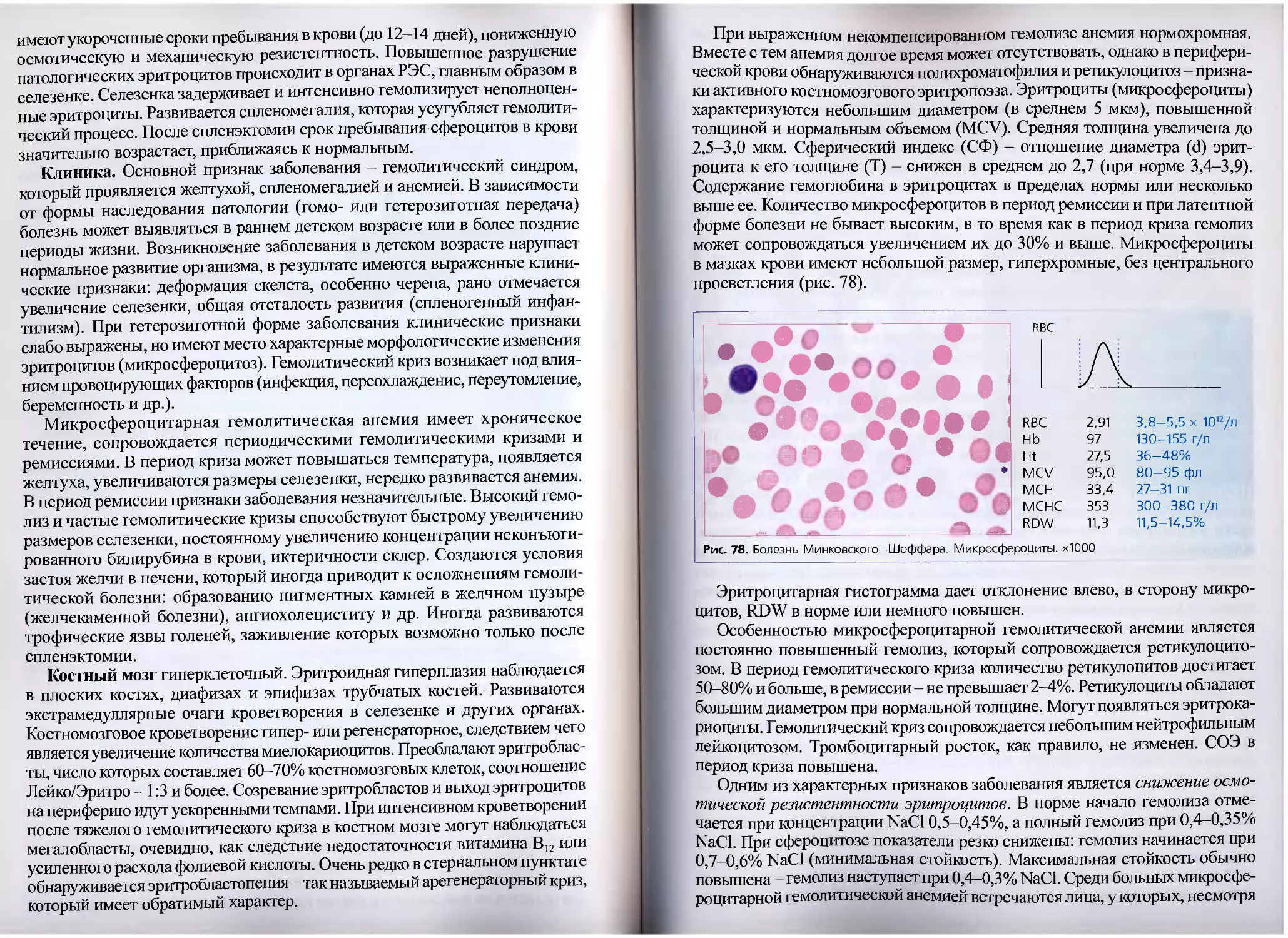
/













































































Author: Луговская С.А. Почтарь М.Е. Долгов В.В. Морозова В.Т.
Tags: патология сердечно-сосудистой системы сердечно-сосудистые заболевания внутренние болезни медицина
ISBN: 978-5-94789-340-3
Year: 2009
Similar




Text
В.В. Долгов, С.А. Луговская, В.Т. Морозова, М.Е. Почтарь
КАФЕДРА
КАД
Лабораторная диагностика
АНЕМИЙ
ВТОРОЕ ИЗДАНИЕ
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ
РОССИЙСКОЙ ФЕДЕРАЦИИ
РОССИЙСКАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ ПОСЛЕДИПЛОМНОГО ОБРАЗОВАНИЯ
В.В. Долгов, С.А. Луговская, В.Т. Морозова, М.Е. Почтарь
Лабораторная диагностика
АНЕМИЙ
ВТОРОЕ ИЗДАНИЕ
КАФЕДРА
Москва 2009
КАД
долгов
Владимир
Владимирович
доктор
медицинских наук,
профессор,
заведующий
кафедрой
клинической
лабораторной
диагностики
РМАПО
ЛУГОВСКАЯ
Светлана
Алексеевна
доктор
медицинских наук,
профессор кафедры
клинической
лабораторной
диагностики
РМАПО
УДК 616.155.194-07
ББК 54.11
Д64
Долгов В.В., Луговская С.А.,
Морозова В.Т., Почтарь М.Е.
Д64 Лабораторная диагностика анемий, -
2-е изд., доп. - М.-Тверь: ООО «Изда-
тельство «Триада», 2009. - 148 с., 92 ил.
ISBN 978-5-94789-340-3
fl
МОРОЗОВА
Виктория
Тазаретовна
заслуженный
деятель науки РФ,
доктор
медицинских наук,
профессор кафедры
клинической
лабораторной
диагностики РМАПО
ПОЧТАРЬ
Маргарита
Евгеньевна
кандидат
медицинских наук,
доцент кафедры
клинической
лабораторной
диагностики
РМАПО
Второе издание руководства переработа-
но с учетом последних достижений в об-
ласти биохимических и гематологических
исследований в лабораторной диагностике.
Книга продолжает серию учебных матери-
алов, подготовленных на основе опыта пре-
подавания клинической лабораторной диа-
гностики врачам циклов переподготовки и
повышения квалификации.
В руководстве большое внимание уде-
лено патогенезу отдельных форм анемий,
на основе которого строятся алгоритмы
обследования больных, их диагностика,
представлены традиционные и новые мето-
ды оценки анемий, в том числе результаты
лабораторного исследования с использова-
нием гематологических анализаторов. При-
ведены справочные материалы и обширное
количество иллюстраций в виде микрофо-
тографий, полученных как при микроско-
пии препаратов крови и костного мозга, так
и при работе на гематологических анализа-
торах.
Книга предназначена для врачей клини-
ческой лабораторной диагностики, широко-
го круга врачей, использующих результаты
лабораторного анализа в своей практике, и
студентов медицинских институтов.
ББК 54.11
© В.В. Долгов. С.А. Луговская,
В,Т. Морозова. М.Е. Почтарь. 2009
© Оформление ООО «Издательство «Триада», 2009
ISBN 978-5-94789-340-3
СОДЕРЖАНИЕ
ОБЩИЕ ПРЕДСТАВЛЕНИЯ О КРОВЕТВОРЕНИИ.......................5
ЭРИТРОПОЭЗ................................................7
Регуляция эритропоэза...............................12
Эритроциты..........................................13
Основные характеристики эритроцитов.................17
Номенклатура клеток эритроидного ряда...............21
Морфология клеток эритропоэза.......................22
ОСНОВНЫЕ ЛАБОРАТОРНЫЕ ПОКАЗАТЕЛИ ОЦЕНКИ
ЭРИТРОПОЭЗА, ПОЛУЧАЕМЫЕ НА ГЕМАТОЛОГИЧЕСКИХ
АНАЛИЗАТОРАХ.............................................25
ОПРЕДЕЛЕНИЕ И КЛАССИФИКАЦИЯ АНЕМИЙ.......................36
АНЕМИИ ВСЛЕДСТВИЕ КРОВОПОТЕРИ............................39
Острая постгеморрагическая анемия...................39
Хроническая постгеморрагическая анемия..............43
АНЕМИИ, ОБУСЛОВЛЕННЫЕ НЕДОСТАТОЧНОСТЬЮ
ЭРИТРОПОЭЗА..............................................43
Обмен железа........................................43
Всасывание железа...............................47
Транспорт железа в крови........................49
Внутриклеточный метаболизм железа...............51
Депонирование железа............................53
Регуляция баланса железа........................54
Выведение железа................................57
Гипохромные анемии..................................58
Железодефицитная анемия.........................58
Анемии, связанные с нарушением синтеза порфиринов.68
Нормохромные анемии.................................72
Анемия хронических заболеваний..................72
Анемия при хронической почечной недостаточности.76
Апластические анемии............................79
11арциальная красноклеточная аплазия................S3
Анемии при злокачественных образованиях.............85
Гиперхромные (мегалобластные) анемии.....................86
Анемии, обусловленные дефицитом витамина В12........86
Фолиеводефицитные анемии............................98
АНЕМИИ ВСЛЕДСТВИЕ УСИЛЕННОГО РАЗРУШЕНИЯ
ЭРИТРОЦИТОВ (ГЕМОЛИТИЧЕСКИЕ АНЕМИИ)..........................100
Гемолиз эритроцитов.....................................100
Внутриклеточный гемолиз............................100
Внутрисосудистый гемолиз...........................102
Дифференциально-диагностические признаки
внутриклеточного и внутрисосудистого гемолиза......104
Анемии, обусловленные внеэритроцитарными
гемолитическими факторами...............................107
Иммунные гемолитические анемии.....................107
Аутоиммунные гемолитические анемии.................111
Пароксизмальная холодовая гемоглобинурия
с двухфазными гемолизинами
(анемия Доната-Ландштейнера).......................116
Гемолитическая анемия, обусловленная соматической
мутацией клеток-предшественников миелопоэза........116
Анемии, обусловленные эритроцитарными факторами.........120
Гемолитические анемии, связанные с нарушением
структуры мембраны эритроцитов (эритроцитопатии)...120
Гемолитические анемии, обусловленные дефицитом
ферментов эритроцитов (эритроцитарные энзимопатии).130
Гемолитические анемии, связанные с нарушением
синтеза глобина (гемоглобинопатии).................137
ОСНОВНАЯ ЛИТЕРАТУРА..........................................146
Гемопоэз - это сбалансированная, непрерывно обновляющаяся система,
подчиняющаяся строгим механизмам регуляции, направленным на поддер-
жание равновесия между образованием клеток и их разрушением. Отли-
чительной чертой гемопоэза является разнообразие как видов клеток, их
функций, морфологии, продолжительности жизни, так и места пребывания
в организме.
Основной закон кроветворения - сохранение постоянства количественного
и качественного состава различных клеточных ростков. Конечной целью
кроветворения является образование зрелых функционально полноценных
гемопоэтических клеток.
Пролиферация, дифференцировка и апоптоз - генетически заложенные
программы, предопределяющие существование и функционирование всех
клеток организма, в том числе и клеток крови. Каждый из этих процессов
имеет свои механизмы, регулируется определенными цитокинами, ростовы-
ми факторами, генами. В регуляции процессов пролиферации и дифферен-
цировки гемопоэтических клеток большую роль играет стромальное мик-
роокружение. К стромальным элементам костного мозга относят клеточный
компонент: фибробласты, жировые клетки, макрофаги, остеобласты, эндо-
телиальные клетки и внеклеточный (экстрацеллюлярный) матрикс, который
составляют продукты секреции стромальных клеток (коллаген, фибронектин,
ламинин, гликозаминогликаны, тенасцин и другие белковые компоненты).
Внеклеточный матрикс обеспечивает специфическое прилипание стволовых
кроветворных клеток (СКК). Стромальные клетки секретируют большое
количество регулирующих факторов, без которых невозможна пролиферация
СКК, дифференцировка и функционирование клеток. Для гранулоцитопоэза
важен непосредственный контакт предшественников с фибробластами и их
производными - жировыми клетками, для эритропоэза - с макрофагами.
Пролиферация и дифференцировка клеток крови в костном мозге про-
исходят параллельно. Пролиферация осуществляется митотическим путем,
который контролируется как внешними (цитокины, ростовые факторы), так
и внутренними факторами (циклины, циклинзависимые киназы, транскрип-
ционные факторы и др.). Созревание клетки идет непрерывно, постепенно
замедляется синтез ДНК вплоть до его прекращения, что делает зрелую
дифференцированную клетку неспособной к делению. Пролиферативный
пул костного мозга представлен исключительно молодыми, способными к
делению кроветворными клетками.
Важным физиологическим регулятором гемопоэза является апоптоз. Учи-
тывая огромную продукцию клеток крови (около 5-7 тонн клеток в течение
всей жизни), для поддержания клеточного равновесия и гомеостаза должен
существовать механизм удаления избыточных, поврежденных и старых
клеток. Этим механизмом является апоптоз. Программа самоуничтожения
клетки, которая осуществляется различными внешними и внутренними
сигналами, уравновешивается программой ее блокирования. Дифференци-
ровка гемопоэтических клеток возможна только при условии их выживания,
цля чего необходимы антиапоптотические факторы. Самыми мощными
антиапоптотическими стимулами для нормального кроветворения являются
ростовые факторы. Диапазон их действия достаточно широк, начиная со
стволовой клетки и заканчивая зрелыми элементами, которым они обеспечи-
вают нормальное функционирование. Любые нарушения в апоптотической
системе могут приводить к нежелательным последствиям, нарушая гомеостаз
любой клеточной системы, что часто лежит в основе патогенеза различных
заболеваний.
Структуру кроветворных органов (костный мозг, тимус, селезенка, лим-
фатические узлы) представляют соединительная ткань, паренхима, сосуды,
которые являются общими составляющими паренхиматозных органов.
Основная масса паренхимы состоит из специализированных клеток. Парен-
хима - субстанция нестабильная, в ней процессы образования новых клеток
сменяются их гибелью (апоптоз). Другими словами, для сохранения клеточ-
ного равновесия постоянно происходят альтернативные процессы - одни
клетки заканчивают свой жизненный цикл, другие приходят им на смену.
Зона наиболее активного кроветворения примыкает к эндосту, связывая
костномозговое микроокружение с костной тканью. Зоны активной клеточ-
ной пролиферации отделены от зон дифференцировки, а по мере созревания
клетки из одних зон перемещаются в другие. Миграция клеток из костного
мозга через стенку синусов в периферическую кровь имеет, очевидно, свой
избирательный специфический механизм.
Особенности кроветворения. Отличительными особенностями эмб-
рионального гемопоэза являются наличие периодов, сопровождающихся
сменой его топографии в органах, в которых осуществляется кроветворение,
неодинаковый клеточный состав кроветворной паренхимы и микроокру-
жения, изменение их по мере развития плода. Одни органы включаются
в кроветворение, другие - выключаются. Этой последовательной смене
кроветворной территории сопутствует появление в крови новых клеточных
форм, вида гемоглобина, антигенов и т. д.
В желточном мешке микроокружение СКК составляет мезенхимальный
эндотелий его мезодермальной стенки. В печени появляются клетки эпи-
телия из энтодермы. Микроокружение СКК в селезенке, костном мозге,
лимфатических узлах представлено ретикулярной тканью мезенхимального
происхождения. До 7-го месяца эмбрионального развития в селезенке и
лимфатических узлах имеет место универсальное кроветворение. Затем по
мере развития структура их усложняется, изменяется микроокружение, они
становятся органами лимфопоэза и теряют способность к миелопоэзу.
Следовательно, существует органная специализация кроветворного
микроокружения (соединительной ткани органа), которая определяет
четкую дифференцировку ткани органов гемопоэза в постнатальном периоде.
Однако при некоторых патологических ситуациях миелоидное кроветворение
может вновь возникнуть в селезенке, печени и лимфатических узлах.
Особенность кроветворения взрослых состоит в территориальной его
раздробленности, вследствие чего СКК перемещаются по определенным
территориям и в зависимости от структуры микроокружения оседают в тех
органах кроветворения, где их дальнейшую дифференцировку определяет
стромальная ткань. Ответ органов кроветворения, которые работают как
один слаженный механизм на различные факторы (естественную убыль
клеток, кровопотерю, гемолиз, инфекции и другие), может быть как за счет
изменения количества клеток, так и их морфологии и функции.
В нормальных условиях кроветворные клетки (миелоидные и лимфоид-
ные) обновляются за счет СКК, ранее заселивших органы, а также за счет
вновь поступающих в него предшественников. От действия местных гемо-
поэтических факторов зависит вхождение СКК в митоз, размеры и число
колоний, а также вид клеточного потомства. Особенность костномозгового
кроветворения состоит также в замещении активного костного мозга на
жировой костный мозг, которое происходит по мере роста и развития орга-
низма.
Территории, пригодные для заселения кроветворными клетками, создает
строма кроветворных органов. Элементы стромы - ретикулярные клетки,
фибробласты, имеющие крайне низкий темп обновления, ретикулиновые
волокна (образующие сеть) - гетерогенны и имеют сложную организацию.
От клеток микроокружения зависит реализация дифференцировочных и про-
лиферативных возможностей гемопоэтических клеток-предшественников.
Регулирующее влияние микроокружения на кроветворение подтверждает
гетеротропная трансплантация гемопоэтической ткани, при которой проис-
ходит перенос клеток микроокружения, т. е. «каркас кроветворного органа
является донорским».
ЭРИТРОПОЭЗ
Процесс образования и созревания эритроидных клеток в костном мозге
называется эритропоэзом. Эритроциты - наиболее многочисленная фракция
крови. Они образуют многофункциональную систему жизнеобеспечения ор-
ганизма, которая, благодаря наличию в эритроцитах гемоглобина, участвует
в транспорте кислорода к тканям и выведении углекислого газа из организ-
ма, обеспечивая стабильность внутренней среды организма. У взрослого
человека в физиологических условиях циркулирующий пул эритроцитов
составляет 25 30 х 1012 клеток (около 2 кг). При продолжительности жизни
эритроцита 90-120 дней костный мозг должен продуцировать в течение часа
порядка 1010 клеток. Число эритроцитов в организме регулируется скоростью
их образования и разрушения.
Система, объединяющая самые ранние предшественники эритроидного
ряда, морфологически идентифицируемые пролиферирующие и непролифе-
рирующие ядросодержащие клетки, ретикулоциты и эритроциты, обознача-
ется термином - эритрон.
Кинетика клеток эритропоэза. В норме количество циркулирующих
эритроцитов поддерживается на постоянном уровне. У взрослого человека с
массой тела 70 кг в костном мозге ежедневно продуцируется 20-25 х 1010 но-
вых эритроцитов. Такое количество клеток необходимо для поддержания
нормального уровня гемоглобина. Развитие эритроидных клеток осуществля-
ется процессами дифференцировки и созревания. Схема дифференцировки
клеток эритропоэза представлена на рис. 1.
Родоначальными клетками эритропоэза являются частично детермини-
рованные миелоидные предшественники (КОЕ-ГЭММ). Они образуются из
стволовой полипотентной клетки, претерпевая 5-10 делений. Коммитирован-
ные монопотентные клетки-предшественники, бурстобразующие единицы
эритропоэза (БОЕ-Э) гетерогенны по своему составу, проходят 11-12 деле-
ний. Их различия определяются степенью дифференцировки клеток. В этот
период 60-65% клеток находится в митозе. По мере созревания клеток число
делений сокращается. КОЕ-Э дифференцируются в проэритробласты - самые
ранние морфологически идентифицируемые костномозговые предшест-
венники эритроцитов, способные к синтезу гемоглобина. Проэритробласт
в течение 3-5 суток подвергается дальнейшей дифференцировке, проходит
стадии базофильного, полихроматофильного и оксифильного эритробласта.
На этом этапе функционирования эритрона клетки проходят до 7 митотичес-
ких делений. Однако число делений может сокращаться, что сопровождается
уменьшением количества эритроцитов, срока их созревания и увеличением
размера клетки. Этот процесс называется «перескок деления». Полихрома-
тофильный эритробласт - последняя делящаяся клетка в эритроидном ряду,
созревающая в оксифильный эритробласт.
Процесс дифференцировки эритроидных клеток характеризуется пос-
тепенным уменьшением размеров клеток, ядра, конденсацией хроматина,
накоплением гемоглобина в цитоплазме. Ядро оксифильного нормобласта
превращается в плотную, пикнотичную массу, выталкивается из цитоплазмы
при прохождении клетки через узкие эндотелиальные отверстия в синусоидах
костного мозга. После удаления ядра из нормобластов образуются костно-
мозговые ретикулоциты, в которых продолжается синтез гемоглобина еще
в течение 3—4 суток.
Ретикулоцит Эритроцит
Стволовая
клетка
БОЕ-Э Проэритробласт
КОЕ-Э ЭРИТРОПОЭТИН
Эритробласты
Нормобласты
Витамин
^12
+ --------► ДНК
фолиевая
кислота
Рис. 1. Дифференцировка и регуляция клеток эритропоэза
Продукция ретикулоцитов в костном мозге составляет 3 х 109 клеток в
сутки. Образовавшиеся ретикулоциты созревают в костном мозге в течение
36 —44 часов, после чего поступают в кровь, где дозревают в течение 24—30 ча-
сов. Незрелые ретикулоциты имеют большое количество РНК-содержащих
структур (рибосомы) и, несмотря на отсутствие ДНК, способны синтезиро-
вать гемоглобин, липиды, пурины. В митохондриях ретикулоцитов синтез
АТФ осуществляется за счет использования кислорода, одновременно в этих
клетках протекает и анаэробный гликолиз. Ретикулоцит имеет на поверхности
те же молекулы, что и зрелый эритроцит, включая гликофорин А, антигены
группы крови и системы резус, абсорбирует молекулы железа благодаря
рецепторам к трансферрину, плотность которых более выражена у менее
зрелых ретикулоцитов.
Диаметр ретикулоцитов составляет 7,7 8,5 мкм. Средний объем рети-
кулоцитов на 24—35% больше эритроцитов (101-128 фл), а концентрация
гемоглобина в них ниже, чем в зрелом эритроците, что объясняет появление
гипохромных макроцитов в периферической крови при состояниях, сопровож-
дающихся ретикулоцитозом. Многие энзимы (пируваткиназа, глюкозо-6-фос-
фатдегидрогеназа, каталаза, ангидраза), а также другие компоненты (креатин)
содержатся в ретикулоцитах в более высоких концентрациях, чем в зрелых
эритроцитах. В процессе созревания ретикулоцитов происходит дезагрегация
полирибосом на отдельные рибосомы. Постепенно клетка освобождается от
рибосом, утрачивает митохондрии, места связывания с трансферрином, и син-
тез гемоглобина прекращается. Характерной морфологической особенностью
ретикулоцитов является наличие в цитоплазме зернисто-сетчатой субстанции,
представляющей собой остатки рибосом, выявляемой при суправитальном
методе окраски. В различных ретикулоцитах она отличается полиморфизмом;
чем клетка моложе, тем субстанция более обильная.
В зависимости от степени зрелости в соответствии с классификацией
Гейльмейера, предложенной более 60 лет назад, выделяют 5 групп рети-
кулоцитов (рис. 2). В норме в периферической крови обнаруживаются
преимущественно ретикулоциты III—IV групп (около 61% ретикулоцитов
относится к IV группе, 32% - к III, 7% - ко II и только около 0,1% - к
I группе). Левый сдвиг ретикулоцитов в сторону незрелых клеток (0-1-
II группы) на фоне ретикулоцитоза имеет место при активации эритро-
поэза. Нормальное количество ретикулоцитов в периферической крови
здорового взрослого человека колеблется в пределах 0,2-1,2%. Ретику-
лоцитоз отражает повышенную регенераторную способность костного
мозга. Сохраняющийся ретикулоцитоз может свидетельствовать о про-
должающемся кровотечении, гемолизе. Ретикулоцитопения - индикатор
угнетения эритропоэза.
Количество ретикулоцитов отражает скорость продукции эритроцитов
в костном мозге, поэтому их подсчет имеет значение для оценки степени
активности эритропоэза.
После трансформации ретикулоцитов в эритроциты клетки сохраняют
набор ферментов, необходимых для поддержания молекул гемоглобина в
форме, способной к обратимой оксигенации.
О группа
I группа
II группа III группа iv группа
Ядросодержащие
эритроидные
клетки с густой
ретикулофила-
ментозной сетью
в центре
(оксифильный
нормобласт)
Ретикулоциты
с грубой
шарообразной
сетью
в центре
Ретикулоциты
с менее густой
сетью,
распространенной
по всей
цитоплазме
Клетки
с обрывками
ретикулофила-
ментозной сети
в разных
участках
цитоплазмы
Ретикулоциты
с единичными
нитями
или гранулами
ретикулофиламен-
тозной сети
в отдельных
участках
цитоплазмы
Рис. 2. Распределение ретикулоцитов по степени зрелости
В целом дифференцировка и созревание эритроидных клеток, начиная с
проэритробласта до эритроцита, осуществляются в течение 9-14 дней.
В 1960-1966 гг. Lajtha и Oliver была предложена кинетическая модель
эритрона, основанная на результатах исследований эритропоэза. Результаты
этих исследований легли в основу концепции Stholmann (1967 г.), согласно
которой определенная концентрация гемоглобина (НЬ) выключает клетку из
митотического цикла. Схема кинетической модели эритрона представлена
на рис. 3.
Синтез гемоглобина в эритрокариоцитах костного мозга начинается
на ранней стадии развития эритробласта и заканчивается в ретикулоците
с исчезновением последней рибосомы. Скорость синтеза гемоглобина в
проэритробластах и базофильных эритробластах составляет 0,5 пг в час в
1 клетке. В делящихся клетках после митоза количество гемоглобина умень-
шается наполовину, в течение интерфазы приближается к исходному уровню.
К концу второго митотического цикла (перед делением) клетки содержат
21,6 пг гемоглобина, а в разделившихся дочерних клетках, которые по своей
морфологии являются базофильными эритробластами, по 10,8 пг. В конце
митоза количество гемоглобина в базофильном эритробласте составляет
25,2 пг, а у образовавшихся из него ранних полихроматофильных эритро-
бластов - 13 пг. После деления раннего полихроматофильного эритробласта
образуются средние полихроматофильные эритробласты с концентрацией
гемоглобина внутри клетки, достигающей критической величины - 13,5 пг.
При этом прекращается синтез ДНК, клетка выключается из митотического
цикла и скорость синтеза гемоглобина замедляется. Дальнейшее созревание
клеток красного ряда происходит без деления.
При нормальном эритропоэзе эритрокариоциты проходят в среднем
5 митозов, в результате чего из 1 эритробласта получается 32 эритроцита с
количеством НЬ 27-31 пг.
10
I
1
Рис. 3. Схема эритропоэза по С.И. Рябову: I - нормальный тип эритропоэза; II — терминаль
ный тип. III — неэффективный эритропоэз; 1 — стволовой предшественник; 2 — эритробласт;
3 — пронормобласт; 4 — базофильные нормобласты; 5 — полихроматофильные нормобласты;
6 - оксифильные нормобласты; 7 — ретикулоциты; 8 - зрелые эритроциты
В небольшой популяции эритроидных клеток синтез гемоглобина осу-
ществляется быстрее, и на стадии раннего полихроматофильного эритро-
бласта клетка подходит к митозу с количеством гемоглобина более 27 пг,
при этом она теряет способность к делению. Дальнейшее развитие этой
тетраплоидпой клетки происходит без деления. Из нее образуется крупный
ретикулоцит и затем макроэритроцит, содержащий более 30 пг гемоглоби-
на. Этот тип деления эритрокариоцитов получил название терминального.
В норме терминальный эритропоэз составляет не более 5%. Наличие его дает
возможность быстро регулировать количество эритроцитов в зависимости
от различных физиологических состояний.
5-10% эритрокариоцитов, достигнув критической массы гемоглобина
(27 пг) на стадии базофильного эритробласта, гибнут в костном мозге,
uv1zj,'innzizivo эсишпам anuiuuou ииниш 1pvii(J33y. Jt5 фИ-З*14-'”
логических условиях неэффективный эритропоэз - один из факторов
регуляции эритрона, поддержания необходимого количества эритроцитов
в крови. Для оценки величины неэффективного эритропоэза может быть
использован цитохимический метод определения количества PAS-положи-
тельных эритрокариоцитов. В костном мозге здорового человека их число
не превышает 3-8%. Увеличение объема неэффективного эритропоэза,
возможно, свидетельствует о накоплении или увеличении клеток с оши-
бочной дифференцировочной или пролиферативной программой. Такие
клетки подлежат элиминации посредством физиологической гибели, поэ-
тому уровень неэффективного эритропоэза отражает интенсивность
апоптоза.
В норме на долю эритрокариоцитов костного мозга приходится 20-30%
всех ядросодержащих клеток. Этот показатель может быть выражен отно-
шением количества лейкоцитов к эритрокариоцитам (или Л/Э), в норме оно
составляет 3 (4) : 1.
Регуляция эритропоэза
Поддержание нормального состава эритрона находится под контролем
различных механизмов, основными из которых являются давление кислорода
в тканях и секреция эритропоэтина. Уровень оксигенации тканей зависит от
интенсивности кровотока, концентрации гемоглобина, степени его насыще-
ния кислородом и сродства гемоглобина к кислороду.
Центральная роль в регуляции эритропоэза принадлежит эритропоэтину
(ЭПО). Уменьшение снабжения тканей кислородом или увеличение пот-
ребности в кислороде стимулирует продукцию ЭПО и, наоборот, избыток
кислорода в тканях (гипероксия) подавляет образование гормона. Ранние
предшественники эритропоэза (БОЕ-Э) характеризуются низкой чувстви-
тельностью к действию ЭПО и требуют для своего роста присутствия спе-
циального стимулятора эритропоэза - бурстстимулирующей активности. По
мере дифференцировки БОЕ-Э появляются более зрелые предшественники,
приобретающие чувствительность к ЭПО. Дифференцированные клетки-
предшественники - колониеобразующие единицы эритропоэза (КОЕ-Э) -
отличаются максимальной чувствительностью к ЭПО. Тканевая гипоксия
приводит к избыточному синтезу ЭПО, который стимулирует пролиферацию
БОЕ-Э, увеличение общего содержания эритрокариоцитов в костном мозге
и ретикулоцитов в периферической крови (рис. 4).
Образование эритробластов происходит только в присутствии достаточной
концентрации эндогенного ЭПО, в противном случае клетки подвергаются
гибели (апоптозу). Воздействие ЭПО осуществляется через эритропоэтин-
чувствительные клеточные рецепторы, количество которых максимально на
клетках КОЕ-Э, проэритробластах и базофильных эритробластах. Связыва-
ние ЭПО с соответствующим рецептором предотвращает апоптоз клеток.
В регуляции эритропоэза помимо ЭПО принимают участие фактор ство-
ловых клеток, ИЛ-3, ГМ-КСФ, витамин В12, фолиевая кислота, витамин В6,
микроэлементы (железо, медь, цинк, селен и др.), гормоны (тироксин, анд-
рогены, кортикостероиды, гормоны роста).
Начальные этапы эритропоэза характеризуются появлением на мембране
клеток HLAI и II классов, CD34, рецептора к трансферрину (CD71), раннего
миелоидного антигена CD33 и Rh-антигенов, а также небольшим количеством
рецепторов к ЭПО. Более зрелые предшественники — КОЕ-Э — отличаются
высокой чувствительностью к ЭПО и экспрессией максимального количества
эритроидных маркеров: рецепторов к ЭПО, специфического протеина - гли-
кофорина А и рецепторов к трансферрину. Гликофорин А - гликопротеин
эритроцитарной мембраны, предотвращает агрегацию эритроцитов.
Эритроциты
Эритроциты - самая многочисленная популяция
клеток крови. Эритроцит в покое имеет форму двоя-
ковогнутого диска, которая увеличивает его площадь
и обеспечивает наибольшую поверхность газообмена
(рис. 5).
Диаметр наибольшего числа эритроцитов составляет
7,2-7,5 мкм, площадь поверхности - 140 мкм2. объем -
90 мкм3 (фл). Высокая пластичность и деформируемость
мембраны позволяют эритроциту проходить через ка-
рие. 5. Эритроцит
пилляры диаметром 2-5 мкм, через стенки синусоидов, диаметр пор которых в
селезенке имеет 0,5-0,7 мкм, и затем возвращаться к исходным параметрам.
13
Клетка покрыта двухслойной полупроницаемой мембраной (плазмолем-
мой), в двойной слой фосфолипидов которой встроены белки гликопротеины,
углеводная часть их образует надмембранный слой - гликокаликс. Клетка
имеет спектрин-актиновый цитоскелет, большая часть белков которого также
составляют фосфолипиды. Клеточная мембрана и цитоскелет определяют
архитектонику эритроцита и его способность к деформации при прохожде-
нии микроциркуляторного русла. Гликопротеиновые комплексы мембраны
организованы таким образом, что озрицательно заряженные участки (чаще
сиаловые группы полисахаридов) обращены наружу, придавая поверхности
эритроцитов отрицательный заряд.
Внутренняя сторона мембраны эритроцитов связана с сетью миофиламент-
ных белков, формирующих спектрин-актиновый цитоскелет, придающий
эритроциту специфическую двояковогнутую форму (рис. 6).
В ячейках цитоскелета находятся молекулы гемоглобина. По данным
Т.С. Истомановой и соавт., число молекул НЬ в 1 эритроците составляет
около 280 млн, по данным А.А. Липаца - 400 млн.
Гемоглобин (НЬ) - дыхательный пигмент, сложный белок - хромопротеид.
Его небелковая часть (простатическая группа), включающая железо, называ-
ется гемом, белковый компонент - глобином. На долю глобина приходится
96% сухого веса НЬ, на долю гема - 4%. Молекула НЬ имеет 4 гема. Благодаря
присутствию в составе гема иона железа, гемоглобин переносит кислород
от легочных альвеол к тканям и осуществляет транспортировку углекислого
газа от тканей к легким.
Синтез гемоглобина начинается на самой ранней стадии развития
эритроидных элементов. При его нарушении содержание НЬ в эритроците
Рис. 6. Схема мембраны эритроцита
снижается, ячейки цитоскелета остаются не заполнены НЬ, что проявляется
гипохромией эритроцитов в мазках крови и повышением в них концентра-
ции неиспользованного на синтез порфирина. Мм гемоглобина - 64-66 кДа.
В эритроцитах взрослых людей 95-98% приходится на Hb A (adult - взрос-
лый), 2-3% - на НЬ А2,1-2% - на HbF (fetus - плод). Гемоглобин F у новорож-
денных составляет 70-90%, но к концу первого года жизни его количество
резко снижается. HbF может присутствовать не во всех эритроцитах.
Эритроцитарные антигены. На мембране эритроцита имеется более
250 антигенов, которые располагаются подобно мозаике. Роль антигенов
заключается в регуляции дифференцировки и созревания клеток. Наибо-
лее изучены антигены систем АВО и резус. Резус-антигены находятся на
мембране эритроцитов независимо от пола и возраста, не связаны с изо-
антигенами А, В и др., равномерно распределяясь во всех группах крови.
Эритроцитарные антигены не меняются в течение жизни человека и могут
тысячелетиями сохраняться у трупов при низких температурах и неопре-
деленно долго при высыхании. Распад клетки приводит к потере ею специ-
фической антигенности. Системы эритроцитарных антигенов наследуются
обычно независимо друг от друга. Ген, определяющий группу крови системы
АВО, обладает тремя аллелями - в виде антигенов А, В и отсутствия этих
антигенов в виде третьей формы - 0. Антигены А и В являются кодоми-
нантными и доминируют над 0-антигеном. Резус-антигены при передаче их
от обоих родителей могут проявляться в двойной дозе (гомозиготы) или в
простой дозе (гетерозиготы) при передаче от одного из родителей. Например,
антигены группы А (II) - в виде АА или АО, резус-антигены - CDE/CDE
или CDE/cDe. Антигены А и В впервые обнаруживаются па 37-й день раз-
вития эмбриона. Формирование резус-антигенов начинается с 3—4-го месяца
внутриутробного развития.
Мембрана эритроцита выполняет разные функции: барьерную, транс-
портную, сорбционную (особенно в отношении микроэлементов), ме-
таболическую благодаря наличию ферментных систем, регулирующих
энергетические и окислительные процессы, транспорт ионов, перекисное
окисление липидов, генерирующих и утилизирующих активные формы
кислорода.
Метаболизм эритроцита. Эритроцит переносит кислород, но сам
его не использует для образования собственной энергии, потому что к
моменту созревания клетки он теряет митохондрии и рибосомы, в нем
исчезают процессы, связанные с образованием энергии путем утилизации
кислорода. АТФ в эритроцитах образуется исключительно в процессе
анаэробного гликолиза, при этом очень активно используется глюкоза.
Эритроциты, составляя около 3% массы тела, в день потребляют при-
близительно 20 г (10%) всего количества глюкозы, поступающей в орга-
низм. Процесс гликолиза ферментативный, в нем участвует более десяти
ферментов. При их недостаточности эритроциты быстро разрушаются.
В процессе гликолиза формируются продукты (соединения), которые
снижают сродство гемоглобина к кислороду, способствуя отдаче его тка-
ням. Следующие метаболические пути обеспечивают функциональную
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
I
активность эритроцитов: 1) гликолиз; 2) пентозофосфатный цикл; 3) путь
Раппопорта-Либеринга - образования 2,3-дифосфоглицерата (2,3-ДФГ);
4) глутатионовый путь; 5) АТФ-зависимый катионный насос на мембране
эритроцита; 7) ферменты, влияющие на состав фосфолипидов эритроци-
тарной мембраны; 8) метгемоглобинредуктаза, превращающая метгемог-
лобин (MetHb) в гемоглобин (НЬ).
Последовательность основных этапов гликолиза и ферменты, катализи-
рующие этот процесс, представлены на рис. 7.
В эритроцитах, в отличие от других тканей, в процессе гликолиза об-
разуется много 2,3-дифосфоглицериновой кислоты, которая, соединяясь с
В-цепями глобина, снижает сродство гемоглобина к кислороду, способствуя
тем отдаче его тканям.
Из всех ферментов, обеспечивающих гликолиз, чаще других встречается
наследственный дефицит пируваткиназы, которая катализирует его энергооб-
разующую стадию, реже глюкозофосфатизомеразы. Нарушения активности
других ферментов очень редки. Конечные продукты гликолиза - молочная
Рис. 7. Гликолиз и сопряженные с ним метаболические процессы, протекающие в эритроци-
тах. ПФП — пентозофосфатный путь (шунт); РЛП — Раппопорта—Либеринга путь образования
2,3-ДФГ
кислота и АТФ. Прекращение гликолиза приводит эритроцит к «метаболи-
ческой смерти» - процессу, который заканчивается гемолизом.
Кроме того, до 30% глюкозы эритроцита используется в пентозофос-
фатном цикле, результатом которого является синтез НАДФН2 — основного
метаболита, обеспечивающего функционирование антиоксидантных систем
в эритроците. Эта система эритроцита предупреждает чрезмерное накопле-
ние свободных радикалов двухвалентного железа и кислорода, способных
вызвать активацию перекисного окисления липидов эритроцитарной мем-
браны и его гемолиз.
Избыточное образование продуктов перекисного окисления и глики-
рование клеточных структур оказывают цитотоксическое повреждающее
действие на мембраны эритроцитов и эндотелий сосудов. Конечные про-
дукты гликирования необратимо связываются с белками клеток, изменяя
их биологические и функциональные свойства, снижая способность эрит-
роцитов к деформации, что в свою очередь приводит к застойным явлениям
в микроциркуляторном русле и, как следствие, к возникновению тканевой
гипоксии с выраженными изменениями гомеостаза и гемодинамики. Глико-
зилирование белковых структур сосудистой стенки усиливает образование
эндотелина-1, обладающего вазоконстрикторным (сосудосуживающим)
действием. Дизрегуляция ригидности сосудистой стенки и увеличение
внутрисосудистого давления обусловлены также дисфункцией мембран
эритроцитов, нарушением их способности сорбировать и транспортировать
в ткани норадреналин и ацетилхолин, участвующие в регуляции тонуса
сосудистой стенки.
Нарушение доставки кислорода крови тканя м рассматривается как по-
казатель тяжести патологического процесса и метаболических изменений
в эритроцитах и в организме, поскольку эритроциты ответственны за
кислородное обеспечение всех энергообразующих в нем процессов.
Основные характеристики эритроцитов
Деформируемость эритроцитов. Эритроциты обладают способностью
изменять свою форму - деформироваться, что обеспечивает им прохожде-
ние через капилляры. На деформируемость мембраны эритроцитов влияют
наличие кислорода в эритроцитах, интенсивность перекисного окисления
липидов, состояние антиоксидантной системы. Способность эритроцитов
к деформации определяется внутренней (цитоплазматической) вязкостью,
вязкостно-эластичными свойствами мембраны и отношением площади
клетки к ее объему.
Деформация эритроцитов сопровождается увеличением проницаемости
мембран для одновалентных катионов и кальция. В эритроцитах влияние
ионов кальция опосредуется через их взаимодействие с кальмодулином - бел-
ком, связывающим кальций, большая часть (85%) которого локализуется в
Цитоплазме, а остальное количество связано с мембраной. При деформации
Клетки повышается внутриклеточная концентрация кальция, значительное
повышение его сопровождается образованием эхиноцитов. Увеличение кон-
17
центрации кальция стимулирует его элиминацию из клетки и последующее
восстановление ее формы. Деформация требует энергии, которая поставля-
ется за счет гликолиза.
Цитоплазматическая вязкость эритроцита существенно зависит от
концентрации гемоглобина. При физиологических концентрациях это вли-
яние невелико, однако при высокой концентрации гемоглобина (предельная
концентрация НЬ 380 г/л) величина цитоплазматической вязкости эритроцита
возрастает, при этом существенно ухудшается его деформируемость. Низ-
кая способность эритроцитов к деформации, в сравнении с эритроцитами
взрослых, отмечается у новорожденных, что объясняют более высоким
содержанием гемоглобина. Внутриэритроцитарная вязкость зависит также
и от вида гемоглобина. Так, при серповидноклеточной анемии для восста-
новленной формы HbS характерно снижение растворимости в десятки раз,
что ведет к образованию геля, имеющего более высокую вязкость. Снижение
деформируемости эритроцитов имеет место и при других гемоглобинопатиях.
В условиях эритроцитоза деформируемость эритроцитов не претерпевает
существенных изменений.
Эритроциты обмениваются с внешней средой липидами и холестери-
ном, необходимыми для поддержания определенного состава мембраны и
деформируемости клеток. При увеличении в составе мембраны холесте-
рина при дис- и гиперлипопротеинемиях уменьшается деформируемость
эритроцитов.
На способность эритроцитов к деформации существенно влияют физико-
химические факторы (pH, осмолярность, газовый состав крови, температура).
Поэтому при задержке эритроцитов в синусоидах селезенки и закислении
окружающего пространства деформируемость их уменьшается, они теряют
способность проникать через выходные отверстия синусоидов и поглощаются
макрофагальными элементами. Деформируемость эритроцитов оптимальна
при pH 7,4.
Способность эритроцитов менять форму и проходить через узкие капилля-
ры повышается с увеличением отношения площади (величины) поверхности
эритроцита к его объему. Эритроциты с аномальной формой характеризуются
повышенной резистентностью к деформации.
При повышении деформируемости эритроцитов увеличивается контакт
их мембраны со стенкой капилляров и перенос кислорода между альвеолами
и эритроцитами в легких и между эритроцитами и тканями на периферии, а
при снижении деформируемости мембраны эритроцитов обмен кислорода
ухудшается.
Способность эритроцитов к деформации обусловлена особенностя-
ми клеточного скелета и структуры мембраны и может меняться при
патологических состояниях.
Эти изменения могут быть зарегистрированы с помощью такого инте-
грального показателя, как скорость оседания эритроцитов (СОЭ).
Электростатический заряд имеет решающее значение для поддержания
эритроцитов во взвешенном состоянии. Электростатические силы определя-
ют отталкивание эритроцитов друг от друга и от стенки сосудов. Эритроциты
заряжены отрицательно. В сосудистом русле они перемещаются в плазме
крови в постоянном вращательном движении, совершая вокруг своей оси
до 90 об/с. Во время движения крови эритроциты находятся в деформиро-
ванном состоянии. Наиболее выражены изменения формы эритроцитов в
микроциркуляторном русле, капилляры которого могут иметь диаметр менее
2 мкм (рис. 8).
Агрегационная способность — свойство эритроцитов создавать линейные
цепочки в виде монетных столбиков (агрегаты) - носит обратимый характер
(физиологическая агрегация). В здоровом организме непрерывно происходит
процесс «агрегации-дезагрегации», что поддерживает нормальную текучесть
крови.
В образовании агрегатов участвуют плазменные факторы, в том числе фак-
торы гемостаза, антитела, электростатические, механические и другие. Ме-
ханизм агрегации эритроцитов объясняют возникновением мостиков между
клетками с участием крупномолекупярных белков. Увеличение концентрации
фибриногена, а,-, а2-, Р-глобулинов, криоглобулинов, иммуноглобулинов в
плазме крови приводит к усилению агрегации эритроцитов. Препятствует
агрегации отрицательный заряд эритроцитов и альбумины, конкурирующие
за сорбционные центры с высокомолекулярными белками, но не создающие
мостики между эритроцитами. Наиболее частой причиной нарушения агре-
гационных свойств эритроцитов является изменение величины соотношения
альбумины/глобулины. Об усиленной агрегации эритроцитов может свиде-
тельствовать ускорение СОЭ.
Особое значение агрегация эритроцитов и их способность к деформации
приобретают в микроциркуляторном звене кровообращения, где имеются
функциональные и анатомические предпосылки к стазу крови.
Микровезикуляция. В течение жизни эритроцита имеет место необрати-
мое уменьшение его поверхности в результате процесса микровезикуляции
(экзовезикуляции), которая обеспечивает удаление поврежденных участков
Рис. 8. Деформация эритроцитов позволяет им проходить сквозь узкие места и увеличивает
способность к газообмену с окружающей тканью и поддерживает оптимальную диффузию
газов
1
цитоплазматической мембраны. Обновление фосфолипидного состава бислоя
происходит в результате выпячивания (наружу) мембраны с образованием
микровезикулы и ее слущивания.
Повреждение мембран в результате активации процессов перекисного
окисления липидов при различных патологических состояниях приводит к
усилению процесса микровезикуляции, который становится патологичес-
ким. На внешнюю поверхность мембраны выходят внутриэритроцитарные
фосфолипиды, обладающие тромбопластиновой активностью. Эритроциты
с разрушенными мембранами образуют в микрососудах эритроцитарные
агрегаты, вокруг которых появляется большое количество тромбоцитов и
пучков фибрина. Появление в кровотоке гемолизированных форм и фраг-
ментов эритроцитарных мембран вызывает усиление процессов скрытого
внутрисосудистого свертывания крови.
Лабораторные критерии патологической микровезикуляции - транс-
формация эритроцитов из дискоцитов в эхиноциты и снижение их
деформируемости.
Старение эритроцита. По мере старения клетки деформируемость эрит-
роцитов претерпевает значительные изменения, что связано с уменьшением
эластичности мембран и увеличением цитоплазматической вязкости клетки,
а также со снижением величины отношения поверхности эритроцита к его
объему. С течением времени объем клетки уменьшается, но содержание
гемоглобина в клетке практически не меняется. Однако его концентрация
соответственно растет и составляет в старых эритроцитах около 380 г/л.
У старых эритроцитов отмечается модификация биохимических свойств мем-
бран, что наряду с другими факторами может вести к повышению активности
фагоцитоза эритроцитов макрофагами селезенки, печени и костного мозга,
в цитоплазме которых эритроцит гемолизируется или дезинтегрируется без
предварительного гемолиза.
Функции эритроцита
• Участие в газообмене благодаря способности эритроцита связывать
кислород и углекислый газ за счет высокого содержания гемоглоби-
на.
• Поддержание кислотно-основного состояния организма, поскольку ге-
моглобин - главная буферная система крови.
• Эритроциты определяют реологию крови, участвуют в гемостазе.
Основными факторами, влияющими на реологические свойства крови,
являются:
• клеточный - количество и состояние форменных элементов крови,
прежде всего эритроцитов, составляющих около 45% объема крови.
Их состояние изменяет Ht и вязкость крови. Нарушение эластичности,
деформируемости и агрегационной активности эритроцитов может
привести к образованию монетных столбиков, сладж-синдрому. Эти
факторы играют ключевую роль в формировании реологических свойств
крови;
• плазменный — высокая концентрация в плазме крупномолекулярных
белков (фибриноген и продукты его деградации, IgM, а2-макроглобу-
лин) усиливает агрегацию эритроцитов. Макромолекулярные вещества
(липопротеиды), а также наличие в крови высокого титра антител,
которые могут вызывать агглютинацию и гемолиз клеток, повышают
вязкость плазмы;
• эндотелиально-сосудистый - эндотелий продуцирует биологически
активные вещества (эндотелины, оксид азота), способные вызывать
спазм и расширение сосудов, обеспечивает повышение или пониже-
ние свертывающего потенциала крови (фактор Виллебранда, актива-
тор плазминогена), регулирует агрегационные и дезагрегационные
свойства клеток крови (простациклин, простагландины и другие).
Гликирование белковых структур эритроцитов и эндотелия сосу-
дов способствует развитию тканевой гипоксии, которая активирует
повышенное образование реактивных оксидантов, инициирующих
патологию.
• гидродинамический - замедление кровотока, турбулентность, за-
вихрение, прерывистый ток крови или другие нарушения кровотока
повышают агрегацию эритроцитов, приводят к образованию монетных
столбиков, стазу, сладж-синдрому. Обезвоживание и кровопотеря усу-
губляют нарушения динамики кровотока (текучести) и способствуют
развитию патологического процесса.
• Эритроциты участвуют в иммунных процессах, взаимодействуя с антите-
лами, циркулирующими иммунными комплексами, благодаря наличию на
мембране Fc-рецепторов к иммуноглобулинам, комплементу и большого
числа поверхностных антигенов.
Изменение функционального состояния эритроцита происходит в основ-
ном вследствие воздействия на него факторов микроокружения и старения
клетки, что сказывается на состоянии липидного бислоя мембраны, белкового
скелета клетки и ее метаболизма. Факторы микроокружения также изменяют
межклеточные взаимодействия эритроцитов, меняя реологические характе-
ристики крови, и, в конечном счете, приводят к разрушению эритроцитов в
клетках РЭС.
Таким образом, осуществляя свои функциональные способности, эрит-
роциты участвуют в поддержании гомеостаза организма.
Номенклатура клеток эритроидного ряда
На сегодняшний день не существует унифицированной номенклату-
ры клеток эритропоэза. В различных странах используются несколько
классификаций морфологически распознаваемых клеток эритроидного
Ряда. В классификации ВОЗ все клетки эритроидного ряда обозначаются
как эритробласты (проэритробласт, базофильный, полихроматофильный
и оксифильный эритробласты). На рис. 9 приведена схема эритропоэза с
Учетом терминологии клеток эритропоэза, используемой в нашей стране и
за рубежом.
21
Рис. 9. Номенклатура клеток эритропоэза в соответствии с различными классификациями
Морфология клеток эритропоэза
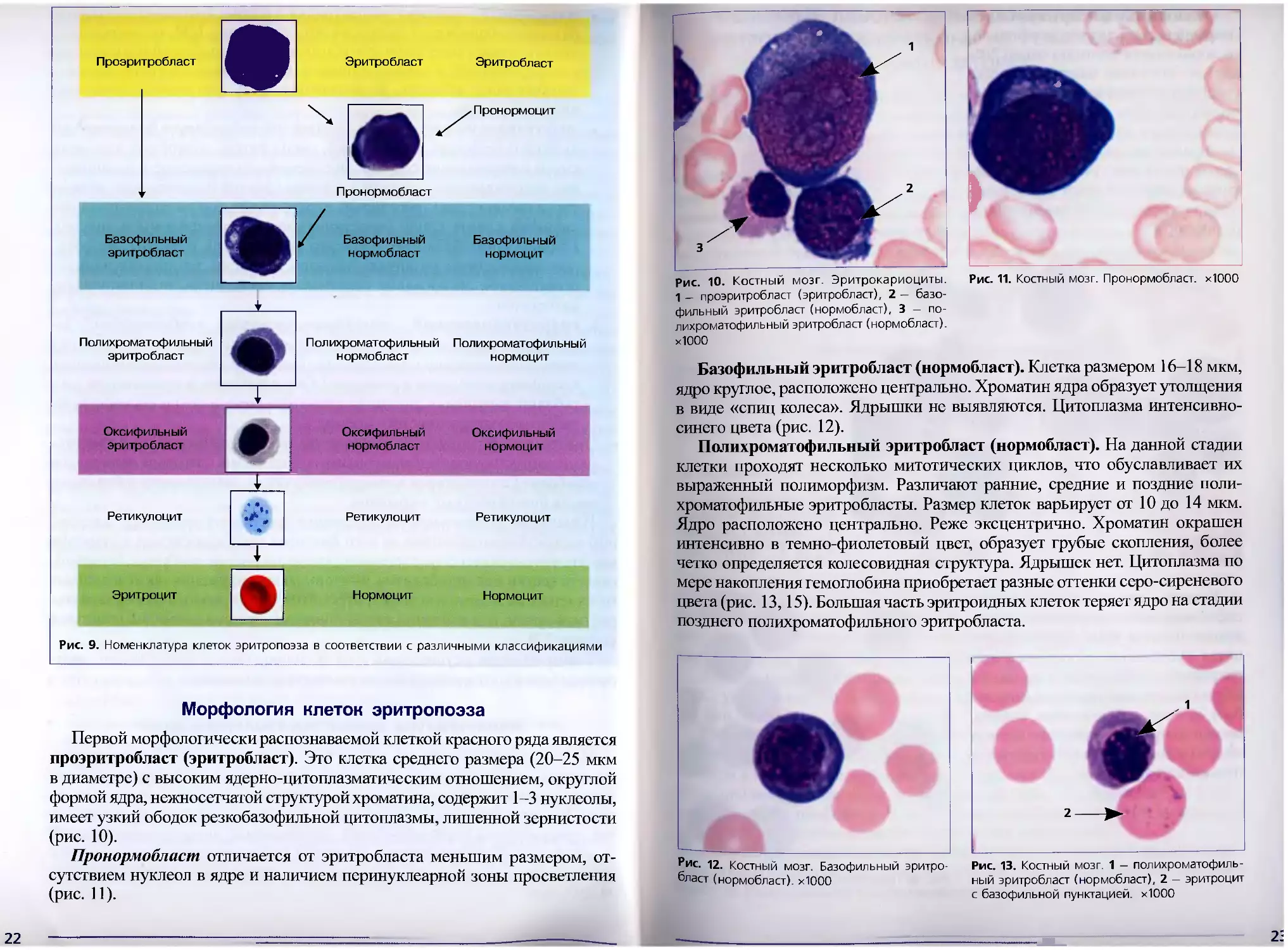
Первой морфологически распознаваемой клеткой красного ряда является
проэритробласт (эритробласт). Это клетка среднего размера (20-25 мкм
в диаметре) с высоким ядерно-цитоплазматическим отношением, округлой
формой ядра, нежносетчатой структурой хроматина, содержит 1-3 нуклеолы,
имеет узкий ободок резкобазофильной цитоплазмы, лишенной зернистости
(рис. 10).
Пронормобласт отличается от эритробласта меньшим размером, от-
сутствием нуклеол в ядре и наличием перинуклеарной зоны просветления
(рис. 11).
22
Рис. 10. Костный мозг. Эритрокариоциты.
1 - проэритробласт (эритробласт), 2 — базо-
фильный эритробласт (нормобласт), 3 - по-
лихроматофильный эритробласт (нормобласт).
хЮОО
Рис. 11. Костный мозг. Пронормобласт. хЮОО
Базофильный эритробласт (нормобласт). Клетка размером 16-18 мкм,
ядро круглое, расположено центрально. Хроматин ядра образует утолщения
в виде «спиц колеса». Ядрышки не выявляются. Цитоплазма интенсивно-
синего цвета (рис. 12).
Полихроматофильный эритробласт (нормобласт). На данной стадии
клетки проходят несколько митотических циклов, что обуславливает их
выраженный полиморфизм. Различают ранние, средние и поздние поли-
хроматофильные эритробласты. Размер клеток варьирует от 10 до 14 мкм.
Ядро расположено центрально. Реже эксцентрично. Хроматин окрашен
интенсивно в темно-фиолетовый цвет, образует грубые скопления, более
четко определяется колесовидная структура. Ядрышек нет. Цитоплазма по
мерс накопления гемоглобина приобретает разные оттенки серо-сиреневого
цвега (рис. 13,15). Большая часть эритроидных клеток теряет ядро на стадии
позднего полихроматофильного эритробласта.
Рис. 12. Костный мозг. Базофильный эритро-
бласт (нормобласт). хЮОО
Рис. 13. Костный мозг. 1 - полихроматофиль-
ный эритробласт (нормобласт), 2 - эритроцит
с базофильной пунктацией. хЮОО
Оксифильный эритробласт
(нормобласт). Стадию оксифильно-
го нормобласта проходят около 20%
клеток. Это самая маленькая клетка
среди ядросодержащих клеток крас-
ного ряда. Она имеет небольшое
пикнотичное ядро, цвет цитоплаз-
мы идентичен цвету окружающих
эритроцитов (рис. 14). После вытал-
кивания ядра из клетки нормобласт
превращается в костномозговой
ретикулоцит.
Рис. 14. Костный мозг. Оксифильный эритро-
бласт (нормобласт). хЮОО
Рис. 15. Костный мозг.
Эритроидный островок.
1 - макрофаг с гемосиде-
рином, 2 — эритробласты.
хЮОО
Ретикулоциты представляют
собой незрелые эритроциты, содер-
жащие остатки РНК. Зернисто-сет-
чатая субстанция выявляется при
суправитальной окраске, в частности
с бриллиантовым крезиловым голу-
бым, вначале в виде глыбок, а затем,
по мере созревания ретикулоцитов, в
виде клубков, сеточки или отдельных
пылинок (рис. 16).
Рис. 16. Периферическая кровь. Ретикулоциты
хЮОО
ОСНОВНЫЕ ЛАБОРАТОРНЫЕ ПОКАЗАТЕЛИ ОЦЕНКИ
ЭРИТРОПОЭЗА, ПОЛУЧАЕМЫЕ НА ГЕМАТОЛОГИЧЕСКИХ
АНАЛИЗАТОРАХ
RBC (red blood cells) - количество эритроцитов крови (х 10|2/л). Опреде-
ление количества эритроцитов осуществляется путем вычитания из общего
числа клеток в цельной крови тромбоцитов и лейкоцитов. Для исключения
из счета тромбоцитов, которые имеют существенно меньшие размеры по
сравнению с эритроцитами и лейкоцитами, используются пороговые значе-
ния. Считаются все частицы размером более 36 фл. Следует отметить, что в
некоторых гематологических анализаторах лейкоциты включаются в подсчет
вместе с эритроцитами, но их влияние в норме незначительно, т. к. количест-
во лейкоцитов существенно меньше (на 3 порядка - несколько тысяч) числа
эритроцитов (несколько миллионов). В случаях гиперлейкоцитоза ошибка
измерения эритроцитов возрастает (табл. 1).
Таблица 1
Возможные ошибки измерения RBC
Ложное завышение Ложное занижение
• Гигантские тромбоциты (с объемом более 30 фл) • Криоглобулинемия • Высокий лейкоцитоз (более 50 х 109/л) • Агглютинация эритроцитов • Выраженный микроцитоз эритроцитов • Гемолизированные образцы крови
Присутствие криоглобулинов может вызывать увеличение показателей
WBC, RBC или PLT и концентрации HGB. В таких случаях следует прогреть
образец крови до 37 °C в течение 30 минут и немедленно провести измере-
ние образца. Криоглобулинемия может наблюдаться у больных миеломой,
макроглобулинемией Вальденстрема, злокачественными новообразования-
ми, лейкозом, лимфопролиферативными и аутоиммунными заболеваниями,
вирусным гепатитом, сахарным диабетом.
Агглютинация эритроцитов может привести к занижению показателей
RBC, увеличению MCV. Это можно проверить по повышенным значениям
МСН и МСНС.
Нормобласты (NRBC): большинство гематологических анализаторов
подсчитывает все ядросодержащие клетки, поэтому при наличии нормо-
оластов в периферической крови они определяются как лейкоциты и могут
быть причиной увеличения WBC и лимфоцитов, т. к. нормобласты имеют
Размер малого лимфоцита. В этих случаях необходим строгий визуальный
контроль и коррекция истинного количества лейкоцитов.
Например', общее количество лейкоцитов при подсчете в анализаторе -
‘ю х 109/л. В лейкоцитарной формуле на 100 лейкоцитов имеется 50 нормо-
оластов. Рассчн аем истинное количество лейкоцитов в крови:
150 клеток (общее количество
лейкоцитов и нормобластов,
полученное при подсчете лейко-
цитарной формулы)
100 клеток (лейкоциты)
45 х Ю’/л (количество клеток
в 1 мкл, полученное при подсчете
в камере или на анализаторе)
X (истинный лейкоцитоз крови)
100x45 х 109/л
= 30х 109/л.
Таким образом, истинное число лейкоцитов в крови составляет 30 х 109/л.
В анализаторах фирмы Sysmex (ХЕ-2100, XT-2000i) и Bayer (ADVIA 120)
при наличии нормобластов в крови коррекция лейкоцитов проводится ав-
томатически. Следует отметить, что порог чувствительности определения
нормобластов в анализаторе Sysmex ХЕ-2100 составляет менее 20/мкл, что
с помощью микроскопического исследования определить представляется
невозможным.
Нормобласты появляются в периферической крови при онкогематологи-
ческих заболеваниях, анемиях (гемолитические, В|2- и фолиеводефицитные),
метастатическом поражении костного мозга, тяжелых септических состояни-
ях и интоксикациях. Появление нормобластов в послеоперационном периоде
является плохим прогностическим признаком, предсказывающим возможный
летальный исход. Наличие в крови нормобластов может расцениваться как
маркер гипоксии и воспаления.
HGB (hemoglobin)', концентрация гемоглобина (г/дл или г/л) в большинс-
тве гематологических анализаторов определяется фотометрически. Коэффи-
циент вариации при этом не превышает 2%. Причины получения ошибочных
результатов приведены в табл. 2.
Таблица 2
Возможные ошибки измерения HGB
Ложное завышение Ложное занижение
• Высокий лейкоцитоз (более 100 х 109/л) • Присутствие нестабильных гемоглобинов (HbS, НЬС) • Гиперлипидемия • Гипербилирубинемия • Криоглобулинемия • Гемолиз (in vivo) • Парапротеинемия • Резистентные к лизису эритроциты • Образование микросгустков в пробе крови
Повышение концентрации гемоглобина наблюдается при реактивных
и опухолевых эритроцитозах, обезвоживании. Снижение концентрации
гемоглобина имеет место при анемиях, гипергидратации.
HCT (hematocrit) - гематокрит. Показатель вычисляется как сумма прямо
измеренных объемов эритроцитов в единице объема крови и выражается в %.
Таким образом, проблемы «остаточной» плазмы (плазмы, оставшейся между
эритроцитами при центрифугировании) в гематологических анализаторах
по сравнению с гематокритной центрифугой не существует. Коэффициент
вариации для автоматического метода - менее 1% в сравнении с 1-2% при
определении показателя методом центрифугирования. В табл. 3 приведены
факторы, которые могут привести к ошибкам измерения НСТ.
Возможные ошибки измерения НСТ
Таблица 3
Ложное завышение Ложное занижение
• Гигантские тромбоциты (с объемом более 30 фл) • Криоглобулинемия • Высокий лейкоцитоз (более 50 х 109/л) • Гипергликемия (>600 мг/дл) • Диабетический кетоацидоз • Агглютинация эритроцитов • Выраженный микроцитоз эритроцитов(<36 фл)
Выраженная агглютинация эритроцитов может привести к получению
неправильных значений НСТ, т. к. агглютинаты эритроцитов могут воспри-
ниматься прибором как лейкоциты и не учитываться при расчете гематокрита.
В таких случаях рекомендуется определение гематокрита на гематокритной
центрифуге.
Повышение гематокрита наблюдается при реактивных и опухолевых
эритроцитозах, уменьшении объема циркулирующей плазмы (ожоговая бо-
лезнь, дегидратация). Снижение гематокритной величины имеет место при
анемиях, беременности (второй триместр), гипергидратации.
При гипергликемии и диабетическом кетоацидозе отмечается гипер-
осмолярность плазмы крови. При разведении крови in vitro изотоническим
раствором происходит быстрое набухание эритроцитов, что и вызывает за-
вышение НСТ. В этих случаях определение гематокрита на гематокритной
центрифуге является более точным.
Показатели гематокрита и гемоглобина являются важными параметрами
общего состояния здоровья, повышение которых, например, у спортсменов
может свидетельствовать о приеме препаратов, вызывающих экзогенную
стимуляцию костного мозга (введение эритропоэтина).
MCV (mean corpuscular volume) - средний объем эритроцита, выражает-
ся в кубических микрометрах (мкм3) или в фемтолитрах (1 фл = 1 мкм3 или
1 х 10 |5/л). MCV определяется большинством гематологических анализа-
торов благодаря прямой зависимости амплитуды электрического импульса
°т объема клетки. Вычисляется MCV делением суммы клеточных объемов
на число эритроцитов.
В то же время MCV - это средний показатель объема всей популяции
эритроцитов, содержащихся в диапазоне 36-360 фл. Поэтому необходимо
иметь в виду, что MCV может иметь нормальное значение при наличии у
пациента одновременно выраженного макро- и микроцитоза, большом коли-
честве аномальных эритроцитов (например, при серповидноклеточной ане-
мии, выраженном пойкилопитозе). В этом случае особую диагностическую
важность приобретает анализ эритроцитарной гистограммы и морфология
клеток в мазках крови. Возможные причины ошибок в измерении MCV
приведены в табл. 4.
Таблица 4
Возможные ошибки измерения МСУ
Ложное завышение Ложное занижение
• Холодовые агглютинины • Диабетический кетоацидоз • Гиперосмолярность плазмы • Гипернатриемия • Высокий лейкоцитоз (более 50 х 109/л) • Длительное хранение крови (более 8 часов) • Ретикулоцитоз • Макротромбоцитоз • Повышенное содержание фрагментов эритроцитов в крови вследствие механического гемолиза • Коагулопатия потребления
При наличии агглютинации эритроцитов прибор воспринимает их как
одну большую клетку, если размер их меньше верхнего порога эритроци-
тарного канала, что приводит к увеличению MCV. Сохранение крови in vitro
и измерение таких проб при 37 °C способствует получению правильных
результатов.
MCV является важным показателем в дифференциальной диагностике
анемий. На основании MCV анемии разделяют на нормоцитарные (MCV 80-
100 фл), микроцитарные (MCV менее 80 фл) и макроцитарные (MCV более
100 фл).
MCV - показатель, отражающий изменения, возникающие в эритроцитах
при длительном хранении крови. Изменения в мембране эритроцитов воз-
никают раньше, чем в лейкоцитах и тромбоцитах, поэтому хранение крови
более 8 часов вызывает увеличение MCV.
MCV меняется в течение жизни: у новорожденных достигает 128 фл,
в первую неделю снижается до 100-112 фл, к году составляет 77-79 фл, в
возрасте 4-3 лет нижняя граница нормы (80 фл) стабилизируется.
МСН (mean corpuscular hemoglobin) - среднее содержание гемоглобина
в эритроците (пг). Рассчитывается по формуле:
Гемоглобин (г/л)
МСН = к--------------------------ДДТ '
Количество эритроцитов х 10
МСН характеризует среднее содержание гемоглобина в отдельном эритро-
ците в абсолютных единицах. В норме МСН составляет 27-31 пг. МСН - бо-
лее объективный параметр, чем устаревший цветовой показатель, который
не отражает синтез гемоглобина и его содержание в эритроците.
Возможные ошибки измерения. Параметр МСН является расчетным, по-
этому к ложноповышенным результатам приводят все факторы, влияющие
на увеличение значений гемоглобина и снижение количества эритроцитов.
Ложнопониженные результаты МСН получаются вследствие ошибок, свя-
занных с неправильным определением числа эритроцитов (завышения их
количества), и занижения концентрации гемоглобина.
Изменения МСН лежат в основе разделения анемий на нормохромные
(МСН - 27-31 пг), гипохромные (МСН менее 27 пг) и гиперхромные (МСН
более 31 пг). Снижение МСН наблюдается при анемиях, обусловленных
нарушением синтеза гемоглобина (железодефицитной анемии, порфирии),
повышение - при макроцитарных и особенно мегалобластных анемиях.
МСНС (mean corpuscular hemoglobin concentration) - средняя концентра-
ция гемоглобина в эритроците (г/дл). Вычисляется по формуле:
Гемоглобин (г/дл)
МСНС = —-------------х W0 (г/дл).
Гематокрит (%)
Различия между двумя последними индексами заключаются в том, что
МСН указывает на массу гемоглобина в одном эритроците и выражается в
долях грамма (пикограммах). МСНС показывает концентрацию гемоглобина
в одном эритроците, т. е. соотношение содержания гемоглобина к объему
клетки. Он отражает насыщение эритроцита гемоглобином и в норме состав-
ляет 30-38 г/дл. В отличие от МСН параметр МСНС не зависит от клеточ-
ного объема и является чувствительным показателем нарушения процессов
гемоглобинообразования.
Возможные ошибки измерения. Поскольку параметр МСНС является
расчетным, то к ложнозавышенным результатам приводят все факторы,
влияющие на завышение значений гемоглобина и занижение гематокрита
(последний связан с измерением объема эритроцитов). Ложнопониженные
результаты МСНС получаются вследствие неправильного определения MCV
(завышения их значения) и занижения концентрации гемоглобина.
Снижение значения МСНС наблюдается при заболеваниях, сопровожда-
ющихся нарушением синтеза гемоглобина.
Повышение МСНС выше 38 г/дл встречается редко (врожденный сфероци-
тоз), т. к. это может закончиться кристаллизацией гемоглобина и гемолизом
эритроцита. Чаще всего увеличение МСНС свидетельствует об ошибках,
Допущенных при измерении пробы (погрешности определения гемогло-
бина или MCV). Поэтому данный параметр часто используется в качестве
индикатора ошибок, допущенных на аналитическом или преаналитическом
этапах работы.
Анализаторы фирмы Siemens (ADVIA) непосредственно измеряют кон-
центрацию гемоглобина в каждом отдельном эритроците и строят гистог-
раммы распределения клеток не только по объему, но и по концентрации
гемоглобина. При этом возрастает точность определения этого параметра и
вводится новый показатель - гетерогенность эритроцитов по концентрации
гемоглобина (HDW-hemoglobin distribution width). Кроме того, в этих же ана-
лизаторах существует ряд дополнительных параметров для характеристики
периферического звена эритрона: LHD, %Нуро, %Hyper, %Micro, %Масто.
LHD (low hemoglobin density), % - гемоглобин низкой плотности - новый
параметр, который коррелирует с процентом гипохромных эритроцитов. При
наличии более 10% гипохромных эритроцитов показатель LHD >5,5%.
Показатель LHD является индикатором железодефицитного состояния.
%Нуро - процент гипохромных (содержащих менее 28 пг гемоглобина)
эритроцитов, имеет значение в диагностике гипохромных анемий и мониторин-
ге терапии эритропоэтином (норма до 4%). Более 10% гипохромных эритроци-
тов является индикатором железодефицитного состояния. Увеличение количест-
ва гипохромных эритроцитов наблюдается при ЖДА, талассемии, АХЗ.
%Нурег - процент гиперхромных (содержащих более 41 пг гемоглобина)
эритроцитов (норма до 4%).
%Micro - процент микроцитов (эритроцитов с объемом менее 60 фл),
имеет значение в диагностике микроцитарных анемий и мониторинге те-
рапии рЭПО.
%Масго - процент макроцитов (эритроцитов с объемом более 120 фл),
имеет значение в диагностике макроцитарных анемий.
RDW (red cell distribution width) - показатель гетерогенности эритроцитов
по объему, характеризует степень анизоцитоза. Этот показатель вычисляется
большинством современных гематологических анализаторов на основании
гистограммы распределения эритроцитов как коэффициент вариации объема
эритроцитов:
SD
RDW-CV (%) =--------х100,
v 7 MCV
где SD - стандартное среднеквадратическое отклонение объема эритроцита
от среднего значения (рис. 17). На этот показатель влияет MCV, поэтому как
при микроцитозе, так и при макроцитозе отмечается тенденция к увеличению
RDW-CV.
В гематологических анализаторах фирмы Sysmex имеется еще один рас-
четный показатель RDW - это RDW-SD, который независим от MCV и пред-
ставляет собой прямое измерение ширины эритроцитарной гистограммы на
уровне 20% от базовой линии по оси Y. При этом высота пика RBC-гистограм-
мы принимается за 100% (рис. 18). Норма RDW-SD - 42 ± 5 фл. Клинически
значимое значение, характеризующее анизоцитоз, RDW-SD >60 фл.
Рис. 17. Схематическое изображе-
ние распределения эритроцитов
по объему (MCV), на основании
которого рассчитывается показатель
анизоцитоза (RDW)
30
Рис. 18. Определение RDW-SD.
Ширина RBC-гистограммы на
уровне 20% от базовой линии
по оси Y
Оба показателя RDW определяют вариабельность эритроцитов по
объему. Повышение RDW предполагает присутствие смешанной попу-
ляции клеток (нормоциты и микроциты или макроциты и нормоциты).
RDW-SD является более чувствительным показателем при наличии ми-
норной популяции макроцитов или микроцитов, т. к. он измеряет ниж-
нюю часть кривой распределения эритроцитов по объему. В то же время
этот показатель будет изменяться при высоком ретикулоцитозе в силу
их большого объема, что расширяет основание кривой распределения
эритроцитов. RDW-CV менее чувствителен к присутствию небольшой
популяции микроцитов или макроцитов или ретикулоцитов, но лучше
отражает общие изменения в размере эритроцитов при макроцитарной
или микроцитарной анемии.
Анизоцитоз улавливается прибором значительно быстрее, чем при
визуальном просмотре мазка крови. Оценка степени анизоцитоза под
микроскопом сопровождается целым рядом ошибок. При высыхании
в мазках диаметр эритроцитов уменьшается на 10-20%. В толстых
препаратах он меньше, чем в тонких. В то же время показатель RDW
характеризует колебания объема клеток внутри популяции и не связан
с абсолютной величиной объема эритроцитов. Поэтому при наличии в
крови популяции эритроцитов с измененным, но достаточно однородным
размером (например, микроциты) значения RDW могут быть в преде-
лах нормы (11,5-14,5%). В то же время при выраженном анизоцитозе
эритроцитов показатель MCV, характеризующий средний объем всей
клеточной популяции, является нормальным, a RDW будет повышенным.
Таким образом, сочетанное использование двух параметров - RDW и
MCV - позволяет точнее характеризовать изменения в периферическом
звене эритрона.
FRC (fragment red cells) (RBC-F) - подсчет фрагментов эритроцитов,
используется для оценки тромботических микроангиопатий.
Гистограмма - это графическое распределение различных видов
клеток по их количеству и объему. Для построения гистограмм гема-
тологические анализаторы подсчитывают миллионы клеток в одном
образце, сортируют импульсы по амплитуде и распределяют частицы
объемом от 24 до 360 фл по 256 каналам, каждый из которых соответс-
твует объему частиц.
Эритроцитарная гистограмма
Как правило, регистрируемая кривая подчиняется закону нормального
(гауссова) распределения. Гистограмма должна начинаться и заканчиваться на
базовой линии и между нижним и верхним дискриминатором. По горизонтали
откладывается объем измеряемой клетки в фл (1 фл = 10 15/л), вертикальная
ось на графике фиксируется как 100% шкала.
Нормальная эритроцитарная гистограмма имеет симметричную (купо-
лообразную) форму (рис. 19). На некоторых анализаторах при уменьшении
количества эритроцитов амплитуда гистограммы уменьшается.
При появлении патологических или нескольких популяций эритроцитов
форма гистограммы меняется. Несмотря на то что пороговое значение для
эритроцитов составляет 36 фл, дополнительная область от 24 до 36 фл по-
зволяет выявить клетки небольших размеров. Аномальное распределение
эритроцитов представлено на рис. 20-22.
Рис. 19. Нормальная эритроцитарная
гистограмма имеет симметричную
(куполообразную) форму. Гисто-
грамма начинается и заканчивается
на базовой линии (пунктирными
линиями указана зона расположе-
ния нормальной RBC-гистограммы,
сплошной — гистограмма конкрет-
ного пациента)
Рис. 20. Кривая распределения RBC
стартует не на базовой линии. В этом
случае тромбоциты и эритроциты не
могут быть четко разделены. Воз-
можные причины: микроэритроциты,
в этом случае MCV 4-, RDW норма
или Т; гигантские тромбоциты, в
этом случае MPV Т, PDWT; агрегация
тромбоцитов, в этом случае PLT 4.
Ошибки, получаемые в подобных си-
туациях, могут существенно исказить
результат исследования тромбоци-
тов и обычно незначительно влияют
на значения эритроцитов, так как
тромбоцитов намного меньше, чем
эритроцитов. LD и UD — нижний и
верхний дискриминаторы для RBC
Рис. 21. Сдвиг эритроцитарной
гистограммы вправо. Возможные
причины: холодовая агглютина-
ция эритроцитов (в этом случае
МСНС >40 г/дл) эритробласты
(в этом случае MCV Т). Для ис-
ключения холодовой агглютинации
образец необходимо подогреть до
37 °C и снова измерить
Рис. 22. Эритроцитарная гисто-
грамма имеет несколько пиков.
Возможные причины: ответ на
терапию препаратами железа у
больных с ЖДА, гемотрансфу-
зии — эритроциты донора и па-
циента имеют разные размеры;
гиперлейкоцитоз более 600 х 109/л.
В последнем случае возможно по-
лучение неправильных результатов
подсчета количества эритроцитов,
результат должен быть уточнен
Ретикулоцитарные параметры
Ретикулоциты представляют собой незрелые эритроциты, содержащие
остатки РНК и образующиеся после потери нормобластами ядер.
В связи с появлением высокотехнологичных гематологических ана-
лизаторов стало возможным получать, помимо классических, дополни-
тельные, информативные ретикулоцитарные параметры. Аббревиатура
ретикулоцитарных показателей в анализаторах различных фирм-произ-
водителей различна. В тексте приводятся обозначения, используемые в
приборах Sysmex (ХЕ-2100, XT-2000i), а также GEN-S, LH750 (Beckman
Coulter).
Классические параметры ретикулоцитов
RET% - относительное количество ретикулоцитов (в %).
RET# - абсолютное количество ретикулоцитов (х 109/л).
Ретикулоцитоз с резким увеличением фракции незрелых ретикулоцитов
на фоне активного эритропоэза отражает повышенную регенераторную
способность костного мозга. Сохраняющийся ретикулоцитоз может свиде-
тельствовать о продолжающемся кровотечении или гемолизе.
Ретикулоцитопения - индикатор угнетения эритропоэза (гипоплазии кро-
ветворения, мегалобластные анемии, анемии при хронических заболеваниях,
тяжелая ЖДА, анемии при ХПН).
Возможные ошибки измерения ретикулоцитов
1аилициj
Ложное завышение
• Включения в эритроцитах (тельца Жолли, малярийные паразиты)
• Высокий лейкоцитоз
• Аномальные формы гемоглобина
• Гипертромбоцитоз
• Гигантские тромбоциты
Нормализация абсолютного количества ретикулоцитов (RET#) - показа-
тель восстановления пролиферативной активности эритрокариоцитов.
Объемные параметры ретикулоцитов
MCVr (Mean Cell Volume Reticulocytes) - средний объем ретикулоци-
тов (фл).
MSCV (Mean Sphered Cell Volume) - средний объем сферических клеток,
включающих эритроциты и ретикулоциты (фл).
• Низкий объем ретикулоцитов объясняет появление микроцитов в пе-
риферической крови.
• Показатели объема ретику)iоцитов мо гут использоваться в диагностике
ЖДА, мониторинге ответа на терапию железосодержащими препара-
тами, фолиевой кислотой, витамином В12.
• Повышение MSCV у спортсменов указывает на злоупотребление пре-
паратами, стимулирующими эритропоэз.
• MSCV меньше MCVr - индикатор наследственного сфероцитоза.
Параметры, характеризующие степень зрелости ретикулоцитов
LFR% - популяция малых зрелых RET (87-99%).
MFR% - популяция средних RET (2-12%).
HFR% (1-2%) - популяция больших незрелых RET.
MFR + HFR определяется как фракция незрелых ретикулоцитов - IRF
(Immature Reticulocyte Fraction) (2-14%). Фракция незрелых ретикулоцитов
может служить индикатором активности эритропоэза.
Увеличение фракции незрелых ретикулоцитов свидетельствует об ускоренном
выбросе незрелых клеток из костного мозга. Фракция незрелых ретикулоцитов
повышается значительно раньше (как правило, на 2 дня), чем процент ретику-
лоцитов, и может служить наиболее чувствительным маркером в мониторинге
за состоянием эритропоэтической активности костного мозга и эффективности
лечения витамином В|2, фолиевой кислотой, препаратами железа и ЭПО.
Содержание НЬ в ретикулоцитах
RET-Y (расчетный показатель размера клеток по среднему значению
FSC), или Ret-He (норма 28,2-36,4 пг), или CHr (Bayer Technicon Н2) (норма
28-32 пг).
При усилении эритропоэза из костного мозга поступают в циркуляцию
незрелые ретикулоциты, период созревания которых в крови удлиняется и
составляет от 1,5 до 2,5 дней (рис. 23). Появление незрелых ретикулоцитов
в крови соответствует явлению полихромазии эритроцитов в окрашенном
мазке крови.
Костный мозг Кровь
Гематокрит (%) Ретикулоциты (дни) Ретикулоциты (дни)
Рис. 23. Изменение длительности созревания ретикулоцитов при анемии
Эти физиологические особенности ретикулоцитов принимаются во вни-
мание при расчете ретикулоцитарных индексов.
Ретикулоцитарные индексы (RPI и CRC)
В случае изменения только гематокрита рассчитывается индекс CRC
(Corrected Reticulocyte Count) - скорректированный подсчет ретикуло-
цитов по формуле:
CRC=RET(%)x-^,
0,45
где Ht - гематокрит больного; RET% - количество ретикулоцитов (%), изме-
ренное в крови при данном гематокрите; 0,45 - идеальный гематокрит.
Если у больного одновременно с низким гематокритом (Ht) в пери-
ферической крови присутствуют незрелые ретикулоциты (MFR и HFR),
то рассчитывается индекс продукции ретикулоцитов RPI (Reticulocyte
production index):
RpI =_________RET(%)xHt____________
0,45 x дни циркуляции RET в крови
Величина RPI широко варьирует в зависимости от степени тяжести
анемии, продукции ЭПО и других факторов. Снижение данного индекса
менее 2 указывает на низкую пролиферативную активность эритрокарио-
цитов.
Расчет данных индексов позволяет дать правильную оценку характера
эритропоэза и таким образом выбрать адекватную программу лечения боль-
ных. На рис. 24 приведен пример, иллюстрирующий обоснованность расчета
индекса продукции ретикулоцитов.
35
3 2 10 12 3
КМ Дни ПК
Рис. 24. Изменение RPI в зависимости от значений гематокрита. Количество ретикулоцитов
при гематокрите 15% в 7,5 раза превышает значение ретикулоцитов при гематокрите 45%, что
предполагает наличие более активного эритропоэза. Однако величина RPI во всех случаях со-
ставляет 1,0, что свидетельствует об одинаковой эритропоэтической активности костного мозга.
КМ — костный мозг, ПК — периферическая кровь, НСТ — гематокрит, RET — ретикулоциты,
CRC - скорректированный подсчет ретикулоцитов, RPI — индекс продукции ретикулоцитов
Исследование ретикулоцитов используется для:
• оценки активности эритропоэза при состояниях, сопровождающихся
гемолизом или кровопотерей;
• детекции нарушения регенераторной способности костного мозга при
дефиците железа, витаминов В|2, В6, фолатов, меди и мониторинга
соответствующей терапии;
• оценки состояния эритропоэза на фоне лечения эритропоэтином;
• оценки способности костного мозга к регенерации после цитотокси-
ческой терапии и трансплантации костного мозга;
• оценки восстановления синтеза ЭПО после трансплантации почки
• допингового контроля у спортсменов (прием ЭПО).
ОПРЕДЕЛЕНИЕ И КЛАССИФИКАЦИЯ АНЕМИЙ
Анемия - это состояние, характеризующееся снижением концентрации ге-
моглобина и, в большинстве случаев, количества эритроцитов и гематокрита
в единице объема крови.
Критериями ВОЗ для диагностики анемии считаются:
• у мужчин число эритроцитов <4,0 млн/мкл, НЬ <130 г/л, Ht <39%;
• у женщин число эритроцитов <3,8 млн/мкл, НЬ <120 г/л, Ht <36%;
• у беременных НЬ <110 г/л, Ht <33%.
Анемия может быть относительная, вызванная увеличением объема
плазмы (гемодилюция, гиперволемия), и абсолютная — обусловленная из-
менением количества циркулирующих эритроцитов. Относительная анемия
наблюдается при беременности, сердечной недостаточности, трансфузии
кровезаменителей, острой кровопотере. Гемодилюция сопровождается
снижением количества эритроцитов и содержания гемоглобина в единице
объема при полном сохранении общей массы эритроцитов. Анемия может
маскироваться при состояниях, сопровождающихся сгущением крови (обиль-
ная рвота, профузная диарея и др.).
Анемии разнообразны по своему генезу и часто имеют смешанный пато-
генез. В большинстве случаев анемия - не самостоятельная нозологическая
форма, а проявление основного заболевания. Она сопутствует диффузным
болезням соединительной ткани (ревматоидный артрит, системная красная
волчанка, системные васкулиты), заболеваниям желудочно-кишечного тракта,
печени, почек (хроническая почечная недостаточность), злокачественным
новообразованиям, хроническим инфекционным заболеваниям и воспали-
тельным процессам.
Большое разнообразие факторов, лежащих в основе развития анемий,
затрудняет их дифференциальную диагностику. На первом этапе диагнос-
тического поиска основной целью является определение патогенетического
варианта анемии, то есть основного механизма, обуславливающего снижение
эритроцитов и гемоглобина. На следующем этапе диагностика заболевания
направлена на установление патологического процесса, лежащего в основе
анемического синдрома, другими словами, выявление причины анемии у
конкретного больного. Эти этапы диагностики анемий базируются на данных
лабораторного исследования и зависят во многом как от уровня и качества
проведенных исследований, так и от правильной интерпретации полученных
результатов.
Существует несколько классификаций анемий, основанных на этиологи-
ческих, патогенетических, гематологических признаках. Не все они имеют
практическое значение в связи с тем, что анемия преимущественно может
быть симптомом какого-то заболевания, а патогенетические механизмы раз-
вития ее при различных заболеваниях разнообразны и часто сложны.
Для практических целей наиболее удобной является классификация
анемий, построенная по патогенетическому принципу. Согласно этой клас-
сификации выделяют 3 основные группы анемий.
Патогенетическая классификация анемий
I.
II.
1.
Анемии вследствие кровопотери
• Острая постгеморрагическая анемия.
• Хроническая постгеморрагическая анемия.
Анемии, обусловленные недостаточностью эритропоэза:
Гипохромные анемии:
• Железодефицитная анемия.
• Анемии, связанные с нарушением синтеза порфиринов.
2. Нормохромные анемии:
• Анемии хронических заболеваний.
• Анемия при хронической почечной недостаточности.
• Апластические анемии.
• Анемии при опухолевых и метастатических поражениях костного
мозга.
3. Гиперхромные (мегалобластные) анемии:
• Анемии, обусловленные дефицитом витамина В12.
• Фолиеводефицитные анемии.
III. Анемии вследствие усиленного разрушения эритроцитов (гемолити-
ческие анемии):
1. Анемии, обусловленные внеэритроцитарными факторами.
• Иммунные гемолитические анемии:
- изоиммунные гемолитические анемии;
— аутоиммунные гемолитические анемии.
• Гемолитические анемии, обусловленные механическим повреждением
эритроцитов.
2. Анемии, обусловленные эритроцитарными факторами.
• Гемолитические анемии, связанные с нарушением структуры мембраны
эритроцитов (эритроцитопатии - наследственные и приобретенные):
- микросфероцитарная гемолитическая анемия;
- овалоцитарная гемолитическая анемия;
- стоматоцитарная гемолитическая анемия;
- гемолитические анемии, обусловленные нарушением структуры
липидов мембраны эритроцитов (акантоцитоз).
• Гемолитические анемии, обусловленные дефицитом ферментов эрит-
роцитов (эритроцитарные энзимопатии):
— гемолитические анемии, связанные с недостаточностью активности
ферментов гликолиза;
- гемолитические анемии, связанные с недостаточностью активности
ферментов пентозофосфатного шунта;
— гемолитические анемии, связанные с недостаточностью активности
ферментов глутатионовой системы.
• Гемолитические анемии, связанные с нарушенным синтезом глобина
(гемоглобинопатии):
- талассемии;
— гемолитические анемии, обусловленные носительством аномального
гемоглобина - HbS, HbC, HbD, НЬЕ и др.;
- гемолитические анемии, обусловленные носительством аномальных
нестабильных гемоглобинов.
3. Гемолитическая анемия, обусловленная соматической мутацией клеток-
предшественников миелопоэза.
• Пароксизмальная ночная гемоглобинурия.
Высокая точность и воспроизводимость результатов, возможность подсче-
та большого количества клеток и расчетных параметров на гематологических
анализаторах позволили вновь вернуться к дифференциально-диагностичес-
кому алгоритму разделения анемий на основании эритроцитарных индексов,
предложенных M.Wintrobe еще в 1929 г. (рис. 25).
Рис. 25. Классификация анемий с использованием эритроцитарных индексов
Функциональная классификация анемий, основанная на исследовании
костного мозга и количества ретикулоцитов, позволяет оценить эффектив-
ность продукции эритроцитов и классифицировать анемии на гипопроли-
феративные и гиперпролиферативные.
АНЕМИИ ВСЛЕДСТВИЕ КРОВОПОТЕРИ
Острая постгеморрагическая анемия
Острая постгеморрагическая анемия - состояние, которое развивается
в результате быстрой потери значительного объема крови. Независимо от
патогенеза заболевания при анемии в организме нарушаются окислительные
процессы и возникает гипоксия. Степень анемии зависит от быстроты и
количества кровопотери и степени адаптации организма к новым условиям
существования.
11ричиной острой кровопотери могут быть нарушение целостности сте-
нок сосуда вследствие ранения, поражения патологическим процессом при
различных заболеваниях (язва желудка и кишечника, опухоль, туберкулез,
инфаркт легкого, варикозное расширение вен пищевода, патологические
роды), нарушение в системе гемостаза (гемофилия). Последствия этих из-
менений независимо от причин, вызвавших их, однотипны.
Ведущие симптомы обширного кровотечения - остро возникающий де-
фицит объема циркулирующей крови (ОЦК) и нарушение гемостаза. В ответ
на развитие дефицита ОЦК включаются адаптационные механизмы, направ-
ленные на его компенсацию.
Сразу же после кровопотери наблюдаются признаки коллапса: резкая
слабость, падение кровяного давления, бледность, головокружение, обмороч-
ное состояние, тахикардия, холодный пот, рвота, цианоз, судороги. Позднее
появляются собственно анемические симптомы.
Нормальная реакция организма на кровопотерю характеризуется акти-
вацией гемопоэза. В ответ на гипоксию увеличивается синтез и секреция
почками ЭПО, что приводит к усилению эритропоэза. Резко увеличивается
количество эритрокариоцитов.
На фоне основных патофизиологических сдвигов различают несколько
фаз течения болезни.
Рефлекторная фаза сопровождается спазмом периферических сосудов,
который приводит к уменьшению объема сосудистого русла. Первоначально
снижается приток крови к сосудам кожи, подкожной клетчатке и мышцам.
Происходит перераспределение крови по органам и системам - осуществляет-
ся централизация кровообращения, что способствует компенсации дефицита
ОЦК. Благодаря выключению периферических сосудов из кровообращения
сохраняется кровоток в жизненно важных органах (головной и спинной мозг,
миокард, надпочечники). В основе компенсаторного механизма, приводящего
к спазму сосудов, лежит дополнительный выброс надпочечниками катехол-
аминов, оказывающих прессорное действие. При недостаточном кровоснаб-
жении почек повышается секреция ренина клетками юкстагломерулярного
аппарата (ЮГА). Под влиянием ренина в печени образуется ангиотензиноген,
который суживает сосуды, стимулирует секрецию альдостерона надпочеч-
никами, активирующего реабсорбцию натрия в проксимальных канальцах
почек. За натрием в плазму крови возвращается вода. Задержка натрия ведет
к усилению реабсорбции воды в канальцах и уменьшению мочеобразова-
ния. Так вода сохраняется в организме. В результате гормональных сдвигов
и снижения почечного кровотока прекращается фильтрация в почечных
клубочках и нарушается образование мочи. Эти изменения ведут к резкому
уменьшению диуреза с последующим падением артериального давления
(АД) в клубочках ниже 40 мм рт. ст.
Рефлекторная фаза может продолжаться до 8-12 ч и редко больше.
Лабораторные показатели. Уменьшение общего объема сосудистого
русла приводит к тому, что, несмотря на абсолютное уменьшение количества
эритроцитарной массы, показатели гемоглобина и эритроцитов в единице
объема крови приближаются к исходным цифрам и не отражают степень
анемизации, не изменяется величина гематокрита, в то время как ОЦК резко
снижен. Непосредственно после кровопотери имеет место скрытая анемия,
число лейкоцитов редко превышает 9,0-10,0 х 109/л. Чаще отмечается лей-
копения, нейтропения. В период кровотечения в связи с большим потреб-
пением тромбоцитов, которые мобилизуются для его остановки, возможно
снижение их содержания.
фаза компенсации (гидремическая) развивается через 2 3 ч после кро-
вопотери, характеризуется мобилизацией межтканевой жидкости и поступ-
лением ее в кровяное русло. Постгеморрагический период сопровождается
выходом эритроцитов из депо и увеличением ОЦК с последующим сниже-
нием вязкости крови и улучшением ее реологии. Этот адаптивный механизм
обозначают термином «реакция аутогемодилюции». Тем самым создаются
условия для восстановления центральной и периферической гемодинамики
и микроциркуляции.
Фаза гемодилюции в зависимости от величины и длительности крово-
потери может продолжаться от нескольких часов до нескольких дней. Она
характеризуется увеличением проницаемости стенок сосудов, что приво-
дит к поступлению в кровяное русло тканевой жидкости. Приток тканевой
жидкости восстанавливает ОЦК, снимает спазм периферических сосудов и
способствует одновременному, равномерному снижению количества гемо-
глобина и эритроцитов в единице объема крови.
Длительный спазм периферических сосудов (недостаточность аутоге-
модилюции) при больших кровопотерях может приводить к нарушению
капиллярного кровотока.
При падении АД ниже 80 мм рт. ст. кровь скапливается в капиллярах,
замедляется скорость ее движения, что приводит к образованию агрегатов из
эритроцитов (сладж-синдром) и к стазу. В результате изменения капиллярного
кровотока происходит образование микросгустков, что ведет к нарушению
микроциркуляции с последующим развитием геморрагического шока и не-
обратимых изменений в органах.
Кровопотерю в 10-15% ОЦК организм переносит легко, до 25% ОЦК -
с незначительными нарушениями гемодинамики. При кровопотере свыше
25% собственные механизмы адаптации оказываются несостоятельными.
Потеря около 50% циркулирующих эритроцитов не является смертельной.
Вместе с тем снижение объема циркулирующей плазмы на 30% не совмес-
тимо с жизнью.
Лабораторные показатели. Развивающаяся спустя 1-2 дня после
кровопотери анемия носит нормохромный нормоцитарный характер
(рис. 26). Насыщение эритроцитов гемоглобином и его концентрация
в одном эритроците зависят от наличия запасов железа в организме.
Наибольшие изменения гематологических показателей периферической
крови наблюдаются обычно через 4-5 дней после кровопотери. Эти
изменения обусловлены активной пролиферацией костномозговых эле-
ментов. Через 3-5 дней после кровотечения развивается ретикулоцитоз
с резким увеличением фракции незрелых ретикулоцитов (IRF), что на
фоне активного эритропоэза отражает регенераторную способность кос-
тного мозга, которая становится максимальной к 7-10-му дню. Размер
эритроцитов после кровотечения несколько возрастает (макроцитоз).
появление полихроматофильных макроцитов приводит к увеличению
MFV, и анемия может стать макроцитарной нормохромной. Могут по-
явиться эритробласты. При сочетании ретикулоцитоза и повышенного
MCV можно ошибочно диагностировать гемолитическую анемию. После
остановки кровотечения нормализация количества ретикулоцитов от-
мечается до 2 недель. Если количество ретикулоцитов к началу второй
недели не снижается, это может свидетельствовать о продолжающемся
кровотечении.
На 5-8-й день после кровотечения обычно наступает умеренный лейко-
цитоз (до 12-20 х 109/л), палочкоядерный сдвиг влево (реже до миелоцитов).
Стойкий лейкоцитоз имеет место при наличии присоединившейся инфекции.
Количество тромбоцитов увеличивается до 500 х 109/л. Иногда в течение
нескольких дней наблюдается тромбоцитоз до 1 млн.
RBC 1,76 3,8-5,5 х 1012/л
НЬ 50 130-155 г/л
Ht 19,4 36-48%
MCV 83,0 80—95 фл
мен 28,4 27-31 пг
мснс 342 300-380 г/л
RDW 14,0 11,5-14,5%
Рис. 26. Периферическая кровь больного
острой постгеморрагической анемией. Но|
мохромная нормоцитарная анемия
При небольших кровопотерях депонированное железо поступает в
костный мозг, где расходуется для синтеза гемоглобина. При однократ-
ной острой кровопотере отмечается преходящее снижение уровня сыво-
роточного железа в плазме. При больших кровопотерях сывороточное
железо остается низким. Дефицит резервного железа сопровождается
сидеропенией и развитием железодефицитной анемии. На степень анемии
оказывают влияние объем и темп кровопотери, время с момента кровоте-
чения, резерв железа в органах депо, исходное количество эритроцитов
и гемоглобина.
Гипоксия тканей, развивающаяся при кровопотере, приводит к накопле-
нию в организме недоокисленных продуктов обмена и к ацидозу, который
первое время носит компенсированный характер. Прогрессирование процесса
сопровождается развитием некомпенсированного ацидоза со снижением
pH крови до 7,2 и ниже. В терминальной стадии к ацидозу присоединяется
алкалоз. Существенно уменьшается напряжение углекислого газа (рСО2) в
результате гипервентиляции легких и связывания бикарбонатов плазмы. Ды-
хательный коэффициент возрастает. Развивается гипергликемия, повышается
активность ферментов ЛДГ и ACT, что может быть связано с поражением
печени и почек. В сыворотке уменьшается концентрация натрия и кальция,
увеличивается содержание калия, магния, неорганического фосфора и хлора,
концентрация последнего зависит от степени ацидоза и может снижаться
при его декомпенсации.
Хроническая постгеморрагическая анемия
Гипохромная нормоцитарная анемия, возникающая при длительной
умеренной кровопотере, например при хронических желудочно-кишечных
кровотечениях (при язве желудка, двенадцатиперстной кишки, геморрое и
т и.), а также при гинекологических и урологических заболеваниях, будет
описана ниже в разделе «Железодефицитная анемия».
АНЕМИИ,
ОБУСЛОВЛЕННЫЕ НЕДОСТАТОЧНОСТЬЮ ЭРИТРОПОЭЗА
Эта группа анемий является наиболее распространенной во всем мире.
Нарушение обмена железа на уровне его поступления в организм, утили-
зации или реутилизации из эритроцитов является ключевым моментом
большинства из них.
Обмен железа
Железо является необходимым элементом многих белков и ферментов,
участвующих в ключевых процессах метаболизма, роста, пролиферации
и регенерации клеток, транспорте кислорода к тканям, тканевом дыхании
и др. Исключительная роль железа определяется биологическими функция-
ми белков, в состав которых входит этот биометалл. К наиболее известным
железосодержащим белкам относятся гемоглобин и миоглобин, в составе
которых железо, доставляя кислород к тканям, обеспечивает тканевое ды-
хание. Дефицит железа приводит к нарушению синтеза гемоглобина и раз-
витию гипоксии. Железо входит также в состав значительного количества
ферментов, участвующих в окислительно-восстановительных реакциях, в
процессах энергообразования (цитохромы, НАДН-дегидрогеназа, сукци-
натдегидрогеназа), биосинтезе ДНК (рибонуклеотидредуктаза) и делении
клеток, детоксикации продуктов эндогенного распада, нейтрализующих
активные формы кислорода (оксидазы, пероксидазы, цитохромоксидазы,
каталазы) (табл. 6).
В последние годы установлена роль железосодержащих белков (ферритин)
в реализации клеточного иммунитета, регуляции кроветворения. Вместе с тем
железо может быть исключительно токсичным элементом, если присутствует
в организме в повышенных концентрациях, превышающих емкость железо-
содержащих белков. Потенциальная токсичность свободного двухвалентного
Железа (Feb) объясняется его способностью запускать цепные свободноради-
кальные реакции, приводящие к перекисному окислению липидов биологи-
ческих мембран, образованию высокореактивных кислородных радикалов,
способных повреждать мембраны клеток, белки, нуклеиновые кислоты и
в целом вызывать гибель клетки. Двойственная сущность функций железа
диктует необходимость формирования жесткой регуляции концентрации
железа в организме человека.
Таблица 6
Основные железосодержащие белки и ферменты, их функции
Железосодержащие белки и ферменты Функции
Гемоглобин Транспорт кислорода
Миоглобин Транспорт и депонирование кислорода в мышцах
Каталаза Расщепление перекиси водорода
Цитохромы Тканевое дыхание
Пероксидаза Окислительные реакции
Трансферрин Транспорт железа
Лактоферрин Транспорт железа, бактерицидная функция
Ферритин Депонирование железа в тканях
Гемосидерин Депонирование железа в тканях
Ксантиноксидаза Образование мочевой кислоты
Рибонуклеотидредуктаза Синтез ДНК
Системный гомеостаз железа регулируется на уровне всасывания железа
в тонком кишечнике. Процесс выведения (экскреции) железа пассивный,
нерегулируемый. В норме баланс железа остается стабильным, и потери
железа уравновешиваются повышением доставки его во время абсорбции.
Транспорт и депонирование железа осуществляются специальными белка-
ми - трансферрином, трансферриновым рецептором 1 и ферритином.
Общее количество железа в организме здорового человека составляет
3,5-5,0 г, примерно 2/3 приходится на гемоглобин эритроцитов и эритроидных
предшественников. Распределение железа в организме взрослого человека
в норме представлено на рис. 27.
В норме из пищи всасывается в тонком кишечнике 1-2 мг железа. Обяза-
тельные суточные потери железа также составляют около 1-2 мг. Повышен-
ные потери железа во время менструации (0,5 -1 мг в день) или повышенные
И
Железо пищи
12-перстная кишка
1-2 мг/день
ФУНКЦИОНАЛЬНОЕ
ЖЕЛЕЗО
ФУНКЦИОНАЛЬНОЕ
ЖЕЛЕЗО
Трансферрин
плазмы
3 мг
Костный мозг
300 м г
МЫШЦЫ
(миоглобин)
300 мг
Печень
1000 мг
ДЕПОНИРОВАННОЕ
ЖЕЛЕЗО
Гемоглобин
эритроцитов
1800 мг
Макрофаги СМФ
600 мг
ФУНКЦИОНАЛЬНОЕ
ЖЕЛЕЗО
Десквамация эпителия
Менструация
Другие потери
1-2 мг/день
Потери железа
Рис. 27. Обмен железа в организме взрослого человека
потребности в железе во время беременности (около 500 мг) компенсируются
увеличением всасывания алиментарного железа (максимальная способность
всасывания - 3 мг в день). У детей потеря железа составляет 0,1-0,3 мг в
сУтки, увеличиваясь до 0,5-1,0 мг в сутки у подростков. Ежедневная по-
требность детскою организма в железе —0,5—1,2 мг в день. У детей раннего
возраста в связи с быстрыми темпами роста и развития наблюдается повы-
шенная потребность в железе. В этот период жизни запасы железа быстро
истощаются из-за усиленного его потребления из депо: у недоношенных
детей - к 3-му месяцу, у доношенных - к 5-6-му месяцам жизни.
Ежедневная потребность в железе превышает его поступление с пищей и
составляет около 20 мг, которые в основном расходуются на синтез гемоглоби-
на. Она удовлетворяется за счетреутилизированного железа, образующегося в
макрофагах селезенки и других органах системы мононуклеарных фагоцитов
(СМФ) при фагоцитозе старых или поврежденных эритроцитов.
Около % общего содержания железа в организме сосредоточено в гепато-
цитах и макрофагах селезенки, печени и костного мозга. Мобилизация железа
из депо осуществляется в период отрицательного баланса железа, например
при недостаточном поступлении его с пищей, избыточной кровопотере. Со-
вокупность гомеостатических и транспортных механизмов используется для
создания динамического равновесия между утилизацией железа (в основном
клетками костного мозга) и поступлением его (абсорбция, мобилизация из
депо, реутилизация). Этот баланс и отражается в относительно узком диа-
пазоне концентрации железа в плазме крови (10-30 мкмоль/л).
Обмен железа в организме состоит из нескольких этапов: всасывание в
желудочно-кишечном тракте, транспорт, внутриклеточный метаболизм и
депонирование, реутилизация, экскреция из организма.
Наиболее простая схема метаболизма железа представлена на рис. 28.
Рис. 28. Схема метаболизма железа в организме
Всасывание железа
Железо в организм человека поступает с пищей в виде неорганического и
гемового железа. При обычном питании неорганическое, или окисное, желе-
зо (Fe3+) составляет в пище 80 90%, на гемовое, или закисное, железо (Fe2')
приходится 10 -20%. Механизм всасывания неорганического железа хорошо
изучен в отличие от абсорбции гемового железа. Fe3' нерастворимо в растворах
с pH выше 3,0 и должно быть растворено в желудке, чтобы в последующем
абсорбироваться в более щелочной среде 12-перстной кишки. Растворенное
в желудке железо хелатируется соединениями, которые сохраняют его в рас-
творе, когда оно поступает в 12-перстную кишку. Хелатирование выполняется
муцином и некоторыми компонентами пищи (аминокислоты, сахара, амины и
др.). Лимонная, аскорбиновая, янтарная, яблочная кислоты, фруктоза, сорбит,
метионин, цистеин, микроэлементы (медь), никотинамид способствуют всасы-
ванию железа в организме. Другие вещества пищи вызывают преципитацию
железа и образуют макромолекулы, которые не всасываются в кишечнике.
Адсорбцию железа ухудшают танины чая и кофе, фосфаты, карбонаты, окса-
латы, ЭДТА, используемая в качестве консервантов, антацидные препараты,
тетрациклины. Нормальному усвоению железа препятствует хеликобактерная
инфекция. Гемовое железо (Fe2+) растворимо в дуоденальном содержимом и
всасывается энтероцитами, на него не влияют компоненты пищи.
Адсорбция железа, как неорганического, так и гемового, происходит в
проксимальных отделах тонкого кишечника, наиболее интенсивно в 12-перст-
ной кишке. При дефиците железа зона всасывания расширяется дистально,
захватывая слизистую верхнего отдела подвздошной кишки, что обеспечивает
усиление его абсорбции.
Всасывание железа в кишечнике является ключевым пунктом в
гомеостазе железа и включает следующие этаны:
• окисление Fe3+ в Fe2+;
• всасывание железа апикальной мембраной энтероцита и внутрикле-
точный транспорт;
• перенос железа через базолатеральную мембрану с помощью транс-
портера железа ферропортина (IREG-1).
В 12-перстной кишке Fe3' превращается в Fe2+ с помощью витамин С-за-
висимой ферриредуктазы (DcytB - дуоденальный цитохром В) щеточной
каймы энтероцита. Предполагается возможное участие и других редуктаз
в этом процессе. При сидеропении синтез ферриредуктазы повышается,
что приводит к увеличению скорости всасывания алиментарного железа.
Свободное яЛлезо (Fe2+) транспортируется через щеточную кайму апикаль-
ной мембраны энтероцита (обращена в просвет кишечника) с помощью
Двухвалентного транспортера металла DMT-1 (divalent metal transporter-1).
Данный белок синтезируется только в криптах двенадцатиперстной кишки.
v '1 транспортирует также и другие двухвалентные металлы (марганец,
ооальт, медь, цинк, кадмий, свинец) из просвета кишечника в энтероцит.
рисутствие высоких концентраций ионов кальция, являющегося конкурент-
ам ингибитором DMT-1, снижает всасывание железа. Экспрессия DMT-J
регулируется запасами железа в организме, а также алиментарным железом
и повышается при ЖДА.
Молекулярные механизмы всасывания и транспорта в энтероцит гемо-
вого железа до конца не изучены. Исследования последних лет позволили
открыть единственный транспортер гемового железа в энтероцитах 12-перс-
тной кишки - НСР-1 (heme carrier protein-1). Наибольшей гем связывающей
активностью обладает 12-перстная кишка. Поступление гемового железа
не конкурирует с негемовым железом. Внутри энтероцита гемоксигеназа-1
высвобождает железо из протопорфирина, который затем распадается, а сво-
бодное железо вместе с негемовым железом выводится из энтероцита. При
ЖДА в клетках обнаруживается повышенное количество гемоксигеназы.
Поступившее в энтероцит Fe2+ может быть депонировано в виде феррити-
на или переносится с транспортным белком (мобилферрином или клеточным
трансферрином) к базолатеральной мембране, где находится трансмемб-
ранный белок ферропортин. Это единственный транспортер железа через
базолатеральную мембрану, известный в настоящее время (рис. 9). Высокая
экспрессия ферропортина обнаружена в клетках, участвующих в экспорте
Ife”, КрОВЬ
Транс .
феррин Т ферропортин-1 (=IREG)
дуктаза 0
Рис. 29. В 12-перстной кишке Fe3+
превращается в Fe2+ с помощью ви-
тамин С-зависимой ферриредуктазы
(дуоденальный цитохром В) щеточ-
ной каймы энтероцита. Свободное
железо (Fe2+) транспортируется через
щеточную кайму апикальной мембра-
ны энтероцита (обращена в просвет
кишечника) с помощью двухвалент-
ного транспортера металла — ДМТ-1
(divalent metal transporter-1). Посту-
пившее в энтероцит Fe2+ может быть
депонировано в виде ферритина
или переносится с транспортным
белком (мобилферрином) к базола-
теральной мембране, где находится
трансмембранный белок ферропор-
тин-1, передающий железо трансфер-
рину плазмы крови
48
железа (дуоденальные энтероциты, макрофаги, гепатоциты, синцитиотро-
(Ьобласты плаценты). Ферропортин играет решающую роль в транспорте
железа от матери плоду. В экспериментальных исследованиях показано,
что дефицит ферропортина у мышей вызывет гибель эмбриона. Подобно
DMT-1 ферропортин связывается с Fe2+, пересекая базолатеральную мембра-
ну где Fe2+ окисляется вновь в Fe3+. Ферропортин с участием ферроксидазы
(гефестин в энтероцитах и церулоплазмин в макрофагах и гепатоцитах) осу-
ществляет экспорт железа из клетки в плазму крови, где оно соединяется с
трансферрином. Функцией церулоплазмина, обладающего ферроксидазной
активностью, является мобилизация железа из депо, в основном из макро-
фагов. Кроме того, при стрессовых ситуациях, затрагивающих метаболизм
железа (хроническая кровопотеря), церулоплазмин способен стимулировать
процесс всасывания железа.
Особенности всасывания железа у новорожденных детей. У детей,
находящихся на грудном вскармливании, всасывание железа из грудного
молока происходит очень активно, хотя содержание железа в грудном молоке
невелико - 1,5 мг в литре. Биодоступность железа составляет 60%, этому
способствует особая его форма - железосодержащий белок лактоферрин.
В молекуле лактоферрина определено два центра связывания ионов Fe3+.
Лактоферрин в грудном молоке находится в двух формах - ненасыщенной
и насыщенной, соотношение которых меняется в зависимости от периода
лактации. В течение 1-3 месяцев жизни превалирует насыщенная железо-
транспортная форма лактоферрина. Наличие специфических рецепторов к
лактоферрину на эпителиальных клетках слизистой кишечника способс-
твует адгезии с ними лактоферрина и более полной его утилизации. Кроме
того, лактоферрин, связывая лишнее, невсосавшееся железо в кишечнике,
лишает условно-патогенную микрофлору необходимого для ее жизнеде-
ятельности микроэлемента и запускает неспецифические бактерицидные
механизмы. Установлено, что бактерицидная функция IgA реализуется
только в присутствии лактоферрина. Интересен тот факт, что грудное молоко
повышает абсорбцию железа из продуктов, употребляемых одновременно
с ним. Интенсивные обменные процессы у грудных детей приводят к тому,
что к 5-6-му месяцу жизни антенатальные запасы железа истощаются даже
У детеле благополучным перинатальным анамнезом и вскармливаемых
грудным молоком.
Транспорт железа в крови
Железо в сосудистом русле находится в связи с трансферрином. Трансфер-
рин - гликопротеид с Мм 88 кДа, синтезируется гепатоцитами в соответствии
с наличием железа в организме. В ответ на дефицит железа повышается
транскрипция трансферриновой мРНК, и, наоборот, при исчезновении не-
достатка железа синтез трансферрина снижается. В крови трансферрин
связывает молекулы Fe3+. Имеется три формы трансферрина: трансферрин,
содержащий два атома железа, трансферрин с одним атомом железа и апо-
трансферрин (не содержит железа) (рис. 30).
Трансферрин
апо 1 атом 2 атома
железа железа
Рис. 30. В организме человека имеется
три формы трансферрина: трансфер-
рин, содержащий два атома железа,
трансферрин с одним атомом железа и
апотрансферрин. Трансферрин с двумя
атомами железа эффективнее отдает
железо клеткам
В норме насыщение трансферрина
железом составляет около 30%, но
оно может достигать 100% при выра-
женной перегрузке железом. Из 30%
приблизительно 10% приходится на
трансферрин, содержащий два атома
железа. Снижение насыщения транс-
феррина железом приводит к увели-
чению содержания трансферрина с
одним атомом железа. Например, при
НТЖ 30% соотношение трансферрина
с двумя атомами железа к трансферри-
ну с одним атомом железа составляет
1:2, при НТЖ 15% -1:5. Трансферрины
плазмы имеют значительные различия
в способности к связыванию с рецеп-
торами к трансферрину. Трансферрин
с двумя атомами железа имеет в 4 раза
большую прочность связывания (аф-
финность) по сравнению с трансфер-
рином, содержащим один атом железа, и в 24 раза большее связывание, чем
апотрансферрин. Эти различия лежат в основе эффективной отдачи железа
клеткам. В физиологических условиях и при дефиците железа только транс-
феррин осуществляет железотранспортную функцию. Неспецифическое свя-
зывание железа с другими транспортными белками, в частности альбумином,
наблюдается при перегрузке железом. Биологическая функция трансферрина
заключается в его способности легко образовывать диссоциирующие ком-
плексы с железом, что обеспечивает создание нетоксического пула железа
в кровотоке, который доступен и позволяет распределять и депонировать
железо в организме. Связывая железо, трансферрин предохраняет клетки
от токсического воздействия супероксидных и гидроксильных радикалов,
перекиси и от инфекции, лишая некоторые микроорганизмы возможности
использовать железо для поддержания своего метаболизма. Металлосвязы-
вающий участок молекулы трансферрина не является строго специфичным
для железа. Трансферрин может связывать также хром, медь, магний, цинк,
кобальт, однако сродство этих металлов ниже, чем железа. Следует отме-
тить, что синтез трансферрина на фоне острого воспаления снижается,
что необходимо учитывать при интерпретации показателей обмена
железа.
Основным источником железа, связанного с трансферрином, является
поступление его из гемоглобина, катаболизированного макрофагами печени
и селезенки. Они фагоцитируют и лизируют старые и измененные эритро-
циты и с помощью гемоксигеназы освобождают железо из протопорфирина,
которое либо депонируется в макрофагах в виде ферритина, либо передается
ферропортином в плазму, где оно связывается с трансферрином.
Нормальная концентрация трансферииа в крови составляет 2-4 г/л.
Внутриклеточный метаболизм железа
Железо, связанное с трансферрином, поступает во все соматические
клетки организма человека. Железо трансферрина очень прочно связано и не
входит в клетку пассивно. Для поступления комплекса Fe3+ - трансферрин в
клетку необходимы специфические механизмы его связывания. Большинство
клеток, в том числе эритрокариоциты и гепатоциты, содержат на мембране
рецепторы к трансферрину, необходимые для поступления железа в клетку.
Трансферриновый рецептор - трансмембранный гликопротеин, состоит из
2 идентичных доменов, связанных дисульфидными мостиками с общей мо-
лекулярной массой 180 кДа. На каждом домене возможно связывание двух
молекул трансферрина. Трансферриновый рецептор (CD71) - это место входа
железа в клетку (рис. 31).
Комплекс Fe3+ - трансферрин связывается с рецептором трансферрина
и поступает в клетку путем эндоцитоза в составе клатринового пузырька
(рис. 32). В сформированной везикуле при низких значениях pH ионы железа
освобождаются от трансферрина и транспортируются во внутриклеточный
лабильный пул железа, а комплекс апотрансферрин - рецептор во внутри-
клеточных везикулах возвращается на наружную поверхность клетки, апо-
трансферрин поступает в плазму крови, а рецептор остается на мембране.
В эритрокариоцитах часть Fe3+ превращается в Fe2', которое используется
на синтез гемоглобина. Эритробласт может одновременно присоединить до
100 000 молекул трансферрина и получить 200 000 молекул железа. Судьба
Fe3‘ в клетке неоднозначна: либо используется на синтез железосодержащих
ферментов, либо откладывается в виде ферритина. Внутриклеточный сво-
бодный пул железа играе т важную роль в регуляции пролиферации клетки,
экспрессии трансферриновых рецепторов и др. Неиспользуемая часть железа
хранится внутриклеточно в молекуле ферритина в нетоксичной форме (Fe3+).
Fjeo-COO-
дн f f TfR
f~<0°
^^sTfR
Рис. 31. Рецептор трансферрина (TfR) и растворимый рецептор трансферрина (sTfR) Стрелкой
показан участок, в котором происходит специфический протеолиз
Растворимый Трансферрин — Fe
рецептор ।
трансферрина V I
в кровотоке I *
Рис. 32. Взаимодействие внутри- и внеклеточного пулов железа. Трансферрин связывается со спе-
цифическими рецепторами на поверхности клеток, после чего комплекс трансферрина с рецептором
погружается в составе эндосомы. При снижении pH в эндосоме (<5,5) железо высвобождается из
комплекса с трансферрином и транспортируется к местам внутриклеточной утилизации, главным
образом в митохондрии. Комплекс апотрансферрин — рецептор возвращается на клеточную поверх-
ность, где апотрансферрин высвобождается в плазму крови и в последующем может вступать в
следующий транспортный цикл. Трансферриновый рецептор возвращается на поверхность клетки,
часть рецепторов сбрасывается клеткой в кровь, формируя растворимый рецептор к трансферрину.
В клетке железо используется или депонируется в виде ферритина. Чем больше внутри клетки будет
Fe, тем активнее образуется ферритин и менее выражена на мембране экспрессия рецепторов к
трансферрину. Железо способно связываться в клетке с железорегуляторными белками, что приводит
к подавлению специфической мРНК и снижению синтеза трансферриновых рецепторов
Увеличение внутриклеточного лабильного пула железа приводит к стиму-
ляции синтеза ферритина и снижению экспрессии трансферриновых рецеп-
торов. Обратный процесс развивается при снижении в клетке пула железа.
Регуляция этих процессов осуществляется железорегуляторными элементами
(IREs - iron regulatory elements), которые находятся в тесном взаимодействии
с железорегуляторными белками (IRPs - iron responsive proteins), реагирую-
щими на изменение внутриклеточного пула железа.
Экспрессия трансферриновых рецепторов зависит от потребности клетки
в железе. Определенная часть рецепторов к трансферрину в виде мономе-
ров сбрасывается клеткой в сосудистое русло, образуя растворимые транс-
ферриновые рецепторы, способные связывать трансферрин. Количество
мембранных рецепторов находится в прямой пропорции с рецепторами,
обнаруженными в плазме крови. При перегрузке железом число клеточных
и растворимых рецепторов к трансферрину снижается. При сидеропении ли-
шенная железа клетка реагирует повышенной экспрессией трансферриновых
рецепторов на своей мембране, увеличением растворимых трансферриновых
рецепторов в плазме крови и снижением количества внутриклеточного фер-
ритина. Установлено, что чем выше плотность экспрессии трансферриновых
рецепторов, тем выраженнее пролиферативная активность клетки. Таким
образом, экспрессия рецепторов трансферрина зависит от двух фак-
торов - количества депонированного железа в составе ферритина и
пролиферативной активности клетки. Количество растворимых транс-
ферриновых рецепторов является чувствительным индикатором как
активности эритропоэза, так и дефицита железа.
Депонирование железа
Основными формами депонированного железа являются ферритин и его
агрегированная форма - гемосидерин, которые связывают «избыточное»
железо и депонируют его практически во всех тканях организма, но особенно
интенсивно в печени, селезенке, мышцах, костном мозге. Железо в виде фер-
ритина представляет лабильный пул, с помощью которого оно доставляется
в клетки тканей для синтеза гемоглобина, миоглобина, ферментов и т. п.
ферритин - комплекс, состоящий из
гидрата закиси Fe3+ и белка апоферритина,
представляет собой полукристаллическую
структуру (рис. 33). Молекулярная масса
ферритина составляет 480 кДа.
Ферритин образуется как сферическая
белковая молекула с полостью, в которой де-
понируется относительно большое количество
фосфатгидроокиси железа (Fe3+). Апоферри-
тин покрывает в виде оболочки ядро из фос-
фатгидроокиси железа (FeOOH). Молекула
ферритина содержит 24 однотипных цилинд-
рических субъединицы, образующие сфери-
ческую структуру с внутренним пространс-
твом диаметром около 70 А, сфера имеет поры
диаметром 10 А. Ионы Fe2’ диффундируют
через поры, окисляются до Fe3+, превращаются
в FeOOH и кристаллизируются.
Рис. 33. Ферритин в виде сфе-
рической белковой молекулы с
полостью, где депонируется фос-
фатгидроокись железа (Fe3+)
Ферритин содержит примерно 15-20% общего железа в организме. Мо-
лекулы ферритина растворимы в воде, каждая из них может аккумулировать
До 4500 атомов Fe3+. Превышение этого предела сопровождается отложением
фосфатных и гидроокисных форм железа (гемосидерин) в печени, мышце
сердца. Железо депонируется и высвобождается из ферритина в двухвалент-
ной форме. Ферритин локализуется преимущественно внутриклеточно,
где играет важную роль в кратковременном и длительном депонировании
Железа, регуляции клеточного метаболизма и детоксикации избытка железа.
Основными источниками сывороточного ферритина являются моноциты
крови, макрофаги печени (клетки Купфера) и селезенки.
Ферритин, циркулирующий в крови, практически не участвует в депони-
ровании железа, однако концентрация ферритина в сыворотке в физиологи-
ческих условиях прямо коррелирует с количеством депонированного железа
в организме. При дефиците железа, который не сопровождается другими
заболеваниями, так же как при первичной или вторичной перегрузке железом,
nvnujai vjm ipvppnгина и сыворотке дают дос таточно точное представление о
количестве железа в организме. Поэтому в клинической диагностике концен-
трация ферритина должна использоваться в первую очередь как параметр,
оценивающий депонированное железо.
Гемосидерин (производное ферритина) по структуре мало отличается от
ферритина. После того как макрофаг поглощает молекулы железа, напри-
мер после фагоцитоза старых эритроцитов, немедленно начинается синтез
апоферритина, который накапливается в цитоплазме, связывает железо, об-
разуя ферритин. Макрофаг насыщается железом в течение 4 ч, после чего в
условиях перегрузки железом в цитоплазме молекулы ферритина агрегируют
в мембрансвязанные частицы, известные как сидеросомы. В сидеросомах
молекулы ферритина кристаллизуются (рис. 34), формируется гемосидерин.
Рис. 34. Часть макрофага из очага организую-
щейся гематомы. Темные участки — несколько
сидеросом, сформированных кристаллами ге-
мосидерина. Множество мелких темных точек
в цитоплазме соответствует макромолекулам
ферритина
Гемосидерин «упакован» в лизосо-
мах и включает комплекс, состоящий
из ферритина, окисленных остатков
липидов и других компонентов. Гра-
нулы гемосидерина представляют
собой внутриклеточные отложения
железа, которые выявляются при
окраске цитологических и гисто-
логических препаратов по Перлсу.
В отличие от ферритина гемосидерин
нерастворим в воде, поэтому железо
гемосидерина с трудом подлежит
мобилизации и практически не ис-
пользуется организмом.
Регуляция баланса железа
Описано три механизма, регулирующих всасывание железа и, таким об-
разом, поддерживающих баланс железа в организме:
• алиментарный регулятор,
• депо-регулятор,
• эритроидный регулятор.
Алиментарный регулятор: при высоком содержании в энтероцитах
внутриклеточного железа подавляется экспрессия переносчика DMT-1 и
клетки становятся резистентными к получению дополнительного железа.
Этот регулятор реагирует только на алиментарное железо.
Депо-регулятор реагирует на общее содержание железа в организме, а не
на алиментарное железо. В случае когда количество железа в депо (печень,
селезенка, скелетные мышцы, эритроциты) падает, депо-регулятор увеличи-
вает поступление железа в энтероциты до того момента, пока не восстано-
вится его депо. Этот регулятор не оказывает значительного воздействия на
всасывание гемового железа, а направлен на накопление негемового железа.
Депо-регулятор также выполняет задачу предотвращения перегрузки железом
после того, как потребности организма в железе обеспечены полностью. Он
перестраивает работу кишечного эпителия таким образом, что всасывание
железа энтероцитами уменьшается в условиях повышенных запасов железа
и, наоборот, повышает всасывание железа, когда потребности организма
снижены. Предполагается, что ферритин, трансферрин и растворимый ре-
цептор к трансферрину являются теми гуморальными факторами, которые
передают сигналы от депо к кишечнику.
Эритроидный регулятор не реагирует на содержание железа в орга-
низме, а модулирует всасывание железа в ответ на потребности железа для
эритропоэза. Эритроидный регулятор имеет более высокую способность для
повышения абсорбции железа по сравнению с депо-регулятором. Так, при
анемии абсорбция железа возрастает до 20-40 мг в день, что значительно
выше тех величин, которые способен индуцировать депо-регулятор. Можно
предположить, что эритрон должен оказывать влияние на всасывание железа
в кишечнике, т. к. большая часть железа используется для нужд эритропо-
эза. Возможно, существует пока еще не выделенный гуморальный фактор,
передающий сигналы из костного мозга в кишечник. Эритропоэтин таким
фактором не является, т. к. в энтероцитах отсутствуют рецепторы к ЭПО.
Повышение всасывания алиментарного железа наблюдается при ЖДА,
талассемиях, сидеробластных анемиях, наследственной дисэритропоэти-
ческой анемии. В то же время при других формах анемий (наследственный
сфероцитоз, ЛИГА, серповидноклеточная анемия), характеризующихся теми
же изменениями эритропоэза, всасывание железа в кишечнике не стимулиру-
ется. Исходя из этого, гиперпролиферативиые анемии разделяют на 2 класса:
стимулирующие и не стимулирующие всасывание железа в кишечнике. При
анемиях первого класса эритроидные клетки разрушаются внутри костного
мозга (неэффективный эритропоэз), при анемиях второго класса эритроциты
разрушаются в периферической крови. Значение места распада эритроцитов
пока не определено.
Указанные регуляторы всасывания железа являются гуморальными фак-
торами, они поддерживают гомеостаз железа в целом организме. Для поддер-
жания гомеостаза железа в одной клетке существуют особые регуляторные
механизмы. Единственной генетической системой регуляции являются
изменения в экспрессии ферритина и рецепторов к трансферрину.
В последние годы уделяется большое внимание белку гепсидину (hepatic
bactericidal protein), который является ключевым регулятором обмена железа.
Это гормон, состоящий из 25 аминокислотных пептидов, синтезируется пре-
имущественно в печени, хотя низкая экспрессия его обнаружена в мышцах,
кишечнике, желудке, легких и сердце, экскретируется с мочой. Синтез и сек-
реция гепсидина контролируется тремя белками: HFE (белок наследственного
гемохроматоза), гемоювелин, рецептор к трансферрину-2. Гепсидин обладает
как антибактериальной активностью к грамотрицательным и грамположи-
тельным бактериям, так и противогрибковой активностью.
Механизм действия гепсидина:
• ингибирует всасывание железа в кишечнике,
• блокирует транспорт железа через плаценту,
• блокирует выход железа из макрофагов.
Молекулярной мишенью гепсидина является ферропортин (трансмемб-
ранный протеин), экспрессирующийся макрофагами, включая клетки
Купфера, гепатоцитами, энтероцитами, клетками плаценты. Выход железа
из клетки ингибируется связыванием гепсидина с ферропортином, послед-
ний погружается в цитоплазму и разрушается в лизосомах клетки. Таким
образом, удаление ферропортина из базолатеральной мембраны приво-
дит к блокаде выведения железа из клетки в плазму крови. Абсорбция
железа энтероцитами осуществляется только в течение 2 дней, после чего
они слущиваются и удаляются. Таким образом, транспорт железа ферро-
портином через базолатеральную мембрану приводит либо к связыванию
с трансферрином плазмы, либо к выведению из организма со слущиваю-
щимся эпителием кишечника. Когда запасы железа адекватны или высокие,
синтезирующийся в печени гепсидин циркулирует в тонком кишечнике
и вызывает интернализацию (погружение) ферропортина в цитоплазму
клетки, блокируя единственный путь транспорта железа из энтероцита в
плазму. При снижении запасов железа продукция гепсидина подавляется,
ферропортин экспрессируется в большей концентрации на базолатеральной
мембране энтероцита и других клеток, тем самым повышается выход железа
из клетки в плазму и связывание его с трансферрином. Сходный механизм
взаимодействия гепсидин - ферропортин объясняет присутствие макрофагов,
перегруженных железом, которые часто встречаются при воспалительных
заболеваниях, сопровождающихся высокой продукцией гепсидина. При вы-
сокой концентрации гепсидина блокируется ферропортин и, следовательно,
выход железа из макрофагов.
Гепсидин быстро синтезируется в ответ на поступление алиментарного
железа. В экспериментальных условиях инъекция одной дозы синтетического
гепсидина мышам вызывала критическое падение концентрации железа в
плазме крови в течение 1 часа. Таким образом, взаимодействие гепсидина
с ферропортином создает нормальную концентрацию внеклеточного же-
леза. Прямое взаимодействие гепсидина с ферропортином обеспечивает
гомеостатический механизм, позволяющий регулировать уровень железа в
плазме и его распределение в тканях. Механизм гиперпродукции гепсидина
обусловлен воздействием провоспалительных цитокинов, преимущественно
ИЛ-6, и наблюдается при воспалительных заболеваниях. Сниженная секреция
гепсидина имеет место при ЖДА, гипоксии, неэффективном эритропоэзе
(рис. 35).
Анемии, характеризующиеся перегрузкой железа, сопровождаются не-
эффективным эритропоэзом и повышением всасывания железа в тонком
кишечнике. Наиболее часто такая анемия регистрируется при талассемиях.
Парадоксальная ситуация возникает с синтезом гепсидина при талассемии.
У этих больных концентрация гепсидина в моче низкая, несмотря на высокое
содержание ферритина в сыворотке крови. Ингибицию гепсидина рассмат-
ривают как нецелесообразный физиологический ответ, который приводит к
ухудшению перегрузки железом в тканях. Данный факт предположительно
интерпретируется влиянием анемии на синтез гепсидина, ассоциированной с
повышенным или неэффективным эритропоэзом. Низкий уровень гепсидина
Низкая
концентрация
гепсидина (ЖДА)
Fe
Fe
Клетки,
участвующие
в транспорте
железа
(энтероцит,
гепатоцит,
макрофаг)
Высокая концентрация
гепсидина
(воспаление, гиперферремия)
Fe
Fe
Высвобождение железа в плазму
Рис. 35. Биологические эффекты взаимодействия гепсидин — ферропортин на транспорт железа
из клетки. При низкой концентрации гепсидина транспорт железа в плазму осуществляется
ферропортином через базолатеральную мембрану. При высокой его концентрации гепсидин
связывает ферропортин, вызывает интернализацию (погружение) его в цитоплазму клетки,
блокируя единственный путь транспорта железа из энтероцита или макрофага в плазму
при наследственных анемиях может быть одним из факторов, приводящих к
гиперабсорбции железа, перегрузке и повреждению тканей, развитию фиб-
роза.
Выведение железа
Физиологические потери железа организмом практически неизменны. За
сутки из организма мужчины теряется около 1 мг железа с мочой, потом при
стрижке ногтей, волос, слущивающимся эпителием кожи. Кал содержит как
невсосавшееся железо, так и железо, выделяющееся с желчью и в составе
слущивающегося эпителия кишечника. У женщин наибольшая потеря железа
происходит с менструацией. В среднем потеря крови за одну менструацию
составляет около 30 мл, что соответствует 15 мг железа (за сутки женщина
теряет от 0,8 до 1,5 мг железа). Исходя из этого, суточная потребность в же-
лезе у женщин детородного возраста увеличивается до 2-4 мг в зависимости
°т объема кровопотери.
Согласно современным представлениям наиболее адекватным тестом для
оценки метаболизма железа в организме является определение концентрации
ферритина и железа в сыворотке крови, гомогенатах печени, трансферрина,
иЖСС, насыщения трансферрина железом, гепсидина, содержания раство-
римых трансферриновых рецепторов в сыворотке крови (табл. 7).
Таблица /
Лабораторные показатели обмена железа в норме
Сывороточное железо Мужчины 0,5-1,7 г/л (11,6-31,3 мкмоль/л) Женщины 0,4-1,6 г/л (9-30,4 мкмоль/л) Дети: до 2 лет 0.4-1.0 г/л (7-18 мкмоль/л) 7-16 лет 0,5-1,2 г/л (9-21,5 мкмоль/л)
Общая железосвязывающая способность (ОЖСС) 2,6-5,0 г/л (46-90 мкмоль/л)
Трансферрин Дети (3 мес. - 10 лет) 2,0-3,6 г/л Взрослые 2-4 г/л (23^45 мкмоль/л) Пожилые (>60 лет) 1,8-3,8 г/л
Насыщение трансферрина железом (НТЖ) 15-45%
Ферритин сыворотки крови Мужчины 15-200 мкг/л Женщины 12-150 мкг/л Дети: 2-5 месяцев 50-200 мкг/л 0,5-16 лет 7-140 мкг/л
Растворимые рецепторы к трансферрину Зависят от фирмы-производителя наборов
Гипохромные анемии
Железодефицитная анемия
Распространенность железодефицитных состояний у женщин детородного
возраста и детей в некоторых регионах России достигает 30 -60%. По данным
ВОЗ, число лиц с латентным дефицитом железа в мире составляет 3,6 млрд,
а ЖДА - 1,8 млрд человек. При оценке заболеваемости болезнями крови и
кроветворных органов ликвидаторов аварии на ЧАЭС, работников атомной
промышленности России выявлено преобладание анемий, среди которых
основную часть занимают железодефицитные анемии (84,9% - у женщин
и 78,4% - у мужчин). Железодефицитные состояния являются причиной
нарушения метаболических процессов в организме, снижения работоспособ-
ности у взрослых, увеличения восприимчивости к острым респираторным
вирусным инфекциям, вызывают задержку роста и развития детей. В связи
с этим большое значение имеет своевременная диагностика нарушений
обмена железа, железодефицитной анемии (ЖДА), мониторинг в процессе
лечения и профилактика дефицита железа у населения. Основные причины
дефицита железа представлены в табл. 8.
юопица а
Наиболее частые причины железодефицитной анемии
Хроническая кровопотеря
Кровопотери у женщин
. меноррагии, метроррагии, роды, лактация
Кровопотери из желудочно-кишечного тракта
• язвенная болезнь
• язвенный колит
• опухоли, полипы
• геморрой
• дивертикулез
• глистные инвазии (анкилостомидоз)
• варикозное расширение вен пищевода
• прием препаратов (нестероидные противовоспалительные средства,
салициллаты, кумарины, глюкокортикоиды)
Кровопотери в замкнутые полости (нарушение реутилизации железа)
• гпомические опухоли
• изолированный легочный сидероз
• наличие эндометриотических полостей, не связанных с полостью матки
Повышенная потребность в железе
• беременность
• лактация
• быстрый рост в пубертатном периоде
Неадекватное поступление железа (растительно-молочная диета)
Причины дефицита железа у детей
• недоношенность
• многоплодная беременность
• искусственное вскармливание
• быстрый рост
• инфекции
Донорство
Нарушение транспорта железа
• наследственная атрансферринемия
• приобретенная гипотрансферринемия (нарушение белоксинтезирующей
функции печени, протеинурии)___________________________________________
Нарушение всасывания _________________
• хронический гастрит, энтерит
• резекция тонкой кишки, желудка
• лямблиоз
• глистные инвазии
* целиакия
Заместительная терапия рекомбинантным эритропоэтином
(хроническая почечная недостаточность, анемии хронических заболеваний,
миелодиспластический синдром и др.)
Хроническая кровопотеря является наиболее частой причиной развитая
ЖДА. В 500 мл крови содержится 250 мг железа (в 1 мл - 0,5 мг), поэтому
при продолжающемся кровотечении происходит значительная потеря желе-
за, несмотря на увеличение всасывания его в кишечнике. При ежедневной
потере 6-12 мл крови развивается отрицательный баланс железа. Наиболее
частыми источниками кровопотерь являются желудочно-кишечный тракт и
мочеполовые органы (миома матки, эндометриоз, функциональные менор-
рагии).
Общая потребность в железе во время беременности значительно выше,
чем у небеременных женщин. ВI триместре потребности в железе несколько
снижены из-за прекращения менструации, что сохраняет 0,56 мг железа в
сутки (160 мг за беременность). Необходимость в железе значительно воз-
растает во II и III триместре вследствие увеличения эритроцитарной массы
и роста плода и плаценты. Количество эритроцитов может увеличиться на
20%, что требует дополнительно 400 мг железа. Ежедневная потребность в
железе к III триместру увеличивается до 6 мг/сутки. Общие потребности в
железе, связанные с беременностью, представлены в табл. 9.
Таблица 9
Общие потребности в железе, связанные с беременностью
(Beaton G.H. Am. J. Clin. Nutr. 2000; 72: 257-264)
Показатели Количество железа, мг
Общие затраты железа при беременности:
• плод 270
• плацента 90
• увеличение эритроцитарной массы 450
• обязательные потери (эпителий кишечника, 230
кожа, моча)
Всего 1040
Кровопотеря в родах 150
Общая потребность, связанная с беременностью 1190
Реутилизация железа при распаде эритроцитов, образовавшихся во время беременности -450
Отсутствие менструации во время беременности -160
Всего -610
«Чистое» увеличение потребности в железе во время беременности 580
.ж?
Диагностика Анемии
J "Алкор Био», 197110, С.-Петербург, а/я 243
"ЛеФон: (812) 596 67 76, 596 67 79, 596 67 81, телефон/факс (812) 596 67 80
KOrbio@peter|jni<,ru www.alkorbio.ru ___________________________
О^Алкор Б»
• Пренатальная диагноста
Диагностика функций
надпочечников
Диагностика репродукп
функции
• Диагностика функций
щитовидной железы
• Неонатальный скрининг)
• Онкомаркеры
• Диагностика анемии
• Диагностика ToRCH-инф
• Диагностика хламидиоз
• Диагностика гепатита С
• Аллергодиагностика
• Программный комплекс
«Пренатальный монитор
синдрома Дауна»
• Программа «ИФА-масте
• Автоматический
ИФА-анализатор «Alisei»
Современная иммуноферментная тест-система
для определения Ферритина в сыворотке крови
• Метод - твердофазный 1/1ФА
• Высо^я чувствительность и специфичность
•Диапазон определения - 5-1000 нг/мл
• Время анализа - 1 час
•Жидкие реагенты, готовые к использованию
• Срок годности - 12 месяцев
• Возможность использования полуавтоматических
и автоматических анализаторов открытого типа
Международный сертификат ISO 9001-2000
^-маркировка - соответствие требованиям
свР°союза по Безопасности
ру №фс 012а2004/5444-06
по каталогу 100-22
все тесты иля иистносп
у одного производителя
К
Полный клинический анализ крови
Подсчет ретикулоцитов, определение параметров ретикулоцитов
Определение ненасыщенной железосвязывающей способности крови
Определение общей железосвязывающей способности крови
Определение концентрации железа в сыворотке
Определение степени насыщения трансферрина
Определение концентрации трансферрина
Определение концентрации гаптоглобина
Определение концентрации ферритина
Определение концентрации фолата
Определение концентрации фолата в эритроцитах
Определение концентрации витамина В12
Определение антител к внутреннему фактору
Определение концентрации эритропоэтина
Определение концентрации растворимого рецептора трансферри
n^irnarv Care
Представительство компании BECKMAN
123056, Москва, ул. Ю. Фучика, д. 6, стр. 2
Тел.: (495) 937 16 64,937 16 63, факс (495) 254 64 07
E-mail: beckman.ru@beckmancoulter.com
BECKMAN
COULTER
В исследовании NHANES III, проведенном в США, показано, что средний
уровень сывороточного ферритина у женщин в возрасте 20-44 лет составляет
36 нг/мл, что соответствует запасам железа около 300 мг (1 нг/мл СФ = 8 мг
железа в тканевых депо взрослого человека). Учитывая тот факт, что во время
беременности дополнительно требуется 580-800 мг железа, в этот период
абсорбция железа должна увеличиться на 280-500 мг (1,2-2 мг/сут) для
поддержания нормального баланса железа в организме. Однако всасывание
в кишечнике строго регулируется запасами железа. I [оэтому, если беремен-
ность наступает при адекватных запасах железа в организме, сначала уровень
кишечной абсорбции достаточно низкий и повышенные потребности в же-
лезе удовлетворяются из тканевых депо. Во второй половине беременности
истощение запасов железа стимулирует всасывание железа в кишечнике,
которое может увеличиться в 5-9 раз к концу П-1 II триместра.
физиологические роды всегда сопровождаются кровопотерей, состав-
ляющей в среднем 250-500 мл, во время операции кесарева сечения сред-
няя кровопотеря - около 900 мл. После родов в течение 6 недель железо
теряется с выделениями из родовых путей. Дефицит железа неизбежен
у женщин, имевших четверо и более родов, так как при каждой бере-
менности, родах, лактации оно расходуется в значительной степени. По
данным ряда авторов, ЖДА не самый частый вид анемии у беременных.
Кроме ЖДА, наиболее часто встречаются АХЗ, развивающиеся на фоне
инфекционно-воспалительных заболеваний (урогенитальные инфекции,
пиелонефрит и др.).
Ц период внутриутробного развития плода концентрация железа во всех
органах и тканях плода остается примерно одинаковой и только в последние
недели беременности происходит его накопление в печени и селезенке в виде
депо. У недоношенных детей депо не успевает образоваться, вследствие
чего дети рождаются с дефицитом железа. У детей в возрасте от 2 месяцев
до 1 года развитию ЖДА способствует наличие скрытого дефицита железа у
матери во время беременности, осложненного течения беременности, искус-
ственное вскармливание неадаптированными молочными смесями, коровьим
и козьим молоком, отсутствие в рационе мясных блюд, интенсивный рост.
Согласно литературным данным, 90% детей раннего и более 30% школьного
возраста страдают дефицитом железа. Быстрый рост в пубертатном периоде
вызывает повышенные потребности в железе, что обусловлено увеличением
количества эритроцитов и мышечной массы. У девочек дефицит железа воз-
никает значительно чаще, чем у мальчиков, что связано с дополнительным
фактором - кровопотерей во время менструации.
Частая сдача крови донорами истощает запасы железа и может привес-
ти к развитию ЖДА. Сдача донором 300 мл крови лишает его организм
150 мг железа. Таким образом, регулярные доноры находятся в группе
Риска развития ЖДА. Мужчинам, сдающим кровь более 4 раз (женщинам
более 2 раз) в год, необходимо проведение обязательного исследования всех
параметров метаболизма железа, а также современных ретикулоцитарных
показателей, отражающих эритропоэтическую активность и состояние
запасов железа.
Дефицит железа возможен в результате нарушения всасывания в кишечни-
ке (после обширной резекции желудка или тонкой кишки, при хроническом
энтерите). У пожилых людей он обычно связан с однообразным питанием и
преобладанием молочно-растительной диеты.
Дефицит железа может наблюдаться у бегунов и пловцов на длинные
дистанции.
Нарушение транспорта железа из депо к эритрону имеет место при сниже-
нии синтеза трансферрина (наследственная атрансферринемия встречается
с частотой 1:100 000 новорожденных).
Больные с хронической почечной недостаточностью, в программу те-
рапии которых входит гемодиализ, предрасположены к дефициту железа,
т. к. потеря крови в процессе диализа и микрокровопотери сопутствуют
уремии. Терапия рекомбинантным эритропоэтином (рЭПО) и витамином В12
приводит к стимуляции эритропоэза и усиленному потреблению железа
эритрокариоцитами, что при наличии истощенного депо железа может спо-
собствовать развитию железодефицитной анемии. К потере железа с мочой
приводит также внутрисосудистый гемолиз, обусловленный пароксизмаль-
ной ночной гемоглобинурией или другими причинами. В каждом случае
дефициту железа предшествует в первую очередь истощение его запасов из
макрофагов печени, селезенки и костного мозга (латентный железодефи-
цит), затем уменьшается транспортное железо, насыщение трансферрина
железом, увеличивается всасывание железа, далее снижается активность
железосодержащих ферментов, и в последнюю очередь нарушается синтез
гемоглобина и развивается ЖДА.
Клинические проявления. Выделяют несколько стадий железодефицит-
ного состояния: предлатентный, латентный дефицит железа и собственно
железодефицитная анемия. В первую очередь снижается содержание депони-
рованного железа в органах и тканях, затем транспортного железа, несколько
позже - железа гемсодержащих ферментов и затем - железа, необходимого
для синтеза гемоглобина.
Предлатентный дефицит железа - состояние, которое сопровождается
увеличением всасывания железа в желудочно-кишечном тракте. Клинические
симптомы отсутствуют. Лабораторные показатели (картина периферической
крови, показателей обмена железа) остаются в пределах нормы. Единствен-
ным методом, позволяющим определить эту стадию, является радиоизотоп-
ный, обнаруживающий присущую железодефициту повышенную абсорбцию
59Fe3+ в кишечнике. Однако его применение ограничено у большинства па-
циентов (дети, женщины фертильного возраста).
Латентный (скрытый) дефицит железа сопровождается сидеропе-
ническим синдромом, обусловленным тканевым дефицитом железа. Он
включает: сухость кожи, изменения ногтей (ломкость, слоистость, исчер-
ченность, «койлонихии» - ногти ложкообразной формы), сглаженность
сосочков языка, ангулярный стоматит («заеды» в углах рта), извращение
вкуса и обоняния, выпадение волос, кариес, мышечную слабость, от-
ставание в физическом и психомоторном развитии детей. Многообразие
клинических симптомов дефицита железа объясняется широтой спектра
метаболических нарушений вследствие снижения активносш железосо-
держащих ферментов.
Лабораторные показатели метаболизма железа на этой стадии характери-
зуются гипоферритинемией, снижением концентрации сывороточного железа,
увеличением содержания трансферрина и растворимых рецепторов для транс-
феррина, а также общей железосвязывающей способности (ОЖСС). В костном
мозге развивается железодефицитный эритропоэз, который характеризуется
снижением количества сидеробластов, увеличением свободного протопорфи-
рина в эритроцитах, отсутствием в макрофагах гемосидерина (отрицательная
реакция Перпса). Однако синтез гемоглобина на этой стадии не нарушен и
эритроцитарные показатели (Hb, RBC, MCV, МСН, МСНС) сохраняются в
пределах нормы. Следует помнить, что в случае незначительного снижения
MCV, МСН и повышения RDW при нормальной концентрации гемоглобина
можно предположить наличие латентного дефицита железа и исследовать
содержание ферритина в сыворотке крови. При трансформации латентного
дефицита железа в железодефицитную анемию повышение ROW (показатель
анизоцитоза эритроцитов) может предшествовать изменениям других эритро-
цитарных параметров.
Железодефицитная анемия (ЖДА) проявляется гипоксическим (слабость,
головокружение, обмороки, тахикардия, одышка) и сидеропеническим
синдромами. Дефицит железа в организме приводит к нарушению функции
иммунокомпетентных клеток и к нарушению клеточных механизмов имму-
норезистентности, что является причиной частых острых респираторных и
вирусных заболеваний, особенно у детей.
В зависимости от состояния эритропоэтической активности костного
мозга различают регенераторную и гипорегенераторную стадии ЖДА,
а в соответствии с лабораторными показателями три степени тяжести
ЖДА:
• легкую - содержание гемоглобина более 90 г/л,
• среднюю - 70-90 г/л,
• тяжелую - менее 70 г/л.
Лабораторные показатели. Регенераторная стадия (гиперпролифера-
тивная) ЖДА характеризуется нормальной клеточностью костного мозга,
умеренной гиперплазией клеток красного ряда (количество их достигает
40-60% от общего количества миелокариоцитов), преобладанием базофиль-
ных и полихроматофильных эритробластов. Гиперплазия эритроидного
ростка обусловлена компенсаторным усилением синтеза эритропоэтина в
ответ на тканевую гипоксию. При недостатке железа клетки эритрона син-
тезируют гемоглобин в меньшей концентрации, что приводит к уменьшению
гемоглобинизации цитоплазмы созревающих эритрокариоцитов. Поскольку
Деление клеток зависит от концентрации гемоглобина, число митозов во
время их созревания может быть повышенным и это приводит к образова-
нию не только гипохромных, но и мелких по размеру эритрокариоцитов и
соответственно ретикулоцитов и эритроцитов (микроцитов). В пунктатах
костного мозга увеличено число микрогенераций полихроматофильных
эритробластов (рис. 36) с узким, чаще неотчетливым ободком цитоплазмы,
Рис. 36. Костный мозг. Ми-
крогенерация полихрома-
тофильных эритробластов
при железодефицитной
анемии. хЮОО
имеющим неровные контуры. В эритрокариоцитах наблюдается неравно-
мерная окраска цитоплазмы.
Другие миелоидные ростки не изменены.
В костном мозге снижено количество сидеробластов - менее 10% (окраска
по Перлсу), а также макрофагов с гранулами гемосидерина (депонированное
железо).
Общий анализ крови. Количество эритроцитов находится в пределах
нормы. Отмечается снижение концентрации гемоглобина, показателей ге-
матокрита, МСН (менее 27 пг), МСНС (менее 30 г/дл), MCV (менее 80 фл).
Показатель анизоцитоза - RDW может оставаться нормальным, что сви-
детельствует о преобладании однородных клеток с малым объемом, либо
немного увеличен (рис. 37). В регенераторной стадии железодефицитной
анемии эритроцитарная гистограмма имеет обычную форму и лишь смещает-
ся влево. Выраженное снижение объема эритроцитов отражается и на форме
тромбоцитарной гистограммы: она не заканчивается на базисной кривой, а
поднимается правой своей частью вверх (рис. 38, а), либо появляется вто-
рой пик в области 30 35 фл, что указывает на наличие в образце популяции
микроэритроцитов (рис. 38, б).
RBC 4,58 3,8—5,5 x 10’2/л
Hb 76 130-155 г/л
Ht 33 36-48%
MCV 60,9 80-95 фл
MCH 16,6 27-31 пг
MCHC 272 300—380 г/л
RDW 19,0 11,5-14,5%
Рис. 37. ЖДА - регенераторная стадия Эрит-
роцитарная гистограмма. Штрихом показано
нормальное распределение эритроцитов
(Sysmex SE-9000)
Рис. 38. Тромбоцитарная гистограмма при ЖДА
Рис. 39. Периферическая кровь. Гипохромия и микроцитоз
эритроцитов, единичные мишеневидные эритроциты (1), ова-
лоциты (2) при железодефицитной анемии. хЮОО
Морфологическим
признаком железодефи-
цитной анемии является
гипохромия эритроцитов
и анизоцитоз со склон-
ностью к микроцитозу.
В мазках крови могут
обнаруживаться еди-
ничные мишеневидные
эрит^Лциты, овалоциты
(рис. 39). Снижено коли-
чество сидероцитов.
Относительное и аб-
солютное количество
ретикулоцитов при ЖДА
в пределах нормы, что
свидетельствует о сохраняющейся регенераторной способности костномоз-
гового кроветворения на фоне дефицита железа, либо несколько повышено
при наличии кровотечения. Количество лейкоцитов и тромбоцитов при
ЖДА остается в пределах нормы. При хронических кровотечениях может
наблюдаться небольшой тромбоцитоз либо тромбоцитопения. СОЭ чаще
бывает нормальной.
По мере дальнейшего нарушения процессов гемоглобинообразования
происходит еще большее снижение MCV, МСН и МСНС. У таких больных
эритроцитарная гистограмма имеет вид одиночного пика, значительно
сдвинутого в левую сторону, RDW (показатель анизоцитоза эритроцитов)
увеличен. Отмечается появление микроретикулоцитов - ретикулоцитов со
сниженным средним объемом (MRV), и наблюдается снижение среднего
объема сферулированных эритроцитов (MSCV). Показатели, оценивающие
степень зрелости ретикулоцитов (IRF, HLR%, HLR#), находятся в пределах
нормальных величин.
Гипорегенераторная стадия ЖДА характеризуется истощением про-
лиферативной активности костного мозга, снижением количества сиде-
робластов, повышением неэффективного эритропоэза, что приводит к
снижению количества эритроцитов, появлению популяции красных клеток
с увеличенным объемом. Эритроцитарная гистограмма уплощается и зна-
чительно растягивается вдоль оси X, указывая на наличие двух популяций
эритроцитов - микро- и макроцитов. MCV может увеличиваться, так как
является усредненным показателем объема эритроцитов. Присутствие мик-
ро- и макроцитов приводит к повышению RDW, что коррелирует с наличи-
ем смешанного анизоцитоза в мазках периферической крови (рис. 40, 41).
Может наблюдаться анизохромия эритроцитов, а также незначительный
пойкилоцитоз. Количество ретикулоцитов снижено, что отражает снижение
пролиферативной активности эритроидных клеток.
При ЖДА отмечается снижение показателя СНг или RetHe (RET-Y) <28 пг,
отражающего концентрацию гемоглобина в ретикулоцитах, который явля-
ется индикатором железодефицитного эритропоэза. Процент гипохромных
эритроцитов (% Hypo) более 10% указывает также на железодефицитное
состояние. Для функционального дефицита железа пороговое значение (cut-
off %) Hypo составляет более 5%.
На фоне приема препаратов железа отмечается незначительное повышение
количества эритроцитов, увеличение концентрации гемоглобина, МСН, МСНС,
MCV. Показатель анизоцитоза (RDW) значительно повышается, что свидетель-
ствует о гетерогенности популяции эритроцитов. Эритроцитарная гистограмма
становится бимодальной, первый пик ее характеризует популяцию с низким
RBC 3,47 3,8-5,5 х 1012/л
НЬ 67 130-155 г/л
Ht 20,2 36-48% ,
MCV 74,1 80—95 фл '
МСН 19,3 27-31 пг
МСНС 261 300-380 г/л
RDW 28,1 11,5-14,5%
Рис. 40. Железодефицитная анемия. Гипоре-
генераторная стадия
Рис. 41. Периферическая кровь при гипорегенераторной стадии ЖДА. Выраженный смешанный
анизоцитоз эритроцитов и гипохромия. Видны анулоциты, шизоциты, овалоциты. хЮОО
RBC
RBC 2,5 3,8-5,5 х 1012/л
НЬ 30 130-155 г/л
Ht 12,6 36-48%
MCV 50,3 80-95 фл
МСН 12,2 27-31 пг
МСНС 242 300-380 г/л
RDW 17,4 11,5-14,5%
объемом (микроциты), второй — по-
явление эритроцитов с нормальным
объемом (нормоциты) (рис. 42).
Максимальный подъем ретикуло-
цитов приходится на 16-18-й день
лечения, в то время как показатель
фракции незрелых ретикулоцитов
(IRF) увеличивается несколько рань-
ше, что позволяет использовать его
для более ранней оценки активации
эритропоэза в мониторинге терапии
больных ЖДА. Отмечается также
250 fl
Рис. 42. Бимодальная эритроцитарная гисто-
грамма б-й с ЖДА на фоне приема препа-
ратов железа
повышение содержания гемоглобина в ретикулоцитах (RET-Y или RET-He)
и снижение % Hypo.
ЖДА характеризуется снижением содержания железа, ферритина в сыво-
ротке крови, % насыщения трансферрина железом, повышением концентра-
ции растворимых рецепторов к трансферрину (sTfR), ОЖСС, трансферрина,
увеличением свободных протопорфиринов эритроцитов.
Информативным показателем оценки метаболизма железа является коэф-
фициент насыщения трансферрина железом (НТЖ), который рассчитывается
по формуле:
т , Железо (мкг/дл) ,лл
Насыщение трансферрина железом (%) = —------------—;—-——- х 100.
Трансферрин (мг/дл) х 141
При ЖДА показатель НТЖ снижается менее 15%. Концентрация ферритина
в сыворотке крови отражает величину запасов железа в организме. Снижение
уровня ферритина (менее 15 мкг/л) наблюдается как при латентном дефиците
железа, так и при ЖДА. Все вышеперечисленные показатели должны ис-
пользоваться только в комплексе с гематологическими параметрами, т. к. их
изменения наблюдаются при самых разнообразных заболеваниях. Содержание
ферритина в сыворотке крови не всегда отражает истинные запасы железа в
организме и часто повышается, независимо от количества депонированного
железа, при воспалении, инфекциях, онкологических заболеваниях, заболе-
ваниях печени и других состояниях. В этих случаях определение высокой
концентрации sTfR позволяет диагностировать ЖДА. Концентрация раство-
римых рецепторов к трансферрину отражает потребности преимущественно
эритроидных клеток в железе, поэтому в случаях повышения эритропоэтичес-
кой активности костного мозга и выраженного дефицита железа отмечается
повышение их содержания в сыворотке крови. Количество sTfR остается
стабильным при воспалительных процессах и беременности.
Величину запасов железа можно определить с помощью десфералового
теста. У здоровых людей после введения 500 мг десферала выводится в сутки
0,8-1,3 мг железа, а при его дефиците - менее 0,4 мг.
ЖДА необходимо дифференцировать с другими гипохромными анемиями:
талассемией, анемией хронических заболеваний, наследственными и приоб-
ретенными анемиями, связанными с нарушением синтеза порфиринов.
Анемии, связанные с нарушением
синтеза порфиринов
Анемии этой группы обусловлены недостаточной или аномальной ути-
лизацией внутриклеточного железа при синтезе гемоглобина, несмотря
на нормальное или даже повышенное содержание железа в митохондриях
эритрокариоцитов. Такие дефекты могут быть связаны с наследственными
нарушениями, главным образом при синтезе порфиринов, или с приобретен-
ным характером поражения, например в результате отравления свинцом или
недостаточности витамина В6. Отличительным признаком этого типа анемий
является насыщение организма железом, в связи с чем ранее использовался
термин «сидероахрестические», т. е. железонасыщенные анемии.
Обмен порфиринов
Порфирины - органические вещества, в основе которых лежит тетрапир-
рольное кольцо с четырьмя пиррольными кольцами или ядрами, соединен-
ными между собой метановыми мостиками или связями (=СН-) (рис. 43).
Рис. 43. Структура гема а — в виде струк-
турной формулы; б — в пространственном
изображении
Порфирины широко распростра-
нены в природе в виде комплексов
с ионами железа - НЬ и Mb, входят
в состав ферментов (каталазы, ок-
сидазы, цитохромоксидазы и др.).
Порфирины синтезируются во всех
клетках организма, но основная
масса - в эритроидных клетках кост-
ного мозга. На основе порфиринов
в эритробластах костного мозга
синтезируется гемоглобин. Синтез
порфиринов происходит также в
клетках печени, они используются
для образования окислительно-вос-
становительных ферментов (катала-
зы, цитохромов, пероксидазы).
Синтез гема начинается в ми-
тохондриях с присоединения сук-
цинил-кофермента-А (СоА) к гли-
цину, в результате чего образуется
8-аминолевулиновая кислота (АЛК).
Этот процесс идет с участием фер-
мента АЛК-синтетазы, в качестве
кофермента которого выступает пи-
ридоксаль-5-фосфат (производное
витамина В6). Активность фермента
может быть угнетена химическими
веществами, в частности свинцом,
алкоголем, снижена глюкозой или гемом. Считается, чтАЛК-синтетаза ограни-
чивает скорость биосинтеза гема в печени. При конденсации двух молекул 8-АЛК
образуется порфобилиноген (ПБГ). Эта реакция катализируегся дегидразой АЛК,
активность которой снижается под действием свинца и гемина. При конденсации
четырех молекул ПБГ синтезируется уропорфириноген (УРО). В этом процессе
участвуют 2 фермента: уропорфириноген-Ш-синтетаза и порфобилиноген-де-
заминаза. Из УРО образуется копропорфириноген III, синтез которого катали-
зируется ферментом УРО-декарбоксилазой. Копропорфириноген III под дейс-
твием копропорфириноген Ill-оксидазы превращается в протопорфириноген IX.
Следующий этап биосинтеза гема - преобразование протопорфириногена IX в
протопорфирин IX - осуществляется ферментом протопорфириногеноксида-
зой. Заканчивается синтез порфиринов включением двухвалентного железа в
протопорфирнн IX. Процесс катализируется митохондриальным ферментом
феррохелатазой (гемсинтетаза), в результате чего образуе тся гем.
Гем одинаков для всех видов гемоглобина, последние различаются между
собой структурой глобина. Гем крайне неустойчив и легко превращается в
гематин с окислением Fe2+ в Fe3' и присоединением к последнему ОН . Он
также легко превращается в гемин, который вместо ОН содержит хлор. Гем
обладает способностью вступать в обратимую комбинацию с кислородом.
При связывании кислорода железо должно быть в активной 2-валентной
форме. Основные этапы синтеза гема показаны на рис. 44. Каждый этап
контролируется специфическим ферментом.
Отрицательная
обратная
связь
Глицин + сукцинат
8-аминолевулиновая кислота (АЛК)
СоА- пи ридоксал ь -
фосфат
Уропорфириноген III
I
Копропорфириноген III
Протопорфириноген IX
Протопорфирин IX
Уропорфириноген I
I
Копропорфириноген I
Рис. 44. Биосинтез гема, 6-аминолевулиновая
кислота (АЛК) образуется путем соединения
глицина и сукцината. В этом процессе учас
твуют пиридоксальфосфат и АЛК-синтетаза
Реакция синтеза гема контролируется меха
низмом обратной связи. 2 молекулы АЛК
конденсируются в монопиррольную форму
порфобилиногена (ПБГ). 4 молекулы ПБГ фор
мируют тетрапиррольное кольцо уропорфи-
риногена. Образуется 2 изомерных формы —
I и III. В основном в синтезе гема участвует
изомер III. Синтез гема включает образование
копропорфириногена и протопорфириногена
с последующим присоединением Fe2*
связи, где конечный продукт гем регулирует синтез АЛК на уровне транс-
крипции и трансляции. Основная часть порфиринов, синтезированных в
костном мозге, идет для образования гемоглобина.
В результате дефицита одного из ферментов синтеза гема развивается
порфирия. При порфирии из-за нарушения синтеза гема снимается механизм
обратной связи, прекращается ингибирование скорость-лимитирующего
фермента АЛК-синтетазы, поэтому при легких формах порфирии удается
поддерживать адекватный синтез гема (анемия не развивается), а происхо-
дит накопление промежуточных продуктов. АЛК, ПБГ и уропорфириноген
водорастворимы, поэтому при накоплении экскретируются с мочой (рис. 45),
при этом моча приобретает розовый или красный оттенок. Копропорфирин
и протопорфирин нерастворимы в воде, они экскретируются с желчью и
определяются в кале.
Отрицательная обратная связь
: АЛК-синтетаза
' Ф (2) ® |
>АЛК-----► ПБГ -----► УРО -----► КОПРО ------► ПРОТО -------► ГЕМ
Экскреция с мочой Экскреция с калом
Рис. 45. Наиболее часто встречающийся дефицит или частичное снижение активности опре-
деленных ферментов синтеза гема, являющиеся причиной развития: Ф - острой перемежаю-
щейся порфирии, ® - наследственной копропорфирин, ® — вариегатной (пестрой) порфирии.
Эритропоэтическая порфирия может возникнуть при дефектах на уровне Фи®
Синтез порфиринов происходит в виде двух изомеров - III и I. Изомер III
порфиринов используется для синтеза гема. При нарушении этого пути
изомер I синтезируется в большем количестве, чем в норме. Эритроциты,
содержащие гемоглобин с изомером I, имеют меньшую продолжительность
жизни и быстро разрушаются в селезенке (болезнь Гюнтера, или врожденная
эритропоэтическая порфирия).
Анемии при нарушении синтеза порфиринов
Развитие анемии обусловлено недостаточной или аномальной утилиза! щей
внутриклеточного железа при синтезе гемоглобина, несмотря на нормальное
или даже повышенное содержание железа в митохондриях эритроидных
предшественников.
Лабораторные исследования выявляют высокую концентрацию железа
и ферритина в сыворотке крови и повышенное насыщение трансферрина
железом. В костном мозге отмечается гиперплазия эритроидных клеток.
Окраска на железо выявляет патогномоничные изменения, обусловленные
тов, — кольцевидные сидеробласты.
Наследственные и приобретенные анемии, связанные с нарушением
синтеза порфиринов, характеризуются гипохромией, высоким содержанием
железа сыворотки и гемосидерозом органов.
Наследственные анемии этого типа встречаются сравнительно редко,
преимущественно у мужчин, так как наследование сцеплено с Х-хромосо-
мой. Относительно чаще наблюдается форма болезни, вызванная дефектами
синтеза S-аминолевулиновой кислоты. Нарушение образования протопор-
фирина обуславливает невозможность связывания железа, вследствие этого
происходит накопление его в организме. Преимущественное поступление
железа в печень приводит к развитию цирроза, в поджелудочной железе - к
сахарному диабету, накопление железа в яичках - к евнухоидизму, в надпо-
чечниках - их недостаточности. Клинические проявления болезни зависят
от выраженности анемии и признаков избыточного отложения железа в
организме. У больных в молодом возрасте анемия в большинстве случаев
невыраженная (80-90 г/л), однако гемоглобин постепенно падает до 50-60 г/л.
Содержание ретикулоцитов нормальное или несколько сниженное. Эрит-
роциты гипохромные, отмечается анизоцитоз, пойкилоцитоз, отдельные
мишеневидные эритроциты.
В костном мозге-гиперплазия красного ростка, увеличен процент базо-
фильных, полихроматофильных и снижено количество оксифильных эритро-
кариоцитов, много кольцевидных сидеробластов (рис. 46). Для этой анемии
характерны признаки неэффективного эритропоэза, который определяется
как анемия с относительной или абсолютной ретикулоцитопенией.
Содержание железа в сыворотке значительно повышено (до 100 мкмоль/л),
трансферрин насыщен железом почти на 100%. При исследовании содержа-
ния порфиринов в эритроцитах у некоторых больных обнаруживается сниже-
ние протопорфирина до 3-9 мкмоль/л (норма 18-90 мкмоль/л) и повышение
Рис. 46. Костный
мозг. Кольцевид-
ные сидеробласты.
хЮОО
Рис. 47. Отравление свинцом. Костный мозг
Кольцевидные сидеробласты. хЮОО
копропорфирина до 60-75 мкмоль/л (норма до 12 мкмоль/л). В отдельных
случаях снижается как протопорфирин, так и конропорфирин. Содержание
5-аминолевулиновой кислоты и копропорфирина в моче нормальное.
Приобретенные анемии обусловлены нарушением синтеза порфиринов,
возникающим чаще при отравлении свинцом или дефиците витамина Вй.
Патогенез анемии при свинцовом отравлении. Свинец блокирует ак-
тивные центры двух ферментов, участвующих в синтезе гема - синтетазы
8-аминолевулиновой кислоты и феррохелатазы, снижает скорость синтеза
a-цепи глобина. Нарушается включение железа в молекулу протопорфирина,
увеличивается содержание железа в сыворотке и отложение его в тканях.
Своеобразен вид больного - землистая бледность с сероватым оттенком,
связанная как с анемией, так и со
спазмом сосудов и отложением в коже
порфиринов, может быть лиловая
кайма на деснах.
В костном мозге отмечается
резкое увеличение кольцевидных
сидеробластов (рис. 47). В перифери-
ческой крови постепенно снижается
содержание гемоглобина до 50 60
г/л, эритроциты с выраженной гипох-
ромией (низкие цветовой показатель,
МСН, МСНС), выявляется анизопой-
килоцитоз, базофильная пунктация
эритроцитов (рис. 48).
Содержание железа в сыворотке
крови повышается до 350-550 мкг/дл,
насыщение трансферрина железом
достигает 100%. В сыворотке крови
отмечается высокая концентрация
ферритина. Самым характерным
биохимическим признаком свинцо-
вого отравления является увеличение
концентрации в моче 8-аминолевули-
новой кислоты и копропорфирина, в
эритроцитах повышено содержание
протопорфирина.
Рис. 4В. Отравление свинцом. Базофильная
пунктация эритроцитов периферической кро-
Нормохромные анемии
Анемия хронических заболеваний
Взаимосвязь анемии с хроническими инфекционными (туберкулез, ге-
патит и др.), ревматическими заболеваниями, другими воспалительными
процессами описана более 150 лет назад. Для характеристики таких анемий
в литературе используются термины: хроническая анемия, анемия воспале-
ния и анемия хронических заболеваний. Наиболее частым и устоявшимся
термином является «анемия хронических заболеваний» (АХЗ). Анемия часто
сопровождает и неинфекционныс воспалительные заболевания кишечника,
соединительной ткани, множественную миелому и другие злокачественные
новообразования. АХЗ занимает по распространенности второе место после
железодефицитной анемии.
В 1945 г. G.E. Cartwright, М.М. Wintrobe провели серию эксперименталь-
ных исследований у животных и человека и охарактеризовали клинические
и биохимические признаки АХЗ. Анемия обычно носит нормохромный
нормоцитарный характер, однако гипохромия встречается так же часто, и
иногда анемия становится микроцитарной гипохромной. АХЗ постоянно со-
провождается нарушением метаболизма железа: гипоферремией, некоторым
снижением концентрации трансферрина в крови и повышением ферритина
как в крови, так и в органах и тканях.
В настоящее время рассматриваются следующие патофизиологические
факторы, участвующие в развитии АХЗ:
• влияние провоспалительных цитокинов,
• воздействие гепсидина,
• низкая продукция эритропоэтина,
• ингибиция эритропоэза,
• укорочение продолжительности жизни эритроцитов.
Активация иммунной системы при воспалительных, инфекционных и не-
которых онкологических заболеваниях антигенными факторами индуцирует
синтез провоспалительных цитокинов (ИЛ-1, ФНО-а, ИЛ-6), интерферонов.
АНЕМИЯ
Снижение
продукции
ЭПО
Снижение
функционального
железа
Супрессия ранних
эритроидных
предшественников
Рис. 49. Схема патогенеза анемии хронических заболеваний
ИЛ-6 является основным индуктором синтеза гепсидина в печени, котоый
значительно повышается независимо от состояния эритропоэтичессой
активности и изменений в обмене железа. Гепсидин является негативным
регулятором как выведения железа из макрофагов, так и всасыванияже-
леза в тонком кишечнике. Результат действия гепсидина - блокада жееза
в клетках СМФ, гепатоцитах и энтсроцитах, нарушение передачи жееза
трансферрину и быстрое развитие гипоферремии. Механизм ингибции
выведения железа из клетки связан с образованием комплекса гепсидш -
ферропоргин (клеточный транспортер железа, участвующий в передач! его
из клетки плазменному трансферрину) и последующим его расщеплешем.
Сходный механизм взаимодействия гепсидин - ферроиортин приводи к
снижению всасывания железа в тонком кишечнике, что может привести к
снижению запасов железа, однако это встречается очень редко, напримерпри
ювенильном ревматоидном артрите, при котором отмечается значитаьно
повышенный синтез ИЛ-6.
В норме ежедневно около 20 мг железа поступает в плазму крови,свя-
зывается с трансферрином и большая часть его доставляется эритрощным
клеткам костного мозга для синтеза гемоглобина. Так как всасывание железа
и мобилизация его из депо нарушены, а клетки в костном мозге продотжа-
ют расходовать железо на свои нужды, то плазменный пул железа бьстро
истощается, вызывая гипоферремию. При продолжительной гипоферр<мии
развивается железодефицитный эритропоэз, одной из характеристик котсрого
является повышение концентрации протопорфирина в эритроцитах.
Таким образом, каскад событий, который приводит к развитию АХЗ, сво-
дится к следующему: действию ИЛ-6 на избыточный синтез гепсидин!, на-
рушению метаболизма железа (гипоферремия, гиперферритинемия, избыток
железа в макрофагах тканей и органов), развитию перераспределителшого
или функционального дефицита железа, снижению доставки железа к эртгро-
кариоцитам костного мозга, нарушению эритропоэза и развитию анемии.
Ряд цитокинов (ИЛ-1, ФНО-а, ТФР-Р) подавляют экспрессию гена
эритропоэтина (ЭПО) в клетках почек и печени, у больных АХЗ отмечается
неадекватно низкая продукция ЭПО. Кроме того, в экспериментальных ис-
следованиях показано, что эритроидные предшественники при АХЗ обтару-
живают в некоторой степени резистентность к пролиферативному действию
ЭПО. Ингибиторный эффект провоспалительных цитокинов на эритропоэз
не ограничивается их действием на продукцию ЭПО и на чувствительность
к этому гормону, доказано прямое супрессирующее воздействие ряда цито-
кинов на пролиферацию эритроидных клеток костного мозга. Кроме того,
экспериментальные исследования in vitro показали прямое ингибирующее
воздействие гепсидина на эритроидное колониеобразование.
Укорочение продолжительности жизни эритроцитов при АХЗ ранее
объясняли воздействием активированных макрофагов, которые избыточно
фагоцитировали и секвестрировали эритроциты. Однако незначительная
деструкция эритроцитов при АХЗ сама по себе недостаточна для объяснения
развития анемии, т. к. костный мозг при необходимости может увеличить
продукцию эритроцитов в 6-8 раз. Эти данные свидетельствуют о том, что
при АХЗ пролиферативная активность костномозговых эритроидных клеток
нарушена. Повышенный гемолиз эритроцитов при АХЗ связывают с дей-
ствием ИЛ-1 и ФНО-а.
Таким образом, патогенез АХЗ достаточно сложный, в котором участвуют
различные механизмы, ведущими из которых являются воздействие гепси-
дина и провоспалительных цитокинов (рис. 49).
Лабораторные показатели. У больных с анемией хронических заболе-
ваний костный мозг характеризуется нормальным или сниженным, реже
повышенным количеством эритрокариоцитов, повышенным содержанием
сидеробластов и макрофагов с гранулами гемосидерина. Возможно обнару-
жение эритрофагоцитоза.
Периферическая кровь. Чаще анемия при АХЗ нормохромная нормо-
цитарная, реже умеренно гипохромная или гипохромно-микроцитарная
(рис. 50).
Число ретикулоцитов нормальное или снижено, ретикулоцитарный индекс
продукции снижен, что отражает сниженную пролиферативную активность
костного мозга. Повышен уровень протопорфирина в эритроцитах.
Изменения метаболизма железа характеризуются перераспредели-
тельным дефицитом железа (снижение сывороточного железа, ОЖСС,
трансферрина, НТЖ и повышение содержания сывороточного ферритина).
Ферритин относится к острофазным белкам, поэтому повышенный уровень
сывороточного ферритина при АХЗ может отражать не только запас железа в
организме, но и явиться проявлением острофазного ответа, что ограничивает
его использование в качестве показателя определения запасов железа. В боль-
шинстве случаев АХЗ повышены и другие белки острой фазы. Количество
sTfR остается стабильным при воспалительных процессах (табл. 10).
Дифференциальная диагностика АХЗ и ЖДА основана на комплексном
исследовании обмена железа, острофазных белков, растворимых рецепторов
к трансферрину и в последнее время - гепсидина в моче, концентрация ко-
торого повышена при АХЗ (табл. 11). Ошибочная диагностика ЖДА может
повлечь за собой назначение препаратов железа (парентерально) с развитием
вторичного гемосидероза.
RBC 2,45 3,8-5,5 х 1012/л
НЬ 74 130-155 г/л
Ht 19,9 36-48%
MCV 81,1 80-95 фл
МСН 30,5 27-31 пг
МСНС 377 300-380 г/л
RDW 16,4 11,5-14,5%
рИс 50. Периферическая кровь при АХЗ. Нормохромная нормоцитарная анемия Видны еди-
ничные мишеневидные эритроциты и стоматоциты. хЮОО
Таблица 10
Лабораторные показатели при некоторых хронических заболеваниях
Показатели Острое и хроническое воспаление Заболевания почек Эндокринные заболевания
MCV (фл) 75-90 80-90 80-105
Полихромазия Нет Нет Нет
Ретикулоцитарный индекс продукции <2 <2 <2
Костномозговой индекс Лейко/Эритро 3:1 3:1 3:1
Запасы железа в КМ Повышены Норма Норма
Ферритин сыворотки Повышен Норма Норма
Примечание. MCV - средний объем эритроцитов, КМ - костный мозг.
Таблица 11
Дифференциальная диагностика анемии хронических заболеваний
и железодефицитной анемии
Анемия при хронической почечной недостаточности
Анемия - один из наиболее характерных синдромов, сопровождающих
течение хронической почечной недостаточности (ХПН). Степень выражен-
ности анемии не всегда коррелирует с уремией, тем не менее при снижении
клиренса креатинина ниже 30 мл/мин величина гематокрита всегда ниже
30%.
Патогенез анемического синдрома при ХПН сложен и до сих пор полно-
стью не раскрыт. В развитии нефрогенной анемии играют роль следующие
факторы: дефицит эндогенного эритропоэтина, ингибиторы эритропоэза,
гемолиз, укорочение продолжительности жизни эритроцитов, токсическое
влияние на эритроциты продуктов азотистого обмена, кровопотери, дефицит
фолиевой кислоты, железа и др. Роль перечисленных факторов в патогенезе
анемии у больных с ХПН не подвергается в настоящее время сомнению,
однако основное значение принадлежит абсолютному или относительному
дефициту эндогенного эритропоэтина (эЭПО). При хронической почечной
недостаточности содержание эЭПО снижено (норма 6-25 МЕд/мл).
Эритропоэтин - гликопротеид с молекулярной массой 40-60 кДа,
продуцируется интерстициальными клетками коры почек. Образовав-
шийся эЭПО не накапливается в клетках, а сразу секретируется в кровь.
Обладая сравнительно небольшой молекулярной массой, он фильтруется
в клубочках и подвергается деградации в ткани почек. Неотличающийся
по строению и биологической активности эЭПО секретируется также
гепатоцитами, интерстициальными клетками печени, макрофагами,
однако суммарная доля этих внепочечных источников эЭПО в норме не
превышает 10-15% общей продукции. Скорость образования эЭПО об-
ратно пропорциональна кислородотранспортной функции крови, уровню
гипоксии, анемии.
ЭПО относится к гемопоэтическим факторам роста, который регули-
рует продукцию эритроцитов в зависимости от потребности организма в
кислороде. Другой важной функцией ЭПО является предотвращение апоп-
тоза клеток-предшественников эритропоэза на поздних стадиях развития,
поэтому недостаточный уровень эЭПО приводит к увеличению объема
неэффективного эритропоэза, что подтверждается повышением количества
PAS-положительных эритроидных клеток в костном мозге. Таким образом,
недостаточная подукция ЭПО в почках является главным фактором развития
нефрогенной анемии. Вместе с тем, хотя и не решающую роль, в патогенезе
анемии при ХПН играют уремические токсины, которые неспецифически
ингибируют пролиферацию и дифференцировку эритрокариоцитов. В качес-
тве предполагаемых ингибиторов эритропоэза выступают паратиреоидный
гормон, спермин и другие факторы.
Важную роль в развитии нефрогенной анемии играют ингибиторы эЭПО,
в частности фактор некроза опухоли (ФНО-а), ИЛ-1, трансформирующий
фактор роста (ТФР-Р), у-интерферон. Провоспалительные цитокины (ИЛ-1
и ФНО-а), уровень которых повышен в почечной ткани больных нефропа-
тиями, дополнительно угнетают синтез эЭПО у больных ХПН.
У больных ХПН одним из факторов, способствующих развитию анемии,
является повышенный гемолиз. У большинства больных период жизни
эритроцитов резко снижен. Показано, что при уремии активность боль-
шинства ферментов в эритроцитах нормальная или высокая и уровень АТФ
повышен, отмечается уменьшение активности Na+-K+-Hacoca, что вызывает
изменение мембраны, ригидность эритроцитов и укорочение продолжи-
тельности их жизни. Острый гемолиз описан как результат воздействия
меди, содержащейся в диализате. Гемолиз может быть также в результате
микроангиопатической деструкции эритроцитов при некоторых системных
заболеваниях, сопровождающихся поражением ночек. Дефицит фолиевой
кислоты всегда развивается у больных, находящихся на гемодиализе, т. к.
фолиевая кислота является легко диализируемой. Кроме того, уремическая
сыворотка ингибирует мембранный транспорт фолатов, что может нарушать
утилизацию фолатов клетками. У больных ХПН, находящихся на диализе,
наблюдается дефицит железа. Пациент теряет от 5-6 мг железа ежедневно
или приблизительно 2 г в год. Эти кровопотери вызывают дополнительные
требования к усилению эритропоэза и в значительной степени способствуют
развитию анемии.
Анемия может быть первым признаком интоксикации алюминием у боль-
ных на гемодиализе. Алюминий вызывает микроцитарную гипопролифера-
тивную анемию. Предполагают, что алюминий может препятствовать синтезу
гема и, возможно, транспорту железа, связанного с трансферрином.
При уремии в плазме крови обнаружено усиление активности протеоли-
тических ферментов, что приводит к десиализации мембран эритроцитов,
снижению поверхностного заряда, предрасполагая эритроциты к разрушению
макрофагами.
Лабораторные показатели. Клеточность костного мозга в большинс-
тве случаев ХПН остается нормальной. В зависимости от степени сниже-
ния продукции эЭПО отмечается нормальное или сниженное содержание
эритрокариоцитов.
В периферической крови наблюдается анемия, которая имеет характер
нормоцитарной нормохромной. Количество ретикулоцитов при нефроген-
ной анемии обычно нормальное или незначительно повышено и зависит от
степени активности костномозгового эритропоэза. На фоне длительного
гемодиализа, вследствие кровопотерь, анемия может стать гипохромной
микроцитарной. В мазках крови наблюдается пойкилоцитоз различной
степени выраженности с преобладанием эхиноцитов (шиповидные эритро-
циты) (рис. 51).
RBC 1,9 3,8—5,5 х 10,2/л
НЬ 60 130-155 г/л
Ht 16,5 36-48%
MCV 84,7 80-95 фл
МСН 31,6 27-31 пг
МСНС 363 300—380 г/л
RDW 14,2 11,5-14,5%
Рис 51. Периферическая кровь при ХПН. Нормохромная нормоцитарная анемия. Эхиноциты. хЮОО
Изучение новых параметров ретикулоцитов показало, что у больных ХПН
увеличение относительного и абсолютного количества ретикулоцитов сопро-
вождается повышением среднего объема ретикулоцитов, что свидетельствует
о преобладании среди них молодых форм. Отмечается повышение фракции
незрелых ретикулоцитов (IRF), их относительного (HLR%) и абсолютного
количества (HLR#), что характеризует наличие активного эритропоэза в
костном мозге, несмотря на дефицит эЭПО. Возможно, это связано с ком-
пенсаторной реакцией костного мозга на постоянные кровопотери при ге-
модиализе. При развитии гипохромной микроцитарной анемии отмечается
снижение Ret-He.
Лечение больных ХПН препаратами рекомбинантного эритропоэтина
приводит к частичной коррекции анемии, однако вследствие стимуляции
эритропоэза может развиться ЖДА, что диктует необходимость исследовать
метаболизм железа в динамике проведения терапии рЭПО.
Апластичесние анемии
Апластическая анемия (АА) - заболевание, характеризующееся резким
угнетением костномозгового кроветворения, торможением процессов про-
лиферации и дифференцировки клеточных элементов с развитием глубокой
панцитопении в периферической крови. Апластическая анемия на протяже-
нии многих десятков лет со времени первого описания, опубликованного в
1888 г. Р. Ehrlich, для большинства больных была прогностически крайне
неблагоприятным заболеванием. Частота встречаемости АА составляет
2,0 на 1 млн в год, при колебании этого показателя в зависимости от страны
от 0,6 до 5,0 на 1 млн в год.
Апластические анемии могут быть наследственными и приобретенными.
Наследственная форма АА (анемия Фанкони) сочетается с другими наследс-
твенными аномалиями. Приобретенные формы АА связаны с действием
различных агентов: ионизирующая радиация, лекарственные препараты - ан-
тибиотики, сульфаниламидные, антитиреоидные, противосудорожные, про-
тивотуберкулезные, противодиабетические препараты, декарис, анальгин,
левомицетин, тетрациклин, бутадион, препараты золота и др., химические
соединения (бензол и его производные, этилированный бензин, пестициды
и др.); вирусы гепатита А, В, С, вирус Эпштейна-Барр, цитомегаловирус, ви-
русы герпеса, ВИЧ, парвовирус В19. Апластическая анемия, развивающаяся
в течение первых 6 месяцев у больного, перенесшего острый вирусный гепа-
тит, называют гепатитассоциированной АА. Доказана способность вирусов
гепатита А, В, С ингибировать рост колоний и дифференцировку клеток-
предшественников. Считается, что гепатитассоциированная АА, скорее всего,
является следствием иммунной агрессии в отношении гемопоэза. Описаны ап-
ластические кризы у больных различными формами гемолитических анемий,
вызванные парвовирусной инфекцией. Мишенью для парвовируса В19 служат
эритроидные клетки-предшественники, поражение которых с врожденным
или приобретенным иммунодефицитом может привести к развитию парциаль-
ной красноклеточной аплазии. Из числа эндогенных факторов, угнетающих
гемопоэз, отмечают нарушение функции яичников, щитовидной и вилочковой
желез. Изменение функции тимуса нередко сопровождается возникновением
парциальной красноклеточной аплазии. В большинстве случаев АА этиоло-
гический фактор остается неизвестным {идиопатические АА).
Представления о патогенезе АА в 1970-80 гг. предполагали связь между
развитием аплазии кроветворения и дефектом стволовых клеток, нарушени-
ем регуляции гемопоэза иммунокомпетентными клетками и повреждением
микроокружения, т. е. стромы костного мозга. Дефектность стволовых клеток
при АА доказана культуральными исследованиями, в которых обнаружено
снижение колониеобразующей способности клеток-предшественников гемо-
поэза. Иммунологическое фенотипирование клеток костного мозга выявило
уменьшение количества клеток, экспрессирующих маркер CD34, характерный
для ранних гемопоэтических клеток-предшественников. Функция стромы
костного мозга у большинства больных А А страдает в меньшей степени, чем
функция гемопоэтических клеток. Обнаружена нормальная или даже повы-
шенная способность стромальных клеток при АА продуцировать гемопоэти-
ческие ростовые факторы. Только у небольшой части больных АА выявлена
сниженная продукция стромальными клетками костного мозга ГМ-КСФ,
ИЛ-3, ИЛ-1, ИЛ-6, при этом концентрация ЭПО и тромбопоэтина, Г-КСФ
оказалась повышенной. Таким образом, многочисленными исследованиями
с использованием культур клеток и молекулярно-биологического подхода нс
удалось доказать наличие при АА выраженного дефицита факторов роста и
их ведущей роли в патогенезе АА.
В последние годы большинство исследователей ведущую роль в развитии
АА придают иммунной деструкции гемопоэза. При А А показано повышение
активности цитотоксических Т-лимфоцитов и натуральных киллеров, оказы-
вающих подавляющее действие на костный мозг. Большинство фундаменталь-
ных исследований связывают развитие костномозговой недостаточности при
АА с появлением в периферической крови и костном мозге активированных
цитотоксических Т-лимфоцитов, продуцирующих фактор некроза опухоли
(ФИО), у-интерферон, которые способны в условиях in vitro ингибировать
нормальный гемопоэз. Вследствие этого происходит значительное снижение
пула гемопоэтических клеток и развитие аплазии костного мозга.
В последние годы большое внимание уделяется изучению апоптоза как
одного из механизмов развития аплазии кроветворения. При АА имеет мес-
то повышенная способность гемопоэтических клеток к апоптозу, т. к. на их
поверхности экспрессируются рецепторы, являющиеся маркерами апоптоза
(CD95). Воздействие на клетки ФИО, у-интерферона вызывает усиление
экспрессии рецепторов апоптоза на клеточной мембране.
Исследования последних лет обнаружили наличие общего для пароксиз-
мальной ночной гемоглобинурии и некоторых больных АА молекулярного
дефекта, возникающего в результате соматической мутации гена PIG-A,
связанного с Х-хромосомой и участвующего в синтезе белкового комплекса,
называемого GP1 (гликозилинозитолфосфолипидный якорь), способного
инактивировать комплемент на поверхности клеток, в частности на поверх-
ности эритроцитов. Высказывается предположение о том, что появление
даже небольшого клона PIG-A дефектных гемопоэтических клеток может
оказаться причиной иммунной атаки, направленной на нормальные крове-
творные клетки и приводящей к развитию аплазии кроветворения.
Различные этиопатогенетические механизмы ведут к нарушению эритро-,
грануло- и тромбоцитопоэза, что служит первоначальным антигенным сигна-
лом, приводящим к активации иммунной системы или срыву толерантности
и аутоиммунной деструкции гемопоэза при АА. В настоящее время АА
рассматривается как гетерогенная по своему происхождению и механизмам
развития группа аплазий кроветворения, для которых ведущими в патогенезе
являются поражение стволовых клеток (первичное или в результате иммун-
ной деструкции) и аутоиммунная агрессия в отношении гемопоэза (первичная
или в ответ на появление клона дефектных стволовых клеток).
В патогенезе АА рассматривают следующие механизмы:
• Дефект стволовых клеток вследствие воздействия неизвестного пускового
агента (уменьшение числа клеток, экспрессирующих маркер CD34, харак-
терный для ранних гемопоэтических клеток-предшественников, снижение
колониеобразующей способности клеток-предшественников гемопоэза).
• Нарушение регуляции гемопоэза иммунокомпетентными клетками
(повышение активности цитотоксических Т-лимфоцитов (CD8+), про-
дуцирующих фактор некроза опухоли, у-интерферон, которые ингиби-
руют нормальный гемопоэз и подавляют образование гемопоэтических
колоний).
• Повреждение микроокружения, т. е. стромы костного мозга.
• Наследственный генетический дефект.
Клиническая картина определяется анемическим, геморрагическим
синдромом и инфекционными осложнениями. Основные проявления А А
обусловлены угнетением нормального кроветворения, гипоксией тканей и
органов (одышка, тахикардия, слабость, головокружение) и резкой тромбо-
цитопенией (кровоподтеки, петехии, носовые кровотечения, меноррагии и
другие кровотечения). В результате выраженной нейтропении развиваются
воспалительные процессы.
Критериями диагноза АА являются:
• трехростковая цитопения: анемия (НЬ <110 г/л), гранулоцитопения (гра-
нулоциты <2,0 х 109/л), тромбоцитопения (тромбоциты <100 х 109/л);
• снижение клеточности костного мозга и отсутствие мегакариоцитов по
данным пунктата костного мозга;
• преобладание жирового костного мозга по данным исследования тре-
панобиоптата.
Костный мозг. Количество миелокариоцитов в костном мозге резко
снижено (менее 40 х 109/л), отмечается замещение гемопоэтической ткани
Жировой тканью. Наблюдается задержка созревания клеток трех ростков
кроветворения. Обычно количество бластных клеток находится в пределах
нормы. На фоне снижения общего числа гранулоцитов повышено отно-
сительное содержание лимфоцитов, плазматических клеток (до 10-12%).
Встречаются макрофаги, липофаги (рис. 52). Эритропоэз характеризуется
абсолютным уменьшением количества эритрокариоцитов и нарушением
Рис. 52. Костный мозг больного с ги-
поплазией кроветворения. Единичные
миелокариоцигы (хЮО), макрофаги (1),
фибробласт (2). хЮОО
их дифференцировки. Количество сидеробластов и сидероцитов в костном
мозге значительно возрастает. Резко снижено количество мегакариоцитов,
а в тяжелых случаях они могут отсутствовать. При гистологическом иссле-
довании треианобиоптатов костная ткань сохраняет нормальную структуру.
Поражение костного мозга имеет очаговый характер. В местах опустошения
активный костный мозг замещается жировой и/или фиброзной тканью.
Однако даже при крайне тяжелом угнетении гемопоэза возможны активные
очаги кроветворения.
Периферическая кровь. Характерны признаки выраженной нормохром-
ной анемии с резким снижением концентрации НЬ (25-80 г/л), количества
эритроцитов (0,7-2,5 х 1012/л), умеренным анизоцитозом с тенденцией к
макроцитозу, пойкилоцитозом (рис. 53). Ретикулоцитопения варьирует от
0,3 до 0,9%, при гемолизе достигает 4-5%. Характерной для АА является
выраженная лейкопения (до 2,5-0,55 х Ю9/л) с абсолютной нейтропенией и
относительным лимфоцитозом. В случае присоединения инфекции может
наблюдаться сдвиг влево до миелоцитов. Резко выражена тромбоцитопения
(2,0-25,0 х 109/л), иногда в мазках периферической крови тромбоциты могут
отсутствовать. В большинстве случаев АА ускорена СОЭ.
Тяжелая АА определяется при наличии двух любых из перечисленных
критериев (гранулоцитопения <0,5 х 109/л, тромбоциты <20 х Ю9/л, рсти-
RBC iia «'*,111 I t 1 1 1 1 1 1 I а а । а —1
250 fl
RBC 1.15 3,8-5,5 x 10,2/л
Hb 37 130-155 г/л
Ht 12,2 36-48%
MCV 100,0 80-95 фл
MCH 32,2 27-31 пг
MCHC 322 300-380 г/л
RDW 25,9 11,5-14,5%
Рис. 53. Кровь больного с гипопластической
анемией
кулоциты <1% с коррекцией по гематокриту или <20 х 109/л) в сочетании с
аплазией КМ по данным трепанобиопсии. Сверхтяжелая АА - гранулоци-
топения составляет <0,2 х Ю9/л.
На фоне частых гемотрансфузий при АА изменения в метаболизме железа
характеризуются повышением содержания сывороточного железа и НТЖ,
что ведет к развитию гемосидероза. Увеличение концентрации эритропоэ-
тина в крови связывают с падением утилизации его клеточными элементами
костного мозга.
Наследственные гипопластические анемии
Наследственные гипопластические анемии у детей составляют около 25%
всех вариантов АА, чаше встречаются у мальчиков.
Конституциональная апластическая анемия Фанкони - аутосомно-
рецессивное заболевание, характеризующееся врожденными соматическими
аномалиями и прогрессирующей костномозговой недостаточностью. Частота
встречаемости 1 :36О—3000 населения, одинаково часто болеют как мальчики,
так и девочки. Заболевание выявляется в возрасте 4-10 лет. Клетки харак-
теризуются гиперчувствительностью к ДНК-повреждающим агентам, что
определяет нестабильность генома.
Клинические проявления - прогрессирующая костномозговая недостаточ-
ность, множественные врожденные аномалии органов и систем, высокий риск
развития злокачественных новообразований (ОМЛ, МДС и рак). Врожден-
ные пороки развития наблюдаются у 70-80% больных. Это пороки развития
костной системы (добавочные пальцы, отсутствие фаланг, отсутствие ребра,
лопатки и др.), зрительного аппарата, строения черепа - микроцефалия, от-
сутствие ушных раковин, сколиоз, добавочные позвонки, пороки строения
мочевыделительной системы, половой системы в виде дисплазии, удвоения
почек, гипогенитализма, аплазии матки, отсутствия яичников, врожденные
пороки сердца, задержка психического развития. Диспигментация кожи
наблюдается у 75-80% больных в виде генерализованной пигментации,
пигментные пятна типа «кофе с молоком» и/или очагов гипопигментации.
Течение заболевания хроническое с периодами обострения и ремиссии.
Выраженность симптомов (анемический, геморрагический, инфекцион-
ные осложнения) определяется степенью угнетения гемопоэза. Основным
гематологическим симптомом является нарушение кроветворения в виде
прогрессирующей гипоплазии костного мозга. В результате развивается пан-
цитопения. Изменения крови и костного мозга аналогичны идиопатической
АА. Длительность жизни в среднем составляет около 7 лет.
Парциальная красноклеточная аплазия
Редкое заболевание, характеризующееся нормохромной анемией, рети-
кулоцитопенией в сочетании с изолированной эритроидной гипоплазией
Костного мозга. Парциальная красноклеточная аплазия (ПККА) может быть
наследственной и приобретенной.
Наследственная парциальная гипопластическая анемия Даймон-
да-Блэкфана. Врожденное заболевание, характеризующееся отсутствием
эритроидных клеток в костном мозге. В 20% случаев имеет семейный ха-
рактер с аутосомно-доминантным или рецессивным наследованием. Частота
встречаемости анемии Даймонда-Блэкфана - 5-7 случаев на 1 млн населения
в год. Изучение клеток костного мозга больных анемией Даймонда-Блэкфана
в клоногенных культурах выявило снижение или отсутствие эритроидных
предшественников. Предполагают, что нарушение клеточной дифференци-
ровки возможно на любом этапе созревания элементов эритропоэза-либо на
конечном этапе, либо на уровне коммитированных клеток-предшественников.
Эритроидные предшественники малочувствительны к ЭПО, а также к другим
факторам роста (ИЛ-3, фактор стволовых клеток). Отсутствие чувствитель-
ности к ЭПО может объяснить высокий апоптоз эритроидных предшествен-
ников по сравнению с нормой. Анемия обычно выявляется на 1-3-м месяце
жизни. Она сочетается с черепно-лицевыми аномалиями или аномалиями
строения большого пальца в 50-60% случаев и отставанием больных в росте.
Психомоторное развитие ребенка нормальное. Спустя 5-6 лет от начала забо-
левания появляется сероватый оттенок кожи (следствие гемосидсроза) из-за
постоянных гемотрансфузий. Наблюдается спленомегалия, гепатомегалия.
Особенностью данного заболевания является отсутствие геморрагического
синдрома. Течение заболевания хроническое. Развитие гемосидероза с присо-
единением инфекционных осложнений может быть причиной летальных ис-
ходов. Имеется высокий риск возникновения острых миелоидных лейкозов.
Костный мозг нормоклеточный. По мере прогрессирования заболевания
наблюдается тенденция к уменьшению клеточности костного мозга. Мие-
лограмма характеризуется изолированной эритроидной гипоплазией уже с
начала развития заболевания. Морфологические изменения эритрокариоци-
тов проявляются в виде мелких клеток с пикнотичными ядрами и скудной
цитоплазмой. Число лимфоцитов увеличено, обнаруживаются фибробласты,
количество плазматических клеток не изменено. Гранулоцитопоэз обычно не
нарушен, иногда наблюдается эозинофилия, задержка созревания на стадии
миелоцита и метамиелоцита. Содержание мегакариоцитов нормальное. На
поздних стадиях заболевания возможна трехростковая костномозговая недо-
статочность в виде апластической анемии. Может быть повышено содержание
сидеробластов. В ремиссии наблюдается увеличение числа эритрокариоцитов
в костном мозге и ретикулоцитов в крови до нормальных цифр.
Периферическая кровь характеризуется нормохромной макроцитарной
анемией, ретикулоцитопснией, появляющейся еще в период новорожден-
ное™. В мазках крови отмечается анизоцитоз с тенденцией к макроцитозу,
пойкилоцитоз. Число лейкоцитов нормальное, хотя у 20% больных может
быть нейтропения. Количество тромбоцитов нормальное или увеличенное,
реже сниженное. В лейкоцитарной формуле иногда выявляется эозинофилия.
Уровень эритропоэтина сыворотки крови значительно повышен. В сыворотке
крови большинства больных определяются антитела к эритрокариоцитам.
Приобретенная форма ПККА рассматривается как синдром или само-
стоятельное заболевание, возникающее в результате спонтанных мутаций,
а также в виде идиопатической формы (более 50% случаев). Хроническая
форма ПККА встречается очень редко и обнаруживается у 30-50% пациентов
с тимомами. В сыворотке крови таких больных обнаруживают антитела к
эритроидным клеткам костного мозга. ПККА ассоциирована с Т-клеточными
лейкозами и лимфомами, острыми вирусными гепатитами, парвовирусной
инфекцией (рис. 54). Острая форма ПККА описана у больных с наследс-
твенной гемолитической анемией после перенесенных вирусных инфекций.
В патогенезе играют роль иммунные механизмы повреждения эритропоэза,
вызывающие его супрессию. Диагностика основана на выявлении гипореге-
нераторной анемии, изолированного дефицита клеток эритроидного ряда в
костном мозге.
Рис. 54. Костный мозг. Парвовирусная инфекция. Гигантские проэритробласты. х1000
Анемии при злокачественных образованиях
У 30-50% онкологических больных имеется в той или иной степени вы-
раженности анемический синдром. Патофизиология хронической анемии
(анемии хронических заболеваний) при злокачественных новообразованиях
включает интенсивное взаимодействие между популяцией опухолевых клеток
и иммунной системой, что проявляется активацией макрофагов и повышен-
ной секрецией различных цитокинов. К развитию анемии приводят и другие
причины, такие, как кровопотери, гемолиз, метастатическое поражение
костного мозга, побочные эффекты химиотерапии и радиотерапии, микро-
ангиопатии, повреждение почек, нарушение питания. Патогенез анемии при
злокачественных новообразованиях во многом сходен с таковым при АХЗ,
при которых ведущую роль придают повышенному синтезу гепсидина и
провоспалительных цитокинов.
Поражение костного мозга при гемобластозах и множественных метаста-
зах солидных опухолей приводит к угнетению нормальных ростков крове-
творения, в том числе и эритроидного, что сопровождается развитием анемии,
которая может занимать основное место в клинической картине. Метастазы
в костный мозг встречаются при опухолях различных локализаций, однако
наиболее характерны для рака молочной железы, предстательной железы,
почек, легкого, щитовидной железы, нейробластомы. Так, при раке молоч-
ной железы они выявляются в 50% случаев, при нейробластоме у детей -
в 50-67%, при мелкоклеточном раке легкого - в 17-45% наблюдений.
Анемия при злокачественных новообразованиях относится так же, как
и анемия хронических заболеваний, к гипопролиферативным анемиям.
Чаще анемия носит нормохромный нормоцитарный характер, количество
ретикулоцитов снижено, нередко развивается панцитопения. В мазках кро-
ви выявляется анизоцитоз, пойкилоцитоз, полихроматофилия, встречаются
эритрокариоциты. В лейкоцитарной формуле может наблюдаться левый сдвиг
до миелоцитов. При морфологическом исследовании пунктатов костного
мозга выявляются комплексы опухолевых клеток (рис. 55).
Рис. 55. Костный
мозг. Метастазы
рака. хЮО (а, в),
хЮОО (б, г)
Гиперхромные (мегалобластные) анемии
Анемии, связанные с нарушением синтеза ДНК, могут быть как наследс-
твенными, так и приобретенными. Общим признаком этих анемий является
наличие в костном мозге мегалобластического кроветворения. Чаще наблю-
дается изолированный дефицит витамина В12, реже - фолиевой кислоты.
Анемии, обусловленные дефицитом витамина В12
Пернициозная анемия описана в 1849 г. Аддисоном. Несколько позже,
в 1872 г., Бирмер дал определение ее как «прогрессирующей пернициозной
анемии». Эрлих обнаружил в костном мозге крупные клетки со своеобразной
структурой хроматина и назвал их «мегалобласты». В 1926 г. Майнот и Мэр-
фи показали возможность излечения пернициозной анемии при назначении
сырой печени, содержащей внешний фактор - витамин В12. Практически
одновременно Каслом (1929 г.) был открыт внутренний желудочный фактор,
а Смитом и Риккесом (1948 г.) - витамин В|2, что способствовало установ-
лению патогенеза пернициозной анемии.
Частота встречаемости В ^-дефицитной анемии увеличивается с возрастом
и составляет у молодых 0,1%, пожилых - 1%, после 75 лет регистрируется
у 4% лиц.
Обмен витамина В12
Структура и механизм всасывания витамина В/2
Витамин В)2-цианкобаламин. Химическая структура витамина В12 напо-
минает гем и представлена в виде: I) корринового ядра, состоящего из тет-
рапиррольного кольца, окружающего атом кобальта (Со3+); 2) нуклеотидной
группы, что в совокупности носит название кобаламин; 3) CN-группы или
других групп, т. е. ОН в гидроксикобаламине, дезоксиаденозил в дезоксиаде-
нозилкобаламине и метиловая группа (СН3) в метилкобаламине, соединенных
с атомом кобальта (рис. 56). Мстилкобаламин является основной формой
витамина В,2в плазме крови человека.
Источником поступления витамина В|2 являются только продукты животного
происхождения: мясо, яйца, сыр, молоко, особенно много его в печени и почках.
При обычной диете человек по-
лучает в среднем от 5 до 15 мкг
витамина ежедневно. Общее
содержание витамина состав-
ляет от 2 до 5 мг (в среднем
4 мг), из них 1 мг депонируется
в печени. Ежедневная потеря
витамина с мочой и калом
составляет от 0,66 до 2,1 мкг
(в среднем 1,3 мкг). Суточная
потребность в витамине В12
составляет от 2 до 5 мкг, что
объясняет развитие анемии в
течение нескольких лет после
прекращения поступления
витамина в организм человека.
В период беременности, роста
и при состояниях, сопровож-
дающихся повышенным ме-
таболизмом цианкобаламина,
Увеличиваются суточные пот-
ребности в витамине В12.
В продуктах животного
Происхождения витамин В12
связан с белками и освобожда-
Рис. 56. Структура молекулы витамина В12. Состоит из
атома кобальта, окруженного тетрапиррольным коль-
цом. нуклеотидной группы, располагающейся перпен-
дикулярно, и СН-группы
стся в желудке под влиянием соляной кислоты и пепсина. Это является пер-
вым важным этапом, необходимым для нормального всасывания витамина.
У многих пожилых лиц дефицит цианкобаламина развивается вследствие
неспособности к его выделению из пищи, например при атрофическом
гастрите, когда имеется снижение секреции соляной кислоты и протеаз.
В желудочном соке имеется два белка, которые конкурируют за связывание с
витамином В|2: внутренний фактор и R-белки (R-rapid - гликопротеины, об-
ладающие быстрой электрофоретической подвижностью), вырабатываемые
слюнными железами. При низком pH желудочного сока R-белки обладают
более высоким аффинитетом к витамину, чем внутренний фактор. Таким об-
разом, после высвобождения в желудке витамин В12 связывается с R-белками.
Ни R-белки, ни внутренний фактор нс перевариваются соляной кислотой и
пепсином. Внутренний фактор является более устойчивым к действию фер-
ментов секрета поджелудочной железы, чем R-белки. После прохождения
желудочного содержимого в 12-псрстную кишку протеазы панкреатического
секрета освобождают витамин В,2из комплекса R - В12, который связыва-
ется с внутренним фактором Касла (ВФ, IF - intrinsic factor). ВФ является
одним из витамин В12-связываюших белков, секретируется париетальными
клетками слизистой оболочки фундального отдела и тела желудка. Секреция
ВФ повышается при наличии пищи в желудке и находится под контролем
п. vagus. Выработка ВФ стимулируется гастрином, гистамином, ингибиру-
ется атропином, соматостатином, блокаторами Н2-гистаминовых рецепторов
и после ваготомии. ВФ абсорбирует более 70% поступающего с пищей
витамина (путем диффузии поступает менее 2%). В присутствии витамина
В|2 две молекулы ВФ быстро образуют димер, связывающий две молекулы
витамина (1 мг ВФ способен связать около 30 мкг витамина В|2).
Всасывание витамина В12 происходит в подвздошной кишке, где имеются
специфические рецепторы. Наибольшая их плотность имеется на микровор-
синках дистальных отделов подвздошной кишки. Количество рецепторов в
подвздошной кишке является фактором, лимитирующим скорость процесса
поступления витамина В12в организм. После присоединения к рецепторам
комплекс витамин В12- ВФ расщепляется в присутствии Са2' и pH 7,0, и
витамин В12 проникает в митохондрии клеток слизистой оболочки кишки.
Внутриклеточно образующийся комплекс витамин В[2- транскобаламин II
(ТК II) поступает в плазму, где быстро удаляется и захватывается многими
клетками, особенно в печени. Связанный комплекс и его рецептор переносятся
в клетку путем пиноцитоза, где витамин В ^освобождается от ТК II, который
распадается. В печени происходит метилирование витамина, после чего ме-
тилкобаламин попадает в плазму крови. Транскобаламин II - глобулин с Мм 38
кДа, синтезируется гепатоцитами, фибробластами, макрофагами и энтероци-
тами. Транскобаламин II связывает только 20-30% витамина, остальная часть
связывается с транскобаламинами I и III, которые преимущественно синтези-
руются гранулоцитами. Большая часть витамина в плазме крови соединена
с транскобаламином I. В отличие от комплекса витамин В|2 - ТК II, который
циркулирует в крови менее одного часа перед поглощением его клетками,
комплекс витамин В12— ТК I находится в крови от 9-10 суток (рис. 57).
Рис. 57. Обмен витамина В12 в организме. После освобождения от пищи витамин В,2 в желудке
связывается с R-белком, а затем в подвздошной кишке с внутренним фактором (ВФ), выраба-
тываемым париетальными клетками слизистой желудка. Комплекс ВФ — В|2 всасывается после
специфического связывания с рецепторами подвздошной кишки. В кровотоке В,2 переносится
специализированными транспортными белками, называемыми транскобаламинами (ТК), кото-
рые доставляют В12 в костный мозг, печень, почки и другие органы
Таким образом, для нормального обмена витамина В12 необходимы сле-
дующие факторы:
- наличие витамина в пище,
- желудочная секреция,
- адекватная панкреатическая секреция,
— интактная подвздошная кишка,
- транскобаламины.
Дефект одного из этих факторов может привести к развитию В ^-дефи-
цитной анемии.
Роль витамина В12
Витамин В|2необходим в двух биохимических реакциях: 1) образование из
Nj-метилтстрагидрофолата тетрагидрофолата, в этом же процессе гомоцисте-
ин превращается в метионин; 2) производное витамина В12 - 5’-дезоксиаде-
нозилкобаламин выполняет роль кофактора при переходе метилмалонил-КоА
в сукцинил-КоА (рис. 58).
Витамин В12 участвует в превращении метилтетрагидрофолата, поступа-
ющего в клетку из крови, в тетрагидрофолат (коферментная форма фолиевой
кислоты). Реакция катализируется метионинсинтазой. Образующиеся ак-
тивные фолаты необходимы для синтеза пуриновых и пирмидиновых осно-
ваний - предшественников ДНК и РНК. Из тетрагидрофолата может вновь
образовываться 5,10-метилентетрагидрофол ат, который участвует в синтезе
пуриновых соединений. В этой же реакции путем реметилирования одновре-
Недостаточное образование 5'-дезокси-
аденозил-В12 при дефиците витамина В12
сопровождается подавлением синтеза жир-
ных кислот в нервной ткани и развитием
тяжелой неврологической симптоматики
Недостаточное образование тетрагидро-
фолата из-за дефицита витамина В12 или
фолиевой кислоты ведет к мегалобластной
анемии в результате снижения синтеза
нуклеиновых кислот
Рис. 58. Биохимические процессы с участием витамина В,2. Тетра гидрофолат, образующийся
при участии витамина В12 и фолата, необходим для синтеза пурина и ДНК
менно синтезируется метионин из гомоцистеина. Донором метильных групп
для синтеза метионина является 5-метилтетрагидрофолат, который образуется
в реакции, катализируемой метилентетрагидрофолатредуктазой (МТГФР).
Этот фермент имеет большое значение в обмене гомоцистеина. Переносчи-
ком метильной группы с 5-метилтетрагидрофолата на гомоцистеин служит
кофермент - метилкобаламин, производное витамина В|2. В результате чего
образуется метионин (рис. 59). Недостаток фолиевой кислоты и витамина В12
приводит к нарушению процессов реметилирования гомоцистеина в метио-
нин и повышению содержания в крови гомоцистеина. Гипергомоцистеинемия
является фактором риска развития сердечно-сосудистых заболеваний.
Вторая реакция, в которой участвует витамин В12, а именно дезоксиаде-
нозилкобаламин, необходима для биосинтеза жирных кислот. При распаде
жирных кислот образуется пропионовая кислота, в процессе метаболизма
которой синтезируется ряд промежуточных продуктов, в частности метил-
малоновая кислота. Последняя под влиянием 5’-дезоксиаденозилкобаламина
превращается в янтарную кислоту (сукцинил-КоА). При отсутствии или не-
достатке дезоксиаденозилкобаламина отмечается метилмалоновая ацидемия
и ацидурия (появление метилмалоновой кислоты в моче), нарушается синтез
жирных кислот, результатом которого являются повреждение нервной ткани
и появление неврологических расстройств.
Таким образом, дефицит витаминов В12 и фолиевой кислоты нарушает
метаболизм нуклеиновых кислот, вызывает ингибирование клеточного
деления. Эти нарушения наиболее выражены со стороны гемопоэтических
клеток и клеток желудочно-кишечного тракта.
ДНК-РНК
Пуриновые основания
Рис. 59. Метаболизм тетра гидрофолата. ТГФ — тетра гидрофолат, МТГФР — метилентетрагид-
рофолатредуктаза
Причины развития дефицита витамина В12
1. Нарушение всасывания.
• Отсутствие внутреннего фактора Касла (атрофический гастрит, тоталь-
ная гастрэктомия, резекция 2/3 желудка, облучение желудка).
• Заболевания тонкой кишки (энтерит, тропическая спру, целиакия,
стриктура, полипоз, резекция подвздошной кишки, синдром Имерс-
лунд-Гресбека).
• Хронические заболевания поджелудочной железы.
• Лекарственные препараты (ПАСК, колхицин, метотрексат, 6-меркап-
топурин, гидроксимочевина, фенобарбитал и др.) препятствуют вса-
сыванию В|2.
2. Конкурентное потребление.
• Широкий лентец (дифиллоботриоз).
• Избыточный рост патологической микрофлоры при наличии диверти-
кулеза или синдрома «слепой петли», стриктур, фистул.
3. Повышенная утилизация витамина В12.
• Злокачественные новообразования.
• Гипертиреоз.
4. Наследственный дефицит транскобаламина II.
5. Недостаточное поступление с пищей.
• Строгая вегетарианская диета.
6. Аутоиммунные антитела к париетальным клеткам желудка и внутрен-
нему фактору.
Наиболее частой причиной развития дефицита витамина В|2 является
атрофический гастрит, частота и выраженность которого увеличива-
ются с возрастом. При этом прекращается или уменьшается секреторная
способность слизистой желудка и синтез внутреннего фактора. Между
нарушением выработки внутреннего фактора и снижением кислото- и
ферментообразующей функции желудка нет параллелизма. Снижение
секреции внутреннего фактора возникает после гастрэктомии или резек-
ции 2/3 желудка. Однако В12-дефицитная анемия развивается не ранее чем
через 3-4 года. Это обусловлено достаточно большими запасами витамина
(3-4 мг).
Существенное значение в развитии дефицита витамина В|2 имеют гене-
тические факторы. Семейная предрасположенность выявляется у 20-30%
больных. В этих семьях заболевание возникает в 20 раз чаще, чем в попу-
ляции. В большинстве случаев семейная предрасположенность связана с
выработкой аутоантител к париетальным клеткам желудка и внутреннему
фактору. Чаще всего обнаруживаются аутоантитела к антигенам париеталь-
ных клеток желудка (более чем у 80% больных В12-дефицитной анемией
и почти у 50% пациентов с гастритом). Антитела также встречаются и в
составе желудочного сока. Их продуцентами являются иммунные клетки
собственной пластинки слизистой оболочки желудка. В настоящее вре-
мя известно два типа антител к париетальным клеткам. Первый из них
реагирует с цитоплазматическим антигеном, второй - с мембранным ан-
тигеном (рис. 60). Антитела второго типа в присутствии комплемента об-
ладают цитотоксичностью
Антитела
II типа
ВФ
ВФ
ВФ
В12
Антитела
I типа
В12
в отношении париетальных
клеток. Антитела к внутрен-
нему фактору встречаются
почти у 50-70% больных
В12-дефицитной анемией.
Антитела I типа блокиру-
ют кобаламин-связываю-
щий участок на молеку-
ле внутреннего фактора,
предотвращая связывание
витамина. Антитела II типа
блокируют сайт на моле-
куле внутреннего фактора,
Рис. 60. Антитела к внутреннему фактору встречаются
почти у 60% больных В|2-дефицитной анемией. Боль-
шинство из них принадлежит к блокирующему типу
антител, которые соединяются с внутренним фактором
на участке присоединения молекулы витамина В,2
участвующего в связывании
с рецепторами энтероцитов,
в результате чего нарушает-
ся всасывание витамина В |2.
Все антитела к внутреннему
фактору относятся к IgG.
Нередко В12-дефицитная анемия сочетается с другими аутоиммунными
заболеваниями (гипертиреоз, гипотиреоз, гипопаратиреоз, недостаточность
надпочечников, аутоиммунная гемолитическая анемия).
В редких случаях наблюдаются наследственные формы заболевания, обус-
ловленные дефицитом внутреннего фактора, селективной мальабсорбцией
витамина В12 (синдром Имерслунд-Гресбека), дефицитом транскобалами-
на И, нарушениями внутриклеточного метаболизма витамина В12.
Причиной развития В12-дефицитной анемии может быть конкурентное
потребление витамина в кишечнике микрофлорой или паразитами (инвазия
широким лентецом, синдром слепой кишки, дивертикулез). Микробная флора
потребляет витамин В)2, идущий для синтеза ДНК бактерий.
Клиническая картина заболевания включает три основных синд-
рома:
• анемический,
• желудочно-кишечный,
• неврологический.
Заболевание обычно регистрируется у лиц в возрасте 50-60 лет. Чаще
всего В12-дефицитная анемия дебютирует анемическим синдромом (утом-
ляемость, общая слабость, сердцебиение, одышка при обычной нагрузке).
Исключение составляют пациенты, у которых нарастание неврологичес-
кой симптоматики опережает развитие анемического синдрома. Жалобы
со стороны желудочно-кишечного тракта предъявляют 40-50% больных
(снижение аппетита вплоть до отвращения к пище, потеря вкуса, боли в
полости рта, жжение языка, расстройства стула), однако нарушения со сто-
роны ЖКТ не являются абсолютно патогномоничным признаком дефицита
витамина В12. Нередко наблюдаются боли и жжение в языке - симптомы
глоссита (ощущение «ошпаренного языка»). Глоссит проявляется в виде
алого языка с участками воспаления, сглаженными, реже атрофированными
сосочками - так называемый «лакированный» язык. У 10-20% больных
встречается увеличение печени и селезенки, желудочная секреция резко
снижена.
В основе неврологических расстройств лежит фуникулярный миелоз
боковых и/или задних столбов спинного мозга, являющийся следствием
демиелинизации, а затем и дегенеративных изменений нервных волокон в
спинном мозге и периферических нервах. Нарушения со стороны нервной
системы выявляются в виде парестезий, ватности ног, онемения пальцев,
изменения тактильной и температурной чувствительности в конечностях,
походки (передвигаются неуверенно, широко расставляя ноги), ощущения
постоянного холода в ногах. Нередко наблюдается мышечная слабость,
возможна атрофия мышц. В первую очередь поражаются симметрично
нижние конечности, затем живот, верхние конечности поражаются реже.
В самых тяжелых случаях развивается арефлексия, стойкие параличи
нижних конечностей, нарушения функции тазовых органов. При В12-
дефицитной анемии возможны судорожные припадки, галлюцинации,
ослабление памяти, потеря ориентации в пространстве, расстройства
психики, психозы с депрессивными или маниакальными состояниями,
нарушения зрения. Тяжелым осложнением является развитие перници-
озной комы, возникающей вследствие быстрой ансмизации и ишемии
головного мозга.
Лабораторные исследования. Дефицит В12 наиболее отчетливо прояв-
ляется в клетках гемопоэтической ткани.
Костный мозг. Костный мозг гиперклеточный за счет увеличения количес-
тва эритрокариоцитов. Соотношение Лейко/Эритро 1:2—1:3 (норма 3:1 -4:1).
Характерен мегалобластический тип кроветворения с высоким уровнем
неэффективного эритропоэза. Клетки не способны синтезировать достаточ-
ное количество ДНК для клеточного деления, клеточный цикл замедляется.
В результате нехватки ДНК для вступления в стадию митоза костный мозг
переполняется клетками, что создает видимость повышенного эритропоэза.
Во время продолжительной фазы покоя хроматин диффузно рассеивается на
всем протяжении ядра, придавая ему мегалобластный вид. Способность ме-
галобластов к делению резко нарушена (они совершают только 1-2 митоза).
При дефиците В|2 РНК исчезает более медленно по сравнению с нормальными
клетками и остается в клетках с выраженной гемоглобинизацией.
В результате нарушения клеточного деления эритроидные клетки ста-
новятся крупными (мегалобласты), качественно изменяется их структура.
Ядра мегалобластов всегда имеют характерное нсжносстчатое распределение
хроматина, асинхронное созревание ядра и цитоплазмы (при выраженной
гемоглобинизации ядро со-
храняет свой незрелый вид)
(рис. 61, 62). Конденсация
хроматина в мегалобластах
начинается при почти закон-
ченной гемоглобинизации
цитоплазмы; конденсиро-
ванный хроматин располага-
ется не радиально, а в виде
глыбок неправильной фор-
мы. Ядро по мере созревания
полихроматофильного и
оксифильного мегалобласта
Рис. 61. Костный мозг. Мегалобластная анемия. хЮОО располагается чаще Эксцен-
трично. Ядра мегалобластов
исчезают путем фрагмента-
ции (кариорексис), часто с
образованием почковидных
отростков и телец Жолли.
Количественные соотноше-
ния между мегалобластами
различной степени зрелости
весьма изменчивы и зависят
от активности костномозго-
вого кроветворения. Преоб-
ладание промегалобластов и
базофильных мегалобластов
Рис. 62. Костный мозг. Мегалобластная анемия хЮОО создает КЭрТИНу «Синего»
костного мозга (рис. 63).
Отмечаются значительные
дегенеративные изменения
в ядрах клеток, уродливость,
многоядерность, митозы.
Несмотря на эритроид-
ную гиперплазию костного
мозга, продукция эритроци-
тов при дефиците витамина
В12 снижена, что приводит
к анемии. Это обусловлено
резким повышением неэф-
фективного эритропоэза и
разрушением эритроидных
Рис. 63. «Синий» костный мозг. Мегалобластная анемия
хЮОО
Рис. 64. Костный мозг. Мегалобластная анемия. Ги-
гантские палочкоядерные нейтрофилы. хЮОО
предшественников в кост-
ном мозге. Кроме того, продолжительность жизни мегалобластов в 2—4 раза
меньше нормальной, поэтому большинство клеток, не созревая, погибают
в костном мозге. Из мегалобластов образуются мегалоциты и макроциты.
Мегалоциты имеют укороченный период жизни (около 40 дней), быстро
подвергаются деформации и разрушению. Впутрикостномозговой гемолиз
эритрокариоцитов (неэффективный гемопоэз) и короткая продолжительность
жизни мегалоцитов приводят к повышению уровня непрямого билирубина и
ЛДГ в сыворотке крови.
Недостаток витамина
В12 влечет за собой изме-
нения со стороны лейко-
поэза, поскольку синтез
ДНК нарушается во всех
клетках. Замедление про-
цессов пролиферации приво-
дит к увеличению размеров
миелоцитов, метамиело-
цитов, палочкоядерных и
сегментоядерных нейтро-
филов (рис. 64), характер-
на гиперсегментация ядер
нейтрофилов, в тяжелых
случаях - нейтропения.
Количество мегакариоцитов обычно нормальное, может нарушаться от-
шцуровка тромбоцитов.
Периферическая кровь. Результатом мегалобластического кроветворения
является развитие макроцитарной гиперхромной анемии (концентрация НЬ
может снижаться до 25—40 г/л). Количество эритроцитов резко снижено
(1,0-1,5 х 1012/л). Отмечается высокий цветовой показатель (1,1—1,4), увели-
чение среднего объема эритроцитов (MCV > 100 фл) и среднего содержания
гемоглобина в эритроците (МСН >32 пг) при нормальных значениях средней
концентрации гемоглобина в одном эритроците (МСНС). Эритроцитарная
гистограмма значительно смещается вправо, уплощается, растягиваясь вдоль
оси X (рис. 65).
Эритроциты отличаются равномерной окраской - гиперхромные вследс-
твие увеличения толщины клеток, без центрального просветления, диамет-
ром более 10 мкм (макроциты и мегалоциты). Им свойственен анизоцитоз,
пойкилоцитоз, шизоцитоз, встречаются эритроциты с остатками ядерной
субстанции (кольца Кебота, тельца Жолли), базофильной пунктацией (ос-
татки РНК), полихроматофильные эритроциты (рис. 66, 67). Нередко при-
сутствуют мегалобласты.
У больных В 12-дефицитной анемией на фоне макроцитарной, гиперхром-
ной анемии отмечается нормальное или сниженное относительное коли-
чество ретикулоцитов (RET%), однако их абсолютное содержание (RET#),
независимо от относительного содержания, всегда уменьшено. Средние
объемы ретикулоцитов (MRV) и сфсрулированных эритроцитов (MSCV)
повышены аналогично MCV эритроцитов. Фракция незрелых ретикулоцитов
(IRF) и относительное количество незрелых ретикулоцитов (HLR%) также
увеличиваются, в то время как абсолютное количество незрелых ретикуло-
цитов (HLR#) снижается. В процессе лечения витамином В|2 отмечается
Рис. 66. Периферическая кровь. Макро- и мегалоциты (1), эритроциты с тельцами Жол-
ли (2), нормобласты (3), полихроматофильные эритроциты (4) при В,2-дефицитной анемии.
1x1000
RBC 2,9 3,8-5,5 х 10,2/л
НЬ 102 130-155 г/л
Ht 33,5 36-48%
MCV 110,0 80-95 фл
МСН 35,0 27-31 пг
МСНС 330 300-380 г/л
RDW 16,5 11,5-14,5%
Рис. 67. Периферическая кровь. Базофильная пунктация эритроцитов (1), кольцо Кебота (2),
тельца Жолли (3), гиперсегментированный нейтрофил (4) при В,?-дефицитной анемии. хЮОО
RBC
RBC 1,51 3,8—5,5 х 1012/л
НЬ 64 130-155 г/л
Ht 17,4 36-48%
MCV 115,0 80-95 фл
МСН 41,1 27-31 пг
МСНС 354 300-380 г/л
RDW 27,1 11,5-14,5%
положительная динамика со стороны эритроцитарных показателей. Ретику-
лоцитарный криз развивается на 6-й день терапии, однако к концу 1 -го месяца
наблюдения абсолютное количество ретикулоцитов может полностью не
нормализоваться, что свидетельствует о еще недостаточно восстановленной
регенераторной способности костного мозга и необходимости продолжения
лечения витамином В12. Фракция незрелых ретикулоцитов (IRF) и количество
незрелых ретикулоцитов (HLR#, HLR%) повышаются значительно раньше
(на 2-3-й день лечения) и опережают подъем ретикулоцитов (RET%). Таким
образом, мониторинг за состоянием эритропоэтической активности костного
мозга и эффективностью лечения витамином В12 можно оценивать по наибо-
лее чувствительным маркерам - IRF, HLR%, HLR#.
Дефицит В|2 влияет на все пролиферирующие гемопоэтические клетки,
поэтому в крови наблюдается панцитопения. Однако гранулоцитопения и
тромбоцитопения выражены не столь резко, как анемия. Механизм развития
нарушений со стороны гранулоцитов и тромбоцитов частично связывают с
неэффективным грануло- и тромбоцитопоэзом.
Для В12-дефицитной анемии характерна лейкопения, нейтропения с от-
носительным лимфоцитозом, моноцитопения, может наблюдаться анэози-
нофилия или абазофилия. Отмечается появление в крови гигантских гипср-
сегментированных нейтрофилов (количество сегментов более 5) (рис. 67),
иногда сдвиг влево до миелоцитов и мстамиелоцитов.
Тромбоцитопения носит умеренный характер, тромбоцитов редко бывает
менее 100 х 109/л, встречаются гигантские формы, но функция их не нарушена
и геморрагический синдром наблюдается редко. В зависимости от степени
выраженности анемии СОЭ ускорена до 50-70 мм/ч. В сыворотке крови имеет
место снижение содержания витамина В12 (менее 80 пмоль/л).
Диагноз В12-дефицитной анемии может быть установлен только при мор-
фологическом исследовании костного мозга, которое следует проводить до
введения витамина В12. Инъекция витамина В|2в течение 1-2 суток изменяет
тип кроветворения в костном мозге. Мегалобласты уменьшаются в размерах,
меняется структура ядра, клетки становятся макронормобластами. Только
по присутствию гигантских форм нейтрофилов можно предположить, что
имело место мегалобластное кроветворение.
Гематологическая ремиссия определяется нормализацией костномозгового
кроветворения и показателей периферической крови. Анемия корригируется
в пределах 6 12 недель. У больных, которым длительно проводят витами-
нотерапию, может со временем развиваться желсзодефицитная анемия при
нарушении метаболизма железа и активизации синтеза гемоглобина. В этих
случаях отмечаются нормо- или умеренно гипохромные эритроциты (МСН
26-32 пг).
Синдром Имерслунд-Грисбека. Редкая наследственная патология (имеется
около 100 наблюдений). Всасывание витамина В 12 нарушено из-за отсутствия
рецепторов в тощей кишке, связывающих комплекс В|2 - внутренний фактор.
Проявляется мегалобластной анемией, нормальной желудочной секрецией,
нормальным содержанием внутреннего фактора, протеинурией без других
изменений в моче и без развития почечной недостаточности.
Фолиеводефицитные анемии
В организме человека фолиевая кислота содержится в количестве 5-10 мг.
Суточная потребность в фолиевой кислоте составляет 50-100 мкг. Запасы ее
истощаются через 3—4 месяца после прекращения поступления в организм.
Фолаты синтезируются растениями и микроорганизмами. Наибольшее ко-
личество фолиевой кислоты содержится в зеленых овощах, фруктах, печени,
почках, дрожжах. Более 50% фолиевой кислоты может разрушаться при
кулинарной обработке пищи, отсюда дефицит ее у лиц, употребляющих ва-
реные продукты. Фолиевая кислота всасывается в двенадцатиперстной кишке
и проксимальном отделе тощей кишки. Способность кишечника всасывать
фолиевую кислоту превышает суточную потребность в витамине. В плазме
крови она связывается с транспортными белками (Р2-макроглобулином,
альбумином). Основным депо фолатов является печень, где она находится в
неактивном состоянии (в виде тетрагидрофолиевой кислоты) и переходит в
активную форму по мере метаболических потребностей клеток. Выведение
фолиевой кислоты из организма происходит преимущественно с мочой, в
меньшей степени с калом.
Фолаты, так же как и витамин В|2, занимают ключевое положение во
многих видах клеточного метаболизма, включая синтез аминокислот и
нуклеиновых кислот, особенно необходимых для пролиферирующих кле-
ток. Коэнзимы фолиевой кислоты необходимы для образования пуриновых
соединений, биосинтеза метионина.
Причины развития дефицита фолиевой кислоты
1. Снижение содержания в пище.
• Алкоголизм, голодание, «чай с бутербродами».
• Длительная кулинарная обработка пищи.
2. Нарушение всасывания.
• Хронический энтероколит, резекция тонкой кишки, диабетическая
энтеропатия.
• Алкоголизм, целиакия, тропический спру, амилоидоз.
• Недоношенные дети, находящиеся на искусственном вскармливании.
3. Повышение потребности.
• Беременность.
• Гемолиз, лейкозы, рак, туберкулез, гипертиреоз.
4. Уменьшение запасов в печени.
• Алкоголизм, цирроз, гепатоцеллюлярный рак.
5. Лекарственные препараты (нарушение утилизации).
• Цитостатики.
• Контрацептивы.
• Противосудорожные.
• Противотуберкулезные препараты.
• Препараты для лечения гиперлипидемии.
Клиника. Болеют чаще лица молодого возраста, беременные женщины.
Преобладают признаки анемии: бледность кожи с легкой субиктеричностью,
тахикардия, слабость. Неврологическая симптоматика несвойственна этим
больным, нарушения со стороны желудочно-кишечного тракта незначитель-
ны: в виде диареи, синдрома мальабсорбции.
У лиц, страдающих эпилепсией и шизофренией, дефицит фолиевой кис-
лоты приводит к учащению приступов и ухудшению течения заболевания.
Дефицит фолиевой кислоты увеличивает риск осложнений беременности и
родов (выкидыши, кровотечения, отслойка плаценты, недоношенность).
Изменения в крови и костном мозге аналогичны В12-дефицитной анемии.
В сыворотке крови отмечается снижение уровня фолата (норма 6-20 нг/мл),
концентрация его уменьшена и в эритроцитах (норма 160-640 нг/мл).
АНЕМИИ ВСЛЕДСТВИЕ УСИЛЕННОГО РАЗРУШЕНИЯ
ЭРИТРОЦИТОВ (ГЕМОЛИТИЧЕСКИЕ АНЕМИИ)
Гемолиз эритроцитов
Гемолиз (от греческого слова Haima кровь, Lysis - разрушение) - разру-
шение клеток гемопоэза. Продолжительность жизни эритроцитов составляет
от 110 до 130 дней, в среднем 120 дней. В течение одной минуты эритроцит
дважды проходит через капилляры меньшего диаметра (2-4 мкм), чем диа-
метр эритроцита (большая часть которых имеет d 7,2-7,5 мкм). За период
жизни эритроцит покрывает расстояние в 150-200 км, из которых около
половины составляют узкие территории. На некоторое время эритроциты
застаиваются в синусах селезенки, где сосредоточена специализированная
система фильтра и удаления состарившихся эритроцитов.
Внутриклеточный гемолиз
В нормальном организме существует постоянное равновесие между про-
дукцией и деструкцией кроветворных клеток. Основная масса эритроцитов
разрушается путем фрагментации (эритрорексиса) с последующим лизисом
и эритрофагоцитозом в органах ретикулоэндотелиальной системы (РЭС),
преимущественно в селезенке, частично в печени. Нормальный эритроцит
проходит синусы селезенки благодаря своему свойству изменять форму. По
мере старения эритроциты теряют способность деформироваться, задержи-
ваются в синусах селезенки и секвестрируются.
Из поступившей в селезенку крови 90% эритроцитов проходит, не задер-
живаясь и не подвергаясь фильтрационному отбору (рис. 68).
Рис. 68. Схематическое изображение прохождения крови через селезенку: а - только около
10% эритроцитов подвергается фильтрации в синусоидах, 90% эритроцитов проходит селезенку
без задержки; б — фильтрация эритроцита через фенестр синусоида селезенки
10% эритроцитов попадает в систему сосудистых синусов и вынуждены
выбираться из них, профильтровываясь через поры (фенестры), размер ко-
торых на порядок меньше (0,5-0,7 мкм), чем диаметр эритроцита. У старых
эритроцитов изменена ригидность мембраны, они застаиваются в синусо-
идах. В синусах селезенки снижен pH и концентрация глюкозы, поэтому
при задержке в них эритроцитов последние подвергаются метаболическому
истощению. Макрофаги расположены по обеим сторонам синусов, их ос-
новная функция - элиминировать старые эритроциты. В макрофагах РЭС
заканчивается разрушение эритроцита (внутриклеточный гемолиз). В нор-
мальном организме с помощью внутриклеточного гемолиза разрушается
почти 90% эритроцитов.
Механизм распада гемоглобина в клетках РЭС начинается с одновре-
менного отщепления от него молекулы глобина и железа. В оставшемся
тетрапиррольном кольце под действием фермента гсмоксигеназы происхо-
дит окисление а-метиновой связи, при этом гем теряет свою цикличность,
образуя линейную структуру - биливердин. На следующем этапе путем
ферментативного восстановления у-метиновой связи биливердин-редуктазой
биливердин превращается в билирубин. Билирубин, образованный в РЭС,
поступает в кровь, связывается с альбумином плазмы и в таком комплексе
поглощается гепатоцитами, которые обладают селективной способностью
захватывать билирубин из плазмы. До поступления в гепатоцит билирубин
носит название неконъюгированный, или непрямой. При высокой гиперби-
лирубинемии небольшая часть может оставаться несвязанной с альбумином
и фильтроваться в почках.
Паренхиматозные клетки печени адсорбируют билирубин из плазмы с
помощью транспортных систем белков мембраны гепатоцита - протеина Y
(лиганлин) и протеина Z. Место пребывания протеина Z в основном в сли-
зистой кишечника, однако при высокой билирубинсмии после насыщения
протеина Y он включается в транспорт пигмента и в печени. В гепатоците
неконъюгированный билирубин подвергается конъюгации главным обра-
зом с глюкуроновой кислотой. Этот процесс катализируется ферментом
уридилдифосфат(УДФ)-глюкуронилтрансферазой с образованием конъюги-
рованного билирубина в виде моно- и дигпюкуронидов. Активность фермента
снижается при поражении гепатоцита. Активность этого фермента, так же
как и лигандина, низкая у новорожденных. Поэтому печень новорожденного
не в состоянии переработать больших количеств билирубина распадающихся
избыточных эритроцитов, и у новорожденных развивается физиологическая
желтуха.
Конъюгированный билирубин выделяется из гепатоцита с желчью в виде
комплексов с фосфолипидами, холестерином и солями желчных кислот.
Дальнейшее преобразование билирубина происходит в желчных путях под
влиянием дегидрогеназ с образованием уробилиногенов, мезобилирубина и
других производных билирубина. Уробилиноген в двенадцатиперстной кишке
всасывается энтероцитом и с током крови воротной вены возвращается в
печень, где окисляется до конечных продуктов - дипирролов.
Остальной билирубин и его производные поступают в кишечник, в
котором последовательно восстанавливаются (3- и о-метиновые связи,
и превращаются в стеркобилиноген. Основная масса стеркобилиногена
в толстой кишке подвергается окислению в стеркобилин и выделяется с
калом. Небольшая часть всасывается в кровь и через геморроидальные
вены и нижнюю полую вену поступает в почки и выводится с мочой.
Следовательно, билирубин экскретируется из организма в виде стерко-
билина кала и уробилина мочи. По концентрации стеркобилина в кале
можно судить об интенсивности гемолиза. От концентрации стеркоби-
лина в кишечнике зависит и степень уробилинурии. Однако генез уро-
билинурии определяется также функциональной способностью печени к
окислению уробилиногена. Поэтому увеличение уробилина в моче может
свидетельствовать не только о повышенном распаде эритроцитов, но и о
поражении гепатоцитов.
Лабораторными признаками повышенного внутриклеточного ге-
молиза являются: увеличение содержания в крови неконъюгированного
билирубина, стеркобилина кала и уробилина мочи.
Патологический внутриклеточный гемолиз может возникнуть вследс-
твие:
• наследственной неполноценности мембраны эритроцита (эритроци-
топатии);
• нарушения синтеза гемоглобина и ферментов (гемоглобинопатии,
энзимопатии);
• изоиммунологического конфликта по групповой и Rh-принадлежности
крови матери и плода;
• избыточного количества эритроцитов (физиологическая желтуха,
эритробластоз новорожденного, эритремия - при количестве эритро-
цитов более 6-7 х 1012/л).
Внутрисосудистый гемолиз
Внутрисосудистый гемолиз - физиологический распад эритроцитов
непосредственно в кровотоке. На его долю приходится около 10% всех
гемолизирующихся клеток (рис. 69). Этому количеству разрушающихся
эритроцитов соответствует от 1 до 4 мг свободного гемоглобина (ферро-
гемоглобин, в котором железо 2-валентное - Fe2+ в 100 мл плазмы крови.
Освобожденный в кровеносных сосудах в результате гемолиза гемоглобин
связывается в крови с белком плазмы - гаптоглобином (hapto - по гречески
«связываю»), который относится к а2-гл°булинам. Образующийся комп-
лекс гемоглобин - гаптоглобин имеет Мм от 140 до 320 кДа, в то время как
фильтр клубочков почек пропускает молекулы Мм меньше 70 кДа. Комплекс
поглощается РЭС и разрушается ее клетками. Способность гаптоглобина
связывать гемоглобин препятствует экстраренальному его выведению. Ге-
моглобинсвязывающая способность гаптоглобина составляет 100 мг в 100 мл
крови (100 мг%). Превышение резервной гемоглобинсвязывающей емкости
гаптоглобина (при концентрации гемоглобина 120-125 г/л) или снижение
его уровня в крови сопровождается выделением свободного гемоглобина
через почки с мочой. Это имеет место при массивном внутрисосудистом
гемолизе (рис. 70).
Поступая в почечные канальцы, гемоглобин адсорбируется клетками
почечного эпителия. Реабсорбированный эпителием почечных каналь-
цев гемоглобин разрушается in situ с образованием ферритина и гемо-
Рис. 69. Внутрисосудистый и внутриклеточный гемолиз. Внутрисосудистый гемолиз является
быстрой реакцией, связанной, как правило, с абсорбцией на мембране эритроцита антител
класса IgM и активацией системы комплемента. Внутриклеточный гемолиз - процесс доста-
точно медленный, сопровождается сначала адгезией на эритроцитарной мембране IgG, затем
фагоцитозом «маркированных клеток»
Рис. 70. Возможные пути утилизации гемоглобина при внутрисосудистом гемолизе. При мас-
сивном внутрисосудистом гемолизе некоторое количество НЬ фильтруется в почках (фильтр в
клубочках пропускает молекулы <60—70 кДа, НЬ имеет Мм около 64 кДа). В почечных канальцах
значительное количество НЬ подвергается эндоцитозу и может вызвать их гемосидероз, а часть
НЬ выделяется с мочой. Основная масса освобождающегося при внутрисосудистом гемолизе
гемоглобина связывается гаптоглобином, комплекс утилизируется в селезенке, печени. Часть
свободного НЬ накапливается в плазме и обратимо окисляется до метгемоглобина (ферри-
гемоглобина, в котором железо трехвалентное — Fe3+). При распаде НЬ до гема и глобина
гем расщепляется или связывается специфическим белком гемопексином или альбумином.
Комплексы затем, так же как гаптоглобин, утилизируются печенью
сидерина. Возникает гемосидероз почечных канальцев. Эпителиальные
клетки почечных канальцев, нагруженные гемосидерином, слущиваются
и выделяются с мочой. При гемоглобинемии, превышающей 125-135 мг
в 100 мл крови, канальцевая реабсорбция оказывается недостаточной, в
моче появляется свободный гемоглобин. Между уровнем гемоглобинемии
и появлением гемоглобинурии не существует четкой зависимости. При
постоянной гемоглобинемии гемоглобинурия может возникать при более
низких цифрах свободного гемоглобина плазмы. Снижение концентрации
гаптоглобина в крови может вызывать гемоглобинурию и гемосидери-
нурию при более низких концентрациях свободного гемоглобина крови.
Уменьшение гаптоглобина возможно при наследственном его дефиците,
нефропатиях вследствие потери (протеинурия) или длительном гемолизе в
результате потребления. При высокой гемоглобинемии часть гемоглобина
окисляется до метгемоглобина. Возможен распад гемоглобина в плазме
до гема и глобина. В этом случае гем связывается альбумином или специ-
фическим белком плазмы - гемопексином. Комплексы затем, так же как
гемоглобин - гаптоглобин, подвергаются фагоцитозу. Строма эритроцитов
поглощается и разрушается макрофагами селезенки или задерживается в
концевых капиллярах периферических сосудов.
Лабораторные признаки внутрисосудистого гемолиза:
• гемоглобинемия,
• гемоглобинурия,
• гемосидеринурия.
Патологический внутрисосудистый гемолиз может возникнуть при
токсических, механических, радиационных, инфекционных, иммуно- и
аутоиммунных повреждениях мембраны эритроцитов, дефиците витаминов,
паразитах крови. Усиленный внутрисосудистый гемолиз наблюдается при
пароксизмальной ночной гемоглобинурии, эритроцитарных энзимопатиях,
паразитозах, в частности малярии, приобретенных аутоиммунных гемоли-
тических анемиях, посттрансфузионных осложнениях при несовместимости
по групповой или резус-принадлежности, переливании донорской крови с
высоким титром антиэритроцитарных антител, которые появляются при
инфекциях, сепсисе, инфекционном гепатите, злокачественном новообра-
зовании, беременности и т. д.
Дифференциально-диагностические признаки
внутриклеточного и внутрисосудистого гемолиза
Видом гемолиза определяется симптоматика и течение заболевания
(табл. 12). Анемии, обусловленные преимущественно внутрисосудистым
гемолизом, имеют, как правило, острое начало болезни, характеризуются
повышением содержания свободного гемоглобина крови, выделением его с
мочой и отложением в канальцах почек. Анемиям с внутриклеточным гемо-
лизом более свойственно хроническое течение с гемолитическими кризами,
ремиссиями и спленомегалией, которая развивается в ответ на длительный
повышенный гемолиз эритроцитов. Каждому виду гемолиза соответствуют
определенные лабораторные показатели. Гемолиз с внутриклеточной локали-
зацией процесса сопровождается изменениями обмена желчных пигментов
с отложением гемосидерина в селезенке. Внутрисосудистое кроверазруше-
ние характеризуется повышением содержания свободного гемоглобина в
сыворотке крови, выделением его с мочой и отложением гемосидерина в
канальцах почек.
Таблица 12
Сравнительная характеристика внутриклеточного
и внутрисосудистого гемолиза
Признаки гемолиза Внутрисосудистый Внутриклеточный
Локализация гемолиза Сосудистая система РЭС
Патогенетический фактор Гемолизины, энзимопатия эритроцитов Аномалия формы эритроцитов
Гепатоспленомегалия Незначительная Значительная
Морфологиче ские изменения эритроцитов Анизоцитоз Микросфероцитоз, овалоцитоз, мишеневидные, серповидноклеточные И др.
Локализация гемосидероза Канальцы почек Селезенка, печень, костный мозг
Лабораторные признаки гемолиза Гемоглобинемия Гемоглобинурия Гемосидеринурия Г ипербилирубинемия Повышение стеркобилина в кале и уробилина в моче
Вместе с тем в некоторых ситуациях, например при наличии в крови двух
видов антиэритроцитарных антител (агглютининов и гемолизинов), могут
обнаруживаться признаки как внутриклеточного, так и внутрисосудистого
гемолиза. Гемосидерин также может присутствовать в канальцах почек и
селезенке у больных гемолитической анемией с внутрисосудистым гемоли-
зом при гемотрансфузиях. Степень гемолиза зависит от активности клеток
РЭС и титра антител.
Сокращение длительности жизни эритроцитов - общая характеристика
всех гемолитических анемий. Если интенсивность гемолиза незначительно
превышает физиологический уровень, то избыточное разрушение эрит-
роцитов компенсируется регенеративной пролиферацией костного мозга.
При этом в крови обнаруживаются признаки активации кроветворения
(ретикулоцитоз и полихроматофилия). Количество ретикулоцитов в крови
может достигать 8-10%, а эритроцитов и гемоглобина оставаться в преде-
лах нормы. Возможны лейкоцитоз и незначительный тромбоцитоз. Другие
признаки гемолиза - повышение концентрации неконъюгированного би-
лирубина и/или гемосидеринурия и гемоглобинемия. При гемотрансфу-
зии эритромассы больным гемолитической анемией с внутрисосудистым
гемолизом гемосидерин будет обнаруживаться и в канальцах почек, и в
селезенке.
При патологическом увеличении разрушения эритроцитов более чем в
5 раз и недостаточной активности гемопоэза развивается анемия, степень
которой зависит от интенсивности гемолиза, исходных гематологических
показателей и состояния эритропоэза. Анемия носит норме-, гиперхромный
характер. Длительный или часто повторяющийся внутрисосудистый гемолиз
приводит к дефициту железа в организме и к развитию железодефицитной
анемии. Между гемолизом и анемией может установиться равновесие. Это
так называемый компенсированный гемолиз. Непрекращающийся гемолиз
при недостаточности кроветворения сопровождается прогрессирующей
анемией, что вызывает декомпенсацию гемолиза.
Костномозговое кроветворение характеризуется преимущественно
реактивными изменениями. Наиболее часто наблюдается эритробластоз,
возможно увеличение гранулоцитов и мегакариоцитов.
В периферической крови - ретикулоцитоз, полихроматофилия, эритро-
нормобластоз. Может быть нормальное количество лейкоцитов, лейкопения
и лейкоцитоз со сдвигом в лейкоцитарной формуле влево до миелоцитов.
При остром гемолизе, обусловленном трансфузией несовместимой крови,
обнаруживаются групповые или антирезус-антитела.
Освободившаяся при гемолизе строма эритроцитов поглощается и разру-
шается макрофагами селезенки или задерживается в концевых капиллярах,
нарушая их микроциркуляцию. Внутрисосудистый гемолиз эритроцитов
сопровождается поступлением в кровоток эритроцитарного тромбопластина,
что может способствовать нарушению гемокоагуляции. Поэтому при остром
внутрисосудистом гемолизе независимо от основного заболевания возможны
изменения гемостаза.
Схема лабораторного обследования больных гемолитической анемией
представлена в табл. 13.
Таблица 13
Схема лабораторного обследования больных гемолитической анемией
Вид гемолитических анемий Общие исследования Дополнительные исследования
Анемии, обусловленные внеэритроцитарными факторами
Токсические, инфекционные, паразитарные, посттрансфузионные, иммунные, аутоиммунные и др. Гемограмма, включая эритроцитарные индексы и тромбоциты Ретикулоциты Мазок и толстая капля на малярию, прямая и непрямая проба Кумбса, свободный гемоглобин крови, гемоглобин и гемосидерин мочи, циркулирующие иммунные комплексы
Анемии, обусловленные эритроцитарными факторами
Эритроцитопатии Гемограмма, включая эритроцитарные индексы и тромбоциты Ретикулоциты Миелограмма, осмотическая резистентность эритроцитов, билирубин и его фракции, уробилин мочи
Энзимопатии эритроцитов То же Ферменты эритроцитов (Г-6-ФДГ, пируваткиназа, глутатионсинтетаза), осмотическая резистентность эритроцитов, свободный гемоглобин крови, гемоглобин и гемосидерин мочи
Гемоглобинопатии То же Электрофорез гемоглобина, проба на серповидность эритроцитов, тельца Гейнца, билирубин и его фракции, уробилин мочи
Пароксизмальная ночная гемоглобинурия То же Проба Хема, сахарозная проба
Анемии, обусловленные внеэритроцитарными
гемолитическими факторами
Иммунные гемолитические анемии
Иммунная гемолитическая анемия (ИГА) изоиммунного или аутоиммун-
ного (ЛИГА) генеза - клинический синдром, проявляющийся некомпенсиро-
ванным гемолизом, который развивается вследствие аберрации иммунных
реакций, направленных против измененных (чужеродных) и неизмененных
антигенов эритроцитов (при патологии лимфоидной ткани).
При ИГА в организме появляются чужеродные антигены, на которые выра-
батываются антитела нормальными клетками иммунологической ткани. При
ЛИГА клетки иммунологической системы синтезируют антитела к собствен-
ным неизмененным антигенам эритроцитов. Причиной иммунизации могут
быть перенесенные инфекции (вирусные, бактериальные), лекарственные
препараты, переохлаждение, вакцинация и другие факторы, под влиянием
которых образуются антитела к клеточным элементам крови. При наличии
двух заболеваний ИГА считается симптоматической или вторичной. Иммун-
ная гемолитическая анемия может предшествовать другим заболеваниям, тем
не менее наличие ее указывает на генерализованное, множественное имму-
нологическое нарушение. Иммунный конфликт происходит с антигенами,
находящимися на поверхности клеток, либо с клеточными структурами, в
результате эритроциты разрушаются - развивается гемолитическая анемия.
Течение болезни, клиническую картину, гематологические и лабораторные
показатели определяют вид антител и их функциональные характеристики.
Приобретенные гемолитические анемии возникают при появлении в
сыворотке крови агглютининов, представляющих по серологическим свойс-
твам неполные и полные холодовые и тепловые антитела. Их отличительной
особенностью является внутриклеточный гемолиз и изменение показателей
пигментного обмена. Гемолитические анемии, обусловленные присутствием
в крови гемолизинов, вызывают разрушение эритроцитов в кровяном русле
при обязательном взаимодействии с комплементом. Для них характерно на-
личие внутрисосудистого гемолиза, показатели которого - гемоглобинемия,
гемоглобинурия и гемосидеринурия.
Локализация лизиса эритроцитов зависит от серологических свойств
антител, подкласса антител, их концентрации на клеточной мембране,
температурного оптимума действия (холодовые, тепловые). Так, холодовые
антитела, которые относятся к IgM, реагируют с эритроцитами при низких
температурах (ниже температуры тела). В эту реакцию обычно вовлекаются
и факторы системы комплемента, поэтому преобладает внутрисосудистый
гемолиз, а секвестрация эритроцитов в селезенке ограничена. Тепловые ан-
титела, принадлежащие к IgG, вызывают реакцию при температуре тела без
вовлечения комплемента. Секвестрация эритроцитов в селезенке является
ведущим механизмом деструкции клеток.
Посттрансфузионные анемии
Наиболее частым внеэритропитарным фактором, вызывающим гемолити-
ческую анемию, являются антитела к антигенам эритроцитов, возникающие
при введении в организм чужеродных антигенов. В основе анемий, развива-
ющихся в результате переливания крови, лежит, как правило, внутрисосудис-
тый гемолиз эритроцитов. Многочисленные причины, которые приводят к
гемотрансфузионным осложнениям, обусловлены несоблюдением правил при
переливании крови. Можно выделить, по крайней мере, шесть групп различ-
ных факторов, способствующих развитию посттрансфузионных реакций.
Причины осложнений при гемотрансфузиях'.
• Несовместимость крови донора и реципиента по антигенам эритроцитов
системы АВО, резус и других систем.
• Недоброкачественность перелитой крови (бактериальное загрязнение,
перегревание, переохлаждение, гемолиз эритроцитов, денатурация
белков вследствие длительного хранения, нарушения температурного
режима хранения и т. п.).
• Погрешности в методике трансфузии (воздушная эмболия, тромбоэмбо-
лия, циркуляторная перегрузка, сердечно-сосудистая недостаточность
и др.).
• Массивные дозы трансфузии (40-50% объема ОЦК). В этом случае
50% перелитых эритроцитов секвестрируется в органах, что приводит
к нарушению реологии крови (синдром гомологичной крови).
• Не учитываются строго противопоказания к переливанию крови.
• Перенос возбудителя инфекционных заболеваний с переливаемой
кровью.
Кровь каждого человека принадлежит к какой-либо одной из 4 групп крови
системы АВО в зависимости от присутствия на эритроцитах антигенов А и В и
соответствующих антител в плазме крови - агглютининов (анти-А и анти-В).
В табл. 14 приведены характеристики основных групп крови системы АВО.
Во избежание несовместимости крови донора и реципиента по эритроцитам
следует учитывать их групповую и резус-принадлежность. Предпочтение
отдается гемотрансфузиям одногруппной крови, совместимой по резус-фак-
тору. В экстренных случаях возможно переливание эритроцитарной массы
0 (I) реципиенту любой группы крови.
Таблица 14
Характеристика групп крови системы АВО
Группа крови системы АВО Эритроциты Сыворотка крови
наличие антигенов реакция с антителами наличие антител реакция с антигенами эритроцитов
анти- А(а) анти- В(Р) анти-А анти-В А-антиген В-антиген
Осф (I) нет — - — анти-А и анти-В + +
Ар (II) А + — + анти-В - +
Ba (III) В — + + анти-А + -
АВО (IV) А и В + + + нет - -
Наиболее частая причина посттрансфузионных осложнений - перелива-
ние несовместимой крови, результатом которого является развитие реакции
антител IgM (несовместимость по АВО) или IgG (несовместимость по резус-
фактору) с антигенами, встроенными в клеточную мембрану эритроцитов
реципиента, связывание с комплементом и последующий гемолиз.
В клинической картине посттрансфузионных осложнений выделяют два пе-
риода-гемотрансфузионный шок и острую почечную недостаточность (ОПН).
Гемотрансфузионный шок развивается в ближайшие минуты или часы. Начи-
нается реакция с появления боли в пояснице, грудине, по ходу вен. Появляется
беспокойство, озноб, одышка, гиперемия кожных покровов. В тяжелых случаях
развивается шок. Обязательным признаком при переливании несовместимой
крови является острый гемолиз. Характер гемолиза определяется видом антител:
при наличии аплютининов имеет место преимущественно внутриклеточный
гемолиз, гемолизины вызывают внутрисосудистый гемолиз. При групповой
несовместимости внутрисосудистый гемолиз определяется наличием высокого
титра иммунных или аутоиммунных анти-А- или анти-В-антител донора, кровь
которого переливают реципиенту. Первые признаки гемолиза выявляются сразу
после переливания несовместимой крови. Тяжесть клинической и гематологи-
ческой симптоматики зависит от дозы перелитой крови.
Костномозговое кроветворение характеризуется резкой гиперплазией
с преимущественной активацией эритропоэза. При острой почечной не-
достаточности в костном мозге обнаруживается угнетение эритропоэза по
гипорегенераторному типу.
Кровь. Анемия, возникающая вследствие повышенного распада эрит-
роцитов, имеет гиперрегенераторный характер, о котором можно судить по
нарастанию в крови ретикулоцитов, наличию полихроматофилии, эритро-
кариоцитов. Другие гематологические признаки гемолиза (изменение ос-
мотической резистентности эритроцитов, их объема, диаметра, цветового
показателя) непостоянны и нетипичны. Изменения лейкопоэза также непос-
тоянны, чаще отмечается лейкоцитоз со сдвигом лейкоцитарной формулы
влево вплоть до миелоцитов.
В сыворотке крови повышена концентрация неконъю тированного билиру-
бина. Лишь в первые часы после переливания несовместимой крови можно
обнаружить гемоглобинемию, поскольку свободный гемоглобин быстро по-
глощается клетками РЭС и выводится почками (гемоглобинурия). Количество
мочи уменьшается, благодаря наличию в ней свободного гемоглобина (гемогло-
бинурия) и гемосидерина (гемосидеринурия) она приобретает бурый цвет.
Анемия - постоянный симптом ОПН. Она характеризуется как макроци-
тарная, нормохромная, гипорсгенераторная. Анемия выявляется с первых
дней гемотрансфузионного осложнения и нс купируется до тех пор, пока не
нормализуется функция почек.
Гемолитическая болезнь новорожденных (эритробластоз плода)
Гемолитическая анемия новорожденных чаще всего связана с несовмес-
тимостью родителей по резус-фактору (Rh): у Rh-отрицательной женщины в
период беременности Rh-положительным плодом, унаследовавшим Rh-поло-
жительный фактор от отца, образуются антирсзус-антитсла. Резус-конфликт
развивается приблизительно у одной из 15 рсзус-отрицательных женщин, и,
возможно, не последнюю роль в этом играет повышенная для эритроцитов
проницаемость плаценты. Антитела, возникающие в организме матери,
проникают в кровь плода, оседают на поверхности клеток и вызывают их
агглютинацию с последующим гемолизом эритроцитов в организме плода.
В результате у новорожденного в первые часы жизни развивается гемолити-
ческая анемия с эритробластозом, желтуха. Развитие эритробластоза плода
объясняют активной реакцией костного мозга на распад эритроцитов в ор-
ганизме плода. Антитела в организме плода образоваться не могут.
Анти-Яй-антитела в крови резус-отрицательной женщины могут сохра-
няться в течение многих лет. Дифференциация Rh-фактора в эритроцитах
плода начинается с 3-4-го мес. внутриутробной жизни, а образование Rh-
антител в организме матери - с 4-5-го мес. беременности. Поэтому при
раннем прерывании беременности иммунизации женщины не происходит.
Титр анти-Яй-антител в организме матери накапливается в основном в кон-
це беременности, в период родов антитела оседают на эритроцитах плода,
вызывая их гемолиз. Титр антител увеличивается с каждой последующей
беременностью, поэтому и вероятность Яй-конфликта увеличивается с
каждой беременностью.
Гемолитическая болезнь новорожденных может зависеть также от несов-
местимости крови матери и плода по групповой системе АВО, когда через
плаценту в кровь плода проходят анти-А- или анти-В-агглютинины матери.
Обычно групповая несовместимость по системе АВО крови матери и плода
наблюдается при первой беременности. При гемолитической болезни чаще
имеет место внутриклеточный гемолиз.
Клиника и лабораторные показатели. У новорожденных отмечаются
резкая желтуха, увеличение селезенки и печени, кожные геморрагии, анемия
со значительным количеством эритробластов, достигающих 100-150 тыс. в
1 мкл, и высоким ретикулоцитозом. Нейтрофильный лейкоцитоз со сдвигом
до миелоцитов, неконъюгированная гипербилирубинемия, повышенное
содержание стеркобилина в кале и уробилина в моче.
Аутоиммунные гемолитические анемии
Под аутоиммунной гемолитической анемией (АИГА) понимают такую,
при которой антитела вырабатываются против собственного неизмененного
антигена. По течению АИГА подразделяются на острые и хронические.
На основании серологической характеристики антител и клинических
проявлений выделяют 4 вида АИГА:
1. Аутоиммунные гемолитические анемии с неполными тепловыми
агглютининами. Они составляют 47-80% от всех АИГА.
2. Аутоиммунные гемолитические анемии с тепловыми гемолизинами.
3. Аутоиммунные гемолитические анемии с полными холодовыми
агглютининами, составляющие 12%.
4. Аутоиммунные гемолитические анемии с двухфазными гемолизинами.
АИГА встречаются преимущественно после 40 лет и у детей до 10 лет, соотно-
шение заболевших женщин и мужчин оценивается как 2:1. В патогенезе гемолиза
имеет значение комплекс факторов: класс, подкласс и титр антиэритроцитарных
антител, температурный оптимум их действия, антигенные особенности эрит-
роцитарной мембраны и направленность иммуноглобулинов к тем или иным
антигенам, система комплемента и активность клеток системы мононуклеарных
фагоцитов. Все агглютинины разделяются на полные и неполные. Неполные
агглютинины отличаются тем, что не дают агглютинации, если эритроциты
находятся в водно-солевой среде. Полные агглютинины дают агглютинацию в
любой среде. Неполные антитела обладают меньшей, по сравнению с полными
антителами, молекулярной массой. В связи с этим клиническая картина этих
форм гемолитической анемии отличается от других форм.
При лабораторном исследовании выявляется снижение гемоглобина
до низких цифр (ниже 50 г/л), повышение уровня непрямого билирубина.
Анемия чаще нормохромная или умеренно гиперхромная. Содержание ре-
тикулоцитов может доходить до 87%. Морфологически при АИГА может
наблюдаться микросфероцитоз. Проведение морфологического анализа
крови является необходимым моментом в диагностике, так как при аппа-
ратном анализе крови макро-, микросфероциты нивелированы. В костном
мозге в большинстве случаев красный росток гиперплазирован. Иногда
количество эритрокариоцитов уменьшается. Вероятно, эти кризы связаны с
очень большим количеством антител, вследствие чего разрушаются не только
эритроциты, но и эритрокариоциты. Возможен мегалобластный характер
кроветворения, что связано с относительным дефицитом фолиевой кислоты
и витамина В12 из-за их быстрого потребления. Выявляется спленомегалия,
также возможно увеличение печени.
АИГА диагностируют по наличию аутоантител, фиксированных на эрит-
роцитах, с помощью пробы Кумбса, при которой антиглобулиновые антитела
вступают во взаимодействие с иммуноглобулинами эритроцитов (прямая ре-
акция Кумбса) и вызывают агглютинацию эритроцитов. Можно выявлять цир-
кулирующие антитела в сыворотке крови непрямой пробой Кумбса, смешивая
сыворотку с эритроцитами донора. Как правило, выраженность прямой реакции
Кумбса тесно коррелирует с количеством IgG, фиксированных на эритроцитах.
Отрицательная проба Кумбса не исключает АИГА. Она может иметь место
при интенсивном гемолизе, массивной гормональной терапии, низком титре
антител. Эритроциты, на которых помимо 1g фиксирован комплемент, быстрее
удаляются из кровотока. В этом процессе принимают участие макрофаги печени
и селезенки. При отсутствии на поверхности эритроцита комплемента реша-
ющую роль в гемолизе играют молекулы 1g и основным местом деструкции
эритроцитов является селезенка. Этим объясняется высокая эффективность
спленэктомии при АИГА с неполными тепловыми агглютининами, когда
имеет место опсонизация эритроцитов 1g, и отсутствие эффекта от операции
при АИГА с полными холодовыми агглютининами, при которой эритроциты
сенсибилизированы комплементом и разрушаются в макрофагах печени.
Аутоиммунная гемолитическая анемия, обусловленная неполными
тепловыми агглютининами
Это наиболее распространенная форма аутоиммунных анемий. Заболе-
вание может быть как идиопатическим, то есть без явной причины, так и
симптоматическим. Симптоматические или вторичные АИГА развиваются
на фоне лимфопролиферативных заболеваний и других злокачественных
опухолей, болезней соединительной ткани, инфекций, аутоиммунных заболе-
ваний (тиреоидит, неспецифический язвенный колит, сахарный диабет I типа,
саркоидоз и др.). Тепловые агглютинины могут появляться при лечении боль-
шими дозами пенициллина или цефалоспоринов, при этом они направлены
против комплекса антибиотика с антигенами мембраны эритроцита. Отмена
антибиотика ведет к прекращению гемолиза эритроцитов.
АИГА с тепловым типом антител характеризуется наличием на поверх-
ности эритроцитов антител, как правило, направленных против антигенов
системы резус. С помощью моноспецифических антисывороток на поверх-
ности эритроцитов удается обнаружить иммуноглобулины класса G, А, М
и компоненты комплемента СЗ и С4. Гибель эритроцитов связана в одних
случаях только с наличием антител на клеточной поверхности, в других -
с присутствием комплемента. Течение болезни может быть острое, хро-
ническое и подострое. Обычно гемолиз развивается постепенно, редко
остро. Острое начало более характерно для детского возраста и всегда
в ассоциации с инфекционным процессом. Разрушение эритроцитов
происходит в селезенке (внутриклеточный гемолиз), поэтому свободный
гемоглобин в сыворотке и моче не выявляется. В клинике наблюдаются
признаки, характерные для анемии (бледность, сердцебиение, головокру-
жение) и внутриклеточного гемолиза (желтуха различной интенсивности,
спленомегалия).
В костном мозге отмечается гиперплазия эритроидного ростка, встречают-
ся клетки с мегалобластоидной структурой ядерного хроматина. Анемия имеет
нормо- или гиперхромный характер и сопровождается, как правило, умерен-
ным, реже высоким ретикулоцитозом. Снижение концентрации гемоглобина
зависит от степени гемолитического криза и достигает 50 г/л. В мазках крови
отмечается анизоцитоз, полихроматофилия, могут присутствовать микроциты,
микросфероциты, макроциты, мегалоциты, эритрокариоциты. При автомати-
ческом подсчете клеток отмечаются высокие RDW (показатель анизоцитоза)
и МСН (среднее содержание гемоглобина в эритроците) (рис. 71, 72).
Количество лейкоцитов зависит от активности костного мозга и основного
заболевания, которое лежит в основе гемолиза: может быть нормальным,
однако при острой форме наблюдается лейкоцитоз со сдвигом влево, иногда
лейкопения.
RBC
250 fl
RBC 2,12 3,8-5,5 х 1012/л
НЬ 82 130-155 г/л
Ht 28 36-48%
MCV 121,7 80—95 фл
МСН 38,7 27-31 пг
мснс 31,8 300-380 г/л
RDW 17,7 11,5-14,5%
Рис. 71. Аутоиммунная гемолитическая анемия
Периферическая кровь
Рис. 72. Периферическая кровь ЛИГА. Выраженный смешанный анизоцитоз эритроцитов,
полихроматофилия. Видны микро-, макро-, мегалоциты, сфероциты, базофильная пунктация
эритроцитов. хЮОО
RBC
RBC 0,95 3,8-5,5 х 1012/л
НЬ 33 130-155 г/л
Ht 8,8 36-48%
MCV 93,5 80-95 фл
МСН 35,0 27-31 пг
МСНС 374 300-380 г/л
RDW 32,0 11,5-14,5%
Содержание тромбоцитов в большинстве случаев нормальное, реже сни-
женное. Однако возможно одновременное вовлечение в процесс двух или
трех ростков.
Решающим диагностическим признаком этого вида ЛИГА является по-
ложительная прямая проба Кумбса. Параллелизма между выраженностью
прямой пробы Кумбса и интенсивностью гемолиза нет. Отрицательная
реакция Кумбса не исключает диагноз ЛИГА. Минимальная разрешающая
способность ее сос тавляет 100-500 молекул IgG на эритроцит, при меньшей
концентрации антител реакция будет отрицательной. Во многих случаях
АИГА в разрушении эритроцитов активно участвует система комплемента,
цменно поэтому прямая проба Кумбса нередко бывает отрицательной. Кро-
ме того, недостаточное отмывание эритроцитов при проведении реакции
приводит к тому, что на поверхности эритроцитов остаются неотмытые
сывороточные иммуноглобулины, которые нейтрализуют антигпобулиновую
сыворотку. Отрицательная проба может быть следствием потери с поверх-
ности эритроцита антител низкой афинности в процессе отмывания.
Агрегат-гемагглютинационная проба, разработанная в 1976 г., во много
раз повысила чувствительность реакции Кумбса, однако в виду трудоемкос-
ти она не применяется в широкой клинической практике. Использование
иммуноферментного анализа позволяет количественно оценить содержание
иммуноглобулинов на поверхности одного эритроцита, а также определить
их класс и тип. Значение этих исследований обусловлено тем, что различные
классы и типы иммуноглобулинов обладают неодинаковой физиологической
активностью in vivo. Показано усиление остроты гемолиза при участии в
процессе одновременно нескольких классов иммуноглобулинов. Кроме того,
подкласс иммуноглобулинов во многом определяет остроту гемолиза и место
преимущественной деструкции эритроцитов. В настоящее время применяется
гелевый тест (фирма «Диамед», Швейцария), аналогичный пробе Кумбса, но
более чувствительный. Проба не требует отмывания эритроцитов, с которым
теряется часть 1g, так как гель разделяет эритроциты от плазмы.
Аутоиммунная гемолитическая анемия, обусловленная полными
холодовыми агглютининами (холодовая гемагглютининовая болезнь)
Холодовые антитела относятся к IgM. Описаны идиопатические формы,
однако наиболее часто процесс является вторичным. В молодом возрасте
холодовая гемагглютининовая болезнь (ХГАБ) обычно осложняет течение
острой микоплазменной инфекции и разрешается по мере купирования
последней. У пожилых больных холодовой гемолиз сопутствует хрони-
ческим лимфопролиферативным заболеваниям, протекающим с секрецией
парапротеина IgM. Наиболее часто ХГАБ сопровождает макроглобулинемию
Вальденстрема и хронический лимфолейкоз с секрецией IgM, а также систем-
ные заболевания соединительной ткани. Для этого вида анемии характерен
преимущественно внутриклеточный гемолиз.
Макроглобулин, обладающий свойствами холодовых агглютининов,
благодаря высокой молекулярной массе вызывает гипервискозный синдром.
Заболевание проявляется синдромом Рейно, развитием акроцианоза, тром-
бофлебита, тромбозов, трофических изменений, вплоть до акрогангрены.
IgM функционирует при низких температурах. Оптимальной для действия
макроглобулина является температура +4 °C. Поэтому весь симптомокомп-
лекс заболевания разыгрывается на холоде, при переохлаждении открытых
частей тела. При переходе в теплое помещение гемолиз прекращается.
Холодовая форма АПГА отличается аутоагглютинацией эритроцитов,
которая происходит после взятия крови и часто мешает определению ко-
личества эритроцитов, группы крови и биохимическому анализу. Эта аг-
глютинация обратима и полностью исчезает при подогреве крови. Анемия
нормохромная (НЬ >75 г/л), ретикулоцитоз. При исследовании крови на
гематологических анализаторах агглютинация часто приводит к увеличе-
нию среднего объема эритроцитов и ложно низким значениям эритроцитов.
Количество лейкоцитов и тромбоцитов в пределах нормальных величин,
ускоренная СОЭ. В сыворотке крови - незначительное увеличение неконъ-
югированного билирубина.
Наличие холодовых агглютининов затрудняет определение количества
эритроцитов, групповой принадлежности эритроцитов и СОЭ. Поэтому оп-
ределение проводят либо с подогретым физиологическим раствором, либо в
термостате при температуре 37 °C. В сыворотке крови таких больных обна-
руживают диагностически значимое повышение титра холодовых антител,
на поверхности эритроцитов - IgM. При использовании поливалентной ан-
тиглобулиновой сыворотки прямая проба Кумбса оказывается в ряде случаев
положительной. Полные холодовые агглютинины имеют специфичность к
системе антигенов И, Рр на поверхности эритроцитов.
Аутоиммунные гемолитические анемии,
обусловленные тепловыми гемолизинами
Этот вариант АИГА встречается гораздо реже. В патогенезе этой формы ане-
мии основную роль играют тепловые гемолизины, оптимум действия которых
проявляется при 37 °C. Заболевание имеет хроническое течение и характеризует-
ся признаками внутрисосудистого гемолиза. Доминирующим диагностическим
критерием является гемоглобинурия и гемосидеринурия, которые окрашивают
мочу, как правило, в черный (бурый) цвет. Интенсивность окраски зависит от
степени гемолиза. При выраженном гемолизе имеет место небольшая сплено-
мегалия и незначительное повышение неконъюгированного билирубина.
В костном мозге отмечается активный эритропоэз. В периферической
крови - анемия нормо- или гипохромного типа, ретикулоцитоз как резуль-
тат постепенной потери организмом железа. Количество лейкоцитов может
быть повышено нередко со сдвигом до миелоцитов. Иногда развивается
тромбоцитоз, осложняющийся тромбозами периферических вен, именно
они обуславливают тяжелую симптоматику. Положительным может быть
непрямой тест Кумбса, особенно в кислой среде, тогда как прямая проба
Кумбса чаще бывает отрицательной.
Пароксизмальная холодовая гемоглобинурия с двухфазными
гемолизинами (анемия Доната-Ландштейнера)
В патогенезе заболевания играет роль переохлаждение тела, перенесенная
вирусная инфекция, в частности грипп, корь, паротит, нельзя полностью ис-
ключить сифилис. Двухфазные гемолизины относятся к классу IgG3. Фикса-
ция гемолизинов на эритроцитах происходит при температуре 0-15 °C (первая
фаза), а внутрисосудистый гемолиз, который идет с участием комплемента,
при температуре 37 °C (вторая фаза). Гемолитический эффект наступает при
переходе человека в теплое помещение.
Заболевание проявляется приступами озноба, лихорадкой, болями в жи-
воте, рвотой, тошнотой, вазомоторными нарушениями, появлением мочи
черного цвета через несколько часов после переохлаждения. Может появиться
иктеричность склер, спленомегалия.
В костном мозге отмечается гиперплазия красного ростка. В перифе-
рической крови содержание гемоглобина вне криза бывает нормальным.
В период криза развивается анемия, ретикулоцитоз, лейкопения, реже тром-
боцитопения. В сыворотке крови имеет место повышение уровня свободного
гемоглобина плазмы, снижение концентрации гаптоглобина. В моче - гемо-
глобинурия, гемосидеринурия.
Гемолитическая анемия, обусловленная соматической мутацией
клеток-предшественников миелопоэза
Пароксизмальная ночная гемоглобинурия (болезнь Маркиафава-Микели)
Относительно редкая форма приобретенной гемолитической анемии.
Возникает болезнь в любом возрасте, но встречается у лиц преимущественно
среднего возраста. У 10% детей с апластической анемией зарегистрированы
признаки пароксизмальной ночной гемоглобинурии.
Патогенез. Причиной повышенного гемолиза эритроцитов является
дефект мембраны эритроцитов, лейкоцитов и тромбоцитов, обусловленный
соматической мутацией в стволовых кроветворных клетках pig-A-гена,
ответственного за синтез гликозилфосфатидилинозитолового (GPI) якоря.
Нарушается трансформация N-ацетилглюкозамина в фосфатидилинозитол,
что приводит к неполному построению GPI-якорных протеинов. В норме
все клетки человека имеют механизм защиты от активных компонентов
комплемента благодаря присутствию на мембране комплементарных регуля-
торных протеинов: фактора, ускоряющего распад комплемента (DAF, CD55),
мембранного ингибитора реактивного лизиса (MIRL, CD59), мембранного
кофакторного протеина (МСР, CD46), комплементного рецептора типа 1
(CR1, CD35). DAF (фактор, ускоряющий распад комплемента) на поверх-
ности эритроцита блокирует формирование СЗ-конвертазных комплексов
(С4Ь2а, СЗЬВЬ) или способствует их разрушению. Именно DAF отсутствует
при ИНГ II и III типов как на эритроцитах, так и на тромбоцитах. При ПНГ
эритроциты также могут быть чувствительны к лизису комплементом из-за
отсутствия или снижения экспрессии CD59.
Клетки крови и костного мозга становятся гиперчувствительными к воз-
действию нормальных компонентов плазмы, в первую очередь к СЗ-компо-
ненту комплемента, глюкозе, некоторым факторам системы свертывания кро-
ви. Гемолизу может способствовать ночной ацидоз, беременность, инфекция
и другие факторы. Признаки аномалии свойственны не только эритроцитам,
но и клеткам лейкопоэза и тромбоцитопоэза. Отсюда редукция гемопоэза и
цитопения. В лимфоузлах, селезенке может обнаруживаться экстрамедул-
лярное кроветворение в виде очагов эритробластной гиперплазии.
Заболевание протекает с внутрисосудистым гемолизом, поэтому гемосиде-
роз обнаруживается в канальцах почек. В случае больших количеств гемотранс-
фузий гемосидерин откладывается также в РЭС, главным образом в селезенке.
Распад эритроцитов стимулирует активность факторов свертывания крови и
играет значительную роль в патогенезе тромбоэмболических осложнений.
Клиника. Отличительные признаки заболевания - ночные гемолитичес-
кие кризы, сопровождающиеся выделением мочи бурого цвета (гемоглобин-
урия, гемосидеринурия). Кризы возникают спонтанно либо провоцируются
интеркуррентными инфекциями, в том числе вирусными, гемотрансфузиями,
стимуляторами эритропоэза (например, витамин В|2), лекарственными пре-
паратами, переутомлением и т. п. Гемоглобинурийные кризы имеют место
часто во время физиологического сна. Интервалы между кризами бывают
различные: могут повторяться ежедневно, еженедельно, раз в год в течение
нескольких лет или быстро приводят больного к гибели. Гепатомегалия и
спленомегалия не характерны для болезни Маркиафава -Микели, однако они
могут наблюдаться в связи с развитием посттрансфузионного гемосидероза
органов, при тромбозах системы селезеночных вен, застойном полнокровии,
инфаркте селезенки. Наклонность к тромбозам сосудов наряду с тромбоци-
топенией являются частыми осложнениями болезни.
Костный мозг. На первом этапе болезни имеет место гиперпластический
тип кроветворения с преобладанием эритробластов и нередко мегакарио-
цитов, другие клеточные элементы сохранены, но не принимают участия
в гиперплазии. При прогрессировании заболевания кроветворная актив-
ность костного мозга снижается: уменьшается количество эритробластов,
мегакариоцитов, элементов лейкопоэза, отмечается задержка их созревания
на стадии миелоцита. Изредка возникает арегенераторный криз. В костном
мозге в этих случаях имеет место либо изолированная (парциальная) апла-
зия эритроидного ростка, либо тотальный миелопарез. Арегенераторный
криз затем может сменяться ремиссией. В конечной стадии заболевания
развивается выраженная диффузная гипоплазия костного мозга. Истощение
костномозгового кроветворения проявляется панцитопенией.
Кровь. Анемия является постоянным симптомом заболевания и носит
чаще нормохромный характер (рис. 73).
RBC 3,58 3,8—5,5 х 1012/л
НЬ 107 130-155 г/л
Ht 31 36-48%
MCV 91,3 80-95 фл
МСН 29,9 27-31 пг
мснс 32,7 30,0-38,0 г/л
RDW И.З 11,5-14,5%
Рис. 73. Болезнь Маркиафава—Микели. Пери
ферическая кровь
По мере развития болезни железо постепенно выводится из организма с мо-
чой, и анемия приобретает гипохромный характер. Концентрация гемоглобина
в период обострения может составлять 30 50 г/л. Анемия сопровождается не-
большим ретикулоцитозом (2-4%), не соответствующим степени малокровия.
В период арегенераторных кризов отмечается ретикулоцитопения. Анемия
носит макроцитарный, реже нормоцитарный характер. Отмечается анизоцитоз,
пойкилоцитоз и полихроматофилия. Могут встречаться нормобласты. Цветовой
показатель определяется характером анемии и состоянием обмена железа в ор-
ганизме. Почти постоянная лейкопения (до 3-1,5 х 109/л и ниже) и нейтропения.
При инфекционных осложнениях может иметь место небольшой лейкоцитоз
(до 10-11 х Ю9/л), палочкоядерный сдвиг. Периодически наблюдается моно-
цитоз, относительный лимфоцитоз, базофилия и эозинофилия. Количество
тромбоцитов несколько понижено, возможна значительная тромбоцитопения
(ниже 30 х Ю9/л). Частым симптомом болезни является панцитопения.
В сыворотке крови в период криза происходит повышение концентрации
неконъюгированного билирубина, свободного гемоглобина, снижение гап-
тоглобина, регистрируется положительный тест Хема (лизис исследуемых
эритроцитов донорской сывороткой, где имеются белки системы комплемен-
та). В моче - гемоглобинурия, гемосидеринурия.
Диагноз подтверждается выявлением мутации pig-A-гена и изучением
экспрессии GPI-связанных протеинов на клетках крови.
Гемолитические анемии, обусловленные механическим повреждением
эритроцитов
Травма (с микроангиопатическим гемолизом). При избыточном воз-
действии сил сдвига и турбулентности в периферической крови появляются
фрагменты эритроцитов необычной формы (треугольные, шлемовидные
и др., рис. 74), они служат основанием для диагноза. Из-за присутствия
подобных фрагментов MCV снижается, a RDW увеличивается (проявле-
ние анизоцитоза). Источник травмирования может быть различным; он
может находиться вне сосудов (маршевая гемоглобинурия), внутри серд-
ца (обызвествление и стеноз аортального клапана или дефекты протезов
клапанов сердца), в артериолах (злокачественная гипертензия), в концевых
артериолах (ДВС-синдром). При микроангиопатической гемолитической
анемии, возникающей из-за травматической фрагментации эритроцитов при
прохождении через искусственные сердечные клапаны или поврежденные
кровеносные сосуды, анемия может быть резкой с фрагментированными
эритроцитами (шизоциты, шлемовидные эритроциты).
Рис. 74. Периферическая кровь. Шизоциты (1), шлемовидные эритроциты (2) у больного с
искусственным клапаном сердца. хЮОО
RBC 3,9 3,8-5,5 х 10’7л
НЬ 119 130-155 г/л
Ht 43,1 36-48%
MCV 87 80-95 фл
МСН 30,5 27-31 пг
мснс 349 300-380 г/л
RDW 14,5 11,5-14,5%
Повышенный гемолиз сопровождается лейкоцитозом с нейтрофильным
сдвигом и выраженной тромбоцитопенией. Выявляется низкий уровень фиб-
риногена, протромбина, факторов VII и X плазмы, которые свидетельствуют
о коагулопатии потребления.
При механическом повреждении эритроцитов в крови появляются
шлемовидные эритроциты.
Маршевая гемоглобинурия. Внутрисосудистый гемолиз в результате
механического повреждения эритроцитов в капиллярах стоп. Часто единс-
твенным симптомом болезни является преходящая гемоглобинурия с появ-
лением мочи черного цвета. Интенсивность гемоглобинурии и интервалы
между приступами зависят от тяжести поражения и нагрузки. Анемии, как
правило, не бывает.
Химические яды. Внутрисосудистый гемолиз эритроцитов могут вы-
зывать яды органического и неорганического происхождения (сапонин,
фенилгидразин, мышьяк, свинец, отравление грибами и др.).
Ожоговая болезнь. Гемолиз эритроцитов при ожоговой болезни наблю-
дается в случае поражения 20% поверхности тела и имеет сложный генез.
Признаки гемолиза выявляются не сразу. Они маскируются сгущением
крови вследствие шокового состояния и потери плазмы непосредственно
после ожога. В послешоковом периоде в фазе компенсированной гидремии
отмечается снижение эритроцитов и гемоглобина. Гемолиз может сопровож-
даться желтухой, а в тяжелых случаях гемоглобинемией и гемоглобинурией
с развитием острой почечной недостаточности.
Гемолиз, вызванный инфекциями. Инфекционные агенты способны
вызывать гемолитическую анемию как в результате воздействия токсинов
(например, Clostridium perfringens, а- и Р-гемолитические стрептококки, ме-
нингококки), так и прямой инвазии и разрушения эритроцитов (Plasmodium,
Bartonella'}.
Малярия. В патогенезе анемии при малярии основным фактором является
инвазия малярийными плазмодиями, вызывающими распад эритроцитов в
процессе эритроцитарной шизогонии. Гемолиз преимущественно внутрисо-
судистый. При хроническом течении инфекции в патогенезе гемолиза акцент
с распада циркулирующих эритроцитов переносится на органы РЭС, где
ведущая роль принадлежит увеличенной селезенке (гиперспленизм).
Гемолитико-уремический синдром. Нередко развивается внезапно после
перенесенной инфекции и вакцинации. Этиологическим фактором могут
быть микроорганизмы, вирусы, которые приводят к выработке антител и
образованию комплексов антиген - антитело. Эти изменения вызывают
внутрисосудистый гемолиз и нарушение гемокоагуляции. В различные
сроки болезни может развиться острая почечная недостаточность. Анемия
сопровождается большими морфологическими изменениями эритроцитов
(шизоциты, эхиноциты, рис. 75), нейтрофильным лейкоцитозом со сдвигом
влево, тромбоцитопенией.
Рис. 75. Периферическая кровь. Эхиноциты. хЮОО
RBC
RBC 1,9 3,8—5,5 х 1012/л
НЬ 60 130-155 г/л
Ht 16,5 36-48%
MCV 84,7 80-95 фл
МСН 31,6 27-31 пг
МСНС 363 300-380 г/л
RDW 14,2 11,5-14,5%
Анемии, обусловленные эритроцитарными факторами
Гемолитические анемии, связанные с нарушением структуры
мембраны эритроцитов (эритроцитопатии)
Мембрана эритроцитов в качестве структурной основы, как и другие
биомембраны, имеет бислой фосфолипидов, в котором встроены белки.
Внутренняя сторона мембраны эритроцитов связана с сетью миофиламент-
ных белков, формирующих цитоскелет и придающих эритроциту в покое
специфическую двояковогнутую форму (рис. 76).
Структурная организация мембраны эритроцита позволяет этой клетке
быть, с одной стороны, высокоэластичной (примерно в 100 раз более эластич-
ной, чем латексная пленка той же толщины), с другой стороны, выдерживать
очень высокие нагрузки без фрагментации. Специальное строение амфи-
фильных липидов (растворимы в полярных и неполярных растворителях)
позволяет встраиваться в мембрану белкам, которые формируют эластичный
цитоскелет, деформируемый в 2 направлениях - трансмембранно и вдоль
мембраны (рис. 77).
Липидный бислой эритроцитарной мембраны содержит в эквивалентных
количествах холестерин и фосфолипиды. Фосфолипиды расположены асим-
метрично: фосфатидилхолин и сфингомиелин предпочтительно представлены
на наружной стороне, тогда как большая часть фосфатидилэтаноламина и
весь фосфатидилсерин и фосфоинозитиды - на внутренней стороне бислоя.
Именно фосфатидилсерин и фосфоинозитиды способны взаимодействовать
с белками цитоскелета - спектрином и белком 4.1R. Нарушение асимметрич-
ности и выход фосфатидилсерина на наружную сторону мембраны приводят
к фагированию эритроцитов макрофагами. Описано более 50 трансмембран-
ных белков, около 25 из которых связаны с групповой принадлежностью.
Анкирин
Спектрин
-----а
----₽
Белок 4.1
Белок 4.2
Анионтранспортирующий белок (белок 3)
Рис. 76. Схематическое изображение внутренней стороны мембраны эритроцита с сетью
миофиламентных белков, формирующих цитоскелет. Фосфолипиды образуют асимметрич-
ную бислойную мембрану, холестерин растворен между хвостами жирных кислот, придавая
определенную жесткость мембране. Гликофорин А и В — трансмембранные гликопротеины,
определяющие антигенные и рецепторные свойства мембраны эритроцитов. Белок 3 — анионт-
ранспортирующий белок, к нему со стороны цитозоля ассоциирован белок 4.2 и НЬ. Спектрин,
актин и тропомиозин формируют цитоскелет на внутренней стороне. Спектрин — гетеродимер,
имеет а- ир-цепи. Анкирин связывает белок 3 с цитоскелетом
Актин
Тропомиазин
В
Фосфолипидный бислой
А
профиламенты
актина
Рис. 77. Мембрана эритроцита (по X. An, N. Mohandas) построена из амфифильных липидов, в
которую встроены белки, формирующие поперечную эластичную ось из p-спектрина, анкирина,
белка 4.2, белка 3 и комплекса Rh. Продольную (латеральную) эластичную ось формируют а- и
p-спектрины и комплекс сократительных белков, сопряженный с гликофорином С. Такое строение
позволяет эритроциту выдерживать разные нагрузки и деформироваться как в продольном, так
и поперечном направлении
Ряд белков определяет проницаемость мембраны для катионов, воды, моче-
вины и др. метаболитов. Структурными белками, обеспечивающими связь
с внутренними белками цитоскелета, являются белок 3, гликофорин С и
Rh-антигены. Основными белками цитоскелета являются а- и 0-спектрины,
актин, белок 4.1R, аддуцин, дематин, тропомиозин и тропомодулин. При-
нципиальным моментом в поддержании специфической формы эритроцита
является взаимодействие белков внутреннего цитоскелета с мембранными
белками и фосфолипидами.
При патологии белков и связей между элементами цитоскелета наруша-
ется форма эритроцита, что может сопровождаться развитием овалоцитоза
(эллиптоцитоза), сфероцитоза, стоматоцитоза, акантоцитоза. Описано до
20 различных аномалий белков мембраны.
Микросфероцитарная гемолитическая анемия
(микросфероцитоз, болезнь Минковского-Шоффара)
Наследственно-семейное заболевание, чаще встречается гетерозиготная
форма. Наследуется по аутосомно-доминантному типу. Распространена
практически повсеместно, во всех расовых группах. Однако в Северной
Европе достаточно высока частота этой патологии, которая достигает
1:3000 населения. Чаще всего болезнь проявляется в возрасте 3-15 лет,
однако нередко клинические признаки выявляются в неонатальном пе-
риоде. Могут наблюдаться спорадические формы микросфероцитарной
анемии.
Патогенез. При микросфероцитозе описаны разнообразные дефекты со-
става или функции белков мембраны эритроцитов. Наследственный дефект
мембраны эритроцита способствует повышению ее проницаемости для
ионов натрия, воды, что в конечном итоге изменяет объем клетки. Наиболее
распространена аутосомно-доминантная форма, связанная с нарушением
взаимодействия спектрина с анкирином и белком 4.2, или дефицитом бел-
ка 4.2, или с комбинированным дефицитом анкирина и спектрина. Кроме
того, описана микросфероцитарная анемия из-за отсутствия спектрина, она
наследуется как рецессивная аутосомная мутация. Слабое взаимодействие
трансмембранных белков (белок 3 и Rh-комплекс) может приводить к фраг-
ментации мембраны, снижению площади поверхности мембраны, повы-
шению ее проницаемости, увеличению содержания в клетке осмотически
активных веществ. Таким образом, наследственный сфероцитоз - результат
дефекта в каком-либо белке, участвующем в формировании вертикального
взаимодействия между внутренним цитоскелетом, сформированным на
спектрине, и трансмембранными белками. В табл. 15 представлена частота
мутаций, определяющих нарушения в синтезе белков, приводящих к изме-
нению формы эритроцитов.
Таблица 15
Частота наследственных мутаций в составе белков эритроцитов,
приводящая к сфероцитозу
Сфероцитоз %
Анкирин 50-60
Спектрин 20
Белок 3 15-20
Белок 4.2 <5
Rh-комплекс <1
Неидентифицированный дефект 10
Формирующийся сфероцит при движении в селезенке испытывает меха-
ническое затруднение, длительно задерживается в красной пульпе, теряет
способность деформироваться в узких участках кровотока в синусах селе-
зенки. Снижение эластичности мембраны приводит к фрагментации при
прохождении эритроцита через капилляры, что сопровождается уменьшением
размеров эритроцита. Через 2-3 пассажа через селезенку сфероцит подвер-
гается лизису и фагоцитозу (внутриклеточный гемолиз). Патология эрит-
роцитов проявляется морфологической аномалией - микросфероцитозом,
который, по данным одних авторов, выявляется в костном мозге на стадии
эритробласта в период гемотлобинизации, по другим - форма эритроцита
изменяется в периферической крови. Морфологическая аномалия эритроци-
тов остается на всю жизнь и в случае удаления селезенки. Микросфероциты
имеют укороченные сроки пребывания в крови (до 12-14 дней), пониженную
осмотическую и механическую резистентность. Повышенное разрушение
патологических эритроцитов происходит в органах РЭС, главным образом в
селезенке. Селезенка задерживает и интенсивно гемолизирует неполноцен-
ные эритроциты. Развивается спленомегалия, которая усугубляет гемолити-
ческий процесс. После спленэктомии срок пребывания сфероцитов в крови
значительно возрастает, приближаясь к нормальным.
Клиника. Основной признак заболевания - гемолитический синдром,
который проявляется желтухой, спленомегалией и анемией. В зависимости
от формы наследования патологии (гомо- или гетерозиготная передача)
болезнь может выявляться в раннем детском возрасте или в более поздние
периоды жизни. Возникновение заболевания в детском возрасте нарушает
нормальное развитие организма, в результате имеются выраженные клини-
ческие признаки: деформация скелета, особенно черепа, рано отмечается
увеличение селезенки, общая отсталость развития (спленогенный инфан-
тилизм). При гетерозиготной форме заболевания клинические признаки
слабо выражены, но имеют место характерные морфологические изменения
эритроцитов (микросфероцитоз). Гемолитический криз возникает под влия-
нием провоцирующих факторов (инфекция, переохлаждение, переутомление,
беременность и др.).
Микросфероцитарная гемолитическая анемия имеет хроническое
течение, сопровождается периодическими гемолитическими кризами и
ремиссиями. В период криза может повышаться температура, появляется
желтуха, увеличиваются размеры селезенки, нередко развивается анемия.
В период ремиссии признаки заболевания незначительные. Высокий гемо-
лиз и частые гемолитические кризы способствуют быстрому увеличению
размеров селезенки, постоянному увеличению концентрации неконъюги-
рованного билирубина в крови, иктеричности склер. Создаются условия
застоя желчи в печени, который иногда приводит к осложнениям гемоли-
тической болезни: образованию пигментных камней в желчном пузыре
(желчекаменной болезни), ангиохолециститу и др. Иногда развиваются
трофические язвы голеней, заживление которых возможно только после
спленэктомии.
Костный мозг гиперклеточный. Эритроидная гиперплазия наблюдается
в плоских костях, диафизах и эпифизах трубчатых костей. Развиваются
экстрамедуллярные очаги кроветворения в селезенке и других органах.
Костномозговое кроветворение гипер- или регенераторное, следствием чего
является увеличение количества миелокариоцитов. Преобладают эритроблас-
ты, число которых составляет 60-70% костномозговых клеток, соотношение
Лейко/Эритро -1:3 и более. Созревание эритробластов и выход эритроцитов
на периферию идут ускоренными темпами. При интенсивном крове творении
после тяжелого гемолитического криза в костном мозге могут наблюдаться
мегалобласты, очевидно, как следствие недостаточности витамина В|2 или
усиленного расхода фолиевой кислоты. Очень редко в стернальном пунктате
обнаруживается эритробластопения - так называемый арегенераторный криз,
который имеет обратимый характер.
При выраженном некомпенсированном гемолизе анемия нормохромная.
Вместе с тем анемия долгое время может отсутствовать, однако в перифери-
ческой крови обнаруживаются полихроматофилия и ретикулоцитоз - призна-
ки активного костномозгового эритропоэза. Эритроциты (микросфероциты)
характеризуются небольшим диаметром (в среднем 5 мкм), повышенной
толщиной и нормальным объемом (MCV). Средняя толщина увеличена до
2,5-3,0 мкм. Сферический индекс (СФ) - отношение диаметра (d) эрит-
роцита к его толщине (Т) - снижен в среднем до 2,7 (при норме 3,4-3,9).
Содержание гемоглобина в эритроцитах в пределах нормы или несколько
выше ее. Количество микросфероцитов в период ремиссии и при латентной
форме болезни не бывает высоким, в то время как в период криза гемолиз
может сопровождаться увеличением их до 30% и выше. Микросфероциты
в мазках крови имеют небольшой размер, гиперхромные, без центрального
просветления (рис. 78).
Рис. 78. Болезнь Минковского-Шоффара. Микросфероциты. хЮОО
RBC 2,91 3,8-5,5 х 10'2/л
НЬ 97 130-155 г/л
Ht 27,5 36-48%
MCV 95,0 80—95 фл
МСН 33,4 27-31 пг
мснс 353 300-380 г/л
RDW 11,3 11,5-14,5%
Эритроцитарная гистограмма дает отклонение влево, в сторону микро-
цитов, RDW в норме или немного повышен.
Особенностью микросфероцитарной гемолитической анемии является
постоянно повышенный гемолиз, который сопровождается ретикулоцито-
зом. В период гемолитического криза количество ретикулоцитов достигает
50-80% и больше, в ремиссии - не превышает 2-4%. Ретикулоциты обладают
большим диаметром при нормальной толщине. Могут появляться эритрока-
риоциты. Гемолитический криз сопровождается небольшим нейтрофильным
лейкоцитозом. Тромбоцитарный росток, как правило, не изменен. СОЭ в
период криза повышена.
Одним из характерных признаков заболевания является снижение осмо-
тической резистентности эритроцитов. В норме начало гемолиза отме-
чается при концентрации NaCl 0,5-0,45%, а полный гемолиз при 0,4-0,35%
NaCl. При сфероцитозе показатели резко снижены: гемолиз начинается при
0,7-0,6% NaCl (минимальная стойкость). Максимальная стойкость обычно
повышена - гемолиз наступает при 0,4—0,3% NaCl. Среди больных микросфе-
роцитарной гемолитической анемией встречаются лица, у которых, несмотря
на явный сфероцитоз, осмотическая резистентность эритроцитов нормальная.
В этих случаях необходимо исследовать резистентность эритроцитов по
отношению к гипотоническим солевым растворам после предварительной
их инкубации в течение двух суток. Спленэктомия не устраняет сниженной
осмотической и механической устойчивости эритроцитов.
Развитию спленомегалии с синдромом гиперспленизма сопутствует лейко-
пения, нейтропения и нередко нерезко выраженная тромбоцитопения. Отме-
чается снижение гаптоглобина. Последствия высокого гемолиза: билируби-
немия с преобладанием неконъюгированного билирубина, в моче увеличено
содержание уробилиногена, моча имеет коричнево-красный оттенок, каловые
массы резко окрашены из-за большого количества стеркобилиногена.
Овалоцитарная гемолитическая анемия
(овалоклеточная, наследственный овалоцитоз, эллиптоцитоз)
Редкая форма болезни, распространена в Западной Африке (2%), насле-
дуется по аутосомно-доминантному типу. В зависимости от гетеро- или го-
мозиготной передачи возможны различные клинические и гематологические
проявления болезни.
Патогенез. В основе заболевания лежит патология мембраны эритроцитов.
Возникает, как правило, из-за молекулярного дефекта белков «цитоскелета»
мембраны. Механическая основа уменьшения стабильности мембраны - ос-
лабление латеральных связей между молекулами спектрина (димер-димерное
взаимодействие) или дефект комплекса спектрин - актин - протеин 4.1R.
Наиболее частой причиной (65% случаев) наследственного овалоцитоза
является мутация, приводящая к замене аминокислот в аминотерминальной
части а-спектрина. Мутации генов, ответственных за синтез [3-спектрина,
встречаются примерно в 30% случаев, гетерозиготное носительство мутации
сопровождается разнообразием клинических проявлений. Продолжитель-
ность жизни овалоцитов в организме укорочена. Заболевание характери-
зуется внутриклеточным гемолизом с преимущественным разрушением
эритроцитов в селезенке.
Клиника. Как аномалия овалоцитоз в большинстве случаев представляет
собой бессимптомное носительство без клинических проявлений, однако
примерно у 10% больных анемия средней тяжести или даже тяжелая. При
гомозиготной форме клинические признаки овалоцитарной анемии практи-
чески не отличаются от микросфероцитоза. Заболевание характеризуется
хроническим нетяжелым течением с гемолитическими кризами, сопровож-
дающимися компенсированным или декомпенсированным гемолизом, жел-
тухой и анемией, уровень которой зависит от компенсаторных возможностей
эритропоэза. Больным свойственны спленомегалия, конституциональные
изменения скелета, в частности черепа, возможны трофические язвы голени
и другие симптомы, которые могут наблюдаться при микросфероцитарной
гемолитической анемии.
В костном мозге характерен регенераторный или гиперрегенераторный
тип кроветворения с преобладанием эритробластов. Соотношение Лей-
ко/Эритро составляет 1:3 и более за счет эритробластов в зависимости от
активности гемолиза и костномозгового кроветворения.
Кровь. Анемия носит нормохромный характер с высоким ретикулоци-
тозом. Овалоциты имеют нормальный средний объем (MCV) и среднее со-
держание гемоглобина (МСН). Наибольший диаметр эритроцитов достигает
12 мкм, наименьший - 2 мкм (рис. 79, 80). Овалоцитоз эритроцитов может
составлять от 10 до 40-50% клеток при гетерозиготном носительстве и до
96% эритроцитов при полном носительстве аномальных генов. Осмоти-
ческая резистентность овалоцитов понижена, повышен аутогемолиз, СОЭ
ускорена.
Рис. 79. Овалоцитоз. Периферическая кровь.
х900
Рис. 80. Овалоцитоз. Сканирующая электронная
микроскопия
Овалоцитоз как симптоматическая форма (с небольшим числом овалоци-
тов) может встречаться при различных патологических состояниях, главным
образом при гемолитических анемиях, заболеваниях печени, миелодисплас-
тическом синдроме. Известно сочетание овалоцитоза с серповидноклеточной
анемией, талассемией, пернициозной анемией. В таких случаях овалоцитоз
носит временный характер и исчезает при эффективной терапии основного
заболевания. Поэтому к истинному овалоцитозу следует относить лишь те
случаи, при которых не менее 10% эритроцитов имеют овальную форму и
патология носит наследственный характер.
Стоматоцитарная гемолитическая анемия (стоматоцитоз)
Редкая форма заболевания, наследуется по аутосомно-доминантному типу.
Патогенез. В основе заболевания лежит изменение функции структурных
белков мембраны эритроцитов, приводящее к нарушению регуляции объема
клетки. Деформируемость эритроцита критически зависит от соотношения
площади поверхности и объема клетки. Дискоидная клетка имеет возмож-
ность изменять форму и преодолевать узкие пространства капилляров, что
облегчает и обмен кислорода в капиллярах легких и периферических тканей.
Шаровидная клетка практически не способна изменить форму, у нее сниже-
на способность в обмене кислорода с тканями. У нормального эритроцита
площадь поверхности около 140 мкм2, объем около 90 фл, концентрация
гемоглобина около 330 г/л. Большие мембранные белки играют определяю-
щую роль в катионном трансмембранном обмене эритроцита и тем самым
регулируют объем клетки. К таким белкам относятся трансмембранные
Na+-K+-Cl -копереносчик, Na -Cl -копереносчик, ионообменный белок 3,
Na'-K -копереносчик, №"-К‘-АТФ-аза, Са2+-АТФ-аза и другие. Нарушение
функционирования этих белков с накоплением катионов внутри эритроцита
приводит к накоплению в нем воды с приобретением сферичности клетки.
Аномалия эритроцитов сопровождается повышенным их разрушением в РЭС,
главным образом в селезенке, вследствие внутриклеточного гемолиза.
Клиника может быть с различными проявлениями - от полной компенса-
ции у носителей патологического гена до тяжелой гемолитической анемии,
напоминающей микросфероцитоз. Внутриклеточный гемолиз эритроцитов
сопровождается увеличением селезенки, желтухой, имеет место склонность
к образованию камней и изменениям скелета.
Костный мозг гиперклеточный за счет расширенного красного ростка.
Показатели костномозгового кроветворения зависят от степени выраженнос-
ти гемолиза и активности эритропоэза. Ремиссия может не сопровождаться
анемией, в период криза анемия носит, как правило, регенераторный или
гиперрегенераторный характер.
Рис. 81. Стоматоциты. Периферическая кровь.
х900
Кровь. Морфологическая осо-
бенность болезни - стоматоцитоз
(рис. 81), который характеризуется
наличием в центре клетки неокра-
шенного участка в виде вытянутой
светлой полосы, напоминающей
форму рта (Stoma - рот) или округ-
лой формы. Объем эритроцитов и
концентрация гемоглобина не отли-
чаются от нормы, резистентность
эритроцитов может быть понижена.
В период тяжелых гемолитических
кризов наблюдается низкий уровень
гемоглобина и эритроцитов. Анемия
сопровождается повышенным содер-
жанием ретикулоцитов и неконъюги-
рованного билирубина.
Гемолитические анемии, обусловленные нарушением структуры
липидов мембраны эритроцитов (акантоцитоз)
Редкое заболевание, наследуется по аутосомно-рецессивному типу.
Патогенез. Заболевание связано с нарушением липидного обмена.
Снижение содержания холестерина, триглицеридов, фосфолипидов в
крови отражается на липидном составе мембраны эритроцитов, в них
снижена концентрация лецитина, фосфатидилхолина, повышено содержа-
ние сфингомиелина; уровень холестерина нормальный либо повышен, а
содержание фосфолипидов нормальное или уменьшено. Все эти нарушения
в эритроцитах способствуют снижению текучести мембраны и изменению
их формы. Эритроциты приобретают зубчатый контур, похожий на листья
аканта, поэтому их называют акантоцитами. Аномальные эритроциты
разрушаются главным образом в селезенке вследствие внутриклеточного
гемолиза.
В клинике имеют место признаки анемии, гемолиза эритроцитов, симп-
томы нарушения обмена липидов: пигментный ретинит, нистагм глаз, тремор
рук, атаксическая походка.
Кровь. Анемия нормохромная
нормоцитарная. Основным морфо-
логическим признаком этой формы
гемолитической анемии являются
эритроциты с зубчатым контуром -
акантоциты (рис. 82), которые могут
составлять до 40-80% эритроцитов.
Отмечается ретикулоцитоз. Осмо-
тическая стойкость эритроцитов
нормальная или сниженная. Коли-
чество лейкоцитов и тромбоцитов в
пределах нормы.
В костном мозге - гиперплазия
клеточных элементов эритропоэза.
Эритроциты такой же формы встре-
чаются при циррозе печени (рис. 83),
у пациентов, находящихся на АИК,
космонавтов после приземления.
Рис. 82. Акантоцитоз. Периферическая кровь.
хЮОО
Рис. 83. Периферическая кровь больного с циррозом печени. Акантоциты. хЮОО
RBC
RBC 2,57 3,8-5,5 х 10’2/л
НЬ 70 130-155 г/л
Ht 21,9 36-48%
MCV 85,2 80-95 фл
МСН 27,2 27-31 пг
МСНС 320 300-380 г/л
RDW 18,6 11,5-14,5%
Гемолитические анемии,
обусловленные дефицитом
ферментов эритроцитов
(эритроцитарные энзимопатии)
Эритроцит переносит кислород, но сам его не использует для образования
собственной энергии, потому что к моменту созревания клетки он теряет
митохондрии и рибосомы, в нем исчезают процессы, связанные с образова-
нием энергии путем утилизации кислорода.
В настоящее время известно более 20 реакций в метаболизме эритроцитов,
нарушение которых приводит к укорочению продолжительности их жизни,
потере клетками резистентности к различным воздействиям (лекарственные
препараты, пыльца цветов и др.) и преждевременному их разрушению.
Основные метаболические процессы (пути) в эритроцитах, обеспечива-
ющие его функциональную активность: 1) гликолиз; 2) пентозофосфатный
цикл; 3) путь Раппопорта-Либеринга - образования 2,3-дифосфоглицерата
(2,3-ДФГ); 4) глутатионовый путь; 5) ферменты катаболизма пуринов и пи-
римидинов; 6) АТФ-зависимый катионный насос на мембране эритроцита;
7) ферменты, влияющие на состав фосфолипидов эритроцитарной мембра-
ны; 8) метгемоглобинредуктаза, превращающая метгемоглобин (MetHb) в
гемоглобин (НЬ).
Гемолитические анемии, обусловленные дефицитом ферментов эрит-
роцитов (несфероцитарные гемолитические анемии), имеют рецессивный
тип наследования. Клинические и гематологические проявления болезни
зависят от локализации наследственного ферментного дефекта в эритроци-
тах. Эритроцитарные энзимопатии связаны с недостаточностью ферментов
гликолиза (пируваткиназа, гексокиназа, глюкозофосфатизомераза, триозо-
фосфатизомераза), пентозофосфатного пути или метаболизма глутатиона
(глюкозо-б-фосфатдегидрогеназа, 6-фосфоглюконатдегидрогеназа и глута-
тионредуктаза). Чаще всего энзимопатии связаны с дефектами глюкозо-6-
фосфатдегидрогеназы, пируваткиназы или глутатионредуктазы. Энзимопатии
с дефектами других метаболических путей редки и не имеют практического
значения в возникновении гемолитических анемий.
Лабораторное подтверждение эритроцитарных энзимопатий основано
на биохимическом определении активности ферментов в гемолизате.
Принцип определения активности ферментов типичный для биохими-
ческих исследований - сопряженные реакции с определением на длине
волны 340 нм кинетики изменения кофакторов НАД/НАДН в реакционной
смеси, в которую добавляется избыток субстратов, так чтобы скорость-
лимитирующей реакцией была бы реакция с тестируемым ферментом.
Референтные значения активности эритроцитарных ферментов представ-
лены в табл. 16.
Таблица 16
Референтные значения активности ферментов гликолиза
[Pekrum A., Schrdter W., 1994]
Ферменты Активность
мкмоль субстрата / г НЬ в 1 мин мкмоль субстрата/ 10" эритроцитов в 1 мин
Гликолиз
Пируваткиназа 20,2 ± 2,2 41 ± 10
Гексокиназа 1,0 ±0,1 2,3 ± 0,5
Глюкозофосфатизомераза 44,7 ± 4,8 124 ± 13
Триозофосфатизомераза 2180 ±254 6055 ± 705
Пентозофосфатный путь и метаболизм глутатиона
Глюкозо-6-фосфатдегидрогеназа 11,0± 1,6 30,6 ±4,5
6-фосфоглкжонатдегидрогеназа 9,5 ± 1,5 26,2 ±4,1
Глутатионредуктаза 4,6 ± 0,8 25,7 ±3,0
Гемолитические анемии, связанные с недостаточностью
активности ферментов гликолиза
Патогенез. Гликолиз, или анаэробное окисление, - основной метаболи-
ческий процесс в эритроцитах, в котором глюкоза превращается в лактат.
Глюкоза - главный источник энергии для жизнедеятельности эритроцита.
Эритроциты, составляя около 3% массы тела, в день потребляют приблизи-
тельно 20 г (10%) всего количества глюкозы, поступающей в организм. Про-
цесс гликолиза ферментативный, в нем участвует более десяти ферментов.
При их недостаточности эритроциты быстро разрушаются. Последователь-
ность основных этапов гликолиза и катализирующие этот процесс ферменты
представлены на рис. 84. Гликолиз завершается восстановлением пирувата
в лактат, при этом одна молекула глюкозы расщепляется на две молекулы
молочной кислоты (лактата) без использования молекулярного кислорода.
Лактат через мембрану эритроцита покидает клетку, поглощается другими
тканями и сгорает в цикле Кребса. В эритроцитах, в отличие от других тканей,
в процессе гликолиза образуется много 2,3-дифосфоглицериновой кислоты,
которая, соединяясь с P-цепями глобина, снижает сродство гемоглобина к
кислороду, способствуя тем самым отдаче его тканям.
Дефицит пируваткиназы. Пируваткиназа на заключительном этапе гли-
колиза катализирует образование АТФ (энергообразующая стадия). Дефицит
активности пируваткиназы может привести к снижению в эритроцитах АТФ
и накоплению промежуточных продуктов гликолиза, которые образуются на
Рис. 84. Гликолиз и сопряженные с ним метаболические процессы, протекающие в эритроци-
тах: Пфп — пентозофосфатный путь (шунт); РЛП — Раппопорта—Либеринга путь образования
2,3-ДФГ
предшествующих этапах. Содержание конечных продуктов гликолиза - пи-
рувата и лактата - снижается. Недостаток АТФ сопровождается нарушением
функции АТФ-азного насоса эритроцита и потерей ионов калия. Снижение
одновалентных ионов в эритроците приводит к дегидратации и сморщива-
нию клетки, что затрудняет оксигенацию и отдачу кислорода гемоглобином.
Вместе с тем накопление промежуточных продуктов гликолиза, в частности
2,3-дифосфоглицерата, понижающего сродство гемоглобина к кислороду,
облегчает отдачу кислорода тканям.
Клинические симптомы болезни наблюдаются у гомозиготных носите-
лей. Заболевание характеризуется умеренной или тяжелой гемолитической
анемией с внутриклеточным гемолизом. Повышенный гемолиз обнаружива-
ется с рождения и сопровождается частыми и тяжелыми гемолитическими
кризами. Появление признаков болезни в 17-30 лет отличается скудной
клинической симптоматикой в виде иктеричности склер и кожных покровов.
Спленомегалия наблюдается почти постоянно, иногда и у гетерозиготных
носителей, хотя анемия у них, как правило, отсутствует. Гемолитический криз
провоцируется инфекцией, тяжелой физической нагрузкой, беременностью,
усиливается гемолиз во время менструаций.
В пунктате костного мозга выраженный эритрокариоцитоз.
Самым главным критерием диагностики является дефицит активности
пируваткиназы. Выраженные клинические эффекты наблюдаются в тех
случаях, когда остаточная активность фермента ниже 30% нормы.
В крови в большинстве случаев имеет место нормохромная несфероци-
тарная анемия с незначительным анизоцитозом и пойкилоцитозом. Коли-
чество гемоглобина и эритроцитов может быть нормальным, пониженным,
возможна и выраженная анемия (НЬ 40-60 г/л), эритроцитарные индексы
приближаются к норме. Нередко в мазках выявляются полихроматофилия
и эритроциты с базофильной пунктацией, иногда мишеневидные эритро-
циты, эритрокариоциты. Ретикулоцитоз в период криза может достигать
70%. Количество лейкоцитов и тромбоцитов обычно нормальное, хотя
в редких случаях бывает сочетанный ферментный дефект эритроцитов,
лейкоцитов и тромбоцитов. СОЭ в отсутствие резкой анемии - в пределах
нормы. Осмотическая резистентность эритроцитов не коррелирует с фор-
мой дефицита фермента и даже при одном и том же дефекте эритроцитов
может быть разной.
В сыворотке крови при гемолитическом кризе повышен неконъюгирован-
ный (непрямой) билирубин.
Приобретенный дефицит пируваткиназы эритроцитов был обнаружен при
остром лейкозе, лимфомах, пароксизмальной ночной гемоглобинурии.
Дефицит глюкозофосфатизомеразы (ГФИ) наследуется по аутосомно-
рецессивному типу и сопровождается развитием несфероцитарной гемоли-
тической анемии. У гетерозигот с незначительным снижением активности
ГФИ (40-60% от нормальной величины) клинических проявлений не на-
блюдается. При гомозиготной передаче с активностью фермента до 10%,
особенно в детском возрасте, развивается тяжелая гемолитическая анемия
с ретикулоцитозом, без морфологических особенностей эритроцитов. Ане-
мия, выявленная у новорожденных, характеризуется затяжным хроническим
течением. По частоте дефицит ГФИ занимает 3-е место среди врожденных
эритроцитарных энзимопатий, сопровождающихся гемолизом (после дефи-
цита глюкозо-6-фосфатдегидрогеназы и пируваткиназы). Дефицит глюкозо-
фосфатизомеразы может иметь приобретенный характер: при остром лейкозе,
цитопении, аплазии костномозгового кроветворения.
Гемолитические анемии, связанные с недостаточностью активности
ферментов пентозофосфатного шунта
Пентозофосфатный путь — второй путь распада глюкозы. Он сопряжен с
гликолизом, около 10% глюкозы метаболизируется через этот путь. В процес-
се пентозофосфатного цикла в эритроците образуется восстановленная форма
НАДФН, необходимого для восстановления глутатиона. Восстановленный
глутатион в эритроците препятствует воздействию окислителей, предуп-
реждает накопление в них перекисных и свободнорадикальных соединений,
которые могут индуцировать активацию перекисного окисления липидов
мембраны эритроцитов и их разрушение.
Глюкозо-6-фосфагдегидрогеназа (Г-6-ФДГ) - единственный фермент
пентозофосфатного пути, первичный дефицит которого ведет к гемолити-
ческой анемии. Это самая распространенная эритроцитарная энзимопатия:
в мире около 200 миллионов человек имеют эту патологию. Она превали-
рует среди жителей бассейна Средиземного моря, Юго-Восточной Азии,
Индии. Ген синтеза Г-6-ФДГ сцеплен с Х-хромосомой, поэтому заболе-
вание проявляется значительно чаще у мужчин. Имеется несколько типов
Г-6-ФДГ-энзимопатии. Г-6-ФДГ-энзимопатия типа А встречается часто у
негров (10-20% мужчин), характеризуется относительно легким течением,
редко сопровождается гемолизом. При дополнительной мутации (Г-6-ФДГ
Mahidol) имеет место тяжелая гемолитическая анемия у новорожденных и
в некоторых случаях хроническая анемия у взрослых. Этот вариант нередко
регистрируется среди жителей Юго-Восточной Азии. Г-6-ФДГ-энзимопа-
тия типа В (средиземноморская) характерна для жителей Ирака, курдов,
сефардских евреев, ливанцев. На территории стран Содружества дефицит
Г-6-ФДГ встречается неравномерно в различных республиках и областях.
Чаще других патология обнаруживается в Азербайджане, Дагестане, реже
у узбеков и таджиков, среди русских она составляет 0-2%.
Провоцирующими факторами гемолитического криза могут быть инфек-
ционные заболевания - грипп, сальмонеллезная инфекция, вирусный гепатит,
употребление в пищу конских бобов (фавизм), вдыхание цветочной пыльцы,
последнее сопровождается обычно более легким гемолитическим кризом, но
возникает через несколько минут после контакта с пыльцой. Особенностью
фавизма является острый гемолиз, наступающий быстрее, чем вызванный
приемом лекарств, и диспептические расстройства. Гемолитический криз
может быть спровоцирован приемом некоторых лекарственных препаратов,
чаще всего противомалярийных (примахин, хинин, акрихин), сульфанил-
амидных (норсульфазол, стрептоцид, сульфадиметоксин, бисептол), нитро-
фурановых (фуразолидон, фурадонин), ПАСК, противоглистных препаратов
и других. Клинические симптомы могут возникать на 2-3-и сутки от начала
приема препарата. Первыми симптомами обычно бывают иктеричность склер
и темная моча. Прекращение приема лекарства исключает развитие тяжелого
гемолитического криза. В противном случае на 4-5-е сутки возникает гемо-
литический криз с выделением мочи черного или бурого цвета - результат
внутрисосудистого гемолиза эритроцитов.
При тяжелом течении болезни повышается температура, появляется го-
ловная боль, рвота, иногда понос. Возникает одышка, часто увеличивается
селезенка. Внутрисосудистый гемолиз провоцирует активацию свертывания
крови, что может приводить к блокаде микроциркуляции в почках и острой
почечной недостаточности.
В костном мозге наблюдается резкое раздражение эритропоэза. В
крови - анемия, в период криза количество гемоглобина снижается до
20-30 г/л, увеличивается количество ретикулоцитов, лейкоцитов со сдви-
гом до миелоцитов. Количество тромбоцитов обычно не меняется. При
тяжелом гемолитическом кризе может выявляться большое количество
телец Гейнца-Эрлиха как результат преципитации цепей глобина и белков
Рис. 85. Тельца Гейнца-Эрлиха. Периферичес-
кая кровь. Суправитальная окраска 1 - ретику-
лоцит, 2 - тельца Гейнца. хЮОО
мембраны эритроцитов (рис. 85).
Отмечается анизоцитоз, пойкило-
цитоз, полихроматофилия, базо-
фильная пунктация, тельца Жолли.
В сыворотке крови повышается
содержание свободного гемогло-
бина (внутрисосудистый гемолиз),
часто увеличивается концентрация
неконъюгированного билирубина,
наблюдается гипогаптоглобинемия.
В моче - гемоглобинурия, гемоси-
деринурия. Диагностика основана
на определении уровня фермента
Г-6-ФДГ.
Раппопорта-Либеринга путь образования 2,3-дифосфоглицерата
(2,3-ДФГ) имеет существенное значение в регуляции сродства гемоглобина с
О2. Концентрация 2,3-ДФГ в эритроцитах часто достигает 5 ммоль/л, что срав-
нимо с концентрацией гемоглобина и выше концентрации АТФ (1-2 ммоль/л).
2,3-ДФГ, уменьшая сродство гемоглобина к кислороду, способствует отдаче
кислорода тканям на периферии. В то же время на насыщение гемоглобина
кислородом в легких он не влияет. Содержание 2,3-ДФГ, как правило, повы-
шается при анемии и гипоксии для облегчения передачи кислорода тканям.
Фетальный гемоглобин (HbF) менее чувствителен к 2,3-ДФГ, чем гемоглобин
взрослых (НЬА), поэтому 2,3-ДФГ материнских эритроцитов способствует
переносу кислорода через плаценту от НЬА к HbF. Интенсивность цикла
Раппопорта-Либеринга меняется в зависимости от pH, поэтому содержание
2,3-ДФГ снижается при ацидозе. Два фермента участвуют в цикле Раппо-
порта-Либеринга: 2,3-ДФГ-мутаза и 2,3-ДФГ-фосфатаза. Наследственные
формы дефицита этих ферментов чрезвычайно редки.
Гемолитические анемии, связанные с недостаточностью активности
ферментов глутатионовой системы
Глутатионовый путь защищает эритроцитарные мембраны и гемогло-
бин от повреждения перекисями. В эритроцитах в силу их специфической
функции содержится большое количество молекул железа и кислорода. При
окислении двухвалентного железа кислородом образуются свободные ради-
калы, которые способны инициировать цепи окисления: Fe2+ + О2 + Н1 —*
Fe3+ + НО2’. Токсический эффект в клетке оказывают свободные Ре2+-ионы,
которые способны вызывать реакции инициации образования свободных
радикалов, индуцировать перекисное окисление липидов и образование ак-
тивного гидроксильного иона СОН). Кислород непосредственно участвует в
образовании перекисей и радикалов (Н2О2, ’ОН, R', RO', RO2’, где R - радикал
длинноцепочечной жирной кислоты), которые вызывают окисление белков
мембран и увеличение проницаемости липидного слоя мембран для Н+, Са2+
и других ионов и метаболитов. В результате эритроциты гемолизируются.
Восстановление гидроперекисей (ROOH) до спиртов (ROH) могут осу-
ществлять нуклеофильные соединения, в эритроцитах это глутатион. Для
защиты от активных Fe24 и радикалов кислорода в эритроцитах постоянно
в процессе пентозофосфатного пути расходуется энергия на образование
НАДФН, который служит источником поддержания высокой концентрации
восстановленной формы глутатиона (Г-SH), используемого для удаления
перекисей и радикалов (рис. 86).
Рис. 86. Сопряжение пентозофосфатного и глутатионового путей. Г-SH - восстановленная форма
глутатиона; T-SS-Г — окисленная форма глутатиона; ROOH - гидроперекись; ROH - спирт
Таким образом, для удаления токсичных гидроперекисей и радикалов в
эритроците не только должен активно функционировать пентозофосфатный
путь образования НАДФН, но и происходить постоянно синтез глутатиона.
В синтезе глутатиона используются 2 фермента: глутатионредуктаза и
глутатионсинтетаза. При дефиците этих ферментов в клинике отмечаются
признаки гемолитической анемии.
Метаболизм пуринов и пиримидинов включает 3 фермента, изменение
активности которых ведет к гемолитической анемии. Аденозинтрифосфа-
таза участвует в обеспечении работы Na-, К-АТФ-азы или Na-насоса на
эритроцитарной мембране. Аденозиндезаминаза превращает АТФ, АДФ и
АМФ в инозин после дефосфорилирования. При редкой врожденной ауто-
сомно-доминантной гемолитической анемии активность фермента повышена
в 35-70 раз, происходит разрушение АТФ, не хватает энергии для удаления
Na из эритроцитов, что приводит к гемолизу. Пиримидин-5 ’-нуклеотидаза
участвует в катаболизме рибосомальной РНК, при снижении ее активности
наступает агрегация РНК, которая выявляется в виде базофильной зернис-
тости в эритроцитах. Агрегация РНК в ретикулоцитах ведет к снижению
эластичности мембраны эритроцитов и их гемолизу. Гемолитическая анемия
сопровождается спленомегалией.
Дефицит пиримидин-5’-нуклеотидазы может быть вызван тяжелым от-
равлением свинцом, имеет место при Р-талассемии.
Ферментами фосфолипидного метаболизма эритроцитарной мембраны
являются лецитин-холестерин-ацилтрансфераза (ЛХАТ) и лизолецитин-ацил-
трансфераза (ЛАТ), они поддерживают оптимальное соотношение лизолеци-
тина с лецитином на внешней стороне эритроцитарной мембраны. Дефицит
этих ферментов приводит к морфологическим изменениям эритроцитов
(мишеневидные эритроциты) и к умеренной гемолитической анемии.
Метгемоглобинредуктаза или НАДН-зависимая метгемоглобинредук-
таза (диафораза) и цитохром-Ь^редуктаза превращают метгемоглобин в
гемоглобин (рис. 87). Метгемоглобин - производное гемоглобина, в котором
Fe2+ переходит в Fe3+.
Рис. 87. Образование и удаление метгемоглобина
При обмене в эритроцитах образуется немного метгемоглобина, кото-
рый восстанавливается обратимо в гемоглобин под воздействием фермента
метгемоглобинредуктазы. В крови метгемоглобин не превышает 2% общего
содержания гемоглобина. Врожденный дефицит фермента метгемогло-
бинредуктазы сопровождается увеличением в крови MetHb, что нарушает
доставку кислорода к тканям и вызывает развитие цианоза. Повреждение
только в эритроцитах не ведет к тяжелым гемолитическим кризам. Однако
недостаточность цитохром-Ь5-редуктазы может быть генерализованным
заболеванием, затрагивающим большинство тканей организма, при этом
развивается прогрессирующая энцефалопатия.
Гемолитические анемии, связанные с нарушением синтеза
глобина (гемоглобинопатии)
Структура глобина
Глобин - белок группы гистонов, в его структуру входит 4 полипептидных
цепи. Известны 5 пептидных цепей (а - 141 аминокислота, р, у, 5, £ - в каж-
дой по 146 аминокислот). Аминокислотный состав полипептидных цепей
глобина определяет вид гемоглобина. В процессе эмбриогенеза происходит
смена типа кроветворения и вида гемоглобина. Так, Hb Gowerl и Gower2
(примитивный гемоглобин - НЬР), наличие которых отмечается в эритро-
идных клетках эмбриона, исчезают к концу 3-го месяца. На смену им при
кроветворении в печени начинает синтезироваться фетальный гемоглобин
(HbF), составляющий примерно 75% НЬ пуповинной крови новорожденных,
но к концу 1 -го года его количество снижается до 1 % НЬА, и НЬА2 начинают
синтезироваться в период костномозгового кроветворения плода. На долю
НЬА, взрослого человека приходится 96-98%, НЬА2 - 2-3%.
HbF (а2 у2) - НЬ плода, в его составе 2 a-цепи и 2 у-цепи глобина. У нею
высокая степень сродства к 02, он устойчив к щелочному денатурированию,
что используется в химическом определении.
НЬА, (н2р?) - основная форма гемоглобина взрослого человека, для кото-
рого характерна относительно высокая скорость денатурации в щелочной и
кислой средах.
НЬА2 (а2б2) - электрофоретическая подвижность значи тельно меньше,
чем у НЬА,, поэтому определяется при электрофорезе как малоподвижный
компонент.
Аномальные НЬ. В основе около 400 описанных в настоящее время
аномальных НЬ заложены мутации их структурных генов. Из этих мутаций
около 100 сопровождаются гемолитическими нарушениями разной степени.
Большинство аномальных НЬ отличается заменой 1 аминокислоты в той
или иной пептидной цепи глобина - это точечная мутация. Исходно нор-
мальный и патологический НЬ обозначали большими буквами алфавита:
нормальный - А, фетальный - F, первый аномальный НЬ назвали S, но так
как аномальных НЬ больше, чем букв алфавита, то решено их называть по
местности, в которой они были описаны. Многие точечные мутации не сопро-
вождаются клиническими симптомами, но некоторые вызывают гемоглоби-
нопатию. С клинической точки зрения аномальные НЬ могут обуславливать
ряд синдромов - гемолитическую анемию, врожденный цианоз, семейную
полицитемию и т. д.
Различают количественные и качественные гемоглобинопатии. При
количественных гемоглобинопатиях происходит нарушение соотношения
обычных цепей глобина. Качественные гемоглобинопатии - заболевания,
при которых генетическая аномалия приводит к синтезу НЬ с измененной
структурой глобина. Основой лабораторной диагностики качественных и
количественных гемоглобинопатий является электрофорез гемоглобина на
ацетате целлюлозы.
Талассемии
Талассемии - гетерогенная группа наследственно обусловленных забо-
леваний, в основе которых лежит нарушение синтеза одной из полипептид-
ных цепей глобина, что приводит к увеличению продукции других цепей и
развитию дисбаланса между ними. Талассемии относят к количественным
гемоглобинопатиям, так как структура цепей гемоглобина не изменена.
Различают а-талассемию, когда нарушается синтез а-непей, и р-талассемию
при блокаде синтеза p-цепей глобина. Описаны также случаи у-, б- и рб-
талассемии с нарушением синтеза соответствующих цепей глобина. Чаще
встречаются Р-талассемии. Цепи, синтезируемые в избыточном количестве,
накапливаются и откладываются в эритрокариоцитах костного мозга и эрит-
роцитах периферической крови, вызывая повреждение клеточной мембраны
и преждевременную гибель клеток. Эритрокариоциты гибнут в селезенке,
костном мозге. Анемия сопровождается небольшим повышением ретикуло-
цитов. Дисбаланс синтеза глобиновых цепей вызывает развитие неэффектив-
ного эритропоэза, внутриклеточный гемолиз эритроцитов периферической
крови, гиперспленизм и развитие гипохромной анемии различной степени
тяжести.
Р-талассемия. Р-талассемия - гетерогенное заболевание. В настоящее
время известно более 100 мутаций, вызывающих Р-талассемию. Обычно
дефект состоит в образовании неполноценной мРНК р-глобина. Разнообразие
молекулярных дефектов приводит к тому, что так называемая гомозиготная
Р-талассемия нередко представляет двойное гетерозиготное состояние по
разным дефектам синтеза Р-глобина. Различают Р°-талассемию, когда у го-
мозигот полностью отсутствует синтез p-цепей глобина, и р+-талассемию,
при частично сохраненном синтезе p-цепей. Среди р+-талассемий выделяют
две основные формы: тяжелую средиземноморскую форму, при которой син-
тезируется около 10% нормальной цепи (большая талассемия, анемия Кули),
и более легкую негритянскую форму, когда сохраняется около 50% синтеза
нормальной p-цепи. В группу р-талассемий относят также 8р-талассемию и
Hb Lepore. В результате этого имеются существенные расхождения в клини-
ческой картине различных форм талассемии, однако для всех Р-талассемий
общим является внутриклеточный гемолиз эритроцитов, неэффективный
эритропоэз в костном мозге и спленомегалия.
Большая талассемия (анемия Кули, thalassemia major). Считается гомо-
зиготной формой талассемии, хотя во многих случаях заболевание является
двойным гетерозиготным состоянием по различным формам р-талассемии.
Клинически заболевание проявляется к концу 1-2-го года жизни ребенка
спленомегалией, желтухой, бледностью кожных покровов, изменением
костей - квадратный череп, уплощенная переносица, выступающие скулы,
сужение глазных щелей. Дети физически плохо развиты.
В костном мозге наблюдается гиперплазия красного ростка, выявляется
значительное количество сидеробластов. В крови - гипохромная микроци-
тарная анемия (снижены MCV, МСН, МСНС), резкий анизоцитоз, встречают-
ся эритроциты с базофильной пунктацией, эритрокариоциты, пойкилоцитоз,
мишеневидные эритроциты, шизоциты (рис. 88). Даже при тяжелой анемии
количество ретикулоцитов не бывает высоким, так как в костном мозге вы-
ражен неэффективный эритропоэз. Отмечается повышение осмотической
резистентности эритроцитов. Характерна лейкопения с относительным лим-
фоцитозом, в период гемолитического криза - нейтрофильный лейкоцитоз
со сдвигом влево. В период криза может наблюдаться левый сдвиг.
В сыворотке крови имеет место гипербилирубинемия за счет неконъ-
югированного билирубина, повышено содержание сывороточного железа.
Избыточное отложение железа приводит к сидерозу органов. Характерным
Рис. 88. р-талассемия. Периферическая кровь — выраженный анизоцитоз, мишеневидные эрит-
роциты, гипохромия, пойкилоцитоз, анизоцитоз, нормобласты. хЮОО
признаком большой талассемии является выраженное увеличение концент-
рации фетального гемоглобина. Количество НЬА варьируег в зависимости от
типа талассемии. У гомозигот с р°-талассемией НЬА практически отсутствует.
При Р'-талассемии (средиземноморский тип) НЬА варьирует от 10 до 25%,
при Р+-талассемии негритянского типа содержание НЬА значительно выше.
Однако тяжесть заболевания не всегда коррелирует с количеством фетального
гемоглобина. Содержание НЬА2 может быть различным, чаще повышенным,
но отношение НЬА2/ НЬА всегда меньше, чем 1:40. Диагноз подтверждается
электрофорезом гемоглобина (уровень HbF - до 70%).
Малая талассемия (thalassemia minor) является гетерозиготной фор-
мой р-талассемии. Клинически малая талассемия характеризуется менее
выраженными симптомами, чем большая талассемия, может протекать
практически бессимптомно.
В костном мозге - гиперплазия эритроидного ростка, количество си-
деробластов повышено или нормальное. В крови наблюдается умеренная
гипохромная микроцитарная анемия: умеренное снижение гемоглобина при
нормальном, а иногда и повышенном количестве эритроцитов, снижение
индексов MCV, МСН, МСНС (рис. 89). В мазках крови отмечается анизоци-
кровь. хЮОО
RBC 2,33 3,8-5,5 х 1012/л
НЬ 65 130-155 г/л
Ht 21,9 36-48%
MCV 94,0 80-95 фл
МСН 26,9 27-31 пг
МСНС 297 300-380 г/л
RDW 38,1 11,5-14,5%
Рис. 89. Талассемия. Периферическая
тоз, пойкилоцитоз, мишеневидность эритроцитов, может быть базофильная
пунктация эритроцитов, выявляется ретикулоцитоз.
В сыворотке крови имеет место умеренная неконъюгированная билиру-
бинемия, содержание железа обычно нормальное или повышенное.
Диагноз устанавливается на основании результатов определения малых
фракций гемоглобина НЬА2 и HbF. Для больных гетерозиготной формой
Р-талассемии характерно повышение содержания фракции НЬА2 до 3,5-8%
и примерно у половины больных - HbF до 2,5-7%.
бр-талассемия (HbF-талассемия) представляет собой заболевание,
обусловленное нарушением синтеза как Р-, так и 5-цепей глобина. Гетерози-
готное состояние характеризуется гипохромной анемией. Содержание НЬА2
нормальное или даже пониженное, содержание HbF значительно повыше-
но и может достигать 5-20%. Клинико-гематологическая картина близка
к Р-талассемии. Гомозиготная форма бр-талассемии протекает легче, чем
гомозиготная форма Р-талассемии. У гомозигот по 5Р-талассемии в крови
отсутствуют НЬА и НЬА2, определяется только HbF.
а-талассемия. а-талассемия возникает при делеции генов, кодирующих
синтез данной цепи. Продукция a-цепей регулируется двумя парами неоди-
наковых по значению генов, располагающихся в 11 -й паре хромосом. При
дефиците a-цепей в крови новорожденных накапливаются тетрамеры НЬ
Bart’s (у4), а в постнатальном периоде (в том числе у взрослых) - НЬ Н (Р4).
Различают 4 основные формы а-талассемии.
Гомозиготная а-талассемия развивается вследствие полной блокады
синтеза a-цепей и характеризуется отсутствием нормальных гемоглобинов
(70-100% составляет Hb Bart’s). Hb Bart’s не способен переносить кислород
из-за аномально повышенного сродства к нему. Вследствие чего наступает
аноксия тканей, приводящая к развитию водянки и внутриутробной гибели
плода.
Н-гемоглобинопатия. Обусловлена значительным угнетением продукции
a-цепей вследствие отсутствия 3 из 4 генов. Избыточный синтез р-цепей
приводит к их накоплению и образованию тетрамеров р4 (НЬН). У новорож-
денных 20—40% приходится на долю Hb Bart’s (у4), который позднее меняется
на НЬН. Гемоглобин Н в функциональном отношении неполноценен, так
как обладает очень высоким сродством к кислороду. НЬН не связывается
с гаптоглобином, является нестабильным, нестойким, легко подвергается
окислению и осаждается в клетке по мере ее старения. При этом заболевании
наблюдается повышенное образование MetHb. Агрегация НЬН вызывает
изменение пластичности мембраны эритроцитов, нарушает метаболизм
клеток, что сопровождается гемолизом.
Клинически гемоглобинопатия-Н протекает в форме промежуточной
талассемии. Заболевание выявляется обычно к концу 1-го года жизни
хронической гемолитической анемией умеренной степени тяжести, из-
редка наблюдается бессимптомное течение. Заболевание характеризуется
сравнительно нетяжелым клиническим течением, гепатоспленомегалией,
желтушностью, анемией. Изменения скелета незначительные. В костном
мозге - умеренная гиперплазия эритроидного ростка, незначительный
неэффективный эритропоэз. В крови - выраженная гипохромия и мишене-
видность эритроцитов, небольшой ретикулоцитоз. После инкубации крови
с крезиловым синим при 55 °C выпадает нестабильный гемоглобин Н в
виде множества мелких фиолетово-синих включений в эритроцитах, что
отличает ее от других форм а-талассемий. После спленэктомии включения
НЬН по внешнему виду начинают напоминать тельца Гейнца. Однако по
химической структуре они отличаются от телец Гейнца тем, что состоят из
преципитированных p-цепей ф4), в то время как тельца Гейнца представляют
собой осажденные молекулы НЬА (сь,р2) и некоторые другие нестабильные
гемоглобины. При электрофорезе сыворотки крови в щелочном буфере
наблюдается дополнительная фракция, движущаяся впереди НЬА (быстро-
двигающаяся фракция). У взрослых людей значения НЬН составляют 5-30%,
до 18% может приходиться на долю НЬ Bart’s, НЬА2 снижен (1-2%), HbF в
норме или слегка повышен (0,3-3%).
Малая а-талассемия (a-thj. Гетерозиготное состояние по гену a-th,.
Синтез a-цепей снижен в умеренной степени. В периферической крови
обнаруживаются легкая степень анемии с характерными для талассемии
морфологическими изменениями эритроцитов. У новорожденных, носителей
этого гена, в пуповинной крови содержание НЬ Ban’s не превышает 5-6%.
Продолжительность жизни эритроцитов на нижней границе нормы.
Бессимптомная форма a-талассемии (a-th2). Гетерозиготное состоя-
ние по гену a-th2 характеризуется незначительным снижением продукции
a-цепей, не приводящим к развитию анемии и морфологическим измене-
ниям эритроцитов. У новорожденных, носителей этого гена, НЬ Bart’s не
превышает 1-2%.
Гемолитические анемии, обусловленные носительством
аномального гемоглобина
Серповидноклеточная анемия (гемоглобин S)
Серповидноклеточная анемия (гемоглобинопатия S) - качественная
гемоглобинопатия. Это наиболее распространенная гемоглобинопатия.
Аномалия структуры гемоглобина при серповидноклеточной анемии за-
ключается в замене в положении 6 P-цепи глутаминовой кислоты на валин,
что приводит к усилению связи одной молекулы гемоглобина с другой.
Гемоглобинопатия S чаще развивается у лиц, проживающих в странах, где
распространена малярия (Средиземноморье, Африка, Индия, Средняя Азия).
Замена одной аминокислоты на другую сопровождается тяжелыми физико-
химическими изменениями гемоглобина и ведет к деполимеризации HbS.
Дезоксигенация вызывает отложение молекул аномального гемоглобина в
виде мононитей, которые агрегируют, превращаясь в кристаллы продолгова-
той формы, изменяя тем самым мембрану и форму эритроцитов в виде сер-
пов (рис. 90). Средняя продолжительность жизни эритроцитов при анемии,
гомозиготной по гемоглобину S, составляет около 17 дней. В то же время
такая аномалия делает эти эритроциты неприемлемыми для жизнедеятель-
Рис. 90. Серповидноклеточная анемия. Периферическая
кровь. хЮОО
ности плазмодий: носители
гемоглобина S не болеют
малярией, что путем естес-
твенного отбора привело
к распространению этой
гемоглобинопатии в странах
«малярийного пояса».
Гомозиготная форма кли-
нически проявляется спустя
несколько месяцев после
рождения. Характерна рез-
кая болезненность суставов,
припухлость кистей рук,
стоп, голеней, связанная с
тромбозом сосудов, костные
изменения (высокий рост, искривленный позвоночник, башенный череп,
измененные зубы). Нередки асептические некрозы головок бедренной и
плечевой костей, инфаркт легких, окклюзии церебральных сосудов. У де-
тей развивается гепатомегалия, спленомегалия. Болезнь характеризуется
гемолитическими кризами с внутрисосудистым гемолизом, поэтому час-
тым осложнением в клинике бывают тромбозы мелких и крупных сосудов
различных органов.
В крови невыраженная нормохромная анемия. При гемолитическом кризе
имеет место резкое падение гемоглобина и гематокрита, ретикулоцитоз, нор-
мобластоз, тельца Жолли, серповидные эритроциты (рис. 91), базофильная
пунктация, мишеневидные эритроциты, пойкилоцитоз, лейкоцитоз, тром-
боцитоз, ускорение СОЭ, повышение неконъюгированного билирубина.
Моча черного цвета: свободный гемоглобин, гемосидерин. Присоединение
инфекций может сопровождаться апластическим кризом - эритроцитопенией,
ретикулоцитопенией, тромбо- и лейкоцитопенией.
Серповидность может быть выявлена в пробе с метабисульфитом натрия
или при наложении жгута на основание пальца (снижение доступа кислоро-
RBC
RBC 2,47 3,8-5,5 х 1012/л
НЬ 84 130-155 г/л
Ht 24,4 36-48%
MCV 96,0 80-95 фл
МСН 34,2 27-31 пг
МСНС 346 300-380 г/л
RDW 13,5 11,5-14,5%
Рис. 91. Серповидноклеточная анемия
Периферическая кровь. хЮОО
да). Окончательный диагноз устанавливается после электрофореза исследу-
емой крови, где наблюдается 90% HbS, 2-10% HbF, НЬА отсутствует.
Гетерозиготная форма (носительство признака серповидноклеточной
анемии) характеризуется доброкачественным течением заболевания.
У некоторых больных единственным симптомом может быть спонтанная
гематурия, связанная с мелкими инфарктами сосудов почек.
Тяжелая гипоксия развивается на больших высотах. В этих случаях могут
быть тромботические осложнения. Во время криза в крови отмечается низкий
гемоглобин, серповидные эритроциты, эритрокариоциты.
Тест на серповидноклеточностъ. Окисленные НЬА и HbS характеризу-
ются одинаковой растворимостью, восстановленный НЬА имеет в 2 раза
худшую растворимость, восстановленный HbS в 50 раз хуже растворим, чем
обычный НЬ. Поэтому при пробе с 2% раствором метабисульфата натрия,
переводящим НЬ в восстановленную форму, или при перетяжке пальца на
5 мин для создания венозного застоя с гипоксией HbS выпадает в виде геля,
в котором образуются кристаллы удлиненной формы, которые сходны с
серповидными эритроцитами.
Электрофорез. У гетерозигот HbS составляет 20-25%, при электрофорезе
он имеет меньшую подвижность, чем НЬА.
Гемолитические анемии, обусловленные носительством аномальных
гемоглобинов С, D, Е
Распространенными формами стабильных гемоглобинов являются С,
D, Е.
В НЬС глутаминовая кислота в положении 6 заменена лизином, что
ведет к его кристаллизации. В НЬЕ лютаминовая кислота в положении 26
заменена лизином, в HbD глутаминовая кислота в положении 121 заменена
на глутамин. Гетерозиготные формы протекают без клиники.
У гомозигот симптоматика обусловлена анемией: характерны легкая
Рис. 92. Гемоглобинопатия С. Периферическая
кровь. Мишеневидные эритроциты. хЮОО
гемолитическая анемия, желтуха,
спленомегалия. Анемия носит нор-
моцитарный характер, в крови от
30 до 100 мишеневидных клеток
(рис. 92). Характерна склонность
к кристаллизации молекул гемо-
глобина. Сочетание всех 3 видов ге-
моглобинопатий с талассемией дает
тяжелую клиническую картину.
НЬС распространен среди амери-
канских негров и негров ЮАР, НЬЕ
чаще всего встречается в Юго-Вос-
точной Азии и среди негров. Специ-
фического лечения не существует,
анемия обычно не достигает степени,
требующей гемотрансфузии.
Гемолитические анемии, обусловленные носительством аномальных
нестабильных гемоглобинов
Замена аминокислот в НЬА в а- или p-цепях вызывает появление ано-
мального нестабильного гемоглобина. Замещение в зоне прикрепления гема
вызывает молекулярную нестабильность, ведущую к денатурации и преци-
питации гемоглобина внутри эритроцита. Преципитированный гемоглобин
прикрепляется к мембране эритроцита, что приводит к разрушению эритро-
цита, появлению телец Гейнца, нарушается эластичность и проницаемость
клеточной мембраны. При прохождении через селезенку эритроциты теряют
часть мембраны, а затем разрушаются.
Клиника. Гемолитическая анемия наблюдается с детства. Кризы могут
быть вызваны лекарствами или инфекцией. В крови отмечается низкий гемо-
глобин, мишеневидные эритроциты, базофильная пунктация, полихромазия,
ретикулоцитоз, тельца Гейнца, повышено содержание эритрокариоцитов.
Осмотическая резистентность эритроцитов нормальная или немного уве-
личена. Исследование первичной структуры патологического гемоглобина
позволяет установить тип нестабильного гемоглобина.
Аномальный гемоглобин составляет ЗСМ0% от общего количества ге-
моглобина.
ОСНОВНАЯ ЛИТЕРАТУРА
1. Анастасевич Л.А., Малкоч А.В. Же-
лезодефицитные анемии у детей ран-
него возраста // Анемия. 2006. № 1-2.
С. 53-58.
2. Гематология / Под ред. О.А. Рукавицы-
на. СПб., 2007. С. 29-146.
3. Демихов В.Г, Морщакова Е.Ф., Деми-
хова Е.В. с соавт. Распространенность
дефицита железа у беременных жен-
щин // Вопросы гематологии/онкологии
и иммунопатологии в педиатрии. 2002.
Т. 1,№2. С. 21-23.
4. Демихов В. Г. Профилактика дефицита
железа у беременных женщин // Воп-
росы гематологии/онкологии и иммуно-
патологии в педиатрии. 2003. Т. 2, № 2,
С. 25-31.
5. Долгов В.В., Луговская С.А., Поч-
тарь М.Е. Лабораторная диагностика
нарушений обмена железа. СПб.: Витал
Диагностике, 2002.
6. Долгов В. В., Луговская С. А., Морозова В. Т,
Почтарь М.Е. Лабораторная диагности-
ка анемий. Тверь: Триада, 2001. 84 с.
7. Исследование системы крови в клини-
ческой практике / Под ред. Г. И. Козинца,
В.А. Макарова. М.: Триада-Х, 1997.
480 с.
8. Кассирский И.А., Алексеев Г.А. Кли-
ническая гематология. М.: Медицина,
1970. 800 с.
9. Козинец Г.И. Физиологические системы
организма человека, основные показате-
ли. М.: Триада-Х, 2000.
10. Козинец Г.И., Погорелов В.М., Шма-
ровД.А., Боев С.Ф., Сазонов В.В. Клетки
крови. Современные технологии их
анализа. М.: Триада-Фарм, 2002. 535 с.
11. Козинец Г.И., Хакимова Я.Х., Быко-
ва И.А. и др. Цитологические особен-
ности эритрона при анемиях. Т.: Меди-
цина, 1988.
12. Козинец Г.И., Погорелов В.М., Дягиле-
ва О.А., Наумова И.Н. Кровь: клини-
ческий анализ, диагностика анемий и
лейкозов. Интерпретация результатов.
М.: Медицина XXI, 2006. 251 с.
13. Козинец Г.И., Сарычева Т.Г., Луговс-
кая С. А., Дягилева О. А., Погорелов В. М„
Проценко Д.Д. Гематологический атлас:
настольная книга врача-лаборанта. М.:
Практическая медицина, 2008. 187 с.
14. Луговская С.А., Морозова В.Т., Поч-
тарь М.Е., Долгов В.В. Лабораторная
гематология. М.: Триада, 2006. 222 с.
15. Луговская С.А., Почтарь М.Е. Ретику-
лоциты. М., 2006. 58 с.
16. Луговская С.А., Почтарь М.Е., Дол-
гов В. В. Гематологические анализаторы.
Интерпретация анализа крови. М.: Три-
ада, 2007. 109 с.
17. Маршалл В.Дж. Клиническая биохимия.
М.; СПб., 1999.
18. Мосягина Е.И., Владимирская Е.Б.,
ТорубароваН.А., Мызина Н.В. Кине-
тика форменных элементов крови. М.:
Медицина, 1976.
19. Петухов В.И., Быкова Е.Я., Бондаре Д. К.
с соавт. Сывороточный ферритин в
диагностике железодефицитных состо-
яний И Гематология и трансфузиология.
2003. Т. 48, № 2. С. 41-44.
20. Руководство по гематологии И Под ред.
А.И. Воробьева. М.: Ньюдиамед, 2002.
21. Румянцев А.Г., Морщакова Е.Ф., Пав-
лов А.Д. Эритропоэтин: биологические
свойства, возрастная регуляция эритро-
поэза, клиническое применение. М.:
ГЭОТАР-МЕД, 2002. 168 с.
22. Рябов С.И.. ШосткаГ.Д. Молекулярно-
генетические аспекты эритропоэза. М.:
Медицина, 1973.
23. Наследственные анемии и гемогло-
бинопатии / Под ред. Ю.Н. Токарева,
С.Р. Холлан, Ф. Корраля-Альмонте. М.:
Медицина, 1983.
24. Погорелов В.М., Козинец Г. И., Кова-
лева Л. Г. Лабораторно-клиническая
диагностика анемий. М.: Медицинское
информационное агентство, 2004.
25. Троицкая О.В., Юшкова Н.М., Волко-
ва Н.В. Гемоглобинопатии. М.: Изд-во
Российского университета дружбы
народов, 1996.
26. Хоффбранд В., Петит Дж. Атлас-
справочник. Гематология. М.: Практика,
2007. 406 с.
27. Хубутия М.Ш., Шевченко О.П. Гомоцисте-
ин при коронарной болезни сердца и сер-
дечного трансплантата. М., 2004. 270 с.
28. Шиффман Ф.Дж. Патофизиология
крови. М.; СПб., 2000.
29. Andrews N.C. Forging a field: the golden
age of iron biology H Blood. 2008. V. 112,
№2. P. 219-230.
30. Atanasiu И, Manolescu B., Stoian I.
Hepcidin - central regulator of iron me-
tabolism// Eur. J. Haematol. 2007. V. 78,
№ l.P. 1-11.
31. Baynes J., Dominiczak M.H. Medical
Biochemistry. L.: Mosby, 1999.
32. Hugman A. Hepcidin: an important
new regulator of iron homeostasis //
Clin. Lab. Haem. 2006. V. 28. P. 75-83.
33. Ganz T. Hepcidin and its Role in Regulating
Systemic Iron Metabolism // Haematology.
2006. V. 4. P. 29- 35.
34. Kushner J. P Anemia of chronic disease //
Hematology. 2006. V. 2, № 1. P. 47^49.
35. Nemeth E. Hepcidin: the principal regulator
of systemic iron metabolism // Hemato-
logy. 2006. V. 2, № 1. P. 36-41.
36. Xiuli An., Mohandas N. Disorders of red
cell membrane // British J. Haematol. 2008.
V. 141. P. 367-373.
37. Weiss G., Goodnough L.T. Anemia of
Chronic Disease // N. Engl. J. Med. 2005.
V. 352. P. 1011-1023.
В.В. Долгов, С.А. Луговская, В.Т. Морозова, М.Е. Почтарь
Лабораторная диагностика
анемий
Второе издание, дополненное
ООО «Издательство «Триада». ИД № 06059 от 16.10.01 г.
170034, г. Тверь, пр. Чайковского, 9, оф. 504, тел./факс: (4822) 42-90-22, 35-41-30
E-mail: triada@stels.tver.ru, http://www.triada.tver.ru
Подписано к печати 23.12.08. Формат 62x94 1/8, обрезной.
Бумага мелованная. Гарнитура Times New Roman.
Печать офсетная. Усл. печ. л. 18,5. Тираж 2000 экз.
Отпечатано в ООО «Тверская фабрика печати».
170006, г. Тверь, Беляковский пер., 46. Тел. (4822) 35-32-13
Заказ № 4527
АВТОМАТИЧЕСКИЕ ГЕМАТОЛОГИЧЕСКИЕ
АНАЛИЗАТОРЫ
HORIBA АВХ
MIX-Rate 10-100
А от 10 до 400 тестов в час
х встроенный миксер
а автоматический контроль качества
А минимальный объем пробы
А от 8 до 45 параметров
х полная дифференциация лейкоцитов и ретикулоцитов
х от 60 до 120 проб в час
А минимальный объем пробы
А автоматическая система подготовки и окрашивания мазков
АВТОМАТИЧЕСКИЕ АНАЛИЗАТОРЫ СОЭ
VITAL DIAGNOSTICS
КОНТРОЛЬНЫЕ МАТЕРИАЛЫ
STRECK LABORATORIES
А для всех типов гематологических анализаторов и анализаторов СОЭ
3DIFF-Nihon Kohden МЕК-6400; Horiba АВХ Micros 60; Sysmex® КХ-21, pocH-100i
Coulter* А'-Т/А'- Г diffAc-T 2diff и др.
5 DIFF - Pentra 60С/ 80/120; Abbott CELL-DYN® 3500/3700/4000, Coulter'” Ac-T 5diff.
ADVIA120/2120 и др.
А максимальный срок годности:
• для гематологических анализаторов - до 6 месяцев
• для анализаторов СОЭ - до 12 месяцев
А максимальная стабильность открытых флаконов
МИКСЕРЫ
VITAL DIAGNOSTICS
Duo-mix
А независимые сменные штативы
А смена штатива нажатием одной кнопки
А штативы для пробирок с разным диаметром
А широкий выбор режимов перемешивания
Капиллярные - КАВЕ GK
Вакуумные - KABEVETTE V
Вакуумно-шприцевые - PRIMAVETTE S
PRIMAVETTE V
KABEVETTE G
ВЗЯТИЯ КРОВИ
КАВЕ LABORTECHNIK
Лабикс
123007, Россия, Москва, ул. Розанова, 10,
тел. в Москве /495/234 8877, факс /495/ 234-8811,
Автоматический гематологический анализатор
ADVIA 2120i с модулем окраски мазков
Совершенная платформа
Гибкая интеграция классических методик и современных технологий
для диагностики анемий
Высокопроизводительный гематологический анализатор ADVIA 21201 предоставляет самые
'современные гематологические показатели для диагностики анемий: расширенный анализ эритроцитов
I и ретикулоцитов, уникальные ретикулоцитарные индексы и гематологические флаги.
Более 45 измеряемых и вычисляемых параметров, в том числе широкий перечень показателей для
скрини нга и мониторинга лечения анемий различной этиологии:
Эритроцитарные параметры
РВС — количество эритроцитов
HGB - концентрация гемоглобина
НСТ - гематокриз
MCV — средний объем эритроцита
МСН — среднее содержание гемоглобина
₽ эритроцитах
V1CHC — средняя концентрация гемоглобина
в эритроците (расчетный)
ZHCM - средняя концентрация гемоглобина
'з эритроците (измеряемый)
1DW — ширина распределения эритроцитов
[ю объему (показатель гетерогенности
|ритроцитов)
IDW - ширина распределения количества
емоглобина в эритроците (из гистограммы)
Ретикулоцитарные параметры
[o#RETIC — абсолютное и относительное количество ретикулоцитов
[ICVr - средний объем ретикулоцитов
'НСМг — средняя концентрация гемоглобина в ретикулоците (расчетный)
"Hr — среднее содержание гемоглобина в ретикулоцитах
kF — индекс зрелости ретикулоцитов
Модуль окраски мазков позволяет подготовить гематологические препараты высочайшего качества
ня проведения микроскопии и уточнения лабораторного диагноза. Аппарат выполнит окраску лю-
^>1м из выбранных методов: Райта-Гимзы, Райта модифицированный, Мая-Грюнвальда-Гимзы.
коростьокраски — 120стекол/час
Производительность в режиме СВС, CBC/Diff — 120; CBC/Diff/Retic, CBC/Retic— 74 тестов/час
%#NRBC — абсолютное и относительное
количество нормобластов
СН — среднее распределение количества
гемоглобина в эритроците (из гистограммы )
CHDW — ширина распределения концентрации
гемоглобина в эритроците (из гистограммы)
%HYPO — % гипохромных эритроцитов
%HYPER — % гиперхромных эритроцитов
%#MICRO — абсолютное и относительное
количество микроцитов
%#MACRO — абсолютное и относительное
количество макроцитов
ANISO - анизоцитоз
НС VAR — вариации гемоглобина
в эритроцитах
ООО «АСТРА-77» — официальный дистрибьютор компании
SIEMENS HEALTHCARE DIAGNOSTICS
Телефон (495) 925-77-59 Адрес в Интернет: http://www.astra77.ru
Факс 64951 926-71-11 —
Ill ДИНА ИНТЕРНЕШНЕЛ
Цитоцентрифуги фирмы Wescor- два в одног
Автоматы для окрашивания клеток по методам:
• Гимзы (Романовского, Мая-Грюнвальда),
• Грама,
• Циля-Нильсена и аурамин-родаминовому
плюс автоматическое приготовление клеточных
монослоев на стеклах — жидкостная цитология
(как дополнительная опция)
Фиксация,
окрашивание,
высушивание
за 4-5 мин!
ЭЛЛ „П1/1ЫД МЫТРОМСШ
► Простота
► Надежность
► Удобство
► Экономичность!
го
го
5
О
о
Sysmex <^воёье>
ГЕМАТОЛОГИЧЕСКИЕ АНАЛИЗАТОРЫ Х-КЛАССА
Гематология сегодня - это проточная
цитофлюориметрия
XT-1800i/2000i
производительность 80 проб/час
ХЕ-2100
производительность 150 проб/час
NEW
РУССКИЙ язык
XS-800i/1000i производительность 60 проб/час
РУССКИЙ ЯЗЫК
• новые приборы Х-класса для лабораторий
с небольшим потоком образцов
самый компактный 5-diff гематологический
анализатор на 26 параметров
лейкоцитарная формула в капиллярной
крови - требуется только 20 мкл образца
для анализа - идеален для педиатрии,
онкологии и поликлиник
Ф. Хоффманн - Ля Рош, ЗАО «Рош-Москва», г. Москва.
Тел. (495) 229-62-50. Факс (495) 229-62-95/96.
Ф. Хоффманн - Ля Рош, ЗАО «Рош-Москва», г. Санкт-Петербург
Тел. (812) 703-52-60. Факс (812) 703-52-61.
Ф. Хоффманн - Ля Рош, ЗАО «Рош-Москва», г. Новосибирск.
Тел. (383) 335-88-04. Факс (383) 335-88-03.
• использование уникальной технологии проточной
цитофлюориметрии дает надежный результат даже
в случае липемии, наличия устойчивых к лизису
эритроцитов и инфекции
Реклама
• надежная система, требующая
минимального обслуживания
• программное обеспечение
на русском языке
9 785947 893403