
этом состоянии он напоминает щслочно-земельныр металлы. В частности, он
сооеаждяется вместе с BaF2 (и RaF2). В состояние окисления +3 он переходит
под действием сильных окислителей. Эти данные говорят о том, что нобелий
является аналогом иттербия.
С использованием современной техники ускорения тяжелых ионов в 1961 г.
осуществлено взаимодействие
25Off I 10R ь 257q 4- Ч'п
Обнаруженный элемент J№ 103 было предложено назвать лоуренсием (Lr) в честь
американского физика Лоуренса — изобретателя циклотрона. В настоящее время
известны изотопы Lr с массовыми числами 255—260. Последний является самым
долгоживущим (Г. =180 с). Установлено, что Lr имеет единственную степень
/2
окисления +3 и является аналогом лютеция.
Таким образом, синтез тяжелых заурановых элементов (кюридов) и изучение
их свойств показали, что наиболее характерной степенью окисления у них явля-
является +3 с некоторыми вариациями (до -j-4 и +2) у ряда элементов, т.е. наблюда-
наблюдается аналогия с элементами второй семерки лантаноидов.
3. Трансактиниды. В соответствии с актинидной гипотезой последним из Ъ{-
элементов, как ожидалось, должен быть элемент № 103 — лоуренсий E/46rf'7s2).
Принципиально важным, с этой точки зрения, должно было оказаться открытие
элемента № 104 и изучение его свойств, что позволило бы подтвердить либо
опровергнуть актинидную гипотезу. Этот элемент был синтезирован в 1964 г. в
Дубне группой ученых под руководством Г.Н. Флерова:
94* U "*" 10 * 104^U "*" ^0П
Элемент был назван курчатовием (Ки) в честь выдающегося советского ученого и
организатора науки акад. И.В. Курчатова. К настоящему времени получены
516
изотопы курчатовия с массовыми числами 257—261. Их периоды полураспада от
11 мин у 258Ки до 70 с у 26IKu. Несмотря на то что в первых опытах было получе-
получено всего 37 атомов этого элемента, чешский ученый И. Звара, работавший в
группе Флерова, идентифицировал новый элемент и исследовал его свойства с
помощью специальных экспрессных методов анализа. Было показано, что курча-
товий резко отличается по свойствам от предыдущих элементов. Так, легколету-
легколетучий хлорид курчатовия подобен тетрахлоридам циркония и гафния. Подобно
гафнию, курчатовий ведет себя и при хроматографическом разделении. Таким
образом, курчатовий является тяжелым аналогом гафния и элементом IVB-rpyn-
пы; его электронная конфигурация [Rn]5/;16(f27s2.
С открытием курчатовия стато ясно, что он, не являясь актинидом, продолжа-
продолжает семейство Grf-элементов в 7-м периоде. Курчатовий и бо.чее тяжелые элементы
можно назвать трансактинидами.
В 1967—1970 гг. в Дубне был синтезирован элемент № 105:
243 Дгп i 22\Р 1 2613 -I- 4>П
Первооткрыватели предложили назвать этот элемент нильсборнем (N.s) в честь
Бора. Американские ученые, также получившие этот элемент на полгода позже,
назвали его ганий (На) в честь Гана. Сейчас известны изотопы 20uNs — -('-Ns.
Последний — самый долгоживущий G" = 40 с).
В 1974 г. параллельно в СССР (май) и США (сентябрь) был синтезирован
элемент № 106 по реакциям
2О'РЬ + «Сг —> 2vp + 2'n (СССР)
252Cf + 160 —> Цр + Ци (США)
И наконец, в 1975 г. вновь в Дубне группой Флерова была осуществлена иденти-
идентификация нескольких ядер элемента N"- 107. полученного по реакции
МП1 + 24^' 107^ + ^0И
Несмотря на то что химические свойства этих элементов пока не изучены,
можно ожидать, что элементы 105—107 являются аналогами тантала, вольфрама и
рения, хотя различие между 6rf-. 7s- и 7р-оболочкамп, как показывает расчет,
невелико и однозначно предсказать их свойства затруднительно.
Успехи в синтезе трансурановых элементов и синтез трансактинидов (Ки.
105—107) поставили впрямую вопрос о верхней границе Периодической системы.
Эта проблема привлекала внимание ученых в течение длительного времени. Еще
Д.И. Менделеев, исправив атомные массы тория и урана, поместил их в IVB- и
VIB-группы. Синтез нептуния и плутония позволил выделить в проблеме конца
системы два аспекта: о естественной границе и о возможном пределе синтеза
искусственных элементов. Первый аспект в земных условиях решается просто:
последним элементом в их естественной последовательности является уран. Одна-
Однако, учитывая возможность самопроизвольного синтеза Np и Ри при воздействии
на природный уран нейтронов (за счет космических лучей, естественных процес-
процессов деления), можно полагать, что на Земле последним природным элементом
является плутоний. Если же рассматривать Периодический закон в космическом
масштабе, то проблема естественного конца системы становится неоднозначной и
517
непосредственно смыкается со вторым аспектом пределом устойчивости атом-
атомных ядер. В самом деле, на определенной стадии эволюции звезд возникают
условия, при которых возможен синтез элементов вплоть до калифорния.
Достижения современной ядерной физики и химии позволяют более опреде-
определенно судить о возможностях синтеза новых искусственных сверхтяжелых элемен-
элементов. Эта проблема также неоднозначна. С одной стороны, последовательное уве-
увеличение числа протонов в ядре приводит к более резкому возрастанию числа
нейтронов (атомная масса растет быстрее, чем атомный номер) и нестабильность
тяжелых ядер должна увеличиваться с ростом числа нейтронов, вплоть до невоз-
невозможности их существования. С другой стороны, оболочечная модель ядра пред-
предполагает наличие полностью завершенных нуклонных слоев. Такие завершенные
нуклонные оболочки обладают повышенной стабильностью. На этом основано
представление о так называемых "островках стабильности" среди сверхтяжелых
элементов, ближайший из которых должен находиться вблизи Z — 126, т.е. соот-
соответствующие ядра должны обладать сравнительно высокой устойчивостью.
В определении верхней границы системы помимо физических проблем, свя-
связанных со стабильностью ядер, существуют и химические проблемы, обусловлен-
обусловленные строением и энергетикой валентных электронных оболочек. Уже у актинидов
была отмечена конкуренция 5/- и б^-орбиталей, что приводит к многообразию и
сравнимой стабильности различных степеней окисления элементов. У элементов
№ 105—107 конкурируют &d-, 7 s (и даже 7р-)-оболочки. Эта тенденция должна
быть еще более ярко выражена в 8-м периоде.
Структура 7-го периода в общих чертах представляется достаточно определен-
определенной. Он, как и 6-й период, должен содержать 32 .элемента и заканчиваться эле-
элементом N° 118 ("экарадон"). Однако уже здесь трудно предсказать химические
свойства и принадлежность вновь открываемых элементов к тем или иным груп-
группам Периодической системы из-за неопределенности порядка заполнения 6rf- и
7р-орбиталей. Можно лишь констатировать, что все эти элементы (за исключени-
исключением, может быть, экарадона) будут металлами.
Еще сложнее обстоит дело с предсказанием свойств элементов 8-го периода.
По правилу Клечковского, начиная с элемента № 121 должны заполняться впер-
впервые появляющиеся кайносимметричные б^-орбитали, вмещающие 18 электронов.
Эта оболочка для элементов 8-го периода является четвертой снаружи, что не
имеет аналогии среди известных элементов. Уже одно это обстоятельство затруд-
затрудняет прогнозирование свойств ^-элементов. Можно лишь ожидать, что они будут
еще более похожи по свойствам между собой, чем лантаноиды. А принимая во
внимание очень небольшое различие в энергиях Ъд-, 6/-, Id-, 8s и т.д. оболочек и
связанную с этим неопределенность в последовательности их заполнения, можно
предположить наличие исключительно сильно выраженной горизонтальной ана-
аналогии. Кроме того, не исключена возможность одновременного существования
элемента в различных степенях окисления и реализации пока не известных степе-
степеней окисления +9, +10 и т.д.
Таким образом, проблема верхней границы Периодической системы многопла-
нова. Здесь химия тесно смыкается с физикой. Решение этой проблемы, крайне
важной с методологической точки зрения, возможно только совместными уси-
усилиями ученых разных специальностей с использованием всех возможностей техни-
техники настоящего и будущего.
ЛИТЕРАТУРА
Ахмегов Н.С. Общая и неорганическая химия. — М.: Высшая школа, 1988.
Годовиков А.А. Периодическая система Д.И. Мендетее^ч и силовые характеристики
элементов. — Новосибирск: Наука. 1981.
Дей М., Селбин Д. Теоретическая неорганическая химия. — М.' Мир. 1976.
Дикерсон Р., Грей Г., Хейт Дж. Основные законы химии. Т. 1, 2. - М.: Мир. 1982.
Кемпбел Дж. Современная общая химия. Т. 1—3. — М.: Мир, 1975.
Коллонг Р. Нестехиометрия. — М.: Мир, 1974.
Коттон Ф., Уилкинсон Дж. Основы неорганической химии. — М.: Мир, 1979.
Кукушкин Ю.Н. Химия координационных соединений. — М.: Высшая школа. 1985.
Полинг Л. Общая химия. — М.: Мир, 1974.
Некрасов Б.В. Основы общей химии. Т.1, 2. — М.: Химия, 1973.
Реми Г. Курс неорганической химии. — М.: Мир. Т. 1. — 1963; Т. 2. — 1966.
Третьяков Ю.Д. Химия нестехиометрических окислов. — М.: Изд-во МГУ, 1974.
Угай Я.А. Обшая химия. — М.: Высшая школа, 1984.
Угай Я.А. Неорганическая химия. — М.: Высшая школа, 1989.
Угай Я.А. Введение в химию полупроводников. — М.: Высшая школа. 1975.
Хьюи Дж. Неорганическая химия (строение вещества и реакционная способность).
— М.: Химия, 1987.
Щукарев С.А. Неорганическая химия. — М.: Высшая школа. Т.1. — 1970; Т. 2. — 1972.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Авогадро закон 11
Автокатализ 460
Автокаталитические реакции 460
Автокомплексы 345
Аквакислоты 497
Аквакомплексы 106, 454, 464
Активность 155, 156
Активный комплекс 136
Акцептор 72
Алюмогель 334
Амфолиты 159, 286, 406
Аналогия групповая 227
- горизонтальная 236
- диагональная 236, 237
- контракционная 233
- слоевая 232
- типовая 228
- электронная 228
Аррениуса уравнение 135
Атомная масса 11, 12
Атомные орбитали 30, 31
Аффинаж 496
Ацидокомплексы 106, 502
Бертоллиды 206, 262
Бора теория строения атома 24
Боразол 330
Боразон 329
Боранаты 329
Бораны 87, 328
Бренстеда теория 158
Бутлерова закон 9
Валентность 56
- координационная 103
Ванадилы 428
Ван-дер-Ваальса силы 98, 245
Вернера координационная теория 103
Вода тяжелая 301
Водка царская 408
Водорода пероксид 301
Водородная связь 100, 298, 460
Водородный показатель 160
Волновые функции 29
Восстановитель 170
Газы благородные 482, 484
- инертные 482
Галогеноводороды 470
Гесса закон 125
Гетеродесмическая структура 100
Гиббса правило фаз 194
Гибридизация орбиталей 80, 83
Гидразин 401
Гидроксиламин 401
Гидразин 401
Гидроксокомплексы 106, 385, 491
Гидропероксиды 303
Гомогенности области 207, 208
Гунда правило 40
Дальтона атомистика 10
Дальтониды 206, 262
Действующих масс закон 131
Дейтерий 293
Диаграмма состояния 193
Диборан 87
Диоксигенил-катион 486, 500
Дисперсные системы 145
Диспропорционирование 172, 254, 404, 405,
486
Донор 72
Закон периодический 8, 9, 225
Зонная теория 188
520
Идеальный раствор 148
Изотопная масса 12
Изоолектронные ряды 258, 259
Икосаэдр 244
Индивид химический 21
Индукция химическая 133
Интергалогениды 462
Ионное произведение воды 160
Ионный потенциал 156, 317, 350, 354
Кайносимметрия 228, 231
Каолин 377, 378
Карбамид 365
Карбин 359
Карбонилы 361, 481, 494
Катализаторы 141
Квантовая механика 27
Квантовые числа 31, 34
Квасцы 336
Кинетика химическая 129
Кислота азотистоводородная 402
- азотноватистая 404
- бромноватая 471
- гремучая 365
- динадсерная 441
- дитионовая 438
- железная 491
- золотохлористоводородная 314
- криптоновая 488
- пиросерная 439
- пиросернистая 457
- пирофосфористая 437
- пирофосфорная 413
- полиборная 327
- поликремниевая 374
- политионовая 438
- синильная 364
- сульфоксиловая 439
- теллуровая 446
- тетраборная 327
- тиосерная 439
- хлорная 466
- хлористая 465
- хлорноватая 466
- циановая 365
Кластеры 453, 479
Клатраты 485
Конпропорционирование 392, 404, 437
Координационное число 103
Кратных отношений закон 19
Криоскопическая константа 150
Крист&шшческого поля теория 115
Кристаллохимическое строение 20, 257
Кюриды 513, 516
Лавуазье - Лапласа закон 125
Лантаноидная контракция 232, 350
Лантаноиды 346
Ле Шателье принцип 138. 140
Лиганды 104
Льюиса теория 159
Металлиды 16, 210
Металлическая связь 94
Металлохимические свойства 210
Металлохимия 208
Метод валентных связей 67, 71, 114
- молекулярных орбиталей 88
Мочевина 365
Музера - Пирсона правило 261
Надкислоты 303
Надоснования 303
Неорганический бензол 330
Неорганические полимеры 15
Нернста уравнение 178
Нестойкости константа 108
Нитрозил-ион 406
Нитрозоний 406
Нитроил-ион 287
Озон 433
Озониды 265, 435
Окислительно-восстановительные реакции
170
- - потенциалы 182
Оксоний-ион 298
Октета правило 260
Ониевые ионы 298
Осмотическое давление 151
Оствальда закон разбавления 154
Паули принцип 38
Периодическая система 23, 39, 230, 240,
292, 357, 401, 502, 518
521
Периодичность внутренняя 233, 351
- вторичная 233, 234
Перксенаты 487
Пниктогены 247, 276
Полисульфиды 442
Полупроводники 188
Принцип непрерывности 200
- соответствия 200
Произведение растворимости 162
Промотирование 71
Проникновения эффект 50
Протолитические реакции 464
Разупорядочение янтиструктурное 264
Растворы твердые 197, 201
Редокс-потенциал 415, 465
Ситаллы 378. 379
Сингулярные точки 205
Слейтера метод 37
Соединения 18. 21
- анионоизбыточные 261
- включения 485
- внутрикомплексные 109
- высшего порядка 103
- гомоатомные 239
- интерметаллические 279, 281
- катионоизбыточные 261
- комплексные 103, 290
- металлические 16
- металлоподобные 298
- переменного состава 255, 262
- постоянного состава 16, 262
- сверхкомплексные 107
- характеристические 256, 265, 268, 272
- химическое 21
Солевой эффект 163
Снектрохимический ряд 117
Сродство к электрону 45, 51
Стандартные электродные потенциалы 176
Стеклообразное состояние 187
Супероксиды 265, 435
Термодинамика химическая 7, 121
Тетраборан 328
Тиокислоты 274
Точечные дефекты 206
Трансактиниды 517
Трансвлияние Ш
Триады горизонтальные 482
Углерод 356
Уран 506
Уранила соли 509
Фаза 15, 185
- внедрения 217
- двусторонняя 264, 285
- Курнакова 208
- Лавеса 217
- линейная 204, 207
- односторонняя 264
- переменного состава 207
- промежуточная 205, 207
Фарадея законы 184
Ферми энергия 97
Ферраты 191
Ферриты 491
Фононы 97, 249
Фосфонитрилхлорид 416
Френкепя дефект 264
Фтороний-ион 298
Фуллерит 359
Фульминаты 366
Хартри - Фока метод 37
Хелатный эффект 109
Хелаты 108
Химия 5
- квантовая 66
- неорганическая 6
- общая 6
Хромил-катион 454
Хромит 452
Циан 363
Цианаты 365
Цинтля граница 210, 242, 257, 261, 277
Цирконил-ион 394
Шениты 319
Ширина запрещенной зоны 191
Шредингера волновое уравнение 28
522
Шоттки дефект 264
Шпинели 334
Эбулиоскопическая константа. 150
Эвтектика 196
Эквивалентов закон 19
- электрохимический 184
Экранирования эффект 49
Электрический момент диполя 61
Электролиз 183
Электронная плотность 33
Электронные облака 33
Электроны пегьязьгаающие 90
- разрыхляющие 89
- связывающие 90
Электроотрицательность 75
- относительная (ОЭО) 76, 267
Элементы химические 8
- редкоземельные 346, 349
- типические 229
- трансурановые 516
Энтальпия 124, 201, 435
Энтропия 128, 202
Юм-Розери правило 242
- электронные соединения 219
ОГЛАВЛЕНИЕ
Предисловие 4
ЧАСТЬ I. ОБЩАЯ ХИМИЯ
Глава I. Фундаментальные законы и теории химии 5
1. Определение химии E). 2. Общая химия. Неорганическая химия F). 3. Фун-
Фундаментальные теории и законы химии G)
Глава II. Химическая атомистика. Основные законы и понятия 10
1. Атомистика Дальтона A0). 2. Газовые законы химии A1). 3. Атомная масса.
Молекулярная масса. Молярная масса A1). 4. Структура неорганических веществ
A2).5. Неорганические полимеры A5). 6. Фаза A5). 7. Постоянный и переменный
состав. Формульная масса A6). 8. Ограниченный характер и границы примени-
применимости стехиометрических законов химии. Современная формулировка стехиомет-
рических законов A8). 9. Закон постоянства свойств. Кристаллохимическое строе-
строение и свойства A9). 10 Химический индивид. Химическое соединение B1)
Глава III. Строение атома и Периодическая система элементов
1. Модели строения атома B3). 2. Строение атома по Бору B4). 3. О квантовой
механике B7). 4. Основы квантово-механического рассмотрения атома водорода.
Орбитали C0). 5. Квантовые числа C4). 6. Многоэлектронные атомы C6). 7. Пе-
Периодическая система ачементов и атектронная структура атомов C9). 8. Строение
электронной оболочки и свойства элементов D5)
Глава IV. Химическая связь
1. Химическая связь и валентность E6). 2. Энергия химической связи E7). 3.
Длина химической связи E8). 4. Электрический момент диполя и направленность
связи F0). 5. О ионной связи F2). 6. Ковалентная связь F5). 7. Понятие о кван-
квантовой химии F6). 8. Метод валентных связей (МВС) F7). 9. Валентность и МВС
G1). 10. Насыщаемость ковалентной связи G4). 11. Поляризация химической свя-
связи G5). 12. Направленность ковалентной связи G9). 13. Кратность химической
связи (84). 14. О связях с избытком и дефицитом валентных электронов (86). 15.
Понятие о методе молекулярных орбиталей (88). 16. Сравнение МВС и ММО (93).
17. Металлическая связь (94). 18. Химическая связь в твердых неорганических
веществах (97)
Глава V. Межмолекулярное взаимодействие и комллексообразование
1. Силы Ван-дер-Ваальса (98). 2. Водородная связь A00). 3. Комплексные соеди-
соединения. Координационная теория Вернера A03). 4. Номенклатура комплексных
23
56
соединений A04). 5. Классификация комплексных соединений A06). 6. Устойчи-
Устойчивость комплексных соединений. Константа нестойкости. Двойные соли A07). 7.
Хелаты и внутри комплексные соединения A08). 8. Изомерия комплексных соеди-
соединений (ПО). 9. Трансвлияние A11). 10. Природа химической связи в комплексных
соединениях (ИЗ)
Глава VI. Учение о химических процессах 121
1. Понятие о химической термодинамике A21). 2. Экзо- и эндотермические реак-
реакции. Основы термохимии A24). 3. Направление химических процессов. Энтропия.
Свободная энергия A27). 4. Понятие о химической кинетике. Скорость химичес-
химических реакций A29). 5. Основной закон химической кинетики A30). 6. Параллель-
Параллельные, последовательные, сопряженные и цепные реакции A31). 7. Зависимость ско-
скорости реакции от температуры. Энергия активации A34). 8. Обратимые химичес-
химические реакции. Химическое равновесие A36). 9- Смещение химического равновесия.
Принцип Ле Шателье A38). 10 Понятие о катализе. Гомогенный и гетерогенный
катализ A41)
Глава VII. Жидкое состояние. Растворы 144
1. Жидкое состояние. Структура жидкости A44). 2. Дисперсные системы. Раство-
Растворы A45). 3. Процесс образования растворов A47). 4. Идеальный раствор. Законы
разбавленных растворов A48). 5. Электролитическая ионизация. Степень и кон-
константа ионизации A52). 6. Понятие о теорий сильных электролитов. Активность
A55). 7. Кислотно-основная ионизация A56). 8. Теории кислот и оснований A58).
9. Водородный показатель. Индикаторы A59). 10. Обменные реакции между иона-
ионами. Произведение растворимости A62). 11. Реакции нейтрализации и гидролиза
A63). 12. Окислительно-восстановительные реакции A70). 13. Гетерогенные реак-
реакции в растворах A74). 14. Химические источники тока. Электролиз A81)
Глава VIII. Твердое состояние. Твердые растворы 185
1. Понятие о твердой фазе A85). 2. Кристаллическое, стеклообразное, аморфное
состояния A86). 3. Представление о зонной теории. Металлы, полупроводники,
изоляторы A88). 4. Основы физико-химического анализа A92). 5. Типы диаграмм
состояния A94). 6. Твердые растворы B01). 7. Соединения постоянного и пере-
менног состава. Дальтониды и бертоллиды B04)
Глава IX. Металлохимия 208
1. Элементохимия B08). 2. Металлохимические свойства элементов B10). 3. При-
Примитивные типы химического взаимодействия в металлических системах B12). 4.
Образование соединений в металлических системах B14). 5. Металлохимические
свойства и диаграммы состояния B21)
ЧАСТЬ II. НЕОРГАНИЧЕСКАЯ ХИМИЯ
Раздел 1. Периодический закон как основа химической систематики 225
Глава X. Структура Периодической системы 225
1. Этапы развития Периодического закона B25). 2 Групповая и типовая аналогии
B27). 3. Электронная аналогия. Кайносимметрия B28). 4. Переходные металлы.
Контракционная аналогия B32). 5. Орбитальные радиусы. Вторичная и внутрен-
внутренняя периодичность B33). 6. Горизонтальная и диагональная аналогии B36)
Глава XI. Простые вещества как гомоатоыные соединения
1. Химическое и кристаллохимическое строение простых веществ B39). 2. Метал-
Металлы и неметаллы в Периодической системе B41). 3. Физические свойства простых
веществ B44). 4. Химические свойства простых веществ B49). 5. Нахождение в
природе и общие принципы получения простых веществ B50). 6. Особо чистые
вещества B54)
239
Глава XII. Бинарные химические соединения
1. Классификация бинарных соединений B56). 2. Кристаллохимическое строение
сиды B65). 5. Водородные соединения B68). 6. Галогениды B71). 7. Халькогени-
ды B73). 8. Пниктогениды B75). 9. Карбиды, силициды, бориды B77), 10. Интер-
Интерметаллические соединения B79)
Глава XIII. Сложные химические соединения
1. Классификация сложных соединений B80). 2. Гидроксиды как характеристи-
характеристические соединения B82). 3. Кислотно-основные свойства. Амфотерность гидрокси-
дов B83). 4. Окислительно-восстановительные свойства гидроксидов B87). 5. Со-
Соли кислородсодержащих кислот B89). 6- Комплексные соединения B90)
255
279
Раздел 2. Химия элементов .
292
292
Глава XIV. Водород
1. Уникальное положение водорода в Периодической системе B92). 2. Изотопы во-
водорода B93). 3 Атомарный и молекулярный водород B94). 4. Физические и хими-
химические свойства водорода B95). 5. Гидриды и летучие водородные соединения
B96). 6. Получение водорода B99). 7. Вода B99). 8. Пероксид водорода C01)
Глава XV. Элементы I группы 303
1. Литий C04). 2. Щелочные металлы C07). 3. Подгруппа меди C10)
Глава XVI. Элементы II группы 315
1. Бериллий C15). 2. Магний C18). 3. Щелочно-земельные металлы C20). 4. Под-
Подгруппа цинка C22)
Глава XVII. Элементы III группы 325
1. Бор C25). 2. Алюминий C31). 3. Подгруппа галлия C37). 4. Подгруппа скандия
и РЗЭ C46)
Глава XVIII. Элементы IV группы * 355
1. Углерод C56). 2. Кремний C69). 3. Подгруппа германия C79). 4. Подгруппа ти-
титана C90)
Глава ХГХ. Элементы V группы 396
1. Азот C97). 2. Фосфор D09). 3. Подгруппа мышьяка D17). 4. Подгруппа вана-
ванадия D26)
Глава XX. Элементы VI группы 432
1. Кислород D32). 2. Сера D35). 3. Подгруппа селена D43). 4. Подгруппа хрома
D48)
Глава XXI. Элементы VII группы 456
1. Фтор D57). 2. Хлор DСЗ). 3. Подгруппа брома D68). 4. Подгруппа марганца
D73)
Глава XXII. Элементы VIII группы 482
1. Элементы VIIIA-группы D83). 2. Металлы триады железа D88). 3. Платиновые
металлы D95)
Глава XXIII. Радиоактивные и синтезированные элементы 501
1. Радиоактивные аналоги стабильных элементов Периодической системы E02). 2.
Металлы семейства актинидов E04). 3. Трансактиниды E16)
Литература 519
Предметный указатель 520
Оглавление 524